Drugs, Health Technologies, Health Systems
Health Technology Review
Drugs for Adults With Spinal Muscular Atrophy: A Focused Review and Critical Appraisal of Recent Evidence (2021 to 2025) on Nusinersen and Risdiplam
Key Messages
What Is the Issue?
Spinal muscular atrophy (SMA) is a rare genetic condition that affects muscle strength and movement in both children and adults. Two treatments, nusinersen (Spinraza) and risdiplam (Evrysdi), are publicly reimbursed in Canada, primarily for use in children. The Canadian Drug Expert Committee (CDEC) reimbursement recommendations for both drugs highlighted serious limitations in the evidence for the effectiveness and safety of these treatments in adults with SMA. In particular, the evidence is limited by the lack of high-quality, comparative data, making it difficult to determine whether observed effects are due to the drugs or to other factors, and whether these effects are clinically meaningful in adults. As a result, reimbursement recommendations issued by CDEC do not support treatment initiation of nusinersen in patients older than aged 18 years or initiation of risdiplam in patients aged older than 25 years.
What Did We Do?
We searched for recently published, peer-reviewed studies using clinical trial and comparative observational study designs to answer these questions:
What is the quality of comparative evidence on the effectiveness and safety of nusinersen and risdiplam in adults with SMA?
What is the quality of comparative evidence on the effectiveness and safety of nusinersen and risdiplam in adults with SMA who were previously treated with gene therapy (onasemnogene abeparvovec)?
Is it feasible and useful to conduct a new, comprehensive review to inform future reimbursement decisions for these treatments?
We searched key resources, including journal citation databases, and conducted a focused internet search for relevant clinical trials and comparative observational studies published since the initial search dates of the previous CADTH reimbursement reviews for nusinersen and risdiplam. We focused on studies that directly compared these treatments with each other, with other therapies, or with standard care in adults with SMA to address concerns raised about the previously reviewed evidence.
What Did We Find?
Only 1 new comparative study (Vázquez-Costa et al.) was found for nusinersen in adults with SMA. This study compared adults treated with nusinersen with those who received no treatment over a follow-up period of up to 30 months.
No eligible comparative studies were found for risdiplam in adults aged older than 25 years with SMA.
No published studies were identified for nusinersen or risdiplam in adults with SMA who were previously treated with gene therapy (onasemnogene abeparvovec).
The Vázquez-Costa et al. study had significant limitations, including a high risk of bias due to differences in patient characteristics between the treated and untreated groups (e.g., ability to sit or to walk at start of treatment), missing data, inconsistent findings within and across motor function outcomes, use of unvalidated thresholds to define clinically meaningful treatment effects, and incomplete outcome reporting. These limitations make it difficult to draw reliable conclusions about the benefits or harms of nusinersen in adults with SMA.
Many additional real-world studies were identified; however, they were noncomparative or did not focus specifically on adults with SMA, included small numbers of adults, and had similar methodological limitations to previously reviewed studies. None provided robust evidence to answer the research questions or to address the methodological limitations and uncertainties of the studies included in previous reviews.
What Does This Mean?
Comparative evidence is required to establish the relative benefits and harms of a therapy versus existing standards of care and to draw meaningful conclusions about their outcomes to inform policy decision-making. The current evidence base for nusinersen and risdiplam in adults with SMA remains very limited and of low quality, which makes it difficult to determine if the drugs achieved their outcomes. There is still no strong comparative evidence to support reimbursement of these treatments for adults with SMA, and due to serious limitations in the available real-world studies, it remains unclear whether either nusinersen or risdiplam have clinically meaningful benefit in adults.
Given the limitations of the available evidence — including the lack of robust comparative studies; methodological weaknesses in existing data; and the absence of consistent, clinically meaningful outcomes — conducting a de novo systematic review at this time is not feasible and is unlikely to yield clearer conclusions. As such, we do not anticipate that such a review would provide sufficient new information to support a reassessment of current reimbursement criteria for nusinersen and risdiplam in adults with SMA.
Abbreviations
5q SMA
chromosome 5q13.2 spinal muscular atrophy
6MWT
6-minute walk test
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale Revised
CDEC
Canadian Drug Expert Committee
CI
confidence interval
EK2
Egen Klassifikation Scale Version 2
HFSME
Hammersmith Functional Motor Scale – Expanded
MID
minimal important difference
OR
odds ratio
ppFVC
percent predicted forced vital capacity
RCT
randomized controlled trial
ROBINS-I
Cochrane Risk of Bias in Nonrandomized Studies of Interventions
RULM
Revised Upper Limb Measure
SD
standard deviation
SE
standard error
SMA
spinal muscular atrophy
SMN2
survival motor neuron 2 gene
Context and Policy Issues
What Is SMA?
SMA is a rare, progressive neuromuscular disorder characterized by degeneration of alpha motor neurons in the anterior horn of the spinal cord, leading to progressive muscle weakness and atrophy.1,2 Most cases of SMA (approximately 95%) are caused by homozygous deletion and/or mutation of the SMN1 gene,3,4 located on chromosome 5q13.2 (5q SMA). This genetic defect results in reduced production of the SMN protein, which is essential for the maintenance of motor neurons. A related gene, SMN2, can produce small amounts of functional SMN protein; however, the extent of this expression is variable and correlates with disease severity.1-3
SMA is often classified into clinical types (types 0 to IV) based on age at symptom onset, the highest motor function achieved, and prognosis (Table 1). These types correspond broadly to SMN2 copy number; however, SMA severity is better understood on a continuum, with considerable symptom overlap and variability in disease severity across individuals.
Table 1: SMA Clinical Characteristics
SMA type | Typical SMN2 copy number | Age of onset | Highest motor function achieved | Common features |
|---|---|---|---|---|
Type 0 | 1 | Before birth | None |
|
Type I | 1 to 2 | < 6 months | None |
|
Type II | 2 to 3 | 6 to 18 months | Sitting |
|
Type III | 3 to 4 | > 18 months to adolescence | Walking independently (may lose ability) |
|
Type IV | ≥ 4 | Adulthood | Walking independently |
|
SMA = spinal muscular atrophy
Sources: Wang et al.,5 Russman,6 Talbot and Tizzano,7 Mercuri et al.8
What Are the Medications for the Treatment of SMA?
Nusinersen (Spinraza) and risdiplam (Evrysdi) are disease-modifying therapies approved by Health Canada for the treatment of 5q SMA. Nusinersen is an intrathecally administered antisense oligonucleotide that promotes the inclusion of exon 7 in SMN2 mRNA transcripts, thereby increasing the production of functional SMN protein.9 Risdiplam is an orally administered small-molecule SMN2 splicing modifier that similarly enhances the production of full-length SMN protein by modulating pre–messenger ribonucleic acid splicing.10
Onasemnogene abeparvovec (Zolgensma) is also available for the treatment of SMA. It is an intravenously administered gene replacement therapy that delivers a functional copy of the SMN1 gene using an adeno-associated virus serotype 9 vector, enabling sustained production of SMN protein directly from the introduced gene.
Why Is It Important to Do This Review?
Reimbursement recommendations for nusinersen and risdiplam in Canada currently include age-based conditions, reflecting significant uncertainty about their benefits in adults with SMA. Evidence appraisals and reimbursement recommendations by CDEC (nusinersen 2019 and 2022 and risdiplam 2021)11-13 and other14 CADTH assessments highlighted several key limitations in the available evidence:
Lack of high-quality comparative studies: Comparative studies are essential for determining whether observed treatment effects can be attributed to the treatment itself, rather than to other influences, like the natural course of the disease or patient-specific factors. Most evidence for nusinersen in adults comes from noncomparative interventional or observational studies, with no randomized controlled trials (RCTs) directly comparing nusinersen to other therapies or to placebo. The evidence for the effects of risdiplam in adults with SMA is based on subgroup data from a single RCT.
Limited adult representation: The pivotal RCT for risdiplam included relatively few adults — approximately 12% of the included patients were aged 18 to 25 years — and none of the RCTs for nusinersen included adults. Although the noncomparative studies evaluating nusinersen included a wider age range, they included few or no patients with later-onset or more slowly progressive forms of SMA, limiting the generalizability of the findings to the broader population of adults with SMA.
Methodological concerns: Studies in adults for nusinersen were characterized by limited or lack of age and functional status stratification; high loss to follow-up and missing data; and unaddressed confounding factors, such as natural disease progression and potential training effects in outcome assessments. The adult subgroup in the RCT for risdiplam was relatively small and the trial was not designed or statistically powered for evaluating treatment effects in subgroups, including no statistical adjustment for imbalances between study groups, which limit the certainty of conclusions.
Uncertain clinical meaningfulness: Reported treatment effects in adults have been modest and evaluated over relatively short time frames (typically shorter than 15 months). Their clinical significance is unclear, especially in the absence of validated thresholds for meaningful change or direct comparative data.
As a result, most jurisdictional drug programs do not reimburse initiation of these therapies in adults aged older than 18 years for nusinersen and older than 25 years for risdiplam, although some funding is granted on a case-by-case basis.
Since the CDEC recommendations, a substantial volume of new, primarily real-world studies has emerged, reporting potential benefits of nusinersen and risdiplam in adults with SMA. However, it remains unclear whether this newer evidence adequately addresses prior concerns about study design, confounding, and lack of comparators. As comparative efficacy or effectiveness and safety data are central to health technology assessment decision-making, a focused review is needed to assess whether this emerging body of evidence can support more confident conclusions about the clinical value of these therapies in adults with SMA and potentially inform future reimbursement criteria.
The objectives of this evidence review are:
to critically appraise recent literature evaluating the comparative beneficial and harmful effects of nusinersen and risdiplam for the treatment of SMA in patients aged older than 18 or 25 years, respectively
to assess the feasibility and potential utility of conducting a review to support the reassessment of jurisdictional reimbursement criteria.
Research Questions
What is the quality of the comparative evidence evaluating the efficacy, effectiveness, and safety of nusinersen and risdiplam in adults with SMA?
For nusinersen in patients aged older than 18 years?
For risdiplam in patients aged older than 25 years?
Within these adult populations, is there comparative evidence that includes patients previously treated with onasemnogene abeparvovec?
What is the feasibility and potential utility of conducting a de novo systematic review to support the reassessment of jurisdictional reimbursement criteria for nusinersen and risdiplam in these adult SMA populations?
Methods
Literature Search Methods
The literature search strategy used in this report is based on searches developed for the previous CADTH reimbursement reviews for nusinersen (Spinraza) and risdiplam (Evrysdi). For the current report, an information specialist conducted a literature search on key resources, including MEDLINE, Embase, the Cochrane Database of Systematic Reviews, the International HTA Database, and the websites of Canadian and major international health technology agencies, as well as a focused internet search. The search approach was customized to retrieve a limited set of results, balancing comprehensiveness with relevancy. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s Medical Subject Headings (MeSH), and keywords. Search concepts were developed based on the elements of the research questions and selection criteria. The main search concepts were nusinersen or risdiplam. The following clinical trials registry was searched: the US National Institutes of Health’s clinicaltrials.gov. The search was completed on March 5, 2025, and limited to any articles published or made available since the initial search dates of the previous reimbursement reviews; December 17, 2020, for risdiplam (Evrysdi) and December 10, 2021, for nusinersen (Spinraza).
No language limits were applied. The search strategy is available on request.
Selection Criteria and Methods
One reviewer screened citations and selected studies. In the first level of screening, titles and abstracts were reviewed and potentially relevant articles were retrieved and assessed for inclusion. As an update to previous CADTH reports conducted for the reimbursement reviews of risdiplam15 and nusinersen,16 articles were included if they were made available since the previous search date and were not included in the 2021 and 2022 CADTH reports, respectively. The final selection of full-text articles was based on the inclusion criteria presented in Table 2.
In addition to the electronic database and grey literature searches, a manual search of reference lists from identified systematic reviews was conducted. The jurisdictional drug programs also provided a list of studies (submitted to them by clinicians who treat SMA) for appraisal alongside the body of evidence identified through the literature search (Appendix 2, Table 8). All of these studies — except for conference abstracts, which were not included in the search strategy — were captured in the literature search and assessed for eligibility.
Criteria | Description |
|---|---|
Population | Adults with SMA who are aged older than 18 years (nusinersen) or older than 25 years (risdiplam) Subgroups:
|
Intervention |
|
Comparator |
|
Outcomes | Efficacy or effectiveness:
Harms:
|
Study designs | Comparative studies: randomized controlled trials, controlled clinical trials, comparative observational studies |
SMA = spinal muscular atrophy.
aStandard of care includes medications or other therapies (e.g., physiotherapy) for the supportive management of SMA via a multidisciplinary approach, including neurology, pulmonology, orthopedics, nutrition, and rehabilitation.
Why Are Comparative Studies Important?
Comparative studies are important because they provide a benchmark that helps distinguish whether observed treatment effects are due to the intervention itself or to other factors, such as natural disease progression or patient characteristics.17-21 In the context of adults with SMA, this is especially important because the natural history in this population is poorly characterized and variable, shaped by many factors, including differences in numbers in SMN2 copies, motor milestones achieved and other aspects of functional status (including respiratory and bulbar), age at symptom onset, disease duration, and prior or concurrent treatments.22-24 While some natural history studies have suggested more consistent natural history with subtypes II and sometimes III,24,25 other studies have shown that SMA type may not be predictive of outcomes.7,22,26 In adults, prolonged disease duration amplifies this variability, as accumulated secondary complications (e.g., contractures, scoliosis) reduce the predictive ability of SMA type alone.26 In addition, SMA type is a clinical characterization of several patient and disease factors without standardized and objective definitions or criteria and thus is more susceptible to subjectivity.7,24,27
This variability in natural history makes it difficult to determine what outcomes might be expected in the absence of treatment, undermining efforts to judge whether reported changes in single-arm studies reflect true clinical benefit. Without a comparator group or reliable natural history data, decision-makers are left with considerable uncertainty about the effectiveness of these treatments in adults, limiting the usefulness of the evidence for informing reimbursement decisions.
Noncomparative real-world evidence is recognized for its contribution to providing contextual information, insights into long-term outcomes, and data on patient experiences — particularly when RCTs are not feasible.17 However, given that the key reimbursement question is what clinical value nusinersen and risdiplam provide compared to existing treatments,17,28,29 comparative evidence is better suited to reduce uncertainty and support confident decision-making. In this review, the emphasis on comparative studies reflects the need identified in CDEC recommendations for more definitive evidence of treatment effect relative to existing approaches to managing SMA — beyond what single-arm studies can provide.
Exclusion Criteria
Articles were excluded if they did not meet the selection criteria outlined in Table 2, were duplicate publications, or were published before April 21, 2021, for risdiplam or April 27, 2022, for nusinersen.
None of the identified studies on risdiplam met the inclusion criteria.
Critical Appraisal of Individual Studies
The included publication was critically appraised by 1 reviewer using the Cochrane Risk of Bias in Nonrandomized Studies of Interventions (ROBINS-I) tool.30 Ratings of the risk of bias — low, moderate, serious, or critical — were reported for 7 of the risk of bias domains using the ROBINS-I algorithms to judge the rating. An overall risk of bias judgment was not reported.
Summary of Evidence
Quantity of Research Available
A total of 621 citations were identified in the literature search. Following screening of titles and abstracts, 566 citations were excluded and 55 potentially relevant reports from the electronic search were retrieved for full-text review. No relevant publications were retrieved from the grey literature search for full-text review. No additional potentially relevant studies were identified through a manual search of reference lists from the identified systematic reviews. Of these potentially relevant articles, 54 publications were excluded for various reasons and 1 publication met the inclusion criteria and was included in this report. This comprised 1 observational study based on real-world data. Appendix 2 summarizes the studies that were excluded. Appendix 3 presents the PRISMA31 flow chart of the study selection.
Summary of Study Characteristics
The study by Vázquez-Costa et al.32 was an observational comparative study, based on real-world data, conducted in Spain. It included patients with 5q SMA types II and III, comparing those who were treated with nusinersen to a comparator group of patients who did not receive the treatment (nonexposed). Patients were eligible for the study if they were aged older than 15 years. Nusinersen was administered according to the approved product monograph in Spain. The outcomes assessed included motor function (using the Hammersmith Functional Motor Scale – Expanded [HFMSE], Revised Upper Limb Module [RULM], and 6-minute walk test [6MWT]), pulmonary function, need for noninvasive ventilation, need for gastrostomy tube placement, and adverse events. Clinician and patient global impression were also measured but these were not eligible outcome measures per the protocol for this review.
Additional details regarding the characteristics and results from the Vázquez-Costa et al. study, as well as the critical appraisal, are provided in Appendix 4.
Limitations
Included Study
The primary limitation of this review is the limited and methodologically weak evidence base regarding the effects of nusinersen and risdiplam in adults with SMA. For nusinersen, only 1 study — Vázquez-Costa et al.32 — met the inclusion criteria, and it was limited by its serious risk of bias from confounding and missing data. For risdiplam, no studies met the inclusion criteria as all identified studies were noncomparative and therefore excluded based on the protocol.
Although the Vázquez-Costa study represents a methodological improvement over earlier real-world evidence33-38 by including a concurrent comparison group, it continues to exemplify key limitations typical of this evidence base. The study used a nonrandomized, observational design with a relatively small sample size (N = 79) and is subject to multiple unresolved sources of bias. These methodological limitations substantially limit the internal validity of the findings and preclude drawing causal conclusions about the effectiveness of nusinersen.
While the study’s eligibility criteria allowed the inclusion of adolescents aged at least 16 years, the mean age of patients exceeded 30 years and the first quartile in the nontreated group (representing the lowest age reported; the full range was not reported) was 18.6 years, suggesting reasonable alignment with this review’s focus on adults with SMA. However, the study was not explicitly designed to assess age-related treatment effects, nor did it stratify or analyse results in a way that would clarify which factors — beyond age — may predict treatment response on motor or other functional outcomes.
The current reimbursement criteria for nusinersen (and risdiplam) incorporate age thresholds, but these function more as proxies for clinically relevant prognostic factors — including SMN2 copy number, symptom onset, disease duration, and functional status. The heterogeneity in these characteristics among the patients included in the Vázquez-Costa et al. study adds some real-world relevance, but this diversity also undermines the internal validity of the results. In an observational, nonrandomized design, this level of heterogeneity introduces confounding — both measured and unmeasured — that cannot be fully adjusted for, limiting the reliability of any observed treatment effects.
Vázquez-Costa et al. attempted to identify prognostic factors associated with improved clinical global impression, which was not an end point of relevance to this review for its lack of disease specificity, sensitivity to changes that may be clinically important to patients, and subjectivity. Moreover, the sample size of 79 patients (and even smaller numbers in certain subsets) restricts the feasibility of robust subgroup analyses, which would be useful to identify whether particular subsets of adults — defined by functional ability or SMN2 copy number — derive benefit from treatment. As a result, the study does not adequately address a key gap in the current evidence base: What prognostic markers may better guide clinical and reimbursement decisions in adults with SMA?
The clinical significance of between-group differences in changes from baseline on functional outcome measures — such as the HFMSE, RULM, 6MWT, Egen Klassifikation Scale Version 2 (EK2), and Amyotrophic Lateral Sclerosis Functional Scale Revised (ALSFRS-R) — is difficult to interpret as none of these measures have empirically validated minimal important differences (MIDs) for adults with SMA. Without validated thresholds, it is unclear whether changes represent improvements that are meaningful from a patient or clinical perspective. This limitation is compounded in the responder analyses, which compared the proportion of patients in each group who achieved specified levels of functional improvement based on MIDs extrapolated from children with SMA or other diseases. In the absence of well-established thresholds for clinically meaningful change, the reported odds ratios (ORs) for responder outcomes lack interpretability and offer limited insight into the actual magnitude or relevance of the treatment effects. Consequently, although P values associated with the responder analyses may indicate statistical significance, the analyses do not meaningfully inform whether the treatments produced functionally important benefits for adults.
In addition, the Vázquez-Costa et al. study was not conceived or designed to answer the review question related to the effect of nusinersen in patients who previously received treatment with onasemnogene abeparvovec.
Excluded Studies
The characteristics of the studies that were excluded from this evidence review (Appendix 2) — including those submitted to the drug programs from clinicians who treat SMA — were assessed to provide the drug programs with additional information about the evidence for nusinersen and risdiplam published since the CDEC recommendations. Table 3 summarizes the key limitations of the studies that were excluded due to their noncomparative design (i.e., not RCTs, controlled trials, or comparative observational studies) based on the ROBINS-I risk of bias domains.
Table 4 summarizes the assessment of the relevance of the included (Vázquez-Costa et al.) and excluded published, original research (i.e., those reporting original findings and not including systematic reviews; conference abstracts; editorials; or studies on nonrelevant outcomes, such as those unrelated to efficacy, effectiveness, or harms) for addressing the limitations identified in reimbursement recommendations. For a complete list of the excluded studies, refer to Appendix 2.
Table 3: Limitations of the Excluded Noncomparative Studies in Adults With SMA Based on the ROBINS-I Domains
ROBINS-I domain | Limitations |
|---|---|
Bias due to confounding | The absence of a comparator arm precludes any control for confounding. Key variables such as baseline motor function (scales) and functional status (e.g., ambulatory), age, SMN2 copy number, disease duration, and prior treatment may influence outcomes but cannot be adjusted for in these uncontrolled designs. This severely limits causal inference. Small sample sizes (including variability within studies in the evaluable population) exacerbate the inability to account for potential confounding factors, further limiting interpretability in the absence of a control arm. |
Bias in selection of participants into the study | The inclusion and exclusion criteria were not clearly specified or were inconsistently applied. Selective inclusion of patients who initiated treatment may have introduced selection bias. |
Bias in classification of interventions | Prior treatment history (e.g., switching from other SMA therapies) occurred in some or all patients in some of the studies. This was variably reported and generally not accounted for and may have affected outcome interpretation. |
Bias due to deviations from intended interventions | There was no comparator group to assess adherence or deviations. Supportive care practices were not consistently documented, potentially contributing to differences in outcomes. It is difficult to determine if the observed effects are due to nusinersen or to risdiplam. |
Bias due to missing data | There was substantial missing outcome data in several studies, with unclear reasons for loss to follow-up or missing assessments. Outcome reporting may have been incomplete or selective. Small and fluctuating sample sizes magnify the impact of missing data and reduce the reliability of outcome estimates. |
Bias in measurement of outcomes | Motor function outcomes were often assessed without blinding and using scales that may have varied in administration across sites. This introduced the potential for observer and reporting bias. |
Bias in selection of the reported result | Outcomes were reported inconsistently across the studies, which introduced the potential for selective reporting of positive findings. |
ROBINS-I = Cochrane Risk of Bias in Nonrandomized Studies of Interventions; SMA = spinal muscular atrophy.
Table 4: Assessment of Study Relevance for Reimbursement Decisions: Nusinersen and Risdiplam in Adults With SMA
Studya,b | Objective | Does it address reimbursement recommendation concerns?c |
|---|---|---|
Nusinersen | ||
Included studies | ||
Vázquez-Costa et al. (2022)32 | Harms and efficacy of nusinersen in adults with SMA treated with nusinersen compared with patients who were not treated |
|
Excluded studies | ||
Alves et al. (2024)39 | Harms and efficacy of nusinersen and nusinersen followed by onasemnogene abeparvovec on motor, respiratory, and bulbar function in children with SMA type I | The study was done solely in children aged < 10 years and does not apply to the assessment. |
Bieniaszewska et al. (2023)40 | Effectiveness of nusinersen and risdiplam on motor function in children with SMA types II or III | The study was done solely in children aged < 15 years and does not apply to the assessment. |
Cavaloiu et al. (2024)41 | Effectiveness of nusinersen on motor function in adults with SMA types II and III |
|
Coratti et al. (2024)42 | Effectiveness of nusinersen on motor function in children and adults with SMA type II |
|
De Albuquerque et al. (2025)43 | Effectiveness of nusinersen, risdiplam, and onasemnogene abeparvovec on motor function in children and adults with SMA types I to IV |
|
Diener HC. (2024)44 | Effectiveness of nusinersen on motor function in adults with SMA |
|
Elsheikh et al. (2021)45 | Harms, tolerability, and effectiveness of nusinersen in adults with SMA |
|
Günther et al. (2024)d,e,46 | Effectiveness and harms of nusinersen in adults with SMA types I to IV |
|
Lefeuvre et al. (2022)47 | Effectiveness and harms of nusinersen in adults with SMA types II or III |
|
Lilien et al. (2024)48 | Assess patient-reported perceptions of nusinersen treatment effects on motor function in children and adults with SMA types I to III |
|
Tachibana et al. (2024)49 Related publication: Tachibana et al. (2024) (early interim analysis)50 | Harms and effectiveness of nusinersen in patients with SMA of all types and ages as part of a postmarketing surveillance program in Japan |
|
Zhuang et al. (2023)51 | Harms of nusinersen, risdiplam, and onasemnogene abeparvovec using global pharmacovigilance data from the FDA Adverse Event Reporting System |
|
Risdiplam | ||
Included studies | ||
None | — | — |
Excluded studies | ||
Ashrafi et al. (2024)52 | Effectiveness of risdiplam and nusinersen in children with SMA types II or III | The study was done solely in children aged < 10 years and does not apply to the assessment. |
Bjelica et al. (2024)d,e,53 | Effectiveness of risdiplam on motor function and patient-reported treatment satisfaction in adults with SMA |
|
Brakemeier et al. (2024)d,e,54 | Effectiveness of risdiplam on swallowing-related quality of life in adults with SMA type II or III who are nonambulatory |
|
Chiriboga et al. (2024)d,e,55 Related publication: Chiriboga et al. (2023) (12-month results)56 | Harms and exploratory efficacy of risdiplam in patients with SMA previously treated with other disease-modifying therapies or who are treatment naive |
|
Hahn et al. (2022)57 | Harms of risdiplam in a compassionate use program for children and adults with SMA in Germany |
|
Jira et al. (2024)58 | Relationship between gait speed and fall risk in adults with SMA who were ambulatory |
|
Kessler et al. (2024)d,e,59 | Efficacy of risdiplam by comparing electrophysiological biomarkers with clinical outcome measures in adults with SMA type II or III |
|
Kwon et al. (2022)60 | Harms and access outcomes from an expanded access program of risdiplam in children and adults with SMA type I or II |
|
McCluskey (2023)d,e,61 | Expanded access assessment in Ireland |
|
Ñungo Garzón et al. (2023)d,e,62 | Harms and effectiveness of risdiplam on functional improvements on motor, bulbar, and respiratory function in adolescents and adults with SMA type II who were nonsitters |
|
Oskoui et al. (2023)63 | Efficacy and safety of risdiplam in patients with SMA type II or nonambulant type III |
|
Severa et al. (2024)d,e,64 | Effectiveness and tolerability of risdiplam in adults with SMA type II or III |
|
Sitas et al. (2024)d,e,65 | Effectiveness of risdiplam on nonmotor symptoms and functional outcomes in adults with SMA type II or III |
|
SMA = spinal muscular atrophy.
aThis column only includes the excluded studies that reported original research findings. It excludes systematic reviews (including meta-analyses and indirect treatment comparisons), previously reviewed studies, studies on outcomes not relevant to the review (i.e., those other than efficacy, effectiveness, or harms), and other reports (e.g., editorials), including conference abstracts. Refer to Appendix 2 for complete excluded studies information.
bStudies that included both nusinersen and risdiplam are positioned in the table based on the primary objective or on the drug listed first in the title or abstract of the article.
cEvidence limitations identified by the Canadian Drug Expert Committee: lack of comparative studies, limited adult representation, methodological concerns, uncertain clinical meaningfulness.
dThe study was provided by drug programs (submitted to them by clinicians who treat SMA) for appraisal alongside the body of evidence identified through the literature search.
eAlso identified in the literature search.
Of the 25 excluded original research studies summarized in Table 4, 4 included only children39,40,52,66 and 10 had a mix of children and adults with SMA.42,43,46,48,49,51,55,57,60,62 Most of the studies (20) were noncomparative,41-49,51,53-55,57,59-62,64,65 1 study enrolled patients who received risdiplam before age 25 years,63 and 1 study did not evaluate an intervention.58 Twelve studies (5 for nusinersen41,44-47 and 7 for risdiplam53,54,59,61,62,64,65) were noncomparative but included a fully or mostly adult population with SMA. Three of these studies followed patients for longer than 14 months (the maximum follow-up duration available from previously reviewed studies), including 1 with follow-up to 54 months; all were studies on nusinersen.41,44,46 However, despite their potential to inform longer-term outcomes, these studies share several methodological limitations that limit their usefulness for assessing comparative clinical effectiveness and harms.
The methodological limitations were similar to those previously identified from previously reviewed noncomparative studies. The lack of a comparison group precludes adjustment for key confounding factors — such as baseline function, age, and prior treatment — thereby limiting causal inference. Although some studies had total sample sizes greater than 100 patients (most were less than 20 patients; range, 6 to 237), the number of patients included in specific outcome analyses (when reported) was often much smaller. For example, in 1 study with a larger sample size of 237 adults, the final analysis at 38 months was based on 120 patients.44 In the JEWELFISH study, which enrolled 174 patients, 63 were adults (aged > 18 years) and only 13 had received risdiplam as their sole treatment for SMA. 55 These small total and reduced sample sizes limit the statistical precision of treatment effect estimates. In the absence of a control group, small and variable sample sizes further increase the risk of confounding and make it difficult to assess whether observed outcomes are due to treatment or underlying patient characteristics. Other concerns included potential selection bias, incomplete or selectively reported outcome data, unblinded outcome assessments, and inconsistent reporting of supportive therapies and intervention history for SMA. Collectively, these limitations reduce the internal validity and generalizability of the findings.
Similar methodological concerns were noted in the studies provided to the drug programs from clinicians who treat SMA as part of their request to CDA-AMC (Table 4).
Two excluded studies warrant specific mention. The preliminary results from the aforementioned JEWELFISH study55 — an open-label, single-arm interventional study — were previously reported in the CADTH reimbursement review of risdiplam.55 This study evaluated the harms, pharmacokinetics, and pharmacodynamics of risdiplam in pediatric and adult patients with SMA (aged 1 to 60 years; 63 aged > 18 years) who had received risdiplam as their sole treatment or had received prior treatment with nusinersen, onasemnogene abeparvovec, or olesoxime (not available in Canada). Although 24-month results were identified in the current literature search, the study was excluded at the full-text screening stage due to its noncomparative design, which means that we could not determine whether individuals receiving the drug fared better than those who did not. Additional limitations included the exploratory nature of the motor function outcomes and the lack of stratified analyses by age and baseline function, which would preclude interpretation of treatment effects in adult populations. While the study included a subgroup of patients previously treated with onasemnogene abeparvovec, this cohort consisted solely of children aged 1 to 5 years (N = 14). Given the small sample size and limited generalizability, the findings do not meaningfully inform the objectives of this evidence review.
The second study of note was 1 based on data from patients in Canada. The study by Côté et al. (2025) is a longitudinal analysis of real-world data evaluating the effects of nusinersen in 17 adults with SMA types II and III over a 36-month period.67 Conducted at the Institut de réadaptation en déficience physique de Québec, the study assessed motor function, pulmonary function, and patient-reported outcomes. The study was excluded from this review at the abstract screening stage due to its noncomparative design. Several important limitations that affect the validity, interpretability, and generalizability of its findings were identified. Selection bias is a potential concern, with 77% of the cohort composed of patients with SMA type III and no information provided on those who were not included, such as individuals who chose not to be treated with nusinersen or may have discontinued nusinersen due to harms. This also raises questions about the representativeness of the study population, particularly given the underrepresentation of patients with type II SMA and exclusion of patients with type IV SMA. As with other noncomparative studies, confounding remains a key limitation (especially from the reported heterogeneity in baseline characteristics), and the lack of a comparator arm precludes causal inference. Missing data were common due to logistical and COVID-19–related issues. The statistical method used to handle missing data (pairwise deletion) assumes data were missing completely at random, which is an assumption that is likely invalid in this context. The number of patients included in specific outcome analyses was often much smaller than the total sample size, as low as 2 patients in some analyses, and never more than 10 for most others. Although the study reports individual-level benefits (e.g., 9 of 17 patients showed motor function improvement), the interpretation may overstate treatment effects because of the small evaluable sample sizes and uncertainty about response thresholds being met. Only 1 outcome — change in 6MWT — met the study-specified target MID of 30 m, and the results across other scales did not consistently exceed specified thresholds for clinical significance. Taken together, the study does not provide robust or generalizable evidence of nusinersen’s effectiveness in adults with SMA.
Systematic reviews,68-75 meta-analyses,76-82 and a single indirect treatment comparison66 were excluded based on the study design inclusion criterion, which limited eligible evidence to RCTs, controlled trials, and comparative observational studies. Although these types of evidence can be informative when based on comparative data, the systematic reviews and meta-analyses identified primarily included noncomparative studies that would have been excluded individually for failing to meet design criteria. The indirect treatment comparison offered potentially relevant comparative evidence between nusinersen and risdiplam; however, it was based on previously reviewed clinical trials that were conducted in a pediatric population (< 10 years of age), limiting its applicability to adults with SMA.
None of the studies identified by clinicians who treat SMA and provided to the jurisdictional drug programs for consideration, and appraised in this review alongside those found through the literature search, met the inclusion criteria for this evidence review (these studies are outlined in Appendix 2, Table 8). Specifically, none met the eligibility criterion for comparative study design. Three studies were available only as conference abstracts, with no corresponding peer-reviewed publications, and were therefore excluded. One study, by Maggi et al.,83 had already been reviewed as part of the CADTH reimbursement reassessment of nusinersen in adults with SMA types II and III, and contributed to the 2022 CDEC recommendation. As a result, it did not constitute new evidence. Overall, the studies shared the same methodological limitations as those identified in the literature search, as summarized in Table 3 and Table 4.
Are RCTs Feasible in Adults With SMA?
Given the limitations of the current evidence base — dominated by small, noncomparative observational studies with high risk of bias — it is important to consider whether more rigorous evidence, such as that from RCTs, could be generated to support decision-making in this population.
RCTs are considered the most reliable method for evaluating treatment effects because they minimize bias through randomization and the use of control groups. Both CDA-AMC and the UK’s National Institute for Health and Care Excellence, among other agencies, emphasize RCTs as the preferred evidence base for assessing comparative clinical effectiveness in their health technology assessment methods guidance, particularly when informing reimbursement decisions for new or costly interventions.
While RCTs in adults with SMA may pose logistical and methodological challenges, they are not inherently infeasible. Adults are estimated to represent approximately 26% of the global SMA population84 and SMA prevalence is comparable to or greater than that of other rare neuromuscular diseases — such as Duchenne muscular dystrophy — in which RCTs have been completed or are under way. For example, several RCTs in Duchenne muscular dystrophy have enrolled adolescent and adult populations, demonstrating that RCTs are achievable even in rare neurologic conditions with small and heterogeneous patient populations.85
Notably, adults were included in the phase III SUNFISH Part 2 trial of risdiplam, which randomized patients aged 2 to 25 years with type II or III SMA. Although only approximately 12% of enrolled patients were aged 18 or older, this inclusion demonstrates that enrolling adults in RCTs is feasible with appropriate design and recruitment strategies.86
The practical implementation of RCTs or other comparative study designs in this population would require careful consideration of several factors, including the heterogeneity of the population, potential influence of previous treatment, and the risk of substantial dropout (already seen in existing observational studies), especially with a 12-month or longer follow-up needed to observe clinically meaningful outcomes.
While RCTs remain the gold standard for evaluating treatment effects, CDA-AMC methods guidance recognizes that real-world evidence — including observational studies and external control arms — may be considered, especially when RCTs are not feasible or ethical.17 This ensures that valuable insights from real-world settings are not overlooked in the assessment of clinical effectiveness.
Conclusions and Implications for Decision- or Policy-Making
Previous reviews by CADTH and CDEC reimbursement recommendations have identified major methodological limitations in the evidence base of nusinersen and risdiplam in adults with SMA. These limitations include noncomparative study designs; small sample sizes; short-term follow-ups; and multiple sources of bias, especially selection bias, confounding, and attrition bias; that precluded causal interpretation of treatment effects. In addition, determining whether observed treatment effects are clinically meaningful has been hindered by the use of outcome measures that are not well suited to adults with SMA and by the lack of validated thresholds to determine clinical meaningfulness.
This evidence review aimed to critically appraise the comparative benefits and harms of nusinersen and risdiplam in adults with SMA, and to assess the feasibility and utility of a more comprehensive review to inform jurisdictional reimbursement decisions. The findings highlight continued significant limitations in the evidence base in the adult population, for whom robust comparative studies remain scarce.
Only 1 new study — Vázquez-Costa et al. — met the inclusion criteria. This multicentre observational cohort study compared 39 patients treated with nusinersen with 40 untreated controls over a mean follow-up of 16 months.32 No eligible studies evaluating risdiplam in adults aged older than 25 years were identified and no comparative studies included patients previously treated with onasemnogene abeparvovec.
The Vázquez-Costa et al. study was limited by serious risk of bias due to confounding as a result of its nonrandomized design and inadequate adjustment for baseline differences in important patient- and SMA-related factors (e.g., functional status). Risk of bias from patient selection was rated moderate to serious, as the treated and untreated groups may have differed in prognosis. Further limitations included moderate to serious risks of bias due to incomplete or missing data, deviations from intended interventions, and variability in outcome measurements. Incomplete reporting of key outcomes also introduced concerns about selective reporting.
Taken together, these limitations suggest that the current comparative evidence for nusinersen in adults is of low quality and insufficient to draw robust conclusions about its effectiveness or harms. The absence of eligible studies for risdiplam in this population further underscores the evidence gap. Moreover, the lack of comparative data on adults previously treated with onasemnogene abeparvovec limits the generalizability of the findings to clinical scenarios in which treatment sequencing is increasingly relevant.
Beyond the limited and low-certainty comparative evidence, the literature search also identified many noncomparative studies evaluating nusinersen and risdiplam in real-world settings. While these studies may offer insights into how these treatments are used in clinical practice, they consistently shared key weaknesses, such as small participant numbers (as few as 6 patients in some studies); inconsistent or incomplete reporting of outcomes; and potential sources of bias, including patient selection and missing data. A critical limitation of these studies is the absence of a comparator group and/or clear natural history of progression, which makes it difficult to determine whether observed changes in function or disease progression are attributable to the treatment or to other factors, which is particularly important in a heterogeneous population like adults with SMA. As such, the findings from these studies are not reliable enough to support decisions about the relative benefits and harms of the treatments, especially when making reimbursement or policy decisions that require a clear understanding of treatment effects.
Given these findings, a de novo systematic review may have limited feasibility and utility at this time, unless new, higher-quality comparative studies become available. The current evidence does not adequately address the research questions posed, nor the limitations in evidence identified in existing reimbursement recommendations, especially regarding comparative effectiveness and harms with nusinersen and risdiplam in adults with SMA.
References
1.D'Amico A, Mercuri E, Tiziano FD, et al. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71. doi:10.1186/1750-1172-6-71 PubMed
2.Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35(23):8691-700. doi:10.1523/jneurosci.0417-15.2015 PubMed
3.Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831-46. doi:10.1016/j.ncl.2015.07.004 PubMed
4.Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124. doi:10.1186/s13023-017-0671-8 PubMed
5.Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-49. doi:10.1177/0883073807305788 PubMed
6.Russman BS. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol. 2007;22(8):946-51. PubMed
7.Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017;24:529. doi:10.1038/gt.2017.52 PubMed
8.Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115. doi:10.1016/j.nmd.2017.11.005 PubMed
9.Biogen Canada Inc. Spinraza (nusinersen): solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium [product monograph]. April 20, 2020.
10.Hoffmann-La Roche Limited. Evrysdi (risdiplam powder for oral solution): 60 mg/bottle (0.75 mg/mL after reconstitution), oral or enteral [product monograph]. April 14, 2021.
11.CADTH. Drug Reimbursement Expert Review Committee final recommendation: nusinersen (Spinraza - Biogen Canada Inc.). February 27, 2019. Accessed 2025 May 15. https://www.cda-amc.ca/sites/default/files/cdr/complete/SR0576-Spinraza-Resubmission-Mar-1-19.pdf
12.CADTH. Reimbursement recommendation: nusinersen (Spinraza). Can J Health Tecnol. 2022;2(8). doi:10.51731/cjht.2022.430
13.CADTH. Reimbursement recommendation: risdiplam (Evrysdi). Can J Health Technol. 2021;1(8). doi:10.51731/cjht.2021.136
14.CADTH. Nusinersen for adolescents and adults with spinal muscular atrophy: a review of clinical effectiveness. September 10, 2020.
15.CADTH. Reimbursement review: risdiplam (Evrysdi) Can J Health Tecnol. 2021;1(11). doi:10.51731/cjht.2021.185
16.CADTH. Reimbursement review: nusinersen (Sprinraza) Can J Health Tecnol. 2022;2(11). doi:10.51731/cjht.2022.496
17.Canada's Drug Agency. Methods guide for health technology assessment. 2025. Accessed May 15, 2025. https://www.cda-amc.ca/methods-guide
18.Guyatt G, Oxman AD, Akl EA, et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol. 2011;64(4):383-94. doi:10.1016/j.jclinepi.2010.04.026 PubMed
19.Guyatt GH, Oxman AD, Sultan S, et al. GRADE guidelines: 9. Rating up the quality of evidence. J Clin Epidemiol. 2011;64(12):1311-6. doi:10.1016/j.jclinepi.2011.06.004 PubMed
20.Schünemann H, Hill S, Guyatt G, et al. The GRADE approach and Bradford Hill's criteria for causation. J Epidemiol Community Health. 2011;65(5):392-5. doi:10.1136/jech.2010.119933 PubMed
21.Reeves BC, Deeks JJ, Higgins JPT, et al. Chapter 24: Including non-randomized studies on intervention effects [last updated October 2019]. In: Higgins JPT TJ, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, ed. Cochrane Handbook for Systematic Reviews of Interventions version 65. Cochrane; 2024.
22.Shimizu-Motohashi Y, Chiba E, Mizuno K, et al. Muscle impairment in MRI affect variability in treatment response to nusinersen in patients with spinal muscular atrophy type 2 and 3: a retrospective cohort study. Brain Dev. 2023;45(3):161-170. doi:10.1016/j.braindev.2022.11.002 PubMed
23.Vill K, Tacke M, König A, et al. 5qSMA: standardised retrospective natural history assessment in 268 patients with four copies of SMN2. J Neurol. 2024;271(5):2787-2797. doi:10.1007/s00415-024-12188-5 PubMed
24.Aponte Ribero V, Martí Y, Batson S, et al. Systematic literature review of the natural history of spinal muscular atrophy: motor function, scoliosis, and contractures. Neurology. 2023;101(21):e2103-e2113. doi:10.1212/wnl.0000000000207878 PubMed
25.Mercuri E, Lucibello S, Pera MC, et al. Long-term progression in type II spinal muscular atrophy: a retrospective observational study. Neurology. 2019;93(13):e1241-e1247. doi:10.1212/wnl.0000000000008166 PubMed
26.Wan HWY, Carey KA, D'Silva A, et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi:10.1186/s13023-020-1339-3 PubMed
27.Wijngaarde CA, Brink RC, de Kort FAS, et al. Natural course of scoliosis and lifetime risk of scoliosis surgery in spinal muscular atrophy. Neurology. 2019;93(2):e149-e158. doi:10.1212/wnl.0000000000007742 PubMed
28.Canada's Drug Agency. Reimbursement reviews. Updated October 29, 2020. Accessed May 24, 2025. https://www.cda-amc.ca/reimbursement-reviews
29.Canada’s Drug Agency. Expert committee deliberation at Canada's Drug Agency. 2025. Accessed May 15, 2025. https://www.cda-amc.ca/sites/default/files/MG%20Methods/expert_committee_deliberation.pdf
30.Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. 2016;355:i4919. doi:10.1136/bmj.i4919 PubMed
31.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700. doi:10.1136/bmj.b2700 PubMed
32.Vázquez-Costa JF, Povedano M, Nascimiento-Osorio AE, et al. Nusinersen in adult patients with 5q spinal muscular atrophy: a multicenter observational cohorts' study. Eur J Neurol. 2022;29(11):3337-3346. doi:10.1111/ene.15501 PubMed
33.Mix L, Winter B, Wurster CD, et al. Quality of life in SMA patients under treatment with nusinersen. Front Neurol. 2021;12:626787. doi:10.3389/fneur.2021.626787 PubMed
34.Elsheikh B, Severyn S, Zhao S, et al. Safety, tolerability, and effect of nusinersen in non-ambulatory adults with spinal muscular atrophy. Front Neurol. 2021;12:650532. doi:10.3389/fneur.2021.650532 PubMed
35.Brakemeier S, Stolte B, Thimm A, et al. Assessment of bulbar function in adult patients with 5q-SMA type 2 and 3 under treatment with nusinersen. Brain Sci. 2021;11(9):20. doi:10.3390/brainsci11091244 PubMed
36.Duong T, Wolford C, McDermott MP, et al. Nusinersen treatment in adults with spinal muscular atrophy. Neurol. 2021;11(3):e317-e327. doi:10.1212/CPJ.0000000000001033 PubMed
37.Moshe-Lilie O, Visser A, Chahin N, et al. Nusinersen in adult patients with spinal muscular atrophy: observations from a single center. Neurology. 2020;95(4):e413-e416. doi:10.1212/WNL.0000000000009914 PubMed
38.Kizina K, Stolte B, Totzeck A, et al. Fatigue in adults with spinal muscular atrophy under treatment with nusinersen. Sci Rep. 2020;10(1):11069. doi:10.1038/s41598-020-68051-w PubMed
39.Alves B, Araujo A, Santos FND, et al. Type-1 spinal muscular atrophy cohort before and after disease-modifying therapies. Arq Neuropsiquiatr. 2024;82(11):1-8. doi:10.1055/s-0044-1791757 PubMed
40.Bieniaszewska A, Sobieska M, Steinborn B, et al. Examination of upper limb function and the relationship with gross motor functional and structural parameters in patients with spinal muscular atrophy. Biomedicines. 2023;11(4):24. doi:10.3390/biomedicines11041005 PubMed
41.Cavaloiu B, Simina IE, Vilciu C, et al. Nusinersen improves motor function in type 2 and 3 spinal muscular atrophy patients across time. Biomedicines. 2024;12(8):06. doi:10.3390/biomedicines12081782
42.Coratti G, Civitello M, Rohwer A, et al. Changes in abilities over the initial 12 months of nusinersen treatment for type II SMA. Neuromuscul Disord. 2024;41:42-50. doi:10.1016/j.nmd.2024.05.003 PubMed
43.de Albuquerque ALA, Chadanowicz JK, Bevilacqua IP, et al. Clinicogenetic characterization and response to disease-modifying therapies in spinal muscular atrophy: real-world experience from a reference center in Southern Brazil. J Pediatr (Rio J). 2025;101(1):38-45. doi:10.1016/j.jped.2024.07.011 PubMed
44.Diener HC. Spinal muscular atrophy: long-term efficacy and safety of nusinersen in adults with 5q-SMA. [German]. Psychopharmakotherapie. 2024;31(3):111-112.
45.Elsheikh B, Severyn S, Zhao S, et al. Safety, tolerability, and effect of nusinersen treatment in ambulatory adults with 5q-SMA. Front Neurol. 2021;12:650535. doi:10.3389/fneur.2021.650535 PubMed
46.Gunther R, Wurster CD, Brakemeier S, et al. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. Lancet Reg Health Eur. 2024;39:100862. doi:10.1016/j.lanepe.2024.100862 PubMed
47.Lefeuvre C, Brisset M, Sarlon M, et al. Nusinersen treatment in adults with severe spinal muscular atrophy: a real-life retrospective observational cohort study. Rev Neurol (Paris). 2022;178(3):234-240. doi:10.1016/j.neurol.2021.10.010 PubMed
48.Lilien C, Vrscaj E, Thapaliya G, et al. Patients' perceptions of nusinersen effects according to their responder status. J Clin Med. 2024;13(12):11. doi:10.3390/jcm13123418 PubMed
49.Tachibana Y, Sato R, Makioka H, et al. Safety and effectiveness of nusinersen, a treatment for spinal muscular atrophy, in 524 patients: results from an interim analysis of post-marketing surveillance in Japan. Int J Neurosci. 2024;134(11):1185-1197. doi:10.1080/00207454.2023.2251662 PubMed
50.Tachibana Y, Takasaki S, Hoshino M, et al. Real-world safety and effectiveness of nusinersen, a treatment for spinal muscular atrophy, in 401 Japanese patients: results from an interim analysis of post-marketing surveillance. Int J Neurosci. 2024;134(2):153-162. doi:10.1080/00207454.2022.2095270 PubMed
51.Zhuang W, Lu M, Wu Y, et al. Safety concerns with nusinersen, risdiplam, and onasemnogene abeparvovec in spinal muscular atrophy: a real-world pharmacovigilance study. Clin Drug Investig. 2023;43(12):949-962. doi:10.1007/s40261-023-01320-4 PubMed
52.Ashrafi MR, Babaee M, Hashemi Nazari SS, et al. Comparative efficacy of risdiplam and nusinersen in Type 2 and 3 spinal muscular atrophy patients: a cohort study using real-world data. J Neuromuscul Dis. 2024;11(6):1190-1199. doi:10.1177/22143602241288087 PubMed
53.Bjelica B, Wohnrade C, Cespedes I, et al. Risdiplam therapy in adults with 5q-SMA: observational study on motor function and treatment satisfaction. BMC Neurol. 2024;24(1):67. doi:10.1186/s12883-024-03562-x PubMed
54.Brakemeier S, Lipka J, Schlag M, et al. Risdiplam improves subjective swallowing quality in non-ambulatory adult patients with 5q-spinal muscular atrophy despite advanced motor impairment. J Neurol. 2024;271(5):2649-2657. doi:10.1007/s00415-024-12203-9 PubMed
55.Chiriboga CA, Bruno C, Duong T, et al. JEWELFISH: 24-month results from an open-label study in non-treatment-naive patients with SMA receiving treatment with risdiplam. J Neurol. 2024;271(8):4871-4884. doi:10.1007/s00415-024-12318-z PubMed
56.Chiriboga CA, Bruno C, Duong T, et al. Risdiplam in patients previously treated with other therapies for spinal muscular atrophy: an interim analysis from the JEWELFISH study. Neurol. 2023;12(2):543-557. doi:10.1007/s40120-023-00444-1 PubMed
57.Hahn A, Gunther R, Ludolph A, et al. Short-term safety results from compassionate use of risdiplam in patients with spinal muscular atrophy in Germany. Orphanet J Rare Dis. 2022;17(1):276. doi:10.1186/s13023-022-02420-8 PubMed
58.Jira K, Jaworek A, Allen M, et al. The association between gait speed and falls in ambulatory adults with spinal muscular atrophy: a retrospective pilot study. Front Neurol. 2024;15:1491466. doi:10.3389/fneur.2024.1491466 PubMed
59.Kessler T, Sam G, Wick W, et al. Evaluation of risdiplam efficacy in 5q spinal muscular atrophy: a systematic comparison of electrophysiologic with clinical outcome measures. Eur J Neurol. 2024;31(1):e16099. doi:10.1111/ene.16099 PubMed
60.Kwon JM, Arya K, Kuntz N, et al. An expanded access program of risdiplam for patients with Type 1 or 2 spinal muscular atrophy. Ann. 2022;9(6):810-818. doi:10.1002/acn3.51560 PubMed
61.McCluskey G, Lamb S, Mason S, et al. Risdiplam for the treatment of adults with spinal muscular atrophy: experience of the Northern Ireland neuromuscular service. Muscle Nerve. 2023;67(2):157-161. doi:10.1002/mus.27755 PubMed
62.Nungo Garzon NC, Pitarch Castellano I, Sevilla T, et al. Risdiplam in non-sitter patients aged 16 years and older with 5q spinal muscular atrophy. Muscle Nerve. 2023;67(5):407-411. doi:10.1002/mus.27804 PubMed
63.Oskoui M, Day JW, Deconinck N, et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J Neurol. 2023;270(5):2531-2546. doi:10.1007/s00415-023-11560-1 PubMed
64.Severa G, Alfaro MDC, Alimi Ichola C, et al. Risdiplam: therapeutic effects and tolerability in a small cohort of 6 adult type 2 and type 3 SMA patients. Orphanet J Rare Dis. 2024;19(1):430. doi:10.1186/s13023-024-03442-0 PubMed
65.Sitas B, Hancevic M, Bilic K, et al. Risdiplam real world data - looking beyond motor neurons and motor function measures. J Neuromuscul Dis. 2024;11(1):75-84. doi:10.3233/jnd-230197 PubMed
66.Ribero VA, Daigl M, Marti Y, et al. How does risdiplam compare with other treatments for Types 1-3 spinal muscular atrophy: a systematic literature review and indirect treatment comparison. J Comp Eff Res. 2022;11(5):347-370. doi:10.2217/cer-2021-0216 PubMed
67.Cote I, Hodgkinson V, Nury M, et al. A real-world study of nusinersen effects in adults with spinal muscular atrophy type 2 and 3. Can J Neurol Sci. 2025;52(1):119-128. doi:10.1017/cjn.2024.49 PubMed
68.Aldukain M, Aldukain A, Hobani A, et al. The impact of nusinersen treatment on respiratory function in patients with spinal muscular atrophy: a systematic review. J Clin Med. 2024;13(21):22. doi:10.3390/jcm13216306 PubMed
69.Dosi C, Masson R. The impact of three SMN2 gene copies on clinical characteristics and effect of disease-modifying treatment in patients with spinal muscular atrophy: a systematic literature review. Front Neurol. 2024;15:1308296. doi:10.3389/fneur.2024.1308296 PubMed
70.Erdos J, Wild C. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: a systematic review of real-world study data. Eur J Paediatr Neurol. 2022;39:1-10. doi:10.1016/j.ejpn.2022.04.006 PubMed
71.Giess D, Erdos J, Wild C. An updated systematic review on spinal muscular atrophy patients treated with nusinersen, onasemnogene abeparvovec (at least 24 months), risdiplam (at least 12 months) or combination therapies. Eur J Paediatr Neurol. 2024;51:84-92. doi:10.1016/j.ejpn.2024.06.004 PubMed
72.McGrattan K, Walsh K, Mehl L, et al. Systematic literature review of the impact of spinal muscular atrophy therapies on bulbar function. J Neuromuscul Dis. 2024:22143602241303373. doi:10.1177/22143602241303373 PubMed
73.Wadman RI, van der Pol WL, Bosboom WM, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2020;1:CD006282. doi:10.1002/14651858.CD006282.pub5 PubMed
74.Yang M, Awano H, Tanaka S, et al. Systematic literature review of clinical and economic evidence for spinal muscular atrophy. Adv Ther. 2022;39(5):1915-1958. doi:10.1007/s12325-022-02089-2 PubMed
75.Zhao X, Liao Y, Zhao J, et al. Motor function and safety of nusinersen and risdiplam in asian patients with types 2-4 spinal muscular atrophy (SMA): a systematic review and meta-analysis. Adv Ther. 2025;17:17. doi:10.1007/s12325-024-03101-7 PubMed
76.Chen B, Gong Y, Zhou T. The impact of nusinersen and risdiplam on motor function for spinal muscular atrophy type 2 and 3: a meta-analysis. J Coll Physicians Surg Pak. 2024;34(8):948-955. doi:10.29271/jcpsp.2024.08.948 PubMed
77.Chongmelaxme B, Yodsurang V, Vichayachaipat P, et al. Gene-based therapy for the treatment of spinal muscular atrophy types 1 and 2: a systematic review and meta-analysis. Gene Ther. 2024;27:27. doi:10.1038/s41434-024-00503-8 PubMed
78.Gavriilaki M, Moschou M, Papaliagkas V, et al. Nusinersen in adults with 5q spinal muscular atrophy: a systematic review and meta-analysis. Neurotherapeutics. 2022;19(2):464-475. doi:10.1007/s13311-022-01200-3 PubMed
79.Hagenacker T, Maggi L, Coratti G, et al. Effectiveness of nusinersen in adolescents and adults with spinal muscular atrophy: systematic review and meta-analysis. Neurol. 2024;13(5):1483-1504. doi:10.1007/s40120-024-00653-2 PubMed
80.Kant-Smits K, Bartels B, van der Heiden L, et al. The effect of disease-modifying therapies on lung function and respiratory muscle strength in spinal muscular atrophy: systematic review and meta-analysis. Respir Care. 2025;70(3):337-348. doi:10.4187/respcare.12378 PubMed
81.Pascual-Morena C, Martinez-Vizcaino V, Cavero-Redondo I, et al. Efficacy of risdiplam in spinal muscular atrophy: a systematic review and meta-analysis. Pharmacotherapy. 2024;44(1):97-105. doi:10.1002/phar.2866 PubMed
82.Qiao Y, Chi Y, Gu J, et al. Safety and efficacy of nusinersen and risdiplam for spinal muscular atrophy: a systematic review and meta-analysis of randomized controlled trials. Brain Sci. 2023;13(10):07. doi:10.3390/brainsci13101419
83.Maggi L, Bello L, Bonanno S, et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. 2020;91(11):1166-1174. doi:10.1136/jnnp-2020-323822 PubMed
84.Verhaart IEC, Robertson A, Leary R, et al. A multi-source approach to determine SMA incidence and research ready population. J Neurol. 2017;264(7):1465-1473. doi:10.1007/s00415-017-8549-1 PubMed
85.ClinicalTrials.gov. Duchenne muscular dystrophy | recruiting, active, not recruiting, completed studies. 2025. Accessed May 15, 2025. https://clinicaltrials.gov
86.Mercuri E, Deconinck N, Mazzone ES, et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022;21(1):42-52. doi:10.1016/s1474-4422(21)00367-7 PubMed
87.HTA glossary. Accessed May 24, 2025. https://htaglossary.net/
88.CADTH, Health Canada. Guidance for reporting real-world evidence. 2023. Accessed May 24, 2025. https://www.cda-amc.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf
89.Vázquez-Costa JF, Povedano M, Nascimiento-Osorio AE, et al. Validation of motor and functional scales for the evaluation of adult patients with 5q spinal muscular atrophy. Eur J Neurol. 2022;29(12):3666-3675. doi:10.1111/ene.15542 PubMed
90.Wijngaarde CA, Stam M, Otto LAM, et al. Muscle strength and motor function in adolescents and adults with spinal muscular atrophy. Neurology. 2020;95(14):e1988-e1998. doi:10.1212/wnl.0000000000010540 PubMed
91.Coratti G, Pera MC, Montes J, et al. Different trajectories in upper limb and gross motor function in spinal muscular atrophy. Muscle Nerve. 2021;64(5):552-559. doi:10.1002/mus.27384 PubMed
92.Pera MC, Coratti G, Forcina N, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017;17(1):39. doi:10.1186/s12883-017-0790-9 PubMed
93.Mazzone ES, Mayhew A, Montes J, et al. Revised upper limb module for spinal muscular atrophy: development of a new module. Muscle Nerve. 2017;55(6):869-874. doi:10.1002/mus.25430 PubMed
94.Pera MC, Coratti G, Mazzone ES, et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve. 2019;59(4):426-430. doi:10.1002/mus.26419 PubMed
95.Hagenacker T, Wurster CD, Gunther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317-325. doi:10.1016/S1474-4422(20)30037-5 PubMed
96.McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48(3):357-68. doi:10.1002/mus.23905 PubMed
97.Dunaway Young S, Montes J, Kramer SS, et al. Six-minute walk test is reliable and valid in spinal muscular atrophy. Muscle Nerve. 2016;54(5):836-842. doi:10.1002/mus.25120 PubMed
98.Steffensen B, Mayhew A, Aloysius A, et al. T.P.1.07. Egen classification revisited in SMA [conference abstract]. Neuromuscul Disord. 2008;18(9):740-741.
99.Walter MC, Wenninger S, Thiele S, et al. Safety and treatment effects of nusinersen in longstanding adult 5q-SMA Type 3 - a prospective observational study. J Neuromuscul Dis. 2019;6(4):453-465. doi:10.3233/JND-190416 PubMed
100.Agencia española de medicamentos y productos sanitarios. Informe de Posicionamiento Terapéutico de nusinersen (Spinraza) en atrofia muscular espinal. 2018. Accessed May 16, 2025. https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-nusinersen-Spinraza-atrofia-muscular-espinal.pdf
101.European Medicines Agency. Product information: Spinraza (nusinersen): solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium. 2022. Accessed Apr 4, 2022. https://www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_en.pdf
Appendix 1: Glossary
Please note that this appendix has not been copy-edited.
Term | Definition |
|---|---|
Appraisal of evidence, critical appraisal | “The process of assessing and interpreting scientific research results by systematically analysing their validity, clinical and statistical significance, and clinical relevance.”87 |
Assessment | “A scientific process used to describe and analyse the properties of a health technology — its safety, efficacy, feasibility and indications for use, cost and cost-effectiveness, as well as social, economic and ethical consequences.”87 |
Comparator | “In clinical research, a technology or intervention that serves as a reference in comparative studies. In [health technology assessment and] health economics, an option with which the technology or intervention evaluated is compared.”87 |
Effectiveness | The effect of a drug (or other technology) observed under routine conditions (in contrast to efficacy). |
Efficacy | The effect of a drug (or other technology) observed under ideal conditions, such as a clinical trial (in contrast to effectiveness). |
Pivotal trial | A study designed to support the efficacy and safety of a drug for a regulatory submission. |
Real-world data (RWD) | Data relating to patient status and/or the delivery of health care collected from a variety of sources, and can include electronic medical records, clinical and disease registries, and administrative databases.88 |
Real-world evidence (RWE) | Evidence on the use, safety, effectiveness, and cost of health technologies that is derived from real-world data.88 |
Reimbursement review | Reimbursement Reviews performed by CDA-AMC are comprehensive assessments of the clinical effectiveness and cost-effectiveness, as well as patient and clinician perspectives, of a drug or drug class. The assessments inform nonbinding recommendations that help guide the reimbursement decisions of Canada's federal, provincial, and territorial governments, with the exception of Quebec.28 |
Single-arm study | “An analysis or evaluation of a study with only one branch, i.e., a trial in which there was no parallel comparison group and all the subjects received the same intervention.”87 |
Appendix 2: Excluded Studies
Please note that this appendix has not been copy-edited.
Table 6: Excluded Studies From the Literature Search
Reference | Reason for Exclusion |
|---|---|
Aldukain M, Aldukain A, Hobani A, et al. The Impact of Nusinersen Treatment on Respiratory Function in Patients with Spinal Muscular Atrophy: A Systematic Review. J Clin Med. 2024; 13(21):6306. |
|
Alhamadani F, Zhang K, Parikh R, et al. Adverse Drug Reactions and Toxicity of the Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab Dispos. 2022; 50(6):879-887. |
|
Alves BKAMF, Araujo APQC, Santos FND, Ribeiro MG. Type-1 spinal muscular atrophy cohort before and after disease-modifying therapies. Coorte de atrofia muscular espinhal tipo 1 antes e depois de terapias modificadoras da doença. Arq Neuropsiquiatr. 2024; 82(11):1-8. doi:10.1055/s-0044-1791757 |
|
Ashrafi MR, Babaee M, Hashemi Nazari SS, et al. Comparative efficacy of risdiplam and nusinersen in Type 2 and 3 spinal muscular atrophy patients: A cohort study using real-world data. J Neuromuscul Dis. 2024;11(6):1190-1199. |
|
Badina M, Bejan GC, Sporea C, et al. Changes in pNFH Levels in Cerebrospinal Fluid and Motor Evolution after the Loading Dose with Nusinersen in Different Types of Spinal Muscular Atrophy. Medicina. 2023; 59(7):1244. |
|
Bahadır Şenol H, Yıldız G, Polat Aİ, et al. Safety and Efficacy of Nusinersen Focusing on Renal and Hematological Parameters in Spinal Muscular Atrophy. Brain Behav. 2025 Jan;15(1):e70221. |
|
Bieniaszewska A, Sobieska M, Steinborn B, Gajewska E. Examination of Upper Limb Function and the Relationship with Gross Motor Functional and Structural Parameters in Patients with Spinal Muscular Atrophy. Biomedicines. 2023; 11(4):1005. |
|
Castellana E. Switching from Nusinersen to Risdiplam in Spinal Muscular Atrophy: A Comparative Analysis of Safety, Efficacy, and Economic Impact. Hosp Pharm. Published online September 22, 2024. |
|
Cavaloiu B, Simina I-E, Vilciu C, et al. Nusinersen Improves Motor Function in Type 2 and 3 Spinal Muscular Atrophy Patients across Time. Biomedicines. 2024; 12(8):1782. |
|
Chen B, Gong Y, Zhou T. The Impact of Nusinersen and Risdiplam on Motor Function for Spinal Muscular Atrophy Type 2 and 3: A Meta-Analysis. J Coll Physicians Surg Pak. 2024;34(8):948-955. |
|
Chiriboga CA, Bruno C, Duong T, et al. JEWELFISH: 24-month results from an open-label study in non-treatment-naïve patients with SMA receiving treatment with risdiplam. J Neurol. 2024;271(8):4871-4884. |
|
Chiriboga CA, Bruno C, Duong T, et al. Risdiplam in Patients Previously Treated with Other Therapies for Spinal Muscular Atrophy: An Interim Analysis from the JEWELFISH Study [published correction appears in Neurol Ther. 2023 Oct;12(5):1799-1801. |
|
Chongmelaxme, B., Yodsurang, V., Vichayachaipat, P. et al. Gene-based therapy for the treatment of spinal muscular atrophy types 1 and 2: a systematic review and meta-analysis. Gene Ther (2024).27:27 |
|
Coratti G, Civitello M, Rohwer A, et al. Changes in abilities over the initial 12 months of nusinersen treatment for type II SMA. Neuromuscul Disord. 2024;41:42 to 50. doi:10.1016/j.nmd.2024.05.003 |
|
Cordts I, Fuetterer C, Wachinger A, et al. Long-Term Dynamics of CSF and Serum Neurofilament Light Chain in Adult Patients With 5q Spinal Muscular Atrophy Treated With Nusinersen. Neurology. 2025 Mar 11;104(5):e213371. |
|
De Albuquerque ALA, Chadanowicz JK, Bevilacqua IP, et al. Clinicogenetic characterization and response to disease-modifying therapies in spinal muscular atrophy: real-world experience from a reference center in Southern Brazil. Jornal de Pediatría. 2025;101(1):38-45. |
|
Diener HC. Spinal muscular atrophy: Long-term efficacy and safety of nusinersen in adults with 5q-SMA. [German]. Psychopharmakotherapie. 2024 31(3):111-112. |
|
Dosi C, Masson R. The impact of three SMN2 gene copies on clinical characteristics and effect of disease-modifying treatment in patients with spinal muscular atrophy: a systematic literature review. Front Neurol. 2024;15:1308296. |
|
Elsheikh B, Severyn S, Zhao S, et al. Safety, Tolerability, and Effect of Nusinersen Treatment in Ambulatory Adults With 5q-SMA. Front Neurol. 2021;12:650535. |
|
Erdos J, Wild C. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: A systematic review of real-world study data. Eur J Paediatr Neurol. 2022;39:1-10. |
|
Freigang M, Steinacker P, Wurster CD, et al. Glial fibrillary acidic protein in cerebrospinal fluid of patients with spinal muscular atrophy. Ann Clin Transl Neurol. 2022;9(9):1437-1448. |
|
Gavriilaki M, Moschou M, Papaliagkas V, et al. Nusinersen in Adults with 5q Spinal Muscular Atrophy: a Systematic Review and Meta-analysis. Neurotherapeutics. 2022;19(2):464-475. |
|
Giess D, Erdos J, Wild C. An updated systematic review on spinal muscular atrophy patients treated with nusinersen, onasemnogene abeparvovec (at least 24 months), risdiplam (at least 12 months) or combination therapies. Eur J Paediatr Neurol. 2024;51:84-92. |
|
Hagenacker T, Maggi L, Coratti G, et al. Effectiveness of Nusinersen in Adolescents and Adults with Spinal Muscular Atrophy: Systematic Review and Meta-analysis. Neurol Ther. 2024;13(5):1483-1504. |
|
Hahn A, Günther R, Ludolph A, et al. Short-term safety results from compassionate use of risdiplam in patients with spinal muscular atrophy in Germany [published correction appears in Orphanet J Rare Dis. 2022 Oct 25;17(1):387. |
|
Jira K, Jaworek A, Allen M, et al. The association between gait speed and falls in ambulatory adults with spinal muscular atrophy: a retrospective pilot study. Front Neurol. 2024;15:1491466. |
|
Kant-Smits K, Bartels B, van der Heiden L, et al. The Effect of Disease-Modifying Therapies on Lung Function and Respiratory Muscle Strength in Spinal Muscular Atrophy: Systematic Review and Meta-Analysis. Respir Care. 2025;70(3):337-348. |
|
Kwon JM, Arya K, Kuntz N, et al. An expanded access program of risdiplam for patients with Type 1 or 2 spinal muscular atrophy. Ann Clin Transl Neurol. 2022;9(6):810-818. |
|
Lefeuvre C, Brisset M, Sarlon M, et al. Nusinersen treatment in adults with severe spinal muscular atrophy: A real-life retrospective observational cohort study. Rev Neurol (Paris). 2022;178(3):234-240. |
|
Leibrock, B., Landfeldt, E., Hussong, J. et al. Areas of improvement in the medical care of SMA: evidence from a nationwide patient registry in Germany. Orphanet J Rare Dis. 2023;18(1):32 |
|
Lilien C, Vrscaj E, Thapaliya G, et al. Patients' Perceptions of Nusinersen Effects According to Their Responder Status. J Clin Med. 2024;13(12):3418. |
|
McGrattan K, Walsh K, Mehl L, Kaur S, Dilly KW. Systematic literature review of the impact of spinal muscular atrophy therapies on bulbar function. Journal of Neuromuscular Diseases. 2024;12(2):195-217. |
|
Mendonça RH, Ortega AB, Matsui C Jr, et al. Gene replacement therapy for spinal muscular atrophy: safety and preliminary efficacy in a Brazilian cohort. Gene Ther. 2024;31(7-8):391-399. |
|
Mercuri E, Baranello G, Boespflug-Tanguy O, et al. Risdiplam in types 2 and 3 spinal muscular atrophy: A randomised, placebo-controlled, dose-finding trial followed by 24 months of treatment. Eur J Neurol. 2023;30(7):1945-1956. |
|
Mercuri E, Deconinck N, Mazzone ES, et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial [published correction appears in Lancet Neurol. 2022 Feb;21(2):e2. |
|
O'Brien K, Nguo K, Yiu EM, Woodcock IR, Billich N, Davidson ZE. Nutrition outcomes of disease modifying therapies in spinal muscular atrophy: A systematic review. Muscle Nerve. 2024;70(5):890-902. |
|
Oskoui M, Day JW, Deconinck N, et al. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA) [published correction appears in J Neurol. 2023 May;270(5):2547-2549. |
|
Osmanovic A, Abu-Fares O, Gotz F, et al. Nusinersen in spinal muscular atrophy (SMA): Clinical application in adult patients with SMA type II. [German]. Nervenheilkunde. 2019;38(6):409-414. |
|
Paik J. Risdiplam: A Review in Spinal Muscular Atrophy. CNS Drugs. 2022;36(4):401 to 410. |
|
Pascual-Morena C, Martínez-Vizcaíno V, Cavero-Redondo I, et al. Efficacy of risdiplam in spinal muscular atrophy: A systematic review and meta-analysis. Pharmacotherapy. 2024;44(1):97-105. |
|
Pickl S. Spinal muscular atrophy: Experiences in the use of nusinersen in children and adults. [German]. Psychopharmakotherapie. 2021;28(5):220-221 |
|
Qiao Y, Chi Y, Gu J, Ma Y. Safety and Efficacy of Nusinersen and Risdiplam for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Brain Sci. 2023;13(10):1419. |
|
Ribero VA, Daigl M, Martí Y, et al. How does risdiplam compare with other treatments for Types 1-3 spinal muscular atrophy: a systematic literature review and indirect treatment comparison. J Comp Eff Res. 2022;11(5):347-370. |
|
Sahashi K, Hashizume A, Kuwatsuka Y, et al. The Japan Registry for Adult Subjects of Spinal Muscular Atrophy (jREACT-SMA): Protocol for a Longitudinal Observational Study. JMIR Res Protoc. 2022;11(12):e38878. |
|
Severa G, Alfaro MDC, Alimi Ichola C, et al. Risdiplam: therapeutic effects and tolerability in a small cohort of 6 adult type 2 and type 3 SMA patients. Orphanet J Rare Dis. 2024;19(1):430. |
|
Sollner B. Risdiplam - The first oral drug for the treatment of spinal muscular atrophy can compensate for the lack of SMN protein. [German]. Journal fur Pharmakologie und Therapie. 2024;33(2):40-43. |
|
Tachibana Y, Sato R, Makioka H, Hoshino M, Jin M. Safety and effectiveness of nusinersen, a treatment for spinal muscular atrophy, in 524 patients: results from an interim analysis of post-marketing surveillance in Japan. Int J Neurosci. 2024;134(11):1185-1197. |
|
Tachibana Y, Takasaki S, Hoshino M, Makioka H, Jin M. Real-world safety and effectiveness of nusinersen, a treatment for spinal muscular atrophy, in 401 Japanese patients: results from an interim analysis of post-marketing surveillance. Int J Neurosci. 2024;134(2):153-162. |
|
Valancius M, Pang H, Zhu J, Cole SR, Jonsson Funk M, Kosorok MR. A causal inference framework for leveraging external controls in hybrid trials. Biometrics. 2024;80(4):ujae095. |
|
Vázquez-Costa JF, Povedano M, Nascimiento-Osorio AE, et al. Validation of motor and functional scales for the evaluation of adult patients with 5q spinal muscular atrophy. Eur J Neurol. 2022;29(12):3666-3675. |
|
Wadman RI, van der Pol WL, Bosboom WM, et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2020 Jan 6;1(1):CD006282. |
|
Yaftian F, Mobinizadeh M, Olyaeemanesh A, et al. Can SMA Innovative Treatments Be Reimbursed? A Rapid Review. Iranian Journal of Pediatrics. 2024;34(5) (no pagination) |
|
Yang M, Awano H, Tanaka S, et al. Systematic Literature Review of Clinical and Economic Evidence for Spinal Muscular Atrophy. Adv Ther. 2022;39(5):1915-1958. |
|
Zhao X, Liao Y, Zhao J, et al. Motor Function and Safety of Nusinersen and Risdiplam in Asian Patients with Types 2-4 Spinal Muscular Atrophy (SMA): A Systematic Review and Meta-Analysis. Adv Ther. 2025;42(4):1611-1626. |
|
Zhuang W, Lu M, Wu Y, et al. Safety Concerns with Nusinersen, Risdiplam, and Onasemnogene Abeparvovec in Spinal Muscular Atrophy: A Real-World Pharmacovigilance Study. Clin Drug Investig. 2023;43(12):949-962. |
|
ITC = indirect treatment comparison; PD = pharmacodynamic; PK = pharmacokinetic; RCT = randomized controlled trial; SMA = spinal muscular atrophy.
aIncluded studies were not eligible for this review.
bIncluded studies previously assessed in CDA-AMC reimbursement reviews of nusinersen and/or risdiplam and not eligible for inclusion in this review.
cThe ITC used data in patients aged < 10 years only.
Table 7: Excluded Studies From the Grey Literature Search (ClinicalTrials.Gov)
Reference | Reason for Exclusion |
|---|---|
Nusinersen | |
An Open-Label Extension Study for Patients With Spinal Muscular Atrophy Who Previously Participated in Investigational Studies of ISIS 396443. National Library of Medicine (US). |
|
A Phase 2, Randomized, Double-blind, Sham-procedure Controlled Study to Assess the Safety and Tolerability and Explore the Efficacy of ISIS 396443 (BIIB058) Administered Intrathecally in Subjects With Spinal Muscular Atrophy Who Are Not Eligible to Participate in the Clinical Studies ISIS 396443-CS3B or ISIS 396443-CS4. National Library of Medicine (US). |
|
A Phase 3, Randomized, Double-blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Later-onset Spinal Muscular Atrophy. National Library of Medicine (US). |
|
A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile-onset Spinal Muscular Atrophy. National Library of Medicine (US). |
|
A Study to Assess the Efficacy, Safety, Tolerability, and Pharmacokinetics of Multiple Doses of ISIS 396443 Delivered Intrathecally to Patients With Infantile-Onset Spinal Muscular Atrophy. National Library of Medicine (US). |
|
An Open-Label, Dose Escalation Study to Assess the Safety, Tolerability and Dose-Range Finding of Multiple Doses of ISIS 396443 Delivered Intrathecally to Patients With Spinal Muscular Atrophy. National Library of Medicine (US). |
|
Risdiplam | |
A Phase I, 2-Part, Open-Label Study to Investigate the Safety, Tolerability, and Pharmacokinetics of Multiple Doses of Risdiplam and the Effect of Risdiplam on the Pharmacokinetics of Midazolam Following Oral Administration in Healthy Patients. National Library of Medicine (US). |
|
An Open-Label, Single-Dose, Parallel-Group, Two-Part Study to Evaluate the Pharmacokinetics and Safety of Risdiplam in Subjects With Mild or Moderate Hepatic Impairment Compared to Subjects With Normal Hepatic Function. National Library of Medicine (US). |
|
An Open-Label Study of Risdiplam in Infants With Genetically Diagnosed and Presymptomatic Spinal Muscular Atrophy. National Library of Medicine (US). |
|
A Two Part Seamless, Open-label, Multicenter Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of Risdiplam (RO7034067) in Infants With Type 1 Spinal Muscular Atrophy. National Library of Medicine (US). |
|
A Two Part Seamless, Multi-Center Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of Risdiplam (RO7034067) in Type 2 and 3 Spinal Muscular Atrophy Patients. National Library of Medicine (US). |
|
CDA-AMC = Canada’s Drug Agency; ITC = indirect treatment comparison; PD = pharmacodynamic; PK = pharmacokinetic; RCT = randomized controlled trial; SMA = spinal muscular atrophy.
Table 8: Excluded Studies Identified in the Request to CDA-AMC
Reference | Reason for Exclusion | Notes and Commentsa |
|---|---|---|
Bjelica, B., Wohnrade, C., Cespedes, I. et al. Risdiplam therapy in adults with 5q-SMA: observational study on motor function and treatment satisfaction. BMC Neurol. 2024;24:67. | Excluded at abstract screening.
|
|
Brakemeier S, Lipka J, Schlag M, et al. Risdiplam improves subjective swallowing quality in non-ambulatory adult patients with 5q-spinal muscular atrophy despite advanced motor impairment. J Neurol. 2024 May;271(5):2649-2657. | Excluded at abstract screening.
|
|
Chiriboga CA, Bruno C, Duong T, et al. JEWELFISH: 24-month results from an open-label study in non-treatment-naïve patients with SMA receiving treatment with risdiplam. J Neurol. 2024;271(8):4871-4884. | Excluded at full-text screening.
|
|
Guittari C, Candrilli S, Miles L, et al. P09 Real-world treatment with risdiplam in adults with spinal muscular atrophy (SMA): a multicenter study. Neuromuscular Disorders. 2023;33(Suppl 1):S164 | Conference abstracts were not eligible for this review and were not identified during literature screening. |
|
Günther R, Wurster CD, Brakemeier S, et al. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. Lancet Reg Health Eur. 2024;6;39:100862. | Excluded at abstract screening.
|
|
Kessler T, Sam G, Wick W, Weiler M. Evaluation of risdiplam efficacy in 5q spinal muscular atrophy: A systematic comparison of electrophysiologic with clinical outcome measures. Eur J Neurol. 2024;31(1):e16099. | Excluded at abstract screening.
|
|
Maggi L, Bello L, Bonanno S, et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. 2020;91(11):1166-1174. | Already reviewed by CDA-AMC (SR0713). Published before 2021. |
|
McCluskey G, Lamb S, Mason S, et al. Risdiplam for the treatment of adults with spinal muscular atrophy: Experience of the Northern Ireland neuromuscular service. Muscle Nerve. 2023;67(2):157-161. | Excluded at abstract screening.
|
|
Nanni AG, Milella G, Piccirilli G, et al. Preliminary data on safety and efficacy of risdiplam treatment in a small cohort of adult 5q spinal muscular atrophy. Acta Myol. 2023;42(1 Suppl 1):41-113. | Conference abstracts were not eligible for this review and were not identified during literature screening. |
|
Ñungo Garzón NC, Pitarch Castellano I, Sevilla T, Vázquez-Costa JF. Risdiplam in non-sitter patients aged 16 years and older with 5q spinal muscular atrophy. Muscle Nerve. 2023 May;67(5):407-411. | Excluded at abstract screening.
|
|
Schön M, Domingues S, Moreno T, Oliveira Santos M. A severely affected adult type 2 spinal muscular atrophy patient treated with risdiplam. Neurol Sci. 2023;44(4):1449-1450. | Excluded at abstract screening.
|
|
Severa G, Alfaro MDC, Alimi Ichola C, et al. Risdiplam: therapeutic effects and tolerability in a small cohort of 6 adult type 2 and type 3 SMA patients. Orphanet J Rare Dis. 2024;19(1):430. | Excluded at full-text screening.
|
|
Sitas B, Hancevic M, Bilic K, et al. Risdiplam Real World Data - Looking Beyond Motor Neurons and Motor Function Measures. J Neuromuscul Dis. 2024;11(1):75-84. | Excluded at abstract screening.
|
|
Zoppi D, Bencivenga RP, Nevano A, et al. Risdiplam in type 2 and 3 Spinal Muscular Atrophy: results of a cohort of adult Italian patients. Acta Myol. 2022;41(3 Suppl 1):31-84. | Conference abstracts were not eligible for this review and were not identified during literature screening. |
|
6MWT = 6-minute walk test; EK2 = Egen Klassifikation Scale Version 2; ppFVC = percentage predicted forced vital capacity; HFMSE = Hammersmith Functional Motor Scale – Expanded; HRQoL = health-related quality of life; IQR = interquartile range; MFM32 = 32-item Motor Function Measure; PD = pharmacodynamic; PK = pharmacokinetic; RCT = randomized controlled trial; RHS = Revised Hammersmith score; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
aAll of the studies had noncomparative designs and was the primary reason for exclusion from the review and the key limitation in study design.
Appendix 3: Selection of Included Studies
Figure 1: PRISMA31 Flow Chart of Study Selection
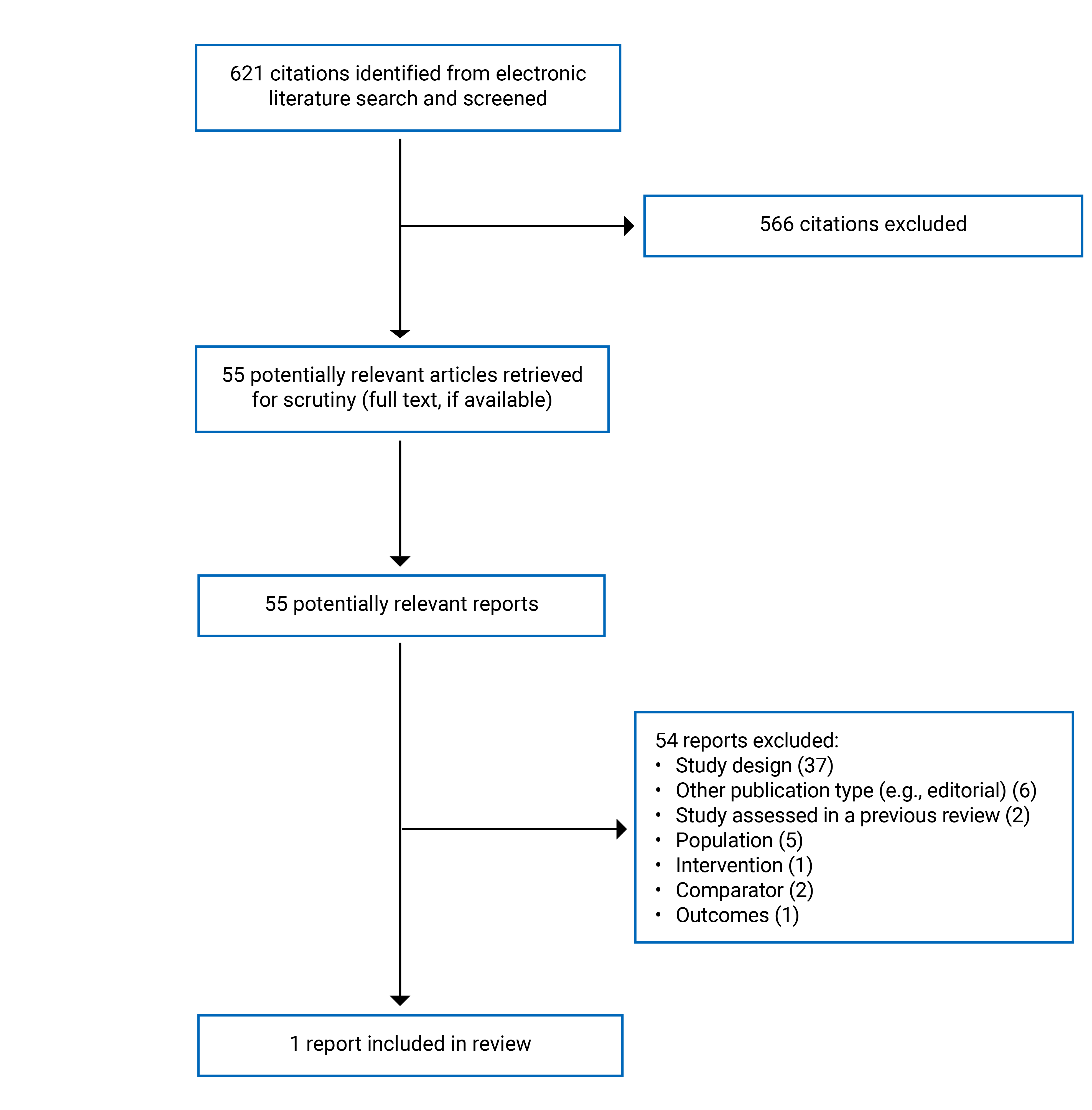
Appendix 4: Detailed Information and Critical Appraisal of the Included Study
Please note that this appendix has not been copy-edited.
Study Design
The Vázquez-Costa et al. study, published in 2022, was a prospective, observational, comparative cohort study conducted at 5 hospitals in Spain. Adolescent and adult patients with genetically confirmed 5q SMA were divided into 2 groups based on treatment status: those who initiated nusinersen and those who remained untreated. Outcomes were assessed at baseline and at 6 months and the end of study visit for each patient using overall function, motor function, and respiratory function measures. The study performed statistical comparisons between groups, adjusting for baseline scores, allowing evaluation of longer-term effectiveness of nusinersen under real-world conditions.
The study characteristics are summarized in Table 9.
Table 9: Characteristics of the Included Study
Study citation, country, funding source | Study design | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
Vázquez-Costa et al. (2022) 5 hospitals in Spain Funding source: Mixed sources: regional and European rare disease organizations and foundations, government research programs of Spain. No pharmaceutical industry funding | Prospective, comparative observational study using real-world data from 5 hospitals. | Eligibility criteria: Individuals older than 15 years of age with genetically confirmed SMA (either homozygous deletion or compound heterozygous mutation in SMN1). Nusinersen treated N = 39 Non-treated N = 40 Baseline characteristics
| Intervention: Nusinersen intrathecal injection: 12 mg loading doses at days 0, 14, 28 and 65 and maintenance doses every 4 months, as per product monograph in Spain. Comparator: No treatment with nusinersen. All patients received the same multidisciplinary care in their respective treatment centres. | Primary Effectiveness Outcomes Change from baseline to 6 months in:
Secondary Effectiveness Outcomes Change from baseline to 6 months in:
Harms Outcomes: Frequency of patients-reported adverse events Follow-up (months):
|
6MWT = 6-minute walk test; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Scale Revised; EK2 = Egen Klassifikation Scale Version 2; ppFVC = percentage predicted forced vital capacity; HFMSE = Hammersmith Functional Motor Scale – Expanded; IQR = interquartile range; NIV = non- invasive ventilation; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
aSMA type IIa: those who were able to sit unsupported, but not to stand or walk with help. SMA type IIb: those who were able to stand or walk with help. SMA type IIIa: those able to walk without help, in whom the disease started before 36 months of age. SMA type IIIb: those able to walk without help, in whom the disease started after 36 months of age.
bWalkers were defined as those able to walk at least 5 steps without assistance. Sitters were defined as those able to sit without assistance and support their head for more than 10 seconds.
cSevere scoliosis was defined as having a greater than 45°Cobb angle and/or previously had surgery for scoliosis.
Source: Vázquez-Costa et al.32
Country of Origin
The Vázquez-Costa et al. study was conducted in Spain.
Patient Population
In the nusinersen-treated group (N = 39), 51% of patients were male, the mean age was 33 (standard deviation [SD] = 13) years, and most patients (74%) had type III SMA at baseline. Approximately 67% were non-ambulatory, and 59% had 3 copies of the SMN2 gene. Just over half of treated patients had severe scoliosis (51%). Baseline mean HFMSE score was 25.9 (SD = 20.1), RULM was 20.6 (SD = 11.0), and among walking patients, the 6MWT mean was 269.8 (SD = 123.4) metres.
In the untreated group (N = 40), 43% of patients were male, with a mean age of 30 (SD = 14) years, and 43% had type III SMA. Seventy-three percent (73%) were non-ambulatory, and 68% had 3 SMN2 copies. Greater than two-thirds (68%) of untreated patients had severe scoliosis. Baseline mean HFMSE was 30.0 (SD = 25.5), RULM was 18.3 (SD = 14.4), and ambulatory patients had a 6MWT mean of 432.4 (SD = 127.6) metres.
Interventions and Comparators
Nusinersen was administered as the intervention in the exposed group, while the comparator group did not receive nusinersen during the study period. The study reported that nusinersen was administered as 12 mg loading doses on days 0, 14, 28, and 65, followed by maintenance doses every 4 months. Specific information regarding concomitant or supportive therapies, or patients’ previous treatment history, was not provided.
Outcomes
Six effectiveness outcome measures relevant to this review were reported in the Vázquez-Costa et al. study:
Hammersmith Functional Motor Scale – Expanded (HFMSE):89-91 The HFMSE assesses motor function in patients with SMA types II and III. It was primarily developed for pediatric patients, especially those least able to sit. It consists of 33 items scored on a 3-point scale (0 to 2), with total scores ranging from 0 to 66. Higher scores indicate better motor function. The HFMSE has shown moderate to strong correlations (Spearman r > 0.40) on measures of validity and reliability in children and adolescents with SMA. In adults, its use is more limited. While some studies suggest it may be feasible and responsive, there is limited empirical evidence on its measurement properties in this population. Moreover, the scale may be subject to floor and ceiling effects, limiting its sensitivity to detect change in adults with either very limited or near-normal function. An MID of greater than 2 points is commonly cited in pediatric populations, but this has not been directly estimated in adults with SMA and is typically extrapolated.92
Revised Upper Limb Module (RULM):89-91,93,94 The RULM evaluates upper limb function in individuals with SMA and includes 20 items scored from 0 to 37. Higher scores indicate better function. The RULM has strong (Spearman r > 0.70) content validity, inter- and intra-rater reliability, and responsiveness in children and adults with SMA. It is especially useful for non-ambulatory adults with SMA types II and III. However, it may also demonstrate ceiling effects in stronger individuals and floor effects in those with severe weakness, reducing its sensitivity to change at the extremes of function. Additionally, performance-based assessments like the RULM may be influenced by fatigue, compensatory movements, or variability in effort. A MID of 2 points has been suggested based on distribution methods and clinical judgment but has not been validated specifically for adults with SMA.95
6-Minute Walk Test (6MWT):89,93,96,97 The 6MWT measures functional exercise capacity and endurance by asking individuals to walk as far as possible in 6 minutes. Higher distances indicate better function. It has demonstrated validity, reliability, and responsiveness in ambulatory individuals with SMA, including adults. However, the test can be limited by fatigue, motivation, comorbidities, and orthopedic deformities such as scoliosis or contractures. It may not capture short-term or subtle treatment effects and is not applicable to non-ambulatory individuals. There is also potential variability in test administration and pacing, especially in community settings. While a 30-metre MID and a 5% to10% change from baseline are commonly cited,16 these thresholds are not empirically derived for adults with SMA.
Egen Klassifikation Scale Version 2 (EK2):89,98 The EK2 is a self-reported functional scale developed for non-ambulatory individuals with SMA, assessing mobility, respiratory, bulbar, and daily function. Scores range from 0 to 51, with lower scores indicating better function. It has shown strong correlations (Spearman r > 0.60) for content validity, internal consistency, and responsiveness in non-ambulatory adolescents and adults. However, as a self-reported measure, it may be subject to recall bias, subjectivity, and ceiling effects in patients with higher function. It may also be less responsive in detecting short-term or subtle treatment-related changes. A validated MID has not been established in any population. A threshold of ≥ 2 points was used in the Vázquez-Costa et al. study for responder analyses.
Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R):89,99 The ALSFRS-R includes 12 items assessing bulbar, fine motor, gross motor, and respiratory function (score range: 0 to 48). Higher scores indicate better function. Though not specific to SMA, it has been used in SMA studies involving adults and has demonstrated moderate feasibility and responsiveness. However, it may not be well suited to the unique progression and distribution of muscle weakness in SMA, especially since it was designed for a different disease (amyotrophic lateral sclerosis [ALS]) that progresses more rapidly. Some items may show limited variability or relevance in SMA populations, and the scale may lack sensitivity to detect small functional changes. There is no validated MID in SMA. A threshold of ≥ 2 points was used in the Vázquez-Costa et al. study for responder analyses.
Percent predicted forced vital capacity (ppFVC):89 ppFVC is a measure of respiratory function expressed as a percentage of the predicted value based on age, sex, height, and race. Higher values indicate better pulmonary function. It is widely used to monitor disease progression in SMA and has good test-retest reliability. However, ppFVC has several limitations, particularly in adults with SMA. First, body position and musculoskeletal complications (e.g., scoliosis) can make consistent positioning difficult and potentially lead to under- or overestimation of respiratory function. Second, prediction equations are based on reference populations (age, sex, height) that assume typical growth and development. The predicted values may be invalid due to short stature or contractures, misrepresenting true respiratory function. Third, patients with bulbar weakness, fatigue, or limitations in coordination may have suboptimal or inconsistent performance. A specific MID has not been defined in SMA, but changes of 5% to 10% are often used based on studies in other neuromuscular conditions. A threshold of was not specified in the Vázquez-Costa et al. study and no responder analyses were conducted for the change in ppFVC.
The number of patients who started noninvasive ventilatory support or required placement of a gastrostomy tube were also reported.
Other effectiveness outcomes of interest to this review — overall survival, event-free survival, hospitalization, symptom severity, health-related quality of life, and caregiver burden — were not reported.
Harms
The frequencies of patient-reported adverse events were recorded and categorized by severity and relationship to treatment according to MedDRA (version 21.1).
Summary of Critical Appraisal
Additional details regarding the strengths and limitations of the included publication are provided in Table 10.
Risk of Bias Due to Confounding
Confounding by disease severity and disease characteristics (e.g., number of SMN2 copies), baseline function, comorbidities, previous and concurrent treatments, and age is highly likely, particularly in the absence of randomization. The authors reported adjusting regression models estimating functional score-based treatment effect at 6 months for baseline scores of the functional scales in their analyses. For the analysis of treatment effects at the last assessment, regression models were further adjusted for time on treatment by including follow-up duration and its interaction with the treatment term. While these statistical adjustments help mitigate confounding from these specific factors, they do not account for other clinically important confounders that were omitted from the models and not addressed through other methodological approaches. No adjustments were made for socioeconomic factors that may influence access to care, or for treatment adherence, both of which could be systematically associated with treatment assignment and outcomes. Furthermore, aside from the inclusion of covariates to account for varying follow-up times at the final assessment, no other time-varying confounding factors were considered. Additionally, there was considerable heterogeneity in baseline characteristics between the treated and untreated groups (e.g., differences in functional status), as well as substantial within-group variability, as evidenced by the large SDs for mean values and wide interquartile ranges for medians. The magnitude of this baseline heterogeneity both between and within groups increases the risk of bias due to confounding and may have meaningfully influenced the observed treatment effects. Thus, there is a serious risk of bias from confounding.
Risk of Bias From Selection of Patients
Patients were recruited from 5 hospital-based referral centres in Spain and were considered eligible for nusinersen treatment based on national reimbursement criteria. However, the study did not clearly define eligibility for nusinersen. It noted that access restrictions excluded individuals with the most severe forms of SMA (SMA types 0 and IA) and the mildest form (type IV). According to the criteria of the Agencia Española de Medicamentos y Productos Sanitarios,100 patients diagnosed with 5q SMA were eligible, excluding types 0, IA, and IV. The study stated that “patients meeting the criteria established by the health department were routinely offered nusinersen treatment. The final decision to start the treatment was made by the patient after discussion with the neurologist….” While this shared decision-making process may reflect real-world clinical practice, it introduces the potential for selection bias, as the included population may have been systematically different from the broader population of individuals with SMA. Patients who were more motivated or who had access to a support system, who had greater preservation of motor functionality, or who were perceived to be more likely to benefit from nusinersen may have been more likely to initiate treatment.
Selection bias is further suggested by the recruitment of patients through specialized referral centres, raising the possibility of referral or survivor bias (i.e., patients in better health being more likely to present for or be considered for treatment). Baseline characteristics support the presence of systematic differences between groups. Compared to treated patients, untreated patients were more frequently classified as SMA type II (50% versus 25%), non-sitters (defined as those unable to sit without assistance nor head support for more than 10 seconds; 50% versus 26%), had severe scoliosis (68% versus 51%), and used noninvasive ventilation (38% versus 23%), despite a shorter mean disease duration (25 versus 29 years). Baseline scores on functional outcome scales also generally indicated poorer function in the untreated group, except for the HFMSE (not assessed in non-sitters) and the 6MWT (none of the patients with SMA type IV were treated). These baseline imbalances suggest that patients treated with nusinersen were in better clinical condition at study entry, which may bias the estimated treatment effects. Overall, the patient selection process raises a moderate to serious risk of bias due to the potential for systematic differences in prognosis between the treated and untreated groups.
Risk of Bias in Classification of Interventions
There is a low risk of bias related to the classification of the intervention (nusinersen). The classification of patients into treated and untreated groups was clear. There is no indication of misclassification.
Bias Due to Deviations From Intended Interventions
Deviations from the intended intervention may have occurred if patients failed to complete the full nusinersen loading or maintenance dosing schedule due to clinical deterioration, adverse events, or barriers to access. The study attempted to address this issue by reporting the mean number of doses received, which was 6. However, this figure was presented without context — such as the SD, range, or the expected total number of doses — given the length of follow-up. Based on a reported mean follow-up of approximately 16 months (median 15 months), patients would be expected to have received at least 7 doses. The lack of detailed information on dosing completeness limits interpretability and raises concerns about differential adherence.
The dosage of nusinersen used in the study was similar to that approved in the EU and Canada;9,101 however, the final loading dose day was 2 days later in the study (day 65) compared to the recommended regimen as per the product monograph (day 63). No rationale for this was reported, although it is not expected to influence results.
Adherence to therapy was not explicitly reported, and the reporting of adverse events was limited. Although the discussion section noted that treatment was “temporarily discontinued in 7.7% of patients” due to adverse events, no further details were provided regarding the timing, duration, or impact of these discontinuations. This omission introduces uncertainty about whether and to what extent deviations from intended treatment occurred, and whether such deviations may have introduced bias — particularly if they were related to patient prognosis.
The study also reported that all patients, whether treated or untreated, received multidisciplinary care within their respective clinics. While this reflects good clinical practice and enhances the generalizability of the findings — given the systemic nature of SMA — no specific information was provided about the content, frequency, or duration of such care. Moreover, this co-intervention was not measured or controlled for in the analysis, which leaves open the possibility of residual confounding due to unbalanced or evolving supportive care over time. Thus, there is a moderate risk of bias due to deviations from the intended intervention (nusinersen).
Bias Due to Missing Data
The study did not report the completeness of outcome data or describe the methods used to handle missing data. The extent of missingness, its distribution across groups, and whether it was associated with patient prognosis (e.g., sicker patients being more likely to drop out) was not addressed. This lack of information limits confidence in the internal validity of the findings, as outcome data that are not missing at random can introduce bias.
The Vázquez-Costa et al. study enrolled 79 patients — 39 in the nusinersen-treated group and 40 in the untreated comparator group. It was reported that 1 patient discontinued nusinersen after the second loading dose due to lack of lumbar access and was excluded from the effectiveness analyses. The patient’s baseline characteristics were not reported, preventing assessment of whether this exclusion may have introduced bias.
Several outcome measures were administered only to applicable subgroups, depending on patients’ physical capabilities. Based on the baseline distribution of sitters, non-sitters, and ambulatory patients, the expected analyzable populations were as follows:
RULM, ALSFRS-R, and ppFVC: up to 78 patients
HFMSE: 48 patients (sitters only)
6MWT: 24 patients (ambulatory only)
EK2: 54 patients (non-ambulatory only)
However, the actual sample sizes used in the analyses were substantially lower in several cases. For instance:
EK2 analyses included only 31 to 40 patients, depending on the analysis, compared with the 54 expected.
ALSFRS-R analyses showed large attrition: 78 patients were eligible, yet only 42 were included in the change-from-baseline to 6 months and responder analyses, and just 5 patients were included in the analysis of change to end of study.
No explanation was provided for these discrepancies, and the numbers of patients per treatment group were not reported. It was reported that, while all centres collected motor scale scores and pulmonary function test results, functional scale scores were missing from some centres. No specific information about the number of centres, the exact functional tests, or the amount of missing data were mentioned. This absence of detail hinders the ability to judge whether missing data were balanced between groups, random, or related to outcomes. The impact of this missing data is especially concerning given the relatively small sample size of the study, which magnifies the risk of biased or imprecise effect estimates. Overall, there is a serious risk of bias related to missing data.
Risk of Bias in Measurement of Outcomes
There is moderate risk of bias in the measurement of outcomes due to several important limitations. The outcome measures used — HFMSE, RULM, 6MWT, EK2, ALSFRS-R, and ppFVC — each have known limitations in adults with SMA:
HFMSE and RULM, while commonly used in SMA, were originally developed for pediatric populations and may have reduced responsiveness and ceiling or floor effects in adults, particularly in those at the extremes of motor function.
6MWT is only applicable to ambulatory patients, which limits its applicability across the heterogeneous population of adults with SMA, and results may be influenced by fatigue or motivation.
EK2 is a self-reported tool for non-ambulatory individuals, and is subject to recall bias.
ALSFRS-R, though validated in ALS, is not disease-specific to SMA and has not been systematically validated in this population.
ppFVC is widely used and generally reliable, but it is influenced by body positioning, which can influence effort and technique on what is a maximal effort test. Additionally, 50% (versus 26%) of untreated patients were non-sitters with moderate to severe respiratory impairment at baseline (mean ppFVC 60% versus 73%), which could seriously compromise their ability to perform pulmonary function testing.
Moreover, baseline scores on these measures varied widely across patients, reflecting significant heterogeneity in disease severity, which may influence observed changes. The study only adjusted for baseline scale scores, leaving residual confounding from other factors (i.e., disease duration, scoliosis, comorbidities, and respiratory support among others) unaccounted for.
It was reported that motor and functional scales were administered by experienced and/or trained neurologists and physiotherapists, and that the same evaluator was used for every patient throughout the study, if possible. However, no details on whether the investigators were successful in achieving this goal were provided, nor was there mention of assessments of consistency between evaluations if a different evaluator administered the scale.
It was also not explicitly stated whether outcome measurement and/or analysis was blinded to clinicians and study investigators to prevent systematic differences that could influence results and result reporting.
Additionally, missing data may further bias results. The handling of missing data was not described in sufficient detail to assess whether the assumptions made (e.g., missing at random) are reasonable. Given that patients with lower functionality may be less likely to complete certain assessments (e.g., 6MWT), data may not be missing at random, increasing the risk of bias.
Taken together, the combination of outcome measure limitations, patient heterogeneity with limited adjustment for confounding, and concerns about missing data suggest a moderate risk of bias in outcome measurement.
Risk of Bias in Selection of the Reported Results
There is concern for bias due to selective reporting of results. The study did not clearly indicate whether a publicly available protocol or a prespecified statistical analysis plan were developed. In the absence of such documentation, it is not possible to determine whether all prespecified outcomes were reported or whether the analytical approaches adhered to a predefined plan. This limits transparency and increases the risk of outcome reporting bias.
Although the authors reported several non-statistically significant findings, which reduces concerns about selective reporting based solely on statistical significance, there remains a risk of selective analysis reporting. Multiple outcome measures were used to assess similar constructs (e.g., motor function), including HFMSE, RULM, and 6MWT. While the use of multiple scales may be justified in adult patients with SMA due to the absence of a single comprehensive measure of motor function, this multiplicity increases the potential for selective emphasis on favourable outcomes.
Furthermore, different statistical approaches were applied across time points and outcomes (e.g., regression analyses at various time points, responder analyses), with no indication that these were prespecified. This analytic flexibility introduces risk of selective reporting of favourable analyses, especially in the context of a small sample size and limited statistical power.
Importantly, no adjustments were made for multiple comparisons, despite the large number of outcomes and statistical tests performed. Interpreting P values (e.g., P < 0.05) without appropriate correction increases the risk of inflated type I error, potentially leading to erroneous conclusions about treatment benefit. This concern is heightened by the small treatment effects, unaddressed confounding, and missing data discussed in other domains.
Finally, as noted elsewhere, incomplete reporting of key methodological details limits a thorough assessment of the validity and robustness of the results. Taken together, these issues indicate serious risk of bias in the selection of the reported results.
Table 10: Critical Appraisal of the Vázquez-Costa et al. Cohort Study Using the ROBINS-I Tool
Domain | Rating | Notes |
|---|---|---|
1. Bias due to confounding | Serious |
|
2. Bias in selection of patients | Moderate to serious |
|
3. Bias in classification of interventions | Low | The classification of patients into treated and untreated groups appears clear and appropriate. There is no indication of misclassification. |
4. Bias due to deviations from intended interventions | Moderate | Deviations could occur if patients did not complete the full loading or maintenance doses due to clinical decline, treatment adverse events, or access barriers. The study attempts to address this by reporting on the mean number of doses of nusinersen were received. It states that a mean of 6 doses was received but without context of the variance (e.g., standard deviation) or what the maximum number of doses were expected over the unspecific maximum follow-up duration. With a mean follow-up duration of approximately 16 months (median 15 months) patients would be expected to receive 7 doses. Adherence to therapy was not explicitly reported. It was not reported clearly in the adverse events results, but the discussion section of the study noted that adverse events “led to short-term treatment discontinuation in 7.7% of patients.”32 No further information about the duration of the treatment discontinuations in these patients was provided. The limited reporting on these scenarios undermines the validity of the results. In addition, the study reported that all patients, regardless of whether they were treated or not, received the same multidisciplinary care in their respective clinics. This improves the generalizability of the results as it is recommended that patients with SMA are cared for in a multidisciplinary manner given the broad systemic effects of the condition. However, no details about this care — nature, intensity, changes over the follow-up period, and so forth — were provided and it was not accounted for in the analysis. |
5. Bias due to missing data | Serious |
|
6. Bias in measurement of outcomes | Moderate |
|
7. Bias in selection of the reported results | Serious |
|
6MWT = 6-minute walk test; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Scale Revised; EK2 = Egen Klassifikation Scale Version 2; ppFVC = percentage predicted forced vital capacity; HFMSE = Hammersmith Functional Motor Scale – Expanded; MID = minimally important difference; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy.
Source: Vázquez-Costa et al.32
Summary of Findings
Clinical Effectiveness of Nusinersen
Vázquez-Costa et al. defined a statistically significant result based on a P value < 0.05.
Hammersmith Functional Motor Scale – Expanded
The difference between groups for the mean change from baseline to 6 months in the HFMSE was 2.0 (SE = 1.1) points higher with nusinersen treatment (P = 0.082) (Table 11). Responder analysis based on a patient’s last study assessment indicated that 25% patients treated with nusinersen compared with 10% of untreated patients improved with greater than 2 points change from baseline (P = 0.300). After adjusting for the baseline values and follow-up time, nusinersen treatment was associated with an improvement in HFMSE compared with no treatment (OR = 1.15; 95% confidence interval [CI], 1.04 to 1.27; P = 0.006).
Revised Upper Limb Module
The study reported a within-group mean change in RULM total score from baseline to 6 months of 1.7 (SD = 3.3) points with nusinersen treatment compared with –0.6 (SD = 2.3) among untreated patients, for a between-group difference of 2 (SE = 0.5) points (P < 0.001) (Table 12). The odds ratio for treatment effect at the last assessment was 1.02 (95% CI, 0.96 to 1.09). A 2-point or greater improvement occurred in 50% of treated and 23% of untreated patients at the last study assessment (P = 0.033).
6-Minute Walk Test
Among a subgroup of ambulatory patients assessed with the 6MWT, the difference in the mean distance walked at 6 months was –6.3 (SE = 46.1) metres shorter in the nusinersen-treated group compared with the untreated group (P = 0.894) (Table 13). The association favoured the treated group at the last study assessment (OR = 1.071; 95% CI, 1.065 to 1.078). A response, defined as a ≥ 30-metre improvement, was achieved by 75% of treated patients compared with 57% of untreated patients at the last assessment.
Egan Klassifikation Scale Version 2
Treated patients’ mean EK2 scores decreased (improved) –2.7 (SD = 2.7) points at 6 months while untreated patients’ mean scores increased (worsened) by 1.1 (SD = 2.8) points, for a between-group difference of –4.0 (SE = 3.3) points (Table 14). The odds ratio for the treatment effect was 0.81 (95% CI, 0.71 to 0.92; P = 0.001) as of the last assessment. A larger percentage of treated patients (80%) achieved the threshold for a clinically meaningful response versus untreated patients (23%; P < 0.001).
Amyotrophic Lateral Sclerosis Functional Rating Scale Revised
The mean difference between the groups in global functional capacity, as measured by the ALSFRS-R, was 3.4 (SD = 3.0) points in favour of the nusinersen-treated group compared with the untreated group at 6 months (Table 15). At the last assessment, treatment was associated with an odds ratio of 1.04 (95% CI, 0.94 to 1.14). None of the untreated patients achieved the clinician-based responder threshold of greater than or equal to 2 points while 26% of treated patients did (P = 0.005).
Percentage Predicted Forced Vital Capacity
The mean change in ppFVC from baseline to 6 months was 2.6% (SD = 8.3%) for treated patients and –1.1% (5.7%) for untreated patients (Table 16). The estimated between-group difference was 3.2% (SE = 2.1%) (P = 0.139). After adjusting for the baseline values and follow-up time, the odds ratio for the treatment effect of nusinersen compared with being untreated was 1.00 (95% CI, 0.90 to 1.12; P = 0.975). No responder analysis was reported for ppFVC.
Need for Noninvasive Ventilation
One patient in the nusinersen-treated group started noninvasive ventilation during follow-up, after a respiratory infection that required hospitalization. No patients in the untreated group required noninvasive ventilation.
Need for Gastrostomy
No patients in either group were reported as newly requiring placement of a gastrostomy tube.
Harms of Nusinersen
Adverse events were only reported for the nusinersen-treated group, of which 77% (30 of 39) had an event over the follow-up period. Most adverse events were related to the intrathecal administration of nusinersen (e.g., postlumbar puncture headache). Two events of urinary retention due to neurogenic bladder postadministration were reported. At least 1 of the events of neurogenic bladder was permanent; it was not explicitly stated if both events were permanent.
Table 11: Summary of Effectiveness Findings in the Included Study — HFMSE Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
HFMSE | ||
Baseline, N = 50 | N = NR | N = NR |
Mean (SD) total score | 25.9 (20.1) | 30.0 (25.5) |
Median (IQR) total score | 21 (7, 47) | 25 (4, 57) |
Change from baseline to 6 monthsa, N = 44 | N = NR | N = NR |
Mean change (SD) | 2.4 (4.5) | 0.2 (1.8) |
Estimated difference (SE) | 2.0 (1.1) | |
P value for the differenceb | 0.082 | |
Responder analysisc, N = 44 | N = NR | N = NR |
Percentage of patients with an improvement ≥ 3 points at the last visit | 25% | 10% |
P value for the differenceb | 0.300 | |
Odds ratio (95% CI) for the difference in treatment effect at the least visitd, e | 1.15 (1.04 to 1.27) | |
P value for the differenceb | 0.006 | |
CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale – Expanded; IQR = interquartile range; NR = not reported; SD = standard deviation; SE = standard error.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cThe percentages of treated and untreated patients who improved by at least the minimal important difference empirically established (or from clinician opinion) for the scale or end point. Chi-square tests were used to calculate the P values for the differences in percentages of responders.
dMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio > 1 favours the nusinersen-treated group.
eThe analysis was based on a sample of 50 patients.
Source: Vázquez-Costa et al.32
Table 12: Summary of Effectiveness Findings in the Included Study — RULM Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
RULM | ||
Baseline, N = 75 | N = NR | N = NR |
Mean (SD) total score | 20.6 (11.0) | 18.3 (14.4) |
Median (IQR) total score | 21 (12, 29) | 14 (5, 36) |
Change from baseline to 6 monthsa, N = 71 | N = NR | N = NR |
Mean change (SD) | 1.7 (3.3) | −0.6 (2.3) |
Estimated difference (SE) | 2.0 (0.5) | |
P value for the differenceb | < 0.001 | |
Responder analysisc, N = 72 | N = NR | N = NR |
Percentage of patients with an improvement ≥ 2 points at the last visit | 50% | 23% |
P value for the differenceb | 0.033 | |
Odds ratio (95% CrI) for the difference in treatment effect at the least visitd, e | 1.02 (0.96 to 1.09) | |
P value for the differenceb, f | NA | |
CrI = credible interval; IQR = interquartile range; RULM = Revised Upper Limb Module; NA = not applicable; NR = not reported; SD = standard deviation; SE = standard error.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cThe percentages of treated and untreated patients who improved by at least the minimal important difference empirically established (or from clinician opinion) for the scale or end point. Chi-square tests were used to calculate the P values for the differences in percentages of responders.
dMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio > 1 favours the nusinersen-treated group.
eThe analysis was based on a sample of 75 patients.
fThe model did not converge due to limited sample size. Bayesian modelling adjustment with a weakly informative prior (N[0, 3]) was used and therefore a P value was produced. Rationale for the informative priors was not provided.
Source: Vázquez-Costa et al.32
Table 13: Summary of Effectiveness Findings in the Included Study — 6MWT Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
6MWT | ||
Baseline, N = 22 | N = NR | N = NR |
Mean (SD) distance (m) | 269.8 (123.4) | 432.4 (127.6) |
Median (IQR) distance (m) | 281 (180, 368) | 461 (390, 499) |
Change from baseline to 6 monthsa, N = 17 | N = NR | N = NR |
Mean change (SD) (m) | 23.2 (62.8) | 19.9 (70.0) |
Estimated difference (SE) (m) | –6.3 (46.1) | |
P value for the differenceb | 0.894 | |
Responder analysisc, N = 17 | N = NR | N = NR |
Percentage of patients with an improvement ≥ 30 m at the last visit | 75% | 57% |
P value for the differenceb | 0.85 | |
Odds ratio (95% CI) for the difference in treatment effect at the least visitd, e | 1.071 (1.065 to 1.078) | |
P value for the differenceb | < 0.001 | |
6MWT = 6-minute walk test; CI = confidence interval; IQR = interquartile range; SD = standard deviation; SE = standard error; NR = not reported.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cThe percentages of treated and untreated patients who improved by at least the minimal important difference empirically established (or from clinician opinion) for the scale or end point. Chi-square tests were used to calculate the P values for the differences in percentages of responders.
dMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio > 1 favours the nusinersen-treated group.
eThe analysis was based on a sample of 22 patients.
Source: Vázquez-Costa et al.32
Table 14: Summary of Effectiveness Findings in the Included Study — EK2 Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
EK2 | ||
Baseline, N = 38 | N = NR | N = NR |
Mean (SD) total score | 14.8 (9.2) | 23.2 (9.5) |
Median (IQR) total score | 9 (8, 23) | 22 (18, 29) |
Change from baseline to 6 monthsa, N = 30 | N = NR | N = NR |
Mean change (SD) | −2.7 (2.7) | 1.1 (2.8) |
Estimated difference (SE) | –4.0 (3.3) | |
P value for the differenceb | 0.221 | |
Responder analysisc, N = 44 | N = NR | N = NR |
Percentage of patients with an improvement ≥ 2 points at the last visit | 80% | 23% |
P value for the differenceb | < 0.001 | |
Odds ratio (95% CI) for the difference in treatment effect at the least visitd, e | 0.81 (0.71 to 0.92) | |
P value for the differenceb | 0.001 | |
CI = confidence interval; EK2 = Egen Klassifikation Scale Version 2; IQR = interquartile range; NR = not reported; SD = standard deviation; SE = standard error.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cThe percentages of treated and untreated patients who improved by at least the minimal important difference empirically established (or from clinician opinion) for the scale or end point. Chi-square tests were used to calculate the P values for the differences in percentages of responders.
dMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio < 1 favours the nusinersen-treated group.
eThe analysis was based on a sample of 40 patients.
Source: Vázquez-Costa et al.32
Table 15: Summary of Effectiveness Findings in the Included Study — ALSFRS-R Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
ALSFRS-R | ||
Baseline, N = 52 | N = NR | N = NR |
Mean (SD) total score | 31.4 (8.4) | 26.6 (11.1) |
Median (IQR) total score | 32 (25, 39) | 29 (20, 31) |
Change from baseline to 6 monthsa, N = 42 | N = NR | N = NR |
Mean change (SD) | 0.8 (1.6) | −0.08 (1.2) |
Estimated difference (SE) | 3.4 (3.0) | |
P value for the differenceb | 0.999 | |
Responder analysisc, N = 42 | N = NR | N = NR |
Percentage of patients with an improvement ≥ 2 points at the last visit | 26% | 0% |
P value for the differenceb | 0.005 | |
Odds ratio (95% CrI) for the difference in treatment effect at the least visitd, e | 1.04 (0.94 to 1.14) | |
P value for the differenceb, f | NA | |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Scale Revised; CrI = credible interval; IQR = interquartile range; NA = not applicable; NR = not reported; SD = standard deviation; SE = standard error.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cThe percentages of treated and untreated patients who improved by at least the minimal important difference empirically established (or from clinician opinion) for the scale or end point. Chi-square tests were used to calculate the P values for the differences in percentages of responders.
dMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio > 1 favours the nusinersen-treated group.
eThe analysis was based on a sample of 5 patients.
fThe model did not converge due to limited sample size. Bayesian modelling adjustment with a weakly informative prior (N[0, 3]) was used and therefore a P value was produced. Rationale for the informative priors was not provided.
Source: Vázquez-Costa et al.32
Table 16: Summary of Effectiveness Findings in the Included Study — ppFVC Outcome
Outcome | Vázquez-Costa et al. | |
|---|---|---|
Nusinersen treated (N = 39) | Untreated (N = 40) | |
ppFVC | ||
Baseline, N = 48 | N = NR | N = NR |
Mean (SD) percentage | 72.9 (37.7) | 59.6 (39.3) |
Median (IQR) percentage | 77 (40, 105) | 45 (29, 88) |
Change from baseline to 6 monthsa, N = 40 | N = NR | N = NR |
Mean change (SD) | 2.6 (8.3) | −1.1 (5.7) |
Estimated difference (SE) | 3.2 (2.1) | |
P value for the differenceb | 0.139 | |
Responder analysis | NE | NE |
Odds ratio (95% CI) for the difference in treatment effect at the least visitc, d | 1.00 (0.90 to 1.12) | |
P value for the differenceb | 0.975 | |
CI = confidence interval; IQR = interquartile range; NE = not estimated; NR = not reported; SD = standard deviation; SE = standard error.
aDerived from rank-based regression models adjusting for baseline scores.
bP values < 0.05 were considered statistically significant.
cMixed ordinal regression models adjusted for follow-up time (in months) and the interaction between time and treatment. An odds ratio > 1 favours the nusinersen-treated group.
dThe analysis was based on a sample of 48 patients.
Source: Vázquez-Costa et al.32
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.