Drugs, Health Technologies, Health Systems
Health Technology Review
Cerliponase Alfa for Pediatric Patients With Neuronal Ceroid Lipofuscinosis Type 2 Disease
Key Messages
What Is the Issue?
Neuronal ceroid lipofuscinosis type 2 (CLN2) disease is an ultrarare, severe, and rapidly progressing lysosomal storage disorder, with a global incidence of approximately 0.15 to 9.0 per 100,000 live births. It has a devastating impact on children and families, leading to rapid functional decline and early death without effective treatment.
Cerliponase alfa is currently the only disease-modifying therapy approved by Health Canada for CLN2 disease.
Drug plans have requested an updated evidence review to support reimbursement decisions for cerliponase alfa, with a focus on evaluating their current discontinuation criteria that is based largely on motor-language score changes.
What Did We Do?
We searched key resources, including journal citation databases, and conducted a focused internet search for relevant studies of benefits and harms, as well as evidence-based clinical practice guidelines published since our previous review in 2019.
One reviewer screened articles for inclusion based on predefined criteria, critically appraised the included studies and guidelines, and summarized the findings.
We identified patients, caregivers, and families in Canada with lived experience with CLN2 disease and cerliponase alfa, and gathered their perspectives through semistructured interviews. The insights and priority treatment outcomes that they shared helped to inform our interpretation of the literature and conclusions.
We engaged 2 clinical experts in the diagnosis and management of CLN2 disease to provide input on the project protocol and draft report, offering clinical context and interpretation of the evidence.
What Did We Find?
Clinical experts identified improved quality of life (QoL), the mitigation of disease progression, and patient safety as key outcomes in treating people with CLN2 disease. Caregivers shared these priorities and emphasized additional outcomes, including patients’ comfort and happiness. Both groups underscored the importance of shared decision-making in initiating and discontinuing treatment with cerliponase alfa based on patients’ specific needs and goals.
Three primary clinical studies (2 observational studies and 1 open-label extension study of a single-arm clinical trial) with untreated historical controls and 2 evidence-based clinical practice guidelines met our preidentified inclusion criteria.
In alignment with our previous review, the 3 identified studies suggested that cerliponase alfa may slow disease progression in patients with CLN2 disease, helping to preserve or delay deterioration of motor-language function and reduce mortality compared to untreated patients. However, methodological limitations (e.g., results presented descriptively without any statistical analysis) limit the ability to make direct inferences to the benefit of cerliponase alfa on these outcomes, and whether these differences are clinically meaningful.
The evidence identified from the single-arm, open-label trial examining the benefit of cerliponase alfa on health-related quality of life (HRQoL) was variable. Some domains such as emotional functioning and seizure control showed improvement (indicating better HRQoL in these domains), while other domains such as physical functioning and daily activities declined (indicating worsening HRQoL in these domains) over time. Due to the noncomparative nature of the available data, causal inferences cannot be drawn. Observed changes in HRQoL, including seizure control, may reflect the natural progression of the disease, unadjusted confounding factors (e.g., use of antiseizure medications, disease severity, and concurrent illnesses), placebo effects, or the effects of cerliponase alfa.
Adverse events (AEs) were reported as common but manageable by the included study authors, with no deaths or treatment withdrawals reported due to AEs.
No studies were identified that met our inclusion criteria on caregiver burden measures.
No studies conducted subgroup analyses based on disease severity at treatment initiation, time of symptom onset, or patient age group.
Evidence-based clinical guidelines support the use of cerliponase alfa in patients with CLN2 disease, including patients with early-stage disease or who are presymptomatic, but note that evidence is limited regarding advanced disease and when to stop treatment.
What Does It Mean?
Findings suggest that early initiation of cerliponase alfa in CLN2 disease may help preserve or delay deterioration of motor-language function, particularly in patients with a motor-language score above 3 (i.e., some functional abilities are still preserved).
The emerging evidence that suggests a potential benefit of cerliponase alfa on QoL, seizure control, and mortality outcomes — which were deemed important to those with lived experience — has methodological limitations (e.g., lack of concurrent control groups; patients being aware of the treatment they received; and residual confounding factors, such as concurrent use of antiseizure medications and concurrent illnesses) that limit the ability to make definitive conclusions on the relative benefit of cerliponase alfa on these outcomes.
New evidence from the identified published clinical studies generally aligns with the previous conclusions of our 2019 clinical review.
The 3 primary studies did not examine the impact of treatment discontinuation on clinical outcomes and provide limited information to inform discontinuation criteria. In addition, 1 evidence-based clinical guideline highlighted a scarcity of data to inform treatment discontinuation.
Abbreviations
AE
adverse event
CDEC
Canadian Drug Expert Committee
CI
confidence interval
CLN2
neuronal ceroid lipofuscinosis type 2
ERT
enzyme replacement therapy
HRQoL
health-related quality of life
HTA
health technology assessment
ICV
intracerebroventricular
OCEBM
Oxford Centre for Evidence-Based Medicine
QoL
quality of life
SAE
serious adverse event
Introduction and Rationale
Background
What Is CLN2 Disease?
CLN2 disease, also known as late infantile neuronal ceroid lipofuscinoses or Jansky-Bielschowsky disease, is classified under the umbrella term of neuronal ceroid lipofuscinoses (NCLS), collectively referred to as Batten disease. CLN2 disease is a rare, severe, and rapidly progressing lysosomal storage disorder caused by mutations in the TPP1 gene (also known as the CLN2 gene).1,2 Mutations in the TPP1 gene lead to a deficiency or complete absence of the TPP1 enzyme, which is essential for normal lysosomal function.2 The severe deficiency or complete absence of the functional TPP1 enzyme, particularly within neurons of the brain and retina, results in the accumulation of toxic materials, including autofluorescent lipopigments, leading to progressive damage to the nervous system and cell death.2,3 CLN2 disease causes a decline in cognitive abilities and motor functions, and also causes drug-resistant epilepsy, ataxia (difficulty coordinating movements), vision loss, and a significantly shortened life expectancy.3
CLN2 disease is an ultrarare disease, with an incidence that ranges from approximately 0.15 to 9.0 per 100,000 live births worldwide.4 In Canada, Newfoundland has historically reported the highest incidences of CLN2 disease (9.0 per 100,000 live births),5 potentially due to founder effects, where certain genetic traits are more common in a population descended from a small number of ancestors.
CLN2 disease most often appears in children between the ages of 2 and 4 years, often beginning with subtle signs, such as delayed language development or the onset of seizures.1,4 The disease then progresses rapidly, with children losing abilities such as walking, talking, and vision.4 By the age of 6 to 7 years, most children with CLN2 disease lose all motor and speech functions, and death usually occurs between the ages of 10 and 15 years.6-8 Some patients with CLN2 disease present with an atypical (nonclassical) later onset (after the age of 4 years) form of the disease, which presents with slower disease progression of functional decline and more varied behavioural, motor, and vision symptoms compared to typical clinical presentations.9,10
What Is the Current Treatment Landscape of CLN2 Disease?
Historically, treatment for CLN2 disease focused on supportive care, which involves a multidisciplinary team of health care professionals — including pediatric neurologists, ophthalmologists, physiotherapists, and palliative care specialists — working together to manage symptoms, improve HRQoL, and prevent disease-related complications.4,8 Supportive care remains essential in managing the complex needs of children with CLN2 disease, including addressing issues such as nutritional difficulties and mobility issues.11 The first disease-modifying therapy for CLN2 disease, cerliponase alfa, was approved by the European Medicines Agency and the FDA in 2017, and by Health Canada in 2018. Cerliponase alfa remains the only approved disease-modifying therapy for people with CLN2 disease. Cerliponase alfa is an enzyme replacement therapy that targets the underlying enzyme deficiency in CLN2 disease, shifting treatment from purely supportive care to care that aims to slow disease progression.8
Clinical studies commonly use the CLN2 Clinical Rating Scale to evaluate the treatment effects of cerliponase alfa. The Clinical Rating Scale includes 4 domains: motor, language, vision, and seizures. Each domain is scored from 0 (complete loss of function) to 3 (normal function), yielding a total score ranging from 0 to 12, with higher scores indicating better function.12 Most studies focus on the combined motor-language score as the primary outcome. This subscore ranges from 0 to 6, where higher values reflect more developed motor and language abilities.12 One study reported a 1-point change in the combined motor-language score of the CLN2 Clinical Rating Scale to be both measurable and clinically meaningful; however, it was unclear whether this referred to a within-group or between-group change.13 The methodology used to establish this threshold remains unclear. It is uncertain whether this value can reliably define the minimal important difference (MID) value, which represents the smallest change in an outcome measure that patients perceive as beneficial or meaningful.
What Is Cerliponase Alfa?
Cerliponase alfa, a recombinant form of the human TPP1 enzyme, is designed to replace the missing or deficient TPP1 enzyme in patients with CLN2 disease.8 Clinicians administer cerliponase alfa directly into the central nervous system via intracerebroventricular (ICV) infusion.8 The standard regimen involves a 300 mg dose (adjusted for younger children) infused over approximately 4 hours, every 2 weeks.8 Cerliponase alfa is associated with potential AEs, such as pyrexia (fever), vomiting, seizures, and hypersensitivity reactions, among others.14 The ICV delivery system also carries potential risks, particularly device-related complications such as infections, which may require antibiotic treatment or device replacement.14 It is important to note that while enzyme replacement therapy aims to slow disease progression, it does not reverse existing brain damage or cure the disease.15 Early diagnosis and treatment of CLN2 disease are therefore vital to preserve function and slow decline for as long as possible.16
Policy Issues
Cerliponase alfa (Brineura) was previously recommended for reimbursement (with conditions) by the Canadian Drug Expert Committee (CDEC) in May 2019 as monotherapy for patients with CLN2 disease.17 Recommended initiation criteria required a confirmed CLN2 disease diagnosis through TPP1 enzyme testing and genetic testing, with the patient scoring a minimum of 1 in both motor and language domains and a combined motor-language score of 3 or more on the CLN2 Clinical Rating Scale, based on the eligibility criteria of the pivotal single-arm trial. Discontinuation was recommended when the combined motor-language score decreased by 2 or more points, or reached zero after 2 consecutive 24-week assessments, indicating functional decline or complete loss of function, based on the stopping criteria of the pivotal single-arm trial.17 Drug plans have requested an updated review of the evidence to inform their discontinuation criteria.
Policy Question
What new evidence exists to inform the discontinuation criteria for cerliponase alfa (Brineura) since the 2019 recommendation by CDEC?
Purpose
The objective of this evidence review is to evaluate studies that have become available since the 2019 recommendation by CDEC on the benefits and harms of cerliponase alfa, compared to placebo or best supportive care, in pediatric patients with CLN2 disease. We also aim to identify and summarize relevant evidence-based recommendations published after 2019 about the use of cerliponase alfa in pediatric patients with CLN2 disease.
Research Questions
To address the policy question about evidence to inform the discontinuation criteria for cerliponase alfa, the ideal approach would involve clinical studies comparing the benefits and harms of discontinuing versus continuing treatment among patients with CLN2 disease who are already receiving the therapy. Specifically, such studies would assess patient-important outcomes between those who stopped treatment and those who continued.
However, our preliminary search did not identify any studies meeting these criteria. In the absence of direct evidence, we reviewed indirect evidence by examining studies on the benefits and harms of cerliponase alfa that have become available since the 2019 recommendation by CDEC, as well as evidence-based clinical practice guidelines. Although clinical studies or guidelines may not directly define discontinuation criteria, information on treatment benefits, harms, and evidence-based recommendations may help support these decisions.
The following research questions were identified to address the policy question:
What is the clinical effectiveness and safety of cerliponase alfa versus best supportive care, placebo, or no treatment (if no comparative evidence) for pediatric patients with CLN2 disease, based on studies published since 2019?
What are the evidence-based recommendations published since 2019 regarding the use and administration of cerliponase alfa for pediatric patients with CLN2 disease?
Methods
Rapid review methodology was used to provide drug programs with timely and pertinent evidence to inform their decision-making.
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE and Embase via Ovid, the Cochrane Database of Systematic Reviews, the International HTA Database, the websites of health technology assessment (HTA) agencies in Canada and major international HTA agencies, as well as a focused internet search. The search approach was customized to retrieve a limited set of results, balancing comprehensiveness with relevance. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. Search concepts were developed based on the elements of the research questions and selection criteria. The main search concept was cerliponase alfa. The search was completed on April 11, 2025, and limited to English-language documents published since January 1, 2019. We limited the review to English-language studies due to resource constraints and published studies since the last 2019 review to align with the scope of the policy question. Only published, peer-reviewed, or publicly accessible studies were included to enhance transparency and reproducibility.
Selection Criteria and Methods
One reviewer screened citations and selected studies. In the first level of screening, titles and abstracts were reviewed and potentially relevant articles were retrieved and assessed for inclusion. The final selection of full-text articles was based on the inclusion criteria presented in Table 1.
Criteria | Description |
|---|---|
Population | Pediatric patients with CLN2 disease Subgroups of interest:
|
Intervention | Cerliponase alfa (Brineura) |
Comparator | Best supportive care Placebo No comparator (if no comparative evidence) |
Outcomes | Efficacy outcomes:
Harms outcomes: SAEs, AEs, WDAEs, deaths due to AEs, and notable harms (administration-related [e.g., infection], cerebrospinal fluid pleocytosis, gastrointestinal [e.g., vomiting], fever, hypersensitivity) |
Study designs | This review used a hierarchical approach to evidence inclusion, prioritizing the most robust and current evidence. We first sought recent, comprehensive, and high-quality systematic reviews, with or without meta-analyses. If we did not identify suitable systematic reviews, or if relevant systematic reviews were not comprehensive of the comparisons and outcomes of interest, we would include RCTs. If RCT evidence was insufficient to answer the research question, we would consider nonrandomized comparative intervention studies (e.g., cohort, case-control). Finally, we would consider single-arm studies only if critical evidence gaps remained after considering comparative studies, particularly for assessing long-term benefits or harms. We focused on the evidence-based guidelines for research question 2. |
AE = adverse event; CLN2 = neuronal ceroid lipofuscinosis type 2; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event.
Note: We considered a review to be systematic if it included the following elements: an objective and research question(s), indications that evidence was searched for in a systematic way (e.g., information on 1 or more of the following: names of databases, search platforms or engines, search date, keywords, or search strategy), and inclusion and exclusion criteria. We considered a guideline to be evidence-based if a systematic search of the literature was undertaken and a guideline panel was involved in informing the recommendations.
Exclusion Criteria
We excluded studies if they did not meet the selection criteria outlined in Table 1, if they were duplicate publications, or if they were published before the 2019 recommendation by CDEC. We excluded all abstracts, including conference abstracts, and did not consider data presented solely in abstract form (e.g., subgroup analyses). We also excluded letters to the editor, commentaries, opinion pieces, narrative reviews, book chapters, publications that were not in English, and guidelines lacking clear methodology.
Critical Appraisal of the Individual Studies
One reviewer extracted data aligned with the research questions and composed a narrative summary of the findings from the included studies. One reviewer critically appraised the methodological quality of included studies using the Downs and Black checklist18 for primary studies and the Appraisal of Guidelines for Research and Evaluation (AGREE) II instrument19 for clinical practice guidelines. Summary scores were not calculated for the included studies; rather, the strengths and limitations of each included publication were described narratively. We considered the findings of the critical appraisals when summarizing study results and drawing conclusions.
Engagement
A call for input on the project scope from interested parties was issued at project initiation through our weekly newsletter and on our website.
Clinical Expert Engagement
Two clinical experts with expertise in the diagnosis and management of patients with CLN2 disease were consulted during our review. Both experts provided input on the project protocol, including the proposed scope, and refinement of the review. The experts also reviewed and provided feedback on the draft report, offering clinical context and interpretation of the evidence, particularly regarding the relevance and applicability of clinical outcomes to the management of CLN2 disease in practice. Their input was used to help inform the interpretation of findings and the identification of key limitations.
Engagement With People With Lived Experience
Semistructured interviews were conducted virtually with people who were willing to share their lived experiences with cerliponase alfa and CLN2 disease. Five caregivers and 1 patient responded to questions about disease history, impact of diagnosis on daily life, treatment decisions, goals of care, and priority health outcomes. Four caregivers and 1 patient had direct experience with cerliponase alfa as a treatment; 1 caregiver’s child had not been treated with cerliponase alfa but the caregiver had extensive knowledge of cerliponase alfa.
Recordings and transcripts generated from the interviews were used by a Canada’s Drug Agency (CDA-AMC) Engagement Officer to summarize patient and caregiver insights. Participants reviewed their interview summaries for accuracy. The summaries were then consolidated and themes were reported. The results of the final report were shared and discussed with participants before the publication of this report.
Summary of Engagement With People With Lived Experience
The people with lived experience who participated in our interviews shared their journeys with CLN2 disease, from the onset of symptoms through to diagnosis and treatment, with parents describing the realities of caring for children with a rare, progressive illness. The long process of obtaining and coming to terms with a CLN2 disease diagnosis was universally portrayed as extremely stressful and emotionally devastating, with life-changing impacts to individuals and families. Parents actively researched and pursued treatment and supportive care options, with many transitioning into full-time caregiving immediately after their child's diagnosis. They expressed that the pursuit of any medical intervention was driven by the desire to optimize their child’s QoL; preserve their dignity; and maintain their happiness, safety, and well-being.
For some families, the ability to promptly begin treatment with cerliponase alfa was constrained by drug availability due to regulatory challenges, limited access to clinical trials, and trial eligibility criteria. The parents of children for whom cerliponase alfa was indicated and initiated noted reduced frequency of seizures and a slower decline in motor skills. While they acknowledged that the medication was not curative, it was credited with extending periods of symptom stability, helping to sustain an enhanced QoL, and prolonging survival. Parents described the financial strain of managing CLN2 disease, as caregiving responsibilities often made paid employment difficult while various medical expenses and treatment costs resulted in substantial out-of-pocket spending. Many made major life adjustments to accommodate the economic burden of the disease.
A summary of the engagement with people with lived experience using the Guidance for Reporting Involvement of Patients and the Public (GRIPP2) framework is provided in Appendix 1, Table 2.
Summary of Input From Patient or Clinician Groups or Industry
No submissions were received from patient or clinician groups. Input was received from BioMarin Pharmaceutical, the manufacturer of cerliponase alfa. The manufacturer agreed with the proposed project scope but suggested its expansion to include real-world evidence sources, citing the rarity of CLN2 disease.
The manufacturer submitted references for consideration, including publications, conference abstracts, and summaries from unpublished clinical study reports. All references were screened against predefined inclusion criteria. Several studies had already been identified through the literature search, while others were excluded for not meeting inclusion criteria. Additionally, some submitted data were not included in the review due to a lack of detailed methodologies. Additionally, the manufacturer provided clinician statements and a summary of select discontinuation criteria used in international jurisdictions.
Summary of Engagement With Clinical Experts
According to 2 clinical experts, disease progression and the preferences of patients, caregivers, or families play a crucial role in decisions about discontinuing treatment. The expert suggested treatment may be discontinued if the Clinical Rating Scale (Hamburg) score reaches 0 (complete loss of function) and there is significant caregiver burden, or if the patient experiences repeated central nervous system infections, requiring prolonged hospital admissions and treatment. Other factors that may lead to discontinuation include the need for major procedures, worsening seizures, or deterioration in cognitive function.
Summary of Evidence
Quantity of Research Available
We identified a total of 183 citations in the literature search. After screening titles and abstracts, we excluded 157 citations and retrieved 26 potentially relevant records from the electronic search for full-text review. We also retrieved 9 potentially relevant records from the grey literature search for full-text review. Of these potentially relevant records (35), we excluded 30 for various reasons (refer to Appendix 3 for excluded studies and reasons for exclusion) and included 3 primary studies with natural history controls and 2 clinical practice guidelines that met the inclusion criteria. Given that we identified primary studies with historical controls, we excluded single-arm studies and case series without a control group. We also excluded HTA reports, as these are typically developed by agencies focused on local resource allocation and cost-effectiveness.
Figure 1 (Appendix 2) presents the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)20 flow chart of the study selection. We also screened potentially relevant findings from ongoing clinical studies. No results were available at the time of review. These ongoing clinical studies are listed in Table 3 under the ongoing studies category. We did not find any systematic reviews or randomized controlled trials (RCTs) that met our inclusion criteria.
Summary of Study Characteristics
Primary Studies
Three primary studies21-23 assessed the effectiveness and safety of cerliponase alfa compared to untreated natural history controls in children with CLN2 disease. Schulz et al. (2025)22 conducted a real-world, multicentre study using the DEM-CHILD registry, comparing prospectively collected data from 21 children treated with cerliponase alfa (whose data were collected prospectively, with a mean follow-up of 106.7 weeks) to 21 matched untreated controls (whose data were collected retrospectively, with a mean follow-up of 76.1 weeks). The study adjusted for baseline combined motor-language score, age, sex, and genotype when analyzing motor-language function outcomes. In a separate study, Schulz et al. (2024)23 reported on the long-term extension (up to 240 weeks) of a multicentre, open-label single-arm study across Germany, Italy, the UK, and the US, including 24 patients treated with cerliponase alfa and 42 untreated patients from the DEM-CHILD database. The study adjusted for baseline combined motor-language score, age, genotype, and sex in assessing both motor-language function and mortality outcomes. Estublier et al. (2021)21 performed a retrospective review of medical records for 8 children treated with cerliponase alfa at 3 hospitals in France (with a mean follow-up of 26 months), compared to historical untreated patients (unclear sample size in this group) from a previous study.7 The study did not perform any adjusted analyses for the reported outcomes. In all studies, treated patients received cerliponase alfa via intracerebroventricular infusion at a dose of 300 mg every 2 weeks.
Two studies22,23 used untreated patients from the DEM-CHILD registry as historical controls. One study21 did not report the key details of its control group such as the functional assessment scales used and the sample size. In 1 study,23 the baseline motor-language score for the natural history cohort was defined as the first score of less than 6 occurring at or after 36 months of age; 2 studies21,22 did not report on the definition of baseline motor-language score. Details regarding the untreated patients were not reported directly but instead referenced in a separate study.7 Based on the external study,7 before the development of the motor-language CLN2 Clinical Rating Scale — which was created specifically for the Brineura clinical trials — the motor-language functions of patients in the historical control group were likely assessed using the Hamburg scale. In that study, disease progression was tracked longitudinally in 67 patients. Among the 41 patients with complete assessments across the disease course, motor-language scores on the Clinical Rating Scale (adapted from the Hamburg scale) declined rapidly, from normal (score of 6) to no function (score of 0), at an average decline rate of 1.81 points per year (95% confidence interval [CI], 1.50 to 2.12) over approximately 30 months.
The assessed outcomes included functional outcomes measured by the CLN2 Clinical Rating Scale and AEs in all 3 included studies.21-23 Schulz et al. (2024)23 also assessed mortality; MRI changes; biomarkers; HRQoL measured by the PedsQL Parent Report for Toddlers, PedsQL Family Impact Module, and CLN2 Disease-Specific Quality of Life dimension scores; and development milestones measured by the Denver II Developmental Scale Age Equivalent Performance. In this evidence review, we summarized the functional outcomes, mortality, HRQoL measures, and AEs.
In 1 study,23 HRQoL was assessed every 24 weeks using the PedsQL Parent Report for toddler assessment (21 items across 4 dimensions) and the PedsQL Family Impact Module (36 items across 5 dimensions), both of which were scored on a 0 to 4 Likert scale and transformed to a 0 to 100 scale, with higher scores indicating better HRQoL or better family functioning.12,24 A CLN2-specific QoL tool developed by the cerliponase alfa manufacturer (BioMarin) included 28 items across 6 dimensions. Scoring for the CLN2-specific QoL tool was performed analogously to that for PedsQL, according to the included study.23 The study did not report MID values and recall periods for all HRQoL measures.23 In another study,24 the clinically important difference for within-group change was defined using a distribution-based method for each domain. The distribution-based clinically important differences were reported as follows: seizures (9.08), sleep (12.94), behaviour (6.79), and daily activities (7.67).
Evidence-Based Guidelines
Two clinical guidelines25,26 provide recommendations for the diagnosis and management of CLN2 disease. In this evidence review, we focus on using enzyme replacement therapy with cerliponase alfa to manage CLN2 disease. Sampaio et al. (2023)25 developed a guideline to inform the management of CLN2 disease in Brazil. The guideline was informed by a literature review using PubMed, Embase, and SciELO, but the search time frame was not clearly reported. Mole et al. (2021)26 developed an international guideline based on 2 independent systematic reviews conducted in 2019; the search was conducted from PubMed, Google Scholar, and internal databases.26 Both guidelines25,26 graded the evidence using the Oxford Centre for Evidence-Based Medicine (OCEBM) levels and used structured or modified Delphi processes to reach consensus among a guideline panel of experts. In the guideline developed in Brazil,25 the development group included 9 child neurologists, 1 of whom was also a medical geneticist. The other guideline26 involved a panel of 41 experts who responded to a questionnaire, comprising pediatric neurologists (54%), metabolic specialists (20%), geneticists (9%), neurosurgeons (2%), pediatricians (6%), and others (9%).
Appendix 4 presents additional details regarding the characteristics of the included studies and guidelines.
Summary of Critical Appraisal
Primary Studies
One study22 demonstrated methodological rigour through the use of matched historical controls and appropriate statistical methods; however, it lacked blinding, had a small sample size (n = 21 in each group), and applied retrospectively collected data for controls with different follow-up durations than the intervention group. The small sample size (n = 21 per group) limits the precision of effect estimates and the ability to generalize the results to the broader population of people with CLN2 disease. The use of retrospectively collected data for the control group, with different follow-up durations compared to the intervention group, raises concerns about bias due to confounding and inconsistent outcome measurement.
The extension23 of the multicentre, open-label, single-arm study was registered and well documented, with detailed outcome reporting. However, it also relied on historical controls with unclear baseline characteristics, lacked blinding, and did not perform a formal sample size calculation. These issues may contribute to bias due to residual confounding and limit the internal validity of the findings. The absence of data on HRQoL and AEs in the control group further restricted the study’s ability to inform on these outcomes, which are important to patients and decision-makers. There was a lack of detailed reporting on missing outcome data. For example, HRQoL scores were missing at study completion for 3 out of 23 participants (13%) to 6 out of 23 participants (26%) across various domains, excluding the “feeding with gastrostomy tube” domain, which was not applicable to all participants. No AE data were reported for the control group. It is also unclear whether patients with more severe disease were excluded from HRQoL assessment, which could potentially lead to an overestimation of the observed improvement and further reduce confidence in the results.
The remaining retrospective study21 had several limitations: it involved a very small sample size (n = 8), lacked clear inclusion and exclusion criteria, did not match controls, and did not apply any formal statistical comparisons. Additionally, the intervention details and AE severity were insufficiently reported.21 These limitations severely limit the interpretability and reliability of the findings. The absence of methodological safeguards increases the risk of bias due to confounding, selection bias, and reporting bias.
All 3 studies21-23 had small sample sizes and lacked concurrent control groups, which limits the precision and generalizability of their findings. The extension study23 of a multicentre, open-label, single-arm study did not implement blinding; in the observational studies, patients were aware of the treatment they received, which may have influenced outcome reporting. Two studies22,23 attempted to reduce confounding resulting from differences in prognostic factors and treatment effect modifiers across the 2 cohorts by matching patients who received treatment with historical controls based on prognostic factors such as age, baseline motor-language scores, and genotype. They applied this matching either within the main analysis or as part of a sensitivity analysis.22,23 Although this approach improved comparability between groups, residual confounding likely remained, such as differences in diagnostic methods or supportive care or differences in any additional characteristics that the authors were unable to match. The historical control groups lacked data on HRQoL measures and AEs,22,23 which limited the external validity of the findings.
All studies did not clearly report how functional scores were assessed for patients in the historical control group.21-23 It is assumed that the control group’s scores were likely evaluated using the Hamburg scale.7 Although the CLN2 Clinical Rating Scale was adapted from the Hamburg scale, the differences in category descriptions between the CLN2 Clinical Rating Scale and Hamburg scale may contribute to bias in the comparison between patients treated with cerliponase alfa and the historical controls.
Evidence-Based Guidelines
Both guidelines25,26 clearly defined their objectives, target populations (patients with CLN2 disease), and intended users (clinicians). Both guidelines25,26 searched for relevant evidence and then reached consensus to formulate recommendations. Both guidelines25,26 conducted literature searches, but the details of the searches were not provided. However, 1 guideline26 did not search Embase, potentially missing relevant evidence due to unclear or incomplete searches.
Both guidelines25,26 were developed by a team of clinical experts. However, it is uncertain whether a methodology expert was involved in the development of the 2 guidelines. One of the guidelines26 included a patient advocate to seek the views or preferences of the target population. It is unclear whether the perspectives or preferences of the target population were sought or had an influence on the recommendations in the other guideline.25 Therefore, the recommendations regarding cerliponase alfa may not adequately reflect the values and preferences of patients or caregivers.
The 2 guidelines25,26 used OCEBM criteria to assess the certainty of the evidence. The 2 guidelines used different labels to indicate the level of evidence. For example, 1 guideline used “1b” or “5,” while the other used “B,” “C,” or “D.” However, neither guideline clearly explained what each level of evidence meant.25,26 The links between the recommendations and the supporting evidence and the strength of recommendations were also unclear in the 2 guidelines.25,26 Both guidelines25,26 disclosed the competing interests of their development group. However, several authors in the guidelines had received research grants from industries (e.g., BioMarin), and it is unclear how they addressed these competing interests.25,26 It is unclear if the competing interests impacted the development of recommendations. One guideline26 provided specific, easily identifiable recommendations, while the other guideline’s recommendations were less clearly presented.25
Appendix 5 presents additional details regarding the strengths and limitations of the included studies and guidelines.
Summary of Findings
Based on the 3 primary studies21-23 and 2 evidence-based clinical practice guidelines25,26 included in this evidence review, we have summarized the reported data available since 2019 on the effectiveness and safety of cerliponase alfa in pediatric patients, and relevant recommendations for its use based on guidelines published since 2019. New evidence from published clinical studies since the 2019 review is consistent with the previous clinical review report.12
One23 of the 3 primary studies extended a previous 48-week, single-arm, open-label, multicentre, dose-escalation study27 that compared outcomes of patients treated with cerliponase alfa with historical controls. The original study reported that cerliponase alfa was associated with less decline in motor and language function scores compared with no treatment.27 The extension study23 suggested that treatment with cerliponase alfa continued to slow CLN2 disease progression over an average of more than 5 years (240 weeks), with safety events that were expected and reported as manageable by the study authors.23
Appendix 6 presents the summary of findings by different outcomes.
Clinical Effectiveness of Cerliponase Alfa Compared With Untreated Natural History Controls
Unreversed Function 2-Point Decline or a Score of Zero in the Motor-Language Domain on the CLN2 Clinical Rating Scale (Indicating a Noticeable Worsening or Complete Loss of the Person's Ability in Motor and Language Domains, Without Any Recovery)
Two studies22,23 reported that cerliponase alfa was associated with a reduced hazard of experiencing an unreversed 2-point decline or a score of zero in the motor-language domain on the CLN2 Clinical Rating Scale compared with untreated historical controls, with a mean follow-up of 106.7 weeks in 1 study22 and up to 240 weeks of follow-up in the other study.23
Two studies22,23 also reported that cerliponase alfa was associated with a reduced hazard of reaching a score of zero in the motor-language domain of the CLN2 Clinical Rating Scale, compared with untreated historical controls, with a mean follow-up of 106.7 weeks in 1 study and up to 240 weeks of follow-up in the other study.
When the motor and language domains were analyzed separately, 1 study23 (with up to 240 weeks of follow-up) reported that cerliponase alfa was associated with a reduced hazard of experiencing an unreversed 2-point decline or a score of zero compared with untreated historical controls for both domains.
Rate of Function Decline
Two studies22,23 reported that patients treated with cerliponase alfa experienced a slower rate of function decline in the CLN2 motor-language score, compared with untreated historical control data, with a mean follow-up of 106.7 weeks in 1 study and up to 240 weeks of follow-up in the other study.
One study (with up to 240 weeks of follow-up)23 observed similar results when analyzing the motor and language domains separately.
Two studies21,23 reported that patients treated with cerliponase alfa had higher mean CLN2 Clinical Rating Scale motor-language scores at various time points (week 145, week 289, or their last evaluation) compared with the untreated historical cohort. However, both studies21,23 presented the data descriptively and did not perform any statistical tests to assess between-group differences.
One study (with up to 240 weeks of follow-up)23 found smaller changes from baseline across 4 individual CLN2 Clinical Rating Scale domains (i.e., motor, language, vision, and seizures), suggesting a slower functional decline over time in the treatment group compared with the untreated historical cohort. However, the study23 presented the data descriptively and did not perform any statistical tests to assess between-group differences.
Mortality
No deaths were reported among patients treated with cerliponase alfa in the 3 included studies,21-23 with up to 240 weeks of follow-up in 1 study.23
One study (with up to 240 weeks of follow-up)23 found that treatment with cerliponase alfa was associated with a reduced hazard of death compared to untreated historical controls. The study adjusted for genotype and sex in the unmatched analysis, and further accounted for baseline motor-language score and baseline age in the matched analysis.
One study (with a mean follow-up of 106.7 weeks)22 reported no deaths in the cerliponase alfa group, while 6 deaths occurred in the untreated historical control group (n = 21 for both intervention and control groups). The study did not perform statistical testing for the mortality outcome.
HRQoL Measures
HRQoL measures were not available for patients in the untreated historical control group.23
The extension study23 of a multicentre, open-label, single-arm study with up to 240 weeks of follow-up reported the HRQoL results for patients treated with cerliponase alfa:
In the PedsQL Parent Report for Toddlers, the largest HRQoL decline was observed in physical functioning, followed by school functioning and social functioning, and a slight improvement in emotional functioning at study completion compared to baseline. A decline in within-group HRQoL scores indicates a worsening QoL over time, while an increase reflects better HRQoL.
In the PedsQL Family Impact Module, slight mean within-group changes were observed at the study completion compared to baseline.23
In the CLN2 disease-specific HRQoL instrument, within-group changes in seizure scores improved,24 while a within-group change in daily activity scores declined at study completion compared to baseline.24 For sleep, behaviour, and feeding domains, slight mean within-group changes were observed at study completion compared to baseline.24
Caregiver Burden Using Validated Scales
No studies were identified that met our inclusion criteria and reported on caregiver burden measures.
Safety of Cerliponase Alfa Compared With Untreated Natural History Controls
Safety data were not available for patients in the untreated historical control group in all 3 included studies.21-23
Any Treatment-Emergent AEs
Two studies22,23 reported treatment-emergent adverse events, with the number of patients who experienced 1 or more AEs reported as 16 out of 24 (66.7%) in 1 study and 24 out of 24 (100%) in the other.
One study22 reported that 7 out of 24 patients (29%) experienced AEs that led to hospitalization.
AEs reported for at least 50% of patients in any of the included studies21-23 involved upper respiratory tract infection, pyrexia or fever, vomiting, seizure, rhinitis, gait disturbance, dysphagia, constipation, device-related infections, and device end of service.
Any Serious AEs
Two studies22,23 reported any serious adverse events (SAEs), with the number of patients who experienced 1 or more SAEs reported as 4 out of 24 (17%) in 1 study and 21 out of 24 (88%) in the other.
One study22 that included 24 patients treated with cerliponase alfa reported 4 SAEs of grade 3 or grade 4 severity, all of which led to hospitalization.
SAEs reported in more than 1 participant included23 product issues (device end of service, device leakage, device malfunction), hypersensitivity, device-related infections, upper respiratory tract infections, dysphagia, gastroenteritis, pleocytosis, dental caries, device deployment issues, epilepsy, bacterial pharyngitis, pyelonephritis, and pyrexia.
Withdrawals Due to AEs
No withdrawals from treatment due to AEs were reported in the 3 included studies.21-23
Deaths Due to AEs
No deaths due to AEs were reported in the 3 included studies.
Subgroups of Interest
No studies conducted subgroup analyses based on disease severity at treatment initiation, time of symptom onset, or patient age group. Some patients from the included studies21-23 that were relevant to the disease severity subgroup included the following:
One study22 reported 5 matched pairs with a baseline motor-language score of 2 or lower. Among them, 1 patient maintained their score, and 2 experienced slower disease progression compared to their matched natural history controls, as measured by motor and language scores. The study also presented patient-level observations of motor and language scores, noting that the results were consistent across different ages at treatment initiation (< 3 years, 3 years to < 4 years, 4 years to < 5 years, 5 years to < 6 years, and ≥ 6 years) as well as across baseline motor and language scores (range, 2 points to 6 points).22
Another study23 included 1 patient with a baseline motor-language score of 1; this patient maintained their score while the matched natural history controls showed a decline.
A third study21 in patients in France (n = 8) included 3 patients with baseline motor-language scores of 2 or lower. All 3 patients experienced a 1-point decline in motor-language score at their final evaluation (mean follow-up: 26 months).
Recommendations for Using Cerliponase Alfa in Patients With CLN2 Disease
Two evidence-based guidelines25,26 provided the recommendations for using enzyme replacement therapy (cerliponase alfa) in patients with CLN2 disease.
Patient Eligibility
Symptomatic patients with CLN2 disease: Both guidelines recommended enzyme replacement therapy.25,26 However, 1 guideline aimed at patients in Brazil25 specified that patient eligibility for treatment with enzyme replacement therapy should be children aged 3 years or younger with a Clinical Rating Scale score of 3 or higher, while the other guideline26 did not impose any age-based or score-based restrictions.
Presymptomatic patients with CNL2 disease: Enzyme replacement therapy is recommended for genetically confirmed cases by the guideline aimed at patients in Brazil.25 However, the optimal timing remains uncertain due to the absence of trial data.25
Advanced CLN2 disease (e.g., Clinical Rating Scale score less than 3): The guideline aimed at patients in Brazil stated that no clear benefit of enzyme replacement therapy has been demonstrated for patients with advanced CLN2 disease (i.e., Clinical Rating Scale below 3).25
Patients with atypical (nonclassical) CLN2 disease: Enzyme replacement therapy may be initiated after diagnosis, exclusion of contraindications, and shared decision-making with the patient or caregivers.26
Dosage and Administration
Setting: Perform infusions in a sterile, isolated room with minimal personnel and continuous monitoring. A hospital ward is acceptable if no infectious patients are present.25
A multidisciplinary team — including a child neurologist, neurosurgeon, pediatrician, and nurse — should oversee the procedure.25
Premedication: Administer an antihistamine 30 minutes before infusion to reduce adverse effects.25
Dose: 300 mg (or age-appropriate dose) every 2 weeks via ICV infusion over 4 hours.25,26
Device handling:
Use a permanent ICV catheter (e.g., Ommaya reservoir) with a syringe infusion pump.25
A very experienced pediatric neurosurgeon should place the catheter under general anesthesia.26
Only trained personnel should access the device to minimize complications.25,26
Treatment Discontinuation Criteria
The evidence-based clinical practice guideline for patients in Brazil was unable to identify high-quality evidence to support a recommendation on discontinuation criteria for cerliponase alfa.25
Most dosing and administration recommendations or statements were from the guideline aimed for patients in Brazil.25
Limitations
The rapid review methodology used in this review has several limitations that may affect the interpretation of our findings. Study selection, data extraction, and critical appraisal were conducted by a single reviewer, which may increase the risk of errors, bias, and the potential omission of relevant studies. The restricted search strategy excluded studies published before 2019, limiting the comprehensiveness of the evidence base. We also did not perform detailed assessments of risk of bias or evaluate the certainty of the evidence, which limits our ability to contextualize the benefits and harms of cerliponase alfa in children with CLN2 disease. For HRQoL and AEs, data were only available from the treatment arms of the included studies challenging the review team’s ability to draw causal conclusions for these outcomes.21-23 We did not comprehensively assess whether other single-arm studies reported these outcomes in this patient population. We also did not conduct a comprehensive search for evidence on the psychometric properties or MID values of the reported outcome measures, as this was beyond the scope of the review. The limited availability of such information may affect the ability to fully contextualize the findings.
In this evidence review, we did not identify any systematic reviews, RCTs, or studies with concurrent controls that evaluated the effectiveness and safety of cerliponase alfa. Although we found 3 primary studies,21-23 1 was an extension of a single-arm trial with up to 240 weeks of follow-up,23 1 was a retrospective study21 with significant reporting limitations that restricted our ability to interpret its findings. For instance, while the body of evidence suggests that cerliponase alfa may contribute to improved seizure control, this conclusion is limited by the lack of seizure frequency data in control groups, absence of statistical analyses, and failure to adjust for key confounders such as antiseizure medication use, disease severity, and concurrent illnesses. However, given the rarity of the disease, it is unlikely that a randomized trial would be feasible.
We did not find any recommendations that directly addressed our policy question (i.e., treatment discontinuation criteria). One guideline25 noted insufficient data to establish discontinuation criteria for cerliponase alfa in CLN2 disease. While 2 evidence-based guidelines provided recommendations for the use of cerliponase alfa, most were based on expert consensus.25,26 Neither guideline clearly reported the evidence base for recommendations and the link between recommendations and supporting evidence was also unclear.25,26 This suggests that the current recommendations may be influenced by clinical opinion in the absence of clear supporting evidence, highlighting the need for cautious interpretation.
We did not identify any evidence-based guidelines developed by Canadian organizations or involving experts from Canadian institutions. Given potential implementation challenges, such as access to treatment and unique patient characteristics in Canada (e.g., higher incidence or specific genotypes in Newfoundland),5 the applicability of these recommendations to Canadian settings remains uncertain.
The engagement of people with lived experience faced several limitations. Targeted outreach through specific patient groups restricted broader participation, potentially excluding other interested individuals. Additionally, time and resource constraints limited the number of participants who were able to take part in the project.
Conclusions and Implications for Decision- or Policy-Making
The people with lived experience who were interviewed identified improved QoL, the mitigation of disease progression, and the importance of ensuring patients’ comfort, safety, and happiness as key treatment outcomes for CLN2 disease. Caregivers stressed the importance of early access to treatment, transparent communication regarding treatment expectations, and patient-centred decision-making with their child’s health care providers regarding starting and stopping medications. The 2 clinical experts consulted highlighted that the preferences of patients, caregivers, and families play a crucial role in their decisions about discontinuing treatment.
The accounts of lived experience highlighted the profound impact of CLN2 disease on children and their families. Collectively, they underscore the critical importance of early and accurate diagnosis, as well as the need for comprehensive and coordinated care frameworks for families affected by CLN2 disease. The emotional toll of a CLN2 diagnosis is immense, with parents describing the devastating reality of caring for a child with a progressive illness while urgently seeking effective treatments. Financial hardship compounds these challenges, forcing many families to make difficult personal and economic sacrifices in their efforts to secure appropriate care and maintain their child's well-being. People with lived experience emphasized that while cerliponase alfa is not curative, it slows disease progression, stabilizes symptoms, prolongs life, and preserves QoL.
We identified 3 primary studies21-23 with natural history controls and 2 evidence-based clinical practice guidelines25,26 to answer our research questions. The evidence reviewed in this report largely aligns with the conclusions from the previous Reimbursement Review conducted in 2019.12 More recent findings suggest that early initiation of cerliponase alfa in the disease course may help preserve or delay deterioration of motor-language function, particularly in patients with a motor-language score above 3 (some functional abilities are still preserved). One extension study23 with up to 240 weeks of follow-up reported slower functional decline in motor, language, vision and seizure among patients treated with cerliponase alfa compared to historical controls. All studies21-23 reported no deaths among patients treated with cerliponase alfa, while 2 studies22,23 reported several deaths in historical control groups, including 1 study23 with up to 240 weeks of follow-up. The emerging evidence on the potential benefit of cerliponase alfa in reducing seizures and mortality outcomes, while promising, has methodological limitations (e.g., results presented descriptively without statistical comparisons, historical controls with unclear baseline characteristics) that limit the ability to make direct inferences on the effects of cerliponase alfa.
Limited evidence from single-arm data23 suggests that patients with CLN2 disease treated with cerliponase alfa experienced varying mean score changes in HRQoL measures, which were reported as important outcomes by families who shared their lived experiences. Some domains — such as emotional functioning and seizure control — showed improvement (indicating better HRQoL in these domains), while other domains — such as physical functioning and daily activities — declined (indicating worsening HRQoL in these domains) over time. Due to the noncomparative nature of the available data, causal inferences cannot be drawn (e.g., changes in HRQoL may be attributed to natural history, placebo effects, concurrent seizure medications, or the effects of cerliponase alfa).
AEs were common but generally manageable (as noted by the study authors) among CLN2 patients treated with cerliponase alfa.21-23 No deaths or treatment withdrawals due to AEs were reported.
No studies were identified that met our inclusion criteria on caregiver burden measures. No studies conducted subgroup analyses based on disease severity at treatment initiation, time of symptom onset, or patient age group.
Two clinical guidelines25,26 support the use of cerliponase alfa in symptomatic and genetically confirmed presymptomatic patients, or patients with atypical (nonclassical) CLN2 disease. The eligibility requirement of being aged at least 3 years, with a Clinical Rating Scale score of 3 or higher was inconsistently reported between the 2 guidelines.25,26 One guideline25 highlighted the lack of evidence for use in advanced disease (i.e., a motor-language score of ≤ 2). In alignment with the findings from the 3 identified primary studies,21-23 1 guideline25 highlighted a scarcity of data to inform treatment discontinuation.
It is important to note that CLN2 disease is an ultrarare disease, and it is unlikely that well-controlled randomized trials would be feasible in this population. While several new studies were identified and reviewed in this report, none directly compared the impacts of cerliponase alfa discontinuation on clinical outcomes or provided additional evidence to inform discontinuation criteria. One evidence-based guideline noted that there are insufficient data to establish such criteria. Given the complexity and progressive nature of CLN2 disease, both clinical experts and people with lived experience emphasized that decisions to initiate, continue, or discontinue cerliponase alfa should be made through a shared decision-making process involving clinicians, patients, and caregivers.
The discontinuation criteria from CDEC in 201917 were informed by the stopping criteria of the pivotal trial, which is defined as a decrease of 2 or more points or a score of zero on the combined motor-language scale across 2 consecutive 24-week assessments, indicating functional decline or complete loss of function. The current review did not identify any new clinical evidence since the 2019 CDEC reimbursement recommendation that directly informs when treatment with cerliponase alfa should be discontinued.
Special thanks: CDA-AMC extends our special thanks to the individuals who shared their lived experience with the research team, as well as to patient organizations supporting those with CLN2 disease and their families. We thank Terri Gortnar, Mark Gortnar, Lori Brown, Leah Brochu, and Mike Brochu for providing input on the direction of the research, drawing from lived experience, and providing valuable contributions to this report. A very special thank you to Claire Gortnar, Josef Gortnar, Daniel Brown, and Neil Brochu.
References
1.UptoDate. Neuronal ceroid lipofuscinosis. 2024. Accessed April 29, 2025. https://www.uptodate.com/
2.Fietz M, AlSayed M, Burke D, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119(1):160-167. doi: 10.1016/j.ymgme.2016.07.011 PubMed
3.Schaefers J, van der Giessen LJ, Klees C, et al. Presymptomatic treatment of classic late-infantile neuronal ceroid lipofuscinosis with cerliponase alfa. Orphanet J Rare Dis. 2021;16(1):221. doi: 10.1186/s13023-021-01858-6 PubMed
4.Williams RE, Adams HR, Blohm M, et al. Management Strategies for CLN2 Disease. Pediatr Neurol. 2017;69:102-112. doi: 10.1016/j.pediatrneurol.2017.01.034 PubMed
5.Moore SJ, Buckley DJ, MacMillan A, et al. The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clin Genet. 2008;74(3):213-22. doi: 10.1111/j.1399-0004.2008.01054.x PubMed
6.Specchio N, Pietrafusa N, Trivisano M. Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy. Ther Clin Risk Manag. 2020;16:213-222. doi: 10.2147/tcrm.S241048 PubMed
7.Nickel M, Simonati A, Jacoby D, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. 2018;2(8):582-590. doi: 10.1016/s2352-4642(18)30179-2 PubMed
8.Lewis G, Morrill AM, Conway-Allen SL, Kim B. Review of Cerliponase Alfa: Recombinant Human Enzyme Replacement Therapy for Late-Infantile Neuronal Ceroid Lipofuscinosis Type 2. J Child Neurol. 2020;35(5):348-353. doi: 10.1177/0883073819895694 PubMed
9.Guelbert N, Espitia Segura OM, Amoretti C, et al. Classic and Atypical Late Infantile Neuronal Ceroid Lipofuscinosis in Latin America: Clinical and Genetic Aspects, and Treatment Outcome with Cerliponase Alfa. Mol Genet Metab Rep. 2024;38(no pagination). doi: 10.1016/j.ymgmr.2024.101060
10.Chang H, Soangra R, Bhola R, GrantBeuttler M, Wang RY. A long-term course of gait assessment in the atypical CLN2 patients along with ICV clerliponase alpha treatment. Mol Genet Metab. 2023;138(2):107048. doi: 10.1016/j.ymgme.2022.107048
11.Schulz A, Jain M, Butt T, et al. The challenges of living with and caring for a child or children affected by neuronal ceroid lipofuscinosis type 2 disease: In-Depth family surveys in the United Kingdom and Germany. J Inborn Errors Metab Screen. 2020;8:e20190013. doi: 10.1590/2326-4594-JIEMS-2019-0013
12.CADTH. Drug Reimbursement Review clinical guidance report: cerliponase alfa (Brineura) for CLN2 disease. 2019. Accessed April 28, 2025. https://www.cda-amc.ca/sites/default/files/cdr/clinical/sr0574-brineura-clinical-review-report.pdf
13.Wyrwich KW, Schulz A, Nickel M, et al. An Adapted Clinical Measurement Tool for the Key Symptoms of CLN2 Disease. J Inborn Errors of Metab Screen. 2018;6:2326409818788382. doi: 10.1177/2326409818788382
14.Ammendolia I, Sframeli M, Esposito E, et al. Adverse Reactions to the Orphan Drug Cerliponase Alfa in the Treatment of Neurolipofuscinosis Type 2 (CLN2). Pharmaceuticals (Basel). 2024;17(11):11. doi: 10.3390/ph17111513 PubMed
15.Radic Nisevic J, Kolic I, Kostanjski M, Kovacevic F, Prpic I. Early Symptoms and Treatment Outcomes in Neuronal Ceroid Lipofuscinosis Type 2: Croatian Experience. J Pers Med. 2024;14(8):24. doi: 10.3390/jpm14080783 PubMed
16.Augustine EF, Adams HR, de Los Reyes E, et al. Management of CLN1 Disease: International Clinical Consensus. Pediatr Neurol. 2021;120:38-51. doi: 10.1016/j.pediatrneurol.2021.04.002 PubMed
17.CADTH. Drug Expert Committee Recommendation: cerliponase alfa (Brineura) for CLN2 disease. May 23, 2019. Accessed April 28, 2025. https://www.cda-amc.ca/cerliponase-alfa
18.Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. doi: 10.1136/jech.52.6.377 PubMed
19.Agree Next Steps Consortium. The AGREE II Instrument. AGREE Enterprise; 2017. Accessed April 29, 2025. https://www.agreetrust.org/wp-content/uploads/2017/12/AGREE-II-Users-Manual-and-23-item-Instrument-2009-Update-2017.pdf
20.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1-e34. doi: 10.1016/j.jclinepi.2009.06.006 PubMed
21.Estublier B, Cano A, Hoebeke C, et al. Cerliponase alfa changes the natural history of children with neuronal ceroid lipofuscinosis type 2: The first French cohort. Europ J Paediat Neurol. 2021;30:17-21. doi: 10.1016/j.ejpn.2020.12.002 PubMed
22.Schulz A, Schwering C, Wibbeler E, et al. Real-world clinical outcomes of patients with CLN2 disease treated with cerliponase alfa. Front Neurol. 2025;16:1516026. doi: 10.3389/fneur.2025.1516026 PubMed
23.Schulz A, Specchio N, de Los Reyes E, et al. Safety and efficacy of cerliponase alfa in children with neuronal ceroid lipofuscinosis type 2 (CLN2 disease): an open-label extension study. Lancet Neurol. 2024;23(1):60-70. doi: 10.1016/S1474-4422(23)00384-8 PubMed
24.Due C, Quinn J, Gissen P, et al. Psychometric Validation of the CLN2 Quality of Life Questionnaire in Participants with CLN2 Disease Treated with Cerliponase Alfa. Healthcare (Basel). 2024;12(22):08. doi: 10.3390/healthcare12222229
25.Sampaio LPB, Manreza MLG, Pessoa A, et al. Clinical management and diagnosis of CLN2 disease: consensus of the Brazilian experts group. Arq Neuropsiquiatr. 2023;81(3):284-295. doi: 10.1055/s-0043-1761434 PubMed
26.Mole SE, Schulz A, Badoe E, et al. Guidelines on the diagnosis, clinical assessments, treatment and management for CLN2 disease patients. Orphanet J Rare Dis. 2021;16(1):185. doi: 10.1186/s13023-021-01813-5 PubMed
27.Schulz A, Ajayi T, Specchio N, et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. 2018;378(20):1898-1907. doi: 10.1056/NEJMoa1712649 PubMed
Appendix 1: Summary of Patient and Caregiver Engagement
Please note that this appendix has not been copy-edited.
Table 2: Reporting of Patient and Caregiver Engagement Using the GRIPP2 Framework
Section and topic | Item |
|---|---|
1. Aim | Six contributors (5 caregivers and 1 patient) shared their experience with and perspectives on cerliponase alfa and CLN2 disease. Their accounts provided insight into lived experiences and care priorities and helped the review team contextualize the findings from the literature. |
2. Methods | People with lived experience were identified through targeted outreach to disease-specific patient groups. The project scope and engagement opportunity were communicated via email and an introductory meeting was conducted by Zoom with interested individuals. Three semistructured interviews were conducted virtually by a CDA-AMC Engagement Officer with 3 families located in different parts of Canada. Interview #1 included a parent/caregiver, interview #2 included 2 parents/caregivers, and interview #3 included 2 parents/caregivers and a person living with CLN2 disease. The interviews were recorded, summarized, and shared back with participants to ensure accuracy and completeness. Interview themes and overall conclusions were derived from the collective insights of participants. The final report was shared and results discussed with participants before report publication. |
3. Results of Engagement | Diagnosis and Impact on Daily Life Families described the diagnostic journey as complex, often beginning with unexplained seizures or developmental regression. The time to diagnosis ranged from several months to more than a year, during which families experienced considerable uncertainty. Receiving a diagnosis of CLN2 disease was life-altering and marked by profound emotional distress. Parents reported a significant shift in personal priorities, career disruption, financial hardship, and changes in family dynamics with several assuming full-time caregiving roles. Treatment Priorities and Outcomes of Importance Across all families, 1 of the most important care goals was maintaining the child’s comfort, dignity, and quality of life. Families emphasized the importance of minimizing suffering while preserving the child’s spirit and individuality. Seizure control was noted by 1 family as an important outcome. Slowing disease progression was also a highly valued outcome, with early treatment noted as being particularly effective in achieving this. Families reported that their expectations of treatment evolved over time as the realities of the condition became more apparent. Experience with Cerliponase Alfa Due to the timing of diagnosis, 1 family shared they were unable to obtain the treatment as they did not meet clinical trial eligibility, or the clinical trial was not recruiting at the time, while another family was able to access it through a clinical trial in the US Where treatment was initiated, experiences varied, even within siblings in the same family unit. Some families observed significant benefits such as improved seizure control or slowed motor and language decline. A few caregivers noted that benefits diminished over time, particularly in later stages of the disease. The treatment regimen was described as burdensome, involving lengthy hospital visits and considerable logistical planning. Parents talked about challenges in not only the administration of therapy but also care coordination, travel, and respite. In spite of this, families emphasized that treatment offered hope and extended their child’s survival, which they considered highly meaningful even if it was not curative. System Challenges and Supports Families highlighted the importance of early diagnosis and access to genetic testing to support timely intervention. They also stressed the need for clear, realistic information to support informed decision-making around treatment initiation and discontinuation. The value of shared, compassionate decision-making with care teams was emphasized, particularly when discussing treatment goals or transitions in care. Participants expressed gratitude for care teams and patient organizations. However, they noted challenges navigating complex care pathways, accessing adequate home care and respite support, and managing out-of-pocket costs for essential therapies. Continuity of care and the anticipated transition to adult services were also identified by some as concerns. |
4. Discussions and Conclusions | Several important points emerged in the interviews. According to people with lived experience who took part: The management of CLN2 can be financially burdensome. Essential therapies and supportive services frequently constitute high out-of-pocket costs. Families navigating CLN2 disease often face financial strain and must make significant lifestyle adjustments to manage expenses. Maintaining steady paid employment can become difficult as 1 or both parents juggle the responsibilities of work and home life. CLN2 requires constant and intensive caregiving. The complex medical needs of children with CLN2 disease demand continuous attention, often requiring 1 parent to step into a full-time caregiving role, thereby completely reshaping family dynamics, daily routines, and plans for the future. There are benefits and limitations to cerliponase alfa in treating CLN2 disease. Cerliponase alfa prolongs life of children with CLN2 disease but is not a cure. The effectiveness of cerliponase alfa depends on when it is introduced in the disease progression and not all children who are diagnosed will be eligible for it. Even when it is available, its effects are not indefinite. Patients, families, and caregivers must be mindful of their expectations of the medication. Understanding both the benefits and limitations of treatment is essential to making informed decisions about care. CLN2 disease treatment choices should be made collaboratively by families and their medical team. The decision to start or stop a medication is an individual 1 and should be based on each patient’s unique circumstances. While cerliponase alfa is expensive and not curative, decisions around initiation and discontinuation should reside with families and their care providers. Optimizing comfort and maximizing quality of life for children with CLN2 disease are among the most important care priorities. Minimizing both immediate and long-term suffering in children with CLN2 disease is a top priority, with a focus on reducing disease- and treatment-related distress while ensuring well-being. Emotional health and overall happiness of their children are integral to caregiving decisions. Families navigating CLN2 disease grapple with its progressive and debilitating nature, particularly the rapid loss of mobility, communication, and independence in their children. Any treatment or intervention that can preserve function even temporarily and extend time together holds immense value. Collectively, these insights underscore the profound challenges that families affected by CLN2 disease face—not only financial burdens, caregiving demands, and complex treatment decisions, but also the deep emotional distress of navigating the relentless progression of an incurable disease that has profoundly impacted their child’s life. People with lived experience who were interviewed noted that cerliponase alfa offers a life-prolonging treatment option for CLN2 disease but is not a cure, and its effectiveness depends on many factors, making informed, collaborative decision-making essential for families and their health care providers. |
5. Reflections and Critical Perspectives | Participants were very willing to share their lived experience with CLN2 disease and cerliponase alfa for the purpose of this review and did so thoughtfully and candidly. Success of the engagement is related to several factors. Participants were briefed on the purpose of the review and the role of input from people with lived experience. They were supported throughout the process by a dedicated Engagement Officer. Calls were scheduled at the convenience of the participants. Relevant materials and questions were shared in advance of the interviews. Supplemental written information was shared after conversations to ensure understanding and clarify details. Participants were acknowledged in the report in the manner in which they were most comfortable, and they were provided with an honorarium as a gesture of thanks for their contribution to the project. E-mails sent to specific groups limited the involvement of other potentially interested individuals. Time and resource constraints limited the number of people who were able to participate in the project. Finally, tight timelines necessitated steps in the review process to happen quickly, which was a challenge. |
Appendix 2: Selection of Included Studies
Please note that this appendix has not been copy-edited.
Figure 1: PRISMA20 Flow Chart of Study Selection
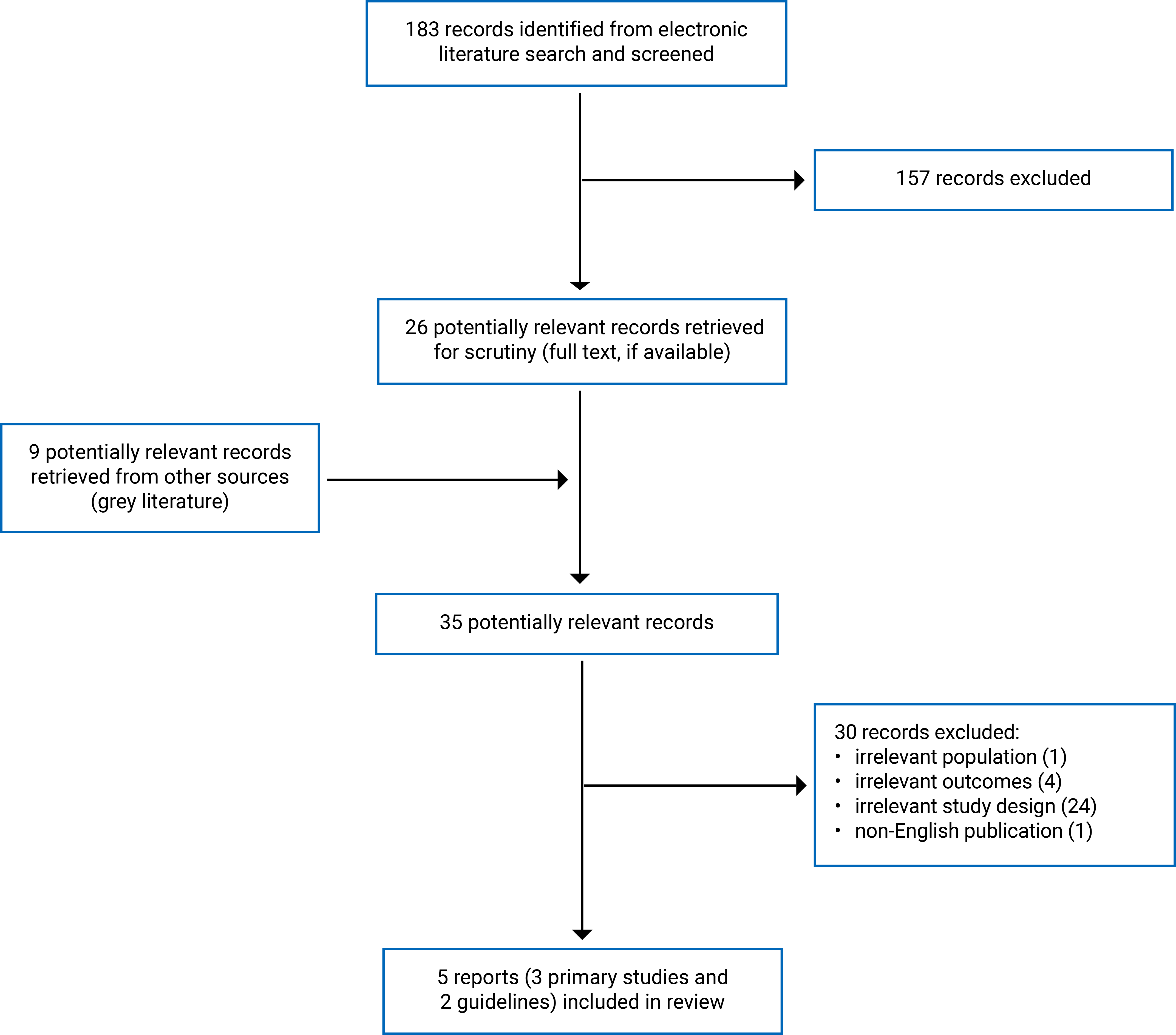
PRISMA = Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Appendix 3: List of Excluded Citations and Potentially Relevant Ongoing Studies
Please note that this appendix has not been copy-edited.
Table 3: Excluded Citations and Potential Relevant Ongoing Studies
Citations | Reasons for exclusion |
|---|---|
PRIGLINGER, C., et al. Enzyme Replacement Therapy in CLN2-Associated Retinopathy. Klin Monatsbl Augenheilkd. 2025;242(3):213 to 218. https://dx.doi.org/10.1055/a-2528-7886 | Irrelevant study design (Narrative review) |
AMMENDOLIA, I., et al. Adverse Reactions to the Orphan Drug Cerliponase Alfa in the Treatment of Neurolipofuscinosis Type 2 (CLN2). Pharmaceuticals. 2024;17(11):11. https://dx.doi.org/10.3390/ph17111513 | Irrelevant study design (single-arm without comparative group) |
BOUSTANY, R. M. Cerliponase alfa and neuronal ceroid lipofuscinosis type 2: long-term outcomes and lessons for future research. Lancet Neurol. 2024;23(1):5 to 7. https://dx.doi.org/10.1016/S1474-4422(23)00467-2 | Irrelevant study design (commentary article) |
GAUR, P., et al. Enzyme Replacement Therapy for CLN2 Disease: MRI Volumetry Shows Significantly Slower Volume Loss Compared with a Natural History Cohort. Am J Neuroradiol. 2024;45(11):1791 to 1797. https://dx.doi.org/10.3174/ajnr.A8408 | Irrelevant outcome (MRI) |
GUELBERT, N., et al. Classic and Atypical Late Ingantile Neuronal Ceroid Lipofuscinosis in Latin America: Clinical and Genetic Aspects, and Treatment Outcome with Cerliponase Alfa. Mol Genet Metab Rep. 2024;38(no pagination). https://dx.doi.org/10.1016/j.ymgmr.2024.101060 | Irrelevant study design (single-arm without a comparative group) |
GUELBERT, N., et al. Corrigendum to “Classic and atypical late infantile neuronal ceroid lipofuscinosis in Latin America: Clinical and genetic aspects, and treatment outcome with cerliponase alfa.” Mol Genet Metab Rep. 2024 41:101081. https://dx.doi.org/10.1016/j.ymgmr.2024.101081 | Irrelevant study design (corrigendum study) |
RADIC NISEVIC, J., et al. Early Symptoms and Treatment Outcomes in Neuronal Ceroid Lipofuscinosis Type 2: Croatian Experience. J Pers Med. 2024;14(8):24. https://dx.doi.org/10.3390/jpm14080783 | Irrelevant study design (single-arm without a comparative group) |
SPAULL, R., et al. Evolution of Movement Disorders in Patients With CLN2-Batten Disease Treated With Enzyme Replacement Therapy. Neurol. 2024;103(3):e209615. https://dx.doi.org/10.1212/WNL.0000000000209615 | Irrelevant study design (single-arm without a comparative group) |
DULZ, S., et al. Ongoing retinal degeneration despite intraventricular enzyme replacement therapy with cerliponase alfa in late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2 disease). Br J Ophthalmol. 2023;107(10):1478 to 1483. https://dx.doi.org/10.1136/bjo-2022-321260 | Irrelevant study design (single-arm without a comparative group) |
HENDERSON, R., et al. Intravitreal Cerliponase alfa slows the rate of retinal thinning in patients with CLN2 type Batten Disease: A first in man report. Invest Ophthalmol Vis Sci. 2023;64(8):788. | Irrelevant study design (meeting abstract) |
ATISKOVA, Y., et al. Progressive retinal degeneration despite intraventricular enzyme replacement therapy with cerliponase alfa in classic late-infantile CLN2 disease. Invest Ophthalmol Vis Sci. 2022;63(7):4526-F0313. | Irrelevant study design (meeting abstract) |
ATISKOVA, Y., et al. Visual perception and macular integrity in non-classical CLN2 disease. Graefes Arch Clin Exp Ophthalmol. 2022;260(11):3693 to 3700. https://dx.doi.org/10.1007/s00417-022-05662-1 | Irrelevant study design (single-arm without a comparative group) |
GUELBERT, G., et al. Neuronal Ceroid Lipofuscinosis Type 2: A Case Series from Argentina. J Inborn Errors Metab Screen. 2022;10(no pagination). https://dx.doi.org/10.1590/2326-4594-JIEMS-2022-0001 | Irrelevant study design (single-arm without comparative group) |
AUGUSTINE, E. F., et al. Management of CLN1 Disease: International Clinical Consensus. Pediatr Neurol. 2021;120:38 to 51. https://dx.doi.org/10.1016/j.pediatrneurol.2021.04.002 | Irrelevant population (CLN1 patients) |
ESPITIA SEGURA, O. M., et al. Real world effectiveness of cerliponase alfa in classical and atypical patients. A case series. Mol Genet Metab Rep. 2021;27:100718. https://dx.doi.org/10.1016/j.ymgmr.2021.100718 | Irrelevant study design (single-arm without comparative group) |
SCHAEFERS, J., et al. Presymptomatic treatment of classic late-infantile neuronal ceroid lipofuscinosis with cerliponase alfa. Orphanet J Rare Dis. 2021;16(1):221. https://dx.doi.org/10.1186/s13023-021-01858-6 | Irrelevant study design (case report) |
SCHWERING, C., et al. Development of the “Hamburg Best Practice Guidelines for ICV-Enzyme Replacement therapy (ERT) in CLN2 Disease” Based on 6 Years Treatment Experience in 48 Patients. J Child Neurol. 2021;36(8):635 to 641. https://dx.doi.org/10.1177/0883073821989154 | Irrelevant outcome (Recommendations for ICV procedure) |
WIBBELER, E., et al. Cerliponase Alfa for the Treatment of Atypical Phenotypes of CLN2 Disease: A Retrospective Case Series. J Child Neurol. 2021;36(6):468 to 474. https://dx.doi.org/10.1177/0883073820977997 | Irrelevant study design (single-arm without comparative group) |
DE LOS REYES, E., et al. Intracerebroventricular Cerliponase Alfa for Neuronal Ceroid Lipofuscinosis Type 2 Disease: Clinical Practice Considerations From US Clinics.Pediatr Neurol. 2020;110:64 to 70. https://dx.doi.org/10.1016/j.pediatrneurol.2020.04.018 | Irrelevant outcome (Recommendations for ICV procedure) |
LEWIS, G., et al. Review of Cerliponase Alfa: Recombinant Human Enzyme Replacement Therapy for Late-Infantile Neuronal Ceroid Lipofuscinosis Type 2. J Child Neurol. 2020;35(5):348 to 353. https://dx.doi.org/10.1177/0883073819895694 | Irrelevant study design (Narrative review) |
ANONYMOUS. Cerliponase alfa for neuronal ceroid lipofuscinosis type 2 disease. Aust Prescr. 2019;42(3):102. https://dx.doi.org/10.18773/austprescr.2019.029 | Irrelevant study design (commentary) |
NICE. Cerliponase alfa for treating neuronal ceroid lipofuscinosis type 2; 2019: https://www.nice.org.uk/guidance/hst12 | Irrelevant study design (HTA) |
German Federal Joint Committee. [Pharmaceutical Directive/Annex XII: Cerliponase Alfa - reassessment after the deadline (type 2 neuronal ceroid lipofuscinosis)]; 2022: https://database.inahta.org/article/22854 | Irrelevant outcome (No formal recommendations) |
Council for Choices in Health Care in Finland. Summary of a recommendation by COHERE Finland on cerliponase alfa in the treatment of neuronal ceroid lipofuscinosis type 2 (CLN2); 2021: https://palveluvalikoima.fi/documents/1237350/89863500/Serliponase_alfa_Summary_EN_+030921.pdf/c5055317-5703-8ea8-4091-77524e843aab/Serliponase_alfa_Summary_EN_+030921.pdf?t=1630677841969 | Irrelevant study design (guidelines with unclear methods) |
IqWiG. [G22 to 25] Cerliponase alfa (neuronal ceroid lipofuscinosis type 2) – Assessment according to §35a (para. 1, sentence 11) Social Code Book V (expiry of the decision); 2022: https://www.iqwig.de/en/projects/g22-25.html | Non-English publication |
EMA. Brineura; risk management plan and product information updated 2023: https://www.ema.europa.eu/en/medicines/human/EPAR/brineura | Irrelevant study design (guidelines with unclear methods) |
Ireland Health Service Executive. HSE Guidelines for the Treatment of Neuronal Ceroid Lipofuscinosis Type 2 (CLN2) with Cerliponase alfa (Brineura®); 2024: https://www.hse.ie/eng/about/who/acute-hospitals-division/drugs-management-programme/protocols/hse-guidelines-for-the-treatment-of-battens-disease-with-cerliponase.pdf | Irrelevant study design (guidelines with unclear methods) |
Drugs for rare diseases: a review of national and international health technology assessment agencies and public payers’ decision-making processes; 2021: https://www.cda-amc.ca/sites/default/files/es/es0355-drugs-for-rare-diseases-pw.pdf | Irrelevant study design (government report) |
cerliponase alfa (SR0574): https://www.cda-amc.ca/cerliponase-alfa | Irrelevant study design (HTA) |
Scottish Medicines Consortium. Medicine: cerliponase alfa (brand name: Brineura®) for neuronal ceroid lipofuscinosis type 2; 2020: https://scottishmedicines.org.uk/media/5453/cerliponase-alfa-brineura.pdf | Irrelevant study design (guidelines with unclear methods) |
Longitudinal Assessment of Atypical Tripeptidyl Peptidase 1 Enzyme Deficiency (Neuronal Ceroid Lipofuscinosis Type 2) Patients. National Library of Medicine (US). https://clinicaltrials.gov/study/NCT04098211 | Ongoing study without results |
Examining Developmental Outcomes of Children Diagnosed With CLN2 Disease. National Library of Medicine (US). https://clinicaltrials.gov/study/NCT03862274 | Ongoing study without results |
Natural History and Long-term Clinical Assessments of All Forms of Neuronal Ceroid Lipofuscinoses - Capturing Key Symptoms and Disease Progression as Part of the Independent, International NCL DEM-CHILD Patient Database. National Library of Medicine (US). https://clinicaltrials.gov/study/NCT04613089 | Ongoing study without results |
Cerliponase Alfa Observational Study. National Library of Medicine (US). https://clinicaltrials.gov/study/NCT04476862 | Ongoing study without results |
Intravitreal Enzyme Replacement Therapy to Prevent Retinal Disease Progression in Children With Neuronal Ceroid Lipofuscinosis Type 2 (CLN2). National Library of Medicine (US). https://clinicaltrials.gov/study/NCT05152914 | Ongoing study without results |
Appendix 4: Characteristics of Included Studies
Please note that this appendix has not been copy-edited.
Table 4: Characteristics of the Included Primary Studies
Study citation, country, funding source | Study design | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
Schulz et al. (2025)22 Germany Funding source: German Federal Ministry of Education and Research, European Union, BioMarin Pharmaceutical Inc. | Real-world, observational study based on the DEM-CHILD database (a multicentre, multinational clinical registry) | Children with CLN2 disease (n = 21) treated with cerliponase alfa between August 26, 2016 and December 31, 2020 (Data were prospectively collected). For untreated natural history controls (n = 21), data were collected retrospectively. Sex: males: n = 10 (48%) in intervention group, n = 16 (76%) in natural history group Mean (SD) age at baseline: 4.7 (1.9) years (range: 0.7 to 9.9 years) for both groups Mean (SD) baseline motor-language score: 3.9 (1.6) (range: 1 to 6) for both groups | Intervention: Cerliponase alfa administered as an intracerebroventricular infusion (300 mg every 2 weeks), outside of the clinical trial setting Comparators: Untreated CLN2 disease (had never received cerliponase alfa) Patients matched 1:1 with untreated natural history controls based on baseline age and motor-language score. | Outcomes:
|
Schulz et al. (2024)23 Germany, Italy, the UK, and the US Funding source: BioMarin Pharmaceutical Inc. | An extension study for a single-arm, open-label, multicentre dose-escalation study (NCT01907087) | Children aged 3 to 16 years with CLN2 disease (n = 24) confirmed by genetic analysis and enzyme testing. Untreated natural history controls consisted of patients with CLN2 disease (n = 42) enrolled in the DEM-CHILD database in Germany and Italy. Sex: males: n = 9 (38%) in intervention group; control group: NR. Mean age at baseline: 4.9 (1.3) years (range: 3.1 to 8.9 years) in the intervention group; control group: NR. Mean baseline motor-language score: 3.5 (1.2) years (range: 1 to 6) in the intervention group; control group: NR. | Intervention: Cerliponase alfa administered as an intracerebroventricular infusion (300 mg every 2 weeks) Comparators: Untreated CLN2 disease patients from the DEM-CHILD database | Outcomes:
Follow-up: up to 240 weeks open-label extension with a 6-month safety follow-up. |
Estublier et al. (2021)21 France Funding source: No specific funding | Retrospective observational study in 3 university hospitals in France | Children with CLN2 disease (n = 8). Untreated natural history controls from another study (Nickel, Schulz et al., Lancet, 2018, likely DEM-CHILD cohort). Sex: males: n = 4 (50%) in intervention group Mean age at baseline: 56 (13) months (range: 33 to 60 months) in the intervention group. Mean baseline motor-language score: 3.1 (NR) years (range: 1 to 5) in the intervention group | Intervention: Cerliponase alfa (300 mg, every 15 days) through an Ommaya ventricular reservoir, which is inserted by a neurosurgeon Comparators: Untreated CLN2 disease patients from Nickel, Schulz et al., 2018 | Outcomes:
Mean Follow-up (from the beginning of treatment to last evaluation): 26 months for the intervention group; control group: NR. |
AEs = adverse event; CLN2 = neuronal ceroid lipofuscinosis type 2; ERT = enzyme replacement therapy; HRQoL = health-related quality of life; NA = not applicable; NR = not reported.
Table 5: Characteristics of Included Guidelines
Intended users, target population | Intervention and practice considered | Major outcomes considered | Evidence collection, selection, and synthesis | Evidence quality assessment | Recommendations development and evaluation | Guideline validation |
|---|---|---|---|---|---|---|
Sampaio et al. (2023)25 | ||||||
Intended users: Clinicians (pediatricians and child neurologists) in Brazil Target population: patients with CLN2 disease or suspected of having CLN2 disease | Clinical management and diagnosis of CLN2 disease: including ERT with cerliponase Alpha and multidisciplinary management | Clinical evaluation of patients with CLN2 disease, such as the Hamburg scale and Clinical Rating Scale: functional decline, HRQoL, and reduction in seizure frequency | A literature search of PubMed, Embase and SCiELO was performed, and publications (systematic reviews, randomized and nonrandomized clinical studies, observational studies, and case series) were considered. | OCEBM Levels of Evidence | The Delphi method was employed for consensus. The multiple-choice questionnaire with 92 questions was used. Virtual meetings for discussion and voting. | The guideline involved 9 Brazilian child neurologists. The guideline was published in a peer-reviewed medical journal. |
Mole et al. (2021)26 | ||||||
Intended users: Health care professionals involved in the management of patients with CLN2 disease. Target population: patients with CLN2 disease | Clinical assessment, management and diagnosis of CLN2 disease, such as ERT with cerliponase alfa, supportive and symptomatic treatments. | Clinical outcomes | Two systematic literature reviews were independently conducted (February and March 2019) to identify current evidence for treatment and management of CLN2 disease. The databases included PubMed, Google scholar and interrogation of internal literature libraries. | OCEBM Levels of evidence | Consensus reached through a modified Delphi process involving international experts. Statements validated by health care professionals through anonymous voting. | Two external health care professionals, independent of the guideline development process, reviewed the manuscript. They identified gaps and areas of confusion, assessed the guideline using the AGREE II instrument, and provided an overall assessment score of 5.93. |
AGREE II = Appraisal of Guidelines for Research and Evaluation; ERT = Enzyme Replacement Therapy; HRQoL = health-related quality of life; OCEBM = Oxford Centre for Evidence-Based Medicine.
Appendix 5: Critical Appraisal of Included Studies
Please note that this appendix has not been copy-edited.
Table 6: Strengths and Limitations of Clinical Studies Using the Downs and Black Checklist18
Strengths | Limitations |
|---|---|
Schulz et al. (2025)22 | |
|
|
Schulz et al. (2024)23 | |
|
|
Estublier et al. (2021)21 | |
|
|
CI = confidence interval.
Table 7: Strengths and Limitations of Guidelines Using AGREE II19
Item | Sampaio et al. (2023)25 | Mole et al. (2021)26 |
|---|---|---|
Domain 1: Scope and purpose | ||
1. The overall objective(s) of the guideline is (are) specifically described. | Yes | Yes |
2. The health question(s) covered by the guideline is (are) specifically described. | Yes | Yes |
3. The population (patients, public, and so on) to whom the guideline is meant to apply is specifically described. | Yes | Yes |
Domain 2: Stakeholder involvement [wording from original source] | ||
4. The guideline development group includes individuals from all relevant professional groups. | Unclear if at least 1 methodology expert was included. | Unclear if at least 1 methodology expert was included. |
5. The views and preferences of the target population (patients, public, and so on) have been sought. | Unclear, probably no | Yes (the guideline committee included input from a patient advocate) |
6. The target users of the guideline are clearly defined. | Yes | Yes |
Domain 3: Rigour of development | ||
7. Systematic methods were used to search for evidence. | Yes | Partially (Embase was not searched) |
8. The criteria for selecting the evidence are clearly described. | No (clear on the study design) | No |
9. The strengths and limitations of the body of evidence are clearly described. | Yes | No |
10. The methods for formulating the recommendations are clearly described. | Yes | Yes |
11. The health benefits, side effects, and risks have been considered in formulating the recommendations. | Yes | Yes |
12. There is an explicit link between the recommendations and the supporting evidence. | No | No |
13. The guideline has been externally reviewed by experts before its publication. | Unclear | Yes |
14. A procedure for updating the guideline is provided. | No | Yes |
Domain 4: Clarity of presentation | ||
15. The recommendations are specific and unambiguous. | No | The strength of recommendation was unclear. |
16. The different options for management of the condition or health issue are clearly presented. | Yes | Yes |
17. Key recommendations are easily identifiable. | No | Yes |
Domain 5: Applicability | ||
18. The guideline describes facilitators and barriers to its application. | Yes | Yes |
19. The guideline provides advice and/or tools on how the recommendations can be put into practice. | No | No |
20. The potential resource implications of applying the recommendations have been considered. | Yes | Yes |
21. The guideline presents monitoring and/or auditing criteria. | No | Yes |
Domain 6: Editorial independence | ||
22. The views of the funding body have not influenced the content of the guideline. | Unclear (BioMarin provided financial support for the writing, and the guideline developers declared that the funder played no role in the content of this publication) | Unclear |
23. Competing interests of guideline development group members have been recorded and addressed. | Conflicts of interest were declared, but it was unclear how they were addressed. Several authors had received honorarium or financial support from BioMarin. | Conflicts of interest were declared, but it was unclear how they were addressed. Several authors had received grants or consultation fees from BioMarin. |
AGREE II = Appraisal of Guidelines for Research and Evaluation II.
Appendix 6: Main Study Findings
Please note that this appendix has not been copy-edited.
Table 8: Summary of Findings by Outcome — Unreversed Function 2-Point Decline or a Score of Zero on CLN2 Clinical Rating Scalea
Study | Group | Sample size (n) | Follow-up, mean (SD), weeks | Median time to event (95% CI) | HR (95% CI)b | P value | Notes |
|---|---|---|---|---|---|---|---|
Motor-language score | |||||||
Schulz et al. (2024)23 | ERT | 23 | 106.7 (64.1) | 272 (199 to NE) weeks | 0.14 (0.06 to 0.33) | < 0.001 | Sensitivity analyses availablec |
NH | 42 | 76.1 (43.1) | 49 (39 to 58) weeks | ||||
Schulz et al. (2025)22 | ERT | 21 | NR | NR | 0.08 (0.02 to 0.26) | < 0.001 | None |
NH | 21 | NR | NR | ||||
Motor domain | |||||||
Schulz et al. (2024)23 | ERT | 23 | 106.7 (64.1) | NR | 0.03 (0.01 to 0.16) | < 0.001 | Sensitivity analyses availablec |
NH | 42 | 76.1 (43.1) | NR | ||||
Language domain | |||||||
Schulz et al. (2024)23 | ERT | 23 | 106.7 (64.1) | NR | 0.17 (0.07 to 0.44) | 0.0002 | Sensitivity analyses availablec |
NH | 41 | 76.1 (43.1) | NR | ||||
CI = confidence interval; CLN2 = neuronal ceroid lipofuscinosis type 2; ERT = enzyme replacement therapy; HR = hazard ratio; NE = not estimable; NH = natural history; NR = not reported.
aA study reported a 1-point change in the CLN2 Clinical Rating Scale motor-language score to be clinically meaningful, although the methodology behind this threshold is unclear and its validity as a minimally important difference remains uncertain.13
bAn HR less than 1 indicates a benefit in favour of ERT.
cSensitivity analyses using 3 matched variables (baseline age, motor-language score, and genotype) yielded results consistent with the primary analysis based on 2 variables (baseline age and motor-language score).
Table 9: Summary of Findings by Outcome — Unreversed Function Score of Zero in the CLN2 Clinical Rating Scale Motor-Language Scorea
Study | Group | Sample size (n) | Follow-up, mean (SD), weeks | Median time to event (95% CI) | HR (95% CI)b | P value |
|---|---|---|---|---|---|---|
Schulz et al. (2024)23 | ERT | 23 | 106.7 (64.1) | NE | 0.02 (0.00 to 0.08) | < 0.001 |
NH | 42 | 76.1 (43.1) | 109 (90 to 129) weeks | |||
Schulz et al. (2025)22 | ERT | 21 | NR | NR | 0.07 (0.01 to 0.40) | 0.003 |
NH | 21 | NR | NR |
CI = confidence interval; CLN2 = neuronal ceroid lipofuscinosis type 2; ERT = enzyme replacement therapy; HR = hazard ratio; NE = not estimable; NH = natural history; NR = not reported.
aA study reported a 1-point change in the CLN2 Clinical Rating Scale motor-language score to be clinically meaningful, although the methodology behind this threshold is unclear and its validity as a minimally important difference remains uncertain.13
bAn HR less than 1 indicates a benefit in favour of ERT.
Table 10: Summary of Findings by Outcome — Rate of Function Decline in CLN2 Clinical Rating Scalea
Study | Group | Sample size (n) | Mean (SD) per 48 weeks | MD (95% CI)b per 48 weeks | P value | Notes |
|---|---|---|---|---|---|---|
Motor-language score | ||||||
Schulz et al. (2024)23 | ERT | NR | 0.64 (0.09) | 1.35 (0.99 to 1.71) | < 0.0001 | Adjusted for baseline covariates: age, baseline motor-language score, sex and genotype |
NH | NR | 1.99 (0.14) | ||||
ERT | NR | 0.38 (0.50) | 1.75 (1.39 to 2.11) | < 0.0001 | Unadjusted | |
NH | NR | 2.13 (0.95) | ||||
Schulz et al. (2025)22 | ERT | 21 | 0.46 (0.43) | 1.42 (0.74 to 2.10) | 0.0003 | Unadjusted |
NH | 21 | 1.88 (1.45) | ||||
Motor score | ||||||
Schulz et al. (2025)22 | ERT | 20 | 0.23 (0.28) | 0.75 (0.38 to 1.12) | 0.0003 | Unadjusted |
NH | 20 | 0.99 (0.75) | ||||
Language score | ||||||
Schulz et al. (2025)22 | ERT | 18 | 0.16 (0.28) | 0.88 (0.40 to 1.36) | 0.001 | Unadjusted |
NH | 19 | 1.04 (0.96) | ||||
CI = confidence interval; CLN2 = neuronal ceroid lipofuscinosis type 2 ; ERT = enzyme replacement therapy; MD = mean difference; NE = not estimable; NH = natural history; NR = not reported, SD = standard deviation.
aA study reported a 1-point change in the CLN2 Clinical Rating Scale motor-language score to be clinically meaningful, although the methodology behind this threshold is unclear and its validity as a minimally important difference remains uncertain.13
bAn MD greater than 0 indicates a benefit in favour of ERT.
Table 11: Summary of Findings by Outcome — Function Scores at Different Time Points in CLN2 Clinical Rating Scalea
Study | Time points | Groups | Sample size (n) | Mean (SD) | Notes |
|---|---|---|---|---|---|
Motor-language score | |||||
Schulz et al. (2024)23 | Baseline | ERT | 23 | 3.5 (1.2) | None |
— | NH | 42 | NR | None | |
Week 145 | ERT | NR | 2.8 (1.43) | None | |
— | NH | NR | Approaching 0 | None | |
Week 313 | ERT | NR | 1.8 (1.07) | None | |
— | NH | NR | NR | None | |
Estublier et al. (2021)21 | Diagnosis | ERT | 7 | 3.6 (NR) | None |
— | NH | NR | NR | None | |
Baseline | ERT | 7 | 3.1 (NR) | The beginning of ERT | |
— | NH | NR | NR | None | |
Last evaluation | ERT | 7 | 2.8 (NR) | Changeb: −0.3 (NR) | |
— | NH | NR | NR | Changeb: −2.5 (NR) | |
Motor score | |||||
Schulz et al. (2024)23 | Baseline | ERT | 23 | 2.0 (0.6) | None |
— | NH | 42 | 2.4 (0.5) | None | |
Week 289 | ERT | 18 | 1.0 (0.5) | Changeb: −1.0 (0.6) | |
— | NH | 17 | 0 (0) | Changeb: −2.5 (0.5) | |
Language score | |||||
Schulz et al. (2024)23 | Baseline | ERT | 23 | 1.4 (0.7) | None |
— | NH | 42 | 2.1 (0.7) | None | |
Week 289 | ERT | 18 | 0.8 (0.7) | Changeb: −0.6 (0.8) | |
— | NH | 16 | 0 (0) | Changeb: −2.3 (0.5) | |
Vision score | |||||
Schulz et al. (2024)23 | Baseline | ERT | 23 | 2.9 (0.3) | None |
— | NH | 39 | 2.5 (0.6) | None | |
Week 289 | ERT | 18 | 1.5 (0.9) | Changeb: −1.4 (0.9) | |
— | NH | 16 | 0.2 (0.6) | Changeb: −2.3 (0.9) | |
Seizure score | |||||
Schulz et al. (2024)23 | Baseline | ERT | 23 | 1.7 (1.2) | None |
— | NH | 35 | 1.7 (1.1) | None | |
Week 289 | ERT | 18 | 2.4 (0.8) | Changeb: 0.8 (1.2) | |
— | NH | 15 | 0.7 (1.2) | Changeb: −1.0 (1.0) | |
CLN2 = neuronal ceroid lipofuscinosis type 2 ; ERT = enzyme replacement therapy; NH = natural history; NR = not reported, SD = standard deviation.
aA study reported a 1-point change in the CLN2 Clinical Rating Scale motor-language score to be clinically meaningful, although the methodology behind this threshold is unclear and its validity as a minimally important difference remains uncertain.13
bMean (SD) change from baseline (the beginning of ERT) to the time point in patients with available measurements at both time points; in 1 study,23 the baseline motor-language score for the NH cohort was defined as the first score of less than 6 occurring at or after 36 months of age; 1 study21 did not report on the definition for baseline motor-language score.
Table 12: Summary of Findings by Outcome — Mortality
Study | Group | Sample size (n) | Follow-up, mean (SD), weeks | Number of events | HR (95% CI)a | P value | Notes |
|---|---|---|---|---|---|---|---|
From birth to the time of death or censoring | |||||||
Schulz et al. (2024)23 | ERT | 23 | 106.7 (64.1) | 0 | 0.04 (0.00 to 0.30) | 0.029 | None |
NH | 42 | 76.1 (43.1) | 22 | None | |||
From the baseline to the time of death or censoring | |||||||
Schulz et al. (2024)23 | ERT | 17 | 106.7 (64.1) | 0 | 0.04 (0.00 to 0.42) | 0.047 | None |
NH | 17 | 76.1 (43.1) | 4 | Median Time to death: 313 weeks | |||
Schulz et al. (2025)22 | ERT | 21 | 106.7 (64.1) | 0 | NR | NR | None |
NH | 21 | 76.1 (43.1) | 6 | Median Time to death: 313 weeks | |||
CI = confidence interval; ERT = enzyme replacement therapy; HR = hazard ratio; NE = not estimable; NH = natural history; NR = not reported.
aAn HR less than 1 indicates a benefit in favour of ERT.
Table 13: Summary of Findings by Outcome — Quality of Life for Patients Treated With ERT: PedsQL Parent Report for Toddlers
Study | Time points | Sample size (n) | Mean (SD) | Mean changes (SD)a |
|---|---|---|---|---|
Physical functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 63.5 (22.2) | −29.6 (26.8) |
Study completion (up to 240-week follow-up) | 18 | 40.2 (23.9) | ||
Emotional functioning | ||||
Schulz et al. (2024)23 | Baseline | 22 | 70.2 (20.7) | 2.9 (21.5) |
Study completion (up to 240-week follow-up) | 18 | 70.6 (12.7) | ||
Social functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 49.8 (15.9) | −10.6 (17.1) |
Study completion (up to 240-week follow-up) | 18 | 40.8 (22.8) | ||
School functioning | ||||
Schulz et al. (2024)23 | Baseline | 22 | 57.0 (19.6) | −16.4 (34.4) |
Study completion (up to 240-week follow-up) | 17 | 45.6 (24.9) | ||
ERT = enzyme replacement therapy; SD = standard deviation.
aMean change from baseline to study completion in patients with available measurements at both time points; higher score indicates better quality of life; minimal important difference: not reported.
Table 14: Summary of Findings by Outcome — Quality of Life for Patients Treated With ERT: PedsQL Family Impact Module
Study | Time points | Sample size (n) | Mean (SD) | Mean changes (SD)a |
|---|---|---|---|---|
Physical functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 63.4 (16.5) | −4.8 (22.3) |
Study completion (up to 240-week follow-up) | 19 | 57.7 (18.2) | ||
Emotional functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 54.3 (20.6) | 0.3 (16.4) |
Study completion (up to 240-week follow-up) | 19 | 55.0 (17.3) | ||
Social functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 63.3 (22.1) | −3.0 (21.1) |
Study completion (up to 240-week follow-up) | 19 | 56.9 (20.6) | ||
Cognitive functioning | ||||
Schulz et al. (2024)23 | Baseline | 23 | 62.4 (21.6) | −1.6 (33.0) |
Study completion (up to 240-week follow-up) | 19 | 58.4 (21.0) | ||
Communication | ||||
Schulz et al. (2024)23 | Baseline | 23 | 64.9 (22.3) | 0.4 (20.1) |
Study completion (up to 240-week follow-up) | 19 | 61.0 (21.0) | ||
ERT = enzyme replacement therapy; SD = standard deviation.
Note: A higher score indicates better family functioning; minimal important difference: not reported.
aMean change from baseline to study completion in patients with available measurements at both time points; HRQoL scores were missing at study completion for 4 out of 23 participants (17%) in various domains.
Table 15: Summary of Findings by Outcome — Quality of Life for Patients Treated With ERT: CLN2 Disease-Specific Quality of Life Dimension Scoresa
Study | Time points | Sample size (n) | Mean (SD) | Mean changes (SD)b |
|---|---|---|---|---|
Seizures domain | ||||
Schulz et al. (2024)23 | Baseline | 22 | 64.2 (18.2) | 12.5 (20.1) |
Study completion (up to 240-week follow-up) | 19 | 81.8 (19.1) | ||
Feeding (no G-tube) | ||||
Schulz et al. (2024)23 | Baseline | 22 | 75.6 (22.6) | −4.9 (29.8) |
Study completion (up to 240-week follow-up) | 17 | 72.2 (19.8) | ||
Feeding (with G-tube) | ||||
Schulz et al. (2024)23 | Baseline | Not measured | Not measured | NA |
Study completion (up to 240-week follow-up) | 6 | 87.5 (13.7) | ||
Sleep | ||||
Schulz et al. (2024)23 | Baseline | 22 | 79.8 (25.9) | −3.4 (24.1) |
Study completion (up to 240-week follow-up) | 20 | 77.5 (18.5) | ||
Behaviour | ||||
Schulz et al. (2024)23 | Baseline | 22 | 73.7 (13.6) | −1.8 (18.8) |
Study completion (up to 240-week follow-up) | 20 | 73.1 (11.7) | ||
Daily activities | ||||
Schulz et al. (2024)23 | Baseline | 22 | 81.7 (15.4) | −18.2 (21.8) |
Study completion (up to 240-week follow-up) | 20 | 64.5 (18.6) | ||
CLN2 = neuronal ceroid lipofuscinosis type 2; ERT = enzyme replacement therapy; G-tube = gastrostomy tube; NA = not applicable; SD = standard deviation.
aA study reported distribution-based clinically important differences for several domains: seizures (9.08), sleep (12.94), behaviour (6.79), and daily activities (7.67).24
bMean change from baseline to study completion in patients with available measurements at both time points; higher score indicates better quality of lie; minimal important difference: not reported.
Table 16: Summary of Findings by Outcome — AEs for Patients Treated With ERT
Outcomes | Schulz et al. (2025),22 n = 24 | Schulz et al. (2024),23 n = 24 | Estublier et al. (2021),21 n = 8 | |
|---|---|---|---|---|
Number (%) patients who experienced 1 or more AEs (%) | AEs resulting in hospitalization (%) | Number (%) patients who experienced 1 or more AEs (%) | Number (%) patients who experienced 1 or more AEs (%) | |
Any treatment-related AEs | 16 (66.7%) | 7 (29.2%) | 24 (100%) | NR |
Grade 2 | 14 (58.3%) | 3 (12.5%) | NR | NR |
Grade 3 | 1 (4.2%) | 1 (4.2%) | 18 (75%) | NR |
Grade 4 | 3 (12.5%) | 3 (12.5%) | 3 (13%) | NR |
Any serious AEs | 4 (17%) | 4 (17%) | 21 (88%) | NR |
Deaths due to AEs | 0 | NA | 0 | 0 |
WDAEs | 0 | NA | 0 | 0 |
AE = adverse event; ERT = enzyme replacement therapy; NA = not applicable; NR = not reported; SD = standard deviation; WDAE = withdrawal due to adverse event.
Table 17: Summary of Findings by Outcome — AEs for Patients Treated With ERT
Outcomes | Schulz et al. (2025),22 n = 24 | Schulz et al. (2024),23 n = 24 | Estublier et al. (2021),21 n = 8 | |
|---|---|---|---|---|
Number (%) patients who experienced 1 or more AEs (%) | AEs resulting in hospitalization (%) | Number (%) patients who experienced 1 or more AEs (%) | Number (%) patients who experienced 1 or more AEs (%) | |
Upper respiratory tract infection | NR | NR | 21 (88%) | NR |
Pyrexia or fever | 12 (50.0%) | 4 (16.7%) | 20 (83%) | NR (indicated common but data were not available) |
Viral upper respiratory tract infection | NR | NR | 19 (79%) | NR |
Vomiting | 8 (33.3%) | 5 (20.8%) | 19 (79%) | NR |
Nausea | 5 (20.8%) | 2 (8.3%) | NR | NR |
Arrythmia | 3 (12.5%) | 0 (0) | NR | NR |
Generalized tonic-clonic seizure | NR | NR | 16 (67%) | NR |
Seizure | 3 (12.5%) | 0 (0) | 14 (58%) | NR |
Constipation | NR | NR | 13 (54%) | NR |
Device end of service | NR | NR | 13 (54%) | NR |
Device leakage | 2 (8.3%) | 2 (8.3%) | NR | NR |
Dysphagia | NR | NR | 13 (54%) | NR |
Epilepsy | NR | NR | 13 (54%) | NR |
Rhinitis | NR | NR | 13 (54%) | NR |
Gait disturbance | NR | NR | 12 (50%) | NR |
Cough | NR | NR | 11 (46%) | NR |
Dystonia | NR | NR | 11 (46%) | NR |
Tremor | NR | NR | 11 (46%) | NR |
Visual impairment | NR | NR | 11 (46%) | NR |
Hypersensitivity | NR | NR | 10 (42%) | NR |
Myoclonus | NR | NR | 10 (42%) | NR |
Device-related infection | 3 (12.5%) | 3 (12.5%) | 9 (38%) | 4 (50%)a |
Diarrhea | NR | NR | 9 (38%) | NR |
Extensor plantar response | NR | NR | 9 (38%) | NR |
Gastroenteritis | NR | NR | 9 (38%) | NR |
Needle issue | NR | NR | 9 (38%) | NR |
Sleep disorder | NR | NR | 9 (38%) | NR |
Viral infection | NR | NR | 9 (38%) | NR |
Flushing | 1 (4.2%) | 0 (0) | NR | NR |
Headache | 1 (4.2%) | 1 (4.2%) | NR | NR |
ERT = enzyme replacement therapy; NR = not reported.
aIncluding suspected infection.
Table 18: Summary of Findings by Outcome — SAEs for Patients Treated With ERT
Outcomes | Schulz et al. (2024),23 n = 24 |
|---|---|
Number (%) patients who experienced 2 or more SAEs (%) | |
Device end of service | 13 (54%) |
Hypersensitivity | 7 (29%) |
Device-related infection | 7 (29%) |
Upper respiratory tract infection | 5 (21%) |
Dysphagia | 4 (17%) |
Gastroenteritis | 4 (17%) |
Pleocytosis | 3 (13%) |
Dental caries | 2 (8%) |
Device deployment issue | 2 (8%) |
Epilepsy | 2 (8%) |
Pharyngitis bacterial | 2 (8%) |
Pyelonephritis | 2 (8%) |
Pyrexia | 2 (8%) |
ERT = enzyme replacement therapy; SAE = serious adverse event.
Table 19: Summary of Recommendations in Included Guidelines
Recommendation | Strength of the recommendation and quality of evidence | Supporting evidence and/or rationale |
|---|---|---|
Sampaio et al. (2023)25 | ||
“The recommended dose for treating individuals with CLN2 is 300 mg every 2 weeks, administered via intracerebroventricular (ICV) infusion, performed with a dilution of 300 mg in 10 mL and infused in 4 hours (consensus; LE: 1b)” (p. 291) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 1b | The guideline cited the following reference to support the recommendation: Schulz A, Ajayi T, Specchio N, et al.; CLN2 Study Group. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med 2018;378(20):1898 to 1907 |
“The essential devices for the proper application are a permanent ICV infusion catheter (Ommaya type) and an infusion pump for a syringe (consensus; LE: 5).” (p. 291) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
“A child neurologist, a neurosurgeon, a pediatrician, and a nurse should be involved in the team for the infusion (consensus; LE: 5).” (p. 291) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
“The minimum necessary conditions for the use of ERT to reduce the risk of infection or other complications are: (a) a room with isolation and strict asepsis; (b) a restriction in the number of people in the room during the infusion, avoiding visitors and other professionals not essential to the procedure; (d) regular monitoring of vital signs; and (e) monitoring with equipment throughout the infusion period (consensus; LE: 4)” (p. 291 and p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 4 | NR |
“The infusion can be conducted in the hospital ward, as long as there are no other patients with infection (consensus; LE: 4)” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 4 | NR |
“After the placement of the infusion catheter, immediate care should avoid contamination of the dressing and trauma at the procedure site (consensus; LE: 4)” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 4 | NR |
The minimum period between the placement of the ventricular intracerebral device by the neurosurgeon until the first ERT infusion is 5 to 7 days or maybe longer, depending on local edema (consensus; LE: 5). | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
The ICV catheter must be changed every 4 years to decrease the infection or CSF leakage risk (consensus; LE: 5) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
“To minimize the side effects of cerliponase alpha, we should infuse an antihistamine drug 30 minutes before the enzyme (consensus; LE: 1b)” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 1b | NR |
“The eligible patients with CLN2 for ERT are children, ≥ 3 years old, with a Clinical Rating Scale ≥ 3 points (consensus; LE: 1b)” (p. 292)” | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 1b | NR |
“There is no current data to support the beneficial effect in patients with a score below three, and the benefits in a more advanced stage of the disease are not clear (consensus; LE: 5)” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
“Children with a confirmed diagnosis of presymptomatic CLN2 should be treated with Cerliponase alpha (consensus; LE: 5). (consensus; LE: 5). However, there is not enough evidence to define the best time to start treatment in those patients as the clinical trial only evaluated symptomatic patients (consensus; LE: 5).” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | The guideline cited the following reference to support the recommendation: Schulz A, Ajayi T, Specchio N, et al.; CLN2 Study Group. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med 2018;378(20):1898 to 1907 |
“There is not enough data to establish the criteria for discontinuing the use of Cerliponase alpha (consensus; LE: 5).” (p. 292) | The recommendation was agreed upon by at least 75% of the panel. Level of evidence: 5 | NR |
Mole, et al. (2021)26a | ||
“Initiation of long-term ERT with Cerliponase alfa at 300 mg (or age-appropriate) dose every other week through intraventricular infusion is suggested in non-classical TPP1 deficiency patients after confirmed diagnosis and agreement between parents and provider, as long as no contraindications to therapy exist. Initiation of long-term ERT with Cerliponase alfa at 300 mg (or age-appropriate) dose every other week through intraventricular infusion is recommended in classical CLN2 patients with the potential to benefit from this therapy.” (p. 9 in table 5) | Among 37 responders, the recommendation received consensus from 84% of experts. Level of evidence: C | NR |
“Disease-modifying treatment with a licensed therapy ideally should be delivered by a team experienced in the management of CLN2 disease and use of any required devices. For current ERT treatment for CLN2 disease, this includes brain intraventricular devices” (p. 9 in table 5) | Among 39 responders, the recommendation received consensus from 93% of experts. Level of evidence: C | NR |
“Intraventricular devices should be placed under general anaesthesia by a very experienced paediatric neurosurgeon” (p. 9 in table 5) | Among 36 responders, the recommendation received consensus from 92% of experts. Level of evidence: C | NR |
“Intraventricular device should only be accessed by a trained individual to limit/minimise complications” (p. 9 in table 5) | Among 39 responders, the recommendation received consensus from 95% of experts. Level of evidence: C | NR |
CLN2 = neuronal ceroid lipofuscinosis type 2; ERT = enzyme replacement therapy; ICV = intracerebroventricular; LE = level of evidence; NR = not reported.
aIn the discussion section of the guideline, the guideline author mentioned that ERT treatment should be discontinued in patients who are affected by another life-limiting condition or experience severe infusion-related adverse reactions that are not preventable or manageable.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.