CADTH Health Technology Review
Design, Rationale, and Preliminary Results of the Canadian Homozygous Familial Hypercholesterolemia Registry: 2008 to 2022 Update
This article has been peer reviewed.
Peer-Reviewed Article
Authors: Leslie Brown, Isabelle Ruel, Alexis Baass, Jean Bergeron, Liam R. Brunham, Lubomira Cermakova, Patrick Couture, Daniel Gaudet, Gordon A. Francis, Robert A. Hegele, Iulia Iatan, G.B. John Mancini, Brian W. McCrindle, Thomas Ransom, Mark H. Sherman, Ruth McPherson, Jacques Genest
Abstract
Homozygous familial hypercholesterolemia (HoFH) is an orphan disease characterized by extreme elevations of low-density lipoprotein cholesterol (LDL-C) in the blood and premature atherosclerotic cardiovascular disease. Untreated, survival beyond 30 years is rare. HoFH is caused by pathogenic variants in the genes that regulate LDL-C clearance, especially the LDL receptor (LDLR) gene, inherited from both parents. The more common form, heterozygous familial hypercholesterolemia (HeFH) has a worldwide prevalence of 1 in 311, but HoFH prevalence varies between 1 in 386,000 and 1 in 1,000,000. There are estimated to be 38 to 100 overall cases in Canada, with an estimated 1 to 3 new cases per year. The Canadian HoFH Registry was created by clinicians from across Canada to understand the burden of disease, current treatments, outcomes, and costs to society. To date, we have identified 79 cases of HoFH across the country and have enrolled 52 patients from 5 provinces, with complete data available on 46 of these patients. The registry will be used to inform decisions for access to specialized therapies, such as LDL apheresis and orphan medications.
Introduction
The association between skin manifestations (tendinous xanthomas), markedly elevated cholesterol (especially low-density lipoprotein cholesterol [LDL-C]), and early onset heart attacks within families was made by Norwegian physician Karl Muller in 1939.1 Subsequently, the mode of inheritance — classified as heterozygous familial hypercholesterolemia (HeFH) or homozygous familial hypercholesterolemia (HoFH) — was characterized in the 1960s by Avedis Kachadurian.2 The definition of HoFH was revised several times, as increased knowledge of metabolic pathways and the genes controlling them developed over time. The genetic basis of HoFH was elucidated by Goldstein and Brown with the discovery of the LDL receptor (LDLR), for which they received the Nobel Prize in 1985.3
The diagnosis of HoFH is based on untreated elevated LDL-C above 13 mmol/L (the 95th percentile for Canadians < 20 years of age is 3.5 mmol/L), the presence of extensor and planar xanthomas, and both parents being affected with HeFH. Since the identification of genes involved in familial hypercholesterolemia (FH) — namely, the LDLR, apolipoprotein B (APOB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes, as well as LDLR adaptor protein 1 (LDLRAP1) gene — the definition has been extended to include severe hypercholesterolemia and bi-allelic (one from each parent) pathogenic variants.4 A more restrictive definition only includes bi-allelic LDLR gene variants. There is therefore considerable overlap between HoFH and severe HeFH (Figure 1). Furthermore, there are more than 2,000 pathogenic variants of the LDLR gene with different phenotypic expressions.5 “Null” variants that completely obliterate expression of LDLR are associated with a more severe clinical course and are less responsive to standard therapies than “defective” variants, for which some function is retained. Patients with HoFH have normal physical, cognitive, and sexual development.6
The worldwide prevalence of HeFH is estimated at 1 in 311, and that of HoFH at 1 in 386,000 to 1 in 1,000,000, which would predict an estimated 38 to 100 cases in Canada, based on the population of 38,000,000.7 The province of Quebec has a higher burden of HoFH due to at least 2 founder effects.8 The variability of pathogenic genetic variants reflects the demographics of Canadian society; thus, the Hardy-Weinberg equilibrium is no longer valid in the estimation of genetic homozygosity prevalence.
Medical care across Canada is under the jurisdiction of the governments of the 10 provinces and 3 territories, spread over 9.98 million km2. The FH Canada Registry was created in 2014 to bring together a network of collaborators to create a registry of patients with FH and other lipoprotein disorders.9 The registry network of collaborators includes more than 200 clinicians, scientists, and other health care professionals across Canada. Physicians include specialists in disorders of lipoproteins, cardiology, endocrinology, internal medicine, and family practice.
Figure 1: Spectrum of Cholesterol Range and Disease Severity in Familial Hypercholesterolemia
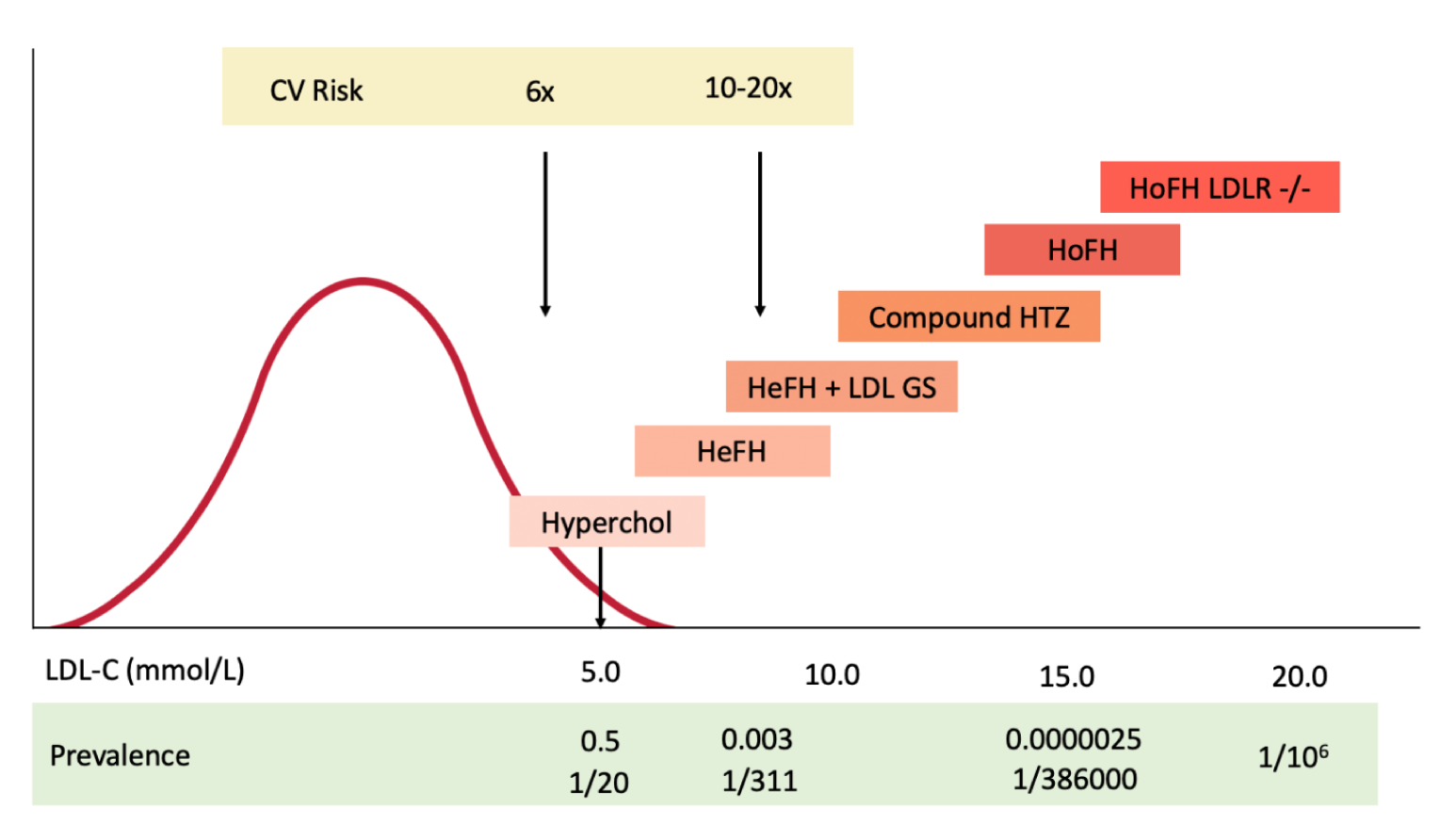
CV = cardiovascular; HeFH = heterozygous familial hypercholesterolemia; HoFH = homozygous familial hypercholesterolemia; HTZ = heterozygote; Hyperchol = hypercholesterolemia; LDL-C = low-density lipoprotein cholesterol; LDL GS = low-density lipoprotein genetic score; LDLR = low-density lipoprotein receptor.
Note: The curve represents the distribution of LDL-C levels, with the 95th percentile for adults living in Canada being 5.0 mmol/L.21 HoFH is defined as bi-allelic pathogenic variants of the LDLR gene. “Null” variants (no LDLR activity) have a more severe phenotype.
Source: Adapted from Bélanger et al.10 with permission.
The diagnosis of HoFH in Canada is made on the basis of cutaneous (skin) manifestations developing in childhood, markedly elevated LDL-C levels from birth, and, less commonly, very early atherosclerotic cardiovascular disease (ASCVD), such as myocardial infarction by age 20. Most patients are referred to a physician specializing in lipoprotein disorders. Treatment is aimed at lowering LDL-C levels. Survival of HoFH patients has doubled since the introduction of statins.10 The standard of care includes maximally tolerated statins and ezetimibe (an inhibitor of intestinal cholesterol absorption). Bile acid binding resins — such as cholestyramine, colestipol, or colesevelam — are used as well, although with very limited efficacy. Other approaches including PCSK9 inhibitors are used, as most patients cannot reach recommended treatment guidelines on currently available medications. The aim of therapy is to lower LDL-C levels according to the Canadian Cardiovascular Society guidelines.11,12 Thresholds for the intensification of therapy are an LDL-C level above 2.5 mmol/L in the primary prevention setting (no evidence of ASCVD) and above 1.8 mmol/L in secondary prevention, although these are rarely met with current therapies.13 Some patients require extracorporeal LDL filtration (LDL apheresis), in which LDL-C particles are selectively removed from the blood. This technique shares similarities with kidney dialysis and, despite being considered standard of care for severe HoFH, is only currently available in Quebec, Ontario (in pediatrics), and Alberta.14 In other provinces, plasmapheresis (a form of plasma exchange) is sometimes used. Because of these approaches, patients live now into their sixth decade of life,15 albeit with a considerable burden of cardiovascular disease (especially cardiovascular interventions)16 and reduced quality of life.17
The response to drug therapy is dependent on the type of genetic variant causing FH. Genetic variants that completely abolish the LDLR (“null” variants) have a poor response to conventional drugs. Novel therapeutics include lomitapide (Juxtapid), an inhibitor of microsomal triglyceride transfer protein used in HoFH in Canada, although tolerance and liver toxicity are major issues.18 Inhibitors of proprotein convertase subtilisin kexin type 9 (PCSK9), including the monoclonal antibodies evolocumab (Repatha) and alirocumab (Praluent) have modest LDL-C lowering efficacy in HoFH patients with 1 or more null variants. A novel compound, evinacumab (Evkeeza), is a human monoclonal antibody directed at angiopoietin-like 3 protein (ANGPTL3) that is being investigated in patients with HoFH, including patients with null mutations of the LDLR gene.19,20 Lastly, gene therapy consisting of adenovirus associated virus-8 mediated gene transfer or somatic genetic modification using genome editing is undergoing active research.
The Canadian HoFH Registry was created to understand the burden of disease of HoFH, current treatments, outcomes, and costs to society, as well as to provide scientific evidence for advocacy for greater access to specialized therapies.
Methods
Registry Development
The first FH registry in Canada was established in the province of British Columbia in 201322 and consolidated into the FH Canada Registry a year later.9 Since that time, the national FH Canada Registry has expanded to include 19 academic centres across Canada, as well as numerous peripheral sites, in a “hub and spoke” model.23 The “hubs” are based in each province in centres recognized at the national level for their expertise in lipoprotein disorders and have advanced laboratory facilities. Radiating from these centres are the clinics that treat patients with FH, the “spokes.” An advisory panel including patient representatives was involved in the registry design, validation of data entry, and initiation. The project is registered at ClinicalTrials.gov (#NCT02009345). Data from patients with rare disorders of lipoprotein metabolism (orphan diseases) are being collected as well, in a portion of the registry called Systems and Molecular Approaches to Severe Hyperlipidemias (SMASH). It includes, for example, HoFH, familial chylomicronemia syndrome, LCAT deficiency, Tangier disease, and sitosterolemia. Using the infrastructure from the FH Canada Registry, and through a collaboration with the lipid specialists actively treating patients with HoFH in Canada, we have subsequently created the Canadian HoFH Registry. A yearly update of vital and clinical status will allow longitudinal follow-up of these patients. Data are entered into the registry by the national coordinator, after capture by the referring centre specialist, using the data dictionary.
Financial Maintenance
The biopharmaceutical industry provided seed money to initiate the FH Canada Registry and helped to put in place the infrastructure to approach provincial and federal granting agencies (e.g., Fonds de la Recherche du Québec – Santé [FRQ-S] in Quebec and the Canadian Institutes of Health Research [CIHR]). The creation of the Canadian HoFH Registry was made possible after leveraging the FH Canada Registry to be granted with the Knowledge Synthesis Grant on Socio-Economic Burden of Inherited Disease from CIHR, entitled Homozygous Familial Hypercholesterolemia (HoFH) in Canada (CIHR SBI-167982). The long-term maintenance of the Canadian HoFH Registry will be dependent upon continued support from biopharmaceutical industry and Canadian peer-reviewed funding agencies. Such registries are in keeping with initiatives from CIHR24 and the Réseau Québecois des Maladies Orphelines (RQMO; https://publications.msss.gouv.qc.ca/msss/fichiers/2022/22-916-01W.pdf).25
Registry Representation
Canada has a long history of excellence in the field of lipoprotein disorders. All collaborators participating in the Canadian HoFH Registry are internationally recognized lipid specialists and have extensive experience in the diagnosis and treatment of HoFH. They have published significantly on clinical outcomes in FH patients, including patients with HoFH,22 and have contributed to the generation of clinical guidelines for the diagnosis and treatment of patients with FH in Canada endorsed by the Canadian Cardiovascular Society.11 Collecting data on a majority of HoFH patients in Canada is therefore highly feasible, with only a minority of unknown or undiagnosed cases in communities lacking access to specialized lipid clinics being overlooked.
Governance, Privacy, and Security
Ownership
The leadership of the registry rests with the members of the steering committee, who are specialists in the diagnosis and treatment of HoFH in Canada, and a national coordinator (please refer to author affiliations for more details). They work as a team to manage the registry and guide decision-making related to funding, operations, and dissemination of information.
Security
A secure database should fulfill the following criteria:
It must comply with privacy regulations in all provinces and use passwords that allow limited access, with access tailored to each site.
It must allow researchers access under strictly controlled conditions after peer-review.
The software must be flexible enough to allow easy data entry.
The James Hogg Research Centre at St. Paul’s Hospital, University of British Columbia, Vancouver is providing the custom-built secure research platform, named iCAPTURE, used to enter the data for the FH Canada Registry and the Canadian HoFH Registry. This centre maintains multi-level security for all database and computer systems. The security measures require physical entry to the computer facility and limit access to the systems within the facility. The data centre is a single-entry, electronic card–accessible room that is equipped with video surveillance. Entry is accessible to authorized personnel only.
The database utilizes an Oracle backend and is firewalled and maintained in a separate Providence Research network, and it is US FDA, Health Canada, PHIA (Personal Health Information Act), and PIPEDA (Canada Personal Information Protection and Electronic Documents Act) compliant. All data are de-identified based on role-based access and redacted automatically within the database. All connections to and from the system are encrypted with 256-bit AES encryption that is updated on an annual basis. Saved records are stored on an encrypted storage array, also with 256-bit AES encryption. Backups are maintained on site and in the British Columbia Government Q9 data centre in Kamloops, British Columbia.
All coordinators are granted usage via role-based access, use two-factor authentication, and have their account passwords updated every 90 days. All user access is logged. The coordinators and their roles are validated on a yearly basis. All coordinator actions while accessing the system are logged, including entry, editing, and viewing of records.
Privacy and Ethics
The national FH Canada Registry was first approved by the Research Ethics Board (REB) of the McGill University Health Centre in November 2013. The project proposal, data questionnaire, and consent forms, adapted to include the specific site information from each Canadian investigator, were then approved by their individual institutional REB. At each site, the consent process includes explanation of the nature of the registry, the type of data to be collected, the risks and benefits, and signature of the consent form after assessment of the patient’s comprehension by the site coordinator.
At each local site, data entry is performed using nominative information and coded specifically to the institution (i.e., with the hospital identification number). Data privacy is made possible by the research platform, which randomly generates a unique identifier for each patient. Since the platform has the flexibility to allow limited access to specific information, no single individual or research group has access to data that should remain confidential. Only completely anonymous data, without any identifier — such as name, hospital identification number, or date of birth — is made available during the data extraction process. Data analysis is performed via a secure privileged remote access (PRA) method that records all tasks and data usage by the data analyst. Data cannot leave the data centre.
Data Structure
Connectivity
The data collected in the Canadian HoFH Registry is in line with the HoFH International Clinical Collaborators initiative (HICC; ClinicalTrials #NCT04815005), the only international registry on HoFH. In fact, because both registries are using similar datasets and have a data transfer agreement, the data on the first 15 patients with HoFH enrolled in the Canadian HoFH Registry has been shared with the HICC consortium.26 At present, the Canadian HoFH Registry has no direct access to other data sources, such as provincial administrative databases, electronic medical records, prescription drug databases, and so forth.
Data Collection
Patients are eligible for inclusion into the Canadian HoFH Registry if they have received a clinical or genetic diagnosis of HoFH by their treating physician.5 When genetic testing is available, patients are considered to have HoFH if they carry bi-allelic FH pathogenic variants (either homozygous, compound heterozygous, or double heterozygous). Little is known regarding the clinical manifestations of pathogenic genetic variants; therefore, patients who have double heterozygosity (i.e., digenic inheritance involving 1 LDLR variant plus 1 APOB or PCSK9 variant) will be entered into the registry, and will also be analyzed separately. If necessary, the treating physician can refer a patient with HoFH to an HoFH specialist who participates in the registry for the data collection. The year 2008 is used as the starting point, as we started collecting data on patients with HoFH at that time and aim to provide major health outcomes for at least 10 years.27 No final date of data collection has been determined. The Canadian HoFH Registry is using an internationally validated data dictionary.26 Briefly, upon consent from the patient, the site coordinator can access the patient’s medical chart to collect data on genetics for FH, baseline lipid levels, physical signs at time of diagnosis, family history, major adverse cardiovascular events, hospitalizations, medications or treatments in time, and treated lipid levels in time. The data on patients with HoFH collected by the Canadian registry sites are to be entered by the national coordinator in the HoFH registry database, using the web-based iCAPTURE platform described previously, as a sub-registry of the larger FH Canada Registry.23 After registration, yearly follow-up data including the same variables can be added when available.
Data Sharing
The data from the Canadian HoFH Registry can be shared with other researchers in the field of lipid disorders, as indicated on the consent form. Each request for research projects using the registry data is required to demonstrate local ethics approval. Data are provided in a de-identified manner and as aggregate data. Specific data analyses can be made available to policy-makers, governments, and industry on a per-need basis after submission of a written request and approval by the steering committee members.
Registry Additions
The data dictionary used in the registry was created and validated by experts in the field of HoFH and is therefore quite complete. If additional variables are collected, an amendment including an updated version of the data dictionary must be submitted and approved by the REB at each individual Canadian HoFH Registry site. The structure of the FH Canada Registry can be adapted to another patient population or data on another disorder of lipids or lipoproteins in humans.
Data Quality
The data dictionary has been validated.26 Data are often numerical with fixed ranges or categories with determined options. Each variable is clearly defined in the data dictionary, and the web-based database uses pre-determined fields and subroutines to ensure the correctness of the information recorded in the registry. Entry errors are displayed automatically. In an orphan disease registry such as the Canadian HoFH Registry, missing data — especially data at the time of diagnosis — cannot be completely avoided. The national coordinator is responsible for making sure missing data are kept to a minimum through regular communication with individual sites.
ReQueST Tool Learnings
The Registry Evaluation and Quality Standards Tool (REQueST) was developed by the European Network for Health Technology Assessment, with the aim of evaluating the suitability and reliability of registries.28 The tool uses criteria and standards published in existing guidelines, frameworks, and projects, and covers all important aspects relating to the quality of registries. We recently validated the Canadian HoFH Registry using the ReQueST tool and found it useful to maximize the quality of the registry data. The tool would be essential when setting up a new registry.
Value of Data
Registry Value and Use in Policy
The Canadian HoFH Registry presents a unique opportunity for funders and stakeholders to have access to a group of experts in HoFH across Canada, with international links, and to a harmonized database for patients with HoFH. With access to a network of established clinics and a unified database, clinically oriented projects can be rapidly implemented. The initiative allows the framework to set up collaborative research on HoFH under the peer review system (HSFC, CIHR). The data collected can be used in health outcomes and health economic studies to determine the burden of the disease in Canada, identify regional disparities, and allow resource allocation. It can allow the development of clinical practice guidelines specific for HoFH aid, with precise clinical, phenotypic, and genetic diagnoses, and will provide personalized medicine for these patients. It provides patients with expert care through the FHCanada network, as well as access to ongoing clinical trials and novel therapies. Any results from data analyses will be published in peer-reviewed journals specializing in lipid disorders and prevention of cardiovascular diseases. These publications will be extremely useful for knowledge transfer and teaching, and to work with the Canadian Organization for Rare Disorders (CORD),29 as well as regulatory agencies and orphan disease advocacy groups in each province to advocate for access to diagnosis and treatment for patients with HoFH.
Initial Results
In 2019, a preliminary survey sent to clinicians across the FH Canada network originally identified 79 potential HoFH patients, in keeping with the estimated prevalence in Canada. A preliminary assessment for the inclusion criteria, and the inclusion of additional eligible patients, narrowed the expected number of patients to 69. The main reasons for exclusion included the inability to confirm HoFH diagnosis unambiguously, mainly due to unfamiliarity of the disorder to the treating physician. Virtual meetings of the members of the steering committee identified another 9 patients (Figure 2). A further 16 patients were excluded because of lack of patient consent, loss of patients to follow-up, and lack of data collection due to various reasons.
Figure 2: Search Strategy of the Registry
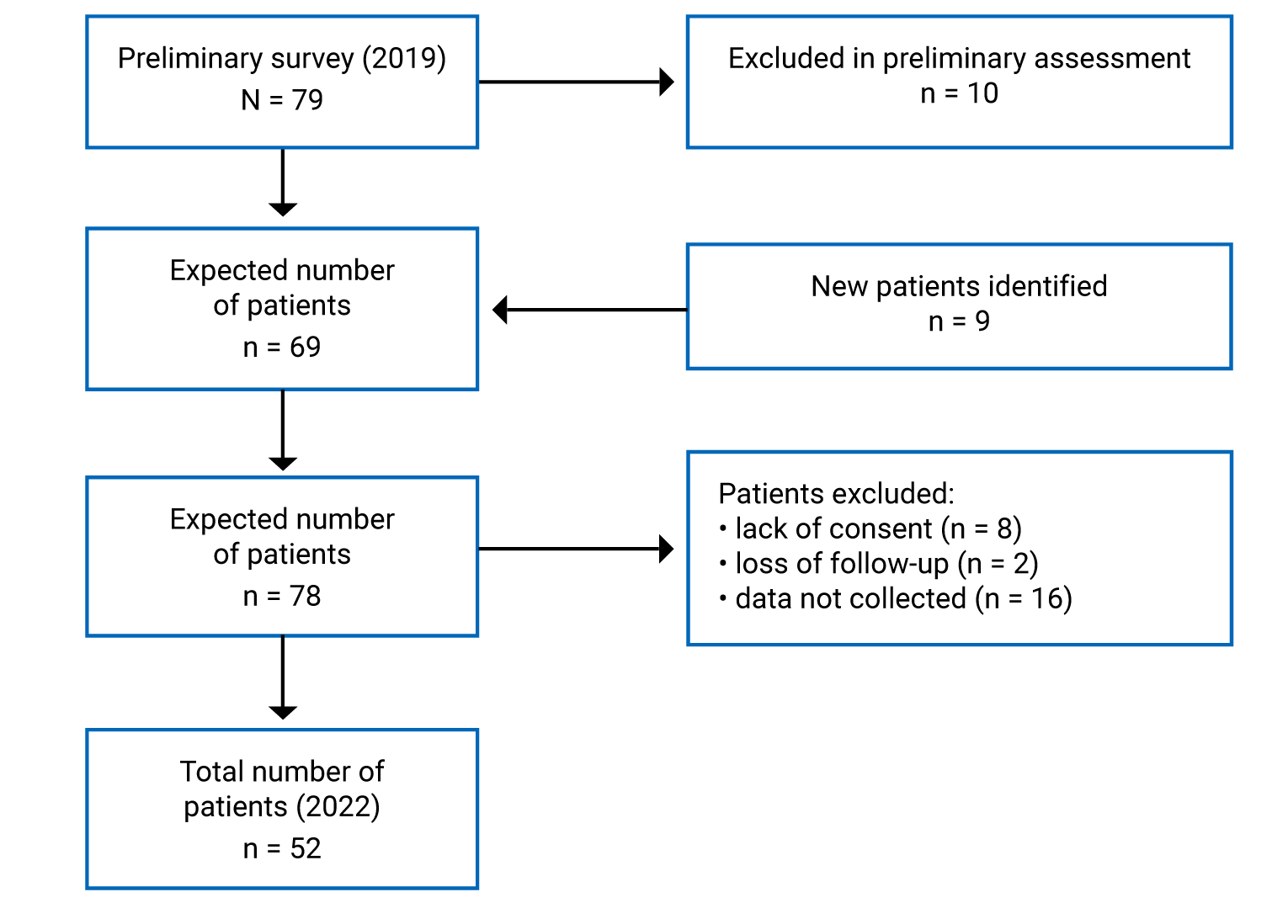
Note: Patients were excluded based on preliminary assessments of their diagnosis. The inclusion of additional patients not captured in the original survey is also shown.
As of April 2022, the Canadian HoFH Registry has enrolled 52 patients across 5 provinces, and holds complete data on 46 of those patients. The provincial distribution of patients is outlined in Figure 3. Based on 52 cases, a majority (69%) were found in Quebec, attributable predominantly to founder effects of 2 distinct variants of the LDLR gene. Of the enrolled individuals, 41 (89%) had a confirmed genetic diagnosis with mutations described. Patient demographics are briefly described in Table 1.
Table 1: Demographics of Patients in the Canadian HoFH Registry
Demographics | Number of patients |
|---|---|
Patients enrolled (n) | 52 |
Patients captured (n) | 46 |
Male, n (%) | 21 (45.6) |
Pediatric cases,a n (%) | 7 (15.2) |
Age, years (mean ± SD) | 44 ± 19 |
Age of diagnosis, years (mean ± SD) | 16 ± 4 |
HoFH = Homozygous familial hypercholesterolemia; SD = standard deviation.
aYounger than 18 years of age.
Figure 3: Geographic Distribution of Patients With HoFH in the Registry
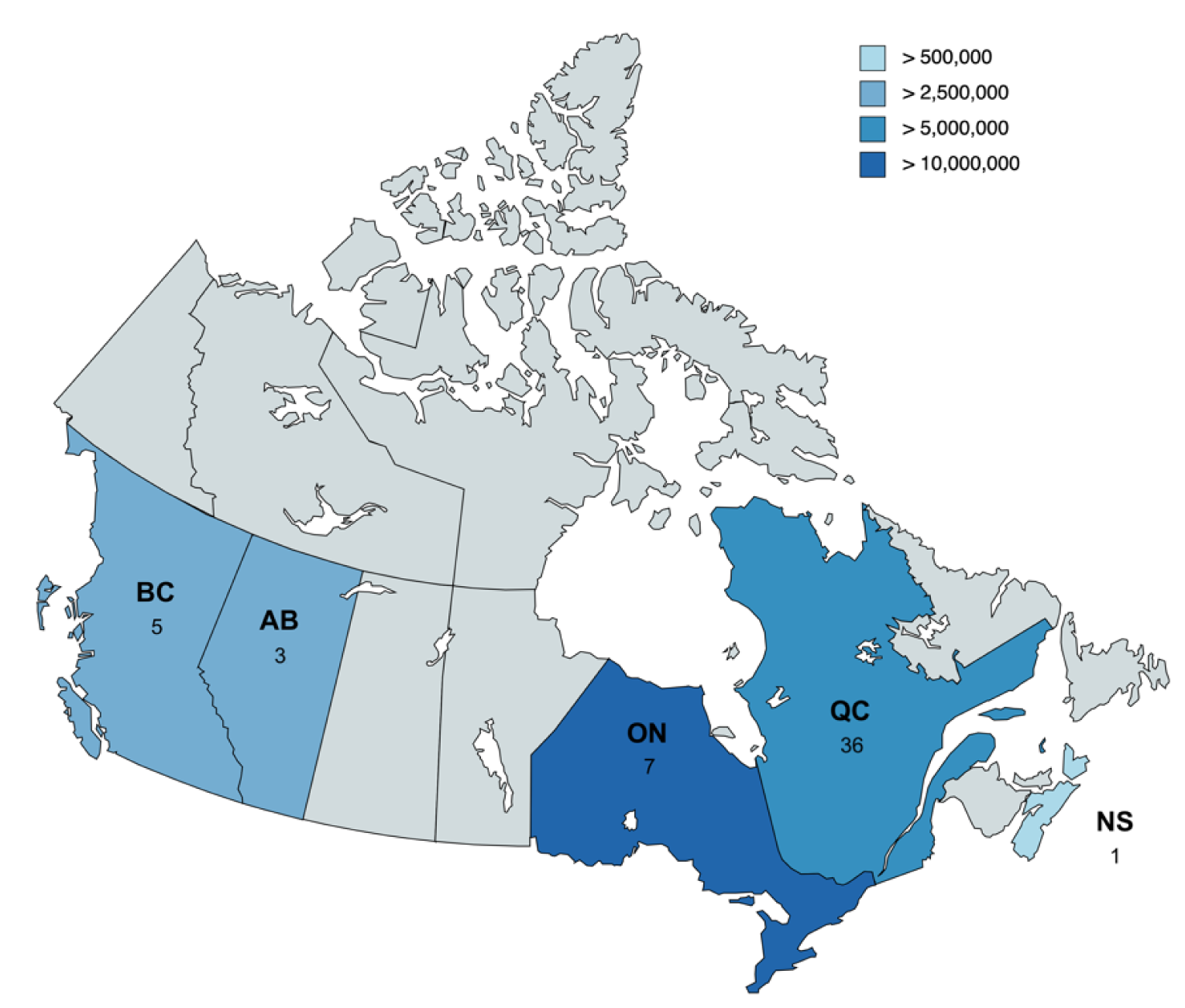
AB = Alberta; BC = British Columbia; HoFH = homozygous familial hypercholesterolemia; NS = Nova Scotia; ON = Ontario; QC = Quebec.
Note: Total number of registered patients per province is shown. Relative provincial populations are shown through colour-coding for reference.7
Discussion
The Canadian HoFH Registry was created by physicians specializing in severe disorders of lipoprotein metabolism, and includes pediatric endocrinologists, internal medicine specialists, lipidologists, endocrinologists, and cardiologists. The FH Canada Registry9 provided the necessary infrastructure in terms of network, standardized data collection, and shared interest in studying the disease. The aims of the registry are to better understand the demographics within Canadian society, the burden of the disease and associated resource utilization, current treatments and outcomes, access to orphan drugs, quality of life, and costs to society. We also plan to improve access to specialized therapies such as LDL apheresis and the appropriate use of orphan medications.
The diagnosis of HoFH used in the registry follows that established by international consensus4 and a molecular diagnosis, now widely available in Canada. We are aware that we have not yet uncovered all the genes that can cause FH, and that deep intronic mutations (currently not sequenced in clinical laboratories) in known genes, especially the LDLR gene, may be pathogenic. HoFH covers a spectrum of extremely severe hypercholesterolemia (Figure 1) and, if untreated, leads to death by a median age of 30 years.10
As discussed previously, there are an estimated 38 to 100 HoFH cases in Canada. Based on a questionnaire sent to members of FHCanada (Figure 2), we identified 52 patients, as of April 2022. The challenges of providing care for patients with HoFH in Canada, which has a population of approximately 38,000,000 spread across 9.98 million km2, are many. Furthermore, the complexity and diversity of provincial and territorial regulatory processes make it difficult to provide a uniform standard of care. This is particularly true for LDL apheresis, currently available only in Quebec City, London, and Edmonton, as well as plasmapheresis in Vancouver and Toronto.
A major strength of the Canadian HoFH Registry is the Canadian health care system in which it operates, and the highly collaborative spirit among clinicians and other health care professionals involved. There are limitations, which merit discussion. The lack of funding for emerging registries from granting agencies means that the initiation and initial functioning often comes in the form of unrestricted grants from the pharmaceutical industry. Another limitation relates to the identification of patients cared for by physicians who may be unfamiliar with the disease or unwilling to participate in data collection. This is particularly important in cases where the patient is not jointly followed by an academic medical centre.
The Canadian HoFH Registry will generate data to better understand the risk of ASCVD in HoFH, the impact of treatment, the development of late complications, clinical outcomes, and survival. We will be able to establish the roles of genotype, sex, and therapies on outcomes and quality of life. Health economics data will be derived from a comprehensive review of a patient’s clinical course and linking administrative databases (such as the Régie de l’Assurance Maladie du Québec). We will become involved in patient advocacy, and we will work with the US-based FH Foundation and the FH Collaborative Studies from the European Atherosclerosis Society (EAS). In addition, we will generate data that will guide decisions for orphan drug treatment and LDL apheresis access in Canada.
The establishment of the registry will create a resource for collaborative research and peer-reviewed funding. Importantly, we will enable collaborations across Canada and internationally to examine the effects of novel therapies and their roles in modifying the clinical course.
References
1.Müller C. Angina pectoris in hereditary xanthomatosis. Arch Int Med. 1939;64(4):675-700.
2.Khachadurian AK. The Inheritance of Essential Familial Hypercholesterolemia. Am J Med. 1964;37:402-407. PubMed
3.Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161-172. PubMed
4.Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146-2157. PubMed
5.Inserm. Orphanet Report Series - prevalence and incidence of rare diseases: bibliographic data. https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Accessed 2022 Apr 2.
6.Al-Shaikh AM, Abdullah MH, Barclay A, Cullen-Dean G, McCrindle BW. Impact of the characteristics of patients and their clinical management on outcomes in children with homozygous familial hypercholesterolemia. Cardiol Young. 2002;12(2):105-112. PubMed
7.Statistics Canada. Table 7-10-0009-01. Population estimates, quarterly. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000901. Accessed 2022 Apr 2.
8.Moorjani S, Roy M, Gagne C, et al. Homozygous familial hypercholesterolemia among French Canadians in Quebec Province. Arteriosclerosis. 1989;9(2):211-216. PubMed
9.FH Canada familial hypercholesterolemia. www.fhcanada.net. Accessed 2022 Apr 2.
10.Belanger AM, Akioyamen L, Alothman L, Genest J. Evidence for improved survival with treatment of homozygous familial hypercholesterolemia. Curr Opin Lipidol. 2020;31(4):176-181. PubMed
11.Brunham LR, Ruel I, Aljenedil S, et al. Canadian Cardiovascular Society Position Statement on Familial Hypercholesterolemia: Update 2018. Can J Cardiol. 2018;34(12):1553-1563. PubMed
12.Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. PubMed
13.Thompson GR, Blom DJ, Marais AD, Seed M, Pilcher GJ, Raal FJ. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur Heart J. 2018;39(14):1162-1168. PubMed
14.Medical Advisory Secretariat. Low-density lipoprotein apheresis: an evidence-based analysis. Ont Health Technol Assess Ser. 2007;7(5):1-101. PubMed
15.Thompson GR, Seed M, Naoumova RP, et al. Improved cardiovascular outcomes following temporal advances in lipid-lowering therapy in a genetically-characterised cohort of familial hypercholesterolaemia homozygotes. Atherosclerosis. 2015;243(1):328-333. PubMed
16.Banerjee A, Alothman L, Couture P, et al. The Lifelong Burden of Homozygous Familial Hypercholesterolemia. Can J Cardiol. 2019;35(10):1419 e1411-1419 e1414.
17.Alothman L, Belanger AM, Ruel I, et al. Health-related quality of life in homozygous familial hypercholesterolemia: A systematic review and meta-analysis. J Clin Lipidol. 2022;16(1):52-65. PubMed
18.Aljenedil S, Alothman L, Belanger AM, et al. Lomitapide for treatment of homozygous familial hypercholesterolemia: The Quebec experience. Atherosclerosis. 2020;310:54-63. PubMed
19.Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N Engl J Med. 2020;383(24):2307-2319. PubMed
20.Jeraj N, Huang SS, Kennedy BA, Hegele RA. Treatment of Homozygous Familial Hypercholesterolemia With Evinacumab. CJC Open. 2022;4(3):347-349. PubMed
21.Ruel I, Brisson D, Aljenedil S, et al. Simplified Canadian Definition for Familial Hypercholesterolemia. Can J Cardiol. 2018;34(9):1210-1214. PubMed
22.Al-Sarraf A, Allard M, Martinka M, Frohlich J. Regional and national familial hypercholesterolemia registries: present international application, importance, and needs for Canada. Can J Cardiol. 2013;29(1):6-9. PubMed
23.Brunham LR, Ruel I, Khoury E, et al. Familial hypercholesterolemia in Canada: Initial results from the FH Canada national registry. Atherosclerosis. 2018;277:419-424. PubMed
24.Canadian Institutes of Health Research. https://cihr-irsc.gc.ca. Accessed 2022 Nov 16.
25.Pour une meilleure reconnaissance et prise en charge des personnes atteintes de maladies rares. Gouvernement du Quebec; 2022: https://publications.msss.gouv.qc.ca/msss/fichiers/2022/22-916-01W.pdf. Accessed 2022 Nov 16.
26.Tromp TR, Hartgers ML, Hovingh GK, et al. Worldwide experience of homozygous familial hypercholesterolaemia: retrospective cohort study. Lancet. 2022;399(10326):719-728. PubMed
27.Awan Z, Alrasadi K, Francis GA, et al. Vascular calcifications in homozygote familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2008;28(4):777-785. PubMed
28.European Network for Health Technology Assessment. The Registry Evaluation and Quality Standards Tool (REQueST). 2021; https://www.eunethta.eu/request-tool-and-its-vision-paper/. Accessed 2022 Apr 2.
29.Canadian Organization for Rare Disorders. https://www.raredisorders.ca. Accessed 2022 Apr 2.
Appendix 1: Author Information
Note that this appendix has not been copy-edited.
Authors
Leslie Brown,1 Isabelle Ruel,1* Alexis Baass,1 Jean Bergeron,2* Liam R. Brunham,3* Lubomira Cermakova,3 Patrick Couture,2 Daniel Gaudet,4* Gordon A. Francis,3 Robert A. Hegele,5* Iulia Iatan,1 G.B. John Mancini,6 Brian W. McCrindle,7* Thomas Ransom,8 Mark H. Sherman,9 Ruth McPherson,10 Jacques Genest1*
*Steering committee
1Research Institute of the McGill University Health Centre, Montreal, QC, Canada
2Endocrinology and Nephrology Unit, CHU de Québec — Université Laval Research Centre, Québec City, QC, Canada
3Centre for Heart Lung Innovation, Providence Health Care Research Institute; Departments of Medicine and Medical Genetics, University of British Columbia, Vancouver, BC, Canada
4Lipidology Unit, Community Genomic Medicine Centre, Department of Medicine, Université de Montréal and ECOGENE-21 Clinical and Translational Research Centre, Chicoutimi, QC, Canada
5Departments of Medicine and Biochemistry, Schulich School of Medicine and Robarts Research Institute, Western University, London, ON, Canada
6Centre for Cardiovascular Innovation, University of British Columbia, Vancouver, BC, Canada
7Department of Pediatrics, The Labatt Family Heart Centre, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
8Queen Elizabeth II Health Sciences Centre, Dalhousie University, Halifax, NS, Canada
9Department of Endocrinology, McGill University Health Centre, Montreal, QC, Canada
10Lipid Clinic and Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON, Canada
Conflicts of Interest
All authors reported their conflicts of interest as follows:
Alexis Baass receives research funding/grants from Sanofi and Amgen.
Liam Brunham received funding/honoraria for speaking engagements on lipid-lowering therapies from Amgen, Novartis, HLS Therapeutics, Ultragenyx, and Medison.
Daniel Gaudet had funding/honoraria for speaking engagements and writing articles/editorials for Akcea, Amgen, Amryt, Astra Zeneca, Cerenis, Esperion, Gemphire, HDL Therapeutics, Ionis, Regeneron, and Sanofi. He is currently employed by the Université de Montreal. He received payments as an advisor/consultant and research funding grants from Akcea, Allergan, Amgen, Amryt, Acasti, Aegerion, Applied Therapetics, Arrowhead, AstraZeneca, Boehringer-Ingelhein, Ceapro, CRISPR-Therapeutics, Dalcor, EliLilly, Esperion, Ionis, Kowa, Novartis, Novonordisk, NRC, Pfizer, Regeneron, Saliogen, Sanofi, The Medicine Company, Uniqurie, and Verve Therapeutics.
Jacques Genest has received funding for speaking engagements from Novartis, FH Canada, Amgen, CIHR, the Canadian Cardiovascular Society, and INESSS. Has also received funding/honoraria to represent inclisiran to Health Canada, is a chair on Familial Hypercholesterolemia Canada, received a CIHR project grant for Familial Hypercholesterolemia, is a Canadian Cardiovascular Society dyslipidemia guideline committee member, and is an INESSS consultant on Familial Hypercholesterolemia and PCSK9 inhibitors.
Robert Hegele had speaking engagements with Amgen for evolocumab, Novartis for inclisiran, and HLS Therapeutics for icosapent ethyl. He received payments as a consultant from Ultragenyx, Novartis, and HLS Therapeutics.
John Mancini received funding for speaking engagements and educational lectures from Amgen, Sanofi, and Novartis. He received funding for research grants with Amgen and Novartis, and undertakes occasional advisory activities for Amgen, Sanofi, and Novartis.
Brian McCrindle received payment as an advisor from Chiesi, Amryt Pharma, Esperion, Ultragenyx, and Janssen.
Ruth McPherson had speaking engagements with Novartis, Amgen, and Pendopharm. She received research funding from Novartis for LPA antisense, and Amgen for evolocumab. She participated in a scientific advice program for Novartis inclisiran.
Thomas Ransom had speaking engagements with Amgen. He was a member of an advisory board meeting with Novartis.
Mark Sherman had speaking engagements/educational lectures with HLS Pharmaceuticals for isosapent ethyl, and Novo Nordisk for semaglutide. He received payments as an advisor with Sanofi, Novonordisk, Akcea, HLS Pharmaceuticals, Astra Zeneca, Gilead, and Janssen. He also participated in projects on icosapent ethyl, semaglutide, and insulin degludec.
Jean Bergeron, Leslie Brown, Lubomira Cermakov, Patrick Couture, Gordon Francis, Iulia Iatan, and Isabelle Ruel completed conflict-of-interest forms with no conflicts to declare.
Funding
This work was supported by a Knowledge Synthesis Grant from the Canadian Institutes of Health Research [SBI-167982 to JG].
ISSN: 2563-6596
This article is subject to copyright and other intellectual property rights and may only be used for non-commercial, personal use, or private research and study. This material is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. This article should not be used as a substitute for professional medical advice or for the application of professional judgment in any decision-making process. Users may use this document at their own risk.
This article – unlike others published in the journal -- was submitted by an author group outside CADTH. It was peer reviewed by 2 external reviewers, revised by the corresponding author, and accepted for publication in the journal. Care has been taken to ensure that the information presented is original and has not been submitted elsewhere.