CADTH Reimbursement Review
Calaspargase Pegol (Asparlas)
Sponsor: Servier Canada Inc.
Therapeutic area: Acute lymphoblastic leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
ALL
acute lymphoblastic leukemia
AYA
adolescent and young adult
BMI
body mass index
CBC
complete blood count
CI
confidence interval
CNS
central nervous system
CNS3
central nervous system status of 3
COG
Children’s Oncology Group
CR
complete remission
CSF
cerebrospinal fluid
CTCAE
Common Terminology Criteria for Adverse Events
DFCI
Dana-Farber Cancer Institute
DFS
disease-free survival
EFS
event-free survival
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ITT
intention to treat
LL
lymphoblastic lymphoma
LLSC
Leukemia & Lymphoma Society of Canada
MAC
multiagent chemotherapeutic
MRD
minimal residual disease
NSAA
nadir serum asparaginase activity
OH-CCO
Ontario Health (Cancer Care Ontario)
OR
odds ratio
OS
overall survival
PAA
plasma asparaginase activity
PCR
polymerase chain reaction
PD
pharmacodynamic
Ph positive
Philadelphia chromosome positive
PK
pharmacokinetic
PP
per protocol
RCT
randomized controlled trial
SAA
serum asparaginase activity
SAE
serious adverse event
SD
standard deviation
SOC
standard of care
TDM
therapeutic drug monitoring
TEAE
treatment-emergent adverse event
WBC
white blood cell
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Calaspargase pegol (Asparlas), 3,750 units/5 mL (750 units/mL), concentrate for solution for IV infusion |
Sponsor | Servier Canada Inc. |
Indication | As a component of a multiagent chemotherapeutic regimen for the treatment of ALL in pediatric and young adult patients aged 1 year to 21 years |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 8, 2023 |
Recommended dosage | The recommended dosage of calaspargase pegol is 2,500 units/m2 administered as IV infusion no more frequently than every 21 days. Dosing: Calaspargase pegol is used as part of combination chemotherapy protocols with other antineoplastic drugs. Calaspargase pegol is not a bioequivalent alternative to pegaspargase. In a multiagent chemotherapeutic regimen, calaspargase pegol at the same dose and frequency of pegaspargase may increase toxicities due to the longer half-life of calaspargase pegol. Dose adjustments: Therapeutic drug monitoring may be considered to assess silent inactivation of asparaginase per institutional guidelines. If premedication is administered, therapeutic drug monitoring may be measured per institutional guidelines or based on trough asparaginase activity levels before the next administration of calaspargase pegol. If asparaginase activity values fail to reach target levels, the use of a different asparaginase preparation could be considered. Premedication: Premedicate patients with acetaminophen, an H1 receptor blocker (such as diphenhydramine), and an H2 receptor blocker (such as famotidine) 30 minutes to 60 minutes before the administration of calaspargase pegol to decrease the risk and severity of both infusion and hypersensitivity reactions. Steroid administration may also be considered in the premedication regimen. |
ALL = acute lymphoblastic leukemia; NOC = Notice of Compliance.
The sponsor’s application was filed with CADTH on a pre-Notice of Compliance basis and the CADTH clinical report is reflective of the proposed Health Canada indication and information incorporated into the draft product monograph that was submitted to Health Canada and CADTH. The proposed Health Canada indication was not limited to a certain age group, whereas the final indication was limited to patients aged 1 year to 21 years.
Introduction
Acute lymphoblastic leukemia (ALL) is the least common type of leukemia diagnosed in adults; however, it is the most common type of leukemia diagnosed in young children.1 It is estimated that ALL represents 75% to 80% of acute leukemias among children and 20% of all leukemias among adults.2 In 2018, the incidence rate of ALL for all ages in Canada (excluding Quebec) was 1.3 patients per 100,000, with the majority of patients being younger than 19 years.3,4 ALL is a heterogeneous group of disorders that results from the clonal proliferation and expansion of malignant lymphoid cells in the bone marrow, blood, and extramedullary sites,5 classified into 2 major subtypes: B-lymphoblastic and T-lymphoblastic leukemia, with further division according to the presence and type of genetic abnormalities.5,6 ALL of the B-cell phenotypes occurs in approximately 80% to 85% of pediatric patients and nearly 75% of adult patients. The frequency of the Philadelphia chromosome in patients with ALL is about 3% to 5% in pediatric patients, and 25% to 30% in adult patients.7 Signs and symptoms of ALL are highly variable, with most patients experiencing bruising, bleeding, dyspnea, dizziness, infections due to neutropenia, anemia, thrombocytopenia, and pain. Multiagent chemotherapeutic (MAC) is the established standard of care (SOC) treatment for newly diagnosed patients with ALL, and asparaginase (e.g., pegaspargase) is an essential component of these regimens. Two different childhood ALL treatment strategies are commonly used across Canada, originating from the Children’s Oncology Group (COG) and the Dana-Farber Cancer Institute (DFCI) consortia.8,9 Both protocols include pegaspargase. According to the clinical experts consulted by CADTH, patients who are unable to receive asparaginase treatment (e.g., they lack access to a consistent supply of treatment drug, they experience allergy intolerance) are less likely to be cured with chemotherapy alone.
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of calaspargase pegol 2,500 units/m2 given intravenously no more frequently than every 21 days as a component of an MAC regimen for the treatment of ALL in children and adults. Calaspargase pegol has not been previously reviewed by CADTH.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 1 patient group submission from the Leukemia & Lymphoma Society of Canada (LLSC), which conducted an online survey of 47 patients or caregivers of patients with ALL from all 10 provinces in Canada during April to May 2023. Patients reported that ALL progresses quickly and aggressively and that to prevent disease progression, the immediate start of treatment upon diagnosis is vital. However, patients’ experience with drug shortages at some point during their ALL treatment were extremely stressful, impacting their mental health, physical health, quality of life, home life, social life, work life, and finances. In addition to secure, readily accessible, and effective treatment, factors important to patients when evaluating new treatments for ALL included side effects, a physician’s recommendation, quality of life, cost, secure supply, and the number of treatments.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical specialists with expertise in the diagnosis and management of ALL reported that the goal of treatment for patients with ALL is curative, aimed at maximizing survival while minimizing short-term and long-term toxicities. Current treatment for ALL in Canada was identified by the clinical experts consulted by CADTH as comprising MAC regimens that include pegylated asparaginase, using pediatric protocols developed by the COG or the DFCI among children or pediatric-inspired protocols among adults, estimated to be 2.5 years to 3.5 years in duration. The clinical experts consulted by CADTH noted that patients who do not respond to treatment require high-dose chemotherapy and/or allogeneic stem cell transplant and experience high rates of treatment failure. According to the clinical experts consulted by CADTH, patients would benefit from a consistent supply of asparaginase, with a shorter frequency of dosing, and treatments with improved tolerability.
Asparaginase is an essential component of front-line ALL therapy, and 2 extended half-life formulations have been developed for clinical use. Pegylated asparaginase has a half-life of 5.7 days and is administered every 14 days. Calaspargase pegol has a half-life of 16.1 days and is administered every 21 days. The clinical experts consulted by CADTH do not expect a shift in the current treatment paradigm with calaspargase pegol; rather, they believe that it will replace pegaspargase, given its prolonged half-life, longer dosing interval, and the need for fewer administrations (10 versus 15) over the course of treatment.
The clinical experts consulted by CADTH indicated that all patients with newly diagnosed ALL would benefit from treatment with calaspargase pegol, since asparaginase has a unique mechanism of action and is considered to comprise an essential component of therapy in ALL. Patients with relapsed ALL were also considered by the clinical experts consulted by CADTH to potentially benefit from calaspargase pegol, if there was no known prior intolerance to other forms of asparaginase. Patients of any age with Philadelphia chromosome–positive (Ph-positive) status (except for adult patients due to potential overlapping toxicities with tyrosine kinase inhibitors) and B-cell or T-cell immunophenotype were considered by the clinical experts consulted by CADTH to be eligible for asparaginase treatment and therefore appropriately targeted for treatment with calaspargase pegol. The clinical experts consulted by CADTH specified that several risk factors are considered before starting treatment to help inform the protocol used, including age older than 10 years, white blood cell (WBC) count below 50 × 109/L at presentation or diagnosis, adverse genetic features including karyotype (e.g., the translocation t(9;22)(q34;q11), hypodiploidy), molecular studies (e.g., BCR-ABL, KMT2A mutations), and gene expression (e.g., IKZF, CRLF2).
The clinical experts consulted by CADTH reported that a clinically meaningful treatment response should be assessed using overall survival (OS), complete remission (CR) postinduction, minimal residual disease (MRD)–negative status postinduction, and serum asparaginase activity (SAA) levels following the administration of asparaginase to monitor adequate asparaginase depletion and clinical reactions (e.g., allergic or infusion-related reaction, silent inactivation). According to the clinical experts consulted by CADTH, treatment with asparaginase including calaspargase pegol should be discontinued in the event of notable adverse events (AEs) (e.g., hypersensitivity reaction including silent inactivation, allergic reaction, development of neutralizing antibodies, severe liver toxicity, severe pancreatitis, severe thrombotic or hemorrhagic event, persistent severe hepatic dysfunction). The clinical experts consulted by CADTH indicated that patients with ALL are often treated in hospital or cancer centres as inpatients or outpatients by hematologists or oncologists.
Clinician Group Input
CADTH received 1 clinician group submission from the Ontario Health (Cancer Care Ontario) (OH-CCO) Hematology Cancer Drug Advisory Committee comprising 2 clinicians. The clinician group noted no significant unmet need among patients who are eligible for standard induction of ALL treatment with pegaspargase; however, patients treated with calaspargase pegol as a component of MAC could benefit from less frequent dosing and should be assessed for treatment response using standard leukemia response criteria, with treatment to be discontinued upon progressive disease or significant intolerance. OH-CCO noted that the appropriate setting for treatment with calaspargase pegol is acute leukemia treatment centres with the presence of leukemia specialists.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for calaspargase pegol: relevant comparators, consideration for the initiation of therapy, consideration for the prescribing of therapy, generalizability, care provision issues, and system and economic issues.
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs. Refer to Table 5 for more details.
Clinical Evidence
Systematic Review
Description of Studies
Two phase II, multicentre, randomized, open-label trials assessed the efficacy and safety of calaspargase pegol 2,500 IU/m2 compared with pegaspargase 2,500 IU/m2. The COG study AALL07P4 (known as the COG AALL07P4 study) enrolled 166 patients aged 1 year to 30 years in 23 study sites, all located in the US, with newly diagnosed high-risk B-cell ALL. The primary objective of the COG AALL07P4 trial was to determine the pharmacokinetic (PK) comparability (asparaginase activity) of the interventions during induction and consolidation while patients were receiving augmented Berlin-Frankfurt-Münster therapy. Secondary objectives of the COG AALL07P4 trial included pharmacodynamic (PD) parameters during induction and consolidation, MRD (day 29), CR rate (day 29), survival (event-free survival [EFS], disease-free survival [DFS] of CR, and OS), and treatment-emergent adverse events (TEAEs). The DFCI study 11 to 001 (known as the DFCI 11-001 study) enrolled 239 patients aged 1 year to 21 years in the US (in 6 sites) and Canada (in 3 sites) with newly diagnosed ALL or lymphoblastic lymphoma (LL). The primary objective of the DFCI 11-001 trial was to determine the PK comparability of the interventions during remission induction and postinduction (i.e., determine SAA levels and assess toxicity). Secondary end points of the DFCI 11-001 trial included MRD (day 32), CR rate (day 32), EFS, DFS from the attainment of CR, and OS.
Across both the COG AALL07P4 and DFCI 11-001 trials, there were notable similarities and differences in baseline demographics. In the COG AALL07P4 trial, most patients were older than 10 years (66.3%). In the DFCI 11-001 trial, most patients were younger than 10 years (75%). All patients in the COG AALL07P4 study and nearly all patients in the DFCI 11-001 study (96%) had ALL. Most patients in the DFCI 11-001 study had B-cell ALL (87%) including patients with T-cell ALL, whereas patients with B-cell immunophenotype were exclusively enrolled in the COG AALL07P4 trial. Most patients had a central nervous system (CNS) status of 1 in both trials with a minority of patients designated as having a central nervous system status of 3 (CNS3) in the COG AALL07P4 trial (fewer than 10% of patients) and the DFCI 11-001 trial (fewer than 2% of patients). Most patients did not have steroid therapy before study treatment in the COG AALL07P4 ||||| or DFCI 11-001 (|||) study. While the majority of patients were older than 10 years in the COG AALL07P4 study (median = 11 years), most patients were younger than 10 years in the DFCI 11-001 study (median = 4 years to 5 years). Approximately 62% of patients were diagnosed when they were 10 years or older in the COG AALL07P4 trial, whereas nearly 75% of patients were diagnosed aged younger than 10 years in the DFCI 11-001 trial. Patients in the COG AALL07P4 study were distributed equally across combined age and WBC categories, whereas more than 70% of patients in the DFCI 11-001 study were younger than 10 years, with WBC count below 50 × 109/L.
Efficacy Results
The key efficacy results from the COG AALL07P4 trial and the DFCI 11-001 trial are summarized in Table 2 and Table 3, respectively. Results in the COG AALL07P4 trial were based on the December 31, 2015, data cut-off date. Results in the DFCI 11-001 trial were based on the October 5, 2016, data cut-off date and, where indicated, from a day 120 follow-up with an updated data cut-off date of June 12, 2017.
Overall Survival
In the COG AALL07P4 study, the median OS was not reached at the data cut-off on December 31, 2015. Patients had been followed for a median of 62.6 months (range = ||| || ||||). The 1-year OS rate (95% confidence interval [CI]) among patients in the full analysis set (FAS) population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol and pegaspargase group, respectively. The 4-year OS rate among patients in the FAS population was ||||| ||||| || ||||| in the calaspargase pegol and ||||| ||||| || ||||| in the pegaspargase group, respectively. The hazard ratio (HR) in the FAS population was |||| ||||| || ||||| in the calaspargase pegol versus pegaspargase group. Findings of the intention-to-treat (ITT) population were consistent with results for the FAS population.
In the DFCI 11-001 study, the median OS was not reached at the data cut-off on October 5, 2016. Patients had been followed for a median of |||| months (range = ||| || ||||). The 1-year OS rate among patients in the FAS ALL population was ||||| |||| ||| |||| || ||||| for calaspargase pegol and |||||| |||| ||| ||||| || |||||| for pegaspargase. At the day 120 cut-off, the median follow-up duration was |||| months and |||| months for the calaspargase pegol and pegaspargase groups, respectively. The 2-year OS rate among patients in the FAS ALL population was ||||| |||| ||| |||| || ||||| for calaspargase pegol and unchanged for pegaspargase. Findings of the ITT ALL population were consistent with results for the FAS population.
DFS From CR
In the COG AALL07P4 study, the 1-year DFS rate among patients in the FAS population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 4-year DFS rate among patients in the FAS population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The HR in the FAS population was |||| ||||| || ||||| in the calaspargase pegol group versus the pegaspargase group. DFS results were identical to the FAS population.
In the DFCI 11-001 study, the 1-year DFS rate among patients in the FAS ALL population who attained CR was ||||| |||| ||| |||| || ||||| in the calaspargase pegol group and ||||| |||| ||| |||| || ||||| in the pegaspargase group. At the day 120 follow-up, the 2-year DFS rate among patients in the FAS ALL population who attained CR was ||||| ||||| || ||||| and ||||| ||||| || ||||| for calaspargase pegol versus pegaspargase, respectively. The results of DFS among patients attaining CR in the ITT ALL population were identical to those of the FAS ALL population.
Event-Free Survival
In the COG AALL07P4 study, the 1-year EFS rate among patients in the FAS population was ||||| ||||| || ||||| and 89.6% (95% CI, 74.0% to 96.1%) in the calaspargase pegol group and the pegaspargase group, respectively. The 4-year EFS rate was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The HR was |||| ||||| || ||||| for the calaspargase pegol group when compared with the pegaspargase group. Findings for EFS in the ITT population were consistent with results for the FAS population.
In the DFCI 11-001 trial, the 1-year EFS rate among patients in the FAS ALL population was ||||| |||| ||| |||| || ||||| and ||||| |||| ||| |||| || ||||| in the calaspargase pegol group and pegaspargase group, respectively. At the day 120 follow-up, the 2-year EFS rate in the FAS population was ||||| |||| ||| |||| || ||||) and ||||| |||| ||| |||| || ||||| in the calaspargase pegol group and pegaspargase group, respectively. Results of EFS among patients in the ITT ALL population were consistent with results for the FAS ALL population.
Complete Remission
In the COG AALL07P4 study, the proportion of patients in the FAS population who attained CR by day 29 was || || || ||||||| ||| ||| |||| || ||||| in the calaspargase pegol group and || || || ||||||| ||| ||| |||| || ||||| in the pegaspargase group. Findings for CR at the end of induction day 29 for the ITT population were consistent with results for the FAS population.
In the DFCI 11-001 study, the proportion of patients in the FAS ALL population who attained CR by day 32 was ||| || ||| ||||||| ||| ||| |||| || ||||| in the calaspargase pegol group and ||| || ||| ||||||| ||| ||| |||| || ||||) in the pegaspargase group. Results for CR by day 32 in the ITT ALL population were consistent with results for the FAS ALL population.
Minimal Residual Disease
In the COG AALL07P4 study, the proportion of patients in the MRD-evaluable FAS population with positive MRD (≥ 0.1% detectable leukemia cells in a bone marrow biopsy or aspirate with validated 6-colour multiparameter flow cytometry) at induction day 29 were ||| || || ||||||| in the calaspargase pegol group and || || || ||||||| in the pegaspargase group. Findings for positive MRD (≥ 0.1%) at the end of induction day 29 in the ITT population were consistent with results in the FAS population.
In the DFCI 11-001 study, the proportion of patients in the FAS ALL population with MRD of 0.01 or greater by PCR assay were |||| ||| |||||| in the calaspargase pegol group and | || ||| in the pegaspargase group. Findings for MRD of 0.01 or greater at the end of induction day 32 in the ITT ALL population were consistent with results in the FAS ALL population.
Serum Asparaginase Activity
SAA levels were not reported in the COG AALL07P4 trial.
In the DFCI 11-001 trial, the proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| |||| ||| |||| || |||||| for calaspargase pegol versus ||||| |||| ||| |||| || |||||| for pegaspargase at 5 minutes to 10 minutes after infusion on induction day 7 (odds ratio [OR] |||||; 90% CI, ||||| || |||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were |||||| (95% CI | |||| || |||||) for calaspargase pegol versus ||||| (95% CI, |||| || |||||) for pegaspargase at 4 days after infusion on day 11 (OR |||||| 90% CI, ||||| || |||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were 100.0% (95% CI, 96.8% to 100.0%) for calaspargase pegol versus 100.0% (95% CI, 95.0% to 100.0%) for pegaspargase at 11 days after infusion on day 18 (OR = 0.479; 90% CI, 0.063% to 3.626%). The proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| (95% CI, |||| || ||||) for calaspargase pegol versus ||||| (95% CI, |||| || ||||) for pegaspargase at 18 days after infusion on day 25 (OR |||||; 90% CI, ||||| || ||||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| (95% CI, |||| || ||||) for calaspargase pegol versus ||||| (95% CI, |||| || ||||) for pegaspargase at 25 days after infusion on day 32 (OR ||||||; 90% CI, |||||| || ||||||). Estimates of treatment effect on SAA levels using adjusted analyses (controlled for age, sex, initial risk group, disease type, and baseline WBC count) were similar to unadjusted analyses.
Harms Results
The analysis population for harms included all patients who received at least 1 dose of any study drug, with patients grouped according to the treatment received. Safety data were from the primary safety analyses for the COG AALL07P4 study (data cut-off date of December 31, 2015) and the DFCI 11-001 study (data cut-off date of October 5, 2016).
In the COG AALL07P4 trial, the percentage of patients reporting any TEAEs was ||||| for calaspargase pegol and ||||| for pegaspargase. In the DFCI 11-001 trial, the percentage of patients who experienced any TEAEs was ||||| for the calaspargase pegol group and |||||| patients in the pegaspargase group. In the COG AALL07P4 trial, the most common TEAEs occurring in at least 25% of patients in either treatment group (calaspargase pegol versus pegaspargase) were hypoalbuminemia (27.9% versus 5.8%); hyperglycemia (79.1% versus 50.0%); blood bilirubin, increased (62.8% versus 50.0%); neutrophil count, decreased (55.8% versus 51.9%); febrile neutropenia (55.8% versus 42.3%); alanine aminotransferase, increased (34.9% versus 38.5%); platelet count, decreased (34.9% versus 25.0%); WBC count, decreased (37.2% versus 28.5%); hypokalemia (27.9% versus 11.5%); anemia (25.6% versus 26.9%); activated partial thromboplastin time, prolonged (30.2% versus 19.2%); peripheral motor neuropathy (27.9% versus 19.2%); and abdominal pain (32.6% versus 11.5%). In the DFCI 11-001 trial, the most common TEAEs occurring in at least 25% of patients in either treatment group (calaspargase pegol versus pegaspargase) were hypoalbuminemia (81.4% versus 82.4%); alanine transaminase, increased (78.8% versus 77.3%); aspartate aminotransferase, increased (53.4% versus 58.8%); blood bilirubin, increased (45.8% versus 43.7%); hypokalemia (45.8% versus 39.5%); febrile neutropenia (33.9% versus 40.3%); hyperglycemia (33.9% versus 28.6%); hypoglycemia (30.5% versus 36.1%); hypertriglyceridemia (28.0% versus 36.1%); and stomatitis (25.4% versus 20.2%).
In the COG AALL07P4 study, the number of patients with at least 1 serious adverse event (SAE) was not reported. The percentage of patients with at least 1 grade 3 or grade 4 TEAE was 97.7% in the calaspargase pegol group and 90.4% in the pegaspargase group. The most common grade 3 or grade 4 TEAEs in the calaspargase pegol group and the pegaspargase group, respectively, were neutrophil count, decreased (55.8% versus 51.9%); febrile neutropenia (55.8% versus 42.3%); WBC count, decreased (37.2% versus 28.8%); and hyperglycemia (37.2% versus 17.3%). In the DFCI 11-001 study, the percentage of patients who experienced grade 3 or grade 4 TEAEs was ||||| in the calaspargase pegol group and ||||| in the pegaspargase group. The most common grade 3 or grade 4 TEAEs in the calaspargase pegol and pegaspargase groups, respectively, were alanine aminotransferase, increased (49.2% versus 60.5%); hypokalemia (43.2% versus 36.1%); febrile neutropenia (33.9% versus 40.3%); and hypoalbuminemia (27.1% versus 27.7%). The percentage of patients who experienced at least 1 SAE was 24.6% and 21.8% in the calaspargase pegol group and the pegaspargase group, respectively. SAEs that occurred in at least 2% of patients in either the calaspargase pegol group or the pegaspargase group, respectively, were lipase, increased (4.2% versus |||||); pancreatitis (5.9% versus ||||); sepsis (3.4% versus |||||| hyperglycemia (2.5% versus ||||); febrile neutropenia (1.7% versus ||||); amylase, increased (0.8% versus ||||); alanine aminotransferase, increased (2.5% versus ||||); aspartate aminotransferase, increased (2.5% versus |||); and neutropenic colitis (2.5% versus |||).
In the COG AALL07P4 study, the percentage of patients who stopped study treatment due to an AE was ||||| and ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The reasons for stopping study treatment due to an AE were not reported. In the DFCI 11-001 study, the percentage of patients who stopped study treatment due to an AE was 28.0% in the calaspargase pegol group and 19.3% in the pegaspargase group. Withdrawals due to adverse events (WDAEs) in the calaspargase pegol group compared with the pegaspargase group, respectively, were due to hypersensitivity (8.5% versus |||||; lipase, increased (6.8% versus ||||); pancreatitis (5.9% || |||| ||||||); drug hypersensitivity (5.1% versus ||||); amylase, increased (4.2% versus ||||); and anaphylactic reaction (1.7% versus |).
|| ||| |||||||| || ||| ||| ||||||||||| | |||||| |||||||| || ||| |||||||||||| ||||| ||||| || ||||||| ||| ||| |||||||| |||||||| ||||||||||| || |||||| | |||||| |||| ||| || ||||| |||||||||| ||||||| |||| ||| ||||||||| | ||| || ||||| ||||||| ||| |||||| |||||||| || ||| |||||||||||| |||||| || |||||| | |||| ||| || |||||||||| ||||||| ||| ||| ||||||||| | |||| ||| || ||||| ||||||| || |||| ||||||| | |||||| ||| | |||||| |||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| |||||| || ||| |||||||||||| ||||| ||||| ||| ||| || ||||||| ||||||| || ||||||| ||| ||||| ||||||||| |||| ||| ||||||||| |||| ||| |||||| || ||| |||||||||||| ||||| |||| ||| || |||||||| ||| |||||||||| ||||||| ||| ||| |||||||||| || ||| |||||||||||| ||||| ||||| ||| ||| ||| ||||||| |||| ||||| |||| |||| ||||||||||| |||| |||||| ||| ||||||
Notable harms identified in the CADTH review included hypersensitivity reactions, anaphylactic reactions, silent inactivation, pancreatitis, thrombosis, hemorrhage, and hepatotoxicity. In the COG AALL07P4 trial, |||| of patients in the calaspargase pegol group and |||| of patients in the pegaspargase group experienced hypersensitivity events. In the DFCI 11-001 trial, ||||| of patients in the calaspargase pegol group and ||||| of patients in the pegaspargase group experienced hypersensitivity events. In the COG AALL07P4 trial, 25.6% of patients in the calaspargase pegol group and 19.2% of patients in the pegaspargase group experienced anaphylactic reactions. In the DFCI 11-001 trial, |||| of patients in each treatment group experienced anaphylactic reactions. In the COG AALL07P4 study, silent inactivation was not reported by any patient. In the DFCI 11-001 study, 1.7% of patients in the calaspargase pegol group experienced silent inactivation and were switched to Erwinia asparaginase treatment. In the COG AALL07P4 study, 18.6% and 7.7% of patients experienced pancreatitis in the calaspargase pegol group and the pegaspargase group, respectively. In the DFCI 11-001 study, 11.9% and 16.8% of patients experienced pancreatitis in the calaspargase pegol group and the pegaspargase group, respectively. In the COG AALL07P4 study, venous thrombosis was ||| |||||||| || ||| |||||||. In the DFCI 11-001 study, |||| of patients experienced venous thrombosis in each of the calaspargase pegol and pegaspargase groups. || ||||||| experienced hemorrhage in the COG AALL07P4 study or the DFCI 11-001 study. In the COG AALL07P4 trial, the percentage of patients who experienced increased blood bilirubin was 62.8% and 50.0% and who experienced increased alanine aminotransferase was 34.9% and 38.5% in the calaspargase pegol group and the pegaspargase group, respectively. In the DFCI 11-001 trial, the percentage of patients who experienced increased blood bilirubin was 45.8% and 43.7% and who experienced increased alanine aminotransferase was 78.8% and 77.3% in the calaspargase pegol group and the pegaspargase group, respectively. ||| ||||||| in the calaspargase pegol group experienced hepatic failure.
Critical Appraisal
The COG AALL07P4 and DFCI 11-001 trials were phase II, randomized, open-label randomized controlled trials (RCTs). Randomization appeared to be adequate in the COG AALL07P4 and DFCI 11-001 studies since treatment groups were generally balanced for key baseline characteristics and therefore likely to be at low risk for selection bias. The open-label study design may have biased outcomes with subjective assessments for harms due to knowledge of the assigned treatment, although the direction for potential bias is unclear. The phase II study design of the COG AALL07P4 and DFCI 11-001 trials did not allow the trials to assess comparative efficacy between calaspargase pegol and pegaspargase. The sample sizes (97 patients in the COG AALL07P4 study and 239 patients in the DFCI 11-001 study) were relatively small and the magnitude of the treatment effect estimates observed in a small study sample may not be replicable in a larger study sample. Findings from the COG AALL07P4 and DFCI 11-001 studies were not controlled for multiple comparisons. There were balanced between-group proportions of patients who were censored or had missing outcome data that were unlikely to substantially impact findings in the COG ALL07P4 study or the DFCI 11-001 study despite lack of imputation. Differences in trial population and backbone treatment protocols in the COG AALL07P4 and DFCI 11-001 trials precluded the ability to combine findings across outcomes. In the COG AALL07P4 and DFCI 11-001 trials, median survival estimates were not reached at the time of data cut-off. Assessments for CR and MRD at day 29 appeared to be appropriate to capture the presence or absence of disease at the end of remission induction. SAA levels were a primary end point in the DFCI 11-001 study and have been reported to serve as an important end point in assessing calaspargase pegol’s ability to maintain asparagine suppression in plasma, its half-life duration, and its ability to be administered with less dosing frequency than pegaspargase. Outcomes that were not assessed included health-related quality of life (HRQoL) (the COG AALL07P4 and DFCI 11-001 studies) and silent inactivation (the COG AALL07P4 study). The comparator used in the COG AALL07P4 and DFCI 11-001 trials was appropriate as pegaspargase is a pegylated formulation of asparaginase (calaspargase pegol uses the same mechanism of action as pegaspargase) and has been a component of current SOC. Calaspargase pegol is intended to substitute for pegaspargase and would be used for patients who would have otherwise received a pegaspargase-containing MAC.
The COG AALL07P4 and DFCI 11-001 studies were phase II trials and enrolled small samples of patients with ALL. Nevertheless, clinical experts consulted by CADTH remarked that a small patient population is expected, given the disease area, and a phase III RCT would neither be feasible nor ethical to conduct. While patients in the COG AALL07P4 and DFCI 11-001 trials were considered representative of patients with ALL, subpopulations of patients excluded from enrolment included pediatric patients with Ph-positive status (excluded from the COG AALL07P4 and DFCI 11-001 trials), patients with T-cell ALL (excluded from the COG AALL07P4 trial), and patients with relapse or refractory disease (excluded from the COG AALL07P4 and DFCI 11-001 trials). According to the clinical experts consulted by CADTH, the effects of treatment with calaspargase pegol could be generalizable to these patients. The clinical experts consulted by CADTH noted that it is anticipated that patients would benefit from treatment with calaspargase pegol based on its similarity to and extrapolation of findings for pegaspargase. Different backbone therapies were employed, with an intermittent asparagine depletion versus continuous asparagine depletion protocol for the COG AALL07P4 study versus the DFCI 11-001 study, respectively. The clinical experts consulted by CADTH observed that both COG-based and DFCI-based protocols were employed by institutions across Canada. The clinical experts consulted by CADTH expected outcomes to be similar regardless of the treatment protocol employed for patients with ALL. Outcomes reported in the COG AALL07P4 and DFCI 11-001 studies appeared to be aligned with outcomes of interest for patients with ALL, according to the clinical experts consulted by CADTH. The clinical experts consulted by CADTH highlighted the importance of SAA levels in therapeutic drug monitoring (TDM) for both efficacy (i.e., adequate asparagine depletion and sustained SAA levels of 0.10 IU/mL or greater) and safety (e.g., hypersensitivity reactions including silent inactivation). In general, the clinical experts consulted by CADTH did not anticipate clinically meaningful differences in efficacy between calaspargase pegol and pegaspargase.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.10,11 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias.
The selection of outcomes for the GRADE assessment was based on the sponsor’s Summary of Clinical Evidence,12 discussions with clinical experts consulted by CADTH, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: survival (OS, DFS from CR, and EFS), CR at the end of induction, MRD at the end of induction, SAA, and harms (WDAEs, hypersensitivity reactions, anaphylactic reactions, and silent inactivation).
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of a clinically important effect for CR at the end of induction based on a threshold informed by the clinical experts consulted by CADTH for this review. The target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for survival rates (OS, DFS from CR, EFS), MRD at the end of induction, SAA during induction, and harms.
For the GRADE assessments, findings from the COG AALL07P4 and DFCI 11-001 studies were assessed individually because the trials were different in terms of enrolled populations (patients with high-risk B-cell ALL in the COG AALL07P4 trial and patients with ALL and LL in the DFCI 11-001 trial) and employed different treatment protocols (intermittent asparagine depletion in the COG AALL07P4 trial and continuous asparagine depletion in the DFCI 11-001 trial).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for calaspargase pegol versus pegaspargase in patients with high-risk B-cell ALL in the COG AALL07P4 trial. Table 3 presents the GRADE summary of findings for calaspargase pegol versus pegaspargase in patients with ALL in the DFCI 11-001 trial.
Table 2: Summary of Findings for Calaspargase Pegol Versus Pegaspargase for Patients With High-Risk B-Cell ALL in COG AALL07P4 Study
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Pegaspargase 2,500 IU/m2 | Calaspargase pegol 2,500 IU/m2 | Difference | |||||
Overall survival: Full analysis set | |||||||
Probability of being alive at 1 year Median follow-up: 62.6 months | || || |||| | || | ||| ||| |||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowa | Calaspargase pegol may result in little to no difference in overall survival at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive at 4 years Median follow-up: 62.6 months | || || |||| | || | ||| ||| |||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowa | Calaspargase pegol may result in little to no difference in overall survival at 4 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Disease-free survival from complete remission: Full analysis set | |||||||
Probability of being alive disease-free from complete remission at 1 year Median follow-up: 62.6 months | || || |||| | || | ||| ||| |||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowb | Calaspargase pegol may result in little to no difference in disease-free survival from complete remission at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive disease-free from complete remission at 4 years Median follow-up: 62.6 months | || || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Moderatec | Calaspargase pegol likely results in little to no difference in disease-free survival from complete remission at 4 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Event-free survival: Full analysis set | |||||||
Probability of being alive event-free at 1 year Median follow-up: 62.6 months | || || |||| | || | ||| ||| ||||| | ||| ||| ||||| ||||| | || ||||| ||| |||||||||| || | Lowa | Calaspargase pegol may result in little to no difference in event-free survival at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive event-free at 4 years Median follow-up: 62.6 months | || || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowa | Calaspargase pegol may result in little to no difference in event-free survival at 4 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Complete remission at end of induction day 29: Full analysis set | |||||||
Complete remission rate at end of induction Follow-up: Day 29 | || || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowd | Calaspargase pegol may result in an increase in complete remission at the end of induction day 29 when compared with pegaspargase. |
Minimal residual disease (positive minimal residual disease, ≥ 0.1%) at end of induction day 29: Full analysis set | |||||||
Positive minimal residual disease (≥ 0.1%) rate at end of induction Follow-up: Day 29 | || || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowa | Calaspargase pegol may result in little to no difference in positive minimal residual disease (≥ 0.1%) at the end of induction day 29 when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Serum asparaginase activity ≥ 0.10 IU/mL during remission induction | |||||||
Serum asparaginase activity ≥ 0.10 IU/mL rate | NA | No data available | No data available | No data available | No data available | NA | There is no evidence for the effect of calaspargase pegol on serum asparaginase activity when compared with pegaspargase. |
HRQoL | |||||||
HRQoL due to treatment | NA | No data available | No data available | No data available | No data available | NA | There is no evidence for the effect of calaspargase pegol on HRQoL when compared with pegaspargase. |
Harms: Safety analysis set | |||||||
WDAEs Follow-up: Throughout study | || || |||| | || | ||| ||| |||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Very lowe, f | The evidence is very uncertain about the effects on WDAEs of calaspargase pegol when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Hypersensitivity reactions Follow-up: Throughout study | || || |||| | || | || ||| |||||| | || ||| |||||||||| | || ||||| ||| |||||||||| || | Lowf | Calaspargase pegol may result in little to no difference in hypersensitivity reactions when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Anaphylactic reactions Follow-up: Throughout study | || || |||| | || | ||| ||| |||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowf | Calaspargase pegol may result in little to no difference in anaphylactic reactions when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Silent inactivation Follow-up: Throughout study | NA | No data available | No data available | No data available | No data available | NA | There is no evidence for the effect of calaspargase pegol on silent inactivation when compared with pegaspargase. |
AE = adverse event; ALL = acute lymphoblastic leukemia; CI = confidence interval; COG = Children’s Oncology Group; HRQoL = health-related quality of life; NA = not applicable; NR = not reported; RCT = randomized controlled trial; WDAE = withdrawal due to adverse event.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 2 levels for very serious imprecision. There is no established minimal important difference and clinical experts consulted by CADTH could not provide a threshold of important difference. In the absence of a known threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the upper and lower bounds of the 95% CI for difference between groups suggested a possibility of both benefit and harm.
bRated down 2 levels for very serious imprecision. No threshold was identified in the literature, but according to the clinical experts consulted by CADTH for the review, any difference in disease-free survival could be considered clinically meaningful, so the null was used as the threshold. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the upper and lower bounds of the 95% CI for difference between groups suggested a possibility of both benefit and harm.
cRated down 1 level for serious imprecision. No threshold was identified in the literature, but according to the clinical experts consulted by CADTH for the review, any difference in disease-free survival could be considered important, so the null was used as the threshold. The CADTH review team judged that the effect estimate was unlikely to include any important effect; however, the upper bound of the 95% CI for difference between groups suggested a possibility of benefit.
dRated down 2 levels for very serious imprecision. No threshold was identified in the literature, but according to the clinical experts consulted by CADTH for the review, a 5% difference between groups in complete remission could be considered clinically meaningful; however, the upper and lower bounds of the 95% CI for difference between groups suggested a possibility of both important benefit and important harm.
eRated down 1 level for risk of bias. The open-label study design may have biased WDAEs from patients’ and assessors’ knowledge of assigned treatment, although the direction of potential bias is unclear. Moreover, the clinical experts consulted by CADTH noted that since asparaginase is given as part of a multiagent chemotherapeutic protocol, it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component.
fRated down 2 levels for very serious imprecision. In the absence of an established threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the effect estimate was based on very few events. Moreover, the clinical experts consulted by CADTH noted that since asparaginase is given as part of a multiagent chemotherapeutic protocol, it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component.
Source: COG AALL07P4 Clinical Study Report.13 Details included in the table were provided from sponsor in response to additional data request.14
Table 3: Summary of Findings for Calaspargase Pegol Versus Pegaspargase for Patients With ALL in DFCI 11-001 Study
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Pegaspargase 2,500 IU/m2 | Calaspargase pegol 2,500 IU/m2 | Difference | |||||
Overall survival: Full analysis set | |||||||
Probability of being alive at 1 year Median follow-up: 26.6 months | ||| || |||| | || | ||||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Moderatea | Calaspargase pegol likely results in little to no difference in overall survival at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive at 2 years Median follow-up: 26.6 months | ||| || |||| | || | ||||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Lowb | Calaspargase pegol may result in little to no difference in overall survival at 2 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Disease-free survival from complete remission: Full analysis set | |||||||
Probability of being alive disease-free from complete remission at 1 year Median follow-up: 26.6 months | ||| || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Lowc | Calaspargase pegol may result in little to no difference in disease-free survival from complete remission at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive disease-free from complete remission at 2 years Median follow-up: 26.6 months | ||| || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Moderated | Calaspargase pegol likely results in little to no difference in disease-free survival at 2 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Event-free survival: Full analysis set | |||||||
Probability of being alive event-free at 1 year Median follow-up: 26.6 months | ||| || |||| | || | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Lowe | Calaspargase may result in little to no difference in event-free survival at 1 year when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Probability of being alive event-free at 2 years Median follow-up: 26.6 months | ||| || |||| | || | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Moderatea | Calaspargase likely results in little to no difference in event-free survival at 2 years when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Complete remission at end of induction: Full analysis set | |||||||
Complete remission rate at end of induction Follow-up: Day 32 | ||| || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | || ||||| ||| |||||||||| || | Moderatef | Calaspargase pegol likely results in little to no difference in complete remission at end of induction day 32 when compared with pegaspargase. |
Minimal residual disease (≥ 0.01) at end of induction: Full analysis set | |||||||
Minimal residual disease ≥ 0.01 rate Follow-up: Day 32 | ||| || |||| | || | | ||| ||||| | || ||| |||||||||| | || ||||| ||| |||||||||| || | Lowb | Calaspargase pegol may result in little to no difference in minimal residual disease (≥ 0.01) at end of induction day 32 when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
SAA ≥ 0.10 IU/mL during remission induction: Pharmacokinetic analysis set | |||||||
SAA ≥ 0.10 IU/mL rate Follow-up: Day 7 (4 minutes to 5 minutes postinfusion) | ||| || |||| | ||| ||| ||||| | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Lowh | Calaspargase pegol may result in little to no difference in SAA ≥ 0.10 IU/mL 4 minutes to 5 minutes postinfusion when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
SAA ≥ 0.10 IU/mL rate Follow-up: Day 11 (4 days after dose) | ||| || |||| | ||| ||| ||||| | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Lowh | Calaspargase pegol may result in little to no difference in SAA ≥ 0.10 IU/mL 4 days postinfusion when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
SAA ≥ 0.10 IU/mL rate Follow-up: Day 18 (11 days after dose) | ||| || |||| | ||| ||| ||||| | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Lowh | Calaspargase pegol may result in little to no difference in SAA ≥ 0.10 IU/mL 11 days postinfusion when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
SAA ≥ 0.10 IU/mL rate Follow-up: Day 25 (18 days after dose) | ||| || |||| | ||| ||| ||||| | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Moderatei | Calaspargase pegol likely results in little to no difference in SAA ≥ 0.10 IU/mL 18 days postinfusion when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
SAA ≥ 0.10 IU/mL rate Follow-up: Day 32 (25 days after dose) | ||| || |||| | ||| ||| ||||| | ||| ||| ||||| | || ||||| ||| ||||||| | || ||||| ||| ||||||| | Moderatej | Calaspargase pegol likely results in a greater proportion of patients with SAA ≥ 0.10 IU/mL 25 days postinfusion when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
HRQoL | |||||||
HRQoL due to treatment | NA | No data available | No data available | No data available | No data available | NA | There is no evidence for the effect of calaspargase pegol on HRQoL when compared with pegaspargase. |
Harms: Safety analysis set | |||||||
WDAEs Follow-up: Throughout study | ||| || |||| | || | ||| ||| ||||| | ||| ||| |||||||||| | ||| ||| |||||||||| | Very lowk, l | The evidence is very uncertain for the effect of calaspargase pegol in WDAEs when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Hypersensitivity reactions Follow-up: Throughout study | ||| || |||| | || | || ||| ||||| | || ||| |||||||||| | || ||| |||||||||| | Lowl | Calaspargase pegol may result in little to no difference in hypersensitivity reactions when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Anaphylactic reactions Follow-up: Throughout study | ||| || |||| | || | || ||| ||||| | || ||| |||||||||| | || ||| |||||||||| | Lowl | Calaspargase pegol may result in little to no difference in anaphylactic reactions when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
Silent inactivation Follow-up: Throughout study | ||| || |||| | || | | ||| ||||| | || ||| |||||||||| | || ||| |||||||||| | Lowl | Calaspargase pegol may result in little to no difference in silent inactivation when compared with pegaspargase. There is some uncertainty about the clinical importance of the estimates. |
AE = adverse event; ALL = acute lymphoblastic leukemia; CI = confidence interval; DFCI = Dana-Farber Cancer Institute; GEE = generalized estimating equation; HRQoL = health-related quality of life; NA = not applicable; NR = not reported; SAA = serum asparaginase activity; WDAE = withdrawal due to adverse event.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. There is no known threshold and clinical experts consulted by CADTH could not provide a threshold of important difference. In the absence of a known threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the lower bound of the 95% CI for the difference between groups suggested a possibility of harm.
bRated down 2 levels for very serious imprecision. In the absence of a known threshold, the CADTH team rated their certainty in a non-0 effect. Although no threshold (i.e., the null) was crossed, the effect estimate was based on very few events in each group.
cRated down 2 levels for very serious imprecision. According to the clinical experts consulted by CADTH for the review, any difference in disease-free survival could be considered clinically meaningful, so the null was used as the threshold. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the effect estimate was based on very few events.
dRated down 1 level for serious imprecision. According to the clinical experts consulted by CADTH for the review, any difference in disease-free survival could be considered clinically meaningful, so the null was used as the threshold. The CADTH review team judged the between-group difference was unlikely to include an important effect; however, the lower bound of the 95% CI for the difference between groups suggested a possibility of harm.
eRated down 2 levels for very serious imprecision. There was no established minimal important difference and clinical experts consulted by CADTH could not provide a threshold of important difference. In the absence of a known threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the upper and lower bounds of the 95% CI for difference between groups suggested a possibility of both benefit and harm.
fRated down 1 level for serious imprecision. No known threshold was identified but according to the clinical experts consulted by CADTH for the review, a 5% difference between groups in complete remission could be considered clinically meaningful. The CADTH review team judged that the effect estimate was unlikely to include an important effect; however, the lower bound of the 95% CI for difference between groups suggested a possibility of important harm.
gOdds ratios of SAA levels were estimated using a GEE model for comparing categorical SAA levels between treatments, with 90% CIs, adjusted for the following: treatment, actual sampling time points, and the interaction of treatment and actual sampling time points as effects.
hRated down 2 levels for very serious imprecision. In the absence of a known threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the effect estimate was based on very few events.
iRated down 1 level for serious imprecision. There was no known threshold and clinical experts consulted by CADTH could not provide a threshold of important difference, so the null was used as the threshold. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the 95% CI for the difference between groups suggested a possibility of benefit.
jRated down 1 level for serious imprecision. There was no known threshold and clinical experts consulted by CADTH could not provide a threshold of important difference, so the null was used as the threshold. The CADTH team judged that the point estimate for the between-group difference was likely to include an important benefit. Both lower and upper boundaries of the 95% CI of the between-group difference suggested a possibility of benefit. Although no threshold (i.e., the null) was crossed, the effect estimate was based on relatively few events in each group.
kRated down 1 level for risk of bias due to open-label study design and patients’ and assessors’ knowledge of assigned treatment. The open-label study design may have biased WDAEs from knowledge of assigned treatment, although the direction of potential bias is unclear. Moreover, the clinical experts consulted by CADTH noted that since asparaginase is given as part of a multiagent chemotherapeutic protocol, it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component.
lRated down 2 levels for very serious imprecision. In the absence of an established threshold, the null was used. The CADTH review team judged that the point estimate for the between-group difference was unlikely to include an important effect; however, the effect estimate was based on very few events. Moreover, the clinical experts consulted by CADTH noted that since asparaginase was given as part of a multiagent chemotherapeutic protocol, it was challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component.
Source: DFCI 11-001 Clinical Study Report.15 Details included in the table were provided from sponsor in response to additional data request.14
Long-Term Extension Studies
No long-term extension studies were submitted in the systematic review evidence.
Indirect Comparisons
No indirect treatment comparisons were submitted in the systematic review evidence.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies addressing important gaps in the systematic review evidence were identified.
Conclusions
Evidence from a randomized phase II, open-label trial (the COG AALL07P4 study) in patients aged 1 year to 30 years with high-risk B-cell ALL suggested that calaspargase pegol likely results in little to no difference in DFS rates from the attainment of CR at 4 years (moderate certainty) and may result in an increase in CR at the end of induction (low certainty) compared with pegaspargase. Furthermore, the COG AALL07P4 trial suggested that compared with pegaspargase, calaspargase pegol may result in little to no difference in OS rates at 1 year and 4 years, DFS rates from the attainment of CR at 1 year, EFS rates at 1 year and 4 years, and MRD at the end of induction (low certainty).
Evidence from a randomized phase II, open-label trial (the DFCI 11-001 trial) in patients aged 1 year to 21 years with ALL suggested that calaspargase pegol likely results in a greater proportion of patients with SAA of at least 0.10 IU/mL at day 32 (moderate certainty) when compared with pegaspargase and likely results in little to no difference in OS rates at 1 year, DFS rates from the attainment of CR at 2 years, EFS rates at 2 years, and CR at the end of induction (moderate certainty) compared with pegaspargase. Furthermore, the DFCI 11-001 study suggested that compared with pegaspargase, calaspargase pegol may result in little to no difference in OS at 2 years, DFS rates from the attainment of CR at 1 year, EFS rates at 1 year, and MRD at the end of induction (low certainty).
SAA is an established surrogate measure of asparagine depletion. SAA levels above 0.10 IU/mL have been shown to be associated with complete asparagine depletion and therefore, therapeutic benefit. The clinical experts consulted by CADTH considered the results for SAA measured during induction to be supportive of calaspargase pegol’s greater biological stability compared with pegaspargase, of calaspargase pegol’s ability to maintain asparagine suppression in plasma, of its longer half-life duration compared to pegaspargase, and of its ability to be administered with less dosing frequency than pegaspargase (i.e., a schedule of every 3 weeks versus every 2 weeks).
No unexpected safety signals were identified with treatment of calaspargase pegol in the COG AALL07P4 or the DFCI 11-001 trial. The clinical experts consulted by CADTH noted that asparaginase is given as part of an MAC protocol and it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component. The COG AALL07P4 and DFCI 11-001 trials did not report data on HRQoL.
Confidence in the effect estimates from the COG AALL07P4 and the DFCI 11-001 studies was limited, primarily due to both studies not being designed to assess comparative efficacy between calaspargase pegol and pegaspargase, relatively small sample sizes, and the open-label design, which may have biased subjective outcomes such as harms.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of calaspargase pegol 2,500 units/m2 given intravenously no more frequently than every 21 days as a component of an MAC regimen in the treatment of ALL in children and adults.
Disease Background
The content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
ALL is a heterogeneous group of disorders that results from the clonal proliferation and expansion of malignant lymphoid cells in the bone marrow, blood, and extramedullary sites.5 WHO classifies ALL into 2 major subtypes: B-lymphoblastic and T-lymphoblastic leukemia, which are further divided according to the presence and type of genetic abnormalities.5,6 The frequency of ALL B-cell phenotypes is approximately 80% to 85% in pediatric patients and close to 75% in adult patients. The frequency of the Philadelphia chromosome in patients with ALL is about 25% to 30% in pediatric patients and about 3% to 5% in adult patients.7
Although ALL primarily starts from the bone marrow and peripheral blood, any organ or tissue may be infiltrated.16 Signs and symptoms of ALL are highly variable. At presentation, patients may have easy bruising, bleeding, dyspnea, dizziness, infections due to anemia, thrombocytopenia, and neutropenia. In some patients, extremity and joint pain present. Twenty percent of patients have lymphadenopathy, splenomegaly, and/or hepatomegaly, and 10% of patients have symptoms of CNS involvement.17 The symptoms burden is high as patients report a median of 9 physical and 2 psychological symptoms, and 61% of patients report 10 or more concurrent symptoms.16
Multiple factors affect prognosis in ALL and are used for risk stratification, which is the basis for treatment planning and management of disease.5 These factors include demographic, clinical, biological, or genetic features of leukemia, and response to treatment.18
The US Surveillance, Epidemiology, and End Results database has shown improvements in survival with 5-year OS rates of 89% for children and 89% for adolescents and young adults (AYAs) (15 years to 39 years). However, 5-year survival rates for the other adult patients remain low at approximately 20% to 40%. Five-year survival rates are especially poor in older adult patients at approximately 20%.2
ALL is the least common type of leukemia diagnosed in adults; however, it is the most common type of leukemia diagnosed in young children, occurring more often in boys than girls.1 It is estimated that ALL represents 75% to 80% of acute leukemias among children and 20% of all leukemias among adults.2 The median age of diagnosis is 17 years with 53.5% of patients diagnosed before the age of 20 years.2 In 2018, the incidence rate of ALL for all ages in Canada (excluding Quebec) was 1.3 per 100,000, with the majority of patients being younger than 19 years.3,4
In general, the diagnosis of ALL requires the demonstration of 20% or greater bone marrow lymphoblasts on bone marrow aspirate and biopsy materials, which includes morphologic assessment of Wright-Giemsa–stained bone marrow aspirate smears, and hematoxylin-stained and eosin-stained core biopsy and clot sections; comprehensive flow cytometric immunophenotyping; baseline flow cytometric and/or molecular characterization of leukemic clone to facilitate subsequent MRD analysis; and karyotyping of G-banded metaphase chromosomes.19
Optimal risk stratification and treatment planning require testing marrow or peripheral blood lymphoblasts for specific recurrent genetic abnormalities using interphase fluorescence in situ hybridization testing, including probes capable of detecting the major recurrent genetic abnormalities; reverse transcriptase-polymerase chain reaction testing for BCR-ABL1 in B-cell ALL (quantitative or qualitative), including the determination of transcript size; and comprehensive testing by next-generation sequencing for gene fusions and pathogenic mutations.19
Standards of Therapy
The content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
Pretreatment Phase
To manage potential hypersensitivity and infusion reactions with any asparaginase treatment, some Canadian institutions have integrated the administration of premedication such as acetaminophen, diphenhydramine, and corticosteroids into their treatment protocols.20,21 Studies have shown that the administration of premedication before pegylated asparaginase treatment results in significant reductions in severe, clinically evident hypersensitivity reactions.20,22,23 Currently, the use of premedication varies across Canada. Some clinicians prefer to avoid masking any occurrence of hypersensitivity, using it as an indicator to further investigate the occurrence of silent inactivation (subclinical asparaginase activity levels). Still, TDM is required to detect subclinical asparaginase activity levels, with or without the use of premedication.20,21,24
Treatment Phase
The goal of first-line treatment of ALL is curative, requiring the attainment of CR.25 Treatment consists of multiple phases of MAC: the induction phase, the intensification and/or consolidation phase, the CNS therapy phase, and the maintenance phase.1,2,18
Escherichia coli–derived asparaginase is a cornerstone component of the induction and intensification and/or consolidation phases of first-line treatment,1,2,18 and for more than 30 years, has been a significant chemotherapeutic addition, ultimately becoming part of the WHO list of essential drugs.26,27 Pegaspargase, a pegylated form of E. coli–derived asparaginase, is the established SOC asparaginase product incorporated across Canada as part of standard clinical practice and clinical trials.
Two different childhood ALL treatment strategies are commonly used across Canada, originating from the COG and DFCI consortia.8,9 Both COG and DFCI protocols include the use of pegaspargase. There is also consensus that AYA patients demonstrate improved outcomes when treated with a pediatric-inspired regimen.28 According to the clinical expert consulted by CADTH, most pediatric patients with ALL are treated using COG protocols in Canada except in Quebec, where they are treated using DFCI protocols. AYA patients are predominantly treated with a modified DFCI regimen or another pediatric-inspired regimen.28 Adult patients (non-AYA) with ALL also commonly receive a dose-modified DFCI pediatric-inspired regimen containing pegaspargase.
There are key differences in COG and DFCI protocols regarding backbone therapies, phases, schedules, and duration and frequency of asparaginase. COG protocols are aimed at intermittent asparagine depletion with rotating phases lasting 1 month to 2 months from induction through delayed intensification. In contrast, DFCI protocols are aimed at continuous asparagine depletion with a prolonged intensification phase that is employed within cycles of MAC. In COG protocols, patients receive a single dose of the study drug at induction (and at extended induction for slow early responders), followed by additional doses of the study drug at each subsequent phase (consolidation, interim maintenance phase I, delayed intensification I, interim maintenance phase II [as applicable], delayed intensification phase II [as applicable], and maintenance). In DFCI protocols, however, patients receive a single dose of the study drug during remission induction, and then every 2 weeks during intensification.
While Erwinia-derived asparaginase is available in Canada (e.g., Rylaze [crisantaspase recombinant]), as per its Health Canada indication, it is reserved for patients who have developed hypersensitivity to E. coli–derived L-asparaginase (such as pegaspargase or calaspargase pegol),29-32 and is not considered for initial first-line treatment.
In general, the goal of the induction phase, which is typically 4 weeks to 6 weeks in duration, is to rid the blood and marrow of visible leukemic blast cells and attain CR. A combination of chemotherapy is used during induction, such as vincristine, an anthracycline, a glucocorticoid, and asparaginase. A CNS therapy phase designed to provide adequate CNS prophylactic or actual treatment is generally planned after induction or consolidation. The goal of the intensification and/or consolidation phase is to reduce or eliminate any remaining leukemic cells. Specifically, for consolidation, several short sequential courses of chemotherapy are employed, usually with cytarabine, high-dose methotrexate, vincristine, asparaginase, mercaptopurine, and glucocorticoids. This sequence is sometimes followed by a late intensification phase (reinduction therapy) that includes a combination of drugs similar to that used during the induction therapy. Altogether, the intensification and/or consolidation phase is 4 months to 6 months in duration.
Though the efficacy of asparaginase products in ALL treatment is dependent on the depletion of asparagine, the direct measurement of asparagine depletion is difficult and not practical as a routine clinical test. As such, asparaginase levels are measured in serum as a surrogate marker for asparagine depletion. This has been validated as a method of quantifying the efficacy of asparaginase formulations.33
“Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation,” an article from international journal Haematologica that includes Canadian clinical expert Dr. James Whitlock as a principal author, outlines how asparaginase activity levels should be used in identifying patients with subtherapeutic asparaginase activity from silent inactivation in clinical practice, which is reflected in pediatric clinical trials in Canada.34 Hypersensitivity reactions to asparaginase can occur with or without clinical symptoms of an allergy, the latter referred to as silent inactivation; in both situations, the drug is completely neutralized. Just as importantly, patients may also develop atypical allergies without inactivation (allergic-like reactions), which does not require a discontinuation. Overall, the importance of TDM in the management of patients treated with any of the asparaginase products is recognized; however, the routine use of TDM may vary in different settings across Canada.
Post-Treatment Phase
Patients concluding the asparaginase-containing phases of an MAC protocol will continue to receive maintenance therapy, typically consisting of daily mercaptopurine and weekly methotrexate, with or without vincristine, and glucocorticoid pulses every 1 month to 3 months over the course of 2 years to 3 years.
Drug Under Review
Key characteristics of calaspargase pegol are summarized in Table 3 with other treatments available for the treatment of patients with ALL.
The recommended dosage under review by Health Canada for calaspargase pegol is 2,500 units/m2 given intravenously no more frequently than every 21 days. Calaspargase pegol is indicated as a component of an MAC regimen for the treatment of patients with ALL. It has not been previously reviewed by CADTH.
Calaspargase is a conjugate of L-asparaginase (L-asparagine amidohydrolase) and monomethoxy polyethylene glycol. L-asparaginase is an enzyme that catalyzes the conversion of the amino acid L-asparagine into aspartic acid and ammonia. The depletion of L-asparagine in blood serum results in the inhibition of protein synthesis, DNA synthesis, and ribonucleic acid synthesis — especially in leukemic blasts that are not able to synthesize L-asparagine, thus undergoing programmed cell death. Normal cells, in contrast, are capable of synthesizing L-asparagine and are less affected by its rapid withdrawal during treatment with the enzyme L-asparaginase. The mechanism of action of calaspargase pegol is the same as that of the established SOC pegaspargase. To mitigate the risk of drug shortages, the sponsor developed calaspargase pegol to extend the shelf life and half-life relative to the sponsor’s original product, pegaspargase. Both calaspargase pegol and pegaspargase contain the same asparagine-specific enzyme derived from E. coli, as a conjugate of L-asparaginase linked to a similar monomethoxy polyethylene glycol. The only difference between the 2 products is the linker connecting the 2 components. Pegaspargase contains a succinimidyl succinate linker, while calaspargase pegol contains a succinimidyl carbonate linker, the latter being less prone to enzymatic hydrolysis and more stable. As a result of the improved stability, calaspargase pegol has a 36-month shelf life compared to 8 months for the pegaspargase formulation.35
Calaspargase pegol is currently under review by Health Canada as a component of an MAC regimen for the treatment of patients with ALL. The Notice of Compliance for this indication was expected on November 8, 2023. The sponsor requested reimbursement as per the indication to be reviewed by CADTH.
The FDA approved calaspargase pegol on December 20, 2018, as a component of an MAC regimen for the treatment of ALL in pediatric and young adult patients aged 1 month to 21 years.
Table 4: Key Characteristics of Calaspargase Pegol and Pegaspargase
Characteristic | Calaspargase pegol | Pegaspargase |
|---|---|---|
Mechanism of action | Depletion of L-asparagine in blood serum results in inhibition of protein synthesis, DNA synthesis, and RNA synthesis, especially in leukemic blasts that are not able to synthesize L-asparagine, thus undergoing apoptosis | Depletion of L-asparagine in blood serum results in inhibition of protein synthesis, DNA synthesis, and RNA synthesis, especially in leukemic blasts that are not able to synthesize L-asparagine, thus undergoing apoptosis |
Indicationa | Under review by Health Canada as a component of a multiagent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia | A component of a multiagent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia |
Route of administration | IV infusion | Intramuscular injection or IV infusion |
Recommended dosage | 2,500 units/m2 no more frequently than every 21 days | 2,500 units/m2 every 14 days in pediatric patients with body surface area > 0.6 m2 and younger than 21 years There are data suggesting that the dosage of pegaspargase could be adjusted to 82.5 units/kg body weight every 14 days in children with a body surface area < 0.6 m2, or to 2,000 units/m2 every 14 days in adult patients older than 21 years. |
Serious adverse effects or safety issues | Hypersensitivity, pancreatitis, thrombosis, hemorrhage, hepatotoxicity Monitor patients at least weekly, with bilirubin, transaminases, glucose, and clinical examinations until recovery from the cycle of therapy. If an adverse reaction should occur, modify treatment according to specified dosage modifications. | Hypersensitivity, central nervous system toxicity, glucose intolerance, hepatotoxicity, infections (myelosuppression), pancreatitis, nephropathy, thrombosis, and/or coagulopathy. Pegaspargase should only be given in a hospital setting where appropriate resuscitation equipment is available. Patients should be closely monitored and carefully observed for any adverse reactions throughout the infusion period. Monitor patients for an hour after administration for hypersensitivity reactions. In addition to the clinical exam, regularly monitor coagulation parameters, liver impairment, serum amylase, uric acid level, peripheral blood count, and hyperglycemia. |
RNA = ribonucleic acid.
aHealth Canada–approved indication.
Sources: Draft Product Monograph for calaspargase pegol36 and Product Monograph for pegaspargase.37
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the Stakeholder section of this report.
CADTH received 1 patient group submission from LLSC. LLSC is a national charitable status organization dedicated to finding a cure for blood cancers and to improving the quality of life of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support.
LLSC conducted an online survey with 74 respondents from 10 provinces in Canada during April to May 2023. The majority of respondents (n = 47) identified as patients with ALL and 20 respondents indicated that they were caregivers of patients with ALL. Respondents who indicated that they were neither a patient with ALL nor a caregiver of a patient with ALL were disqualified from the survey. About half of the patients with ALL indicated that they were older than 30 years at the time of their ALL diagnosis while the other half was younger than 30 years.
LLSC stated that the questions in this survey were not intended to measure the efficacy of the drug under review because it was assumed to be as effective as current treatment options and was also budget-neutral. The questions in this survey were aimed at highlighting the importance of safeguarding the health care system to ensure that treatment medications are supply-secure for those experiencing ALL.
Survey respondents reported their experience with drug shortages at some point during their ALL treatment. Reponses highlighted extremely stressful conditions, fear that the treatment would be unsuccessful, the feeling of powerlessness and being let down by the system, poor quality of life, and lack of sleep because of stress, anxiety, mood swings, and spending time trying to find an alternative solution. Some respondents noted financial impacts, as they had to pay for alternative therapies and buy products to help them cope.
Respondents indicated that they needed to feel included in decision-making as their treatment plan for ALL would have effects on many areas of their lives. In addition to the effectiveness of the treatment, there were other factors that are important to patients when evaluating new treatments for ALL, including side effects, a physician’s recommendation, quality of life, cost, secure supply, and number of treatments.
LLSC highlighted that ALL progresses quickly and aggressively and that to prevent disease progression, the immediate start of treatment upon diagnosis is vital. Survey respondents expressed that delaying the start of treatment may lead to cancer progression, to their bodies being less receptive and tolerable to treatment, and potentially to death.
LLSC noted the importance of having alternative treatments available to ensure treatment can continue should manufacturers run into supply issues. Having supply-secure treatment options would provide comfort and peace of mind to patients and their caregivers during an already difficult and challenging time. Most of the survey respondents indicated that they would be supportive of a government providing public funding for an alternative treatment option for ALL that would work equally well with similar costs to the health care system and would provide treatment assurance in case an alternative ALL medication became unavailable.
Most of the survey respondents reported that the availability of treatment throughout the entire course of therapy is extremely important to them. Some of the respondents commented that ensuring a secure and readily available supply of medications for treatment is something that can be done and would remove stress from patients and caregivers and improve patients’ outcomes.
LLSC asked survey respondents to rate the level of impact that a treatment shortage due to supply issues would have on their lives. Respondents noted the following areas as being most impacted (listed in order of importance to survey respondents): mental health, physical health, quality of life, home life, social life, work life, and finances.
Respondents were asked by LLSC to describe in 3 words their emotional response to being told that a drug that was part of their treatment regimen was suddenly not available due to supply issues. Survey respondents mostly reported words associated with fear, stress, despair, and defeat.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of ALL.
Unmet Needs
The clinical experts consulted by CADTH reported that the goal of treatment for patients with ALL is curative, aimed at maximizing survival while minimizing short-term and long-term toxicities. The clinical experts consulted by CADTH also noted the growing number of clinical-, genetic-, and response-based risk stratification factors that support better survival and/or reduce toxicities.
Current treatment for ALL in Canada was identified by the clinical experts consulted by CADTH as comprising MAC regimens that include pegylated asparaginase, using pediatric protocols developed by COG or DFCI in centres treating children, or pediatric-inspired protocols in centres that treat adults, respectively. The clinical experts consulted by CADTH estimated that the total duration of MAC treatment for patients with ALL who respond is approximately 2.5 years to 3.5 years. Asparaginase is incorporated into the first phases of MAC treatment during the first year. The clinical experts consulted by CADTH noted that patients who do not respond to treatment (e.g., patients who become refractory to current treatment options, patients who receive lower than 80% of the recommended dose of asparaginase) require high-dose chemotherapy and/or allogeneic stem cell transplant and experience high rates of treatment failure. According to the clinical experts consulted by CADTH, current suppliers of L-asparaginase (pegaspargase) in Canada have been unable to provide a consistent supply that sufficiently meet the needs of all patients due to the drug’s relatively short half-life (2 weeks). The clinical expert consulted by CADTH noted that new treatments with a prolonged half-life may make treatments more convenient for patients and care teams by leading to reduced frequency of administration. Pegaspargase’s half-life requires a relatively high frequency of dosing (e.g., 15 treatments during the intensification phase). The clinical experts consulted by CADTH agreed that patients with ALL would benefit from treatments that are better tolerated and more convenient, which may lead to improved compliance.
Place in Therapy
The clinical experts consulted by CADTH anticipated that calaspargase pegol would become the new standard for prolonged asparaginase therapies and would replace the current SOC due to its incorporation of a more stable linker, which in turn is expected to lead to a prolonged shelf life, more stable supply chain, and comparatively shorter frequency of dosing (e.g., approximately 10 treatments during the intensification phase). The clinical experts consulted by CADTH did not expect calaspargase pegol to cause a shift in the current treatment paradigm.
Patient Population
The clinical experts consulted by CADTH indicated that all patients with newly diagnosed ALL would benefit from treatment with calaspargase pegol, since asparaginase has a unique mechanism of action playing an essential part in adequate asparagine depletion and is considered to comprise an essential component of therapy in ALL. Patients with relapsed ALL were also considered by the clinical experts consulted by CADTH to potentially benefit from calaspargase pegol, if there was no known prior intolerance to other forms of asparaginase given that such intolerance applies to any patient receiving an E. coli–derived enzyme. The clinical experts consulted by CADTH noted that misdiagnosis of ALL is unlikely. The clinical experts consulted by CADTH reported that all patients with ALL are tested for the presence of the BCR-ABL fusion gene (gene sequence; Philadelphia chromosome) by nested polymerase chain reaction (PCR), where patients who test positive for this gene are treated with tyrosine kinase inhibitors; therefore, treatment with L-asparaginase should be avoided due to overlapping toxicities. Patients with Ph-positive ALL were excluded from the COG AALL07P4 and DFCI 11-001 studies. The clinical experts consulted by CADTH noted that currently, pediatric patients and adult patients with Ph-positive ALL are on distinct treatment protocols that include tyrosine kinase inhibitor therapy. The clinical experts noted that while adult patients would not receive asparaginase therapy concurrently with tyrosine kinase inhibitor therapy due to overlapping toxicities, they considered asparaginase therapy an option for pediatric patients with Ph-positive ALL and would not expect PK and safety outcomes to differ in these patients compared to pediatric patients with Ph-negative ALL. Other patient subgroups excluded from clinical trial populations were infants aged up to 1 year (in the COG AALL07P4 and DFCI 11-001 trials), adults (patients older than 21 years in the DFCI 11-001 trial and older than 30 years in the COG AALL07P4 trial), and patients with disease immunophenotype (T-cell ALL in the COG AALL07P4 trial), who were considered by the clinical experts consulted by CADTH to be eligible for asparaginase treatment and therefore appropriately targeted for treatment with calaspargase pegol. The clinical experts consulted by CADTH specified that several risk factors are considered before starting treatment, including age older than 10 years, WBC count below 50 × 109/L at presentation or diagnosis, adverse genetic features including karyotype (e.g., the translocation t(9;22)(q34;q11), hypodiploidy), molecular studies (e.g., BCR-ABL, KMT2A mutations), and gene expression (e.g., IKZF, CRLF2). Regardless of risk classification, the clinical experts consulted by CADTH highlighted MRD response as a key on-treatment prognostic factor such that patients at standard risk or high risk but with inadequate MRD response are treated using high-risk or very high-risk protocols incorporating asparaginase, respectively.
Assessing the Response Treatment
The clinical experts consulted by CADTH reported that a clinically meaningful treatment response should be assessed using OS, CR postinduction, MRD-negative status postinduction, and SAA levels following the administration of asparaginase to monitor adequate asparagine depletion and clinical reactions (e.g., allergic or infusion-related reaction, silent inactivation).
Discontinuing Treatment
According to the clinical experts consulted by CADTH, treatment with asparaginase including calaspargase pegol should be discontinued in the event of notable AEs (e.g., hypersensitivity reaction including silent inactivation, allergic reaction, development of neutralizing antibodies, severe liver toxicity, severe pancreatitis, severe thrombotic or hemorrhagic event, persistent severe hepatic dysfunction).
Prescribing Considerations
The clinical experts consulted by CADTH indicated that patients with ALL are treated in specialized treatment centres as inpatients or outpatients by hematologists or oncologists with significant training and experience. The clinical experts consulted by CADTH also noted that ALL treatment has to be administered in a setting where the urgent recognition of, and intervention for, serious immediate AEs such as anaphylaxis or other hypersensitive reactions can occur. The relevant specialists would be oncologists and critical care staff.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the Stakeholder section of this report.
CADTH received 1 clinician group submission from the OH-CCO Hematology Cancer Drug Advisory Committee. OH-CCO’s cancer drug advisory committees provide evidence-based clinical and health system guidance on drug-related issues in support of OH-CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. Information for this review was collected through videoconferencing and 2 clinicians contributed to this submission.
According to the OH-CCO, the current standard treatment for patients with ALL is pegaspargase, which is potentially curative. The clinicians providing input noted that there is no large unmet need as pegaspargase is available; however, less frequent dosing may be beneficial.
OH-CCO stated that calaspargase pegol is a standard component of an MAC for ALL. Patients who are eligible for the standard induction of ALL treatment would be best suited for treatment with calaspargase pegol. The clinicians providing input noted that to determine the treatment response, the standard leukemia response criteria should be applied, and treatment should be discontinued upon progressive disease or significant intolerance. OH-CCO noted that the appropriate setting for treatment with calaspargase pegol is acute leukemia treatment centres with the presence of leukemia specialists.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Two studies, 1 in patients newly diagnosed with ALL or LL (patients aged 1 year to 21 years) and the other in patients newly diagnosed with high-risk B-cell ALL (patients aged 1 year to 30 years) compared calaspargase pegol to pegaspargase. Pegaspargase is funded by several jurisdictions and would be a relevant comparator. Of note, pegaspargase has not been reviewed by CADTH pCODR. Other potentially relevant comparators are crisantaspase and Erwinia-derived asparaginase (Erwinase) — although this is for the setting of hypersensitivity or silent inactivation of the E. coli–derived asparaginase. Are there any data for comparators other than pegaspargase? | The clinical experts consulted by CADTH reported that pegaspargase is currently used as front-line treatment for patients with newly diagnosed ALL, and therefore considered to be an appropriate comparator for calaspargase pegol. The clinical experts consulted by CADTH were not aware of data or clinical trials to date that use other comparators. |
Considerations for initiation of therapy | |
The clinical trials were completed in patients 1 year to 21 years in the DFCI 11-001 study and 1 year to 30 years in the COG AALL07P4 study. Should patients older than 30 years be eligible for calaspargase? The clinical trials were completed in patients with newly diagnosed disease. Is there evidence to support use in relapsed or refractory ALL or LL? | The clinical experts consulted by CADTH noted that while the trials included patients 1 year up to 30 years, they agreed that the results of the trials could be generalized to patients younger than 1 year and older than 30 years. The clinical experts consulted by CADTH felt that these patients could be treated with an asparaginase-based treatment, including calaspargase pegol. While the COG AALL07P4 trial excluded patients with LL, 9 patients with LL participated in the DFCI 11-001 trial. The clinical experts felt that it would be reasonable to generalize the results of the DFCI 11-001 trial to pediatric and adult patients with LL as they anticipated that treatment outcomes in these patients would be very similar to patients with ALL. The DFCI 11-001 and COG AALL07P4 trials did not enrol patients with relapsed or refractory ALL. The clinical experts consulted by CADTH noted that calaspargase pegol may also be used for patients with relapsed or refractory ALL, if there was no evidence of prior allergic reaction or hypersensitivity to asparaginase. |
Considerations for prescribing of therapy | |
According to the US product monograph, the standard dose of calaspargase pegol is 2,500 units/m2 in 100 mL normal saline or dextrose 5% in water IV over 1 hour every 21 days. Calaspargase pegol comes as a 3,750 units single-use vial, which means that for patients over 1.5 m2, there will be wastage. However, this is the same dose and vial size as pegaspargase, which is given every 14 days, so there is an advantage to the calaspargase pegol in terms of number of infusions as well as wastage to prepare those doses. In some protocols, pegaspargase is capped at 3,750 units as a maximum dose. Would capping also apply to calaspargase pegol? Can pERC clarify the dosing schedule for calaspargase pegol as there may be differences in the frequency of administration with pegaspargase? | According to the clinical experts consulted by CADTH, while the capping of calaspargase pegol as well as pegaspargase at 3,750 units is permitted according to institutional preference using COG protocols, capping is unlikely for adult patients where dose intensity for asparaginase is considered directly tied to outcome. Moreover, the clinical experts consulted reported that the difference in dosing schedule for calaspargase pegol as compared to pegaspargase is less notable for COG-based protocols, which use a discontinuous asparaginase depletion strategy, than for DFCI-based protocols, which use a continuous asparaginase depletion strategy. |
Alignment of the existing funding for pegaspargase would need to be considered (e.g., some jurisdictions fund pegaspargase on a per vial basis and also fund inpatient use). | This is a comment from the drug plans to inform pERC deliberations. |
Generalizability | |
Should there be any consideration for switching patients receiving pegaspargase to calaspargase pegol? | The clinical experts consulted by CADTH did not anticipate any issues switching patients who were receiving pegaspargase to calaspargase pegol since the asparaginase component is the same for both drugs. They anticipated that patients would be switched from pegaspargase to calaspargase pegol once the former product is withdrawn. The clinical experts agreed that they would not switch from pegaspargase to calaspargase for toxicity or inactivation. |
Care provision issues | |
Pegaspargase may be given by an intramuscular or IV route of administration. Calaspargase pegol is only indicated for IV administration. According to the US product monograph, patients should be monitored for hypersensitivity for 1 hour after administration. The product is stored in the refrigerator; therefore, sufficient fridge space is required. | This is a comment from the drug plans to inform pERC deliberations. |
System and economic issues | |
Consideration for pricing should take into account the usual cost-effectiveness analyses, but also should not exceed the per-cycle drug program cost of treatment with the least costly comparator reimbursed. | This is a comment from the drug plans to inform pERC deliberations. |
There tend to be ongoing shortages of asparaginase products, so having alternatives is helpful. PAG is interested in knowing if pegaspargase will be available once calaspargase pegol is Health Canada–approved (i.e., will both products be available)? | The clinical experts noted that they were unable to comment. The sponsor has noted in submission materials received by CADTH for this review that “(…) North American COG and DFCI consortia are currently transitioning and amending existing protocols to include calaspargase pegol, as a replacement to pegaspargase. (…) Following regulatory approval of calaspargase pegol by Health Canada, Servier will also be transitioning to calaspargase pegol in the near future, to securely provide a more reliable supply of a high-quality standard pegylated E. coli–derived asparaginase.”35 |
ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; LL = lymphoblastic lymphoma; PAG = Provincial Advisory Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of calaspargase pegol 2,500 units/m2 given intravenously no more frequently than every 21 days as a component of an MAC regimen for the treatment of patients with ALL. The focus will be placed on comparing calaspargase pegol to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of calaspargase pegol is presented in 4 sections with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows critical appraisal of the evidence. The sponsor did not include long-term extension studies, indirect evidence, or additional studies addressing important gaps in the pivotal and RCT evidence.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal phase II study and 1 phase II study identified in the systematic review.
Systematic Review
The content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | COG AALL07P4 study (pivotal) | DFCI 11-001 study |
|---|---|---|
Designs and populations | ||
Study design | Phase II, multicentre, randomized, open-label, active-controlled trial | Phase II, multicentre, randomized, open-label, active-controlled trial |
Locations | 23 study sites in the US | 9 study sites in total, in the US (6) and Canada (3) |
Patient enrolment dates | Start date: July 2008 End date: March 2021 | Start date: June 2012 End date: July 2021 |
Randomized (N) | 166 | 239a |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Calaspargase pegol 2,500 IU/m2 (for patients starting treatment before protocol amendment 2, September 9, 2009) or 2,100 IU/m2 (for patients starting treatment after amendment 2) administered by IV infusion as a single dose over 1 hour on day 4 of induction, on day 4 of extended induction (i.e., if applicable, for patients with M2 or M1 morphology with MRD ≥ 0.1% at day 29), on day 15 and day 43 of consolidation, on day 2 and day 22 of both interim maintenance periods, and on day 4 and day 43 of both delayed intensification periods | Calaspargase pegol 2,500 IU/m2 administered by IV infusion as a single dose over 1 hour on day 7 of the remission induction phase, and every 3 weeks for 30 consecutive weeks (10 doses) starting with CNS therapy phase for standard-risk and high-risk patients or consolidation IC for very high-risk patients |
Comparator(s) | Pegaspargase 2,500 IU/m2 administered by IV infusion as a single dose over 1 hour on day 4 of induction, on day 4 of extended induction (i.e., if applicable, for patients with M2 or M1 morphology with MRD ≥ 0.1% at day 29), on day 15 and day 43 of consolidation, on day 2 and day 22 of both interim maintenance periods, and on day 4 and day 43 of both delayed intensification periods | Pegaspargase 2,500 IU/m2 administered by IV infusion as a single dose over 1 hour on day 7 of the remission induction phase, and every 2 weeks for 30 consecutive weeks (15 doses) starting with CNS therapy phase for standard-risk and high-risk patients or consolidation for very high-risk patients |
Study duration | ||
Screening phase | NR | NR |
Run-in phase | NA | NA |
Treatment phase |
|
|
Follow-up phase | Follow-up ongoing | Follow-up ongoing |
Outcomes | ||
Primary end point | Primary end point
| Primary end points
|
Secondary and exploratory end points | Secondary efficacy end points
Secondary PK end point
Secondary PD end points
Exploratory end points
| Secondary efficacy end points
Exploratory end points
|
Publication status | ||
Publications | Silverman et al. (2016)38 Schore et al. (2016)39 Angiolillo et al. (2014)40 Angiolillo et al. (2012)41 | Vrooman et al. (2021)42 Vrooman et al. (2019)43 |
Trial Registration | ClinicalTrials.gov Identifier: NCT00671034 | ClinicalTrials.gov Identifier: NCT01574274 |
AE = adverse event; AESI = adverse event of special interest; ALL = acute lymphoblastic leukemia; AUC0–25d = area under the curve plasma asparaginase activity from time 0 to 25 days; AUC 0-inf = area under the curve plasma asparaginase activity from time 0 to infinity; Cmax = maximum asparaginase activity exposure peak concentration; CNS = central nervous system; COG = Children’s Oncology Group; CR = complete remission; CSF = cerebrospinal fluid; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; MRD = minimal residual disease; NA = not applicable; NR = not reported; NSAA = nadir serum asparaginase activity; OS = overall survival; PD = pharmacodynamic; PEG = pegaspargase; PK = pharmacokinetic; SAA = serum asparaginase activity; WBC = white blood cell.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
aOne additional patient in the DFCI 11-001 study, who had an initial pathology report indicative of precursor B-cell ALL, had a subsequent pathology report indicative of leukemia of ambiguous lineage. Therefore, this patient was no longer eligible to participate in the study and did not receive any doses of the study drug.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14
Two phase II, multicentre, randomized, open-label trials (the COG AALL07P4 and DFCI 11-001 trials) assessed the efficacy and safety of calaspargase pegol in pediatric and AYA patients. According to the sponsor, the clinical safety and efficacy of calaspargase pegol was built on pegaspargase. The primary objectives of the clinical development program for calaspargase pegol were focused on the feasibility (i.e., feasibility referring to the evaluation of PK end points) of use and safety, as well as PD comparability versus pegaspargase in anticipation of this formulation soon becoming the only available commercial pegaspargase-asparaginase preparation.35,44
The COG AALL07P4 study enrolled 166 patients aged 1 year to 30 years with newly diagnosed high-risk B-cell ALL in 23 US sites. Patients were considered high risk as defined by the WHO 2008 classification if they had precursor B-cell ALL. Patients were randomized in a 2:1 ratio to calaspargase pegol (2,500 IU/m2 or 2,100 IU/m2) or pegaspargase 2,500 IU/m2. As the proposed Health Canada indication recommends a dose for calaspargase pegol of 2,500 IU/m2, this CADTH clinical report will present data for patients who were randomized to receive calaspargase pegol 2,500 IU/m2 and will exclude data that refer to patients who were first randomized to receive calaspargase pegol 2,100 IU/m2 and subsequently switched to receive pegaspargase 2,500 IU/m2. Details regarding the cohort of patients who were randomized to receive calaspargase pegol 2,100 IU/m2 and later switched to pegaspargase 2,500 IU/m2 are provided in the Interventions section of this clinical report (refer to protocol amendments in this report in the COG AALL07P4 study).
The primary objective of the COG AALL07P4 trial was to determine the PK comparability (asparaginase activity) of the interventions during induction and consolidation while patients were receiving augmented Berlin-Frankfurt-Münster therapy. Secondary objectives of the COG AALL07P4 study included PD parameters during induction and consolidation, MRD (day 29), CR rate (day 29), survival (EFS, DFS of CR, and OS), and TEAEs. The Clinical Study Report with a data cut-off date of December 31, 2015, was the primary data source for the COG AALL07P4 study.
The DFCI 11-001 study enrolled 239 patients in the US (in 6 sites) and Canada (in 3 sites) with newly diagnosed ALL or LL. Patients were randomized in a 1:1 ratio to calaspargase pegol 2,500 IU/m2 or pegaspargase 2,500 IU/m2 and stratified into 4 risk groups based on disease type, age, and initial risk-group classification: 1) patients with ALL younger than 10 years and standard risk, 2) patients with ALL younger than 10 years and initial high risk, 3) patients with ALL 10 years and older (and by definition initial high risk), and 4) patients with LL. Three final risk groups (standard risk, high risk, very high risk) were assigned after the completion of the remission induction phase for patients who attained CR (refer to Table 7 for risk group criteria). The DFCI 11 to 011 trial assigned patients in the final very high-risk group to receive more intensive treatment (refer to the Intervention section as follows and to Figure 2). There were 2 primary objectives of the DFCI 11-001 trial. One primary objective was to assess the toxicity associated with calaspargase pegol administration and determine if it was different from the toxicity associated with pegaspargase 2,500 IU/m2 given as a single dose during remission induction and then every 2 weeks for 30 weeks during postinduction therapy. Another primary objective was to determine the SAA levels associated with calaspargase pegol administered as a single dose during remission induction and then every 2 weeks for 30 weeks during postinduction therapy, and to compare them to the SAA associated with pegaspargase as a single dose during remission induction and then every 2 weeks for 30 weeks during postinduction therapy. Primary end points included AEs and laboratory AEs, adverse events of special interest (AESIs), SAA levels and nadir serum asparaginase activity (NSAA) levels, the SAA level of 0.10 IU/mL or greater at induction phase day 7, day 11, day 18, day 25, and day 32, the NSAA level of 0.10 IU/mL or greater during the postinduction phase overall and by visit day, and the SAA level at week 7. Secondary end points of the DFCI 11-001 trial included MRD (day 32), CR rate (day 32), EFS, DFS of CR, OS, and TEAEs. The Clinical Study Report with a data cut-off date of October 5, 2016, was the primary data source for the DFCI 11-001 study, with an additional Day 120 Report that reported updated survival data (data cut-off date of June 12, 2017) and safety data (data cut-off date of February 1, 2017).
Populations
Inclusion and Exclusion Criteria
In the COG AALL07P4 trial, patients first needed to be eligible for, and enrolled in, the COG study AALL03B1 (Classification of Acute Lymphoblastic Leukemia) or any successor classification study. Eligible patients were aged between 1 year and 30 years at the time of diagnosis and had newly diagnosed high-risk B-precursor ALL with no prior cytotoxic chemotherapy except for steroids and intrathecal cytarabine. For patients younger than 10 years, WBC count had to be 50,000/μL or greater. Patients who were older than 10 years or had received prior steroids could have any WBC count. Key exclusion criteria included patients with Down syndrome, testicular leukemia, prior cytotoxic chemotherapy, pregnancy, and those who were breastfeeding.
In the DFCI 11-001 trial, eligible patients were aged 1 year to 21 years, with newly diagnosed ALL or LL and no prior therapy (except for short courses of corticosteroid, a single dose of intrathecal cytarabine at the time of a diagnostic lumbar puncture, and/or emergent radiation to the mediastinum or other life-threatening masses). Key exclusion criteria included patients who had received more than 7 days of corticosteroids in the preceding 4 weeks or more than 28 days of corticosteroids in the preceding 6 months; any chemotherapy or radiotherapy for a previous malignancy; any antineoplastic drug (except for intrathecal cytarabine); and any other investigational drugs.
Table 7: Final Risk Group Classification in DFCI 11-001 Study
Standard risk | High risk | Very high risk |
|---|---|---|
All criteria must be met | At least 1 criterion must be met | At least 1 criterion must be met |
|
And all of the following criteria must be met:
|
|
ALL = acute lymphoblastic leukemia; CNS = central nervous system; CSF = cerebrospinal fluid; DFCI = Dana-Farber Cancer Institute; LL = lymphoblastic lymphoma; MRD = minimal residual disease; WBC = white blood cell.
Note: MRD was not to be used to change risk group of patients with T-ALL. Patients with LL were to be treated as standard risk, high risk, or very high risk based on age, immunophenotype, CNS status, and cytogenetics, as specified in Table 7. Presenting WBC count and end of induction MRD were not to be used in LL for risk-group classification. Patients with t(9;22) were removed from protocol therapy at day 15 and not assigned a final risk group.
Source: DFCI 11-001 Clinical Study Report.15
There were key similarities and differences in eligibility criteria between the COG AALL07P4 and DFCI 11-001 trials. Notably, the DFCI 11-001 trial was inclusive of most patients with any ALL or LL (except Burkitt lymphoma) while the COG AALL07P4 trial focused on patients with high-risk ALL (B-precursor ALL). While both trials included children and adolescents, a higher age limit was used in the COG AALL07P4 study (30 years) compared with the DFCI 11-001 study (21 years). Due to the potential teratogenic effects of the study drugs, people who were pregnant were excluded from the COG AALL07P4 and DFCI 11-001 trials. The trials differed in populations that were ineligible. Patients with Down syndrome and testicular leukemia were excluded from the COG AALL07P4 study. Patients with B-cell (Burkitt lymphoma) ALL, an HIV-positive status, or a history of previous malignancy were excluded from the DFCI 11-001 study. With respect to laboratory measures, the COG AALL07P4 study stipulated WBC count as a criterion, whereas the DFCI 11-001 study used direct bilirubin for eligibility. In both trials, patients who had had prior chemotherapy were not eligible but patients with intrathecal cytarabine were allowed before registration in both trials. In the COG AALL07P4 trial, patients who received prior steroid therapy (duration of therapy and time frame not specified) were eligible whereas in the DFCI 11-001 trial, patients with short courses of steroids (nonchronic steroid therapy) were eligible for enrolment.
Interventions
The treatment schemas for the COG AALL07P4 study and the DFCI 11-001 study are depicted in Figure 1 and Figure 2, respectively, and the full treatment protocols for the COG ALL074 study and the DFCI 11-001 study are depicted in Figure 10 and Figure 11, respectively, in Appendix 1.
There were key differences in treatment protocols between the COG AALL07P4 trial and the DFCI 11-001 trial in backbone therapies, phases, schedules, and duration and frequency of asparaginase (calaspargase pegol or pegaspargase). Notably, the COG AALL07P4 study group protocol was adapted from Berlin-Frankfurt-Münster group protocols and contained rotating phases lasting 1 month to 2 months from induction through delayed intensification. In contrast, the DFCI protocol employed a prolonged intensification phase of 20 weeks to 30 weeks (CNS therapy phase and consolidation phase II) aimed at continuous asparagine depletion within 3-week cycles of MAC. In the COG AALL07P4 trial, patients received a single dose of the study drug at induction (and at extended induction for slow early responders), followed by 2 doses of the study drug at each subsequent phase (consolidation, interim maintenance phase I, delayed intensification, and interim maintenance phase II [as applicable], delayed intensification phase II [as applicable], and maintenance), for a total of up to 12 doses of asparaginase. In the DFCI 11-001 trial, however, patients received a single dose of a study drug (calaspargase pegol or pegaspargase) during remission induction, and then calaspargase pegol every 3 weeks or pegaspargase every 2 weeks during intensification, for a total of 11 doses of calaspargase pegol or 16 doses of pegaspargase.
COG AALL07P4 Study
In the COG AALL07P4 study, the treatment periods included remission induction (5 weeks), extended induction (2 weeks; for patients with M1 or M2 morphology with MRD ≥ 0.1% at day 29), consolidation (8 weeks), interim maintenance (up to two 8-week periods), delayed intensification (up to two 8-week periods), and maintenance therapy (12-week cycles for a total of 2 years from the start of interim maintenance phase I for females and 3 years from the start of interim maintenance phase I for males). Patients in the study received the randomly assigned asparaginase product (i.e., calaspargase pegol or pegaspargase) on the same schedule. Prior to randomization, patients were permitted to receive intrathecal cytarabine. After randomization, remission induction was initiated with vincristine (day 1, day 8, day 15, and day 22), daunorubicin (day 1, day 8, day 15, and day 22), prednisone or dexamethasone (day 1 through day 14 for patients aged younger than 10 years, or day 1 through day 28 for patients aged 10 years and older), asparaginase (day 4), and methotrexate (day 8, day 15, day 22, and day 29; patients with CNS3 were dosed on day 15 and day 22). Following induction, extended induction was employed based on the results of the day 29 bone marrow evaluation. A further treatment course following extended induction was based on early response using a cut-off of MRD of 0.1% for treatment stratification. Patients with M1 bone marrow status (< 5% blasts by morphologic bone marrow analysis) on day 8 or day 15, and day 29, and MRD of less than 0.1% were considered rapid early responders, with all other patients considered slow early responders. Following induction, patients classified as rapid early responders received 1 interim maintenance course followed by 1 delayed intensification phase. Patients classified as slow early responders and/or CNS3-positive received the same therapy plus a second interim maintenance and delayed intensification phase before maintenance therapy.
Figure 1: Treatment Schema for the COG AALL07P4 Study
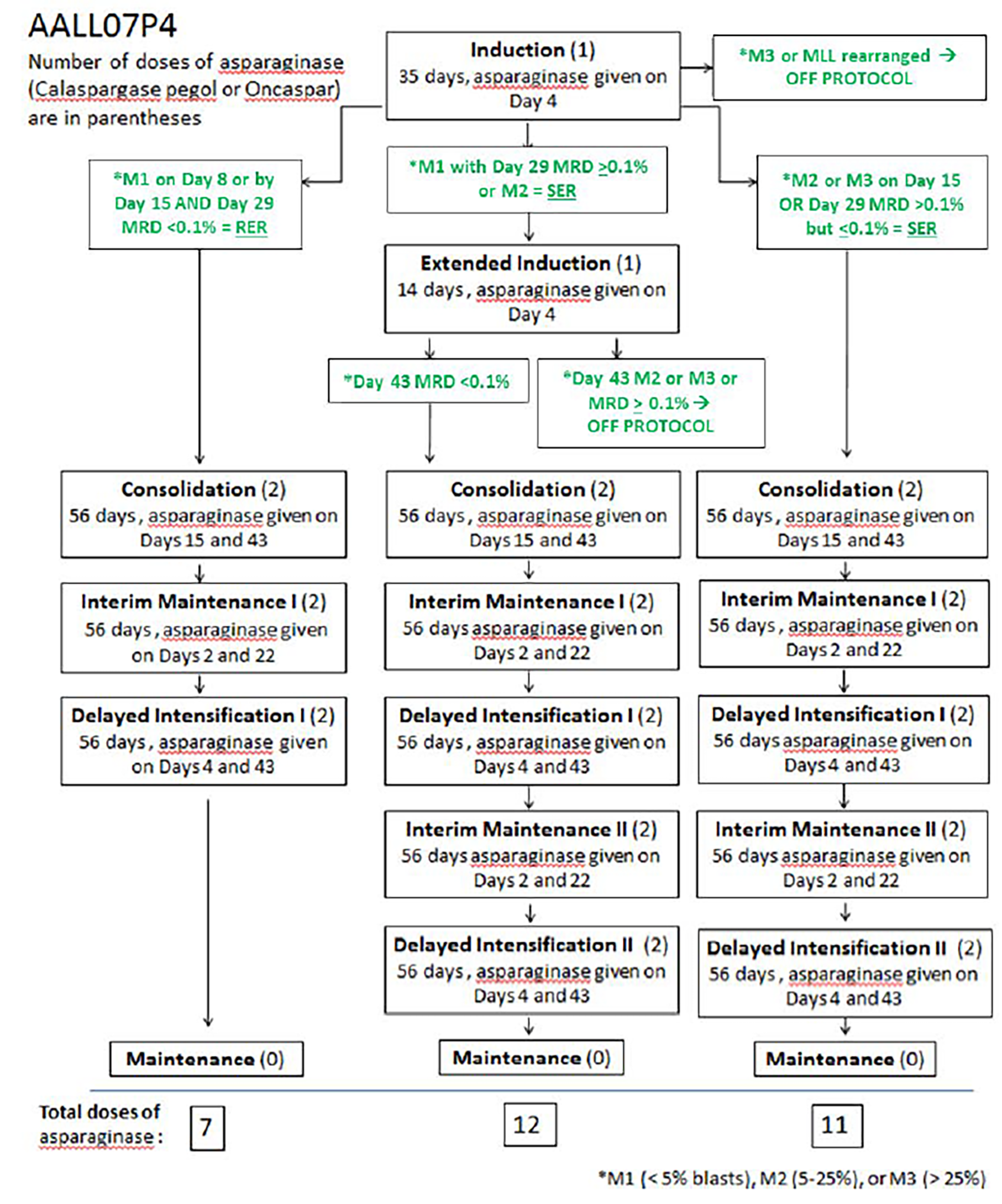
MRD = minimal residual disease; RER = rapid early responder; SER = slow early responder.
Source: COG AALL07P4 Clinical Study Report.13
![In the DFCI 11-001 study, treatment phases included a steroid prophase (3 days), induction (4 weeks), consolidation I (3 weeks), central nervous system therapy (3 weeks), consolidation II (27 weeks), and continuation (approximately 70 weeks, until 104 weeks [24 months] of continuous complete remission). Treatment was stratified based on patients’ risk status at randomization. Patients were considered as standard risk when the following criteria were met: age younger than 10 years, WBC count below 50,000/mm3, absence of central nervous system-leukemia at diagnosis, and with B-cell phenotype; all other patients were considered as high risk. Patients received a single dose of a study drug (calaspargase pegol or pegaspargase) during remission induction, and then calaspargase pegol every 3 weeks or pegaspargase every 2 weeks during intensification, for a total of 11 doses of calaspargase pegol or 16 doses of pegaspargase.](https://www.canjhealthtechnol.ca/index.php/cjht/article/download/PC0321r/version/867/1869/6950/PC0321CL-fig02.png)
Protocol Amendments in COG AALL07P4 Study
There were ||| protocol amendments (||||| ||||| |||||||| ||| ||||) in the COG AALL07P4 study. A key change (|||||||| ||||||||| ||) was for patients randomized to calaspargase pegol at 2,100 IU/m2 dose who were redirected to receive pegaspargase 2,500 IU/m2, as described in more detail as follows. All other amendments were minor and administrative in nature.
Patients in the COG AALL07P4 study were initially randomized to calaspargase pegol 2,500 IU/m2 or pegaspargase 2,500 IU/m2 as a 1-hour IV infusion on day 4 of induction, on day 4 of extended induction, on day 15 and day 43 of consolidation, on day 2 and day 22 of both interim maintenance periods, and on day 4 and day 43 of both delayed intensification periods. Patients enrolled before protocol amendment 2 (September 9, 2009) were randomized to 2,500 IU/m2 of calaspargase pegol or pegaspargase. A planned analysis of preliminary PK data from an initial cohort of 18 patients suggested that the 2,500 IU/m2 dose of calaspargase pegol was associated with prolonged exposure to asparaginase. Therefore, patients newly enrolled after amendment 2 were randomized in a 2:1 ratio to calaspargase pegol 2,100 IU/m2 or pegaspargase 2,500 IU/m2; patients who were already randomized to calaspargase pegol 2,500 IU/m2 continued on this dose for the remainder of the study. Regardless of the pegylated asparaginase formulation or dose, dosing schedules (i.e., intervals between doses) were the same in the calaspargase pegol and pegaspargase treatment groups. The number of total scheduled asparaginase doses in the first 7 months to 11 months of therapy was dependent on whether the patient was a rapid early responder receiving 7 doses, a slow early responder receiving 11 doses, or a slow early responder with extended induction receiving 12 doses.
After a planned interim analysis of rapid early responder and MRD data by the COG independent Data Safety Monitoring Committee in late 2010 || ||| |||||||| |||||||| ||| |||||||| || |||||||||||| ||||| ||||| |||||| || |||||||| || |||||||||||| ||||| ||||| |||||| ||| || |||||||| || |||||||||||| ||||| ||||||| it was determined that patients receiving 2,100 IU/m2 calaspargase pegol had a lower rate of rapid early responders and higher rate of positive MRD at the end of induction compared to patients in the other groups. As such, this met predefined protocol criteria for halting enrolment on December 22, 2010, with 166 patients enrolled (69 patients on calaspargase pegol 2,100 IU/m2, 42 patients on calaspargase pegol 2,500 IU/m2, and 55 patients on pegaspargase 2,500 IU/m2). Subsequently, patients in the calaspargase pegol 2,100 IU/m2 group were nonrandomly assigned to receive pegaspargase 2,500 IU/m2 for the remaining duration of therapy. Of note, at the time of this interim analysis, all participants who had been randomized to receive calaspargase pegol 2,500 IU/m2 had completed study drug treatment.
As the proposed Health Canada indication recommends a dose for calaspargase pegol of 2,500 IU/m2, this clinical report presents data for patients who were randomized to receive calaspargase pegol 2,500 IU/m2 and excludes data that refers to patients who were randomized to receive calaspargase pegol 2,100 IU/m2 and were subsequently switched to pegaspargase 2,500 IU/m2. Since data for the calaspargase pegol 2,100 IU/m2 group are excluded, any analyses related to this dose — including sensitivity analyses for patients who were switched from calaspargase pegol 2,100 IU/m2 to pegaspargase 2,500 IU/m2 — is also excluded from this clinical report.
DFCI 11-001 Study
In the DFCI 11-001 study, treatment periods included a steroid prophase (3 days), remission induction (29 days), consolidation phase I (3 weeks for standard-risk or high-risk patients, 9 weeks for very high-risk patients), CNS therapy phase (3 weeks), consolidation phase II (27 weeks), and continuation phase (until 24 months from the date when CR was attained). Remission induction was initiated following a diagnostic lumbar puncture and intrathecal cytarabine (plus IV methylprednisolone therapy unless a patient had already received steroid pretreatment before beginning study treatment) and included a 4-week multiagent regimen of vincristine (day 4, day 11, day 18, and day 25), prednisone (day 4 to day 32), doxorubicin (day 4 and day 5), and methotrexate (day 6). On day 7 of remission induction, patients in the DFCI 11-001 study received a single dose of the assigned asparaginase treatment as a 1-hour infusion. Beginning at week 7 of treatment, patients in the calaspargase pegol group received 1 dose every 3 weeks for a total of 10 doses and patients in the pegaspargase group received 1 dose every other week for a total of 15 doses. In both groups, treatment started with CNS therapy for standard-risk and high-risk patients or consolidation IC for very high-risk patients.
Remission assessment was performed on day 32. For patients with ALL, CR was defined as less than 1% marrow malignant lymphoblasts with peripheral blood count recovery and the absence of extramedullary disease. For patients with LL, CR required a 70% or greater reduction in the size of the largest nodes and/or masses, normalization of physical examination, and no cerebrospinal fluid (CSF) or marrow disease. Patients with persistent morphologic leukemia (≥ 1% leukemic blasts in marrow) or less than 70% reduction in the size of the largest nodes and/or masses at the end of induction were classified as induction failure and removed from the study protocol. Patients who did not meet these criteria (e.g., had 5% or greater blasts among patients with leukemia, or lower than 70% reduction in size of largest nodes or masses among patients with LL) were considered to have induction failure, and were removed from the protocol. Patients with greater than 5% lymphoblasts identified morphologically in bone marrow aspirate or biopsy as leukemic by flow cytometry, cytogenetics, fluorescence in situ hybridization, immunohistochemistry, or other tests, or who developed biopsy-proven extramedullary site of disease (e.g., CSF, testicle, lymph node) were considered to have met the criteria for relapse. For all others, treatment continued through the consolidation and continuation phases, with a total planned duration of treatment of 24 months from the CR date. In total, patients in the pegaspargase 2,500 IU/m2 group received a total of 16 doses, while patients who were part of the calaspargase pegol 2,500 IU/m2 group received a total of 11 doses.
Protocol Amendments in DFCI 11-001 Study
There were ||| protocol amendments (final dated February 24, 2016) in the DFCI 11-001 study. Key changes to the planned analyses (||||||||| ||||||| |||| ||| ||||) were the inclusion of an ITT analysis set for efficacy data, summarizing demographic and baseline characteristics using the FAS population, specifying the definition for TEAEs, revising the definitions of EFS and OS, adding and defining DFS, and clarifying subgroup analyses.
Prior Therapy
In the COG AALL07P4 study, patients were not to receive prior cytotoxic chemotherapy with the exception of steroids and intrathecal cytarabine. Patients who were receiving prior steroid therapy were monitored for the dose and duration (refer to the Prior Therapy for Determining Patient Eligibility and Risk Stratification section in Appendix 1).
In the DFCI 11-001 study, prior therapies of corticosteroids, intrathecal cytarabine, and emergent radiation were permitted. Short courses of corticosteroids (defined as up to 7 days of corticosteroids in the 4 weeks before study registration) were permitted before registration; patients with corticosteroid use of more than 7 days within the same time frame or more than 28 days over the preceding 6 months were not eligible for the study. Patients who received pretreatment with corticosteroids were not to receive steroid prophase. A single dose of intrathecal cytarabine at the time of a diagnostic lumbar puncture was allowed before registration; if this was done, a patient was not to receive day 1 intrathecal cytarabine thereafter.
Concomitant Therapy
In the COG AALL07P4 trial, calaspargase pegol and pegaspargase were considered study drugs, and the other chemotherapy drugs administered as part of the study protocol were considered concomitant therapy. These medications included cytarabine, vincristine, daunorubicin, methotrexate, steroids (prednisone and dexamethasone), cyclophosphamide, mercaptopurine, thioguanine, and doxorubicin. Administration of the protocol-specified chemotherapy was documented in the patient’s medical records and recorded in the case report form. No other concomitant medications were recorded in the case report form. Drugs with known potential interactions with antileukemic drugs were avoided. Anticonvulsants (e.g., phenytoin, phenobarbital, carbamazepine) were to be avoided due to induction of drug-metabolizing enzymes and being associated with decreased EFS. The use of azole antifungals may have required dose reductions of vincristine, anthracyclines, etoposide, and steroids due to induction of drug-metabolizing enzymes. Macrolide antibiotics were to be avoided due to the induction of drug-metabolizing enzymes. Rifampin was not to be used.
In the DFCI 11-001 trial, supportive care at diagnosis and during treatment (e.g., blood transfusions, growth factors, the use of corticosteroids other than as antileukemic drugs, folinic acid as methotrexate rescue, management of febrile neutropenia) was permitted. Antibiotic prophylaxis was permitted during the steroid prophase and remission induction for the management of fever.
Removal of Patients From Therapy or Assessment
In the COG AALL07P4 study, patients who discontinued protocol therapy were to be followed until they met the criteria for being off study (i.e., death, lost to follow-up, enrolment in another COG study with tumour therapeutic intent, withdrawal of consent, or the 10th anniversary of study entry). Patients were removed from protocol therapy if any of the following criteria were met: M3 bone marrow status on day 29, the identification of very high-risk ALL features, patients requiring extended induction (i.e., patients with M2 or M3 bone marrow status or who had MRD of 1% or greater following extended induction), recurrent leukemia following CR, the refusal of further protocol therapy by the patient (or parent or guardian), the completion of planned therapy, physician determination in a patient’s best interest, the development of a second malignancy, patient off-treatment for an unrelated disease, disease progression before active treatment, alternative therapy, the patient becoming pregnant, or the patient not being evaluable for postinduction therapy. Patients who withdrew consent for any further data submission, enrolled in another COG study with tumour therapeutic intent, were lost to follow-up, or died were removed from the study.
In the DFCI 11-001 study, patients who were removed from protocol therapy but continued to be followed up included patients who had failed to attain remission at the end of the remission induction phase, patients with Ph-positive (BCR/ABL) ALL, patients with disease recurrence, patients with the development of a second malignancy, patients who withdrew from the study, and patients with general or specific changes in their condition or compliance with protocol therapy, rendering the patient unacceptable for further treatment according to the opinion of the treating investigator. Patients who withdrew consent, were lost to follow-up, or died were removed from the study.
Outcomes
Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected the end points considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points in Table 8 were assessed using the GRADE tool. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Overall Survival
In the COG AALL07P4 trial, OS was defined as the time from randomization to death. OS by responder status (rapid early responders, slow early responders, extended induction at day 43) were conducted for the FAS population.
In the DFCI 11-001 trial, OS was defined as the time from randomization to death.
DFS From CR
In the COG AALL07P4 study, DFS was defined as the time from the attainment of CR to the documented relapse, diagnosis of a second malignant neoplasm, or death for patients who attain CR.
In the DFCI 11-001 study, DFS was defined as the time from the attainment of CR to the documented relapse, diagnosis of a second malignant neoplasm, or death for patients who attain CR.
Event-Free Survival
In the COG AALL07P4 trial, EFS was defined as the time from randomization to date of the documented relapse, induction failure (induction day 29 or day 43), the diagnosis of a second malignant neoplasm, or death from any cause, whichever came first. EFS was assessed during augmented postinduction intensification therapy among patients with high-risk ALL.
In the DFCI 11-001 trial, EFS was defined as the time from randomization to the date of the documented relapse, including induction failure, diagnosis of a second malignant neoplasm, or death from any cause, whichever comes first.
Complete Remission
Patients aged 1 year to 18 years with ALL can expect CR rates in about 98% of cases at 5 years postdiagnosis. For patients with standard-risk average disease, the 6-year continuous CR for standard 4-week consolidation versus 8-week intensified augmented Berlin-Frankfurt-Münster consolidation was 87.8% (standard deviation [SD] = 1.3%) versus 89.1% (SD = 1.2%) (P = 0.52).45 The clinical experts consulted by CADTH indicated that a clinically meaningful difference in CR would be a 5% difference between groups without overlapping CIs, with patients matched between groups, and where all confounders and molecular subtypes have been accounted for.
In the COG AALL07P4 trial, CR was defined as attaining M1 bone marrow (< 5% lymphoblasts) on day 29 of induction.
In the DFCI 11-001 trial, among patients with leukemia, CR was defined as an interpretable bone marrow with fewer than 1% lymphoblasts and peripheral blood without lymphoblasts, and an absolute phagocyte count of at least 1,000/mm3 and no evidence of extramedullary leukemia (no blasts in spinal fluid, at least a 70% reduction in the size of masses noted on imaging at diagnosis as defined for patients with LL) on day 32. Among patients with LL, CR was defined as having at least a 70% reduction in the size of the largest nodes or masses at diagnosis on day 32, where reduction could be measured as a decrease in a sum of the products of the 2 greatest diameters of the largest dominant nodes or nodal masses.
Minimal Residual Disease
MRD is considered to be 1 of the most significant prognostic factors in ALL, independent of patient age, B-cell or T-cell origin, or genetic subtype.46 An MRD level of 0.1% has been accepted by the FDA to identify patients with ALL with a high risk of relapse.46
MRD assessed by the COG AALL07P4 trial using flow cytometry at the end of 4 weeks of induction therapy among patients with newly diagnosed ALL has been considered to be strongly associated with outcome in children and AYAs with B-precursor ALL.47,48 In the COG AALL07P4 trial, MRD was measured in bone marrow biopsy or aspirate at day 29 of induction using a 6-colour multiparameter flow cytometry method. The COG AALL07P4 trial assigned patients with high levels of MRD to receive more intensive treatment. Flow cytometric detection of leukemia-associated immunophenotypes and PCR amplification of antigen-receptor genes appear to be the most reliable methods for detecting MRD in patients with ALL.49 MRD-positive was defined as 0.1% or greater and MRD-negative as less than 0.1% detectable leukemia cells.
In the DFCI 11-001 trial, MRD was measured using a PCR technique targeting lymphoblast-specific immunoglobulin gene rearrangements.
In the DFCI 11-001 trial, patients with high end-of-induction MRD (≥ 0.001) were assigned to the very high-risk final risk group (refer to Figure 7). MRD at the end of induction status was 1 of the efficacy outcomes in the DFCI 11-001 trial, measured as high end-of-induction MRD (defined as ≥ 0.001 by PCR assay) and low end-of-induction MRD (< 0.001). The end of induction MRD of 0.01 or greater was also reported for both groups.
Serum Asparaginase Activity
The clinical effectiveness of asparaginase is considered to be based upon the adequate depletion of asparagine.34 The cornerstone of PK assessment in the clinical pharmacology program of calaspargase pegol is the measurement of plasma asparagine.50 The FDA’s multidisciplinary review report of calaspargase pegol in combination with chemotherapy for ALL reported that asparaginase depletion is considered a valid surrogate end point for clinical efficacy of asparaginase treatment.51 However, it is not feasible to accurately measure plasma asparagine outside the context of a clinical trial due to rapid ex vivo breakdown.9 SAA is more easily measured using surrogate assays and is well correlated with asparagine depletion and clinical effectiveness.34 The current consensus guidelines have advocated for a threshold of 0.1 IU/mL. This cut-off of 0.1 IU/mL or greater measured either after a single dose or at steady state has been used in many research and treatment protocols to determine therapeutic asparaginase activity and levels above this cut-off have been shown to result in complete asparagine depletion.34,51 The FDA has approved previous asparaginase products in combination with chemotherapy, including calaspargase pegol for ALL, on the basis of accepting evidence of asparaginase activity as a surrogate for clinical efficacy in these asparaginase treatments.50,51
In the DFCI 11-001 study, SAA was measured predose and after a single dose of calaspargase pegol during remission induction at 5 minutes to 10 minutes, and then once on day 7, day 11, day 18, day 25, and day 32. The proportion of patients with an SAA level of 0.10 IU/mL or greater (and the proportion of patients with an SAA level of 0.40 IU/mL or greater) was assessed for each treatment group.
Harms
In the COG AALL07P4 study, safety information included AEs and was recorded throughout the course of the study. AEs were any event that resulted in persistent or significant disabilities or incapacities, congenital anomalies, medically important events or experiences, or birth defects following protocol treatment. SAEs were not specifically defined in the study protocol; rather, categories of events relevant to the safety profile assessment were considered serious: all grade 3 and higher hyperbilirubinemia, systemic infections and pneumonias, osteonecrosis, all grade 2 and higher pancreatitis or convulsions, and all anaphylactic reactions. The grading of AEs was categorized according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE), version 3.0, and per amendment 5, switched to version 4.0 to retrospectively apply to all previously reported AE data, including the following adjudicated as serious: CTCAE grade 4 life-threatening consequences, CTCAE grade 3 medical intervention or hospitalization indicated, CTCAE grade 3 with laboratory findings and bleeding, CTCAE grade 3 limiting self-care, and transfusion, radiologic, endoscopic, or elective operative intervention indicated. AEs were coded into system organ classes and preferred terms using Medical Dictionary for Regulatory Activities, version 19.0.
In the DFCI 11-001 study, toxicities included AEs, SAEs, and AESIs reported by patients, discovered during questioning, directly observed, or detected by physical examination, laboratory test, or other means. AEs were any undesirable sign, symptom, or medical condition or experience that developed or worsened in severity after a patient started the first dose of study treatment or any procedure specified in the protocol (including abnormal laboratory values or diagnostic test results if they induced clinical signs or symptoms requiring treatment or further diagnostic tests), even if the event was not considered related to the study. SAEs were any AE, regardless of causality, that resulted in death, was life-threatening, required intensive inpatient medical interventions, resulted in persistent or significant disability or incapacity, was a congenital anomaly or birth defect, or was considered to be an important medical event based on medical judgment that could jeopardize the patient and require medical or surgical intervention. The descriptions and grading of AEs were categorized according to CTCAE version 4.0.
Table 8: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | COG AALL07P4 study | DFCI 11-001 study |
|---|---|---|---|
OS | From randomization to the last contact or death | Secondary efficacy end point | Secondary efficacy end point |
DFS | Time from attainment of CR to the documented relapse, diagnosis of a second malignant neoplasm, or death for patients who attained CR | Secondary efficacy end point | Secondary efficacy end point |
EFS | Time from randomization to the date of the documented relapse, induction failure, diagnosis of a second malignant neoplasm, or death from any cause, whichever occurs first | Secondary efficacy end point | Secondary efficacy end point |
CR rate | COG AALL07P4 study: Day 29 (i.e., 25 days after the induction dose of study drug) DFCI 11-001 study: Day 32 (i.e., 25 days after the induction dose of study drug) | Secondary efficacy end point | Secondary efficacy end point |
MRD | COG AALL07P4 study: Day 29 of induction and, if applicable, at the end of extended induction (study day 43) DFCI 11-001 study: Day 32 of induction | Secondary efficacy end point | Secondary efficacy end point |
SAA levels | DFCI 11-001 study: Predose and at 5 minutes to 10 minutes postdose on day 4, day 11, day 18, and day 25 | Not assessed | Primary end point |
HRQoLa | Not assessed | Not assessed | Not assessed |
TEAE | COG AALL07P4 study: AEs starting or worsening upon treatment of study medication to starting no later than 60 days after the last dose of study medication or Erwinia substitution. AEs were recorded during all treatment periods. DFCI 11-001 study: AEs starting or worsening upon treatment of study medication to starting no later than 30 days after the last dose of study medication or Erwinia asparaginase substitution. AEs were monitored during induction and every 3 weeks for 30 consecutive weeks during postinduction (for calaspargase pegol) or every 2 weeks for 30 consecutive weeks during postinduction (for pegaspargase). | Secondary end point | Primary end point |
AE = adverse event; CR = complete remission; COG = Children’s Oncology Group; CSF = cerebrospinal fluid; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; HRQoL = health-related quality of life; MRD = minimal residual disease; OS = overall survival; SAA = serum asparaginase activity; TEAE = treatment-emergent adverse event.
Statistical testing for all end points were not adjusted for multiple comparisons (e.g., hierarchical testing).
aOutcome identified of importance according to patient group input.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE included the following.
Survival outcomes were identified by the clinician group input and specified by the clinical experts consulted by CADTH to include OS, DFS from CR, and EFS. EFS and OS were also key inputs in the sponsor’s pharmacoeconomic model.
CR at the end of induction was identified as important by the clinical experts consulted by CADTH and by clinician group input (as response to treatment).
MRD at the end of induction was identified as important by the clinical experts consulted by CADTH and by clinician group input (as a measurement of progressive disease). Specifically, the clinical experts consulted by CADTH highlighted MRD levels of 0.1% or greater as well as 0.01 or greater as most clinically relevant; these were included in GRADE assessments. Results for high end-of-induction MRD (≥ 0.001) and low end-of-induction MRD (< 0.001) are included in Appendix 1 in Table 18.
SAA levels were measured during induction in the DFCI 11-001 trial to assess calaspargase pegol’s ability to maintain an SAA level of 0.1 UI/mL or greater 3 weeks to 4 weeks after a single dose and to assess its half-life duration compared to pegaspargase.44 According to the clinical experts consulted by CADTH, calaspargase pegol was developed with greater biological stability than pegaspargase and SAA levels during induction compared to pegaspargase could provide an indication of calaspargase pegol’s ability to maintain asparagine suppression in plasma, its half-life duration, and its ability to be administered with less dosing frequency than pegaspargase (i.e., a schedule of every 3 weeks versus every 2 weeks). Therefore, SAA levels during induction were identified as a relevant outcome by the clinical experts consulted by CADTH. However, the clinical experts consulted by CADTH noted that SAA assessment during induction as performed in the DFCI 11-001 trial is not routinely performed in clinical practice, where SAA assessments are done according to a different schedule for the purpose of identifying and managing asparaginase hypersensitivity and silent inactivation.34
The assessments of SAA every 3 weeks for 30 consecutive weeks during postinduction therapy in the DFCI 11-001 trial was done to further determine how best to dose calaspargase pegol during postinduction therapy.44 SAA assessments during postinduction in the DFCI 11-001 trial were not reported to be 1 of the most important outcomes to routinely guide treatment selection in clinical practice and was not included in GRADE assessments. The SAA assessment during postinduction served to assess the appropriate dosage of calaspargase pegol but may have provided less relevant information about the comparative efficacy between drugs in clinical practice. In the COG AALL07P4 trial, plasma asparaginase activity (PAA) levels after an induction dose (measured on day 4, day 15, day 22, day 29, and day 43 of induction; this included the proportion of patients with a PAA level of 0.10 IU/mL or greater [and the proportion of patients with a PAA level of 0.40 IU/mL or greater] for each study group) were measured rather than SAA levels. Since PAA levels were specified by consensus guidelines to be difficult to reliably ascertain, and reported by clinical experts consulted by CADTH to be unfeasible to measure in clinical practice and not used to make treatment selection in clinical practice, data for this outcome were not included in the GRADE assessment.34 Results for PAA after induction dose are displayed in ||||| || in Appendix 1.
HRQoL due to treatment and HRQoL due to drug shortage were identified as important by patient input. No data on HRQoL were collected in either the COG AALL07P4 trial or the DFCI 11-001 trial.
Harms of treatment were identified as important by patient input (as side effects of treatment), clinician group input (as significant treatment intolerance), and the clinical experts consulted by CADTH (including hypersensitivity reactions, anaphylactic reactions, silent inactivation, and WDAEs). Hypersensitivity reactions, anaphylactic reactions, silent inactivation, and WDAEs were highlighted by the clinical experts consulted by CADTH as specifically relevant AEs for the management of patients in clinical practice.
Statistical Analysis
Sample Size and Power Calculation
COG AALL07P4 Study
The aims of the COG AALL07P4 study were to assess the PK (including maximum asparaginase activity exposure [Cmax], area under the curve from time 0 to 25 days[AUC0–25d], and area under the curve from time 0 to infinity [AUC0–inf]) and PD properties (asparagine depletion from plasma and CSF) of calaspargase pegol and pegaspargase during induction and consolidation.13 Cmax and AUC0–25d were assessed by the ratio of geometric means and corresponding 2-sided 90% CIs. If a 2-sided 90% CI was totally contained within the conventional bioequivalence limits ranging from 0.8 to 1.25, PK comparability in treatment exposure between calaspargase pegol and pegaspargase could be claimed. The 2-step approach as outlined in the protocol was not required as a lower limit greater than 80% of a 2-sided 90% CI for the ratio was equal to rejecting the null hypothesis of an exposure of 80% or lower compared to pegaspargase 2,500 IU/m2 at the 5% level of statistical significance (Schuirman [1987]52). The original study design was based on a planned preliminary PK evaluation of 18 patients (11 patients in the calaspargase pegol 2,500 IU/m2 group and 7 patients in the pegaspargase group) as well as MRD and other safety end points of pegaspargase 2,500 IU/m2 with a contemporary benchmark of calaspargase pegol 2,500 IU/m2. A sample size of 90 patients was based on the precision analysis of the estimation of the true induction day 29 MRD rate for patients receiving the calaspargase pegol regimen. Based on a blinded review of MRD data from previous studies (the COG P9900 study and the COG AALL0232 study), baseline MRD negativity (defined as < 0.01% detectable disease in bone marrow on day 29 of induction) was estimated to be 70%. Assuming similar treatment effect between the calaspargase pegol 2,500 IU/m2 regimen and pegaspargase 2,500 IU/m2 regimen, 60 evaluable patients in the calaspargase pegol group would result in approximately 12% of patients attaining a negative MRD with 95% CI.13 The COG AALL07P4 study was not adequately powered to evaluate clinical efficacy end points (i.e., day 29 end-of-induction MRD status, CR, EFS, DFS from the attainment of CR, and OS).51
In total, 123 patients were randomized in a 2:1 ratio between calaspargase pegol 2,500 IU/m2 and pegaspargase 2,500 IU/m2. Eighteen (15%) patients were expected to be removed from therapy during induction for reasons such as having very high-risk features or being unevaluable for postinduction therapy. Therefore, a total of 105 patients was available to meet study end points, with 90% of patients evaluable for MRD, and 75% to 80% of patients considered rapid early responders, and 2% of patients requiring extended induction therapy. Thus, it was expected that, with 105 patients, there would be at least 90 patients evaluable for MRD who had not received extended induction therapy.
DFCI 11-001 Study
Sample size calculations in the DFCI 11-001 trial were based on NSAA levels for having 112 evaluable patients in each group from 240 enrolled patients. Assuming 99% of patients in the pegaspargase group had at least 1 NSAA level of 0.10 IU/mL or greater (considered therapeutic) during the postinduction asparaginase consolidation, it was expected to detect a 10% reduction in the proportion of patients who had at least 1 NSAA level of 0.10 IU/mL or greater with 89% power during the postinduction asparaginase consolidation, using a 1-sided Fisher exact test at the 0.025 significance level. The rationale for a 10% reduction in the proportion of patients with 1 NSAA level of 0.10 IU/mL or greater was not reported. A total of 224 evaluable patients (112 in each group) would provide 96% power to detect a mean difference of 0.15 IU/mL in the asparaginase activity level at the week-7 time point (estimated to be steady state) based on data from DFCI protocol 05 to 001 (Place et al. [2015]53), using a 2-sided t test and assuming an SD of 0.30. The rationale for a mean difference of 0.15 IU/mL in asparaginase activity level was not reported.
Statistical Testing
COG AALL07P4 Study
The COG AALL07P4 trial was not designed to assess efficacy (i.e., MRD at end of induction day 29, CR at end of induction day 29, EFS, DFS from the attainment of CR, and OS) of calaspargase pegol in terms of superiority or noninferiority to pegaspargase. In the COG AALL07P4 study, the number of patients with negative MRD (< 0.1% detectable disease) with 95% CI were reported for each treatment group. Adjusted analyses were conducted using logistic regression for covariates of age, baseline WBC count, baseline body mass index (BMI), and asparaginase activity (by time point). The number of patients with CR with 95% CI were reported for each treatment group after patients completed induction therapy. Patients were to be followed for a minimum of 3 years for survival estimates (EFS, DFS, and OS). The product-limited Kaplan-Meier method was used to estimate survival probabilities by treatment with curves presented for calaspargase pegol and pegaspargase groups, and HRs were estimated using the Cox model with 95% CIs. Adjusted analyses of survival estimates were conducted using the Cox model for covariates of age, sex, baseline WBC count, and baseline BMI. For OS, the duration of follow-up since randomization to last contact or death was tabulated for each treatment group. The number and proportion of patients experiencing TEAEs were summarized by treatment group.
No adjustment for multiple testing or control of type I error was conducted for any end point, and there was no imputation for missing data.
The Clinical Study Report with a data cut-off date of December 31, 2015, was the most recent one available for the COG AALL07P4 trial and was the data source used for all efficacy and safety end points in this clinical report.
DFCI 11-001 Study
No prespecified criteria were defined to determine comparative efficacy between trial groups. The trial was not designed to assess the efficacy of calaspargase pegol in terms of superiority or noninferiority to pegaspargase. In the DFCI 11-001 study, the number and proportion of patients with high end-of-induction MRD (≥ 0.001 by PCR assay) and low end-of-induction MRD (< 0.001) with 2-sided 95% exact binomial CIs were reported for each treatment group. The number and proportion of patients with MRD (≥ 0.01 by PCR assay) were also reported for each treatment group. The number and proportion of patients attaining CR were summarized by treatment. Patients were to be followed for 5-year survival estimates of EFS, DFS, and OS. The Kaplan-Meier method was used to estimate the survival probabilities by treatment. One-year estimates were provided with 95% CIs estimated using the Greenwood formula. Additional time point estimates were to be considered when data are more mature. The number and proportion of patients with an SAA level of 0.10 IU/mL or greater were summarized by treatment and visit with 2-sided Clopper-Pearson CIs. Linear mixed effects and generalized estimating equation models were used to compare SAA levels longitudinally between treatment groups, and estimates were reported using geometric means and ORs. Models were fitted with and without covariates (included individually as fixed effects) for age, sex, initial risk classification, disease type (ALL or LL), and baseline WBC count. The number and proportion of patients experiencing TEAEs were reported, including AESIs with 80% CIs.
No adjustment for multiple testing was conducted for any end point, and there was no imputation for missing data.
The Clinical Study Report for the DFCI 11-001 study with a data cut-off date of October 5, 2016, for PK, PD, and efficacy analyses was the primary data source. A Day 120 Report provided additional data with a data cut-off date of June 12, 2017, for survival data and of February 1, 2017, for safety data. The Vrooman et al. (2021)42 publication reported a median 5-year follow-up for EFS and OS but did not report a cut-off date for these data. The Clinical Study Report (data cut-off date of October 5, 2016) was the data source for all efficacy (MRD, CR, 1-year EFS rate, 1-year DFS, 1-year OS, and SAA levels) and safety end points in this clinical report. Data from the Day 120 Report provided updated survival data (2-year rates for EFS, DFS, and OS with a CCOD of June 12, 2017) and safety data (CCOD of February 1, 2017).
Subgroup Analyses
COG AALL07P4 Study
In the COG AALL07P4 study, subgroup analyses were prespecified for MRD, CR, EFS, OS, DFS, and TEAEs. Subgroup analyses of interest for this clinical report focused on primary end points; therefore, subgroup analyses for these secondary end points were not included.
DFCI 11-001 Study
In the DFCI 11-001 study, subgroup analyses were prespecified for MRD, CR, EFS, OS, DFS, and TEAEs. Subgroup analyses of interest for this clinical report focused on primary end points of SAA levels and TEAEs. Subgroup analyses compared SAA levels longitudinally between treatment groups for age (< 10 years, ≥ 10 years), sex, initial risk-group classification (standard risk, high risk), disease type (ALL, precursor B-cell ALL), and baseline WBC count (< 50,000 × 109/L, ≥ 50,000 × 109/L). Subgroup analysis of TEAEs was conducted for initial risk classification. No tests of interaction were conducted for subgroup analyses.
Sensitivity Analyses
COG AALL07P4 Study
In the COG AALL07P4 study, a sensitivity analysis for MRD was conducted for patients with low end-of-induction MRD versus high end-of-induction MRD among the ITT and per-protocol (PP) populations. For EFS, OS, and DFS, sensitivity analyses were conducted for induction failure as an event at time 0, including data after Erwinia substitution, and including data after other new anticancer therapies.
DFCI 11-001 Study
In the DFCI 11-001 study, a sensitivity analysis for MRD was conducted for patients with high end-of-induction MRD (≥ 0.001 by PCR assay) among the PP population. For EFS and DFS, sensitivity analyses were conducted for the PP population, initial diagnosis (ALL, precursor B-cell ALL), induction failure as an event at time 0, including data after Erwinia substitution, and including data after other new anticancer therapies.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
COG AALL07P4 study | ||||
OS | OS estimates using product-limit method with KM curves for each treatment group; HR estimated with corresponding 95% CI using Cox model Correlation of end of induction MRD and OS using Cox model, controlling for treatment effect The duration of follow-up from randomization to the last contact or death was tabulated by treatment. | Not adjusted for multiple testing | If the patient was alive at the date of the analysis cut-off or lost to follow-up, then OS was to be censored at the last contact date before data cut-off date. Patients who died before the end of induction were not included in the analysis. |
|
DFS | DFS estimates using product-limit method with KM curves for each treatment group; HR estimated with corresponding 95% CI using Cox model after a minimum of 3 years of follow-up The correlation between end of induction therapy MRD and DFS was explored. | Not adjusted for multiple testing | Patients were censored at the last date of follow-up visit before the start of anticancer therapy. |
|
EFS | EFS estimates using product-limit method with KM curves for each treatment group; HR estimated with corresponding 95% CI using Cox model after a minimum of 3 years of follow-up The correlation between end of induction therapy MRD and EFS was explored. | Not adjusted for multiple testing | Patients were censored at the last date of follow-up visit in the absence of a documented EFS event, or before the start of anticancer therapy (including Erwinia substitution). |
|
CR | Observed CR rate at the end of induction with 95% CI per treatment group after all patients completed their induction therapy | Not adjusted for multiple testing | No imputation for missing data | None |
MRD | Observed negative MRD (< 0.1%) rate on day 29 of induction with corresponding 95% CI per treatment group | Not adjusted for multiple testing | If day 29 MRD was not evaluable, the results of day 8 and/or day 15 marrow morphology were used to determine the response definition as RER or SER. No imputation for missing data | Low end-of-induction MRD vs. high end-of-induction MRD (ITT and PP populations) |
Harms | The number and proportion of patients experiencing TEAEs summarized by treatment group | Not adjusted for multiple testing | No imputation for missing data | None |
DFCI 11-001 study | ||||
OS | KM method with 1-year estimates using the Greenwood formula with 95% CIs; additional OS time point estimates were to be reported when data were more mature The duration of follow-up from randomization to the last contact or death was tabulated by treatment. | Not adjusted for multiple testing | If the participant was alive at the date of the analysis cut-off or lost to follow-up, then OS was to be censored at the last contact date before the data cut-off date. | None |
DFS | KM method was used to estimate the survival probabilities. 1-year estimates were to be provided along with the 95% CIs estimated using the Greenwood formula; additional time points were to be considered when data were more mature. | Not adjusted for multiple testing | In the absence of a documented DFS event, DFS was censored on the date of last follow-up; if a patient started new anticancer therapy, including Erwinia asparaginase substitution, without events, the DFS was to be censored at the last follow-up before the start of anticancer therapy. Patients with Ph-positive were censored at the date of the last dose of protocol therapy because they started a new treatment regimen, including imatinib, after discontinuing study treatment. |
|
EFS | KM method with 1-year estimates using the Greenwood formula with 95% CIs; additional OS time point estimates were to be reported when data more mature | Not adjusted for multiple testing | In the absence of a documented EFS event, EFS was censored on the date of the last follow-up; if a patient started new anticancer therapy, including Erwinia asparaginase substitution, without events, the EFS was to be censored at the last follow-up before the start of anticancer therapy. Patients with Ph-positive were censored at the date of the last dose of protocol therapy because they started a new treatment regimen, including imatinib, after discontinuing study treatment. |
|
CR | The number and proportion of patients attaining CR summarized by treatment | Not adjusted for multiple testing | No imputation for missing data | None |
MRD | Frequency and proportion of patients with a 2-sided 95% exact binomial CI of high end-of-induction MRD (≥ 0.001) and low end-of-induction MRD (< 0.001) summarized by treatment | Not adjusted for multiple testing | Patients with indeterminate MRD values were excluded from analysis. No imputation for missing data | Proportion of patients in the PP population with high end-induction MRD (≥ 0.001 by PCR assay); patients with baseline high MRD were not counted as high end-of-induction MRD |
SAA levels | Number and proportion of patients (along with 2-sided 95% Clopper-Pearson CIs) summarized by treatment and visit SAA levels also tabulated with descriptive statistics by treatment and induction day | Not adjusted for multiple testing Linear mixed effects and GEE models were used to compare SAA levels longitudinally between treatment groups; models were fitted with and without the following covariates: age, sex, initial risk-group classification, disease type, and baseline WBC count. Covariates were included individually in the models as fixed effects. | Analyses were based on complete cases (no imputation for missing data). | None |
Harms | The number and proportion of patients experiencing TEAEs summarized by treatment group. The number and proportion of patients experiencing asparaginase-related toxicity were reported with 80% CIs for each group, and compared to historical controls (the pegaspargase group on the DFCI protocol 05 to 001). | Not adjusted for multiple testing Subgroup analyses of TEAEs for initial risk classification | No imputation for missing data | None |
CI = confidence interval; COG = Children’s Oncology Group; CR = complete remission; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; GEE = generalized estimating equation; HR = hazard ratio; ITT = intention-to-treat; KM = Kaplan-Meier; MRD = minimal residual disease; OS = overall survival; Ph-positive = Philadelphia chromosome positive; PP = per protocol; RER = rapid early responder; SAA = serum asparaginase activity; SER = slow early responder; TEAE = treatment-emergent adverse event; vs. = versus; WBC = white blood cell.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Analysis Populations
Analysis sets for each study included in the systematic review are summarized in Table 10.
Table 10: Analysis Populations of COG AALL07P4 Study and DFCI 11-001 Study
Study | Population | Definition | Application |
|---|---|---|---|
COG AALL07P4 study | ITT | All randomized patients classified and analyzed according to the treatment they were randomized to receive | Efficacy analyses (MRD, CR, EFS, DFS, OS) |
FAS | All randomized patients who received at least 1 administration of study medication calaspargase pegol or pegaspargase. Patients were analyzed according to the intended or planned treatment group. | Demographics, baseline characteristics, disposition, and efficacy analyses (MRD, CR, EFS, DFS, and OS) | |
PPAS | All patients in the FAS population who did not have any major protocol violations | Efficacy analyses (MRD, CR, EFS, DFS, and OS) | |
SAS | Randomized patients who had received any dose of calaspargase pegol or pegaspargase. Patients were classified based on the actual treatment received. Treatment received was defined as the treatment assigned (dose-level and schedule planned) if it was received at least once, or as the first treatment received when starting therapy with study medication if the assigned treatment was never received. Each patient was classified into and analyzed consistently within 1 (and only 1) treatment group. | Safety analyses | |
PKAS | All patients who had received 94% to 106% of the nominal dose of calaspargase pegol or pegaspargase on induction day 4 and were evaluable for PK. Patients were PK-evaluable if they had had at least 3 quantifiable measurements after starting infusion and before the asparaginase activity level decreased to below the limit of quantification. If a patient randomized to calaspargase pegol 2,100 IU/m2 later switched to pegaspargase per protocol amendment 4A, the data after the switch were to be excluded from the PKAS. Patients who were inadvertently administered calaspargase pegol 2,500 IU/m2 instead of pegaspargase (randomized treatment) were included in the PKAS based on actual treatment received. | PK analyses | |
DFCI 11-001 study | ITT | All randomized patients classified and analyzed according to the treatment they were randomized to receive | Efficacy analyses (MRD, CR, EFS, DFS, OS) |
ITT ALL | All randomized patients with ALL | Efficacy analyses (MRD, CR, EFS, DFS, OS) | |
FAS | All randomized patients who received at least 1 administration of study medication calaspargase pegol or pegaspargase. Patients were analyzed according to the intended or planned treatment group. | Demographics, baseline characteristics, disposition, and efficacy analyses (MRD, CR, EFS, DFS, OS) | |
FAS ALL | All patients in the FAS with ALL | Demographics, baseline characteristics, disposition, and efficacy analyses (MRD, CR, EFS, DFS, OS) | |
PPAS | All patients in the FAS population who did not have major protocol deviations or violations; all decisions to exclude patients from the PPAS were to be made before the database lock for the final analysis | Sensitivity analysis of the FAS population for selected efficacy end points (MRD, CR, EFS, DFS, OS) | |
SAS | All patients who had received at least 1 administration of study medication. Patients were to be classified according to treatment received, where the treatment received was defined as the treatment assigned if it was received at least once, or as the first treatment received when starting therapy with study medication if the assigned treatment was never received. Each patient was to be classified into and analyzed consistently within 1 (and only 1) treatment group. | Safety analyses | |
PKAS | All patients who had received at least 1 dose of calaspargase pegol or pegaspargase on induction day 7 and were evaluable for PK. Patients were included in the PKAS if they had had at least 1 postinfusion measurement above the lower limit of quantification.a | PK analyses |
ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; CR = complete remission; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; FAS = full analysis set; ITT = intention-to-treat; MRD = minimal residual disease; NSAA = nadir serum asparaginase activity; OS = overall survival; PK = pharmacokinetic; PKAS = pharmacokinetic analysis set; PPAS = per-protocol analysis set; SAS = safety analysis set.
aPatients were considered to have valid PK assessments, and evaluable for PKAS, if their NSAA levels were below the limit of quantification set to half of the lower limit of quantification (0.025 IU/mL) for statistical analyses.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Results
Patient Disposition
Patient disposition in the COG AALL07P4 trial (CCOD of December 31, 2015) and the DFCI 11-001 trial (CCOD of October 5, 2016) is summarized in Table 11.
In the COG AALL07P4 trial, || || || ||||||| patients in the calaspargase pegol ||| || || || ||||||| patients in the pegaspargase group discontinued the study drug, all due to an AE. Similar proportions of patients between treatment groups discontinued protocol therapy (i.e., randomized treatment). Twenty-nine (70.7%) patients in the calaspargase pegol group and 37 (68.5%) patients in the pegaspargase group were continuing on study protocol at the time of data cut-off.
In the DFCI 11-001 trial, 49 of 118 (41.5%) patients in the calaspargase pegol group and 36 of 199 (30.3%) patients in the pegaspargase group discontinued the study drug; of these, 17.8% and 13.4% of patients discontinued due to an AE in the calaspargase pegol group and the pegaspargase group, respectively. A total of 69 (58.5%) patients in the calaspargase pegol group and 83 (69.7%) patients in the pegaspargase group completed study drug treatment.
A higher proportion of patients discontinued the study drug in the COG AALL07P4 study than in the DFCI 11-001 study. More patients in the COG AALL07P4 study (44.2%) compared with the DFCI 11-001 study (12.2%) discontinued protocol therapy. |||||| |||||||||| of patients in the DFCI 11-001 study completed protocol therapy compared with approximately 56% of patients in the COG AALL07P4 study. Finally, a comparatively higher proportion of patients discontinued the study in the COG AALL07P4 trial (24.2%) compared with the DFCI 11-001 trial (|||||| ).
In the DFCI 11-001 study, some patients’ risk classification shifted from initial risk at time of diagnosis to final risk at the end of induction using the final risk classification tool (refer to Table 7 in this report). Among || || ||| ||||||| patients in the calaspargase group who were designated as standard risk at the time of diagnosis, the number of patients who shifted to high risk, very high risk, Ph-positive, and unknown status were || | ||||||| | ||||||| and | ||||||, respectively. Among || ||||||| patients in the calaspargase pegol group who were designated as high risk at the time of diagnosis, the number of patients who shifted to very high risk, Ph-positive, and unknown status were | ||||||| | ||||||| and | ||||||| Among || || ||| ||||||| patients in the pegaspargase group who were designated as standard risk at the time of diagnosis, the number of patients who shifted to high risk, very high risk, Ph-positive, and unknown status were | ||||||| ||||||| | ||||||| and | ||||||| respectively. Among || ||||||| patients in the pegaspargase group who were designated as high risk at the time of diagnosis, the number of patients who shifted to very high risk, Ph-positive, and unknown status were | |||||||| | ||||||| and |||, respectively.
Table 11: Summary of Patient Disposition From the Studies Included in the Systematic Review — FAS
Patient disposition | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 41) | Pegaspargase 2,500 IU/m2 (N = 54) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Screened, N | NA | NA | NA | NA |
Randomized, N | 166 | 239 | ||
Treated, N | 41a | 54a | 118 | 119 |
Discontinued study drug, N (%) | || |||||| | || |||||| | 49 (41.5) | 36 (30.3) |
During induction | NA | NA | 13 (11.0) | 7 (5.9) |
During PI | NA | NA | 36 (30.5) | 29 (24.4) |
AE experienced at time of study drug discontinuation | || |||||| | || |||||| | 21 (17.8) | 16 (13.4) |
Discontinued protocol therapy, N (%) | 19 (36.3) | 23 (32.6) | 16 (13.6) | 13 (10.9) |
Reason for discontinuing protocol therapy, n (%) | ||||
AEs | 7 (17.1) | 1 (1.9) | NA | NA |
Alternative therapy | 0 (0.0) | 1 (1.9) | NA | NA |
VHR ALL features | 3 (7.3) | 5 (9.3) | NA | NA |
M3 marrow on day 43 | 1 (2.4) | 2 (3.7) | NA | NA |
Recurrent leukemia | 0 (0.0) | 3 (5.6) | NA | NA |
Death | 2 (4.9) | 1 (1.9) | NA | NA |
Refused further therapy | 3 (7.3) | 5 (9.3) | NA | NA |
Entered another COG study | 0 (0.0) | 0 (0.0) | NA | NA |
Nonevaluable for PI therapy | 0 (0.0) | 0 (0.0) | NA | NA |
Physician decision | 3 (7.3) | 5 (9.3) | NA | NA |
Induction failure | NA | NA | 4 (3.4) | 1 (0.8) |
Patients who were Ph-positive | NA | NA | 3 (2.5) | 4 (3.4) |
Relapse | NA | NA | 1 (0.9) | 4 (3.4) |
Induction death | NA | NA | 1 (0.9) | 0 (0.0) |
Remission death | NA | NA | 1 (0.9) | 0 (0.0) |
Withdrawal from treatment | NA | NA | 2 (1.7) | 0 (0.0) |
Other | NA | NA | 4 (3.4) | 4 (3.4) |
Completed protocol therapy, N (%) | 22 (53.7) | 31 (57.4) | || |||||| | || |||||| |
Protocol therapy ongoing, N (%) | 29 (70.7) | 37 (68.5) | || |||||| | || |||||| |
Completed study drug treatment, N (%) | NR | NR | || |||||| | || |||||| |
Discontinued from study, n (%) | 8 (19.5) | 15 (27.8) | | ||||| | | ||||| |
Reason for discontinuation from study, n (%) | ||||
Death | 2 (4.9) | 5 (9.3) | | ||||| | | ||||| |
Entered another COG study | 2 (4.9) | 1 (1.9) | || | || |
Lost to follow-up | 4 (9.8) | 9 (16.7) | || | || |
Withdrawal of consent and follow-up | NA | NA | | ||||| | | ||||| |
ITT, N | 42 | 55 | ||| | ||| |
ITT ALL only, N | NA | NA | ||| | ||| |
FAS, N | 41 | 54 | ||| | ||| |
FAS ALL only, N | NA | NA | ||| | ||| |
PP, N | 37 | 42 | || | || |
PKAS, N | 43 | 47 | ||| | ||| |
Safety, N | 43 | 52 | ||| | ||| |
AE = adverse event; ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; FAS = full analysis set; ITT = intention-to-treat; NA = not applicable; NR = not reported; Ph-positive = Philadelphia chromosome–positive; PI = postinduction; PKAS = pharmacokinetic analysis set; PP = per protocol; VHR = very high risk.
Note: The data cut-off date was December 31, 2015, in the COG AALL07P4 study (Clinical Study Report) and June 12, 2017, in the DFCI 11-001 study (Day 120 Report).
aThree patients were randomized to receive pegaspargase 2,500 IU/m2 but were inadvertently administered calaspargase pegol 2,500 IU/m2.
bIn the event of a severe systemic or recurrent local allergic reaction to the study drug, Erwinia asparaginase was to be substituted.
Sources: COG AALL07P4 Clinical Study Report12 and DFCI 11-001 Day 120 Report.13,54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Baseline Characteristics
Baseline characteristics for the COG AALL07P4 and DFCI 11-001 trials are summarized in Table 12. The baseline characteristics outlined in Table 12 are limited to those that were most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
In the COG AALL07P4 study, patients were all newly diagnosed with high-risk B-precursor ALL. The groups were generally similar in baseline characteristics. Most patients were older than 10 years (108 of 163 patients; 66.3%), male (50.9%), and white (82.2%). More patients in the calaspargase pegol group were younger than 10 years at diagnosis (43.9%) compared with the pegaspargase group (33.3%). A lower proportion of patients was older than 10 years presenting with WBC counts below 50,000/μL at baseline in the calaspargase pegol group (43.9%) compared with the pegaspargase group (55.6%). Patients had a higher median baseline WBC count in the calaspargase pegol group (59 × 109/L) compared with the pegaspargase group (27 × 109/L). Steroid use before protocol therapy was similar between groups, with only 5.5% of patients having received steroids in the week preceding their diagnosis. Concomitant antileukemia medications administered per protocol were similarly dosed across the groups and data for other concomitant medications were not collected.
In the DFCI 11-001 study, patients’ baseline characteristics were balanced overall between the calaspargase pegol group and the pegaspargase group. Most patients were younger than 10 years (178 of 237 patients; 75%), male (61.6%), and white (70.5%). Most patients (84.0%) did not have steroid use before protocol treatment, with similar between-group proportions.
Table 12: Summary of Baseline Characteristics From the Studies Included in the Systematic Review — FAS
Characteristic | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 41) | Pegaspargase 2,500 IU/m2 (N = 54) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Age, years | ||||
Mean (SD) | 9.4 (6.2) | 10.4 (5.6) | 6.4 (4.7) | 6.2 (4.7) |
Median (range) | 11.0 (1 to 26) | 11.0 (1 to 23) | 5.0 (1 to 20) | 4.0 (1 to 18) |
< 10 | 18 (43.9) | 18 (33.3) | 89 (75.4) | 89 (74.8) |
≥ 10 | 23 (56.1) | 36 (66.7) | 29 (24.6) | 30 (25.2) |
10 to < 16 | 18 (43.9) | 28 (51.9) | || |||||| | || |||||| |
≥ 16 | 5 (12.2) | 8 (14.8) | | ||||| | | ||||| |
Sex, n (%) | ||||
Female | 27 (65.9) | 23 (42.6) | 43 (36.4) | 48 (40.3) |
Male | 14 (34.1) | 31 (57.4) | 75 (63.6) | 71 (59.7) |
BMI (kg/m2) | ||||
Mean (SD) | 19.9 (5.8) | 21.0 (7.4) | NR | NR |
Median (range) | 17.9 (14.1 to 39.3) | 17.6 (13.6 to 50.0) | |||||||||| || ||||| | |||||||||| || ||||| |
Diagnosis, n (%) | ||||
ALL | 41 (100.0) | 54 (100.0) | 113 (95.8) | 115 (96.6) |
LL | 0 | 0 | 5 (4.2) | 4 (3.4) |
Immunophenotype, n (%) | ||||
B-cell immunophenotype | 41 (100.0) | 54 (100.0) | 103 (87.3) | 103 (86.6) |
T-cell immunophenotype | 0 | 0 | 15 (12.7) | 16 (13.4) |
FISH translocation testing: MLL gene rearrangement, n (%) | ||||
Negative | 40 (97.6) | 104 (95.4) | NA | NA |
Positive | 1 (2.4) | 5 (4.6) | NA | NA |
WBC count at diagnosis × 109/L, n (%) | ||||
< 50 | 19 (46.3) | 33 (61.1) | 113 (95.8) | 118 (99.2) |
≥ 50 | 22 (53.7) | 21 (38.9) | 5 (4.2) | 1 (0.8) |
Age (years) and baseline WBC count × 109/L, n (%) | ||||
Age < 10 and WBC count < 50 | | ||||| | | ||||| | || |||||| | || |||||| |
Age < 10 and WBC count ≥ 50 | || |||||| | || |||||| | | ||||| | | ||||| |
Age ≥ 10 and WBC count < 50 | || |||||| | || |||||| | || |||||| | || |||||| |
Age ≥ 10 and WBC count ≥ 50 | | |||||| | | |||||| | | ||||| | ||| |
CNS status at diagnosis, n (%) | ||||
CNS1 | 35 (85.4) | 46 (85.2) | 90 (76.3) | 98 (82.4) |
CNS2 | 5 (12.2) | 8 (14.8) | 18 (15.3) | 15 (12.6) |
CNS3 | 5 (12.2) | 4 (7.4) | 1 (0.8) | 3 (2.5) |
Initial DFCI risk group,a n (%) | ||||
SR | NA | NA | 70 (59.3) | 70 (58.8) |
HR | NA | NA | 48 (40.7) | 49 (41.2) |
Steroids before protocol treatment, n (%) | ||||
Yes | 3 (7.3) | 1 (1.9) | || |||||| | || |||||| |
No | 38 (92.7) | 101 (92.7) | ||| |||||| | || |||||| |
ALL = acute lymphoblastic leukemia; BMI = body mass index; CNS = central nervous system; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; FAS = full analysis set; FISH = fluorescence in situ hybridization; HR = high risk; LL = lymphoblastic lymphoma; NA = not applicable; NR = not reported; SD = standard deviation; SR = standard risk; WBC = white blood cell.
aPatients were categorized as standard risk if they were younger than 10 years, presenting with a WBC count of less than 50,000/mm3, with B-cell phenotype, and without CNS leukemia (defined as no blast cells in spinal fluid or below 5 WBCs in spinal fluid with blasts present) at diagnosis. All other patients were categorized as high risk.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Across both the COG AALL07P4 and DFCI 11-001 trials, there were notable similarities and differences in baseline demographics. All patients in the COG AALL07P4 trial and nearly all patients in the DFCI 11-001 trial (96%) had ALL. The majority of patients in the DFCI 11-001 study had B-cell ALL (87%) and 13% of patients had T-cell ALL, whereas patients with B-cell immunophenotype were exclusively enrolled in the COG AALL07P4 trial. Most patients had a CNS status of 1 in both trials, with a minority of patients designated as CNS3 in the COG AALL07P4 study (fewer than 10%) and the DFCI 11-001 study (fewer than 2%). Most patients did not have steroid therapy before study treatment in the COG AALL07P4 study (|||) or the DFCI 11-001 study (|||). While the majority of patients were older than 10 years in the COG AALL07P4 trial (median = 11 years), most were younger than 10 years in the DFCI 11-001 trial (median = 4 years to 5 years). Approximately 62% of patients were diagnosed when they were 10 years or older in the COG AALL07P4 trial, whereas nearly 75% of patients were diagnosed at younger than 10 years in the DFCI 11-001 trial. Patients in the COG AALL07P4 trial were distributed equally across combined age and WBC categories, whereas more than 70% of patients in the DFCI 11-001 trial were younger than 10 years, with a WBC count below 50 × 109/L.
Exposure to Study Treatments
Patient exposure to study treatments in the COG AALL07P4 trial and the DFCI 11-001 trial is presented in Table 13.
In the COG AALL07P4 study, median (range) exposure time to the study drug was similar across treatment groups, with an exposure time of 6.9 months (range = 0.0 months to 12.7 months) in the calaspargase pegol group and 6.5 months (range = 0.0 months to 11.8 months) in the pegaspargase group. ||| |||||||||| || |||||||| ||| |||||||| || ||||| ||| || ||||| ||||||| ||||| |||| ||||| ||| || || || ||||||| || ||| |||||||||||| ||||| ||| || || || ||||||| || ||| |||||||||||| ||||| ||||||||| ||||||
In the DFCI 11-001 study, the last planned dose of calaspargase pegol was 1 week later than the last planned dose of pegaspargase due to the 3 times weekly versus twice weekly dosing of the 2 respective study drugs. The median (range) duration of exposure to study drug was 7.79 months (range = 0.0 months to 10.2 months) for calaspargase pegol and 8.05 months (range = 0.0 months to 11.4 months) for pegaspargase. ||| |||||| ||||||| |||||||||| |||| ||| |||||||||||| ||||| ||| |||||||| ||||| |||||| || ||||||| ||||| ||| ||||| |||| |||| ||| |||||||||||| || |||||||| ||||| |||||| || ||||||| ||| || ||||||||||| || |||||| ||||||||| ||| ||||| || |||||||||||| ||||| |||||| || ||||| || |||||||||||| |||| ||| |||| || ||||||||| ||||||| |||| |||||||||||||| ||||||
Concomitant Medications and Cointerventions
In the COG AALL07P4 and DFCI 11-001 trials, calaspargase pegol and pegaspargase were the study drugs, and the other chemotherapy drugs administered as part of the study protocol were considered concomitant therapy. In both studies, other concomitant medications were not required to be collected and are therefore not summarized.
Subsequent Treatment
In the COG AALL07P4 trial || || || ||||||| in calaspargase pegol and || || || ||||||| in pegaspargase and 85 patients in the DFCI 11-001 trial discontinued the study drug; among these patients, || ||||||| and || ||||||| switched to Erwinia in the COG AALL07P4 study and the DFCI 11-001 study, respectively. In the COG AALL07P4 study, patients who experienced severe systemic or recurrent local allergic reactions to the study drug were permitted to substitute the study drug with Erwinia asparaginase (protocol amendment 7a section 5.1, page 1,522) || ||| ||||||||| | ||||||| |||||||||| || |||||||||||| ||| ||||||||||||| |||||||||||| |||||||||||| ||||| ||| ||| ||||| |||| ||| ||||||||||| | ||||| | |||||||| |||||||| |||||||| |||| |||||||||||| | ||||||| |||| || |||||||||||| |||| ||| ||| ||| |||||| |||| |||||||| || ||||||| ||||||||||||||| |||| ||||||| ||| |||||||| || ||| |||||||||||| ||||| ||||| ||| ||| |||| ||| || ||| |||||||||||| ||||| ||| ||| ||| ||| |||||||| ||||| In the DFCI 11-001 study, 21 of 118 (17.8%) patients in the calaspargase pegol group switched, mostly due to hypersensitivity (19 patients). All of the patients who switched to Erwinia in the pegaspargase group did so due to hypersensitivity.
Table 13: Summary of Patient Exposure From the Studies Included in the Systematic Review — Safety Analysis Set
Characteristic | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 43) | Pegaspargase 2,500 IU/m2 (N = 52) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
|||||||||| ||||| |||||| |||||| ||||||| | |||||| |||||| || |||||| | |||||||||||||| || |||||||| | ||||||| |||||| || |||||| | ||||||| |||||| || |||||| |
Duration, months, median (range) | 6.93 (0.0 to 12.7) | 6.52 (0.0 to 11.8) | 7.79 (0.0 to 10.2) | 8.05 (0.0 to 11.4) |
Total number of doses administered, median (range) | 4.0 (1 to 11) | 4.5 (1 to 12) | 11.0 (1 to 11) | 16.0 (1 to 17) |
| || |||||||| |||| |||||| |||||| || ||||| ||||||||||||| |||||| ||||||| | ||||| ||||| || |||||| | |||||| ||||| || |||||| | ||||| ||| || |||| | ||||| ||| || |||| |
|||||||| | ||| || ||||| ||||||||||||| | ||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||||| | ||| || ||||| ||||||||||||| | ||| | || |||||| | || |||||| | || |||||| | || |||||| |
COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Table 14: Summary of Subsequent Treatment From the Studies Included in the Systematic Review — FAS
Subsequent treatment | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 41) | Pegaspargase 2,500 IU/m2 (N = 54) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Switched to Erwinia,a n (%) | 8 (19.5) | 9 (16.7) | 21 (17.8) | 16 (13.4) |
Reason for switching to Erwinia, n (%) | ||||
Hypersensitivity | NA | NA | 19 (NR) | 16 (NR) |
Silent activation | NA | NA | 2 (1.7) | 0 |
Administration of the incorrect PEG-asparaginase, n (%) | 0 | | ||||| | NA | NA |
COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; FAS = full analysis set; NA = not applicable; PEG = pegaspargase.
aPatients who experienced severe systemic or recurrent local allergic reaction to the study drug were switched to Erwinia asparaginase as a substitute.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Efficacy
Findings for key efficacy outcomes in the COG AALL07P4 and DFCI 11-001 trials are summarized in Table 15.
Table 15: Summary of Key Efficacy Results From the Studies Included in the Systematic Review
Outcome | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 42) | Pegaspargase 2,500 IU/m2 (N = 55) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Data cut-off date | December 31, 2015 | October 5, 2016a | ||
Median follow-up time, months (range) | 62.6 (0.5 to 86.3) | 26.6 (0.5 to 45.6) | ||
OS: Full analysis set | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Deaths at DCO,a n (%) | | |||||| | | |||||| | | ||||| | | ||||| |
1-year OS, % (95% CI) | |||| |||||| || ||||| | |||||||||| || ||||| | |||| |||||| || ||||| | |||||||||||| || |||||| |
2-year OS, % (95% CI) | |||| |||||| || ||||| | |||||||||| || ||||| | |||||||||| || |||||| | |||||||||||| || ||||||| |
4-year OS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | || | || |
HR (95% CI)b | |||||||||| || ||||| | ||||||||| | || | || |
DFS from CR: Full analysis set | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Events, n (%) | | ||||| | | ||||| | | ||||| | | ||||| |
1-year DFS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| |
2-year DFS, % (95% CI) | |||| |||||| || ||||| | |||||||||| || ||||| | |||||||||| || |||||| | |||||||||| || |||||| |
4-year DFS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | || | || |
HR (95% CI)b | |||| |||||| || ||||| | ||||||||| | || | || |
EFS: Full analysis set | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Events, n (%) | | ||||| | | |||||| | | ||||| | | ||||| |
1-year EFS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| |
2-year EFS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||| |||||| || |||||| | |||| |||||| || |||||| |
4-year EFS, % (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | || | || |
HR (95% CI)b | |||||||||| || ||||| | ||||||||| | || | || |
CR at end of induction: Full analysis set | ||||
End of induction day 29 | ||||
Number of patients contributing to the analysis, n | || | || | || | || |
Number of patients attaining CR, n (%) | || |||||| | || |||||| | || | || |
Number of patients attaining CR, 95% CI | |||| || |||| | |||| || |||| | || | || |
End of induction day 32 | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Number of patients attaining CR, n (%) | || | || | ||| |||||| | ||| |||||| |
Number of patients attaining CR, 95% CI | || | || | |||| || |||| | |||| || |||| |
MRD at end of induction: Full analysis set | ||||
MRD at induction day 29 | ||||
Number of patients contributing to the analysis, n (MRD-evaluable) | || | || | || | || |
Positive MRD (≥ 0.1%), n (%) | | |||||| | || |||||| | || | || |
Negative MRD (< 0.1%), n (%) | || |||||| | || |||||| | || | || |
Negative MRD (< 0.1%), 95% CI | |||| || |||| | |||| || |||| | || | || |
MRD at induction day 29 | ||||
Number of patients contributing to the analysis, n (MRD-evaluable) | || | || | || | || |
MRD ≥ 0.01, n (%) | | ||||| | | ||||| | || | || |
MRD at induction day 32 | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
MRD ≥ 0.01, n (%) | || | || | | ||||| | || |
SAA levels during induction: PKASc | ||||
Day 7 (5 minutes to 10 minutes postinfusion) | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
SAA ≥ 0.10 IU/mL, % (95% CI) | || | || | |||| |||||| || |||||| | |||| |||||| || |||||| |
Difference, OR (90% CI)d | || | ||||||||| | ||||| ||||||| || |||||| | ||||||||| |
Day 11 (4 days after dose) | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
SAA ≥ 0.10 IU/mL, % (95% CI) | || | || | ||||| |||||| || |||||| | |||| |||||| || |||||| |
Difference, OR (90% CI)d | || | ||||||||| | ||||| ||||||| || |||||| | ||||||||| |
Day 18 (11 days after dose) | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
SAA ≥ 0.10 IU/mL, % (95% CI) | || | || | ||||| |||||| || |||||| | |||||||||| || |||||| |
Difference, OR (90% CI)d | || | ||||||||| | ||||| ||||||| || |||||| | ||||||||| |
Day 25 (18 days after dose) | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
SAA ≥ 0.10 IU/mL, % (95% CI) | || | || | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference, OR (90% CI)d | || | ||||||||| | ||||| ||||||| || ||||||| | ||||||||| |
Day 32 (25 days after dose) | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
SAA ≥ 0.10 IU/mL, % (95% CI) | || | || | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference, OR (90% CI)d | || | ||||||||| | |||||| |||||||| || ||||||| | ||||||||| |
CI = confidence interval; COG = Children’s Oncology Group; CR = complete remission; DCO = data cut-off; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; HR = hazard ratio; ITT = intention-to-treat; MRD = minimal residual disease; NSAA = nadir serum asparaginase activity; OR = odds ratio; OS = overall survival; PKAS = pharmacokinetic analysis set; SAA = serum asparaginase activity.
aSurvival rates for OS, DFS from CR, and EFS for the 2-year time point were based on an updated data cut-off date of June 12, 2017, with a median follow-up time of 34.73 months and 34.04 months for the calaspargase pegol group and the pegaspargase group, respectively.
bThe HR was derived from a Cox proportional hazards model. An HR below 1 favours the calaspargase pegol treatment and an HR greater than 1 favours pegaspargase.
cSAA levels were reported for the PKAS (patients who received at least 1 dose of calaspargase pegol or pegaspargase on induction day 7 and had at least 1 postinfusion measurement above the lower limit of quantification) in the DFCI 11-001 trial.
dThe OR (90% CI) was estimated using a generalized estimating equation model for comparing categorical SAA levels between treatment, actual sampling time points, and the interaction of treatment and actual sampling time points as effects.
Sources: COG AALL07P4 Clinical Study Report13,15 and DFCI 11-001 Clinical Study Report.14Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Overall Survival
COG AALL7P4 Study
In the COG AALL07P4 study, the median OS was not reached at the data cut-off date on December 31, 2015 (refer to |||||| |). Patients had been followed for a median of 62.6 months (range = 0.5 months to 86.3 months). |||| || || ||||||| patients in the calaspargase pegol group and | || || ||||||| patients in the pegaspargase group had died. The proportion of patients who were censored (alive at the time of data cut-off) ||| || ||||||| ||| || ||||||| in the calaspargase pegol group and the pegaspargase group, respectively. Overall, ||||| |||||| for calaspargase pegol and ||||| for pegaspargase) of patients had a minimum of 4 years of follow-up for survival. The 1-year OS rate among patients in the FAS population was ||||| ||||| || |||||, ||| ||||| ||||| || ||||) in the calaspargase pegol group and the pegaspargase group, respectively. The 4-year OS rate among patients in the FAS population was ||||| ||||| || |||||, ||| ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 2-year and 3-year OS rates were consistent with minimal differences to the 4-year OS rates. The HR in the FAS population was |||| ||||| || ||||| in the calaspargase pegol group versus the pegaspargase group. Findings of the ITT population (refer to Appendix 1, Table 17) were consistent (minimal difference) with results for the FAS population.
DFCI 11-001 Study
In the DFCI 11-001 study, at CCOD of October 5, 2016, patients had been followed for a median of 26.6 months (range = 0.5 months to 45.6 months) (|||||| |)|||| ||| |||||| patients in the calaspargase pegol group and ||| ||| |||||| patients in the pegaspargase group had died. The proportion of patients who were censored (alive at the date of analysis or lost to follow-up) were ||| ||||||| and ||| ||||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 1-year OS rate among patients in the FAS ALL population was ||||| |||| ||| ||||| || |||||| for the calaspargase pegol group and |||||| |||| ||| |||||| || ||||||| for the pegaspargase group. At the day 120 cut-off, the median follow-up duration was |||| months and |||| months for the calaspargase pegol group and the pegaspargase group, respectively. The number of patients who died during the day 120 follow-up was ||||||||| || ||| |||||||||||| ||||| |||||| ||| | |||||||||| ||||||| ||| |||| || ||| |||||||||||| |||||. The 2-year OS rate among patients in the FAS ALL population was ||||| |||| ||| ||||| || |||||| for the calaspargase pegol group and ||||||||| for the pegaspargase group. The findings of the ITT ALL population (refer to Appendix 1, Table 17) were consistent (minimal difference) with the results for the FAS population.
Sensitivity analyses for OS among patients with ALL and patients with precursor B-cell in the FAS population were consistent with the primary analysis for OS.


DFS From CR
COG AALL7P4 Study
In the COG AALL07P4 study, median DFS was not reached at the data cut-off date of December 31, 2015 (refer to |||||| |). Patients had been followed for a median of 62.6 months (range = 0.5 months to 86.3 months). The number of patients with events were | || || |||||| in the calaspargase pegol group and | || || |||||| in the pegaspargase group. The proportion of patients who were censored and remained event-free were || ||||||| and || ||||||| in the calaspargase pegol group and the pegaspargase group, respectively. ||| |||||| || |||||||| ||| |||| |||||||| ||| || || |||||||||| |||||||| ||||||||| |||||||| |||||| ||||||||| ||||||||| || ||||| ||| || ||||||| ||| || ||||||| || ||| |||||||||||| ||| |||||||||||| |||||| ||||||||||||| ||| |||||| || |||||||| ||| |||| |||||||| ||| ||||||||| ||||||| ||| || ||||||| ||| || ||||||| ||||||||| ||||||| |||||||||||| ||||| | ||||||| |||||||| ||| | ||||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| The 1-year DFS rate among patients in the FAS population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 4-year DFS rate among patients in the FAS population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 2-year and 3-year DFS rates were consistent with minimal differences to the 4-year DFS rates. The HR of DFS in the FAS population was |||| ||||| || ||||| in the calaspargase pegol group versus the pegaspargase group. Findings for the ITT population were identical to those of the FAS population.
DFCI 11-001 Study
In the DFCI 11-001 study, at the data cut-off date of October 5, 2016, patients were followed up for a median time of 26.6 months (range = 0.5 months to 45.6 months) (|||||| |). The number of patients with events were | || ||| |||||| in the calaspargase pegol group and | || ||| |||||| in the pegaspargase group. A total of ||| ||||||| patients and ||| ||||||| patients in the calaspargase pegol group and the pegaspargase group, respectively, were censored. ||| |||||| || |||||||| ||| |||| |||||||| ||| || || |||||||||| |||||||| ||||||||| |||||||| |||||| ||||||||| ||||||||| || ||||| ||| || ||||||| ||| || ||||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| |||||| ||||||| |||||||| ||| || ||||||| |||||||| |||| |||||||| || ||||||| |||||||||||| ||||||| || ||| |||||||||||| ||||| ||| |||||||||||| ||||||| ||||||||||||. The 1-year DFS rate among patients in the FAS ALL population who attained CR was ||||| |||| ||| |||| || ||||| in the calaspargase pegol group and ||||| |||| ||| |||| || ||||| in the pegaspargase group. At day 120 follow-up, | |||||| patients in the calaspargase pegol group and | |||||| patients in the pegaspargase group had experienced a DFS event. At day 120 follow-up, the 2-year DFS rate among patients in the FAS ALL population who attained CR was ||||| ||||| || ||||| versus ||||| ||||| || ||||| for calaspargase pegol versus pegaspargase, respectively (|||||| |). The results of DFS among patients attaining CR in the ITT ALL population were identical to the results for the FAS ALL population.
Sensitivity analyses for DFS from CR among patients with ALL and patients with precursor B-cell ALL in the FAS population were consistent with the primary analysis for DFS from CR. Similarly, sensitivity analyses by initial risk diagnosis among patients in the FAS population were consistent with the primary analysis for DFS for CR.


Event-Free Survival
COG AALL7P4 Study
In the COG AALL07P4 study, the median EFS rate was not reached at the data cut-off on December 31, 2015 (refer to |||||| |). Patients had been followed for a median of 62.6 months (range = 0.5 months to 86.3 months). ||||| ||| || || |||||| patients in the calaspargase pegol group and | ||| || || ||||||| patients in the pegaspargase group experienced an event. A total of || ||||||| |||||||| and || ||||||| patients in the calaspargase pegol group and the pegaspargase group, respectively, were censored. ||| |||||| || |||||||| ||| |||| |||||||| ||| || || |||||||||| |||||||| ||||||||| |||||||| |||||| ||||||||| ||||||||| || ||||| |||| || ||||||| ||| || ||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| ||| |||||| || |||||||| ||| |||| |||||||| ||| ||||||||||| ||||||| |||| || ||||||| ||| || |||||||| ||||||||| ||||||| |||||||||||| ||||| | ||||||| |||||||| ||| | ||||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||. The 1-year EFS rate among patients in the FAS population was ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 4-year EFS rate among patients in the FAS population ||| ||||| ||||| || ||||| and ||||| ||||| || ||||| in the calaspargase pegol group and the pegaspargase group, respectively. The 2-year and 3-year EFS rates were consistent with minimal differences to the 4-year EFS rates. The HR was |||| ||||| || ||||| for the calaspargase pegol group when compared with the pegaspargase group. The findings for EFS in the ITT population were consistent with the results for the FAS population.
DFCI 11-001 Study
In the DFCI 11-001 study, at the data cut-off date of October 5, 2016, patients were followed up for a median time of 26.6 months (range = 0.5 months to 45.6 months) (|||||| |). ||||| ||| || ||| |||||| patients in the calaspargase pegol group and | ||| || ||| |||||| patients in the pegaspargase group had experienced an event. A total of ||| ||||||| patients and ||| ||||||| patients in the calaspargase pegol group and the pegaspargase group, respectively, were censored. ||| |||||| || |||||||| ||| |||| |||||||| ||| || || |||||||||| |||||||| ||||||||| |||||||| |||||| ||||||||| ||||||||| || ||||| |||| || ||||||| ||| || ||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| ||| |||||| || |||||||| ||| |||| |||||||| ||| ||||||||||| ||||||| |||||||| ||||||||||||| |||| |||||||| ||| || ||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||. The 1-year EFS rate among patients in the FAS ALL population was ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || |||||| in the calaspargase pegol group and the pegaspargase group, respectively. At the day 120 follow-up with the data cut-off date of June 12, 2017, | |||||||||| ||||||| || |||| ||||| ||||||||||| || |||||| The 2-year EFS rate among patients in the FAS ALL population at the day 120 update was ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || |||||| in the calaspargase pegol group and the pegaspargase group, respectively. The results of EFS among patients in the ITT ALL population were consistent with the results for the FAS ALL population.
Sensitivity analyses for EFS among patients with ALL and patients with precursor B-cell ALL in the FAS population were consistent with the primary analysis for EFS. Sensitivity analyses for initial risk diagnosis among patients in the FAS population were consistent with the primary analysis for EFS.


CR at End of Induction
COG AALL7P4 Study
In the COG AALL07P4 study, the proportion of patients in the FAS population who attained CR by day 29 was || || || ||||||| ||| ||| ||||| || |||||| in the calaspargase pegol group, and || || || ||||||| ||| ||| ||||| || |||||| in the pegaspargase group. The findings for CR at the end of induction day 29 for the ITT population were consistent with the results for the FAS population.
DFCI 11-001 Study
In the DFCI 11-001 study, the proportion of patients in the FAS ALL population who attained CR by day 32 was ||| || ||| ||||||| ||| ||| ||||| || |||||| in the calaspargase pegol group and ||| || ||| ||||||| ||| ||| ||||| || |||||| in the pegaspargase group. The results for CR at the end of induction day 32 in the ITT ALL population were consistent with the results for the FAS ALL population.
Sensitivity analyses for CR for initial risk diagnosis, age group classification (< 10 years, ≥ 10 years, 10 to < 16 years, and ≥ 16 years), and BMI category (< 50th percentile versus ≥ 50th percentile) were consistent with the primary analysis for CR.
MRD at End of Induction
COG AALL7P4 Study
In the COG AALL07P4 study, the proportion of patients in the MRD-evaluable FAS population with positive MRD (≥ 0.1% detectable leukemia cells in bone marrow biopsy or aspirate with validated 6-colour multiparameter flow cytometry) at induction day 29 |||| | || || ||||||| in the calaspargase pegol group and || || || ||||||| in the pegaspargase group. Conversely, the proportion of patients in the MRD-evaluable FAS population with negative MRD (< 0.1%) at induction day 29 were || ||||||| and || ||||||| in the calaspargase pegol group and the pegaspargase group, respectively. The proportion of patients in the FAS population with MRD of 0.01 or greater at induction day 29 were | |||||| in the calaspargase pegol group and | |||||| in the pegaspargase group. The findings for positive MRD (≥ 0.1%) at the end of induction day 29 in the ITT population were consistent with the results for the FAS population.
DFCI 11-001 Study
In the DFCI 11-001 study, results for MRD were identical for the FAS and ITT populations. In the DFCI 11-001 study, the proportion of patients in the FAS ALL population with MRD of 0.01 or greater were | || ||| |||||| in the calaspargase pegol group and ||| in the pegaspargase group. ||| ||||||| |||| ||||||| ||| || ||||||| ||| || ||||||| || |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||. The findings for MRD of 0.01 or greater at the end of induction day 32 in the ITT ALL population were consistent with the results for the FAS ALL population.
SAA During Induction
COG AALL7P4 Study
In the COG AALL07P4 study, SAA levels were not reported.
DFCI 11-001 Study
In the DFCI 11-001 study, at the data cut-off date of October 5, 2016, arithmetic mean SAA levels were between |||| ||| |||| ||||| |||||| in the pegaspargase treatment group than in the calaspargase pegol treatment group at the first (5 minutes to 10 minutes), second (4 days), and third (11 days) measurement time points after administration (|||||| |). At the fourth (18 days) and fifth (25 days) measurement time points, arithmetic mean SAA levels were |||| ||| |||| ||||| |||||| with calaspargase pegol than with pegaspargase, respectively. The arithmetic mean SAA level at day 32 (25 days after drug administration) was |||||| ||| | ||||||| in the calaspargase pegol group || | |||| and |||||| ||| | ||||||| in the pegaspargase group || | ||||. The results for geometric means were consistent with the results for arithmetic means.
The proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| |||| ||| |||| || |||||| for the calaspargase pegol group versus ||||| |||| ||| |||| || |||||| for the pegaspargase group at 5 minutes to 10 minutes after infusion on induction day 7 (OR |||||| ||| ||| ||||| || ||||||. The proportion of patients with SAA levels of 0.10 IU/mL or greater were |||||| (95% CI, |||| || |||||| for the calaspargase pegol group versus ||||| (95% CI, |||| || |||||) for the pegaspargase group at 4 days after infusion on day 11 (OR |||||; 90% CI, ||||| || |||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were |||||| (95% CI, |||| || |||||) for the calaspargase pegol group versus |||||| (95% CI, |||| || |||||) for the pegaspargase group at 11 days after infusion on day 18 (OR |||||; 90% CI, ||||| || |||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| (95% CI, |||| || ||||) for the calaspargase pegol group versus ||||| (95% CI, |||| || ||||) for the pegaspargase group at 18 days after infusion on day 25 (OR |||||; 90% CI, ||||| || ||||||). The proportion of patients with SAA levels of 0.10 IU/mL or greater were ||||| (95% CI, |||| || ||||) for the calaspargase pegol group versus ||||| (95% CI, |||| || ||||) for the pegaspargase group at 25 days after infusion on day 32 (OR ||||||; 90% CI, |||||| || ||||||).
Estimates of treatment effect on SAA levels using adjusted analyses (controlled for age, sex, initial risk group, disease type, and baseline WBC count) were similar to unadjusted analyses for each time point.

Health-Related Quality of Life
HRQoL was not assessed in the COG AALL07P4 trial or the DFCI 11-001 trial.
Harms
Harms data for the COG AALL07P4 and DFCI 11-001 trials are summarized in Table 16. Safety data for the COG AALL07P4 trial were from the primary safety analyses (CCOD of December 31, 2015). Safety results for the DFCI 11-001 trial were available at the CCOD of October 5, 2016, and day 120 follow-up (CCOD of February 1, 2017). ||| ||||||| || ||| |||||||||||| ||||| ||| |||| |||||| ||| ||| ||| |||||||||| ||| || ||||||| |||||||| Findings from the updated safety data (CCOD of February 1, 2017) were ||||||||| consistent with the primary safety analyses (CCOD of October 5, 2016) and are therefore not presented.
Table 16: Summary of Harms Results From the Studies Included in the Systematic Review
Harm | COG AALL07P4 study | DFCI 11-001 study | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 43) | Pegaspargase 2,500 IU/m2 (N = 52) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Most common AEs, n (%): Safety analysis set | ||||
Data cut-off dates | December 31, 2015 | October 5, 2016 | ||
Number of patients contributing to analysis, n | 43 | 52 | 118 | 119 |
Patients with ≥ 1 TEAE | || |||||| | || |||||| | ||| |||||| | ||| ||||||| |
TEAEs in ≥ 25% patients in any group, n (%) | ||||
Hypoalbuminemia | 12 (27.9) | 3 (5.8) | 96 (81.4) | 98 (82.4) |
ALT, increased | 15 (34.9) | 20 (38.5) | 93 (78.8) | 92 (77.3) |
Hyperglycemia | 34 (79.1) | 26 (50.0) | 40 (33.9) | 34 (28.6) |
AST, increased | 10 (23.3) | 11 (21.2) | 63 (53.4) | 70 (58.8) |
Hypokalemia | 12 (27.9) | 6 (11.5) | 54 (45.8) | 47 (39.5) |
Febrile neutropenia | 24 (55.8) | 22 (42.3) | 40 (33.9) | 48 (40.3) |
Hypertriglyceridemia | 7 (16.3) | 6 (11.5) | 33 (28.0) | 43 (36.1) |
Hypocalcemia | | ||||| | | |||||| | || | || |
Blood fibrinogen, decreased | 6 (14.0) | 3 (5.8) | 26 (22.0) | 32 (26.9) |
Stomatitis | 6 (14.0) | 6 (11.5) | 30 (25.4) | 24 (20.2) |
Activated partial thromboplastin time, prolonged | 13 (30.2) | 10 (19.2) | 14 (11.9) | 18 (15.1) |
Neutrophil count, decreased | 24 (55.8) | 27 (51.9) | NA | NA |
WBC count, decreased | 16 (37.2) | 15 (28.8) | NA | NA |
Platelet count, decreased | 15 (34.9) | 13 (25.0) | NA | NA |
Anemia | 11 (25.6) | 14 (26.9) | NA | NA |
Peripheral motor neuropathy | 12 (27.9) | 10 (19.2) | NA | NA |
Peripheral sensory neuropathy | 8 (18.6) | 5 (9.6) | | ||||| | ||| |
Bilirubin conjugated, increased | NA | NA | 26 (22.0) | 38 (31.9) |
SAEs, n (%): Safety analysis set | ||||
Number of patients contributing to analysis, n | 43 | 52 | 118 | 119 |
Patients with ≥ 1 SAE | NA | NA | 29 (24.6) | 26 (21.8) |
Patients with grade 3 or grade 4 TEAE, n (%) | 42 (97.7) | 47 (90.4) | ||| |||||| | ||| |||||| |
Grade 3 or grade 4 TEAEs in ≥ 25% of patients in any group, n (%) | ||||
Neutrophil count, decreased | 24 (55.8) | 27 (51.9) | NR | NR |
Febrile neutropenia | 24 (55.8) | 22 (42.3) | 40 (33.9) | 48 (40.3) |
ALT, increased | 14 (32.6) | 19 (36.5) | 58 (49.2) | 72 (60.5) |
Platelet count, decreased | 15 (34.9) | 13 (25.0) | NR | NR |
WBC count, decreased | 16 (37.2) | 15 (28.8) | NR | NR |
Hyperglycemia | 16 (37.2) | 9 (17.3) | NR (< 25) | NR (< 25) |
Hypokalemia | 12 (27.9) | 6 (11.5) | 51 (43.2) | 43 (36.1) |
Anemia | 11 (25.6) | 14 (26.9) | NR | NR |
AST, increased | NA (< 25) | NA (< 25) | 31 (26.3) | 36 (30.3) |
Anaphylactic reaction | 11 (25.6) | 10 (19.2) | NR | NR |
Hypoalbuminemia | 11 (25.6) | 2 (3.8) | 32 (27.1) | 33 (27.7) |
Stomatitis | NA (< 25) | NA (< 25) | 30 (25.4) | 24 (20.2) |
Withdrawals due to AEs, n (%): Safety analysis set | ||||
Number of patients contributing to analysis, n | 43 | 52 | 118 | 119 |
Patients who stopped study treatment | 13 (30.2) | 14 (26.9) | 33 (28.0)a | 23 (19.3)a |
Patients who stopped study treatment, risk difference (95% CI) | |||| ||||||| || ||||| | ||||||||| | ||||||||||| || |||| | ||||||||| |
Withdrawals due to AEs in ≥ 2% of patients in any group, n (%) | ||||
Hypersensitivity | NA | NA | 10 (8.5) | | ||||| |
Lipase, increased | NA | NA | 8 (6.8) | | ||||| |
Pancreatitis | NA | NA | 7 (5.9) | | ||||| |
Drug hypersensitivity | NA | NA | 6 (5.1) | | ||||| |
Amylase, increased | NA | NA | 5 (4.2) | | ||||| |
Anaphylactic reaction | NA | NA | 2 (1.7) | ||| |
Deaths, n (%): Full analysis set (COG AALL07P4 trial) and intention-to-treat population (DFCI 11-001 trial) | ||||
Number of patients contributing to analysis, n | 41 | 54 | 118 | 119 |
Patients who diedd | | |||||| | | |||||| | || |||| | | ||||| |
Due to this disease | || | | ||||| | || | || |
Due to other cause | | |||||| | | ||||| | || | || |
Relapse | || | || | || | | |||| |
Induction death | | ||||| | || | | |||| | || |
Remission death | || | || | | |||| | || |
Primary disease | || | || | | |||| | || |
Unknown | || | || | || | || |
AESIs, n (%): Safety analysis set | ||||
Number of patients contributing to analysis, n | 43 | 52 | 118 | 119 |
Patients with ≥ 1 AESI | NA | NA | || |||||| | || |||||| |
Hypersensitivity | | ||||| | | ||||| | 11 (9.3) | 7 (5.9) |
Anaphylactic reaction | 11 (25.6) | 10 (19.2) | | ||||| | | ||||| |
Silent inactivation | NA | NA | 2 (1.7) | 0 |
Pancreatitis | 8 (18.6) | 4 (7.7) | 14 (11.9) | 20 (16.8) |
Venous thrombosis | NA | NA | | ||||| | | ||||| |
Blood bilirubin, increased | 27 (62.8) | 26 (50.0) | 54 (45.8) | 52 (43.7) |
ALT, increased | 15 (34.9) | 20 (38.5) | 93 (78.8) | 92 (77.3) |
Hepatic failure | NA | NA | | ||||| | || |
AE = adverse event; AESI = adverse event of special interest; ALT = alanine transaminase; AST = aspartate aminotransferase; CI = confidence interval; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; NA = not applicable; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WBC = white blood cell.
aIn the DFCI 11-001 study, the investigators did not designate some of the hypersensitivity AEs that resulted in a switch to Erwinia as AEs leading to study drug discontinuation for 6 patients in the pegaspargase 2,500 IU/m2 group.
bDeaths were reported for the full analysis set in the COG AALL07P4 trial and for the intention-to-treat population in the DFCI 11-001 trial.
cOne patient randomized to calaspargase pegol 2,500 IU/m2 who died during induction was not treated with the study drug.
dReasons for deaths may not add up to the total number of patients who died because cause of death may be attributed to more than 1 reason (e.g., a patient who died due to primary disease and the death occurred during induction).
Sources: COG AALL07P4 Clinical Study Report12 and DFCI 11-001 Clinical Study Report.13,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Adverse Events
In the COG AALL07P4 trial, the percentage of patients reporting any TEAEs was ||||| ||| || ||| for the calaspargase pegol group and ||||| ||| || ||| for the pegaspargase group. The most common TEAEs occurring in at least 25% of patients in the calaspargase pegol group and the pegaspargase group, respectively, were hyperglycemia (79.1% versus 50.0%); blood bilirubin, increased (62.8% versus 50.0%); neutrophil count, decreased (55.8% versus 51.9%); febrile neutropenia (55.8% versus 42.3%); alanine aminotransferase, increased (34.9% versus 38.5%); platelet count, decreased (34.9% versus 25.0%); WBC count, decreased (37.2% versus 28.5%); hypokalemia (27.9% versus 11.5%); anemia (25.6% versus 26.9%); activated partial thromboplastin time, prolonged (30.2% versus 19.2%); peripheral motor neuropathy (27.9% versus 19.2%); and abdominal pain (32.6% versus 11.5%).
In the DFCI 11-001 trial, |||||| ||| |||||||| |||| || |||| |||||| experienced at least 1 TEAE. The number of patients who experienced TEAEs were ||| ||||||| patients in the calaspargase pegol group and ||| |||||||| patients in the pegaspargase group. The most common TEAEs occurring in at least 25% of patients in the calaspargase pegol group and the pegaspargase group, respectively, were hypoalbuminemia (81.4% versus 82.4%); alanine transaminase, increased (78.8% versus 77.3%); aspartate aminotransferase, increased (53.4% versus 58.8%); blood bilirubin, increased (45.8% versus 43.7%); hypokalemia (45.8% versus 39.5%); febrile neutropenia (33.9% versus 40.3%); hyperglycemia (33.9% versus 28.6%); hypoglycemia (30.5% versus 36.1%); hypertriglyceridemia (28.0% versus 36.1%); and stomatitis (25.4% versus 20.2%).
The most common TEAEs in the calaspargase pegol group in the COG AALL07P4 trial and the DFCI 11-001 trial, respectively, were hypoalbuminemia (27.9% and 81.4%); hyperglycemia (79.1% and 33.9%); alanine transaminase, increased (34.9% and 78.8%); blood bilirubin, increased (62.8% and 45.8%); and febrile neutropenia (55.8% and 33.9%). The most common TEAEs in the pegaspargase group were hyperglycemia (50.0% and 82.4%); alanine transaminase, increased (38.5% and 77.3%); blood bilirubin, increased (50.0% and 43.7%); and febrile neutropenia (42.3% and 40.3%).
Serious Adverse Events
In the COG AALL07P4 trial, the number of patients with at least 1 SAE was not reported. The number of patients with at least 1 grade 3 or grade 4 TEAE was 42 (97.7%) patients in the calaspargase pegol group and 47 (90.4%) patients in the pegaspargase group. The most common grade 3 or grade 4 TEAEs in the calaspargase pegol group were neutrophil count, decreased (55.8%); febrile neutropenia (55.8%); WBC count, decreased (37.2%); and hyperglycemia (37.2%). The most common grade 3 or grade 4 TEAEs in the pegaspargase group were neutrophil count, decreased (51.9%); febrile neutropenia (42.3%); alanine transaminase, increased (36.5%); and WBC count, decreased (28.8%).
In the DFCI 11-001 trial, a total of ||| ||||||| patients experienced grade 3 or grade 4 TEAEs. The number of patients who experienced grade 3 or grade 4 TEAEs were ||| ||||||| in the calaspargase pegol group and ||| ||||||| in the pegaspargase group. The most common grade 3 or grade 4 TEAEs in the calaspargase pegol group were alanine transaminase, increased (49.2%); hypokalemia (43.2%); febrile neutropenia (33.9%); and hypoalbuminemia (27.1%). The most common grade 3 or grade 4 TEAEs in the pegaspargase group were alanine transaminase, increased (60.5%); febrile neutropenia (40.3%); and hypokalemia (36.1%). The percentage of patients who experienced at least 1 SAE was 24.6% and 21.8% in the calaspargase pegol group and the pegaspargase group, respectively. SAEs that occurred in at least 2% of patients in either the calaspargase pegol group or the pegaspargase group, respectively, were lipase, increased (4.2% |||||| |||||); pancreatitis (5.9% |||||| |||||| sepsis (3.4% |||||| ||||); hyperglycemia (2.5% |||||| |||||| febrile neutropenia (1.7% |||||| |||||| amylase, increased (0.8% |||||| ||||); alanine transaminase, increased (2.5% |||||| ||||); aspartate aminotransferase, increased (2.5% |||||| |); and neutropenic colitis (2.5% |||||| |).
Withdrawals Due to Adverse Events
In the COG AALL07P4 trial, the number of patients who stopped study treatment due to an AE was || ||||||| and || ||||||| in the calaspargase pegol group and the pegaspargase group, respectively. The reasons for stopping study treatment due to an AE were not reported.
In the DFCI 11-001 trial, the number of patients who stopped study treatment due to an AE was 33 (28.0%) patients in the calaspargase pegol group and 23 (19.3%) patients in the pegaspargase group. WDAEs in the calaspargase pegol group compared with pegaspargase were due to hypersensitivity (8.5% versus ||||); lipase, increased (6.8% versus ||||); pancreatitis (5.9% in both groups); drug hypersensitivity (5.1% versus ||||); amylase, increased (4.2% versus ||||); and anaphylactic reaction (1.7% versus ||||).
Mortality
|| ||| |||||||| || ||| ||| ||||||||||| | |||||| |||||||| || ||| |||||||||||| ||||| ||||| || ||||||| ||| ||| ||||||| |||||||| ||||||||||| || |||||| | |||||| |||| ||| || ||||||||| |||||||||| ||||||| |||| ||| ||||||||| | ||| || ||||| ||||||| ||| |||||| |||||||| || ||| |||||||||||| |||||| || |||||| | |||| ||| || |||||||||| ||||||| ||| ||| ||||||||| | |||| ||| || ||||| ||||||. Table 16 reports the number of patients who died in the FAS population.
|| |||| ||||||| | |||||| ||| | |||||| |||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| ||||| |||||| || ||| |||||||||||| ||||| |||||| | ||| ||| || ||||||| ||||||| |||||||| ||| ||||| | |||| ||||||||| ||||||| ||| | ||| || ||||||||| |||||| ||| |||||| || ||| |||||||||||| ||||| |||| ||| || |||||||| ||| |||||||||| ||||||| ||| ||| |||||||||| || ||| |||||||||||| ||||| ||||| ||| ||| ||| ||||||| |||| ||||| |||| |||| ||||||||||| |||| |||||| ||| |||||.
Notable Harms
Hypersensitivity Reactions
In the COG AALL07P4 trial, | |||||| patients in the calaspargase pegol group and | |||||| patients in the pegaspargase group experienced hypersensitivity events | || |||||||| || |||||| ||||||||| ||||| |||| |||||||||||||||| ||||||||| |||| |||||||| ||| ||||||||||||| ||||||||| || |||||||| ||||||||||||||||.
In the DFCI 11-001 trial, || ||||||| patients in the calaspargase pegol group and || ||||||| patients in the pegaspargase group experienced hypersensitivity events. ||| ||||||| |||||||| ||| | |||||| ||||||| |||||| |||||||| ||| ||||||||| |||||| |||||||| |||||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| ||||| ||||||| ||| | ||||||| |||||||| || ||| |||||||||||| ||||| ||| |||||||||||| |||||| ||||||||||||| ||| ||| |||| |||||||| ||||||||||||||||||.
Anaphylactic Reactions
In the COG AALL07P4 study, 11 (25.6%) patients in the calaspargase pegol group and 10 (19.2%) patients in the pegaspargase group experienced anaphylactic reactions.
In the DFCI 11-001 study, | |||||| patients in each treatment group experienced anaphylactic reactions.
Silent Inactivation
Silent inactivation was not reported by any patient in the COG AALL07P4 trial. In the DFCI 11-001 trial, 2 (1.7%) patients in the calaspargase pegol group experienced silent inactivation and were switched to Erwinia asparaginase treatment.
Pancreatitis
In the COG AALL07P4 trial, 8 (18.6%) patients in the calaspargase pegol group and 4 (7.7%) patients in the pegaspargase group experienced pancreatitis.
In the DFCI 11-001 trial, 14 (11.9%) patients in the calaspargase pegol group and 20 (16.8%) patients in the pegaspargase group experienced pancreatitis.
Venous Thrombosis
In the COG AALL07P4 study, venous thrombosis was ||| |||||||| || ||| |||||||.
In the DFCI 11-001 study, | |||||| patients in the calaspargase pegol group and | |||||| patients in the pegaspargase group experienced venous thrombosis.
Hemorrhage
|| |||||||| || |||||| ||||||||| ||||| experienced hemorrhage in the COG AALL07P4 study or the DFCI 11-001 study.
Hepatotoxicity
In the COG AALL07P4 study, the number of patients in the calaspargase pegol group and pegaspargase group, respectively, who experienced increased blood bilirubin were 27 (62.8%) patients and 26 (50.0%) patients. There were 15 (34.9%) patients and 20 (38.5%) patients who experienced increased alanine aminotransferase in the calaspargase pegol group and pegaspargase group, respectively.
In the DFCI 11-001 study, 54 (45.8%) patients and 52 (43.7%) patients experienced increased blood bilirubin in the calaspargase pegol group and pegaspargase group, respectively. There were 93 (78.8%) patients and 92 (77.3%) patients who experienced increased alanine aminotransferase in the calaspargase pegol group and pegaspargase group, respectively. ||| ||||||| || ||| |||||||||||| ||||| ||||| ||||||||||| ||||||| |||||||.
Critical Appraisal
Internal Validity
COG AALL07P4 Study
The COG AALL07P4 study was a phase II, randomized, open-label RCT. Methods of randomization appeared to be adequate. In the COG AALL07P4 study, patients were randomized 2:1 to calaspargase pegol or pegaspargase based on permuted blocks (of 6) using an electronic remote data entry system. Treatment groups were generally balanced for baseline characteristics including prognostic factors and medical history, indicating that randomization was likely appropriate, and risk of selection bias was likely low. The open-label study design may have biased outcomes with subjective assessments for measurement of harms due to knowledge of the assigned treatment, although the direction of potential bias is unclear. AEs highlighted by the clinical experts consulted by CADTH to be especially relevant for the management of patients in clinical practice included hypersensitivity reactions, anaphylactic reactions, silent inactivation, and WDAEs. The open-label study design may have biased WDAEs due to knowledge of assigned treatment, although the direction of potential bias is unclear.
The COG AALL07P4 trial (N = 97) was not designed or powered to formally assess comparative efficacy outcomes (i.e., day 29 end-of-induction MRD, CR, EFS, DFS from the attainment of CR, and OS), making assessments of relative therapeutic efficacy of pegaspargase challenging. The protocol did not prespecify a degree of difference from which to formally conclude noninferiority or similarity between calaspargase pegol and pegaspargase. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect and end points were not adjusted for multiple comparisons. The sample size of 97 patients was relatively small and the magnitude of the treatment effect estimates observed in a small study sample may not be replicable in a larger study sample. The clinical experts consulted by CADTH did not anticipate clinically meaningful differences in efficacy between calaspargase pegol and pegaspargase due to the products’ similar mechanisms of action and use of the same asparagine-specific enzyme (i.e., derived from E. coli).
There was no imputation for missing outcome data. However, there were few patients missing in the calaspargase pegol group and the pegaspargase group, respectively, for CR at the end of induction day 29 (1 and 0) and MRD at the end of induction day 29 (up to 3 and 5), and the proportion of patients who were censored for survival outcomes appeared to be balanced between groups.
The comparator used in the COG AALL07P4 study was appropriate as pegaspargase is a pegylated formulation of asparaginase (calaspargase pegol uses the same mechanism of action as pegaspargase) and has been a component of current SOC. Calaspargase pegol is intended to substitute for pegaspargase and would be used for patients who would have otherwise received a pegaspargase-containing MDC. In addition to asparaginase, the MDC components that were included in the study protocol (cytarabine, vincristine, daunorubicin, mercaptopurine, thioguanine, and doxorubicin) were concomitant therapy and reported to be similarly dosed between treatment groups.
At the time of the data cut-off date, December 31, 2015, and at a median follow-up time of 62.6 months, median survival estimates (OS, DFS from the attainment of CR, and EFS) had not yet been reached in either treatment group, which is not unexpected for a patient population with 5-year OS rates of 89% for children and 89% for AYAs (15 years to 39 years).2 The timing of assessments for CR and MRD at day 29 appears to be appropriate to capture the presence or absence of disease at the end of remission induction. Among treatment response assessments, MRD was measured using flow cytometry employed in the COG AALL07P4 study, which was reported in published guidelines as a valid method to detect disease.34
DFCI 11-001 Study
The DFCI 11-001 study was a phase II, randomized, open-label RCT. Treatment groups appeared to be balanced overall for baseline characteristics and the risk of bias arising from issues related to randomization or allocation concealment is likely low. The open-label study design may have biased outcomes with subjective assessments for the measurement of harms due to knowledge of the assigned treatment, although the direction of potential bias is unclear. AEs highlighted by the clinical experts consulted by CADTH that were especially relevant for the management of patients in clinical practice included hypersensitivity reactions, anaphylactic reactions, silent inactivation, and WDAEs. The open-label study design may have biased WDAEs due to knowledge of the assigned treatment, although the direction of potential bias is unclear.
The DFCI 11-001 trial was not designed or powered to formally assess comparative efficacy (i.e., SAA, day 29 end-of-induction MRD, CR, EFS, DFS from the attainment of CR, and OS), making assessments of the relative therapeutic benefit of pegaspargase challenging. The protocol did not prespecify a degree of difference from which to formally conclude noninferiority or similarity between calaspargase pegol and pegaspargase. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect and end points were not adjusted for multiple comparison. The clinical experts consulted by CADTH did not anticipate clinically meaningful differences in efficacy between calaspargase pegol and pegaspargase due to the products’ similar mechanisms of action and use of the same asparagine-specific enzyme (i.e., derived from E. coli).
SAA measured after a single dose was 1 of the primary end points and used the established therapeutic threshold of at least 0.1 IU/mL, which was previously set as an important limit of clinical significance and is considered a valid surrogate for complete asparagine depletion.34,51 The FDA has approved previous asparaginase products in combination with chemotherapy, including calaspargase pegol for the present target population, on the basis of accepting evidence of asparaginase activity as a “valid surrogate for clinical efficacy.”46,51 SAA levels were measured during induction in the DFCI 11-001 trial to assess calaspargase pegol’s ability to maintain an SAA level of 0.1 UI/mL or greater 3 weeks to 4 weeks after a single dose and to assess its half-life duration compared to pegaspargase. The mechanism of action of calaspargase pegol is the same as that of the established SOC pegaspargase; however, calaspargase pegol was developed with greater biological stability than pegaspargase. The clinical experts consulted by CADTH agreed that this end point serves to support calaspargase pegol’s ability to maintain asparagine suppression in plasma, its half-life duration, and its ability to be administered with less dosing frequency than pegaspargase (i.e., a schedule of every 3 weeks versus every 2 weeks).
The assessments of SAA every 3 weeks for 30 consecutive weeks during postinduction therapy in the DFCI 11-001 trial was another primary end point, with the objective of further determining how best to dose calaspargase pegol during postinduction therapy.44 As noted earlier in this report, this outcome to determine an appropriate dose was not assessed in this report as it was not identified as being 1 of the most important outcomes to guide treatment selection in clinical practice by the clinical experts consulted by CADTH. The FDA in its assessment of calaspargase pegol for this indication noted that a large amount of PK data to assess asparaginase activity in the DFCI 11-001 trial was not evaluable. Therefore, the FDA used an imputation method based on the population PK modelling and simulation that included patients with valid PK observations from the COG AALL07P4 and DFCI 11-001 trials, which indicated sufficient levels of asparaginase activity at steady state during postinduction to support the proposed dosing of every 3 weeks for calaspargase pegol.50,51
There were balanced between-group proportions of patients who were censored for survival outcomes and a low number of patients missing in the calaspargase pegol and pegaspargase groups, respectively, for CR at the end of induction day 32 (5 and 4), MRD at the end of induction at day 32 (5 and 4), and SAA levels (up to 8 and 11); overall, missing outcome data were unlikely to substantially impact findings despite the lack of imputation.
At the time of the data cut-off date, October 5, 2016, and at a median follow-up time of 26.6 months, median survival estimates (OS, DFS from the attainment of CR, and EFS) had not yet been reached in either treatment group. At the time of the updated data cut-off date of June 12, 2017, with a median follow-up time of ||||| months for the calaspargase pegol group and ||||| months for the pegaspargase group, median survival estimates had also not been reached yet in either treatment group, which is not unexpected for a patient population with 5-year OS rates of 89% for children and 89% for AYAs (15 years to 39 years).2
The timing of assessments for CR and MRD at day 32 appears to be appropriate to capture the presence or absence of disease at the end of remission induction.
In both the COG AALL07P4 study and the DFCI 11-001 study, the FAS data were used for survival outcomes, CR at the end of induction, and MRD at the end of induction. While it is preferable to rely on ITT analyses to ensure patients are included in statistical analyses according to the group they were randomized to, rather than according to the treatment they received, there were minimal differences between the FAS and ITT populations, and the analyses based on FAS or ITT were minimally different.
CR and MRD assessments using flow cytometry in the COG AALL07P4 study and a PCR assay in the DFCI 11-001 study were reported by the clinical experts to be appropriate and in line with observations in clinical practice. The time points of assessment for CR and MRD on day 29 (the COG AALL07P4 study) and day 32 (the DFCI 11 to 011 study) of the end of induction were appropriate according to the clinical experts, given the differences in duration, frequency, and phases among available treatment protocols.
Outcomes identified as important by patients and clinical experts consulted by CADTH that were not assessed included HRQoL (the COG AALL07P4 and DFCI 11-001 trials) and silent inactivation (the COG AALL07P4 trial).
External Validity
There were notable limitations for generalizability in the COG AALL07P4 and DFCI 11-001 studies. Both were phase II trials with small samples of patients with ALL enrolled. The clinical experts consulted by CADTH remarked that given the disease area and a relatively small patient population therein, a phase III RCT would neither be feasible due to challenges in accruing a sufficient sample nor ethical due to pegaspargase-asparaginase as current front-line treatment for ALL.
While patients in both trials were representative of patients with ALL, subpopulations of patients were missing in each trial. The clinical experts consulted by CADTH considered the results from the COG AALL07P4 and DFCI 11-001 studies to be generalizable to patients younger than 1 year (excluded from the COG AALL07P4 and DFCI 11-001 trials) and patients older than 21 years (excluded from the DFCI 11-001 trial) or 30 years (excluded from the COG AALL07P4 trial). According to the clinical experts consulted by CADTH, patients with T-cell immunophenotype (excluded from the COG AALL07P4 trial) would also be expected to benefit from treatment with calaspargase pegol, provided there was no known prior hypersensitivity to asparaginase. While the trials only enrolled patients with newly diagnosed ALL, the clinical experts consulted by CADTH felt that it would be reasonable to generalize the results of the COG AALL07P4 and DFCI 11-001 studies to patients with relapse or refractory disease and noted an emerging consensus treatment protocol for first relapse among pediatric patients with B-lineage ALL.2,55 Similarly, the clinical experts consulted by CADTH considered the results of the COG AALL07P4 and DFCI 11-001 studies to be generalizable to pediatric patients with Ph-positive status who were excluded from these trials. The clinical expert treating pediatric patients who was consulted by CADTH reported no concerns with using asparaginase-based treatment in pediatric patients who are given tyrosine kinase inhibitors, should current treatment protocols for ALL be expanded to include patients with Ph-positive status. However, the clinical expert consulted by CADTH who was treating adults reported avoidance of asparaginase concurrently with tyrosine kinase inhibitors due to the potential for greater toxicities. Overall, the clinical experts consulted by CADTH indicated that there was a lack of data from prospective clinical trials to inform optimal treatment for subgroups of patients and, therefore, it is anticipated that patients may benefit from treatment with calaspargase pegol based on its similarity to and extrapolation of findings for pegaspargase. In general, the populations enrolled in the COG AALL07P4 and DFCI 11-001 trials were considered by the clinical experts consulted by CADTH to be representative of patients with ALL. The clinical experts consulted by CADTH anticipated that calaspargase pegol would be used in patients who would otherwise receive a pegaspargase-containing MAC.
Different backbone therapies were employed, with an intermittent asparagine depletion versus continuous asparagine depletion protocol for the COG AALL07P4 study versus the DFCI 11-001 study, respectively. Differences in treatment protocols between the trials were echoed by the clinical experts consulted by CADTH to be observed in clinical practice, acknowledging that ALL is the most common malignancy in pediatrics with commonly adopted protocols but lacking formal established national standards. The clinical expert consulted by CADTH treating pediatric patients with ALL reported that COG-based protocols are employed by institutions across Canada except in Quebec, where DFCI-based protocols are used. A modified DFCI protocol was reported by the clinical expert consulted by CADTH treating adults with ALL to be used at a centre in Alberta. The clinical experts consulted by CADTH agreed that they expected outcomes to be similar across all treatment protocols for ALL.
Outcomes reported in the COG AALL07P4 and DFCI 11-001 trials appeared to be aligned with outcomes of interest for patients with ALL according to the clinical experts consulted by CADTH. In the COG AALL07P4 trial, attaining M1 bone marrow status (fewer than 5% malignant lymphoblasts) was used to define CR at the end of induction. In the DFCI 11-001 trial, fewer than 1% malignant lymphoblasts was used to define CR at the end of induction. According to the clinical experts consulted, an M1 bone marrow status is used to determine clinically meaningful remission among children and adults with ALL. As such, the use of 1% as a cut-off for disease detection appears to be conservative when compared with a 5% detection cut-off. The clinical experts consulted by CADTH reported that the total duration of MAC treatment for ALL is approximately 2.5 years to 3.5 years for patients who respond, with asparaginase being used within the first year of treatment. Therefore, survival outcomes assessed at 1 year are likely insufficient for follow-up. There was an absence of established MIDs for all outcomes, and the clinical experts consulted by CADTH did not provide expert opinion–based MIDs for most outcomes. According to the clinical experts consulted by CADTH, any decrease in DFS from the attainment of CR is clinically important. With respect to CR at the end of induction, the clinical experts consulted by CADTH reported a 5% difference between groups to be clinically meaningful. Evaluations of SAA levels were not reported by the clinical experts consulted by CADTH to be mandated in practice, with variable adoption across centres. The clinical experts highlighted the importance of SAA levels in TDM for both efficacy (i.e., adequate asparagine depletion and sustained SAA levels of 0.10 IU/mL or greater) and safety (e.g., hypersensitivity reactions including silent inactivation).
In general, the clinical experts consulted by CADTH commented that they did not anticipate clinically meaningful differences in efficacy between calaspargase pegol and pegaspargase.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:10,11
“High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word ‘likely’ for evidence of moderate certainty (e.g., ‘X intervention likely results in Y outcome’).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word ‘may’ for evidence of low certainty (e.g., ‘X intervention may result in Y outcome’).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as ‘very uncertain.’”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and/or publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of a clinically important effect for CR at the end of induction based on a threshold informed by the clinical experts consulted by CADTH for this review. The target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for survival rates (OS, DFS from CR, EFS), MRD at the end of induction, SAA during induction, and harms.
For the GRADE assessments, findings from the COG AALL07P4 and DFCI 11-001 studies were assessed individually because the trials enrolled different patient populations (patients with high-risk B-cell ALL in the COG AALL07P4 study and patients with ALL and LL in the DFCI 11-001 study) and employed different treatment protocols (intermittent asparagine depletion in the COG AALL07P4 study and continuous asparagine depletion in the DFCI 11-001 study).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for calaspargase pegol versus pegaspargase in patients with high-risk B-cell ALL in the COG AALL07P4 trial. Table 3 presents the GRADE summary of findings for calaspargase pegol versus pegaspargase in patients with ALL in the DFCI 11-001 trial.
Discussion
Summary of Available Evidence
Two phase II, multicentre, randomized, open-label trials assessed the efficacy and safety of calaspargase pegol 2,500 IU/m2 compared with pegaspargase 2,500 IU/m2. The COG AALL07P4 trial enrolled 166 patients aged 1 year to 30 years in 23 study sites, all located in the US, with newly diagnosed high-risk B-cell ALL. The primary objective of the COG AALL07P4 trial was to determine the PK comparability (asparaginase activity) of the interventions during induction and consolidation while patients were receiving augmented Berlin-Frankfurt-Münster therapy. Secondary objectives of the COG AALL07P4 study included PD parameters during induction and consolidation, MRD (day 29), CR rate (day 29), survival (EFS, DFS of CR, and OS), and TEAEs. The DFCI 11-001 study enrolled 239 patients aged 1 year to 21 years in the US (in 6 sites) and Canada (in 3 sites) with newly diagnosed ALL or LL. The primary objective of the DFCI 11-001 study was to determine the PK comparability of the interventions during remission induction and postinduction (i.e., determine SAA levels and assess toxicity). Secondary objectives of the DFCI 11-001 study included MRD (day 32), CR rate (day 32), EFS, DFS from the attainment of CR, and OS.
In the COG AALL07P4 trial, most patients were older than 10 years (66.3%), male (50.9%), and white (82.2%). In the DFCI 11-001 trial, most patients were younger than 10 years (75%), male (61.6%), and white (70.5%). Across both the COG AALL07P4 trial and the DFCI 11-001 trial, there were notable similarities and differences in baseline demographics. All patients in the COG AALL07P4 trial and nearly all patients in the DFCI 11-001 trial (96%) had ALL. Most patients in the DFCI 11-001 study had B-cell ALL (87%), including patients with T-cell ALL (13%), whereas the COG AALL07P4 study was limited to patients with B-cell immunophenotype. Most patients had a CNS status of 1 in both trials with a minority of patients designated as CNS3 in the COG AALL07P4 study (fewer than 10%) and the DFCI 11-001 study (fewer than 2%). Most patients did not have steroid therapy before study treatment in the COG AALL07P4 trial (93%) or the DFCI 11-001 trial (84%). While the majority of patients were older than 10 years in the COG AALL07P4 trial (median = 11 years), most were younger than 10 years in the DFCI 11-001 trial (median = 4 years to 5 years). Approximately 62% of patients were diagnosed when they were 10 years or older in the COG AALL07P4 study, whereas nearly 75% of patients were diagnosed younger than 10 years in the DFCI 11-001 study. Patients in the COG AALL07P4 study were distributed equally across combined age and WBC categories, whereas more than 70% of patients in the DFCI 11-001 study were younger than 10 years with a WBC count below 50 × 109/L.
Interpretation of Results
Efficacy
All outcomes in the COG AALL07P4 trial and most outcomes in the DFCI 11-001 trial were secondary end points without adjustment for multiplicity, and therefore considered as supportive evidence for assessments of certainty in effect estimates. Additionally, the relatively small samples of patients enrolled in these phase II trials resulted in wide CIs across most outcome effect estimates.
As the aim of treatment for patients with ALL is curative, survival was highlighted as an important outcome by patients, the clinical experts consulted by CADTH, and clinicians. Prevention of or delayed disease progression was also identified as important to patients. As such, the survival outcomes of OS, DFS from CR, and EFS appear appropriate and were captured in both the COG AALL07P4 trial and the DFCI 11-001 trial. However, the duration of follow-up for OS, DFS from CR, and EFS at 1 year was insufficient to adequately capture the full impact of treatment on patients. While asparaginase is incorporated into the first phases of MAC treatment over the first year, the clinical experts consulted by CADTH estimated that the total duration of MAC treatment for patients with ALL who respond is approximately 2.5 years to 3.5 years. |||||| ||||||||| || |||||||| ||| |||||| || | ||||| || |||| |||||| ||| | ||||| || ||| |||||||| ||| ||| ||||||| |||||| ||| ||| |||| ||| || ||| || ||| ||||| |||||| According to a publication by Vrooman et al. (2021),42 5-year OS was estimated at 94.0% (standard error = 2.2) for calaspargase pegol compared with 95.8% (standard error = 1.9) for pegaspargase (P value for difference between groups = 0.72); however, there was significant uncertainty for these findings, which lacked numerical values for the number of OS events and a date for data cut-off.42 The review team was unable to rigorously assess the conduct and reporting of the 5-year OS analyses, due to the limited data available in the publication.
CR and MRD assessments using flow cytometry in the COG AALL07P4 study and a PCR assay in the DFCI 11-001 study were reported to be appropriate by the clinical experts consulted by CADTH and in line with observations in clinical practice. The time points of assessment for CR and MRD on day 29 (the COG AALL07P4 study) and day 32 (the DFCI 11 to 011 study) of the end of induction were appropriate according to the clinical experts consulted by CADTH, given the differences in duration, frequency, and phases among available treatment protocols.
SAA measurements are considered to be a surrogate for the efficacy of asparaginase formulations.46 The use of 0.10 IU/mL as a marker of therapeutic asparaginase activity (levels above 0.10 IU/mL have been shown to be associated with complete asparagine depletion and therefore, therapeutic benefit) aligns with consensus expert recommendations34 and input from clinical experts consulted by CADTH. The consensus panel noted that increased access to real-time validated asparaginase measurements (e.g., via regulatory-approved assays) has enabled more reliable asparaginase activity monitoring in clinical practice, highlighting SAA assessment as important in detecting allergic reactions, silent inactivation, and the potential for modifying dosing of asparaginase treatment.34
The results of calaspargase pegol compared with pegaspargase appeared generally similar for survival, including OS, DFS from CR, EFS, and MRD at the end of induction. The findings suggested that calaspargase pegol may result in an increase in CR at the end of induction compared with pegaspargase. There was uncertainty in the effect estimates for which the majority of outcomes suggested little to no difference between treatment groups because estimates were based on small sample sizes and few events, and CIs that suggested the possibility of benefit, harm, or both. There was a potential risk of bias arising from patient-reported and assessor-reported WDAEs due to the subjective nature of assessments and knowledge of treatment assignment in the open-label study design.
An MRD level of 0.1% has been accepted by the FDA to identify patients with ALL with a high risk of relapse.46 The clinical experts consulted by CADTH agreed that the end of induction MRD is 1 of the most clinically meaningful predictors of outcomes in ALL and is used as part of standard-risk stratification strategies for patients with ALL. The clinical experts consulted by CADTH considered the results for the end of induction MRD to be relevant and indicative of calaspargase pegol resulting in no decline in the percentage of patients who attain an early response compared with pegaspargase.
In the COG AALL07P4 trial, PAA levels were measured (measured on day 4, day 15, day 22, day 29, and day 43 of induction; this included the proportion of patients with a PAA level of 0.10 IU/mL or greater [and the proportion of patients with a PAA level of 0.40 IU/mL or greater] for each study group). PAA results were displayed in ||||| || in Appendix 1. According to the clinical experts consulted by CADTH, PAA is not currently measured in clinical practice and not used to make treatment selection in clinical practice. Generally, it is not feasible to accurately measure PAA outside the context of a clinical trial. Therefore, a surrogate measure, SAA, has been developed, which is an accepted and valid therapeutic drug assay. PAA is the most direct way to determine if a patient attained an adequate response to asparaginase therapy and as such, may also serve to assess a drug’s ability to maintain asparagine suppression in plasma.14 In the COG AALL07P4 study, the majority of patients in each treatment group had PAA of 0.1 IU/mL or greater through 18 days following the induction dose. By 25 days following the induction dose, there was a slight decrease in the proportion of patients with PAA of 0.1 IU/mL or greater for calaspargase pegol ||||||| and a larger decrease for patients in the pegaspargase ||||||| group. The results appear in line with the SAA results in the DFCI 11-001 study, which are supportive of calaspargase pegol’s ability to maintain asparaginase activity above 0.1 IU/mL with a longer half-life compared to pegaspargase.
The effects of calaspargase pegol were similar to pegaspargase for OS, DFS from CR, EFS, CR at the end of induction, MRD at the end of induction, and SAA levels. SAA levels at day 32 during induction suggested that calaspargase pegol likely resulted in a greater proportion of patients with an SAA level of at least 0.10 IU/mL compared to pegaspargase. Overall, there was uncertainty in the effect estimates for which the majority of outcomes suggested little to no difference between treatment groups, based on small sample sizes and few events, and CIs that suggested the possibility of benefit, harm, or both.
The clinical experts consulted by CADTH considered the results for SAA measured during induction to be relevant and indicative of calaspargase pegol’s greater biological stability compared with pegaspargase, calaspargase pegol’s ability to maintain asparagine suppression in plasma, its longer half-life duration compared to pegaspargase, and its ability to be administered with less dosing frequency than pegaspargase (i.e., a schedule of every 3 weeks versus every 2 weeks).
Though the DFCI 11-001 study was not designed to formally assess comparative treatment effects in SAA levels between calaspargase pegol and pegaspargase, the mechanism of action of calaspargase pegol is equivalent to that of pegaspargase, in addition to calaspargase pegol being designed with greater biological stability, which supports the plausibility of the link between the drug and the outcome.
In the DFCI 11-001 study, the results for MRD at the end of induction for 0.01 or greater were supportive of calaspargase pegol resulting in no decline in the percentage of patients who attain an early response compared with pegaspargase. In addition, the results for high end-of-induction MRD (defined as ≥ 0.001 by PCR assay) and low end-of-induction MRD (defined as < 0.001) are included in Table 18 in Appendix 1 as they were not identified by the clinical experts as being 1 of the most important end points. The results of the proportion of patients who attained low end-of-induction MRD further support the findings observed for MRD at the end of induction in the COG AALL07P4 trial that calaspargase pegol does not result in a decline in the proportion of patients who attain an early response compared with pegaspargase. The proportion of participants in the FAS with evaluable MRD was similar between the calaspargase pegol and pegaspargase groups: ||||| |||||| ||||| ||| ||| ||||| |||||| ||||| |||||
The clinical experts consulted by CADTH noted the similarities between calaspargase pegol and pegaspargase in moiety and within the context of COG-based treatment protocols; they did not anticipate a clinically meaningful difference in efficacy outcomes overall between the asparaginase drugs. Given that the body of evidence across the COG AALL07P4 study and the DFCI 11-001 study demonstrated similarities in outcomes between treatment groups, the clinical experts consulted by CADTH agreed that there is potential for a more consistent supply of asparaginase (due to the increased shelf life of calaspargase pegol) and reduced frequency of drug administration, which was identified as important to patients.
It is challenging to adequately interpret the findings considering the differences in patient populations and backbone treatment protocols, where findings presented in the COG AALL07P4 trial and the DFCI 11-001 trial were mainly applicable to patients aged 1 year to 30 years with high-risk B-cell ALL with augmented Berlin-Frankfurt-Münster treatment and to patients aged 1 year to 21 years with ALL with an intermittent asparagine depletion treatment protocol, respectively. Findings from the COG AALL07P4 and DFCI 11-001 trials may be generalized to patients with ALL of all ages, patients with relapsed or refractory ALL, patients with T-cell immunophenotype, and patients with LL based on input from clinical experts consulted by CADTH. However, according to the clinical experts consulted by CADTH for the review, there is uncertainty in whether specific subgroups of patients may derive greater benefit from treatment with calaspargase pegol in the absence of data.
HRQoL was identified as an important outcome by patients and clinical experts consulted by CADTH but was not assessed in the COG AALL07P4 study or the DFCI 11-001 study. Input from patients who experienced a drug shortage at some point during their ALL treatment highlighted poor HRQoL and financial impacts arising from the requirement to pay for alternative therapies and products to help them cope. According to patients, having alternative treatments available to ensure continued treatment was important to patients and their caregivers during an already difficult and challenging time. The clinical experts consulted by CADTH agreed that they did not expect significant differences between calaspargase pegol and pegaspargase in terms of HRQoL outcomes.
Harms
The safety profile of calaspargase pegol and pegaspargase showed similar proportions of patients with TEAEs, SAEs including grade 3 or grade 4 SAEs, and deaths among patients aged 1 year to 30 years with high-risk B-cell ALL in the COG AALL07P4 study and among patients aged 1 year to 21 years with ALL in the DFCI 11-001 study. While the number of patients who stopped study treatment due to an AE was similar between treatment groups in the COG AALL07P4 trial, there was a higher rate of WDAEs in the calaspargase pegol group compared with pegaspargase in the DFCI 11-001 trial (28.0% versus 19.3%). WDAEs were due to higher rates of hypersensitivity, increased amylase, and anaphylactic reactions in the calaspargase pegol group, whereas higher rates of increased lipase and drug hypersensitivity led to withdrawals by patients in the pegaspargase group. AESIs included hypersensitivity reactions, anaphylactic reactions, silent inactivation, pancreatitis, venous thrombosis, hemorrhage, and hepatotoxicity. The frequency of hypersensitivity reactions for calaspargase pegol compared with pegaspargase was lower in the COG AALL07P4 study ||||| |||||| |||||| and similar between groups in the DFCI 11-001 study |||||| |||||| ||||||| Anaphylactic reactions were reported by more patients in the COG AALL07P4 trial (25.6% versus 19.2%) but with equal frequency in the DFCI 11-001 trial (|||| ||| |||| ||||||) in the calaspargase pegol group and the pegaspargase group, respectively. Silent inactivation was not reported by any patient in the COG AALL07P4 study but was reported by 2 patients (1.7%) in the calaspargase pegol group and 0 patient in the pegaspargase group in the DFCI 11-001 study. Pancreatitis was reported by more patients in the COG AALL07P4 study (18.6% versus 7.7%) but by fewer patients in the DFCI 11-001 study (11.9% versus 16.8%) in the calaspargase pegol group and the pegaspargase group, respectively. With respect to thrombosis events, there were higher rates of increased blood bilirubin in both the COG AALL07P4 trial (62.8% versus 50.0%) and the DFCI 11-001 trial (45.8% versus 43.7%), and increased alanine aminotransferase in both the COG AALL07P4 trial (34.9% versus 38.5%) and the DFCI 11-001 trial (78.8% versus 77.3%) in the calaspargase pegol group and the pegaspargase group, respectively. | |||||| ||||||| ||||||||||| ||||||| ||||||| || |||| |||||| || ||| |||||||||||| ||||| |||||||||||.
The draft product monograph for calaspargase pegol included serious warnings and precautions for hypersensitivity, pancreatitis, thrombosis, hemorrhage, and hepatotoxicity;36 these were therefore included as AESIs in the review. No patient experienced venous thrombosis in the COG AALL07P4 trial or hemorrhage in the COG AALL07P4 trial or the DFCI 11-001 trial.
Patients expressed side effects as 1 of the factors that was important to them when evaluating new treatments for ALL. Overall, there were small sample sizes of enrolled patients in the COG AALL07P4 and DFCI 11-001 studies with similar rates of TEAEs overall and differing rates of notable harms between groups and across studies. The clinical experts consulted by CADTH noted that since asparaginase is given as part of an MAC protocol, it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component. The clinical experts consulted by CADTH acknowledged that several AEs captured by the COG AALL07P4 and DFCI 11-001 studies were not specific to asparaginase and may be attributed to an intensive (versus intermittent) treatment strategy (e.g., hypoalbuminemia), a specific MAC component (e.g., stomatitis may be attributed to anthracycline treatment), or to the disease itself (e.g., febrile neutropenia is commonly experienced by patients with ALL). According to the clinical experts consulted by CADTH, hyperglycemia during induction and late hyperbilirubinemia were the only AEs that occurred at a higher rate with calaspargase pegol; these were AEs that were expected to occur with asparaginase preparations and were considered to be manageable. Overall, the clinical experts consulted by CADTH did not anticipate significant differences in harms between calaspargase pegol and pegaspargase. The FDA assessment report for calaspargase pegol in combination with chemotherapy for ALL did not identify any unexpected or unacceptable safety issues.51
Conclusion
Evidence from a randomized phase II, open-label trial (the COG AALL07P4 study) in patients aged 1 year to 30 years with high-risk B-cell ALL suggested that calaspargase pegol likely results in little to no difference in DFS rates from the attainment of CR at 4 years (moderate certainty) and may result in an increase in CR at the end of induction (low certainty) compared with pegaspargase. Furthermore, the COG AALL07P4 trial suggested that compared with pegaspargase, calaspargase pegol may result in little to no difference in OS rates at 1 year and 4 years, DFS rates from the attainment of CR at 1 year, EFS rates at 1 year and 4 years, and MRD at the end of induction (low certainty).
Evidence from a randomized phase II, open-label trial (the DFCI 11-001 trial) in patients aged 1 year to 21 years with ALL suggested that calaspargase pegol likely results in a greater proportion of patients with SAA during the induction phase of at least 0.10 IU/mL at day 32 (moderate certainty) when compared with pegaspargase and likely results in little to no difference in OS rates at 1 year, in DFS rates from the attainment of CR at 2 years, EFS rates at 2 years, and CR at the end of induction (moderate certainty) compared with pegaspargase. Furthermore, the DFCI 11-001 study suggested that compared with pegaspargase, calaspargase pegol may result in little to no difference in OS at 2 years, DFS from the attainment of CR at 1 year, EFS at 1 year, and MRD at the end of induction (low certainty).
SAA is an established surrogate measure of asparagine depletion. SAA levels above 0.10 IU/mL have been shown to be associated with complete asparagine depletion and therefore, therapeutic benefit. The clinical experts consulted by CADTH considered the results for SAA measured during induction to be supportive of calaspargase pegol’s greater biological stability compared with pegaspargase, calaspargase pegol’s ability to maintain asparagine suppression in plasma, its longer half-life duration compared to pegaspargase, and its ability to be administered with less dosing frequency than pegaspargase (i.e., a schedule of every 3 weeks versus every 2 weeks).
No unexpected safety signals were identified with treatment of calaspargase pegol in the COG AALL07P4 trial or the DFCI 11-001 trial. The clinical experts consulted by CADTH noted that asparaginase is given as part of an MAC protocol and it is challenging to attribute differences in AEs observed in small numbers of patients to a single treatment protocol component. The COG AALL07P4 and DFCI 11-001 studies did not report data on HRQoL.
Confidence in the effect estimates from the COG AALL07P4 and DFCI 11-001 studies was limited, primarily due to both studies not being designed to assess comparative efficacy between calaspargase pegol and pegaspargase, relatively small sample sizes, and the open-label design, which may have biased subjective outcomes such as harms.
References
1.Canadian Cancer Society. What is acute lymphoblastic leukemia? [2023]; https://cancer.ca/en/cancer-information/cancer-types/acute-lymphoblastic-leukemia-all. Accessed 2023 May 2.
2.Brown PA, Shah B, Advani A, et al. Acute Lymphoblastic Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19(9):1079-1109. PubMed
3.Statistics Canada. Number and rates of new cases of primary cancer, by cancer type, age group and sex [accessed by sponsor]. https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310011101. Accessed 2022 Jan 5.
4.Statistics Canada. Population estimates on July 1st, by age and sex [accessed by sponsor]. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2022 Jan 5.
5.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6):e577. PubMed
6.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. PubMed
7.Zuckerman T, Rowe JM. Pathogenesis and prognostication in acute lymphoblastic leukemia. F1000Prime Rep. 2014;6:59. PubMed
8.Gupta S, Wang C, Raetz EA, et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group. J Clin Oncol. 2020;38(17):1897-1905. PubMed
9.Maese L, Rau RE. Current Use of Asparaginase in Acute Lymphoblastic Leukemia/Lymphoblastic Lymphoma. Front Pediatr. 2022;10:902117. PubMed
10.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
11.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
12.Asparlas (calaspargase pegol) as a component of a multi-agent chemotherapeutic (MAC) regimen for the treatment of patients with acute lymphoblastic leukemia (ALL): clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Asparlas (calaspargase pegol), 3,750 units/5 mL (750 units/mL), solution for IV infusion. Laval (QC): Servier Canada Inc.; 2023 May 12.
13.COG AALL07P4 Clinical Study Report: A Pilot Study of Intravenous EZN-2285 (SC PEG E. coli L asparaginase, IND 100594) or Intravenous Oncaspar in the Treatment of Patients with High-Risk Acute Lymphoblastic Leukemia (ALL): A Limited Institution Pilot Study [internal sponsor's report]. Westlake Village (CA): Baxalta US Inc.; 2017 Oct 30.
14.Servier Canada Inc. response to June 23, 2023 CADTH request for additional information regarding calaspargase pegol CADTH review [internal additional sponsor's information]. Laval (QC): Servier Canada Inc.; 2023.
15.DFCI 11-001 Clinical Study Report: Randomized Study of Intravenous Calaspargase Pegol (SC-PEG-L-asparaginase) and Intravenous Oncaspar in Children and Adolescents with Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma [internal sponsor's report]. Boston (MA): Baxalta US Inc.; 2017 Nov 29.
16.Zimmermann C, Yuen D, Mischitelle A, et al. Symptom burden and supportive care in patients with acute leukemia. Leuk Res. 2013;37(7):731-736. PubMed
17.Alberta Health Services. Clinical Practice Guideline LYHE-005 – Version 2. Acute Lymphoblastic Leukemia in Adults. Effective date: June, 2022 [accessed by sponsor]. 2022; https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe005-all.pdf. Accessed 2022 Jun.
18.Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146-1162. PubMed
19.National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Acute Lymphoblastic Leukemia Version 1.2022 — April 4, 2022 [accessed by sponsor]. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1410.
20.Cooper SL, Young DJ, Bowen CJ, Arwood NM, Poggi SG, Brown PA. Universal premedication and therapeutic drug monitoring for asparaginase-based therapy prevents infusion-associated acute adverse events and drug substitutions. Pediatr Blood Cancer. 2019;66(8):e27797. PubMed
21.Juluri KR, Siu C, Cassaday RD. Asparaginase in the Treatment of Acute Lymphoblastic Leukemia in Adults: Current Evidence and Place in Therapy. Blood Lymphat Cancer. 2022;12:55-79. PubMed
22.Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood. 2015;125(24):3702-3710. PubMed
23.Stock W, Luger SM, Advani AS, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood. 2019;133(14):1548-1559. PubMed
24.Marini BL, Brown J, Benitez L, et al. A single-center multidisciplinary approach to managing the global Erwinia asparaginase shortage. Leuk Lymphoma. 2019;60(12):2854-2868. PubMed
25.Institut national d’excellence en santé et en services sociaux (INESSS). Oncaspar – Leucémie lymphoblastique aigüe. Avis transmis au ministre en janvier 2023. 2023; https://www.inesss.qc.ca/thematiques/medicaments/medicaments-evaluation-aux-fins-dinscription/extrait-davis-au-ministre/oncaspar-leucemie-lymphoblastique-6187.html. Accessed 2023 Feb 16.
26.Baruchel A, Brown P, Rizzari C, et al. Increasing completion of asparaginase treatment in childhood acute lymphoblastic leukaemia (ALL): summary of an expert panel discussion. ESMO Open. 2020;5(5):e000977. PubMed
27.World Health Organization. WHO Model Lists of Essential Medicines [accessed by sponsor]. 2022; https://www.who.int/groups/expert-committee-on-selection-and-use-of-essential-medicines/essential-medicines-lists. Accessed 2022 Jan 8.
28.Siegel SE, Stock W, Johnson RH, et al. Pediatric-Inspired Treatment Regimens for Adolescents and Young Adults With Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia: A Review. JAMA Oncol. 2018;4(5):725-734. PubMed
29.Berger F, Pileri SA, Harris NL, Jaffe ES, Stein H. World Health Organization Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon (FR): International Agency for Research on Cancer; 2008.
30.Buske C, Leblond V. How to manage Waldenstrom's macroglobulinemia. Leukemia. 2013;27(4):762-772. PubMed
31.Pieters R, Hunger SP, Boos J, et al. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011;117(2):238-249. PubMed
32.Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(3):185-196. PubMed
33.Bleyer A, Asselin BL, Koontz SE, Hunger SP. Clinical application of asparaginase activity levels following treatment with pegaspargase. Pediatr Blood Cancer. 2015;62(6):1102-1105. PubMed
34.van der Sluis IM, Vrooman LM, Pieters R, et al. Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica. 2016;101(3):279-285. PubMed
35.Drug Reimbursement Review sponsor submission: Asparlas (calaspargase pegol), 3,750 units/mL (750 units/mL) injection for intravenous use [internal sponsor's package]. Laval (QC): Servier Canada Inc.; 2023 May 12.
36.Asparlas (calaspargase pegol): 3,750 units/5 mL (750 units/mL) injection [draft product monograph]. Laval (QC): Servier Canada Inc.; 2023.
37.Oncaspar (pegaspargase): 3,750 U/5 mL (750 U/mL) injection [product monograph]. Laval (QC): Servier Canada Inc.; 2017 Feb 24: https://pdf.hres.ca/dpd_pm/00038396.PDF. Accessed 2023 August 8.
38.Silverman LB, Blonquist TM, Hunt SK, et al. Randomized Study of Pegasparagase (SS-PEG) and Calaspargase Pegol (SC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001. Blood. 2016;128(22):175.
39.Schore RJ, Devidas M, Bleyer A, et al. Anti-Pegaspargase, Anti-Calaspargase Pegol, and Anti-Polyethelene Glycol Antibody Incidence in High Risk Acute Lymphoblastic Leukemia Patients Receiving Pegaspargase or Calaspargase Pegol and Associated Anaphylactic or Hypersensitivity Reaction Rates: Results from Children's Oncology Group (COG) Study AALL07P4. Blood. 2016;128(22):3965.
40.Angiolillo AL, Schore RJ, Devidas M, et al. Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children's Oncology Group Study AALL07P4. J Clin Oncol. 2014;32(34):3874-3882. PubMed
41.Angiolillo AL, Schore RJ, Reaman GH, et al. Pharmacokinetic (PK) and pharmacodynamics (PD) properties of SC-PEG e. coli L-asparaginase (EZN-2285) in the treatment of patients with acute lymphoblastic leukemia (ALL): Results from Children's Oncology Group (COG) study AALL07P4. J Clin Oncol. 2012;30(15_suppl):9543-9543.
42.Vrooman LM, Blonquist TM, Stevenson KE, et al. Efficacy and Toxicity of Pegaspargase and Calaspargase Pegol in Childhood Acute Lymphoblastic Leukemia: Results of DFCI 11-001. J Clin Oncol. 2021;39(31):3496-3505. PubMed
43.Vrooman LM, Blonquist TM, Supko JG, et al. Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia/lymphoma: Results of DFCI 11-001. J Clin Oncol. 2019;37(15_suppl):10006-10006.
44.DFCI 11-001 Protocol: Randomized Study of Intravenous Calaspargase Pegol (SC-PEG asparaginase) and Intravenous Oncaspar in Children and Adolescents with Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma [internal sponsor's report]. Boston (MA): Sigma-Tau Pharmaceuticals Inc.; 2016 Feb 24.
45.Maloney KW, Devidas M, Wang C, et al. Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0331. J Clin Oncol. 2020;38(6):602-612. PubMed
46.Center for Drug Evaluation Research. Hematologic Malignancies: Regulatory Considerations for Use of Minimal Residual Disease in Development of Drug and Biological Products for Treatment, Guidance for Industry. Silver Spring (MD): U. S. Food and Drug Administration (FDA); 2020 Jan: https://www.fda.gov/media/134605/download. Accessed 2023 Aug 28.
47.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111(12):5477-5485. PubMed
48.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood. 2015;126(8):964-971. PubMed
49.Coustan-Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96(8):2691-2696. PubMed
50.Li RJ, Jin R, Liu C, et al. FDA Approval Summary: Calaspargase Pegol-mknl For Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults. Clin Cancer Res. 2020;26(2):328-331. PubMed
51.Center for Drug Evaluation Research. Multi-discipline review(s). Asparlas (Calaspargase pegol-mknl), 3,750 units/mL (750 units/mL) injection for intravenous use. Company: Servier Pharmaceutical, LLC. Application No.: BLA 761102. Approval date: 12/19/2018 (FDA drug approval package). Silver Spring (MD): U. S. Food and Drug Administration (FDA); 2018: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761102Orig1s000MultidisciplineR.pdf. Accessed 2023 Aug 28.
52.Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15(6):657-680. PubMed
53.Place AE, Stevenson KE, Vrooman LM, et al. Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol. 2015;16(16):1677-1690. PubMed
54.SC-PEG-L-asparaginase (Calaspargase Pegol) Study DFCI 11-001 Day 120 Report [internal sponsor's report]. Boston (MA): Baxalta US Inc.; 2018 Apr 16.
55.Hogan LE, Brown PA, Ji L, et al. Children's Oncology Group AALL1331: Phase III Trial of Blinatumomab in Children, Adolescents, and Young Adults With Low-Risk B-Cell ALL in First Relapse. J Clin Oncol. 2023;41(25):4118-4129. PubMed
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Figure 10: Treatment Protocol for the COG AALL07P4 Study
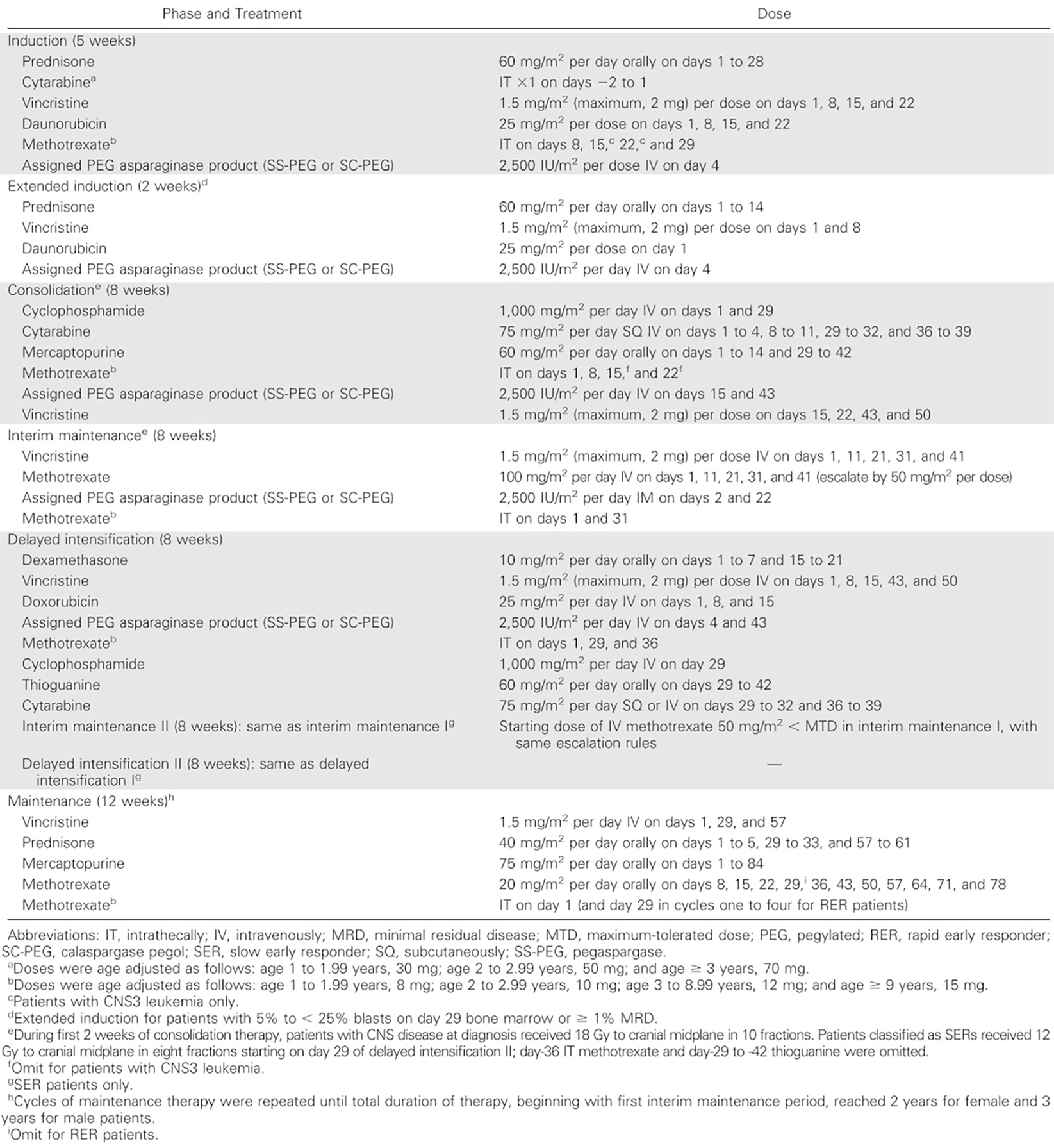
Sources: The sponsor’s Summary of Clinical Evidence12 and Angiolillo et al. (2014).40 Reproduced with permission from Wolters Kluwer Health, Inc.: Anne L. Angiolillo, Reuven J. Schore, Meenakshi Devidas, Michael J. Borowitz, Andrew J. Carroll, Julie M. Gastier-Foster, Nyla A. Heerema, Taha Keilani, Ashley R. Lane, Mignon L. Loh, Gregory H. Reaman, Peter C. Adamson, Brent Wood, Charlotte Wood, Hao W. Zheng, Elizabeth A. Raetz, Naomi J. Winick, William L. Carroll, and Stephen P. Hunger. Pharmacokinetic and Pharmacodynamic Properties of Calaspargase Pegol Escherichia coli L-Asparaginase in the Treatment of Patients With Acute Lymphoblastic Leukemia: Results From Children’s Oncology Group Study AALL07P4, Journal of Clinical Oncology, Volume 32, Issue 34, page 3874 to 3882. Available at: https://ascopubs.org/doi/10.1200/JCO.2014.55.5763
Figure 11: Treatment Protocol for the DFCI 11-001 Study

6-MP = 6-mercaptopurine; ALL = acute lymphoblastic leukemia; ASP = asparaginase, calaspargase, or calaspargase pegol; CR = complete remission; DEX = dexamethasone; DFCI = Dana-Farber Cancer Institute; DOX = doxorubicin; HD-MTX = high-dose methotrexate; HR = high risk; IM = intramuscular; IT = intrathecal; IT-M = IT methotrexate; IT-MAH = IT methotrexate/cytarabine/hydrocortisone; MTX = methotrexate; PO = orally; SR = standard risk; VCR = vincristine; VHR = very high risk.
a Patients with CNS leukemia at diagnosis (CNS-2 and CNS-3) received intrathecal cytarabine 2 times per week until CSF was clear of malignant cells on 3 consecutive examinations.
b Not administered if high-dose methotrexate started within 72 hours of intrathecal methotrexate given on day 32 of induction.
c According to random assignment: calaspargase pegol 2,500 IU/m2/dose IV every 3 weeks (10 total doses) or pegaspargase 2,500 IU/m2/dose IV every 2 weeks (15 total doses). Continued at designated interval across treatment phases through consolidation phase II once started (within CNS therapy phase for SR and HR patients, within consolidation IC for VHR patients).
d IT methotrexate/cytarabine/hydrocortisone administered every 18 weeks in consolidation phase II and continuation phases for those who received previous cranial radiation.
Sources: The sponsor’s Summary of Clinical Evidence12 and Vrooman et al. (2021).42 Reproduced with permission from Wolters Kluwer Health, Inc.: Lynda M. Vrooman, Traci M. Blonquist, Kristen E. Stevenson, Jeffrey G. Supko, Sarah K. Hunt, Sarah M. Cronholm, Victoria Koch, Samantha Kay-Green, Uma H. Athale, Luis A. Clavell, Peter D. Cole, Marian H. Harris, Kara M. Kelly, Caroline Laverdiere, Jean-Marie Leclerc, Bruno Michon, Andrew E. Place, Marshall A. Schorin, Jennifer J. G. Welch, Donna S. Neuberg, Stephen E. Sallan, and Lewis B. Silverman. Efficacy and Toxicity of Pegaspargase and Calaspargase Pegol in Childhood Acute Lymphoblastic Leukemia: Results of DFCI 11-001, Journal of Clinical Oncology, Volume 39, Issue 31, page 3496 to 3505. Available at: https://ascopubs.org/doi/full/10.1200/JCO.20.03692
Prior Therapy for Determining Patient Eligibility and Risk Stratification
In the COG AALL07P4 study, prior steroid therapy determined eligibility and risk stratification in the following manner:
For patients receiving steroids within the week preceding diagnosis:
Patients who had received oral or IV steroids for less than 48 hours during the week immediately before diagnosis were eligible for classification and randomization if the results of a complete blood count (CBC) obtained before the initiation of steroid therapy (≤ 72 hours before steroids) were available, and the necessary fluorescence in situ hybridization, cytogenetic, and molecular test results were interpretable. The presteroid CBC and age of the patient were used to determine National Cancer Institute-Rome risk classification (standard-risk versus high-risk ALL).
If a patient had received oral or IV steroids for 48 hours or more during the week immediately before diagnosis (and the results of a presteroid CBC were available to assign National Cancer Institute risk group), the patient was treated as a slow early responder and was nonrandomly assigned to the augmented regimen (early response as judged by day 8 or day 15 marrow may be influenced by the steroid pre-treatment).
In the absence of a presteroid CBC, patients who had received less than 48 hours of steroids were assigned to the high-risk protocol because the presenting WBC count may have been influenced by steroid pretreatment. These patients were eligible for randomization in the high-risk study. As expected, patients with a slow early response were assigned to the full augmented BFM treatment group.
In the absence of a presteroid CBC, patients who had received more than 48 hours of steroids were treated as a slow early responder on the high-risk study and were assigned to the full augmented group.
Inhalational steroids were not considered pre-treatment.
Patients receiving steroids within 1 month of diagnosis (e.g., week –4 to week –1):
Patients who received less than 48 hours of steroids did not have their risk assignment changed.
Patients who received more than 48 hours of steroids in week –4 to week –1 were assigned to the high-risk protocol and were eligible for randomization.
Table 17: Summary of Key Efficacy Results From the Studies Included in the Systematic Review
Outcome | COG AALL07P4 study (ITT population) | DFCI 11-001 study (ITT ALL population) | ||
|---|---|---|---|---|
Calaspargase pegol 2,500 IU/m2 (N = 42) | Pegaspargase 2,500 IU/m2 (N = 55) | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) | |
Data cut-off date | December 31, 2015 | October 5, 2016 | ||
Median follow-up time, months (range) | 62.6 (0.5 to 86.3) | 26.6 (0.5 to 45.6) | ||
OS | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Deaths at DCO,a n (%) | | |||||| | | |||||| | | ||||| | | ||||| |
1-year OS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||||||||||| || |||||| |
2-year OS, % (95% CI) | || | || | |||| |||||| || ||||| | |||||||||||| || |||||| |
4-year OS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | || | || |
HR (95% CI) | |||| |||||| || ||||| | ||||||||| | || | || |
DFS from CR | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Events, n (%) | | ||||| | | ||||| | | ||||| | | ||||| |
1-year DFS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
2-year DFS, % (95% CI) | || | || | |||| |||||| || ||||| | |||| |||||| || ||||| |
4-year DFS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | || | || |
HR (95% CI) | |||| |||||| || ||||| | ||||||||| | || | || |
EFS | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Events, n (%) | | ||||| | | |||||| | || ||||| | | ||||| |
1-year EFS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
2-year EFS, % (95% CI) | || | || | |||| |||||| || ||||| | |||| |||||| || ||||| |
4-year EFS, % (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | || | || |
HR (95% CI) | |||| |||||| || ||||| | ||||||||| | || | || |
CR at end of induction | ||||
End of induction day 29 | ||||
Number of patients contributing to the analysis, n | || | || | || | || |
Number of patients attaining CR, n (%) | || |||||| | || |||||| | || | || |
Number of patients attaining CR, 95% CI | |||| || |||| | |||| || |||| | || | || |
End of induction day 32 | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
Number of patients attaining CR, n (%) | || | || | ||| |||||| | ||| |||||| |
Number of patients attaining CR, 95% CI | || | || | |||| || |||| | |||| || |||| |
MRD at end of induction | ||||
MRD at induction day 29 | ||||
Number of patients contributing to the analysis, n (MRD-evaluable ITT population) | || | || | || | || |
Positive MRD (≥ 0.1%), n (%) | | |||||| | || |||||| | || | || |
Positive MRD rate, 95% CI | || | || | || | || |
Negative MRD (< 0.1%), n (%) | || |||||| | || |||||| | || | || |
Negative MRD rate, 95% CI | |||| || |||| | |||| || |||| | || | || |
MRD at induction day 29 | ||||
Number of patients contributing to the analysis, n (ITT population) | || | || | || | || |
MRD ≥ 0.01, n (%) | | ||||| | | |||||| | || | || |
MRD at induction day 32 | ||||
Number of patients contributing to the analysis, n | || | || | ||| | ||| |
MRD ≥ 0.01, n (%) | || | || | | ||||| | || |
ALL = acute lymphoblastic leukemia; CI = confidence interval; COG = Children’s Oncology Group; CR = complete remission; DCO = data cut-off; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; HR = hazard ratio; ITT = intention-to-treat; MRD = minimal residual disease; OR = odds ratio; OS = overall survival; PKAS = pharmacokinetic analysis set.
Notes: Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
In the DFCI 11-001 study, all efficacy outcomes including survival (OS, EFS, and DFS) and response to treatment (CR at the end of induction, and MRD at the end of induction) were conducted using the ITT ALL population.
aSerum asparaginase levels were reported for the PKAS (patients who received at least 1 dose of calaspargase pegol or pegaspargase on induction day 7 and had at least 1 postinfusion measurement above the lower limit of quantification) in the DFCI 11-001 trial.
Sources: COG AALL07P4 Clinical Study Report12 and DFCI 11-001 Clinical Study Report.13,15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.12
Table 18: Summary of MRD Results From DFCI 11-001 Study — FAS ALL
Outcome | Calaspargase pegol 2,500 IU/m2 (N = 118) | Pegaspargase 2,500 IU/m2 (N = 119) |
|---|---|---|
MRD at induction day 32 | ||
Number of patients contributing to the analysis, n (MRD-evaluable) | || | || |
Low (< 0.001), n (%) | || |||||| | || |||||| |
Low (< 0.001), 95% CIa | |||| || |||| | |||| || |||| |
High (≥ 0.001), n (%) | || |||||| | || |||||| |
High (≥ 0.001), 95% CIa | ||| || |||| | ||| || |||| |
ALL = acute lymphoblastic leukemia; CI = confidence interval; DFCI = Dana-Farber Cancer Institute; FAS = full analysis set; MRD = minimal residual disease.
aThe 2-sided 95% CI is based on the Clopper-Pearson method.
Source: DFCI 11-001 Clinical Study Report.15

Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALL
acute lymphoblastic leukemia
BIA
budget impact analysis
BSA
body surface area
COG
Children’s Oncology Group
CUA
cost-utility analysis
DFCI
Dana-Farber Cancer Institute
EFS
event-free survival
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
LY
life-year
MAC
multiagent chemotherapeutic
OS
overall survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Calaspargase pegol (Asparlas), 3,750 units/5 mL (750 units/mL), solution for IV infusion |
Submitted price | Calaspargase pegol 3,750 units/5 mL (750 units/mL) vial: $7,441.88 per vial |
Indication | As a component of an MAC regimen for the treatment of ALL in pediatric and young adult patients aged 1 year to 21 years |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 8, 2023 |
Reimbursement request | As per indication |
Sponsor | Servier Canada Inc. |
Submission history | Not previously reviewed |
ALL = acute lymphoblastic leukemia; MAC = multiagent chemotherapeutic; NOC = Notice of Compliance.
Note: The sponsor’s application was filed on a pre-NOC basis and the pharmacoeconomic submission is reflective of the proposed indication and information incorporated into the draft product monograph that was submitted to Health Canada and CADTH. The sponsor’s submission included a broader age range than the final indication; however, other details incorporated in the final product monograph were not considered in the pharmacoeconomic review, which suggested the potential for increased resource use associated with calaspargase pegol.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Combination decision tree and PSM |
Target population | Patients with ALL receiving an asparaginase-containing MAC regimen |
Treatment | Calaspargase pegol as a component of an MAC regimen |
Comparator | Pegaspargase (Oncaspar) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, Lys |
Time horizon | Lifetime (89 years) |
Key data sources | COG AALL07P4 trial DFCI 11 to 001 trial |
Submitted results | Calaspargase pegol is dominant when compared to pegaspargase (i.e., more QALYs [0.23] and less incremental costs [–$999]). CADTH noted that calaspargase pegol was associated with fewer incremental LYs (–0.44) when compared to pegaspargase. |
Key limitations |
|
CADTH reanalysis results |
|
AE = adverse event; ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; DFS = disease-free survival; EFS = event-free survival; LY = life-year; MAC = multiagent chemotherapeutic; OS = overall survival; PSM = partitioned survival model; QALY = quality-adjusted life-year.
Conclusions
Based on the sponsor-submitted clinical evidence from the Children’s Oncology Group (COG) study known as the COG AALL07P4 trial, the effects of calaspargase pegol compared with pegaspargase may result in little to no difference in overall survival (OS), event-free survival (EFS), hypersensitivity reactions, and anaphylactic reactions. Using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool, CADTH categorized this evidence as having low certainty, due to limitations associated with the small sample sizes, wide confidence intervals, and the absence of clinically meaningful thresholds. Additionally, there was no evidence for the effect of calaspargase pegol compared with pegaspargase on health-related quality of life (HRQoL) and the evidence is very uncertain about the effects of calaspargase pegol on withdrawals due to adverse events (AEs) (very low certainty). However, clinical expert feedback obtained by CADTH indicated that the relative efficacy and safety of calaspargase pegol compared with pegaspargase is likely to be comparable in Canadian clinical practice.
To address some of the identified limitations, CADTH assumed equivalent efficacy and safety inputs for calaspargase pegol and pegaspargase. Additionally, CADTH assumed equivalent AE management costs between treatments and applied the patient population characteristics described in the Dana-Farber Cancer Institute (DFCI) trial known as the DFCI 11 to 001 trial. In the CADTH base case, calaspargase pegol is associated with higher total costs (an incremental cost of $4,088) and equivalent quality-adjusted life-years (QALYs). These results were driven by the higher drug cost of calaspargase pegol in comparison to pegaspargase and by the assumption of no difference in clinical outcomes.
Based on the CADTH review, there is no robust evidence to suggest calaspargase pegol is associated with clinical benefits that are different from pegaspargase. As a result, there is no evidence to support a price premium for calaspargase pegol compared with pegaspargase. CADTH was unable to address the uncertainty regarding the price of pegaspargase paid by CADTH-participating drug plans.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Leukemia and Lymphoma Society of Canada, which conducted an online survey with 74 respondents from all 10 provinces in Canada. The majority of the respondents (n = 47) identified as patients with acute lymphoblastic leukemia (ALL) and 20 respondents indicated they were caregivers of patients with ALL. All other respondents were disqualified. The questions in the survey did not attempt to measure the efficacy of calaspargase pegol as it was assumed to be as effective as current treatment options. Respondents reported their experience with drug shortages during their ALL treatment and noted experiencing feelings of powerlessness, poor quality of life, lack of sleep due to stress, anxiety, and mood swings, along with devoting time attempting to find an alternative solution. Some respondents noted financial impacts, as they had to pay for alternative therapies and buy products to help cope with the situation. The Leukemia and Lymphoma Society of Canada noted the importance of having alternative treatments available to reduce the impact, should manufacturers run into supply issues. Most respondents reported that the availability of treatment throughout the course of therapy is extremely important and ensuring a secure supply of medications would remove stress from patients and caregivers.
CADTH received 1 registered clinician group input submission for this review from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee. The input indicated that the current standard treatment for patients with ALL is pegaspargase, which they believed has the potential to be curative. Although the clinician input noted that there is no large unmet need as pegaspargase is available, less frequent dosing would be beneficial from a patient perspective.
Drug plan input for this review indicated pegaspargase, crisantaspase, and Erwinia-derived asparaginase may be relevant comparators for calaspargase pegol. The public drug plans also sought input on the relevance of patients older than 30 years, as the 2 trials used in the sponsor submission — the COG AALL07P4 study and the DFCI 11 to 001 study — included patients under 30 years. The drug plans sought input on whether calaspargase pegol would also be capped at 3,750 units, in line with pegaspargase use. Additionally, the plans noted that due to ongoing shortages of asparaginase products (i.e., Erwinia asparaginase, pegaspargase, and crisantaspase), having alternative therapies available would be helpful.
Several of these concerns were addressed in the sponsor’s economic submission.
The sponsor included pegaspargase as a relevant comparator in the cost-utility analysis (CUA).
The sponsor assumed patients switched to Erwinia asparaginase, or crisantaspase in the scenario analysis, to ensure treatment completion if hypersensitivity occurred.
The calaspargase pegol dosage was capped at 3,750 units.
CADTH was unable to address the following concerns raised from stakeholder input:
Concerns regarding the available supply of asparaginase products were noted. The implications of introducing calaspargase pegol based on the availability of other asparaginase products was not addressed in the sponsor submission. CADTH noted that the sponsor’s budget impact analysis (BIA) suggested that within 3 years of calaspargase pegol being reimbursed, pegaspargase would have a 0% market share (i.e., not be used) in CADTH-participating drug programs.
Calaspargase pegol has a longer shelf life than pegaspargase (36 months for calaspargase pegol compared with 8 months for pegaspargase), thus impacting the uptake of calaspargase pegol and future use of pegaspargase. It is unclear what impact the introduction of calaspargase pegol would have on drug shortages for asparaginase products.
Economic Review
The current review is for calaspargase pegol (Asparlas) as a component of a multiagent chemotherapeutic (MAC) regimen for treatment of patients with ALL.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a CUA comparing the costs and outcomes for calaspargase pegol and pegaspargase, as determined in the COG AALL07P4 and DFCI 11 to 001 trials for the treatment of patients with ALL.1 The modelled population was in line with the COG AALL07P4 trial. The target population is aligned with the Health Canada–indicated population.
Calaspargase pegol is available in a vial containing 3,750 units/5 mL (750 units/mL) of solution for IV infusion at a submitted price of $7,441.88 per vial. According to the draft product monograph, the recommended dosage is 2,500 units/m2 given intravenously no more frequently than every 21 days.2 No duration of therapy is recommended in the product monograph. However, there are 2 commonly used treatment protocols in Canadian practice: the protocol used in the COG AALL07P4 trial and the protocol used in the DFCI 11 to 001 trial. In the COG AALL07P4 trial, treatment of either calaspargase pegol (2,500 units/m2) or pegaspargase (2,500 units/m2) was administered during the induction and consolidation phases and the total scheduled asparaginase doses in the 7 months to 11 months of therapy was dependent on whether the patient was a rapid early responder receiving 7 doses, a slow early responder receiving 11 doses, or in extended induction receiving 12 doses.3 In the DFCI 11 to 001 trial, patients received either a total of 11 doses of calaspargase pegol (2,500 units/m2) or a total of 16 doses of pegaspargase (2,500 units/m2) over a single induction phase followed by a 30-week postinduction period of asparaginase therapy. In the sponsor’s model, the dosages used for calaspargase pegol and pegaspargase were derived from the COG AALL07P4 trial. In the sponsor’s submission, 7 doses (1 dose in the induction phase, 2 doses in the consolidation phase, 2 doses in the delayed intensification phase, and 2 doses in the second interim maintenance phase) were used to derive the cost per therapy. No extended duration of therapy was considered.
At the sponsor’s assumed dose of 2,500 units/m2, using a body surface area (BSA) of 1.34 m2 and assuming wastage of unused drug product, the cost of calaspargase pegol was $7,442 per dose; over the 7-dose course of treatment as per the COG AALL07P4 trial protocol, the cost of calaspargase pegol was $52,093 per patient. At a cost of $6,615 per 3,750 unit vial of pegaspargase, using the same dosing regimen as calaspargase pegol, pegaspargase cost $6,615 per dose; over the 7-dose course of treatment as per the COG AALL07P4 trial, the cost of pegaspargase was $46,305.3
The clinical outcomes modelled were radiological progression-free survival and OS. The economic outcomes of interest were QALYs and life-years (LYs). The patient age at the model entry was 11 years and the economic analysis was undertaken over a lifetime time horizon (assumed to be 89 years) from the perspective of the Canadian publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum.
Model Structure
The sponsor submitted a hybrid model structure that consisted of a decision tree to model patients completing their full course of treatment with calaspargase pegol or pegaspargase, followed by a partitioned survival model (PSM) representing patients’ long-term outcomes in the following 3 health states: EFS, relapsed disease (active or progressed disease), and death (Figure 1).
All patients began an asparaginase-containing MAC regimen in either treatment arm (calaspargase pegol or pegaspargase), with their treatment course modelled by a decision-tree structure.1 While receiving treatment, silent inactivation or hypersensitivity reactions could result in either a switch to Erwinia asparaginase or an application of the desensitization protocol. Patients in the Erwinia asparaginase state could also transition to the desensitization protocol as they could experience hypersensitivity reactions but would not experience silent inactivation. The duration for patients to move through the short-term decision tree was approximately 38 weeks, determined through the model trace. Subsequently, all surviving patients entered the PSM phase, and all patients were assumed to begin in the EFS (remission) state. Patients then could remain in the EFS state, transition to relapse, or die. General population mortality rates were assumed after a period of 5 years in the EFS health state.
Model Inputs
The modelled population generally reflected the baseline characteristics of the enrolment population in the COG AALL07P4 trial. Pediatric as well as adolescent and young adult patients entered the model at a starting age of 11 years, with a mean BSA of 1.34 m2 and a mean weight of ||||| ||.3 In the base case, the sponsor compared calaspargase pegol to pegaspargase, which is the only asparaginase currently marketed in Canada for the initial treatment of patients with ALL. In response to hypersensitivity reactions, desensitization protocols could be administered for either treatment and patients were switched to Erwinia asparaginase in the event of silent inactivation or a high-grade hypersensitivity reaction.
As there are different protocols for treating patients with ALL, the sponsor chose to use the protocol from the COG AALL1732 trial in the base case for high-risk pediatric patients.4 Standard-risk pediatric patients were treated using a regimen from the COG AALL1731 protocol and were considered as part of a scenario analysis.5 Based on the COG AALL07P4 trial, which was used to determine the dose of calaspargase pegol in pediatric patients, patients received either calaspargase pegol or pegaspargase in 4 of the 6 phases. In the induction phase (5 weeks), a single dose of either treatment was given. Following this, 2 doses were given in the consolidation phase (8 weeks), after which no doses were given in the interim maintenance phase (9 weeks). In both the delayed intensification phase (8 weeks) and the interim maintenance phase (8 weeks), 2 additional doses of the treatments were given in each phase, followed by no additional doses in the maintenance phase (104 weeks).3 Movement through the phases was modelled by a decision tree and involved 2 key pathways. In the first pathway, patients completed the full course of treatment with either calaspargase pegol or pegaspargase. In the second pathway, patients switched to Erwinia asparaginase at some point before their asparaginase treatment course had finished, either due to a high-grade hypersensitivity reaction or due to silent inactivation. Hypersensitivity rates for calaspargase pegol and pegaspargase were derived from the COG AALL07P4 and DFCI 11 to 001 studies.3,6 Silent inactivation rates were derived from the Marini et al. review where the midpoint value for the rate was used in cases where a range of rates was provided.7 Surviving patients entered the PSM phase of the model, which was informed by time-to-event analysis.
In the base case, EFS data were derived from the COG AALL07P4 study’s Clinical Study Report.3 Both the COG AALL07P4 and DFCI 11 to 001 trials found no significant difference in EFS between calaspargase pegol and pegaspargase, despite a numerical difference between calaspargase pegol and pegaspargase in terms of EFS.3,8 Additionally, no significant difference in outcomes existed between patients who switched to Erwinia asparaginase and patients who remained on pegaspargase. Therefore, the sponsor assumed patients who switched from calaspargase pegol to Erwinia asparaginase exhibited no change in efficacy as well. OS data were also derived from the COG AALL07P4 Clinical Study Report.9 The sponsor noted that premature discontinuation of asparaginase treatment has been demonstrated to significantly decrease EFS and OS in high-risk patients, with a numeric decrease in EFS for standard-risk patients.9-12 In the sponsor’s model, patients were assumed to receive a full course of asparaginase therapy, with or without the use of desensitizing drugs or switching to Erwinia asparaginase to ensure the completion of therapy. AE management costs were applied as a 1-time cost at the first model cycle. The rates for each treatment arm were calculated based on the COG AALL07P4 trial.6
Health state utility values were based on published literature with EFS utility specific to high-risk patients.13 Relapsed disease utilities were derived from a Canadian cost-effectiveness analysis of high-risk relapsed pediatric patients based on the Pediatric Quality of Life Inventory, while EFS utilities were derived from a review reporting values based on the Health Utilities Index.14,15 These values were derived from a published HRQoL study that examined pediatric patients with ALL from hospitals in Canada and the US, with a mean age of 6.1 years.16
Costs included drug acquisition and administration costs, AE management costs, hypersensitivity costs, asparaginase activity monitoring costs, desensitization protocol costs, and monitoring and disease management costs. Unit costs for premedications were sourced from the Ontario Drug Benefit Formulary and the Newfoundland and Labrador Prescription Drug Program as a proxy for the rest of Canada.17,18 Unit costs for calaspargase pegol and pegaspargase were sourced from the manufacturer and from published literature for Erwinia asparaginase. Treatment administration costs were reflective of the dosing guidance in the Ontario Schedule of Benefits: Physician Services Under the Health Insurance Act.19
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 5,000 iterations. Deterministic and probabilistic results were consistent. The probabilistic findings are presented as follows.
Base-Case Results
In the sponsor’s base-case analysis over an 89-year time horizon, calaspargase pegol was dominant (i.e., less costly and associated with more QALYs) compared to pegaspargase. At a willingness-to-pay threshold of $50,000 per QALY, the probability of calaspargase pegol being cost-effective was 55.9% due to the small incremental differences in costs and QALYs.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. pegaspargase ($/QALY) |
|---|---|---|---|---|---|
Pegaspargase | $133,444 | Reference | 33.35 | Reference | Reference |
Calaspargase pegol | $132,444 | –$999 | 33.58 | 0.23 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Notes: Costs are rounded to the nearest dollar.
Calaspargase pegol is dominant in the sponsor’s economic evaluation (more effective and less costly) than pegaspargase.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor performed scenario analyses by changing discount rates, presenting the societal perspective, removing asparaginase activity monitoring (including asparaginase activity monitoring only in the case of hypersensitivity), presenting the adult patient population and the standard-risk population, using DFCI 11 to 001 trial protocol and clinical data, using desensitization for Erwinia asparaginase only, removing Erwinia asparaginase due to shortages, using crisantaspase recombinant instead of Erwinia asparaginase, changing functional cure time to 7.8 years, and assuming equivalent comparative efficacy. The scenario analyses indicated that the sponsor’s findings were sensitive to the cure time, patient population, and assumptions regarding comparative efficacy.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Comparative clinical efficacy between calaspargase pegol and pegaspargase is uncertain: In its economic analysis, the sponsor used data from the COG AALL07P4 study to derive EFS and OS distributions to inform the effectiveness of calaspargase pegol and pegaspargase over the modelled 89-year time horizon,6 which resulted in calaspargase pegol being associated with fewer Lys but greater QALYs than pegaspargase. Thus, although the sponsor’s results suggested calaspargase pegol was associated with incremental QALYs compared with pegaspargase, these were derived from fewer overall Lys. Although the findings from the COG AALL07P4 and DFCI 11 to 001 trials indicated that the effects of calaspargase pegol and pegaspargase were similar for EFS and OS, as noted in the GRADE tables in the clinical review, CADTH’s appraisal identified uncertainty in the available clinical evidence. The uncertainty identified was due to small sample sizes, wide confidence intervals, and the absence of clinically meaningful thresholds. Despite the methodological uncertainty identified, the CADTH clinical review concluded that calaspargase pegol may result in little to no difference in OS, disease-free survival, EFS, and minimal residual disease compared with pegaspargase. This aligned with clinical expert feedback obtained by CADTH, which indicated that the clinical effects of calaspargase pegol and pegaspargase were expected to be similar in Canadian clinical practice.
CADTH conducted a reanalysis assuming equivalent comparative efficacy between calaspargase pegol and pegaspargase.
Age of patient population entering model is inappropriate: In the sponsor’s submitted economic analysis, patient characteristics were assumed to be comparable with the population in the COG AALL07P4 trial. As a result, patients entered the model at a starting age of 11 years, with a mean BSA of 1.34 m2 and a mean weight of ||||| ||. However, clinical expert feedback obtained by CADTH indicated that the patient population in the COG AALL07P4 trial is not representative of the pediatric population initiating treatment as observed in Canadian clinical practice, and that the population included in the DFCI trial more accurately represents the age of the patient population with ALL in Canada at treatment initiation.
CADTH conducted a reanalysis to align with the patient population in the DFCI 11 to 001 trial. In this trial, patients entered the model at the age of 5 years with a BSA of |||| || and a weight of ||||| ||.
Treatment-related AEs are uncertain: The sponsor applied AE management costs as a 1-time cost in the first model cycle. Rates for each treatment arm were calculated based on data from the COG AALL07P4 trial. In this trial, the results reported less frequent and fewer AEs in the calaspargase pegol treatment arm in comparison to the pegaspargase treatment arm. However, results of the CADTH clinical appraisal found that frequencies of AEs were similar between calaspargase pegol and pegaspargase in both the COG AALL07P4 trial and the DFCI 11 to 001 trial and thus, the use of calaspargase pegol could result in little to no difference in hypersensitivity reactions and anaphylactic reactions compared with pegaspargase. Therefore, the differing AE management costs applied in the sponsor submission are uncertain.
CADTH conducted a reanalysis to assume equivalent AE management costs for each treatment arm.
Duration of treatment is uncertain: The sponsor assumed both calaspargase pegol and pegaspargase treatment would be provided for 38 weeks, after which time costs associated with these treatments were not considered in the model. Clinical expert feedback indicated that the total duration of MAC treatment for patients with ALL who respond is approximately 2.5 years to 3.5 years, while time on an asparaginase treatment is limited to the first year (although duration of treatment may vary). As a result, total costs assumed in the sponsor’s model may differ to what is considered in clinical practice.
Due to the programming of the sponsor’s modelling approach, CADTH could not address this limitation. Extending the duration of treatment could result in a greater incremental treatment cost associated with calaspargase pegol relative to pegaspargase due to the higher drug cost (refer to Table 7).
Comparator pricing is not publicly available: Pegaspargase acquisition costs were provided by Servier Canada and could not be validated by CADTH. CADTH reviewed the IQVIA DeltaPA database and pegaspargase pricing had an end date (December 16, 2019). It was unclear how accessible the treatment is across Canada. The sponsor indicated that once introduced, calaspargase pegol would only be used for patients who would have otherwise received pegaspargase and would not produce any incremental utilization of asparaginase-containing MAC. CADTH noted that pegaspargase may not be available on all Canadian drug formularies; the sponsor noted that it was available in only Ontario, Alberta, British Columbia, and Saskatchewan. As a result, accessibility of pegaspargase may vary based on jurisdiction. Therefore, it is uncertain if the reimbursement of calaspargase pegol would result in fewer incremental costs than pegaspargase.
CADTH could not address the uncertainty regarding the price of pegaspargase paid by drug plans.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The patients included in the COG AALL07P4 and DFCI 11 to 001 trials were representative of the indicated patient population. | Uncertain. The CADTH clinical appraisal noted that the trial evidence was likely representative of patients with ALL, but did not include all patients who may be considered eligible for treatment with calaspargase pegol in Canadian clinical practice. |
Patients in the analysis were assumed to have received a full course of asparaginase therapy, with or without the use of desensitizing drugs and/or a switch to Erwinia asparaginase to ensure completion. | Appropriate. According to clinical experts consulted by CADTH, it is reasonable to assume desensitizing drugs would be used, or a switch to Erwinia asparaginase might have been made to ensure treatment completion. |
It was assumed that patients on Erwinia asparaginase do not experience silent inactivation and that silent inactivation is assumed to occur by the same dose by which hypersensitivity reactions occur. | Appropriate and validated by clinical experts consulted by CADTH. |
ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes include applying baseline patient characteristics in the model for age, BSA, and weight from the DFCI 11 to 001 trial, assuming equivalent efficacy between calaspargase pegol and pegaspargase, and assuming equivalent AE management costs. Table 5 details the changes made to derive the CADTH reanalysis and the summary results of that reanalysis are presented in Table 6. Treatment with calaspargase pegol was more costly than pegaspargase (incremental cost of $4,088) while total QALYs were equal.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Patient starting age | Patients entered the model at 11 years, derived from the COG AALL07P4 trial. | Patients entered the model at 5 years as per the DFCI 11 to 001 trial, which better represents the starting age of treatment in patients with ALL. |
2. Patient BSA and weight | Patients entered the model with a mean BSA of 1.34 m2 and a mean weight of ||||| || as per the COG AALL07P4 trial. | Patients entered the model with a median BSA of |||| || and a mean weight of ||||| || as per the DFCI 11 to 001 trial. |
3. Assuming equivalent efficacy between calaspargase pegol and pegaspargase | EFS and OS are not equivalent between treatments. | EFS and OS are equivalent between treatments. |
4. Assuming equivalent AE management costs between calaspargase pegol and pegaspargase | Calaspargase pegol is associated with higher AE management costs in comparison to pegaspargase. Calaspargase pegol: $8,580.01 Pegaspargase: $6,253.64 | AE management costs are equivalent between the treatments ($8,580.01). |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
AE = adverse event; ALL = acute lymphoblastic leukemia; BSA = body surface area; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; EFS = event-free survival; OS = overall survival.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Pegaspargase | 133,443.69 | 33.35 | Reference |
Calaspargase pegol | 132,444.48 | 33.58 | Dominant | |
CADTH reanalysis 1 (patient age) | Pegaspargase | 134,599.36 | 34.84 | Reference |
Calaspargase pegol | 133,167.06 | 35.12 | Dominant | |
CADTH reanalysis 2 (patient BSA and weight) | Pegaspargase | 118,231.29 | 33.34 | Reference |
Calaspargase pegol | 116,771.64 | 33.61 | Dominant | |
CADTH reanalysis 3 (equivalent efficacy) | Pegaspargase | 123,830.54 | 34.58 | Reference |
Calaspargase pegol | 130,246.08 | 34.58 | More costly, equal QALYs | |
CADTH reanalysis 4 (equivalent AE management costs) | Pegaspargase | 135,857.53 | 33.33 | Reference |
Calaspargase pegol | 132,696.39 | 33.59 | Dominant | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) | Pegaspargase | 121,639.69 | 34.84 | Reference |
Calaspargase pegol | 125,727.55 | 34.84 | More costly, equal QALYs |
AE = adverse event; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH did not undertake any scenario analyses. CADTH noted that based on the submitted price of calaspargase pegol, and the sponsor-reported price of pegaspargase, an 11% price reduction would be required for calaspargase pegol to be no more costly than pegaspargase. CADTH could not validate the price of pegaspargase paid by CADTH-participating drug plans. As such, the magnitude of price reduction required to ensure no additional costs are incurred is uncertain.
Issues for Consideration
Shortage of asparaginase products in Canada: Stakeholders and clinical experts consulted by CADTH highlighted that while introducing an additional asparaginase product will assist with the shortage, the sponsor-submitted BIA reported that pegaspargase may be phased out, with calaspargase pegol expected to capture all market shares within 3 years. In the sponsor’s submission, the sponsor indicated that due to the relatively short shelf life of pegaspargase (8 months, compared with 36 months for calaspargase pegol), the reimbursement of calaspargase pegol could provide a more consistent supply of asparaginase products across Canada. If actualized, this may meet an unmet need that was noted in both the stakeholder input and in clinical expert feedback obtained by CADTH. Although the difference in shelf life was commented on by the sponsor in its Pharmacoeconomic report, it was not adequately incorporated into the sponsor’s economic evaluation, and thus could not be addressed by CADTH within reanalysis.
Overall Conclusions
Based on the sponsor-submitted clinical evidence from the COG AALL07P4 trial, the effects of calaspargase pegol compared with pegaspargase may result in little to no difference in OS, EFS, hypersensitivity reactions, and anaphylactic reactions. Using GRADE, CADTH categorized this evidence as having low certainty, due to limitations associated with the small sample sizes, wide confidence intervals, and the absence of clinically meaningful thresholds. Additionally, there was no evidence for the effect of calaspargase pegol compared with pegaspargase on HRQoL and the evidence is very uncertain about the effects of calaspargase pegol on withdrawals due to AEs (very low certainty). However, clinical expert feedback obtained by CADTH indicated that the relative efficacy and safety of calaspargase pegol compared with pegaspargase is likely to be comparable in Canadian clinical practice.
In addition to concerns regarding the comparative clinical efficacy and safety of the treatments, CADTH identified limitations associated with the patient population characteristics and treatment costs in the model. To address some of the identified limitations, CADTH assumed equivalent efficacy and safety inputs for calaspargase pegol and pegaspargase. Additionally, CADTH assumed equivalent AE management costs between treatments and applied the patient population characteristics described in the DFCI 11 to 001 trial. As a result of these changes, the findings of the CADTH base-case analysis indicate that calaspargase pegol is associated with higher costs (an incremental cost of $4,088) and equivalent QALYs. These results were driven by the higher cost of calaspargase pegol in comparison to pegaspargase and by assuming no difference in clinical outcomes. These results differ from the sponsor’s base case, in which a QALY benefit is assumed due to more time in the EFS state despite fewer overall Lys, and cost savings are driven by less time alive for patients on calaspargase pegol.
Based on the CADTH review, there is no robust evidence to suggest calaspargase pegol is associated with additional clinical benefit compared with pegaspargase. As a result, there is no evidence to support a price premium for calaspargase pegol over pegaspargase. CADTH was unable to address the uncertainty regarding the price of pegaspargase paid by CADTH-participating drug plans.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: calaspargase pegol, 3,750 units/5 mL (750 units/mL), injection for intravenous use. Laval (QC): Servier Canada Inc.; 2023 May 12. In.
2.Asparlas (calaspargase pegol): 3,750 units/5 mL (750 units/mL), injection for intravenous use product monograph]. In: Laval (QC): Servier Canada Inc.; May 3 2023.
3.Baxalta USI. Randomized Study of Intravenous Calaspargase Pegol (SC-PEG-L-asparaginase) and Intravenous Oncaspar® in Children and Adolescents with Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma (Study Report: DFCI 11-001). 2017.
4.University of Iowa Health C. AALL1732: A study to test if the addition of Inotuzumab Ozogamicin (InO) to standard chemotherapy treatment in patients diagnosed with National Cancer Institute (NCI) High-Risk (HR) B cell Acute Lymphoblastic Leukemia (B-ALL) or NCI Standard-Risk (SR) …. https://clinicaltrials.uihealthcare.org/studies/aall1732-study-test-if-addition-inotuzumab-ozogamicin-ino-standard-chemotherapy-treatment. Published 2019. Accessed.
5.University of Iowa Health C. AALL1731: A study to compare the addition of Blinatumomab in combination with chemotherapy in patients diagnosed with standard risk B-cell Acute Lymphoblastic Leukemia (B-ALL), Down syndrome B ALL and the treatment of patients with localized B-cell Lym…. https://clinicaltrials.uihealthcare.org/studies/aall1731-study-compare-addition-blinatumomab-combination-chemotherapy-patients-diagnosed. Published 2019. Accessed.
6.Baxalta USI. A Pilot Study of Intravenous EZN-2285 (SC PEG E. coli L asparaginase, IND 100594) or Intravenous Oncaspar® in the Treatment of Patients with High-Risk Acute Lymphoblastic Leukemia (ALL): A Limited Institution Pilot Study (Clinical Study Report). 2017.
7.Marini BL, Brown J, Benitez L, et al. A single-center multidisciplinary approach to managing the global Erwinia asparaginase shortage. Leuk Lymphoma. 2019;60(12):2854-2868. PubMed
8.Vrooman LM, Blonquist TM, Stevenson KE, et al. Efficacy and Toxicity of Pegaspargase and Calaspargase Pegol in Childhood Acute Lymphoblastic Leukemia: Results of DFCI 11-001. J Clin Oncol. 2021;39(31):3496-3505. PubMed
9.Gupta S, Wang C, Raetz EA, et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group. J Clin Oncol. 2020;38(17):1897-1905. PubMed
10.Dos Santos AC, Dos Santos JMB, da Costa Lima E, Land MGP. L-asparaginase doses number as a prognostic factor in childhood acute lymphoblastic leukemia: A survival analysis study. Cancer Rep (Hoboken). 2021:e1533. PubMed
11.Burke MJ, Zalewska-Szewczyk B. Hypersensitivity reactions to asparaginase therapy in acute lymphoblastic leukemia: immunology and clinical consequences. Future Oncol. 2022. PubMed
12.Schmidt MP, Ivanov AV, Coriu D, Miron IC. L-Asparaginase Toxicity in the Treatment of Children and Adolescents with Acute Lymphoblastic Leukemia. J Clin Med. 2021;10(19). PubMed
13.McCormick M, Lapinski J, Friehling E, Smith K. Premedication prior to PEG-asparaginase is cost-effective in pediatric patients with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2021;68(8):e29051. PubMed
14.Furzer J, Gupta S, Nathan PC, et al. Cost-effectiveness of Tisagenlecleucel vs Standard Care in High-risk Relapsed Pediatric Acute Lymphoblastic Leukemia in Canada. JAMA Oncol. 2020;6(3):393-401. PubMed
15.Barr RD, Sala A. Quality-adjusted survival: a rigorous assessment of cure after cancer during childhood and adolescence. Pediatr Blood Cancer. 2005;44(3):201-204. PubMed
16.Furlong W, Rae C, Feeny D, et al. Health-related quality of life among children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2012;59(4):717-724. PubMed
17.Government of O. Ontario Drug Benefit Formulary. https://www.formulary.health.gov.on.ca/formulary/. Published 2023. Accessed.
18.Newfoundland, Labrador Department of H, Community S. Prescription Drug Program (NLPDP). https://www.health.gov.nl.ca/health/prescription/newformulary.asp. Published 2022. Accessed.
19.Government of O. Schedule of Benefits: Physician Services Under the Health Insurance Act. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Published 2022. Accessed.
20.DeltaPA. In: [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 1800 Jan 1.
21.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: calaspargase pegol, 3,750 units/5 mL (750 units/mL), injection for intravenous use. Laval (QC): Servier Canada Inc.; 2023 May 12. In.
22.PM-DFCI < 60 ALL Protocol_Pegaspargase (with Therapeutic Drug Monitoring [TDM]). Version v24; Version date: 02.06.2021.
23.PM-DFCI ≥ 60 ALL Protocol_Pegaspargase (with Therapeutic Drug Monitoring [TDM]). Version v19; Version date: 02.06.2021.
24.Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868-1876. PubMed
25.Statistics C. Number and rates of new cases of primary cancer, by cancer type, age group and sex. https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310011101. Accessed January 5, 2022.
26.Statistics C. Population estimates on July 1st, by age and sex. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed January 5, 2022.
27.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535-1548. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for the Treatment of ALL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Average daily cost | Course cost (38 weeks, 7 doses) |
|---|---|---|---|---|---|---|
Calaspargase pegol (Asparlas) | 3,750 units / 5 mL | Injection for IV infusion | $7,441.8800a | 7 doses of 2,500 units / m2 over treatment course. 1 dose during induction (5 weeks), 2 doses during consolidation (8 weeks), 0 doses during interim maintenance (9 weeks), 2 doses during delayed intensification (8 weeks) and 2 doses during the second interim maintenance (8 weeks). | $195.84 | $52,093 |
Asparaginase | ||||||
Pegaspargase (Oncaspar) | 3,750 units / 5 mL | Solution for IM injection or IV infusion | $6,615.0000a | 7 doses of 2,500 units / m2 over treatment course. 1 dose during induction (5 weeks), 2 doses during consolidation (8 weeks), 0 doses during interim maintenance (9 weeks), 2 doses during delayed intensification (8 weeks) and 2 doses during the second interim maintenance (8 weeks). | $174.08 | $46,305 |
IM = intramuscular.
Note: Treatment course and recommended dosage is reflective of the COG AALL07P4 trial.6
aTreatment price from sponsor submission.1 CADTH searched the IQVIA DeltaPA database for the wholesale pricing for pegaspargase and treatment was listed with an end date of December 16, 2019. The last available price for pegaspargase was $6,615.9700.20 It is unclear if treatment is still available in Canada and at what price.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal section. Starting age of population is closer to DFCI 11 to 001 trial, according to clinical experts consulted by CADTH. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Calaspargase pegol and pegaspargase share equivalent efficacy. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
In the sponsor-submitted base case, patients treated with calaspargase pegol are associated with fewer LYs (–0.44) but more QALYs (0.23) than patients treated with pegaspargase.
Table 9: Disaggregated Results of the Sponsor's Base Case
Parameter | Calaspargase pegol | Pegaspargase | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 37.78 | 38.22 | –0.44 |
By health state | |||
Induction | 0.08 | 0.08 | 0.00 |
Consolidation | 0.15 | 0.15 | 0.00 |
Interim maintenance | 0.16 | 0.16 | 0.00 |
Delayed intensification | 0.15 | 0.15 | 0.00 |
Interim maintenance (second) | 0.15 | 0.15 | 0.00 |
Maintenance | 0.00 | 0.00 | 0.00 |
Event-free survival | 32.30 | 28.63 | 3.68 |
Relapsed disease | 4.78 | 8.89 | –4.11 |
Discounted QALYs | |||
Total | 33.58 | 33.35 | 0.23 |
By health state | |||
Induction | 0.06 | 0.06 | 0.00 |
Consolidation | 0.13 | 0.13 | 0.00 |
Interim maintenance | 0.13 | 0.13 | 0.00 |
Delayed intensification | 0.13 | 0.13 | 0.00 |
Interim maintenance (second) | 0.13 | 0.13 | 0.00 |
Maintenance | 0.00 | 0.00 | 0.00 |
Event-free survival | 29.40 | 26.07 | 3.33 |
Relapsed disease | 3.58 | 6.67 | –3.09 |
Discounted costs ($) | |||
Total | 132,444 | 133,443,69 | –999 |
Drug acquisition and administration | 90,197.54 | 86,045.68 | 4,151.86 |
Hypersensitivity | $308.05 | $352.68 | –$44.63 |
Asparaginase activity monitoring | $1,488.78 | $1,495.81 | –$7.03 |
Desensitization protocol | $4,736.02 | $4,766.66 | –$30.64 |
Additional medication | $47.34 | $47.60 | –$0.27 |
Tests and medical visits | $27,088.60 | $34,494.41 | –$7,405.81 |
Adverse events | $8,578.15 | $6,240.84 | $2,337.31 |
Lost productivity | 0 | 0 | 0 |
ICER ($/QALY) | Calaspargase pegol was dominant | ||
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Calaspargase pegol | Pegaspargase | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 39.91 | 39.91 | 0.00 |
By health state | |||
Induction | 0.08 | 0.08 | 0.00 |
Consolidation | 0.15 | 0.15 | 0.00 |
Interim maintenance | 0.17 | 0.17 | 0.00 |
Delayed intensification | 0.15 | 0.15 | 0.00 |
Interim maintenance (second) | 0.15 | 0.15 | 0.00 |
Maintenance | 0.00 | 0.00 | 0.00 |
Event-free survival | 29.99 | 29.99 | 0.00 |
Relapsed disease | 9.23 | 9.23 | 0.00 |
Discounted QALYs | |||
Total | 34.84 | 34.84 | 0.00 |
By health state | |||
Induction | 0.06 | 0.06 | 0.00 |
Consolidation | 0.13 | 0.13 | 0.00 |
Interim maintenance | 0.16 | 0.16 | 0.00 |
Delayed intensification | 0.13 | 0.13 | 0.00 |
Interim maintenance (second) | 0.14 | 0.14 | 0.00 |
Maintenance | 0.00 | 0.00 | 0.00 |
Event-free survival | 27.32 | 27.32 | 0.00 |
Relapsed disease | 6.92 | 6.92 | 0.00 |
Discounted costs ($) | |||
Total | $125,727.55 | $121,639.69 | $4,087.86 |
Drug acquisition and administration | $75,481.45 | $71,347.21 | $4,134.24 |
Hypersensitivity | $308.80 | $353.76 | –$44.97 |
Asparaginase activity monitoring | $1,494.01 | $1,494.01 | $0.00 |
Desensitization protocol | $4,749.89 | $4,749.89 | $0.00 |
Additional medication | $47.60 | $47.60 | $0.00 |
Tests and medical visits | $35,058.43 | $35,058.43 | $0.00 |
Adverse events | $8,587.37 | $8,588.79 | –$1.42 |
Lost productivity | $0.00 | $0.00 | $0.00 |
ICER ($/QALY) | Calaspargase pegol is associated with higher costs and the same amount of QALYs when compared to pegaspargase. | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 11: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed expected budgetary impact resulting from reimbursing calaspargase pegol as a component of MAC for the treatment of patients with ALL.21 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). The sponsor did not evaluate the impact on the Non-Insured Health Benefits program population. The analysis was performed using jurisdiction-specific values by summing up individual provincial results to obtain consolidated results. Key inputs to the BIA are documented in Table 12.
The following key assumptions were made by the sponsor:
For the pediatric population, the sponsor used a BSA of |||| ||, derived from averaging the pediatric BSA in trials DFCI 11 to 001 and COG AALL07P4.6,8
For the pediatric population, a ||||||| split was assumed between the COG AALL07P4 and DFCI 11 to 001 trials. For the adult population, an ||||||| split between the modified DFCI and GMALL trials.22-24
The sponsor assumed calaspargase pegol would capture 100% of the market shares by year 2.
Table 12: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
ALL Incidence Pediatric Adult | 3.25 per 100,00025 0.70 per 100,00026 |
Ph chromosome-negative Pediatric Adult | 97%27 75%27 |
Proportion treated Pediatric Adult | 100% 100% |
Proportion treated with asparaginase-containing MAC regimen Pediatric Adult | |||| |||| |
Number of patients eligible for drug under review Pediatric Adult | ||| | ||| | || |||| | || | || |
Market uptake (3 years) | |
Uptake (reference scenario) Pegaspargase | 100% / 100% / 100% |
Uptake (new drug scenario) Calaspargase pegol Pegaspargase | 50% / 100% / 100% 50% / 0% / 0% |
Cost of treatment (per patient) | |
Cost of treatment over treatment protocol Calaspargase pegol Pediatric Adult Pegaspargase Pediatric Adult | $55,070 $76,800 $52,259 $68,267 |
ALL = acute lymphoblastic leukemia; MAC = multiagent chemotherapeutic; Ph = Philadelphia.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of calaspargase pegol as a component of MAC for the treatment of ALL would lead to an incremental budget impact of $612,476 in year 1, $1,237,537 in year 2 and $1,250,314 in year 3. The total 3-year incremental cost of calaspargase pegol was $3,100,327. Sensitivity analyses were completed by the sponsor to include lymphoblastic lymphoma and relapsed/refractory patients, exclude wastage, extend dosing frequency for adults, as well as change asparaginase dosing by +/− 20%. The results of the sensitivity analyses demonstrated that the 3-year total incremental budget impact may vary from –$534,253 up to $3,364,435.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have implications on the results of the BIA:
Patient characteristics in BIA do not align with CUA: In the sponsor-submitted BIA, a BSA of |||| || and 1.85 m2 were used for the pediatric and adult populations, respectively, to determine the cost of pegaspargase and calaspargase pegol. The pediatric BSA was derived by averaging the BSAs reported in the DFCI 11 to 001 and COG AALL07P4 trials. The adult patient BSA was derived from guidance from Institut national d’excellence en santé et en services sociaux. Alternatively, in the sponsor-submitted CUA, patients entered the model at a BSA of 1.34 m2, derived from the COG AALL07P4 trial population. However, feedback from clinical experts consulted by CADTH highlighted that the DFCI 11 to 001 trial population has characteristics that are more representative of the indicated population in clinical practice. Due to this, the BSA in the CUA and was ultimately altered to |||| || for the CADTH base case to align with the DFCI trial population. Therefore, the incremental budget impact is uncertain.
While the BSA populations do not align in the BIA and CUA, CADTH did not alter the pediatric BSA in the BIA to align with the CADTH base case. As the BIA represents the incremental budget impact over a 3-year horizon, an average BSA is comparable to the population of interest, while using base information reflective of a younger demographic increasing over time was considered more reflective of the eligible population for the CUA.
Use of DFCI 11 to 001 trial protocol is not representative of pan-Canadian clinical use: In the sponsor’s submission, the costs by treatment option were determined by weighting the costs of each treatment by the protocol distribution. For the pediatric population, the protocol distribution was ||| and ||| for the COG AALL07P4 and DFCI 11 to 001 trials, respectively. Feedback from clinical experts obtained by CADTH reported that DFCI 11 to 001 trial protocol was exclusively used for the pediatric population in Quebec. As the pan-Canadian BIA does not include Quebec, the use of the protocol is not appropriate in determining the cost of treatment. The clinical experts, however, agreed that the protocol distribution of ||| and ||| for the modified DFCI trial and GMALL protocols for the adult population, respectively, was reasonable.
To address this limitation, CADTH used a 100% distribution for the COG AALL07P4 trial protocol for the pediatric population.
Pricing, availability, and accessibility of pegaspargase are uncertain: As noted in the appraisal of the economic evaluation, pegaspargase acquisition costs at the time of submission to CADTH were provided by Servier Canada and could not be validated by CADTH. Furthermore, it is unclear how accessible pegaspargase is across Canada; the sponsor noted that it was available in only Ontario, Alberta, British Columbia, and Saskatchewan yet assumed in the BIA that pegaspargase was used and publicly reimbursed across all jurisdictions. The sponsor subsequently indicated that calaspargase pegol would only be used for patients who would have otherwise received pegaspargase, although this does not align with the submitted analysis.
CADTH was unable to address this limitation. If pegaspargase is not publicly reimbursed in all jurisdictions, and calaspargase pegol becomes reimbursed in all jurisdictions, then the estimated incremental cost to the CADTH-participating drug programs of funding calaspargase pegol is underestimated.
CADTH Reanalyses of the BIA
Table 13: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None. | — | — |
Changes to derive the CADTH base case | ||
1. Distribution of DFCI 11 to 001 trial for pediatric population | Trial distribution: COG AALL07P4: ||| DFCI 11 to 001: ||| | Trial distribution: COG AALL07P4: 100% DFCI 11 to 001: 0% |
CADTH base case | Reanalysis 1 | |
The results of the CADTH step-wise reanalysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15. Based on the CADTH base case, the budget impact associated with calaspargase pegol’s reimbursement in the indicated target population is expected to be $913,376 in year 1, $1,841,318 in year 2, and $1,856,090 in year 3, with a 3-year total of $4,610,784.
Table 14: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $3,100,327 |
CADTH base case | $4,610,784 |
BIA = budget impact analysis.
CADTH conducted a scenario analysis to explore the impact of a 11% price reduction in the price of calaspargase pegol, consistent with that required for calaspargase pegol to cost no-more-than pegaspargase.
Table 15: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $15,698,663 | $15,817,521 | $15,938,020 | $16,060,185 | $47,815,725 |
New drug | $15,698,663 | $16,429,997 | $17,175,557 | $17,310,498 | $50,916,052 | |
Budget impact | $0 | $612,476 | $1,237,537 | $1,250,314 | $3,100,327 | |
CADTH base case | Reference | $14,449,004 | $14,613,922 | $14,730,456 | $14,848,631 | $44,193,010 |
New drug | $14,449,004 | $15,527,298 | $16,571,775 | $16,704,721 | $48,803,794 | |
Budget impact | $0 | $913,376 | $1,841,318 | $1,856,090 | $4,610,784 | |
CADTH scenario analysis 1: 11% price reduction | Reference | $14,499,004 | $14,613,922 | $14,730,456 | $14,848,631 | $44,193,010 |
New drug | $14,499,004 | $14,613,922 | $14,730,456 | $14,848,631 | $44,193,010 | |
Budget impact | $0 | $0 | $0 | $0 | $0 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Leukemia and Lymphoma Society of Canada
About the Leukemia and Lymphoma Society of Canada
LLSC (bloodcancers.ca) is a national charitable status organization dedicated to finding a cure for blood cancers and its ability to improve the quality of life of people affected by blood cancers and their families by funding life-enhancing research and providing educational resources, services, and support. The Leukemia and Lymphoma Society of Canada is the largest charitable organization in Canada dedicated to blood cancer, our focus includes:
Funding research from bench to bedside
Rethinking how a person navigates their blood cancer experience
Providing targeted blood cancer information
Offering tools for psychological and emotional support
Empowering Canadians to take charge of their blood cancer experience through practical support and advocacy.
Information Gathering
One online survey was created through SurveyMonkey. Information was gathered from April to May 2023. The survey was developed and distributed by LLSC, in English only. The survey was distributed through various social media channels and directly by email.
The survey asked for input from patients and caregivers of patients who were treated for ALL. 74 respondents participated in this survey. The majority of respondents indicated that they were the ALL patient (47 responses). 20 respondents were a caregiver of an ALL patient. Respondents who indicated that they were not an ALL patient or a caregiver of an ALL patient were disqualified from the survey (7 respondents).
The following is a breakdown of the ages of the ALL patients at their time of diagnosis:
1-4 years old (7)
5-14 years old (7)
15-18 years old (3)
19-29 years old (14)
Age 30+ (35)
(1 respondent did not answer this question or continue with the survey after this question.)
This survey was answered by respondents from all 10 provinces of Canada. The majority of respondents identified the location of their primary residence as Ontario (29 responses), followed by British Columbia (15 responses), Alberta (10 responses), Nova Scotia (7 responses), Saskatchewan (3 responses), Newfoundland and Labrador (3 responses), New Brunswick (2 responses), PEI (2 responses), Manitoba (2 responses) and Quebec (1 response). No respondents indicated that they lived in any of the Canadian Territories, the United States or internationally.
Open-ended responses that reflected the sentiment of participants are included verbatim to provide a deeper understanding of patient perspectives. Patient perspectives and responses to survey questions were consistent among all demographic groups.
The questions respondents were asked in this survey were not intended to measure the efficacy of this treatment. We understand that this treatment is equally as effective as current treatment options and is also budget neutral. The questions in this survey were aimed at highlighting the importance of safeguarding our healthcare system to ensure that treatment medications are supply secure for those experiencing ALL. The respondent answers speak to the value to ALL patients and their families of knowing that treatment options will be available to patients throughout their ALL experience and that healthcare systems will provide treatment options that will ensure the prevention of delays and interruptions to treatment regimens due to drug shortages.
The survey results explained below reflect the feelings and opinions of patients and their families who have experienced treatment for ALL. The responses recorded reflect the experiences, fears, and real-life impact that illness, treatment, treatment issues and delays, and potential drug shortages can cause families to experience when a loved one, or they themselves, are going through ALL. These issues are not limited to their physical health but include themes regarding effects on their well-being as a whole, also considering their emotional and mental health.
4 of our survey respondents made comments in answer to survey questions that reflected that they each were affected by a drug shortage at some point throughout their ALL-treatment experience. They each expressed how this made them feel:
“To be assured the whole protocol can be followed is huge - we ran into a supply a drug supply issue, not fun at all!”
Knowing that all components of ALL treatment are supply secure and will remain available to you throughout the entirety of your treatment:
“It’s extremely valuable, there was a time where my dasatinib was low stock and it was extremely stressful not knowing if I would have the medication, I required the next month.”
“It would be a huge mental setback. Everything seemed to be so scheduled that I felt the healthcare team had everything under control and that was very reassuring for me. I did have a couple of medications that were delayed in being available for a day.”
“During my treatment, for 1 week my protocol was modified because there was a shortage in the drug. I was quite stressed that this would impact the success of the treatment, but the pharmacist reassured me that 1 week without this injection would not be so harmful. However, this raised great concerns for me at the time. I was feeling let down by the system and quite powerless that I was the one having to deal with the consequences. I think if this would have happened for a longer period (more than a week), this would have had a very negative impact on my stress level, quality of life (lack of sleep because of stress, anxiety, feeling powerless), home life (mood swings, spending time trying to find an alternative solution), social life (not wanted to see anyone having to hear tones of cancer advice). Financially, this can have also an impact as people can feel the need to consult side therapies and buy products to help them with trauma, PTSD, energy... and all this could add a significant stress as they are costly temporary fixes. I spent far too much money on omega-3 oils, melatonin tablets, milk powders, B11 vitamins.”
Disease Experience
Without many treatment options available currently, patients are generally limited in having the ability to make choices when it comes to their treatment path.
Patients want to feel that they have a say in their care plans and feel included in decision making as their ALL treatment plan will have an effect on many areas of their lives.
Respondents were asked, “In addition to effectiveness, when you are making decisions about a new cancer treatment, what are the most important factors that you consider? (Select all that apply)” 54 Respondents answered this question. The top answers are as follows.
Table 1: Factors That Patients Would Consider in Decisions For New Cancer Treatment
Side effects | Physician recommendation | Quality of life | Financial cost | Secure supply of the treatment drug | Number of treatments |
|---|---|---|---|---|---|
41/54 (76%) | 40/54 (74%) | 38/54 (70%) | 23/54 (43%) | 22/54 (41%) | 21/54 (39%) |
ALL is an illness that progresses quickly and aggressively and requires immediate treatment upon diagnosis.
In order to try to prevent the progression of ALL or the spread of ALL throughout other areas of the body, ALL patients need treatment drugs to remain readily available so that they can start treatment as quickly as possible.
We presented respondents with an open-ended question asking what they thought the outcomes/implications could have been had they not been able to get started on an ALL treatment regimen right away. The following are some of the direct quotes from respondents in answer to this question:
“The cancer could have spread into my brain”
“Cardiac arrest”
“Death”
“Life threatening”
“I would have died”
“I would have succumbed to my illness”
“death.......he was weeks away!”
“I was told without treatment or delayed treatment I would’ve died within 30 days.”
“I’ve been told that had I not started treatment right away the cancer was progressing quickly, and my body would have been less receptive to the treatment, and I could have died in a matter of weeks.”
“Becoming weaker, meaning I would have less of a chance of withstanding all the treatments. Could have led to incomplete remission, relapse, need for bone marrow transplant, or death.”
“I was told that had I waited a week to come to the hospital I would have probably died. Scary stuff.”
“More complex treatment, more/worse side effects, more painful symptoms.”
“A poorer prognosis, significant symptoms, poorer quality of life and an earlier death.”
Experiences With Currently Available Treatments
Currently available treatments have the benefit of being effective, safe, and made from quality ingredients. Unfortunately, none of these are useful factors for patients if the available drugs are not accessible for their use due to supply issues. Offering alternative treatments that are equally as effective, safe, quality assured and cost effective, ensures that a treatment option will be available for patients should manufacturers of currently available treatments run into supply issues. In turn, knowing that treatment options that are supply secure will remain available to ALL patients, offers these patients and their families’ comfort and peace of mind during an already difficult and challenging time.
Respondents were asked, “Would you be supportive of government provided public funding for a second option for treatment of ALL that works equally well, doesn’t cost the healthcare system more money and provides assurance as an option in case of essential medications becoming unavailable?” 51 Respondents answered this question. 98% of respondents (50/51) answered that they would be supportive, and 2% (1/51) respondent answered that they would have more questions.
Some comments left by those surveyed in response to this question:
“I am supportive of any treatment of the disease. I was lucky not to encounter that issue, but it should be available, whether it costs the same or more.”
“Absolutely! Marketplace diversity is an important element to protect against drug shortages. Alternatives being available and publicly funded could save the system money as well, as patients don't get sicker and require hospitalization.”
Improved Outcomes
Having alternative supply secure treatment options available in case of manufacturing issues leading to a shortage of available treatment drugs could have an extremely significant impact on the overall wellbeing of ALL patients and their families in many ways. Not having this added stress could be exponentially helpful to those experiencing ALL.
Respondents were asked, “What is the value to you of knowing that all components of ALL treatment are supply secure and will remain available to you throughout the entirety of your treatment?” Respondents were asked to rate the level of importance from “Not Important” to “Extremely Important”. 53 respondents answered this question. Respondents’ answers were as follows.
Extremely Important: 49/53 (92.45%)
Neutral:1/53 (1.89%)
Somewhat important: 3/53 (5.66%)
“Having a child undergo treatment for cancer is an experience that is worse than humanly imaginable. To compound that experience because of lack of supply would take this experience to an entirely different level. There are already so many studies of parents of childhood cancer requiring long term therapy and pharmaceutical treatments for PTSD. To not have a regular supply will compound that issue. A person can only process so much when they are in the trenches. There is no acceptable excuse to not have required treatment medications available in a timely manner. This is Canada. The value to me is maximizing the likelihood of my child’s survival and maintaining the care takers own mental health. Everything already feels out of your control as you experience this rollercoaster ride - you shouldn't be forced off the track because of a supply issue.”
“Knowing that you will receive the best possible treatment is paramount to proper mental outlook and treatment outcomes.”
“Cancer is terrible enough to deal with a parent of a child with cancer. Every single thing that complicates treatment and quality of life has an incremental and negative effect on the patient and their family. Some complications cannot be prevented - fevers, nausea etc. Ensuring a secure and readily available supply of medications for treatment is something that can be done and would remove stress that otherwise would add to the patient and carer's lives.”
“We rely on the treatment being available in order to survive. Without it the fear alone is unimaginable.”
“It is extremely valuable and important to be able to access the medications required in order to attempt to stay alive and be with your family, and it’s equally important to feel security in that drug supply chain. Having a positive outlook makes the world of difference to a cancer patients’ outcome so to take away that sense of security and possible hope would be horrible.”
“You need to know that you will be able to complete the treatment from start to finish. If not, this could prolong the amount of time you’re in treatment and also risk your life.”
Experience With Drug Under Review
ALL patients and their families deserve to have the peace of mind of knowing that there are alternative therapies that will remain available to them should drug shortages become a barrier with current treatment options.
Those going through ALL should not have to worry that their ALL treatment will be interrupted or delayed due to manufacturing issues. Treatment interruptions and/or delays can have an impact on ALL patients and their families in many aspects of their lives and not just make a difference in their physical health and disease outcomes.
Survey respondents were asked, “As a patient or caregiver, imagine that your healthcare team tells you that a drug that is a part of your treatment regimen is not available right now due to a supply issue. How would you rate the level of impact that this could have on you and/or your support system in the following areas?” Respondents were asked to rate from 1-No Impact/Not Applicable to 5-Extremely Large Impact. We used weighted measurement to record respondent’s answers. 54 respondents answered this question. Collective ratings were as follows…
Table 2: Level of Impact of the Unavailability of the Treatment Regimen Due to Supply Issue
Mental health | Physical health | Quality of life | Home life | Social life | Work life | Finances |
|---|---|---|---|---|---|---|
4.69/5 | 4.21/5 | 4.16/5 | 3.87/5 | 3.61/5 | 3.04/5 | 2.91/5 |
Some respondents’ comments in response to this question:
“There would have been higher anxiety which would have led to overall mental and physical illness, taking away energy needed to heal.”
“Mentally I would’ve been terrified of death due to drug availability. I would be very angry and I'm pretty sure it would consume my way of thinking.”
“The feeling of despair would be overwhelming.”
“The stress of having cancer is all consuming. And to add the stress of a drug - that you feel may save your life - is not available, would be devastating. Trying to stay optimistic and positive is so difficult and when roadblocks such as this pop up it can take days to rebound from. It's also an issue of control. When you are sick you are at the doctor's mercy. Having no control to help yourself is a hard thing to accept. And in this situation the feeling of helplessness would be crushing.”
“The mental impact of the stress of worrying about when the drug would be available would potentially impact my physical health, my mental well-being and my caregiver’s mental well-being as well. It could also have a substantial impact on your finances if you were living away from home for treatment. Overall, it would be a significant negative impact for my quality of life.”
The mental wellbeing of patients, as well as caregivers, is an integral part of healing when it comes to any illness. Patients’ abilities to understand treatment regimens, participate in their care, have a positive outlook, and follow through with their ALL treatment plans can be greatly affected by poor mental health and negative feelings.
Supply issues with much needed cancer treatment drugs, interruptions, delays, or sudden changes to cancer care plans can lead to major setbacks for patients as well as caregivers. The effects of such a thing do not just have physical effects on patients but mental effects as well. Patients are often already in a fragile mental state due to the worry and stress of their cancer experience. A sudden change or interruption can have a grave effect on patients’ outlooks, feelings, and the level of confidence that patients and caregivers have in their healthcare team, healthcare systems and treatment plans.
Respondents were asked to describe in 3 words how they would feel if their healthcare team told them that a drug that was a part of their treatment regimen was suddenly not available due to a supply issue. The following are the words they used in their answers: afraid, terrified, scared, fearful, angry, anxious, hopeless, devastated, upset, distraught, worried, discouraged, depressed, alone, emotional, despair, frustration, panic, exasperated, disappointment, defeated, shock, helpless, lost, desperate, stressed, sad, misunderstood.
Some additional comments from these respondents:
“Absolutely heartbreaking!”
“Go to USA!”
“Very poor outlook.”
Companion Diagnostic Test
Not applicable.
Anything Else?
Treatment options that will remain readily available for patient use and have less risk of being affected by drug shortages should be considered necessary as alternative treatment options for ALL patients.
Calaspargase Pegol, as an alternative treatment option for ALL has proven to be effective, safe, is made from quality ingredients and is comparably as cost effective to healthcare systems as currently available treatment. Knowing that all aspects of their ALL treatment regimen will remain available to patients throughout the entire course of their treatment will aid in minimizing the difficulties, significant burden and trauma of illness and treatment, and make these unfortunate experiences more manageable for patients and their families.
Calaspargase Pegol in comparison with Oncaspar (pegaspargase) requires that patients receive less does (11vs 16), less often (every 3 weeks, rather than every 2 weeks).
Calaspargase Pegol has a shelf life of 36 months in comparison with Oncaspar’s 8-month shelf life.
These differences mean more time for manufacturers to meet supply demands. These differences could also minimize disruptions to quality-of-life and lessen negative psychosocial aspects of illness and treatment for ALL patients and their families.
We would strongly advise CADTH to recommend reimbursement of Calaspargase Pegol treatment as a component of a multi-agent chemotherapeutic (MAC) regimen for the treatment of patients with acute lymphoblastic leukemia (ALL).
Conflict of Interest Declaration — Leukemia and Lymphoma Society of Canada
Did you receive help from outside your patient group to complete this submission?
No response.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No response.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for Leukemia and Lymphoma Society of Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Servier | X | — | — | — |
Clinician Input
Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
About the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
OH-CCO’s Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information was gathered via videoconferencing.
Current Treatments and Treatment Goals
Current standard treatment is Pegaspargase. These are potentially curative therapies.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
There is no large unmet need as pegaspargase is available; however less frequent dosing may be beneficial.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
This is a standard component of multiagent chemotherapy for ALL.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients eligible for standard induction ALL treatment.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Standard leukemia response criteria.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Progressive disease or significant intolerance.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Acute leukemia treatment centers with leukemia specialists
Additional Information
No.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Lead, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 30-03-2023
Table 4: COI Declaration for OH-CCO’s Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Pierre Villeneuve
Position: Member, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
Date: 20-04-2023
Table 5: COI Declaration for OH-CCO’s Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.