Drugs, Health Technologies, Health Systems
Reimbursement Review
Epcoritamab (Epkinly)
Sponsor: AbbVie Corporation
Therapeutic area: Relapsed or refractory diffuse large B-cell lymphoma
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
aNHL
aggressive B-cell non-Hodgkin lymphoma
ASCT
autologous stem cell transplant
BR
bendamustine plus rituximab
C1D1
cycle 1, day 1
C1D8
cycle 1, day 8
C1D15
cycle 1, day 15
C1D22
cycle 1, day 22
CAR
chimeric antigen receptor
CD3
cluster of differentiation 3
CD20
cluster of differentiation 20
CI
confidence interval
CIT
chemoimmunotherapy
CNS
central nervous system
CR
complete response
CRS
cytokine release syndrome
CTLS
clinical tumour lysis syndrome
DLBCL
diffuse large B-cell lymphoma
DOCR
duration of complete response
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EQ VAS
EQ visual analogue scale
ESS
effective sample size
FACT-G
Functional Assessment of Cancer Therapy–General
FACT-Lym
Functional Assessment of Cancer Therapy–Lymphoma
FACT-LymS
Functional Assessment of Cancer Therapy–Lymphoma Subscale
FAS
full analysis set
FL
follicular lymphoma
FLG3B
follicular lymphoma grade 3B
GCB
germinal centre B cell
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HAS
Haute Autorité de Santé
HGBCL
high-grade B-cell lymphoma
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplant
HTA
Health Technology Assessment
ICANS
immune effector cell–associated neurotoxicity syndrome
IL-6R
interleukin-6 receptor
IPI
International Prognostic Index
IRC
independent review committee
ITC
indirect treatment comparison
LBCL
large B-cell lymphoma
LC
Lymphoma Canada
LLSC
Leukemia & Lymphoma Society of Canada
LYRIC
lymphoma response to immunomodulatory therapy criteria
MAIC
matching-adjusted indirect comparison
NHL
non-Hodgkin lymphoma
NICE
National Institute for Health and Care Excellence
NOC/c
Notice of Compliance with conditions
ORR
objective response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PMBCL
primary mediastinal B-cell lymphoma
pola-B/R
polatuzumab vedotin plus rituximab, with or without bendamustine
pola-BR
polatuzumab vedotin plus bendamustine and rituximab
pola-R
polatuzumab vedotin plus rituximab
PR
partial response
PRO
patient-reported outcome
PT
preferred term
R/R
relapsed or refractory
R-CHOP
rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone
R-CIT
rituximab-based chemoimmunotherapy
RCT
randomized controlled trial
R-GemOx
rituximab, gemcitabine, and oxaliplatin
RP2D
recommended phase II dose
RWE
real-world evidence
SC
subcutaneous
SCT
stem cell therapy
SD
standard deviation
SLR
systematic literature review
SNDS-C
Supplement to a New Drug Submission – Confirmatory
TEAE
treatment-emergent adverse event
TLR
time-limited reimbursement
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Epcoritamab (Epkinly)
|
Sponsor | AbbVie Corporation |
Indication | Treatment of adult patients with relapsed or refractory DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, high-grade B-cell lymphoma, primary mediastinal B-cell lymphoma, or follicular lymphoma grade 3B after 2 or more lines of systemic therapy who have previously received, or are unable to receive, CAR T-cell therapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | October 13, 2023 |
Recommended dose | Cycle 1: 0.16 mg (priming dose) on day 1; 0.8 mg (intermediate dose) on day 8; 48 mg (full dose) on day 15 Cycles 2 and 3: 48 mg once per week Cycles 4 to 9: 48 mg once every 2 weeks (days 1 and 15 only of each cycle) Cycle 10 onward: 48 mg once every 4 weeks (day 1 only of each 28-day cycle) |
Eligible for consideration as a time-limited recommendation | Yes |
CAR chimeric antigen receptor = DLBCL = diffuse large B-cell lymphoma; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; SC = subcutaneous.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma (NHL), accounting for approximately 30% to 40% of all NHL cases in Canada.1 DLBCLs are a heterogeneous group of aggressive B-cell malignancies that differ in clinical presentation, molecular features, prognosis, and treatment options.1,2
The Canadian Cancer Society estimates that 11,400 people in Canada were diagnosed with NHL in 2022, with 3,000 dying from the disease.3 International studies have estimated the incidence of DLBCL in the US and England at approximately 7 cases per 100,000 persons per year.4 Based on statistics from 1975 to 2017, the estimated 5-year relative survival rate at diagnosis was 63.8% in US.5 For patients who are not chemosensitive and are ineligible for autologous stem cell transplant (ASCT), or who relapse after stem cell therapy (SCT) or chimeric antigen receptor (CAR) T-cell therapy, the prognosis is poor, and there is no standard approach to treatment. Available options are currently limited to palliative chemotherapies, including rituximab, gemcitabine, and oxaliplatin (R-GemOx), polatuzumab vedotin with bendamustine and rituximab (pola-BR), and clinical trials with novel drugs.1,6
Epcoritamab is a humanized immunoglobulin gamma 1–bispecific antibody that binds to a specific extracellular epitope of cluster of differentiation 20 (CD20) on B-cells and to cluster of differentiation 3 (CD3) on T-cells. The activity of epcoritamab is dependent upon the simultaneous engagement of CD20-expressing cells and CD3-expressing endogenous T-cells by epcoritamab that induces specific T-cell activation and T-cell–mediated killing of CD20-expressing cells.7
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of epcoritamab for the treatment of adult patients with relapsed or refractory (R/R) DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, high-grade B-cell lymphoma (HGBCL), primary mediastinal B-cell lymphoma (PMBCL), or follicular lymphoma grade 3B (FLG3B) after 2 or more lines of systemic therapy who have previously received or are unable to receive CAR T-cell therapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, Lymphoma Canada (LC) and the Leukemia and Lymphoma Society of Canada (LLSC), responded to CADTH’s call for patient input for the current review of epcoritamab. LC is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. LLSC is a national organization dedicated to finding a cure for blood cancers and to supporting patients and their families by funding life-enhancing research and providing educational resources, services, and support.
LC gathered information for this submission through a survey conducted from October 3, 2023 to November 20, 2023, targeting patients living with large B-cell lymphoma (LBCL). The LC survey data include information from 33 respondents. In addition, LC included a submission from France that was based on a survey regarding the use of epcoritamab for DLBCL conducted by Ensemble Leucémie Lymphomes Espoir with 9 survey respondents, supported by results of the Lymphoma Coalition's 2022 survey that included the experience of patients with DLBCL (n = 171). LLSC conducted 4 1-on-1 interviews in November 2023; 2 interviewees were patients with DLBCL, and 2 were caregivers. Three interviewees resided in Canada and 1 interviewee resided in the US.
According to the input from both groups, living with LBCL is associated with extreme fatigue, body aches, nausea, shortness of breath, lack of energy, and stress and worry, all of which have significant impacts on day-to-day activities and quality of life.
Patient groups identified a need for additional second- and third-line treatment options and described difficulties managing treatment regimens and side effects. Currently available treatments take significant mental and psychological tolls on patients and loved ones, are associated with immense financial burdens, and negatively affect the ability to work, travel long distances, and participate in daily activities. According to both inputs, patients expect new treatments to be more effective and less invasive, with fewer side effects. Patients are seeking choice in their treatment decisions and a variety of options that offer a longer life span, lengthier remission, and better quality of life.
Patients indicated that epcoritamab could offer hope and relief to those with LBCL who require a third-line treatment option; the subcutaneous (SC) administration route could mean less time in hospitals per visit, which can improve the quality of life of patients and caregivers.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH stated that the goal of treatment at this stage is palliative and generally includes maintaining health-related quality of life (HRQoL) by relieving lymphoma-related symptoms, delaying disease progression, and balancing the toxicities of therapy. There is no standard of care in this setting, but options include chemotherapy (e.g., pola-BR or rituximab-based chemoimmunotherapy [R-CIT]), radiation, and potential enrolment in clinical trials. The clinical experts stated that there is an unmet need for safe and effective treatments for patients who are palliative and not eligible for curative treatment, or for whom second-line treatment consisting of SCT or CAR T-cell therapy has not been effective, given that there are limited treatment options for disease control and that currently available options are often associated with significant toxicity that limits these options’ usefulness and applicability. Additionally, patients who relapse after transplant or CAR T-cell therapy often have poor prognoses and very poor bone marrow function, which prevents them from receiving or tolerating further cytotoxic therapy. The clinical experts also noted that there is a significant group of patients who may be eligible for intensive treatments but are unable to access them due to barriers based on location. For example, many patients are unable to travel with caregivers to specialized cellular therapy sites and choose not to have this treatment because they wish to be treated closer to home. As such, there is an additional unmet need for treatments that patients can access and receive closer to home.
After the failure of first-line therapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) (curative intent), treatment in the second line consists of salvage R-CIT and ASCT for patients who are transplant-eligible and chemosensitive (curative intent). Third-line therapy consists of CAR T-cell therapy (curative intent). There is no standard of care following these treatment options; patients who are transplant-ineligible in the second and third lines tend to receive either palliative R-CIT (e.g., pola-BR or R-GemOx) with noncurative intent or radiation or enrol in clinical trials. The clinical experts highlighted that there is a planned shift to the use of CAR T-cell therapy as second-line therapy for primary refractory or early relapsed DLBCL, pending funding, in Canada. The clinical experts emphasized that cytopenias are a major problem associated with palliative cytotoxic treatment options.
Acknowledging that the Health Canada–approved indication for epcoritamab limits usage to patients “who have previously received or are unable to receive CAR T-cell therapy,” the clinical experts consulted by CADTH highlighted that epcoritamab could be a beneficial treatment option in the following circumstances:
for use after patients have received CAR T-cell therapy
for patients who are ineligible to receive CAR T-cell therapy
for patients who are eligible to receive CAR T-cell therapy, but did not (e.g., due to logistical challenges or choice).
The experts noted that these patients would be identified in routine practice by clinicians familiar with the treatment of lymphoma patients undergoing surveillance for relapse (clinical and/or imaging). The experts could not identify a specific subgroup of patients who would demonstrate an enhanced or reduced benefit from epcoritamab treatment. The experts highlighted that repeat biopsy is not always required in cases of suspected relapse of DLBCL unless it is a remote relapse or the patient has a history of indolent lymphoma and it is unclear which lymphoma has relapsed.
The clinical experts stated that patients’ response to treatment would be assessed using the Lugano criteria. Patients would undergo interim imaging every 3 months to confirm response, after which they would either continue or discontinue treatment. Patients are also assessed for lymphoma-related symptoms at each visit; however, the clinical experts noted that while the assessed outcomes are more subjective, these do factor into patients’ decisions to continue therapy. The experts also noted that the frequency of these assessments, and the collection of data, may vary across Canada. In terms of meaningful response to treatment, the clinical experts stated that a response lasting 6 months or longer with improved symptoms can be considered meaningful. The experts did not consider temporary shrinking of tumours beneficial to patients and believed that a meaningful partial response (PR) or complete response (CR) should have a duration of at least 6 months; otherwise, the treatment should be discontinued. Additionally, with a current median overall survival (OS) of 6 months in this population, the experts considered a benefit of at least 6 months and 3 months over the current standard of care to be clinically meaningful for OS and progression-free survival (PFS), respectively.
The clinical experts suggested that treatment with epcoritamab should be discontinued upon overt disease progression or lack of response to treatment. The experts noted that adverse events (AEs) may vary, and resolution of severe AEs can allow for the resumption of therapy; due to this variability, discontinuation should be based on physician judgment and patient request.
The clinical experts indicated that patients with R/R DLBCL are typically under the care of hematologists or oncologists who are familiar with the treatment of lymphoma patients. They also noted that the monitoring and treatment of these patients must be conducted at tertiary centres that have the means to monitor and treat cytokine release syndromes (CRSs), which may require some initial training of site staff before implementation.
Clinician Group Input
Three clinician groups — LC (3 clinicians contributing), the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee (7 clinicians contributing), and the LLSC Nurses Network (6 clinicians contributing) — responded to CADTH’s call for clinician group input.
According to these groups, there are poor and limited treatment options for patients with R/R DLBCL. LC indicated that for patients who are eligible for intensive curative therapies, options such as ASCT and CAR T-cell therapy are considered. Patients who have disease progression after CAR T-cell therapy, or who are unfit for this therapy for medical and/or social reasons, have the greatest unmet need for treatment because no other curative-intent therapy is readily available to them.
In contrast, those who are not eligible for curative ASCT-based or CAR T-cell therapy approaches are managed with palliative approaches, such as with pola-BR or tafasitamab (an anti–cluster of differentiation 19 antibody) in combination with lenalidomide. CADTH notes that tafasitamab in combination with lenalidomide received a do not reimburse recommendation and is not currently reimbursed by participating drug programs.8 A small percentage of patients might pursue allogeneic stem cell transplant, but the vast majority of patients in this setting are managed with a variety of palliative chemotherapy regimens, radiation therapy, or clinical trials. Multiple novel drugs (ibrutinib, lenalidomide, tafasitamab, and selinexor) do not have Health Canada approvals or provincial funding for R/R DLBCL.
The clinician groups noted that there is an unmet need for safe and effective treatments for patients who are not eligible for CAR T-cell therapy or for whom second-line treatment has not been effective. LC and the LLSC Nurses Network added that there are limited treatment options for disease control, and that currently, the available options are often associated with significant toxicity, side effects, and mental and physical treatment fatigue. LC stated that while ASCT or CAR T-cell therapy are considered effective for some patients, others are unable to access them due to location barriers. Only select sites are equipped to offer CAR T-cell therapy.
The most important goals of treatment for DLBCL, according to clinician groups, are to prolong survival, delay disease progression, and improve symptoms, which in turn can improve quality of life of patients and caregivers. Clinician groups agreed, in regard to the indication, that epcoritamab can be used in the third line or beyond if the patient was previously treated with CAR T-cell therapy or is ineligible for CAR T-cell therapy. LC and the LLSC Nurses Network stated that as an off-the-shelf product, this treatment could alleviate regional access issues, and that the SC injection could become a more feasible, well-favoured option than currently available treatments.
According to the clinician groups, improved survival (PFS, OS), blood work, decreased presence of cancer cells in bone marrow, and improvement in disease symptoms are outcomes used to determine whether a patient is responding to treatment. LC added that a clinically meaningful response would be PR or CR, typically determined using CT and or PET scans.
The clinician groups agreed that discontinuation of therapy should be considered in patients with disease progression or toxicity, and that epcoritamab can be given in any inpatient and outpatient setting that has expertise in managing CRS and neurotoxicity as well as the ability to admit and monitor patients receiving anticancer therapy.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, funding algorithm, care provision issues, and system and economic issues. Refer to Table 7 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, phase I and II, open-label, single-arm study, the EPCORE NHL-1 study, was included in this review. The review for epcoritamab was based on the dose expansion phase of the study, which consisted of 157 patients with R/R LBCL who had relapsed after or had not responded to at least 2 prior systemic treatment regimens. Patients were excluded if they had a known primary central nervous system (CNS) lymphoma or known CNS involvement or had CAR T-cell therapy within 30 days or ASCT within 100 days before the first dose of epcoritamab, or any prior allogeneic hematopoietic stem cell transplant (HSCT). Eligible patients received treatment with epcoritamab monotherapy at the step-up recommended doses: a priming dose of 0.16 mg (cycle 1, day 1 [C1D1]), an intermediate dose of 0.8 mg (cycle 1, day 8 [C1D8]), and a full dose of 48 mg (cycle 1, day 5 [C1D15], cycle 1, day 22 [C1D22], and every 4 weeks thereafter until unacceptable toxicity or disease progression). The primary end point was objective response rate (ORR), with secondary or exploratory end points of CR, duration of response (DOR), PFS, OS, HRQoL, and safety.
The majority of patients in the intention-to-treat population of patients with LBCL had DLBCL (88.5%), with smaller subgroups who had HGBCL (5.7%), PMBCL (2.5%), or FLG3B (3.2%). Patients had received 2 (29.9%), 3 (30.6%), or 4 or more prior lines of antilymphoma therapy (39.5%), and a majority were refractory to their last prior therapy (82.8%). Prior CAR T-cell therapy was reported for 38.9% of patients, and 19.7% had received prior stem cell transplant.
Efficacy Results
Table 2 summarizes results for the efficacy end points from the EPCORE NHL-1 trial using the most recent data cut-off date (April 21, 2023).
OS: || ||||||| patients died, resulting in a median OS of 18.5 months (95% confidence interval [CI], 11.7 to ||||). The estimated proportions of patients who remained alive at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || ||||||| respectively.
PFS: ||| ||||||| patients experienced a PFS event (disease progression or death) based on the Lugano criteria. The median PFS duration was 4.4 months (95% CI, 3.0 months to 8.8 months). The estimated percentages of patients remaining progression-free at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || ||||||| respectively. Overall results for the 18 patients in the cohort of patients with other LBCL subtypes were similar to those for the cohorts of patients with LBCL and DLBCL.
CR: The CR rate, based on Lugano criteria, was ||||| |||| ||| |||| || ||||| when determined by the independent review committee (IRC) and ||||| |||| ||| |||| || ||||| when determined by the investigator. The median duration of complete response (DOCR) was |||| |||||| |||| ||| |||| || ||| when assessed by IRC and |||| |||||| |||| ||| |||| || ||| when assessed by the investigators.
ORR: The ORR (CR plus PR) in patients with LBCL was 63.1% (95% CI, 55.0% to 70.6%), with || ||||||| and || ||||||| patients achieving best response of CR and PR, respectively.
DOR: For patients who had achieved PR or CR || | |||| the median DOR was |||| |||||| |||| ||| ||| || ||||| when assessed by IRC using Lugano criteria. The estimated percentages of patients still responding at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || ||||||| respectively. The median DOR was |||| |||||| (95% CI, 13.0 to 26.5) when assessed by the investigators using Lugano criteria.
Functional Assessment of Cancer Therapy–Lymphoma (FACT-Lym) total score: 140 patients completed the FACT-Lym, and the mean score at baseline was 118.4 (standard deviation [SD] = 25.47). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| ||||||| and |||| |||||||| respectively. At the end-of-treatment assessment (n = 54), the mean change from baseline in total score was |||| |||||||.
Functional Assessment of Cancer Therapy–General (FACT-G) total score: 140 patients completed the FACT-G, and the mean score at baseline was 76.2 (SD = 16.86). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| ||||||| and ||| |||||||, respectively. At the end-of-treatment assessment || | |||, the mean change from baseline in total score was |||| ||||||||.
Functional Assessment of Cancer Therapy–Lymphoma Subscale (FACT-LymS): The sponsor evaluated 6 questions from the FACT-Lym that were related to the symptoms of lymphoma: body pain (P2), severe fever (BRM3), night sweats (ES3), lack of energy (GP1), tires easily (BMT6), and weight loss (C2). One hundred and 40 patients completed the FACT-LymS, and the mean score at baseline was 42.2 (SD = 9.98). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| |||||| and ||| ||||||| respectively.
Harms Results
As of the data cut-off date (April 21, 2023), ||| ||||||| patients with LBCL had experienced at least 1 treatment-emergent adverse event (TEAE). A total of ||| ||||||| patients experienced grade 3 or higher TEAEs. The most frequent TEAEs (experienced by at least 20% of patients) by preferred term (PT) were CRS (80 patients [51.0%]), pyrexia (not attributed to CRS; || |||||||), fatigue (|| |||||||), neutropenia (|| |||||||||), nausea) (|| |||||||||), anemia (|| |||||||||), and diarrhea (|| |||||||). The most common grade 3 or 4 TEAEs (experienced by ≥ 5% of patients) by PT in patients with LBCL (N = 157) were neutropenia (|| ||||||| patients), anemia (|| ||||||| patients), neutrophil count decrease (|| |||||| patients), COVID-19 (|| |||||| patients), and thrombocytopenia (| |||||| patients). Serious TEAEs were reported in ||| ||||||| patients. The most frequent serious TEAEs (experienced by ≥ 2% of patients) by PT in patients with LBCL were CRS (|| |||||||||); COVID-19 (|| |||||| patients); COVID-19 pneumonia (| |||||| patients); pleural effusion (| ||||||||); pneumonia (| |||||| patients); pyrexia (not attributed to CRS); sepsis; immune effector cell–associated neurotoxicity syndrome (ICANS); and febrile neutropenia (| |||||| patients each). ||||| patients experienced at least 1 TEAE that led to treatment discontinuation, and ||||| of patients had at least 1 TEAE that led to delayed dosing.
CRS: Eighty patients (51.0%) had at least 1 CRS event. The majority of these were grade 1 (50 patients out of 80 patients) and occurred most frequently after the first full dose of epcoritamab (65 patients out of 80 patients). Grade 2 and grade 3 events occurred in 25 patients and 5 patients, respectively, out of 80 patients. There were no grade 4 or 5 events. The CRS symptoms resolved in || || || ||||||| patients.
ICANS: ICANS events were reported in 10 patients (6.4%); of these, 7 patients (4.5%) had grade 1 ICANS, 2 patients (1.3%) had grade 2 ICANS, and 1 patient (0.6%) had grade 5 (fatal) ICANS. | ||||||| patients experienced ICANS events that led to dose delay, and in | ||||||| patient, this event led to treatment discontinuation.
Clinical tumour lysis syndrome (CTLS): Two patients (1.3%) experienced CTLS events. Both were grade 3 in severity. ||||||| ||||| ||| |||||||| ||||| || ||| ||||||||| |||||| ||||| ||| || ||||||| ||||||||||||||.
Critical Appraisal
The EPCORE NHL-1 trial is an ongoing, phase I and II, multicentre, open-label, single-arm study of epcoritamab. The trial is being conducted as part of a clinical trial program, including the ongoing comparative phase III trial, EPCORE DLBCL-1. The single-arm trial was justified considering that the study was designed as an early-phase I and II study in which an internal comparator group is not required; it is also justified given the severity of illness for patients at this stage (i.e., those with refractory or relapsed illness after at least 2 lines of prior systemic therapy). However, the decision to conduct a single-arm study has implications for the overall strength and interpretability of the results. There is an increased risk of bias in the estimation of treatment effects due to the potential for confounding related to natural history and prognostic factors. The potential influence of selection bias is also difficult to ascertain in a single-arm study. Additionally, time-to-event end points cannot be adequately assessed in a single-arm trial because all patients receive the same treatment. As such, the effect of epcoritamab on time-to-event end points, such as PFS, OS, and DOR, is uninterpretable, and can be considered as exploratory and supportive only.
Health Canada issued a Notice of Compliance with conditions (NOC/c) for epcoritamab based on promising results from the EPCORE NHL-1 trial. In the absence of a comparator group in the trial, assessing the comparative clinical value of epcoritamab relies on indirect treatment comparisons (i.e., unanchored, matching-adjusted indirect comparisons [MAICs]), which rely on numerous assumptions about the comparability of treatment groups, thereby increasing the uncertainty related to the comparative efficacy. The uncertainty in the comparative efficacy of epcoritamab versus relevant comparators was acknowledged by Health Canada, which has specified that the sponsor must provide phase III trial results showing that epcoritamab improves the OS of patients with DLBCL compared to investigator's choice of either bendamustine plus rituximab (BR) or R-GemOx. In addition to the single-arm design, the study was administered in an open-label manner: the investigator and study participants were aware of their treatment status, potentially increasing the risk of detection and performance biases. As such, the open-label trial design limits the interpretability of the subjective study outcomes, such as tumour response, patient-reported outcomes (PROs) (including HRQoL), and AEs. To mitigate the impact of this bias, PFS and ORR were assessed by both the IRC and investigator using the Lugano classification criteria for the response.
The EPCORE NHL-1 study was an international, multicentre study, and the clinical experts consulted by CADTH had no concerns regarding the generalizability of its results to the Canadian setting. The experts noted that the baseline characteristics were a reasonable reflection of the patient population for whom epcoritamab could be considered an appropriate treatment in clinical practice in Canada. The proportion of patients with an Eastern Cooperative Oncology Group (ECOG) Performance Status of 2 was relatively low (3.2%); the clinical experts noted that this could be greater in clinical practice, especially given that the indication is limited to those who are ineligible for CAR T-cell therapy. The clinical experts noted that 40% of patients with prior CAR T-cell therapy exposure is a reasonable reflection of the target population in Canada (although percentages would vary across jurisdictions), and that the overall proportion of patients with stem cell transplant could be slightly lower than might be anticipated in routine practice in Canada for patients for whom 2 or more lines of systemic therapy have not been effective. The treatment regimen used in the EPCORE NHL-1 trial aligns with the recommendations on the Health Canada–approved product monograph for epcoritamab (i.e., a priming dose of 0.16 mg, an intermediate dose of 0.8 mg, and a full dose of 48 mg thereafter).7 The clinical experts consulted by CADTH noted that the dosages of epcoritamab and the medications used to manage AEs during the treatment period are reflective of the regimen that would be administered in practice in Canada.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.9,10 Although GRADE guidance is not available for noncomparative studies, the CADTH review team assessed pivotal single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty, with no opportunity for rating up.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: median OS, median PFS, change from baseline in HRQoL, and clinical response (i.e., CR, ORR, and median DOR). For time-to-event outcomes, landmark analyses at 12 months and 18 months were also of interest.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The target of the certainty of evidence assessment was the presence of a clinically important improvement in survival (OS and PFS) and HRQoL, which were considered the most important outcomes of treatment by the clinical experts consulted by CADTH, the clinician group, and patient groups. According to the clinical experts consulted by CADTH, clinically important thresholds for the outcomes of OS and PFS were a benefit of at least 6 months and 3 months over current standard of care for OS and PFS, respectively. Additionally, response to treatment (CR, ORR, and DOR) was included in the certainty of evidence assessment based on the potential translation to long-term survival outcomes.
Table 2: Summary of Findings on Epcoritamab for Patients With R/R DLBCL
Outcome and follow-up | Patients (studies), N | Effect | Certaintya | What happens |
|---|---|---|---|---|
Survival | ||||
OS Follow-up (median): 25.1 months (95% CI, 24.0 months to 26.0 months) | 157 (1 single-arm trial) | Median (95% CI) OS: 18.5 months (11.7 months to ||||) 12-month OS rate (95% CI): ||||| |||||| || |||||| 18-month OS rate (95% CI): ||||| |||||| || |||||| | Very lowb, c | The evidence about the effects of epcoritamab on OS vs. any comparator is very uncertain. |
PFS (IRC-assessed) Follow-up (median): 22.3 months (95% CI, 22.0 months to 23.0 months) | 157 (1 single-arm trial) | Median (95% CI) PFS: 4.4 months (3.0 months to 8.8 months) 12-month PFS rate (95% CI): ||||| |||||| || |||||| 24-month PFS rate (95% CI): ||||| |||||| || |||||| | Very lowb, c | The evidence about the effects of epcoritamab on PFS vs. any comparator is very uncertain. |
HRQoL | ||||
FACT-Lym Follow-up (median): NR | 157 (1 single-arm trial) | Total score: Mean (SD) CFB to cycle 5: ||| ||||||| Mean (SD) CFB to cycle 7: |||| ||||||| | Very lowb, c, d, e | The evidence about the effects of epcoritamab on FACT-Lym vs. any comparator is very uncertain. |
FACT-G total score Follow-up (median): NR | 157 (1 single-arm trial) | Total score: Mean (SD) CFB to cycle 5: ||| ||||||| Mean (SD) CFB to cycle 7: ||| ||||||| | Very lowb, c, d, e | The evidence about the effects of epcoritamab on FACT-G vs. any comparator is very uncertain. |
FACT-Lym symptoms Follow-up (median): NR | 157 (1 single-arm trial) | Total score: Mean (SD) CFB to cycle 5: ||| |||||| Mean (SD) CFB to cycle 7: ||| |||||| | Very lowb, c, d, e | The evidence about the effects of epcoritamab on FACT-Lym symptoms vs. any comparator is very uncertain. |
Clinical response to treatment | ||||
CR (95% CI) (IRC-assessed) Follow-up (median): 20.8 months (95% CI, 20.4 months to 21.1 months) | 157 (1 single-arm trial) | ||||| |||| ||| |||| || ||||| | Lowe | Epcoritamab may result in a large CR rate, but the evidence is still uncertain. |
ORR (IRC-assessed) Follow-up (median): 20.8 months (95% CI, 20.4 months to 21.1 months) | 157 (1 single-arm trial) | 63.1% (95% CI, 55.0% to 70.6%) | Lowe | Epcoritamab may result in a large ORR, but the evidence is still uncertain. |
DOR (IRC-assessed) Follow-up (median): 20.8 months (95% CI, 20.4 months to 21.1 months) | 157 (1 single-arm trial) | Median (95% CI) DOR: |||| |||||| |||| || ||||| 12-month event-free rate (95% CI): ||||| |||||| || |||||| 18-month event-free rate (95% CI): ||||| |||||| || |||||| | Very lowb, c | The evidence about the effects of epcoritamab on DOR vs. any comparator is very uncertain. |
CFB = change from baseline; CI = confidence interval; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HRQoL = health-related quality of life; IRC = independent review committee; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; R/R = relapsed or refractory; SD = standard deviation.
Note: All serious concerns with respect to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias are documented in the table footnotes.
aIn the absence of a comparator group, conclusions about efficacy relative to any comparator cannot be drawn, and the certainty of evidence is started at very low.
bIn the EPCORE NHL-1 trial, statistical testing for this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
cRated down 1 level for serious imprecision due to the low number of events and the small sample size.
dRated down 1 level for serious risk of bias due to potential for bias arising from the open-label nature of the study and the subjective nature of the outcome.
eDespite the study limitations resulting in the certainty of evidence starting as “very low,” the outcomes of CR and ORR are demonstrative of an antitumour effect, which is supported by regulatory authorities (FDA, Health Canada, and European Medicines Agency). As such, given the effect size, which was believed to be large and clinically important, the CADTH review team considered the certainty of this evidence to be higher. Note that the outcome could be rated down 1 level for serious indirectness because a surrogate outcome (ORR) was used as the primary outcome in the place of OS and PFS.
Source: Sponsor’s clinical submission.11
Indirect Treatment Comparisons
Description of Studies
One sponsor-submitted, indirect treatment comparison (ITC) was summarized and critically appraised by CADTH. The MAICs focused on 3 patient populations: the overall population of patients with LBCL; the population of patients with LBCL with no prior CAR T-cell therapy; and the population of patients with LBCL without prior CAR T-cell therapy who were considered eligible to receive CAR T-cell therapy. The ITCs of interest for the CADTH review were epcoritamab versus pola-BR and R-CIT. The sponsor-submitted ITC included comparisons against 3 CAR T-cell therapy regimens: axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel. Given that the Health Canada–approved indication for epcoritamab states that the drug is approved for use only in patients “who have previously received or are unable to receive CAR T-cell therapy,” CADTH does not consider CAR T-cell therapies to be relevant comparators for the current review. The approach is consistent with applications that have been filed in the same therapeutic area. Outcomes evaluated in the MAIC included OS, PFS, ORR, and CR.
Efficacy Results
Epcoritamab Versus Polatuzumab Vedotin With Rituximab, With or Without Bendamustine (Overall Population of Patients With LBCL)
In the adjusted overall population of patients with LBCL, the sponsor reported that epcoritamab was associated with significant improvements in both PFS |||| |||||| ||| ||| ||||| || |||||| | | |||||| and OS |||| |||||| ||| ||| ||||| || |||||| | | |||||| compared to polatuzumab vedotin with rituximab, with or without bendamustine (pola-B/R). The sponsor also reported a significant improvement with epcoritamab versus pola-B/R in both CR rate ||||||| ||| ||| ||||| || |||||| |||||||| and ORR ||||||| ||| ||| |||| || |||||| | | ||||||.
Epcoritamab Versus Pola-BR (Patients Without Prior CAR T-Cell Therapy)
Compared with pola-BR in the analysis for patients with prior CAR T-cell therapy, the sponsor reported no significant difference in PFS |||| |||||| ||| ||| ||||| || |||||| | | ||||||, OS |||| |||||| ||| ||| ||||| || |||||| | | ||||||, or CR rate ||||||| ||| ||| |||||| || |||||| | | ||||||. The sponsor reported that epcoritamab was associated with an improvement in ORR versus pola-BR ||||||| ||| ||| |||| || |||||| | | ||||||.
Epcoritamab Versus Chemoimmunotherapy (Patients Without Prior CAR T-Cell Therapy)
PFS could not be reported for the comparison versus R-CIT in the population of patients with LBCL with no prior CAR T-cell therapy. Compared to chemoimmunotherapy (CIT), the sponsor reported that epcoritamab was associated with significant improvements in OS |||| |||||| ||| ||| ||||| || |||||| | | ||||||; CR rate ||||||| ||| ||| ||||| || |||||| | | ||||||; and ORR ||||||| ||| ||| ||||| || |||||| | | ||||||.
Critical Appraisal
Given the lack of direct evidence comparing epcoritamab to relevant treatments in the R/R DLBCL third-line setting, the sponsor’s decision to conduct an ITC (i.e., unanchored MAIC) was justified. There were important differences in the design of the included studies and the cohorts evaluated that limit the ability to draw strong conclusions about the efficacy of epcoritamab compared with pola-BR and R-CIT. The EPCORE NHL-1 study of epcoritamab was a phase I and II, single-arm study, whereas the GO29365 study was a comparative, phase Ib and II randomized, open-label study; the SCHOLAR-1trial was a retrospective research study; and Liebers et al. (2021) was a real-world study. In addition, all the comparisons involved the use of subgroup data from 1 or both of the studies included in the ITC.
In addition to differences in study design, there were notable differences in the eligibility criteria of the included studies, resulting in heterogeneity in baseline characteristics across populations. The sponsor provided a comprehensive list of likely prognostic factors and treatment-effect modifiers (identified through consultation with clinical experts). However, adjustment of all these factors could not be achieved due to differences in reporting across the various studies and a lack of access to patient-level data (other than for those enrolled in the EPCORE NHL-1 trial). It is unclear if the lack of adjustment for differences in baseline characteristics (particularly those that may be prognostic. such as primary refractory disease) would have an impact on the results of the MAIC. A key limitation of the sponsor-submitted MAICs, which is a limitation inherent to all unanchored MAICs, is that it assumes that all effect modifiers and prognostic factors are accounted for in the model. This assumption is largely considered impossible to meet, according to the National Institute for Health and Care Excellence (NICE) Decision Support Unit Technical Guidance report on the methods for population-adjusted indirect treatment comparisons (ITCs).
Overall, CADTH concluded that there were multiple limitations in the sponsor-submitted MAICs, including differences in inclusion and exclusion criteria, heterogeneity in baseline characteristics across studies, and notable reductions in sample sizes due to matching and weighting. There was also significant uncertainty about the overall generalizability of the results to the population in Canada. Additionally, wide 95% CIs led to imprecision and uncertainty in the results.
CADTH notes that the Haute Autorité de Santé (HAS) in France similarly concluded that no formal conclusions can be drawn from the sponsor’s MAICs, citing methodological limitations that include uncertainty regarding the quality of the data (particularly the real-world evidence [RWE]), significant heterogeneity between the populations included in the different studies, and residual differences across the various treatments after weighting. However, NICE acknowledged that, despite the uncertainty associated with the sponsor’s MAICs, epcoritamab was likely to be more effective than R-CIT based on the sponsor’s MAIC. Clinical experts consulted by NICE noted that epcoritamab could plausibly be more effective than pola-BR; however, the NICE expert committee noted that there was too much uncertainty with the ITC and concluded that an assumption of equal efficacy would be more appropriate to inform the economic evaluation.
Long-Term Extension Studies
Not applicable.
Studies Addressing Gaps in the Evidence From the Systematic Review
Not applicable.
Consideration for a Time-Limited Recommendation
A time-limited reimbursement (TLR) recommendation is a recommendation by the CADTH expert committee to publicly fund a drug or drug regimen for a certain period of time based on the condition that the sponsor will conduct 1 or more clinical studies that address uncertainty in the clinical evidence. CADTH would subsequently conduct a reassessment of the additional evidence and issue a final reimbursement recommendation within a defined period of time. Based on the preliminary assessment by CADTH, epcoritamab meets the criteria to be considered by the expert committee for a TLR recommendation.
The basis for the TLR recommendation and subsequent reassessment would be the ongoing EPCORE DLBCL-1 phase III study, which is comparing epcoritamab monotherapy with investigator’s choice of either BR or R-GemOx for improvement in the OS of patients with DLBCL. The primary end point is OS, and the NOC/c Qualifying Notice from Health Canada states that the sponsor should acknowledge that authorization may be revoked if the trial fails to show an OS benefit for epcoritamab over BR or R-GemOx. The clinical experts consulted by CADTH expressed concerns regarding the choice of comparator in the EPCORE DLBCL-1 trial (i.e., BR or R-GemOx) because it was felt that the efficacy data from the EPCORE NHL-1 trial were compelling and that BR and R-GemOx would be associated with significant toxicities for patients. The experts noted that patients at this stage of disease would likely have already received R-CIT in the course of disease and been shown to be refractory to the treatment; as such, the experts expressed concerns regarding clinical equipoise in the trial, with a belief that those randomized to BR or R-GemOx would be receiving an inferior treatment option. The clinical experts noted that more appropriate comparators would be newer therapies that have recently emerged in the second- and third-line setting, such as pola-BR and CAR T-cell therapy. CADTH noted that the curative potential of CAR T-cell therapy would typically make this the preferred option for many patients. This could pose challenges in the design and conduct of a comparative clinical trial.
In its comments on the draft report, the sponsor clarified that at the start of the EPCORE DLBCL-1 study (January 2021), neither CAR T-cell therapy nor pola-BR were widely used. Therefore, R-CIT was considered the most appropriate comparator, and CIT remains a treatment option in practice in Canada for the treatment of R/R LBCL. CADTH agrees with the sponsor’s assessment regarding the choice of comparator at the time of initiating the EPCORE DLBCL-1 trial. (The final CADTH recommendation for pola-BR was issued in April 2021, 4 months after the trial began.)
Conclusions
One phase I and II, single-arm, open-label trial, the EPCORE NHL-1 trial, provided evidence for the efficacy and safety of epcoritamab in adult patients with R/R LBCL who have relapsed after or have not responded to at least 2 prior systemic therapies. Clinicians and patients highlighted the need for accessible, alternative treatment options for patients in this treatment setting. Improvements in survival were considered the most important outcomes of treatments by patients and clinicians. Although OS and PFS were evaluated in the EPCORE NHL-1 trial, the single-arm design precludes the ability to accurately evaluate the impact of epcoritamab treatment on these important end points. Nonetheless, the study demonstrated that ||||| of patients achieved CR, which was considered a clinically important result by the clinical experts consulted by CADTH. HRQoL is an outcome that is important to patients, and many patients in the EPCORE NHL-1 trial demonstrated improvements from baseline after initiating the treatment; however, due to the noncomparative design, high patient attrition rates, and open-label administration of the treatment, the effect of epcoritamab on HRQoL remains uncertain. Overall, the clinical experts consulted by CADTH believe the results demonstrated that epcoritamab offers clinically meaningful improvements for this heavily pretreated patient population and may help address an unmet medical need for a treatment that may extend life, improve symptoms, and be considered tolerable by patients.
Harms associated with epcoritamab were largely consistent with the mechanism of action, including a high frequency of patients experiencing CRS (50.1%) and serious infections (|||||||). All patients received pretreatment with standardized medications (i.e., prednisolone, diphenhydramine, and acetaminophen) to mitigate the risk of CRS. The majority of patients recovered from the CRS events, and the product monograph provides detailed guidance on grading and managing these events in practice. The clinical experts consulted by CADTH noted that patients will likely require hospitalization for a 24-hour monitoring period after the first full dose of epcoritamab (because outpatient monitoring may be challenging) and that this requirement may pose important challenges for the health system and could limit the adoption of the treatment.
There were important limitations with the sponsor-submitted ITCs that were used to inform the comparative effectiveness of epcoritamab versus R-CIT and pola-BR (which were the comparators considered most relevant for this review, given the Health Canada–approved indication for epcoritamab). Results for the MAICs varied across the comparisons for epcoritamab versus pola-B/R, with the sponsor claiming significant improvements in PFS, OS, CR rate, and ORR in the overall population of patients with LBCL and no significant difference in PFS, OS, or CR rate in the population without prior CAR T-cell therapy. CADTH considered the analyses of epcoritamab versus pola-BR to be associated with significant uncertainty due to small sample sizes and heterogeneity across the studies and patient populations. The sponsor’s MAIC suggested that epcoritamab was superior to CIT for patients with no prior exposure to CAR T-cell therapy. The clinical experts consulted by CADTH felt that it was plausible that treatment with epcoritamab could offer greater clinical benefits for patients in comparison to R-CIT for the target patient population on the basis that these patients have already demonstrated disease progression and drug resistance following exposure to their initial R-CIT regimen (typically R-CHOP) and that the potential toxicity of R-CIT regimens at this stage of disease can limit the regimens’ clinical utility. However, important limitations with the MAIC make it challenging to quantify the magnitude of potential added benefit and preclude the ability to draw evidence-based conclusions regarding the comparative effectiveness of epcoritamab versus R-CIT. Given that the sponsor has been mandated by Health Canada to conduct a head-to-head trial against R-CIT in the relevant patient population, there will be direct evidence to inform the comparative clinical benefit. This evidence could be included in a reassessment application as part of a TLR.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of epcoritamab for the treatment of adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, HGBCL, PMBCL, or FLG3B after 2 or more lines of systemic therapy who have previously received or are unable to receive CAR T-cell therapy.
Disease Background
The contents of this section were informed by materials submitted by the sponsor and by clinical expert input. These have been summarized and validated by the CADTH review team.
DLBCL is the most common type of NHL, accounting for 30% to 40% of all NHL cases in Canada.1 DLBCLs are a heterogeneous group of aggressive B-cell malignancies that differ in clinical presentation, molecular features, prognosis, and treatment options. Some types of indolent B-cell lymphomas (e.g., follicular lymphoma) can transform into DLBCL.1,2
Patients with DLBCL typically present with enlarged symptomatic masses in the lymph nodes, typically in the neck, chest, or abdomen; however, widespread DLBCL can also arise in tissues outside the lymph nodes (i.e., extranodal involvement), such as in the bone marrow, bones, brain, and gastrointestinal tract, among others. DLBCL can also cause systemic B symptoms (i.e., unexplained fever, weight loss, and night sweats) and elevated serum lactate dehydrogenase.4
DLBCL is an aggressive disease that is typically diagnosed in more advanced stages; however, 30% to 40% of patients are diagnosed when the disease is localized (stage I or II).2 It is diagnosed through surgical or core needle biopsy, usually of an involved lymph node or extranodal site. Histological evaluation is performed in accordance with the WHO classification of lymphoid neoplasms, which categorizes lymphomas on the basis of cytology, immunophenotype, and genetic and clinical features.12 A morphological diagnosis of the cell of origin to distinguish between activated B-cell type (25% to 30% of DLBCL cases) and germinal centre B-cell (GCB) type (approximately 60% of DLBCL cases) is generally confirmed by immunohistochemistry or flow cytometry and has been considered a major prognostic factor in the first-line treatment of DLBCL.6,13-16 Other molecular subtypes with prognostic implications have been identified, including double-hit lymphoma (with concurrent translocations of MYC and either BCL2 or BCL6), a particularly aggressive, high-risk subtype with poor prognosis.12,17 Double-expressor lymphoma (with overexpression of MYC and BCL2) is not considered a separate entity, and has also been associated with a worse prognosis.18,19 However, the prognostic significance of these biomarkers remains controversial, and optimal clinical management has not been established.19-22
Disease staging is crucial for determining the appropriate treatment and assessing the prognosis. The gold standard for staging patients with DLBCL is PET or CT scan. Patients are initially divided into 2 groups by stage: limited or advanced. The extent of DLBCL is determined using the Ann Arbor staging classification system, which further categorizes the disease into 4 stages according to the extent of lymph node and extranodal sites involvement. For prognostic purposes, the International Prognostic Index (IPI) is calculated to assign a prognosis to patients undergoing treatment with chemotherapeutic regimens.23
There are limited estimates of DLBCL incidence and prevalence in Canada. The Canadian Cancer Society estimated that 11,400 people in Canada were diagnosed with NHL in 2022 (with approximately 40% of cases being DLBCL), with 3,000 dying from the disease.1,3 International studies have estimated the incidence of DLBCL in the US and UK at approximately 7 cases per 100,000 persons per year.4
Although the cure rate of DLBCL is high, 30% to 50% of patients will relapse after or be refractory to treatment with standard, first-line R-CHOP or a similar regimen.16,24 The estimated 5-year relative survival at diagnosis with DLBCL was 63.8% in the US, based on statistics from 1975 through 2017.5 Until the recent approval of CAR T-cell therapy, treatment for patients not eligible for SCT or who relapsed after SCT had been largely palliative, with median survival being approximately 6 months.25
Standards of Therapy
The contents of this section were informed by materials submitted by the sponsor and clinical expert input. These have been summarized and validated by the CADTH review team.
First-line treatment for DLBCL is relatively standardized across Canada, with most patients receiving R-CHOP once every 3 weeks.6,26 While most patients respond well to R-CHOP, 30% to 50% of patients will either be refractory to or relapse following first-line therapy. Patients who relapse early (within 12 months)1 and patients with refractory disease have worse prognoses than those who do not relapse within 12 months, even with second-line therapy.26,27
Patients requiring second-line treatment for R/R DLBCL are classified based on their eligibility to receive SCT. Based on the Canadian Evidence-Based Guideline for the Treatment of R/R DLBCL, for patients who are refractory to R-CHOP or who relapse after 12 months of R-CHOP, the standard approach consists of salvage, platinum-based chemotherapy followed by high-dose chemotherapy and ASCT (for patients with chemosensitive disease who meet the eligibility criteria for transplant). In patients who are ineligible for SCT, second-line treatment options include pola-BR or chemotherapy, with or without rituximab.1,6
Currently, CAR T-cell therapy is approved in Canada for patients with R/R DLBCL following 2 or more lines of therapy. As such, CAR T-cell therapy is the standard treatment approach for patients with R/R DLBCL who do not respond to salvage chemotherapy (i.e., who are transplant-ineligible) or who relapse after SCT.1,6 Though currently not adopted, CAR T-cell therapy may be offered as second-line treatment to eligible patients.28
For patients who are not chemosensitive and are ineligible for ASCT and relapse after SCT or CAR T-cell therapy, the prognosis is poor, and there is no standard treatment approach. Available options are currently limited to palliative chemotherapies — including R-GemOx, pola-BR, and tafasitamab with lenalidomide — or clinical trials with novel drugs.1,6 Tafasitamab with lenalidomide received a “do not reimburse” recommendation from CADTH and is not currently reimbursed by participating drug programs.8
Based on input from the clinician groups, novel drugs — including ibrutinib, lenalidomide, tafasitamab, and obinutuzumab — are available through compassionate access programs in Canada, but generally do not have Health Canada approvals or provincial funding for the treatment of R/R DLBCL.
Drug Under Review
Mechanism of Action
Epcoritamab is a humanized immunoglobulin gamma-1–bispecific antibody that binds to a specific extracellular epitope of CD20 on B-cells and to CD3 on T-cells. The activity of epcoritamab is dependent upon the simultaneous engagement of CD20-expressing cells and CD3-expressing endogenous T-cells by epcoritamab, which induces specific T-cell activation and T-cell–mediated killing of CD20-expressing cells.7
Dosing and Administration
Epcoritamab is administered through SC injection at step-up doses of 0.16 mg, 0.8 mg, and 48 mg according to the following schedule:
Cycle 1: 0.16 mg (priming dose) on day 1; 0.8 mg (intermediate dose) on day 8; 48 mg (full dose) on day 15
Cycles 2 and 3: 48 mg once per week over a 28-day period (days 1, 8, 15, and 22 of a 28-day cycle)
Cycles 4 through 9: 48 mg once every 2 weeks (days 1 and 15 only of each cycle)
Cycle 10 and onward: 48 mg once every 4 weeks (day 1 only of each 28-day cycle)7
Table 3: Dosing Schedule for Epcoritamab
Schedule detail | Cycle 1 | Cycles 2 and 3 | Cycles 4 through 9 | Cycle 10 and onward | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Day of cycle | 1 | 8 | 15 | 22 | 1 | 8 | 15 | 22 | 1 | 15 | 1 |
Epcoritamab dose (mg)a | 0.16 | 0.8 | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 |
Source: Epcoritamab product monograph.7
Recommended Premedication for CRS
The product monograph recommends that prophylaxis be initiated against Pneumocystis jirovecii pneumonia (PJP) and herpes virus infections before starting treatment with epcoritamab. Epcoritamab should be administered only under the supervision of a health professional experienced in the treatment of cancer patients who has access to appropriate medical support to manage severe reactions, such as CRS and ICANS. Recommended premedications for CRS are summarized in Table 4.7
Table 4: Recommended Premedications for CRS
Cycle | Patient requiring premedication | Premedication | Administration |
|---|---|---|---|
Cycle 1 | All patients |
|
|
|
| ||
Cycle 2 and beyond | Patients who experienced grade 2 or grade 3a CRS with previous dose |
|
|
CRS = cytokine release syndrome.
aPatients will be permanently discontinued from epcoritamab after a grade 4 CRS event.
Source: Epcoritamab product monograph.7
Approved Indication and Reimbursement Request
Epcoritamab has been approved by Health Canada for the treatment of adult patients with R/R DLBC not otherwise specified, DLBCL transformed from indolent lymphoma, HGBCL, PMBCL, or FLG3B after 2 or more lines of systemic therapy who have previously received, or are unable to receive, CAR T-cell therapy. The sponsor has requested that epcoritamab be reimbursed in accordance with the indication approved by Health Canada. Figure 1 summarizes the sponsor’s proposed place in therapy for epcoritamab.
Figure 1: Sponsor’s Proposed Place in Therapy for Epcoritamab for LBCL
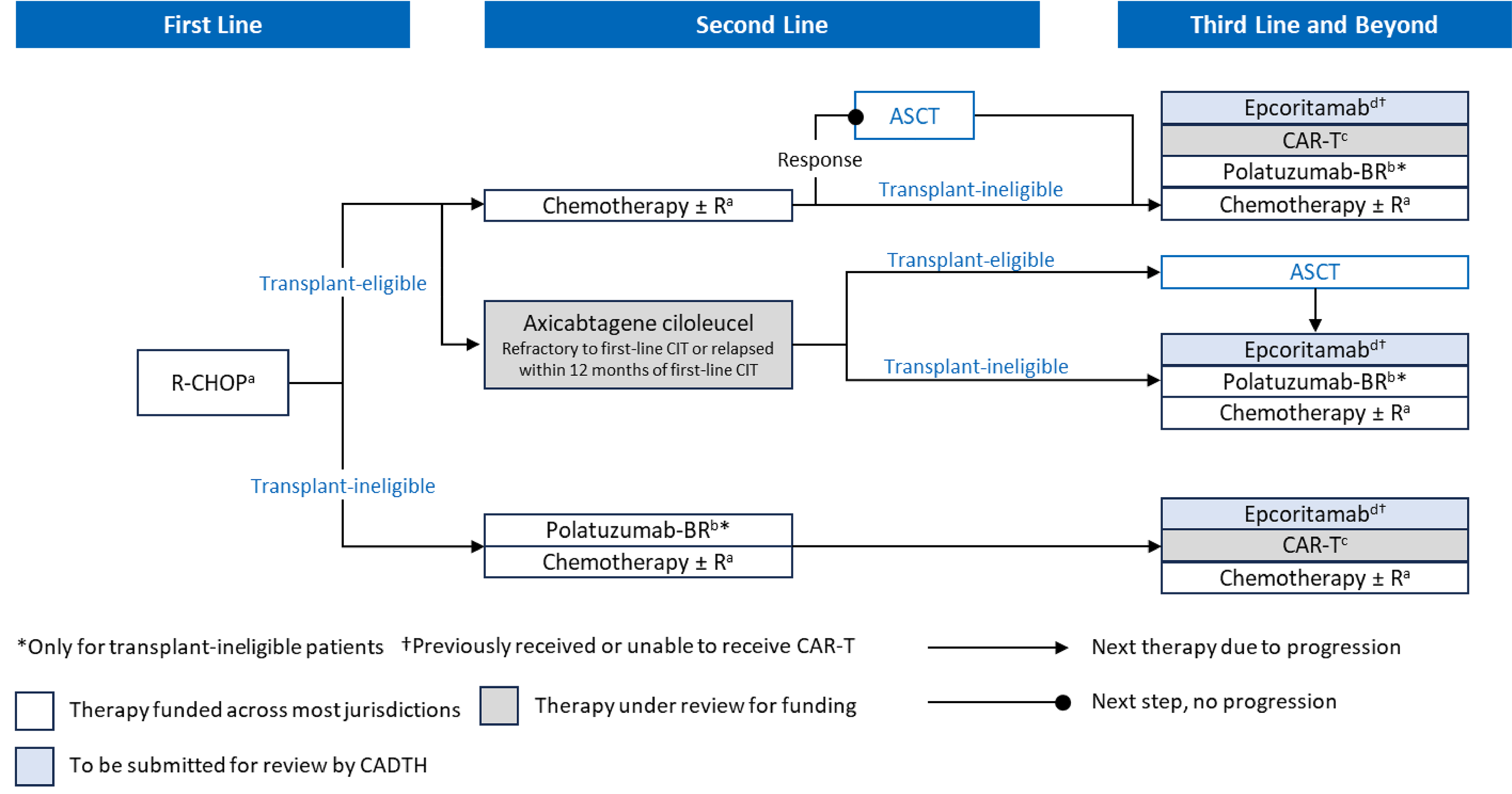
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; CAR = chimeric antigen receptor; CIT = chemoimmunotherapy; LBCL = large B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CHOP = rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone.
a Alternate rituximab-based chemotherapy regimens.
b Pola-BR is funded only for patients with DLBCL who are ineligible for ASCT and have received at least 1 prior therapy.
c CAR T-cell therapy options include lisocabtagene maraleucel, tisagenlecleucel, and axicabtagene ciloleucel.
d For patients who have previously received or are unable to receive CAR T-cell therapy.
Source: Sponsor submission materials.
CAR T-Cell Therapy Considerations
The Health Canada–approved indication for both epcoritamab and glofitamab limits the approved usage to patients “who have previously received or are unable to receive CAR T-cell therapy.” This indication is more restrictive than the approved labels from the FDA, European Medicines Agency, and UK Medicines and Health care products Regulatory Agency29-31 and the proposed indication currently under review by the Therapeutic Goods Administration in Australia. None of these organizations has included the condition that patients have previously received CAR T-cell therapy or are unable to receive CAR T-cell therapy.
CADTH sought clarification from the sponsor regarding the rationale for the wording in the indication because patients in the EPCORE NHL-1 trial were not required to have previously received CAR T-cell therapy or be unable to receive CAR T-cell therapy (i.e., this was not stated in the eligibility criteria for the trial). The sponsor stated that Health Canada provided advance feedback indicating that, to qualify for the NOC/c review process, AbbVie should modify the proposed indication to focus on populations with high unmet medical needs (e.g., patients who have received CAR T-cell therapy). Following the feedback from Health Canada, AbbVie proposed wording for the indication that met the NOC/c criteria of unmet medical need (i.e., “…where patient is unable to receive or has previously received CAR T-cell therapy.”)
The sponsor noted that from a clinical perspective, the current Health Canada–approved indication allows for use by patients:
after CAR T-cell therapy
who are ineligible to receive CAR T-cell therapy
who are eligible to receive CAR T-cell therapy, but did not (e.g., due to logistical challenges or choice).
The clinical experts consulted by CADTH agreed with the sponsor’s interpretation of the indication, identified these patient groups as potential candidates for epcoritamab, and similarly noted that the approved indication is not reflective of the trial population for the EPCORE NHL-1 study. In addition, the clinical experts noted that some patients who are ineligible to receive CAR T-cell therapy at the time of initiating treatment with epcoritamab may subsequently become eligible for CAR T-cell therapy if their disease status changes or other barriers to CAR T-cell therapy access cease to apply.
The clinical experts noted that the treatment landscape for R/R LBCL in Canada may shift toward earlier usage of CAR T-cell therapy because axicabtagene ciloleucel (Yescarta) received a favourable recommendation from CADTH for use as a second-line option based on the results of the ZUMA-7 trial. Axicabtagene ciloleucel was under review by the pan-Canadian Pharmaceutical Alliance at the time of this review and was not funded by any of the participating drug programs (refer to Figure 1).
As part of the NOC/c requirements, AbbVie will file confirmatory evidence of efficacy in the setting of R/R DLBCL in the form of a Supplement to a New Drug Submission – Confirmatory (SNDS-C). Upon approval of this SNDS-C, conditions associated with the NOC would be removed by Health Canada.
Non-DLBCL Forms of Aggressive NHL
Health Canada considered the small number of patients enrolled in the EPCORE NHL-1 trial who had forms of aggressive B-cell non-Hodgkin lymphoma (aNHL) other than DLBCL (i.e., those with HGBCL, PMBCL, and FLG3B) and noted that adequately powered clinical studies would be problematic to conduct given the rarity of these subtypes. After an examination of response rates in these patients, Health Canada concluded that epcoritamab demonstrated efficacy and that these patients are treated in the same manner as those with DLBCL in clinical practice.32 This included patients living with HGBCL, a more aggressive form of aNHL than DLBCL. The approved indication for epcoritamab specifically identifies approval for use in HGBCL, which is different from the indications for the other Health Canada–approved regimens for R/R LBCL (i.e., glofitamab, polatuzumab vedotin, pembrolizumab, and tafasitamab).7,33-36 Keytruda (pembrolizumab) has been approved for use in the treatment of adult and pediatric PMBCL and in patients who have relapsed after 2 or more lines of therapy. The sponsor recently declined to file a submission to CADTH for this indication. The clinical experts consulted by CADTH noted that the evidence supports the efficacy of epcoritamab for patients with rarer forms of lymphoma.
Table 5: Health Canada–Approved Indications for Use in R/R LBCL
R/R LBCL subtypes | Epcoritamab | Glofitamab | Pola-BR | Pembrolizumab |
|---|---|---|---|---|
DLBCL NOS | Approved | Approveda | Approvedb | Not Approved |
DLBCL transformed from indolent lymphoma | Approved | Approveda | Not Approvedc | Not Approved |
HGBCL | Approved | Not Approved | Not Approvedd | Not Approved |
PMBCL | Approved | Approveda | Not Approved | Approvede |
FLG3B | Approved | Not Approved | Not Approved | Not Approved |
DLBCL = diffuse large B-cell lymphoma; FLG3B = follicular lymphoma grade 3B; HGBCL = high-grade B-cell lymphoma; LBCL = large B-cell lymphoma; NOS = not otherwise specified; PMBCL = primary mediastinal B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R//R = relapsed or refractory.
aReceived a draft recommendation from CADTH in favour of reimbursement.
bReceived a final recommendation from CADTH in favour of reimbursement.
cSome participating jurisdictions provide reimbursement for pola-BR for this population.
dPolatuzumab vedotin with rituximab, cyclophosphamide, doxorubicin, and prednisone is indicated for the treatment of adult patients with previously untreated LBCL, including diffuse DLBCL NOS, HGBCL, Epstein-Barr virus–positive DLBCL NOS, and T-cell or histiocyte-rich LBCL.
eSponsor declined to file a submission with CADTH for this indication.
Conditional Regulatory Approval
Health Canada issued an NOC/c for epcoritamab with the following key confirmatory requirements regarding efficacy:
The sponsor should commit to the submission of a clinical trial for the purposes of providing confirmatory evidence of efficacy in the setting of R/R DLBCL. Specifically, the primary analyses of Study GCT3013 to 05: A Randomized, Open-Label, Phase 3 Trial of Epcoritamab vs Investigator’s Choice Chemotherapy in Relapsed/Refractory Diffuse Large B-cell Lymphoma (EPCORE DLBCL-1) should be submitted to Health Canada as an SNDS-C.
The primary efficacy objective of the EPCORE DLBCL-1 phase III study is to demonstrate that epcoritamab monotherapy improves the OS of patients with DLBCL compared to investigator’s choice of either BR or R-GemOx. The sponsor should acknowledge that authorization may be revoked if the trial fails to show an OS benefit for epcoritamab over investigator’s choice of therapy. The sponsor should provide an estimated date of completion of the primary analyses for the study as well as an estimated date for the submission of the study to Health Canada.
Characteristics of Drugs for Patients With R/R LBCL
Epcoritamab has not been previously reviewed by CADTH. Its key characteristics are summarized in Table 6 along with those of other treatments available for R/R LBCL.
Table 6: Key Characteristics of Epcoritamab, Glofitamab, and Combination Chemotherapy
Characteristic | Epcoritamab | Glofitamab | Combination chemotherapy | |
|---|---|---|---|---|
Pola-BR | Rituximab-based chemotherapy (e.g., R-GDP, R-ICE, R-GemOx) | |||
Mechanism of action | Epcoritamab is a humanized immunoglobulin gamma-1–bispecific antibody that binds to a specific extracellular epitope of CD20 on B-cells and to CD3 on T-cells. The activity of epcoritamab is dependent upon simultaneous engagement of CD20-expressing cells and CD3-expressing endogenous T-cells by epcoritamab that induces specific T-cell activation and T-cell–mediated killing of CD20-expressing cells.7 | Glofitamab is a bispecific mAb that simultaneously binds to CD20 on the B-cell and CD3 on the T-cell to mediate the formation of an immunological synapse, with subsequent potent T-cell activation and proliferation, secretion of cytokines, and the release of cytolytic proteins that result in the lysis of CD20-expressing B-cells. | Polatuzumab vedotin is a CD79b-targeted ADC that preferentially delivers an antimitotic drug, MMAE, to B-cells, which results in the killing of malignant B-cells. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis. | Rituximab is a chimeric mAb that binds to the antigen CD20, a transmembrane protein found on the surface of normal and malignant B lymphocytes. CD20 regulates an early step in the activation of cell cycle initiation and differentiation. |
Indicationa | For the treatment of adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, HGBCL, PMBCL, or FLG3B after 2 or more lines of systemic therapy and who have previously received or are unable to receive CAR T-cell therapy | For the treatment of adult patients with R/R DLBCL not otherwise specified, DLBCL arising from trFL, or PMBC who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T-cell therapy | For the treatment of adult patients with R/R DLBCL, not otherwise specified, who are not eligible for autologous stem cell transplant and have received at least 1 prior therapy | Not approved in Canada in the R/R setting of DLBCL |
Route of administration | SC injection | IV infusion | IV infusion | IV infusion |
Recommended dose | Dosing begins with a step-up dosing schedule to minimize the risk of CRS. The recommended dose after step-up is 48 mg. | Dosing begins with a step-up dosing schedule to minimize the risk of CRS. The recommended dose after step-up is 30 mg. | Polatuzumab vedotin, bendamustine, and rituximab can be administered in any order on day 1 of each cycle.
| Not approved in Canada in the R/R setting of DLBCL. |
Inpatient hospitalization required at time of drug administration | For 24 hours following administration of the first full dose, patients should remain within proximity of a health care facility and be monitored for signs and symptoms of CRS and ICANS, or alternatively consider hospitalization. | All patients must be monitored for signs and symptoms of potential CRS during infusion and for at least 10 hours after completion of the first dose (i.e., 2.5 mg on cycle 1, day 8). | Not required | Not required |
Duration of therapy | No limit on the number of cycles; can be given until disease progression or unacceptable toxicity | 12 cycles or until disease progression or unmanageable toxicity | 6 cycles (21 days per cycle) | Various (depends on regimen) |
Serious adverse effects or safety issues | CRS ICANS | CRS | Polatuzumab vedotin: Infections and myelosuppression | Rituximab: Infusion reactions, PML, TLS, HBV reactivation, mucocutaneous reactions, infections, cardiovascular events |
Other | Premedication should be administered according to the product monograph to reduce the risk of CRS. | All patients must receive a single 1,000 mg dose of obinutuzumab on cycle 1, day 1 (7 days before initiation of glofitamab treatment). Premedication should be administered according to the product monograph to reduce the risk of CRS. | NA | NA |
ADC = antibody-drug conjugate; CAR = chimeric antigen receptor; CD3 = cluster of differentiation 3; CD20 = cluster of differentiation 20; CD79b = cluster of differentiation 79b; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; FLG3B = follicular lymphoma grade 3B; HBV = hepatitis B virus; HGBCL = high-grade B-cell lymphoma; ICANS = immune effector cell–associated neurotoxicity syndrome; mAb = monoclonal antibody; MMAE = monomethyl auristatin E; NA = not applicable; PMBCL = primary mediastinal B-cell lymphoma; PML = progressive multifocal leukoencephalopathy; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-GDP = rituximab plus gemcitabine, dexamethasone, cisplatin; R-GemOx = rituximab, gemcitabine, and oxaliplatin; R-ICE = rituximab plus ifosfamide, carboplatin, etoposide; R/R = relapsed or refractory; SC = subcutaneous; TLS = tumour lysis syndrome; trFL = follicular lymphoma.
Source: Product monographs for epcoritamab,7 glofitamab (Columvi) for injection,33 polatuzumab vedotin (Polivy) for injection,34 and rituximab (Rituxan) for injection.37
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the Stakeholder Perspectives section of this report.
Two patient groups, LC and LLSC, responded to CADTH’s call for patient input for the current review of epcoritamab. LC is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. LLSC is a national organization dedicated to finding a cure for blood cancers and supporting patients and their families by funding life-enhancing research and providing educational resources, services, and support.
LC gathered information for this submission through a survey from October 3, 2023 to November 20, 2023, targeting patients living with LBCL. The LC survey data included 33 respondents. The LC information included a submission from France that was based on a survey regarding the use of epcoritamab for DLBCL conducted by Ensemble Leucémie Lymphomes Espoir, with 9 survey respondents; this was supported by the results of the Lymphoma Coalition's 2022 survey, which included the experiences of patients with DLBCL (n = 171). LLSC conducted 4 1-on-1 interviews in November 2023; 2 interviewees were patients with DLBCL, and 2 were caregivers. Three interviewees resided in Canada, and 1 resided in the US.
According to both inputs, living with LBCL is associated with extreme fatigue, body aches, nausea, shortness of breath, lack of energy, and stress and worry, all of which have a significant impact on day-to-day activities and patients’ quality of life.
From the LC input, the majority of 15 patients indicated that they had received 1 to 3 or more lines of treatment, such as R-CHOP as first-line treatments; rituximab-based chemotherapy and radiation as second-line treatments; and CAR T-cell therapy and pola-BR as a third-line therapy. While most patients (67%) in the LC survey indicated that they were satisfied with their front-line treatment options, respondents from both surveys stated that there is a lack of second- and third-line treatment options and described difficulties managing treatment regimens and side effects. Currently available treatments take significant mental and psychological tolls on patients and their loved ones, are associated with immense financial burdens, and negatively affect people’s ability to work, travel long distances, and participate in daily activities.
Both inputs indicated that patients expect new treatments to be more effective and less invasive with fewer side effects. Patients are seeking choice in their treatment decisions and would like a variety of options that offer a longer life span, lengthier remission, and better quality of life.
In the LC input, 1 patient from the survey and 8 patients from the French Health Technology Assessment (HTA) submission indicated they were treated with epcoritamab. In the LLSC input, 1 caregiver spoke about their experience with epcoritamab. According to both inputs, patients experienced fewer side effects, and only 1 patient discontinued the treatment due to side effects. Patients indicated that this treatment option could offer hope and relief to many third-line patients. In addition, the SC administration of the drug results in less time spent in hospital per visit, which can improve the quality of life of patients and caregivers.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of LBCL.
Unmet Needs
R/R LBCL is associated with a poor prognosis, and in patients who are not eligible for or who did not respond to cellular therapy (i.e., CAR T-cell therapy and/or ASCT), the outcomes are poor (i.e., OS < 6 months). The clinical experts indicated that the goal of treatment at this stage is palliative and generally includes maintaining HRQoL by relieving lymphoma-related symptoms, delaying disease progression, and balancing the toxicities of therapy. There is no standard of care in this setting, but options include chemotherapy (e.g., pola-BR), radiation, and clinical trials. The experts noted that there is an unmet need for safe, effective treatments for palliative patients who are not eligible for curative treatment (including those who are clinically ineligible or unable to receive treatments for other reasons) or who have not responded to second-line treatment consisting of SCT or CAR T-cell therapy, given that there are limited treatment options for disease control and that currently available options are often associated with significant toxicity, limiting these options’ usefulness and applicability. Additionally, after transplant and CAR T-cell therapy, patients often have a poor prognosis and very poor bone marrow function, which prevents them from receiving or tolerating further cytotoxic therapy.
Input from the experts suggested that there is also a significant group of patients who may be eligible for intensive treatments but unable to access them due to barriers based on geographic location and capacity limitations within the health care system. Many patients are unable to travel with caregivers to specialized cellular therapy sites and choose not to have this treatment because they wish to be treated closer to home. As such, there is an additional unmet need for treatments that patients can access and receive closer to home.
Place in Therapy
First-line treatment for patients with LBCL is administered with curative intent and consists of R-CHOP (or a reduced dose of R-CHOP for patients aged 75 years or older or who have significant comorbid illnesses). Second-line treatment consists of salvage R-CIT and ASCT for transplant-eligible, chemosensitive patients (i.e., curative intent). Third-line therapy can include CAR T-cell therapy (i.e., curative intent). There is no standard of care following these treatment options, and transplant-ineligible patients in the second- and third-line settings tend to receive palliative R-CIT (e.g., pola-BR, rituximab plus gemcitabine, dexamethasone, and cisplatin, or R-GemOx) with noncurative intent, radiation, and/or consideration for enrolment in clinical trials for investigative therapies. The clinical experts highlighted that there is a planned shift in Canada to use CAR T-cell therapy as second-line therapy for primary refractory or early relapsed LBCL, pending funding. The clinical experts emphasized that cytopenias are an important problem with currently available palliative treatment options.
The experts highlighted that epcoritamab should be restricted to patients who are not eligible for curative therapies, have already received CAR T-cell therapy, or would not be able to receive epcoritamab later (i.e., whether as third-line therapy after CAR T-cell therapy or because they are ineligible); they envisioned epcoritamab occupying the same therapeutic space as pola-BR. The clinical experts noted that some patients could be considered for CAR T-cell therapy after receiving treatment with epcoritamab and subsequently experiencing disease progression, including:
patients who were considered eligible for CAR T-cell therapy and had previously chosen not to receive the treatment, but remain candidates for the therapy after receiving epcoritamab
patients whose clinical status made them borderline candidates for CAR T-cell therapy due to tumour burden or underlying comorbidity and whose condition improves after receiving epcoritamab to a point where they can be considered candidates for CAR T-cell therapy.
The clinical experts noted that the fixed-dose SC administration of epcoritamab would offer efficiencies for both patients and health care providers compared with the IV administration required for the existing comparator options. In addition, epcoritamab is given as monotherapy, whereas all comparator regimens are administered as combinations. This may reduce the time required for patients to spend at cancer treatment centres, offering improvements in quality of life for patients and their caregivers as well as reductions in the time needed for health care providers to administer the treatments.
Patient Population
The experts noted that these patients would be identified in routine practice by clinicians familiar with the treatment of lymphoma patients undergoing surveillance for relapse (clinical and/or imaging).
In accordance with the indication approved by Health Canada, the clinical experts noted that patients with R/R LBCL requiring third-line or later treatment who are not eligible for or who did not respond adequately to intensive cellular therapies (i.e., SCT or CAR T-cell therapy) would be considered for epcoritamab. The experts could not identify a specific subgroup of patients who would demonstrate an enhanced or reduced benefit from epcoritamab treatment based on available evidence. However, it was noted that patients who cannot receive CAR T-cell therapy because of logistical or geographic reasons could be candidates.
Patients who may not be suitable for epcoritamab could include those who do not want continuous treatment (given that epcoritamab does not have a limited number of cycles and would be administered until disease progression or unacceptable toxicity); those who develop CRS that does not resolve following the first cycle of treatment; and those who are able and willing to receive CAR T-cell therapy. The experts also noted that patients with impaired B-cell or T-cell immunity (e.g., those with hypogammaglobulinemia from multiple previous lines of immune therapies) may have exhausted their T-cells, making a response to epcoritamab less likely.
The experts highlighted that repeat biopsy is generally not required in cases of suspected relapse of LBCL unless it is a remote relapse or the patient had a history of indolent lymphoma and it was unclear which lymphoma had relapsed. Similar to glofitamab, no companion diagnostic test is required for epcoritamab.
The clinical experts noted that epcoritamab could help address an unmet need for patients who may be ineligible for or unwilling to receive additional chemotherapeutic regimens in the third-line setting (e.g., either pola-B/R or R-CIT). The experts noted that third-line CIT would not be expected to have significant response rates among patients who have been heavily pretreated, including with prior exposures to R-CIT in the first-line setting, as well as salvage chemotherapy before ASCT and/or bridging therapy before CAR T-cell therapy. As such, the different mechanism of action offered by bispecific T-cell engagers (i.e., epcoritamab or glofitamab) would be an attractive option for these patients. However, the experts noted that the potential need for hospital admission to monitor for CRS and ICANS could lessen some of the enthusiasm for these new treatment options.
Assessing the Response to Treatment
The clinical experts stated that response to treatment would include standard assessment of lymphoma response using the Lugano criteria. Patients would undergo interim imaging every 3 months to confirm response, which would lead either to ongoing treatment or to discontinuation. Patients are also assessed for lymphoma-related symptoms at each visit; the clinical experts noted that these outcomes are more subjective but do factor into patients’ decisions about continuation of therapy. The experts also noted that the frequency of these assessments and collection of data may vary across Canada.
In terms of meaningful response to treatment, the clinical experts stated that a response of 6 months or more with improved symptoms can be considered meaningful. The experts did not consider temporary shrinking of tumours beneficial to patients. They believed that initial responses (PR or CR) should exceed 6 months; otherwise, treatment should be discontinued.
Additionally, with a current median OS of 6 months in this population, the experts considered a benefit of at least 6 months and 3 months over current standard of care to be clinically meaningful for OS and PFS, respectively.
The experts noted the need to ensure careful monitoring for serious adverse reactions, given that these could negate the potential benefits of the treatment.
Discontinuing Treatment
The clinical experts suggested that treatment with epcoritamab should be discontinued upon disease progression, lack of response, or unacceptable toxicity.
Prescribing Considerations
The clinical experts indicated that patients with R/R LBCL are typically under the care of hematologists or oncologists who are familiar with the treatment of lymphoma patients. They also noted that the monitoring and treatment of these patients must initially be conducted at tertiary centres that have the means to monitor and treat CRS, which may require some initial training of site staff before implementation. Following the first few cycles, the clinical experts noted that treatment may continue at regional centres because the risk of CRS decreases.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the Stakeholder section of this report.
Three clinician groups — LC (3 clinicians contributing), the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee (7 clinicians contributing), and the LLSC Nurses Network (6 clinicians contributing to the input) — responded to CADTH’s call for clinician group input.
According to these groups, there are poor and limited treatment options for patients with R/R DLBCL. LC indicated that for patients who are eligible for aggressive curative intensive therapies, options such as ASCT and CAR T-cell therapy are considered. Those who have disease progression after CAR T-cell therapy, or who are unfit for this therapy for medical and/or social reasons, have the greatest unmet need for treatment because no other curative-intent therapy is readily available for them.
In contrast, those who are not eligible for curative ASCT-based or CAR T-cell therapy approaches are managed with palliative approaches, such as pola-BR or tafasitamab (an anti–cluster of differentiation 19 antibody) in combination with lenalidomide. CADTH notes that tafasitamab in combination with lenalidomide received a do not reimburse recommendation and is not currently reimbursed by participating drug programs.8 A small percentage of patients might pursue allogeneic stem cell transplant, but the vast majority of patients in this setting are managed with a variety of palliative chemotherapy regimens, radiation therapy, or clinical trials. Multiple novel drugs (ibrutinib, lenalidomide, tafasitamab, and selinexor) do not have Health Canada approvals or provincial funding for R/R DLBCL.
The clinician groups noted that there is an unmet need for safe and effective treatments for patients who are not eligible for CAR T-cell therapy or for whom second-line treatment has not been effective. LC and the LLSC Nurses Network added that there are limited treatment options for disease control, and that currently, the available options are often associated with significant toxicity, side effects, and mental and physical treatment fatigue. LC stated that while ASCT or CAR T-cell therapy are considered effective for some patients, others are unable to access them due to location barriers. Only a select sites are equipped to offer CAR T-cell therapy.
The most important goals of treatment for DLBCL, according to clinician groups, are to prolong survival, delay disease progression, and improve symptoms, which in turn can improve the quality of life of patients and caregivers. Clinician groups agreed, in regard to the indication, that epcoritamab can be used in the third line or beyond if the patient was previously treated with CAR T-cell therapy or is ineligible for CAR T-cell therapy. LC and the LLSC Nurses Network stated that as an off-the-shelf product, this treatment could alleviate regional access issues, and that the SC injection could become a more feasible, well-favoured option than currently available treatments.
According to the clinician groups, improved survival (PFS, OS), blood work, decreased presence of cancer cells in bone marrow, and improvement in disease symptoms are outcomes used to determine whether a patient is responding to treatment. LC added that a clinically meaningful response would be PR or CR, typically determined using CT and or PET scans.
The clinician groups agreed that discontinuation of therapy should be considered in patients with disease progression or toxicity and that epcoritamab can be given in any inpatient and outpatient setting that has the ability to admit and monitor patients who are receiving anticancer therapy and has expertise in managing CRS and neurotoxicity.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 7.
Table 7: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Based on the preliminary evidence provided by the sponsor (e.g., the phase II EPCORE NHL-1 trial), please comment on how the efficacy and safety of epcoritamab compares to:
| The clinical experts consulted by CADTH noted that there was heterogeneity across the different clinical studies for each of these regimens, making it challenging to draw conclusions regarding the comparative clinical benefit of each. However, the clinical experts also noted the following:
|
Considerations for initiation of therapy | |
The EPCORE NHL-1 trial included patients who had received prior CAR T-cell therapy (38.9% of the population of patients with LBCL), but it was not a requirement for inclusion in the trial. The Health Canada–approved indication for epcoritamab states that patients must have received prior CAR T-cell therapy or be unable to receive it. Could the clinical experts please comment on what scenarios make a patient “unable to receive CAR T-cell therapy”? | The clinical experts consulted by CADTH noted that eligibility for CAR T-cell therapy is determined by patient factors (e.g., age, cardiac function, renal function, and liver function), tumour factors (e.g., rate of tumour progression and extent of extranodal involvement), and issues related to manufacturer and health system capacity. The clinical experts emphasized that patients who have not received CAR T-cell therapy and receive treatment with epcoritamab should become eligible for CAR T-cell therapy should their clinical condition and/or personal circumstances change such that CAR T-cell therapy would be the preferred option. |
In a scenario with access to either CAR T-cell therapy or epcoritamab in a patient who has already received 2 prior lines of systemic therapy, what would guide treatment selection? | The clinical experts consulted by CADTH noted that CAR T-cell therapy would generally be the preferred option for patients who are sufficiently fit to receive the treatment, given that it can be a curative regimen and because there are longer-term follow-up data available in comparison with bispecific therapies, such as epcoritamab. As noted, treatment with CAR T-cell therapy is highly invasive, and health system resource discrepancies can lead to equity and access issues, depending on capacity and other logistical considerations. Additional treatment options, such as epcoritamab, are required for patients who are not candidates for CAR T-cell therapy. |
The programs noted that glofitamab (Columvi) is undergoing review by CADTH for a similar indication (i.e., treatment of adult patients with R/R DLBCL NOS, DLBCL arising from follicular lymphoma, or PMBCL who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T-cell therapy). Drug programs noted that consistency with the initiation criteria in the same therapeutic space can be beneficial from a formulary management perspective. | For consideration by the CADTH expert committee. |
Considerations for continuation or renewal of therapy | |
No issues identified | Not applicable |
Considerations for discontinuation of therapy | |
No issues identified | Not applicable |
Considerations for prescribing of therapy | |
Depending on the last dose given and the length of any treatment interruptions, the priming schedule may need to be given again once treatment is resumed. Could the clinical experts please comment on the complexity of the dosage schedule and the potential need to repeat the priming and intermediate doses in the event of an interruption? | The clinical experts consulted by CADTH noted that monotherapy with epcoritamab could be considered less complicated than many of the alternative regimens, which often involve the IV administration of multiple drugs. The main issue with epcoritamab is the potential need to hospitalize patients at the time of administering the first full dose of the drug (given that hospital capacity issues can be on ongoing challenge within the health care system). |
Epcoritamab is administered through SC injection, which may offer efficiencies for health care providers and improved quality of life for patients; however, IV access is still required during treatment if supportive care may be required following administration. | The clinical experts noted that the fixed-dose SC administration of epcoritamab would offer efficiencies for both patients and health care providers compared with the IV administration required for the existing comparator options. In addition, epcoritamab is given as monotherapy, whereas all comparator regimens are as administered as combinations. This may reduce the time patients need to spend at cancer treatment centres, offering potential improvements in quality of life for patients and their caregivers as well as reductions in the time needed for health care providers to administer treatments. |
Generalizability | |
Please comment on whether the EPCORE NHL-1 trial data can be generalized to the following patients:
| The clinical experts consulted by CADTH noted that these patients were excluded from the EPCORE NHL-1 trial; as a result, there is no evidence to inform the use of epcoritamab in these patients. However, the clinical experts noted the following:
|
The EPCORE NHL-1 trial included patients who had received prior CAR T-cell therapy, but it was not a required inclusion criterion. Only 38.9% of the EPCORE NHL-1 patient cohort received prior CAR T-cell therapy, and of those, 75% experienced progressive disease within the first 6 months. The Health Canada–approved indication for epcoritamab indicates that patients must have received prior CAR T-cell therapy or be unable to receive it. Could the clinical experts please comment on whether there is a clinical rationale for why patients should be required to have prior CAR T-cell therapy or be unable to receive CAR T-cell therapy to be eligible for epcoritamab? | The clinical experts consulted by CADTH do not believe there is a clinical rationale for why patients should be required to have prior CAR T-cell therapy or be unable to receive CAR T-cell therapy to be eligible for epcoritamab. The clinical experts noted that epcoritamab has been shown to be clinically beneficial for patients who could be considered candidates for CAR T-cell therapy. Acknowledging the absence of studies directly comparing epcoritamab against CAR T-cell therapies, the clinical experts consulted by CADTH noted that CAR T-cell therapy would generally be the preferred option for patients who are sufficiently fit to receive the treatment, given that it can be a curative regimen and that longer-term follow-up data are available. |
Care provision issues | |
Due to the risk of CRS, patients require close monitoring, appropriate supportive care interventions, and hospital admission for certain doses of epcoritamab (i.e., first full dose on week 3). This represents an increase in the use of health care resources (i.e., inpatient facilities) and administrative efforts to coordinate inpatient and outpatient settings on a weekly basis once patients initiate epcoritamab treatment. Use of T-cell engager therapies increases the risk of infections that can be serious and complex. Additional resources may be required to address infectious complications. | For consideration by the expert committee regarding organizational feasibility of adoption by the health system. The clinical experts consulted by CADTH noted that reserving a hospital bed for patients who are going to receive treatment with epcoritamab is logistically difficult. In a typical Canadian hospital setting, the wards are likely to be fully or nearly fully occupied. In addition, the clinical experts noted that the planned hospitalization would require coordination between the outpatient infusion clinics and the inpatient hospital wards. The clinical experts consulted by CADTH noted that patients treated with epcoritamab may also require immunoglobulin infusion support. |
Patients experiencing CRS (usually grade 2 or higher) will require supportive treatment with tocilizumab. If there is concurrent ICANS, the product monograph recommends alternatives to tocilizumab “if possible” (such as anakinra or siltuximab) to manage the toxicity, and potentially further treatment with anakinra. The impact of the costs and acquisition of these therapies adds budget impact and logistical complexities. The funding of these therapies needs to be incorporated as part of any implementation to ensure that sites can manage CRS and/or ICANS. | For consideration by the expert committee regarding organizational feasibility of adoption by the health system. The clinical experts consulted by CADTH noted that access to these drugs is essential for the safe administration of epcoritamab. |
System and economic issues | |
Drug programs noted that the product monograph recommends patients be monitored for signs and symptoms of CRS and ICANS for 24 hours after the first full dose of epcoritamab (i.e., 48 mg administered on day 15 of cycle 1). The monograph recommends that patients remain within the proximity of a health care facility and be monitored for signs and symptoms of CRS and ICANS or consider hospitalization. | For consideration by expert committee regarding organizational feasibility of adoption by the health system. The clinical experts consulted by CADTH noted that, in the event that there are no options for 24-hour outpatient monitoring available (e.g., insufficient treatment facility availability), patients will likely require short-term hospital admission while the treatment is administered. This would likely be the case until criteria are available that would allow for the proactive identification of those who are at high risk of adverse events or until better prophylactic regimens are developed to minimize the risk of CRS and/or ICANS. |
Epcoritamab is available in 2 vial strengths:
The drug programs noted that these are single-use vials and that wastage will be incurred during the priming and intermediate dosing, given the fixed vial size. Would the clinical experts agree that there is likely to be wastage, given the vial sizes for epcoritamab? | The clinical experts consulted by CADTH agreed that wastage would occur within pharmacies preparing the drug for administration. The clinical experts noted that wastage is an inefficient use of health care resources and that the sponsor could consider marketing alternative dosage strengths that would limit wastage. |
The budget impact to anticancer and supportive care budgets is of concern. The approved indication indicates that prior CAR T-cell therapy is required (or that patients are “unable” to receive such therapy). PAG notes that prior CAR T-cell therapy was not a required inclusion criterion in the EPCORE NHL-1 trial. The affordability of requiring CAR T-cell therapy (or being “unable” to receive CAR T-cell therapy) before epcoritamab is of significant concern. | For consideration by expert committee regarding economic feasibility of adoption. |
The intensive monitoring required with early doses of epcoritamab presents increased resource-use costs. Not all jurisdictions will have the capacity to admit patients. Additionally, drug costs for inpatient vs. ambulatory use may be borne by different drug budgets, depending upon jurisdiction. | For consideration by expert committee regarding organizational feasibility of adoption by the health system. The clinical experts consulted by CADTH noted that the lack of availability of the required admissions or ambulatory monitoring facilities may limit uptake of this treatment. |
Commentary on time-limited recommendation | |
The phase III EPCORE DLBCL-1 trial is comparing epcoritamab to investigator’s choice of chemoimmunotherapy (either BR or R-GemOx). The drug programs have indicated that BR is currently reimbursed in most jurisdictions for relapsed indolent lymphomas. R-GemOx is not a common regimen in jurisdictions in Canada for relapsed LBCL. Please comment on the clinical relevance of R-GemOx as a comparator for the phase III trial. | The clinical experts consulted by CADTH expressed concerns regarding the choice of comparator in the EPCORE DLBCL-1 trial (i.e., BR or R-GemOx). They felt that the efficacy data from the EPCORE NHL-1 trial were compelling and that BR and R-GemOx would be associated with significant toxicities for patients. The experts noted that patients at this stage of disease would likely have received R-CIT earlier in the course of disease and shown to be refractory to it; as such, they expressed concerns regarding clinical equipoise in the trial and noted that patients randomized to BR or R-GemOx would be receiving an inferior treatment option. They also noted that more appropriate comparators would be the newer therapies that have recently emerged in the second- and third-line setting, such as pola-BR and CAR T-cell therapy. In comments on the draft report, the sponsor clarified that at the start of the EPCORE DLBCL-1 study (January 2021), neither CAR T-cell therapy nor pola-BR were widely used. Therefore, R-CIT was considered the most appropriate comparator, and chemoimmunotherapy remains a treatment option used in practice in Canada for R/R LBCL. |
BR = bendamustine plus rituximab; CAR = chimeric antigen receptor; CNS = central nervous system; CRS = cytokine release syndrome; DLBCL = diffuse large B-cell lymphoma; ICANS = immune effector cell–associated neurotoxicity syndrome; LBCL = large B-cell lymphoma; NOS = not otherwise specified; PAG = Provincial Advisory Group; PMBCL = primary mediastinal B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CIT = rituximab-based chemoimmunotherapy; R-GemOx = rituximab, gemcitabine, and oxaliplatin; R/R = relapsed or refractory; SC = subcutaneous.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of epcoritamab (5 mg/mL and 60 mg/mL) for SC injection in the treatment of adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, HGBCL, PMBCL, or FLG3B after 2 or more lines of systemic therapy who have previously received or are unable to receive CAR T-cell therapy. The focus will be on comparing epcoritamab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of epcoritamab is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The next section includes indirect evidence from the sponsor. No sponsor-submitted long-term extension studies or additional studies that were considered by the sponsor to address important gaps in the systematic review evidence were submitted.
Included Studies
Clinical evidence from the following has been included in the CADTH review and appraised in this document:
1 pivotal study identified in the systematic review (the EPCORE NHL-1 study)
1 ITC (a MAIC).
Systematic Review
The contents of this section were informed by materials submitted by the sponsor. The following information has been validated by the CADTH review team.
Description of Studies
Characteristics of the EPCORE NHL-1 study are summarized in Table 8.
Table 8: Details of Studies Included in the Systematic Review
Detail | EPCORE NHL-1 study (dose expansion parta) |
|---|---|
Designs and populations | |
Study design | Phase I and II, open-label, multicentre trial including a dose escalation part and an expansion part |
Locations | In the aNHL cohort of the expansion part, 157 patients were administered epcoritamab across 54 sites in Australia (9 sites), South Korea (9 sites), US (9 sites), France (4 sites), the Netherlands (4 sites), Spain (4 sites), Denmark (3 sites), Germany (3 sites), UK (3 sites), Poland (2 sites), Singapore (2 sites), Canada (1 site), and Italy (1 site). |
Patient enrolment dates | First patient enrolled (dose expansion part): June 19, 2020 End date: Trial is ongoing |
Key data cut-offs | Date of last observation for last patient recorded:
|
Enrolled (N) | 219 |
Received study treatment (N) | 157 patients received epcoritamab |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | All patients in the expansion part of the trial received the epcoritamab RP2D regimen, consisting of a priming dose of 0.16 mg (C1D1), an intermediate dose of 0.8 mg (C1D8), and a full dose of 48 mg (C1D15, C1D22, and thereafter) administered by SC injection. In this part of the trial, epcoritamab was administered in 28-day cycles as follows:
|
Comparator(s) | Not applicable |
Study duration | |
Screening phase | ≤ 30 days before the first dose of epcoritamab |
Treatment phase | For each patient, the treatment period continued until disease progression unless the patient fulfilled 1 of the discontinuation criteria. The trial will run for a maximum of 5 years after the last patient’s first dose. |
Follow-up phase | Until withdrawal of consent, loss to follow-up, or death |
Outcomes | |
Primary end point | ORR determined by Lugano criteria as assessed by IRC, defined as the proportion of patients with BOR of PR or CR |
Secondary and exploratory end points | Secondary:
Exploratory:
Safety outcomes: AEs, serious AEs, mortality, AEs of special interest, hospitalizations, cytokine measures |
Publication status | |
Publications | Thieblemont et al. (2023)39 (publication based on January 2022 data cut-off) Karimi et al. (2023)40 (abstract based on November 2022 data cut-off) Thieblemont et al. (2021)41 (abstract for trial design) Hutchings et al. (2021)42 (publication for dose escalation part) Clinicaltrials.gov identifier: NCT03625037 |
AE = adverse event; ASCT = autologous stem cell transplant; AST = aspartate aminotransferase; ALT = alanine transaminase; aNHL = aggressive B-cell non-Hodgkin lymphoma; BOR = best objective response; CAR = chimeric antigen receptor; CD3 = cluster of differentiation 3; CD20 = cluster of differentiation 20; C1D1 = cycle 1, day 1; C1D8 = cycle 1, day 8; C1D15 = cycle 1, day 15; C1D22 = cycle 1, day 22; CNS = central nervous system; CR = complete response; DH = double hit; DLBCL = diffuse large B-cell lymphoma; DOCR = duration of complete response; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FDG = fluorodeoxyglucose; FLG3B; HGBCL = high-grade B-cell lymphoma; HSCT = hematopoietic stem cell transplant; IRC = independent review committee; LBCL = large B-cell lymphoma; mAb = monoclonal antibody; MRD = minimum residual disease; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PK = pharmacokinetic; PMBCL = primary mediastinal B-cell lymphoma; PR = partial response; q.w. = once weekly; q.2.w. = once every 2 weeks; RP2D = recommended phase II dose; TH = triple hit; TTCR = time to complete response; TTR = time to response; ULN = upper limit of normal.
aThe EPCORE NHL-1 trial was a phase I and II trial with a dose escalation part and a dose expansion part. Unless otherwise specified, the details provided are for the dose expansion part (aNHL cohort), given that this is the focus of the CADTH submission.
Source: Sponsor’s Summary of Clinical Evidence.11
One study was included in the review. The EPCORE NHL-1 study is an ongoing, phase I and II, multicentre, open-label, single-arm study of epcoritamab monotherapy in patients with R/R with DLBCL (de novo or transformed from all indolent subtypes, including Richter’s transformation), including patients diagnosed with double-hit or triple-hit DLBCL, with MYC and BCL2 and/or BCL6 rearrangements, or other LBCL (including PMBCL, HGBCL, or FLG3B). The study was divided into 2 parts: part 1 (a phase I dose escalation study) and part 2 (a phase II dose expansion study). The primary objective of the EPCORE NHL-1 study was to evaluate the efficacy, safety, and tolerability of escalating doses of epcoritamab monotherapy. The overall study design of the EPCORE NHL-1 trial is shown in Figure 2.
Results from the January 2022 data cut-off formed the basis for approval by Health Canada and were published by Thieblemont et al. (2023).39 The final analysis was recently completed with a data cut-off of April 21, 2023, and this analysis is the focus of the CADTH submission.
The epcoritamab recommended phase II dose (RP2D) regimen was selected based on results from the dose escalation part of the trial and consisted of an initial priming dose of 0.16 mg (C1D1), an intermediate dose of 0.8 mg (C1D8), and a full dose of 48 mg at C1D15, C1D22, and thereafter. In the expansion part of the trial, epcoritamab was administered as monotherapy by SC injection weekly during cycles 1 to 3, every 2 weeks during cycles 4 to 9, and every 4 weeks during cycle 10 and beyond (until unacceptable toxicity, PD, or withdrawal of consent).44
The aNHL cohort in the expansion part of the trial was conducted in 2 stages. In stage 1, patients with DLBCL were enrolled. Following an interim futility analysis, additional patients with DLBCL and other LBCL subtypes were enrolled for stage 2 as summarized here:44 The aNHL cohort consisted of 128 to (a planned maximum size of) 158 patients with R/R aNHL (also referred to as LBCL). In stage 1, there were 28 patients with DLBCL. In stage 2, there were 100 additional patients with DLBCL and less than or equal to 30 patients with other LBCL subtypes (HGBCL, PMBCL, FLG3B). The planned maximum sample size for the aNHL expansion cohort was 158 patients. A total of 157 patients, including 139 with DLBCL and 18 patients with other LBCL subtypes, were treated with the epcoritamab RP2D regimen.44
The primary end point was ORR by the IRC using Lugano criteria.45 Secondary end points included DOR, CR rate, DOCR, PFS, time to response per IRC, and OS. In addition, minimum residual disease and PROs for HRQoL were assessed as exploratory end points.39
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria for the EPCORE NHL-1 study are summarized in Table 4. Briefly, patients eligible for the cohort of interest to this review (i.e., dose expansion) consisted of adults (i.e., at least 18 years of age) diagnosed with DLBCL or other aggressive NHL (including PMBCL, HGBCL, or FLG3B) R/R disease who had previously been treated with at least 2 prior lines of systemic therapy, including at least 1 anti-CD20–containing regimen, and who had not responded to, or were ineligible for, ASCT. Patients were required to have an ECOG Performance Status of 0, 1, or 2. Relapsed disease was defined as recurrence at least 6 months after completion of therapy, and refractory disease was defined as progression either during therapy or within 6 months of completion of therapy. Patients with prior CAR T-cell therapy were eligible if it had been 30 days or more since their last treatment. There were no requirements related to minimum life expectancy or absolute leukocyte count.39,44
Interventions
The EPCORE NHL-1 trial was an open-label, single-arm trial in which patients received epcoritamab until unacceptable toxicity, PD, or withdrawal of consent.44
Epcoritamab
A step-up dosing approach was employed, primarily to mitigate the development of serious CRS. Planned doses for the dose escalation part ranged from 0.0128 mg to 60 mg. Patients received a priming dose on day 1 of cycle 1 and an intermediate dose on day 8 of cycle 1 before receiving full doses for the remainder of the treatment period. Epcoritamab was administered SC in 28-day cycles starting with once-weekly dosing, then moving to biweekly dosing and finally once every 4 weeks.46
From the dose escalation part, a dose of 48 mg was identified as the RP2D based on the safety data, together with pharmacokinetic and pharmacodynamic modelling and exposure-response and exposure-safety analyses. Therefore, this dose was investigated in the phase II dose expansion part.39
In the dose expansion part, epcoritamab was administered by SC injection in treatment cycles of 4 weeks (i.e., every 28 days). The preferred injection site was the lower part of the abdomen or thigh. Changing the injection site from the left to right side or vice versa was recommended, especially during weekly administration.
During the expansion part of the trial, the RP2D regimen, which included a priming dose of 0.16 mg (C1D1), an intermediate dose of 0.8 mg (C1D8), and a full dose of 48 mg (C1D15, C1D22, and thereafter), was administered according to the following schedule:44
Cycles 1 to 3: days 1, 8, 15, and 22 (weekly)
Cycles 4 to 9: days 1 and 15 (every 2 weeks)
Cycle 10 and beyond until unacceptable toxicity, PD, or withdrawal of consent: day 1 (every 4 weeks)
During the expansion part, hospitalization was only required for a minimum of 24 hours after the first full dose of epcoritamab in cycle 1.44 Epcoritamab was taken according to the schedule shown in Figure 3 until disease progression or unacceptable toxicity.
Figure 3: Epcoritamab Dosing Schedule
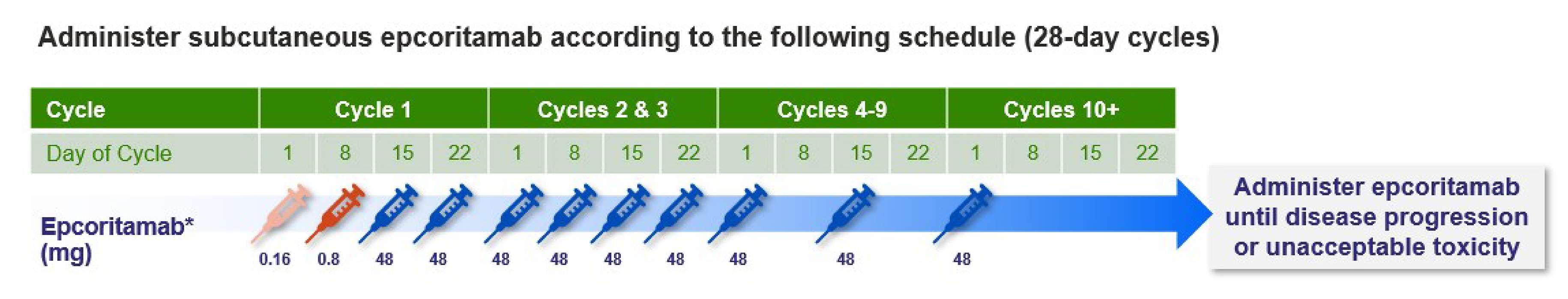
Note: Epcoritamab doses in cycle 1 were as follows: day 1, 0.16 mg (priming dose); day 8, 0.8 mg (intermediate dose); day 15, 48 mg (full dose); day 22, 48 mg. Epcoritamab doses in cycle 2 and onward were 48 mg.
Source: Product monograph.7
Premedication Prior to Epcoritamab Administration
Patients were premedicated with corticosteroids, antihistamines, and antipyretics 30 minutes to 120 minutes before the first 4 doses of epcoritamab. For subsequent doses of epcoritamab, premedication and CRS prophylaxis were optional.44
Prophylactic Corticosteroids Following Epcoritamab Administration
Corticosteroids were administered following epcoritamab administration on day 2, day 3, and day 4 in conjunction with all 4 doses of epcoritamab in cycle 1 (i.e., priming, intermediate, and first 2 full doses). If CRS greater than or equal to grade 2 occurred following the fourth administration of epcoritamab on day 22 of cycle 1, corticosteroid administration on the day of and for 3 days following epcoritamab administration was continued for subsequent epcoritamab doses until a dose was given after which no CRS occurred. Otherwise, 4-day consecutive corticosteroids were administered following epcoritamab dosing only for cycle 1 and for any repriming cycles.44 Based on the investigator’s evaluation, the corticosteroid daily dose requirement could be reduced to mitigate possible side effects from high-dose steroid administration.44
Permitted Concomitant Therapy
Concomitant medications were allowed to provide adequate patient care and were given as clinically indicated, except for antilymphoma therapy. All concomitant medications were recorded except for vitamins and nutrient supplements. Supportive medications, such as premedication, antiviral medication, and anti–interleukin-6 receptor (IL-6R) were provided at the trial site.44 For treatment of CRS, patients were recommended to receive supportive care, including infusion of saline, systemic glucocorticoids, antihistamines, antipyretics, support for blood pressure (vasopressin, vasopressors), support for low-flow and high-flow oxygen, and positive pressure ventilation and/or monoclonal antibodies against IL-6R (e.g., IV tocilizumab).44 Patients considered to be at increased risk for CTLS were recommended to receive hydration and prophylactic treatment with a uric acid-lowering drug. If signs of CTLS occurred, supportive therapy, including rasburicase, was allowed.44 Prophylactic antibiotic, antiviral, and antifungal therapies were allowed unless medically contraindicated. The use of growth factors for neutropenia, such as granulocyte colony-stimulating factor, was allowed during treatment with epcoritamab. In case of recurring greater than or equal to grade 3 neutropenia, use of growth factors was mandated.44
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 9, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted by CADTH and in the stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 9: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | EPCORE NHL-1 study |
|---|---|---|
OS | Median, 12 months and 18 months | Secondary |
PFS | Median, 12 months and 18 months | Secondary |
CR | Any | Secondary |
ORR | Any | Primary |
DOR | Median, 12 months and 18 months | Secondary |
HRQoL (FACT-Lym) | Baseline, cycle 3, cycle 5, cycle 7, and cycle 9 | Secondary and/or exploratory |
Notable Harms (CRS, ICANS, serious infections) | Any | — |
CR = complete response; CRS = cytokine release syndrome; DOR = duration of response; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HRQoL = health-related quality of life; ICANS = immune effector cell–associated neurotoxicity syndrome; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s Summary of Clinical Evidence.
Efficacy Outcomes
According to the clinical expert input, patient group input, and clinician group input received by CADTH, patients for whom second- and third-line treatments have not been effective have few safe and effective treatment options, and currently available therapies are palliative. Patients typically have poor survival (e.g., OS less than 6 months). Therefore, outcomes related to survival were included as outcomes to be assessed using GRADE. In the EPCORE NHL-1 study, these included OS and PFS.
The clinical experts consulted by CADTH noted that clinical response, particularly CR rate in R/R LBCL, is an important outcome of treatment; thus, CR and ORR were included in the GRADE assessment. Further to CR and ORR alone, the clinical experts suggested that DOR may demonstrate greater treatment effect than response rates alone; thus, this was included in the GRADE assessment.
HRQoL was identified as an important outcome in patients with R/R LBCL; thus, the change from baseline in HRQoL outcomes, including FACT-Lym total score, were included in the GRADE assessment.
Primary Efficacy Outcome
The primary efficacy end point of the EPCORE NHL-1 trial was ORR rate, defined as the proportion of patients whose best overall response was CR or PR based on IRC assessment of PET and/or CT scans using Lugano criteria. Patients who achieved a PR or CR after a PD event had been reported were included in the analysis of ORR || | |||.
Secondary Efficacy Outcomes
Secondary end points are summarized in Table 8 and included additional response outcomes (e.g., ORR and CR assessed according to lymphoma response to immunomodulatory therapy criteria [LYRIC] criteria), time-to-event outcomes (OS and PFS), response durations (DOR and DOCR), and HRQoL outcomes.
Assessment of Disease Response and PD
Response assessments according to the imaging assessment were performed by the site investigators to inform decisions regarding continuation of treatment for the patient. Response was assessed according to both Lugano and LYRIC criteria (with LYRIC including an assessment of indeterminate response). The response assessments and disease progression for the primary and secondary study end points were determined by an IRC according to the study protocol. The IRC workflow included the multistep evaluation presented here.
Radiology review:
Time point-by-time point radiology review: Each imaging time point for a patient was assessed by 2 independent reviewers, who assessed tumour burden at baseline and determined an overall tumour assessment at each postbaseline time point according to the Lugano criteria and LYRIC. Both PET-CT or CT-MRI and fluorodeoxyglucose PET 5-point scoring assessments per both criteria were performed when both modalities were available.
Global radiology review: The same independent reviewers globally assessed the reviewed time points and confirmed or updated their previous overall tumour assessments according to the Lugano criteria and separately according to LYRIC.
Radiology adjudication review: This was required if the independent radiologists’ results for a global radiology review conflicted. An independent radiologist who did not participate in the earlier reviews for the patient chose the independent radiologist with whom they agreed most as the final assessment and provided justifying comments separately for the Lugano criteria-based and LYRIC-based overall tumour assessment s.
Oncology review: Following the radiology review, an independent oncologist (single read) reviewed the final radiology review assessments and available pertinent clinical data and provided the final overall tumour assessment per visit per Lugano criteria, and separately by LYRIC.
Secondary radiology review: The time point-by-time point radiology review and global radiology review for a subset of patients was repeated. The secondary radiology review was used for the determination of intrareader agreement for that subset of patients and did not alter the original read.
Patient-Reported Outcomes
PROs in the EPCORE NHL-1 trial included the FACT-Lym, FACT-G, and EQ-5D-3L.
FACT-Lym: A questionnaire used to assess quality of life in lymphoma patients. It consists of a general quality of life instrument (i.e., FACT-G) and a condition-specific module called “Lym.” The sponsor reports that FACT-Lym is a validated quality of life questionnaire for patients with lymphoma. FACT-G consists of 27 statements categorized by 5 subscales (Physical Well-Being, Social/Family Well-Being, Emotional Well-Being, Functional Well-Being, and Additional Concerns). Patients score each statement using a 5-point scale, with responses ranging from “not at all” to “very much.” The Lym module consists of 15 statements that score using an identical 5-point scale as that applied the FACT-G.47
FACT-Lym symptoms: The sponsor evaluated 6 questions from the FACT-Lym that were related to the symptoms of lymphoma: P2 (body pain), BRM3 (fever), ES3 (night sweats), GP1 (lack of energy), BMT6 (tires easily), and C2 (weight loss). Changes from baseline in these questions were evaluated as secondary end points in the EPCORE NHL-1 trial. The sponsor reported that compliance was |||| at most study time points, and lower than this at |||| |||||||| ||||| |||||||| ||| ||| |||||||| |.
EQ-5D-3L: The EQ-5D-3L consists of a descriptive system and EQ visual analogue scale (EQ VAS). The descriptive system consists of 5 dimensions (mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression), with 3 levels (no health problems, moderate health problems, and extreme health problems). The EQ VAS records self-rated health on a scale ranging from 0 (worst imaginable health state) to 100 (best imaginable health state).47 The sponsor reported that the minimal important difference can be approximated at 0.08 for change from baseline in EQ-5D-3L health utility scores and a change from baseline in EQ VAS score of 7 points (citing a retrospective analysis from Pickard et al. [2007] conducted in patients living with advanced cancer).48
Table 10: Summary of Outcome Measures and Corresponding Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
FACT-LymS | The FACT-Lym consists of the 27-item FACT-G and the 15-item LymS.49 The FACT-G questionnaire assesses 4 dimensions of HRQoL: physical, social and family, emotional, and functional well-being.49 The LymS is an NHL-specific, patient-reported questionnaire used to assess HRQoL, specifically disease-specific symptoms and concerns.49 Items are rated on a 5-point Likert scale, with higher scores indicating better HRQoL.49 | In a study49 of 84 adult patients with NHL, measurements were taken at baseline, 3 days to 7 days, and 8 weeks to 12 weeks. Validity: Known-groups (construct) validity was demonstrated by LymS scores, which differentiated between patients with ECOG PS of 0, 1, or 2 and between patients on or off active treatment (e.g., radiation and chemotherapy), but did not differentiate between patient groups defined by NHL grade.49 Concurrent validity was demonstrated based on correlations between LymS and SF-36 PCS (r = 0.62) and MCS (r = 0.48) and POMS total score (r = 0.60).49 Divergent validity was demonstrated based on the near 0 association between the LymS and the Marlowe-Crowne Social Desirability Scale-Short Form (r = 0.15).49 Reliability: LymS demonstrated good internal consistency, with Cronbach alpha ranging from 0.79 to 0.85 at each assessment time point.49 LymS demonstrated good test-retest reliability based on an ICC of 0.84 (retested at 3 days to 7 days from baseline; n = 74).49 Responsiveness: FACT-Lym differentiated patients in each of the 3 groups defined by retrospective ratings of change at the final assessment and defined by change over 3 months in PS (worse, unchanged, better; effect sizes > 0.50).49 | Using distribution- and anchor-based methods, the investigators suggested that the likely MID range for LymS in patients with NHL is approximately 3 points to 5 points or 5% to 8% of the scale range (0 to 60).49 |
ECOG = Eastern Cooperative Oncology Group; FACT-G = Functional Assessment of Cancer Treatment–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; LymS = lymphoma subscale; MCS = Mental Component Summary; MID = minimal important difference; NHL = non-Hodgkin lymphoma; PCS = Physical Component Summary; POMS = Profile of Mood States; PS = Performance Status; SF-36 = Short Form (36) Health Survey.
Harms Outcomes
All safety analyses were conducted using the safety analysis set, which included 157 patients with LBCL who received at least 1 dose of epcoritamab in the EPCORE NHL-1 study. The adverse events of special interest (AESIs) for the EPCORE NHL-1 trial were CRS, CTLS, and ICANS. All AEs were graded by the investigator according to National Cancer Institute Common Terminology Criteria for Adverse Events, Version 5.0, except for CRS, ICANS, and CTLS. Events of CRS and ICANS were graded according to American Society for Transplantation and Cellular Therapy criteria,50 and CTLS events were graded according to Cairo-Bishop.51
Statistical Analysis
Clinical Trial End Points
The statistical analyses of the trial end points are presented in Table 11.52
Table 11: Statistical Analysis of Efficacy End Points in the EPCORE NHL-1 Trial Dose Expansion Part
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
ORR determined by Lugano criteria as assessed by IRC | Primary analysis of ORR was IRC-assessed per Lugano criteria in the FAS. ORR, disease control rate (BOR of SD and better), and the corresponding 95% exact CI were provided for DLBCL, other subtypes, and overall LBCL. | NA | Not applicable (calculated from FAS) | Sensitivity analyses of ORR were performed in a manner similar to the primary analysis for the following:
|
DORa | The DOR, estimated using the KM product-limit method, was displayed graphically. A summary table was produced presenting numbers of events and censored, minimum, maximum, median, first and third quantile, and landmark DOR rates at 3 months, 6 months, 9 months, and 12 months (and beyond, if available). Median, first and third quartiles along with 2-sided 95% CIs were computed based on log-log transformation. | NA | Patients with incomplete or no baseline tumour assessments were censored at day 1. Patients without documented progression or death were censored at the date of the last evaluable tumour assessment. | Sensitivity analyses were performed based on IRC disease assessment in the RES. Similar analyses based on investigator disease assessment in the FAS and RES were also conducted. |
CRa | In the FAS, CR rate and corresponding 95% exact CIs were provided for DLBCL, other subtypes, and overall LBCL. | NA | Not applicable (calculated from FAS) | Sensitivity analyses were performed based on IRC disease assessment in the RES. Similar analyses based on investigator disease assessment in the FAS and RES were also conducted. |
DOCRa | DOCR was derived for patients reaching CR. DOCR analyses were conducted using similar methods for DOR in the FAS. | NA | Patients with incomplete or no baseline tumour assessments were censored at day 1. Patients without documented progression or death were censored at the date of the last evaluable tumour assessment. | Analyses were presented based on investigator assessment (Lugano criteria). |
PFSa | PFS was derived for all patients and analyzed using methods similar to those used for DOR. PFS rate at landmark time T was defined as the probability that a patient had not progressed and was alive at time T following day 1 of cycle 1. PFS rates at 6 months, 9 months, and 12 months (or later times, if available) were presented along with 95% CIs. | NA | Patients with incomplete or no baseline tumour assessments were censored at day 1. Patients without documented progression or death (or new antilymphoma therapy, for the primary definition only) were censored at the date of the last evaluable tumour assessment. | Analyses were presented based on investigator assessment (Lugano criteria). |
OS | OS was derived for all patients and analyzed in the FAS using methods similar to those used for the DOR. | NA | If a patient was not known to have died, then OS was censored at the latest date on which the patient was known to be alive. Survival status was to be assessed at least every 3 months after the last administration of epcoritamab and to continue until the patient died or withdrew from the trial. | NA |
Changes in lymphoma symptoms, well-being, and general health status from baseline as measured by the FACT-Lym | Descriptive statistics for FACT-Lym assessment along with changes from baseline at each assessment time point were presented for FACT-Lym TOI, FACT-G total scores, FACT-Lym total scores, subscale scores, and 6 items of special interest (P2 [body pain], BRM3 [fever], ES3 [night sweats], GP1 [lack of energy], BMT6 [tired easily], and C2 [losing weight]). A line graph summarizing the mean change from baseline with standard error bar, with a reference line on the MID, was also produced for the FACT-Lym TOI, FACT-G total scores, FACT-Lym total scores, and FACT-LymS. | NA | Assuming that more than 50% of the items comprising a subscale are answered, a subscale score is calculated as the prorated sum of the item responses for that subscale (i.e., replacing missing values with the mean of the completed items for that subscale). If more than half of the item results within a subscale domain were missing, the subscale score was set to missing. If any subscale score was missing, the relevant overall scores that were based on that subscale were also reported as missing. | NA |
Changes in well-being and general health status from baseline as measured by EQ-5D-3L | Descriptive statistics were presented for EQ-5D-3L index score and EQ VAS along with changes from baseline at each assessment time point. A line graph summarizing the mean change from baseline with standard error bar was also produced for the EQ-5D-3L index score and EQ VAS, with a reference line on MID. | NA | EQ-5D-3L completion and compliance rates were calculated for each assessment time point. Distribution for noncompletion reason (i.e., did not complete, disease progression, discontinued due to AE or other reason, or death) were also included. | NA |
AE = adverse event; BOR = best overall response; CI = confidence interval; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; DOCR = duration of complete response; EQ VAS = EQ visual analogue scale; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FAS = full analysis set; IRC = independent review committee; KM = Kaplan-Meier; LBCL = large B-cell lymphoma; LYRIC = lymphoma response to immunomodulatory therapy criteria; MID = minimum important difference; mRES = modified response-evaluable set; NA = not applicable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PP = per protocol; RES = response-evaluable set; SD = stable disease; TOI = Trial Outcome Index.
aSecondary response end points included determination by both Lugano criteria and LYRIC as assessed by IRC.
Source: EPCORE NHL-1 Statistical Analysis Plan52 and Clinical Study Report (April 2023 data cut-off).43
Sample Size and Power Calculation
In total, up to 158 patients were planned for enrolment.52 Assuming a nonevaluable rate of 10%, a sample size of 128 patients in the DLBCL group would provide approximately 90% power to detect the alternative hypothesis of at least 50% ORR while ensuring a 2-sided significance level of 0.05 using a 1-sample exact binomial test under the null hypothesis of at most 35% ORR. The probability of futility at the end of stage 1 was approximately 30% under the null and 2.1% under the alternative.52
Statistical Testing
For continuous variables, descriptive statistics included the number of nonmissing values (n), mean, SD, median, and minimum and maximum values. In addition, 25th percentile and 75th percentile were provided for some outcomes.52 Time-to-event variables were analyzed using Kaplan-Meier estimates (median time, first and third quartiles), with the number and percentage of patients with event or censoring reported. If specified, 95% CIs were provided using the Brookmeyer and Crowley method with log-log transformation. Landmark event-free rates were also presented together with the 95% CIs for some end points.52
The primary analysis was conducted based on a data cut-off of January 31, 2022. The April 21, 2023 analysis is the most recent analysis and is the focus of the CADTH submission.
Subgroup Analyses
As per the prespecified statistical analysis plan, ORR and CR rates based on IRC assessment per Lugano criteria in the full analysis set (FAS) were summarized within the subgroups listed along with 95% exact CIs in a forest plot for DLBCL and LBCL. For any subgroup that included fewer than 20 patients, either the analysis for was not carried out or combining of subgroups was considered.52 The subgroups were:
age (< 65 years, 65 years to < 75 years, ≥ 75 years)
gender (male, female)
race (white, Asian, Black, or other)
region (North America, Europe, other)
baseline ECOG Performance Score (0, 1, 2, or higher)
baseline weight (< 65 kg, 65 kg to < 85 kg, ≥ 85 kg)
number of prior antilymphoma therapies (2, 3, 4, or more than 4)
time from last anti-CD20 therapy until first dose of epcoritamab (< median, ≥ median)
prior CAR T-cell therapy experience (yes, no)
prior ASCT (yes, no)
prior antilymphoma therapy status (primary refractory, other)
most recent prior anti-CD20 containing therapy (refractory, relapse)
chromosomal abnormality (double hit, triple hit, other)
Ann Arbor staging (I or II; III or IV)
IPI (0 to 2; ≥ 3)
DLBCL disease state (de novo, transformed)
molecular classification (GCB, non-GCB)
molecular classification (activated B-cell)
overall presence of antidrug antibodies (positive or nonpositive)
As per the prespecified statistical analysis plan, forest plots of median PFS (both primary and secondary definitions) based on IRC assessment per Lugano criteria along with 95% CIs were also produced for each level of each of these subgroups for the FAS.52 OS was also assessed.
In addition to the prespecified analyses previously listed, key analyses of ORR, CR, PFS, and OS were conducted for the following subgroups: primary refractory disease; refractory to prior CAR T-cell therapy; and elevated lactate dehydrogenase.43
Analysis Populations
Analysis populations in the EPCORE NHL-1 trial are summarized in Table 12.
Table 12: Analysis Populations of the EPCORE NHL-1 Trial
Population | Definition | Application |
|---|---|---|
Full analysis set | All enrolled patients who received at least 1 dose of epcoritamab | All primary and secondary efficacy analyses were conducted using the full analysis set |
Safety analysis set | All enrolled patients who received at least 1 dose of epcoritamab, which is the same as the full analysis set | Harms outcomes were assessed based on the safety analysis set |
Response-evaluable set | All patients in the full analysis set who had measurable disease at baseline and who had at least 1 postbaseline disease evaluation or died within 60 days of the first dose without postbaseline disease assessment | Sensitivity analyses for ORR and DOR were conducted in the response-evaluable set |
Patient-reported outcome analysis set | All patients in the full analysis set with a baseline and at least 1 postbaseline patient-reported outcome score | For analyses relating to changes in baseline, the patient-reported outcome analysis was used |
DOR = duration of response; ORR = overall response rate.
Sources: EPCORE NHL-1 Statistical Analysis Plan;52 sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
The disposition of patients in the EPCORE NHL-1 trial dose expansion part is summarized in Table 13. A total of ||| patients were screened; 157 patients were enrolled and received at least 1 dose of epcoritamab. At the time of the April 21, 2023, data cut-off, || ||||||| patients were still receiving epcoritamab treatment and ||| ||||||| patients had discontinued. The most frequently reported reasons for discontinuation were disease progression (|| |||||||) and AEs (|| |||||||). The sponsor reported that | |||||| patients discontinued epcoritamab after deciding to proceed to transplant after having CR (| | |) or PR (| | |) to the treatment. A total of || ||||||| permanently discontinued the trial, with death being the most common reason (|||||).
Table 13: Patient Disposition in the EPCORE NHL-1 Trial Dose Expansion Part (FAS)
Patient disposition | EPCORE NHL-1 study patients with LBCL |
|---|---|
Epcoritamab (N = 157) | |
Patients enrolled, N | ||| |
Patients not treated, N | || |
Not eligible, n | || |
Other, n | ||| |
Patients treated, N | 157 |
Ongoing study treatment | || ||||||| |
Discontinued study treatment | ||| ||||||| |
Primary reason for treatment discontinuation | |
Progressive diseasea | || ||||||| |
Clinical progression | || |||||| |
Disease progression according to response criteria | || ||||||| |
Adverse event | || ||||||| |
Death | ||| |
Withdrawal by patientb | | |||||| |
Decision to proceed with transplant | | |||||| |
Maximum clinical benefit | | |||||| |
Otherc | | |||||| |
Patients remain on trial | || ||||||| |
Discontinued from trial | || ||||||| |
Death | || ||||||| |
Lost to follow-up | | |||||| |
Patient withdrew consent from trial | || |||||| |
aNHL = aggressive B-cell non-Hodgkin lymphoma; DLBCL = diffuse large B-cell lymphoma; LBCL = large B-cell lymphoma.
aProgressive disease includes both clinical progression and documented radiographic disease progression.
b“Withdrawal by patient” includes: 2 patients who refused to come to the site for the end-of-treatment visit before completing cycle 1 and died due to disease progression on day 46 and day 91; 1 patient who was in complete response following cycle 14 and did not want to come into the hospital frequently anymore; 1 patient who opted for hospice and died from disease progression the day after discontinuing treatment; and 1 patient whose treatment discontinuation occurred 5 days after their last lymphoma assessment showing disease progression (the specific reason for the patient’s decision to discontinue treatment cannot be established).
c“Other” includes: 2 patients due to investigator decision (not further specified); 1 patient who proceeded to chimeric antigen receptor T-cell therapy following a partial response to epcoritamab; 1 patient due to adverse events and good response to treatment; and 1 patient who was too frail to continue further therapy and likely approaching end of life.
Source: Sponsor’s Summary of Clinical Evidence.11
Baseline Characteristics
The baseline characteristics are outlined in Table 14. The median age was 64.0 years, with 48 patients (30.6%) aged 65 years to under 75 years and 29 patients (18.5%) aged greater than or equal to 75 years. Most patients had a baseline ECOG Performance Status of 0 (47.1%) or 1 (49.7); 3.2% had an ECOG Performance Status of 2. Most (139 patients [88.5%]) had DLBCL histology, with 97 patients (69.8%) with DLBCL having de novo disease and 40 patients (28.8%) having transformed disease. Patients with other LBCL subtypes included 9 patients (5.7%) with HGBCL, 5 patients (3.2%) with FLG3B, and 4 patients (2.5%) with PMBCL.
Table 15 summarizes the treatment history for those enrolled in the EPCORE NHL trial. The median number of prior lines of antilymphoma therapy was 3.0 (range, 2 to 11), with 47 patients (29.9%) having received 2 prior lines of therapy, 48 patients (30.6%) having received 3 lines, and 62 patients (39.5%) having received greater than or equal to 4 lines. Overall, 61 patients (38.9%) had received prior CAR T-cell therapy and 31 patients (19.7%) had prior ASCT.
Table 14: Summary of Baseline Characteristics From the EPCORE NHL Trial Dose Expansion Part (FAS)
Characteristic | EPCORE NHL-1 study patients with LBCL (N = 157) |
|---|---|
Age (years) | |
Mean (standard deviation) | |||| ||||||| |
Median (range, minimum to maximum) | 64.0 (20 to 83) |
Age category (years) | |
< 65 years | 80 (51.0%) |
65 years to < 75 years | 48 (30.6%) |
≥ 75 years | 29 (18.5%) |
Sex (at birth) | |
Male | 94 (59.9%) |
Female | 63 (40.1%) |
Race | |
White | || ||||||| |
Asian | || ||||||| |
Other | | |||||| |
Not reported | || ||||||| |
Ethnic origin | |
Hispanic or Latino | ||| |
Not Hispanic or Latino | || ||||||| |
Weight (kg) at baseline | |
Mean (standard deviation) | |||| ||||||| |
Median (range, minimum to maximum) | |||| |||||| |||||| |
ECOG Performance Status | |
0 | 74 (47.1%) |
1 | 78 (49.7%) |
2 | 5 (3.2%) |
Baseline renal function (CrCl, mL/min) | |
Normal (90) | || ||||||| |
Mildly impaired (60 to < 9 0) | || ||||||| |
Moderately impaired (30 to < 60) | || ||||||| |
Severe impaired (15 to < 30) | ||| |
Baseline hepatic function per NCI criteria | |
Normal | ||| ||||||| |
Mild dysfunction | || ||||||| |
Moderate dysfunction | | |||||| |
Severe dysfunction | ||| |
Missing | ||||||| |
Disease type at trial entry | |
DLBCL | 139 (88.5%) |
HGBCL | 9 (5.7%) |
PMBCL | 4 (2.5%) |
FL grade 3B | 5 (3.2%) |
DLBCL type | |
De novo | 97 (61.8%) |
Transformed | 40 (25.5%) |
Disease type at initial diagnosis | |
FL | || ||||||| |
MZL | | |||||| |
SLL | | |||||| |
Other | | |||||| |
Unknown | | |||||| |
Not applicable | || ||||||| |
DLBCL cell of origin classification, per local laboratoryb | |
GCB | 65 (41.4%) |
ABC/non-GCB | 56 (35.7%) |
Unknown | || ||||||| |
Not applicable | || ||||||| |
Median time from initial diagnosis to first dosec (minimum to maximum), years | 1.6 (0.0 to 28.4) |
MYC and BCL2 and/or BCL6 rearrangements, per local laboratory | |
Double-hit lymphoma | 11 (7.0%) |
Triple-hit lymphoma | 6 (3.8%) |
Other | 20 (12.7%) |
MYC and BCL2 and/or BCL6 rearrangements, per central laboratory FISH analysis | |
Number evaluated | 99 |
Double-hit lymphoma | || ||||||| |
Triple-hit lymphoma | | |||||| |
Other | || ||||||| |
Ann Arbor stage at screening | |
I | | |||||| |
IE | | |||||| |
II | || ||||||| |
IIE | | |||||| |
III | || ||||||| |
IIIE | | |||||| |
IIIS | | |||||| |
IV | || ||||||| |
IPI (at study entry) | |
0 to 2 | 55 (35.0%) |
≥ 3 | || ||||||| |
Unknown | | |||||| |
Not applicable | 18 (11.5%) |
Presence of constitutional symptoms | || ||||||| |
Night sweats | || |||||| |
Weight loss (> 10% over last 6 months) | | |||||| |
Fever | | |||||| |
Extreme fatigue | | |||||| |
ABC = activated B-cell; CrCl = creatinine clearance; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; FISH = fluorescence in situ hybridization; FL = follicular lymphoma; GCB = germinal centre B-cell; HGBCL = high-grade B-cell lymphoma; IPI = International Prognostic Index; LBCL = large B-cell lymphoma; MZL = marginal zone lymphoma; NCI = National Cancer Institute; PMBCL = primary mediastinal large B-cell lymphoma; SLL = small lymphocytic lymphoma.
aOther includes HGBCL (n = 9), FL grade 3B (n = 5), and PMBCL (n = 4).
bPatients who had results from local laboratory analysis collected as medical history.
Source: EPCORE NHL-1 Clinical Study Report (April 21, 2023)43 and sponsor’s Summary of Clinical Evidence.11
Table 15: Summary of Treatment History in the EPCORE NHL Trial Dose Expansion Part (FAS)
Treatment history | aNHL cohort | ||
|---|---|---|---|
DLBCL (N = 139) | Other subtypes (N = 18)a | LBCL (N = 157) | |
Prior radiotherapy | || ||||||| | | ||||||| | || ||||||| |
Prior stem cell transplant | || ||||||| | | ||||||| | 31 (19.7%) |
ASCT | || ||||||| | | ||||||| | 31 (19.7%) |
Relapsed ≤ 12 months after ASCT | || ||||||| | | ||||||| | 18 (11.5%) |
Allogeneic SCT | | |||||| | ||| | | |||||| |
Prior systemic therapy received | |||
Anti-CD20 | ||| |||||| | || |||||| | ||| |||||| |
Anti-CD19 | | |||||| | ||| | | |||||| |
Alkylating-containing drugs | ||| |||||| | || |||||| | ||| |||||| |
Anthracyclines | ||| ||||||| | || ||||||| | ||| ||||||| |
Nucleotide | ||| ||||||| | || ||||||| | ||| ||||||| |
Topo inhibitor | || ||||||| | || ||||||| | ||| ||||||| |
PI3K inhibitor | | |||||| | ||| | | |||||| |
BCL2 inhibitor | | |||||| | ||| | | |||||| |
PolyV | || |||||| | | ||||||| | || ||||||| |
CAR T-cell therapy | || ||||||| | | ||||||| | 61 (38.9%) |
Other | ||| |||||| | || |||||| | ||| |||||| |
Prior lines of antilymphoma therapy, median (minimum to maximum) | ||| ||| ||| | ||| ||| || | ||| ||| ||| |
1 | ||| | ||| | ||| |
2 | || ||||||| | | ||||||| | || ||||||| |
3 | || ||||||| | | ||||||| | || ||||||| |
≥ 4 | || ||||||| | || ||||||| | || ||||||| |
Time from end of last-line antilymphoma therapy to first dose of epcoritamab (months), median (minimum to maximum) | ||| ||| |||| | ||| ||| ||| | 2.4 (0 to 153) |
Primary refractory diseaseb | || ||||||| | || ||||||| | || ||||||| |
Refractory to ≥ 2 consecutive lines of prior antilymphoma therapy,c n (%) | ||| ||||||| | || ||||||| | ||| ||||||| |
Last-line systemic antineoplastic therapy | |||
Refractoryc | ||| ||||||| | || ||||||| | ||| ||||||| |
No response | || ||||||| | || ||||||| | || ||||||| |
Relapsed within 6 months after therapy completion | || ||||||| | | ||||||| | || ||||||| |
Relapsedd | || ||||||| | | ||||||| | || ||||||| |
aNHL = aggressive B-cell non-Hodgkin lymphoma; ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CD19 = cluster of differentiation 19; CD20 = cluster of differentiation 20; DLBCL = diffuse large B-cell lymphoma; FL = follicular lymphoma; HGBCL = high-grade B-cell lymphoma; LBCL = large B-cell lymphoma; PMBCL = primary mediastinal B-cell lymphoma; SCT = stem cell transplant.
aOther includes HGBCL (n = 9), FL grade 3B (n = 5), and PMBCL (n = 4).
bConsidered primary refractory if refractory to front-line antilymphoma therapy.
cConsidered refractory if experienced disease progression or stable disease as best response or disease progression within 6 months after therapy completion.
dConsidered relapsed if experienced disease progression more than 6 months after last treatment.
Source: Clinical Study Report for the EPCORE NHL-1 study.43
Exposure to Study Treatments
Patients with LBCL in the study received epcoritamab for a mean duration of ||| months and ||| cycles, with an average of |||| doses administered. Epcoritamab exposure and treatment compliance are summarized in Table 16. The median number of cycles initiated was ||| ||||||| | || ||| and the median duration of treatment was ||| months ||||||| | || ||||.43
A total of || ||||||| patients with LBCL initiated cycle 4 treatment, providing a close estimation of patients who received at least 3 months of treatment. Similarly, || ||||||| patients with LBCL initiated cycle 7, approximating at least 6 months of treatment; || ||||||| patients with LBCL initiated cycle 10, approximating at least 9 months of treatment; and || ||||||| patients with LBCL initiated cycle 13, approximating at least 12 months of treatment. || |||||||| ||||| initiated cycle 26, approximating at least 2 years of treatment. As of the data cut-off date of 21 April 2023, || ||||||| patients with LBCL were continuing on epcoritamab treatment.43
Overall, || ||||||| patients with LBCL required at least 1 dose delay during the trial, including || ||||||| patients who required a dose delay due to an AE and || |||||| patients who required a dose delay for another reason, including COVID-19 control measures (Table 16). Dose reductions were not allowed in this trial.43
The median relative dose intensity for patients with LBCL was |||| ||||||| ||| |||| in cycle 1 to cycle 3 (weekly dosing), |||| ||||||| ||| |||| in cycle 4 to cycle 9 (with dosing every 2 weeks), and ||||| ||||||| ||| |||| in cycle 10 and beyond (dosing every 4 weeks). For cycle 1 to cycle 3, relative dose intensity was greater than or equal to 90% for ||| ||||||| patients; for cycle 4 to cycle 9, relative dose intensity I was greater than or equal to 90% for more than ||| of patients; and for cycle 10 and beyond, relative dose intensity was greater than or equal to 90% for ||| of patients. The exposure to the study drug allowed adequate assessment of safety and efficacy in the trial population.43
A dose delay referred to a delay in the administration of epcoritamab relative to the per-protocol schedule per investigator. A dose repriming cycle consists of a weekly schedule of a priming dose, an intermediate dose, and 2 full doses.43
As of the data cut-off date, || |||||| patients required epcoritamab repriming. For 9 of these patients, the reason for repriming was dose delay (due to AEs in ||| patients, ||| of which were related to COVID-19 infection and ||| of which were due to COVID-19 control measures). No specified reason was given for the remaining patient who was reprimed. In all ||| patients, the repriming occurred late in the treatment cycle (i.e., after cycle 11), and no patient experienced CRS after repriming.43
Table 16: Epcoritamab Exposure and Compliance in the EPCORE NHL-1 Study Dose Expansion Part (Safety Analysis Set)
Exposure | EPCORE NHL-1 study patients with LBCL |
|---|---|
Epcoritamab (N = 157) | |
Number of cycles | |
Median number of cycles initiated (minimum to maximum) | | || ||| ||| |
Mean number of cycles initiated (SD) | ||| ||||||| |
Median number of doses received (minimum to maximum) | |||| ||| ||| |
Mean number of doses received initiated (SD) | |||| ||||||| |
Duration of treatment | |
Median duration of treatment (minimum to maximum), monthsb | ||||| |||||| |||||| |
Mean (SD), months | ||||| |||||||| |
Relative dose intensity (%)c | |
Cycle 1 to cycle 3 | |
N | ||| |
Mean (SD) | |||| |||||| |
Median | ||||| |
Minimum to maximum | ||| ||| |
50% to < 70% | | ||||| |
70% to < 90% | || |||||| |
90% to < 110% | ||| |||||| |
Cycle 4 to cycle 9 | |
n | || |
Mean (SD) | |||| |||||| |
Median | ||||| |
Minimum to maximum | ||| ||| |
50% to < 70% | || |
70% to < 90% | | |||||| |
90% to < 110% | || ||||||| |
Cycle 10 and beyond | |
n | || |
Mean (SD) | |||| ||||||| |
Median | |||| |
Minimum to maximum | ||| ||| |
50% to < 70% | | |||||| |
70% to < 90% | | |||||| |
90% to < 110% | || ||||||| |
Number of patients experiencing dose delay | || ||||||| |
Reason for dose delayd | |
Adverse event | | ||||||| |
Othere | || ||||||| |
Number of patients with repriming | || |||||| |
FL = follicular lymphoma; HGBCL = high-grade B-cell lymphoma; LBCL = large B-cell lymphoma; PMBCL = primary mediastinal large B-cell lymphoma; SD = standard deviation.
aOther includes 9 patients with HGBCL, 5 patients with FL grade 3B, and 4 patients with PMBCL.
bDuration of treatment was calculated as last dose date minus first dose date plus 1.
cActual dose intensity is calculated as the actual dose administered on and after the first full dose divided by duration of dosing period in a 28-day cycle. Relative dose intensity is calculated as actual dose intensity divided by planned full dose intensity in the analysis period.
dPatients may experience multiple times of dose delay.
eIncludes patients who had dose delays due to COVID-19 control measures.
Source: EPCORE NHL-1 Clinical Study Report (April 21, 2023)43 and sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
Concomitant medications used by greater than or equal to 20% of patients in the aNHL expansion cohort are summarized in Table 17.43 For treatment of CRS, patients were recommended to receive supportive care, including saline infusion, systemic glucocorticoids, antihistamines, antipyretics, support for blood pressure (vasopressin, vasopressors), support for low-flow and high-flow oxygen and positive pressure ventilation, and/or monoclonal antibodies against IL-6R (e.g., IV administration of tocilizumab).43 Patients considered to have an increased risk of CTLS were recommended to receive hydration and prophylactic treatment with a uric acid-lowering drug. If signs of CTLS occurred, supportive therapy, including rasburicase, was allowed.43
Prophylactic antibiotic, antiviral, and antifungal therapies were allowed unless medically contraindicated. The use of growth factors for neutropenia, such as granulocyte colony-stimulating factor, was allowed during treatment with epcoritamab. In case of recurring neutropenia of greater than or equal to grade 3, use of growth factors was mandated.43 All 157 patients received corticosteroids before and after epcoritamab administration, per the criteria specified in the protocol. Overall, || ||||||| patients received concomitant allopurinol, and || ||||||| patients received concomitant rasburicase; these medications were used mostly as prophylactic measures in patients with high risk factors for CTLS.43
Table 17: Concomitant Medications in at Least 20% of Patients Overall in the EPCORE NHL-1 Study Expansion Cohort (Safety Analysis Set)
ATC code level 2 generic name | EPCORE NHL-1 study patients with LBCL |
|---|---|
Epcoritamab (N = 157) | |
Antibacterials for systemic use | ||| ||||||| |
Sulfamethoxazole; trimethoprim | ||| ||||||| |
Piperacillin sodium; tazobactam sodium | || ||||||| |
Amoxicillin trihydrate; clavulanate potassium | || ||||||| |
Analgesics | ||| ||||||| |
Paracetamol | ||| ||||||| |
Antivirals for systemic use | ||| ||||||| |
Aciclovir | || ||||||| |
Valaciclovir | || ||||||| |
Drugs for acid-related disorders | ||| ||||||| |
Pantoprazole | || ||||||| |
Omeprazole | || ||||||| |
Antithrombotic drugs | || ||||||| |
Antigout preparations | ||||||||| |
Allopurinol | || ||||||| |
Blood substitutes and perfusion solutions | || ||||||| |
Sodium chloride | || ||||||| |
Corticosteroids for systemic use | || ||||||| |
Dexamethasone | || ||||||| |
Psycholeptics | || ||||||| |
Mineral supplements | || ||||||| |
Potassium chloride | || ||||||| |
Drugs for constipation | ||||||||| |
Vaccines | || ||||||| |
Antiemetics and antinauseants | || ||||||| |
All other therapeutic products | || ||||||| |
Vitamins | || ||||||| |
Antihistamines for systemic use | || ||||||| |
Antimycotics for systemic use | || ||||||| |
Beta-blocking drugs | || ||||||| |
Drugs for functional gastrointestinal disorders | || ||||||| |
Anti-inflammatory and antirheumatic drugs | || ||||||| |
Immune sera and immunoglobulins | || ||||||| |
ATC = anatomic therapeutic chemical; LBCL = large B-cell lymphoma.
Note: ATC code level 2 IS presented only where at least 1 generic name met the cut-off of 20% in that category.
aOther includes 9 patients with high-grade B-cell lymphoma, 5 patients with follicular lymphoma grade 3B, and 4 patients with primary mediastinal B-cell lymphoma.
Source: EPCORE NHL-1 Clinical Study Report (April 21, 2023).43
Subsequent Treatment
Subsequent antilymphoma therapies used by patients with LBCL are summarized in Table 18. A total of || ||||||| patients with LBCL went on to receive a subsequent antilymphoma therapy. The most common subsequent systemic therapy received was rituximab (||| ||||||| patients). In addition, || ||||||| patients received subsequent radiotherapy and || |||||| patients received subsequent CAR T-cell therapy.43 A total of | |||||| patients received a subsequent allogeneic HSCT, and | |||||| patient received a subsequent ASCT. ||||| of these patients had a best overall response of CR (including the patient who received ASCT); ||| patient had PR; and ||| patient had stable disease per IRC using Lugano criteria. ||||| patients were alive as of the data cut-off date; ||| patient died after coding (i.e., had a cardiac arrest) in the emergency department; ||| patient died of respiratory failure; and ||| ||||||| ||| |||||||| |||| |||| ||| || ||||||| |||||||||||.43
Table 18: Subsequent Antilymphoma Therapies in the EPCORE NHL-1 Study Dose Expansion Part (FAS)
Number of treated patients, n (%) | EPCORE NHL-1 study patients with LBCL |
|---|---|
Epcoritamab (N = 157) | |
Patients with any subsequent antilymphoma therapy | || ||||||| |
Subsequent radiotherapy | || ||||||| |
Subsequent CAR T-cell therapy | || |||||| |
Subsequent stem cell transplant | || |||||| |
Autologous | | |||||| |
Allogeneic | | |||||| |
Patients received subsequent systemic drug therapy | || ||||||| |
Rituximab | || ||||||| |
Lenalidomide | || |||||| |
Polatuzumab vedotin | || |||||| |
Bendamustine | || |||||| |
ATC = anatomic therapeutic chemical; CAR = chimeric antigen receptor; LBCL = large B-cell lymphoma.
Note: Patients are counted at most 1 time within each generic name, and at most 1 time per ATC level.
aWhere incidence for LBCL was greater than 5.0%.
Source: EPCORE NHL-1 Clinical Study Report (April 21, 2023).43
Efficacy
Efficacy results from the April 2023 cut-off date for the EPCORE NHL-1 study are summarized in Table 19.
Table 19: Summary of Key Efficacy Results From the EPCORE NHL-1 Study
End points | Epcoritamab N = 157 |
|---|---|
Best overall response based on IRC assessment, Lugano criteria | |
ORR,a n (%) | 99 (63.1) |
(95% CI)b | (55.0 to 70.6) |
CR rate, n (%) | || ||||||| |
(95% CI)b | |||||| ||||| |
Best overall response, n (%) | |
CR | || ||||||| |
PR | || ||||||| |
Stable disease | 5 (3.2%) |
PD | 37 (23.6%) |
Not evaluable | || ||||||| |
Duration of response based on IRC assessment, Lugano criteria | |
Number of responders (PR or CR) | || |
Number (%) of events | || ||||||| |
Duration of response, median months (95% CI)b | |||| ||||| ||||| |
Estimated percentage of patients remaining in response (95% CI)c | |
3 months | ||||| ||||||| |||||| |
6 months | ||||| ||||||| |||||| |
9 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
Duration of complete response based on IRC assessment, Lugano criteria | |
Number of complete responders | 63 |
Number (%) of events | || ||||||| |
Duration of complete response, median months (95% CI)b | |||| |||||| ||| |
Estimated percentage of patients remaining in complete response (95% CI)c | |
3 months | ||||| ||||||| |||||| |
6 months | ||||| ||||||| |||||| |
9 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
PFS (primary definitiond) based on IRC assessment, Lugano criteria | |
Number (%) of events | ||| ||||||| |
PFS, median months (95% CI)b | 4.4 (3.0 to 8.8) |
Estimated percentage of patients remaining progression-free (95% CI)c | |
6 months | ||||| ||||||| |||||| |
9 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
Time to response based on IRC assessment, Lugano criteria | |
Number of responders | || |
Mean time to response, months (SD) | ||| |||||| |
Median time to response, months (minimum, maximum) | ||| ||||| |||| |
Time to complete response based on IRC assessment, Lugano criteria | |
Number of complete responders | 63 |
Mean time to complete response, months (SD) | ||| |||||| |
Median time to complete response, months (minimum, maximum) | ||| ||||| ||||| |
Overall survival | |
Number (%) of events | || ||||||| |
Overall survival, median months (95% CI)b | 18.5 (11.7 to ||||) |
Estimated percentage of patients remaining alive (95% CI)c | |
6 months | ||||| ||||||| |||||| |
9 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
15 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
Time to next antilymphoma therapy | |
Number (%) of events | || ||||||| |
Median time to next antilymphoma therapy, months (95% CI)b | ||| ||||| ||||| |
Estimated percentage of patients not initiating next line of therapy (95% CI)b | |
3 months | ||||| ||||||| |||||| |
6 months | ||||| ||||||| |||||| |
9 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
15 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
Rate and duration of MRD negativity per ctDNA assay | |
MRD-evaluable population, n | ||| |
Number (%) of events | || ||||||| |
Median duration of MRD negativity, months (95% CI)b | |||| |||||| ||| |
Estimated percentage of patients remaining MRD-negative (95% CI)b | |
6 months | ||||| ||||||| |||||| |
12 months | ||||| ||||||| |||||| |
18 months | ||||| ||||||| |||||| |
CI = confidence interval; CR = complete response; ctDNA = circulating tumour DNA; IRC = independent review committee; MRD = minimum residual disease; ORR = overall response rate; PD = progressive disease; PFS = progression-free disease; PR = partial response; SD = standard deviation.
aORR is equal to CR plus PR.
bBased on the Clopper-Pearson method.
cBased on Kaplan-Meier estimate.
dThe primary definition of PFS accounted for the initiation of subsequent antilymphoma therapy and censored PFS at the last evaluable tumour assessment on or before the date of subsequent antilymphoma therapy. The subsequent antilymphoma therapies for PFS censoring consist of systemic antilymphoma therapy and curative-intent radiotherapy on 1 and only target lesion.
Source: EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off).43
Overall Survival
OS was a secondary end point of the EPCORE NHL-1 study. Results for OS at the clinical cut-off date (April 21, 2023) in the primary efficacy population are summarized in Table 19 and displayed visually in Figure 4. By the clinical cut-off date, || ||||||| patients had died, resulting in a median OS of 18.5 months (95% CI, 11.7 months to |||| months). The estimated proportions of patients who remained alive at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || ||||||| respectively.
Figure 4: Kaplan-Meier Plot of OS — Patients With LBCL, EPCORE NHL-1 Study Dose Expansion Part (FAS) [Redacted]

CI = confidence interval; CR = complete response; LBCL = large B-cell lymphoma; PR = partial response.
Source: Adapted from data in the EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off)43
Progression-Free Survival
Results for PFS based on IRC assessment (Lugano criteria) are summarized in Table 19 and displayed visually in Figure 5. ||| ||||||| patients experienced a PFS event (i.e., disease progression or death) according to the primary definition. The median PFS (primary definition) was 4.4 months (95% CI, 3.0 months to 8.8 months). The estimated percentages of patients remaining progression-free at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| ||| ||||| |||| ||| ||||| || ||||||| Overall results for the 18 patients in the cohort of other LBCL subtypes were similar to those for the LBCL and DLBCL cohorts. Similar PFS rates were observed using the secondary definition (i.e., median PFS was ||| |||||| |||| ||| ||| || ||||).
Similar to DOR, for PFS based on IRC assessment (primary definition), there was a higher number of censored patients when evaluated using LYRIC compared with Lugano criteria. Consequently, median PFS was longer using LYRIC (|||| |||||| for LBCL) than with Lugano criteria (||| |||||| for LBCL).
Figure 5: Kaplan-Meier Plot of PFS (Primary Definition) Based on IRC Assessment, Lugano Criteria — Patients With LBCL, EPCORE NHL-1 Study Dose Expansion Part (FAS) [Redacted]

CI = confidence interval; IRC = independent review committee; LBCL = large B-cell lymphoma.
Source: Adapted from data in the EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off).43
Figure 6: EPCORE NHL-1 Study — Kaplan-Meier Plot, Stratified by Response, for PFS in Patients with LBCL [Redacted]

LBCL = large B-cell lymphoma.
Source: EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off).43
Clinical Response
Results for all clinical response outcomes of interest, including CR, PR, ORR, and DOR, are summarized in Table 16.
CR Rate
The CR rates, based on Lugano criteria, were ||||| |||| ||| |||| || ||||| when determined by the IRC and ||||| |||| ||| |||| || ||||| when determined by the investigator. The median DOCRs were |||| |||||| |||| ||| |||| || ||| when assessed by IRC and |||| |||||| |||| ||| |||| || ||| when assessed by the investigators. The CR rate was ||||| |||| ||| |||| || ||||| when assessed by the IRC using LYRIC, with a median DOCR of |||| |||||| |||| ||| |||| || |||||.
Overall Response Rate
The ORR (CR plus PR) in patients with LBCL (N = 157) was 63.1% (95% CI, 55.0% to 70.6%), with || ||||||| and || ||||||| patients achieving best responses of CR and PR, respectively. Patients with a response in the primary analysis included || patients with a PR or CR (using Lugano criteria) after assessment of PD. These responses were also included in the analysis of the secondary end points, such as CR rate, DOR, DOCR, TTR, time to CR, and PFS.
Duration of Response
For patients who had achieved PR or CR || | |||| the median DOR was |||| |||||| |||| ||| ||| || ||||| when assessed by IRC using Lugano criteria. The estimated percentages of patients remaining in response at 12 months and 18 months were ||||| |||| ||| ||||| || |||||| and ||||| |||| ||| ||||| || ||||||| respectively. The median DORs were |||| |||||| |||| ||| |||| || ||||| when assessed by the investigators using Lugano criteria and |||| |||||| |||| ||| |||| || ||||||.
Table 20: Summary of Subgroup Analyses From the EPCORE NHL-1 Study
||||||||| | | || ||||||||||| |||||||| | |||||| || |||| ||| | ||| |||| ||| | || |||| ||| |
|---|---|---|---|---|
||| |||||||| | |||||| | 18.5 (11.7 to ||||| | ||
||| | ||||
||| ||||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
|| || ||| ||||| | ||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| ||||| | ||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
|||||||| |||| ||||||||||| ||||| | ||||
||| | ||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| | ||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
|||||| || ||||| ||||||||||||| ||||||||| | ||||
||| | ||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
|||| | ||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
||||| ||||| |||||||||| | ||||
|||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| | ||||| | |||| |||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
|||||||||| || ||||| ||||| |||||||||| | ||||
||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||||| |||| | ||||
|||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| | |||||| | |||| |||||| ||||| | ||| ||||| ||||| | ||| ||||| |||| |
||||| ||||||||||||| ||||||| |||||| | ||||
||||||| |||||||||| | ||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
||||| | ||||| | || |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
|||| |||||| ||||| ||||||||| |||||||||| ||||||| | ||||
|||||||||| | |||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
||||||| | |||| | || |||||| ||| | |||| ||||| ||||| | ||| ||||| |||| |
||||||||||| ||||||||||| ||||||| | ||||
||||||||||||||||||||| | ||||| | ||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||||| | ||||| | ||| ||||| ||||| | ||| ||||| |||| | ||| |||| |||| |
||| |||| | |||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| ||||| ||||||| | ||||
|||| | ||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
|||||| | |||||| | |||| ||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
||||| | ||||
| || | | ||||| | |||| |||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
||||| ||||||| ||||| | ||||
|| |||| | ||||| | |||| |||||| ||||| | ||| ||||| |||| | ||| ||||| |||| |
||||||||||| | ||||| | |||| ||||| ||| | ||| ||||| |||| | ||| ||||| |||| |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CI = confidence interval; CR = complete response; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; ORR = objective response rate; OS = overall survival; R-IPI = revised International Prognostic Index.
Source: EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off).43
Patient-Reported Outcomes
PROs from the EPCORE NHL-1 trial are summarized in Table 21, Table 22, and Table 23. The collection of PROs (i.e., FACT-Lym and EQ-5D-3L) was prespecified up to cycle 9. PRO data at the end of treatment assessment refer to patients who progressed and discontinued therapy.43
FACT-Lym Total Score
At baseline, a total of 140 patients completed the FACT-Lym, and the mean score at baseline was 118.4 (SD = 25.47). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| ||||||) and |||| |||||||| respectively. At the end-of-treatment assessment (n = 54), the mean change from baseline in total score was |||| |||||||.43
FACT-G Total Score
At baseline, a total of 140 patients completed the FACT-G, and the mean score at baseline was 76.2 (SD = 16.86). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| ||||||| and ||| |||||||, respectively. At the end-of-treatment assessment (| | ||), the mean change from baseline in total score was |||| ||||||||43
FACT-LymS
The sponsor evaluated 6 questions from the FACT-Lym that were related to the symptoms of lymphoma: P2 (body pain), BRM3 (fever), ES3 (night sweats), GP1 (lack of energy), BMT6 (tires easily), and C2 (weight loss). At baseline, a total of 140 patients completed the FACT-LymS, and the mean score at baseline was 42.2 (SD = 9.98). At cycle 5, day 1 (n = 66) and cycle 7, day 1 (n = 52), the mean changes from baseline in total score were ||| |||||| and ||| ||||||, respectively. At the end-of-treatment assessment (n = |||), the mean change from baseline in total score was ||| ||||||. Table 21 summarizes the proportion of patients who reported at least a 1-category improvement in any of the 6 key lymphoma symptoms (i.e., body pain, fever, night sweats, lack of energy, tires easily, and weight loss, with each assessed using a 5-point severity response scale ranging from “not at all” to “very much”) without worsening in the other 5 symptoms, from baseline through cycle 9, day 1.43
Table 21: Summary of FACT-Lym Total Score, FACT-G Total Score, and FACT-LymS
Time point | FACT-Lym total score | FACT-G total score | FACT-Lym S | |||
|---|---|---|---|---|---|---|
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
Baseline C1, D1 | 140 | 118.4 (25.47) | 140 | 76.2 (16.86) | 140 | 42.2 (9.98) |
On-treatment C3, D1 | || | ||||| ||||||| | || | |||| ||||||| | || | |||| |||||| |
Change from BL | || | ||| ||||||| | || | ||| |||||| | || | ||| |||||| |
C5, D1 | || | ||||| ||||||| | || | |||| ||||||| | || | |||| |||||| |
Change from BL | || | ||| ||||||| | || | ||| ||||||| | || | ||| |||||| |
C7, D1 | || | ||||| ||||||| | || | |||| ||||||| | || | |||| |||||| |
Change from BL | || | |||| ||||||| | || | ||| ||||||| | || | ||| |||||| |
C9, D1 | || | ||||| ||||||| | || | |||| ||||||| | || | |||| |||||| |
Change from BL | || | |||| ||||||| | || | ||| ||||||| | || | ||| |||||| |
EOT | || | ||||| ||||||| | || | |||| ||||||| | || | |||| |||||| |
Change from BL | || | |||| ||||||| | || | |||| ||||||| | || | ||| |||||| |
BL = baseline; C1 = cycle 1; C3 = cycle 3; C5 = cycle 5; C7 = cycle 7; C9 = cycle 9; D1 = day 1; EOT = end of treatment; FACT-G = Functional Assessment of Cancer Therapy–General; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FACT-LymS = Functional Assessment of Cancer Therapy–Lymphoma Subscale; SD = standard deviation.
Sources: EPCORE NHL-1 Clinical Study Report (April 21, 2023)43 and sponsor’s Summary of Clinical Evidence.11
Table 22: Summary of FACT-Lym — Lymphoma Symptoms From the EPCORE NHL-1 Study
Symptom | Patients reporting symptom at BL | Proportion of patients with ≥ a 1-point category improvement without worsening in any other symptoms | ||||
|---|---|---|---|---|---|---|
C2, D1 | C3, D1 | C5, D1 | C7, D1 | C9, D1 | ||
Body pain | || | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||||| |
Fever | || | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||| |
Night sweats | || | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||||| |
Lack of energy | || | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||||| |
Tires easily | ||| | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||||| |
Weight loss | || | |||||||||||||||| | ||||| | ||||| | ||||| | |||||||||||||||| |
BL = baseline; C2 = cycle 2; C3 = cycle 3; C5 = cycle 5; C7 = cycle 7; C9 = cycle 9; D1 = day 1; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma.
Sources: EPCORE NHL-1 Clinical Study Report (April 21, 2023)43 and sponsor’s Summary of Clinical Evidence.11
EQ-5D-3L
At baseline, a total of ||| patients completed the EQ-5D-3L. The mean (SD) utility score at baseline was ||||| ||||||||. At cycle 5, day 1 (n = |||) and cycle 7, day 1 (n = |||), the mean changes from baseline in total score were ||||| |||||||| and ||||| ||||||||| respectively. At the end-of-treatment assessment (n = |||), the mean change from baseline in total score was |||||| |||||||||43 The mean (SD) score in EQ VAS at baseline was |||||| |||||||||. At cycle 5, day 1 (n = |||) and cycle 7, day 1 (n = |||), the mean changes from baseline in total score were ||||| ||||||||| and |||||| |||||||||, respectively. At the end-of-treatment assessment (n = |||), the mean change from baseline in total score was |||||| ||||||||||
Table 23: Summary of EQ-5D-3L From the EPCORE NHL-1 Study
Time point | EQ-5D utility score | EQ VAS | ||
|---|---|---|---|---|
n | Mean (SD) | n | Mean (SD) | |
Baseline C1D1 | ||| | ||||| |||||||| | ||| | |||||| ||||||||| |
On-treatment C3D1 | || | ||||| |||||||| | || | |||||| ||||||||| |
Change from BL | || | ||||| |||||||| | || | ||||| ||||||||| |
C5D1 | || | ||||| |||||||| | || | |||||| ||||||||| |
Change from BL | || | ||||| |||||||| | || | ||||| ||||||||| |
C7D1 | || | ||||| |||||||| | || | |||||| ||||||||| |
Change from BL | || | ||||| |||||||| | || | |||||| ||||||||| |
C9D1 | || | ||||| |||||||| | || | |||||| ||||||||| |
Change from BL | || | ||||| |||||||| | || | |||||| ||||||||| |
EOT | || | ||||| |||||||| | || | |||||| ||||||||| |
Change from BL | || | |||||| |||||||| | || | |||||| ||||||||| |
BL = baseline; C1 = cycle 1; C3 = cycle 3; C5 = cycle 5; C7 = cycle 7; C9 = cycle 9; D1 = day 1; EOT = end of treatment; EQ VAS = EQ visual analogue scale; SD = standard deviation.
Source: EPCORE NHL-1 Clinical Study Report (April 21, 2023)43 and sponsor’s Summary of Clinical Evidence.11
Harms
Refer to Table 24 for harms data. As of the data cut-off date (April 21, 2023), ||| ||||||| patients with LBCL had experienced at least 1 TEAE. Serious TEAEs were reported in ||| ||||||| patients. A total of ||| ||||||| patients experienced grade 3 or higher TEAEs, and ||| ||||||| patients had grade 3 or higher TEAEs considered related to epcoritamab by the investigator.43
Table 24: Summary of Harms Results From Studies Included in the Systematic Review
Adverse events | Epcoritamab (N = 157) |
|---|---|
Most common (≥ 10%) TEAEs by SOC and PT, n (%) | |
Patients with ≥ 1 TEAE | ||| ||||||| |
General disorders and administration-site conditions | ||| ||||||| |
Pyrexia | || ||||||| |
Fatigue | || ||||||| |
Injection-site reaction | || ||||||| |
Edema peripheral | || ||||||| |
Injection-site erythema | || |||||| |
Gastrointestinal disorders | || ||||||| |
Nausea | || ||||||| |
Diarrhea | || ||||||| |
Abdominal pain | || ||||||| |
Constipation | || ||||||| |
Vomiting | || ||||||| |
Infections and infestations | || ||||||| |
COVID-19 | || ||||||| |
Immune system disorders | || ||||||| |
Cytokine release syndrome | 80 (51.0%) |
Blood and lymphatic system disorders | || ||||||| |
Neutropenia | || ||||||| |
Anemia | ||||||||| |
Thrombocytopenia | || ||||||| |
Musculoskeletal and connective tissue disorders | || ||||||| |
Back pain | ||||||||| |
Metabolism and nutrition disorders | || ||||||| |
Decreased appetite | || ||||||| |
Hypokalemia | || |||||| |
Nervous system disorders | || ||||||| |
Headache | || ||||||| |
Respiratory, thoracic, and mediastinal disorders | || ||||||| |
Cough | || |||||| |
Psychiatric disorders | || ||||||| |
Insomnia | || ||||||| |
Serious TEAEs by SOC and PT (≥ 2% of patients), n (%) | |
Patients with ≥ 1 serious TEAE | ||| ||||||| |
Immune system disorders | || ||||||| |
Cytokine release syndrome | || ||||||| |
Infections and infestations | || ||||||| |
COVID-19 | || |||||| |
COVID-19 pneumonia | | |||||| |
Pneumonia | | |||||| |
Sepsis | | |||||| |
Upper respiratory tract infection | | |||||| |
Nervous system disorders | || |||||| |
Immune effector cell–associated neurotoxicity syndrome | | |||||| |
Respiratory, thoracic, and mediastinal disorders | || |||||| |
Pleural effusion | | |||||| |
Blood and lymphatic system disorders | | |||||| |
Febrile neutropenia | | |||||| |
General disorders and administration-site conditions | | |||||| |
Pyrexia | | |||||| |
Patients who discontinued epcoritamab (all) due to TEAES, by SOC and PT, n (%) | |
Patients with ≥ 1 serious TEAE | || ||||||| |
Infections and infestations | || |||||| |
COVID-19 pneumonia | | |||||| |
COVID-19 | | |||||| |
Pneumonia | | |||||| |
Pneumonia, bacterial | | |||||| |
Progressive multifocal leukoencephalopathy | | |||||| |
Neoplasms, benign, malignant, and unspecified (including cysts and polyps) | | |||||| |
Myelodysplastic syndrome | | |||||| |
Anogenital warts | | |||||| |
Lung neoplasm malignant | | |||||| |
Cardiac disorders | | |||||| |
Myocardial infarction | | |||||| |
Myocarditis | | |||||| |
Nervous system disorders | | |||||| |
Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids | | |||||| |
Immune effector cell–associated neurotoxicity syndrome | | |||||| |
General disorders and administration-site conditions | | |||||| |
Fatigue | | |||||| |
Immune system disorders | | |||||| |
Cytokine release syndrome | | |||||| |
Fatal TEAEs (all) by PT, n (%) | |
Patients with fatal TEAEs | || ||||||| |
COVID-19 pneumoniaa | | |||||| |
COVID-19 | | |||||| |
Pneumonia | | |||||| |
Pneumonia, bacterial | | |||||| |
Progressive multifocal leukoencephalopathy | | |||||| |
Immune effector cell–associated neurotoxicity syndrome | | |||||| |
Myocardial infarction | | |||||| |
Myocarditisa | | |||||| |
General physical health deterioration | | |||||| |
Hepatoxicity | | |||||| |
Pulmonary embolism | | |||||| |
Adverse events of special interest, n (%) | |
Patients with ≥ 1 cytokine release syndrome event | 80 (51.0%) |
Grade 3 | || ||||||| |
Patients with ≥ 1 immune effector cell–associated neurotoxicity syndrome event | 10 (6.4%) |
Grade 3 | ||| |
Grade 4 | ||| |
Grade 5 | | |||||| |
Patients with ≥ 1 clinical tumour lysis syndrome event | 2 (1.3%) |
Grade 3 | | |||||| |
PT = preferred term; SOC = system organ class; TEAE = treatment-emergent adverse event.
aOne patient died due to fatal TEAEs related to COVID-19 pneumonia and myocarditis.
Source: EPCORE NHL-1 Clinical Study Report (April 2023 data cut-off).43
Adverse Events
In patients with LBCL (N = 157), the most frequent (i.e., noted in at least 20% of patients) TEAEs by PT were CRS (80 patients [51.0%]), pyrexia (not attributed to CRS; || |||||||), fatigue (|| |||||||), neutropenia (|| |||||||), nausea (|| |||||||), anemia (|| ||||||| patients), and diarrhea (|| |||||||). Grade 3 or 4 TEAEs were reported for ||| ||||||| patients with LBCL. The most common grade 3 or grade 4 TEAEs (≥ 5%) by PT in patients with LBCL (N = 157) were neutropenia (|| |||||||), anemia (|| |||||||), neutrophil count decrease (||| ||||||), COVID-19 (|| ||||||), and thrombocytopenia (| ||||||).43
Serious Adverse Events
Serious TEAEs were reported in ||| ||||||| patients with LBCL. The most frequent (≥ 2%) serious TEAEs by PT in patients with LBCL were CRS (|| |||||||); COVID-19 (|| ||||||); COVID-19 pneumonia (| ||||||); pleural effusion (| ||||||); pneumonia (| ||||||); and pyrexia (not attributed to CRS), sepsis, ICANS, and febrile neutropenia (| |||||| each).43
Withdrawal or Interruption Due to Adverse Events
In patients with LBCL (N = 157), || ||||||| patients experienced at least 1 TEAE that led to treatment discontinuation. The most common of these were COVID-19 pneumonia (| ||||||), COVID-19 (| ||||||), and myelodysplastic syndrome (| ||||||). Other TEAEs that led to treatment discontinuation in this group, occurring in 1 patient each, were pneumonia, bacterial pneumonia, progressive multifocal leukoencephalopathy, anogenital warts, malignant lung neoplasm, myocardial infarction, myocarditis, chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids, ICANS, fatigue, and CRS.43
There were || ||||||| patients with LBCL who had TEAEs leading to dose delay, and for || |||||||| In patients with LBCL (N = 157), the most common (≥ 2% overall) TEAEs leading to dose delay were COVID-19 (19 patients [12.1%]); CRS (11 patients [7.0%]); neutropenia (| |||||||| thrombocytopenia (5 patients [3.2%]); and pyrexia (not attributed to CRS), upper respiratory tract infection, and pleural effusion (| |||||| each).43
Mortality
Table 25 provides a summary of deaths reported in the EPCORE NHL-1 trial for the population of patients with LBCL. Deaths were reported for || ||||||| patients with LBCL, || ||||||| patients with DLBCL, and || ||||||| patients with other LBCL subtypes. The majority of deaths were attributed to disease progression (||||| of patients).43
Table 25: Summary of Mortality From the EPCORE NHL-1 Study
Deaths | DLBCL (N = 139) | Other Subtypes (N = 18) | LBCL (N = 157) |
|---|---|---|---|
Deaths | || ||||||| | || ||||||| | || ||||||| |
Primary causes of death | |||
Disease progression | || ||||||| | | ||||||| | || ||||||| |
Adverse event | || ||||||| | ||| | || |||||| |
Other | | |||||| | | |||||| | | |||||| |
Unknown | ||| | | |||||| | | |||||| |
DLBCL = diffuse large B-cell lymphoma; LBCL = large B-cell lymphoma.
Source: Clinical Study Report.43
Notable Harms
Table 39 provides a summary of the notable harms from the EPCORE NHL-1 trial, including CRS events, ICANS events, and serious infections.
Cytokine Release Syndrome
The AESI of CRS was analyzed and summarized at the patient level and event level. In the patient-level analysis, patients with multiple CRS events were counted only once, but may have been counted in more than 1 dosing period. In the event-level analysis, all CRS events are counted, including multiple episodes experienced by the same patient. In patients with LBCL, 80 patients (51.0%) had at least 1 CRS event. The majority of these were grade 1 events (|| | of 80 patients) and occurred most frequently after the first full dose of epcoritamab on C1D15 (||| of 80 patients). Grade 2 and grade 3 events occurred in ||| and ||| of 80 patients, respectively. There were no grade 4 or 5 events. The most common symptom was fever (||| of 80 patients). The most common treatment reported for CRS was anticytokine therapy using tocilizumab (||| of 80 patients); no other anticytokines were used. Events of CRS were treated with corticosteroids (beyond those scheduled for mandatory CRS prophylaxis) for ||| of 80 patients who experienced CRS. Oxygen was used to treat ||| of 80 patients who experienced CRS.
The median time to first CRS onset was ||||| days (range, || ||) correlating with the first full dose of epcoritamab on C1D15. The median time to CRS resolution was |||| days (range, || ||). Of the | | CRS events that occurred for the priming doses, most (|||) were grade 1, and only 2 were grade 3; no grade 3 events were observed with the priming doses. The CRS symptoms resolved in ||| of 80 patients (||||||). For most dosing periods, CRS resolved for all patients. The exception was the second full dose: CRS was unresolved for 2 patients of 7 patients within that dosing period.
Immune Effector Cell–Associated Neurotoxicity
ICANS symptoms were monitored at every visit and daily for patients who were hospitalized because of CRS. All but 1 of the reported ICANS events occurred in patients with DLBCL. ICANS events of were reported in 10 patients (6.4%); | ||||||| patients had grade 1 ICANS, | ||||||| patients had grade 2 ICANS; and | ||||||| patient had grade 5 (fatal) ICANS.
The fatal episode of ICANS, in a 72-year-old female patient with DLBCL, was an on-treatment event with onset on day 12, which was 4 days after the patient’s most recent dose of the study drug. The episode was considered related to the study drug. This event was confounded with long-standing cardiovascular comorbidities, grade 3 pancreatitis, and multifocal cerebral and splenic infarcts. The patient had received only the priming and intermediate doses.
Three patients (|||||) experienced ICANS events that led to dose delays, and in | ||||||| patient, the event led to treatment discontinuation. The median time to first onset was |||| |||| ||||||| || ||||, correlating with the period shortly following the first full dose of epcoritamab on C1D15. As of the data cut-off date, ICANS events had resolved for | ||||||| patients, with the median time to resolution of ||| |||| ||||||| || ||. Of the 10 patients with LBCL with an ICANS event, ||| patients received at least 1 concomitant medication as treatment. The only medications used to treat ICANS for more than | |||||||| were dexamethasone (||| patients) and levetiracetam (||| patients).
Clinical Tumour Lysis Syndrome
Among patients with LBCL, there were 2 patients (1.3%) who experienced CTLS events; both events were considered treatment-related in the disease progression setting. Both were grade 3 in severity. Neither had resolved before the patients’ deaths (both due to disease progression).
Serious Infections
Forty-six patients (29.3%) experienced at least 1 serious infection during the EPCORE NHL-1 trial. The majority of patients who had serious infections had at least 1 event that was classified as grade 3 (|||||), with grade 4 and 5 events reported for ||| and ||| patients, respectively.
Critical Appraisal
Internal Validity
Trial design: The EPCORE NHL-1 phase I and II trial was the only study included in this review. The study is an ongoing phase I and II, multicentre, open-label, single-arm study of epcoritamab. The choice to conduct a single-arm trial was justified by the sponsor on the basis that the study was designed as an early-phase I and II study in which an internal comparator group is not required; the severity of illness for patients at this stage (i.e., those with R/R illness following at least 2 lines of prior systemic therapy) was also a justification. However, the decision to conduct a single-arm study has implications for the overall strength and interpretability of the results. As a single-arm study, there is an increased risk of bias in the estimation of treatment effects due to the potential for confounding related to natural history and prognostic factors. The potential influence of selection bias is also difficult to ascertain in a single-arm study. Additionally, time-to-event end points cannot be adequately assessed in a single-arm trial because all patients receive the same treatment. As such, the effects of epcoritamab on time-to-event end points, such as PFS, OS, and DOR, are uninterpretable, and can only be considered as exploratory and supportive.
Lack of comparator: In the absence of a comparator group in the EPCORE NHL-1 study, an assessment of the comparative clinical value of epcoritamab relies on ITCs (i.e., unanchored MAICs), which rely on numerous assumptions about the comparability of treatment groups, thereby increasing the uncertainty related to the comparative effectiveness (refer to the Indirect Evidence section for details). The uncertainty in the comparative efficacy of epcoritamab versus relevant comparators was acknowledged by Health Canada, which has specified that the sponsor must provide phase III trial results showing that epcoritamab improves the OS of patients with LBCL compared to investigator's choice of either BR or R-GemOx.
Open label: In addition to the single-arm design, the EPCORE NHL-1 study was administered in an open-label manner whereby the investigator and study participants were aware of their treatment status, potentially increasing the risk of detection bias and performance bias. As such, the open-label trial design limits interpretability of the subjective study outcomes, such as tumour response and PROs, including HRQoL and AEs. However, to mitigate the impact of this bias, PFS and ORR were assessed by both IRC and the investigator using the Lugano classification criteria for the response. For response outcomes, there was generally a low discordance rate between IRC- and investigator-assessed responses in the EPCORE NHL-1 trial. Discordance between IRC- and investigator-assessed responses and the potential influence on outcomes are well documented, including guidance from the FDA to industry.53 Given the open-label design of the trial, it is possible that the IRC provided less potentially biased tumour assessments compared with investigator’s assessments.
Sample size: A limited number of patients were included in the primary efficacy population (N = 157). Although the EPCORE NHL-1 study was powered for the primary end point, the magnitude of the treatment-effect estimates observed in a relatively small study sample may not be replicable in a larger study sample.
Outcomes: The clinical experts consulted by CADTH and the regulatory authorities indicated that the outcomes studied in the EPCORE NHL-1 trial were those typically assessed for the target population. Health Canada reviewers noted that the median follow-up times for patients enrolled in the EPCORE NHL-1 trial are similar to those of other therapies that have been studied in the population of patients with R/R LBCL. Despite PFS and OS results that were considered clinically meaningful by the clinical experts consulted by CADTH, the results for survival end points should only be considered supportive of the overall antitumour effect of epcoritamab due to the single-arm design and the secondary nature of the outcomes. Quality of life outcomes, which were identified as important to patients in their input to CADTH, were also secondary end points of the EPCORE NHL-1 study.
The primary end point of ORR in the EPCORE NHL-1 study does not appear to be aligned with regulatory guidance from the FDA53 regarding hematologic cancers, which cites CR as opposed to ORR as a direct measure of the drug’s antitumour activity in oncology clinical trials. Health Canada similarly questioned the choice of ORR as the primary efficacy outcome rather than CR rate, which their review team considered to be the more informative outcome measure in the population of patients with R/R LBCL. Health Canada noted that the DOR was relatively short for those with a best overall response of PR (refer to PFS Figure 6).32 In response to Health Canada’s question regarding the use of ORR as the primary end point, the sponsor provided the following as justification that ORR is a clinically meaningful surrogate end point in the context of single-arm trials that are intended to support market authorization for treatment that is intended to fulfill the unmet medical needs of patients with R/R LBCL:
ORR was chosen as the primary efficacy outcome based on benchmark studies that led to recent approvals in R/R LBCL that also used ORR as the primary efficacy end point: Minjuvi (tafasitamab); Kymriah (tisagenlecleucel); and Breyanzi (lisocabtagene maraleucel).
CR was a secondary end point in the EPCORE NHL-1 study, with a CR rate of ||||||.
Response-stratified analyses demonstrated that those who achieved a CR had the highest PFS and OS benefit and those who achieved a PR (|||||| achieved longer median PFS ||| |||||| and OS |||| ||||||) than those of nonresponders (||| months PFS and ||| months OS).54
In a retrospective analysis of the GOYA trial55 and a meta-analysis of studies of LBCL,56 the prognostic value of PET in assessing CR with respect to PFS and OS was evaluated. Although the results of these studies suggest that end-of-treatment CR was a predictor of PFS and OS, and that CR could be an effective surrogate end point for survival, these studies were conducted in previously untreated patients; thus, it remains unclear whether there is an association between CR rate and survival in patients receiving third-line treatment for LBCL. Recent literature has highlighted that the correlation between response rates and survival is variable and that there are gaps in understanding the strength of response as a surrogate outcome in hematologic malignancy trials.57-59 Overall, the clinical experts consulted by CADTH felt that the clinical trial end points were appropriate for the target population. In addition, data from the phase II trials of glofitamab and epcoritamab demonstrated that CR responses were durable and that patients who experienced CR survived longer than those who did not respond to treatment or whose best response was PR. The sponsor also noted that an analysis of patients in Denmark (N = 130) who received treatment with a bispecific T-cell engager (glofitamab or epcoritamab) in phase I and II clinical trials reported that 73% of patients with CR remained in complete remission after 3 years.60
Statistical approach: There were no methods incorporated to account for multiple testing; thus, all secondary end points were considered supportive and should be interpreted with respect to type I error. There was also no control for multiplicity across the cohorts selected for inclusion in the review.
External Validity
Trial location and setting: The EPCORE NHL-1 study was an international, multicentre study that included sites in Australia, South Korea, US, France, Netherlands, Spain, Denmark, Germany, UK, Poland, Singapore, Italy, and Canada (1 site with 1 patient enrolled). The clinical experts consulted by CADTH had no concerns regarding the generalizability of the study results to the setting in Canada. Patients in the EPCORE NHL-1 trial were hospitalized for at least 24 hours after receiving the first full dose of epcoritamab. The clinical experts consulted by CADTH noted that this is reflective of how this drug will be used in practice in Canada.
Intervention: The treatment regimen used in the EPCORE NHL-1 trial aligns with the recommendations shown on the Health Canada–approved product monograph for epcoritamab (i.e., a priming dose of 0.16 mg; an intermediate dose of 0.8 mg; and full doses of 48 mg thereafter).44 The pretreatment and posttreatment medications (e.g., prednisolone) used to reduce the risk of CRS in the EPCORE NHL-1 trial are reflective of those recommended in the product monograph, as were the therapies used to manage the more severe CRS events (i.e., tocilizumab or additional corticosteroids). The clinical experts consulted by CADTH noted that dosing of epcoritamab and the medications used for the management of AEs throughout the peritreatment period are reflective of the regimen that would be administered in practice in Canada.
Outcomes: Outcomes included in the EPCORE NHL-1 trial were relevant in the management of R/R LBCL and were identified as important to patients and clinicians. For tumour response and disease progression, measurement using the Lugano criteria is standardized across jurisdictions. However, it was noted in the clinician group input that PET and/or CT are not available in all jurisdictions. While the experts considered response outcomes to be important in the treatment of R/R LBCL and believed that the responses observed in the EPCORE NHL-1 trial study were better than they would expect to observe from other currently available treatments, they noted that survival and the prevention of progression are of greatest importance to patients at this stage. As previously mentioned, the estimates of PFS and OS may be overestimated due to the relatively small information fraction and overall immaturity of the data, which may affect the generalizability of the results to the population of patients with R/R LBCL in Canada. The patient group input also highlighted the importance of treatments that improve HRQoL and provide the ability to engage in usual activities. However, as identified for PFS and OS, the EPCORE NHL-1 trial was not adequately designed to assess the effects of epcoritamab on HRQoL and related outcomes.
Population: The clinical experts consulted by CADTH noted that the inclusion and exclusion criteria used in the EPCORE NHL-1 trial were a reasonable reflection of the patient population for whom epcoritamab could be considered an appropriate treatment in clinical practice in Canada. Although patients who had received ASCT and were refractory or experienced relapse relatively rapidly were excluded from the EPCORE NHL-1 trial, the clinical experts noted that these patients could be considered candidates for epcoritamab. (That is, the exclusion from the EPCORE NHL-1 trial of patients who had received ASCT within 100 days before the first dose of epcoritamab may not be reflective of clinical practice, and these patients could be considered for treatment.) Overall, the clinical experts concluded that the baseline characteristics of those enrolled in the EPCORE NHL-1 trial were reflective of the target population in Canada.
The proportion of patients with an ECOG Performance Status of 2 included in the EPCORE NHL-1 trial was relatively low (3.2%). The clinical experts noted that the proportion could be greater in clinical practice, especially given that the indication is limited to those who are ineligible for CAR T-cell therapy.
Prior CAR T-cell therapy: The clinical experts noted that 40% of patients with prior CAR T-cell therapy exposure is a reasonable reflection of the target population for epcoritamab in Canada (while noting that the figure would vary across jurisdictions). They also noted that the overall proportion of patients with a history of stem cell transplant could be slightly lower than might be anticipated in routine practice in Canada for patients for whom 2 or more lines of systemic therapy have not been effective. Both the clinical experts consulted by CADTH and the participating drug programs noted that the treatment landscape for R/R LBCL in Canada may shift toward earlier usage of CAR T-cell therapy, given that axicabtagene ciloleucel (Yescarta) received a recommendation to reimburse with conditions from CADTH for use as a second-line option, based on the results of the ZUMA-7 trial. Axicabtagene ciloleucel was under review by the pan-Canadian Pharmaceutical Alliance at the time of this review and was not funded by any of the participating drug programs for any line of therapy (refer to Figure 1).
Concomitant treatments: The clinical experts noted that the type and distribution of concomitant medications were a reasonable reflection of what would be administered in practice in Canada in the R/R LBCL setting.
Subsequent Treatments: The clinical experts consulted by CADTH noted that the distribution of subsequent antilymphoma therapies received after the discontinuation of epcoritamab was a reasonable reflection of what could be expected in routine practice in Canada. The clinical experts noted that ||| of the patients in the EPCORE NHL-1 trial received no subsequent antilymphoma therapy, which is an indication of how severe the baseline level of disease was in this trial (i.e., epcoritamab was the final therapy before patients died). A total of |||| of patients (n = |||) received subsequent CAR T-cell therapy in the EPCORE-1 NHL trial. The clinical experts noted that they would anticipate similar rates in practice in Canada of patients receiving potentially curative therapies after treatment with epcoritamab (e.g., CAR T-cell therapy or stem cell transplant), and they emphasized that epcoritamab could provide a useful option as a bridging therapy for those awaiting CAR T-cell infusion (acknowledging that such usage would be off-label, based on the current indication approved by Health Canada). Similarly, the clinical experts consulted by CADTH noted that patients who do not respond or progress following epcoritamab could be considered candidates for CAR T-cell therapy (acknowledging that the Health Canada–approved indication for epcoritamab would suggest that the treatment should be initiated only in those who could not receive CAR T-cell therapy at the time of initiating therapy with epcoritamab).
Tolerability: The clinical experts noted that similar rates of discontinuation and the reasons for discontinuation were aligned with the expectations for the target patient population in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and final certainty ratings were determined as outlined by the GRADE Working Group:9,10
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. This means the true effect is likely to be close to the estimate of the effect, but there is a possibility that it may be substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. This means the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. This means the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Although GRADE guidance is not available for noncomparative studies, the CADTH review team assessed pivotal, single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty, with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The target of the certainty of evidence assessment was the presence of a clinically important improvement in survival (PFS and OS) and HRQoL, which were considered the most important treatment outcomes by the clinical experts consulted by CADTH as well as the clinician and patient group inputs. According to the clinical experts consulted by CADTH, clinically important thresholds for the outcomes of PFS and OS were a benefit of at least 6 months and 3 months over current standard of care for OS and PFS, respectively. Additionally, response to treatment (CR, ORR, DOR) was included in the certainty of evidence assessment based on the potential translation to long-term survival outcomes.
The EPCORE NHL-1 trial, a phase I and II, single-arm, open-label study of epcoritamab monotherapy, was the only study included in the GRADE assessment.
Results of GRADE Assessments
Table 2 presents the narrative GRADE summary of findings for epcoritamab monotherapy from the EPCORE NHL-1 study.
Long-Term Extension Studies
No long-term extension studies were submitted to CADTH or identified in the literature.
Indirect Evidence
The contents of this section were informed by materials submitted by the sponsor. The following information has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
Given the single-arm nature of the EPCORE NHL-1 trial, there is no direct head-to-head evidence comparing epcoritamab against the following comparators for patients with R/R LBCL or R/R DLBCL with exposure to at least 2 prior therapies:
CIT
pola-BR
pola-B/R
axicabtagene ciloleucel
tisagenlecleucel
lisocabtagene maraleucel.
ITC Design
Objectives
The objective of the sponsor-provided ITC was to compare epcoritamab against comparator drug therapies (i.e., CIT, pola-BR, axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel) for patients with R/R LBCL or R/R DLBCL with exposure to at least 2 prior therapies.
Study Selection Methods
Eligibility criteria for the systematic literature review (SLR) are summarized in Table 26. The sponsor conducted the SLR to identify, extract, and analyze relevant clinical evidence for greater than or equal to third-line therapies in R/R LBDC or R/R DLBCL. The technical report notes that the studies were selected in accordance with the approved indication for epcoritamab; however, this appears more accurate for the international context, in which the approved indication has not been restricted to patients who have already received or are unable to receive CAR T-cell therapy. Specifically, the sponsor included CAR T-cell therapies as comparators in the MAICs, but these cannot be considered comparators in the context of the Health Canada–approved indication.
Table 26: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Population of patients with ≥ third-line R/R LBCL or DLBCL (i.e., as full study population or subgroup population) |
Intervention | Epcoritamab, dosed as in the EPCORE NHL-1 study |
Comparator | R-CIT, pola-B/R (including both pola-BR and pola-R), axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel |
Outcome |
|
Study designs |
|
Publication characteristics | Only publications reporting on clinical studies (i.e., RCTs, non-RCTs, and observational studies) in the R/R LBCL and R/R DLBCL setting were included. |
Exclusion criteria | Studies that offered evidence only for a mixed population (i.e., the population of patients with R/R LBCL or R/R DLBCL population who had received 2 or more lines of therapy), or that had an overall sample size of < 20 patients, were not eligible for data extraction. |
Databases searched | MEDLINE, Embase, the Cochrane Library, clinical conference proceedings, clinical trial registries |
Selection process | The SLR was conducted in concert with the guidelines set out by Cochrane and the CRD and the 27-item PRISMA Statement checklist. This means the selection of publications was 2-phased: first, the titles and abstracts of all identified records were screened; subsequently, included publications were checked for inclusion based on the full texts. Screening was performed by 2 independent reviewers, and inclusions and exclusions were based on prespecified eligibility criteria. |
Data extraction process | A total of 14,301 publications were identified from the search of the electronic databases on October 11, 2022 (phase I), December 8, 2022 (phase II), and April 4, 2023 (phase III).a After title and abstract and full-text screening, a total of 268 peer-reviewed publications were deemed eligible for inclusion in the SLR. From the conference proceeding searches, another 92 relevant abstracts were identified, and 2 additional publications were included from citation review. This resulted in a total of 362 publications eligible for inclusion based on the prespecified criteria (i.e., clinical evidence in R/R LBCL and R/R DLBCL). Of these 362, a total of 158 publications were relevant for reporting because they presented clinical evidence in the population of patients with ≥ third-line LBCL or DLBCL (≥ 20 patients) in a European, North American, or global perspective. |
Quality assessment | QA was performed for all RCT and non-RCT publications except conference proceedings, given that there would be insufficient methodological data to assess the study quality. The QA was done by 1 researcher and checked by a second.
|
CR = complete response; CRD = Centre for Reviews and Dissemination; DLBCL = diffuse large B-cell lymphoma; HR = hazard ratio; IRC = independent review committee; ITC = indirect treatment comparison; LBCL = large B-cell lymphoma; MAIC = matching-adjusted indirect comparison; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; pola-B/R = polatuzumab vedotin with rituximab, with or without bendamustine; pola-BR = polatuzumab vedotin with bendamustine and rituximab; pola-R = polatuzumab vedotin with rituximab; QA = quality assessment; R-CIT = rituximab-based chemoimmunotherapy; R/R = relapsed or refractory; RCT = randomized controlled trial; RWE = real-world evidence; SLR = systematic literature review.
aAn exception is the 5-year follow-up from the ZUMA-1 trial of axicabtagene ciloleucel, which was included in the MAIC of the epcoritamab April 2023 data cut-off.
Source: Sponsor’s indirect comparison.11
ITC Analysis Methods
Unanchored MAICs were determined to be the most appropriate analysis method, given that network meta-analysis generation was determined infeasible due to the single-arm nature of the EPCORE NHL-1 study. For each MAIC, patient-level data from the EPCORE NHL-1 trial data were reweighted using a propensity scoring approach to match the comparator trial data based on the availability of common patient characteristics. Under the counterfactual conditions of the comparator trial characteristics, the unanchored MAIC for survival were formed by calculating the logarithm of the hazard ratio of the reweighted epcoritamab survival curve versus the comparator’s digitized survival curve. The log estimates were then exponentiated and presented on the natural scale of the hazard ratio. Similarly, the mean difference between the predicted response for the reweighted epcoritamab arm and the comparator’s simulated patient-level data were calculated.
Analysis Populations for the ITC
To account for differing data availability across epcoritamab and the comparators, the sponsor identified 3 distinct populations (refer to Table 27).
Table 27: ITC Analysis Populations
EPCORE NHL-1 trial population | Number of patients | Application within the sponsor’s MAIC |
|---|---|---|
Overall trial population | 157 |
|
No prior CAR T-cell therapy | 96 |
|
No prior CAR T-cell therapy, but eligible for CAR T-cell therapy | 57 |
|
CAR = chimeric antigen receptor; ITC = indirect treatment comparison; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CIT = rituximab-based chemoimmunotherapy; RCT = randomized controlled trial.
Source: Sponsor’s indirect comparison.11
Characteristics Included in Propensity Matching
The sponsor stated that variables for adjustment in the propensity matching were selected using the following approaches: literature review, empirical testing of prognostic status in the EPCORE NHL-1 trial, and clinical expert input as to whether patient characteristics are important to adjust for in the population of patients with R/R LBCL and R/R DLBCL. The sponsor selected the following final characteristics:
Age (≥ 65 years)
Gender
DLBCL histology (including transformed follicular lymphoma) versus not DLBCL
Primary refractoriness
Refractory to greater than or equal to 2 consecutive lines of therapy
Refractory to last prior anti-CD20 drug
Refractoriness to last treatment when information on last prior anti-CD20 or primary refractoriness is not available
Prior CAR T-cell therapy
Prior ASCT
Relapse within 12 months of ASCT
ECOG Performance Status greater than 1
Disease stage III to IV
It was considered unnecessary to adjust for IPI score if the stage was included, given that IPI score correlates with disease stage
Characteristics Not Included in Propensity Matching
The sponsor reported that the following core adjustment characteristics were considered, but not included: bridging therapy use, secondary CNS involvement, lactate dehydrogenase blood level before treatment, and the exact number of prior therapy lines. Additional characteristics adjusted for in scenario analyses included tumour burden, bulky disease, IPI score, extranodal disease, and creatinine clearance. Characteristics were excluded due to being inapplicable and/or not present in the EPCORE NHL-1 trial data, not being prognostic as per the analysis of the data, or not being relevant, as per clinical expert opinion. The exact number of prior therapy lines was specifically not adjusted for due to variability in the number of earlier lines of therapy in each trial.
Propensity Score Weighting
Propensity score weighting was performed for the EPCORE NHL-1 trial data: the data were reweighted to match the baseline characteristics of those reported in the comparator trial using the method of moments as described by Signorovitch et al.61 such that the weighted means of key baseline characteristics from the EPCORE NHL-1 study exactly matched those in the study being compared and such that each individual patient’s weight was equal to their estimated odds of being in the comparator trial versus the EPCORE NHL-1 trial. Whenever indicated — and to improve the accuracy and precision of final parameter estimates — adjustment weights were incrementally truncated at the following thresholds of the weights’ distribution to reduce the occurrence of extreme weights (i.e., generally higher than 5 or 6 or lower than 0.1) while preserving the resulting balance in adjusted baseline characteristics, as previously recommended:
|| and |||
|||| and |||||
|| and |||
The final decision concerning which distribution cut-off point was appropriate for the particular ITC was driven by whether the next broader cut-off selection substantially imbalanced the baseline characteristics of epcoritamab and the comparator. Therefore, for different ITC analyses, different cut-off points were deemed optimal. The impact of reweighting is that there is less statistical information in the reweighted trial data, which is reflected in the effective sample size (ESSs). The maximum ESS is equal to the original trial size and occurs when the patient characteristics of the EPCORE NHL-1 trial and the comparator are identical.
Table 28: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Unanchored comparisons were conducted following the recommendations of the NICE guidance on population-adjusted ITCs based on propensity score reweighting methods. The sponsor reported that this method was preferred over simulated treatment comparisons (i.e., outcome regressions) due to the suspected possible small number of events available. |
Outcomes | PFS, OS, ORR, CR |
Subgroup analysis | As described, 3 distinct EPCORE NHL-1 trial populations were assessed to account for differing comparator data availability. |
CR = complete response; NICE = National Institute for Health and Care Excellence; ORR = overall response rate; OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s Summary of Clinical Evidence.11
Pola-BR has been previously reviewed by CADTH and received a recommendation to reimburse with conditions as a second- or third-line option. The recommendation from CADTH was based on the results of the GO29365 study, a phase Ib and II, open-label RCT in which pola-BR demonstrated improvements in CR rate, ORR, PFS, and OS compared with BR alone. The sponsor for epcoritamab elected not to include the GO29365 study in the submitted MAIC estimating the comparative efficacy of epcoritamab versus pola-BR for the overall population of patients with LBCL, opting instead to use RWE from Liebers et al. (2021). The sponsor cited the following reasons for concluding that the GO29365 and EPCORE NHL-1 trials were not sufficiently similar for the ITC:
Lines of therapy: Approximately 30% of patients in the GO29365 trial had prior exposure to only 1 prior line of therapy; the sponsor suggested that the patients enrolled in the GO29365 study were less ill than those enrolled in the EPCORE NHL-1 trial (who were more heavily pretreated at the time of enrolment). CADTH notes that the sponsor is correct with respect to the number of prior lines of therapy that patients received in the GO29365 study compared with the EPCORE NHL-1 trial. Specifically, those in the EPCORE NHL-1 trial had received a median of 3 prior lines of therapy compared with a median of 2 lines of prior therapy in the GO29365 study.
Prior exposure to CAR T-cell therapy: No patients enrolled in the GO29365 study had received prior treatment with CAR T-cell therapy, whereas 39% of those enrolled in the EPCORE NHL-1 trial had. CADTH notes that prior exposure to CAR T-cell therapy is an important prognostic factor and would make the GO29365 study inappropriate for comparisons with the overall cohort of patients with LBCL in the EPCORE NHL-1 trial or with the cohort of patients with prior CAR T-cell exposure.
Real-world efficacy: The sponsor stated that clinical experts had stated that the efficacy of pola-BR does not seem as promising in the real world versus the GO29365 trial. CADTH notes that evaluating efficacy in short-term clinical trials can overestimate the clinical benefit of drugs versus the real-world setting. However, this potential bias would apply to all early-phase clinical trial settings, not just the GO29365 trial. As with other HTA agencies (i.e., NICE and HAS), CADTH considered the approach of comparing data derived from a clinical trial for 1 drug against data derived from a real-world world setting for the comparator to be associated with uncertainty, given the differences in patient selection and data collection.
The sponsor did use subgroup data from the GO29365 trial for the comparison of epcoritamab versus pola-BR for patients with no prior CAR T-cell exposure (specifically extracting data for patients who had not responded to 2 prior lines of therapy).
Comparisons Versus CIT
The sponsor was able to conduct an ITC of epcoritamab versus CIT only for patients with no prior CAR T-cell therapy exposure (due to an absence of data for CIT in the target population). Hence, the subgroup of patients from the EPCORE NHL-1 trial (n = 96) was compared against a subgroup of patients from the SCHOLAR-1 study. The SCHOLAR-1 study was a retrospective cohort analysis with data drawn from 4 sources: observational cohorts from the MD Anderson Cancer Center; the Molecular Epidemiology Resource of the University of Iowa/Mayo Clinic Lymphoma Specialized Program of Research Excellence; the phase III Canadian Cancer Trials Group study; and the phase III CORAL study. The specific subgroup of patients from the SCHOLAR-1 study used by the sponsor was derived from a published ITC of axicabtagene ciloleucel versus CIT in which patients from the ZUMA-1 trial were matched against those from the SCHOLAR-1 dataset.
Comparisons Versus CAR T-cell Therapies
The sponsor-submitted ITC included comparisons against 3 CAR T-cell therapy regimens: axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel. Given that the Health Canada–approved indication for epcoritamab states that the drug is approved for use only in patients “who have previously received or are unable to receive CAR T-cell therapy,” CADTH does not consider CAR T-cell therapies to be relevant comparators for the current review. The approach is consistent with applications that have been filed in the same therapeutic area. The sponsor’s results are presented in this section of the CADTH report but are not otherwise appraised or interpreted by CADTH.
ITC Results
Summary of Included Studies
Table 29 summarizes selected patient and study characteristics for the trials included in the sponsor’s MAICs.
Table 29: Characteristics of the Trials Included in the ITC
Characteristics | EPCORE NHL-1 study | SCHOLAR-1 study, Neelapu (2021) | GO29365 study (subgroup) | Liebers (2021) |
|---|---|---|---|---|
Treatment | Epcoritamab | CIT | Pola-BR | Pola-B/Ra |
N | 157 | 340 | 29 | 54b |
Trial design | OL, phase I and II | RWE | Phase II RCT | RWE |
Main patient selection criteria | ≥ 2 LOT | ≥ 1 LOT | ≥ 2 LOT | ≥ 2 LOT |
Median follow-up, months | 22.3 (PFS) 25.1 (OS) | 5.4 | 22.3 | 7.5 |
Median PFS, months (IRC) | 4.4 | NA | 7.4 | NR |
Median PFS, months (investigator) | ||| | NA | 6.3 | 3.3 |
Comparator in trial | None | None | BR | None |
HR for PFS (95% CI) | NA | NA | 0.42 (0.22 to 0.78) | NA |
Median OS, months | 18.5 | 5.4 | 11.5 | 5.4 |
HR for OS for main treatment in trial (95% CI) | NA | NA | 0.47 (0.25 to 0.89) | NA |
ORR, % (IRC) | 63.1 | NR | NR | NR |
ORR, % (investigator) | |||| | 34 | 50.0 | 48.1 |
CR, % (IRC) | |||| | NR | NR | NR |
CR, % (investigator) | |||| | 12 | 42.2 | 14.8 |
DLBCL histology, % | 88.5 | NR | 96.5 | 90.7 |
Median age, years | 64 | 55a | 65 | 73.5 |
Age ≥ 65 years, % | 49 | 16 | 52 | NR |
Male, % | 59.9 | 68 | 72 | 68.5 |
ECOG < 2, % | 96.8 | 100 | 89.3 | NR |
Prior Tx, median (range) | | || || ||| | NR | 3 (2 to 7) | 3 (2 to 8) |
≥ 3 prior Tx, % | |||| | 29 | 62.1 | NR |
Primary refractory, % | |||| | 37 | NR | NR |
Prior ASCT, % | 19.8 | NR | 34.5 | 9.3 |
Prior CAR T-cell therapy, % | 38.9 | 0 | 0 | 13.7 |
Stage III or IV, % | |||| | 65 | 86.2 | NR |
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; CAR = chimeric antigen receptor; CIT = chemoimmunotherapy; CR = complete response; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; HR = hazard ratio; IRC = independent review committee; LOT = lines of therapy; NA = not applicable; NR = not reported; OL = open label; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; pola-B = polatuzumab vedotin with bendamustine; pola-B/R = polatuzumab vedotin with rituximab, with or without bendamustine; pola-BR = polatuzumab vedotin with bendamustine and rituximab; pola-R = polatuzumab vedotin with rituximab; RWE = real-world evidence; Tx = treatment.
aThe sponsor included the results for all 54 patients included in the salvage chemotherapy group of Liebers et al., which required pooling of patients who received pola-BR (n = 32 [59.3%]), pola-R (n = 20 [37.0%]), pola-B (n = 1 [1.85%]), and pola-R with gemcitabine (n = 1 [1.85%]).
bFifty-four patients were included in the salvage treatment cohort.
Table 30: Assessment of Homogeneity for MAIC Comparing Epcoritamab to Pola-B/R for Overall Population of Patients With LBCL
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Prior therapy | Refractory disease: The proportions of patients who were considered refractory to their last lines of antilymphoma therapy were 82.8% and 87.0% in the EPCORE NHL-1 trial and Liebers et al. (2021), respectively, and the definitions of refractoriness were consistent across the 2 studies (i.e., no response or disease progression within 6 months of the last treatment). The proportion of patients who were considered primary refractory was an adjustment factor in the MAIC; however, this information was not reported in the publication by Liebers et al. (2021). Lines of therapy: Patients in both the EPCORE NHL-1 trial and Liebers et al. (2021) were heavily pretreated, with both studies reporting a median number of 3 prior lines of antilymphoma therapy (range, 2 to 11 and range, 2 to 8, respectively). Prior CAR T-cell therapy and/or ASCT: There are differences across the populations in the EPCORE NHL-1 trial and Liebers et al. (2021) trial in the proportion of patients with prior CAR T-cell therapy (38.9% vs. 9.3%, respectively) and prior ASCT (19.7% vs. 9.3%, respectively). CADTH identified an error in the sponsor’s data extraction sheet with respect to the proportion of patients who received prior CAR T-cell therapy. (This figure was reported as 13.7% in the sponsor’s sheet, which refers to the patients from the pola-R bridging therapy cohort, not the target population of those in salvage therapy cohort.) |
ECOG PS | Patients enrolled in the EPCORE NHL-1 trial generally had good ECOG PS; only 5 patients (3.2%) had a baseline ECOG PS of 2. The clinical experts consulted by CADTH noted that this is common in oncology clinical trials and that a higher proportion of patients with an ECOG PS of 2 or more could be considered candidates for epcoritamab in clinical practice (depending on reimbursement status). The Liebers et al. (2021) publication did not report baseline PS for either the overall cohort or the subpopulations of interest for the current MAIC. As such, it is not possible to evaluate the potential heterogeneity of patients in Liebers et al. (2021) and the EPCORE NHL-1 trial with respect to baseline PS. Of particular interest could be the 37.0% of patients who received the pola-R regimen, given that these patients may have had a poorer PS, such that additional cytotoxic chemotherapy was deemed to be inappropriate. The sponsor notes that the MAICs were adjusted for the proportion of patients with an ECOG PS > 1; however, the sponsor reports that this information was not available for the patients included in the Liebers et al. (2021) study. |
Patient age | The median age in the FAS set of the EPCORE NHL-1 trial was 64.0 years (range, 20 years to 83 years), and the median age of those in the salvage chemotherapy group of the Liebers et al. trial was notably older, at 73.5 years (range, 37 years to 87 years). The sponsor notes that the MAICs were adjusted for the proportion of patients aged 65 years or older; however, they report that this information was not available for the patients included in the Liebers et al. (2021) study. |
Trial eligibility criteria | Patients enrolled in the EPCORE NHL-1 trial were selected according to trial inclusion and exclusion criteria, whereas those in the Liebers et al. (2021) study were drawn from a real-world sample in Germany. |
Intervention (regimen and dosing) | Epcoritamab was administered in accordance with defined study protocol and is aligned with recommendations in the Canadian and international product labels. In contrast, patients in the salvage chemotherapy group of the Liebers et al. (2021) study could have received 1 of several polatuzumab-containing regimens. The sponsor included the results for all 54 patients included in the salvage chemotherapy group of Liebers et al., which required pooling patients who received pola-BR (n = 32 [59.3%]), pola-R (n = 20 [37.0%]), pola-B (n = 1 [1.85%]), and pola-R with gemcitabine (n = 1 [1.85%]). The appropriateness of pooling comparator data for patients who received treatment with and without a chemotherapy backbone for the purposes of evaluating the comparative efficacy of treatments is uncertain. This may bias the results in favour of epcoritamab, given that Liebers et al. (2021) reported that those who received the chemotherapy backbone had a more favourable outcome (i.e., an OR rate of 52.9% vs. 40%, respectively; P = 0.4). In addition, Health Canada has not approved pola-R (i.e., without bendamustine) for use in the treatment of R/R DLBCL, and the 2021 CADTH recommendation in favour of reimbursement included only the pola-BR regimen. |
Evaluation of end points | Response to epcoritamab was evaluated under the controlled conditions of a registration trial protocol (IRC and investigator assessments) using standardized radiological assessments for patients and a multiple-step process for determining response and/or disease progression. In contrast, the evaluations in Liebers et al. were conducted under real-world conditions, which could include physician judgment in the absence of a CT scan. Liebers et al. reported that response assessment with CT scans was available for 41 of 54 patients (75.9%) in the salvage chemotherapy group. No analysis was reported with a breakdown of those who received pola-BR or pola-R. |
Definitions of end points | Response to treatment was evaluated according to preplanned study protocol in the EPCORE NHL-1 trial and solely by the treating clinicians in Liebers et al. (2021). This, coupled with differences in the approaches to imaging, may make it challenging to compare results for CR and PR across the 2 studies. |
Timing of end point evaluation | Liebers et al. (2021) reported a median time of first CT response assessment as 50 days (range, 12 days to 193 days) and a median time to best CT response of 68 days (range, 12 days to 217 days). These ranges underscore the variation that can be anticipated when evaluating response in a real-world setting in comparison with the controlled conditions of the EPCORE NHL-1 trial. |
Clinical trial setting | The EPCORE NHL-1 trial was a multinational trial. Liebers et al. (2021) exclusively used data from a compassionate use program in Germany. |
Study design | The EPCORE NHL-1 trial was a phase I and II, single-arm registration trial, whereas Liebers et al. (2021) was based on a retrospective chart review from real-world clinical practice in Germany. |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CR = complete response; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; IRC = independent review committee; LBCL = diffuse large B-cell lymphoma; MAIC = matching-adjusted indirect comparison; pola-B = polatuzumab vedotin with bendamustine; pola-B/R = polatuzumab with rituximab, with or without bendamustine; pola-BR = polatuzumab vedotin with bendamustine and rituximab; pola-R = polatuzumab vedotin with rituximab; PR = partial response; PS = Performance Status; R/R = relapsed or refractory.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 31: Assessment of Homogeneity for MAIC Comparing Epcoritamab to Pola-BR for Population Without Prior CAR T-Cell Therapy
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Prior therapy | Refractory disease: The proportion of patients who were considered primary refractory was an adjustment factor in the MAIC; however, this information was not reported for the subgroup analysis by Sehn et al. (2020). Lines of therapy: Patients in both the EPCORE NHL-1 trial and Sehn et al. (2021) were heavily pretreated, with both studies reporting a median number of 3 prior lines of antilymphoma therapy (range, 2 lines to 11 lines and range, 2 lines to 7 lines, respectively). However, a greater proportion of patients in the Sehn et al. (2020) subgroup had received more than 3 lines of prior therapy compared with those in the EPCORE NHL-1 trial (62.1% vs. ||||||| respectively). The sponsor’s MAIC was not adjusted for this difference. Prior ASCT: A greater proportion of those in the Sehn et al. (2020) subgroup analysis had received prior ASCT than in the EPCORE NHL-1 trial (34.5% vs. ||||||||). |
Performance Status | The subgroup of patients from the EPCORE NHL-1 trial generally had a good ECOG PS, with only | |||||||| ||||||| who had a baseline ECOG PS of 2. The clinical experts consulted by CADTH noted that this is common in oncology clinical trials and that a higher proportion of patients with ECOG PS of 2 or more could be considered candidates for epcoritamab in actual clinical practice (depending on reimbursement status). The subgroup analysis from Sehn et al. (2020) included a greater proportion of patients with an ECOG PS of 2 (10.7%). The sponsor notes that the MAICs were adjusted for the proportion of patients with an ECOG PS > 1. |
Patient characteristics | Patient age: The proportion of patients aged 65 years and older was greater in the subgroup of patients in the EPCORE NHL-1 trial than in the subgroup of patients from Sehn et al. (2020) (61.63% vs. 52%, respectively). The sponsor reported that the MAIC was adjusted for this difference. Disease stage: A greater proportion of patients from the Sehn et al. (2020) subgroup were classified as having stage III or IV disease at baseline compared with those in the subgroup from the EPCORE NHL-1 trial (86.2% vs. |||||| respectively). The sponsor reported that the MAIC was adjusted for differences in baseline disease stage. |
Trial eligibility criteria | The analysis required the extraction of subgroup data from both the EPCORE NHL-1 trial (n = |||) and the Sehn et al. trial (n = 29). |
Intervention (regimen and dosing) | Epcoritamab was administered in accordance with defined study protocol and is in alignment with recommendations in the Canadian and international product labels. |
Evaluation of end points: | Response to epcoritamab was evaluated under the controlled conditions of a registration trial protocol (IRC and investigator assessments) using standardized radiological assessments for patients and a multiple-step process for determining response and/or disease progression. |
Definitions of end points | Response to treatment was evaluated according to a preplanned study protocol in the EPCORE NHL-1 trial. Patients in Sehn et al. (2020) were evaluated using modified IRC Lugano criteria. |
Clinical trial setting | Both the EPCORE NHL-1 trial and Sehn et al. (2020) were multinational clinical trials. |
Study design | The EPCORE NHL-1 trial was a phase I and II, single-arm trial. Sehn et al. (2020) was a phase II RCT comparing pola-BR vs. BR alone. |
ASCT = autologous stem cell transplant; BR = bendamustine and rituximab; CAR = chimeric antigen receptor; ECOG = Eastern Cooperative Oncology Group; IRC = independent review committee; MAIC = matching-adjusted indirect comparison; pola-BR = polatuzumab vedotin with bendamustine and rituximab; PS = Performance Status; RCT = randomized controlled trial.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Table 32 summarizes the association strength of several patient characteristics of interest with 12-month PFS and OS in the EPCORE NHL-1 trial. Across the literature and results from the empirical test for whether patient characteristics were prognostic, the following features came up as possible variables that need to be adjusted for: age, sex, ECOG Performance Status, histology, IPI score, disease stage, primary refractoriness, response to recent prior therapy, number of prior lines of therapy, prior ASCT, and prior CAR T-cell therapy.
Table 32: Association of Patient Characteristics With PFS and OS in the EPCORE NHL-1 Study
Characteristic | Level | PFS by IRC (LUGANO criteria) at 12 months | P value for difference across levels | OS at 12 months | P value for difference across levels |
|---|---|---|---|---|---|
Age group | < 65 years | ||||| | |||| | ||||| | |||| |
65 years to < 75 years | ||||| | ||||| | |||
≥ 75 years | ||||| | ||||| | |||
Sex | Female | ||||| | |||| | ||||| | |||| |
Male | ||||| | ||||| | |||
DLBCL type | De novo | ||||| | |||| | ||||| | |||| |
Transformed | ||||| | ||||| | |||
Unknown | || | || | |||
ECOG PS | 0 | ||||| | |||| | ||||| | ||||| |
1 | ||||| | ||||| | |||
2 | || | ||||| | |||
Histology | DLBCL | ||||| | |||| | ||||| | |||| |
FL | || | ||||| | |||
HGBCL | || | ||||| | |||
PMBCL | || | ||||| | |||
IPI score | 0 to 2 | ||||| | |||| | ||||| | ||||| |
≥ 3 | ||||| | ||||| | |||
Not applicable | || | ||||| | |||
Primary refractory | No | ||||| | ||||| | ||||| | ||||| |
Yes | ||||| | ||||| | |||
Prior ASCT | No | ||||| | |||| | ||||| | |||| |
Yes | ||||| | ||||| | |||
Prior CAR T-cell therapy | No | ||||| | |||| | ||||| | |||| |
Yes | ||||| | ||||| | |||
Prior lines of therapy | 2 | ||||| | |||| | ||||| | |||| |
3 | ||||| | ||||| | |||
> 3 | ||||| | ||||| | |||
Response to recent prior therapy | Refractory | ||||| | |||| | ||||| | ||||| |
Relapsed | ||||| | ||||| | |||
Disease stage | Stage I | ||||| | |||| | ||||| | |||| |
Stage II | ||||| | ||||| | |||
Stage III | ||||| | ||||| | |||
Stage IV | ||||| | ||||| |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; ECOG = Eastern Cooperative Oncology Group; EUnetHTA = European Network for Health Technology Assessment; FL = follicular lymphoma; HGBCL = high-grade B-cell lymphoma; IPI = International Prognostic Index; IRC = independent review committee; NE = not estimated; OS = overall survival; PFS = progression-free survival; PMBCL = primary mediastinal B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; PS = Performance Status.
Note: Given that the EUnetHTA submission did not report best response in the population of patients on pola-BR with 2 or more prior therapies (PICO 1B), the estimate for best response among those on 2 or more prior therapies (N = 102) in the extension of the Sehn et al. trial was used.62This was deemed to be a justified assumption for the analysis, given that authors of the extension study concluded that “the baseline characteristics in the extension cohort were similar to the [original] randomized pola-BR cohort,” which is where the PICO 1b population was derived from in the EUnetHTA submission.
Source: Sponsor’s indirect comparison.11
Evidence Networks
Figure 7: Evidence Network Diagram for Unanchored MAICs
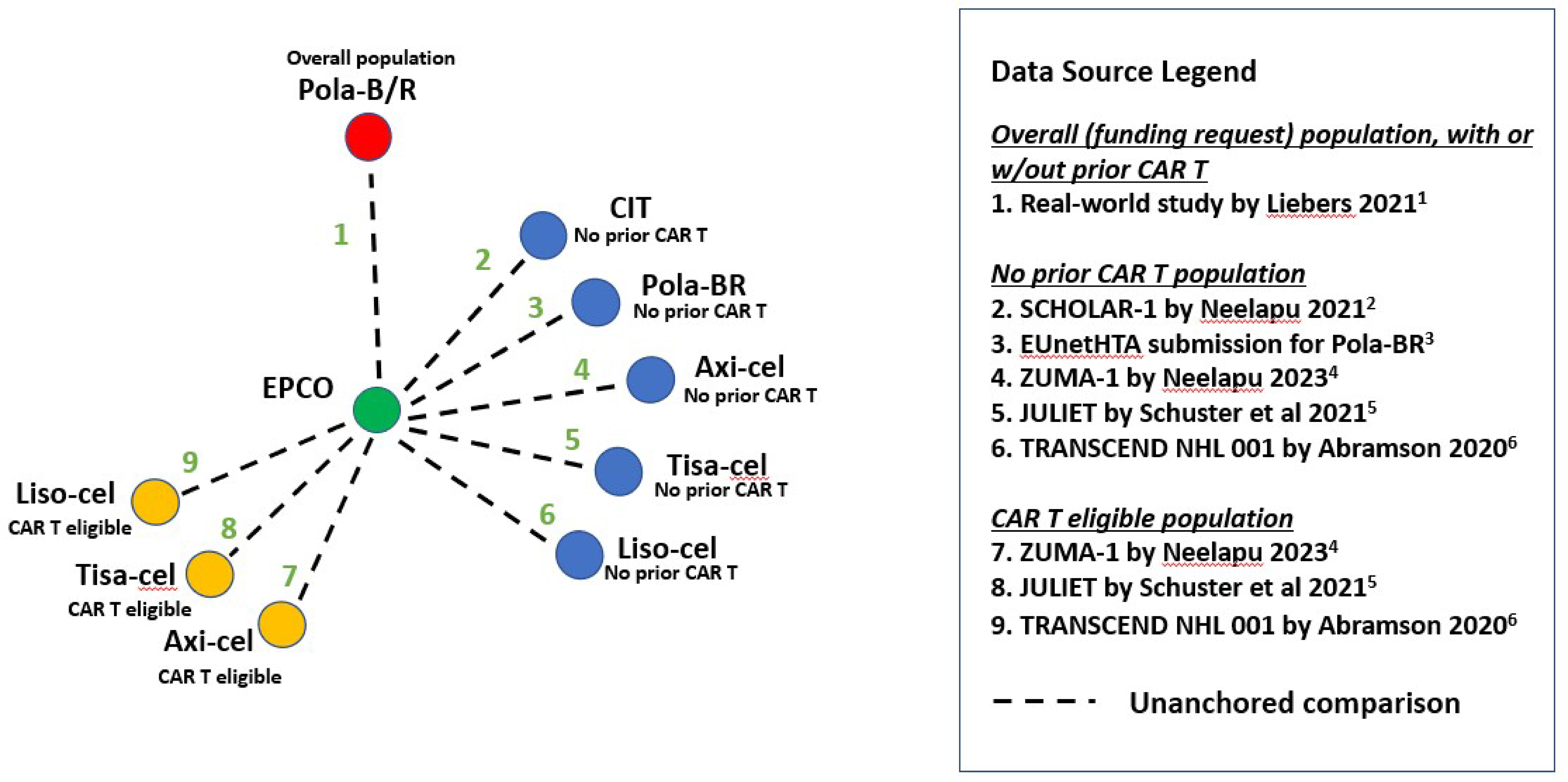
Axi-cel = axicabtagene ciloleucel; CAR = chimeric antigen receptor; CIT = chemoimmunotherapy; Liso-cell = lisocabtagene maraleucel; MAIC = matching-adjusted indirect comparison; pola-B/R = real-world polatuzumab-based regimens, including polatuzumab vedotin with bendamustine and rituximab as well as polatuzumab vedotin with rituximab; pola-BR = polatuzumab vedotin with bendamustine and rituximab; Tisa-cel = tisagenlecleucel.
Sources: Liebers et al. (2021);63 Neelapu et al. (2021);64 Neelapu et al. (2023);65 EUnetHTA (2020);66 Schuster et al. (2021);67 Abramson et al. (2020).68
ESSs for MAICs
Weight truncation was assessed individually for each ITC to determine the most optimal cut-off. For each of the key comparisons described previously, a summary of weight truncation and ESS is provided.
Table 33: Weight Truncation and ESS for Each Pairwise, Unanchored MAIC
Epcoritamab vs. comparator | Weight truncation | ESS (n, % of set) |
|---|---|---|
Overall population of patients with LBCL (unadjusted sample size: n = 157) | ||
vs. pola-B/R | || |||||||||| | || ||||||| |
Population with LBCL, no prior CAR T-cell therapy (unadjusted sample size: n = 96) | ||
vs. R-CIT | || ||| ||| | || ||||||| |
vs. pola-BR | || |||||||||| | || ||||||| |
vs. axicabtagene ciloleucel | || ||| ||| | || ||||||| |
vs. tisagenlecleucel | |||| ||| ||||| | || ||||||| |
vs. lisocabtagene maraleucel | || |||||||||| | || ||||||| |
Population with LBCL, no prior CAR T-cell therapy, eligible for CAR T-cell therapy (unadjusted sample size: n = 57) | ||
vs. axicabtagene ciloleucel | || ||| ||| | || ||||||| |
vs. tisagenlecleucel | |||| ||| ||||| | || ||||||| |
vs. lisocabtagene maraleucel | || |||||||||| | || ||||||| |
CAR = chimeric antigen receptor; ESS = effective sample size; LBCL = large B-cell lymphoma; MAIC = matching-adjusted indirect comparison; pola-B/R = polatuzumab vedotin with rituximab, with or without bendamustine; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CIT = rituximab-based chemoimmunotherapy.
Source: Sponsor’s indirect comparison.11
Patient Characteristics
Epcoritamab Versus Pola-B/R (Overall Population of Patients With LBCL)
Patient characteristics for the MAIC comparing epcoritamab versus pola-B/R in the overall population of patients with LBCL are summarized in Table 34.
Epcoritamab Versus Pola-BR (Patients Without Prior CAR T-Cell Therapy)
Patient characteristics for the MAIC comparing epcoritamab versus pola-BR for patients with LBCL without prior CAR T-cell therapy are summarized in Table 35.
Epcoritamab Versus CIT (Patients Without Prior CAR T-Cell Therapy)
Patient characteristics for the MAIC comparing epcoritamab versus CIT in patients without prior CAR T-cell therapy are summarized in Table 36.
Table 34: Adjusted Patient Characteristics for Epcoritamab Versus Pola-B/R for the Overall Population of Patients With LBCL
Characteristics | Epcoritamab | Pola-B/R (N = 54) | |
|---|---|---|---|
Unadjusted (N = 157) | Adjusted (ESS = 96) | ||
Age (median) | 64.0 | |||| | |||| |
Age ≥ 73.5 years | 22.9% | ||||| | ||| |
Male (%) | 59.9% | ||||| | ||||| |
DLBCL (%) | 88.5% | ||||| | ||||| |
Time from diagnosis, years (median) | 1.58 | |||| | |||| |
Prior treatment lines (median) | 3 | ||| | ||| |
Did not respond to ASCT (relapse within 12 months) | 11.5% | |||| | |||| |
Prior CAR T-cell therapy (mean) | 9.3% | |||| | |||| |
Refractoriness to last treatment (mean) | 82.8% | ||||| | ||||| |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; ESS = effective sample size; LBCL = large B-cell lymphoma; pola-B/R = polatuzumab vedotin with rituximab, with or without bendamustine.
Source: Sponsor’s indirect comparison.11
Table 35: Adjusted Patient Characteristics for Epcoritamab Versus Pola-BR for Patients With LBCL Without Prior CAR T-Cell Therapy
Characteristics | Epcoritamab | Pola-BR (N = 29) | |
|---|---|---|---|
Unadjusted (N = 96) | Adjusted (ESS = 56) | ||
Age, median (years) | |||| | |||| | |||| |
Age ≥ 65 years (%) | ||||| | ||||| | ||||| |
Male (%) | ||||| | ||||| | ||||| |
DLBCL (%) | ||||| | |||||| | |||||| |
ECOG PS 0 to 1 (%) | ||||| | ||||| | ||||| |
Disease stage III to IV (%) | ||||| | ||||| | ||||| |
IPI score ≥ 3 (%) | ||||| | ||||| | ||||| |
2 lines of prior therapy (%) | ||||| | ||||| | ||||| |
≥ 3 lines of prior therapy (%) | ||||| | ||||| | ||||| |
Refractory to last anti-CD20 drug (%) | ||||| | ||||| | ||||| |
Refractory to last antilymphoma therapy (%) | ||||| | ||||| | ||||| |
Prior ASCT (%) | ||||| | ||||| | ||||| |
ASCT = autologous stem cell transplant; CD20 = cluster of differentiation 20; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; IPI = International Prognostic Index; LBCL = large B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab.
Source: Sponsor’s indirect comparison.11
Table 36: Adjusted Patient Characteristics for Epcoritamab Versus CIT for Patients With LBCL Without Prior CAR T-Cell Therapy
Characteristics | Epcoritamab | CIT (N = 340) | |
|---|---|---|---|
Unadjusted (N = 96) | Adjusted (ESS = ||) | ||
Age, median (years) | |||| | |||| | |||| |
Male (%) | ||||| | ||||| | ||||| |
Age ≥ 65 years (%) | ||||| | ||||| | ||||| |
≥ 3 lines of chemo and ASCT (%) | ||||| | ||||| | ||||| |
Primary refractory (%) | ||||| | ||||| | ||||| |
Refractory to ≥ 2 consecutive lines of therapy (%) | ||||| | ||||| | ||||| |
SCT any time after refractory disease (%) | ||||| | ||||| | ||||| |
Relapse within 12 months of ASCT (%) | ||||| | ||||| | ||||| |
ECOG PS 0 or 1 (%) | ||||| | |||||| | |||||| |
Disease stage III or IV (%) | ||||| | ||||| | ||||| |
IPI score ≥ 3 (%) | ||||| | ||||| | ||||| |
ASCT = autologous stem cell transplant; CAR = chimeric antigen receptor; CIT = chemoimmunotherapy; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; IPI = International Prognostic Index; LBCL = large B-cell lymphoma; SCT = stem cell therapy.
Source: Sponsor’s indirect comparison.11
Efficacy
Table 37 summarizes the efficacy results for the sponsor’s unanchored MAICs.
Table 37: Summary of Efficacy Results From Unanchored MAICs
Epcoritamab vs. comparator | PFS HR (95% CI); P value | OS HR (95% CI); P value | ORR RD (95% CI); P value | CR RD (95% CI); P value |
|---|---|---|---|---|
Overall population of patients with LBCL (unadjusted sample size: n = 157) | ||||
Vs. pola-B/R | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| ||||| || |||||||||||| | ||||| |||||| || ||||||||||||| |
Population of patients with LBCL and no prior CAR T-cell therapy (unadjusted sample size: n = 96) | ||||
Vs. R-CIT | ||| | ||||| |||||| || ||||||||||||| | ||||| |||||| || ||||||||||||| | ||||| |||||| || ||||||||||||| |
Vs. pola-BR | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| ||||| || |||||||||||| | ||||| ||||||| || |||||||||||| |
Vs. axicabtagene ciloleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | |||| ||||||| || |||||||||||| | ||||| ||||||| || |||||||||||| |
Vs. tisagenlecleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| ||||| || |||||||||||| | ||||| |||||| || |||||||||||| |
Vs. lisocabtagene maraleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| ||||||| || ||||||||||| | ||||| ||||||| || ||||||||||| |
Population of patients with LBCL without prior CAR T-cell therapy who were eligible for CAR T-cell therapy (unadjusted sample size: n = 57) | ||||
Vs. axicabtagene ciloleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || ||||||||||||| | |||| ||||||| || |||||||||||| | ||||| ||||||| || |||||||||||| |
Vs. tisagenlecleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| ||||| || |||||||||||| |
Vs. lisocabtagene maraleucel | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| | ||||| |||||| || |||||||||||| |
CAR = chimeric antigen receptor; CI = confidence interval; CR = complete response; LBCL = large B-cell lymphoma; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; pola-B/R = polatuzumab vedotin with rituximab, with or without bendamustine; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CIT = rituximab-based chemoimmunotherapy; RD = risk difference; vs. = versus.
Source: Sponsor’s indirect comparison.11
Epcoritamab Versus Pola-B/R (Overall Population of Patients With LBCL)
In the adjusted overall population of patients with LBCL, the sponsor reported that epcoritamab was associated with significant improvements in both PFS (||| |||||| ||| ||| ||||| || |||||| | | |||||) and OS (||| |||||| ||| ||| ||||| || |||||| | | |||||) compared to pola-B/R. The sponsor also reported a significant improvement with epcoritamab versus pola-B/R in both CR rate (|||||| ||| ||| ||||| || |||||| |||||||) and ORR (|||||| ||| ||| |||| || |||||| | | |||||).
Epcoritamab Versus Pola-BR (Patients Without Prior CAR T-Cell Therapy)
Compared with pola-BR in the analysis of patients with prior CAR T-cell therapy, the sponsor reported no significant difference in PFS (||| |||||| ||| ||| ||||| || |||||| | | |||||), OS (||| |||||| ||| ||| ||||| || |||||| | | |||||), or CR rate ||||||| ||| ||| |||||| || |||||| | | |||||). The sponsor reported that epcoritamab was associated with an improvement in ORR versus pola-BR (|||||| ||| ||| |||| || |||||| | | |||||).
Epcoritamab Versus CIT (Patients Without Prior CAR T-Cell Therapy)
PFS was not reported in the SCHOLAR-1 study;25 as such, it could not be reported for the comparison versus R-CIT in the population of patients with LBCL without prior CAR T-cell therapy. Compared to CIT, the sponsor reported that epcoritamab was associated with significant improvements in OS (||| |||||| ||| ||| ||||| || |||||| | | |||||); CR rate (|||||| ||| ||| ||||| || |||||| | | |||||); and ORR (|||||| ||| ||| ||||| || |||||| | | |||||).
Harms
Harms outcomes were not assessed as part of the ITCs.
Critical Appraisal of the MAICs
Given the lack of direct evidence comparing epcoritamab to relevant treatments in the R/R LBCL third-line setting, the sponsor’s decision to conduct an ITC (i.e., an unanchored MAIC) was justified.
Systematic Literature Review
The sponsor-submitted MAICs were informed by an SLR that included planned searches of multiple databases and conference proceedings up to April 2023. The sponsor’s search strategy and the eligibility criteria for study selection were clearly reported within the application documents. The sponsor reported that a formal quality assessment using the Appraisal of RCT checklist by the Centre for Reviews and Dissemination and the quality assessment checklist for nonrandomized clinical trials from the centre’s Guidance for Undertaking Reviews in Health Care (2009) was applied during the conduct of the SLR for the purpose of selecting studies for the MAICs.
Study Selection
There were important differences in the design of the included studies and the cohorts evaluated that limit the ability to draw strong conclusions about the efficacy of epcoritamab compared with pola-BR and CIT. The EPCORE NHL-1 study of epcoritamab was a phase I and II, single-arm study, whereas the GO29365 study was a comparative phase Ib and II randomized, open-label study; SCHOLAR-1 was a retrospective research study; and Liebers et al. (2021) was a retrospective cohort, real-world study. In addition, all the comparisons involved the use of subgroup data from 1 or both of the studies included in the indirect comparison, thereby reducing the ESS and increasing the likelihood of the results being biased from a selective population.
In addition to differences in study design, notable differences in the eligibility criteria of the included studies resulted in heterogeneity in baseline characteristics across populations. The sponsor provided a list of likely prognostic factors, confounding factors, and treatment-effect modifiers (identified through consultation with clinical experts). However, adjustment of all these factors could not be achieved due to differences in reporting across the various studies and a lack of access to patient-level data (other than for those enrolled in the EPCORE NHL-1 trial). It is unclear if the lack of adjustment for differences in baseline characteristics (particularly those that may be prognostic factors, such as primary refractory disease) would have an impact on the results of the MAIC because sensitivity analyses with a full model were not provided. A key limitation of the sponsor-submitted MAICs, which is a limitation inherent to all unanchored MAICs, is that it assumes that all effect modifiers and prognostic factors are accounted for in the model. This assumption is largely considered impossible to meet, according to the NICE Decision Support Unit Technical Guidance report on the methods for population-adjusted indirect comparisons.69 Thus, the certainty of the evidence generated by unanchored MAICs that are based on partially adjusted models is typically low or very low (particularly in the absence of adequate sensitivity analyses testing choices made related to the model assumptions).
Appraisal of ITC Methods and Results
Overall, CADTH concluded that there were multiple limitations in the sponsor-submitted MAICs, including differences in inclusion and exclusion criteria, heterogeneity in baseline characteristics across studies, and notable reductions in sample sizes following weighting. There was significant uncertainty about the overall generalizability of the results to patients living in Canada. Additionally, wide 95% CIs led to imprecision and uncertainty in the results.
Specific commentaries for each MAIC considered relevant to the CADTH assessment are reported here. As previously noted, the comparisons for epcoritamab versus CAR T-cell therapies have not been appraised by CADTH because they cannot be considered relevant comparators in the context of the current restricted indication that has been approved by Health Canada.
Epcoritamab Versus Pola-B/R (Overall Population of Patients With LBCL)
Important limitations with the sponsor’s MAIC for epcoritamab versus pola-B/R for the overall population of patients with LBCL are as follows:
Small sample size: The analysis started with the FAS set from the epcoritamab trial (n = 157) but required the extraction of subgroup data from Liebers et al. (2021) (i.e., only those in the salvage pola-B/R treatment group; n = 54). Following adjustment of selected baseline characteristics, the ESS was reduced to ||| patients. This is a small sample size with which to evaluate differences in outcomes that are important to patients (for example, the ongoing, phase III EPCORE DLBCL-1 trial has a target enrolment of 470 patients, ||| of whom have not responded adequately to 2 or more lines of therapy).
Unadjusted patient characteristics: CADTH identified several baseline characteristics that were not adjusted for in the sponsor’s MAIC (due to the absence of data reported for the comparator group derived from the Liebers et al. [2021] publication), including the proportion of patients with primary refractory disease; the proportion of patients who were refractory to their last antilymphoma therapy; baseline ECOG Performance Status; and the baseline ages of patients. It is unclear if the lack of adjustment for differences in baseline characteristics (particularly those that may be prognostic factors, such as primary refractory disease) would have an impact on the results of the MAIC.
Differences in study design: The EPCORE NHL-1 trial was a phase I and II, single-arm registration trial, whereas Liebers et al. (2021) was based on a retrospective chart review from real-world clinical practice of patients in Germany who received polatuzumab through special access. Thus, the inherent differences in study design, study conduct, patient selection, and analysis increase the uncertainty in the comparison.
Differences in outcome collection: Response to epcoritamab was evaluated under the controlled conditions of a registration trial protocol (IRC and investigator assessments) using standardized radiological assessments for patients (i.e., PET-CT and/or CT-MRI) and a multiple-step process for determining response and/or disease progression. In contrast, the evaluations in Liebers et al. were conducted under real-world conditions, which could include physician judgment in the absence of a CT scan. Liebers et al. reported that response assessment with CT scans was available for 41 patients of 54 patients (75.9%) in the salvage chemotherapy group. (No analysis was reported with a breakdown by those who received pola-BR or polatuzumab vedotin with rituximab [pola-R].)
Differences in interventions: Patients in the salvage chemotherapy group of the Liebers et al. (2021) study could have received 1 of several polatuzumab-containing regimens. The sponsor included the results for all 54 patients included in the salvage chemotherapy group, which required the pooling of patients who received pola-BR (n = 32 [59.3%]), pola-R (n = 20 [37.0%]), pola-B (n = 1 [1.85%]), and pola-R with gemcitabine (n = 1 [1.85%]). The appropriateness of pooling comparator data for patients who received treatment with and without a chemotherapy backbone for the purposes of evaluating the comparative efficacy of treatments cannot be determined without information about the similarity of effects of these and sensitivity analyses testing the effects of this assumption on the model. This approach may bias the results in favour of epcoritamab, given that Liebers et al. (2021) reported that those who received the chemotherapy backbone had a more favourable outcome (i.e., OR rate of 52.9% versus 40%, respectively [P = 0.4]). In addition, Health Canada has not approved pola-R (i.e., without bendamustine) for use in the treatment of R/R DLBCL, and the 2021 CADTH recommendation in favour of reimbursement included only the pola-BR regimen.
Epcoritamab Versus Pola-BR (Patients Without Prior CAR T-Cell Therapy)
Important limitations with the sponsor’s MAIC for epcoritamab versus pola-BR for patients without prior CAR T-cell therapy are as follows:
Small sample size: The analysis required the extraction of subgroup data from both the EPCORE NHL-1 trial (n = 96) and the Sehn et al. trial (n = 29). Following adjustment of selected baseline characteristics, the ESS was reduced to ||| patients. As with the other MAIC reported for epcoritamab versus pola-BR, this is a small sample size to evaluate differences in outcomes that are important to patients. For example, the ongoing, phase III EPCORE DLBCL-1 trial has a target enrolment of 480 patients; ||| ||| |||| have not responded adequately to 2 or more lines of therapy.
Unadjusted patient characteristics: CADTH identified several baseline characteristics for which the sponsor’s MAIC did not adjust (typically due to the absence of data reported for the comparator groups), including the proportion of patients with primary refractory disease and the proportion of patients who had received more than 3 lines of prior antilymphoma therapy. It is unclear if the lack of adjustment for differences in baseline characteristics (particularly those that may be prognostic factors, such as primary refractory disease) would have an impact on the results of the MAIC.
The clinical experts consulted by CADTH noted that pola-BR would typically be used as a second-line therapy in patients without prior CAR T-cell therapy; thus, it would be offered as an earlier line of therapy compared with the currently approved indication for epcoritamab (i.e., after at least 2 lines of systemic therapy).
Epcoritamab Versus R-CIT (Patients Without Prior CAR T-Cell Therapy)
Important limitations in the sponsor’s MAIC for epcoritamab versus pola-BR for patients without prior CAR T-cell therapy are as follows:
Small sample size: The analysis required the extraction of subgroup data from both the EPCORE NHL-1 trial (n = 96) and the SCHOLAR-1 trial (n = 340). After adjusting for selected baseline characteristics, the ESS was reduced to only ||| patients (i.e., ||||% of the FAS population from the EPCORE NHL-1 trial or ||||% of the subgroup of patients with no prior CAR T-cell therapy).
Lines of prior systemic therapy: The SCHOLAR-1 trial included patients who had at least 1 prior line of antilymphoma therapy. There was no breakdown regarding those who had received 2 or more lines of therapy (as per the indication under review and the trial population of the EPCORE NHL-1 trial). Reviewers from NICE noted the sponsor’s assertion that the subgroup of patients from Neelapu et al. (2021) limits inclusion to those with at least 2 prior lines of antilymphoma therapy, but they were unable to confirm that assumption.
Eligibility for CAR T-cell therapy: The sponsor used a subgroup of patients from the SCHOLAR-1 trial (n = 340) that was reported in the publication by Neelapu et al. (2021). This was a cohort of patients selected to inform an indirect comparison versus a CAR T-cell therapy (axicabtagene ciloleucel); as such, the population may represent those who are specifically eligible to receive CAR T-cell therapy. This does not necessarily limit comparability versus the EPCORE NHL-1 trial population (because, as previously noted, the trial was not limited to those who are ineligible to receive CAR T-cell therapy); however, it does not represent the target population based on the approved indication from Health Canada.
Differences in study design: The EPCORE NHL-1 trial was a multinational trial, whereas the SCHOLAR-1 trial was a retrospective cohort analysis with data drawn from 4 sources: observational cohorts from the MD Anderson Cancer Center; the Molecular Epidemiology Resource of the University of Iowa/Mayo Clinic Lymphoma Specialized Program of Research Excellence; the phase III Canadian Cancer Trials Group study; and the phase III CORAL study.
Differences in disease: SCHOLAR-1 includes only patients with refractory disease rather than a mix of patients with R/R disease. NICE reviewers noted that this could underestimate the survival outcomes for R-CIT versus using a mixed population. In response to this comment, the sponsor compared survival estimates between the SCHOLAR-1 study and other published studies for R-CIT in patients with R/R DLBCL. The sponsor’s analysis demonstrated that the survival estimates from the SCHOLAR-1 trial are within the ranges reported in the other studies (Figure 8).
Differences in outcome collection: Response to epcoritamab was evaluated under the controlled conditions of a registration trial protocol (with IRC and investigator assessments) using standardized radiological assessments for patients (i.e., PET-CT and/or CT-MRI) and a multiple-step process for determining response and/or disease progression. In contrast, the data from the SCHOLAR-1 trial were drawn from multiple different sources and from evaluations of patients’ responses to treatment by clinicians.
Inconsistency in interventions: The types of CIT used in the SCHOLAR-1 trial and the proportion of patients who received each type of chemotherapy are not reported. This means it is unclear whether all or most patients received R-CIT.
Censoring: The Kaplan-Meier curves for OS that were reported in the publication by Neelapu et al. (2021) do not contain information on censoring; hence, the sponsor assumed that the pattern of censoring for the subgroup in the SCHOLAR-1 trial would be the same as that for the full population reported in the publication by Crump et al. (2017). Reviewers from NICE noted that this introduces additional uncertainty in the sponsor’s MAIC.
Figure 8: Survival Curves for R-CIT Studies in R/R DLBCL [Redacted]

DLBCL = diffuse large B-cell lymphoma; R-CIT = rituximab-based chemoimmunotherapy; R/R = relapsed or refractory.
Source: Sponsor’s submission to CADTH.11
Summary
Epcoritamab Versus Pola-B/R (Overall Population of Patients With LBCL)
The sponsor claimed that epcoritamab is associated with significant improvements compared with pola-B/R in PFS (||| |||||| ||| ||| ||||| || |||||| | | ||||||), OS (HR = 0.386; 95% CI, 0.246 to 0.606; P < 0.001), CR rate (|||||| ||| ||| ||||| || |||||| | | ||||||| and ORR (|||||| ||| ||| |||| || |||||| | | |||||). CADTH considers the analysis of epcoritamab versus pola-B/R to be associated with significant uncertainty, with the potential to overestimate the survival outcomes in favour of epcoritamab versus pola-B/R. NICE similarly concluded that this analysis had significant limitations, considering it to be even more limited than the analysis that leveraged the subgroup analysis data from Sehn et al. (2020) (i.e., the analysis for epcoritamab versus pola-BR in patients without prior CAR T-cell therapy).
Epcoritamab Versus Pola-BR (Patients Without Prior CAR T-Cell Therapy)
The sponsor reported no significant difference between pola-BR and epcoritamab in PFS (||| |||||| ||| ||| ||||| || |||||| | | |||||), OS (||| |||||| ||| ||| ||||| || |||||| | | |||||), or CR rate (||||| ||| ||| |||||| || |||||| | | |||||). The sponsor reported that epcoritamab was associated with an improvement in ORR versus pola-BR (|||||| ||| ||| |||| || |||||| | | |||||). CADTH agreed with the assessment from NICE, which concluded that the uncertainty regarding the comparative efficacy of epcoritamab versus pola-BR remained unresolved and that the analysis presented for the subgroup of patients who were naive to CAR T-cell therapy (i.e., those derived from the subgroup of patients reported for Sehn et al. (2020) [n = 29]) may overestimate survival outcomes in favour of epcoritamab versus pola-BR.
Epcoritamab Versus R-CIT (Patients Without Prior CAR T-Cell Therapy)
The sponsor’s analysis suggested that treatment with epcoritamab was superior to CIT for patients with no prior exposure to CAR T-cell therapy; however, there are important limitations with the MAIC that pose challenges when it comes to evaluating and quantifying the potential added benefit of the treatment — specifically, the small ESS (n = |||) and the heterogeneity across the study populations (e.g., different study designs, lack of reporting and adjustment for potentially relevant patient characteristics, and differences in the CIT regimens used in SCHOLAR-1). The clinical experts consulted by CADTH felt that it was plausible that treatment with epcoritamab could be superior to R-CIT for the target patient population on the basis that these patients have already demonstrated disease progression following exposure to an R-CIT regimen (typically R-CHOP), and that the potential toxicity of R-CIT regimens at this stage of disease can limit the regimens’ clinical utility. However, quantifying any additional benefit remains challenging due to the limitations of the available indirect comparisons. In addition, the sponsor has been mandated by Health Canada to conduct a head-to-head trial against R-CIT in the relevant patient population, which suggests that the regulatory authority has similar concerns about uncertainty in the benefit of epcoritamab versus R-CIT. Overall, CADTH considers the submitted MAICs to be associated with too much uncertainty to conclude on the magnitude of improvement conferred by epcoritamab versus R-CIT or to be certain that the phase III EPCORE DLBCL-1 trial would address this uncertainty for the health system.
CADTH notes that HAS similarly concluded that no formal conclusions could be drawn from the sponsor’s MAIC citing methodological limitations that include uncertainty regarding the quality of the data (particularly the RWE), significant heterogeneity between the populations included in the different studies, and residual differences across the various treatments after weighting. However, NICE acknowledged that, despite the uncertainty associated with the sponsor’s MAICs, epcoritamab was likely to be more effective than R-CIT, based on the sponsor’s MAIC. Clinical experts consulted by NICE noted that epcoritamab could plausibly be more effective than pola-BR; however, the NICE expert committee noted that there was too much uncertainty in the indirect comparison and concluded that an assumption of equal efficacy would be more appropriate to inform the economic evaluation.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Consideration for TLR Recommendation
A TLR recommendation is a recommendation by the CADTH expert committee to publicly fund a drug or drug regimen for a certain period of time based on the condition that the sponsor will conduct 1 or more clinical studies that address uncertainty in the clinical evidence. CADTH would subsequently conduct a reassessment of the additional evidence and issue a final reimbursement recommendation within a defined period of time. Based on the preliminary assessment by CADTH (Table 40), epcoritamab meets the criteria to be considered by the expert committee for a TLR recommendation. In accordance with the CADTH Procedures for Time-Limited Reimbursement Recommendations, this section of the report provides an assessment of the existing gaps in the evidence and the sponsor’s evidence-generation plans.
Eligibility Criteria for a TLR Recommendation
Regulatory Status
Health Canada issued an NOC/c on October 13, 2023 for epcoritamab with the following key confirmatory requirements regarding efficacy:
The sponsor should commit to submitting a clinical trial for the purposes of providing confirmatory evidence of efficacy in the setting of R/R DLBCL. Specifically, the primary analyses of Study GCT3013 to 05: A Randomized, Open-Label, Phase 3 Trial of Epcoritamab vs Investigator’s Choice Chemotherapy in Relapsed/Refractory Diffuse Large B-cell Lymphoma (EPCORE DLBCL-1) should be submitted to Health Canada as an SNDS-C.
The primary efficacy objective of the EPCORE DLBCL-1 phase III study is to demonstrate that epcoritamab monotherapy improves the OS of patients with DLBCL compared to investigator’s choice of either BR or R-GemOx. The sponsor should acknowledge that the authorization may be revoked if the trial fails to show an OS benefit of epcoritamab over the investigator’s choice of therapy. The sponsor should provide an estimated date of completion of the primary analyses for the study as well as an estimated date for the submission of the study to Health Canada.
Commitment to File for Reassessment
The sponsor has expressed a commitment to file a reassessment application with CADTH in accordance with the time frames specified in the procedures for TLR recommendations. The phase III trial will be completed within a time frame that will not exceed 3 years from the target expert committee meeting date. ||| ||||||| ||| ||||| |||| ||| ||||||||| |||| |||||||| || |||||||||||||||||| || ||| |||||||||| || ||| ||||||| |||||||| ||||| |||| ||| ||| |||||||| ||||| |||||| ||| ||| ||||| ||| |||||||||||||| ||||| ||||| || |||| |||| ||| ||| |||||| ||||| |||| |||||| |||||| || |||||| ||||| This is within the 3-year period described in the CADTH procedures for TLR recommendations.
Evidence-Generation Plans (EPCORE DLBCL-1 Trial)
The EPCORE DLBCL-1 trial meets the eligibility criteria for a TLR recommendation because it is a phase III clinical trial conducted using epcoritamab as monotherapy in the target population for this review. Specifically, the following was noted by CADTH:
Study design: The trial is a phase III trial that will be reported within the time frame specified in CADTH’s procedures.
Intervention: Epcoritamab will be administered as monotherapy at the dosages recommended in the Canadian product monograph.
Patient population: Generally, the same population (i.e., DLBCL) is included, with acceptable differences, specifically:
Histology: Patients with PMBCL are included in the phase II trial, but not in the phase III trial. Patients with T-cell and/or histiocyte-rich LBCL are included in the phase III trial, but not in the phase II trial.
Disease status: Disease status is the same across the phase II and phase III clinical trials (i.e., R/R).
Lines of therapies: These are not identical because the phase III trial will also include patients who have received 1 prior line of therapy. However, the sponsor provided the following justification that the criteria for a TLR recommendation should be met: enrolment for patients with 1 prior line of therapy will be capped at ||| patients (out of a total N = 470); randomization will be stratified based on number of prior lines of therapy (1 or ≥ 1); a prespecified subgroup analysis for the target population (i.e., those with at least 2 prior lines of therapy) will be provided.
Comparator: Epcoritamab will be directly compared against the investigator’s choice of R-GemOx or BR.
Outcomes: The primary end point is OS. Secondary end points include PFS, ORR, CR, DOR, TTR, time to next antilymphoma therapy, rate and duration of minimum residual disease negativity, FACT-Lym, and antiepcoritamab antibody response.
Table 38: Pending Phase III Trial (EPCORE DLBCL-1 Study)
Detail | EPCORE DLBCL-1 study |
|---|---|
Designs and populations | |
Study design | Open-label, randomized study of epcoritamab monotherapy vs. prespecified investigator’s choice of R-GemOx or BR |
Locations | Australia, Austria, Belgium, Canada, China, Denmark, Finland, France, Germany, Hungary, Israel, Italy, Japan, Republic of Korea, Netherlands, Norway, Poland, Russian Federation, Singapore, Spain, Sweden, Taiwan, Turkey, UK, US |
Patient enrolment dates | Start date: January 13, 2021 Primary completion date: December 2024 (final data collection date for primary outcome measure) Study completion date: April 2028 ||||||| ||| ||||| |||| ||| |||||||| ||||| |||||| |||| || ||||| || |||| |||| ||| ||| |||||| ||||| |||| |||||| |||||| || |||||| ||||| |
Enrolled (N) | N = 480
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | All patients in the expansion part of the trial received the epcoritamab RP2D regimen, consisting of a priming dose of 0.16 mg (C1D1), an intermediate dose of 0.8 mg (C1D8), and a full dose of 48 mg (C1D15, C1D22, and thereafter) administered by SC injection. In this part of the trial (i.e., dose expansion), epcoritamab was administered in 28-day cycles as follows:
|
Comparators | Investigator’s choice will be 1 of the following:
|
Study duration | |
Screening phase | ≤ 30 days before the first dose of epcoritamab |
Treatment phase | For each patient, the treatment period continued until disease progression unless the patient fulfilled 1 of the discontinuation criteria. The trial will run for a maximum of 5 years after the last patient’s first dose. |
Follow-up phase | Until withdrawal of consent, loss to follow-up, or death |
Outcomes | |
Primary end point | OS |
Secondary and exploratory end points |
|
Publication status | |
Publications | ClinicalTrials.gov (NCT04628494) |
ALT = alanine transaminase; ASCT = autologous stem cell transplant; AST = aspartate aminotransferase; BR = bendamustine plus rituximab; C1D1 = cycle 1, day 1; C1D8 = cycle 1, day 8; C1D15 = cycle 1, day 15; C1D22 = cycle 1, day 22; CAR = chimeric antigen receptor; CD20 = cluster of differentiation 20; CNS = central nervous system; CR = complete response; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FL = follicular lymphoma; FLG3B = follicular lymphoma grade 3B; HGBCL = high-grade B-cell lymphoma; HSCT = hematopoietic stem cell transplant; LYRIC = lymphoma response to immunomodulatory therapy criteria; mAb = monoclonal antibody; MRD = minimum residual disease; NOS = not otherwise specified; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; R-CHOP = rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; R-GemOx = rituximab, gemcitabine, and oxaliplatin; R/R = relapsed or refractory; RP2D = recommended phase II dose; SC = subcutaneous; TTNT = time to next antilymphoma therapy; TTR = time to response; ULN = upper limit of normal.
aAssessed by IRC and investigator assessment based on Lugano and LYRIC criteria.
bRelapsed disease is defined as disease that has recurred greater than or equal to 6 months after completion of therapy. Refractory disease is defined as disease that either progressed during therapy or progressed within 6 months (< 6 months) of completion of therapy.
Source: Sponsor’s submission materials.11
Assessment of Gaps in the Evidence
The basis for the TLR and subsequent reassessment would be the ongoing EPCORE DLBCL-1 phase III study that is evaluating whether epcoritamab monotherapy improves the OS of patients with DLBCL compared to investigator’s choice of either BR or R-GemOx. The primary end point is OS, and the NOC/c Qualifying Notice from Health Canada states that the sponsor should acknowledge that authorization may be revoked if the trial fails to show an OS benefit for epcoritamab over BR or R-GemOx. The clinical experts consulted by CADTH expressed concerns regarding the choice of comparator in the EPCORE DLBCL-1 study (i.e., BR or R-GemOx): it was felt that the efficacy data from the EPCORE NHL-1 trial were compelling, and that BR and R-GemOx would be associated with significant toxicities for patients. The experts noted that patients at this stage of disease would likely have received R-CIT earlier in the course of disease and been shown to be refractory to the treatment. As such, they expressed concerns regarding clinical equipoise in the trial, with a belief that those randomized to BR or R-GemOx would be receiving an inferior treatment option. The clinical experts noted that more appropriate comparators would be the newer therapies that have emerged in the second- and third-line settings, such as pola-BR and CAR T-cell therapy. CADTH noted that the curative potential of CAR T-cell therapy would typically make this the preferred option for many patients, which could pose challenges for the design and conduct of a comparative clinical trial.
In comments on the draft report, the sponsor clarified that at the start of the EPCORE DLBCL-1 study (January 2021), neither CAR T-cell therapy nor pola-BR were widely used. Therefore, R-CIT was considered the most appropriate comparator, and CIT remains an option in practice in Canada for the treatment of R/R LBCL. CADTH agrees with the sponsor’s assessment regarding the choice of comparator at the time of initiating the EPCORE DLBCL-1 trial. (The final CADTH recommendation for pola-BR was issued in April 2021, 4 months after the trial had initiated.)
Discussion
Summary of Available Evidence
One ongoing, phase I and II, open-label, single-arm study (the EPCORE NHL-1 study) was included in this review of epcoritamab. The review was based on the dose expansion phase of the study, which consisted of 157 patients with R/R LBCL who had relapsed after or not responded adequately to at least 2 prior systemic treatment regimens. Patients were excluded if they had a known primary CNS lymphoma or known CNS involvement, had received CAR T-cell therapy within 30 days or ASCT within 100 days before the first dose of epcoritamab, or had received any prior allogeneic HSCT. Eligible patients received treatment with epcoritamab monotherapy at the step-up recommended doses: a priming dose of 0.16 mg (C1D1), an intermediate dose of 0.8 mg (C1D8), and a full dose of 48 mg (C1D15, C1D22, and every 4 weeks thereafter until unacceptable toxicity or disease progression). The primary end point was ORR, with secondary end points of CR, DOR, PFS, OS, HRQoL, and safety.
The majority of patients in the LBCL intention-to-treat population had DLBCL (88.5%), with smaller subgroups who had HGBCL (5.7%), PMBCL (2.5%), or FLG3B (3.2%). Patients in the EPCORE NHL-1 trial were heavily pretreated at the time of enrolment, with 29.9%, 30.6%, and 39.5% having received 2, 3, or greater than or equal to 4 prior lines of antilymphoma therapy, respectively. A majority (82.8%) were refractory to their last prior therapy. Prior CAR T-cell therapy was reported in 38.9% of patients, and 19.7% had received prior stem cell transplant.
One sponsor-submitted ITC was summarized and critically appraised by CADTH. The MAICs focused on 3 patient populations: the overall population of patients with LBCL; the population of patients with LBCL without prior CAR T-cell therapy; and the population of patients with LBCL without prior CAR T-cell therapy who were considered eligible to receive CAR T-cell therapy. The indirect comparisons of interest for the CADTH review were epcoritamab versus pola-BR and R-CIT. The sponsor-submitted ITC included comparisons against 3 CAR T-cell therapy regimens: axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel. Given that the Health Canada–approved indication for epcoritamab states that the drug is approved only for use in patients “who have previously received or are unable to receive CAR T-cell therapy,” CADTH does not consider CAR T-cell therapies to be relevant comparators for the current review. The approach is consistent with applications that have been filed in the same therapeutic area. Outcomes evaluated in the MAICs included OS, PFS, ORR, and CR.
No long-term extension studies or studies addressing gaps in the systematic review were included.
Interpretation of Results
Efficacy
After refractoriness or relapse following first- and second-line treatments in LBCL, prognosis is poor due to the aggressive and quickly progressing nature of the disease. Beyond established first-line therapies (e.g., R-CHOP), treatment for R/R LBCL is an evolving landscape, with novel CAR T-cell therapies emerging in both the second- and third-line settings. Pola-BR has been recommended by CADTH for use as a second- or third-line option for patients who are ineligible to receive ASCT.28,70 The clinical experts consulted by CADTH — and the clinician group input — highlighted that other than pola-BR, R-CIT are the only remaining option; however, these are associated with significant toxicities, limiting the use of these options. The clinical experts consulted by CADTH suggested that treatment at this stage would likely be palliative, although epcoritamab may replace pola-BR or other R-CIT as the preferred option in Canada. Patients highlighted a considerable need for alternative treatments that can extend life and improve HRQoL.
The efficacy of epcoritamab was assessed in the EPCORE NHL-1 phase I and II trial, which enrolled 157 patients with R/R LBCL (88.5% with DLBCL). The overall interpretation of the efficacy results from the study was limited, given the internal and external validity issues identified, led primarily by the trial’s single-arm, open-label design, which precludes the ability to attribute results to treatment with epcoritamab. As noted in the GRADE assessment, conclusions about efficacy relative to any comparator cannot be drawn from single-arm studies; thus, the certainty of evidence starts at “very low” and cannot be rated up. Furthermore, the nonrandomized, early-phase study design of the EPCORE NHL-1 trial makes the trial susceptible to selection bias. The clinical experts consulted by CADTH agreed; they also noted that there is more certainty around the primary response end points of ORR and CR, given that these were assessed by independent and investigator reviews and included objective measurements, such as fluorodeoxyglucose uptake, as measured by PET scans.
The clinical experts consulted by CADTH, patient groups, and clinician groups noted that improvement in survival is the most important outcome of treatment, with the clinical experts noting that landmark analyses (i.e., 12-month and 18-month event-free rates) were potentially more suggestive of benefit than median survival. No established thresholds of clinical importance for survival have been identified; however, it was the clinical experts’ opinion that the observed OS and PFS potentially represented improvements over currently used treatments for R/R LBCL (i.e., 3 months for PFS, 6 months for OS). Overall, due to the single-arm nature of the EPCORE NHL-1 trial, the ability to interpret the results for OS and PFS was significantly limited. Similar conclusions were drawn by Health Canada, as reflected by the issuance of an NOC/c for this indication.
The primary end point of the EPCORE NHL-1 trial was ORR (composite of patients whose best overall response was CR or PR), and the response rate was 63.1% (95% CI, 55.0% to 70.6%), surpassing the prespecified threshold of 50% reported in the trial protocol. The clinical experts consulted by CADTH noted that a threshold of 50% for ORR was appropriate and could be suggestive of a clinically important response for the target population. The primary end point of ORR in the EPCORE NHL-1 study was questioned by Health Canada, given that CR has been cited as providing a direct measure of the drug’s antitumour activity in oncology clinical trials. The sponsor noted that ORR has been used in pivotal trials for other drugs approved in Canada for R/R LBCL, including Minjuvi (tafasitamab), Kymriah (tisagenlecleucel), and Breyanzi (lisocabtagene maraleucel), and that CR was a prespecified secondary end point in the EPCORE NHL-1 trial. The proportion of patients with CR in the EPCORE NHL-1 trial was ||||| |||| ||| |||| || ||||| which was considered clinically relevant by regulatory authorities and the clinical experts consulted by CADTH. The CR rate for epcoritamab in the EPCORE NHL-1 trial is similar to the rate reported in the pivotal trial for glofitamab (i.e., 35.2%; 95% CI, 26.24% to 44.96%). As previously noted, the current literature suggests some correlation between response and improved survival; however, the results are highly variable and have not been studied specifically in the population under review.
The input from the clinical experts consulted by CADTH for this review was consistent with the input received for other R/R LBCL drugs and noted that CRs are considered more important than PRs in LBCL. However, it was noted that this was particularly relevant for earlier lines of therapy, during which the intent of treatment is curative, and that PR can be clinically important in the palliative care setting. The experts noted that PR with a sufficiently long DOR could be clinically meaningful in the case of drugs such as epcoritamab that are taken by patients who have been heavily pretreated and for whom the intention is to extend life and improve quality of life (especially given that epcoritamab can be administered continuously, provided the patient is benefiting and tolerating the therapy). Health Canada noted that DORs were relatively short for those with a best overall response of PR (refer to PFS Figure 6). The clinical experts consulted by CADTH noted that PR could be considered successful for this patient population, particularly older patients, given the otherwise poor outcomes for those who have exhausted other available treatment options. The absence of a CR would not necessarily warrant consideration of alternative options for these patients until disease progression or unacceptable toxicity.
HRQoL was also considered an important outcome by patients, and improvement in HRQoL was identified as a treatment goal by clinicians. End points for HRQoL in the EPCORE NHL-1 trial were secondary (i.e., FACT-LymS) or exploratory (i.e., FACT-Lym Trial Outcome Index, FACT-G total score, and FACT-Lym total score). These outcomes had high attrition rates (e.g., fewer than 50% of patients remaining at cycle 5). As such, the evidence for the effects of epcoritamab on HRQoL were considered by CADTH reviewers to have very low certainty; as with OS and PFS, no firm conclusions could be drawn about the observed results. Health Canada reviewers similarly noted that the HRQoL end points reported for the EPCORE NHL-1 trial are not readily interpretable from
Health Canada considered the small number of patients enrolled in the EPCORE NHL-1 trial who had forms of aNHL other than DLBCL (i.e., those with HGBCL, PMBCL, and FLG3B) and noted that adequately powered clinical studies would be problematic to conduct, given the rarity of these subtypes. After examining response rates in these patients, Health Canada concluded that epcoritamab demonstrated efficacy and that these patients are treated in the same manner as those with DLBCL in clinical practice.32 This includes patients living with HGBCL, a more aggressive form of aNHL than DLBCL. The approved indication for epcoritamab specifically identifies approval for use in HGBCL, which is different from the indications for the other Health Canada–approved regimens for R/R LBCL (i.e., glofitamab, polatuzumab vedotin, pembrolizumab, and tafasitamab).33-36,44 The clinical experts consulted by CADTH noted that the evidence supports the efficacy of epcoritamab for patients with rarer forms of lymphoma.
The CADTH clinical review noted that there is no evidence of a clear mechanism by which epcoritamab would provide clinical benefit to patients with PD, which is in alignment with the clinical expert feedback received by CADTH.
Results for the MAICs varied across the comparisons for epcoritamab versus pola-BR. The sponsor claimed significant improvements in PFS, OS, CR rate, and ORR in the overall population of patients with LBCL, and no significant difference in PFS, OS, or CR rate in the population of patients without prior CAR T-cell therapy. CADTH considered the analyses of epcoritamab versus pola-BR or pola-B/R to be associated with significant uncertainty due to small sample sizes and heterogeneity across the studies and patient populations.
The sponsor’s analysis suggested that epcoritamab was superior to CIT for patients with no prior exposure to CAR T-cell therapy; however, there are important limitations with the MAIC that pose challenges when it comes to evaluating and quantifying the potential added benefit of the treatment — specifically, the small ESS (n = 36) and the heterogeneity across the study populations (e.g., different study designs, lack of reporting and adjustment for potentially relevant patient characteristics, and differences in the CIT regimens used in SCHOLAR-1). The clinical experts consulted by CADTH felt that it was plausible that treatment with epcoritamab could be superior to R-CIT for the target patient population on the basis that these patients have already demonstrated disease progression following exposure to an R-CIT regimen (typically R-CHOP) and that the potential toxicity of R-CIT regimens at this stage of disease can limit the regimens’ clinical utility. However, quantifying any additional benefit remains challenging due to the limitations of the available indirect comparisons. In addition, the sponsor has been mandated by Health Canada to conduct a head-to-head trial against R-CIT in the relevant patient population, which suggests that the regulatory authority has similar concerns about uncertainty in the benefit of epcoritamab versus R-CIT. Overall, CADTH considers the submitted MAICs to be associated with too much uncertainty to conclude on the magnitude of improvement conferred by epcoritamab versus R-CIT or to be certain that the phase III EPCORE DLBCL-1 trial would address this uncertainty for the health system.
CADTH notes that other HTA agencies, including NICE and HAS, similarly concluded that no formal conclusions can be drawn from the sponsor’s MAICs, citing methodological limitations, including uncertainty regarding the quality of the data (particularly the RWE), significant heterogeneity between the populations included in the different studies, and residual differences across the various treatments after weighting.71 However, NICE acknowledged that, despite the uncertainty associated with the sponsor’s MAICs, epcoritamab was likely to be more effective than R-CIT. Clinical experts consulted by NICE noted that epcoritamab could plausibly be more effective than pola-BR; however, the NICE expert committee noted that there was too much uncertainty with the indirect comparison, and concluded that an assumption of equal efficacy would be more appropriate to inform the economic evaluation.
Harms
The product monograph for epcoritamab contains black box warnings for the risks of CRS and ICANS, both of which can be serious, life-threatening AEs. To reduce the risk of CRS, epcoritamab is administered using a step-up dosing schedule; the product monograph provides detailed recommendations for premedication (e.g., prednisolone [100 mg oral or IV] 30 minutes to 120 minutes before administration and then for 3 consecutive days after administration for the first cycle). The clinical experts consulted by CADTH noted that the recommended premedications that are specified in the product monograph would be applied in clinical practice, with the goal of preventing or limiting the severity of CRS.
The clinical experts consulted by CADTH noted that the most important AE for epcoritamab is CRS, a prespecified AESI in the EPCORE NHL-1 trial, and that 80 patients (51.0%) had at least 1 CRS event during the trial. The majority of events were mild (grade 1; ||||| |||||||) with grade 2 and grade 3 events occurring in ||||| ||||||| ||| |||| |||||| of patients, respectively. These events were managed in accordance with the clinical trial protocol, which is the same as the recommendations outlined in the product monograph (e.g., withhold further dosing until resolution of the event; treat with tocilizumab ||||||| |||||| and additional corticosteroids ||||||| |||||| as required).
The product monograph recommends that patients be monitored for signs and symptoms of CRS and ICANS for 24 hours after the first full dose of epcoritamab (i.e., 48 mg administered on day 15 of cycle 1). The product monograph recommends that patients remain near a health care facility and be monitored for signs and symptoms of CRS and ICANS or consider hospitalization. The clinical experts noted that patients will likely require hospitalization for 24-hour monitoring after the first full dose of epcoritamab (given that outpatient monitoring may be challenging). The clinical experts and participating drug programs noted that this requirement may pose important challenges for the health system and could limit adoption of the treatment. This may lead to challenges with equity and access across the country for this patient population. The clinical experts noted that some other cancer therapies, such as venetoclax (Venclexta), can also require hospitalization during initial treatment(s), although this is less common. The clinical experts noted that, following the first few cycles, treatment may continue at regional centres, given that the risk of CRS decreases.
The clinical experts consulted by CADTH noted that the risk of CRS and ICANS means epcoritamab must initially be administered at tertiary centres that have the means to monitor and treat these events. This may require initial training of site staff before implementation. The experts emphasized that every centre starting this treatment will need to have internal processes to manage CRS and ICANS as well as the ability to admit patients and administer tocilizumab, anakinra, or siltuximab. As in the case of the need for hospitalization, the clinical experts and participating drug programs noted that these requirements may pose challenges for the health system and could limit the adoption of the treatment (e.g., the hospitals best suited to administer epcoritamab would likely be FACT-accredited centres that have experience with CRS- or ICANS-related treatments).
Patient groups indicated a need for new treatments to have fewer side effects than currently available ones. There is no direct or indirect evidence evaluating the comparative safety of epcoritamab versus the relative comparators for this review. The product monograph for polatuzumab vedotin (Polivy) lists black box warnings regarding the risk of fatal, life-threatening, or serious infections and serious and severe myelosuppression, including neutropenia, febrile neutropenia, thrombocytopenia, and anemia. Epcoritamab has not been directly or indirectly compared with R-CIT at the time of this review (the ongoing, phase III EPCORE DLBCL-1 trial will provide such a comparison). The clinical experts noted that epcoritamab will require much more monitoring than would be expected for R-CIT during the first cycle due to the risk of CRS and ICANS. However, after the first full dose of epcoritamab, if the CRS and/or ICANS is resolved or was not significant, then the monitoring will likely be similar to that required for R-CIT.
The clinical experts noted that the comparative safety of epcoritamab versus the R-CIT regimens is favourable in the longer-term (e.g., after initial management and resolution of CRS and/or ICANS). However, they emphasized that cytotoxic therapies would not be considered ideal comparators for the target patient population. Specifically, they noted that patients who have already progressed with early R-CIT treatment may have bone marrow suppression; as noted previously, there would be a preference to administer a treatment with a different mechanism.
The product monograph for glofitamab (Columvi) also contains a black box warning regarding the risk of CRS, but the pretreatment regimen for all patients includes the IV administration of a single, 1,000 mg dose of obinutuzumab on cycle 1, day 1 (i.e., 7 days before the initiation of glofitamab treatment), with the purpose of depleting circulating and lymphoid tissue B-cells and minimizing the risk of CRS. The clinical experts noted that the pretreatment regimen for glofitamab could have the potential to reduce CRS events, but that IV infusion of obinutuzumab could pose additional access challenges in comparison with the premedication regimen for epcoritamab. Overall, the available data and clinical experience are insufficient to allow comment on the comparative safety of epcoritamab and glofitamab at the time of this review.
Health Canada mandates enhanced postmarket surveillance procedures for all products authorized under the NOC/c policy, including mandatory reporting of all serious adverse reactions that occur in Canada and all serious unexpected adverse reactions that occur outside of Canada.72
Consideration for a TLR Recommendation
A TLR recommendation is a recommendation by the CADTH expert committee to publicly fund a drug or drug regimen for a certain period of time based on the condition that the sponsor will conduct 1 or more clinical studies that address uncertainty in the clinical evidence. CADTH would subsequently conduct a reassessment of the additional evidence and issue a final reimbursement recommendation within a defined period of time. Based on the preliminary assessment by CADTH (Table 40), epcoritamab meets the criteria to be considered by the expert committee for a TLR recommendation.
The basis for the TLR and subsequent reassessment would be the ongoing EPCORE DLBCL-1 phase III study that is evaluating whether epcoritamab monotherapy improves OS in patients with DLBCL compared to investigator’s choice of either BR or R-GemOx. The primary end point is OS, and the NOC/c Qualifying Notice from Health Canada states that the sponsor should acknowledge that authorization may be revoked if the trial fails to show an OS benefit for epcoritamab over BR or R-GemOx. The clinical experts consulted by CADTH expressed concerns regarding the choice of comparator in the EPCORE DLBCL-1 trial (i.e., BR or R-GemOx); it was felt that the efficacy data from the EPCORE NHL-1 trial were compelling and that BR and R-GemOx would be associated with significant toxicities for patients. The experts noted that patients at this stage of disease would likely have received R-CIT earlier in the course of disease and been shown to be refractory to the treatment. As such, they expressed concerns regarding clinical equipoise in the trial, believing that patients randomized to BR or R-GemOx would be receiving an inferior treatment option. The clinical experts noted that more appropriate comparators would be the newer therapies that have emerged in the second- and third-line settings, such as pola-BR and CAR T-cell therapy. CADTH noted that the curative potential of CAR T-cell therapy would typically make this the preferred option for many patients, which could pose challenges for the design and conduct of a comparative clinical trial.
In comments on the draft report, the sponsor clarified that at the start of the EPCORE DLBCL-1 study (January 2021), neither CAR T-cell therapy nor pola-BR were widely used. Therefore, R-CIT was considered the most appropriate comparator, and CIT remains a treatment option in practice in Canada for the treatment of R/R LBCL. CADTH agrees with the sponsor’s assessment regarding the choice of comparator at the time of initiating the EPCORE DLBCL-1 trial. (The final CADTH recommendation for pola-BR was issued in April 2021, 4 months after the trial had initiated.)
Other Considerations
Epcoritamab is administered SC as monotherapy. This may offer efficiencies for the health care system relative to the appropriate comparators identified for this review (e.g., R-CIT or pola-BR). All the comparators require IV administration of multiple drugs. Patient groups noted that the SC route of administration could mean less time in hospital per visit, which can improve the quality of life of patients and caregivers.
The sponsor is currently conducting a phase III trial of epcoritamab in combination with R-CHOP versus R-CHOP alone in patients with newly diagnosed DLBCL (the EPCORE DLBCL-2 study).
Conclusion
One phase I and II, single-arm, open-label trial (EPCORE NHL-1) provided evidence for the efficacy and safety of epcoritamab in adult patients with R/R LBCL who have relapsed after or have not responded adequately to at least 2 prior systemic therapies. Clinicians and patients highlighted the need for accessible, alternative treatment options for patients in this treatment setting. Improvements in survival were considered the most important outcomes by patients and clinicians. Although OS and PFS were evaluated in the EPCORE NHL-1 trial, the single-arm design precludes the ability to accurately evaluate the impact of epcoritamab treatment on these important end points. Nonetheless, the study demonstrated that ||||| of patients achieved CR, which was considered a clinically important result by the clinical experts consulted by CADTH. HRQoL is an important outcome for patients, and many patients in the EPCORE NHL-1 trial demonstrated improvements from baseline after initiating the treatment; however, due to the noncomparative design, high patient attrition rates, and open-label administration of the treatment, the effect of epcoritamab on HRQoL remains uncertain. Overall, the clinical experts consulted by CADTH believe the results demonstrated that epcoritamab offers clinically meaningful improvements for this heavily pretreated patient population and may help address an unmet medical need for a treatment that may extend life, improve symptoms, and be tolerable for patients.
Harms associated with epcoritamab were largely consistent with the mechanism of action, including a high frequency of patients who experienced CRS (50.1%) and serious infections (29.3%). All patients received pretreatment with standardized medications to mitigate the risk of CRS (i.e., prednisolone, diphenhydramine, acetaminophen). The majority of patients recovered from the CRS events. The product monograph provides detailed guidance on grading and managing these events in practice. The clinical experts consulted by CADTH noted that patients will likely require hospitalization for a 24-hour monitoring period after the first full dose of epcoritamab (given that outpatient monitoring may be challenging) and that this requirement may pose important challenges for the health system and could limit adoption of the treatment.
There were important limitations with the sponsor-submitted ITCs that were used to inform the comparative effectiveness of epcoritamab versus R-CIT and pola-BR (the comparators considered most relevant for this review, given the Health Canada–approved indication for epcoritamab). Results for the MAICs varied across the comparisons of epcoritamab versus pola-B/R, with the sponsor claiming significant improvements in PFS, OS, CR rate, and ORR in the overall population of patients with LBCL and no significant differences in PFS, OS, or CR rate in the population of patients without prior CAR T-cell therapy. CADTH considered the analyses of epcoritamab versus pola-BR to be associated with significant uncertainty due to small sample sizes and heterogeneity across the studies and patient populations. The sponsor’s MAIC suggested that epcoritamab was superior to CIT for patients with no prior exposure to CAR T-cell therapy. The clinical experts consulted by CADTH felt that treatment with epcoritamab could plausibly offer greater clinical benefits for patients compared to R-CIT for the target patient population on the basis that these patients have already demonstrated disease progression and drug resistance following exposure to their initial R-CIT regimen (typically R-CHOP), and because the potential toxicity of R-CIT regimens at this stage of disease can limit the regimens’ clinical utility. However, important limitations with the MAIC make it challenging to quantify the magnitude of the potential added benefit and preclude the drawing of evidence-based conclusions regarding the comparative effectiveness of epcoritamab versus R-CIT. Because the sponsor has been mandated by Health Canada to conduct a head-to-head trial against R-CIT in the relevant patient population, there will be direct evidence to inform the comparative clinical benefit; this evidence could be included in a reassessment application as part of a TLR recommendation.
References
1.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian evidence-based guideline for the treatment of relapsed/refractory diffuse large B-cell lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Relapsed_Refractory_DLBCL_VF_Digital.pdf. Accessed 2023 Nov 22.
2.Canadian Cancer Society. Diffuse large B-cell lymphoma. 2023; https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/treatment/treatment-by-type/diffuse-large-b-cell-lymphoma. Accessed 2023 Sep 06.
3.Canadian Cancer Society. Non-Hodgkin lymphoma statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/statistics. Accessed 2023 Sep 06.
4.Freedman AS, Aster JC. Epidemiology, clinical manifestations, pathologic features, and diagnosis of diffuse large B cell lymphoma. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2023: http://www.uptodate.com. Accessed 2023 Aug 16.
5.Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2017. Bethesda (MD): National Cancer Institute; 2020: https://seer.cancer.gov/csr/1975_2017/. Accessed 2023 Aug 16.
6.Clinical practice guideline on lymphoma, version 19. Edmonton (AB): Cancer Care Alberta, Alberta Health Services,; 2023: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe002-lymphoma.pdf. Accessed 2023 Sep 06.
7.Epkinly (epcoritamab for injection): 4 mg in 0.8 mL (5 mg/mL) concentrate for solution for subcutaneous injection; or Epkinly (epcoritamab injection): 48 mg in 0.8 mL (60 mg/mL) solution for subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2023 Oct 13.
8.Drug Reimbursement Expert Review Committee final recommendation: tafasitamab (Minjuvi -Incyte Biosciences Canada Corporation). Can J Health Technol. 2022;2(10). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PC0266/PC0266. Accessed 2024 May 28.
9.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
10.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
11.Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection [internal sponsor's package]. Pointe-Claire (QC): AbbVie Corporation; 2023 Nov 14.
12.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. PubMed
13.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100(17):9991-9996. PubMed
14.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520-13525. PubMed
15.Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123(8):1214-1217. PubMed
16.Sehn LH, Salles G. Diffuse large B-cell lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
17.Sarkozy C, Sehn LH. Management of relapsed/refractory DLBCL. Best Pract Res Clin Haematol. 2018;31(3):209-216. PubMed
18.Riedell PA, Smith SM. Double hit and double expressors in lymphoma: definition and treatment. Cancer. 2018;124(24):4622-4632. PubMed
19.Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26 Suppl 5:v116–125. PubMed
20.Savage KJ, Johnson NA, Ben-Neriah S, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009;114(17):3533-3537. PubMed
21.Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol. 2010;28(20):3360-3365. PubMed
22.Lin P, Dickason TJ, Fayad LE, et al. Prognostic value of MYC rearrangement in cases of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. Cancer. 2012;118(6):1566-1573. PubMed
23.International Non-Hodgkin's Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin's lymphoma. N Engl J Med. 1993;329(14):987-994. PubMed
24.Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematology Am Soc Hematol Educ Program. 2016;2016(1):366-378. PubMed
25.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800-1808. PubMed
26.Shafey M, Savage KJ, Skrabek P, et al. Frontline treatment of diffuse large B-cell lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Frontline_DLBCL_VF_Digital.pdf. Accessed 2023 Nov 22.
27.Bachanova V, Perales MA, Abramson JS. Modern management of relapsed and refractory aggressive B-cell lymphoma: a perspective on the current treatment landscape and patient selection for CAR T-cell therapy. Blood Rev. 2020;40:100640. PubMed
28.Provisional funding algorithm: large B-cell lymphoma. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/PH0027-LBCL.pdf. Accessed 2023 Nov 22.
29.Product information: Tepkinly (epcoritamab). (European public assessment report). Amsterdam (NL): European Medicines Agency; 20 July 2023: https://www.ema.europa.eu/en/documents/product-information/tepkinly-epar-product-information_en.pdf. Accessed 2023 Nov 25.
30.Genmab US, Inc. Prescribing information: Epkinly (epcoritmab) injection. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2023: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/761324s000lbl.pdf. Accessed 2023 Nov 25.
31.Summary of product characteristics - Tepkinly 48 mg solution for injection. London (UK): Medicines and Healthcare products Regulatory Agency; 2023: https://mhraproducts4853.blob.core.windows.net/docs/415e06e94836ee51838fcc9d5ba62713e492e206. Accessed 2023 Nov 25.
32.Health Canada reviewer's report: Epkinly (epcoritimab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): Abbvie Corporation; 2023 Oct 13.
33.Columvi (glofitamab): 1 mg/mL concentrate for solution for intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2023 Mar 24: https://pdf.hres.ca/dpd_pm/00070059.PDF. Accessed 2023 Nov 25.
34.Polivy (polatuzumab vedotin for injection): 30 mg or 140 mg, lyophilized powder for solution in single-use vial, for intravenous infusion only [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2023 Jan 20: https://pdf.hres.ca/dpd_pm/00069254.PDF. Accessed 2023 Aug 14.
35.Minjuvi (tafasitamab for injection): 200 mg lyophilized powder for solution in single-use vial, for infusion [product monograph]. Oakville (ON): Innomar Strategies; 2021 Aug 19: https://pdf.hres.ca/dpd_pm/00062585.PDF. Accessed by sponsor, no date provided.
36.Keytuda (pembrolizumab): 100 mg/4 mL solution in a single-use vial, solution for intravenous infusion [product monograph]. Kirkland (QC): Merck Canada Inc.; 2023 Dec 12: https://pdf.hres.ca/dpd_pm/00073783.PDF. Accessed by sponsor, no date provided.
37.Rituxan (rituximab for injection): 10 mg/mL intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2023 Jun 02: https://pdf.hres.ca/dpd_pm/00071131.PDF. Accessed 2023 Aug 17.
38.Kita K, Arai S, Nishiyama A, et al. In vivo imaging xenograft models for the evaluation of anti-brain tumor efficacy of targeted drugs. Cancer Med. 2017;6(12):2972-2983. PubMed
39.Thieblemont C, Phillips T, Ghesquieres H, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell-engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. 2023;41(12):2238-2247. PubMed
40.Karimi Y, Ghesquieres H, Jurczak W, et al. Effect of follow-up time on the ability of subcutaneous epcoritamab to induce deep and durable complete remissions in patients with relapsed/refractory large B-cell lymphoma: updated results from the pivotal EPCORE NHL-1 trial. J Clin Oncol. 2023;41(16_suppl):7525-7525.
41.Thieblemont C, Clausen MR, Balari AS, et al. Phase 3 trial (GCT3013-05) of epcoritamab versus standard of care in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Hematol Oncol. 2021;39(S2):341.
42.Hutchings M, Mous R, Clausen MR, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398(10306):1157-1169. PubMed
43.Clinical Study Report: GCT3013-01, version 2. A phase 1/2, open-label, dose-escalation trial of GEN3013 in patients with relapsed, progressive or refractory b-cell lymphoma [internal sponsor's report]. Plainsboro (NJ): Genmab US, Inc.; 2023 Oct 03.
44.Genmab/AbbVie. Data on File: Outcomes in R/R DLBCL Patients Treated with Standard of Care. COTA (US EHR) Database Analysis to Contextualize aNHL Data from EPCORE-NHL1. 2022 [sponsor provided reference].
45.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068. PubMed
46.Hutchings M, Mous R, Clausen MR, et al. Subcutaneous epcoritamab induces complete responses with an encouraging safety profile across relapsed/refractory B-cell non-Hodgkin lymphoma subtypes, including patients with prior CAR-T therapy: updated dose escalation data. Blood. 2020;136(Supplement 1):45-46.
47.Clinical Study Report: GCT3013-01, protocol v9.0. A phase 1/2, open-label, dose-escalation trial of GEN3013 in patients with relapsed, progressive or refractory b-cell lymphoma [internal sponsor's report]. Plainsboro (NJ): Genmab US, Inc.; 2020 Sep 23.
48.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
49.Hlubocky FJ, Webster K, Cashy J, Beaumont J, Cella D. The development and validation of a measure of health-related quality of life for non-Hodgkin’s lymphoma: the Functional Assessment of Cancer Therapy—Lymphoma (FACT-Lym). Lymphoma. 2013;2013:147176.
50.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625-638. PubMed
51.Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26(16):2767-2778. PubMed
52.Statistical Analysis Plan: GCT3013-01. A phase 1/2, open-label, dose-escalation trial of GEN3013 in patients with relapsed, progressive or refractory b-cell lymphoma [internal sponsor's report]. Genmab US, Inc.: Plainsboro (NJ); 2022 Mar 17.
53.Clinical trial endpoints for the approval of cancer drugs and biologics: guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018: https://www.fda.gov/media/71195/download. Accessed 2023 Sep 29.
54.Health Canada clarifax: Epkinly (epcoritamab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): Abbvie Corporation; 2023.
55.Kostakoglu L, Martelli M, Sehn LH, et al. End-of-treatment PET/CT predicts PFS and OS in DLBCL after first-line treatment: results from GOYA. Blood Adv. 2021;5(5):1283-1290. PubMed
56.Broglio K, Kostakoglu L, Ward C, et al. PET-CR as a potential surrogate endpoint in untreated DLBCL: meta-analysis and implications for clinical trial design. Leuk Lymphoma. 2022;63(12):2816-2831. PubMed
57.Mohyuddin GR, Koehn K, Sborov D, et al. Quality of control groups in randomised trials of multiple myeloma enrolling in the USA: a systematic review. Lancet Haematol. 2021;8(4):e299-e304. PubMed
58.Prasad V, Kim C, Burotto M, Vandross A. The strength of association between surrogate end points and survival in oncology: a systematic review of trial-level meta-analyses. JAMA Intern Med. 2015;175(8):1389-1398. PubMed
59.Sengar M, Hopman WM, Mohyuddin GR, et al. Randomised controlled trials evaluating anticancer therapies in haematological cancers: an overview of global research activity. Ecancermedicalscience. 2023;17:1558. PubMed
60.Kyvsgaard ER, Clausen MR, Hasselbalch Riley AC, et al. CD3xCD20 bispecific antibodies yields long-term survival in relapsed/refractory B cell lymphoma: a follow-up study of patients treated in pivotal phase 1/2 trials. Blood. 2023;142(Supplement 1):618-618.
61.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947. PubMed
62.Sehn LH, Hertzberg M, Opat S, et al. Polatuzumab vedotin plus bendamustine and rituximab in relapsed/refractory DLBCL: survival update and new extension cohort data. Blood Adv. 2022;6(2):533-543. PubMed
63.Liebers N, Duell J, Fitzgerald D, et al. Polatuzumab vedotin as a salvage and bridging treatment in relapsed or refractory large B-cell lymphomas. Blood Adv. 2021;5(13):2707-2716. PubMed
64.Neelapu SS, Locke FL, Bartlett NL, et al. Comparison of 2-year outcomes with CAR T cells (ZUMA-1) vs salvage chemotherapy in refractory large B-cell lymphoma. Blood Adv. 2021;5(20):4149-4155. PubMed
65.Neelapu SS, Jacobson CA, Ghobadi A, et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. 2023;141(19):2307-2315. PubMed
66.EUnetHTA PTJA06, Authoring Team. Polatuzumab vedotin in combination with bendamustine and rituximab for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who are not candidates for haematopoietic stem cell transplant. Joint Assessment. Diemen (NL): EUnetHTA; 2020: https://www.eunethta.eu/ptja06/. Accessed 2023 Mar 30.
67.Schuster SJ, Tam CS, Borchmann P, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22(10):1403-1415. PubMed
68.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839-852. PubMed
69.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
70.Drug Reimbursement Expert Review Committee final recommendation: polatuzumab vedotin (Polivy - Hoffmann-La Roche Limited). Ottawa (ON): CADTH; 2021 Apr 21: https://www.cadth.ca/sites/default/files/pcodr/Reviews2021/10227PolatuzumabDLBCL_fnRec_EC21Apr2021_final.pdf. Accessed by sponsor, no date provided.
71.Transparency Committee. Tepkinly (epcoritamab) - diffuse large B-cell lymphoma. Saint-Denis (FR): Haute Autorité de Santé; 2023: https://www.has-sante.fr/upload/docs/application/pdf/2023-07/epcoritamab_ap1_decision_et_avisct_ap218.pdf Accessed 2023 Dec 06.
72.Guidance document: Notice of Compliance with Conditions (NOC/c). Ottawa (ON): Health Canada; 2016: https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/compli-conform/noccg_accd-eng.pdf. Accessed 2023 Nov 30.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 39: Summary of Notable Harms From the EPCORE NHL-1 Study
Summary of AESI | LBCL (N = 157) |
|---|---|
Summary of CRS events | |
≥ 1 CRS event, n (%) | 80 (51.0%) |
Grade 1 | || ||||||| |
Grade 2 | || ||||||| |
Grade 3 | | |||||| |
Requiring oxygen | || ||||||| |
Requiring vasopressora | | |||||| |
Requiring vasopressin | | |||||| |
Treated with anticytokine therapy | || ||||||| |
Tocilizumab | || ||||||| |
Treated with corticosteroid for CRS | || ||||||| |
Leading to dose delay | || ||||||| |
Leading to treatment discontinuation | | |||||| |
Time to first CRS onset (days) | |
Mean (SD) | |||| |||||| |
Median (range) | |||| ||| ||| |
Time to CRS resolution (days) | |
Patients with resolved CRS | || ||||||| |
Mean (SD) | ||| |||||| |
Median (range) | ||| ||| ||| |
Summary of ICANS events | |
≥ 1 ICANS event, n (%) | 10 (6.4%) |
Grade 1 | | |||||| |
Grade 2 | | |||||| |
Grade 3 | ||| |
Leading to dose delay | | ||||||| |
Leading to treatment discontinuation | | ||||||| |
Time to first ICANS onset (days) | |
Mean (SD) | |||| ||||||| |
Median (range) | |||| ||| |||| |
Time to ICANS resolution (days) | |
Patients with resolved ICANS | | ||||||| |
Mean (SD) | ||| |||||| |
Median (range) | ||| ||| || |
Summary of serious infections | |
Patients with at least 1 event | 46 (29.3%) |
Grade 1 | | |||||| |
Grade 2 | | |||||| |
Grade 3 | || ||||||| |
Grade 4 | | |||||| |
Grade 5 | || |||||| |
Number of episodes per patient | |
1 event | || ||||||| |
2 events | || ||||||| |
3 events | | |||||| |
≥ 4 events | | |||||| |
Patients with treatment required [a] | || ||||||| |
Time from first dose to first onset (days) | |
Mean (SD) | ||||| |||||||| |
Median | ||||| ||| |||| |
Time to resolution (days) | |
Patients with resolved event [a] | || ||||||| |
Mean (SD) | |||| ||||||| |
Median | |||| ||| |||| |
AESI = adverse event of special interest; CRS = cytokine release syndrome; ICANS = immune effector cell–associated neurotoxicity syndrome; LBCL = large B-cell lymphoma; SD = standard deviation.
aExcluding midodrine/midodrine hydrochloride, milrinone, vasopressin.
Appendix 2: CADTH Preliminary Assessment for TLR Eligibility
Please note that this appendix has not been copy-edited.
Table 40: Preliminary CADTH Assessment of Eligibility for a TLR Recommendation
Eligibility criteria | CADTH assessment |
|---|---|
Regulatory status | |
The drug has been issued an NOC/c by Health Canada or is undergoing review through Health Canada’s advance consideration process under the NOC/c policy. | Criterion that has been met: NOC/c was issued on October 13, 2023. |
Evidence generation | |
A phase III clinical trial is being planned and/or conducted at the time of the submission to CADTH. | Criterion has been met: Sponsor is currently conducting the phase III EPCORE DLBCL-1 trial as part of the NOC/c postmarket requirements for the indication that will be reviewed by CADTH. |
The phase III trial is being or will be conducted in a patient population that is reflective of the indication being reviewed by CADTH | Criterion has been met:
Overall, CADTH concludes that is acceptable and meets the criterion for a time-limited recommendation. |
The phase III trial is being or will be conducted using the intervention at dosages and/or combination regimens that are reflective of the intervention that will be reviewed by CADTH. | Criterion has been met: The sponsor has noted that the dosage regimen for epcoritamab studied in the EPCORE DLBCL-1 phase III trial is the same as the EPCORE NHL-1 phase II trial that will be reviewed in the initial application to CADTH (i.e., use as monotherapy [0.16 mg SC priming dose on day 1, 0.8 mg SC intermediate dose on day 8, and 48 mg SC full dose on days 15 and 22 of cycle 1]; 48 mg SC on days 1, 8, 15, 22 of cycles 2 and 3; 48 mg SC on days 1 and 15 of cycles 4 through 9; and 48 mg SC on day 1 of cycle 10 and onward). |
The phase III trial will be completed within a time frame that will not exceed 3 years from the target expert committee meeting date. | Criterion has been met: || ||||||||||| |||| |||||| |||||||| || |||||| ||| ||||||| ||| ||||| |||| ||| ||||||||| |||| |||||||| || |||||||||||||||||| || ||| |||||||||| || ||| ||||||| |||||||| ||||| |||| ||| ||| |||||||| ||||| |||||| ||| ||| ||||| ||| |||||||||||||| ||||| ||||| || |||| |||| ||| ||| |||||| ||||| |||| |||||| |||||| || |||||| ||||.
|
Commitment to file for reassessment | |
Sponsor is willing to commit to file a reassessment application with CADTH in accordance with the time frames specified in the procedures for time-limited recommendations. | Criterion has been met: Sponsor has expressed commitment to file the reassessment within the required timelines. |
Pharmacoeconomic_Review
Abbreviations
ASCT
autologous stem cell transplant
BIA
budget impact analysis
CAR
chimeric antigen receptor
DLBCL
diffuse large B-cell lymphoma
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LBCL
large B-cell lymphoma
LY
life year
MAIC
matching-adjusted indirect comparison
OS
overall survival
PFS
progression-free survival
PMBCL
primary mediastinal B-cell lymphoma
pola-BR
polatuzumab vedotin with bendamustine and rituximab
PSM
partitioned survival model
QALY
quality-adjusted life-year
RDI
relative dose intensity
R-CIT
rituximab-based chemoimmunotherapy
R-GDP
rituximab plus gemcitabine, dexamethasone, and cisplatin
R-GemOx
rituximab, gemcitabine, and oxaliplatin
R/R
relapsed or refractory
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Epcoritamab (Epkinly), 4 mg in 0.8 mL (5 mg/mL) concentrate for solution for subcutaneous injection and 48 mg in 0.6 mL (60 mg/mL) concentrate for solution for subcutaneous injection |
Indication | For the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), not otherwise specified, DLBCL transformed from indolent lymphoma, high grade B-cell lymphoma (HGBCL), primary mediastinal B-cell lymphoma (PMBCL) or follicular lymphoma grade 3B (FLG3b) after 2 or more lines of systemic therapy and who have previously received or are unable to receive chimeric antigen receptor (CAR) T-cell therapy |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | October 13, 2023 |
Reimbursement request | As per indication |
Sponsor | AbbVie Corporation |
Submission history | Previously reviewed: No |
Eligible for consideration as a time-limited recommendation | Yes |
CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; FLG3B = follicular lymphoma grade 3B; HGBCL = high grade B-cell lymphoma; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; PMBCL = primary mediastinal B-cell lymphoma.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partition survival model |
Target population | Adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, HGBCL, PMBCL, or FLG3B after 2 or more lines of systemic therapy who have previously received or are unable to receive CAR T-cell therapy |
Treatment | Epcoritamab |
Dose regimen | Epcoritamab:
|
Submitted price | Epcoritamab:
|
Submitted treatment cost (per 28-day cycle) | Cycle 1: $14,320 Cycles 2 and 3: $26,436 Cycles 4 to 9: $13,218 Cycle 10 and beyond: $6,609 |
Comparators | Pola-BR Key scenario analyses:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs and LYs |
Time horizon | Lifetime (30 years) |
Key data sources | Data from the EPCORE NHL-1 trial were used to inform PFS and OS for epcoritamab, with MAICs informing the comparative efficacy of relevant comparators. |
Submitted results | ICER = $11,938 per QALY gained (incremental costs = $40,529; incremental QALYs = 3.40) compared with pola-BR |
Key limitations |
|
CADTH reanalysis results |
|
CAR = chimeric antigen receptor; DLBCL = diffuse large B-cell lymphoma; FLG3B = follicular lymphoma grade 3B; HGBCL = high grade B-cell lymphoma; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = matching-adjusted indirect comparison; OS = overall survival; PFS = progression-free survival; PMBCL = primary mediastinal B-cell lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; QALY = quality-adjusted life-year; R-CIT = rituximab-based chemoimmunotherapy; RDI = relative dose intensity; R/R = relapsed or refractory.
Conclusions
The CADTH clinical review noted that evidence from a phase I and II, single-arm, open-label trial (EPCORE NHL-1) in adult patients with relapsed or refractory (R/R), large B-cell lymphoma (LBCL) who have relapsed after or failed to respond to at least 2 prior systemic therapies showed that a clinically important proportion of patients treated with epcoritamab (|||||) achieved a complete response. This is aligned with clinical expert feedback received by CADTH, which noted that the evidence from the EPCORE NHL-1 study demonstrated that epcoritamab offers clinically meaningful improvements for this heavily pretreated patient population. While progression-free survival (PFS) and overall survival (OS) were evaluated in the EPCORE NHL-1 study, due to the study design, CADTH could not accurately evaluate the impact of epcoritamab on these outcomes. Due to the lack of direct evidence, the comparative effectiveness of epcoritamab with relevant comparators (e.g., polatuzumab with bendamustine and rituximab [pola-BR] and rituximab-based chemoimmunotherapy [R-CIT]) was assessed based on sponsor-submitted indirect treatment comparisons (ITCs) (i.e., unanchored matching-adjusted indirect comparisons [MAICs]). CADTH identified important limitations with the ITCs such that the results were considered uncertain. The clinical experts consulted by CADTH felt that it was plausible that treatment with epcoritamab could be superior to treatment with R-CIT for the target patient population, but that the magnitude of impact was uncertain. The clinical experts agreed that, given the uncertainty in the comparative evidence, there is no robust evidence that epcoritamab is more effective than pola-BR.
In line with the clinical evaluation, CADTH considered separate pairwise analyses in which epcoritamab was compared with pola-BR, and with R-CIT, due to the different populations on which the information was based.
In the absence of robust evidence to support an incremental benefit for epcoritamab, CADTH assumed equivalent efficacy between epcoritamab when compared with pola-BR in the intention-to-treat population of the EPCORE NHL-1 trial. This resulted in similar quality-adjusted life-years (QALYs) and an incremental cost of $27,294 for epcoritamab compared with pola-BR. The key drivers in the difference in costs were treatment acquisition costs and the treatment-specific monitoring associated with epcoritamab.
When compared with R-CIT in a population with no prior chimeric antigen receptor (CAR) T-cell therapy use, epcoritamab was more costly and associated with increased life-years (LYs) and QALYs in the CADTH reanalysis. The magnitude of the incremental benefit is associated with uncertainty due to the limitations in the available clinical evidence and the sponsor’s modelling approach. Based on CADTH deterministic scenario analyses assessing alternate effect estimates, the incremental benefit may range from 1.54 QALYs (if the epcoritamab versus R-CIT hazard ratio [HR] equals ||||) to 0.50 QALYs (if the HR equals 0.8). In these instances, the incremental cost-effectiveness ratio (ICER) for epcoritamab ranges from $96,804 to $270,752 per QALY gained compared with R-CIT in patients who had not previously received CAR T-cell therapy. The key drivers affecting the ICER were the treatment acquisition costs associated with epcoritamab and the postprogression survival benefit for patients receiving epcoritamab. Using the |||| HR from the sponsor-submitted ITC for epcoritamab versus R-CIT, and assuming a postprogression benefit for epcoritamab, a price reduction of at least 45% is required for epcoritamab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained compared to R-CIT. When considering alternate HRs, or when removing the postprogression survival benefit — in line with CADTH scenario analyses — a greater price reduction is required.
Compared to pola-BR, there is insufficient clinical evidence to justify a price premium for epcoritamab relative to the total cost of pola-BR. CADTH notes that a price reduction may be required for epcoritamab to be no more costly than pola-BR, given that the prices informing pola-BR were based on the public list price and that epcoritamab does not have a set treatment duration, unlike other available treatments for this indication.
This review of epcoritamab was conducted as a time-limited recommendation by CADTH pending additional clinical studies to address uncertainty in the clinical evidence. When this information becomes available, CADTH will subsequently conduct a reassessment of the additional evidence (which may affect the assessment of comparative effectiveness and cost-effectiveness) and issue a final recommendation.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Leukemia and Lymphoma Society of Canada and Lymphoma Canada through 1-on-1 interviews and an online survey, respectively. Feedback was received from 37 participants (33 survey responders and 4 interviewees), 15 of whom were in Canada. Patients with LBCL reported that fatigue, body aches and pains, enlarged lymph nodes, bodily swelling, neutropenia, and shortness of breath were the most common physical symptoms. They also reported psychosocial impacts. When asked about their experiences with currently available treatments, the majority said they had received 3 or more lines of treatment, with side effects remaining a significant issue. Patient input indicated a desire for longer disease remission, longer survival, overall improved quality of life outcomes, and fewer side effects. One survey respondent and 1 interviewee indicated that they had experience with epcoritamab. Overall, the feedback on epcoritamab was positive, with respondents noting there were some side and psychological effects.
Clinician input was provided by the Leukemia and Lymphoma Society of Canada Nurses Network, Lymphoma Canada, and the Ontario Health Hematology Cancer Drug Advisory Committee. The input noted that current treatment options for the third line of therapy and beyond include pola-BR, R-CIT, chemotherapy, and radiation. However, there remains an unmet need for third-line treatments of diffuse large B-cell lymphoma (DLBCL), particularly for patients who are ineligible for CAR T-cell therapy or for whom this therapy did not work. The treatment goals include prolonging life, delaying disease progression, and improving quality of life by alleviating symptoms. Clinician input noted that the subcutaneous injection administration route of epcoritamab may be less resource-intensive than infusion treatments. Concerns regarding adverse events (particularly cytokine release syndrome) are of concern and may require monitoring and management.
CADTH participating drug plans noted the discrepancy between the Health Canada indication and trial inclusion criteria and inquired about what defines a patient who is unable to receive CAR T-cell therapy. The plans further noted that glofitamab is currently undergoing a CADTH review with a similar reimbursement request. The drug plans commented on the complexity of the dosing schedule of epcoritamab and noted that while it is administered subcutaneously, there may be instances where IV access is still required for supportive care following epcoritamab administration. Concerns about potential cytokine release syndrome associated with inpatient administration for monitoring and managing were raised.
Several of these concerns were addressed in the sponsor’s model:
Adverse events associated with epcoritamab were included within the analysis.
The impact of R/R DLBCL on patients’ quality of life was captured through utility values.
CADTH was unable to address the following concern raised in the stakeholder input:
The comparative clinical efficacy of glofitamab versus epcoritamab for the indicated population was not included in the sponsor’s submission.
Economic Review
The current review is for epcoritamab (Epkinly) for adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, high grade B-cell lymphoma, primary mediastinal B-cell lymphoma (PMBCL), or follicular lymphoma grade 3B after 2 or more lines of systemic therapy who have previously received, or are unable to receive, CAR T-cell therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of epcoritamab compared to pola-BR. The modelled population was adult patients with R/R DLBCL not otherwise specified, DLBCL transformed from indolent lymphoma, high grade B-cell lymphoma, PMBCL, or follicular lymphoma grade 3B after 2 or more lines of systemic therapy who have previously received or are unable to receive CAR T-cell therapy.1 This aligned with the Health Canada–indicated population. The sponsor also included comparisons with R-CIT and CAR T-cell therapies (i.e., lisocabtagene maraleucel, axicabtagene ciloleucel, and tisagenlecleucel) in a key scenario analysis assessing a population with no prior CAR T-cell therapy use. The sponsor also included comparisons of epcoritamab with each of the CAR T-cell therapies in a population of patients with no prior CAR T-cell therapy use but who were eligible for CAR T-cell therapy.1
The intervention under review is epcoritamab, which is given through subcutaneous injection in 28-day cycles. There are unique dosing schedules for cycle 1, cycles 2 and 3, and cycles 4 to 9. In cycle 1, a priming dose of 0.16 mg is given on day 1, followed by a 0.8 mg intermediate dose on day 8, and 48 mg doses on days 15 and 22. For cycles 2 and 3, 48 mg doses of epcoritamab are provided on days 1, 8, 15, and 22. For cycles 4 to 9, 48 mg doses are administered on days 1 and 15. A 48 mg dose is administered on day 1 of each cycle thereafter until disease progression or unacceptable toxicity. In addition to epcoritamab, patients will receive premedication with 650 mg to 1,000 mg of acetaminophen, 50 mg of diphenhydramine, and 100 mg of prednisolone as single doses on day 1.2 The cost of epcoritamab is $6,557 per administration in the 10th cycle and beyond (based on a ||||% dose intensity for epcoritamab, as informed by the EPCORE NHL trial), with the cost per cycle varying within the first 10 cycles (from $6,609 to $26,436).1 Epcoritamab is injected in the lower part of the abdomen or thigh.
Pola-BR is assumed to consist of no more than 6 21-day cycles comprising 1.8 mg/kg of polatuzumab vedotin (total dose not recommended to exceed 240 mg), 90 mg/m2 of bendamustine on days 1 and 2, and 375 mg/m2 IV rituximab on day 1. The cost for pola-BR is $18,995 per cycle, based on the sponsor’s assumptions relating to weighted dosing and vial sharing.1
In key scenario analyses, R-CIT is represented by rituximab, gemcitabine, and oxaliplatin (R-GemOx), which can be given for up to 8 14-day cycles and comprises rituximab at a dose of 375 mg/m2 IV on day 1 of each 14-day cycle, gemcitabine at a dose of 1,000 mg/m2 IV on day 1 of each 14-day cycle, and oxaliplatin at a dose of 100 mg/m2 on day 1 of each 14-day cycle. The cost for R-GemOx is $3,053 per cycle, based on the sponsor’s assumptions relating to vial-sharing.1 The sponsor also included CAR T-cell therapies, with the 1-time drug acquisition costs for the 3 included therapies obtained from prior CADTH reviews.3,4
Outcomes are modelled in terms of the proportion of patients each week who are progression-free on treatment, progression-free off treatment, and in progressive disease. Analysis is conducted from a Canadian health care payer perspective, with a lifetime horizon (30 years) and a discount rate of 1.5% applied to both costs and benefits.1
Model Structure
The submitted model takes the form of a partitioned survival model (PSM) that independently models the proportions of the patient population who are progression-free on treatment, progression-free off treatment, in progressive disease, and dead.1
PSMs model the proportions of patients in each state that are independent at specific times, rather than modelling the transition from 1 state to another. This requires the assumption that the proportion of the population that is progression-free is independent of the proportion who remain on treatment. Similarly, PSMs also require the assumption that the proportion of patients who remain alive is independent of the proportion of alive patients who are progression-free.
The sponsor’s submitted model also included a functional cure state, used to model long-term remission. The time point at which functional cure is assumed in the model is user-modifiable. A functional cure was assumed to occur at 3 years, based on clinical expert opinion received by the sponsor.1
Model Input
Baseline characteristics in the included population were obtained from the EPCORE NHL-1 trial. This included the proportion of the population who are female (40.1%) and the population’s mean age (62 years), body weight (73.6 kg), and body surface area (1.86 m2).1
The sponsor fitted parametric survival curves to patient-level survival data from the EPCORE NHL-1 trial to derive PFS and OS estimates for epcoritamab for the entire model time horizon. The sponsor chose the parametric survival distribution used in the base case based on fit statistics, visual inspection, and clinical and external validity. The sponsor selected the generalized gamma distribution for PFS and the lognormal distribution for OS. The sponsor then derived PFS and OS curves for relevant comparators within each analysis through HR adjustment. The HRs were obtained from pairwise MAICs conducted by the sponsor for this review and applied to the epcoritamab reference curves.5 This process was applied to the sponsor’s base case as well as 2 key scenario analyses.
The mortality rate used in the sponsor’s model for a given cycle was the maximum of the hazard of the extrapolated OS curve for a given treatment and the hazard of general population mortality. To derive a functional cure state, the sponsor assumed that at 3 years, patients in PFS were considered long-term remitters.1 After this point, patients were no longer at risk of progression. Instead, they experienced general population mortality that was adjusted to account for increased risk of death due to long-term cancer complications, in alignment with the value used in the National Institute for Health and Care Excellence review of pola-BR for the treatment of DLBCL.6 Additionally, the sponsor set the OS curve to equal PFS after the curves crossed.1
The sponsor derived health state utility values from linear mixed-model analyses of EQ-5D data obtained in the EPCORE NHL-1 trial.1 The sponsor also assumed that patients considered functionally cured had a utility equivalent to that of the age-adjusted general population.1 Utility values were adjusted for age deterioration, and the sponsor considered adverse event–related utility decrements for the first model cycle.1
Time on treatment, used to estimate drug acquisition costs for epcoritamab, was obtained from the EPCORE NHL-1 trial, whereas for pola-BR and R-CIT, it was set to equal PFS.1 The analysis also includes drug acquisition costs, drug administration costs, treatment-specific resource utility costs, health state–specific resource utilization costs, therapy postdiscontinuation of treatment costs, and adverse event management costs.1 The sponsor assumed drug wastage in its base case.1 Drug acquisition costs for included CAR T-cell therapies considered in key scenario analyses were obtained from prior CADTH reimbursement review reports.3,4
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted probabilistic analyses with 1,000 replications. The deterministic and probabilistic results were similar. The probabilistic results are presented in the following section.
Base-Case Results
In the sponsor’s probabilistic base-case analysis, epcoritamab was associated with $267,749 in total costs and 4.18 total QALYs, whereas pola-BR was associated with $227,220 in total costs and 0.79 in total QALYs. Therefore, compared with pola-BR, epcoritamab was associated with an additional 3.40 QALYs at an additional cost of $40,529, for an ICER of $11,938 per QALY gained.
The sponsor reported that at a willingness-to-pay threshold of $25,000 per QALY gained, epcoritamab was the most cost-effective intervention for 100% of the iterations. The QALY gains were derived from greater time spent in the progression-free health state, whereas the incremental costs associated with epcoritamab were primarily derived from drug acquisition, with the only notable cost offsets obtained by avoiding subsequent treatments with epcoritamab.
Based on the deterministic results, approximately 87% of incremental QALYs for epcoritamab were found to be accrued during the extrapolation period (i.e., median follow up of 25.1 months from the EPCORE NHL-1 trial data).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. pola-BR ($ per QALY) |
|---|---|---|---|---|---|
Pola-BR | 227,220 | Reference | 0.79 | Reference | Reference |
Epcoritamab | 267,749 | 40,529 | 4.18 | 3.40 | 11,938 |
ICER = incremental cost-effectiveness ratio; pola-BR = polatuzumab vedotin with bendamustine rituximab; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted 2 key scenario analyses considering important subpopulations. The first considered a population of patients with no prior CAR T-cell therapy, including patients both eligible and ineligible for CAR T-cell therapy, and included CAR T-cell therapies, pola-BR, and R-CIT as comparators. In this scenario, R-CIT, pola-BR, epcoritamab, and lisocabtagene maraleucel were on the cost-effectiveness frontier. Axicabtagene ciloleucel and tisagenlecleucel were dominated by epcoritamab. Epcoritamab was associated with a sequential ICER of $54,145 per QALY gained versus pola-BR. Given that efficacy inputs were informed by MAIC analyses in which patient-level data from the EPCORE NHL-1 trial were reweighted to match the comparator trial, a sequential analysis may not be appropriate. The pairwise comparison between epcoritamab and R-CIT found that epcoritamab was associated with an ICER of $42,687 per QALY gained.
In the second key scenario analysis, the sponsor assessed the cost-effectiveness of epcoritamab in a population of patients with no prior CAR T-cell therapy who are CAR T-cell therapy eligible. This analysis compared epcoritamab with 3 CAR T-cell therapies. In this scenario, epcoritamab dominated all 3 therapies.
The sponsor conducted a number of additional sensitivity and scenario analyses and noted that the ICER was most sensitive to changes in perspective, time horizon, and efficacy HR for pola-BR in comparison with epcoritamab. The sponsor also conducted a scenario analysis from a societal perspective. This analysis included lost work productivity for patients and caregivers as well as travel costs borne by the patient. In this analysis, relative to pola-BR, epcoritamab was associated with an ICER of $50,910 per QALY gained. This result differed notably from the sponsor’s base-case analysis using a health care payer perspective.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations in the sponsor’s analysis that have notable implications for the economic analysis.
Relevant comparators were not included in the base-case analysis. The sponsor included pola-BR as the only comparator in the submitted base case. While clinical expert feedback received by CADTH agreed that pola-BR is a relevant comparator, it was noted that due to the restricted public funding status of pola-BR in jurisdictions across Canada, R-CITs are additional comparators that should be considered in the population eligible for epcoritamab.
Due to differences in the populations of the EPCORE NHL-1 and SCHOLAR-1 trials, the sponsor included R-CIT in a key scenario assessing a population of adult patients with LBCL with no prior CAR T-cell therapy (including both patients who are clinically ineligible for CAR T-cell therapy and those who were clinically eligible for, but did not receive, CAR T-cell therapy). R-GemOx was used as a proxy to inform the cost of R-CIT in the analysis; however, clinical expert feedback received by CADTH noted that R-GemOx is not commonly used in Canadian clinical practice. The experts stated that because it is a more aggressive chemoimmunotherapy, it is used in a minority of patients who are younger, with good performance status and few comorbid illnesses. More commonly used chemoimmunotherapies include rituximab plus gemcitabine, dexamethasone, and cisplatin (R-GDP); rituximab plus dexamethasone, cytarabine, and cisplatin; and rituximab plus ifosfamide, carboplatin, etoposide.
In the CADTH reanalysis, R-CIT and pola-BR were included as comparators. Due to differences in patient populations informing the efficacy data analyses comparing epcoritamab to R-CIT and pola-BR, analyses were conducted as pairwise analyses in separate populations (i.e., the subgroup of patients with LBCL with no prior CAR T-cell therapy and the overall intention-to-treat population, respectively).
CADTH conducted a scenario analysis in which the cost of R-CIT was informed by R-GDP with a maximum of 3 cycles.
The long-term survival assumptions are uncertain and may be overestimated. In the sponsor’s submitted analysis, efficacy for epcoritamab was informed by extrapolating patient-level survival data from the open-label, single-arm EPCORE NHL-1 trial. In the sponsor’s base case and key scenario analysis, PFS was informed by the generalized gamma distribution (except for key scenario 1, in which lognormal was selected), and OS was informed by the lognormal distribution. The sponsor assumed that patients who remained progression-free at 3 years were functionally cured. While functionally cured patients maintained an increased risk of mortality versus the general population due to long-term cancer complications, it was assumed that these patients were no longer at risk of progression for the remainder of the modelled time horizon. Clinical expert feedback received by CADTH stated that functional cure in clinical practice may be defined as patients who are progression-free after several years in the absence of ongoing treatment (i.e., no longer receiving treatment) with a negative PET scan. Given that epcoritamab is an ongoing treatment until disease progression or unacceptable toxicity, there remains significant uncertainty as to whether it has a curative effect on patients with R/R DLBCL. However, clinical expert feedback received by CADTH commented that the complete response rate based on Lugano criteria from the EPCORE NHL-1 trial is clinically meaningful. They further stated that while patients may experience a reduced rate of progression after remaining progression-free for 1 to 2 years, it is unreasonable to assume that a patient would not have any risk of progression. In the absence of clinical evidence informing the rate of progression after being “functionally cured,” the natural history of patients who were modelled to be functionally cured at 3 years remains unknown.
Additionally, the comparative efficacies of pola-BR and comparators in the key scenario analyses (i.e., R-CIT and CAR T-cell therapies) were derived by applying HRs obtained from the pairwise MAIC conducted by the sponsor to the survival curves of epcoritamab. Based on this method, the sponsor estimated that approximately 19% of patients receiving R-CIT would be alive at 1 year. Clinical expert feedback received by CADTH noted that the sponsor’s survival estimates for R-CIT are likely underestimated, given that the survival results from the SCHOLAR-1 trial reported a median OS of 6.3 months, with a 1-year survival rate of 28%.7 Although it was acknowledged that patients in the SCHOLAR-1 trial may differ slightly from the subpopulation of adult patients with LBCL with no prior CAR T-cell therapy in the EPCORE NHL-1 trial, the clinical expert feedback received by CADTH noted that there would be overlap between the 2 populations and that outcomes are likely to be more aligned with the SCHOLAR-1 trial results than with the sponsor’s estimates. However, due to the modelling method utilized by the sponsor, in which the survival curves of comparators are derived from the survival curves of epcoritamab through HRs, any curve adjustments made to model the anticipated survival of patients on epcoritamab would underestimate the survival of patients with R/R DLBCL on comparators or vice versa.
CADTH removed the assumption that patients were functionally cured if they remained progression-free 3 years after treatment initiation.
CADTH was unable to address the limitations with the sponsor’s HRs, estimated from the MAIC, for R-CIT relative to epcoritamab. In the CADTH reanalysis, the PFS and OS curves for epcoritamab were set to the lognormal and Weibull distributions, respectively. Due to the dependent nature of the R-CIT survival curves on the survival curves of epcoritamab, and the lack of alternate HRs, CADTH was unable to fully address the comparative efficacy limitation. Despite CADTH’s reanalysis changes, the survival estimates for R-CIT are underestimated. This is likely to bias the results in favour of epcoritamab.
The model fails to capture causal relationships appropriately. The sponsor’s model does not adequately capture the causal relationships between patient characteristics or the probability of progression and death. Specifically, the sponsor modelled a postprogression survival benefit for patients receiving epcoritamab such that patients are expected to live approximately half a year longer (i.e., 0.48 LYs) after they experience progression and discontinue epcoritamab, relative to patients receiving pola-BR who experience progression and discontinue pola-BR. Similar findings were identified for epcoritamab and R-CIT in the sponsor’s key scenario analysis, which was performed in patients with no prior CAR T-cell exposure, although epcoritamab was associated with a larger postprogression survival increment (i.e., 0.58 LYs). The CADTH clinical review noted that there is no evidence of a clear mechanism by which epcoritamab would provide clinical benefit to patients with progressive disease; this aligns with the clinical expert feedback received by CADTH. The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that likely do not accurately reflect casual relationships within the disease pathway. The structural assumptions of a PSM may produce a postprogression survival bias that favours epcoritamab.
CADTH was unable to determine the extent to which the implied postprogression benefit was due to the effect of treatment with epcoritamab, structural bias in the PSM, or limitations in the comparator efficacy evidence. To explore the impact of the postprogression analysis on the cost-effectiveness of epcoritamab, CADTH conducted a scenario analysis in which the postprogression benefit of epcoritamab was removed.
The comparative clinical evidence of epcoritamab versus comparators is uncertain. In the absence of direct head-to-head evidence comparing epcoritamab to relevant treatments in the R/R DLBCL third-line setting, the comparative treatment efficacy of epcoritamab versus pola-BR and R-CIT was informed by sponsor-conducted MAICs for each comparison. While a relevant randomized controlled trial for pola-BR was identified (i.e., the GO29365 study), the sponsor elected to inform the clinical efficacy of pola-BR in the submitted MAIC from an observational real-world evidence study by Liebers et al. (2021).8 This was done due to the difference in pretreatment and CAR T-cell exposure of the GO29365 study population versus patients in the EPCORE NHL-1 trial. However, as noted in the CADTH clinical report, usage of the real-world evidence study was associated with limitations due to the difference in study design and outcomes collected compared to the EPCORE NHL-1 trial. While the clinical expert feedback received by CADTH stated that epcoritamab may be more effective than pola-BR, it was noted that in the absence of direct, head-to-head comparative evidence, this statement is associated with uncertainty. The CADTH clinical review team reported that no definitive conclusion could be drawn from the sponsor-submitted MAIC due to several methodological limitations, including differences in inclusion and exclusion criteria, heterogeneity in baseline characteristics across studies, and notable reductions in sample size due to matching and weighing. Given the use of HRs derived from the MAIC to inform clinical efficacy, substantial uncertainty exists in the comparison of epcoritamab versus pola-BR in the economic analysis. This is further exemplified in the contrasting results between the sponsor’s base case and the first key scenario analysis conducted in a subpopulation of adult patients with LBCL with no prior CAR T-cell therapy (including both patients who are clinically ineligible for CAR T-cell and those who are clinically eligible, but did not receive the therapy), in which the adjusted HRs versus pola-BR were ||||| (95% confidence interval, ||||| to |||||) and ||||| (95% confidence interval, ||||| to |||||), respectively. As noted in the CADTH clinical review, Haute Autorité de Santé in France similarly concluded that no formal conclusions could be drawn from the sponsor’s MAICs. Furthermore, while the clinical experts consulted by the National Institute for Health and Care Excellence in the UK noted that it was plausible that epcoritamab was more effective than pola-BR, the institute’s expert committee noted that there was too much uncertainty with the indirect evidence, and concluded that an assumption of equal efficacy would be more appropriate to inform the economic evaluation.
Additionally, there remains uncertainty in the sponsor’s MAIC comparing epcoritamab versus R-CIT for patients without prior CAR T-cell therapy due to the small effective sample size (i.e., n = ||||) and heterogeneity across the study population. Although clinical expert feedback received by CADTH noted that it is plausible that epcoritamab could be more effective that R-CIT, the magnitude of that benefit is unknown due to limitations in the available indirect comparisons.
Given the uncertainty in the conclusions from the MAIC, CADTH assumed equal efficacy of epcoritamab versus pola-BR in its reanalysis.
CADTH additionally conducted scenario analyses using alternative HRs in an attempt to explore the comparative clinical efficacy of epcoritamab versus R-CIT.
Time on treatment for epcoritamab is uncertain. To account for treatment discontinuation for reasons such as toxicity without progression, time on treatment with epcoritamab was informed by time to treatment discontinuation data from the EPCORE NHL-1 trial in the sponsor’s base-case analysis. The clinical expert feedback received by CADTH noted that while it is reasonable to consider reasons other than progression for treatment discontinuation, very few patients in clinical practice do so because patients who discontinue treatment without progression will often experience progression shortly afterward. Should patients in clinical practice largely not discontinue epcoritamab until progression, then the use of time to treatment discontinuation from the trial may underestimate the total cost of epcoritamab, biasing the results in favour of epcoritamab.
To explore the impact of time on treatment for epcoritamab, CADTH conducted a scenario analysis in which epcoritamab time on treatment was equal to PFS.
The sponsor underestimated drug costs. In the sponsor’s base-case analysis, the mean relative dose intensity (RDI) observed in the EPCORE NHL-1 trial was used to derive the drug acquisition cost for epcoritamab. Additionally, RDI values informed from the National Institute of Health and Care Excellence were used to inform the cost for pola-BR.6 The inclusion of RDI may underestimate the total cost of treatment in clinical practice because the dose received by a patient may be different from the planned dose for several reasons (i.e., missed doses, dose reductions, and so on).
In the CADTH reanalysis, RDI was set to 100%.
Poor modelling practices were employed. The sponsor’s submitted model included numerous IFERROR and ISERROR statements, leading to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical; it remains unclear whether the model is running inappropriately by overriding errors.
CADTH was unable to address this limitation and notes that a thorough validation of the sponsor’s model was not possible.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
R-GemOx is a reasonable proxy for chemoimmunotherapy costs. | The clinical expert feedback received by CADTH noted that R-GemOx is not generally used in Canada. Furthermore, as a more aggressive chemoimmunotherapy treatment, it would typically be used in younger patients with good performance status and few comorbid illnesses. However, given that R-GemOx–specific inputs were primarily used as a proxy for chemotherapy costs in the model, the use of R-GemOx as a simplifying assumption was considered reasonable. |
Patients experiencing functional cure have a lower mortality rate than noncured patients, but a higher mortality rate than the general population (i.e., a 41% increase) due to long-term cancer complications. | Reasonable. |
The efficacy of subsequent treatment is implicitly captured in PFS and OS data and extrapolations. | The clinical expert feedback received by CADTH noted that the efficacy of subsequent treatments may be implicitly captured by the OS; however, there may be discrepancies for PFS. The impact on the cost-effectiveness of epcoritamab is unknown. |
CAR T-cell therapies are excluded as comparators in the base-case analysis. | Reasonable, based on the Health Canada indication. However, clinical expert feedback received by CADTH noted that CAR T-cell therapies may still be relevant comparators for some patients who did not receive such therapy previously due to logistical reasons (i.e., access issues) or choice. In the sponsor’s first key scenario analysis considering a subpopulation of patients with no prior CAR T-cell therapy, R-CIT, pola-BR, epcoritamab, and lisocabtagene maraleucel were on the cost-effectiveness frontier. Axicabtagene ciloleucel and tisagenlecleucel were dominated (i.e., were more costly and less effective) by epcoritamab. Compared to lisocabtagene maraleucel, epcoritamab was less costly and less effective (incremental QALYs = –0.53; incremental costs = –$351,238); therefore, the ICER for lisocabtagene maraleucel vs. epcoritamab was $667,197 per QALY gained. In the sponsor’s second key scenario assessing the cost-effectiveness of epcoritamab in a population of patients with no prior CAR T-cell therapy but who are eligible for CAR T-cell therapy, epcoritamab dominated (i.e., more QALYs, less costly) all 3 CAR T-cell therapies. |
CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin with bendamustine and rituximab; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH reanalysis case was derived by making changes in model parameter values and assumptions in consultation with clinical experts. The following changes were made to address several limitations: conducting pairwise analyses for epcoritamab versus pola-BR and R-CIT; removing the 3-year functional cure assumption; using the Weibull distribution to inform OS in the analysis versus R-CIT; assuming equal efficacy between epcoritamab versus pola-BR; and setting the RDI of drugs to 100%.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH reanalysis | ||
1. Comparators | Pola-BR | Pola-BR and R-CIT (pairwise analyses) |
2. Functional cure | After 3 years | Excluded |
3. Epcoritamab long-term survival (R-CIT analysis) | PFS: Lognormal OS: Lognormal | PFS: Lognormal OS: Weibull |
4. Clinical efficacy epcoritamab vs. pola-BR | OS HR = ||||| PFS HR = ||||| | OS HR = 1 PFS HR = 1 |
5. RDI | Epcoritamab: ||% Polatuzumab vedotin: 97% Bendamustine: 95% Rituximab: 95% | 100% for all |
CADTH reanalysis | ― | Reanalysis 2 + 4 + 5 (vs. pola-BR) Reanalysis 2 + 3 + 5 (vs. R-CIT) |
HR = hazard ratio; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-CIT = rituximab-chemotherapy; RDI = relative dose intensity; vs. = versus.
The CADTH reanalysis comparing epcoritamab to pola-BR found that epcoritamab was more costly (i.e., $278,990 versus $251,696) and associated with similar QALYs (i.e., 3.07 versus 3.07). Therefore, the ICER of epcoritamab was $4,989,538 per QALY gained compared to pola-BR. The difference in QALYs was due to modelled differences in adverse events. However, in the absence of direct or indirect comparative evidence, there is uncertainty as to whether the estimated differences in adverse events between epcoritamab and pola-BR would be expected in clinical practice.
In the CADTH reanalysis comparing epcoritamab to R-CIT, epcoritamab was more costly (i.e., $300,784 versus $150,374) and more effective (i.e., 2.21 versus 0.50 QALYs). Therefore, the ICER of epcoritamab compared to R-CIT is $87,735 per QALY gained based on the probabilistic analysis. The probability of cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY was 1%. Based on this pairwise analysis, patients receiving epcoritamab are expected to live approximately half a year longer (i.e., 0.83 LYs) after experiencing progression and discontinuing epcoritamab relative to patients receiving R-CIT. This benefit is due to the sponsor’s modelling approach; there is no robust evidence to support the validity of this postprogression benefit. CADTH noted that the probabilistic ICER differed from the deterministic ICER estimate (i.e., $87,735 versus $96,804 per QALY gained).
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Pairwise analysis 1 (epcoritamab vs. pola-BR; population of patients in the EPCORE NHL-1 trial) | ||||
Sponsor’s base case (probabilistic) | Pola-BR | 227,220 | 0.79 | Reference |
Epcoritamab | 267,749 | 4.18 | 11,938 | |
CADTH reanalysis 2 | Pola-BR | 228,522 | 0.65 | Reference |
Epcoritamab | 277,353 | 3.06 | 20,330 | |
CADTH reanalysis 4 | Pola-BR | 239,004 | 4.12 | Reference |
Epcoritamab | 268,127 | 4.13 | 5,281,469 | |
CADTH reanalysis 5 | Pola-BR | 230,389 | 0.76 | Reference |
Epcoritamab | 269,147 | 4.13 | 11,503 | |
CADTH reanalysis (reanalysis 2 + 4 + 5) (deterministically) | Pola-BR | 251,221 | 3.05 | Reference |
Epcoritamab | 278,373 | 3.06 | 4,923,900 | |
CADTH reanalysis (reanalysis 2 + 4 + 5) (probabilistic) | Pola-BR | 251,696 | 3.07 | Reference |
Epcoritamab | 278,990 | 3.07 | 4,989,538 | |
Pairwise analysis 2 (epcoritamab vs. R-CIT; subpopulation of patients with no prior CAR T-cell therapy) | ||||
Sponsor’s base case (probabilistic) | R-CIT | 150,297 | 0.52 | Reference |
Epcoritamab | 288,030 | 3.74 | 42,687 | |
CADTH reanalysis 2 | R-CIT | 150,811 | 0.48 | Reference |
Epcoritamab | 305,224 | 2.92 | 63,183 | |
CADTH reanalysis 3 | R-CIT | 150,512 | 0.50 | Reference |
Epcoritamab | 288,557 | 3.53 | 45,482 | |
CADTH reanalysis 5 | R-CIT | 150,506 | 0.50 | Reference |
Epcoritamab | 290,601 | 3.63 | 44,672 | |
CADTH reanalysis (reanalysis 2 + 3 + 5) (deterministically) | R-CIT | 150,680 | 0.46 | Reference |
Epcoritamab | 299,739 | 2.00 | 96,804 | |
CADTH reanalysis (reanalysis 2 + 3 + 5) (probabilistic) | R-CIT | 150,374 | 0.50 | Reference |
Epcoritamab | 300,784 | 2.21 | 87,735 | |
CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; pola-BR = polatuzumab vedotin with bendamustine and rituximab; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
Note: R-CIT was represented by R-GemOx.
Scenario Analysis Results
Several scenario analyses were conducted on the CADTH reanalysis to investigate the impact of specific drivers, such as using R-GDP to inform R-CIT costs, alternative epcoritamab time on treatment, alternative HRs for epcoritamab relative to R-CIT, the inclusion of CAR T-cell therapies as comparators, and the removal of postprogression benefit associated with epcoritamab. Results of these scenario analyses are presented in Appendix 4 (Table 15 and Table 16).
A scenario analysis in which the cost of R-CIT was informed by the R-GDP regimen resulted in an ICER of $95,805 per QALY gained for epcoritamab versus R-CIT. In the analysis in which the time on treatment for epcoritamab was set to be equal to PFS, the ICER was $109,940 per QALY gained for epcoritamab versus R-CIT. The scenario analysis in which the postprogression benefit of epcoritamab was removed (i.e., the QALYs accrued during the progressed health state for epcoritamab were set to be equal to the QALYs accrued during the progressed health state for R-CIT) resulted in an ICER of $120,435 per QALY gained for epcoritamab versus R-CIT. As noted previously, while clinical expert feedback received by CADTH noted that it is plausible that epcoritamab could be more effective that R-CIT, the magnitude of that benefit is unknown due to limitations in the available indirect comparisons. To explore the uncertainty around the HR used to inform R-CIT efficacy, CADTH conducted scenario analyses using HRs of 0.5 and 0.8 for OS and PFS, respectively, which increased the ICERs for epcoritamab versus R-CIT from $96,804 per QALY gained to $119,064 and $270,752, respectively.
CADTH undertook price reduction analysis based on the sponsor’s key scenario 1 results and CADTH’s reanalysis versus R-CIT. The CADTH reanalysis suggests that a price reduction of approximately 45% would be required to achieve cost-effectiveness of epcoritamab at a willingness-to-pay threshold of $50,000 per QALY gained versus R-CIT. When considering the scenario analysis in which the postprogression benefit of epcoritamab was removed, a price reduction of approximately 60% would be required for epcoritamab to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained versus R-CIT.
There is insufficient clinical evidence to justify a price premium for epcoritamab relative to the total cost of pola-BR. Based on the CADTH reanalysis, a price reduction of approximately 21% would be required for epcoritamab to achieve cost parity with pola-BR. CADTH notes that a price reduction may be required for epcoritamab to be no more costly than pola-BR, given that the prices informing pola-BR were based on public list prices and that epcoritamab does not have a set treatment duration, unlike other available treatments for this indication.
Table 7: CADTH Price Reduction Analyses
Analysis Price reduction | Unit drug costa ($) $ per 48 mg in 0.8 mL vial | ICERs for epcoritamab vs. R-CIT ($ per QALY) | |
|---|---|---|---|
Sponsor key scenario 1 | CADTH reanalysis | ||
No price reduction | 6,609 | 42,687 | 87,735 |
10% | 5,978 | Dominant | 79,291 |
20% | 5,287 | Dominant | 70,848 |
30% | 4,626 | Dominant | 62,404 |
40% | 3,965 | Dominant | 53,961 |
50% | 3,305 | Dominant | 45,517 |
60% | 2,644 | Dominant | 37,073 |
70% | 1,983 | Dominant | 28,630 |
80% | 1,322 | Dominant | 20,186 |
90% | 661 | Dominant | 11,743 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; R-CIT = rituximab-based chemoimmunotherapy; vs. = versus.
aEpcoritamab is available in 2 dosages. The 4 mg/0.8 mL vial costs $550.75 per vial; the 48 mg/0.8 mL vial costs $6,609.00 per vial. A similar price reduction would be required for the 4 mg unit cost.
Issues for Consideration
The presented results are based on publicly listed prices. Polatuzumab vedotin was recommended with the condition of a price reduction; the CADTH pan-Canadian Oncology Drug Review Expert Review Committee noted that the ICERs estimated by the sponsor and CADTH likely underestimated the ICER. CADTH identified substantial limitations in the sponsor’s economic evaluation that precluded a base case but noted that a price reduction of 35% to 84% may be required for polatuzumab vedotin to be considered cost-effective at a threshold of $50,000 per QALY gained.
Glofitamab recently underwent review by CADTH for the treatment of adult patients with R/R DLCBL not otherwise specified, DLBCL arising from follicular lymphoma, or PMBCL who have received 2 or more lines of systemic therapy and are ineligible to receive or cannot receive CAR T-cell therapy or have previously received CAR T-cell therapy following obinutuzumab pretreatment. The cost-effectiveness of epcoritamab versus glofitamab in this patient population is unknown. Note that glofitamab has a set treatment duration of 12 cycles, with a 28-day cycle cost ranging from $14,345 to $16,694. This includes the cost of pretreatment with obinutuzumab and any required premedications.
Study GCT3013 to 05, A Phase III Trial of Epcoritamab vs Investigator’s Choice Chemotherapy in R/R DLBCL (EPCORE DLBCL-1) is currently under way to address uncertainty in the clinical evidence and is expected to be completed within a time frame that will not exceed 3 years from the target expert committee date. Thus, CADTH conducted this review of epcoritamab as a time-limited recommendation on the basis that the sponsor has expressed a commitment to filing a reassessment application with CADTH within the time frames specified in the procedures for time-limited recommendations. CADTH notes that the studied population for the confirmatory trial differs slightly from the population for epcoritamab currently under review.
Overall Conclusions
The CADTH clinical review noted that the evidence from a phase I and II, single-arm, open-label trial (EPCORE NHL-1) in adult patients with R/R LBCL who have relapsed after or who did not respond to at least 2 prior systemic therapies showed that a clinically important proportion of patients treated with epcoritamab (||||%) achieved complete response. This aligns with clinical expert feedback received by CADTH that noted that the evidence from the EPCORE NHL-1 trial demonstrated that epcoritamab offers clinically meaningful improvements for this heavily pretreated patient population. While PFS and OS were evaluated in the EPCORE NHL-1 trial, due to the study design, CADTH could not accurately evaluate the impact of epcoritamab on these outcomes. Due to the lack of direct evidence, the effectiveness of epcoritamab versus relevant comparators (e.g., pola-BR and R-CIT) was assessed based on the sponsor-submitted ITCs, which were unanchored MAICs. CADTH identified important limitations with the ITCs, such that the results were considered uncertain. The clinical experts consulted by CADTH felt that it was plausible that treatment with epcoritamab could be superior to treatment with R-CIT for the target patient population, but that the magnitude of impact was uncertain. The clinical experts agreed that, given the uncertainty in the comparative evidence, there is no robust evidence that epcoritamab is more effective than pola-BR.
In line with the clinical evaluation, due to the different populations upon which the information was based, CADTH considered separate pairwise analyses comparing epcoritamab with pola-BR and with R-CIT.
In the absence of robust evidence to support an incremental benefit for epcoritamab, CADTH assumed equivalent efficacy between epcoritamab and pola-BR in the intention-to-treat population of the EPCORE NHL-1 trial. This resulted in similar QALYs and an incremental cost of $27,294 for epcoritamab compared with pola-BR. The key drivers in this cost difference were related to treatment acquisition and treatment-specific monitoring associated with epcoritamab.
When compared with R-CIT in a population with no prior CAR T-cell therapy use, epcoritamab was more costly and associated with increased LYs and QALYs in the CADTH reanalysis. The magnitude of the incremental benefit is associated with uncertainty due to limitations in the available clinical evidence and the sponsor’s modelling approach. Based on CADTH deterministic scenario analyses assessing alternate effect estimates, the incremental benefit may range from 1.54 QALYs (if the epcoritamab versus R-CIT HR equals ||||) to 0.50 QALYs (if the HR equals 0.8). In these instances, the ICER for epcoritamab ranges from $96,804 to $270,752 per QALY gained compared with R-CIT in patients who had not previously received CAR T-cell therapy. The key drivers affecting the ICER were the treatment acquisition costs associated with epcoritamab and the postprogression survival benefit for patients receiving epcoritamab. Using the |||| HR from the sponsor-submitted ITC for epcoritamab versus R-CIT and assuming a postprogression benefit for epcoritamab, a price reduction of at least 45% is required for epcoritamab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained compared to R-CIT. When considering alternate HRs, or when removing the postprogression survival benefit — in line with CADTH scenario analyses — a greater price reduction is required.
There is insufficient clinical evidence to justify a price premium for epcoritamab relative to the total cost of pola-BR. Based on the CADTH reanalysis, a price reduction of approximately 21% is required for epcoritamab to be no more costly than pola-BR. However, given that the prices informing pola-BR were based on the public price lists, and that epcoritamab does not have a set treatment duration compared to other available treatments for this indication, a greater price reduction may be required.
This review of epcoritamab was conducted as a time-limited recommendation by CADTH pending additional clinical studies to address uncertainty in the clinical evidence. When this information becomes available, CADTH will conduct a reassessment of the additional evidence (which may affect the assessment of comparative effectiveness and cost-effectiveness) and issue a final recommendation.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): AbbVie Corporation; 2023 Nov 14.
2.Epkinly (epcoritamab for injection): 4 mg in 0.8 mL (5 mg/mL) concentrate for solution for subcutaneous injection; or Epkinly (epcoritamab injection): 48 mg in 0.8 mL (60 mg/mL) solution for subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2023 Oct 13.
3.Drug Reimbursement Review clinical review, pharmacoeconomic review, ethics review, and stakeholder input: axicabtagene ciloleucel (Yescarta) for large B-cell lymphoma. Can J Health Technol. 2023;3(6). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0293r/PG0293r. Accessed 2024 Jan 14.
4.Drug Reimbursement Review clinical review, pharmacoeconomic review, ethics review, and stakeholder input: lisocabtagene maraleucel (Breyanzi) for relapsed or refractory large B-cell lymphoma. Can J Health Technol. 2022;2(10). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PG0258r/PG0258r. Accessed 2024 Jan 14.
5.Indirect treatment comparison for epcoritamab in patients with relapsed or refractory large B-cell lymphoma based on EPCORE NHL-1 trial [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): AbbVie Corporation; 2023 Mar 10.
6.National Institute for Health and Care Excellence. Polatuzumab vedotin with rituximab and bendamustine for treating relapsed or refractory diffuse large B-cell lymphoma. (Technology appraisal guidance TA649) 2020; https://www.nice.org.uk/guidance/ta649. Accessed 2024 Jan 15.
7.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800-1808. PubMed
8.Liebers N, Duell J, Fitzgerald D, et al. Polatuzumab vedotin as a salvage and bridging treatment in relapsed or refractory large B-cell lymphomas. Blood Adv. 2021;5(13):2707-2716. PubMed
9.Cancer Care Ontario (CCO). Drug formulary: Bend+Pola+Ritu. 2024; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/62866. Accessed 2024 Feb 01.
10.Columvi (glofitamab for injection): 1 mg/mL concentrate for solution for intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2023 Mar 24: https://pdf.hres.ca/dpd_pm/00070059.PDF. Accessed 2024 Feb 01.
11.Government of Alberta. Interactive drug benefit list. 2024; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2024 Jan 20.
12.Drug Reimbursement Expert Review Committee final recommendation: glofitamab (Columvi- Hoffmann-La Roche Limited). Can J Health Technol. 2024;4(2). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/PC0320/PC0320. Accessed 2024 Jan 18.
13.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2024 Jan 14.
14.GDP+RITU regimen: gemcitabine-dexamethasone-Platinol (CISplatin)-rituximab [regimen monograph]. Toronto (ON): Cancer Care Ontario (CCO); 2024: https://www.cancercareontario.ca/sites/ccocancercare/files/GDPRITU_HEM_NHL_HI_INT_A.pdf. Accessed 2024 Feb 01.
15.Cancer Care Ontario (CCO). Drug formulary: ICE+RITU. 2024; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/68001. Accessed 2024 Feb 01.
16.DHAP+RITU regimen: dexamethasone - high dose Ara-C (Cytarabine) - Platinol (CISplatin) - riTUXimab [regimen monograph]. Toronto (ON): Cancer Care Ontario (CCO); 2020: https://www.cancercareontario.ca/en/system/files_force/DHAPRITU_HEM_NHLINTHI_A.pdf?download=1. Accessed 2024 Feb 01.
17.Cancer Care Ontario (CCO). Drug formulary: GEMOX+RITU. 2024; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/67996. Accessed 2024 Feb 01.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for R/R DLBCL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | 28-day Cycle cost ($) |
|---|---|---|---|---|---|---|
Epcoritamab | ||||||
Epcoritamab (Epkinly) | 5 mg/ mL 60 mg/ mL | Vial Vial | 550.7500a 6,609.000a | Cycle 1: 0.16 mg on day 1, 0.8 mg on day 2, and 48 mg on day 15 and 22 Cycles 2 to 3: 48 mg on Day 1, 8, 15, and 22 Cycles 4 to 9: 48 mg once on Day 1 and 15 Cycles 10+: 48 mg on Day 1 | Cycle 1: 511.41 Cycles 2 to 3: 944.14 Cycles 4 to 9: 472.07 Cycles 10+: 236.04 | Cycle 1: 14,320 Cycles 2 to 3: 26,436 Cycles 4 to 9: 13,218 Cycles 10+: 6,609 |
Prednisone (generic) | 5 mg 50 mg | Tab | 0.0220 0.1735 | Premedication Cycle 1: 100 mg in before and for 3 consecutive days following each epcoritamab administration Cycle 2+: 100 mg before and for 3 consecutive days following each epcoritamab administrationb | Cycle 1: 0.20 Cycles 2 to 3: 0.20b Cycle 4 to 9: 0.10b Cycle 10+: 0.05b | Cycle 1: 6 Cycles 2+: 6b Cycles 4 to 9: 3b Cycle 10+: 1b |
Diphenhydramine | 50 mg/ mL | Vial | 4.0400c | Premedication Cycle 1: 50 mg before the epcoritamab administration | Cycle 1: 2.77 | Cycle 1:162 |
Epcoritamab (regimen cost) | Cycle 1: 517.38 Cycle 2 to 3: 944.34 Cycle 4 to 9: 472.17 Cycle 10+: 236.09 | Cycle 1: 14,487 Cycle 2 to 3: 26,442 Cycle 4 to 9: 13,221 Cycle 10+: 6,610 | ||||
Glofitamabd | ||||||
Glofitamab (Columvi) | 2.5 mg/ mL 10 mg/ mL | Vial Vial | 1,040.0000e 4,160.0000e | Cycle 1: 2.5 mg on day 8 and 10 mg on day 15 Cycles 2 to 12: 30 mg on day 1 | Cycle 1: 247.62 Cycles 2 to 12: 594.29 | Cycle 1: 6,933 Cycles 2 to 12: 16,640 |
Obinutuzumab (Gazyva) | 25 mg/ mL | Vial | 5,477.8400f | Premedication Cycle 1: 1,000 mg on day 1 | Cycle 1: 260.85 | Cycle 1: 7,304 |
Diphenhydramine | 50 mg/ mL | Vial | 4.0400c | Premedication Cycle 1: 50 mg on day 8 and 15 Cycles 2 to 3: 50 mg on day 1 Cycles 4 to 12: 50 mg before glofitamab infusion | Cycle 1: 3.85 Cycles 2 to 12: 1.92 | Cycle 1: 108 Cycles 2 to 12: 54 |
Glofitamab regimen | Cycle 1: 512.31 Cycle 2 to 3: 596.21 Cycle 4 to 12: 594.29 | Cycle 1: 14,345 Cycle 2 to 3: 16,694 Cycle 4 to 12: 16,640 | ||||
Pola-BR | ||||||
Bendamustine (generic) | 25 mg vial 100 mg vial | Lyophilized powder | 250.0000f 1,000.0000f | 90 mg/ m2on day 1 and 2 every 21 days | 166.67 | 4,667 |
Polatuzumab Vedotin (Polivy) | 140 mg/ mL | Vial | 14,750.0000f | 1.8 mg/ kg on day 1 every 21 days | 702.38 | 19,667 |
Rituximab (biosimilar) | 100 mg/ 10 mL 500 mg/ 50 mL | Vial Vial | 297.0000 1,485.0000 | 375 mg/ m2 on day 1 every 21 days | 99.00 | 2,772 |
Pola-BR regimen | 968.05 | 27,105 | ||||
Note: All prices are from the Ontario Drug Benefit Formulary (accessed January 2024), unless otherwise indicated, and do not include dispensing fees. Calculations assume patient weight and body surface area of 75 kg and 1.8 m2, respectively. Recommended dosage informed from respective product monographs, unless otherwise indicated.9,10
aSponsor’s pharmacoeconomic submission.1
bFor patients who experienced grade 2 or 3 CRS with previous dose.
cAlberta Blue Cross formulary (access January 2024).11
dAdditional premedication associated with Glofitamab include prednisone and acetaminophen. Costs associated with these products are not presented above due to per 28-day costs being < $1.
eCADTH review of glofitamab.12
fCost informed from Delta PA (accessed January 2024).13
Table 9: CADTH Cost Comparison Table for R/R DLBCL (Salvage Chemotherapy)
Treatment | Strength / concentration | Form | Price | Recommended dosagea | Daily cost | 28-day cycle cost |
|---|---|---|---|---|---|---|
R-GDP | ||||||
Rituximab (biosimilar) | 100 mg/ 10 mL 500 mg/ 50 mL | Vial Vial | 297.0000 1,485.0000 | 375 mg/m2 on day 1 every 21 days | 99.00 | 2,772 |
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000b 540.0000b | 1,000 mg/m2 on days 1 and 8 every 21 days | 51.43 | 1,440 |
Dexamethasone (generics) | 4 mg | Tab | 0.6112 | 40 mg PO days 1 to 4 every 21 days | 1.16 | 33 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/ mL IV solution | 135.0000b 270.0000b | 75 mg/m2 IV on day 1 every 21 days | 19.29 | 540 |
R-GDP regimen | 170.88 | 4,785 | ||||
R-ICE | ||||||
Rituximab (biosimilar) | 100 mg/ 10 mL 500 mg/ 50 mL | Vial Vial | 297.0000 1,485.0000 | 375 mg/m2 IV on day 1 every 21 days | 99.00 | 2,772 |
Ifosfamide (Ifex) | 1,000 mg vial 3,000 mg vial | Powder for solution | 143.8700b 440.5899b | 1,667 mg/m2 days 1 to 3 every 21 days | 61.66 | 1,726 |
Carboplatin (generic) | 50 mg 150 mg 450 mg 600 mg | 10 mg/mL vial for injection | 70.0000b 210.0000b 600.0000b 775.0020b | AUC 5 on day 1; maximum dose for AUC 5 is 750 mg every 21 days | 46.90 | 1,313 |
Etoposide (generic) | 100 mg 200 mg 500 mg 1,000 mg | 20 mg/mL IV solution | 75.0000b 150.0000b 375.0000b 750.0000b | 100 mg/m2 days 1 to 3 every 21 days | 21.43 | 600 |
R-ICE regimen | 228.99 | 6,412 | ||||
R-DHAP | ||||||
Rituximab (biosimilar) | 100 mg/ 10 mL 500 mg/ 50 mL | Vial Vial | 297.0000 1,485.0000 | 375 mg/ m2 on day 1 every 21 or 28 days | 99.00 | 2,772 |
Dexamethasone (generics) | 4 mg | Tab | 0.6112 | 40 mg PO days 1 to 4 every 21 or 28 days | 0.87 | 24 |
Cytarabine (generic) | 500 mg vial 2,000 mg vial | 100 mg/mL IV solution | 76.8500b 306.5000b | 2,000 mg/ m2 IV every 12 hours on day 2 every 21 or 28 days | 43.85 | 1,228 |
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/ mL IV solution | 135.0000b 270.0000b | 100 mg/ m2 on day 1 every 21 or 28 days | 19.29 | 540 |
R-DHAP regimen | 138.26 | 3,871 | ||||
R-GemOx | ||||||
Gemcitabine (generics) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000b 540.0000b | 1,000 mg/m2 on day 1 every 14 days | 38.57 | 1,080 |
Oxaliplatin (generics) | 50 mg 100 mg 200 mg | 5 mg/ mL IV solution | 45.0000b 90.0000b 180.0000b | 100 mg/m2 on day 1 every 14 days | 12.86 | 360 |
Rituximab (biosimilar) | 100 mg/ 10 mL 500 mg/ 50 mL | Vial Vial | 297.0000 1,485.0000 | 375 mg/m2 on day 1 every 14 days | 148.50 | 4,158 |
R-GemOx regimen | 199.93 | 5,598 | ||||
All costs are from the Ontario Drug Benefit Formulary (accessed January 2024), unless otherwise indicated. Dispensing fees are not included. Calculations assume patient weight and body surface area of 75 kg and 1.8 m2, respectively.
aRecommended dosing informed by Ontario Cancer Care dosing regimens, unless otherwise stated.14-17
bCost informed from Delta PA (accessed January 2024).
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH appraisal. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CADTH appraisal. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CADTH appraisal. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Base Case Versus Pola-BR
Parameter | Epcoritamab | Pola-BR |
|---|---|---|
Discounted LYs | ||
Total | 5.32 | 1.05 |
Progression-free | 4.62 | 0.82 |
Progressed | 0.71 | 0.22 |
Discounted QALYs | ||
Total | 4.18 | 0.79 |
Progression-free | 3.70 | 0.65 |
Progressed | 0.49 | 0.16 |
Adverse events | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 267,749 | 227,220 |
Drug related | 134,359 | 87,115 |
Total drug acquisition | 126,016 | 78,061 |
Total drug administration | 1,138 | 1,131 |
Treatment-specific monitoring | 7,206 | 7,923 |
Disease management | 6,574 | 2,414 |
Progression-free | 2,064 | 991 |
Progressed | 4,510 | 1,423 |
Subsequent treatment | 105,067 | 116,413 |
Drug acquisition | 104,665 | 115,999 |
Drug administration | 402 | 415 |
Drug monitoring | 0 | 0 |
Other costs | 21,749 | 21,278 |
One-time drug, admin, monitoring | 3,042 | 501 |
Disease progression | 2,666 | 2,922 |
Terminal care | 13,775 | 15,241 |
Adverse events | 2,266 | 2,614 |
LY = life year; pola-BR = polatuzumab vedotin with bendamustine and rituximab; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Key Scenario 1 Versus R-CIT
Parameter | Epcoritamab | R-CIT |
|---|---|---|
Discounted LYs | ||
Total | 4.81 | 0.71 |
Progression-free | 3.96 | 0.38 |
Progressed | 0.85 | 0.33 |
Discounted QALYs | ||
Total | 3.74 | 0.52 |
Progression-free | 3.17 | 0.29 |
Progressed | 0.58 | 0.23 |
Adverse events | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 288,030 | 150,319 |
Drug related | 151,747 | 22,724 |
Total drug acquisition | 142,136 | 13,546 |
Total drug administration | 1,268 | 907 |
Treatment-specific monitoring | 8,343 | 8,272 |
Disease management | 7,524 | 2,714 |
Progression-free | 2,125 | 588 |
Progressed | 5,399 | 2,126 |
Subsequent treatment | 106,618 | 103,755 |
Drug acquisition | 106,210 | 103,654 |
Drug administration | 408 | 101 |
Drug monitoring | 0 | 0 |
Other costs | 22,142 | 21,104 |
One-time drug, admin, monitoring | 3,042 | 364 |
Disease progression | 2,759 | 2,989 |
Terminal care | 14,057 | 15,427 |
Adverse events | 2,284 | 2,323 |
LY = life year; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 13: Disaggregated Summary of the CADTH Reanalysis — Versus Pola-BR
Parameter | Epcoritamab | Pola-BR |
|---|---|---|
Discounted LYs | ||
Total | 4.23 | 4.23 |
Progression-free | 3.26 | 3.26 |
Progressed | 0.97 | 0.97 |
Discounted QALYs | ||
Total | 3.07 | 3.07 |
Progression-free | 2.41 | 2.42 |
Progressed | 0.67 | 0.67 |
Adverse events | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 278,990 | 251,696 |
Drug related | 135,560 | 109,514 |
Total drug acquisition | 127,166 | 98,314 |
Total drug administration | 1,140 | 1,376 |
Treatment-specific monitoring | 7,254 | 9,823 |
Disease management | 12,445 | 12,445 |
Progression-free | 6,224 | 6,224 |
Progressed | 6,221 | 6,221 |
Subsequent treatment | 108,624 | 109,595 |
Drug acquisition | 108,211 | 109,207 |
Drug administration | 413 | 388 |
Drug monitoring | 0 | 0 |
Other costs | 22,361 | 20,143 |
One-time drug, admin, monitoring | 3,042 | 501 |
Disease progression | 2,794 | 2,794 |
Terminal care | 14,245 | 14,245 |
Adverse events | 2,280 | 2,603 |
LY = life year; pola-BR = polatuzumab vedotin with bendamustine and rituximab; QALY = quality-adjusted life-year.
Table 14: Disaggregated Summary of the CADTH Reanalysis — Versus R-CIT [R-GemOx], No Prior CAR T-Cell Therapy Population
Parameter | Epcoritamab | R-CIT |
|---|---|---|
Discounted LYs | ||
Total | 3.05 | 0.70 |
Progression-free | 1.83 | 0.31 |
Progressed | 1.22 | 0.39 |
Discounted QALYs | ||
Total | 2.21 | 0.50 |
Progression-free | 1.38 | 0.24 |
Progressed | 0.84 | 0.27 |
Adverse events | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 300,784 | 150,374 |
Drug related | 152,988 | 22,277 |
Total drug acquisition | 143,366 | 13,256 |
Total drug administration | 1,271 | 890 |
Treatment-specific monitoring | 8,350 | 8,132 |
Disease management | 11,265 | 3,060 |
Progression-free | 3,486 | 591 |
Progressed | 7,779 | 2,469 |
Subsequent treatment | 113,500 | 103,939 |
Drug acquisition | 113,062 | 103,839 |
Drug administration | 437 | 101 |
Drug monitoring | 0 | 0 |
Other costs | 23,032 | 21,098 |
One-time drug, admin, monitoring | 3,042 | 364 |
Disease progression | 2,915 | 2,984 |
Terminal care | 14,802 | 15,434 |
Adverse events | 2,273 | 2,310 |
LY = life year; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
Scenario Analyses
Table 15: Summary of CADTH’s Economic Evaluation Results — Alternative HR Scenario Analyses for Epcoritamab Versus R-CIT
Drug | Total costs ($) | Total QALYs ($) | ICER ($ per QALY gained) |
|---|---|---|---|
CADTH base case (HR = |||||) | |||
R-CIT | 150,680 | 0.46 | — |
Epcoritamab | 299,739 | 2.00 | 96,804 |
Epcoritamab vs. R-CIT HR alternative 1 (HR = 0.5) | |||
R-CIT | 157,732 | 0.81 | Reference |
Epcoritamab | 299,739 | 2.00 | 119,064 |
Epcoritamab vs. R-CIT HR alternative 2 (HR = 0.8) | |||
R-CIT | 164,762 | 1.50 | Reference |
Epcoritamab | 299,739 | 2.00 | 270,752 |
HR = hazard ratio; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy.
Note: analyses were conducted deterministically.
Table 16: Summary of CADTH’s Economic Evaluation Results — Scenario Analysis for Epcoritamab Versus R-CIT, No Prior CAR T-Cell Therapy Population
Drug | Total costs ($) | Total QALYs ($) | ICER ($ per QALY gained) |
|---|---|---|---|
R-CIT Costs informed by R-GDP | |||
R-CIT | 145,254 | 0.48 | Reference |
Epcoritamab | 300,856 | 2.10 | 95,805 |
Epcoritamab time on treatment equal to PFS | |||
R-CIT | 150,561 | 0.49 | Reference |
Epcoritamab | 337,812 | 2.19 | 109,940 |
Epcoritamab postprogression benefit removeda | |||
R-CIT | 150,680 | 0.46 | Reference |
Epcoritamab | 299,739 | 1.70 | 120,435 |
CAR = chimeric antigen receptor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; R-CIT = rituximab-chemotherapy.
Note: analyses were conducted probabilistically.
aconducted on the deterministic analysis, where the QALYs for epcoritamab in the progressed health state were set equal to QALYs in the progressed health state of R-CIT.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Please note that this appendix has not been copy-edited.
Table 17: Summary of Key Takeaways
Key Takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing epcoritamab for the treatment of adult patients with LBCL after 2 or more lines of systemic therapy and who have previously received or unable to receive CAR T-cell therapy. The BIA was undertaken from the perspective of a Canadian public drug plan over a 3-year time horizon (2024 to 2026) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. Data informing the model were obtained from various sources, including the published literature, the sponsor’s internal data, and input from clinical experts consulted by the sponsor.
The sponsor stratified their population based on CAR T-cell access and eligibility, given access to CAR T-cell varies by province. Furthermore, the sponsor’s BIA accounted for attrition due to following CAR T-cell access. The comparators included in the reference scenario with market share were pola-BR and R-GemOx. A scenario which included market shares for CAR T-cell therapies was also conducted by the sponsor. Key inputs to the BIA are documented in Table 19.
Key assumptions included:
It was assumed that if a province funds CAR T-cell therapy, 70% of eligible patients will receive CAR T-cell therapy, and 50% of eligible patients residing in provinces where CAR T-cell is not funded will receive access through out of province funding mechanisms.
Provincial coverage was assumed to be 100% given epcoritamab is anticipated to be administered in the hospital setting initially.
It was assumed R-GemOx was representative of available rituximab-chemoimmunotherapy treatment options.
Epcoritamab is expected to capture market share from all comparators, but primarily from pola-BR.
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Estimated population size | 30,514,175 |
Annual NHL incidence | 0.036% |
Proportion with LBCL | 39.5% |
LBCL patients receiving first-line therapy | ||% |
Proportion of patients who relapse and receive second-line therapy | ||||% |
Proportion of patients who relapse and receive third-line therapy | ||||% |
Proportion of patients medically eligible for CAR T-cell therapy in the third-line setting | 70% |
Provincial funding (weighted average based on provinces with and without CAR T-cell therapy funding) | 69% |
Proportion of patients relapsing after CAR T-cell therapy | 50% |
Proportion of patients receiving fourth line plus therapy post CAR T-cell therapy | 50% |
Proportion not able to receive CAR T-cell that receive a third-line plus therapy | 100% |
Drug plan eligibility for epcoritamab | 100% |
Number of patients eligible for drug under review | 337 / 347 / 352 |
Market uptake (3 years) | |
Uptake (reference scenario) Pola-BR R-GemOx | ||% / ||% / ||% ||% / ||% / ||% |
Uptake (new drug scenario) Epcoritamab Pola-BR R-GemOx | 25% / 50% / 70% ||||% / ||% / ||% ||||% / ||% / ||% |
Cost of treatment (per patient) | |
Cost of treatment over expected duration of treatmenta Epcoritamab Pola-BR R-GemOx | $165,294 $110,338 $11,508 |
CAR = chimeric antigen receptor; LBCL = large B-cell lymphoma; NHL = non-Hodgkin lymphoma; pola-BR = polatuzumab vedotin with bendamustine and rituximab; R-GemOx = rituximab, gemcitabine, and oxaliplatin.
aDuration of treatment were set to predicted mean time to treatment discontinuation from the sponsor submitted pharmacoeconomic model.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing epcoritamab for the treatment of adult patients with LBCL after 2 or more lines of systemic therapy and who have previously received or unable to receive CAR T-cell therapy would be $28,316,967 (year 1: $2,217,979; year 2: $9,485,790; year 3: $16,613,198).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The proportion of patients who receive third-line therapy is underestimated. The sponsor estimated that ||||% of patients receive third-line therapy after relapsing based on an ONCO-CAPPS analysis. However, clinical expert feedback received by CADTH noted this does not align with clinical expectation and is expected to be higher as it is anticipated that approximately 50% of patients who relapse after second-line salvage chemotherapy will proceed to autologous stem cell transplant (ASCT), with the remaining being eligible for other third-line treatments. And of the patients who proceed to ASCT, the majority of patients who undergo ASCT will relapse.
CADTH address this limitation by revising the proportion of patients who relapse and receive third-line therapy to 50%.
Uncertainty in the proportion of patients who relapse after CAR T-cell therapy. The sponsor estimated that 50% of patients would relapse following CAR T-cell therapy. Clinical expert feedback received by CADTH noted that there is uncertainty in the estimate which could be as high as 70%.
CADTH conducted a scenario analysis where the proportion of patients who relapse after CAR T-cell therapy is set to 70%.
Exclusion of premedication drug costs associated with epcoritamab is not appropriate. Costs included in the sponsor’s BIA included only drug acquisition costs associated with epcoritamab, pola-BR, R-GemOx, and CAR T-cell therapies. However, as noted in the product monograph of epcoritamab, epcoritamab is associated with premedication where the cost which would be incurred by the public drug plans. Therefore, exclusion of such costs may underestimate the total treatment costs associated with epcoritamab biasing results in favour of epcoritamab.
CADTH included premedication costs associated with epcoritamab.
R-GemOx as a proxy for all chemoimmunotherapies costs may not be reflective of Canadian clinical practice. In the sponsor’s submitted BIA, the sponsor assumed that there is no difference in the effectiveness of the common chemotherapy regimens in Canada. As such R-GemOx was used as a proxy to inform chemoimmunotherapies in the base case. While clinical expert feedback received by CADTH agreed that assuming no difference in common chemotherapy regimens in Canada was a reasonable simplifying assumption, it was noted that R-GemOx is not generally used in Canada as it is a more aggressive chemoimmunotherapy that is minorly used in younger patients with good performance status and few comorbidities and thus R-GDP may be more appropriate to consider.
CADTH addressed this limitation by incorporating R-GDP where it was assumed that the salvage chemotherapy distribution consisted of 50% R-GDP and 50% R-GemOx.
CADTH Reanalyses of the BIA
Table 19: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Proportion of patients who relapse and receive third-line therapy | ||||% | 50% |
2. Epcoritamab premedication costs | Excluded | Included |
3. Salvage chemotherapy costs | Informed by R-GemOx | Informed by 50% R-GemOx and 50% R-GDP |
CADTH base case | reanalysis 1 + 2 + 3 | |
R-GemOx = rituximab, gemcitabine, and oxaliplatin; R-GDP = rituximab plus gemcitabine, dexamethasone, and cisplatin.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 20 and a more detailed breakdown is presented in Table 21.
Based on the CADTH base case, the estimated incremental budget impact of reimbursing epcoritamab is $3,478,047 in year 1, $14,752,278 in year 2, and $25,799,166 in year 3. Therefore, the 3-year total budget impact is $44,029,491. The scenario analysis where the proportion of patients who relapse after CAR T-cell therapy was set to 70% resulted in a 3-year budget impact of $47,363,841.
Table 20: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $28,316,967 |
CADTH reanalysis 1 | $43,699,023 |
CADTH reanalysis 2 | $28,401,141 |
CADTH reanalysis 3 | $28,446,936 |
CADTH base case | $44,029,491 |
BIA = budget impact analysis.
Table 21: Detailed Breakdown of the CADTH Reanalyses of the BIA
Analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $16,712,340 | $18,730,123 | $22,381,153 | $24,455,509 | $65,566,785 |
New drug | $16,712,340 | $20,948,102 | $31,866,943 | $41,068,707 | $93,883,752 | |
Budget impact | $0.00 | $2,217,979 | $9,485,790 | $16,613,198 | $28,316,967 | |
CADTH base case | Reference | $25,524,957 | $28,658,653 | $34,327,168 | $37,548,596 | $100,534,417 |
New drug | $25,524,957 | $32,136,700 | $49,079,446 | $63,347,762 | $144,563,908 | |
Budget impact | $0.00 | $3,478,047 | $14,752,278 | $25,799,166 | $44,029,491 | |
CADTH scenario analysis: proportion of patients who relapse after CAR T-cell therapy | Reference | $27,457,816 | $30,828,872 | $36,926,783 | $40,392,239 | $108,147,894 |
New drug | $27,457,816 | $34,570,299 | $52,796,228 | $68,145,208 | $155,511,735 | |
Budget impact | $0.00 | $3,741,427 | $15,869,445 | $27,752,969 | $47,363,841 | |
CADTH scenario analysis: 45% price reduction | Reference | $25,524,957 | $28,658,653 | $34,327,168 | $37,548,596 | $100,534,417 |
New drug | $25,524,957 | $25,932,654 | $32,914,178 | $38,221,187 | $97,068,019 | |
Budget impact | $0.00 | -$2,725,999 | -$1,412,990 | $672,590 | -$3,466,399 |
BIA = budget impact analysis; CAR = chimeric antigen receptor.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.