Drugs, Health Technologies, Health Systems
Reimbursement Review
Capivasertib (Truqap)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Hormone receptor–positive, human epidermal growth factor receptor 2–negative, locally advanced or metastatic breast cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Testing Procedure Assessment Review
Clinical Review
Abbreviations
AE
adverse event
AI
aromatase inhibitor
BICR
blinded independent central review
CBCN
Canadian Breast Cancer Network
CDA-AMC
Canada’s Drug Agency
CDK4/6
cyclin-dependent kinase 4 and 6
CI
confidence interval
CrI
credible interval
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-BR23
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ER
estrogen receptor
ET
endocrine therapy
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HER2
human epidermal growth factor receptor 2
HR
hormone receptor
HRQoL
health-related quality of life
IHC
immunohistochemistry
IQR
interquartile range
ITT
intention to treat
KM
Kaplan-Meier
MID
minimal important difference
NGS
next-generation sequencing
NMA
network meta-analysis
OH-CCO
Ontario Health Cancer Care Ontario
OS
overall survival
PH
proportional hazards
PFS
progression-free survival
PFS2
time from randomization to second progression on next-line treatment or death because of any cause
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
REAL
Research Excellence Active Leadership
RCT
randomized controlled trial
SAE
serious adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Capivasertib (Truqap) 400 mg (2 tablets of 200 mg each) taken orally twice daily for 4 days followed by 3 days off treatment in combination with fulvestrant 500 mg, administered intramuscularly on days 1, 15, and 29, then monthly thereafter |
Sponsor | AstraZeneca Canada Inc. |
Indication | Capivasertib is indicated in combination with fulvestrant for the treatment of adult females with hormone receptor–positive, human epidermal growth factor receptor 2–negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing, adjuvant therapy. |
Reimbursement request | Capivasertib in combination with fulvestrant for the treatment of adult patients with hormone receptor–positive, human epidermal growth factor receptor 2–negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy |
Health Canada approval status | Approved |
Health Canada review pathway | Standard and Project Orbis Type A |
NOC date | January 26, 2024 |
Recommended dose | Capivasertib: until disease progression or unacceptable toxicity |
NOC = Notice of Compliance.
Introduction
Breast cancer was the second most-diagnosed cancer in Canada in 2023 and the most prevalent among females, with projected estimates of about 29,700 new cases in the overall population in 2023 (29,400 in females and 260 in males).1 The 5-year prevalence of breast cancer in females reported in Canada in 2018 was 110,955 patients,2 equating to a 5-year prevalence rate of 0.73%.3 Breast cancer is a heterogeneous disease,4,5 classified into subtypes based on specific cell types affected, gene expression, and receptors expressed on the surface of or inside tumour cells. Hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative breast cancer subtypes are the most prevalent in North America, accounting for at least 60% to 70% of all breast cancer cases.6 Disease staging follows the American Joint Committee on Cancer tumour, node, metastasis system.7 Tumour biopsy with pathology review and biomarker assessment (e.g., including HR and HER2 status) are completed for confirmatory diagnosis and to determine disease subtype and guide treatment decision-making.7,8
Signs and symptoms vary by disease stage and may include swelling in the surrounding lymph nodes, nipple changes (e.g., discharges), skin changes (e.g., erythema, skin ulcers, eczema), breast pain or heaviness, or other persistent changes in the breast.9,10 Metastatic, HR-positive, HER2-negative breast cancer also negatively affects patient quality of life because the symptoms that manifest are the result of disease progression and administered treatments. Common symptoms reported include pain, fatigue, nausea, vomiting, cognitive problems, depression, hair loss, lymphedema, sleep disturbances, loss of appetite, anxiety, and sexual dysfunction.11-13
Five percent to 10% of genetic alterations are inherited from a parent.14 Genetic alterations can also be acquired during tumour development; these are often known as somatic alterations. Somatic alterations of interest to this review are in the PI3K, AKT, or mTOR pathway, which is a cell-signalling pathway regulating cell proliferation and survival. Alterations in the PI3K, AKT, or mTOR signalling axis are observed in up to 48% of all patients with HR-positive, HER2-negative breast cancer.15,16 In HR-positive, HER2-negative breast cancers, PI3K, AKT, or mTOR pathway activation most frequently arises from PIK3CA alterations, occurring in approximately 30% of patients.17-21 A further approximately 4% of advanced breast cancers harbour AKT1-activating alterations or amplifications, and approximately 5% have inactivating alterations in PTEN.17,22,23 Survival outcomes following progression on endocrine-based therapies diminish significantly with later lines of single-drug chemotherapy, with median progression-free survival (PFS) and overall survival (OS) estimated to be as low as 3 months and 7 months, respectively, for patients treated with 5 lines of chemotherapy, while median PFS and OS are estimated to be around 7.5 months and 13.5 months, respectively, after the initiation of a second line of chemotherapy.24
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of capivasertib (Truqap) 400 mg, taken orally twice daily for 4 days, followed by 3 days off treatment, in combination with fulvestrant 500 mg, administered intramuscularly every 14 days after the first 3 injections and every 28 days thereafter for the treatment of adults with locally advanced or metastatic breast cancer.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the call for input from Canada’s Drug Agency (CDA-AMC) and by clinical expert(s) we consulted for the purpose of this review.
Patient Input
Two patient groups, the Canadian Breast Cancer Network (CBCN) and Rethink Breast Cancer, provided input for this review. Information from the CBCN group was sourced from 3 online surveys: the CBCN 2022 Triple Negative Breast Cancer Patient Survey (981 participants, 31 of whom had metastatic, HR-positive breast cancer), the CBCN 2017 Metastatic Breast Cancer Patient Survey (180 metastatic patients, 38 of whom had metastatic, HR-positive breast cancer), and the CBCN 2012 Metastatic Breast Cancer Patient and Caregiver Survey Report (71 patients and 16 caregivers). No patients taking the drug under review participated in these surveys.
Information from Rethink Breast Cancer was gathered through programming and meetings with patients with breast cancer and an online survey of 78 patients living with metastatic breast cancer, which ran from September 2018 to April 2019. Rethink Breast Cancer also conducted interviews with 5 patients (4 from the US and 1 from Canada) living with HR-positive, HER2-negative, metastatic breast cancer. The 4 patients in the US had experience taking capivasertib for HR-positive, HER2-negative, metastatic breast cancer. The patient in Canada reported taking a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor and having a PIK3CA mutation.
The 2 groups highlighted that metastatic disease poses a significant or debilitating impact on patients’ quality of life. Rethink Breast Cancer stated that breast cancer may have greater emotional effects and lifestyle impacts on younger patients, especially those diagnosed in their twenties, thirties, and early forties, because women in these age groups are more likely to face fertility or family-planning challenges, diagnosis during pregnancy, demands of childcare, and impacts on relationships, body image, dating, and sexuality. These impacts can leave them feeling isolated from peers who do not have cancer. They may also experience career hiatuses and financial insecurity. The CBCN noted similar issues.
The CBCN highlighted that current treatment goals for patients with metastatic breast cancer include controlling the progression of the disease (i.e., extending life) and reducing cancer-related symptoms (i.e., extending or stabilizing quality of life). They further noted that patients diagnosed with HR-positive, HER2-negative, metastatic breast cancer have limited options for targeted treatments in addition to poor prognoses and poor survival outcomes.
Rethink Breast Cancer stated that patients go to great lengths to avoid standard chemotherapy and suffer both emotionally and physically for this reason. The group added that patients on standard chemotherapy have a lot of difficulty managing their illnesses. Rethink Breast Cancer indicated that the primary improvement that patients with metastatic breast cancer seek is to extend their lives beyond what is expected with the help of currently available, publicly funded therapies and to enjoy better quality of life.
Rethink Breast Cancer noted that all 4 patients who had taken the drug under review highlighted the importance of having access to new therapies that have the possibility of extending their lives. Three of these patients shared that they are experiencing good quality of life while taking capivasertib, continuing to work, enjoy time with loved ones, and live their lives.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts indicated that, because the treatment goal for patients is palliative, the unmet needs of patients are for new treatments that would delay progression, prolong OS, and improve quality of life while exposing them to minimal toxicity. The experts noted that patients become refractory to current treatment options, and subsequent therapy is limited to chemotherapy, which has significant impacts on quality of life and resource utilization. The clinical experts agreed that capivasertib plus fulvestrant would be used in the second-line setting, and that it would alter the current treatment paradigm because there are currently no targeted treatments in the second-line setting for most patients. The clinical experts indicated that the patients best suited for capivasertib plus fulvestrant would be those eligible for second-line therapy following treatment with an aromatase inhibitor (AI) and CDK4/6 inhibitor. The experts highlighted that, in their local practices, they rarely test for PIK3CA, AKT1, or PTEN alterations (outside of clinical trials) because testing is not funded, given that no publicly funded treatments require this companion diagnostic. The clinical experts indicated that in clinical practice, a combination of radiography (approximately every 3 months) and biochemical and clinical parameters are used to determine whether a patient is responding to or progressing on treatment. The experts agreed that a clinically meaningful response includes radiological response or stabilization, improvement in patient symptoms, and maintenance of health-related quality of life (HRQoL). The clinical experts indicated that treatment with capivasertib plus fulvestrant should be discontinued if a patient experiences disease progression (defined radiologically or clinically), cannot tolerate treatment, or prefers to discontinue. The clinical experts noted that patients receiving capivasertib plus fulvestrant should be under the care of a medical oncologist in their community who can manage the toxicities associated with the therapy. They noted that it would be reasonable for patients to receive the therapy at a distributed oncology centre where day-to-day follow-up is with a general practitioner in oncology.
Clinician Group Input
Input for the review of capivasertib was received from 2 clinician groups: the Research Excellence Active Leadership (REAL) Canadian Breast Cancer Alliance and the Ontario Health Cancer Care Ontario (OH-CCO) Breast Cancer Drug Advisory Committee. A total of 13 clinicians (8 from the REAL alliance and 5 from the OH-CCO committee) provided input for this submission.
Both emphasized that the primary goals of systemic treatment for advanced breast cancer are to improve or prolong survival, maintain or improve quality of life, manage or minimize toxicities associated with treatment, alleviate symptoms, and delay the initiation of chemotherapy. The REAL group emphasized that treatment options with survival benefit and good tolerability are limited for patients in the second-line setting (i.e., those who have relapsed on first-line therapy in the metastatic setting) and patients who relapse while on, or within 12 months of completing, adjuvant endocrine therapy (ET). Similarly to the clinical experts consulted by CDA-AMC, the group further indicated that treatment goals that are not being met by currently available treatments in this population are improving OS, maintaining of quality of life, minimizing toxicities, and delaying the start of chemotherapy. They also noted that not all patients respond to available treatments, and that patients may become refractory to current treatment options; thus, additional treatment options might be needed for these patients.
While the OH-CCO Breast Cancer Drug Advisory Committee indicated that the drug under review would add an additional line of endocrine-based therapy, the REAL group recommended it as a treatment option for all patients (males and premenopausal, perimenopausal, and postmenopausal females) who have HR-positive, HER2-negative, metastatic breast cancer and have progressed on first-line, standard of care treatment in the metastatic setting or have progressed while on, or within 12 months of completing, adjuvant ET and have 1 or more PIK3, AKT, or PTEN alterations.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CDA-AMC recommendation for capivasertib plus fulvestrant:
relevant comparators
consideration for initiation of therapy
consideration for discontinuation of therapy
considerations for prescribing of therapy
generalizability
care provision issues
system and economic issues.
The clinical experts we consulted provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, phase III, randomized controlled trial (RCT), the CAPItello-291 trial (N = 708), met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the CAPItello-291 trial was to assess the efficacy and safety of capivasertib plus fulvestrant compared with matched placebo plus fulvestrant in adults with locally advanced (inoperable) or metastatic, HR-positive, HER2-negative breast cancer. The trial enrolled patients who had disease recurrence or progression during or after AI therapy with or without a CDK4/6 inhibitor. The trial included 2 populations, which were analyzed separately: the overall population (all enrolled patients [N = 708]) and the altered population (N = 289). The altered population included patients who had tested positive for tumours with 1 or more PIK3CA, AKT1, or PTEN alterations. This population is the focus of the reimbursement request. Enrolled patients were randomly assigned in a 1-to-1 ratio to receive capivasertib 400 mg (taken orally twice daily) in combination with fulvestrant 500 mg (administered intramuscularly every 14 days after the first 3 injections and every 28 days thereafter) or matching placebo plus fulvestrant. Randomization was stratified by liver metastases (yes or no), prior use of CDK4/6 inhibitors (yes or no), and geographic location (region 1, 2, or 3). The outcomes relevant to the CDA-AMC review included the primary outcome of PFS per Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1), as assessed by the investigators, and secondary outcomes of OS and safety. HRQoL — a secondary outcome in the trial, measured using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module (EORTC QLQ-BR23) — was also considered relevant. At the request of the sponsor, time from randomization to second progression on next-line treatment or death because of any cause (PFS2) and time to first subsequent chemotherapy were included for the altered population. These outcomes are included in Appendix 1. The trial population was predominately white (58%) and female (99%), with a mean age of 58 years (range, 26 years to 90 years). Overall, key baseline characteristics were generally balanced between the treatment groups in both populations. Most patients were postmenopausal females (77.0%), had previously received a CDK4/6 inhibitor (70%), and had an Eastern Cooperative Oncology Group (ECOG) Performance Status of 0 (66.0%), indicating good overall performance. A similar proportion of patients in both groups (approximately 41%) had an altered tumour status. In the altered population, the group receiving placebo plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 0 (72.4% versus 60.0%) and a lower proportion of patients with an ECOG Performance Status of 1 (26.9% versus 40.0%) than the group receiving capivasertib plus fulvestrant. Further, the group receiving placebo plus fulvestrant had a higher proportion of patients who had received no prior lines of therapy for advanced or metastatic cancer (14.9% versus 7.7%) and a lower proportion of patients who had received 1 prior line of therapy for advanced or metastatic cancer (59.0% versus 69.0%) compared with the group receiving capivasertib plus fulvestrant.
Efficacy Results
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. Efficacy and safety data were evaluated at the planned primary analysis for PFS, with a data cut-off date of August 15, 2022. An interim analysis for OS was conducted on this date. This section includes data from both the overall population and the altered population. The focus of the Health Canada indication and reimbursement request is the altered population; however, given that the overall population also included a proportion of patients with known AKT-altered status, the results for the overall population have also been included. It should be noted that 59% of patients in the overall population do not meet the criteria for the reimbursement request (i.e., they were of known nonaltered status or unknown alteration status).
Progression-Free Survival
In the overall population, PFS events had been reported for 258 patients (72.7%) in the group receiving capivasertib plus fulvestrant and for 293 patients (83.0%) in the group receiving placebo plus fulvestrant at the data cut-off. In the altered population, PFS events occurred in 121 patients (78.1%) in the group receiving capivasertib plus fulvestrant and in 115 patients (85.8%) in the group receiving placebo plus fulvestrant. The median durations of follow-up in all patients in the capivasertib plus fulvestrant group and the placebo plus fulvestrant group were 14.9 months and 14.3 months, respectively (range not reported). In the overall population, the median PFS was 7.2 months (95% confidence interval [CI], 5.5 months to 7.4 months) in the group receiving capivasertib plus fulvestrant versus 3.6 months (95% CI, 2.8 months to 3.7 months) in the group receiving placebo plus fulvestrant (log-rank test P < 0.001), with a between-group hazard ratio of 0.60 (96.5% CI, 0.50 to 0.72). In the altered population, the median PFS was 7.3 months (95% CI, 5.5 months to 9.0 months) in the group receiving capivasertib plus fulvestrant versus 3.1 months (95% CI, 2.0 months to 3.7 months) in the group receiving placebo plus fulvestrant (log-rank test P < 0.001), with a between-group hazard ratio of 0.50 (95% CI, 0.38 to 0.65). The results of sensitivity analyses were consistent with the primary analysis, and the results were consistent across the exploratory subgroup analysis by previous use of a CDK4/6 inhibitor in favour of capivasertib plus fulvestrant. For the exploratory subgroup analysis by AKT pathway status (nonaltered) in the overall population, the hazard ratio was 0.70 (95% CI, 0.56 to 0.88) in favour of capivasertib plus fulvestrant. This subgroup included patients of both known nonaltered and unknown alteration status. Among patients of known nonaltered status, the hazard ratio was 0.79 (95% CI, 0.61 to 1.02), and among patients of unknown alteration status, the hazard ratio was 0.52 (95% CI, 0.32 to 0.83). The point estimate for the hazard ratio for the known nonaltered subgroup (i.e., 0.79) falls outside of the 95% CI for the hazard ratio for both the overall population and the altered population. As noted by Health Canada, the effect observed in the overall population was likely driven by patients in the altered population, and the effect observed in the nonaltered population was likely driven by the population with unknown or no results.25
In the overall population, the Kaplan-Meier (KM)–estimated probabilities of PFS at 6 months and 12 months were 51.8% (95% CI, 46.4% to 57.0%) versus 32.0% (95% CI, 27.0% to 37.0%) and 28.5% (95% CI, 23.7% to 33.5%) versus 18.4% (95% CI, 14.4% to 22.8%) in the capivasertib plus fulvestrant and placebo plus fulvestrant groups, respectively. In the altered population, the KM-estimated probabilities of PFS at 6 months and 12 months were 53.4% (95% CI, 45.1% to 60.9%) versus 29.6% (95% CI, 21.9% to 37.7%) (between-group difference = █████ [95% CI, ████ to ████]) and 28.2% (95% CI, 21.2% to 35.6%) versus 15.8% (95% CI, 10.0% to 22.7%) (between-group difference = █████ [95% CI, ███ to ████]), respectively.
Overall Survival
By the August 15, 2022, data cut-off date, the median OS had not been reached in either group, with 25% and 31% of patients experiencing an event in the capivasertib plus fulvestrant and placebo plus fulvestrant groups, respectively. In the overall population, the hazard ratios were 0.74 (95% CI, 0.56 to 0.98) and 0.69 (95% CI, 0.45 to 1.05) in the altered population. In the overall population, the KM-estimated probabilities of being alive at 18 months and 24 months were 73.9% (95% CI, 68.3% to 78.7%) versus 65.0% (95% CI, 58.7% to 70.6%) and 64.3% (95% CI, 55.5% to 71.8%) versus 56.5% (95% CI, 48.3% to 63.9%) in the capivasertib plus fulvestrant and placebo plus fulvestrant groups, respectively. In the altered population subgroup, the KM-estimated probabilities of being alive at 18 months and 24 months were 73.2% (95% CI, 64.8% to 80.0%) versus 62.9% (95% CI, 53.1% to 71.2%) (between-group difference = █████ [95% CI, ████ to ████]) and █████ (95% CI, ████ to ████) versus █████ (95% CI, ████ to ████) (between-group difference = ████ [95% CI, ████ to ████]) in the capivasertib plus fulvestrant and placebo plus fulvestrant groups, respectively.
Health-Related Quality of Life
In the altered population, baseline global health status scores were similar in both treatment groups. At cycle 10, the between-group least squares mean difference from baseline was ████ (95% CI, █████ to ████; total sample = ██). For the EORTC QLQ-BR23, baseline scale scores were similar in both treatment groups and suggested intermediate to high functioning (median scores ≥ 55) and low symptomatology (median scores < 20), except for future perspective and feeling upset by hair loss. At cycle 17, the between-group mean differences in change from baseline were ████ for body image (95% CI, █████ to ████; total sample = ██); ███ for sexual functioning (95% CI, –███ to ████; total sample = ██); not estimable for sexual enjoyment (total sample = █); ███ for future perspective (95% CI, █████ to ████; total sample = ██); ███ for systemic therapy side effects (95% CI, ████ to ████; total sample = ██); ███ for breast symptoms (95% CI, ████ to ████; total sample = ██); ████ for arm symptoms (95% CI, ███ to █████; total sample = ██); and ████ for feeling upset by hair loss (█████ to ████; total sample = █). The HRQoL results were generally consistent across the cycles and reflected those of the overall population (data not shown).
Harms Results
The harms data reported in this section are from the data cut-off date of August 15, 2022. Given that the sample size of the overall population was larger than the altered population, the harms data summarized in this section are for the overall population; this approach was considered appropriate by the CDA-AMC review team. The safety profile of capivasertib plus fulvestrant in the altered population reflected the overall population. Most patients in the trial reported at least 1 adverse event (AE) (96.6% of patients receiving capivasertib plus fulvestrant and 82.3% of patients receiving placebo plus fulvestrant). The most frequently reported AEs of any grade in the group receiving capivasertib plus fulvestrant were diarrhea (experienced by 72.4% of patients versus 20.0% of patients receiving placebo plus fulvestrant), rash (38.0% versus 7.1%, respectively), and nausea (34.6% versus 15.4%, respectively). The most frequently reported AEs in the group receiving placebo plus fulvestrant were also diarrhea and nausea. A numerically higher proportion of serious adverse events (SAEs) were reported in patients taking capivasertib plus fulvestrant (16.1%) than in those taking placebo plus fulvestrant (8.0%). The most common SAE with capivasertib plus fulvestrant was diarrhea (1.7% versus 0.3% for those taking placebo plus fulvestrant). Study treatment discontinuation because of AEs was numerically higher in the group receiving capivasertib plus fulvestrant (9.3%) than in the group receiving placebo plus fulvestrant (0.6%). The most common AE leading to discontinuation of capivasertib or placebo was rash (████ versus ██ with placebo). Deaths were reported in 24.5% of patients in the group receiving capivasertib plus fulvestrant and in 30.6% of patients in the group receiving placebo plus fulvestrant. The majority of deaths in both groups were attributed to disease progression, which occurred in 22.3% of patients in the group receiving capivasertib plus fulvestrant and 28.9% of patients in the group receiving placebo plus fulvestrant. A higher proportion of notable AEs were reported in patients receiving capivasertib plus fulvestrant (█████) than in those receiving placebo plus fulvestrant (█████). The most common notable harms among the group receiving capivasertib plus fulvestrant were noninfectious diarrhea (experienced by 72.4% of patients versus 20.3% of patients receiving placebo plus fulvestrant), rash (38.0% versus 7.1%, respectively), and stomatitis (20.0% versus 5.7%, respectively).
Critical Appraisal
The CAPItello-291 trial randomization procedures, including the stratification factors, were appropriate and conducted by interactive response technology. In the altered population, the group receiving placebo plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 0 (72.4% versus 60.0%) and a lower proportion of patients with an ECOG Performance Status of 1 (26.6% versus 40.0%) than the group receiving capivasertib plus fulvestrant. Further, the group receiving placebo plus fulvestrant had a higher proportion of patients who had received no prior lines of therapy for advanced or metastatic cancer (14.9% versus 7.7%) and a lower proportion of patients who had received 1 prior line of therapy for advanced or metastatic cancer (59.0% versus 69.0%) compared with the group receiving capivasertib plus fulvestrant. These imbalances were likely because of chance, given that all other baseline characteristics of patients appeared balanced between groups and, as a result, unlikely to have resulted in bias. To minimize the risk of bias in the measurement of the outcome, the trial performed tumour assessments using RECIST 1.1 criteria; radiographic scans were assessed by blinded independent central review (BICR) as a sensitivity analysis. The PFS BICR results were similar to the primary investigator-assessed results. Sample size and power calculations were based on PFS and OS in the overall population and on PFS in the altered population; the trial was powered to detect significant differences for both outcomes. Prespecified analyses of OS and PFS in the overall and altered populations were appropriately controlled for multiple comparisons. All other analyses were descriptive. These included the 2 HRQoL outcomes, EORTC QLQ-C30 and EORTC QLQ-BR23, which were deemed clinically important. The sample sizes for the subgroup analyses of PFS were small. The trial may not have been powered to detect subgroup differences. While the trial met its primary objective of assessing PFS, the median OS was not reached in either treatment group, and there was imprecision in the estimates for between-group differences in survival probability at 18 months and 24 months (i.e., the 95% CIs were wide and included the potential for no difference between the 2 treatment groups). In addition, there is uncertainty as to whether the PFS benefits (as a surrogate outcome for OS) will translate into survival benefits. Given that the results at the data cut-off date represent an interim analysis for OS, and the results were based on few events, longer follow-up is needed to inform the true effect of capivasertib plus fulvestrant compared with placebo plus fulvestrant on survival. The certainty of evidence for many HRQoL outcomes was limited because of the risk of bias stemming from imprecision and missing outcomes data, both at baseline and at the selected follow-up times. Based on visual inspection of the KM plots for PFS and OS, it does not appear that there was any major violation of the proportional hazards (PH) assumption. However, the results of the PH assessment in the sponsor-submitted network meta-analysis (NMA) showed evidence of non-PHs across most studies, including the CAPItello-291 trial. As such, the hazard ratios for PFS and OS may not fully reflect the true effects.
In general, the population requested for reimbursement aligns with the Health Canada indication, except that the reimbursement request is not limited to female patients. Enrolment in the CAPItello-291 trial was open to both male and female patients, and 7 males were enrolled. The clinical experts consulted by CDA-AMC agreed that including males in the reimbursement request is appropriate because the proportion of included patients reflects the low prevalence of breast cancer in males, and that management of breast cancer in both males and females is similar. Given the small proportion of males in the trial, it was not possible to ascertain from the data whether males would experience different treatment outcomes compared with females. However, the clinical experts agreed that they would expect similar efficacy and harms among both males and females. The dosing and administration of capivasertib plus fulvestrant was consistent with the Health Canada–approved product monograph. Patients with PIK3CA, AKT1, or PTEN tumour alterations (i.e., the altered population, which is the focus of the Health Canada–approved indication) were identified by postrandomization central testing of tumour tissue collected before randomization based on a prespecified list of molecular alterations using a validated assay. The CDA-AMC team considered this diagnostic approach appropriate, although the clinical experts noted that testing for PIK3CA, AKT1, or PTEN tumour alterations is not part of routine clinical practice, and access to testing varies across Canada. According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the CAPItello-291 trial were generalizable to adults with HR-positive, HER2-negative, advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations in the Canadian setting. The trial included outcomes that were important to patients and clinicians. The patient group indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development, and Evaluations (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CDA-AMC expert committee deliberations. A final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for PFS and OS were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference points for the certainty of the evidence assessment for EORTC QLQ-C30 global health status score and EORTC QLQ-BR23 functional and symptom scales scores were set according to the presence or absence of an important effect based on a threshold suggested by the sponsor that was informed by the literature. Because of the lack of a formal minimal important difference (MID) estimate for SAEs, the target of the certainty of evidence assessment was set according to the presence or absence of any (nonnull) effect. The selection of outcomes for the GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
survival outcomes (PFS and OS)
HRQoL outcomes (i.e., EORTC QLQ-C30 global health status and EORTC QLQ-BR23 functional and symptom scales scores)
harms outcome (SAEs).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for patients receiving capivasertib plus fulvestrant versus those receiving placebo plus fulvestrant.
Table 2: Summary of Findings for Capivasertib Plus Fulvestrant Versus Placebo Plus Fulvestrant for Patients With HR-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer in the Altered Population
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Capivasertib plus fulvestrant | Placebo plus fulvestrant | Difference | |||||
PFS in the FAS, August 15, 2022 data cut-off | |||||||
Probability of PFS at 6 months Median follow-ups:
| 289 (1 RCT) | NA | ███ per 1,000 (███ ██ ███) | ███ per 1,000 | ███ more per 1,000 (███ ████ ██ ███ ████) | Higha | Capivasertib plus fulvestrant results in a clinically important increase in the probability of PFS at 6 months when compared with placebo plus fulvestrant. |
Probability of PFS at 12 months Median follow-ups:
| 289 (1 RCT) | NA | ███ per 1,000 (███ ██ ███) | ███ per 1,000 | ███ more per 1,000 (██ ████ ██ ███ ████) | Moderateb | Capivasertib plus fulvestrant likely results in a clinically important increase in the probability of PFS at 12 months when compared with placebo plus fulvestrant. |
OS in the FAS, August 15, 2022 data cut-off | |||||||
Probability of survival at 18 months Median follow-ups:
| 289 (1 RCT) | NA | ███ per 1,000 (███ ██ ███) | ███ per 1,000 | ███ more per 1,000 (██ █████ ██ ███ ████) | Lowc | Capivasertib plus fulvestrant may result in a clinically important increase in the probability of survival at 18 months when compared with placebo plus fulvestrant. |
Probability of survival at 24 months Median follow-ups:
| 289 (1 RCT) | NA | ███ per 1,000 (███ ██ ███) | ███ per 1,000 | ██ more per 1,000 (██ █████ ██ ███ ████) | Lowd | Capivasertib plus fulvestrant may result in a clinically important increase in the probability of survival at 24 months when compared with placebo plus fulvestrant. |
EORTC QLQ-C30 global health status in the FAS, August 15, 2022 data cut-off | |||||||
LS mean change from baseline in global health status; scores range from 0 to 100, with higher scores indicating better health status Time point: cycle 10 | ██ (1 RCT) | NA | █████ ███████ ██ █████ | █████ | ████ ███████ ██ ████) | Lowe | Capivasertib plus fulvestrant may result in little to no clinically important difference in global health status at cycle 10 when compared with placebo plus fulvestrant. |
EORTC QLQ-BR23 scales in the FAS, August 15, 2022, data cut-off | |||||||
Mean change from baseline in body image score; scores range from 0 to 100, with higher scores indicating better body image Time point: cycle 17 | ██ (1 RCT) | NA | █████ (SD = ████) | NR | ████ (█████ ██ ████) | Very lowf | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on body image at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in sexual functioning score; scores range from 0 to 100, with higher scores indicating better sexual functioning Time point: cycle 17 | ██ (1 RCT) | NA | ████ (SD = ███) | NR | ███ (████ ██ ████) | Very lowg | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on sexual functioning at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in sexual enjoyment score; scores range from 0 to 100, with higher scores indicating better sexual enjoyment Time point: cycle 17 | ██ (1 RCT) | NA | NE | NE | NE | NAh | There is no evidence for the effect of capivasertib plus fulvestrant on sexual enjoyment at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in future perspective score; scores range from 0 to 100, with higher scores indicating better future perspective Time point: cycle 17 | ██ (1 RCT) | NA | █████ (SD = ████) | NR | ███ (█████ ██ ████) | Very lowf | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on future perspective at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in systemic therapy side effects score; scores range from 0 to 100, with higher scores indicating greater level of side effects Time point: cycle 17 | ██ (1 RCT) | NA | ███ (SD = ████) | NR | ███ (████ ██ ████) | Very lowi | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on systemic therapy side effects at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in breast symptoms score; scores range from 0 to 100, with higher scores indicating greater level of symptoms Time point: cycle 17 | ██ (1 RCT) | NA | █████ (SD = ████) | NR | ███ (████ ██ ████) | Very lowi | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on breast symptoms at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in arm symptoms score; scores range from 0 to 100, with higher scores indicating greater level of symptoms Time point: cycle 17 | ██ (1 RCT) | NA | ████ (SD = █████ | NR | ████ (███ ██ ████) | Very low i | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on arm symptoms at cycle 17 when compared with placebo plus fulvestrant. |
Mean change from baseline in feeling upset by hair loss score; scores range from 0 to 100, with higher scores indicating greater level of being upset Time point: cycle 17 | | (1 RCT) | NA | NR | NR | ████ (█████ ██ █████ | Very lowf | The evidence is very uncertain about the effect of capivasertib plus fulvestrant on feeling upset by hair loss at cycle 17 when compared with placebo plus fulvestrant. |
Harms in the safety population, August 15, 2022 data cut-off | |||||||
SAEs Median follow-ups:
| 289 (1 RCT) | NR | ███ per 1,000 (NR) | ███ per 1,000 | ██ more per 1,000 (█████ ██ ███ ████) | Moderatej | Capivasertib plus fulvestrant likely results in an increase in the proportion of patients who experience SAEs, compared with placebo plus fulvestrant. The clinical importance of the increase is uncertain. |
CI = confidence interval; EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; LS = least squares; NA = not applicable; NE = not estimable; NR = not reported; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The between-group absolute effects at the time point were requested by CDA-AMC to facilitate the GRADE assessment (i.e., for PFS, OS, EORTC QLQ-BR23 scales, and SAEs).
aA between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) at 6 months was clinically important, according to the clinical experts. The point estimate and entire confidence exceeded the threshold.
bRated down 1 level for serious imprecision because of the 95% CI for the between-group difference including the possibility of an important benefit and a trivial effect when compared with placebo plus fulvestrant; a between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) was clinically significant at 12 months, according to the clinical experts.
cRated down 2 levels for very serious imprecision because of the 95% CI for the between-group difference including the possibility of an important benefit, little to no difference, and possible harm when compared to placebo plus fulvestrant; a between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) was clinically significant at 18 months, according to the clinical experts.
dRated down 2 levels for very serious imprecision because of the 95% CI for the between-group difference including the possibility of an important benefit and important harm when compared to placebo plus fulvestrant; a between-group absolute risk difference of 5% (50 fewer or more events per 1,000 patients) was clinically significant at 24 months, according to the clinical experts.
eRated down 2 levels for risk of bias because of missing outcomes data. There is no imprecision in the estimate (the point estimate and entire 95% CI for the between-group difference show little to no difference). Based on the sponsor’s suggestion and informed by ranges identified in the literature, a 10-point change from baseline in EORTC QLQ-C30 global health status score was considered clinically important.
fRated down 2 levels for very serious imprecision because of the 95% CI for the between-group difference including the possibility of both benefit and harm when compared with placebo plus fulvestrant; based on the sponsor’s suggestion (which was informed by ranges identified in the literature), a 10-point change from baseline in EORTC QLQ-BR23 scale score was considered clinically important. Rated down 2 levels for risk of bias due missing outcomes data.
gRated down 1 level for serious imprecision because of the 95% CI for the between-group difference including the possibility of both benefit and little to no difference when compared with placebo plus fulvestrant; based on the sponsor’s suggestion (which was informed by ranges identified in the literature), a 10-point change from baseline in in EORTC QLQ-BR23 scale score was considered clinically important. Rated down 2 levels for risk of bias because of missing outcomes data.
hNot estimable because of missing outcomes data.
iRated down 1 level for serious imprecision because of the 95% CI for the between-group difference including the possibility of both harm and little to no difference when compared with placebo plus fulvestrant; based on the sponsor’s suggestion (which was informed by ranges identified in the literature), a 10-point change from baseline in in EORTC QLQ-BR23 scale score was considered clinically important. Rated down 2 levels for risk of bias because of missing outcomes data.
jRated down 1 level for serious imprecision because of the 95% CI for the between-group absolute risk difference including the possibility of both benefit and harm. There was no known MID; therefore, the target of the certainty appraisal was any effect. Sources: CAPItello-291 Clinical Study Report;26 sponsor’s Summary of Clinical Evidence; sponsor’s response to requested additional information.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
One sponsor-submitted NMA was included in the submission to inform the pharmacoeconomic model and identify indirect comparisons that fill gaps in the comparative evidence for other treatments of interest for HR-positive, HER2-negative, advanced or metastatic breast cancer. The objective of the NMA was to indirectly compare the treatment effects of capivasertib versus other relevant comparators for the treatment of adult patients with HR-positive, HER2-negative, advanced breast cancer with AKT pathway–altered tumours after progression on, or during or after treatment with, endocrine-based regimens. The protocol of the systematic review and NMA was registered a priori in the International Prospective Register of Systematic Reviews.
Description of Studies
The systematic literature review identified 33 studies that informed the feasibility assessment, of which 10 were included in the NMA. The base-case network was plotted to compare capivasertib 400 mg plus fulvestrant 500 mg to fulvestrant 250 mg or 500 mg, exemestane 25 mg, everolimus 10 mg plus exemestane 25 mg, and capecitabine 1,250 mg/m2 monotherapy. The comparison across studies suggested differences for menopausal status, prior CDK4/6 use, HER2 status, AKT pathway alteration status, and line of therapy. Fixed- and random-effects NMAs were conducted for PFS and OS using a Bayesian framework, and results were summarized as hazard ratios and 95% credible intervals (CrIs). The NMA used the altered population data from the CAPItello-291 and FAKTION trials, whereas the other included studies did not report on AKT pathway–altered tumours. An assessment of the PH assumption was performed for PFS and OS that included visual inspection of the log-cumulative hazards and the scaled Schoenfeld residual plots, and by evaluating the Grambsch-Therneau nonproportionality test.
Efficacy Results
The results for both PFS and OS favoured capivasertib plus fulvestrant versus exemestane 25 mg, fulvestrant 500 mg, and fulvestrant 250 mg. For both PFS and OS, the results comparing capivasertib plus fulvestrant to both everolimus 10 mg plus exemestane 25 mg and capecitabine 2,500 mg/m2 favoured capivasertib plus fulvestrant; however, the 95% CrIs included the possibility of no difference or of the comparator being favoured (i.e., the CrIs crossed the null). The results of the PH assessment showed evidence of non-PHs across most studies.
Harms Results
Harms were not assessed in the NMA.
Critical Appraisal
The methods used to conduct the systematic literature review and NMA were prespecified with an a priori protocol and used appropriate criteria to search databases, select studies, extract data, and assess risk of bias in the included studies. Selection bias is expected to be low, given the comprehensiveness of the searches and the methods for study selection. The NMA included relevant outcomes identified by the CDA-AMC team (PFS and OS); however, important outcomes, such as HRQoL and harms, were not included in the comparisons. Overall, the network was sparse (i.e., there were many comparisons, but few studies). The results of the inconsistency analysis indicated that the consistency assumption was met for PFS; however, the only closed loop in the network did not include capivasertib plus fulvestrant. It was not possible to assess for inconsistency across direct and indirect evidence in the OS NMA because of the absence of loops in the network (i.e., there was no direct evidence). The PH assumption was violated in almost all comparisons for PFS and OS; as such, the hazard ratios may not be fully reflective of the true effects. The exchangeability assumption was violated because there were several notable sources of heterogeneity for potential effect modifiers across the included studies. Identified variables of concern included AKT pathway alterations, prior CDK4/6 inhibitor treatment, HER2 status, region of enrolment, line of therapy, and menopausal status. Specifically, of the 10 included studies, only 2 reported results for patients with AKT pathway alterations (the CAPItello-291 and FAKTION trials); both involved capivasertib. For other treatments, there was no evidence in the population with altered AKT pathway. Only 1 of the 10 included studies (the CAPItello-291 study) reported subgroup data based on prior CDK4/6 inhibitor treatment, which is recognized as a prognostic factor. Although the authors provided evidence for treatment-effect modifiers, it was not clear how these were identified (i.e., whether a literature review or expert consensus was performed). As such, it is not clear whether all treatment-effect modifiers were accounted for in the feasibility assessment. In addition, the median follow-up times across the included trials were not reported. In general, the magnitude and direction of potential bias because of heterogeneity and lack of proportionality on outcome estimates cannot be predicted. Because of these limitations in the NMA, no definitive conclusions could be drawn on the relative treatment effects of capivasertib plus fulvestrant versus other relevant comparators.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The FAKTION trial (N = 140) was an investigator-initiated, multicentre, randomized, double-blind, placebo-controlled, biomarker-adaptive, phase II trial that enrolled patients from 19 hospitals in the UK. The sponsor submitted this study because it contained longer follow-ups for OS compared to the pivotal trial. Eligible patients were postmenopausal females with locally advanced or metastatic, HR-positive, HER2-negative breast cancer who were not suitable for surgical resection. Patients were considered suitable for ET but had received no more than 3 previous lines of ET and up to 1 line of chemotherapy for advanced breast cancer. They had also experienced progressive disease during treatment with a third-generation AI or relapsed on an AI in the adjuvant setting. Patients were randomized 1 to 1 to receive fulvestrant 500 mg with either capivasertib 400 mg twice daily or placebo until disease progression, unacceptable toxicity, withdrawal of consent, or loss to follow-up. Allocation was balanced by minimization according to PIK3CA mutation status (mutated versus wild type), PTEN expression status (null versus detected in ≥ 1% of tumour cells at a moderate or strong intensity or in ≥ 10% of cells at a weak intensity), measurable versus nonmeasurable disease, and primary versus secondary resistance to a third-generation AI. The outcomes relevant to the CDA-AMC review included the primary outcome of investigator-assessed PFS and the secondary outcomes of OS and safety.
The FAKTION trial included an overall population, with both expanded pathway–altered and –nonaltered subgroups. The expanded pathway–altered subpopulation included patients who tested positive for tumours with 1 or more PIK3CA, AKT1, or PTEN alterations and is the focus of the indication and reimbursement request under review. Test results were considered positive if either of 2 assays (the Foundation One CDx Clinical Trial next-generation sequencing [NGS] assay testing of tumour biopsy samples and/or the GuardantOMNI Research Use Only assay testing of plasma) detected 1 or more PIK3CA, AKT1, or PTEN alterations. Because the clinical experts consulted by CDA-AMC indicated that NGS is the preferred assay to test for PIK3CA, AKT1, or PTEN alterations, this section included efficacy outcomes for the NGS-identified, pathway-altered analysis set as well. In the expanded pathway–altered subpopulation, the median ages were 60 years (interquartile range [IQR], 55 years to 69 years) in the capivasertib plus fulvestrant group and 62 years (IQR, 56 years to 68 years) in the placebo plus fulvestrant group. Some notable imbalances were observed between the treatment groups in the patient characteristics for the expanded pathway–altered subpopulation. The group receiving capivasertib plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 1 (36% versus 24%) than the group receiving placebo plus fulvestrant. Most patients had metastatic disease (96%); the sites of metastases were largely imbalanced between the treatment groups. Visceral disease was present in 30 patients (77%) in the group receiving capivasertib plus fulvestrant and in 24 patients (65%) in the group receiving placebo plus fulvestrant. The group receiving capivasertib plus fulvestrant had a higher proportion of patients with primary AI resistance (38% versus 27%), but a lower proportion of patients with secondary AI resistance (62% versus 73%). By the data cut-off date of November 25, 2021, the median follow-up for the expanded pathway–altered subpopulation was 58.5 months (IQR, 45.9 months to 64.1 months) for patients treated with fulvestrant plus capivasertib and 62.3 months (IQR, 62.1 months to 70.3 months) for patients treated with fulvestrant plus placebo. For the expanded pathway–altered subgroup, the median follow-up durations were 54.3 months (IQR, 45.5 months to 61.2 months) for the group receiving capivasertib and fulvestrant and 62.3 months (IQR, 62.1 months to not reached) for the group receiving placebo and fulvestrant.
Efficacy Results
Progression-Free Survival
A PFS event was recorded for 66 patients out of 76 patients (87%) in the expanded pathway–altered subgroup: 30 patients out of 39 patients (77%) received capivasertib plus fulvestrant, and 36 patients out of 37 patients (97%) received placebo plus fulvestrant. Median PFS was 12.8 months (95% CI, 6.6 months to 18.8 months) in the group receiving capivasertib plus fulvestrant versus 4.6 months (95% CI, 2.8 months to 7.9 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.44; 95% CI, 0.26 to 0.72).
Similar results were observed in the NGS-identified, pathway-altered analysis set, where a PFS event was recorded for 25 patients out of 34 patients (74%) who received capivasertib and all 29 patients (100%) who received placebo. Median PFS was 13.4 months (95% CI, 6.6 months to 20.7 months) in the group receiving capivasertib plus fulvestrant versus 3.1 months (95% CI, 2.8 months to 7.1 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.36; 95% CI, 0.20 to 0.65).
Overall Survival
At the time of analysis, 57 patients out of 76 patients (75%) in the expanded pathway–altered subgroup had died. Of these, 25 patients of the 39 patients (64%) had received capivasertib plus fulvestrant and 32 patients of the 37 patients (86%) had received placebo plus fulvestrant. Median OS in the expanded pathway–altered subgroup of patients receiving capivasertib plus fulvestrant was 38.9 months (95% CI, 23.3 months to 50.7 months) compared with 20.0 months (95% CI, 14.8 months to 31.4 months) for those receiving placebo plus fulvestrant (adjusted hazard ratio = 0.46; 95% CI, 0.27 to 0.79).
Similar results were observed in the post hoc analysis involving the NGS-identified, pathway-altered subgroup, in which an OS event was recorded for 21 patients out of 34 patients (61%) who had received capivasertib plus fulvestrant and 25 patients out of 29 patients (86%) who had received placebo plus fulvestrant. Median OS was 39.0 months (95% CI, 22.3 months to 50.7 months) in the group receiving capivasertib plus fulvestrant versus 20.9 months (95% CI, 14.1 months to 35.4 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.44; 95% CI, 0.24 to 0.81).
Harms Results
Safety analyses included all patients who had received at least 1 dose of the assigned study drug. All randomly assigned patients were included in the safety analyses. The most commonly reported AEs were diarrhea, nausea, hyperglycemia, fatigue, vomiting, decreased appetite, and rash (maculo-papular). The proportions of participants experiencing grade 3 to 5 AEs (irrespective of causality) were 45 patients out of 69 patients (65%) in the group receiving capivasertib plus fulvestrant and 35 patients out of 70 patients (50%) in the group receiving placebo plus fulvestrant. The most common grade 3 to 4 AEs experienced by patients were hypertension (22 patients out of 69 patients [32%] in the group receiving capivasertib plus fulvestrant versus 18 patients out of 71 patients [25%] in the group receiving placebo plus fulvestrant), diarrhea (10 patients [14%] versus 3 patients [4%]), rash (14 patients [20%] versus 0 patients), infection (4 patients [6%] versus 2 patients [3%]), and fatigue (1 patient [1%] versus 3 patients [4%]). Although serious adverse reactions were reported (only in the group receiving capivasertib plus fulvestrant), the total number of SAEs, irrespective of causality, was not reported in the publication. The most commonly reported SAEs experienced by patients were dyspnea, back pain, lower respiratory tract infection, pain, abdominal pain, and noncardiac chest pain. As of the data cut-off date, 21 patients (30%) in the capivasertib group and 31 patients (44%) in the placebo group had died. A total of 2 deaths occurred among patients with AEs.
Critical Appraisal
The FAKTION trial was a randomized, double-blind, placebo-controlled, phase II trial. The randomization and masking procedures were appropriate. Because it was a phase II trial that included fewer patients and aimed to provide preliminary evidence about the efficacy and harms of the study drug, the results cannot be considered confirmatory. Despite randomization, imbalances were observed at baseline in patients’ disease characteristics (e.g., ECOG Performance Status, histopathological subtype, visceral disease, AI given as last treatment before registration, previous ET, and PIK3CA and PTEN results). Because of the small sample size, there is an increased risk that prognostic balance was not achieved, as evidenced by imbalances in patients’ baseline disease and demographic characteristics. As such, it is possible that the observed effects were either overestimated or underestimated and may have been driven by prognostic differences between the 2 groups (i.e., may not be reflective of the true treatment effect). Results of the Schoenfeld tests for the PH assumption were not statistically significant; however, these may not have been powered to detect a violation. No major violations of the PH assumption were noted through visual inspection of the KM plots. The differences in PFS and OS between the treatment groups observed in the FAKTION trial for the altered patient population were considered clinically meaningful by the clinical experts consulted for this review. Both patients and investigators were blinded to the treatment assignments (i.e., capivasertib plus fulvestrant or placebo plus fulvestrant). PFS was assessed by the investigator, without adjudication through BICR. It is possible that patients and investigators may have become unblinded because of imbalances in notable harms across the 2 treatment groups (e.g., more patients experienced diarrhea and rash in the group receiving capivasertib plus fulvestrant). As such, there may be an increased risk of bias in the measurement of PFS and subjective harms; however, the presence and direction of bias is uncertain. Censoring reasons seemed balanced between the treatment groups.
The population enrolled in the FAKTION trial consisted of postmenopausal females with histological confirmation of HR-positive, HER2-negative, locally advanced or metastatic, inoperable breast cancer that was not amenable to curative surgical resection. This was a subset of the Health Canada–indicated population (i.e., premenopausal and postmenopausal adult females). The narrower patient population may affect the generalizability of the trial results in the Canadian setting. In addition, male patients and patients with prior CDK4/6 inhibitor treatment were not enrolled. Male patients would be included in the patient population of the sponsor’s reimbursement request; however, they are not included in the Health Canada indication. The clinical experts consulted by CDA-AMC noted that all patients in Canada who are candidates for treatment with capivasertib plus fulvestrant will have been treated with a CDK4/6 inhibitor because these are now part of the usual first-line treatment in combination with ET; males would also be considered candidates for treatment. HRQoL was not measured but is considered important by both patients and clinicians. No data on the race or ethnicity of patients were available, which made it difficult to contextualize the results in the Canadian setting. The dosing and administration of capivasertib plus fulvestrant were consistent with the Health Canada–approved product monograph.
Conclusions
Evidence from 1 ongoing, phase III, double-blind RCT (the CAPItello-291 trial) reported on outcomes that were important to both patients and clinicians. The trial showed high and moderate certainty of evidence that treatment with capivasertib plus fulvestrant results in a clinically meaningful increase in PFS at 6 months and 12 months, respectively, compared to placebo plus fulvestrant in adults with locally advanced or metastatic, HR-positive, HER2-negative breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations. At the time of the interim analysis, median OS had not been reached in either group, and no definitive conclusions can be drawn with respect to HRQoL because of concerns about imprecision and missing outcomes data. Although the FAKTION study reported a longer duration of follow-up for OS, the trial had important methodological limitations (e.g., imbalances in important baseline characteristics) and limited generalizability (e.g., it enrolled only postmenopausal females and excluded patients with prior CDK4/6 inhibitor treatment) that made it difficult to draw firm conclusions. There were no new safety signals identified. The safety of capivasertib plus fulvestrant was consistent with the known safety profiles of the individual drugs; however, the trial showed that treatment with capivasertib plus fulvestrant likely results in an increase in the proportion of patients who experience SAEs when compared with placebo plus fulvestrant. Because of limitations in the indirect treatment comparison, no conclusions can be drawn about the relative efficacy and safety of capivasertib plus fulvestrant compared to fulvestrant 250 mg or 500 mg, exemestane 25 mg, everolimus 10 mg plus exemestane 25 mg, or capecitabine 1,250 mg/m2.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of capivasertib (Truqap) 400 mg, taken orally twice daily for 4 days, followed by 3 days off treatment, in combination with fulvestrant 500 mg, administered intramuscularly every 14 days after the first 3 injections and every 28 days thereafter for the treatment of adults with locally advanced or metastatic breast cancer.
Disease Background
The contents of this section were informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
Breast cancer was the second most-diagnosed cancer in Canada in 2023 and the most prevalent among females, with projected estimates of about 29,700 new cases in the overall population in 2023 (29,400 in females and 260 in males).1 The 5-year prevalence of breast cancer in females reported in Canada in 2018 was 110,955 patients,2 equating to a 5-year prevalence rate of 0.73%.3 Breast cancer is a heterogeneous disease4,5 that is classified into subtypes based on the specific cell types affected, gene expression, and receptors expressed on the surface of or inside tumour cells. For instance, the presence or absence of the expression of the HER2, estrogen receptors (ERs), or progesterone receptors affects the proliferation of the cancer cells, the patient’s prognosis and response to treatment, and recurrence.5,27,28 HR-positive, HER2-negative breast cancer subtypes are the most prevalent in North America, accounting for at least 60% to 70% of all breast cancer cases.6 An HR-positive, HER2-negative breast cancer is defined — according to American Society of Clinical Oncology and College of American Pathologists criteria — as a tumour having more than 1% immunohistochemistry (IHC) expression of ER and/or progesterone receptors and a lack of HER2 expression (which includes HER2-low expression [i.e., IHC score of 1+ or 2+], confirmed as negative by in situ hybridization and HER2 IHC-0 expression).29-32
Diagnosis is based on clinical presentation of lesions at mammographic screening, radiological imaging (such as ultrasound or CT), and/or physical examination.33,34 Disease staging follows the American Joint Committee on Cancer system.7 Tumour biopsy with pathology review and biomarker assessment (e.g., including HR and HER2 status) are completed for confirmatory diagnosis and to determine disease subtype and guide treatment decision-making.7,8 HR and HER2 status testing are routinely conducted at initial diagnosis,35 and IHC or fluorescence in situ hybridization testing is widely available across jurisdictions in Canada.8
Signs and symptoms vary by disease stage and may include swelling in the surrounding lymph nodes, nipple changes (e.g., discharges), skin changes (e.g., erythema, skin ulcers, eczema), breast pain or heaviness, and/or other persistent changes in the breast.9,10 Metastatic, HR-positive, HER2-negative breast cancer also negatively affects patient quality of life, given that the symptoms that manifest are the result of disease progression and treatments administered. Commonly reported symptoms include pain, fatigue, nausea, vomiting, cognitive problems, depression, hair loss, lymphedema, sleep disturbance, loss of appetite, anxiety, and sexual dysfunction.11-13
At least 5% to 10% of genetic alterations are inherited from a parent.14 Genetic alterations can also be acquired during tumour development; these are often known as somatic alterations. Somatic alterations of interest to this review are in the PI3K, AKT, or mTOR pathway, which is a cell-signalling pathway regulating cell proliferation and survival. Alterations in the PI3K, AKT, or mTOR signalling axis are observed in up to 48% of all patients with HR-positive, HER2-negative breast cancer.15,16 In HR-positive, HER2-negative breast cancers, PI3K, AKT, or mTOR pathway activation most frequently arises from PIK3CA alterations, occurring in approximately 30% of patients.17-21 A further approximately 4% of advanced breast cancers harbour AKT1-activating alterations or amplifications, and approximately 5% have inactivating alterations in PTEN.17,22,23 Survival outcomes following progression on endocrine-based therapies decline with later lines of single-drug chemotherapy, with median PFS and OS estimated to be as low as 3 months and 7 months, respectively, for patients treated with 5 lines of chemotherapy, while the median PFS and OS are estimated to be around 7.5 months and 13.5 months, respectively, after the initiation of a second line of chemotherapy.24
Standards of Therapy
The contents of this section were informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
Treatment priorities for patients with metastatic breast cancer remain focused on prolonging survival, reducing tumour burden, and extending time on ETs while delaying the use of toxic drugs, such as chemotherapies, to maintain or improve HRQoL. According to Canadian guidelines,36 there is currently no specific standard of care for patients with HR-positive, HER2-negative breast cancer who harbour 1 or more PIK3CA, AKT1, or PTEN alterations. Therefore, these patients are currently treated the same as any patient with HR-positive, HER2-negative breast cancer. According to the clinical experts consulted by CDA-AMC, the current treatment paradigm for locally advanced or metastatic, HR-positive, HER2-negative breast cancer in Canada does not differ between females and males; the established first-line treatment is ET with a CDK4/6 inhibitor.37 Additional therapies available to these patients are based on ET, targeted therapies combined with ET, chemotherapies, and antibody-drug conjugates involving:37
ET with selective ER modulators (e.g., tamoxifen), AIs (e.g., anastrozole, letrozole, or exemestane), and a selective ER degrader (e.g., fulvestrant)
targeted therapy with CDK4/6 inhibitors ribociclib, abemaciclib, and palbociclib combined with ET; everolimus combined with exemestane in patients who progress on CDK4/6 inhibitors
chemotherapy with capecitabine, docetaxel, paclitaxel, nab-paclitaxel, doxorubicin, epirubicin, vinorelbine, gemcitabine, 5 fluorouracil-epirubicin-cyclophosphamide, fluorouracil-adriamycin-cytoxan, adriamycin-cytoxan, gemcitabine plus cisplatin, or cyclophosphamide-methotrexate-fluorouracil
trastuzumab deruxtecan, an antibody-drug conjugate, used as monotherapy for patients who have received a prior line of chemotherapy and are no longer eligible for ET.
Most of these treatment options are funded with restrictions across the majority of Canadian provincial and territorial drug programs (excluding Quebec) and included in the CDA-AMC Provisional Funding Algorithm for HR-positive, HER2-negative breast cancer.37 However, everolimus with exemestane and fulvestrant monotherapy are not consistently funded across provinces in patients who have progressed on a prior CDK4/6 inhibitor. Additionally, ribociclib and palbociclib are the only CDK4/6 inhibitors that are publicly available in the metastatic setting across Canada.
Drug Under Review
Capivasertib, in combination with fulvestrant, has been approved by Health Canada for the treatment of adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy.38 The current reimbursement request is for capivasertib in combination with fulvestrant for the treatment of adult patients with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy. This reimbursement request is aligned with the approved Health Canada indication except that it is not limited to females with breast cancer. Capivasertib has not been previously reviewed by CDA-AMC.38
The recommended dose of capivasertib is 400 mg (2 oral tablets of 200 mg), taken twice daily for 4 consecutive days, followed by 3 days off treatment. The recommended dose of fulvestrant is 500 mg, administered intramuscularly on days 1, 15, and 29, and then once monthly thereafter.38
Capivasertib is an inhibitor of the kinase activity of all 3 isoforms of serine and threonine kinase AKT (AKT1, AKT2, and AKT3). In tumours, AKT activation happens as a result of upstream activation by other signalling pathways, and also through genetic alterations in AKT1, PTEN, and/or PIK3CA.38
The key characteristics of capivasertib in combination with fulvestrant are summarized in Table 3 with other treatments available for adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy.
Table 3: Key Characteristics of Capivasertib Plus Fulvestrant, Fulvestrant, Everolimus Plus Exemestane, Capecitabine, and Paclitaxel
Characteristic | Capivasertib plus fulvestrant | Fulvestrant | Everolimus plus exemestane | Capecitabine | Paclitaxel |
|---|---|---|---|---|---|
Mechanism of action | Capivasertib is an inhibitor of the kinase activity of all 3 isoforms of serine and threonine kinase AKT (AKT1, AKT2, and AKT3). The combination of capivasertib and fulvestrant reduces the growth of ER-positive breast cancer cell lines and tumour models with and without alterations in PIK3CA, PTEN, or AKT. | Fulvestrant is a nonagonist ER antagonist that blocks the trophic actions of estrogens without itself having any partial agonist (estrogen-like) activity. | Everolimus reduces cell proliferation by inhibiting mTORC1, glycolysis, and angiogenesis in solid tumours in vivo, both through direct antitumour cell activity and inhibition of the tumour stromal compartment. Exemestane is a potent competitive human placental aromatase inhibitor that lowers circulating estrogen concentrations in postmenopausal females. | Capecitabine is a tumour-activated, antineoplastic drug (antimetabolite), that is selectively activated to the cytotoxic moiety, 5-FU, by thymidine phosphorylase in tumours. This leads to cell injury by both DNA- and RNA-derived mechanisms through further metabolization of 5-FU to FdUMP and FUTP. | Paclitaxel disrupts the dynamic equilibrium within the microtubule system and blocks cells in the late G2 phase and M phase of the cell cycle, inhibiting cell replication and impairing the function of nervous tissue. |
Indicationa | Capivasertib is indicated in combination with fulvestrant for the treatment of adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy. | Fulvestrant is indicated for the:
| Everolimus in combination with exemestane is indicated for the treatment of postmenopausal females with HER2-negative, advanced breast cancer after recurrence or progression following treatment with an NSAI (letrozole or anastrozole). Exemestane is indicated for the treatment of females with advanced breast cancer with naturally or artificially induced postmenopausal status whose disease has progressed from antiestrogen therapy. | Capecitabine is indicated as monotherapy for the treatment of advanced or metastatic breast cancer after the failure of standard therapy, including a taxane, unless therapy with a taxane is clinically contraindicated. | Paclitaxel is indicated in breast cancer as a second-line therapy for metastatic breast cancer. |
Route of administration | Oral | Intramuscular injection | Oral | Oral | IV |
Recommended dose | 400 mg (2 tablets of 200 mg) taken twice daily for 4 days followed by 3 days off treatment | 500 mg administered as 2 of the 5 mL injections of | Everolimus is administered in 2.5 mg, 5 mg, or 10 mg tablets once daily. Exemestane is administered in 25 mg tablets once daily. | Available in 150 mg and 500 mg tablets, with a recommended dosage of 1,250 mg/m2 twice daily | 260 mg/m2 administered over 30 minutes every 3 weeks |
Serious adverse effects or safety issues | Cutaneous adverse reactions, including DRESS, EM, and palmar-plantar erythrodysesthesia; hyperglycemia, including diabetic ketoacidosis; severe diarrhea associated with dehydration and acute kidney injury | Elevated transaminase, bilirubin, and alkaline phosphatase levels; hypersensitivity reactions, including angioedema and urticaria | Everolimus: noninfectious pneumonitis, infections, and renal failure; hypercholesterolemia and hypertriglyceridemia; hyperglycemia; stomatitis, including mouth ulceration; decreased hemoglobin, lymphocytes, neutrophils, and platelets; all grades of hemorrhage Exemestane: Not recommended for use in premenopausal females or females diagnosed with osteoporosis. May increase the risk of ischemic cardiovascular diseases, gastric ulcer, and hypercholesterolemia; may cause a reduction in BMD with a possible consequent increased risk of fracture; may cause arthralgias and/or myalgias. | Acute renal failure secondary to dehydration; cardiotoxicity; severe skin reactions, such as hand-and-foot syndrome, Stevens-Johnson syndrome, and toxic epidermal necrolysis; severe toxicity (e.g., stomatitis, diarrhea, mucosal inflammation, neutropenia, and neurotoxicity); altered coagulation parameters and/or bleeding | Bone marrow suppression (primarily neutropenia); sepsis with or without neutropenia; pneumonitis |
5-FU = 5-fluorouracil; BMD = bone mineral density; DRESS = drug reaction with eosinophilia and systemic symptoms; EM = erythema multiforme; ER = estrogen receptor; FdUMP = 5-fluoro-2’-deoxyuridine monophosphate; FUTP = 5-fluorouridine triphosphate; G2 phase = gap 2 phase; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; M phase = mitosis phase; mTORC1 = mammalian target of rapamycin complex 1; NSAI = Nonsteroidal aromatase inhibitor; RNA = ribonucleic acid.
aHealth Canada–approved indication.
Source: Product monographs38-43 and sponsor’s Summary of Clinical Evidence.
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups.
Two patient groups, the CBCN and Rethink Breast Cancer, provided input for this review. Information from the CBCN group was sourced from 3 online surveys: the CBCN 2022 Triple Negative Breast Cancer Patient Survey (981 participants, 31 of whom had metastatic, HR-positive breast cancer), the CBCN’s 2017 Metastatic Breast Cancer Patient Survey (180 metastatic patients, 38 of whom had metastatic, HR-positive breast cancer), and the CBCN’s 2012 Metastatic Breast Cancer Patient and Caregiver Survey Report (71 patients and 16 caregivers). The CBCN’s 2012 Survey was conducted in collaboration with Rethink Breast Cancer. No patients taking the drug under review participated in these surveys.
Information from Rethink Breast Cancer was gathered through programming and meetings with patients with breast cancer and an online survey of 78 patients living with metastatic breast cancer, which ran from September 2018 to April 2019. Rethink Breast Cancer also conducted interviews with 5 patients (4 from the US and 1 from Canada) living with HR-positive, HER2-negative, metastatic breast cancer. The 4 patients in the US had experience taking capivasertib for HR-positive, HER2-negative, metastatic breast cancer. The patient in Canada reported taking a CDK4/6 inhibitor and having a PIK3CA mutation.
The 2 groups highlighted that metastatic disease poses a significant or debilitating impact on patients’ quality of life. Rethink Breast Cancer stated that breast cancer may have greater emotional effects and lifestyle impacts on younger patients, especially those diagnosed in their twenties, thirties, and early forties, because women in these age groups are more likely to face fertility or family-planning challenges, diagnosis during pregnancy, demands of childcare, and impacts on relationships, body image, dating, and sexuality. These impacts can leave them feeling isolated from peers who do not have cancer. They may also experience career hiatuses and financial insecurity. The CBCN also noted similar issues: the disease may restrict patients’ ability to care for children and dependents and their ability to be social and participate meaningfully in their communities.
The CBCN highlighted that current treatment goals for patients with metastatic breast cancer include controlling the progression of the disease (i.e., extending life) and reducing cancer-related symptoms (i.e., extending or stabilizing quality of life). They further noted that patients diagnosed with HR-positive, HER2-negative, metastatic breast cancer have limited options for targeted treatments in addition to poor prognoses and poor survival outcomes. Respondents in the CBCN’s 2017 survey indicated some key factors that influenced their decisions about treatments. These included their effectiveness, ability to prolong quality of life, side effects, costs, and accessibility. Patients with metastatic breast cancer who responded to the CBCN 2012 and 2017 surveys also expressed the need for personal choice and autonomy in choosing treatments.
Rethink Breast Cancer stated that patients go to great lengths to avoid standard chemotherapy and suffer both emotionally and physically for this reason. The group added that patients on standard chemotherapy have a lot of difficulty managing their illnesses. Rethink Breast Cancer indicated that the primary improvement that patients with metastatic breast cancer seek is to extend their life beyond what is expected with the help of currently available, publicly funded therapies and to enjoy a better quality of life. In the 2018 to 2019 survey conducted by Rethink Breast Cancer, patients rated controlling their disease and extending their life expectancy as the most important outcomes for treatment. This finding suggests that patients value long-term health outcomes over immediate concerns, such as reducing symptoms or managing side effects.
Rethink Breast Cancer noted that all 4 patients who had taken the drug under review highlighted the importance of having access to new therapies that have the possibility of extending their lives. Three of these patients shared that they are experiencing a good quality of life while taking capivasertib, continuing to work, enjoy time with loved ones, and live their lives.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of locally advanced or metastatic breast cancer.
Unmet Needs
The clinical experts indicated that because the treatment goal for patients is palliative, patients’ unmet needs are for new treatments that would delay progression, prolong OS, and improve quality of life while exposing patients to minimal toxicity. The experts noted that patients become refractory to current treatment options, and subsequent therapy is limited to chemotherapy, which has significant impacts on quality of life and resource utilization. All 3 experts highlighted that the balance between treatment efficacy and quality of life would be important.
Place in Therapy
The clinical experts agreed that capivasertib plus fulvestrant would be used in the second-line setting. They highlighted that although this drug combination targets the PI3K, AKT, PTEN signalling pathway, in their opinion, it may be used more widely in clinical practice (i.e., in patients without 1 of these alterations). The experts indicated that capivasertib plus fulvestrant would alter the current treatment paradigm because there are no targeted treatments in the second-line setting for most patients (not including the occasional patient who accesses alpelisib or takes part in a clinical trial).
Patient Population
The clinical experts agreed that the patients best suited for capivasertib plus fulvestrant would be those eligible for second-line therapy following treatment with an AI and CDK4/6 inhibitor. As per the CAPItello-291trial protocol, the experts noted that patients previously exposed to fulvestrant or to AKT, PI3K, or mTOR inhibitors would neither be eligible; nor would patients with diabetes who are receiving insulin or have a baseline glycated hemoglobin of at least 8.0%. The experts highlighted that in their local practice, they rarely test for PIK3CA, AKT1, or PTEN alterations (outside of clinical trials) because testing is not publicly funded, given that no publicly funded treatments require this companion diagnostic.
Assessing the Response Treatment
The clinical experts indicated that, in clinical practice, a combination of radiography (approximately every 3 months) and biochemical and clinical parameters are used to determine whether a patient is responding to or progressing on treatment; however, they noted that the frequency of radiographic scans may be different across regions and that access to repeat scans can be challenging. The experts also noted that they use CA15 to 3, CEA, and CA 125 tumour markers to monitor patients’ progress on treatment. The experts agreed that a clinically meaningful response includes radiological response or stabilization, improvement in patient symptoms, and maintenance of HRQoL.
Discontinuing Treatment
The clinical experts indicated that treatment with capivasertib plus fulvestrant should be discontinued if the patient experiences disease progression (whether defined radiologically or clinically), find treatment intolerable, or choose.
Prescribing Considerations
The clinical experts indicated that patients receiving capivasertib plus fulvestrant should be under the care of a medical oncologist in the community who can manage the toxicities associated with the therapy. They noted that it would be reasonable for patients to receive the therapy at a distributed oncology centre where day-to-day follow-up is with a general practitioner in oncology.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups.
Input on the review of capivasertib was received from 2 clinician groups: the REAL Canadian Breast Cancer Alliance and the OH-CCO Breast Cancer Drug Advisory Committee. A total of 13 clinicians (8 from the REAL alliance and 5 from the OH-CCO committee) provided input for this submission.
Both the REAL alliance and the OH-CCO committee emphasized that the primary goals of systemic treatment for advanced breast cancer are to improve or prolong survival, maintain or improve quality of life, manage or minimize toxicities associated with treatment, alleviate symptoms, and delay the initiation of chemotherapy (the last point was added by the REAL alliance). While discussing the unmet needs of patients, the OH-CCO committee highlighted that advanced breast cancer is a common and incurable disease, and improved treatments are needed. The group also added that it might be useful to have oral therapies available, given that there are many lines of treatment. The REAL alliance emphasized that treatment options with survival benefit and good tolerability are limited for patients in the second-line setting (i.e., those who have relapsed on first-line therapy in the metastatic setting) and patients who relapse while on, or within 12 months of completing, adjuvant ET. The group further indicated that treatment goals that are not being met by currently available treatments in this population are improving OS, maintaining quality of life, minimizing toxicities, and delaying the start of chemotherapy. The group also noted that not all patients respond to available treatments, and patients may become refractory to current treatment options; thus, additional treatment options might be needed for these patients.
While the OH-CCO committee indicated that the drug under review would add a line of endocrine-based therapy, the REAL alliance recommended it as a treatment option for all patients (i.e., males and premenopausal, perimenopausal, and postmenopausal females) who have HR-positive, HER2-negative, metastatic breast cancer and have progressed on first-line, standard of care treatment in the metastatic setting or have progressed while on, or within 12 months of completing, adjuvant ET and have 1 or more PIK3CA, AKT1, or PTEN alterations.
The OH-CCO Cancer Drug Advisory Committee indicated that in Ontario, patients with metastatic breast cancer have access to funded testing for PIK3CA mutations; however, testing for PTEN and AKT1 mutations is not currently funded. For access to the drug under review, patients would require testing for AKT pathway alterations.
The REAL Canadian Breast Cancer Alliance noted that when determining whether a patient is responding to treatment in clinical practice, the following outcomes are considered to be clinically meaningful responses: stabilization or reduction in the frequency or severity of symptoms (e.g., pain, dyspnea); maintenance or improvement of performance status; ability to maintain or increase activities of daily living; and tumour radiographic response, with either stabilization of disease or response as measured by RECIST 1.1 criteria. Both the REAL alliance and OH-CCO committee agreed that treatment discontinuation is determined based on disease progression or the occurrence of severe or unacceptable toxicity.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The CAPItello-291 phase III study evaluated capivasertib plus fulvestrant vs. placebo plus fulvestrant in patients with HR-positive, HER2-negative advanced breast cancer who were experiencing recurrence or progression on or after an ET-containing regimen. There is no funded standard of care for HR-positive, HER2-negative, advanced breast cancer targeting PIK3CA, AKT1, or PTEN alterations in patients who have progressed following at least 1 endocrine-based regimen in the metastatic setting. In the first-line setting, most patients receive a CDK4/6 inhibitor plus aromatase inhibitor. As per the CDA-AMC provisional funding algorithm, patients in second or later lines are treated with available funded therapies, including endocrine monotherapy (e.g., fulvestrant); a CDK4/6 inhibitor plus fulvestrant (only if no CDK4/6 inhibitor was administered in the first line) or everolimus plus exemestane (not yet funded in the majority of provinces if there is previous exposure to CDK4/6 inhibitors); various chemotherapy drugs (capecitabine, docetaxel, paclitaxel, nab-paclitaxel, doxorubicin, epirubicin, vinorelbine, gemcitabine, 5 fluorouracil-epirubicin-cyclophosphamide); or, in the case of HER2-low, trastuzumab deruxtecan (for patients who have received a prior line of chemotherapy and are no longer eligible for ET). | Comment from the drug plans to inform pERC deliberations. |
Considerations for initiation of therapy | |
Are patients with stable brain metastases eligible for capivasertib plus fulvestrant? | The clinical experts indicated that patients with stable brain metastases should be eligible for capivasertib plus fulvestrant. They noted that concurrent use of steroid therapy may increase the risk of hyperglycemia, but that it would be clinically appropriate with monitoring. |
Considerations for discontinuation of therapy | |
In the CAPItello-291 study, treatment was discontinued for objective radiologic disease progression or for clinical disease progression and/or worsening of disease. What are the criteria to discontinue capivasertib plus fulvestrant in real-world clinical practice? If there is radiologic disease progression, but no clinical deterioration or worsening of disease, can treatment be continued beyond radiologic progression? | The clinical experts noted that treatment with capivasertib plus fulvestrant should continue until disease progression or unacceptable toxicities if patients are clinically responding. |
Can capivasertib be continued as a single drug if fulvestrant is discontinued because of toxicity, and vice versa? | The clinical experts noted that if fulvestrant is discontinued, treatment with capivasertib as a single drug should not be continued, but that if capivasertib is discontinued, fulvestrant monotherapy can be continued. |
Generalizability | |
Eligibility in the CAPItello-291 study included an ECOG PS of 0 or 1. Should patients with an ECOG PS > 1 be eligible? | The clinical experts indicated that it would be reasonable to extend the eligibility to patients with an ECOG PS of 2 or less. |
Adult females (premenopausal and/or postmenopausal) and adult males with metastatic breast cancer were eligible for the CAPItello-291 study. Premenopausal or perimenopausal females were required to be rendered postmenopausal through surgical or chemical means. Should male patients with breast cancer use a GnRH agonist in combination with fulvestrant and capivasertib? | The clinical experts indicated that male patients should receive a GnRH agonist in combination with fulvestrant and capivasertib. The clinical experts also noted that, because management of breast cancer is similar in males and females, the reimbursement request for the inclusion of male patients is appropriate and ensures equitable access to capivasertib plus fulvestrant for males and individuals transitioning to males. |
Should patients currently receiving an alternate second or later line of therapy be switched to capivasertib plus fulvestrant at the time of implementation if the therapy is recommended and considered superior? | The clinical experts indicated that patients receiving an alternate second or later line of therapy who are clinically stable or responding to treatment should not be switched to capivasertib plus fulvestrant but should be eligible to receive capivasertib plus fulvestrant if they experience disease progression or intolerance, with no prior exposure to fulvestrant. |
Care provision issues | |
Fulvestrant is administered as a monthly injection (i.e., 500 mg IM on days 1, 15, and 29, and monthly thereafter). Capivasertib is an oral therapy dosed as 400 mg twice a day (2 tablets of 200 mg taken twice a day for a total daily dose 800 mg) on an empty stomach, given on an intermittent weekly dosing schedule. Patients are dosed on days 1 to 4 in each week (4 days on, 3 days off) of a 28-day treatment cycle. The dosing schedule may be confusing for some patients. Capivasertib is a substrate of CYP3A4. However, data suggest that glucuronidation may be the major metabolic route. Coadministration of some CYP3A4 inhibitors may increase exposure to capivasertib, potentially affecting toxicity, while CYP3A4 inducers may decrease the exposure to capivasertib, potentially affecting efficacy. Drug-drug interaction checking should be performed before initiating therapy and whenever any other therapies are being considered. | Comment from the drug plans to inform pERC deliberations. |
There is a relatively high frequency of adverse effects associated with capivasertib, including diarrhea, rash, nausea, hyperglycemia, and potential hypersensitivity. These require careful monitoring, assessment, and intervention as needed. | Comment from the drug plans to inform pERC deliberations. |
PIK3CA, AKT1, or PTEN testing is not a currently funded standard of care. What are the methods or assays that can test for PIK3CA, AKT1, or PTEN alterations? What is the optimal timing for biomarker testing (e.g., at time of diagnosis, or as part of eligibility assessment before initiation)? Is the PIK3CA, AKT1, or PTEN alteration or mutation stable, or does testing need to be repeated periodically? | The clinical experts indicated that NGS is the preferred assay to test for PIK3CA, AKT1, or PTEN alterations. They also noted that there are other technologies available, such as polymerase chain reaction and Sanger sequencing. However, NGS is superior because it can test for multiple mutations at the same time. The clinical experts indicated that the optimal time for biomarker testing could be at the time of metastatic diagnosis; if the alteration is stable, there is no need for repeat testing. Additional information regarding NGS testing for PIK3CA, AKT1, or PTEN alterations is available in the Testing Procedure Assessment Report. |
What percentage of patients with HR-positive, HER2-negative, metastatic breast cancer harbour PIK3CA, AKT1, or PTEN alterations? | The clinical experts noted that approximately 40% of patients with HR-positive, HER2-negative, metastatic breast cancer harbour PIK3CA, AKT1, or PTEN alterations. |
A previous phase II study (the FAKTION trial) suggested that benefit was limited to tumours with PIK3CA, AKT1, or PTEN mutations. Is there a difference between PIK3CA, AKT1, or PTEN pathway alterations and mutations, and are any different outcomes expected in these groups? | The clinical experts noted that PIK3CA, AKT1, or PTEN alterations and mutations are terms that are used interchangeably in the literature and generally have the same meaning. |
System and economic issues | |
The sponsor estimates that 214 patients in year 1, 308 patients in year 2, and 393 patients in year 3 would receive treatment with capivasertib plus fulvestrant. These numbers yield direct drug costs for capivasertib plus fulvestrant of $16.1 million, $23.2 million, and $29.6 million in years 1 to 3, respectively. This results in an incremental budget impact of $8.9 million in year 1, $12.8 million in year 2, and $16.4 million in year 3, amounting to a 3-year incremental budget impact of $38.1 million. What are the CDA-AMC–estimated patient numbers and budget impact analysis? | This is addressed in the pharmacoeconomic report. |
The sponsor estimates that the total pan-Canadian, 3-year incremental budget impact of PIK3CA, AKT1, or PTEN alteration testing would be $3.9 million. What are the CDA-AMC–estimated testing costs? | This is addressed in the pharmacoeconomic report. |
Generic fulvestrant is commercially available. Confidential prices are available for all CDK4/6 inhibitors and trastuzumab deruxtecan. There are generics commercially available for aromatase inhibitors, everolimus, and all chemotherapy comparators. | Comment from the drug plans to inform pERC deliberations. |
CDA-AMC = Canada’s Drug Agency; CDK4/6 = cyclin-dependent kinase 4 and 6; CYP3A4 = cytochrome P450 3A4; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ET = endocrine therapy; GnRH = gonadotropin-releasing hormone; HR = hormone receptor; HER2 = human epidermal growth factor receptor 2; IM = intramuscularly; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; NGS = next-generation sequencing.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of capivasertib 400 mg, taken orally twice daily, plus fulvestrant 500 mg, administered intramuscularly every 28 days, in the treatment of adults with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy. The focus will be placed on comparing capivasertib plus fulvestrant to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of capivasertib plus fulvestrant is presented in 4 sections, with our critical appraisal of the evidence included at the end of each. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section would usually include long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section includes 1 additional study that was considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
One pivotal trial identified in the systematic review
One indirect treatment comparison
One additional study addressing gaps in the evidence.
Systematic Review
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included study are summarized in Table 5. The trial design is shown in Figure 1.
The CAPItello-291 trial26 is an ongoing, phase III, randomized, placebo-controlled, multicentre trial that aims to assess the efficacy and safety of capivasertib 400 mg, taken orally, plus fulvestrant 500 mg, administered through intramuscular injection, compared with matched placebo plus fulvestrant in adults with locally advanced (inoperable) or metastatic, HR-positive, HER2-negative breast cancer. The trial enrolled patients who had disease recurrence or progression during or after AI therapy, with or without a CDK4/6 inhibitor. Patients who tested positive for tumours with 1 or more PIK3CA, AKT1, or PTEN alterations were assigned to an altered population subgroup. The objective of the trial was to assess efficacy and safety in both the overall population (i.e., all enrolled patients) and the altered population subgroup. The focus of the Health Canada indication and reimbursement request is aligned with the altered population; however, given that the overall population also included a proportion of patients with known AKT-altered status, the results for the overall population were also included. It should be noted that 59% of patients in the overall population do not meet the reimbursement request (i.e., are of known nonaltered or unknown alteration status).
Table 5: Details of Study Included in the Systematic Review
Detail | CAPItello-291 trial |
|---|---|
Design and population | |
Study design | Phase III, double-blind, placebo-controlled, parallel-group, randomized trial |
Locations | This study was conducted at 181 sites across 3 geographic regions. Region 1 included the US, Canada, Western Europe, Australia, and Israel (12 sites); region 2 included Latin America, Eastern Europe, and Russia (23 sites); and region 3 included Asia (46 sites). Across geographic regions, the study was conducted in 19 countries (Argentina, Australia, Belgium, Canada, China, France, Germany, Hungary, Israel, Italy, Japan, Peru, Poland, Russian Federation, Spain, Taiwan, the UK, and the US). |
Patient enrolment dates | Start date: April 16, 2020 End date: October 13, 2021 |
Randomized (N) | Total: N = 708
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Capivasertib 400 mg (2 tablets of 200 mg) orally twice daily (b.i.d. on days 1 to 4 in each week of a 28-day treatment cycle) plus fulvestrant 500 mg (2 intramuscular injections of 250 mg/5 mL solution) on day 1 of weeks 1 and 3 of cycle 1, and then on day 1, week 1 of each 28-day treatment cycle thereafter until disease progression, unacceptable toxicity, or treatment discontinuation |
Comparator(s) | Matched placebo plus fulvestrant 500 mg |
Study duration | |
Screening phase | Up to 42 days before first treatment dose |
Treatment phase | From time of first dose until discontinuation criteria were met |
Follow-up phase | Through 28 days after last dose of study treatment for safety follow-up. Follows the imaging schedule until radiographic progression for long-term follow-up. |
Outcomes | |
Primary outcome | Investigator-assessed PFS (primary PFS analysis conducted after a median follow-up of 13 months [database lock on 03 October 2022]) in the overall population and in the subgroup with PIK3CA, AKT1, or PTEN alterations |
Secondary and exploratory outcomes | Secondary (in overall and altered populations):
Exploratory:
|
Publication status | |
Publications | ClinicalTrials.gov number NCT04305496 Turner et al. (2023) |
AI = aromatase inhibitor; b.i.d. = twice daily; CBR = clinical benefit rate; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-BR23 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; ER = estrogen receptor; ET = endocrine therapy; FFPE = formalin-fixed paraffin-embedded; HbA1C = hemoglobin A1C; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; LHRH = luteinizing hormone–releasing hormone; mTOR = mammalian target of rapamycin; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PFS2 = time from randomization to second progression or death; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SERD = selective estrogen receptor degrader; VAS = visual analogue scale.
Source: CAPItello-201 Clinical Study Report [Details included in the table are from the sponsor’s Summary of Clinical Evidence].26
Enrolled patients were randomly assigned through an interactive web response system in a 1-to-1 ratio (N = 708 from 181 sites) to receive capivasertib plus fulvestrant (N = 355) or placebo plus fulvestrant (N = 353). Sixteen patients were randomized across 5 sites in Canada. Randomization was stratified by liver metastases (yes or no), prior use of CDK4/6 inhibitors (yes or no), and geographic location (region 1, 2, or 3). The patients and investigators were blinded to treatments. The trial included a screening phase of up to 28 days and a treatment phase that lasted until discontinuation criteria were met: these included disease progression, unacceptable toxicity, withdrawal of consent, or death. All patients were followed up 28 days after treatment discontinuation and until radiographic progression for long-term follow-up.
The outcomes relevant to the CDA-AMC review included the primary outcome of PFS per RECIST 1.1, as assessed by the investigators, and the secondary outcomes of OS and safety. HRQoL — a secondary outcome in the trial that was also considered relevant — was measured using the EORTC QLQ-C30 and EORTC QLQ-BR23. At the request of the sponsor, PFS2 and time to first subsequent chemotherapy (defined as time from randomization to the earlier start date of subsequent chemotherapy after discontinuation of randomized treatment or death from any cause) were included for the altered population and are included as an appendix (outcome measures and results are summarized in Appendix 1).
Efficacy and safety data were evaluated at the planned primary analysis for PFS, with a data cut-off date of August 15, 2022. An interim analysis for OS was also conducted on this date.
Figure 1: Study Design of CAPItello-291 Trial
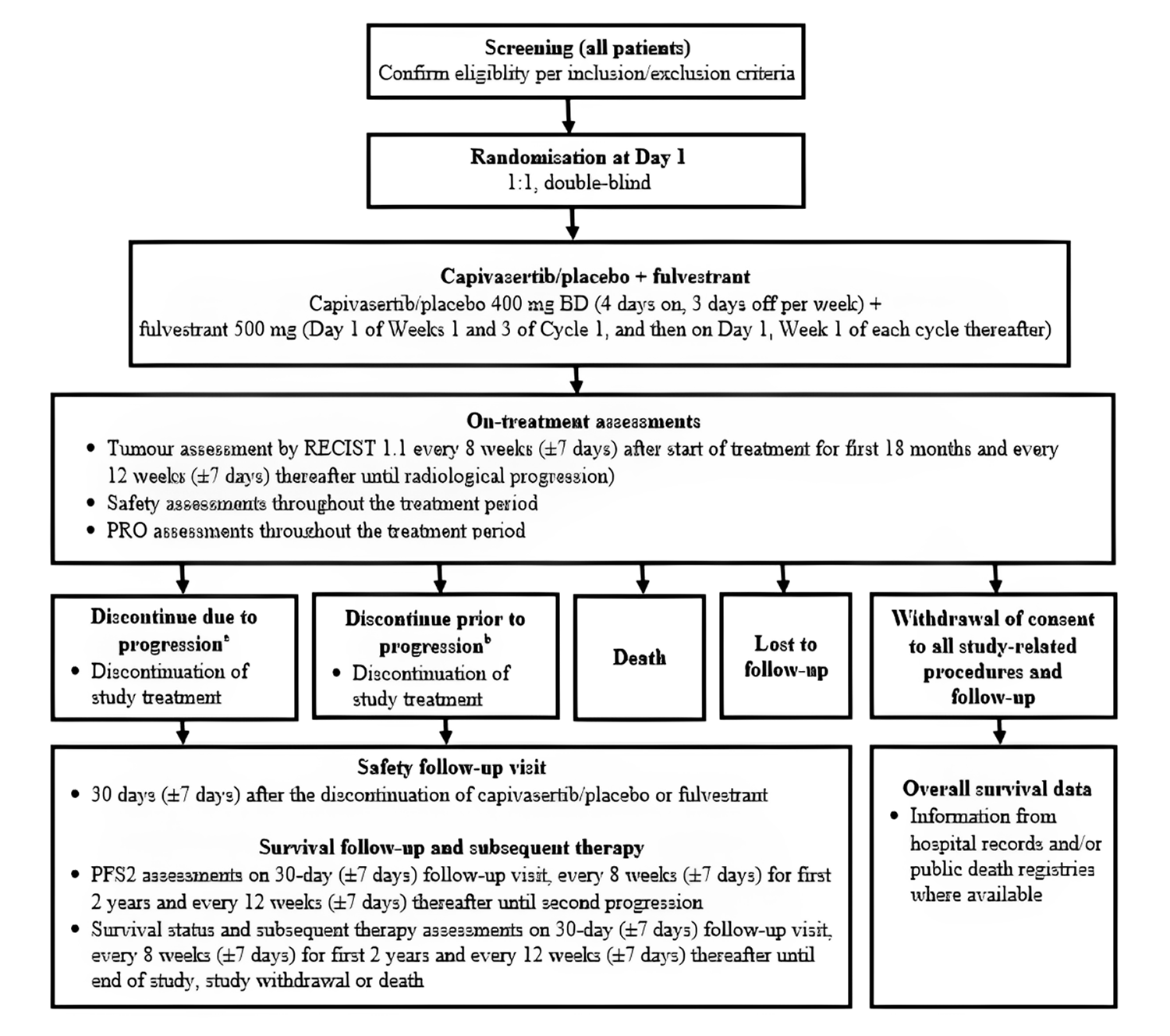
BD = twice daily; PFS2 = time from randomization to second progression or death; PGI-TT = Patient's Global Impression of Treatment Tolerability; PRO = patient-reported outcome; PRO-CTCAE = patient-reported outcome version of the Common Terminology Criteria for Adverse Events; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aIf a patient discontinued because of progression, PROs (not including PGI-TT and PRO-CTCAE) were assessed at progression and every 4 weeks (± 3 days) postdiscontinuation until PFS2.
bPatients who discontinued treatment before progression continued to be scanned by RECIST 1.1 every 8 weeks for the first 18 months and every 12 weeks thereafter until progression, regardless of the reason for treatment discontinuation. If the patient discontinued because of toxicity, but did not progress, then PROs (not including PGI-TT and PRO-CTCAE) were assessed every 4 weeks until progression, at progression, and every 4 weeks postprogression until PFS2. Note: Follow-up visit should read 30 days.
Source: CAPItello-201 Clinical Study Report; details included in the figure are from the sponsor’s Summary of Clinical Evidence.26
Populations
Inclusion and Exclusion Criteria
A detailed description of the key inclusion and exclusion criteria for the CAPItello-291 trial is provided in Table 5. Eligible patients were adult, premenopausal, perimenopausal, or postmenopausal females or males with HR-positive, HER2-negative, locally advanced (i.e., inoperable) or metastatic breast cancer. Patients had to have relapse or disease progression during or after treatment with an AI, with or without previous CDK4/6 inhibitor therapy. Patients were allowed to have received up to 2 previous lines of ET and 1 previous line of chemotherapy in the context of advanced disease. The trial protocol required the enrolment of a minimum of 51% of patients with previous CDK4/6 inhibitor treatment. Patients were required to have an ECOG Performance Status of 0 or 1 with no deterioration over the preceding 2 weeks and life expectancy of greater than or equal to 12 weeks. Patients with previous exposure to fulvestrant or another selective ER degrader, or to AKT, PI3K, or mTOR inhibitors, were excluded, as were patients with diabetes who were receiving insulin or had a baseline hemoglobin A1C level of at least 8.0% (i.e., 63.9 mmol per mole). Mandatory baseline tissue samples, derived from the primary or recurrent cancer site, were required from all patients at screening and analyzed postrandomization to determine PIK3CA, AKT1, or PTEN alteration status. Activating mutations in PIK3CA and AKT1 genes and inactivating alterations in PTEN genes were determined centrally by means of NGS with the use of the FoundationOneCDx assay in all countries except China, where OncoScreen Plus was performed. Patients whose tumours had at least 1 qualifying alteration in these 3 genes were included in the altered population subgroup. Patients with tumours that did not have a qualifying alteration detected in any of these 3 genes, or with an unknown test result, were included in the AKT pathway–nonaltered population.
Interventions
Patients received capivasertib 400 mg, taken orally twice daily for 4 days followed by 3 days off, plus fulvestrant 500 mg, administered intramuscularly every 14 days for the first 3 injections and every 28 days thereafter, or matched placebo plus fulvestrant. One cycle was defined as 4 weeks of capivasertib or placebo. The capivasertib and placebo film-coated tablets were identical in appearance and presented in the same packaging to ensure blinding of capivasertib. Patients were to continue treatment until disease progression (assessed according to RECIST 1.1), unacceptable toxicity, withdrawal of consent, or death. Dose reduction of capivasertib or placebo was allowed: from 400 mg twice daily to 320 mg twice daily, and then to 200 mg twice daily, as clinically appropriate. Dose reduction of fulvestrant was not allowed. Patients who discontinued capivasertib or fulvestrant for reasons other than disease progression could continue to receive the other drug. Premenopausal and perimenopausal females were allowed to receive a luteinizing hormone–releasing hormone agonist for the duration of the trial treatment period.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CDA-AMC as well as the input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform its expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Serious, treatment-emergent AEs were considered important for informing the expert committee deliberations and were also assessed using GRADE.
Table 6: Efficacy Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | CAPItello-291 trial |
|---|---|---|
Progression-free survival | 6 months and 12 months | Primarya |
Overall survival | 18 months and 24 months | Secondarya |
EORTC QLQ-C30 global health status | At cycle 10 | Secondary |
EORTC QLQ-BR23 functional and symptom scalesb | At cycle 17 | Secondary |
EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30.
aStatistical testing for these outcomes were adjusted for multiple comparisons.
bThe EORTC QLQ-BR23 functional and symptom scales included body image, sexual functioning, sexual enjoyment, future perspective, systemic therapy side effects, breast symptoms, arm symptoms, and feeling upset by hair loss.
Source: CAPItello-201 Clinical Study Report; details included are from the sponsor’s Summary of Clinical Evidence.26
Progression-Free Survival
The primary outcome for the CAPItello-291 trial was investigator-assessed PFS in both in the overall population and altered population subgroup. PFS was defined as the time from randomization until disease progression per RECIST 1.1, or death because of any cause, regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy before progression. Tumour assessments were done with CT or MRI of the chest, abdomen, and pelvis at screening every 8 weeks for the first 18 months, and then every 12 weeks until disease progression. Radiographic bone scans were performed at screening and repeated as clinically indicated. Patients who discontinued capivasertib or fulvestrant for reasons other than disease progression continued to undergo scans every 8 weeks until disease progression.
Overall Survival
The secondary outcome of OS was defined as the time from randomization until death because of any cause regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy. Assessments for survival were conducted every 8 weeks for the first 2 years following objective disease progression or treatment discontinuation and then every 12 weeks.
Health-Related Quality of Life
The secondary outcome of HRQoL was measured by change in baseline in patient-reported global health status as well as disease-specific functioning and symptom scales using the EORTC QLQ-C30 and EORTC QLQ-BR23 questionnaires, respectively.
The EORTC QLQ-C30 consists of 30 questions that can be combined to produce 5 functional domains (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and/or vomiting), and a 2-item global health status scale along with 5 individual item symptom scores (for appetite loss, dyspnea, insomnia, constipation, and diarrhea). An outcome variable consisting of a score from 0 to 100 was derived for the global measure of health status scale, with higher scores indicating better health status. The validity, reliability, and MID of the EORTC QLQ-C30 are summarized in Table 7.
The EORTC QLQ-BR23 is a breast cancer–specific module. The self-administered instrument includes 23 items and yields 5 multi-item scores (body image, sexual functioning, arm symptoms, breast symptoms, and systemic therapy side effects). Items are scored on a 4-point verbal rating scale, where the possible responses are not at all, a little, quite a bit, and very much. Scores are converted to a scale from 0 to 100, where higher scores indicate better functioning, better HRQoL, or greater levels of symptoms. The validity and reliability of the EORTC QLQ-BR23 instrument has been assessed among Dutch, Spanish, and American patients with breast cancer; the instrument is summarized in Table 7.44 For both questionnaires, no data were identified in the literature for responsiveness; the sponsor suggested an absolute change greater than or equal to 10 points from baseline, which was informed by the literature and used in other trials, to define a clinically meaningful change.45,46
Safety Outcomes
The assessment of safety was based on the proportion of patients experiencing 1 or more AEs, SAEs, notable AEs, AEs leading to discontinuation, AEs leading to dose modification, and deaths. AEs were reported at each study visit and coded using the Medical Dictionary for Regulatory Activities, Version 25.0. An independent data and safety monitoring committee assessed the progress of the trial approximately every 6 months and reviewed unblinded safety data.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A 30-item, patient-reported, cancer-specific HRQoL questionnaire using 4-point and 7-point Likert scales.47 There are 15 domains for the EORTC QLQ-C30. Functional scales ranging from 0 to 100 (with higher scores indicating higher functioning) include global health status or QoL, physical functioning, role functioning, emotional functioning, cognitive functioning, and social functioning. Symptom scales ranging from 0 to 100 (with higher scores indicating a greater degree of symptoms or worse condition) include fatigue, pain, nausea and/or vomiting, dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties.47 | Content validity: When mapping to WHO’s ICF framework, 25 of the 30 items in the EORTC QLQ-C30 were endorsed by 21 health care professionals using the Delphi technique (≥ 70% agreement).48 Discriminant validity: Spearman’s rank correlations with external parameters, such as ECOG Performance Status, ranged from 0.02 to 0.56 among 150 patients in Canada with metastatic breast cancer.49 Convergent validity: Spearman’s rank correlations with scores on the Profile of Mood States and Psychosocial Adjustment to Illness Scale ranged from 0.02 to 0.76 among 150 patients in Canada with metastatic breast cancer.49 Reliability: Interrater reliability: The median kappa coefficient for patient-observer agreement across the 30 items in the EORTC QLQ-C30 was 0.86, with a range of 0.48 to 1.00, in patients with metastatic breast cancer, representing substantial to near-perfect agreement for most items.50,51 Responsiveness: No literature was identified that assessed responsiveness in patients with breast cancer. | In a paper synthesizing data from 21 published EORTC QLQ-C30 phase III trials enrolling 13,015 patients across 9 cancer types, the anchor-based MID for the global health status scale for between-group change over time in patients with breast cancer ranged from –13 to –6 for deterioration, and from 8 to 11 for improvement.52 The sponsor suggested an absolute change greater than or equal to 10 points from baseline, which was informed by the literature and used in other trials, to define a clinically meaningful change.45,46 |
EORTC QLQ-BR23 | A self-administered instrument including 23 items and yielding 5 multi-item scores (body image, sexual functioning, arm symptoms, breast symptoms, and systemic therapy side effects). Items are scored on a 4-point verbal rating scale as not at all, a little, quite a bit, and very much. Scores are converted to a 0 to 100 scale in the same way as specified for EORTC QLQ-C30, in which higher scores indicate better functioning, better HRQoL, or greater level of symptoms.44 | The validity and reliability of the EORTC QLQ-BR23 instrument were assessed among 170 Dutch, 168 Spanish, and 158 American patients with breast cancer in Amsterdam, Pamplona, and Houston, Texas, respectively. These patients were receiving treatments with chemotherapy or radiotherapy (Dutch and Spanish patients) or regular care (American patients) in their respective care centres.44 Validity: The clinical validity was assessed by the known-group comparison method, where the instrument demonstrated the ability to discriminate between subgroups of patients known to differ in clinical status.44 Reliability: The Cronbach alpha coefficient value ranged from 0.46 to 0.94 in the Spanish patient population, 0.57 to 0.89 in the Dutch patient population, and 0.70 to 0.91 in the American patient population, demonstrating that all scales in the American patient group demonstrated acceptable internal consistency by exceeding the accepted Cronbach alpha threshold of > 0.70, defined by Nunnally et al.44,53 Responsiveness: No literature was identified that assessed responsiveness in patients with breast cancer. | The observed MIDs ranged from 0.4 to 4 at 6 months and from 7 to 20 at 3 months for deterioration. In case of improvement, the observed MIDs ranged from 0.7 to 2 at 6 months and from 2 to 15 at 3 months.54 The sponsor suggested an absolute change greater than or equal to 10 points from baseline, which was informed by the literature and used in other trials, to define a clinically meaningful change.45,46 |
ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; ICF = International Classification of Functioning, Disability and Health; MID = minimal important difference; QoL = quality of life.
Statistical Analysis
A summary of the statistical analysis of efficacy outcomes is provided in Table 8.
Sample Size and Power Calculation
Assuming a significance level of 5%, a total of 492 OS events were required to achieve 90% power to detect a treatment effect of an average hazard ratio of 0.74 in the overall population, assuming a 12-month delay to a treatment effect, and a hazard ratio of 0.64 after the delay. Assuming 70% maturity at the time of the final analysis, approximately 700 patients were needed for randomization. Of these 700 randomized patients, it was expected that a minimum of 280 patients will have a tumour harbouring an eligible PIK3CA, AKT1, or PTEN alteration, based on a prevalence rate of approximately 40% to 45%, and that a minimum of approximately 224 patients will be in the altered population, assuming a test failure rate of 20%. The PFS primary analysis took place after PFS reached approximately 77% maturity (542 events) in the overall population and approximately 77% of PFS events had occurred in patients whose tumours harboured an eligible PIK3CA, AKT1, or PTEN alteration, based on a prevalence rate of approximately 40% to 45% (174 events would have been observed if the test failure rate was 20%). Assuming a significance level of 3.5%, a total of 542 PFS events would provide greater than 99% power to detect a treatment-effect hazard ratio of 0.64 in the overall population. Given the estimated sample size of the altered population, and assuming a significance level of 5% following recycling of the remaining 3.5% alpha, a total of 217 PFS events (approximately 77% maturity) would provide 90.8% power to detect a treatment-effect hazard ratio of 0.64 in the altered population. After all the predefined PFS outcomes had been tested, the remaining alpha was used for testing OS in the altered population and subsequently for OS in overall population. The OS interim analysis was to occur when approximately 394 OS events had been observed in the overall population (56% maturity, 80% information fraction). The OS final analysis will take place when approximately 70% maturity has been observed in both the overall population and altered population. The exact significance level will be determined according to the O’Brien and Fleming method based on the actual number of events observed at the OS interim analysis.
Statistical Testing
The primary outcome of PFS was analyzed using a 1-sided log-rank test, stratified by the presence or absence of liver metastases, previous use of a CDK4/6 inhibitor (yes or no), and geographic area (assessed in the overall population only) for generation of the P value and using a method that corresponds to the Breslow approach for handling ties. To estimate the effect of treatment, the hazard ratio, associated 95% CI, and CI adjusted for multiplicity were estimated from a stratified Cox PH model with the Efron method to control for ties. OS was analyzed in the same way as PFS. To control the family-wise error rate at 5% (2-sided) for the treatment comparisons in OS and PFS (in both the overall and altered populations), a predefined method for multiplicity control with an alpha-exhaustive recycling strategy accounting for intrinsic correlation between test statistics was applied. According to alpha (test mass) splitting and alpha recycling, if the higher-level hypothesis in the methods for multiplicity control is rejected for superiority, then the next lower-level hypothesis is tested. The test mass that becomes available after each rejected hypothesis is recycled to the lower-level hypotheses not yet rejected.
For the HRQoL outcomes of EORTC QLQ-C30 and EORTC QLQ-BR23, analyses included patients with an evaluable baseline assessment and at least 1 evaluable postbaseline assessment and were reported as changes from baseline. For EORTC QLQ-C30 global health status, data were reported up to cycle 10; beyond this, the data were excluded from the analysis because there were fewer than 20 observations in the group receiving placebo plus fulvestrant. For the EORTC QLQ-BR23 scales score, data were reported at cycle 17. Although postbaseline data for most scales were limited at this time point, the data were consistent with cycles closer to the median duration of follow-up for all patients. A mixed-effects model for repeated measures analysis for randomized patients with an evaluable baseline assessment and at least 1 evaluable postbaseline assessment was performed. Analyses were not controlled for multiplicity. Safety data were summarized descriptively.
Subgroup Analysis
For the prespecified subgroups of interest, the stratified hazard ratios of PFS were performed according to AKT pathway–altered status and previous use of a CDK4/6 inhibitor. The subgroup analysis was conducted using a Cox PH model and presented as forest plots.
Data Imputation Methods and Censoring
For PFS, if patients progressed or died immediately after 2 or more consecutive missed visits, they were censored at the time of the latest evaluable RECIST 1.1 assessment before the missed visits. Patients who had not progressed, or who had died by the time of analysis, were censored at the time of the latest date of assessment from their last evaluable RECIST 1.1 assessment. For OS, if patients were known to have died, but only a partial death date was available, the date of death was imputed as the latest of the last date known to be alive plus 1 from the database. Patients not known to have died at the time of analysis were censored based on the last recorded date on which they were known to be alive. Data imputation for the HRQoL outcomes consisted of the following: if less than 50% of the subscale items were missing, then the subscale score was divided by the number of nonmissing items; if at least 50% of the items were missing, that subscale was treated as missing; and missing single items were treated as missing.
Sensitivity Analyses
The key sensitivity analysis for PFS was assessment by BICR. In addition, sensitivity analyses were conducted to assess the potential impact of COVID-19 deaths on PFS and OS. Evaluation-time bias and attrition bias were assessed by analyzing site investigator data. To assess the possibility of ascertainment bias, PFS was analyzed based on BICR data.
Table 8: Statistical Analysis of Efficacy Outcomes
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS | Stratified log-rank test providing a P value and stratified Cox proportional hazard model providing a hazard ratio (95% CI and alpha-adjusted CI) for formal primary analysis using RECIST 1.1 based on investigator assessments for the FAS PIK3CA, AKT1, or PTEN֪–altered subgroup | Stratified by geographic region (region 1: US, Canada, Western Europe, Australia, and Israel; region 2: Latin America, Eastern Europe and Russia; region 3: Asia), liver metastases (yes vs. no), and prior use of CDK4/6 inhibitors (yes vs. no) | If the patient progresses or dies immediately after 2 or more consecutive missed visits, the patient is censored at the time of the latest evaluable RECIST 1.1 assessment before the 2 missed visits. Patients who have not progressed or died at the time of analysis are censored at the time of the latest date of assessment from their last evaluable RECIST 1.1 assessment. |
|
OS | Stratified log-rank test using similar methodology as described for the primary PFS end point | Stratified by randomization stratification variables | If a patient is known to have died, but only a partial death date is available, then the date of death will be imputed as the latest of the last date on which they were known to be alive plus 1 from the database and the death date using the available information provided:
If there is evidence of death, but the date is entirely missing, it is treated as missing (i.e., censored at the last known alive date). |
|
EORTC QLQ-C30 | Change from baseline derived using an MMRM analysis of all the postbaseline scores for each visit. The model includes treatment, visit, treatment-by-visit interaction, and stratification factors. Baseline score, baseline score by visit as covariates, and patient will be included as a random effect. | Same as for PFS | For each subscale, if < 50% of the subscale items are missing, then the subscale score will be divided by the number of nonmissing items. If at least 50% of the items are missing, then that subscale will be treated as missing. Missing single items are treated as missing. The reason for any missing questionnaire will be identified and recorded. | None |
EORTC QLQ-BR23 | Same as for EORTC QLQ-C30 | Same as for PFS | Same as for EORTC QLQ-C30 | None |
BICR = blinded independent central review; CI = confidence interval; EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; MMRM = mixed-effects model for repeated measures; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: CAPItello-201 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Analysis Populations
The analysis populations of the CAPItello-291 trial are provided in Table 9. The efficacy outcomes, including HRQoL, were analyzed based on the full analysis set (FAS). The safety outcomes were analyzed using the safety analysis set, defined as patients who received at least 1 dose of any study medication.
Table 9: Analysis Populations in the CAPItello-29 Trial
Population | Definition | Application |
|---|---|---|
FAS (overall population) | This population comprised all patients who were randomized into the study, excluding those randomized in China after the first visit of the last patient in the global cohort. The FAS was analyzed according to randomized treatment regardless of the treatment received (i.e., using the intent-to-treat principle). Patients who were randomized, but did not subsequently receive treatment, are included in the FAS. | OS, PFS, EORTC QLQ-C30, and EORTC QLQ-BR23 analyses in the overall population |
SAS (overall population) | The SAS comprised all patients included in the FAS who received at least 1 dose of study drug (fulvestrant, capivasertib, or placebo) and were analyzed according to the treatment received. If a patient received at least 1 dose of capivasertib, they were summarized in the capivasertib groupfor safety summaries (e.g., the capivasertib groupincluded patients randomized to capivasertib who received at least 1 dose of capivasertib, or placebo patients who received at least 1 dose of capivasertib in error at any time). If a patient who was randomized to capivasertib received only placebo, then they were summarized as part of the placebo group. Patients who received only fulvestrant were also included in the safety analysis set and were included in the treatment groupto which they were randomized. | Safety analyses in the overall population |
Altered population subgroup FAS (altered population) | This population comprised all patients included in the FAS who had a PIK3CA, AKT1, or PTEN-altered tumour determined by central testing. | OS, PFS, EORTC QLQ-C30, and EORTC QLQ-BR23 analyses in the altered population |
Altered population subgroup SAS (altered population) | This population comprised all patients included in the SAS with a PIK3CA, AKT1, or PTEN-altered tumour determined by central testing. | Safety analyses in the altered population |
EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; OS = overall survival; PFS = progression-free survival; SAS = safety analysis set.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Results
This section includes data from both the overall population and altered population. The focus of the reimbursement request is the altered population; however, given that the overall population also included a proportion of patients with known AKT-altered status, the results for the overall population were included for PFS, OS, and harms. It should be noted that 59% of patients in the overall population do not meet the reimbursement request criteria. For brevity, only the results for the HRQoL outcomes in the altered population are reported; these results reflect those of the overall population.
Patient Disposition
A summary of patient disposition for the data cut-off date of August 15, 2022, is in Table 10. In total, 901 patients were screened, of whom 708 patients (79%) were randomized to capivasertib plus fulvestrant (n = 355) or placebo plus fulvestrant (n = 353). One hundred and 93 patients were screened out because they did not meet 1 or more of the eligibility criteria. Three patients in the group assigned to placebo plus fulvestrant did not receive treatment; 1 died before their first dose, 1 withdrew consent, and the reason for the third not receiving treatment was unknown. In the overall population, capivasertib was discontinued in 292 patients (82.3%), and placebo was discontinued in 307 patients (87.7%). The main reason for discontinuation of capivasertib or placebo was disease progression, which occurred in 209 patients (58.9%) receiving capivasertib plus fulvestrant and in 273 patients (78.0%) receiving placebo plus fulvestrant. The disposition of the patients in the altered population was consistent with that of the overall population. In the altered population, capivasertib treatment was discontinued in ███ patients (█████), and placebo was discontinued in ███ (██████ patients. The main reason for discontinuation of capivasertib or placebo was disease progression, which occurred in ██ patients (█████) receiving capivasertib plus fulvestrant and in ███ (██████ patients receiving placebo plus fulvestrant. Numerically, a larger proportion of patients discontinued capivasertib or placebo because of AEs (12.4% versus 1.7%, respectively, in the overall population and ████ versus █████ respectively, in the altered population). Numerically, a larger proportion of patients terminated the study in the group receiving placebo plus fulvestrant (█████ versus █████, respectively, in the overall population and █████ versus █████, respectively, in the altered population). In total, important protocol deviations were reported in 62 patients (8.8%), primarily because of restricted prior concomitant medications. The types and frequencies of these deviations were comparable between the treatment groups.
Table 10: Summary of Patient Disposition in the CAPItello-291 Trial
Patient disposition | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant | Placebo plus fulvestrant | Capivasertib plus fulvestrant | Placebo plus fulvestrant | |
Screened, Na | 901 | |||
Screened out, Nb | 193 | |||
Randomized, N (%) | 355 (100) | 353 (100) | 155 (100) | 134 (100) |
Treated, N | 355 (NR) | 350 (NR) | 155 (NR) | 133 (NR) |
Discontinued capivasertib, n (%)c | 292 (82.3) | 307 (87.7) | ███ ██ | ███ ██ |
Reason for capivasertib or placebo discontinuation, n (%)c | ||||
Condition under investigation worsenedd | 209 (58.9) | 273 (78.0) | ██ ███ | ███ ██ |
Adverse event | 44 (12.4) | 6 (1.7) | ██ ███ | █████ |
Subjective disease progressione | 16 (4.5) | 17 (4.9) | █████ | █████ |
Patient decision | 13 (3.7) | 6 (1.7) | █████ | █████ |
Other | 7 (2.0) | 4 (1.1) | █████ | █████ |
Severe non-compliance with protocol | 2 (0.6) | 0 | █████ | █████ |
Investigator decision | 1 (0.3) | 1 (0.3) | █████ | █████ |
Discontinued fulvestrant, n (%)c | 287 (80.8) | 308 (88.0) | ███ ███ | ███ ██ |
Reason for fulvestrant discontinuation, n (%)c | ||||
Condition under investigation worsenedd | 230 (64.8) | 275 (78.6) | ███ ███ | ███ ██ |
Subjective disease progressione | 19 (5.4) | 17 (4.9) | █████ | █████ |
Patient decision | 15 (4.2) | 7 (2.0) | █████ | █████ |
Adverse event | 12 (3.4) | 2 (0.6) | █████ | █████ |
Other | 7 (2.0) | 5 (1.4) | █████ | █████ |
Severe non-compliance with protocol | 1 (0.3) | 0 | █████ | █████ |
Investigator decision | 3 (0.8) | 2 (0.6) | █████ | █████ |
Terminated study, n (%) | 105 (29.6) | 137 (38.8) | █████ | █████ |
Deathf | 85 (23.9) | 105 (29.7) | █████ | █████ |
Withdrawal by patient | 17 (4.8) | 27 (7.6) | █████ | █████ |
Lost to follow-up | 3 (0.8) | 3 (0.8) | █████ | █████ |
Other | 0 | 2 (0.6) | █████ | █████ |
Ongoing study, n (%) | 250 (70.4) | 216 (61.2) | █████ | █████ |
Ongoing treatment at DCO 1, n (%)c | 71 (20.0) | 43 (12.3) | █████ | █████ |
Ongoing capivasertib or placebo treatment | 63 (17.7) | 43 (12.3) | █████ | █████ |
Ongoing fulvestrant treatment | 68 (19.2) | 42 (12.0) | █████ | █████ |
FAS, n (%)g | 355 (100) | 353 (100) | 155 (43.7) | 134 (38.0) |
SAS, n (%)h | 355 (100) | 350 (99.2) | 155 (43.7) | 133 (37.7) |
DCO = data cut-off; FAS = full analysis set; NR = not reported; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAS = safety analysis set.
aInformed consent received.
bPercentages are calculated from the number of patients not randomized. Includes patients enrolled and still in screening.
cPercentages are calculated from the number of patients who received treatment.
dUsed for radiological progression, as assessed by RECIST 1.1.
eUsed for clinical disease progression (i.e., progression with symptoms only, without radiological evidence of progression).
fObtained from public records or survival follow-up.
gOverall population: all randomized patients, regardless of the treatment actually received. Patients who were randomized, but did not subsequently go on to receive study treatment, are included in the analysis in the treatment group to which they were randomized. Altered population: all patients in the FAS with a PIK3CA, AKT1, or PTEN-altered tumour.h Overall population: all patients randomized into the study who receive at least 1 dose of study drug. Altered population: all patients included in the SAS with a PIK3CA, AKT1, or PTEN-altered tumour.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Baseline Characteristics
A summary of baseline demographic and disease characteristics among patients in the FAS population is in Table 11. The characteristics outlined in the table are limited to those most relevant to this review or that were expected to affect the outcomes or interpretation of the study results. Overall, key baseline characteristics were generally balanced between the treatment groups in both populations, and were similar between the overall and altered populations. The trial population was predominately white (58%) and female (99%), with a mean age of 58 years (range, 26 years to 90 years). Most patients (66.0%) had an ECOG Performance Status of 0 (indicating good overall performance), were postmenopausal females (77.0%), and had previously received a CDK4/6 inhibitor (70%). A similar proportion of patients in both groups had an altered tumour status (approximately 41%). Among the 419 patients included in the nonaltered population subgroup, 25.3% (n = 106) had an unknown alteration status.
Table 11: Summary of Baseline Characteristics for the FAS in the CAPItello-291 Trial
Patient disposition | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 355) | Placebo plus fulvestrant (n = 353) | Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
Mean age (SD), year | 58.6 (11.25) | 57.4 (11.91) | 58.8 (10.26) | 59.8 (11.61) |
Sex, n (%) | ||||
Male | 3 (0.8) | 4 (1.1) | 2 (1.3) | 0 |
Female | 352 (99.2) | 349 (98.9) | 153 (98.7) | 134 (100) |
Race, n (%) | ||||
White | 201 (56.6) | 206 (58.4) | 75 (48.4) | 76 (56.7) |
Asian | 95 (26.8) | 94 (26.6) | 48 (31.0) | 35 (26.1) |
Black or African American | 4 (1.1) | 4 (1.1) | 2 (1.3) | 1 (0.7) |
American Indian or Alaska Native | 2 (0.6) | 2 (0.6) | 1 (0.6) | 1 (0.7) |
Native Hawaiian or other Pacific Islander | 1 (0.3) | 0 | 0 | 0 |
Other | 52 (14.6) | 47 (13.3) | 29 (18.7) | 21 (15.7) |
ECOG Performance Status, n (%) | ||||
0 (normal activity) | 224 (63.1) | 241 (68.3) | 93 (60.0) | 97 (72.4) |
1 (restricted activity) | 131 (36.9) | 111 (31.4) | 62 (40.0) | 36 (26.9) |
2 (in bed less than or equal to 50% of the time) | 0 | 1 (0.3) | 0 | 1 (0.7) |
Site of metastases, n (%) | ||||
Bone only | 51 (14.4) | 52 (14.7) | 25 (16.1) | 16 (11.9) |
Liver | 156 (43.9) | 150 (42.5) | 70 (45.2) | 53 (39.6) |
Visceral | 237 (66.8) | 241 (68.3) | 103 (66.5) | 98 (73.1) |
AJCC stage IV, n (%) | 116 (32.7) | 118 (33.4) | 50 (32.3) | 44 (32.8) |
Menopausal status (females only), n (%) | ||||
Premenopausal or perimenopausal | 65 (18.3) | 89 (25.2) | 23 (14.8) | 29 (21.6) |
Postmenopausal | 287 (80.8) | 260 (73.7) | 130 (83.9) | 105 (78.4) |
Prior CDK4/6 inhibitor, n (%) | ||||
Yes | 247 (69.6) | 249 (70.5) | 113 (72.9) | 93 (69.4) |
No | 108 (30.4) | 104 (29.5) | 42 (27.1) | 41 (30.6) |
Prior chemotherapy, n (%) | ||||
(Neo)adjuvant treatment only | 145 (40.8) | 148 (41.9) | 62 (40.0) | 61 (45.5) |
Locally advanced (inoperable) or metastatic treatment | 65 (18.3) | 64 (18.1) | 30 (19.4) | 23 (17.2) |
Prior lines of therapy for advanced or metastatic disease (including endocrine therapy or chemotherapy), n (%) | ||||
0 | 37 (10.4) | 52 (14.7) | 12 (7.7) | 20 (14.9) |
1 | 235 (66.2) | 208 (58.9) | 107 (69.0) | 79 (59.0) |
2 | 73 (20.6) | 77 (21.8) | 31 (20.0) | 29 (21.6) |
3 | 10 (2.8) | 16 (4.5) | 5 (3.2) | 6 (4.5) |
PIK3CA, AKT1, or PTEN alteration status, n (%) | ||||
Altered | 155 (43.7) | 134 (38.0) | 155 (100) | 134 (100) |
PIK3CA only | 110 (31.0) | 92 (26.1) | — | — |
AKT1 only | 18 (5.1) | 15 (4.2) | — | — |
PTEN only | 21 (5.9) | 16 (4.5) | — | — |
PIK3CA and AKT1 | 2 (0.6) | 2 (0.6) | — | — |
PIK3CA and PTEN | 4 (1.1) | 9 (2.5) | — | — |
Nonaltered | 200 (56.3) | 219 (62.0) | — | — |
Known nonaltered (confirmed non altered)a | 142 (40.0) | 171 (48.4) | — | — |
No result (unknown) | 58 (16.3) | 48 (13.6) | — | — |
AJCC = American Joint Committee of Cancer; CDK4/6 = cyclin-dependent kinase 4/6; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; SD = standard deviation.
aAll patients included in the overall population with no qualifying alterations in PIK3CA, AKT1, or PTEN in their tumour, as determined by central testing. Patients with unknown PIK3CA, AKT1, or PTEN alteration status were excluded from this subgroup.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
In the altered population, the group receiving placebo plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 0 (72.4% versus 60.0%) and a lower proportion of patients with an ECOG Performance Status of 1 (26.9% versus 40.0%) than the group receiving capivasertib plus fulvestrant. Further, the group receiving placebo plus fulvestrant had a higher proportion of patients who had received no prior lines of therapy for advanced or metastatic cancer (14.9% versus 7.7%) and a lower proportion of patients who had received 1 prior line of therapy for advanced or metastatic cancer (59.0% versus 69.0%), compared with the group receiving capivasertib plus fulvestrant.
Exposure to Study Treatments
By the data cut-off date of August 15, 2022, in the overall population, the median duration of treatment with capivasertib was 5.4 months (range, 0.1 months to 26.3 months); for treatment with placebo, it was 3.58 months (range, 0.1 months to 25.0 months). The median duration of treatment with fulvestrant in the group receiving capivasertib plus fulvestrant was 5.75 months (range, 0.5 months to 26.3 months); in the group receiving placebo plus fulvestrant, it was 3.68 months (range, 0.5 months to 25.1 months). The median percentages of the actual dose delivered relative to the intended dose (i.e., relative dose intensity) were 95.3% (IQR, 78.0% to 100.0%) for capivasertib, 99.7% (IQR, 96.6% to 100.0%) for placebo, and 100% (IQR, 100.0% to 100.0%) for fulvestrant in both treatment groups. The median treatment durations in the overall population safety analysis set were similar to those of the altered population subgroup (data not shown).
In the overall population, there were more dose interruptions and reductions because of AEs in the group receiving capivasertib plus fulvestrant than in the group receiving placebo plus fulvestrant. AEs leading to a dose interruption occurred in 124 patients (34.9%) receiving capivasertib, and in 36 patients (10.3%) receiving placebo. AEs leading to dose reduction occurred in 70 patients (19.7%) receiving capivasertib, and in 6 patients (1.7%) receiving placebo. The most common AEs leading to dose interruptions or reductions with capivasertib were diarrhea, maculo-papular rash, and vomiting. AEs leading to dose interruptions and reductions across the treatment groups in the altered population were similar to those of the overall population.
In the overall population, most patients (███) received at least 1 approved concomitant medication during the trial. This rate was comparable between treatment groups. The most commonly used, permitted concomitant medication classes were anilides (█████; primarily paracetamol), antipropulsives (██████ primarily loperamide), and proton pump inhibitors (█████; primarily omeprazole). The following classes of concomitant medication were used more frequently in the group receiving capivasertib plus fulvestrant than in the group receiving placebo plus fulvestrant: anilides (█████ versus █████), antipropulsives (█████ versus █████), proton pump inhibitors (█████ versus █████), glucocorticoids (█████ versus █████), other antihistamines for systemic use (█████ versus ████), potent (group III) corticosteroids (█████ versus ████), and piperazine derivatives (█████ versus ████). Concomitant medication use in the altered population was consistent with use in the overall population.
Subsequent Treatment
By the data cut-off date of August 15, 2022, █████ of all randomized patients had received subsequent anticancer treatment after discontinuing the study treatment (Table 12). The proportion of patients receiving subsequent anticancer treatments was lower in the group receiving capivasertib plus fulvestrant (█████ in the overall population and █████ in the altered population) than in the group receiving placebo plus fulvestrant (█████ in the overall population and █████ in the altered population); the most common type of therapy used was cytotoxic chemotherapy (█████ versus ██████ respectively in the overall population and █████ versus ██████ respectively in the altered population).
Table 12: Summary of Subsequent Treatment for the FAS in the CAPItello-291 Trial
Subsequent anticancer treatmenta | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 355) | Placebo plus fulvestrant (n = 353) | Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
Postdiscontinuation anticancer therapy, n (%) | ███ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
Cytotoxic chemotherapy | ███ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
Hormonal therapy | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Targeted therapy | ██ █████ | ██ █████ | ██ █████ | ██ ██████ |
Antiangiogenic therapy | ██ █████ | ██ █████ | █████ | █████ |
PARP inhibitor | █████ | █████ | █████ | █████ |
Biologic therapy | █████ | █████ | █████ | █████ |
Experimental therapy | █████ | █████ | █████ | █████ |
Immunotherapy | █████ | █████ | █████ | █████ |
Radiopharmaceuticals | █████ | █████ | █████ | █████ |
Other | █████ | █████ | █████ | █████ |
PARP = poly(adenosine diphosphate–ribose) polymerase; FAS = full analysis set.
aTherapies used after discontinuing study treatment. Patients may have more than 1 anticancer therapy.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Efficacy
Only those efficacy outcomes and analyses of subgroups identified as important to this review are reported. The main findings for the efficacy outcomes for the CAPItello-291 trial are from the August 15, 2022 data cut-off.
PFS by Investigator Assessment
Table 13 provides a summary of results for PFS by investigator assessment using RECIST 1.1. In the overall population, PFS events had been reported for 258 patients (72.7%) in the group receiving capivasertib plus fulvestrant and for 293 patients (83.0%) in the group receiving placebo plus fulvestrant. The most common censoring reason for patients in both groups was being progression-free at the data cut-off (█████ and █████, respectively). In the altered population subgroup, PFS events had been reported for 121 patients (78.1%) in the group receiving capivasertib plus fulvestrant and for 115 patients (85.8%) in the group receiving placebo plus fulvestrant. The most common censoring reason for patients in both groups was being progression-free at the data cut-off (█████ and █████ respectively). The median duration of follow-up (defined as time to censoring or death) in all patients in both groups was 14.9 months and 14.3 months (ranges not reported), respectively. The median duration of follow-up for PFS among censored patients in the altered population was 16.4 months (range, 0.0 months to 24.9 months) in the group receiving capivasertib plus fulvestrant and ████ months (range, ███ ██ ████) in the group receiving placebo plus fulvestrant.
In the overall population, the median PFS was 7.2 months (95% CI, 5.5 months to 7.4 months) in the group receiving capivasertib plus fulvestrant versus 3.6 months (95% CI, 2.8 months to 3.7 months) in the group receiving placebo plus fulvestrant (log-rank test P < 0.001), with a between-group hazard ratio of 0.60 (96.5% CI, 0.50 to 0.72). In the altered population subgroup (Figure 2), the median PFS was 7.3 months (95% CI, 5.5 months to 9.0 months) in the group receiving capivasertib plus fulvestrant versus 3.1 months (95% CI, 2.0 months to 3.7 months) in the group receiving placebo plus fulvestrant (log-rank test P < 0.001), with a between-group hazard ratio of 0.50 (95% CI, 0.38 to 0.65). The results of the sensitivity analyses were consistent with those of the primary analysis for both populations.
In the overall population, the KM-estimated probabilities of PFS at 6 months and 12 months were 51.8% (95% CI, 46.4% to 57.0%) versus 32.0% (95% CI, 27.0% to 37.0%) and 28.5% (95% CI, 23.7% to 33.5%) versus 18.4% (95% CI, 14.4% to 22.8%) in the capivasertib plus fulvestrant and placebo plus fulvestrant groups, respectively. In the altered population, the KM-estimated probabilities of PFS at 6 months and 12 months were 53.4% (95% CI, 45.1% to 60.9%) versus 29.6% (95% CI, 21.9% to 37.7%) (between-group difference, █████ [95% CI, ████ ██████]) and 28.2% (95% CI, 21.2% to 35.6%) versus 15.8% (95% CI, 10.0% to 22.7%) (between-group difference, █████ [95% CI, ███ ██ ████]), respectively.
PFS Subgroup Analyses
The efficacy results for PFS were consistent across the exploratory subgroup analyses by previous use of a CDK4/6 inhibitor in favour of capivasertib plus fulvestrant. In the overall population, the hazard ratio was 0.59 (95% CI, 0.48 to 0.72) in favour of capivasertib plus fulvestrant in patients who had previously used CDK4/6 inhibitors, and 0.64 (95% CI, 0.45 to 0.90) in patients who had not previously used CDK4/6 inhibitors. In the altered population subgroup, the hazard ratio was ████ (95% CI, ████ █ ████) in favour of capivasertib plus fulvestrant in patients who had previously used CDK4/6 inhibitors, and ████ (95% CI, ████ ██ ████) in patients who had not previously used CDK4/6 inhibitors. For the exploratory subgroup analysis by AKT pathway–nonaltered status in the overall population, the hazard ratio was 0.70 (95% CI, 0.56 to 0.88) in favour of capivasertib plus fulvestrant. This subgroup included patients of both known nonaltered and unknown alteration status. Among patients of known nonaltered status, the hazard ratio was 0.79 (95% CI, 0.61 to 1.02), and among patients of unknown alteration status, the hazard ratio was 0.52 (95% CI, 0.32 to 0.83). The point estimate for the hazard ratio for the known nonaltered subgroup (i.e., 0.79) falls outside the 95% CI for the hazard ratio for both the overall population and the altered population.25 As noted by Health Canada, the effect observed in the overall population was likely driven by patients in the altered population, and the effect observed in the nonaltered population was likely driven by the population with unknown or no results.25
Table 13: PFS in the FAS of the CAPItello-291 Trial
PFS by investigator assessment | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 355) | Placebo plus fulvestrant (n = 353) | Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
August 15, 2022, data cut-off | ||||
Patients with events, n (%)a | ||||
Total | 258 (72.7) | 293 (83.0) | 121 (78.1) | 115 (85.8) |
Progressive disease | 249 (70.1) | 281 (79.6) | 115 (74.2) | 108 (80.6) |
Death | 9 (2.5) | 12 (3.4) | 6 (3.9) | 7 (5.2) |
Patients censored, n (%) | 97 (27.3) | 60 (17.0) | 34 (21.9) | 19 (14.2) |
Median PFS, months (95% CI)b | 7.2 (5.5 to 7.4) | 3.6 (2.8 to 3.7) | 7.3 (5.5 to 9.0) | 3.1 (2.0 to 3.7) |
Hazard ratio (96.5% CI for overall population; 95% CI for altered population)c | 0.60 (0.50 to 0.72) | 3E0.50 (0.38 to 0.65) | ||
Log-rank test P value | < 0.001 | < 0.001 | ||
Probability of being event-free at 6 months, % (95% CI)b | 51.8 (46.4 to 57.0) | 32.0 (27.0 to 37.0) | 53.4 (45.1 to 60.9) | 29.6 (21.9 to 37.7) |
Difference, % (95% CI)d | NR | ████ █████ ██ █████ | ||
Probability of being event-free at 12 months, % (95% CI)b | 28.5 (23.7 to 33.5) | 18.4 (14.4 to 22.8) | 28.2 (21.2 to 35.6) | 15.8 (10.0 to 22.7) |
Difference, % (95% CI)d | NR | ████ ████ ██ █████ | ||
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; PFS = progression-free survival; NR = not reported; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aDoes not include RECIST 1.1 progression events that occur after 2 or more missed visits or death after 2 visits after baseline where the patient has no evaluable visits or does not have a baseline assessment.
bKM estimate.
cStratified Cox proportional hazards model. A hazard ratio of less than 1 favours capivasertib plus fulvestrant. For the overall population, the log-rank test and Cox model are stratified by the presence of liver metastases (yes vs. no), prior use of CDK4/6 inhibitors (yes vs. no), and geographic region (region 1: US, Canada, Western Europe, Australia, and Israel; region 2: Latin America, Eastern Europe and Russia; region 3: Asia). For the altered population, the log-rank test and Cox model are stratified by the presence of liver metastases (yes vs. no) and prior use of CDK4/6 inhibitors (yes vs. no).
dBetween-group differences were requested from the sponsor to facilitate the certainty of evidence appraisals.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Figure 2: KM Plot of PFS, Altered Population in the FAS of the CAPItello-291 Trial
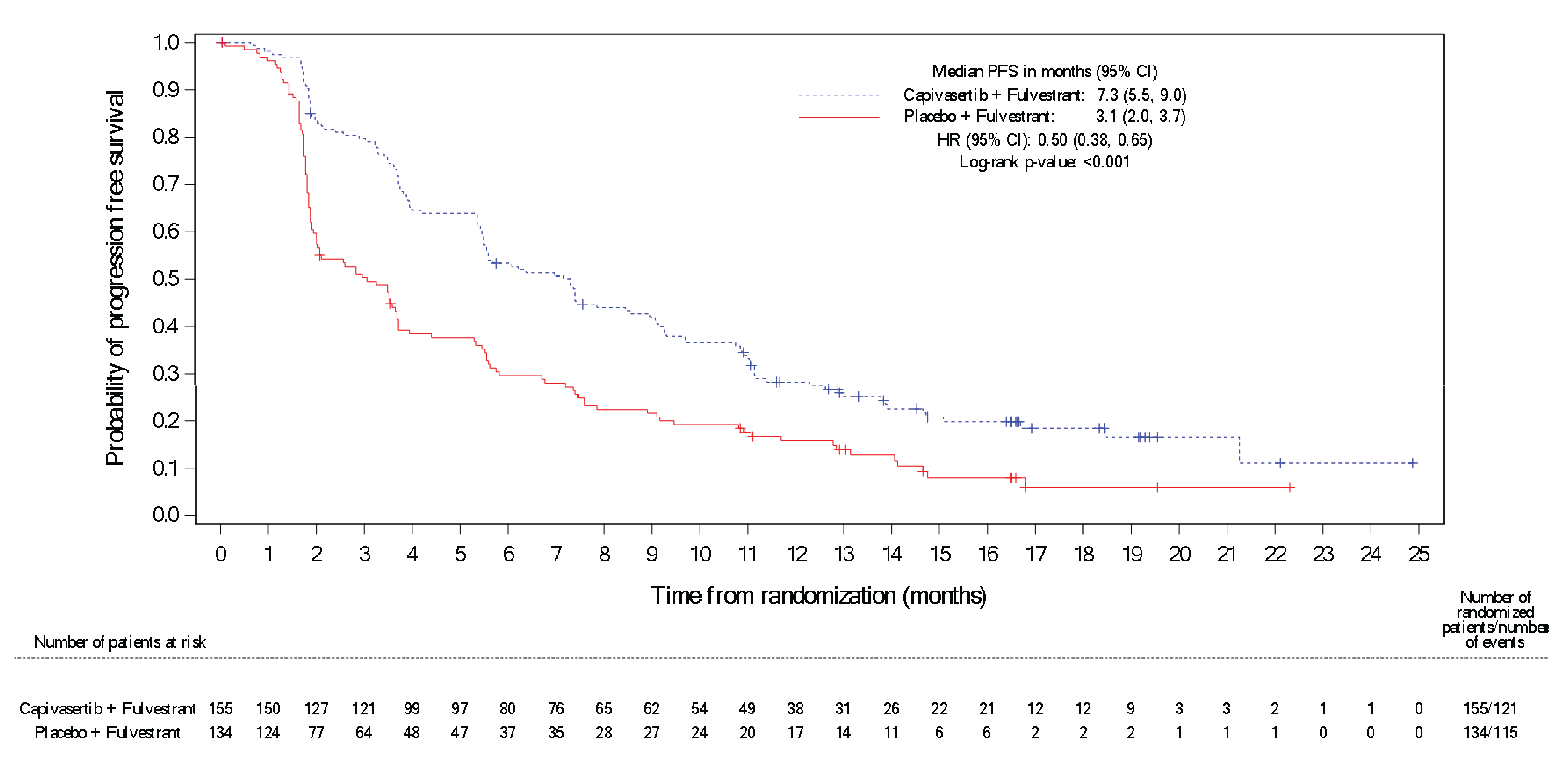
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; KM = Kaplan-Meier; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Notes: A + indicates censored observation.
Does not include RECIST 1.1 progression events that occur after 2 or more missed visits or within 2 visits after baseline where the patient has no evaluable visits or does not have a baseline assessment.
Two-sided P value.
The hazard ratio was calculated using a stratified Cox proportional hazards model. The log-rank test and Cox model were stratified by the presence of liver metastases (yes versus no) and prior use of CDK4/6 inhibitors (yes versus no). A hazard ratio of less than 1 favours capivasertib plus fulvestrant.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Overall Survival
Table 14 provides a summary of OS findings for the first interim analysis. The median OS had not been reached by the August 15, 2022 data cut-off date, with 25% and 31% of patients in the overall population experiencing an event in the capivasertib plus fulvestrant group and placebo plus fulvestrant group, respectively. The median OS was also not reached in the altered population, with 26.5% and 34.3% of patients in the respective groups experiencing an event. In the overall population and altered population, the estimated hazard ratios were 0.74 (95% CI, 0.56 to 0.98) and 0.69 (95% CI, 0.45 to 1.05), respectively (refer to Figure 3).
In the overall population, the KM-estimated probabilities of being alive at 18 months and 24 months were 73.9% (95% CI, 68.3% to 78.7%) versus 65.0% (95% CI, 58.7% to 70.6%) and 64.3% (95% CI, 55.5% to 71.8%) versus 56.5% (95% CI, 48.3% to 63.9%) in the groups receiving capivasertib plus fulvestrant and placebo plus fulvestrant, respectively. In the altered population subgroup, the KM-estimated probabilities of being alive at 18 months and 24 months were 73.2% (95% CI, 64.8% to 80.0%) versus 62.9% (95% CI, 53.1% to 71.2%) (between-group difference, █████ [95% CI, ████ ██ ████]), and █████ (95% CI, ████ ██ ████) versus █████ (95% CI, ████ ██ ████) (between-group difference, ████ [95% CI, ████ ██ ████]) in the groups receiving capivasertib plus fulvestrant and placebo plus fulvestrant, respectively.
Table 14: Overall Survival in the FAS of the CAPItello-291 Trial
Overall survival | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 355) | Capivasertib plus fulvestrant (n = 155) | Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
August 15, 2022, data cut-off | ||||
Death, n (%) | 87 (24.5) | 108 (30.6) | 41 (26.5) | 46 (34.3) |
Patients censored, n (%) | 268 (75.5) | 245 (69.4) | ███ ██████ | ██ ██████ |
Still in survival follow-up | 249 (70.1) | 215 (60.9) | ███ ██████ | ██ ██████ |
Terminated before death | 19 (5.4) | 30 (8.5) | █████ | █████ |
Lost to follow-up | 4 (1.1) | 3 (0.8) | █████ | █████ |
Withdrawn consent | 15 (4.2) | 25 (7.1) | █████ | █████ |
Discontinued study | 0 (0) | 2 (0.6) | █████ | █████ |
Death with no recorded death date | 0 (0) | 0 (0) | █████ | █████ |
Overall survival, months | ||||
Median (95% CI)a | NE (NE to NE) | NE (21.7 to NE) | NE (NE to NE) | NE (20.3 to NE) |
Hazard ratio (95% CI)b | 0.74 (0.56 to 0.98) | 0.69 (0.45 to 1.05) | ||
Log-rank test P value | NA | NA | ||
Probability of being event-free at 18 months, % (95% CI)b | 73.9 (68.3 to 78.7) | 65.0 (58.7 to 70.6) | 73.2 (64.8 to 80.0) | 62.9 (53.1 to 71.2) |
Between-group difference, % (95% CI)c | NR | ████ █████ ██ █████ | ||
Probability of being event-free at 24 months, % (95% CI)b | 64.3 (55.5 to 71.8) | 56.5 (48.3 to 63.9) | ████ █ | ████ █ |
Between-group difference, % (95% CI)c | NR | ███ █████ ██ █████ | ||
Median duration of follow-up in censored patients, months (range) | 15.9 (0.5 to 26.4) | 15.4 (0.5 to 26.0) | ████ █ | ████ █ |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; NA = not applicable; NE = not estimable; NR = not reported.
aKM estimate. The CI for median overall survival is derived based on the Brookmeyer-Crowley method.
bStratified Cox proportional hazards model. A hazard ratio of less than 1 favours capivasertib plus fulvestrant. A 0.01% alpha penalty was assigned to the OS analyses of no detriment. Formal analysis not prespecified. Log-rank test and Cox model stratified by presence of liver metastases (yes vs. no) and prior use of CDK4/6 inhibitors (yes vs. no).cBetween-group differences were requested from the sponsor to facilitate the certainty of evidence appraisals.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Figure 3: KM Plot of OS, Altered Population in the FAS of the CAPItello-291 Trial
![In the altered population, the median OS was not reached, with 26.5% (n = 41) and 34.3% (n = 46) of patients in the groups receiving capivasertib plus fulvestrant and the group receiving placebo plus fulvestrant experiencing an event, respectively. The estimated between-group hazard ratio was 0.69 (95% CI, 0.45 to 1.05). The KM-estimated probabilities of being alive at 18 months and 24 months were 73.2% (95% CI, 64.8% to 80.0%) versus 62.9% (95% CI, 53.1% to 71.2%) and [redacted] (95% CI, [redacted]) versus [redacted] (95% CI, [redacted]) in the groups receiving capivasertib plus fulvestrant and placebo plus fulvestrant, respectively.](https://www.canjhealthtechnol.ca/index.php/cjht/article/download/PC0341r/version/1056/2317/9059/PC0341-Clinical_Review-fig03.png)
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; HR = hazard ratio; KM = Kaplan-Meier; OS = overall survival.
Notes: A + indicates censored observation.
A 0.01% alpha penalty was assigned to the OS analyses of no detriment. Formal analysis not prespecified.
Patients not known to have died at the time of analysis are censored at the last recorded date on which the patient was last known to be alive.
The hazard ratio was calculated using a stratified Cox proportional hazards model. Log-rank test and Cox model were stratified by presence of liver metastases (yes versus no) and prior use of CDK4/6 inhibitors (yes versus no). A hazard ratio of less than 1 favours capivasertib plus fulvestrant.
Source: CAPItello-291 Clinical Study Report; details included are from the sponsor’s Summary of Clinical Evidence.26
HRQoL by EORTC QLQ-C30 and QLQ-BR23
The EORTC QLQ-C30 global health status and EORTC QLQ-BR23 functional and symptom scales scores for the altered population are summarized in Table 15. At baseline, global health status scores were similar in both treatment groups, and there were no clinically meaningful changes (defined by the sponsor as a change in score from baseline of ≥ 10 points) observed in either group over the 10 cycles of treatment. The least squares mean difference in the change from baseline was ████ (95% CI, █████ ██ ████).
For the EORTC QLQ-BR23, the number of patients completing questionnaires in which at least 1 subscale could be determined was greater than or equal to 20 in both treatment groups up to cycle 17. At baseline, scale scores were similar in both treatment groups and suggested intermediate to high functioning (median scores ≥ 55) and low symptomatology (median scores < 20), except for future perspective and feeling upset by hair loss. At cycle 17, the between-group mean differences in change from baseline were ████ for body image (95% CI, █████ ██ ████; total sample = ██), ███ for sexual functioning (95% CI, ████ ██ ████; total sample = ██), not estimable for sexual enjoyment (total sample = ██), ███ for future perspective (95% CI, █████ ██ ████; total sample = ██), ███ for systemic therapy side effects symptoms (95% CI, ████ ██ ████; total sample = ██), ███ for breast symptoms (95% CI, ████ ██ ████; total sample = ██), ████ for arm symptoms (95% CI, ███ ██ █████; total sample = ██), and ████ ██████ ██ ████; total sample = |) for feeling upset by hair loss. The HRQoL results were generally consistent across the cycles and reflected those of the overall population (data not shown).
Table 15: Mean Changes in EORTC QLQ-C30 and EORTC QLQ-C30 in the FAS of the CAPItello-291 Trial
Scales | Altered population | |
|---|---|---|
Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
August 15, 2022 data cut-off | ||
EORTC QLQ-C30 global health status score (higher score indicates better health status) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 10, mean (SD), n | ████ ████████████ | ████ ████████████ |
Change from baseline, LS mean (95% CI), n | █████ █████ ██ | █████ █████ ██ |
Between-group difference, LS mean (95% CI) | ███ █████ ██ ████ | |
EORTC QLQ-BR23 body image (higher score indicates better body image) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 17, mean (SD), n | ████ ███████ █████ | NR |
Change from baseline, mean (SD), n | █████ ████████████ | NR |
Between-group difference, mean (95% CI), n | ████ ██████ ██ █████████ ███ ████ | |
EORTC QLQ-BR23 sexual functioning (higher score indicates better sexual functioning) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 17, mean (SD), n | ████ ███████ █████ | NR |
Change from baseline, mean (SD), n | ████ ███████████ | NR |
Between-group difference, mean (95% CI), n | ███ █████ ██ ████████████ ████ | |
EORTC QLQ-BR23 sexual enjoyment (higher score indicates better sexual enjoyment) | ||
Baseline, mean (SD), n | ████ ████████████ | ████ ████████████ |
Cycle 17, mean (SD), n | NR | NR |
Change from baseline, mean (SD), n | NR | NR |
Between-group difference, mean (95% CI), n | Not estimable, ████ ███ | |
EORTC QLQ-BR23 future perspective (higher score indicates better future perspective) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 17, mean (SD), n | ████ ████████████ | NR |
Change from baseline, mean (SD), n | █████ ████████████ | NR |
Between-group difference, mean (95% CI), n | ███ ██████ ██ ████████████ ████ | |
EORTC QLQ-BR23 systemic therapy side effects (higher score indicates greater level of side effects) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 17, mean (SD), n | ████ ████████████ | NR |
Change from baseline, mean (SD), n | ███ ████████████ | NR |
Between-group difference, mean (95% CI), n | ███ █████ ██ ████████████ ████ | |
EORTC QLQ-BR23 breast symptoms (higher score indicates greater level of symptoms) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ █████████████ |
Cycle 17, mean (SD), n | ████ ████████████ | NR |
Change from baseline, mean (SD), n | █████ ████████████ | NR |
Between-group difference, mean (95% CI), n | ███ █████ ██ ████████████ ████ | |
EORTC QLQ-BR23 arm symptoms (higher score indicates greater level of symptoms) | ||
Baseline, mean (SD), n | ████ █████████████ | ████ ████████████ |
Cycle 17, mean (SD), n | ████ ████████████ | NR |
Change from baseline, mean (SD), n | ████ ████████████ | NR |
Between-group difference, mean (95% CI), n | ████ ████ ██ ███████████ ████ | |
EORTC QLQ-BR23 feeling upset by hair loss (higher score indicates greater level of being upset) | ||
Baseline, mean (SD), n | ████ ████████████ | ████ ████████████ |
Cycle 17, mean (SD), n | NR | NR |
Change from baseline, mean (SD), n | NR | NR |
Between-group difference, mean (95% CI), n | ████ ██████ ██ ██████████ ███ | |
CI = confidence interval; EORTC QLQ-BR23 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire – Breast Cancer Module; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; LS = least squares; NR = not reported; SD = standard deviation.
Note: Summary statistics were presented only when the number of observations was at least 20 and greater than one-third of patients were dosed at a time point within at least 1 treatment group.
Source: CAPItello-291 Clinical Study Report; details included are from the sponsor’s Summary of Clinical Evidence.26
Harms
Harms data reported in this section are from the data cut-off date of August 15, 2022. The key harm results for the safety analysis population are summarized in Table 16. Because the sample size of the overall population was larger than that of the altered population, the harms data summarized in text in the following section are for the overall population. It should be noted that the overall population includes a proportion of patients who are not relevant to the reimbursement request (i.e., 59% are of PIK3CA, AKT1, or PTEN nonaltered status or unknown alteration status). The safety profile of capivasertib plus fulvestrant in the altered population subgroup was similar to that in the overall population.
Adverse Events
Most patients in the trial reported at least 1 AE (96.6% with capivasertib plus fulvestrant and 82.3% with placebo plus fulvestrant in the overall population). The most frequently reported AEs of any grade in the group receiving capivasertib plus fulvestrant were diarrhea (72.4% versus 20.0% with placebo plus fulvestrant), rash (38.0% versus 7.1% with placebo plus fulvestrant), and nausea (34.6% versus 15.4 with placebo plus fulvestrant). The most frequently reported AEs in the group receiving placebo plus fulvestrant were also diarrhea and nausea.
Serious AEs
A numerically higher proportion of SAEs was reported in patients taking capivasertib plus fulvestrant (16.1%) than placebo plus fulvestrant (8.0%). The most common SAE in the group receiving capivasertib plus fulvestrant was diarrhea (experienced by 1.7% of patients versus 0.3% of patients receiving placebo plus fulvestrant).
Withdrawal Due to AEs
Study treatment discontinuation because of AEs was numerically higher in the group receiving capivasertib plus fulvestrant (9.3%) than in the group receiving placebo plus fulvestrant (0.6%). The most common AE leading to discontinuation of capivasertib or placebo was rash (████ versus ██ with placebo).
Mortality
Deaths were reported in 24.5% of patients in the group receiving capivasertib plus fulvestrant and in 30.6% of patients in the group receiving placebo plus fulvestrant. The majority of deaths in both groups (22.3% in the group receiving capivasertib plus fulvestrant and 28.9% in the group receiving placebo plus fulvestrant) were attributed to disease progression. No deaths in the trial were attributed to the study treatment.
Notable Harms
A higher proportion of notable AEs was reported in patients taking capivasertib plus fulvestrant (█████) than in patients taking placebo plus fulvestrant (█████). The most common notable harms in both groups were noninfectious diarrhea (72.4% and 20.3%, respectively) rash (38.0% and 7.1%, respectively), and stomatitis (20.0% and 5.7%, respectively).
Table 16: Summary of Harms Results in the Safety Population of the CAPItello-291 Trial
Harms | Overall population | Altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 355) | Placebo plus fulvestrant (n = 350) | Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
Most common (> 10%) AEs (any grade), n (%) | ||||
Any AE | 343 (96.6) | 288 (82.3) | ███ ██████ | ██ █ |
Diarrhea | 257 (72.4) | 70 (20.0) | 119 (76.8) | 25 (18.8) |
Nausea | 123 (34.6) | 54 (15.4) | 54 (34.8) | 18 (13.5) |
Rash | 78 (22.0) | 15 (4.3) | 31 (20.0) | 8 (6.0) |
Fatigue | 74 (20.8) | 45 (12.9) | 35 (22.6) | 18 (13.5) |
Vomiting | 73 (20.6) | 17 (4.9) | 32 (20.6) | 9 (6.8) |
Headache | 60 (16.9) | 43 (12.3) | 27 (17.4) | 16 (12.0) |
Decreased appetite | 59 (16.6) | 22 (6.3) | 27 (17.4) | 10 (7.5) |
Hyperglycemia | 58 (16.3) | 13 (3.7) | 26 (16.8) | 5 (3.8) |
Maculo-papular rash | 57 (16.1) | 9 (2.6) | 32 (20.6) | 2 (1.5) |
Stomatitis | 52 (14.6) | 17 (4.9) | 27 (17.4) | 6 (4.5) |
Asthenia | 47 (13.2) | 36 (10.3) | 22 (14.2) | 17 (12.8) |
Pruritus | 44 (12.4) | 23 (6.6) | 24 (15.5) | 6 (4.5) |
Anemia | 37 (10.4) | 17 (4.9) | 14 (9.0) | 7 (5.3) |
Urinary tract infection | 36 (10.1) | 23 (6.6) | 16 (10.3) | 7 (5.3) |
Arthralgia | 33 (9.3) | 38 (10.9) | 17 (11.0) | 17 (12.8) |
Common (> 1%) SAEs (any grade), n (%)a | ||||
Any SAE | 57 (16.1) | 28 (8.0) | 28 (18.1) | 14 (10.5) |
Diarrhea | 6 (1.7) | 1 (0.3) | █████ | █████ |
Vomiting | 4 (1.1) | 2 (0.6) | █████ | █████ |
Maculo-papular rash | 5 (1.4) | 0 (0) | █████ | █████ |
Hyperglycemia | 3 (0.8) | 0 (0) | █████ | █████ |
Hypercalcemia | 1 (0.3) | 2 (0.6) | █████ | █████ |
Acute kidney injury | 3 (0.8) | 0 (0) | █████ | █████ |
Asthenia | 2 (0.6) | 0 (0) | █████ | █████ |
Patients (> 1%) who discontinued treatment because of AEs, n (%)b | ||||
Total all-cause AEs leading to discontinuation of capivasertib or placebo only | 33 (9.3) | 2 (0.6) | 10 (6.5) | 1 (0.8) |
Rash | ██ █████ | █████ | █████ | █████ |
Vomiting | █████ | █████ | █████ | █████ |
Diarrhea | █████ | █████ | █████ | █████ |
Maculo-papular rash | █████ | █████ | █████ | █████ |
Pyrexia | █████ | █████ | █████ | █████ |
Deaths, n (%) | ||||
Patients who died | 87 (24.5) | 108 (30.6) | 41 (26.5) | 46 (34.3) |
Disease progressionc | 79 (22.3) | 102 (28.9) | ██ ██████ | ██ ██ |
AE with outcome of death only | 4 (1.1) | 1 (0.3) | █████ | █████ |
AE with outcome of death and death related to disease | 0 (0) | 0 (0) | █████ | █████ |
AE with outcome of death and AE onset date falling more than 30 days (± 7 days) after the last dose | 1 (0.3) | 0 (0) | █████ | █████ |
Otherd | 3 (0.8) | 5 (1.4) | █████ | █████ |
Notable harms, n (%) | ||||
AEs of any grade | ███ ██████ | ███ ██ | ███ ██████ | ██ ██ |
Noninfectious diarrhea | 257 (72.4) | 71 (20.3) | ███ ██████ | ██ ███ |
Rash | 135 (38.0) | 25 (7.1) | ██ ██████ | ██ ███ |
Stomatitis | 71 (20.0) | 20 (5.7) | ██ ██████ | █████ |
Hyperglycemia | 60 (16.9) | 14 (4.0) | ██ ██████ | █████ |
Urinary tract infection | 50 (14.1) | 24 (6.9) | ██ ██████ | █████ |
QT prolongation | 11 (3.1) | 0 (0) | █████ | █████ |
Infective pneumonia | 8 (2.3) | 9 (2.6) | █████ | █████ |
AE = adverse event; SAE = serious adverse event.
Note: AEs with an onset date on or after the date of the first dose, AEs with onset date before dosing that worsen after dosing, and AEs occurring up to 30 days (± 7 days) following the date of the last dose are reported. Percentages are based on the total number of patients in the treatment group.
aPatients with multiple SAEs are counted once for each system organ class or preferred term.
bPatients with multiple AEs leading to discontinuation of capivasertib or placebo only are counted once for each system organ class or preferred term.
cDeath related to disease under investigation only (determined by the investigator).
dPatients who died and are not captured in the earlier categories.
Source: CAPItello-291 Clinical Study Report; details are from the sponsor’s Summary of Clinical Evidence.26
Critical Appraisal
Internal Validity
The CAPItello-291 trial was a randomized, double-blind, phase III trial. Randomization procedures, including stratification by the presence or absence of liver metastases, previous use of a CDK4/6 inhibitor, and geographic region, were appropriate and conducted by interactive response technology. In the altered population, the group receiving placebo plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 0 (72.4% versus 60.0%) and a lower proportion of patients with an ECOG Performance Status of 1 (26.6% versus 40.0%) than the group receiving capivasertib plus fulvestrant. Further, the group receiving placebo plus fulvestrant had a higher proportion of patients who had received no prior lines of therapy for advanced or metastatic cancer (14.9% versus 7.7%) and a lower proportion of patients who had received 1 prior line of therapy for advanced or metastatic cancer (59.0% versus 69.0%) compared with the group receiving capivasertib plus fulvestrant. These imbalances were likely the result of chance, given that all other baseline characteristics of patients appeared balanced between groups, so are unlikely to have resulted in bias.
In total, important protocol deviations were reported in 62 patients (8.8%). These primarily included the use of restricted prior concomitant medications. The type and frequency of these deviations were comparable between the treatment groups; therefore, the presence, magnitude, and direction of potential bias is unclear.
Although patients and investigators were blinded to treatment assignment, there was potential for unblinding because of the imbalances in notable harms across treatment groups (i.e., higher in the group receiving capivasertib plus fulvestrant). Knowledge of treatment assignment could increase the risk of bias in the measurement of subjective outcomes (including HRQoL) and subjective harms, although the extent and direction of bias cannot be predicted. Objective outcomes like OS are unlikely to be affected by bias because of unblinding. To minimize the risk of bias in the measurement of PFS, the trial performed tumour assessments using RECIST 1.1 criteria, and radiographic scans were assessed by BICR as a sensitivity analysis. The PFS BICR results were similar to the primary investigator-assessed results.
Sample sizes and power calculations were based on PFS and OS in the overall population and on PFS in the altered population, and the trial was powered to detect significant differences for both outcomes. The prespecified analyses of OS and PFS in the overall and altered populations were appropriately controlled for multiple comparisons. All other analyses were descriptive. This included the HRQoL outcomes EORTC QLQ-C30 and EORTC QLQ-BR23, which were deemed clinically important outcomes for the disease. The sample sizes for the subgroup analyses of PFS were small. The trial may not have been powered to detect subgroup differences.
Sensitivity analyses were conducted for the primary outcome of PFS, and the findings were consistent with the primary analysis in the FAS. While the trial met its primary objective of assessing PFS, the median OS was not reached in either treatment group, and there was imprecision in the estimates for between-group differences in survival probability at 18 months and 24 months (i.e., the 95% CIs were wide and included the potential for no clinically important difference between the 2 treatment groups). In addition, there is uncertainty as to whether the PFS benefits (as a surrogate outcome for OS) will translate into survival benefits. There are systematic literature reviews of RCTs investigating other treatments for HR-positive, HER2-negative, metastatic breast cancer that have reported a correlation between PFS and OS.55,56 However, these correlations do not mean that PFS results as a surrogate can predict the final OS outcome for a specific treatment. Given that the results at the data cut-off date represent an interim analysis for OS and that the results were based on few events, longer-term follow-up is needed to inform the true effect of capivasertib plus fulvestrant compared with placebo plus fulvestrant on survival. The certainty of evidence for many HRQoL outcomes was limited because of risk of bias — because of missing outcomes data, both at baseline and at the selected follow-up times — and because of imprecision. In addition, patients were permitted to receive posttreatment anticancer medications after the study treatment had been discontinued (approximately 49% of all patients), and this may influence the assessment of OS. As such, the estimated effect is a combination of treatment with capivasertib plus fulvestrant compared with placebo plus fulvestrant plus concomitant treatments postprogression. The subsequent treatments were not balanced between groups and were not reflective of Canadian clinical practice.
The trial authors stated that the PH assumption was assessed by examining plots of complementary log-log (event times) versus log (time); however, the assessment results were not reported. Based on visual inspection of the KM plots for PFS and OS, it does not appear that there was any major violation of the PH assumption. However, the results of the PH assessment in the sponsor-submitted NMA showed evidence of non-PHs across most studies, including the CAPItello-291 trial. As such, the hazard ratios for PFS and OS may not be fully reflective of the true effects.
The EORTC QLQ-C30 and EORTC QLQ-BR23 have been validated in patients with cancer and breast cancer, respectively, with evidence of reliability and MID ranges. Based on the MID ranges identified in the literature, the sponsor suggested a 10-point change from baseline score as a clinically meaningful change; this was considered reasonable by the CDA-AMC review team. No evidence was identified in the literature for responsiveness. Additionally, the results of these outcomes were subject to potential bias because of missing data, although the direction and extent of bias are unclear. Therefore, the potential impacts on patients’ HRQoL remain very uncertain.
External Validity
In general, the population requested for reimbursement aligns with the Health Canada indication except that the reimbursement request is not restricted to adult females. Enrolment in the CAPItello-291 trial was open to both male and female patients, and 7 males were enrolled. The clinical experts consulted by CDA-AMC agreed that including males in the reimbursement request is appropriate because the proportion of included patients reflected the low prevalence of breast cancer in males and the fact that management of breast cancer is similar in males and females. Given the small proportion of males in the trial, it was not possible to ascertain from the data whether males would experience different treatment outcomes compared with females. However, the clinical experts agreed that they would expect similar efficacy and harms among both. The clinical experts also noted that, although patients with diabetes who were receiving insulin were excluded from the trial, they could be candidates for receiving capivasertib plus fulvestrant if their diabetes was controlled. The dosing and administration of capivasertib plus fulvestrant were consistent with the Health Canada–approved product monograph.
Patients with PIK3CA, AKT1, or PTEN-altered tumours (i.e., the altered population, which is the focus of the Health Canada–approved indication) were identified through postrandomization central testing of tumour tissue collected before randomization based on a prespecified list of molecular alterations, using a validated assay. The CDA-AMC team considered this diagnostic approach appropriate, although the clinical experts noted that testing for PIK3CA, AKT1, or PTEN tumour alterations is not part of routine clinical practice, and access to testing varies across Canada.
According to the clinical experts consulted by CDA-AMC, the eligibility criteria and baseline characteristics of the CAPItello-291 trial were generalizable to adults in the Canadian setting with HR-positive, HER2-negative, advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations. However, the trial did not include patients with a poor ECOG Performance Status. The clinical experts noted that limiting enrolment to patients with an ECOG Performance Status of 0 or 1 is not entirely representative of patients with HR-positive, HER2-negative, advanced or metastatic breast cancer in Canada; they expect to encounter patients with higher scores in their practices.
Dose adjustments were allowed in the trial, and the methods for making adjustments were outlined in the protocol. The clinical experts noted that dose adjustments or modifications are anticipated in a clinical practice setting to manage AEs while maintaining drug benefit.
The trial included outcomes that were important to patients and clinicians. The patient group indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform our expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:57,58
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — that is, the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — that is, the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — that is, the true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessments for PFS and OS were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of the evidence assessment for the EORTC QLQ-C30 global health status score and EORTC QLQ-BR23 functional and symptom scales scores were set according to the presence or absence of an important effect based on a threshold suggested by the sponsor that was informed by the literature. Because of the lack of a formal MID estimate for SAEs, the target of the certainty of evidence assessment was set according to the presence or absence of any (nonnull) effect.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for capivasertib plus fulvestrant versus placebo plus fulvestrant.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
The aim of this section is to summarize and critically appraise 1 sponsor-submitted NMA59 used to inform the pharmacoeconomic model and to fill gaps in the comparative evidence for other treatments of interest for HR-positive, HER2-negative, advanced or metastatic breast cancer.
Description of the NMA
The systematic literature search and study selection criteria for the NMA are summarized in Table 17.
Table 17: Study Selection Criteria and Methods for the NMA Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients with HR-positive, HER2-negative, or HER2-mixed, unreported, or unknown status, unresectable and/or metastatic breast cancer who were previously treated with endocrine therapy in the (neo)adjuvant or advanced setting |
Intervention | Capivasertib in combination with fulvestrant, including the following dosages:
|
Comparator |
|
Outcome | Studies must report at least 1 of the following outcomes:
|
Study designs |
|
Publication characteristics | None |
Exclusion criteria |
|
Databases searched | Relevant studies were identified by searching (search date: March 28, 2023; update: August 8, 2023) the following databases from inception through the Ovid platform:
Hand searches for relevant materials from the following scientific conferences were conducted from 2020 to 2023:
Manual searches of the following clinical trial registries were performed:
|
Selection process | Study selection followed a 2-stage screening process based on the review of titles and abstracts (stage 1) and then full-text articles (stage 2). During both stages, each report was assessed by 2 independent investigators. Any disagreements were resolved by consensus, if needed. |
Data extraction process | Data extraction for the included studies was conducted by 2 independent analysts. Any disputes were resolved by consensus or by a third reviewer. Data from the included studies were extracted into a standardized table template developed in Microsoft Excel. |
Risk of bias assessment | Two independent researchers assessed the risk of bias in the included trials. Following reconciliation between the 2 researchers, a third researcher was included to reach consensus for any remaining discrepancies. The Cochrane Collaboration’s Risk of Bias tool (Version 2) was used to assess the risk of bias in included clinical trial. |
CTCAE = Common Terminology Criteria for Adverse Events; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; NMA = network meta-analysis; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumours.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Indirect Treatment Comparison Design
Objectives
The objective of the sponsor-submitted NMA was to indirectly compare the treatment effects of capivasertib plus fulvestrant versus other relevant comparators for the treatment of adult patients with HR-positive, HER2-negative, advanced breast cancer with AKT pathway–altered tumours who had progressed during or after treatment with endocrine-based regimens.59 The protocol of the systematic review and NMA was a priori registered in the International Prospective Register of Systematic Reviews.
Study Selection Methods
A systematic literature search was conducted initially on March 28, 2023 and updated on August 8, 2023 using the criteria described in Table 17. The review included RCTs and single-arm trials investigating treatments in adults with HR-positive, HER2-negative or HER2 of mixed, unreported, or unknown status, unresectable or metastatic breast cancer who had been previously treated with an ET in the (neo)adjuvant or advanced setting. The following efficacy outcomes of interest for this CDA-AMC review were reported: PFS and OS. There were no search restrictions. Identified citations were assessed for eligibility against predefined inclusion and exclusion criteria. Two reviewers independently screened titles and abstracts and relevant full-text citations for inclusion. Discrepancies were resolved through consensus. Data extraction and quality assessment of included studies were performed by 2 reviewers, and discrepancies were resolved by a third reviewer. Risk of bias assessment of included studies was performed using the Cochrane Collaboration’s Risk of Bias tool (Version 2) at the study level.
NMA Analysis Methods
Details of the NMA analysis methods are in Table 18. A NMA feasibility assessment was undertaken to summarize the potential outcome-specific networks for comparing capivasertib with relevant comparators of interest. The feasibility assessment included the following 3 steps: explore the connectivity of the identified trials from the systematic literature review based on the interventions of the trials to form a best-case scenario evidence network; explore the comparability of the trials of the best-case scenario evidence network by study designs and patient characteristics and assess for heterogeneity; and generate outcome-specific evidence networks using each outcome of interest across each trial of the best-case network. Although the authors provided evidence for heterogeneity in potential treatment-effect modifiers, it was not clear how these were identified (i.e., whether through a literature review or by expert consensus).
NMAs were performed within a Bayesian framework. For each end point, fixed- and random-effects NMAs were performed using capivasertib plus fulvestrant as the reference treatment in the network. The treatment effects for PFS and OS were modelled in terms of the log hazard ratio and its standard error. The NMA was performed using a treatment difference model, assuming a normal likelihood and identity link function for the log hazard ratios. The NMA results were summarized as hazard ratios and 95% CrIs for treatment versus capivasertib plus fulvestrant 500 mg. In both instances, a hazard ratio of less than 1 indicated that a treatment was more efficacious than its comparator, and vice versa for values higher than 1. The results were presented as forest plots.
An assessment of the PH assumption was performed for PFS and OS with a KM plot and treatment-effect estimate. This included visual inspection of the log-cumulative hazards and the scaled Schoenfeld residual plots and evaluation of the Grambsch-Therneau nonproportionality test. The following conditions indicated non-PH: a Grambsch-Therneau test statistic of P value less than 0.05; a nonhorizontal line for beta(t) on the Schoenfeld plot; and/or evidence of nonparallel log-cumulative hazard curves between arms. The NMA used the altered population data from the CAPItello-291 and FAKTION trials.
Table 18: NMA Analysis Methods
Methods | Description |
|---|---|
Analysis methods | NMAs were performed within a Bayesian framework in accordance with the methods outlined in the NICE Decision Support Unit and published by Dias et al.60 For each end point, fixed- and random-effects NMAs were performed using fulvestrant 500 mg as the reference treatment in the network. All analyses were performed using R Version 4.0.2 with the multinma (Version 0.5.1), survival (Version 3.1.12), and rstan (Version 2.26.23) packages. |
Priors | All priors for treatment effect were considered noninformative and given as logHR for OS and PFS. For all models, vague priors were used for the trial-specific baseline effects and the basic relative-effect parameters. To estimate the between-study heterogeneity in the random-effects model, there must exist several links in a network with more than 1 study. Because of the limited number of studies in the network, the random-effects NMAs were conducted using informative priors for the between-study heterogeneity, based on Turner et al.:61
As a sensitivity analysis, models were also fitted using a vague prior for between-study heterogeneity. |
NMA model selection | No model comparison was required. (Only fixed-effects NMA models were applied because there was insufficient evidence available to estimate the between-study heterogeneity required to run random-effects models.) |
Assessment of model fit | Model fit was assessed by comparing the total residual deviance to the number of data points contributing to the model. In a well-fitting model, the number of data points and total residual deviance will be similar. A meaningful difference in model fit was determined by a 3-point or greater difference in DIC score. |
Assessment of heterogeneity | The presence of clinical and methodological heterogeneity was assessed through a feasibility assessment comparing the population, interventions and/or comparators, outcome, and study characteristics. The feasibility assessment process aligned with ISPOR, NICE, and PRISMA guidelines.60,62-64 |
Assessment of consistency | An assessment of consistency between the direct and indirect effect estimates from the NMA was performed. Consistency was assessed using the node-splitting methodology. For each comparison, evidence of inconsistency was judged based on the level of agreement in results across direct and indirect data in the NMA and assessed using Bayesian P values for inconsistency. |
Assessment of convergence | Convergence was assessed using trace plots, density plots, Gelman-Rubin-Brooks plots, and autocorrelation plots. All model estimates were based on a sample of 10,000 iterations after a warm-up of 10,000 iterations across 4 chains. Parameter estimates from every second iteration of the sampling procedure were discarded (i.e., “thinning” was set to 2), and convergence was assessed using the potential scale reduction factor, “r hat,” for each parameter. An r hat of approximately 1.0 and less than 1.01 demonstrated convergence (i.e., that stable estimates had been reached for all parameters). |
Outcomes |
|
Follow-up time points | Latest result or follow-up time point |
Construction of nodes | Treatment nodes distinguished between monotherapies and combination therapies. Different dosages of the same drugs were given separate nodes. |
Sensitivity analyses | Assessment of the assumption of model fit for between-study heterogeneity; models also fitted using a vague prior. |
DIC = deviance information criterion; ISPOR = International Society for Pharmacoeconomics and Outcomes Research; logHR = log hazard ratio; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; OS = overall survival; PFS = progression-free survival; PRISMA = Preferred Reporting Items for Systematic reviews and Meta-Analyses; RECIST = Response Evaluation Criteria in Solid Tumours.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Results of the NMA
Summary of Included Studies
A summary of the included studies is in Table 19. The systematic literature review identified 33 studies that informed the feasibility assessment for inclusion in the NMA. The base-case network was plotted to compare capivasertib plus fulvestrant 500 mg to other approved treatments for HR-positive, HER2-negative or HER2 of mixed, unreported, or unknown status, unresectable and/or metastatic breast cancer previously treated with ET in the (neo)adjuvant or advanced setting.
Of the 33 included studies, 15 could not be connected to the main network with capivasertib, mostly because the comparators were not relevant. In addition, 2 studies were excluded because of irrelevant dosing regimens for fulvestrant. The remaining 16 studies were considered in the base-case network assessment; however, 6 studies were ultimately excluded because the studies did not include relevant comparators of interest for this CDA-AMC review. In total, 10 studies were included in the NMA (Table 19), with the following interventions:
capivasertib 400 mg plus fulvestrant 500 mg (monthly with loading dose)
fulvestrant 250 mg or 500 mg
exemestane 25 mg
everolimus 10 mg plus exemestane 25 mg
capecitabine monotherapy 1,250 mg/m2.
Figure 4: NMA Base-Case Network
Source: NMA technical report; details included are from the sponsor’s Summary of Clinical Evidence.59
Sources of Heterogeneity
The assessment of homogeneity for the included studies is in Table 20. The comparison across studies suggested limited variation in age, ECOG Performance Status, and prevalence of visceral disease. Key differences across studies were menopausal status, prior CDK4/6 use (stratification factor in the CAPItello-291 trial), HER2 status, AKT pathway alteration status, and line of therapy.
Table 19: Summary of Studies Included in the NMA
Study | PFS | OS | Intervention | Comparator | Sample size | Region | HR status | HER2 status | AKT-altered status | Prior treatment |
|---|---|---|---|---|---|---|---|---|---|---|
CAPItello-291 trial26 | Included | Included | Capivasertib plus fulvestrant 500 mg | Placebo plus fulvestrant 500 mg | 289 | Multinational | Positive | Negative | Altered subgroup | ET ± prior CDK4/6 inhibitors |
Included | Included | Capivasertib plus fulvestrant 500 mg | Fulvestrant 500 mg | 140 | UK | Positive | Negative | Altered subgroup | ET ± prior CDK4/6 inhibitors | |
BOLERO-2 trial67 | Included | Included | Everolimus plus exemestane | Exemestane | 724 | Multinational | Positive | Negative | NA | ET, no prior CDK4/6 inhibitors |
BOLERO-5 trial68 | Included | Not included | Everolimus plus exemestane | Exemestane | 159 | China | Positive | Negative | NA | ET, no prior CDK4/6 inhibitors |
EFECT trial69 | Included | Not included | Fulvestrant 250 mg | Exemestane | 693 | Multinational | Positive | Mixed or unknown | NA | ET, no prior CDK4/6 inhibitors |
SOFEA trial70 | Included | Included | Fulvestrant 250 mg | Exemestane | 723 | Multinational | Positive | Mixed or unknown | NA | ET, no prior CDK4/6 inhibitors |
Included | Included | Fulvestrant 500 mg | Fulvestrant 250 mg | 736 | Multinational | Positive | Mixed or unknown | NA | ET, no prior CDK4/6 inhibitors | |
FRIEND trial73 | Included | Not included | Fulvestrant 500 mg | Exemestane | 144 | China | Positive | Negative | NA | ET, no prior CDK4/6 inhibitors |
NCT01300351 trial74 | Included | Not included | Fulvestrant 500 mg | Fulvestrant 250 mg | 221 | China | Positive | Mixed or unknown | NA | ET, no prior CDK4/6 inhibitors |
BOLERO-6 trial75 | Included | Included | Everolimus plus exemestane | Capecitabine | 309 | Multinational | Positive | Negative | NA | ET, no prior CDK4/6 inhibitors |
HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; NA = not available; NMA = network meta-analysis; ET = endocrine therapy; CDK4/6 = cyclin-dependent kinase 4 and 6.
Note: The SOFEA trial reported data for additional nonapproved treatment groups not included within the base-case network: SOFEA trial administeredfulvestrant plus anastrozole 2.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.
Results
Progression-Free Survival
The network diagram for the investigator-assessed PFS NMA (N = 10 studies) is shown in Figure 5. Based on the goodness-of-fit statistics for the NMAs, according to the deviance information criterion, a fixed-effects model followed by a random-effects model with informative and vague priors, respectively, were conducted. The network contained 1 closed loop, comprising fulvestrant 250 mg, fulvestrant 500 mg, and exemestane. The NMA was performed using data from the AKT pathway–altered subgroup of the only studies reporting these subpopulations, which were the CAPItello-291 and FAKTION trials. In the absence of data, the treatment effects for all other interventions were based on data from the biomarker-unselected, intention-to-treat (ITT) populations of comparator studies. The NMA assumed that AKT pathway alteration was a treatment-effect modifier for capivasertib. For comparator treatments, because of the different mechanism of action, there was no a priori expectation of treatment-effect modification.
Table 20: Assessment of Homogeneity for the NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Prior CDK4/6 inhibitor therapy | Only 1 of 10 studies (the CAPItello-291 trial) reported subgroup results by history of CDK4/6 inhibitor treatment. In line with current practice, most patients enrolled in the CAPItello-291 trial (70.1%) had previously received a CDK4/6 inhibitor. The remaining 9 studies were conducted before the introduction of CDK4/6 inhibitors. |
HER2 status | Four studies reported a mixed or unknown HER2 status: the SOFEA, CONFIRM, EFECT, and NCT01300351 trials. Of these, only the SOFEA trial reported subgroup results according to HER2 status. The subgroup analysis of the SOFEA trial suggests that HER2 status may not be an effect modifier for PFS when comparing fulvestrant 250 mg with exemestane 25 mg. Subgroup results were not available for the CONFIRM, EFECT, or NCT01300351 trials. |
AKT pathway alteration status | AKT pathway–altered subgroup results were reported by only 2 studies (the CAPItello-291 trial76 and FAKTION trial66) because testing for PIK3CA, AKT1, or PTEN alterations was not routine practice when the other trials in the network were conducted. In these 2 studies, patients with HR-positive, HER2-negative breast cancer who have PIK3CA or PTEN alterations exhibit worse overall PFS and OS compared with those without these alterations. For consistency across studies, the NMA used data from the NGS-identified subgroup of the FAKTION trial,66 which was considered the closest matching subgroup to the NGS-identified, AKT pathway–altered subgroup in the CAPItello-291 trial. AKT pathway alterations were found to be a treatment-effect modifier in the CAPItello-291 and FAKTION trials, based on a comparison of the results between the AKT pathway–altered subpopulations and nonaltered subpopulations. This is also reflected in capivasertib’s licensed indication in Canada for the AKT pathway–altered population. Therefore, the NMA was performed using the AKT pathway–altered subgroup results from the CAPItello-291 and FAKTION trials only, and it utilized biomarker-unselected population data (i.e., intention to treat) for comparator trials. |
Menopausal status | Most studies (9 out of 10) included only patients who were postmenopausal. The CAPItello-291 study reported a mixed cohort of patients who were premenopausal, perimenopausal, or postmenopausal; however, the majority of studies (80% or greater) were postmenopausal. |
Region | Six studies were multinational. The BOLERO-5, FRIEND, and NCT01300351 trials were conducted in China. The FAKTION trial was conducted in the UK. |
Line of therapy | The majority of patients in the CAPItello-291 trial (62.6%) and FAKTION trial (median 1 prior line) had received treatment in a second-line setting. With the notable exceptions of the CONFIRM and FRIEND studies, both of which had a greater share of patients in the first-line setting (approximately 50% and 100%, respectively), most studies, on average, recruited patients who were at similar treatment lines to those enrolled in the CAPItello-291 and FAKTION trials. |
ROB | Four studies were assessed as having a high ROB, according to the Cochrane ROB 2 tool. These include the BOLERO-6 and FRIEND studies, which were judged to high risk for reasons of baseline imbalance (the BOLERO-6 trial) and inadequate reporting of randomization and blinding (the FRIEND trial). There were some concerns about ROB in the remaining studies; these were attributed to lack of reporting for randomization and concealment methodology as well as open-label design. No study was considered to have low ROB. |
CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; NGS = next-generation sequencing; NMA = network meta-analysis; OS = overall survival; PFS = progression-free survival; ROB = risk of bias.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
The NMA results for the fixed- and random-effects models are shown in Figure 6. The point estimates for the hazard ratios and associated 95% CrIs for PFS favoured capivasertib plus fulvestrant versus exemestane 25 mg, fulvestrant 500 mg, and fulvestrant 250 mg. The point estimates for the hazard ratios comparing capivasertib plus fulvestrant to everolimus 10 mg plus exemestane 25 mg and capecitabine 2,500 mg/m2 favoured capivasertib plus fulvestrant; however, the 95% CrIs included the possibility of no difference or that the comparator was favoured (i.e., crossed the null). The results of the random-effects model had wider 95% CrIs than those of the fixed-effects model. The results of the PH assessment showed evidence of non-PHs across most studies, including the CAPItello-291 study (i.e., ITT and AKT pathway–altered populations) and the BOLERO-5 study.
Figure 5: NMA Evidence Network for PFS
NMA = network meta-analysis; PFS = progression-free survival.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Figure 6: NMA Forest Plot Comparison with Capivasertib Plus Fulvestrant for PFS [Redacted]

CrI = credible interval; NMA = network meta-analysis; PFS = progression-free survival.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Overall Survival
The network diagram for the OS NMA (N = 6 studies) is shown in Figure 7. The goodness-of-fit statistics for the OS NMA were the same as for PFS. The network contained no closed loops. The NMA results for the fixed- and random-effects models are shown in Figure 8. The point estimates for the hazard ratios and associated 95% CrIs for OS favoured capivasertib plus fulvestrant versus exemestane 25 mg, fulvestrant 500 mg, and fulvestrant 250 mg. The point estimates for the hazard ratios comparing capivasertib plus fulvestrant to everolimus 10 mg plus exemestane 25 mg and capecitabine 2,500 mg/m2 favoured capivasertib plus fulvestrant; however, the 95% CrIs included the potential for no difference or that the comparator could be favoured (i.e., 95% CrIs crossed the null). The results of the random-effects model had wider 95% CrIs than those of the fixed-effects model. The results of the PH assessment showed evidence of non-PHs across all studies.
Figure 7: NMA Evidence Network for OS
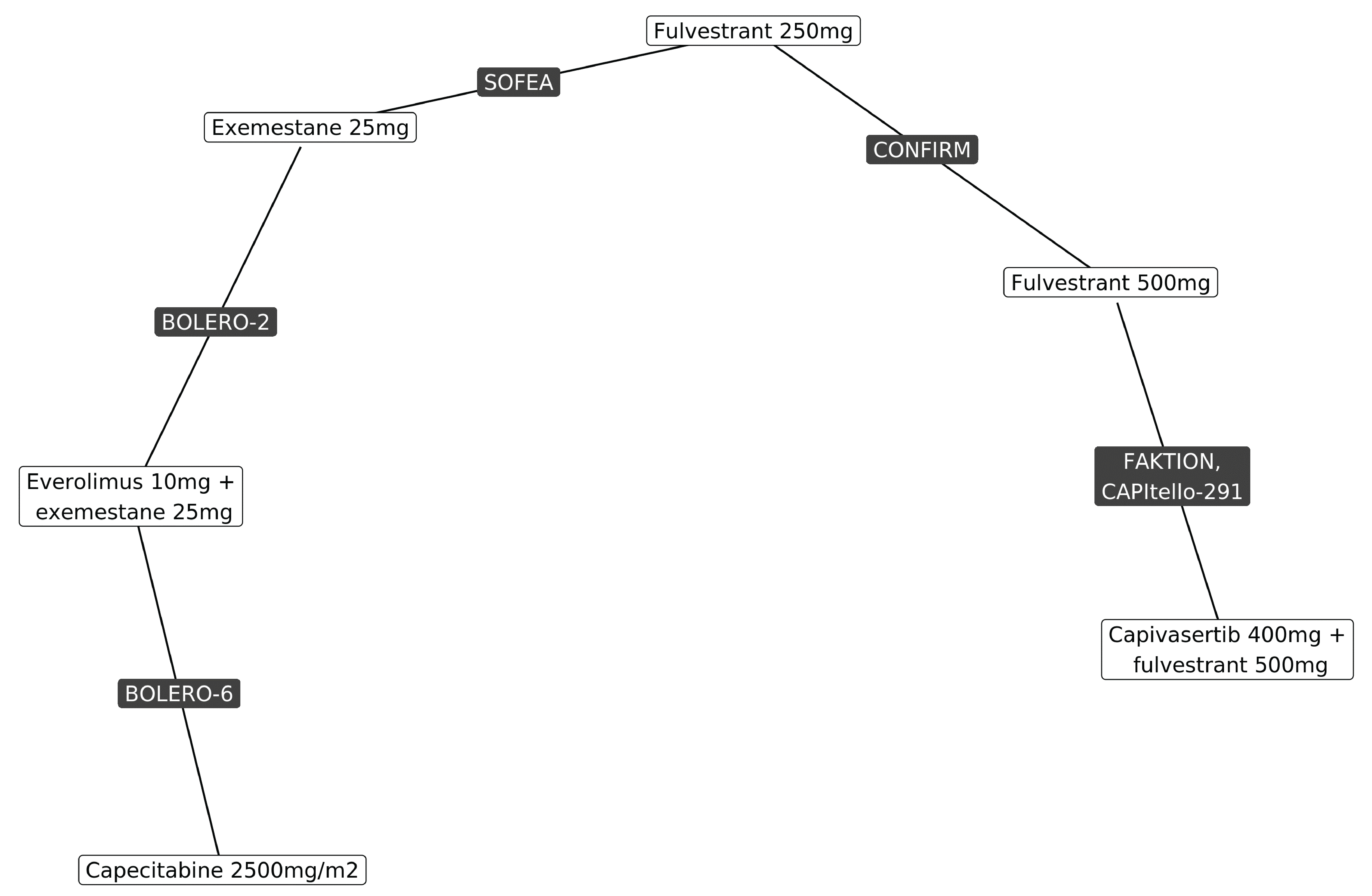
NMA = network meta-analysis; OS = overall survival.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Figure 8: NMA Forest Plot Comparison With Capivasertib Plus Fulvestrant for OS [Redacted]

CrI = credible interval; NMA = network meta-analysis; OS = overall survival.
Source: NMA technical report; details are from the sponsor’s Summary of Clinical Evidence.59
Critical Appraisal of the NMA
The methods used to conduct the systematic literature review and NMA were prespecified with an a priori protocol and used appropriate criteria to search databases, select studies, extract data, and assess risk of bias in the included studies. Selection bias is expected to be low, given the comprehensiveness of the searches and methods for study selection.
The NMA included relevant outcomes identified by the CDA-AMC team (PFS and OS); however, important outcomes, such HRQoL and harms, were not included in the comparisons. Overall, the network was sparse (i.e., many comparisons, but few studies). The results of the inconsistency analysis indicated that the consistency assumption was met for PFS, although the only closed loop in the network did not include capivasertib plus fulvestrant. It was not possible to assess for inconsistency across direct and indirect evidence in the OS NMA because of the absence of loops in the network (i.e., no direct evidence). Several comparisons in the NMA were based on the results of more than 2 studies linked in the network (e.g., the BOLERO-2 trial to the SOFEA trial to the CONFIRM trial to the CAPItello-291 trial), which led to increased uncertainty in the relative treatment effects. These included comparisons between capivasertib plus fulvestrant, everolimus plus exemestane, and capecitabine monotherapy. The PH assumption was violated in almost all comparisons for PFS and OS; as such, the hazard ratios may not be fully reflective of the true effects.
The exchangeability assumption was violated because there were several notable sources of heterogeneity for potential effect modifiers across the included studies. Identified variables of concern included AKT pathway alterations, prior CDK4/6 inhibitor treatment, HER2 status, region of enrolment, line of therapy, and menopausal status. Specifically, of the 10 included studies, only 2 reported results on patients with AKT pathway alterations (the CAPItello-291 and FAKTION trials), both involving capivasertib. For other treatments, there was no evidence in the AKT pathway–altered population. Only 1 of the 10 included studies (the CAPItello-291 study) reported subgroup data based on prior CDK4/6 inhibitor treatment, which is recognized as a prognostic factor. Although the authors provided evidence for treatment-effect modifiers, it was not clear how these were identified (i.e., whether a literature review or expert consensus was performed). As such, it is not clear whether all treatment-effect modifiers were accounted for in the feasibility assessment. In addition, the median follow-up times across the included trials were not reported.
The risk of bias assessment at the individual study level and at the outcome level — and their potential impact on the NMA effect estimates — were not explicitly assessed or discussed, and no sensitivity analyses were conducted to examine the influence of high-risk-of-bias studies on relative treatment effects. All the included studies were judged by the authors of the NMA to have some potential for (or to be at high risk of) bias, which reduces the certainty in the effect estimates.
In general, the magnitude and direction of potential bias because of heterogeneity and lack of proportionality on outcome estimates cannot be predicted. Because of these limitations in the NMA, no definitive conclusions could be drawn about the relative treatment effects of capivasertib plus fulvestrant versus other relevant comparators.
Studies Addressing Gaps in the Systematic Review Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
To supplement the evidence available for patients with HR-positive, HER2-negative, advanced breast cancer harbouring PIK3CA, AKT1, or PTEN alterations who have progressed on ETs without prior CDK4/6 inhibitors, the sponsor summarized and provided publications for the FAKTION trial. The sponsor’s rationale for including this study was that it included longer follow-up times for OS compared to the pivotal trial.
The sponsor also submitted the results of a real-world study using the Oncology Outcomes database in Alberta, which evaluated survival, treatment patterns, and health care resource use in patients in Canada with HER2-negative, advanced breast cancer who have progressed on prior ET. However, because the study did not report any data specific to the efficacy or harms of capivasertib plus fulvestrant that were relevant to the indication under review, the results of this study have not been summarized in the current report.
Description of Studies
The FAKTION study was an investigator-initiated, multicentre, randomized, double-blind, placebo-controlled, biomarker-adaptive, phase II trial in which patients were enrolled from 19 hospitals in the UK. Study details are provided in Table 21.
Table 21: Details of Study Addressing Gaps in the Systematic Review Evidence
Detail | FAKTION trial |
|---|---|
Design and population | |
Study design | Phase II, randomized, multicentre, double-blind, placebo-controlled trial |
Locations | UK (19 hospitals) |
Patient enrolment dates | Start date: May 2014 End date: March 2019 |
Enrolled, N | 149 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Intramuscular fulvestrant 500 mg (day 1) every 28 days (plus a loading dose of 500 mg on day 15 of cycle 1) with oral capivasertib 400 mg twice daily on an intermittent weekly schedule of 4 days on and 3 days off |
Comparator(s) | Intramuscular fulvestrant 500 mg (day 1) every 28 days (plus a loading dose of 500 mg on day 15 of cycle 1) with oral placebo twice daily on an intermittent weekly schedule of 4 days on and 3 days off |
Study duration | |
Screening phase | 4 weeks |
Treatment phase | Until progressive disease according to RECIST 1.1, development of unacceptable toxicities, loss to follow-up, or withdrawal of consent |
Follow-up phase | Every 3 months |
Outcomes | |
Primary end point | PFS |
Secondary end points |
|
Notes | |
Publications | |
AI = aromatase inhibitor; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: Howell et al. (2022),65 Jones et al. (2020);66 details are from the sponsor’s Summary of Clinical Evidence.
Populations
Eligible patients were postmenopausal females with locally advanced or metastatic, ER-positive breast cancer not suitable for surgical resection. Patients were considered suitable for ET, but had received no more than 3 previous lines of ET and up to 1 line of chemotherapy for advanced breast cancer. They also had progressive disease during treatment with a third-generation AI or relapsed on an AI in the adjuvant setting. The FAKTION trial included an overall population that included both expanded pathway–altered and pathway-nonaltered subgroups. The expanded pathway–altered subpopulation included patients who tested positive for tumours with 1 or more PIK3CA, AKT1, or PTEN alterations and is the focus of the reimbursement request. Test results were considered positive if either assay (i.e., the Foundation One CDx Clinical Trial NGS assay testing of tumour biopsy samples and/or the GuardantOMNI RUO assay testing of plasma) detected 1 or more PIK3CA, AKT1, or PTEN alterations.65 Given that the clinical experts consulted by CDA-AMC indicated that NGS is the preferred assay to test for PIK3CA, AKT1, or PTEN alterations, this section included efficacy outcomes for the NGS-identified, pathway-altered analysis set as well.
Interventions
Patients were randomized 1 to 1 to receive fulvestrant 500 mg (on day 1 and day 15 of cycle 1 and on day 1 only of subsequent 28-day cycles) with either capivasertib 400 mg twice daily or placebo (4 days on and 3 days off, starting on cycle 1, day 15) until disease progression, unacceptable toxicity, withdrawal of consent, or loss to follow-up. Allocation was balanced by minimization according to PIK3CA mutation status (mutated versus wild type), PTEN expression status (null versus detected in ≥ 1% of tumour cells at moderate or strong intensity or ≥ 10% of cells at weak intensity), measurable versus nonmeasurable disease, and primary versus secondary resistance to a third-generation AI. Fulvestrant was given as 250 mg in a 5 mL solution for intramuscular injection. Dose reductions for fulvestrant were not permitted.65
Outcomes
The primary end point was investigator-assessed PFS, defined as the time from randomization to either the first documented progression confirmed by RECIST 1.1 (regardless of whether the participant withdrew from the study or received another anticancer therapy before progression) or death from any cause in the ITT population.
Secondary end points were OS (defined as the time from randomization to death from any cause), objective response (defined as the proportion of patients with a complete or partial response, according to RECIST 1.1), clinical benefit (defined as the proportion of patients with an objective response or stable disease lasting ≥ 24 weeks), and safety in the ITT population. The assessment of safety was based on the incidence of AEs, SAEs, notable AEs, AEs leading to discontinuation, AEs leading to dose modification, and deaths. Objective response and clinical benefit were not relevant to the current report; therefore, these were not summarized.65
Statistical Analysis
As of March 2020 (primary analysis), the sample size was calculated for a phase II screening design, based on a primary outcome of PFS. The sample size calculation assumed a time-to-event hazard ratio of 0.65, 90% power, a 1-sided alpha of 0.20, and an overall loss to follow-up of 10%. A total of 98 events were required in 138 participants with 18-month accrual and a 6-month minimum follow-up period for the analysis.66
The primary and updated analyses were performed in the FAS, which comprised all randomly assigned participants, on an ITT basis. An interim analysis of change in tumour size 8 weeks after randomization in the first 40 participants without pathway alteration had been planned. This analysis was done to allow adaptation of recruitment according to participants’ pathway alteration status. Time-to-event distributions were estimated using the KM method. The significance threshold was set at 0.05. Participants were censored at day 1 if they had no follow-up RECIST 1.1 assessment unless they died within 2 visits of baseline, in which case they were censored at their death date. Participants without disease progression confirmed by RECIST 1.1, and those who died or progressed after missing the last 2 RECIST 1.1 assessments, were considered censored for PFS at the date of the last RECIST 1.1 assessment or at the point of withdrawal of consent. Cox regression was performed to measure hazard ratios. Multivariable Cox regression was done to adjust the estimates for the randomization minimization variables. Hazard ratios were adjusted for pathway status as measured at randomization, primary or secondary AI resistance, and measurable or nonmeasurable disease. This adjustment was prespecified in the original as well as the updated statistical analysis plan.66
PFS was measured from enrolment to any disease progression and/or any death, defined according to strict RECIST 1.1. Lesions were compared to baseline measurements to assess progression. PFS was described using KM curves in both arms of the trial. The median PFS was calculated for each arm of the trial. The log-rank test was used to formally test the equality of the survivor functions. Cox regression was also performed to adjust the hazard ratio for the stratification factors for the PHs.66
OS was measured from randomization to death, with participants still alive censored at the date last assessed. The OS data were summarized and analyzed in the same way as PFS. The PH assumption was checked using Cox-Snell residuals and Schoenfeld’s global test.66
An updated analysis was available as of June 2022. Following the primary analysis, biomarker profiling was expanded to use NGS assays to interrogate the relevant genetic alterations more comprehensively. It also aimed to identify alterations in the PI3K, AKT, and/or PTEN pathways accurately.65
The updated statistical analysis plan was developed and approved before the planned data cut-off date. It specified that OS would be analyzed after 98 deaths in the ITT population. This plan also defined the prespecified exploratory end point of analyzing PFS and OS outcomes in subgroups in which the identification of PI3K, AKT, and/or PTEN pathway alteration status versus pathway–nonalteration status was expanded to include AKT1 testing (expanded testing panel) and results from NGS assays. For the original secondary end point, the hypothesis that the combination of fulvestrant and capivasertib would show greater benefit in participants whose tumours carried PI3K, AKT, and/or PTEN pathway alterations was examined by analyzing PFS and OS outcomes in expanded pathway–altered and nonaltered subgroups. OS and PFS were analyzed using the same statistical tests as described for the ITT population in this section earlier. There was no adjustment for multiplicity of testing. Two-sided P values were reported, with P ≤ 0·05 being considered significant. Schoenfeld’s tests for OS and PFS in the ITT population, the expanded pathway–altered and –nonaltered subgroups, remained consistent with the PH assumption. The exploratory end points assessing the benefit of fulvestrant plus capivasertib versus fulvestrant plus placebo in the pathway-altered and pathway-nonaltered subgroups identified by NGS alone were defined post hoc.65
Results
Patient Disposition
Between March 16, 2015, and March 6, 2018, 183 patients were screened for eligibility, and 140 patients (77%) were randomly assigned to receive either fulvestrant plus capivasertib (n = 69 [49%]) or fulvestrant plus placebo (n = 71 [51%]). All randomly assigned patients were included in the efficacy and safety analyses. Participants were followed up until all had at least 6 months’ follow-up and the minimum 98 disease progression events required for analysis had been confirmed. The disposition of patients in the overall population is shown in Table 22. Patients in the expanded pathway–altered subpopulation were considered of relevance to this indication under review, and results for this subgroup are the sole focus in this section. The disposition of patients in the expanded pathway–altered subpopulation is shown in Table 23.
Baseline Characteristics
A summary of baseline demographic and disease characteristics of patients in the overall population and expanded pathway–altered subgroup are in Table 24. In the expanded pathway–altered subpopulation, the median ages were 60 years (IQR, 55 years to 69 years) in the capivasertib plus fulvestrant arm and 62 years (IQR, 56 years to 68 years) in the placebo plus fulvestrant group. All patients were postmenopausal females; the FAKTION study did not collect data on race or ethnicity. Most patients (66.0%) had an ECOG Performance Status of 0, indicating good overall performance.
Table 22: Summary of Patient Disposition From the FAKTION Study (Overall Population)
Patient disposition | Overall population | |
|---|---|---|
Capivasertib plus fulvestrant | Placebo plus fulvestrant | |
Screened, N | 183 | |
Screened out, N | 38 | |
Reason for being screened out | ||
Not meeting inclusion criteria | 34 | |
Declined to participate | 2 | |
Other | 2 | |
Consented and registered | 145 | |
Not included | 5 | |
Reason for not including | ||
Poor cardiac function | 2 | |
Abnormal liver function | 2 | |
HER2-positive breast cancer | 1 | |
Randomized, N | 140 | |
Randomized, N (%) | 69 (49) | 71 (51) |
Discontinued capivasertib or placebo, n (%) | 68 (49) | 71 (51) |
Reason for capivasertib or placebo discontinuation, n (%) | ||
Clinical disease progression or death | 57 (84) | 68 (96) |
Intolerance to treatment because of toxicity and serious adverse events | 8 (12) | 0 (0) |
Patient decision | 0 (0) | 2 (3) |
Other | 3 (4) | 1 (1) |
ITT analysis and safety analysis, N | 69 | 71 |
HER2 = human epidermal growth factor receptor 2; ITT = intention to treat.
Source: Adapted from Howell SJ, et al., copyright 2022.65 This work is licensed under the CC BY 4.0 DEED Attribution 4.0 International. Full text is available here: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00284-4/fulltext.
Table 23: Summary of Patient Disposition From the FAKTION Study (Expanded Pathway–Altered Population)
Patient disposition | Altered population | |
|---|---|---|
Capivasertib plus fulvestrant | Placebo plus fulvestrant | |
ITT analysis and analyses, N | 39 | 37 |
Progression-free survival analysis | ||
Cases of RECIST 1.1 progression | 28 | 34 |
Deaths (counted as event) | 2 | 2 |
Deaths (censored) | 3 | 1 |
Active RECIST 1.1 follow-up at data lock (censored) | 2 | 0 |
Lost to follow-up (censored) | 4 | 0 |
Overall survival analysis | ||
Deaths | 25 | 32 |
Alive on active follow-up (censored) | 10 | 2 |
Lost to follow-up (censored) | 3 | 2 |
Withdrawn consent (censored) | 1 | 1 |
ITT = intention to treat; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: Adapted from Howell SJ, et al., copyright 2022.65 This work is licensed under the CC BY 4.0 DEED Attribution 4.0 International. Full text is available here: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00284-4/fulltext.
Table 24: Summary of Baseline Characteristics of Patients From the FAKTION Study (Expanded Pathway–Altered Population)
Patient disposition | Overall population | Expanded pathway–altered population | ||
|---|---|---|---|---|
Capivasertib plus fulvestrant (n = 69) | Placebo plus fulvestrant (n = 71) | Capivasertib plus fulvestrant (n = 39) | Placebo plus fulvestrant (n = 37) | |
Median age, years (IQR); range | 62 (55 to 68); 42 to 81 | 61 (53 to 68); 40 to 82 | 60 (55 to 69); 46 to 81 | 62 (56 to 68); 47 to 73 |
Sex, n (%) | ||||
Male | 0 | 0 | 0 | 0 |
Female | 69 (100) | 71 (100) | 39 (100) | 37 (100) |
ECOG Performance Status, n (%) | ||||
0 (normal activity) | 42 (61) | 49 (69) | 25 (64) | 25 (68) |
1 (restricted activity) | 25 (36) | 17 (24) | 14 (36) | 9 (24) |
2 (in bed less than or equal to 50% of the time) | 1 (1) | 2 (3) | 0 | 1 (3) |
Missing | 1 (1) | 3 (4) | 0 | 2 (5) |
Stage, n (%) | ||||
III inoperable | 0 | 1 (1) | 0 | 1 (3) |
IV | 68 (99) | 68 (96) | 38 (97) | 35 (95) |
Missing | 1 (1) | 2 (3) | 1 (3) | 1 (3) |
Number of disease sites, n (%) | ||||
Median (IQR); range | 2 (2 to 3); 1 to 5 | 2 (1 to 3); 1 to 5 | 2 (2 to 3); 1 to 5 | 2 (1 to 3); 1 to 5 |
1 | 13 (19) | 19 (27) | 8 (21) | 11 (30) |
2 | 56 (81) | 52 (73) | 31 (79) | 26 (70) |
Site of metastases,a n (%) | ||||
Bone | 59 (86) | 55 (77) | 34 (87) | 28 (76) |
Liver | 32 (46) | 29 (41) | 22 (56) | 12 (32) |
Lung | 30 (43) | 28 (39) | 17 (44) | 17 (46) |
Lymph | 28 (41) | 31 (44) | 14 (36) | 19 (51) |
Pericardial or pleural | 5 (7) | 3 (4) | 2 (5) | 0 |
Brain | 1 (1) | 1 (1) | 1 (3) | 1 (3) |
Chest wall or skin | 1 (1) | 3 (4) | 0 | 2 (5) |
Other visceral | 2 (3) | 1 (1) | 2 (5) | 0 |
Visceral disease, n (%) | 49 (71) | 47 (66) | 30 (77) | 24 (65) |
Measurable disease,b n (%) | 49 (71) | 50 (70) | 27 (69) | 26 (70) |
Primary or secondary AI resistance,b n (%) | ||||
Primary | 25 (36) | 26 (37) | 15 (38) | 10 (27) |
Secondary | 44 (64) | 45 (63) | 24 (62) | 27 (73) |
Previous adjuvant endocrine therapy, n (%) | 60 (87) | 65 (92) | 34 (87) | 35 (95) |
Previous adjuvant chemotherapy, n (%) | 36 (52) | 42 (59) | 20 (51) | 21 (57) |
Prior lines of endocrine treatment for metastatic or locally advanced disease, n (%) | ||||
0 | 9 (13) | 6 (8) | 6 (15) | 2 (5) |
1 | 39 (57) | 45 (63) | 22 (56) | 26 (70) |
≥ 2 | 20 (29) | 20 (28) | 11 (28) | 9 (24) |
Missing | 1 (1) | 0 | 0 | 0 |
Metastatic chemotherapy for advanced breast cancer, n (%) | 17 (25) | 20 (28) | 9 (23) | 9 (24) |
AI = aromatase inhibitor; ECOG = Eastern Cooperative Oncology Group; IQR = interquartile range.
aSites are not mutually exclusive.
bRandomization minimization factor.
Source: Adapted from Howell SJ, et al., copyright 2022.65 This work is licensed under the CC BY 4.0 DEED Attribution 4.0 International. Full text is available here: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00284-4/fulltext.
Some notable imbalances were observed between the treatment groups in the patient characteristics for the expanded pathway–altered subpopulation. The group receiving capivasertib plus fulvestrant had a higher proportion of patients with an ECOG Performance Status of 1 (36% versus 24%) than the group receiving placebo plus fulvestrant. Most patients had metastatic disease (96%). The sites of metastases were largely imbalanced between the capivasertib plus fulvestrant group and the placebo plus fulvestrant group, involving bone sites (87% versus 76%, respectively), liver (56% versus 32%), lung (44% versus 46%), lymph (36% versus 51%), pericardial or pleural (5% versus 0), chest wall or skin (0 versus 5%), and other visceral (5% versus 0). The number of metastatic sites ranged from 0 to 7. The group receiving capivasertib plus fulvestrant had a higher proportion of patients with metastatic disease in 2 sites (79% versus 70%), but a lower proportion of patients with metastatic disease in 1 site (21% versus 30%) than the group receiving placebo plus fulvestrant. Visceral disease was present in 30 patients (77%) in the capivasertib plus fulvestrant arm and in 24 patients (65%) in the placebo plus fulvestrant arm, indicating imbalances between the treatment groups. The group receiving capivasertib plus fulvestrant had a higher proportion of patients with primary AI resistance (38% versus 27%), but a lower proportion of patients with secondary AI resistance (62% versus 73%). The group receiving placebo plus fulvestrant had a higher proportion of patients with previous adjuvant ET (95% versus 87%) as well as previous adjuvant chemotherapy (57% versus 51%) than the group receiving capivasertib plus fulvestrant. The proportions of patients with prior lines of ET for metastatic or locally advanced disease had notable imbalances as well, with 15% versus 5% of patients having 0 lines, 56% versus 70% having 1 line, and 28% versus 24% having greater than or equal to 2 lines of treatment in the group receiving capivasertib plus fulvestrant versus the group receiving placebo plus fulvestrant.
In the expanded testing panel using advances in genetic testing assays, PIK3CA, AKT1 and PTEN alterations were identified in tumours from 76 patients (54%) of the 140 patients in the ITT population (in 39 patients receiving capivasertib and 37 receiving placebo). Tumour alterations were identified in 20 patients (25%) of the 81 patients whose tumours had been originally considered as pathway-nonaltered (10 of whom had been assigned to the capivasertib group and 10 to the placebo group).65
Exposure to Study Treatments
Patients assigned to each group received study treatment until disease progression, development of unacceptable AEs, loss to follow-up, or withdrawal of consent. By the data cut-off date of November 25, 2021, the median follow-up durations for the expanded panel were 58.5 months (IQR, 45.9 months to 64.1 months) for patients treated with fulvestrant plus capivasertib and 62.3 months (IQR, 62.1 months to 70.3 months) for patients treated with fulvestrant plus placebo. For the expanded pathway–altered subgroup, the median follow-up durations were 54.3 months (IQR, 45.5 months to 61.2 months) for the group treated with fulvestrant and capivasertib and 62.3 months (IQR, 62.1 months to not reached) for the group treated with fulvestrant and placebo.66
The PFS analysis in the ITT population was updated after 118 progression events had occurred (in 54 patients [78%] of the 69 patients assigned to capivasertib and in 64 patients [90%] of the 71 patients assigned to placebo). The Schoenfeld’s tests were consistent with the PH assumption; thus, the assumption was adequately met.66
Efficacy
Progression-Free Survival
A PFS event was recorded for 66 patients of 76 patients (87%) in the expanded pathway–altered subgroup, with 30 patients of 39 patients (77%) receiving capivasertib plus fulvestrant and 36 patients of 37 patients (97%) receiving placebo plus fulvestrant. Median PFS durations were 12.8 months (95% CI, 6.6 months to 18.8 months) in the group receiving capivasertib plus fulvestrant versus 4.6 months (95% CI, 2.8 months to 7.9 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.44; 95% CI, 0.26 to 0.72).65
Similar results were observed in the NGS-identified, pathway-altered analysis set, in which a PFS event was recorded for 25 patients of 34 patients (74%) who received capivasertib plus fulvestrant and all 29 patients (100%) who received placebo plus fulvestrant. Median PFS was extended in those who received capivasertib plus fulvestrant compared to those who received placebo plus fulvestrant: 13.4 months (95% CI, 6.6 months to 20.7 months) in the group receiving capivasertib plus fulvestrant versus 3.1 months (95% CI, 2.8 months to 7.1 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.36; 95% CI, 0.20 to 0.65). Results of the Schoenfeld’s tests for PHs were not statistically significant. PFS in the expanded pathway–altered subgroup is presented in Figure 9.
Overall Survival
At the time of analysis, 57 patients of 76 patients (75%) in the expanded pathway–altered subgroup had died. Of these, 25 patients of the 39 patients (64%) had received capivasertib plus fulvestrant and 32 patients of the 37 patients (86%) had received placebo plus fulvestrant. Median OS in the expanded pathway–altered subgroup of patients receiving capivasertib plus fulvestrant was 38.9 months (95% CI, 23.3 months to 50.7 months) compared with 20.0 months (95% CI, 14.8 months to 31.4 months) for those receiving placebo plus fulvestrant (adjusted hazard ratio = 0.46; 95% CI, 0.27 to 0.79).65 Results for the Schoenfeld’s tests for PHs were not statistically significant. OS in the expanded pathway–altered subgroup is presented in Figure 10.
Similar results were observed in the post hoc analysis involving the NGS-identified, pathway-altered subgroup, where an OS event was recorded for 21 patients of 34 patients (61%) who received capivasertib plus fulvestrant and 25 patients of 29 patients (86%) who received placebo plus fulvestrant. Median OS durations were 39.0 months (95% CI, 22.3 months to 50.7 months) in the group receiving capivasertib plus fulvestrant versus 20.9 months (95% CI, 14.1 months to 35.4 months) in the group receiving placebo plus fulvestrant (adjusted hazard ratio = 0.44; 95% CI, 0.24 to 0.81).65
Figure 9: KM Plot of PFS for the Expanded Pathway–Altered Population in the FAKTION Trial
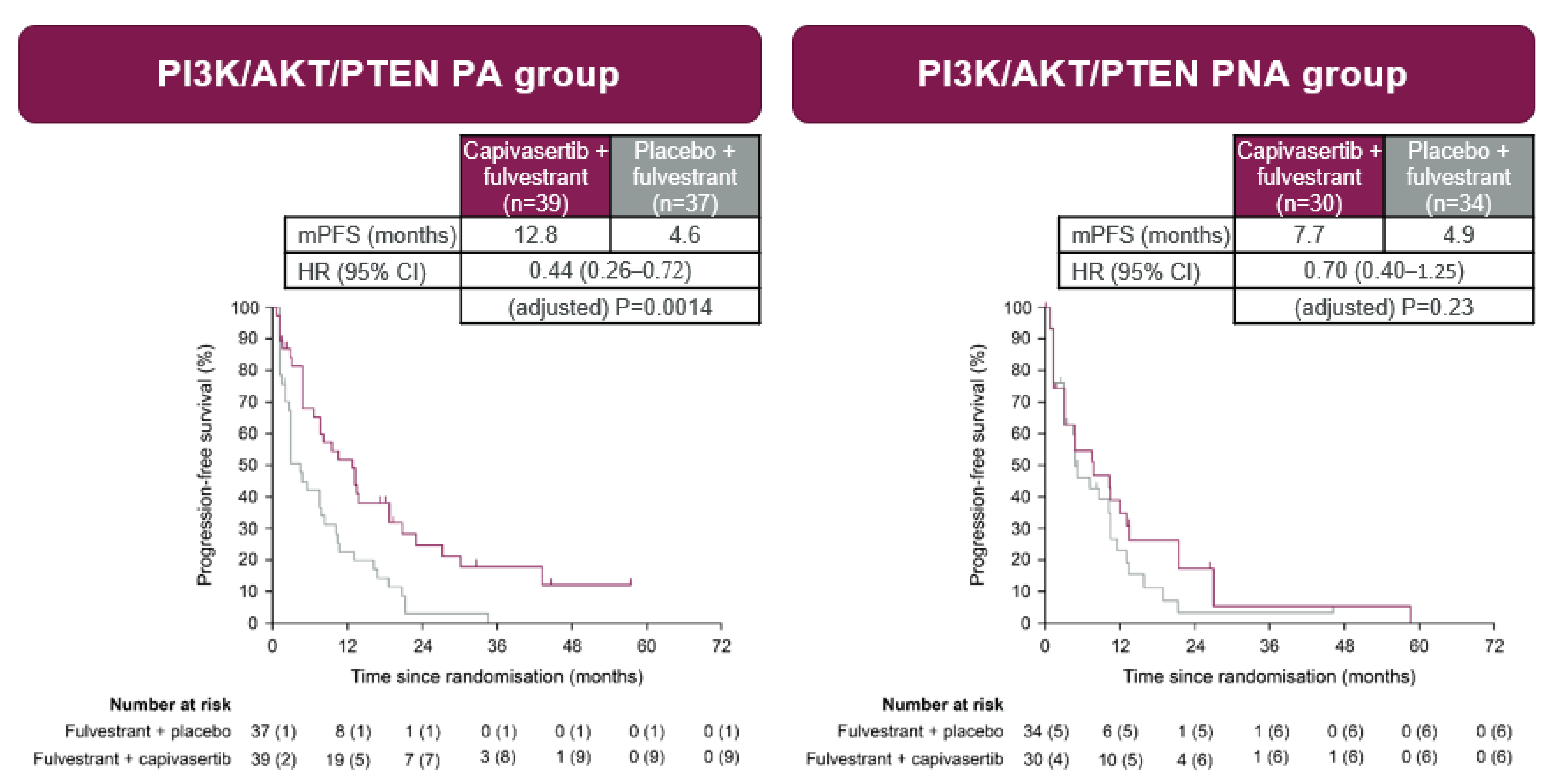
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; mPFS = median progression-free survival; NGS = next-generation sequencing; PFS = progression-free survival.
Note: Pathway alteration status was assessed using NGS.
Source: Adapted from Howell SJ, et al., copyright 2022.65 This work is licensed under the CC BY 4.0 DEED Attribution 4.0 International. Full text is available here: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00284-4/fulltext.
Harms
Safety analyses included all patients who had received at least 1 dose of the assigned study drug. All randomly assigned patients were included in the safety analyses. The most commonly reported AEs, regardless of dose, schedule, or causality, were diarrhea, nausea, hyperglycemia, fatigue, vomiting, decreased appetite, and maculo-papular rash. The proportions of participants experiencing grade 3 to 5 AEs (irrespective of causality) were 45 (65%) in the group receiving capivasertib plus fulvestrant and 35 (50%) in the group receiving placebo plus fulvestrant. One patient in the group receiving placebo plus fulvestrant experienced a grade 5 hemorrhage related to disease progression. All cases of severe diarrhea, rash, hyperglycemia, and vomiting in both groups were grade 3, except for 1 grade 4 event of diarrhea in the group receiving placebo plus fulvestrant. The most common grade 3 or 4 AEs experienced by patients were hypertension (in 22 patients of 69 patients [32%] in the group receiving capivasertib plus fulvestrant versus 18 patients of 71 patients [25%] in the group receiving placebo plus fulvestrant), diarrhea (in 10 patients [14%] versus 3 patients [4%]), rash (in 14 patients [20%] versus 0 patients), infection (in 4 patients [6%] versus 2 patients [3%]), and fatigue (in 1 patient [1%] versus 3 patients [4%]).
Figure 10: KM Plot of OS in the Expanded Pathway–Altered Population of the FAKTION Trial
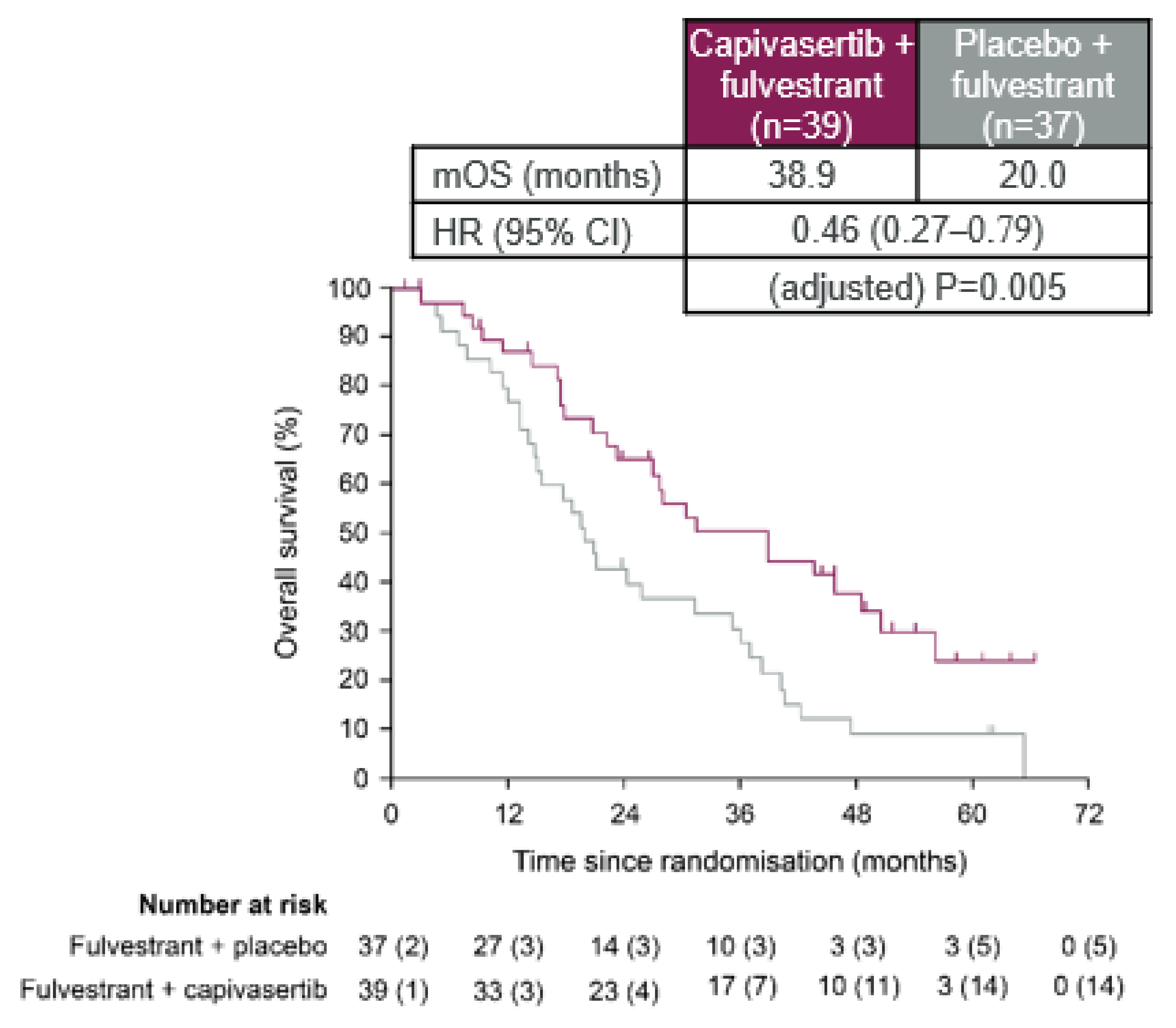
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; mOS = median overall survival; OS = overall survival.
Source: Adapted from Howell SJ, et al., copyright 2022.65 This work is licensed under the CC BY 4.0 DEED Attribution 4.0 International. Full text is available here: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00284-4/fulltext.
Although serious adverse reactions (reported only in the capivasertib plus fulvestrant group) were reported, the total number of SAEs, irrespective of causality, was not reported in the publication. The most commonly reported SAEs experienced by patients were dyspnea, back pain, lower respiratory tract infection, pain, abdominal pain, and noncardiac chest pain. As of the data cut-off date, 21 patients (30%) patients in the capivasertib group and 31 patients (44%) in the placebo group had died. A total of 2 deaths occurred among patients with AEs.65,66
Critical Appraisal
Internal Validity
The FAKTION trial was a randomized, double-blind, placebo-controlled, phase II trial. The randomization and masking procedures were appropriate. Because it was a phase II trial that included fewer patients and aimed to provide preliminary evidence about the efficacy and harms of the study drug, the results cannot be considered confirmatory.
Despite randomization, imbalances were observed at baseline in patients’ disease characteristics (e.g., ECOG PS, histopathological subtype, visceral disease, AI given as last treatment before registration, previous ET, and PIK3CA and/or PTEN results). Because of the small sample size, there is an increased risk that prognostic balance was not achieved, as evidenced by these imbalances. As such, it is possible that the observed effects were either overestimated or underestimated and may have been driven by prognostic differences between the 2 groups (i.e., may not be reflective of the true treatment effect). Cox regression was used to estimate hazard ratios with CIs and P values for both PFS and OS outcomes, and multivariable Cox regression was used to adjust the estimates for the randomization minimization variables. Hazard ratios were adjusted for pathway status as determined at randomization, primary or secondary AI resistance, and measurable or nonmeasurable disease. Results of the Schoenfeld’s tests for the PH assumption were not statistically significant, although these may have not been powered to detect a violation. No major violations of the PH assumption were noted through visual inspection of the KM plots. The differences in PFS and OS between the treatment groups observed in the FAKTION trial for the altered patient group were considered clinically meaningful by the clinical experts consulted for this review.
Both patients and investigators were blinded to the capivasertib plus fulvestrant or placebo plus fulvestrant assignment. PFS was assessed by the investigator, without BICR adjudication. It is possible that patients and investigators may have become unblinded because of imbalances in notable harms across the 2 treatment groups (e.g., more patients experienced diarrhea and rash in the capivasertib plus fulvestrant group). As such, there may be an increased risk of bias in the measurement of PFS and subjective harms; however, the presence and direction of bias is uncertain. Censoring reasons seemed balanced between the treatment groups.
External Validity
The FAKTION trial population was limited to postmenopausal females with histological confirmation of HR-positive, HER2-negative, locally advanced or metastatic, inoperable breast cancer that was not amenable to curative surgical resection; this was a subset of Health Canada–indicated population (i.e., premenopausal and postmenopausal adult females). The narrower patient population may affect the generalizability of the trial results in the Canadian setting. In addition, male patients and patients with prior CDK4/6 inhibitor treatment were not enrolled. Male patients would be included in the patient population of the sponsor’s reimbursement request, although they are not included in the Health Canada indication. The clinical experts noted that all patients in Canada who are candidates for treatment with capivasertib plus fulvestrant will have been treated with a CDK4/6 inhibitor because these are now part of usual first-line treatment in combination with ET; males would also be considered candidates for treatment. HRQoL, which is considered important by both patients and clinicians, was not measured. No data on race or ethnicity of patients were available, which makes it difficult to contextualize the results in the Canadian treatment setting. The dosing and administration of capivasertib plus fulvestrant were consistent with the Health Canada–approved product monograph.
Discussion
Summary of Available Evidence
One pivotal, phase III, double-blind RCT, 1 NMA, and 1 study addressing gaps in the pivotal RCT evidence submitted by the sponsor were summarized in this report.
One ongoing trial, the CAPItello-291 trial (N = 708), met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the CAPItello-291 trial was to assess the efficacy and safety of capivasertib plus fulvestrant compared with matched placebo plus fulvestrant in adults with locally advanced (inoperable) or metastatic, HR-positive, HER2-negative breast cancer. The trial enrolled patients who had disease recurrence or progression during or after AI therapy, with or without a CDK4/6 inhibitor. The trial included 2 populations that were analyzed separately: the overall population (all enrolled patients) and the altered population (N = 289), which included patients who tested positive for tumours with 1 or more PIK3CA, AKT1, or PTEN alterations. The Health Canada indication and reimbursement request align with the altered population. The outcomes most relevant to the CDA-AMC review included the primary outcome of PFS per RECIST 1.1 assessed by the investigators and the secondary outcomes of OS, HRQoL, and safety. The HRQoL outcomes included EORTC QLQ-C30 global health status score and EORTC QLQ-B23 functional and symptom scales scores. The trial population was predominately white (58%) and female (99%), with a mean age of 58 years (range, 26 years to 90 years). Most patients were postmenopausal females (77.0%), had previously received a CDK4/6 inhibitor (70%), and had an ECOG Performance Status of 0 (66.0%), indicating good overall performance.
In the absence of direct comparative evidence of capivasertib plus fulvestrant versus other relevant comparators, an NMA was conducted by the sponsor. The objective of the NMA was to provide evidence for the efficacy of capivasertib plus fulvestrant relative to fulvestrant 250 mg or 500 mg, exemestane 25 mg, everolimus 10 mg plus exemestane 25 mg, and capecitabine 1,250 mg/m2 in adult patients with HR-positive, HER2-negative, advanced breast cancer with AKT pathway–altered tumours who progressed during or after treatment with endocrine-based regimens. Fixed- and random-effects NMAs were conducted for PFS and OS using a Bayesian framework, and the results were summarized as hazard ratios and 95% CrIs.
The sponsor also submitted the FAKTION trial, which was a phase II, multicentre, double-blind RCT that compared capivasertib plus fulvestrant versus placebo plus fulvestrant among postmenopausal adult females with HR-positive, HER2-negative, metastatic or locally advanced, inoperable breast cancer to address gaps in the pivotal RCT evidence. The sponsor’s rationale for including this study was that it included longer follow-up durations for OS compared to the pivotal trial. The outcomes relevant to the CDA-AMC review included the primary outcome of investigator-assessed PFS and secondary outcomes of OS and safety. In the expanded pathway–altered subpopulation, the median ages were 60 years (IQR, 55 years to 69 years) in the capivasertib plus fulvestrant and 62 years (IQR, 56 years to 68 years) in the placebo plus fulvestrant arms.
Interpretation of Results
The evidence from the pivotal trial, CAPItello-291, addressed treatment outcomes noted as important by both patients and clinicians. The patient group input indicated that stopping disease progression, prolonging life, improving HRQoL, and reducing treatment side effects are important to them. Similarly, the clinical experts consulted by CDA-AMC indicated that because the treatment goal for patients is palliative, the unmet needs of patients are for new treatments that can delay progression, prolong survival, and improve quality of life while exposing patients to minimal toxicity. The FAKTION study attempted to fill the gap in the CAPItello-291 trial by reporting a longer duration of follow-up for OS in the altered subpopulation; however, the trial had important methodological limitations. A key limitation was that the study enrolled only postmenopausal females with histological confirmation of ER-positive breast cancer, which was a subset of the patients included in the Health Canada indication and reimbursement request; in addition, it had a small sample size, likely contributing to the imbalances in key patient baseline disease and demographic characteristics. This narrow patient population may affect the generalizability of the trial results in the Canadian setting. It is possible that the observed effects were either overestimated or underestimated and may have been driven by prognostic differences between the treatment groups. In addition, male patients and patients with prior CDK4/6 inhibitor treatment were not enrolled; and HRQoL, which is considered important by both patients and clinicians, was not measured. For these reasons, the FAKTION study was deemed insufficient to support definitive conclusions.
Efficacy
The CAPItello-291 trial supported a clinically meaningful improvement of capivasertib plus fulvestrant over placebo plus fulvestrant for PFS in adults with locally advanced or metastatic, HR-positive, HER2-negative breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations at the final PFS analysis (August 15, 2022 data cut-off). In the altered population, the between-group differences in probabilities of PFS at 6 months and 12 months were █████ (95% CI, ████ ██ ████) and █████ (95% CI, ███ ██ ████), respectively. For the GRADE assessment, the clinical experts consulted by CDA-AMC suggested a clinically important threshold of 5% absolute risk difference between groups for both 6 months and 12 months. Based on this threshold, there was high- and moderate-certainty evidence for a clinically important increase in the probability of PFS at 6 months and 12 months, respectively. The PFS findings were consistent across the subgroup analysis by prior exposure to CDK4/6 inhibitors; however, the analysis was exploratory and potentially not powered to detect differences between groups.
By the August 15, 2022 data cut-off date, the median OS had not been reached in either group, and the between-group differences in probabilities of OS at 18 months and 24 months were █████ (95% CI, ████ ██ ████) and ████ (95% CI, ████ ██ ████), respectively. For the GRADE assessment, the clinical experts suggested a clinically important threshold of a 5% absolute risk difference for 18 months and 24 months. Using this threshold, there was low-certainty evidence for a clinically important increase in the probability of OS at 18 months and 24 months compared to placebo plus fulvestrant. Because patients were permitted to receive various posttreatment, anticancer medications after study treatment had been discontinued (approximately 49% of all patients), the potential treatment benefit on OS would have been subject to a degree of uncertainty. It is also unclear if all the subsequent therapies were relevant to Canadian practice. Given the importance of this outcome to patients and clinicians, longer follow-up durations for the OS analysis would have been preferred to determine the clinical value of treatment with capivasertib plus fulvestrant. In addition, there is uncertainty as to whether the PFS benefits (as a surrogate outcome for OS) will translate into survival benefits. There are systematic literature reviews of RCTs investigating other treatments for HR-positive, HER2-negative, metastatic breast cancer that have reported a correlation between PFS and OS.55,56 However, these correlations do not mean that PFS results as a surrogate can predict the final OS outcome for a specific treatment. As such, only direct OS results would confirm if a clinically relevant difference in survival time is because of treatment with capivasertib plus fulvestrant.
For the EORTC QLQ-C30 global health status score, the certainty of evidence was low for capivasertib plus fulvestrant, resulting in little to no clinically important difference at cycle 10, based on the sponsor’s suggested threshold (informed by the literature) of a 10-point change from baseline score. The low certainty of evidence was attributed to risk of bias because of missing outcomes data. For the EORTC QLQ-BR23 functional and symptom scale scores, the certainty of evidence for all outcomes was very low for the effect of capivasertib plus fulvestrant at cycle 17 compared to placebo plus fulvestrant. The very low certainty of evidence was attributed to serious or very serious imprecision because of the 95% CI for the between-group difference including the possibility of either benefit, little to no difference or harm, and risk of bias because of missing outcomes data. For the sexual enjoyment scale, there was no evidence for comparison at cycle 17. Although postbaseline data for most scales were limited at cycle 17, the data were consistent with cycles closer to the median duration of follow-up for all patients.
In general, the population requested for reimbursement aligns with the Health Canada indication, except that the reimbursement request is not limited to female patients. Enrolment in the CAPItello-291 trial was open to both male and female patients, and 7 males were enrolled. The clinical experts consulted by CDA-AMC agreed that including males in the reimbursement request is appropriate because the proportion of included patients reflected the low prevalence of breast cancer in males, and because the management of breast cancer in males and females is similar. Given the small proportion of males in the trial, it was not possible to ascertain from the data whether males would experience different treatment outcomes compared with females. However, the clinical experts agreed that they would expect similar efficacy and harms among both males and females.
Based on the sponsor-submitted NMA, the results for both PFS and OS favoured capivasertib plus fulvestrant over exemestane 25 mg, fulvestrant 500 mg, and fulvestrant 250 mg. For both PFS and OS, the results comparing capivasertib plus fulvestrant to everolimus 10 mg plus exemestane 25 mg and capecitabine 2,500 mg/m2 favoured capivasertib plus fulvestrant; however, the 95% CrIs included the possibility of no difference or of the comparator being favoured (i.e., crossed the null). The results of the PH assessment showed evidence of non-PHs across most studies, including the CAPItello-291, BOLERO-5, and PEARL trials. As such, the hazard ratios may not be reflective of the true effects. There were several notable sources of heterogeneity for potential effect modifiers across the included studies. Identified variables of concern included AKT pathway alterations, prior CDK4/6 inhibitor treatment, HER2 status, region of enrolment, line of therapy, and menopausal status. Specifically, of the 10 included studies, only 2 studies reported results on patients with AKT pathway alterations (the CAPItello-291 and FAKTION trials); both involved capivasertib. For other treatments, there was no evidence in the AKT pathway–altered population. Only 1 of the 10 included studies (the CAPItello-291 trial) reported subgroup data based on prior CDK4/6 inhibitor treatment. In addition, the median follow-up times across the included trials were not reported. Although the authors provided evidence for heterogeneity in potential treatment-effect modifiers, it was not clear how these were identified (i.e., whether a literature review or expert consensus was performed). As such, it is not clear whether all treatment-effect modifiers were accounted for in the feasibility assessment. The magnitude and direction of potential bias on outcome estimates because of heterogeneity cannot be predicted. Because of these limitations in the NMA, no definitive conclusions could be drawn about the relative treatment effects of capivasertib plus fulvestrant versus fulvestrant 250 mg or 500 mg, exemestane 25 mg, everolimus 10 mg plus exemestane 25 mg, or capecitabine 1,250 mg/m2.
Harms
The safety profile of capivasertib plus fulvestrant in the altered population was similar to the safety profile in the overall population; because the sample size of the overall population was larger, the harms data summarized here are for the overall population. Most patients in the trial reported at least 1 AE. The most frequently reported AEs of any grade in the group receiving capivasertib plus fulvestrant were diarrhea, rash, and nausea. These AEs were numerically higher in the group receiving capivasertib plus fulvestrant than in the group receiving placebo plus fulvestrant. The clinical experts indicated that the higher incidence of AEs is expected with a combination treatment compared to a single-drug treatment, and that with appropriate care, the AEs would be manageable for many patients. The most frequently reported AEs in the group receiving placebo plus fulvestrant were diarrhea and nausea. A higher proportion of patients in the group receiving capivasertib plus fulvestrant experienced SAEs, with diarrhea being the most common. Based on the GRADE assessment, in the altered population, the between-group absolute risk difference of experiencing SAEs was █████ Using the null as a threshold (given that no clinically important threshold was determined), CDA-AMC judged that there is moderate certainty of evidence for capivasertib plus fulvestrant resulting in an increase in the proportion of patients who experience SAEs compared with placebo plus fulvestrant. This moderate certainty was attributed to serious imprecision because of the 95% CI for the between-group absolute risk difference including the possibility of both benefit and harm. Study treatment discontinuation because of AEs was numerically higher in the group receiving capivasertib plus fulvestrant than in the group receiving placebo plus fulvestrant, with rash being the most common AE leading to discontinuation of capivasertib or placebo. The incidence of death was similar between groups, with the majority of deaths attributed to disease progression. A higher proportion of notable AEs were reported in patients taking capivasertib plus fulvestrant than in those taking placebo plus fulvestrant, with noninfectious diarrhea, rash, and stomatitis being the most common.
The sponsor-submitted NMA did not include harms; therefore, no conclusions could be drawn about the safety of capivasertib plus fulvestrant relative to other relevant comparators.
Conclusion
Evidence from 1 ongoing, phase III, double-blind RCT (the CAPItello-291 trial) reported on outcomes that were important to both patients and clinicians. The trial showed high and moderate certainty of evidence that treatment with capivasertib plus fulvestrant results in a clinically meaningful increase in PFS at 6 months and 12 months, respectively, compared to placebo plus fulvestrant in adults with locally advanced or metastatic, HR-positive, HER2-negative breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations. At the time of the interim analysis, median OS had not been reached in either group. No definitive conclusions can be drawn about HRQoL because of concerns related to imprecision and missing outcomes data. Although the FAKTION study reported a longer follow-up duration for OS, it had important methodological limitations (e.g., imbalances in important baseline characteristics) and limited generalizability (e.g., it enrolled only postmenopausal females and excluded patients with prior CDK4/6 inhibitor treatment) that made it difficult to draw firm conclusions. There were no new safety signals identified; the safety of capivasertib plus fulvestrant was consistent with the known safety profiles of the individual drugs. However, the trial showed that treatment with capivasertib plus fulvestrant likely results in an increase in the proportion of patients who experience SAEs versus treatment with placebo plus fulvestrant. Because of limitations in the indirect treatment comparison, no conclusions can be drawn about the relative efficacy and safety of capivasertib plus fulvestrant compared to fulvestrant 250 mg or 500 mg, exemestane 25 mg, everolimus 10 mg plus exemestane 25 mg, or capecitabine 1,250 mg/m2.
References
1.Canadian cancer statistics: a 2023 special report on cancer prevalence. Toronto (ON): Canadian Cancer Statistics Advisory Committee; 2023: https://cancer.ca/Canadian-Cancer-Statistics-2023-EN. Accessed 2023 Nov 30.
2.Agenzia Italiana del Farmaco. Valutazione dell’innovativita’ - Medicinale: Kisqali (ribociclib). 2020; https://www.aifa.gov.it/documents/20142/1220805/KISQALI_14265_INNOV._v.1.0.pdf. Accessed 2023 Jun 12.
3.Statistics Canada. Population estimates on July 1st, by age and sex. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2023 Jul 19.
4.Alaklabi S, Roy AM, Attwood K, et al. Real world outcomes with alpelisib in metastatic hormone receptor-positive breast cancer patients: A single institution experience. Front Oncol. 2022;12:1012391. PubMed
5.National Cancer Institute. Cancer stat facts: female breast cancer subtypes. 2022; https://seer.cancer.gov/statfacts/html/breast-subtypes.html. Accessed 2022 May 25.
6.Khan NAJ, Abdallah M, Tirona MT. Hormone Receptor Positive/HER2 Negative Breast Cancer With Isolated Bladder Metastasis: A Rare Case. J Investig Med High Impact Case Rep. 2021;9:23247096211022186. PubMed
7.NCCN Clinical Practice Guidelines: breast. Version 4.2023 Plymouth Meeting (PA): National Comprehensive Cancer Network; 2023: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 2023 Aug 8.
8.BC Cancer. Breast cancer clinical guidelines. 6.9.0 metastatic breast cancer. 2017; http://www.bccancer.bc.ca/books/breast/management/metastatic-breast-cancer. Accessed 2021 Feb 8.
9.NCCN Clinical Practice Guidelines: breast cancer screening and diagnosis. Version 1.2020. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2020: https://www.nccn.org/professionals/physician_gls/pdf/breast-screening.pdf. Accessed 2023 Aug 8.
10.American Cancer Society. Breast cancer signs and symptoms. 2022; https://www.cancer.org/cancer/breast-cancer/about/breast-cancer-signs-and-symptoms.html. Accessed 2022 Jul 11.
11.Irvin W, Jr., Muss HB, Mayer DK. Symptom management in metastatic breast cancer. Oncologist. 2011;16(9):1203-1214. PubMed
12.Mokhtari-Hessari P, Montazeri A. Health-related quality of life in breast cancer patients: review of reviews from 2008 to 2018. Health Qual Life Outcomes. 2020;18(1):338. PubMed
13.Ades AE, Caldwell DM, Reken S, Welton NJ, Sutton AJ, Dias S. NICE DSU Technical Support Document 7: Evidence synthesis of treatment efficacy in decision making: a reviewer’s checklist 2012 [sponsor submitted reference].
14.Feng Y, Spezia M, Huang S, et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5(2):77-106. PubMed
15.Andrikopoulou A, Chatzinikolaou S, Panourgias E, et al. “The emerging role of capivasertib in breast cancer”. Breast. 2022;63:157-167. PubMed
16.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncology. 2016;2(12):1565-1573. PubMed
17.AstraZeneca. Clinical Study Report for CAPItello-291: A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative (HR+/HER2−) Breast Cancer Following Recurrence or Progression On or After Treatment with an Aromatase Inhibitor (CAPItello-291) [CONFIDENTIAL internal manufacturer's report]. 2022 [sponsor submitted reference].
18.Martorana F, Motta G, Pavone G, et al. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front Pharmacol. 2021;12:662232-662232. PubMed
19.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084-6091. PubMed
20.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61-70. PubMed
21.Vasan N, Toska E, Scaltriti M. Overview of the relevance of PI3K pathway in HR-positive breast cancer. Ann Oncol. 2019;30(Suppl_10):x3-x11. PubMed
22.Hortobagyi GN, Chen D, Piccart M, et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J Clin Oncol. 2016;34(5):419-426. PubMed
23.Turner NC, Kingston B, Kilburn LS, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020;21(10):1296-1308. PubMed
24.Waks AG, Gharaibeh M, Sjekloca N, et al. 253P Unmet need in heavily pre-treated patients with HR+/HER2- metastatic breast cancer (mBC) in the US: a ConcertAI analysis. Ann Oncol. 2022;33:S653.
25.Health Canada reviewer's report: Truqap (capivasertib) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Truqap (capivasertib) tablets, 160 mg and 200 mg, oral. Cambridge (UK): AstraZeneca Canada Inc.; 2024 Jan 25.
26.Clinical Study Report: D3615C00001. A phase III double-blind randomised study assessing the efficacy and safety of capivasertib plus fulvestrant versus placebo plus fulvestrant as treatment for locally advanced (inoperable) or metastatic hormone receptor positive, human epidermal growth factor receptor 2 negative breast cancer following recurrence or progression on or after treatment with an aromatase inhibitor (CAPItello-291) [internal sponsor's report]. Cambridge (UK): AstraZeneca Inc; 2023 Feb 27.
27.Breast cancer facts and figures 2019-2020. Atlanta (GA): American Cancer Society; 2019: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2019-2020.pdf. Accessed 2023 Aug 8.
28.Simon J, Chaix M, Billa O, et al. Survival in patients with HR+/HER2− metastatic breast cancer treated with initial endocrine therapy versus initial chemotherapy. A French population-based study. Br J Cancer. 2020;123(7):1071-1077. PubMed
29.Allison KH, Hammond MEH, Dowsett M, et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J Clin Oncol. 2020;38(12):1346-1366. PubMed
30.Tarantino P, Hamilton E, Tolaney SM, et al. HER2-low breast cancer: pathological and clinical landscape. J Clin Oncol. 2020;38(17):1951-1962. PubMed
31.Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. Arch Pathol Lab Med. 2018;142(11):1364-1382. PubMed
32.Zhang H, Moisini I, Turner BM, et al. Significance of HER2 in microinvasive breast carcinoma. Am J Clin Pathol. 2021;156(1):155-165. PubMed
33.Bhushan A, Gonsalves A, Menon JU. Current state of breast cancer diagnosis, treatment, and theranostics. Pharmaceutics. 2021;13(5). PubMed
34.BC Cancer. Breast cancer mangement guidelines. 2017; http://www.bccancer.bc.ca/books/breast. Accessed 2023 Feb 1.
35.Seung SJ, Traore AN, Pourmirza B, Fathers KE, Coombes M, Jerzak KJ. A population-based analysis of breast cancer incidence and survival by subtype in Ontario women. Curr Oncol. 2020;27(2):e191-e198. PubMed
36.Canadian Cancer Society. Treatments for breast cancer. https://cancer.ca/en/cancer-information/cancer-types/breast/treatment. Accessed 2023 Aug 31.
37.CADTH. Provisional funding algorithm - Breast cancer with hormone receptor (HR) positive, human epidermal growth factor receptor 2 (HER2) negative, or low; triple negative breast cancer, including HR negative, HER2 negative and HR negative, HER2 low. 2023; https://www.cadth.ca/sites/default/files/DRR/2023/PH0033-HR%2BHER2-BC-and-TNBC-with-HER2-Low-Inclusion.pdf. Accessed 2023 Dec 13 [sponsor submitted reference].
38.Truqap® (capivasertib): 160 mg and 200 mg tablets [product monograph]. Mississauga (ON): AstraZeneca; 2024 Jan 24.
39.Faslodex (fulvestrant injection): 50mg/mL [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2020: https://www.astrazeneca.ca/content/dam/az-ca/downloads/productinformation/faslodex-product-monograph-en.pdf. Accessed 2024 Apr 30.
40.Teva-everolimus: 2.5 mg, 5 mg, 7.5 mg and 10 mg tablets [product monograph]. Toronto (ON): Teva Canada Limited 2019: https://pdf.hres.ca/dpd_pm/00054349.PDF. Accessed 2024 Apr 30.
41.PrAromasin (exemestane): tablets, 25 mg, oral [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021: https://pdf.hres.ca/dpd_pm/00064219.PDF. Accessed 2024 Apr 30.
42.Teva-Capecitabine: 150 mg and 500 mg tablets [product monograph]. Toronto (ON): Teva Canada Limited; 2018: https://pdf.hres.ca/dpd_pm/00046666.PDF. Accessed 2024 Apr 30.
43.Abraxane (paclitaxel): powder for suspension, 100 mg paclitaxel/vial, intravenous [product monograph]. Saint-Laurent (QC): Bristol-Myers Squibb Canada; 2024: https://www.bms.com/assets/bms/ca/documents/productmonograph/ABRAXANE_EN_PM.pdf. Accessed 2024 Apr 30.
44.Sprangers MA, Groenvold M Fau - Arraras JI, Arraras Ji Fau - Franklin J, et al. The European Organization for Research and Treatment of Cancer breast cancer-specific quality-of-life questionnaire module: first results from a three-country field study. 1996(0732-183X (Print) [sponsor submitted reference]).
45.AstraZeneca. Statistical Analysis Plan for CAPItello-291: A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative (HR+/HER2−) Breast Cancer Following Recurrence or Progression On or After Treatment with an Aromatase Inhibitor (CAPItello-291) [CONFIDENTIAL internal manufacturer's report]. 2022 [sponsor submitted reference].
46.AstraZeneca. Clinical Study Protocol for CAPItello-291: A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative (HR+/HER2−) Breast Cancer Following Recurrence or Progression On or After Treatment with an Aromatase Inhibitor (CAPItello-291) [CONFIDENTIAL internal manufacturer's report]. 2022 [sponsor submitted reference].
47.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
48.Letellier ME, Dawes D, Mayo N. Content verification of the EORTC QLQ-C30/EORTC QLQ-BR23 with the International Classification of Functioning, Disability and Health. Qual Life Res. 2015;24(3):757-768. PubMed
49.McLachlan SA, Devins GM, Goodwin PJ. Validation of the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (QLQ-C30) as a measure of psychosocial function in breast cancer patients. Eur J Cancer. 1998;34(4):510-517. PubMed
50.Groenvold M, Klee MC, Sprangers MA, Aaronson NK. Validation of the EORTC QLQ-C30 quality of life questionnaire through combined qualitative and quantitative assessment of patient-observer agreement. J Clin Epidemiol. 1997;50(4):441-450. PubMed
51.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159-174. PubMed
52.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: a synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. PubMed
53.Nunnally JC, Bernstein IH. Psychometric theory. 3rd ed ed. New York: McGraw-Hill; 1994.
54.Ousmen A, Conroy T, Guillemin F, et al. Impact of the occurrence of a response shift on the determination of the minimal important difference in a health-related quality of life score over time. (1477-7525 (Electronic) [sponsor submitted reference]).
55.Forsythe A, Chandiwana D, Barth J, Thabane M, Baeck J, Tremblay G. Progression-free survival/time to progression as a potential surrogate for overall survival in HR+, HER2- metastatic breast cancer. Breast Cancer (Dove Med Press). 2018;10:69-78. PubMed
56.Lux MP, Böhme S, Hücherig S, Jeratsch U, Kürschner N, Lüftner D. Surrogate threshold effect based on a meta-analysis for the predictive value of progression-free survival for overall survival in hormone receptor-positive, HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2019;176(3):495-506. PubMed
57.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
58.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
59.Network meta-analysis of capivasertib plus fulvestrant versus alternative treatments for HR+/HER2- advanced breast cancer after prior endocrine therapy [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Truqap (capivasertib) tablets, 160 mg and 200 mg, oral. Cambridge (UK): AstraZeneca Canada Inc.; 2024 Jan 25.
60.Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Making. 2013;33(5):607-617. PubMed
61.Turner RM, Dominguez-Islas CP, Jackson D, Rhodes KM, White IR. Incorporating external evidence on between-trial heterogeneity in network meta-analysis. Stat Med. 2019;38(8):1321-1335. PubMed
62.Hoaglin DC, Hawkins N, Jansen JP, et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 2. Value Health. 2011;14(4):429-437. PubMed
63.Jansen JP, Fleurence R, Devine B, et al. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 1. Value Health. 2011;14(4):417-428. PubMed
64.Hutton B, Salanti G, Caldwell DM, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med. 2015;162(11):777-784. PubMed
65.Howell SJ, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 2022;23(7):851-864. PubMed
66.Jones RH, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21(3):345-357. PubMed
67.Baselga J, Campone M, Sahmoud T, et al. Everolimus in combination with exemestane for postmenopausal women with advanced breast cancer who are refractory to letrozole or anastrozole: results of the BOLERO-2 phase III trial. Eur J Cancer. 2011;47:6-7.
68.Shao Z, Cai L, Wang S, et al. BOLERO-5: A phase II study of everolimus and exemestane combination in Chinese post-menopausal women with ER+/HER2- advanced breast cancer. Ann Oncol. 2021;32(Supplement 5):S463.
69.Chia S, Gradishar W, Mauriac L, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26(10):1664-1670. PubMed
70.Johnston Sr, Kilburn LS, Ellis P, et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol. 2013;14(10):989-998. PubMed
71.Di Leo A, Jerusalem G, Petruzelka L, et al. CONFIRM: a Phase III, Randomized, Parallel-Group Trial Comparing Fulvestrant 250 mg vs Fulvestrant 500 mg in Postmenopausal Women with Estrogen Receptor-Positive Advanced Breast Cancer. Cancer Res. 2010;69(24 Supplement).
72.Di Leo A, Jerusalem G, Petruzelka L, et al. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst. 2014;106(1). PubMed
73.Wang J, Cai L, Song Y, et al. Clinical efficacy of fulvestrant versus exemestane as first-line therapies for Chinese postmenopausal oestrogen-receptor positive /human epidermal growth factor receptor 2 -advanced breast cancer (FRIEND study). Eur J Cancer. 2023;184:73-82. PubMed
74.Zhang Q SZ, Shen K, et al. Fulvestrant 500 mg vs 250 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer: a randomized, double-blind registrational trial in China. Oncotarget. 2016;7(35):57301-57309. PubMed
75.Jerusalem G, De Boer RH, Hurvitz S, et al. Everolimus Plus Exemestane vs Everolimus or Capecitabine Monotherapy for Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer: The BOLERO-6 Randomized Clinical Trial. JAMA Oncology. 2018;4(10):1367-1374. PubMed
76.Turner NC, Oliveira M, Howell SJ, et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2023;388(22):2058-2070. PubMed
Appendix 1: Detailed Outcomes Data for PFS2 and Time to Chemotherapy (CAPItello-291 Trial)
Please note that this appendix has not been copy-edited.
Following discontinuation of study treatment because of disease progression, as determined by investigator-based by RECIST 1.1 assessment, patients who started on subsequent cancer therapy postprogression were continued to be followed at the 30-day follow-up visit, every 8 weeks (± 7 days) for the first 2 years, and every 12 weeks (± 7 days) thereafter for documentation of progression on second-line therapy. Patients alive and for whom a second disease progression had not been observed were censored at the date last known alive and without a second disease progression (i.e., censored at the PFS or PFS2 assessment date, whichever was later, if the patient had not had a second progression or death). Time from randomization to second progression or death in the Overall Population and Altered Populations were analyzed using a stratified log-rank test, using the same methodology as described for the primary PFS outcome for the overall population. The PFS2 analysis in the overall population was stratified by the stratification factors. The effect of treatment was estimated by the hazard ratio together with its corresponding 95% CI. KM plots were presented by treatment group. Results for the altered population are summarized in Table 25.
For time to first subsequent chemotherapy or death (i.e., date of first subsequent chemotherapy, death or censoring-date of randomization + 1), patients alive and not known to have had a first subsequent chemotherapy were censored at the earliest of: date of study termination, date last known alive, data cut-off date of August 15, 2022, or the last date that the patient was known not to have received a first subsequent chemotherapy. This outcome was analyzed using the same methodology and model as that used for the analysis of PFS, except no formal comparisons were made and multiplicity adjustment was not applied. Results for the altered population are summarized in Table 26.
Table 25: PFS 2 — FAS, CAPItello-291 Trial
PFS2 by investigator assessment | Altered population | |
|---|---|---|
Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
August 15, 2022 data cut-off | ||
Patients with events, n (%) | ||
Total | 79 (51.0) | 87 (64.9) |
Second progressive disease | 57 (36.8) | 62 (46.3) |
Death in absence of second progression | 22 (14.2) | 25 (18.7) |
Patients censored, n (%)a | 76 (49.0) | 47 (35.1) |
Median PFS2, months (95% CI)b | 15.5 (13.2 to 17.6) | 10.8 (8.1 to 12.7) |
Hazard ratio (95% CI)c | 0.52 (0.38 to 0.71) | |
Log-rank test two-sided P-valued | < 0.001 | |
Probability of being event-free at 6 months, % (95% CI)b | 86.7 (80.2 to 91.2) | 72.2 (63.5 to 79.2) |
Probability of being event-free at 12 months, % (95% CI)b | 64.4 (56.0 to 71.6) | 44.6 (35.6 to 53.2) |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; PFS = progression-free survival.
aPatients alive and for whom a second disease progression has not been observed censored at the date last known alive and without a second disease progression.
bKM estimate.
cCalculated using stratified Cox proportional hazards model. A hazard ratio < 1 favours capivasertib plus fulvestrant. Log-rank test and Cox model stratified by presence of liver metastases (yes vs. no), prior use of CDK4/6 inhibitors (yes vs. no).
dP value was not adjusted for multiple comparisons.
Source: CAPItello-291 Clinical Study Report [Details included in the table are from the sponsor’s Summary of Clinical Evidence].
Table 26: Time to First Subsequent Chemotherapy — FAS, CAPItello-291 Trial
Time to first subsequent chemotherapy or death | Altered population | |
|---|---|---|
Capivasertib plus fulvestrant (n = 155) | Placebo plus fulvestrant (n = 134) | |
August 15, 2022 data cut-off | ||
Total number of patients with events, n (%) | 103 (66.5) | 100 (74.6) |
Patients censored, n (%)a | 52 (33.5) | 34 (25.4) |
Median time to event, months (95% CI)b | 11.0 (9.1 to 13.6) | 6.0 (4.4 to 8.0) |
Hazard ratio (95% CI)c | 0.56 (0.42 to 0.74) | |
Log-rank test two-sided P value | < 0.001 | |
Probability of being event-free at 6 months, % (95% CI)b | ████ ████ | ████ ███ |
Probability of being event-free at 12 months, % (95% CI)b | ████ ████ | ████ ███ |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier.
aPatients not known to have had a first subsequent chemotherapy or died.
bKM estimate.
cCalculated using stratified Cox proportional hazards model. A hazard ratio < 1 favours capivasertib plus fulvestrant.
dP value was not adjusted for multiple comparisons.
Source: CAPItello-291 Clinical Study Report [Details included in the table are from the sponsor’s Summary of Clinical Evidence].
Pharmacoeconomic Review
Abbreviations
BCC
Breast Cancer Canada
BIA
budget impact analysis
BSA
body surface area
CBCN
Canadian Breast Cancer Network
CDA-AMC
Canada’s Drug Agency
CDK4/6
cyclin-dependent kinase 4 and 6
ET
endocrine therapy
HER2
human epidermal growth factor receptor 2
HR
hormone receptor
ICER
incremental cost-effectiveness ratio
NGS
next-generation sequencing
NMA
network meta-analysis
OH-CCO
Ontario Health Cancer Care Ontario
OS
overall survival
PD
progressed disease
PF
progression-free
PFS
progression-free survival
QALY
quality-adjusted life-year
REAL
Research Excellence Active Leadership
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Capivasertib (Truqap) 160 mg and 200 mg oral tablets |
Indication | In combination with fulvestrant for treatment of adult female patients with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA/AKT1/PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on, or within 12 months of completing, adjuvant therapy. |
Health Canada approval status | Approved (NOC) |
Health Canada review pathway | Standard and Project Orbis Type A |
NOC date | January 26, 2024 |
Reimbursement request | In combination with fulvestrant for treatment of adult patients with HR-positive, HER2-negative, locally advanced, or metastatic breast cancer with 1 or more PIK3CA/AKT1/PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on, or within 12 months of completing, adjuvant therapy. |
Sponsor | AstraZeneca Canada Inc. |
Submission history | First submission to Canada’s Drug Agency |
HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target populations | Adult female patients with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on, or within 12 months of completing, adjuvant therapy in Canada. |
Treatment | Capivasertib used in combination with fulvestrant |
Dose regimen | The recommended dose of capivasertib in combination with fulvestrant is 400 mg (2 tablets of 200 mg) taken orally twice daily approximately 12 hours apart (for a total daily dose of 800 mg) for 4 days followed by 3 days off treatment until disease progression or unacceptable toxicity occurs. |
Submitted price | Capivasertib, 160 mg: $147.60 per tablet Capivasertib, 200 mg: $147.60 per tablet |
Submitted treatment cost | The per-patient cost for 28 days of capivasertib is $9,446. When used in combination with fulvestrant, the per-patient, 28-day cost for capivasertib plus fulvestrant in the first 28 days is $10,612 and in subsequent 28-days is $10,029. |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (20 years) |
Key data source | CAPItello-291 trial |
Submitted results | Based on the submitted sequential analysis, the cost-effectiveness of capivasertib plus fulvestrant vs. endocrine monotherapy was $145,131 per QALY gained (incremental QALYs = 1.02; incremental cost = $148,032). Other comparators were either dominated (everolimus plus exemestane) or extendedly dominated (chemotherapy). |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; HER2 = human epidermal growth factor receptor 2; HR = hormone receptor; ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year.
Conclusions
One ongoing, phase III, double-blind, randomized controlled trial (the CAPItello-291 trial) compared capivasertib plus fulvestrant with placebo plus fulvestrant in adult patients with locally advanced or metastatic, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations. Using the Grading of Recommendations, Assessment, Development, and Evaluations framework, the clinical review by Canada’s Drug Agency (CDA-AMC) showed high and moderate certainty of evidence that treatment with capivasertib plus fulvestrant likely results in a clinically meaningful increase in progression-free survival (PFS) at 6 months and 12 months, respectively, compared to placebo plus fulvestrant. Overall survival (OS) data were immature, and no definitive conclusions could be drawn about health-related quality of life because of concerns related to imprecision and the large quantity of missing outcomes data. Because of limitations in the indirect treatment comparison, no conclusions could be drawn about the relative efficacy of capivasertib plus fulvestrant versus chemotherapy or everolimus plus exemestane.
The CDA-AMC base case incremental cost-effectiveness ratio (ICER) for capivasertib plus fulvestrant versus endocrine monotherapy was $221,165 per quality-adjusted life-year (QALY) gained (incremental cost = $118,477; incremental benefit = 0.54 QALYs). Higher drug costs associated with capivasertib were responsible for $112,025 in additional costs. Higher life expectancy associated with capivasertib (0.63 incremental life-years) drove the increase in QALYs. Most of the benefit (72%) was incurred after 2 years. Therefore, cost-effectiveness is heavily influenced by long-term survival benefit, for which the evidence is uncertain because of data immaturity in the trial.
The main difference between the CDA-AMC and sponsor’s base cases is the assumption of the long-term (beyond 2 years) impact of capivasertib plus fulvestrant on OS. The results of the sponsor’s base-case analysis estimate a mean 1.26 life-year extension for patients receiving capivasertib plus fulvestrant versus endocrine monotherapy, whereas the CDA-AMC base case estimates a smaller mean life-year extension of 0.63 years. The CDA-AMC base case may overestimate survival benefit because it assumes treatment benefit beyond treatment discontinuation; a scenario analysis shows that removing the posttreatment discontinuation benefit decreases mean life extension to 0.46 years.
The cost-effectiveness of capivasertib plus fulvestrant compared to chemotherapy and everolimus plus exemestane is uncertain, given the lack of direct comparative evidence and methodological limitations with the indirect evidence. Given the small number of adult male patients in the CAPItello-291 trial, cost-effectiveness in the sponsor’s reimbursement request (all adults as opposed to just females) is dependent on the extrapolation of results from female patients.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Three patient groups, the Canadian Breast Cancer Network (CBCN), Rethink Breast Cancer, and Breast Cancer Canada (BCC), provided input for this review. CBCN collected information in 2012 (in collaboration with Rethink Breast Cancer), 2017, and 2022 through online surveys comprising responses from 69 patients with HR-positive, HER2-negative, metastatic breast cancer in Canada in addition to a key informant interview (although CBCN was not able to speak with patients taking capivasertib for the treatment of HR-positive, HER2-negative metastatic breast cancer). Rethink Breast Cancer drew results from an online survey of 78 patients living with metastatic breast cancer from completed from September 2018 to April 2019. Rethink Breast Cancer also conducted interviews in February 2024 with 4 patients in the US living with metastatic breast cancer who are currently taking capivasertib. BCC collected information in 2023 through electronic surveys comprising responses from 171 patients with recurrent, metastatic breast cancer and their caregivers. BCC also conducted surveys in 2024 and identified 5 patients with capivasertib experience. Overall, patients’ disease experiences were influenced by the physical symptoms associated with metastatic breast cancer (e.g., fatigue, insomnia, pain, nausea), negative psychosocial effects (e.g., associated with restrictions to their employment and careers, ability to care for dependents, and ability to be social in their communities), and adverse side effects associated with chemotherapy (e.g., fatigue, nausea, depression, problems with concentration, memory loss, diarrhea, and insomnia). Patients noted that important outcomes of treatment include PFS, OS, treatment effectiveness, and minimal side effects to allow productivity, mobility, and quality of life. The 4 patients with capivasertib experience described mild to moderate side effects, including diarrhea, rash, and nausea. They also noted the lack of treatment options and reported being grateful to be eligible for another line of treatment — especially if they had been on multiple lines of treatment — that allowed them to live a quality life.
Registered clinician input was received from the Ontario Health Cancer Care Ontario (OH-CCO) Breast Cancer Drug Advisory Committee and the Research Excellence Active Leadership (REAL) Canadian Breast Cancer Alliance. Clinicians indicated that the current pathway of care for patients with HR-positive, HER2-negative, metastatic breast cancer with 1 or more PIK3, AKT1, or PTEN alterations in the second-line setting — and in the first-line setting, for patients who relapse while on or within 12 months of completing adjuvant endocrine therapy (ET) — varies depending on clinical factors and tumour biology. The REAL Canadian Breast Cancer Alliance clinicians outlined that adjuvant and first-line standard of care is ET combined with a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor for HR-positive, HER2-negative metastatic breast cancer. ET alone in the first-line setting should be reserved for the small group of patients with comorbidities or an Eastern Cooperative Oncology Group Performance Status that prevents treatment with a CDK4/6 inhibitor. In the adjuvant setting, clinicians note growing evidence that ET plus a CDK4/6 inhibitor increases benefits for patients with HR-positive, HER2-negative, early breast cancer who are at higher risk of relapse. Currently, abemaciclib plus ET is indicated for select high-risk patients in Canada. Strategies for second-line treatment are guided by tumour genomics (including PIK3CA mutations, estrogen receptor 1 mutations, and germline BRACA1 and BRACA2 mutations). Evidence-based, second-line therapy options include exemestane plus everolimus, tamoxifen plus everolimus, fulvestrant plus everolimus, or chemotherapy. Later-line treatment options for women who progress after 2 lines of ET include chemotherapy and antibody drug conjugates (i.e., sacituzumab govitecan and trastuzumab deruxtecan). For patients with asymptomatic, slowly progressive disease, treatment options include ET continuation and tamoxifen. The OH-CCO Breast Cancer Drug Advisory Committee references the provisional funding algorithm, in which first-line therapy for patients who have not received an adjuvant CDK4/6 inhibitor would consist of ribociclib or palbociclib with an aromatase inhibitor. Second-line treatment would be endocrine monotherapy, everolimus plus exemestane, or chemotherapy, depending on the clinical status of the patient. Clinicians noted that treatment is palliative in intent. Overall, OH-CCO and REAL clinicians noted that the goals are to prolong PFS and OS, alleviate symptoms, maintain or improve quality of life, manage or minimize the toxicities associated with treatment, and delay the initiation of chemotherapy, including the use of antibody drug conjugates. Clinicians also noted that they expected the indication for capivasertib to be in line with the CAPItello-291 trial criteria, and that capivasertib would complement other available treatments.
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. Drug plans noted several uncertainties. First, it is unclear whether patients with stable brain metastases would be eligible. Second, the drug plans wondered whether, in real-world clinical practice, in the case of radiologic disease progression without clinical deterioration or disease worsening, treatment could be continued beyond radiologic progression. Third, the drug plans queried whether capivasertib could be continued as a single drug if fulvestrant was discontinued because of toxicity, and vice versa. Fourth, the plans asked whether male patients with breast cancer should use a gonadotropin-releasing hormone agonist in combination with fulvestrant and capivasertib. Finally, the plans asked whether patients currently receiving a comparator are eligible to switch to capivasertib plus fulvestrant at the time of implementation.
Participating drug plans expressed further concerns about the potential of the drug under review to change the place in therapy of comparator drugs, especially given the complex therapeutic space involving multiple lines of therapy and subpopulations. In addition, the drug plans are concerned about potential toxicity because of the coadministration of certain CYP3A4 inhibitors that may increase exposure to capivasertib; the management of adverse effects, such as tumour lysis; PIK3CA, AKT1, or PTEN companion diagnostic testing methods, timing, and different alterations causing different expected outcomes. Further, the drug plans anticipated budget impacts and sustainability issues, and noted confidential negotiated prices for comparators.
Several of these concerns were addressed in the sponsor’s report and model:
Capivasertib monotherapy was not assessed.
Next-generation sequencing (NGS) for PIK3CA, AKT1, and PTEN is required before treatment with capivasertib, and is captured as a 1-time cost in the sponsor’s model.
Additional costs, such as those related to administration, resource use, adverse events, and terminal care, were included in the sponsor’s model.
The impact of adverse events and PFS on quality of life were assessed.
CDA-AMC was unable to address the following concern raised in the stakeholder input:
The sponsor’s modelling approach calculates subsequent therapy as a 1-time cost when a patient progresses for the entire time horizon, and it does not capture patients who go on to potentially receive multiple lines of subsequent therapy after the initial subsequent therapy does not work for them.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of capivasertib in combination with fulvestrant compared with endocrine monotherapy, everolimus plus exemestane, and chemotherapy. The model population was based on the CAPItello-291 trial, which comprised adult patients (99% female) with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence while on or within 12 months of completing adjuvant therapy. The Health Canada–indicated population is for female patients and excludes male patients. The sponsor is seeking reimbursement for both female and male patients.1
Capivasertib is available as 160 mg and 200 mg oral tablets. The recommended dosage of capivasertib, when taken in combination with fulvestrant, is 400 mg (2 tablets of 200 mg) taken orally twice daily approximately 12 hours apart (for a total daily dose of 800 mg) for 4 days, followed by 3 days off treatment until disease progression or unacceptable toxicity occurs. At the sponsor’s submitted price of $147.60 per 200 mg tablet, the 28-day cost of capivasertib is $9,446.40 per patient. In combination with fulvestrant, the first 28-day cost is $11,195.09 per patient, and the subsequent 28-day cost is $10,029.30 per patient. The first 28-day cost and subsequent 28-day cost per patient of endocrine monotherapy ($1,315.11 and $440.76, respectively) are determined by considering which ET is given. The sponsor’s internal market share data derive the following distribution: fulvestrant (███), tamoxifen (███), anastrozole (████), letrozole (████), and exemestane (██). The 28-day cost per patient of everolimus plus exemestane is $1,454.83. The 28-day cost per patient of chemotherapy, amounting to $1,491.37, is determined by considering the weighted average of capecitabine (███) and paclitaxel (███). In the base-case analysis, the relative dose intensity was set at 100%, and wastage was included (i.e., the cost of the entire vial was incurred per administration).1
The economic outcomes of interest were QALYs and life-years. The economic evaluation was conducted over a lifetime horizon (i.e., 20 years) from the perspective of the Canadian publicly funded health care payer. Costs and effects were discounted at 1.5% per annum.1
Model Structure
The sponsor submitted a partitioned survival model with 3 health states: progression-free (PF), progressed disease (PD), and death. The death health state was the absorbing state for deaths from any cause. The proportion of patients in each mutually exclusive health state at any time over the time horizon was derived from independently modelled survival curves of PFS and OS using data from the CAPItello-291 trial. The proportion of patients alive was based on the OS curve. OS was partitioned into the PF and PD states using the PFS curve. All patients entered the model in the PF health state. Patients who progressed moved to the PD state, derived from subtracting PFS from OS. Patients in the PD state cannot move back to the PF state and are assumed to receive a basket of subsequent therapy. The state occupancy for death was calculated using the OS curve. The time to treatment discontinuation was set to equal to PFS over time.1
Model Inputs
Baseline patient characteristics in the model were reflective of the CAPItello-291 trial intention-to-treat population,2 which included female and male patients from the altered population (i.e., for all adults with AKT pathway alteration; average age = 59.3 years; average weight = 68.4 kg; body surface area [BSA] = 1.75 m2). When male patients were removed from the baseline (n = 2), the baseline characteristics differed by less than 1% (i.e., for female patients with AKT pathway alteration, average age = 59.2 years; average weight = 68.3 kg; BSA = 1.75 m2). Baseline characteristics were not available for CDK4/6 inhibitor–naive or –experienced, AKT pathway–altered subgroups, and was assumed to be the same.1
The efficacy inputs (i.e., PFS and OS) for the reference arm (i.e., placebo plus fulvestrant) were based on the results from the CAPItello-291 trial.2 Kaplan-Meier estimates of the reference-arm PFS and OS were used to fit parametric survival curves to extrapolate the treatment effect beyond the observed trial data over the entire model time horizon (i.e., 20 years). From the reference arm, capivasertib plus fulvestrant, everolimus plus exemestane, and chemotherapy survival curves were estimated by applying the hazard ratios of the treatment from the network meta-analysis (NMA). In the model, endocrine monotherapy efficacy was modelled using the efficacy of placebo plus fulvestrant monotherapy (from the reference arm of the CAPItello-291 trial),2 and chemotherapy efficacy was modelled after capecitabine (from the NMA).1 A series of parametric survival functions were fitted to the PFS and OS patient-level data of the reference arm to determine the best-fitting distribution based on goodness-of-fit statistics, visual inspection, assessment of underlying hazard functions, and clinical plausibility regarding long-term progression and survival. The model used all-cause mortality life table data from Statistics Canada so that the risk of death was greater than or equal to the background risk of death by age and gender. The standard parametric survival models were fitted to the CAPItello-291 trial patient-level data. All parametric curves in the model base case were separated into CDK4/6 inhibitor–naive and –experienced. The sponsor’s chosen parametric survival distribution for PFS fulvestrant data in the CDK4/6 inhibitor–experienced and –naive altered population was the lognormal distribution. The sponsor’s chosen parametric survival distribution for OS fulvestrant data in the CDK4/6 inhibitor–experienced altered population was the gamma distribution. The sponsor’s chosen parametric distribution for OS fulvestrant data in the CDK4/6 inhibitor–naive altered population was the Weibull distribution.1
Health state utility values applied in the economic model were based on the CAPItello-291 trial population. These patients were administered the EQ-5D-5L questionnaire at baseline and every 4 weeks (± 3 days) until “progression free survival 2” (defined as the time from randomization until second progression on next-line treatment [as assessed by the investigator at the local site] or death because of any cause). Utility decrements for treatment-related adverse events included in the model were based on non-Canadian published literature on various disease areas (i.e., metastatic breast cancer, non–small cell lung cancer, and type 1 diabetes).1
The sponsor’s reference case included costs related to drug acquisition and administration, disease management, adverse events, subsequent treatment, biomarker testing, and end of life. The dosing regimen for capivasertib was sourced from the CAPItello-291 trial.2 All other dosing regimens were based on the OH-CCO monographs and relevant clinical trials.3 Drug prices were based on the lowest dispensable unit price among provincial formularies3-9 (excluding Quebec), with the exception of capivasertib, which was based on the sponsor-submitted price. Drug doses were weight-dependent or calculated based on BSA, and drug wastage was accounted for in the model. Subsequent treatment costs were informed by subsequent treatments received in the CAPItello-291 trial and in consultation with clinicians in Canada. The durations of subsequent treatments were informed by clinician feedback. Administration costs were applied to intramuscular and IV therapies and sourced from the Ontario Ministry of Health Schedule of Benefits.10 Health care resource use and costs for disease monitoring were based on values from the altered population in the CAPItello-291 trial, clinician opinion in Canada, and an Alberta study to inform real-world practices in Canada. The unit costs associated with ongoing disease monitoring were informed by the Ontario Schedule of Benefits for Physician Services.11 Adverse events occurring in at least 2% of the CAPItello-291 study population of grade 3 or higher were included where a 1-time cost in the economic model was informed by the Canadian Institute for Health Information patient cost estimator.12 NGS to confirm PIK3CA, AKT1, and/or PTEN alteration and a dihydropyrimidine dehydrogenase test to assess the risk of severe toxicity from fluoropyrimidine drugs (such as capecitabine) were 1-off costs in the economic model informed by the CDA-AMC reimbursement review of alpelisib and Ontario Health, respectively. A 1-off, end-of-life cost was included upon entry into the death state, based on the Statistics Canada Consumer Price Index.13
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (more than 10,000 Monte Carlo iterations for the base-case and scenario analyses). The deterministic results for incremental life-years, QALYs, and cost were all similar, but slightly higher than the probabilistic results. The probabilistic findings are presented here.
Base-Case Results
In the sponsor’s reference base case, capivasertib plus fulvestrant was more costly and more effective than endocrine monotherapy (incremental cost = $148,032; incremental QALYs = 1.02), everolimus plus exemestane (incremental cost = $139,038; incremental QALYs = 1.17), and chemotherapy (incremental cost = $83,388; incremental QALYs = 0.86), resulting in ICERs of $145,131, $119,217, and $96,956 per QALY gained over a 20-year time horizon, respectively (refer to the results in Table 3). Based on a sequential analysis, everolimus plus exemestane and chemotherapy did not appear on the cost-effectiveness frontier because they were extendedly dominated and dominated, respectively. The sponsor reported that the results were unchanged when adult male patients were added to the population, given that they made up less than 1% of the CAPItello-291 trial. Based on a willingness-to-pay (WTP) threshold of $50,000 per QALY gained, there is close to 0% probability that capivasertib plus fulvestrant would be the most cost-effective strategy.1
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Endocrine monotherapy | 88,743 | 1.81 | Reference |
Capivasertib plus fulvestrant | 236,775 | 2.83 | 145,129 |
Dominated treatments | |||
Everolimus plus exemestane | 97,737 | 1.66 | Dominated by endocrine monotherapy |
Chemotherapy | 153,387 | 1.97 | Extendedly dominated by endocrine monotherapy and capivasertib plus fulvestrant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
The sponsor conducted various scenario analyses encompassing a variety of considerations, such as alternative discount rates, the proportion of prior CDK4/6 inhibitor–experienced versus –naive patients in the altered population, time horizon, CDK4/6 inhibitor–naive data sources, testing costs, parametric curve selections for CDK4/6 inhibitor–experienced patients, trastuzumab deruxtecan use as a subsequent therapy, societal perspective, and CDK4/6 inhibitor–experienced patients only. The sponsor’s societal perspective scenario analysis resulted in an ICER of $190,587 per QALY gained relative to endocrine monotherapy. This is higher than the sponsor’s base case ICER using a public health care payer perspective. All other resulting ICERs in the scenario analysis were similar to the sponsor’s base-case analysis. All of the sponsor’s scenario analyses resulted in capivasertib plus fulvestrant being more costly and more effective, which was aligned with the base case.1
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The proportional hazards assumption for PFS and OS between capivasertib plus fulvestrant and endocrine monotherapy is improbable. The sponsor selected a dependent lognormal model to extrapolate PFS and a dependent gamma model to extrapolate OS for capivasertib plus fulvestrant versus endocrine monotherapy (i.e., a single parametric model with a treatment coefficient). In using a dependent proportional hazards model to characterize the comparative efficacy of capivasertib plus fulvestrant versus the comparators, the sponsor assumed that the treatment effect on OS was constant over time, regardless of progression and treatment discontinuation. Given that most patients have progressed and discontinued treatment after 30 months, the proportional hazards assumption would indicate that there is a substantial treatment benefit after treatment discontinuation. It would also indicate that time on treatment and progression have little to no impact on OS.
In the sponsor’s base case, patients live for an additional 6 months in the postprogression state if they receive capivasertib plus fulvestrant instead of endocrine monotherapy. The sponsor notes that data on time from randomization to second progression on next-line treatment or death because of any cause indicate a continued treatment effect of capivasertib beyond progression, and attributes this effect to the drug’s potential ability to restore cancer cells’ sensitivity to other therapies. However, there were no robust data to indicate that mortality rates postprogression were superior for patients who progressed on capivasertib plus fulvestrant versus endocrine monotherapy.
The sponsor assessed the proportional hazards assumption for both PFS and OS in the CAPItello-291 trial using several statistical tests and found some evidence that this assumption may not hold for both PFS and OS. These tests apply only to the available data; uncertainty remains as to whether the assumption would hold for the extrapolated period for which no data exist. In most cases, data have shown that the proportional hazards assumption does not hold in the long-term.14 In the most recent data cut for the CAPItello-291 trial (August 15, 2022), there were 72 OS events with 34.6% data maturity. This period was approximately 2 years, whereas the modelled time horizon was extrapolated to 20 years. Given the length of the extrapolated period, it is unlikely that the proportional hazards assumption would hold for such an extended period. For the proportional hazards assumption to hold, capivasertib would have to continue to reduce the rate of mortality after treatment discontinuation and progression indefinitely. Insufficient data were provided to indicate this to be the case. Clinical experts consulted by CDA-AMC also noted that, in the absence of evidence, the likelihood is for treatment impact on OS to wane over time, as is expected for most cancers.15
The CDA-AMC base case applied independent parametric models to estimate the PFS and OS of capivasertib plus fulvestrant compared to endocrine monotherapy.
The gamma model was selected for extrapolating OS data for both capivasertib plus fulvestrant and endocrine monotherapy. This led to a diminishing impact on OS over time (refer to Figure 2).
The selection of the gamma curves still leads to a postprogression survival benefit for patients receiving capivasertib, as shown in Figure 2; the relative risk never hits 1, which would indicate no further treatment benefit relative to endocrine monotherapy. For a scenario analysis, the Weibull curve was selected for capivasertib, which removes the postprogression treatment benefit.
The NMA for chemotherapy and everolimus plus exemestane is uncertain. Chemotherapy and everolimus plus exemestane were not used as comparators in the CAPItello-291 trial. Because of the lack of head-to-head, randomized clinical trials evaluating capivasertib plus fulvestrant versus chemotherapy and everolimus plus exemestane, an NMA was used to estimate the relative efficacy. The hazard ratio from the NMA was applied to the extrapolated placebo plus fulvestrant arm from the CAPItello-291 trial to estimate the survival curves for chemotherapy and everolimus plus exemestane. The efficacy of chemotherapy was informed by capecitabine. However, the CDA-AMC clinical review noted several important limitations with the indirect comparison, such as evidence of nonproportional hazard being present across most studies, as well as several notable sources of heterogeneity for potential effect modifiers. The magnitude and direction of potential bias because of heterogeneity and lack of proportionality on outcome estimates cannot be predicted. Because of this, no definitive conclusions could be drawn from the NMA. The results of the NMA differ substantially from those of the CAPItello-291 trial. Based on the NMA, the OS hazard ratio for capivasertib plus fulvestrant endocrine monotherapy was ████ ████ ███ ████ ██ ████ relative to 0.69 (95% CI, 0.45 to 1.05), as reported in the CAPItello-291 trial. Given the methodological limitations, the results from the NMA may be highly influenced by bias; as such, the results may provide misleading conclusions.
In the CDA-AMC base case, evidence to inform the relative efficacy of capivasertib plus fulvestrant versus endocrine monotherapy was taken directly from the CAPItelo-291 trial. The cost-effectiveness of capivasertib plus fulvestrant versus chemotherapy or everolimus plus exemestane was unassessed; therefore, it is unknown.
The impact of subsequent therapies on costs and health outcomes are uncertain. The sponsor’s model estimates the costs associated with subsequent therapy received by the patient as a 1-time cost. The CDA-AMC clinical experts stated that, in clinical practice, although the next line of subsequent therapy can be estimated, patients can continue to be unresponsive to therapy and receive further lines of therapy, which would be challenging to estimate, given the model structure. For example, in the sponsor’s model, ██% of patients who do not respond to capivasertib plus fulvestrant go on to receive trastuzumab deruxtecan, and ██% of patients who do not respond to chemotherapy go on to receive trastuzumab deruxtecan. Although this may occur for the first line of subsequent therapy, patients who do not respond to capivasertib plus fulvestrant and then do not respond to additional subsequent therapies will likely go on to receive trastuzumab deruxtecan, but this scenario is not captured in the sponsor’s model. A higher rate of trastuzumab deruxtecan use among patients receiving chemotherapy would improve OS in these patients, based on evidence.16 The sponsor’s model captures the costs associated with subsequent therapy use, but does not capture the impact of these therapies on long-term survival.
Additionally, the sponsor estimates that patients who receive capivasertib plus fulvestrant will spend longer in the postprogression health state. However, in the sponsor’s model, subsequent therapy costs are not linked to time spent in the postprogression state. If patients spend more time in the postprogression state, then more drug costs will be incurred.
Given the high degree of uncertainty regarding the differential rates of subsequent therapies postprogression, CDA-AMC removed the cost of subsequent therapy from the base case. This assumes that subsequent therapy costs will be similar across all comparators.
As a scenario analysis, subsequent therapy costs were linked to time spent in the progressed state. The same distribution of therapies used by the sponsor was used by CDA-AMC to determine what the per-cycle drug cost would be in the progressed state (██% no treatment, ██% trastuzumab deruxtecan, ███% sacituzumab govitecan, ██% capecitabine, ██% paclitaxel).
A modelling error was identified when estimating the cost of testing. When estimating the cost of testing, the sponsor divided the cost by the number of patients needed to test to identify 1 patient. These 2 numbers should be multiplied to identify the cost of testing. For example, if 3 patients are tested, and only 1 receives a positive result, then the cost of 3 tests is incurred for each positive test. In the sponsor’s analysis, only a third of the cost of 1 test is incurred per identified case.
CDA-AMC updated the model to multiply the cost of testing by the number needed to identify 1 positive case.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Baseline characteristics in the AKT pathway–altered population of the CAPItello-291 trial are not differentiated between CDK4/6 inhibitor–naive and –experienced, AKT pathway–altered subgroups. | Appropriate, according to the clinical experts consulted for this review. |
The biomarker testing was a 1-off cost. | Appropriate, according to the clinical experts consulted for this review. |
The results for adult patients (including female and male) were assumed to be the same as the results for female adult patients. | Appropriate, according to the clinical experts consulted for this review. |
CDA-AMC = Canada’s Drug Agency; CDK4/6 = cyclin-dependent kinase 4 and 6.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook the reanalyses outlined in Table 5 to address, where possible, the limitations within the sponsor’s submitted economic model. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Testing cost | $231 | $2,430 |
Changes to derive the CDA-AMC base case | ||
1. Proportional hazards assumption | Dependent model
| Independent model
|
2. Subsequent therapy cost | Only 1 additional line of therapy modelled | Subsequent therapy cost set to $0, assuming no difference in subsequent therapy costs across comparators |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency; CDK4/6 = cyclin-dependent kinase 4 and 6; HR = hazard ratio; OS = overall survival; PFS = progression-free survival.
The results of these stepwise analyses can be found in Table 6. Results from the probabilistic analysis of the CDA-AMC base case found that capivasertib plus fulvestrant was associated with an incremental benefit of 0.54 QALYs and an incremental cost of $118,477 compared with endocrine monotherapy. The ICER for capivasertib plus fulvestrant versus endocrine monotherapy was $221,165 per QALY gained. Based on a WTP threshold of $50,000, there was a 0% probability that capivasertib plus fulvestrant would be the most cost-effective strategy.
The results were primarily driven by the drug acquisition cost of capivasertib plus fulvestrant and the treatment effects on OS in the extrapolated period.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Capivasertib plus fulvestrant | 244,716 | 2.97 | Reference |
Endocrine monotherapy | 89,342 | 1.92 | 148,978 | |
Sponsor’s base case (corrected) | Capivasertib plus fulvestrant | 246,915 | 2.97 | Reference |
Endocrine monotherapy | 89,342 | 1.92 | 151,086 | |
CDA-AMC reanalysis 1 | Capivasertib plus fulvestrant | 207,418 | 2.45 | Reference |
Endocrine monotherapy | 89,342 | 1.92 | 223,770 | |
CDA-AMC reanalysis 2 | Capivasertib plus fulvestrant | 223,898 | 2.97 | Reference |
Endocrine monotherapy | 66,457 | 1.92 | 150,960 | |
CDA-AMC base case (reanalyses 1 + 2), deterministic | Capivasertib plus fulvestrant | 183,980 | 2.45 | Reference |
Endocrine monotherapy | 66,457 | 1.92 | 222,722 | |
CDA-AMC base case (reanalyses 1 + 2), probabilistic | Capivasertib plus fulvestrant | 184,998 | 2.50 | Reference |
Endocrine monotherapy | 66,521 | 1.97 | 221,165 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated. The cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s results and the CDA-AMC base case. The CDA-AMC base case suggested that an 85% price reduction of capivasertib would be required to achieve cost-effectiveness for capivasertib plus fulvestrant relative to endocrine monotherapy at a $50,000 per QALY threshold (Table 7).
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost, capivasertib (per 28 days) | ICERs for capivasertib plus fulvestrant vs. endocrine monotherapy ($/QALY) | |
|---|---|---|---|
Price reduction | $ | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 147.60 (9,446) | 145,129 | 221,165 |
10% | 132.84 (8,502) | 131,059 | 203,217 |
20% | 103.32 (7,557) | 116,988 | 182,892 |
30% | 103.32 (6,612) | 102,918 | 162,568 |
40% | 88.56 (5,668) | 88,847 | 142,243 |
50% | 73.80 (4,723) | 74,776 | 121,919 |
60% | 59.04 (3,778) | 60,706 | 101,594 |
70% | 44.28 (2,834) | 46,635 | 81,270 |
80% | 29.52 (1,889) | 32,565 | 60,945 |
90% | 14.76 (945) | 18,494 | 40,621 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Additional analyses were performed on the CDA-AMC base case to determine the impact of alternative assumptions on the cost-effectiveness of capivasertib plus fulvestrant compared with endocrine monotherapy. These included:
Changing the independently fitted parametric distribution of OS for capivasertib plus fulvestrant to the Weibull distribution.
Increasing the cost of subsequent therapy usage associated with capivasertib plus fulvestrant.
The results of these analyses are presented in Appendix 4, tables 12 to 14. The ICER was most sensitive to the alternate parametric distribution of OS for capivasertib plus fulvestrant (ICER = $296,104 per QALY gained).
Issues for Consideration
NGS is required before initiating treatment with capivasertib plus fulvestrant to confirm PIK3CA, AKT1, and/or PTEN alteration status. However, this is currently not part of routine practice in many places across Canada.
Overall Conclusions
One ongoing, phase III, double-blind, randomized controlled trial (the CAPItello-291 trial) compared capivasertib plus fulvestrant with placebo plus fulvestrant in adult female patients with locally advanced or metastatic, HR-positive, HER2-negative breast cancer with 1 or more PIK3CA, AKT1, or PTEN alterations. Using the Grading of Recommendations, Assessment, Development, and Evaluations framework, the CDA-AMC clinical review showed high and moderate certainty of evidence that treatment with capivasertib plus fulvestrant likely results in a clinically meaningful increase in PFS at 6 months and 12 months, respectively, compared to placebo plus fulvestrant. The OS data were immature, and no definitive conclusions can be drawn about health-related quality of life because of concerns related to imprecision and a large quantity of missing outcomes data. Because of limitations in the indirect treatment comparison, no conclusions could be drawn about the relative efficacy of capivasertib plus fulvestrant versus chemotherapy or everolimus plus exemestane.
The CDA-AMC base-case reanalysis included correcting the cost of testing; using different assumptions when extrapolating PFS and OS; and removing subsequent therapy cost. Given the limitations in the NMA, the CDA-AMC base case focused on the comparison of capivasertib plus fulvestrant versus endocrine monotherapy only. The CDA-AMC base case ICER for capivasertib plus fulvestrant versus endocrine monotherapy was $221,165 per QALY gained (with an incremental cost of $118,477 and incremental benefit of 0.54 QALYs). Higher drug costs associated with capivasertib were responsible for $112,025 in additional costs. Higher life expectancy associated with capivasertib (0.63 incremental life-years) drove the increase in QALYs. Most of the benefit (72%) was incurred after 2 years. Therefore, cost-effectiveness is heavily influenced by long-term survival benefit, for which evidence is uncertain because of data immaturity from the trial.
The main difference between the CDA-AMC and sponsor base cases was the assumption of long-term impact (beyond 2 years) of capivasertib plus fulvestrant on OS. The sponsor’s base-case analysis estimates a mean 1.26 life-year extension for patients receiving capivasertib plus fulvestrant versus endocrine monotherapy, whereas the CDA-AMC base case estimates a smaller mean life-year extension of 0.63 years. The CDA-AMC base case may overestimate survival benefit because it assumes treatment benefit beyond treatment discontinuation; a scenario analysis shows that removing this benefit results in 0.46 years of life extension. If treatment benefit is sustained after treatment discontinuation, then this would likely extend time on subsequent therapies. A scenario analysis shows that including these extra costs increases the ICER to $233,905 per QALY gained. In the CDA-AMC base case, capivasertib would require a price reduction of at least 85% for capivasertib plus fulvestrant to be considered cost-effective at a WTP threshold of $50,000 per QALY gained.
The cost-effectiveness of capivasertib plus fulvestrant compared to chemotherapy and everolimus plus exemestane is uncertain, given the lack of direct comparative evidence. The clinical experts noted that, unlike chemotherapy, everolimus plus exemestane is rarely used. The cost per 28 days associated with chemotherapy ranges from $205 (oral) to $4,400 (IV), whereas the 28-day cost of endocrine monotherapy ranges from $9 (oral) to $583 (subcutaneous). The cost-effectiveness of capivasertib plus fulvestrant versus chemotherapy will be influenced by how different patients perform on chemotherapy relative to endocrine monotherapy, as well as by the proportion of patients who receive IV treatment (paclitaxel) versus oral treatment (capecitabine). If the efficacy of chemotherapy is considered similar to that of endocrine monotherapy, then the cost-effectiveness of capivasertib plus fulvestrant versus chemotherapy may be similar to the cost versus endocrine monotherapy.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Truqap (capibasertib), tablets, 160 mg and 200 mg, oral. Mississauga (ON): Sponsor AstraZeneca Canada Inc; 2024 Feb 14.
2.AstraZeneca. Clinical Study Report for CAPItello-291: A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative (HR+/HER2−) Breast Cancer Following Recurrence or Progression On or After Treatment with an Aromatase Inhibitor (CAPItello-291) [CONFIDENTIAL internal manufacturer's report]. 2023 [sponsor submitted reference].
3.Cancer Care Ontario: funded evidence-informed regimens. 2024; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2024 Mar 1.
4.Saskatchewan Drug Plan: search formulary. 2024; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2024 Mar 1.
5.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/ [sponsor submitted reference].
6.Government of New Brunswick. New Brunswick Drug Plans Formulary. https://www2.gnb.ca/content/gnb/en/departments/health/MedicarePrescriptionDrugPlan/NBDrugPlan/ForHealthCareProfessionals/NewBrunswickDrugPlansFormulary.html [sponsor submitted reference].
7.Government of Nova Scotia. Nova Scotia Pharmacare formulary. 2021; https://novascotia.ca/dhw/pharmacare/formulary.asp [sponsor submitted reference].
8.B. C. Government. BC PharmaCare formulary search. 2024; https://pharmacareformularysearch.gov.bc.ca. Accessed 2024 Mar 1.
9.Cancer Care Ontario. Drug formulary. 2023; https://www.cancercareontario.ca/en/cancer-treatments/chemotherapy/drug-formulary. Accessed 2024 Mar 1.
10.Ontario Ministry of Health. Schedule of benefits for Laboratory Services - Laboratories and Genetic Branch. Lab. Med. 2023 [sponsor submitted reference].
11.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf [sponsor submitted reference].
12.Patient cost estimator. 2024; https://www.cihi.ca/en/patient-cost-estimator [sponsor submitted reference].
13.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf [sponsor submitted reference].
14.Guyot P, Ades AE, Beasley M, Lueza B, Pignon JP, Welton NJ. Extrapolation of Survival Curves from Cancer Trials Using External Information. Med Decis Making. 2017;37(4):353-366. PubMed
15.Guyot P, Ades Ae Fau - Ouwens MJNM, Ouwens Mj Fau - Welton NJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. (1471-2288 (Electronic) [sponsor submitted reference]).
16.Modi S, Jacot W, Yamashita T, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med. 2022;387(1):9-20. PubMed
17.pan-Canadian Pharmaceutical Alliance. Current generic categories report February 2024. 2024; https://www.pcpacanada.ca/generic-drug-framework. Accessed 2024 Mar 1.
18.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Verzenio (abemaciclib), tablets, 50 mg, 100 mg, 150 mg, 200 mg oral. Toronto (ON): Eli Lilly Canada Inc; 2024 Feb 9.
19.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Truqap (capibasertib), tablets, 160 mg and 200 mg, oral. Mississauga (ON): Sponsor AstraZeneca Canada Inc; 2024 Feb 14.
20.CDA-AMC. abemaciclib. https://www.cadth.ca/abemaciclib-0. Accessed 2024 May 7.
21.Pan H, Gray R, Braybrooke J, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med. 2017;377(19):1836-1846. PubMed
22.Statistics Canada. Table 13-10-0751-01 Number of prevalent cases and prevalence proportions of primary cancer, by prevalence duration, cancer type, attained age group and sex. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310075101. Accessed 2024 Mar 1.
23.Canadian cancer statistics: a 2022 special report on cancer prevalence. Toronto (ON): Canadian Cancer Society: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2022-statistics/2022-special-report/2022_prevalence_report_final_en.pdf?rev=7755f9f350e845d58e268a59e3be608e&hash=3F3F30CADD8CAF0049636B5A41EDBB13&_gl=1*x1rkvv*_gcl_au*MTI4NjAxNjcxLjE3MTIwODY. Accessed 2024 May 23.
24.Statistics Canada. Population estimates on July 1, by age and gender. 2024; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=2.1&cubeTimeFrame.startYear=2018&cubeTimeFrame.endYear=2023&referencePeriods=20180101%2C20230101. Accessed 2024 May 23.
25.Canadian Centre for Cancer Information. Breast cancer. https://cancer-data.canada.ca/type/breast-sein-eng.htm. Accessed 2024 May 23.
26.Cossetti RJ, Tyldesley SK, Speers CH, Zheng Y, Gelmon KA. Comparison of breast cancer recurrence and outcome patterns between patients treated from 1986 to 1992 and from 2004 to 2008. (1527-7755 (Electronic) [sponsor submitted reference]).
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison for HR-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day Cost ($)a |
|---|---|---|---|---|---|---|
Capivasertib (Truqap) | 200 mg | Tablet | 147.6000 | 400 mg, twice daily for 4 days followed by 3 days off | 337.37 | 9,446.40 |
Fulvestrant (Faslodex) | 50 mg/mL | 250 mg / 5 mL Prefilled syringe injection | 58.2895 | 500 mg on days 1,15, and 29, and then once monthly thereafter | 582.90 | First 28 days: 1,165.79 Thereafter: 582.90 |
Capivasertib plus Fulvestrant | 920.27 | First 28 days: 10,612.19 Thereafter: 10,029.30 | ||||
Endocrine monotherapy | ||||||
Anastrozole (Arimidex) | 1 mg | Tablet | 0.9522 | 1 mg daily | 0.95 | 26.66 |
Exemestane (Aromasin) | 25 mg | Tablet | 1.3263 | 25 mg daily | 1.33 | 37.14 |
Fulvestrant (Faslodex) | 50 mg/mL | Prefilled syringe injection | 58.2895 | 500 mg on days 1, 15, and 29, and then every 28 days | 582.90 | First 28 days: 1,165.79 Thereafter: 582.90 |
Letrozole (Femara) | 2.5 mg | Tablet | 1.3780 | 2.5 mg daily | 1.38 | 38.58 |
Tamoxifen (Nolvadex) | 20 mg | Tablet | 0.3500 | 20 mg daily | 0.35 | 9.80 |
Targeted therapy | ||||||
Everolimusa (Afinitor) | 10 mg | Tablet | 50.6636 | 10 mg daily | 50.66 | 1,418.58 |
Exemestane (Aromasin) | 25 mg | Tablet | 1.3263 | 25 mg daily | 1.33 | 37.14 |
Everolimus plus Exemestane | 51.99 | 1,455.71 | ||||
Chemotherapy | ||||||
Capecitabine (Xeloda) | 150 mg 500 mg | Tablet | 0.4575 1.5250 | 1,000 mg/m2 to 1,250 mg/m2 twice daily on days 1 to 14, repeat every 21 days | 7.32 to 8.74 | 204.96 to 244.81 |
Paclitaxel | 6 mg/mL | 1 mL | 60 | 175 mg/m2 IV on day 1, repeat every 21 days | 204.96 | 4,400 |
CDA-AMC = Canada’s Drug Agency; HR = hormone receptor.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2024), unless otherwise indicated, and do not include dispensing fees. A body surface area of 1.75 square meter is assumed.
aSource: Pan-Canadian Pharmaceutical Alliance generics categories report.17
Appendix 2: Submission Quality
Please note that this table has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | The cost and impact of subsequent therapies could not be robustly explored. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.

Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Capivasertib plus fulvestrant | Endocrine monotherapy |
|---|---|---|
Discounted LYs | ||
Total | 3.44 | 2.21 |
By health state or data source | ||
Progression free | 1.07 | 0.40 |
Progressed disease | 2.37 | 1.81 |
Discounted QALYs | ||
Total | 2.83 | 1.81 |
By health state or data source | ||
Progression free | 0.90 | 0.34 |
Progressed disease | 1.93 | 1.47 |
Discounted costs ($) | ||
Total | 236,775 | 88,743 |
Drug acquisition | 146,345 | 3,503 |
Administration | 67 | 25 |
Resource use (excluding test cost) | 14,225 | 9,414 |
Testing cost | 233 | 0 |
Terminal care | 51,391 | 52,512 |
Subsequent therapy | 22,951 | 22,801 |
Adverse events | 1,562 | 489 |
LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Capivasertib plus fulvestrant | Endocrine monotherapy |
|---|---|---|
Discounted LYs | ||
Total | 3.05 | 2.41 |
By health state | ||
Progression free | 0.84 | 0.43 |
Progressed disease | 2.21 | 1.99 |
Discounted QALYs | ||
Total | 2.50 | 1.97 |
By health state | ||
Progression free | 0.71 | 0.36 |
Progressed disease | 1.80 | 1.61 |
Discounted costs ($) | ||
Total | 184,998 | 66,521 |
Drug acquisition | 116,574 | 3,639 |
Administration | 55 | 26 |
Resource use (excluding test cost) | 12,759 | 10,304 |
Testing cost | 2,435 | 0 |
Terminal care | 51,613 | 52,062 |
Subsequent therapy | 0 | 0 |
Adverse events | 1,561 | 490 |
CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
Figure 2: Relative Risk of Death Over Time, Given Different Assumptions Regarding Long-Term Extrapolation of OS

PH = proportional hazards
Source: Sponsor’s pharmacoeconomic submission.1
Scenario Analyses
Table 12: Summary of the CDA-AMC Scenario Analysis 1 Extrapolating OS With the Weibull Parametric Distribution
Drug | Total costs ($) | Incremental cost ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Capivasertib plus Fulvestrant | 184,799 | Reference | 2.37 | Reference | Reference |
Endocrine monotherapy | 66,676 | 118,123 | 1.97 | 0.40 | 296,104 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; OS = overall survival; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.18
Table 13: Summary of the CDA-AMC Scenario Analysis 2 Linking Subsequent Therapy Costs to Time in the Progressed State
Drug | Total costs ($) | Incremental cost ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Capivasertib plus Fulvestrant | 267,976 | Reference | 2.50 | Reference | Reference |
Endocrine monotherapy | 142,138 | 125,838 | 1.96 | 0.54 | 233,905 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.18
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take Aways
Key take aways of the budget impact analysis |
|---|
CDA-AMC identified the following key limitations with the sponsor’s analysis:
CDA-AMC reanalysis:
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) to assess the expected budgetary impact resulting from introducing capivasertib for the Health Canada indication, for use in combination with fulvestrant for the treatment of adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more alterations in PIK3CA, AKT1, or PTEN following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy, from the perspective of the public drug plan in the Canadian setting (excluding Quebec) over a 3-year time horizon. The sponsor’s submission considered the following costs: primary therapy drug acquisition, subsequent therapy drug acquisition, drug administration, adverse event, pharmacy, and testing. In the reference scenario, the sponsor assumed that patients would be eligible to receive a basket of endocrine monotherapy, everolimus plus exemestane, or a basket of chemotherapy. In the new drug scenario, capivasertib plus fulvestrant was assumed to proportionally displace market shares.19
The sponsor estimated the eligible population size using an epidemiological approach that begins with 3 different population groups: newly diagnosed metastatic breast cancer, late relapse to metastatic breast cancer, and early relapse to metastatic breast cancer (i.e., progressed while on or within 12 months of completing adjuvant therapy).19 Key inputs to the BIA are documented in Table 15.
Key assumptions included:
Among newly diagnosed or late relapse to metastatic breast cancer patients, it was assumed that ██% would receive an ET-based regimen in the first-line setting. Following this treatment, it was assumed that ████% of patients would continue to a subsequent therapy (i.e., second line or later).
The base year testing rate for PIK3CA, AKT1, or PTEN was assumed to be 0% in all jurisdictions except Ontario, where the base year testing rate was ██%. The testing rates were assumed to be 50%, 60%, and 70% for years 1 to 3, respectively, across all drug plans in the new drug scenario. In the reference scenario, it was assumed only Ontario would have a testing rate, which was held at ██%.
Because of the absence of targeted therapies for patients with PIK3CA, AKT1, or PTEN alterations, the sponsor assumed a highly accelerated uptake of capivasertib plus fulvestrant, and that it will displace existing regimens equally from all comparators based on their respective baseline market shares.
Market shares are assumed to capture patients eligible for capivasertib plus fulvestrant in the second-line setting, as well as the smaller proportion who are eligible in the first- and third-line setting.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Newly diagnosed mBC Incidence of breast cancer Proportion of advanced or mBC Proportion of HR-positive, HER2-negative, advanced or mBC | Province-specific (excluding Quebec) 4.87% 64.78% |
Late relapse to mBC Prevalence of BC Proportion early BC Proportion HR-positive, HER2-negative Proportion relapse to eBC to mBC Proportion received 1L ET-based regimen Proportion eligible for subsequent treatment | Province-specific (excluding Quebec) 94.00% 64.78% 2.76% █████% █████% |
Early relapse to metastatic breast cancer (I.e., progressed during or within 12 months of completing adjuvant therapy) Proportion at high risk of recurrence Proportion tested Ki-67 high (≥ 20%) Proportion early relapse to mBC | 12.00% 24.9% 4.00% |
AKT pathway Proportion with PIK3CA, AKT1, or PTEN alterations | 40.80% |
Number of eligible patients under reference scenario | 0/0/0 |
Number of eligible patients under new drug scenario | 212/305/390 |
Market Uptake (3 years) | |
Uptake (reference scenario) Capivasertib plus fulvestrant Endocrine monotherapy Everolimus plus Exemestane Chemotherapy | 0%/0%/0% 40%/40%/40% 10%/10%/10% 50%/50%/50% |
Uptake (new drug scenario) Capivasertib plus fulvestrant Endocrine monotherapy Everolimus plus Exemestane Chemotherapy | 55% / 65% / 70% 18% / 14% / 12% 5% / 4% / 3% 23% / 18% / 15% |
Cost of treatment (per patient, year) | |
Capivasertib plus fulvestrant Endocrine monotherapy Everolimus plus Exemestane Chemotherapy | $75,301.98 $2,503.39 $8,698.13 $9,078.71 |
mBC = metastatic breast cancer; HR = hormone receptor; HER2 = human epidermal growth factor receptor 2; BC = breast cancer; eBC = early breast cancer; ET = endocrine therapy; Ki-67 = city of Kiel original clone 67; AKT = protein kinase B signalling pathway.
Source: Sponsor’s pharmacoeconomic submission.19
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population
Source: Sponsor’s pharmacoeconomic submission.19
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of capivasertib for use in combination with fulvestrant for the treatment of adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more alterations in PIK3CA, AKT1, or PTEN following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy will be associated with a 3-year cost of $37,798,688 (Year 1: $8,835,648; Year 2: $12,725,519; Year 3: $16,237,522).19
CDA-AMC Appraisal of the Sponsor’s BIA
Market share for capivasertib is underestimated. The sponsor’s BIA assumes that the market uptake for capivasertib was 55%, 65%, and 70% in years 1, 2, and 3 respectively. This would mean in year 1, 45% of eligible patients would not receive capivasertib (this decreases to 35% in years 2 and 30% in year 3). This market share applies to patients who received testing and tested positive for PIK3CA, AKT1, or PTEN alteration. In consultation with clinical experts, it was noted that it would be unlikely for patients who have a confirmed PIK3CA, AKT1, or PTEN alteration not to receive treatment with capivasertib. Testing uptake was seen as the main limiting factor to market uptake. It was noted that because of patient and clinician preference not everyone who tests positive will receive capivasertib, but this number will likely be small.
CDA-AMC base-case reanalysis increased the market uptake for capivasertib to 70%, 80%, and 90% for years 1, 2, and 3 respectively.
Uptake and duration of treatment for subsequent therapy are uncertain. In the sponsor’s analysis uptake and duration of treatment for subsequent therapies were informed mainly by clinical expert feedback as opposed to data. Clinical experts consulted by CDA-AMC noted that the proportion of patients who do not receive subsequent therapy is overestimated.
Furthermore, the sponsor’s model estimates the subsequent therapy received by the patient for the entire duration of the lifetime time horizon as a 1-time cost. CDA-AMC clinical experts state that although the next line of subsequent therapy can be estimated, patients will likely fail and receive further lines of subsequent therapies in clinical practice. In the sponsor’s model for example, ██% of patients who fail capivasertib plus fulvestrant go on to receive trastuzumab deruxtecan and ██% of patients who fail chemotherapy go on to receive trastuzumab deruxtecan. Although this may occur for the next line of therapy, patients who fail capivasertib plus fulvestrant and then fail multiple subsequent therapies will eventually go on to receive trastuzumab deruxtecan, which is not captured in the sponsor’s model. The sponsor’s model therefore predicts that funding capivasertib will reduce uptake of trastuzumab deruxtecan in many patients. Although the rate of uptake for trastuzumab may be slightly lower for those who receive capivasertib plus fulvestrant it is unlikely to be as dramatically reduced as predicted by the sponsor. A more sophisticated analysis would be required that allows for multiple lines of therapy considering attrition rates over time.
Finally, the sponsor reports that capivasertib may extend time on subsequent therapies as it may restore cancer cells’ sensitivity to other therapies. Although this is uncertain, this could increase subsequent therapy costs for those who initially receive capivasertib plus fulvestrant.
In the CDA-AMC base case subsequent therapy costs were removed. Given that subsequent therapy use may not deviate significantly across treatment arms it is unlikely this will have a substantial impact on the budget in the long run. A more sophisticated analysis that allows for multiple lines of therapies would be required to measure the potential impact subsequent therapy use could have on the budget.
Ki-67 testing for early breast cancer patients with a high risk of recurrence to receive abemaciclib is no longer required. In January 2022, abemaciclib received Notice of Compliance status from Health Canada for the adjuvant treatment of adult patients with HR-positive, HER2-negative, node-positive early breast cancer at high risk of disease recurrence based on clinicopathologic features and a high Ki-67 test score (≥ 20%). However, in December 2023, abemaciclib received an updated Notice of Compliance status from Health Canada where the high Ki-67 test score is no longer required for this population of patients.20
Furthermore, the sponsor estimated that 12% of adult patients with HR-positive, HER2-negative, node-positive early breast cancer will have a high risk of disease recurrence based on clinicopathological features. This estimate is likely an underestimate because according to Pan et al., risk of distant recurrence was strongly correlated with the original nodal status. Pan et al. reports that breast cancer patients with ER-positive, T1 and T2 patients with 1 to 3 positive lymph nodes have an approximately 30% probability of having a distant recurrence.21 Although the Pan et al. study population does not include T3 patients, who are a subset of patients considered for abemaciclib, patients who are considered for abemaciclib because of the high risk of recurrence based on clinicopathological features would likely have a higher probability of having a distant recurrence. Therefore the 12% value is likely an underestimate.
Finally, when calculating the number of patients who receive abemaciclib the sponsor assumes every year 12% of the prevalent cohort (patients with a breast cancer diagnosis for up to 10 years) are considered for abemaciclib based on Ki-67 testing. Abemaciclib is only given in the adjuvant setting and has only been publicly funded in Canada since 2023. Patients with a breast cancer diagnosis for more than 1 year will have either not received abemaciclib or received it and have already relapsed meaning they are currently receiving a first-line therapy now anyway. Therefore, when considering who relapses early on abemaciclib the BIA should only focus on new diagnoses (incident patients).
The CDA-AMC base case removes the Ki-67 testing requirement to receive abemaciclib. As a scenario analysis the testing requirement was retained.
The CDA-AMC base case only considers incident patients when estimating the size of the population who have received abemaciclib. For each jurisdiction, the size of the abemaciclib population was calculated as follows:
(Incident population) * (probability not diagnosed with metastatic disease) * (probability HR+/HER2-) * (probability of high recurrence)
For example, for British Columbia the calculation is as follows: 4,533 * (95.13%) * (64.78%) * (12%) = 335
The same rate of relapse (4% after 12 months) was assumed, so for BC 13 patients (335 * 4%) would be considered for capivasertib plus fulvestrant if they had altered PIK3CA, AKT1, or PTEN.
The prevalent breast cancer patient population for the late relapse to metastatic subgroup was miscalculated: The sponsor used the province-specific, 2018 prevalence of breast cancer from Statistics Canada,22 and divided this number by the average population in years 2022 to 2031 for the corresponding province to calculate the prevalence rate for each province. Using future year population in the denominator to calculate the prevalence rate will miscalculate the prevalence rate if the population size changes. To calculate the prevalence rate the number of diagnoses must be divided by the size of the population in the year the diagnoses occurred. Otherwise keeping the number of diagnoses fixed but allowing the population to change assumes prevalence rates will change over time. As the population grows the sponsor therefore underestimates the prevalence rate.
Second, the sponsor applied the prevalence rate to a population over the age of 15 years only, likely to account for the indication specifying adults only. However, the prevalence number cited by the sponsor was calculated by taking the total number of prevalent patients in the whole population, not just those over the age of 15 years. The Canadian cancer statistics noted on January 1 2018 there were 111,795 patients living in Canada who had been diagnosed with breast cancer in the past 5 years.23 On January 1 2018 the population of Canada was 37,072,620.24 As a percentage of the population this therefore equates to 0.304% (111,795 / 37,072,620). This closely matches the 5-year prevalence rate reported in the Canadian cancer statistics (299 cases per 100,000 or 0.299%).25 Given the size of the nonadult breast cancer population is so small; recalculating the prevalence rate to attempt to remove them without access to granular data introduces unnecessary uncertainty into the BIA.
The CDA-AMC base-case reanalysis recalculated the prevalence rate by using the rate reported by the data with no modifications.22 These prevalence estimates are reported as cases per 100,000. To derive a percentage the number of cases was divided by 100,000.
The prevalence rate was applied to the full population of Canada, not just those over the age of 15 years. The population of each jurisdiction was updated to reflect the most recent data with gender breakdown (1 July 2023).24 Population growth estimates were left unchanged. This assumes the prevalence rate is made up exclusively of adult patients.
The proportion of early breast cancer patients with HR-positive, HER2-negative who relapse to metastatic breast cancer was overestimated. The sponsor referenced table 3 from the paper by Cossetti et al. and used the data for ER-negative, HER2-negative patients to inform the proportion of breast cancer relapse to metastatic disease. However, it would be more appropriate to use the HR-positive HER2-negative data as this pertains to the indicated population for capivasertib. Second, the data provided gives relapse rates based on the number of years the patient has had their breast cancer diagnosis. The sponsor takes a simple mean across the years assuming an even distribution of time spent diagnosed with breast cancer.26 However, data shows that when looking at a cohort of breast cancer patients who have been diagnosed from 0 to 10 years the distribution across the years is not even. Data shows that female prevalence of breast cancer per 100,000 people is 256 for people who have been diagnosed for less than 2 years, 589 for people who have been diagnosed for less than 5 years, and 1,030 for people who have been diagnosed for less than 10 years. Using this data, it can be estimated that when looking at a 10-year prevalent cohort 25% are in their first 2 years of diagnosis, 33% have been diagnosed for 2 to 5 years and 43% have been diagnosed for 5 to 10 years. This can be used to derive a weighted average of relapse rates over 10 years.
CDA-AMC base-case reanalysis recalculates the relapse rate using HR-positive, HER2-negative cohort 2 data by weighting the yearly mean rates according to the population of the corresponding year. Table 17 outlines how the weighted relapse rate was calculated.
The costs of drug acquisition are underestimated. The sponsor’s BIA model used the median annual treatment time to calculate the cost of each treatment for all patients entering the BIA. For example, patients on capivasertib plus fulvestrant were assumed to be on treatment for an average of 6.8 months and patients on endocrine monotherapy were on treatment for an average of 3.4 months. The cost of treatment per year was constant each year. For example, the annual cost of capivasertib plus fulvestrant was $75,302 and the annual cost of endocrine monotherapy was $2,503 for each year of the BIA. However, this does not account for any skewness in the data. For example, if a large enough proportion of patients remained on therapy for more than 1 year these additional costs are missed if the median is used. Using the data on time to treatment discontinuation evidence shows that using the median underestimates costs associated with capivasertib plus fulvestrant.
Using the submitted economic evaluation with CDA-AMC base-case changes (see Table 6), for patients receiving capivasertib plus fulvestrant CDA-AMC estimated:
average drug costs 1 year after initiating therapy (model was run for 1 year with 0% discounting)
average drug costs 2 years after initiating therapy (model was run for 2 years with 0% discounting)
average drug costs 3 years after initiating therapy (model was run for 3 years with 0% discounting).
For patients entering the BIA in year 1 and receiving capivasertib plus fulvestrant: $79,574 was applied in year 1, $20,246 was applied in year 2 and $7,681 was applied in year 3.
For patients entering the BIA in year 2 and receiving capivasertib plus fulvestrant $79,574 was applied to the first year in the BIA, $20,246 was applied to the second year.
For patients entering the BIA in year 3 and receiving capivasertib plus fulvestrant only $79,574 was applied.
The average time on therapy for endocrine monotherapy was increased to 5 months to better reflect costs seen in the economic evaluation which accounts for data skewness. Unlike capivasertib plus fulvestrant nearly all patients have discontinued endocrine monotherapy before 1 year and therefore it was assumed all costs are incurred in the first year on therapy.
Current metastatic/locally advanced patients are excluded. The sponsor’s BIA only looks at new relapses of breast cancer, which is relapses that occur after capivasertib plus fulvestrant is funded. However, some patients with a current metastatic diagnosis would be eligible for capivasertib plus fulvestrant if they received testing and tested positive for PIK3CA, AKT1, or PTEN alteration. Exclusion of these patients underestimates the BIA in the short-term. In the long-term, all new relapses are considered and therefore the cohort of metastatic/locally advanced patients who were not tested upon relapse will decrease over time.
CDA-AMC was unable to address this limitation though notes exclusion of these patients underestimates the BIA in the short-term.
CDA-AMC Reanalyses of the BIA
Table 16: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Market share of capivasertib plus fulvestrant for patients who tested positive for PIK3CA, AKT1, or PTEN year 1/ year 2/ year 3 | 55% / 65% / 70% | 70% / 80% / 90% |
2. Subsequent therapy costs | Included | Excluded |
3. Estimating the number of patients eligible for capivasertib plus fulvestrant who relapsed early (< 12 months) on abemaciclib | Assumed all HR+/HER2- patients diagnosed in past 10 years at high risk of recurrence and tested Ki-67 high received abemaciclib each year | Only included HR+/HER2- patients diagnosed each year from 2024 onwards (incident patients) at high risk of recurrence when looking at who relapses early on abemaciclib |
4. Prevalence calculations updated | Number of diagnosed patients from 2018 were divided by average population size over the age of 15 years old from 2022 to 2031:
Prevalence rates applied to population over the age of 15 years. | Prevalence rates taken directly from Statistics Canada:
Prevalence rates applied to full population using July 1 2023 estimates of female population in Canada.24 |
5. Proportion of early breast cancer patients who relapse to metastatic breast cancer | 2.76% | 1.79% |
6. Drug cost for: capivasertib plus fulvestrant | Capivasertib plus fulvestrant: Every patient incurs $75,301 in drug costs Endocrine monotherapy: Assume patients are on therapy for 3.4 months | Capivasertib plus fulvestrant: Patients who enter the BIA in year 1 have 3 years of costs tracked, patients who enter the BIA in year 2 have 2 years of costs tracked, patients who enter the BIA in year 3 have 1 year of costs tracked.
Endocrine monotherapy: Assume patients are on therapy for 5 months |
CDA-AMC base case | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 | |
NIBH = Non-Insured Health Benefits Program for First Nations and Inuit.
aAssumed to equal to Ontario.
Table 17: Calculations Used to Derive the Annualized Relapse Rate
Year(s) since diagnosis | Annual relapse rate (%)b | % of patientsa |
|---|---|---|
0 to 1 | 1.4 | 12.5% |
1 to 2 | 2.6 | 12.5% |
2 to 3 | 3.1 | 10.8% |
3 to 4 | 2.3 | 10.8% |
4 to 5 | 2.1 | 10.8% |
5 to 6 | 1.6 | 8.6% |
6 to 7 | 1.7 | 8.6% |
7 to 8 | 1 | 8.6% |
8 to 9 | 0.7 | 8.6% |
9 to 10 | 0.7c | 8.6% |
Weighted average annual relapse rate | 1.79 | |
aSource: Statistics Canada for 0 to 2 years; 2 to 5 years and 6 to 10 years. Assumed even distribution of patients for individual years within these ranges. For example, 24.5% of patients are within their first 2 years since diagnosis so it is assumed 12.5% are in their first year and 12.5% are in their second year.
bSource: Cossetti et al.26
cAssumed the same rate as 8 to 9 years,
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. In the CDA-AMC base case, the 3-year budget impact is expected to be $81,103,794 (year 1: $16,102,743; year 2: $26,971,824; year 3: $38,029,227) should capivasertib be reimbursed as per the Health Canada indication (i.e., for use in combination with fulvestrant for the treatment of adult females with HR-positive, HER2-negative, locally advanced or metastatic breast cancer with 1 or more alterations in PIK3CA, AKT1, or PTEN following progression on at least 1 endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy).
Table 18: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | 37,798,688 |
CDA-AMC reanalysis 1 | 47,784,360 |
CDA-AMC reanalysis 2 | 62,498,512 |
CDA-AMC reanalysis 3 | 37,248,401 |
CDA-AMC reanalysis 4 | 42,434,446 |
CDA-AMC reanalysis 5 | 28,041,909 |
CDA-AMC reanalysis 6 | 53,465,158 |
CDA-AMC base case (1 + 2 + 3 + 4 + 5 + 6) | 81,103,794 |
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | 3-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 3,183,227 | 21,943,400 | 26,741,216 | 31,683,376 | 80,367,993 |
New drug | 3,183,227 | 30,779,048 | 39,466,735 | 47,920,897 | 118,166,681 | |
Budget impact | — | 8,835,648 | 12,725,519 | 16,237,522 | 37,798,688 | |
CDA-AMC base case | Reference | 308,061 | 2,124,304 | 2,596,741 | 3,086,138 | 7,807,183 |
New drug | 308,061 | 18,227,047 | 29,568,565 | 41,115,365 | 88,910,977 | |
Budget impact | — | 16,102,743 | 26,971,824 | 38,029,227 | 81,103,794 | |
CDA-AMC scenario analysis 1 (reimbursement request, including males and females) | Reference | 313,177 | 2,148,911 | 2,626,432 | 3,120,960 | 7,896,302 |
New drug | 313,177 | 18,438,210 | 29,907,373 | 41,580,821 | 89,926,403 | |
Budget impact | — | 16,289,299 | 27,280,941 | 38,459,861 | 82,030,101 | |
CDA-AMC scenario analysis 2 (testing costs only)a | Reference | 205,173 | 208,619 | 212,123 | 215,686 | 636,429 |
New drug | 205,173 | 1,038,837 | 1,268,907 | 1,506,908 | 3,814,652 | |
Budget impact | — | 830,217 | 1,056,784 | 1,291,221 | 3,178,223 | |
CDA-AMC scenario analysis 3 (100% testing uptake) | Reference | 308,061 | 4,248,608 | 4,327,902 | 4,408,768 | 12,985,278 |
New drug | 308,061 | 36,454,095 | 50,772,728 | 61,195,099 | 148,421,922 | |
Budget impact | — | 32,205,487 | 46,444,826 | 56,786,331 | 135,436,643 | |
CDA-AMC scenario analysis 4 (Ki-67 required for abemaciclib) | Reference | 299,043 | 2,060,425 | 2,518,636 | 2,993,291 | 7,572,352 |
New drug | 299,043 | 17,678,973 | 28,679,278 | 39,878,530 | 86,236,781 | |
Budget impact | — | 15,618,548 | 26,160,642 | 36,885,239 | 78,664,429 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
aThis analysis only looks at the cost of testing as drug costs are the same as the CDA-AMC base case.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 19:
Sponsor’s reimbursement request population: adult population including both males and females.
Analyzing the cost of testing assuming:
Currently in years 1, 2 and 3: 278 / 283 / 288 individuals receive PIK3CA, AKT1, or PTEN alteration testing respectively.
If capivasertib is reimbursed, the number of individuals who receive PIK3CA, AKT1, or PTEN alteration testing increases to 1,385 / 1,692 / 2,009 in years 1, 2, and 3, respectively.
The cost of the test is $750.
Assuming 100% testing uptake of PIK3CA, AKT1, or PTEN alteration.
Assuming Ki-67 testing is required for patients to receive abemaciclib. Of the early breast cancer patients with HR-positive/HER2-negative and a high risk of recurrence, 24.90% of patients are assumed to take the Ki-67 test and test high (≥ 20%) to receive abemaciclib.
Testing Procedure Assessment Review
Abbreviations
FFPE
formalin-fixed, paraffin-embedded
HER2
human epidermal growth factor receptor 2
HR
hormone receptor
NGS
next-generation sequencing
OCA
Oncomine Comprehensive Assay
Objective
The objective of the testing procedure assessment is to identify and describe important health system implications of testing for PIK3CA, AKT1, and PTEN alterations in patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer to determine their eligibility for capivasertib.
Methods
The contents of this section have been informed by materials submitted by the sponsor, literature search, and clinical expert input.
The materials submitted by the sponsor that were related to the diagnostic test were validated and summarized by the review team.
An information specialist conducted a literature search of key resources, including MEDLINE, the Cochrane Database of Systematic Reviews, the International Health Technology Assessment Database, and the websites of Canadian and major international health technology agencies. A focused internet search was also conducted. The search approach was customized to retrieve a limited set of results, balancing comprehensiveness with relevancy. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were next-generation sequence (NGS) testing and breast cancer. Secondary searches were conducted using search filters developed by Canada’s Drug Agency to limit retrieval to citations related to economic and equity considerations. The search was completed on March 12, 2024 and limited to English-language documents published since January 1, 2019.
The clinical expert input was provided by 3 clinical specialists with expertise in the diagnosis and management of locally advanced or metastatic breast cancer.
Context
What Are PIK3CA, AKT1, and PTEN Alterations?
PIK3CA, AKT, and PTEN mutations collectively represent a complex network of genetic alterations that are implicated in various cancers, including breast cancer. These are somatic mutations acquired during tumour development that can affect disease outcomes and patients’ responses to drugs.1
The PI3K/AKT1/mTOR pathway is a key signalling cascade involved in regulating various cellular processes, such as cell proliferation and survival.2 Dysregulation of this pathway leads to aberrant cell signalling and is associated with tumour progression and drug resistance in breast cancer.3 PIK3CA, AKT1, and PTEN genes are closely linked to the PI3K/AKT1/mTOR pathway. Mutations to these genes can contribute to cancer development and progression.2,3
Alterations in the PI3K/AKT/mTOR signalling axis pathway are observed in up to 48% of all patients with HR-positive, HER2-negative breast cancer.4,5 In these breast cancers, PI3K/AKT/mTOR pathway activation most frequently arises from PIK3CA alterations, which occur in approximately 30% of patients.6-10 A further approximately 4% of advanced breast cancers harbour AKT1-activating alterations or amplifications, and approximately 5% have inactivating alterations in PTEN.10-12 Alterations in certain higher-risk genes can also influence breast cancer survival. In patients with HR-positive breast cancer, the presence of 1 or more PIK3CA, AKT, or PTEN alterations has been associated with accelerated disease progression and worse clinical outcomes.13-16
How Are PIK3CA, AKT1, and PTEN Alterations Identified?
PIK3CA, AKT, and PTEN alterations can be detected by conducting genomic testing of the tumour cells. Tissue samples of the tumour collected using minimally invasive procedures as part of routine diagnostic care (e.g., tissue biopsy or liquid biopsy) can be used to detect alterations. Multiple techniques for testing are available, such as polymerase chain reaction, NGS, and Sanger sequencing.17 While polymerase chain reaction assays are rapid, low-cost tests, these target specific mutations and cannot provide a comprehensive analysis.18 NGS is the preferred technology because of its greater sensitivity and ability to test for multiple genes simultaneously.18 Sanger sequencing, while considered the “gold standard” for DNA sequencing, allows the sequencing of only 1 DNA fragment at a time, has low scalability, and is considered less suitable for complex samples, such as tumour tissue.19,20
For patients who may be candidates to receive capivasertib plus fulvestrant, confirmation of a PIK3CA, AKT, and/or PTEN alteration through NGS testing of biopsy tissue could be carried out at the time of metastatic diagnosis.21,22 Clinical experts agreed that the optimal time for testing would be at the time of metastatic diagnosis, and that NGS is the method of choice.
What Is NGS?
NGS is a method of testing for germline and somatic genetic mutations that involves sequencing several genes and gene fragments simultaneously.23 DNA extracted from the tissue samples (e.g., formalin-fixed, paraffin-embedded [FFPE] samples, liquid biopsy) is prepared for use on a sequencer by a method known as library preparation followed by target enrichment. Then, clinical sequencing is performed using platforms such as Illumina or Thermo Fisher.23 Each DNA fragment is immobilized, clonally amplified, and sequenced by fluorescent detection or ion-based sequencing. Thus, the prepared raw data go through a bioinformatics pipeline to deliver what is known as a variant call file. Lastly, variants are interpreted and validated before a final report is produced.23
NGS is increasingly used in Canada to detect somatic and/or germline alterations for risk stratification as well as to guide therapeutic decisions where targeted therapies are available. Several NGS testing panels are available across Canada that can detect alterations in PIK3CA, AKT, and PTEN, such as the Oncomine Comprehensive Assay (OCA) and AmpliSeq Focus Panel. In Canada, according to the information provided by the sponsor, the most commonly used somatic testing panel is the OCA v3 or Plus (Thermo Fisher).24 The OCA is a multibiomarker NGS assay that covers PIK3CA, AKT, and PTEN, and it has undergone performance validation that demonstrated its accuracy, high specificity, sensitivity, and low sample input requirement, which enables the analysis of even small and challenging FFPE samples.25,26 This requirement is a significant advantage because primary samples from breast cancer patients can be limited.
In advanced breast cancer, NGS is best placed to detect somatic and/or germline alterations at metastatic diagnosis to provide prognostic information, identify therapeutic targets, and monitor treatment response.21 Studies have confirmed the utility of NGS to guide targeted, next-line therapy for metastatic breast cancer.
What Is the Current Testing Practice for Breast Cancer in Canada?
In Canada, tissue biopsy is standard of care for predicted breast cancer patients to confirm diagnosis as well as estrogen receptor, progesterone receptor, and HER2 status. The biopsy is typically conducted in an outpatient setting and is not associated with any prolonged recovery time or complications. Tissue samples collected through tissue biopsy are sent to pathology for processing. Collected tissue samples prepared as FFPE are saved for up to 20 years27 and could be used for future testing.
If NGS assay for PIK3CA, AKT, and PTEN to determine eligibility for capivasertib is warranted, previously collected FFPE tissue or newly collected samples are typically sent to in-house molecular labs or a centralized laboratory. There is no finite period during which results of the NGS testing would be valid; in addition, the use of archived patient biopsy tissue for testing is accepted. Testing results are to be reviewed by a registered oncologist and pathologist.
Testing Procedure Considerations
What Are the Health System Considerations?
What Is the Availability of NGS Testing Panels in Canada?
Availability
NGS testing panels are widely available across Canadian provinces; however, the clinical experts indicated that these are typically available to patients through clinical trials only. The clinical experts also indicated that most large clinical centres across the provinces have in-house capability for NGS testing. There are no publicly funded or private genetic testing facilities in the territories.28 Samples from the jurisdictions that do not have testing facilities are sent to provincial testing centres. A summary of each province’s testing panel and genomic capability is provided in Table 1.
According to the sponsor, provinces that currently have smaller panels, including Nova Scotia and Quebec, are working to validate and upgrade to broader test offerings. These upgrades may include comprehensive panels, such as Illumina’s TruSight Oncology 500 panel, which covers all 3 genes (PIK3CA, AKT1, and PTEN).
Funding
NGS testing for patients with advanced (unresectable or metastatic) breast cancer is subject to various funding arrangements within Canada.29 While most laboratories in Canada include PIK3CA, AKT1, and PTEN on their NGS panels, funded testing options that target all 3 alterations are limited or not available.30-32 Public funding of these tests varies between provinces, with Ontario funding NGS testing for the PIK3CA gene in advanced or metastatic breast cancer where directed therapy is under consideration.33 According to the sponsor, Quebec provides funding for an NGS testing panel that includes PIK3CA and AKT1. Other than Quebec, Ontario is the only jurisdiction that offers provincial-level funding for testing of any of the 3 genes of interest. This variability in funding underscores the variability in care currently received by patients across different regions.
Table 1: Available NGS Testing Panels for the Relevant Genes per Canadian Province
Province | Panel | Genes covered |
|---|---|---|
OncoPanel (custom panel; Illumina sequencing) | PIK3CA, AKT1, and PTEN | |
Alberta36 | Cancer Biomarker Comprehensive DNA Panel | PIK3CA, AKT1, and PTEN |
Saskatchewan | Oncomine Comprehensive Assay Plus (Thermo Fisher) | PIK3CA, AKT1, and PTEN |
Manitoba37 | QIAseq Targeted DNA Panel (Qiagen) | PIK3CA, AKT1, and PTEN |
Ontario33 | Variable, but mostly Oncomine Comprehensive Assay v3 (Thermo Fisher) | PIK3CA, AKT1, and PTEN |
AmpliSeq Focus Panel (Illumina) | PIK3CA, AKT1 | |
New Brunswick | Oncomine Comprehensive Assay Plus (Thermo Fisher) | PIK3CA, AKT1, and PTEN |
Nova Scotia | AmpliSeq Focus Panel (Illumina) | PIK3CA, AKT1 |
Newfoundland and Labrador | Out-of-province testing in Nova Scotia | PIK3CA, AKT1 |
Prince Edward Island | Out-of-province testing in New Brunswick | PIK3CA, AKT1, and PTEN |
NGS = next-generation sequencing.
Note: The data presented in this table reflect the availability of testing at the time of the sponsor’s submission. NGS testing panels and their availability may change over time.
Source: Sponsor’s clinical evidence.
Funding by Sponsor
███ ███████ ███ ████████ █████████ ███████ ██ ███████ ███ ███████ ███████ ██████████ ████████ ████████ ████ ████████ ███████ ███████████ ██████ █ ████████ ████████ ████████ ████████ ███████████ ████ ███████ ███████ ██ ████████ ██ ██ ████ █████████ ██ ███ ████████ ████████ ███████ ███ ████ ███ ████ ███████ ██████ ███ ███ █████ ██ █████ ███ ███████ █████ ██████ █████ ████ █████████ █████ ██████ ██ ████ ███████ ████████████ ███████ ███ █████████ ████ ████ █████ ████████ ███████████████████ █████████████████ ████ ████████ ██████████ ████████████ █████████████ ███ █████████ ████████████ ██ ███████ █████████ ██ ███ ████████████ ███ ████████ ███████ ██ ███████ ███████ █████ █████████ ██████████ ██████ ██████ █████████ ██ ████ ██ ████████ █████████████ ██ ████████ ███ ███████████████ ████████ ██ █████ █████████████ █████████████ ███ ███████ █████ ████ ████ █████ ████████████ ██ ███████ ███ ██████████ ██ ████████ ███████ ██ █████ ██████████ ██ ████████ ████ ██ ███████ ████ █████ ████ █ ████ ████ ██████ ███████ ███ ███████ ███ ██ ████████████ ███ ███████ ████ █████████ ████ ████ ███ ███████ ████ ████████ ██████████ █████████ ██ ████████ █████████ ████ ███ ██████ █████████ █████████ ██ ███████ ███ ███████ ███████████ ██ ███████ ███████ ████ ███ ████ ██ ██████████ █████████ ██████ ██████ ████████ ██ ██████ ███ ████ ██ █████ ████ ███ ███████ ████████████
How Many Individuals in Canada Would Be Expected to Require the Testing Procedure?
Breast cancer is the most common type of cancer among female individuals in Canada. In 2022, an estimated 28,900 women and 270 men were diagnosed with breast cancer.40-42 Among people diagnosed with breast cancer, 64.8% have HR-positive, HER-negative disease. Patients with HR-positive, HER2-negative, locally advanced or metastatic breast cancer would be expected to require the testing procedure to determine their eligibility for capivasertib. The population eligible for testing is estimated to be 2,756 patients per year. Using an epidemiological approach, the sponsor estimated that █████ patients would be eligible for testing, with █████ female patients and | | male patients. However, the pharmacoeconomic reanalysis identified several issues with the approach and amended derive a base case for the budget impact analysis. Addressing these errors reduced the size of the population eligible for testing to 2,756 patients (2,722 females and 35 males). The sponsor noted that testing uptake would reach only 70% of patients within the next 3 years; not all those eligible for testing would receive it.
What Is the Expected Timing and Frequency of Testing for PIK3CA, AKT, and PTEN Alterations?
According to the clinical experts, for most patients, the testing would be conducted at the time of metastatic diagnosis. NGS testing for PIK3CA, AKT, and PTEN alterations can be done using previously collected tissue samples, in most cases. There is evidence suggesting that there is limited evolution of patients’ tumour genetics following initial testing.43 Given that PIK3CA, AKT, and PTEN alterations are considered stable, repeat testing is likely not required. It is possible that a new sample might be required in specific circumstances, such as when a previously collected tissue sample is not available or accessible (e.g., when patients move from other jurisdictions or countries).
The turnaround time for tests is 1 week to 2 weeks for liquid biopsy and up to 6 weeks for FFPE specimens.17 According to the OCA manufacturer, it could be as little as 5 days from sample to annotated results.24
What Are the Expected Human Health Resource Impacts of Testing for PIK3CA, AKT, and PTEN Alterations in Breast Cancer?
Implementation of NGS testing for PIK3CA, AKT, and PTEN alterations will have significant health system impacts, according to the clinical experts consulted for this work. First, there would be an impact on personnel, such as increased workloads for pathologists, lab technicians, bioinformaticians, and oncologists. Incorporating routine testing for PIK3CA, AKT, and PTEN alterations for all patients with a metastatic breast cancer diagnosis would also necessitate conversations between patients and their providers to ensure understanding of the nature of any test results; in some cases, it may lead a need for focused genetic counselling services.44 Increased demand for testing could necessitate the expansion of genetic counselling services to ensure that patients receive comprehensive counselling and support. There could also be an impact on currently available testing infrastructure. For example, the laboratory workforce may need additional capacity to meet the increase in demand. There are no publicly funded or private genetic testing facilities in the territories.28 In Prince Edward Island and Newfoundland and Labrador, sending samples for out-of-province testing is the current practice. Measures would need to be put in place to meet these new workloads, including tasks related to the collection, preparation, and shipping of samples to the central laboratory or testing facility. ██ ██ ███████ ███████ ███ ████████ ███████ ████ ███ ███████ █████ █████ █████ ██████████████ █████ ████ ██ ████ ██ ███████████ ███ ████████ ██ ███████ ████ ███ ████████ █████████ ██ ███ ███████ ███ █████████ ███ █████ ██████████ ████ ██████████████ ███ █████████ ██ ███ ████████
Centralized testing may have some advantages compared to in-house testing, such as standardization, timely bioinformatical updates, and scalability. However, issues related to invalid or problematic samples may not be easily resolved in a centralized facility. Lack of individual clinical patient data may also affect interpretation by pathologists in a central lab.43
What Are Some Patient-Related Considerations?
Patient-related considerations in genomic testing for breast cancer encompass informed decision-making regarding testing, understanding the possible psychological impacts of positive results, facilitating communication with patients, and other barriers. Based on the experiences of the clinical experts, financial burden is the main barrier to testing, given that the current model involves patients paying out of pocket for NGS testing. ██ ██ ███████ ██ ███ ████████ ███████ ██ ███ ███████ █████ ████ █████████ ████ ██ ████ ███████ ███ ██████ ██ █████ ███████ ██ ██ ██████████.
With centralized testing or testing at major centres, patients living in rural or remote areas may face barriers to accessing testing. However, given that in most situations, NGS testing would be done on previously collected samples (and would require shipping of samples to the centralized testing centres), the impacts on patients and their families could be low.
Cancer patient advocacy groups have identified several barriers and challenges to genomic testing in Canada across tumour groups. The identified barriers include clinician factors (e.g., lack of awareness), perceived challenges related to timely access to testing and availability of results, interjurisdictional differences in access, capacity, and funding, and social determinants (e.g., patients with lower health literacy may be less likely to advocate for testing because of lack of awareness).45
What Are the Clinical Considerations?
Clinical Utility
In advanced breast cancer, NGS is best placed to detect somatic and/or germline alterations at metastatic diagnosis to provide prognostic information, identify therapeutic targets, and monitor treatment response.21 Studies have confirmed the utility of NGS in guiding targeted next-line therapy for metastatic breast cancer. For example, in 1 study evaluating alterations in this pathway for patients with metastatic, HR-positive, HER2-negative breast cancer, NGS supported the decision for the most promising treatment option in 58.5% of patients.22
Diagnostic Accuracy
NGS testing is considered an accurate diagnostic method for detecting mutations in metastatic breast cancer. While we have not critically appraised the evidence, targeted NGS has been reported to be a reliable option to identify PIK3CA mutations using tumour FFPE samples.46
Other clinical considerations include a need for standardized reporting within and between testing facilities and issues related to data sharing, data ownership, and privacy. █████ █ ███████████ ███████ ██████ ████████ ███ ████ ████ ███ ███████ the latter remain considerations for implementation.47 However, these aspects are not specific to testing for PIK3CA, AKT, and PTEN alterations for breast cancer.
What Are the Cost Considerations?
What Is the Cost of Testing for PIK3CA, AKT, and PTEN Alterations?
According to the materials provided by the sponsor, the pricing for the OCA v3 or comparable NGS assay is $750 per test. The number of patients needed to evaluate to identify 1 patient who would be eligible for capivasertib was calculated by the sponsor as ███ ███████. Thus, the cost of NGS per eligible patient was estimated as ███ ███████ ██████████. This value was used in the pharmacoeconomic reanalysis.
At the time of authoring this report, there is inconsistent access to testing for PIK3CA, AKT, and PTEN alterations across jurisdictions. Most patients currently access testing through clinical trials, special programs, or private payment options17,28
References
1.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008;68(15):6084-6091. PubMed
2.Papa A, Pandolfi PP. The PTEN−PI3K Axis in Cancer. Biomolecules. 2019;9(4). PubMed
3.Skolariki A, D'Costa J, Little M, Lord S. Role of PI3K/Akt/mTOR pathway in mediating endocrine resistance: concept to clinic. Explor Target Antitumor Ther. 2022;3(2):172-199. PubMed
4.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncology. 2016;2(12):1565-1573. PubMed
5.Andrikopoulou A, Chatzinikolaou S, Panourgias E, et al. “The emerging role of capivasertib in breast cancer”. Breast. 2022;63:157-167. PubMed
6.Martorana F, Motta G, Pavone G, et al. AKT inhibitors: New weapons in the fight against breast cancer? Front Pharmacol. 2021;12:662232-662232. PubMed
7.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084-6091. PubMed
8.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61-70. PubMed
9.Vasan N, Toska E, Scaltriti M. Overview of the relevance of PI3K pathway in HR-positive breast cancer. Ann Oncol. 2019;30(Suppl_10):x3-x11. PubMed
10.AstraZeneca. Clinical Study Report for CAPItello-291: A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative (HR+/HER2−) Breast Cancer Following Recurrence or Progression On or After Treatment with an Aromatase Inhibitor (CAPItello-291) [CONFIDENTIAL internal manufacturer's report]. 2022 [sponsor submitted reference].
11.Hortobagyi GN, Chen D, Piccart M, et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J Clin Oncol. 2016;34(5):419-426. PubMed
12.Turner NC, Kingston B, Kilburn LS, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. The Lancet Oncology. 2020;21(10):1296-1308. PubMed
13.Li S, Shen Y, Wang M, et al. Loss of PTEN expression in breast cancer: association with clinicopathological characteristics and prognosis. Oncotarget. 2017;8(19):32043-32054. PubMed
14.Mosele F, Stefanovska B, Lusque A, et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann Oncol. 2020;31(3):377-386. PubMed
15.Mollon LE, Anderson EJ, Dean JL, et al. A Systematic Literature Review of the Prognostic and Predictive Value of PIK3CA Mutations in HR(+)/HER2(-) Metastatic Breast Cancer. Clin Breast Cancer. 2020;20(3):e232-e243. PubMed
16.Gerratana L, Davis AA, Polano M, et al. Understanding the organ tropism of metastatic breast cancer through the combination of liquid biopsy tools. Eur J Cancer. 2021;143:147-157. PubMed
17.Alpelsib (Piqray) CADTH reimbursement review. Ottawa (ON): CADTH; 2020.
18.Toppmeyer DL, Press MF. Testing considerations for phosphatidylinositol-3-kinase catalytic subunit alpha as an emerging biomarker in advanced breast cancer. Cancer Med. 2020;9(18):6463-6472. PubMed
19.Arteche-López A, Ávila-Fernández A, Romero R, et al. Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes. Sci Rep. 2021;11(1):5697. PubMed
20.Cheng C, Fei Z, Xiao P. Methods to improve the accuracy of next-generation sequencing. Front Bioeng Biotechnol. 2023;11:982111. PubMed
21.Jones TE, Zou J, Tseng GC, Roy S, Bhargava R. The Utility of Next-Generation Sequencing in Advanced Breast and Gynecologic Cancers. Am J Clin Pathol. 2021;156(3):455-460. PubMed
22.Hempel D, Ebner F, Garg A, et al. Real world data analysis of next generation sequencing and protein expression in metastatic breast cancer patients. Sci Rep. 2020;10(1):10459. PubMed
23.Yohe S, Thyagarajan B. Review of Clinical Next-Generation Sequencing. Arch Pathol Lab Med. 2017;141(11):1544-1557. PubMed
24.Thermofisher Scientific. Oncomine comprehensive assays. 2024; https://www.thermofisher.com/ca/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/oncomine-cancer-research-panel-workflow.html. Accessed 2024 May 2.
25.ThermoFisher Scientific. Oncomine comprehensive assay v3. 2024; https://www.thermofisher.com/uk/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/oncomine-cancer-research-panel-workflow/oncomine-comprehensive-assay.html. Accessed 2024 May 1.
26.ThermoFisher Scientific. Performance verification of oncomine assays and NCI-MATCH trial assay. 2024; https://www.thermofisher.com/uk/en/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/analytical-performance-oncomine-nci-match-trial-assay.html. Accessed 24 May 1.
27.Canadian Association of Pathologists. The retention and use of human biologic material. 2014; https://www.cap-acp.org/guide_retention-human-biologic-material.php. Accessed 2024 May 2.
28.Canadian Cancer Survivor Network. Genetic testing. https://survivornet.ca/learn/health-concerns-for-cancer-patients/genetic-testing/. Accessed 2024 May 2.
29.Weymann D, Dragojlovic N, Pollard S, Regier DA. Allocating healthcare resources to genomic testing in Canada: latest evidence and current challenges. J Community Genet. 2022;13(5):467-476. PubMed
30.Cancer Genetics and Genomics Laboratory-BC Cancer. Mainstreamed hereditary cancer testing. 2023; http://cancergeneticslab.ca/hereditary/mainstreamed-testing/#INDICATIONS. Accessed 2023 Nov 25.
31.Cancer Care Ontario. Comprehensive Cancer Biomarker Testing Program. In: Health O, ed2023.
32.AstraZeneca. Breast cancer specialist interview for Canadian Reimbursement Submissions of TruqapTM [conducted 15 May 2023; CONFIDENTIAL internal manufacturer's report]. 2023 (ad board). 2023 [sponsor submitted reference].
33.Cancer Care Ontario. Comprehensive cancer biomarker testing program. 2023; https://www.cancercareontario.ca/sites/ccocancercare/files/assets/ComprehensiveCancerTestingIndications.pdf. Accessed 2024 May 1.
34.Cancer Genetics and Genomics Laboratory-BC Cancer. OncoPanel. 2024; http://cancergeneticslab.ca/genes/oncopanel/. Accessed 2024 May 1.
35.van de Ven M, Jzerman M, Retel V, van Harten W, Koffijberg H. Developing a dynamic simulation model to support the nationwide implementation of whole genome sequencing in lung cancer. BMC Med Res Methodol. 2022;22(1):83. PubMed
36.Alberta Precision Laboratories. Cancer biomarker comprehensive DNA panel. 2024; https://www.albertahealthservices.ca/webapps/labservices/indexAPL.asp?id=9362&tests=&zoneid=1&details=true. Accessed 2024 May 1.
37.Wasney D. Deciphering anticancer drugs: the increasing role of pharmacogenomics and pharmacogenetics in treatment optimization. Winnipeg (MB): CancerCare Manitoba: https://www.cancercare.mb.ca/export/sites/default/For-Health-Professionals/.galleries/files/Provincial-Cancer-Care-Conference-/Deciphering-Anticancer-Drugs-Danica-Wasney-November-18-2022.pdf. Accessed 2024 May 2.
38.AmpliSeq Focus Panel – Préparation et envoi des échantillons Montreal (QC): McGill University Health Centre: https://cusm.ca/sites/default/files/docs/m-Labs/ampliseq-focus-panel-test-information-instructions-fr.pdf. Accessed 2024 May 2.
39.Test compagnon de Piqraymc. Québec City (QC): Institut national d’excellence en santé et en services sociaux 2022: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Janvier_2022/TC_Piqray_2022_01.pdf. Accessed 2024 Jun 7.
40.Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Statistics Advisory Committee; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2023 Aug 31.
41.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. PubMed
42.Canadian Cancer Society. Breast cancer statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/breast/statistics. Accessed 2023 May 10.
43.Colomer R, Miranda J, Romero-Laorden N, et al. Usefulness and real-world outcomes of next generation sequencing testing in patients with cancer: an observational study on the impact of selection based on clinical judgement. EClinicalMedicine. 2023;60:102029. PubMed
44.Borle K, Kopac N, Dragojlovic N, et al. Where is genetic medicine headed? Exploring the perspectives of Canadian genetic professionals on future trends using the Delphi method. Eur J Hum Genet. 2022;30(5):496-504. PubMed
45.Snow S, Brezden-Masley C, Carter MD, et al. Barriers and Unequal Access to Timely Molecular Testing Results: Addressing the Inequities in Cancer Care Delays across Canada. Curr Oncol. 2024;31(3):1359-1375. PubMed
46.Venetis K, Pepe F, Munzone E, et al. Analytical Performance of Next-Generation Sequencing and RT-PCR on Formalin-Fixed Paraffin-Embedded Tumor Tissues for PIK3CA Testing in HR+/HER2− Breast Cancer. Cells. 2022;11(22):3545. PubMed
47.Yip S, Christofides A, Banerji S, et al. A Canadian Guideline on the Use of Next-Generation Sequencing in Oncology. Curr Oncol. 2019;26(2):241-254. PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.