Drugs, Health Technologies, Health Systems
Reimbursement Review
Tarlatamab (Imdelltra)
Sponsor: Amgen Canada
Therapeutic area: Extensive-stage small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BICR
blinded independent central review
BiTE
bispecific T-cell engager
BOI
burden of illness
CAS
Cancer Analysis System
CAV
cyclophosphamide, doxorubicin, and vincristine
CDA-AMC
Canada's Drug Agency
CFI
chemotherapy-free interval
CI
confidence interval
CNS
central nervous system
CR
complete response
CRS
cytokine release syndrome
DCO
data cut-off date
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module
ESS
effective sample size
ES-SCLC
extensive-stage small cell lung cancer
ECOG PS
Eastern Cooperative Oncology Group Performance Status
GRADE
Grading of Recommendations Assessment, Developments Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ICANS
immune effector cell-associated neurotoxicity syndrome
IPD
individual patient data
ITC
indirect treatment comparison
LOT
line of treatment
LS-SCLC
limited-stage small cell lung cancer
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
NE
not estimable
OOS
Oncology Outcomes Study
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PR
partial response
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
RWE
real-world evidence
SAE
serious adverse event
SCLC
small cell lung cancer
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TTD
time to treatment discontinuation or death
TTNTD
time to next treatment discontinuation or death
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Drug product: Tarlatamab Strength: 1 mg or 10 mg Dosage: Initial dose of 1 mg on day 1, followed by 10 mg on day 8 and day 15, and q.2.w. thereafter Route of administration: IV infusion |
Sponsor | Amgen Canada |
Indication | Tarlatamab is indicated for the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy. |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c; Project Orbis Type B |
NOC date | September 11, 2024 |
Recommended dose | Initial dose of 1 mg IV on day 1 followed by 10 mg on day 8 and day 15, and q.2.w. thereafter |
ES-SCLC = extensive-stage small cell lung cancer; NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions; q.2.w. = every 2 weeks.
Introduction
Lung cancer is one of the most frequently diagnosed cancers and the leading cause of cancer-related death in Canada.1 Small cell lung cancer (SCLC) is a highly aggressive and rapidly progressive lung cancer subtype, accounting for approximately 10% to 15% of all lung cancer cases, and predominantly occurs in smokers.2 Between 2012 to 2016 in Canada, the age-standardized incidence rate of SCLC was estimated to be 6.9 per 100,000 patients for both sexes combined and 7.3 patients and 6.6 patients per 100,000 patients for males and females, respectively.3 SCLC often presents with a large hilar mass with bulky mediastinal adenopathy and is characterized by a rapid doubling time, high growth fraction, propensity to metastasize, and transient responses to conventional chemotherapy and radiotherapy.4 SCLC has a substantial impact on patients’ physical, psychological, and social well-being.5,6 Common symptoms of SCLC include persistent cough, chest pain that gets worse when coughing, laughing, or taking a deep breath, hemoptysis, and hoarseness and/or wheezing.7 Some patients may also experience a loss of appetite, unintended weight loss, fatigue, and recurrent episodes of lung infections such as pneumonia or bronchitis.7 Additionally, patient health is further compromised by toxicities during chemotherapy and side effects with current therapies (e.g., neutropenia, infection, mucositis, myelosuppression, alopecia, nausea, vomiting, fatigue, sore mouth, or diarrhea). Patient’s health-related quality of life (HRQoL) is also significantly impaired due to anxiety, depression, and distress associated with the disease and treatments.8
SCLC can be classified as limited-stage SCLC (LS-SCLC) and extensive-stage SCLC (ES-SCLC).9 At the extensive stage, tumours spread beyond the boundaries of limited disease and include distant metastases, malignant pericardial or pleural effusions, and contralateral supraclavicular and contralateral hilar involvement.4,10 At the time of diagnosis, approximately 70% of patients present with extensive-stage disease. Prognosis for these patients is poor with a median overall survival (OS) of 12 months to 13 months from the time of diagnosis,11-13 and 5-year survival rates ranging from 1% to 10%.2,14,15
In Canada, etoposide with platinum-based chemotherapy in combination with the PD-L1 inhibitors, either durvalumab or atezolizumab are the typical first-line treatment options.10,16,17 Second-line treatment options in Canada include topotecan (only indicated for platinum-sensitive SCLC after failure of 1L chemotherapy), cyclophosphamide, doxorubicin, and vincristine (CAV), or rechallenge with etoposide and platinum-based chemotherapy (only if progression occurs after an interval of approximately ≥ 3 months from the last dose of 1L chemotherapy).18,19 The prognosis for relapsed SCLC in the 2L setting is very poor, with response rates to current treatments less than 25%, short-lived disease control, as well as short progression-free survival (PFS) (4 months to 6 months) and OS (< 9 months).17,20 Currently in the third-line setting, there are no Health Canada–approved treatments. Available options in this setting include rechallenging with the initial regimen and other single-agent or combination chemotherapy regimens, enrolling in clinical trials, or best supportive care.1,18,21
Tarlatamab is a novel bispecific delta-like ligand–directed CD3 T-cell engager that binds to DL33 expressed on the surface of cells, including tumour cells, and CD3 expressed on the surface of T cells. It triggers T-cell activation, production of inflammatory cytokines, and lysis of delta-like ligand 3–expressing cells.22 The recommended dose of tarlatamab is an initial dose of 1 mg on day 1, followed by 10 mg on day 8 and day 15, and every 2 weeks thereafter.22 Tarlatamab has been issued a Notice of Compliance with Conditions by Health Canada on September 11, 2024, and the approved indication is for the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.22 The sponsor’s reimbursement request aligns with the Health Canada indication.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tarlatamab (lyophilized powder for solution for IV infusion, 1 mg and 10 mg per vial) in the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
A joint patient group submission from Lung Cancer Canada, Lung Health Foundation, and the Canadian Cancer Survivor Network was received for this review. Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research, and advocacy. The Lung Health Foundation is a registered charity that assists and empowers people living with or caring for others with lung disease. The Canadian Cancer Survivor Network is a national network of patients, families, survivors, and community partners, who take collaborative action to promote the best standards of care for patients with cancer. Information provided for this submission was collected through virtual interviews with 3 patients and their caregivers between July 2024 and August 2024. Two patients were living in Canada, and 1 was in the US. All patients had ES-SCLC and had experience with tarlatamab which was used as third line or later (third line or further) therapy.
The patient groups emphasized that SCLC is an aggressive type of cancer with a high symptom burden, rapid disease progression, and poor health outcomes. Current treatments for ES-SCLC (chemotherapy and immunotherapy) are associated with limited duration of response, harsh side effects, increased dependence on caregivers in daily activities, and an impact on the patients’ functionality. As such, the patient groups highlighted an urgent need for a new treatment beyond the 1L setting which should be effective in controlling the disease and symptoms, minimizing side effects of the treatments, allowing patients to maintain a meaningful quality of life, minimizing caregiver burden, delaying disease progression, and offering patients an additional treatment option upon disease progression or when other treatments are exhausted. All 3 patients who received tarlatamab indicated that this drug was effective in treating the disease and improving their quality of life, compared to their previous therapies. The patients also reported significant side effects when receiving the first dose, which improved over time.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review emphasized that once a patient with ES-SCLC becomes platinum-resistant, systemic therapy options become very limited with poorer response rates and reduced OS. The clinical experts indicated that the most important goals of treatment for patients with ES-SCLC are to prolong life, delay disease progression, reduce the severity of cancer-related symptoms, and improve patients’ HRQoL, as well as balancing the toxicities of therapy. There are currently no Health Canada–approved treatments for ES-SCLC in the 3L+ setting. As such, the clinical experts identified the need for new safe and effective treatment options in patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy, particularly those who are platinum refractory. Relevant for all lines of therapy for SCLC, the clinical experts also noted that there is a need for treatments that can effectively control central nervous system (CNS) metastasis, given the increased risk of CNS metastasis requiring whole-brain radiotherapy in patients with ES-SCLC.
In line with the Health Canada indication, the clinical experts indicated that tarlatamab would become the preferred treatment option for patients who progress on or after 2 lines of therapy (i.e., 3L+). Both clinical experts noted that tarlatamab could be an option for patients with platinum-refractory disease, which would be 2L or later; however, they noted that this is not within the scope of this review and the use of tarlatamab in the 2L setting is not an approved Health Canada indication. The clinical experts also highlighted that because only a limited number of patients are likely able to receive treatment in the 3L setting due to severe functional decline or disease progression, tarlatamab is not expected to cause a major shift in the current treatment paradigm.
Per the Health Canada indication, patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy would be considered for treatment with tarlatamab. The clinical experts noted that there is currently no biomarker to determine the patients who will respond best to tarlatamab and therefore suggested that tarlatamab should be offered to all patients who are eligible to receive this treatment. However, based on the clinical experts’ experience, patients may be more likely to benefit from tarlatamab if they are younger, have fewer comorbidities or lower burden of disease, previously responded to treatment with etoposide and a platinum-based agent, or had a longer duration of response to previous treatments. Meanwhile, the clinical experts acknowledged that the challenges of administering tarlatamab (e.g., hospital admission in a specialized centre is required) will limit the uptake of this treatment in practice. In addition, concerns about the adverse events (AEs) related to the use of tarlatamab (such as higher risk of cytokine release syndrome [CRS], particularly in patients with cardiac comorbidities) may also limit clinicians from administering this drug to patients who do not meet the eligibility criteria of the DeLLphi-301 trial.
The clinical experts noted that in general, outcomes used in clinical practice align with those seen in clinical trials of ES-SCLC, which include OS, PFS, CNS metastasis–free survival, HRQoL, and symptom relief. Both clinical experts agreed that response to treatment should be assessed every 2 months to 3 months with imaging examinations to determine if response or stable disease is observed. The clinical experts stated that in the 3L setting, any improvement of 2 months or greater in OS or PFS over the standard treatment options would be clinically meaningful. The experts also highlighted that there is likely variation in how clinicians would measure response or success, and clinicians may also include stability or improvement of symptoms in their assessment.
The clinical experts noted that treatment with tarlatamab will be discontinued if there is evidence of disease progression, intolerable or unmanageable toxicities including grade 3 or higher CRS or immune effector cell-associated neurotoxicity syndrome (ICANS), or deterioration of quality of life, as well as patient preferences. The clinical experts also noted that if clinical benefits from treatment with tarlatamab are maintained despite evidence of radiographic progression, patients may be allowed to continue this therapy. Decisions regarding whether or not patients should continue treatment with tarlatamab beyond radiographic progression are at the discretion of the treating physician.
The clinical experts indicated that patients with ES-SCLC are under the care of medical oncologists and/or oncologists with experience administering and managing systemic therapy. In line with the DeLLphi-301 trial, the clinical experts noted that patients receiving treatment with tarlatamab should be hospitalized for 24 hours during cycle 1 day 1 and cycle 1 day 8 due to the increased risk of CRS and ICANS. Additionally, treatment centres must have immediate access to an onsite intensive care unit capable of managing these potentially fatal AEs. However, the clinical experts noted that, according to clinical trial guidelines, while patients should initiate and receive tarlatamab in an inpatient setting, transition to an outpatient setting would be reasonable after 3 to 4 treatments. Furthermore, the clinical experts noted that supervision of later treatment cycles may be conducted by other practitioners in the treatment team, such as nurse practitioners or general practitioners in oncology.
Clinician Group Input
Two clinician groups provided input for the review of tarlatamab: the Lung Cancer Canada Medical Advisory Committee and the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee. A total of 29 clinicians from the Lung Cancer Canada Medical Advisory Committee and 5 clinicians from the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee provided input for this submission.
In general, the clinician group inputs were consistent with the input provided by the clinical experts consulted for this review. The clinician groups indicated that there are limited treatment options available for patients with ES-SCLC in the 2L or later-line setting, and they are usually associated with suboptimal treatment effect and significant toxicities. As such, the clinician groups highlighted a pressing need for 3L+ treatment options that can prolong life, maintain quality of life, and minimize toxicities. Based on the results from clinical trials, the clinician groups suggested that tarlatamab is best suited for patients with progressive SCLC, who have exhausted 2 or more lines of therapy, who have an adequate performance status, who cannot tolerate further cytotoxic therapy, and who can manage potential side effect from tarlatamab. The clinician groups also highlighted that tarlatamab has a unique mechanism of action (i.e., a bispecific T-cell engager [BiTE] agent), and speculated that tarlatamab may offer a viable treatment option for patients who have only had 1 prior treatment and are not candidates for further chemotherapy.
When assessing treatment response to tarlatamab, the clinician groups indicated that both clinical evaluations and radiologic assessments are essential, and treatment should be discontinued if there is evidence of disease progression and/or significant toxicity. Additionally, the clinician groups indicated that tarlatamab should be administered in specialized centres with an inpatient setting to handle any potential toxicities from the treatment.
Drug Program Input
Input was obtained from the drug programs that participate in reimbursement review process of Canada’s Drug Agency (CDA-AMC). Please refer to Table 4 for further information. The following were identified as key factors that could potentially impact the implementation of tarlatamab:
considerations for initiation of therapy
considerations for discontinuation of therapy
generalizability
systems and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, phase II, open-label, single-arm trial, DeLLphi-301 (N = 222), was included in this review. The purpose of this study was to evaluate the efficacy and safety of tarlatamab (10 mg) in adult patients with histologically or cytologically confirmed relapsed or refractory SCLC who progressed or recurred following 1 platinum-based regimen and at least 1 other prior line of treatment (LOT). Patients must have had measurable lesions as defined per the Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1) within 21 days before the first dose of tarlatamab and must have had adequate organ function. Patients were excluded if they had symptomatic or untreated brain metastases. The primary efficacy end point of this study was objective response rate (ORR), with secondary end points of OS and PFS, and exploratory end points of HRQoL and safety. At baseline, of the 99 patients in the 10 mg group, most were men (71.1%) and the median age was 64 years (range, 35 years to 82 years). Overall, patients had a median of 2 (range, 1 to 6) prior lines of therapy, including 73.7% of patients with prior pr PD-1 or PD-L1 inhibitors. Time to progression after 1L platinum therapy was less than 90 days for 27 patients (27.3%), between 90 days and 179 days for 22 patients (22.2%), longer than 180 days for 20 patients (20.2%), and unknown for 30 patients (30.3%). Most patients had metastatic disease (98.0%), no brain metastases (77.8%) or liver metastases (61.6%), and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 1 (73.7%).
The primary Clinical Study Report and 3 associated addenda were provided for the DeLLphi-301 trial. The primary data cut-off (DCO) was June 27, 2023, and the latest DCO was May 16, 2024.
Efficacy Results
Primary and secondary analyses on the efficacy outcomes were conducted in the investigator full analysis set and blinded independent central review (BICR) full analysis set.
Overall Survival
At the DCO of June 27, 2023, in the tarlatamab 10 mg group (part 1 and part 2), median OS was 14.3 months (95% confidence interval [CI], 10.8 months to not estimable [NE]), with a median follow-up time of 10.6 months (95% CI, 9.2 months to 11.5 months). The 12-month OS rate was 57.7% (95% CI, 45.0% to 68.4%).
At the DCO of May 16, 2024, the median OS was 15.2 months (95% CI, 10.8 months to NE), with a median follow-up time of 20.7 months (95% CI, 19.6 months to 21.7 months). The 12-month OS rate was 56.6% (95% CI, 45.6% to 66.3%).
Progression-Free Survival
At the DCO of June 27, 2023, in the 10 mg group (part 1 and part 2), median PFS by BICR was 4.3 months (95% CI, 3.0 months to 5.6 months) with a median follow-up time of 9.7 months (95% CI, 8.3 months to 10.9 months).
At the DCO of October 2, 2023, median PFS by BICR was 4.3 months (95% CI, 3.0 months to 5.6 months) with a median follow-up time of 13.6 months (95% CI, 11.0 months to 13.8 months).
Health-Related Quality of Life
Analyses of HRQoL end points were conducted for part 1 and part 2. As of June 27, 2023, the results for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module (EORTC QLQ-LC13) showed that the least squares mean changes from baseline up to cycle 12 were −4.5 (95% CI, −11.4 to 2.4) for cough, −6.5 (95% CI, −10.9 to −2.1) for chest pain, and −10.2 (95% CI, −16.4 to −4.0) for dyspnea composite score in the 10 mg group. The least squares mean change from baseline up to cycle 12 for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) subscales of global health status/quality of life was 11.5 (95% CI, 6.6 to 16.4) in the tarlatamab 10 mg group.
Objective Response Rates
As of June 27, 2023, the ORR in the BICR full analysis set for part 1 and part 2 was 41.4% (97.5% CI, 30.3% to 53.2%) for patients in the 10 mg group.
As of the DCO of January 12, 2024, the ORR in the BICR full analysis set for part 1 and part 2 was 40.4% (95% CI, 30.7% to 50.7%) for patients in the 10 mg group.
The vast majority of patients achieved partial response (PR) instead of complete response (CR) in the ORR assessment.
Harms Results
Safety analyses were conducted in the safety analysis set, which included all patients who received at least 1 dose of tarlatamab.
As of the DCO June 27, 2023, 99% of patients experienced treatment-emergent AEs (TEAEs) at the tarlatamab 10 mg dose. The most commonly reported all-grade TEAEs (≥ 20% of patients) in the 10 mg group were CRS (51.5%), decreased appetite (30.3%), pyrexia (39.4%), constipation (31.3%), anemia (28.3%), dysgeusia (24.2%), fatigue (23.2%), and asthenia (21.2%). Grade 3 or higher AEs were reported for 60 patients (60.6%). The most frequently reported grade 3 or higher TEAEs (≥ 5% of patients) in the 10 mg group were anemia, lymphopenia, lymphocyte count decreased, hyponatremia, fatigue, and asthenia. Of note, CRS was the most frequently reported AE in the tarlatamab 10 mg group, but no patients in this group had grade 3 or higher CRS events.
Serious AEs (SAEs) were reported for 58 patients (58.6%) in the 10 mg dose group (part 1 and part 2) and 15 patients (44.1%) in the modified safety monitoring 10 mg group as of the DCO of June 27, 2023. The most frequently reported (≥ 2 patients) SAEs by preferred term in the 10 mg group (part 1 and part 2) were CRS (26.3%), pyrexia (6.1%), pneumonia (4.0%), device-related infection (3.0%), respiratory tract infection (3.0%), ICANS (2.0%), and hyponatremia (4.0%). In the part 3 modified safety monitoring 10 mg target dose group, the most frequently reported (≥ 2 patients) SAEs by preferred term were CRS (14.7%) and ICANS and respiratory failure (5.9% each). The incidence of SAEs reported as of October 2, 2023, was similar to the DCO of June 27, 2023.
AEs leading to discontinuation of tarlatamab were reported for 7 patients (7.1%) in the 10 mg group and 4 patients (11.8%) in the 10 mg modified safety monitoring group, when assessed at the DCO of October 2, 2023.
Fatal AEs were reported for 2 patients (2.0%) in the 10 mg group in part 1 and part 2 and 3 patients (8.8%) in the part 3 modified safety monitoring 10 mg group. None of the deaths were considered by the investigator to be related to tarlatamab.
In the 10 mg group (part 1, part 2, and part 3), CRS occurred in 70 patients (52.6%). Note that 1 patient in the 10 mg modified safety monitoring group had a grade 3 or higher CRS event. AE data are pooled for the 10 mg group and the 10 mg modified safety monitoring group to summarize the occurrence of any grade 3 or higher events in either group. In the 10 mg group (part 1 and part 2), 9 patients (9.1%) had ICANS events and none of the patients had grade 3 or higher events.
Critical Appraisal
The DeLLphi-301 study is an ongoing, phase II, open-label, single-arm trial evaluating the efficacy and safety of tarlatamab in patients with ES-SCLC. The potential influence of selection bias is difficult to ascertain in a single-arm trial. One of the key limitations of the DeLLphi-301 trial was the absence of a comparator group. In a single-arm trial, treatment effect of the study drug cannot be directly assessed because the trial design is not able to distinguish what proportion of the estimated treatment response can be attributed to the study drug, placebo effects, a patient’s natural history, or other prognostic factors. As a single-arm trial, patients in the DeLLphi-301 trial were aware of the intervention that they were taking, which potentially increased the risk of detection bias and performance bias and limited the interpretability of the subjective study outcomes such as patient-reported outcomes including HRQoL and AEs. The primary end point of the DeLLphi-301 trial was ORR which is directly attributable to the antitumour activity of tarlatamab despite the single-arm design. Clinically meaningful outcomes for this review included OS and PFS; however, time-to-event end points cannot be adequately assessed in a single-arm trial, thus, the effect of tarlatamab on these end points can only be considered as exploratory and supportive. Despite OS and PFS results that were considered clinically meaningful by the clinical experts consulted for this review, because of the combination of the single-arm design, secondary nature of the outcomes, and short follow-up duration, the results for these end points should only be considered supportive of the overall antitumour effect of tarlatamab. In this trial, patients’ HRQoL was assessed using both disease-specific and generic questionnaires. However, due to the large amount of missing data, the effect of tarlatamab on patients’ quality of life remains uncertain.
Based on the feedback from the clinical experts consulted for this review, the eligibility criteria and baseline characteristics of patients enrolled in the DeLLphi-301 trial generally reflected a patient population in Canadian clinical practice that would receive treatment with tarlatamab for ES-SCLC, although in practice, this treatment may be used in a broader population than the DeLLphi-301 trial. The experts also confirmed that the use of concomitant therapies and subsequent anticancer therapies were generally consistent with the Canadian practice. The outcome measures in the DeLLphi-301 trial are clinically relevant in clinical trials of ES-SCLC.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.23,24
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed pivotal single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The target of the certainty of evidence assessment was the presence of a clinically important improvement in survival (PFS and OS) and HRQoL, which were considered the most important outcomes to treatment by the clinical experts consulted for this review, and the clinician group and patient group inputs. According to the clinical experts, clinically importance thresholds for the outcomes of OS and PFS were a benefit of at least 2 months over current standard of care for OS and PFS. Additionally, response to treatment (ORR) was included in the certainty of evidence assessment based on the potential translation to long-term survival outcomes.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for tarlatamab from the DeLLphi-301 trial, for the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS
PFS
HRQoL measured with EORTC QLQ-C30 and EORTC QLQ-LC13
ORR
risk of SAE
risk of CRS
risk of ICANS.
Table 2: Summary of Findings for the Efficacy and Safety of Tarlatamab for Patients With ES-SCLC (No Comparator)
Outcome and follow up | Patients (studies), N | Effect | Certaintya | What happens |
|---|---|---|---|---|
Survival | ||||
OS (months) Median follow up (as of June 27, 2023): 10.6 months | 99 (1 single-arm trial) | Median = 14.3 months (95% CI, 10.8 months to NE) 12-month rate = 57.7% (95% CI, 45.0% to 68.4%) | Very lowb | The evidence is very uncertain about the effect of tarlatamab on OS vs. any comparator |
OS (months) Median follow up (as of May 16, 2024): 20.7 months | 99 (1 single-arm trial) | Median = 15.2 months (95% CI, 10.8 months to NE) 12-month rate = 56.6% (95% CI, 45.6% to 66.3%) | Very lowb | The evidence is very uncertain about the effect of tarlatamab on OS vs. any comparator |
PFS (months) Median follow up (as of June 27, 2023): 9.7 months | 99 (1 single-arm trial) | Median = 4.3 months (95% CI, 3.0 months to 5.6 months) 12-month rate = 25.7% (95% CI, 16.7% to 35.8%) | Very lowc | The evidence is very uncertain about the effect of tarlatamab on PFS vs. any comparator |
PFS (months) Median follow up (as of October 2, 2023): 13.6 months | 99 (1 single-arm trial) | Median = 4.3 months (95% CI, 3.0 months to 5.6 months) 12-month rate = 25.2% (95% CI, 16.6% to 34.7%) | Very lowc | The evidence is very uncertain about the effect of tarlatamab on PFS vs. any comparator |
HRQoL | ||||
EORTC QLQ-C30 score, mean CFB (95% CI) Median follow up (as of June 27, 2023): 10.6 months | 14 (1 single-arm trial) | Global health status/QoL: Mean CFB to cycle 12 = 11.50 (95% CI, 6.63 to 16.37) | Very lowc,d | The evidence is very uncertain about the effect of tarlatamab on global health status/QoL score in EORTC QLQ-C30 vs. any comparator |
EORTC QLQ-LC13 scores, mean CFB (95% CI) Median follow up (as of June 27, 2023): 10.6 months | 14 (1 single-arm trial) | Dyspnea composite score: Mean CFB to cycle 12 = −10.21 (95% CI, −16.41 to −4.02) Cough: Mean CFB to cycle 12 = −4.48 (95% CI, −11.35 to 2.39) Chest pain: Mean CFB to cycle 12 = −6.50 (95% CI, −10.94 to −2.06) | Very lowc,d | The evidence is very uncertain about the effect of tarlatamab on dyspnea, cough, and chest pain scores in EORTC QLQ-LC13 vs. any comparator |
Response to treatment | ||||
ORR (CR + PR) (97.5% CI) Median follow up (as of June 27, 2023): NR | 99 (1 single-arm trial) | N (%): 41 (41.4%) (97.5% CI, to 30.3 to 53.2) | Very lowe | The evidence is very uncertain about the effect of tarlatamab on ORR vs. any comparator |
ORR (CR + PR) (95% CI) Median follow up (as of January 12, 2024): NR | 99 (1 single-arm trial) | N (%): 40 (40.4%) (95% CI, 30.7 to 50.7) | Very lowe | The evidence is very uncertain about the effect of tarlatamab on ORR vs. any comparator |
Harms | ||||
Patients with ≥ 1 SAE Median follow up (as of October 2, 2023): NR | 99 (1 single-arm trial) | 586 per 1,000 | Very lowf | The evidence is very uncertain about the effect of tarlatamab on the risk of SAE vs. any comparator |
Patients with CRS Median follow up (as of October 2, 2023): NR | 99 (1 single-arm trial) | 515 per 1,000 | Very lowf | The evidence is very uncertain about the effect of tarlatamab on the risk of CRS vs. any comparator |
Patients with ICANS Median follow up (as of October 2, 2023): NR | 99 (1 single-arm trial) | 91 per 1,000 | Very lowg | The evidence is very uncertain about the effect of tarlatamab on the risk of ICANS vs. any comparator |
CFB = change from baseline; CI = confidence interval; CR = complete response; CRS = cytokine release syndrome; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; ES-SCLC = extensive-stage small cell lung cancer; HRQoL = health-related quality of life; ICANS = immune effector cell-associated neurotoxicity syndrome; NE = not estimable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; QoL = quality of life; RECIST = Response Evaluation Criteria in Solid Tumours; SAE = serious adverse event; vs. = versus.
Note: PFS and ORR were assessed with RECIST Version 1.1 by blinded independent central review; All serious concerns with study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias are documented in the table footnotes.
aIn absence of a comparator arm, conclusions about efficacy relative to any comparator cannot be drawn and certainty of evidence started at very low.
bIn the DeLLphi-301 trial, statistical testing for this outcome was not adjusted for multiplicity. However, despite the study limitations resulting in the certainty of evidence starting as “very low,” the effect size of improvement in OS (median OS was 14 months to 15 months) was considered large in the third-line setting by the clinical experts consulted for this review.
cIn the DeLLphi-301 trial, statistical testing for this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
dRated down 2 levels for very serious study limitations due to the very low completion rate for HRQoL questionnaires (data were available for only 14% of the study population at cycle 12). Rated down 1 level because statistical testing for this outcome was not adjusted for multiplicity in the DeLLphi-301 trial and should be considered as supportive evidence.
eDespite the study limitations resulting in the certainty of evidence starting as “very low,” the effect size of change in response rate was considered large (compared to ORR of approximately 15% for conventional treatments) by the clinical experts consulted for this review. In the DeLLphi-301 trial, this was a primary efficacy outcome. However, the outcome could be rated down 1 level for serious indirectness as a surrogate outcome of ORR was used as the primary outcome in the place of OS and PFS, and the clinical experts consulted for this review noted that ORR is not a clinically meaningful outcome in clinical trials of SCLC, unless it is interpreted with other efficacy outcomes.
fDespite the relatively high incidence rate for SAEs and CRS (in > 50% of patients), conclusions about the effect of tarlatamab relative to any comparator cannot be drawn in absence of a comparator arm, and certainty of evidence started at very low.
gRated down 1 level for serious imprecision, due to the low event rate in a small patient population.
Sources: Primary Clinical Study Report for DeLLphi-301,25 Addendum 01 to the Primary Clinical Study Report for DeLLphi-301,26 Addendum 02 to the Primary Clinical Study Report for DeLLphi-301,27 and Addendum 03 to the Primary Clinical Study Report for DeLLphi-301.28 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No relevant long-term extension studies were submitted by the sponsor for this review.
Indirect Comparisons
Description of Studies
In the absence of head-to-head evidence comparing tarlatamab to other relevant therapies used to manage ES-SCLC, the sponsor submitted 1 analysis using propensity score weighting (the DeLLphi-301 study versus the Flatiron study)29 and 2 matching-adjusted indirect comparisons (MAICs; the DeLLphi-301 study versus the Cancer Analysis System [CAS] study and the DeLLphi-301 study versus the Oncology Outcomes Study [OOS])30 which indirectly compared OS and PFS of tarlatamab with currently available treatments in patients with ES-SCLC in the 3L+ setting. The DeLLphi-301 trial versus the Flatiron study analysis assessed additional outcomes of interest, including ORR and time to treatment discontinuation or death (TTD). The comparator studies (Flatiron, CAS, and OOS) were conducted based on registry data collected by the sponsor.
Efficacy Results
In the DeLLphi-301 study versus the Flatiron study, 97 patients were included in the tarlatamab cohort. The effective sample size (ESS) for the comparator therapies cohort was 62 (53.4% of the original sample size) after weighting. The ESS for the tarlatamab cohort after match adjustment was 27.05 (27.9% of the original sample size) for the DeLLphi-301 study versus the CAS study analysis and 59.65 (61.5% of the original sample size) for the DeLLphi-301 study versus the OOS analysis. The comparator therapies cohort in the DeLLphi-301 study versus the CAS study analysis and the DeLLphi-301 study versus the OOS analysis consisted of 540 and 71 patients, respectively.
Overall Survival
In the DeLLphi-301 study versus the Flatiron study, the median OS was 14.3 months (95% CI, 10.5 months to NE) for the tarlatamab cohort versus 6.6 months (95% CI, 4.8 months to 10.2 months) for the comparator therapies cohort after weighting. The hazard ratio (HR) of OS (with postprogression adjustment) between the tarlatamab cohort and the weighted comparator therapies cohort was 0.47 (95% CI, 0.30 to 0.79; P = 0.003), in favour of tarlatamab. Results were consistent with the OS analysis that was not adjusted for postprogression use of tarlatamab. Results of the sensitivity analyses were consistent with the primary analysis.
Following match adjustment, the HR of OS in the base case was in favour of tarlatamab over comparator therapies for the DeLLphi-301 study versus the CAS study MAIC (HR = █████; 95% CI, █████ to █████; P = 0.0001) and the DeLLphi-301 study versus the OOS MAIC (HR = 0.291; 95% CI, 0.184 to 0.459; P = 0.0000). Results of the scenario analyses were consistent with the analysis.
Progression-Free Survival
In the DeLLphi-301 study versus the Flatiron study, the median PFS was 4.9 months (95% CI, 2.9 months to 6.7 months) for the tarlatamab cohort versus 3.0 months (95% CI, 1.9 months to 3.9 months) for the comparator therapies cohort after weighting, with a HR of 0.61 (95% CI, 0.41 to 0.94; P = 0.021), in favour of tarlatamab. Results of the sensitivity analyses were consistent with the primary analysis.
Following match adjustment, the HR of PFS in the base case was in favour of tarlatamab over comparator therapies for the DeLLphi-301 study versus the CAS study MAIC (HR = █████; 95% CI, █████ to █████; P = 0.0000) and the DeLLphi-301 study versus the OOS MAIC (HR = 0.326; 95% CI, 0.215 to 0.493; P = 0.0000). Results of the scenario analyses were consistent with the base-case analysis.
Objective Response Rate
In the DeLLphi-301 study versus the Flatiron study, the ORR was 40% for the tarlatamab cohort versus 23% for the comparator therapies cohort after weighting, with an odds ratio of 2.29 (95% CI, 1.05 to 5.58; P = 0.05) in favour of tarlatamab. Results of the sensitivity analyses were in general consistent with the primary analysis, except for the sensitivity analysis where prognostic factors of low importance were adjusted for, in addition to factors of high and medium importance, and results did not favour either intervention.
This outcome was not assessed in the DeLLphi-301 study versus the CAS study and the DeLLphi-301 study versus the OOS analyses.
Time to Treatment Discontinuation or Death
In the DeLLphi-301 study versus the Flatiron study, the median time to TTD was 3.65 months (95% CI, 2.37 months to 5.36 months) for the tarlatamab cohort versus 2.33 months (95% CI, 1.41 months to 3.25 months) for the comparator therapies cohort after weighting. The HR of TTD (with postprogression adjustment) between the tarlatamab cohort and the weighted comparator therapies cohort was 0.60 (95% CI, 0.42 to 0.90; P = 0.010), in favour of tarlatamab. Results were consistent with the TTD analysis that was not adjusted for postprogression use of tarlatamab.
This outcome was not assessed in the DeLLphi-301 study versus the CAS study and the DeLLphi-301 study versus the OOS analyses.
Harms Results
Harms outcomes were not assessed in the studies.
Critical Appraisal
There is a risk of selection bias given that the comparator studies (Flatiron, CAS and OOS studies) selected for inclusion into the analysis were identified in the absence of prespecified methods. Other limitations included the heterogeneity between included studies regarding patient population, specifically with respect to life expectancy at baseline (in all 3 analyses), definition of PFS, and temporal discordance in data collection period (during which a major change in treatment pattern occurred), as well as inability to adjust for potential prognostic factors (e.g., number of prior lines of treatment, previous use of PD-1 or PD-L1 inhibitor). In addition, there remained an imbalance in several baseline patient characteristics that were identified as potential prognostic factors following weighting in the DeLLphi-301 study versus the Flatiron study analysis. These limitations likely introduce bias due to confounding in the relative treatment effect estimates. A sizable reduction in ESS after the weighting process was observed in all studies, suggesting that there was a poor population overlap between studies and that the results may be heavily influenced by a subset of the sample in the trials who may not be representative of the full sample. HRQoL and harms outcomes, which are important to patients and clinicians, were not assessed in the analyses, representing a gap in evidence.
Studies Addressing Gaps in the Evidence From the Systematic Review
No relevant studies addressing gaps in the evidence from the systematic review were submitted by the sponsor.
Conclusions
One phase II, single-arm, open-label trial (DeLLphi-301) provided evidence for the efficacy and safety of tarlatamab in adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
Clinicians and patients highlighted the need for new treatment options that prolong life (survival being considered the most important outcomes), delay disease progression, reduce the severity of symptoms, have a reduced AE profile, and improve patients’ HRQoL. Although interpretation of the OS and PFS results evaluated in the study were limited by the single-arm design and immaturity from limited follow up, precluding the ability to attribute the study results to treatment with tarlatamab, the clinical experts consulted for this review considered the median OS and PFS, as well as the 12-month OS and PFS rates clinically meaningful. Results from the DeLLphi-301 trial also suggested that some patients (41.4%) will experience a response to tarlatamab, although the evidence is still uncertain. Although HRQoL was an important outcome for patients with ES-SCLC, due to the noncomparative design and high attrition rates in the DeLLphi-301 trial, the effect of tarlatamab on HRQoL remains uncertain. In terms of harms, although nearly all patients reported an AE, the safety profile of tarlatamab was consistent with its mechanism of action, including a large proportion of patients who experienced CRS. Despite the high frequency of many AEs, most of the reported AEs are manageable according to the clinical experts consulted for this review. However, CRS and ICANS in any grades were considered clinically significant AEs for tarlatamab, and remain a concern for clinicians, given that these are new AEs for thoracic oncology.
There is a lack of direct comparative evidence between tarlatamab and relevant treatments for patients with ES-SCLC in the 3L setting. Results from the sponsor-submitted indirect treatment comparisons (ITCs) suggested that tarlatamab was associated with improved OS, PFS, and ORR compared with the other active treatments used in clinical practice, such as platinum-based therapy, PD-L1–based therapy, topotecan, CAV, or irinotecan. However, the evidence was very uncertain about the effects of tarlatamab on any outcomes versus any comparator, and the ability to draw firm conclusions on the magnitude of clinical benefit of tarlatamab was hindered by the limitations in the evidence, including a risk of selection bias in study inclusion, a sizable reduction in ESS, the heterogeneity in patient population and study design, and inadequate or lack of adjustment for potential prognostic factors that may introduce unmeasurable confounding in the relative treatment effect estimates.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tarlatamab (lyophilized powder for solution for IV infusion, 1 mg and 10 mg per vial) in the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Lung cancer is one of the most commonly diagnosed cancers and the leading cause of cancer-related death in Canada.1 In 2024, it is estimated that more than 32,100 people living in Canada will be diagnosed with lung and bronchus cancer.31 The age-standardized incidence rate of lung and bronchus cancer in Canada (excluding Quebec) was 58.9 per 100,000 persons in 2023 and is predicted to be 63.8 per 100,000 persons in 2024.3 SCLC is a highly aggressive and rapidly progressive lung cancer subtype with neuroendocrine differentiation, accounting for 10% to 15% of all lung cancer cases and predominantly occurs in smokers.2 Between 2012 to 2016 in Canada, the age-standardized incidence rate of SCLC was estimated to be 6.9 per 100,000 persons for both sexes combined and 7.3 and 6.6 per 100,000 persons for males and females, respectively.3 SCLC often presents with a large hilar mass with bulky mediastinal adenopathy and is characterized by a rapid doubling time, high growth fraction, propensity to metastasize, and transient responses to conventional chemotherapy and radiotherapy.4 These factors contribute toward the disease being labelled as “difficult to treat” with a poor prognosis.
SCLC has a substantial impact on patients’ physical, psychological, and social well-being.5,6 While the symptoms and signs of SCLC vary from person to person, common symptoms include persistent cough, chest pain that gets worse when coughing, laughing, or taking a deep breath, hemoptysis (coughing up blood), and hoarseness and/or wheezing. Some patients may also experience a loss of appetite, unintended weight loss, fatigue, and recurrent episodes of lung infections such as pneumonia or bronchitis.7 Additionally, patient health is further compromised by toxicities during chemotherapy and side effects with current therapies (e.g., neutropenia, infection, mucositis, myelosuppression, alopecia, nausea, vomiting, fatigue, sore mouth, or diarrhea). Patients’ HRQoL is also significantly impaired due to anxiety, depression, and distress associated with the disease and treatments.8
Treatment selection for patients with SCLC are determined by histology, cancer stage, and patients’ general health status and comorbidities.32 SCLC can be classified as LS-SCLC and ES-SCLC as per the Veterans’ Administration Lung Study Group staging system.2 LS-SCLC is defined as disease that is limited to the ipsilateral hemithorax and regional lymph nodes and can be encompassed in a safe radiotherapy field.9 Patients with tumours that have spread beyond the supraclavicular areas are considered having ES-SCLC, which includes distant metastases, malignant pericardial or pleural effusions, and/or contralateral supraclavicular and contralateral hilar lymph node involvement.4,10 At the time of diagnosis, approximately 30% of patients present with limited-stage disease, which is associated with a median OS of less than 2 years. Approximately 70% of patients present with extensive-stage disease at the initial diagnosis. Although ES-SCLC is highly responsive to chemotherapy with overall response rates between 60% to 70%, many patients relapse or develop drug resistance eventually.16,33,34 Prognosis for these patients continues to remain poor for those with relapsed disease, with a median OS of 12 months to 13 months from the time of diagnosis,11-13 and 5-year survival rates ranging from 1% to 10%.2,14,15 Important prognostic factors in SCLC include disease stage, performance status, age, sex, and markers of excessive bulk disease.10,35-37
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
As previously noted, high response rates are obtained with initial treatment as SCLC is highly sensitive to chemotherapy and radiotherapy, though it usually relapses within months despite treatment,2 and prognosis remains very poor, with most patients eventually dying of recurrent disease.17,38 Because ES-SCLC is generally considered incurable, the most important treatment goals for patients with ES-SCLC are prolonging survival, tumour shrinkage (reflected by response rates), reducing disease-related symptoms, and maintaining or improving patients’ HRQoL,17,19,38,39 which were corroborated by the clinical experts consulted for this review and the patient and clinician group inputs.
The conventional therapy for the initial systemic therapy in patients with SCLC is platinum-based chemotherapy combinations (cisplatin or carboplatin), based upon their clinical activity and toxicity profile.2 In Canada, a platinum agent and etoposide in combination with PD-L1 inhibitors, either durvalumab or atezolizumab, are the typical 1L treatment options. Irinotecan and platinum-based chemotherapy can be used as an alternative for etoposide and platinum-based chemotherapy.18,33,34,38,40,41 2L treatment options in Canada include topotecan (only indicated for platinum-sensitive SCLC after failure of 1L chemotherapy, as per the clinical experts’ opinions), irinotecan, CAV, or rechallenge with etoposide and platinum-based chemotherapy (only if progression occurs after an interval of approximately ≥ 3 months from the last dose of 1L chemotherapy).18,19 The prognosis for relapsed SCLC in the 2L setting is very poor, with response rates to current treatments less than 25%, short-lived disease control, as well as short PFS (4 months to 6 months) and OS (< 9 months).17,20 Currently in the 3L setting, there are no Health Canada–approved treatments. Available options in this setting include rechallenging with the initial regimen and other single-agent or combination chemotherapy regimens.1 Canadian treatment guidelines also recommend enrolment in clinical trials or best supportive care as additional options in later lines.18,21 Most studies in the 3L+ setting are single-arm, given the low number of available patients and attrition across lines of therapy, as well as the lack of established standard of care therapy.
The clinical experts consulted for this review confirmed that these approaches are adopted in Canadian clinical practice. Treatments in this setting (3L+) are palliative in nature, and the choice of drugs varies across the country. The experts also noted that platinum-based chemotherapy and etoposide, topotecan alone, irinotecan with or without platinum chemotherapy, and CAV are relevant comparators for tarlatamab.
Drug Under Review
Tarlatamab is a novel bispecific delta-like ligand 3–directed CD3 T-cell engager that binds to delta-like ligand 3 expressed on the surface of cells, including tumour cells, and CD3 expressed on the surface of T cells. It triggers T-cell activation, production of inflammatory cytokines, and lysis of delta-like ligand 3–expressing cells.22
Tarlatamab is provided as lyophilized powder for solution for IV infusion, 1 mg and 10 mg per vial. The recommended dose of tarlatamab is an initial dose of 1 mg on day 1, followed by 10 mg on day 8 and day 15, and every 2 weeks thereafter. It is administered as an IV infusion over 1 hour. Treatment with tarlatamab can be continued until disease progression or unacceptable toxicity.22 Serious or life-threatening AEs such as CRS and neurologic toxicity including ICANS have been reported in patients receiving tarlatamab.22
Tarlatamab has been issued a Notice of Compliance with Conditions by Health Canada on September 11, 2024, and the approved indication is for the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy. The sponsor’s reimbursement request aligns with the Health Canada–approved indication.
Key characteristics of tarlatamab and other treatments available for ES-SCLC in Canada are summarized in Table 3.
Table 3: Key Characteristics of Tarlatamab and Comparators
Characteristic | Tarlatamab | Topotecan | CAV | Irinotecan (with or without cisplatin or carboplatin) | Etoposide (with or without cisplatin or carboplatin) |
|---|---|---|---|---|---|
Mechanism of action | BiTE molecules bring T cells into close proximity of cancer cells inducing lysis of cancer cells | Inhibits topoisomerase I by stabilizing the covalent complex of enzyme and strand-cleaved DNA, which is an intermediate of the catalytic mechanism, thereby inducing breaks in the protein-associated DNA single strands, resulting in cell death | Cyclophosphamide: bifunctional alkylating drug that crosslinks DNA Vincristine: microtubule inhibitor Doxorubicin: DNA synthesis inhibition | Cisplatin and carboplatin: interferes with DNA intrastrand and interstrand crosslinks in cells Irinotecan: topoisomerase I inhibition | Cisplatin and carboplatin: interferes with DNA intrastrand and interstrand crosslinks in cells Etoposide: topoisomerase II inhibition |
Indicationa | Treatment of ES-SCLC with disease progression on or after at least 2 prior lines of therapy, including platinum-based chemotherapy | For sensitive SCLC after failure of first-line chemotherapy (defined as recurrence at least 60 days after first-line chemotherapy) | Cyclophosphamide: malignant lymphomas (various), multiple myeloma, leukemias (various), mycosis fungoides Doxorubicin: various neoplasms Vincristine: various neoplasms | Cisplatin: metastatic testicular tumours, metastatic ovarian tumours, advanced bladder cancer Carboplatin: advanced ovarian carcinoma of the epithelial region Irinotecan: metastatic carcinoma of colon or rectum | Cisplatin: metastatic testicular tumours, metastatic ovarian tumours, advanced bladder cancer Carboplatin: advanced ovarian carcinoma of the epithelial region Etoposide: SCLC, malignant lymphoma, NSCLC, testicular malignancies |
Route of administration | IV | IV | Cyclophosphamide: IV and oral Doxorubicin: IV Vincristine: IV | Carboplatin and cisplatin: IV Irinotecan: IV | Carboplatin and cisplatin: IV Etoposide: IV and oral |
Recommended dose | Day 1: 1 mg Day 8: 10 mg Day 15 and 2 weeks after thereafter: 10 mg (1-hour IV infusion) | 1.5 mg/m2 over 30 minutes q.d. for 5 consecutive days, starting on day 1 of a 21-day course | Cyclophosphamide IV: 40 to 50 mg/kg (1.5 g/m2 to 1.8 g/m2) administered as 10 mg/kg to 20 mg/kg q.d. for 2 days to 5 days (adult loading); 10 mg/kg to 15 mg/kg (350 mg/m2 to 550 mg/m2) every 7 days to 10 days (adult maintenance), or 3 mg/kg to 5 mg/kg (110 mg/m2 to 185 mg/m2) twice weekly (adult maintenance) Cyclophosphamide oral: 1 mg/kg to 5 mg/kg q.d. Doxorubicin: 60 mg/m2 to 75 mg/m2 every 21 days or 20 mg/m2 IV q.1.w. Vincristine: 1.4 mg/m2 q.1.w. (adults) | Irinotecan: 350 mg/m2 q.3.w. or 125 mg/m2 q.1.w. for 4 weeks followed by a 2-week rest Cisplatin: 50 mg/m2 to 75 mg/m2 as a single IV dose q.3.w. or q.4.w., or 15 mg/m2 to 20 mg/m2 IV q.d. for 5 days, q.3.w., or q.4.w. Carboplatin: 400 mg/m2 given as a single infusion over 15 minutes to 60 minutes | Cisplatin: refer to column 4 Carboplatin: refer to column 4 Etoposide IV: 50 mg/m2 to 100 mg/m2 q.d. for 5 days q.4.w. Etoposide oral: 100 mg/m2 to 200 mg/m2 q.d. for 5 days |
Serious adverse effects or safety issues | CRS, neurologic toxicity such as ICANS | Neutropenia, neutropenic colitis, and interstitial lung disease | Cyclophosphamide: secondary malignancy, acute cardiac toxicity, severe QT prolongation and ventricular tachyarrhythmia, hepatotoxicity, severe myelosuppression, urotoxicity, nephrotoxicity, acute pulmonary toxicity, infusion reactions, drug–drug interactions, fulminating anaphylaxis, vaccine-induced infection Doxorubicin: chemical cystitis, cardiomyopathy, decreased LVEF, congestive heart failure, secondary malignancies, tissue necrosis, myelosuppression, hepatotoxicity Vincristine: leukopenia, alopecia, neuromuscular changes including sensory impairment, paresthesia, neuropathic pain, motor difficulties, constipation | Irinotecan: severe early and late diarrhea, typhlitis, ulcerative and ischemic colitis, ileus, and intestinal perforation, myelosuppression, infections, thromboembolic events, hyperglycemia, hepatoxicity, infusion reactions Carboplatin: myelosuppression, peripheral neuropathy, hepatotoxicity, nephrotoxicity, cardiovascular toxicity, infusion reactions Cisplatin: myelosuppression, infusion reactions, infections, neurotoxicity, nephrotoxicity, cardiovascular toxicity | Carboplatin: myelosuppression, peripheral neuropathy, hepatotoxicity, nephrotoxicity, cardiovascular toxicity, infusion reactions Cisplatin: myelosuppression, infusion reactions, infections, neurotoxicity, nephrotoxicity, cardiovascular toxicity Etoposide: myelosuppression, cardiovascular toxicity, nausea and vomiting, alopecia, infusion reactions |
BiTE = bispecific T-cell engager; CAV = cyclophosphamide, doxorubicin, and vincristine; CRS = cytokine release syndrome; ES-SCLC = extensive-stage small cell lung cancer; ICANS = immune effector cell-associated neurotoxicity syndrome; LVEF = left ventricular ejection fraction; NSCLC = non–small cell lung cancer; q.d. = once daily; q.1.w. = once a week; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; SCLC = small cell lung cancer.
aHealth Canada–approved indication.
Sources: Product monographs for tarlatamab,22 topotecan,42 cyclophosphamide,43 doxorubicin,44 vincristine,45 irinotecan,46 cisplatin,47 carboplatin,48 and etoposide.49,50
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website: tarlatamab | CDA-AMC.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received a joint patient group submission from Lung Cancer Canada, the Lung Health Foundation, and the Canadian Cancer Survivor Network. Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research, and advocacy. The Lung Health Foundation is a registered charity that assists and empowers people living with or caring for others with lung disease. The Canadian Cancer Survivor Network is a national network of patients, families, survivors, and community partners, who take collaborative action to promote the best standards of care for patients with cancer.
Information provided for this submission consisted of thoughts and experiences of 3 patients with ES-SCLC and their caregivers. Data were collected by Lung Cancer Canada through virtual interviews with patients conducted. All interviews were conducted between July 2024 and August 2024. All patients (2 males, 1 female) with ES-SCLC had experience with tarlatamab, 2 of whom resided in Canada (1 in Ontario and the other in New Brunswick) and 1 in the US.
The patient groups emphasized that SCLC is an aggressive type of lung cancer, with a high symptom burden, rapid disease progression, and poor health outcomes. Given the lack of developments in new treatment options for patients with SCLC beyond 1L therapy, the patient groups highlighted an urgent need for a new treatment beyond the 1L setting for patients with ES-SCLC. The patient groups noted that new treatments should be effective in controlling the disease and symptoms, minimizing side effects of the treatments, allowing patients to maintain a meaningful quality of life, minimizing caregiver burden, delaying disease progression, and offering patients an additional treatment option upon disease progression or when other treatments are exhausted.
Regarding currently available treatment options for ES-SCLC, patients noted that chemotherapy is associated with limited duration of response, harsh side effects, increased dependence on caregivers in daily activities, and an impact on the patients’ functionality. Two patients also had experience with immunotherapy either in combination with chemotherapy, or after successful completion of chemotherapy. However, 1 patient stopped treatment with immunotherapy due to neutropenia and the other due to disease progression.
As noted, all patients had experience with tarlatamab as a 3L+ therapy. Two patients accessed tarlatamab through a clinical trial, and 1 through Health Canada’s Special Access Program. All patients indicated that they had significant side effects when received the first dose of tarlatamab; however, these side effects improved over time. Two patients experienced CRS during their first infusion, which improved over time. One patient continued to work during treatment with tarlatamab. Overall, patients indicated that tarlatamab was effective in treating their disease and delaying disease progression. They also indicated that tarlatamab significantly improved their quality of life, similar to before their diagnosis, and they had a better experience with tarlatamab than with previous therapies.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of SCLC.
Unmet Needs
The clinical experts emphasized that once a patient with ES-SCLC become resistant to systemic platinum chemotherapy, treatment options become very limited with poorer response rates and reduced OS. The clinical experts indicated that the most important goals of treatment for patients with ES-SCLC are to prolong life, delay disease progression, reduce the severity of cancer-related symptoms, and improve patients’ HRQoL, as well as balance the toxicities of therapy. The 2 clinical experts consulted for this review noted that there have been no new funded treatments in the 2L setting or beyond in approximately 25 years, and there are currently no Health Canada–approved treatments for ES-SCLC in the 3L+ setting. As such, the clinical experts identified the need for new safe and effective treatment options for patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy, particularly those who are platinum refractory. Relevant for all lines of therapy for SCLC, the clinical experts noted that there is also a need for treatments that can effectively control CNS metastasis, given the increased risk of CNS metastasis requiring whole-brain radiotherapy in patients with ES-SCLC.
Place in Therapy
The clinical experts noted that patients with ES-SCLC would likely have received etoposide with platinum-based chemotherapy at least once, and likely another type of chemotherapy (or rechallenge of etoposide with platinum-based chemotherapy) before tarlatamab could be considered. In line with the Health Canada–approved indication, the clinical experts indicated that tarlatamab would become the preferred treatment option for patients who progress on or after 2 lines of therapy (i.e., 3L+). The clinical experts also highlighted that because only a limited number of patients are likely able to receive treatment in the 3L setting due to severe functional decline or disease progression, tarlatamab is not expected to cause a major shift in the current treatment paradigm.
Patient Population
The clinical experts consulted for this review noted that patients are identified by a combination of clinical examination, laboratory review, and imaging examinations, and would generally already be under the care of oncologists. The clinical experts indicated that the patients with ES-SCLC are often stigmatized due to the heavy association with tobacco use. Per the Health Canada–approved indication, patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy would be considered for treatment with tarlatamab. The clinical experts noted that there is currently no biomarker to determine which patients will respond best to tarlatamab, and they therefore suggested that tarlatamab should be offered to all patients who are eligible to receive this treatment. However, based on the clinical experts’ experience, patients who are younger, have fewer comorbidities or lower burden of disease, previously responded to etoposide with platinum-based chemotherapy, or had a longer duration of response to previous treatments may be more likely to benefit from tarlatamab.
The clinical experts acknowledged the challenges of administering tarlatamab, which requires hospital admission in a specialized centre, which may limit the uptake of this treatment in practice. In addition, the clinical experts noted the concerns about the AEs associated with tarlatamab (such as higher risk of CRS, particularly in patients with cardiac comorbidities) which may also limit clinicians in administering this drug to patients who do not meet the eligibility criteria of the DeLLphi-301 trial.
Assessing the Response to Treatment
The clinical experts consulted for this review noted that in general, outcomes used in clinical practice align with those seen in clinical trials of ES-SCLC. Important outcomes for patients with ES-SCLC include OS, PFS, CNS metastasis–free survival, HRQoL, and symptom relief. The clinical experts indicated that CNS disease progression (for patients with or without baseline CNS disease) would also be of particular interest to clinicians.
Both clinical experts agreed that response to treatment should be assessed every 2 months to 3 months with imaging examinations to determine if response or stable disease is observed. In terms of meaningful response to treatment, the clinical experts stated that in the 3L setting, any improvement of 2 months or greater in OS or PFS over the standard treatment options would be clinically meaningful. The experts also highlighted that there is likely variation in how clinicians would measure response or success and may also include stability or improvement of symptoms in their assessment.
Discontinuing Treatment
According to the clinical experts consulted for this review, treatment with tarlatamab will be discontinued if there is evidence of disease progression, intolerable or unmanageable toxicities including grade 3 or higher CRS or ICANS, or deterioration of quality of life, as well as patient preferences. However, the clinical experts noted that if clinical benefits from treatment with tarlatamab are maintained despite evidence of radiographic progression, patients may be allowed to continue this therapy in certain circumstances (e.g., no worsening of symptoms, quality of life is not declining, or the patient’s overall condition remains stable). Decisions regarding whether or not patients should continue treatment with tarlatamab beyond radiographic progression would be at the discretion of the treating physician.
Prescribing Considerations
The clinical experts indicated that patients with ES-SCLC are under the care of medical oncologists and/or oncologists with experience administering and managing systemic therapy. In line with the DeLLphi-301 trial, the clinical experts noted that patients receiving treatment with tarlatamab should be hospitalized for 24 hours during cycle 1 day 1 and cycle 1 day 8 due to the increased risk of CRS and ICANS. Additionally, treatment centres must have immediate access to an onsite intensive care unit capable of managing these potentially fatal AEs. However, the clinical experts noted that, according to clinical trial guidelines, while patients should initiate and receive tarlatamab in an inpatient setting, transition to an outpatient setting would be reasonable after 3 to 4 treatments. Furthermore, the clinical experts noted that supervision of later treatment cycles may be conducted by other practitioners in the treatment team, such as nurse practitioners or general practitioners in oncology.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups.
Two clinician groups, the Lung Cancer Canada Medical Advisory Committee and the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee provided input for this review. The Lung Cancer Canada Medical Advisory Committee consists of clinicians and key opinion leaders who have been providing input for submissions of new lung cancer drugs to the health technology assessment process for many years. The Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee provides evidence-based clinical and health system guidance on drug-related issues to support the mandate of Cancer Care Ontario including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. A total of 29 clinicians from the Lung Cancer Canada Medical Advisory Committee and 5 clinicians from the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee provided input for this submission. The submission from the Lung Cancer Canada Medical Advisory Committee was informed by data and information from publicly available sources, primarily published manuscripts and conference presentations, together with the clinical experience of the Medical Advisory Committee members. Information from the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee was gathered via email.
The Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee noted that current treatment options for patients with ES-SCLC are mainly palliative care or best supportive care. The Lung Cancer Canada Medical Advisory Committee noted that available single-agent chemotherapy options can provide palliation of rapidly progressing symptoms but have significant toxicities, and their impact on survival remains uncertain. The clinician groups indicated that for patients with disease progression or recurrence despite platinum-based chemotherapy, treatment options are limited. The clinician group highlighted that lurbinectedin, a newer alkylating agent, has Health Canada approval but is not publicly funded in Canada, limiting its use. Patients who have undergone 2 lines of chemotherapy often have compromised hematologic reserves, making them less suitable for additional systemic chemotherapies. Both clinician groups agreed that the goals of treatment for patients with SCLC who have progressed after 2 previous lines of therapy should include: improved OS, prolonged PFS, reasonable toxicity with low incidence of dose modification, and improved HRQoL. Thus, both clinician groups indicated that there is a pressing need for novel 3L+ treatment options, particularly chemotherapy-free regimens, especially for patients whose conventional therapy has failed.
Per the clinician groups, tarlatamab represents the first regulatory-approved BiTE agent in thoracic malignancies and second in all solid tumours. The clinician groups indicated that there is an increasing understanding and comfort among medical oncologists in managing the unique toxicities (i.e., CRS and ICANS) associated with BiTEs, and the clinician groups considered this therapy one of the novel immunotherapeutic approaches for patients with cancer with poor prognosis.
The clinician groups suggested that the most suitable candidates for treatment with tarlatamab are in line with the clinical trial and include patients with progressive SCLC (most likely patients with stage IV disease), who have exhausted 2 or more lines of therapy, as well as patients with an adequate performance status (e.g., ECOG PS of 0 to 1), those who cannot tolerate further cytotoxic therapy, and those who can manage potential side effects (CRS and ICANS).
The clinician groups indicated that assessing both clinical (reduction in symptoms and maintenance of quality of life) and radiologic parameters, with scans performed every 6 weeks to 12 weeks during the first year or as clinically indicated, would ensure that patients receive personalized optimal care according to their evolving needs. The clinician groups and clinical expert consulted for this review agreed that treatment with tarlatamab should be discontinued if significant toxicity or unequivocal disease progression are observed. However, the Lung Cancer Canada Medical Advisory Committee suggested that radiographic and clinical responses should be considered before treatment discontinuation. Both clinician groups indicated that tarlatamab should be administered in specialized centres with an inpatient setting to handle any potential toxicities from the treatment, and patients should only be discharged if all symptoms resolve. The clinician groups highlighted the concerns regarding CRS, where patients require hospitalization for the initial dose of tarlatamab, which can strain hospital resources, especially in facilities already facing pressure on inpatient beds.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There was no comparator for tarlatamab in the phase II DeLLphi-301 trial. Generic chemotherapy (i.e., CAV or single-agent chemotherapy) would be used in patients with good performance status after 2 lines of therapy. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
One of the eligibility criteria in the DeLLphi-301 trial was ECOG PS 0 to 1. Should patients with ECOG PS score ≥ 2 be considered for treatment with tarlatamab? | The clinical experts indicated that in clinical practice, patients with an ECOG PS score of 2 may be eligible for treatment with tarlatamab. However, those with an ECOG PS score > 2 should not be treated with tarlatamab. |
If a patient was treated with platinum-based chemotherapy in the LS-SCLC setting and then progressed to ES-SCLC and was treated with a line of treatment, does the line of therapy received in the LS-SCLC setting count as 1 of the 2 prior treatments required before tarlatamab? | The clinical experts noted that in this particular situation, the platinum-based chemotherapy received in the LS-SCLC setting can be counted as 1 of the 2 prior treatments required before tarlatamab therapy. |
Considerations for discontinuation of therapy | |
As per the trial protocol for the DeLLphi-301 study, patients with ES-SCLC were allowed to continue tarlatamab beyond radiographic progression, if the investigator thought that tarlatamab provided continued clinical benefit. In this case, when should treatment with tarlatamab be discontinued? | The clinical experts indicated that if clinical benefits from treatment with tarlatamab are maintained despite evidence of radiographic progression, patients may be allowed to continue therapy in certain circumstances (e.g., no worsening of symptoms, quality of life is not declining, or the patients’ overall condition remains stable). The clinical experts highlighted that in these cases radiation therapy can be delivered to particular metastatic lesions that are growing as determined by imaging. Decisions regarding whether or not patients should continue treatment with tarlatamab beyond radiographic progression is at the discretion of the treating physician. However, the experts noted that if the disease is truly progressing and the patient becomes symptomatic, the clinical benefit from tarlatamab would decline, and treatment would be discontinued. |
Considerations for prescribing of therapy | |
Tarlatamab is administered as an IV infusion on day 1, day 8, and day 15 of cycle 1 and then day 1 and day 15 of subsequent cycles. | Comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Should patients currently on other systemic therapies be switched to tarlatamab? | The clinical experts indicated that in clinical practice, it is unlikely for patients with ES-SCLC to be switched to another treatment when they respond well to the current treatment. Usually in the 3L setting, the treatment effect of a drug is not durable; therefore, the physician can quickly find out whether the patient responds well to a treatment or not. If the patient has progressive disease and meets the eligibility criteria of tarlatamab, they can be offered this treatment; however, this should be considered another line of therapy, not treatment switching. |
Patients are required to be within a 1-hour drive from a specialized centre (i.e., emergency department) for cycle 1 of tarlatamab in the event of the occurrence of CRS. | Comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
A stabilizer is required for the compounding of tarlatamab, to prevent absorption of the drug to the IV bags and tubing. The product monograph includes the stabilizer within the tarlatamab package. | Comment from the drug programs to inform pERC deliberations. |
CRS, ICANS, and infections can occur with tarlatamab. Monitoring of these AEs (particularly for CRS) is required, especially in cycle 1 of treatment. | Comment from the drug programs to inform pERC deliberations. |
Tarlatamab may require inpatient administration during the ramp-up stage. Additional therapies may be required during treatment with tarlatamab to manage the AEs (e.g., tocilizumab may be required for the management of CRS, IV steroids may be required for the management of ICANS, other drugs such as anakinra may be needed in patients who are not responding to steroids for ICANS). To ensure equitable access, the costs of these therapies, especially in the inpatient setting, need to be incorporated as part of any implementation. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
The incremental budget impact of tarlatamab to current standard of care used in Canada is substantial. The drug plans noted other system or economic issues over 3 years compared to current expenditures in the 3L setting that jurisdictions are paying for. | Comment from the drug programs to inform pERC deliberations. |
Per the product monograph, patients must be monitored for 24 hours after cycle 1, day 1 and cycle 1, day 8 doses of tarlatamab in an appropriate health care setting. Would patients be required to be treated with tarlatamab in the inpatient setting (potential additional costs to be considered include the costs of hospitalization for monitoring of CRS)? Would patients be able to start treatment with tarlatamab in an outpatient setting? If yes, which patients would be eligible to be treated in the outpatient setting, and which patients would start in inpatient setting? | The clinical experts stated that patients must be monitored for 24 hours in an inpatient setting, at least for the first 2 doses of tarlatamab (as recommended in the clinical trials). The clinical experts indicated that patients are likely to experience CRS on their first cycle of treatment and their chance of CRS on the second cycle reduces. Per the product monograph, after cycle 1, day 1, and cycle 1, day 8 patients would be monitored postinfusion in the outpatient setting. |
Current chemotherapy used in the 3L setting for ES-SCLC includes generic chemotherapy, although prices are confidential. | Comment from the drug programs to inform pERC deliberations. |
3L = third line; AE = adverse event; CAV = cyclophosphamide, doxorubicin, and vincristine; CRS = cytokine release syndrome; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ES-SCLC = extensive-stage small cell lung cancer; ICANS = immune effector cell-associated neurotoxicity syndrome; LS-SCLC = limited-stage small cell lung cancer; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of tarlatamab (lyophilized powder for solution for IV infusion, 1 mg and 10 mg per vial) in the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy. The focus will be placed on comparing tarlatamab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of tarlatamab is presented in 4 sections with the critical appraisal of the evidence by CDA-AMC included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, no studies addressing gaps were submitted by the sponsor.
Included Studies
Clinical evidence from the following are included in this review and appraised in this document:
1 pivotal study (DeLLphi-301) identified in systematic review25
3 ITCs including 2 MAICs30 and 1 observational study using propensity score weighting of individual patient data (IPD).29
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
The DeLLphi-301 trial is an ongoing, multinational, phase II, open-label, single-arm trial to evaluate the efficacy and safety of tarlatamab in adult patients with recurrent SCLC who have progressed or recurred following 1 platinum-based regimen (with or without immune checkpoint inhibitor) and at least 1 other line of therapy.
Between December 2021 and May 2023, a total of 222 patients were recruited from 56 sites in 17 countries. No patients from Canada were enrolled. The study was conducted in 3 parts, and the study design is illustrated in Figure 1. Part 1 (n = 176) was a dose comparison and selection phase which randomized patients in a 1:1 ratio to receive 1 of the 2 target dose levels of tarlatamab, either 10 mg (n = 88) or 100 mg (n = 88). Enrolment continued in a randomized fashion until dose selection was finalized. Following a prespecified interim analysis that identified 10 mg as the target dose for subsequent parts of the trial, patient enrolment continued at the selected 10 mg dose in part 2 (n = 12) of the trial, a dose expansion phase, until a total of 100 patients across part 1 and part 2 were treated at the 10 mg dose. Part 3 (n = 34) was initiated after completing enrolment of part 1 and part 2, to enrol up to approximately 30 additional patients at the 10 mg dose, focused on safety of tarlatamab with reduced inpatient monitoring.
The study involved a screening period (up to 21 days) to assess eligibility for participation. Long-term follow up was to be conducted (through clinic visit, telephone, or chart review) every 3 months (± 2 weeks) for 1 year after the last patient’s last dose of tarlatamab or 5 years from the first patient enrolled, whichever occurs first. The DeLLphi-301 trial is ongoing with an estimated study completion date of October 31, 2026. The data presented in this submission are based on:
the primary Clinical Study Report25 with a primary DCO of June 27, 2023
Addendum 01 to the primary Clinical Study Report26 which includes updated efficacy (response and survival) and safety data reflecting an additional 90 days of follow up from the primary analysis (DCO of October 2, 2023)
Addendum 02 to the primary Clinical Study Report27 which includes updated response data (ORR, disease control rate, and duration of response only per BICR) (DCO of January 12, 2024)
Addendum 03 to the primary Clinical Study Report28 which includes updated survival data (OS only) (DCO of May 16, 2024).
A clinical safety monitoring and mitigation strategy with emphasis on CRS, neutropenia, neurologic toxicities, pituitary dysfunction, tumour lysis syndrome, and sucrose toxicity was implemented in this study which included, but was not limited to prevention, ongoing monitoring, early recognition, and prompt management.
Figure 1: Study Design of DeLLphi-301
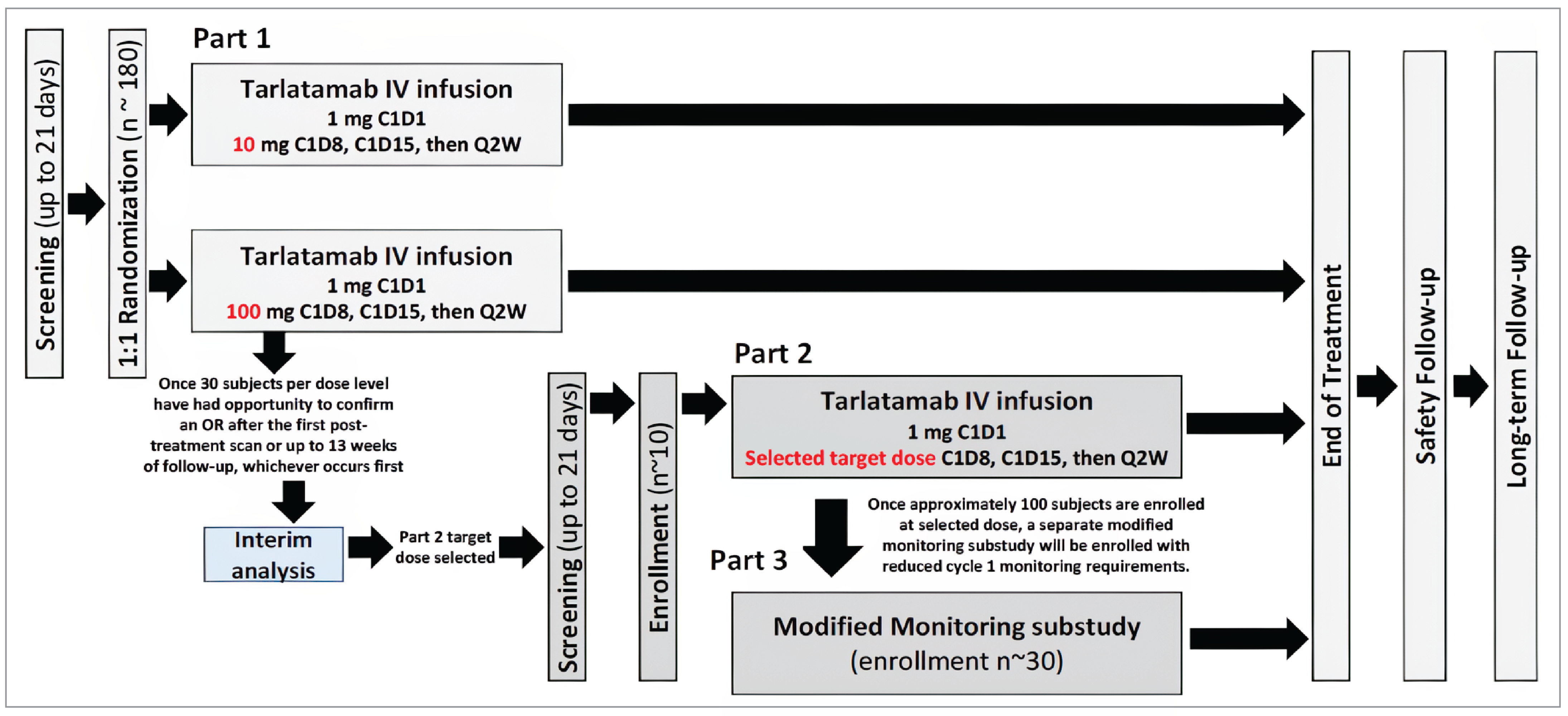
C = cycle; D = day; n = total number of patients; OR = objective response [rate]; Q2W = every 2 weeks.
Note: Characteristics of the included studies are summarized in Table 5.
Source: Primary Clinical Study Report for DeLLphi-301.25
Table 5: Details of Studies Included in the Systematic Review
Detail | DeLLphi-301 |
|---|---|
Designs and populations | |
Study design | Phase II, open-label, single-arm study of tarlatamab in patients with recurrent SCLC who have progressed or recurred following 1 platinum-based regimen (with or without checkpoint inhibitor) and at least 1 other LOT (re-treatment with a platinum-based regimen is considered a second LOT) |
Locations | The study was conducted at 56 sites in 17 countries: Austria, Belgium, Denmark, France, Germany, Greece, Italy, Japan, Netherlands, Poland, Portugal, South Korea, Spain, Switzerland, Taiwan, UK, and the US |
Patient enrolment dates | Start date: December 1, 2021 End date: Study is ongoing (estimated study completion date of October 31, 2026) |
Randomized (N) | Total: N = 222 Tarlatamab 10 mg: n = 100 (part 1 and part 2) Tarlatamab 100 mg: n = 88 (part 1) Tarlatamab 10 mg (modified safety monitoring): n = 34 (part 3) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Tarlatamab: 1 mg step dose on cycle 1 day 1, followed by 10 mg or 100 mg starting cycle 1 day 8, cycle 1 day 15, then every 2 weeks IV thereafter, until disease progression |
Comparator(s) | None |
Study duration | |
Screening phase | Up to 21 days |
Treatment phase | Treatment was given until disease progression, unacceptable toxicity, withdrawal of consent, or end of study. Data presented are based on a data snapshot of July 18, 2023, (primary DCO: June 27, 2023), after a median follow up of 10.6 months in the 10 mg group, and 10.3 months in the 100 mg group. |
Follow-up phase | Upon permanent discontinuation from the study treatment for any reason, a safety follow-up visit was performed approximately 42 days (± 5 days) after the last dose of tarlatamab, even if subsequent anticancer therapy had been initiated within that period. After the safety follow-up visit, patients entered LTFU to assess survival and/or the initiation of subsequent cancer therapy. LTFU was to be conducted every 3 months (± 2 weeks) for 1 year after the last patient’s last dose of tarlatamab or 5 years from first patient enrolled, whichever occurred first. |
Outcomes | |
Primary end point | Part 1: ORR (CR and PR); incidence of TEAEs; serum concentrations of tarlatamab Part 1 and part 2: ORR as assessed by BICR per RECIST 1.1 Part 3: incidence of TEAEs |
Secondary end points (all parts) |
|
Exploratory end points (all parts) |
|
Publication status | |
Publications | Ahn et al.51 Paz-Ares et al. (2023)52 |
AE = adverse event; BICR = blinded independent central review; CNS = central nervous system; CR = complete response; CRS = cytokine release syndrome; DCO = data cut-off date; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; FACT-G = Functional Assessment of Cancer Therapy–General; GP5 = The single item “I am bothered by side effects of treatment,” rated on a 5-point Likert scale and part of the FACT-G; LOT = line of treatment; LTFU = long-term follow up; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; PRO-CTCAE = Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.; SCLC = small cell lung cancer; TEAE = treatment-emergent adverse event; VAS = visual analogue scale.
Sources: Study protocol for DeLLphi-301,53 Primary Clinical Study Report for DeLLphi-301,25 Addendum 01 to the Primary Clinical Study Report for DeLLphi-301,26 Addendum 02 to the Primary Clinical Study Report for DeLLphi-301,27 Addendum 03 to the Primary Clinical Study Report for DeLLphi-301,28 and statistical analysis plan for DeLLphi-301.54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Patients included in the DeLLphi-301 study were aged 18 years or older (or legal adult age within their country) with histologically or cytologically confirmed relapsed or refractory SCLC who progressed or recurred following 1 platinum-based regimen and at least 1 other prior LOT. Patients must have had measurable lesions defined per RECIST 1.1 within 21 days before the first dose of tarlatamab and must have had adequate organ function. Patients with symptomatic or untreated brain metastases and who had prior anticancer therapy within 28 days before the first dose of tarlatamab were excluded.
Interventions
In part 1 of the DeLLphi-301 trial, tarlatamab was administered as an IV infusion for 60 minutes followed by a flush. A 1 mg step dose was administered on cycle 1 day 1. A 10 mg or 100 mg target dose was administered starting on cycle 1 day 8, cycle 1 day 15, and every 2 weeks thereafter. Premedication included dexamethasone 8 mg IV (or equivalent dose of other corticosteroid) administered 1 hour before tarlatamab infusion on day 1 and day 8 of cycle 1 only. Prophylaxis with IV hydration (1 L normal saline over 4 to 5 hours) was administered immediately following all tarlatamab doses in cycle 1.
All sites were required to ensure that CRS rescue medications were available on site, including corticosteroids, and tocilizumab (for sites in regions where tocilizumab was approved and available) or siltuximab (if tocilizumab was not available) for potential treatment of CRS. Dose modification of tarlatamab was permitted in case of AEs.
In countries where standard of care 1L systemic treatment includes platinum-containing chemotherapy in combination with PD-L1 inhibitor, it was required that patients had failed PD-L1 inhibitor therapy as part of their 1L systemic treatment or were ineligible to receive PD-L1 inhibitor therapy. Additionally, a platinum-based regimen followed by checkpoint inhibitor or anti-PD-L1 drug as maintenance therapy was considered 1 LOT. Re-treatment with a platinum-based regimen was considered a second LOT.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform the CDA-AMC expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the CDA-AMC expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | DeLLphi-301 |
|---|---|---|
OS | Median, OS rates at 3, 6, 9, and 12 months | Secondary |
PFS | Median, PFS rates at 3, 6, 9, and 12 months | Secondary |
HRQoL | ||
EORTC QLQ-C30 | Cycle 1 day 1, cycle 1 day 8, cycle 1 day 22, cycle 2 day 1, cycle 2 day 15, cycle 3 day 1, and every 6 weeks until week 48, then every 12 weeks until EOT, SFU; completed before the patient was informed of their disease status | Exploratory |
EORTC QLQ-LC13 | ||
ORR (CR and PR) | At individual DCOs (June 27, 2023; October 2, 2023; January 12, 2024) | Primary interim analysis (part 1) for dose selection for part 2a Primary: part 1 and part 2 (BICR FAS)a Sensitivity: part 1 and part 2 (investigator FAS) Secondary: part 3 |
Safety | Safety interim analyses occurred approximately every 3 months | Primary (for part 1 only and part 3) and secondary (all parts) |
BICR = blinded independent central review; CR = complete response; DCO = data cut-off date; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; EOT = end of treatment; FAS = full analysis set; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; SFU = safety follow up.
aTo adjust for multiplicity for an interim analysis to select a dose and a superiority test of part 1 and part 2, a 97.5% 2-sided CI was used for the latter primary analysis. The 97.5% CI was compared to the 15% target proportion. Statistical testing for adjusting for multiple comparisons was not performed for other efficacy and safety end points in the DeLLphi-301 study.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Overall Survival
OS was defined as the interval from the date of first dose of tarlatamab to the event of death due to any cause. Patients still alive were censored at the date last known to be alive. If the date last known to be alive was after the date that triggered the analysis (i.e., the DCO), the patient was censored at the analysis trigger date.
Progression-Free Survival
PFS per RECIST 1.1 was defined as the interval from the date of first dose of tarlatamab to the earlier of progressive disease per RECIST 1.1 or death due to any cause. Patients who started a new treatment before this date were censored at the last visit before new treatment.
Health-Related Quality of Life
Analyses of patient-reported outcomes were exploratory in part 1 and part 2. Analysis of HRQoL was not conducted in part 3.
EORTC QLQ-C30
The EORTC QLQ-C30 is a self-reporting 30-item generic instrument which assesses 5 functional domains (physical, role, emotional, cognitive, and social), 9 symptom scales (fatigue, nausea and vomiting, pain, dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and a global health status/quality of life scale, over the past week.55 Most items are reported on a 1 to 4 verbal response scale with response options of “not at all,” “a little,” “quite a bit,” and “very much,” while the 2 global health status/quality of life items are reported on a 1 to 7 numeric response scale anchored at the ends with “very poor” and “excellent.” All responses are linearly transformed to produce domain scores on a 0 to 100 range, where higher scores represent a greater amount of the concept being measured.56 Between-group minimal important differences (MIDs) for improvement (5 points) and deterioration (−5 points) on the “global health status” were identified for patients with lung cancer for this questionnaire.57 Another study demonstrated the appropriateness of the 10-point EORTC score threshold used in practice in patients with non-SCLC.56
EORTC-QLC-LC13
The EORTC QLQ-LC1358 is a disease-specific supplement to the EORTC QLQ-C30. The lung cancer questionnaire module comprises 1 multi-item scale to assess dyspnea, and a series of single items assessing lung cancer-related symptoms (i.e., coughing, hemoptysis, dyspnea, and pain) and side effects from conventional chemotherapy and radiotherapy (i.e., hair loss, neuropathy, sore mouth, and dysphagia). The EORTC QLQ-LC13 items use the same 1 to 4 verbal response scale as the EORTC QLQ-C30 items, and domain scores are also transformed to a 0 to 100 metric,56 with higher scores for the scales and single items representing a high level of symptomatology or problems.59
Objective Response Rate
The primary end point (for part 1 and part 2) was ORR as assessed by BICR using the RECIST 1.1 criteria. ORR was defined as the proportion of patients with a best overall response of either CR or PR.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.60 The core questionnaire consists of 30 items that make 5 multi-item functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning), 3 multi-item symptom scales (fatigue [3 items], nausea/vomiting [2 items], and pain [2 items]), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item global QoL scale. Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert-type scale (1 = not at all, 2 = a little, 3 = quite a bit, 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert-type scale (1 = very poor, 7 = excellent).61 Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas an increase in the function and QoL scale scores reflects an improvement.61 According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale, the score for that scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, missing items are ignored.61 | In studies with lung cancer patients. Validity: 184 patients with several cancer types (breast [n = 66], lung [n = 61], and colorectal [n = 57]) completed the EORTC QLQ-C30 and FACT-G.62 Moderate to strong correlations were observed between the 5 EORTC QLQ-C30 functioning scales (r = 0.41 to 0.77), and FACT-G and EORTC QLQ-C30 scales (r = 0.64 to 0.76).62 In another study of 111 patients with lung cancer (n = 101) or pleural mesothelioma (n = 11), a strong correlation was observed between EORTC QLQ-C30 emotional functioning and the HADS anxiety scale (r = −0.75).63 The HADS depression scale correlated substantially with all functioning scales (ranging from r = −0.40 to −0.55), fatigue (r = 0.52) and appetite loss (r = 0.48).63 Overall, the HADS scale correlated well with all EORTC QLQ-C30 scales (r = 0.28 to 0.75), excluding nausea/vomiting.54 Similarly, BPI scales correlated with all EORTC QLQ-C30 scales (r = 0.20 to 0.72), excluding nausea/vomiting and financial difficulties.54 These results support convergent validity. Strong correlations with both clinical measures of severity and self-reported health status (r > 0.50) for key domains such as physical functioning and fatigue, EORTC QLQ-C30 functioning scales (r = 0.41 to 0.77),62 HADS with all EORTC QLQ-C30 scales (r = 0.28 to 0.75; excluding nausea/vomiting),63 and BPI scales with all EORTC QLQ-C30 scales (r = 0.20 to 0.72; except nausea/vomiting and financial difficulties),63 supporting convergent validity. Reliability: Cronbach alpha coefficients were used to measure internal consistency. For functional scales, the Cronbach alpha values ranged from 0.70 to 0.85 showing good internal consistency. For symptoms scales, the Cronbach alpha values ranged from 0.70 to 0.90, suggesting high reliability.55 Additionally, a cluster-based analysis of several cancer types showed correlations of 0.91 for physical function-related measures (role, physical social functions and fatigue, pain, and global health status), 0.68 for psychological function (emotional functioning, cognitive functioning, and insomnia), and 0.63 for gastrointestinal symptoms (nausea, appetite loss, and vomiting).64 Responsiveness: The EORTC QLQ-C30 was administered before treatment and once during treatment to 305 patients with nonresectable lung cancer from centres in 13 countries.55 Over a 28-day period, there was a statistically significant difference in the global QoL scores (P < 0.01) between patients whose condition improved or worsened based on ECOG PS during pre-treatment and on-treatment periods. No significant difference was observed among patients whose ECOG PS remained stable or unchanged.55 | Between-group MIDs for improvement (5 points) and deterioration (−5 points) on the global health status scale were identified for patients with lung cancer.57 The MID estimates in patients with SCLC and breast cancer who reported “a little” change in the SSQ had corresponding changes in the EORTC QLQ-C30 of 5 points to 10 points, those who reported a “moderate” change had corresponding changes of approximately 10 points to 20 points, and those who reported “very much” change had corresponding changes of > 20 points.65 In a study of patients with advanced cancer, MID estimates of the EORTC QLQ-C30 ranged from a meaningful change for improvement of 9.1 units (cognitive functioning) to 23.5 units (pain), and a meaningful change for deterioration ranging from 7.2 units (physical functioning) to 13.5 units (role functioning). Distribution-based estimates were closest to 0.5 SD.66 In another study of patients with multiple myeloma (n = 239), MID estimates for EORTC QLQ-C30 mean scores ranged from 7.6 (improved global QoL) to –12.1 (deteriorated global QoL).67 |
EORTC QLQ-LC13 | The EORTC QLQ-LC13 is a lung cancer-specific questionnaire used to supplement the EORTC QLQ-C30 and contains 13 items related to lung cancer symptoms and treatment side effects including: a 3-item scale assessing dyspnea; and 9 single items including pain in chest, pain in arm or shoulder, pain in other parts, coughing, hemoptysis, sore mouth or tongue, dysphagia, peripheral neuropathy, and alopecia.55 All the scales range in score from 0 to 100. Higher scores on the symptom scales reflect worse symptoms.55 | Validity: Construct validity has been established between pain score and disease type (P < 0.001). Also, based on ECOG PS, construct validity was confirmed in dyspnea, coughing, and pain (P < 0.001) scores.58 Correlation between spirometry result and dyspnea score was found to be weak (r = 0.24). Moderate correlation was observed between BPI intensity score and EORTC QLQ-LC13 pain score (r > 0.4).63 Reliability: Good internal consistency with Cronbach alpha values ranging from 0.52 to 0.89 in functioning and symptom scales.55,58,65 Internal consistency was found to be unacceptable for pain scores (alpha = 0.53 to 0.54) when EORTC QLQ-LC13 was used alone without the EORTC QLQ-C30 questionnaire pain items.58 In another study, Cronbach alpha for dyspnea has been confirmed as acceptable (i.e., alpha = 0.76).63 Responsiveness: Dyspnea, coughing, and pain score improved over time between pretreatment and during treatment (statistically significant, P < 0.001).58 | No relevant studies on MID in patients with SCLC were identified. No studies with an MID were available in the overall lung cancer population or for NSCLC. |
BPI = Brief Pain Inventory; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; FACT-G = Functional Assessment of Cancer Therapy–General; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; QoL = quality of life; SCLC = small cell lung cancer; SD = standard deviation; SSQ = subjective significance questionnaire.
Harms
Harms were assessed by review of AEs, SAEs, study drug discontinuation due to AEs, and notable harms. AEs were defined as any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related. An AE were considered an SAE if it resulted in any of the following events: fatal; immediately life-threatening; inpatient hospitalization or prolongation of existing hospitalization; persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions; congenital anomaly or birth defect in a neonate or infant born to a mother or father exposed to study treatment; or other medically important serious event as assessed by the investigator or sponsor. Notable harms of interest, which were identified by the clinical experts consulted for this review included CRS and ICANS. CRS was defined as a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.68,69 ICANS used the criteria referenced in the publication by the Lee et al. study.69 ICANS grade is determined by the most severe event (e.g., depressed level of consciousness, seizure, motor findings, or raised intracranial pressure or cerebral edema) not attributable to any other cause. Results of harms as of October 2, 2023, were available.
Statistical Analysis
Sample Size and Power Calculation
On the basis of an analysis of the published literature, an ORR of 15% was prespecified in the protocol as the historical control benchmark among patients with previously treated SCLC. Assuming that an ORR would occur with tarlatamab in 30% of the patients, 100 patients receiving tarlatamab at the target dose determined during part 1 and part 2 of the trial would be needed to provide a probability of approximately 0.92 that the lower limit of the 97.5% CIs in the analysis of an ORR would exceed 15%. In total, approximately 220 patients were to be enrolled in the study, with approximately 100 patients enrolled at the selected dose in part 1 and part 2 — approximately 90 patients from part 1 and 10 patients from part 2. Part 3 was conducted after completing enrolment of part 1 and part 2 and enrolled up to approximately 30 additional patients, with modified cycle 1 monitoring criteria.
In part 1, data from 30 patients per arm were used to perform an interim analysis. There was no enrolment pause for the interim analysis. Based on an estimated enrolment rate of 25 patients per month, it was estimated that an additional 60 patients per arm would be enrolled by the time the dose decision was made. The remaining 10 patients were to be enrolled at the selected target dose in part 2. With a sample size of approximately 100 patients at the selected target dose, the probability that the expected lower boundary of the 97.5% 2-sided CI of observed ORR was anticipated to be greater than 15%. A sample size of 100 patients at the selected target dose provides a 63% probability of observing at least 1 AE with a true 1% incidence rate and 99% probability of observing at least 1 AE with a true 5% incidence rate.
For part 3, a sample size of up to approximately 30 patients at the selected target dose will provide a 26% probability of observing at least 1 AE with a true 1% incidence rate and 79% probability of observing at least 1 AE with a true 5% incidence rate.
Statistical Testing
An interim analysis occurred in part 1 when 30 patients per dose level had confirmed an objective response after the first posttreatment scan or up to 13 weeks of follow up, whichever occurred first. The analysis to inform the selection of the dose to be evaluated in part 2 of the study occurred in parallel to the futility analysis. Based on the totality of safety and efficacy data, a decision to stop the enrolment in 1 of the investigational arms for the remainder of the study was made by the dose selection committee. The primary analysis was planned when all patients were enrolled in part 1 and part 2 and had at least 24 weeks of follow up from the first scheduled postbaseline tumour assessment. Primary analysis was based on the disease response assessment by BICR per RECIST 1.1. The final analysis was to occur when enrolment was complete and each patient completed the study, including long-term follow up. The relationship of covariates to efficacy end points will be explored if appropriate.
CIs for proportions were estimated using the Clopper-Pearson exact method.70 Kaplan-Meier methods were used to estimate the median and percentiles for time-to-event end points with CI calculated using the Brookmeyer and Crowley method.71 Kaplan-Meier methods were also used to estimate landmarks for time-to-event end points (e.g., 1-year OS) with the Greenwood formula used to estimate the standard error used in CI calculation.72 Mixed models for repeated measurements were used to assess the change from baseline over time in HRQoL. Data from part 3 were analyzed both separately and combined with part 1 and part 2, where applicable.
Multiplicity Control
The sponsor noted that in an interim analysis for dose selection and a superiority test of part 1 and part 2, multiplicity adjustment was performed in the analysis of ORR, and a 97.5% 2-sided CI was used for the latter primary analysis. With the enrolment of 100 patients, the lower boundary of the 97.5% 2-sided CI for ORR greater than 15% was considered clinically meaningful change based on the results from previous open-label studies of pembrolizumab and nivolumab. Multiplicity was not adjusted for other efficacy and safety outcomes.
Handling of Missing Data
Handling of missing data in outcome measures is provided in Table 8. Imputation rules for missing data (start and stop dates and death dates) in the DeLLphi-301 trial were provided in the statistical analysis plan of this trial.
Sensitivity Analyses
A summary of the sensitivity analyses for select end points is provided in Table 8.
Subgroup Analyses
The following baseline covariates were used as appropriate to evaluate ORR in prespecified subgroups:
age (< 65 years versus ≥ 65 years)
region (North America versus Europe versus Asia versus rest of world)
race (American Indian or Alaska Native, Asian, Black or African American, Native Hawaiian or Other Pacific Islander, white, multiple, other)
delta-like ligand 3 cut points: < 75% versus 75% and < 25% versus 25% at moderate (2+) and strong (3+) staining intensities; other delta-like ligand 3 cut points may be explored
number of prior lines of anticancer therapy (2 versus 3)
prior PD-1 or PD-L1 inhibitor therapy (yes versus no)
sum of diameters of target lesions at baseline (< median versus median)
platinum sensitivity (< 90 days versus 90 and < 180 days versus 180 days)
brain metastasis at baseline (yes versus no)
liver metastasis at baseline (yes versus no).
If there were insufficient number of patients in the subgroup (i.e., < 10% of the whole population), relevant subgroups were combined.
Table 8: Statistical Analysis of Efficacy End Points in DeLLphi-301
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | OS was analyzed using the same method as described for the PFS end points. | NA | If the date last known to be alive is after the date that triggers the analysis (i.e., the DCO), the patient will be censored at the analysis trigger date. | For OS censoring up to the DCO, if there were deaths reported after the DCO before data snapshot date, the data available after the DCO may be used to derive the OS as sensitivity analysis. |
PFS | The distribution of PFS, including median and selected quantiles, were estimated using the Kaplan-Meier method with 95% CI calculated using the Brookmeyer and Crowley method. PFS rates at the selected time points (e.g., 6 months and 12 months) were reported along with 95% CI using the Greenwood formula to estimate the standard error in the CI calculation. | NA | Patients with no PD or death before DCO or EOS are censored at their last visit before DCO or new treatment start. | None |
EORTC QLQ-C30 EORTC QLQ-LC13 | The change from baseline over time in disease-related symptoms of dyspnea, physical functioning, and global health status was estimated for treatment dose levels using a REML MMRM under the assumption of MAR and with a UN or secondarily CS covariance structure. Similarly, the change from baseline over time for symptoms of cough and chest pain, the remaining subscales of EORTC QLQ-LC13, EORTC QLQ-C30, and the symptom burden score measured by the GP5 question from FACT- G was analyzed using an MMRM approach. The median time to deterioration for the subscales of EORTC QLQ-LC13 and EORTC QLQ-C30 along with selected quartiles was analyzed as PFS. | MMRM covariates include:
| Using MAR assumption in MMRMs. | None |
ORR (CR and PR) | The number and percentage of patients with a BOR of CR or PR were summarized along with a Clopper-Pearson exact 97.5% CI. | NA | Patients without a postbaseline tumour assessment were considered as nonresponders. | BICR FAS analysis is primary. Investigator FAS analysis is sensitivity. |
BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; CS = compound symmetry (covariance structure); DCO = data cut-off date; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; EOS = end of study; FACT-G = Functional Assessment of Cancer Therapy–General; FAS = full analysis set; GP5 = The single item “I am bothered by side effects of treatment,” rated on a 5-point Likert scale and part of the FACT-G; MAR = missing at random; MMRM = mixed effect model for repeated measures; NA = not available; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; REML = restricted maximum likelihood-based; UN = unstructured (covariance structure).
Sources: Primary Clinical Study Report for DeLLphi-301,25 protocol for DeLLphi-301,53 and statistical analysis plan for DeLLphi-301.54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The analysis populations in the DeLLphi-301 study are presented in Table 9.
Results
Patient Disposition
Of the 2 dose levels assessed in part 1 of the DeLLphi-301 study, the 10 mg every 2 weeks regimen was selected at the prespecified interim analysis for further evaluation in part 2.
Patient disposition in the DeLLphi-301 study is summarized in Table 10. Overall, 134 patients were randomized to receive tarlatamab 10 mg in part 1, or enrolled into part 2 and part 3, and 133 patients received at least 1 dose of tarlatamab, including 99 patients in the 10 mg target dose group across part 1 and part 2, and 34 patients in the part 3 modified safety monitoring 10 mg group.
Table 9: Analysis Populations in DeLLphi-301
Population | Definition | Application |
|---|---|---|
Investigator full analysis set | All patients who were enrolled in part 1 and part 2, received at least 1 dose of tarlatamab, and had ≥ 1 measurable lesion at baseline as assessed by investigator using RECIST 1.1 criteria. | Primary interim analysis on part 1 only of ORR. Sensitivity analysis for part 1 and part 2 of ORR. Secondary analysis for part 1 and part 2 of DCR, DOR, DoDC, and PFS. |
BICR full analysis set | All patients who were enrolled in part 1 and part 2, received at least 1 dose of tarlatamab, and had ≥ 1 measurable lesion at baseline as assessed by BICR using RECIST 1.1 criteria. | Primary analysis for part 1 and part 2 of ORR. Secondary analysis for part 1 and part 2 of DCR, DOR, DoDC, and PFS. |
Safety analysis set | All patients who received at least 1 dose of tarlatamab. | Analysis of all safety end points, unless noted otherwise, were conducted on the safety analysis set. Analyses of OS and concordance in assessment of BOR by investigator versus BICR for part 1 and part 2. |
Interim efficacy analysis set | All patients in the safety analysis set who were followed at least up to 12 weeks starting from day 1. Patients who stopped disease assessments before 12 weeks were included in this analysis set if the DCO was at least 12 weeks after their first dose date. | Interim analyses. |
Investigator interim RECIST 1.1 analysis set | All part 3 patients in the safety analysis set who had an opportunity to be followed for at least 7 weeks starting from day 1 and had 1 or more measurable lesions at baseline as assessed by investigator using RECIST 1.1 criteria. Patients who stopped disease assessments before 7 weeks were included in this analysis set if the DCO was at least 7 weeks after their first dose date. | Primary analysis for part 3 of ORR, DCR, DOR, DoDC, and PFS. |
BICR interim RECIST 1.1 analysis set | All part 3 patients in the safety analysis set who had an opportunity to be followed for at least 7 weeks starting from day 1 and had ≥ 1 or more measurable lesions at baseline as assessed by BICR using RECIST 1.1 criteria. Patients who stopped disease assessments before 7 weeks were included in this analysis set if the DCO was at least 7 weeks after their first dose date. | Primary analysis for part 3 of ORR, DCR, DOR, DoDC, and PFS. |
ITT analysis set | All patients who were randomized (part 1) and enrolled (part 2) according to assigned treatment dose levels during randomization and enrolment of the study. | Analyses of PRO end points were conducted on the ITT analysis set for part 1 and part 2. |
BICR = blinded independent central review; BOR = best overall response; DCO = data cut-off date; DCR = disease control rate; DoDC = duration of disease control; DOR = duration of response; ITT = intent to treat; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The main reasons for screen failures included “untreated or symptomatic brain metastases and leptomeningeal disease” (32 patients), “inadequate baseline organ function” (20 patients), “brain metastases treated less than 14 days before first dose of treatment” (14 patients), ECOG PS greater than 1” (13 patients), “consent not provided before initiation of study activities” (8 patients), “had not received at least 2 prior lines of therapy including platinum-based chemotherapy” (5 patients), and “unlikely and/or unable to complete all study activities” (5 patients).51
As of the June 27, 2023, DCO, 70 patients (70%) in part 1 and part 2 of the DeLLphi-301 study discontinued treatment with tarlatamab. The main reasons for treatment discontinuation were disease progression (52%), AEs (7%), patient request (6%), and death (3%). Forty-three patients (43%) in part 1 and part 2 of the DeLLphi-301 study discontinued the study.
Baseline Characteristics
Baseline characteristics for patients in the 10 mg group and the 10 mg modified safety monitoring group are summarized in Table 11. Of the 99 patients in the 10 mg group, most were male (71.1%). The median age was 64 years (range, 35 years to 82 years). Overall, patients had a median of 2 (range, 1 to 6) prior lines of therapy, including prior PD-1 or PD-L1 inhibitors (73.7%). The majority of the patients (65.7%) received 2 prior lines of therapy. Time to progression after 1L platinum therapy was less than 90 days for 27 patients (27.3%), between 90 days and 179 days for 22 patients (22.2%), longer than 180 days for 20 patients (21.4%), and unknown for 30 patients (30.3%). Most patients had metastatic disease (98.0%), no brain metastases (77.8%), or liver metastases (61.6%), and an ECOG PS score of 1 (73.7%).
Table 10: Summary of Patient Disposition in DeLLphi-301 (DCO of June 27, 2023)
Patient disposition | Tarlatamab 10 mg n = 100 | Tarlatamab 10 mg modified safety monitoring n = 34 |
|---|---|---|
Randomized | 100 (100.0) | 34 (100.0) |
V | 88 (88.0) | 0 (0.0) |
Patients enrolled in part 2 | 12 (12.0) | 0 (0.0) |
Patients enrolled in part 3 | 0 (0.0) | 34 (100.0) |
Tarlatamab accounting | ||
Patients who never received tarlatamab | 1 (1.0) | 0 (0.0) |
Patients who received tarlatamab | 99 (99.0) | 34 (100.0) |
Continuing tarlatamab | 29 (29.0) | 20 (58.8) |
Discontinued tarlatamab | 70 (70.0) | 14 (41.2) |
Disease progression | 52 (52.0) | 8 (23.5) |
AE | 7 (7.0) | 3 (8.8) |
Death | 3 (3.0) | 2 (5.9) |
Patient request | 6 (6.0) | 0 (0.0) |
Requirement for alternative therapy | 0 (0.0) | 1 (2.9) |
Other | 2 (2.0) | 0 (0.0) |
Study completion accounting | ||
Patients continuing study | 57 (57.0) | 25 (73.5) |
Discontinued from study | 43 (43.0) | 9 (26.5) |
Death | 35 (35.0) | 8 (23.5) |
Withdrawal of consent from study | 7 (7.0) | 0 (0.0) |
Lost to follow up | 1 (1.0) | 1 (2.9) |
Decision by sponsor | 0 (0.0) | 0 (0.0) |
Fulla/interimb analysis set, n | 99a | 33b |
Safety analysis set, n | 99c | 34c |
AE = adverse event; BICR = blinded independent central review; DCO = data cut-off date; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: Values are n (%) unless otherwise indicated.
aBICR full analysis set for part 1 and part 2: all patients who were randomized (part 1) or enrolled (part 2), received at least 1 dose of tarlatamab, and had ≥ 1 measurable lesion at baseline as assessed by BICR using RECIST 1.1 criteria.
bBICR interim RECIST analysis set for part 3: all part 3 patients in the safety analysis set who had an opportunity to be followed for at least 7 weeks starting from day 1 and had ≥ 1 measurable lesion at baseline as assessed by BICR using RECIST 1.1 criteria. For primary analysis, all efficacy analyses for part 3 were based on interim RECIST analysis sets.
cAll patients who received at least 1 dose of tarlatamab.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 11: Summary of Baseline Characteristics of DeLLphi-301, Safety Analysis Set
Characteristic | Tarlatamab 10 mg n = 99 | Tarlatamab 10 mg modified safety monitoring n = 34 |
|---|---|---|
Age (years) | ||
Median (range) | 64.0 (35 to 82) | 65.5 (49 to 80) |
Mean (SD) | 63.6 (8.6) | 65.5 (8.1) |
Age group, n (%) | ||
18 to 64 years | 51 (51.5) | 14 (41.2) |
65 to 74 years | 38 (38.4) | 14 (41.2) |
75 to 84 years | 10 (10.1) | 6 (17.6) |
≥ 85 years | 0 (0.0) | 0 (0.0) |
Sex, n (%) | ||
Male | 71 (71.7) | 24 (70.6) |
Female | 28 (28.3) | 10 (29.4) |
Race, n (%) | ||
American Indian or Alaska Native | 0 (0.0) | 0 (0.0) |
Asian | 41 (41.4) | 2 (5.9) |
Black or African American | 0 (0.0) | 1 (2.9) |
Native Hawaiian or Other Pacific Islander | 0 (0.0) | 0 (0.0) |
White | 57 (57.6) | 31 (91.2) |
ECOG status at baseline, n (%)a | ||
0 | 26 (26.3) | 10 (29.4) |
1 | 73 (73.7) | 24 (70.6) |
Weight, kg | ||
Mean (SD) | 73.96 (18.42) | 77.49 (16.26) |
Median (range) | 71.35 (41.3 to 123.4) | 79.75 (44.7 to 107.9) |
Smoking history, n (%) | ||
Never | 8 (8.1) | 1 (2.9) |
Current | 18 (18.2) | 5 (14.7) |
Former | 73 (73.7) | 28 (82.4) |
Prior lines of therapy, n (%) | ||
1 | 2 (2.0) | 0 (0.0) |
2 | 65 (65.7) | 22 (64.7) |
3 | 18 (18.2) | 6 (17.6) |
> 3 | 14 (14.1) | 6 (17.6) |
Mean (SD) | 2.5 (1.0) | 2.6 (1.0) |
Median (range) | 2.0 (1 to 6) | 2.0 (2 to 6) |
Time from initial cancer diagnosis to randomization or enrolment, months | ||
Mean (SD) | 19.10 (10.09) | 20.09 (11.62) |
Median (range) | 15.90 (5.5 to 61.5) | 17.10 (7.1 to 60.9) |
Prior PD-1 or PD-L1 inhibitor, n (%) | ||
Yes | 73 (73.7) | 28 (82.4) |
No | 26 (26.3) | 6 (17.6) |
Prior radiotherapy for current malignancy, n (%) | ||
Yes | 77 (77.8) | 24 (70.6) |
No | 22 (22.2) | 10 (29.4) |
Prior surgery for current malignancy, n (%) | ||
Yes | 12 (12.1) | 7 (20.6) |
No | 87 (87.9) | 27 (79.4) |
Disease stage at initial diagnosis, n (%) | ||
Stage 0 | 0 (0.0) | 0 (0.0) |
Stage I | 0 (0.0) | 0 (0.0) |
Stage II | 3 (3.0) | 3 (8.8) |
Stage III | 18 (18.2) | 4 (11.8) |
Stage IV | 66 (66.7) | 22 (64.7) |
Unknown/missing | 12 (12.1) | 5 (14.7) |
Disease stage at screening, n (%) | ||
Stage 0 | 0 (0.0) | 0 (0.0) |
Stage I | 0 (0.0) | 0 (0.0) |
Stage II | 1 (1.0) | 2 (5.9) |
Stage III | 5 (5.1) | 1 (2.9) |
Stage IV | 87 (87.9) | 28 (82.4) |
Unknown/missing | 6 (6.1) | 3 (8.8) |
Metastatic at baseline, n (%) | ||
Yes | 97 (98.0) | 32 (94.1) |
No | 2 (2.0) | 2 (5.9) |
Platinum sensitivity, n (%)b | ||
< 90 days | 27 (27.3) | 7 (20.6) |
≥ 90 and < 180 days | 22 (22.2) | 7 (20.6) |
≥ 180 days | 20 (20.2) | 9 (26.5) |
Unknown/missing | 30 (30.3) | 11 (32.4) |
Brain metastases at baseline, n (%) | ||
Yes | 22 (22.2) | 4 (11.8) |
No | 77 (77.8) | 30 (88.2) |
Liver metastases at baseline, n (%) | ||
Yes | 38 (38.4) | 12 (35.3) |
No | 61 (61.6) | 22 (64.7) |
DLL3 cut points, n (%) | ||
< 75% at moderate (2+) and strong (3+) staining intensity | 46 (46.5) | 10 (29.4) |
≥ 75% at moderate (2+) and strong (3+) staining intensity | 36 (36.4) | 6 (17.6) |
< 25% at moderate (2+) and strong (3+) staining intensity | 14 (14.1) | 5 (14.7) |
≥ 25% at moderate (2+) and strong (3+) staining intensity | 68 (68.7) | 11 (32.4) |
ECOG = Eastern Cooperative Oncology Group; SD = standard deviation.
a0 = fully active, able to carry on all predisease performance without restriction; 1 = restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature (e.g., light housework, office work).
bPlatinum sensitivity is calculated as end of first-line platinum therapy to date of first progression.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
As of the DCO of June 27, 2023, at least 1 dose of tarlatamab was administered to 99 patients in the 10 mg group across part 1 and part 2, and 34 patients in the 10 mg group with modified safety monitoring in part 3. Patient exposure in the DeLLphi-301 study is summarized in Table 12. In the 10 mg group (part 1 and part 2), patients were treated with tarlatamab for a median of 22.1 weeks (range, 0.1 weeks to 66.1 weeks). The median relative dose intensity was 92.9% (range, 3.2% to 114.8%), and the mean relative dose intensity was 85.5% (standard deviation = 21.9%). In the 10 mg group with modified safety monitoring (part 3), patients were treated with tarlatamab for a median of 6.1 weeks (range, 0.1 weeks to 16.4 weeks). The median relative dose intensity was 100.0% (range, 3.2% to 100.0%) and the mean relative dose intensity was 92.6% (standard deviation = 19.0%).
Concomitant medications used in at least 10% of the total population are summarized in Table 13. In the 10 mg group, the majority of patients (97%) received concomitant medications during the treatment period. All patients (100%) in the 10 mg modified safety monitoring group received concomitant medications. The most commonly prescribed concomitant medications in the DeLLphi-301 trial were paracetamol (78.8%) and dexamethasone (36.4%) in the 10 mg group. The proportion of patients who received these concomitant medications in the 10 mg modified safety monitoring group was similar to the 10 mg group.
Table 12: Patient Exposure in DeLLphi-301, Safety Analysis Set (DCO June 27, 2023)
Detail | Tarlatamab 10 mg n = 99 | Tarlatamab 10 mg modified safety monitoring n = 34 |
|---|---|---|
Number of doses received | ||
Mean (SD) | 13.4 (9.0) | 5.4 (2.5) |
Median (min, max) | 13.0 (1, 34) | 5.0 (1, 10) |
Cumulative dose, mg | ||
Mean (SD) | 124.3 (89.4) | 44.8 (24.7) |
Median (min, max) | 121.1 (1.0, 331.0) | 41.0 (1.0, 91.0) |
Relative dose intensity, %a | ||
Mean (SD) | 85.5 (21.9) | 92.6 (19.0) |
Median (min, max) | 92.9 (3.2, 114.8) | 100.0 (3.2, 100.0) |
Treatment duration (weeks) | ||
Mean (SD) | 24.17 (18.30) | 7.32 (4.68) |
Median (min, max) | 22.14 (0.1, 66.1) | 6.14 (0.1, 16.4) |
DCO = data cut-off date; max = maximum; min = minimum; SD = standard deviation.
aRelative dose intensity (%) = (actual cumulative dose/planned cumulative dose) × 100.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 13: Concomitant Medication Use in 10% or More of the Total Population in DeLLphi-301, Safety Analysis Set (DCO June 27, 2023)
Concomitant medication, n (%) | Tarlatamab 10 mg n = 99 | Tarlatamab 10 mg modified safety monitoring n = 34 |
|---|---|---|
Number of patients reporting use of concomitant medications | 96 (97.0) | 34 (100.0) |
Paracetamol | 78 (78.8) | 27 (79.4) |
Dexamethasone | 36 (36.4) | 11 (32.4) |
Sodium chloride | 32 (32.3) | 5 (14.7) |
Magnesium oxide | 24 (24.2) | 9 (26.5) |
Omeprazole | 19 (19.2) | 13 (38.2) |
Lactulose | 24 (24.2) | 4 (11.8) |
Metoclopramide | 17 (17.2) | 3 (8.8) |
Potassium chloride | 14 (14.1) | 10 (29.4) |
Oxycodone hydrochloride | 12 (12.1) | 3 (8.8) |
Ibuprofen | 15 (15.2) | 5 (14.7) |
Furosemide | 17 (17.2) | 5 (14.7) |
Pantoprazole | 14 (14.1) | 4 (11.8) |
Levothyroxine sodium | 13 (13.1) | 2 (5.9) |
Ondansetron | 10 (10.1) | 11 (32.4) |
Prednisolone | 9 (9.1) | 1 (2.9) |
Fentanyl | 10 (10.1) | 4 (11.8) |
Megestrol acetate | 13 (13.1) | 1 (2.9) |
Metamizole | 11 (11.1) | 7 (20.6) |
Morphine sulphate | 7 (7.1) | 4 (11.8) |
Pregabalin | 13 (13.1) | 2 (5.9) |
Salbutamol | 8 (8.1) | 10 (29.4) |
Dexamethasone sodium phosphate | 10 (10.1) | 2 (5.9) |
Famotidine | 8 (8.1) | 5 (14.7) |
Levofloxacin | 10 (10.1) | 2 (5.9) |
Metoclopramide hydrochloride | 13 (13.1) | 1 (2.9) |
Morphine | 14 (14.1) | 6 (17.6) |
Tocilizumab | 6 (6.1) | 3 (8.8) |
Piperacillin sodium and tazobactam sodium | 15 (15.2) | 1 (2.9) |
Acetylcysteine | 8 (8.1) | 2 (5.9) |
Erdosteine | 11 (11.1) | 0 (0.0) |
Oxygen | 11 (11.1) | 6 (17.6) |
Acetylsalicylic acid | 7 (7.1) | 5 (14.7) |
Enoxaparin sodium | 8 (8.1) | 9 (26.5) |
Lorazepam | 8 (8.1) | 6 (17.6) |
Pantoprazole sodium sesquihydrate | 7 (7.1) | 4 (11.8) |
DCO = data cut-off date.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
A post hoc analysis of postprogression therapies showed that 19 patients received 26 subsequent therapies.73 As shown in Table 14, the postprogression regimens were ones used commonly in real-world practice in the 3L+ setting, including platinum-containing regimens, CAV, irinotecan, and topotecan. Only 1 patient received an investigational drug.
Table 14: Postprogression Therapies in DeLLphi-301
Regimen | n |
|---|---|
Carboplatin | 1 |
Carboplatin and irinotecan | 1 |
Doxorubicin, cyclophosphamide, and vincristine | 1 |
Investigational new drug | 1 |
Irinotecan | 5 |
Lurbinectedin | 1 |
PD-1 inhibitor | 1 |
Platinum | 1 |
Platinum and etoposide | 1 |
Platinum, etoposide, and PD-L1 inhibitor | 1 |
Platinum and taxane | 1 |
Other | 1 |
Sacituzumab govitecan | 1 |
Topotecan | 5 |
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Overall Survival
OS was defined as the interval from the date of first dose of tarlatamab to the event of death due to any cause. In the 10 mg group (part 1 and part 2), the median OS was 14.3 months (95% CI, 10.8 months to NE), with a median follow-up time of 10.6 months (95% CI, 9.7 months to 11.3 months) at the DCO of June 27, 2023. The 12-month OS rate was 57.7% (95% CI, 45.0% to 68.4%). At later DCOs (October 2, 2023, and May 16, 2024), the results of OS were consistent with the primary analysis (Table 15).
Table 15: OS Assessment in DeLLphi-301 Part 1 and Part 2, Safety Analysis Set
Detail | DCO: June 27, 2023a | DCO: October 2, 2023 | DCO: May 16, 2024 |
|---|---|---|---|
Part 1 and part 2: Tarlatamab (10 mg) n = 99 | |||
Events (death), n (%) | 35 (35.4) | 44 (44.4) | 54 (54.5) |
Censored, n (%) | 64 (64.6) | 55 (55.6) | 45 (45.5) |
Alive at last follow up | 57 (57.6) | 48 (48.5) | 37 (37.4) |
Withdrawal of consent from study | 6 (6.1) | 6 (6.1) | 7 (7.1) |
Lost to follow up | 1 (1.0) | 1 (1.0) | 1 (1.0) |
Median OS, months (95% CI)b | 14.3 (10.8 to NE) | 15.2 (10.8 to NE) | 15.2 (10.8 to NE) |
Median follow-up time of OS, months, (95% CI)b,c | 10.6 (9.2 to 11.5) | 13.8 (12.9 to 14.7) | 20.7 (19.6 to 21.7) |
KM estimate at 3 months, % (95% CI)d | 88.7 (80.5 to 93.6) | 88.7 (80.5 to 93.6) | 88.7 (80.5 to 93.6) |
KM estimate at 6 months, % (95% CI)c | 73.4 (63.2 to 81.2) | 73.4 (63.2 to 81.2) | 73.4 (63.2 to 81.2) |
KM estimate at 9 months, % (95% CI)c | 68.0 (57.1 to 76.6) | 67.9 (57.4 to 76.4) | 67.9 (57.4 to 76.4) |
KM estimate at 12 months, % (95% CI)c | 57.7 (45.0 to 68.4) | 56.7 (45.6 to 66.3) | 56.6 (45.6 to 66.3) |
CI = confidence interval; DCO = data cut-off date; KM = Kaplan-Meier; NE = not estimable; OS = overall survival.
aDCO of June 27, 2023, corresponded to the primary analysis.
bMedian was estimated using the KM method and 95% CIs of the median were estimated using log-log transformation of KM survival estimate by the Brookmeyer and Crowley (1982) method.
cThe follow-up time was measured by reversing the status indicator for censored and events.
d95% CIs were estimated using the Kalbfleisch and Prentice (1980) method.
Sources: Primary Clinical Study Report for DeLLphi-301,25 Addendum 01 to the Primary Clinical Study Report for DeLLphi-301,26 Addendum 02 to the Primary Clinical Study Report for DeLLphi-301,27 and Addendum 03 to the Primary Clinical Study Report for DeLLphi-301.28 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Progression-Free Survival
Results for PFS are summarized in Table 16. At the DCO of June 27, 2023, in the 10 mg group (part 1 and part 2), median PFS by BICR was 4.3 months (95% CI, 3.0 months to 5.6 months) with a median follow-up time of 9.7 months (95% CI, 8.3 months to 10.9 months). At the DCO of October 2, 2023, median PFS by BICR was 4.3 months (95% CI, 3.0 months to 5.6 months) with a median follow-up time of 13.6 months (95% CI, 11.0 months to 13.8 months). The percentage of patients with event of disease progression or death was 65.7% at the DCO of June 27, 2023, and 68.7% at the DCO of October 2, 2023.
Table 16: PFS Assessment by BICR in DeLLphi-301, Full Analysis Set for Part 1 and
Part 2
Detail | Part 1 and part 2: Tarlatamab 10 mg N = 99a | |
|---|---|---|
DCO: June 27, 2023 | DCO: October 2, 2023 | |
Events, n (%) | 65 (65.7) | 68 (68.7) |
Death | 8 (8.1) | 8 (8.1) |
Disease progression | 57 (57.6) | 60 (60.6) |
Censored, n (%) | 34 (34.3) | 31 (31.3) |
On study without disease progression or death | 23 (23.2) | 19 (19.2) |
No evaluable postbaseline disease assessment | 2 (2.0) | 2 (2.0) |
Missed ≥ 2 consecutive assessments | 3 (3.0) | 3 (3.0) |
Started new anticancer therapy | 3 (3.0) | 4 (4.0) |
Withdrawal of consent from study | 3 (3.0) | 3 (3.0) |
Median PFS (KM), months (95% CI)b | 4.3 (3.0 to 5.6) | 4.3 (3.0 to 5.6) |
Median follow-up time (KM), months (95% CI)b,c | 9.7 (8.3 to 10.9) | 13.6 (11.0 to 13.8) |
KM estimate at 3 months, % (95% CI)d | 58.8 (48.1 to 68.1) | 58.8 (48.1 to 68.1) |
KM estimate at 6 months, % (95% CI)d | 39.7 (29.6 to 49.6) | 39.7 (29.6 to 49.6) |
KM estimate at 9 months, % (95% CI)d | 27.6 (18.4 to 37.5) | 27.9 (19.0 to 37.5) |
KM estimate at 12 months, % (95% CI)d | 25.7 (16.7 to 35.8) | 25.2 (16.6 to 34.7) |
BICR = blinded independent central review; CI = confidence interval; DCO = data cut-off date; KM = Kaplan-Meier; PFS = progression-free survival.
aBICR full analysis set for part 1 and part 2.
bMedian was estimated using the KM method and 95% CI of the median was estimated using log-log transformation of KM survival estimate by the Brookmeyer and Crowley (1982) method.
cThe follow-up time was measured by reversing the status indicator for censored and events.
d95% CIs were estimated using the Kalbfleisch and Prentice (1980) method.
Sources: Primary Clinical Study Report for DeLLphi-301,25 Addendum 01 to the Primary Clinical Study Report for DeLLphi-301,26and Addendum 02 to the Primary Clinical Study Report for DeLLphi-301.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Health-Related Quality of Life
Analyses of HRQoL end points were only conducted for part 1 and part 2. As of the June 27, 2023, DCO, the results for EORTC QLQ-LC13 showed that the least squares mean changes from baseline up to cycle 12 were −4.5 (95% CI, −11.4 to 2.4) for cough, −6.5 (95% CI, −10.9 to −2.1) for chest pain, and −10.2 (95% CI, −16.4 to −4.0) for dyspnea composite score. For the EORTC QLQ-C30 global health status/quality of life subscale, the least squares mean change from baseline up to cycle 12 was 11.5 (95% CI, 6.6 to 16.4) (Table 17).
Table 17: Summary of HRQoL Outcomes — ITT Analysis Set for Part 1 and Part 2 (DCO June 27, 2023)
HRQoL | DeLLphi-301 Part 1 and part 2: Tarlatamab 10 mg N = 100 |
|---|---|
EORTC QLQ-LC13 | |
Number of patients with observed data at cycle 12, day 1, n | 14 |
Dyspnea composite scorea | |
Baseline, mean (SD) | 26.01 (21.58) |
Change from baseline up to cycle 12, LS mean (95% CI)b | −10.21 (−16.41 to −4.02) |
Cough | |
Baseline, mean (SD) | 26.74 (27.32) |
Change from baseline up to cycle 12, LS mean (95% CI)b | −4.48 (−11.35 to 2.39) |
Chest pain | |
Baseline, mean (SD) | 11.36 (21.78) |
Change from baseline up to cycle 12, LS mean (95% CI)b | −6.50 (−10.94 to −2.06) |
EORTC QLQ-C30 | |
Number of patients with observed data at cycle 12, day 1, n | 14 |
Global health status or QoL | |
Baseline, mean (SD) | 59.24 (19.43) |
Change from baseline up to cycle 12, LS mean (95% CI)b | 11.50 (6.63 to 16.37) |
CI = confidence interval; DCO = data cut-off date; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; HRQoL = health-related quality of life; ITT = intention to treat; LS = least squares; N = number of all randomized or enrolled patients; n = number of patients with observed data; QoL = quality of life; SD = standard deviation.
aDyspnea composite score represents the composite score of item 3, 4, and 5 from EORTC QLQ-LC13 and item 8 from EORTC QLQ-C30 questionnaires.
bMixed model for repeated measures analysis is based on change from baseline up to cycle 12 day 1 with the dependent variable, time, baseline score, and treatment dose levels as fixed effects, and subject intercept and slope of time for change from baseline as random effects.
Sources: Primary Clinical Study Report for DeLLphi-301.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Objective Response Rates
ORR was a primary efficacy end point for part 1 and part 2. ORR was defined as the proportion of patients with a best overall response of CR or PR as defined by RECIST 1.1. As of June 27, 2023, the ORR in the BICR full analysis set for part 1 and part 2 was 41.4% (97.5% CI, 30.3% to 53.2%) for patients in the 10 mg group. Tumour shrinkage, as assessed by BICR, was observed in 72 patients (72.7%) in the BICR full analysis set for part 1 and part 2. The results of ORR were consistent with the primary analysis at later DCOs (October 2, 2023, and January 12, 2024). Tumour shrinkage as assessed by BICR was observed in 72 patients (72.7%) in the BICR full analysis set for part 1 and part 2, at all 3 DCOs. Most patients (40.4%) achieved PR in the ORR assessment (Table 18).
Table 18: ORR Assessment in DeLLphi-301, Part 1 and Part 2 BICR
Detail | DCO: June 27, 2023, in FAS | DCO: October 2, 2023, in SAS | DCO: January 12, 2024, in SAS |
|---|---|---|---|
Part 1 and part 2: Tarlatamab 10 mg N = 99 | |||
BOR as assessed by BICRa, n (%) | |||
CR, confirmed | 1 (1.0) | 2 (2.0) | 3 (3.0) |
PR, confirmed | 40 (40.4) | 39 (39.4) | 37 (37.4) |
Stable disease | 29 (29.3) | 29 (29.3) | 30 (30.3) |
Progressive disease | 19 (19.2) | 19 (19.2) | 19 (19.2) |
Not evaluable | 3 (3.0) | 3 (3.0) | 3 (3.0) |
No postbaseline scan | 7 (7.1) | 7 (7.1) | 7 (7.1) |
ORR as assessed by BICR | |||
ORR, n (%) | 41 (41.4) | 41 (41.4) | 40 (40.4) |
97.5% CIa | 30.3 to 53.2 | 31.6 to 51.8b | 30.7 to 50.7b |
Any tumour shrinkage as assessed by BICR, n (%) | |||
Yesc | 72 (72.7) | 72 (72.7) | 72 (72.7) |
At least 30% tumour shrinkage | 47 (47.5) | 47 (47.5) | 47 (47.5) |
No | 19 (19.2) | 19 (19.2) | 19 (19.2) |
Missing | 8 (8.1) | 8 (8.1) | 8 (8.1) |
BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; DCO = data cut-off date; FAS = full analysis set; ORR = objective response rate; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAS = safety analysis set.
Note: The BICR full analysis set for part 1 and part 2 consists of all patients who are randomized (part 1) or enrolled (part 2), received at least 1 dose of tarlatamab, and have ≥ 1 measurable lesions at baseline as assessed by BICR using RECIST 1.1 criteria; the safety analysis set for part 1 and part 2 includes all patients who received at least 1 dose of tarlatamab in part 1 and part 2 of the study.
aExact CI was calculated using the Clopper-Pearson method. ORR was defined as the proportion of patients with a BOR of CR or PR as defined by RECIST 1.1.
b95% CI.
cIncludes patients who had any tumour shrinkage in the target lesions at postbaseline assessment.
Sources: Primary Clinical Study Report for the DeLLphi-301 trial, DCO June 27, 2023,25 Addendum 01 to the Clinical Study Report for the DeLLphi-301 trial, DCO October 2, 2023,26 and Addendum 02 to the Clinical Study Report for the DeLLphi-301 trial, DCO January 12, 2024.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Subgroup analyses were conducted for ORR to explore the consistency of the treatment effect across subgroups as assessed by BICR. Response was seen across all subgroups for patients with and without prior treatment with PD-1 or PD-L1 inhibitors and for patients whose disease progressed within 90 days, between 90 days to 179 days, 180 days or more, or had an unknown response after platinum therapy. Results from analyses across all relevant subgroups were generally consistent with the primary analyses for ORR.
Figure 2: Forest Plot of Subgroup Analysis of ORR — DCO June 27, 2023
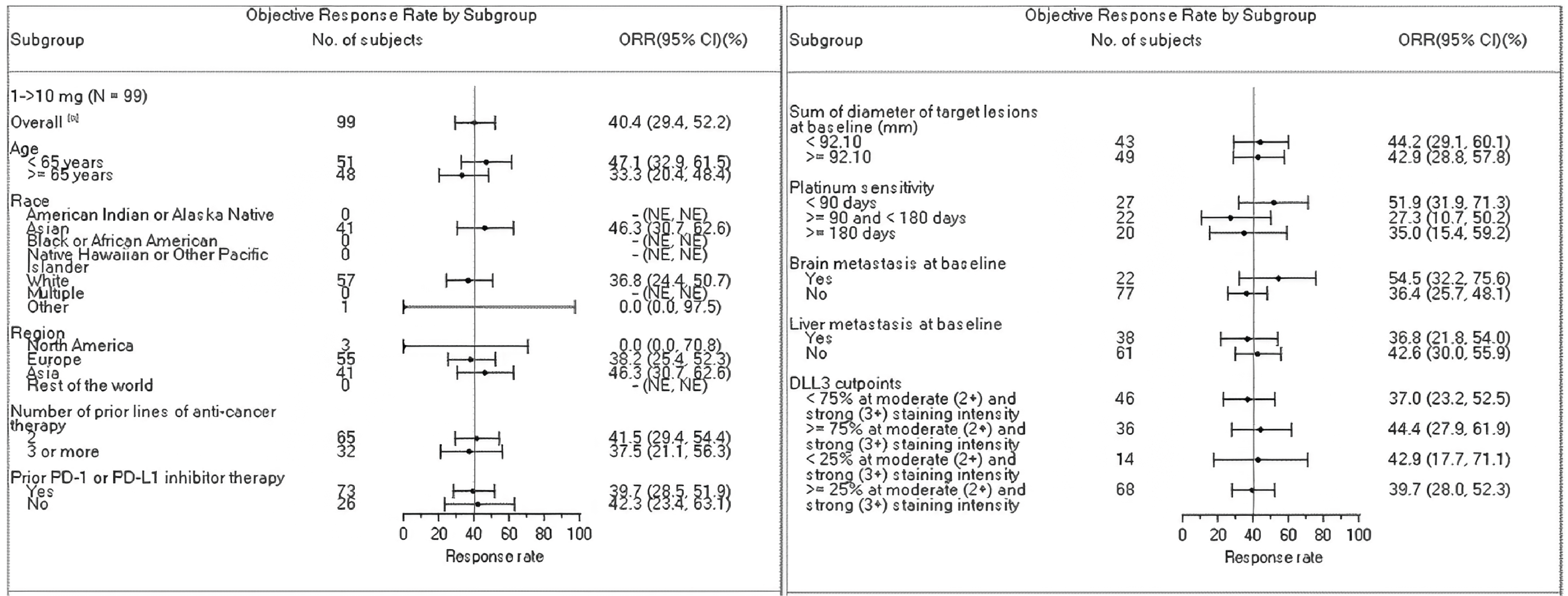
1 - > 10 mg = 1 mg step dose to 10 mg target dose; BICR = blinded independent central review; CI = confidence interval; DCO = data cut-off date; FAS = full analysis set; NE = not estimable; ORR = objective response rate.
Notes: The median of sum of diameter of target lesions at baseline was 92.10, based on the BICR full analysis set for part 1 and part 2 using BICR tumour assessments. Patients with missing or unknown values were not included in the analysis.
Platinum sensitivity is calculated as end of first-line platinum therapy to date of first progression. Patients with missing or unknown values were not included in the analysis.
aThe exact 97.5% CI was calculated using the Clopper-Pearson method. The exact 95% CI was calculated using the Clopper-Pearson method.
Source: Primary Clinical Study Report for the DeLLphi-301 trial.25
Harms
Safety analyses were conducted using the safety analysis set that consists of all patients who received at least 1 dose of tarlatamab. The latest DCO for AEs was October 2, 2023.
Adverse Events
The most frequently reported all-grade TEAEs and grade 3 or higher TEAEs are summarized in Table 19.
As of the June 27, 2023 DCO, 99% of patients experienced TEAEs at the 10 mg dose. The most commonly reported TEAEs (≥ 20% of patients) were CRS (51.5%), decreased appetite (30.3%), pyrexia (39.4%), constipation (31.3%), anemia (28.3%), dysgeusia (24.2%), fatigue (23.2%), and asthenia (21.2%).
Grade 3 or higher AEs were reported for 60 patients (60.6%). The most frequently reported grade 3 or greater TEAEs (≥ 5% of patients) in the 10 mg group were anemia, lymphocyte count decreased, lymphopenia, hyponatremia, fatigue, and asthenia. Of note, CRS was the most frequently reported AE in the 10 mg group, but no patients in this group had grade 3 or higher CRS events. One patient in the 10 mg modified safety monitoring group had a grade 3 and higher CRS event.
Serious Adverse Events
SAEs were reported for 58 patients (58.6%) in the 10 mg target dose group (part 1 and part 2) and 15 patients (44.1%) in the modified safety monitoring 10 mg group as of the June 27, 2023, DCO.
The most frequently reported (≥ 2 patients) SAEs in the 10 mg group (part 1 and part 2) were CRS (26.3%), pyrexia (6.1%), pneumonia (4.0%), respiratory tract infection (3.0%), ICANS (2.0%), and hyponatremia (4.0%). In the part 3 modified safety monitoring 10 mg target dose group, the most frequently reported (≥ 2 patients) SAEs by preferred term were CRS (14.7%), ICANS (5.9%), and respiratory failure (5.9%). The incidence of SAEs reported as of October 2, 2023, was similar to the DCO of June 2023.
Withdrawal Due to AEs
As of the June 27, 2023, DCO, 7 patients (7.1%) in the 10 mg group and 3 patients (8.8%) in the 10 mg modified safety monitoring group had AEs leading to discontinuation of tarlatamab. Reasons for treatment discontinuation included anemia (1.0%), thrombocytopenia (1.0%), cholestasis (1.0%), COVID-19 pneumonia (1.0%), abnormal ECOG PS (1.0%), tumour lysis syndrome (1.0%), and muscular weakness (1.0%). The results of withdrawals due to AEs at the October 2, 2023, DCO were similar to the primary analysis.
Mortality
Fatal AEs were reported for 2 patients (2.0%; 1 pneumonia and 1 respiratory acidosis) in the 10 mg group in part 1 and part 2, and 3 patients (8.8%; 1 aspiration, 1 pulmonary embolism, and 1 respiratory failure) in the part 3 modified safety monitoring 10 mg group, as of the June 27, 2023, DCO and October 2, 2023, DCO. None of the deaths were considered by the investigator to be related to tarlatamab.
Notable Harms
In part 1, part 2, and part 3, CRS occurred in 70 patients (52.6%). One patient in the 10 mg modified safety monitoring group had a grade 3 and higher CRS event.
In the 10 mg group (part 1 and part 2), 9 patients (9.1%) had ICANS events, though none were grade 3 or higher.
Table 19: Summary of Harms Results in DeLLphi-301 — Safety Analysis Set
Harms | DCO: June 27, 2023 | DCO: October 2, 2023 | ||
|---|---|---|---|---|
Part 1 and part 2: Tarlatamab 10 mg n = 99 | Part 3: Tarlatamab 10 mg MSM n = 34 | Part 1 and part 2: Tarlatamab 10 mg n = 99 | Part 3: Tarlatamab 10 mg MSM n = 34 | |
All TEAEs | 98 (99.0) | 34 (100.0) | 99 (100.0) | 34 (100.0) |
Most frequent AEs (> 20%) | ||||
Anemia | 28 (28.3) | 10 (29.4) | 29 (29.3) | (32.4) |
Decreased appetite | 30 (30.3) | 15 (44.1) | — | — |
CRS | 51 (51.5) | 19 (55.9) | 51 (51.5) | 19 (55.9) |
Pyrexia | 39 (39.4) | 10 (29.4) | 40 (40.4) | 10 (29.4) |
Constipation | 31 (31.3) | 8 (23.5) | 31 (31.3) | 8 (23.5) |
Dysgeusia | 24 (24.2) | 16 (47.1) | 25 (25.3) | 17 (50.0) |
Fatigue | 23 (23.2) | 9 (26.5) | 23 (23.2) | 9 (26.5) |
Asthenia | 21 (21.2) | 12 (35.3) | 21 (21.2) | 12 (35.3) |
Nausea | 16 (16.2) | 6 (17.6) | 16 (16.2) | 9 (26.5) |
Dyspnea | 15 (15.2) | 6 (17.6) | 15 (15.2) | 7 (20.6) |
Hypomagnesemia | 10 (10.1) | 7 (20.6) | 12 (12.1) | 7 (20.6) |
Headache | 9 (9.1) | 5 (14.7) | 9 (9.1) | 7 (20.6) |
Diarrhea | 7 (7.1) | 8 (23.5) | 7 (7.1) | 9 (26.5) |
Grade ≥ 3 AEs | 60 (60.6) | 22 (64.7) | 60 (60.6) | 24 (70.6) |
SAEs | 58 (58.6) | 15 (44.1) | 58 (58.6) | 18 (52.9) |
AEs leading to discontinuation | 7 (7.1) | 3 (8.8) | 7 (7.1) | 4 (11.8) |
Death | 2 (2.0) | 3 (8.8) | 2 (2.0) | 3 (8.8) |
Notable harms | ||||
CRS | 51 (51.5) | 19 (55.9) | 51 (51.5) | 19 (55.9) |
CRS, grade ≥ 3 | 0 | 1 (2.9) | 0 | 1 (2.9) |
ICANS | 9 (9.1) | 4 (11.8) | 9 (9.1) | 4 (11.8) |
ICANS, grade ≥ 3 | 0 | 0 | 0 | 0 |
AE = adverse event; ASTCT = American Society for Transplantation and Cellular Therapy; CRS = cytokine release syndrome; CTCAE = Common Terminology Criteria for Adverse Events; DCO = data cut-off date; ICANS = immune effector cell-associated neurotoxicity syndrome; MSM = modified safety monitoring; SAE = serious adverse event; SCLC = small cell lung cancer; TEAE = treatment-emergent adverse event.
Notes: Values are n (%).
For patients with multiple events, only the worst severity grade is reported.
AEs were coded using MedDRA version 26.0. CRS and ICANS events were graded using ASTCT.69 Other AEs were graded using CTCAE version 5.0.
Events of SCLC or disease progression are excluded.
Sources: Primary Clinical Study Report for the DeLLphi-301 trial, DCO June 27, 202325 and Addendum 01 to the Clinical Study Report for the DeLLphi-301 trial, DCO October 2, 2023.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The DeLLphi-301 study is an ongoing, phase II, open-label, single-arm trial evaluating the efficacy and safety tarlatamab in patients with ES-SCLC. The potential influence of selection bias is difficult to ascertain in a single-arm trial. Another key limitation of the DeLLphi-301 trial is the absence of a comparator group to assess the efficacy and harms of tarlatamab compared to other relevant active treatments. In Canada, currently there are no authorized treatments in the 3L setting for patients with ES-SCLC, and the available treatment options are limited. Thus, it is difficult to compare tarlatamab with relevant comparators in a clinical trial. In a single-arm trial, treatment effect of the study drug cannot be directly assessed because the trial design is not able to distinguish what proportion of the estimated treatment response can be attributed to the study drug, placebo effects, a patient’s natural history, or other prognostic factors.
Because it was a single-arm trial, patients in the DeLLphi-301 trial were aware of the intervention that they were taking, which potentially increased the risk of detection bias and performance bias. Moreover, the open-label design limited the interpretability of the subjective study outcomes such as patient-reported outcomes including HRQoL and AEs. To mitigate the impact of this bias, all outcomes included in this review were assessed by both BICR and the investigator using RECIST 1.1. Concordance between assessments by BICR and the investigators was examined for best overall response, and the results showed that the concordance rate between the BICR and investigator for responder versus nonresponder was 89%. The concordance rates for other efficacy outcomes are not provided. Given the open-label design of the trial, it is likely that the BICR would be able to provide less biased tumour assessments compared with the investigator’s assessments.
The primary end point of the DeLLphi-301 trial was ORR. Response rate as a primary study end point in oncology trials has been supported by regulatory guidance (e.g., FDA) for market access, as ORR is directly attributable to the antitumour activity of the study drug. Additionally, ORR is generally based on objective and quantitative assessment and is evaluable with a single-arm design, even though this end point may not always correlate with survival.74 The clinical experts consulted for this review echoed that results of ORR should be interpreted with other efficacy outcomes, such as OS, which remains the most clinically relevant efficacy end point in cancer research. OS and PFS, which were secondary efficacy end points in the DeLLphi-301 study were considered clinically meaningful outcomes for this review. However, time-to-event end points cannot be adequately assessed in a single-arm trial as all patients received the same treatment. As such, the effect of tarlatamab on time-to-event end points can only be considered as exploratory and supportive. At the DCO of May 16, 2024, with a median follow-up time of 15.2 months, 54 patients (54.5%) in part 1 and part 2 had died; while the study is currently ongoing, the 95% CI for OS was still NE, suggesting the immaturity of the survival data. Despite OS and PFS results that were considered clinically meaningful by the clinical experts consulted for this review, because of the combination of the single-arm design, the secondary nature of the outcomes, and the short follow-up duration, the results for survival end points should only be considered supportive of the overall antitumour effect of tarlatamab. The clinical experts consulted for this review also identified brain metastasis–free survival as important, though this was not assessed in the DeLLphi-301 study.
In this trial, HRQoL was assessed using both disease-specific and generic questionnaires. These are validated instruments and MIDs for the scale score have been reported for various cancer types including lung cancer, except for EORTC QLQ-LC13, where an MID was not identified from the literature. According to the available data, results for HRQoL were missing for a large proportion of the study participants. For example, at cycle 12, data were available in only 14 patients for EORTC QLQ-C30 and EORTC QLQ-LC13. Due to the large amount of missing data for HRQoL, results for these end points have a high level of uncertainty and a high risk of bias such that definitive conclusions cannot be drawn for these end points.
In this trial, the sponsor noted that in an interim analysis for dose selection and a superiority test of part 1 and part 2, multiplicity adjustment was performed in the analysis of ORR, and a 97.5% 2-sided CI was used. The lower boundary of the 97.5% 2-sided CI for ORR greater than 15% was considered a clinically meaningful change based on the results from previous open-label studies of pembrolizumab and nivolumab. A detailed description of the methods used to account for multiple testing for response rate was not provided; therefore, it is unclear whether a true multiplicity adjustment has been conducted. There was no control for multiplicity for other efficacy or safety outcomes in the DeLLphi-301 study, thus all secondary end points were considered supportive and should be interpreted with consideration for any increased risk of type I error.
External Validity
In the DeLLphi-301 trial, the median number of prior therapies was 2, all patients received prior platinum-based chemotherapy, and 74% of the patients received prior PD-L1 inhibitor therapy. Almost all patients (98%) had metastatic disease at baseline, and most patients had an ECOG PS of 1 (74%). Based on the feedback from the clinical experts consulted for this review, the eligibility criteria and baseline characteristics of patients enrolled in the DeLLphi-301 trial generally reflected a patient population in clinical practice in Canada that would receive treatment with tarlatamab for ES-SCLC, although in practice, this treatment may be used in a broader population than the DeLLphi-301 trial, such as those with an ECOG PS of 2, or those who had major surgery within 28 days of the first dose of tarlatamab. Although there were no sites in Canada included in the trial, the clinical experts noted that the study findings of the DeLLphi-301 study can be generalized to a population of patients living in Canada.
The experts also confirmed that the use of concomitant therapies and subsequent anticancer therapies were generally consistent with practice in Canada and that the outcome measures in the DeLLphi-301 trial are clinically relevant in clinical trials of ES-SCLC.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.23,24
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed pivotal single-arm trials for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The target of the certainty of evidence assessment was the presence of a clinically important improvement in survival (PFS and OS) and HRQoL, which were considered the most important outcomes to treatment by the clinical experts consulted for this review, and the clinician group and patient group inputs. According to the clinical experts, clinically importance thresholds for the outcomes of OS and PFS were a benefit of at least 2 months over current standard of care for OS and PFS, respectively. Additionally, response to treatment (ORR) was included in the certainty of evidence assessment based on the potential translation to long-term survival outcomes.
The DeLLphi-301 trial, a phase II, single-arm, open-label study of tarlatamab was the only study included in the GRADE assessment.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for tarlatamab from the DeLLphi-301 trial, for the treatment of adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
Long-Term Extension Studies
There were no relevant long-term extension studies submitted for this review.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
No direct comparative evidence between tarlatamab and relevant comparators was identified by the sponsor-conducted systematic review. In an effort to address this evidence gap, the sponsor conducted 1 analysis using propensity score weighting of IPD29 and 2 MAICs,30 which indirectly compared tarlatamab with currently available treatments in patients with ES-SCLC in the 3L+ setting. The objective of this section is to summarize and critically appraise the methods and findings of these studies.
Description of Studies
The sponsor submitted 1 observational study comparing patients in the tarlatamab group of the DeLLphi-301 study to patients who received available therapies for ES-SCLC in the 3L+ setting from the Flatiron Health Research Database (referred to as Flatiron hereafter). Flatiron is a database of electronic health records comprising IPD collected from oncology practices and cancer centres in the US. The DeLLphi-301 study versus the Flatiron study was not randomized, and propensity score weighting was applied in an attempt to adjust for confounding between the cohorts. In addition, the sponsor submitted 2 MAICs that compared the DeLLphi-301 study with sponsor-conducted retrospective observational cohort studies based on aggregate registry data, including the CAS study conducted in the UK and the OOS conducted in Canada.
These studies aimed to assess the relative treatment effects of tarlatamab versus relevant comparator therapies on patients with ES-SCLC who had progressed or recurred following 1 platinum-based regimen and at least 1 other LOT. All studies assessed OS and PFS. TTD, time to next treatment discontinuation or death (TTNTD), and ORR were also assessed in the DeLLphi-301 study versus the Flatiron study.
Of note, the sponsor considered the DeLLphi-301 study versus the Flatiron study to have provided the primary evidence for assessment of comparative effectiveness of tarlatamab versus comparator therapies due to the availability of IPD from the Flatiron study which allowed application of inclusion and exclusion criteria of the DeLLphi-301 study to the Flatiron study to promote similarity and for more complete adjustment of differences in prognostic factors between the tarlatamab and comparator therapy cohorts. According to the sponsor, the Flatiron study comparison also allowed for analyses of the broadest set of clinical outcomes, had high-quality clinical data and sufficient sample size, and was reflective of a contemporaneous population of people with SCLC.
The MAICs were considered by the sponsor as supporting evidence because matching of patient populations and adjustment for baseline differences were more limited in the MAICs relative to the DeLLphi-301 study with the Flatiron study given the availability of only aggregate data from these sources and that only analyses of OS and PFS were feasible. The sponsor’s justification for submitting the MAICs was that the DeLLphi-301 study versus CAS study analysis offered a larger sample size than available in the Flatiron study and reflected use of available 3L+ therapies in the UK, which do not entirely overlap with available therapies in the US reflected in the Flatiron study. The DeLLphi-301 study versus OOS analysis provided evidence from a cohort reflective of the 3L+ ES-SCLC treatment landscape in Canada.
Study Selection Methods
The sponsor conducted a systematic literature review (SLR) to identify clinical trial studies reporting efficacy and safety of interventions for patients with SCLC in the 3L+ setting (referred to as the clinical SLR hereafter). Studies identified by the clinical SLR were further evaluated in a feasibility assessment to select studies for inclusion. Table 20 presents a summary of the selection criteria and methods applied in the process. No studies met the selection criteria. The clinical SLR confirmed that there were no available head-to-head trials between tarlatamab and relevant comparators; therefore, evidence for tarlatamab would be based solely on the single-arm, phase II DeLLphi-301 trial. An external control would be required to create a comparator arm on which to perform unanchored ITC.
The sponsor subsequently conducted a SLR of published real-world observational studies (referred to as the burden of illness [BOI] SLR hereafter) of relevant comparator therapies to identify real-word evidence (RWE) for inclusion into the ITC based on the same selection criteria as the clinical SLR, except that the study design of interest was observational cohort studies as opposed to RCTs and nonrandomized clinical trials. Studies identified by the BOI SLR were further evaluated in a feasibility assessment for their suitability as sources of external control data by comparing key study design elements and reliability of the datasets.
The clinical and BOI SLRs were conducted using multiple literature databases (MEDLINE, Embase, and Cochrane Central Register of Controlled Clinical Trials) and study selection was conducted by 2 independent reviewers, with discrepancies being resolved by a third reviewer. Data extraction and risk of bias assessment was conducted by 1 reviewer and validated by a second reviewer. Risk of bias assessment was performed in the clinical SLR only, and the Cochrane Risk of Bias 2 tool for RCTs and the Downs and Black tool for single-arm trials were used. Reasons for study exclusion were documented.
Table 20: Study Selection Criteria and Methods for Indirect Evidence Submitted by the Sponsor
Characteristics | Study selection criteria |
|---|---|
Population | Adult patients (aged ≥ 18 years) with SCLC with disease progression on or after front-line therapy and at least 1 other prior line of therapy Subgroups of particular interest including but not limited to:
|
Intervention and comparators |
|
Outcome | Treatment pattern (i.e., distribution by real-world 3L+ regimens) and any of the following efficacy outcomes:
|
Study designsa |
|
Exclusion criteria | Studies that did not include interventions or comparators of interest, for example, in a 2L+ population without reporting 3L+ subgroup results and the 3L+ subgroup is < 80% of the overall population |
Databases searched | MEDLINE, Embase, and Cochrane Central Register of Controlled Trials |
Selection process | Articles screened independently by 2 reviewers, with discrepancies being resolved by a third reviewer |
Data extraction process | Conducted by 1 reviewer and validated by a second reviewer |
Quality assessment | Conducted by 1 reviewer using the Cochrane Risk of Bias 2 tool for RCTs, and the Downs and Black checklist for single-arm trials; and validated by a second reviewer |
2L+ = second-line or later; 3L+ = third line or later; CAV = cyclophosphamide, doxorubicin, and vincristine; ES-SCLC = extensive-stage small cell lung cancer; RCT = randomized controlled trial; SCLC = small cell lung cancer.
aIn the burden of illness SLR, observational cohort studies, as opposed to RCTs and nonrandomized clinical trials, were of interest.
Sources: Sponsor’s Evidence Synthesis Feasibility Assessment Report.75 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Comparative Evidence Analysis Methods
IPD Propensity Score Weighting (DeLLphi-301 Versus Flatiron)
Sample selection: Key patient eligibility criteria from the DeLLphi-301 trial were applied to patients from the Flatiron study to create an external control arm of patients receiving comparator therapies in the 3L+ setting for ES-SCLC. Key inclusion and exclusion criteria for patients initiating tarlatamab in the DeLLphi-301 study are described in Table 21, along with the application of these criteria in the Flatiron study data to select the comparator therapies cohort.
The following trial inclusion and exclusion from the DeLLphi-301 trial could not be applied to the Flatiron study cohort due to lack of collection, missingness, or inapplicability in the Flatiron study:
measurable lesions as per RECIST 1.1 within 21 days before the first dose of tarlatamab
willingness to provide archived tumour tissue samples or undergo pretreatment tumour biopsy (exceptions allowed)
minimum life expectancy of 12 weeks.
Table 21: Inclusion and Exclusion Criteria in Tarlatamab and Comparator Therapies Cohorts
Tarlatamab (DeLLphi-301) | Comparator therapies (Flatiron) |
|---|---|
Inclusion criteria | |
Initiated tarlatamab 10 mg | Initiated comparator therapies currently available in Canada in the 3L+ settings, including CAV, topotecan, irinotecan, platinum-based regimens without PD-1 or PD-L1 inhibitor immunotherapy, taxanes, and others |
Aged ≥ 18 years at screening | Aged ≥ 18 years at initiation of 3L+ treatment |
Histologically or cytologically confirmed SCLC |
|
ES-SCLC; progressed or recurred following 1 platinum-based regimen and at least 1 other prior LOT before tarlatamab initiation (i.e., patients are 3L+ at tarlatamab initiation) |
|
ECOG PS of 0 or 1 at screening | ECOG PS of 0 or 1 within a window of 28 days before or 7 days after initiation of 3L+ treatment |
|
|
Adequate organ function, defined based on laboratory/test values for markers of hematological, coagulation, renal, hepatic, pulmonary, and cardiac function | Proxied by patients initiating 3L+ treatment, given that adequate organ function is required before initiating any anticancer therapies in clinical practice |
Exclusion criteria | |
Untreated or symptomatic CNS metastases or leptomeningeal disease | Untreated brain or CNS metastases as proxied by neither a record of treatment for brain metastases nor initiation of 3L+ ES-SCLC treatment |
Evidence of interstitial lung disease or active, noninfectious pneumonitis | Not applied to the Flatiron study cohort Initiating 3L+ treatment was considered to reflect that active management was in place or a justification for not actively treating interstitial lung disease or active noninfectious pneumonitis was made before initiating anticancer treatment |
History of other malignancies within the past 2 years | Evidence of other malignancies except carcinoma in situ within 2 years prior |
History of MI, symptomatic CHF, or arterial thrombosis within the 12 months prior | History of MI, CHF, or arterial thrombosis within 12 months prior |
| Not applied to the Flatiron study cohort as this was not considered prognostic for outcomes in this population |
Prior therapy with tarlatamab | Not applicable |
Currently receiving or recently ended treatment with another investigational device or drug study | Received any investigative agents as part of a clinical trial during any LOT (including 3L+) |
3L+ = third line or later; CAV = cyclophosphamide, doxorubicin, and vincristine; CHF = congestive heart failure; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ES-SCLC = extensive-stage small cell lung cancer; LOT = line of treatment; MI = myocardial infarction; NSCLC = non–small cell lung cancer; SCLC = small cell lung cancer; vs. = versus.
aIncluding paclitaxel and docetaxel.
Sources: DeLLphi-301 vs. Flatiron Technical Report.29
Index line selection in the comparator therapies cohort: Some patients in the comparator therapies cohort met eligibility criteria at more than 1 LOT. For these patients, 1 eligible LOT was identified and included in the analysis using a propensity score-based approach as follows. First, all eligible LOTs for comparator therapies cohort were identified. A propensity score model at the level of LOT was fit using all eligible LOTs for the comparator therapies cohort and all LOTs for patients in the tarlatamab cohort. The dependent variable in the model was the treatment indicator (i.e., whether a LOT was treated with tarlatamab or not), while the independent variables included age at index, sex, ECOG PS at index, disease stage at diagnosis, number of previous LOTs at index, chemotherapy-free interval (CFI) after 1L therapy, presence of brain metastases at index, time from SCLC diagnosis to index date, and smoking status. The model was fit using a generalized estimating equation approach with a logistic link function to account for any correlation across multiple eligible LOTs for the same patient. A propensity score for each LOT was estimated as the model-predicted probability of the patient in that LOT being treated with tarlatamab conditional on the adjusted baseline prognostic factors. For patients in the comparator therapies cohort with multiple eligible LOTs, their LOT with the highest propensity score was selected as their index LOT for use in all subsequent analytic steps.
Use of this approach intends to allow for selection of a single LOT for the patients who were eligible at multiple LOTs that most reduced imbalances of prognostic factors across the tarlatamab and available treatment option cohorts. All subsequent analyses used a single LOT for all patients and were conducted at the patient level.
Propensity score adjustment: Prognostic factors for outcomes in SCLC were identified by the sponsor through a multistep approach, incorporating literature review, empirical analyses of available data, and expert input. Propensity score weighting was implemented at the patient level to reduce the imbalances of prognostic factors between the tarlatamab and comparator therapies cohorts. In the primary analysis, confounding variables considered by the sponsor to be of high (age, sex, ECOG PS, disease stage at diagnosis, number of previous LOTs, smoking status) and medium importance (CFI after 1L therapy, presence of brain metastasis, time from SCLC diagnosis to baseline) were adjusted for.
Standardized mortality weighting was used to reweight patients so that the distributions of baseline characteristics of patients treated with comparator therapies matched that of patients treated with tarlatamab. This approach is consistent with estimating the average treatment effect in the treated approach. Density plots of the propensity score distribution were generated to visually confirm adequate distributional overlap of propensity score scores between the 2 treatment groups.
To assess whether balance in confounding factors was achieved by the propensity score adjustment, the distributions of baseline characteristics both preweighting and postweighting were summarized. Standardized mean differences for the differences between tarlatamab and comparator therapy cohorts were also calculated. E-values, defined as the minimum strength of association that an unmeasured confounder would need to have with both the exposure and the outcome, conditional on adjusted covariates, to fully explain observed tarlatamab outcome HRs were calculated for OS, TTD, and PFS outcomes.
Outcome analyses: Efficacy outcomes including OS, OS adjusted for postprogression use of tarlatamab, TTD, TTD adjusted for postprogression use of tarlatamab, TTNTD, PFS, and ORR were compared between the tarlatamab and comparator therapies cohorts before and after propensity score weighting. Note that results for TTNTD are not of interest to this review and will not be further summarized.
OS adjusted for postprogression use of tarlatamab was estimated through the 2-stage method in a post hoc analyses of the DeLLphi-301 study.76,77 TTD adjusted for postprogression use of tarlatamab was defined as the TTD that would have been observed had tarlatamab only been used until disease progression. In this analysis, patients with postprogression use of tarlatamab were instead treated as having discontinued at time of progression, with their event or censoring status and times updated accordingly.
For OS, TTD, and PFS, comparisons were conducted using unweighted and weighted Kaplan-Meier analyses and log rank tests. In addition, HRs were estimated before and after weighting using unweighted and weighted Cox proportional hazards models, respectively. The 95% CIs for HRs were estimated using bootstrapping. The proportional hazards assumption was assessed using log cumulative hazard plots and Schoenfeld residuals tests.
For ORR, the proportion of patients achieving CR or PR was summarized and compared between the tarlatamab and available treatment option cohorts before and after weighting. Odds ratios were estimated before and after weighting using unweighted and weighted logistic regression models, respectively. The 95% CIs for odd ratios were estimated based on bootstrapping.
Sensitivity analyses: Three sensitivity analyses were conducted. First, PFS analyses were repeated considering only patients in the Flatiron study datasets whose progression was determined based on radiographic evidence alone as PFS events.
Second, in addition to the adjustment of high and medium importance confounding variables, adjustment of the lower importance confounding variables (i.e., prior PD-1 or PD-L1 inhibitor use, liver metastases, race) were included in propensity score models as well to assess robustness of the relative treatment effect to these additional adjustments.
Third, to reflect the entire range of comparator therapies received in 3L+ settings in the Flatiron study data, analyses were repeated including all eligible index therapies in the available treatment cohort. PFS analyses based on radiographic evidence alone and adjustment of lower importance confounding variables were also conducted for this overall Flatiron study analysis to assess the robustness of the results.
MAICs (DeLLphi-301 Versus CAS and DeLLphi-301 Versus OOS)
An unanchored MAIC approach was selected by the sponsor for the indirect comparison analyses owing to the noncomparative nature of the DeLLphi-301 study and the lack of a common comparator. IPD were available from the DeLLphi-301 study while aggregate patient data were available from the comparator studies (the CAS study and OOS). The covariates used for match adjustment were identified using a multistep approach, incorporating literature review, empirical analyses of available data (including multivariable regressions based on the DeLLphi301 trial data and meta-regression of published studies of 2L and 3L+ treatment for SCLC), and expert input. The adjustment factors were prioritized by level of prognostic importance as follows based on clinical expert opinion:
highest importance — ECOG PS, age, disease stage, and number of previous LOTs
medium importance — CFI, sex, brain metastases, and time from SCLC diagnosis
lowest importance — previous use of PD-1 or PD-L1 inhibitor, liver metastases, and race.
In the base-case analysis of the DeLLphi-301 study versus the CAS study, match adjustments were applied to covariates including age at index, sex, ECOG PS at index, brain metastases (at index for tarlatamab versus at 1L for comparator), liver metastases (at index for tarlatamab versus at 1L for comparator), disease stage at diagnosis, CFI, and time from diagnosis to index. Two scenario analyses were conducted to assess the impact of excluding CFI (scenario analysis 1) and disease stage at diagnosis (scenario analysis 2) from match adjustment on study results.
In the base-case analysis of the DeLLphi-301 study versus the CAS study, match adjustments were applied to covariates including age at index, sex, disease stage at diagnosis, and time from diagnosis to index. Two scenario analyses were conducted to assess the impact of excluding time from diagnosis to index (scenario analysis 1) and disease stage at diagnosis (scenario analysis 2) from match adjustment on study results.
In each MAIC, patients in the tarlatamab group were assigned weights such that after weighting, average characteristics of patients in the tarlatamab group matched those reported for patients in the comparator therapies cohort, and each patient’s weight was equal to their estimated odds of being in the comparator therapies cohort versus the tarlatamab group. Weights were obtained based on a logistic regression model for the odds of enrolment in the comparator therapies group versus the tarlatamab group, with all matched-on baseline characteristics included as independent variables in the model. Because only summary statistics for baseline characteristics were available for the aggregate population, a method of moments estimator was used to estimate the parameters of this logistic regression model. The weights obtained were used to calculate the ESS achieved after weighting patients.78,79 Baseline patient characteristics before and after weighting were presented.
Kaplan-Meier curves of OS and PFS reported for the comparator therapies cohort were digitized and pseudo-IPD were created. The pseudo-IPD were then combined with the tarlatamab IPD, and unadjusted OS and PFS for each group were generated based on Kaplan-Meier analyses. HRs and 95% CIs were estimated from Cox proportional hazards models comparing these 2 cohorts.
Adjusted survival functions were based on weighted Kaplan-Meier analyses. Adjusted HRs and 95% CIs were based on weighted Cox proportional hazards models. The 95% CIs for adjusted HRs were estimated using a robust sandwich estimator to account for the uncertainty in the estimation of the underlying MAIC weights.
A summary of the analysis methods for IPD propensity score weighting analysis and the MAICs is shown in Table 22.
Table 22: Propensity Score Weighting and MAIC Analysis Methods
Methods | DeLLphi-301 vs. Flatiron | DeLLphi-301 vs. CAS | DeLLphi-301 vs. OOS |
|---|---|---|---|
Analysis methods | IPD propensity score weighting | Unanchored MAIC | |
Covariates used for match adjustment (base case) | Age, sex, ECOG PS, disease stage at diagnosis, number of previous LOTs, smoking status, CFI after 1L therapy, presence of brain metastasis, time from SCLC diagnosis to baseline | Age at index, sex, ECOG PS at index, brain metastases (at index for tarlatamab vs. at 1L for comparator), liver metastases (at index for tarlatamab vs. at 1L for comparator), disease stage at diagnosis, CFI, time from diagnosis to index | Age at index, sex, disease stage at diagnosis, time from diagnosis to index, liver metastases at diagnosis |
Outcomes of interest | OS, PFS, ORR, TTD | OS, PFS | |
Scenario analyses | Analysis 1: Considering only patients whose progression was determined based on radiographic evidence alone as PFS events Analysis 2: Adjusting for confounding variables of high, medium, and low prognostic importance Analysis 3: Includes all eligible index therapies in the Flatiron study cohort | Scenario 1: Same match adjustment covariates as the base case, excluding CFI Scenario 2: Same match adjustment covariates as the base case, excluding disease stage at diagnosis | Scenario 1: Same match adjustment covariates as the base case, excluding time from diagnosis to index Scenario 2: Same match adjustment covariates as the base case, excluding disease stage at diagnosis |
1L = first-line; CAS = Cancer Analysis System; CFI = chemotherapy-free interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IPD = individual patient data; LOT = line of treatment; MAIC = matching-adjusted indirect comparison; OOS = Oncology Outcomes Study; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SCLC = small cell lung cancer; TTD = time to treatment discontinuation or death; vs. = versus.
Sources: DeLLphi-301 vs. Flatiron Technical Report,29 and Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30
Results
Summary of Included Studies
Seven published RWE studies were identified by the BOI SLR. These studies, along with 3 sponsor-conducted RWE registry studies with unpublished data, were considered by the sponsor as potential sources for external control data and were assessed in a feasibility assessment. All 7 published RWE studies were identified by the BOI SLR but were subsequently excluded due to small sample sizes (the Steffens et al.,80 Aktas et al.,81 and Inomata et al.82 studies); limited reporting of all efficacy end points that are relevant for ITCs supporting reimbursement or health technology assessment submissions (the Simos et al.83 and Nagy-Mignotte et al.84 studies); inclusion of a sizable portion of patients with ECOG PS of 2 or higher; or patients who were only eligible or received best supportive care, which were not included in the DeLLphi-301 study (the Coutinho et al.85 and Saruwatari et al.86 studies). As well, the study periods for the Coutinho et al.85 and Saruwatari et al.86 studies were before 2016, with no data that reflect treatment pattern changes (e.g., lurbinectedin’s approval in 2L in 2016) and improved supportive care over time.
The 3 sponsor-conducted RWE studies (the CAS, OOS, and Flatiron studies) were eventually selected by the sponsor as the sources for external control data. The Flatiron and CAS studies were considered by the sponsor to be the most suitable sources of evidence given reasonable sample size, comparable eligibility criteria versus the DeLLphi-301 study, and availability of baseline characteristics for potential adjustments in ITCs, IPD (Flatiron study only), and relevant efficacy end points. The sponsor noted that while the OOS had a smaller sample size and limited availability of baseline characteristics for adjustments in ITCs, it reflected treatment patterns and outcomes for a population of people requiring 3L treatment for ES-SCLC in a setting in Canada and was thus deemed valuable for exploratory analyses by the sponsor.
The characteristics of the DeLLphi-301 study, Flatiron study, CAS study, and OOS are summarized in Table 23. The DeLLphi-301 study was a phase II, single-arm, open-label trial of tarlatamab conducted in Europe, East Asia, and North America, while the Flatiron study, CAS study, and OOS were retrospective observational cohort studies using registry data of patients receiving available treatment for SCLC in the US, UK, and Canada, respectively. Data collection occurred between 2021 and 2023 in the DeLLphi-301 study and started earlier in the Flatiron study (2013 to 2021), CAS study (2013 to 2022), and OOS (2011 to 2022). The CAS study and OOS included adult patients receiving therapies for ES-SCLC in the 3L setting, while the DeLLphi-301 study and the Flatiron study included patients in the 3L+ setting. Patients in the DeLLphi-301 study, Flatiron study, and CAS study were required to have previously received 1 line of platinum-based chemotherapy and have an ECOG PS of 0 or 1 but such requirements did not exist for OOS. In the DeLLphi-301 study, Flatiron study, and CAS study, the majority of patients had ES-SCLC at diagnosis (80%, 68%, and 95%, respectively) and all patients had an ECOG PS score of 0 or 1 and had previously received platinum-based chemotherapy at baseline, while these data were not reported in OOS. The studies also differed by sample sizes (the DeLLphi-301 study, N = 97; Flatiron study, N = 184; CAS study, N = 540; and OOS, N = 71) and proportion of patients with brain metastases at baseline (the DeLLphi-301 study, 22%; Flatiron study, 21%; CAS study, 5%; and not reported in OOS). As well, the definition of PFS differed. In the DeLLphi-301 and Flatiron studies, PFS was defined as time to disease progression or death; disease progression was assessed based on radiographic progression as per RECIST 1.1 criteria in the DeLLphi-301 study and based on radiographic imaging, pathology reports, or clinical examination in the Flatiron study. In the CAS study and OOS, PFS was proxied by time to TTD as data on progression were not available. Tarlatamab was administered as the study intervention in the DeLLphi-301 study. In the Flatiron study, patients received the following 3L+ therapy, including PD-L1–based therapy, lurbinectedin, topotecan, platinum-based chemotherapy, irinotecan, CAV, and other 3L+ therapies. In the CAS study, patients received topotecan (40%), CAV or other chemotherapy regimens (35%), platinum and etoposide regimens without PD-L1 inhibitor therapy (20%), and other platinum-containing regimens (5%). In OOS, patients received topotecan (20%), CAV (30%), platinum and etoposide regimens (24%), and other therapies (27%).
Table 23: Summary of Included Studies in Propensity Score Weighting Analysis and MAICs
Characteristics | DeLLphi-301 (index study) | Flatiron (comparator study) | CAS (comparator study) | OOS (comparator study) |
|---|---|---|---|---|
Study design | Phase II, single-arm, open-label trial | Retrospective observational cohort study (registry data–based) | Retrospective observational cohort study (registry data–based) | Retrospective observation cohort study (registry data–based) |
Study site | Europe, East Asia, North America (0 sites in Canada) | US | UK | Canada |
IPD available | Yes | Yes | No | No |
Period of data collection | December 2021 to June 2023 | January 2013 to October 2021 | July 2013 to May 2022 | January 2011 to December 2022 |
Population | 3L+ ES-SCLC (N = 97) | 3L+ ES-SCLC (N = 184) | 3L ES-SCLC (N = 540) | 3L ES-SCLC (N = 71) |
Key inclusion or exclusion criteria |
|
|
|
|
Intervention (%) | Tarlatamab (100%) |
|
|
|
Comparator | None | None | None | None |
Outcomes of interest | OS, PFS | OS, PFS, ORR, and TTD | OS, PFS | OS, PFS |
Definition of OS | Time from index treatment initiation to death due to any cause. An “adjusted OS” accounting for patients’ postprogression use of tarlatamab was measured. | Time from index date to death. | Time from index treatment initiation to death due to any cause. | Time from index treatment initiation to death due to any cause. |
Definition of PFS | Time from randomization to the earlier of disease progression per RECIST 1.1 or death. | Defined as time from index date to disease progression or death, whichever occurred first. Disease progression was retrospectively captured and identified based on clinical documentation of progression from different sources of evidence, which may have included radiographic imaging, pathology reports, or clinical examination. | Proxied by time from treatment initiation to the earlier of discontinuation or death. | Proxied by time from treatment initiation to the earlier of discontinuation or death. |
Definition of ORR | Defined as the proportion of patients with CR or PR per RECIST 1.1 before initiation of a subsequent anticancer treatment, if any. | Defined as the proportion of all treated patients with CR or PR. Response assessments were identified based on clinical documentation of progression from various sources of evidence, including interpretation of a radiology or pathology report, or clinical examination. | NA | NA |
Definition of TTD | Defined as time from index date to TTD, whichever occurred first. TTD adjusted for postprogression use of tarlatamab was defined as the TTD that would have been observed had tarlatamab only been used until disease progression. In this analysis, patients with postprogression use of tarlatamab were instead treated as having discontinued at time of progression, with their event/censoring status and times updated accordingly. | Defined as time from index date to TTD, whichever occurred first. | NA | NA |
Baseline disease characteristics and treatment history | ||||
Disease stage at diagnosis | LS-SCLC (22%) ES-SCLC (78%) | LS-SCLC (32%) ES-SCLC (68%) | LS-SCLC (15%) ES-SCLC (85%) | NR |
ECOG PS | 0 or 1: 100% | 0 or 1: 100% | 0 or 1: 100% | NR |
Brain metastases at baseline | 22% | 21% | 5% | NR |
Prior platinum-based chemotherapy | 100% | 100% | 100% | NR |
Number of prior LOTs | 2 (66%) ≥ 3 (32%) | 2 (68%) 3 (32%) | 2 (100%) | 2 (100%) |
3L = third line; 3L+ = third line or later; CAS = Cancer Analysis System; CAV = cyclophosphamide, doxorubicin, and vincristine; CR = complete response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ES = extensive stage; ES-SCLC = extensive-stage small cell lung cancer; IPD: individual patient data; LOT = line of treatment; LS-SCLC = limited-stage small cell lung cancer; NA = not applicable; NR = not reported; NSCLC = non–small cell lung cancer; OOS = Oncology Outcomes Study; ORR = objective response rate; OS = overall survival; PD-L1 = programmed cell death 1 ligand 1; PFS = progression-free survival; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SCLC = small cell lung cancer; TTD = time to treatment discontinuation or death; vs. = versus.
Sources: DeLLphi-301 vs. Flatiron Technical Report,29 and Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Study Results (DeLLphi-301 Versus Flatiron)
Analyses in this study were based on the October 2, 2023, DCO date of the DeLLphi-301 trial.
Sample Selection
Of the 99 patients who received at least 1 dose of tarlatamab in the DeLLphi-301 trial, 2 patients had only 1 prior LOT and were excluded, resulting in a total of 97 patients in the analytical sample. Of the 331 patients in the Flatiron study cohort who initiated 3L+ treatments, 184 patients met the selection criteria as outlined in Table 21 and were included for index line selection. To reflect treatment patterns in Canada, 68 out of 184 patients (37%) were excluded as their 3L+ treatments contained lurbinectedin or PD-1 or PD-L1 inhibitors, which were not approved or reimbursed in Canada. Therefore, a total of 116 patients with 145 eligible LOTs were included in the comparator therapies (Flatiron study cohort) for the index line selection in the next step. Among them, 93 patients had a unique eligible LOT, while 23 patients had multiple eligible LOTs.
Index Treatment and Line Distribution
In terms of line distribution, the most common index treatment line of tarlatamab was 3L (67.0%), followed by fourth line (18.6%), and others (fifth line to seventh line, 14.4%). For comparator therapies, before line selection, the most common index treatment line was 3L (60.7%), followed by fourth line (29.7%), and others (fifth line and sixth line, 9.7%). After line selection, the most common index line was 3L (70.7%), followed by fourth line (20.7%), and others (fifth line and sixth line, 8.6%). The most common index treatment was topotecan (30.2%), followed by paclitaxel (15.5%), carboplatin and etoposide combination therapy (6.9%), carboplatin and irinotecan combination therapy (6.9%), irinotecan (6.0%), cisplatin and irinotecan combination therapy (3.4%), and docetaxel (3.4%).
Baseline Patient Characteristics Before and After Propensity Score Adjustment
The baseline characteristics for tarlatamab and comparator therapies cohorts prepropensity score weighting and postpropensity score weighting are presented in Table 24. Comparing the baseline characteristics between the tarlatamab cohort and the comparator therapies cohort after weighting, the tarlatamab cohort had a smaller proportion of patients who were white (56.7%), had ECOG PS score of 0 (25.8%), had CFI of 180 days or more after 1L therapy (38.1%), had previously received PD-1 or PD-L1 inhibitors (73.2%), and had liver metastasis (38.1%) compared with the weighted comparator therapies cohort (74.9%, 33.2%, 44.7%, 80.0%, and 45.9%, respectively). Ninety-seven patients were included in the tarlatamab cohort. The ESS for the comparator therapies cohort was 62 (53.4% of the original sample size) after weighting.
Table 24: Baseline Patient Characteristics for the Tarlatamab and Flatiron Cohorts, Before and After Weighting
Baseline characteristics | Tarlatamab (DeLLphi-301) N = 97 | Comparator therapies (Flatiron) | |
|---|---|---|---|
Before weighting N = 116 | After weighting ESS = 62 | ||
Age at index (years), mean (SD) | 63.5 (8.7) | 65.8 (8.4) | 63.0 |
Sex | |||
Female | 26 (26.8) | 61 (52.6) | (28.8) |
Male | 71 (73.2) | 55 (47.4) | (71.2) |
Race or ethnicity | |||
White | 55 (56.7) | 88 (75.9) | (74.9) |
Non-white | 42 (43.3) | 28 (24.1) | (25.1) |
Smoking history before index | |||
Ever smoking | 89 (91.8) | 112 (96.6) | (92.0) |
Never smoking | 8 (8.2) | 4 (3.4) | (8.0) |
ECOG PS at index | |||
0 | 25 (25.8) | 39 (33.6) | (33.2) |
1 | 72 (74.2) | 77 (66.4) | (66.8) |
Number of previous lines of therapies | |||
2 | 65 (67.0) | 82 (70.7) | (71.4) |
3 | 18 (18.6) | 24 (20.7) | (16.8) |
> 3 | 14 (14.4) | 10 (8.6) | (11.8) |
CFI after 1L therapy | |||
< 90 days | 19 (19.6) | 31 (26.7) | (14.1) |
≥ 90 and < 180 days | 41 (42.3) | 41 (35.3) | (41.2) |
≥ 180 days | 37 (38.1) | 44 (37.9) | (44.7) |
Time from SCLC diagnosis to index (years), mean (SD) | 1.6 (0.8) | 1.5 (0.8) | 1.6 (NR) |
TNM stage at diagnosis | |||
Stage I to II | 3 (3.1) | 6 (5.2) | (5.1) |
Stage III | 18 (18.6) | 18 (15.5) | (18.5) |
Stage IV | 65 (67.0) | 77 (66.4) | (64.4) |
Unknown | 11 (11.3) | 15 (12.9) | (12.0) |
Previous use of PD-1 or PD-L1 inhibitors | 71 (73.2) | 95 (81.9) | (80.0) |
Brain metastases before or on index | 22 (22.7) | 40 (34.5) | (24.6) |
Liver metastases before or on index | 37 (38.1) | 55 (47.4) | (45.9) |
1L = first line; CFI = chemotherapy-free interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; SCLC = small cell lung cancer; SD = standard deviation; TNM = tumour, node, metastasis; vs. = versus.
Values are n (%) unless otherwise indicated.
Sources: DeLLphi-301 vs. Flatiron Technical Report.29
The comparisons of OS (with postprogression adjustment), PFS, ORR, and TTD (with postprogression adjustment) between the tarlatamab (the DeLLphi-301 study) and comparator therapy (the Flatiron study) cohorts before and after weighing are shown Table 25.
OS Adjusted for Postprogression Use of Tarlatamab
The median follow-up time for OS (with postprogression adjustment) was 13.4 months for the tarlatamab cohort, and 16.7 months for the comparator therapies cohort. The median OS was 14.3 months (95% CI, 10.5 months to NE) for the tarlatamab cohort versus 6.6 months (95% CI, 4.8 months to 10.2 months) for the comparator therapies cohort after weighting. The HR of OS (with postprogression adjustment) between the tarlatamab cohort and the weighted comparator therapies cohort was 0.47 (95% CI, 0.30 to 0.79; P = 0.003), in favour of tarlatamab. The E-value for the observed association between tarlatamab and OS (with postprogression adjustment) was 2.75. Results were consistent with the OS analysis that was not adjusted for postprogression use of tarlatamab (tarlatamab versus weighted comparator therapies cohort, HR = 0.45 [95% CI, 0.28 to 0.74; P = 0.001]). Results of the sensitivity analyses were consistent with the primary analysis.
Progression-Free Survival
The median PFS was 4.9 months (95% CI, 2.9 months to 6.7 months) for the tarlatamab cohort versus 3.0 months (95% CI, 1.9 months to 3.9 months) for the comparator therapies cohort after weighting. The HR of PFS between the tarlatamab cohort and the weighted comparator therapies cohort was 0.61 (95% CI, 0.41 to 0.94; P = 0.021), in favour of tarlatamab. The E-value for the observed association between tarlatamab and PFS was 2.15. Results of the sensitivity analyses were consistent with the primary analysis.
Objective Response Rate
The ORR was 40% for the tarlatamab cohort versus 23% for the comparator therapies cohort after weighting. The odds ratio between the tarlatamab cohort and the weighted comparator therapies cohort was 2.29 (95% CI, 1.05 to 5.58; P = 0.05), in favour of tarlatamab. Results of the sensitivity analyses were in general consistent with the primary analysis, except for the sensitivity analysis where prognostic factors of low importance were adjusted for, in addition to factors of high and medium importance, and results did not favour either intervention.
TTD Adjusted for Postprogression Use of Tarlatamab
The median TTD was 3.65 months (95% CI, 2.37 months to 5.36 months) for the tarlatamab cohort versus 2.33 months (95% CI, 1.41 months to 3.25 months) for the comparator therapies cohort after weighting. The HR of TTD (with postprogression adjustment) between the tarlatamab cohort and the weighted comparator therapies cohort was 0.60 (95% CI, 0.42 to 0.90; P = 0.010), in favour of tarlatamab. The E-value for the observed association between tarlatamab and TTD (with postprogression adjustment) was 1.81. Results were consistent with the TTD analysis that was not adjusted for postprogression use of tarlatamab (tarlatamab versus weighted comparator therapies cohort, HR = 0.47 [95% CI, 0.32 to 0.70; P < 0.001]).
Table 25: Results of the DeLLphi-301 vs. Flatiron Study
Outcomes | Tarlatamab (DeLLphi-301) | Comparator therapies (Flatiron) | |
|---|---|---|---|
Before weighting | After weighting | ||
OS adjusted for postprogression use of tarlatamab | |||
N | 97 | 116 | ESS = 62 |
Median follow-up time (months)a | 13.37 | 16.69 | |
Events of total (%) | 43 of 97 (44.33) | 82 of 116 (70.69) | |
Median OS (months) (95% CI) | 14.29 (10.51 to NE) | 5.39 (4.50 to 6.37) | 6.57 (4.76 to 10.18) |
HR (95% CI) of tarlatamab vs. comparator therapies | Reference | 0.39 (0.26 to 0.56) | 0.47 (0.30 to 0.79) |
P value | Reference | < 0.001 | 0.003 |
PFS | |||
N | 97 | 115 | ESS = 62 |
Median follow-up time (months)a | 13.60 | 13.34 | |
Events of total (%) | 66 of 97 (68.04) | 94 of 115 (81.74) | |
Median PFS (months) (95% CI) | 4.93 (2.86 to 6.74) | 2.73 (2.10 to 3.22) | 2.96 (1.94 to 3.88) |
HR (95% CI) of tarlatamab vs. comparator therapies | Reference | 0.55 (0.40 to 0.76) | 0.61 (0.41 to 0.94) |
P value | Reference | < 0.001 | 0.021 |
ORR | |||
N | 97 | 116 | ESS = 62 |
ORR (%) | 40 | 20 | 23 |
Odds ratio (95% CI) of tarlatamab vs. comparator therapies | Reference | 2.72 (1.49 to 5.06) | 2.29 (1.05 to 5.58) |
P value | Reference | 0.001 | 0.05 |
TTD adjusted for postprogression use of tarlatamab | |||
N | 97 | 116 | ESS = 62 |
Median follow-up time (months)a | 13.83 | 10.58 | |
Events of total (%) | 78 of 97 (80.41) | 95 of 116 (81.90) | |
Median TTD (months) (95% CI) | 3.65 (2.37 to 5.36) | 2.07 (1.41 to 2.33) | 2.33 (1.41 to 3.25) |
HR (95% CI) of tarlatamab vs. comparator therapies | Reference | 0.58 (0.42 to 0.78) | 0.60 (0.42 to 0.90) |
P value | Reference | < 0.001 | 0.010 |
CI = confidence interval; ESS = effective sample size; HR = hazard ratio; NE = not estimable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TTD = time to treatment discontinuation or death; vs. = versus.
aMedian follow-up time was estimated using the reversed Kaplan-Meier approach.
Sources: Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30
Study Results (DeLLphi-301 Versus CAS and DeLLphi-301 Versus OOS)
Baseline Patient Characteristics of Included Studies
The ESS for the tarlatamab cohort after match adjustment was 27.05 (27.9% of the original sample size) for the DeLLphi-301 study versus CAS study analysis and 59.65 (61.5% of the original sample size) for the DeLLphi-301 study versus OOS. The comparator therapies cohort in the DeLLphi-301 study versus CAS study analysis and the DeLLphi-301 study versus OOS analysis consisted of 540 and 71 patients, respectively.
Overall Survival
Following match adjustment, the HR of OS in the base case was in favour of tarlatamab over comparator therapies for the DeLLphi-301 study versus CAS study MAIC (HR = █████; 95% CI, █████ to █████; P = 0.0001) and the DeLLphi-301 study versus OOS MAIC (HR = 0.291; 95% CI, 0.184 to 0.459; P = 0.0000). Results of the scenario analyses were consistent with the base-case analysis (Table 26). Kaplan-Meier curves for OS are shown in Figure 3 and Figure 4.

Figure 4: Kaplan-Meier Curves of OS (DeLLphi-301 vs. OOS)
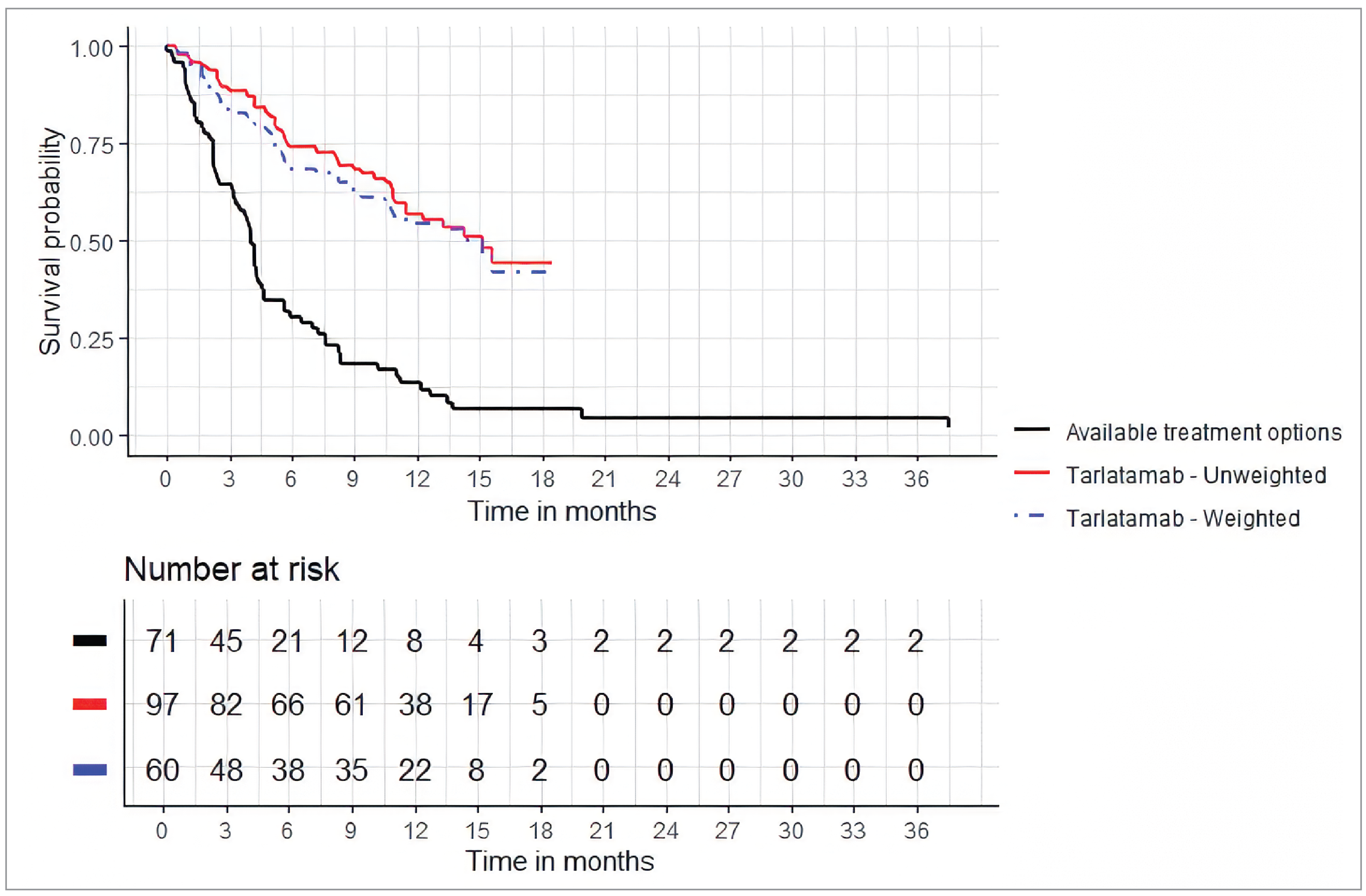
OOS = Oncology Outcomes Study; OS = overall survival; vs. = versus.
Sources: Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30
Progression-Free Survival
Following match adjustment, the HR of PFS in the base case was in favour of tarlatamab over comparator therapies for the DeLLphi-301 study versus CAS study MAIC (HR = █████; 95% CI, █████ to █████; P = 0.0000) and the DeLLphi-301 study versus OOS MAIC (HR = 0.326; 95% CI, 0.215 to 0.493; P = 0.0000). Results of the scenario analyses were consistent with the base-case analysis (Table 26). Kaplan-Meier curves for PFS are shown in Figure 5 and Figure 6.

Figure 6: Kaplan-Meier Curves of PFS (DeLLphi-301 vs. OOS)
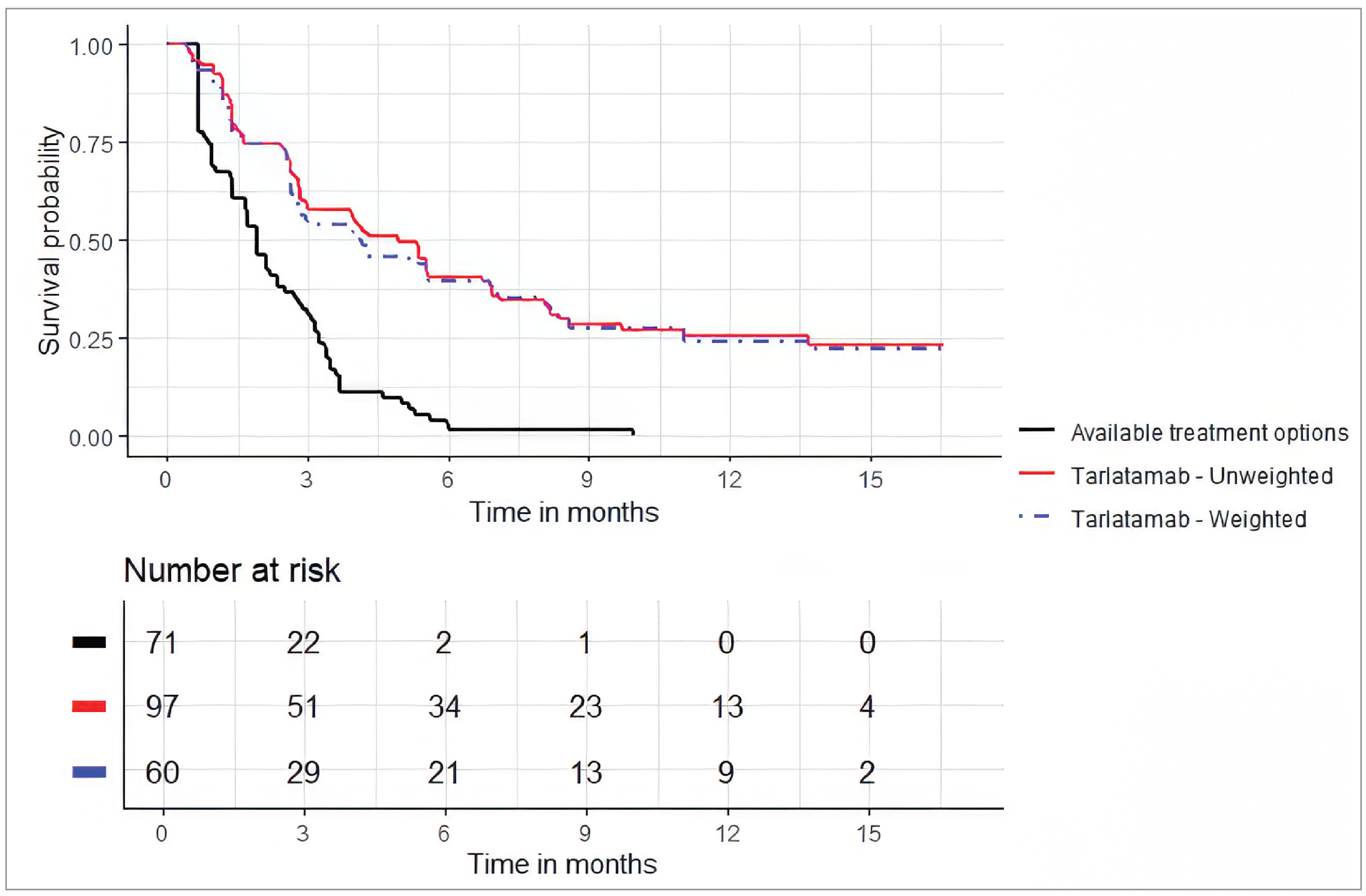
OOS = Oncology Outcomes Study; PFS = progression-free survival; vs. = versus.
Sources: Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30
Analysis | DeLLphi-301 vs. CAS study | DeLLphi-301 vs. OOS study | ||||
|---|---|---|---|---|---|---|
ESS | HR (95% CI) | P value | ESS | HR (95% CI) | P value | |
OS — HR of tarlatamab vs. comparator therapies | ||||||
Unadjusted | ██ | █████ ██████ ██ ██████ | 0.0000 | 97 | 0.254 (0.171 to 0.378) | 0.0000 |
Adjusted (base case) | ██ | █████ ██████ ██ ██████ | 0.0001 | 59.65 | 0.291 (0.184 to 0.459) | 0.0000 |
Adjusted (scenario 1) | ██ | █████ ██████ ██ ██████ | 0.0001 | 59.68 | 0.288 (0.182 to 0.456) | 0.0000 |
Adjusted (scenario 2) | ██ | █████ ██████ ██ ██████ | 0.0001 | 80.64 | 0.257 (0.167 to 0.393) | 0.0000 |
PFS — HR of tarlatamab vs. comparator therapies | ||||||
Unadjusted | ██ | █████ ██████ ██ ██████ | 0.0000 | 97 | 0.304 (0.215 to 0.430) | 0.0000 |
Adjusted (base case) | ██ | █████ ██████ ██ ██████ | 0.0000 | 59.65 | 0.326 (0.215 to 0.493) | 0.0000 |
Adjusted (scenario 1) | ██ | █████ ██████ ██ ██████ | 0.0000 | 59.68 | 0.323 (0.213 to 0.489) | 0.0000 |
Adjusted (scenario 2) | ██ | █████ ██████ ██ █████ | 0.0000 | 80.64 | 0.311 (0.214 to 0.453) | 0.0000 |
CAS = Cancer Analysis System; CI = confidence interval; ESS = effective sample size; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; OOS = Oncology Outcomes Study; OS = overall survival; PFS = progression-free survival; vs. = versus.
Sources: Technical Report for DeLLphi-301 vs. CAS and DeLLphi-301 vs. OOS.30
Critical Appraisal of Propensity Score Weighting Analysis and MAICs
SLRs were conducted by the sponsor with appropriate study selection methods to inform study inclusion for the ITCs, although all identified studies were subsequently excluded in the feasibility assessment. The Flatiron, CAS, and OOS studies selected for inclusion into the analyses were identified in the absence of prespecified methods; rather, they were sponsor-conducted registry studies that were deemed by the sponsor to be appropriate for the ITCs. While reasons for exclusion of the published studies identified by the SLR were documented, it should be noted that the Flatiron, CAS, and OOS studies were also subject to similar concerns which could have led to these studies being excluded. Data collection period for both studies were initiated far in the past (2013 for the CAS and Flatiron studies; 2011 for OOS). PFS was not a measured outcome in either the CAS study or OOS and was proxied by time to discontinuation or death in the ITCs. The OOS had a small sample size (N = 71) and limited reporting of baseline characteristics (ECOG PS and history of platinum-based chemotherapy not reported). There is a risk of selection bias due to inconsistent application of selection criteria in the feasibility assessment.
Selection criteria were applied to the Flatiron study to promote similarity between the tarlatamab and comparator therapy cohorts. Both patient cohorts had ES-SCLC, previously received 1 platinum-based chemotherapy and at least 1 other prior LOT (i.e., 3L+), and had an ECOG PS score of 0 or 1. However, it should be noted that the DeLLphi-301 trial included patients with a life expectancy of at least 12 weeks at baseline, but such criterion was not applied to the Flatiron study cohort. Thus, the OS results could potentially be biased due to possible differences in the likelihood of survival at baseline between the cohorts. As well, patients with brain metastases were included in the DeLLphi-301 study cohort only if brain metastasis was treated, asymptomatic, or had no evidence of pseudoprogression, while patients with brain metastases were included in the Flatiron study cohort regardless of treatment history and stability of disease. The sponsor assumed that in the Flatiron study cohort, initiation of 3L+ treatment for SCLC reflects that brain metastasis was either treated or reasons for not treating brain metastasis had been considered before initiating treatments. This assumption was considered to be appropriate and not expected to introduce bias, as per clinical expert input.
An important limitation of the DeLLphi-301 study versus OOS is that the patient population in the index DeLLphi-301 trial was narrower than the comparator OOS. While both studies required patients to have previously received at least 2 LOTs for SCLC, the DeLLphi-301 study required 1 of the LOTs to be platinum-based chemotherapy; however, such inclusion criterion was absent in OOS. The proportion of patients with history of platinum-based chemotherapy was also not reported in OOS. While it was not possible to account for such difference in adjustment process due to unavailable data from the OOS cohort, it is unlikely to introduce meaningful bias in OS given the expected widespread use of platinum-based chemotherapy before 3L, in accordance with treatment guidelines in Canada. With respect to the analysis of the DeLLphi-301 study versus the CAS study, the patient population of included studies also differed in that the DeLLphi-301 study included patients eligible for 3L+ therapy while the CAS study included patients eligible for 3L only; no adjustments were made in the analyses to account for such difference in LOT. While there may be a risk of bias in comparing the DeLLphi-301 cohort receiving 3L+ therapy with the CAS study cohort receiving 3L therapy, this bias would likely underestimate the relative benefit of tarlatamab, as patients in the fourth-line or later-line setting are generally expected to have a worse prognosis. In addition, the DeLLphi-301 study included patients who had a life expectancy of at least 12 weeks while the CAS study and OOS did not have such selection criterion. It is unclear if the likelihood of survival at baseline differed between the patient cohorts, which could potentially introduce bias to the OS results.
Definition of PFS differed between the DeLLphi-301 study and the Flatiron study. In the DeLLphi-301 study, disease progression was assessed based on radiographic imaging. In the Flatiron study, disease progression was retrospectively identified based on clinical documentation of radiographic imaging, pathology reports, or clinical examination, which could have contributed to variability in PFS adjudication. A sensitivity analysis, which used radiographic evidence alone to determine PFS events in the Flatiron study, was conducted and showed results consistent with the base case. In the CAS study and OOS, PFS was not a measured outcome. The use of TTD as a proxy for PFS could potentially introduce bias because treatment discontinuation could be led by factors other than radiographic tumour progression, such as AEs and consent withdrawal. This could result in a seemingly shorter PFS in the comparator therapies cohort, leading to potential bias in favour of tarlatamab. In the clinical experts’ opinion, the impact on PFS results is likely to be minor. Also, differential use of subsequent anticancer treatment between patient cohorts could potentially bias the OS results. The sponsor assumed that subsequent treatments after the 3L would typically consist of chemotherapies with similar efficacy, based on input from a clinical expert they consulted. However, in the absence of supporting data on subsequent anticancer treatment use from the comparator studies, the review team considered the impact on OS results to be uncertain.
There is also temporal discordance in data collection period between studies where the Flatiron study (2013 to 2021), CAS study (2013 to 2022), and OOS (2011 to 2022) were initiated much earlier than the DeLLphi-301 study (2021 to 2023). The changes in treatment patterns and supportive care over time are potential sources of heterogeneity between studies. The clinical experts noted that between 2011 and 2013, immunotherapy (PD-L1 inhibitors: atezolizumab and durvalumab) was introduced and was commonly used in addition to platinum doublet therapy in the 1L setting. This represents a major change in treatment practice. PD-L1 inhibitor use was identified as a potential prognostic factor of low relevance by the sponsor. A sensitivity analysis of the DeLLphi-301 study versus the Flatiron study ITC, which adjusted for prior PD-1 or PD-L1 inhibitor use, was conducted and showed results consistent with the primary analysis; however, no adjustment for prior PD-1 or PD-L1 inhibitor use was made in the DeLLphi-301 study versus OOS and DeLLphi-301 study versus CAS study ITCs (both primary and sensitivity analyses), which could be a potential source of bias. Furthermore, the studies had a different composition of study site locations (the DeLLphi-301 study in Europe, East Asia, and North America; Flatiron study in the UK; CAS study in the UK; OOS in Canada). The clinical experts did not expect the treatment approach to be notably different between these countries.
The choice to conduct propensity score weighting analysis and MAICs was appropriately justified. Prognostic factors that were adjusted for in the weighting process were identified based on targeted literature review, empirical analyses of data from the DeLLphi-301 trial, and expert input. Of note, the study by Austin suggested that the identification of potentially prognostically important covariates should be based on clinical expertise or an SLR of available literature, rather than statistical testing in the study sample.87 The identified factors were considered appropriate by the clinical experts consulted by CDA-AMC. In the base case of the DeLLphi-301 study versus the Flatiron study, factors considered of low prognostic importance by the sponsor, including previous use of PD-1 or PD-L1 inhibitors, presence of liver metastasis, and race were not adjusted for in the weighting process. A sensitivity analysis, which adjusted for all identified prognostic factors (high, medium, and low importance), was conducted and results were consistent with the base case for OS and PFS outcomes (in favour of tarlatamab) but showed a wide 95% CI for ORR indicating imprecision in results. In regard to the MAICs, several of the potential prognostic factors identified by the sponsor were not adjusted for (DeLLphi-301 study versus CAS study analysis: number of prior LOTs, previous use of PD-1 or PD-L1 inhibitor, and race; DeLLphi-301 study versus OOS: ECOG PS, number of prior LOTs, CFI, presence of brain metastasis, previous use of PD-1 or PD-L1 inhibitor, and race) due to unavailability of data. As well, the clinical experts consulted by CDA-AMC noted that baseline weight loss, prior prophylactic cranial irradiation, and prior thoracic radiotherapy were potential prognostic factors or treatment effect modifiers; these factors were not adjusted for in any of the analyses. The lack of adjustment for potential prognostic factors or treatment effect modifiers could potentially lead to confounding and introduce bias to the results. Of note, E-values were computed by the sponsor for the DeLLphi-301 study versus Flatiron study analysis to help assess the likelihood of results being biased due to unmeasured confounding; however, methods used for estimating these values were not outlined in the submitted materials. Due to variability in the computation of this metric, without a review of the methods used, the review team was unable to assess the likelihood of results being biased due to unmeasured confounding based on the reported E-values.
In the analysis of the DeLLphi-301 study versus the Flatiron study, following weighting, there remained an imbalance in several baseline patient characteristics that were identified as potential prognostic factors (race, ECOG PS, CFI, history of PD-1 or PD-L1 inhibitor treatment, and presence of liver metastasis) between the tarlatamab cohort and the adjusted comparator therapies cohort. Inadequate adjustment for these factors suggests that the study results were likely biased due to confounding. This bias is in addition to the aforementioned potential bias that results from a lack of adjustment for potential prognostic factors or treatment effect modifiers. There was a sizable reduction in ESS after the weighting process in all studies (reduced by 46.6% in the comparator therapies cohort of the Flatiron study, reduced by 72.1% and 38.5% in the tarlatamab cohorts of the CAS study and OOS, respectively), which suggested poor population overlap. A significant reduction in sample size can contribute to imprecision, increasing uncertainty of the results. A notable reduction in ESS also suggests that results may be heavily influenced by a subset of the sample in the trials who may not be representative of the full sample. Given that the population from the analyses of the DeLLphi-301 study versus the CAS study and the DeLLphi-301 study versus OOS were weighted to the DeLLphi-301 study, the results may be driven by a subset of patients in the tarlatamab group, limiting generalizability to the full population presented by the DeLLphi-301 study. Conversely, as the population from the analysis of the DeLLphi-301 study versus the Flatiron study was weighted to the Flatiron study comparator, the results may be driven by a subset of patients in the comparator therapies group, limiting generalizability to the full population presented by the comparator studies. Of note, comparator therapies were pooled and assessed as a single intervention group in the studies. The clinical experts considered this approach to be reasonable given that there is currently no standard of care for ES-SCLC in the 3L+ setting and that treatment choice is largely based on physician’s choice in clinical practice. The clinical experts noted that the distribution of comparator therapies in the comparator studies was reasonably similar to clinical practice in Canada.
The studies assessed OS and PFS, which are outcomes important to patients and clinicians. The comparative effects of tarlatamab and comparator therapies on other efficacy outcomes of interest, including HRQoL and harms, were not investigated, and thus, unknown.
Studies Addressing Gaps in the Systematic Review Evidence
There were no relevant studies addressing the gaps in the systematic review evidence submitted for this review.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 ongoing, phase II, open-label, single-arm trial, the DeLLphi-301 study (N = 222). The purpose of this study was to evaluate the efficacy and safety of tarlatamab (10 mg) in adult patients with histologically or cytologically confirmed relapsed or refractory SCLC who progressed or recurred following 1 platinum-based regimen and at least 1 other prior LOT. Patients must have had measurable lesions as defined per RECIST 1.1 within 21 days before the first dose of tarlatamab and must have had adequate organ function. Patients were excluded if they had symptomatic or untreated brain metastases. The primary efficacy end point of this study was ORR, with secondary end points of OS and PFS, and exploratory end points of HRQoL and safety. At baseline, of the 99 patients in the 10 mg group, most were men (71.1%), and the median age was 64 years (range, 35 years to 82 years). Overall, patients had a median of 2 (range, 1 to 6) prior lines of therapy, including 72.7% of patients with prior PD-1 or PD-L1 inhibitors. Time to progression after 1L platinum therapy was less than 90 days for 27 patients (27.3%), between 90 days and 179 days for 22 patients (22.2%), longer than 180 days for 20 patients (21.4%), and unknown for 30 patients (30.3%). Most patients had metastatic disease (98.0%), no brain metastases (77.8%) or liver metastases (61.6%), and an ECOG PS score of 1 (73.7%).
In the absence of head-to-head evidence comparing tarlatamab to other relevant therapies used to manage ES-SCLC, the sponsor submitted 1 analysis using propensity score weighting (DeLLphi-301 study versus Flatiron study)29 and 2 MAICs (DeLLphi-301 study versus CAS study and DeLLphi-301 study versus OOS)30 which indirectly compared OS and PFS of tarlatamab (data sourced from the DeLLphi-301 study) with currently available treatments (data sourced from registry studies) in patients with ES-SCLC in the 3L+ setting. The analysis of the DeLLphi-301 study versus the Flatiron study assessed additional outcomes of interest, including ORR and TTD.
Interpretation of Results
Efficacy
After refractoriness or relapse from 1L and 2L treatments in ES-SCLC, prognosis is very poor due to the aggressive and quickly progressing nature of the disease. Currently in the 3L setting, there are no Health Canada–approved treatments for ES-SCLC, and the available treatments are limited. In addition, the existing treatments are associated with significant side effects. The clinical experts consulted for this review indicated that the most important treatment goals for this patient population are to prolong life, delay disease progression, reduce the severity of symptoms, minimize adverse effects from the treatments, and improve patients’ HRQoL.
The efficacy of tarlatamab (10 mg) in patients with ES-SCLC in the 3L setting was assessed in the DeLLphi-301 trial, which consisted of 3 parts: dose selection phase, dose expansion phase, and a small substudy of safety using modified monitoring criterion. As per the input from patient groups, clinical experts consulted for this review, and clinician groups, improved survival was considered the most important outcome of treatment, although there were not established thresholds of clinical importance for survival identified in the target population. In the DeLLphi-301 trial, the OS rate was 57.7% and the PFS rate was 25.7% at 12 months, suggesting after 1 year of treatment, more than half of the patients who received tarlatamab treatment were still alive and a quarter of them were free of disease progression. The results for OS were difficult to interpret due to the lack of comparator, and the data at the latest DCO (May 16, 2024) suggested that the OS data were still immature. However, in the clinical experts’ opinions, the improvement in survival in patients treated with tarlatamab was promising, especially for a patient population that has a very poor prognosis with current treatments (median OS of 12 months to 13 months from the time of diagnosis, and the 5-year survival rates ranging from 1% to 10%). In general, the interpretation of the efficacy results from this study was limited given the internal and external validity issues identified in the critical appraisal section of this report, such as potential for selection bias and challenges in interpreting the impact of tarlatamab on time-to-event end points (e.g., survival benefit). As noted in the GRADE assessment (Table 2), conclusions about efficacy relative to any comparator cannot be drawn from single-arm studies. Health Canada also noted that “an improvement in survival has not been established” in the Health Canada Reviewer Report.88 Similarly, the GRADE certainty of evidence assessment for the outcome of PFS was rated as “very low.” Overall, due to the single-arm study design of the DeLLphi-301 study, the ability to interpret the results for OS and PFS was very limited, and this was reflected by the issuance of a Notice of Compliance with Conditions for this indication. Health Canada stated that the benefit of tarlatamab in terms of OS will be evaluated in an ongoing randomized phase III clinical trial which is required as a commitment to the Notice of Compliance with Conditions.88
HRQoL was also considered an important outcome by the patients, and improvement of HRQoL was identified as 1 of the treatment goals by clinicians. However, the quality of these outcomes was compromised due to high attrition rates and potential bias due to patients’ awareness of their treatment, and definitive conclusion on patients’ quality of life cannot be drawn based on the data available in this study.
The primary efficacy end point, ORR, although an effective measure of antitumour activity, was considered less valuable in assessing the treatment effect of tarlatamab in patients with ES-SCLC, if it is not considered along with the other efficacy outcomes. The result of ORR as evaluated by BICR using RECIST 1.1 was 41.4% (97.5% CI, 30.3% to 53.2%) and met the protocol-defined threshold for clinically meaningful benefit (lower bound of 97.5% CI > 15%). The clinical experts considered the reported response rate promising in the context of 3L treatment for ES-SCLC in which there are no authorized treatments in Canada. However, due to the study design of the DeLLphi-301 study, these results were considered very uncertain for supporting conclusions. Note that among the 40 patients who responded to the treatment, only 1 achieved CR, while 40 achieved PR.
Results from the submitted propensity score weighting analysis and MAICs suggested that tarlatamab was associated with improved OS, PFS, ORR, and TTD compared with comparator therapies in patients with ES-SCLC in the 3L+ setting. However, there was notable uncertainty in the results due to limitations of the studies, including a risk of selection bias in study inclusion and a sizable reduction in ESS. As well, the heterogeneity in patient population and study design, temporal discordance in the data collection period during which a major change in treatment pattern occurred, and inadequate adjustment for, or lack of adjustment for, potential prognostic factors may introduce unmeasurable confounding in the relative treatment effect estimates.
Harms
In the DeLLphi-301 trial, safety analyses were conducted for all patients who received at least 1 dose of tarlatamab. Nearly all patients (99%) experienced an AE with tarlatamab, and 59% of patients reported an SAE. The most commonly reported AEs occurring in at least 20% of patients included CRS, anemia, decreased appetite, pyrexia, constipation, dysgeusia, fatigue, and asthenia. Sixty patients in part 1 and part 2 of the DeLLphi-301 trial developed grade 3 or higher AEs. The clinical experts consulted for this review noted that most AEs are manageable and there was no unusual safety signals identified.
CRS and ICANS were notable harms of interest for tarlatamab and to this review, and while not novel for oncology in general, are new AEs for thoracic oncology treatments due to the unique mechanism of action for tarlatamab. CRS was reported in 51.5% of patients in part 1 and part 2, while ICANS was reported in 2 patients. It is worth noting that even though the GRADE certainty of evidence assessment for the outcome of CRS and SAE was rated as “very low” — mainly because of the study design of the DeLLphi-301 study — the rates of these harms outcomes in this study were greater than 50%, suggesting a clear harm related to the treatment with tarlatamab. In the DeLLphi-301 trial, most of the AEs were low grade and no fatal events of CRS or ICANS occurred. The clinical experts consulted for this review indicated that CRS and ICANS at any grades should be considered clinically significant AEs.
Harms outcomes were not assessed in the submitted propensity score weighting analysis and MAICs, and thus, the comparative safety of tarlatamab and currently available treatments in the population of interest are unknown.
Conclusion
One phase II, single-arm, open-label trial (DeLLphi-301) provided evidence for the efficacy and safety of tarlatamab in adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy.
Clinicians and patients highlighted the need for new treatment options that prolong life (survival being considered the most important outcomes), delay disease progression, reduce the severity of symptoms, have a reduced AE profile, and improve patients’ HRQoL. Although interpretation of the OS and PFS results evaluated in the study were limited by the single-arm design and immaturity from limited follow up, precluding the ability to attribute the study results to treatment with tarlatamab, the clinical experts consulted for this review considered the median OS and PFS, as well as the 12-month OS and PFS rates, clinically meaningful. Results from the DeLLphi-301 trial also suggested that some patients (40.4%) will experience a response to tarlatamab, although the evidence is still uncertain. Although HRQoL was an important outcome for patients with ES-SCLC, due to the noncomparative design and high attrition rates in the DeLLphi-301 trial, the effect of tarlatamab on HRQoL remains uncertain. In terms of harms, although nearly all patients reported an AE, the safety profile of tarlatamab was consistent with its mechanism of action, including a large proportion of patients who experienced CRS. Despite the high frequency of many AEs, most of the reported AEs are manageable according to the clinical experts consulted for this review. However, CRS and ICANS in any grades were considered clinically significant AEs for tarlatamab, and remains a concern for clinicians, given that these are new AEs for thoracic oncology.
There is a lack of direct comparative evidence between tarlatamab and relevant treatments for patients with ES-SCLC in the 3L setting. Results from the sponsor-submitted ITCs suggested that tarlatamab was associated with improved OS, PFS, and ORR compared with the other active treatments used in clinical practice, such as platinum-based therapy, PD-L1–based therapy, topotecan, CAV, or irinotecan. However, the evidence was very uncertain about the effects of tarlatamab on any outcomes versus any comparator, and the ability to draw firm conclusions on the magnitude of clinical benefit of tarlatamab was hindered by the limitations in the evidence, including a risk of selection bias in study inclusion, a sizable reduction in ESS, the heterogeneity in patient population and study design, and inadequate or lack of adjustment for potential prognostic factors that may introduce unmeasurable confounding in the relative treatment effect estimates.
References
1.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [sponsor-supplied reference]. Canadian Cancer Society; 2020. https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf
2.Kelly K. Post TW, ed. Extensive-stage small cell lung cancer: Initial management. UpToDate; 2024. Accessed July 2024. http://www.uptodate.com
3.Amgen Canada Inc. Drug Reimbursement Review sponsor submission: Imdelltra (tarlatamab), powder for solution for intravenous infusion, 1 mg, 10 mg [internal sponsor's package]. August 14, 2024.
4.Byers LA, Gay CM. Post TW, ed. Pathobiology and staging of small cell carcinoma of the lung. UpToDate; 2024. Accessed July 2024. http://www.uptodate.com
5.Goldman JW, Garassino MC, Chen Y, et al. Patient-reported outcomes with first-line durvalumab plus platinum-etoposide versus platinum-etoposide in extensive-stage small-cell lung cancer (CASPIAN): a randomized, controlled, open-label, phase III study. Lung Cancer. 2020;149:46-52. doi:10.1016/j.lungcan.2020.09.003 PubMed
6.Vedadi A, Shakik S, Brown MC, et al. The impact of symptoms and comorbidity on health utility scores and health-related quality of life in small cell lung cancer using real world data. Qual Life Res. 2021;30(2):445-454. doi:10.1007/s11136-020-02615-1 PubMed
7.Qin A. Small Cell Lung Cancer. National Organization for Rare Disorders; 2023. Accessed October 31, 2023. https://rarediseases.org/rare-diseases/small-cell-lung-cancer/
8.Chambers SK, Baade P, Youl P, et al. Psychological distress and quality of life in lung cancer: the role of health-related stigma, illness appraisals and social constraints. Psychooncology. 2015;24(11):1569-77. doi:10.1002/pon.3829 PubMed
9.Midthun DE. Post TW, ed. Overview of the initial treatment and prognosis of lung cancer. UpToDate; 2024. Accessed July 2024. http://www.uptodate.com
10.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: small cell lung cancer (Version 3.2023) [sponsor-supplied reference]. December 21, 2022.
11.Tariq S, Kim SY, Monteiro de Oliveira Novaes J, Cheng H. Update 2021: Management of Small Cell Lung Cancer. Lung. 2021;199(6):579-587. doi:10.1007/s00408-021-00486-y PubMed
12.Paz-Ares L, Dvorkin M, Chen Y, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394(10212):1929-1939. doi:10.1016/S0140-6736(19)32222-6 PubMed
13.Horn L, Mansfield AS, Szczesna A, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379(23):2220-2229. doi:10.1056/NEJMoa1809064 PubMed
14.PDQ® Adult Treatment Editorial Board. Small cell lung cancer treatment [sponsor-supplied reference]. National Cancer Institute; March 2, 2023. Accessed May 11, 2023. https://www.cancer.gov/types/lung/hp/small-cell-lung-treatment-pdq
15.Blackhall F, Girard N, Livartowski A, et al. Treatment patterns and outcomes among patients with small-cell lung cancer (SCLC) in Europe: a retrospective cohort study. BMJ Open. 2023;13(2):e052556. doi:10.1136/bmjopen-2021-052556 PubMed
16.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. doi:10.1038/s41572-020-00235-0 PubMed
17.Ganti AKP, Loo BW, Bassetti M, et al. Small Cell Lung Cancer, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19(12):1441-1464. doi:10.6004/jnccn.2021.0058 PubMed
18.Nicholson AG, Chansky K, Crowley J, et al. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: Proposals for the Revision of the Clinical and Pathologic Staging of Small Cell Lung Cancer in the Forthcoming Eighth Edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2016;11(3):300-11. doi:10.1016/j.jtho.2015.10.008
19.Mieras A, Pasman HRW, Klop HT, et al. What Goals Do Patients and Oncologists Have When Starting Medical Treatment for Metastatic Lung Cancer? Clin Lung Cancer. 2021;22(3):242-251 e5. doi:10.1016/j.cllc.2020.06.014 PubMed
20.Khurshid H, Ismaila N, Bian J, et al. Systemic therapy for small-cell lung cancer: ASCO-Ontario Health (Cancer Care Ontario) Guideline. J Clin Oncol. 2023:JCO2301435. doi:10.1200/JCO.23.01435 PubMed
21.AstraZeneca Canada Inc. Imfinzi® (durvalumab): concentrate for solution for infusion, 50 mg / mL, intravenous [product monograph; sponsor-supplied reference]. 2022. Accessed May 2022. https://www.astrazeneca.ca/content/dam/az-ca/downloads/productinformation/imfinzi-product-monograph-en.pdf
22.Amgen Canada Inc. Imdelltra (tarlatamab for injection): lyophilized powder for solution for intravenous infusion, 1 mg and 10 mg per vial [product monograph; sponsor-supplied reference]. 2024. Accessed September 11, 2024. https://pdf.hres.ca/dpd_pm/00077062.PDF
23.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
24.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
25.Amgen Inc. Primary Analysis for the Clinical Study Report for DeLLphi-301: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects with Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment [CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. March 28, 2024.
26.Amgen Inc. Addendum 01 to the Primary Clinical Study Report for Study DeLLphi-301: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects With Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment [CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. 2023.
27.Amgen Inc. Addendum 02 to the Primary Clinical Study Report for Study DeLLphi-301: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects With Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment [CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. 2024.
28.Amgen Inc. Addendum 03 to the Primary Clinical Study Report for Study DeLLphi-301: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects With Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment [CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. 2024.
29.Amgen Canada Inc. Tarlatamab vs. real-world physicians’ choice of therapies in patients with relapsed or refractory small cell lung cancer after two or more prior lines of treatment: patient-level indirect treatment comparison (ITC) of DeLLphi-301 vs. Flatiron real-world data [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imdelltra (tarlatamab), powder for solution for intravenous infusion, 1 mg, 10 mg. August 5, 2024.
30.Amgen Canada Inc. Matching adjusted indirect treatment comparisons of tarlatamab vs. available treatment options in patients with relapsed or refractory small cell lung cancer after two prior lines of treatment [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imdelltra (tarlatamab), powder for solution for intravenous infusion, 1 mg, 10 mg. June 12, 2024.
31.Canadian Cancer Society. Lung and bronchus cancer statistics. 2024. Accessed September 12, 2024. https://cancer.ca/en/cancer-information/cancer-types/lung/statistics
32.PDQ® Adult Treatment Editorial Board. Small Cell Lung Cancer Treatment (PDQ®)–Health Professional Version. National Cancer Institute; 2024. Accessed November 1, 2024. https://www.cancer.gov/types/lung/hp/small-cell-lung-treatment-pdq
33.Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur Respir J. 2010;35(1):202-15. doi:10.1183/09031936.00105009 PubMed
34.Johnson BE, Janne PA. Basic treatment considerations using chemotherapy for patients with small cell lung cancer. Hematol Oncol Clin North Am. 2004;18(2):309-22. doi:10.1016/j.hoc.2003.12.008 PubMed
35.Dingemans AC, Fruh M, Ardizzoni A, et al. Small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Main article. Ann Oncol. 2021;32(7):839-853. doi:10.1016/j.annonc.2021.03.207 PubMed
36.Jeon DS, Kim HC, Kim SH, et al. Five-Year Overall Survival and Prognostic Factors in Patients with Lung Cancer: Results from the Korean Association of Lung Cancer Registry (KALC-R) 2015. Cancer Res Treat. 2023;55(1):103-111. doi:10.4143/crt.2022.264 PubMed
37.National Institute for Health and Care Excellence. Atezolizumab with carboplatin and etoposide for untreated extensive-stage small-cell lung cancer [ID1504] (Technology Appraisal TA638). Committtee papers (company submission) [sponsor-supplied reference]. 2019. Accessed May 15, 2022. https://www.nice.org.uk/guidance/ta638/documents/committee-papers
38.CADTH. Lurbinectedin (Zepzelca) - CADTH Clinical, Pharmacoeconomic and Stakeholder Review [sponsor-supplied reference]. 2023.
39.Farago AF, Keane FK. Current standards for clinical management of small cell lung cancer. Transl Lung Cancer Res. 2018;7(1):69-79. doi:10.21037/tlcr.2018.01.16 PubMed
40.Borghaei H, Pundole X, Anderson E, et al. Abstract #EP13.07-03: Treatment patterns and outcomes in recent US clinical practice for SCLC patients after two prior lines of therapy. Paper presented at: 2023 World Conference on Lung Cancer (WCLC); September 9-12, 2023; Singapore [sponsor-supplied reference]. 2023.
41.Simon M, Argiris A, Murren JR. Progress in the therapy of small cell lung cancer. Crit Rev Oncol Hematol. 2004;49(2):119-33. doi:10.1016/S1040-8428(03)00118-5 PubMed
42.Novartis Pharmaceuticals Canada Inc. Hycamtin® (topotecan) [Canadian product monograph; sponsor-supplied reference]. 2019. Accessed May 2022.
43.Bristol-Myers Squibb Canada. Cytoxan (cyclophosphamide): 100, 750, 1000 and 2000 mg, U.S.P. sterile powder; 25 and 50 mg, U.S.P. tablets [product monograph; sponsor-supplied reference]. 2023. https://pdf.hres.ca/dpd_pm/00001000.PDF
44.Pfizer Canada ULC. Doxorubicin (doxorubicin) [product monograph; sponsor-supplied reference]. 2021. Accessed May 2022.
45.Pfizer Canada ULC. Vincristine (vincristine sulfate): 1 mg/mL injection, U. S. P. [product monograph; sponsor-supplied reference]. 2021. Accessed May 2022.
46.Pfizer Canada ULC. Irinotecan (irinotecan hydrochloride injection, U. S. P.): solution, 20 mg / mL, intravenous [product monograph; sponsor-supplied reference]. 2019. Accessed May 2022.
47.Pfizer Canada ULC. Cisplatin Injection BP (cisplatin injection): 1 mg / mL (50 mg and 100 mg cisplatin per vial) sterile solution [product monograph; sponsor-supplied reference]. 2022. https://pdf.hres.ca/dpd_pm/00072950.PDF
48.Pfizer Canada ULC. Carboplatin Injection BP (carboplatin injection): 10 mg / mL (50 mg, 150 mg, 450 mg, 600 mg of carboplatin per vial) sterile solution [product monograph; sponsor-supplied reference]. 2023. https://pdf.hres.ca/dpd_pm/00072214.PDF
49.Teva Canada Limited. Vepesid (etoposide injection, Teva standard): 20 mg/mL [product monograph; sponsor-supplied reference]. 2022. https://pdf.hres.ca/dpd_pm/00034660.PDF
50.Xediton Pharmaceuticals Inc. Vepesid (etoposide capsules): 50 mg capsule [product monograph; sponsor-supplied reference]. 2022. https://pdf.hres.ca/dpd_pm/00054405.PDF
51.Ahn MJ, Cho BC, Felip E, et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N Engl J Med. 2023;doi:10.1056/NEJMoa2307980 PubMed
52.Paz-Ares L, Ahn MJ, Felip E, et al. 508MO Tarlatamab for patients (pts) with previously treated small cell lung cancer (SCLC): Primary analysis of the phase II DeLLphi-301 study. Ann Oncol. 2023;34:S1664-S1665. doi:10.1016/j.annonc.2023.10.587
53.Amgen Inc. Clinical Trial Protocol. A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects with Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment [sponsor-supplied reference]. 2021.
54.Amgen Inc. Statistical analysis plan for CSR: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects with Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment (DeLLphi-301) V6.0 [amendment no. 5; CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. June 28, 2023.
55.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi:10.1093/jnci/85.5.365 PubMed
56.Coon CD, Schlichting M, Zhang X. Interpreting Within-Patient Changes on the EORTC QLQ-C30 and EORTC QLQ-LC13. Patient. 2022;15(6):691-702. doi:10.1007/s40271-022-00584-w PubMed
57.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi:10.1016/j.ejca.2023.04.027 PubMed
58.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30a(5):635-42. doi:10.1016/0959-8049(94)90535-5 PubMed
59.EORTC. EORTC QLQ- LC13 Scoring Manual. Accessed October 31, 2024. https://multimedia.elsevier.es/PublicationsMultimediaV1/item/multimedia/S1807593222022931:mmc1.pdf?idApp=UINPBA00004N
60.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. doi:10.1016/j.jclinepi.2015.08.007 PubMed
61.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLC-C30 scoring manual (3rd Edition). European Organisation for Research and Treatment of Cancer; 2001. Accessed May 25, 2024. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf
62.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9(106):106. doi:10.1186/1477-7525-9-106 PubMed
63.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi:10.1007/s11136-007-9210-8 PubMed
64.Machingura A, Taye M, Musoro J, et al. Clustering of EORTC QLQ-C30 health-related quality of life scales across several cancer types: Validation study. Eur J Cancer. 2022;170:1-9. doi:10.1016/j.ejca.2022.03.039 PubMed
65.Osoba D, Zee B, Pater J, Warr D, Kaizer L, Latreille J. Psychometric properties and responsiveness of the EORTC quality of Life Questionnaire (QLQ-C30) in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353-64. doi:10.1007/BF00451727 PubMed
66.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-17. doi:10.1111/ajco.12070 PubMed
67.Kvam AK, Fayers PM, Wisloff F. Responsiveness and minimal important score differences in quality-of-life questionnaires: a comparison of the EORTC QLQ-C30 cancer-specific questionnaire to the generic utility questionnaires EQ-5D and 15D in patients with multiple myeloma. Eur J Haematol. 2011;87(4):330-7. doi:10.1111/j.1600-0609.2011.01665.x PubMed
68.Porter DL, Maloney DG. Post TW, ed. Cytokine release syndrome (CRS). UpToDate; 2024. Accessed November 11, 2024. https://www.uptodate.com/
69.Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25(4):625-638. doi:10.1016/j.bbmt.2018.12.758 PubMed
70.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26(4):404-413. doi:10.1093/biomet/26.4.404
71.Brookmeyer R, Crowley J. A Confidence Interval for the Median Survival Time. Biometrics. 1982;38(1):29-41.
72.Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data, second edition. Wiley-Interscience; 2002. Accessed November 11, 2024. https://onlinelibrary.wiley.com/doi/pdf/10.1002/9781118032985.fmatter?msockid=26f7f9e1c38f69b53f81ea0ac2596879
73.Amgen Inc. Cost-effective analysis of tarlatamab for the treatment of adult patients with extensive stage small cell lung cancer after two or more lines of treatment including platinum-based chemotherapy. Technical Report [CONFIDENTIAL internal manufacturer's report; sponsor-supplied reference]. 2024.
74.U.S. Food and Drug Administration. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. 2018. Accessed November 12, 2024. https://www.fda.gov/media/71195/download
75.Amgen Canada Inc. Evidence Synthesis Feasibility Assessment Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imdelltra (tarlatamab), powder for solution for intravenous infusion, 1 mg, 10 mg. July 19, 2024.
76.Latimer NR, White IR, Tilling K, Siebert U. Improved two-stage estimation to adjust for treatment switching in randomised trials: g-estimation to address time-dependent confounding. Stat Methods Med Res. 2020;29(10):2900-2918. doi:10.1177/0962280220912524 PubMed
77.Skaltsa K, Ivanescu C, Naidoo S, Phung D, Holmstrom S, Latimer NR. Adjusting overall survival estimates after treatment switching: a case study in metastatic castration-resistant prostate cancer. Target Oncol. 2017;12:111-121. doi:10.1007/s11523-016-0472-3 PubMed
78.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE [sponsor-supplied reference]. NICE Decision Support Unit; 2016. http://www.nicedsu.org.uk
79.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28:935-945. doi:10.2165/11538370-000000000-00000 PubMed
80.Steffens CC, Elender C, Hutzschenreuter U, et al. Treatment and outcome of 432 patients with extensive-stage small cell lung cancer in first, second and third line - Results from the prospective German TLK cohort study. Lung Cancer. 2019;130:216-225. doi:10.1016/j.lungcan.2019.02.026 PubMed
81.Aktas G, Kus T, Kalender ME, Sevinc A, Camci C, Kul S. Survival analysis in second-line and third-line chemotherapy with irinotecan followed by topotecan or topotecan followed by irinotecan for extensive-stage small-cell lung cancer patients: a single-center retrospective study. Onco Targets Ther. 2016;9:1921-6. doi:10.2147/OTT.S101390 PubMed
82.Inomata M, Hayashi R, Tokui K, et al. Outcome and prognostic factors in patients with small cell lung cancer who receive third-line chemotherapy. Tumori. 2014;100(5):507-11. doi:10.1700/1660.18164 PubMed
83.Simos D, Sajjady G, Sergi M, et al. Third-line chemotherapy in small-cell lung cancer: an international analysis. Clin Lung Cancer. 2014;15(2):110-8. doi:10.1016/j.cllc.2013.11.003 PubMed
84.Nagy-Mignotte H, Guillem P, Vignoud L, et al. Outcomes in recurrent small-cell lung cancer after one to four chemotherapy lines: a retrospective study of 300 patients. Lung Cancer. 2012;78(1):112-20. doi:10.1016/j.lungcan.2012.06.006 PubMed
85.Coutinho AD, Shah M, Lunacsek OE, Eaddy M, Willey JP. Real-world treatment patterns and outcomes of patients with small cell lung cancer progressing after 2 lines of therapy. Lung Cancer. 2019;127:53-58. doi:10.1016/j.lungcan.2018.11.009 PubMed
86.Saruwatari K, Umemura S, Nomura S, et al. Prognostic Factor Analysis in Patients With Small-Cell Lung Cancer Treated With Third-Line Chemotherapy. Clin Lung Cancer. 2016;17(6):581-587. doi:10.1016/j.cllc.2016.05.022 PubMed
87.Austin PC. The use of propensity score methods with survival or time-to-event outcomes: reporting measures of effect similar to those used in randomized experiments. Stat Med. 2014;33(7):1242-58. doi:10.1002/sim.5984 PubMed
88.Amgen Canada Inc. Health Canada reviewer's report: Imdelltra (tarlatamab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imdelltra, 1 mg and 10 mg per vial, lyophilized powder for intravenous infusion. 2024.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAV
cyclophosphamide, doxorubicin, and vincristine
CDA-AMC
Canada’s Drug Agency
CRS
cytokine release syndrome
ES-SCLC
extensive-stage small cell lung cancer
ICANS
immune effector cell-associated neurotoxicity syndrome
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LY
life-year
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
SCLC
small cell lung cancer
TTD
time to discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tarlatamab (Imdelltra), 1 mg and 10 mg, lyophilized powder for solution for IV infusion |
Indication | For the treatment of adult patients with extensive-stage small cell lung cancer with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard (Project Orbis Type B) |
NOC date | September 11, 2024 |
Reimbursement request | As per indication |
Sponsor | Amgen Canada |
Submission history | No |
NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult patients with ES-SCLC after platinum-based chemotherapy and at least 1 other treatment |
Treatment | Tarlatamab |
Dose regimen | IV infusion with a step-up dose of 1 mg on day 1, followed by a 10 mg dose on day 8 and day 15, and every 2 weeks thereafter in a 28-day cycle until disease progression |
Submitted price | $1,545.00 per 1 mg vial $15,450.00 per 10 mg vial |
Submitted treatment cost | $27,740 for first 28-day cycle and $26,420 for subsequent cycles |
Comparator | Basket comparator, consisting of topotecan; a platinum agent and etoposide; combination therapy of cyclophosphamide, doxorubicin, and vincristine; etoposide; and irinotecan |
Perspective | Canadian publicly funded health care payer |
Outcomes | LYs, QALYs |
Time horizon | Lifetime (20 years) |
Key data sources | DeLLphi-301: A phase II, open-label, single-arm, multinational trial for tarlatamab Comparative efficacy of tarlatamab with basket comparator informed by sponsor’s submitted patient-level ITC based on the Flatiron study external control cohort |
Submitted results | ICER = $178,286 per QALY (incremental cost: $189,132; incremental QALYs: 1.06) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; ES-SCLC = extensive-stage small cell lung cancer; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Conclusions
The clinical review by Canada’s Drug Agency (CDA-AMC) found that the evidence within the DeLLphi-301 trial precludes the ability to attribute the study results to treatment given the single-arm design and immature survival data. Nearly all patients reported adverse events (AEs), including a large proportion of patients who experienced cytokine release syndrome (CRS), although the safety profile was consistent with the mechanism of action of tarlatamab and most AEs were manageable. Due to the lack of direct comparative evidence with relevant treatments for patients with extensive-stage small cell lung cancer (ES-SCLC) in the third-line setting, the sponsor submitted an indirect treatment comparison (ITC). The CDA-AMC clinical review noted that the ability to draw firm conclusions on tarlatamab's comparative clinical benefit was hindered by limitations in the evidence including a risk of selection bias, the sizable reduction in effective sample size, heterogeneity in patient population and study design, and an inadequate or lack of adjustment for potential prognostic factors. In light of this, the comparative clinical effects of tarlatamab remain very uncertain.
CDA-AMC undertook reanalyses to address some of the key limitations in the sponsor’s analysis which included selecting alternative parametric functions to extrapolate overall survival (OS) and progression-free survival (PFS) for tarlatamab and the basket comparator, setting relative dose intensity (RDI) to be identical across all treatments, and applying an alternative set of health utility values. The CDA-AMC reanalysis results were largely consistent with the sponsor. In adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy; tarlatamab remained more costly but more effective compared with a basket comparator consisting of topotecan, a platinum agent and etoposide, combination therapy of cyclophosphamide, doxorubicin, and vincristine (CAV), etoposide, and irinotecan. The incremental cost-effectiveness ratio (ICER) of tarlatamab compared to the basket comparator was $559,946 per quality-adjusted life-year (QALY) gained (incremental costs: $222,201; incremental QALYs: 0.40). A price reduction of 85.6% (from $1,545 to $222 per 1 mg vial and $15,450 to $2,220 per 10 mg vial) was necessary for tarlatamab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
The cost-effectiveness of tarlatamab is driven by assumptions pertaining to the extrapolation of OS for tarlatamab and its price. In the CDA-AMC reanalysis, fewer of the incremental QALY benefits (30%) were derived from the period beyond which there are observed trial data (i.e., extrapolated period) compared to the sponsor’s base-case analysis (57%). However, given both the sponsor and CDA-AMC analyses were limited by the lack of head-to-head clinical evidence to inform relative treatment effects, the cost-effectiveness of tarlatamab and price-reduction estimates are highly uncertain. Further price reductions may be warranted to address this uncertainty.
Perspectives of Patients, Clinicians, and Drug Programs
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
The patient input was provided jointly by Lung Cancer Canada, the Canadian Cancer Survivor Network, and the Lung Health Foundation. The information was collected through virtual interviews with 3 patients diagnosed with ES-SCLC. Two patients were from Canada and 1 was from the US. Current treatment options beyond first-line therapy are chemotherapy, immunotherapy, and radiation for specific metastases. Treatment goals for ES-SCLC include symptom relief, managing disease progression, and prolonging life. All patients interviewed had experience with tarlatamab. Two patients experienced CRS during initial tarlatamab infusions, but the side effects decreased over time. Overall, patients reported that tarlatamab improved their quality of life, allowing them to regain independence and return to daily activities, and they experienced better symptom control compared to previous therapies.
The clinician inputs were received from the Ontario Health–Cancer Care Ontario Lung Cancer Drug Advisory Committee and Lung Cancer Canada Medical Advisory Committee. The current pathway of care for patients with small cell lung cancer (SCLC) typically involves first-line treatment with chemotherapy, often combined with immunotherapy for ES-SCLC. Despite initial high response rates, most patients develop drug resistance and face poor outcomes, with limited treatment options after disease progression, particularly for those with platinum-resistant or refractory disease. Clinicians noted that the goal of treatment would be to improve survival, rapidly reduce tumour burden, have reasonable toxicity, and improve quality of life. Clinicians noted that tarlatamab would be used as the last line of treatment and would be most suitable in patients with extensively pretreated SCLC with higher physiologic reserve and Eastern Cooperative Oncology Group Performance Status of 0 to 1 who have the capacity to self-manage. The first cycle would be administered in an inpatient setting given the risk of CRS and immune effector cell-associated neurotoxicity (ICANS) with at least a 24-hour to 48-hour observation period post administration, and all subsequent outpatient infusion would require monitoring of patients 0 hours to 4 hours post infusion with ongoing vigilance by caregivers at home.
The drug plans highlighted concerns about the DeLLphi-301 trial being a single-arm trial with no comparators. They further questioned whether patients with Eastern Cooperative Oncology Group Performance Status greater than 1 could receive tarlatamab, and if prior treatment in the limited-stage SCLC setting would count toward the 2 required treatments in patients with ES-SCLC. Drug plans similarly noted that additional health care resources (e.g., hospitalization, pharmacotherapy) would be required for the first 2 doses of tarlatamab to monitor and manage CRS and ICANS. Questions remained on the potential discontinuation criteria given the DeLLphi-301 trial permitted patients to continue tarlatamab beyond radiographic progression.
Several of the concerns were addressed in the sponsor's model.
The clinical efficacy of tarlatamab was modelled based on OS and PFS.
The utility and resource impacts of grade 3 or higher AEs that occurred in more than 3% of patients in the DeLLphi-301 trial along with grade 1 or 2 CRS and ICANS were included in the submitted economic model.
The first 2 administrations of tarlatamab (i.e., day 1 and day 8 of cycle 1) were assumed to be administered in the inpatient setting.
Within the sponsor’s budget impact analysis, it was assumed that patients who originally had limited-stage SCLC and who received treatment would be eligible for tarlatamab if the cancer relapses or recurs to ES-SCLC and if they have disease progression on or after at least 2 prior lines of therapy.
In addition, CDA-AMC addressed some of these concerns as follows.
The sponsor assumed that tarlatamab would be discontinued upon disease progression, aligned with its product monograph.1 CDA-AMC conducted a scenario analysis to explore the potential cost-effectiveness if patients were permitted to continue treatment postprogression with the duration of tarlatamab use reflecting the time on treatment that is reported in the tarlatamab trial.
CDA-AMC was unable to address the following concerns raised by input: absence of head-to-head clinical evidence to inform the comparative clinical effectiveness of the sponsor’s model.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of tarlatamab for the treatment of adult patients with ES-SCLC after platinum-based chemotherapy and at least 1 other treatment. In the model, the comparator was a basket of therapies consisting of topotecan, etoposide with platinum-based chemotherapy, CAV, etoposide, and irinotecan. The modelled population reflects its Health Canada indication.
Tarlatamab is available as lyophilized powder in 1 mg or 10 mg vials and is packaged with 2 vials of 7 mL IV solution stabilizer to reconstitute for IV infusion.2 The recommended dosing regimen starts with 1 mg on day 1, followed by a 10 mg dose on day 8 and day 15, then 10 mg every 2 weeks in subsequent 28-day cycles.1 At the submitted price of $1,545 per 1 mg vial and $15,450 per 10 mg vial, the sponsor estimated that the drug acquisition costs for tarlatamab in the first 28-day cycle are $27,740, with subsequent cycles costing $26,420 per patient based on an RDI of 85.5%.2 To reduce the risk of CRS, concomitant medications should be administered as recommended before and after cycle 1 (e.g., 8 mg dexamethasone on day 1 and day 8).1 The distribution of regimens forming the basket comparator was based on an Albertan retrospective cohort study.3 The sponsor estimated the 28-day cost of the basket comparator to be $2,216 per patient based on an RDI ranging from 100% to 76%.2 Drug wastage was considered for both IV and oral therapies in the model.
The clinical outcomes of interest were life-years (LYs) and QALYs. The economic analysis was conducted over a lifetime (20 years; weekly cycles) time horizon from the perspective of a public health care payer in Canada. Discounting (1.5% per annum) was applied for both costs and outcomes.2
Model Structure
The sponsor submitted a partitioned survival model with 3 health states: progression-free, postprogression, and death (Figure 1). The proportion of patients who were progression-free, postprogression, or were dead at any time over the model’s horizon was derived from non—mutually exclusive survival curves based on the area under the survival curve. All patients entered the model in the progression-free health state. The proportion of patients in the progression-free health state during each model cycle was estimated based on the PFS curve. OS was partitioned to estimate the proportion of patients in the dead state and the postprogression state, reflecting alive patients who had progressed. The proportion of patients in the postprogression health state was based on the difference between the OS and the PFS curves.2
Model Inputs
The baseline characteristics used to inform the model were derived from the DeLLphi-301 study,4,5 a phase II, open-label, single-arm trial assessing tarlatamab's efficacy and safety in adult patients with ES-SCLC after platinum-based chemotherapy and at least 1 other treatment (mean age = 63.5 years; 26.8% female; mean weight = 73.96 kg; mean body surface area = 1.78 m2).2
Key clinical inputs (PFS, OS, and time to discontinuation [TTD]) for tarlatamab were generated from the DeLLphi-301 study (data cut-off: October 2, 2023; n = 97), with adjustments on OS and TTD to remove the impact of postprogression treatment. Extrapolation curves were chosen based on clinical plausibility and statistical fit.2 Data to inform the basket comparator were derived from the Flatiron study health research database (January 1, 2013, to October 31, 2021), which includes longitudinal real-world data of patients from the US. Patients with third-line or later ES-SCLC treated according to American Society of Clinical Oncology–Ontario Health guidelines6 were selected based on the inclusion and exclusion criteria of the DeLLphi-301 study with additional propensity score and standardized mortality ratio weighting. The weighted PFS, OS, and TTD curves for the basket comparator were derived according to the same statistical distributions selected for the corresponding tarlatamab curves.
Health state utility values were derived from patient-level EQ-5D-5L data collected in the DeLLphi-301 trial (data cut-off: June 27, 2023), with tariffs from Canada applied.7 Disutility inputs were sourced from the literature and applied to grade 3 or higher AEs occurring in more than 3% of patients in any treatment arm. Disutilities for grade 1 or 2 CRS and ICANS were also included, which were specific to tarlatamab. AE disutilities were applied during the first treatment cycle, with AE rates based on the DeLLphi-301 trial for tarlatamab and literature for the basket comparator.2
Costs in the model included drug acquisition, drug administration, subsequent treatment, medical resource utilization and monitoring, AE management, and terminal care costs. Drug acquisition costs were calculated by the sponsor as a function of unit drug costs, dosing schedules, RDI, and the proportion of patients on treatment. The cost of tarlatamab was based on the sponsor’s submitted price while all other drug acquisition costs were obtained from the CDA-AMC submission of lurbinectedin or the Ontario Drug Benefit Formulary.2,8 The sponsor further included tarlatamab pretreatment costs with dexamethasone. Administration costs for IV therapies were included for both arms, including delivery, nursing workload, pharmacy workload, and infusion chair time, with unit prices obtained from the Ontario Schedule of Benefits and literature.9-12 Cost of inpatient monitoring was included for the initiation of tarlatamab treatment according to the DeLLphi-301 trial and assumed patients would be hospitalized for 24 hours on day 1 and day 8 of cycle 1.2,4 After disease progression, a portion of patients received subsequent treatments (assumed to be same as third-line treatment distribution of the basket comparator) based on the DeLLphi-301 trial and the UK Cancer Analysis System study for tarlatamab and the basket comparator, respectively.13-16 The frequency of medical resource utilization (i.e., emergency, ambulatory, and nonemergency ambulatory visits) and monitoring (i.e., chest X-ray, electrocardiogram) were based on literature, with unit prices from the Ontario Schedule of Benefits.9 AE management costs, including CRS and ICANS, were applied during the first cycle. A one-time terminal care cost was applied when patients transitioned to death. All costs were adjusted to 2024 values in Canadian dollars.2
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following. The submitted analysis was based on the submitted price for tarlatamab.
Base-Case Results
In the sponsor’s base-case analysis, tarlatamab was associated with estimated cost of $221,531 and 1.93 QALYs over a 20-year time horizon. When compared with the basket comparator, tarlatamab had an incremental cost of $189,132 and a gain of 1.06 QALYs, resulting in an estimated ICER of $178,286 per QALY gained (Table 3). In addition, the sponsor’s model estimated that there would be an incremental LY gain of 0.42 associated with tarlatamab. At a willingness-to-pay threshold of $50,000 per QALY gained, tarlatamab had a 0% probability of being considered the optimal treatment.
The key cost driver within the sponsor’s submitted model was the price of tarlatamab while the key QALY driver was the utility value for progression-free state. More than half of the incremental QALY benefits (57%) for tarlatamab were derived from the period beyond which there are observed trial data (i.e., extrapolated period) and, at the end of the 20-year time horizon, 2.0% of modelled patients in the tarlatamab arm and 0.3% in the basket comparator arm remained alive.
Additional results from the sponsor’s submitted economic base are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. basket comparator ($/QALY) |
|---|---|---|---|---|---|
Basket comparator | 32,400 | Reference | 0.87 | Reference | Reference |
Tarlatamab | 221,531 | 189,132 | 1.93 | 1.06 | 178,286 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted probabilistic scenario analysis to account for methodological and structural uncertainties related to key assumptions, including no adjustment of tarlatamab OS and TTD to account for postprogression tarlatamab use and alternative data sources to inform the efficacy of the basket comparator. The ICERs ranged from $121,226 per QALY gained (using Oncology Outcomes Study data to inform the efficacy of the basket comparator) to $215,756 per QALY gained (using treatment duration and OS of tarlatamab as observed in the DeLLphi-301 trial). Deterministic scenario analyses were conducted to evaluate different efficacy and model setting parameters, such as alternative parametric functions for OS, PFS, and TTD extrapolation. The results were largely aligned with the sponsor’s base-case analysis.
No scenario analysis was conducted using a perspective other than the health care payer.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Uncertainty in the comparative clinical efficacy and safety: The DeLLphi-301 study was a single-arm trial with no head-to-head studies comparing tarlatamab to the basket comparator. In a single-arm trial, there is a risk of selection bias; therefore, the treatment effect of the study drug cannot be directly assessed because what proportion of the estimated treatment response can be attributed to a patient’s natural history and other prognostic factors cannot be determined. As such, the sponsor conducted an ITC using patient-level data from the Flatiron study external control cohort to estimate comparator efficacy for the economic model. The CDA-AMC Clinical Review Report identified multiple limitations in the sponsor's ITC, including a risk of selection bias in study inclusion, a sizable reduction in effective sample size, the heterogeneity in patient population and study design, and inadequate or lack of adjustment for potential prognostic factors. Consequently, there is significant uncertainty in interpreting the clinical comparison between tarlatamab and the basket comparator.
The PFS and OS curves for tarlatamab were derived from the DeLLphi-301 trial, while those for the basket comparator were based on the Flatiron study external control cohort. Parametric survival functions were used to extrapolate the Kaplan-Meier curves over the model’s time horizon. The sponsor chose a log-logistic distribution to extrapolate OS for both tarlatamab and the basket comparator, resulting in 5-year OS rates of 12.2% and 3.0%, respectively. However, clinical expert input received by CDA-AMC for this review indicated that the proportion of patients alive at 5 years, as predicted from the selected OS curve, was highly optimistic. Indeed, a real-world study on patients with ES-SCLC in Canada found that the 5-year OS rate for 3 types of second-line chemotherapy was close to 0% and, according to clinical expert feedback, the prognosis of patients in the third-line setting would be worse.17 For PFS, the sponsor selected a generalized gamma distribution to extrapolate survival outcomes for both arms, resulting in 2-year PFS rates of 15.2% for tarlatamab and 3.4% for the basket comparator. Clinical experts consulted by CDA-AMC suggested these figures were likely overestimated, given the poor prognosis and rapid progression of ES-SCLC in the third-line setting.
CDA-AMC reanalyses employed alternative parametric functions for both arms to ensure that PFS and OS estimates were more closely aligned with expert expectations and with the published literature. Specifically, CDA-AMC selected a Weibull distribution for tarlatamab OS, resulting in a 5-year OS rate of 2.2%, and a gamma distribution for the basket comparator OS, with a 5-year OS rate of 0.1%. For PFS in both arms, a gamma distribution was applied, yielding 2-year PFS rates of 4.8% for tarlatamab and 0.3% for the comparator.
Uncertainty in drug acquisition costs: The sponsor applied an RDI of 85.50% for tarlatamab, which was multiplied by the sponsor’s submitted price for tarlatamab to derive drug acquisition costs.2 CDA-AMC submitted an additional information request to the sponsor to clarify how the RDI was derived and received clarification that this was calculated from the DeLLphi-301 trial as the actual cumulative dose divided by the planned dose.18 The sponsor’s approach to estimate drug acquisition costs does not consider other factors that may affect dosage adjustments. Dosage may vary based on delays in dosing due to toxicity (tarlatamab should be withheld until ICANS or CRS are resolved as per the product monograph) and subsequently stepped up to restart therapy after a dose delay.1 Furthermore, the sponsor's approach to incorporating RDI into the basket comparator biased the sponsor’s results in favour of tarlatamab as the RDI of comparator treatments were higher (range, 93% to 100%) with the exception of topotecan where an RDI of 76% was selected based on a study published in 1999.19
While clinical expert feedback sought by CDA-AMC noted that, across both the intervention and the comparators informing the basket comparator, the RDI is unlikely to be 100%, there is uncertainty to what the real-world RDI would be to inform drug acquisition costs. To remain consistent across tarlatamab and its comparators in light of the limited real-world evidence available, an RDI of 100% was assumed for tarlatamab and all basket comparator treatments in the CDA-AMC reanalysis. A scenario analysis was further conducted in which the original RDI was kept for tarlatamab and topotecan while the treatments within the basket comparator had their RDI lowered to the same value as tarlatamab.
Inappropriate health utilities for progression-free and postprogression health states: The health utilities for the progression-free and postprogression health states used in the model were 0.834 and 0.755, respectively, estimated from patient-level EQ-5D-5L data collected in the DeLLphi-301 trial.2 The expected utility for the general population of people living in Canada at the same age range as studied in the model would be 0.839.20 The utility values selected by the sponsor would suggest that patients with ES-SCLC in the progression-free state would have a health-related quality of life similar to their general population counterpart. Given the aggressiveness of ES-SCLC and that the indication of tarlatamab is for those with disease progression on or after at least 2 prior lines of therapy, there is limited face validity to support the sponsor’s claim. In fact, a published literature review found that health utilities for patients with stable and progressive ES-SCLC in Canada were 0.72 and 0.52, respectively.21
In its reanalysis, CDA-AMC applied health utilities of 0.72 for the progression-free state and 0.52 for the postprogression state.21 The sponsor’s original health utilities were included in a scenario analysis.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
For the basket comparator, the distribution of comparator regimens informing treatment cost was based on a real-world study from Canada, while efficacy was sourced from the Flatiron study database. | Inappropriate but acceptable. While this limits one’s ability to address the cost-effectiveness of tarlatamab for individual treatment regimens used in this patient population, input from clinical experts obtained by CDA-AMC noted that treatment options would mainly be palliative or best supportive care for this patient population. While the inconsistency between the sources for comparator regimens in cost and efficacy could introduce uncertainty in the cost-effectiveness results, the variation in the 28-day cycle costs across comparator regimens were small ($103 to $2,835) relative to the costs of tarlatamab and unlikely to have a substantial impact on the overall results. |
The first 2 dose of tarlatamab was assumed to be administered an inpatient setting. For those with CRS or ICANS, resumption of the administration of tarlatamab treatment may not always be conducted in an inpatient setting. | Inappropriate but unlikely to be key model driver. According to the product monograph, patients with grade 2 CRS require 24-hour monitoring in an appropriate health care setting when resuming tarlatamab treatment.1 |
Disutilities and costs associated with grade 1 or 2 CRS and grade 1 or 2 ICANS were included. Specifically, hospitalization due to CRS was assumed to be 1 day. | Inappropriate but unlikely to be influential. Results of the DeLLphi-301 study indicated the median duration of CRS was 4 days with a median duration of hospitalization for serious CRS events ranging from 2.5 days to 4 days depending on the patient cohort.4,5 While the sponsor's assumptions may underestimate the AE-related costs for tarlatamab, the proportion of AE management costs relative to total costs in the tarlatamab arm is minimal (1.4%) and is unlikely to significantly impact the results if the hospitalization duration increased. |
A proportion of patients was assumed to receive subsequent active treatments in each treatment arm. | Uncertain but unlikely to impact the model given the proportion of total costs that are related to subsequent treatment is low. It is reasonable, according to clinical experts input sought by CDA-AMC, to assume that the majority of patients who subsequently progress after treatment would not receive an active treatment. |
Patients would discontinue tarlatamab upon disease progression. | Potentially reasonable. The product monograph states that patients should be treated (with tarlatamab) until disease progression. However, clinical expert feedback received by CDA-AMC noted that some clinicians may continue to administer tarlatamab beyond disease progression similar to the approach taken in the DeLLphi-301 trial if no reimbursement criteria restrict its use due to the limited third-line treatment options available. This could lead to ongoing costs for tarlatamab, leaving the impact on cost-effectiveness uncertain. CDA-AMC conducted a scenario analysis to explore the potential impact if patients remained on tarlatamab even upon progression, with time-to-treatment duration reflecting the DeLLphi-301 trial. |
AE = adverse event; CDA-AMC = Canada's Drug Agency; CRS = cytokine release syndrome; ICANS = immune effector cell-associated neurotoxicity syndrome.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. Changes to the sponsor’s analyses are summarized in Table 5 and include alterations to the OS and PFS extrapolation in both arms, adjustment to RDIs and revising the health utilities. CDA-AMC was unable to fully address limitations regarding the uncertainty in comparative efficacy due to the DeLLphi-301 study being a single-arm trial.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1a. Tarlatamab OS extrapolation | Log-logistic | Weibull |
1b. Basket comparator OS extrapolation | Log-logistic | Gamma |
1c. Tarlatamab PFS extrapolation | Generalized gamma | Gamma |
1d. Basket comparator PFS extrapolation | Generalized gamma | Gamma |
2. RDI | 85.5% for tarlatamab, 76.0% for topotecan, 93.0% for carboplatin, 90.0% for etoposide, and 100% for others | 100% for all treatments |
3. Health utilities | 0.834 for progression-free and 0.755 for postprogression | 0.720 for progression-free and 0.520 for postprogression |
CDA-AMC base case | Reanalysis 1a + 1b + 1c + 1d + 2 + 3 | |
CDA-AMC = Canada's Drug Agency; OS = overall survival; PFS = progression-free survival; RDI = relative dose intensity.
In the CDA-AMC base case, tarlatamab was associated with a total cost of $255,623 and 0.93 QALYs, compared to $33,422 and 0.53 QALYs for the basket comparator. Approximately 30% of the incremental QALYs were gained from the period beyond which there are observed trial data (reduced from 57% in the sponsor’s base case). The CDA-AMC model estimated a small gain in LYs (0.65) associated with tarlatamab compared to the basket comparator. Similar to the sponsor’s base case, drug costs accounted for most of tarlatamab’s total expected costs (91%). The resulting ICER for tarlatamab compared to the basket comparator was $559,964 per QALY gained (incremental costs: $222,201; incremental QALYs: 0.40), with a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Further details and disaggregated outcomes are available in Table 11.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER vs. basket comparator ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Basket comparator | 32,400 | 0.87 | Reference |
Tarlatamab | 221,531 | 1.93 | 178,286 | |
CDA-AMC reanalysis 1a: tarlatamab OS | Basket comparator | 32,400 | 0.87 | Reference |
Tarlatamab | 221,814 | 1.23 | 531,705 | |
CDA-AMC reanalysis 1b: basket comparator OS | Basket comparator | 32,451 | 0.69 | Reference |
Tarlatamab | 221,531 | 1.93 | 152,305 | |
CDA-AMC reanalysis 1c: tarlatamab PFS | Basket comparator | 32,400 | 0.87 | Reference |
Tarlatamab | 223,171 | 1.88 | 190,049 | |
CDA-AMC reanalysis 1d: Basket comparator PFS | Basket comparator | 32,513 | 0.86 | Reference |
Tarlatamab | 221,531 | 1.93 | 176,873 | |
CDA-AMC reanalysis 2: RDI | Basket comparator | 33,239 | 0.87 | Reference |
Tarlatamab | 253,403 | 1.93 | 207,538 | |
CDA-AMC reanalysis 3: health utilities | Basket comparator | 32,400 | 0.67 | Reference |
Tarlatamab | 221,531 | 1.53 | 219,962 | |
CDA-AMC base case (reanalyses 1a + 1b + 1c + 1d + 2 + 3) | Basket comparator | 33,422 | 0.53 | Reference |
Tarlatamab | 255,623 | 0.93 | 559,946 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; RDI = relative dose intensity; vs. = versus.
Scenario Analysis Results
CDA-AMC conducted price analyses based on the sponsor’s base case and the base-case reanalysis by CDA-AMC. Based on the base case by CDA-AMC, a price reduction of approximately 85.6% would be required to achieve cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY (Table 7).
Table 7: CDA-AMC Price-Reduction Analyses
Analysis | Unit drug costs | ICERs for tarlatamab vs. basket comparator ($/QALY) | |
|---|---|---|---|
Price reduction | Sponsor base case | CDA-AMC reanalysis | |
No price reduction | $1,545 | $178,425 | $555,503 |
10% | $1,391 | $159,528 | $496,473 |
20% | $1,236 | $140,631 | $437,443 |
30% | $1,082 | $121,734 | $378,413 |
40% | $927 | $102,837 | $319,383 |
50% | $773 | $83,940 | $260,353 |
60% | $618 | $65,042 | $201,323 |
68% | $494 | $50,000 | — |
70% | $464 | $46,145 | $142,293 |
80% | $309 | $27,248 | $83,263 |
86% | $222 | — | $50,000 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CDA-AMC conducted a scenario analysis to assess the impact of an alternative RDI assumption on the cost-effectiveness of tarlatamab. In this analysis, both the RDI of tarlatamab and its comparator were lowered to 85.5%. CDA-AMC also conducted a scenario analysis to evaluate the impact of extended tarlatamab use beyond progression on the cost-effectiveness results.
CDA-AMC conducted a scenario analysis to assess the impact of an alternative RDI assumption on the cost-effectiveness of tarlatamab. In this analysis, both the RDI of tarlatamab and its comparator were lowered to 85.5%. CDA-AMC also conducted a scenario analysis to evaluate the impact of extended tarlatamab use beyond progression on the cost-effectiveness results.
Issues for Consideration
Tarlatamab administration requires hospitalization and must be overseen by health care professionals equipped to manage CRS and neurologic toxicity. According to clinical expert feedback received by CDA-AMC, these specialized requirements may limit the number of treatment sites capable of initiating tarlatamab therapy.
Overall Conclusions
The CDA-AMC clinical review found that the evidence within the DeLLphi-301 trial precludes the ability to attribute the study results to treatment given the single-arm design and immature survival data. Nearly all patients reported AEs, including a large proportion of patients who experienced CRS, although the safety profile was consistent with the mechanism of action of tarlatamab and most AEs were manageable. Due to the lack of direct comparative evidence with relevant treatments for patients with ES-SCLC in the third-line setting, the sponsor submitted an ITC. The CDA-AMC clinical review noted that the ability to draw firm conclusions on tarlatamab's comparative clinical benefit was hindered by limitations in the evidence including a risk of selection bias, the sizable reduction in effective sample size, heterogeneity in patient population and study design, and an inadequate or lack of adjustment for potential prognostic factors. In light of this, the comparative clinical effects of tarlatamab remain very uncertain.
CDA-AMC identified several limitations with the submitted economic evaluation, including uncertainty in comparative efficacy and safety, uncertainty in treatments costs due to the biased application of RDI, and health utilities that lacked face validity. CDA-AMC undertook reanalyses to address some of the key limitations in the sponsor’s analysis which included selecting alternative parametric functions to extrapolate OS and PFS for tarlatamab and the basket comparator, setting RDI to be identical across all treatments, and applying an alternative set of health utility values. The CDA-AMC reanalysis results were largely consistent with the sponsor. In adult patients with ES-SCLC with disease progression on or after at least 2 prior lines of therapy including platinum-based chemotherapy, tarlatamab remained more costly but more effective compared with a basket comparator consisting of topotecan, a combination therapy of a platinum agent and etoposide, combination therapy of CAV, etoposide, and irinotecan. The ICER of tarlatamab compared to the basket comparator was $559,946 per QALY gained (incremental costs: $222,201; incremental QALYs: 0.40). A price reduction of 85.6% (from $1,545 to $222 per 1 mg vial and $15,450 to $2,220 per 10 mg vial) was necessary for tarlatamab to be considered cost-effective at willingness-to-pay-threshold of $50,000 per QALY gained threshold.
The reanalysis revealed that the cost-effectiveness results were sensitive to the selection of the parametric functions to inform the OS curves for tarlatamab and basket comparator Specifically, with the Weibull function applied to inform the OS curve for tarlatamab (i.e., lower long-term OS rates), the ICER increased significantly. The selection of this statistical distribution to inform the OS curves alongside revisions to more conservative OS curves for the basket comparator resulted in fewer of the incremental QALY benefits (30%) derived from the period beyond which there are observed trial data (i.e., extrapolated period) compared to the sponsor’s base-case analysis (57%). Additionally, similar to the sponsor’s results, the CDA-AMC reanalysis indicated that the price of tarlatamab comprised the majority (91%) of the total cost in the tarlatamab arm.
Given both the sponsor and CDA-AMC analyses were limited by the lack of head-to-head clinical evidence to inform relative treatment effects, the cost-effectiveness of tarlatamab and price-reduction estimates are highly uncertain. Further price reductions may be warranted to address this uncertainty.
References
1.Amgen Canada Inc. Imdelltra (tarlatamab): lyophilized powder for solution for intravenous infusion, 1 mg and 10 mg per vial [product monograph]. July 19, 2024. Updated September 11, 2024.
2.Amgen Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imdelltra (tarlatamab), lyophilized powder for solution for intravenous infusion, 1 mg and 10 mg per vial. July 19, 2024.
3.Suri G, Boyne DJ, Caveen M, et al. CO37 Real-World Treatment Patterns, Healthcare Resource Utilization (HCRU) and Clinical Outcomes of Patients with Extensive-Stage Small-Cell Lung Cancer (ES-SCLC) in Alberta, Canada. Value in Health. 2024;27(6):S23-S23. doi:10.1016/j.jval.2024.03.126
4.Ahn MJ, Cho BC, Felip E, et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N Engl J Med. 2023;389(22):2063-2075. doi:10.1056/NEJMoa2307980 PubMed
5.Amgen Inc. Data on file; Protocol 20200491: A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects with Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment (DeLLphi-301) [internal sponsor's report; sponsor-supplied reference].
6.Khurshid H, Ismaila N, Bian J, et al. Systemic Therapy for Small-Cell Lung Cancer: ASCO-Ontario Health (Cancer Care Ontario) Guideline. J Clin Oncol. 2023;41(35):5448-5472. doi:10.1200/jco.23.01435 PubMed
7.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi:10.1097/MLR.0000000000000447 PubMed
8.Ontario Ministry of Health. Ontario Drug Benefit Formulary/Comparative Drug Index [sponsor-supplied reference]. Accessed May 8, 2024. https://www.formulary.health.gov.on.ca/formulary/
9.Ontario Ministry of Health. Schedule of Benefits. Physician Services Under the Health Insurance Act (effective July 24, 2023) [sponsor-supplied reference]. 2023. Accessed May 8, 2024. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-2024-01-24.pdf
10.Government of Canada. Job Bank. Registered Nurse (R.N.) in Canada [sponsor-supplied reference]. 2022. Accessed May 8, 2024. https://www.jobbank.gc.ca/marketreport/wages-occupation/993/CA
11.Government of Canada. Job Bank. Pharmacist in Canada [sponsor-supplied reference]. 2022. Accessed May 8, 2024. https://www.jobbank.gc.ca/marketreport/wages-occupation/18196/ca
12.Pettigrew M, Kavan P, Surprenant L, Lim HJ. Comparative net cost impact of the utilization of panitumumab versus cetuximab for the treatment of patients with metastatic colorectal cancer in Canada. J Med Econ. 2016;19(2):135-47. doi:10.3111/13696998.2015.1105230 PubMed
13.Amgen Inc. Data on file; Study 20220199: Real-world natural history study of patients with SCLC in England (CAS) [internal sponsor's report; sponsor-supplied reference].
14.Amgen Inc. Data on file; Trial analysis of DeLLphi-301 (Data cutoff date: 27 Jun 2023) [internal sponsor's report; sponsor-supplied reference].
15.Amgen Inc. Data on file; Trial analysis of DeLLphi-301 (Data cutoff date: 16 May 2024) [internal sponsor's report; sponsor-supplied reference].
16.Amgen Inc. Data on file; Trial analysis of DeLLphi-301 (Data cutoff date: 02 Oct 2023) [internal sponsor's report; sponsor-supplied reference].
17.O'Sullivan DE, Cheung WY, Syed IA, et al. Real-World Treatment Patterns, Clinical Outcomes, and Health Care Resource Utilization in Extensive-Stage Small Cell Lung Cancer in Canada. Curr Oncol. 2021;28(4):3091-3103. doi:10.3390/curroncol28040270 PubMed
18.Amgen Canada Inc. Amgen response to October 21, 2024 request for additional information regarding Imdelltra (tarlatamab) CDA review: rationale for RDI weighting [internal additional sponsor's information]. October 22, 2024.
19.von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol. 1999;17(2):658-67. doi:10.1200/JCO.1999.17.2.658 PubMed
20.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2024;25(1):147-155. doi:10.1007/s10198-023-01570-1 PubMed
21.Labbé C, Leung Y, Silva Lemes JG, et al. Real-World EQ5D Health Utility Scores for Patients With Metastatic Lung Cancer by Molecular Alteration and Response to Therapy. Clin Lung Cancer. 2017;18(4):388-395.e4. doi:10.1016/j.cllc.2016.12.015 PubMed
22.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed November 13, 2024. https://www.formulary.health.gov.on.ca/formulary/
23.Borghaei H, Pundole X, Sangaré L, et al. Natural history of SCLC patients treated in third-line and beyond: A retrospective real world study. Lung Cancer. 2024;193:107819. doi:10.1016/j.lungcan.2024.107819 PubMed
24.Amgen Inc. Clinical Study Report: 20200491. A Phase 2 Study Evaluating the Efficacy, Safety, Tolerability, and Pharmacokinetics of Tarlatamab in Subjects With Relapsed/Refractory Small Cell Lung Cancer After Two or More Prior Lines of Treatment (DeLLphi-301) [internal data; sponsor-supplied reference]. 2023.
25.Statistics Canada. Table 13-10-0111-01. Number and rates of new cases of primary cancer, by cancer type, age group and sex [sponsor-supplied reference]. 2024. https://doi.org/10.25318/1310011101-eng
26.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [sponsor-supplied reference]. Canadian Cancer Society; 2020. https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf
27.CADTH. Drug Reimbursement Review: lurbinectedin (Zepzelca) [sponsor-supplied reference]. Can J Health Technol. 2023;3(4). https://cadth.ca/sites/default/files/DRR/2023/PC0281-Zepzelca_combined.pdf
28.Elegbede AA, Gibson AJ, Fu H, et al. Real-World Adherence to Guideline-Recommended Treatment for Small Cell Lung Cancer. Am J Clin Oncol. 2020;43(4):236-242. doi:10.1097/COC.0000000000000657 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following Table 8 have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Extensive-Stage SCLC
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost | 28-day cycle cost |
|---|---|---|---|---|---|---|
Tarlatamab | 1 mg 10 mg | Lyophilized Powder for IV infusion | $1,545.0000a $15,450.0000a | Cycle 1: 1 mg on day 1, followed by 10 mg on day 8 and 15 Subsequent cycles: 10 mg on day 1 and 15 of each 28-day cycle | Cycle 1: $1,158.75 Cycle 2+: $1,103.57 | Cycle 1: $32,445 Cycle 2+: $30,900 |
Dexamethasone | 4 mg/mL | IV | $1.6922 | Cycle 1: 8 mg on day 1 and 8 | $0.24 | $6.76 |
Tarlatamab | Cycle 1: $1,158.99 Cycle 2+: $1,103.57 | Cycle 1: $32,452 Cycle 2+: $30,900 | ||||
CAV | ||||||
Cyclophosphamide | 500 mg 1,000 mg 2,000 mg | Dry powder for IV infusion | $107.8100b $195.4200b $359.4000b | 800 mg/m2 on day 1 of each 21-day cycle | $14.44 | $404 |
Doxorubicin | 10 mg 50 mg 200 mg | Lyophilized powder for IV infusion | $50.0000b $250.5000b $770.0000b | 50 mg/m2 on day 1 of 21-day cycle | $23.83 | $667 |
Vincristine | 1 mg 2 mg 5 mg | Powder for IV infusion | $30.6000b $61.2000b $153.0000b | 1.4 mg/m2 on day 1 of each 21-day cycle | $4.37 | $122 |
CAV | $42.64 | $1,194 | ||||
Platinum-based regimens | ||||||
Carboplatin + etoposide | ||||||
Carboplatin | 50 mg 150 mg 450 mg 600 mg | Solution for IV infusion | $70.0000b $210.0000b $600.0000b $775.0000b | AUC 5 on day 1 of each 21-day cycle | $46.90 | $1,313 |
Etoposide | 100 mg 200 mg 400 mg 500 mg 1,000 mg | IV infusion | $75.0000b $150.0000b $300.0000b $375.0000b $750.0000b | 100 mg/m2 daily on day 1 to 3 of each 21-day cycle | $21.43 | $600 |
Regimen Cost | $68.33 | $1,913 | ||||
Monotherapies | ||||||
Etoposide | ||||||
Etoposide | 50 mg | Tablet | $32.6400b | 50 mg/m2 on day 1 to 14 for each cycle of 21 or 28 days | $32.64 | $914 |
Topotecan | ||||||
Topotecan | 1 mg 4 mg | Lyophilized powder for IV injection | $138.7500b $555.0000b | 1.5 mg/m2 on day 1 to 5 of each 21-day cycle | $99.11 | $2,775 |
Off-label treatment | ||||||
Irinotecan | ||||||
Irinotecan | 40 mg 100 mg 300 mg 500 mg | Solution for IV injection | $208.3500b $520.8500b $1,560.0000b $2,600.0000b | 50 to 100 mg/m2 on day 1 of each 21-day cycle | $24.80 to | $694 to |
CDA-AMC = Canada's Drug Agency.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed September 2000), unless otherwise indicated, and do not include dispensing fees. For the purposes of dosage calculation, the average patient was assumed to weigh 75 kg, have a body surface area of 1.85 mg2 and included wastage of unused medication in vials.
aSponsor’s submitted price.
bIQVIA Delta Price Advisor wholesale price. Accessed September 2024.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to key limitations discussed elsewhere in this report. Because the DeLLphi-301 study is a single-arm trial, the sponsor conducted an ITC to inform comparative efficacy in the economic model. Issues were noted regarding the uncertainty in the comparative effectiveness. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key assumptions table. The sponsor’s model lacks flexibility to adjust the duration of hospitalization due to CRS during tarlatamab treatment, and the frequency of hospitalizations required when resuming tarlatamab treatment after CRS. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
ITC = Indirect treatment comparison; CRS = cytokine release syndrome.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
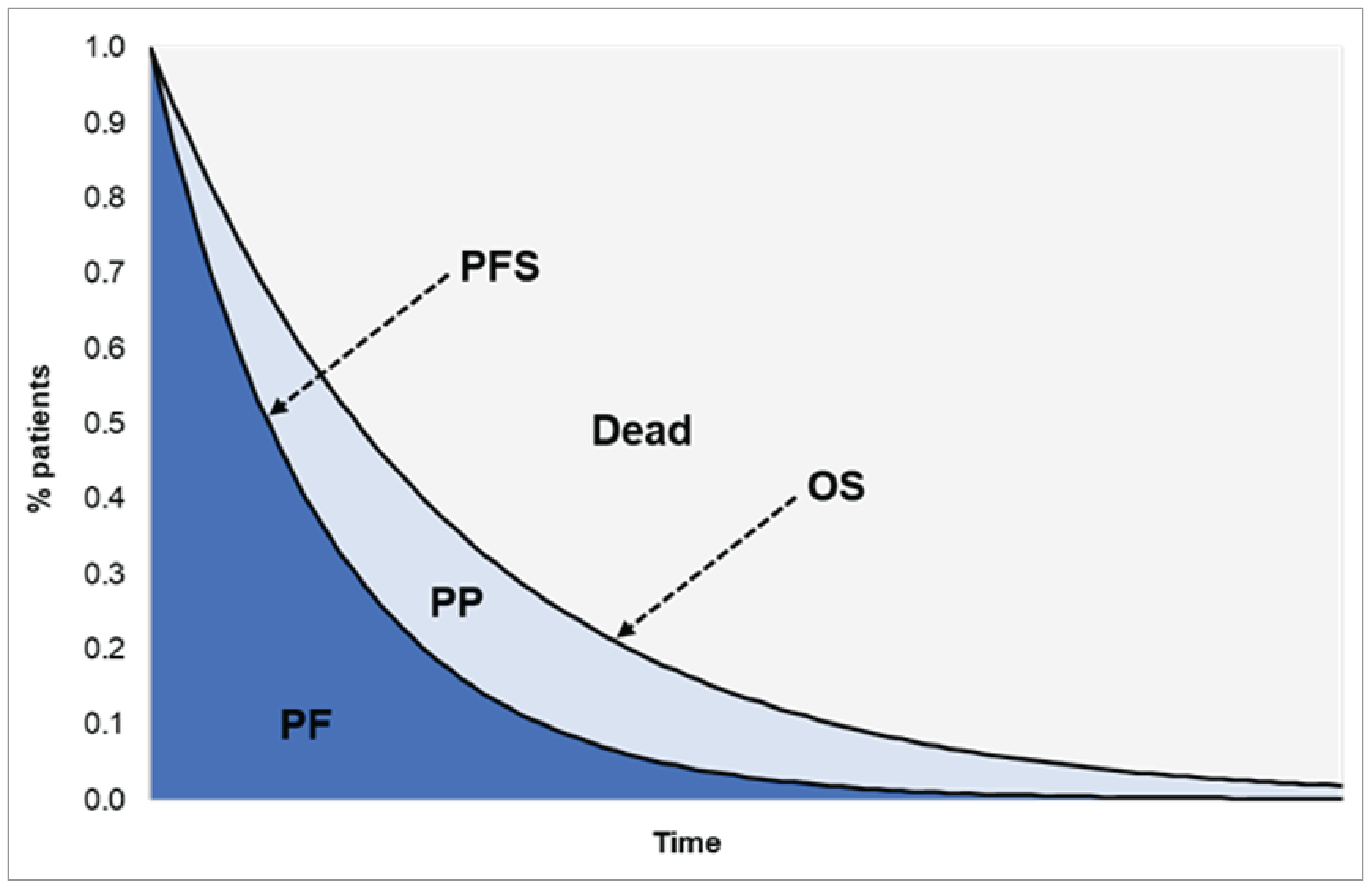
OS = overall survival; PF = progression-free; PFS = progression-free survival; PP = post progression.
Source: Sponsor’s pharmacoeconomic submission.2
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of Sponsor’s Economic Evaluation Results — Base Case
Parameter | Tarlatamab | Basket comparator | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 2.42 | 1.11 | 1.31 |
Progression-free | 1.41 | 0.52 | 0.88 |
Progressed | 1.01 | 0.58 | 0.42 |
Discounted QALYs | |||
Total | 1.93 | 0.87 | 1.06 |
Progression-free | 1.17 | 0.44 | 0.74 |
Progressed | 0.76 | 0.44 | 0.32 |
Decrements due to AEs | −0.003 | −0.005 | 0.002 |
Discounted costs ($) | |||
Total | 221,531 | 32,400 | 189,132 |
Drug costs | 199,338 | 10,986 | 188,352 |
Administration costs | 3,351 | 1,781 | 1,570 |
Medical resource use and monitoring costs | 1,193 | 649 | 544 |
Hospitalization costs at treatment initiation | 3,306 | 0 | 3,306 |
Subsequent treatment costs (one-off) | 706 | 570 | 137 |
Terminal care costs | 10,562 | 10,942 | −380 |
AE management costs | 3,075 | 7,472 | −4,397 |
AE = adverse event; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of Economic Evaluation Results by CDA-AMC
Parameter | Tarlatamab | Basket comparator | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.53 | 0.87 | 0.65 |
Progression-free | 0.70 | 0.42 | 0.27 |
Progressed | 0.83 | 0.45 | 0.38 |
Discounted QALYs | |||
Total | 0.93 | 0.53 | 0.40 |
Progression-free | 0.50 | 0.31 | 0.19 |
Progressed | 0.43 | 0.23 | 0.20 |
Decrement due to AEs | −0.003 | −0.005 | 0.002 |
Discounted costs ($) | |||
Total | 255,623 | 33,422 | 222,201 |
Drug costs | 232,951 | 11,860 | 221,090 |
Administration costs | 3,374 | 1,793 | 1,581 |
Medical resource use and monitoring costs | 1,203 | 653 | 549 |
Hospitalization costs at treatment initiation | 3,303 | 0 | 3,303 |
Subsequent treatment costs (one-off) | 812 | 629 | 183 |
Terminal care costs | 10,905 | 11,015 | −109 |
AE management costs | 3,075 | 7,472 | −4,397 |
ICER ($/QALY) | 559,946 | ||
AE = adverse event; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Table 12: Summary of CDA-AMC Scenario Analysis
Scenarios | Treatment | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CDA-AMC base case | Basket comparator | 33,422 | 0.53 | Reference |
Tarlatamab | 255,623 | 0.93 | 559,964 | |
CDA-AMC Scenario Analysis 1: Alternate RDI values | Basket comparator | 31,515 | 0.53 | Reference |
Tarlatamab | 223,453 | 0.93 | 483,682 | |
CDA-AMC Scenario Analysis 2: Extended tarlatamab treatment postprogression | Basket comparator | 33,489 | 0.53 | Reference |
Tarlatamab | 322,965 | 0.97 | 659,115 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada's Drug Agency.
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) to estimate the three-year budget impact of reimbursing tarlatamab as a third-line treatment for patients with ES-SCLC that has progressed after at least 2 prior lines of treatment. The analyses were done from the perspective of the public drug plan in Canada over a 3-year time horizon from 2026 to 2028, with only drug acquisition costs included in the base-case analysis.
The sponsor utilized an incidence-based approach to define the eligible population. The duration of treatment for the comparator therapies was informed by a US real-world evidence study while treatment duration for tarlatamab,23 this was informed by the DeLLphi-301 study.24 Key inputs to the BIA are documented in Table 14.
Key model assumptions included:
The incidence rate of lung cancer is constant over the 3-year time horizon that is analyzed.
For tarlatamab and the comparator regimens, the sponsor assumed 100% RDI.
Cost captured within the BIA included drug acquisition. Drug cost relating to the management of AEs (e.g., tocilizumab to manage CRS, IV steroids for the management of ICANS) and hospitalization costs associated with tarlatamab (e.g., the first 2 doses would be administered in a hospital inpatient setting and may require additional days of hospitalization in patients with CRS and ICANS AEs) were not considered within the sponsor’s BIA.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Adult population living in Canada | 33,440,808 / 34,054,246 / 34,679,21225 |
Annual lung cancer incidence | 63.9 / 100,00025 |
Proportion of lung cancer that is SCLC | 12%26 |
Proportion of SCLC that is extensive stage | 75%27 |
ES – Receiving 1L therapy | 90.4%28 |
ES – Receiving 2L therapy | 32.3%28 |
ES – Receiving 3L therapy | 32.9%28 |
Proportion of SCLC that is limited stage | 25%27 |
LS – Receiving treatment | 96.7%28 |
LS – Relapsing/recurred to extensive stage | 80.8%28 |
LS – Receiving 2L therapy | 30.2%28 |
LS – Receiving 3L therapy | 28.6%28 |
Number of patients eligible for drug under review | 227 / 232 / 236 |
Market uptake (3 years) | |
Uptake (reference scenario) CAV Carboplatin + Etoposide Oral Etoposide Irinotecan IV Topotecan | 30% / 30% / 30% 25% / 25% / 25% 15% / 15% / 15% 10% / 10% / 10% 20% / 20% / 20% |
Uptake (new drug scenario) Tarlatamab CAV Carboplatin + Etoposide Oral Etoposide Irinotecan IV Topotecan | 25% / 30% / 40% 20% / 15% / 10% 25% / 25% / 25% 15% / 15% / 10% 10% / 10% / 10% 5% / 5% / 5% |
Cost of treatment (per patient) | |
Monthly cost of treatment Tarlatamab- first cycle Tarlatamab-subsequent cycle CAV Carboplatin + Etoposide Oral Etoposide Irinotecan IV Topotecan | $35,269.45 $33,589.96 $1,138.07 $2,125.19 $886.92 $1,461.36 $2,850.66 |
Median duration of treatment (months) | |
Tarlatamab CAV Carboplatin + Etoposide Oral Etoposide Irinotecan IV Topotecan | 5.1 1.7 1.7 1.7 1.7 1.7 |
CAV = cyclophosphamide + doxorubicin + vincristine; ES = extensive stage; LS = limited stage; SCLC = small cell lung cancer.
Summary of the Sponsor’s BIA Results
The sponsor estimated the budget impact of reimbursing tarlatamab as a third-line therapy for ES-SCLC to be $9,628,556 in year 1, $11,785,113 in year 2, and $16,039,263 in year 3, for a 3-year total budget impact of $37,452,931.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The rate of uptake of tarlatamab is uncertain: The sponsor projected that 25% of patients would receive tarlatamab in year 1, with uptake increasing to 40% by year 3, based on real-world evidence from the US. As these data were based on the US, this may not be generalizable to a publicly financed health care system in Canada. Clinical expert feedback sought by CDA-AMC indicated that initial uptake in the first year may be lower due to logistical challenges (i.e., limited inpatient hospital bed spaces) but that uptake in the third year would be higher (ranging from 50% to 80%) given the lack of alternative treatment options and as clinicians acquire more experience with tarlatamab.
Given uncertainties to how tarlatamab would penetrate the market in Canada if reimbursed publicly, CDA-AMC conducted a scenario analysis testing different uptake rates based on the feedback of clinical expert opinion sought by CDA-AMC.
Potential for broader health care system impacts: A narrow public payer perspective was considered with only direct treatment costs captured (e.g., exclusion of concomitant medication and subsequent treatment). Tarlatamab is initially administered in a hospital setting, requiring health care resources beyond the scope of public drug plans’ coverage. There is further an increased risk of CRS and ICANS events that would require management in an inpatient setting. Such costs were not considered in the sponsor’s submitted BIA and it remains uncertain the extent to which the funding of tarlatamab would have a budgetary impact on the broader health care system.
No reanalyses could be conducted as such costs were not included in the sponsor’s submitted BIA.
Duration on treatment does not align with the submitted economic model: The sponsor used data from a global randomized control trial of 17 countries to estimate the treatment duration for tarlatamab, while relying on a US-based real-world evidence database to inform the treatment duration of the comparators. The median time on treatment did not align with the estimates contained within the submitted economic model that were adjusted to account for discontinuation upon progression (4.6 months for tarlatamab and 2.5 months for the basket comparator). With a lower median duration of treatment for tarlatamab, the budget impact of tarlatamab is expected to be reduced.
In the reanalysis by CDA-AMC, the median time on treatment was adjusted to align with the values reported within the sponsor’s economic model.
Wastage was not accounted for in the base case: For IV medications, unused portions of vials are often discarded due to fixed dosing and patient-specific requirements. This is especially pertinent to the comparator regimens given there is a greater potential for wastage given the dosing schedule. Incorporating wastage in the base case more accurately represents the financial burden on the health care system.
In the reanalysis by CDA-AMC, wastage was included to improve the accuracy of the BIA.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by modifying the duration on treatment to align with the economic evaluation and assumed drug wastage would occur. The changes to the derived CDA-AMC base case are described in Table 15.
Table 15: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Duration on treatment (months) | Tarlatamab – 5.1 Comparators – 1.7 | Tarlatamab – 4.6 Comparators – 2.5 |
2. Wastage costs | Excluded | Included |
CDA-AMC base case | 1 + 2 | |
BIA = budget impact analysis; CDA-AMC = Canada's Drug Agency.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in The estimated budget impact of reimbursing tarlatamab is sensitive to change in duration on treatment. The modifications to the sponsor’s base case reduced the estimated budget impact by 11%.
Table 16 and a more detailed breakdown is presented in Table 18. Based on the CDA-AMC reanalysis, the budget impact of the reimbursement of tarlatamab for the treatment of ES-SCLC is expected to be $8,530,040 in year 1, $10,458,714 in year 2, $14,270,385 in year 3, with a three-year total of $33,259,139.
The estimated budget impact of reimbursing tarlatamab is sensitive to change in duration on treatment. The modifications to the sponsor’s base case reduced the estimated budget impact by 11%.
Table 16: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $37,452,931 |
CDA-AMC reanalysis 1 | $33,395,606 |
CDA-AMC reanalysis 2 | $37,361,468 |
CDA-AMC base case | $33,259,139 |
BIA = budget impact analysis; CDA-AMC = Canada's Drug Agency.
Table 17: Scenario Analyses Market Share (New Drug Scenario)
Scenario | Treatment | Year 1 | Year 2 | Year 3 |
|---|---|---|---|---|
Scenario Analysis 1: 20% uptake year 1 and 50% uptake in year 3 | Tarlatamab | 20.0% | 30.0% | 50.0% |
CAV | 24.0% | 15.0% | 6.0% | |
Carboplatin + Etoposide | 25.0% | 25.0% | 25.0% | |
Oral Etoposide | 15.0% | 15.0% | 6.0% | |
Irinotecan | 10.0% | 10.0% | 10.0% | |
IV Topotecan | 6.0% | 5.0% | 3.0% | |
Scenario Analysis 2: 20% uptake year 1 and 80% uptake in year 3 | Tarlatamab | 20.0% | 30.0% | 80.0% |
CAV | 24.0% | 15.0% | 2.0% | |
Carboplatin + Etoposide | 25.0% | 25.0% | 10.7% | |
Oral Etoposide | 15.0% | 15.0% | 2.0% | |
Irinotecan | 10.0% | 10.0% | 4.3% | |
IV Topotecan | 6.0% | 5.0% | 1.0% |
CAV = cyclophosphamide + doxorubicin + vincristine.
CDA-AMC also conducted sensitivity analyses to address remaining uncertainty, using the CDA-AMC base case. The results are provided in Table 18. These included:
Tarlatamab’s market share was projected at 20% in year 1, 30% in year 2, and 50% in year 3 (Table 17).
Tarlatamab’s market share was projected at 20% in year 1, 30% in year 2, and 80% in year 3 (Table 17).
Treatment duration was assumed to continue postdisease progression, with TTD estimated at 6 months for tarlatamab and 2.5 months for the comparators.
Table 18: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $644,406 | $656,162 | $668,140 | $680,343 | $2,649,053 |
New drug | $644,406 | $10,284,718 | $12,453,253 | $16,719,606 | $40,101,984 | |
Budget impact | $0 | $9,628,566 | $11,785,113 | $16,039,262 | $37,452,931 | |
CDA-AMC base case | Reference | $1,038,221 | $1,057,163 | $1,076,460 | $1,096,120 | $4,267,964 |
New drug | $1,038,221 | $9,587,203 | $11,535,174 | $15,366,506 | $37,527,103 | |
Budget impact | $0.00 | $8,530,040 | $10,458,714 | $14,270,385 | $33,259,139 | |
CDA-AMC scenario analysis: 20% uptake year 1 and 50% uptake in year 3 | Reference | $1,038,221 | $1,057,163 | $1,076,460 | $1,096,120 | $4,267,964 |
New drug | $1,038,221 | $7,857,745 | $11,535,174 | $18,960,856 | $39,391,996 | |
Budget impact | $0 | $6,800,582 | $10,458,714 | $17,864,736 | $35,124,032 | |
CDA-AMC scenario analysis: 20% uptake year 1 and 80% uptake in year 3 | Reference | $1,038,221 | $1,057,163 | $1,076,460 | $1,096,120 | $4,267,964 |
New drug | $1,038,221 | $7,857,745 | $11,535,174 | $29,684,707 | $50,115,847 | |
Budget impact | $0 | $6,800,582 | $10,458,714 | $28,588,587 | $45,847,883 | |
CDA-AMC scenario analysis: Continued treatment post disease progression | Reference | $1,038,221 | $1,057,163 | $1,076,460 | $1,096,120 | $4,267,964 |
New drug | $1,038,221 | $12,260,755 | $14,802,001 | $19,801,827 | $47,902,804 | |
Budget impact | $0.00 | $11,203,593 | $13,725,541 | $18,705,707 | $43,634,840 |
BIA = budget impact analysis; CDA-AMC = Canada's Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.