Drugs, Health Technologies, Health Systems
Reimbursement Review
Blinatumomab (Blincyto)
Sponsor: Amgen Canada Inc.
Therapeutic area: B-cell precursor acute lymphoblastic leukemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
Ac2orn
Advocacy for Canadian Childhood Oncology Research Network
AE
adverse event
ALL
acute lymphoblastic leukemia
B-LLy
B-lymphoblastic lymphoma
CAR
chimeric antigen receptor
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CLSG-GCEL
Canadian Leukemia Study Group — Groupe Canadien d’Étude sur la Leucémie
CR
complete remission
CRi
complete remission with incomplete peripheral blood count recovery
CNS
central nervous system
CSF
cerebrospinal fluid
CTCAE
Common Terminology Criteria for Adverse Events
DCO
data cut-off
DFCI
Dana-Farber Cancer Institute
DFS
disease-free survival
DT
double trisomies of chromosomes 4 and 10
ECOG
Eastern Cooperative Oncology Group
FAS
full analysis set
FISH
fluorescent in situ hybridization
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
HTS
high-throughput screening
IQR
interquartile range
ITT
intention-to-treat
KM
Kaplan-Meier
LLSC
Leukemia & Lymphoma Society of Canada
MID
minimal important difference
MNAR
missing not at random
MRD
minimal residual disease
NCI
National Cancer Institute
NE
not evaluable
OH-CCO
Ontario Health — Cancer Care Ontario
OPACC
Ontario Parents Advocating for Children with Cancer
OS
overall survival
Ph
Philadelphia chromosome
POGO
Pediatric Oncology Group of Ontario
PS
performance status
RCT
randomized controlled trial
RFS
relapse-free survival
RSMT
restricted mean survival time
SAE
serious adverse event
SCT
stem cell transplantation
SE
standard error
SR
standard risk
SR-average
standard risk acute lymphoblastic leukemia with average risk of relapse
SR-favourable
standard risk acute lymphoblastic leukemia with favourable risk of relapse
SR-high
standard risk with high risk of relapse
TEAE
treatment-emergent adverse event
TLS
tumour lysis syndrome
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Blinatumomab (Blincyto), 28 mcg/day for patients weighing 45 kg and more and 15 mcg/m2 per day for patients weighing less than 45 kg (up to a maximum of 28 mcg/day), lyophilized powder for solution, IV infusion by infusion pump |
Sponsor | Amgen Canada Inc. |
Indication | For the treatment of patients with Philadelphia chromosome-negative CD19 positive B-cell precursor acute lymphoblastic leukemia in the consolidation phase of multiphase chemotherapy. |
Reimbursement request | Treatment of adult and pediatric patients with Philadelphia chromosome–negative CD19-positive B-cell precursor acute lymphoblastic leukemia in the consolidation phase of multiphase chemotherapy in the frontline setting |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | December 27, 2024 |
Recommended dose |
|
NOC = Notice of Compliance.
Introduction
Acute lymphoblastic leukemia (ALL) is a rare and aggressive hematologic malignancy of undifferentiated lymphoid precursors characterized by the proliferation of immature and abnormal lymphoid cells in the bone marrow and peripheral blood.1 In 2019 alone, 440 new cases of ALL were diagnosed in Canada (excluding Quebec).2 From 2015 to 2017, the 5-year net survival rate for patients in Canada aged 15 to 99 years was 47%.3 While ALL is the least common type of leukemia in adults, it is the most prevalent cancer among children and young adults.4 B-lymphoblastic (B-cell) phenotype is the most common type of ALL, accounting for approximately 85% of pediatric ALL cases and around 75% of adult ALL cases.5,6 The leukemia cells in many patients with B-cell ALL have chromosomal abnormalities, with 1 of the most prevalent abnormalities occurring in the Philadelphia chromosome (Ph) (occurring in 1% to 3% of childhood ALL cases and 11% to 29% of adult ALL cases).7 Determining the ALL subtype and presence of chromosomal abnormalities is critical for understanding the disease status, risk factors, and treatment planning. In addition, minimal residual disease (MRD) assessments provide information on the prognosis and chance of relapse, with higher MRD levels indicating greater chances of relapse.8 MRD testing by flow cytometry is widely available in Canada and recommended for ALL. In the pediatric ALL population, MRD testing is part of the standard of care and is publicly funded. In the adult ALL population, while it is routinely performed, funding for MRD testing is not uniform across Canada.
While conventional regimens differ in terms of specific drug selection, dosing, and duration, they all typically include 3 phases: induction, consolidation (sometimes called “intensification”), and maintenance; during these phases, intensive multidrug chemotherapy protocols are used.4 However, not all patients respond to available conventional treatments, relapse remains a substantial risk, and many patients experience toxicity-related adverse events (AEs). Further, patients with ALL experience a variety of symptoms, including fatigue, dry mouth, lack of appetite, irritability, and nervousness.9 These symptoms, coupled with disease outlook, treatment modalities, side effects, and other comorbidities common in ALL highlight the substantial unmet need. The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of blinatumomab, 28 mcg/day for adult patients weighing 45 kg or more and 15 mcg/m2 per day for adult patients weighing less than 45 kg, and pediatric patients, lyophilized powder for solution, IV infusion by infusion pump in the treatment of Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in the frontline setting.
Blinatumomab has previously been reviewed by Canada’s Drug Agency (CDA-AMC) for the treatment of pediatric and adult patients with Ph-negative B-cell precursor ALL in the relapse or refractory setting, adult patients with relapsed or refractory Ph-positive B-cell precursor ALL, and patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic complete remission (CR) with MRD greater than or equal to 0.1%. All final CDA-AMC recommendations were to reimburse with clinical criteria and/or conditions.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical expert(s) consulted for the purpose of this review.
Patient Input
CDA-AMC received 2 inputs for this submission. The first input, provided by the Leukemia & Lymphoma Society of Canada (LLSC), included survey results of adult patients with ALL and their caregivers. The second input was a joint submission provided by LLSC, Advocacy for Canadian Childhood Oncology Research Network (Ac2orn), Ontario Parents Advocating for Children with Cancer (OPACC), and Childhood Cancer Canada and was based on interviews with 3 caregivers of pediatric patients with B-cell ALL who received blinatumomab treatment. The patient group input for pediatric patients was supplemented with information on disease experience and experience with currently available treatments, gathered from previous patient group submissions to CDA-AMC for blinatumomab for pediatric patients with Ph-negative B-cell precursor ALL in the relapsed or refractory setting (pan-Canadian Oncology Drug Review [pCODR] 10099 Blinatumomab ALL for pediatrics in 2017 and PX0367 Blinatumomab for ALL for pediatrics in 2024).
LLSC is a national charitable organization dedicated to finding a cure for blood cancers and improving the quality of life of people affected by blood cancers and their families by funding research and providing educational resources, services, and support. Ac2orn is a national organization committed to advocating translational research and effective treatments to realize the goal of curing cancers in children, adolescents, and young adults. OPACC advocates for families and organizations navigating the childhood cancer journey. Childhood Cancer Canada supports children diagnosed with cancer and their families.
Input received for adults with ALL is summarized first, followed by patient input from caregivers of pediatric patients with ALL.
LLSC conducted a survey in October and November 2024. The respondents were patients with ALL (69%) and their caregivers (28%). Out of 103 respondents providing their age, 9% were aged 0 to 17 years and were disqualified from the survey. The survey respondents included 49% aged 18 to 39 years, 32% aged 40 to 64 years, 8% aged 65 to 74 years, and 3% aged 75 years or older. Among 82 adults providing responses, most were from Canada, 1 was from the US, and 1 was international. A negative to very negative impact on personal or home life due to ALL was reported by 82% of respondents. When considering their social life, 75% of respondents reported negative to very negative impacts due to ALL. Low energy, fear of infections, frequent hospital visits, depression and/or anxiety, ALL symptom burden, and inadequate nutrition were reported as factors contributing to negative impacts of ALL. Eighteen respondents had experience with blinatumomab. Among all other types of treatment, chemotherapy was the most commonly reported type of ALL treatment (98% of respondents), followed by stem cell transplantation (SCT) (48% of respondents), radiation therapy (45% of respondents), immunotherapy (27% of respondents), targeted therapy (7% of respondents), chimeric antigen receptor (CAR) T-cell therapy (5% of respondents), and other types (16% of respondents; included natural medicine, Chinese medicine, sound baths, meditation, transfusions steroids, and antiemetics). Fatigue and neutropenia were reported by the respondents as the most severe side effects of current treatments (excluding blinatumomab), followed by thrombocytopenia, infections, nausea, vomiting, diarrhea, anemia, fever, peripheral edema, headaches, infusion reactions, neurologic symptoms, and cytokine release syndrome. These side effects had substantial impacts on patients’ lives, including frequent hospitalizations and lower functionality. LLSC added that the most important outcomes for patients include longer remission with consideration of side effects, quality of life, and financial costs.
According to LLSC, 18 respondents stated that they or the person they care or cared for were treated with blinatumomab for ALL. The methods of access were primarily through clinical trials (33%) and compassionate use programs (27%); the remainder of patients accessed blinatumomab through private insurance, Régie de l’assurance maladie du Québec (RAMQ) — Quebec socialized health care, government funding, or they did not know. Out of 15 respondents, 67%, 20%, and 13% of respondents believed that their ALL completely, partially, or did not respond to blinatumomab, respectively. Respondents were asked to rate the severity of the side effects of blinatumomab (from 1 — did not experience to 4 — severe). Among 15 respondents, the highest-rated side effects, as measured by weighted averages, were neutropenia (2.29), fatigue or weakness (2.27), and fever (2), followed by anemia (1.71), thrombocytopenia (1.71), infections (1.64), headaches (1.57), neurologic symptoms (i.e., confusion, seizures, difficulty speaking; 1.53), cytokine release syndrome (1.5), diarrhea (1.47), peripheral edema (1.47), nausea or vomiting (1.4), and infusion reactions (i.e., chills, rash, difficulty breathing; 1.27). In terms of comparing blinatumomab to other treatments, among 15 respondents, 40% felt it was as difficult to tolerate as other treatments, 27% felt it was less or much less difficult, and 7% felt it was more difficult to tolerate. Additionally, out of 15 respondents, 40% strongly agreed, 20% agreed, 33% felt neutral, and 7% disagreed that blinatumomab improved their quality of life compared to other treatments. Most patients indicated that they were likely to take blinatumomab again or recommend it to other patients.
The joint patient group input for pediatric patients gathered information through interviews with 3 caregivers of pediatric patients with B-cell ALL. According to this patient group submission, to avoid repetitive questioning and to minimize emotional strain and undue harm, the interviews focused on experiences and quality of life associated with blinatumomab treatment. For all other information, this patient group input referred to previous LLSC-Ac2orn-OPACC joint submissions to CDA-AMC for relapsed or refractory ALL in children. First, the following section presents the disease experience and experience with currently available treatments gathered from previous patient input submissions. Second, it summarizes patients’ and caregivers’ experience with blinatumomab treatment and patients’ quality of life during blinatumomab treatment compared to their experience with prior treatments for pediatric ALL, based on the joint patient group submission for this current review.
Based on input gathered in previous patient input submissions to CDA-AMC for pediatric patients with relapsed or refractory ALL, children may experience a variety of symptoms, including severe fatigue, pain, high fever, bleeding, bruising, bone pain, and swollen lymph nodes. However, the impacts of pediatric cancer relapse on the child and their family extend beyond physical symptoms. It was highlighted in the patient group submissions that relapse and immunosuppression severely limit children’s ability to engage in normal activities, placing a significant emotional and physical burden on both the child and their family. Families experience intense stress, financial strain, and disrupted daily routines, with caregivers often facing severe impacts on their mental health. It was explained in the patient group submissions that the standard of care for treating relapsed or refractory pediatric ALL typically involves a combination of strategies, including drug therapy and radiation. For patients with refractory disease, these aggressive treatments often result in serious side effects, such as immunosuppression, severe pain, infections, anemia, and organ damage, which can significantly impact the child's quality of life. There is a significant unmet need for more effective, less toxic therapies that improve quality of life by reducing treatment burden and minimizing the need for frequent hospital visits. Outpatient treatment options that decrease hospital stays was also cited as crucial for maintaining normalcy and reducing stress for both patients and their families. In terms of improved outcomes, it was noted in the patient group submissions that patients and their families are looking for options that are not only effective but also gentle on their children. They seek innovative therapies that can provide significant benefits without causing undue harm or severe side effects. Additionally, having these treatments covered by drug plans is crucial, as coverage alleviates the financial burden on families.
In the 3 caregiver interviews conducted by the joint patient group submission for this present review, 2 of the caregivers had children who were 2 years old, and 1 had a child who was 10 years old at diagnosis. Two caregivers were residents of Ontario, and 1 resided in British Columbia. The input focused on patients’ and caregivers’ experiences with administering blinatumomab treatment at home, as well as on comparing their quality of life during blinatumomab treatment to their experiences with prior treatments for pediatric ALL. According to the joint submission, the current conventional therapy for pediatric patients with B-cell ALL living in Canada is chemotherapy infusions, which are often accompanied by serious side effects, negative impact on patients’ physical health, and long hospital stays. Based on the joint input, all 3 caregivers mentioned that they and the patients had overall positive experiences with blinatumomab, especially in comparison to chemotherapy infusion. Because blinatumomab is an outpatient treatment with “gentler” effects, the patients were able to live with family, play with peers, and stay out of the hospital. The 3 caregivers reported that they felt nervous, the child experienced fever, and the child experienced side effects at the start of blinatumomab treatment. According to the patient group input, cognitive testing was done routinely at every blinatumomab bag change to check for neurologic AEs; none of the caregivers reported neurologic AEs as a side effect in the children. Caregivers reported an extra burden because they had to receive a new medication bag of blinatumomab every 4 days and had to visit the medical centre for any glitches in the blinatumomab delivery system. Caregivers noted that, because blinatumomab is a relatively new treatment method, knowledge of blinatumomab’s delivery method was limited among health care professionals. Some caregivers emphasized the need for greater variety for the backpack delivery system for blinatumomab that would be able to accommodate patients of all ages. According to the joint submission, all 3 caregivers related that blinatumomab made a big difference to the quality of life for their patients, themselves, and the rest of their family compared to traditional chemotherapy infusion. Financial stress was experienced by caregivers due to the high cost and lack of public funding for blinatumomab. Finally, the joint submission made the following suggestions for improved outcomes with new therapies:
Provide caregivers with tips and tools on what to expect with the blinatumomab delivery system and some at-home solutions to help to alleviate practical challenges such as driving to cancer centres at unexpected times.
Train nurses at local hospitals to administer blinatumomab (to mitigate the requirement of driving to cancer centres).
Improve blinatumomab knowledge and skills of the nurses at the cancer centres.
Provide more choices in the delivery system backpacks based on body size and strength of the patient.
Clinician Input
Input From Clinical Experts Consulted for This Review
Two clinical specialists with expertise in the diagnosis and management of Ph-negative B-cell precursor ALL provided input for this review. One of the clinical experts has specific expertise in the diagnosis and management of adults with Ph-negative B-cell precursor ALL, while the other has expertise in pediatric patients with Ph-negative B-cell precursor ALL.
Both clinical experts felt the overarching goal of Ph-negative B-cell precursor ALL treatment is to achieve CR, prevent relapses, and cure the disease (thus providing long-term survival), while minimizing acute and late toxicities. Experts highlighted that not all patients respond to available conventional treatments, relapse remains a substantial risk, and many patients experience toxicity-related AEs. The clinical experts consulted for this review noted that unplanned inpatient admissions due to complications associated with the current conventional treatments are common. Further, as ALL is the most common childhood cancer, the clinical expert with experience treating pediatric patients with ALL highlighted that decreasing relapse rates in ALL would have a significant impact on childhood cancer mortality rates and obviate the need for additional expensive and/or toxic therapies.
Both clinical experts consulted for this review felt that blinatumomab would add an alternative first-line treatment option for patients with Ph-negative B-cell precursor ALL. The experts recommended using 1 to 4 cycles of blinatumomab in sequence with conventional consolidation chemotherapy. Compared to current treatment options, clinical experts noted that blinatumomab’s unique mechanism of action, blocking CD19 antigen expressed on the leukemic blasts, has the potential to improve overall response rate, relapse-free survival (RFS), and overall survival (OS), while decreasing AEs associated with the frequent use of intensive chemotherapy, the potentially improving quality of life for patients with Ph-negative B-cell precursor ALL. If blinatumomab reduces the use of allogeneic SCT by preventing relapse, clinical experts noted that the risk of treatment-related mortality may also decrease.
The clinical experts consulted for this review noted any adult or pediatric patient with Ph-negative B-cell precursor ALL who achieves a CR following induction therapy, regardless of risk (MRD and standard-risk acute lymphoblastic leukemia [SR] with or without high-risk or very high–risk status) and age, would be best suited for first-line consolidation treatment with blinatumomab.
The clinical expert with experience treating pediatric patients noted that the 1 exception may be to exclude pediatric patients from receiving blinatumomab if their expected event-free survival on a particular conventional chemotherapy backbone is expected to be greater than 95% (i.e., those with SR and a favourable risk of relapse [SR-favourable]). However, experts flagged that the molecular characterization of leukemia is rapidly evolving, and the identification of a new high-risk somatic change may alter how patients are currently classified.
The expert felt that adult patients with Ph-negative B-cell precursor ALL who have a poor performance status (PS) (i.e., Eastern Cooperative Oncology Group [ECOG] PS greater than 2) should not be eligible for blinatumomab, while no PS restriction should be made in children, as blinatumomab has been shown to be well tolerated and potentially life-saving. Clinical experts felt there were insufficient data to recommend blinatumomab treatment in patients with acute undifferentiated leukemia and those with Burkitt’s leukemia.
The clinical experts consulted for this review noted that treatment response is based on bone marrow evaluation (including morphological evaluation and MRD assessment) at the end of induction and consolidation treatment. RFS, disease-free survival (DFS), OS, and other survival parameters, such as relapse incidence, are long-term parameters used to assess treatment response in both clinical trials and clinical practice. In both clinical trials and clinical practice, AEs are used to assess treatment benefit. The clinical expert with experience treating adults with ALL noted that, following the completion of maintenance treatment, relapse is assessed every 3 months during the first year, every 6 months during the second year, and then annually between years 3 and 5. The clinical expert with experience treating pediatric patients with ALL noted that, following the completion of maintenance treatment, follow-up practices vary by centre. However, in their clinical practice, relapse is assessed monthly for the first 3 months, then every 3 months during the first year, every 4 months in the second year, every 6 months in the third year, and then annually for the rest of the patient’s life.
The clinical trial, blinatumomab’s label, and the clinical experts consulted for this review noted that blinatumomab should be discontinued in the event of grade 4 cytokine release syndrome, grade 4 neurotoxicity and/or psychiatric events, and grade 4 thrombosis. Other grade 4 toxicities that are deemed clinically significant may require discontinuation of blinatumomab. Toxicities that require dose interruption of blinatumomab and that do not return to grade 1 or less by 14 days (or, in the case of neurotoxicity, by 7 days) necessitate discontinuation of blinatumomab. Recurrent toxicities leading to dose interruption of blinatumomab also require permanent discontinuation of blinatumomab.
The clinical expert with experience treating adult patients with ALL noted that patients who develop any debilitating conditions (such as ECOG PS of greater than 2) should discontinue blinatumomab treatment.
While the clinical experts agreed with these discontinuation criteria, the benefits of treatment, availability of alternative treatments, risks of discontinuation, and patients’ willingness to continue treatment must also be taken into account before making a final discontinuation decision. In adult patients, the expert felt that blinatumomab should be discontinued in the event of disease progression and relapse or upon the patient’s request.
Both clinical experts indicated that treatment with blinatumomab should be initiated on an inpatient basis (3 days for the first infusion and 2 days for subsequent infusions) and, if the drug is well tolerated, that subsequent infusions can be performed in an outpatient clinic.10 Given the pharmacy, nursing, and monitoring requirements for blinatumomab infusions, specialist care at an oncology centre is required. Both clinical experts consulted for this review felt that any conventional chemotherapy used for ALL treatment can be combined with blinatumomab. They also felt that clinicians should have the ability to adjust the dosage, number of cycles, and cycle length based on toxicity and individual patient’s needs.
The clinical expert with experience treating adults noted that at least 2 cycles of blinatumomab should be given before SCT to deepen the patient’s remission. SCT would likely be required in adult patients in their second CR (regardless of MRD status) or in first CR with continuous MRD positivity and would be considered in adult patients in first CR with MRD-negative status after initially presenting with the highest risk disease of relapse.
Clinician Group Input
Three clinician groups each submitted inputs to CDA-AMC: Ontario Health — Cancer Care Ontario (OH-CCO) Hematology Cancer Drug Advisory Committee (with input from 6 clinicians); Pediatric Oncology Group of Ontario (POGO) (input from 11 clinicians); and Canadian Leukemia Study Group (CLSG) — Groupe Canadien d’Étude sur la Leucémie (GCEL) (input from 7 clinicians). One group provided input relevant to pediatric patients (POGO) and another for adults (CLSG-GCEL), while the third group did not specify an age group of patients represented.
The clinician groups were generally in agreement with the feedback received from the clinical experts consulted for this review. In terms of the goals of therapy, the clinical experts and clinician groups indicated that the main goals are cure of the disease while minimizing toxicity. Other goals include achieving CR, preventing relapse, and avoiding need for second-line therapies, such as allogenic SCT, or CAR T-cell therapy. In pediatric patients, the POGO group stated that relapse therapies carry significant toxicity that leaves young patients with a wide variety of potential lifelong late effects. In adults, the clinician groups noted the substantial risk of relapse, even in patients who are MRD-negative. Thus, additional treatments are needed to improve survival, CR, and relapse rate, and to minimize or reduce toxicity of treatments (during consolidation and/or later therapies).
All clinician groups agreed that the patient population suitable to receive blinatumomab during consolidation include those newly diagnosed with Ph-negative CD19-positive B-cell precursor ALL, regardless of MRD status. Both the pediatric clinical expert consulted for this review and the POGO group indicated that patients who meet the SR-average or high-risk stratification criteria have less favourable outcomes with chemotherapy alone and are expected to benefit from the addition of blinatumomab during consolidation therapy. Those who meet the SR-favourable stratification criteria have excellent response rates (event-free survival greater than 95%) with chemotherapy alone and may not require blinatumomab therapy. The POGO group noted an unmet need for augmentation therapies that have a tolerable AE profile and may allow for cytotoxic “breaks” in therapy to facilitate recovery from cytotoxic-associated complications, such as fungal infections. According to input from POGO, patients with Ph-positive B-cell precursor ALL may also benefit from blinatumomab therapy. The clinician groups agree that standard response assessment for ALL would be relevant to monitoring the effects of blinatumomab, including MRD status, duration of response, relapse, and OS. The adult clinician groups noted the MRD threshold of 0.1% (≥ 103) is obsolete, and the current standard is greater than or equal to 0.01% (≥ 104).
The experts consulted and the clinician groups agreed that blinatumomab would be discontinued if the patient experienced disease progression or significant toxicity and that specialist care in an ALL treatment centre is warranted. The POGO group expressed that reimbursement strategies should account for all forms of drug wastage that may occur when pediatric doses are prepared.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. Key factors identified that could potentially impact the implementation of the CDA-AMC recommendation for blinatumomab used in the consolidation phase of multiphase chemotherapy were use among those outside of the pivotal trials age range (i.e., among those less than 1 year of age and older than 70 years), use among specific risk categories (including those with ECOG PS of greater than 2), and whether those patients with undifferentiated leukemia and those with Burkitt leukemia should be eligible.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing, phase III, open-label, multicentre, international study (5.9% of the randomized sample was from Canada), the E1910 trial, met the inclusion criteria for the systematic review conducted by the sponsor. A second phase III, open-label, multicentre, international study (6.1% of the randomized sample was from Canada), the AALL1731 trial, was included to inform the reimbursement request among pediatric patients. Throughout the systematic review section, details for the E1910 trial are summarized first, followed by those for the AALL1731 trial.
The E1910 trial was aimed at evaluating the efficacy and safety of blinatumomab used in the consolidation phase of multiphase chemotherapy for the treatment of adult patients with Ph-negative B-cell precursor ALL and MRD-negative status. Secondary outcomes included assessing the efficacy and safety of blinatumomab in patients with MRD-positive status, while post hoc analyses were conducted among an MRD-agnostic cohort. Following confirmation of eligibility, all patients completed induction and intensification treatment. Those achieving a CR or CR with incomplete peripheral blood count recovery (CRi) were then randomized 1:1 using stratified randomization to receive consolidation treatment with blinatumomab plus conventional chemotherapy (blinatumomab plus chemotherapy) or conventional chemotherapy alone (chemotherapy). CR was defined as neutrophil count of 1.0 × 109/L or greater, platelet count of 100 × 109/L or greater, no leukemic blasts present in the peripheral blood, adequate bone marrow cellularity with trilineage hematopoiesis, 5% or less blasts in the bone marrow, and no extramedullary leukemia (e.g., CNS or soft tissue involvement). CRi had the same definition, with the exception of incomplete platelet recovery (i.e., platelets greater than 75 and less than 100 × 109/L) or incomplete neutrophil count recovery (i.e., greater than 0.75 and less than 1 × 109/L). Stratified randomization was based on MRD-positive versus MRD-negative status, aged 30 to 54 years versus 55 years or older, CD20-positive versus CD20-negative status, rituximab use versus no use, and intention to receive allogeneic SCT versus no plan for SCT. Following a protocol amendment related to the approval of blinatumomab therapy for patients with MRD-positive status, these patients were assigned (rather than randomized) to blinatumomab plus chemotherapy. Overall, 286 eligible patients were randomized or assigned to either the blinatumomab plus chemotherapy arm (n = 152) or the chemotherapy arm (n = 134) and included in the Step 3 analysis set. The outcomes relevant to this review include OS, RFS, and harms data collected at the June 23, 2023, data cut-off (DCO) date for the primary analysis.
In the E1910 trial, slightly more patients were female (51.4% versus 48.6% male), the mean age at enrolment was 49.9 years (standard deviation = 11.5 years), and patients were primarily white (79.4%), followed by Black or African American (5.9%), Asian (2.1%), American Indian or Alaska Native (1.0%), Native Hawaiian or other Pacific Islander (0.3%) (groupings from original study), and either not reported or unknown (4.5% and 6.6%, respectively). Most patients had an ECOG PS of 1 (58.7%), and 37.1% of randomized or assigned patients had an ECOG PS of 0. Overall, 224 (78.3%) of the randomized or assigned patients had MRD-negative status and were included in the full analysis set (FAS) (112 [73.7%] in the blinatumomab plus chemotherapy arm and 112 [83.6%] in the chemotherapy arm) and 62 (21.7%) had MRD-positive status and were included in the Step 3 MRD-positive analysis set (40 [26.3%] in the blinatumomab plus chemotherapy arm and 22 [16.4%] in the chemotherapy arm).
The AALL1731 trial was aimed at evaluating the efficacy and safety of blinatumomab for the treatment of pediatric patients with SR B-cell ALL. The primary end point was DFS, and the primary objective was to determine whether the addition of blinatumomab (administered as nonsequential cycles) to conventional chemotherapy would improve DFS in all randomized patients with SR B-cell ALL (patients either had an average or high risk of relapse on the basis of clinical features). A post hoc objective was to determine whether the addition of blinatumomab to conventional chemotherapy would improve OS in all randomized patients. All eligible patients received induction treatment before undergoing assessments for relapse risk and receiving 1 cycle of consolidation treatment. Only those with average (SR-average) and high risk (SR-high) of relapse were randomized 1:1 using stratified randomization (based on SR-average versus SR-high and a diagnosis versus no diagnosis of Down syndrome among those with SR-average risk) to blinatumomab plus conventional chemotherapy or conventional chemotherapy alone. Overall, 1,440 eligible patients were randomized to either the blinatumomab plus chemotherapy arm (n = 718) or the chemotherapy arm (n = 722). The outcomes relevant to this review include OS, DFS, and harms data collected at the interim analysis, with DCO date June 30, 2024.
At the planned interim analysis with the DCO date June 30, 2024, the Data Safety Monitoring Committee recommended that the blinatumomab randomization be permanently closed based on the 3-year DFS estimates of 96.0% (standard error [SE] = 1.2%) for the blinatumomab plus chemotherapy arm versus 87.6% (SE = 2.1%) for the chemotherapy alone arm. The associated hazard ratio (HR) was 0.39 (95% confidence interval [CI], 0.24 to 0.64), which exceeded the prespecified interim efficacy stopping criteria.
In the AALL1731 trial, the median age at randomization was 4.3 years (interquartile range [IQR], 2.8 to 6.4). Slightly more patients were male (52.6% [758 of 1,440] versus 47.5% female) and patients were primarily non-Hispanic white (50.4%), followed by Hispanic (25.8%), non-Hispanic Black (5.6%), non-Hispanic Asian (4.3%), or other or unknown (13.9%). Overall, 835 (58.0%) of randomized patients had SR-average (417 in the blinatumomab plus chemotherapy arm and 417 in the chemotherapy arm) and 605 (42.0%) had SR-high (304 in the blinatumomab plus chemotherapy arm and 301 in the chemotherapy arm).
Efficacy Results
Overall Survival
E1910 Trial
In the E1910 trial, the median duration of OS follow-up was 4.5 years in both arms of the Step 3 analysis set (i.e., the overall cohort randomized or assigned to 1 of the treatment arms), at which point 30 (of 152 patients; 19.7%) deaths had occurred in the blinatumomab plus chemotherapy arm and 53 (of 134 patients 39.6%) in the chemotherapy arm. The Kaplan-Meier (KM) estimate for median OS was NE in both arms, with a stratified HR of 0.47 (95% CI, 0.30 to 0.74). The between-group difference in the probability of survival for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 3 and 5 years was 16.9% (95% CI, 5.5% to 28.3%) and 20.8% (95% CI, 8.5% to 33.0%), respectively.
In the FAS (i.e., the MRD-negative cohort) of the E1910 trial, the median duration of OS follow-up was 4.5 years in both arms, at which point 19 (of 112 patients; 17.0%) deaths had occurred in the blinatumomab plus chemotherapy arm and 40 (of 112 patients; 35.7%) in the chemotherapy arm. The KM estimate for median OS was NE in both arms, with a stratified HR of 0.44 (95% CI, 0.25 to 0.76). The between-group difference in the probability of survival for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 3 and 5 years was 15.6% (95% CI, 3.0% to 28.2%) and 19.9% (95% CI, 6.3% to 33.5%), respectively.
In the Step 3 MRD-positive analysis set (i.e., the MRD-positive cohort) of the E1910 trial, the median duration of OS follow-up was 4.6 years in the blinatumomab plus chemotherapy arm and 5.0 years in the chemotherapy arm, at which point 11 (of 40 patients; 27.5%) deaths had occurred in the blinatumomab plus chemotherapy arm and 13 (of 22 patients; 59.1%) in the chemotherapy arm. The KM estimate for median OS was NE in the blinatumomab plus chemotherapy arm and was 1.9 months (95% CI, 0.6 months to NE) in the chemotherapy arm; the stratified HR was 0.40 (95% CI, 0.14 to 1.12). The between-group difference in the probability of survival for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 3 and 5 years was 31.1% (95% CI, 4.1% to 58.0%) and 32.3% (95% CI, 5.4% to 59.3%), respectively.
AALL1731 Trial
In the AALL1731 trial, the median duration of follow-up was 2.5 years (IQR, 1.6 to 3.2 years). Neither the KM estimate for median OS nor the HR were reported. The probability of survival from randomization to 3 years was 98.4% (SE = 0.8%) in the blinatumomab plus chemotherapy arm and 97.1% (SE = 1.1%) in the chemotherapy arm. Between-group differences were not reported. Subgroup analyses among the cohort with an average risk of relapse (probability of survival from randomization to 3 years was 100.0% [SE = not applicable] in the blinatumomab plus chemotherapy arm and 98.4% [SE = 1.0%] in the chemotherapy arm) and high risk of relapse (probability of survival from randomization to 3 years was 96.1% [SE = 2.0%] and 95.3% [SE = 2.2%], respectively) showed similar results.
Relapse-Free Survival
E1910 Trial
In the E1910 trial, the median duration of RFS follow-up was 4.5 years in both arms of the Step 3 analysis set, at which point 36 patients (of 152; 23.7%) had a relapse event in the blinatumomab plus chemotherapy arm and 56 patients (of 134; 41.8%) in the chemotherapy arm. The KM estimate for median RFS was NE in both arms, with a stratified HR of 0.53 (95% CI, 0.35 to 0.81). The between-group difference in the probability of RFS for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 1, 3, and 5 years was 12.2% (95% CI, 1.8% to 22.7%), 17.3% (95% CI, 5.6% to 28.9%), and 18.4% (6.3% to 30.6%), respectively.
In the FAS in the E1910 trial, the median duration of RFS follow-up was 4.5 years in both arms, at which point 25 patients (of 112; 22.3%) had a relapse event in the blinatumomab plus chemotherapy arm and 43 patients (of 112; 38.4%) in the chemotherapy arm. The KM estimate for median OS was NE in both arms, with a stratified HR of 0.53 (95% CI, 0.32 to 0.88). The between-group difference in the probability of RFS for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 1, 3, and 5 years was 8.2% (95% CI, –3.0% to 19.4%), 15.4% (95% CI, 2.3% to 28.4%), and 16.5% (95% CI, 2.6% to 30.3%), respectively.
In the Step 3 MRD-positive analysis set in the E1910 trial, the median duration of RFS follow-up was 4.6 years in the blinatumomab plus chemotherapy arm and 5.0 years in the chemotherapy arm, at which point 11 patients (of 40; 27.5%) had a relapse event in the blinatumomab plus chemotherapy arm and 13 patients (of 22; 59.1%) in the chemotherapy arm. The KM estimate for median OS was NE in the blinatumomab plus chemotherapy arm and was 0.6 months (95% CI, 0.2 months to NE) in the chemotherapy arm; the stratified HR was 0.37 (95% CI, 0.13 to 1.03). The between-group difference in the probability of RFS for the blinatumomab plus chemotherapy arm versus the chemotherapy arm at 1, 3, and 5 years was 38.0% (95% CI, 11.6% to 64.5%), 32.4% (95% CI, 6.1% to 58.7%), and 32.4% (95% CI, 6.1% to 58.7%), respectively.
AALL1731 Trial
RFS was not assessed in the AALL1731 trial.
Disease-Free Survival
E1910 Trial
DFS was not assessed in the E1910 trial.
AALL1731 Trial
In the AALL1731 trial, the median duration of follow-up was 2.5 years (IQR, 1.6 to 3.2 years). The probability of remaining disease-free from randomization to 3 years was 96.0% (SE = 1.2%) in the blinatumomab plus chemotherapy arm and 87.9% (SE = 2.1%) in the chemotherapy arm, and the associated HR was 0.39 (95% CI, 0.24 to 0.64). The between-group differences in the probability of DFS at 3 years and the KM estimate for median DFS were not reported. Subgroup analyses among the cohort with an average risk of relapse (probability of DFS from randomization to 3 years was 97.5% [SE = 1.3%] in the blinatumomab plus chemotherapy arm and 90.2% [SE = 2.3%] in the chemotherapy arm, with an associated HR of 0.33 [95% CI, 0.15 to 0.69]) and high risk of relapse (probability of survival from randomization to 3 years 94.1% [SE = 2.5%] and 84.8% [SE = 3.8%], respectively, with an associated HR of 0.45 [95% CI, 0.24 to 0.85]) showed similar results.
Harms Results
Safety outcomes for the E1910 trial were thoroughly reported in the data provided to the CDA-AMC review team and are summarized in this section. For the AALL1731 trial, only the occurrence of select grade 3 or greater treatment-emergent AEs (TEAEs) was available.
In the E1910 trial, harms data are summarized among the Step 3 safety analysis set, which includes all patients in the Step 3 analysis set who took at least 1 dose of protocol-specified therapies. Similarly, in the AALL1731 trial, harms data are summarized among all patients who underwent randomization, started postconsolidation protocol therapy, and had data submitted.
Treatment-Emergent AEs
E1910 Trial
In the E1910 trial, by the June 23, 2023, DCO, a similar percentage of patients in both treatment arms of the Step 3 safety analysis set experienced a TEAE (145 of 147 [98.6%] in the blinatumomab plus chemotherapy arm and 125 of 128 [97.7%] in the chemotherapy arm). The 3 most common TEAEs in the blinatumomab plus chemotherapy arm were investigations (e.g., blood cell counts; 91.8%), blood and lymphatic system disorders (e.g., anemia; 62.6%), and nervous system disorders (e.g., headache; 57.8%). In the chemotherapy arm, the 3 most common TEAEs were investigations (96.9%), blood and lymphatic system disorders (70.3%), and gastrointestinal disorders (e.g., diarrhea; 44.5%).
AALL1731 Trial
In the AALL1731 trial, TEAEs of any grade were not provided in the materials reviewed by the CDA-AMC review team.
Grade 3 or Greater TEAE
E1910 Trial
In the E1910 trial, a similar percentage of patients in both treatment arms of the Step 3 safety analysis set experienced a grade 3 or greater TEAE (141 of 147 [95.9%] in the blinatumomab plus chemotherapy arm and 125 of 128 [97.7%] in the chemotherapy arm). In the blinatumomab plus chemotherapy arm, the 3 most common grade 3 or higher TEAEs were the same as the 3 most common TEAEs of any grade (i.e., investigations [89.8%], blood and lymphatic system disorders [39.5%], and nervous system disorders [22.4%]). In the chemotherapy arm, the 3 most common grade 3 or higher TEAEs were investigations (96.1%), blood and lymphatic system disorders (57.0%), and infection and infestation (e.g., sepsis; 24.2%).
AALL1731 Trial
In the AALL1731 trial, the reporting of TEAEs was restricted to selected grade 3 or higher ones. Generally, it was reported that the percentage of selected grade 3 or higher TEAEs was well balanced across treatment arms. Notable differences included a higher percentage of patients in the blinatumomab plus chemotherapy arms of both the SR-average (47.0% [165 of 351]) and SR-high (57.1% [156 of 273]) cohorts who experienced febrile neutropenia than in the chemotherapy arm alone (39.6% [149 of 376] and 50.5% [140 of 277], respectively) and a higher percentage of patients in the blinatumomab plus chemotherapy arm in the SR-average cohort who experienced sepsis or catheter-related infection (14.8%) and other infections (32.8%) than in the chemotherapy arm alone (5.1% and 26.3%, respectively). In both SR-average and SR-high cohorts, the 3 most common selected grade 3 or higher TEAEs in the blinatumomab plus chemotherapy arms were febrile neutropenia (47.0% and 57.1%, respectively), other infection (32.8% and 35.2%, respectively), and sepsis or catheter-related infection (14.8% and 20.9%, respectively). In both SR-average and SR-high cohorts, the 3 most common selected grade 3 or higher TEAEs in the chemotherapy arms were febrile neutropenia (39.6% and 50.5%, respectively), other infections (26.3% and 37.9%, respectively), and mucositis (15.4% and 17.7%, respectively).
In both SR-average and SR-high subgroups, 1 patient in the blinatumomab plus chemotherapy group and no patient in the chemotherapy group experienced cytokine release syndrome of grade 3 or higher. In the SR-average subgroup, 3 (0.9%) and 4 (1.1%) patients experienced pancreatis of grade 3 or higher in the blinatumomab plus chemotherapy and chemotherapy groups, respectively. Patients in this subgroup experienced neurotoxic events of grade 3 or higher (seizure: 4 [1.1%] and 7 [1.9%]; all other CNS events: 2 [0.6%] and 3 [0.8%]; and peripheral neuropathy: 2 [0.6%] and 9 [2.4%] in the blinatumomab plus chemotherapy and chemotherapy groups, respectively). In the SR-high subgroup, 11 (4.0%) and 8 (2.9%) of patients experienced pancreatis of grade 3 or higher in the blinatumomab plus chemotherapy and chemotherapy groups, respectively. Patients in this subgroup experienced neurotoxic events of grade 3 or higher (seizure: 10 [3.7%] and 7 [2.5%]; all other CNS events: 3 [1.1%] and 4 [1.4%]; peripheral neuropathy: 6 [2.2%] and 2 [0.7%] in the blinatumomab plus chemotherapy and chemotherapy groups, respectively).
Withdrawals Due to AEs
E1910 Trial
In the E1910 trial, withdrawals due to AEs were not reported among the Step 3 safety analysis set; however, they were available for the Step 3 analysis set. Among patients included in the Step 3 analysis set, 14 of 152 patients (9.2%) in the blinatumomab plus chemotherapy arm discontinued treatment due to AEs, side effects, or complications, compared to 5 of 134 patients (3.7%) discontinuing due to AEs, side effects, or complications in the chemotherapy arm.
AALL1731 trial
In the AALL1731 trial, withdrawals due to AEs were not provided in the materials reviewed by the CDA-AMC team.
Treatment-Emergent Serious AEs
E1910 Trial
In the E1910 trial, a higher percentage of patients in the blinatumomab plus chemotherapy arm of the Step 3 safety analysis set experienced treatment-emergent serious AEs (SAEs) (82 of 147 [55.8%]) than in the chemotherapy arm (36 of 128 [28.1%]). The 3 most common treatment-emergent SAEs in the blinatumomab plus chemotherapy arm were infections and infestations (22.4%), investigations (15.6%), and nervous system disorders (15.0%). In the chemotherapy arm, the 3 most common treatment-emergent SAEs were infections and infestations (14.8%), blood and lymphatic system disorders (11.7%), and investigations (4.7%).
AALL1731 Trial
In the AALL1731 trial, treatment-emergent SAEs were not provided in the materials reviewed by the CDA-AMC team.
Fatal TEAEs
E1910 Trial
In the E1910 trial, a similar percentage of patients in both treatment arms of the Step 3 safety analysis set had a fatal TEAE (3 of 147 [2.0%] in the blinatumomab plus chemotherapy arm and 2 of 128 [1.6%] in the chemotherapy arm). In the blinatumomab plus chemotherapy arm, 2 of the fatal TEAEs were due to sepsis (1.4%) and 1 was due to intracranial hemorrhage (0.7%). In the chemotherapy arm, 1 of the fatal TEAEs was due to sepsis (0.8%) and 1 was due to cardiac arrest (0.8%).
AALL1731 Trial
In the AALL1731 trial, 5 patients had a fatal TEAE while in remission; all of these patients were classified as having high risk of relapse. Two of the patients were in the chemotherapy arm (both deaths were sepsis-related) and 3 patients were in the blinatumomab plus chemotherapy arm (1 death was due to sepsis, 1 was due to multiorgan failure, and 1 was due to hypoxic ischemic encephalopathy). None of the deaths occurred during blinatumomab cycles.
Notable Harms
E1910 Trial
In the E1910 trial, among TEAEs of special interest identified in the blinatumomab product monograph and highlighted as important by clinical experts consulted for this review, tumour lysis syndrome was not reported for the Step 3 safety analysis set. Cytokine release syndrome occurred in a higher percentage of patients in the blinatumomab plus chemotherapy arm (23 of 147 [15.6%]) than in the chemotherapy arm (0 of 128 [0.0%]). While infections were not stratified according to which were deemed serious, any infections and infestations occurred in a higher percentage of patients in the blinatumomab plus chemotherapy (34.7%) than in the chemotherapy arm (27.3%). However, most subcategories of infections of special interest (including Fusarium infection, fungal pneumonia, septic shock, Aspergillus infection, bronchopneumonia, Candida infection, enterococcal bacteremia, Escherichia sepsis, and lung infection) were not reported. Finally, relatively few patients in both treatment arms experienced neurotoxicity (2.4% versus 0.0%, respectively) or pancreatitis (0.0% and 0.8%, respectively).
AALL1731 Trial
In the AALL1731 trial, some of the TEAEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review are covered in the reporting of grade 3 or greater TEAEs. However, data on notable harms of any grade were not available.
Critical Appraisal
The E1910 and AALL1731 trials were both randomized, open-label, phase III studies. The open-label nature of both studies poses a risk of bias from lack of blinding. The risk of bias due to lack of blinding is minimal for objective outcomes such as OS. However, it remains for more subjectively assessed outcomes such as RFS, DFS, and AEs. While central laboratories reviewed and confirmed relapses in both studies to mitigate potential bias for RFS and DFS outcomes, assessment bias remains a risk for AEs.
As none of the analyses in the E1910 and AALL1731 trials were adjusted for multiple testing, there is an increased risk of type I error for statistically significant results.
All results from the E1910 and AALL1731 trials should be interpreted in light of the fact that they are based on interim analyses, which may overestimate the observed treatment effects for blinatumomab plus chemotherapy.11 Additionally, data from both trials remained immature (59 of 94 planned OS events had occurred in the FAS of the E1910 trial and 81 of 194 planned DFS events had occurred in the overall cohort of the AALL1731 trial). Nonetheless, as the clinical experts consulted for this review with experience treating patients with ALL felt the chemotherapy arms in both studies performed as expected, bias resulting from the interim analysis effect and immature data were deemed to be minimal. Further, OS results in the AALL1731 trial and OS and RFS results in the Step 3 analysis set in the E1910 trial must be interpreted in light of the fact that these are from post hoc analyses, which are at risk of data manipulation. However, the results of the post hoc analyses were consistent with those observed for primary and secondary outcomes as well as with the clinical experts’ expectations regarding the performance of the control and intervention arms.
The outcomes measured in the E1910 and AALL1731 trials addressed the key treatment goals identified by patient and clinician group input submitted to CDA-AMC and were deemed to be relevant by the consulted clinical experts.
E1910 Trial
As a result of a protocol amendment in the E1910 trial, patients who were MRD-positive were assigned, rather than randomized, to the blinatumomab plus chemotherapy arm; this occurred as a result of updated evidence suggesting blinatumomab plus chemotherapy should be the new standard of care for MRD-positive patients.12,13 As a result, a higher percentage of patients in the blinatumomab plus chemotherapy arm were MRD-positive. MRD-positive patients have a higher chance of relapse; however, the direction and magnitude of this potential selection bias is unclear.8
Baseline characteristics were generally similar in the Step 3 analysis set and the FAS. However, the distribution of most characteristics was not similar for patients in the Step 3 MRD-positive analysis set. The absence of randomization for the Step 3 MRD-positive analysis set increases the risk of imbalance in measured and unmeasured confounders, although, as mentioned, the magnitude and direction of the potential selection bias is hard to determine.
Stratified Cox proportional hazards models, adjusted for stratification factors, were used to estimate the HRs and CIs for OS and RFS. These models assume proportional hazards across treatment arms. Visual inspection of the KM curves by the CDA-AMC review team revealed the OS and RFS curves for the intervention and comparator treatment arms crossed multiple times and did not separate until approximately 4 and 6 months, respectively. While this suggests that the HRs may not reflect the treatment effect over time, it is more likely a result of variation in effects between the treatment and an active control during the early stages of treatment initiation. The KM curves remained separate for the remainder of the observation period, suggesting that the proportional hazards assumption was adequately met. Additionally, sensitivity analyses in the FAS using restricted mean survival time (RMST), which does not rely on the proportional hazards assumption, supported the results of the Cox proportional hazards models for OS and RFS.
The OS analysis in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set indicated a survival benefit for the blinatumomab plus chemotherapy arm compared to the chemotherapy arm. However, its internal validity may have been influenced by the potential impact of postrelapse therapies. The OS analysis was based on the intention-to-treat (ITT) approach, which assumes that postrelapse therapies are nondifferentially distributed between groups — a condition that may not hold, given the observed disparities in postrelapse therapy use — and the OS analysis did not control or adjust for subsequent postrelapse therapy. While this approach improves generalizability of the OS results, there is potential for confounding by postrelapse therapy, especially considering the noted differences in the use of efficacious postrelapse therapies. Although the observed OS effect represents the combined impact of frontline blinatumomab plus chemotherapy plus subsequent treatments, the overall effect of the differences in use of postrelapse therapy was more likely to favour the chemotherapy alone arm.
The clinical experts consulted for this review with experience treating adult patients with ALL noted that the modified Dana-Farber Cancer Institute (DFCI) protocol is currently the most commonly used protocol to treat Ph-negative B-cell precursor ALL in Canadian clinical practice. The chemotherapy regimen used in E1910 is built upon the UKALLXII/E2993 chemotherapy regimen with dosing modifications based on the C10403 AYA trial, and no indirect treatment comparison was submitted, thus limiting the generalizability to the Canadian setting. However, the clinical expert consulted for this review with experience treating adult patients with ALL felt the efficacy, based on CR and OS, of the regimen used in E1910 was similar to the modified DFCI regimen.
AALL1731 Trial
Baseline characteristics were generally similar between treatment arms, except for MRD in peripheral blood on day 8 in both the SR-average and SR-high subgroups and for cytogenic risk group in the SR-high subgroup. More patients in the blinatumomab plus chemotherapy arm (41.5% and 40.5%) than patients in the chemotherapy arm (35.2% and 35.2%) in the SR-average and SR-high strata had 1% and greater MRD in peripheral blood on day 8. A smaller percentage of patients with SR-high risk in the blinatumomab plus chemotherapy arm had favourable cytogenetics (23.9% versus 31.6%), and a larger percentage had neutral cytogenetics (54.8% versus 48.7%). Both MRD and cytogenetic risk are well-established prognostic factors in ALL and influence treatment response and outcomes.14-16 The highlighted differences indicate that a larger percentage of patients in the blinatumomab plus chemotherapy arm would be at higher risk of relapse and reduced survival. These could contribute to bias in the comparative outcomes between the treatment arms, potentially favouring chemotherapy.
Among randomized patients, more patients assigned to the blinatumomab plus chemotherapy arm did not start postconsolidation treatment (n = 55 of patients with SR-average risk and n = 20 of patients with SR-high risk) than in the chemotherapy arm (n = 27 of SR-average patients and n = 13 of SR-high risk). These patients were included in the ITT analyses, which are widely used in clinical trials to preserve randomization and provide an unbiased estimate of the treatment effect. The ability of ITT analyses to handle attrition bias depends on the context and the mechanisms of missing data. ITT analyses typically produce unbiased treatment estimates in situations where data are missing completely at random or missing at random, if appropriate methods are used to impute missing data (e.g., multiple imputation). However, in the case of data missing not at random, the ITT analysis may produce biased estimates. It was reported for the study that substantial or informative missingness was neither anticipated nor planned for, based on experience from a previous Children’s Oncology Group ALL trial. However, this assumption may be unrealistic, as the reasons for not initiating postconsolidation therapy vary across the risk strata and treatment groups, potentially leading to data that is not missing at random. Without additional information or results from analyses to determine whether data were missing not at random, it is not possible to determine the impact of attrition bias, if any. No data on treatment completion, discontinuation, dose modifications, and the use of off-protocol treatments were submitted to CDA-AMC for review. Therefore, a comprehensive assessment of attrition and adherence and their potential impact on outcomes could not be done.
The AALL1731 trial planned to use stratified Cox proportional hazards models, adjusted for stratification factors, to test DFS. Tests of the proportional hazards assumption, including the Schoenfeld residuals test (P = 0.031), a Wald test of time-varying interaction (P = 0.048), and a Kolmogorov-type supremum test (P = 0.068), suggested the assumption may not hold. Visual inspection of the KM curves showed no converging or crossing of the DFS curves but indicated that the treatment effect could have a delay of about 2 months before achieving a separation of the curves. In a sensitivity analysis, DFS was analyzed using the RMST method, which does not rely on the proportional hazards assumption and is an appropriate alternative survival analysis approach. Results for the RMST method, which does not rely on the proportional hazards assumption, supported the results of the Cox proportional hazards models for DFS.
Data completeness and transparency of reporting were limitations of the AALL1731 trial. Detailed patient disposition among randomized patients, the receipt of on- and off-protocol therapy, any TEAEs, withdraws due to AEs, treatment-emergent SAEs, and notable harms were not reported.
External Validity
While the comparators in the E1910 and AALL1731 trials were deemed to be acceptable by the clinical experts consulted for this review, blinatumomab is already publicly reimbursed for patients who are MRD-positive (per PC0204 CDA-AMC reimbursement review). No direct or indirect evidence was provided assessing the comparative efficacy of blinatumomab for the currently funded versus the currently being reviewed indication. The clinical experts consulted for this review indicated that, for MRD-positive patients, it is expected that blinatumomab for indications currently funded and currently being reviewed would have a similar efficacy. According to the clinicians consulted for this review, clinicians would likely prefer prescribing blinatumomab per the current review because of the ability to incorporate blinatumomab with chemotherapy and the lack of a requirement to complete 3 intensive chemotherapy blocks. In addition, clinical experts consulted for this review noted that the current review of blinatumomab adds value for patients who have lower levels of MRD (MRD of 0.01% to less than 0.1%, constituting an estimated 5% to 10% of patients as per clinical experts), who are excluded from the currently funded blinatumomab indication. On the other hand, in situations where pediatric patients cannot tolerate additional cycles of consolidation chemotherapy, blinatumomab per the currently funded indication allows for receipt of multiple cycles of blinatumomab to manage leukemia without requiring alternating treatment with consolidation chemotherapy. According to the clinical experts, the choice between both blinatumomab indications should be left to the treating clinician and the patient.
The evidence under review was restricted to a narrower population than the reimbursement request:
Age groups (E1910 and AALL1731 trials): While both trials restricted enrolment based on age (aged 30 to 70 years in the E1910 trial and aged 1 to < 10 years in the AALL1731 trial), the clinical experts consulted for this review felt the results of the AALL1731 trial could be generalized to those younger than 1 year and between 10 and 18 years, while the results of the E1910 trial could be generalized to those aged between 18 and 30 years and older than 70 years.
Risk groups (AALL1731 trial): The AALL1731 trial results were restricted to patients with SR B-cell ALL (defined as patients aged 1 year to less than 10 years at diagnosis and a white cell count of less than 50,000/µL) with an average or high risk of relapse after induction therapy. The clinical expert consulted for this review with experience treating pediatric patients with ALL noted that the definition of SR B-cell ALL is well established and felt the results among the cohort with a SR B-cell ALL and high risk of relapse (i.e., SR-high) could be generalized to patients with high-risk and very high–risk B-cell ALL. While no data were available among the cohort with a favourable risk of relapse, the clinical expert noted that these patients may not require treatment with blinatumomab, as the efficacy of chemotherapy in these patients is high. However, the clinical expert consulted for this review felt the decision to treat pediatric patients with SR-favourable risk should be left up to the treating physician.
MRD status (AALL1731 trial): Patients with SR-high risk after induction therapy who had MRD of 0.1% or greater were reassessed for MRD at the end of consolidation therapy, which is common practice, according to the clinical expert treating pediatric patients. Patients with an end-of-consolidation MRD of less than 0.1% were randomized; those with MRD of 0.1% to less than 1.0% (n = 14) were nonrandomly assigned to receive blinatumomab plus chemotherapy (results are not available); and those with MRD greater than 1% (n = 7) were removed from the trial. The clinical expert treating children agreed to generalize the results of patients with SR-high risk and MRD of less than 0.1% at the end of consolidation therapy to patients with MRD of 0.1% to less than 1.0%. The clinical expert treating children noted that the patient population with SR-high risk and end-of-consolidation MRD of 1% or greater (n = 7) was removed from the trial, as these patients reflect a rare and very high–risk subgroup with disease that is considered refractory and would be managed differently.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.17,18 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. While literature-based thresholds were unavailable for the current review, the clinical experts consulted for this review provided estimates for clinically meaningful thresholds for all outcomes assessed using GRADE.
Findings from the E1910 trial were included in the GRADE assessments. Results from the AALL1731 trial were not appraised using GRADE, as only published results, rather than a formal Clinical Study Report, was provided to CDA-AMC, and the data were based on publicly available sources.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS (probabilities at 3 and 5 years) and RFS (probabilities at 1, 3, and 5 years) in the overall population (i.e., regardless of MRD status), MRD-negative, and MRD-positive populations
harms (any grade 3 or greater TEAEs, treatment-emergent SAEs, and fatal AEs) in the overall population.
Table 2 presents the GRADE summary of findings for blinatumomab plus chemotherapy versus chemotherapy for adults with Ph-negative B-cell precursor ALL.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Comparisons
No indirect evidence was submitted.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies to address gaps within the systematic review evidence were submitted.
Table 2: Summary of Findings for Blinatumomab Plus Chemotherapy vs. Chemotherapy for Adults With Ph-Negative CD19-Positive B-Cell Precursor Acute Lymphoblastic Leukemia
Outcome, population, and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Chemotherapy | Blinatumomab plus chemotherapy | Difference | |||||
Overall survival | |||||||
Probability of OS at 3 years | |||||||
Step 3 analysis seta (MRD-negative and MRD-positive patients) Follow-up: median 4.5 years in both armsb | 286 (1 RCT) | NR | 65.7 per 100 | 82.6 per 100 (75.5 to 87.8) | 16.9 more per 100 (5.5 more to 28.3 more per 100) | Highc,d | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being alive at 3 years compared to chemotherapy. |
Full analysis sete (MRD-negative patients) Follow-up: median 4.5 years in both armsb | 224 (1 RCT) | NR | 70.0 per 100 | 85.5 per 100 (77.5 to 90.9) | 15.6 more per 100 (3.0 more to 28.2 more per 100) | Moderatec,f | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being alive at 3 years compared to chemotherapy in MRD-negative patients. |
Step 3 MRD-positive analysis setg (MRD-positive patients) Follow-up: median 4.6 years in blinatumomab arm and median 5.0 years in chemotherapy alone armb | 62 (1 RCT) | NR | 43.2 per 100 | 74.2 per 100 (57.4 to 85.2) | 31.1 more per 100 (4.1 more to 58.0 more per 100) | Moderatec,h | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being alive at 3 years compared to chemotherapy in MRD-positive patients. |
Probability of OS at 5 years | |||||||
Step 3 analysis seta (MRD-negative and MRD-positive) Follow-up: median 4.5 years in both armsb | 286 (1 RCT) | NR | 58.3 per 100 | 79.1 per 100 (71.4 to 85.0) | 20.8 more per 100 (8.5 more to 33.0 more per 100) | Highc,d | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being alive at 5 years compared to chemotherapy. |
Full analysis sete (MRD-negative patients) Follow-up: median 4.5 years in both armsb | 224 (1 RCT) | NR | 62.5 per 100 | 82.4 per 100 (73.7 to 88.4) | 19.9 more per 100 (6.3 more to 33.5 more per 100) | Highc,i | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being alive at 5 years compared to chemotherapy in MRD-negative patients. |
Step 3 MRD-positive analysis setg (MRD-positive patients) Follow-up: median 4.6 years in blinatumomab arm and median 5.0 years in chemotherapy alone armb | 62 (1 RCT) | NR | 37.8 per 100 | 70.1 per 100 (52.0 to 82.5) | 32.3 more per 100 (5.4 more to 59.3 more per 100) | Highc,j | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being alive at 5 years compared to chemotherapy in MRD-positive patients. |
Relapse-free survival | |||||||
Probability of RFS at 1 year | |||||||
Step 3 analysis seta (MRD-negative and MRD-positive) Follow-up: median 4.5 years in both armsb | 286 (1 RCT) | NR | 75.8 per 100 | 88.0 per 100 (81.7 to 92.3) | 12.2 more per 100 (1.8 more to 22.7 more per 100) | Moderatec,k | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being relapse-free at 1 years compared to chemotherapy. |
Full analysis sete (MRD-negative patients) Follow-up: median 4.5 years in both armsb | 224 (1 RCT) | NR | 81.9 per 100 | 90.1 per 100 (82.8 to 94.4) | 8.2 more per 100 (3.0 less to 19.4 more per 100) | Moderatec,l | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being relapse-free at 1 years compared to chemotherapy in MRD-negative patients. |
Step 3 MRD-positive analysis setg (MRD-positive patients) Follow-up: median 4.6 years in blinatumomab arm and median 5.0 years in chemotherapy alone armb | 62 (1 RCT) | NR | 44.3 per 100 | 82.4 per 100 (66.5 to 91.2) | 38.0 more per 100 (11.6 more to 64.5 more per 100) | Highc,m | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being relapse-free at 1 years compared to chemotherapy in MRD-positive patients. |
Probability of RFS at 3 years | |||||||
Step 3 analysis seta (MRD-negative and MRD-positive) Follow-up: median 4.5 years in both armsb | 286 (1 RCT) | NR | 61.4 per 100 | 78.7 per 100 (71.2 to 84.4) | 17.3 more per 100 (5.6 more to 28.9 more per 100) | Highc,n | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being relapse-free at 3 years compared to chemotherapy. |
Full analysis sete (MRD-negative patients) Follow-up: median 4.5 years in both armsb | 224 (1 RCT) | NR | 65.7 per 100 | 81.1 per 100 (72.5 to 87.2) | 15.4 more per 100 (2.3 more to 28.4 more per 100) | Moderatec,l | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being relapse-free at 3 years compared to chemotherapy in MRD-negative patients. |
Step 3 MRD-positive analysis setg (MRD-positive patients) Follow-up: median 4.6 years in blinatumomab arm and median 5.0 years in chemotherapy alone armb | 62 (1 RCT) | NR | 39.4 per 100 | 71.8 per 100 (54.8 to 83.3) | 32.4 more per 100 (6.1 more to 58.7 more per 100) | Highc,o | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being relapse-free at 3 years compared to chemotherapy in MRD-positive patients. |
Probability of RFS at 5 years | |||||||
Step 3 analysis seta (MRD-negative and MRD-positive) Follow-up: median 4.5 years in both armsb | 286 (1 RCT) | NR | 57.2 per 100 | 75.6 per 100 (67.8 to 81.8) | 18.4 more per 100 (6.3 more to 30.6 more per 100) | Highc,n | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being relapse-free at 5 years compared to chemotherapy. |
Full analysis sete (MRD-negative patients) Follow-up: median 4.5 years in both armsb | 224 (1 RCT) | NR | 60.5 per 100 | 77.0 per 100 (67.8 to 83.8) | 16.5 more per 100 (2.6 more to 30.3 more per 100) | Moderatec,l | Blinatumomab plus chemotherapy likely results in a clinically important increase in the probability of being relapse-free at 5 years compared to chemotherapy in MRD-negative patients. |
Step 3 MRD-positive analysis setg (MRD-positive patients) Follow-up: median 4.6 years in blinatumomab arm and median 5.0 years in chemotherapy alone armb | 62 (1 RCT) | NR | 39.4 per 100 | 71.8 per 100 (54.8 to 83.3) | 32.4 more per 100 (6.1 more to 58.7 more per 100) | Highc,o | Blinatumomab plus chemotherapy results in a clinically important increase in the probability of being relapse-free at 3 years compared to chemotherapy in MRD-positive patients. |
Harms | |||||||
Incidence of any grade 3 or greater TEAE Step 3 safety analysis setp (MRD-negative and MRD-positive) Follow-up: by June 23, 2023, DCO | 275 (1 RCT) | NR | 97.7 per 100 | 95.9 per 100 | 1.74 less per 100 (5.87 less to 2.40 more per 100) | Moderatec,q | Blinatumomab plus chemotherapy likely results in little to no in the incidence of any grade 3 or greater TEAEs compared to chemotherapy. |
Incidence of treatment-emergent SAE Step 3 safety analysis setp (MRD-negative and MRD-positive) Follow-up: by June 23, 2023, DCO | 275 (1 RCT) | NR | 28.1 per 100 | 55.8 per 100 | 27.66 more per 100 (16.47 more to 38.84 more per 100) | Moderatec,r | Blinatumomab plus chemotherapy likely results in a clinically important increase in the incidence of treatment-emergent SAEs compared to chemotherapy. |
Incidence of fatal TEAEs Step 3 safety analysis setp (MRD-negative and MRD-positive) Follow-up: by June 23, 2023, DCO | 275 (1 RCT) | NR | 1.6 per 100 | 2.0 per 100 | 0.48 more per 100 (2.66 less to 3.62 more per 100) | Very lowc,s | The evidence is very uncertain about the effect of blinatumomab plus chemotherapy on fatal TEAEs when compared to chemotherapy. |
CI = confidence interval; DCO = data cut-off; MRD = minimal residual disease; NR = not reported; OS = overall survival; RCT = randomized controlled trial; RFS = relapse-free survival; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aStep 3 analysis set: All 286 Step 3 randomized or registered patients combined, regardless of MRD status (152 patients in the blinatumomab plus chemotherapy arm and 134 patients in the chemotherapy arm).
bEstimated via median time to KM censoring.
cThe between-group difference in survival probability was requested from the sponsor to aid in the interpretation of the results for this end point.
dCertainty was not rated down for indirectness or imprecision. Although limitations regarding internal validity were identified (open-label design, potential selection bias, results from interim analyses, and OS was conducted as a post hoc analysis), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated minimal important difference (MID) was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful, and this value was used as the threshold.
eFAS: All 224 Step 3 randomized patients who are assessed as MRD-negative centrally after induction and intensification chemotherapy (112 patients in the blinatumomab plus chemotherapy arm and 112 patients in the chemotherapy arm).
fCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, results from interim analyses, and OS was conducted as a post hoc analysis), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. Rated down 1 level for imprecision. No empirically derived and validated MID was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful, and this value was used as the threshold. The point estimate suggests clinically meaningful increases in OS at 3 years, while the lower bound of the 95% CI crosses the between-group difference threshold of 5%.
gStep 3 MRD-positive analysis set: All 62 patients from Step 3 analysis set who are MRD-positive at Step 3 using the protocol-specified cut-off of 0.01% or less (40 patients in the blinatumomab plus chemotherapy arm and 22 patients in the chemotherapy arm).
hCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, results from interim analyses, and OS was conducted as a post hoc analysis), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. Rated down 1 level for imprecision. No empirically derived and validated MID was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful, and this value was used as the threshold. The point estimate suggests clinically meaningful increases in OS at 3 years, while the lower bound of the 95% CI crosses the between-group difference threshold of 5%. While results are from a relatively small sample (n = 62), certainty was not further rated down due to imprecision as this limitation was determined to have a small or no impact on the results.
iCertainty was not rated down for indirectness or imprecision. Although limitations regarding internal validity were identified (open-label design, results from interim analyses, and OS was conducted as a post hoc analysis), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful, and this value was used as the threshold.
jCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, results from interim analyses, and OS was conducted as a post hoc analysis), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. While results are from a relatively small sample (n = 62), certainty was not rated down due to imprecision as the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful, and this value was used as the threshold.
kCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, and results from interim analyses), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of remaining relapse-free. The clinical experts consulted for this review suggested that a 5% to 7% between-group difference would be clinically meaningful and this range was used as the threshold. Rated down 1 level for imprecision. The point estimate suggests clinically meaningful increases in RFS at 1 year, while the lower bound of the 95% CI crosses the between-group difference threshold of 5% to 7%.
lCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design and results from interim analyses), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of remaining relapse-free. The clinical experts consulted for this review suggested that a 5% to 10% between-group difference would be clinically meaningful, and this range was used as the threshold. Rated down 1 level for imprecision. The point estimate suggests clinically meaningful increases in RFS at 1, 3, and 5 years while the lower bound of the 95% CI crosses the between-group difference threshold of 5% to 10%.
mCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, and results from interim analyses), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of remaining relapse-free. The clinical experts consulted for this review suggested that a 5% to 10% between-group difference would be clinically meaningful, and this range was used as the threshold. While results are from a relatively small sample (n = 62), certainty was not rated down due to imprecision as this limitation was determined to have a small or no impact on the results.
nCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, and results from interim analyses), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of remaining relapse-free. The clinical experts consulted for this review suggested that a 5% to 7% between-group difference would be clinically meaningful, and this range was used as the threshold. The point estimate and upper bound of the 95% CI suggests clinically meaningful increases in RFS at 3 and 5 years. The lower bound of the 95% CI is higher than the lower limit of the between-group difference threshold (5%), and, thus, certainty was not rated down; however, if the upper limit of the threshold (7%) is used certainty would be rated down due to imprecision.
oCertainty was not rated down for indirectness. Although limitations regarding internal validity were identified (open-label design, potential selection bias, and results from interim analyses), certainty was not rated down for risk of bias because the limitations were determined to have a small or no impact on the results. No empirically derived and validated MID was identified for the between-group difference in the probability of remaining relapse-free. The clinical experts consulted for this review suggested that a 5% to 10% between-group difference would be clinically meaningful and this range was used as the threshold. The point estimate and upper bound of the 95% CI suggests clinically meaningful increases in RFS at 3 and 5 years. The lower bound of the 95% CI is higher than the lower limit of the between-group difference threshold (5%), and, thus, certainty was not rated down; however, if the upper limit of the threshold (10%) is used certainty would be rated down due to imprecision. While results are from a relatively small sample (n = 62), certainty was not rated down due to imprecision as this limitation was determined to have a small or no impact on the results.
pStep 3 safety analysis set: All 275 patients in the Step 3 analysis set who took at least 1 dose of protocol-specified therapies (147 patients in the blinatumomab plus chemotherapy arm and 128 patients in the chemotherapy arm).
qCertainty was not rated down for indirectness or imprecision. Rated down 1 level for risk of bias. Due to the open-label nature of the study, there is substantial risk of bias for subjective outcomes. No empirically derived and validated MID was identified for the between-group difference in the incidence of any grade 3 or greater TEAE. The clinical experts consulted for this review suggested that a 10% between-group difference would be clinically meaningful, and this value was used as the threshold.
rCertainty was not rated down for indirectness or imprecision. Rated down 1 level for risk of bias. Due to the open-label nature of the study there is substantial risk of bias for subjective outcomes. No empirically derived and validated MID was identified for the between-group difference in the incidence of treatment-emergent SAE. The clinical experts consulted for this review suggested that a 10% to 15% between-group difference would be clinically meaningful, and this range was used as the threshold.
sCertainty was not rated down for indirectness. Rated down 1 level for risk of bias. Due to the open-label nature of the study there is substantial risk of bias for subjective outcomes. Rated down 2 levels for imprecision. No empirically derived and validated MID was identified for the between-group difference in the incidence of fatal TEAEs. The clinical experts consulted for this review suggested that a greater than 0% between-group difference would be clinically meaningful and this value was used as the threshold. The point estimate suggests clinically meaningful increases in fatal TEAEs, while the upper and lower bounds of the 95% CI cross the between-group difference threshold of 0% suggested by clinical experts. Given the small number of events, there is substantial uncertainty in the between-group difference.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Conclusions
The E1910 (adult-only population) and the AALL1731 (pediatric-only population) trials were phase III, open-label, multicentre, international studies aimed at evaluating the efficacy and safety of blinatumomab used in the consolidation phase of multiphase chemotherapy for the treatment of patients with Ph-negative B-cell precursor ALL.
A post hoc analysis in the E1910 trial aligned with the target patient population under review, which consists of patients with MRD-agnostic, Ph-negative, B-cell precursor ALL. The results of the post hoc analysis showed that blinatumomab plus chemotherapy demonstrated clinically meaningful improvements in OS and RFS compared to chemotherapy, with high to moderate certainty of evidence across different time points. The E1910 trial met its primary end point, with statistically significant improvements in OS favouring the blinatumomab plus chemotherapy group in MRD-negative patients. Secondary analyses for OS in MRD-positive patients and RFS in MRD-negative or MRD-positive patients were supportive of the benefit observed with blinatumomab plus chemotherapy over chemotherapy in the primary analyses, with high to moderate certainty of evidence across different time points. Post hoc analyses are at risk of data manipulation; however, results of the post hoc analyses were consistent with those for the primary (OS) and secondary (RFS) outcomes in patients with MRD-negative disease, mitigating concerns about data validity.
The primary analysis in the AALL1731 trial for the overall trial population aligned with the present target patient population and suggested clinically meaningful improvements in DFS with blinatumomab plus chemotherapy compared to chemotherapy. Post hoc analyses for OS suggested little to no difference between blinatumomab plus chemotherapy compared to chemotherapy in the target patient population. Post hoc analyses are at risk of data manipulation; however, results of the OS post hoc analyses were consistent with clinical expectations, based on input from the clinical experts consulted by CDA-AMC, mitigating concerns about data validity. DFS and OS results for populations with SR-average and SR-high risk category were overall consistent with the results observed in the overall trial population. GRADE assessment was not conducted for results of the AALL1731 trial, as only limited public data were available.
According to clinical experts consulted for this review, the harms profile of blinatumomab plus chemotherapy was consistent with their expectations, given the known AEs.
While blinatumomab is currently reimbursed for patients with Ph-negative CD19-positive B-cell precursor ALL with MRD-positive disease, the clinical experts consulted for this review anticipated similar efficacy for the indications currently funded and being reviewed. According to the clinical experts, both options provide unique advantages, and, therefore, the choice between these 2 options should be left to the treating physician and patient.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of blinatumomab, 28 mcg per day for adult patients weighing 45 kg or more and 15 mcg/m2 per day for adult patients weighing less than 45 kg and pediatric patients, lyophilized powder for solution, IV infusion by infusion pump in the treatment of Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in the frontline setting.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
ALL is a rare and aggressive hematologic malignancy of undifferentiated lymphoid precursors characterized by the proliferation of immature and abnormal lymphoid cells in the bone marrow and peripheral blood.1 The proliferation of these immature and abnormal lymphoid cells in the bone marrow subsequently prevails over the production of normal bone marrow elements, ultimately resulting in decreased red blood cells, white blood cells, and platelet counts.20 Common symptoms of ALL include fatigue, reduced energy, dry mouth, lack of appetite, and sweating, along with psychological symptoms such as irritability, nervousness, and sadness.9
In 2019 alone, 440 new cases of ALL were diagnosed in Canada (excluding Quebec), resulting in an age-standardized incidence rate of 1.5 per 100,000 Canadians.2 Between 2015 to 2017, the 5-year net survival for patients in Canada aged 15 to 99 years was 47%,3 with improved survival rates among younger patients.21 While ALL is the least common type of leukemia in adults, it is the most prevalent cancer among children and young adults.4 In Canada, ALL accounts for 75% of all childhood cancers and over half of the new ALL cases in 2019 occurred in Canadians aged 19 years of age and under.3 The annual incidence rate of ALL among children under the age of 15 years is 3 to 4 new cases per 100,000 children.22
WHO classifies ALL based on the type of lymphocytes affected and specific gene or chromosomal changes in the leukemia cells.23 The 2 major subtypes of ALL based on lymphocytes are B-lymphoblastic (B-cell) and T-lymphoblastic leukemia.24,25 B-cell phenotypes are the most common, accounting for approximately 85% of pediatric ALL cases and around 75% of adult ALL cases.5,6 The leukemia cells in many patients with B-cell ALL have chromosomal abnormalities with 1 of the most prevalent abnormalities occurring in the Philadelphia chromosome (occurring in 1% to 3% of childhood ALL cases and 11% to 29% of adult ALL cases).7 According to the Surveillance, Epidemiology, and End Results (SEER) database in the US, the 5-year OS between 2010 and 2017 for adult and pediatric patients was 72% for ALL (overall) and 73% for patients with Ph-negative ALL; when focusing only on pediatric patients aged 0 to 14 years, the corresponding estimates were 93% in both cohorts, with an inverse relationship observed between age and survival age categories.21
While there is no standard staging system, ALL is commonly classified as untreated (newly diagnosed patients who have not been treated yet), remission (patients who are treated and are in remission), relapsed (leukemia has come back after treatment), or refractory (ALL did not respond to treatment).4 The Toronto Pediatric Cancer Stage Guidelines describe ALL using a 2-tier staging system, with Tier 1 categorizing patients based on central nervous system (CNS)–positive and CNS-negative status and Tier 2 categorizing patients based on no CNS involvement and no blasts in cerebrospinal fluid (CSF) (CNS-1), no CNS involvement and blasts in CSF (CNS-2), and CNS involvement and blasts in CSF (CNS-3).26
Determining the ALL subtype and presence of chromosomal abnormalities is critical for understanding the disease status, risk factors, and treatment planning. According to the Canadian Cancer Society, the prognostic factors for ALL include age, white blood cell count, ALL classification, chromosome changes, and presence of leukemia cells in the CNS and early relapse.4 ALL diagnoses typically begin with a review of the patient’s health history, followed by a physical exam.27 Alberta Health Services Guidelines recommend that leukemia diagnoses be based on the results of bone marrow studies incorporating morphological assessment, immunophenotyping, cytogenetic, fluorescent in situ hybridization (FISH), and molecular evaluation.8 MRD assessment provides information on the prognosis and chance of relapse, with higher MRD levels indicating greater chance of relapse.8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The treatment of Ph-negative B-cell precursor ALL typically includes 3 phases: induction, consolidation (sometimes called “intensification”), and maintenance.4 While treatment regimens differ in terms of specific drug selection, dosing, and duration, most regimens include vincristine, asparaginase, corticosteroids (dexamethasone is often preferred to prednisone, as it penetrates the blood-brain barrier and acts on resting leukemic blast cells), immune modulators (methotrexate), and anthracycline (daunorubicin, doxorubicin, idarubicin), with or without cyclophosphamide or cytarabine.28 Details of specific regimens identified by clinical experts consulted for this review as commonly used in Canada for ALL are summarized in Table 3. Patients are stratified according to risk characteristics (i.e., age, white blood cell count at diagnosis, CNS and/or testicular disease, genetic abnormalities, and MRD after induction therapy) to ensure that the appropriate intensity of treatment is administered to patients with high risk of relapse, while avoiding unnecessary toxicity.25,29,30 At diagnosis, pediatric patients with ALL are categorized into 3 risk groups:31
SR, defined as children aged 1 to less than 10 years who have a white blood cell count less than 50,000/µL of blood at the time of diagnosis and absence of CNS or testicular disease
high risk, defined as children aged 10 and older and/or children who have a white blood cell count of 50,000/µL of blood or more at the time of diagnosis
very high risk, defined as children under 1 year of age, children identified by certain genetic changes in their leukemia, children who have a slow improvement from initial treatment, and children who have signs of leukemia after the first 4 weeks of treatment.
Throughout all phases, CNS prophylaxis is given to prevent ALL from spreading to the CNS.25 The entire Ph-negative B-cell ALL treatment may take more than 2 years.
Induction Therapy
Induction is the first phase of treatment and is typically initiated in the hospital.25 During this phase, a multidrug combination of chemotherapies and steroids are typically used with the goal of achieving a CR, defined as less than 5% of blasts remaining.4
At the completion of induction therapy, bone marrow aspirates and biopsies are used to determine whether CR has been achieved, and, if it has been achieved, MRD status is assessed.13,25 MRD quantification is an essential component of patient evaluation over the course of sequential ALL therapy, as previous studies have demonstrated a strong correlation between the presence of MRD and the risk of relapse.32
Consolidation Therapy
The objectives of consolidation treatment for ALL are to enhance and solidify the initial response achieved through induction chemotherapy while minimizing the risk of relapse.4 High doses of chemotherapy are the primary consolidation treatment, and many of the same drugs received during induction are given during consolidation.
Children with SR are assigned to 3 risk-of-relapse groups based on tumour genetics, CNS status, and MRD:
SR-favourable includes those with favourable cytogenetics (ETV6: RUNX1 or double trisomies of chromosomes 4 and 10 [DT]), and end-of-induction bone marrow MRD less than 0.01%.
SR-high includes those with unfavourable cytogenetics (iAMP21, KMT2A rearrangement, t(17;19), hypodiploidy), end-of-induction MRD of 0.01% or greater, or neutral cytogenetics with CNS-2 status.
All others are classified as SR-average.
Children with SR-favourable and SR-average disease typically receive conventional consolidation treatment with some combination of vincristine, mercaptopurine, methotrexate, doxorubicin, dexamethasone, pegaspargase, cyclophosphamide, cytarabine, thioguanine, and intrathecal methotrexate. Children assigned to SR-high, high risk, and very high–risk groups receive consolidation treatment with the same drugs, albeit at a stronger dose and for a longer period of time.
In adult patients with MRD-positive disease, Alberta Health Services recommends consolidation treatment with immunotherapy using 1 to 2 cycles of blinatumomab.8 MRD-positive adult patients may be considered for allogeneic SCT.8,13
Blinatumomab is currently Health Canada–approved and publicly funded for the treatment of patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic CR with MRD greater than or equal to 0.1%. Under the currently funded indication, adult and pediatric patients can receive treatment with up to 4 sequential cycles of blinatumomab if they have previously received at least 3 intensive chemotherapy blocks of treatment that are age-appropriate. The indication currently funded for blinatumomab partially overlaps with the indication currently under review. Differences between the indication currently funded versus that currently being reviewed for blinatumomab in patients who are MRD-positive include remission status (first and second remission versus first remission), MRD restrictions (great than or equal to 0.1% versus no MRD restriction), administration schedule (sequential cycles versus alternating with consolidation chemotherapy), and number of cycles of blinatumomab treatment for pediatric patients (maximum of 4 versus 2 cycles of blinatumomab).
Maintenance Therapy
Maintenance therapy is the final and usually the longest phase of treatment. The objective of this phase is to kill more cancer cells and stop any remaining leukemia cells from growing.4 Regimens are typically less intensive than prior chemotherapy and are given in an outpatient setting with the goal of reducing the risk of relapse.
Maintenance therapy for pediatric patients with ALL typically include vincristine, dexamethasone, mercaptopurine, and methotrexate.
Table 3: Chemotherapy Regimens Used in Canada for Ph-Negative B-Cell Precursor ALL Treatment
Protocol name | Used to treat pediatric vs. adult patients | Regimen |
|---|---|---|
COG AALL1731/1732/1631 | Standard-risk pediatrics | Doxorubicin, vincristine, pegaspargase or calaspargase, cytarabine, methotrexate, mercaptopurine, cyclophosphamide, and IT therapy |
COG AALL 1131 | High-risk pediatrics | IT cytosine arabinoside, vincristine, daunorubicin, pegaspargase, methotrexate, cyclophosphamide, mercaptopurine, doxorubicin, and thioguanine |
Modified DFCI protocol 91 to 01 | Pediatrics and adults | Doxorubicin, vincristine, methotrexate, cytarabine, PEG-asparaginase, mercaptopurine, and IT therapy (and cyclophosphamide or cytarabine and etoposide for pediatric patients at very high risk) |
COG = Children’s Oncology Group; ALL = acute lymphoblastic leukemia; DFCI = Dana-Farber Cancer Institute; IT = intrathecal; Ph = Philadelphia chromosome.
Source: National Comprehensive Cancer Network (NCCN) guidelines, 2024.33 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Drug Under Review
Key characteristics of blinatumomab are summarized in Table 4 with other treatments available for the treatment of patients with Ph-negative CD19-positive B-cell precursor ALL.
Blinatumomab 28 mcg/day for patients weighing 45 kg or more and 15 mcg/m2 per day (not to exceed 28 mcg/day) for patients weighing less than 45 kg as a continuous IV infusion delivered at a constant flow rate using an infusion pump received Health Canada authorization for the treatment of patients with Ph-negative CD19 positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy. A single cycle of treatment is 28 days (4 weeks) of continuous IV infusion followed by a 14-day (2-week) treatment-free interval. As per the blinatumomab product monograph, the safety and efficacy of blinatumomab have been established in pediatric patients (as young as 1 month to less than 18 years of age) with Ph-negative relapsed or refractory B-cell ALL. The safety and efficacy of blinatumomab have not been established in pediatric patients under 1 month of age.34 A Notice of Compliance (NOC) was issued on December 27, 2024.
The Health Canada–approved indication is for blinatumomab as consolidation treatment for Ph-negative CD19 B-cell ALL, regardless of age (pediatric and adult patients), MRD status, or line of treatment. The sponsor’s reimbursement request is for the treatment of adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in the frontline setting. The sponsor’s reimbursement request differs from the Health Canada indication in that it is limited to patients in the frontline setting, while remaining consistent with the Health Canada indication regarding age and MRD status. In Canada, blinatumomab is currently Health Canada–authorized for the treatment of:
patients with Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy
patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic CR with MRD greater than or equal to 0.1%; to be selected for treatment based on detection of MRD as determined by an accredited laboratory using validated assay methods
adult patients with relapsed or refractory B-cell precursor ALL
pediatric patients with Ph-negative relapsed or refractory B-cell precursor ALL.
Blinatumomab has been previously reviewed by CDA-AMC for the treatment of:
patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic CR with MRD greater than or equal to 0.1% (final recommendation to reimburse for adult and pediatric patients with clinical criteria and/or conditions, issued on October 29, 2020)
adult patients (i.e., 18 years of age or older) with Ph-positive B-cell precursor ALL, who have relapsed after or are refractory to at least 1 second-generation or later tyrosine kinase inhibitor, or are intolerant to second-generation or later tyrosine kinase inhibitors and intolerant or refractory to imatinib (final recommendation to reimburse with clinical criteria and/or conditions, issued on April 4, 2019)
pediatric patients with Ph-negative relapsed or refractory B-cell precursor ALL (final recommendation to reimburse for those who are in second or later relapse, who relapsed after allogeneic hematopoietic SCT, or who have refractory disease with clinical criteria and/or conditions, issued on August 23, 2017)
patients with Ph-negative relapsed or refractory B-cell precursor ALL (final recommendation to reimburse for adult patients with clinical criteria and/or conditions, issued on August 31, 2017)
patients with Ph-negative relapsed or refractory B-cell precursor ALL (final recommendation to not reimburse for adults who have only had 1 prior systemic chemotherapy and to reimburse with clinical criteria and/or conditions for adults those who have had at least 2 prior lines of systemic therapy, issued on April 1, 2016).
Blinatumomab is a bispecific T-cell engager molecule that binds specifically to CD19 expressed on the surface of cells of B-lineage origin and CD3 expressed on the surface of T-cells. It activates endogenous T-cells by connecting CD3 in the T-cell receptor complex with CD19 on benign and malignant B-cells, including B-cell precursor ALL cells. The antitumour activity of blinatumomab immunotherapy is not dependent on T-cells bearing a specific T-cell receptor or on peptide antigens presented by cancer cells but is polyclonal in nature and independent of human leukocyte antigen molecules on target cells. Blinatumomab mediates the formation of a cytolytic synapse between the T-cell and the tumour cell, releasing proteolytic enzymes (such as perforin and granzymes) to kill both proliferating and resting target cells, which closely resembles a natural cytotoxic T-cell reaction. Blinatumomab is associated with transient upregulation of cell adhesion molecules, production of cytolytic proteins, release of inflammatory cytokines, and proliferation of T-cells, resulting in elimination of CD19-positive cells.
In B-cell ALL, CD19 is typically present on leukemic cells, making it a valuable target for immunotherapies like blinatumomab. Clinical trials for blinatumomab may not require patient selection based on CD19 status, as CD19 expression is generally assumed in B-cell ALL cases. Instances of CD19-negative B-cell ALL are extremely rare.35
Testing Procedure Considerations
The current standard of care in Canada for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic CR is to determine eligibility for blinatumomab as a consolidation therapy based on MRD positivity.8,49,50 MRD refers to the presence of subclinical disease detected after a patient with ALL has reached CR morphologically.33,51 MRD positivity is defined by the sponsor as greater than or equal to 0.1% mononuclear cells.34 If blinatumomab becomes funded regardless of the MRD status, as per the new indication under review, MRD testing would no longer be needed as a companion diagnostic. The sponsor mentioned that, although blinatumomab use regardless of the MRD status is anticipated to become the preferred approach (i.e., all patients who reach CR after induction should be offered blinatumomab), blinatumomab use based on MRD positivity, as per the previous indication, should still be an option, with the choice between treatment approaches up to the clinician and patient.52 In addition, the clinical experts consulted for this review noted that MRD testing should still be conducted before and during treatment with blinatumomab to risk stratify, to prognosticate outcomes, and to guide disease management, including as part of multifactorial decisions around discontinuing treatment to proceed with allogeneic stem cell transplant.33,51,53-56
Table 4: Key Characteristics of Blinatumomab and Regimens Used in the Modified DFCI Protocol and COG Protocol
Treatment | Pediatric or adult patients | Mechanism of action | Indicationa | Route of administration | Recommended doseb | Serious adverse effects or safety issues | |
|---|---|---|---|---|---|---|---|
Used to treat | Indicateda | ||||||
Blinatumomab | Both | Both | Mediates the formation of a cytolytic synapse between the T-cell and the tumour cell, releasing proteolytic enzymes to kill both proliferating and resting target cells |
| IV | Body weight ≥ 45 kg: fixed dose of 28 mcg/day on days 1 to 28 Body weight < 45 kg: 15 mcg/m2 per day (not to exceed 28 mcg/day) on days 1 to 28 | Cytokine release syndrome, tumour lysis syndrome, neurologic toxicities, serious infections, pancreatitis, benzyl alcohol toxicity (gasping syndrome) Relapse of CD19-negative B-cell precursor ALL, lineage switch from ALL to AML |
Vincristine | Both | Both | Interferes with microtubule formation, preventing mitosis | Treatment of acute leukemia In combination with other oncolytic agents for various tumour types | IV | Pediatrics:
Adults:
| Fetal harm, acute uric acid nephropathy, acute shortness of breath, severe bronchospasm |
Cyclophosphamide | Pediatrics | Pediatrics | Alkylating drug that is cytotoxic by damaging DNA and RNA strands, as well as inhibition of DNA synthesis | Frequently responsive myeloproliferative and lymphoproliferative disorders (e.g., leukemias) Frequently responsive solid malignancies (e.g., neuroblastoma) Malignant neoplasms of the lung | IV and oral | Pediatrics: 1,000 mg/m2
| Secondary malignancy, acute cardiac toxicity, ventricular tachyarrhythmia, infection, hepatotoxicity, severe myelosuppression, urotoxicity, acute pulmonary toxicity, fulminating anaphylaxis Live vaccines may lead to vaccine-induced infection |
Cytarabine | Both | Both | Inhibits DNA synthesis | Newly diagnosed children with acute lymphocytic leukemia Primarily for induction and maintenance of remission in acute leukemia in both adults and children Other: acute myelocytic leukemia, chronic myelocytic leukemia (blast phase), acute erythroleukemia, and non-Hodgkin lymphoma in children | IV | Pediatrics:
Adults:
| Cardiomyopathy with subsequent death, GI toxicity, acute pancreatitis, CNS toxicity, infection, pulmonary toxicity, myelosuppression |
Dexamethasone | Both | Both | Inhibits cell growth and induces apoptosis; anti-inflammatory, immunosuppressive, antiemetic | For palliative management of leukemias and lymphomas | Oral | Pediatrics:
Adults:
| Contraindicated in patients with systemic fungal infections. Administration of live-virus vaccines is contraindicated. |
Daunorubicin | Pediatrics | Both | Inhibits DNA replication and transcription | Initial treatment of myeloblastic and acute lymphoblastic leukemias; also to induce a remission in patients with chronic myeloid leukemia, reticulosarcoma, Ewing or Wilms tumours and lymphosarcoma | IV | Pediatrics:
| Contraindicated in patients who exhibit myocardial lesions or to those older than 75 years. Infections should be treated before the start. Adverse effects: hyperuricemia, colitis, tissue necrosis |
Doxorubicin | Both | Both | Inhibits DNA and RNA synthesis | Alone or in combination with other anticancer agents to treat numerous neoplastic conditions such as acute lymphoblastic leukemia | IV | Pediatrics:
Adults:
| Cardiomyopathy, secondary malignancies, extravasation and tissue necrosis, myelosuppression and sequelae, hepatic impairment |
Etoposide | Pediatrics | Both | Inhibits topoisomerase II and interferes with the synthesis and repair of DNA | Approved for small cell and non–small cell carcinomas of the lung, malignant lymphoma, and testicular malignancies Other use: leukemias, small cell carcinomas outside the lungs, adrenal cancer, gynecological cancers, sarcomas, CNS cancers, thymoma, and Merkel cell carcinoma | IV | 50 to 100 mg/m2 consolidation days 3, 4, and 5 | Severe myelosuppression, liver and renal failure, pulmonary deterioration, thrombocytopenia, and ascites Contraindicated in patients with severe leukopenia, thrombocytopenia, and severe hepatic and/or renal impairment The use of live vaccines should be avoided. |
Methotrexate | Both | Both | Folate antagonist, prevents DNA synthesis, repair, and cellular replication | Numerous neoplastic diseases, including ALL As a disease-modifying antirheumatic drug Treatment of psoriasis | Oral | Pediatrics:
Adults:
| Contraindicated in:
|
6-mercaptopurine | Both | Both | Interferes with purine metabolism, inhibiting DNA and RNA synthesis | The maintenance therapy of acute lymphatic (lymphocytic, lymphoblastic) leukemia, as part of a combination regimen | Oral | Pediatrics:
Adults:
| Immunizations with live-organism vaccines is contraindicated. Myelosuppression, hepatotoxicity, teratogenesis |
Pegaspargase | Both | Both | Enzymatic break down of the amino acid L-asparagine inhibits protein-synthesis, DNA synthesis, and RNA synthesis, especially in leukemic blasts | A component of a multidrug chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia | IM, IV | Pediatrics:
Adults:
| Contraindicated in patients with:
|
Prednisone | Both | Both | Inhibits cell growth and induces apoptosis; anti-inflammatory, immunosuppressive, antiemetic | Indicated in conjunction with appropriate specific antineoplastic disease therapy for acute or chronic lymphocytic leukemia, Hodgkin or non-Hodgkin lymphoma and related problems | Oral | Pediatrics:
Adults:
| Contraindicated in:
|
Thioguanine | Pediatric | Both | An antimetabolite that blocks purine metabolism, inhibits DNA and RNA synthesis | Treatment of acute leukemia; not recommended for maintenance therapy or similar long-term continuous treatments due to the high risk of liver toxicity | Oral |
| Myelosuppression, liver toxicity, potential severe infection after immunization using a live-organism vaccine |
ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; b.i.d. = twice daily; CNS = central nervous system; COG = Children’s Oncology Group; DFCI = Dana-Farber Cancer Institute; GI = gastrointestinal; IM = intramuscular; MRD = minimal residual disease; Ph = Philadelphia; p.o. = per oral; RNA = ribonucleic acid; SR = standard risk.
aHealth Canada–approved indication.
bThe protocols for the modified DFCI were informed by Alberta Health Services for the adult population25 and Vrooman et al. for the pediatric population.36 The clinical experts consulted for this review confirmed both DFCI protocols matched those currently used in Canadian clinical practice. The Children’s Oncology Group protocol was informed by the clinical expert consulted for this review with experience treating children in all.
Source: Draft product monograph for blinatumomab (Blincyto)34 and product monographs for vincristine,37 cyclophosphamide,38 cytarabine,39 dexamethasone,40 daunorubicin,41 doxorubicin,42 etoposide,43 methotrexate,44 6-mercaptopurine (Purinethol),45 pegaspargase (Oncaspar),46 prednisone,47 thioguanine.48
According to the clinical experts consulted for this review, multiparameter flow cytometry is the only method currently available in Canada for MRD testing. Flow cytometry analyzes cells as they flow in a stream through a beam of light. Individual cell types can be identified by conjugating a fluorescent dye to a monoclonal antibody. Multiparameter flow cytometry uses multiple fluorescent dyes to detect a variety of markers that may be found on leukemic cells.57 MRD testing by flow cytometry requires fresh cellular materials obtained through a bone marrow biopsy.53 Samples need to be tested by the receiving laboratory within 24 to 48 hours of collection to ensure viability of the cells.53 MRD testing using flow cytometry is not standardized in Canada, and, therefore, policies, procedures, and equipment sensitivity can differ among leukemia or cancer centres.53 MRD testing by flow cytometry requires the expertise of a highly trained pathologist to correctly interpret the results.53,58 Laboratories in Canada that perform flow cytometry for MRD testing can detect the presence of residual cancer cells with a sensitivity threshold of 10–3 cells (i.e., 1 cancer cell among 1,000 bone marrow cells or 0.1%), although some laboratories are able to detect up to 10–4 cells (i.e., 1 cancer cell among 10,000 bone marrow cells or 0.01%).53 As well, the panel of antibodies used to detect leukemia cell markers may differ between laboratories.53 The clinical experts, however, confirmed that the panels should be relatively equal in their ability to detect leukemia cells. Molecular methods used for testing the MRD status, such as real time quantitative polymerase chain reaction and next-generation sequencing, are not yet available in Canada, although they are in use and standardized in other countries.33,51,53,58-60 Molecular methods can detect MRD at higher sensitivities than flow cytometry can, but they are more expensive, have longer turnaround times, and require an index pretreatment analysis using the diagnostic biopsy sample.58,59
CDA-AMC considered the potential impacts of MRD testing in adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL if blinatumomab becomes funded in the consolidation phase of multiphase chemotherapy in the frontline setting, regardless of the MRD status, including impacts to health systems, patients (including families and caregivers), and costs. While MRD testing is widely available and routinely performed, it might not be consistently funded across jurisdictions in Canada. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Table 5.
Table 5: Considerations for MRD Testing in Adult and Pediatric Patients With Ph-Negative CD19-Positive B-Cell Precursor ALL in the Consolidation Phase of Multiphase Chemotherapy in the Frontline Setting
Consideration | Criterion | Available information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | The sponsor estimates about 110 adult patients and 130 pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in Canada, excluding Quebec, will receive MRD testing in 2025.61 The clinical experts agreed with this estimate and confirmed that all patients with Ph-negative CD19-positive B-cell precursor ALL would routinely be tested for the MRD status if they reached morphological CR at the end of induction chemotherapy. Since MRD testing is already part of the recommended standard of care for adult and pediatric patients with Ph-negative CD19 positive B-cell precursor ALL, the number of individuals expected be tested is not anticipated to increase if blinatumomab becomes funded under the new indication. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | According to the clinical experts, flow cytometry for MRD testing is widely available through leukemia or cancer centres across Canada. However, they also mentioned that, for adult patients, funding for this testing is inconsistent across and within jurisdictions. The experts confirmed that initial MRD testing and, if indicated, repeat testing are funded across Canada for all pediatric patients with ALL and with CR at the end of induction chemotherapy. Other methods of MRD testing, such as PCR or NGS, are not currently available or funded in Canada. The experts predicted that these other methods may become available in Canada in the next couple of years. | |
Testing procedure as part of routine care | In line with the previous indication of blinatumomab use specific to MRD-positive cases, MRD testing is currently being done in adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL who attain morphological CR at the end of induction chemotherapy. The clinical experts confirmed that MRD testing is routinely performed to guide the management of both adult and pediatric patient populations. In both populations, MRD testing is conducted at the end of induction chemotherapy in accordance with guidelines published in Canada and internationally.33,53,54,60 | |
Repeat testing requirements | According to the clinical experts, MRD testing frequency for adult patients with Ph-negative CD19-positive B-cell precursor ALL is not standardized. The timing of subsequent MRD testing can vary based on the treatment protocol, risk category, and institutional policies and procedures.8,33,53 The experts remarked that MRD testing should not be repeated more often than every 3 months, as it could be burdensome to patients and clinicians. According to the clinical experts, in pediatric patients with positive MRD at the end of induction, MRD testing is typically repeated only once at the end of consolidation chemotherapy. The experts noted that in rare instances (i.e., less than 5%), a child may need additional MRD testing after consolidation. No additional repeat testing in pediatric patients is anticipated from the new indication. | |
Impacts on human and other health care resources by provision of the testing procedure | MRD testing is currently part of the standard of care for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL. Therefore, no additional impact to human and other health care resources testing is anticipated from the new indication. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | The clinical experts noted that funding for MRD testing in adult patients with Ph-negative CD19-positive B-cell precursor ALL is inconsistent across and within jurisdictions in Canada. It is unclear whether access to testing would be affected in locations without existing funding. Since MRD testing is currently part of the standard of care and funded for pediatric patients with Ph-negative CD19-positive B-cell precursor ALL, no barriers to access were identified or anticipated. |
Expected turnaround times for the testing procedure | The clinical experts estimated that the turnaround time for flow cytometry-based MRD testing is no more than 72 hours for both adult and pediatric patients with ALL. If MRD testing by NGS becomes available in Canada, the turnaround time is expected to be around 2 weeks.53 The clinical expert confirmed this would be acceptable based on the frequency of patient visits. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | The clinical experts highlighted that the invasive and repetitive bone marrow biopsies required for MRD testing may be painful and affect the emotional well-being and quality of life for adult patients. Similarly, repeat bone marrow biopsies may be burdensome to clinicians by increasing their workload. Pediatric patients are sedated for invasive procedures such as bone marrow biopsies to minimize stress and pain. Since MRD testing is currently part of the standard of care for pediatric patients with Ph-negative CD19-positive B-cell precursor ALL, there is no additional burden to patients, families, and/or caregivers anticipated from the new indication. | |
Clinical | Clinical utility and validity of the testing procedure | There is evidence to demonstrate the clinical utility and validity of MRD testing for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL.32,55,56,a It is important to note, however, that a sensitivity threshold of 10–4 is the recommended minimum needed to assess MRD by flow cytometry.33,51,53,54,60 The clinical experts did not think the lower sensitivity threshold (10–3) currently used in some laboratories in Canada would affect clinical practice. |
Risks of harm associated with the testing procedure | The clinical experts stated that complications arising from bone marrow biopsies are very rare. Since MRD testing is currently part of the standard of care for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL, there is no additional risk of harm associated with the testing anticipated from the new indication. | |
Cost | Projected cost of the testing procedure | According to the clinical experts, estimated costs for flow cytometry-based MRD testing are $1,000 to $1,200 per test for adult patients and $450 per test for pediatric patents. Since MRD testing is already part of the standard of care for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL, there is no additional cost anticipated from the new indication. |
ALL = acute lymphoblastic leukemia; CR = complete remission; MRD = minimal residual disease; NGS = next-generation sequencing; PCR = polymerase chain reaction; Ph = Philadelphia chromosome.
aCanada’s Drug Agency has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 2 inputs for this submission. The first input, provided by LLSC, included survey results of adult patients with ALL and their caregivers. The second input was a joint submission provided by LLSC, Ac2orn, OPACC, and Childhood Cancer Canada and was based on interviews with 3 caregivers of pediatric patients with B-cell ALL who received blinatumomab treatment. The patient group input for pediatric patients was supplemented with information on disease experience and experience with currently available treatments, gathered from previous patient group submissions to CDA-AMC for blinatumomab for pediatric patients with Ph-negative B-cell precursor ALL in the relapsed or refractory setting (pCODR 10099 Blinatumomab ALL for pediatrics in 2017 and PX0367 Blinatumomab for ALL for pediatrics in 2024).
LLSC is a national charitable organization dedicated to finding a cure for blood cancers and improve the quality of life of people affected by blood cancers and their families by funding research and providing educational resources, services, and support. Ac2orn is a national organization committed to advocating for translational research and effective treatments to realize the goal of curing cancers in children, adolescents, and young adults. OPACC advocates for families and organizations navigating the childhood cancer journey. Childhood Cancer Canada supports children diagnosed with cancer and their families.
Input received for adults with ALL is summarized first followed by patient input from caregivers of pediatric patients with ALL.
LLSC conducted a survey in October and November 2024. The respondents were patients with ALL (69%) and their caregivers (28%). Out of 103 respondents providing their age, 9% were aged 0 to 17 years and were disqualified from the survey. The survey respondents included 49% aged 18 to 39 years, 32% aged 40 to 64 years, 8% aged 65 to 74 years, and 3% aged 75 years or older. Among 82 adults providing responses, most were from Canada, 1 was from the US, and 1 was international. A negative to very negative impact on personal or home life due to ALL was reported by 82% of respondents. When considering their social life, 75% of respondents reported negative to very negative impacts due to ALL. Low energy, fear of infections, frequent hospital visits, depression and/or anxiety, ALL symptom burden, and inadequate nutrition were reported as factors contributing to negative impacts of ALL. Eighteen respondents had experience with blinatumomab. Among all other types of treatment, chemotherapy was the most commonly reported type of ALL treatment (98% of respondents) followed by SCT (48% of respondents), radiation therapy (45% of respondents), immunotherapy (27% of respondents), targeted therapy (7% of respondents), CAR T-cell therapy (5% of respondents), and other types (16% of respondents; included natural medicine, Chinese medicine, sound baths, meditation, transfusions steroids, and antiemetics). Fatigue and neutropenia were reported by the respondents as the most severe side effects of current treatments (excluding blinatumomab), followed by thrombocytopenia, infections, nausea, vomiting, diarrhea, anemia, fever, peripheral edema, headaches, infusion reactions, neurologic symptoms, and cytokine release syndrome. These side effects had substantial impacts on patients’ lives, including frequent hospitalizations and lower functionality. LLSC added that the most important outcomes for patients include gaining longer remission with consideration of side effects, quality of life, and financial costs.
According to LLSC, 18 respondents stated that they or the person they care or cared for were treated with blinatumomab for ALL. The methods of access were primarily through clinical trials (33%) and compassionate use programs (27%); the remainder of patients accessed blinatumomab through private insurance, Régie de l'assurance maladie du Québec (RAMQ) — Quebec socialized health care, government funding, or they did not know. Out of 15 respondents, 67%, 20%, and 13% of respondents believed that their ALL completely, partially, or did not respond to blinatumomab, respectively. Respondents were asked to rate the severity of the side effects of blinatumomab (from 1 — did not experience to 4 — severe). Among 15 respondents, the highest-rated side effects, as measured by weighted averages, were neutropenia (2.29), fatigue or weakness (2.27), and fever (2), followed by anemia (1.71), thrombocytopenia (1.71), infections (1.64), headaches (1.57), neurologic symptoms (i.e., confusion, seizures, difficulty speaking; 1.53), cytokine release syndrome (1.5), diarrhea (1.47), peripheral edema (1.47), nausea or vomiting (1.4), and infusion reactions (i.e., chills, rash, difficulty breathing; 1.27). In terms of comparing blinatumomab to other treatments, among 15 respondents, 40% felt it was as difficult to tolerate as other treatments, 27% felt it was less or much less difficult, and 7% felt it was more difficult to tolerate. Additionally, out of 15 respondents, 40% strongly agreed, 20% agreed, 33% felt neutral, and 7% disagreed that blinatumomab improved their quality of life compared to other treatments. Most patients indicated that they were likely to take blinatumomab again or recommend it to other patients.
The joint patient group input for pediatric patients gathered information through interviews with 3 caregivers of pediatric patients with B-cell ALL. According to this patient group submission, to avoid repetitive questioning and to minimize emotional strain and undue harm, the interviews were focused on experiences and quality of life associated with blinatumomab treatment. For all other information, this patient group input referred to previous LLSC-Ac2orn-OPACC- joint submissions to CDA-AMC for relapsed or refractory ALL in pediatric patients. First, the following section presents the disease experience and experience with currently available treatments gathered from previous patient input submissions. Second, it summarizes patients’ and caregivers’ experience with blinatumomab treatment and patients’ quality of life during blinatumomab treatment compared to their experience with prior treatments for pediatric ALL, based on the joint patient group submission for this current review.
Based on input gathered in previous patient input submissions to CDA-AMC for pediatric patients with relapsed or refractory ALL, children may experience a variety of symptoms, including severe fatigue, pain, high fever, bleeding, bruising, bone pain, and swollen lymph nodes. However, the impacts of pediatric cancer relapse on the child and their family extend beyond physical symptoms. It was highlighted in the patient group submissions that relapse and immunosuppression severely limit children’s ability to engage in normal activities, placing a significant emotional and physical burden on both the child and their family. Families experience intense stress, financial strain, and disrupted daily routines, with caregivers often facing severe impacts on their mental health. It was explained in the patient group submissions that the standard of care for treating relapsed or refractory pediatric ALL typically involves a combination of strategies, including drug therapy and radiation. For patients with refractory disease, these aggressive treatments often result in serious side effects, such as immunosuppression, severe pain, infections, anemia, and organ damage, which can significantly impact the child's quality of life. There is a significant unmet need for more effective, less toxic therapies that improve quality of life by reducing treatment burden and minimizing the need for frequent hospital visits. Outpatient treatment options that decrease hospital stays was also cited as crucial for maintaining normalcy and reducing stress for both patients and their families. In terms of improved outcomes, it was noted in the patient group submissions that patients and their families are looking for options that are not only effective but also gentle on their children. They seek innovative therapies that can provide significant benefits without causing undue harm or severe side effects. Additionally, having these treatments covered by drug plans is crucial, as coverage alleviates the financial burden on families.
In the 3 caregiver interviews conducted by the joint patient group submission for this present review, 2 of the caregivers had children who were 2 years old, and 1 had a child who was 10 years old at diagnosis. Two caregivers were residents of Ontario, and 1 resided in British Columbia. The input focused on patients' and caregivers' experiences with administering blinatumomab treatment at home, as well as on comparing their quality of life during blinatumomab treatment to their experiences with prior treatments for pediatric ALL. According to the joint submission, the current conventional therapy for pediatric patients with B-cell ALL living in Canada is chemotherapy infusions, which are often accompanied by serious side effects, negative impact on patients’ physical health, and long hospital stays. Based on the joint input, all 3 caregivers mentioned that they and the patients had overall positive experiences with blinatumomab, especially in comparison to chemotherapy infusion. Because blinatumomab is an outpatient treatment with “gentler” effects, the patients were able to live with family, play with peers, and stay out of the hospital. The 3 caregivers reported that they felt nervous, the child experienced fever, and the child experienced side effects at the start of blinatumomab treatment. According to the patient group input, cognitive testing was done routinely at every blinatumomab bag change to check for neurologic AEs; none of the caregivers reported neurologic AEs as a side effect in the children. Caregivers reported an extra burden because they had to receive a new medication bag of blinatumomab every 4 days and had to visit the medical centre for any glitches in the blinatumomab delivery system. Caregivers noted that, because blinatumomab is a relatively new treatment method, knowledge of blinatumomab’s delivery method was limited among health care professionals. Some caregivers emphasized the need for greater variety for the backpack delivery system for blinatumomab that would be able to accommodate pediatric patients of all ages. According to the joint submission, all 3 caregivers related that blinatumomab made a big difference to the quality of life for their patients, themselves, and the rest of their family compared to traditional chemotherapy infusion. Financial stress was experienced by caregivers due to the high cost and lack of public funding for blinatumomab. Finally, the joint submission made the following suggestions for improved outcomes with new therapies:
Provide caregivers with tips and tools on what to expect with the blinatumomab delivery system, and some at-home solutions to help alleviate practical challenges such as driving to cancer centres at unexpected times.
Train nurses at local hospitals to administer blinatumomab (to mitigate the requirement of driving to cancer centres).
Improve blinatumomab knowledge and skills among the nurses at the cancer centres.
Provide more choices in the delivery system backpacks based on body size and strength of the patient.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of Ph-negative B-cell precursor ALL. One of the clinical experts has specific expertise in the diagnosis and management of adults with Ph-negative B-cell precursor ALL, while the other has expertise in pediatrics with Ph-negative B-cell precursor ALL.
Unmet Needs
The clinical experts consulted for this review felt the overarching goal of Ph-negative B-cell precursor ALL treatment is to achieve CR, prevent relapses, and cure the disease (thus providing long-term survival), while minimizing acute and late toxicities. Current conventional therapy remains suboptimal, as not all patients respond to available treatments, relapse remains a substantial risk, and many patients experience toxicity-related AEs. The clinical experts consulted for this review noted that unplanned inpatient admissions due to complications associated with current conventional therapy are common. Further, as ALL is the most common childhood cancer, the clinical expert with experience treating pediatric patients with ALL highlighted that decreasing relapse rates in ALL would have a significant impact on childhood cancer mortality rates and obviate the need for additional expensive and/or toxic therapies.
Place in Therapy
The clinical experts consulted for this review felt that blinatumomab would add an alternative first-line treatment option for patients with Ph-negative B-cell precursor ALL. The experts recommended using 1 to 4 cycles of blinatumomab in sequence with conventional consolidation chemotherapy. Compared to current treatment options, clinical experts noted that blinatumomab’s unique mechanism of action blocking CD19 antigen expressed on the leukemic blasts, has the potential to improve overall response rate, RFS, and OS, while decreasing AEs associated with the frequent use of intensive chemotherapy, potentially improving quality of life for patients with Ph-negative B-cell precursor ALL. If blinatumomab reduces the use of allogeneic SCT by preventing relapse, clinical experts noted that the risk of treatment-related mortality may also decrease.
Patient Population
The clinical experts consulted for this review noted any adult or pediatric patient with Ph-negative B-cell precursor ALL who achieves a CR following induction therapy, regardless of risk (i.e., MRD and pediatric risk groups) and age, would be best suited for first-line consolidation treatment with blinatumomab.
The clinical expert with experience treating pediatric patients noted that the sole exception may be to exclude pediatric patients from receiving blinatumomab if their expected event-free survival on a particular chemotherapy backbone is expected to be greater than 95% (i.e., those with SR-favourable risk category). However, experts flagged that the molecular characterization of leukemia is rapidly evolving, and the identification of a new high-risk somatic change may alter how patients are currently classified.
The experts felt that adults with Ph-negative B-cell precursor ALL who have a poor PS (i.e., ECOG PS greater than 2) should not be eligible for blinatumomab, while no PS restriction should be made in children, as blinatumomab has been shown to be well tolerated and potentially life-saving. Similar to the pivotal trial exclusion criteria, clinical experts consulted for this review felt there were insufficient data to recommend blinatumomab treatment in patients with acute undifferentiated leukemia and those with Burkitt leukemia.
As BCR-ABL reverse transcriptase-polymerase chain reaction molecular testing to determine Ph status and assessment of CR following induction therapy are standard clinical practice in Canada, clinical experts noted that no additional testing would be required before initiating blinatumomab treatment.
Assessing the Response to Treatment
Outcome metrics for Ph-negative B-cell precursor ALL used in clinical practice are typically aligned with those used in clinical trials. The clinical experts consulted for this review noted that treatment response in clinical practice and in clinical trials is based on bone marrow evaluation (including morphological evaluation and MRD assessment) at the end of induction and consolidation treatment. In both clinical practice and in clinical trials, MRD negativity is defined as less than 0.1% (or 0.01% if flow-based MRD testing can reach 10–4) in adults and less than 0.01% in children (except for pediatric patients with SR and DT, in whom it is defined as less than 0.1%). Clinical experts consulted for this review felt that definitions of relapse were well established and aligned across centres and clinicians and were similar between clinical practice and trials. RFS, DFS, OS, and other survival parameters, such as relapse incidence, are long-term parameters used to assess treatment response in both clinical trials and clinical practice. In both clinical practice and clinical trials, AEs are used to assess treatment benefit.
The clinical expert with experience treating adults with ALL noted that, following the completion of maintenance treatment, relapse is assessed every 3 months during the first year, every 6 months during the second year, and then annually between years 3 and 5.
The clinical expert with experience treating pediatric patients with ALL noted that, following the completion of maintenance treatment, relapse is assessed monthly for the first 3 months, every 3 months for the first year, every 4 months in the second year, every 6 months in the third year, and then annually for the rest of the patient’s life; however, follow-up practices vary by centre.
Discontinuing Treatment
The clinical trial, blinatumomab’s label, and the clinical experts consulted for this review noted that blinatumomab should be discontinued in the event of grade 4 cytokine release syndrome, grade 4 neurotoxicity and/or psychiatric events, and grade 4 thrombosis. Other grade 4 toxicities that are deemed clinically significant may require discontinuation of blinatumomab. Toxicities that require dose interruption of blinatumomab and that do not return to grade 1 or less by 14 days (or in the case of neurotoxicity, by 7 days) necessitate discontinuation of blinatumomab. Recurrent toxicities leading to dose interruption of blinatumomab also require permanent discontinuation of blinatumomab. One of the clinical experts consulted for this review also noted that patients who develop any debilitating conditions (such as ECOG PS of greater than 2) should discontinue blinatumomab treatment.
The clinical expert with experience treating adults noted that, while they were in agreement with these discontinuation criteria, the benefits of treatment, availability of alternative treatments, risks of discontinuation, and patients’ willingness to continue treatment must also be taken into account. The clinical expert with experience treating adults also noted that blinatumomab should be discontinued in the event of disease progression and relapse or upon the patient’s request.
Prescribing Considerations
Clinical experts indicated that treatment with blinatumomab should be initiated on an inpatient basis (3 days for the first infusion and 2 days for subsequent infusions); if it is well tolerated, subsequent infusions can be performed in an outpatient clinic.10 Given the pharmacy, nursing, and monitoring requirements for blinatumomab infusions, specialist care at an oncology centre is required. Both clinical experts consulted for this review felt that any conventional chemotherapy used for ALL treatment can be combined with blinatumomab. They also felt that clinicians should have the ability to adjust the dosage, number of cycles, and cycle length of blinatumomab based on toxicity and individual patient’s needs.
The clinical expert with experience treating adults noted that at least 2 cycles of blinatumomab should be given before SCT to deepen the patient’s remission. SCT would likely be required in patients in their second CR (regardless of MRD status) or in first CR with continuous MRD positivity and would be considered in patients in first CR with MRD-negative status after initially presenting with the highest risk disease for relapse.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Three clinician groups each submitted inputs to CDA-AMC: OH-CCO Hematology Cancer Drug Advisory Committee (with input from 6 clinicians); POGO (input from 11 clinicians); and CLSG-GCEL (input from 7 clinicians). One group provided input relevant to pediatric patients (POGO) and another for adults (CLSG-GCEL), while the third group did not specify an age group of patients represented.
The clinician groups were generally in agreement with the feedback received from the clinical experts consulted for this review. In terms of the goals of therapy, the clinical experts and clinician groups indicated that the main goals are cure of the disease while minimizing toxicity. Other goals include achieving CR, preventing relapse, and avoiding need for second-line therapies, such as allogenic SCT or CAR T-cell therapy. In pediatric patients, the POGO group stated that relapse therapies carry significant toxicity that leave young patients with a wide variety of potential lifelong late effects. In adults, the clinician groups noted the substantial risk of relapse, even in patients who are MRD-negative. Thus, additional treatments are needed to improve survival, CR and relapse rate, and to minimize or reduce toxicity of treatments (during consolidation and/or later therapies).
All clinician groups agreed that the patient population suitable to receive blinatumomab during consolidation include those newly diagnosed with Ph-negative CD19-positive B-cell precursor ALL, regardless of MRD status. Both the pediatric clinical expert consulted for this review and the POGO group indicated that patients who meet the SR-average or high-risk stratification criteria, have less favourable outcomes with chemotherapy alone and are expected to benefit from the addition of blinatumomab during consolidation therapy. Those who meet the SR-favourable stratification criteria have excellent response rates (event-free survival greater than 95%) with chemotherapy alone and may not require blinatumomab therapy. The POGO group noted an unmet need for augmentation therapies that have a tolerable adverse effect profile and may allow for “breaks” in therapy to facilitate recovery from cytotoxic-associated complications, such as fungal infections. According to input from POGO, patients with Ph-positive B-cell precursor ALL may also benefit from blinatumomab therapy. The clinician groups agree that standard response assessment for ALL would be relevant to monitoring the effects of blinatumomab, including MRD status, duration of response, relapse, and OS. The adult clinician groups noted the MRD threshold of 0.1% (≥ 103) is obsolete, and the current standard is greater than or equal to 0.01% (≥ 104).
The experts consulted and clinician groups agreed that blinatumomab would be discontinued if the patient experienced disease progression or significant toxicity, and that specialist care in an ALL treatment centre is warranted. The POGO group expressed that reimbursement strategies should account for all forms of drug wastage that may occur when pediatric doses are prepared.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
| This was a comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
| The clinical expert with experience treating adult patients with ALL noted that multiparameter flow cytometry is used to test MRD and has a sensitivity of up to 10–4. The clinical expert stated that not all centres in Canada can achieve a sensitivity of 10–4; however, at a minimum, most centres in Canada can achieve a sensitivity 10–3 and, as a consequence, MRD negativity should be defined as < 0.1%. |
| Clinical experts felt that all adult and pediatric patients who are eligible for conventional chemotherapy should also be eligible for blinatumomab reimbursement, regardless of age. The clinical expert with experience treating adults with ALL noted that patients with an ECOG performance status of 2 or lower are eligible for conventional chemotherapy, and a similar criterion should be used for determining blinatumomab eligibility in adults. The same clinical expert also felt that, across risk categories, there was sufficient evidence that blinatumomab consolidation treatment provides benefit and thus should be reimbursed without restriction to a specific risk. The clinical expert with experience treating children with ALL noted that, other than patients who present with very severe toxicities from leukemia, such as sepsis and multiorgan failure, all patients are eligible for conventional chemotherapy, and a similar criterion should be used for determining blinatumomab eligibility in pediatric patients. The same clinical expert noted that the 1 exception may be to exclude pediatric patients from receiving blinatumomab if their expected event-free survival on a conventional chemotherapy backbone is expected to be greater than 95% (i.e., those with SR-favourable status). However, the expert flagged that the molecular characterization of leukemia is rapidly evolving, and the identification of a new high-risk somatic change may alter how patients are currently classified. |
Considerations for discontinuation of therapy | |
Discontinuation of blinatumomab at consolidation was due to disease progression or intolerable toxicity. | This was a comment from the drug programs to inform pERC deliberations. |
Treatment is for 4 doses in consolidation for adults. Treatment is for 2 blocks of consolidation in pediatrics. | This was a comment from the drug programs to inform pERC deliberations. |
Considerations for prescribing of therapy | |
| This was a comment from the drug programs to inform pERC deliberations. |
Generalizability | |
| Both clinical experts consulted for this review felt that there was insufficient data to recommend blinatumomab treatment in patients with acute undifferentiated leukemia and those with Burkitt leukemia. The clinical expert with experience treating adults with ALL felt that adults with Ph-negative B-cell precursor ALL who have a poor performance status (i.e., ECOG greater than 2) should not be eligible for blinatumomab. In the pediatric trial (AALL1731), enrolment was not restricted by performance status. The clinical expert with experience treating children with ALL felt that no performance status restriction should be made in children, as blinatumomab has been shown to be well tolerated and potentially life-saving. |
Funding algorithm (oncology only) | |
Request an initiation of a rapid provisional funding algorithm. Note that, if the final reimbursement recommendation for this drug under review is “do not reimburse,” the project will be suspended indefinitely. | This was a comment from the drug programs to inform pERC deliberations. |
Drug may change place in therapy of drugs reimbursed in subsequent lines. | This was a comment from the drug programs to inform pERC deliberations. |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products | This was a comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
A single cycle of treatment is 28 days (4 weeks) of continuous infusion followed by a 14 day (2 week) treatment-free interval. Patients may receive up to 4 cycles of blinatumomab consolidation treatment (adults) or 2 blocks of blinatumomab (pediatric). Blinatumomab infusion bags should be admixed to infuse over 24 hours, 48 hours, 72 hours, 96 hours, or 7 days. Patients weighing greater than or equal to 45 kg receive a fixed dose, and, for patients weighing less than 45 kg, the dose is calculated using the patient’s body surface area. Dependent on drug preparation, more than a single vial may be required for a dose and there may be drug wastage. Patients will start therapy in hospital and then take the pump home. Patients will return to the facility for bag changes (duration varies based on jurisdiction procedures). | This was a comment from the drug programs to inform pERC deliberations. |
Blinatumomab is associated with adverse effects and will require monitoring by oncologist, nurse, and others. | This was a comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
Provision of blinatumomab for newly diagnosed patients with Ph-negative ALL may translate into substantial budget impact. | This was a comment from the drug programs to inform pERC deliberations. |
This therapy will be administered in centres that provide treatment for adult and pediatric patients with ALL and may not be available in all provinces and territories. Hospitalization is required for the first 3 days of the first cycle and the first 2 days of the second cycle. All subsequent cycles and reinitiation require supervision for the first 4 hours. Centres are familiar with the administration and preparation of blinatumomab. | This was a comment from the drug programs to inform pERC deliberations. |
If funded, the upfront cost will have significant budget impacts for the first couple of years, as jurisdictions treat newly diagnosed patients, as well as provide blinatumomab for those who have relapsed or refractory ALL (previously funded indications). | This was a comment from the drug programs to inform pERC deliberations. |
ALL = acute lymphoblastic leukemia; AUL = acute undifferentiated leukemia; COG = Children’s Oncology Group; ECOG = Eastern Cooperative Oncology Group; FISH = fluorescence in situ hybridization; MRD = minimal residual disease; PCR = polymerase chain reaction; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; Ph = Philadelphia chromosome; SR = standard risk.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of blinatumomab, 28 mcg/day for adult patients weighing 45 kg of more and 15 mcg/m2 per day for adult patients weighing less than 45 kg and pediatric patients, lyophilized powder for solution, IV infusion by infusion pump in the treatment of Ph-negative B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in the frontline setting. The focus will be placed on comparing blinatumomab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of blinatumomab is presented in this section, with the CDA-AMC critical appraisal of the evidence included at the end of the section. The systematic review section includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this section using the GRADE approach follows the critical appraisal of the evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 studies: 1 pivotal RCT in adult patients identified in the sponsor’s systematic review and a second RCT provided by the sponsor in a pediatric population of patients with Ph-negative B-cell precursor ALL.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 7.
Table 7: Details of Studies Included in the Systematic Review
Detail | E1910 trial | AALL1731 trial |
|---|---|---|
Designs and populations | ||
Study design | Phase III, randomized, controlled, open-label, multicentre, international trial | Phase III, randomized, controlled, open-label, multicentre, international trial |
Locations | A total of 77 centres in 3 countries (Canada [6 sites in Canada comprising 5.9% of patients], Israel [3.1% of patients], and the US [90.9% of patients]) | A total of 228 centres in 4 countries (Australia [4.1% of patients], Canada [16 sites including 6.8% of patients], New Zealand [1.7% of patients], and the US [87.4% of patients]) |
Patient enrolment dates | Start date: May 19, 2014 End date: October 18, 2019 | Start date: June 28, 2019 End date: June 30, 2024 |
Randomized (N) | Total: N = 286 randomized or assigned
| Total: N = 1,440 randomized
|
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Blinatumomab 28 mcg/day for patients weighing 45 kg or more and 15 mcg/m2 per day (not to exceed 28 mcg/day) for patients weighing less than 45 kg, by continuous IV infusion for a 28-day period per cycle, given after treatment intensification (2 cycles with 2-week interval in between) and during treatment consolidation (2 cycles) | Blinatumomab 15 mcg/m2 per day (not to exceed 28 mcg/day) by continuous IV infusion for a 28-day period for 2 nonsequential cycles added to chemotherapy |
Comparator(s) | Chemotherapy for each 28-day cycle during treatment consolidation (cycles 1, 2, and 4):
Chemotherapy for each 42-day cycle (cycle 3 only) during treatment consolidation:
| SR-average: Interim maintenance I (56 days):
Delayed intensification (56 days)
Interim maintenance II (56 days)
SR-high: Interim maintenance I (56 days):
Delayed intensification (63 days)
Interim maintenance II (56 days)
|
Study duration | ||
Treatment phase | Treatment induction:
Treatment intensification:
Treatment consolidation:
Treatment maintenance:
| Treatment induction:
Treatment consolidation:
Treatment maintenance:
|
Follow-up phase | All patients, including those who discontinue protocol therapy early, will be followed for 10 years from the start of treatment. | All patients, including those who discontinue protocol therapy early, will be followed for 10 years from study enrolment. |
Outcomes | ||
Primary end point | OS (measured as time from randomization until death due to any cause) in patients with MRD-negative disease |
|
Secondary, exploratory, and post hoc end points | Secondary:
Exploratory: Anti-blinatumomab antibody count Post hoc:
| Secondary:
Exploratory:
Post hoc:
|
Publication status | ||
Publications | Clinicaltrials.gov (2023)62 Litzow et al. (2024)63 Litzow et al. (2022)64 Litzow et al. (2023)65 Luger et al. (2023)66 | ClinicalTrials.gov (2024)67 Gupta et al. (2024)68 |
ALL = acute lymphoblastic leukemia; BCR = B-cell receptor; B-LLy = B-lymphoblastic lymphoma, CNS = central nervous system; CR = complete remission; CRi = complete remission with incomplete peripheral blood count recovery; DFS = disease-free survival; DS = Down syndrome; ECOG = Eastern Cooperative Oncology Group; EOI = end of induction; MRD = minimal residual disease; OS = overall survival; PS = performance status; RFS = relapse-free survival; SCT = stem cell transplant; SR = standard risk; TRM = treatment-related mortality.
aEnd-of-consolidation MRD assessed via flow cytometry.
Source: BLINCYTO E1910 Clinical Study Report, Gupta et al., and Clinicaltrials.gov.19,62,67,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The E1910 trial was a randomized, open-label, controlled, phase III study aimed at evaluating the efficacy and safety of blinatumomab for the treatment of adult patients with Ph-negative B-cell precursor ALL. The E1910 trial was sponsored by the National Cancer Institute (NCI) and administered by the Eastern Cooperative Oncology Group–American College of Radiology Imagine Network (ECOG-ACRIN Cancer Research Group). Patients with Ph-negative B-cell precursor ALL were recruited from 77 centres in the US, Israel, and Canada. All patients initially received induction chemotherapy for up to 3 months (defined as Step 1, Arm A), and those who achieved CR or CRi continued on study and received intensification treatment (defined as Step 2, Arm B; Figure 1). MRD status was then determined and, initially, patients with both MRD-negative and MRD-positive status were then randomized (Step 3) to receive either blinatumomab plus chemotherapy (Arm C) or chemotherapy (Arm D). Randomization was stratified by MRD status, age (30 to 54 years versus 55 years or older), CD20 status (positive versus negative), rituximab use (yes versus no), and intention to receive allogeneic SCT (yes versus no). Following a protocol amendment related to the FDA accelerated approval of blinatumomab therapy for patients with MRD-positive status (March 2018), these patients were assigned (rather than randomized) to blinatumomab plus chemotherapy (Arm C). Before the protocol amendment, 44 patients with MRD-positive status were randomized (22 of 286 [7.7%] to each treatment arm) and, following the protocol amendment, 18 (6.3%) patients were assigned to the blinatumomab plus chemotherapy arm. Patients in both treatment groups received maintenance therapy for 2.5 years from the start of the intensification phase (Step 4, Arm E). The chemotherapy regimen in the E1910 trial builds upon the UKALLXII/E2993 chemotherapy regimen, with dosing modifications based on C10403 AYA trial.69-71 Details of the regimen used in the E1910 trial are summarized in Table 9.
The primary objective of the E1910 trial was to compare the OS of patients treated with blinatumomab plus chemotherapy to chemotherapy in the FAS (i.e., patients with MRD-negative status following induction and intensification chemotherapy). Secondary objectives relevant to this review were to compare RFS of patients treated with blinatumomab plus chemotherapy to chemotherapy in the FAS, to compare OS and RFS of patients treated with blinatumomab plus chemotherapy to chemotherapy in the Step 3 MRD-positive analysis set, and to assess toxicities of blinatumomab and the chemotherapy regimen. Post hoc objectives were to compare OS and RFS of blinatumomab plus chemotherapy to chemotherapy in the Step 3 analysis set. The primary analysis of the E1910 trial results presented in this report are from the June 23, 2023, DCO date. The study design of the E1910 trial is depicted in Figure 1.
Similarly, the AALL1731 trial was a randomized, open-label, controlled, phase III study aimed at evaluating the efficacy and safety of blinatumomab for the treatment of pediatric patients with B-cell ALL. The AALL1731 trial was supported by grants from the NCI of the National Institutes of Health, St. Baldrick’s Foundation, and in part by Amgen. Pediatric patients with SR B-cell ALL were recruited from 228 centres in Australia, Canada, New Zealand, and the US. All patients initially received induction treatment before being stratified into 3 categories based on risk of relapse (SR-favourable, SR-average, and SR-high) and receiving consolidation treatment (Figure 2). Patients with average or high risk of relapse were randomized to either blinatumomab plus chemotherapy or chemotherapy arms. The 1:1 randomization was stratified according to risk groups (SR-average versus SR-high) and Down syndrome among those with SR-average (yes versus no). In both treatment groups of the AALL1731 trial, patients received 1 56-day cycle of interim maintenance II and then proceeded to receive 84-day cycles of maintenance therapy to continue for 2 years after the start of postconsolidation therapy. Patients classified as having high risk of relapse in the AALL1731 trial received an augmented Berlin-Frankfurt-Münster chemotherapy backbone, while those classified as having average risk of relapse received the standard therapy. Details of the regimen used in the AALL1731 trial are summarized in Figure 3 and Figure 4, Table 22, and Table 23.
In the AALL1731 trial, the primary end point was DFS and the primary objective was to determine whether the addition of blinatumomab (administered as nonsequential cycles) to conventional chemotherapy would improve DFS in all randomized patients with SR B-cell ALL (patients either had an average or high risk of relapse on the basis of clinical features). None of the secondary or exploratory objectives (Table 7) in the AALL1731 trial were relevant to this review. A post hoc objective was to determine whether the addition of blinatumomab to conventional chemotherapy would improve OS in all randomized patients.
The final analysis for DFS was planned when 194 DFS events were observed. Interim efficacy and futility monitoring were planned for when 20% (futility only), 40%, 60%, 80%, and 100% of the expected information (i.e., 194 DFS events) was observed, using the alpha-t2 spending function for efficacy monitoring and the futility monitoring method described by Anderson and High. Results were reviewed by the Data Safety Monitoring Committee. Details of the study design of the AALL1731 trial can be found in Figure 2. The results from the AALL1731 trial are from an interim analysis with a DCO of June 30, 2024.
At the planned interim analysis (June 30, 2024, DCO), the Data Safety Monitoring Committee recommended that the blinatumomab randomization be permanently closed based on the 3-year DFS estimates of 96.0% (SE = 1.2%) for the blinatumomab plus chemotherapy arm versus 87.6% (SE = 2.1%) for the chemotherapy alone arm. The associated HR was 0.39 (95% CI, 0.24 to 0.64), which exceeded the prespecified interim efficacy stopping criteria. (Data for the AALL1731 trial were sourced from published data only, and the exact stopping criteria were not publicly reported.)
The clinical expert with experience treating pediatric patients indicated that, while the nomenclature for treatment phases used in the AALL1731 trial differ from the historical 3-phase terminology (i.e., remission or induction, consolidation, and maintenance), the treatment phases during which blinatumomab is prescribed (i.e., consolidation, interim maintenance, and delayed intensification) can be viewed as the historical consolidation phase.”
Induction Phase
In both the E1910 and AALL1731 trials, all patients initially received induction chemotherapy.
In the E1910 trial, patients initially received 2 cycles of combination induction chemotherapy, with the addition of pegaspargase for patients younger than 55 years of age and the addition of rituximab for patients with CD20-positive ALL (Figure 1). The induction phase in the E1910 trial was defined as Step 1 and took up to 3 months. In the AALL1731 trial, a common 3-drug dexamethasone-based induction treatment was used. Patients with CNS-2 disease also received twice weekly cytarabine in addition to their initial dose (except on days 8 and 29, when methotrexate is administered) until 3 consecutive cerebrospinal samples were clear of blasts. At the end of induction, all eligible patients were stratified into 3 risk groups: SR-favourable, SR-average, and SR-high (Table 8).
Those with SR-average risk category with undetectable MRD assessed by high-throughput screening (HTS) were nonrandomly assigned to receive the AALL1731 trial chemotherapy, and their results are not reported here. Patients who had Down syndrome and in whom ALL was classified as SR-high were nonrandomly assigned to receive blinatumomab with lower toxicity chemotherapy, and their results are not reported here.
Intensification Phase
After the E1910 trial induction, patients in hematologic CR or CRi continued on study and received 1 intensification cycle of high-dose methotrexate with pegaspargase for CNS prophylaxis (Figure 1). In the E1910 trial, the intensification phase was defined as Step 2. Remission status was assessed after intensification treatment. CR was defined as neutrophil count of 1.0 × 109/ L or greater, platelet count of 100 × 109/L or greater, no leukemic blasts present in the peripheral blood, adequate bone marrow cellularity with trilineage hematopoiesis, 5% or less blasts in the bone marrow, and no extramedullary leukemia (e.g., CNS or soft tissue involvement). The definition of CRi is the same as CR with the exception of incomplete platelet recovery (i.e., platelets greater than 75 × 109/L and less than 100 × 109/L) or incomplete neutrophil count recovery (i.e., greater than 0.75 × 109/L and less than 1 × 109/L). MRD status was determined centrally by 6-colour flow cytometry, with MRD negativity defined as ≤ 1 × 10–4 (0.01%).
In the AALL1731 trial, patients proceeded directly to consolidation chemotherapy.
Table 8: Definitions of Cohorts in the AALL1731 Trial
Risk of relapse category | CNS stage | Favourable genetics | Neutral cytogeneticsa | Unfavourable cytogeneticsb | Peripheral blood MRD on day 8 | End-of-induction bone marrow MRD | End-of-induction IF-EM | |
|---|---|---|---|---|---|---|---|---|
ETV6-RUNX1 | DT | |||||||
SR-favourablec | 1, 2 | Yes to either | No | No | < 1% | < 0.01% | No | |
SR-average | 1, 2 | Yes to either | No | No | ≥ 1% | < 0.01% | No | |
1, 2d | Yes to either | No | No | < 1% | < 0.01% | No | ||
1, 2 | No | Yes | No | No | Any | ≥ 0.01 to < 0.1% | No | |
1 | No | No | Yes | No | Any | < 0.01% | No | |
SR-high | 1, 2 | Yes | No | No | No | Any | ≥ 0.01% | No |
1, 2 | No | Yes | No | No | Any | ≥ 0.1% | No | |
1 | No | No | Yes | No | Any | ≥ 0.01% | No | |
2 | No | No | Yes | No | Any | Any | No | |
1, 2 | No | No | No | Yes | Any | Any | No | |
1, 2 | Any | Any | Any | Any | Any | Any | Yes | |
CNS = central nervous system; DT = double trisomies 4 and 10; IF-EM = extramedullary disease induction failure; MRD = minimal residual disease; SR = standard risk.
aNeutral cytogenetics is defined as the absence of favourable and unfavourable cytogenetics.
bPresence of iAMP21, KMT2A-R (formerly MLL-R), hypodiploidy (modal chromosome number less than 44, DNA index < 0.81, or other clear evidence of a hypodiploid clone), or t(17;19).
cNo steroid pretreatment within 1 month before diagnosis.
dReceived steroid pretreatment within 1 month before diagnosis.
Source: Gupta et al.68
Consolidation Phase
Patients enrolled in the E1910 trial with MRD-negative status proceeded to randomization of consolidation treatment. Those with MRD-positive status initially underwent randomization. However, after the approval of blinatumomab therapy for patients with MRD-positive status, a protocol amendment assigned all subsequent patients with MRD-positive status to the blinatumomab group.
Those randomized to chemotherapy in the E1910 trial received three 28-day cycles of chemotherapy and one 42-day cycle of chemotherapy (Figure 1). Those randomized or assigned to the blinatumomab plus chemotherapy arm initially received two 28-day cycles of blinatumomab with a 2-week treatment-free break between the cycles, followed by chemotherapy (three 28-day cycles of chemotherapy and one 42-day cycle of chemotherapy), with one 28-day cycle of blinatumomab before the final chemotherapy cycle, and then 1 final 28-day cycle of blinatumomab. In the E1910 trial, the consolidation phase was defined as Step 3.
All remaining patients in the AALL1731 trial with SR-average and SR-high received a 28-day cycle of consolidation chemotherapy with slightly different regimens for those with SR-average and SR-high. Patients with SR-high after induction therapy who had MRD of 0.1% or greater were reassessed for MRD at the end of 56-day consolidation phase, which is common practice according to the clinical expert with experience treating pediatric patients. Patients with end-of-consolidation MRD of less than 0.1% were randomized, those with MRD of 0.1% to less than 1.0% (N = 14) were nonrandomly assigned to receive blinatumomab plus chemotherapy (results are not available), and those with MRD greater than 1% (N = 7) were removed from the trial. The clinical expert treating children agreed to generalize the results of patients with SR-high and MRD of less than 0.1% at the end of consolidation phase to patients with MRD of 0.1% to less than 1.0%. The clinical expert treating children noted that the patient population with SR-high and end-of-consolidation MRD of 1% or greater (N = 7) was removed from the trial, as these patients reflect a rare and very high–risk subgroup that is considered refractory and would be managed differently. Following consolidation treatment, all remaining patients were randomized to received two 28-day cycles of nonsequential blinatumomab in combination with chemotherapy or chemotherapy alone. Those assigned to the blinatumomab plus chemotherapy arm proceeded to receive 1 28-day cycle of blinatumomab followed by a 56-day interim maintenance cycle, another 28-day blinatumomab cycle, and then a 56-day (if SR-average) or 63-day (if SR-high) delayed intensification cycle. Those assigned to the chemotherapy arm proceeded directly to the first 56-day interim maintenance cycle, followed directly by the 56-day (if SR-average) or 63-day (if SR-high) delayed intensification cycle.
Maintenance Phase
In both studies, all patients received maintenance therapy regardless of the treatment group assignment.
In the E1910 trial, maintenance therapy continued for 2.5 years from the start of the intensification phase (Figure 1). In the E1910 trial, the maintenance phase was defined as Step 4.
In both treatment groups of the AALL1731 trial, patients received one 56-day cycle of interim maintenance II treatment and then proceeded to receive 84-day cycles of maintenance therapy to continue for 2 years after the start of postconsolidation therapy (defined as either the second cycle of blinatumomab or delayed intensification) (Figure 2).
Figure 1: Schematic of the E1910 Trial’s Study Design
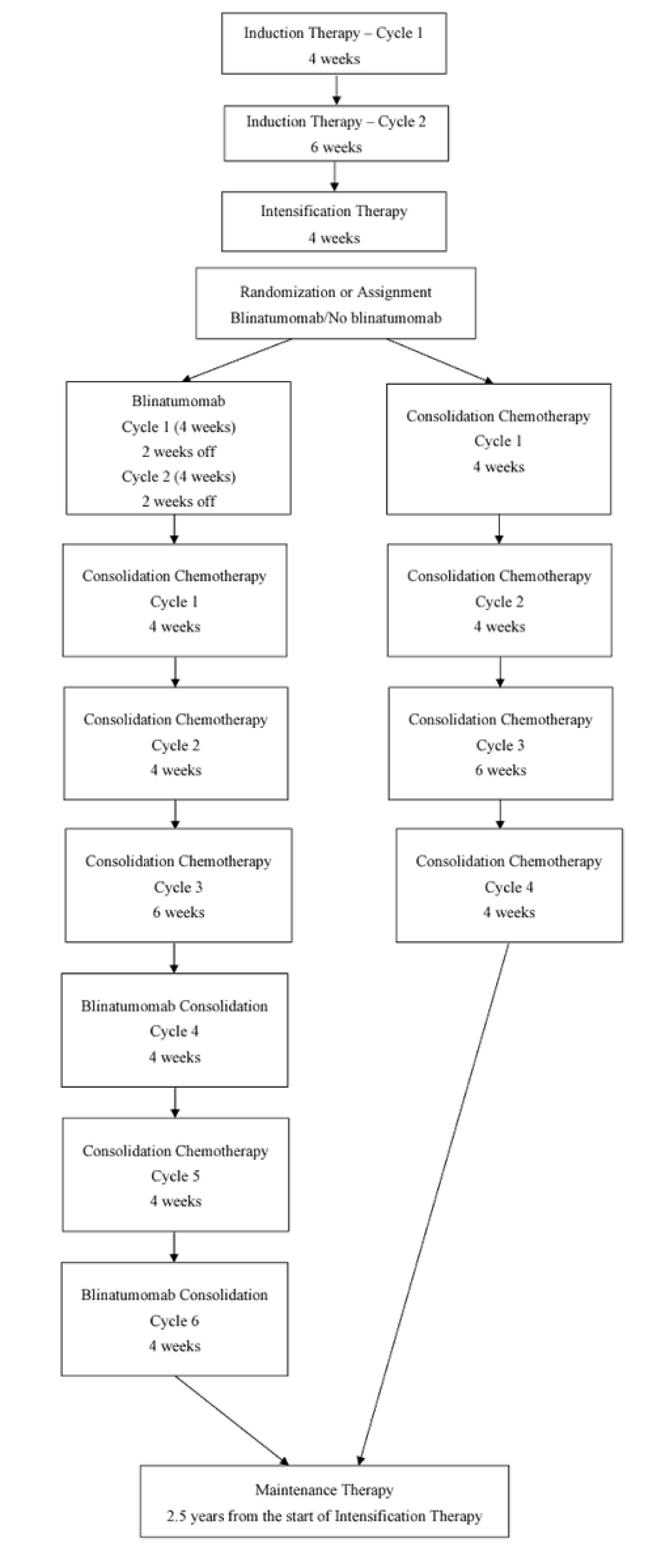
Note: In the E1910 trial, the induction phase was called Step 1, the intensification phase was called Step 2, the consolidation phase was called Step 3, and the maintenance phase was called Step 4.
Source: BLINCYTO Clinical Study Report.19
Figure 2: Schematic of the AALL1731 Trial’s Study Design
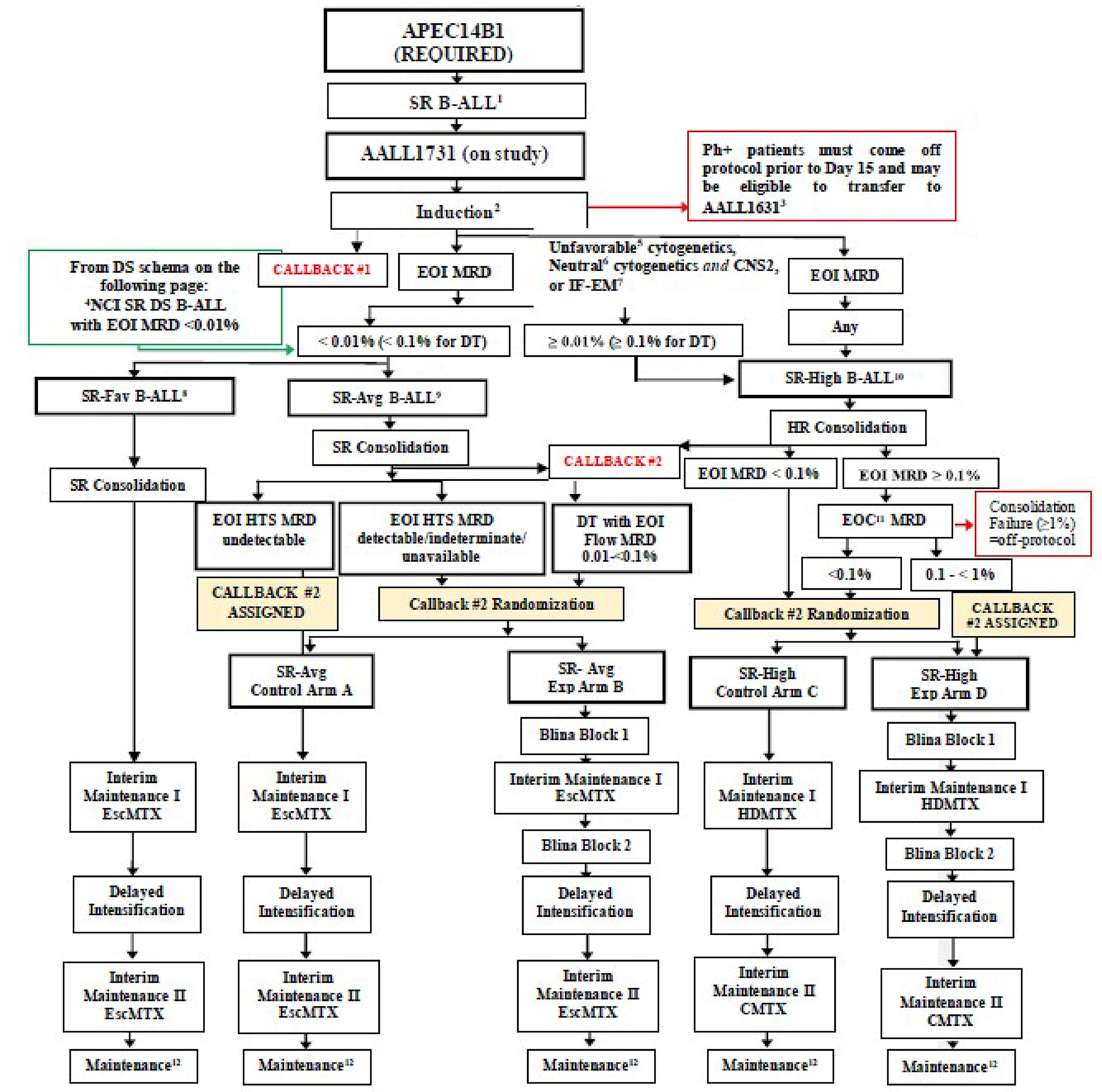
ALL = acute lymphoblastic leukemia; CMTX = Capizzi methotrexate; DS = Down syndrome; DT = double trisomy of chromosomes 4 and 10; EOC = end of consolidation; EOI = end of induction; EscMTX = escalating-dose methotrexate; Exp = experimental; HDMTX = high-dose methotrexate; HTS = high-throughput screening; MRD = minimal residual disease; NCI = National Cancer Institute; Ph = Philadelphia chromosome; SR-Avg = standard-risk average; SR-Fav = standard-risk favourable; SR-high = standard-risk high.
1NCI SR with steroid pretreatment, CNS-3, or testicular leukemia transfer to AALL1732 at beginning of induction.
2Four week, 3 drug induction.
3Patients with Ph-positive ALL go off-study before day 15 of induction and, if eligible, enrol on AALL1631.
4Patients with DS SR B-cell ALL lacking high-risk features and with EOI MRD < 0.01% receive treatment on the SR-Fav or SR-Avg B-cell ALL arm.
5Unfavourable cytogenetics include KMT2A-R, iAMP21, hypodiploidy, t(17;19).
6Neutral cytogenetics: lacking favourable and unfavourable cytogenetic features.
7Refer to the definition of extramedullary disease induction failure (IF-EM) in Section 3.3.5 of the AALL1731 protocol.
8SR-favourable: NCI SR (non-Down syndrome and Down syndrome) B-cell ALL, CNS-1 or CNS-2, with favourable cytogenetics (ETV6-RUNX1 fusion or DT), day 8 PB MRD < 1%, and end-of-induction bone marrow MRD < 0.01%.
9Meet features for therapy on the SR chemotherapy backbone.
10Meet features for therapy on the high-risk chemotherapy backbone.
11End-of-consolidation bone marrow MRD evaluation is required for patients with SR-high risk category with end-of-induction bone marrow MRD ≥ 0.1% and is recommended but not required for patients with SR-high risk category with end-of-induction bone marrow MRD 0.01% to 0.099%. Patients with treatment failure during the consolidation phase received off-protocol therapy.
12Timed from the start of the phase following consolidation phase for a total of 2 years for both females and males.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Populations
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria are provided in Table 7.
Briefly, in the E1910 trial, patients between the ages of 30 and 70 years with newly diagnosed Ph-negative B-cell precursor ALL and an ECOG PS of 0 to 3 were eligible for the induction phase (Step 1). New diagnoses of B-cell precursor ALL were based upon bone marrow or peripheral blood immunophenotyping and Ph-negativity was established by conventional cytogenetics, FISH, and/or polymerase chain reaction. Patients with Ph-positive ALL, Burkitt leukemia or lymphoma, mature B-cell leukemia, T-cell ALL, T-cell lymphoblastic lymphoma, or B-cell lymphoblastic lymphoma were ineligible. Patients were excluded if they had an antecedent hematologic disorder, history of recent myocardial infarction, or uncontrolled heart failure. Patients could not have a concurrent active malignancy for which they were receiving treatment, and those with pre-existing significant CNS pathology or uncontrollable seizure disorders were excluded. Following induction therapy, only those with an ECOG PS of 0 to 2, who achieved a CR or CRi and were CNS-negative for leukemia were eligible for postinduction intensification (Step 2). Patients eligible for consolidation treatment (Step 3) randomization or assignment to the blinatumomab plus chemotherapy or chemotherapy arm must have had an ECOG PS of 0 to 2, have maintained peripheral blood evidence of a remission, and have a CR or CRi.
In the AALL1731 trial, patients who were older than 1 year and younger than 10 years of age with newly diagnosed SR B-cell ALL and who were enrolled on Project: Every Child (APEC14B1) were eligible for enrolment. Newly diagnosed B-cell ALL was based on bone marrow samples or peripheral blood immunophenotyping. Those with acute undifferentiated leukemia, CNS-3 leukemia, or testicular leukemia were ineligible for enrolment. Treatment was discontinued in the event of Ph-positive identification. Data for patients who were enrolled in the AALL1731 trial and deemed eligible but did not undergo randomization (including those with Down syndrome and a high risk of relapse, or those with localized B-cell lymphoblastic lymphoma) are not reported here.
Interventions
In both the E1910 and AALL1731 trials, the main study intervention was blinatumomab administered as a continuous IV infusion over 28-day cycles.
In the E1910 trial, a fixed dose of 28 mcg/day of blinatumomab was administered after the completion of induction and intensification therapy for 4 cycles (2 cycles immediately following induction and intensification therapy, 1 cycle during the consolidation phase, and 1 cycle immediately following chemotherapy consolidation treatment; Table 9). Patients weighing less than 45 kg were given 15 mcg/m2 per day of blinatumomab at the same interval. In both treatment arms, patients received consolidation chemotherapy followed by maintenance therapy (Table 21). The chemotherapy regimen in the E1910 trial builds upon the UKALLXII/E2993 chemotherapy regimen with dosing modifications based on C10403 AYA trial.
In the AALL1731 trial, 15 mcg/m2 per day up to a maximum of 28 mcg/day of blinatumomab was administered after the completion of consolidation treatment for 2 cycles (1 cycle immediately following consolidation therapy and 1 cycle following interim maintenance I) (Figure 2). Each cycle was followed by 7 days of rest. In both treatment arms, patients received standard of care chemotherapy interim maintenance, delayed intensification, interim maintenance II, and maintenance therapy (Table 22 and Table 23).
In both studies, it was recommended that patients be hospitalized for at least the first 2 to 3 days of the first cycle of blinatumomab and the first 1 to 2 days of each subsequent cycle. However, the decision to hospitalize was at the discretion of the investigator. In the outpatient setting, either the patient returned to the study site for all changes of infusion bags or an ambulatory or home care service performed infusion bag changes in the patient’s home. Treatment continued until disease progression, relapse, treatment discontinuation, unacceptable toxicity, withdrawal of consent, or end of study treatment. In the E1910 trial, treatments were delayed if the absolute neutrophil count was less than 0.75 × 109/L and the platelet count was 75 × 109/L or less. Blinatumomab dose modifications were allowed in both studies in the event of AEs deemed to be possibly, probably, or definitely related to treatment. Generally, in the event of a grade 3 AE, treatment was stopped immediately and withheld until the AE resolved to grade 1 or lower. Treatment was discontinued in the event of grade 4 AEs.
In lieu of consolidation and maintenance treatment in the E1910 trial, patients could proceed to allogeneic SCT at the discretion of the treating physician (immediately following intensification treatment in the chemotherapy arm and immediately following the initial 2 cycles of blinatumomab for those in the blinatumomab plus chemotherapy arm).
In the E1910 trial, reasons for early removal from protocol treatment were categorized as:
treatment completed per protocol criteria
disease progression relapse during active treatment
AEs, side effects, or complications
death on study
patient withdrawal or refusal after beginning protocol therapy
alternative therapy
patient off treatment for other complicating disease
other.
In the AALL1731 trial, reasons for removal from protocol treatment included:
progressive disease at the end of Induction (B-lymphoblastic lymphoma [B-LLy] only)
consolidation failure (B-cell ALL only)
failure to achieve CR at the end of consolidation treatment (B-LLy only)
definitive relapse
identified as Ph-positive (BCR-ABL1)
incomplete induction phase data for risk stratification (B-cell ALL only)
patient found at any point after enrolment to be unevaluable (e.g., major deviations from protocol therapy, found to have MYC translocation associated with mature [Burkitt] B-cell ALL or B-LLy)
AE requiring removal from protocol therapy
refusal of further protocol therapy by patient, parent, or guardian
completion of planned therapy
physician determination that removal is in the patient’s best interest
development of a second malignancy
receiving radiation therapy other than protocol-defined cranial and testicular radiation for patients with Down syndrome.
Table 9: Regimens Used in the Consolidation Phase of the E1910 Trial
Study treatment | Unit dose strengths or dosage levels | Dosage formulation or administration frequency | Route of administration |
|---|---|---|---|
Blinatumomab (cycles 1 and 2, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
Consolidation therapy (cycles 1 and 2, 28-day cycle) | |||
Cytarabine | 75 mg/m2 | Day 1 to 5 | IV or SC injection |
Etoposide | 100 mg/m2 | Day 1 to 5 | IV injection |
Methotrexate | 12.5 mg | Day 1 (± 1) | IT injection |
Pegaspargase | 2,000 IU/m2 (1,000 IU/m2 if age ≥ 55 years; maximum 1 vial or 3,750 IU) | Day 5 (cycle 1 only) | IV or IM injection |
Rituximab | 375 mg/m2 | Day 5 (if CD20-positive) | IV injection |
Consolidation therapy (cycle 3, 42-day cycle) | |||
Daunorubicin | 25 mg/m2 | Day 1, 8, 15, 22 | IV injection |
Vincristine | 1.4 mg/m2 | Day 1, 8, 15, 22 | IV injection |
Dexamethasone | 10 mg/m2 (cap at 20 mg/day) | Day 1 to 7, 15 to 21 (days 1 to 7 only if age ≥ 55 years) | Oral |
Methotrexate | 12.5 mg | Day 2 (± 1) | IT injection |
Cyclophosphamide | 650 mg/m2 | Day 29 | IV injection |
Cytarabine | 75 mg/m2 | Day 30 to 33, 37 to 40 | IV or SC injection |
6-mercaptopurine | 60 mg/m2 | Day 29 to 42 | Oral |
Rituximab | 375 mg/m2 | Day 8 (optional if CD20-positive) | IV injection |
Blinatumomab consolidation therapy (cycle 4, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
Consolidation therapy (cycle 5, 28-day cycle) | |||
Cytarabine | 75 mg/m2 | Day 1 to 5 | IV or SC injection |
Etoposide | 100 mg/m2 | Day 1 to 5 | IV injection |
Methotrexate | 12.5 mg | Day 1 (± 1) | IT injection |
Rituximab | 375 mg/m2 | Day 8 (if CD20-positive) | IV injection |
Blinatumomab consolidation therapy (cycle 6, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
IM = intramuscular; IT = intrathecal; SC = subcutaneous.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 3: Regimens Used in the AALL1731 Trial in Patients With SR-Average
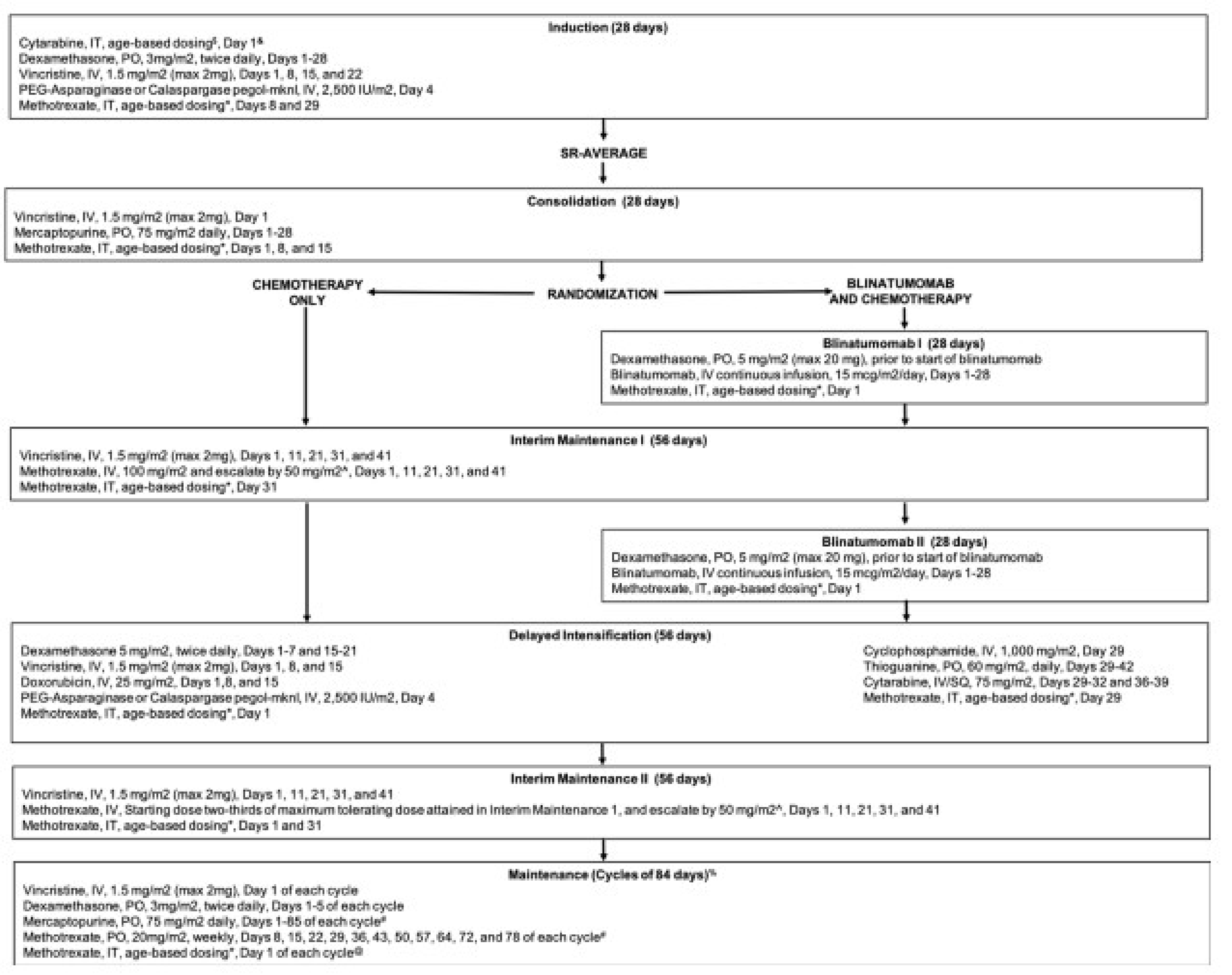
IT = intrathecal; max = maximum; PEG = pegaspargase; PO = orally; SR-avg = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse; SQ = subcutaneous.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Figure 4: Regimens Used in the AALL1731 Trial in Patients With SR-High
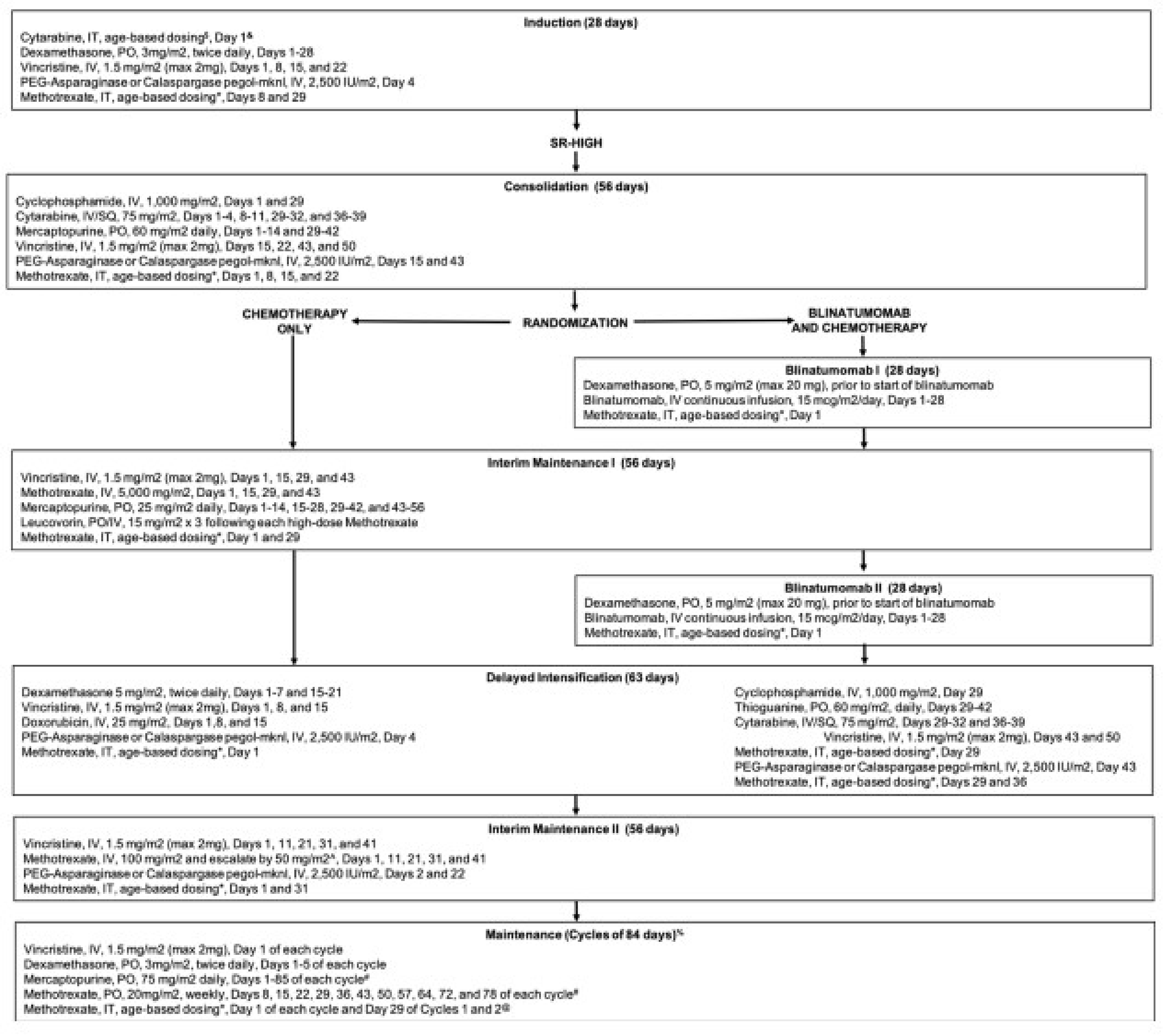
IT = intrathecal; max = maximum; PEG = pegaspargase; PO = orally; SR-avg = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse; SQ = subcutaneous.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 10, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Selected efficacy end points and notable harms outcomes considered important for informing expert committee deliberations were assessed using GRADE. The results of the pediatric trial were not appraised using GRADE, as only limited published results were provided. Additionally, the pediatric data became available closer to the end of the clinical review process, which precluded an evaluation using the GRADE framework. In summary, OS and RFS from the E1910 trial for the Step 3 analysis set and stratified by MRD status (i.e., FAS and Step 3 MRD-positive analysis set) and grade 3 and greater TEAEs, treatment-emergent SAEs, and fatal TEAEs from the E1910 trial’s Step 3 analysis set were assessed using GRADE.
Table 10: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | E1910 | AALL1731 | Outcome included in the CDA-AMC GRADE assessment |
|---|---|---|---|---|
OS in overall population | Time from reference date to latest DCOa and at 0.5, 1, 2, 3, 4, 5, 6, and 7 years in E1910 and 3 years in AALL1731 | Post hoc analysis | Post hoc analysis | At 3 and 5 years for E1910 |
OS in patients who are MRD-negative | Primary | NR | At 3 and 5 years for E1910 | |
OS in patients who are MRD-positive | Secondary | NR | At 3 and 5 years for E1910 | |
RFS in the overall population | Time from reference date to latest DCOa and at 0.5, 1, 2, 3, 4, 5, 6, and 7 years | Post hoc analysis | NR | At 1, 3, and 5 years for E1910 |
RFS in patients who are MRD-negative | Key secondary | NR | At 1, 3, and 5 years for E1910 | |
RFS in patients who are MRD-positive | Secondary | NR | At 1, 3, and 5 years for E1910 | |
DFS in overall population | Time from reference data to latest DCOa and at 3 years | NR | Primary | No |
Harms in overall population | Through to the latest DCOa | Secondary safety | Safety outcomes | GRADE ≥ 3 TEAE, treatment-emergent SAE, fatal TEAE for E1910 |
DCO = data cut-off; DFS = disease-free survival; MRD = minimal residual disease; NR = not reported; OS = overall survival; RFS = relapse-free survival; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: Statistical testing was not performed on any of the end points in the E1910 trial to adjust for multiple comparisons.
aPrimary analysis DCO in E1910 was on June 23, 2023, and in AALL1731 was on June 30, 2024.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the E1910 trial, treatment response was assessed before the start of each cycle of treatment (i.e., induction, intensification, consolidation, SCT), during the postintensification period, and after treatment every 3 months for the first 2 years from study entry, then every 6 months between 2 to 5 years from study entry, and once annually during 6 to 10 years following study entry. The frequency at which treatment response was assessed in the AALL1731 trial was not reported in documentation received by the CDA-AMC review team.
For survival outcomes in the E1910 trial, the reference date for analyses was defined as the randomization date for analyses of OS and RFS among patients who were MRD-negative and was either the randomization date (for those who were randomized before the protocol amendment) or registration date (for patients who were assigned to the blinatumomab plus chemotherapy arm following the protocol amendment) for patients who were MRD-positive. In the AALL1731 trial, the reference date was the end of the consolidation phase (i.e., when patients were randomized).
Overall Survival
In the E1910 trial, OS was defined as time from the reference date to death due to any cause. In the AALL1731 trial, OS was defined as the time from randomization to death or censoring at the date of last contact.
Clinical experts consulted for this review advised that an OS between-group difference of 5% in adults with Ph-negative B-cell precursor ALL and greater than 0% in the pediatric patients with Ph-negative B-cell precursor ALL is considered clinically meaningful. As a result, in the E1910 trial, a 5% between-group difference in OS in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set would be considered clinically meaningful differences. In the AALL1731 trial, a between-group difference of greater than 0% in OS in the overall cohort and subgroups stratified by risk of relapse (i.e., SR-average and SR-high) would be considered clinically meaningful.
Relapse-Free Survival
In the E1910 trial, RFS was defined as time from randomization to relapse or death due to any cause. Relapse was defined as the reappearance or persistence of blasts in the blood, presence of greater than 5% blasts (not attributable to another cause), or, in the case of isolated CNS relapse, the study chair was consulted and a bone marrow biopsy was performed to confirm the presence or lack of medullary relapse. Specimens were assessed centrally. RFS was not estimated in the AALL1731 trial.
Clinical experts consulted for this review advised an RFS between-group difference of 5% to 7% in the overall adult population with Ph-negative B-cell precursor ALL and 5% to 10% in the populations stratified by MRD status is considered clinically meaningful. In the E1910 trial, a 5% to 7% between-group difference in OS in the Step 3 analysis set and a 5% to 10% between-group difference in OS in the FAS and the Step 3 MRD-positive analysis set would be considered clinically meaningful differences.
Disease-Free Survival
In the AALL1731 trial, DFS was defined as the time from randomization to the date of relapse, a second malignant neoplasm, or death, whichever occurred first. Relapse included bone marrow relapse, CNS relapse, testicular relapse, or other extramedullary relapse. Specimens were assessed centrally. DFS was not estimated in the E1910 trial.
Clinical experts consulted for this review advised a DFS between-group difference of 3% to 5% in both the overall population and risk-of-relapse stratified populations is considered clinically meaningful. In the AALL1731 trial, a 3% to 5% between-group difference in DFS in the overall cohort and subgroups stratified by risk of relapse (i.e., SR-average and SR-high) would be considered clinically meaningful.
Harms End Points
To assess the toxicities of blinatumomab plus chemotherapy compared to chemotherapy, TEAEs, treatment-emergent SAEs, grade 3 and higher TEAE, fatal TEAE, and TEAEs of special interest were collected in the E1910 trial for all patients who received at least 1 dose of protocol-specified therapies.
SAEs were defined as an SAE requiring expedited reporting via the Cancer Therapy Evaluation Program Adverse Event Reporting System (CTEP AERS).
AEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review included:
cytokine release syndrome
tumour lysis syndrome (TLS)
neurologic toxicities, including immune effector cell-associated neurotoxicity syndrome
serious infections, including sepsis, pneumonia, Fusarium infection, fungal pneumonia, septic shock, Aspergillus infection, bronchopneumonia, Candida infection, enterococcal bacteremia, Escherichia sepsis, and lung infection
pancreatitis.
In the AALL1731 trial, AEs were assessed with the use of the NCI Common Terminology Criteria for Adverse Events (CTCAE), version 5.0. The occurrence of selected AEs of grade 3 or higher were summarized. No further details of how AEs were analyzed in the AALL1731 trial were available.
The estimated threshold at which clinical experts consulted for this review deemed fatal TEAEs to be clinically meaningful in both adults and pediatric patients with Ph-negative B-cell precursor ALL was greater than 0%. Thus, a between-group difference of greater than 0% fatal TEAEs in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set of the E1910 trial and in the overall cohort, the SR-average cohort, and the SR-high cohort of the AALL1731 trial would be considered clinically meaningful. For any grade 3 or greater TEAE, the estimated clinically significant threshold suggested by clinical experts consulted for this review was 10% and greater and, for treatment-emergent SAEs, it was 10% to 15%. A between-group difference of greater than 10% for grade 3 or greater TEAE and 10% to 15% in treatment-emergent SAEs in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set of the E1910 trial and in the overall cohort and subgroups stratified by risk of relapse (i.e., SR-average and SR-high) of the AALL1731 trial would be considered clinically meaningful.
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE include the following.
Survival outcomes identified by the patient and clinician group input and specified by the clinical experts consulted for this review were OS and RFS. OS was a key input for the pharmacoeconomic model and was deemed relevant by the clinical experts consulted for this review. For OS, the clinical experts noted that assessing survival at 3 years would allow enough events to have occurred while still showing any early benefit. Clinical experts noted that relapse events would be expected to occur before survival events. For both OS and RFS, a later time point with enough patients at risk are necessary to assess any sustained benefit. As such OS at year 3 and 5 and RFS at year 1, 3, and 5 in the E1910 trial were selected for GRADE. OS and RFS outcomes were assessed in the Step 3 analysis set (i.e., the MRD-agnostic population) to align with the unrestricted reimbursement request under review. OS and RFS in the FAS and the Step 3 MRD-positive analysis set were also assessed in GRADE to align with the primary and secondary outcomes of the E1910 trial and allow for any differences across populations to be highlighted.
Safety and toxicity outcomes were identified as important outcomes by patient and clinical group input. Any grade 3 or greater TEAEs, treatment-emergent SAEs, and fatal TEAEs were identified as the most important safety outcomes by the clinical experts consulted for this review. Safety and toxicity outcomes were only assessed in the Step 3 analysis set of the E1910 trial for GRADE, as this is the population that aligns with the reimbursement request under review, and clinical experts consulted for this review advised to prioritize this population.
Statistical Analysis
The statistical analyses for all study end points included in this review are presented in Table 11.
Table 11: Statistical Analysis of Efficacy End Points From Studies Included in the Systematic Review
End point | Statistical model | Adjustment factors | Handling of missing data | Additional analyses |
|---|---|---|---|---|
E1910 trial | ||||
OS | Primary analysis:
Subgroup analysis:
| Adjusted for sequential monitoring Stratification factors included age (30 to 54 vs. ≥ 55 years), CD20 status (positive vs. negative), rituximab use (yes vs. no), and whether patients intend to receive allogeneic SCT or not. |
| Supplemental analysis:
Sensitivity analyses:
Post hoc analysis in Step 3 analysis set:
|
RFS | Primary analysis:
Subgroup analysis:
| As for OS | As for OS | As for OS |
AALL1731 trial | ||||
DFS | Primary analysis:
| Stratification factors included: SR-average with and without Down syndrome, SR-average with Down syndrome, and SR-high. | The primary analysis was performed according to the intention-to-treat principle and thus included all eligible patients who had undergone randomization. On the basis of the previous COG ALL trial, it was not anticipated or planned for substantial or informative missingness. | Sensitivity analyses:
Post hoc analysis:
|
OS | NA | NA | As for DFS | Post hoc analysis:
|
ALL = acute lymphoblastic leukemia; CI = confidence interval; COG = Children’s Oncology Group; DCO = data cut-off; DFS = disease-free survival; FAS = full analysis set; HR = hazard ratio; KM = Kaplan-Meier; NA = not applicable; OS = overall survival; PPAS = per-protocol analysis set; RFS = relapse-free survival; RMST = restricted mean survival time; SCT = stem cell transplant; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Source: BLINCYTO Clinical Study Report,19 BLINCYTO Statistical Analysis Plan v3.0,72 and Gupta et al. (2024).68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The E1910 trial planned to enter a total of 488 patients with Ph-negative B-cell precursor ALL aged 30 to 70 years. It was assumed that a total of 190 (39% of the 488) patients would be MRD-negative and be randomized to receive either blinatumomab plus chemotherapy or chemotherapy alone. It was assumed the survival function of this patient population with ALL could be described by a cure rate model. For patients who were MRD-negative, a 35% long-term cure rate and 13-month median OS in the noncured group in the control arm were assumed. After adjustment for sequential monitoring with 190 patients who were MRD-negative, the study would have 80% power to detect a 45% reduction in hazard rate in the blinatumomab plus chemotherapy arm relative to the chemotherapy arm, using 1-sided log-rank test at the significance level of 0.025 (assuming 2 years of follow-up, which is equivalent to detecting an improvement in the 3-year OS rate from 45% to 64%). The number of events needed was 94.
In the AALL1731 trial, a 5-year baseline DFS rate of 90.1% was assumed (excluding patients with undetectable MRD by HTS). As a result, the AALL1731 trial planned to enrol between 6,420 and 6,720 patients to randomize approximately 2,245 patients and consequently to have about 81% power (2-sided alpha 5%, minimum follow-up of 3 years) to detect (via group sequential log-rank tests) an improvement in 5-year postconsolidation DFS from 90.1% to 93.4% (HR = 0.655; total expected events = 194). Interim DFS analyses were planned when 20% (for nonbinding futility only), 40%, 60%, and 80% of the anticipated 194 DFS events had occurred, with the use of alpha-t2 spending functions. A 1-sided P value of 0.025 was used for the interim analyses of DFS; all other analyses used 2-sided P values.
Statistical Test or Model
The preplanned comparison of OS between the E1910 trial treatment arms in the ITT population was conducted using the 1-sided stratified log-rank test with age (30 to 54 versus 55 or older years of age), CD20 status (positive versus negative), rituximab use (yes versus no), and whether patients intend to receive allogeneic SCT or not. Postrelapse systemic anticancer therapies were allowed at the discretion of the treating physician to reflect real-world clinical practice, with no restriction or control over their use and no adjustment in the OS analysis for their potential differential distribution between arms. Critical values for the primary OS comparison were determined using a truncated version of the Lan-DeMets error spending rate function, corresponding to the O’Brien-Fleming boundary, taking into account the errors spent at the interim efficacy analyses such that the overall 1-sided type I error is maintained at 0.025.
Time to events for OS and RFS in the E1910 trial were summarized with HRs using stratified Cox regression models adjusted for sequential monitoring and age (30 to 54 versus 55 or older years of age), CD20 status (positive versus negative), rituximab use (yes versus no), and whether patients intend to receive allogeneic SCT or not.
Additionally, in the E1910 trial, OS and RFS KM plots were presented by treatment arm. Summaries of the proportion alive and/or relapse-free at select time points, KM quartiles (when estimable), the number of patients with events, and the number of patients censored were presented.
While the primary analyses in the AALL1731 trial were stratified log-rank tests (stratified by SR-average with and without Down syndrome, SR-average with Down syndrome, and SR-high), descriptive DFS and OS rates were estimated using the KM method and SE and CIs estimated by the method of Peto. All survival time analyses assume a Weibull distribution with shape parameter of 0.6 (based on historical data) unless otherwise noted.
Multiple Testing Procedure
No multiple testing procedures were used in the E1910 or AALL1731 trials.
Data Imputation Methods
In the E1910 trial, missing or incomplete data for AEs (onset date for late AEs) and death were imputed. Date of AEs was imputed if at least the year of the event was known. The date of death was imputed if at least the year of death is known and if the known date of death is earlier than or the same as the date last known to be alive. If the date of death was fully missing, or if the last known date alive was later than the date of death, the patient’s survival time was censored.
Based on results of similar previous Children’s Oncology Group ALL trials, the AALL1731 trial did not anticipate or plan for substantial or informative missingness.
Post Hoc Analyses
In the E1910 trials, a post hoc analysis was performed comparing the OS and RFS of blinatumomab plus chemotherapy to chemotherapy alone in the Step 3 analysis set.
In the AALL1731 trial, post hoc analyses included evaluating the duration of OS, relapse (overall and according to site of relapse), and DFS in subgroups defined according to patient-related and disease-related factors (described in the Subgroup Analyses section).
Subgroup Analyses
The following prespecified subgroup analyses were performed in the E1910 trial:
gender (female or male)
race (groupings from original study) (American Indian or Alaska Native, Asian, Black, or African American, Native Hawaiian or Other Pacific Islander, or white)
ethnicity (Hispanic or Latino or not Hispanic or Latino)
age (≥ 18 years to < 35 years [in practicality, the lower bound of this bracket aligns with the enrolment criteria of ≥ 30 years], ≥ 35 years to < 55 years, ≥ 55 years to < 65 years, or ≥ 65 years).
In the AALL1731 trial, the DFS analysis was repeated among those with SR-average and SR-high risk category. Additionally, the following post hoc subgroup analyses of DFS were performed, but were not formally powered:
sex (female or male)
race or ethnic group (Hispanic, non-Hispanic Asian, non-Hispanic Black, non-Hispanic white, or other or unknown)
CNS status (CNS-1 [defined by no leukemic blasts as assessed by cytologic evaluation] or CNS-2 [defined by < 5 white cells per microlitre and the presence of blasts or traumatic lumbar puncture with the presence of blasts as assessed by cytologic evaluation but the absence of blasts as calculated by means of the Steinherz-Bleyer algorithm])
cytogenetic risk group (favourable, neutral, or unfavourable)
MRD in bone marrow on day 29, as assessed by multiparameter flow cytometry (less than 0.01% or greater than and equal to 0.01%)
MRD in bone marrow on day 29, as assessed by HTS (detectable less than 1 × 10–5 or detectable greater than and equal to 1 × 10–5).
Subgroup analyses in both the E1910 and AALL1731 trials are reported as supportive evidence.
Sensitivity Analyses
In the E1910 trial, sensitivity analyses on primary efficacy analyses (OS and RFS) in the population that was MRD-negative were repeated:
using the per-protocol analysis set (includes subjects in FAS excluding the following protocol deviations: noncompliant treatment administration at Step 3 and subjects randomized but ineligible for Step 3 treatment)
where OS or RFS is censored at the time of allogeneic SCT, using the FAS
where OS or RFS is censored at the time of allogeneic SCT or start of nonprotocol systemic anticancer therapy, whichever is earlier, using the FAS
RMST by Step 3 arm and difference in RMST with 95% CI will be presented using the FAS.
In the AALL1731 trial, a post hoc per-protocol sensitivity analysis that included only randomly assigned patients who started postconsolidation therapy was performed. In the event of strong evidence of nonproportionality of the DFS KM survival curves, a RMST sensitivity analysis was used with follow-up truncated at the time of the last follow-up visit. No other information on sensitivity analysis was available.
Analysis Populations
The analysis populations are defined in Table 12.
Table 12: Analysis Populations of the E1910 and AALL1731 Trials
Study | Population | Definition | Application |
|---|---|---|---|
E1910 trial | Full analysis set (FAS) | All Step 3 randomized patients who are assessed as MRD-negative centrally after induction and intensification chemotherapy | All efficacy analyses |
Step 3 analysis set | All Step 3 randomized or registered patients, regardless of MRD status at Step 3 | Post hoc efficacy analyses | |
Step 3 MRD-positive analysis set | All patients from Step 3 analysis set that are MRD-positive at Step 3 using the protocol-specified 10–4 cut-off | Secondary end point efficacy analyses | |
Step 3 safety analysis set | All patients in the Step 3 analysis set who take at least 1 dose of protocol-specified therapies | Safety analyses | |
Per-protocol analysis set (PPAS) | All patients in FAS except for those with the following protocol deviations:
| Sensitivity analyses | |
AALL1731 trial | Overall cohort | Patients with standard-risk B-cell ALL who had an average or high risk of relapse and underwent randomization (ITT sample) | All efficacy analyses |
SR-average | Patients with standard-risk B-cell ALL who had an average risk of relapse and underwent randomization | All efficacy analyses | |
SR-high | Patients with standard-risk B-cell ALL who had a high risk of relapse and underwent randomization | All efficacy analyses | |
Safety analysis set | Patients with standard-risk B-cell ALL who had an average or high risk of relapse, underwent randomization, started postconsolidation protocol therapy, and had data submitted | Safety analyses |
ALL = acute lymphoblastic leukemia; FAS = full analysis set; ITT = intention-to-treat; MRD = minimal residual disease; PPAS = per-protocol analysis set; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Source: BLINCYTO E1910 Clinical Study Report and Gupta et al. (2024).19,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Throughout the Results section, a summary of the E1910 trial is initially presented followed by a summary of the AALL1731 trial.
Unless otherwise specified, patient disposition results from the E1910 trial are presented among those included in the Step 3 analysis set; efficacy data are presented among those included in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set; and harm results are presented among the Step 3 safety analysis set.
In the AALL1731 trial, patient disposition is presented among those with SR-average and SR-high risk categories, efficacy data are presented among the overall cohort, and those with SR-average and SR-high risk categories, and harm results are presented among those with SR-average and SR-high risk categories who started postconsolidation protocol therapy.
Patient Disposition
Of 488 patients enrolled in the E1910 trial for induction therapy, 147 discontinued treatment primarily due to other reasons (n = 41) and disease progression relapse (n = 21), and 8 patients did not initiate treatment (Table 24). While 333 patients registered for intensification treatment, 2 patients did not initiate treatment and 45 discontinued treatment primarily due to disease progression relapse during active treatment (n = 12). As a result, 286 eligible patients were randomized or assigned to a treatment arm and included in the Step 3 analysis set (152 patients in the blinatumomab plus chemotherapy arm and 134 patients in the chemotherapy arm; Table 13). A higher percentage of patients in the blinatumomab plus chemotherapy arm completed Step 3 (consolidation) treatment (94 of 152 [61.8%]) compared to those in the chemotherapy arm (70 of 134 [52.2%]). The primary reason for discontinuation of consolidation treatment was due to AEs or complications in the blinatumomab plus chemotherapy arm (9.2%) and in the chemotherapy arm it was primarily due to disease progression or relapse during active treatment (15.7%). In both arms, the primary reason for discontinuing the study was due to death (19.7% in the blinatumomab plus chemotherapy arm and 39.6% in the chemotherapy arm).
In the AALL1731 trial, 4,264 patients with SR B-cell ALL were screened for enrolment and 246 patients were excluded before risk stratification at the end of induction (Table 24). An additional 2,578 patients were ineligible or excluded from randomization, ultimately resulting in 1,440 patients randomized to either blinatumomab plus chemotherapy (n = 718) or chemotherapy (n = 722). Among those randomized, 835 had SR-average risk category (n = 417 in the blinatumomab plus chemotherapy arm and n = 418 in the chemotherapy arm) and 605 had SR-high risk category (n = 301 in the blinatumomab plus chemotherapy arm and n = 304 in the chemotherapy arm). No data were provided on the percentage of patients completing or discontinuing the intervention treatment in the AALL1731 trial.
Table 13: Summary of Patient Disposition Postrandomization From Studies Included in the Systematic Review
Patient disposition | E1910 trial (Step 3 analysis set) | AALL1731 trial | ||||
|---|---|---|---|---|---|---|
Blinatumomab plus chemotherapy (N = 152) | Chemotherapy (N = 134) | SR-average (N = 835) | SR-high (N = 605) | |||
Blinatumomab plus chemotherapy (N = 417) | Chemotherapy (N = 418) | Blinatumomab plus chemotherapy (N = 301) | Chemotherapy (N = 304) | |||
Did not receive any Step 3 or consolidation treatment, n (%) | 5 (3.3) | 6 (4.5) | 55 | 27 | 20 | 13 |
Had definitive relapse | NR | NR | NR | 1 | NR | NR |
Were not evaluable | NR | NR | 16 | 17 | 6 | 8 |
Withdrawn | NR | NR | 35 | 3 | 11 | 3 |
Withdrew consent | NR | NR | 2 | NR | 1 | NR |
Pending start of therapy at DCO | NR | NR | 2 | 6 | 2 | 2 |
Completed treatment, n (%) | 94 (61.8) | 70 (52.2) | NR | NR | NR | NR |
Discontinued from treatment, n (%) | 53 (34.9) | 58 (43.3) | NR | NR | NR | NR |
Disease progression or relapse during active treatment | 9 (5.9) | 21 (15.7) | NR | NR | NR | NR |
Adverse events or complications | 14 (9.2) | 5 (3.7) | NR | NR | NR | NR |
Death on study | 6 (3.9) | 4 (3.0) | NR | NR | NR | NR |
Patient withdrawal or refusal before beginning protocol therapy | 8 (5.3) | 9 (6.7) | NR | NR | NR | NR |
Alternative therapy | 5 (3.3) | 4 (3.0) | NR | NR | NR | NR |
Patient off treatment for other complicating disease | 3 (2.0) | 2 (1.5) | NR | NR | NR | NR |
Other | 8 (5.3) | 13 (9.7) | NR | NR | NR | NR |
Discontinued from study, n (%) | 37 (24.3) | 63 (47.0) | NR | NR | NR | NR |
Consent withdrawn | 6 (3.9) | 7 (5.2) | NR | NR | NR | NR |
Lost to follow-up | 1 (0.7) | 3 (2.2) | NR | NR | NR | NR |
Death | 30 (19.7) | 53 (39.6) | NR | NR | NR | NR |
Induction or intensification safety analysis set, N | 480 | NA | NA | NR | NR | |
FAS, N | 112 | 112 | NR | NR | NR | NR |
Safety analysis set, N | 111 | 112 | NR | NR | NR | NR |
Per-protocol analysis set, N | 72 | 73 | NR | NR | NR | NR |
Step 3 analysis set, N | 152 | 134 | NR | NR | NR | NR |
SR-average | NA | NA | 417 | 418 | NA | NA |
SR-high | NA | NA | NA | NA | 301 | 304 |
Step 3 safety analysis set, N | 147 | 128 | NA | NA | NR | NR |
Blinatumomab safety analysis set, N | 147 | 0 | NA | NA | NR | NR |
Step 3 MRD-positive analysis set, N | 40 | 22 | NA | NA | NR | NR |
DCO = data cut-off; FAS = full analysis set; MRD = minimal residual disease; NA = not applicable; NR = not reported; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Source: BLINCYTO E1910 Clinical Study Report and Gupta et al. (2024).19,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics for the Step 3 analysis set are outlined in Table 14 and are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Additional baseline characteristics from the E1910 trial that were deemed to be less clinically relevant by the clinical experts consulted for this review are presented in Table 25 for completeness.
The baseline demographics and characteristics in the E1910 trial were generally well balanced between the 2 treatment arms. For the blinatumomab plus chemotherapy arm (N = 152) and chemotherapy arm (N = 134), 45.4% and 52.2% of patients, respectively, were male (versus 54.6% and 47.8% were female, respectively), and the median age at enrolment was 51 years (range, 30 to 69 years) and 50.5 years (range, 30 to 70 years), respectively. The majority of patients were younger than 55 years (60.5% in the blinatumomab plus chemotherapy arm and 56.7% in the chemotherapy arm). Fewer patients in the blinatumomab plus chemotherapy arm were aged 65 years or older (7.9%) than in the chemotherapy arm (14.9%). The majority of patients were white (77.0% in the blinatumomab plus chemotherapy arm and 82.1% in the chemotherapy arm) and were not of Hispanic or Latino ethnicity (82.2% in the blinatumomab plus chemotherapy arm and 82.8% in the chemotherapy arm). Most patients had an ECOG PS of 0 (37.5% and 36.6%, respectively) or 1 (57.2% and 60.4%, respectively), and the primary country of residence was the US (92.1% and 89.6%, respectively), with relatively few patients coming from Canada (6.6% and 5.2%, respectively). Baseline characteristics stratified by MRD status were generally similar between treatment arms for patients who were MRD-negative but not for those who were MRD-positive (Table 26).
In both those with SR-average and SR-high risk categories, the baseline demographics and characteristics in the AALL1731 trial were generally well balanced between the 2 treatment arms. Slightly fewer patients were female (range from 45.1% [137 of 304] in the SR-high chemotherapy arm to 49.6% [207 of 417] in the SR-average blinatumomab plus chemotherapy arm) than male, and median age ranged from 4.0 years (range, 1.0 to 9.9 years) in the SR-average blinatumomab plus chemotherapy arm to 4.6 years (range, 1.0 to 10.0 years) in the SR-high blinatumomab plus chemotherapy arm. The majority of patients were non-Hispanic white (range from 46.1% [140 of 304] in the SR-high chemotherapy arm to 51.8% [156 of 301] in the SR-high blinatumomab plus chemotherapy arm). Just 3.8% (32 of 835) of the SR-average cohort had Down syndrome, while patients with SR-high risk category who had Down syndrome were not included in the randomized population. Patients were primarily from the US (87.4% [730 of 835] in the SR-average cohort and 87.4% [529 of 605] in the SR-high cohort), with just 5.6% and 6.8% of patients in the SR-average and SR-high cohorts, respectively, coming from Canada.
Table 14: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | E1910 trial (Step 3 analysis set) | AALL1731 trial | ||||
|---|---|---|---|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | SR-average | SR-high | |||
Blinatumomab plus chemotherapy | Chemotherapy | Blinatumomab plus chemotherapy | Chemotherapy | |||
Sex, n (%) | ||||||
Female | 83 (54.6) | 64 (47.8) | 207 (49.6) | 195 (46.7) | 143 (47.5) | 137 (45.1) |
Male | 69 (45.4) | 70 (52.2) | 210 (50.4) | 223 (53.3) | 158 (52.5) | 167 (54.9) |
Ethnicity, n (%) | ||||||
Hispanic or Latino | 21 (13.8) | 15 (11.2) | NR | NR | NR | NR |
Not Hispanic or Latino | 125 (82.2) | 111 (82.8) | NR | NR | NR | NR |
Not reported | 2 (1.3) | 3 (2.2) | NR | NR | NR | NR |
Unknown | 4 (2.6) | 5 (3.7) | NR | NR | NR | NR |
Race, n (%) | ||||||
American Indian or Alaska Native | 2 (1.3) | 1 (0.7) | NR | NR | NR | NR |
Asian | 4 (2.6) | 2 (1.5) | NR | NR | NR | NR |
Black or African American | 12 (7.9) | 5 (3.7) | NR | NR | NR | NR |
Native Hawaiian or other Pacific Islander | 1 (0.7) | 0 (0.0) | NR | NR | NR | NR |
White | 117 (77.0) | 110 (82.1) | NR | NR | NR | NR |
Not reported | 7 (4.6) | 6 (4.5) | NR | NR | NR | NR |
Unknown or other | 9 (5.9) | 10 (7.5) | NR | NR | NR | NR |
Race or ethnic group, n (%) | ||||||
Hispanic | NR | NR | 100 (24.0) | 104 (24.9) | 84 (27.9) | 84 (27.6) |
Non-Hispanic Asian | NR | NR | 20 (4.8) | 19 (4.5) | 13 (4.3) | 10 (3.3) |
Non-Hispanic Black | NR | NR | 26 (6.2) | 20 (4.8) | 16 (5.3) | 18 (5.9) |
Non-Hispanic white | NR | NR | 217 (52.0) | 213 (51.0) | 156 (51.8) | 140 (46.1) |
Other or unknown | NR | NR | 54 (12.9) | 62 (14.8) | 32 (10.6) | 52 (17.1) |
Age at enrolment (years) | ||||||
Mean (SD) | 49.6 (11.0) | 50.2 (12.0) | NR | NR | NR | NR |
Median (range) | 51.0 (30 to 69) | 50.5 (30 to 70) | 4.0 (1.0 to 9.9) | 4.3 (1.0 to 10.0) | 4.6 (1.0 to 10.0) | 4.2 (1.1 to 9.9) |
IQR | 41.0 to 58.5 | 40.0 to 61.0 | NR | NR | NR | NR |
Age group, n (%) | ||||||
≥ 18 and < 35 years | 18 (11.8) | 19 (14.2) | NR | NR | NR | NR |
≥ 35 and < 55 years | 74 (48.7) | 57 (42.5) | NR | NR | NR | NR |
< 55 years | 92 (60.5) | 76 (56.7) | NR | NR | NR | NR |
≥ 55 and < 65 years | 48 (31.6) | 38 (28.4) | NR | NR | NR | NR |
≥ 55 years | 60 (39.5) | 58 (43.3) | NR | NR | NR | NR |
≥ 65 years | 12 (7.9) | 20 (14.9) | NR | NR | NR | NR |
Country of residence, n (%) | ||||||
Australia | NA | NA | 25 (6.0) | 24 (5.7) | 10 (3.3) | 15 (4.9) |
Canada | 10 (6.6) | 7 (5.2) | 20 (4.8) | 27 (6.5) | 18 (6.0) | 23 (7.6) |
New Zealand | NA | NA | 4 (1.0) | 5 (1.2) | 4 (1.3) | 6 (2.0) |
US | 140 (92.1) | 120 (89.6) | 368 (88.2) | 362 (86.6) | 269 (89.4) | 260 (85.5) |
Israel | 2 (1.3) | 7 (5.2) | NA | NA | NA | NA |
Down syndrome, n (%) | ||||||
Yes | NR | NR | 16 (3.8) | 16 (3.8) | NA | NA |
No | NR | NR | 401 (96.2) | 402 (96.2) | 301 (100.0) | 304 (100.0) |
ECOG PS, n (%) | ||||||
0 | 57 (37.5) | 49 (36.6) | NR | NR | NR | NR |
1 | 87 (57.2) | 81 (60.4) | NR | NR | NR | NR |
2 | 8 (5.3) | 4 (3.0) | NR | NR | NR | NR |
3 | 0 (0.0) | 0 (0.0) | NR | NR | NR | NR |
4 | 0 (0.0) | 0 (0.0) | NR | NR | NR | NR |
White cell count (× 10−9/L), median (range) | NR | NR | 7.7 (0.3 to 49.7) | 7.5 (0.0 to 49.7) | 8.8 (0.4 to 47.8) | 7.4 (0.6 to 49.8) |
CNS status, n (%)a | ||||||
CNS-1 | NR | NR | 399 (95.7) | 407 (97.4) | 242 (80.4) | 245 (80.6) |
CNS-2 | NR | NR | 18 (4.3) | 11 (2.6) | 59 (19.6) | 59 (19.4) |
Cytogenetic risk group, n (%) | ||||||
Favourable | NR | NR | 127 (30.5) | 114 (27.3) | 72 (23.9) | 96 (31.6) |
Neutral | NR | NR | 290 (69.5) | 304 (72.7) | 165 (54.8) | 148 (48.7) |
Unfavourable | NR | NR | NA | NA | 64 (21.3) | 60 (19.7) |
Bone marrow biopsy results, n (%) | ||||||
Involved | 10 (6.6) | 1 (0.7) | NR | NR | NR | NR |
Not involved | 137 (90.1) | 269 (94.1) | NR | NR | NR | NR |
Indeterminate | 5 (3.3) | 1 (0.7) | NR | NR | NR | NR |
Bone marrow cellularity, % | ||||||
N | 151 | 134 | ||||
Mean (SD) | 43.6 (19.8) | 41.4 (18.4) | NR | NR | NR | NR |
Median (IQR) | 40.0 (30.0 to 60.0) | 40.0 (30.0 to 50.0) | NR | NR | NR | NR |
Minimum to maximum | 0.0 to 100.0 | 10.0 to 90.0 | NR | NR | NR | NR |
Bone marrow blasts, % | ||||||
N | 152 | 134 | NR | NR | NR | NR |
Mean (SD) | 1.3 (1.1) | 1.3 (1.3) | NR | NR | NR | NR |
Median (IQR) | 1.0 (0.0 to 2.0) | 1.0 (0.0 to 2.0) | NR | NR | NR | NR |
Minimum to maximum | 0 to 5 | 0 to 6 | NR | NR | NR | NR |
MRD in peripheral blood on day 8 (per MPFC), n (%) | ||||||
< 1% | NR | NR | 242 (58.0) | 271 (64.8) | 177 (58.8) | 196 (64.5) |
≥ 1% | NR | NR | 173 (41.5) | 147 (35.2) | 122 (40.5) | 107 (35.2) |
Unknown | NR | NR | 2 (0.5) | 0 | 2 (0.7) | 1 (0.3) |
MRD in bone marrow on day 29 (per MPFC), n (%) | ||||||
< 0.01% | NR | NR | 375 (89.9) | 374 (89.5) | 75 (24.9) | 78 (25.7) |
0.01% to < 1.00% | NR | NR | 42 (10.1) | 44 (10.5) | 195 (64.8) | 192 (63.2) |
≥ 1.00% | NR | NR | NA | NA | 31 (10.3) | 34 (11.2) |
MRD in bone marrow on day 29 (per HTS), n (%)b | ||||||
Detectable, ≥ 1 × 10−5 | NR | NR | 151 (40.3) | 154 (41.2) | NA | NA |
Detectable, < 1 × 10−5 | NR | NR | 201 (53.6) | 196 (52.4) | NA | NA |
Indeterminate | NR | NR | 21 (5.6) | 17 (4.5) | NA | NA |
Unavailable | NR | NR | 2 (0.5) | 7 (1.9) | NA | NA |
CNS = central nervous system; ECOG = Eastern Cooperative Oncology Group; IQR = interquartile range; HTS = high-throughput sequencing; MPFC = multiparameter flow cytometry; MRD = minimal residual disease; NA = not applicable; NR = not reported; PS = performance status; SD = standard deviation; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Notes: Step 3 analysis set includes all Step 3 randomized or registered patients.
MRD-positive before Step 3: MRD value ≥ 0.01%; MRD-negative before Step 3: MRD value ≤ 0.01%.
Baseline is the last assessment taken on or before the date of first protocol-specified therapy administration for Step 3. If no protocol-specified therapy is administered for Step 3, then baseline is the last assessment taken on or before the date of randomization or Step 3 registration. For baseline, body surface area and weight consider the latest nonmissing assessment taken before postrandomization or Step 3 registration.
DCO date: June 23, 2023.
aCNS-1 was defined by no leukemic blasts, as assessed by cytologic evaluation. CNS-2 was defined by fewer than 5 white cells per microlitre and the presence of blasts or traumatic lumbar puncture with the presence of blasts as assessed by cytologic evaluation but the absence of blasts as calculated by means of the Steinherz-Bleyer algorithm.
bPatients with DT and MRD of 0.01% to less than 0.1% in the bone marrow at the end of induction therapy as assessed by multiparameter flow cytometry were categorized as having SR B-cell ALL with an average risk of relapse but did not undergo MRD testing by HTS.
Source: BLINCYTO E1910 Clinical Study Report and Gupta et al. (2024).19,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
No data on exposure to treatment in the AALL1731 trial were provided.
Exposure to Protocol Treatments
Of the 147 patients in the Step 3 safety analysis set of the E1910 trial who received blinatumomab plus chemotherapy, 37 patients (25.2%) received on-protocol allogeneic SCT compared to 28 of 128 (21.9%) in the chemotherapy arm (Table 15). All patients in the chemotherapy arm had observed data for exposure to consolidation chemotherapy compared to just 92 patients in the blinatumomab plus chemotherapy arm. A similar number of patients in both treatment arms had observed data for exposure to maintenance chemotherapy (n = 78 and n = 71, respectively). The relative dose intensity of consolidation and maintenance chemotherapy was similar in both treatment arms.
In the E1910 trial, all 147 patients in the blinatumomab plus chemotherapy arm received at least 1 dose of blinatumomab. The median relative dose intensity of 0.84 (IQR 0.73 to 0.99) indicates that more than three-quarters of patients received lower doses of blinatumomab than planned.
Table 15: Summary of Patient Exposure to Treatments From Studies Included in the Systematic Review
Protocol treatments | E1910 trial (Step 3 safety analysis set) | |
|---|---|---|
Blinatumomab plus chemotherapy (N = 147) | Chemotherapy (N = 128) | |
Receipt of on-protocol allogeneic SCT, n (%) | 37 (25.2) | 28 (21.9) |
RDI of blinatumomaba | ||
N | 147 | NA |
Mean (SD) | 0.80 (0.24) | NA |
Median (IQR) | 0.84 (0.73 to 0.99) | NA |
Minimum to maximum | 0.2 to 1.5 | NA |
RDI chemotherapy during consolidationa,b | ||
N | 92 | 128 |
Mean (SD) | 0.91 (0.12) | 0.86 (0.27) |
Median (IQR) | 0.93 (0.87 to 0.99) | 0.98 (0.81 to 1.00) |
Minimum to maximum | 0.4 to 1.1 | 0.0 to 1.2 |
RDI chemotherapy during maintenancea,b | ||
n | 78 | 71 |
Mean (SD) | 0.84 (0.19) | 0.86 (0.18) |
Median (IQR) | 0.84 (0.75 to 0.93) | 0.86 (0.77 to 0.97) |
Minimum to maximum | 0.3 to 1.3 | 0.1 to 1.2 |
IQR = interquartile range; NA = not applicable; RDI = relative dose intensity; SCT = stem cell transplant; SD = standard deviation.
Note: DCO date: June 23, 2023.
aRegimen-specific relative dose intensity is calculated as (actual dose intensity/planned dose intensity).
bRelative dose intensity is reported for all chemotherapy regimens used during the phase of treatment.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Nonprotocol Treatments
By the June 23, 2023, DCO of the E1910 trial, a similar percentage of patients in both arms of the E1910 trial received nonprotocol therapy for ALL before a protocol-defined relapse (4 of 152 [2.6%] in the blinatumomab plus chemotherapy and 6 of 134 [4.5%] in the chemotherapy arm), with the most common type being nonprotocol chemotherapy in both arms (1.3% and 2.2%, respectively; Table 16).
Table 16: Summary of Exposure to Nonprotocol Therapy for ALL From Studies Included in the Systematic Review
Nonprotocol therapy | E1910 trial (Step 3 analysis set) | |
|---|---|---|
Blinatumomab plus chemotherapy (N = 152) | Chemotherapy (N = 134) | |
Patients receiving nonprotocol therapy for ALL (only if treatment started before protocol-defined relapse), n (%) | 4 (2.6) | 6 (4.5) |
Chemotherapy | 2 (1.3) | 3 (2.2) |
Hormonal therapy | 0 (0.0) | 1 (0.7) |
Immunotherapy | 0 (0.0) | 1 (0.7) |
High-dose chemotherapy or stem cell transplant (autologous or allogeneic) | 1 (0.7) | 3 (2.2) |
Surgery | 1 (0.7) | 0 (0.0) |
Radiation therapy | 0 (0.0) | 0 (0.0) |
Other therapy | 0 (0.0) | 0 (0.0) |
ALL = acute lymphoblastic leukemia.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Use of Subsequent Therapies
Following a protocol-defined relapse in the E1910 trial, a higher percentage of patients in the chemotherapy arm received off-protocol allogeneic SCT (12 of 134 [12.7%] versus 12 of 152 [7.9%] in the blinatumomab plus chemotherapy arm) and off-protocol blinatumomab (25.4% versus 0.0% in the blinatumomab plus chemotherapy arm; Table 17). Receipt of CAR T-cell therapy was █████████████ in the blinatumomab plus chemotherapy arm (████) than in the chemotherapy arm (████).
Table 17: Summary of Exposure to Off-Protocol Therapy for ALL From Studies Included in the Systematic Review
Off-protocol therapya | E1910 trial (Step 3 analysis set) | |
|---|---|---|
Blinatumomab plus chemotherapy (N = 152) | Chemotherapy (N = 134) | |
Patients receiving off-protocol allogeneic SCT, n (%) | 12 (7.9) | 17 (12.7) |
Patients receiving off-protocol blinatumomab, n (%) | 0 (0.0) | 34 (25.4) |
Patients receiving CAR T-cell therapy, n (%)b | ██████ | | █████ |
ALL = acute lymphoblastic leukemia; CAR = chimeric antigen receptor; SCT = stem cell transplantation.
aSponsor clarified off-protocol therapies were those given following a protocol-defined relapse.
bCAR T-cell therapy information was collected from sites in the first quarter of 2024 in response to an FDA query.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Efficacy Results From the E1910 Trial
OS in the Step 3 Analysis Set
OS results for the Step 3 analysis set of the E1910 trial at the June 23, 2023, DCO are summarized in Table 18. The median follow-up time for OS was 4.5 years for both treatment arms, and the median OS was NE in either arm. The OS stratified HR was 0.47 (95% CI, 0.30 to 0.74, P < 0.001) favouring the blinatumomab plus chemotherapy arm. The difference in survival probabilities for blinatumomab plus chemotherapy versus chemotherapy was 16.9% in year 1 and 20.8% in year 5. The KM plot for OS in the Step 3 analysis set is presented in Figure 5.
Figure 5: Kaplan-Meier Analysis for OS — E1910 Trial Step 3 Analysis Set
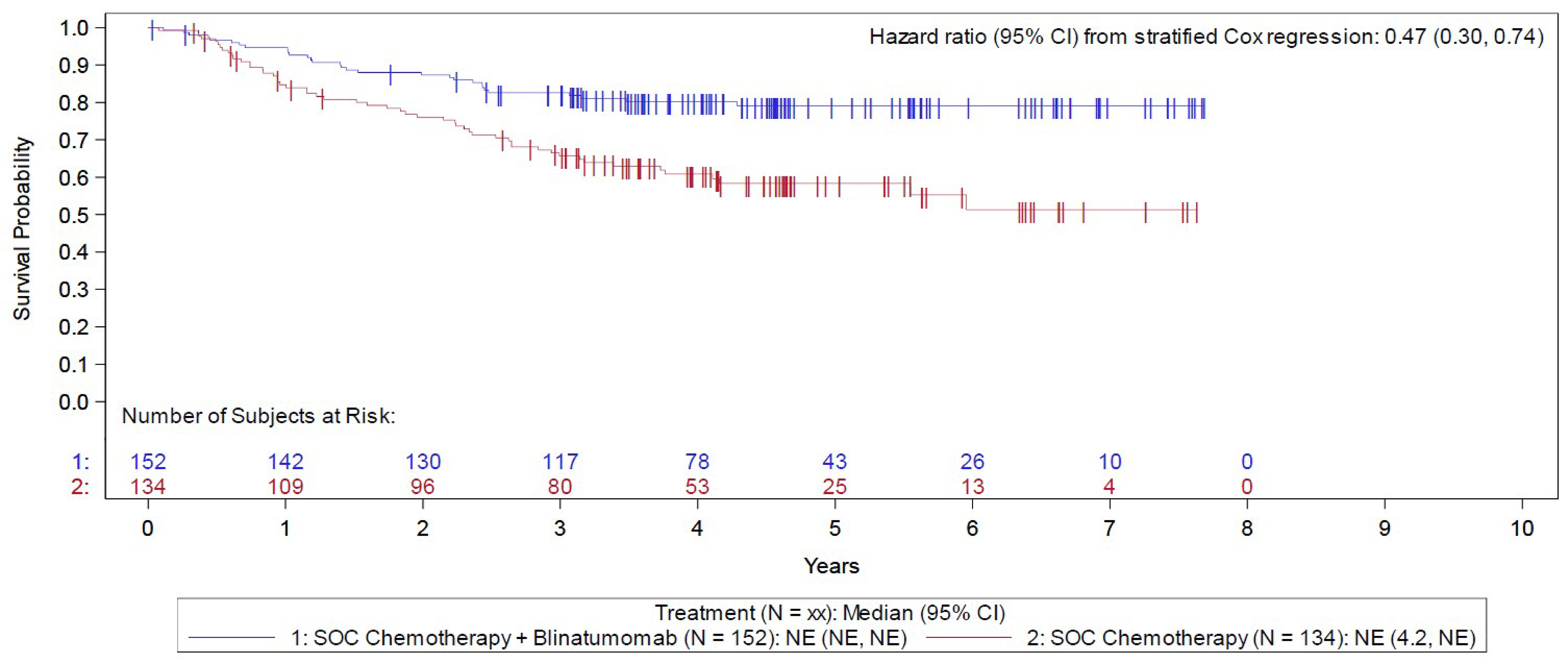
CI = confidence interval; NE = not evaluable; OS = overall survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization or registration date is the reference date for analysis of the descriptive OS analyses.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
OS in the FAS
OS results for the FAS of the E1910 trial at the June 23, 2023, DCO are summarized in Table 19. The median follow-up time for OS was 4.5 years for both treatment arms, and a higher percentage of deaths occurred in the chemotherapy arm (40 of 112 [35.7%]) compared to the blinatumomab plus chemotherapy arm (19 of 112 [17.0%]). The median OS was NE in both arms. The difference in survival probabilities for blinatumomab plus chemotherapy versus chemotherapy was 15.6% in year 1 and 19.9% in year 5. The KM plot for OS in the FAS is presented in Figure 6.
Subgroup and sensitivity analyses were generally consistent with those of the main analysis (Table 27, Table 30, Table 31, and Table 32).
Figure 6: Kaplan-Meier Analysis for OS — E1910 Trial FAS
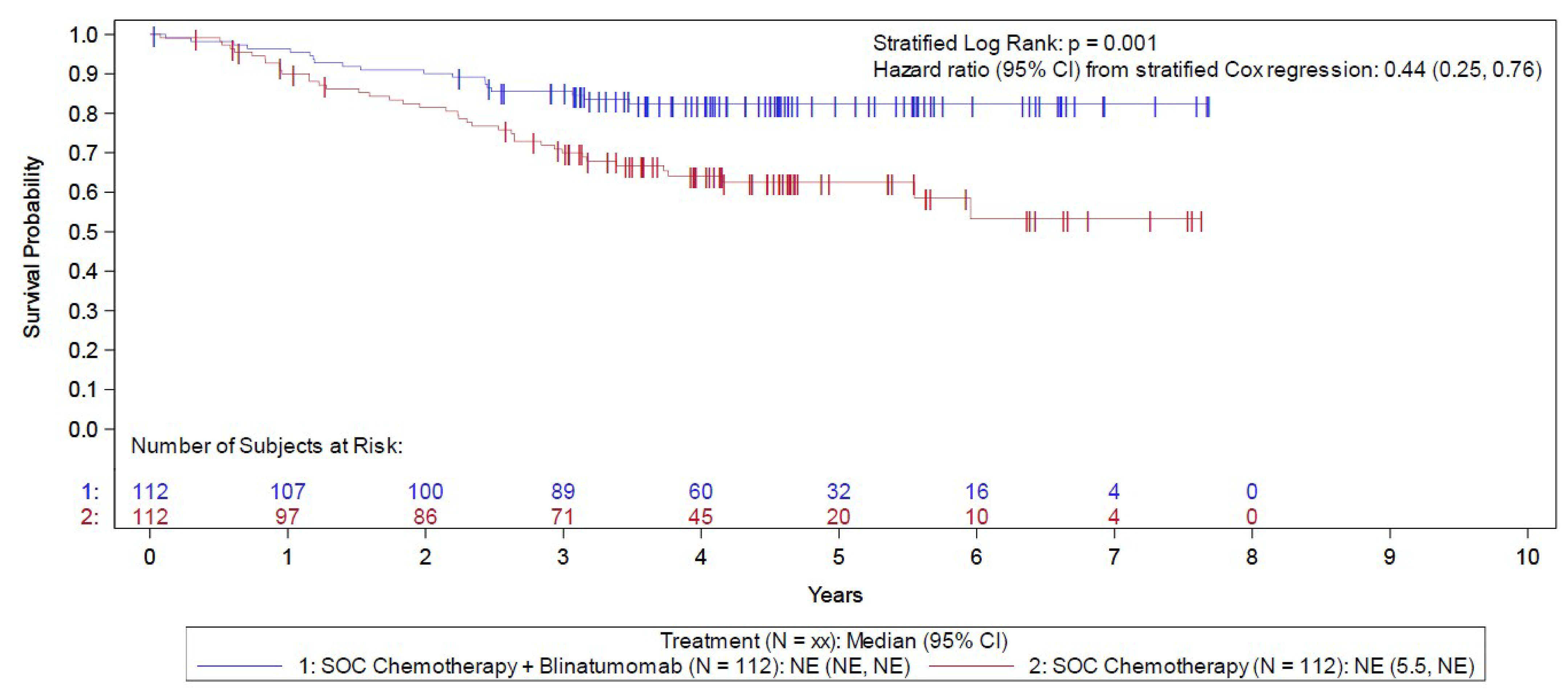
CI = confidence interval; FAS = full analysis set; NE = not evaluable; OS = overall survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization date is the reference date for analysis of the primary OS end point.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
OS in the Step 3 MRD-Positive Analysis Set
OS results for the Step 3 MRD-positive analysis set of the E1910 trial at the June 23, 2023, DCO are summarized in Table 19. The median follow-up time for OS was 4.6 years for the blinatumomab plus chemotherapy arm and 5.0 years for the chemotherapy arm. A higher percentage of deaths occurred in the chemotherapy arm (13 of 22 [59.1%]) compared to the blinatumomab plus chemotherapy arm (11 of 40 [27.5%]). Median OS was NE in the blinatumomab plus chemotherapy arm and was 1.9 (95% CI, 0.6 to NE) years in the chemotherapy arm. The difference in survival probabilities for blinatumomab plus chemotherapy versus chemotherapy was 31.3% in year 1 and 32.3% in year 5. The KM plot for OS in the Step 3 analysis set is presented in Figure 7.
Figure 7: Kaplan-Meier Analysis for OS — E1910 Trial Step 3 MRD-Positive Analysis Set
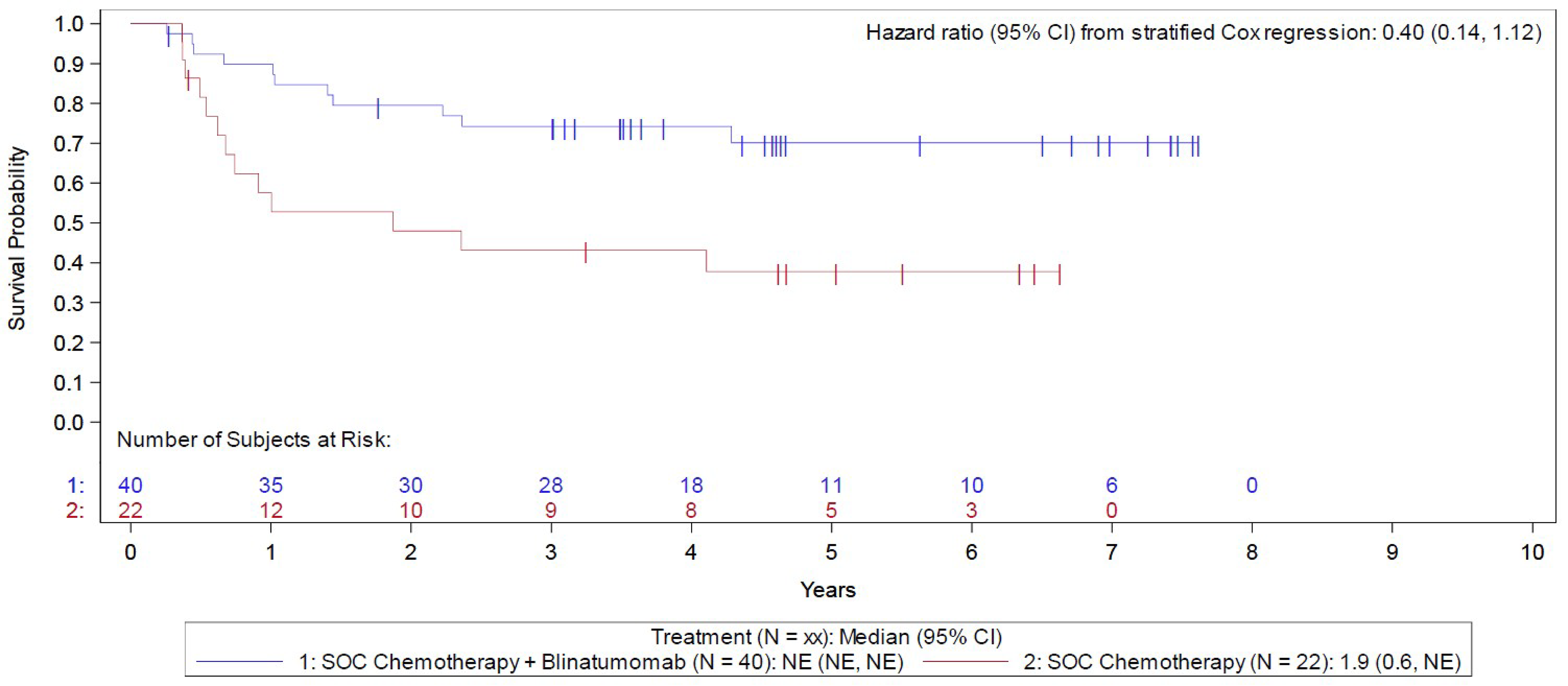
CI = confidence interval; MRD = minimal residual disease; NE = not evaluable; OS = overall survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization or registration date is the reference date for descriptive OS analyses using MRD-positive patients at Step 3.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
RFS in the Step 3 Analysis Set
The RFS results for those in the Step 3 analysis set of the E1910 trial are reported in Table 18. The median follow-up time for RFS was 4.5 years for both treatment arms, and median time to a relapse event was NE in both arms. The difference in the probability of remaining relapse-free for blinatumomab plus chemotherapy versus chemotherapy was 12.2% in year 1, 17.3% in year 3, and 18.4% in year 5. The KM plot for RFS in the Step 3 analysis set is presented in Figure 8.
Figure 8: Kaplan-Meier Analysis for RFS — E1910 Trial Step 3 Analysis Set
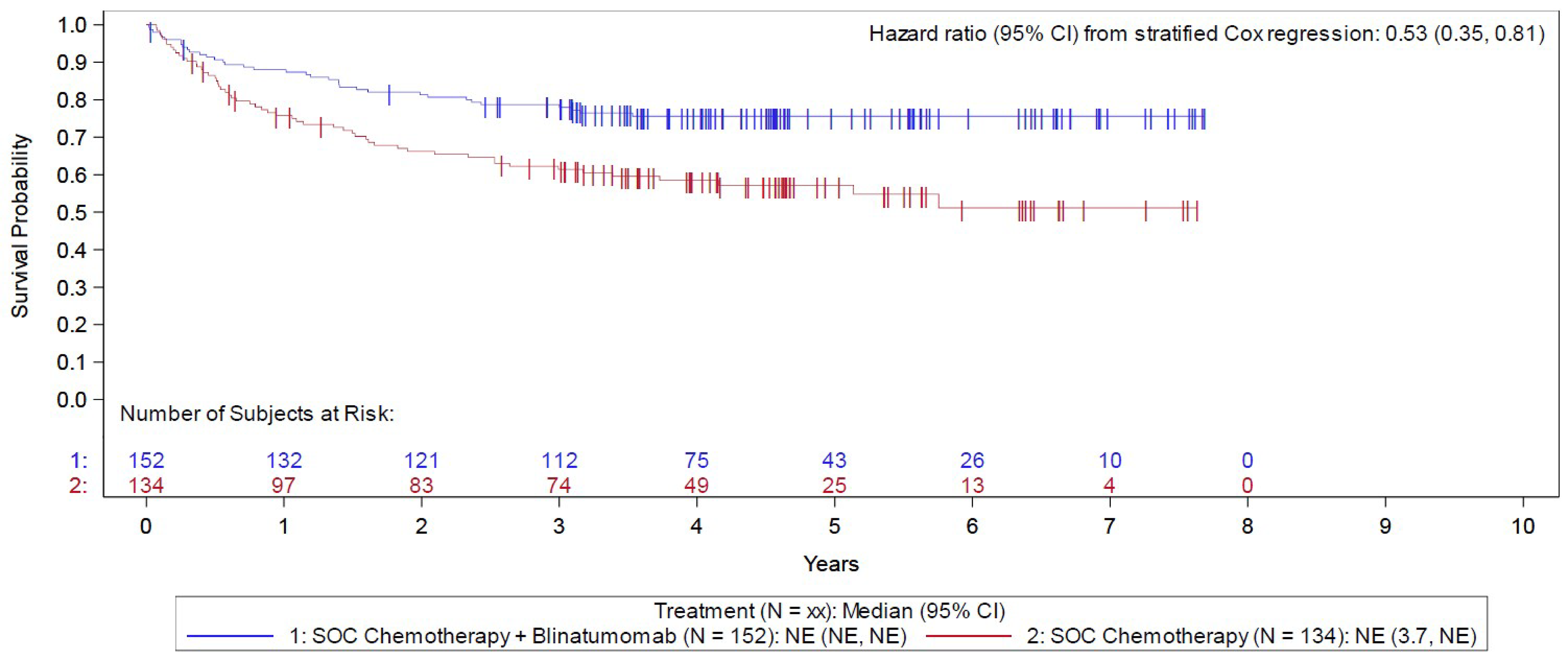
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; NE = not evaluable; RFS = relapse-free survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization or registration date is the reference date for analysis of the descriptive RFS analyses.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
RFS in the FAS
The RFS results for those in the FAS of the E1910 trial are reported in Table 19. The median follow-up time for RFS was 4.5 years for both treatment arms. Median time to a relapse event was NE in both arms. The difference in the probability of remaining relapse-free for blinatumomab plus chemotherapy versus chemotherapy was 8.2% in year 1, 15.4% in year 3, and 16.5% in year 5. The KM plot for OS in the Step 3 analysis set is presented in Figure 9.
Subgroup analyses were generally consistent with those of the main analysis (Table 27).
Figure 9: Kaplan-Meier Analysis for RFS — E1910 Trial FAS
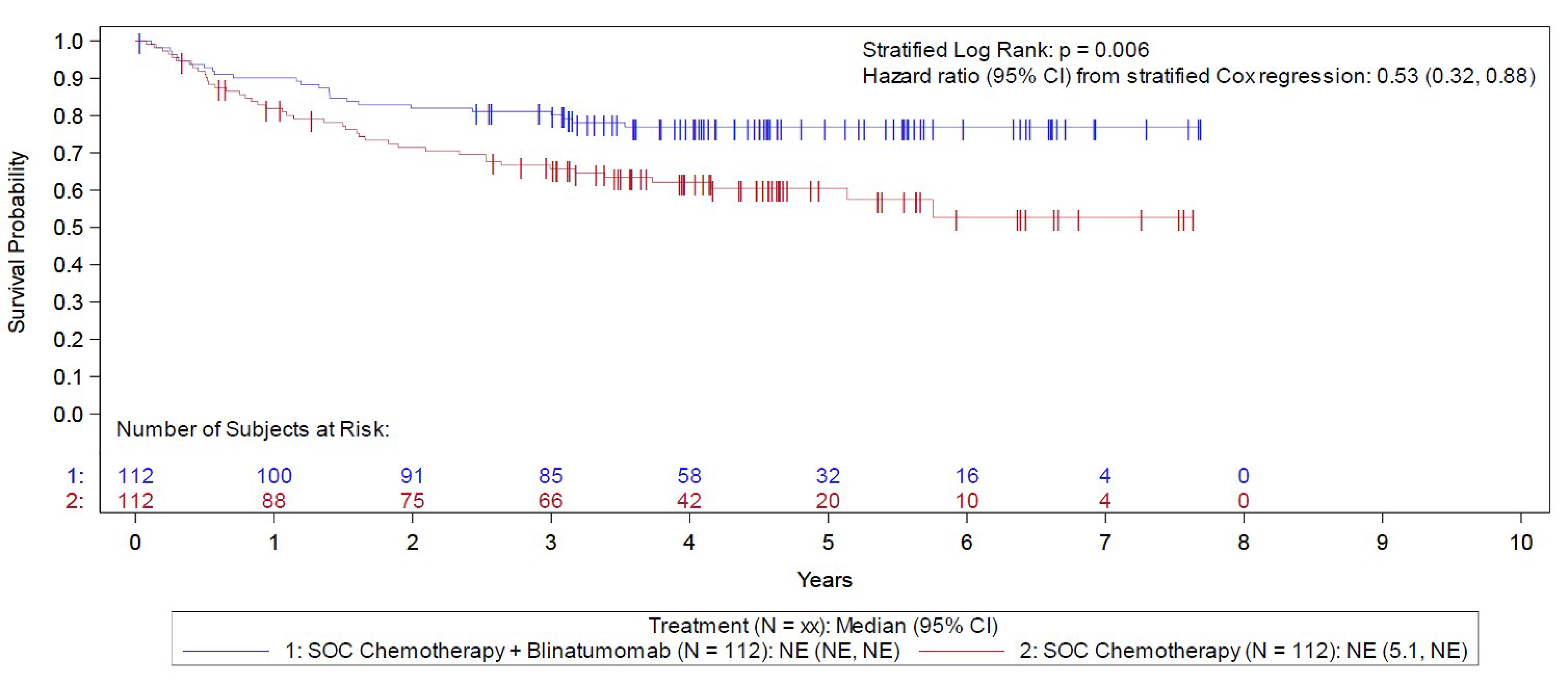
CI = confidence interval; FAS = full analysis set; NE = not evaluable; RFS = relapse-free survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization date is the reference date for the RFS analysis of the secondary end point examining RFS in MRD-negative patients at Step 3.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
RFS in the Step 3 MRD-Positive Analysis Set
The RFS results for those in the Step 3 MRD-positive analysis set of the E1910 trial are reported in Table 19. The median follow-up time for RFS was 4.6 years in the blinatumomab plus chemotherapy arm and 5.0 years in the chemotherapy arm. Median time to a relapse event was NE (95% CI, NE to NE) in the blinatumomab plus chemotherapy arm and 0.6 (95% CI, 0.2 to NE) years in the chemotherapy arm. The difference in the probability of remaining relapse-free for blinatumomab plus chemotherapy versus chemotherapy was 38.0% in year 1 and 32.4% in years 3 and 5. The KM plot for OS in the Step 3 analysis set is presented in Figure 10.
Figure 10: Kaplan-Meier Analysis for RFS — E1910 Trial Step 3 MRD-Positive Analysis Set
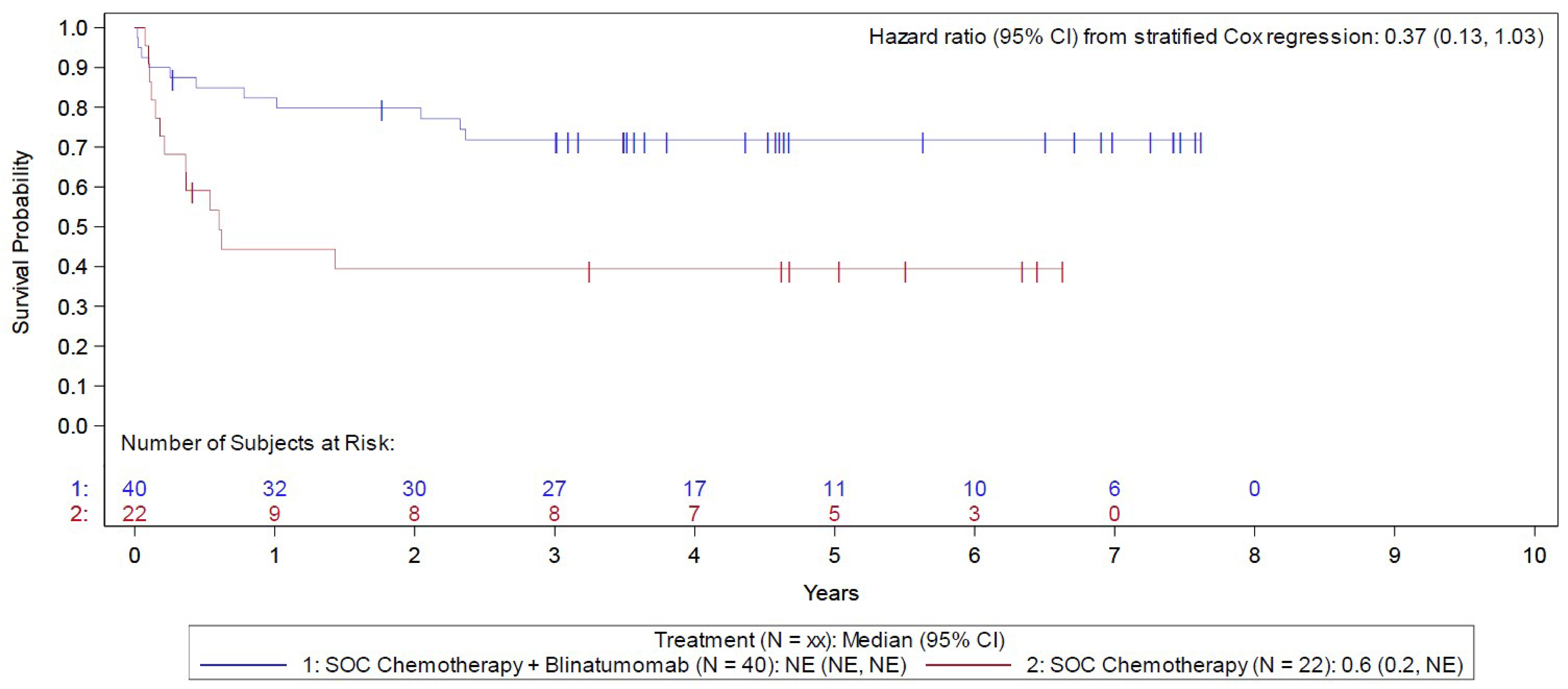
CI = confidence interval; MRD = minimal residual disease; NE = not evaluable; RFS = relapse-free survival; SOC = standard of care.
Notes: Censor indicated by vertical bar I. Step 3 randomization or registration date is the reference date for descriptive RFS analyses using MRD-positive patients at Step 3.
DCO date: June 23, 2023.
Source: BLINCYTO Clinical Study Report.19
Efficacy Results From the AALL1731 Trial
Overall Survival
OS results in the overall cohort of the AALL1731 trial at the June 30, 2024, DCO are summarized in Table 18. The median duration of follow-up was 2.5 years (IQR, 1.6 to 3.2 years). The probability of survival from randomization to 3 years was 98.4% (SE = 0.9%) for those in the blinatumomab plus chemotherapy arm and was 97.1% (SE = 1.1) for those in the chemotherapy arm. The KM plot is presented in Figure 11.
Subgroup analyses stratified by patients with SR-average and SR-high risk categories were consistent with those of the main analysis (Figure 11 and Table 28).
Figure 11: Kaplan-Meier Analysis for OS — AALL1731 Trial
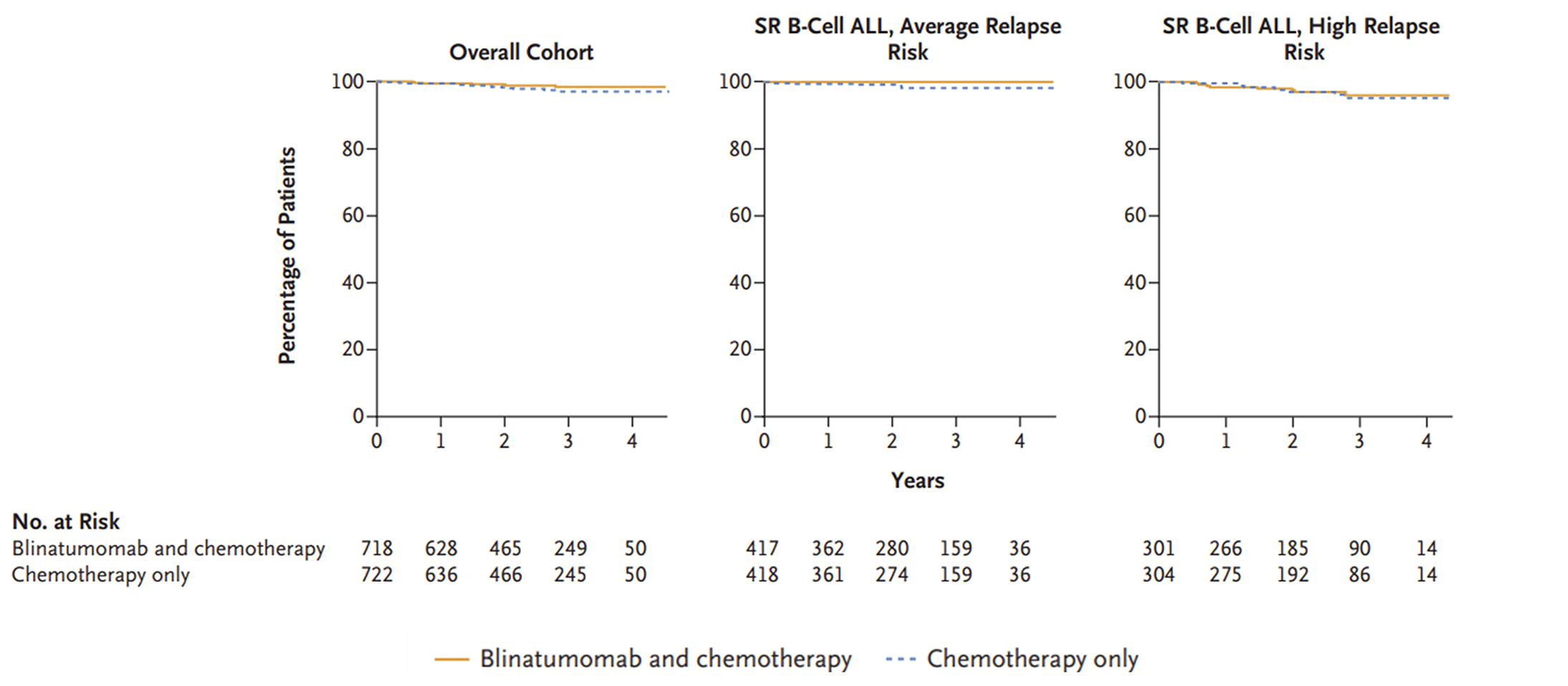
ALL = acute lymphoblastic leukemia; ITT = intention-to-treat; OS = overall survival; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Notes: OS was not a protocol-specified formal end point.
DCO date: June 30, 2024.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Disease-Free Survival
DFS results in the overall cohort of the AALL1731 trial at the June 30, 2024, DCO are summarized in Table 18. The median duration of follow-up was 2.5 years (IQR, 1.6 to 3.2 years). The probability of survival from randomization to 3 years was 96.0% (SE = 1.2%) for those in the blinatumomab plus chemotherapy arm and was 87.9% (SE = 2.1%) for those in the chemotherapy arm. The DFS stratified HR was 0.39 (95% CI, 0.24 to 0.64), favouring the blinatumomab plus chemotherapy arm. The KM plot for DFS in the overall cohort is presented in Figure 12.
Subgroup analyses stratified by patients with SR-average and SR-high risk categories were consistent with those of the main analysis (Figure 12 and Table 28). Post hoc subgroup analyses were generally consistent with those of the main analysis (Figure 13). Of note, when stratifying results of the overall cohort only by MRD status by multiparameter flow cytometry at day 29 (i.e., at the end of induction), results were similar to the overall cohort in those with a MRD less than 0.01% (i.e., MRD-negative) and 0.01% and greater (i.e., MRD-positive).
Sensitivity analyses using RMST indicated that the addition of blinatumomab to chemotherapy improved DFS, as measured by the difference in RMST by 72 days (95% CI, 36 to 108 days; P < 0.001 by stratified log-rank test; Table 33). Sensitivity analyses using RMST among patients with SR-average and SR-high were consistent with those of the main analysis (Table 33).
Figure 12: Kaplan-Meier Analysis for DFS — AALL1731 Trial
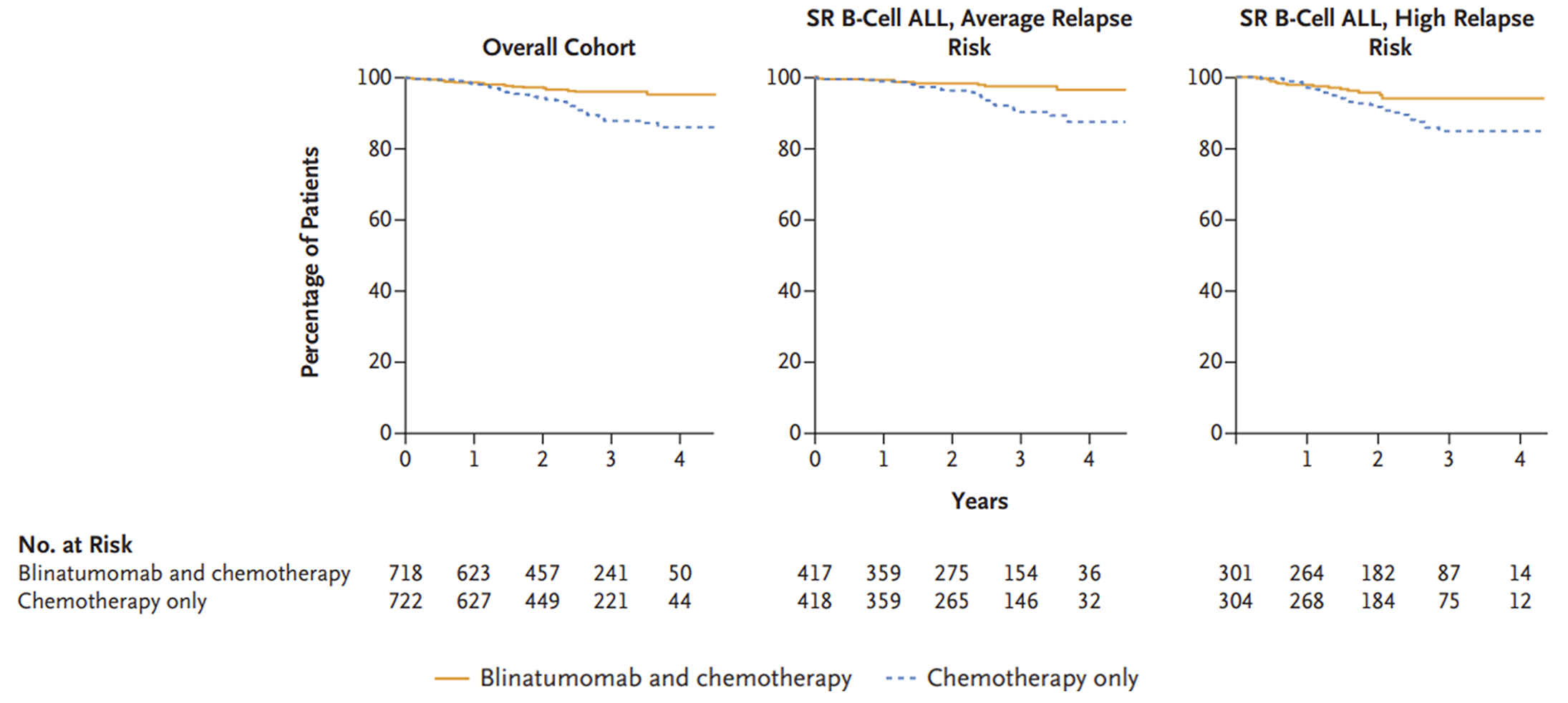
ALL = acute lymphoblastic leukemia; DFS = disease-free survival; KM = Kaplan-Meier; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Note: DCO date: June 30, 2024.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Table 18: E1910 and AALL1731 Trials — Summary of Key Efficacy Results in the Overall Trial Populations
Variable | E1910 trial (Step 3 analysis set) | AALL1731 trial overall cohort | ||
|---|---|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | Blinatumomab plus chemotherapy | Chemotherapy | |
OS | ||||
Patients included in the analysis, n | 152 | 134 | 718 | 722 |
Patients with OS events, n (%) | 30 (19.7) | 53 (39.6) | NR | NR |
Patients censored, n (%) | 122 (80.3) | 81 (60.4) | NR | NR |
Completed study without event | 0 (0.0) | 0 (0.0) | NR | NR |
Continues on study | 115 (75.7) | 71 (53.0) | NR | NR |
Discontinued study | 7 (4.6) | 10 (7.5) | NR | NR |
Consent withdrawn | 6 (3.9) | 7 (5.2) | NR | NR |
Lost to follow-up | 1 (0.7) | 3 (2.2) | NR | NR |
OS (months), median (95% CI) | NE (NE to NE) | NE (4.2 to NE) | NR | NR |
Stratified HR (95% CI)a | 0.47 (0.30 to 0.74) | NR | ||
P valueb | < 0.001 | NR | ||
Duration of follow-up (years), median (95% CI) | 4.5 (4.1 to 4.6)c,d,e | 4.5 (4.1 to 4.7)c,d,e | 2.5 (IQR, 1.6 to 3.2) | |
OS probability at 0.5 years, % (95% CI) | 96.7 (92.2 to 98.6) | 96.2 (91.2 to 98.4) | NR | NR |
OS probability at 1 year, % (95% CI) | 94.7 (89.6 to 97.3) | 84.7 (77.3 to 89.9) | NR | NR |
OS probability at 2 years, % (95% CI) | 87.3 (80.9 to 91.7) | 76.1 (67.7 to 82.5) | NR | NR |
OS probability at 3 years, % (95% CI) | 82.6 (75.5 to 87.8) | 65.7 (56.7 to 73.2) | 98.4 (SE = 0.8) | 97.1 (SE = 1.1) |
Difference in survival probability at 3 years, % (95% CI) | 16.9 (5.5 to 28.3) | NR | ||
OS probability at 4 years, % (95% CI) | 80.3 (72.8 to 85.9) | 60.9 (51.6 to 68.9) | NR | NR |
OS probability at 5 years, % (95% CI) | 79.1 (71.4 to 85.0) | 58.3 (48.8 to 66.7) | NR | NR |
Difference in survival probability at 5 years, % (95% CI) | 20.8 (8.5 to 33.0) | NR | ||
OS probability at 6 years, % (95% CI) | 79.1 (71.4 to 85.0) | 51.3 (38.6 to 62.6) | NR | NR |
OS probability at 7 years, % (95% CI) | 79.1 (71.4 to 85.0) | 51.3 (38.6 to 62.6) | NR | NR |
RFS | ||||
Patients included in the analysis, n | 152 | 134 | NR | NR |
Patients with RFS events, n (%) | 36 (23.7) | 56 (41.8) | NR | NR |
Relapse | 23 (15.1) | 42 (31.3) | NR | NR |
Death due to any cause | 13 (8.6) | 14 (10.4) | NR | NR |
P valueb | NE | NR | ||
Patients censored, n (%) | 116 (76.3) | 78 (58.2) | NR | NR |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) | NR | NR |
Completed study without event | 0 (0.0) | 0 (0.0) | NR | NR |
Continues on study | 111 (73.0) | 68 (50.7) | NR | NR |
Discontinued study | 5 (3.3) | 10 (7.5) | NR | NR |
Consent withdrawn | 5 (3.3) | 7 (5.2) | NR | NR |
Lost to follow-up | 0 (0.0) | 3 (2.2) | NR | NR |
RFS (months), median (95% CI) | NE (NE to NE) | NE (3.7 to NE) | NR | NR |
Stratified HR (95% CI)a | 0.53 (0.35 to 0.81) | NR | ||
P valueb | 0.003 | NR | ||
Duration of follow-up, median (95% CI) yearsc,d,e | 4.5 (4.1 to 4.6) | 4.5 (4.0 to 4.6) | NR | NR |
RFS probability at 0.5 years, % (95% CI) | 90.7 (84.8 to 94.4) | 86.5 (79.5 to 91.3) | NR | NR |
RFS probability at 1 year, % (95% CI) | 88.0 (81.7 to 92.3) | 75.8 (67.5 to 82.2) | NR | NR |
Difference in survival probability at 1 year, % (95% CI) | 12.2 (1.8 to 22.7) | NR | ||
RFS probability at 2 years, % (95% CI) | 81.4 (74.2 to 86.7) | 66.2 (57.4 to 73.7) | NR | NR |
RFS probability at 3 years, % (95% CI) | 78.7 (71.2 to 84.4) | 61.4 (52.4 to 69.2) | NR | NR |
Difference in survival probability at 3 years, % (95% CI) | 17.3 (5.6 to 28.9) | NR | ||
RFS probability at 4 years, % (95% CI) | 75.6 (67.8 to 81.8) | 58.5 (49.3 to 66.5) | NR | NR |
RFS probability at 5 years, % (95% CI) | 75.6 (67.8 to 81.8) | 57.2 (47.9 to 65.4) | NR | NR |
Difference in survival probability at 5 years, % (95% CI) | 18.4 (6.3 to 30.6) | NR | ||
RFS probability at 6 years, % (95% CI) | 75.6 (67.8 to 81.8) | 51.1 (39.3 to 61.8) | NR | NR |
RFS probability at 7 years, % (95% CI) | 75.6 (67.8 to 81.8) | 51.1 (39.3 to 61.8) | NR | NR |
DFS | ||||
Patients included in the analysis, n | NR | NR | 718 | 722 |
DFS probability at 3 years, % (SE) | NR | NR | 96.0 (1.2) | 87.9 (2.1) |
Stratified HR (95% CI)a | NR | 0.39 (0.24 to 0.64) | ||
P value | NR | < 0.0001f | ||
CI = confidence interval; DFS = disease-free survival; HR = hazard ratio; IQR = interquartile range; KM = Kaplan-Meier; NE = not evaluable; NR = not reported; OS = overall survival; RFS = relapse-free survival; SE = standard error.
Notes: OS analyses in E1910 and AALL1731, RFS analysis in the Step 3 analysis set of E1910, and DFS analysis in the overall cohort of AALL1731 were performed post hoc.
DCO dates: E1910 = June 23, 2023; AALL1731 = June 30, 2024.
aThe HR estimates are obtained from a stratified Cox regression model. An HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
bOne-sided stratified log-rank test P value is provided.
cYears are calculated as days from randomization or registration date to event or censor date, divided by 365.25.
dDefined as median KM time to censoring in the E1910 trial.
eTime to censoring measures follow-up time by reversing the status indicator for censored events.
fP values are presented from interim analyses; however, thresholds for efficacy or futility were not reported in the trial’s statistical analysis plan. As a result, these P values should not be interpreted as evidence of statistical significance.
Source: BLINCYTO E1910 Clinical Study Report and Gupta et al. (2024).19,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 19: E1910 Trial — Summary of Key Efficacy Results Stratified by MRD status, FAS and Step 3 MRD-Positive Analysis Set
Variable | Full analysis set | Step 3 MRD-positive analysis set | ||
|---|---|---|---|---|
Blinatumomab plus chemotherapy (N = 112) | Chemotherapy (N = 112) | Blinatumomab plus chemotherapy (N = 40) | Chemotherapy (N = 22) | |
OS | ||||
Patients with OS events, n (%) | 19 (17.0) | 40 (35.7) | 11 (27.5) | 13 (59.1) |
Patients censored, n (%) | 93 (83.0) | 72 (64.3) | 29 (72.5) | 9 (40.9) |
Completed study without event | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Continues on study | 88 (78.6) | 64 (57.1) | 27 (67.5) | 7 (31.8) |
Discontinued study | 5 (4.5) | 8 (7.1) | 2 (5.0) | 2 (9.1) |
Consent withdrawn | 4 (3.6) | 6 (5.4) | 2 (5.0) | 1 (4.5) |
Lost to follow-up | 1 (0.9) | 2 (1.8) | 0 (0.0) | 1 (4.5) |
OS (months), median (95% CI) | NE (NE to NE) | NE (5.5 to NE) | NE (NE to NE) | 1.9 (0.6 to NE) |
Stratified HR (95% CI)a | 0.44 (0.25 to 0.76) | 0.40 (0.14 to 1.12) | ||
P valueb | 0.003 | 0.082 | ||
Duration of follow-up (years), median (95% CI)c,d,e | 4.5 (4.1 to 4.6) | 4.5 (4.0 to 4.6) | 4.6 (3.6 to 6.5) | 5.0 (3.2 to 6.4) |
OS probability at 0.5 years, % (95% CI) | 98.2 (93.0 to 99.5) | 99.1 (93.8 to 99.9) | 92.4 (78.2 to 97.5) | 81.6 (58.0 to 92.7) |
OS probability at 1 year, % (95% CI) | 96.4 (90.7 to 98.6) | 90.0 (82.6 to 94.3) | 89.8 (75.1 to 96.0) | 57.6 (34.2 to 75.3) |
OS probability at 2 years, % (95% CI) | 90.1 (82.8 to 94.4) | 81.5 (72.8 to 87.6) | 79.5 (63.2 to 89.2) | 48.0 (26.0 to 67.0) |
OS probability at 3 years, % (95% CI) | 85.5 (77.5 to 90.9) | 70.0 (60.3 to 77.7) | 74.2 (57.4 to 85.2) | 43.2 (22.2 to 62.6) |
Difference in survival probability at 3 years, % (95% CI) | 15.6 (3.0 to 28.2) | 31.1 (4.1 to 58.0) | ||
OS probability at 4 years, % (95% CI) | 82.4 (73.7 to 88.4) | 64.1 (53.9 to 72.7) | 74.2 (57.4 to 85.2) | 43.2 (22.2 to 62.6) |
OS probability at 5 years, % (95% CI) | 82.4 (73.7 to 88.4) | 62.5 (52.0 to 71.3) | 70.1 (52.0 to 82.5) | 37.8 (17.8 to 57.7) |
Difference in survival probability at 5 years, % (95% CI) | 19.9 (6.3 to 33.5) | 32.3 (5.4 to 59.3) | ||
OS probability at 6 years, % (95% CI) | 82.4 (73.7 to 88.4) | 53.3 (37.8 to 66.5) | 70.1 (52.0 to 82.5) | 37.8 (17.8 to 57.7) |
OS probability at 7 years, % (95% CI) | 82.4 (73.7 to 88.4) | 53.3 (37.8 to 66.5) | 70.1 (52.0 to 82.5) | NE (NE to NE) |
RFS | ||||
Patients with RFS events, n (%) | 25 (22.3) | 43 (38.4) | 11 (27.5) | 13 (59.1) |
Relapse | 15 (13.4) | 32 (28.6) | 8 (20.0) | 10 (45.5) |
Death due to any cause | 10 (8.9) | 11 (9.8) | 3 (7.5) | 3 (13.6) |
Patients censored, n (%) | 87 (77.7) | 69 (61.6) | 29 (72.5) | 9 (40.9) |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Completed study without event | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Continues on study | 84 (75.0) | 61 (54.5) | 27 (67.5) | 7 (31.8) |
Discontinued study | 3 (2.7) | 8 (7.1) | 2 (5.0) | 2 (9.1) |
Consent withdrawn | 3 (2.7) | 6 (5.4) | 2 (5.0) | 1 (4.5) |
Lost to follow-up | 0 (0.0) | 2 (1.8) | 0 (0.0) | 1 (4.5) |
RFS (months), median (95% CI) | NE (NE to NE) | NE (5.1 to NE) | NE (NE to NE) | 0.6 (0.2 to NE) |
Stratified HR (95% CI)a | 0.53 (0.32 to 0.88) | 0.37 (0.13 to 1.03) | ||
P valueb | 0.013 | 0.056 | ||
Duration of follow-up (years), median (95% CI)c,d,e | 4.5 (4.1 to 4.7) | 4.5 (4.0 to 4.6) | 4.6 (3.5 to 6.5) | 5.0 (3.2 to 6.4) |
RFS probability at 0.5 years, % (95% CI) | 92.8 (86.1 to 96.3) | 91.9 (85.1 to 95.7) | 84.9 (69.5 to 92.9) | 59.1 (36.1 to 76.2) |
RFS probability at 1 year, % (95% CI) | 90.1 (82.8 to 94.4) | 81.9 (73.4 to 87.9) | 82.4 (66.5 to 91.2) | 44.3 (23.2 to 63.6) |
Difference in survival probability at 1 year, % (95% CI) | 8.2 (–3.0 to 19.4) | 38.0 (11.6 to 64.5) | ||
RFS probability at 2 years, % (95% CI) | 82.0 (73.5 to 88.0) | 71.5 (61.9 to 79.0) | 79.8 (63.6 to 89.3) | 39.4 (19.3 to 59.0) |
RFS probability at 3 years, % (95% CI) | 81.1 (72.5 to 87.2) | 65.7 (55.9 to 73.8) | 71.8 (54.8 to 83.3) | 39.4 (19.3 to 59.0) |
Difference in survival probability at 3 years, % (95% CI) | 15.4 (2.3 to 28.4) | 32.4 (6.1 to 58.7) | ||
RFS probability at 4 years, % (95% CI) | 77.0 (67.8 to 83.8) | 62.1 (52.0 to 70.7) | 71.8 (54.8 to 83.3) | 39.4 (19.3 to 59.0) |
RFS probability at 5 years, % (95% CI) | 77.0 (67.8 to 83.8) | 60.5 (50.1 to 69.4) | 71.8 (54.8 to 83.3) | 39.4 (19.3 to 59.0) |
Difference in survival probability at 5 years, % (95% CI) | 16.5 (2.6 to 30.3) | 32.4 (6.1 to 58.7) | ||
RFS probability at 6 years, % (95% CI) | 77.0 (67.8 to 83.8) | 52.7 (38.5 to 65.0) | 71.8 (54.8 to 83.3) | 39.4 (19.3 to 59.0) |
RFS probability at 7 years, % (95% CI) | 77.0 (67.8 to 83.8) | 52.7 (38.5 to 65.0) | 71.8 (54.8 to 83.3) | NE (NE to NE) |
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; MRD = minimal residual disease; NE = not evaluable; OS = overall survival; RFS = relapse-free survival.
Note: DCO date: June 23, 2023.
aThe HR estimates are obtained from a stratified Cox regression model. A HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
bOne-sided stratified log-rank test P value is provided.
cYears are calculated as days from randomization or registration date to event or censor date, divided by 365.25.
dDefined as median KM time to censoring in the E1910 trial.
eTime to censoring measures follow-up time by reversing the status indicator for censored events.
Note: OS analyses in the MRD-negative cohort (FAS) was the primary end point of E1910; OS in the MRD-positive cohort (Step 3 MRD-positive analysis set) and RFS in the MRD-negative and MRD-positive cohorts were secondary end points in E1910.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Refer to Table 20 for harms data.
Treatment-Emergent AEs
By June 23, 2023, a similar percentage of patients in both treatment arms of the Step 3 safety analysis set in the E1910 trial experienced a TEAE (145 of 147 [98.6%] in the blinatumomab plus chemotherapy arm and 125 of 128 [97.7%] in the chemotherapy arm). The 3 most common TEAEs in the blinatumomab plus chemotherapy arm were investigations (e.g., blood cell counts; 91.8%), blood and lymphatic system disorders (e.g., anemia; 62.6%), and nervous system disorders (e.g., headache; 57.8%). In the chemotherapy arm, the 3 most common TEAEs were investigations (96.9%), blood and lymphatic system disorders (70.3%), and gastrointestinal disorders (e.g., diarrhea; 44.5%).
TEAEs of any grade in the AALL1731 trial were not provided in the materials received by the CDA-AMC review team.
Grade 3 or Higher TEAEs
A similar percentage of patients in both treatment arms of the Step 3 safety analysis set in the E1910 trial experienced a grade 3 or greater TEAE (141 of 147 [95.9%] in the blinatumomab plus chemotherapy and 125 of 128 [97.7%] in the chemotherapy arm). In the blinatumomab plus chemotherapy arm, the 3 most common grade 3 or higher TEAEs were the same as the 3 most common TEAEs of any grade (i.e., investigations [89.8%], blood and lymphatic system disorders [39.5%], and nervous system disorders [22.4%]). In the chemotherapy arm, the 3 most common grade 3 or higher TEAEs were investigations (96.1%), blood and lymphatic system disorders (57.0%), and infection and infestation (e.g., sepsis; 24.2%).
The reporting of TEAEs in the AALL1731 trial were restricted to selected grade 3 or higher ones stratified by patients with SR-average or SR-high. Generally, it was reported that the percentage of select grade 3 of higher TEAEs were well balanced across treatment arms in the AALL1731 trial. Notable differences included a higher percentage of patients in the blinatumomab plus chemotherapy arms of both the SR-average (47.0% [165 of 351]) and SR-high (57.1% [156 of 273]) risk categories who experienced febrile neutropenia than in the chemotherapy arm alone (39.6% [149 of 376] and 50.5% [140 of 277], respectively), and in the SR-average risk category, a higher percentage of patients in the blinatumomab plus chemotherapy arm who experienced sepsis or catheter-related infection (14.8%) and other infections (32.8%) than in the chemotherapy arm alone (5.1% and 26.3%, respectively). In both SR-average and SR-high risk categories, the 3 most common grade 3 or higher TEAEs in the blinatumomab plus chemotherapy arms were febrile neutropenia (47.0% and 57.1%, respectively), other infection (32.8% and 35.2%, respectively), and sepsis or catheter-related infection (14.8% and 20.9%, respectively). In both SR-average and SR-high risk categories, the 3 most common grade 3 or higher TEAEs in the chemotherapy arms were febrile neutropenia (39.6% and 50.5%, respectively), other infections (26.3% and 37.9%, respectively), and mucositis (15.4% and 17.7%, respectively).
In both SR-average and SR-high subgroups, 1 patient in the blinatumomab plus chemotherapy group and no patient in the chemotherapy group experienced cytokine release syndrome of grade 3 or higher. In the SR-average subgroup, 3 (0.9%) and 4 (1.1%) patients experienced pancreatis of grade 3 or higher in the blinatumomab plus chemotherapy and chemotherapy groups, respectively. Patients in this subgroup experienced neurotoxic events of grade 3 or higher (seizure: 4 [1.1%] and 7 [1.9%]; all other CNS events: 2 [0.6%] and 3 [0.8%]; and peripheral neuropathy: 2 [0.6%] and 9 [2.4%] in the blinatumomab plus chemotherapy and chemotherapy groups, respectively). In the SR-high subgroup, 11 (4.0%) and 8 (2.9%) of patients experienced pancreatis of grade 3 or higher in the blinatumomab plus chemotherapy and chemotherapy groups, respectively. Patients in this subgroup experienced neurotoxic events of grade 3 or higher (seizure: 10 [3.7%] and 7 [2.5%]; all other CNS events: 3 [1.1%] and 4 [1.4%]; peripheral neuropathy: 6 [2.2%] and 2 [0.7%] in the blinatumomab plus chemotherapy and chemotherapy groups, respectively).
Withdrawals Due to AEs
Withdrawals due to AEs were not reported among the Step 3 safety analysis set of the E1910 trial; however, they were available for the Step 3 analysis set. Among patients included in the Step 3 analysis set, 14 of 152 patients (9.2%) in the blinatumomab plus chemotherapy arm discontinued treatment due to AEs, side effects, or complications compared to 5 of 134 patients (3.7%) discontinuing due to AEs, side effects, or complications in the chemotherapy arm.
Withdrawals due to AEs in the AALL1731 trial were not provided in the materials received by the CDA-AMC review team.
Treatment-Emergent SAEs
A higher percentage of patients in the blinatumomab plus chemotherapy arm of the Step 3 safety analysis set in the E1910 trial experienced treatment-emergent SAEs (82 of 147 [55.8%]) than in the chemotherapy arm (36 of 128 [28.1%]). The 3 most common treatment-emergent SAEs in the blinatumomab plus chemotherapy arm were infections and infestations (22.4%), investigations (15.6%), and nervous system disorders (15.0%). In the chemotherapy arm, the 3 most common treatment-emergent SAEs were infections and infestations (14.8%), blood and lymphatic system disorders (11.7%), and investigations (4.7%).
Treatment-emergent SAEs in the AALL1731 trial were not provided in the materials received by the CDA-AMC review team.
Fatal TEAEs
A similar percentage of patients in both treatment arms of the Step 3 safety analysis set in the E1910 trial had a fatal TEAE (3 of 147 [2.0%] in the blinatumomab plus chemotherapy arm and 2 of 128 [1.6%] in the chemotherapy arm). In the blinatumomab plus chemotherapy arm, 2 of the fatal TEAEs were due to sepsis (1.4%) and 1 was due to intracranial hemorrhage (0.7%). In the chemotherapy arm, 1 of the fatal TEAEs was due to sepsis (0.8%) and 1 was due to cardiac arrest (0.8%).
In AALL1731, 5 patients had a fatal TEAE while in remission, all of whom were classified as having high risk of relapse. Two of the patients were in the chemotherapy arm (both deaths were sepsis-related) and 3 patients were in the blinatumomab plus chemotherapy arm (1 death was due to sepsis, 1 was due to multiorgan failure, and 1 was due to hypoxic ischemic encephalopathy). None of the deaths occurred during blinatumomab cycles.
Notable Harms
Among TEAEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review, TLS was not reported for the Step 3 safety analysis set in the E1910 trial. Cytokine release syndrome occurred in a higher percentage of patients in the blinatumomab plus chemotherapy arm (23 of 147 [15.6%]) than in the chemotherapy arm (0 of 128 [0.0%]). While infections were not stratified by those deemed serious, any infections and infestations occurred in a higher percentage of patients in the blinatumomab plus chemotherapy arm (34.7%) than in the chemotherapy arm (27.3%). However, most subcategories of infections of special interest, including Fusarium infection, fungal pneumonia, septic shock, Aspergillus infection, bronchopneumonia, Candida infection, enterococcal bacteremia, Escherichia sepsis, and lung infection, were not reported. Finally, relatively few patients in both treatment arms experienced neurotoxicity (2.4% in the blinatumomab plus chemotherapy arm versus 0.0% in the chemotherapy arm) or pancreatitis (0.0% and 0.8%, respectively).
Some of the TEAEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review are covered in the reporting of grade 3 or greater TEAEs. However, data on notable harms of any grade were not available.
Notable harms in the AALL1731 trial were not provided in the materials received by the CDA-AMC review team.
Table 20: E1910 and AALL1731 Trials — Summary of Harms Results
Adverse events | E1910 trial (Step 3 safety analysis set) | AALL1731 (safety analysis set) | ||||
|---|---|---|---|---|---|---|
Blinatumomab plus chemotherapy (N = 147) | Chemotherapy (N = 128) | SR-average | SR-high | |||
Blinatumomab plus chemotherapy (N = 351) | Chemotherapy (N = 376) | Blinatumomab plus chemotherapy (N = 273) | Chemotherapy (N = 277) | |||
Most common treatment-emergent adverse events (occurring in ≥ 10% in either study arm), n (%) | ||||||
≥ 1 adverse event | 145 (98.6) | 125 (97.7) | NR | NR | NR | NR |
Blood and lymphatic system disorders | 92 (62.6) | 90 (70.3) | NR | NR | NR | NR |
Anemia | 84 (57.1) | 73 (57.0) | NR | NR | NR | NR |
Febrile neutropenia | 32 (21.8) | 37 (28.9) | NR | NR | NR | NR |
Gastrointestinal disorders | 81 (55.1) | 57 (44.5) | NR | NR | NR | NR |
Diarrhea | 49 (33.3) | 30 (23.4) | NR | NR | NR | NR |
Vomiting | 45 (30.6) | 30 (23.4) | NR | NR | NR | NR |
Abdominal pain | 32 (21.8) | 20 (15.6) | NR | NR | NR | NR |
Nausea | 24 (16.3) | 9 (7.0) | NR | NR | NR | NR |
General disorders and administration site conditions | 44 (29.9) | 19 (14.8) | NR | NR | NR | NR |
Fatigue | 23 (15.6) | 12 (9.4) | NR | NR | NR | NR |
Pyrexia | 19 (12.9) | 6 (4.7) | NR | NR | NR | NR |
Immune system disorders | 25 (17.0) | 4 (3.1) | NR | NR | NR | NR |
Cytokine release syndrome | 23 (15.6) | 0 (0.0) | NR | NR | NR | NR |
Infections and infestations | 51 (34.7) | 35 (27.3) | NR | NR | NR | NR |
Sepsis | 15 (10.2) | 13 (10.2) | NR | NR | NR | NR |
Device-related infection | 15 (10.2) | 8 (6.3) | NR | NR | NR | NR |
Investigations | 135 (91.8) | 124 (96.9) | NR | NR | NR | NR |
Neutrophil count decreased | 129 (87.8) | 119 (93.0) | NR | NR | NR | NR |
Platelet count decreased | 117 (79.6) | 107 (83.6) | NR | NR | NR | NR |
White blood cell count decreased | 76 (51.7) | 82 (64.1) | NR | NR | NR | NR |
Lymphocyte count decreased | 44 (29.9) | 37 (28.9) | NR | NR | NR | NR |
Alanine aminotransferase increased | 19 (12.9) | 10 (7.8) | NR | NR | NR | NR |
Aspartate aminotransferase increased | 15 (10.2) | 5 (3.9) | NR | NR | NR | NR |
Metabolism and nutrition disorders | 40 (27.2) | 30 (23.4) | NR | NR | NR | NR |
Hyperglycemia | 20 (13.6) | 12 (9.4) | NR | NR | NR | NR |
Musculoskeletal and connective tissue disorders | 34 (23.1) | 12 (9.4) | NR | NR | NR | NR |
Nervous system disorders | 85 (57.8) | 48 (37.5) | NR | NR | NR | NR |
Headache | 61 (41.5) | 42 (32.8) | NR | NR | NR | NR |
Tremor | 30 (20.4) | 5 (3.9) | NR | NR | NR | NR |
Psychiatric disorders | 27 (18.4) | 7 (5.5) | NR | NR | NR | NR |
Respiratory, thoracic, and mediastinal disorders | 24 (16.3) | 13 (10.2) | NR | NR | NR | NR |
Vascular disorders | 39 (26.5) | 17 (13.3) | NR | NR | NR | NR |
Embolism | 15 (10.2) | 8 (6.3) | NR | NR | NR | NR |
Hypertension | 16 (10.9) | 6 (4.7) | NR | NR | NR | NR |
Grade ≥ 3 treatment-emergent adverse events (occurring in ≥ 10% in either study arm), n (%) | ||||||
Patients with Grade ≥ 3 adverse events | 141 (95.9) | 125 (97.7) | NR | NR | NR | NR |
Difference (95% CI) | –1.74 (–5.87 to 2.40) | NR | NR | |||
Blood and lymphatic system disorders | 58 (39.5) | 73 (57.0) | NR | NR | NR | NR |
Anemia | 44 (29.9) | 54 (42.2) | NR | NR | NR | NR |
Febrile neutropenia | 32 (21.8) | 37 (28.9) | 165 (47.0) | 149 (39.6) | 156 (57.1) | 140 (50.5) |
Gastrointestinal disorders | 18 (12.2) | 22 (17.2) | NR | NR | NR | NR |
General disorders and administration site conditions | 15 (10.2) | 7 (5.5) | NR | NR | NR | NR |
Infections and infestations | 42 (28.6) | 31 (24.2) | NR | NR | NR | NR |
Sepsisa | 15 (10.2) | 13 (10.2) | 52 (14.8) | 19 (5.1) | 57 (20.9) | 47 (17.0) |
Other infectionb | NR | NR | 115 (32.8) | 99 (26.3) | 96 (35.2) | 105 (37.9) |
Investigations | 132 (89.8) | 123 (96.1) | NR | NR | NR | NR |
Neutrophil count decreased | 125 (85.0) | 119 (93.0) | NR | NR | NR | NR |
Platelet count decreased | 102 (69.4) | 101 (78.9) | NR | NR | NR | NR |
White blood cell count decreased | 74 (50.3) | 81 (63.3) | NR | NR | NR | NR |
Lymphocyte count decreased | 41 (27.9) | 35 (27.3) | NR | NR | NR | NR |
Metabolism and nutrition disorders | 24 (16.3) | 23 (18.0) | NR | NR | NR | NR |
Nervous system disorders | 33 (22.4) | 13 (10.2) | NR | NR | NR | NR |
Vascular disorders | 19 (12.9) | 11 (8.6) | NR | NR | NR | NR |
Mucositis | NR | NR | 46 (13.1) | 58 (15.4) | 41 (15.0) | 49 (17.7) |
Withdrawal due to adverse events, n (%) | ||||||
Patients who discontinued treatment due to adverse events, side effects, or complications | 14 of 152 (9.2)c | 5 of 134 (3.7)c | NR | NR | NR | NR |
Treatment-emergent serious adverse events (occurring in ≥ 10% in either study arm), n (%) | ||||||
Patients with ≥ 1 treatment-emergent adverse events requiring expedited reporting | 82 (55.8) | 36 (28.1) | NR | NR | NR | NR |
Difference (95% CI) | 27.66 (16.47 to 38.84) | NR | NR | |||
Blood and lymphatic system disorders | 20 (13.6) | 15 (11.7) | NR | NR | NR | NR |
Febrile neutropenia | 18 (12.2) | 15 (11.7) | NR | NR | NR | NR |
General disorders and administration site conditions | 20 (13.6) | 3 (2.3) | NR | NR | NR | NR |
Infections and infestations | 33 (22.4) | 19 (14.8) | NR | NR | NR | NR |
Investigations | 23 (15.6) | 6 (4.7) | NR | NR | NR | NR |
Nervous system disorders | 22 (15.0) | 0 (0.0) | ||||
Treatment-emergent fatal adverse events, n (%) | ||||||
Patients who died, n (%) | 3 (2.0) | 2 (1.6) | 0 (NR) | 0 (NR) | 3 (NR) | 2 (NR) |
Difference (95% CI) | 0.48 (–2.66 to 3.62) | NR | NR | |||
Cardiac disorders | 0 (0.0) | 1 (0.8) | 0 | 0 | 0 | 0 |
Cardiac arrest | 0 (0.0) | 1 (0.8) | 0 | 0 | 0 | 0 |
Infections and infestations | 2 (1.4) | 1 (0.8) | 0 | 0 | 0 | 0 |
Sepsis | 2 (1.4) | 1 (0.8) | 0 | 0 | 1 (NR) | 2 (NR) |
Nervous system disorders | 1 (0.7) | 0 (0.0) | 0 | 0 | NR | NR |
Intracranial hemorrhage | 1 (0.7) | 0 (0.0) | 0 | 0 | 0 | 0 |
Hypoxic ischemic encephalopathy | 0 | 0 | 0 | 0 | 1 (NR) | 0 |
Multiorgan failure | 0 | 0 | 0 | 0 | 1 (NR) | 0 |
Treatment-emergent adverse events of special interest, n (%) | ||||||
Cytokine release syndrome | 23 (15.6) | 0 (0.0) | NR | NR | NR | NR |
Tumour lysis syndrome | NR | NR | NR | NR | NR | NR |
Neurotoxicity | 4 (2.7) | 0 (0.0) | NR | NR | NR | NR |
Immune effector cell-associated neurotoxicity syndrome | NR | NR | NR | NR | NR | NR |
Infections and infestations | 51 (34.7) | 35 (27.3) | NR | NR | NR | NR |
Sepsis | 15 (10.2) | 13 (10.2) | NR | NR | NR | NR |
Pneumonia | 5 (3.4) | 2 (1.6) | NR | NR | NR | NR |
Fusarium infection | NR | NR | NR | NR | NR | NR |
Fungal pneumonia | NR | NR | NR | NR | NR | NR |
Septic shock | NR | NR | NR | NR | NR | NR |
Aspergillus infection | NR | NR | NR | NR | NR | NR |
Bronchopneumonia | NR | NR | NR | NR | NR | NR |
Candida infection | NR | NR | NR | NR | NR | NR |
Enterococcal bacteremia | NR | NR | NR | NR | NR | NR |
Escherichia sepsis | NR | NR | NR | NR | NR | NR |
Lung infection | NR | NR | NR | NR | NR | NR |
Pancreatitis | 0 (0) | 1 (0.8) | NR | NR | NR | NR |
CI = confidence interval; NR = not reported; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Note: DCO date: June 23, 2023.
aCategory called “sepsis or catheter-related infection” in AALL1731. Sepsis of CTCAE grade 3 was defined by the presence of a positive blood culture and signs or symptoms, along with an indication for treatment. Sepsis of CTCAE grade 4 was defined by having life-threatening consequences and an indication for urgent intervention.
bThis category includes all other infection-related CTCAE, version 5.0, terms other than febrile neutropenia and sepsis or catheter-related infection.
cEstimated among the Step 3 analysis set.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
E1910 Trial
Following the FDA’s accelerated approval of blinatumomab for patients with MRD-positive B-cell precursor ALL in March 2018, patients in the E1910 trial who were MRD-positive after induction and intensification therapy were no longer randomized but rather assigned to the blinatumomab plus chemotherapy group, comprising 18 (29% of the overall Step 3 MRD-positive group). This protocol amendment occurred because of updated evidence indicating blinatumomab plus chemotherapy should be the standard of care for individuals with MRD-positive status.12,13 As a result, a higher percentage of patients in the blinatumomab plus chemotherapy arm were MRD-positive (14.0%) than in the chemotherapy arm (7.7%). Patients with MRD-positive status have a higher chance of relapse; however, the direction and magnitude of this potential selection bias is unclear.8
Baseline characteristics for the Step 3 analysis set were generally similar between treatment arms, with exceptions including a lower percentage of males (45.4% in the blinatumomab plus chemotherapy arm versus 52.2% in the chemotherapy arm) and fewer patients aged 65 or older (7.9% versus 14.9%, respectively) in the blinatumomab plus chemotherapy arm. Baseline characteristics were also generally similar between treatment arms for those who were MRD-negative. However, the distribution of most characteristics was not similar for patients who were MRD-positive (Table 26; e.g., in the blinatumomab plus chemotherapy arm 35.0% versus 63.6% in the chemotherapy arm were male, 75.0% versus 95.5% were white, respectively, and 75.0% versus 95.5% did not have bone marrow involvement at baseline, respectively). This dissimilar distribution may be due to the relatively small sample sizes per treatment group among those with MRD-positive status (N = 40 for blinatumomab plus chemotherapy and N = 22 for chemotherapy alone) or the impact of the switch from randomized to assigned patient allocation in this subgroup, or both. Nonetheless, the absence of randomization for patients MRD-positive status increases the risk of imbalance in measured and unmeasured confounders, although, as mentioned, the magnitude and direction of this potential selection bias is hard to determine.
The percentage of patients with consolidation chemotherapy dose modifications was similar across treatment arms, indicating low risk of bias related to deviations in intended treatment protocols.
The open-label nature of the E1910 trial poses a risk of bias from lack of blinding. This risk is minimal for objective outcomes such as OS; however, it remains for more subjectively assessed outcomes, such as RFS and AEs. While a central laboratory reviewed and confirmed relapses to mitigate potential bias, assessment bias remains a risk for AEs.
As none of the analyses in the E1910 trial were adjusted for multiple testing, there is an increased risk of type I error for statistically significant results.
An independent Data Safety Monitoring Committee reviewed the results of the third interim analysis and determined that the efficacy threshold for the primary end point was met (the threshold was not reported), based on the “positive efficacy results reported in the blinatumomab plus chemotherapy arm.”19 Consequently, the third interim analysis results were designated as the primary analysis at the June 23, 2023, DCO. Given that all results from the E1910 trial were based on interim analyses, observed treatment effects may be overestimated for blinatumomab plus chemotherapy.11 Additionally, data in the E1910 trial remained immature at the June 23, 2023, DCO (59 of the planned 94 OS events had occurred in the FAS), with the median time to OS and RFS events not reached in all arms and cohorts other than the chemotherapy arm of the Step 3 MRD-positive analysis set. Nonetheless, as the clinical expert with experience treating adults with ALL felt the chemotherapy arm performed as expected, bias resulting from the interim analysis effect and immature data were deemed to be minimal.
OS and RFS results from the Step 3 analysis set (MRD-positive and MRD-negative combined) must be interpreted in light of the fact that these are from post hoc analyses, which are at risk of data manipulation. However, comparison of these post hoc analyses to the primary (OS) and key secondary (RFS) outcomes in the FAS revealed similar treatment effects for the blinatumomab plus chemotherapy arm, mitigating concerns about data validity.
Stratified Cox proportional hazards models, adjusted for stratification factors, were used to estimate the HRs and CIs for OS and RFS. These models assume proportional hazards across treatment arms. Visual inspection of the KM curves by the CDA-AMC review team revealed that, for OS and RFS, the curves for the intervention and comparator treatment arms crossed multiple times and did not separate until approximately 6 and 4 months, respectively. While this suggests that the HRs may not reflect the treatment effect over time, it is more likely a result of variation in effects between the treatment and an active control during the early stages of treatment initiation. The KM curves remained separate for the remainder of the observation period, suggesting that the proportional hazards assumption was adequately met. Additionally, sensitivity analyses in the MRD-negative FAS using RMST, which does not rely on the proportional hazards assumption, supported the results of the Cox proportional hazards models for OS and RFS.
The OS analyses in the Step 3 analysis set, the FAS, and the Step 3 MRD-positive analysis set indicated a survival benefit for the blinatumomab plus chemotherapy arm compared to chemotherapy alone. However, its internal validity may have been influenced by the potential impact of postrelapse therapies. The OS analysis was based on the ITT approach, which assumes that postrelapse therapies are nondifferentially distributed between groups — a condition that may not hold, given the observed disparities in postrelapse therapy used (Table 17). A higher percentage of patients in the chemotherapy arm received off-protocol blinatumomab (25.4% versus 0% in the blinatumomab plus chemotherapy arm) and allogeneic SCT (12.7% versus 7.9%, respectively), whereas receipt of CAR T-cell therapy was █████████████ in the blinatumomab plus chemotherapy arm (████ versus ████). In response to questions from CDA-AMC reviewers, the sponsor stated that the study protocol did not specify the types or timing of postrelapse therapies to better reflect real-world clinical practice. Additionally, the OS analyses did not control or adjust for subsequent postrelapse therapy. CDA-AMC reviewers agree that this approach would improve generalizability of the OS results, but there is potential for confounding by postrelapse therapy, especially considering the noted differences in the use of efficacious postrelapse therapies. Sensitivity analyses were conducted (primarily on the MRD-negative subgroup OS analyses), with some adjusting for allogeneic SCT or censoring at the start of nonprotocol therapies. These analyses, while yielding consistent directionality of benefit with blinatumomab plus chemotherapy, revealed attenuation or nonstatistical significance of the treatment effect in specific scenarios (e.g., sensitivity analysis that censored patients at allogeneic SCT or start of nonprotocol systemic anticancer therapy). Although the observed OS effect represents the combined impact of frontline blinatumomab plus chemotherapy plus subsequent treatments, the overall effect of the differences in use of postrelapse therapy was more likely to favour the chemotherapy alone arm.
The FDA considers OS the gold-standard primary outcome in oncology, and its definition in the E1910 trial aligns with FDA guidance.73 RFS is considered to be an important cancer end point by the FDA in situations where survival may be prolonged, making an OS end point impractical.73 RFS is a composite end point that not only accounts for survival but also includes relapse, which is desired in patients with acute leukemias. RFS can also be assessed before a survival benefit is demonstrated.73 No data were submitted by the sponsor supporting the correlation of RFS with OS in adults with Ph-negative B-cell precursor ALL. Thus, the validity of RFS as a surrogate for OS is unclear in the current context.
The date of AEs and deaths with partially missing dates was imputed. Imputations of dates at which AEs or death occurred with only the known year relies on potentially unrealistic assumptions of noninformative missingness (i.e., missing completely at random or missing at random), which may introduce bias. These approaches introduce bias if missing not at random (MNAR) mechanisms, such as unrecorded events following specific treatments, are present. Further, this imputation approach fails to capture the uncertainty in the imputed values, resulting in an underestimation in the variability and potentially affecting the resulting CIs. Without information on the degree of missingness that lead to imputations in AE and death, it was not possible to assess the impact of bias from censoring.
AALL1731 Trial
The AALL1731 trial used stratified randomization by risk of relapse (average versus high) and the presence of Down syndrome among those with SR-average (yes versus no) risk category. Baseline characteristics were generally similar between treatment arms, except for MRD in peripheral blood on day 8 in both the SR-average and SR-high subgroups and for cytogenic risk group in the SR-high subgroup. More patients in the blinatumomab plus chemotherapy arms (41.5% in SR-average and 40.5% in SR-high) than in the chemotherapy arms (35.2% in both SR-average and SR-high) had 1% and greater MRD in peripheral blood on day 8. A smaller percentage of SR-high patients in the blinatumomab plus chemotherapy arm had favourable cytogenetics (23.9% versus 31.6%), and a larger percentage had neutral cytogenetics (54.8% versus 48.7%). Both MRD and cytogenetic risk are well-established prognostic factors in ALL and influence treatment response and outcomes.14-16 The highlighted differences indicate that a larger percentage of patients in the blinatumomab plus chemotherapy arm would be at higher risk of relapse and reduced survival. These could contribute to bias in the comparative outcomes between the treatment arms, potentially favouring chemotherapy. The analyses of OS and DFS based on Cox proportional hazards models were stratified by SR-average with and without Down syndrome, SR-average with Down syndrome, and SR-high risk categories. Stratification by SR-average and SR-high risk categories would not directly adjust for imbalances in other prognostic factors, such as MRD and cytogenetic risk, unless these are included as covariates in the model.74,75
Among randomized patients, more patients assigned to the blinatumomab plus chemotherapy arm did not start postconsolidation treatment (n = 55 of those with SR-average risk and n = 20 of those with SR-high risk) than in the chemotherapy arm (n = 27 of patients with SR-average risk and n = 13 of those with SR-high risk). The primary reason for patients not starting postconsolidation therapy was being withdrawn from the blinatumomab plus chemotherapy arms, while in the chemotherapy arms, the primary reason for not starting postconsolidation treatment was due to patients not being able to be evaluated. These patients were included in the ITT analyses. ITT analyses are widely used in clinical trials to preserve randomization and provide an unbiased estimate of the treatment effect. The ability of ITT analyses to handle attrition bias depends on the context and the mechanisms of missing data. ITT analyses will typically produce unbiased treatment estimates in situations where data are missing completely at random or missing at random if appropriate methods are used to impute missing data (e.g., multiple imputation). However, in the case of data MNAR, the ITT may produce biased estimates. It was reported for the study that substantial or informative missingness was neither anticipated nor planned for, based on experience from the previous Children’s Oncology Group ALL trial. However, given that reasons for not starting postconsolidation therapy across the risk strata and treatment groups included those that may result in data MNAR (e.g., patients not able to be evaluated), this assumption is potentially unrealistic. Without additional information or results from analyses to determine whether data were MNAR, it is not possible to determine the impact of attrition bias, if any. The clinical expert consulted for this review with experience treating pediatric patients with ALL noted the imbalance in not starting postconsolidation treatment would likely have resulted in underestimation of the true between-group differences. No data on treatment completion, discontinuation, dose modifications, and the use of off-protocol treatments were publicly available for the AALL1731 trial. Therefore, a comprehensive assessment of attrition and adherence and their potential impact on outcomes could not be done.
Similar to the E1910 trial, the open-label nature of the AALL1731 trial could introduce risk of bias. As mentioned, the risk of bias due to the absence of blinding is minimal for objective outcomes, such as OS. However, there is substantial risk for more subjectively assessed outcomes such as DFS and AEs. As in the E1910 trial, while relapse events were confirmed via central pathology review to mitigate potential assessment bias for DFS events, bias due to unblinding remains for AEs.
None of the analyses in the AALL1731 trial were adjusted for multiple testing, thereby increasing the risk of type I error for statistically significant results.
All results from the AALL1731 trial should be interpreted in light of the fact that these are based on interim analyses, which may overestimate treatment effects.11 Data in the AALL1731 trial remained immature at the June 30, 2024, DCO (81 of 194 DFS events had occurred in the overall cohort). Nonetheless, as the clinical expert with experience treating pediatric patients with ALL felt the chemotherapy arm performed as expected, bias resulting from the interim analysis effect and immature data were deemed to be minimal.
OS results must be interpreted in light of the fact that these are from post hoc analyses, which are at risk of data manipulation. OS results in the AALL1731 trial revealed little to no differences across treatment arms, which the clinical expert with experience treating children with ALL indicated was expected, due to highly effective salvage therapy in this population. The clinical expert indicated that a longer follow-up would likely be needed to observe noticeable differences across the blinatumomab plus chemotherapy arm versus the chemotherapy arm.
The AALL1731 trial planned to use stratified Cox proportional hazards models, adjusted for stratification factors, to test DFS. Tests of the proportional hazards assumption, including the Schoenfeld residuals test (P = 0.031), a Wald test of time-varying interaction (P = 0.048), and a Kolmogorov-type supremum test (P = 0.068), suggested the assumption may not hold. Visual inspection of the KM curves showed no converging or crossing of the DFS curves but indicated the treatment effect could have a delay of about 2 months before achieving a separation of the curves. In a sensitivity analysis, DFS was analyzed using the RMST method, which does not rely on the proportional hazards assumption and is an appropriate alternative survival analysis approach. RMST analyses require selection of an appropriately long time horizon to truncate the analysis, which could bias results if the time horizon is not sufficiently long. In the AALL1731 trial, the RMST analysis was truncated at the maximum follow-up time (1,652 days or 4.52 years). As the median follow-up was 2.5 years (IQR, 1.6 to 2.3 years), and visual inspection of the KM curves showed little risk of the curves crossing beyond 2 years, the CDA-AMC review team deemed the RMST time horizon appropriate in the context of the trial’s follow-up period. The results of the RMST method, which do not rely on the proportional hazards assumption, supported the results of the Cox proportional hazards models for DFS.
As previously mentioned, the FDA considers OS the gold-standard primary outcome in oncology, and its definition in the AALL1731 trial aligned with FDA guidance.73 Like RFS, DFS is considered to be an important cancer end point by the FDA in situations where survival may be prolonged, making an OS end point impractical.73 DFS can also provide insight into the durability of a treatment response.73 A previous study has shown a significantly positive correlation between DFS (evaluated as leukemia-free survival) and OS outcomes in acute myeloid leukemia.76 However, no data were submitted by the sponsor supporting the correlation of DFS with OS in pediatrics with Ph-negative B-cell precursor ALL. Thus, the validity of DFS as a surrogate for OS in the current context is unknown.
Based on previous ALL trials, the AALL1731 trial did not anticipate or plan for missing data, and no data imputations were performed. It was only reported that no data were missing from the primary DFS analyses. As no information was provided on the degree of missingness for other outcomes, the CDA-AMC review team cannot determine the accuracy of this assumption.
Data completeness and transparency of reporting were limitations in the AALL1731 trial. Detailed patient disposition among randomized patients, the receipt of on- and off-protocol therapy, any TEAEs, withdraws due to AEs, treatment-emergent SAEs, and notable harms were not available.
External Validity
E1910 Trial
The clinical experts with expertise in the diagnosis and management of adults with Ph-negative B-cell precursor ALL felt the inclusion and exclusion criteria used in the E1910 trial were generally aligned with those typically used in ALL clinical trials. While the E1910 trial restricted inclusion to only those aged 30 to 70 years, the clinical expert consulted for this review felt the results could be generalized to all adults eligible for chemotherapy, regardless of age. This same clinical expert consulted for this review stated that the percentage of adult patients completing and discontinuing treatment during each phase of the trial aligned with clinical practice in Canada.
While only 5.9% of patients randomized in the E1910 trial were from Canada, which may lower the generalizability of the results to the Canadian setting, the clinical expert consulted for this review who treats adult patients indicated the baseline characteristics of the study population were representative of patients seen in clinical practice in Canada.
The outcomes measured in the E1910 trial addressed the key treatment goals identified by patient and clinician group inputs submitted to CDA-AMC and were deemed to be relevant by the consulted clinical experts.
The clinical expert consulted for this review with expertise in the diagnosis and management of adults with Ph-negative B-cell precursor ALL noted that the modified DFCI regimen is the most commonly used protocol to treat Ph-negative B-cell precursor ALL in Canada. As the E1910 trial used a chemotherapy regimen based on the UKALLXII/E2993 chemotherapy protocol, with dosing modifications based on the C10403 AYA trial, the generalizability of the trial results to clinical practice in Canada may be limited. The 1 clinical expert consulted for this review with experience treating adult patients with ALL stated that the efficacy of the chemotherapy regimen used in the E1910 trial, as measured by OS and RFS (including CR), can be expected to be similar to that of the modified DFCI regimen.
AALL1731 Trial
The clinical experts with expertise in the diagnosis and management of children with Ph-negative B-cell precursor ALL felt the inclusion and exclusion criteria used in the AALL1731 trial were generally aligned with those typically used in ALL clinical trials. Nonetheless, the AALL1731 trial imposed various restrictions that limits the generalizability to the pediatric population of interest for the reimbursement request under review. This includes the AALL1731 trial restriction to those aged 1 to 10 years; however, the clinical experts consulted for this review felt the results of the AALL1731 trial could be generalized to patients under 1 year old and those aged 10 to 18 years.
While the AALL1731 trial restricted enrolment to patients with SR ALL and randomization to those with an average or high risk of relapse, the clinical expert consulted for this review with experience in the diagnosis and management of pediatric patients with ALL felt those with SR-favourable (including patients with SR-average and undetectable MRD assessed by HTS at the end of induction) may not require treatment with blinatumomab, as their expected event-free survival on a conventional chemotherapy backbone is anticipated to be greater than 95%. However, the clinical expert consulted for this review felt the decision to treat pediatric patients with SR-favourable status should be left up to the treating physician. The clinical expert consulted for this review with experience treating pediatric patients with ALL noted that the definition of SR B-cell ALL is well established and felt the results among the cohort with a standard-risk B-cell ALL and high risk of relapse (i.e., SR-high) could be generalized to patients with high-risk and very high–risk B-cell ALL. In addition, based on the AALL1731 data and in the context of the E1910 data, the Children’s Oncology Group halted its study for high-risk B-cell ALL (COG AALL1732) and recommended that these patients also receive 2 nonsequential blocks of blinatumomab as part of their standard treatment.
The clinical expert consulted for this review felt the percentage of patients completing and discontinuing treatment during each phase of the trial was in line with clinical practice in Canada.
While only 6.1% of patients randomized in the AALL1731 trial were from Canada, which may lower the generalizability of the results to the Canadian setting, the clinical expert consulted for this review who treats pediatric patients indicated the baseline characteristics of the study population were representative of those for patients seen in clinical practice in Canada.
The outcomes measured in the AALL1731 trial addressed the key treatment goals identified by patient and clinician group inputs submitted to CDA-AMC for this review and were deemed to be relevant by the consulted clinical experts.
Patients with Down syndrome who were high risk were nonrandomly assigned to receive blinatumomab plus a less intensive chemotherapy regimen, and no data were received for this cohort (n = 49). In contrast, patients with Down syndrome who met the criteria for inclusion into the SR-average cohort proceeded to randomization. Of the 835 patients included in the SR-average cohort, 36 (3.8%) had Down syndrome. By the June 30, 2024, DCO it was reported that none of the 16 patients with SR-average and Down syndrome assigned to the blinatumomab plus chemotherapy arm experienced a DFS event compared to 2 of the 16 patients with SR-average and Down syndrome assigned to the chemotherapy arm experienced a DFS event. While no data were available for patients with Down syndrome who had high-risk disease, the clinical expert with experience treating pediatric patients with ALL noted that results from the SR-high cohort could be generalized to these patients.
Patients with SR-high risk category after induction therapy and who had MRD of 0.1% or greater were reassessed for MRD at the end of consolidation, which is common practice according to the clinical expert with experience treating pediatric patients. Patients with end-of-consolidation MRD of less than 0.1% were randomized, those with MRD of 0.1% to less than 1.0% (N = 14) were nonrandomly assigned to receive blinatumomab plus chemotherapy (results are not available), and those with MRD greater than 1% (N = 7) were removed from the trial. The clinical expert treating children agreed to generalize the results of patients with SR-high and MRD of less than 0.1% at the end of consolidation to patients with MRD of 0.1% to less than 1.0%. The clinical expert treating children noted that the patient population with SR-high risk category and end-of-consolidation MRD of 1% or greater (N = 7) was removed from the trial, as these patients reflect a rare and very high–risk subgroup that is considered refractory and would be managed differently.
Pediatric patients with SR B-cell ALL and average risk of relapse after induction (MRD less than 0.01% by flow cytometry and neutral cytogenetics) were assessed for their MRD status via HTS at the end of consolidation. If MRD by HTS assessment was undetectable, patients were nonrandomly assigned to chemotherapy alone, and results are not available. The clinical expert with experience treating pediatric patients with ALL noted that HTS is not widely available and most centres in Canada do not have access to this methodology. Nonetheless, the clinical expert noted that patients with undetectable MRD by HTS would be considered to have SR-favourable ALL, and, as such, may not require treatment with blinatumomab as their expected event-free survival on a particular chemotherapy backbone is expected to be greater than 95%. The clinical experts cautioned against any restrictions by relapsing risk as the molecular characterization of leukemia is rapidly evolving, and the identification of a new high-risk somatic change may alter how patients are currently classified.
The clinical experts consulted for this review noted that the chemotherapy regimen used in the AALL1731 trial and the modified DFCI regimen are the most commonly used protocols to treat Ph-negative B-cell precursor ALL in Canada. The chemotherapy regimen used in the AALL1731 trial is representative of practice in Canada.
E1910 and AALL1731 Trials
While the comparators in the E1910 and AALL1731 trials were deemed to be acceptable by the clinical experts consulted for this review, blinatumomab is already publicly reimbursed for patients who are MRD-positive (per PC0204 CDA-AMC reimbursement review). No direct or indirect evidence was provided assessing the comparative efficacy of blinatumomab for the indication currently funded versus that currently being reviewed.
The indication currently funded for blinatumomab partially overlaps with the indication currently under review. Differences between the indication currently funded versus that currently being reviewed for blinatumomab in patients who are MRD-positive include remission status (first and second remission versus first remission), MRD restrictions (greater than or equal to 0.1% versus no MRD restriction), administration schedule (sequential cycles versus alternating with consolidation chemotherapy), and number of cycles of blinatumomab treatment for pediatric patients (maximum of 4 versus 2 cycles of blinatumomab). The clinical experts consulted for this review indicated that, under the currently funded indication, blinatumomab is typically used to achieve an MRD-negative status before SCT or maintain disease control in patients who cannot proceed to SCT. In comparison, the incorporation of blinatumomab alternating with consolidation chemotherapy (per the current review) offers patients a chance at prolonged remission or cure while avoiding SCT. The dosage of blinatumomab (28 mcg/day for patients 45 kg or greater; 15 mcg/m2 per day [maximum 28 mcg/day] for patients less than 45 kg) is applicable to both indications. The clinical experts consulted for this review indicated that, for patients who are MRD-positive, it is expected that blinatumomab per the indication currently funded and that currently being reviewed would have a similar efficacy. According to the clinicians consulted for this review, clinicians would likely prefer prescribing blinatumomab per the current review because they can incorporate blinatumomab with chemotherapy and do not need to complete 3 intensive chemotherapy blocks. In addition, clinical experts consulted for this review noted that the current review of blinatumomab adds value for patients who have lower levels of MRD (MRD of 0.01% to less than 0.1%, constituting an estimated 5% to 10% of patients as per clinical experts), who are excluded from the currently funded blinatumomab indication. The clinical experts agreed that blinatumomab as per current funding should continue to be available to patients, as adoption of the blinatumomab as per the current review into B-cell ALL treatment centres across Canada may take time, requiring training and building standardized medical protocols and workflows. Further, in situations where pediatric patients cannot tolerate additional cycles of consolidation chemotherapy, blinatumomab per the currently funded indication allows for receipt of multiple cycles of blinatumomab to manage leukemia without requiring alternating treatment with consolidation chemotherapy. According to the clinical experts, the choice between these blinatumomab indications should be left to the treating clinician and the patient.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:17,18
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. While literature-based thresholds were unavailable for the current review, the clinical experts consulted for this review provided estimates for clinically meaningful thresholds for all outcomes assessed using GRADE.
Findings from the E1910 trial were included in the GRADE assessments. Results from the AALL1731 trial were not appraised using GRADE because only published results, rather than a formal Clinical Study Report, were provided to CDA-AMC, and the data were based on publicly available sources.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS (probabilities at 3 and 5 years) and RFS (probabilities at 1, 3, and 5 years) in the overall population (i.e., regardless of MRD status) and MRD-negative and MRD-positive populations
harms (any grade 3 or greater TEAEs, treatment-emergent SAEs, and fatal AEs) in the overall population.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for blinatumomab plus chemotherapy versus chemotherapy for adults with Ph-negative B-cell precursor ALL.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Evidence
No indirect evidence was submitted.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies to address gaps within the systematic review evidence were submitted.
Discussion
Summary of Available Evidence
One ongoing, phase III, open-label, multicentre, international study, the E1910 trial, met the inclusion criteria of the systematic review conducted by the sponsor, and a second phase III, open-label, multicentre, international study, the AALL1731 trial, was included to inform the reimbursement request among pediatric patients. The E1910 trial enrolled adult patients with newly diagnosed Ph-negative B-cell precursor ALL, while the AALL1731 trial enrolled pediatric patients with newly diagnosed SR Ph-negative B-cell precursor ALL. In both studies, patients were randomized to receive either 15 mcg/m2 per day of blinatumomab for adult and pediatric patients weighing less than 45 kg (up to a maximum of 28 mcg/day) or 28 mcg/day for adult patients weighing 45 kg or more in combination with chemotherapy, or chemotherapy alone. Patients randomized to the blinatumomab plus chemotherapy arms received 4 cycles of blinatumomab in the E1910 trial compared to 2 cycles of blinatumomab in the AALL1731 trial. The primary objectives of both studies were to compare the efficacy and safety of blinatumomab for the treatment of Ph-negative B-cell precursor ALL with the E1910 trial specifically designed to evaluate comparative efficacy and safety among those with MRD-negative disease. The outcomes relevant to this review include OS and harms data from both trials, RFS in the E1910 trial, and DFS in the AALL1731 trial.
Interpretation of Results
The Health Canada–approved indication is for blinatumomab as consolidation treatment for Ph-negative CD19 B-cell ALL, regardless of age (pediatric and adult patients), MRD status, or line of treatment.77 The sponsor’s reimbursement request is for the treatment of adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in the frontline setting. The sponsor’s reimbursement request differs from the Health Canada indication in that it is limited to patients in the frontline setting, while remaining consistent with the Health Canada indication regarding age and MRD status. No data were submitted by the sponsor for the setting of relapsed ALL.
Efficacy
The patient group input collected for this review highlighted that the key treatment goals for patients and their caregivers are to reduce relapses, prolong life, and reduce AEs associated with therapies. The effect of blinatumomab plus chemotherapy at achieving these goals was evaluated for adult patients in the E1910 trial by the assessment of OS, RFS, and AEs and for pediatric patients in the AALL1731 trial by the assessment of OS, DFS, and AEs.
E1910 Trial
Clinical experts consulted for this review considered OS to be the most clinically important outcome for adults. The clinical experts considered a 5% between-group difference in the probability of survival in the overall and MRD-stratified populations to be clinically meaningful. Patients in the Step 3 analysis set aligned with the sponsor’s reimbursement request, which is MRD-agnostic, and the clinical experts consulted for this review agreed the results could be generalized to all adult patients regardless of age. Based on the identified threshold, blinatumomab plus chemotherapy, compared to chemotherapy, results in clinically important increases in the probability of being alive at 3 and 5 years in the Step 3 analysis set (high certainty per GRADE assessment). At a median OS follow-up of 4.5 years in both arms, 83 deaths had occurred in the Step 3 analysis set and the median OS was NE in both arms. Blinatumomab plus chemotherapy demonstrated clinically important improvements in survival rates compared to chemotherapy at 3 and 5 years in patients with MRD-negative disease (i.e., FAS; moderate and high certainty per GRADE assessment, respectively) and in patients with MRD-positive disease (i.e., Step 3 MRD-positive analysis set; moderate and high certainty per GRADE assessment, respectively). The E1910 trial met its primary end point, with statistically significant improvements in OS favouring the blinatumomab plus chemotherapy group in the FAS. Proportionally more deaths occurred in the Step 3 MRD-positive analysis set than in the FAS — which is not surprising based on the poorer prognosis with MRD-positive status — and the median OS was NE in the blinatumomab plus chemotherapy arms of both the FAS and the Step 3 analysis set.
RFS was a secondary outcome in the E1910 trial and considered a clinically important outcome for patients with Ph-negative B-cell precursor ALL by the clinical experts consulted for this review. The clinical experts consulted for this review indicated a 5% to 7% between-group difference in the overall population and a 5% to 10% between-group difference in the MRD-positive and MRD-negative populations to be clinically meaningful. At a median RFS follow-up of 4.5 years in both arms, 92 relapse events had occurred in the Step 3 analysis set and the median RFS was NE in both arms. In the Step 3 analysis set, the between-group differences in RFS at 1, 3, and 5 years were 12.2% (95% CI, 1.8 to 22.7), 17.3% (95% CI, 5.6 to 28.9), and 18.4% (95% CI, 6.3 to 30.6), respectively. These point estimates suggested clinically meaningful increases in the probability of being relapse-free at each time point. While the certainty of evidence at year 1 was rated as moderate because the lower bound of the 95% CI crossed the significance threshold, certainty of evidence at year 3 and 5 was rated as high as per the GRADE approach. Similar results were observed at all time points in the FAS and the Step 3 MRD-positive analysis set, although proportionally more relapse events occurred in the Step 3 MRD-positive analysis set than in the FAS. The median RFS was NE in the blinatumomab plus chemotherapy arms of the Step 3 MRD-positive analysis set and the FAS.
AALL1731 Trial
Similar to the adult population, clinical experts consulted for this review considered OS to be the most clinically important outcome for pediatric patients. Clinical experts consulted for this review indicated that any between-group difference in OS probability greater than 0% is clinically meaningful for children with ALL, both in the overall population and in the subgroups with average and high risk of relapse. As previously noted, the AALL1731 trial imposed various restrictions that limited the generalizability (e.g., age, SR B-cell ALL, and an average or high risk of relapse). However, the clinical experts consulted for this review indicated that results of the overall cohort could be generalized to the Health Canada indication and the sponsor’s reimbursement request, while results from the SR-high cohort could be generalized to those with high-risk or very high–risk B-cell ALL, an end-of-consolidation MRD greater than 0.1% to less than 1%, and those with Down syndrome. In the overall cohort, the probability of survival from randomization to 3 years was 98.4% (SE = 0.8%) in the blinatumomab plus chemotherapy arm and 97.1% (SE = 1.1%) in the chemotherapy arm; the associated probabilities in the subgroup of patients with SR-average risk category were 100.0% (SE = not applicable) and 98.4% (SE = 1.0%), respectively, and in the SR-high subgroup were 96.1% (SE = 2.0%) and 95.3% (SE = 2.2%), respectively. As the precise between-group differences in the proportion of patients surviving from randomization to 3 years were not available for the AALL1731 trial, it was not possible to conclusively determine whether the between-group difference included the expert-defined clinically meaningful threshold of 0%. Visual inspection of the OS KM curves suggested little to no difference between the blinatumomab plus chemotherapy arms compared to the chemotherapy alone arms. The clinical expert with experience treating pediatric patients with ALL indicated this observation is as expected due to the established effectiveness of chemotherapy and salvage therapies in this population. The clinical expert also indicated that a longer follow-up is needed to observe meaningful differences in OS between treatment arms.
DFS was the primary outcome in the AALL1731 trial and considered to be a clinically important outcome for patients with Ph-negative B-cell precursor ALL by the clinical experts consulted for this review. The clinical experts consulted for this review indicated a 3% to 5% between-group difference in the overall population and in the populations at average and high risk of relapse to be clinically meaningful. In the overall population, the probability of DFS from randomization to 3 years was 96.0% (SE = 1.2%) in the blinatumomab plus chemotherapy arm and 87.9% (SE = 2.1%) in the chemotherapy arm. The associated probabilities in the subgroup of patients with SR-average were 97.5% (SE = 1.3%) and 90.2% (SE = 2.3%), respectively, and in the SR-high subgroup were 94.1% (SE = 2.5%) and 84.8% (SE = 3.8%), respectively. Visual inspection of the DFS curves suggested that the differences in DFS probabilities across all groups exceeded the expert-defined clinically meaningful threshold range of 3% to 5%. While the proportion of patients disease-free at 3 years was assessed, between-group differences were not available for the AALL1731 trial. Assessments of the proportional hazards assumption revealed evidence that the proportionality assumption may not hold; consequently, DFS sensitivity analyses were performed using RMST. The unadjusted difference in disease-free RMST was 72 days (95% CI, 30 to 108 days) in the overall population, 67 days (95% CI, 24 to 110 days) in the SR-average cohort, and 79 days (95% CI, 17 to 140 days) in the SR-high cohort. Across all cohorts, results from the DFS RMST analyses supported the results of the Cox proportional hazards models for DFS. The clinical expert consulted by CDA-AMC for this review with experience treating pediatric patients with ALL felt it would be difficult to determine an exact threshold at which the difference in disease-free RMST would be clinically important. However, it was their opinion that the observed mean differences indicate a clinically meaningful treatment benefit associated with blinatumomab plus chemotherapy compared to chemotherapy alone.
In populations where a large percentage of patients achieve CR with chemotherapy or when survival may be prolonged, such as pediatric patients with ALL, longer follow-up times may be required to detect benefits in OS end points.73 This was particularly relevant in the AALL1731 trial, where only small differences in OS were observed 3 years after randomization. In such situations, the FDA recognizes RFS and DFS as important outcomes for assessing the durability of a treatment response.73 However, no evidence was submitted supporting the correlation of RFS or DFS with OS in adult and pediatric patients with Ph-negative B-cell precursor ALL. Consequently, the validity of RFS and DFS as surrogate end points for OS in this context remains uncertain.
The chemotherapy used in the E1910 trial was based on the UKALLXII/E2993 chemotherapy regimen, with dosing modifications based on C10403 AYA trial. While the clinical expert with experience treating adults with ALL noted that the modified DFCI regimen is more commonly used in Canada, the efficacy of both regimens is similar and not anticipated to impact generalizability to the Canadian setting.
The open-label nature of both the E1910 trial and the AALL1731 trial raises concerns of bias due to lack of blinding. As OS is an objective outcome, the risk of bias due to lack of blinding is minimal. Both the E1910 trial and the AALL1731 trial used central laboratories to review and confirm relapses to mitigate potential bias due to unblinding. However, assessment bias remains a risk for AEs.
While health-related quality of life (HRQoL) outcomes were identified as an important outcome for both adult and pediatric patients with B-cell precursor ALL, the E1910 trial did not collect any HRQoL outcomes, and none of the protocol-specified HRQoL data collected in the AALL1731 trial were available from published data sources. Clinical experts consulted for this review highlighted that the practice around the collection and assessment of HRQoL outcomes in patients with ALL are evolving and that there are currently no standard patient-reported outcomes in ALL clinical trials.
While the study design of trials informing the evidence under review imposed restrictions that raised concerns regarding the generalizability of the results (e.g., age restriction in adult and pediatric patients; risk group and MRD restriction in pediatric patients), clinical experts consulted for this review felt the results could be generalized to the target population of the reimbursement request under review (i.e., adult and pediatric patients with Ph-negative B-cell precursor ALL). The clinical experts noted an exception for pediatric patients with an SR-favourable risk category, as conventional chemotherapy regimens are highly effective in this group; however, the clinical experts consulted for this review felt the decision to treat pediatric patients with SR-favourable risk should be left up to the treating physician.
For patients with MRD-positive status, consolidation monotherapy with blinatumomab is currently funded for MRD-positive patients. However, no direct or indirect comparative evidence was submitted for the blinatumomab indication currently funded versus that currently being reviewed. It was the opinion of clinical experts consulted for this review that, for patients with MRD-positive status, the efficacy of blinatumomab per the currently funded indication and that currently being reviewed would be similar.
Harms
A similar percentage of patients in both treatment arms of the Step 3 safety analysis set in the E1910 trial experienced a TEAE (145 of 147 [98.6%] in the blinatumomab plus chemotherapy arm and 125 of 128 [97.7%] in the chemotherapy arm). In the blinatumomab plus chemotherapy arm of the E1910 trial, 95.9% of patients experienced a grade 3 or greater TEAE, compared to 97.7% in the chemotherapy arm (based on the GRADE assessment, blinatumomab plus chemotherapy likely results in little to no difference in the incidence of grade 3 or greater TEAEs compared to chemotherapy). A higher percentage of patients in the blinatumomab plus chemotherapy arm of the Step 3 safety analysis set in the E1910 trial experienced treatment-emergent SAEs (55.8%) than in the chemotherapy arm (28.1%; based on the GRADE assessment, blinatumomab plus chemotherapy likely results in a clinically important increase in the incidence of treatment-emergent SAEs compared to chemotherapy). A similar percentage of patients in both treatment arms of the Step 3 safety analysis set experienced a fatal TEAE (2.0% in the blinatumomab plus chemotherapy arm and 1.6% in the chemotherapy arm; based on the GRADE assessment, the evidence is very uncertain about the effect of blinatumomab plus chemotherapy on fatal TEAEs when compared to chemotherapy).
In the AALL1731 trial, the incidence of selected grade 3 or higher TEAEs was well balanced across treatment arms. Selected grade 3 or higher TEAEs were the only the AALL1731 trial harm data submitted for this review, thus limiting the ability to fully appraise harms in the pediatric population.
For the E1910 and AALL1731 trials, the clinical experts consulted for this review felt the data presented did not raise any new concerns about the safety profile of blinatumomab, and the observed events were consistent with what is expected with the drug.
The product monograph indicated that patients treated with blinatumomab are at increased risk of cytokine release syndrome, TLS, neurologic toxicities, infections, and pancreatitis. Premedication with dexamethasone is recommended, and patients should be observed closely for infusion reactions, particularly during the first infusion of the first and second cycle. Management of these events and reactions may require either temporary interruption or discontinuation of blinatumomab.
The clinical experts consulted for this review expected that a higher percentage of patients treated with blinatumomab plus chemotherapy would have TEAEs due to receiving more treatment than those in the chemotherapy arm. The clinical experts consulted by the review team determined that, overall, the safety profile of blinatumomab plus chemotherapy was consistent with their expectations for this drug and deemed it acceptable.
Conclusion
The E1910 (adult-only population) and the AALL1731 (pediatric-only population) trials were phase III, open-label, multicentre, international studies aimed at evaluating the efficacy and safety of blinatumomab used in the consolidation phase of multiphase chemotherapy for the treatment of patients with Ph-negative B-cell precursor ALL.
A post hoc analysis in the E1910 trial aligned with the target patient population under review, which consists of patients with MRD-agnostic, Ph-negative, B-cell precursor ALL. The results of the post hoc analysis showed that blinatumomab plus chemotherapy demonstrated clinically meaningful improvements in OS and RFS compared to chemotherapy, with high to moderate certainty of evidence across different time points. The E1910 trial met its primary end point, with statistically significant improvements in OS favouring the blinatumomab plus chemotherapy group in patients with MRD-negative status. Secondary analyses for OS in patients with MRD-positive status and RFS in patients with MRD-negative or MRD-positive status were supportive of the benefit observed with blinatumomab plus chemotherapy over chemotherapy in the primary analyses, with high to moderate certainty of evidence across different time points. Post hoc analyses are at risk of data manipulation; however, results of the post hoc analyses were consistent with those for the primary and secondary outcomes, mitigating concerns about data validity.
The primary analysis in the AALL1731 trial for the overall trial population aligned with the present target patient population and suggested clinically meaningful improvements in DFS with blinatumomab plus chemotherapy compared to chemotherapy. Post hoc analyses for OS suggested little to no difference between blinatumomab plus chemotherapy compared to chemotherapy in the target patient population. Post hoc analyses are at risk of data manipulation; however, results of the OS post hoc analyses were consistent with clinical expectations, based on input from the clinical experts consulted by CDA-AMC, mitigating concerns about data validity. DFS and OS results for SR-average and SR-high populations were overall consistent with the results observed in the overall trial population. GRADE assessment was not conducted for results of the AALL1731 trial, as only limited public data were available.
According to clinical experts consulted for this review, the harms profile of blinatumomab plus chemotherapy was consistent with their expectations, given the known AEs.
While blinatumomab is currently reimbursed for patients with Ph-negative CD19-positive B-cell precursor ALL with MRD-positive disease, the clinical experts consulted for this review anticipated similar efficacy between the indication currently funded and that currently being reviewed. According to the clinical experts, both options provide unique advantages and therefore the choice between these 2 options should be left to the treating physician and patient.
References
1.Puckett Y, Chan O. Acute Lymphocytic Leukemia. StatPearls; 2023. Accessed November 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459149/
2.Statistics Canada. Number and rates of new cases of primary cancer, by cancer type, age group and sex [Table: 13-10-0111-01]. 2024. Accessed July 15, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101
3.Canadian Cancer Society. Canadian Cancer Statistics. 2021. Accessed January 22, 2025. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf?rev=2b9d2be7a2d34c1dab6a01c6b0a6a32d&hash=01DE85401DBF0217F8B64F2B7DF43986
4.Canadian Cancer Society. Acute Lymphoblastic Leukemia Statistics. 2024. Accessed March 1, 2024. https://cancer.ca/en/cancer-information/cancer-types/acute-lymphoblastic-leukemia-all/statistics
5.Inaba H, Pui CH. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J Clin Med. 2021;10(9). doi: 10.3390/jcm10091926 PubMed
6.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6):e577. doi: 10.1038/bcj.2017.53 PubMed
7.Mrózek K, Harper DP, Aplan PD. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2009;23(5):991-1010, v. doi: 10.1016/j.hoc.2009.07.001
8.Alberta Health Services. Guideline Resource Unit. Acute Lymphoblastic Leukemia in Adults. Clinical Practice Guideline LYHE-005 – Version 3. 2023. Accessed October 10, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe005-all.pdf
9.Lan X, Wu J, Liao Z, Wu Y, Hu R. Prevalence of symptoms in children with acute lymphoblastic leukaemia: a systematic review and meta-analysis. BMC Cancer. 2023;23(1):1113. doi: 10.1186/s12885-023-11581-z PubMed
10.Withycombe JS, Kubaney HR, Okada M, et al. Delivery of Care for Pediatric Patients Receiving Blinatumomab: A Children's Oncology Group Study. Cancer Nurs. 2024;47(6):451-459. doi: 10.1097/ncc.0000000000001309 PubMed
11.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-7. doi: 10.1001/jama.2010.310 PubMed
12.Amgen Inc. Study E1910 - Clinical Study Report (Primary Analysis) - A Phase III Randomized Trial of Blinatumomab (IND117467, NSC765986) for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults. 2024 [sponsor submitted reference].
13.NCCN. NCCN Clinical Practice Guidelines in Oncology: Acute Lymphoblastic Leukemia Version 2.2024. 2024. Accessed November 22, 2024. https://www.nccn.org/professionals/physician_gls/pdf/all.pdf
14.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood. 2015;126(8):964-71. doi: 10.1182/blood-2015-03-633685 PubMed
15.O'Connor D, Enshaei A, Bartram J, et al. Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol. 2018;36(1):34-43. doi: 10.1200/jco.2017.74.0449 PubMed
16.Rau RE, Dai Y, Devidas M, et al. Prognostic impact of minimal residual disease at the end of consolidation in NCI standard-risk B-lymphoblastic leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer. 2021;68(4):e28929. doi: 10.1002/pbc.28929 PubMed
17.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
18.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
19.Amgen Inc. Study E1910 - Clinical Study Report (Primary Analysis) - A Phase III Randomized Trial of Blinatumomab (IND117467, NSC765986) for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults 2024:123.
20.NCCN. NCCN Guidelines for Patients: Acute lymphoblastic leukemia. 2023. Accessed November 11, 2024. https://www.nccn.org/patients/guidelines/content/PDF/all-patient.pdf
21.Sasaki K, Jabbour E, Short NJ, et al. Acute lymphoblastic leukemia: A population-based study of outcome in the United States based on the surveillance, epidemiology, and end results (SEER) database, 1980–2017. Am J Hematol. 2021;96(6):650-658. doi: 10.1002/ajh.26156 PubMed
22.Bader P, Locatelli F, Peters C. Paediatric Acute Lymphoblastic Leukaemia (ALL). In: Kroger N, Gribben J, Chabannon C, Yakoub-Agha I, Einsele H, eds. The EBMT/EHA CAR-T Cell Handbook. 2022:57-9.
23.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-405. doi: 10.1182/blood-2016-03-643544 PubMed
24.Leukemia & Lymphoma Society. Acute Lymphoblastic Leukemia in Adults. 2024. Accessed November 4, 2024. https://www.lls.org/booklet/acute-lymphoblastic-leukemia-all-adults
25.Cancer Care Alberta. Acute Lymphoblastic Leukemia in Adults. 2023. Accessed November 4, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lyhe005-all.pdf
26.Gupta S, Aitken JF, Bartels U, et al. Paediatric cancer stage in population-based cancer registries: the Toronto consensus principles and guidelines. The Lancet Oncology. 2016;17(4):e163-e172. doi: 10.1016/S1470-2045(15)00539-2 PubMed
27.Canadian Cancer Society. Acute lymphoblastic leukemia. Accessed November 4, 2024. https://cancer.ca/en/cancer-information/cancer-types/acute-lymphoblastic-leukemia-all
28.Hoelzer D, Bassan R, Dombret H, Fielding A, Ribera JM, Buske C. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v69-v82. doi: 10.1093/annonc/mdw025 PubMed
29.Moricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood. 2008;111(9):4477-89. doi: 10.1182/blood-2007-09-112920 PubMed
30.Abou Dalle I, Jabbour E, Short NJ. Evaluation and management of measurable residual disease in acute lymphoblastic leukemia. Ther Adv Hematol. 2020;11:2040620720910023. doi: 10.1177/2040620720910023 PubMed
31.National Cancer Institute. Childhood Acute Lymphoblastic Leukemia (PDQ®)–Patient Version. 2024. Accessed November 17, 2024. https://www.cancer.gov/types/leukemia/patient/child-all-treatment-pdq#top
32.Berry DA, Zhou S, Higley H, et al. Association of Minimal Residual Disease With Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-analysis. JAMA Oncol. 2017;3(7):e170580. doi: 10.1001/jamaoncol.2017.0580 PubMed
33.NCCN. Acute Lymphoblastic Leukemia, Version 4.2023. 2023. Accessed February 20, 2024. https://www.nccn.org/professionals/physician_gls/pdf/all.pdf
34.Amgen Inc. BLINCYTO® (blinatumomab): Product Monograph. 2024 [sponsor submitted reference].
35.Ghodke K, Bibi A, Rabade N, et al. CD19 negative precursor B acute lymphoblastic leukemia (B-ALL)-Immunophenotypic challenges in diagnosis and monitoring: A study of three cases. Cytometry B Clin Cytom. 2017;92(4):315-318. doi: 10.1002/cyto.b.21373 PubMed
36.Vrooman LM, Blonquist TM, Stevenson KE, et al. Efficacy and Toxicity of Pegaspargase and Calaspargase Pegol in Childhood Acute Lymphoblastic Leukemia: Results of DFCI 11-001. J Clin Oncol. 2021;39(31):3496-3505. doi: 10.1200/jco.20.03692 PubMed
37.Pfizer Canada ULC. Vincristine sulfate injection USP: 1 mg/mL [product monograph]. 2021. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00062283.PDF
38.Accord Healthcare Inc. Cyclophosphamide for injection: 500 mg, 1000 mg and 2000 mg per vial [product monograph]. 2022. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00067461.PDF
39.Pfizer Canada ULC. Cytarabine injection: 100 mg/mL (100 mg/mL, 1 g / 10 mL, 2 g / 20 mL) [product monograph]. 2020. Accessed December 23, 2024. https://webfiles.pfizer.com/file/2ce1cb4a-d488-4e8a-8bd3-8d4249f5d9f8?referrer=ccb731e5-4f2d-4f4a-b2dc-e5e912145fc6
40.Apotex Inc. Apo-Dexamethasone (dexamethasone tablets): tablet, 0.5 mg and 4 mg, oral [product monograph]. 2022. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00065302.PDF
41.Generic Medical Partners Inc. Daunorubicin injectable solution (daunorubicin hydrochloride):20 mg/4 mL and 50 mg/10 mL solution for injection [product monograph]. 2020. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00057229.PDF
42.Accord Healthcare Inc. Doxorubicin Injection, BP (doxorubicin hydrochloride injection): 2 mg/mL, 10 mg (5 mL), 50 mg (25 mL) and 200 mg (100 mL) vials BP [product monograph]. 2018. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00048371.PDF
43.Teva Canada Limited. Etoposide injection: (etoposide injection, Teva standard): 20 mg/mL [product monograph]. 2016. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00034660.PDF
44.Pfizer Canada Inc. APO-Methotrexate (methotrexate tablets USP): 2.5 mg (as methotrexate) [product monograph]. 2017. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00043044.PDF
45.Teva Canada Limited. Purinethol (mercaptopurine): tablet 50 mg [product monograph]. 2014. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00025910.PDF
46.Baxalta Canada Corporation. Oncaspar (pegaspargase injection): pegaspargase injection solution, 3750 U / 5 mL [product monograph]. 2017. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00038396.PDF
47.Apotex Inc. Product monograph: APO-Prednisone [product monograph]. 2015. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00030893.PDF
48.Aspen Pharmacare Canada Inc. Lanvis (thioguanine): tablets, 40 mg, oral [product monograph]. 2019. Accessed December 23, 2024. https://pdf.hres.ca/dpd_pm/00050561.PDF
49.BC Cancer. BC Cancer Protocol Summary for Treatment of Pre-B-Cell Acute Lymphoblastic Leukemia with Minimal Residual Disease using Blinatumomab. 2022. Accessed January 10, 2025. http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Leukemia-BMT/ULKMRDBLIN_protocol.pdf
50.Cancer Care Ontario. Drug Formulary: blinatumomab. 2024. Accessed January 10, 2025. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44426
51.NCCN. Guidelines for Pediatric Acute Lymphoblastic Leukemia, Version 2.2025. 2025. Accessed January 10, 2025.
52.Amgen Inc. Reponse to additional information request-2025-01-03. Data on file. 2025 [sponsor submitted reference].
53.Tierens A, Stockley TL, Campbell C, et al. Consensus Recommendations for MRD Testing in Adult B-Cell Acute Lymphoblastic Leukemia in Ontario. Current Oncology. 2021;28(2):1376-1387. doi: 10.3390/curroncol28020131 PubMed
54.Sabloff M FH, Sivajohanathan D, Howlett C, Ross C, Schuh A, et al. Minimal residual disease testing in acute leukemia. Ontario Health - Cancer Care Ontario; 2022. Accessed January 10, 2025. https://www.cancercareontario.ca/fr/file/74401/download?token=sjakR6lc
55.Health Quality Ontario. Minimal Residual Disease Evaluation in Childhood Acute Lymphoblastic Leukemia: A Clinical Evidence Review. Ont Health Technol Assess Ser. 2016;16(7):1-52. PubMed
56.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111(12):5477-85. doi: 10.1182/blood-2008-01-132837 PubMed
57.McKinnon KM. Flow Cytometry: An Overview. Curr Protoc Immunol. 2018;120:5.1.1-5.1.11. doi: 10.1002/cpim.40
58.Meleveedu KS, Litzow M. Advances in measurable residual disease monitoring for adult acute lymphoblastic leukemia. Adv Cell Gene Ther. 2019;2(4):e67. doi: 10.1002/acg2.67
59.Gökbuget N, Boissel N, Chiaretti S, et al. Diagnosis, prognostic factors, and assessment of ALL in adults: 2024 ELN recommendations from a European expert panel. Blood. 2024;143(19):1891-1902. doi: 10.1182/blood.2023020794 PubMed
60.Gokbuget N, Boissel N, Chiaretti S, et al. Management of ALL in adults: 2024 ELN recommendations from a European expert panel. Blood. 2024;143(19):1903-1930. doi: 10.1182/blood.2023023568 PubMed
61.Amgen Inc. BIA report DEC2024. Data on file. 2024 [sponsor submitted reference].
62.National Cancer Institute. NCT02003222: A Phase III Randomized Trial of Blinatumomab for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults. Clinical trial registration. clinicaltrials.gov; 2023. Accessed December 14, 2023. https://clinicaltrials.gov/study/NCT02003222
63.Litzow MR, Sun Z, Mattison RJ, et al. Blinatumomab for MRD-Negative Acute Lymphoblastic Leukemia in Adults. N Engl J Med. 2024;391(4):320-333. doi: 10.1056/NEJMoa2312948 PubMed
64.Litzow MR, Sun Z, Paietta E, et al. Consolidation Therapy with Blinatumomab Improves Overall Survival in Newly Diagnosed Adult Patients with B-Lineage Acute Lymphoblastic Leukemia in Measurable Residual Disease Negative Remission: Results from the ECOG-ACRIN E1910 Randomized Phase III National Cooperative Clinical Trials Network Trial. Blood. 2022;140(Supplement 2):LBA-1. doi: 10.1182/blood-2022-171751
65.Litzow M, Sun Z, Mattison R, et al. S115: Consolidation with blinatumomab improves overall and relapse-free survival in patients with newly diagnosed b-cell acute lymphoblastic leukemia: impact of age and MRD level in ECOG-ACRIN E1910. Hemasphere. 2023;7(Suppl):e1944062. doi: 10.1097/01.HS9.0000967372.19440.62
66.Luger SM, Sun Z, Mattison RJ, et al. Assessment of Outcomes of Consolidation Therapy By Number of Cycles of Blinatumomab Received in Newly Diagnosed Measurable Residual Disease Negative Patients with B-Lineage Acute Lymphoblastic Leukemia: In the ECOG-ACRIN E1910 Randomized Phase III National Clinical Trials Network Trial. Blood. 2023;142(Supplement 1):2877. doi: 10.1182/blood-2023-189648
67.National Cancer Institute. NCT03914625: A Study to Investigate Blinatumomab in Combination With Chemotherapy in Patients With Newly Diagnosed B-Lymphoblastic Leukemia (AALL1731). Clinicaltrials.gov; 2024. Accessed November 10, 2024. https://clinicaltrials.gov/study/NCT03914625
68.Gupta S, Rau RE, Kairalla JA, et al. Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. N Engl J Med. 2024;0(0). doi: doi:10.1056/NEJMoa2411680 PubMed
69.Patel B, Rai L, Buck G, et al. Minimal residual disease is a significant predictor of treatment failure in non T-lineage adult acute lymphoblastic leukaemia: final results of the international trial UKALL XII/ECOG2993. Br J Haematol. 2010;148(1):80-9. doi: 10.1111/j.1365-2141.2009.07941.x PubMed
70.Rowe JM, Buck G, Burnett AK, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106(12):3760-7. doi: 10.1182/blood-2005-04-1623 PubMed
71.Stock W, Luger SM, Advani AS, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood. 2019;133(14):1548-1559. doi: 10.1182/blood-2018-10-881961 PubMed
72.Amgen Inc. Study E1910 - Statistical Analysis Plan v3.0. 2024 [sponsor submitted reference].
73.FDA. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. 2018. Accessed 15 April 2024. https://www.fda.gov/media/71195/download
74.European Medicines Agency. Guideline on adjustment for baseline covariates in clinical trials. 2015. Accessed January 8, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-adjustment-baseline-covariates-clinical-trials_en.pdf
75.Holmberg MJ, Andersen LW. Adjustment for Baseline Characteristics in Randomized Clinical Trials. JAMA. 2022;328(21):2155-2156. doi: 10.1001/jama.2022.21506 PubMed
76.Buyse M, Michiels S, Squifflet P, et al. Leukemia-free survival as a surrogate end point for overall survival in the evaluation of maintenance therapy for patients with acute myeloid leukemia in complete remission. Haematologica. 2011;96(8):1106-12. doi: 10.3324/haematol.2010.039131 PubMed
77.Health Canada. Notice of Compliance Information. 2024. Accessed January 13, 2025. https://health-products.canada.ca/noc-ac/nocInfo?no=34618
Appendix 1: Description of Regimen Used in Studies Included in the Systematic Review
Please note this appendix has not been copy-edited.
Table 21: Summary Treatment Protocol Used in the E1910 Trial
Study treatment | Unit dose strengths/ dosage levels | Dosage formulation/ administration frequency | Route of administration |
|---|---|---|---|
Induction Therapy (cycle 1, 28-day cycle) | |||
Cytarabine | 70 mg | Day 1 | IT injection |
Daunorubicin | 25 mg/m2 | Days 1, 8,15, 22 | IV push |
Vincristine | 1.4 mg/m2 (max 2 mg) | Days 1, 8,15, 22 | IV injection |
Dexamethasone | 10 mg/m2 (max 20 mg) | Days 1 to 7, 15 to 21 (days 1 to 7 only if age ≥ 55 years) | Oral |
Methotrexate | 12.5 mg | Day 14 (± 1) | IT injection |
Pegaspargase | 2,500 IU/m2 (max 1 vial or 3,750 IU) | Day 18 (omit if age ≥ 55 years) | IV or IM injection |
Rituximab | 375 mg/m2 | Days 8, 15 (if CD20 positive) | IV injection |
Induction Therapy (cycle 2, 28-day cycle) | |||
Cyclophosphamide | 1,000 mg/m2 (800 mg/m2 if age > 60 years) | Day 1, 29 | IV injection |
Cytarabine | 75 mg/m2 | Day 1 to 4, 8 to 11, 29 to 32, 36 to 39 | IV or SC injection |
6-mercaptopurine | 60 mg/m2 | Day 1 to 14, 29 to 42 | Oral |
Pegaspargase | 2,000 IU/m2 (max 1 vial or 3,750 IU) | Day 15 (omit if age ≥ 55 years) | IV or IM injection |
Methotrexate | 12.5 mg | Day 1, 8, 15, 22 (± 1) | IT injection |
Rituximab | 375 mg/m2 | Days 8, 15 (if CD20 positive) | IV injection |
Intensification Therapy (cycle 1, 28-day cycle) | |||
Methotrexate | 3 g/m2 | Day 1, 8 | IV injection |
Pegaspargase | 2,000 IU/m2 (1,000 IU/m2 if age ≥ 55 years; max 1 vial or 3,750 IU) | Day 9 | IV or IM injection |
Leucovorin rescue | 10 mg/m2 | IV every 6 hours × 4 doses beginning 22 to 24 hours after MTX; then 10 mg/m2 orally every 6 hours × 72 hours | IV injection and oral |
Blinatumomab (cycles 1 and 2, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
Consolidation Therapy (cycles 1 and 2, 28-day cycle) | |||
Cytarabine | 75 mg/m2 | Day 1 to 5 | IV or SC injection |
Etoposide | 100 mg/m2 | Day 1 to 5 | IV injection |
Methotrexate | 12.5mg | Day 1 (± 1) | IT injection |
Pegaspargase | 2,000 IU/m2 (1,000 IU/m2 if age ≥ 55 years; max 1 vial or 3,750 IU) | Day 5 (Cycle 1 only) | IV or IM injection |
Rituximab | 375 mg/m2 | Day 5 (if CD20 positive) | IV injection |
Consolidation Therapy (cycle 3, 42-day cycle) | |||
Daunorubicin | 25 mg/m2 | Day 1, 8, 15, 22 | IV injection |
Vincristine | 1.4 mg/m2 | Day 1, 8, 15, 22 | IV injection |
Dexamethasone | 10 mg/m2 (cap at 20 mg/day) | Day 1 to 7, 15 to 21 (days 1 to 7 only if age ≥ 55 years) | Oral |
Methotrexate | 12.5 mg | Day 2 (± 1) | IT injection |
Cyclophosphamide | 650 mg/m2 | Day 29 | IV injection |
Cytarabine | 75 mg/m2 | Day 30 to 33, 37 to 40 | IV or SC injection |
6-mercaptopurine | 60 mg/m2 | Day 29 to 42 | Oral |
Rituximab | 375 mg/m2 | Day 8 (optional if CD20 positive) | IV injection |
Blinatumomab Consolidation Therapy (cycle 4, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
Consolidation Therapy (cycle 5, 28-day cycle) | |||
Cytarabine | 75 mg/m2 | Day 1 to 5 | IV or SC injection |
Etoposide | 100 mg/m2 | Day 1 to 5 | IV injection |
Methotrexate | 12.5mg | Day 1 (± 1) | IT injection |
Rituximab | 375 mg/m2 | Day 8 (if CD20 positive) | IV injection |
Blinatumomab Consolidation Therapy (cycle 6, 28-day cycle) | |||
Blinatumomab | 28 mcg/day | Continuous infusion for 28 days | IV injection |
Maintenance Therapy (up to 2.5 years after treatment intensification) | |||
Vincristine | 1.4 mg/m2 | Day 1, then every 3 months (max 2 mg/dose) with prednisone | IV injection |
Prednisone | 60 mg/m2 | Day 1 to 5 every 3 months | Oral |
Methotrexate | 20 mg/m2 | Once every week | Oral or IV injection |
6-mercaptopurine | 75 mg/m2 | Every day, continuous | Oral |
Methotrexate | 12.5 mg | Day 1 (± 3) every 3 months | IT injection |
IM = intramuscular; IT = intrathecal; MTX = methotrexate; SC = subcutaneous.
Source: BLINCYTO Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 22: Summary Treatment Protocol Used in Patients With SR-Average in the AALL1731 Trial
Study treatment | Patient age | Unit dose strengths/dosage levels (per day) | Dosage formulation/ Administration frequency | Route of administration |
|---|---|---|---|---|
Induction Therapy (Arms A and B) | ||||
Cytarabine | 1 to 1.99 years | 30 mg | Time of diagnosis or Day 1 | IT injection |
2 to 2.99 years | 50 mg | |||
≥ 3 years | 70 mg | |||
Vincristine | ≥ 365 days to < 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 8, 15, 22 | IV injection |
Dexamethasone | ≥ 365 days to < 10 years | 3 mg/m2 × 2 | Day 1 to 28 (no taper) | Oral |
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to < 10 years | 2500 IU/m2 | Day 4 | IV or IM injection |
Methotrexate | 1 to 1.99 years | 8 mg | Day 8 and 29 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Consolidation Therapy (Arms A and B) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1 | IV injection |
Mercaptopurine | ≥ 365 days to ≤ 10 years | 75 mg/m2 | Day 1 to 28 | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1, 8, 15 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 2, 9, 16 (DS patients only) | Oral or IV injection |
Blinatumomab Block I (Arm B only) | ||||
Blinatumomab | ≥ 365 days to ≤ 10 years | 15 mcg/m2 (max 28 mcg/day) | Day 1 to 28 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 5 mg/m2 | Day 1 (before blinatumomab therapy) | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 2 (DS patients only) | Oral or IV injection |
Interim Maintenance Part I (Arms A and B) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 11, 21, 31, 41 | IV injection |
Methotrexate | ≥ 365 days to ≤ 10 years | 100 mg/m2 escalating by 50 mg/m2/dose | Day 1, 11, 21, 31, 41 | IV injection |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 31 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 32 (DS patients only) | Oral or IV injection |
Blinatumomab Block II (Arm B only) | ||||
Blinatumomab | ≥ 365 days to ≤ 10 years | 15 mcg/m2 (max 28 mcg/day) | Day 1 to 28 | IV injection |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 32 (DS patients only) | Oral or IV injection |
Delayed Intensification Therapy (Arms A and B) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 8, 15 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 5 mg/m2 × 2 | Day 1 to 7, 15 to 21 | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 31 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Doxorubicin | ≥ 365 days to ≤ 10 years | 25 mg/m2 | Day 1, 8, 15 | IV injection |
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 2 (DS patients only) | Oral or IV injection |
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to ≤ 10 years | 2500 IU/m2 | Day 4 | IV or IM injection |
Interim Maintenance Part II (Arms A and B) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 11, 21, 31, 41 | IV injection |
Methotrexate | ≥ 365 days to ≤ 10 years | 100 mg/m2 escalating by 50 mg/m2/dose | Day 1, 11, 21, 31, 41 | IV injection |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 1, 31 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Leucovorin | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 2, 32 (DS patients only) | Oral or IV injection |
Maintenance (Arms A and B) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 3 mg/m2 × 2 | Day 1 to 5 | Oral |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Oral Methotrexate | ≥ 365 days to ≤ 10 years | 20 mg/m2 | Day 8, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78 | IV injection |
Mercaptopurine | ≥ 365 days to ≤ 10 years | 75 mg/m2 | Day 1 to 84 | Oral |
Leucovorin (Arm B only) | ≥ 365 days to ≤ 10 years | 5 mg/m2 q6h × 2 | Day 2 (DS patients only) | Oral or IV injection |
IM = intramuscular; IT = intrathecal; IU = international unit.
Source: Gupta et al. 2024.68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 23: Summary Treatment Protocol Used in Patients With SR-Average in the AALL1731 Trial
Study treatment | Patient age | Unit dose strengths/dosage levels (per day) | Dosage formulation/ Administration frequency | Route of administration |
|---|---|---|---|---|
Induction Therapy (Arms C and D) | ||||
Cytarabine | 1 to 1.99 years | 30 mg | Time of diagnosis or Day 1 | IT injection |
2 to 2.99 years | 50 mg | |||
≥ 3 years | 70 mg | |||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 8, 15, 22 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 3 mg/m2 × 2 | Day 1 to 28 (no taper) | Oral |
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to ≤ 10 years | 2500 IU/m2 | Day 4 | IV or IM injection |
Methotrexate | 1 to 1.99 years | 8 mg | Day 8 and 29 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Consolidation Therapy (Arms C and D) | ||||
Cytarabine | ≥ 365 days to ≤ 10 years | 75 mg/m2 | Day 1 to 4, 8 to 11, 29 to 32, 36 to 39 | IV injection |
Cyclophosphamide | ≥ 365 days to ≤ 10 years | 1000 mg/m2 | Day 1, 29 | IV injection |
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 15, 22, 43, 50 | IV injection |
Mercaptopurine | ≥ 365 days to ≤ 10 years | 60 mg/m2 | Day 1 to 14, 29 to 42 | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1, 8, 15, 22 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to ≤ 10 years | 2500 IU/m2 | Day 15, 43 | IV or IM injection |
Blinatumomab Block I (Arm D only) | ||||
Blinatumomab | ≥ 365 days to ≤ 10 years | 15 mcg/m2 (max 28 mcg per day) | Day 1 to 28 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 5 mg/m2 | Day 1 (before blinatumomab therapy) | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Interim Maintenance Part I (Arms C and D) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 15, 29, 43 | IV injection |
Methotrexate | ≥ 365 days to ≤ 10 years | 5,000 mg/m2 | Day 1, 15, 29, 43 | IV injection |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 1, 29 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Mercaptopurine | ≥ 365 days to ≤ 10 years | 25 mg/m2 | Day 1 to 14, 15 to 28, 29 to 42, 43 to 56 | Oral |
Leucovorin | ≥ 365 days to ≤ 10 years | 15 mg/m2 × 3 | Day 3 to 4, 17 to 18, 31 to 32, 45 to 46 | Oral or IV injection |
Blinatumomab Block II (Arm D only) | ||||
Blinatumomab | ≥ 365 days to ≤ 10 years | 15 mcg/m2 (max 28 mcg per day) | Day 1 to 28 | IV injection |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Delayed Intensification Therapy (Arms C and D) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 8, 15 | IV injection |
Dexamethasone | ≥ 365 days to ≤ 10 years | 5 mg/m2 × 2 | Day 1 to 7, 15 to 21 | Oral |
Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Doxorubicin | ≥ 365 days to ≤ 10 years | 25 mg/m2 | Day 1, 8, 15 | IV injection |
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to ≤ 10 years | 2,500 IU/m2 | Day 4 | IV or IM injection |
Interim Maintenance Part II (Arms C and D) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1, 11, 21, 31, 41 | IV injection |
Methotrexate | ≥ 365 days to ≤ 10 years | 100 mg/m2 escalating by 50 mg/m2/dose | Day 1, 11, 21, 31, 41 | IV injection |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 1, 31 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Pegaspargase (or Calaspargase pegol) | ≥ 365 days to ≤ 10 years | 2,500 IU/m2 | Day 2, 22 | IV or IM injection |
Maintenance (Arms C and D) | ||||
Vincristine | ≥ 365 days to ≤ 10 years | 1.5 mg/m2 (max 2 mg) | Day 1 | IV injection |
Prednisone or Prednisolone | ≥ 365 days to ≤ 10 years | 20 mg/m2 | Day 1 to 5 | Oral |
IT Methotrexate | 1 to 1.99 years | 8 mg | Day 1 | IT injection |
2 to 2.99 years | 10 mg | |||
3 to 8.99 years | 12 mg | |||
≥ 9 years | 15 mg | |||
Oral Methotrexate | ≥ 365 days to ≤ 10 years | 20 mg/m2 | Day 8, 15, 22, 29, 36, 43, 50, 57, 64, 71, 78 | IV injection |
Mercaptopurine | ≥ 365 days to ≤ 10 years | 75 mg/m2 | Day 1 to 84 | Oral |
Abbreviations: IM = intramuscular; IT = intrathecal; IU = international unit; SC = subcutaneous.
Source: Gupta et al., 2024.68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 2: Patient Disposition
Please note this appendix has not been copy-edited.
Table 24: Summary of Patient Disposition Prior to Randomization From Studies Included in the Systematic Review
Patient disposition | E1910 | AALL1731 | ||||
|---|---|---|---|---|---|---|
Blinatumomab plus chemotherapy (N = 152) | Chemotherapy (N = 134) | SR-average (N = 835) | SR-high (N = 605) | |||
Blinatumomab plus chemotherapy (N = 417) | Chemotherapy (N = 418) | Blinatumomab plus chemotherapy (N = 301) | Chemotherapy (N = 304) | |||
Screened, N | 488 | 4,264 | ||||
Reason for Step 1 (induction) failure, n (%) | ||||||
Did not receive any Step 1 treatment | 8 (1.6) | 1 | ||||
Were ineligible | NR | 27 | ||||
Discontinued Step 1 treatment | 147 (30.1) | 218 | ||||
Treatment completed per-protocol criteria | 24 (4.9) | NR | ||||
Death on study | 14 (2.9) | 20 | ||||
Disease progression relapse during active treatment | 21 (4.3) | NR | ||||
Adverse event/side effects/complications | 13 (2.7) | 1 | ||||
Patient withdrawal/refusal after beginning protocol therapy | 16 (3.3) | 19 | ||||
Alternative therapy | 16 (3.3) | NR | ||||
Patient off treatment for other complicating disease | 2 (0.4) | NR | ||||
Were Ph+ | NR | 28 | ||||
Had inadequate information | NR | 41 | ||||
Were enrolled in another COG study | NR | 5 | ||||
Were not able to be evaluated | NR | 9 | ||||
Withdrawn consent | NR | 5 | ||||
Had pending risk stratification at DCO | NR | 90 | ||||
Other | 41 (8.4) | NR | ||||
Reason for Step 2 (intensification) failure, n (%) | NA | |||||
Did not receive any Step 2 treatment | 2 (0.4) | NA | ||||
Discontinued Step 2 treatment | 45 (9.2) | NA | ||||
Treatment completed per-protocol criteria | 23 (4.7) | NA | ||||
Death on study | 0 (0.0) | NA | ||||
Disease progression relapse during active treatment | 12 (2.5) | NA | ||||
Adverse event/side effects/complications | 1 (0.2) | NA | ||||
Patient withdrawal/refusal after beginning protocol therapy | 2 (0.4) | NA | ||||
Alternative therapy | 1 (0.2) | NA | ||||
Other | 6 (1.2) | NA | ||||
Underwent risk stratification at EOI | NA | NA | 4,018 | |||
Ineligible for randomization | NA | NA | 1,684 | |||
SR-favourable relapse risk | NA | NA | 1,635 | |||
SR-high with Down syndrome | NA | NA | 49 | |||
Eligible based of risk stratification | NA | NA | 1,418 | 916 | ||
Ineligible for randomization | NA | NA | 187 | 55 | ||
Discontinue due to adverse event | NA | NA | 1 | 1 | ||
Lost to follow-up | NA | NA | 1 | 1 | ||
Had Ph+ | NA | NA | NR | 4 | ||
Consolidation treatment failure | NA | NA | NR | 7 | ||
Died | NA | NA | NR | 2 | ||
Enrolled in another COG study | NA | NA | 1 | 4 | ||
Not evaluable | NA | NA | 34 | 22 | ||
Undetectable HTS | NA | NA | 150 | NR | ||
End-of-consolidation MRD 0.1 to < 1% | NA | NA | NR | 14 | ||
Excluded | NA | NA | 396 | 256 | ||
Withdrawn | NA | NA | 343 | 203 | ||
Withdrew consent | NA | NA | 21 | 12 | ||
Pending randomization at DCO | NA | NA | 32 | 41 | ||
ALL = acute lymphoblastic leukemia; COG = Children’s Oncology Group; DCO = data cut-off; EOI = end of induction; FAS = full analysis set; HTS = high-throughput sequencing; MRD = minimal residual disease; NA = not applicable; NR = not reported; Ph+ = Philadelphia chromosome-positive; SR-average = standard risk with average risk of relapse; SR-high = standard risk with high risk of relapse.
Source: BLINCYTO E1910 Clinical Study Report and Gupta, 2024.19,68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 3: Baseline Demographics
Please note this appendix has not been copy-edited.
Table 25: Additional Baseline Characteristics From the E1910 Trial
Characteristic | E1910 trial (Step 3 analysis set) | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
Height, cm | ||
n | 152 | 133 |
Mean (SD) | 169.47 (13.03) | 170.00 (10.82) |
Median (IQR) | 170.20 (162.60 to 177.80) | 170.00 (163.00 to 177.80) |
Minimum to maximum | 62.7 to 193.00 | 117.90 to 193.00 |
Weight, kg | ||
n | 152 | 134 |
Mean (SD) | 86.44 (22.47) | 87.38 (21.90) |
Median (IQR) | 85.60 (70.10 to 101.95) | 83.20 (72.00 to 103.30) |
Minimum to maximum | 35.50 to 157.40 | 49.20 to 182.40 |
Body surface area (m2) | ||
n | 152 | 134 |
Mean (SD) | 1.995 (0.287) | 2.010 (0.269) |
Median (IQR) | 2.000 (1.800 to 2.195) | 1.995 (1.800 to 2.200) |
Minimum to maximum | 1.430 to 2.830 | 1.440 to 2.750 |
Prior surgeryb, n (%) | ||
Yes | 6 (3.9) | 7 (5.2) |
No | 146 (96.1) | 127 (94.8) |
Prior radiation therapy, n (%) | ||
Yes | 4 (2.6) | 4 (3.0) |
No | 148 (97.4) | 130 (97.0) |
IQR = interquartile range; SD = standard deviation.
Notes: Step 3 analysis set includes all Step 3 randomized or registered patients.
Baseline is the last assessment taken on or before the date of first protocol-specified therapy administration for Step 3. If no protocol-specified therapy is administered for Step 3 then baseline is the last assessment taken on or before the date of randomization/Step 3 registration. For baseline, body surface area and weight consider the latest non-missing assessment taken prior or postrandomization/Step 3 registration.
DCO date: June 23, 2023.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 26: Summary of Baseline Characteristics From the E1910 Trial by MRD Status
Characteristic | E1910 trial (Step 3 analysis set) | |||
|---|---|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |||
MRD-positive | MRD-negative | MRD-positive | MRD-negative | |
Sex, n (%) | ||||
Male | 14 (35.0) | 55 (49.1) | 14 (63.6) | 56 (50.0) |
Female | 26 (65.0) | 57 (50.9) | 8 (36.4) | 56 (50.0) |
Ethnicity, n (%)a | ||||
Hispanic or Latino | 8 (20.0) | 13 (11.6) | 5 (22.7) | 10 (8.9) |
Not Hispanic or Latino | 30 (75.0) | 95 (84.8) | 16 (72.7) | 95 (84.8) |
Not reported | 1 (2.5) | 1 (0.9) | 1 (4.5) | 2 (1.8) |
Unknown | 1 (2.5) | 3 (2.7) | 0 (0.0) | 5 (4.5) |
Race, n (%)a | ||||
American Indian or Alaska Native | 0 (0.0) | 2 (1.8) | 0 (0.0) | 1 (0.9) |
Asian | 1 (2.5) | 3 (2.7) | 0 (0.0) | 2 (1.8) |
Black or African American | 3 (7.5) | 9 (8.0) | 1 (4.5) | 4 (3.6) |
Native Hawaiian or Other Pacific Islander | 0 (0.0) | 1 (0.9) | 0 (0.0) | 0 (0.0) |
White | 30 (75.0) | 87 (77.7) | 21 (95.5) | 89 (79.5) |
Not reported | 2 (5.0) | 5 (4.5) | 0 (0.0) | 6 (5.4) |
Unknown or Other | 4 (10.0) | 5 (4.5) | 0 (0.0) | 10 (8.9) |
Age at enrolment (years) | ||||
Mean (SD) | 48.5 (11.0) | 50.1 (11.0) | 51.5 (12.5) | 50.0 (11.9) |
Median (range) | 49.0 (30 to 68) | 51.5 (30 to 69) | 54.5 (30 to 69) | 50.0 (30 to 70) |
IQR | 39.5, 57.5 | 41.0, 59.0 | 39.0, 61.0 | 40.0, 60.5 |
Age group, n (%) | ||||
≥ 18 and < 35 years | 5 (12.5) | 13 (11.6) | 2 (9.1) | 17 (15.2) |
≥ 35 and < 55 years | 21 (52.5) | 53 (47.3) | 9 (40.9) | 48 (42.9) |
< 55 years | 26 (65.0) | 66 (58.9) | 11 (50.0) | 65 (58.0) |
≥ 55 and < 65 years | 11 (27.5) | 37 (33.0) | 7 (31.8) | 31 (27.7) |
≥ 55 years | 14 (35.0) | 46 (41.1) | 11 (50.0) | 47 (42.0) |
≥ 65 years | 3 (7.5) | 9 (8.0) | 4 (18.2) | 16 (14.3) |
Country of residence, n (%) | ||||
Canada | 3 (7.5) | 7 (6.3) | 0 (0.0) | 7 (6.3) |
Israel | 0 (0.0) | 2 (1.8) | 1 (4.5) | 6 (5.4) |
US | 37 (92.5) | 103 (92.0) | 21 (95.5) | 99 (88.4) |
ECOG PS, n (%) | ||||
0 | 18 (45.0) | 39 (34.8) | 9 (40.9) | 40 (35.7) |
1 | 20 (50.0) | 67 (59.8) | 12 (54.5) | 69 (61.6) |
2 | 2 (5.0) | 6 (5.4) | 1 (4.5) | 3 (2.7) |
3 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
4 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Bone marrow biopsy results, n (%) | ||||
Involved | 7 (17.5) | 3 (2.7) | 1 (4.5) | 0 (0.0) |
Not involved | 30 (75.0) | 107 (95.5) | 21 (95.5) | 111 (99.1) |
Indeterminate | 3 (7.5) | 2 (1.8) | 0 (0.0) | 1 (0.9) |
Bone marrow cellularity, % | ||||
n | 40 | 111 | 22 | 112 |
Mean (SD) | 42.9 (18.6) | 43.9 (20.3) | 37.7 (18.6) | 42.2 (18.4) |
Median (IQR) | 45.0 (30.0 to 57.5) | 40.0 (30.0 to 60.0) | 40.0 (30.0 to 50.0) | 40.0 (30.0 to 50.0) |
Minimum to maximum | 5 to 90 | 0 to 100 | 10 to 75 | 10 to 90 |
Bone marrow blasts, % | ||||
n | 40 | 112 | 22 | 112 |
Mean (SD) | 1.0 (1.0) | 1.3 (1.2) | 1.5 (1.1) | 1.3 (1.3) |
Median (IQR) | 1.0 (0.0 to 1.0) | 1.0 (0.0 to 2.0) | 1.0 (1.0 to 2.0) | 1.0 (0.0 to 2.0) |
Minimum to maximum | 0 to 5 | 0 to 5 | 0 to 4 | 0 to 6 |
ECOG = Eastern Cooperative Oncology Group; IQR = interquartile range; MRD = minimal residual disease; PS = performance status; SD = standard deviation.
Notes: Step 3 analysis set includes all Step 3 randomized or registered patients.
MRD-positive before Step 3: MRD value ≥ 0.01%. MRD-negative before Step 3: MRD value ≤ 0.01%.
Baseline is the last assessment taken on or before the date of first protocol-specified therapy administration for Step 3. If no protocol-specified therapy is administered for Step 3 then baseline is the last assessment taken on or before the date of randomization/Step 3 registration. For baseline, body surface area and weight consider the latest non-missing assessment taken prior or postrandomization/Step 3 registration.
DCO date: June 23, 2023.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 4: Subgroup Analyses
Please note this appendix has not been copy-edited.
Table 27: Subgroup Analyses in the E1910 Trial
Variable | E1910 trial | |||
|---|---|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | Hazard ratio (95% CI) | P value | |
OS in the Full Analysis Set | ||||
No. of patients included in the analysis | 112 | 112 | NR | NR |
No. of all patients with OS events, n (%) | 19 of 112 (17.0) | 40 of 112 (35.7) | 0.41 (0.24 to 0.71) | NE |
Sex | 0.868 | |||
Female | 9 of 57 (15.8) | 20 of 56 (35.7) | 0.40 (0.18 to 0.88) | — |
Male | 10 of 55 (18.2) | 20 of 56 (35.7) | 0.43 (0.20 to 0.91) | — |
Race | 0.994 | |||
American Indian or Alaska Native | 0 of 2 (0.0) | 0 of 1 (0.0) | NE | — |
Asian | 0 of 3 (0.0) | 0 of 2 (0.0) | NE | — |
Black or African American | 4 of 9 (44.4) | 2 of 4 (50.0) | 0.82 (0.15 to 4.52) | — |
Native Hawaiian or Other Pacific Islander | 0 of 1 (0.0) | 0 of 0 (0.0) | NE | — |
White | 15 of 87 (17.2) | 30 of 89 (33.7) | 0.44 (0.23 to 0.81) | — |
Unknown | 0 of 5 (0.0) | 5 of 10 (50.0) | NE | — |
Not Reported | 0 of 5 (0.0) | 3 of 6 (50.0) | NE | — |
Ethnicity | 1.000 | |||
Hispanic or Latino | 0 of 13 (0.0) | 4 of 10 (40.0) | NE | — |
Not Hispanic or Latino | 18 of 95 (18.9) | 32 of 95 (33.7) | 0.49 (0.28 to 0.88) | — |
Unknown | 1 of 3 (33.3) | 3 of 5 (60.0) | 0.65 (0.06 to 7.23) | — |
Not Reported | 0 of 1 (0.0) | 1 of 2 (50.0) | NE | — |
Age | 0.032 | |||
≥ 18 and < 35 years | 0 of 13 (0.0) | 1 of 17 (5.9) | NE | — |
≥ 35 and < 55 years | 5 of 53 (9.4) | 20 of 48 (41.7) | 0.19 (0.07 to 0.51) | — |
≥ 55 and < 65 years | 9 of 37 (24.3) | 13 of 31 (41.9) | 0.49 (0.21 to 1.15) | — |
≥ 65 years | 5 of 9 (55.6) | 6 of 16 (37.5) | 1.72 (0.51 to 5.80) | — |
RFS in the Full Analysis Set | ||||
No. of patients included in the analysis | 112 | 112 | — | — |
No. of all patients with RFS events, n (%) | 25 of 112 (22.3) | 43 of 112 (38.4) | 0.51 (0.31 to 0.84) | — |
Sex | 0.192 | |||
Female | 9 of 57 (15.8) | 22 of 56 (39.3) | 0.36 (0.17 to 0.79) | |
Male | 16 of 55 (29.1) | 21 of 56 (37.5) | 0.69 (0.36 to 1.32) | |
Race | 0.976 | |||
American Indian or Alaska Native | 0 of 2 (0.0) | 0 of 1 (0.0) | NE | — |
Asian | 0 of 3 (0.0) | 0 of 2 (0.0) | NE | — |
Black or African American | 4 of 9 (44.4) | 2 of 4 (50.0) | 0.88 (0.16 to 4.87) | — |
Native Hawaiian or Other Pacific Islander | 0 of 1 (0.0) | 0 of 0 (0.0) | NE | — |
White | 19 of 87 (21.8) | 33 of 89 (37.1) | 0.50 (0.28 to 0.88) | — |
Unknown | 1 of 5 (20.0) | 5 of 10 (50.0) | 0.32 (0.04 to 2.75) | — |
Not Reported | 1 of 5 (20.0) | 3 of 6 (50.0) | 1.11 (0.10 to 12.48) | — |
Ethnicity | 1.000 | |||
Hispanic or Latino | 3 of 13 (23.1) | 4 of 10 (40.0) | 0.55 (0.12 to 2.46) | — |
Not Hispanic or Latino | 21 of 95 (22.1) | 35 of 95 (36.8) | 0.53 (0.31 to 0.91) | — |
Unknown | 1 of 3 (33.3) | 3 of 5 (60.0) | 0.65 (0.06 to 7.23) | — |
Not Reported | 0 of 1 (0.0) | 1 of 2 (50.0) | NE | — |
Age | 0.164 | |||
≥ 18 and < 35 years | 0 of 13 (0.0) | 2 of 17 (11.8) | NE | — |
≥ 35 and < 55 years | 9 of 53 (17.0) | 20 of 48 (41.7) | 0.35 (0.16 to 0.77) | — |
≥ 55 and < 65 years | 11 of 37 (29.7) | 15 of 31 (48.4) | 0.54 (0.25 to 1.18) | — |
≥ 65 years | 5 of 9 (55.6) | 6 of 16 (37.5) | 1.62 (0.48 to 5.48) | — |
OS = overall survival; RFS = relapse-free survival.
Notes: FAS includes all MRD-negative patients who were randomized. MRD-negative before Step 3: MRD value ≤ 0.01%.
DCO date: June 23, 2023.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 28: Summary of Key Efficacy Results From the AALL1731 Trial Stratified by Risk of Relapse
Variable | SR-average | SR-high | ||
|---|---|---|---|---|
Blinatumomab plus chemotherapy (N = 417) | Chemotherapy (N = 418) | Blinatumomab plus chemotherapy (N = 301) | Chemotherapy (N = 304) | |
OS | ||||
OS probability at 3 years, % (SE) | 100 (NA) | 98.4 (1.0) | 96.1 (2.0) | 95.3 (2.2) |
DFS | ||||
DFS probability at 3 years, % (SE) | 97.5 (1.3) | 90.2 (2.3) | 94.1 (2.5) | 84.8 (3.8) |
Stratified HR (95% CI)a | 0.33 (0.15 to 0.69) | 0.45 (0.24 to 0.85) | ||
DFS = disease-free survival; HR = hazard ratio; NA = not applicable OS = overall survival; SE = standard error.
Notes: OS analyses in AALL1731 were performed post hoc while DFS analyses in the SR-average and SR-high cohorts were the prespecified subgroup analyses of the primary outcome.
DCO date: June 30, 2024
aThe HR estimates are obtained from a stratified Cox regression model. An HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
Source: Gupta, 2024.68
Figure 13: DFS in Various Subgroups as Assessed by RMST Estimates in the AALL1731 Trial
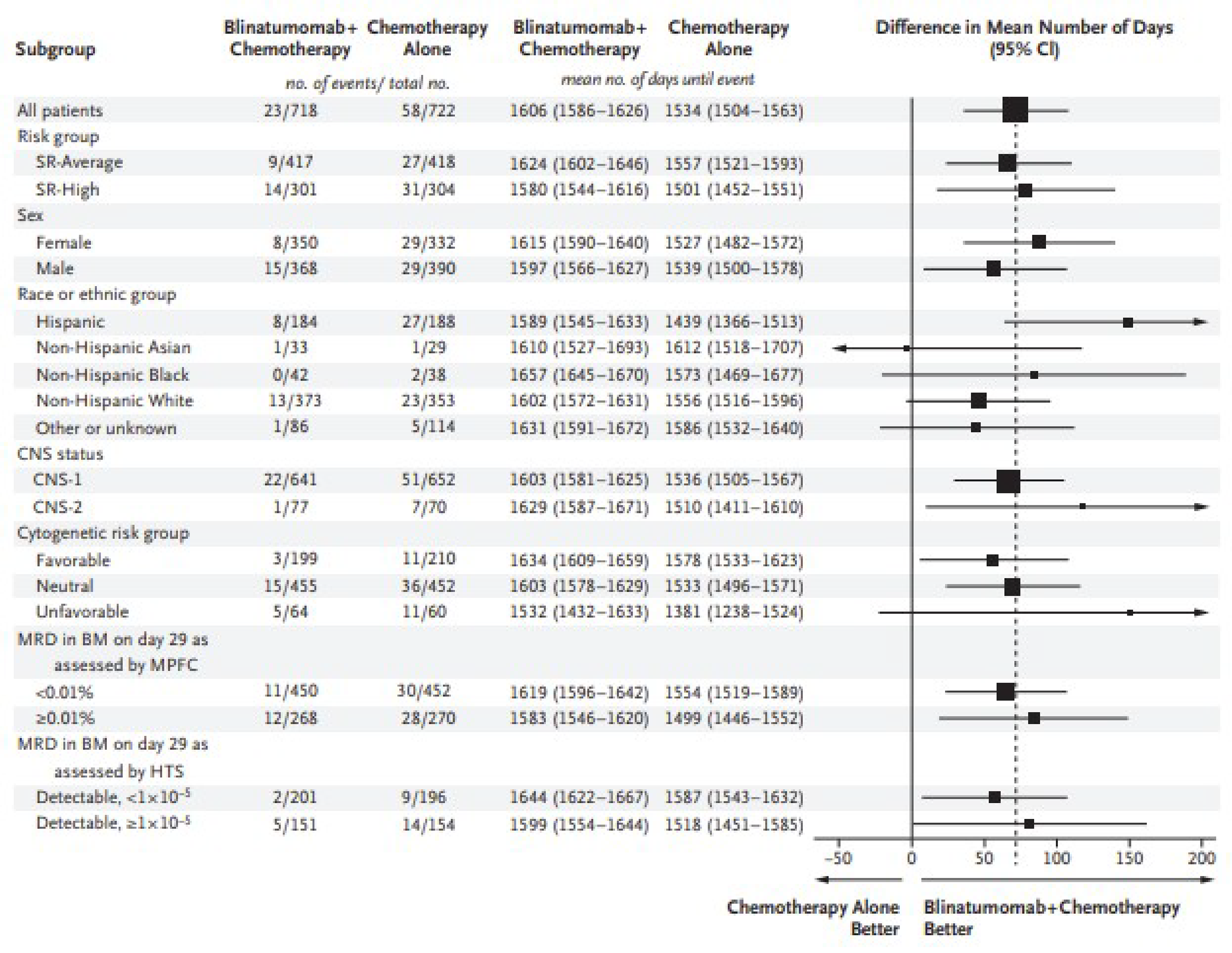
BM = bone marrow; CI = confidence interval; CNS = central nervous system; MPFC = multiparameter flow cytometry; MRD = minimal residual disease.
Notes: Risk group (SR-average and SR-high) was a stratification factor; all other subgroup analyses were post hoc. SR-Average refers to patients with standard-risk B-cell ALL and an average risk of relapse, and SR-high refers to patients with standard-risk B-cell ALL and a high risk of relapse.
The size of each black square is proportional to the total number of patients in the subgroup, and the arrows indicate CIs that exceed the size of the graph.
The widths of the confidence intervals were not adjusted for multiplicity and thus not for analyses other than the primary analysis and should not be used in place of hypothesis testing.
CNS status was classified as CNS1 (defined by no leukemic blasts as assessed by cytologic evaluation) or CNS2 (defined by < 5 white cells per microlitre and the presence of blasts or traumatic lumbar puncture with the presence of blasts as assessed by cytologic evaluation but the absence of blasts as calculated by means of the Steinherz-Bleyer algorithm).
The analysis of the subgroup defined according to MRD as assessed in the bone marrow by HTS on day 29 included only patients categorized as SR-average who had detectable MRD by HTS. Patients with undetectable MRD by HTS were not eligible for randomization. Patients with either unavailable or indeterminate MRD by HTS were eligible for randomization and were included in the primary analysis.
Source: From New Engl J Med, Gupta S et al., Blinatumomab in Standard-Risk B-Cell Acute Lymphoblastic Leukemia in Children. Copyright © (2025) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
Appendix 5: Sensitivity Analyses
Please note this appendix has not been copy-edited.
Table 29: Sensitivity Analysis 1 in the E1910 Trial — Per-Protocol Analysis Set
Variable | E1910 | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
OS | ||
No. of patients included in the analysis | 72 | 73 |
No. of patients with OS events, n (%) | 16 (22.2) | 31 (42.5) |
No. of patients censored, n (%) | 56 (77.8) | 42 (57.5) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 53 (73.6) | 38 (52.1) |
Discontinued study | 3 (4.2) | 4 (5.5) |
Consent withdrawn | 3 (4.2) | 3 (4.1) |
Lost to follow-up | 0 (0.0) | 1 (1.4) |
OS (months), median (95% CI) | NE (NE to NE) | 6.0 (3.4 to NE) |
Stratified HR (95% CI)a,b | 0.48 (0.26 to 0.90) | |
P value | 0.022 | |
Duration of follow-up, median (95% CI) yearsc | 4.5 (4.0 to 4.7) | 4.2 (3.0 to 4.7) |
OS probability at 0.5 years, % (95% CI) | 97.2 (89.3 to 99.3) | 98.6 (90.7 to 99.8) |
OS probability at 1 year, % (95% CI) | 94.4 (85.9 to 97.9) | 86.2 (75.8 to 92.3) |
OS probability at 2 years, % (95% CI) | 86.1 (75.7 to 92.3) | 81.9 (70.9 to 89.1) |
OS probability at 3 years, % (95% CI) | 80.5 (69.4 to 88.0) | 65.9 (53.5 to 75.7) |
OS probability at 4 years, % (95% CI) | 77.3 (65.5 to 85.4) | 57.0 (44.0 to 68.0) |
OS probability at 5 years, % (95% CI) | 77.3 (65.5 to 85.4) | 57.0 (44.0 to 68.0) |
OS probability at 6 years, % (95% CI) | 77.3 (65.5 to 85.4) | 44.0 (25.3 to 61.2) |
OS probability at 7 years, % (95% CI) | 77.3 (65.5 to 85.4) | 44.0 (25.3 to 61.2) |
RFS | ||
No. of patients included in the analysis | 72 | 73 |
No. of patients with RFS events, n (%) | 18 (25.0) | 33 (45.2) |
Relapse | 9 (12.5) | 24 (32.9) |
Death due to any cause | 9 (12.5) | 9 (12.3) |
No. of patients censored, n (%) | 54 (75.0) | 40 (54.8) |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 52 (72.2) | 36 (49.3) |
Discontinued study | 2 (2.8) | 4 (5.5) |
Consent withdrawn | 2 (2.8) | 3 (4.1) |
Lost to follow-up | 0 (0.0) | 1 (1.4) |
RFS (months), median (95% CI) | NE (NE to NE) | 5.8 (2.5 to NE) |
Stratified HR (95% CI)a,b | 0.50 (0.28 to 0.91) | |
P value | 0.023 | |
Duration of follow-up, median (95% CI) yearsc | 4.5 (4.0 to 5.1) | 4.2 (3.7 to 4.6) |
RFS probability at 0.5 years, % (95% CI) | 91.7 (82.4 to 96.2) | 90.4 (80.9 to 95.3) |
RFS probability at 1 year, % (95% CI) | 87.5 (77.4 to 93.3) | 78.0 (66.6 to 85.9) |
RFS probability at 2 years, % (95% CI) | 79.2 (67.8 to 86.9) | 67.9 (55.7 to 77.4) |
RFS probability at 3 years, % (95% CI) | 79.2 (67.8 to 86.9) | 60.6 (48.2 to 70.9) |
RFS probability at 4 years, % (95% CI) | 74.4 (62.4 to 83.0) | 55.2 (42.5 to 66.2) |
RFS probability at 5 years, % (95% CI) | 74.4 (62.4 to 83.0) | 55.2 (42.5 to 66.2) |
RFS probability at 6 years, % (95% CI) | 74.4 (62.4 to 83.0) | 44.3 (27.3 to 60.0) |
RFS probability at 7 years, % (95% CI) | 74.4 (62.4 to 83.0) | 44.3 (27.3 to 60.0) |
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; OS = overall survival; RFS = relapse-free survival; SE = standard error.
Note: DCO date: E1910 = June 23, 2023, and AALL1731 = June 30, 2024
aStratification factors: age (< 55 years vs. ≥ 55 years), CD20 status (positive vs. negative vs. not collected), rituximab use (yes vs. no vs. not collected), intent to receive allogeneic SCT (yes vs. no).
bThe HR estimates are obtained from a stratified Cox regression model. An HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
cDefined as median KM time to censoring in the E1910 trial. Yea Chemotherapy rs are calculated as days from randomization or registration date to event/censor date, divided by 365.25. Time to censoring measures follow-up time by reversing the status indicator for censored and events.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 30: Sensitivity Analysis 2 in the E1910 Trial Censored at Allogeneic SCT for MRD-Negative at Step 3 (FAS)
Variable | E1910 | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
OS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with OS events, n (%) | 13 (11.6) | 27 (24.1) |
No. of patients censored, n (%) | 99 (88.4) | 85 (75.9) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 66 (58.9) | 47 (42.0) |
Received allogeneic SCT | 28 (25.0) | 33 (29.5) |
Discontinued study | 5 (4.5) | 5 (4.5) |
Consent withdrawn | 4 (3.6) | 4 (3.6) |
Lost to follow-up | 1 (0.9) | 1 (0.9) |
OS (months), median (95% CI) | NE (NE to NE) | NE (6.0 to NE) |
Stratified HR (95% CI)a,b | 0.46 (0.24 to 0.90) | |
P value | 0.024 | |
Duration of follow-up, median (95% CI) yearsc | 3.8 (3.2 to 4.2) | 3.6 (3.1 to 4.1) |
OS probability at 0.5 years, % (95% CI) | 98.1 (92.7 to 99.5) | 99.1 (93.8 to 99.9) |
OS probability at 1 year, % (95% CI) | 95.8 (89.1 to 98.4) | 89.4 (80.6 to 94.4) |
OS probability at 2 years, % (95% CI) | 91.0 (82.8 to 95.4) | 84.3 (74.4 to 90.6) |
OS probability at 3 years, % (95% CI) | 87.3 (78.2 to 92.8) | 72.2 (60.8 to 80.8) |
OS probability at 4 years, % (95% CI) | 84.3 (74.3 to 90.6) | 66.8 (54.6 to 76.4) |
OS probability at 5 years, % (95% CI) | 84.3 (74.3 to 90.6) | 64.4 (51.7 to 74.6) |
OS probability at 6 years, % (95% CI) | 84.3 (74.3 to 90.6) | 53.7 (30.6 to 72.1) |
OS probability at 7 years, % (95% CI) | 84.3 (74.3 to 90.6) | 53.7 (30.6 to 72.1) |
RFS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with RFS events, n (%) | 21 (18.8) | 34 (30.4) |
Relapse | 13 (11.6) | 26 (23.2) |
Death due to any cause | 8 (7.1) | 8 (7.1) |
No. of patients censored, n (%) | 91 (81.3) | 78 (69.6) |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 63 (56.3) | 45 (40.2) |
Received allogeneic SCT | 25 (22.3) | 28 (25.0) |
Discontinued study | 3 (2.7) | 5 (4.5) |
Consent withdrawn | 3 (2.7) | 4 (3.6) |
Lost to follow-up | 0 (0.0) | 1 (0.9) |
RFS (months), median (95% CI) | NE (NE to NE) | 5.8 (4.2 to NE) |
Stratified HR (95% CI)a,c | 0.60 (0.34 to 1.04) | |
P value | 0.068 | |
Duration of follow-up, median (95% CI) yearsc | 4.0 (3.3 to 4.3) | 3.6 (3.2 to 4.1) |
RFS probability at 0.5 years, % (95% CI) | 92.0 (84.5 to 95.9) | 91.4 (83.6 to 95.6) |
RFS probability at 1 year, % (95% CI) | 90.8 (83.0 to 95.1) | 83.0 (73.4 to 89.4) |
RFS probability at 2 years, % (95% CI) | 82.5 (73.0 to 89.0) | 73.1 (62.2 to 81.3) |
RFS probability at 3 years, % (95% CI) | 81.4 (71.6 to 88.0) | 65.3 (54.0 to 74.5) |
RFS probability at 4 years, % (95% CI) | 75.8 (65.1 to 83.6) | 61.9 (50.1 to 71.7) |
RFS probability at 5 years, % (95% CI) | 75.8 (65.1 to 83.6) | 59.6 (47.4 to 69.9) |
RFS probability at 6 years, % (95% CI) | 75.8 (65.1 to 83.6) | 47.2 (28.3 to 63.9) |
RFS probability at 7 years, % (95% CI) | 75.8 (65.1 to 83.6) | 47.2 (28.3 to 63.9) |
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; OS = overall survival; RFS = relapse-free survival; SE = standard error.
Note: DCO date: E1910 = June 23, 2023, and AALL1731 = June 30, 2024
aStratification factors: age (< 55 years vs. ≥ 55 years), CD20 status (positive vs. negative vs. not collected), rituximab use (yes vs. no vs. not collected), intent to receive allogeneic SCT (yes vs. no).
bThe HR estimates are obtained from a stratified Cox regression model. An HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
cDefined as median KM time to censoring in the E1910 trial. Years are calculated as days from randomization or registration date to event/censor date, divided by 365.25. Time to censoring measures follow-up time by reversing the status indicator for censored and events.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 31: Sensitivity Analysis 3 in the E1910 Trial Censored at Allogeneic SCT or Start of Nonprotocol Systemic Anticancer Therapy for MRD-Negative at Step 3 (FAS)
Variable | E1910 | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
OS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with OS events, n (%) | 13 (11.6) | 16 (14.3) |
No. of patients censored, n (%) | 99 (88.4) | 96 (85.7) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 64 (57.1) | 43 (38.4) |
Received allogeneic SCT | 28 (25.0) | 27 (24.1) |
Start of nonprotocol systemic anticancer therapy | 2 (1.8) | 21 (18.8) |
Discontinued study | 5 (4.5) | 5 (4.5) |
Consent withdrawn | 4 (3.6) | 4 (3.6) |
Lost to follow-up | 1 (0.9) | 1 (0.9) |
OS (months), median (95% CI) | NE (NE to NE) | NE (6.0 to NE) |
Stratified HR (95% CI)a,b | 0.74 (0.35 to 1.55) | |
P value | 0.420 | |
Duration of follow-up, median (95% CI) yearsc | 3.7 (3.1 to 4.2)c,d | 3.0 (1.7 to 3.5)c,d |
OS probability at 0.5 years, % (95% CI) | 98.1 (92.6 to 99.5) | 99.1 (93.8 to 99.9) |
OS probability at 1 year, % (95% CI) | 95.7 (88.9 to 98.4) | 94.9 (86.9 to 98.1) |
OS probability at 2 years, % (95% CI) | 90.8 (82.4 to 95.3) | 92.0 (82.9 to 96.4) |
OS probability at 3 years, % (95% CI) | 87.1 (77.8 to 92.6) | 83.9 (72.6 to 90.8) |
OS probability at 4 years, % (95% CI) | 84.0 (73.8 to 90.4) | 76.9 (63.4 to 86.0) |
OS probability at 5 years, % (95% CI) | 84.0 (73.8 to 90.4) | 73.7 (59.0 to 83.8) |
OS probability at 6 years, % (95% CI) | 84.0 (73.8 to 90.4) | 59.0 (27.7 to 80.5) |
OS probability at 7 years, % (95% CI) | 84.0 (73.8 to 90.4) | 59.0 (27.7 to 80.5) |
RFS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with RFS events, n (%) | 21 (18.8) | 30 (26.8) |
Relapse | 13 (11.6) | 23 (20.5) |
Death due to any cause | 8 (7.1) | 7 (6.3) |
No. of patients censored, n (%) | 91 (81.3) | 82 (73.2) |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 61 (54.5) | 42 (37.5) |
Received allogeneic SCT | 25 (22.3) | 25 (22.3) |
Start of nonprotocol systemic anticancer therapy | 2 (1.8) | 10 (8.9) |
Discontinued study | 3 (2.7) | 5 (4.5) |
Consent withdrawn | 3 (2.7) | 4 (3.6) |
Lost to follow-up | 0 (0.0) | 1 (0.9) |
RFS (months), median (95% CI) | NE (NE to NE) | 5.8 (4.2 to NE) |
Stratified HR (95% CI)a,b | 0.67 (0.38 to 1.18) | |
P value | 0.168 | |
Duration of follow-up, median (95% CI) yearsc | 3.9 (3.3 to 4.3) | 3.5 (3.0 to 4.0) |
RFS probability at 0.5 years, % (95% CI) | 91.8 (84.2 to 95.9) | 92.4 (84.7 to 96.3) |
RFS probability at 1 year, % (95% CI) | 90.6 (82.6 to 95.0) | 86.1 (76.7 to 91.9) |
RFS probability at 2 years, % (95% CI) | 82.2 (72.4 to 88.7) | 75.5 (64.5 to 83.5) |
RFS probability at 3 years, % (95% CI) | 81.0 (71.0 to 87.8) | 68.6 (57.0 to 77.7) |
RFS probability at 4 years, % (95% CI) | 75.3 (64.5 to 83.3) | 64.7 (52.4 to 74.5) |
RFS probability at 5 years, % (95% CI) | 75.3 (64.5 to 83.3) | 62.0 (49.0 to 72.6) |
RFS probability at 6 years, % (95% CI) | 75.3 (64.5 to 83.3) | 47.0 (25.6 to 65.8) |
RFS probability at 7 years, % (95% CI) | 75.3 (64.5 to 83.3) | 47.0 (25.6 to 65.8) |
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; OS = overall survival; RFS = relapse-free survival; SE = standard error.
Note: DCO date: E1910 = June 23, 2023, and AALL1731 = June 30, 2024
aStratification factors: age (< 55 years vs. ≥ 55 years), CD20 status (positive vs. negative vs. not collected), rituximab use (yes vs. no vs. not collected), intent to receive allogeneic SCT (yes vs. no).
bThe HR estimates are obtained from a stratified Cox regression model. An HR < 1.0 indicates a lower average death rate and a longer survival for patients in the blinatumomab plus chemotherapy arm relative to patients in the chemotherapy-only arm.
cDefined as median KM time to censoring in the E1910 trial. Years are calculated as days from randomization or registration date to event/censor date, divided by 365.25. Time to censoring measures follow-up time by reversing the status indicator for censored and events.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 32: Sensitivity Analysis 4 in the E1910 Trial — RMST (FAS)
Variable | E1910 | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
OS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with OS events, n (%) | 19 (17.0) | 40 (35.7) |
No. of patients censored, n (%) | 93 (83.0) | 72 (64.3) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 88 (78.6) | 64 (57.1) |
Discontinued study | 5 (4.5) | 8 (7.1) |
Consent withdrawn | 4 (3.6) | 6 (5.4) |
Lost to follow-up | 1 (0.9) | 2 (1.8) |
RMST estimate (years) | ||
At 3 years (95% CI) | 2.8 (2.7 to 2.9) | 2.6 (2.4 to 2.7) |
Treatment difference | 0.2 (0.0, 0.4) | |
P value | 0.047 | |
At 5 years (95% CI) | 4.4 (4.2 to 4.7) | 3.9 (3.6 to 4.2) |
Treatment difference | 0.5 (0.2, 0.9) | |
P value | 0.006 | |
RFS | ||
No. of patients included in the analysis | 112 | 112 |
No. of patients with OS events, n (%) | 25 (22.3) | 43 (38.4) |
Relapse | 15 (13.4) | 32 (28.6) |
Death due to any cause | 10 (8.9) | 11 (9.8) |
No. of patients censored, n (%) | 87 (77.7) | 69 (61.6) |
Relapsed before start of RFS assessment | 0 (0.0) | 0 (0.0) |
Completed study without event | 0 (0.0) | 0 (0.0) |
Continues on study | 84 (75.0) | 61 (54.5) |
Discontinued study | 3 (2.7) | 8 (7.1) |
Consent withdrawn | 3 (2.7) | 6 (5.4) |
Lost to follow-up | 0 (0.0) | 2 (1.8) |
RMST estimate (years) | ||
At 3 years (95% CI) | 2.6 (2.4 to 2.8) | 2.4 (2.2 to 2.5) |
Treatment difference | 0.2 (0.0, 0.5) | |
P value | 0.049 | |
At 5 years (95% CI) | 4.2 (3.9 to 4.5) | 3.6 (3.3 to 4.0) |
Treatment difference | 0.5 (0.1, 1.0) | |
P value | 0.021 | |
CI = confidence interval; FAS = Full Analysis Set; OS = overall survival; RFS = relapse-free survival; RMST = restricted mean survival time.
Source: BLINCYTO E1910 Clinical Study Report.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 33: Sensitivity Analysis in the AALL1731 Trial — DFS RMST
Variable | AALL1731 | |
|---|---|---|
Blinatumomab plus chemotherapy | Chemotherapy | |
DFS — overall cohort | ||
No. of patients included in the analysis | 718 | 722 |
Difference in restricted mean survival time (95% CI), days | 72 (36 to 108) | |
P valuea | < 0.001 a | |
Adjusted difference in restricted mean survival time (95% CI), dayse | 74 (38 to 110) | |
DFS — SR-average | ||
No. of patients included in the analysis | 417 | 418 |
Difference in restricted mean survival time (95% CI), days | 67 (24 to 110) | |
DFS — SR-high | ||
No. of patients included in the analysis | 301 | 304 |
Difference in restricted mean survival time (95% CI), days | 79 (17 to 140) | |
CI = confidence interval; DFS = disease-free survival.
Notes: OS analyses in AALL1731 were performed post hoc while DFS analyses in the SR-average and SR-high cohorts were the prespecified subgroup analyses of the primary outcome.
DCO date: June 30, 2024
aP values are presented from interim analyses; however, thresholds for efficacy or futility were not reported in the trial’s statistical analysis plan. As such, these P values should not be interpreted as evidence of statistical significance.
Source: Gupta, 2024.68
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALL
acute lymphoblastic leukemia
BIA
budget impact analysis
CAR
chimeric antigen receptor
CDA-AMC
Canada’s Drug Agency
CIHI
Canadian Institute for Health Information
DFCI
Dana-Farber Cancer Institute
DFS
disease-free survival
ECOG
Eastern Cooperative Oncology Group
HCRU
health care resource utilization
HSCT
hematopoietic stem cell transplantation
ICER
incremental cost-effectiveness ratio
KM
Kaplan-Meier
LLSC
Leukemia & Lymphoma Society of Canada
LY
life-year
MCM
mixture-cure model
MRD
minimal residual disease
NICE
National Institute for Health and Care Excellence
OS
overall survival
Ph
Philadelphia chromosome
PRS
postrelapse survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
RFS
relapse-free survival
SE
standard error
SOC
standard of care
SR
standard risk
SR-average
standard-risk B-cell acute lymphoblastic leukemia with average risk of relapse
SR-high
standard-risk B-cell acute lymphoblastic leukemia with high risk of relapse
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Blinatumomab (BLINCYTO), lyophilized powder for solution, IV infusion by infusion pump. |
Indication | For the treatment of patients with Philadelphia chromosome-negative CD19 positive B-cell precursor acute lymphoblastic leukemia in the consolidation phase of multiphase chemotherapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | December 27, 2024 |
Reimbursement request | Treatment of adult and pediatric patients with Philadelphia chromosome—negative CD19-positive B-cell precursor acute lymphoblastic leukemia in the consolidation phase of multiphase chemotherapy in the frontline setting |
Sponsor | Amgen Canada Inc. |
Submission history | Previously reviewed: Yes Indication: Pediatric patients with Philadelphia chromosome—negative (Ph-negative) relapsed or refractory B-cell precursor ALL Recommendation date: August 23, 2017 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Adult patients with Ph-negative relapsed or refractory B-cell precursor ALL, including those who have had 1 prior line of therapy Recommendation date: August 31, 2017 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Adult patients with Ph-positive B-cell precursor ALL that has relapsed or is refractory to at least 1 second-generation or later tyrosine kinase inhibitor (TKI) or patients who are intolerant to second-generation or later TKIs and intolerant or refractory to imatinib Recommendation date: April 4, 2019 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Patients with Ph-negative CD19-positive B-cell precursor ALL in first or second hematologic complete remission with minimal residual disease greater than or equal to 0.1% Recommendation date: October 29, 2020 Recommendation: Reimburse with clinical criteria and/or conditions |
ALL = acute lymphoblastic leukemia; NOC = Notice of Compliance; Ph = Philadelphia chromosome; TKI = tyrosine kinase inhibitor.
Pagebreak
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival with a mixture-cure component for adults; partitioned survival model for pediatric patients |
Target population | Adult and pediatric patients with Philadelphia chromosome–negative CD19-positive B-cell precursor acute lymphoblastic leukemia in the consolidation phase of multiphase chemotherapy in frontline setting |
Treatment | Blinatumomab in combination with SOC |
Dose regimen | A single cycle of blinatumomab treatment is 28 days of continuous infusion followed by a 14-day treatment-free interval 28 mcg/day for patients weighing 45 kg or more; 15 mcg/m2 per day based on body surface area (not to exceed 28 mcg/day) for patients weighing less than 45 kg |
Submitted price | Blinatumomab: $2,978.26 per 38.5 mcg vial |
Submitted treatment cost | $83,391 per cycle for adults; $46,289 per cycle for pediatric patients |
Comparators | Adult SOC: A multidrug chemotherapy that consists of daunorubicin, vincristine, methotrexate, cyclophosphamide, cytarabine, etoposide, pegaspargase, dexamethasone, and 6-mercaptopurine Pediatric SOC: Varied by standard risk groups, may include vincristine, methotrexate, dexamethasone, pegaspargase, mercaptopurine, leucovorin, doxorubicin, cyclophosphamide, thioguanine, and cytarabine |
Perspective | Canadian publicly funded health care payer |
Outcomes | Life-years, QALYs |
Time horizon | Lifetime (50 years for adults and 92 years for pediatric patients) |
Key data sources | E1910 trial to inform the adult model AALL1731 trial to inform the pediatric model |
Submitted results | ICER = $33,368 per QALY gained (incremental cost: $90,222; incremental QALYs: 2.70) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; HCRU = health care resource utilization; ICER = incremental cost-effectiveness ratio; KM = Kaplan-Meier; OS = overall survival; QALY = quality-adjusted life-year; RDI = relative dose intensity; SOC = standard of care.
Conclusions
According to the Canada’s Drug Agency (CDA-AMC) clinical review, in the adult population, the post hoc analysis of the E1910 trial showed that blinatumomab plus standard of care (SOC) demonstrated clinically meaningful improvements in overall survival (OS) and relapse-free survival (RFS) compared to SOC alone, with high to moderate certainty of evidence across different time points. In the pediatric population, the results of the AALL1731 trial showed clinically meaningful improvements in disease-free survival (DFS) with blinatumomab plus SOC compared to SOC alone. Post hoc analyses for OS in the pediatric population suggested little to no difference between blinatumomab plus SOC compared to SOC alone.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that blinatumomab plus SOC would be considered cost-effective at a willingness-to-pay threshold of $50,000 per quality-adjusted life-year (QALY) gained. The CDA-AMC reanalysis resulted in an incremental cost-effectiveness ratio (ICER) for blinatumomab plus SOC versus SOC alone of $37,111 per QALY gained (incremental cost = $95,728; incremental QALYs = 2.58; incremental life-years [LYs] = 3.04). This analysis represents the combined results of the adult and pediatric populations, for which the predicted costs and outcomes for blinatumomab plus SOC and SOC alone differed. The overall results were influenced largely by the predicted survival benefit in the adult population, in which blinatumomab plus SOC was estimated to result in 6.66 additional LYs (QALYs = 5.54) compared to SOC alone. Subgroup analyses demonstrated that the ICER in the adult population was $27,682 per QALY gained, compared to $508,738 per QALY gained in the pediatric population.
The CDA-AMC base-case results are dependent upon patients treated with blinatumomab plus SOC surviving 3 additional years of life (or 2.58 QALYs) in the overall population. The majority (88%) of the incremental QALYs gained by patients receiving blinatumomab plus SOC in the model occurred beyond the clinical trial time frame, for which we have no comparative clinical data, and were based on extrapolation. In the absence of long-term clinical evidence, the extent of this survival benefit remains uncertain. Should the OS benefit of blinatumomab plus SOC be less than predicted in CDA-AMC analysis, the ICER would be higher, which may influence the conclusions on cost-effectiveness.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from several groups: The Leukemia & Lymphoma Society of Canada (LLSC) conducted a survey for the adult population, and LLSC, Advocacy for Canadian Childhood Oncology Research Network, Ontario Parents Advocating for Child with Cancer, and Childhood Cancer Canada conducted interviews with 3 caregivers of patients who have received blinatumomab for the pediatric population. Adult survey respondents with lived experience (106 respondents, consisting of 75 patients with acute lymphoblastic leukemia [ALL] and 31 caregivers) indicated that ALL had a significant negative impact on quality of life, with low energy, fear of infections, and frequent hospital visits as major contributors. Respondents reported receiving multiple treatments, including chemotherapy, stem cell transplants, radiation therapy, immunotherapy, targeted therapy, chimeric antigen receptor (CAR) T-cell therapy, and others (natural medicine, Chinese medicine, sound baths, meditation, transfusions, steroids, and antiemetics). Caregivers of patients in the pediatric population who were interviewed highlighted the negative impacts of chemotherapy on the patients’ physical and emotional health. The input noted that patients struggled with side effects, including fatigue, neutropenia, thrombocytopenia, infections, nausea or vomiting, and anemia as factors that significantly impacted quality of life. Respondents expressed the desire for alternative treatments with fewer side effects and longer remission. Patients previously treated with blinatumomab reported full or partial clinical response, with fewer side effects, greater tolerability of treatment, and better quality of life.
Clinician input was received from Canadian Leukemia Study Group, Ontario Health — Cancer Care Ontario Hematology Cancer Drug Advisory Committee, and Pediatric Oncology Group of Ontario. Clinicians indicated that the current SOC for patients is risk-stratified, multidrug chemotherapy with the goals of improving survival and achieving complete remission. Additional treatments for patients with relapsed ALL included CAR T-cell therapy and allogeneic stem cell transplantation. Clinician input suggested that blinatumomab would complement current chemotherapy regimens and is expected to lower toxicity and shorten duration of treatment. Clinician groups noted that the availability of minimal residual disease (MRD) assessments varies across Canada, and experience with blinatumomab administration is currently limited to leukemia care centres.
The drug plans raised several questions concerning blinatumomab treatment, including the definition of MRD-negative and the threshold values for MRD status. Concerns were also noted regarding the eligibility of patients outside the clinical trial age range, as well as those with Burkitt leukemia, acute undifferentiated leukemia, or an Eastern Cooperative Oncology Group (ECOG) performance status greater than 2. Additionally, the criteria for discontinuing treatment were questioned, particularly the definitions of loss of response, absence of clinical benefit, disease progression, and fixed-duration therapy. The plans also addressed health care resource needs during blinatumomab administration, such as pumps, hospitalization, and adverse event monitoring, and highlighted the concern about the limited availability of treatment centres for patients with ALL.
Several of those concerns were addressed in the sponsor’s model:
The clinical efficacy and adverse events (AEs) of blinatumomab were modelled based on OS, RFS, and AE data from the E1910 trial and the AALL1731 trial.
The disutilities related to AEs, blinatumomab treatment, and hematopoietic stem cell transplantation (HSCT) were included in the submitted economic model.
The submitted model included administration costs associated with treatment, such as hospitalization, bag changes, pumps, and other related expenses, as well as costs for additional health care resource use (HCRU) and AE management.
CDA-AMC was unable to address the following concern raised from the input from patients, clinicians, and drug programs:
The eligibility of patients outside of the trial’s inclusion criteria for blinatumomab treatment.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of blinatumomab plus SOC compared to SOC alone, in adult and pediatric patients with Philadelphia chromosome–negative (Ph-negative) CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy.1 SOC was defined as a multidrug chemotherapy, following the UKALLXII/ECOG 2993 protocol for adults and AALL1731 protocol for pediatric patients. The modelled population reflects the Health Canada indication.
Blinatumomab is available as lyophilized powder in a 38.5 mcg vial and is packaged with a vial of liquid to prepare the dose of blinatumomab. A single cycle of blinatumomab treatment is 28 days of continuous infusion followed by a 14-day treatment-free interval. Patients weighing greater than 45 kg receive a fixed dose of 28 mcg/day; patients weighing less than 45 kg receive a dose of 15 mcg/m2 per day based on body surface area.2 At the submitted price of $2,978.26 per 38.5 mcg vial, the sponsor estimated that that the drug acquisition cost for blinatumomab in a 28-day cycle was $83,391.28 for adults and $46,288.72 for children, assuming a relative dose intensity (RDI) of 100%.1 The regimen of SOC varies at different treatment time points, and the associated costs per cycle range from $1,321.91 to $8,295.74, assuming an RDI of 100%.1 Drug wastage was considered for both IV and oral therapies in the model.
The clinical outcomes of interest were LYs and QALYs. The economic analysis was conducted over a lifetime (50 years for adults and 92 years for pediatric patients; weekly cycles) time horizon from the perspective of a Canadian public health care payer. Discounting (1.5% per annum) was applied for both costs and outcomes.1
Model Structure
The sponsor submitted a partitioned survival model incorporating a mixture-cure component (MCM) with 3 health states for adult population: RFS, postrelapse survival (PRS), and death (Figure 1). Patients enter the model in the RFS state. Within the RFS state, it is assumed that a patient’s disease is either in a stable state or not actively progressing. Patients can thereafter transition to either the PRS or death states, the latter of which is an absorbing health state. Patients in the PRS state are assumed to have relapsed and therefore move on to second-line treatment. The proportion of patients in the RFS state was calculated based on the RFS curve. The proportion of patients in the PRS state was estimated based on the difference between the OS and the RFS curves, while the proportion of patients in the death state was estimated by subtracting the proportion of living patients from the total cohort.1
To consider the possibility of clinical remission, an MCM was included in the model for the adult cohort to estimate the proportion of patients who are “cured.” Patients whose disease was considered cured were defined as those who remain in remission for 5 years and followed a survival function that was in line with the general population with an additional risk of excess mortality. Survival of patients whose disease was not considered cured was extrapolated based on parametric functions.1
For the pediatric cohort, a partitioned survival model was used without incorporating an MCM, due to immature survival data from the AALL1731 study. OS and DFS were modelled using Kaplan-Meier (KM) estimates up to year 4, followed by general population data with adjustments for excess mortality. Because patients aged 11 to 17 years were excluded from the study, the model assumed their survival outcomes aligned with patients with standard-risk (SR) ALL with high risk of relapse (SR-high).1
Model Inputs
The baseline characteristics and clinical inputs used to inform the model for adult population were derived from a post hoc analysis of the E1910 study, a phase III, randomized control trial assessing the blinatumomab’s efficacy and safety in adult patients with B-cell precursor ALL.3 This post hoc analysis combined patients who were MRD-positive with the patients in the primary analysis of E1910 (n = 286; mean age = 49.9 years; 51.4% female; 48.6% male; mean body mass = 86.64 kg; mean body surface area = 2.00 m2).1 Extrapolation of survival data using the E1910 patient level data were applied to obtain the data beyond the study period. To reflect the possibility for long-term remission and cure, MCMs were fitted. The survival function was selected based on clinical plausibility and statistical fit, and the same distribution was applied across both arms.1
The baseline characteristics for the pediatric cohort were informed by the AALL1731 trial, publications, and assumptions.4,5 Of the pediatric cohort, 59% were aged 0 to 10 years (mean age = 4.3 years; mean body mass = 17.5 kg; body surface area = 0.75 m2) and 41% were aged 11 to 17 years (mean age = 13.8 years; mean body mass = 42.7 kg; body surface area = 1.41 m2).
Health state utility values for relapse-free health state were derived from EQ-5D-5L data collected in the BLAST study, using Canadian tariffs. Health utility values for postrelapse were sourced from the TOWER trial, where values were mapped from European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) to the EQ-5D.6,7 To reflect the change in quality of life of patients due to aging, age-adjusted factors were applied. Additionally, disutility inputs were sourced from the literature and past health technology assessment agency submissions. These were applied to patients who experienced AEs, underwent allogeneic HSCT, or were within 6 months of death.
Costs in the model included drug acquisition, drug administration, HSCT, maintenance therapy, subsequent therapy, HCRU, AE management, and terminal care. For adults, the dosing regimens for both arms followed the E1910 trial.3 After induction therapy, the blinatumomab plus SOC arm included 2 cycles of blinatumomab for intensification, followed by 2 cycles of blinatumomab and 4 cycles of chemotherapy given in various time points in the consolidation phase. Patients in the SOC arm received 4 cycles of chemotherapy during the consolidation phase. For pediatric patients, the dosing regimens were based on the AALL1731 trial.4 Specifically, treatment was stratified by risk category (i.e., SR with average risk of relapse [SR-average] and SR-high), and blinatumomab phases were administered in conjunction with multidrug chemotherapy cycles.
Drug acquisition costs were calculated as a function of unit drug costs, dosing schedules, RDI, and the proportion of patients on treatment in each cycle. The unit cost of blinatumomab was based on the sponsor’s submitted price, while all other drug acquisition costs were obtained from Ontario Drug Benefit Formulary and prior CDA-AMC submissions.8-15 Administration costs were included for both arms, including infusion bag change, pump cost, IV and intrathecal administration, and physician visits, with unit prices obtained from the Schedule of Benefits for Physician Services, Job Bank Canada, and Ontario Schedule of Health Benefits.16-18 For both blinatumomab and chemotherapy treatment, costs for hospital inpatient stay in each cycle were assumed, with the daily cost of hospitalization sourced from Canadian Institute for Health Information (CIHI).19
Adult patients in the blinatumomab plus SOC arm could undergo HSCT after 2 cycles of treatment, while those in the SOC arm could receive HSCT anytime during consolidation chemotherapy. The proportion of patients who received HSCT was sourced from the E1910 trial.3 For pediatric patients, 2% were assumed to undergo HSCT before relapse. The cost of HSCT was obtained from the Interprovincial Billing Rates for Designated High Cost Transplants (2024).20 After consolidation therapy, adult patients in both treatment arms received maintenance therapy for up to 2.5 years (2 years for pediatric patients) or until relapse. The dosing regimen followed the E1910 trial for adults and the AALL1731 trial for children. Upon relapse, adult patients could receive subsequent treatment with either blinatumomab, inotuzumab ozogamicin, chemotherapy, CAR T-cell therapy, or HSCT, while pediatric patients could receive blinatumomab, chemotherapy, CAR T-cell therapy, or HSCT. The proportion of patients receiving these therapies was based on clinical expert feedback and assumptions.21 The dosing regimens and costs for subsequent treatment were sourced from clinical trials, past CDA-AMC and UK National Institute for Health and Care Excellence (NICE) submissions, and clinical guidelines.6,22-26
Inpatient and outpatient HCRU were informed by a prior CDA-AMC submission for blinatumomab and a clinical expert survey, with costs sourced from CIHI and Ontario Schedule of Benefits.7,17,19 AE management costs were included with the unit cost sourced from CIHI.19 A one-off terminal care cost was applied to patients that transition to the death health state. All costs were adjusted to 2024 Canadian values.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented. The submitted analysis was based on the submitted price for blinatumomab.
Base-Case Results
In the sponsor’s base-case analysis in the overall population, blinatumomab plus SOC was associated with an estimated cost of $399,169 and 23.41 QALYs over a lifetime time horizon. When compared with the SOC alone, blinatumomab plus SOC generated 3.25 incremental 3.25 LYs, 2.70 incremental QALYs, and $90,222 incremental costs, resulting in an estimated ICER of $33,638 per QALY gained (Table 3). At a willingness-to-pay threshold of $50,000 per QALY gained, blinatumomab had a 91.3% probability of being considered cost-effective.
The drug cost (43% of total cost) was the key cost driver for the blinatumomab plus SOC arm, while the HCRU costs (24% of total cost) were the key cost driver for SOC alone arm. Approximately 82% of the incremental LYs and 88% of the incremental QALYs were derived from the period beyond the period covered by trial data.
Additional results from the sponsor’s submitted economic base are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 308,947 | Reference | 20.70 | Reference | Reference |
Blinatumomab plus SOC | 399,169 | 90,222 | 23.41 | 2.70 | 33,368 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted deterministic scenario analyses to address structural and parametric uncertainties. These included using an alternative discount rate for costs and efficacy, excluding MCMs (for adults) for survival modelling, narrowing the target population to patients with MRD-negative status (for adults), shortening the time horizon, applying alternative parametric functions for OS and RFS (for adults), excluding drug wastage, reducing health utility values for cured patients, and adjusting standardized mortality ratios. The results were similar to the base-case analysis across key scenarios.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
The long-term comparative clinical efficacy is uncertain: For adult patients in the sponsor’s base case, the RFS and OS curves for both arms were derived from the E1910 trial, and parametric survival functions were used to extrapolate the KM curves over the model’s time horizon. The sponsor’s base case predicts a survival advantage for patients treated with blinatumomab plus SOC compared with SOC alone (adult incremental LYs = 6.74; pediatric incremental LYs = 0.44; overall = 3.32). The sponsor assumed that patients who remained in a relapse-free health state for 5 years would be considered cured and applied a log-normal distribution to extrapolate RFS for both blinatumomab plus SOC and SOC alone arms. However, with the parametric function selected by the sponsor, 82% of the incremental LYs and 88% of the incremental QALYs gained by blinatumomab plus SOC were accrued during the extrapolated period, which contributes to the uncertainty in the long term clinical benefits.
For the pediatric population, the sponsor used KM data directly for the first 4 years of the modelled time horizon without conducting survival analysis for DFS and OS. This approach was inappropriate, as survival analysis provides a more robust approach to account for censoring and variability, allowing for better estimation of long-term outcomes beyond the observed data. The use of KM data directly did not incorporate parameter uncertainty, a requirement for probabilistic analysis, and thus assumed that the point estimates observed in the trial were not associated with any uncertainty. Results of the AALL1731 trial found that the probability of survival from randomization to 3 years was 98.4% (standard error [SE] = 0.8%) in the blinatumomab plus SOC arm and 97.1% (SE = 1.1%) in the SOC only arm. In the cohort with a high risk of relapse, the probability of survival from randomization to 3 years was 96.1% (SE = 2.0%) in the blinatumomab plus SOC arm and 95.3% (SE = 2.2%) in the SOC only arm. It is likely that, based on these point estimates, in probabilistic analysis there would be iterations in which SOC alone arm would be associated with higher OS than the blinatumomab plus SOC arm. However, the submitted model was not programmed to account for such uncertainty and, as a result, it is not possible to establish the impact of this uncertainty on the results of the model. In addition to this, visual inspection of the OS KM curves suggested little difference between blinatumomab plus SOC arms compared to SOC alone arms. Clinical experts consulted by CDA-AMC indicated that this is expected due to the established effectiveness of chemotherapy and salvage therapies in this population.
In reanalysis, CDA-AMC assumed that there would be no OS benefit in the pediatric population. As a result of model inflexibility to account for uncertainty, the lack of an appropriate survival analysis, results of the AALL1731 trial, and clinical expert opinion received by CDA-AMC, CDA-AMC was unable to address other limitations with the sponsor’s approach to modelling the pediatric population (including uncertainty around the use of KM data directly for DFS).
CDA-AMC was unable to resolve additional uncertainty in the comparative efficacy data for the adult population. Using the sponsor’s “uninformed” approach to their mixture-cure model, there was minimal variation in the predicted cure fractions and long-term extrapolations when testing alternative parametric distributions.
HCRU is likely overestimated: In the submitted model, the sponsor assumed that pediatric patients who experienced disease progression would be hospitalized for 2.5 days per month (totalling an average of 30 days per year for reasons unrelated to treatment) for up to 10 years in the model time horizon. Pediatric patients in the relapse-free health state were assumed to be hospitalized for reasons other than receiving treatment for 12 days per year for 5 years due to reasons other than receiving treatment. However, clinical experts consulted by CDA-AMC noted that, as most children achieve cure within 3 years in subsequent lines of therapy, the assumed length of hospitalization for both relapse-free and postrelapse health states in children is likely overestimated. Similarly, for adults in the relapse-free health state, clinical experts consulted by CDA-AMC agreed that the sponsor’s assumption that patients with MRD-positive status would be hospitalized for 21 days per year was likely overestimated and that they would not anticipate differences in hospitalization by MRD status in this health state.
In reanalysis, CDA-AMC assumed that pediatric patients who relapsed following first-line treatment were assumed to be hospitalized for 15 days per year up to 10 years in the model time horizon, after which no additional HCRU costs were applied in the progressed disease health state. For children in the relapse-free health state, hospitalization was assumed to be 6 days per year for up to 5 years, due to reasons unrelated to disease treatment. For adults in the relapse-free health state, hospitalization was assumed to be 1 day per year for up to 5 years, regardless of MRD status.
Uncertainty in drug acquisition costs: Results from the E1910 trial demonstrate that the RDI for blinatumomab ranged from 88% to 93%, while for SOC, it ranged from 86% to 91%. In the economic model, the RDI for blinatumomab aligned with the clinical trial values, but a 100% RDI was assumed for SOC in both arms. Clinical experts consulted by CDA-AMC indicated that this assumption was inappropriate due to potential dose modifications caused by AEs. Assuming a higher RDI for SOC than observed in the trial could overestimate drug acquisition costs in both arms.
In addition to incorporating RDI in the drug cost estimates, the sponsor also accounted for the proportion of patients assumed to start each cycle of blinatumomab and each cycle of consolidation therapy, based on the number of patients beginning each cycle observed in the E1910 trial, which ranged from 100% of patients beginning the first blinatumomab cycle to 50% receiving the fourth cycle of blinatumomab. In the SOC alone arm of the model, 93% of patients start the first cycle of consolidation chemotherapy, and 59% of patients start the final (fourth) cycle of SOC. Because drug acquisition accounts for 46% of the total cost for patients treated with blinatumomab plus SOC, the model is sensitive to changes in drug acquisition costs. However, potential differences in safety and efficacy resulting from higher uptake of blinatumomab are unknown.
In reanalysis, CDA-AMC applied the RDIs observed in the E1910 trial for SOC in both arms.
Poor modelling practices were employed: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors. Additionally, when calculating the HCRU costs unrelated to treatment in relapse-free health states for pediatric population, the formula failed to correctly reference the cells containing the proportion of patients starting treatment for the corresponding cycle. When calculating the utility decrements for pediatric patients who received blinatumomab treatment, the formula linked to an incorrect value for proportion of patients starting the treatment. Finally, the model offered limited flexibility for user input on key parameters. This limitation makes it more challenging for users to effectively compare the results of their own inputs with the original outputs.
CDA-AMC corrected the formula for HCRU costs in the relapse-free health state for the pediatric population and the calculation of utility decrements for pediatric patients who received blinatumomab. CDA-AMC was unable to address the use of IFERROR statements in the model and notes that a thorough validation of the sponsor’s model was not possible. CDA-AMC was unable to modify the model structure to enable dedicated user input fields.
The included comparator does not reflect clinical practice in Canada: For the adult population, the SOC regimen used in the model was built upon the UKALLXII/E2993 chemotherapy regimen, with dosing modifications based on C10403 AYA trial. However, the clinical experts consulted for this review with experience treating adult patients with ALL noted that the modified Dana-Farber Cancer Institute (DFCI) protocol is currently the most used protocol to treat Ph-negative B-cell precursor ALL in clinical practice in Canada. The clinical expert consulted for this review with experience treating adult patients with ALL indicated that the efficacy of the regimen used in the model was likely comparable to that of the modified DFCI regimen. However, if there were differences in comparative efficacy between the SOC used in the E1910 trial and the DFCI regimen, the cost-effectiveness of blinatumomab plus the DFCI regimen compared to the DFCI regimen alone is unknown.
Additionally, the sponsor did not provide evidence assessing the cost-effectiveness of blinatumomab plus SOC compared to blinatumomab as monotherapy, which has an existing indication and reimbursement recommendation for adult and pediatric patients with MRD-positive status. The dosage of blinatumomab is the same across both the current reimbursement request and the currently funded monotherapy. For adults with MRD-positive status, the number of blinatumomab cycles will also remain the same (i.e., 4). However, for pediatric patients, a maximum of 2 cycles are used in the current reimbursement request versus 4 cycles in the already funded version. As a result, the drug acquisition costs for blinatumomab across the 2 indications are likely similar in adults and less costly in the pediatric population because there are 2 fewer cycles of blinatumomab than in the current request.
CDA-AMC could not address this limitation in reanalysis. The cost-effectiveness of blinatumomab plus SOC versus SOC alone when using the DFCI regimen for adults is unknown. Additionally, the cost-effectiveness of blinatumomab plus SOC versus blinatumomab monotherapy in patients with MRD-positive status is unknown.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Pagebreak
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The overall analysis represents both the adult and pediatric populations. | Potentially appropriate. The ICER for the overall population was calculated using weighted averages of costs and QALYs based on the assumed proportion of patients in each health state (54.3% for pediatric and 45.7% for adult patients). If the proportions of adult and pediatric patients expected to be treated with blinatumomab plus SOC in clinical practice in Canada is different, the results of this analysis would change. |
Patients who remained in remission for 5 years were assumed to be cured. | Appropriate. The assumption aligns with clinical practice based on insights provided by clinical experts consulted by CDA-AMC. |
For the first 2 cycles of blinatumomab treatment, adult patients were assumed to not have 14-day treatment-free interval between these 2 cycles. | Inappropriate but unlikely to be a key model driver. This assumption was inconsistent with the description in the product monograph that a single cycle of treatment is 28 days of continuous infusion followed by a 14-day treatment-free interval. |
Due to lack of clinical data for pediatric patients aged 11 to 17 years, it was assumed that the survival for this group of population was similar to those of the patients in the SR-high-risk group in the AALL1731 trial. | Potentially appropriate. The assumption aligns with the clinical experts’ expectations of clinical outcomes in older pediatric patients. |
After 4 years, the survival for pediatric patients was modelled based on the general population, with adjustment for excess mortality (SMR of 1.09). | Potentially appropriate. It lacks supporting references and may introduce uncertainty in long-term survival estimates, especially given the significantly longer extrapolation period compared to the trial period (88 years vs. 4 years). CDA-AMC conducted a scenario analysis to explore the potential impact of assuming an SMR of 2. |
CDA-AMC = Canada’s Drug Agency; SOC = standard of care; SR = standard risk; SMR = standardized mortality ratio.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. Corrections and changes to the sponsor’s analyses are summarized in Table 5 and include corrections to proportion of patients starting treatment, change on the assumption of OS benefit for blinatumomab in pediatric population, and adjustment to inpatient days and RDIs. CDA-AMC was unable to address the limitations regarding the uncertainty in long-term comparative efficacy.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Proportion of patients starting treatment used in relapse-free HCRU cost in pediatric population | Referenced incorrect cells for value of proportion of starting treatment in both arms | Revised with correct value for proportion of starting treatment in both arms |
2. Proportion of patients starting treatment used in blinatumomab utility decrement in pediatric population | Incorporated proportion of starting treatment in adult population | Incorporated proportion of starting treatment in pediatric population |
Changes to derive the CDA-AMC base case | ||
1. a. OS in the pediatric population of SR-average-risk group | Assumed OS benefit for blinatumomab in the pediatric population of SR-average | Assumed no OS benefit for blinatumomab in the pediatric population of SR-average |
1. b. OS in the pediatric population of SR-high-risk group | Assumed OS benefit for blinatumomab in the pediatric population of SR-high | Assumed no OS benefit for blinatumomab in the pediatric population of SR-high |
2. a. Inpatient days for pediatric population in postrelapse health state | 30 days per year for 10 years | 15 days per year for 10 years |
2. b. Inpatient days due to reasons unrelated to treatment for pediatric population in relapse-free health state | 12 days per year for 5 years | 6 days per year for 5 years |
2. c. Inpatient days due to reasons unrelated to treatment for adult population in relapse-free health state | 21 days per year for patients with MRD-negative status; 0.72 days per year for patients with MRD-positive status | 1 day per year regardless of MRD status |
3. RDI for SOC in adult population | 100% for both arms | 91% for blinatumomab plus SOC arm and 86% for SOC alone arm |
CDA-AMC base case | Reanalysis 1a + 1b + 2a + 2b + 2c + 3 | |
CDA-AMC = Canada’s Drug Agency; HCRU = health care resource use; MRD = minimal residual disease; OS = overall survival; RDI = relative dose intensity; SOC = standard of care; SR = standard risk.
In the CDA-AMC base case for overall population, blinatumomab plus SOC was associated with a total cost of $343,489 and 23.72 QALYs, compared to $247,761 and 20.84 QALYs for SOC alone (Table 6). The resulting ICER for blinatumomab plus SOC compared to SOC alone was $37,111 per QALY, with 83.7% probability of cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY.
Results of the CDA-AMC reanalysis were driven by the estimated clinical benefits of blinatumomab plus SOC in the adult population, where the incremental QALYs were 5.54 compared to SOC alone (resulting in an ICER of $27,682 per QALY gained in this subgroup). Conversely, the ICER in the pediatric subgroup for blinatumomab plus SOC compared to SOC alone was $508,738 per QALY gained (incremental costs: $47,350; incremental QALYs: 0.09). Further details and disaggregated outcomes are available in Table 12.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 308,947 | 20.70 | Reference |
Blinatumomab plus SOC | 399,169 | 23.41 | 33,368 | |
Sponsor’s corrected base case | SOC | 303,074 | 20.70 | Reference |
Blinatumomab plus SOC | 391,887 | 23.39 | 33,117 | |
CDA-AMC reanalysis 1a: OS in the pediatric population of SR-average-risk group | SOC | 299,747 | 20.81 | Reference |
Blinatumomab plus SOC | 389,983 | 23.42 | 34,618 | |
CDA-AMC reanalysis 1b: OS in the pediatric population of SR-high-risk group | SOC | 298,823 | 20.74 | Reference |
Blinatumomab plus SOC | 389,983 | 23.42 | 34,087 | |
CDA-AMC reanalysis 2a: Inpatient days for pediatric population in postrelapse health state | SOC | 285,999 | 20.71 | Reference |
Blinatumomab plus SOC | 385,789 | 23.42 | 36,839 | |
CDA-AMC reanalysis 2b. Inpatient days due to reasons unrelated to treatment for pediatric population in relapse-free health state | SOC | 267,142 | 20.71 | Reference |
Blinatumomab plus SOC | 368,019 | 23.42 | 33,918 | |
CDA-AMC reanalysis 2c: Inpatient days due to reasons unrelated to treatment for adult population in relapse-free health state | SOC | 277,501 | 20.71 | Reference |
Blinatumomab plus SOC | 369,874 | 23.42 | 34,101 | |
CDA-AMC reanalysis 3: RDI for SOC in adult population | SOC | 296,519 | 20.71 | Reference |
Blinatumomab plus SOC | 389,404 | 23.42 | 34,290 | |
CDA-AMC base case (reanalyses 1a + 1b + 2a + 2b + 2c + 3) | SOC | 244,034 | 20.84 | Reference |
Blinatumomab plus SOC | 343,094 | 23.42 | 38,475 | |
CDA-AMC base case (reanalyses 1a + 1b + 2a + 2b + 2c + 3; probabilistic) | SOC | 247,761 | 20.84 | Reference |
Blinatumomab plus SOC | 343,489 | 23.42 | 37,111 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The CDA-AMC reanalysis is based on publicly available prices of comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC conducted a scenario analysis to assess the impact of an alternative SMR assumption on the cost-effectiveness of blinatumomab, where SMR was assumed to be 2. Results of this scenario analysis found that the ICER for blinatumomab plus SOC versus SOC alone was $44,498 per QALY gained.
Issues for Consideration
Blinatumomab has been previously reviewed by CDA-AMC for multiple indications, all with recommendations to reimburse with clinical criteria and/or conditions.8,27-30 Additionally, negotiations for blinatumomab with the pan-Canadian Pharmaceutical Alliance concluded with letters of intent.31,32 As a result, a confidential price agreement was established, and the price of blinatumomab paid by drug plans may be different than the price used in the economic evaluation and budget impact analysis.
Overall Conclusions
According to the CDA-AMC clinical review, in the adult population, the post hoc analysis of the E1910 trial showed that blinatumomab plus SOC demonstrated clinically meaningful improvements in OS and RFS compared to SOC alone, with high to moderate certainty of evidence across different time points. In the pediatric population, the results of the AALL1731 trial showed clinically meaningful improvements in DFS with blinatumomab plus SOC compared to SOC alone. Post hoc analyses for OS in the pediatric population suggested little to no difference between blinatumomab plus SOC compared to SOC alone. According to clinical experts consulted for this review, the safety profile of blinatumomab plus SOC was consistent with their expectations and deemed acceptable.
The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that blinatumomab plus SOC would be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. The CDA-AMC reanalysis resulted in an ICER for blinatumomab plus SOC versus SOC alone of $37,111 per QALY gained (incremental cost = $95,728; incremental QALYs = 2.58; incremental LYs = 3.04). This analysis represents the combined results of the adult and pediatric populations, for which the predicted costs and outcomes for blinatumomab plus SOC and SOC alone differed. The overall results were influenced largely by the predicted survival benefit in the adult population, in which blinatumomab plus SOC was estimated to result in 6.66 additional LYs (5.54 QALYs) compared to SOC alone. Subgroup analyses demonstrated that the ICER in the adult population was $27,682 per QALY gained, compared to $508,738 per QALY gained in the pediatric population.
The CDA-AMC base-case results are dependent upon the 3 additional LYs (QALYs = 2.58) in the overall population accrued by patients who were treated with blinatumomab plus SOC. The majority (88%) of the incremental QALYs gained by blinatumomab plus SOC was accrued during the time for which there are no comparative clinical data and were based on extrapolation. In the absence of long-term clinical evidence, the extent of this survival benefit remains uncertain. Should the OS benefit of blinatumomab plus SOC be less than predicted in the CDA-AMC analysis, the ICER would be higher, which may influence the conclusions on cost-effectiveness.
References
1.Amgen Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: blinatumomab, lyophilized powder for solution for infusion, 38.5 mcg. April 3, 2025.
2.Amgen Canada Inc. Product monograph - including patient medication information - Blincyto®. 2024 [sponsor submitted reference].
3.Amgen Inc. Study E1910 - Clinical Study Report (Primary Analysis) - A Phase III Randomized Trial of Blinatumomab (IND117467, NSC765986) for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults. 2024 [sponsor submitted reference].
4.Gupta S, Aitken JF, Bartels U, et al. Paediatric cancer stage in population-based cancer registries: the Toronto consensus principles and guidelines. Lancet Oncol. 2016;17(4):e163-e172. doi: 10.1016/S1470-2045(15)00539-2 PubMed
5.PedMed. Average Weights and Surface Areas. https://www.pedmed.org/DrugApp/Supplementary/SurfaceAreaTable.pdf [sponsor submitted reference]
6.Canada's Drug Agency. BLINCYTO for the treatment of adults with Philadelphia chromosome-negative relapsed or refractory B precursor acute lymphoblastic leukemia- TOWER. 2017 [sponsor submitted reference].
7.Canada's Drug Agency. BLINCYTO® (blinatumomab) for the treatment of MRD+ BCP-ALL. - BLAST. 2020 [sponsor submitted reference].
8.Canada's Drug Agency. Pan-Canadian Oncology Drug Review Final Economic Guidance Report - Blinatumomab (Blincyto) Resubmission for Acute Lymphoblastic Leukemia. 2017. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_blinatumomab_blincyto_all_resub_fn_egr.pdf [sponsor submitted reference]
9.Canada's Drug Agency. Pan-Canadian Oncology Drug Review Final Economic Guidance Report - Inotuzumab Ozogamicin (Besponsa) for Acute Lymphoblastic Leukemia. 2018. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_inotuzumab_ozogamicin_besponsa_all_fn_egr.pdf [sponsor submitted reference]
10.Canada's Drug Agency. CADTH Drug Reimbursement Review - Pharmacoeconomic Report - Eculizumab (Soliris). 2020. https://www.cda-amc.ca/sites/default/files/cdr/pharmacoeconomic/sr0605-soliris-mg-pharmacoeconomic-review-report.pdf [sponsor submitted reference]
11.Canada's Drug Agency. CADTH Drug Reimbursement Review - Crisantaspase Recombinant (Rylaze). 2023. https://www.cda-amc.ca/sites/default/files/DRR/2023/PC0301-Rylaze.pdf [sponsor submitted reference]
12.Canada's Drug Agency. CADTH Reimbursement Review - Brexucabtagene Autoleucel (Tecartus). 2023. https://www.cda-amc.ca/sites/default/files/DRR/2023/PG0304-Tecartus-ALL.pdf [sponsor submitted reference]
13.Canada's Drug Agency. CADTH Drug Reimbursement Review - Polatuzumab Vedotin (Polivy). 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PC0313-Polivy.pdf [sponsor submitted reference]
14.Canada's Drug Agency. CADTH Drug Reimbursement Review - Calaspargase Pegol (Asparlas). 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PC0321-Asparlas-Combined-Report.pdf [sponsor submitted reference]
15.Ministry of Health Long-Term Care. Ontario Drug Benefit Formulary/Comparative Drug Index. 2019. https://www.formulary.health.gov.on.ca/formulary/ [sponsor submitted reference]
16.Government of Canada. Job Bank Canada - Wages - Registered Nurse. 2023. https://www.jobbank.gc.ca/marketreport/wages-occupation/993/QC [sponsor submitted reference]
17.Ontario Ministry of Health. Schedule of Benefits. 2024. Accessed July 1 2024. https://www.ontario.ca/files/2024-04/moh-schedule-benefit-2024-02-20.pdf
18.Ontario Ministry of Health. Schedule of Benefits, Physician Services Under the Health Insurance Act. 2024. https://www.ontario.ca/files/2024-04/moh-schedule-benefit-2024-02-20.pdf [sponsor submitted reference]
19.CIHI. CIHI patient cost estimator. 2023. Accessed July 1 2024. https://www.cihi.ca/en/patient-cost-estimator [sponsor submitted reference]
20.Interprovincial Health Insurance Agreements Coordinating Committee. Interprovincial Billing Rates for Designated High Cost Transplants Effective for Discharges on or after April 1, 2024. 2024. https://www.ontario.ca/files/2024-04/moh-interprovincial-billing-rates-high-cost-transplants-en-2024-04-08.pdf [sponsor submitted reference]
21.Amgen Inc. UK Clinical validation meeting - Data on file. 2024 [sponsor submitted reference].
22.Canada's Drug Agency. Final Economic Guidance Report 2018 - Inotuzumab Ozogamicin (Besponsa) for Acute Lymphoblastic Leukemia. 2018 [sponsor submitted reference].
23.Canada's Drug Agency. CADTH Reimbursement Recommendation: Ciltacabtagene Autoleucel (CARVYKTI). 2023. https://www.cda-amc.ca/sites/default/files/DRR/2023/PG0302-Carvykti.pdf [sponsor submitted reference]
24.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med. 2016;375(8):740-53. doi: 10.1056/NEJMoa1509277 PubMed
25.National Institute for Health Care Excellence. TA541 Inotuzumab ozogamicin for treating relapsed or refractory B-cell acute lymphoblastic leukaemia. 2016. Accessed 10 November 2023. https://www.nice.org.uk/guidance/ta541/documents/committee-papers-2
26.University Health Network. Princess Margaret Cancer Center Clinical Practice Guidelines. Acute Lymphoblastic Leukemia. 2015. https://www.uhn.ca/PrincessMargaret/Health_Professionals/Programs_Departments/Leukemia/Documents/CPG_Leukemia_AcuteMyeloidLeukemia.pdf [sponsor submitted reference]
27.Canada's Drug Agency. Blinatumomab (Blincyto) for Ph- ALL. 2016 [sponsor submitted reference].
28.Canada's Drug Agency. Blinatumomab (Blincyto) for Acute Lymphoblastic Leukemia (pediatric) – Details. 2017 [sponsor submitted reference].
29.Canada's Drug Agency. Blincyto for Philadelphia Chromosome positive B-cell precursor Acute Lymphoblastic Leukemia – Details. 2019 [sponsor submitted reference].
30.Canada's Drug Agency. Blincyto for MRD-positive B-cell precursor ALL — Details. 2020 [sponsor submitted reference].
31.pan-Canadian Pharmaceutical Alliance. Acute Lymphoblastic Leukemia (ALL). 2019 [sponsor submitted reference].
32.pan-Canadian Pharmaceutical Alliance. Minimal Residual Disease (MRD)-Positive B-Cell Precursor Acute Lymphoblastic Leukemia (BCP ALL). 2021 [sponsor submitted reference].
33.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. 2024. Accessed January 15 2025. https://www.formulary.health.gov.on.ca/formulary/
34.Alberta Health Services. Acute Lymphoblastic Leukemia in Adults. 2023 [sponsor submitted reference].
35.IQVIA. DeltaPA. 2024. Accessed October 22, 2024. https://www.iqvia.com/
36.BC Government. BC PharmaCare formulary search. 2024. Accessed January 15, 2025. https://pharmacareformularysearch.gov.bc.ca
37.Amgen Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: blinatumomab, lyophilized powder for solution for infusion, 38.5 mcg. April 3, 2025.
38.Deepa Bhojwani JJY, Ching-Hon P. Biology of Childhood Acute Lymphoblastic Leukemia. Pediatr Clin North Am. 2016. doi: 10.1016/j.pcl.2014.09.004 PubMed
39.Amgen Inc. Study E1910 - Clinical Study Report (Primary Analysis) - A Phase III Randomized Trial of Blinatumomab (IND117467, NSC765986) for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults. 2024 [sponsor submitted reference]:123.
40.Statistics Canada. Canadian Cancer Statistics. 2023. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf [sponsor submitted reference]
41.Canada's Drug Agency. CADTH Drug Reimbursement Review - Polatuzumab Vedotin (Polivy). 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PC0313-Polivy.pdf [sponsor submitted reference]
42.DeAngelo DJ, Stevenson KE, Dahlberg SE, et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia. 2015;29(3):526-34. doi: 10.1038/leu.2014.229 PubMed
43.Amgen Canada Inc. Canadian Clinical experts validation meeting - Data on file. 2024 [sponsor submitted reference].
44.Canada's Drug Agency. CADTH Provisional Funding Algorithm: Adult B-cell precursor acute lymphoblastic leukemia, Philadelphia chromosome negative and positive. 2023. Accessed October 24, 2024. https://www.cadth.ca/adult-b-cell-precursor-acute-lymphoblastic-leukemia-philadelphia-chromosome-negative [sponsor submitted reference]
45.Statistics Canada. Table 13-10-0111-01 Number and rates of new cases of primary cancer, by cancer type, age group and sex. 2024. 10.25318/1310011101-eng [sponsor submitted reference]
46.Statistics Canada. Table 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). 2024. 10.25318/1710005701-eng [sponsor submitted reference]
47.Amgen. Internal forecasting and market research [data on file]. 2024 [sponsor submitted reference].
48.Statistics Canada. Table 13-10-0840-01 Cancer incidence trends, by sex and cancer type. 2023 [sponsor submitted reference].
Appendix 1: Cost Comparison Tables
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC participating drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CDA-AMC Cost Comparison Table for ALL Adult Population
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | 28-day cost ($)a |
|---|---|---|---|---|---|---|
Blinatumomab | ||||||
Blinatumomab (Blincyto) | 0.0385 mg per vial | Lyophilized powder for solution for infusion | $2,978.2600c | Four cycles of blinatumomab: 0.028 mg IV daily for days 1 to 28, followed by a 14-day treatment-free period (42-day cycles) | All cycles: $1,985.51 | All cycles: $55,594 |
UKALLXII/ECOG 2993b | ||||||
Cytarabine | 100 mg/mL | 5 mL vial 20 mL vial Solution for Injection | $76.8500d $306.5000d | Consolidation cycles: 1, 2, 4: 100 mg/m2 IV from days 1 to 5 of a 28-day cycle Consolidation cycle 3: 75 mg/m2 IV day 30 to 33 and 37 to 40 of 42-day cycle | Consolidation cycle 1, 2, 5: $13.72 Consolidation cycle 3: $14.64 | Consolidation cycle 1, 2, 5: $384 Consolidation cycle 3: $410 |
Etoposide | 20 mg/mL | 5 mL vial 10 mL vial 25 mL vial 50 mL vial Solution for injection | $75.0000d $150.0000d $375.0000d $750.0000d | Consolidation cycles: 1, 2, 4: 100 mg/m2 IV from days 1 to 5 of a 28-day cycle | Consolidation cycle 1, 2, 5: $26.79 | Consolidation cycle 1, 2, 5: $750 |
Methotrexate | 10 mg/mL 25 mg/mL | 2 mL vial 2 mL vial Solution for injection | $12.5000 $8.9200 | Consolidation cycle 1, 2, 4: 12.5 mg IT on day 1 of a 28-day cycle Consolidation cycle 3: 12.5 mg IT on day 2 of a 42-day cycle | Consolidation cycle 1, 2, 5: $0.45 Consolidation cycle 3: $0.30 | Consolidation cycle 1, 2, 5: $13 Consolidation cycle 3: $8 |
Pegaspargase (Oncaspar) | 750 IU/mL | 5 mL vial Solution for injection | $6,115.9700d | Consolidation cycle 1: 2000 IU/m2 IV on day 5 (maximum dosage: 3,750 IU) of a 28-day cycle | Consolidation cycle 1: $218.43 | Consolidation cycle 1: $6,116 |
Daunorubicin (Cerubidine) | 5 mg/mL | 4 mL vial Solution for injection | $91.0000 | Consolidation cycle 3: 30 mg/m2 IV days for 1, 8, 15, 22 of a 42-day cycle | Consolidation cycle 3: $26.00 | Consolidation cycle 3: $728 |
Vincristine | 1 mg/mL | 1 mL vial Solution for injection | $30.6000 | Consolidation cycle 3: 1.4 mg/m2 IV days for 1, 8, 15, 22 of a 42-day cycle | Consolidation cycle 3: $8.74 | Consolidation cycle 3: $245 |
Dexamethasone | 0.5 mg 4 mg | Tablet | $0.1564 0.6112 | Consolidation cycle 3: 10 mg/m2 PO, max dose of 20 mg, on days 1 to 7 and 15 to 21 for a 42-day cycle | Consolidation cycle 3: $1.02 | Consolidation cycle 3: $79 |
Cyclophosphamide (Procytox) | 500 mg/mL 1,000 mg/mL 2,000 mg/mL | Powder for solution for injection | $107.8100d $195.4200d $359.4000d | Consolidation cycle 3: 650 mg/m2 IV on day 29 of a 42-day cycle | Consolidation cycle 3: $0.30 | Consolidation cycle 3: $8 |
6-mercaptopurine (Purinethol) | 50 mg | Tablet | $2.8610 | Consolidation cycle 3: 60 mg/ m2 orally on days 29 to 42 of a 42-day cycle | Consolidation cycle 3: $2.66 | Consolidation cycle 3: $74 |
UKALLXII/ECOG 2993 | Consolidation cycle 1: $259.22 Consolidation cycle 2, 4: $40.96 Consolidation cycle 3: $53.65 | Consolidation cycle 1: $7,258 Consolidation cycle 2, 4: $1,147 Consolidation cycle 3: $1,502 | ||||
Modified DFCI 91-01/Al.4 Protocol — CNS therapy and Intensification aged < 60e | ||||||
Vincristine | 1 mg/mL | 1 mL vial Solution for injection | $30.6000 | CNS therapy: 2 mg IV on day 1 of 21 days Intensification: 2 mg IV on day 1 of 21-day cycles for 30 weeks | CNS therapy: $2.91 Intensification: $2.91 | All cycles: $82 |
6-mercaptopurine (Purinethol) | 50 mg | Tablet | $2.8610 | CNS therapy: 50 mg/m2 PO daily for 14 days Intensification: 50 mg/m2 PO daily for 14 consecutive days of 21-day cycles for 30 weeks | CNS therapy: $3.81 Intensification: $3.81 | CNS therapy: $107 Intensification: $107 |
Doxorubicin (Caelyx) | 2 mg/mL | 5 mL vial 25 mL vial 100 mL vial Solution for injection | $50.4500d $252.2500d $973.0000d | CNS therapy: 30 mg/m2 IV on day 1 of 21 days Intensification: 30 mg/m2 IV on day 1 from cycles 1 to 7 of 21-day cycles for 30 weeks | CNS therapy: $24.02 Intensification: $24.02 | CNS therapy: $673 Intensification: $673 |
Methotrexate | 10 mg/mL 25 mg/mL | 2 mL vial 2 mL vial Solution for injection | $12.5000 $8.9200 | CNS therapy: Four doses of 12 mg IT twice weekly for 21-days Intensification: 30 mg/m2 IV, IM, or PO weekly on days 2, 9, and 16 each 21-day cycle for cycles 8 to 10. 12 mg IT at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $14.29 Intensification cycles 1, 6: $0.60 Intensification cycles 8 to 10: $2.55 | CNS therapy: $400 Intensification cycles 1, 6: $17 Intensification cycles 8 to 10: $71 |
Cytarabine | 100 mg/mL | 5 mL vial 20 mL vial Solution for Injection | $76.8500d $306.5000d | CNS therapy: Four doses of 50 mg IT twice weekly for 21-days Intensification: 50 mg IT at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $87.83 Intensification: $3.66 | CNS therapy: $2,459 Intensification: $102 |
Hydrocortisone (Cortef) | 50 mg/mL 125 mg/mL 125 mg/mL | 2 mL vial 2 mL vial 4 mL vial Solution for injection | $5.4400f $9.2059f $19.0296f | CNS therapy: Four doses of 15 mg IT twice weekly for 21 days Intensification: 15 mg at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $6.22 Intensification: $0.26 | CNS therapy: $174 Intensification: $7 |
Dexamethasone | 0.5 mg 4 mg | Tablet | $0.1564 0.6112 | Intensification: 9 mg/m2 PO twice per day on days 1 to 5 of 21-day cycles for 30 weeks | Intensification: $1.46 | Intensification: $41 |
Pegaspargase (Oncaspar) | 750 IU/mL | 5 mL vial Solution for injection | $6,115.9700d | Intensification: 2000 IU/ m2 IV on day 1 of 21-day cycles for 30 weeks | Intensification: $582.47 | Intensification: $16,309 |
Modified DFCI 91-01/Al.4 Protocol — CNS therapy and Intensification aged < 60e | CNS therapy: $139 Intensification cycles 1 to 7: $615 Intensification cycles 8 to 10: $593 | CNS therapy: $3,894 Intensification cycles 1 to 7: $17,211 Intensification cycles 8 to 10: $16,610 | ||||
Modified DFCI 91-01/Al.4 Protocol — CNS therapy and Intensification aged > 60e | ||||||
Vincristine | 1 mg/mL | 1 mL vial Solution for injection | $30.6000 | CNS therapy: 2 mg IV on day 1 of 21 days Intensification: 2 mg IV on day 1 of 21-day cycles for 30 weeks | CNS therapy: $2.91 Intensification: $2.91 | All cycles: $82 |
6-mercaptopurine (Purinethol) | 50 mg | Tablet | $2.8610 | CNS therapy: 50 mg/m2 PO daily for 14 days Intensification: 50 mg/m2 PO daily for 14 consecutive days of 21-day cycles for 30 weeks | CNS therapy: $3.81 Intensification: $3.81 | CNS therapy: $107 Intensification: $107 |
Doxorubicin (Caelyx) | 2 mg/mL | 5 mL vial 25 mL vial 100 mL vial Solution for injection | $50.4500d $252.2500d $973.0000d | CNS therapy: 30 mg/m2 IV on day 1 of 21 days Intensification: 30 mg/m2 IV on day 1 from cycles 1 to 7 of 21-day cycles for 30 weeks | CNS therapy: $24.02 Intensification: $24.02 | CNS therapy: $673 Intensification: $673 |
Methotrexate | 10 mg/mL 25 mg/mL | 2 mL vial 2 mL vial Solution for injection | $12.5000 $8.9200 | CNS therapy: Four doses of 12 mg IT twice weekly for 21-days Intensification: 12 mg IT at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $14.29 Intensification: $0.60 | CNS therapy: $400 Intensification: $17 |
Cytarabine | 100 mg/mL | 5 mL vial 20 mL vial Solution for Injection | $76.8500d $306.5000d | CNS therapy: Four doses of 50 mg IT twice weekly for 21-days Intensification: 50 mg IT at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $87.83 Intensification: $3.66 | CNS therapy: $2,459 Intensification: $102 |
Hydrocortisone (Cortef) | 50 mg/mL 125 mg/mL 125 mg/mL | 2 mL vial 2 mL vial 4 mL vial Solution for injection | $5.4400f $9.2059f $19.0296f | CNS therapy: Four doses of 15 mg IT twice weekly for 21 days Intensification: 15 mg at the start of a cycle every 18 weeks for 30 weeks | CNS therapy: $6.22 Intensification: $0.26 | CNS therapy: $174 Intensification: $7 |
Dexamethasone | 0.5 mg 4 mg | Tablet | $0.1564 $0.6112 | Intensification: 6 mg/m2 PO twice per day on days 1 to 5 of 21-day cycles for 30 weeks | Intensification: $87 | Intensification: $24 |
Pegaspargase (Oncaspar) | 750 IU/mL | 5 mL vial Solution for injection | $6,115.9700d | Intensification: 1,000 IU/ m2 IV on day 1 of 21-day cycles for 30 weeks | Intensification: $290.24 | Intensification: $8,155 |
Modified DFCI 91-01/Al.4 Protocol — CNS therapy and Intensification aged > 60e | CNS therapy: $139 Intensification: $327 | CNS therapy: $3,894 Intensification: $9,167 | ||||
DFCI = Dana-Farber Cancer Institute: IM = intramuscular; IT = Intrathecal; PO = per oral.
Notes All prices are from the Ontario Drug Benefit Formulary (accessed December 2024), unless otherwise indicated, and do not include dispensing fees.33
BSA and weight used to calculate drug dosages are: 2.00 m2 and 86 kg.
aCourse duration: 294 days for blinatumomab + SOC;1 126 for SOC;1 210 days for Modified DFCI.34
bSOC is based on the UKALLXII/ECOG 2993 regimen from the E1910 trial.1
cSponsor submitted price for blinatumomab.1
dDrug price obtained from DeltaPA. Pegaspargase price expired in 2019, last available price shown.35
eDFCI protocol is sourced from Alberta Health Services34 and refers to the CNS and intensification phase of therapy.
fDrug price from BC Pharmacare formulary.36
Table 8: CDA-AMC Cost Comparison Table for ALL Pediatric Population
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | 28-day cost ($)a |
|---|---|---|---|---|---|---|
Blinatumomab | ||||||
Blinatumomab (Blincyto) | 0.0385 mg per vial | Lyophilized powder for solution for infusion | $2,978.2600c | Blinatumomab block 1, 2c: 0.015 mg/m2 IV from days 1 to 28 of 35-day cycles (max 0.028 mg/day) | Blinatumomab block 1, 2: $1,021.12 to $2,382.61 | Blinatumomab block 1, 2: $28,591to $66,713 |
COG AALL1731b | ||||||
Vincristine | 1 mg/mL | 1 mL Solution for injection | $30.6000 | SR-Average Consolidation: 1.5 mg/m2 IV on day 1 of a 28-day cycle Interim Maintenance I, II: 1.5 mg/m2 IV on day 1, 11, 21, 31, and 41 of a 56-day cycle Delayed intensification part 1: 1.5 mg/m2 IV on day 1, 8, 15 of a 56-day cycle SR-High consolidation: 1.5 mg/m2 IV on days 15, 22, 43, and 50 of a 56-day cycle Interim Maintenance I: 1.5 mg/m2 IV on days 1, 15, 29, and 43 of a 56-day cycle Delayed intensification part 1: 1.5 mg/m2 IV on day 1, 8, 15 of a 63-day cycle Delayed intensification part 2: 1.5 mg/m2 IV on day 43, 50 of a 63-day cycle Interim Maintenance II: 1.5 mg/m2 IV on day 1, 11, 21, 31, and 41 of a 56-day cycle | SR-Average Consolidation: $2.19 to $3.28 Interim Maintenance I, and II: $5.46 to $8.20 Delayed intensification part 1: $3.28 to $4.92 SR-High consolidation: $4.37 to $6.56 Interim Maintenance I: $4.37 to $6.56 Delayed intensification part 1: $2.91 to $4.37 Delayed intensification part 2: $1.94 to $2.91 Interim Maintenance II: $4.86 to $7.29 | SR-Average Consolidation: $61 to $92 Interim Maintenance I and II: $153 to $230 Delayed intensification part 1: $92 to $138 SR-High consolidation: $122 to $184 Interim Maintenance I: $122 to $184 Delayed intensification part 1: $82 to $122 Delayed intensification part 2: $54 to $82 Interim Maintenance II: $136 to $204 |
6-mercaptopurine (Purinethol) | 50 mg | Tablet | $2.8610 | SR-average consolidation: 75 mg/m2 per oral on days 1 to 28 of a 28-day cycle SR-high consolidation: 60 mg/m2 per oral on days 1 to 14 and 29 to 42 of a 56-day cycle Interim Maintenance I: 25 mg/m2 per oral on days 1 to 14, 15 to 28, 29 to 42, and 43 to 56 of a 56-day cycle | SR-average consolidation: $5.72 to $8.58 SR-high consolidation: $1.43 to $2.86 Interim Maintenance I: $2.86 to $2.86 | SR-average consolidation: $160 to $240 SR-high consolidation: $40 to $80 Interim Maintenance I: $80 to $80 |
Methotrexate | 10 mg/mL 25 mg/mL | 2 mL vial 2 mL vial Solution for Liquid injection | $12.5000 $8.9200 | SR-average Consolidation, Interim Maintenance I, 2, and Delayed Intensification Part 1, 2: age-based dosing IT on day 1 (1 to 1.99 years: 8 mg; 2 to 2.99 years: 10 mg; 3 to 8.99 years: 12 mg; > = 9 years: 15 mg) Interim Maintenance I, 2: 100 + 150 + 200 + 250 + 300 mg/m2 IV on days 1, 11, 21, 31, 41 of a 56-day cycle. (start at 100 mg/m2 and escalate by 50 mg/m2) SR-high Consolidation: age-based dosing IT on days 1, 8, 15, and 22 (1 to 1.99 years: 8 mg; 2 to 2.99 years: 10 mg; 3 to 8.99 years: 12 mg; > = 9 years: 15 mg) Delayed Intensification Part 1, 2: age-based dosing IT on day 1 (1 to 1.99 years: 8 mg; 2 to 2.99 years: 10 mg; 3 to 8.99 years: 12 mg; > = 9 years: 15 mg) Interim Maintenance 1: 5000 mg/m2 IV on days 1, 15, 29, 43 Interim Maintenance 2: age-based dosing IT on day 1, and 29 (1 to 1.99 years: 8 mg; 2 to 2.99 years: 10 mg; 3 to 8.99 years: 12 mg; > = 9 years: 15 mg) Interim Maintenance 2: 100 + 150 + 200 + 250 + 300 mg/m2 IV on days 1, 11, 21, 31, 41 of a 56-day cycle. (start at 100 mg/m2 and escalate by 50 mg/m2) | SR-average consolidation, Interim Maintenance I, 2, and Delayed Intensification Part 1, 2: $1.34 Interim Maintenance I, 2: $2.39 to $4.62 SR-high Consolidation: $0.89 Delayed Intensification Part 1, 2: $0.36 Interim Maintenance 1: $47.79 to $89.84 Interim Maintenance 2: $0.20 Interim Maintenance 2: $2.12 to $4.11 | SR-average consolidation, Interim Maintenance I, 2, and Delayed Intensification Part 1, 2: $38 Interim Maintenance I, 2: $67 to $129 SR-high Consolidation: $25 Delayed Intensification Part 1, 2: $10 Interim Maintenance 1: $1,338 to $2,515 Interim Maintenance 2: $6 Interim Maintenance 2: $59 to $115 |
Dexamethasone | 0.5 mg 4 mg | Tablet | $0.1564 $0.6112 | SR-average and high Delayed Intensification Part 1: 5 mg/m2 per oral on days 1 to 7, 15 to 21 of a 56-day cycle | Delayed Intensification Part 1: $0.31 to $0.61 | Delayed Intensification Part 1: $9 to $17 |
Doxorubicin (Caelyx) | 2 mg/mL | 5 mL vial 25 mL vial 100 mL vial Solution for injection | $50.4500d $252.2500d $973.0000d | SR-average and high Delayed Intensification Part 1: 25 mg/m2 IV on days 1, 8, 15 of a 56-day cycle | Delayed Intensification Part 1: $12.01 to $13.51 | Delayed Intensification Part 1: $336 to $378 |
Pegaspargase (Oncaspar) | 750 IU/mL | 5 mL vial Solution for injection | $6,115.9700d | SR-average and high Delayed Intensification Part 1: 2,500 IU/m2 IV on day 4 of a 56-day cycle SR-high Consolidation: 2,500 IU/m2 IV on days 15, and 43 of a 56-day cycle Delayed Intensification Part 2: 2,500 IU/m2 IV on day 43 of a 63-day cycle Interim Maintenance II: 2,500 IU/m2 IV on day 2, 22 of a 63-day cycle | SR-average and high Delayed Intensification Part 1: $65.53 to $109.21 SR-high Consolidation: $131.06 to $218.43 Delayed Intensification Part 2: $58.25 to $97.08 Interim Maintenance II: $116.49 to $194.16 | SR-average and high Delayed Intensification Part 1: $1,835 to $3,058 SR-high Consolidation: $3,670 to $6,116 Delayed Intensification Part 2: $1,631 to $2,718 Interim Maintenance II: $3,262 to $5,436 |
Cyclophosphamide (Procytox) | 500 mg/mL 1,000 mg/mL 2,000 mg/mL | Powder for solution for injection | $107.8100d $195.4200d $359.4000d | SR-average: Delayed Intensification Part 2: 1,000 mg/m2 IV on day 29 of a 56-day cycle SR-high Consolidation: 1,000 mg/m2 IV on days 1 and 29 of a 56-day cycle Delayed Intensification Part 2: 1,000 mg/m2 IV on day 4 of a 63-day cycle | SR-average: Delayed Intensification Part 2: $3.49 to $6.98 SR-high Consolidation: $6.98 to $13.96 Delayed Intensification Part 2: $3.10 to $6.20 | SR-average: Delayed Intensification Part 2: $98 to $195 SR-high Consolidation: $195 to $391 Delayed Intensification Part 2: $87 to $174 |
Thioguanine | 50 mg | Tablet | $6.2030 | SR-average and high Delayed Intensification Part 2: 60 mg/m2 per oral on days 29 to 42 of a 56- or 63-day cycle | SR-average and high Delayed Intensification Part 2: $1.38 to $3.10 | SR-average and high Delayed Intensification Part 2: $39 to $87 |
Cytarabine | 100 mg/mL | 5 mL vial 20 mL vial Solution for Injection | $76.8500d $306.5000d | SR-average and high Delayed Intensification Part 2: 75 mg/m2 per oral on days 29 to 32 and 36 to 39 of a 56- or 63-day cycle SR-high Consolidation: 75 mg/m2 IV on days 1 to 4, 8 to 11, 29 to 32, and 36 to 39 of a 56-day cycle | SR-average and high Delayed Intensification Part 2: $10.98 to $21.96 SR-high Consolidation: $9.76 | SR-average and high Delayed Intensification Part 2: $307 to $615 SR-high Consolidation: $273 |
Leucovorin | 5 mg | Tablet | $3.6776 | SR-high Interim Maintenance I: 15 mg/m2 per oral on days 3 to 4, 17 to 18, 31 to 32, and 45 to 46 | SR-high Interim Maintenance I: $1.58 to $2.63 | SR-high Interim Maintenance I: $44 to $74 |
COG AALL1731 | SR-average: $168 to $189 SR-High: $738 to $805 | SR-average: $4,715 to $5,299 SR-High: $20,658 to $22,534 | ||||
aCourse duration: SR-average: 322 days for blinatumomab + SOC chemotherapy; SR-High: 364 days for blinatumomab + SOC chemotherapy. SR-average: 252 for SOC chemotherapy; SR-High: 294 days for SOC chemotherapy. Patients aged < 10 years old have an assumed weight of 17.5 kg, BSA = 0.75 m2; patients aged > 10 years old have an assumed weight of 42.7 kg, BSA = 1.41 m2.1,37
bSOC is based on the AALL1731 trial protocol.1
cFor blinatumomab, if patient weight < 45kg, 0.015mg/m2; if ≥ 45kg, 0.028mg/day.2 Daily and 28-day cycle cost of blinatumomab varied if 1-, 2-, 3-, 4-, or 7-day bags are used.2
dDrug price obtained from DeltaPA. Pegaspargase price expired in 2019, last available price shown.35
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to limitations above. The SOC regimen used for adult population in the model does not reflect clinical practice in Canada. Additionally, the sponsor did not provide evidence assessing the cost-effectiveness of blinatumomab plus SOC compared to blinatumomab as monotherapy, |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitations above. Numerous IFERROR statements were used in the model, making thorough auditing of the model impractical. Moreover, the model offered limited flexibility for user input on key parameters. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to key limitations above. For the pediatric population, KM data were directly used for the first 4 years of the modelled time horizon without conducting survival analysis, making it impossible to assess the impact of the comparator uncertainty on the results of the model. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
KM = Kaplan-Meier; SOC = standard of care.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Blinatumomab plus SOC | SOC |
|---|---|---|
Discounted LYs | ||
Total | 32.98 | 29.73 |
Relapse-free | 31.02 | 26.07 |
Postrelapse | 1.95 | 3.66 |
Discounted QALYs | ||
Total | 23.41 | 20.70 |
Relapse-free | 22.10 | 18.40 |
Postrelapse | 1.30 | 2.30 |
Discounted costs ($) | ||
Total | 399,169 | 308,947 |
Relapse-free | ||
Medication | 170,680 | 21,519 |
Administration | 31,562 | 21,726 |
Maintenance therapy | 6,902 | 10,169 |
HSCT | 30,307 | 33,451 |
AE management | 22,248 | 24,350 |
HCRU | 77,712 | 72,734 |
Postrelapse | ||
Subsequent treatment | 19,847 | 50,724 |
Subsequent administration | 3,656 | 6,965 |
HSCT | 9,771 | 17,573 |
HCRU | 23,823 | 44,147 |
Terminal care | 2,661 | 5,589 |
AE = adverse event; HCRU = health care resource utilization; HSCT = hematopoietic stem cell transplantation; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.
Table 11: Subgroup Results From Sponsor’s Base-Case Analysis
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
Pediatric population | |||||
SOC | 284,243 | Reference | 28.31 | Reference | Reference |
Blinatumomab plus SOC | 323,509 | 39,266 | 28.70 | 0.38 | 102,327 |
Adult population | |||||
SOC | 338,327 | Reference | 11.65 | Reference | Reference |
Blinatumomab plus SOC | 489,153 | 150,826 | 17.11 | 5.46 | 27,607 |
SOC = standard of care; QALY = quality-adjusted life-year; ICER = incremental cost-effectiveness ratio.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Blinatumomab plus SOC | SOC |
|---|---|---|
Discounted LYs | ||
Total | 33.00 | 29.95 |
Relapse-free | 31.03 | 25.99 |
Postrelapse | 1.97 | 3.96 |
Discounted QALYs | ||
Total | 23.42 | 20.84 |
Relapse-free | 22.11 | 18.38 |
Postrelapse | 1.30 | 2.45 |
Discounted costs ($) | ||
Total | 343,489 | 247,761 |
Relapse-free | ||
Medication | 170,332 | 20,856 |
Administration | 31,134 | 20,864 |
Maintenance therapy | 6,939 | 10,171 |
HSCT | 30,109 | 33,255 |
AE management | 22,267 | 24,398 |
HRU | 38,948 | 36,844 |
Postrelapse | ||
Subsequent treatment | 19,768 | 50,962 |
Subsequent administration | 3,638 | 6,988 |
HSCT | 9,708 | 17,695 |
HRU | 7,999 | 20,233 |
Terminal care | 2,647 | 5,494 |
AE = adverse event; HCRU = health care resource utilization; HSCT = hematopoietic stem cell transplantation; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Table 13: Subgroup Results From CDA-AMC Reanalysis
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | Total LYs | Incremental LYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|---|---|
Pediatric population | |||||||
SOC | 213,004 | Reference | 28.57 | Reference | 43.01 | Reference | Reference |
Blinatumomab plus SOC | 260,354 | 47,350 | 28.67 | 0.09 | 43.01 | 0.00 | 508,738 |
Adult population | |||||||
SOC | 289,099 | Reference | 11.64 | Reference | 14.42 | Reference | Reference |
Blinatumomab plus SOC | 442,363 | 153,264 | 17.17 | 5.54 | 21.08 | 6.66 | 27,682 |
SOC = standard of care; QALY = quality-adjusted life-year; ICER = incremental cost-effectiveness ratio.
Note: For the pediatric subgroup, a price reduction of 56% would be required to achieve cost-effectiveness at a threshold of $50,000 per QALY gained.
Scenario Analyses
Table 14: Summary of CDA-AMC Scenario Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case | SOC | 247,761 | 20.84 | Reference |
Blinatumomab plus SOC | 343,489 | 23.42 | 37,111 | |
CDA-AMC Scenario analysis | SOC | 248,874 | 11.95 | Reference |
Blinatumomab plus SOC | 344,804 | 22.11 | 44,498 |
SOC = standard of care; QALY = quality-adjusted life-year; ICER = incremental cost-effectiveness ratio.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the introduction of blinatumomab in the consolidation phase of multiphase chemotherapy of Philadelphia chromosome–negative CD19-positive B-cell precursor ALL for adult and pediatric patients.2 The BIA was undertaken from the perspective of a public drug plan payer in Canada over a 3-year time horizon (2025 to 2027). An epidemiological approach with incidence rates was taken to determine the number of patients eligible for blinatumomab using data from Statistics Canada, published literature, and the E1910 clinical trial.38-44 Drug acquisition, administration, subsequent treatment, and HSCT costs were included in the base case.
The reference case scenario included consolidation phase of multiphase chemotherapy UKALLXII/ECOG 2993 regimen only for the adult population and the AALL1731 trial protocol only for the pediatric population.37 The new drug scenario included the same comparator along with four 42-day cycles of blinatumomab for the adult population, and the AALL1731 trial protocol along with 2 35-day cycles of blinatumomab for the pediatric population.2 Key inputs to the BIA are documented in Table 16.
Key assumptions include:
The eligible population was calculated from age-specific incidence data from Statistics Canada.45
RFS and OS data sourced from clinical trial data to determine maintenance therapy costs and number of alive patients.
Market shares were informed by the sponsor’s internal forecasting and market research.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Target population | Adult population | Pediatric population |
26,890,420 / 27,158,140 / 27,428,480 | 5,974,405 / 5,994,186 / 6,003,747 | |
Baseline incidence of ALL in Canada excluding Quebec per 100,000 population40,45 | 0.76 | 3.01 |
–1.8% | ||
Proportion of patients with ALL with B-cell lineage38 | 85.0% | 85% |
Proportion of patients with B-cell ALL who are Philadelphia chromosome–negative38 | 75.0% | 90% |
Proportion of patients completing induction therapy and beginning consolidation therapy42,43 | 85% | 95% |
Number of patients eligible for drug under review37 | 109 / 108 / 108 | 128 / 126 / 124 |
Market Uptake (3 years) | ||
Uptake (reference scenario) SOC | 100% / 100% / 100% | |
Uptake (new drug scenario) Blinatumomab + SOC47 SOC chemotherapy47 | 75% / 85% / 90% 25% / 15% / 10% | 85% / 90% / 95% 20% / 10% / 5% |
Cost of treatment (per patient)a | ||
Blinatumomab + SOC SOC | $226,996.48 $10,594.26 | SR-average: $92,725.88; SR-high: $174,758.55 SR-average: $9,315.89; SR-high: $49,652.37 |
ALL = acute lymphoblastic leukemia; SOC = standard of care; SR = standard risk
Note: SOC chemotherapy is based on the UKALLXII/ECOG 2993 protocol.
aRDI and cycle-dependent discontinuation was assumed.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing blinatumomab for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in frontline setting to be $87,307,370 (Year 1; $26,861,572; Year 2: $29,480,666; Year 3: $30,965,132). When separated into the adult and pediatric populations, the estimated budget impact was $55,995,499 and $31,311,699, respectively. Although the pediatric subgroup represents a greater proportion of the patient population, the observed subgroup difference in budget impact can be attributed to the number of blinatumomab cycles each subgroup is treated with, as the adult population is treated with more cycles of blinatumomab resulting in higher drug costs.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The selected comparator does not reflect clinical practice in Canada. The sponsor based the comparator SOC regimen off the UKALLXII/ECOG 2993 regimen for the adult population.39 As noted in the Economic Evaluation section, the Clinical Review Report, and the clinical expert feedback, the DFCI protocol is the most commonly used regimen used to treat Ph-negative B-cell precursor ALL in Canada. The DFCI protocol was not included as a comparator in the sponsor’s base-case analysis, limiting the validity of the budget impact as SOC is expected to be displaced by blinatumomab plus SOC. Additionally, the sponsor did not include blinatumomab as monotherapy as a comparator, which has an existing indication and reimbursement recommendation for MRD-positive adult and pediatric patients. However, the drug acquisition costs for blinatumomab across the 2 indications are likely similar in adults, and less costly in the pediatric population because of using 2 fewer cycles of blinatumomab in the current request.
In reanalysis, CDA-AMC adjusted the price of SOC in the adult population to reflect the DFCI protocol.
Postrelapse HSCT costs were inappropriately calculated. The sponsor assumed that patients would be able to receive HSCT transplantation pre- and postrelapse based on results of the E1910 trial (adults) and clinical expert opinion (pediatrics). For prerelapse transplantation, the sponsor assumed that a proportion of all alive patients would be able to receive HSCT. For postrelapse transplantation, the sponsor assumed that 20% of the adult and 45% of the pediatric patients regardless of treatment receive HSCT; however, the calculation was misapplied to the total alive population of each subgroup, rather than alive patients that have experienced a relapse in disease. As a result, the sponsor likely overestimated the costs associated with HSCT in the submitted BIA.
In reanalysis, CDA-AMC removed the postrelapse HSCT costs as the sponsor’s model applied the calculations inappropriately and the model itself limited CDA-AMC ability to perform corrections.
The market uptake of blinatumomab in the pediatric population is likely underestimated. Based on internal forecasting, the sponsor assumed that blinatumomab plus SOC would achieve a market uptake of 85%, 90%, and 95% in the new drug scenario for the pediatric population. Clinical expert feedback agreed that the blinatumomab plus SOC market uptake would reach 95% or greater by year 3 after its initial listing, however, felt that the uptake would be faster than estimated by the sponsor. Clinical experts consulted by CDA-AMC indicated that blinatumomab would be widely adopted across Canadian care centres and would become the preferred treatment over current SOC alone.
CDA-AMC conducted a reanalysis by adjusting the market uptake of blinatumomab for the pediatric population to 95%, 95%, and 95%, for years 1 to 3, respectively.
The average annual change in ALL incidence was likely overestimated. The sponsor cited an annual change in ALL incidence rate of −1.8% based on Statistics Canada cancer incidence data.45 However, between 2010 and 2020, Statistics Canada reported that the annual percent change of age-standardized ALL incidence was 0.5 (95% CI, −0.6% to 1.6%).48 These data suggest that there may be an increase in ALL incidence between 2010 and 2020, however, the estimate is associated with uncertainty. Clinical expert feedback agreed that there was uncertainty regarding a decreasing ALL incidence rate.
In reanalysis, CDA-AMC assumed that the incidence of ALL was constant over time.
The OS benefit for pediatric patients treated with blinatumomab is uncertain. As noted in the Economic Evaluation section of this report, the sponsor used the KM data to represent the OS of the pediatric population was considered inappropriate and likely inaccurately estimated the potential presence and magnitude of an OS benefit in the pediatric population.
CDA-AMC conducted a reanalysis by assuming equivalent OS estimates for SOC alone and blinatumomab plus SOC, using estimates from the blinatumomab plus SOC arm of the AALL1731 trial.
The application of RDI to estimate actual drug costs is not appropriate. For the adult population, the sponsor assumed RDI only for the blinatumomab cycles of blinatumomab plus SOC and assumed 100% RDI for the SOC cycles.1 However, as noted by the economic evaluation of this report, clinical expert feedback indicated this approach to be inappropriate because RDI is unlikely to be 100% for SOC. due to potential dose modifications.
In reanalysis, to demonstrate the impact of RDI, CDA-AMC set RDI to 100% for all blinatumomab cycles to align with the RDI assumed for SOC.
To address uncertainty associated with RDI, CDA-AMC set RDI of the blinatumomab plus SOC arm and SOC arm for the adult population to the observed values from the E1910 trial.
CDA-AMC Reanalyses of the BIA
Table 17: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Adult SOC cost | Applied the cost of UKALLXII/ECOG 2993 | Applied the cost of the DFCI protocol |
2. Postrelapse HSCT cost | Included | Excluded |
3. Pediatric blinatumomab market uptake | Year 1: 85% Year 2: 90% Year 3: 95% | Year 1: 95% Year 2: 95% Year 3: 95% |
4. Pediatric OS | Assumed OS benefit for pediatric patients treated with blinatumomab | Assumed no OS benefit patients treated with blinatumomab |
5. Annual change in incidence | −1.8% | 0% |
6. RDI | RDI was assumed for the blinatumomab cycles of the adult population, and 100% for SOC | RDI was set to 100% for all treatments. |
CDA-AMC base case | 1 + 2 + 3 + 4 + 5 + 6 | |
DFCI = Dana-Farber Cancer Institute; HSCT = hematopoietic stem cell transplantation; OS = overall survival; RDI = relative dose intensity; SOC = standard of care.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19.
Based on CDA-AMC reanalyses, the expected budget impact for funding blinatumomab for adult and pediatric patients with Ph-negative CD19-positive B-cell precursor ALL in the consolidation phase of multiphase chemotherapy in frontline setting to be $102,505,910 over the 3-year time horizon (Year 1: $31,603,539; Year 2: $34,583,830; Year 3: $36,318,540). The increase in budget impact was primarily driven by the alternative blinatumomab market uptake proportions, the removal of the RDI adjustment, and assuming no annual change for ALL incidence rate.
For the adult and pediatric populations, the budget impact is estimated to be $66,257,182 and $36,248,727 over the 3-year time horizon, respectively. The adult population is estimated to represent 45% of the patient population but consists of 65% of the total budget impact. This is expected as the adult population is treated with 4 cycles of blinatumomab at a higher dosage compared to the pediatric population’s 2 cycles. This results in higher costs associated with the adult population of this indication.
Table 18: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 87,307,370 |
CDA-AMC reanalysis 1 | 88,069,431 |
CDA-AMC reanalysis 2 | 84,576,476 |
CDA-AMC reanalysis 3 | 89,066,977 |
CDA-AMC reanalysis 4 | 87,308,251 |
CDA-AMC reanalysis 5 | 95,906,360 |
CDA-AMC reanalysis 6 | 93,544,636 |
CDA-AMC base case | 102,505,910 |
BIA = budget impact analysis; DFCI = Dana-Farber Cancer Institute; HSCT = hematopoietic stem cell transplantation; OS = overall survival; RDI = relative dose intensity; SOC = standard of care.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 19:
E1910 trial RDI for blinatumomab plus SOC and SOC.
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 34,971,160 | 34,590,474 | 34,974,781 | 34,814,175 | 104,379,430 |
New drug | 34,971,160 | 61,452,046 | 64,455,448 | 65,779,306 | 191,686,800 | |
Budget impact | 0 | 26,861,572 | 29,480,666 | 30,965,132 | 87,307,370 | |
CDA-AMC base case | Reference | 17,069,972 | 17,234,762 | 18,220,319 | 18,654,532 | 54,109,612 |
New drug | 17,069,972 | 48,838,301 | 52,804,149 | 54,973,072 | 156,615,522 | |
Budget impact | 0 | 31,603,539 | 34,583,830 | 36,318,540 | 102,505,910 | |
CDA-AMC base case (adults) | Reference | 11,943,733 | 12,091,722 | 12,732,008 | 13,121,290 | 37,945,021 |
New drug | 11,943,733 | 31,635,704 | 35,226,020 | 37,340,479 | 104,202,203 | |
Budget impact | 0 | 19,543,982 | 22,494,012 | 24,219,189 | 66,257,182 | |
CDA-AMC base case (pediatric) | Reference | 5,126,240 | 5,143,039 | 5,488,311 | 5,533,241 | 16,164,591 |
New drug | 5,126,240 | 17,202,597 | 17,578,129 | 17,632,593 | 52,413,318 | |
Budget impact | 0 | 12,059,558 | 12,089,818 | 12,099,352 | 36,248,727 | |
CDA-AMC Scenario Analysis 1: RDI | Reference | 17,069,972 | 17,234,762 | 18,220,319 | 18,654,532 | 54,109,612 |
New drug | 17,069,972 | 46,814,085 | 50,475,616 | 52,468,210 | 149,757,912 | |
Budget impact | 0 | 29,579,323 | 32,255,297 | 33,813,679 | 95,648,299 |
ALL = acute lymphoblastic leukemia; BIA = budget impact analysis; RDI = relative dose intensity.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.