Drugs, Health Technologies, Health Systems
Reimbursement Review
Durvalumab (Imfinzi)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Resectable non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AEPI
adverse event of potential interest
AESI
adverse event of special interest
AJCC
American Joint Committee on Cancer
BICR
blinded independent central review
CCSN
Canadian Cancer Survivor Network
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
CRT
chemoradiotherapy
DCO
data cut-off
DFS
disease-free survival
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EFS
event-free survival
EM
effect modifier
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IA
interim analysis
IA1
interim analysis 1
IA2
interim analysis 2
ICI
immune checkpoint inhibitor
imAE
immune-mediated adverse event
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
LCC
Lung Cancer Canada
LHF
Lung Health Foundation
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
mITT
modified intention to treat
MPR
major pathological response
MTP
multiple testing procedure
NICE DSU TSD 18
National Institute for Health and Care Excellence Decision Support Unit–Technical Support Document 18
NMA
network meta-analysis
NR
not reached
NSCLC
non–small cell lung cancer
OH-CCO
Ontario Health (Cancer Care Ontario)
OS
overall survival
pCR
pathological complete response
PFS
progression-free survival
PORT
postoperative radiotherapy
PRO
patient-reported outcome
QoL
quality of life
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
RECIST 1.1
Response Evaluation Criteria in Solid Tumours version 1.1
SAE
serious adverse event
SLR
systematic literature review
TNM
tumour-nodes-metastasis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 120 mg/2.4 mL and 500 mg/10 mL single-use vials for IV infusion |
Sponsor | AstraZeneca Canada Inc. |
Indication | In combination with platinum-containing chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery, for the treatment of patients with resectable Stage II, IIIA, or IIIB (T3-4 N2) non–small cell lung cancer and no known epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) rearrangements |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 20, 2026 |
Recommended dosage | The recommended dosage of Imfinzi is 1,500 mg in combination with platinum-containing chemotherapy every 3 weeks before surgery (neoadjuvant period), followed by 1,500 mg monotherapy every 4 weeks after surgery (adjuvant period). Before surgery, treatment continues for up to 4 cycles or until disease is deemed unresectable or of unacceptable toxicity. After surgery, treatment continues for up to 12 cycles or until recurrence or unacceptable toxicity. Patients with a body weight of 30 kg or less must receive weight-based dosing of Imfinzi at 20 mg/kg. In combination with chemotherapy, dose at 20 mg/kg every 3 weeks (21 days) is administered before surgery, followed by monotherapy at 20 mg/kg every 4 weeks after surgery until weight increases to greater than 30 kg. |
NOC = Notice of Compliance.
Sources: Sponsor’s Summary of Clinical Evidence and Imfinzi product monograph.45
Introduction
In 2024 alone, an estimated 32,100 Canadians were expected to be diagnosed with lung cancer and around 20,700 deaths were expected to be attributed to the disease — making lung cancer the most commonly diagnosed cancer and leading cause of cancer-related deaths in Canada.1 Lung cancer predominantly affects older adults, with 98% of cases occurring in individuals aged 50 years and older.2 Non–small cell lung cancer (NSCLC) accounts for approximately 88% of all lung cancer cases in Canada (excluding Quebec) and approximately 25% to 30% of patients with NSCLC present with resectable early-stage disease (stage II to stage III).3,4 The standard of care for early-stage NSCLC has historically been surgical resection with curative intent followed by adjuvant chemotherapy. However, given that an estimated 30% to 55% of patients develop postoperative recurrence,5 there remains an unmet need for treatments that reduce recurrence and improve overall survival (OS). Further, patients with NSCLC experience a variety of symptoms, including dyspnea, malaise, depression, anxiety, worsening cough, hemoptysis, pain, weight loss, and hoarseness.6,7 These symptoms, coupled with poor disease outlook, treatment modalities and side effects, and other comorbidities common in patients with NSCLC, significantly impact patients’ quality of life (QoL).8-11 Patients with NSCLC also face significant productivity losses and stigma; notably, people who do not smoke experience negative perceptions associated with lung cancer.12-14 The adjusted 5-year and 10-year net survival rates for all forms and stages of lung cancer are estimated to be 22% and 15%, respectively.15 Recently, immune checkpoint inhibitors (ICIs), such as neoadjuvant nivolumab with chemotherapy, have shown promising results such as improved survival and tumour response.16,17 The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of perioperative durvalumab, 1,500 mg administered as an IV infusion once every 3 weeks in combination with neoadjuvant chemotherapy before surgery and once every 4 weeks as monotherapy after surgery for the treatment of resectable NSCLC in patients without any known EGFR mutations or ALK rearrangements.
Durvalumab has been previously reviewed by Canada’s Drug Agency (CDA-AMC) for the treatment of patients with locally advanced, unresectable NSCLC following curative-intent, platinum-based chemoradiation therapy, for up to a maximum of 12 months. The final recommendation, issued on May 3, 2019, was to reimburse with conditions.18 CDA-AMC has also reviewed durvalumab in extensive-stage small cell lung cancer, biliary tract cancer, unresectable hepatocellular carcinoma, endometrial cancer, limited-stage small cell lung cancer, and metastatic NSCLC, all of which received conditional positive reimbursement recommendations.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the CDA-AMC call for input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
A joint submission was received from the Canadian Cancer Survivor Network (CCSN), Lung Cancer Canada (LCC), and Lung Health Foundation (LHF) in response to the call by CDA-AMC for patient input on the current review of durvalumab.
Information for this submission was collected via an online survey conducted by CCSN from June 11 to July 17, 2024, with input from LCC and LHF. A total of 5 respondents completed the survey, all of whom were patients with lung cancer living in Canada; 4 patients were female and 1 was male. Three of the 5 survey respondents had prior experience with durvalumab.
Respondents included patients with various stages of lung cancer, primarily stage IV. Common symptoms affecting their QoL were fatigue, pain, and shortness of breath. Chemotherapy was the most common treatment received by survey respondents, followed by immunotherapy, targeted therapy, radiation, and surgery.
According to the survey respondents, key aspects of their disease to control included managing tumour growth, shortness of breath, pain, and chemotherapy-related side effects; a lack of mental health support and difficulties accessing counselling and managing travel costs to access treatment were identified as unmet needs.
Respondents experienced a range of side effects such as joint and/or muscle pain, fatigue, nausea, and diarrhea, with 3 of 5 respondents considering these side effects intolerable. In terms of treatment goals, maintaining QoL, finding a cure, and prolonging life were prioritized, followed by delaying symptoms and improving ease of use.
Durvalumab was reported as beneficial by the 3 respondents with prior experience using it. They rated durvalumab significantly better for symptom management and disease progression compared to other therapies, with side effects being similar. All 3 respondents recommended making durvalumab available to eligible patients.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review felt the overarching goal of NSCLC treatment is to cure the disease and in doing so, improve OS, reduce recurrence rates, improve QoL, and reduce the symptom burden. While recent treatment advancements have improved disease outlook, survival rates and disease burden remain suboptimal. According to the clinical experts, ICIs have achieved better results compared to chemotherapy alone;19 however, atezolizumab remains the only reimbursed ICI regimen in the adjuvant setting for NSCLC in Canada and more than half of patients are ineligible for it due to low PD-L1 expression status. In the neoadjuvant setting, neoadjuvant nivolumab in combination with chemotherapy is currently funded. At the time of submission for this review in 2024, no perioperative ICI regimens were approved for resectable NSCLC in Canada. Note that since April 2025, CDA-AMC has issued a positive reimbursement recommendation for perioperative pembrolizumab for resectable NSCLC. The clinical experts noted that patient selection (e.g., based on histology, PD-L1 expression, disease stage, patient preference) is key when determining which patients will best respond to treatment regimens including the use of neoadjuvant, adjuvant, and perioperative approaches; currently, there are no robust head-to-head data to compare these treatment regimens.
The clinical experts noted that perioperative durvalumab with neoadjuvant chemotherapy would add an alternative first-line treatment option for patients with resectable NSCLC. The experts felt that patients with resectable NSCLC who are fit for systemic chemotherapy and ICI without any EGFR mutations or ALK rearrangements would be best suited for perioperative durvalumab with neoadjuvant chemotherapy. Clinical experts highlighted the importance of patient preferences when selecting treatment and that those with questionable resectable disease or who are not fit for platinum-based chemotherapy may be less suited for this approach. Experts emphasized that a short time to treatment initiation is crucial to achieve the best treatment results and the proposed requirement for EGFR and ALK testing is 1 potential area associated with treatment delay.
Outcome metrics for resectable NSCLC used in clinical practice are typically aligned with those used in clinical trials. The clinical experts stated that in clinical trials, patient response to treatment is assessed at the time of surgery using pathological complete response (pCR) and major pathological response (MPR), using survival measures such as OS and disease-free survival (DFS) (i.e., recurrence-free) according to the Response Evaluation Criteria in Solid Tumours (RECIST) criteria, and by disease symptoms. In clinical practice, response at the time of surgery using pCR is the first step to assess benefit from therapy, followed by survival, QoL outcomes (such as patient-reported outcomes [PROs]), and adverse events (AEs). In clinical practice, as in trials, clinical assessments are performed before each cycle of therapy. However, assessments of disease response and recurrence usually occur every 3 months to 6 months during treatment, every 6 months in the first 2 years following the last treatment cycle, and then annually thereafter, which is generally less frequent than in clinical trials.
Clinical experts consulted for this review indicated that perioperative durvalumab with neoadjuvant chemotherapy should be discontinued in the event of disease progression (including the tumour no longer being resectable, metastasis, and/or tumour growth) or recurrence, significant side effects such as immune-related side effects that can be life-threatening or permanent, or upon a patient’s request.
Clinical experts consulted for this review indicated that treatment with perioperative durvalumab with neoadjuvant chemotherapy should be prescribed and managed in outpatient clinics or specialty clinics by a multidisciplinary thoracic team of thoracic surgeons, medical oncologists, and supportive staff such as nursing and allied health.
Clinician Group Input
Two clinician groups provided input for this review: LCC Drug Advisory Committee (18 clinicians contributed to the input) and Ontario Health (Cancer Care Ontario) (OH-CCO) (5 clinicians contributed to the input). Overall, the input was aligned with that of the clinical experts consulted by CDA-AMC.
Both clinician groups and the clinical experts consulted agreed that the treatment paradigm for patients with resectable NSCLC who do not have EGFR mutations or ALK rearrangements consists of 2 main phases: neoadjuvant and adjuvant. Neoadjuvant therapy, administered before surgery, includes chemotherapy and ICI. OH-CCO and LCC recommend neoadjuvant platinum-based chemotherapy combined with ICI (nivolumab) for 3 cycles. Additionally, LCC noted an alternative involving chemotherapy with ICI (durvalumab) for 4 cycles. Following surgery, adjuvant therapy involves chemotherapy and/or ICI, while radiation therapy is reserved for specific cases such as positive margins, as noted by the clinical experts consulted for this review.
The common treatment goals identified across clinician groups included achieving a cure, as measured by OS, and improving DFS. LCC and the clinical experts emphasized reducing recurrence rates and enhancing QoL.
The most significant treatment gap identified by the 2 clinician groups and the clinical experts is the lack of data comparing the efficacy and role of adjuvant ICI following neoadjuvant therapy, including which patients might benefit from it. This uncertainty makes it challenging to determine the optimal strategy for improving survival outcomes, especially given that not all patients respond to current treatments. Presently, ICI in the adjuvant setting is only available to those with high PD-L1 expression.
Both clinician groups and the clinical experts stated that perioperative durvalumab combined with neoadjuvant chemotherapy would offer an alternative treatment approach to neoadjuvant nivolumab in combination with chemotherapy for resectable NSCLC. The clinical experts emphasized its importance, especially for patients who do not achieve a complete pathological response to neoadjuvant therapy, highlighting the potential value of adjuvant ICI in such cases.
According to the clinician groups, the best-suited patients for perioperative durvalumab in combination with neoadjuvant chemotherapy are those with resectable NSCLC, no EGFR mutations or ALK rearrangements, and no contraindications to ICI. They emphasized the role of CT scans for monitoring, with imaging recommended before and after surgery and at regular intervals during and after treatment to check for disease recurrence. In clinical practice, treatment response is assessed through clinical symptoms, survival outcomes (OS and DFS), and pathological response at surgery (pCR and MPR), as highlighted by the clinical expert.
Both clinician groups indicated that factors for discontinuing treatment primarily include disease progression and significant side effects, with patient preferences also playing a key role. For treatment with perioperative durvalumab with neoadjuvant chemotherapy, patients should be treated in an outpatient setting under the supervision of a medical oncologist, or a pulmonologist experienced in the management of thoracic malignancies.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. Key factors identified that could potentially impact the implementation of the CDA-AMC recommendation for perioperative durvalumab with neoadjuvant chemotherapy were the use of alterative chemotherapies in the neoadjuvant phase, identifying which patients should be eligible for reimbursement, and the acceptability of switching treatments.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing trial, a phase III, double-blind, placebo-controlled, randomized, multicentre, international study (including 5 sites in Canada) — the AEGEAN study — met the inclusion criteria for the systematic review conducted by the sponsor. The AEGEAN study was aimed at comparing the efficacy and activity of perioperative durvalumab with neoadjuvant chemotherapy to placebo with neoadjuvant chemotherapy. Following confirmation of eligibility, a total of 802 patients (including 8 patients in Canada) were randomized in a 1:1 ratio (intention-to-treat [ITT] population) using stratified randomization into the 2 treatment arms. Stratified randomization was conducted according to disease stage II versus stage III (AJCC [American Joint Committee on Cancer] Cancer Staging Manual, eighth edition, for tumour-nodes-metastasis [TNM] classification) and PD-L1 expression tumour cells of less than 1% versus tumour cells of 1% or more. Following a protocol amendment, 62 patients with known EGFR mutations or ALK rearrangements were excluded, leaving 740 patients in the modified intention-to-treat (mITT) population: 366 patients included in the perioperative durvalumab with neoadjuvant chemotherapy arm and 374 patients included in the placebo with neoadjuvant chemotherapy arm. The outcomes relevant to this review included the final pCR and MPR analyses performed at the November 10, 2022, data cut-off (DCO) date, and event-free survival (EFS), DFS, OS, PROs, and harms data collected at the May 10, 2024, DCO.
In the AEGEAN study, patients identified primarily as Asian (307 of 740 patients [41.5%]) or white (397 of 740 patients [53.6%]); all other races (including American Indian or Alaska Native, Black or African American, and other) accounted for 36 of 740 patients (4.9%). Patients were also primarily aged 50 years or older (703 of 740 patients [95.0%]) with a median age of 65.0 years (range, 39 years to 85 years); whereas 210 of 740 patients (28.4%) were female, most patients were male (530 of 740 patients [71.6%]). Most patients also either currently or formerly smoked (633 of 740 patients [85.5%]). Overall, at baseline, patients tended to have an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 (506 of 740 patients [68.4%]), present with stage III disease (525 of 740 patients [70.9%]), and have a baseline PD-L1 expression status greater than or equal to 1% (493 of 740 patients [66.6%]). In total, | patients had unknown EGFR mutation status and ██ patients had unknown ALK translocation status; these patients were balanced across both treatment arms.
Efficacy Results
Efficacy outcomes presented as follows are from the most recent DCOs (November 10, 2022, for pCR, MPR, and neoadjuvant QoL outcomes and May 10, 2024, for all other outcomes).
Pathological Complete Response
pCR was tested at the pCR IA (the DCO date was January 14, 2022), where it was deemed statistically significant, and thus, statistical significance was not retested in the final analysis.
At the November 10, 2022, DCO, the final pCR analysis was completed. The percentage of patients achieving pCR was 17.21% (95% confidence interval [CI], 13.49% to 21.48%) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 4.28% (95% CI, 2.46% to 6.85%) in the placebo with neoadjuvant chemotherapy arm. The between-group difference in the percentage of patients achieving pCR was 12.96% (95% CI, 8.67% to 17.57%). Results of the sensitivity and subgroup analysis were consistent with the primary analysis.
Major Pathological Response
MPR statistical significance was tested at the pCR IA (the DCO date was January 14, 2022) where it was deemed statistically significant and thus, statistical significance was not retested at the final analysis.
At the November 10, 2022, DCO, the percentage of patients achieving MPR was 33.33% (95% CI, 28.52% to 38.42%) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 12.30% (95% CI, 9.15% to 16.06%) in the placebo with neoadjuvant chemotherapy arm. The between-group difference in the percentage of patients achieving MPR was 21.03% (95% CI, 15.14% to 26.93%).
Event-Free Survival
At the May 10, 2024, DCO, the median duration of EFS follow-up was █████ months (range, ███ to ████), at which point 124 EFS events (33.9%) had occurred in the perioperative durvalumab with neoadjuvant chemotherapy arm and 165 EFS events (44.1%) in the placebo with neoadjuvant chemotherapy arm. The Kaplan-Meier (KM) estimate for median EFS was not reached (NR) (95% CI, 42.3 months to NR) in the perioperative durvalumab with neoadjuvant chemotherapy arm and was 30.0 months (95% CI, 20.6 months to NR) in the placebo with neoadjuvant chemotherapy arm, with a hazard ratio (HR) of 0.69 (95% CI, 0.55 to 0.88; P = █████). The between-group difference in the probability of EFS at 12 months and 36 months was 9.2% (95% CI, ███ ██ ████) and 12.2% (95% CI, ███ ██ ████), respectively. Results of the sensitivity and subgroup analysis were consistent with the primary analysis.
Disease-Free Survival
At the May 10, 2024, DCO, DFS was tested and did not meet the prespecified boundary for declaring statistical significance. The median of DFS follow-up was █████ months (range, ███ to ████), at which point 60 DFS events (24.8%) had occurred in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 81 DFS events (35.1%) in the placebo with neoadjuvant chemotherapy arm. The KM estimate for median DFS was NR (95% CI, NR to NR) in the perioperative durvalumab with neoadjuvant chemotherapy arm and NR (95% CI, 41.5 months to NR) in the placebo with neoadjuvant chemotherapy arm, with a stratified HR of 0.66 (95% CI, 0.47 to 0.92). The between-group difference in the probability of DFS at 12 months and 36 months was 6.9% (95% CI, ████ ██████) and 9.8% (95% CI, ████ ██ ████), respectively.
Overall Survival
At the EFS interim analysis 2 (IA2) DCO, OS was not eligible for statistical testing (this is because DFS was not statistically significant and therefore, OS was not formally tested based on the multiple testing procedure [MTP]).
At the May 10, 2024, DCO, the median duration of OS follow-up was █████ months (range, ███ to ████), at which point 121 deaths (33.1%) had occurred in the perioperative durvalumab with neoadjuvant chemotherapy arm and 140 deaths (37.4%) in the placebo with neoadjuvant chemotherapy arm. The KM estimate for median OS was NR (95% CI, NR to NR) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 53.2 months (95% CI, 44.3 months to NR) in the placebo with neoadjuvant chemotherapy arm, with a stratified HR of 0.89 (95% CI, 0.70 to 1.14). The between-group difference in the probability of survival at 12 months and 48 months was −1.0% (95% CI, ████ ██ ███) and 4.7% (95% CI, ████ ██ ████), respectively.
EORTC QLQ-C30 Global Health Status and QoL
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) questionnaire-based outcomes were secondary end points and not adjusted for multiplicity in the AEGEAN trial.
The change from baseline to week 12 in the EORTC QLQ-C30 global health status and QoL score was evaluated in the mITT population (n = 740) while the change from adjuvant baseline to week 44 was evaluated in the modified resected set (n = 473). The between-group difference for EORTC QLQ-C30 at week 12 of the neoadjuvant period was █████ (95% CI, █████ ██ █████. The between-group difference for EORTC QLQ-C30 at week 44 of the adjuvant period was █████ (95% CI, █████ ██ █████).
Harms Results
Safety outcomes presented as follows are from the May 10, 2024, DCO.
An overview of safety data is initially presented for the modified safety analysis set (consisting of all patients in the AEGEAN trial who received at least 1 dose of the study treatment and excluding those whose tumours had EGFR mutations or ALK gene rearrangements [N = 367 for the perioperative durvalumab with neoadjuvant chemotherapy arm and N = 370 for the placebo with neoadjuvant chemotherapy arm]). Because granular safety data were not available for the modified safety analysis set, additional data are presented for the safety analysis set, which consists of all randomized patients who received at least 1 dose of study treatment, with treatment arm allocation in accordance with the treatment actually received. The safety analysis set consisted of 799 patients (N = 401 in the perioperative durvalumab with neoadjuvant chemotherapy arm and N = 398 in the placebo with neoadjuvant chemotherapy arm).
Adverse Events
In the modified safety analysis set, a ███████ percentage of patients in both treatment arms experienced any AE over the course of treatment (█████ ████████ in the perioperative durvalumab with neoadjuvant chemotherapy arm versus █████ ████████ in the placebo with neoadjuvant chemotherapy arm). The 3 most frequently reported AEs in both treatment arms of the safety analysis set were anemia (34.9% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 32.2% in the placebo with neoadjuvant chemotherapy arm), nausea (25.7% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 29.9% in the placebo with neoadjuvant chemotherapy arm), and constipation (25.9% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 29.9% in the placebo with neoadjuvant chemotherapy arm).
Serious AEs
████████ ████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm of the modified safety analysis set experienced a serious adverse event (SAE) (█████ ██████████ than in the placebo with neoadjuvant chemotherapy arm (█████ █████████). In the safety analysis set, the 4 most frequently reported SAEs in the perioperative durvalumab with neoadjuvant chemotherapy arm were pneumonia (5.7%), anemia (1.7%), COVID-19 (1.7%), and pneumonitis (1.7%) while in the placebo with neoadjuvant chemotherapy arm, it was pneumonia (4.5%), pneumothorax (2.3%), anemia (1.3%), and COVID-19 (1.3%).
Withdrawals Due to AEs
████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm of the modified safety analysis set prematurely stopped study treatment (i.e., durvalumab, placebo, or neoadjuvant chemotherapy) due to AEs than in the placebo with neoadjuvant chemotherapy arm (█████ ███████ versus █████ ███████, respectively). In the safety analysis set, the 3 most common AEs leading to treatment discontinuation were blood and lymphatic system disorders (██████ respiratory, thoracic, and mediastinal disorders ███████ and nervous system disorder ██████ in the perioperative durvalumab with neoadjuvant chemotherapy arm compared to respiratory, thoracic, and mediastinal disorders (████), investigations (████), and blood and lymphatic system disorders (████) and nervous system disorder █████) in the placebo with neoadjuvant chemotherapy arm.
Mortality
As of May 10, 2024, in the modified resected set ████ ████████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and ████ ████████ of patients in the placebo with neoadjuvant chemotherapy arm experienced an AE resulting in death. In the safety analysis set, the 2 most common AEs resulting in death in the perioperative durvalumab with neoadjuvant chemotherapy arm were infections and infestations (3.2%) and respiratory, thoracic, and mediastinal disorders (1.5%) compared to infections and infestations (1.8%) and cardiac disorders (0.8%) in the placebo with neoadjuvant chemotherapy arm.
Immune-Mediated AEs
Immune-mediated AEs (imAEs) occurred in █████ ████████) of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and in █████ ███████) of patients in the placebo with neoadjuvant chemotherapy arm.
Critical Appraisal
The AEGEAN study is a double-blind, placebo-controlled, phase III, randomized controlled trial (RCT). Randomization and allocation concealment methods were adequate. The postrandomization exclusion of those with known EGFR mutations or ALK rearrangements were judged not to be related to treatment group assignment and therefore, are not expected to introduce bias.
To mitigate the potential bias arising from the subjective interpretation of disease progression and tumour response, a blinded independent review committee (IRC) reviewed all available radiographic tumour assessments to determine tumour response based on Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST 1.1) criteria. As such, the risk of bias resulting from unblinding due to unbalanced harms across treatment arms is lower for pCR, MPR, and survival outcomes; however, there are some concerns for health-related quality of life (HRQoL) outcomes. While pCR and MPR are based on final analyses, EFS, DFS, OS, and HRQoL results should be interpreted in light of the fact that these are based on interim analyses (IAs) that may overestimate treatment effects.20 At the most recent AEGEAN study DCO, results of the coprimary end points, pCR and EFS, were adjusted using the Lan-DeMets alpha spending function with O’Brien-Fleming boundaries that accounted for the actual number of patients at the time of analyses at an overall alpha of 0.5% and 4.5%, respectively. DFS and OS were included in the AEGEAN study’s MTP; OS was not eligible for statistical testing because DFS was not statistically significant. While there was an adequate number of patients at risk in the AEGEAN trial for earlier DFS and OS time points, only about 11% of patients in both treatment arms remained at risk for OS at 48 months and 13% to 19% remained at risk for DFS at 36 months, resulting in substantial uncertainty in results at later time points.
Biases resulting from the self-reporting nature of HRQoL outcomes were deemed to be minimal given that participants were blinded to the intervention received. In the EORTC QLQ-C30 analyses, aimed at estimating the impact to patients’ HRQoL, █████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ of patients in the placebo with neoadjuvant chemotherapy arm were available to provide assessments at week 12 of the neoadjuvant period. In the modified resected set, by week 44 of the adjuvant period █████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ of patients in the placebo with neoadjuvant chemotherapy arm were available to provide EORTC QLQ-C30 assessments. No data imputations were involved in these analyses, so there is a risk of bias due to missing outcomes data. Further, because analyses of the HRQoL end points were not adjusted for multiple testing, there is an increased risk of type I error for statistically significant results.
Given that the analysis of EORTC QLQ-C30 in the adjuvant period, as well as the DFS analyses, were limited to the modified resected set (n = 473) and because this is a subgroup of the mITT population (n = 740), randomization is not maintained and results are not measuring the effect of assignment to the intervention.
The outcomes measured in the AEGEAN trial evaluated the key treatment goals identified by patients’ input collected for this review and were deemed to be relevant by clinical experts consulted for this review. The generalizability of outcomes in the AEGEAN trial to the context in Canada is limited by the fact that only 8 patients included in the ITT population were from Canada. However, this limitation was deemed minimal by the clinical experts consulted for this review, who noted that the baseline characteristics of patients enrolled in the AEGEAN trial were aligned with those seen in clinical practice in Canada. The clinical experts consulted for this review noted that currently, the 2 most commonly used regimens in clinical practice in Canada are neoadjuvant nivolumab with chemotherapy and adjuvant chemotherapy. According to the clinical experts consulted for this review, adjuvant atezolizumab after surgery and adjuvant chemotherapy for patients with PD-L1 of 50% or greater was the least relevant comparator because the treatment decision for perioperative durvalumab has to be made before surgery, whereas the approach for adjuvant atezolizumab would be considered after surgery and adjuvant chemotherapy. In addition to the standard of care chemotherapy regimens used in the AEGEAN trial, clinical experts indicated that in Canada, carboplatin and gemcitabine could also be used for squamous cell NSCLC and cisplatin and vinorelbine may also sometimes be used for both squamous and nonsquamous cell NSCLC. The lack of these regimens used for standard chemotherapy in the AEGEAN trial further limits the generalizability to the setting in Canada; however, clinical experts consulted for this review noted that they would feel comfortable combining perioperative durvalumab with these regimens.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool was used to assess the certainty of the evidence for outcomes considered most relevant to inform deliberations by the CDA-AMC expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group.21,22 Following the GRADE approach, evidence from RCTs started as high certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS (probabilities at 12 months and 48 months), EFS (probabilities at 12 months and 36 months), and pCR
HRQoL (change in EORTC QLQ-C30 global health status and QoL score from adjuvant baseline to week 12 and neoadjuvant baseline to week 44)
harms (imAEs and SAEs).
Table 2 presents the GRADE summary of findings for perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy for adults with resectable NSCLC without any EGFR mutations or ALK rearrangements.
Table 2: Summary of Findings for Perioperative Durvalumab With Neoadjuvant Chemotherapy vs. Placebo With Neoadjuvant Chemotherapy
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo + neoadjuvant chemotherapy | Perioperative durvalumab + neoadjuvant chemotherapy | Difference | |||||
EFS | |||||||
Probability of EFS at 12 months Median (range) follow-up: █████ months (███ to ████) | 740 (1 RCT) | NR | 641 per 1,000 | 733 per 1,000 (681 to 777 per 1,000) | 92 more per 1,000 (██ to ███ ████ per 1,000) | Moderatea | Perioperative durvalumab with neoadjuvant chemotherapy likely results in a clinically important increase in the probability of being event-free and alive at 12 months compared to placebo with neoadjuvant chemotherapy. |
Probability of EFS at 36 months Median (range) follow-up: █████ months (███ to ████) | 740 (1 RCT) | NR | 479 per 1,000 | 601 per 1,000 (539 to 658 per 1,000) | 122 more per 1,000 (██ to ███ ████ per 1,000) | Moderateb | Perioperative durvalumab with neoadjuvant chemotherapy likely results in a clinically important increase in the probability of being event-free and alive at 36 months compared to placebo with neoadjuvant chemotherapy. |
OS | |||||||
Probability of OS at 12 months Median (range) follow-up: █████ months (███ to ████) | 740 (1 RCT) | NR | 853 per 1,000 | 843 per 1,000 (801 to 877 per 1,000) | 10 fewer per 1,000 (██ ████ to ██ ████ per 1,000) | Highc | Perioperative durvalumab with neoadjuvant chemotherapy results in little to no difference in the probability of being alive at 12 months compared to placebo with neoadjuvant chemotherapy. |
Probability of OS at 48 months Median (range) follow-up: █████ months (███ to ████) | 740 (1 RCT) | NR | 529 per 1,000 | 576 per 1,000 (501 to 643 per 1,000) | 47 more per 1,000 (██ ████ to ███ ████ per 1,000) | Lowd | Perioperative durvalumab with neoadjuvant chemotherapy may result in little to no difference in the probability of being alive at 48 months compared to placebo with neoadjuvant chemotherapy. |
EORTC QLQ-C30 global health status and quality of life | |||||||
Change from baseline to end of neoadjuvant period (week 12) | ███ (1 RCT) | NA | █████ | █████ (█████ to █████) | █████ (█████ to ████) | Lowe | Perioperative durvalumab with neoadjuvant chemotherapy may result in little to no difference in quality of life during the neoadjuvant period compared to placebo with neoadjuvant chemotherapy. |
Change from adjuvant baseline to end of adjuvant period (week 44) | ███ (1 RCT) | NA | ████ | █████ (█████ to ████) | █████ (█████ to █████) | Very lowf | The evidence is very uncertain about the effect of perioperative durvalumab with neoadjuvant chemotherapy on quality of life during the adjuvant period compared to placebo with neoadjuvant chemotherapy. |
pCR | |||||||
pCR | 740 (1 RCT) | NR | 43 per 1,000 | 172 per 1,000 (135 to 215 per 1,000) | 130 more per 1,000 (87 to 176 more per 1,000) | Moderateg | Perioperative durvalumab with neoadjuvant chemotherapy likely results in a clinically important increase in the probability of achieving pCR compared to placebo with neoadjuvant chemotherapy. |
Harms | |||||||
Immune-mediated AEs Median (range) follow-up: █████ months (███ to ████) | 737 (1 RCT) | NR | ███ per 1,000 | ███ per 1,000 (NR) | ███ ████ per 1,000 (███ to ███ ████ per 1,000) | Highh | Perioperative durvalumab with neoadjuvant chemotherapy results in a clinically important increase in the proportion of patients who experience ≥ 1 immune-mediated AE compared to placebo with neoadjuvant chemotherapy. |
Serious AEs Median (range) follow-up: █████ months (███ to ████) | 737 (1 RCT) | NR | ███ per 1,000 | ███ per 1,000 (NR) | ██ ████ per 1,000 (||| to ███ ████ per 1,000) | Moderatei | Perioperative durvalumab with neoadjuvant chemotherapy likely results in little to no clinically important difference in the proportion of patients who experience ≥ 1 serious AE compared to placebo with neoadjuvant chemotherapy. |
AE = adverse event; CI = confidence interval; EFS = event-free survival EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; IA1 = interim analysis 1; IA2 = interim analysis 2; KM = Kaplan-Meier; NA = not applicable; NSCLC = non–small cell lung cancer; NR = not reported; OS = overall survival; pCR = pathological complete response; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aCertainty was not rated down for study limitations or indirectness. Rated down 1 level for imprecision. The point estimate suggests a clinically important difference in EFS at 12 months based on a clinically important between-group difference threshold of 5% to 10% suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold. The between-group difference in the probability of EFS was requested from the sponsor to aid in the interpretation of the results for this end point.
bCertainty was not rated down for study limitations or indirectness. Rated down 1 level for imprecision. The point estimate suggests a clinically important difference in EFS at 36 months based on a clinically important between-group difference threshold of 5% to 10% suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold. The between-group difference in the probability of EFS was requested from the sponsor to aid in the interpretation of the results for this end point.
cCertainty was not rated down for study limitations, indirectness, or imprecision. The clinical expert consulted for this review suggested that a 5% to 10% between-group difference could be considered clinically meaningful. OS was not eligible for statistical testing at EFS IA2. The results are considered as supportive evidence. The between-group difference in survival probability was requested from the sponsor to aid in the interpretation of the results for this end point.
dCertainty was not rated down for indirectness. Rated down 1 level due to serious study limitations. Visual inspection of the KM curves suggests substantial censoring after 24 months in both groups and few patients at risk at 48 months, contributing to uncertainty in the OS results. Rated down 1 level for imprecision. The point estimate suggests little to no difference while the upper bound of the 95% CI indicates benefits based on a clinically important between-group difference threshold of 5% to 10% suggested by clinical experts. OS was not eligible for statistical testing at EFS IA2. The results are considered as supportive evidence. The between-group difference in survival probability was requested from the sponsor to aid in the interpretation of the results for this end point.
eCertainty was not rated down for indirectness or imprecision. Rated down 2 levels due to missing data in both arms at week 12 (█████ missing in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ in the placebo with neoadjuvant chemotherapy arm). EORTC QLQ-C30 was not adjusted for multiplicity in the trial and should be considered as supportive evidence.
fCertainty was not rated down for indirectness. Rated down 2 levels due to missing data in both arms at week 44 (█████ missing in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ in the placebo with neoadjuvant chemotherapy arm) and given the modified resected set was analyzed, there may not be prognostic balance between the treatment groups. Rated down a third level for imprecision. ███ █████ ████████ ████████ ███████████ ██████ ██ █████ █████ ███ █████ █████ ██ ███ ███ ██ ███████ ███ █████████ █████████ ██████████ █████████ ██ ██ ██████████ ██ ███ ██████████. EORTC QLQ-C30 was not adjusted for multiplicity in the trial and should be considered as supportive evidence.
gCertainty was not rated down for study limitations or indirectness. Rated down 1 level for imprecision. The point estimate suggests a clinically important difference in pCR based on a clinically important between-group difference threshold of 10% suggested by clinical experts; however, the lower bound of the 95% CI crosses this threshold.
hCertainty was not rated down for study limitations, indirectness, or imprecision. The clinical expert consulted for this review suggested that a 10% between-group difference could be considered clinically meaningful. The between-group difference was requested from the sponsor to aid in the interpretation of the results.
iCertainty was not rated down for study limitations or indirectness. Rated down 1 level for imprecision. The point estimate suggests little to no difference while the upper bound of the 95% CI indicates a clinically important increase in serious AEs based on a clinically important between-group difference threshold of 10% suggested by clinical experts. The between-group difference was requested from the sponsor to aid in the interpretation of the results.
Sources: AEGEAN study EFS IA1 Clinical Study Report24 and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
Due to the lack of direct evidence comparing perioperative durvalumab (durvalumab in combination with chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery) with relevant comparator regimens, including neoadjuvant nivolumab, perioperative pembrolizumab, adjuvant chemotherapy, and surgery only in the treatment of patients with resectable (tumours ≥ 4 cm and/or node positive) NSCLC and no known EGFR mutations or ALK rearrangements, the sponsor conducted indirect treatment comparisons (ITCs), including 2 matching-adjusted indirect comparisons (MAICs) and a network meta-analysis (NMA).
Efficacy Results
The MAIC analyses were insufficient to inform whether perioperative durvalumab or the comparator therapies were favoured for EFS. The 95% CIs (or credible intervals [CrIs] for the random-effects NMA) were wide and included the potential that either of the treatments being compared could be favoured. No other efficacy outcomes were assessed in the ITCs.
Harms Results
No ITCs were done for harms outcomes.
Critical Appraisal
Overall, the ITCs (2 MAICs and 1 NMA) were conducted according to accepted methodological guidance. The potential key limitations of the MAIC analyses included the fact that not all important effect modifiers (EMs) could be adjusted in the analysis. Furthermore, the MAIC methods reduced the effective sample size in both comparisons (████ reduction in the base-case analysis for the comparison versus neoadjuvant nivolumab while a smaller reduction [15% to 17%] occurred in the pembrolizumab comparison). Such reductions in effective sample size may indicate limited overlap in baseline characteristics between the trial populations and increase uncertainty in the weighted estimates. Imprecision (wide 95% CIs) in the effect estimates precluded conclusions as to whether perioperative durvalumab or the comparator therapies were favoured for EFS.
The potential key limitation of the NMA was the heterogeneity (in EMs) across the included studies in terms of the study designs and patient characteristics; notably, some included studies were conducted starting before 1990 and the standard of care has changed since that time. Given differences across studies in staging and tumour assessment criteria, the use of second-generation chemotherapy agents, and other unmeasured characteristics potentially impacting outcomes (e.g., smoking status was infrequently reported), the plausibility of the transitivity assumption is uncertain. In addition, several studies met the inclusion criteria for the ITC but were excluded due to the absence of EFS KM curves and HRs. The impact of the exclusion of these studies on the NMA results was unknown; however, there is a risk of bias due to missing evidence in the synthesis. Finally, all evidence networks were sparse, with no closed loops formed by multiple studies. The CrIs for all effect estimates (per the random-effects model) were wide, precluding conclusions as to whether perioperative durvalumab or the comparator therapies were favoured for EFS.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
The AEGEAN study — a phase III, double-blind, placebo-controlled, international RCT — compared the efficacy and safety of perioperative durvalumab with neoadjuvant chemotherapy with placebo and neoadjuvant chemotherapy in adult patients with resectable NSCLC and no known EGFR mutations or ALK rearrangements. The AEGEAN trial demonstrated that there were added clinical benefits of perioperative durvalumab in pCR and EFS. Compared to placebo with neoadjuvant chemotherapy, perioperative durvalumab with neoadjuvant chemotherapy likely results in a clinically important increase in the number of patients achieving pCR (moderate certainty evidence) and a clinically important increase in EFS rates at 12 months and 36 months (moderate certainty evidence). Results for DFS, a key secondary outcome, were supportive of the primary EFS analyses. Per EFS IA2, perioperative durvalumab with neoadjuvant chemotherapy may result in little to no difference in the probability of being alive at 48 months; however, survival data remained immature (IA2 DCO indicated 35% of patients had died). Results for EFS from the ITCs comparing perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant nivolumab plus chemotherapy, perioperative pembrolizumab plus neoadjuvant chemotherapy followed by adjuvant pembrolizumab, adjuvant chemotherapy, and surgery only were inconclusive, owing to methodological limitations and imprecision. Based on the AEGEAN trial, perioperative durvalumab with neoadjuvant chemotherapy, compared to neoadjuvant chemotherapy, likely results in little to no difference in QoL during the neoadjuvant period (moderate certainty evidence) and may result in clinically meaningful deterioration in HRQoL in the adjuvant period; however, these results had a high degree of missing data and adjuvant results were limited to only those achieving complete resection. According to clinical experts consulted for this review, the safety profile of perioperative durvalumab with neoadjuvant chemotherapy was consistent with their expectations and deemed acceptable. Harms were not investigated in the ITCs, so whether perioperative durvalumab with neoadjuvant chemotherapy results in an increase or decrease in harms compared with other therapies used in Canada is unknown.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of perioperative durvalumab, 1,500 mg administered as an IV infusion once every 3 weeks in combination with neoadjuvant chemotherapy before surgery and once every 4 weeks as monotherapy after surgery for the treatment of resectable NSCLC in patients without any known EGFR mutations or ALK rearrangements.
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Lung cancer is the most frequently diagnosed cancer in Canada and the leading cause of cancer-related deaths. In 2024 alone, approximately 32,100 Canadians were expected to be diagnosed with lung cancer, and 20,700 annual deaths were expected to be attributable to lung cancer.1 Lung cancer predominantly affects older adults, with 98% of cases occurring in individuals aged 50 years and older.2 The adjusted 5-year and 10-year net survival rates for all forms and stages of lung cancer are estimated to be 22% and 15%, respectively.15 In Canada, Indigenous Peoples face significant lung cancer disparities. First Nations adults have a 35% lower 5-year survival rate despite similar incidence rates; Métis adults are more likely to be diagnosed and have a 30% lower 5-year survival rate, and Inuit living in Inuit Nunangat (the Arctic homeland of Inuit in Canada) are more than twice as likely to be diagnosed.25
Lung cancer is classified as either NSCLC or small cell lung cancer, with NSCLC accounting for approximately 88% of all lung cancer cases in Canada, excluding Quebec. NSCLC can be further categorized into adenocarcinoma, which is the most common type of lung cancer accounting for 48% of cases, followed by squamous cell carcinoma (20% of cases), and NSCLC not otherwise specified (20% of cases).26 To date, several molecular alterations have been identified in NSCLC, with the 2 prominent ones being EGFR mutations and ALK rearrangements. EGFR mutations are particularly common in adenocarcinomas, resulting in abnormal signalling pathways that drive tumour growth.27 Similarly, ALK rearrangements occur through gene fusions, leading to abnormal cell proliferation.28 Driver alterations affect a small proportion of patients with NSCLC.29 EGFR mutations are identified in about 10% to 30% of patients with nonsquamous NSCLC, while ALK and ROS1 rearrangements occur in about 2% to 5% and in about 1% to 4%, respectively, of nonsquamous NSCLC tumours. BRAF mutations are observed in 2% of patients with NSCLC.29
NSCLC diagnosis begins with a comprehensive medical history, smoking assessment, and standard tests, including evaluations of lung, renal, and liver function, along with blood and biochemistry tests.7 Imaging techniques such as X-rays, CT scans, PET scans of the chest and abdomen, and MRI of the central nervous system are then used before tissue biopsy is performed to confirm the diagnosis.30,31 To determine the prognosis and best course of treatment, NSCLC is staged using the AJCC staging criteria, which involves TNM classification based on the size and spread of the primary tumour (“T”), lymph node involvement (“N”), and distant metastasis (“M”).7
Approximately 25% to 30% of patients with NSCLC present with resectable early-stage disease (stage II to stage III),3,4 and are usually treated with surgery and chemotherapy.32 However, an estimated 30% to 55% of patients continue to develop postoperative recurrence and ultimately die due to NSCLC.5
Patients with NSCLC experience a variety of symptoms, including dyspnea, malaise, depression, anxiety, worsening cough, hemoptysis, pain, weight loss, and hoarseness.6,7 These symptoms, coupled with disease outlook, treatment modalities and side effects, and other comorbidities common among patients with NSCLC, significantly impact patients’ QoL.8-11 Patients with NSCLC also face significant productivity losses and stigma; notably, those who do not smoke experience negative perceptions associated with lung cancer.12-14
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The treatment goal for patients with early-stage NSCLC is to achieve a complete cure, improve OS and DFS, reduce disease recurrence rates, reduce symptoms, and improve QoL.
Before establishing a treatment plan, tumours should receive molecular testing to identify the presence of any oncogenic drivers and identify their PD-L1 status.33 In patients without EGFR mutations or ALK rearrangements with stage II to stage IIIB NSCLC, the standard of care is surgery with curative intent.32,34,35 Given that surgery alone is suboptimal at eliminating and/or eradicating micrometastases, guidelines recommend additional systemic therapy with platinum-based chemotherapy alone or in combination with ICIs, either before or after surgery.32,36 While postoperative radiotherapy (PORT) is not recommended in patients with completely resected early-stage NSCLC, PORT may be considered in the case of R1 (microscopic residual tumour) resection.37 This treatment pathway excludes patients who have EGFR or ALK alterations because these patients have limited response to immunotherapy and are treated with targeted treatment options.38
Neoadjuvant Therapies
In Canada, the standard of care in the neoadjuvant setting has historically been surgery followed by systemic therapy or neoadjuvant chemoradiation followed by surgery; however, guidelines now support the use of neoadjuvant ICI treatment options. Neoadjuvant treatment with nivolumab (a PD-1 inhibitor) in combination with platinum-based chemotherapy followed by surgery and optional adjuvant platinum-based chemotherapy is recommended and widely funded by provincial drug plans for patients with stage IB and stage IIIA (AJCC Cancer Staging Manual, seventh edition, TNM classification) resectable (tumours ≥ 4 cm or node positive) NSCLC. Neoadjuvant nivolumab in combination with platinum-based chemotherapy received a positive conditional CDA-AMC final recommendation in 2023. In April 2025, CDA-AMC issued a conditional positive final recommendation for pembrolizumab for the treatment of adult patients with resectable stage II, stage IIIA, or stage IIIB (T3-4N2) NSCLC in combination with platinum-containing chemotherapy as neoadjuvant treatment and then continued as monotherapy in the adjuvant setting.
Adjuvant Therapies
Curative-intent surgery followed by adjuvant chemotherapy is the established standard of care in Canada for patients with resectable NSCLC. Adjuvant chemotherapy regimens include up to 4 cycles of platinum-based chemotherapy, where regimen selection is based on a patient’s histology (squamous versus nonsquamous).39
For patients who did not receive treatment with an ICI drug in the neoadjuvant setting, ICI treatment with either atezolizumab or pembrolizumab (PD-L1 inhibitors) can be considered following surgical resection and completion of adjuvant chemotherapy.40,41 In the setting in Canada, up to 16 cycles of atezolizumab following complete surgical resection and adjuvant chemotherapy is funded by drug plans participating in the CDA-AMC reimbursement review process for tumours with PD-L1 of 50% or greater. In February 2025, CDA-AMC issued a conditional positive final recommendation for pembrolizumab for the adjuvant treatment of adult patients with stage IB (tumour stage IIA [T2a] ≥ 4 cm), stage II, or stage IIIA NSCLC who have undergone complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 tumour proportion score of less than 50%. However, because this current file for perioperative durvalumab was initiated before the final recommendation for adjuvant pembrolizumab was issued, adjuvant pembrolizumab was not considered a relevant comparator for this review. It is important to note that there are no data to support a switch from the use of a neoadjuvant ICI treatment to an alternative adjuvant ICI treatment; as such, the back-to-back use of ICI therapies is not currently funded by drug plans participating in the CDA-AMC reimbursement review process or by cancer agencies.42
Drug Under Review
Key characteristics of durvalumab are summarized in Table 3 with other treatments available for resectable NSCLC without any known EGFR mutations or ALK rearrangements.
Durvalumab 1,500 mg, single-use IV infusion, as neoadjuvant treatment in combination with platinum-containing chemotherapy, followed by durvalumab as monotherapy after surgery, for the treatment of patients with resectable stage II, stage IIIA, or stage IIIB (T3-4N2) NSCLC and no known EGFR mutations or ALK rearrangements received Health Canada authorization on February 20, 2026. While the sponsor’s reimbursement request is the same as the Health Canada indication, the wording between the 2 differs slightly (i.e., the Health Canada indication specifies the use of platinum-containing chemotherapy and includes specific NSCLC stages while the reimbursement request does not include these 2 items).
Durvalumab has been previously reviewed by CDA-AMC for the treatment of patients with locally advanced, unresectable NSCLC following curative-intent, platinum-based chemoradiation therapy, for up to a maximum of 12 months. The final recommendation, issued on May 3, 2019, was to reimburse with conditions.18 CDA-AMC has also reviewed durvalumab in extensive-stage small cell lung cancer, biliary tract cancer, unresectable hepatocellular carcinoma, endometrial cancer, limited-stage small cell lung cancer, and metastatic NSCLC, all of which received conditional reimbursement recommendations. The FDA and European Medicines Agency have approved durvalumab for the present indication under review.43,44
Durvalumab is a fully human, high-affinity, immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and cluster of differentiation 80 (B7.1) while leaving PD-1 and/or PD-L2 interaction intact. Durvalumab does not induce antibody-dependent cell-mediated cytotoxicity. The selective blockade of PD-L1 and/or PD-1 and PD-L1 and/or cluster of differentiation 80 interactions releases the inhibition of immune responses and enhances antitumour immune responses.45
Table 3: Key Characteristics of Durvalumab, Nivolumab, and Atezolizumab, and Platinum-Based Chemotherapies
Drug | Mechanism of action | Indicationa | Route of administration | Recommended dosage | Safety issues |
|---|---|---|---|---|---|
Perioperative durvalumab | Humanized monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80 | In combination with platinum-containing chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery, is indicated for the treatment of patients with resectable stage II, IIIA, or IIIB (T3-4N2) NSCLC and no known EGFR mutation or ALK rearrangements | IV | 1,500 mg in combination with platinum-containing chemotherapy every 3 weeks before surgery (neoadjuvant period), followed by 1,500 mg as monotherapy every 4 weeks after surgery (adjuvant period) Patients weighing ≤ 30 kg must receive weight-based dosing of the drug at 20 mg/kg. In combination with chemotherapy, dose at 20 mg/kg every 3 weeks (21 days) is administered before surgery, followed by monotherapy at 20 mg/kg every 4 weeks after surgery until weight increases to > 30 kg. | Immune-mediated pneumonitis, hepatitis, colitis, and endocrinopathies Severe or life-threatening infections Infusion-related reactions Embryo-fetal toxicity |
Perioperative pembrolizumab | IgG4 monoclonal antibody against PD-1 receptors | For the treatment of adult patients with resectable stage II, IIIA, or IIIB (T3-4N2) NSCLC in combination with platinum-containing chemotherapy as neoadjuvant treatment, and then continued as monotherapy as adjuvant treatment after surgery | IV | 4 doses of 200 mg every 3 weeks or 2 doses of 400 mg every 6 weeks in combination with platinum-containing chemotherapy before surgery (neoadjuvant period), followed by up to 13 doses of 200 mg every 3 weeks or 7 doses of 400 mg every 6 weeks after surgery (adjuvant period) | Pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and severe skin reactions |
Neoadjuvant nivolumab | IgG4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, releasing PD-1 pathway–mediated inhibition of the immune response, including the antitumour immune response. In syngeneic mouse tumour models, blocking PD-1 activity resulted in decreased tumour growth. | In combination with platinum-doublet chemotherapy, it is indicated for the neoadjuvant treatment of adult patients with resectable NSCLC (tumours ≥ 4 cm or node positive). | IV | 240 mg administered intravenously over 30 minutes every 2 weeks in combination with fluoropyrimidine and platinum-containing chemotherapy every 4 weeks 480 mg nivolumab administered intravenously over 30 minutes in combination with fluoropyrimidine and platinum-containing chemotherapy every 4 weeks | Severe and fatal immune-mediated adverse reactions, including pneumonitis, interstitial lung disease, encephalitis, myocarditis, SJS, TEN, and autoimmune hemolytic anemia There are no data to inform the use in pregnant people, those breastfeeding, and pediatric patients (aged < 18 years). |
Adjuvant atezolizumab | An Fc-engineered humanized IgG1 monoclonal antibody that directly binds to PD-L1 and blocks interactions with PD-1 and B7.1 receptors, releasing PD-L1 and PD-1 pathway–mediated inhibition of the immune response, including reactivation of the antitumour immune response. Atezolizumab leaves the PD-L1 and PD-1 interaction intact. | As monotherapy for adjuvant treatment following complete resection and no progression after platinum-based adjuvant chemotherapy for adults with stage II to IIIA (according to UICC and AJCC [7th edition]1 staging criteria) NSCLC whose tumours have PD-L1 expression on ≥ 50% of TCs | IV | 840 mg every 2 weeks, 1,200 mg every 3 weeks, or 1,680 mg every 4 weeks | Immune-mediated adverse reactions that include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, and solid organ transplant rejection Infusion reactions (e.g., hypersensitivity, anaphylaxis) Ocular inflammatory toxicity Embryo-fetal toxicity Fetal harm Fatal and other serious complications can occur in patients who receive allogeneic HSCT before or after treatment with a PD-1– and PD-L1–blocking antibody. |
Paclitaxel | Antimicrotubule agent that promotes abnormal microtubule assembly, disrupting the cell’s ability to divide by blocking the G2 and M phases of the cell cycle | First-line treatment of advanced NSCLC | IV | 200 mg/m2 IV infusion over 3 hours followed by either carboplatin or cisplatin every 3 weeks | Should only be administered under the supervision of a physician experienced in the use of cancer chemotherapeutic agents Patients should be pretreated with corticosteroids, antihistamines, and histamine2 receptor antagonists. Should not be administered to patients with baseline neutrophil counts of less than 1,500 cells/mm3 |
Pemetrexed | Antifolate antineoplastic agent that exerts its action by disrupting crucial folate-dependent metabolic processes essential for cell replication | First-line treatment: in combination with cisplatin for patients with good performance status and locally advanced or metastatic disease Maintenance therapy: Monotherapy for patients without disease progression after 4 cycles of first-line platinum-based chemotherapy Second-line treatment: Monotherapy for patients with locally advanced or metastatic NSCLC after prior chemotherapy, showing similar efficacy to docetaxel | IV | 500 mg/m2 administered as an IV infusion over 10 minutes followed by cisplatin or carboplatin on day 1 of each 21-day cycle | Use caution in patients with a history of SJS or TEN. Should be used with caution in patients with pre-existing bone marrow suppression It may also cause nephrotoxicity. It should only be administered by, or under the supervision of, a physician who is experienced in cancer chemotherapy and in the management of related toxicities. |
Vinorelbine | Semisynthetic vinca alkaloid; exerts its antitumour activity by binding to tubulin and inhibiting microtubule assembly, thereby preventing cell mitosis and causing cell death. It is cell cycle phase–specific. | For the re-treatment of advanced NSCLC in combination with cisplatin | IV | 30 mg/m2 over 6 minutes to 10 minutes administered weekly, followed by cisplatin (or carboplatin if there is clinical reason) | Use with extreme caution in patients with compromised marrow reserve. May result in radiosensitizing effects with prior or concomitant radiation therapy Patients with pre-existing neuropathy or prior treatment with other neurotoxic drugs may have increased potential for neurotoxicity. |
Gemcitabine | A cell cycle–dependent antimetabolite and deoxycytidine analogue, it is metabolized intracellularly into active nucleosides that inhibit DNA synthesis and induce apoptosis through incorporation into DNA. | Treatment of patients with locally advanced or metastatic NSCLC in combination with cisplatin | IV | 1,250 mg/m2 over 30 minutes on day 1 and day 8 followed by cisplatin (or carboplatin if there is clinical reason) on day 1 of each 21-day cycle | Bone marrow suppression Pulmonary toxicity Hepatotoxicity Hemolytic uremic syndrome Capillary leak syndrome |
Neoadjuvant or adjuvant platinum-based chemotherapy (e.g., cisplatin, carboplatin) | Antineoplastic agents form crosslinks with the DNA in cancer cells, preventing proper DNA replication and transcription, triggering apoptosis (cell death), and inhibiting tumour growth. | While there is no formal Health Canada indication for any of the platinum-based chemotherapy regimens for resectable NSCLC, these regimens, such as neoadjuvant cisplatin, were recommended as the standard of care for completely resectable stage IIA or IIB and stage IIIA NSCLC, according to joint guidelines from the American Society of Clinical Oncology and OH-CCO,46 and other cancer care agencies such as Cancer Care Alberta,39 and were confirmed with clinical experts consulted for this review. | IV | Either vinorelbine, pemetrexed, gemcitabine, or paclitaxel in combination with either 75 mg/m2 (80 mg/m2 when used in combination with vinorelbine) cisplatin on day 1 over 1 hour or carboplatin AUC 5 (AUC 6 when used in combination with paclitaxel) on day 1 over 30 minutes. All regimens repeated every 21 days for 4 cycles | Nephrotoxicity, neurotoxicity, ocular toxicity and/or retinopathy, ototoxicity, severe nausea and vomiting, gonadotoxicity, and myelosuppression |
AJCC = American Joint Committee on Cancer; AUC 5 = area under the serum drug concentration-time curve 5; AUC 6 = area under the serum drug concentration-time curve 6; CD80 = cluster of differentiation 80; HSCT = hematopoietic stem-cell transplant; IgG1 = immunoglobulin G1; IgG4 = immunoglobulin G4; NSCLC = non–small cell lung cancer; OH-CCO = Ontario Health (Cancer Care Ontario); SJS = Stevens-Johnson syndrome, TC = tumour cell; TEN = toxic epidermal necrolysis; UICC = Union for International Cancer Control.
aHealth Canada–approved indication.
Sources: Kris (2017),46 Cancer Care Alberta,39 and product monographs for durvalumab (Imfinzi),45 pembrolizumab (Keytruda),47 nivolumab (Opdivo),48 atezolizumab (Tecentriq),49 paclitaxel,50 pemetrexed,51 vinorelbine,52 gemcitabine,53 cisplatin,54 and carboplatin.55 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
A joint submission was received from CCSN, LCC, and LHF in response to the call by CDA-AMC for patient input on the current review of durvalumab.
Information for this submission was collected via an online survey conducted by CCSN from June 11 to July 17, 2024, with input from LCC and LHF. The survey was disseminated across various platforms and organizations to collect responses.
A total of 5 respondents, all of whom were patients with lung cancer living in Canada, completed the survey; 4 respondents were female and 1 was male.
Respondents reported various stages of lung cancer, including stage IVA (n = 2), stage IVB (n = 1), and other stage IV classifications (n = 2). Common symptoms affecting their QoL included fatigue (n = 5), pain (n = 3), and shortness of breath (n = 2). Current treatments varied, with chemotherapy being the most common (n = 4), followed by immunotherapy (n = 3), targeted therapy (n = 2), radiation (n = 2), and surgical therapy (n = 1).
When asked about the most important aspects of their disease to control, respondents prioritized managing tumour growth, shortness of breath, and pain and side effects associated with chemotherapy. Survey respondents also identified the following unmet needs: a lack of mental health support, difficulties accessing counselling, and managing travel costs to access treatment. Respondents emphasized the importance of tumour control, managing symptoms, enhancing research and support for lung cancer, and the availability of alternative treatments in their cancer journey.
Respondents reported experiencing a range of adverse effects from their treatments, with joint and muscle pain being the most common, followed by fatigue and diarrhea, and, less frequently, neuropathy, weight loss, anemia, nausea, vomiting, and constipation. Three of the 5 patients considered these side effects intolerable, while 2 managed them with daily acetaminophen.
When asked to rate the importance of issues a new drug should address on a scale of 1 to 7 (with 1 being most important and 7 being least important), respondents prioritized maintaining QoL, providing a cure, and prolonging life, followed by delaying symptoms and improving ease of use. Reducing side effects and the importance of access to new treatment options were also valued, albeit to a lesser degree.
Respondents were asked to rate the level of side effects they would tolerate to extend survival by 2 months, 6 months, or 12 months, with side effects including nausea, fatigue, vomiting, and diarrhea. They reported varying tolerances, from mild effects for short-term extensions to severe effects for longer extensions. When asked about considerations for balancing the advantages and disadvantages of treatment, respondents mentioned factors such as the desire to extend life, maintaining comfort, keeping tumours stable, QoL, time with family, and the potential for new treatments.
Three of the 5 survey respondents had prior experience with durvalumab; these patients reported fatigue as the main adverse effect of durvalumab, with 1 experiencing hives. Respondents described the drug as beneficial, noting it helped keep tumours stable while allowing them to feel normal. Durvalumab was rated much better for symptom management and disease progression compared to other therapies, with little or no difference in side effects and ease of use. All 3 respondents noted that they would recommend durvalumab be available to all patients who qualify for it.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NSCLC.
Unmet Needs
The clinical experts consulted for this review felt the overarching goal of NSCLC treatment is to cure the disease and in doing so, improve OS, reduce recurrence rates, improve QoL, and reduce the symptom burden. While recent treatment advancements have improved disease outlook, survival rates and disease burden remain suboptimal. According to the clinical experts, ICIs have achieved better results compared to chemotherapy alone; however, atezolizumab following adjuvant chemotherapy remains the only approved ICI regimen in the adjuvant resectable NSCLC setting in Canada and more than half of patients are ineligible for it due to low PD-L1 expression status. In the neoadjuvant setting, neoadjuvant nivolumab in combination with chemotherapy is currently funded. At the time of submission for this review in 2024, no perioperative ICI regimens had been approved for resectable NSCLC in Canada. Note that since April 2025, CDA-AMC has issued a positive reimbursement recommendation for perioperative pembrolizumab for resectable NSCLC. The clinical experts noted that patient selection (e.g., based on histology, PD-L1 expression, disease stage, patient preference) is key when determining which patients will best respond to treatment regimens including the use of neoadjuvant, adjuvant, and perioperative approaches; currently, there are no robust head-to-head data to compare these treatment regimens.
Place in Therapy
The experts felt that perioperative durvalumab with neoadjuvant chemotherapy would add an alternative first-line treatment option for patients with resectable NSCLC. Compared to current treatment options, the clinical experts noted that perioperative durvalumab with neoadjuvant chemotherapy would address the underlying disease process in addition to potentially improving symptoms. The clinical experts anticipated that this additional treatment option would be especially important for those patients identified as potentially not achieving a complete response at the time of surgery; however, currently, there is no evidence to support this.
Patient Population
The clinical experts noted that patients with resectable NSCLC who are fit for systemic chemotherapy and immunotherapy without any EGFR mutations or ALK rearrangements would be best suited for perioperative durvalumab with neoadjuvant chemotherapy. Patients’ preferences should be taken into consideration and patients with questionable resectable disease or who are not fit for platinum-based chemotherapy may be less suited for this approach. A short time to treatment initiation is essential to achieve the best treatment results, and the requirement for EGFR and ALK testing may contribute to perioperative durvalumab with neoadjuvant chemotherapy treatment delay. The experts stated that while recent perioperative durvalumab with neoadjuvant chemotherapy studies have not suggested any specific subgroup differences, exploratory evidence suggests that patients with stage III disease may benefit more from a perioperative immunotherapy approach. Further, it is possible that a subset of patients with specific markers may respond better to perioperative durvalumab with neoadjuvant chemotherapy; however, validated markers are not currently available.
Assessing the Response to Treatment
Outcome metrics for resectable NSCLC used in clinical practice are typically aligned with those used in clinical trials. The clinical experts noted that in clinical trials, patient response to treatment is assessed at the time of surgery using pCR, using survival measures such as OS and DFS according to the RECIST criteria, and by disease symptoms. In clinical practice, response at the time of surgery using pCR is the first step to assess benefit from therapy followed by survival, QoL outcomes (such as PROs), and AEs. In clinical practice, as in trials, clinical assessments are performed before each cycle of therapy. However, assessments of disease response and recurrence usually occur every 3 months to 6 months during treatment, every 6 months in the first 2 years following the last treatment cycle, and then annually thereafter, which is generally less frequent than in clinical trials.
Discontinuing Treatment
Clinical experts consulted for this review indicated that perioperative durvalumab with neoadjuvant chemotherapy should be discontinued in the event of disease progression (including the tumour no longer being resectable, metastasis, and/or tumour growth) or recurrence, significant side effects such as immune-related side effects that can be life-threatening or permanent, or upon a patient’s request.
Prescribing Considerations
Clinical experts indicated that treatment with perioperative durvalumab with neoadjuvant chemotherapy should be prescribed and managed in outpatient clinics or specialty clinics by a multidisciplinary thoracic team of thoracic surgeons, medical oncologists, and supportive staff such as nursing and allied health.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Two clinician groups provided input for this review: LCC Drug Advisory Committee (18 clinicians contributed to the input) and OH-CCO (5 clinicians contributed to the input). Overall, the input was aligned with that of the clinical experts consulted by CDA-AMC.
Both clinician groups and the clinical experts consulted agreed that the treatment paradigm for patients with resectable NSCLC who do not have EGFR mutations or ALK rearrangements consists of 2 main phases: neoadjuvant and adjuvant. Neoadjuvant therapy, administered before surgery, includes chemotherapy and ICI. OH-CCO and LCC recommend neoadjuvant platinum-based chemotherapy combined with ICI (nivolumab) for 3 cycles. Additionally, LCC noted an alternative involving chemotherapy with ICI (durvalumab) for 4 cycles. Following surgery, adjuvant therapy involves chemotherapy and/or ICI, while radiation therapy is reserved for specific cases such as positive margins, as noted by the clinical experts consulted for this review.
The common treatment goals identified across clinician groups included achieving a cure, as measured by OS, and improving DFS. LCC and the clinical experts emphasized reducing recurrence rates and enhancing QoL.
The most significant treatment gap identified by the 2 clinician groups and the clinical experts is the lack of data comparing the efficacy and role of adjuvant ICI following neoadjuvant therapy, including which patients might benefit from it. This uncertainty makes it challenging to determine the optimal strategy for improving survival outcomes, especially given that not all patients respond to current treatments. Presently, ICI in the adjuvant setting is only available to those with high PD-L1 expression.
Both clinician groups and the clinical experts stated that perioperative durvalumab combined with neoadjuvant chemotherapy would offer an alternative treatment approach to neoadjuvant nivolumab in combination with chemotherapy for resectable NSCLC. The clinical experts emphasized its importance, especially for patients who do not achieve a complete pathological response to neoadjuvant therapy, highlighting the potential value of adjuvant ICI in such cases.
According to the clinician groups, the best-suited patients for perioperative durvalumab in combination with neoadjuvant chemotherapy are those with resectable NSCLC, no EGFR mutations or ALK rearrangements, and no contraindications to ICI. They emphasized the role of CT scans for monitoring, with imaging recommended before and after surgery and at regular intervals during and after treatment to check for recurrence. In clinical practice, treatment response is assessed through clinical symptoms, survival outcomes (OS and DFS), and pathological response at surgery (pCR and MPR), as highlighted by the clinical expert.
Both clinician groups indicated that factors for discontinuing treatment primarily include disease progression and significant side effects, with patient preferences also playing a key role. For treatment with perioperative durvalumab with neoadjuvant chemotherapy, patients should be treated in an outpatient setting under the supervision of a medical oncologist, or pulmonologist experienced in the management of thoracic malignancies.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation question | Clinical expert response |
|---|---|
Relevant comparators | |
In the phase III AEGEAN trial, durvalumab in combination with chemotherapy was administered as neoadjuvant treatment, followed by treatment with durvalumab monotherapy after surgery, in patients with resectable NSCLC and no EGFR mutations or ALK rearrangements. In Canada, the following options are funded for neoadjuvant and adjuvant treatment in patients with resectable NSCLC and no EGFR mutations or ALK rearrangements:
| This was a comment from the drug programs to inform pERC deliberations. The CDA-AMC review team notes that in February 2025, CDA-AMC issued a conditional positive final recommendation for pembrolizumab for the adjuvant treatment of adult patients with stage IB (tumour stage IIA [T2a] ≥ 4 cm), stage II, or stage IIIA NSCLC who have undergone a complete resection and platinum-based chemotherapy and whose tumours have a PD-L1 tumour proportion score of less than 50%. In April 2025, CDA-AMC issued a conditional positive final recommendation for pembrolizumab for the treatment of adult patients with resectable stage II, stage IIIA, or stage IIIB (T3-4N2) NSCLC in combination with platinum-containing chemotherapy as neoadjuvant treatment, and then continued as monotherapy as adjuvant treatment. |
Considerations for initiation of therapy | |
In the AEGEAN study protocol, patients were eligible with resectable stage IIA to stage IIIB NSCLC, according to version 8 of the IASLC Staging Manual in Thoracic Oncology. In the 2023 NEJM publication, it noted the AJCC eighth edition was used to determine funding eligibility for the trial. In practice in Canada, the eighth edition of AJCC staging is commonly used. What staging system should be used, and are there any meaningful differences between the IASLC and AJCC staging systems? | The clinical experts noted that the IASLC Staging Manual in Thoracic Oncology and the eighth edition of AJCC staging can be considered interchangeable. |
Under what clinical circumstances might perioperative durvalumab be preferred over neoadjuvant nivolumab, adjuvant atezolizumab, or adjuvant pembrolizumab? | Given the lack of robust head-to-head comparisons, the clinical experts noted that at this time, there are no specific criteria to choose perioperative durvalumab with neoadjuvant chemotherapy over neoadjuvant nivolumab, adjuvant atezolizumab, or adjuvant pembrolizumab. The clinical experts noted that patient selection (e.g., based on histology, PD-L1 expression, disease stage, patient preference) is key when determining which patients will best respond to treatment regimens, including the use of neoadjuvant, adjuvant, and perioperative approaches. |
Can durvalumab be administered with alternate chemotherapy if a patient cannot receive or tolerate platinum-based chemotherapy in the neoadjuvant phase? | The clinical experts noted that there are no data to support the use of durvalumab in combination with alternative chemotherapy instead of the platinum-based chemotherapy in the neoadjuvant phase. |
Patients with mixed small cell and NSCLC histology were excluded from the AEGEAN study. Should these patients also be excluded from public funding eligibility? Should patients with large cell neuroendocrine tumours be eligible? | The AEGEAN study excluded patients with mixed small cell and NSCLC histology and included in total 5 patients with large cell carcinoma (subtypes unspecified). Both experts agreed that large cell neuroendocrine tumours may be treated with regimens used in small cell lung cancer and NSCLC and if a patient is on an NSCLC regimen, they should be eligible for perioperative durvalumab. The patients should also be considered eligible to undergo surgery. One of the clinical experts agreed that results of the AEGEAN study could also be generalized to mixed small cell and NSCLC, if NSCLC is the dominant histology being treated. The other clinical expert noted that results of the AEGEAN study should not be generalized to mixed small cell and NSCLC, regardless of whether NSCLC is the dominant histology being treated. |
Consider alignment of eligibility criteria with nivolumab (April 18, 2023, pERC recommendation). | This was a comment from the drug programs to inform pERC deliberations. |
Considerations for prescribing of therapy | |
PAG would like to inform pERC that most jurisdictions use weight-based dosing up to a cap for durvalumab (e.g., 15 mg/kg to 20 mg/kg [up to a maximum of 1,500 mg] every 3 weeks in combination with chemotherapy, then 20 mg/kg [up to a maximum of 1,500 mg] every 4 weeks). | This was a comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Patients with ECOG PS > 1 were excluded from the AEGEAN trial. Should patients with ECOG PS > 1 be eligible? Note: For use of nivolumab plus platinum-double chemotherapy in the neoadjuvant setting, the clinical experts emphasized the need for patients to have robust performance status to be eligible to receive the treatment, given that patients with less clinical reserve will be susceptible to adverse events that may render them ineligible for curative-intent surgery. | The clinical experts noted that individuals with an ECOG PS of 2 may benefit from perioperative durvalumab with neoadjuvant chemotherapy. |
Should patients currently on neoadjuvant nivolumab plus chemotherapy be eligible for a switch to perioperative durvalumab? | Clinical experts noted that it is preferable to decide on the treatment approach upfront to ensure adherence to protocols; however, if perioperative approaches, such as perioperative durvalumab, become funded therapy, the clinical experts agreed that patients should have the option to switch treatment approaches in a time-limited fashion. Both clinical experts suggested that patients already receiving treatment who are less than 3 months postsurgery should have the option to switch therapy to a perioperative approach. |
Funding algorithm (oncology only) | |
Drug may change place in therapy of comparator drugs | This was a comment from the drug programs to inform pERC deliberations. |
Complex therapeutic space with multiple lines of therapy, subpopulations, or competing products | This was a comment from the drug programs to inform pERC deliberations. |
Under what circumstances will perioperative durvalumab be chosen over alternative PD-1 or PD-L1 inhibitors in the curative setting? | Given the lack of robust head-to-head comparisons, the clinical experts noted that at this time, there are no specific criteria to choose perioperative durvalumab with neoadjuvant chemotherapy over alternative PD-1 or PD-L1 inhibitors in the curative setting. However, the clinical experts noted that preliminary results from indirect treatment comparisons suggest that perioperative therapy might offer greater benefit compared to neoadjuvant treatment.52 |
Can pERC confirm that patients would be eligible for downstream PD-1 or PD-L1 inhibitors provided that disease recurrence occurs more than 6 months from the last dose of adjuvant durvalumab? | While there are no data to support the use of downstream PD-1 or PD-L1 inhibitors following disease recurrence more than 6 months from the last dose of adjuvant durvalumab, the clinical experts noted that this would likely occur in clinical practice. The selection of downstream PD-1 or PD-L1 inhibitors would be based on tumour characteristics, disease-free interval, and clinical judgment. |
System and economic issues | |
The sponsor estimates 208, 244, and 264 patients would receive treatment with perioperative durvalumab in year 1 to year 3, respectively, with a 3-year incremental budget impact of $65.2 million ($18.6 million in year 1, $22.3 million in year 2, and $24.3 million in year 3). PAG is concerned that if these estimates are low, there would be a resulting higher budget impact. | This was a comment from the drug programs to inform pERC deliberations. |
Nivolumab, atezolizumab, and pembrolizumab have confidential prices negotiated. | This was a comment from the drug programs to inform pERC deliberations. |
AJCC = American Joint Committee on Cancer; CDA-AMC = Canada’s Drug Agency; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IASLC = International Association for the Study of Lung Cancer; NEJM = New England Journal of Medicine; NSCLC = non–small cell lung cancer; PAG = Provincial Advisory Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of perioperative durvalumab, 1,500 mg administered as an IV infusion every 3 weeks in combination with neoadjuvant chemotherapy before surgery and every 4 weeks as monotherapy after surgery for the treatment of resectable NSCLC in patients without any known EGFR mutations or ALK rearrangements. The focus will be placed on comparing durvalumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of durvalumab is presented in 2 sections with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal study identified in the systematic review
3 ITCs.
Systematic Review
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | AEGEAN study |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind, placebo-controlled, randomized, multicentre, international study |
Locations | A total of 183 sites (across 28 countries) randomized at least 1 patient into the global cohort:
|
Patient enrolment dates | First patient enrolled: December 6, 2018 Last patient enrolled: March 18, 2022 |
Randomized (N) | Randomized, N = 802 (ITT population)
mITT population, N = 740 (excluded patients with known EGFR mutations or ALK rearrangements)
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Patients received durvalumab before and following surgery, in combination with a standard of care platinum-based doublet chemotherapy regimen before surgery. Durvalumab (perioperative) Patients were to receive 1,500 mg durvalumab every 3 weeks for 4 cycles before surgery and every 4 weeks for up to 12 cycles following surgery, until there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion that was met. Neoadjuvant chemotherapy (standard of care, by tumour histology)
|
Comparator(s) | Patients received placebo before and following surgery, in combination with a standard of care platinum-based doublet chemotherapy regimen before surgery. Placebo Patients were to receive placebo every 3 weeks for 4 cycles before surgery and every 4 weeks for 12 cycles following surgery until there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion that was met. Neoadjuvant chemotherapy (standard of care, by tumour histology)
|
Study duration | |
Screening phase | 28 days |
Treatment phase | Neoadjuvant period: 12 weeks followed by surgery or treatment discontinuation, whichever occurred first Adjuvant period: 48 weeks (treatment starting within 10 weeks of surgery, except for patients receiving postoperative radiation therapy, which must have been started within 8 weeks of surgery; durvalumab or placebo must have been started within 3 weeks from the end of postoperative radiation therapy) or treatment discontinuation, whichever occurred first |
Follow-up phase | RECIST 1.1 assessments took place once every 12 weeks ± 1 week (relative to the date of surgery or preplanned surgery) until week 192 (approximately 4 years), and then once every 47 weeks thereafter until RECIST 1.1–defined radiological PD, consent withdrawal, or death. |
Outcomes | |
Primary end point |
|
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
Safety:
|
Publication status | |
Publications | Heymach JV, Harpole D, Mitsudomi T, et al. Perioperative Durvalumab for Resectable Non-Small-Cell Lung Cancer. N Engl J Med. Nov 2 2023;389(18):1672 to 1684.56 |
AE = adverse event; AESI = adverse event of special interest; AJCC = American Joint Committee on Cancer; AUC 5 = area under the serum drug concentration-time curve 5; AUC 6 = area under the serum drug concentration-time curve 6; CTCAE = Common Terminology Criteria for Adverse Events; DFS = disease-free survival; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; EQ VAS = EQ visual analogue scale; HRQoL = health-related quality of life; IASLC = International Association for the Study of Lung Cancer; ITT = intention to treat; mITT = modified intention to treat; MPR = major pathological response; NSCLC = non–small cell lung cancer; OS = overall survival; pCR = pathological complete response; PD = progressive disease; PGI-S = Patient Global Impression of Severity; PRO = patient-reported outcome; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours 1.1; TC = tumour cell.
aIASLC is the group responsible for conducting research to inform future versions of the AJCC. Version 8 of the IASLC Staging Manual in Thoracic Oncology57 was endorsed and published by the AJCC as the AJCC Cancer Staging Manual, eighth edition.58 The 2 are considered interchangeable.
Sources: AEGEAN trial clinical study protocol and AEGEAN trial statistical analysis plan.59,60 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The AEGEAN trial is an ongoing, randomized, double-blind, placebo-controlled, multicentre, phase III study aimed at evaluating the efficacy, activity, and safety of perioperative durvalumab with neoadjuvant chemotherapy in patients with resectable NSCLC. Patients were recruited from 183 international sites, including 5 sites in Canada. Patients were randomized 1:1 to receive either durvalumab and platinum-based chemotherapy before surgery, followed by durvalumab postsurgery (arm 1) or placebo and platinum-based chemotherapy before surgery, followed by placebo postsurgery (arm 2) (Figure 1). Randomization was stratified by disease stage (stage II versus stage III) and PD-L1 expression status (< 1% versus ≥ 1%).
The primary objectives were to compare pCR and EFS in patients treated with perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant chemotherapy with placebo. The DCO dates for the results presented include the following:
the first IA (interim analysis 1 [IA1]) of pCR (DCO date of January 14, 2022)
the final analysis of pCR (DCO date of November 10, 2022)
the first IA of EFS (EFS IA1 DCO date of November 10, 2022)
the second IA of EFS (EFS IA2 DCO date of May 10, 2024).
At Screening
Baseline screening assessments were performed in the 28 days before randomization. During this period, informed signed consent was obtained, tumour samples were collected and sent for centralized PD-L1, EGFR, and ALK testing, patient eligibility was determined, and demographic and characteristic data were collected.
Neoadjuvant Phase
Treatment was to begin on day 1 of the study. Surgery was to occur within and including 40 days of the last neoadjuvant treatment administration or it was considered a delay.
Adjuvant Phase
PORT was allowed for patients in which it was indicated according to local guidance. The initiation of adjuvant treatment was scheduled as soon as clinically feasible and within 10 weeks after surgery or within 3 weeks after the completion of PORT.
Discontinuation and Lost to Follow-Up
Patients discontinued the study treatment if they withdrew consent, if they had an AE that contraindicated further dosing (according to the study investigator), if they had an AE that met the criteria for discontinuation, if they were or intended to become pregnant, if they were noncompliant with the study, if they underwent the initiation of alternative anticancer therapy, if they had a local or distant recurrence, if they had clinical progression, or if they were unable to undergo or complete surgery for reasons other than progressive disease.
Patients were considered lost to follow-up only if no contact had been established by the time the study was completed, such that there was insufficient information to determine the patient’s status at that time.
Figure 1: Schematic of AEGEAN Study Design
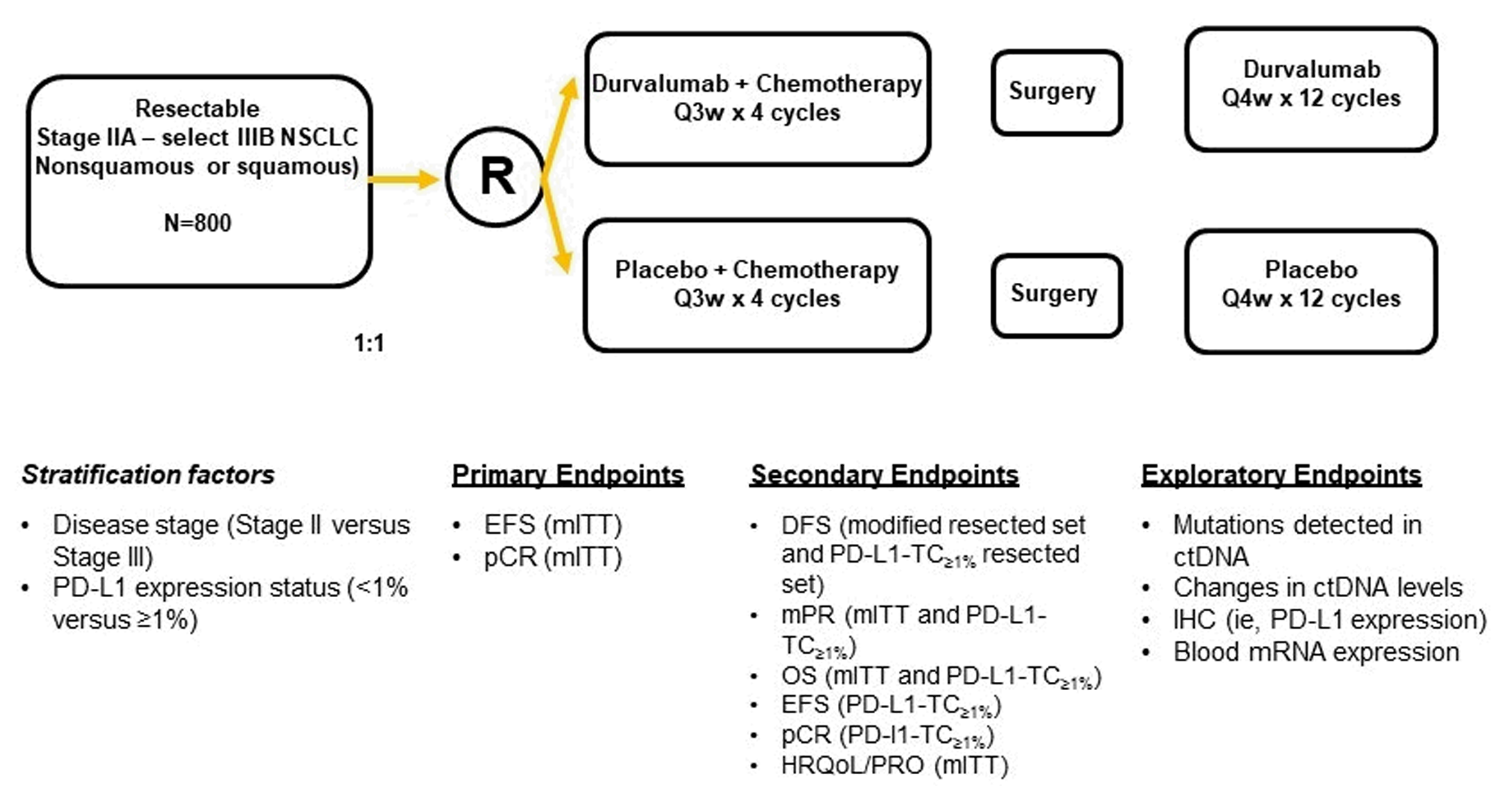
ctDNA = circulating tumour DNA; DFS = disease-free survival; EFS = event-free survival; HRQoL = health-related quality of life; IHC = immunohistochemistry; mITT = modified intention to treat; mPR = major pathological response; mRNA = messenger ribonucleic acid; NSCLC = non–small cell lung cancer; OS = overall survival; pCR = pathological complete response; Q3w = every 3 weeks; Q4w = every 4 weeks; R = randomize; TC = tumour cell.
Note: PD-L1 TC ≥ 1% = the expression of PD-L1 on tumour membrane, at any intensity, in ≥ 1% of tumour cells.
Source: AEGEAN study protocol.60
Populations
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria are provided in Table 5. Briefly, the trial enrolled patients with newly diagnosed, previously untreated, resectable NSCLC (stage IIA to stage IIIB [N2 node stage] disease, either squamous or nonsquamous). At enrolment, patients had to be aged 18 years or older and be candidates for planned surgical treatment with lobectomy, sleeve resection, or bilobectomy. Enrolment was restricted to those with a WHO and/or ECOG PS score of 0 or 1, adequate organ and marrow function, and no prior exposure to immune-mediated therapies. To confirm patient eligibility, a central laboratory was used to analyze EGFR (unless known KRAS mutation), ALK (unless known KRAS mutation or squamous cell carcinoma), and PD-L1 status. With enrolment ongoing, the protocol was amended to exclude patients with tumours classified as T4 for any reason other than size (> 7 cm), whose planned surgery at enrolment was pneumonectomy, or who had documented test results that confirmed the presence of an EGFR mutation (confirmed by central testing) or ALK rearrangements (confirmed by local or central testing). Additional key exclusion criteria were previous exposure to anti–PD-L1, anti–PD-1, or anticytotoxic T-lymphocyte antigen 4 antibodies, uncontrolled intercurrent illness, previously documented autoimmune or inflammatory disorders, the existence of more than 1 primary tumour, and sublobar resections as planned surgery at the time of enrolment.
Interventions
The main study intervention was 1,500 mg fixed-dose durvalumab or placebo (i.e., sterile saline or dextrose solution) administered as a 1-hour infusion (up to a maximum of 8 hours in the case of interruptions) every 3 weeks up to a maximum of 4 cycles before surgery and every 4 weeks for 12 cycles following surgery. If a patient’s weight fell to 30 kg or less, a weight-based dosing equivalent to 20 mg/kg of durvalumab was given at the same interval until the patient’s weight improved to greater than 30 kg. In both treatment arms, chemotherapy was given in combination with the main study intervention during the neoadjuvant treatment phase. When chemotherapy was given on the same day as the main study intervention, the main study intervention was administered before administering chemotherapy. The choice of platinum-doublet chemotherapy regimen included 4 options and was selected based on the patient’s histology and at the discretion of the investigator:
squamous tumour histology — carboplatin plus paclitaxel: carboplatin area under the serum drug concentration-time curve 6 and paclitaxel 200 mg/m2 via IV infusion on day 1 of each 3-week cycle, for 4 cycles
squamous tumour histology — cisplatin plus gemcitabine: cisplatin 75 mg/m2 via IV infusion on day 1 of each 3-week cycle, for 4 cycles, and gemcitabine 1,250 mg/m2 via IV infusion on day 1 and day 8 of each 3-week cycle, for 4 cycles
nonsquamous tumour histology — pemetrexed plus cisplatin: pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 via IV infusion on day 1 of each 3-week cycle, for 4 cycles
nonsquamous tumour histology — pemetrexed plus carboplatin: pemetrexed 500 mg/m2 and carboplatin area under the serum drug concentration-time curve 5 via IV infusion on day 1 of each 3-week cycle, for 4 cycles.
Dose delays were permitted for durvalumab with chemotherapy and placebo with chemotherapy; however, dose reductions were only permitted for chemotherapy regimens.
The standardized course of PORT included but was not limited to doses ranging from 50 Gy to 60 Gy, 1.8 Gy to 2 Gy per fraction, 5 fractions a week; for patients with positive margins of disease (R1), PORT included but was not limited to doses ranging from 60 Gy to 66 Gy, 1.8 Gy to 2 Gy per fraction, 5 fractions a week.
Approved concomitant medications or treatments included those deemed necessary to provide adequate prophylactic or supportive care (e.g., acetaminophen, diphenhydramine), those considered best supportive care (including antibiotics or nutritional support), and inactive viruses (such as the influenza vaccine), or those deemed necessary by the investigator for the patient’s safety and well-being. Rescue medications (steroids and other immunosuppressants) were made available to manage imAEs that could potentially be experienced by patients on durvalumab. Permitted immunosuppressants were infliximab (e.g., for colitis) and mycophenolate (e.g., for hepatitis).
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted by CDA-AMC and the perspectives of patients, clinician groups, and drug programs. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform deliberations by the CDA-AMC expert committee and finalized this list of end points in consultation with members of the expert committee. In summary, EFS, OS, pCR, and HRQoL (measured with the EORTC QLQ-C30 tool) were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE. Results of MPR and DFS are presented in the Clinical Review Report to aid in clinical decision-making but were not appraised using GRADE. pCR was deemed to be the most relevant response outcome because it is commonly used in clinical practice; MPR was noted by the clinical experts consulted for this review to be more commonly used in clinical trials than in clinical practice (while MPR results have been summarized in this Clinical Review Report, they have not been assessed in the GRADE assessment). While EFS reflected the whole study period for all randomized patients (i.e., events occurring in the neoadjuvant, adjuvant, and follow-up periods of the study), DFS was a less comprehensive assessment reflecting the period after complete surgical resection for patients in which this was achieved; EFS was included in the GRADE assessment while DFS was summarized in this Clinical Review Report but not assessed using the GRADE tool. Similarly, results from the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13 (EORTC QLQ-LC13), a 13-item module about lung cancer, are presented to aid in clinical decision-making; however, they were not appraised using GRADE because the EORTC QLQ-C30 was deemed to be more relevant by clinical experts consulted for this review.
Table 6: Outcomes Summarized From the AEGEAN Trial
Outcome measure | Time point | End point | Outcome included in the CDA-AMC GRADE assessment? |
|---|---|---|---|
pCR | At the time of surgical resection from pCR FA | Coprimary end pointa | Yes |
MPR | At the time of surgical resection from pCR FA | Key secondary end pointa | No |
EFS | At EFS IA2 DCO (May 10, 2024) and at 12 months, 24 months, and 36 months | Coprimary end pointa | At 12 months and 36 months |
DFS | At EFS IA2 DCO (May 10, 2024) and at 12 months, 24 months, and 36 months | Key secondary end pointa | No |
OS | At EFS IA2 DCO (May 10, 2024) and at 12 months, 24 months, 36 months, and 48 months | Key secondary end pointa | At 12 months and 48 months |
EORTC QLQ-C30 | From baseline to week 12 and from adjuvant baseline to week 44 | Secondary end point | Change in global health status and QoL during both the neoadjuvant and adjuvant periods |
EORTC QLQ-LC13 | Baseline to EFA IA1 DCO | Secondary end point | No |
Harms | Through EFS IA2 DCO | Safety outcome | Immune-mediated AEs and serious AEs |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; DCO = data cut-off; DFS = disease-free survival; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FA = final analysis; GRADE = Grading of Recommendations Assessment, Development and Evaluation; IA1 = interim analysis 1; IA2 = interim analysis 2; MPR = major pathological response; OS = overall survival; pCR = pathological complete response; QoL = quality of life; SoF = summary of findings.
aStatistical testing for these end points was adjusted for multiple comparisons (hierarchical testing).
Sources: AEGEAN study EFS IA1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Tumour evaluation using RECIST 1.1 was conducted at screening (within 28 days before randomization), after completion of the neoadjuvant period (before surgery), then postsurgery every 12 weeks (± 1 week) until week 48, then every 24 weeks (± 2 weeks) until week 192 (approximately 4 years), and then every 48 weeks (± 2 weeks) thereafter, until local or distant recurrence (as determined by the investigator using RECIST 1.1), consent withdrawal, or death.
Patient-monitoring data, including the reporting of AEs, were collected at screening, at the same time as neoadjuvant and adjuvant durvalumab or placebo administration, after completion of the neoadjuvant period (before surgery), postsurgery (within 10 weeks of surgery), and at month 1, month 2, and month 3 following the last dose of durvalumab or placebo.
PROs followed the same schedule of assessments as patient monitoring, with 1 additional assessment taken 6 months following the last dose of durvalumab or placebo.
Pathological Complete Response
pCR was a coprimary outcome in the AEGEAN study and was defined as the absence of any residual viable tumour at the time of surgical resection in the primary lung lesion and all sampled lymph nodes. The analysis of pCR was based on a blinded assessment per central pathology review. The estimated between-group difference threshold at which clinical experts consulted for this review deemed the pCR difference to be clinically meaningful in resectable NSCLC was 10%.
Major Pathological Response
MPR, defined as 10% or less residual viable tumour tissue in the lung primary tumour at the time of resection, was a key secondary end point in the AEGEAN study. The analysis of MPR was based on assessment per central pathology review and recorded as yes, no, or not evaluable. Clinical experts consulted for this review advised that a between-group difference of 10% in MPR is considered clinically meaningful.
Event-Free Survival
EFS was the second coprimary outcome in the AEGEAN study. EFS was defined as the time from randomization to the first of any of the following: local or distant recurrence; death due to any cause (the event date is the date of death); or progressive disease that precludes surgery (the event date is the date of this determination) or progressive disease discovered and reported by the investigator upon attempting surgery that prevents completion of surgery (the event date is the date of the first attempt at surgery). The EFS analysis was based on a blinded independent central review (BICR) per RECIST 1.1. Clinical experts consulted for this review advised that a between-group difference of 5% to 10% is considered clinically meaningful.
Disease-Free Survival
DFS was a key secondary outcome and was defined as the time from the date of surgery until the first of either of the following: the date of disease recurrence (local or distant), or the date of death due to any cause. The analysis of DFS was based on BICR per RECIST 1.1. Clinical experts consulted for this review advised that a DFS between-group difference of 5% to 10% is considered clinically meaningful.
Overall Survival
The final key secondary outcome in the AEGEAN trial was OS. OS was defined as the time from randomization until death due to any cause, regardless of whether the patient withdraws from randomized therapy or receives another anticancer therapy. Clinical experts consulted for this review advised that an OS between-group difference of 5% to 10% is considered clinically meaningful.
HRQoL End Points
To compare disease-related symptoms and HRQoL, data from the EORTC QLQ-C30 and EORTC QLQ-LC13 measures were collected as other secondary outcomes in the AEGEAN trial.61,62 Descriptions of each measure are as follows (further details are in Table 7).
The EORTC QLQ-C30 is a multidimensional, cancer-specific, self-administered measure of HRQoL consisting of 30 questions across 5 multi-item functional scales (physical, role, emotional, cognitive, and social); 3 multi-item symptom scales (fatigue, pain, and nausea and vomiting); a 2-item global QoL scale; 5 single items assessing additional symptoms commonly reported by patients with cancer (dyspnea, loss of appetite, insomnia, constipation, and diarrhea), and 1 item on the financial impact of the disease. Each item is evaluated using 4-point and 7-point Likert scales; raw scores for each scale are computed as the average of the items that contribute to a particular scale, and each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better HRQoL on the global QoL scale.61
The EORTC QLQ-LC13 is a 13-item lung cancer–specific questionnaire. The module comprises both multi-item and single-item measures of lung cancer–associated symptoms (i.e., coughing, hemoptysis, dyspnea, and pain) and side effects from conventional chemotherapy and radiotherapy (i.e., hair loss, neuropathy, sore mouth, and dysphagia).62 A high score represents a high level of symptom burden.
Changes in scores compared with baseline were evaluated for the scales and items from EORTC QLQ-C30 and EORTC QLQ-LC13 in the neoadjuvant period and for scales and items from EORTC QLQ-C30 in the adjuvant period. Baseline in the neoadjuvant period was defined as the last predose assessment (i.e., neoadjuvant cycle 1, day 1, predose assessment; in the absence of the cycle 1, day 1, predose assessment, the latest assessment before treatment can be used — namely, screening). Baseline in the adjuvant period was defined as the latest measurement after surgery but before the first dose of adjuvant treatment.
A between-group minimal important difference (MID) in the EORTC QLQ-C30 global health status and QoL score has previously been estimated for patients with lung cancer as an increase or decrease of 4 points for improvement and deterioration, respectively.63 No MID for the EORTC QLQ-LC13 in patients with NSCLC was identified.
Harms End Points
To compare the safety and tolerability profile of study treatments, data on AEs were collected from the date of informed consent until 90 days after the last dose of study treatment. AEs were coded based on the Medical Dictionary for Regulatory Activities (version 25.1) and graded for severity according to Common Terminology Criteria for Adverse Events (version 5.0). AE data were evaluated according to the following categories: all AEs (including those causality-related to study treatment), AEs of Common Terminology Criteria for Adverse Events grade 3 or higher, AEs leading to dose modification, AEs with an outcome of death, SAEs, and discontinuation due to AEs.
Adverse events of special interest (AESIs) and adverse events of potential interest (AEPIs) included those from the durvalumab clinical development program:
AESIs were defined as AEs with a likely inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab and requiring more frequent monitoring and/or interventions such as corticosteroids, immunosuppressants, and/or endocrine therapy.
AEPIs were defined as AEs that could have a potential inflammatory or immune-mediated pathophysiological basis resulting from the mechanism of action of durvalumab but were more likely to have occurred due to other pathophysiological mechanisms; thus, the likelihood of the event being inflammatory or immune-mediated in nature is not high and/or is most often explained by the other causes.
ImAEs were identified from both AESIs and AEPIs based on programmatic rules that consider interventions involving systemic steroid therapy, immunosuppressant use, and/or endocrine therapy.
AEs were further categorized based on the time point in which they occurred:
overall period — includes AEs occurring while receiving treatment, a period which is defined as occurring between the first dose of study treatment until the earliest of the maximum of (last dose of study treatment or surgery) plus 90 days, the date of the DCO, or the date of the first dose of subsequent anticancer therapy (which does not include radiation therapy)
neoadjuvant period — includes AEs between the date of the first dose and the day before surgery, or for patients without surgery, up to the earliest of either 90 days after the date of the last dose of neoadjuvant treatment, or the first dose of subsequent anticancer therapy
postsurgery period — includes AEs between the date of surgery (including the day of surgery) and the earliest of either the date of surgery plus 90 days, or the first dose of subsequent anticancer therapy
adjuvant period — includes AEs between the date of the first dose of study treatment postsurgery and the earliest of either 90 days following the date of the last dose of adjuvant treatment, or the first dose of subsequent anticancer therapy.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusion about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | Cancer-specific, self-reported measure of HRQoL A 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, cognitive, and social); 3 multi-item symptom scales (fatigue, pain, and nausea and vomiting); and 5 single items (i.e., dyspnea, appetite loss, sleep disturbance, constipation, and diarrhea); these are measured on a 4-point response scale. The instrument also contains an item assessing the perceived financial impact of the disease and treatment and two 7-point response scales pertaining to global health and QoL. A higher score for functional scales and for global health status represents better functioning ability or HRQoL. A higher score for symptom scales represents a worsening of symptoms.61 | In studies with patients with lung cancer Validity: Moderate to strong correlations between the 5 EORTC QLQ-C30 functioning scales (r, 0.41 to 0.77), between FACT-G and EORTC QLQ-C30 scales (r, 0.64 to 0.76), between HADS and all EORTC QLQ-C30 functioning scales (r, 0.28 to 0.75), and between the BPI scales and all EORTC QLQ-C30 scales except for nausea and vomiting (r, 0.20 to 0.72),64 supporting convergent validity. Known-groups approach: Able to differentiate across different measures of cancer severity — cancer stages (effect size = 0.49), ECOG PS (effect size = 0.65), and self-reported health status (effect size = 1.39).65 Reliability: The Cronbach alpha ranging from 0.56 to 0.93 with 7 scales having acceptable internal consistency (alpha > 0.70)66 Responsiveness: Group differences (improved vs. deteriorated based on ECOG PS) over 28 days between periods of pretreatment and while receiving treatment showed a statistically significant difference in global QoL (P < 0.01) scale. No such difference was identified in patients whose ECOG PS remained unchanged.62 | A recent study63 analyzed MIDs for the EORTC QLQ-C30 using data from 21 phase III trials involving 13,015 patients across 9 cancer types (brain, colorectal, advanced breast, head and/or neck, lung, mesothelioma, melanoma, ovarian, and prostate). It found that anchor-based MIDs for global health status among patients with lung cancer was 4 points for improvement and −4 points for deterioration. |
EORTC QLQ-LC13 | The EORTC QLQ-LC13 is a 13-item lung cancer–specific questionnaire. It is typically used in conjunction with the core questionnaire, the EORTC QLQ-C30. The module comprises a multi-item scale to assess dyspnea, and a series of single items assessing pain, coughing, sore mouth, dysphagia, peripheral neuropathy, alopecia, and hemoptysis. All of the scales and single-item measures range in score from 0 to 100. A high score for the scales and single items represents a high level of symptomatology or problems.61,62 | Validity: Construct validity has been established between pain score and disease type (P < 0.001). Also, based on ECOG PS, construct validity was confirmed in dyspnea, coughing, and pain scores (P < 0.001).62 Correlation between spirometry result and dyspnea score was found to be weak (r = 0.24). The BPI intensity score and EORTC QLQ-LC13 pain score were found to be modestly correlated (r > 0.4).64 Reliability: There was good internal consistency reliability for the dyspnea multi-item scale (alpha = 0.81).62 Responsiveness: Dyspnea, coughing, and pain scores improved significantly over time between periods of pretreatment and while receiving treatment (P < 0.001 for all except for extra thoracic pain, which showed P < 0.05). Responsiveness of chest pain (P < 0.01), dyspnea (P < 0.001), and coughing (P < 0.001) to change in ECOG PS was also noted.62 | No relevant studies on MID in patients with NSCLC were identified. |
BPI = Brief Pain Inventory; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FACT-G = Functional Assessment of Cancer Therapy–General; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; QoL = quality of life; vs. = versus.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Considerations that informed the selection of efficacy outcomes to be summarized and assessed using GRADE include the following.
Survival outcomes were identified by the patient and clinician group input and specified by the clinical experts consulted for this review to include OS. EFS was a key input for the pharmacoeconomic model and was deemed relevant by the clinical experts consulted for this review. The clinical experts noted that outcomes at the longest follow-up were critical to informing long-term benefits or harms of the treatment and those at earlier time points were necessary to assess any sustained benefit or harm. As such, the earliest time point (12 months for both OS and EFS) and latest time point (48 months for OS and 36 months for EFS) were selected for GRADE.
HRQoL outcomes were identified by the patient and clinician groups as an important outcome and the clinical experts consulted for this review further specified the EORTC QLQ-C30 global health status and QoL to be the most informative scale.
pCR was identified as important by the clinical experts consulted for this review and is routinely used in clinical practice.
Safety and toxicity outcomes were identified as important outcomes by patient and clinical group input; imAEs and SAEs were identified as the most important safety outcomes by the clinical experts consulted for this review.
Statistical Analysis
The statistical analyses for all study end points included in this review are presented in Table 8.
Table 8: Statistical Analysis of Efficacy End Points in the AEGEAN Study
End point | Population | Statistical model | Adjustment factor | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|---|
pCR | mITT | Analysis performed using CMH test The effect of treatment was estimated by the difference in proportions between treatment arms, together with its corresponding 95% CI, a CI adjusted for the relevant alpha level, and P value. The CIs for the difference in proportions between arms was computed using stratified Miettinen-Nurminen’s confidence limits. | CMH test stratified by stratification factors (disease stage [stage II vs. stage III] and PD-L1 expression status at baseline [TC < 1% vs. TC ≥ 1%]) | Patients who were not evaluable per central pathology assessment or who did not have a surgical specimen were assigned as not having a response. | Sensitivity analyses and interaction testing were performed to assess the presence of quantitative interactions, and possible bias. |
MPR | mITT | Analyzed using the same statistical methodology as for pCR | CMH test stratified by stratification factors (disease stage [stage II vs. stage III] and PD-L1 expression status at baseline [TC < 1% vs. TC ≥ 1%]) | Same as for pCR | Sensitivity and subgroup analyses, as described for pCR, were repeated for MPR. |
EFS | mITT | Analysis performed using a stratified log-rank test. The effect of treatment was estimated by the HR together with its corresponding 95% CI, a CI adjusted for the relevant alpha level, and P value. The HR and CI were estimated from the stratified Cox proportional hazards model stratified log-rank test. | Adjusted for stratification factors (disease stage [stage II vs. stage III] and PD-L1 expression status at baseline [TC < 1% vs. TC ≥ 1%]) | Patients who had not experienced an EFS event at the time of analysis were censored at the time of the last disease assessment. If any of these events occurred after 2 or more consecutive missed visits after the first postsurgery RECIST 1.1 scan, then the patient was censored at the time of the latest evaluable disease assessment before the 2 missed visits. | Sensitivity analyses and interaction testing were performed to assess the presence of quantitative interactions, and possible bias. |
DFS | Modified resected set | Analyzed using the same statistical methodology as for the analysis of EFS | Adjusted for stratification factors (disease stage [stage II vs. stage III] and PD-L1 expression status at baseline [TC < 1% vs. TC ≥ 1%]) | Same as for EFS | Sensitivity analyses, as described for EFS, were repeated for DFS. |
OS | mITT | Analyzed using the same statistical methodology as for the analysis of EFS | Adjusted for stratification factors (disease stage [stage II vs. stage III] and PD-L1 expression status at baseline [TC < 1% vs. TC ≥ 1%]) | If only a partial date of death was known, the full date was imputed as the latest of the last date known to be alive + 1 day. If there was evidence of death but the date was entirely missing, it was treated as missing and censored to the last known alive date. Patients who had not experienced an OS event at the time of analysis were censored at the date last known alive. | A sensitivity analysis to assess the impact of COVID-19 deaths was performed. |
Change from baseline in symptoms (EORTC QLQ-C30 and EORTC QLQ-LC13) | Neoadjuvant period = mITT Adjuvant period = modified resected set | For the adjuvant period, a second summary was produced for adjuvant period visits where baseline was defined as the latest measurement after surgery but before the first dose of adjuvant treatment. MMRM models for global health status, fatigue, appetite loss, physical functioning, role functioning, dyspnea, chest pain, and cough were used to estimate changes from baseline and difference between treatment arms, by visit and on average during the neoadjuvant period (for EORTC QLQ-C30 and EORTC QLQ-LC13) and the adjuvant period (for EORTC QLQ-C30), with covariate adjustment for baseline score. An assessment of differences between treatment arms was performed descriptively. | NA | No data imputation | Not performed |
CI = confidence interval; CMH = Cochran Mantel-Haenszel; DFS = disease-free survival; EFS = event-free survival; EOTRC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EOTRC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; FA = final analysis; HR = hazard ratio; IA = interim analysis; IA1 = interim analysis 1; mITT = modified intention to treat; MMRM = mixed model of repeated measures; MPR = major pathological response; MTP = multiple testing procedure; NA = not applicable; OS = overall survival; pCR = pathological complete response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; TC = tumour cell; vs. = versus.
Note: The AEGEAN trial is an ongoing study. Per the MTP, DFS and OS will be formally assessed at subsequent IA and/or FA.
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study statistical analysis plan.24,59 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The study planned to screen approximately 1,333 patients to randomize approximately 800 eligible patients in the ITT population and approximately 740 patients in the mITT population. The study was sized to characterize the EFS and pCR benefit of the perioperative durvalumab with neoadjuvant chemotherapy arm versus the placebo with neoadjuvant chemotherapy arm in patients with resectable stage IIA to stage IIIB NSCLC. Enrolment of patients occurred over approximately 4 years, with the first patient enrolled on December 6, 2018, and the last patient enrolled on March 18, 2022.
IAs were planned within the study for the primary end points of pCR (1 planned IA) and EFS (2 planned IAs). The alpha level allocated to pCR was controlled at the interim and final time points by the Lan-DeMets67 spending function with the O'Brien-Fleming boundary, where the significance level applied at the interim depends upon the proportion of information available.
A total of 0.5% alpha was allocated to the pCR end point. The final analysis for pCR was planned to occur when all patients (i.e., approximately 800 patients in the ITT population and 740 patients in the mITT population) had the opportunity to undergo surgery and complete the pathology assessment (where applicable). The planned IA of pCR (based on the mITT population of 400 patients) had a 55% power to detect a between-group difference of 12 percentage points at a 2-sided significance level of 0.008%.
A total of 4.5% alpha was initially allocated to the EFS end point. The first planned IA of EFS (based on 740 patients in the mITT population with 224 events) had a 50% power to show an HR for disease progression, recurrence, or death of 0.69 with a 2-sided significance level of 0.665%. The second planned IA of EFS (based on 740 patients in the mITT population with 296 events) had an 84% power to show an HR for disease progression, recurrence, or death of 0.68 with a 2-sided significance level of 1.9243%. The final EFS analysis is planned to occur when approximately 371 EFS events have been reported (approximately 50% maturity in the mITT population).
Statistical Test or Model
Both pCR and MPR were analyzed using the Cochran-Mantel-Haenszel test stratified by disease stage (stage II versus stage III) and PD-L1 expression status (< 1% versus ≥ 1%) in the mITT population. The effect of treatment was estimated by the difference in proportions between treatment groups, together with their corresponding CI and P values. The CIs for the difference in proportions between groups were estimated using Miettinen-Nurminen’s confidence limits.
For survival outcomes EFS, DFS, and OS, the effects of treatment were estimated by HRs together with their corresponding 95% CIs estimated from the stratified Cox proportional hazards model adjusted for disease stage (stage II versus stage III) and PD-L1 expression status (< 1% versus ≥ 1%). EFS and OS analyses were performed in the mITT population and DFS in the modified resected set. The Cox models were fitted using PROC PHREG with the Efron method to control for ties and the CI was calculated using a profile likelihood approach.
Additionally, KM plots of EFS, DFS, and OS were presented by treatment arm. Summaries of the number and percentage of patients experiencing an event and the type of event were provided along with the median EFS, DFS, and OS and associated 95% CI for each treatment. EFS, DFS, and OS rates at a landmark (12 months, 24 months, 36 months, 48 months, and 60 months, as relevant) along with 95% CIs were estimated using the KM method for each treatment arm.
PRO instruments (EORTC QLQ-C30 and EORTC QLQ-LC13) were summarized descriptively using change from baseline presented separately for the neoadjuvant period (in the mITT population) and the adjuvant period (in the modified resected set population). In the absence of the cycle 1, day 1, predose assessment, the latest assessment before treatment was used (i.e., screening). For the adjuvant period (EORTC QLQ-C30 only), a second summary was produced on the modified resected set for adjuvant period visits where the baseline was defined as the latest measurement after surgery but before the first dose of adjuvant treatment. Change from baseline was also derived using a mixed model of repeated measures analysis for the following scales and items:
EORTC QLQ-C30 symptoms — fatigue and appetite loss
EORTC QLQ-C30 function — physical and role
EORTC QLQ-C30 global health status and QoL
EORTC QLQ-LC13 symptoms — dyspnea, chest pain, and cough.
When fewer than 20 patients were present at a time point in either arm, that time point was excluded from the mixed model of repeated measures analysis. The adjusted mean change from baseline estimates per treatment group and corresponding 95% CIs were presented along with an overall estimate of the treatment difference, 95% CI, and P value.
Multiple Testing Procedure
To strongly control the 2-sided type I error rate at 0.05, a hierarchical MTP that included a gatekeeping strategy was used across the primary end points (EFS and pCR in the mITT population) and alpha-controlled secondary end points (MPR and OS in the mITT population, and DFS in the modified resected population), with alpha allocation and recycling between end points at the interim and final analyses. The overall 2-sided 5% type I error was split between the 2 primary end points, with an alpha level of 0.5% allocated to the pCR analysis and an alpha level of 4.5% allocated to the EFS analysis. If statistical significance was demonstrated for both pCR and MPR, EFS would be tested with an alpha level of 5.0% due to alpha recycling (Figure 2).
Figure 2: Multiple Testing Procedure and Alpha Recycling in the AEGEAN Study
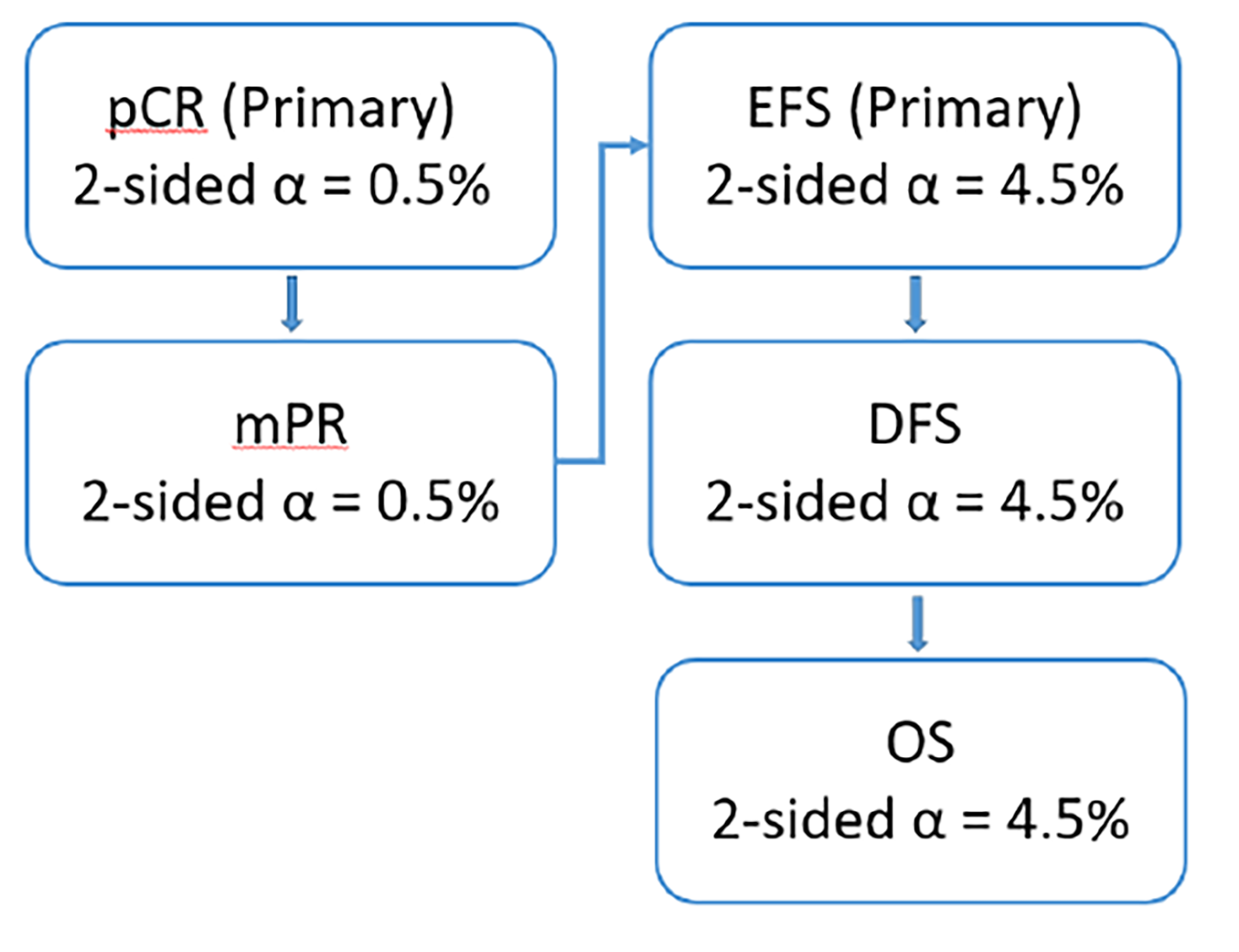
DFS = disease-free survival; EFS = event-free survival; mPR = major pathological response; OS = overall survival; pCR = pathological complete response.
Source: AEGEAN trial statistical analysis plan.59
At the preplanned pCR IA (with a DCO date of January 14, 2022), the analysis of the interim mITT cohort (N = 402 patients) demonstrated that the prespecified boundary for declaring statistical significance of pCR was met (2-sided interim significance boundary = 0.0082%, based on a total 0.5% 2-sided alpha for pCR across interim and final analysis). Consequently, the test mass was recycled to test MPR (key secondary end point), which was also statistically significant at this DCO date. This resulted in a total alpha level of 5% allocated to the EFS analyses, due to alpha recycling.
Based on the analysis of the mITT population (N = 740 patients) at the preplanned EFS IA1 (with a DCO date of November 10, 2022), EFS was also declared statistically significant (2-sided interim significance boundary = 0.9899%, based on a total 5% 2-sided alpha for EFS across interim and final analyses). Data maturity for EFS was 31.9% (i.e., 236 patients had an EFS event among all 740 patients included in the mITT population) at EFS IA1. While the final analysis of pCR and MPR was also performed at this time (with a DCO date of November 10, 2022; N = 740), statistical significance was not retested due to these end points meeting the threshold for declaring significance at the pCR IA.
Per the MTP, DFS was tested at EFS IA1; however, it did not meet the prespecified boundary for declaring statistical significance (the interim significance boundary was based on a total 5% 2-sided alpha for DFS across interim and final analyses). Because DFS was not statistically significant at EFS IA1, OS was not formally tested based on the MTP. Due to the importance of OS, a descriptive summary of OS was provided at EFS IA1. The overall maturity of OS data at EFS IA1 was 22.1% (i.e., 163 mortality events occurred among all 740 patients included in the mITT population).
At the EFS IA2 DCO (May 10, 2024), DFS was tested again and did not meet the prespecified boundary for declaring statistical significance (defined as 30% maturity; adjusted significance boundary = 0.012303, based on an information fraction of 67.8% in the modified resected set). As such, OS was not formally tested at EFS IA2. Because EFS met the prespecified boundary for declaring statistical significance at EFS IA1, statistical significance was not retested at EFS IA2 nor will it be retested at the final analysis.
PROs were not included in the MTP.
Subgroup Analyses
Prespecified subgroup analyses were performed to assess the consistency of treatment effect across expected prognostic and/or predictive factors in the following subgroups:
sex (female or male)
age at randomization (aged < 65 years or ≥ 65 years)
PD-L1 expression status at baseline (< 1% or ≥ 1%) as defined by integrated web response system
PD-L1 expression status at baseline (< 1% or ≥ 1%) as defined by source data
PD-L1 expression status at baseline (< 50% or ≥ 50%) as defined by source data
PD-L1 expression status at baseline (< 1%, 1% to 49%, or ≥ 50%) as defined by source data
PD-L1 expression status at baseline (< 25% or ≥ 25%) as defined by source data
histology (squamous or nonsquamous)
disease stage (stage II or stage III) as defined by integrated web response system
disease stage (stage II or stage III) as defined by source data
smoking (currently, previously, or never)
race (Asian versus not Asian)
region (Asia, Europe, North America, or South America)
ECOG PS (0 versus 1)
chemotherapy doublet at baseline (cisplatin versus carboplatin)
lymph node station (N2 single station versus N2 multistation)
PORT received (yes or no).
Sensitivity Analyses
For all efficacy outcomes, a sensitivity analysis was performed on the primary treatment comparison using PD-L1 expression status at baseline (< 1% versus ≥ 1%) and disease stage (stage II versus stage III) as defined by source data (instead of integrated web response system) as stratification factors. In the instance of missing source data, the integrated web response system value was imputed.
For pCR and MPR, a sensitivity analysis was performed that only included patients with an evaluable pathology sample.
The following sensitivity analyses were run for EFS (mITT population) and DFS (modified resected set):
The primary analyses were rerun based on investigator assessment using RECIST 1.1.
To assess the possible evaluation-time bias that may have been introduced if scans were not performed at the protocol-scheduled time points, the midpoint between the time of progression and the previous evaluable disease assessment (using the final date of the assessment) was analyzed using a stratified log-rank test. Note that midpoint values resulting in noninteger values were rounded down. For patients whose death was treated as an EFS event, the date of death was used to derive the EFS time used in the analysis. To support this analysis, the mean of patient-level average interassessment times was tabulated for each treatment. This approach used BICR tumour assessments.
Attrition bias was assessed by repeating the EFS analysis, except that the actual EFS event times, rather than the censored times, of patients with an EFS event immediately following 2 or more nonevaluable disease assessments was included. In addition, and within the same sensitivity analysis, for patients who took subsequent therapy (note that for this analysis, radiotherapy is not considered a subsequent anticancer therapy) before their last evaluable RECIST assessment, progression that precludes surgery or results in incomplete surgery or death was censored at their last evaluable assessment before taking the subsequent therapy. This analysis was supported by a KM plot of the time to censoring using the EFS data, where the censoring indicator of the EFS analysis is reversed.
Ascertainment bias was assessed by analyzing the site investigator data.
Because the OS end point does not include RECIST assessments, the following OS sensitivity analyses were performed on the mITT:
An analysis was performed examining the censoring patterns to rule out attrition bias using a KM plot of time to censoring where the censoring indicator of OS is reversed.
Patients who took subsequent therapy before their last known alive date were censored at the start date of subsequent therapy (note that for this analysis, radiotherapy is not considered a subsequent anticancer therapy).
Analysis Populations
The analysis populations are defined in Table 9.
Table 9: Analysis Populations of the AEGEAN Study
Population | Definition | Application |
|---|---|---|
ITTa | The ITT population includes all randomized patients. | Demography |
mITT | The mITT population includes all randomized patients, excluding those whose tumours have known EGFR mutations or ALK gene rearrangements. | Demography, all efficacy analyses other than DFS, and HRQoL (neoadjuvant period analyses) |
Modified resected set | Consists of all patients in the resected set,b excluding those whose tumours have EGFR mutation or ALK gene rearrangements. If EGFR mutation or ALK status was unknown for any patient in the resected set, they were included in the modified resected set. | Demography, DFS, and HRQoL (adjuvant period analyses) |
Safety analysis set | The safety analysis set includes all randomized patients in the global cohort who received at least 1 dose (any amount) of any study treatment. | All safety analyses |
DFS = disease-free survival; EFS = event-free survival; HRQoL = health-related quality of life; IA1 = interim analysis 1; ITT = intention to treat; mITT = modified intention to treat; MPR = major pathological response; OS = overall survival; pCR = pathological complete response; RECIST = Response Evaluation Criteria in Solid Tumours.
aThe primary and key secondary efficacy analyses for pCR, EFS, MPR, and OS were repeated on the ITT population, to allow comparison between the ITT population, mITT population, and EGFR mutation and ALK gene rearrangements subgroups.
bThe resected set consists of all patients in the ITT population who had surgical resection following the neoadjuvant period, who did not have R2 (macroscopic residual tumour) margins, and whose first scan following surgery showed no evaluable disease, defined as no postsurgery R2 margins and no RECIST evidence of disease based on the investigator data (i.e., no target lesion, nontarget lesion, or new lesions, unless they were pathologically confirmed to be a new primary malignancy).
Sources: AEGEAN study EFS IA1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition is summarized in Table 10. Of 1,480 patients enrolled in the AEGEAN study, 802 patients were included in the ITT analysis set. Due to EGFR mutation–positive or ALK gene rearrangement–positive tumours, 62 randomized patients (8%) were excluded from the mITT analysis set, leaving 366 patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and 374 patients in the placebo with neoadjuvant chemotherapy arm. Four cycles of neoadjuvant chemotherapy agents were completed by a similar percentage of patients in both treatment arms (84.7% of patients [310 of 366] in the perioperative durvalumab with neoadjuvant chemotherapy arm and 87.2% of patients [326 of 374] in the placebo with neoadjuvant chemotherapy arm). Slightly ████ patients randomized to perioperative durvalumab with neoadjuvant chemotherapy discontinued neoadjuvant durvalumab compared to placebo (█████ versus ██████ respectively). Across both arms, the primary reason for neoadjuvant durvalumab or placebo discontinuation was AEs. A similar percentage of patients in both arms underwent surgery (80.6% of patients in the durvalumab with neoadjuvant chemotherapy arm versus 80.7% of patients in the placebo with neoadjuvant chemotherapy arm) and completed surgery (77.6% of patients in the durvalumab with neoadjuvant chemotherapy arm versus 76.7% of patients in the placebo with neoadjuvant chemotherapy arm).
As of the most recent DCO, the EFS IA2 DCO on May 10, 2024, a similar percentage of patients in both arms of the mITT analysis set underwent PORT (7.1% of patients [26 of 366] in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 6.4% of patients [24 of 374] in the placebo with neoadjuvant chemotherapy arm). Slightly more patients in the perioperative durvalumab with neoadjuvant chemotherapy arm had completed adjuvant treatment (i.e., 12 cycles of durvalumab or placebo) by the EFS IA2 DCO (45.4% of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 40.4% of patients in the placebo with neoadjuvant chemotherapy arm). In the durvalumab with neoadjuvant chemotherapy arm, 20.8% of patients discontinued adjuvant treatment compared to 23.0% of patients in the placebo with neoadjuvant chemotherapy arm; the primary reason for discontinuation in both arms was radiological progression.
Table 10: AEGEAN Study Summary of Patient Disposition in mITT Population
Patient disposition | Durvalumab + neoadjuvant chemotherapy | Placebo + neoadjuvant chemotherapy | ||
|---|---|---|---|---|
EFS IA1 | EFS IA2 | EFS IA1 | EFS IA2 | |
Enrolled, N | 1,480 | |||
Did not advance past screening, n | 678 | |||
Randomized, n | 400 | 402 | ||
Excluded from mITT population, n | 34 | 28 | ||
Included in mITT analysis set, n (%) | 366 (100) | 366 (100) | 374 (100) | 374 (100) |
Neoadjuvant phase, n (%) | ||||
Received neoadjuvant chemotherapy plus durvalumab or placebo | 366 (100) | 366 (100) | 371 (99.2) | 371 (99.2) |
Completed 4 cycles of both neoadjuvant chemotherapy agents | 310 (84.7) | 310 (84.7) | 326 (87.2) | 326 (87.2) |
Completed 4 cycles of neoadjuvant durvalumab or placebo | 318 (86.9) | 318 (86.9) | 331 (88.5) | 331 (88.5) |
Discontinued neoadjuvant durvalumab or placebo | NA | ██ ██████ | NA | ██ ██████ |
Adverse event | NA | ██ █████ | NA | ██ █████ |
Clinical progression | NA | █████ | NA | █████ |
Death | NA | █████ | NA | █████ |
Development of study-specific discontinuation criteria | NA | █████ | NA | █████ |
Other | NA | ██ █████ | NA | █████ |
Radiological progression according to RECIST 1.1 | NA | █████ | NA | █████ |
Patient decision | NA | █████ | NA | █████ |
Surgerya | ||||
Underwent surgery, n (%) | 295 (80.6) | 295 (80.6) | 302 (80.7) | 302 (80.7) |
Did not undergo surgery, n (%)b | 71 (19.4) | 71 (19.4) | 72 (19.3) | 72 (19.3) |
Patient decision | NA | 12 (3.3) | NA | 17 (4.5) |
Unfit for surgery | NA | █████ | NA | █████ |
Adverse event | NA | 5 (1.4) | NA | 4 (1.1) |
Disease progression | NA | 25 (6.8) | NA | 30 (8.0) |
Death | NA | 9 (2.5) | NA | 2 (0.5) |
Investigator decision | NA | █████ | NA | █████ |
Inadequate lung function | NA | █████ | NA | █████ |
Inadequate cardiac function | NA | █████ | NA | █████ |
Surgical resection with curative intent that was performed outside the protocol | NA | █████ | NA | █████ |
Other | NA | █████ | NA | █████ |
Missing | NA | || | NA | █████ |
Completed surgery, n (%) | 284 (77.6) | 284 (77.6) | 287 (76.7) | 287 (76.7) |
R0 resection, n of N (%)c | 269 of 284 (94.7) | NA | 262 of 287 (91.3) | NA |
R1 resection, n of N (%)c | 12 of 284 (4.2) | NA | 22 of 287 (7.7) | NA |
R2 resection, n of N (%) | 1 of 284 (0.7) | NA | 2 of 287 (0.7) | NA |
Completed surgery with missing margins | 1 of 285 (0.4) | NA | 1 (0.3) | NA |
Did not complete surgery, n (%) | 11 (3.0) | 11 (3.0) | 15 (4.0) | 15 (4.0) |
Postsurgery | ||||
Underwent postoperative radiation therapy, n (%) | 26 (7.1) | 26 (7.1) | 21 (5.6) | 24 (6.4) |
Adjuvant phase, ongoing, n (%) | ||||
Started adjuvant durvalumab or placebod | 241 (65.8) | 242 (66.1) | 237 (63.4) | 237 (63.4) |
Completed adjuvant durvalumab or placebo | 88 (24.0) | 166 (45.4) | 79 (21.1) | 151 (40.4) |
Discontinued adjuvant durvalumab or placebo | 68 (18.6) | 76 (20.8) | 70 (18.7) | 86 (23.0) |
Adverse event | NA | ██ █████ | NA | ██ █████ |
Clinical progression | NA | █████ | NA | ██ █████ |
Death | NA | █████ | NA | || |
Other | NA | █████ | NA | █████ |
Radiological progression according to RECIST 1.1 | NA | ██ █████ | NA | ██ ██████ |
Patient decision | NA | █████ | NA | █████ |
Ongoing adjuvant durvalumab or placebo | 85 (23.2) | 0 | 88 (23.5) | 0 |
ITT, N | 400 | 402 | ||
mITT, N | 366 | 374 | ||
Modified resected set, N | 242 | 231 | ||
Modified safety analysis set, N | 367 | 370 | ||
Safety analysis set, N | 401 | 398 | ||
DCO = data cut-off; EFS = event-free survival; IA1 = interim analysis 1; IA2 = interim analysis 2; ITT = intention to treat; mITT = modified intention to treat; NA = not available; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; vs. = versus.
Note: The EFS IA1 DCO date was November 10, 2022; the EFS IA2 DCO date was May 10, 2024.
aAll percentages were calculated from the number of patients randomized, with the exception of rows with reference to footnote “c.”
bExcludes patients with surgery done outside of study.
cPercentages were calculated from the number of patients who received treatment in the neoadjuvant period.
dIncludes 4 patients who did not complete surgery (1 patient for durvalumab + chemotherapy vs. 3 patients for placebo + chemotherapy).
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 24,23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics of the mITT population are outlined in Table 11 and are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. In the AEGEAN study, patients were primarily Asian (41.5% of patients [307 of 740]) or white (53.6% of patients [397 of 740]) with just 1.4% of patients (10 of 740) identifying as American Indian or Alaska Native, 0.9% of patients (7 of 740) identifying as Black or African American, and 2.6% of patients (19 of 740) identifying with other races. Patients were primarily aged 50 years or older (95.0% of patients [703 of 740]) with a median age of 65.0 years (range, 39 years to 85 years), and either currently or formerly smoked (85.5% of patients [633 of 740]). In the perioperative durvalumab with neoadjuvant chemotherapy arm, 68.9% of patients (252 of 366) were male compared to 74.3% of patients (278 of 374 patients) in the placebo with neoadjuvant chemotherapy arm. Overall, at baseline, patients tended to have an ECOG PS score of 0 (68.4% of patients [506 of 740]), present with stage III disease (70.9% of patients [525 of 740]), and have a baseline PD-L1 expression status greater than or equal to 1% (66.6% of patients [493 of 740]). A slightly higher percentage of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm had nonsquamous histology (53.6% of patients [196 of 366]) compared to those in the placebo with neoadjuvant chemotherapy arm (47.9% of patients [179 of 374]). In total, || patients had unknown EGFR mutation status (|| in the perioperative durvalumab with neoadjuvant chemotherapy arm and || in the placebo with neoadjuvant chemotherapy arm) and ██ patients had unknown ALK translocation status (|| in the perioperative durvalumab with neoadjuvant chemotherapy arm and || in the placebo with neoadjuvant chemotherapy arm).
Baseline demographics in the mITT population were generally similar to those in the ITT population (Appendix 1, Table 39).
Exposure to Study Treatments
Exposure to Durvalumab or Placebo
In the modified safety analysis set (N = 737) at the EFS IA2 DCO (May 10, 2024), the median total duration of treatment exposure for the overall period was longer for the durvalumab arm (█████ weeks) than for the placebo arm (█████ weeks) (Table 12). During the neoadjuvant period, the median total duration of treatment exposure was similar in both arms (█████ weeks in the perioperative durvalumab with neoadjuvant chemotherapy arm versus █████ weeks in the placebo with neoadjuvant chemotherapy arms). The median total duration of treatment exposure during the adjuvant period was the same in both arms (█████ weeks).
After accounting for any dose delays, there were no notable differences across arms in the actual duration of exposure in the neoadjuvant and adjuvant periods (█████ weeks and █████ weeks in both arms, respectively). The median actual duration of exposure to the perioperative durvalumab with neoadjuvant chemotherapy arm versus the placebo with neoadjuvant chemotherapy arm for the overall period was similar in each arm (█████ versus █████ weeks, respectively).
Table 11: AEGEAN Study Summary of Baseline Characteristics in mITT Population
Characteristic | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 |
|---|---|---|
Age (years)a | ||
Mean (SD) | 64.1 (9.02) | 63.9 (8.59) |
Median (minimum to maximum) | 65.0 (30 to 88) | 65.0 (39 to 85) |
Age group (years), n (%) | ||
< 50 | 17 (4.6) | 20 (5.3) |
≥ 50 to < 65 | 158 (43.2) | 163 (46.3) |
≥ 65 to < 75 | 147 (40.2) | 155 (41.4) |
≥ 75 | 44 (12.0) | 36 (9.6) |
Sex, n (%) | ||
Female | 114 (31.1) | 96 (25.7) |
Male | 252 (68.9) | 278 (74.3) |
ECOG PS, n (%) | ||
(0) Normal activity | 251 (68.6) | 255 (68.2) |
(1) Restricted in physically strenuous activity | 115 (31.4) | 119 (31.8) |
Race,b n (%) | ||
American Indian or Alaska Native | 6 (1.6) | 4 (1.1) |
Asian | 143 (39.1) | 164 (43.9) |
Black or African American | 4 (1.1) | 3 (0.8) |
White | 206 (56.3) | 191 (51.1) |
Other | 7 (1.9) | 12 (3.2) |
Ethnic group, n (%) | ||
Hispanic or Latino | 63 (17.2) | 56 (15.0) |
Not Hispanic or Latino | 303 (82.8) | 318 (85.0) |
Smoking history, n (%) | ||
Never smoked | 51 (13.9) | 56 (15.0) |
Smoked | 315 (86.1) | 318 (85.0) |
Disease stage at baseline,c n (%) | ||
IIA | 18 (4.9) | 24 (6.4) |
IIB | 86 (23.5) | 86 (23.0) |
III (NOS) | 0 | 1 (0.3) |
IIIA | 173 (47.3) | 165 (44.1) |
IIIB | 88 (24.0) | 98 (26.2) |
IV (NOS) | 1 (0.3) | 0 |
EGFR mutation status, n (%)d | ||
Not detected | ███ ██████ | ███ ██████ |
Not tested (KRAS)e | █████ | █████ |
Not tested (squamous histology)e | ██ ██████ | ██ ██████ |
Unknown (other) | █████ | █████ |
ALK translocation status, n (%)d | ||
Not detected | ███ ██████ | ███ ██████ |
Not tested (squamous histology)e | ██ ██████ | ██ ██████ |
Unknown (other) | █████ | █████ |
Histology type, n (%) | ||
Squamous | 169 (46.2) | 191 (51.1) |
Nonsquamous | 196 (53.6) | 179 (47.9) |
TNM stage at baseline, regional lymph nodes, n (%)f | ||
N0 | 110 (30.1) | 102 (27.3) |
N1 | 75 (20.5) | 87 (23.3) |
N2 | 181 (49.5) | 185 (49.5) |
Single station | 141 (38.5) | 132 (35.3) |
Multistation | 34 (9.3) | 40 (10.7) |
Missing | 6 (1.6) | 13 (3.5) |
PD-L1 expression status at baseline, n (%) | ||
TC < 1% (per IXRS and source data) | 122 (33.3) | 125 (33.4) |
TC ≥ 1% (per IXRS and source data) | 244 (66.7) | 249 (66.6) |
TC of 1% to 49% (per source data) | 135 (36.9) | 142 (38.0) |
TC ≥ 50% (per source data) | 109 (29.8) | 107 (28.6) |
AJCC = American Joint Committee on Cancer; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EFS = event-free survival; IA1 = interim analysis 1; IXRS = interactive web response system; mITT = modified intention to treat; NOS = not otherwise specified; SD = standard deviation; TC = tumour cell; TNM = tumour-nodes-metastasis.
Notes: Baseline is defined as the last observation before randomization if available or, otherwise, an observation after randomization but before the first dose of randomized treatment.
Percentages were calculated based on the number of patients in the analysis set of N.
The DCO date was November 10, 2022.
aAge was calculated using date of randomization.
bRacial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
cStages according to AJCC Cancer Staging Manual, eighth edition.
dCentral laboratory EGFR and ALK data are considered to determine EGFR and ALK status if available; otherwise, local laboratory data may be used. Data from all assessments, including postbaseline data, are considered.
ePer protocol, patients with KRAS gene mutation do not require EGFR or ALK testing; patients with squamous histology do not require ALK testing.
fTNM classification is based on the AJCC Cancer Staging Manual, eighth edition.
Sources: AEGEAN study EFS IA1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 12: AEGEAN Study Summary of Patient Exposure to Durvalumab or Placebo in the Modified Safety Analysis Set
Exposure | Durvalumab + neoadjuvant chemotherapy N = 367 | Placebo + neoadjuvant chemotherapy N = 370 |
|---|---|---|
EFS IA2 | ||
Neoadjuvant period | █████ | █████ |
Total neoadjuvant treatment duration (weeks)a | ||
Duration, mean (SD) | █████ ███████ | █████ ███████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | ████ | ████ |
Actual neoadjuvant treatment duration (weeks)b | ||
Duration, mean (SD) | █████ ███████ | █████ ███████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | ████ | ████ |
Adjuvant period | █████ | █████ |
Total adjuvant treatment duration (weeks)c | ||
Duration, mean (SD) | █████ ████████ | █████ ████████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | █████ | █████ |
Actual adjuvant treatment duration (weeks)b | ||
Duration, mean (SD) | █████ ████████ | █████ ████████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | █████ | █████ |
Total overall | n = 367 | n = 370 |
Total overall treatment duration (weeks)d | ||
Duration, mean (SD) | █████ ████████ | █████ ████████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | █████ | █████ |
Actual overall treatment duration (weeks)e | ||
Duration, mean (SD) | █████ ████████ | █████ ████████ |
Duration, median (minimum to maximum) | █████ ████ ██ █████ | █████ ████ ██ █████ |
Total treatment years | █████ | █████ |
Treatment cycles received | ||
Number of cycles, mean (SD) | ████ ████ | ████ ████ |
Number of cycles, median (Q1 to Q3) | ████ ████ ██ █████ | ████ ████ ██ █████ |
DCO = data cut-off; EFS = event-free survival; IA2 = interim analysis 2; NR = not reported; Q1 = quantile 1; Q3 = quantile 3; SD = standard deviation.
Notes: One patient randomized to placebo + chemotherapy received 1 cycle of adjuvant durvalumab. This patient is reported in the durvalumab + chemotherapy arm with 0 neoadjuvant exposure.
The DCO date was May 10, 2024.
aTotal treatment duration = ([minimum {last dose date where dose > 0 + 20, date of death, or date of DCO} – first dose date] + 1) ÷ 7, where minimum is whichever occurred first.
bActual treatment duration = total treatment duration minus the total duration of dose delays for each period.
cTotal treatment duration = ([minimum {last dose date where dose > 0 + 27, date of death, or date of DCO} – first dose date postsurgery] + 1) ÷ 7, where minimum is whichever occurred first.
dTotal treatment duration of both phases, as the addition of total neoadjuvant duration and total adjuvant duration.
eActual treatment duration = total treatment duration minus the total duration of dose delays for both periods.
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Duration of Exposure to Chemotherapy
In the safety analysis set (n = 799) at the EFS IA1 DCO (November 10, 2022), the exposure to any chemotherapy and to individual chemotherapy agents was similar across both arms, with a median total duration of exposure to any chemotherapy of 12.14 weeks in each arm (Table 13). The exposure to chemotherapy, which was only given in the neoadjuvant portion of treatment, remained unchanged as of the EFS IA2 DCO (May 10, 2024).
The median total duration of exposure to individual chemotherapy treatments was similar across both treatment arms (ranging from 12.00 weeks to 12.14 weeks in each arm) (Table 28). Overall, 26 patients switched platinum-based chemotherapy (██ patients switched from cisplatin to carboplatin as permitted by the protocol and || patients had off-protocol switching from carboplatin to cisplatin). Switching occurred in ████████ ████ patients randomized to perioperative durvalumab with neoadjuvant chemotherapy (████ █████████ than those randomized to placebo with neoadjuvant chemotherapy (████ ██████). In addition, 6 patients had off-protocol switching of non–platinum chemotherapy (|| patients switched from paclitaxel to gemcitabine and || patient switched from pemetrexed to paclitaxel) with ██████████ occurring in the perioperative durvalumab with neoadjuvant chemotherapy arm █████ ████████ and █████████ occurring in the placebo with neoadjuvant chemotherapy arm (████ ██████).
Table 13: AEGEAN Study Summary of Exposure to Neoadjuvant Chemotherapy in the Safety Analysis Set Population
Exposure | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 |
|---|---|---|---|---|
EFS IA1 | EFS IA2 | |||
Any chemotherapy | n = 401 | n = 398 | n = 401 | n = 398 |
Total treatment duration (weeks)a | ||||
Duration, mean (SD) | 12.14 (2.071) | 12.03 (2.103) | 12.14 (2.071) | 12.03 (2.103) |
Duration, median (minimum to maximum) | 12.14 (2.0 to 20.7) | 12.14 (3.0 to 22.7) | 12.14 (2.0 to 20.7) | 12.14 (3.0 to 22.7) |
Total treatment years | — | — | ████ | ████ |
Switched to platinum agent of chemotherapy, n (%) | ██ █████ | ██████ | ██ █████ | ██████ |
Cisplatin to carboplatin | ██ █████ | ██████ | ██ █████ | ██████ |
Carboplatin to cisplatin | ██████ | || | ██████ | || |
Switched to a non–platinum agent of chemotherapy, n (%) | ██████ | ██████ | ██████ | ██████ |
Paclitaxel to gemcitabine | ██████ | ██████ | ██████ | ██████ |
Pemetrexed to paclitaxel | ██████ | || | ██████ | || |
DCO = data cut-off; EFS = event-free survival; IA1 = interim analysis 1; IA2 = interim analysis 2; SD = standard deviation.
Note: The EFS IA1 DCO date was November 10, 2022; the EFS IA2 DCO date was May 10, 2024.
aTotal treatment duration = (minimum [death, DCO, last non-0 dose of chemotherapy {cycle X, day 1} + 20] – first dose date of chemotherapy) + 1 ÷ 7, where minimum is whichever occurred first.
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 24,23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications
At the EFS IA2 DCO, ██████ ███ patients had received ██ █████ ███ concomitant medication during the study (███████ ████████ Table 14). The use of concomitant medications was generally well balanced across treatment arms ████ ███ █████████ ██ █████ ████ ███ ███████████ ██████ █████████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm versus █████ █████████ in the placebo with neoadjuvant chemotherapy arm) ███ ███████ ███ ██████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm versus █████ in the placebo with neoadjuvant chemotherapy arm) in which a ████████ ██████ ██████████ of those randomized to perioperative durvalumab with neoadjuvant chemotherapy use said treatments than those randomized to placebo with neoadjuvant chemotherapy.
The disallowed concomitant medications administered in the AEGEAN trial did not raise any concerns on the outcome measures of the study. Overall (EFS IA2, mITT population), 1 or more disallowed concomitant medication during study treatment was reported for ███████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and ███████ patients in the placebo with neoadjuvant chemotherapy arm (ITT population); most frequently, these were ██████ ██████████ ████████ ██ ████████ █████; data not shown).
Table 14: AEGEAN Study Summary of Concomitant Medication Used in the mITT Population
Concomitant medication | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 |
|---|---|---|---|---|
EFS IA1 | EFS IA2 | |||
Number of patients with concomitant medication, n (%) | ███ ██████ | ███ █████ | ███ ██████ | ███ █████ |
Most common concomitant medication used in ≥ 25% of either treatment arm | ||||
█████████ ██████ ███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
█████████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
██████ ████ ██████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
█████ ███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
██████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
███████ █████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████ ███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
██████████ | ██ ██████ | ██ ██████ | ██ ██████ | ███ ██████ |
██████ ███████████ ███████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████ ███ ███████████████ ███ ██████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
███████████ █████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
█████ ████ ███ ███████████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
█████ ████ | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
█████████ ████ ███████████ | ██ ██████ | ██ ██████ | ███ ██████ | ███ ██████ |
███████████ ██████ █████████ | ██ ██████ | ██ ██████ | ██ ██████ | ███ ██████ |
███████ █████████ ██████████ | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
███████████ ███ █████████████ | ██████ | ██████ | ██ ██████ | ██ ██████ |
███████ ██ ███████████ ████ ██████████ ██████████ | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
ATC = Anatomical Therapeutic Chemical; DCO = data cut-off; EFS = event-free survival; HMG-CoA = hydroxymethylglutaryl-CoA; IA1 = interim analysis 1; IA2 = interim analysis 2; mITT = modified intention to treat.
Notes: A patient could have 1 or more generic term reported under a given ATC classification text. Concomitant medication is defined as started, ended, or ongoing between the first dose date of study treatment and last dose of study treatment + 90 days, as per WHO Drug Dictionary, version September 2022.
The EFS IA1 DCO date was November 10, 2022; the EFS IA2 DCO date was May 10, 2024.
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 24,23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Subsequent Treatment
Overall, the percentage of patients in the mITT population who had received a subsequent anticancer therapy was 17.2% (127 of 740) at the EFS IA1 DCO (November 10, 2022) and █████ █████████ at the EFS IA2 DCO (May 10, 2024) (Table 15). While at the EFS IA1 the use of subsequent anticancer therapy was generally well balanced across the mITT arms — with the exception of the use of immunotherapy treatment, which was slightly higher in the placebo with neoadjuvant chemotherapy arm — ███████ ███████████ were observed at the EFS IA2 DCO. At the EFS IA2 DCO, a ██████ percentage of patients in the placebo with neoadjuvant chemotherapy arm used any form of subsequent anticancer therapy (█████ █████████ versus █████ ████████ in the perioperative durvalumab with neoadjuvant chemotherapy arm). The use of both systemic therapy and radiotherapy was ██████ in the placebo with neoadjuvant chemotherapy arm than in the perioperative durvalumab with neoadjuvant chemotherapy arm (█████ versus █████ and █████ versus ██████ respectively). Across treatment groups, subsequent anticancer therapy tended to be most commonly used for palliative care.
Table 15: AEGEAN Study Summary of Subsequent Anticancer Therapy in the mITT Population
Subsequent anticancer therapy | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 |
|---|---|---|---|---|
EFS IA1 | EFS IA2 | |||
Number of patients with postdiscontinuation anticancer therapy, n (%) | 55 (15.0) | 72 (19.3) | ██ ██████ | ███ ██████ |
Systemic therapy, n (%) | 53 (14.5) | 72 (19.3) | ██ ██████ | ███ ██████ |
Cytotoxic chemotherapy | 37 (10.1) | 37 (9.9) | ██ ██████ | ██ ██████ |
Single agent | 10 (2.7) | 8 (2.1) | ██ █████ | ██ █████ |
Platinum doublet | 26 (7.1) | 24 (6.4) | ██ █████ | ██ █████ |
Other combination | 7 (1.9) | 7 (1.9) | ██████ | ██████ |
Immunotherapy | 17 (4.6) | 39 (10.4) | ██ █████ | ██ ██████ |
Immunotherapy only | 7 (1.9) | 25 (6.7) | ██████ | ██ █████ |
Immunotherapy + chemotherapy | 10 (2.7) | 13 (3.5) | ██ █████ | ██ █████ |
Immunotherapy + other | 1 (0.3) | 1 (0.3) | ██████ | ██████ |
Targeted therapy | 3 (0.8) | 6 (1.6) | ██████ | ██████ |
Other | 2 (0.5) | 3 (0.8) | ██████ | ██████ |
Radiotherapy, n (%) | 37 (10.1) | 48 (12.8) | ██ ██████ | ██ ██████ |
Concomitant chemoradiotherapy | 18 (4.9) | 16 (4.3) | ██ █████ | ██ █████ |
Line of subsequent therapy, n (%) | ||||
First subsequent therapy | 47 (12.8) | 67 (17.9) | ██ ██████ | ███ ██████ |
Second subsequent therapy | 16 (4.4) | 19 (5.1) | ██ █████ | ██ █████ |
≥ third subsequent therapy | 3 (0.8) | 5 (1.3) | ██████ | ██████ |
Type of subsequent therapy, n (%) | ||||
Neoadjuvant | 4 (1.1) | 4 (1.1) | ██████ | ██████ |
Adjuvant | 6 (1.6) | 10 (2.7) | ██████ | ██ █████ |
Definitive | 6 (1.6) | 3 (0.8) | ██████ | ██████ |
Maintenance | 3 (0.8) | 6 (1.6) | ██ █████ | ██████ |
Palliative | 34 (9.3) | 48 (12.8) | ██ ██████ | ██ ██████ |
Not applicable | 5 (1.4) | 4 (1.1) | ██████ | ████ |
DCO = data cut-off; EFS = event-free survival; IA1 = interim analysis 1; IA2 = interim analysis 2; mITT = modified intention to treat.
Notes: Therapies postdiscontinuation of study treatment. Regimen categories and the line of subsequent therapy have been identified from a medical review of preferred terms using treatment start dates.
Patients with therapies in more than 1 category were counted once in each of those categories. Percentages were calculated from the number of patients in the mITT population in that treatment arm.
The EFS IA1 DCO date was November 10, 2022; the EFS IA2 DCO date was May 10, 2024.
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 24,23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
The median duration of follow-up from randomization to the November 10, 2022, DCO was 9.61 months (range, 0.0 months to 46.1 months) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 8.94 months (range, 0.0 months to 32.4 months) in the placebo with neoadjuvant chemotherapy arm. At the May 10, 2024, DCO, the median duration of follow-up was 20.70 months (range, 0.0 months to 58.6 months) and 13.86 months (range, 0.0 months to 58.5 months), respectively.
Pathological Complete Response
As of the November 10, 2022, DCO, the percentage of patients achieving pCR by an IRC was 17.21% (95% CI, 13.49% to 21.48%) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 4.28% (95% CI, 2.46% to 6.85%) in the placebo with neoadjuvant chemotherapy arm, as detailed in Table 16. The difference in pCR for the comparison of perioperative durvalumab with neoadjuvant chemotherapy to placebo with neoadjuvant chemotherapy was 12.96% (95% CI, 8.67% to 17.57%). Results from the sensitivity and subgroup analyses were generally consistent with those of the main analysis. Forest plots of the prespecified subgroup analyses are presented in Figure 9 in Appendix 2.
A similar magnitude of effect and 95% CI was observed at the pCR IA1 DCO. Per protocol, pCR statistical significance was tested at the first IA and because it was statistically significant (P = 0.000036), pCR was not retested for statistical significance at the final analysis.
Major Pathological Response
As of the November 10, 2022, DCO, the percentage of patients achieving MPR was 33.33% (95% CI, 28.52% to 38.42%) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 12.30% (95% CI, 9.15% to 16.06%) in the placebo with neoadjuvant chemotherapy arm (Table 16). The difference with MPR for the comparison of perioperative durvalumab with neoadjuvant chemotherapy to placebo with neoadjuvant chemotherapy was 21.03% (95% CI, 15.14% to 26.93%).
A similar magnitude of effect and 95% CI was observed at the pCR IA1 DCO. Per protocol, MPR statistical significance was tested and deemed statistically significant at the first IA (P = 0.000002); MPR was not retested for statistical significance at the final analysis.
Table 16: Summary of Key Tumour Response Efficacy Results From the AEGEAN Study
Outcome | Durvalumab + neoadjuvant chemotherapy N = 196 | Placebo + neoadjuvant chemotherapy N = 206 | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 |
|---|---|---|---|---|
pCR IA1 (DCO date: January 14, 2022) | pCR FA (DCO date: November 10, 2022) | |||
Primary end point: pCRa | ||||
Number of patients whose disease had a responseb | 35 | 10 | 63 | 16 |
Response rate, % (95% CI)c | 17.86 (12.76 to 23.95) | 4.85 (2.35 to 8.75) | 17.21 (13.49 to 21.48) | 4.28 (2.46 to 6.85) |
Difference, % (95% CI)d | 13.03 (7.11 to 19.52) | 12.96 (8.67 to 17.57) | ||
Adjusted 99.9918% CI | 0.68 to 26.85a | NAe | ||
2-sided P value | 0.000036a | NAe | ||
Secondary end point: MPRa | ||||
Number of patients whose disease had a responsef | 67 | 29 | 122 | 46 |
Response rate, % (95% CI)d | 34.18 (27.57 to 41.28) | 14.08 (9.64 to 19.59) | 33.33 (28.52 to 38.42) | 12.30 (9.15 to 16.06) |
Difference, % (95% CI)d | 20.07 (11.85 to 28.26) | 21.03 (15.14 to 26.93) | ||
Adjusted 99.9918% CI | 3.40 to 36.42a | NAe | ||
2-sided P value | 0.000002a | NAe | ||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DCO = data cut-off; FA = final analysis; IA1 = interim analysis 1; IXRS = interactive web response system; MPR = major pathological response; MTP = multiple testing procedure; NA = not applicable; pCR = pathological complete response; TC = tumour cell; vs. = versus.
aAdjusted for multiplicity. Based on a Lan-DeMets alpha spending function with the O’Brien-Fleming boundary calculated using the actual number of patients at the interim vs. the final analysis, the boundary for declaring statistical significance is 0.0082% for a total 0.5% 2-sided alpha. Corresponding CIs are shown.
bResponse is defined as having 0% residual viable tumour cells within all resected tissue, without R1 (microscopic residual tumour) or R2 (macroscopic residual tumour) margins, and without carcinoma in any examined lymph nodes at the time of resection as assessed by the central pathology laboratory.
cCIs for response rate are calculated using the Clopper-Pearson exact method.
dThe analysis was performed using a CMH test stratified by disease stage (stage II vs. stage III) and by PD-L1 expression status at baseline (TC < 1% vs. ≥ 1%), using the stratification factors from the IXRS. A difference > 0 favours perioperative durvalumab + neoadjuvant chemotherapy. CIs for difference in proportions are computed using the Miettinen and Nurminen’s stratified method.
eBecause IA1 was statistically significant per the MTP, pCR and MPR were not retested for statistical significance at the FA.
fResponse is defined as having 10% or less residual viable tumour tissue in the lung primary tumour at the time of resection as assessed by the central pathology laboratory.
Sources: AEGEAN study EFS IA1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Event-Free Survival
As of the EFS IA2 DCO on May 10, 2024, a total of 289 patients had experienced an EFS event (per RECIST 1.1) in the mITT population, with a lower percentage of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm experiencing an event than in the placebo with neoadjuvant chemotherapy arm (33.9% of patients [124 of 366] versus 44.1% of patients [165 of 374], respectively) (as detailed in Table 17). The median EFS was NR in the perioperative durvalumab with neoadjuvant chemotherapy arm (95% CI, 42.3 months to NR) and was 30.0 months (95% CI, 20.6 months to NR) in the placebo with neoadjuvant chemotherapy arm. The overall HR was 0.69 (95% CI, 0.55 to 0.88) (Figure 3). For patients in the perioperative durvalumab with neoadjuvant chemotherapy arm, the probability of remaining event-free from randomization to 12 months was 73.3% (95% CI, 68.1% to 77.7%), to 24 months was 65.0% (95% CI, 59.4% to 70.0%), and to 36 months was 60.1% (95% CI, 53.9% to 65.8%). For patients in the placebo with neoadjuvant chemotherapy arm, the corresponding probability of remaining event-free was 64.1% (95% CI, 58.7% to 69.0%) for randomization to 12 months, 54.4% (95% CI, 48.7% to 59.6%) to 24 months, and 47.9% (95% CI, 41.8% to 53.8%) to 36 months. Across time points, the difference in EFS probabilities for perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy ranged from 9.2% (95% CI, ███ ██ ████) at 12 months to 12.2% (95% CI, ███ ██ ████) at 36 months. Results from the sensitivity and subgroup analyses were generally consistent with those of the main analysis. Forest plots of the subgroup analyses are presented in Figure 10 in Appendix 2.
A similar magnitude of effect and 95% CI was observed at the EFS IA1 DCO. Per protocol, EFS statistical significance was tested and deemed statistically significant at IA1 (P = 0.003902); EFS was not retested for statistical significance at EFS IA2.
Figure 3: KM Plot of EFS (Using BICR per RECIST 1.1) (mITT Population, EFS IA2)
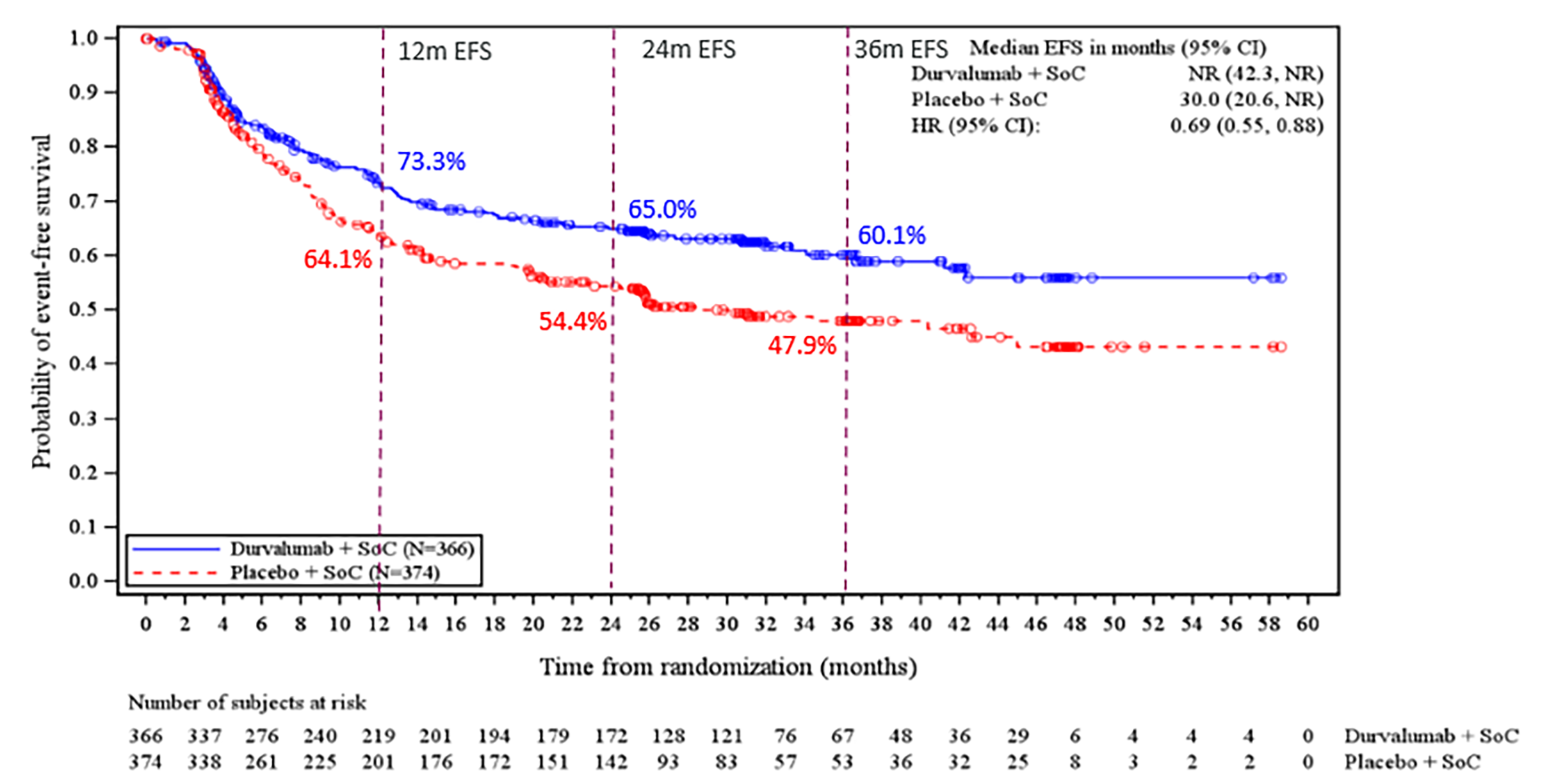
BICR = blinded independent central review; CI = confidence interval; EFS = event-free survival; HR = hazard ratio; IA2 = interim analysis 2; mITT = modified intention to treat; NR = not reached; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; SoC = standard of care.
Notes: EFS is defined as the time from randomization to the earliest progression of disease that precludes surgery or progression with surgery not completed, local or distant disease recurrence, or death due to any cause. Patients who have not experienced an EFS event at the time of analysis are censored at the time of the last disease assessment. If the event occurs after 2 or more consecutive missed visits, the patient is censored at the last disease assessment before the 2 missed visits. A new malignancy that is not NSCLC, as confirmed by pathology, is not considered an EFS event, as per RECIST 1.1.
The data cut-off date of the EFS IA2 was May 10, 2024.
Source: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23
Disease-Free Survival
At both the EFS IA1 and IA2 DCOs, DFS was tested but did not meet the prespecified boundary for declaring statistical significance (at EFS IA2, P = 0.013652 and with a P value of < 0.012303 was required to declare statistical significance at this IA) (Table 17). DFS remains eligible for formal statistical testing per the MTP and will be re-evaluated at the final analysis.
By May 10, 2024, 141 patients had experienced a DFS event (based on a BICR per RECIST 1.1) in the modified resected set, with more patients in the placebo with neoadjuvant chemotherapy arm experiencing an event than in the perioperative durvalumab with neoadjuvant chemotherapy arm (35.1% of patients [81 of 231] versus 24.8% of patients [60 of 242], respectively). While median DFS was NR in both arms, the overall HR was 0.66 (95% CI, 0.47 to 0.92) (Figure 4). For patients in the perioperative durvalumab with neoadjuvant chemotherapy arm, the probability of remaining disease-free from surgical resection to 12 months was 81.0% (95% CI, 75.2% to 85.5%), to 24 months was 75.1% (95% CI, 68.7% to 80.4%), and to 36 months was 71.2% (95% CI, 63.8% to 77.3%). For patients in the placebo with neoadjuvant chemotherapy arm, the corresponding probability of remaining disease-free to 12 months was 74.1% (95% CI, 67.8% to 79.3%), to 24 months was 62.4% (95% CI, 55.2% to 68.8%), and to 36 months was 61.4% (95% CI, 54.0% to 68.0%). Across time points, the difference in survival probabilities for perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy ranged from 6.9% (95% CI, ████ ██ ████) at 12 months to 12.7% (95% CI, ███ ██ ████) at 24 months.
Figure 4: KM Plot of DFS (Using BICR per RECIST 1.1) (Modified Resected Set, EFS IA2)
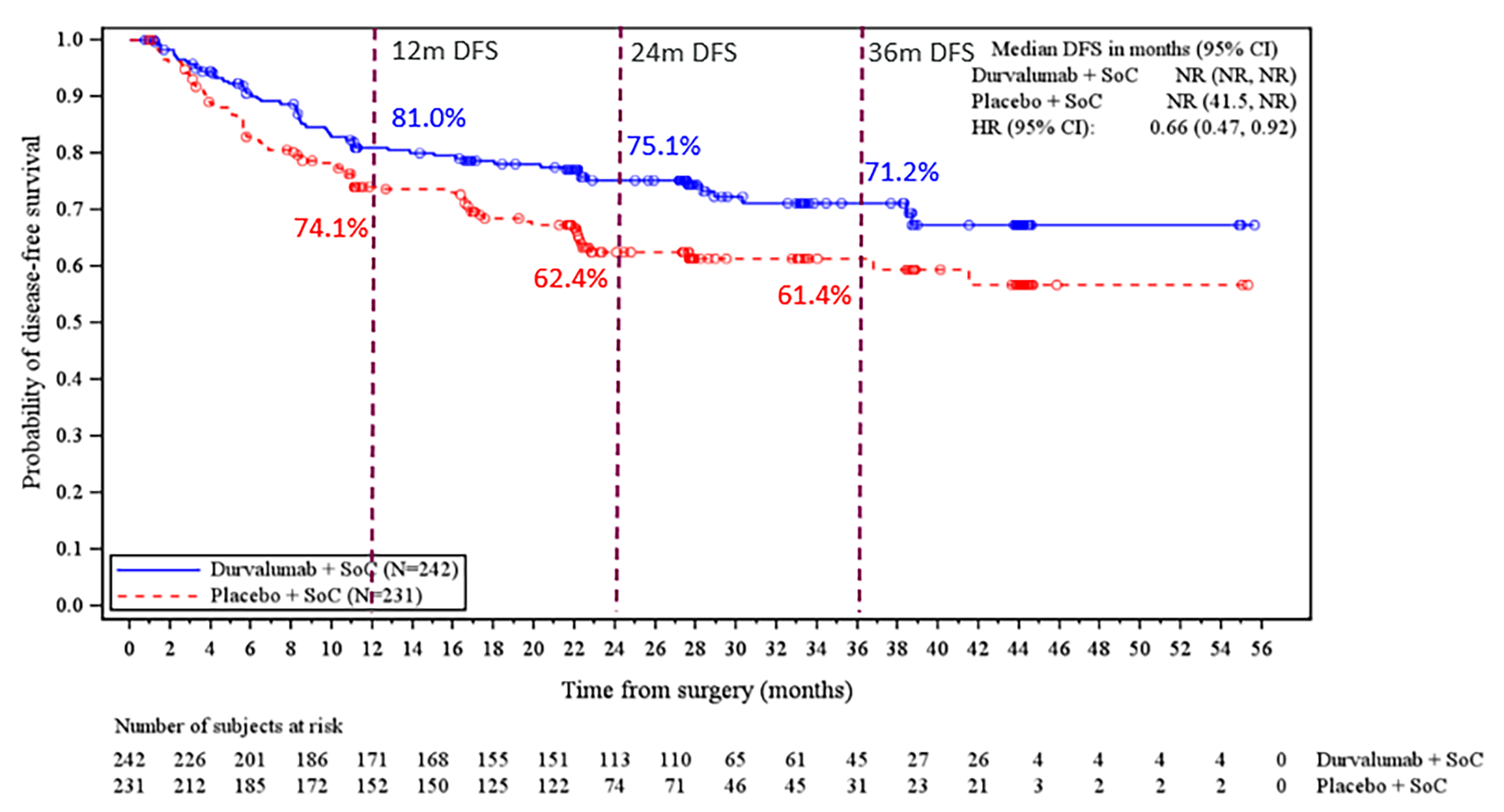
BICR = blinded independent central review; CI = confidence interval; DFS = disease-free survival; EFS = event-free survival; HR = hazard ratio; IA2 = interim analysis 2; NR = not reported; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; SoC = standard of care; vs. = versus.
Notes: The HR was calculated using a stratified Cox proportional hazards model. Stratification factors included disease stage (stage II vs. stage III) and PD-L1 (< 1% vs. ≥ 1%). An HR < 1 favours durvalumab + chemotherapy.
The data cut-off date was May 10, 2024.
Source: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23
Overall Survival
At both the EFS IA1 and EFS IA2 DCOs, OS was not eligible for statistical testing (this is because DFS was not statistically significant; therefore, OS was not formally tested based on the MTP).
At the time of EFS IA2 (May 10, 2024), 261 deaths had occurred in the mITT population, with a similar percentage occurring across treatment arms (33.1% of patients [121 of 366] in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 37.4% of patients [140 of 374] in the placebo with neoadjuvant chemotherapy arm); this was described in Table 17. The median OS was NR (95% CI, NR to NR) in the perioperative durvalumab with neoadjuvant chemotherapy arm and was 53.2 months (95% CI, 44.3 months to NR) in the placebo with neoadjuvant chemotherapy arm; the HR was 0.89 (95% CI, 0.70 to 1.14) (Figure 5). For patients in the perioperative durvalumab with neoadjuvant chemotherapy arm, the probability of survival from randomization to 12 months was 84.3% (95% CI, 80.1% to 87.7%), to 24 months was 74.4% (95% CI, 69.5% to 78.6%), to 36 months was 67.1% (95% CI, 61.6% to 71.9%), and to 48 months was 57.6% (95% CI, 50.1% to 64.3%). For patients in the placebo with neoadjuvant chemotherapy arm, the corresponding probability of survival from randomization to 12 months was 85.3% (95% CI, 81.2% to 88.5%), to 24 months was 72.2% (95% CI, 67.3% to 76.5%), to 36 months was 63.9% (95% CI, 58.4% to 69.0%), and to 48 months was 52.9% (95% CI, 45.7% to 59.7%). Across time points, the difference in survival probabilities for perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy ranged from −1.0% (95% CI, ████ ██ ████ at 12 months to 4.7% (95% CI, ████ ██ ████) at 48 months.
As of the EFS IA1 DCO, the median OS was NR in both arms; the HR was 1.02 (95% CI, 0.75 to 1.39).
Figure 5: KM Plot of OS (mITT Population, EFS IA2)
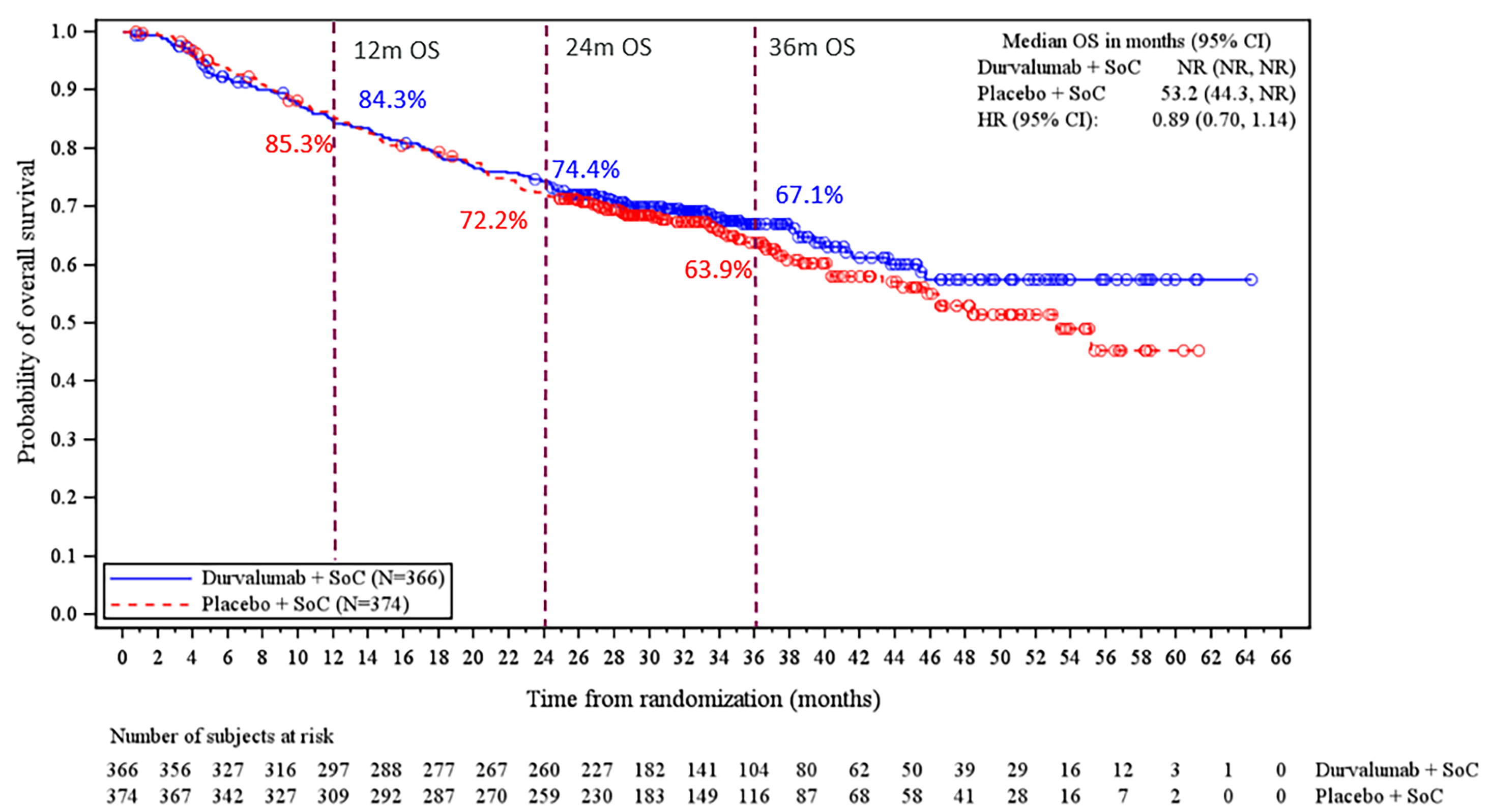
CI = confidence interval; EFS = event-free survival; HR = hazard ratio; IA2 = interim analysis 2; mITT = modified intention to treat; NR = not reported; OS = overall survival; SoC = standard of care.
Notes: A circle indicates a censored observation; 1 month = 30.4375 days.
OS is defined as the time from date of randomization until death due to any cause. Patients who had not experienced an OS event at the time of analysis were censored at the date last known alive.
HR is calculated using a stratified Cox proportional hazards model adjusted for disease stage and PD-L1 expression status at baseline. An HR < 1 favours durvalumab + chemotherapy, to be associated with longer OS than placebo + chemotherapy.
The data cut-off date was May 10, 2024.
Source: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23
Table 17: Summary of Key Survival Efficacy Results From the AEGEAN Study
Outcome | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 | Durvalumab + neoadjuvant chemotherapy N = 366 | Placebo + neoadjuvant chemotherapy N = 374 |
|---|---|---|---|---|
EFS IA1 (DCO date: November 10, 2022) | EFS IA2 (DCO date: May 10, 2024) | |||
Primary end point: EFS (using BICR assessment per RECIST 1.1) | ||||
Number of events, n (%)a | 98 (26.8) | 138 (36.9) | 124 (33.9) | 165 (44.1) |
RECIST 1.1 recurrence | 38 (10.4) | 60 (16.0) | 53 (14.5) | 83 (22.2) |
Progression that precludes surgery | 26 (7.1) | 35 (9.4) | 28 (7.7) | 36 (9.6) |
PD after surgery not undertaken for reasons other than progression | 1 (0.3) | 4 (1.1) | 3 (0.8) | 5 (1.3) |
PD discovered upon attempting surgery | 5 (1.4) | 13 (3.5) | 5 (1.4) | 13 (3.5) |
PD after surgery not completed for reasons other than progression | 0 (0) | 5 (1.3) | 0 (0) | 5 (1.3) |
Death due to any cause | 29 (7.9) | 30 (8.0) | 38 (10.4) | 33 (8.8) |
Number of censored patients, n (%) | 268 (73.2) | 236 (63.1) | 242 (66.1) | 209 (55.9) |
Censored RECIST 1.1 recurrence or PD | 2 (0.5) | 1 (0.3) | 2 (0.5) | 1 (0.3) |
Censored death | 12 (3.3) | 7 (1.9) | 25 (6.8) | 25 (6.7) |
Event-free at time of analysis | 239 (65.3) | 214 (57.2) | 199 (54.4) | 169 (45.2) |
No postbaseline disease assessments or no baseline data | 4 (1.1) | 6 (1.6) | 4 (1.1) | 6 (1.6) |
Lost to follow-up | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Withdrawn consent | 11 (3.0) | 8 (2.1) | 12 (3.3) | 8 (2.1) |
Discontinued study | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Median EFS, months (95% CI)b | Not reached (31.9 to not reached) | 25.9 (18.9 to not reached) | Not reached (42.3 to not reached) | 30.0 (20.6 to not reached) |
HR (95% CI)c | 0.68 (0.53 to 0.88) | 0.69 (0.55 to 0.88) | ||
Adjusted 99.0101% CIc,e | 0.48 to 0.96 | NR | ||
2-sided P valued, f | 0.003902 | █████g | ||
EFS at 12 months, % (95% CI)b | 73.4 (67.9 to 78.1) | 64.5 (58.8 to 69.6) | 73.3 (68.1 to 77.7) | 64.1 (58.7 to 69.0) |
Difference in survival probability, % (95% CI) | 8.9 (NR) | 9.2 ████ ██ █████ | ||
EFS at 24 months, % (95% CI)b | 63.3 (56.1 to 69.6) | 52.4 (45.4 to 59.0) | 65.0 (59.4 to 70.0) | 54.4 (48.7 to 59.6) |
Difference in survival probability, % (95% CI) | 10.9 (NR) | 10.6 ████ ██ █████ | ||
EFS at 36 months, % (95% CI)b | NR | NR | 60.1 (53.9 to 65.8) | 47.9 (41.8 to 53.8) |
Difference in survival probability, % (95% CI) | NR | 12.2 ████ ██ █████ | ||
Secondary end point: DFSh | NR | NR | N = 242 | N = 231 |
Number of events, n (%)i | NR | NR | 60 (24.8) | 81 (35.1) |
Disease recurrence | NR | NR | 44 (18.2) | 69 (29.9) |
Death due to any cause | NR | NR | 16 (6.6) | 12 (5.2) |
Number of censored patients, n (%) | NR | NR | 182 (75.2) | 150 (64.9) |
Censored disease recurrence | NR | NR | 1 (0.4) | 0 (0) |
Censored death | NR | NR | 8 (3.3) | 10 (4.3) |
Disease-free at time of analysis | NR | NR | 170 (70.2) | 138 (59.7) |
Lost to follow-up | NR | NR | 0 (0) | 0 (0) |
Withdrawn consent | NR | NR | 3 (1.2) | 2 (0.9) |
Discontinued study | NR | NR | 0 (0) | 0 (0) |
Median DFS, months (95% CI)c | NR | NR | Not reached (not reached to not reached) | Not reached (41.5 to not reached) |
HR (95% CI)c | NR | 0.66 (0.47 to 0.92) | ||
2-sided P valued | NR | 0.013652j | ||
DFS rate at 12 months, % (95% CI)b | NR | NR | 81.0 (75.2 to 85.5) | 74.1 (67.8 to 79.3) |
Difference in survival probability, % (95% CI) | NR | 6.9 █████ ██ █████ | ||
DFS rate at 24 months, % (95% CI)b | NR | NR | 75.1 (68.7 to 80.4) | 62.4 (55.2 to 68.8) |
Difference in survival probability, % (95% CI) | NR | 12.7 ████ ██ █████ | ||
DFS rate at 36 months, % (95% CI)b | NR | NR | 71.2 (63.8 to 77.3) | 61.4 (54.0 to 68.0) |
Difference in survival probability, % (95% CI) | NR | 9.8 █████ ██ █████ | ||
Secondary end point: OS | ||||
Death events, n (%) | 81 (22.1) | 82 (21.9) | 121 (33.1) | 140 (37.4) |
Number of censored patients, n (%) | 285 (77.9) | 292 (78.1) | 245 (66.9) | 234 (62.6) |
Still in survival follow-up | 271 (74.0) | 279 (74.6) | 230 (62.8) | 222 (59.4) |
Terminated study before death | 14 (3.8) | 13 (3.5) | 15 (4.1) | 12 (3.2) |
Lost to follow-up | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Withdrawal by patient | 14 (3.8) | 12 (3.2) | 15 (4.1) | 12 (3.2) |
Other | 0 (0) | 1 (0.3) | 0 (0) | 0 (0) |
Median OS, months (95% CI)b | Not reached (not reached to not reached) | Not reached (not reached to not reached) | Not reached (not reached to not reached) | 53.2 (44.3 to not reached) |
HR (95% CI)c | 1.02 (0.75 to 1.39) | 0.89 (0.70 to 1.14) | ||
2-sided P valuek | NC | NC | ||
OS probability at 12 months, % (95% CI)b | 83.6 (79.2 to 87.2) | 85.9 (81.7 to 89.1) | 84.3 (80.1 to 87.7) | 85.3 (81.2 to 88.5) |
Difference in survival probability, % (95% CI) | −2.3 (NR) | −1.0 █████ ██ ████ | ||
OS probability at 24 months, % (95% CI)b | 71.7 (65.2 to 77.2) | 72.0 (65.5 to 77.5) | 74.4 (69.5 to 78.6) | 72.2 (67.3 to 76.5) |
Difference in survival probability, % (95% CI) | −0.3 (NR) | 2.2 █████ ██ ████ | ||
OS probability at 36 months, % (95% CI)b | NR | NR | 67.1 (61.6 to 71.9) | 63.9 (58.4 to 69.0) |
Difference in survival probability, % (95% CI) | NR | 3.2 █████ ██ █████ | ||
OS probability at 48 months, % (95% CI)b | NR | NR | 57.6 (50.1 to 64.3) | 52.9 (45.7 to 59.7) |
Difference in survival probability, % (95% CI) | NR | 4.7 █████ ██ █████ | ||
BICR = blinded independent central review; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DCO = data cut-off; DFS = disease-free survival; EFS = event-free survival; HR = hazard ratio; IA1 = interim analysis 1; IA2 = interim analysis 2; IXRS = interactive web response system; mITT = modified intention to treat; MTP = multiple testing procedure; NA = not applicable; NC = not calculated; NR = not reported; NSCLC = non–small cell lung cancer; OS = overall survival; PD = progressive disease; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1.
aTwo missed visits rule applied.
bCalculated using Kaplan-Meier technique.
cCalculated using a stratified Cox proportional hazards model adjusted for disease stage and PD-L1 expression status at baseline. An HR < 1 favours perioperative durvalumab + neoadjuvant chemotherapy over placebo + neoadjuvant chemotherapy.
dP value has been adjusted for multiple testing.
eBased on a Lan-DeMets alpha spending function with an O'Brien-Fleming boundary with the observed number of events, the boundary for declaring statistical significance is 0.9899% for a 5% overall alpha, as per RECIST 1.1.
fCalculated using a stratified log-rank test adjusting for disease stage and PD-L1 expression status at baseline.
gAs IA1 was statistically significant per the MTP, this updated analysis is considered exploratory and a nominal P value has been included for information only rather than for formal hypothesis testing.
hEstimated among the modified resected set.
iDoes not include events occurring after 2 missed RECIST 1.1 assessments. A new malignancy that is not NSCLC, as confirmed by pathology, is not considered a DFS event.
jA P value of < 0.012303 was required to declare statistical significance at IA2.
kAt the DCO date of EFS IA1 and EFS IA2, OS was not eligible for statistical testing (because DFS was not statistically significant, OS was not formally tested based on the MTP).
Sources: AEGEAN study EFS IA1 Clinical Study Report and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures. 24,23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Health-Related Quality of Life
At the EFS IA1 DCO (November 10, 2022), HRQoL was evaluated in the mITT population for the neoadjuvant period using EORTC QLQ-C30 and EORTC QLQ-LC13; at the EFS IA2 DCO (May 10, 2024), HRQoL was reassessed in the modified resected set for the adjuvant period using just the EORTC QLQ-C30 measure.
At the EFS IA1 neoadjuvant baseline, the adherence rate for EORTC QLQ-C30 and EORTC QLQ-LC13 was █████ █████████ for the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ █████████ for the placebo with neoadjuvant chemotherapy arm; by week 12, these rates were █████ ████ evaluable forms/███ patients still under PRO follow-up at week 12) and █████ ████ evaluable forms/███ patients still under PRO follow-up at week 12). At neoadjuvant week 12, there were █████ of patients (███ out of ███ patients at baseline) available to provide assessments in the perioperative durvalumab with neoadjuvant chemotherapy group and █████ of patients (███ out of ███ patients at baseline) available to provide assessments in the placebo with neoadjuvant chemotherapy group.
At the EFS IA2 adjuvant baseline, the adherence rate was █████ █████████ and █████ █████████, respectively, and at week 44, they were █████ ████ evaluable forms/███ patients still under PRO follow-up at week 44) and █████ ████ evaluable forms/███ patients still under PRO follow-up at week 44). At adjuvant week 44, there were █████ of patients (███ out of ███ patients at adjuvant baseline) available to provide assessments in the perioperative durvalumab with neoadjuvant chemotherapy group and █████ of patients (███ out of ███ patients at adjuvant baseline) available to provide assessments in the placebo with neoadjuvant chemotherapy group.
Baseline EORTC QLQ-C30 and EORTC QLQ-LC13 were similar across both treatment arms (Appendix 3, Table 50 and Table 52). At adjuvant baseline, all scores other than ████ ███████████ (mean [standard deviation] perioperative durvalumab with neoadjuvant chemotherapy arm = ████ ███████ versus ████ ███████ in the placebo with neoadjuvant chemotherapy arm) and ██████ ███████████ (mean [standard deviation] perioperative durvalumab with neoadjuvant chemotherapy arm = ████ ███████ versus ████ ███████ in the placebo with neoadjuvant chemotherapy arm) were similar (Appendix 3, Table 51).
Given that the between-group differences in EORTC QLQ-C30 global health status and QoL, physical functioning, role functioning, fatigue, and appetite loss, and in EORTC QLQ-LC13 dyspnea, coughing, and pain in chest were estimated in the AEGEAN trial and deemed to be important by the clinical experts consulted for this review, these outcomes are reported in this section.
During the neoadjuvant period, the point estimate for the between-group difference for the change in EORTC QLQ-C30 global health status and QoL score from baseline to week 12 ███ ███ meet the MID threshold of 4 points (estimated mean difference [95% CI] = █████ ██████ ██ ██████ Table 18).63 However, a between-group difference in the change in EORTC QLQ-C30 global health status and QoL score from adjuvant baseline to week 44 of █████ ████ ████████ ██ ██████ was observed during the adjuvant period (Table 19)
During the neoadjuvant period, the largest between-group difference for the change in scores from baseline to week 12 was observed in the ███████ domain of the EORTC QLQ-C30 (estimated mean difference [95% CI] = ████ █████ ██ ██████ and the smallest was for ████ ██ █████ measured by EORTC QLQ-LC13 (estimated mean difference [95% CI] = █████ ██████ ██ ██████ Table 18). During the adjuvant period, only the EORTC QLQ-C30 was measured and the largest between-group difference for the change in scores from adjuvant baseline to week 44 was ████ ███████████ (estimated mean difference [95% CI] = █████ ███████ ██ ███████ and the smallest was for ████████ ████ (estimated mean difference [95% CI] = ████ ██████ ██ ████; Table 19).
Table 18: Change From Neoadjuvant Baseline to Week 12 in EORTC QLQ-C30 Version 3 and EORTC QLQ-LC13 Scores by MMRM Analysis (mITT Population)
Variable | Durvalumab + neoadjuvant chemotherapy (N = 366) | Placebo + neoadjuvant chemotherapy (N = 374) |
|---|---|---|
EORTC QLQ-C30 version 3: Global measure of health status and quality of life | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ █████ ██ ██████ | █████ █████ ██ ██████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-C30 version 3 function: Physical | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ █████ ██ ██████ | █████ █████ ██ ██████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-C30 version 3 function: Role | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ ██████ ██ ██████ | █████ ██████ ██ ██████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-C30 version 3 symptom: Fatigue | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ █████ ██ █████ | ████ █████ ██ █████ |
Estimated difference (95% CI) | ████ █████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-C30 version 3 symptom: Appetite loss | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | ████ ████ ██ █████ | ████ ████ ██ ██████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-LC13 symptom: Dyspnea | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | ████ ████ ██ █████ | ████ ████ ██ █████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-LC13 symptom: Coughing | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ ██████ ██ ██████ | ██████ ██████ ██ ██████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valuea | █████ | |
EORTC QLQ-LC13 symptom: Pain in chest | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from neoadjuvant baseline to week 12 (95% CI) | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
CI = confidence interval; EFS = event-free survival; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; IA1 = interim analysis 1; mITT = modified intention to treat; MMRM = mixed model of repeated measures.
Notes: The analysis was performed using an MMRM analysis of change from baseline for all postbaseline adjuvant visits, with treatment, visit, and treatment by visit interaction included as explanatory variables and the baseline score as a covariate. Neoadjuvant baseline was the last predose assessment or screening if this is missing. Time points are only included if at least 20 patients are present in each treatment group.
A higher score on the global measure of health status and quality of life and functioning scales indicates better health status and function; thus, a positive change score reflects improvement. Higher scores on symptom scales and items represent greater symptom severity; thus, a negative change score reflects improvement.
The data cut-off date was November 10, 2022.
aNot adjusted for multiple testing.
Sources: AEGEAN study EFS IA1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 19: Change From Adjuvant Baseline to Week 44 in EORTC QLQ-C30 Version 3 Scores by MMRM Analysis (Modified Resected Set)
Outcome | Durvalumab + neoadjuvant chemotherapy (N = 242) | Placebo + neoadjuvant chemotherapy (N = 231) |
|---|---|---|
Global measure of health status and quality of life | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from adjuvant baseline to week 44 (95% CI) | █████ █████ ██ █████ | ████ █████ ██ █████ |
Estimated difference (95% CI) | █████ ██████ ██ ██████ | |
P valuea | █████ | |
Function: Physical | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from adjuvant baseline to week 44 (95% CI) | ████ ████ ██ █████ | ████ █████ ██ █████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valuea | █████ | |
Function: Role | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from adjuvant baseline to week 44 (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ |
Estimated difference (95% CI) | █████ ███████ ██ ██████ | |
P valuea | █████ | |
Symptom: Fatigue | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from adjuvant baseline to week 44 (95% CI) | █████ █████ ██ █████ | █████ ██████ ██ █████ |
Estimated difference (95% CI) | ████ █████ ██ █████ | |
P valuea | █████ | |
Symptom: Appetite loss | ||
Number of patients contributing to the analysis, n | ███ | ███ |
Adjusted mean change from adjuvant baseline to week 44 (95% CI) | █████ ██████ ██ ██████ | ██████ ██████ ██ ██████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valuea | █████ | |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; MMRM = mixed model of repeated measures.
Notes: The analysis was performed using an MMRM analysis of change from baseline for all postbaseline adjuvant visits, with treatment, visit, and treatment by visit interaction included as explanatory variables and the baseline score as a covariate. Adjuvant baseline is the latest measurement after surgery but before the first dose of adjuvant treatment. Time points are only included if at least 20 patients were present in each treatment group.
A higher score on the global measure of health status and quality of life and functioning scales indicates better health status and function; thus, a positive change score reflects improvement. Higher scores on symptom scales and items represent greater symptom severity; thus, a negative change score reflects improvement.
The data cut-off date was May 10, 2024.
aNot adjusted for multiple testing.
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Safety Evaluation Plan
Data presented in this section are from the EFS IA2 DCO (May 10, 2024). An overview of safety data is initially presented for the modified safety analysis set (consisting of all patients in the AEGEAN trial who received at least 1 dose of the study treatment and excluding those whose tumours had EGFR mutations or ALK gene rearrangements; N = 367 for the perioperative durvalumab with neoadjuvant chemotherapy arm and N = 370 for the placebo with neoadjuvant chemotherapy arm). As granular safety data were not available for the modified safety analysis set, additional data are presented for the safety analysis set, which consists of all randomized patients who received at least 1 dose of study treatment, with treatment arm allocation in accordance with the treatment actually received. The safety analysis set consisted of 799 patients (N = 401 in the perioperative durvalumab with neoadjuvant chemotherapy arm and N = 398 in the placebo with neoadjuvant chemotherapy arm).
Table 20 presents data for the modified safety analysis set and Table 21 presents safety data from the overarching safety analysis set; both tables present data from the EFS IA2 DCO. Table 22 presents summaries of AEs for the safety analysis set from the EFS IA2 DCO by study period (i.e., neoadjuvant period, postsurgery period, adjuvant period, and overall study period).
Adverse Events
At the EFS IA2 DCO of May 10, 2024, a ███████ percentage of patients in both treatment arms of the modified safety analysis set experienced 1 or more AE over the course of treatment (█████ ████████ in the perioperative durvalumab with neoadjuvant chemotherapy arm versus █████ ████████ in the placebo with neoadjuvant chemotherapy arm).
Similar results were observed in the safety analysis set (96.5% of patients [387 of 401] in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 95.2% of patients [379 of 398] in the placebo with neoadjuvant chemotherapy arm). The 3 most frequently reported AEs in both treatment arms of the safety analysis set were anemia (34.9% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 32.2% in the placebo with neoadjuvant chemotherapy arm), nausea (25.7% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 29.9% in the placebo with neoadjuvant chemotherapy arm), and constipation (25.9% in the perioperative durvalumab with neoadjuvant chemotherapy arm and 29.9% in the placebo with neoadjuvant chemotherapy arm).
Grade 3 or grade 4 AEs occurred in a ███████ percentage of patients in both arms of the modified safety analysis set arms (██████████████ in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ ████████ in the placebo with neoadjuvant chemotherapy arm). Similarly, in the overall safety analysis set, 43.6% of patients (175 of 401) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 43.2% of patients (172 of 398) in the placebo with neoadjuvant chemotherapy arm experienced a grade 3 or grade 4 AE.
Serious AEs
There were ████████ ████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm of the modified safety analysis set who experienced 1 or more SAE (█████ █████████) than in the placebo with neoadjuvant chemotherapy arm (█████ █████████).
Similar results were observed over the same time period in the overall safety analysis set (39.2% of patients [157 of 401] in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 31.7% of patients [126 of 398] in the placebo with neoadjuvant chemotherapy arm experienced 1 or more SAE). In the safety analysis set, the 4 most frequently reported SAEs in the perioperative durvalumab with neoadjuvant chemotherapy arm were pneumonia (5.7%), anemia (1.7%), COVID-19 (1.7%), and pneumonitis (1.7%) while in the placebo with neoadjuvant chemotherapy arm, they were pneumonia (4.5%), pneumothorax (2.3%), anemia (1.3%), and COVID-19 (1.3%).
Withdrawals Due to AEs
████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm of the modified safety analysis set prematurely stopped study treatment (i.e., durvalumab, placebo, or chemotherapy) due to AEs than in the placebo with neoadjuvant chemotherapy arm (█████ ████████ versus █████ ███████, respectively).
Similar results were observed in the safety analysis set (19.5% of patients [78 of 401] in the perioperative durvalumab with neoadjuvant chemotherapy arm versus 9.8% of patients [39 of 398] in the placebo with neoadjuvant chemotherapy arm discontinued treatment due to AEs), with the most common AEs leading to treatment discontinuation being blood and lymphatic system disorders and respiratory, thoracic, and mediastinal disorders in the perioperative durvalumab with neoadjuvant chemotherapy arm compared to respiratory, thoracic, and mediastinal disorders and investigations in the placebo with neoadjuvant chemotherapy arm.
Mortality
As of May 10, 2024, in the modified resected set ████ ████████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and ████ ███████) of patients in the placebo with neoadjuvant chemotherapy arm experienced an AE resulting in death.
Results were similar in the safety analysis set, with 5.7% of patients (23 of 401) in the perioperative durvalumab with neoadjuvant chemotherapy arm and 3.8% of patients (15 of 398) in the placebo with neoadjuvant chemotherapy arm who experienced an AE resulting in death. In this analysis set, the most common AE in both arms resulting in death was infections and infestations.
Notable Harms
By May 10, 2024, in the modified safety analysis set, ████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm had experienced an AESI or AEPI ██████ ████████) than in the placebo with neoadjuvant chemotherapy arm (█████ █████████). imAEs occurred in █████ ████████) of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and in █████ ███████) of patients in the placebo with neoadjuvant chemotherapy arm.
Similar results were observed in the safety analysis set over the same time period (66.8% of patients [268 of 401] in the perioperative durvalumab with neoadjuvant chemotherapy arm and 52.5% of patients [209 of 398] in the placebo with neoadjuvant chemotherapy arm experienced an AESI or AEPI), with ███████████████ events being the most common type in both arms. ImAEs occurred in 25.4% of patients [102 of 401] in the perioperative durvalumab with neoadjuvant chemotherapy arm and 10.3% of patients [41 of 398] in the placebo with neoadjuvant chemotherapy arm.
Table 20: Overview of AEs by Category (Modified Safety Analysis Set — Overall Study Period at EFS IA2)
AEa | Neoadjuvant period | |
|---|---|---|
Durvalumab + neoadjuvant chemotherapy N = 367 | Placebo + neoadjuvant chemotherapy N = 370 | |
Any AE, n (%) | ███ ██████ | ███ ██████ |
Any AE of grade 3 or grade 4, n (%) | ███ ██████ | ███ ██████ |
Any AE with outcome of death, n (%) | ██ █████ | ██ █████ |
Any AE leading to discontinuation of any study treatment, n (%)b | ██ ██████ | ██ ██████ |
Any AE leading to dose interruption or reduction of study treatment, n (%)b | ███ ██████ | ███ ██████ |
Any AE leading to surgery not done, n (%)c | ██████ | ██████ |
Any AE leading to a delay in surgery, n (%)c,d | ██ █████ | ██ █████ |
Any AE leading to a delay in adjuvant treatment, n (%) | ██████ | ██████ |
Any AE leading to adjuvant treatment not given, n (%) | ██ █████ | ██ █████ |
Any SAE (including events with outcome of death), n (%) | ███ ██████ | ███ ██████ |
Any AESI or AEPIe | ███ ██████ | ███ ██████ |
Immune-mediated AEs | ███ ██████ | ██ ██████ |
Infusion-reaction AEs | ██ █████ | ██ █████ |
AE = adverse event; AEPI = adverse event of potential interest; AESI = adverse event of special interest; EFS = event-free survival; IA2 = interim analysis 2; PT = preferred term; SAE = serious adverse event.
Note: The data cut-off date was May 10, 2024.
aPatients with multiple events in the same category are counted only once in that category. Patients with events in more than 1 category are counted once in each of those categories.
bStudy treatment refers to durvalumab and/or placebo or any chemotherapy and does not include surgery.
cTaken from the SURG module.
dDue to either an AE or unresolved toxicity.
eAn AESI or AEPI is of scientific and medical interest to the study treatment. An AESI or AEPI may be serious or nonserious. Incudes AEs between the date of the first dose and the earliest of the following: the maximum date of (last dose or surgery) + 90 days, or the date of the first dose of subsequent anticancer therapy. Includes AEs with an onset date during this period and AEs with an onset date before dosing that worsen during this period.
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 21: Summary of Key Harms Data in the AEGEAN Study (Safety Analysis Set — Overall Study Period at EFS IA2)
AE | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 |
|---|---|---|
EFS IA2 | ||
Most common AEs (occurring in ≥ 10% of patients in either study arm), n (%)a | ||
≥ 1 adverse event | 387 (96.5) | 379 (95.2) |
Anemia | 140 (34.9) | 128 (32.2) |
Nausea | 103 (25.7) | 119 (29.9) |
Constipation | 104 (25.9) | 90 (22.6) |
Decreased appetite | 74 (18.5) | 73 (18.3) |
Alopecia | 69 (17.2) | 64 (16.1) |
Neutropenia | 68 (17.0) | 72 (18.1) |
Neutrophil count, decreased | 66 (16.5) | 59 (14.8) |
Rash | 63 (15.7) | 33 (8.3) |
Fatigue | 56 (14.0) | 47 (11.8) |
COVID-19 | 54 (13.5) | 45 (11.3) |
Diarrhea | 53 (13.2) | 51 (12.8) |
Pruritus | 51 (12.7) | 26 (6.5) |
Asthenia | 50 (12.5) | 58 (14.6) |
Hypothyroidism | 47 (11.7) | 12 (3.0) |
Procedural pain | 46 (11.5) | 49 (12.3) |
Vomiting | 45 (11.2) | 44 (11.1) |
Arthralgia | 44 (11.0) | 51 (12.8) |
Insomnia | 43 (10.7) | 48 (12.1) |
Cough | 36 (9.0) | 46 (11.6) |
SAEs (occurring in ≥ 1.5% of patients in either study arm), n (%)a | ||
Patients with ≥ 1 SAE | 157 (39.2) | 126 (31.7) |
Pneumonia | 23 (5.7) | 18 (4.5) |
Anemia | 7 (1.7) | 5 (1.3) |
COVID-19 | 7 (1.7) | 5 (1.3) |
Pneumothorax | 4 (1.0) | 9 (2.3) |
Pneumonitis | 7 (1.7) | 1 (0.3) |
Myelosuppression | 6 (1.5) | 2 (0.5) |
Patients who stopped treatment due to AEs (occurring in ≥ 1.5% of patients in either study arm), n (%)b | ||
Any AE leading to discontinuation of any study treatment | 78 (19.5) | 39 (9.8) |
Blood and lymphatic system disorders | ██ █████ | ██████ |
Respiratory, thoracic, and mediastinal disorders | ██ █████ | ██████ |
Nervous system disorders | ██ █████ | ██████ |
Investigations | ██████ | ██████ |
Skin and subcutaneous tissue disorders | ██████ | █ |
Deaths (occurring in ≥ 1 patient in either study arm), n (%) | ||
Patients with any AE with an outcome of death | 23 (5.7) | 15 (3.8) |
Infections and infestations | 13 (3.2) | 7 (1.8) |
Respiratory, thoracic, and mediastinal disorders | 6 (1.5) | 2 (0.5) |
Cardiac disorders | 1 (0.2) | 3 (0.8) |
Metabolism and nutrition disorders | 1 (0.2) | 1 (0.3) |
General disorders and administration site conditions | 1 (0.2) | 1 (0.3) |
Vascular disorders | 1 (0.2) | 0 (0) |
Injury, poisoning, and procedural complications | 1 (0.2) | 0 (0) |
Nervous system disorders | 0 (0) | 1 (0.3) |
Gastrointestinal disorders | 0 (0) | 1 (0.3) |
AESIs or AEPIs (by category), n (%) | ||
Patients with any AESI or AEPI | 268 (66.8) | 209 (52.5) |
███████████████ ██████ | ███ ██████ | ██ ██████ |
███████ ██████ | ██ ██████ | ██ ██████ |
████████████████ | ██ ██████ | ██ ██████ |
█████ ██████████████████ ███ | ██ ██████ | ██ ██████ |
████████████ ██████ | ██ ██████ | ██ █████ |
███████████ | ██ █████ | ██ █████ |
██████████ ██████ | ██ █████ | ██ █████ |
███████████ ██████ | ██ █████ | ██ █████ |
█████ ██████ | ██ █████ | ██ █████ |
█████████████████████████ █████████ | ██ █████ | ██████ |
███████ █████████████ | ██████ | █ |
████████ ██████ | ██████ | ██████ |
███████████ | ██████ | ██████ |
███████████ ██████ | ██████ | ██████ |
██████████ ██████ | ██████ | █ |
████ █ ████████ ████████ | ██████ | █ |
██████████ ████████████ | █ | █ |
████████████ | █ | █ |
██████████████ ████████ | █ | █ |
imAEs, n (%) | ||
Any imAE-related AE | 102 (25.4) | 41 (10.3) |
AE = adverse event; AEPI = adverse event of potential interest; AESI = adverse event of special interest; EFS = event-free survival; IA2 = interim analysis 2; imAE = immune-mediated adverse event; MedDRA = Medical Dictionary for Regulatory Activities; SAE = serious adverse event.
Notes: Includes AEs between the date of the first dose and the earliest of the following: the maximum date of (last dose or surgery) + 90 days or the date of the first dose of subsequent anticancer therapy. Includes AEs with an onset date during this period and AEs with an onset date before dosing that worsen during this period.
Reported by preferred term per MedDRA version 25.1.
The data cut-off date was May 10, 2024.
aPatients with multiple events are counted once per preferred term.
bAction taken: Drug permanently discontinued. Study treatment includes durvalumab, placebo, and chemotherapy.
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 22: Overview of AEs by Category and by Study Treatment Period (Safety Analysis Set at EFS IA2)
AEa | Neoadjuvant period | Postsurgery period | Adjuvant period | Overall period | ||||
|---|---|---|---|---|---|---|---|---|
Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 | Durvalumab + neoadjuvant chemotherapy N = 325 | Placebo + neoadjuvant chemotherapy N = 326 | Durvalumab + neoadjuvant chemotherapy N = 266 | Placebo + neoadjuvant chemotherapy N = 254 | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 | |
Any AE, n (%) | 365 (91.0) | 357 (89.7) | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ | 387 (96.5) | 379 (95.2) |
Any AE of grade 3 or grade 4, n (%) | 131 (32.7) | 145 (36.4) | ██ █████ | ██ █████ | ██ █████ | ██ █████ | 175 (43.6) | 172 (43.2) |
Any AE with outcome of death, n (%) | 8 (2.0) | 4 (1.0) | ██ █████ | █████ | █████ | █████ | 23 (5.7) | 15 (3.8) |
Any AE leading to discontinuation of study treatment, n (%)b | 54 (13.5) | 30 (7.5) | ██ | ██ | ██ █████ | ██ █████ | 78 (19.5) | 39 (9.8) |
Any AE leading to dose interruption or reduction, n (%) | 148 (36.9) | 126 (31.7) | ██ | ██ | ██ █████ | ██ █████ | 194 (48.4) | 148 (37.2) |
Any AE leading to surgery not done, n (%)c | 7 (1.7) | 4 (1.0) | ██ | ██ | ██ | ██ | 7 (1.7) | 4 (1.0)c |
Any AE leading to a delay in surgery, n (%)d | 15 (3.7) | 16 (4.0) | ██ | ██ | ██ | ██ | 15 (3.7) | 16 (4.0) |
Any AE leading to a delay in adjuvant treatment, n (%) | NA | NA | ██████ | ██████ | ██ | ██ | 2 (0.5) | 1 (0.3) |
Any AE leading to adjuvant treatment not given, n (%) | NA | NA | ██ █████ | ██ █████ | ██ | ██ | 28 (7.0) | 20 (5.0) |
Any SAE (including events with outcome of death), n (%) | 83 (20.7) | 66 (16.6) | ██ █████ | ██ █████ | ██ █████ | ██ █████ | 157 (39.2) | 126 (31.7) |
Any AESIs or AEPIs, n (%) | 191 (47.6) | 152 (38.2) | ██ █████ | ██ █████ | ██ █████ | ██ █████ | 268 (66.8) | 209 (52.5) |
Immune-mediated AEs, n (%) | 33 (8.2) | 19 (4.8) | ██ █████ | ██████ | ██ █████ | ██ █████ | 102 (25.4) | 41 (10.3) |
Infusion-reaction AEs, n (%) | ██ █████ | ██ █████ | █ | ██████ | █ | █ | ██ █████ | ██ █████ |
AE = adverse event; AEPI = adverse event of potential interest; AESI = adverse event of special interest; EFS = event-free survival; IA2 = interim analysis 2; NA = not applicable; PT = preferred term; SAE = serious adverse event.
Note: The data cut-off date was May 10, 2024.
aPatients with multiple events in the same category are counted only once in that category. Patients with events in more than 1 category are counted once in each of those categories.
bStudy treatment refers to durvalumab, placebo, and any chemotherapy and does not include surgery.
cTaken from the SURG form. Note that 1 additional patient in the placebo + neoadjuvant chemotherapy arm had an AE leading to surgery not done (5 patients in total), without an AE number (identifying the PT) specified on the SURG form.
dTaken from the SURG form (including AE and unresolved toxicity). Note that 1 additional patient in the durvalumab + chemotherapy arm had an AE (unresolved toxicity) leading to a delay in surgery, without an AE number (identifying the PT) specified on the SURG form (16 patients in total) and 1 additional patient in the placebo + neoadjuvant chemotherapy arm had an AE leading to a delay in surgery, without an AE number (identifying the PT) specified on the SURG form (17 patients in total).
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The AEGEAN study is a double-blind, placebo-controlled, phase III RCT. The study used computer-generated blocked randomization with stratification based on PD-L1 status and disease stage. Allocation concealment methods were adequate. With the exception of a higher percentage of males and a slightly higher percentage of squamous histology in the perioperative durvalumab with neoadjuvant chemotherapy arm — which was deemed to be likely attributable to chance — the study generally reports balanced baseline characteristics suggesting that randomization was successful. Further, the postrandomization exclusion of those with known EGFR mutations or ALK rearrangements were judged not to be related to treatment group assignment, and therefore were not expected to introduce bias. The patients, investigators, and study centre staff were all blinded to the durvalumab or placebo allocation while the study centre pharmacists were unblinded. In the case of a medical emergency, investigators could request to be unblinded.
Patient disposition was similar across both mITT arms, with a similar percentage of patients in each arm completing neoadjuvant treatment, receiving and completing surgery, receiving PORT, and completing adjuvant treatment. As of the most recent DCO on May 10, 2024, no patients had been lost to follow-up (defined as no contact established by the time of the DCO such that there was insufficient information to determine the patient’s status at that time). However, 20 patients had withdrawn consent (i.e., withdrawn from further study treatment and declined to allow any further follow-up information to be obtained: 12 of 366 patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and 8 of 374 patients in the placebo with neoadjuvant chemotherapy arm). The disallowed concomitant medications administered in the AEGEAN study did not raise any concerns on the outcome measures of the study.
pCR, MPR, EFS, DFS, and OS end points were addressed using the multiplicity hierarchical testing procedure, which controlled for type I error. The criteria used for disease progression and tumour response are based on radiographic images and clinical assessment, with the potential for subjective interpretation. To mitigate this potential bias, a blinded IRC reviewed all available radiographic tumour assessments to determine tumour response based on RECIST 1.1 criteria. As such, the risk of bias resulting from unblinding due to unbalanced harms across treatment arms is lower for pCR, MPR, and survival outcomes; however, there are some concerns for HRQoL outcomes.
While pCR and MPR are based on final analyses, EFS, DFS, OS, and HRQoL results should be interpreted in light of the fact that these are based on IAs, which may overestimate treatment effects.20 The HRs and CIs for survival outcomes were estimated from the stratified Cox proportional hazards model adjusted for stratification factors, which relies on the assumption of proportional hazards in both treatment arms. Statistically, there was insufficient evidence to suggest that the nonproportional hazards assumption had been violated from the Grambsch-Therneau proportional hazards test (at EFS IA2, P value for EFS = █████, P value for OS = █████, P value for DFS = █████). However, for EFS and OS results, there was some evidence from visual inspection of log-log plots and a Schoenfeld residuals plot that the proportional hazards assumption may have been violated. Visual inspection of the KM plots by the CDA-AMC review team revealed that for EFS and DFS, the curves for the intervention and comparator treatment did not separate until approximately 4 months. In the KM plot for OS, the curves crossed multiple times and did not begin to separate until approximately 34 months. As such, it is possible that the estimated HRs for these end points may be misleading, given that HRs may not remain constant over time. Nevertheless, all relevant information contributed to the evaluation of these end points, and the overall certainty of evidence was not affected because it was informed by absolute between-group differences in event-free probabilities at clinically relevant time points. While subgroup analyses were planned a priori and generally showed consistent results with those of the main analysis, they may not have been powered to identify differences.
While there was an adequate number of patients at risk in the AEGEAN trial for earlier DFS and OS time points, only about 11% of patients in both treatment arms remained at risk for OS at 48 months and 13% to 19% remained at risk for DFS at 36 months, resulting in substantial uncertainty in results at later time points. At the time of the EFS IA2 DCO, the median OS had not yet been reached in the perioperative durvalumab with neoadjuvant chemotherapy group. Across the treatment groups, 35% of patients had died. Visual inspection of the KM curves suggests substantial censoring after 24 months in both groups, contributing to uncertainty in the OS results, particularly at longer follow-up times. OS was not eligible for statistical testing at this IA because DFS was not statistically significant.
At the most recent DCO on May 10, 2024, DFS and OS data remained statistically nonsignificant, with the median time to a DFS event NR in either treatment group and the median time to a survival event only reached in the placebo with neoadjuvant chemotherapy group. Further, results for DFS and adjuvant HRQoL were estimated among the modified resected set (n = 473) and, because this is a subgroup of the mITT population (n = 740), randomization was not maintained, so there is an increased risk of prognostic imbalances across the treatment arms in this analysis population.
While previous systematic literature reviews (SLRs) and meta-analyses have found positive correlations between EFS and DFS with OS,68,69 results are limited to treatment with neoadjuvant and adjuvant chemotherapy, which cannot be generalized to perioperative durvalumab. As such, the validity of EFS or DFS as a surrogate for OS in the current context is unknown.
While a ███████ percentage of patients in both treatment arms of the modified safety analysis set experienced any AE over the course of treatment, ████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm stopped treatment early due to AEs and had an imAE. Clinical experts consulted for this review indicated the ██████ percentage of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm experiencing an imAE was expected and deemed the safety profile to be acceptable.
A higher percentage of patients in the placebo with neoadjuvant chemotherapy arm received subsequent anticancer therapy than in the perioperative durvalumab with neoadjuvant chemotherapy arm. This differential could potentially introduce a confounding effect on OS because the survival results might be partially attributable to treatments administered after disease progression rather than the study treatment itself. However, the risk of bias due to deviations from the intended interventions in the AEGEAN trial was deemed to be low and clinical experts confirmed that the subsequent anticancer treatments used in the AEGEAN trial were in line with those used in clinical practice in Canada. Further, given that subsequent treatments are expected to be used in real-world clinical practice, this limitation is not expected to impact the generalizability of the results.
Biases resulting from the self-reporting nature of HRQoL outcomes were deemed to be minimal given that patients were blinded to the intervention received. In the EORTC QLQ-C30 analyses, █████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ of patients in the placebo with neoadjuvant chemotherapy arm were available to provide assessments at week 12 of the neoadjuvant period. In the modified resected set, by week 44 of the adjuvant period █████ of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ of patients in the placebo with neoadjuvant chemotherapy arm were available to provide EORTC QLQ-C30 assessments. No data imputations were involved in these analyses, so there is a risk of bias due to missing outcomes data. Further, given that analyses of the HRQoL end points were not adjusted for multiple testing, there is an increased risk of type I error for statistically significant results.
External Validity
With the exception of excluding those with an ECOG PS score of 2, the clinical experts consulted for this review felt the inclusion and exclusion criteria used in the AEGEAN study were generally aligned with what would be expected in NSCLC clinical trials. While the AEGEAN trial only enrolled 8 Canadians in the ITT population (4 in each arm), which may lower the generalizability of the results to the setting in Canada, clinical experts consulted for this review deemed the baseline characteristics to align with those seen in clinical practice in Canada. Nonetheless, there were a few generalizability concerns. For example, the study primarily included Asian and white patients (704 of 740 patients [95.1%] in the mITT population), potentially limiting the generalizability of findings to Canadians of different racial groups. There were relatively few patients who were younger than 50 years (37 of 740 patients [5.0%] in the mITT population) and who were aged 75 years or older (80 of 740 patients [10.8%] in the mITT population). The clinical experts consulted on this review indicated that the age distribution was in line with clinical practice in Canada, where NSCLC is relatively rare in people aged younger than 50 years. Finally, there were relatively few patients who had stage IIA NSCLC (42 of 740 patients [5.7%] in the mITT population); however, the clinical experts consulted for this review noted that the low percentage of patients with stage IIA disease is expected because these patients are typically treated with surgery followed by adjuvant treatment. The distribution of the remaining patients across NSCLC stage IIB (23.1% of patients [172 of 740]), stage IIIA (45.7% of patients [338 of 740]), and stage IIIB (25.1% of patients [186 of 740]) was in line with what clinical experts would expect to see in the clinical setting in Canada.
The outcomes measured in the AEGEAN trial evaluated the key treatment goals identified by patient input collected for this review and were deemed to be relevant by clinical experts consulted for this review. Further, clinical experts consulted for this review deemed the efficacy and harms outcomes of the AEGEAN trial’s comparator arm, placebo with neoadjuvant chemotherapy, to be in line with their expectations. When the AEGEAN study initially started recruiting patients, neoadjuvant chemotherapy followed by surgery was common practice for the treatment of resectable NSCLC; however, clinical experts consulted for this review noted that this treatment strategy is now rarely used in clinical practice and surgery followed by adjuvant chemotherapy may be a more relevant comparator. The clinical experts consulted for this review noted that currently, the 2 most commonly used regimens in clinical practice in Canada are neoadjuvant nivolumab with chemotherapy and adjuvant chemotherapy. According to the clinical experts consulted for this review, adjuvant atezolizumab monotherapy after surgery and adjuvant chemotherapy for patients with PD-L1 of 50% or greater was the least relevant comparator because the treatment decision for perioperative durvalumab has to be made before surgery, whereas the approach for adjuvant atezolizumab would be considered after surgery and adjuvant chemotherapy. In addition to the standard of care chemotherapy regimens used in the AEGEAN study, clinical experts indicated that in Canada, carboplatin and gemcitabine could also be used for squamous cell NSCLC and cisplatin and vinorelbine may also sometimes be used for both squamous and nonsquamous cell NSCLC. The lack of these regimens used for standard chemotherapy in the AEGEAN study further limits the generalizability to the setting in Canada; however, clinical experts consulted for this review noted that they would feel comfortable combining perioperative durvalumab with these regimens.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.21,22
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy for adults with resectable NSCLC without any EGFR mutations or ALK rearrangements. The selected outcomes for GRADE assessment include OS, EORTC QLQ-C30 global health score and QoL, EFS, pCR, imAEs, and SAEs.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
In the absence of head-to-head trial data of durvalumab in combination with chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery (also termed as perioperative durvalumab with neoadjuvant chemotherapy throughout this section) compared with relevant comparators for the treatment of patients with resectable (tumours ≥ 4 cm and/or node positive) NSCLC and no known EGFR mutations or ALK rearrangements, the sponsor conducted 3 ITCs: 2 MAICs that assessed the comparative efficacy (i.e., EFS) of perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant nivolumab with chemotherapy and versus perioperative pembrolizumab with chemotherapy, and an NMA that assessed the efficacy (i.e., EFS) of perioperative durvalumab with neoadjuvant chemotherapy versus adjuvant chemotherapy (i.e., surgery + adjuvant chemotherapy), and surgery only. The objective of this section is to summarize and critically appraise the sponsor-submitted ITCs.
Description of Indirect Comparisons
The objective of the ITCs (1 NMA and 2 MAICs) was to compare the treatment effect on EFS of perioperative durvalumab with neoadjuvant chemotherapy with relevant comparators in resectable NSCLC, including nivolumab in combination with platinum-based doublet chemotherapy in the neoadjuvant setting (the CheckMate 816 study,16,70-74 also termed as neoadjuvant nivolumab with chemotherapy throughout this section), pembrolizumab in combination with platinum-based chemotherapy in the perioperative setting, adjuvant chemotherapy, and surgery only.
Study Selection Methods
An SLR was conducted in July 2022 to identify clinical evidence (efficacy and safety) from RCTs that enrolled adults with stage I to stage III NSCLC who were candidates for surgical resection and had received any or no treatment before surgery. The SLR was supplemented with a targeted literature review in September 2022 to identify RCTs of immunotherapies (atezolizumab, durvalumab, nivolumab, and pembrolizumab) in the adjuvant setting for patients with completely resected NSCLC. Updates to both the SLR and targeted literature review were conducted in October 2023. Published data for the AEGEAN study were identified as part of the SLR update.56,75-77 No other new studies relevant to the EFS networks were identified in the October 2023 search. Following the October 2023 search, the clinical trial landscape and literature were monitored for new data relevant to comparators considered in the ITC until the time of submission to CDA-AMC in August 2024. Results from the AEGEAN study EFS IA2 became available and more recent EFS data were presented at clinical congresses from the CheckMate 816 trial.17 The SLR was not updated, but the new data cuts were assessed in an ITC addendum.78 Study selection criteria and methods are summarized in Table 23.
Table 23: Study Selection Criteria and Methods for Indirect Comparisons
Characteristic | Indirect comparison |
|---|---|
Population | Adult patients (aged ≥ 18 years) with stage I to stage III NSCLC who are candidates for surgical resection of the primary NSCLC |
Intervention | Perioperative durvalumab with neoadjuvant chemotherapy The recommended dosage of durvalumab is 1,500 mg in combination with chemotherapy every 3 weeks before surgery (neoadjuvant period), followed by 1,500 mg as monotherapy every 4 weeks after surgery (adjuvant period). Before surgery, treatment continues for up to 4 cycles or until disease is deemed unresectable or of unacceptable toxicity. After surgery, treatment continues for up to 12 cycles or until recurrence or unacceptable toxicity. Patients weighing ≤ 30 kg must receive weight-based dosing of the drug at 20 mg/kg. In combination with chemotherapy, dose at 20 mg/kg every 3 weeks (21 days) is administered before surgery, followed by monotherapy at 20 mg/kg every 4 weeks after surgery until weight increases to > 30 kg. |
Comparator | The SLR did not restrict studies based on intervention or comparator, with the AEGEAN study clinical SLR report specifying “any or no treatment for stage I–III NSCLC before surgical resection of the primary NSCLC.” Comparators in the ITC included neoadjuvant nivolumab in combination with PBC, neoadjuvant PBC, surgery followed by adjuvant therapy, surgery only, and perioperative pembrolizumab. Dosing information for systemic therapies is provided as follows based on respective product monographs and local dosing protocols, as applicable. Note that study selection for the ITC was not limited to specific (neo)adjuvant chemotherapy dosing protocols. Neoadjuvant nivolumab in combination with PBC The recommended dosage is 240 mg nivolumab administered intravenously over 30 minutes every 2 weeks in combination with fluoropyrimidine- and platinum-containing chemotherapy every 4 weeks or 480 mg nivolumab administered intravenously over 30 minutes in combination with fluoropyrimidine and platinum-containing chemotherapy every 4 weeks. Treatment is recommended until disease progression or unacceptable toxicity. The maximum treatment duration for nivolumab is 2 years. Neoadjuvant pembrolizumab in combination with PBC, followed by adjuvant pembrolizumab monotherapy following surgery The recommended dosage is 200 mg pembrolizumab administered intravenously over 30 minutes every 3 weeks with cisplatin-based chemotherapy for 4 cycles, followed by surgery and adjuvant pembrolizumab 200 mg every 3 weeks for up to 13 cycles. Neoadjuvant PBC, and adjuvant PBC Cisplatin + vinorelbine
Cisplatin + pemetrexed
Cisplatin + gemcitabine
Paclitaxel + carboplatin
|
Outcome | Efficacy outcomes included in the SLR featured EFS, DFS, OS, MPR, pCR, PFS, RFS, and recurrence rate and type. Safety outcomes included AEs, treatment discontinuation or patient withdrawals, and mortality. Surgical outcomes included resection rates, complete resection rates and resection margin, surgical complications, and timing and duration of surgery. HRQoL outcomes included HSUV and HRQoL. ITCs were performed to estimate the relative efficacy of perioperative durvalumab with neoadjuvant chemotherapy vs. relevant comparators in terms of EFS. EFS, DFS, and PFS were used interchangeably across studies from the SLR to describe time-to-event outcomes that included progression and/or recurrence and death as events of interest. Therefore, studies were considered within scope if they reported EFS, PFS, and DFS for outcomes with definitions broadly corresponding to how EFS was defined in the AEGEAN study (i.e., included progression and/or recurrence events and death as events, and assessed time-to-event from the time of randomization [and therefore before surgery or neoadjuvant therapy]). |
Study designs | RCTs and interventional non-RCTsa |
Publication characteristics | Published literature, grey literature, and conference proceedings |
Exclusion criteria | Animal studies, articles not in the English language, noninterventional studies, nonprimary research publications |
Databases searched | MEDLINE, Embase, the Cochrane Library, and the University of York CRD database were searched to identify relevant published literature for the clinical SLR. PubMed was searched to identify relevant published literature for the TLR. Grey literature searches were conducted through ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform, in addition to searching the proceedings from 9 conferences held between 2020 and 2023. Conference proceedings for 2020 to 2022 in the original SLR and 2023 for the update from the following congresses were hand-searched to identify any relevant abstracts for inclusion:
|
Selection process | Electronic databases:
Conference proceedings:
|
Data extraction and quality assessment | Trials identified in the TLR were extracted directly into the clinical SLR data extraction grid and appraised using the same quality assessment checklists as used in the clinical SLR: the University of York CRD checklist for RCTs and the ROBINS-I tool for non-RCTs and observational studies. Because only RCTs were captured during the TLR, only the University of York CRD checklist was ultimately used.79,80 Both data extraction and quality appraisals were undertaken by 1 reviewer with verification by a second reviewer. |
AE = adverse event; ASCO = American Society of Clinical Oncology; CRD = Centre for Reviews and Dissemination; DFS = disease-free survival; EFS = event-free survival; ESMO = European Society for Medical Oncology; HRQoL = health-related quality of life; HSUV = health state utility value; IASLC = International Association for the Study of Lung Cancer; ITC = indirect treatment comparison; MPR = major pathological response; non-RCT = non–randomized controlled trial; NSCLC = non–small cell lung cancer; OS = overall survival; PBC = platinum-based chemotherapy; pCR = pathological complete response; PFS = progression-free survival; RCT = randomized controlled trial; RFS = relapse-free survival; ROBINS-I = Risk Of Bias In Non-Randomized Studies - of Interventions; SLR = systematic literature review; TLR = targeted literature review; vs. = versus.
aStudies with a non-RCT study design were formally included in the initial stages of the original and update review but were deprioritized before the extraction stage as being of less relevance to the ITC feasibility assessment, given the availability of evidence from RCTs.
Source: AEGEAN study clinical SLR report and ITC technical report.81
A subgroup analysis in the submitted ITC for patients with stage III (T1-T3) N2 disease (where N2 disease is treated with neoadjuvant chemoradiotherapy [CRT] + surgery) was not included in this review document because it is not relevant to clinical practice in Canada. In the sponsor’s clinical evidence template, it is stated that Canadian clinical experts consulted for this submission indicated that neoadjuvant CRT is used minimally in early-stage resectable NSCLC. Furthermore, results from the CheckMate 77T study (perioperative nivolumab) were published in May 2024.82 However, the CheckMate 77T study was not included in the ITCs given that perioperative nivolumab is not a relevant comparator for this submission because it has not been reviewed or recommended for funding by CDA-AMC. Therefore, the relevant information of the CheckMate 77T study82 is not presented in this CDA-AMC ITC summary.
ITC Analysis Methods
Overview of Methodology Plan and Feasibility Assessment
A feasibility assessment was conducted to explore potential ITC methods. Because individual patient data were available for the AEGEAN study (an RCT),56,75 both NMA and MAIC methods were considered as part of the ITC feasibility recommendations. The type of analysis and the feasibility of an NMA was assessed according to the flow chart adapted by the sponsor from National Institute for Health and Care Excellence Decision Support Unit–Technical Support Document 18 (NICE DSU TSD 18)83 as recommended by Cope et al. (2014).84 Selected studies of interest were evaluated based on comparability with the AEGEAN study56,75 in terms of study designs, treatment characteristics (e.g., type, frequency, dosing, administration, duration), patient characteristics (particularly, those that may be possible EMs), and outcome definitions and availability. The choice of methodology for the ITC was also made based on assessments of the proportional hazards assumption in each of the studies of interest.
Recommendations on the feasibility of performing an ITC for the AEGEAN study56,75 were primarily informed by the following:
the comparability and availability of outcomes (with studies excluded if no published EFS HR or KM plot was available)
the validity of assumptions relating to homogeneity and similarity and/or transitivity; where differences between studies were identified, the ability to account for these differences via population-adjusted indirect comparison and/or subgroup ITCs was considered.
Anchored ITCs were conducted for all comparisons, using neoadjuvant chemotherapy (± placebo) as a common comparator between studies.
For comparisons versus immunotherapy-based regimens evaluated in separate trials, including neoadjuvant nivolumab plus chemotherapy (the CheckMate 816 study),16,70-74 and perioperative pembrolizumab plus chemotherapy (the KEYNOTE-671 study), imbalances between the AEGEAN trial and the comparator trials in patient characteristics that were considered to be possible EMs (such as stage and PD-L1 expression) were identified. In line with recommendations from NICE DSU TSD 18, MAICs were conducted for these comparisons (Table 24). Because the KEYNOTE-671 study only permitted cisplatin as planned platinum-based chemotherapy, the comparison versus perioperative pembrolizumab was conducted within the planned cisplatin subgroup of the AEGEAN trial.
In the base-case MAICs (scenario 1), all possible EMs were included in the weighting (as per recommendations in NICE DSU TSD 18).83 A scenario analysis (scenario 2) was also conducted in which only those possible EMs that were imbalanced (≥ 5% difference) between trials were included, to assess the impact of different factors on the ITC results.
For comparisons versus adjuvant chemotherapy, and versus surgery only, an NMA was conducted using evidence identified from the multiple studies included in the SLR (Table 24).
Several differences between studies within each of the NMAs were identified (e.g., with respect to stage, type of chemotherapy, and region). However, to include evidence from the multiple studies identified in the SLR, and in the absence of clear candidates among these trials and the lack of clinical and patient characteristics (patient-level data) for conducting pairwise MAICs, NMAs were conducted for these comparisons. Sensitivity analyses to assess the impact of differences between studies on the results of the ITC (via the inclusion or exclusion of studies) were instead proposed for each of the NMAs.
Due to the delayed separation of EFS KM curves in the AEGEAN trial (and other trials), which occurred approximately 3 months after randomization and corresponded to the timing of the first planned tumour assessment in the AEGEAN trial, a piecewise approach for the ITCs (with time intervals of 0 to 3 months and 3+ months) was also explored.
Table 24: Summary of Intervention, Comparators, and ITC Methods Presented in the Submission
Intervention | Comparator | ITC method | Result |
|---|---|---|---|
Comparisons included in the ITCs in the submission | |||
Perioperative durvalumab with neoadjuvant chemotherapy vs…. | Neoadjuvant nivolumab with chemotherapy | MAIC | |
Perioperative durvalumab with neoadjuvant chemotherapy vs…. | Perioperative pembrolizumab with chemotherapy | MAIC | |
Perioperative durvalumab with neoadjuvant chemotherapy vs…. | Neoadjuvant chemotherapy | NMA | Direct evidence: Refer to pivotal AEGEAN trial in the main systematic review section ITC: Table 45 |
Perioperative durvalumab with neoadjuvant chemotherapy vs…. | Surgery only | NMA | |
ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NMA = network meta-analysis; vs. = versus.
Source: Information extracted from the sponsor’s submission.
MAIC Methods
Overview of Statistical Methods
Anchored MAIC analyses were performed to compare EFS of perioperative durvalumab plus neoadjuvant platinum-based chemotherapy from the AEGEAN trial56,75 versus immunotherapy-based regimens evaluated in separate randomized trials, including neoadjuvant nivolumab plus platinum-based chemotherapy from the CheckMate 816 trial16,70-74 and perioperative pembrolizumab plus platinum-based chemotherapy from the KEYNOTE-671 trial, leveraging the common comparator arm of neoadjuvant platinum-based chemotherapy (± placebo).
The first step in conducting the MAIC involved deriving weights such that the average baseline characteristics in the AEGEAN study (postweighting)56,85 matched the published aggregate characteristics of the comparator population in the respective comparator trial (i.e., the CheckMate 816 study and the KEYNOTE-671 study). For this, a propensity score-type logistic regression equation to estimate weights was used. Specifically, weights were estimated by the odds of being enrolled in the target population (i.e., the respective comparator trial population) versus the AEGEAN study, which was calculated as
where  was the vector of baseline variables that were included for weighting. The
was the vector of baseline variables that were included for weighting. The  coefficients were determined by the method of moments because only aggregate data for the
coefficients were determined by the method of moments because only aggregate data for the 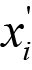 s were available for the comparator trial populations, as described in Signorovitch et al.86 Patients who had missing values for any of the variables included in the MAIC were excluded from the analysis. Once the coefficients were estimated, the equation was applied to the patients from the AEGEAN study56,85 to calculate the individual patient weights.
s were available for the comparator trial populations, as described in Signorovitch et al.86 Patients who had missing values for any of the variables included in the MAIC were excluded from the analysis. Once the coefficients were estimated, the equation was applied to the patients from the AEGEAN study56,85 to calculate the individual patient weights.
Next, the distribution of weights was assessed to identify any overly influential observations. The effective sample size was calculated by
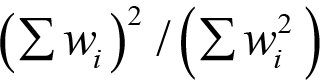
Anchored Comparisons
Based on the individual patient weights for the AEGEAN trial’s population56,75 in the MAIC, the (weighted) EFS HRs for perioperative durvalumab plus neoadjuvant platinum-based chemotherapy versus perioperative placebo plus neoadjuvant platinum-based chemotherapy were derived from weighted Cox proportional hazards models. Given that the weights were derived from the data, robust sandwich estimators were used to compute the standard error of the weighted log-HR in the AEGEAN trial, as recommended in NICE DSU TSD 18.87
The anchored indirect comparison of perioperative durvalumab with neoadjuvant chemotherapy versus a comparator treatment was then calculated on the log-scale, using the weighted AEGEAN study EFS HR and the EFS HR reported from the comparator trial (both HRs versus the common comparator), in accordance with Bucher et al.88
The output from the indirect comparisons included the EFS HR and 95% CI for the comparison of perioperative durvalumab with neoadjuvant chemotherapy versus comparator treatment  . In the MAICs, this HR provided an estimate of the relative effect on EFS of perioperative durvalumab with neoadjuvant chemotherapy versus comparator treatment
. In the MAICs, this HR provided an estimate of the relative effect on EFS of perioperative durvalumab with neoadjuvant chemotherapy versus comparator treatment  in a population matching the characteristics of the comparator trial population. Anchored indirect comparisons using the unweighted HR from the AEGEAN study were also conducted.
in a population matching the characteristics of the comparator trial population. Anchored indirect comparisons using the unweighted HR from the AEGEAN study were also conducted.
Piecewise ITCs
Piecewise ITCs with intervals of 0 to 3 months and 3-plus months were also conducted. In these analyses, HRs from the (weighted) AEGEAN trial’s population and each comparator trial (based on pseudopatient-level data derived from the digitization of the comparator trial KM curves) were generated for each interval. Specifically, the AEGEAN trial (weighted) patient-level data and pseudopatient-level data derived from digitized KM curves for each comparator trial were separated into the respective interval of 0 to 3 months and of 3-plus months using the survSplit function in R, through the creation of an indicator variable denoting the time point. A Cox regression model with an interaction between the time point indicator variable and treatment was then used to obtain an estimate of the piecewise HRs for each trial within the intervals of interest.
The piecewise HRs for each interval were then used in an anchored indirect comparison (i.e., Bucher ITC), as described earlier, to estimate EFS HRs in 0 to 3 months and 3-plus months for perioperative durvalumab with neoadjuvant chemotherapy versus the comparator regimen.
Characteristics Included in Weighting
Subgroup analyses and interaction tests for the AEGEAN study were reviewed for empirical evidence to suggest that a baseline characteristic might be a potential EM in the indirect comparisons with the comparator trials. The following characteristics were considered by the sponsor to be possible EMs: disease stage, PD-L1 expression, region (Asia versus non-Asia), sex, histology, smoking status, and planned platinum chemotherapy. In the base-case analysis (scenario 1), all possible EMs were included in the weighting, regardless of whether imbalances exist between the AEGEAN trial and the comparator trials in these baseline characteristics, as per recommendation 4 in NICE DSU TSD 18.87 An additional analysis (scenario 2) was also conducted to explore the impact on results of only weighting for those characteristics that were imbalanced (≥ 5% difference) between trials.
Key information on the MAIC methods is summarized in Table 25.
Table 25: Indirect Comparison Analysis Methods — Matching-Adjusted Indirect Comparison
Method | Description |
|---|---|
Analysis methods | Anchored MAIC analyses were performed to compare EFS of perioperative durvalumab + neoadjuvant platinum-based chemotherapy from the AEGEAN trial vs. immunotherapy-based regimens evaluated in separate randomized trials, including neoadjuvant nivolumab + platinum-based chemotherapy from the CheckMate 816 trial and perioperative pembrolizumab + platinum-based chemotherapy from the KEYNOTE-671 trial, leveraging the common comparator arm of neoadjuvant platinum-based chemotherapy (± placebo). |
Outcome | EFS (HR and 95% CI) |
Scenarios | AEGEAN study mITT population for comparisons with the CheckMate 816 and KEYNOTE-671 trials
|
CI = confidence interval; EFS = event-free survival; EM = effect modifier; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; mITT = modified intention to treat; vs. = versus.
Source: Indirect treatment comparison technical report.78
NMA Methods
Key information on the NMA methods is summarized in Table 26.
EFS HR data were analyzed using NMAs. A piecewise NMA with intervals of 0 to 3 months and 3-plus months was conducted, in addition to a conventional NMA, to account for the delayed separation of EFS curves in the AEGEAN trial.
Table 26: Indirect Comparison Analysis Methods — Network Meta-Analyses
Method | Description |
|---|---|
Analysis methods | All NMAs were conducted in a Bayesian framework, using Markov Chain Monte Carlo simulation methods, and using R version 4.0.289 with the following packages: Rstan (version 2.19.3), multinma (version 0.5.1), and survival (version 3.1.12). The models were run with 4 chains of 10,000 iterations, of which every second iteration was kept; 5,000 were burn-in iterations (i.e., thinning = 2) to generate the posteriors for the defined parameters. Because EFS is a time-to-event outcome, the log HRs were analyzed using a normal likelihood and identity link (Program 7 of NICE DSU TSD 2).90 |
Priors | Noninformative normal (0 to 100)2 priors were assigned to the treatment effect parameters. Both fixed-effects and random-effects models were conducted, but there were limited data to estimate between-study heterogeneity for random-effects models, so informative priors based on a log-normal distribution (“subjective outcomes [various]” prior, log-normal approximately [–2.93 to 1.582]) were used for random-effects models based on Turner et al.91 |
Assessment of model fit | Given the level of heterogeneity identified in the feasibility assessment, the random-effects model was preferred but the fixed-effects models were also presented for completeness.92 When both fixed-effects and random-effects models were fitted to the data, their DIC were derived. Lower values represent the more parsimonious model, and differences of 3 points were considered meaningful.92 The model goodness-of-fit was assessed by comparing the posterior mean residual deviance to the number of data points in the network.92 The number of data points was calculated as the sum of arms across studies reporting arm-level data, and the sum of studies reporting contrast-based data. Model fit was considered good if the posterior mean residual deviance was similar to the number of data points. |
Assessment of consistency | Assessment of consistency was not feasible due to the sparse evidence networks, with no closed loops formed by multiple studies. |
Assessment of convergence | Convergence of the chains (refer to the first row in this table called Analysis methods) was assessed using the Rhat statistic.93 |
Outcomes | EFS (HR and 95% CI) |
Follow-up time points | The median follow-up times were up to 70 months (the GLCCG01/95 study).94,95 Median follow-up at AEGEAN study EFS IA2 was ████ months. |
Construction of nodes | In NMA, there were multiple trials informing the same comparison (surgery only vs. neoadjuvant chemotherapy). Statistical heterogeneity, which refers to variation in treatment effect between trials, was therefore measured using |
Sensitivity analyses | Sensitivity analyses were also carried out to assess the impact on NMA results of differences between the comparator studies and vs. the AEGEAN study (e.g., by excluding studies that included 2G chemotherapy regimens, excluding Asia-only trials, excluding studies with clear differences in disease stage). |
Subgroup analysis | A network was assessed corresponding to the AEGEAN study N2 subgroup,56,75 which included neoadjuvant CRT + surgery as the additional comparator of interest (and was based on the trial populations for neoadjuvant CRT + surgery). Note: This subgroup analysis is not relevant to this review; therefore, the information of the subgroup is not presented in this document. |
 . The
. The  represents the percentage of variation in effect estimates between trials that is due to heterogeneity rather than sampling error (i.e., chance).
represents the percentage of variation in effect estimates between trials that is due to heterogeneity rather than sampling error (i.e., chance).2G = second generation; CI = confidence interval; CRT = chemoradiotherapy; DIC = deviance information criteria; EFS = event-free survival; HR = hazard ratio; IA1 = interim analysis 1; IA2 = interim analysis 2; NICE DSU TSD 2 = National Institute for Health and Care Excellence Decision Support Unit–Technical Support Document 2; NMA = network meta-analysis; vs. = versus.
Source: Indirect treatment comparison technical report.78
Results of ITCs
Summary of Included Studies
Included Studies
An overview of the AEGEAN study and comparator studies considered for the ITCs is provided in Table 27, including the CheckMate 816 and KEYNOTE-671 trials used in the MAIC analyses and studies included in the NMA. EFS, DFS, and progression-free survival (PFS) were used interchangeably across studies from the SLR to describe time-to-event outcomes that included progression or recurrence and death as events of interest. Chen (2013),96 Roth (1994),97 EFS, DFS, and PFS were used interchangeably across studies from the SLR to describe time-to-event outcomes that included progression or recurrence and death as events of interest. The Chen (2013),96 Roth (1994),97 JCOG 9209,98 and Depierre (2002)99 studies were excluded due to the absence of EFS KM curves and HRs.
Table 27: Overview of Studies Included in the ITCs
Study | Intervention vs. comparator (randomized n) | Setting | Analysis set informing ITC base case | Reported outcome |
|---|---|---|---|---|
Perioperative durvalumab + neoadjuvant PDC (n = 366) vs. neoadjuvant placebo + PDC (n = 374) | Perioperative | mITT (excludes EGFR mutation and ALK+) | EFS | |
Neoadjuvant nivolumab + PDC (n = 179) vs. PDC (n = 179) | Neoadjuvant | ITT (patients with baseline disease stage IB to stage II, patients with baseline disease stage IIIA, PD-L1 < 1%, PD-L1 expression ≥ 1, PD-L1 expression of 1% to 49%, PD-L1 expression ≥ 50%, squamous, nonsquamous, ctDNA CL, no ctDNA CL) | EFS | |
Neoadjuvant pembrolizumab + PDC followed by adjuvant pembrolizumab (n = 397) vs. neoadjuvant placebo + PDC followed by adjuvant placebo (n = 400) | Perioperative | ITT (all randomized patients) | EFS | |
Surgery only (n = 210) vs. neoadjuvant chemotherapy + surgery (n = 199) vs. surgery + adjuvant chemotherapy (N = 210) | Neoadjuvant and adjuvant | ITT (all randomized patients with follow-up data available) | DFS | |
Neoadjuvant chemotherapy + surgery (n = 30) vs. surgery only (n = 30) | Neoadjuvant | ITT (all randomized patients) | EFS | |
CHEST study105 | Neoadjuvant chemotherapy + surgery (n = 129) vs. surgery only (n = 141) | Neoadjuvant | ITT (all randomized patients) | PFS |
MRC LU22/NVALT 2/EORTC 08012 study106 | Neoadjuvant chemotherapy + surgery (n = 258) vs. surgery only (n = 261) | Neoadjuvant | ITT (all randomized patients with follow-up data available) | PFS |
SWOG S9900 study98 | Neoadjuvant paclitaxel and carboplatin (n = 169) + surgery vs. surgery only (n = 168) | Neoadjuvant | ITT (all randomized eligible patients [some patients were randomized but found ineligible and so are not included in this analysis]) | PFS |
Li (2009) study107 | Neoadjuvant cisplatin and vinorelbine (n = 28) vs. surgery only (n = 28) | Neoadjuvant | ITT (all randomized patients) | DFS |
Neoadjuvant CRT (n = 117) + surgery vs. neoadjuvant chemotherapy + surgery (n = 115) | Neoadjuvant | ITT (all randomized patients) | EFS | |
Neoadjuvant cisplatin and etoposide followed by chemoradiation + surgery (n = 264) vs. cisplatin plus etoposide + surgery (n = 260) | Neoadjuvant | ITT (all randomized patients) | PFS | |
WJTOG9903 study110 | Neoadjuvant carboplatin + docetaxel (n = 29) plus surgery vs. neoadjuvant carboplatin + docetaxel + radiotherapy plus surgery (n = 31) | Neoadjuvant | mITT (all patients who received induction chemotherapy) | PFS |
CRT + surgery (n = 14) vs. chemotherapy + surgery (n = 17 to 15) | Neoadjuvant | ITT (all randomized patients) | PFS |
ALK+ = ALK-positive; CL = clearance; CRT = chemoradiotherapy; ctDNA = circulating tumour DNA; DFS = disease-free survival; EFS = event-free survival; ITC = indirect treatment comparison; ITT = intention to treat; mITT = modified intention to treat; PDC = platinum-doublet chemotherapy; PFS = progression-free survival; vs. = versus.
Source: Indirect treatment comparison technical report.78
Comparison of Study Characteristics and Selection of EMs
The following approach was taken by the sponsor to identify potential EMs for the AEGEAN ITCs.
Review stratification factors in the AEGEAN study, and relevant comparator trials evaluating immunotherapy-based perioperative or neoadjuvant regimens (including the CheckMate 816 and KEYNOTE-671 studies), given that these might be indicative of potential EMs and prognostic variables for immunotherapy-based regimens in resectable NSCLC, and seek internal AstraZeneca clinical opinion on factors that might be EMs and prognostic variables (i.e., identify potential EMs and prognostic variables based on biological and clinical plausibility):
stages (stage II versus stage III in the AEGEAN study and stage IB or stage II versus stage III in the CheckMate 816 study) and PD-L1 expression (< 1 versus ≥ 1% in the AEGEAN and CheckMate 816 studies) were included as stratification factors across all studies
region (East Asia versus others) and sex (the CheckMate 816 study only) also were included as stratification factors but not considered as relevant by the AstraZeneca clinical team
histology (nonsquamous versus squamous).
Review EFS forest plots of the AEGEAN study and the subgroup analyses of relevant comparator trials (including the CheckMate 816 and KEYNOTE-671 studies), and interaction tests for the AEGEAN study, for empirical evidence to suggest that a baseline characteristic might be a potential EM:
Forest plots were inspected to provide an initial guide on what might be possible EMs based on nonoverlapping 95% CIs for subgroups and whether the subgroup 95% CI included the overall treatment effect point estimate (subgroups with a nonoverlapping 95% CI or 95% CI that did not include the overall treatment effect were considered to indicate evidence of EM).
In the AEGEAN study, a post hoc sensitivity analysis was conducted to assess the consistency of EFS treatment effect across subgroups using interaction tests for each subgroup variable (sex, age, PD-L1 expression, histology, disease stage, smoking status, race, baseline ECOG PS, and baseline chemotherapy) (1 covariate at a time) and their interaction with the treatment group. Tests for effect modification were conducted by comparing models with and without the interaction term (i.e., [{treatment + covariate} + treatment] × covariate interaction versus [treatment + covariate]) using likelihood ratio tests.
For the AEGEAN study, no factors met the sponsor’s criteria outlined for EM identification based on inspection of forest plots. For each subgroup, the 95% CI included the overall treatment effect (HR for all patients). Within each subgroup analysis, 95% CIs overlapped between subgroups. There were also no significant interactions (5% significance level) between treatment group and each baseline subgroup variable for EFS in the AEGEAN trial’s subgroup interaction tests (post hoc analysis). However, some differences (which may be putative) in factors of interest were observed in the AEGEAN study EFS forest plot, with EFS treatment benefit appearing greater in:
sex — males versus females
regions — Asia versus other regions
PD-L1 expression — 50% or more, followed by 1% to 49%, and lowest in less than 1%
stage — stage III versus stage II and specifically, stage IIIA versus stage II or stage IIIB
smoking status — currently smoke versus formerly or never smoked
planned platinum chemotherapy at baseline — cisplatin versus carboplatin.
Similar EFS treatment benefits were observed in histology subgroups.
For the CheckMate 816 study, no factors met the criteria outlined for EM identification based on inspection of the forest plots. However, according to the study publication authors, EFS benefit was greater in:
PD-L1 expression — 1% or more versus less than 1%
stage — stage IIIA versus stage IB or stage II
histology — nonsquamous versus squamous.
For the KEYNOTE-671 study, no factors met the criteria outlined for EM identification based on inspection of forest plots. According to the study publication authors, EFS benefit was consistent across all subgroups examined, but some subgroups were small and had a low number of events. However, some differences (which may be putative) in factors of interest were observed, including subgroups of sex, PD-L1 expression, stage, smoking status, and region. Similar EFS treatment benefits were observed in histology subgroups.
In addition, based on factors of interest, EFS benefit was different for sex, smoking status, region, and type of platinum therapy subgroups (although differences may be putative).70
Based on biological and clinical plausibility (e.g., inclusion as stratification factors in the phase III trials of perioperative and neoadjuvant immunotherapy-based regimens) and inspection of the EFS forest plots, the following factors were considered by the sponsor as potential EMs when assessing the comparability of population and patient characteristics between studies included in the feasibility assessment: disease stage, PD-L1 expression, region (Asia versus non-Asia), sex, histology, smoking status, and planned platinum chemotherapy.
The sponsor noted that there is only limited empirical evidence to suggest that these characteristics might have an impact on the treatment effect in each trial, partly owing to the challenges of interpreting subgroup analyses (given that any differences on visual inspection may be putative), and the reduced power for assessing any differences between subgroups.
The assessment of homogeneity between identified studies is also summarized in Table 28.
Table 28: Assessment of Homogeneity and Description of Potential EMs Considered
Characteristic | Description of potential EMs |
|---|---|
Study design | The trial designs of the AEGEAN study and comparator trials were similar. Differences in the aspects that may threaten the transitivity assumption were inspected. |
Disease stage | The AEGEAN study included patients with stage II, stage IIA, or stage IIIB (N2 only) as defined by the AJCC Cancer Staging Manual, eighth edition. Disease staging criteria in the other studies were based on earlier AJCC Cancer Staging Manual editions (or were not reported), making comparisons across populations challenging. Similarly, definitions for tumour status and, to a lesser extent, nodal status (unchanged in the eighth, seventh, and sixth editions of the manual) (as part of TNM classification) differed across studies, according to the AJCC Cancer Staging Manual edition used. The CheckMate 816 study included patients with stage IB (≥ 4 cm), stage II, and stage IIIA disease, according to the seventh edition, while the KEYNOTE-671 study enrolled patients with stage II, stage IIIA, or stage IIIB disease, according to the AJCC Cancer Staging Manual, eighth edition. Patients with T2aN0 (4 cm) (stage IB in the seventh edition) would be included in the CheckMate 816 trial but not in the AEGEAN trial, and patients with T4N2 (stage IIIB in both the seventh and eighth editions) would be included in the AEGEAN trial but not the CheckMate 816 trial. Evaluating the differences in baseline disease stage between the AEGEAN and CheckMate 816 studies at an aggregate trial level may, however, be possible by estimating the proportion of patients in the CheckMate 816 study who might be reclassified to different stages according to the eighth edition of the AJCC Cancer Staging Manual. Differences in staging criteria aside, the inclusion and exclusion criteria differed across “adjuvant chemotherapy” and “surgery only” studies, with some studies including only stage IIIA patients (the Roth [1994],97,113 Rosell [1994],103,104 JCOG 9209,98 and Li [2009] trials107) and other studies including patients with stage I, stage II, and stage III disease (e.g., the NATCH [only stage T3N1 in addition to stage I], Chen [2013],96 CHEST,106 MRC LU22/NVALT 2/EORTC 08012,105 SWOG S9900,106 and Depierre [2002] trials).99 “Neoadjuvant CRT + surgery” studies included only patients with stage IIIA disease and, with the exception of the GLCCG01/95 study,94,95 only included patients with stage IIIA N2. |
PD-L1 expression | Distribution across PD-L1 expression levels varied between the AEGEAN, CheckMate 816, and KEYNOTE-671 trials. Compared with the CheckMate 816 trial, a lower proportion of patients with PD-L1 < 1% were included in the AEGEAN trial’s mITT population; compared with the KEYNOTE-671 trial, a lower proportion of patients with PD-L1 ≥ 50% were included in the AEGEAN trial’s cisplatin subgroup (differences between trials ≥ 5%). PD-L1 expression was not reported for any of the adjuvant chemotherapy, surgery only, or neoadjuvant CRT + surgery studies. For any ITC vs. these comparators, the sponsor assumed that there were no differences in treatment effect based in PD-L1 expression within these studies. |
Region | Where region as a baseline characteristic was reported, patients in other studies were enrolled from a single region (e.g., Asia only for the Chen [2013] “adjuvant chemotherapy,” JCOG 9209, and Li [2009] “surgery only” studies, and the WJTOG9903 “neoadjuvant CRT + surgery” study; or Europe and/or North America only for the NATCH “adjuvant chemotherapy,” Roth [1994], CHEST, MRC LU22/NVALT 2/EORTC 08012, and SWOG S9900 “surgery only” studies, and the GLCCG01/95 and IFCT-0101 “neoadjuvant CRT + surgery” studies). |
Sex, histology, and smoking status | Baseline characteristics in the AEGEAN study were similar to those reported in the CheckMate 816 and KEYNOTE-671 studies, with differences between trials < 5%. In each study, the majority of patients were male, approximately half of patients had nonsquamous histology, and the majority of patients currently or formerly smoked. That the majority of patients were male in the AEGEAN study was consistent with the other studies for “adjuvant chemotherapy,” “surgery only,” and “neoadjuvant CRT + surgery.” However, there was variation in tumour histology reported across studies, with the proportion of patients with squamous histology being similar between the AEGEAN and NATCH (“adjuvant chemotherapy”) trials, but not when compared with the CHEST or SWOG S9900 (“surgery only”) trials or the SAKK 16/00 SWS-SAKK-16/00 EU-20138 (“neoadjuvant CRT + surgery”) study. Smoking status was very sparsely reported across these studies. |
Treatment characteristics and planned platinum chemotherapy | All studies considered platinum-containing chemotherapy regimens in the neoadjuvant chemotherapy arm, which represents the common comparator for ITCs with the AEGEAN study. The percentage of cisplatin use was considerably higher in the CheckMate 816 study (78% overall) compared with the AEGEAN study (27% overall). With the exception of the MRC LU22/NVALT 2/EORTC 08012 trial (which included 6 possible chemotherapy regimens, including both cisplatin- and carboplatin-based regimens), adjuvant chemotherapy, surgery only, and neoadjuvant CRT + surgery studies tended to only include 1 chemotherapy regimen rather than investigator’s choice of regimen. Carboplatin-based regimens were used in the NATCH, SWOG S9900, and WJTOG9903 trials, with cisplatin-based regimens used in all other studies. As per the ITCs conducted by the sponsor for the NICE appraisal of neoadjuvant nivolumab (TA876), sensitivity analyses conducted by the sponsor explored for ITCs including adjuvant chemotherapy and surgery to assess the impact of only including trials that include 3G chemotherapy regimens, and excluding those trials that include 2G chemotherapy regimens.114 |
Outcome definitions and reporting | Differences in the following aspects relating to how EFS was assessed in each study may threaten the transitivity assumption. Definitions
Tumour assessment and criteria
For EFS data availability, 7 studies did not report an HR for EFS. Three studies did not report KM data and 1 study (the JCOG 9209 trial98) reported a KM plot that was not considered digitizable (based on overlapping and indistinct curves), meaning that the creation of pseudo-IPD was not feasible for these studies. Three of these studies did not report HRs (the JCOG 9209,98 Roth [1994],97,113 and Depierre [2002] trials)99 and the fourth study (Chen [2013])96 did not report clearly what comparison was being made for the HR reported, which impacted the ability to derive common estimates of relative EFS efficacy for ITCs. |
2G = second generation; 3G = third generation; AJCC = American Joint Committee on Cancer; CRT = chemoradiotherapy; DFS = disease-free survival; EFS = event-free survival; EM = effect modifier; HR = hazard ratio; IPD = individual patient-level data; ITC = indirect treatment comparison; KM = Kaplan-Meier; mITT = modified intention to treat; NICE = National Institute for Health and Care Excellence; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours version 1.1; TNM = tumour-nodes-metastasis; vs. = versus.
Source: Indirect treatment comparison technical report.78
Potential risk of bias — that is, the differences in the aspects that may threaten the transitivity assumption — were inspected by the sponsor and they are provided as follows. Results of the risk of bias appraisals for each contributing study were not explicitly reported.
Study design characteristics: All studies except 2 trials (the Roth [1994]97,113 and IFCT-0101111,112 studies) were phase III RCTs, and there were also 2 studies that did not report the study phase. Based on the risk of bias assessment, these 2 studies did not report any information on treatment allocation concealment. All studies had a multicentre design, except for the Roth [1994] study,97,113 which was a single-centre study. For 3 studies, the institutional design could not be identified (i.e., the Rosell [1994],103,104 Depierre [2002],99 and Li [2009]107 studies).
Geographical settings: Most studies were global and/or European studies; 4 studies — 1 “neoadjuvant CRT + surgery” study (the WJTOG9903 study11), 1 “adjuvant chemotherapy” study (the Chen [2013]96 study), and 2 “surgery only” studies (the JCOG 920998 and Li [2009]107 studies) were conducted in sites in Asia only. Two studies did not report the geographical location (the Rosell [1994]103,104 and Depierre [2002]99,115 studies).
Study start date: The AEGEAN, CheckMate 816, and KEYNOTE-671 studies began in 2017 or later. In contrast, all studies for “adjuvant chemotherapy,” “surgery only,” and “neoadjuvant CRT plus surgery” were started before 2004, with some studies (the Rosell [1994]103,104 and Roth [1994]97,113 trials) starting before 1990. The timing of studies likely impacts the comparability of studies (i.e., staging and tumour assessment criteria, the use of second-generation chemotherapy agents [refer to subsequent sections], which were assessed subsequently, and other unmeasured characteristics potentially impacting outcomes).
Evidence Networks
In the base case, similarity between the AEGEAN and comparator trials evaluating immunotherapy-based regimens was assumed (e.g., with respect to outcome definitions and common comparator characteristics). Anchored indirect comparisons were conducted using neoadjuvant platinum-based doublet chemotherapy (with or without placebo) as the common comparator between trials. The anchored MAIC networks informing the comparisons of perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant nivolumab with chemotherapy (the CheckMate 816 trial) and perioperative pembrolizumab with neoadjuvant chemotherapy followed by adjuvant pembrolizumab (the KEYNOTE-671 trial) are illustrated in Figure 6 and Figure 7, respectively.
For the comparison with perioperative pembrolizumab, an indirect comparison using the AEGEAN trial’s cisplatin subgroup was conducted to align with the KEYNOTE-671 study, in which only cisplatin-based neoadjuvant platinum-doublet chemotherapy regimens were permitted.
Imbalances between the AEGEAN trial (cisplatin subgroup) and comparator trials were identified in baseline patient characteristics considered potential EMs, including PD-L1 expression, disease stage (based on reclassification from AJCC seventh edition to eighth edition), region, and planned platinum chemotherapy. Therefore, population-adjusted indirect comparisons using the MAIC method, as well as anchored indirect comparisons without population adjustment, were conducted.
Figure 6: Anchored MAIC Diagram for Neoadjuvant Durvalumab + PDC + Adjuvant Durvalumab (AEGEAN Study) vs. Neoadjuvant Nivolumab + Chemotherapy (CheckMate 816 Study, ITT Population), Base Case
Adj = adjuvant; durva = durvalumab; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; neoadj = neoadjuvant; PDC = platinum-doublet chemotherapy; vs. = versus.
Source: Indirect treatment comparison technical report.78
Figure 7: Anchored MAIC Diagram for Neoadjuvant Durvalumab + PDC + Adjuvant Durvalumab (AEGEAN Study) vs. Neoadjuvant Pembrolizumab + Cisplatin PDC + Adjuvant Pembrolizumab
Adj = adjuvant; durva = durvalumab; MAIC = matching-adjusted indirect comparison; neoadj = neoadjuvant; PDC = platinum-doublet chemotherapy; pembro = pembrolizumab; vs. = versus.
Source: Indirect treatment comparison technical report.78
In the base case, an NMA for the AEGEAN study and identified trials including adjuvant chemotherapy and surgery were explored, as shown in Figure 8.
However, there were several differences between studies (versus each other and versus the AEGEAN study) that may impact relative treatment effects, including variation in disease stage, type of platinum-doublet chemotherapy regimen (second-generation and third-generation chemotherapy), and region. Options to account for differences (e.g., subgroup ITCs or MAICs), as done for comparisons versus neoadjuvant nivolumab plus platinum-doublet chemotherapy, were limited and therefore, the impact of differences between comparator studies on the NMA results were explored as part of sensitivity analyses (refer to Table 29). Heterogeneity (I2) was tested and/or reported at the time of analyses to demonstrate whether the inclusion or exclusion of studies impacted outcomes. To include evidence from the multiple studies identified in the SLR, and in the absence of clear candidates among these trials for conducting a pairwise MAIC, NMAs were considered to be the preferred approach by the sponsor for these comparisons.
Note that the Chen (2013),96 Roth (1994),97,113 JCOG 9209,98 and Depierre (2002)99 studies were excluded from the network due to the absence of EFS KMs and HRs. The CheckMate 816 study, which could also be included as part of this network (the AEGEAN study mITT population), was, however, included in a separate pairwise analysis to account for imbalances in possible EMs between the AEGEAN and CheckMate 816 studies via a population-adjusted indirect comparison (as described previously).
Figure 8: Network Diagram of Neoadjuvant Durvalumab + PDC + Adjuvant Durvalumab (AEGEAN Study) vs. Adjuvant Chemotherapy, and Surgery Only for EFS
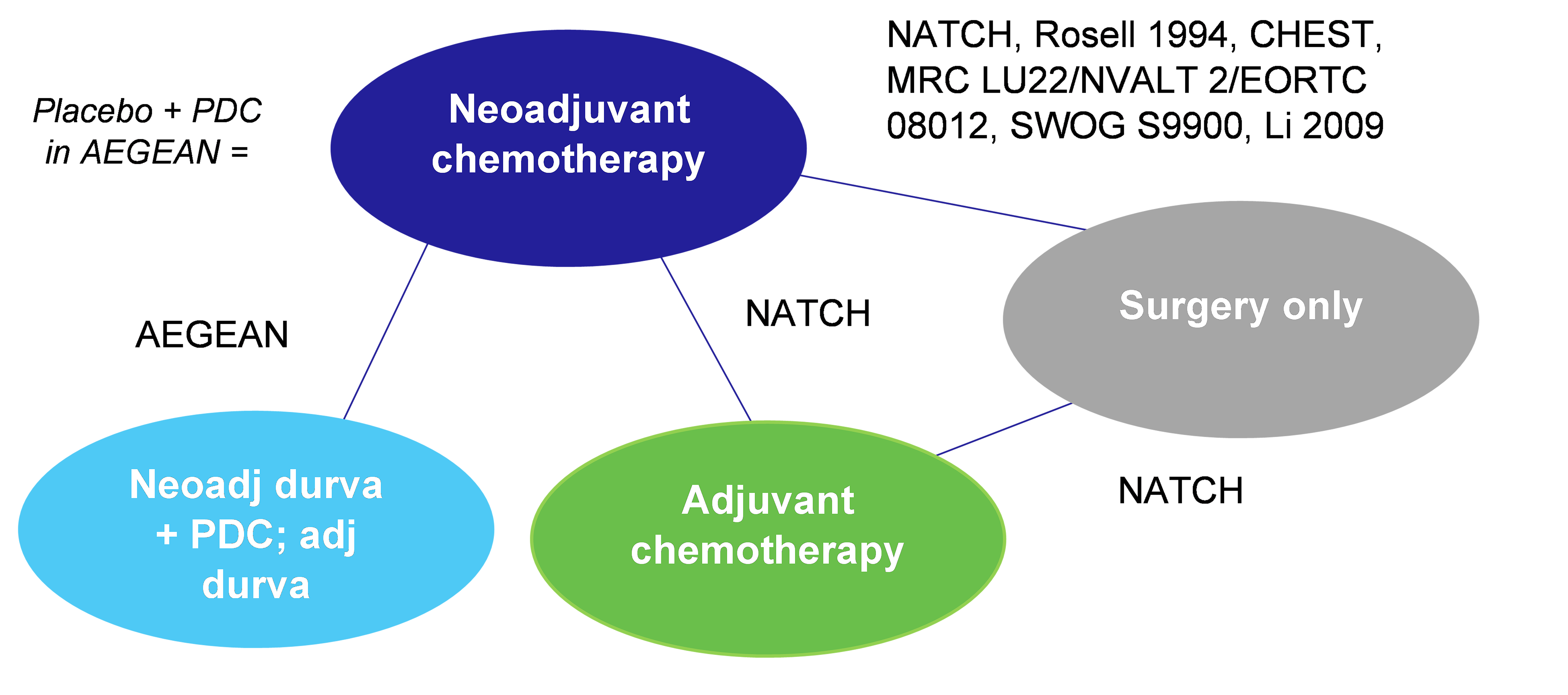
Adj = adjuvant; durva = durvalumab; EFS = event-free survival; neoadj = neoadjuvant; PDC = platinum-doublet chemotherapy; vs. = versus.
Source: Indirect treatment comparison technical report.78
Table 29: Studies Included and Excluded for the Sensitivity Analysis of the AEGEAN Study vs. Adjuvant Chemotherapy and Surgery
SA # | Studies included | Studies excluded | |
|---|---|---|---|
Studies excluded | Reason for exclusion in SA | ||
SA #1 | NATCH, CHEST, SWOG S9900, and Li (2009) studies | MRC LU22/NVALT 2/EORTC 08012 and Rosell (1994) studies | Exclude studies with 2G chemotherapy |
SA #2 | NATCH, CHEST, MRC LU22/NVALT 2/EORTC 08012, and SWOG S9900 studies | Rosell (1994) and Li (2009) studies | Exclude studies with stage III patients only |
SA #3 | NATCH, Rosell (1994), CHEST, MRC LU22/NVALT 2/EORTC 08012, and SWOG S9900 studies | Li (2009) study | Exclude Asia-only studies |
SA #4 | NATCH, CHEST, and SWOG S9900 studies | Rosell (1994), MRC LU22/NVALT 2/EORTC 08012, and Li (2009) studies | Exclude studies for any of the aforementioned reasons |
2G = second generation; SA = sensitivity analysis; vs. = versus.
Source: Indirect treatment comparison technical report.78
Efficacy
Event-Free Survival
MAIC: AEGEAN Study Versus CheckMate 816 Study
An anchored MAIC analysis was performed to compare the efficacy of perioperative durvalumab plus chemotherapy from the AEGEAN study versus neoadjuvant nivolumab plus chemotherapy. The summary of event numbers, and the HRs, for the AEGEAN study (before weighting) and the CheckMate 816 study are shown in Table 30.
Table 30: Trial Summaries of EFS in the AEGEAN and CheckMate 816 Studies
Arm | Trial | N | Events |
|---|---|---|---|
Durvalumab + PDC | AEGEAN study | 366 | 124 |
Placebo + PDC | AEGEAN study | 374 | 165 |
Nivolumab + PDC | CheckMate 816 study | 179 | 75 |
PDC | CheckMate 816 study | 179 | 97 |
DCO = data cut-off; EFS = event-free survival; PDC = platinum-doublet chemotherapy.
Sources: The DCO date in the AEGEAN study was May 10, 2024; the 4-year update database lock date in the CheckMate 816 study was February 23, 2024.17
A comparison of baseline characteristics between the 2 studies (overall trial — namely, both arms) before and after weighting is shown in Table 31 (weighting in scenario 1) and Table 32 (weighting in scenario 2).
There were imbalances in baseline characteristics between the AEGEAN and CheckMate 816 studies, including the following:
the proportion of patients who received cisplatin at baseline (higher in the CheckMate 816 study)
the proportion of patients with stage IIIA at baseline (higher in the CheckMate 816 study)
the proportion of patients with stage IIIB at baseline (lower in the CheckMate 816 study)
the proportion of patients enrolled in Asia (region) (higher in the CheckMate 816 study)
the proportion of patients with PD-L1 of less than 1% (higher in the CheckMate 816 study).
Each of these characteristics is considered by the sponsor to be a possible EM.
After weighting patients, the baseline characteristics in the AEGEAN trial matched those in the CheckMate 816 trial for those variables that were included in the weighting except for ███ ██████████ ██ ████████ ████ ██ ████ ███████████ ██████ ██ ██ which was ██████ in the AEGEAN trial after weighting. Patient characteristics not included in the weighting were not presented before or after weighting. In scenario 2, weighting resulted in an ████████ ██ ███ ██████████ ██ ████████ ████ ████████████ █████████ ███ ███ ██████████ ██ ████████ ███ ███ █████ ███████ introducing imbalances between the AEGEAN and CheckMate 816 studies in these baseline characteristics, both of which are considered possible EMs by the sponsor (Table 32). There were also remaining imbalances by ███ ██████ ██ ███ ████████ ██████ ███████████ and ████ ███████████ ██████ ███████ ██████████ ██ ████ ███████████ ██████ ██ █ ██ ███ ████████ ██████ ████████████ Patient characteristics not included in the weighting were not presented before or after weighting.
Weighting based on planned chemotherapy assigned ██████ weights for cisplatin patients and resulted in ██ █████████ proportion of patients with ██ ████ ██ █████ █ ███ ███ ███.
Table 31: Baseline Characteristics in the CheckMate 816 and AEGEAN Studies (Unweighted and After Weighting to Match CheckMate 816 Study in Scenario 1 [Base Case])
Characteristic | CheckMate 816 study (N = 358) | Unweighted (N = 740) | AEGEAN study Scenario 1 (ESS = █████) | ||
|---|---|---|---|---|---|
n | % | n | % | % | |
Aged < 65 years | 176 | 49.2 | 358 | 48.4 | ████ |
ECOG PS: 0 | 241 | 67.3 | 506 | 68.4 | ████ |
Planned platinum chemotherapy: Cisplatin | 258 | 78.2 | 196 | 26.5 | ████ |
Histology: Nonsquamous | 176 | 49.1 | 375 | 50.7 | ████ |
PD-L1 expression: < 1% | 155 | 46.5 | 247 | 33.4 | ████ |
PD-L1 expression: ≥ 50% | 80 | 24.0 | 216 | 29.2 | ████ |
Region: Asia | 177 | 49.4 | 305 | 41.2 | ████ |
Sex: Female | 103 | 28.8 | 210 | 28.4 | ████ |
Smoking status: Never | 39 | 10.9 | 107 | 14.5 | ████ |
Stage: IIIA | NA | 57.4 | 338 | 45.7 | ████ |
Stage: IIIB | NA | 12.1 | 187 | 25.3 | ████ |
AJCC = American Joint Committee on Cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EM = effect modifier; ESS = effective sample size; NA = not applicable.
Notes: Characteristics included in the weighting to match the CheckMate 816 study: planned platinum chemotherapy, histology, PD-L1 expression, region, sex, smoking status, and stage (all possible EMs) in scenario 1.
For the CheckMate 816 study, the percentage of PD-L1 expression was calculated using the PD-L1 evaluable population as the denominator (N = 333; approximately 7% not evaluable for PD-L1 expression) and the percentage of patients per stage was based on the reclassification of patients according to AJCC Cancer Staging Manual, eighth edition.
Source: Indirect treatment comparison technical report.78
Table 32: Baseline Characteristics in the CheckMate 816 and AEGEAN Studies (Unweighted and After Weighting to Match the CheckMate 816 Study in Scenario 2)
Characteristic | CheckMate 816 study (N = 358) | AEGEAN study Unweighted (N = 740) | AEGEAN study Scenario 2 (ESS = █████) | ||
|---|---|---|---|---|---|
n | % | n | % | % | |
Aged < 65 years | 176 | 49.2 | 358 | 48.4 | ████ |
ECOG PS: 0 | 241 | 67.3 | 506 | 68.4 | ████ |
Planned platinum chemotherapy: Cisplatin | 258 | 78.2 | 196 | 26.5 | ████ |
Histology: Nonsquamous | 176 | 49.1 | 375 | 50.7 | ████ |
PD-L1 expression: < 1% | 155 | 46.5 | 247 | 33.4 | ████ |
PD-L1 expression: ≥ 50% | 80 | 24.0 | 216 | 29.2 | ████ |
Region: Asia | 177 | 49.4 | 305 | 41.2 | ████ |
Sex: Female | 103 | 28.8 | 210 | 28.4 | ████ |
Smoking status: Never | 39 | 10.9 | 107 | 14.5 | ████ |
Stage: IIIA | NA | 57.4 | 338 | 45.7 | ████ |
Stage: IIIB | NA | 12.1 | 187 | 25.3 | ████ |
AJCC = American Joint Committee on Cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EM = effect modifier; ESS = effective sample size; NA = not applicable.
Notes: Characteristics included in the weighting to match the CheckMate 816 study: planned platinum chemotherapy, PD-L1 expression, region, and stage (possible EMs that are imbalanced) in scenario 2.
For the CheckMate 816 study, the percentage of PD-L1 expression was calculated using the PD-L1 evaluable population as the denominator (N = 333; approximately 7% not evaluable for PD-L1 expression) and the percentage of patients per stage was based on the reclassification of patients according to AJCC Cancer Staging Manual, eighth edition.
Source: Indirect treatment comparison technical report.78
Cox regression analysis results on the weighted AEGEAN study’s population (after weighting to match the CheckMate 816 study) are provided in Table 33. In both scenarios, weighting to match the CheckMate 816 study’s population improved the relative treatment benefit of perioperative durvalumab versus placebo compared with the unweighted HR.
The results of the MAIC for both scenarios are shown in Table 34.
MAIC: Piecewise Analysis on AEGEAN and CheckMate 816 Studies
Event numbers before weighting for intervals of 0 to 3 months and 3-plus months are presented in Table 35, and the HRs for each intervention versus the neoadjuvant chemotherapy control arm (before and after weighting in the AEGEAN study) are presented in Table 36.
The results of the piecewise MAICs for both scenarios are shown in Table 36.
Table 33: Cox Regression Analysis of EFS for Durvalumab vs. Placebo in the AEGEAN Study (Unweighted and After Weighting in Scenario 1 and Scenario 2, Overall Trial Period)
Comparison | Scenario | EFS HR | LCL (95%) | UCL (95%) |
|---|---|---|---|---|
Durvalumab + PDC vs. placebo + PDC | Unweighted | ████ | ████ | ████ |
Scenario 1 (base case) | ████ | ████ | ████ | |
Scenario 2 | ████ | ████ | ████ |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: Based on unstratified Cox proportional hazards model.
For scenario 1, weighting was based on all possible EMs: planned platinum chemotherapy, PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: planned platinum chemotherapy, PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
Table 34: MAIC EFS HRs for Perioperative Durvalumab vs. Neoadjuvant Nivolumab (Unweighted and After Weighting in Scenario 1 and Scenario 2, Overall Trial Period)
Comparison | Scenario | EFS HR | LCL (95%) | UCL (95%) |
|---|---|---|---|---|
Durvalumab + PDC vs. nivolumab + PDC | Unweighted | ████ | ████ | ████ |
Scenario 1 (base case) | ████ | ████ | ████ | |
Scenario 2 | ████ | ████ | ████ |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; MAIC = matching-adjusted indirect comparison; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: For scenario 1, weighting was based on all possible EMs: planned platinum chemotherapy, PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: planned platinum chemotherapy, PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
Table 35: Event Numbers in the AEGEAN and CheckMate 816 Trials for Intervals of 0 to 3 Months and 3+ Months
Arm | Trial | Time point | Number at risk | Events |
|---|---|---|---|---|
Durvalumab + PDC | AEGEAN trial | 0 to 3 months | ███ | ██ |
Durvalumab + PDC | AEGEAN trial | 3+ months | ███ | ███ |
Placebo + PDC | AEGEAN trial | 0 to 3 months | ███ | ██ |
Placebo + PDC | AEGEAN trial | 3+ months | ███ | ███ |
PDC | CheckMate 816 trial | 0 to 3 months | ███ | ██ |
PDC | CheckMate 816 trial | 3+ months | ███ | ██ |
Nivolumab + PDC | CheckMate 816 trial | 0 to 3 months | ███ | ██ |
Nivolumab + PDC | CheckMate 816 trial | 3+ months | ███ | ██ |
EFS = event-free survival; KM = Kaplan-Meier; PDC = platinum-doublet chemotherapy.
Note: The number at risk and the number of events in the CheckMate 816 study were based on pseudopatient-level data generated from the digitization of the EFS KM curves.
Source: Indirect treatment comparison technical report.78
Table 36: MAIC Piecewise EFS HRs (Intervals of 0 to 3 Months and 3+ Months) for Perioperative Durvalumab vs. Neoadjuvant Nivolumab (Unweighted and After Weighting in Scenario 1 and Scenario 2)
Comparison | Scenario | 0 to 3 months | 3+ months | ||||
|---|---|---|---|---|---|---|---|
EFS HR | LCL (95%) | UCL (95%) | EFS HR | LCL (95%) | UCL (95%) | ||
Durvalumab + PDC vs. nivolumab + PDC | Unweighted | ████ | ████ | ████ | ████ | ████ | ████ |
Scenario 1 (base case) | ████ | ████ | ████ | ████ | ████ | ████ | |
Scenario 2 | ████ | ████ | ████ | ████ | ████ | ████ | |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; MAIC = matching-adjusted indirect comparison; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: For scenario 1, weighting was based on all possible EMs: planned platinum chemotherapy, PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: planned platinum chemotherapy, PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
MAIC: AEGEAN Study Versus KEYNOTE-671 Study
An anchored MAIC analysis was performed to compare the efficacy of perioperative durvalumab plus chemotherapy from the AEGEAN study versus perioperative pembrolizumab plus chemotherapy followed by adjuvant pembrolizumab from the KEYNOTE-671 study. For this comparison, the AEGEAN trial’s cisplatin subgroup was used to align with the KEYNOTE-671 trial, in which only cisplatin-based neoadjuvant platinum-doublet chemotherapy regimens were permitted. The summary of event numbers and HRs for the AEGEAN trial (before weighting) and the KEYNOTE-671 trial are shown in Table 37.
Table 37: Trial Summaries of EFS in the AEGEAN and KEYNOTE-671 Studies
Arm | Trial | N | Events |
|---|---|---|---|
Durvalumab + PDC | AEGEAN trial | 100 | 28 |
Placebo + PDC | AEGEAN trial | 96 | 42 |
Pembrolizumab + cisplatin-based PDC | KEYNOTE-671 trial | 397 | 172 |
Cisplatin-based PDC | KEYNOTE-671 trial | 400 | 245 |
EFS = event-free survival; PDC = platinum-doublet chemotherapy.
Source: Indirect treatment comparison technical report.78
A comparison of baseline characteristics between the 2 studies (overall trial — namely, both arms) before and after weighting are shown in Table 38 (weighting in scenario 1) and Table 39 (weighting in scenario 2). There were imbalances in baseline characteristics between the AEGEAN and KEYNOTE-671 studies, including the following:
the proportion of patients enrolled in Asia (region) (lower in the KEYNOTE-671 study)
the proportion of patients with PD-L1 of 50% or more (higher in the KEYNOTE-671 study)
the proportion of patients with stage IIIB disease (lower in the KEYNOTE-671 study) and stage IIIA disease (higher in the KEYNOTE-671 study)
the proportion of patients with an ECOG PS score of 0 (lower in the KEYNOTE-671 study).
With the exception of ECOG PS, which the sponsor did not consider to be a possible EM (and was not therefore factored into the weighting), these characteristics were all considered by the sponsor to be possible modifiers of treatment effect. After weighting patients, the baseline characteristics in the AEGEAN trial matched those in the KEYNOTE-671 trial for those variables that were included in the weighting.
There was a modest reduction in the effective sample size (< 20%) in both scenarios, indicating that there was reasonable overlap between studies, and there was no evidence of extreme weights (> 10).
Table 38: Baseline Characteristics in the KEYNOTE-671 and AEGEAN Studies Cisplatin Subgroup (Unweighted and After Weighting to Match the KEYNOTE-671 Study in Scenario 1 [Base Case])
Characteristic | KEYNOTE-671 study (N = 797) | AEGEAN study, unweighted (N = 196) | AEGEAN study, scenario 1 (ESS = 165.5) | ||
|---|---|---|---|---|---|
n | % | n | % | % | |
Aged < 65 years | 435 | 54.6 | 109 | 55.6 | 52.9 |
ECOG PS score: 0a | 499 | 62.6 | 146 | 74.5 | 75.9 |
Planned platinum chemotherapy: Cisplatin | 797 | 100 | 196 | 100 | 100 |
Histology: Nonsquamousb | 453 | 56.8 | 104 | 53.1 | 56.9 |
PD-L1 expression: < 1%b | 289 | 36.3 | 63 | 32.1 | 36.3 |
PD-L1 expression: ≥ 50%a,b | 266 | 33.4 | 45 | 23 | 33.2 |
Region: Asiaa,b | 244 | 30.6 | 73 | 37.8 | 30.7 |
Sex: Femaleb | 234 | 29.4 | 56 | 28.6 | 29.3 |
Smoking status: Neverb | 101 | 12.7 | 31 | 15.8 | 12.7 |
Stage: IIIAa,b | 442 | 55.5 | 95 | 48.5 | 55.5 |
Stage: IIIBa,b | 116 | 14.6 | 49 | 25 | 14.6 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EM = effect modifier; ESS = effective sample size.
aCharacteristics with imbalance (≥ 5% difference) between KEYNOTE-671 and AEGEAN studies.
bCharacteristics included in the weighting to match the KEYNOTE-671 trial: histology, PD-L1 expression, region, sex, smoking status, and stage (all possible EMs) in scenario 1.
Table 39: Baseline Characteristics in the KEYNOTE-671 and AEGEAN Studies Cisplatin Subgroup (Unweighted and After Weighting to Match the KEYNOTE-671 Study in Scenario 2)
Characteristic | KEYNOTE-671 study (N = 797) | AEGEAN study, unweighted (N = 196) | AEGEAN study, scenario 1 (ESS = 165.5) | ||
|---|---|---|---|---|---|
n | % | n | % | % | |
Aged < 65 years | 435 | 54.6 | 109 | 55.6 | 53.7 |
ECOG PS score: 0a | 499 | 62.6 | 146 | 74.5 | 75.8 |
Planned platinum chemotherapy: Cisplatin | 797 | 100 | 196 | 100 | 100 |
Histology: Nonsquamous | 453 | 56.8 | 104 | 53.1 | 55.8 |
PD-L1 expression: < 1%b | 289 | 36.3 | 63 | 32.1 | 36.3 |
PD-L1 expression: ≥ 50%a,b | 266 | 33.4 | 45 | 23 | 33.2 |
Region: Asiaa,b | 244 | 30.6 | 73 | 37.8 | 30.7 |
Sex: Female | 234 | 29.4 | 56 | 28.6 | 28.6 |
Smoking status: Never | 101 | 12.7 | 31 | 15.8 | 15.9 |
Stage: IIIAa,b | 442 | 55.5 | 95 | 48.5 | 55.5 |
Stage: IIIBa,b | 116 | 14.6 | 49 | 25 | 14.6 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EM = effect modifier; ESS = effective sample size.
aCharacteristics with imbalance (≥ 5% difference) between the KEYNOTE-671 and AEGEAN studies.
bCharacteristics included in the weighting to match the KEYNOTE-671 trial: PD-L1 expression, region, and stage (possible EMs that were imbalanced) in scenario 2.
Results from the Cox regression analysis on the weighted AEGEAN study’s population (after weighting to match the KEYNOTE-671 study) are provided in Table 40. In both scenarios, weighting to match the KEYNOTE-671 study’s population improved the relative treatment benefit of perioperative durvalumab versus placebo compared with the unweighted HR.
The results of the MAIC for both scenarios are shown in Table 41.
Table 40: Cox Regression Analysis of EFS for Durvalumab vs. Placebo in the AEGEAN Study (Unweighted and After Weighting in Scenario 1 and Scenario 2)
Comparison | Scenario | EFS HR | LCL (95%) | UCL (95%) |
|---|---|---|---|---|
Durvalumab + PDC vs. placebo + PDC | Unweighted | 0.58 | 0.36 | 0.93 |
Scenario 1 (base case) | 0.47 | 0.28 | 0.79 | |
Scenario 2 | 0.49 | 0.29 | 0.83 |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: Based on unstratified Cox proportional hazards model.
For scenario 1, weighting was based on all possible EMs: PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
Table 41: MAIC EFS HRs for Perioperative Durvalumab vs. Perioperative Pembrolizumab (Cisplatin Only) (Unweighted and After Weighting in Scenario 1 and Scenario 2)
Comparison | Scenario | EFS HR | LCL (95%) | UCL (95%) |
|---|---|---|---|---|
Durvalumab + PDC vs. pembrolizumab + PDC | Unweighted | 0.98 | 0.59 | 1.65 |
Scenario 1 (base case) | 0.80 | 0.46 | 1.39 | |
Scenario 2 | 0.83 | 0.47 | 1.46 |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; MAIC = matching-adjusted indirect comparison; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: For scenario 1, weighting was based on all possible EMs: PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
MAIC: Piecewise Analysis on AEGEAN and KEYNOTE-671 Studies
The event numbers before weighting for the intervals of 0 to 3 months and 3-plus months are presented in Table 42, and the HRs for each intervention versus placebo (before and after weighting in the AEGEAN study) are presented in Table 43.
Table 42: Event Numbers in the AEGEAN Study (Cisplatin Subgroup) and KEYNOTE-671 Study for Intervals of 0 to 3 Months and 3+ Months
Arm | Trial | Time point | Number at risk | Events |
|---|---|---|---|---|
Durvalumab + PDC | AEGEAN study | 0 to 3 months | 100 | 3 |
Durvalumab + PDC | AEGEAN study | 3+ months | 95 | 25 |
Placebo + PDC | AEGEAN study | 0 to 3 months | 96 | 6 |
Placebo + PDC | AEGEAN study | 3+ months | 86 | 36 |
Pembrolizumab + cisplatin-based PDC | KEYNOTE-671 study | 0 to 3 months | 397 | 23 |
Pembrolizumab + cisplatin-based PDC | KEYNOTE-671 study | 3+ months | 369 | 149 |
Cisplatin-based PDC | KEYNOTE-671 study | 0 to 3 months | 400 | 15 |
Cisplatin-based PDC | KEYNOTE-671 study | 3+ months | 378 | 230 |
EM = effect modifier; KM = Kaplan-Meier; PDC = platinum-doublet chemotherapy.
Note: The number at risk and the number of events in the KEYNOTE-671 trial were based on pseudopatient-level data generated from the digitization of the EFS KM curves.
Source: Indirect treatment comparison technical report.78
Table 43: MAIC Piecewise EFS HRs (Intervals of 0 to 3 Months and 3+ Months) for Perioperative Durvalumab vs. Perioperative Pembrolizumab (Cisplatin Only) (Unweighted and After Weighting in Scenario 1 and Scenario 2)
Comparison | Scenario | 0 to 3 months | 3+ months | ||||
|---|---|---|---|---|---|---|---|
EFS HR | LCL (95%) | UCL (95%) | EFS HR | LCL (95%) | UCL (95%) | ||
Durvalumab + PDC vs. pembrolizumab + PDC | Unweighted | 0.30 | 0.06 | 1.41 | 1.13 | 0.66 | 1.96 |
Scenario 1 (base case) | 0.25 | 0.05 | 1.24 | 0.91 | 0.50 | 1.65 | |
Scenario 2 | 0.28 | 0.06 | 1.36 | 0.94 | 0.52 | 1.70 | |
EFS = event-free survival; EM = effect modifier; HR = hazard ratio; LCL = lower confidence limit; MAIC = matching-adjusted indirect comparison; PDC = platinum-doublet chemotherapy; UCL = upper confidence limit; vs. = versus.
Notes: For scenario 1, weighting was based on all possible EMs: planned platinum chemotherapy, PD-L1 expression, region, stage, histology, sex, and smoking status.
For scenario 2, weighting was based on possible EMs that were imbalanced between trials: planned platinum chemotherapy, PD-L1 expression, region, and stage.
Source: Indirect treatment comparison technical report.78
NMA: Durvalumab Versus Neoadjuvant Chemotherapy, Adjuvant Chemotherapy, and Surgery (mITT Population)
The EFS NMA (base case) in the mITT population is shown as a network diagram in Figure 8. Due to the low number of events in the 0- to 3-month interval across studies (with some studies reporting 0 events), the models did not converge, and so this piecewise NMA was not feasible. Therefore, only results for the piecewise NMA using the interval of 3-plus months are presented.
Statistical heterogeneity in the mITT network (base-case and sensitivity analyses) was quantified as 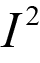 (refer to Table 44). For the NMA in the overall period, the
(refer to Table 44). For the NMA in the overall period, the 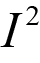 value indicated that there was ████████████ heterogeneity (> 75%)116██████116 between studies informing the surgery versus the neoadjuvant chemotherapy comparison in the base-case analysis and also sensitivity analysis 3 (both of which include Rosell [1994]). Heterogeneity was reduced in other sensitivity analyses, with the lowest
value indicated that there was ████████████ heterogeneity (> 75%)116██████116 between studies informing the surgery versus the neoadjuvant chemotherapy comparison in the base-case analysis and also sensitivity analysis 3 (both of which include Rosell [1994]). Heterogeneity was reduced in other sensitivity analyses, with the lowest 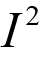 in sensitivity analysis 2, which excluded both Rosell (1994) and Li (2009).
in sensitivity analysis 2, which excluded both Rosell (1994) and Li (2009).
Table 44: Heterogeneity (I2) in the mITT Population Network
Population | Comparison | Analysis | I2 overall period | I2 3+ months piecewise |
|---|---|---|---|---|
mITT | Surgery vs. neoadjuvant chemotherapy | Base case | █████ | █████ |
Sensitivity analysis 1 | █████ | █████ | ||
Sensitivity analysis 2 | █████ | █████ | ||
Sensitivity analysis 3 | █████ | █████ | ||
Sensitivity analysis 4 | █████ | █████ |
mITT = modified intention to treat; vs. = versus.
Note: No assessment of statistical heterogeneity was performed for perioperative durvalumab vs. adjuvant chemotherapy and perioperative durvalumab vs. surgery alone.
Source: Indirect treatment comparison technical report.78
Random-effects models were preferred given the level of heterogeneity identified in the feasibility assessment. With the exception of the base-case analysis and sensitivity analysis 3 (both of which included Rosell [1994]103,104) (deviance information criteria was lower for random-effects models), the deviance information criteria values were similar between fixed-effects and random-effects models for NMAs in the overall period, with differences in deviance information criteria of less than || points. Comparisons of posterior mean residual deviance and the number of data points indicated that the preferred random-effects models provided an adequate model fit in sensitivity analysis 1, sensitivity analysis 2, and sensitivity analysis 4, with residual deviance similar to the number of data points.
The HRs (including 95% CrIs) for comparisons of perioperative durvalumab with neoadjuvant chemotherapy versus each comparator are presented in Table 45 (base case). Within the table, HRs are presented for both random-effects and fixed-effects models for the overall period (0+ months). Table 46 presents the results of the piecewise NMA (intervals of 3+ months) for perioperative durvalumab with neoadjuvant chemotherapy versus adjuvant platinum-based chemotherapy, perioperative durvalumab with neoadjuvant chemotherapy versus surgery alone, and perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant platinum-based chemotherapy.
In the base-case analysis and all sensitivity analyses, results of the analysis of the random-effects model showed point estimates for the HRs that favoured perioperative durvalumab with neoadjuvant chemotherapy versus each of the comparators; however, the 95% CrIs were wide, including the potential that either treatment being compared could be favoured. The results of the fixed-effects model analysis were more precise, and all estimates favoured durvalumab with neoadjuvant chemotherapy over the comparators.
Greater precision (a narrower 95% CrI) compared with the base-case analysis was achieved in NMAs for the overall period in the sensitivity analyses, which excluded Rosell (1994).103,104 In sensitivity analysis 2, which excluded both Rosell (1994)103,104 and Li (2009)107 (studies that included stage III patients only), the EFS HRs (95% CrI) from the random-effects NMA were ████ (95% CrI, █████ ████), ████ (95% CrI, █████ ████), and ████ (95% CrI, █████ ████) for perioperative durvalumab with neoadjuvant chemotherapy versus adjuvant chemotherapy, neoadjuvant chemotherapy, and surgery only, respectively.
Table 45: EFS HR Durvalumab vs. Adjuvant Chemotherapy, Neoadjuvant Chemotherapy, and Surgery Only — mITT Population (Base Case)
Comparison | HR (95% Crl) | |
|---|---|---|
REM analysis | FEM analysis | |
Durvalumab vs. adjuvant chemotherapy | ████ ██████ █████ | ████ ██████ █████ |
Durvalumab vs. surgery only | ████ ██████ █████ | ████ ██████ █████ |
Durvalumab vs. neoadjuvant chemotherapy | ████ ██████ █████ | ████ ██████ █████ |
Crl = credible interval; EFS = event-free survival; FEM = fixed-effects model; HR = hazard ratio; mITT = modified intention to treat; REM = random-effects model; vs. = versus.
Source: Indirect treatment comparison technical report.78
Table 46: NMA Piecewise EFS HRs (Intervals of 3+ Months) for Perioperative Durvalumab vs. Adjuvant PBC, Perioperative Durvalumab vs. Surgery Alone, and Perioperative Durvalumab vs. Neoadjuvant PBC
Comparison | Interval of 3+ months, HR (95% Crl) | |
|---|---|---|
REM analysis | FEM analysis | |
Durvalumab vs. adjuvant chemotherapy | ████ ██████ █████ | ████ ██████ █████ |
Durvalumab vs. surgery only | ████ ██████ █████ | ████ ██████ █████ |
Durvalumab vs. neoadjuvant chemotherapy | ████ ██████ █████ | ████ ██████ █████ |
Crl = credible interval; EFS = event-free survival; FEM = fixed-effects model; HR = hazard ratio; mITT = modified intention to treat; NMA = network meta-analysis; PBC = platinum-based chemotherapy; REM = random-effects model; vs. = versus.
Source: Indirect treatment comparison technical report.78
The sensitivity analysis 2 results for piecewise (3+ months) EFS comparator efficacy (surgery alone, and adjuvant platinum-based chemotherapy) were of particular interest because these are incorporated into the accompanying pharmacoeconomic analysis (Table 47).
Table 47: Piecewise (3+ Months) EFS Comparator Efficacy (Surgery Alone and Adjuvant PBC)
Treatment | Piecewise HR | Lower 95% CI | Upper 95% CI | AEGEAN study reference arm |
|---|---|---|---|---|
Surgery alone | ████ | ████ | ████ | PBO (i.e., neoadjuvant PBC) |
Adjuvant PBC | ████ | ████ | ████ | PBO (i.e., neoadjuvant PBC) |
CI = confidence interval; EFS = event-free survival; HR = hazard ratio; PBC = platinum-based chemotherapy; PBO = placebo.
Source: Indirect treatment comparison technical report.78
The results of these NMAs were largely consistent across base-case and sensitivity analyses.
Harms
Harms end points were not evaluated.
Critical Appraisal of ITCs
Overall, the ITCs (2 MAICs and 1 NMA) were conducted according to accepted methodological guidance.89 The search for potentially eligible studies was adequately comprehensive and methods used to select relevant studies, extract data, and appraise risk of bias were adequate to limit the potential for error and bias. However, given that the included studies were limited to those with freely available full-text articles, there is a risk of bias due to missing evidence. Further, the last search date for this review was in 2024; given that it has taken longer than expected for the deliberation to occur in 2026, newly available evidence may have emerged but not been screened or considered for this review. The extent of this bias is unknown because there is no information on how many articles were excluded because they required payment to access the full text. Further, the results of the risk of bias appraisals for each study included in the ITCs were not explicitly reported. As such, the potential impact of study-level risk of bias on the results of the ITCs could not be assessed. Other potential limitations of the MAICs and the NMA are discussed as follows.
The key limitations of the MAIC analyses include potential imbalances in baseline characteristics considered to be potential EMs between the AEGEAN trial and comparator trials.
For the comparison versus neoadjuvant nivolumab, imbalances exist between the AEGEAN study versus the CheckMate 816 study in baseline characteristics that are considered to be potential EMs (e.g., stage, PD-L1, region, planned chemotherapy). To account for these imbalances in possible EMs, anchored MAICs were conducted in which the AEGEAN study’s population was weighted to more closely match the CheckMate 816 study’s population with respect to possible EMs. However, it is not clear that all the potential EMs were used in the adjustment. The sponsor attempted to identify potential EMs by identifying stratification factors in the included trials, and by investigating the potential for effect modification via the results of their subgroup analyses. However, these subgroup analyses were not likely powered to identify subgroup differences. As such, this method was likely inadequate to identify all potential EMs. Further, as noted by the sponsor, there is limited evidence for EMs in this disease area, raising uncertainty as to whether all EMs were accounted for in the analyses. In addition, the AEGEAN study was a double-blind RCT; however, the CheckMate 816 study was an open-label trial. The open-label nature of the trial had the potential to impact the outcome assessments, although the magnitude and the direction of the impact remain unknown. Such differences in trial design cannot be accounted for via the MAIC methodology.
For the comparison versus perioperative pembrolizumab, the MAIC was conducted using the AEGEAN study cisplatin subgroup to align with the KEYNOTE-671 study, which permitted only cisplatin-based neoadjuvant platinum-doublet chemotherapy regimens. Restricting the AEGEAN study’s population to this subgroup reduced the overall sample size, limiting the generalizability of the MAIC results to patients receiving cisplatin-based neoadjuvant chemotherapy and increasing uncertainty in the estimated treatment effects. As with the comparison versus neoadjuvant nivolumab, uncertainty remains regarding whether all relevant EMs were accounted for in the population-adjustment process.
In the piecewise analysis, the low number of events in this interval should be considered when interpretating results. All effect sizes were associated with high uncertainty, and the 95% CIs crossed the threshold of no difference for both MAICs.
Furthermore, substantial reductions in effective sample size were observed in the base-case MAIC analysis (i.e., ████ reduction), while smaller reductions were observed for the comparison versus perioperative pembrolizumab (15% to 17%). Such reductions may indicate limited overlap in baseline characteristics between the trial populations. The reduced effective sample size contributed to imprecision and may suggest that weighted estimates were influenced by a relatively small subset of patients in the AEGEAN study’s population. The piecewise comparative effect estimates of EFS (at intervals of 0 to 3 months and 3+ months) for perioperative durvalumab with neoadjuvant chemotherapy compared with neoadjuvant nivolumab with chemotherapy were associated with considerable uncertainty, precluding conclusions regarding which treatment may be favoured. The 95% CIs for the comparative effect estimates (HRs) were wide, including the potential for both comparative benefit and harm. Further, as per the pivotal trial evidence, no literature was identified investigating the validity of EFS as a surrogate for OS specific to this treatment type and patient population. As such, the results do not inform on the comparative efficacy between perioperative durvalumab with neoadjuvant chemotherapy versus the comparators with respect to OS.
The key limitation of the NMA was the heterogeneity across the included studies in terms of the study designs (e.g., the trials were phase III RCTs, but 2 studies did not report the study phase; all studies had a multicentre design, but 1 study was a single-centre study; most studies were global and/or European studies, but 4 studies were conducted in sites in Asia only). In addition, 2 studies did not report the geographical location. The AEGEAN and CheckMate 816 studies began in 2017 or later. In contrast, all studies for adjuvant chemotherapy and for surgery only were started before 2004, with some studies (Rosell [1994]103,104 and Roth [1994]97,113) starting before 1990. The timing of studies likely impacts the comparability of studies, given that the standard of care has changed since these studies were undertaken; for example, staging and tumour assessment criteria and the use of second-generation chemotherapy agents have changed. There were differences across the included trials in some patient characteristics. For example, the trials enrolled patients with variable stages of NSCLC, the distribution of PD-L1 expression levels varied across trials, and other characteristics like smoking status were infrequently reported, limiting a thorough homogeneity appraisal. Methodological differences across the trials were noted in outcome definitions and reporting, and tumour assessment and criteria, including how EFS was assessed in each study. The assumption that EFS, DFS, and PFS could be analyzed interchangeably could not be verified but may not be appropriate. The noted differences across trials in patient characteristics, trial design, and trial timing threaten the plausibility of the transitivity assumption.
Several studies (Chen [2013],96 Roth [1994],97 JCOG 9209,98 and Depierre [2002]99) met the inclusion criteria for the ITC but were excluded due to the absence of EFS KM curves and HRs. The impact of the exclusion of these studies on the NMA results was unknown and there is a potential for a risk of bias due to missing evidence.
Direct comparative efficacy of perioperative durvalumab with neoadjuvant chemotherapy versus placebo with neoadjuvant chemotherapy in terms of EFS was established in the AEGEAN RCT. Given the limitations of the NMA, the direct evidence for the pivotal trial presented herein is preferred for this comparison. In all comparisons, the analyses of the random-effects model were preferred based on the model fit analysis. As expected, the random-effects model produced more conservative estimates of the precision of the between-group effects (i.e., had wider CrIs that spanned the null), whereas those for the fixed-effects model had narrower CrIs. In addition to being a poorer fit to the data, the fixed-effects model assumes that differences in the populations enrolled, the implementation of interventions, and outcome measurement had no impact on the magnitude of effect, which is not likely plausible. As per the MAICs, effect estimates for the random-effects NMAs were imprecise, precluding any conclusions as to whether perioperative durvalumab with neoadjuvant chemotherapy or any of the comparator treatments were favoured.
Finally, all evidence networks were sparse, with no closed loops formed by multiple studies.
Together, all the aforementioned limitations increase the potential for biased treatment effect estimates, which limited the robustness of the NMA and MAIC.
No NMAs or MAICs were done for safety outcomes, or for any other outcomes that are important to patients (e.g., OS, HRQoL).
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies to address gaps within the systematic review evidence were submitted.
Discussion
Summary of Available Evidence
One ongoing, phase III, double-blind, placebo-controlled, international RCT (N = 740 [mITT population]), the AEGEAN study, met the inclusion criteria for the sponsor-conducted systematic review. The AEGEAN study enrolled adult patients with newly diagnosed and previously untreated resectable NSCLC. At enrolment, patients were required to have an ECOG PS score of 0 or 1, no prior exposure to immune-mediated therapies, and adequate organ and marrow function. Following a protocol amendment, those with known EGFR mutations or ALK rearrangements were excluded postrandomization. Patients were randomized to receive either 1,500 mg durvalumab every 3 weeks for 4 cycles before surgery in combination with neoadjuvant platinum-based doublet chemotherapy and 1,500 mg durvalumab every 4 weeks for up to 12 cycles following surgery or placebo every 3 weeks for 4 cycles before surgery in combination with neoadjuvant platinum-based doublet chemotherapy and placebo every 4 weeks for 12 cycles following surgery. The primary objectives of the AEGEAN trial were to compare the efficacy and activity of perioperative durvalumab with neoadjuvant chemotherapy compared to placebo with neoadjuvant chemotherapy. The outcomes relevant to this review included the coprimary outcomes of pCR and EFS, and the secondary outcomes of OS, MPR, DFS, HRQoL assessed via EORTC QLC-C3 and EORTC QLC-LC13, and harms. The trial population had a median age of 65.0 years (range, 39 years to 85 years), had fewer female participants (28.4% of patients [210 of 740]) than male participants (71.6% of patients [530 of 740]), and tended to have either currently or formerly smoked (8.8.% of patients [633 of 740]). The population was primarily Asian (41.5% of patients [307 of 740]) and white (53.6% of patients [397 of 740]), with just 1.4% of patients (10 of 740) identifying as American Indian or Alaska Native, 0.9% of patients (7 of 740) identifying as Black or African American, and 2.6% of patients (19 of 740) identifying with other races. At baseline, patients tended to have an ECOG PS score of 0 (68.4% of patients [506 of 740]), presented with stage III disease (70.9% of patients [525 of 740]), and had a baseline PD-L1 expression status greater than or equal to 1% (66.6% of patients [493 of 740]).
In the absence of direct evidence from randomized trials comparing perioperative durvalumab with neoadjuvant chemotherapy to other relevant immunotherapy-based regimens, the sponsor conducted 3 ITCs. Two anchored MAIC analyses were performed to assess the comparative efficacy, based on EFS, of perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant nivolumab with chemotherapy and versus perioperative pembrolizumab with neoadjuvant chemotherapy followed by adjuvant pembrolizumab. One NMA assessed the efficacy of perioperative durvalumab with neoadjuvant chemotherapy versus adjuvant chemotherapy and surgery only on EFS.
Interpretation of Results
Efficacy
The patient group input collected for this review highlighted that the key treatment goals for patients are to prolong life, stop or delay disease symptoms, maintain or improve HRQoL, and reduce AEs. The effect of perioperative durvalumab with neoadjuvant chemotherapy was assessed in the AEGEAN trial by the assessment of OS, pCR, EFS, EORTC QLQ-C30 global health score and QoL, and AEs.
Clinical experts consulted for this review considered OS to be the most clinically important outcome. Clinical experts considered that a 5% to 10% or greater between-group difference in OS is clinically meaningful. Using this threshold, at 48 months, perioperative durvalumab with neoadjuvant chemotherapy may result in little to no difference in the probability of being alive compared to placebo with neoadjuvant chemotherapy. At a median OS follow-up time of █████ months, 261 deaths had occurred in the mITT population and the median OS was NR in the perioperative durvalumab with neoadjuvant chemotherapy arm. Given the median length of follow-up and number of patients at risk, OS results at 12 months were considered to have high certainty of evidence. However, at 48 months, there were relatively few patients at risk (approximately 11% in both treatment arms), resulting in substantial uncertainty in results at later time points, and the CIs crossed the clinically meaningful threshold, resulting in a low certainty of evidence. Of note, the OS data in the AEGEAN trial were immature (261 deaths [35.3%] had occurred in the mITT population) and not formally tested for statistical significance due to the failure of DFS to reach statistical significance; as such, the OS results are considered only as supportive evidence.
EFS and pCR were coprimary end points in the AEGEAN study and also considered by clinical experts consulted for this review as clinically important outcomes for patients with resectable NSCLC. Results showed at 36 months that the difference in EFS rates was 12.2% (95% CI, ███ to ████) in favour of perioperative durvalumab with neoadjuvant chemotherapy. As the clinical experts consulted for this review considered that a 5% to 10% between-group difference in EFS is clinically meaningful, the point estimate and upper bound of the 95% CI are clinically important; however, the lower bound of the 95% CI is below the 5% to 10% threshold, indicating some uncertainty. Further, EFS results must be interpreted in light of the fact that they are from an IA, which may overestimate treatment effects.20 At the final analysis, there was a 12.96% (95% CI, 8.67% to 17.57%) between-group difference in the percentage of patients achieving pCR in favour of perioperative durvalumab with neoadjuvant chemotherapy. The point estimate and upper bound of the 95% CI are clinically important at the 10% threshold suggested by clinical experts, and the lower bound of the 95% CI crosses the threshold, indicating some uncertainly.
Given that the currently available OS results from the AEGEAN study are immature, it is of interest to determine how well the benefits observed in EFS would translate into improvements in OS. A previous SLR and meta-analysis found a positive linear association between EFS and OS based on 8 RCTs among those with resectable NSCLC, indicating that improvements in EFS are likely predictive of improvements in OS in the neoadjuvant setting.69 However, this literature is specific to chemotherapy treatment in the neoadjuvant setting and cannot be generalized to the use of durvalumab in the perioperative setting. As such, the validity of EFS as a surrogate for OS in this setting is not known and it is unclear whether improved EFS will result in a clinically important OS benefit at longer follow-up.
DFS, 1 of the key secondary outcomes of the AEGEAN trial, was tested but did not meet the prespecified boundary for declaring statistical significance at EFS IA1 and EFS IA2. Results for DFS were overall supportive of the primary EFS analyses. At EFS IA2, the between-group difference in DFS rates at 12 months and 36 months was within the threshold of 5% to 10%, suggesting that perioperative durvalumab with neoadjuvant chemotherapy may result in a clinically meaningful increase in DFS compared with neoadjuvant chemotherapy. At both time points, the lower bounds of the 95% CIs crossed the null, indicating serious imprecision in the results. Similarly to OS, there was an adequate number of patients at risk at 12 months; however, relatively few patients remained at risk at 36 months (approximately 19% of patients in the perioperative durvalumab with neoadjuvant chemotherapy arm and 13% of patients in the neoadjuvant chemotherapy arm), resulting in substantial uncertainty in results at later time points. Additionally, there is a risk of bias due to DFS analyses performed using the modified resected set population, which is a subpopulation of the mITT population, and therefore it is likely that prognostic balance across the treatment group has been lost.
Patient input highlighted that maintaining or improving HRQoL is an important treatment goal and clinical experts noted the EORTC QLQ-C30 global health status and QoL scores in the neoadjuvant and adjuvant period are important for clinical decision-making. EORTC QLQ-C30 is a cancer-specific HRQoL questionnaire designed to measure QoL and has been widely used in resectable NSCLC studies.117 This questionnaire was originally tested in a cross-cultural sample of patients with lung cancer in 13 countries (including Canada) and was found to be a reliable and valid measure of QoL for patients with cancer.118 A between-group MID in the EORTC QLQ-C30 global health status and QoL score among patients with lung cancer has previously been defined as a 4-point increase or decrease, for improvement and deterioration, respectively.63 At the end of the neoadjuvant period in the AEGEAN trial, a between-group difference of █████ (95% CI,█████ ██ ████) in the EORTC QLQ-C30 health status and QoL score indicated that perioperative durvalumab with neoadjuvant chemotherapy likely results in little to no difference in QoL during the neoadjuvant period compared to placebo with neoadjuvant chemotherapy. However, results were limited by a substantial amount of missing data at week 12 in both arms (█████ missing in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ in the placebo with neoadjuvant chemotherapy arm). The between-group difference in the EORTC QLQ-C30 health status and QoL score at the end of the adjuvant period was █████ ████ ███ █████ ██ █████), indicating clinically important decreases in QoL during the adjuvant period among those receiving perioperative durvalumab with neoadjuvant chemotherapy compared to placebo with neoadjuvant chemotherapy. While the █████ ████████ ███ █████ ███ ██ ███████ ███ █████████ ███ █ ██████████ █████████ ████████ ██ ███, certainty was limited by the █████ █████ ██ ███ ███ ██ ███ ████████ ███ ███ █████████, results in the adjuvant period being limited to only those included in the modified resected set, and a substantial amount of missing data at week 44 ██████ missing in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ in the placebo with neoadjuvant chemotherapy arm). As such, it is not clear that, compared with placebo with neoadjuvant chemotherapy, perioperative durvalumab with neoadjuvant chemotherapy meets patients’ desire to maintain or improve HRQoL.
Clinical experts consulted for this review highlighted that the use of neoadjuvant chemotherapy on its own for resectable NSCLC has become less common in clinical practice in Canada. In addition to the standard of care neoadjuvant chemotherapy regimens used in the AEGEAN study, clinical experts indicated that in Canada, carboplatin and gemcitabine could also be used for squamous cell NSCLC and cisplatin and vinorelbine may also sometimes be used for both squamous and nonsquamous cell NSCLC. The clinical experts consulted for this review noted that currently, the 2 most commonly used regimens in clinical practice in Canada are neoadjuvant nivolumab with chemotherapy and adjuvant chemotherapy. The sponsor submitted ITCs aimed at comparing perioperative durvalumab with neoadjuvant chemotherapy against several comparators, including neoadjuvant nivolumab with platinum-based chemotherapy, perioperative pembrolizumab with neoadjuvant chemotherapy followed by adjuvant pembrolizumab, adjuvant platinum-based chemotherapy, and surgery only. The point estimates for the difference in EFS from the ITCs (2 MAICs and 1 NMA) favoured perioperative durvalumab with neoadjuvant chemotherapy over neoadjuvant nivolumab, perioperative pembrolizumab, adjuvant chemotherapy, and surgery only, in patients with resectable (tumours ≥ 4 cm and/or node positive) NSCLC and no known EGFR mutations or ALK rearrangements; however, the 95% CIs (or CrIs) for all estimates were too wide to draw any conclusion as to whether perioperative durvalumab with neoadjuvant chemotherapy results in better or worse EFS relative to any of the comparators. Other important limitations to the interpretation of the ITC evidence included important heterogeneity across the included trials in terms of study designs and outcome measures, networks that were sparse and not well connected, and the likelihood that not all the potential important EMs (such as variation of the staging and tumour assessment criteria over time) were matched and adjusted for in the MAICs. None of the ITCs investigated other outcomes that are important to patients, including OS and HRQoL.
Harms
A ███████ percentage of patients in both treatment arms of the modified safety analysis set experienced any AE over the course of treatment (█████ ████████ in the perioperative durvalumab with neoadjuvant chemotherapy arm and █████ ████████ in the placebo with neoadjuvant chemotherapy arm). In the perioperative durvalumab with neoadjuvant chemotherapy arm █████ of patients experienced an SAE compared with █████ in the placebo with neoadjuvant chemotherapy arm (based on the GRADE assessment, perioperative durvalumab with neoadjuvant chemotherapy likely results in little to no clinically important difference in the proportion of patients who experience ≥ 1 SAE, compared to placebo with neoadjuvant chemotherapy). As expected, a ██████ percentage of patients receiving perioperative durvalumab with neoadjuvant chemotherapy experienced imAEs than those receiving placebo with neoadjuvant chemotherapy (█████ versus █████, respectively). Similarly, ████ patients receiving perioperative durvalumab with neoadjuvant chemotherapy prematurely stopped treatment due to AEs than in the placebo with neoadjuvant chemotherapy arm (█████ versus ██████ respectively). ████████████ ██████ patients in the perioperative durvalumab with neoadjuvant chemotherapy arm experienced an AE resulting in death compared to ██ ██████ in the placebo with neoadjuvant chemotherapy arm. Harms were not investigated in the ITCs, so whether perioperative durvalumab with neoadjuvant chemotherapy results in an increase or decrease in harms relative to the comparator therapies is unknown.
The proposed product monograph suggests patients with resectable NSCLC treated with perioperative durvalumab with neoadjuvant chemotherapy should be monitored for signs and symptoms of imAEs. Depending on the severity of the imAEs, durvalumab should be withheld or discontinued. Infections and infusion-related reactions should also be monitored and treated if suspected.
The clinical experts consulted for this review expected that a higher percentage of patients treated with perioperative durvalumab with neoadjuvant chemotherapy would have AEs and immune-mediated side effects compared to those treated with placebo and neoadjuvant chemotherapy. The clinical experts consulted by the review team determined that overall, the safety profile of perioperative durvalumab with neoadjuvant chemotherapy was consistent with their expectations for this drug and deemed it acceptable.
Conclusion
The AEGEAN phase III, double-blind, placebo-controlled, international RCT compared the efficacy and safety of perioperative durvalumab with neoadjuvant chemotherapy against placebo with neoadjuvant chemotherapy in adult patients with resectable NSCLC and no known EGFR mutations or ALK rearrangements. The AEGEAN trial demonstrated added clinical benefits of perioperative durvalumab in pCR and EFS. Compared to placebo with neoadjuvant chemotherapy, perioperative durvalumab with neoadjuvant chemotherapy likely results in a clinically important increase in the number of patients achieving pCR (moderate certainty evidence) and a clinically important increase in EFS rates at 12 months and 36 months (moderate certainty evidence). Results for DFS, a key secondary outcome, were supportive of the primary EFS analyses. Based on EFS IA2 analysis, perioperative durvalumab with neoadjuvant chemotherapy may result in little to no difference in the probability of being alive at 48 months; however, survival data remained immature. Results for EFS from the indirect comparisons of perioperative durvalumab with neoadjuvant chemotherapy versus neoadjuvant nivolumab plus chemotherapy, perioperative pembrolizumab plus neoadjuvant chemotherapy followed by adjuvant pembrolizumab, adjuvant chemotherapy, and surgery only were inconclusive, owing to methodological limitations and imprecision. Based on the AEGEAN trial, perioperative durvalumab with neoadjuvant chemotherapy, compared to neoadjuvant chemotherapy, likely results in little to no difference in QoL during the neoadjuvant period (moderate certainty evidence) and may result in clinically meaningful deterioration in HRQoL in the adjuvant period; however, these results had a high degree of missing data and adjuvant results were limited to only those achieving complete resection. According to clinical experts consulted for this review, the safety profile of perioperative durvalumab with neoadjuvant chemotherapy was consistent with their expectations and deemed acceptable. Harms were not assessed in the indirect comparisons, so whether perioperative durvalumab with neoadjuvant chemotherapy results in an increase or decrease in harms compared with other therapies used in Canada is unknown.
References
1.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024 [sponsor supplied reference] CMAJ. 2024;196(18):E615-e623. doi:10.1503/cmaj.240095 PubMed
2.Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer SS, Canada the Public Health Agency of Canada, Canadian Cancer Statistics: A 2022 special report on cancer prevalence. Toronto, ON: Canadian Cancer Society; 2022. Available at: cancer.ca/Canadian-Cancer-Statistics-2022-EN [sponsor supplied reference]
3.Cagle PT, Allen TC, Olsen RJ. Lung cancer biomarkers: present status and future developments [sponsor supplied reference] Arch Pathol Lab Med. 2013;137(9):1191-8. doi:10.5858/arpa.2013-0319-CR PubMed
4.Le Chevalier T. Adjuvant chemotherapy for resectable non-small-cell lung cancer: where is it going? [sponsor supplied reference] Ann Oncol. 2010;21 Suppl 7:vii196-8. doi:10.1093/annonc/mdq376 PubMed
5.Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3(4):242-9. doi:10.3978/j.issn.2218-6751.2013.12.05 PubMed
6.Doherty M, Sit C, Leighl N, Wheatley-Price P. P2.08-001 Giving a Voice to Patients and Caregivers through the Lung Cancer Canada ‘Faces of Lung Cancer’ Survey: Topic: Patient’s Voice, Patient’s Information. J Thorac Oncol. 2017;12(1):S1109. doi:10.1016/j.jtho.2016.11.1551
7.Canadian Cancer Society. Lung cancer. 2020. Accessed June 23, 2024. https://cancer.ca/en/cancer-information/cancer-types/lung
8.Grant M, Sun V, Fujinami R, et al. Family caregiver burden, skills preparedness, and quality of life in non-small cell lung cancer. Oncol Nurs Forum. 2013;40(4):337-46. doi:10.1188/13.Onf.337-346 PubMed
9.Gridelli C, Perrone F, Nelli F, Ramponi S, De Marinis F. Quality of life in lung cancer patients. Ann Oncol. 2001;12 Suppl 3:S21-5. doi:10.1093/annonc/12.suppl_3.s21 PubMed
10.Sullivan DR, Eden KB, Dieckmann NF, et al. Understanding patients' values and preferences regarding early stage lung cancer treatment decision making. Lung Cancer. 2019;131:47-57. doi:10.1016/j.lungcan.2019.03.009 PubMed
11.Buzaglo J, Gayer C, Mallick R, et al. Understanding the experience of living with non-small-cell lung cancer (NSCLC): a qualitative study. J Community Support Oncol. 2014;12(1):6-12. doi:10.12788/jcso.0010 PubMed
12.Chiu K, MacEwan JP, May SG, et al. Estimating Productivity Loss from Breast and Non-Small-Cell Lung Cancer among Working-Age Patients and Unpaid Caregivers: A Survey Study Using the Multiplier Method. MDM Policy Pract. 2022;7(2):23814683221113846. doi:10.1177/23814683221113846 PubMed
13.Hamann HA, Ostroff JS, Marks EG, Gerber DE, Schiller JH, Lee SJ. Stigma among patients with lung cancer: a patient-reported measurement model. Psychooncology. 2014;23(1):81-92. doi:10.1002/pon.3371 PubMed
14.Iragorri N, de Oliveira C, Fitzgerald N, Essue B. The Indirect Cost Burden of Cancer Care in Canada: A Systematic Literature Review. Appl Health Econ Health Policy. 2021;19(3):325-341. doi:10.1007/s40258-020-00619-z PubMed
15.Canadian Cancer Statistics Advisory in collaboration with the Canadian Cancer SS, Canada the Public Health Agency of Canada, Canadian Cancer Statistics 2023. Toronto, ON: Canadian Cancer Society; 2023. Available at: cancer.ca/Canadian-Cancer-Statistics-2023-EN [sponsor supplied reference]
16.Provencio-Pulla M, Spicer J, Taube JM, et al. Neoadjuvant nivolumab (NIVO) + platinum-doublet chemotherapy (chemo) versus chemo for resectable (IB–IIIA) non-small cell lung cancer (NSCLC): Association of pathological regression with event-free survival (EFS) in CheckMate 816 [sponsor supplied reference] J Clin Oncol. 2022;40(17_suppl):LBA8511-LBA8511. doi:10.1200/JCO.2022.40.17_suppl.LBA8511
17.Spicer J, Girard N, Provencio M, et al. Neoadjuvant nivolumab (NIVO) + chemotherapy (chemo) vs chemo in patients (pts) with resectable NSCLC: 4-year update from CheckMate 816 [sponsor supplied reference] J Clin Oncol. 2024;42(17_suppl):LBA8010-LBA8010. doi:10.1200/JCO.2024.42.17_suppl.LBA8010
18.CADTH. CADTH Drug Reimbursement Expert Review Committee final recommendation: Durvalumab (Imfinzi - AstraZeneca Canada). CADTH; 2019. Accessed 19 September, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10131DurvalumabNSCLC_fnRec_02May2019_approvedbyChair_Post_03May2019_final.pdf
19.Li MSC, Mok KKS, Mok TSK. Developments in targeted therapy & immunotherapy-how non-small cell lung cancer management will change in the next decade: a narrative review. Ann Transl Med. 2023;11(10):358. doi:10.21037/atm-22-4444 PubMed
20.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-7. doi:10.1001/jama.2010.310 PubMed
21.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
22.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
23.AstraZeneca. EFA IA2 Clinical Study Report Appendix 14 (Tables and Figures) [internal sponsor's report]. 2024. Accessed 19 September, 2024.
24.AstraZeneca. Clinical Study Report: Durvalumab (MEDI4736)-D9106C00001. A Phase III, Double-blind, Placebo-controlled, Multi-center International Study of Neoadjuvant/Adjuvant Durvalumab for the Treatment of Patients with Resectable Stages II and III Non-small Cell Lung Cancer (AEGEAN) [internal sponsor's report]. AstraZeneca; 2023. Accessed 19 September, 2024.
25.Canadian Partnership Against Cancer. Lung Cancer and Equity: A Focus on Income and Geography. 2020. Accessed 19 September, 2024. https://www.partnershipagainstcancer.ca/topics/lung-cancer-equity/
26.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer. Toronto, ON: Canadian Cancer Society; 2020. Available from: cancer.ca/Canadian-Cancer-Statistics-2020-EN [sponsor supplied reference]
27.O'Leary C, Gasper H, Sahin KB, et al. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals (Basel). 2020;13(10). doi:10.3390/ph13100273 PubMed
28.Yang X, Zhong J, Yu Z, et al. Genetic and treatment profiles of patients with concurrent Epidermal Growth Factor Receptor (EGFR) and Anaplastic Lymphoma Kinase (ALK) mutations. BMC Cancer. 2021;21(1):1107. doi:10.1186/s12885-021-08824-2 PubMed
29.CADTH. CADTH Reimbursement Review: Provisional Funding Algorithm. 2023. Accessed October 12, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2023/PH0029-Algorithm_secured.pdf
30.B. C. Cancer Agency. Cancer Management Manual -- Lung. Available from: https://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/lung/lung [sponsor supplied reference]
31.Cancer Care Ontario. Lung Cancer Diagnosis Pathway Map, Version 2023.04. Available from: https://www.cancercareontario.ca/sites/ccocancercare/files/assets/LungCancerDiagnosisPathwayMap.pdf [sponsor supplied reference]
32.Postmus PE, Kerr KM, Oudkerk M, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up [sponsor supplied reference] Ann Oncol. 2017;28(suppl_4):iv1-iv21. doi:10.1093/annonc/mdx222 PubMed
33.Melosky B, Blais N, Cheema P, et al. Standardizing biomarker testing for Canadian patients with advanced lung cancer [sponsor supplied reference] Curr Oncol. 2018;25(1):73-82. doi:10.3747/co.25.3867 PubMed
34.Remon J, Soria JC, Peters S. Early and locally advanced non-small-cell lung cancer: an update of the ESMO Clinical Practice Guidelines focusing on diagnosis, staging, systemic and local therapy [sponsor supplied reference] Ann Oncol. 2021;32(12):1637-1642. doi:10.1016/j.annonc.2021.08.1994 PubMed
35.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Non-Small Cell Lung Cancer Version 3.2024 — March 12, 2024.
36.Burdett S, Pignon JP, Tierney J, et al. Adjuvant chemotherapy for resected early-stage non-small cell lung cancer [sponsor supplied reference] Cochrane Database Syst Rev. 2015;2015(3):Cd011430. doi:10.1002/14651858.Cd011430 PubMed
37.Vansteenkiste J, Crinò L, Dooms C, et al. 2nd ESMO Consensus Conference on Lung Cancer: early-stage non-small-cell lung cancer consensus on diagnosis, treatment and follow-up. Ann Oncol. 2014;25(8):1462-74. doi:10.1093/annonc/mdu089 PubMed
38.Calles A, Riess JW, Brahmer JR. Checkpoint Blockade in Lung Cancer With Driver Mutation: Choose the Road Wisely [sponsor supplied reference] Am Soc Clin Oncol Educ Book. 2020;40:372-384. doi:10.1200/edbk_280795 PubMed
39.Cancer Care Alberta. Recommendations Summary: Non-Small Cell Lung Cancer, Small Cell Lung Cancer, Mesothelioma. 2023. Accessed September 23, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-lu-nsclc-summary.pdf
40.Felip E, Altorki N, Zhou C, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial [sponsor supplied reference] Lancet. 2021;398(10308):1344-1357. doi:10.1016/s0140-6736(21)02098-5 PubMed
41.O'Brien M, Paz-Ares L, Marreaud S, et al. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB-IIIA non-small-cell lung cancer (PEARLS/KEYNOTE-091): an interim analysis of a randomised, triple-blind, phase 3 trial [sponsor supplied reference] Lancet Oncol. 2022;23(10):1274-1286. doi:10.1016/s1470-2045(22)00518-6 PubMed
42.CADTH. Provisional Funding Algorithm for Non-Small Lung Cancer without Actionable Oncogenic Alterations. June 2024 [sponsor supplied reference]
43.European Medicines Agency. Imfinzi - opinion on variation to marketing authorisation. 2025. Accessed April 11, 2025. https://www.ema.europa.eu/en/medicines/human/variation/imfinzi-2
44.U.S. Food and Drug Administration. Highlights of Prescribing Information: IMFINZI® (durvalumab). 2024. Accessed April 11, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/761069s043lbledt.pdf
45.IMFINZI (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous. Product Monograph. AstraZeneca Canada Inc., Mississauga, Ontario. 2024 (draft) [sponsor supplied reference]
46.Kris MG, Gaspar LE, Chaft JE, et al. Adjuvant Systemic Therapy and Adjuvant Radiation Therapy for Stage I to IIIA Completely Resected Non-Small-Cell Lung Cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update [sponsor supplied reference] J Clin Oncol. 2017;35(25):2960-2974. doi:10.1200/jco.2017.72.4401 PubMed
47.Merck Canada Inc. Product Monograph: Keytruda. 2026. Accessed March 10, 2026. https://pdf.hres.ca/dpd_pm/00083378.PDF
48.Bristol-Myers Squibb Canada. Product Monograph: OPDIVO®. 2024. Accessed September 24, 2024. https://www.bms.com/assets/bms/ca/documents/productmonograph/OPDIVO_EN_PM.pdf
49.Hoffmann-La Roche Limited. Produce Monograph: TECENTRIQ® 2024. Accessed September 23, 2024. https://assets.roche.com/f/173850/x/54a5473032/tecentriq_pm_e.pdf
50.Accord Healthcare Inc. Produce Monograph: Paclitaxel Injection USP. 2020. Accessed September 23, 2024. https://pdf.hres.ca/dpd_pm/00057982.PDF
51.APOTEX INC. Produce Monograph: Pemetrexed. 2016. Accessed September 23, 2024. https://pdf.hres.ca/dpd_pm/00035165.PDF
52.Pfizer Canada Inc. Product Monograph: Vinorelbine Tartrate for Injection. 2017. Accessed September 23, 2024. https://pdf.hres.ca/dpd_pm/00040265.PDF
53.Accord Healthcare Inc. Product Monograph: Gemcitabine for Injection. 2020. Accessed September 23, 2024. https://pdf.hres.ca/dpd_pm/00055544.PDF
54.Pfizer Canada ULC. Product Monograph: Cisplatin Injection BP. 2023. Accessed September 23, 2024. https://webfiles.pfizer.com/file/e5683fa1-443c-4e3e-8b56-a0b6926b8329?referrer=ccb731e5-4f2d-4f4a-b2dc-e5e912145fc6
55.Pfizer Canada ULC. Product Monograph: Carboplatin Injection BP. 2023. Accessed September 23, 2024. https://webfiles.pfizer.com/file/f75e1537-0508-4f9f-8dc7-cd9c1b40006c?referrer=ccb731e5-4f2d-4f4a-b2dc-e5e912145fc6
56.Heymach JV, Harpole D, Mitsudomi T, et al. Perioperative Durvalumab for Resectable Non-Small-Cell Lung Cancer [sponsor supplied reference] N Engl J Med. 2023;389(18):1672-1684. doi:10.1056/NEJMoa2304875 PubMed
57.Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer [sponsor supplied reference] J Thorac Oncol. 2016;11(1):39-51. doi:10.1016/j.jtho.2015.09.009 PubMed
58.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The eighth edition lung cancer stage classification [sponsor supplied reference] Chest. 2017;151(1):193-203.
59.AstraZeneca. Statistical Analysis Plan: A Phase III, Double-blind, Placebo-controlled, Multi-center International Study of Neoadjuvant/Adjuvant Durvalumab for the Treatment of Subjects with Resectable Stages II and III Non-small Cell Lung Cancer (AEGEAN) [internal sponsor's report]. 2022. Accessed 19 September, 2024.
60.AstraZeneca. Clinical Study Report Appendix 16.1.1: Protocol and Protocol Amendments [internal sponsor's report]. 2021. Accessed 19 September, 2024.
61.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology [sponsor supplied reference] J Natl Cancer Inst. 1993;85(5):365-76. doi:10.1093/jnci/85.5.365 PubMed
62.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life [sponsor supplied reference] Eur J Cancer. 1994;30a(5):635-42. doi:10.1016/0959-8049(94)90535-5 PubMed
63.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types [sponsor supplied reference] Eur J Cancer. 2023;188:171-182. doi:10.1016/j.ejca.2023.04.027 PubMed
64.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi:10.1007/s11136-007-9210-8 PubMed
65.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. doi:10.1186/1477-7525-9-106 PubMed
66.Ozturk A, Sarihan S, Ercan I, Karadag M. Evaluating quality of life and pulmonary function of long-term survivors of non-small cell lung cancer treated with radical or postoperative radiotherapy. Am J Clin Oncol. 2009;32(1):65-72. doi:10.1097/COC.0b013e31817e6ec2 PubMed
67.Lan K, DeMets D. Discrete sequential boundaries for clinical trials. Biometrika. 1983;7(3):659-63.
68.Mauguen A, Pignon JP, Burdett S, et al. Surrogate endpoints for overall survival in chemotherapy and radiotherapy trials in operable and locally advanced lung cancer: a re-analysis of meta-analyses of individual patients' data [sponsor supplied reference] Lancet Oncol. 2013;14(7):619-26. doi:10.1016/s1470-2045(13)70158-x PubMed
69.Ostoros G, Hettle R, Georgoulia N, et al. Association between event-free survival and overall survival after neoadjuvant treatment for non-small cell lung cancer: a systematic review and meta-analysis [sponsor supplied reference] Expert Rev Anticancer Ther. 2023;23(12):1305-1313. doi:10.1080/14737140.2023.2272645 PubMed
70.Forde PM, Spicer J, Lu S, et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer [sponsor supplied reference] N Engl J Med. 2022;386(21):1973-1985. doi:10.1056/NEJMoa2202170 PubMed
71.Deutsch JS, Cimino-Mathews A, Thompson ED, et al. LBA50 Analysis of pathological features and efficacy outcomes with neoadjuvant nivolumab (N) plus platinum-doublet chemotherapy (C) for resectable non-small cell lung cancer (NSCLC) in CheckMate 816 [sponsor supplied reference] Ann Oncol. 2022;33:S1415-S1416. doi:10.1016/j.annonc.2022.08.050
72.Felip E, Wang C, Ciuleanu TE, et al. 932MO Nivolumab (NIVO) plus platinum-doublet chemotherapy (chemo) versus chemo as neoadjuvant treatment for resectable non-small cell lung cancer (NSCLC): Health-related quality of life (HRQoL) outcomes from CheckMate 816 [sponsor supplied reference] Ann Oncol. 2022;33:S973-S974. doi:10.1016/j.annonc.2022.07.1058
73.Girard N, Spicer J, Provencio M, et al. Abstract CT012: Nivolumab (NIVO) + platinum-doublet chemotherapy (chemo) vs chemo as neoadjuvant treatment for resectable (IB-IIIA) non-small cell lung cancer (NSCLC): Event-free survival (EFS) results from the phase 3 CheckMate 816 trial [sponsor supplied reference] Cancer Res. 2022;82(12_Supplement):CT012-CT012. doi:10.1158/1538-7445.Am2022-ct012
74.Spicer J, Wang C, Tanaka F, et al. Surgical outcomes from the phase 3 CheckMate 816 trial: Nivolumab (NIVO) + platinum-doublet chemotherapy (chemo) vs chemo alone as neoadjuvant treatment for patients with resectable non-small cell lung cancer (NSCLC) [sponsor supplied reference] J Clin Oncol. 2021;39(15_suppl):8503-8503. doi:10.1200/JCO.2021.39.15_suppl.8503
75.Heymach JV, Harpole D, Mitsudomi T, et al. Abstract CT005: AEGEAN: A phase 3 trial of neoadjuvant durvalumab + chemotherapy followed by adjuvant durvalumab in patients with resectable NSCLC [sponsor supplied reference] Cancer Res. 2023;83(8_Supplement):CT005-CT005. doi:10.1158/1538-7445.Am2023-ct005
76.Wakelee HA, Liberman M, Kato T, et al. KEYNOTE-671: Randomized, double-blind, phase 3 study of pembrolizumab or placebo plus platinum-based chemotherapy followed by resection and pembrolizumab or placebo for early stage NSCLC [sponsor supplied reference] J Clin Oncol. 2023;41(17_suppl):LBA100-LBA100. doi:10.1200/JCO.2023.41.17_suppl.LBA100
77.Wakelee H, Liberman M, Kato T, et al. Perioperative Pembrolizumab for Early-Stage Non-Small-Cell Lung Cancer [sponsor supplied reference] N Engl J Med. 2023;389(6):491-503. doi:10.1056/NEJMoa2302983 PubMed
78.Cytel. Technical Report for the Indirect Treatment Comparison of Perioperative Durvalumab (AEGEAN) as Treatment for Resectable Stages II and III Non-small Cell Lung Cancer. Unpublished [sponsor supplied reference]
79.Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions [sponsor supplied reference] BMJ. 2016;355:i4919. doi:10.1136/bmj.i4919 PubMed
80.Centre for Reviews and Dissemination. Systematic Reviews: CRD's guidance for undertaking reviews in health care. York: Centre for Reviews and Dissemination, University of York, 2008.
81.Costello Medical. Clinical Systematic and Targeted Literature Reviews in Support of Durvalumab for the Treatment of Resectable NSCLC. Technical Report. [Internal sponsor's report]. 2024. Accessed September 23, 2024.
82.Cascone T, Awad MM, Spicer JD, et al. Perioperative Nivolumab in Resectable Lung Cancer [sponsor supplied reference] N Engl J Med. 2024;390(19):1756-1769. doi:10.1056/NEJMoa2311926 PubMed
83.Phillippo D, Ades T, Dias S, Palmer S, Abrams K, Welton N. Unit NDS, ed. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. 2016. Accessed September 23, 2024. https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf
84.Cope S, Zhang J, Saletan S, Smiechowski B, Jansen JP, Schmid P. A process for assessing the feasibility of a network meta-analysis: a case study of everolimus in combination with hormonal therapy versus chemotherapy for advanced breast cancer [sponsor supplied reference] BMC Med. 2014;12:93. doi:10.1186/1741-7015-12-93 PubMed
85.Heymach JV, Harpole D, Mitsudomi T, Taube JM, Galffy G, Hochmair M, et al. AEGEAN: A phase 3 trial of neoadjuvant durvalumab + chemotherapy followed by adjuvant durvalumab in patients with resectable NSCLC [abstract]. 2023:
86.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept [sponsor supplied reference] Pharmacoeconomics. 2010;28(10):935-45. doi:10.2165/11538370-000000000-00000 PubMed
87.White H. A Heteroskedasticity-Consistent Covariance Matrix Estimator and a Direct Test for Heteroskedasticity [sponsor supplied reference] Econometrica. 1980;48(4):817-838. doi:10.2307/1912934
88.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials [sponsor supplied reference] J Clin Epidemiol. 1997;50(6):683-691. PubMed
89.The Comprehensive R Archive Network. The R Project for Statistical Computing: language and environment for statistical computing. R Foundation for Statistical Computing. Available from: https://www.r-project.org/.
90.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE Decision Support Unit Technical Support Documents. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. National Institute for Health and Care Excellence (NICE) Copyright © 2014 National Institute for Health and Clinical Excellence, unless otherwise stated. All rights reserved.; 2014.
91.Turner RM, Domínguez-Islas CP, Jackson D, Rhodes KM, White IR. Incorporating external evidence on between-trial heterogeneity in network meta-analysis [sponsor supplied reference] Stat Med. 2019;38(8):1321-1335. doi:10.1002/sim.8044 PubMed
92.Spiegelhalter DJ, Best NG, Carlin BP, Van Der Linde A. Bayesian measures of model complexity and fit [sponsor supplied reference] Journal of the royal statistical society: Series b (statistical methodology). 2002;64(4):583-639.
93.Gelman A, Rubin D. Inference from Iterative Simulation Using Multiple Sequences. Statistical Science. 1992;7(4):457-472. doi:10.1214/ss/1177011136
94.ClinicalTrials.gov. Preoperative Twice Daily Chemoradiation in Addition to Chemotherapy Prior to Surgery in Stage III Non-Small Cell Lung Cancer (NSCLC). Available from: https://clinicaltrials.gov/ct2/show/NCT00176137 [sponsor supplied reference]
95.Thomas M, Rübe C, Hoffknecht P, et al. Effect of preoperative chemoradiation in addition to preoperative chemotherapy: a randomised trial in stage III non-small-cell lung cancer [sponsor supplied reference] Lancet Oncol. 2008;9(7):636-48. doi:10.1016/s1470-2045(08)70156-6 PubMed
96.Chen Z, Luo Q, Jian H, et al. Long-term results of a randomized controlled trial evaluating preoperative chemotherapy in resectable non-small cell lung cancer [sponsor supplied reference] Onco Targets Ther. 2013;6:645-50. doi:10.2147/ott.S44503 PubMed
97.Roth JA, Fossella F, Komaki R, et al. A randomized trial comparing perioperative chemotherapy and surgery with surgery alone in resectable stage IIIA non-small-cell lung cancer [sponsor supplied reference] J Natl Cancer Inst. 1994;86(9):673-80. doi:10.1093/jnci/86.9.673 PubMed
98.Nagai K, Tsuchiya R, Mori T, et al. A randomized trial comparing induction chemotherapy followed by surgery with surgery alone for patients with stage IIIA N2 non-small cell lung cancer (JCOG 9209) [sponsor supplied reference] J Thorac Cardiovasc Surg. 2003;125(2):254-60. doi:10.1067/mtc.2003.15 PubMed
99.Depierre A, Milleron B, Moro-Sibilot D, et al. Preoperative chemotherapy followed by surgery compared with primary surgery in resectable stage I (except T1N0), II, and IIIa non-small-cell lung cancer [sponsor supplied reference] J Clin Oncol. 2002;20(1):247-53. doi:10.1200/jco.2002.20.1.247 PubMed
100.Spicer J, Girard N, Provencio M, et al. Neoadjuvant nivolumab (NIVO) + chemotherapy (chemo) vs chemo in patients (pts) with resectable NSCLC: 4-year update from CheckMate 816. Journal of Clinical Oncology. 2024;42(17_suppl):LBA8010-LBA8010. doi:10.1200/JCO.2024.42.17_suppl.LBA8010
101.ClinicalTrials.gov. Neoadjuvant or Adjuvant Chemotherapy in Patients With Operable Non-small-cell Lung Cancer (NATCH). Available from: https://clinicaltrials.gov/ct2/show/NCT00913705 [sponsor supplied reference]
102.Felip E, Rosell R, Maestre JA, et al. Preoperative chemotherapy plus surgery versus surgery plus adjuvant chemotherapy versus surgery alone in early-stage non-small-cell lung cancer [sponsor supplied reference] J Clin Oncol. 2010;28(19):3138-45. doi:10.1200/jco.2009.27.6204 PubMed
103.Rosell R, Gómez-Codina J, Camps C, et al. A randomized trial comparing preoperative chemotherapy plus surgery with surgery alone in patients with non-small-cell lung cancer [sponsor supplied reference] N Engl J Med. 1994;330(3):153-8. doi:10.1056/nejm199401203300301 PubMed
104.Rosell R, Maestre J, Font A, et al. A randomized trial of mitomycin/ifosfamide/cisplatin preoperative chemotherapy plus surgery versus surgery alone in stage IIIA non-small cell lung cancer [sponsor supplied reference] Semin Oncol. 1994;21(3 Suppl 4):28-33. PubMed
105.Scagliotti GV, Pastorino U, Vansteenkiste JF, et al. Randomized phase III study of surgery alone or surgery plus preoperative cisplatin and gemcitabine in stages IB to IIIA non-small-cell lung cancer [sponsor supplied reference] J Clin Oncol. 2012;30(2):172-8. doi:10.1200/jco.2010.33.7089 PubMed
106.Gilligan D, Nicolson M, Smith I, et al. Preoperative chemotherapy in patients with resectable non-small cell lung cancer: results of the MRC LU22/NVALT 2/EORTC 08012 multicentre randomised trial and update of systematic review [sponsor supplied reference] Lancet. 2007;369(9577):1929-37. doi:10.1016/s0140-6736(07)60714-4 PubMed
107.Li J, Yu L-C, Chen P, Shi S-B, Dai C-H, Wu J-R. Randomized controlled trial of neoadjuvant chemotherapy with cisplatin and vinorelbine in patients with stage IIIA non-small cell lung cancer in China [sponsor supplied reference] Asia Pac J Clin Oncol. 2009;5(2):87-94. doi:https://doi.org/10.1111/j.1743-7563.2009.01196.x
108.Pless M, Stupp R, Ris HB, et al. Induction chemoradiation in stage IIIA/N2 non-small-cell lung cancer: a phase 3 randomised trial [sponsor supplied reference] Lancet. 2015;386(9998):1049-56. doi:10.1016/s0140-6736(15)60294-x PubMed
109.ClinicalTrials.gov. Preoperative Chemoradiotherapy vs. Chemotherapy Alone in NSCLC Patients. Accessed July 2023, https://clinicaltrials.gov/study/NCT00030771 [sponsor supplied reference]
110.Katakami N, Tada H, Mitsudomi T, et al. A phase 3 study of induction treatment with concurrent chemoradiotherapy versus chemotherapy before surgery in patients with pathologically confirmed N2 stage IIIA nonsmall cell lung cancer (WJTOG9903) [sponsor supplied reference] Cancer. 2012;118(24):6126-35. doi:10.1002/cncr.26689 PubMed
111.ClinicalTrials.gov. Three Modalities of Treatment in Operable and Resectable Stage IIIA (T1-3, N2) Non-Small Cell Lung Cancer (NSCLC). Accessed July 2023, https://ClinicalTrials.gov/show/NCT00198367 [sponsor supplied reference]
112.Girard N, Mornex F, Douillard JY, et al. Is neoadjuvant chemoradiotherapy a feasible strategy for stage IIIA-N2 non-small cell lung cancer? Mature results of the randomized IFCT-0101 phase II trial [sponsor supplied reference] Lung Cancer. 2010;69(1):86-93. doi:10.1016/j.lungcan.2009.10.003 PubMed
113.Roth JA, Atkinson EN, Fossella F, et al. Long-term follow-up of patients enrolled in a randomized trial comparing perioperative chemotherapy and surgery with surgery alone in resectable stage IIIA non-small-cell lung cancer [sponsor supplied reference] Lung Cancer. 1998;21(1):1-6. doi:10.1016/s0169-5002(98)00046-4 PubMed
114.National Institute for Health and Care Excellence. Nivolumab with chemotherapy for neoadjuvant treatment of resectable non-small-cell lung cancer (TA876). 2023. Accessed September 23, 2024. https://www.nice.org.uk/guidance/ta876
115.ClinicalTrials.gov. S9900: Surgery With or Without Combination Chemotherapy in Treating Patients With Non-small Cell Lung Cancer. Accessed July 2023, https://classic.clinicaltrials.gov/ct2/show/NCT00004011 [sponsor supplied reference]
116.Higgins J, Page M, Elbers R, Sterne J. Chapter 8: Assessing risk of bias in a randomized trial. . Cochrane Handbook for Systematic Reviews of Interventions. 2023. https://training.cochrane.org/handbook
117.Claassens L, van Meerbeeck J, Coens C, et al. Health-related quality of life in non-small-cell lung cancer: an update of a systematic review on methodologic issues in randomized controlled trials. J Clin Oncol. 2011;29(15):2104-20. doi:10.1200/jco.2010.32.3683 PubMed
118.Fayers P, Aaronson N, Bjordal K, Groenvold M, Curran D, Bottomley A. European Organisation for Research and Treatment of Cancer, ed. The EORTC QLQ-C30 Scoring Manual (3rd ed.). 2021. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf
Appendix 1: Patient Disposition and Demographics From AEGEAN Study
Please note that this appendix has not been copy-edited.
Table 48: AEGEAN Study Summary of Baseline Characteristics in the ITT Population
Characteristic | Durvalumab + neoadjuvant chemotherapy N = 400 | Placebo + neoadjuvant chemotherapy N = 402 |
|---|---|---|
Age (years)a | ||
Mean (SD) | 63.9 (9.20) | 63.8 (8.70) |
Median (min, max) | 65.0 (30, 88) | 65.0 (39, 85) |
Age group (years), n (%) | ||
< 50 | 22 (5.5) | 22 (5.5) |
≥ 50 to < 65 | 170 (42.5) | 177 (44.0) |
≥ 65 to < 75 | 160 (40.0) | 165 (41.0) |
≥ 75 | 48 (12.0) | 38 (9.5) |
Sex, n (%) | ||
Female | 138 (34.5) | 111 (27.6) |
Male | 262 (65.5) | 291 (72.4) |
ECOG PS, n (%) | ||
(0) Normal activity | 278 (69.5) | 277 (68.9) |
(1) Restricted in physically strenuous activity | 122 (30.5) | 125 (31.1) |
Race,b n (%) | ||
American Indian or Alaska Native | 7 (1.8) | 4 (1.0) |
Asian | 165 (41.3) | 187 (46.5) |
Black or African American | 5 (1.3) | 3 (0.7) |
White | 216 (54.0) | 196 (48.8) |
Other | 7 (1.8) | 12 (3.0) |
Ethnic group, n (%) | ||
Hispanic or Latino | 68 (17.0) | 58 (14.4) |
Not Hispanic or Latino | 332 (83.0) | 344 (85.6) |
Smoking history, n (%) | ||
Never smoked | 72 (18.0) | 74 (18.4) |
Smoked | 328 (82.0) | 328 (81.6) |
Disease stage at baseline,c n (%) | ||
IIA | 19 (4.8) | 28 (7.0) |
IIB | 100 (25.0) | 92 (22.9) |
III (NOS) | 0 | 1 (0.2) |
IIIA | 186 (46.5) | 178 (44.3) |
IIIB | 94 (23.5) | 103 (25.6) |
IV (NOS) | 1 (0.3) | 0 |
Histology type, n (%) | ||
Squamous | 173 (43.3) | 192 (47.8) |
Nonsquamous | 226 (56.5) | 206 (51.2) |
TNM stage at baseline, regional lymph nodes, n (%)d | ||
N0 | 118 (29.5) | 110 (27.4) |
N1 | 83 (20.8) | 94 (23.4) |
N2 | 199 (49.8) | 198 (49.3) |
Single station | 151 (37.8) | 140 (34.8) |
Multistation | 38 (9.5) | 45 (11.2) |
Missing | 10 (2.5) | 13 (3.2) |
PD-L1 expression status at baseline, n (%) | ||
TC < 1% (per IXRS and source data) | 133 (33.3) | 134 (33.3) |
TC ≥ 1% (per IXRS and source data) | 267 (66.8) | 268 (66.7) |
TC 1 to 49% (per source data) | 151 (37.8) | 158 (39.3) |
TC ≥ 50% (per source data) | 116 (29.0) | 110 (27.4) |
AJCC = American Joint Committee on Cancer; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention to treat; IXRS = interactive web response system; M = metastases; mITT = modified intention to treat; N = node; NOS = not otherwise specified; SD = standard deviation; t = tumour; TC = tumour cell.
Notes: Baseline is defined as the last observation before randomization if available, or otherwise, an observation after randomization but before the first dose of randomized treatment.
Percentages are calculated based on the number of patients in the analysis set.
The DCO date was November 10, 2022.
aAge was calculated using date of randomization.
bRacial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
cStages according to AJCC Cancer Staging Manual, eighth edition.
dTNM classification is based on the AJCC Cancer Staging Manual, eighth edition.
Sources: AEGEAN study event-free survival interim analysis 1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 49: AEGEAN Study Summary of Exposure to Individual Neoadjuvant Chemotherapy in the Safety Analysis Set Population
Exposure | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 | Durvalumab + neoadjuvant chemotherapy N = 401 | Placebo + neoadjuvant chemotherapy N = 398 |
|---|---|---|---|---|
EFS IA1 | EFS IA2 | |||
Carboplatin | n = 309 | n = 301 | n = 309 | n = 301 |
Total treatment duration (weeks)a | ||||
Duration, mean (SD) | 11.68 (2.604) | 11.93 (2.257) | 11.68 (2.604) | 11.93 (2.257) |
Duration, median (min, max) | 12.14 (2.0, 19.0) | 12.00 (3.0, 22.7) | 12.14 (2.0, 19.0) | 12.00 (3.0, 22.7) |
Total treatment years | — | — | ████ | ████ |
Actual treatment duration (weeks)b | ||||
Duration, mean (SD) | 11.17 (2.283) | 11.47 (1.982) | 11.17 (2.283) | 11.47 (1.982) |
Duration, median (min, max) | 12.00 (2.0, 13.3) | 12.00 (3.0, 13.4) | 12.00 (2.0, 13.3) | 12.00 (3.0, 13.4) |
Total treatment years | — | — | ████ | ████ |
Cisplatin | n = 111 | n = 104 | n = 111 | n = 104 |
Total treatment duration (weeks)a | ||||
Duration, mean (SD) | 11.09 (3.131) | 11.42 (2.830) | 11.09 (3.131) | 11.42 (2.830) |
Duration, median (min, max) | 12.00 (3.0, 15.3) | 12.07 (3.0, 17.0) | 12.00 (3.0, 15.3) | 12.07 (3.0, 17.0) |
Total treatment years | — | — | ████ | ████ |
Actual treatment duration (weeks)b | ||||
Duration, mean (SD) | 10.65 (2.869) | 11.00 (2.672) | 10.65 (2.869) | 11.00 (2.672) |
Duration, median (min, max) | 12.00 (3.0, 13.0) | 12.00 (3.0, 13.0) | 12.00 (3.0, 13.0) | 12.00 (3.0, 13.0) |
Total treatment years | — | — | ████ | ████ |
Gemcitabine | n = 60 | n = 54 | n = 60 | n = 54 |
Total treatment duration (weeks)c | ||||
Duration, mean (SD) | 12.05 (2.361) | 11.33 (2.833) | 12.05 (2.361) | 11.33 (2.833) |
Duration, median (min, max) | 12.14 (3.0, 19.0) | 12.14 (1.0, 15.0) | 12.14 (3.0, 19.0) | 12.14 (1.0, 15.0) |
Total treatment years | — | — | ████ | ████ |
Actual treatment duration (weeks)d | ||||
Duration, mean (SD) | 11.01 (1.987) | 10.64 (2.750) | 11.01 (1.987) | 10.64 (2.750) |
Duration, median (min, max) | 12.00 (3.0, 13.0) | 12.00 (1.0, 12.7) | 12.00 (3.0, 13.0) | 12.00 (1.0, 12.7) |
Total treatment years | — | — | ████ | ████ |
Paclitaxel | n = 124 | n = 140 | n = 124 | n = 140 |
Total treatment duration (weeks)d | ||||
Duration, mean (SD) | 11.52 (2.790) | 11.92 (2.510) | 11.52 (2.790) | 11.92 (2.510) |
Duration, median (min, max) | 12.14 (2.0, 17.4) | 12.14 (3.0, 22.7) | 12.14 (2.0, 17.4) | 12.14 (3.0, 22.7) |
Total treatment years | — | — | ████ | ████ |
Actual treatment duration (weeks)e | ||||
Duration, mean (SD) | 11.11 (2.489) | 11.44 (2.154) | 11.11 (2.489) | 11.44 (2.154) |
Duration, median (min, max) | 12.00 (2.0, 13.3) | 12.00 (3.0, 13.0) | 12.00 (2.0, 13.3) | 12.00 (3.0, 13.0) |
Total treatment years | — | — | ████ | ████ |
Pemetrexed | n = 221 | n = 206 | n = 221 | n = 206 |
Total treatment duration (weeks)a | ||||
Duration, mean (SD) | 12.16 (1.921) | 12.07 (1.840) | 12.16 (1.921) | 12.07 (1.840) |
Duration, median (min, max) | 12.14 (2.3, 20.7) | 12.00 (3.0, 17.0) | 12.14 (2.3, 20.7) | 12.00 (3.0, 17.0) |
Total treatment years | — | — | ████ | ████ |
Actual treatment duration (weeks)b | ||||
Duration, mean (SD) | 11.61 (1.554) | 11.66 (1.643) | 11.61 (1.554) | 11.66 (1.643) |
Duration, median (min, max) | 12.00 (2.3, 12.9) | 12.00 (3.0, 13.0) | 12.00 (2.3, 12.9) | 12.00 (3.0, 13.0) |
Total treatment years | — | — | ████ | ████ |
DCO = data cut-off; EFS = event-free survival; IA = interim analysis; SD = standard deviation.
Note: The EFS IA1 DCO date was November 10, 2022; the EFS IA2 DCO date was May 10, 2024.
aTotal treatment duration = (min (death, DCO, last non-0 dose of chemotherapy (cycle x, day 1) + 20) - first dose date of chemotherapy + 1)/7, where minimum is whichever occurred first.
bActual treatment duration = total treatment duration - total duration of dose delays, where delays = sum of (date of dose - date of previous dose - 21 days).
cTotal treatment duration = (min (death, DCO, last non-0 dose of gemcitabine + W) - first dose date of gemcitabine + 1)/7, where minimum is whichever occurred first and W = 6 if the last dose was scheduled on day 1 and W = 13 if the last dose was scheduled on day 8.
dActual treatment duration = total treatment duration - total duration of dose delays, where delays = sum of (date of gemcitabine dose - date of previous gemcitabine dose - X days), where X = 7 if previous dose was day 1 of a cycle and X = 14 if previous dose was day 8 of a cycle.
Sources: AEGEAN study EFS IA1 Clinical Study Report24 and AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 2: Efficacy Results From the AEGEAN Study Subgroup Analyses
Please note that this appendix has not been copy-edited.
Figure 9: pCR in Subgroups (mITT Population, pCR FA)
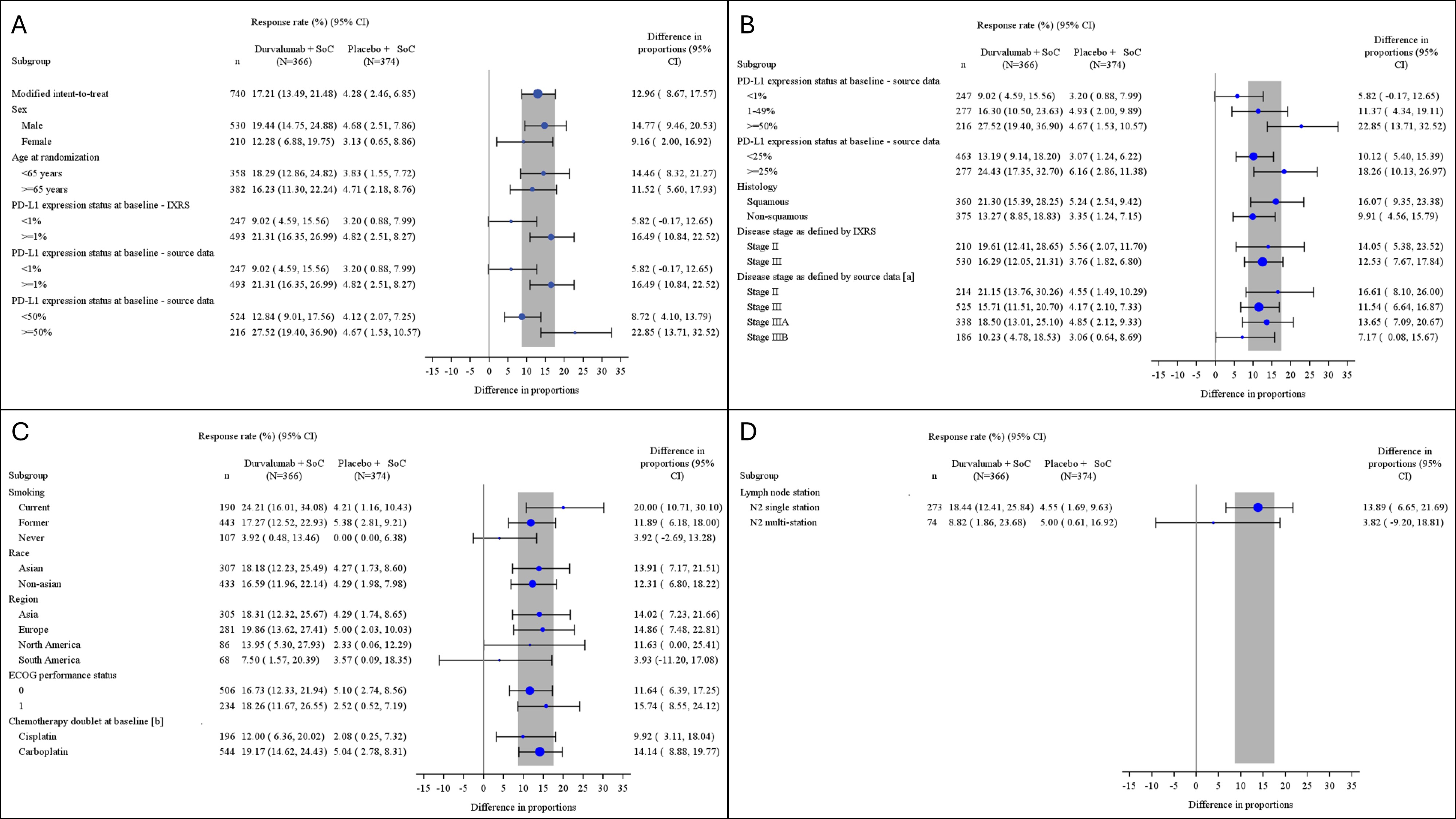
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; FA = final analysis; IXRS = integrated web response system; mITT = modified intention to treat; pCR = pathological complete response; SoC = standard of care.
Notes: The difference in proportions (%) and 95% CI. A difference > 0 favours perioperative durvalumab with neoadjuvant chemotherapy.
The data cut-off date was November 10, 2022.
aOne participant has disease stage IV at ; they are being excluded from analysis.
bRefers to the platinum base of the doublet (planned treatment).
Source: AEGEAN study event-free survival interim analysis 1 Clinical Study Report.24
Figure 10: EFS in Subgroups (mITT Population, EFS IA2)
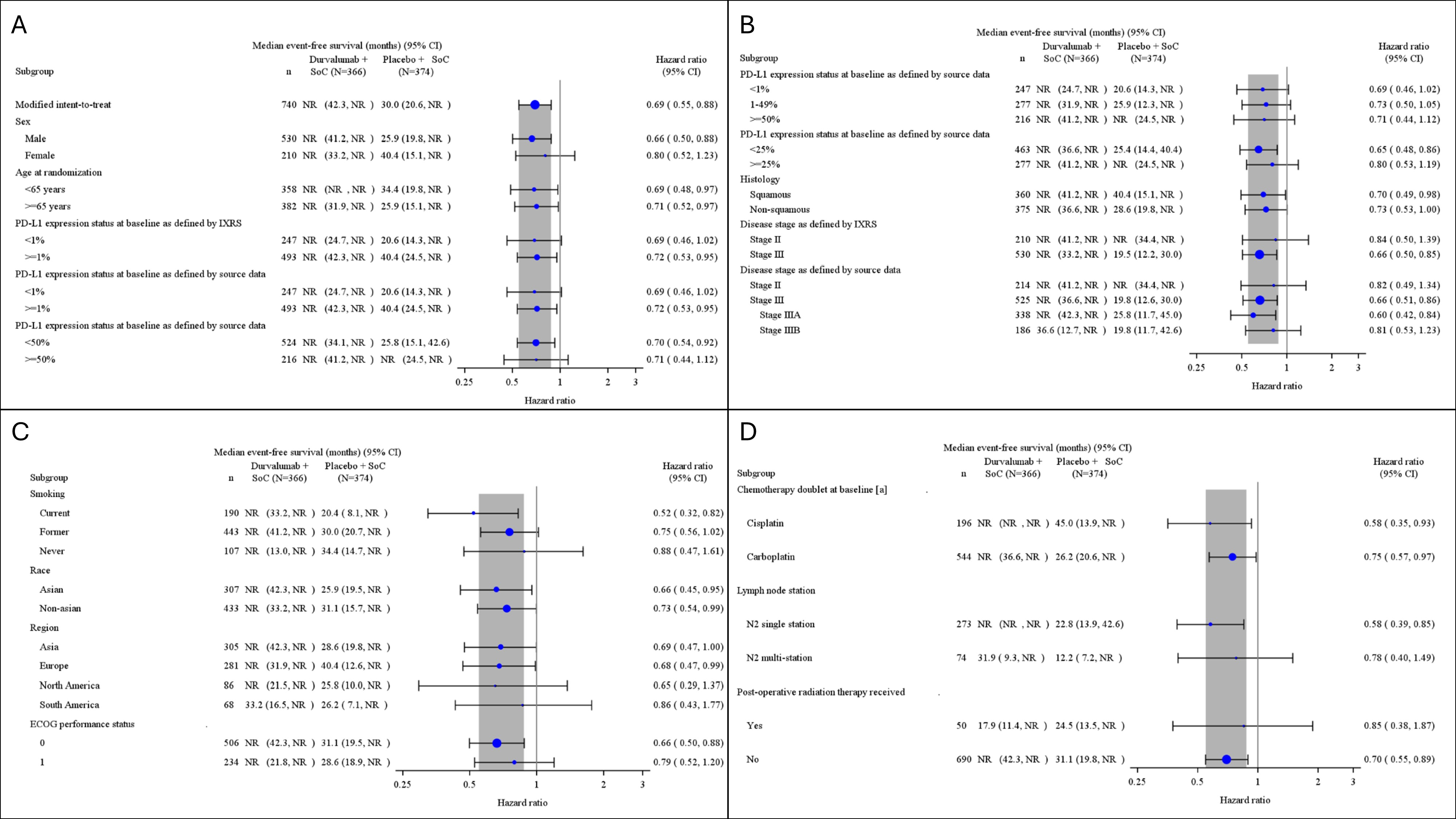
BICR = blinded independent central review; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; IXRS = integrated web response system; NR = not reached; mITT = modified intention to treat; PORT = postoperative radiotherapy; SoC = standard of care.
Notes: The difference in proportions (%) and 95% CI. A difference > 0 favours perioperative durvalumab with neoadjuvant chemotherapy.
The data cut-off date was May 10, 2024.
aRefers to the platinum base of the doublet (planned treatment).
Source: AEGEAN study EFS interim analysis 2 Clinical Study Report Appendix 14 data tables and figures.23
Appendix 3: HRQoL Measures
Table 50: Summary of Baseline and Week 12 EORTC QLQ-C30 Version 3 Scores — Neoadjuvant Period (mITT Population)
Outcome | Absolute score | Change from baseline | ||||||
|---|---|---|---|---|---|---|---|---|
Durvalumab + chemotherapy (N = 366) | Placebo + chemotherapy (N = 374) | Durvalumab + chemotherapy (N = 366) | Placebo + chemotherapy (N = 374) | |||||
N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | |
Global measure of health status and quality of life | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Function: Physical | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Function: Role | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | █████ ███████ | ███ | ████ ███████ |
Function: Emotional | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Function: Cognitive | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Function: Social | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Fatigue | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ |
Symptom: Nausea and vomiting | ||||||||
Baseline | ███ | ███ ██████ | ███ | ███ ██████ | — | — | — | — |
Week 12 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Symptom: Pain | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Symptom: Dyspnea | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Symptom: Insomnia | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ████ ███████ |
Symptom: Appetite loss | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Symptom: Constipation | ||||||||
Baseline | ███ | ███ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Symptom: Diarrhea | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Financial difficulties | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; SD = standard deviation.
Note: This table has not been copy-edited.
Notes: Neoadjuvant baseline is last predose assessment.
Higher score on global measure of health status/quality of life and functioning scales indicates better health status/function (i.e., positive change score reflects improvement). Higher scores on symptom scales/items represent greater symptom severity thus a negative change score reflects improvement.
The data cut-off date was November 10, 2022.
Sources: AEGEAN study event-free survival interim analysis 1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 51: Summary of Adjuvant Baseline and Week 44 EORTC QLQ-C30 Version 3 Scores – Adjuvant Period (Modified Resected Set)
Outcome | Absolute score | Change from baseline | ||||||
|---|---|---|---|---|---|---|---|---|
Durvalumab + chemotherapy (N = 242) | Placebo + chemotherapy (N = 231) | Durvalumab + chemotherapy (N = 242) | Placebo + chemotherapy (N = 231) | |||||
N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | |
Global measure of health status and quality of life | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Function: Physical | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Function: Role | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Function: Emotional | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ |
Function: Cognitive | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ |
Function: Social | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ |
Symptom: Fatigue | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Nausea and vomiting | ||||||||
Adjuvant baselinea | ███ | ███ ██████ | ███ | ███ ███████ | — | — | — | — |
Week 44 | ███ | ███ ███████ | ███ | ███ ██████ | ███ | ███ ███████ | ███ | ████ ███████ |
Symptom: Pain | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Dyspnea | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ████ ███████ |
Symptom: Insomnia | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Appetite loss | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ████ ███████ | ███ | █████ ███████ |
Symptom: Constipation | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Diarrhea | ||||||||
Adjuvant baselinea | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 44 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Financial difficulties | ||||||||
Adjuvant baselinea | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 44 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ████ ███████ |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; SD = standard deviation.
Note: This table has not been copy-edited.
Note: The data cut-off date was May 10, 2024.
aBaseline measurement is the last measurement after surgery but before first dose of adjuvant treatment. A higher score on the global measure of health status/QoL and functioning scales indicates better health status/function thus a positive change score reflects improvement. Higher scores on symptom scales/items represent greater symptom severity thus a negative change score reflects improvement. Only time points with at least 20 patients in either arm are presented.
Sources: AEGEAN study EFS IA2 Clinical Study Report Appendix 14 data tables and figures.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 52: Summary of EORTC QLQ-LC13 Scores — Neoadjuvant Period (mITT Population)
Outcome | Absolute score | Change from baseline | ||||||
|---|---|---|---|---|---|---|---|---|
Durvalumab + chemotherapy (N = 366) | Placebo + chemotherapy (N = 374) | Durvalumab + chemotherapy (N = 366) | Placebo + chemotherapy (N = 374) | |||||
N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | N | Mean (SD) | |
Single item: Coughing | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | █████ ███████ | ███ | █████ ███████ |
Single item: Hemoptysis | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ███ ██████ | ███ | ███ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Symptom: Dyspnea | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Single item: Pain in chest | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Single item: Pain in arm or shoulder | ||||||||
Baseline | ███ | ████ ███████ | ███ | ████ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ████ ███████ |
Single item: Pain in other parts | ||||||||
Baseline | ███ | ████ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Single item: Sore mouth | ||||||||
Baseline | ███ | ███ ██████ | ███ | ███ ██████ | — | — | — | — |
Week 12 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Single item: Dysphagia | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ██████ | — | — | — | — |
Week 12 | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ | ███ | ███ ███████ |
Single item: Peripheral neuropathy | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
Single item: Alopecia | ||||||||
Baseline | ███ | ███ ███████ | ███ | ███ ███████ | — | — | — | — |
Week 12 | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ | ███ | ████ ███████ |
EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; mITT = modified intention to treat; SD = standard deviation.
Note: This table has not been copy-edited.
Notes: Neoadjuvant baseline is last predose assessment.
Higher scores on symptom scales/items represent greater symptom severity thus a negative change score reflects improvement.
The data cut-off date was November 10, 2022.
Sources: AEGEAN study event-free survival interim analysis 1 Clinical Study Report.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AJCC
American Joint Committee on Cancer
BIA
budget impact analysis
DM
distant metastasis
DM1
distant metastasis first line
DM2
distant metastasis second line
EF
event-free
EFS
event-free survival
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
KM
Kaplan-Meier
LRR
locoregional recurrence
MAIC
matching-adjusted indirect comparison
mITT
modified intention to treat
NMA
network meta-analysis
NSCLC
non–small cell lung cancer
OS
overall survival
QALY
quality-adjusted life-year
TDT
time to discontinuation of treatment
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), 120 mg/2.4 mL and 500 mg/10 mL single-use vials for IV infusion |
Indication | In combination with platinum-containing chemotherapy as neoadjuvant treatment, followed by durvalumab as monotherapy after surgery, for the treatment of patients with resectable Stage II, IIIA, or IIIB (T3-4 N2) non–small cell lung cancer and no known epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) rearrangements |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 20, 2026 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Durvalumab (Imfinzi) has previously been reviewed for the following indications: Durvalumab for the treatment of patients with locally advanced, unresectable NSCLC following curative intent platinum-based chemoradiation therapy, for up to a maximum of 12 months. Recommendation date: May 3, 2019 Recommendation: Reimburse with clinical criteria and/or conditions. Durvalumab in combination with etoposide and either carboplatin or cisplatin is indicated for the first-line treatment of adult patients with extensive-stage small cell lung cancer. Recommendation date: July 27, 2021 Recommendation: Reimburse with clinical criteria and/or conditions. Tremelimumab for injection in combination with durvalumab is indicated for the first-line treatment of adult patients with unresectable hepatocellular carcinoma who require systemic therapy. Recommendation date: November 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions. Durvalumab in combination with gemcitabine-based chemotherapy is indicated for the treatment of patients with locally advanced or metastatic biliary tract cancer. Recommendation date: February 3, 2023 Recommendation: Reimburse with clinical criteria and/or conditions. |
CDA-AMC = Canada’s Drug Agency; NOC = Notice of Compliance; NSCLC = non–small cell lung cancer.
Note: The sponsor’s application was filed on a pre-NOC basis and the pharmacoeconomic submission (economic evaluation and budget impact analysis) is reflective of the time at which the initial submission was received. The review was subsequently suspended and, while this review was suspended, perioperative pembrolizumab became a relevant comparator in this population. The sponsor’s pharmacoeconomic evaluation and budget impact analysis were not updated to include pembrolizumab as a relevant comparator. Therefore, the CDA-AMC appraisal does not address the comparative cost-effectiveness of durvalumab versus pembrolizumab, and the budget impact analysis assumed no usage of pembrolizumab. The wording of the indication was revised to refer to stage of NSCLC as opposed to tumour size and/or node positive.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Target population | Adult patients with rNSCLC (stage IIA to stage IIIB [N2 only], according to AJCC staging, eighth edition) whose tumours have no EGFR mutations or ALK aberrations |
Treatment | Perioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery. |
Dosage regimen |
|
Submitted price | Durvalumab: $938.67 per 120 mg/2.4 mL single-use vial for IV infusion Durvalumab: $3,911.11 per 500 mg/10 mL single-use vial for IV infusion |
Submitted treatment cost | The average 21-day cycle cost of durvalumab is $11,733 before surgery and $8,700 after surgery. The total cost of perioperative durvalumab plus neoadjuvant chemotherapy (including the cost of neoadjuvant chemotherapy) is $198,250 ($207,642 including the cost of surgery).b |
Comparators |
|
Perspective | Publicly funded health care payer in Canada |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (36 years) |
Key data sources | Sponsor-submitted 2 MAICs and an NMA in which efficacy inputs for perioperative durvalumab plus neoadjuvant chemotherapy were informed by the AEGEAN trial (data cut-off date = May 10, 2024) |
Submitted results | Results using MAIC-adjusted HR:
Results using unadjusted HR:
|
Key limitations |
|
CDA-AMC reanalysis results |
|
AJCC = American Joint Committee on Cancer; CDA-AMC = Canada’s Drug Agency; EF = event-free; EFS = event-free survival; GPM = general population mortality; HR = hazard ratio; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LRR = locoregional recurrence; LY = life-year; MAIC = matching-adjusted indirect comparison; NMA = network meta-analysis; QALY = quality-adjusted life-year; rNSCLC = resectable non–small cell lung cancer; SMR = standardized mortality ratio; vs. = versus; WTP = willingness to pay.
Note: Platinum-based chemotherapy in the neoadjuvant setting includes cisplatin plus pemetrexed, cisplatin plus gemcitabine, cisplatin plus paclitaxel, carboplatin plus paclitaxel, carboplatin plus pemetrexed, or carboplatin plus gemcitabine. In the adjuvant setting, cisplatin plus vinorelbine and carboplatin plus vinorelbine are assumed to be used in addition to the aforementioned neoadjuvant chemotherapy regimens.
aPatients with a body weight of 30 kg or less must receive weight-based dosing of durvalumab, in combination with chemotherapy, at 20 mg/kg 21 days before surgery and durvalumab monotherapy at 20 mg/kg every 28 days after surgery until weight increases to greater than 30 kg.
bAssumes durvalumab is administered for the maximum number of cycles (i.e., four 21-day cycles in the neoadjuvant period and twelve 28-day cycles in the adjuvant period).
cNeoadjuvant nivolumab consists of neoadjuvant nivolumab plus chemotherapy.
Conclusions
Evidence from the phase III, randomized, placebo-controlled, AEGEAN trial comparing the efficacy and safety of perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy in adult patients with resectable non–small cell lung cancer (NSCLC) and no known EGFR mutations or ALK rearrangements demonstrated that perioperative durvalumab plus neoadjuvant chemotherapy confers added clinical benefit in event-free survival (EFS). The clinical review assessed that perioperative durvalumab plus neoadjuvant chemotherapy likely results in a clinically important increase in EFS at 36 months compared to neoadjuvant chemotherapy, with moderate certainty of evidence. The clinical review further noted that, compared to neoadjuvant chemotherapy, perioperative durvalumab plus neoadjuvant chemotherapy may result in little to no difference in health-related quality of life (HRQoL) during the neoadjuvant period (with low certainty of evidence). Additionally, the evidence is very uncertain about the effect on HRQoL in the adjuvant period. Notably, as of the second interim analysis (data cut-off date = May 10, 2024; overall survival [OS] median duration of follow-up = ████ months), perioperative durvalumab plus neoadjuvant chemotherapy may result in little to no difference on the probability of being alive at 48 months; however, OS data remained immature (per interim analysis 2 data cut-off ██% of patients had died). Finally, the clinical review concluded that, due to the absence of direct evidence and methodological limitations in the indirect treatment comparisons (ITCs) comparing perioperative durvalumab plus neoadjuvant chemotherapy with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, or surgery alone, no definitive conclusions could be drawn regarding the relative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy against these comparators.
Given the limitations with the ITCs, the Canada’s Drug Agency (CDA-AMC) base case focused on the comparison of perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy. In the CDA-AMC base case, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an incremental cost-effectiveness ratio (ICER) of $68,988 per quality-adjusted life-year (QALY) gained compared with neoadjuvant chemotherapy (incremental costs = $76,327; incremental QALYs = 1.11). The CDA-AMC estimated ICER was higher than the sponsor’s base-case value, primarily due to the selection of alternative distributions for extrapolating EFS, as well as assumptions about the cure landmark and smoking-related excess mortality. In the CDA-AMC base case, a price reduction of at least 19% for durvalumab would be required for perioperative durvalumab plus neoadjuvant chemotherapy to be considered cost-effective compared with neoadjuvant chemotherapy at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. This would reduce the price of durvalumab from $938.67 to $760.32 (per 2.4 mL vial), and from $3,911.11 to $3,168.00 (per 10 mL vial). With this price reduction, the per-patient, 21-day drug acquisition costs for durvalumab would be $9,504 in the neoadjuvant setting and $7,128 in the adjuvant setting.
The CDA-AMC base case relies on a sustained OS benefit for perioperative durvalumab plus neoadjuvant chemotherapy, where patients receiving perioperative durvalumab plus neoadjuvant chemotherapy gain 1.28 additional life-years compared to those treated with neoadjuvant chemotherapy. In the absence of long-term clinical evidence, the extent of this survival benefit is highly uncertain. Should the long-term effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy be lower than predicted, the ICER would be higher than the CDA-AMC base case, requiring larger price reductions to achieve cost-effectiveness.
Given the lack of head-to-head comparative data and limitations associated with the ITCs, clinical evidence on the relative clinical efficacy of perioperative durvalumab plus neoadjuvant chemotherapy compared with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, or surgery alone is uncertain. The cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to these comparators is, therefore, unknown. To ensure cost-effectiveness, durvalumab should also be priced no more than the least costly immunotherapy that is publicly funded for adult patients with stage IIA to stage IIIB (American Joint Committee on Cancer [AJCC], eighth edition) resectable NSCLC and no known EGFR mutations or ALK rearrangements.
Finally, the sponsor did not consider adjuvant atezolizumab as a relevant comparator in the economic analysis; therefore, the cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to adjuvant atezolizumab is unknown.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was collaboratively provided to the Canadian Cancer Survivor Network, Lung Cancer Canada, and the Lung Health Foundation via a patient survey. The survey included 5 patients with lung cancer in Canada, 3 of whom had experience with durvalumab. Patients were asked to compare their experience with durvalumab for lung cancer treatment to other therapies on a scale of “much better,” “little or no difference,” or “much worse.” Responses indicated that durvalumab was rated as much better or showed little to no difference in managing symptoms and slowing disease progression, with similar ratings for adverse events (AEs) and ease of use. Patients highlighted fatigue as the primary AE associated with durvalumab. They also noted that current treatment options include radiation, surgery, targeted therapy, immunotherapy, chemotherapy, and clinical trials, each accompanied by side effects such as fatigue, neuropathy, anemia, nausea, digestive issues, weight loss, joint and muscle pain, migraines, hair loss, vision and hearing changes, and memory issues. Patients expressed a strong preference for treatments that allow them to maintain HRQoL, reduce AEs, delay symptom onset, improve ease of use, prolong life, and ideally, offer a cure.
Clinician group input was received from Lung Cancer Canada and Ontario Health (Cancer Care Ontario). Input noted that currently, funded treatment options include neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery. Patients with stage II to stage III NSCLC and a high PD-L1 status may be treated with adjuvant chemotherapy followed by atezolizumab. Clinician group input commented that perioperative durvalumab with chemotherapy would represent an alternative treatment approach within the current therapeutic landscape, specifically as an option to neoadjuvant nivolumab with chemotherapy.
Drug plans participating in the CDA-AMC reimbursement review process commented that neoadjuvant nivolumab plus chemotherapy, adjuvant atezolizumab plus chemotherapy (i.e., stage II to stage IIIA with a PD-L1 tumour proportion score of greater than or equal to 50%), and adjuvant chemotherapy are currently funded for patients with resectable NSCLC and no EGFR mutations or ALK rearrangements. Additionally, it was noted that adjuvant pembrolizumab plus chemotherapy is currently being reviewed by CDA-AMC for a subpopulation of the indicated population (i.e., stage IB to stage IIIA with a PD-L1 tumour proportion score of less than 50%). Drug plans highlighted that most jurisdictions use weight-based dosing for durvalumab (e.g., 15 mg/kg to 20 mg/kg, up to a maximum of 1,500 mg, every 3 weeks, in combination with chemotherapy, then 20 mg/kg, up to a maximum of 1,500 mg, every 4 weeks). Finally, drug plans remarked that the sponsor’s estimated budget impact for reimbursing perioperative durvalumab plus neoadjuvant chemotherapy for the indication under review may be underestimated.
Several of these concerns were addressed in the sponsor’s model:
EFS and OS, outcomes that are valued by patients and clinicians, were included in the model.
The impact of disease and treatment on a patient’s HRQoL was captured with utility values.
The sponsor considered the impact of AEs.
Neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery were included as comparators.
In addition, CDA-AMC addressed the following concern:
A weight-based dosing approach was applied for durvalumab in a scenario, in line with input received from the drug plans.
CDA-AMC was unable to address the following concerns raised from the input received:
The cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to adjuvant atezolizumab in patients whose tumours have PD-L1 expression of greater than or equal to 50% of tumour cells remains unknown.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing perioperative durvalumab plus neoadjuvant chemotherapy (i.e., neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery) compared with adjuvant chemotherapy, neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, and surgery alone in adult patients with resectable NSCLC (stage IIA to stage IIIB [N2 only], according to AJCC staging, eighth edition) whose tumours have no EGFR mutations or ALK aberrations.1 The target population is aligned with the modified intention-to-treat (mITT) population enrolled in the AEGEAN phase III trial and the proposed Health Canada indication.2,3
Durvalumab is available as 2.4 mL and 10 mL (50 mg/mL) single-use vials for IV infusion at the submitted prices of $938.67 and $3,911.11, respectively.1,2 The recommended dosage for NSCLC is 1,500 mg in combination with chemotherapy every 21 days before surgery (neoadjuvant period), followed by 1,500 mg as monotherapy every 28 days after surgery (adjuvant period). Before surgery, treatment continues for up to 4 cycles or until the disease is deemed unresectable or the patient experiences unacceptable toxicity. After surgery, treatment continues for up to 12 cycles or until disease recurrence or the patient experiences unacceptable toxicity.2 The sponsor assumed the following doses for neoadjuvant chemotherapy, in combination with perioperative durvalumab: pemetrexed is administered at a dose of 500 mg/m2, cisplatin is administered at a dose of 75 mg/m2, paclitaxel is administered at a dose of 200 mg/m2, gemcitabine is administered at a dose of 1,250 mg/m2, and carboplatin is administered at a dose of 5 mg/mL per minute in combination with pemetrexed or gemcitabine and 6 mg/mL per minute in combination with paclitaxel. All chemotherapies are assumed to be administered for a maximum of four 21-day cycles in combination with perioperative durvalumab. Sourced from the AEGEAN trial, the sponsor assumed the following relative use estimates (indicated in parentheses) for neoadjuvant chemotherapy in combination with perioperative durvalumab: carboplatin plus pemetrexed (39.4%), cisplatin plus pemetrexed (15.7%), cisplatin plus paclitaxel (30.7%), cisplatin plus gemcitabine (11.5%), and carboplatin plus gemcitabine (2.7%). Assuming vial-sharing for chemotherapy IV treatments, the sponsor estimated the average 21-day cycle cost of durvalumab to be $11,733 before surgery and $8,700 after surgery. The total cost of perioperative durvalumab plus neoadjuvant chemotherapy, as estimated by the sponsor, was $191,321. Based on the aforementioned relative use estimates for chemotherapy, the sponsor estimated the total cost of perioperative durvalumab plus neoadjuvant chemotherapy to be $198,250, or $207,642 including the cost of surgery.
The sponsor assumed the following doses for neoadjuvant nivolumab plus chemotherapy: nivolumab is administered at a dose of 4.5 mg/kg for a maximum of three 21-day cycles. Neoadjuvant nivolumab is assumed to be administered with cisplatin plus pemetrexed (39.6%), cisplatin plus gemcitabine (38.6%), and carboplatin plus paclitaxel (21.8%). The dosages for chemotherapy were aligned with those described previously; however, carboplatin was assumed to be administered at a dose of 5 mg/mL per minute in combination with paclitaxel and nivolumab. All chemotherapies are assumed to be administered for a maximum of three 21-day cycles in combination with neoadjuvant nivolumab. Accounting for the aforementioned relative use estimates for chemotherapy, the sponsor estimated the average 21-day cycle cost of nivolumab to be $6,291 with the total cost of neoadjuvant nivolumab plus chemotherapy to be $34,487 (nivolumab costs = $27,355), or $44,181 including the cost of surgery.
The sponsor assumed that neoadjuvant chemotherapy consisted of the following: carboplatin plus pemetrexed (36.7%), cisplatin plus pemetrexed (15.1%), carboplatin plus paclitaxel (34.9%), cisplatin plus gemcitabine (10.8%), cisplatin plus paclitaxel (0.3%), and carboplatin plus gemcitabine (2.3%). Modelled dosages and the maximum duration of treatment were aligned with those described for chemotherapy, in combination with perioperative durvalumab. The sponsor estimated the average 21-day cycle cost of neoadjuvant chemotherapy to be $1,656 with a total treatment cost of $7,200, or $16,919 including the cost of surgery.
For adjuvant chemotherapy, the sponsor assumed that cisplatin plus vinorelbine and carboplatin plus vinorelbine would be used in addition to the chemotherapy regimens described in the neoadjuvant setting (cisplatin plus pemetrexed, cisplatin plus gemcitabine, cisplatin plus paclitaxel, carboplatin plus paclitaxel, carboplatin plus pemetrexed, and carboplatin plus gemcitabine). All adjuvant chemotherapy regimens were assumed to have equal relative use (i.e., 12.5% each). Vinorelbine was assumed to be administered at a dose of 25 mg/m2 every 21 days for a maximum of 4 cycles. All other treatment dosages and maximum durations of treatment were aligned with those described for chemotherapy, in combination with perioperative durvalumab. The sponsor estimated the average 21-day cycle cost of adjuvant chemotherapy to be $1,663 with a total treatment cost of $4,820, or $16,863 including the cost of surgery. The sponsor estimated that surgery alone costs $12,043.
The clinical outcomes modelled were OS, EFS, and time to discontinuation of treatment (TDT). The model simulated life-years, QALYs, and costs for each treatment over a lifetime time horizon (36 years), discounted at a rate of 1.5% per annum. The analysis was undertaken from the perspective of the public health care payer in Canada.
Model Structure
The sponsor submitted a semi-Markov model with 4 mutually exclusive health states (event-free [EF], locoregional recurrence [LRR], distant metastasis [DM], and death), whereby transitions between states occurred on a monthly (4.35 weeks) cycle length (Figure 1). The DM health state was split between DM without progression (patients receiving distant metastasis first-line [DM1] treatment) and DM with progression (patients receiving distant metastasis second-line [DM2] treatment), and transitions between these states and death were modelled using a nested partitioned survival model. All patients enter the model in the EF health state. In each cycle, patients can remain EF, transition to death, or experience 1 of 2 types of progression: LRR or DM. Patients who transition to LRR may receive active treatment or no treatment (i.e., best supportive care). Patients receiving active treatment in LRR can develop metastases and transition to the DM or death state. Patients receiving no treatment (i.e., best supportive care) in LRR can only transition to death. Patients in the DM state can only transition to death.
Model Inputs
Baseline patient characteristics in the model were reflective of the mITT population from the AEGEAN trial (N = 740).3 The average patient in the modelled cohort, which the sponsor assumed reflected the patient population in Canada, was aged 64 years, weighed ████ kg, had a mean body surface area of ████ m2, and was more likely to be male (71.6%).3 These characteristics were used to inform the drug dosage regimens, as well as the age- and sex-specific distribution of the general population mortality risk, which the sponsor used to cap the lower bound for all-cause mortality in the model.
Clinical efficacy parameters used to characterize OS, EFS, and TDT for perioperative durvalumab plus neoadjuvant chemotherapy, adjuvant chemotherapy, neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, and surgery alone were derived from various data sources. The sponsor used a parametric multistate modelling approach to estimate health state transition probabilities between health states. Transition probabilities from EF to LRR and EF to DM relied on the results of the 2 independent ITCs submitted by the sponsor and data from the AEGEAN trial.4 The sponsor estimated EFS for neoadjuvant chemotherapy based on the observed data from the second interim analysis data cut-off date (May 10, 2024) of the mITT population in the AEGEAN trial and extrapolated the treatment effect beyond the observed trial data (median duration of EFS follow-up = ████ months; maximum follow-up = ████ months). Furthermore, the sponsor used a piecewise extrapolation approach, modelling survival using the observed Kaplan-Meier (KM) data from the AEGEAN trial up to 3 months (approximately the time of surgery) and parametric extrapolation thereafter. The hybrid approach chosen to model EFS for neoadjuvant chemotherapy was KM plus log-logistic. Candidate distributions were assessed based on diagnostic plots, goodness-of-fit statistics, visual inspection, and clinical plausibility of long-term projections as determined by clinical experts in Canada.
To derive the EFS curves for perioperative durvalumab plus neoadjuvant chemotherapy, adjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, and surgery alone, the sponsor assumed proportional hazards and applied the hazard ratios (HRs) derived from anchored ITCs using neoadjuvant chemotherapy as the common comparator between studies. The sponsor submitted 2 independent ITCs because it deemed it infeasible to simultaneously generate relative effect measures for perioperative durvalumab plus neoadjuvant chemotherapy compared to all comparators within 1 connected network. A matching-adjusted indirect comparison (MAIC)–adjusted HR of ████ was used to estimate the comparative effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to neoadjuvant chemotherapy. The MAIC-adjusted HR was specifically employed when comparing perioperative durvalumab plus neoadjuvant chemotherapy to neoadjuvant nivolumab plus chemotherapy because it accounts for baseline imbalances in patient characteristics between the AEGEAN and CheckMate 816 trials. Hence, the relative efficacy of neoadjuvant nivolumab plus chemotherapy versus neoadjuvant chemotherapy (HR = 0.66) was derived from an MAIC. Additionally, an unadjusted HR of ████ was used to estimate the comparative effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to neoadjuvant chemotherapy. The unadjusted HR for perioperative durvalumab plus neoadjuvant chemotherapy was employed when comparing perioperative durvalumab plus neoadjuvant chemotherapy to adjuvant chemotherapy and surgery alone. Hence, the relative efficacy of adjuvant chemotherapy versus neoadjuvant chemotherapy (HR = ████) and the relative efficacy of surgery alone versus neoadjuvant chemotherapy (HR = ████) were derived from a network meta-analysis (NMA).
The transition probability from EF to death for all modelled comparators was informed by pooled data on time-to-death at the first EFS event from both arms of the AEGEAN trial. The sponsor assumed that 40% of patients alive in the EF health state would transition to LRR and 60% would transition to DM. Data from the NSCLC literature were used to inform the remaining transition probabilities; the PACIFIC trial informed the transition probabilities from LRR to DM and LRR to death,5 and multiple KEYNOTE trials informed the transition probabilities from DM to death.6-8 The risk of death after metastatic cancer was assumed to differ based on the subsequent treatment received. Patients receiving best supportive care in the LRR and DM state were assumed to not incur any further treatment.
TDT for perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy were modelled using the KM data from the relevant treatment arms in the AEGEAN trial,9 up to a maximum of 16 treatment cycles for perioperative durvalumab and 4 treatment cycles for neoadjuvant chemotherapy. Given that there are no TDT data available for non–trial comparators, assumptions were made to model treatment discontinuation. TDT for neoadjuvant nivolumab plus chemotherapy was assumed to be the same as for perioperative durvalumab plus neoadjuvant chemotherapy, based on observations from the AEGEAN trial. Similarly, the TDT for adjuvant chemotherapy was assumed to match that of neoadjuvant chemotherapy, as observed in the AEGEAN trial. TDT data were recalibrated and applied from the start of adjuvant chemotherapy (i.e., beginning in month 3, rather than at the start of the model). This recalibration accounted for the differences in timing related to surgery duration and postsurgery recovery between the TDT for neoadjuvant and adjuvant chemotherapy. The proportion of patients receiving adjuvant chemotherapy was based on those who remained in the EF health state and had not experienced progression or death before the initiation of adjuvant treatment. The sponsor assumed the duration of adjuvant chemotherapy was equivalent to the 3 cycles observed in the NATCH study.10
The sponsor did not incorporate treatment effectiveness waning in the submitted model and therefore implied that the predicted benefit associated with perioperative durvalumab plus neoadjuvant chemotherapy would be maintained indefinitely throughout the lifetime horizon of the model. Moreover, the sponsor applied a cure assumption among patients who achieved long-term EFS. The sponsor assumed that 95% of patients who remained EF at year 5 would be considered cured and follow the same risk of death as the age- and sex- adjusted general population of Canada.11
A health state utility value of █████ was applied to the EF health state based on the results of the EQ-5D-5L questionnaire administered to the mITT population in the neoadjuvant phase of the AEGEAN trial.9 The utility value for the LRR health state (0.810) was sourced from the PACIFIC trial, and the utility values for the DM health states (DM1 = 0.759; DM2 = 0.662) were sourced from the KEYNOTE-189 trial, as reported in previous technical appraisals by the National Institute for Health and Care Excellence.12,13
Grade 3 or grade 4 AEs that occurred in more than 5% of patients during the neoadjuvant and/or adjuvant treatment phases of the AEGEAN trial were included in the economic model. For perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy, AE frequencies were obtained from the AEGEAN trial.9 For adjuvant chemotherapy, AE frequencies were assumed to be the same as neoadjuvant chemotherapy. For neoadjuvant nivolumab plus chemotherapy, AE frequencies were informed by the CheckMate 816 trial.14 For surgery alone, AE frequencies were assumed to be 0%. The duration of each AE was assumed to be 1 month, irrespective of the therapy received. Utility decrements for neutropenia, decreased neutrophil count, anemia, dysphagia, decreased white cell count, and decreased platelet count were sourced from the literature.15-17
Costs considered in the submitted economic model included those associated with drug acquisition and administration, radiotherapy, surgery, treatment monitoring, health care resource use, AE management, and terminal care. Drug acquisition costs were sourced from IQVIA DeltaPA.18 The price for durvalumab was aligned with the sponsor-submitted prices.1 Drug administration costs were sourced from the Ontario Schedule of Benefits for Physician Services.19 The sponsor assumed that all IV treatments either cost $54.25 or $75.00 to administer.19 Radiotherapy costs were derived based on unit costs from the Ontario Schedule of Benefits for Physician Services and multiplied by the frequency reported in Albain et al.19,20 The proportion of patients who received surgery was sourced from the AEGEAN trial for perioperative durvalumab plus neoadjuvant chemotherapy (81%) and neoadjuvant chemotherapy (81%),21 and from the CheckMate 816 trial for neoadjuvant nivolumab plus chemotherapy (83%).14 It was assumed that all patients treated with adjuvant chemotherapy or surgery alone incurred the cost of surgery. Surgery costs were sourced from Cressman et al. and weighted according to the surgery type (i.e., thoracotomy versus minimally invasive surgery) observed in the AEGEAN trial for perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy.21,22 It was assumed that patients receiving neoadjuvant nivolumab plus chemotherapy would have the same distribution of surgery types as those treated with perioperative durvalumab plus neoadjuvant chemotherapy. Similarly, patients undergoing surgery alone or receiving adjuvant chemotherapy were assumed to follow the same distribution of surgery types as those treated with neoadjuvant chemotherapy. Treatment monitoring costs were sourced from the Ontario Schedule of Benefits for Laboratory Services,23 multiplied by the frequency of use reported in a previous CDA-AMC reimbursement review.24 Health care resource use costs associated with clinical visits, hospitalizations, and imaging were sourced from the ICES registry.25 AE unit costs were derived from the Canadian Institute for Health Information Patient Cost Estimator.26 The sponsor assumed all AEs (grade 3 or higher) were treated in an inpatient setting. Finally, a 1-time terminal care cost sourced from the Ontario Case Costing Initiative database was included, encompassing expenses related to end-of-life care.27 All costs were inflated to 2024 Canadian dollars when appropriate.
Summary of Sponsor’s Economic Evaluation Results
The base-case analysis was run probabilistically (1,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented as follows.
Base-Case Results
The submitted analysis was based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3 and Table 4. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
In the sponsor’s submitted pairwise comparisons, perioperative durvalumab plus neoadjuvant chemotherapy was more costly and more effective than neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone. Perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $42,978 per QALY gained relative to neoadjuvant chemotherapy (incremental costs = $66,215; incremental QALYs = 1.54). Compared with neoadjuvant nivolumab plus chemotherapy, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $94,904 per QALY gained (incremental costs = $69,360; incremental QALYs = 0.73). Compared with adjuvant chemotherapy, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $49,444 per QALY gained (incremental costs = $76,325; incremental QALYs = 1.54). Finally, compared with surgery alone, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $19,771 per QALY gained (incremental costs = $54,318; incremental QALYs = 2.75).
Given the duration of follow-up in the AEGEAN trial (median EFS follow-up = ████ months; maximum follow-up = ████ months) in contrast to the model’s lifetime horizon of 36 years, it is important to highlight that the majority of the incremental QALYs gained by patients receiving perioperative durvalumab plus neoadjuvant chemotherapy relative to neoadjuvant chemotherapy (88%), neoadjuvant nivolumab plus chemotherapy (88%), adjuvant chemotherapy (88%), and surgery alone (87%) was derived from the period beyond which there is observed trial data (i.e., extrapolated period).
in the sponsor’s analyses, perioperative durvalumab plus neoadjuvant chemotherapy showed an 88%, 44%, 52%, and 92% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained when compared to neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone, respectively. Pairwise results were primarily influenced by treatment acquisition costs and QALYs gained in the EF health state.
Table 3: Summary of the Sponsor’s Economic Evaluation Results Using MAIC-Adjusted HR, Pairwise
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference, as indicated ($/QALY) |
|---|---|---|---|---|---|
Perioperative durvalumab plus neoadjuvant chemotherapya vs. neoadjuvant nivolumab plus chemotherapyb | |||||
Neoadjuvant nivolumab plus chemotherapy | 136,800 | Reference | 9.41 | Reference | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 206,160 | 69,360 | 10.14 | 0.73 | 94,904 |
HR = hazard ratio; ICER = incremental cost-effectiveness ratio; MAIC = matching-adjusted indirect comparison; QALY = quality-adjusted life-year; vs. = versus.
Note: The sponsor’s pairwise comparisons include neoadjuvant chemotherapy as a comparator because it enabled the indirect comparison of treatments. The ICER for perioperative durvalumab plus neoadjuvant chemotherapy vs. neoadjuvant chemotherapy using the MAIC-adjusted HR is $23,431 per QALY gained (incremental costs = $55,578; incremental QALYs = 2.37).
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
bNeoadjuvant nivolumab plus chemotherapy consists of neoadjuvant nivolumab plus chemotherapy.
Source: Sponsor’s pharmacoeconomic submission.1
Table 4: Summary of the Sponsor’s Economic Evaluation Results Using Unadjusted HR, Pairwise
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference, as indicated ($/QALY) |
|---|---|---|---|---|---|
Perioperative durvalumab plus neoadjuvant chemotherapya vs. neoadjuvant chemotherapy | |||||
Neoadjuvant chemotherapy | 150,582 | Reference | 7.76 | Reference | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 216,797 | 66,215 | 9.30 | 1.54 | 42,978 |
Perioperative durvalumab plus neoadjuvant chemotherapya vs. adjuvant chemotherapy | |||||
Adjuvant chemotherapy | 140,472 | Reference | 7.76 | Reference | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 216,797 | 76,325 | 9.30 | 1.54 | 49,444 |
Perioperative durvalumab plus neoadjuvant chemotherapya vs. surgery alone | |||||
Surgery alone | 162,479 | Reference | 6.56 | Reference | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 216,797 | 54,318 | 9.30 | 2.75 | 19,771 |
HR = hazard ratio; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The sponsor’s pairwise comparisons include neoadjuvant chemotherapy as a comparator because it enabled the indirect comparison of treatments. The ICER for perioperative durvalumab plus neoadjuvant chemotherapy vs. neoadjuvant chemotherapy using the unadjusted HR is consistently reported as $42,978 per QALY gained.
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted deterministic scenario analyses encompassing considerations such as alternative time horizons, discount rates, assumptions pertaining to EFS, wastage, utility values, and immunotherapy re-treatment (i.e., not permitting immunotherapy re-treatment, and allowing immunotherapy re-treatment at 12 months without progression, instead of 6 months). Additionally, the sponsor explored applying a standardized mortality rate to general population mortality, and applying a cure assumption at 5.5 years, with 5 months of warm-up starting from 5 years. The sponsor’s results for all scenario analyses were aligned with the submitted base case, in that perioperative durvalumab plus neoadjuvant chemotherapy was more costly and more effective than neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone.
No scenario analysis was conducted using a perspective other than that of the health care payer.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative clinical efficacy of perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone is highly uncertain: There is no direct head-to-head evidence comparing perioperative durvalumab plus neoadjuvant chemotherapy with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone. The comparative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy relative to neoadjuvant chemotherapy (i.e., placebo with neoadjuvant chemotherapy) was evaluated in the AEGEAN trial but did not inform the sponsor’s base case.3 To estimate EFS in the submitted pharmacoeconomic model, the sponsor conducted separate ITCs to derive HRs. These HRs were then applied to a reference curve for neoadjuvant chemotherapy, which was modelled using observed data from the mITT population in the AEGEAN trial (data cut-off date = May 10, 2024; median EFS follow-up = ████ months; maximum follow-up = ████ months). An MAIC-adjusted HR was applied for neoadjuvant nivolumab plus chemotherapy, while unadjusted HRs from the NMA were used for comparisons with neoadjuvant chemotherapy and surgery alone. Depending on the comparator, different HRs were applied to perioperative durvalumab plus neoadjuvant chemotherapy: an unadjusted HR of ████ or an MAIC-adjusted HR of ████ (specifically for the perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant nivolumab plus chemotherapy comparison). As highlighted in the CDA-AMC Clinical Review Report, although the HR point estimates from the ITCs (MAIC and NMA) favoured perioperative durvalumab plus neoadjuvant chemotherapy over neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone in terms of EFS, the 95% confidence intervals were wide, indicating the potential for both benefit and harm. Although the CDA-AMC Clinical Review Report noted that the ITCs were conducted according to accepted methodological guidance, it also emphasized that owing to imprecision and methodological limitations, no definitive conclusions could be drawn from the ITCs in regard to which treatment may be favoured. Key limitations included significant heterogeneity across the included trials, poorly connected networks, and the likelihood that important effect modifiers (e.g., variations in staging and tumour assessment criteria over time) were not fully matched or adjusted for in the MAIC. Moreover, the CDA-AMC Clinical Review Report concluded that because the comparative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant chemotherapy was established in the AEGEAN trial, direct evidence should be preferred over HR estimates derived from the ITCs.
In the CDA-AMC base case, direct head-to-head evidence from the AEGEAN trial was used to estimate the relative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant chemotherapy. However, due to the absence of direct evidence and inconclusive ITC results for comparisons of perioperative durvalumab plus neoadjuvant chemotherapy with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, or surgery alone, its cost-effectiveness relative to these comparators remains unknown.
Long-term EFS is highly uncertain: The sponsor used parametric modelling to extrapolate long-term EFS for neoadjuvant chemotherapy beyond the observable time points in the AEGEAN trial (median EFS follow-up = ████ months; maximum follow-up = ████ months) to a lifetime horizon of 36 years. HRs derived from anchored ITCs were applied to the reference neoadjuvant chemotherapy EFS curve to estimate each of the 3 transitions originating from the EF health state — transitioning to LR, DM, or death — while accounting for competing risks. As a result, the projected long-term EFS curve in each treatment strategy was influenced by the combination of distributions applied to these transitions. The parametric models selected by the sponsor for transitions originating from the EF health state, along with other modelling assumptions — such as the inclusion of a cure assumption (refer to subsequent limitation) — resulted in an incremental gain of 0.96 QALYs to 3.66 QALYs in the EF health state for patients treated with perioperative durvalumab plus neoadjuvant chemotherapy relative to neoadjuvant nivolumab plus chemotherapy, neoadjuvant chemotherapy, adjuvant chemotherapy, and surgery alone. Hence, nearly 90% of the total incremental QALYs gained by patients treated with perioperative durvalumab plus neoadjuvant chemotherapy was accrued through extrapolation. Furthermore, because the sponsor’s approach to modelling EFS for all pairwise comparisons relied on HRs derived from indirect evidence, the incremental benefit attributed to perioperative durvalumab plus neoadjuvant chemotherapy in the EF health state remains highly uncertain (refer to previous limitation).
CDA-AMC evaluated parametric models for neoadjuvant chemotherapy based on both their statistical fit to the observed EFS data during the trial period and the clinical plausibility of the long-term EFS estimates derived through extrapolation. The log-normal distribution was selected to extrapolate the transition from the EF to LRR state and the EF to DM state because of the following: it ranked highest in statistical fit among candidate distributions, matching the sponsor’s log-logistic distribution to the second decimal; it preserved clinical plausibility for long-term survival estimates; and it offered an improved visual alignment between predicted and observed values from the AEGEAN trial compared to the sponsor’s log-logistic selection.
In the CDA-AMC base case, the log-normal distribution was selected to extrapolate the transition from the EF to LRR state and the EF to DM state for patients treated with neoadjuvant chemotherapy.
The cure assumption is uncertain: The sponsor applied a cure assumption proposing that 95% of patients who remained EF at 5 years would be considered cured and would then experience the same mortality risk as the age- and sex-adjusted general population in Canada.11 The sponsor derived their point estimate from a clinician survey; however, CDA-AMC could not validate this estimate because the sponsor was unable to provide a reference when requested.28 Additionally, the sponsor cited literature suggesting that the risk of recurrence peaks shortly after surgery, gradually declining over time, with a reduction in recurrence risk 5 years after surgery.29-36 Clinical experts consulted for this review acknowledged that, while the sponsor’s assumption may be reasonable given that recurrence typically occurs within the first 5 years, there is limited robust evidence to support it. In the absence of definitive evidence, CDA-AMC recognizes the uncertainty regarding the risk of late recurrence that patients with NSCLC may experience beyond the 5-year landmark point for EFS. However, Maeda et al. provide some evidence indicating that the probability of remaining recurrence-free among patients with NSCLC is heterogeneous, varying with lymph node involvement, and is not constant beyond 5 years following complete surgical resection.34 Specifically, Maeda et al. point to the magnitude of late recurrence risk in patients who remain recurrence-free 10 years postresection, which demonstrates that the recurrence-free probability may vary between 89%, 84%, and 65% for patients with N0, N1, and N2 cancers, respectively.34
In addition, the cure assumption implemented by the sponsor equated the long-term survival outcomes of current or former tobacco users with those of the average person in Canada, an assumption lacking face validity. The model presumed that cured patients would experience the same mortality outcomes as the general age- and sex-matched population in Canada, implying no excess smoking-related mortality risk, despite 25% of the AEGEAN trial population being current smokers and 61% being former smokers at baseline.3 Clinical expert input indicated that it is unlikely that cured patients would experience the same long-term health outcomes as the general population in Canada and it is not reasonable to assume no excess smoking-related mortality in a patient population consisting predominantly of current and former smokers. Clinical experts further highlighted that cancer is not the only prognostic indicator of increased mortality in this patient population because current and former tobacco users often face significant comorbidities, including chronic obstructive pulmonary disease, making them more prone to increased mortality from cardiovascular and respiratory diseases.
In the CDA-AMC base case, it was assumed that 76% of patients remaining EF would be cured after 10 years based on the recurrence-free probabilities from Maeda et al. and the N status distribution observed in the AEGEAN trial.3,34
Additionally, in the CDA-AMC base case, it was assumed that cured patients have a relative risk of death of 1.74 compared with the general population in Canada. This estimate was derived from the relative risk of death from all causes reported by Carter et al.37 for current smokers (2.8), former smokers (1.5), and never-smokers (1.0) aged 55 years and older, alongside the distribution of smokers reported in the AEGEAN trial.3
Health state utility values are uncertain: The sponsor used utility values from multiple sources, applying a value of █████ to the EF health state based on EQ-5D-5L questionnaire results from the mITT population in the AEGEAN trial.9 This value, as noted by the sponsor, exceeds the age-adjusted utility for the general population in Canada (0.842),38 suggesting that patients who are EF and are undergoing treatment for NSCLC would have a higher HRQoL than the average person in Canada — an assumption clinical experts disputed for its lack of face validity. To address this, CDA-AMC adopted utility values for the EF and LRR health states from the CheckMate 816 trial, adjusted to reflect utility scores from the general population of patients in Canada (EF = 0.842; LRR = 0.794). However, CDA-AMC acknowledges that these utility values carry uncertainty because they imply patients who are EF and are undergoing treatment for NSCLC would experience an HRQoL on par with the average person in Canada, a notion that clinical experts regarded with some reservation. This likely overestimates the QALYs accrued in the EF health state.
In the CDA-AMC base case, utility values for the EF and LRR health states were sourced from the CheckMate 816 trial.
Drug acquisition costs are uncertain: In the AEGEAN trial, durvalumab was administered in combination with chemotherapy at a fixed dosage of 1,500 mg every 21 days before surgery, and as monotherapy at a fixed dosage of 1,500 mg every 28 days after surgery. These dosing regimens were consistent with those used in the economic model. Participating drug plans indicated that most jurisdictions use weight-based dosing up to a cap for durvalumab (e.g., 15 mg/kg to 20 mg/kg [up to a maximum of 1,500 mg] every 21 days in combination with chemotherapy, then 20 mg/kg [up to a maximum of 1,500 mg] every 4 weeks). Drug plan input further noted that patients in Ontario receive durvalumab using fixed dosage. CDA-AMC notes that in general, using a weight-based dosing approach reduces drug acquisition costs. The sponsor similarly assumed that patients in the DM1 health state receiving pembrolizumab, either as monotherapy or combined with chemotherapy, would receive a fixed dosage of 200 mg every 21 days. Participating drug plans, however, noted that a weight-based dosing approach, capped at 200 mg (2 mg/kg, up to 200 mg, every 21 days), would likely be adopted for pembrolizumab. In addition, the sponsor assumed that patients with nonsquamous histology who are treated with nivolumab in the DM1 health state receive nivolumab at a fixed dosage of 360 mg every 21 days while in other parts of the economic model, a fixed dosage of 4.5 mg/kg every 21 days was assumed. However, according to recommendations from Ontario Health (Cancer Care Ontario) and feedback from participating drug plans, weight-based dosing would more accurately reflect clinical practice.39 Finally, the sponsor sourced all unit drug acquisition costs from IQVIA DeltaPA.18 CDA-AMC notes that there were numerous discrepancies between the unit costs calculated by CDA-AMC and the unit costs calculated by the sponsor for select chemotherapeutic agents. These differences were attributed to factors such as updated pricing data (i.e., for vinorelbine and paclitaxel), reliance on only the most expensive vial sizes in cost estimates (i.e., for docetaxel), and incorrect assumptions about product size or availability in Canada (i.e., for gemcitabine).
In the CDA-AMC base case, weight-based dosing was adopted for pembrolizumab and nivolumab across the pharmacoeconomic model. Additionally, unit drug costs for docetaxel, gemcitabine, paclitaxel, and vinorelbine were updated using cost data from IQVIA DeltaPA.40
A scenario analysis was conducted in which a weight-based dosing for durvalumab was adopted (20 mg/kg).
A relevant comparator was omitted: The sponsor’s analysis excluded adjuvant atezolizumab, which is indicated for patients with resected stage II to stage IIIA NSCLC who received platinum-based chemotherapy and whose tumours have PD-L1 expression on greater than or equal to 50% of tumour cells and do not have EGFR or ALK mutations. Participating drug plans, clinician group feedback, and clinical experts consulted for this review noted that adjuvant atezolizumab following chemotherapy is a relevant treatment option for a subgroup of patients within the reimbursement request population, although clinical experts consulted for this review noted that adjuvant atezolizumab may not be the most relevant comparator. The sponsor justified this exclusion by explaining that atezolizumab, available only after treatment with adjuvant chemotherapy, is considered a subsequent rather than a primary adjuvant treatment. Consequently, indirect comparisons between perioperative durvalumab plus neoadjuvant chemotherapy and adjuvant atezolizumab were deemed unfeasible and inappropriate. Thus, the cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to adjuvant atezolizumab in patients with NSCLC whose tumours have PD-L1 expression on greater than or equal to 50% of tumour cells remains unknown.
CDA-AMC could not address this limitation owing to model structure and a lack of data.
Poor modelling practices were employed: During the review process, numerous calculation errors were identified in the submitted economic model. Although these errors had a minor impact and only affected adjuvant chemotherapy, it remains uncertain whether additional errors went undetected. This uncertainty is due to the model’s extensive use of IFERROR statements, which can replace parameter values with alternate values without alerting the user, potentially masking errors. The frequent use of these statements complicates the thorough auditing of the sponsor’s model because it becomes challenging to determine if errors are being inappropriately overridden.
In the CDA-AMC base case, the identified errors were not corrected because they only impacted adjuvant chemotherapy, which was not included in reanalyses. The extensive use of IFERROR statements prevented a complete validation of the sponsor’s model.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patient demographics from the AEGEAN trial were assumed to be generalizable to the patient population in Canada. | Reasonable. Clinical experts consulted for this review indicated that the baseline and demographic characteristics of patients in the AEGEAN trial reasonably reflect the target patient population in Canada. However, they noted a higher prevalence of nonsquamous histology (60%) compared to squamous histology (40%) among patients they typically see, differing from the distribution observed in the AEGEAN trial. |
The incidence rate of AEs is expected to reflect those observed in clinical trials. | Not appropriate. The incidence rate of grade 3+ AEs for perioperative durvalumab plus neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone were based on naive comparisons, without adjustments for differences in patient characteristics. These AE rates were used to calculate costs in the sponsor's base case. Due to the direct use of clinical trial data, it is unclear whether any observed differences between treatments are attributable to the therapies themselves or to potential biases or confounding factors. Additionally, no safety end point was assessed in the NMA, preventing any conclusions about the safety of perioperative durvalumab plus neoadjuvant chemotherapy in comparison with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone. |
It was assumed that 40% of patients experience a local event and 60% of patients experience a distant event. | Uncertain. The AEGEAN trial reported that 61.2% of patients experienced a local event, while 38.8% experienced a distant event. Clinical experts noted that the timing of recurrence is often correlated with the type of event, meaning these proportions may shift over time. Additionally, some patients may experience both a local and distant event simultaneously. |
To be eligible for re-treatment, the disease of patients who received immunotherapy in the neoadjuvant or adjuvant resectable setting should not have progressed within 6 months after completion of treatment. | Reasonable. Clinical experts agreed that it is reasonable to assume patients must not have experienced disease progression for at least 6 months after completing treatment to be eligible for immunotherapy re-treatment. |
Patients receiving BSC (i.e., no treatment) in the LRR health state could not transition to the DM health state. These patients were assumed to not incur any further treatment and were assigned transition probabilities to death. | Not appropriate. The assumption that patients receiving BSC in the LRR health state cannot transition to the DM health state lacks face validity. Clinical experts noted that patients with LRR can indeed develop DM. Additionally, clinical expert input noted that patients with LRR may not receive active treatment for various reasons, and these reasons may not apply if the patient progresses to DM. |
AE = adverse event; BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; DM = distant metastasis; LRR = locoregional recurrence; NMA = network meta-analysis.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook the reanalyses outlined in Table 6 to address, where possible, the limitations within the sponsor’s submitted economic model. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 6: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. EFS modelling approach for perioperative durvalumab plus neoadjuvant chemotherapy | HR applied to neoadjuvant chemotherapy reference curve | Log-normal parametric distribution fit to KM data from the AEGEAN trial |
2. EFS distribution for neoadjuvant chemotherapy | Log-logistic | Log-normal |
3. Cure assumption |
|
|
4. SMR applied to the GPM | None. Cured patients have the same risk of death as the age- and sex-adjusted general population of Canada. | Cured patients have a relative risk of death of 1.74, accounting for the increased risk of death among current and former smokers. |
5. Utility values in EF and LRR |
|
|
6. Drug dosages in DM1 |
|
|
7. Unit drug costs |
|
|
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 |
CDA-AMC = Canada’s Drug Agency; DM1 = distant metastasis first line; EF = event-free; EFS = event-free survival; GPM = general population mortality; HR = hazard ratio; KM = Kaplan-Meier; LRR = locoregional recurrence; q.3.w. = every 3 weeks; SMR = standardized mortality ratio.
In the CDA-AMC base case, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $68,988 per QALY gained compared with neoadjuvant chemotherapy (incremental costs = $76,327; incremental QALYs = 1.11) (Table 7). There was a 10% probability that perioperative durvalumab plus neoadjuvant chemotherapy was cost-effective at a WTP threshold of $50,000 per QALY gained.
The CDA-AMC estimated ICER was higher than the sponsor’s base-case value, primarily due to the selection of alternative distributions for extrapolating EFS, as well as assumptions about the cure landmark and smoking-related excess mortality. Consistent with the sponsor’s analysis, the CDA-AMC reanalysis estimates that the majority of incremental QALYs (86%) was accrued in the posttrial period of the model on the basis of extrapolation.
The CDA-AMC base case is based on publicly available prices for all drug treatments. A detailed breakdown of the disaggregated results is available in Appendix 4.
Table 7: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Neoadjuvant chemotherapy | 135,608 | 7.51 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapya | 201,973 | 9.01 | 44,105 | |
CDA-AMC reanalysis 1 — EFS modelling approach for perioperative durvalumab | Neoadjuvant chemotherapy | 135,608 | 7.51 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 201,150 | 9.02 | 43,375 | |
CDA-AMC reanalysis 2 — EFS distribution for neoadjuvant chemotherapy | Neoadjuvant chemotherapy | 132,082 | 7.74 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 198,551 | 9.22 | 44,773 | |
CDA-AMC reanalysis 3 — Cure assumption | Neoadjuvant chemotherapy | 142,092 | 7.26 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 208,495 | 8.81 | 42,777 | |
CDA-AMC reanalysis 4 — SMR applied to the GPM | Neoadjuvant chemotherapy | 134,055 | 6.75 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 201,469 | 7.99 | 54,390 | |
CDA-AMC reanalysis 5 —Health state utilities in EF and LRR | Neoadjuvant chemotherapy | 135,608 | 7.29 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 201,973 | 8.73 | 46,098 | |
CDA-AMC reanalysis 6 — Drug dosages in DM1 | Neoadjuvant chemotherapy | 129,615 | 7.51 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 200,706 | 9.01 | 47,246 | |
CDA-AMC reanalysis 7 — Unit drug costs | Neoadjuvant chemotherapy | 138,861 | 7.51 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 205,063 | 9.01 | 43,997 | |
CDA-AMC base case (Reanalyses 1 to 7) | Neoadjuvant chemotherapy | 133,656 | 6.62 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 208,695 | 7.65 | 72,341 | |
CDA-AMC base case (Reanalyses 1 to 7) (Probabilistic) | Neoadjuvant chemotherapy | 145,909 | 6.97 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 222,236 | 8.07 | 68,988 |
CDA-AMC = Canada’s Drug Agency; DM1 = distant metastasis first line; EF = event-free; EFS = event-free survival; GPM = general population mortality; ICER = incremental cost-effectiveness ratio; LRR = locoregional recurrence; QALY = quality-adjusted life-year; SMR = standardized mortality rate.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s base case and the CDA-AMC base case. The CDA-AMC base case suggested that a 19% price reduction for durvalumab would be required for perioperative durvalumab plus neoadjuvant chemotherapy to achieve cost-effectiveness relative to neoadjuvant chemotherapy at a WTP threshold of $50,000 per QALY gained (Table 8).
Table 8: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | ICER for perioperative durvalumab plus neoadjuvant chemotherapya vs. neoadjuvant chemotherapy ($/QALY) | ||
|---|---|---|---|---|
Price reduction | 2.4 mL vial | 10 mL vial | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 938.67 | 3,911.11 | 42,978 | 68,988 |
10% | 844.80 | 3,520.00 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 58,953 |
20% | 750.94 | 3,128.89 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 48,919 |
30% | 657.07 | 2,737.78 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 38,884 |
40% | 563.20 | 2,346.67 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 28,850 |
50% | 469.34 | 1,955.56 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 18,815 |
60% | 375.47 | 1,564.44 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | 8,781 |
70% | 281.60 | 1,173.33 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant |
80% | 187.73 | 782.22 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant |
90% | 93.87 | 391.11 | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant | Perioperative durvalumab plus neoadjuvant chemotherapy is dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
CDA-AMC conducted scenario analyses to assess the impact of using weight-based dosing for durvalumab because most participating drug plans apply weight-based dosing with a cap for durvalumab. The results, presented in Appendix 4, Table 13, aligned with the CDA-AMC base case, showing that perioperative durvalumab plus neoadjuvant chemotherapy is more costly but also more effective than neoadjuvant chemotherapy. In this scenario, the ICER decreased to $64,348 per QALY gained due to decreased total drug acquisition costs for durvalumab.
Issues for Consideration
CDA-AMC has previously reviewed neoadjuvant nivolumab plus chemotherapy for adult patients with resectable NSCLC when used in combination with platinum-doublet chemotherapy.41 The cost-effectiveness results of the evaluation may not be directly comparable to those in the current review, owing to differences in model structure, clinical effectiveness parameters, health state utility values, and cost inputs.
Perioperative pembrolizumab (the KEYNOTE-671 trial)42 and perioperative nivolumab (the CheckMate 77T trial)43 were not considered relevant comparators at the time of writing this report because they are not currently approved by Health Canada for the indicated population and have not been reviewed or recommended for funding by CDA-AMC. The cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy compared to perioperative pembrolizumab and perioperative nivolumab is unknown.
Overall Conclusions
Evidence from the phase III, randomized, placebo-controlled, AEGEAN trial comparing the efficacy and safety of perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy in adult patients with resectable NSCLC and no known EGFR mutations or ALK rearrangements demonstrated that perioperative durvalumab plus neoadjuvant chemotherapy confers added clinical benefit in EFS. The clinical review assessed that perioperative durvalumab plus neoadjuvant chemotherapy likely results in a clinically important increase in EFS at 36 months compared to neoadjuvant chemotherapy, with moderate certainty of evidence. The clinical review further noted that, compared to neoadjuvant chemotherapy, perioperative durvalumab plus neoadjuvant chemotherapy may result in little to no difference in HRQoL during the neoadjuvant period (with low certainty of evidence). Additionally, the evidence is very uncertain about the effect on HRQoL in the adjuvant period. Notably, as of the second interim analysis (data cut-off date = May 10, 2024; OS median duration of follow-up = ████ months), perioperative durvalumab plus neoadjuvant chemotherapy may result in little to no difference on the probability of being alive at 48 months; however, OS data remained immature. Finally, the clinical review concluded that, due to the absence of direct evidence and methodological limitations in the ITCs comparing perioperative durvalumab plus neoadjuvant chemotherapy with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, or surgery alone, no definitive conclusions could be drawn regarding the relative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy against these comparators.
In addition to the limitations with the clinical evidence, CDA-AMC identified several limitations with the sponsor’s economic submission, including uncertainty regarding long-term EFS, uncertainty regarding the assumption of cure among patients who remain EF, the use of health state utility values reflecting improbably high estimates compared to the general population in Canada, and the omission of adjuvant atezolizumab from the economic analysis. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These included using observed EFS data from the AEGEAN trial to estimate the relative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant chemotherapy, adopting a log-normal distribution to extrapolate EFS for neoadjuvant chemotherapy, assuming that 76% of patients who are EF at 10 years posttreatment initiation would be considered cured, assuming cured patients may face an increased smoking-related mortality risk by applying a standardized mortality ratio of 1.74 to the general population mortality in Canada, and using alternative sources to inform utility values for the EF and LRR health states.
Given the limitations with the ITCs, the CDA-AMC base case focused on the comparison of perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy. In the CDA-AMC base case, perioperative durvalumab plus neoadjuvant chemotherapy was associated with an ICER of $68,988 per QALY gained compared with neoadjuvant chemotherapy (incremental costs = $76,327; incremental QALYs = 1.11). When compared to the sponsor’s analysis, the CDA-AMC base case estimated a reduced QALY benefit with perioperative durvalumab plus neoadjuvant chemotherapy (i.e., incremental QALYs = 1.11 [the CDA-AMC base case] versus incremental QALYs = 1.54 [the sponsor’s analysis]) at an increased cost (i.e., incremental costs = $76,327 [the CDA-AMC base case] versus incremental costs = $66,215 [the sponsor’s analysis]). The ICER estimated by CDA-AMC was higher than the sponsor’s base-case value, primarily due to the selection of alternative distributions for extrapolating EFS, as well as assumptions about the cure landmark and smoking-related excess mortality. In the CDA-AMC base case, a price reduction of 19% for durvalumab would be required for perioperative durvalumab plus neoadjuvant chemotherapy to be considered cost-effective compared with neoadjuvant chemotherapy at a WTP threshold of $50,000 per QALY gained. This would reduce the price of durvalumab from $938.67 to $760.32 per 2.4 mL vial, and from $3,911.11 to $3,168.00 per 10 mL vial. With this price reduction, the per-patient, 21-day drug acquisition costs for durvalumab would be $9,504 in the neoadjuvant setting and $7,128 in the adjuvant setting.
The CDA-AMC base case relies on a sustained OS benefit for perioperative durvalumab plus neoadjuvant chemotherapy, where patients receiving perioperative durvalumab gain 1.28 additional life-years compared to those treated with neoadjuvant chemotherapy. In the absence of long-term clinical evidence, the extent of this survival benefit is highly uncertain. Should the long-term effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy be lower than predicted, the ICER would be higher than that of the CDA-AMC base case, requiring larger price reductions to achieve cost-effectiveness. Moreover, when comparing the duration of follow-up in the AEGEAN trial to the model’s time horizon (59 months [approximately 5 years] versus 36 years), it is important to note that most of the QALY and life-year benefit predicted by the model for patients treated with perioperative durvalumab plus neoadjuvant chemotherapy are accrued on the basis of extrapolation, representing model-generated outcomes rather than trial-based evidence.
Given the lack of head-to-head comparative data and limitations associated with the ITCs, clinical evidence on the relative clinical efficacy of perioperative durvalumab plus neoadjuvant chemotherapy compared with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, or surgery alone is uncertain. The cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to these comparators is, therefore, unknown. To ensure cost-effectiveness, durvalumab should also be priced no more than the least costly immunotherapy that is publicly funded for adult patients with stage IIA to stage IIIB (AJCC, eighth edition) resectable NSCLC and no known EGFR mutations or ALK rearrangements.
Finally, the sponsor did not consider adjuvant atezolizumab as a relevant comparator in the economic analysis; therefore, the cost-effectiveness of perioperative durvalumab plus neoadjuvant chemotherapy relative to adjuvant atezolizumab is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Aug 13.
2.Imfinzi (durvalumab): 50 mg/mL, concentrate for solution for intravenous infusion [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Jan 12.
3.AstraZeneca. A Phase III, Double-blind, Placebo-controlled, Multi-center International Study of Neoadjuvant/Adjuvant Durvalumab for the Treatment of Patients with Resectable Stages II and III Non-small Cell Lung Cancer (AEGEAN): Clinical Study Report. 2023.
4.Technical report for the indirect treatment comparison of perioperative durvalumab (AEGEAN) as treatment for resectable stages II and III non-small cell lung cancer: addendum – AEGEAN EFS IA2 update, version 1.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab), 50 mg/mL, concentrate for solution for intravenous infusion. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Aug 02.
5.Spigel DR, Faivre-Finn C, Gray JE, et al. Five-Year Survival Outcomes From the PACIFIC Trial: Durvalumab After Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. J Clin Oncol. 2022;40(12):1301-1311. PubMed
6.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50. J Clin Oncol. 2021;39(21):2339-2349. PubMed
7.Rodriguez-Abreu D, Powell SF, Hochmair MJ, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: protocol-specified final analysis from KEYNOTE-189. Ann Oncol. 2021;32(7):881-895. PubMed
8.Paz-Ares L, Vicente D, Tafreshi A, et al. A Randomized, Placebo-Controlled Trial of Pembrolizumab Plus Chemotherapy in Patients With Metastatic Squamous NSCLC: Protocol-Specified Final Analysis of KEYNOTE-407. J Thorac Oncol. 2020;15(10):1657-1669. PubMed
9.AstraZeneca. Durvalumab in Combination with Chemotherapy as Neoadjuvant Treatment Followed by Durvalumab as Monotherapy After Surgery in Patients with Resectable Stage II or III Non-Small Cell Lung Cancer (AEGEAN). Event-free Survival Second Interim Analysis (EFS IA2) – Tables and Figures. 2024.
10.ClinicalTrials.gov. Neoadjuvant or Adjuvant Chemotherapy in Patients With Operable Non-small-cell Lung Cancer (NATCH). https://clinicaltrials.gov/study/NCT00913705. Accessed July, 2024.
11.Statistics C. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401.
12.National Institute for H, Care E. Durvalumab for maintenance treatment of unresectable non-small-cell lung cancer after platinum-based chemoradiation. Technology appraisal guidance. Reference number: TA798. Published: 22 June 2022. https://www.nice.org.uk/guidance/ta798. Accessed July, 2024.
13.National Institute for H, Care E. Pembrolizumab with pemetrexed and platinum chemotherapy for untreated, metastatic, non-squamous non-small-cell lung cancer. Technology appraisal guidance. Reference number: TA683. Published: 10 March 2021. https://www.nice.org.uk/guidance/ta683/chapter/3-Committee-discussion. Accessed July, 2024.
14.Forde PM, Spicer J, Lu S, et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med. 2022;386(21):1973-1985. PubMed
15.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health Qual Life Outcomes. 2008;6:84. PubMed
16.Aldenhoven L, Ramaekers B, Degens J, et al. Cost-effectiveness of proton radiotherapy versus photon radiotherapy for non-small cell lung cancer patients: Exploring the model-based approach. Radiother Oncol. 2023;183:109417. PubMed
17.Doyle S, Lloyd A, Walker M. Health state utility scores in advanced non-small cell lung cancer. Lung Cancer. 2008;62(3):374-380. PubMed
18.DeltaPA I. Canadian Drug Costs. 2022.
19.Ministry of Health O. Schedule of Benefits. Physician Services. Under the Health Insurance Act (February 20, 2024 (effective April 1, 2024)). 2024.
20.Albain KS, Swann RS, Rusch VW, et al. Radiotherapy plus chemotherapy with or without surgical resection for stage III non-small-cell lung cancer: a phase III randomised controlled trial. The Lancet. 2009;374(9687):379-386. PubMed
21.Clinical Study Report: D9106C00001, AEGEAN. A phase III, double-blind, placebo-controlled, multi-center international study of neoadjuvant/adjuvant durvalumab for the treatment of patients with resectable stages II and III non-small cell lung cancer (AEGEAN) [internal sponsor's report]. Cambridge (UK): AstraZeneca; 2023 Jul 10.
22.Cressman S LSTMCEWKLNBRDABCSFATMS, Pan-Canadian Early Detection of Lung Cancer Study T. Resource Utilization and Costs during the Initial Years of Lung Cancer Screening with Computed Tomography in Canada. J Thorac Oncol. 2014.
23.Ministry of Health O. SCHEDULE OF BENEFITS FOR LABORATORY SERVICES (Ontario Health Insurance Plan Laboratories and Diagnostics Branch). 2023.
24.Canadian Agency for D, Technologies in H. CADTH Reimbursement Review Nivolumab (Opdivo) Non–small cell lung cancer. July 2023.
25.ICES. HOPE-ICES: Survival and costing of patients with resected stage I-III NSCLC diagnosed in Ontario between 2010 and 2019. Accessed March 23, 2021.
26.Cihi. Canadian Institute for Health Information. National Prescription Drug Utilization Information System — Plan Information Document, July 31, 2021. Ottawa, ON: CIHI. 2021.
27.Ontario. Ontario Case Costing Initiative (OCCI).
28.Response to Additional Information Request dated September 23, 2024.
29.Demicheli R, Fornili M, Ambrogi F, et al. Recurrence dynamics for non-small-cell lung cancer: effect of surgery on the development of metastases. J Thorac Oncol. 2012;7(4):723-730. PubMed
30.Lou F, Sima CS, Rusch VW, Jones DR, Huang J. Differences in patterns of recurrence in early-stage versus locally advanced non-small cell lung cancer. Ann Thorac Surg. 2014;98(5):1755-1760; discussion 1760-1751. PubMed
31.Saw SPL, Zhou S, Chen J, et al. Association of Clinicopathologic and Molecular Tumor Features With Recurrence in Resected Early-Stage Epidermal Growth Factor Receptor-Positive Non-Small Cell Lung Cancer. JAMA Netw Open. 2021;4(11):e2131892. PubMed
32.Yamauchi Y, Muley T, Safi S, et al. The dynamic pattern of recurrence in curatively resected non-small cell lung cancer patients: Experiences at a single institution. Lung Cancer. 2015;90(2):224-229. PubMed
33.Yun JK, Kwon Y, Kim J, et al. Clinical impact of histologic type on survival and recurrence in patients with surgically resected stage II and III non-small cell lung cancer. Lung Cancer. 2023;176:24-30. PubMed
34.Maeda R, Yoshida J, Hishida T, et al. Late recurrence of non-small cell lung cancer more than 5 years after complete resection: incidence and clinical implications in patient follow-up. Chest. 2010;138(1):145-150. PubMed
35.Shin DW, Cho JH, Yoo JE, et al. Conditional Survival of Surgically Treated Patients with Lung Cancer: A Comprehensive Analyses of Overall, Recurrence-free, and Relative Survival. Cancer Res Treat. 2021;53(4):1057-1071. PubMed
36.Sonoda D, Matsuura Y, Ichinose J, et al. Ultra-late recurrence of non-small cell lung cancer over 10 years after curative resection. Cancer Manag Res. 2019;11:6765-6774. PubMed
37.Carter BD, Abnet CC, Feskanich D, et al. Smoking and mortality--beyond established causes. N Engl J Med. 2015;372(7):631-640. PubMed
38.Guertin JR, Feeny D, Tarride J-E. Age- and sex-specific Canadian utility norms, based on the 2013–2014 Canadian Community Health Survey. Can Med Assoc J. 2018;190(6):E155. PubMed
39.CCO. nivo dose. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/71636.
40.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed September 24 2024.
41.nivo. https://www.cda-amc.ca/nivolumab-2.
42.Spicer J, Girard N, Provencio M, et al. Neoadjuvant nivolumab (NIVO) + chemotherapy (chemo) vs chemo in patients (pts) with resectable NSCLC: 4-year update from CheckMate 816. Journal of Clinical Oncology. 2024;42(17_suppl):LBA8010-LBA8010.
43.Cascone T, Awad MM, Spicer JD, et al. Perioperative Nivolumab in Resectable Lung Cancer. N Engl J Med. 2024;390(19):1756-1769. PubMed
44.CCO CARBPAC. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46006. Accessed October 15, 2024.
45.Patient cost estimator. 2024; https://www.cihi.ca/en/patient-cost-estimator. Accessed October 30, 2024.
46.Canadian Cancer Statistics Advisory C. Canadian Cancer Statistics: A 2020 special report on lung cancer. Toronto, ON: Canadian Cancer Society; 2020. Available from: cancer.ca/Canadian-Cancer-Statistics-2020-EN.
47.Snee M, Cheeseman S, Thompson M, et al. Treatment patterns and survival outcomes for patients with non-small cell lung cancer in the UK in the preimmunology era: a REAL-Oncology database analysis from the I-O Optimise initiative. BMJ open. 2021;11(9). PubMed
48.Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and Genomic Profiling of Non–Small-Cell Lung Cancer: Implications for Current and Future Therapies. J Clin Oncol. 2013;31(8):1039-1039. PubMed
49.Statistics C. Population estimates on July 1st, by age and sex. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed August 2024.
50.Statistics C. Projected population, by projection scenario, age and sex, as of July 1. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.1&cubeTimeFrame.startYear=2021&cubeTimeFrame.endYear=2028&referencePeriods=20210101%2C20280101. Accessed August 2024.
51.KOL Interview [data on file]. In: AstraZeneca C, ed2024.
52.Canada's Drug A. Nivolumab (Opdivo). Nivolumab (Opdivo): CADTH Reimbursement Review. 2023.
53.Canadian Cancer S. Lung and bronchus cancer statistics. https://cancer.ca/en/cancer-information/cancer-types/lung/statistics. Accessed August 1, 2024.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CDA-AMC Cost Comparison Table for Resectable Non–Small Cell Lung Cancer
Treatment | Strength | Forma | Price ($) | Recommended dosage | Daily or 1-time cost ($) | 21-day cycle cost ($) | |
|---|---|---|---|---|---|---|---|
Durvalumab (Imfinzi) | 50 mg/mL | 2.4 mL (120 mg) 10 mL (500 mg) | 938.6700b 3,911.1100b | Neoadjuvant period: 1,500 mg in combination with chemotherapy every 21 days for up to 4 cyclesc Adjuvant period: 1,500 mg as monotherapy every 28 days for up to 12 cyclesc | Neoadjuvant period: 558.73 Adjuvant period: 419.05 | Neoadjuvant period: 11,733 Adjuvant period: 8,800 | |
Perioperative durvalumab plus chemotherapy (neoadjuvant period)d | |||||||
CRBPGEMC + DURV | 973.67 to 999.39 | 14,115 to 14,655 | |||||
CRBPPACL + DURV | 922.24 to 932.24 | 13,035 to 13,245 | |||||
CRBPPEME + DURV | 629.44 | 14,783 | |||||
CISPGEMC + DURV | 946.05 to 971.77 | 13,535 to 14,075 | |||||
CISPPEME + DURV | 601.83 | 12,638 | |||||
PD-1 inhibitors | |||||||
Atezolizumab (Tecentriq) | 60 mg/mL | 14 mL (840 mg) 20 mL (1,200 mg) | 4,473.2000 6,776.0000 | 840 mg every 14 days, 1,200 mg every 21 days, or 1,680 mg every 28 days, up to 48 weeks | 169.40 to 338.80 | 3,557 to 7,115 | |
Nivolumab (Opdivo) | 10 mg/mL | 4 mL (40 mg) 10 mL (100 mg) | 782.2200 1,955.5600 | 4.5 mg/kg, up to a maximum of 360 mg, every 21 days for up to 3 cycles | 316.61 | 6,649 | |
Neoadjuvant nivolumab plus chemotherapy | |||||||
CRBPGEMC + NIVL | 414.94 to 440.66 | 2,382 to 2,922 | |||||
CRBPPACL + NIVL | 363.51 to 373.51 | 1,302 to 1,512 | |||||
CRBPPEME + NIVL | 387.33 | 8,134 | |||||
CISPGEMC + NIVL | 387.32 to 413.04 | 1,802 to 2,342 | |||||
CISPPEME + NIVL | 359.71 | 7,554 | |||||
CISPVINO + NIVL | 354.95 | 7,454 | |||||
Chemotherapies | |||||||
Carboplatin (generic) | 10 mg/mL | 5 mL (50 mg) 15 mL (150 mg) 45 mL (450 mg) 60 mL (600 mg) | 70.0000 210.0000 599.9985 775.0020 | AUC 5 every 21 dayse | 46.90 | 985 | |
Cisplatin (generic) | 1 mg/mL | 50 mL (50 mg) 100 mL (100 mg) | 135.0000 270.0000 | 75 mg/m2 every 21 days | 19.29 | 405 | |
Docetaxel (generic) | 10 mg/mL | 8 mL (80 mg) 16 mL (160 mg) | 970.2000 1,850.0000 | 75 mg/m2 every 21 days | 47.14 | 990 | |
20 mg/mL | 4 mL (80 mg) 8 mL (160 mg) | 497.0000 990.0000 | |||||
Gemcitabine (generic) | 40 mg/mL | 25 mL (1,000 mg) 50 mL (2,000 mg) | 270.0000 540.0000 | 1,000 mg/m2 to 1,250 mg/m2 on days 1 and 18 every 21 days | 51.43 to 77.14 | 1,080 to 1,620 | |
Paclitaxel (generic) | 6 mg/mL | 5 mL (30 mg) 16.7 mL (100 mg) 25 mL 150 mg 50 mL (300 mg) | 300.0000 1,002.0000 1,500.0000 3,000.0000 | 175 to 200 mg/m2 every 21 days | 157.14 to 185.71 | 3,300 to 3,900 | |
Pemetrexed (generic) | 10 mg/mL | 10 mL (100 mg) 50 mL (500 mg) | 50.0000 250.0000 | 500 mg/m2 every 21 days | 23.81 | 500 | |
Vinorelbine (generic) | 10 mg/mL | 1 mL (10 mg) 5 mL (50 mg) | 80.0000 400.0000 | 25 mg/m2 every 21 days | 19.05 | 400 | |
CRBPDOCE | 94.05 | 1,975 | |||||
CRBPGEMC | 98.33 to 124.05 | 2,065 to 2,605 | |||||
CRBPPACL | 46.90 to 56.90 | 985 to 1,195 | |||||
CRBPPEME | 70.71 | 1,485 | |||||
CRBPVINO | 65.95 | 1,385 | |||||
CISPDOCE | 66.43 | 1,395 | |||||
CISPGEMC | 70.71 to 96.43 | 1,485 to 2,025 | |||||
CISPPEME | 43.10 | 905 | |||||
CISPVINO | 38.33 | 805 | |||||
Non–drug interventions | |||||||
Lung resection | $21,590 to $25,140 per patientf | NA | |||||
AUC = area under the curve; CDA-AMC = Canada’s Drug Agency; CISPDOCE = cisplatin plus docetaxel; CISPGEMC = cisplatin plus gemcitabine; CISPPEME = cisplatin plus pemetrexed; CISPVINO = cisplatin plus vinorelbine; CRBPDOCE = carboplatin plus docetaxel; CRBPGEMC = carboplatin plus gemcitabine; CRBPPACL = carboplatin plus paclitaxel; CRBPPEME = carboplatin plus pemetrexed; CRBPVINO = carboplatin plus vinorelbine; DURV = durvalumab; NIVL = nivolumab.
All prices are wholesale from IQVIA DeltaPA (accessed September 2024),40 unless otherwise indicated, and do not include dispensing fees. Calculations assume a patient body weight of 70 kg, a body surface area of 1.85 m2, and a glomerular filtration rate of 125 mL per minute. Vial wastage was assumed (i.e., no vial-sharing). All recommended dosages are retrieved from Cancer Care Ontario Drug Formulary Regimens.
aAll drug interventions are vials for IV infusion or infusion bags.
bSponsor-submitted price.1
cPatients with a body weight of 30 kg or less must receive a dose of 20 mg/kg every 21 days before surgery, followed by monotherapy at 20 mg/kg every 28 days after surgery until weight increases to greater than 30 kg.2
dChemotherapy backbones are reflective of those used in the economic model.
eThe recommended dose of carboplatin when used in combination with paclitaxel is AUC 5 to 6 every 21 days.44
fRetrieved from the Canadian Institute for Health Information’s Patient Cost Estimator: Case mix group: open lung resection; Fiscal year: 2021 to 2022; Adults (Accessed October 30, 2024).45
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comment |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to the ‘Omission of relevant comparator’ limitation in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation. |
Model has been adequately programmed and has sufficient face validity | No | Refer to the ‘Poor modelling practices were employed’ limitation in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to the ‘Poor modelling practices were employed’ limitation in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Refer to the ‘Poor modelling practices were employed’ limitation in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
DM = distant metastasis; EF = event-free; LRR = locoregional recurrence; TP1 = transition probability 1; TP2 = transition probability 2; TP3 = transition probability 3; TP4 = transition probability 4; TP5 = transition probability 5; TP6 = transition probability 6.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results, Pairwise
Parameter | MAIC-adjusted HR | Unadjusted HR | ||||
|---|---|---|---|---|---|---|
Perioperative durvalumab plus neoadjuvant chemotherapya | Neoadjuvant nivolumab plus chemotherapyb | Perioperative durvalumab plus neoadjuvant chemotherapy | Neoadjuvant chemotherapy | Adjuvant chemotherapy | Surgery alone | |
Discounted LYs | ||||||
Total | 12.21 | 11.37 | 11.24 | 9.47 | 9.48 | 8.09 |
EF | 11.51 | 10.35 | 10.27 | 7.81 | 7.73 | 5.88 |
LRR | 0.21 | 0.30 | 0.29 | 0.45 | 0.49 | 0.62 |
PF with DM | 0.26 | 0.37 | 0.36 | 0.61 | 0.63 | 0.79 |
PD with DM | 0.23 | 0.35 | 0.32 | 0.61 | 0.63 | 0.80 |
Discounted QALYs | ||||||
Total | 10.14 | 9.41 | 9.30 | 7.76 | 7.76 | 6.56 |
EF | 9.62 | 8.66 | 8.60 | 6.55 | 6.48 | 4.94 |
LRR | 0.17 | 0.24 | 0.23 | 0.36 | 0.39 | 0.50 |
PF with DM | 0.20 | 0.28 | 0.27 | 0.46 | 0.48 | 0.60 |
PD with DM | 0.15 | 0.23 | 0.21 | 0.39 | 0.41 | 0.52 |
AEs | −0.00017 | −0.00012 | −0.00017 | −0.00020 | −0.00020 | 0.00000 |
Discounted costs ($) | ||||||
Total | 206,160 | 136,800 | 216,797 | 150,582 | 140,472 | 162,479 |
EF | ||||||
Tx acquisition | 124,072 | 40,472 | 124,072 | 15,359 | 12,059 | 12,059 |
Administration | 472 | 219 | 472 | 177 | 0 | 0 |
HCRU | 36,932 | 34,354 | 34,196 | 28,495 | 28,238 | 23,596 |
Monitoring | 586 | 231 | 565 | 146 | 49 | 49 |
AEs | 2,520 | 1,713 | 2,520 | 2,904 | 2,885 | 0 |
LRR | ||||||
Tx acquisition | 4,050 | 4,580 | 5,553 | 8,307 | 6,960 | 8,772 |
Administration | 25 | 29 | 35 | 54 | 44 | 56 |
HCRU | 4,101 | 5,874 | 5,603 | 8,635 | 9,526 | 12,004 |
Monitoring | 376 | 585 | 565 | 932 | 1,026 | 1,341 |
PF with DM | ||||||
Tx acquisition | 5,289 | 12,938 | 7,660 | 28,508 | 23,947 | 36,086 |
Administration | 36 | 66 | 50 | 129 | 110 | 163 |
HCRU | 8,203 | 11,638 | 11,248 | 19,176 | 19,959 | 25,156 |
Monitoring | 618 | 877 | 848 | 1,446 | 1,505 | 1,897 |
PD with DM | ||||||
Tx acquisition | 3,786 | 4,765 | 5,182 | 8,048 | 6,303 | 7,944 |
Administration | 28 | 36 | 38 | 59 | 50 | 63 |
HCRU | 7,308 | 11,030 | 10,065 | 19,227 | 20,101 | 25,340 |
Monitoring | 618 | 130 | 847 | 1,450 | 181 | 228 |
Terminal care | 7,142 | 7,262 | 7,280 | 7,530 | 7,529 | 7,725 |
AE = adverse event; DM = distant metastasis; EF = event-free; HCRU = health care resource use; HR = hazard ratio; LRR = locoregional recurrence; LY = life-year; MAIC = matching-adjusted indirect comparison; PD = progressed-disease; PF = progression-free; QALY = quality-adjusted life-year; Tx = treatment.
Note: In all pairwise analyses using the unadjusted HRs, the results for perioperative durvalumab plus neoadjuvant chemotherapy remain consistent.
aNeoadjuvant nivolumab plus chemotherapy consists of neoadjuvant nivolumab plus chemotherapy.
bPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Perioperative durvalumab plus neoadjuvant chemotherapya | Neoadjuvant chemotherapy |
|---|---|---|
Discounted LYs | ||
Total | 9.89 | 8.61 |
EF | 8.98 | 7.08 |
LRR | 0.27 | 0.40 |
PF with DM | 0.34 | 0.57 |
PD with DM | 0.30 | 0.56 |
Discounted QALYs | ||
Total | 8.07 | 6.97 |
EF | 7.40 | 5.85 |
LRR | 0.21 | 0.32 |
PF with DM | 0.26 | 0.44 |
PD with DM | 0.19 | 0.36 |
AEs | −0.00017 | −0.00020 |
Discounted costs ($) | ||
Total | 222,236 | 145,909 |
EF | ||
Drug acquisition | 126,917 | 17,744 |
Administration | 474 | 178 |
HCRU | 39,384 | 32,963 |
Treatment monitoring | 559 | 146 |
AEs | 2,522 | 2,905 |
LRR | ||
Drug acquisition | 5,738 | 8,046 |
Administration | 35 | 50 |
HCRU | 5,211 | 7,690 |
Treatment monitoring | 487 | 773 |
PF with DM | ||
Drug acquisition | 6,490 | 21,486 |
Administration | 47 | 122 |
HCRU | 10,852 | 17,975 |
Treatment monitoring | 816 | 1,355 |
PD with DM | ||
Drug acquisition | 4,927 | 7,721 |
Administration | 37 | 57 |
HCRU | 9,395 | 17,666 |
Treatment monitoring | 816 | 1,333 |
Terminal care | 7,529 | 7,699 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; DM = distant metastasis; EF = event-free; HCRU = health care resource use; LRR = locoregional recurrence; LY = life-year; PF = progression-free; QALY = quality-adjusted life-year.
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Scenario Analyses
Table 13: Scenario Analyses Conducted on the CDA-AMC Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case | Neoadjuvant chemotherapy | 145,909 | 6.97 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapya | 222,236 | 8.07 | 68,988 | |
CDA-AMC scenario 1 – Durvalumab is administered using weight-based dosing | Neoadjuvant chemotherapy | 145,576 | 6.90 | Reference |
Perioperative durvalumab plus neoadjuvant chemotherapy | 216,500 | 8.00 | 64,348 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. Results are presented probabilistically.
aPerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
AJCC = American Joint Committee on Cancer; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; EFS = event-free survival; NSCLC = non–small cell lung cancer.
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing perioperative durvalumab plus neoadjuvant chemotherapy for adult patients with resectable NSCLC (stage IIA to stage IIIB [N2 only], according to AJCC staging, eighth edition) whose tumours have no EGFR mutations or ALK aberrations. The BIA was undertaken from the perspective of a public payer in Canada over a 3-year time horizon (2025 to 2027) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. The sponsor’s analysis included drug acquisition costs, assuming wastage. Data informing the model were obtained from various sources, including published literature,46-48 Statistics Canada,49,50 a previous CDA-AMC Pharmacoeconomic Review Report,24 assumption, and input from clinical experts consulted by the sponsor.51 Key inputs to the BIA are documented in Table 15.
The sponsor’s BIA also included the following key assumptions:
Introduction of perioperative durvalumab plus neoadjuvant chemotherapy would displace most neoadjuvant and adjuvant therapies in resectable NSCLC.
Proportions of patients transitioning to LRR, DM, and death following an EFS event are 24%, 40%, and 36%, respectively.
Market shares for perioperative durvalumab plus neoadjuvant chemotherapy would be ██%, ██% and ██%, in year 1 to year 3, respectively.
Patients receiving immunotherapy as first-line therapy for DM would not proceed to second-line therapy for DM.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Pan-Canadian population (excluding Quebec)49 | 25,676,360 |
Annual population growth rate (2024 onward)50 | 1.13% |
Lung cancer incidence ratea | 0.097% |
Proportion of patients with NSCLC46 | 88.0% |
Proportion of patients with NSCLC stage IIA/IIB47 | 10.4% |
Proportion of patients with NSCLC stage IIIA47 | 14.9% |
Proportion of patients with NSCLC stage IIIB47 | 8.0% |
Proportion of patients with resectable disease | |
Proportion of patients with resectable NSCLC stage IIA/IIB51,52 | 50.0% |
Proportion of patients with resectable NSCLC stage IIIA51,52 | 30.0% |
Proportion of patients with resectable NSCLC stage IIIB51 | 5.0% |
Proportion of patients without EGFR/ALK mutations48 | 81.0% |
Patients eligible for neoadjuvant, adjuvant, or perioperative therapyb | 100% |
Patients treated with neoadjuvant, adjuvant, or perioperative therapyc | 45.9% |
Number of patients eligible for drug under review | 833 / 842 / 852 |
Market uptake (3 years) | |
Uptake (reference scenario) Neoadjuvant chemotherapy Neoadjuvant nivolumab plus chemotherapyd Surgery only Adjuvant chemotherapy Adjuvant atezolizumab | 3% / 2% / 1% 38% / 39% / 40% 20% / 20% / 20% 30% / 30% / 30% 9% / 9% / 9% |
Uptake (new drug scenario) Perioperative durvalumab plus neoadjuvant chemotherapye Neoadjuvant chemotherapy Neoadjuvant nivolumab plus chemotherapy Surgery only Adjuvant chemotherapy Adjuvant atezolizumab | ██% / ██% / ██% ||% / ||% / ||% ██% / ██% / ██% ██% / ██% / ██% ██% / ██% / ██% ||% / ||% / ||% |
Total cost of treatment (per patient, excluding surgery) | |
Perioperative durvalumab plus neoadjuvant chemotherapy Neoadjuvant chemotherapy Neoadjuvant nivolumab plus chemotherapy Surgery only Adjuvant chemotherapy Adjuvant atezolizumab | $139,724 $8,417 $26,296 $0 $5,217 $143,448 |
CDA-AMC = Canada’s Drug Agency; NSCLC = non–small cell lung cancer.
aDerived from published literature.53
bBased on sponsor assumption.
cAssumption derived from a previous CDA-AMC reimbursement review.52
dNeoadjuvant nivolumab plus chemotherapy consists of neoadjuvant nivolumab plus chemotherapy.
ePerioperative durvalumab plus neoadjuvant chemotherapy consists of neoadjuvant durvalumab plus chemotherapy before surgery and adjuvant durvalumab monotherapy following surgery.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing perioperative durvalumab plus neoadjuvant chemotherapy for adult patients with resectable NSCLC (stage IIA to stage IIIB [N2 only], according to AJCC staging, eighth edition) whose tumours have no EGFR mutations or ALK aberrations is expected to result in a cost of $65,193,469 (year 1 = $18,566,320; year 2 = $22,298,502; year 3 = $24,328,646).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
EFS is highly uncertain: The sponsor’s BIA incorporates the EFS HRs obtained from the submitted ITCs to estimate the postprogression transition probabilities. As noted in the CDA-AMC document Appraisal of the Sponsor’s Economic Evaluation owing to imprecision and methodological limitations, no definitive conclusions could be drawn from the ITCs on which treatment may be favoured (i.e., perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, and surgery alone). The comparative efficacy of perioperative durvalumab plus neoadjuvant chemotherapy versus neoadjuvant chemotherapy was established in the AEGEAN trial and direct evidence should be preferred over HR estimates derived from the ITCs. CDA-AMC explored the impact of adopting the EFS values for perioperative durvalumab plus neoadjuvant chemotherapy and neoadjuvant chemotherapy at year 1, year 2, and year 3 from the CDA-AMC cost-utility analysis base case, assuming that the proportion of patients remaining EF and treated with neoadjuvant nivolumab plus chemotherapy, adjuvant chemotherapy, adjuvant atezolizumab, or surgery would be equal to the proportion of patients remaining EF and treated with perioperative durvalumab plus neoadjuvant chemotherapy. This adjustment had minimal impact, increasing the 3-year budget impact by less than 1.5%.
CDA-AMC did not adjust for this limitation in reanalyses.
Drug costs are uncertain: As noted in the CDA-AMC document Appraisal of the Sponsor’s Economic Evaluation, while the sponsor assumed that durvalumab and pembrolizumab were administered at fixed dosages, CDA-AMC participating public drug plans noted that most jurisdictions use weight-based dosing up to a cap. Additionally, while the sponsor sourced all unit drug acquisition costs from IQVIA DeltaPA, numerous discrepancies were identified between the unit costs calculated by CDA-AMC and the unit costs calculated by the sponsor. This adjustment had minimal impact, resulting in a decrease of less than 2.5% on the 3-year total budget impact estimate.
A scenario analysis was conducted adopting weight-based dosing for durvalumab and pembrolizumab.
Market shares are uncertain: The sponsor derived market shares in the reference scenario based on internal forecasting, market research, and clinical expert input. Clinical experts consulted by CDA-AMC for this review noted that there are large variations across jurisdictions pertaining to the relative use of neoadjuvant chemotherapy, neoadjuvant nivolumab plus chemotherapy, surgery only, adjuvant chemotherapy, and adjuvant atezolizumab. It was noted that the market share attributed to neoadjuvant nivolumab plus chemotherapy is uncertain with the use of the treatment growing with time due to the shift in practice. Clinical expert input suggested that neoadjuvant nivolumab plus chemotherapy’s market share may be too high in year 1. Furthermore, clinical expert input noted that the 20% market share assigned to surgery alone may be overestimated as this treatment option is primarily for patients who are considered unfit for treatment or decline treatment.
CDA-AMC did not adjust for this limitation in reanalyses.
Market uptake of perioperative durvalumab plus neoadjuvant chemotherapy is uncertain: The sponsor’s submitted base case assumed that ██%, ██%, and ██% of eligible patients would receive perioperative durvalumab plus neoadjuvant chemotherapy in year 1, year 2, and year 3, respectively, based on internal estimates and clinician input elicited by the sponsor. Clinical expert input elicited by CDA-AMC for this review suggested that while the sponsor’s estimates are reasonable, it may take more time to achieve the estimated market shares (i.e., uptake may be slower than modelled). Additionally, clinician input noted that should other treatment options be approved by Health Canada and become publicly reimbursed (i.e., perioperative pembrolizumab and adjuvant pembrolizumab) for the indicated population, the uptake of perioperative durvalumab plus neoadjuvant chemotherapy will likely be lower than estimated by the sponsor.
CDA-AMC did not adjust for this limitation in reanalyses.
CDA-AMC Reanalyses of the BIA
The limitations identified by CDA-AMC had minimal impact on the results of the BIA. Furthermore, in the absence of more reliable estimates to inform the key parameters of the BIA (i.e., market shares and market uptake), the sponsor’s submitted base case was maintained. CDA-AMC conducted a scenario analysis adopting weight-based dosing for durvalumab (20 mg/kg up to 1,500 mg) and pembrolizumab (2 mg/kg up to 200 mg) using the sponsor’s base case (results are provided in Table 16).
Table 16: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 31,647,528 | 31,386,469 | 35,611,461 | 38,517,375 | 137,162,834 |
New drug | 31,647,528 | 49,952,789 | 57,909,964 | 62,846,022 | 202,356,302 | |
Budget impact | 0 | 18,566,320 | 22,298,502 | 24,328,646 | 65,193,469 | |
CDA-AMC scenario analysis 1: Weight-based dosing | Reference | 29,725,165 | 29,454,868 | 32,990,624 | 35,472,799 | 127,643,456 |
New drug | 29,725,165 | 50,189,697 | 57,883,246 | 62,635,152 | 200,433,260 | |
Budget impact | 0 | 20,734,829 | 24,892,622 | 27,162,353 | 72,789,804 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.