Drugs, Health Technologies, Health Systems
Reimbursement Review
Belzutifan (Welireg)
Sponsor: Merck Canada Inc.
Therapeutic area: Advanced renal cell carcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BICR
blinded independent central review
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CR
complete response
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EMA
European Medicines Agency
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FKSI-DRS
Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms
GHS
global health status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HIF
hypoxia-inducible factor
HR
hazard ratio
HRQoL
health-related quality of life
IMDC
International Metastatic Renal Cell Carcinoma Database Consortium
IO
immuno-oncology
ITC
indirect treatment comparison
ITT
intention to treat
KCC
Kidney Cancer Canada
KCRNC
Kidney Cancer Research Network of Canada
KM
Kaplan-Meier
KPS
Karnofsky Performance Status
LS
least squares
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
NCCN
National Comprehensive Cancer Network
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PR
partial response
QoL
quality of life
RCC
renal cell carcinoma
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
TKI
tyrosine kinase inhibitor
TTD
time to deterioration
VEGF
vascular endothelial growth factor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Belzutifan (Welireg), 40 mg, tablets, oral |
Sponsor | Merck Canada Inc. |
Indication | For the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 17, 2024 |
Recommended dose | 120 mg (three 40 mg tablets) administered once daily |
NOC = Notice of Compliance; RCC = renal cell carcinoma; VEGF-TKI = vascular endothelial growth factor tyrosine kinase inhibitor.
Introduction
Kidney cancer is the eighth most common malignancy in Canada. In 2024, the incidence of kidney cancer was estimated to be 9,000 cases in Canada and the mortality rate was estimated to be 3.9 per 100,000 people.1,2 Renal cell carcinoma (RCC) accounts for approximately 90% of all kidney cancers and is classified into various histologic subtypes.3 Of these, the clear cell subtype is the most common, representing approximately 75% of all RCC cases.3,4 Approximately 25% of patients are diagnosed with locally advanced or metastatic RCC and 20% to 40% of patients with localized primary RCC will develop metastatic disease.5 The prognosis of RCC is heavily influenced by cancer stage at diagnosis; the 5-year relative survival rate for patients with localized RCC is 93% compared with 18% for patients with stage IV disease.4 Patients with RCC can experience a wide range of symptoms, although many show no symptoms until the disease is advanced.6 Flank pain, hematuria, and abdominal renal mass are symptoms that strongly suggest locally advanced RCC.6 Patients with RCС can also present with, or subsequently develop, systemic symptoms and paraneoplastic syndromes.6
First-line treatment options for untreated advanced RCC are guided by the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk group classification status.3,7-9 The standard first-line treatment for patients with advanced RCC in any IMDC risk category consists of an immuno-oncology (IO) drug in combination with a tyrosine kinase inhibitor (TKI) or TKI monotherapy.3,7-9 Among patients with advanced RCC in the IMDC intermediate risk or poor risk categories, nivolumab-ipilimumab is also a recommended first-line treatment option.3,7,8 The strategy for subsequent lines of treatment is contingent on the treatments administered in the first-line setting for advanced RCC.3,7-9 Patients who have received first-line IO-based therapy are eligible for TKI monotherapy (sunitinib, pazopanib, axitinib, or cabozantinib). Recommended second-line treatments for patients with RCC naive to IO drugs include nivolumab or TKI monotherapy (axitinib or cabozantinib).3,7-9 Although everolimus was considered as a treatment of advanced RCC after failure of a vascular endothelial growth factor (VEGF)-TKI, the clinical experts consulted by Canada’s Drug Agency (CDA-AMC) agreed that it has been superseded by the emergence of newer or more effective therapies (e.g., cabozantinib and axitinib) and is currently not funded by most provinces after treatment with these monotherapies.10-16 Third-line options for advanced RCC include belzutifan (recommended by National Comprehensive Cancer Network [NCCN] and European Society for Medical Oncology guidelines),3,8 TKI monotherapy,3,7,8 nivolumab (for patients with no prior exposure to IO drugs, although the clinical experts consulted by CDA-AMC suggested this is rarely administered in practice),7 and everolimus.3,10-18 There is an unmet need for effective later-line treatments for patients with advanced RCC, especially among those whose disease has progressed after treatment with an IO drug and a VEGF-TKI.19,20 Moreover, available treatment options for heavily pretreated RCC are often associated with side effects that are difficult to manage and negatively affect quality of life (QoL).21-24
Belzutifan is an inhibitor of hypoxia-inducible factor 2 (HIF-2) alpha, a key oncogenic driver in clear cell RCC. Belzutifan binds to HIF-2 alpha, and in conditions of hypoxia or impairment of VHL protein function, blocks the HIF-2 alpha and HIF-1 beta interaction, leading to a reduced expression of downstream genes, such as those associated with cellular proliferation, angiogenesis, and tumour growth. On December 17, 2024, Belzutifan received a Notice of Compliance by Health Canada for the treatment of advanced RCC in adult patients following treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The sponsor’s reimbursement request is the same as the proposed Health Canada indication.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of belzutifan, 40 mg oral tablets, in the treatment of advanced RCC in adult patients following treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. Belzutifan was previously reviewed by CADTH. On September 20, 2023, a recommendation for reimbursement was issued for belzutifan for the treatment of adult patients with von Hippel-Lindau disease who require therapy for an associated nonmetastatic RCC, central nervous system hemangioblastoma, or nonmetastatic pancreatic neuroendocrine tumour not requiring immediate surgery.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to the CDA-AMC call for input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Group Input
Input was received from Kidney Cancer Canada (KCC) for this submission. KCC is a national community of patients, caregivers, and health professionals who advocate and support patients with kidney cancer. KCC gathered the information through an international online survey done in affiliation with the International Kidney Cancer Coalition in 2022. The respondents were 2,213 patients and caregivers from 39 countries, including 139 respondents (111 patients and 28 caregivers) from Canada. Further, KCC conducted a survey in December 2024 and gathered information from 2 patients — 1 with kidney cancer and 1 with von Hippel-Lindau disease — and 1 caregiver of a patient with kidney cancer, all of whom had experience with belzutifan. The 2 patients also provided consent to have a telephone conversation. KCC noted that approximately one-quarter of patients reported that their current treatments were difficult to tolerate. The most commonly experienced barriers reported by respondents in Canada were wait times to treatment (16%), lack of access to local specialty doctors (10%), lack of access to up-to-date treatment or equipment (9%), cost of treatment (7%), and lack of personal support (5%). KCC explained that there is a general need for more effective therapies with manageable side effects, as well as better predictive and prognostic biomarkers to guide treatment and better early detection of disease. Based on the patient group input, unmet needs include treatments that provide a cure, durable remission, disease stability, long-term duration of response (DOR), improved tolerability, improved disease-specific QoL, and innovative medicines with new mechanisms of action. Additionally, according to KCC, a new treatment needs to address the resistance to existing treatment; not all patients respond to currently available treatments, and patients who do respond to currently funded treatments often become resistant to therapy over time. All 3 individuals with direct experience with belzutifan reported positive experiences with the drug, noting treatment was effective with tolerable side effects.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted by CDA-AMC agreed that there is an unmet need for third-line and fourth-line treatments with novel mechanisms of action for advanced RCC. The importance of improving treatment options for heavily pretreated RCC was emphasized because disease progression occurs in almost all patients who receive treatment and because available treatment options are associated with side effects that are difficult to manage and negatively affect QoL.
The clinical experts stated that belzutifan would be administered in the third-line or fourth-line treatment setting for patients with advanced RCC, after prior treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. One expert agreed that belzutifan would cause a shift in the treatment paradigm for RCC because of the introduction of a new treatment option in the third-line and fourth-line settings and a novel mechanism of action. However, the magnitude of this shift remains unknown because of the small proportion of patients who are eligible for third-line and fourth-line treatments for advanced RCC in Canada. The experts indicated that it is not possible to identify which subgroups of patients would receive more or less treatment benefit from belzutifan.
One clinical expert indicated that the assessment of response to treatment for RCC consisted of CT scans performed every 3 months. One clinical expert stated that clinically meaningful responses to treatment for RCC included radiologic response, symptom status, stable disease, and adequate tolerance of the drug. The clinical experts agreed that discontinuation criteria for belzutifan consisted of disease progression and intolerable toxicities. Both experts also agreed that belzutifan should be prescribed by a medical oncologist experienced in managing advanced RCC. It was also noted that belzutifan can be administered at a patient’s home. One expert noted that transfusions and dose reductions are preferred for the management of anemia and hypoxia related to belzutifan. Although the expert did not note variation in the management of adverse events (AEs) across jurisdictions in Canada, they noted there may be variations in the coverage of home oxygen and erythropoietin, if required.
Clinician Group Input
CDA-AMC received 2 clinician inputs: 1 from the Ontario Health (OH) (Cancer Care Ontario [CCO]) Genitourinary Cancers Drug Advisory Committee (DAC) with 2 clinicians’ contribution, and 1 from the Kidney Cancer Research Network of Canada (KCRNC) with contribution from 10 clinicians. The OH (CCO) DACs provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. KCRNC is a virtual and inclusive national network of clinicians and researchers who treat kidney cancer in Canada. KCRNC is a federally registered not-for-profit organization with a commitment to enhance the knowledge of kidney cancer and its treatment.
Both clinician groups agreed that the first-line systemic treatment for advanced kidney cancer considers the IMDC risk groups. Based on the inputs, treatment goals include improvements in overall survival (OS) and progression-free survival (PFS), with a reduction in the size of metastatic lesions (i.e., objective response rate [ORR]), and a better QoL by controlling symptoms of the disease. The clinician groups noted that the main gap is resistance to the currently available treatment modalities. The clinician groups agreed that belzutifan will be used in the later-line setting, either as a second-line drug (if the patient’s disease progresses after a first-line combination regimen) or as a third-line drug. Based on the inputs, radiological response and symptom improvement are used to determine treatment response.
Drug Program Input
Input was obtained from the drug programs that participated in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for belzutifan:
relevant comparators
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
funding algorithm
care provision issues
system and economic issues.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs (refer to Table 4 for details).
Clinical Evidence
Systematic Review
Description of Studies
One pivotal phase III, open-label, active-controlled randomized trial (LITESPARK-005) evaluated the efficacy and safety of belzutifan (n = 374) compared to everolimus (n = 372) in patients with advanced RCC who were previously treated with a PD-1 or PD-L1 inhibitor and a VEGF-TKI.25 The trial enrolled adult patients with unresectable, locally advanced or metastatic clear cell RCC who had disease progression during or after treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI.
Moreover, patients were required to have adequate organ function, a Karnofsky Performance Status (KPS) score of at least 70%, and no more than 3 prior systemic regimens for locally advanced or metastatic RCC. Randomization of patients in the trial were stratified according to IMDC prognostic score (0 [favourable] versus 1 or 2 [intermediate] versus 3 to 6 [poor]) and the number of prior VEGF- or VEGF receptor–targeted therapies for advanced RCC (1 versus 2 or 3). The primary outcomes of the LITESPARK-005 trial were PFS based on blinded independent central review (BICR) per Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1), and OS; the secondary outcomes of the trial were ORR based on BICR per RECIST 1.1, DOR, health-related quality of life (HRQoL), and safety. The baseline demographic and disease characteristics were balanced between treatment groups. The median age of patients in the trial was 63 years (range, 22 to 90 years), and most patients were male (77.9%; 22.1% were female).25
The trial enrolled patients of the following races or ethnicities: American Indian or Alaska Native (0.7%), Asian (12.1%), Black or African American (1.1%), Native Hawaiian or other Pacific Islander (0.1%), and white (78.8%) [categories are as reported in study]; 2.3% of patients in the trial identified as having multiple races or ethnicities and 5.0% of patients had missing data on race and ethnicity.25 Most patients had a KPS score that ranged between 90 and 100 (64.1%) and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 1 (55.1%).25 Most patients had an intermediate or poor IMDC prognostic risk score (78.3%) and had stage IV RCC at diagnosis (57.0%).25 Most patients had 2 or more organs involved with the disease at baseline (91.2%), with the lung being the most common site of metastatic disease (64.6%). Most patients had a prior nephrectomy (69.7%), received 2 (43.3%) or 3 (42.8%) prior lines of therapy, and received 1 prior VEGF- or VEGF receptor–targeted therapy (50.5%).25
Efficacy Results
PFS Based on BICR per RECIST 1.1
At the time of the first interim analysis (data cut-off: November 1, 2022), the median duration of follow-up was 13.6 months (range, 0.2 to 31.1 months) in the belzutifan group and 13.3 months (range, 0.8 to 31.8 months) in the everolimus group.26 At the time of the data cut-off for this analysis, 519 PFS events (82.9% of the 626 total expected events at the final analysis) had occurred (257 patients in the belzutifan group and 262 patients in the everolimus group).26 The corresponding hazard ratio (HR) was 0.75 (95% confidence interval [CI], 0.63 to 0.90; one-sided P = 0.00077), representing a 25% reduction in the risk of disease progression or death with belzutifan compared with everolimus.26 The predefined success criterion for superiority based on PFS was met at the first interim analysis. Median PFS was similar in the belzutifan and everolimus groups: 5.6 months (95% CI, 3.9 to 7.0 months) for the belzutifan group compared with 5.6 months (95% CI, 4.8 to 5.8 months) for the everolimus group.26 In addition, PFS rates based on Kaplan-Meier (KM) estimates were higher in the belzutifan group than in the everolimus group at all specified time points.26 The estimated PFS rate at 24 months was █████ ████ ███ ████ ██ █████ in the belzutifan group compared with 0% (95% CI, not applicable) in the everolimus group (between-group difference for belzutifan compared with everolimus: █████ ████ ███ ███ ███████████). The PFS results in additional sensitivity analyses were consistent with those of the overall intention-to-treat (ITT) population, and results across all prespecified subgroups favoured belzutifan over everolimus.26
At the time of the final analysis (data cut-off: April 15, 2024), the median duration of follow-up was 21.4 months (range, 0.2 to 47.6 months) in the belzutifan group and 18.3 months (range, 0.8 to 49.2 months) in the everolimus group.27 At the time of the data cut-off for this analysis, 587 PFS events had occurred (308 in the belzutifan group and 279 in the everolimus group).27 The HR for PFS was 0.75 (95% CI, 0.63 to 0.88; one-sided P = 0.00034).27 As was observed for the first interim analysis, median PFS measured at the final analysis was similar in the belzutifan and everolimus groups.27 Moreover, PFS rates based on KM estimates continued to be higher in the belzutifan group than in the everolimus group at all specified time points.27 The estimated PFS rate at 30 months was 14.2% (95% CI, 10.7% to 18.2%) in the belzutifan group compared with 2.7% (95% CI, 1.1% to 5.5%) in the everolimus group (between-group difference for belzutifan compared with everolimus = 11.5%; 95% CI, 7.2% to 15.8%).
Overall Survival
The corresponding statistical hypothesis-testing P value boundary for OS was not met at any of the prespecified analyses (interim and final analyses) of the trial. At the time of the final analysis (data cut-off: April 15, 2024), the median duration of follow-up was 21.4 months (range, 0.2 to 47.6 months) in the belzutifan group and 18.3 months (range, 0.8 to 49.2 months) in the everolimus group.27 There were 513 OS events (approximately 106% of the 483 events planned for the final analysis; 254 occurring in the belzutifan group and 259 occurring the everolimus group) observed.27 The corresponding HR was 0.92 (95% CI, 0.77 to 1.10; one-sided P = 0.17644), which was not statistically significant.27 The median OS was 21.4 months (95% CI 18.2 to 24.3 months) in the belzutifan group and 18.2 months (95% CI, 15.8 to 21.8 months) in the everolimus group.27 The OS rates based on KM estimations were numerically higher in the belzutifan group compared with the everolimus group at all specified time points.27 The estimated OS rate at 36 months was █████ ████ ███ ████ ██ █████ in the belzutifan group compared with 28.0% (95% CI, 23.1% to 33.1%) in the everolimus group (between-group difference for belzutifan compared with everolimus: ████ ████ ███ ████ ██ █████). The OS results in all prespecified subgroups were consistent with those of the overall ITT population.27
ORR Based on BICR per RECIST 1.1
At the time of the first interim analysis (data cut-off: November 1, 2022), the ORR based on BICR per RECIST 1.1 in the belzutifan group was █████ ██████ ███ ████ ██ █████ compared ██ ████ ██████ ███ ███ ██ ████.26 The estimated difference in the percentage of patients with confirmed ORR for belzutifan versus everolimus was █████ ██████ ███ ████ ██ █████ ███████ ██████████.26 The P value crossed the prespecified boundary for statistical significance of 0.001 at the time of the first interim analysis.26 A higher proportion of patients had a confirmed complete response (CR) or partial response (PR) in the belzutifan group (CR = 2.7%; PR = 19.3%) than in the everolimus group (CR = 0%; PR = 3.5%).26 At the time of the second interim analysis (data cut-off: June 13, 2023), the ORR was █████ ██████ ███ ████ ██ █████ in the belzutifan group compared to 3.5% (99.9% CI, 1.2% to 7.8%) in the everolimus group.25 The estimated difference in the percentage of patients with a confirmed ORR for belzutifan versus everolimus was █████ ██████ ███ ████ ██ █████ ███████ ███████ ██████████.25 At the time of the final analysis (data cut-off: April 15, 2024), the ORRs for belzutifan compared to everolimus remained consistent with those of the second interim analysis.27
Duration of Response
At the time of the first interim analysis (data cut-off: November 1, 2022), the median DOR based on RECIST 1.1 by BICR was not yet reached (range, 1.7+ to 23.2+ months [plus signs indicate an ongoing response at the time of data cut-off]) in the belzutifan group and was 17.2 months (range, 3.8 to 18.0+ months) in the everolimus group.26 The proportion of patients who had a response was higher in the belzutifan group than in the everolimus group at each response duration time point (i.e., 6 months through 21 months), based on KM estimates; 74.2% of patients who had a response in the belzutifan group and 68.4% of patients who had a response in the everolimus group had a DOR lasting 12 months or longer.26
At the time of the final analysis (data cut-off: April 15, 2024), the median DOR was 19.3 months (range, 1.9+ to 40.1+ months) in the belzutifan group and 13.7 months (range, 3.8 to 29.5+ months) in the everolimus group. Similar to what was observed at the first interim analysis, the proportion of responders was higher in the belzutifan group compared with the everolimus group at all measured time points, based on KM estimates, during the second interim and final analyses.
Time to Response
At the time of the first interim analysis (data cut-off: November 1, 2022), the median time to response was 3.7 months (range, 1.7 to 16.6 months) in the belzutifan group and 3.7 months (range, 1.8 to 5.4 months) in the everolimus group.26 At the time of the final analysis (data cut-off: April 15, 2024), the median time to response was 3.8 months (range, 1.7 to 22.0 months) in the belzutifan group and 3.7 months (range, 1.8 to 5.7 months) in the everolimus group.27
Health-Related Quality of Life
The key HRQoL outcomes of interest identified in the LITESPARK-005 trial were the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms (FKSI-DRS), and both were measured at the first26 and second25 interim analyses. Results for HRQoL outcomes of interest were not assessed at the final analysis. These outcomes were not controlled for multiplicity; thus, all reported P values and CIs are nominal and descriptive. At the time of the first analysis (data cut-off: November 1, 2022), there was no change in EORTC QLQ-C30 global health status (GHS)/QoL score from baseline to week 17 in the belzutifan group (least squares [LS]: ████ ██████ ████ ███ █████ ██ ████]), whereas, there was a numerical decrease (worsening) in score from baseline in the everolimus group (LS mean, −6.13; 95% CI, −8.51 to −3.75).26 The difference in LS mean change in EORTC QLQ-C30 GHS/QoL score from baseline to week 17 between the 2 groups was ████ ██████ ████ ███ ████ ██ █████ ███████ █████████.26 Both groups reported a numerical decrease (worsening) in FKSI-DRS score from baseline to week 17, with a larger decrease in score noted for patients in the everolimus group (LS mean change, 1.61 points; 95% CI, −2.17 to −1.06) compared with those in the belzutifan group (LS mean change, −0.07 points; 95% CI, −0.59 to 0.45).26 The difference in LS mean change in FKSI-DRS score from baseline between the belzutifan and everolimus groups was 1.54 points (95% CI, 0.81 to 2.28 points; nominal P < 0.0001).26 Moreover, the time to deterioration (TTD) in HRQoL was longer for those in the belzutifan group than for those in the everolimus group in terms of the EORTC QLQ-C30 GHS/QoL and FKSI-DRS scores.26 The results of the second interim analysis (data cut-off: June 13, 2023) were consistent with those reported at the first interim analysis.25
Harms Results
At the time of the final analysis (data cut-off: April 15, 2024), almost all patients in the LITESPARK-005 trial reported at least 1 AE (99.2% in both the belzutifan and everolimus treatment groups).27 The most common AE in both treatment groups was anemia (83.1% in the belzutifan group and 57.2% in the everolimus group), followed by fatigue (32.3%) and nausea (18.5%), in the belzutifan group and stomatitis (38.1%) and fatigue (25.8%) in the everolimus group.27 At least 1 serious adverse event (SAE) was reported in █████ of patients in the belzutifan group and 38.6% of patients in the everolimus group, with the most common SAEs reported being hypoxia (7.3% in the belzutifan group and none in the everolimus group) and anemia (5.4% in the belzutifan group and 2.2% in everolimus group).27 Higher rates of discontinuation of study treatment due to AEs was noted among patients in the everolimus group (15.3%) than those in the belzutifan group (████).27 The most common AEs that led to treatment discontinuation in both of the treatment groups were related to respiratory, thoracic, and mediastinal disorders (1.9% in the belzutifan group and 6.4% in the everolimus group).27 Deaths due to AEs were slightly higher in the everolimus group (5.3%) than in the belzutifan group (3.8%).27 The most common AEs that led to death in both groups were related to infections and infestations (0.8% in the belzutifan group versus 3.1% in the everolimus group).27
Anemia, hypoxia, and dyspnea were identified as clinical AEs of interest because they have been noted to be associated with treatment with belzutifan. At the final analysis, a higher rate of anemia-related events was reported in patients in the belzutifan group compared with patients in the everolimus group (83.3% in the belzutifan group compared with 57.5% in the everolimus group).27 In both groups, most events related to anemia were grade 2 (41.9% in the belzutifan group compared with 29.7% in the everolimus group).27 Moreover, a higher rate of hypoxia was reported in patients in the belzutifan group compared with patients in the everolimus group (14.2% in the belzutifan group compared with 1.1% in the everolimus group).27 In both groups, most events related to hypoxia were grade 3 (10.5% in the belzutifan group versus 0.8% in the everolimus group).27 Similar rates of dyspnea were reported in patients receiving belzutifan (15.3%) and everolimus (14.4%).27 For all 3 clinical AEs of interest, the rates of discontinuation among both treatment groups were low.27
Critical Appraisal
Notable strengths of the trial included the use of an ITT analysis and the stratification of randomization according to IMDC prognostic scores and the number of prior VEGF-targeted therapies for advanced RCC. The randomization process in the trial was deemed to be appropriate, although there was limited detail provided on how randomization numbers allocated to patients were obtained. Moreover, the LITESPARK-005 trial had an open-label study design, indicating that patients and investigators were aware of treatment allocation. Although PFS and ORR were assessed based on BICR, the lack of blinding of treatment allocation to patients may have contributed to performance bias in the results for patient-reported outcomes. The 2 treatment groups were balanced in terms of baseline patient and disease characteristics. Prior and concomitant non-oncologic medications were, overall, balanced between the 2 treatment groups. Some imbalances in the categories of concomitant medications were deemed to have minimal impact on the treatment effect by the clinical experts consulted by CDA-AMC.
It was also noted that ████ patients in the everolimus group received ██████████ █████████ █████████ compared with patients in the belzutifan group. This difference could potentially introduce a confounding effect on OS, as the survival results might be partially attributable to treatments administered after disease progression, rather than the study treatment itself. Although this difference could favour the ██████████ group, the risk of bias due to deviations from the intended interventions was deemed to be low by the clinical experts consulted by CDA-AMC. The clinical experts consulted by CDA-AMC also confirmed that the oncologic therapies used in the LITESPARK-005 trial were largely reflective of those used in clinical practice in Canada, although some differences in treatment sequence were observed. For instance, █████ of patients in the LITESPARK-005 trial received ████████████ ██ █ ██████████ ███████ █████ ██████████ ██ ██████████, whereas the clinical experts consulted by CDA-AMC anticipated that cabozantinib would be used first, followed by belzutifan. The clinical experts noted that if belzutifan were publicly reimbursed, most patients in clinical practice would be expected to receive belzutifan as third-line treatment or, to a lesser extent, as fourth-line treatment, whereas cabozantinib would mainly remain as a second-line treatment. After treatment with belzutifan, the clinical experts anticipated there would be no further approved treatment options in clinical practice in Canada.
In the LITESPARK-005 trial, approximately 13%, 43%, 43%, and 1% of patients received 1, 2, 3, or 4 prior lines of therapy, respectively. Exploratory subgroup results for OS and PFS by line of therapy (i.e., 1, 2, or 3 prior lines of therapy) were consistent with those for the overall trial population. Although some uncertainty remains regarding the impact of differences in prior lines of therapy between the trial and expected clinical practice on the generalizability of results, the clinical experts consulted by CDA-AMC felt it would be reasonable to generalize the trial results to anticipated clinical practice and did not raise concerns regarding the applicability of the LITESPARK-005 trial results in the Canadian clinical context.
Visual inspection of the KM curves by the CDA-AMC review team revealed that the PFS curves for the belzutifan and everolimus treatment arms crossed multiple times and did not separate until approximately 6 months. Although this suggests that the HRs, which were based on proportional hazards models, may not accurately reflect the treatment effect over time, they are likely a result of the variation in effects between the treatment and an active control during the early stages of treatment initiation. The clinical experts consulted by CDA-AMC suggested that belzutifan may have a longer duration of efficacy than everolimus, which may explain why the benefit of belzutifan is observed at time points beyond those corresponding to median PFS. The KM-estimated between-group differences in the probability of PFS at clinically relevant follow-up times were not affected by this limitation.
The primary analysis for PFS and ORR was assessed at the first interim analysis, which may potentially result in an overestimation of the treatment effect for belzutifan. However, 519 PFS events had occurred by the first interim analysis, which constituted 82.9% of the total expected events for PFS at the final analysis. Moreover, the results for PFS and ORR at the first interim analysis were, overall, consistent with those measured at the final analysis. Thus, the review team determined that the risk of overestimation was small. For the assessment of PFS by BICR per RECIST 1.1, a larger proportion of patients in the everolimus group (18.3%) were censored due to the initiation of new anticancer therapy before a PFS event than those in the belzutifan group (5.6%). However, the trial performed sensitivity analyses that counted the initiation of a new anticancer therapy as a PFS event. The results of these analyses were consistent with those of the primary analysis for the ITT population, which suggested that the between-group imbalances in patients starting new anticancer therapy had little impact on the results for PFS.
Key HRQoL outcomes in the LITESPARK-005 trial were measured with the EORTC QLQ-C30 and FKSI-DRS instruments. The interpretation of the results for HRQoL is limited by the lack of adjustments for multiple testing, the low completion rates at later time points, and the imbalances in missing data between the 2 groups.
The LITESPARK-005 trial assessed the safety and efficacy of belzutifan compared with everolimus. Although once considered a standard treatment for pretreated, advanced RCC, the clinical experts consulted by CDA-AMC agreed that everolimus is a less relevant comparator for later-line, advanced RCC than axitinib or cabozantinib and is rarely used in Canadian clinical practice. Thus, the stand-alone results of the trial may not provide a full assessment of the efficacy and safety of belzutifan compared to existing treatments for advanced RCC in Canadian clinical practice. The clinical experts consulted by CDA-AMC suggested that everolimus is similar to axitinib in terms of efficacy, based on how these drugs perform in clinical practice. Apart from AEs related to the mechanism of action of belzutifan (i.e., anemia and hypoxia), the clinical experts did not suggest any additional safety concerns with belzutifan compared to other key drug comparators for RCC in Canada. Of note, the pan-Canadian Oncology Drug Review Expert Review Committee (pERC) previously discussed that everolimus is similar to axitinib in terms of efficacy and safety as a part of the recommendation for funding cabozantinib for the treatment of advanced RCC.20 The clinical experts noted there is currently a lack of head-to-head randomized controlled trials (RCTs) and there is no evidence to suggest that belzutifan performs substantially better than cabozantinib. According to the clinical experts consulted by CDA-AMC, treatment options for third-line or later-line settings for advanced RCC remain limited, and there is a lack of standard treatment options for advanced RCC in the fourth line. The experts emphasized that a treatment’s ability to delay progression and achieve response in these treatment settings would be highly valued in clinical practice.
Based on sponsor-submitted studies assessing PFS as a surrogate for OS, it remains unclear whether PFS could be interpreted as a surrogate outcome for OS for the target population of this review.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.28,29
With the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessments for PFS, OS, ORR, SAEs, and discontinuation due to AEs were set according to the presence or absence of an important effect, based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of evidence assessment for the EORTC QLQ-C30 GHS/QoL score was sourced from the literature.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
clinical outcomes (PFS, OS, ORR)
HRQoL (EORTC QLQ-C30 GHS/QoL)
harms (SAEs, discontinuation due to AEs).
Table 2: Summary of Findings for Belzutifan Versus Everolimus in Patients With Advanced RCC
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Everolimus | Belzutifan | Difference | |||||
PFS in the ITT population, interim analysis 1 (data cut-off: November 1, 2022) | |||||||
Probability of being alive and progression-free at 12 months Follow-up (median)
| 746 (1 RCT) | NR | 171 per 1,000 | ███ ███ █████ █████ ██ ███ ███ ██████ | ███ ████ ███ █████████ ██ ███ ████ ███ ██████ | Higha,b | Belzutifan results in a clinically important increase in the probability of patients being alive and progression-free at 12 months compared with everolimus. |
Probability of being alive and progression-free at 18 months Follow-up (median)
| 746 (1 RCT) | NR | 83 per 1,000 | ███ ███ ██████████ ██ ███ ███ ██████ | ███ ████ ███ █████████ ██ ███ ████ ███ ██████ | Higha,b | Belzutifan results in a clinically important increase in the probability of patients being alive and progression-free at 18 months compared with everolimus. |
Probability of being alive and progression-free at 24 months Follow-up (median)
| 746 (1 RCT) | NR | 0 per 1,000 | ███ ███ ██████████ ██ ███ ███ ██████ | ███ ████ ███ █████ ████ | Moderatea,c | Belzutifan likely results in a clinically important increase in the probability of patients being alive and progression-free at 24 months compared with everolimus. |
OS in the ITT population, final analysis (data cut-off: April 15, 2024) | |||||||
Probability of being alive at 18 months Follow-up (median)
| 746 (1 RCT) | NR | 507 per 1,000 | ███ ███ ██████████ ██ ███ ███ ██████ | ██ ████ ███ █████ ████ █████ ██ ███ ████ ███ ██████ | Moderated | Belzutifan likely results in little to no difference in the probability of patients being alive at 18 months compared with everolimus. |
Probability of being alive at 36 months Follow-up (median)
| 746 (1 RCT) | NR | 280 per 1,000 | ███ ███ ██████████ ██ ███ ███ ██████ | ██ ████ ███ █████ ████ █████ ██ ███ ████ ███ ██████ | Moderated | Belzutifan likely results in little to no clinically important difference in the probability of patients being alive 36 months compared with everolimus. |
ORR in the ITT population, interim analysis 1 (data cut-off: November 1, 2022) | |||||||
Proportion of patients with CR or PR Follow-up (median)
| 746 (1 RCT) | NR | 35 per 1,000 | ███ ███ █████ █████ ██ ███ ███ ██████| | ███ ████ ███ ██████████ ██ ███ ████ ███ ██████| | Higha,f | Belzutifan results in a clinically important increase in ORR compared with everolimus. |
HRQoL in the PRO FAS population, interim analysis 1 (data cut-off: November 1, 2022) | |||||||
LS mean change from baseline in EORTC QLQ-C30 (GHS/QoL scale) at week 17 Follow-up (median)
| 724 (1 RCT) | NR | −6.13 | ████ ███████ ██ █████ | ██████████ ██ █████ | Lowa,g,h | Belzutifan may result in little to no clinically important difference in LS mean change from baseline in EORTC QLQ-C30 (GHS/QoL scale) at week 17 compared with everolimus. |
Harms in the APaT population, final analysis (data cut-off: April 15, 2024) | |||||||
Proportion of patients with SAEs Follow-up (median)
| 732 (1 RCT) | NR | 386 per 1,000 | ███ ███ █████ | ██ ████ ███ █████ ███ ████ ██ ███ ████ ███ ██████ | Moderateg,i | Belzutifan likely results in little to no difference in the proportion of patients with ≥ 1 SAEs compared with everolimus. |
Proportion of patients with AEs leading to discontinuation Follow-up (median):
| 732 (1 RCT) | NR | 153 per 1,000 | ██ ███ █████ | ██ ████ ███ █████ ████ ████ ██ ██ ████ ███ ██████ | Moderateg,j | Belzutifan likely results in a clinically important decrease in the proportion of patients with ≥ 1 AEs leading to discontinuation compared with everolimus. |
AE = adverse event; APaT = all patients as treated; CDA-AMC = Canada's Drug Agency; CI = confidence interval; CR = complete response; EORTC QLQ C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; GHS = global health status; GRADE = Grading of Recommendations, Assessment, Development and Evaluation; HRQoL = health-related quality of life; ITT = intention to treat; LS = least squares; MID = minimal important difference; NC = not calculated; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; PRO = patient-reported outcome; QoL = quality of life; RCC = renal cell carcinoma; RCT = randomized controlled trial; SAE = serious adverse event.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The between-group differences of PFS rates, ORR rate, LS mean change in EORTC QLQ-C30 (GHS/QoL) score from baseline to week 17, proportion of patients with SAEs, and proportion of patients with AEs leading to discontinuation were requested from the sponsor.
aValues for the GRADE assessment of this outcome were sourced from an interim analysis. Although limitations regarding internal validity were identified in the interim analyses, certainty was not rated down for risk of bias, because this limitation was determined to have a small or no impact on the results.
bNot rated down for imprecision. There is no established between-group MID for PFS rates at 12 months or 18 months, but the clinical experts consulted by CDA-AMC considered that a 10% difference between groups in the probability of patients who were alive and progression-free at this time point could be considered a threshold of clinical importance. The point estimate for the between-group difference suggested a clinically important difference between the 2 groups, and the CDA-AMC review team determined that the lower bound of the 95% CI did not appreciably cross the 10% threshold.
cRated down 1 level for serious imprecision. There is no established between-group MID for PFS rates at 24 months, but the clinical experts consulted by CDA-AMC considered that a 10% difference between groups in the probability of patients who were alive and progression-free at this time point could be considered a threshold of clinical importance. A 95% CI could not be calculated for the between-group difference in the probability of patients who were alive and progression-free at 24 months. This was because the 95% CI corresponding to the PFS rate of the everolimus group was not evaluable at 24 months, as no patient in the everolimus group survived to this time point.
dRated down 1 level for serious imprecision. There is no established between-group MID for OS rates at 18 months or 36 months, but the clinical experts consulted by CDA-AMC considered that a 5% difference between groups in the probability of patients who were alive at this time point could be considered a threshold of clinical importance. The point estimate and lower bound of the 95% CI for the between-group difference suggested no clinically important difference between the 2 groups, whereas the upper bound of the 95% CI suggested a clinically important difference for belzutifan versus everolimus based on a 5% threshold.
eBased on the procedure for testing for multiplicity in the trial, the ultimate alpha used for testing ORR was determined to be 0.001. Thus, 99.9% CIs were used for the GRADE assessment of ORR.
fNot rated down for imprecision based on a threshold of 10%. There is no established between-group MID for ORR, but the clinical experts consulted by CDA-AMC considered that a 10% difference between groups in the proportion of patients with an ORR could be considered a threshold of clinical importance. The point estimate and both upper and lower bounds of the 95% CI for the between-group difference suggested a clinically important difference between the belzutifan and everolimus groups based on a 10% threshold.
gThe statistical testing for this end point was not adjusted for multiplicity in the LITESPARK-005 trial and should be considered to be supportive evidence.
hNot rated down for imprecision. Although no literature was identified that estimated MIDs specifically in patients with advanced RCC, a change of 10 points in the EORTC QLQ-C30 score and the summary score is conventionally considered to be an MID.26 The point estimate and both upper and lower bounds of the 95% CI for the between-group difference did not suggest a clinically important difference between the belzutifan and everolimus groups based on a 10 point threshold. Rated down 2 levels for risk of bias due to reporting of outcome was affected by the open-label study design and the low completion rates of the assessment at week 17.
iRated down 1 level for serious imprecision. There is no established between-group MID for the proportion of patients with SAEs, but the clinical experts consulted by CDA-AMC considered that a 10% difference between groups in the proportion of patients with SAEs could be considered a threshold of clinical importance. The point estimate and lower bound of the 95% CI for the between-group difference suggested little to no difference between the 2 groups, whereas the upper bound of the 95% CI suggested important harm for belzutifan versus everolimus based on a 10% threshold.
jRated down 1 level for serious imprecision. There is no established between-group MID for the proportion of patients with AEs leading to discontinuation, but the clinical experts consulted by CDA-AMC considered that a 5% difference between groups in the proportion of patients with AEs leading to discontinuation could be considered a threshold of clinical importance. Although the 95% CI did not appreciably cross the 5% threshold for a clinically important effect, the effect estimate is based on few events.
Sources: LITESPARK-005 Clinical Study Report interim analysis 126 LITESPARK-005 Statistical Report final analysis27 and addendum.30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Long-Term Extension Studies
No long-term extension studies in the evidence from the systematic review were included in this submission.
Indirect Comparisons
In the absence of direct evidence comparing belzutifan to cabozantinib for the treatment of advanced RCC, the sponsor conducted a feasibility assessment for indirect treatment comparisons (ITCs) of belzutifan and cabozantinib in patients with advanced RCC after prior treatment with an immune checkpoint inhibitor and an antiangiogenic therapy. The assessment was conducted using data from the pivotal trials: LITESPARK-005 (belzutifan), and METEOR (cabozantinib). Of note, the METEOR trial had subgroup data available for patients who had received prior treatment with an IO and VEGF-TKI.
Both trials were phase III, open-label RCTs that evaluated patients with advanced clear cell RCC and a minimum KPS score of 70, and both had everolimus as a comparator to their respective interventions. However, the trials significantly differed in terms of ECOG PS and prior therapy. Heterogeneity between the trials in terms of ECOG PS, prior lines of therapy, and type of prior therapy would have the potential to introduce bias into indirect comparisons if differences in these factors were not accounted for. Moreover, the subgroup of patients from the METEOR trial was noted to have a small sample size (n = 32 patients) and was expected to introduce significant uncertainty into the analyses.
To minimize bias from heterogeneity in patient baseline characteristics, the feasibility of conducting alternative methods of ITCs was also assessed, which included unadjusted (i.e., Bucher method) and adjusted (i.e., matching-adjusted indirect comparison [MAIC]) approaches. The Bucher method was deemed to be infeasible because of compromised randomization in the METEOR trial’s subgroup of patients previously treated with IOs and VEGF-TKIs, whereas MAICs were infeasible because of the lack of reporting of key effect modifiers (i.e., IMDC risk classification and number of prior lines of therapy) for this subgroup. The small sample size of the subgroup was also a key limitation for both approaches, given its potential to introduce uncertainty in the analysis. Based on the results of the feasibility assessment, the CDA-AMC review team agreed that neither network meta-analyses (i.e., Bucher ITC) nor alternative methods of MAICs (i.e., anchored MAICs) were likely to provide unbiased treatment effect estimates for the comparison of belzutifan and cabozantinib for the treatment of adult patients with RCC.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the evidence from the systematic review were included in this submission.
Conclusions
According to patients with advanced RCC and clinicians, there remains an unmet need for later-line treatments with novel mechanisms of action to improve survival, response, and HRQoL. One phase III, open-label, active-controlled RCT was included in this review. The LITESPARK-005 trial evaluated the efficacy and safety of belzutifan compared to everolimus in patients with advanced RCC following treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The LITESPARK-005 trial met the predefined success criterion for superiority based on PFS, 1 of the dual primary outcomes, and on ORR, the key secondary end point. With the GRADE approach, results demonstrated that, compared to everolimus, belzutifan demonstrates a clinically important benefit for PFS and ORR. However, belzutifan likely demonstrates little to no difference in OS, the other primary outcome, compared to everolimus. Although belzutifan may result in little to no difference in HRQoL outcomes, the interpretation of these results is limited by the open-label design of the trial, the low completion rates at week 17, and an imbalance of attrition between the belzutifan and everolimus treatment groups. Overall, no new safety signals were observed with belzutifan in the LITESPARK-005 trial. Compared to everolimus, belzutifan likely results in a clinically important decrease in AEs leading to treatment discontinuation but little to no difference in the incidence of SAEs. There was a lack of studies directly comparing the efficacy and safety of belzutifan to currently available treatment options, especially axitinib and cabozantinib. The clinical experts consulted by CDA-AMC suggested that everolimus may have similar efficacy as that of axitinib based on how these drugs perform in clinical practice. They also noted that there is currently no evidence to suggest that belzutifan performs substantially better than cabozantinib.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of belzutifan, 120 mg administered orally, in the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Kidney cancer is the eighth most common malignancy in Canada. In 2024, the incidence of kidney cancer was estimated to be 9,000 cases (5,900 in males and 3,100 in females) in Canada, and the mortality rate was estimated to be 3.9 per 100,000 people (5.7 per 100,000 males and 2.4 per 100,000 females) .1,2 RCC accounts for approximately 90% of all kidney cancers.3 RCC is classified into various histologic subtypes, with the clear cell subtype being the most common, representing approximately 75% of all RCC cases, and the remaining consisting of subtypes such as papillary RCC.3,4 The cancer stage at diagnosis determines the prognosis. The 5-year relative survival rate for localized RCC is 93%, which drops to 18% for patients with distant disease.4 Approximately 20% to 40% of patients with localized primary RCC will develop metastatic disease.5
At presentation, approximately 25% of patients with RCC either have distant metastases or advanced locoregional disease. Many patients with localized disease are asymptomatic and are diagnosed incidentally. Patients with RCC can experience a wide range of symptoms, although many patients show no symptoms until the disease becomes advanced.6 In patients who are not diagnosed incidentally, symptoms and signs are related to the invasion site or distant mеtаstаѕis.6 Flank pain, hematuria, and abdominal renal mass are symptoms that strongly suggest locally advanced RCC.6 Patients with RCС can also present with or subsequently develop systemic symptoms and paraneoplastic syndromes, such as anemia, hepatic dysfunction, fever, hypercalcemia, cachexia, erythrocytosis, secondary amyloidosis, thrombocytosis, and polymyalgia rheumatica.6
Some of the RCC risk factors include smoking, hypertension, obesity, acquired cystic disease of the kidney and chronic kidney disease, occupational exposure to toxic compounds, prolonged ingestion of analgesics, genetic factors, cytotoxic chemotherapy, chronic hepatitis C infection, sickle cell disease, and kidney stones.32
The natural clinical course of RCC and the prognostic models used to assess individual risk varies among patients.33 The IMDC scoring system is based on 6 risk factors: a KPS score of less than 80%, a hemoglobin level of less than the lower limit of normal, a time from initial diagnosis to initiation of treatment of less than 1 year, a corrected serum calcium level of more than the upper limit of normal, a platelet count of more than the upper limit of normal, and an absolute neutrophil count of more than upper limit of normal. The risk category is favourable for those with a risk factor of 0, intermediate for those with a risk factor of 1 or 2, and unfavourable (poor) for those with a risk factor of 3 to 6. These risk categories can be applied for prognosis estimations and treatment decisions in first-line therapy and beyond.33
Patients with suggestive signs and symptoms of possible RCC must undergo CT, ultrasonography, or MRI evaluations for the detection of a mass. If systemic therapy is indicated (i.e., for metastatic disease or locally advanced unresectable disease), histological confirmation of RCC is mandatory before commencing systemic therapy.3 In the patients with localized disease, tissue obtained from nephrectomy or partial nephrectomy is used for diagnosis. For patients with disease in whom cytoreductive nephrectomy is not contemplated, biopsy of a metastasis or, as noted by the clinical experts, biopsy of the primary tumour establishes the diagnosis.6
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
The clinical experts consulted by CDA-AMC indicated that the goals of treatment for advanced RCC among patients include improvements in survival, response, and QoL. According to recent treatment guidelines and the CDA-AMC provisional funding algorithm for RCC, the options for first-line treatment for advanced RCC are guided by IMDC risk group classification status.3,7-9 The CDA-AMC provisional funding algorithm for RCC further classifies recommended treatment options according to prior use of pembrolizumab in the adjuvant setting and length of disease-free survival after completion of adjuvant immunotherapy.7 A summary of the CDA-AMC provisional funding algorithm for RCC is presented in Figure 1.
Regardless of IMDC risk category, the standard first-line treatment for patients with advanced RCC with de novo metastatic disease or a disease-free interval of at least 6 months after pembrolizumab treatment consists of either an IO drug in combination with a TKI or TKI monotherapy with either sunitinib or pazopanib.3,7-9 Recommended IO plus TKI combinations for first-line treatment of RCC include pembrolizumab-axitinib (the KEYNOTE-426 trial34), pembrolizumab-lenvatinib (the CLEAR trial35), and nivolumab-cabozantinib (the CheckMate 9ER trial36). Of these options, the clinical experts consulted by CDA-AMC noted that pembrolizumab-axitinib is the more common option in Canadian clinical practice. Among patients with a disease-free interval of less than 6 months after adjuvant pembrolizumab treatment, the first-line treatment options include TKI monotherapy with either sunitinib or pazopanib.7
Among patients in the IMDC intermediate risk or poor risk category with de novo metastatic disease or a disease-free interval of at least 6 months, nivolumab-ipilimumab is also a recommended first-line treatment option for patients with advanced RCC.3,7,8 Of note, treatment guidelines from the European Society for Medical Oncology3 and the NCCN8 recommend nivolumab-ipilimumab for all IMDC risk categories, but with a weaker level of evidence for the favourable risk category. However, nivolumab-ipilimumab is only funded in Canada for patients in the IMDC intermediate risk or poor risk categories.7 Among patients with a disease-free interval of less than 6 months after pembrolizumab treatment or a contraindication to IO drugs, TKI monotherapy with sunitinib or pazopanib would be the first-line treatment for patients with advanced RCC.7 Of note, the NCCN,8 European Society for Medical Oncology,3 and KCRNC9 guidelines also recommend cabozantinib as an alternative first-line option for patients in the intermediate risk or poor risk category with contraindications to IO drugs. However, at the time of this clinical report, this treatment is not yet funded for this indication in Canada.
The strategy for subsequent lines of treatment is contingent on the treatments administered in the first-line setting for advanced RCC.3,7-9 Patients who receive first-line IO-based therapy are eligible for TKI monotherapy (sunitinib, pazopanib, axitinib, or cabozantinib).3,7,8 The clinical experts consulted by CDA-AMC agreed that patients who receive pembrolizumab-axitinib as a first-line therapy receive cabozantinib monotherapy in the second-line setting, whereas patients who receive pembrolizumab-lenvatinib can receive monotherapy with cabozantinib or axitinib in the second-line setting. Funded second-line treatment options for patients who are naive to IO drugs (i.e., who received sunitinib or pazopanib as first-line treatment) include nivolumab or TKI monotherapy (axitinib or cabozantinib). Of note, nivolumab as a second-line treatment is included in the treatment algorithm for patients who received TKI monotherapy as first-line therapy.7 Everolimus is also considered for the treatment of advanced RCC after the failure of a VEGF-TKI (sunitinib or sorafenib).17,37 However, the clinical experts consulted by CDA-AMC agreed that everolimus has been superseded by newer or more effective therapies (e.g., cabozantinib and axitinib) and is currently not funded in most provinces after treatment with these monotherapies.10-16 Although available and occasionally used, everolimus is not included in the CDA-AMC provisional funding algorithm because it is no longer considered a standard of care in previously treated patients.
Funded third-line treatment options include nivolumab or TKI monotherapy with cabozantinib or axitinib. The clinical experts consulted by CDA-AMC noted that nivolumab is not commonly administered in the third-line setting for advanced RCC in Canada, citing that patients who have not received an IO drug as a first-line or second-line treatment would be unlikely to be prescribed an IO drug in later-line settings.
The clinical experts consulted by CDA-AMC suggested that belzutifan would likely be administered after treatment with cabozantinib in the treatment paradigm for RCC, not replace cabozantinib. Therefore, they anticipated that belzutifan’s place in therapy for RCC would be in the third-line (among patients who received TKI in combination with an IO drug as first-line treatment) or fourth-line (among patients who received IO drug in combination with an IO drug as first-line treatment) treatment setting. However, the clinical experts agreed that belzutifan may be offered instead of cabozantinib for patients who are ineligible for second-line cabozantinib due to underlying health conditions, clinical parameters, or safety concerns. The clinical experts consulted by CDA-AMC also suggested that although some clinicians may prefer to offer belzutifan as a second-line treatment (instead of axitinib) to patients who had received nivolumab-cabozantinib as a first-line treatment, other clinicians may choose to offer belzutifan after second-line treatment with axitinib to avoid the elimination of a line of treatment for advanced RCC. Of note, the clinical experts consulted by CDA-AMC suggested that nivolumab-cabozantinib is not commonly administered as a first-line treatment for advanced RCC in Canadian clinical practice.
Figure 1: Provisional Funding Algorithm Diagram for RCC
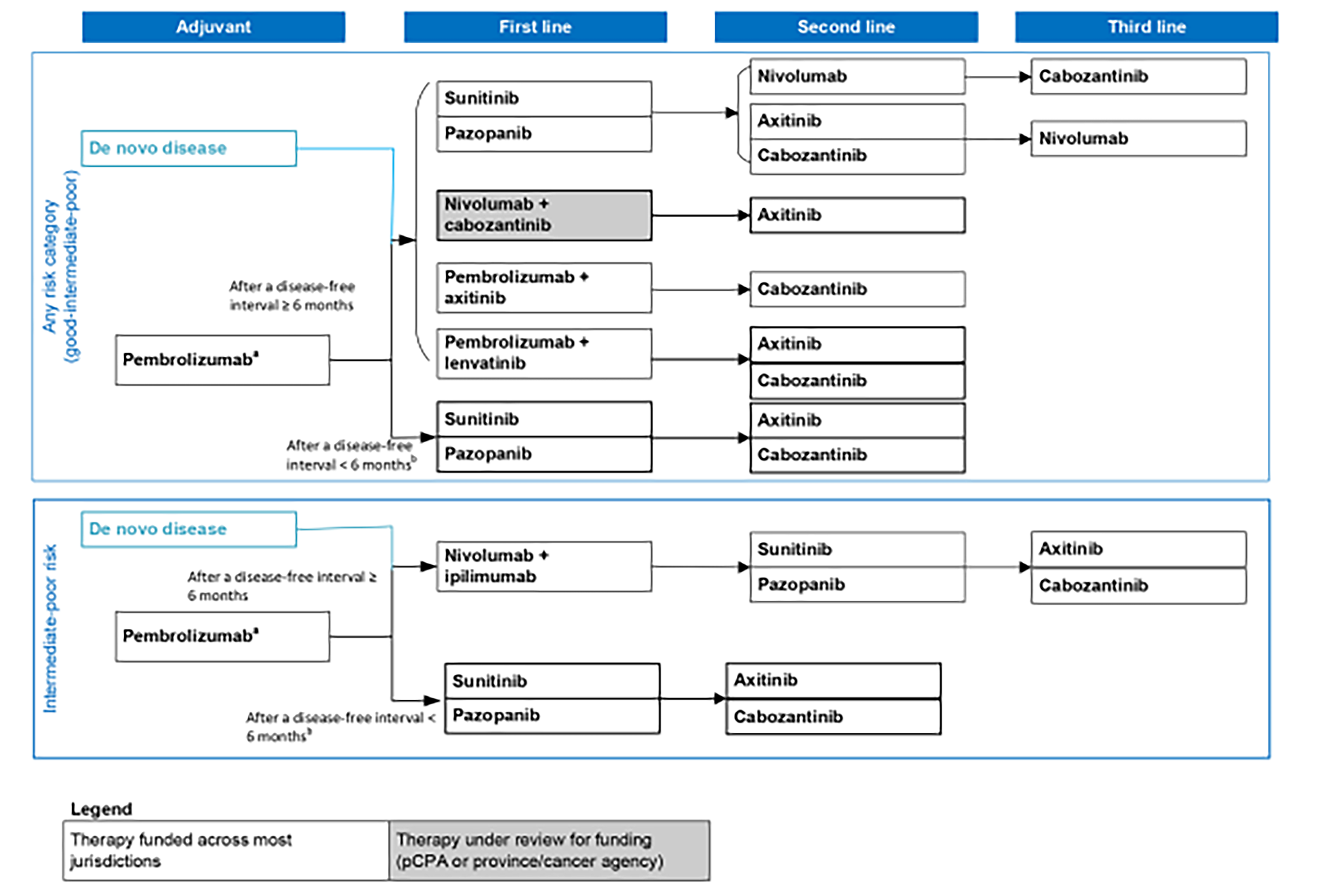
pCPA = pan-Canadian Pharmaceutical Alliance; RCC = renal cell carcinoma.
Note: The provisional funding algorithm (except for the adjuvant setting) applies to all renal cell carcinoma histologies.
aClear cell RCC with an intermediate to high risk or high risk of recurrence after nephrectomy or following nephrectomy and resection of metastatic lesions.
bPatients who experience disease progression less than 6 months after the completion of adjuvant pembrolizumab do not qualify for any further immunotherapy in the metastatic setting.
Source: Canada’s Drug Agency provisional funding algorithm for RCC.7
Drug Under Review
The key characteristics of belzutifan are summarized in Table 3 with other treatments available for the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI.
Belzutifan is supplied as 40 mg tablets for oral administration. The recommended dose is 120 mg (three 40 mg tablets) administered orally, once daily, with or without food.
Belzutifan has been previously reviewed by CDA-AMC, and a positive recommendation was issued on September 1, 2023, for the treatment of adult patients with von Hippel-Lindau disease who require therapy for associated nonmetastatic RCC, central nervous system hemangioblastomas, or nonmetastatic pancreatic neuroendocrine tumours, not requiring immediate surgery.
Belzutifan is an inhibitor of HIF-2 alpha, a transcription factor that plays a role in oxygen-sensing by regulating genes that promote adaptation to hypoxia. Under normal oxygen levels, HIF-2 alpha is targeted for ubiquitin-proteasomal degradation by the VHL protein. A lack of functional VHL protein results in stabilization and the accumulation of HIF-2 alpha. Upon stabilization, HIF-2 alpha translocates into the nucleus and interacts with HIF-1 beta to form a transcriptional complex that regulates the expression of downstream genes, including genes associated with cellular proliferation, angiogenesis, and tumour growth. Belzutifan binds to HIF-2 alpha and, in conditions of hypoxia or the impairment of VHL protein function, belzutifan blocks the HIF-2 alpha and HIF-1 beta interaction, leading to the reduced expression of HIF-2 alpha target genes.
The sponsor’s reimbursement request is the same as the proposed Health Canada indication: for the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI. A Notice of Compliance was issued on December 17, 2024.
Table 3: Key Characteristics of Belzutifan, Axitinib, Cabozantinib, and Everolimus
Characteristic | Belzutifan | Axitinib | Cabozantinib | Everolimus |
|---|---|---|---|---|
Mechanism of action | Inhibition of HIF-2 alpha. Belzutifan binds to HIF-2 alpha and, in conditions of hypoxia or the impairment of VHL protein function, belzutifan blocks the HIF-2 alpha and HIF-1 beta interaction, leading to the reduced expression of HIF-2 alpha target genes | Inhibition of the phosphorylation of VEGF receptor-2 in xenograft tumour vasculature that expresses the target in vivo and produces tumour growth delay, regression, and inhibited metastases in many experimental models of cancer | Inhibition of multiple receptor tyrosine kinases implicated in tumour growth and angiogenesis, pathologic bone remodelling, drug resistance, and metastatic progression of cancer | Inhibition of mTORC1, which reduces cell proliferation, glycolysis, and angiogenesis in solid tumours in vivo, both through direct antitumour cell activity and the inhibition of the tumour stromal compartment |
Indicationa under review | For the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI | For the treatment of patients with metastatic RCC of clear cell histology after failure of prior systemic therapy with either a cytokine or the VEGFR-TKI sunitinib In combination with pembrolizumab, is indicated for the treatment of adult patients with advanced or metastatic RCC with no prior systemic therapy for metastatic RCC | For the treatment of advanced RCC in treatment-naive adults with intermediate risk or poor risk, and in adult patients who have received prior VEGF-targeted therapy. In combination with nivolumab, is indicated for the first-line treatment of adult patients with advanced (not amenable to curative surgery or radiation therapy) or metastatic RCC | For the treatment of patients with metastatic RCC of clear cell morphology, after failure of initial treatment with either of the VEGF receptor TKIs sunitinib or sorafenib |
Route of administration | Oral | Oral | Oral | Oral |
Recommended dose | 120 mg (three 40 mg tablets), orally, once daily | Starting dose of 5 mg twice daily | As a single drug, 60 mg once daily As in combination with nivolumab, 40 mg once daily | 10 mg once daily |
Serious adverse effects or safety issues | Has not been studied in patients with moderate or severe hepatic insufficiency, severe renal insufficiency, embryo-fetal toxicity, severe anemia, or severe hypoxia | Has not been studied in patients with severe hepatic impairment. Clinically significant adverse events include hypertension and hypertensive crisis, arterial thromboembolism, venous thromboembolism, hemorrhage (including gastrointestinal, cerebral, and respiratory tract), gastrointestinal perforation, gastrointestinal fistulas, reversible posterior leukoencephalopathy syndrome, and congestive heart failure and/or cardiomyopathy. | Has not been studied in patients with cardiac impairment, severe renal impairment, or severe hepatic impairment. Clinically significant adverse events include thromboembolism, hypertension and hypertensive crisis, gastrointestinal perforations and fistulas, hemorrhage, hepatotoxicity, reversible posterior leukoencephalopathy syndrome, and wound complications. | Noninfectious pneumonitis, infections, renal failure |
Other | — | Axitinib should be prescribed by a qualified health care professional who is experienced in the use of antineoplastic therapy. | Treatment with cabozantinib should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. | Everolimus should be prescribed by a qualified health care professional who is experienced in the use of antineoplastic therapy |
HIF-1 = hypoxia-inducible factor 1; HIF-2 = hypoxia-inducible factor 2; RCC = renal cell carcinoma; TKI = tyrosine kinase inhibitor; VEGF = vascular endothelial growth factor.
aHealth Canada–approved indication.
Sources: Belzutifan product monograph,38 axitinib product monograph,39 cabozantinib product monograph,40 everolimus product monograph.37
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 1 submission of patient group input from KCC. KCC is a national community of patients, caregivers, and health professionals who advocate and support patients with kidney cancer. KCC gathered the information through an international online survey, in affiliation with the International Kidney Cancer Coalition, in 2022. The respondents were 2,213 patients and caregivers from 39 countries, including 139 respondents (111 patients and 28 caregivers) from Canada. Further, KCC conducted a survey in December 2024, and gathered information from 2 patients and 1 caregiver who had experience with belzutifan. The 2 patients also provided consent to have a telephone conversation.
KCC noted that approximately one-quarter of patient respondents reported that their current treatments were difficult to tolerate. KCC added that, based on the International Kidney Cancer Coalition survey report for Canada, 63% of respondents (n = 79) reported experiencing no barriers to treatment. The most commonly experienced barriers reported by respondents in Canada were wait time to treatment (16%), lack of access to specialty doctors locally (10%), lack of access to up-to-date treatment or equipment (9%), cost of treatment (7%), and lack of personal support (5%). In Canada, the percentage of respondents who agreed or strongly agreed that they understood various treatment options was 77% for surgical options, 67% for active surveillance, 23% for adjuvant therapy (treatment aimed at reducing the chance of recurrence after surgery), and 35% for ablative therapy options (cryoablation or radiofrequency ablation).
KCC explained that there is a general need for more effective therapies with manageable side effects, as well as better predictive and prognostic biomarkers to guide treatment and enhanced early detection of disease. Additionally, new treatments need to address treatment resistance, as not all patients respond to currently available treatments, and patients who do respond to currently funded treatments often become resistant to therapy after some time.
Based on the patient group input, unmet needs include the following:
treatments that provide a cure, durable remission, disease stability, long-term DOR, improved tolerability, improved disease-specific QoL, and innovative medicines with new mechanisms of action
biomarkers to reliably guide treatment selection
clinical trials to provide access to better treatments
improved and informed shared decision-making to ensure the discussion of all multidisciplinary treatment options, including surgery, radiation, ablation, and palliative care
evidence and guidance supporting best sequencing strategies, including postadjuvant therapy
increased access to genetic screening for hereditary syndromes, per guidelines
reduced barriers to quality care, including reduced financial toxicity
increased research to meet the needs of underserved populations and populations with a higher incidence of RCC
improved psychosocial support for patients and their caregivers
survivorship care plans to manage surveillance strategies, late-term effects, and QoL
clinical trials for specific rare-variant RCCs, with the goal of evidence-based treatment guidelines for specific variants
access to therapies for rare-variant RCCs, many of which qualify as rare cancers.
In December 2024, KCC also conducted a survey through Canadian physician investigators who had patients enrolled in the LITESPARK-005 trial. There were 3 respondents, including 2 patients and 1 caregiver, who had experience with belzutifan.
Based on the report, the first of the 2 patients reported that they took 120 mg of belzutifan orally, once daily, for their kidney cancer and after 3 months was told that the treatment was working very well. They had been on belzutifan for 4 years and was still on trial. This patient rated belzutifan as extremely effective. This patient was asked to rate their QoL while taking belzutifan on a scale of 1 to 5, with 1 indicating low to seriously impacted and 5 indicating high to normal living. The patient rated their QoL while on belzutifan as a 4. Regarding side effects, the most obvious appeared to be neuropathy, which started before belzutifan but, at the time of interview, had become worse, per the patient respondent. This patient was also experiencing a rash and a tightening in chest, shallow breathing, and elevated blood pressure. The patient reported that all the side effects were manageable, and they could still do routine activities, such as going grocery shopping and riding their bike. Their tumour remained barely detectable, and their disease was reported as stable. This patient had concerns about their ability to continue to receive belzutifan.
According to KCC, the second patient, who was diagnosed with von Hippel-Lindau disease, had been participating in a belzutifan trial for 2 years and 10 months. This patient rated belzutifan as extremely effective. In terms of side effects, the patient reported that they had some fatigue, low energy, anemia, and dizziness, but rated them as tolerable. After a lengthy journey of dealing with von Hippel-Lindau disease and kidney cancer that included surgeries and multiple failed treatments, this patient was extremely satisfied with belzutifan and was still working full-time.
KCC reported the third respondent was the caregiver of a patient with kidney cancer who had been on belzutifan for over 2 years through participation in a clinical trial. The caregiver rated belzutifan as extremely effective, the side effects as very tolerable, and the QoL as high to normal living. The caregiver reported low energy level as a side effect that was particularly difficult for the patient to tolerate. The caregiver was also asked about the specific side effects experienced with belzutifan and rate them from 1 as “completely intolerable” to 5 as “very tolerable.” The caregiver rated fatigue, lack of energy, and anemia as a 3 and nausea as a 5.
KCC added that not all patients respond to currently available treatments, and often patients who do respond to currently funded treatments become resistant to therapy after some time. There is a need for novel therapies that target different pathways in advanced kidney cancer.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of RCC.
Unmet Needs
The clinical experts consulted by CDA-AMC agreed that there is an unmet need for the availability of treatments with novel mechanisms of action for patients with advanced RCC in the third line and fourth line. The clinical experts noted the need for treatments to improve OS, PFS, and response outcomes in these patients. One clinical expert emphasized the importance of improving treatment options for heavily pretreated RCC, as disease progression occurs in almost all patients receiving treatment. The clinical experts noted that the available treatment options for heavily pretreated RCC are associated with side effects that are difficult to manage and negatively affect QoL. One clinical expert highlighted that patients with autoimmune diseases may not be able to receive immunotherapy, which further underlines the need for additional treatment options for advanced RCC.
Place in Therapy
Belzutifan targets the underlying disease process of RCC. According to the clinical experts consulted by CDA-AMC, belzutifan would be administered as a later line of therapy (e.g., in the third-line or fourth-line treatment setting) for patients with advanced RCC after prior treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. Moreover, the experts noted that belzutifan should be administered as monotherapy after disease progression or in the case of intolerance to other approved drugs for this patient population. One clinical expert stated that they expected belzutifan to cause a shift in the treatment paradigm for advanced RCC because it has a new mechanism of action, compared to existing treatments in this disease area, and it provides an additional treatment option in third-line and fourth-line settings. However, 1 clinical expert noted that the magnitude of this shift remains uncertain due to a relatively small proportion of patients who are eligible for third-line and fourth-line treatment for advanced RCC in Canada.
Patient Population
According to clinical experts consulted by CDA-AMC, belzutifan should be administered as a third-line or fourth-line therapy to patients with advanced RCC. They noted that an estimated 20% of patients with RCC receive treatment in the third-line setting, and 7% receive treatment in the fourth-line setting. The experts felt that would be reasonable for belzutifan to be available for patients with early relapses (i.e., within 6 months of completion of treatment) after treatment with adjuvant pembrolizumab or subsequent progression on a TKI (these patients were not included in the LITESPARK-005 trial). However, they noted that belzutifan should only be administered to patients with clear cell component histology. The clinical experts stated that the diagnosis of advanced RCC is overseen by a medical oncologist or a general practitioner specializing in oncology, although the diagnosis would have been established by the time a patient receives third-line or fourth-line treatment for RCC. One expert noted that belzutifan may be chosen over another VEGF inhibitor if a patient had experienced toxicity related to VEGF therapies in a prior line. One expert noted that patients deemed eligible for belzutifan would be identified based on disease progression (shown on imaging and blood tests) or intolerance to existing treatment options. The experts indicated that it is not possible to identify which subgroup of patients would receive more or less treatment benefit from belzutifan.
The pivotal LITESPARK-005 trial enrolled adult patients with advanced RCC whose disease had progressed after treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI and had a good performance status, indicated by a KPS score of 70% or more. Moreover, the trial listed several exclusion criteria, including (but not limited to) the presence of active brain metastases, the presence of hypoxia or requiring supplemental oxygen, and select comorbidities. Although the clinical experts agreed that the patient population of the LITESPARK-005 trial was more restrictive than patients treated in clinical practice, they noted that patients receiving treatment in clinical practice would be expected to have a good performance status and no hypoxia, due to the safety profile of belzutifan.
Assessing the Response to Treatment
One clinical expert indicated that the assessment of response to treatment for RCC consisted of CT scans performed every 3 months. One clinical expert stated that clinically meaningful responses to treatment for RCC included radiologic response, symptom status, stable disease, and adequate tolerance of the drug. The expert added that the consideration of these parameters for a clinically meaningful response is likely to be consistent across physicians in Canada.
Discontinuing Treatment
According to the clinical experts consulted by CDA-AMC, disease progression and unmanageable toxicities are the main factors in deciding to discontinue treatment with belzutifan. Of note, these criteria align with the discontinuation criteria for belzutifan in the pivotal LITESPARK-005 trial.
Prescribing Considerations
Both clinical experts consulted by CDA-AMC agreed that belzutifan should be prescribed by a medical oncologist with experience in managing patients with advanced RCC. Given that belzutifan is administered in the form of oral pills, it can be administered in a patient’s home. One expert noted that the management of AEs related to belzutifan consists of dose interruption and dose reduction, and that transfusions and dose reductions are preferred for the management of anemia and hypoxia related to belzutifan. That expert also noted that management of AEs would be similar to that used in the LITESPARK-005 trial. Although the expert did not note variations in the management of AEs related to belzutifan across jurisdictions in Canada, there may be variations in the coverage of home oxygen and erythropoietin, if required.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
CDA-AMC received 2 clinician inputs: 1 from the OH (CCO) Genitourinary DAC, with contributions from 2 clinicians; and 1 from the KCRNC, with contributions from 10 clinicians.
The OH (CCO) DACs provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. The KCRNC is a virtual and inclusive national network of clinicians and researchers who treat kidney cancer in Canada. The KCRNC is a federally registered not-for-profit organization with a commitment to enhance the knowledge of kidney cancer and its treatment.
Both clinician groups agreed that the first-line systemic treatment for advanced kidney cancer considers the IMDC risk groups. For all IMDC risk groups, treatment includes combination regimens, such as pembrolizumab-axitinib, pembrolizumab-lenvatinib, and nivolumab-cabozantinib. Alternatives to these treatments include sunitinib and pazopanib. For patients in intermediate risk or poor risk groups, treatment includes nivolumab-ipilimumab, and alternatives include sunitinib and pazopanib. Subsequent-line options include therapies that the patient has not already received in the advanced RCC setting, such as sunitinib, pazopanib, cabozantinib, axitinib, and nivolumab. The TKIs are mainly antiangiogenic and modify the biology of RCC. The PD-L1 checkpoint inhibitors leverage the immune system and allow it to target the cancer cells more effectively. RCC is known to be driven by angiogenesis and is thought to thrive by immune escape, to some degree. Thus, therapies that inhibit angiogenesis and activate the immune system would be of benefit.
Based on the clinician group inputs, treatment goals include an improvement in OS and PFS, a reduction in the size of metastatic lesions (i.e., ORR), and the control of symptoms, which improves QoL.
The clinician groups noted that the main gap in treatment is the resistance to currently available treatments; there is a need for novel drugs that target different pathways in advanced kidney cancer to address this gap. The KCRNC input added that there is a need for treatments that offer better disease control with better side effect profiles, which allows for more treatment individualization and provides an alternative for patients who develop toxicities to TKIs.
The clinician groups agreed that belzutifan will be used in the later-line setting, either as a second-line drug, if the patient’s disease progresses after a first-line combination regimen, or as a third-line drug. Patients with advanced RCC who have progressed on 1 or more immune checkpoint inhibitors and 1 or more TKI therapies would be best suited for treatment with belzutifan. The KCRNC input added that belzutifan could be considered in instances in which there are contraindications to immune checkpoint inhibitor therapy, such as renal transplant or severe autoimmune disease.
Based on the clinician group inputs, radiological response and symptom improvement are used to determine treatment response. Treatment response is assessed every 3 to 4 months with imaging, and the format of additional clinical assessments is determined per patient and oncologist discretion (i.e., in person or virtual). The outcomes assessed include response to treatment, shrinkage of disease on radiographic imaging, OS, and QoL. A clinically meaningful response to treatment includes a CR, a PR, or stable disease. Both clinician groups noted that disease progression or unacceptable toxicity should be considered when deciding to discontinue treatment with belzutifan. The KCRNC added that special consideration should be given to patients with anemia and hypoxia. Both inputs noted that it is appropriate to administer belzutifan in an outpatient clinic setting under the care of a medical oncologist, and the KCRNC input specified medical oncologists with specialization in the treatment of kidney cancer.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The comparator in the phase III LITESPARK-005 trial was everolimus, which is no longer routinely used in Canada and not considered a relevant comparator. Jurisdictions follow the CDA-AMC provisional funding algorithm for metastatic RCC from a funding perspective, and patients are eligible for 1 immune checkpoint inhibitor and 2 TKI therapies. Depending on the choice of first-line therapy, there may be 2 or 3 lines of therapy that are funded. Potential relevant comparators to belzutifan for the target patient population include cabozantinib and axitinib. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Are patients with an early relapse after adjuvant pembrolizumab (i.e., relapse within 6 months of completion) who later receive and progress on a TKI eligible for belzutifan? | Although a lack of data for these patients was noted, the clinical experts consulted by CDA-AMC agreed that belzutifan should be available to these patients due to limited options for treatment. |
Most patients in the LITESPARK-005 trial had clear cell RCC. Are other histological subtypes eligible for belzutifan? Note: Jurisdictions do not currently restrict metastatic RCC regimens by histology in the metastatic setting. | The clinical experts consulted by CDA-AMC agreed that patients with histologic subtypes other than clear cell RCC should not be eligible for belzutifan at this time. One expert noted that the rest of the treatment algorithm for RCC should remain histology agnostic. They also noted that FH-mutated RCC has increased HIF-1 alpha overexpression, which could be targeted by belzutifan. |
Should all IMDC risk categories be eligible? Are all IMDC risk categories expected to benefit similarly? | Both clinical experts consulted by CDA-AMC agreed that patients in any IMDC risk category should be eligible for belzutifan. The LITESPARK-005 trial enrolled patients in favourable risk (21.7%), intermediate risk (66.1%), and poor risk (12.2%) categories. |
Considerations for discontinuation of therapy | |
What are the discontinuation criteria for belzutifan? | Both clinical experts consulted by CDA-AMC agree that disease progression and unmanageable toxicity are key factors in the decision to discontinue treatment with belzutifan. |
Considerations for prescribing of therapy | |
The recommended dose of belzutifan is 120 mg (three 40 mg tablets) administered orally, once daily, with or without food. | Comment from the drug programs to inform pERC deliberations. |
Generalizability | |
Should patients who have received an PD-1 or PD-L1 inhibitor and a VEGF-TKI who are on alternate second-line or third-line therapies be switched to belzutifan, or should belzutifan be used as the next line of therapy? | The clinical experts consulted by CDA-AMC agreed that belzutifan should be reserved for the next line of therapy, citing a lack of current data to suggest that belzutifan performs substantially better than key comparators (e.g., cabozantinib and axitinib). |
Funding algorithm (oncology only) | |
An initiation of a rapid provisional funding algorithm is requested. | Comment from the drug programs to inform pERC deliberations. |
The drug may change the place in therapy of drugs reimbursed in subsequent lines. | Comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Belzutifan is provided as a 40 mg tablet (120 mg is the starting daily dose), provided in bottles of 90 tablets. | Comment from the drug programs to inform pERC deliberations. |
Dispensing will require a discussion of the reproductive risks to patients (all genders), contraception, and the avoidance of pregnancy during therapy and for at least 1 week after the last dose. Patients should be advised of the following:
Patients should be advised of these risks and the need for effective nonhormonal contraception. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
Confidential pCPA pricing exists for nivolumab, pembrolizumab, and many of the TKIs and generic everolimus. | Comment from the drug programs to inform pERC deliberations. |
Drug programs are concerned about the potential large budget impact, given the volume of metastatic RCC patients. Under what clinical circumstances would belzutifan be preferred over other funded options, if there is a choice? | One clinical expert noted that such clinical circumstances have not yet been defined and stated that belzutifan would likely be offered when other treatment options have been exhausted. Another expert noted that belzutifan provides access to another line of therapy for RCC, and that only 20% and 7% of patients, respectively, go on to receive third-line and fourth-line treatment. |
CDA-AMC = Canada’s Drug Agency; HIF-1 = hypoxia-inducible factor 1; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; pCPA = pan-Canadian Pharmaceutical Alliance; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; RCC = renal cell carcinoma; TKI = tyrosine kinase inhibitor; VEGF = vascular endothelial growth factor.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of belzutifan (40 mg tablet for oral administration) in the treatment of adult patients with advanced RCC after treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The focus will be placed on comparing belzutifan to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of belzutifan is presented in 4 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in the first section, using the GRADE approach, follows the critical appraisal of the evidence. Of note, no long-term extension studies, indirect evidence, or additional studies that were considered to address important gaps in the systematic review evidence were submitted by the sponsor.
Included Studies
Clinical evidence from the following source is included in the review and appraised in this document:
1 pivotal study identified in the systematic review.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included study is summarized in Table 5.
Table 5: Details of the Study Included in the Systematic Review
Detail | LITESPARK-005 |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, open-label, efficacy, safety, parallel-assignment, active-controlled intervention |
Locations | 147 sites in 23 countries (Brazil, Canada, Chile, Colombia, Czechia, Denmark, Finland, France, Germany, Hong Kong, Hungary, Italy, Japan, South Korea, Norway, Russia, Spain, Sweden, Taiwan, Türkiye, Ukraine, US, UK) |
Patient enrolment dates | Start date: February 27, 2020 End date: January 19, 2022 |
Randomized (N) | Total: N = 746 Belzutifan = 374 Everolimus = 372 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Belzutifan: 120 mg, orally, once daily until disease progression is radiographically documented per RECIST 1.1, unacceptable AEs, intercurrent illness that prevents the further administration of treatment, investigator’s decision to discontinue the participant, participant withdrawal of consent, pregnancy of the participant, or administrative reasons requiring the cessation of treatment. |
Comparator(s) | Everolimus: 10 mg, orally, once daily until disease progression is radiographically documented per RECIST 1.1, unacceptable AEs, intercurrent illness that prevents the further administration of treatment, investigator’s decision to discontinue the participant, participant withdrawal of consent, pregnancy of the participant, or administrative reasons requiring the cessation of treatment. |
Study duration | |
Screening phase | The 28 days before treatment randomization. |
Treatment phase | Treatment continued until disease progression radiographically documented per RECIST 1.1, unacceptable AEs, intercurrent illness that prevents the further administration of treatment, investigator’s decision to discontinue the participant, or administrative reasons requiring the cessation of treatment. |
Follow-up phase | Efficacy and survival follow-up: Week 9, then every 8 weeks (± 7 days) for the first 49 weeks, and then every 12 weeks (± 7 days) until 1 of the following occurs: BICR-verified disease progression, initiation of new anticancer treatment, withdrawal of consent, pregnancy, death, or end of study. Safety follow-up: AEs occurring up to 30 days (± 7 days) after the last dose of treatment or before the initiation of new anticancer treatment. Serious AEs occurring up to 90 days after the end of treatment or 30 days if the participant initiates new anticancer therapy. |
Outcomes | |
Primary end point | Progression-free survival and overall survival |
Secondary and exploratory end points | Secondary:
Exploratory and/or tertiary:
|
Publication status | |
Publications | Choueiri et al. (2024)41 Albiges et al. (2023)42 Albiges et al. (2024)43 Powles et al. (2024)46 Clinicaltrials.gov identifier: NCT0419575047 EudraCT number: 2019-003444-72E48 |
AE = adverse event; BICR = blinded independent committee review; CNS = central nervous system; DBP = diastolic blood pressure; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; HIF-2 = hypoxia-inducible factor 2; HRQoL = health-related quality of life; KPS = Karnofsky Performance Status; mAb = monoclonal antibody; PD = progressive disease; RCC = renal cell carcinoma; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SBP = systolic blood pressure.
Source: Details included in the table are from the Sponsor’s Summary of Clinical Evidence.31
One pivotal trial (LITESPARK-005) was included in the systematic review. LITESPARK-005 was designed as a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of belzutifan compared to everolimus in patients with advanced RCC after previous treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The primary objective of the LITESPARK-005 trial was to compare the efficacy of belzutifan and everolimus with respect to PFS and OS. The secondary objectives of the trial were to compare belzutifan to everolimus with respect to ORR, DOR, safety, and HRQoL. The primary end points of the LITESPARK-005 trial were PFS based on BICR per RECIST 1.1 and OS. The key secondary end point of the trial was ORR based on BICR per RECIST 1.1. Additional secondary end points of the trial were DOR based on BICR per RECIST 1.1, safety, TTD, and change from baseline in HRQoL. HRQoL was measured using the EORTC QLQ-C30, the FKSI-DRS, and the 5-Level EQ-5D visual analogue scale. For the purposes of this review, the statistical methods and results for EORTC QLQ-C30 and FKSI-DRS will be discussed herein.
The LITESPARK-005 trial enrolled patients with advanced RCC who had been previously treated with a PD-1 or PD-L1 inhibitor and a VEGF-TKI and was conducted at 147 sites in 23 countries (Brazil, Canada, Chile, Colombia, Czechia, Denmark, Finland, France, Germany, Hong Kong, Hungary, Italy, Japan, Norway, Russia, South Korea, Spain, Sweden, Taiwan, Türkiye, Ukraine, UK, and US). Of the 147 sites, 6 were based in Canada. A total of 746 patients were randomized in a 1:1 ratio to receive either belzutifan (n = 374) or everolimus (n = 372). Randomization was stratified according to IMDC prognostic score (0 versus 1 to 2 versus 3 to 6) and the number of prior VEGF- or VEGF receptor–targeted therapies for advanced RCC (1 versus 2 or 3).
The LITESPARK-005 trial consisted of a screening phase, treatment phase, and follow-up phase (Figure 2). During the screening phase, patients provided written consent for study participation and were screened for eligibility within the 28 days before randomization. During the treatment phase, randomized patients received either belzutifan or everolimus. Patients were evaluated radiologically at week 9, every 8 weeks thereafter for the first 49 weeks, and then every 12 weeks thereafter. Treatment was allowed to continue after radiographic progression of RCC per RECIST 1.1, as long as the investigator believed that the patient was still receiving clinical benefit from treatment and that the potential clinical benefit would outweigh the potential risk. Treatment beyond confirmed disease progression based on BICR was allowed but required consultation with and approval by the sponsor. Administration of the assigned intervention continued until documented disease progression verified by BICR, unacceptable toxicity, or a decision to stop treatment. The posttreatment follow-up period included a posttreatment safety follow-up visit that occurred 30 days after the discontinuation of the study intervention. Patients who discontinued study treatment for reasons other than BICR-verified disease progression continued with imaging assessments, per the protocol-defined schedule. After the verification of disease progression, all patients were followed for survival until death, withdrawal of consent, loss to follow-up, or until the end of the study.
Two interim analyses (data cut-off for first interim analysis: November 1, 2022;26 data cut-off for second interim analysis: June 13, 202325) were planned, in addition to the final analysis (data cut-off: April 15, 2024),27 in the LITESPARK-005 trial. This review is based on results from all 3 data cut-off points (refer to Results section). The LITESPARK-005 trial was funded by Merck Sharp and Dohme, a subsidiary of Merck.
Figure 2: LITESPARK-005 Clinical Trial Design
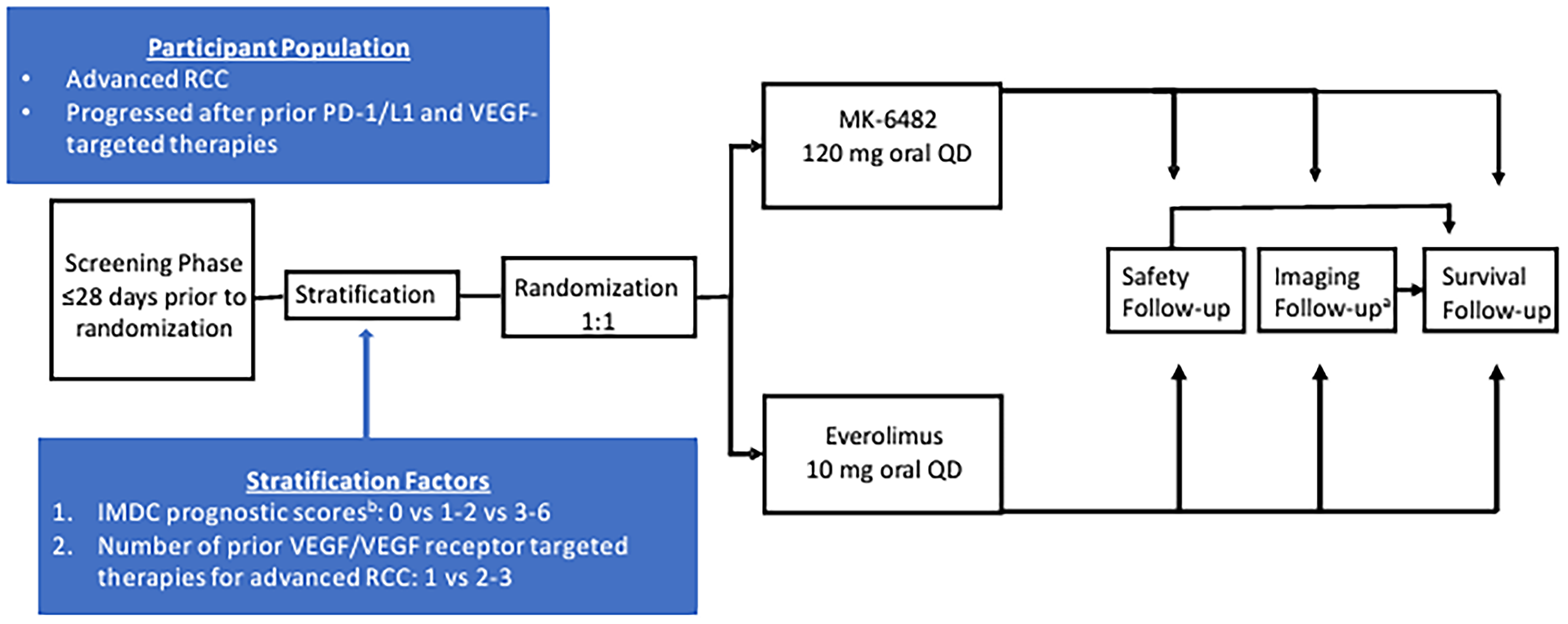
IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; MK-6482 = belzutifan; QD = once daily; RCC = renal cell carcinoma; VEGF = vascular endothelial growth factor; vs = versus.
aPatients who discontinued the study treatment for reasons other than disease progression verified by blinded independent central review continued with imaging assessments, per the protocol-defined schedule, until disease progression verified by blinded independent central review, initiation of a new anticancer treatment, pregnancy, death, withdrawal of consent, or end of the study, whichever occurred first.
Source: LITESPARK-005 Clinical Study Report interim analysis 2.25
Populations
Inclusion and Exclusion Criteria
Patients eligible to participate in the LITESPARK-005 trial were required to be at least 18 years of age and be diagnosed with unresectable, locally advanced, or metastatic clear RCC. Moreover, patients were required to have had disease progression on or after having received treatment with a PD-1 or PD-L1 inhibitor and antiangiogenic therapy (treatments could be administered in sequence or in combination). Eligible patients were also required to have adequate organ function, a KPS score of at least 70%, and no more than 3 prior systemic regimens for locally advanced or metastatic RCC. Patients who required intermittent or supplemental oxygen or had hypoxia, defined as a pulse oximeter reading of less than 92% at rest, were excluded. Moreover, patients who received prior treatment with an HIF-2 alpha inhibitor or a TORC1/PI3K/AKT inhibitor were excluded.
Interventions
In the LITESPARK-005 trial, patients were randomized to receive either belzutifan at a dose of 120 mg (in three 40 mg tablets) or everolimus at a dose of 10 mg. Dose reductions and interruptions for patients receiving belzutifan were allowed in the case of unacceptable toxicity or lower-grade toxicity at the discretion of the investigator, with the study permitting 2 dose reduction levels. Among patients receiving everolimus, dose adjustments were performed as described in the Afinitor (everolimus) package insert.49 Both treatments were administered orally, once daily, until disease progression documented per RECIST 1.1, unacceptable toxicity, intercurrent illness that prevents further administration of treatment, investigator’s decision to discontinue the participant, or administrative reasons requiring the cessation of treatment. Treatment was allowed to continue after radiographic progression of RCC per RECIST 1.1, as long as the investigator believed that the patient was still receiving clinical benefit from the treatment and that the potential clinical benefit would outweigh the potential risk. Treatment beyond confirmed disease progression based on BICR was allowed but required consultation with and approval by the sponsor. Treatment after disease progression according to the BICR per RECIST 1.1 occurred in 117 patients (79 patients in the belzutifan group and 38 patients in the everolimus group). Of note, 1 patient in the belzutifan group received treatment beyond progression without prior approval from the sponsor, which has been recorded as a protocol deviation. In addition, both treatments could be administered at home, although belzutifan was required to be administered during clinic visit days after the completion of blood collection on day 1 of week 1, week 3, and week 5 of the study.
During the screening and treatment phases of the trial, patients were prohibited from receiving additional treatment with other antineoplastic therapies or investigational drugs. Radiation therapy for disease control was also prohibited, although palliative radiation therapy for symptomatic lesions or to the brain was allowed after sponsor consultation. Throughout the study, patients were also prohibited from receiving treatment with systemic glucocorticoids for any purpose other than to modulate AE-related symptoms, strong inhibitors of CYP3A4, and live vaccines. Patients randomized to belzutifan were not prohibited from treatment with P-glycoprotein substrates or inhibitors during the treatment phase. There were no prohibited therapies during the survival follow-up phase. Patients should have received appropriate supportive care measures as deemed necessary by the investigator.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as any outcomes identified as important to this review by the clinical experts consulted and input from patient and clinician groups and public drug plans. The same considerations were used to select end points that were deemed to be most relevant to expert committee deliberations, and the list of end points was finalized in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important to expert committee deliberations were also assessed using GRADE.
Table 6: Summary of Outcomes From the LITESPARK-005 Trial
Outcome measure | Time point | End point status |
|---|---|---|
PFS | From randomization to the first documented disease progression based on RECIST 1.1 by BICR or death from any cause, whichever occurs first. Measured until data cut-off (April 15, 2024). | Primarya |
OS | From randomization to death from any cause. Measured until data cut-off (April 15, 2024). | Primarya |
ORR | Measured until data cut-off (April 15, 2024). | Key secondarya |
DOR | Time from the first documented evidence of confirmed CR or PR until the first documented date of disease progression or death from any cause, whichever occurs first. Measured until data cut-off (April 15, 2024). | Secondarya |
AEs | Any reported event from randomization until data cut-off (April 15, 2024). | Secondary |
SAEs | Any reported event from randomization until data cut-off (April 15, 2024). | Secondary |
Discontinuation due to AEs | Any reported event from randomization until data cut-off (April 15, 2024). | Secondary |
Death due to AEs | Any reported event from randomization until data cut-off (April 15, 2024). | Secondary |
EORTC QLQ-C30 | Randomization until data cut-off (April 15, 2024). Key time point of interest: Week 17. | Secondaryb |
FKSI-DRS | Randomization until data cut-off (April 15, 2024). Key time point of interest: Week 17. | Secondaryc |
EQ-5D-5L VAS | Randomization until data cut-off (April 15, 2024). Key time point of interest: Week 17. | Secondary |
AE = adverse event; BIRC = blinded independent central review; CR = complete response; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAE = serious adverse event; VAS = visual analogue scale.
aStatistical testing for these end points was adjusted for multiple comparisons.
bPertains to items 1 to 5, 29, and 30 of the questionnaire.
cRefers to items 1 to 9 of the questionnaire.
Sources: LITESPARK-005 study protocol.50 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Efficacy Outcomes
Progression-Free Survival
PFS was assessed as a primary end point in the LITESPARK-005 trial, which was defined as the time from randomization to the first documented disease progression per RECIST 1.1 based on BICR or death from any cause, whichever occurred first. The true date of disease progression was approximated by the earlier of the date of the first assessment at which disease progression was objectively documented per RECIST 1.1 by BICR and the date of death. For the primary analysis, patients who experienced a PFS event (e.g., disease progression or death) immediately after 2 or more missed disease assessments were censored at the last disease assessment before the missed visits. Patients who initiated new anticancer therapy were censored at the last disease assessment before the initiation of new anticancer therapy. Patients who did not initiate new anticancer therapy and did not experience a PFS event were censored at the last disease assessment. The patient group input highlighted the need for treatments that provide durable remission and disease stability. Of note, PFS was used to inform the pharmacoeconomic model submitted to CDA-AMC.
Overall Survival
OS was assessed as a primary end point in the LITESPARK-005 trial, which was defined as the time from randomization to the date of death from any cause. Patients without documented death at the time of analysis were censored at the date the patient was last known to be alive. OS was considered important to patients and clinicians, according to patient and clinician group inputs, and was used to inform the pharmacoeconomic model submitted to CDA-AMC.
Objective Response Rate
ORR was assessed as the key secondary end point in the LITESPARK-005 trial, which was defined as the proportion of patients in the analysis population with a best overall response of either confirmed CR or confirmed PR per RECIST 1.1 as assessed by BICR. Of note, ORR was considered a meaningful response to treatment, according to clinician group inputs.
Duration of Response
DOR was assessed as a secondary end point in the LITESPARK-005 trial. Among patients with a confirmed CR or PR, DOR was defined as the time from the first documented evidence of confirmed CR or PR to the first documented date of disease progression or death from any cause, whichever occurred first. Response and progression were assessed using RECIST 1.1 by BICR. Patients who experienced disease progression or died immediately after 2 or more missed disease assessments were censored at the last disease assessment before the missed visits. Patients who initiated new anticancer therapy were censored at the last disease assessment before the initiation of new anticancer therapy. Responding patients who were still alive, had not progressed, had not initiated new anticancer treatment, were not lost to follow-up, and who had a disease assessment within the 5 months before the data cut-off date were considered ongoing responders at the time of analysis. Patient groups highlighted the need for treatments that provide a long-term DOR.
Health-Related Quality of Life
HRQoL was assessed as a secondary end point in the LITESPARK-005 trial. Key HRQoL measures of the trial included the EORTC QLQ-C30 and FKSI-DRS instruments. A summary of their measurement properties is provided in Table 7. HRQoL was considered important to clinicians and patients, according to the submitted input from patient and clinician groups. The change in EORTC QLQ-C30 score from baseline was selected for the GRADE approach, as the clinical experts consulted by CDA-AMC considered the EORTC QLQ-C30 to be the most relevant instrument for assessing HRQoL in patients with advanced RCC.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
The EORTC QLQ-C30 was developed to assess the QoL of patients with cancer. It contains 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, nausea and vomiting, and pain), and 6 single symptom items (dyspnea, sleep disturbance, appetite loss, constipation, diarrhea, and financial impact).51 For each scale, the EORTC QLQ-C30 instrument is scored on a 4-point scale (1 = not at all, 2 = a little, 3 = quite a bit, 4 = very much). The EORTC QLQ-C30 instrument also contains 1 GHS/QoL scale, which uses a 7-point scale, with 1 indicating very poor status and 7 indicating excellent status.
The LITESPARK-005 trial assessed the change from baseline in EORTC QLQ-C30 GHS/QoL score, physical functioning score, and role functioning score. Moreover, the trial assessed the time to confirmed deterioration, measured by these scores. A change of 10 points in EORTC QLQ-C30 score is conventionally considered to be a minimum important difference (MID).52,53 No literature was identified that estimated MIDs specifically in patients with RCC.
Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms
The FKSI-DRS is a patient-reported instrument that measures whether the patient has experienced any of the following 9 kidney cancer-related symptoms: lack of energy, fatigue, weight loss, pain (general), bone pain, shortness of breath, cough, fever, or blood in the urine.54 Each item is scored using 5 response categories (0 = not at all, 1 = a little bit, 2 = somewhat, 3 = quite a bit, 4 = very much). According to the scoring manual, the score for each item must be reversed by subtracting the response from 4. Responses to all FKSI-DRS items are summed to generate a summary symptom score that ranges from 0 to 36, with higher scores indicating improved symptom status.
The LITESPARK-005 trial assessed the change in FKSI-DRS score from baseline. Moreover, the trial assessed TTD, measured by the FKSI-DRS. A decrease in score of 3 points or greater from baseline was considered a clinically relevant deterioration.54
Safety Outcomes
In the LITESPARK-005 trial, AEs were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (Version 5.0). Notable harms associated with treatment with belzutifan included anemia, dyspnea, and hypoxia.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Instrument | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A 30-item instrument meant to assess some of the different aspects that define the QoL of patients with cancer Of the 30 items, 28 are based on a 4-point Likert scale ranging from 1 = “not at all” to 4 = “very much,” or a 7-point numerical rating scale with anchors of 1 (very poor) to 7 (excellent) | The validity, reliability, or responsiveness in patients with advanced RCC have not been evaluated EORTC QLQ-C30 is a validated instrument used to assess HRQoL in oncology trials and studies51 Validity Although all interscale correlations were statistically significant, the correlation was moderate, indicating that the scales assess distinct components of the HRQoL construct Reliability All but the role functioning scale met the minimal standards for reliability (Cronbach alpha coefficient > 0.70) either before or during treatment Responsiveness There were statistically significant changes, in the expected direction, in physical and role functioning, global QoL, fatigue, and nausea and vomiting for patients whose performance status had improved or worsened during treatment | The MID was estimated to be 10 points, based on evidence generated in studies of tumour types55 The sponsor’s CSR26 defined a score of 10 or greater as clinically meaningful No MID has been evaluated in patients with advanced RCC |
FKSI-DRS | A kidney cancer–specific symptom measure Uses a 5-point Likert scale, which ranges from 0 (not at all) to 4 (very much) | The FKSI-DRS is a reliable, valid, and responsive measure of the most important symptoms of advanced kidney cancer54 Validity Evidence of convergent, divergent, and known-group validity has been reported, and the instrument s able to differentiate patients who reported improvement, no change, or worsening in their status in FACT-G domains Reliability Internal consistency reliability coefficients (range, 0.75 to 0.78) are shown at all time points Responsiveness The FKSI-DRS identifies responsiveness to change in clinical status over time | The MID was estimated to be between 2 and 3 points54 |
CSR = Clinical Study Report; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-G = Functional Assessment of Cancer Therapy–General; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; MID = minimal important difference; QoL = quality of life; RCC = renal cell carcinoma.
Source: Additional information provided by the sponsor on January 29, 2025.
Statistical Analysis
Sample Size and Power Calculation
The LITESPARK-005 trial was designed to enrol approximately 736 patients and randomize them in a 1:1 ratio to the belzutifan and everolimus arms. For PFS, the calculated sample size achieved 97.1% power to detect an HR of 0.7 at a one-sided alpha of 0.005, based on approximately 634 progressions or deaths at the final analysis and 87% of the final target number of events at the first interim analysis.
For OS, the calculated sample size achieved 85.2% power to detect an HR of 0.75 at a one-sided alpha of 0.019, based on approximately 483 deaths at the final analysis and approximately 48%, 68%, and 85% of the final target number of events at the 3 interim analyses, respectively.
For ORR, the calculated sample size achieved 99.9% power to detect a difference of 15%, with respect to the percentage of patients with an ORR, between the belzutifan group and the everolimus group, at a one-sided alpha of 0.001, assuming an underlying 20% ORR with belzutifan and a 5% ORR with everolimus.
Statistical Tests and Models
For PFS and OS, the nonparametric KM method was used to estimate the survival curve in each treatment group. Between-group differences in PFS and OS were assessed with the stratified log-rank test, and a stratified Cox proportional hazards model was used to assess the magnitude of the treatment difference between groups. In addition, the HRs and the corresponding 95% CIs from these models were reported. For both outcomes, the stratification factors used for randomization were applied to both the stratified log-rank test and the stratified Cox model. If crossing of the KM curves occurred, the proportional hazards assumption would be tested retrospectively. In the case of a departure from proportionality, an analysis of the restricted mean survival time was performed.
For ORR, the stratified Miettinen and Nurminen method was used to compare the 2 treatment groups. The difference in ORR and its 95% CI from the stratified Miettinen and Nurminen method, with strata weighting by sample size, were reported. The stratification factors used for randomization were applied to the analysis. The point estimate of the ORR was provided by treatment group, along with its corresponding 95% CI, using the exact binomial method.56 DOR was summarized descriptively using KM medians and ranges. Only patients who showed a confirmed CR or PR were included in the analysis.
For the change from baseline in key HRQoL measures (i.e., EORTC QLQ-C30 and FKSI-DRS), a constrained longitudinal data analysis model was applied, with the HRQoL score as the response variable, and treatment, time, the treatment-by-time interaction, and stratification factors used for randomization as covariates. The treatment difference in terms of LS mean change from baseline was estimated from this model, along with its corresponding 95% CI. The model-based LS mean, with 95% CI, was presented for each treatment group for HRQoL scores at the baseline and postbaseline time points. Line plots for the empirical mean change from baseline in EORTC QLQ-C30 and FKSI-DRS scores were provided for all time points, and the model-based LS mean change from baseline to the specified postbaseline time point, along with the corresponding 95% CI, were plotted in bar charts for the EORTC QLQ-C30 and FKSI-DRS. For TTD assessed with the EORTC QLQ-C30 and FKSI-DRS instruments, the KM method was used to estimate the TTD curve for each treatment group. The estimate of median TTD and its corresponding 95% CI were obtained from the KM estimates. The treatment difference in TTD was assessed with the stratified log-rank test, and a two-sided P value was reported. A stratified Cox proportional hazards model was used to assess the magnitude of the treatment difference as an HR and its corresponding 95% CI. The same stratification factors used for randomization were used as the stratification factors in both the stratified log-rank test and the stratified Cox model.
The analysis of safety outcomes followed a tiered approach, and tiers differed with respect to the analyses that were performed. AEs and events that met predefined limits of change in laboratory and vital-sign parameters were either prespecified as tier 1 end points or were classified as belonging to tier 2 or tier 3, based on the number of events observed. Tier 1 events comprised safety parameters or AEs of special interest that were identified a priori and were subject to inferential testing for statistical significance. Of note, there were no known AEs associated with patients who had RCC for which determination of a P value was expected to impact the safety assessment. Thus, no tier 1 events were included for the study protocol. Tier 2 parameters were assessed using point estimates, with corresponding 95% CIs, provided for differences in the position of patients with events, using the Miettinen and Nurminen method. Tier 2 events comprised AEs occurring in at least 10% of patients in either treatment group, grade 3 to grade 5 AEs occurring in at least 5% of patients in either treatment group, and SAEs occurring in at least 5% of patients in either treatment group. Safety end points that belonged to tier 1 or tier 2 were considered to belong to tier 3. Only point estimates by treatment group were provided for tier 3 end points.
Multiple Testing Procedures
The graphical method of Maurer and Bretz was used to control for multiplicity in multiple hypotheses and interim analyses.57 Study hypotheses were tested more than once, and when a particular null hypothesis was rejected, the alpha allocated to that hypothesis could be reallocated to other hypothesis tests. Details of the initial one-sided alpha allocation for each hypothesis are presented in Figure 3.
Figure 3: Multiplicity Diagram for Type I Error Control (One-Sided) in the LITESPARK-005 Trial
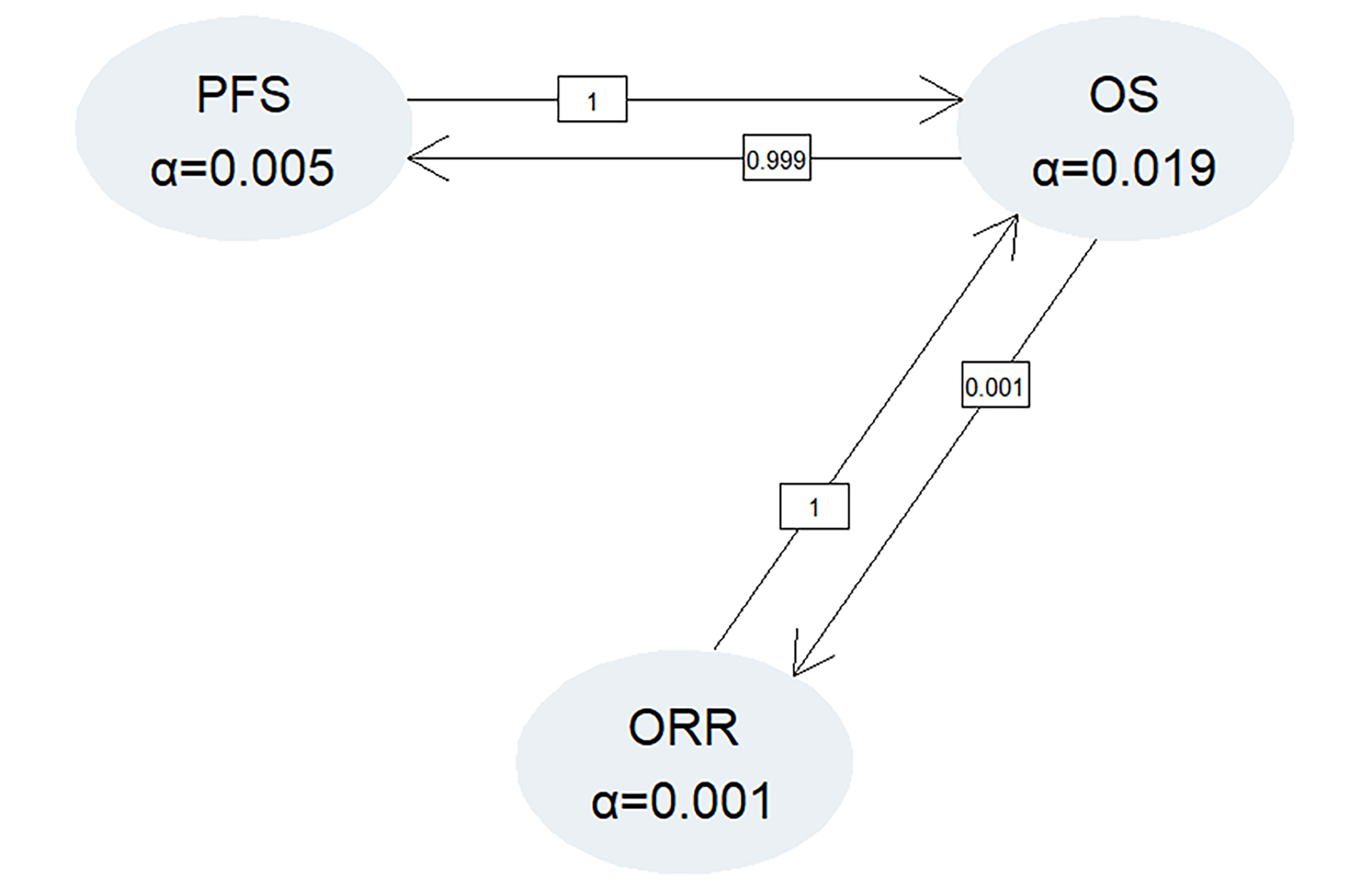
ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Source: Statistical analysis plan for LITESPARK-005.
For PFS, the initial alpha level for testing the hypothesis was 0.005. If the null hypothesis for OS was rejected, the alpha of 0.019 from the OS hypothesis could almost fully be reallocated to PFS hypothesis-testing (Figure 3). Thus, the PFS null hypothesis could be tested at an alpha of 0.005, an alpha of 0.024 (if the OS hypothesis was rejected and the ORR hypothesis was not rejected), or an alpha of 0.025 (if both the OS and ORR null hypotheses were rejected) (Figure 3). A Lan-DeMets O’Brien-Fleming spending function was used to derive the bounds and boundary properties of the PFS hypothesis at each analysis, based on the estimated number of events. The final analysis for PFS at the second interim analysis used the remaining type I error not spent at the first interim analysis, regardless of the actual number of PFS events observed. The P value bound at the final PFS analysis was calculated by considering the correlation between the test statistics determined by the actual number of PFS events at the first interim analysis and the final PFS analysis at the second interim analysis.
For OS, the hypothesis could be tested at an alpha of 0.019 (initially allocated), an alpha of 0.02 (if only the ORR null hypothesis is rejected), an alpha of 0.024 (if only the PFS null hypothesis is rejected), or an alpha of 0.025 (if both the PFS and ORR null hypotheses are rejected). The final analysis used the remaining type I error not spent at the earlier analyses. The event counts for all analyses were used to compute correlations.
For ORR, the primary test of the hypothesis occurred at the first interim analysis, at an initial one-sided alpha level of 0.001, and was based on all randomized patients in the study. If the null hypothesis for ORR was not rejected at the first interim analysis at the initially allocated alpha level and if the null hypothesis for OS and PFS were rejected at any analysis, the P value from the ORR test at the first interim analysis was compared with that of an updated alpha level of 0.025.
Data Imputation Methods
For PFS, patients who experienced a PFS event immediately after 2 or more missed disease assessments were censored at the last disease assessment before the missed visits. For ORR, patients with missing data were considered to be nonresponders.
Subgroup Analyses
Subgroup analyses were performed on the primary end points of PFS and OS and the key secondary end point ORR in the ITT analysis set. The following subgroups were prespecified in the statistical analysis plan:
age (< 65; ≥ 65 years)
sex (female; male)
race (white; all others)
region (North America; Western Europe; rest of the world)
IMDC risk category (favourable; intermediate, poor)
IMDC risk category (favourable; intermediate-poor)
number of prior VEGF-TKI therapies (1; 2 or 3)
number of prior lines of therapy (1; 2; 3).
The consistency of the treatment effect was assessed using descriptive statistics for each category of the subgroups listed. If the number of patients in a given category of a subgroup was less than 10% of the ITT analysis set, the subgroup analysis was not performed for the category of the subgroup variable, and the category of the subgroup variable was not shown in the forest plot. The subgroup analyses for PFS and OS were conducted using an unstratified Cox model, and the subgroup analysis for ORR was conducted using the unstratified Miettinen and Nurminen method.
Sensitivity Analyses
To evaluate the robustness of the PFS end point, assessed by BICR per RECIST 1.1, the following sensitivity analyses, which investigated different sets of censoring rules, were performed:
Sensitivity analysis 1, in which disease progressions and deaths are counted as events, regardless of missed study visits or the initiation of new anticancer therapy.
Sensitivity analysis 2, in which discontinuation of treatment for reasons other than CR or the initiation of new anticancer treatment (whichever occurs last) is considered to be a disease progression event for patients without documented disease progression or death.
In addition, sensitivity analyses were performed for PFS, ORR, and DOR, based on the investigator’s assessment.
Table 8: Statistical Analysis of Efficacy End Points in the LITESPARK-005 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS as assessed by BICR per RECIST 1.1 | Test: stratified log-rank test Estimation: stratified Cox model with the Efron tie handling method |
| Patients who experienced a PFS event immediately after 2 or more missed disease assessments were censored at the last disease assessment before the missed visits Patients who initiated new anticancer therapy were censored at the last disease assessment before the initiation of new anticancer therapy Patients who did not initiate new anticancer therapy and did not experience a PFS event were censored at the last disease assessment | Sensitivity analyses are performed to assess PFS based on investigator’s assessment Sensitivity analyses using different censoring rules:
|
OS | Test: stratified log-rank test Estimation: stratified Cox model with the Efron tie handling method |
| Censored at last known alive date | NR |
ORR as assessed by BICR per RECIST 1.1 | Testing and estimation: stratified Miettinen and Nurminen method |
| Patients with missing data are considered nonresponders | Sensitivity analyses are performed to assess ORR based on investigator’s assessment |
DOR | NR |
| Censored at the last adequate disease assessment or at the last known date of disease progression or death | Sensitivity analyses will be performed to assess DOR based on investigator’s assessment |
Mean change from baseline in EORTC QLQ-C30 and FKSI-DRS | A cLDA model was applied, with the PRO score as the response variable, and treatment, time, the treatment-by-time interaction, and stratification factors used for randomization as covariates |
| The cLDA model implicitly treats missing data as missing at random | NR |
TTD in EORTC QLQ-C30 and FKSI-DRS scores | Test: stratified log-rank test Estimation: stratified Cox model with the Efron tie handling method |
| Patients with no baseline assessments were right censored at the treatment start date. Patients who were still in the study without deterioration or discontinued the study without deterioration were right censored at the time of last assessment | NR |
BICR = blinded independent committee review; cLDA = constrained longitudinal data analysis; CR = complete response; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; RCC = renal cell carcinoma; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; TTD = time to deterioration; vs. = versus.
Source: LITESPARK-005 Clinical Study Report interim analysis 2.25
Analysis Populations
A summary of analysis populations used in the LITESPARK-005 trial that are relevant to this review is provided in Table 9.
Table 9: Analysis Populations in the LITESPARK-005 Trial
Population | Definition | Application |
|---|---|---|
ITT | All randomized patients. Patients were included in the treatment group to which they were randomized. | Efficacy outcomes |
APaT | All randomized patients who received at least 1 dose of the study treatment. Patients were included in the treatment group corresponding to the study treatment they actually received. | Safety outcomes |
PRO FASa | All randomized patients who had at least 1 PRO assessment available and received at least 1 dose of the study medication. | PROs |
APaT = all patients as treated; FAS = full analysis set; ITT = intention to treat; PRO = patient-reported outcome.
aNumber of patients in the PRO FAS refers to the patients with a PRO assessment at baseline; the number of patients at follow-up varies by follow-up time point and PRO assessment.
Sources: LITESPARK-005 Clinical Study Report interim analysis 2.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Results
Patient Disposition
A summary of patient disposition in the LITESPARK-005 trial is provided in Table 10. Of the 996 screened patients, 250 patients (25.1%) were classified as screening failures. Of these 250 patients, 241 patients failed to meet the eligibility criteria and 9 patients were from the safety run-in cohort based in Japan.27 After the screening phase, the remaining 746 patients were randomized to receive either belzutifan (n = 374) or everolimus (n = 372) and were included in the ITT analysis.27 At the time of the final analysis (data cut-off: April 15, 2024), 120 (32.1%) patients in the belzutifan group remained in the study compared to 110 (29.6%) patients in the everolimus group.27 The most common reason for study discontinuation in both groups was death (65.5% for belzutifan group and 68.0% for everolimus group).27 Fewer patients in the belzutifan group discontinued study treatment (85.5%) compared with those in the everolimus group (98.6%).27 The most common reason for the discontinuation of study treatment were progressive disease (68.5% versus 68.9%) and AEs (████ versus 15.3%); discontinuation due to AEs was reported less frequently in the belzutifan group than the everolimus group.27
Table 10: Summary of Patient Disposition in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
Patient disposition | Belzutifan (n = 374) | Everolimus (n = 372) |
|---|---|---|
Screened, N | 996 | |
Reason for screening failure, n (%) | 250 (25.1) | |
Did not meet inclusion criteria or did meet exclusion criteria | 241 (24.1) | |
Japan safety run-in cohorta | 9 (0.90) | |
Randomized, N (%) | 374 | 372 |
Treated | 372 | 360 |
Not treated | 2 | 12 |
Status of study medication in the trial, n (%) | ||
Started | 372 (100.0) | 360 (100.0) |
Discontinued | 318 (85.5) | 355 (98.6) |
Adverse event | ██ █████ | 55 (15.3) |
Clinical progression | 22 (5.9) | 26 (7.2) |
Nonstudy anticancer therapy | 5 (1.3) | 10 (2.8) |
Physician decision | 1 (0.3) | 2 (0.6) |
Progressive disease | 255 (68.5) | 248 (68.9) |
Withdrawal by parent or guardian | 1 (0.3) | 0 (0.0) |
Withdrawal by patient | 11 (3.0) | 14 (3.9) |
Patients ongoing | 54 (14.5) | 5 (1.4) |
Patient status in the trial, n (%) | ||
Discontinued | 254 (67.9) | 262 (70.4) |
Death | 245 (65.5) | 253 (68.0) |
Associated with COVID-19 | 2 (0.5) | 9 (2.4) |
Physician decision | 0 (0.0) | 1 (0.3) |
COVID-19 association unspecified; patient subsequently died | 0 (0.0) | 1 (0.3) |
Withdrawal by patient | 9 (2.4) | 8 (2.2) |
COVID-19 association unspecified; patient subsequently died | 9 (2.4) | 5 (1.3) |
Patients ongoing | 120 (32.1) | 110 (29.6) |
PRO FAS (EORTC QLQ-C30)b | 351 | 333 |
PRO FAS (FKSI-DRS)b | 352 | 334 |
Safety (APaT) | 372 | 360 |
APaT = all patients as treated; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analysis set; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; ITT = intention to treat; PRO = patient-reported outcome.
aNine patients were enrolled in the Japan safety run-in cohort. These patients were not randomized to treatment in the study; all received belzutifan. These patients are not included in the ITT population.
bNumber of patients refers to the patients with a PRO assessment at baseline; the number of patients at follow-up varies by follow-up time point and PRO assessment.
Sources: LITESPARK-005 Clinical Study Report interim analysis 2;25 LITESPARK-005 Statistical Report final analysis.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Baseline demographic and disease characteristics were well balanced between the treatment groups in the LITESPARK-005 trial. The median age of patients in the trial was 63 years (range, 22 to 90 years) and most patients were male (77.9%; female: 22.1%).25 The trial enrolled patients of the following races or ethnicities: American Indian or Alaska Native (0.7%), Asian (12.1%), Black or African American (1.1%), Native Hawaiian or other Pacific Islander (0.1%), and white (78.8%) [categories are as reported in study]; 2.3% of patients in the trial identified as having multiple races or ethnicities and 5.0% of patients had missing data on race and ethnicity.25 Most patients had a KPS score that ranged between 90% and 100% (64.1%) and an ECOG PS of 1 (55.1%).25 Most patients had an intermediate or poor IMDC prognostic risk score (78.3%) and most had stage IV RCC at diagnosis (57.0%).25 The vast majority of patients had 2 or more organs involved with disease at baseline (912%), with the lung (64.6%) being the most common site of metastatic disease.25 Most patients had a prior nephrectomy (69.7%), received 2 (43.3%) or 3 (42.8%) prior lines of therapy, and received 1 prior VEGF- or VEGF receptor–targeted therapy (50.5%).25
Table 11: Summary of Baseline Characteristics in the LITESPARK-005 Trial — ITT Population (Interim Analysis 2; Data Cut-Off of June 13, 2023)
Characteristic | Belzutifan (n = 374) | Everolimus (n = 372) |
|---|---|---|
Demographic characteristics | ||
Sex, n (%) | ||
Female | 77 (20.6) | 88 (23.7) |
Male | 297 (79.4) | 284 (76.3) |
Age (years), median (range) | 62.0 (22.0 to 90.0) | 63.0 (33.0 to 87.0) |
Race, n (%)a | ||
American Indian or Alaska Native | 3 (0.8) | 2 (0.5) |
Asian | 43 (11.5) | 47 (12.6) |
Black or African American | 4 (1.1) | 4 (1.1) |
Native Hawaiian or Other Pacific Islander | 0 (0.0) | 1 (0.3) |
White | 297 (79.4) | 291 (78.2) |
Multiple | 6 (1.6) | 11 (3.0) |
Missing | 21 (5.6) | 16 (4.3) |
Geographic region of enrolling site, n (%) | ||
North America | 75 (20.1) | 89 (23.9) |
Western Europe | 191 (51.1) | 182 (48.9) |
Rest of the world | 108 (28.9) | 101 (27.2) |
Karnofsky Performance Status, n (%) | ||
90 or 100 | 238 (63.6) | 240 (64.5) |
70 or 80 | 135 (36.1) | 131 (35.2) |
Missing | 1 (0.3) | 1 (0.3) |
ECOG PS, n (%) | ||
0 | 161 (43.0) | 163 (43.8) |
1 | 208 (55.6) | 203 (54.6) |
2 | 5 (1.3) | 6 (1.6) |
Disease characteristics | ||
IMDC risk categories, n (%) | ||
Favourable | 79 (21.1) | 83 (22.3) |
Intermediate | 249 (66.6) | 244 (65.6) |
Poor | 46 (12.3) | 45 (12.1) |
IMDC risk categories 2, n (%) | ||
Favourable | 79 (21.1) | 83 (22.3) |
Intermediate or poor | 295 (78.9) | 289 (77.7) |
Site of metastatic disease,b n (%) | ||
Lung | 246 (65.8) | 236 (63.4) |
Lymph node | 232 (62.0) | 215 (57.8) |
Bone | 187 (50.0) | 181 (48.7) |
Liver | 89 (23.8) | 103 (27.7) |
Adrenal gland | 73 (19.5) | 71 (19.1) |
Number of organs involved with disease at baseline, n (%) | ||
1 | 34 (9.1) | 32 (8.6) |
≥ 2 | 340 (90.9) | 340 (91.4) |
RCC histology, n (%) | ||
Clear cell | 347 (92.8) | 351 (94.4) |
Clear cell component | 26 (7.0) | 21 (5.6) |
Missing | 1 (0.3) | 0 (0.0) |
Sarcomatoid feature, n (%) | ||
Yes | 42 (11.2) | 31 (8.3) |
No | 255 (68.2) | 248 (66.7) |
Unknown | 77 (20.6) | 92 (24.7) |
Missing | 0 (0.0) | 1 (0.3) |
RCC stage at initial diagnosis, n (%) | ||
I | 39 (10.4) | 46 (12.4) |
II | 34 (9.1) | 32 (8.6) |
III | 84 (22.5) | 80 (21.5) |
IV | 216 (57.8) | 209 (56.2) |
Missing | 1 (0.3) | 5 (1.3) |
Prior oncologic radiation, n (%) | ||
Yes | 146 (39.0) | 152 (40.9) |
No | 228 (61.0) | 220 (59.1) |
Prior nephrectomy, n (%) | ||
Yes | 261 (69.8) | 259 (69.6) |
No | 113 (30.2) | 113 (30.4) |
Number of prior antiangiogenic (TKI) therapies, n (%) | ||
1 | 187 (50.0) | 190 (51.1) |
2 or 3 | 187 (50.0) | 182 (48.9) |
Number of prior lines of therapy, n (%) | ||
1 | 46 (12.3) | 52 (14.0) |
2 | 157 (42.0) | 166 (44.6) |
3 | 169 (45.2) | 150 (40.3) |
4 | 2 (0.5) | 4 (1.1) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; ITT = intention to treat; RCC = renal cell carcinoma; TKI = tyrosine kinase inhibitor.
aCategories are as reported in study.
bThese sites are the 5 most common metastatic sites and are ordered decreasingly by the total number of patients in the 2 treatment arms.
Sources: LITESPARK-005 Clinical Study Report interim analysis 2.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Exposure to Study Treatments
Patient Exposure
Details on patient exposure to treatments in the LITESPARK-005 trial are summarized in Table 12. At the time of the final analysis (data cut-off: April 15, 2024), patients in the belzutifan group had a longer median duration of treatment than patients in the everolimus group (median duration: ███ ██████ versus ███ ██████ for belzutifan versus everolimus).27 Patients in the belzutifan group received treatment at a median dose of ███ ██ ███ ███, whereas patients in the everolimus group received treatment at a median dose of ██ ██ ███ ███.27
Table 12: Summary of Patient Exposure in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
Exposure | Belzutifan (n = 372) | Everolimus (n = 360) |
|---|---|---|
Duration of therapya (months) | ||
Mean (SD) | ████ █ | ████ █ |
Median (range) | ███ ████ █ | ███ ████ █ |
Total dose administered (mg)b | ||
Mean (SD) | ████ █ | ████ █ |
Median (range) | ████ ████ █ | ███ ████ █ |
Average daily dose administered (mg)c | ||
Mean (SD) | ████ █ | ████ █ |
Median (range) | ███ ████ █ | ███ ████ █ |
ITT = intention to treat; SD = standard deviation.
aDuration on therapy is calculated as the time between the first dose date and the last dose date in each treatment arm.
bTotal dose administered for a participant is the sum of all doses taken by the participant.
cAverage daily dose administered for a participant is calculated as the total dose administered divided by the total number of days when the participant took a nonzero dose.
Sources: LITESPARK-005 Statistical Report final analysis.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Prior and Concomitant Non-Oncologic Medications
Details on exposure to prior non-oncologic medications and concomitant medications in the LITESPARK-005 trial are summarized in Table 13 and Table 14, respectively. At baseline, reported prior and concomitant non-oncologic medications were generally comparable in the belzutifan and everolimus groups. Prior non-oncologic medications reported for at least 10% of patients in either group were levothyroxine sodium (45.7% for the belzutifan group compared with 45.0% for the everolimus group), amlodipine (13.7% for the belzutifan group compared with 21.4% for the everolimus group), paracetamol (16.7% for the belzutifan group compared with 20.0% for the everolimus group), omeprazole (9.9% for the belzutifan group compared with 12.2% for the everolimus group), and acetylsalicylic acid (9.9% for the belzutifan group compared with 11.1% for the everolimus group).25
The most frequently reported categories of concomitant medications in both groups were analgesics (71.5% for the belzutifan group compared with 69.7% for the everolimus group), ophthalmologicals (66.4% for the belzutifan group compared with 78.3% for the everolimus group), and stomatological preparations (62.4% for the belzutifan group compared with 72.8% for the everolimus group).25
Table 13: Summary of Prior Non-Oncologic Medications in the LITESPARK-005 Trial — ITT Population (Interim Analysis 2; Data Cut-Off of June 13, 2023)
Concomitant medication | Belzutifan (n = 372) | Everolimus (n = 360) |
|---|---|---|
Patients with ≥ 1 prior medications, n (%) | 353 (94.9) | 337 (93.6) |
Commonly reported prior non-oncologic medications (received by ≥ 10% of patients in either group), n (%) | ||
Acetylsalicylic acid | 37 (9.9) | 40 (11.1) |
Amlodipine | 51 (13.7) | 77 (21.4) |
Levothyroxine sodium | 170 (45.7) | 162 (45.0) |
Omeprazole | 37 (9.9) | 44 (12.2) |
Paracetamol | 62 (16.7) | 72 (20.0) |
ITT = intention to treat.
Source: LITESPARK-005 Clinical Study Report interim analysis 2.25
Table 14: Summary of Concomitant Non-Oncologic Medications in the LITESPARK-005 Trial — ITT Population (Interim Analysis 2; Data Cut-Off of June 13, 2023)
Concomitant medication | Belzutifan (n = 372) | Everolimus (n = 360) |
|---|---|---|
Patients with ≥ 1 concomitant medications, n (%) | 369 (99.2) | 358 (99.4) |
Commonly reported concomitant medications (received by ≥ 40% of patients in either group), n (%) | ||
Analgesics | 266 (71.5) | 251 (69.7) |
Antianemic preparations | 154 (41.4) | 96 (26.7) |
Antibacterials for systemic use | 146 (39.2) | 161 (44.7) |
Antidiarrheals, intestinal inflammatory and/or anti-infective drugs | 151 (40.6) | 196 (54.4) |
Antithrombotic drugs | 174 (46.8) | 152 (42.2) |
Blood substitutes and perfusion solutions | 167 (44.9) | 164 (45.6) |
Corticosteroids, dermatological preparations | 180 (48.4) | 236 (65.6) |
Corticosteroids for systemic use | 166 (44.6) | 225 (62.5) |
Drugs for acid-related disorders | 172 (46.2) | 214 (59.4) |
Drugs for obstructive airway disease | 118 (31.7) | 158 (43.9) |
Nasal preparations | 150 (40.3) | 200 (55.6) |
Ophthalmologicals | 247 (66.4) | 282 (78.3) |
Ophthalmological and otological preparations | 121 (32.5) | 161 (44.7) |
Otologicals | 175 (47.0) | 215 (59.7) |
Psycholeptics | 144 (38.7) | 155 (43.1) |
Stomatological preparations | 232 (62.4) | 262 (72.8) |
Thyroid therapy | 174 (46.8) | 168 (46.7) |
Vasoprotectives | 166 (44.6) | 195 (54.2) |
ITT = intention to treat.
Source: LITESPARK-005 Clinical Study Report interim analysis 2.25
Prior Systemic Oncologic Treatments
Details on exposure to prior systemic oncologic treatments in the LITESPARK-005 trial are summarized in Table 15. At baseline, most patients in the belzutifan and everolimus groups received no more than 3 prior lines of oncologic treatments (99.5% for the belzutifan group compared with 98.9% for the everolimus group).25 The most reported first-line treatments received in either group were sunitinib malate (31.3% for the belzutifan group compared with 31.5% for the everolimus group), nivolumab (23.5% for the belzutifan group compared with 23.1% for the everolimus group), ipilimumab (17.1% for the belzutifan group compared with 17.2% for the everolimus group), and pazopanib (18.2% for the belzutifan group compared with 16.4% for the everolimus group).25
The most reported second-line treatments received in either group were nivolumab (44.7% for the belzutifan group compared with 38.7% for the everolimus group), cabozantinib (18.4% for the belzutifan group compared with 20.2% for the everolimus group), and axitinib (9.4% in both groups).25
Reported third-line treatments in either group were cabozantinib (23.3% for the belzutifan group compared with 13.7% for the everolimus group), nivolumab (13.6% for the belzutifan group compared with 17.7% for the everolimus group), and axitinib (4.5% for the belzutifan group compared with 5.1% for the everolimus group).25
Table 15: Summary of Prior Systemic Oncologic Treatments in the LITESPARK-005 Trial — ITT Population (Interim Analysis 2; Data Cut-Off of June 13, 2023)
Exposurea | Belzutifan (n = 374) | Everolimus (n = 372) |
|---|---|---|
Patients in population with 1 or more prior systemic oncologic therapies, n (%) | 374 (100.00) | 372 (100.00) |
Patients in population with 2 or more prior systemic oncologic therapies, n (%) | 328 (87.7) | 320 (86.0) |
Patients in population with 3 or more prior systemic oncologic therapies, n (%) | 171 (45.7) | 154 (41.4) |
Commonly reported first-line systemic oncologic therapies (received by ≥ 5% of patients in either group), n (%) | ||
Axitinib | 44 (11.8) | 58 (15.6) |
Cabozantinib | 29 (7.8) | 32 (8.6) |
Ipilimumab | 64 (17.1) | 64 (17.2) |
Nivolumab | 88 (23.5) | 86 (23.1) |
Pazopanib | 68 (18.2) | 61 (16.4) |
Pazopanib hydrochloride | 19 (5.1) | 19 (5.1) |
Pembrolizumab | 39 (10.4) | 48 (12.9) |
Sunitinib malate | 117 (31.3) | 117 (31.5) |
Commonly reported second-line systemic oncologic therapies (received by ≥ 5% of patients in either group), n (%) | ||
Axitinib | 34 (9.1) | 34 (9.1) |
Cabozantinib | 69 (18.4) | 75 (20.2) |
Nivolumab | 167 (44.7) | 144 (38.7) |
Sunitinib malate | 30 (8.0) | 34 (9.1) |
Commonly reported third-line systemic oncologic therapies (received by ≥ 5% of patients in either group), n (%) | ||
Axitinib | 17 (4.5) | 19 (5.1) |
Cabozantinib | 87 (23.3) | 51 (13.7) |
Nivolumab | 51 (13.6) | 66 (17.7) |
ITT = intention to treat.
Note: Every participant is counted a single time for each applicable row and column.
aOf note, 2 patients (0.5%) and 4 patients (1.1%) received 4 prior lines of therapy in the belzutifan and everolimus groups, respectively. Both patients in the belzutifan group received cabozantinib in the fourth-line setting; in the everolimus group, fourth-line treatments were axitinib (1 patient [0.3%]), cabozantinib (2 patients [0.5%]), and nivolumab (1 patient [0.3%]).
Source: LITESPARK-005 Clinical Study Report interim analysis 2.25
Subsequent Oncologic Treatments
Details on subsequent systemic oncologic therapies received by patients in the LITESPARK-005 trial are summarized in Table 16. A ████ proportion of patients in the everolimus group received ██████████ ████████ ██ compared with patients in the belzutifan group (████).27 The most reported ██████████ ████████ therapies among both groups were ███████ ██████, with treatment use being ████ ███ among patients in the belzutifan group ██████ ██.27 Of these, the ████ ██████ ███████ ██ patients received in both groups were ████████████ ██████ ██ ██████ ███ █████, with treatment use being ████ ██ among patients in the belzutifan group.27
Table 16: Summary of Subsequent Systemic Oncologic Treatments in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024) [Redacted]
█████████████████ | █████████████████ |
|---|---|
█████████████████ | █████████████████ |
█████████████████ | █████████████████ |
█████████████████ | █████████████████ |
Note: This table was redacted per the sponsor’s request.
Sources: LITESPARK-005 Statistical Report final analysis.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Efficacy
PFS Based on BICR per RECIST 1.1
A summary of results for PFS at the first interim and final analyses in the LITESPARK-005 trial is provided in Table 17.
At the time of the first interim analysis (data cut-off: November 1, 2022), the median duration of follow-up was 13.6 months (range, 0.2 to 31.1 months) in the belzutifan group and 13.3 months (range, 0.8 to 31.8 months) in the everolimus group.26 At the time of the data cut-off for this analysis, 519 PFS events (82.9% of the 626 total expected events at the final analysis) had occurred (257 patients in the belzutifan group and 262 patients in the everolimus group).26 The corresponding HR was 0.75 (95% CI, 0.63 to 0.90; one-sided P = 0.00077), representing a 25% reduction in the risk of disease progression or death with belzutifan compared with everolimus.26 At the first interim analysis, the predefined success criterion for superiority based on PFS was met.26 Median PFS was similar for both the belzutifan and everolimus groups (5.6 months [95% CI, 3.9 to 7.0 months] for the belzutifan group compared with 5.6 months [95% CI, 4.8 to 5.8 months]) for the everolimus group.26 In addition, PFS rates based on KM estimates were higher in the belzutifan group compared with the everolimus group at 6 months, 9 months, 12 months, 18 months, and 24 months (Figure 4); estimated PFS rates at 24 months were █████ ████ ███ ████ ██ █████ in the belzutifan group and 0% (95% CI, not applicable) in the everolimus group (between-group difference for belzutifan compared with everolimus: █████ ████ ███ ███ ███████████).26 The KM curves for PFS measured at the first interim analysis are shown in Figure 4.
At the time of the second interim analysis (data cut-off: June 13, 2023), the median duration of follow-up was 18.5 months (range, 0.2 to 37.5 months) in the belzutifan group and 17.2 months (range, 0.8 to 39.1 months) in the everolimus group.25 Results for PFS for this analysis were consistent with those of the first interim analysis; the full results for PFS for the second interim analysis are presented in Appendix 1.
At the time of the final analysis (data cut-off: April 15, 2024), the median duration of follow-up was 21.4 months (range, 0.2 to 47.6 months) in the belzutifan group and 18.3 months (range, 0.8 to 49.2 months) in the everolimus group.27 At the time of the data cut-off for this analysis, 587 PFS events had occurred (308 patients in the belzutifan group and 279 patients in the everolimus group).27 The HR for PFS was 0.75 (95% CI, 0.63 to 0.88; one-sided P = 0.00034).27 As was observed in the first interim analysis, the median PFS measured at the final analysis remained similar in both groups (5.6 months [95% CI, 1.9 to 16.5 months] for the belzutifan group compared with 5.6 months [95% CI, 4.8 to 5.8 months] for the everolimus group).27 PFS rates based on KM estimates continued to be higher in the belzutifan group than in the everolimus group at 6 months, 9 months, 12 months, 18 months, 24 months, and 30 months; estimated PFS rates at 30 months were 14.2% (95% CI, 10.7% to 18.2%) in the belzutifan group and 2.7% (95% CI, 1.1% to 5.5%) in the everolimus group (between-group difference for belzutifan compared with everolimus was 11.5% [95% CI, 7.2% to 15.8%]).27 The KM curves for PFS measured at the final analysis are shown in Figure 5.
At the time of the first interim analysis, results from the planned sensitivity analyses for PFS were consistent with those from the primary analysis.26 Results from the sensitivity analysis that evaluated PFS based on investigator assessment per RECIST 1.1 were consistent with those from the analysis based on BICR (HR = 0.68; 95% CI, 0.58 to 0.81; one-sided nominal P < 0.00001). Concordance of the assessment for progressive disease between the BICR and investigator assessments was 90% and 78% in the belzutifan and everolimus groups, respectively.26
Other sensitivity analyses included disease progression and death, regardless of missed study visits or the initiation of new anticancer therapy (HR = 0.75; 95% CI, 0.63 to 0.88; one-sided nominal P < 0.00001), and the discontinuation of treatment (for reasons other than CR or for the initiation of new anticancer treatment) as a disease progression event for patients without documented disease progression or death (HR = 0.68; 95% CI, 0.58 to 0.80; one-sided nominal P < 0.00001).26 Subgroup analyses of PFS in the first interim analysis favoured belzutifan in all prespecified subgroups, including those stratified by IMDC risk category.26 At the final analysis, results from sensitivity and subgroup analyses continued to be consistent with those from the primary analysis.27 Full details of the subgroup analyses for PFS are presented in Appendix 1.
Table 17: Summary of Results for PFS Based on BICR Assessment per RECIST 1.1 in the LITESPARK-005 Trial — ITT Population (Interim Analysis 1 and Final Analysis)
Variable | Interim analysis 1 (data cut-off of November 1, 2022) | Final analysis (data cut-off of April 15, 2024) | ||
|---|---|---|---|---|
Belzutifan (n = 374) | Everolimus (n = 372) | Belzutifan (n = 374) | Everolimus (n = 372) | |
Median follow-up time, months (range) | 13.6 (0.2 to 31.1) | 13.3 (0.8 to 31.8) | 21.4 (0.2 to 47.6) | 18.3 (0.8 to 49.2) |
Number of events (%) | 257 (68.7) | 262 (70.4) | 308 (82.4) | 279 (75.0) |
Death | 23 (6.1) | 40 (10.8) | 25 (6.7) | 41 (11.0) |
Documented progression | 234 (62.6) | 222 (59.7) | 283 (75.7) | 238 (64.0) |
Patients censored, n (%) | 117 (31.3) | 110 (29.6) | 66 (17.6) | 93 (25.0) |
Last assessment before 2 or more consecutive missed assessments | 1 (0.3) | 4 (1.1) | 3 (0.8) | 6 (1.6) |
Last assessment showing no progression | 91 (24.3) | 23 (6.2) | 37 (9.9) | 2 (0.5) |
Last radiologic assessment before new anticancer therapy showing no progression | 21 (5.6) | 68 (18.3) | 22 (5.9) | 70 (18.8) |
Randomization | 4 (1.1) | 15 (4.0) | 4 (1.1) | 15 (4.0) |
KM estimates (months)a | ||||
Median (95% CI) | 5.6 (3.9 to 7.0) | 5.6 (4.8 to 5.8) | 5.6 (3.8 to 6.5) | 5.6 (4.8 to 5.8) |
Q1 to Q3 | 1.9 to 16.7 | 2.1 to 9.2 | 1.9 to 16.5 | 2.1 to 9.2 |
HR vs. everolimus (95% CI)b | 0.75 (0.63 to 0.90) | 0.75 (0.63 to 0.88) | ||
P valuec | 0.00077 | 0.00034d | ||
PFS rate at month 6, % (95% CI) | 46.6 (41.3 to 51.7) | 42.5 (36.8 to 48.0) | 46.1 (40.9 to 51.2) | 42.8 (37.1 to 48.3) |
Difference at 6 months vs. everolimus (95% CI) | 4.1 (−3.5 to 11.7) | 3.3 (−4.3 to 10.9) | ||
PFS rate at month 9, % (95% CI) | 40.4 (35.2 to 45.4) | 28.4 (23.2 to 33.8) | 39.9 (34.8 to 45.0) | 28.7 (23.5 to 34.2) |
Difference at 9 months vs. everolimus (95% CI) | 12.0 (4.6 to 19.4) | 11.2 (3.8 to 18.6) | ||
PFS rate at month 12, % (95% CI) | ████ █████ ██ █████ | 17.1 (12.7 to 22.1) | 33.7 (28.8 to 38.7) | 17.6 (13.2 to 22.4) |
Difference at 12 months vs. everolimus (95% CI) | ████ ████ ██ █████ | 16.1 (9.3 to 22.9) | ||
PFS rate at month 18, % (95% CI) | ████ ██████ █████ | 8.3 (4.9 to 12.7) | 22.1 (17.8 to 26.6) | 9.0 (5.8 to 13.0) |
Difference at 18 months vs. everolimus (95% CI) | ████ ████ ██ █████ | 13.1 (7.4 to 18.8) | ||
PFS rate at month 24, % (95% CI) | ████ █████ ██ █████ | 0.0 (NA to NA) | 17.5 (13.7 to 21.7) | 4.1 (2.0 to 7.2) |
Difference at 24 months vs. everolimus (95% CI) | ████ ████ | 13.4 (8.6 to 18.2) | ||
PFS rate at month 30, % (95% CI) | NR | 14.2 (10.7 to 18.2) | 2.7 (1.1 to 5.5) | |
Difference at 30 months vs. everolimus (95% CI) | NR | 11.5 (7.2 to 15.8) | ||
BICR = blinded independent central review; CI = confidence interval; HR = hazard ratio; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; ITT = intention to treat; KM = Kaplan-Meier; NA = not applicable; NC = not calculated; NR = not reported; PFS = progression-free survival; Q = quartile; RCC = renal cell carcinoma; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; vs. = versus.
aFrom the product-limit (KM) method for censored data.
bBased on Cox regression model with the Efron method of tie handling, with treatment as a covariate, stratified by IMDC risk group (favourable vs. intermediate vs. poor) and number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 or 3).
cOne-sided P value, based the on log-rank test stratified by IMDC risk group (favourable vs. intermediate vs. poor) and number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 to 3).
dThis P value was from a supplemental analysis to the primary analysis and, thus, cannot be used to make inferences about PFS.
Sources: LITESPARK-005 Clinical Study Report interim analysis 1,26 LITESPARK-005 Statistical Report FA27 and Addendum.30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Figure 4: Kaplan-Meier Analysis for PFS per RECIST 1.1 by BICR — ITT Population (Interim Analysis 1; Data Cut-Off of November 1, 2022)
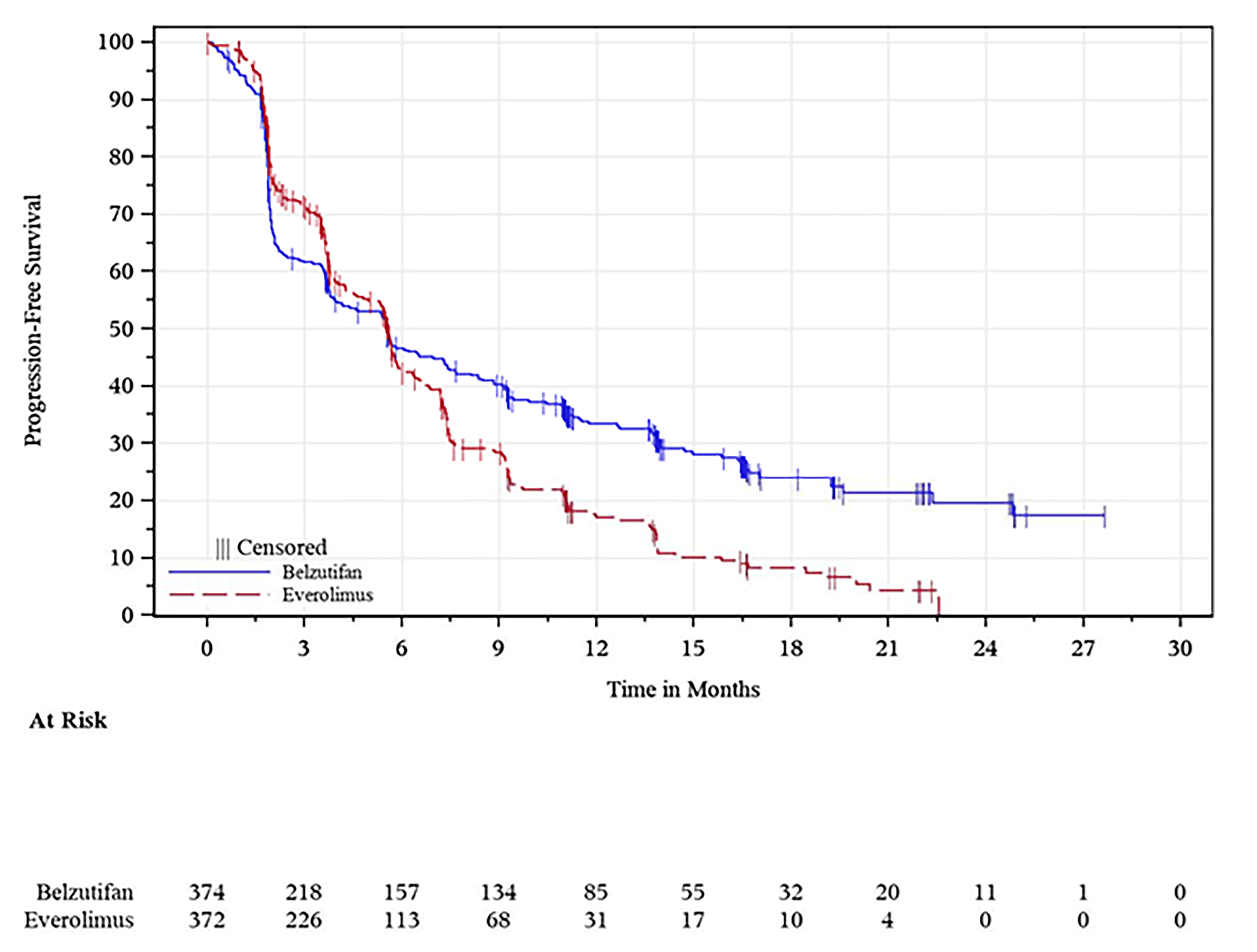
BICR = blinded independent central review; ITT = intention to treat; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: LITESPARK-005 Clinical Study Report interim analysis 1.26
Figure 5: Kaplan-Meier Analysis for PFS per RECIST 1.1 by BICR — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
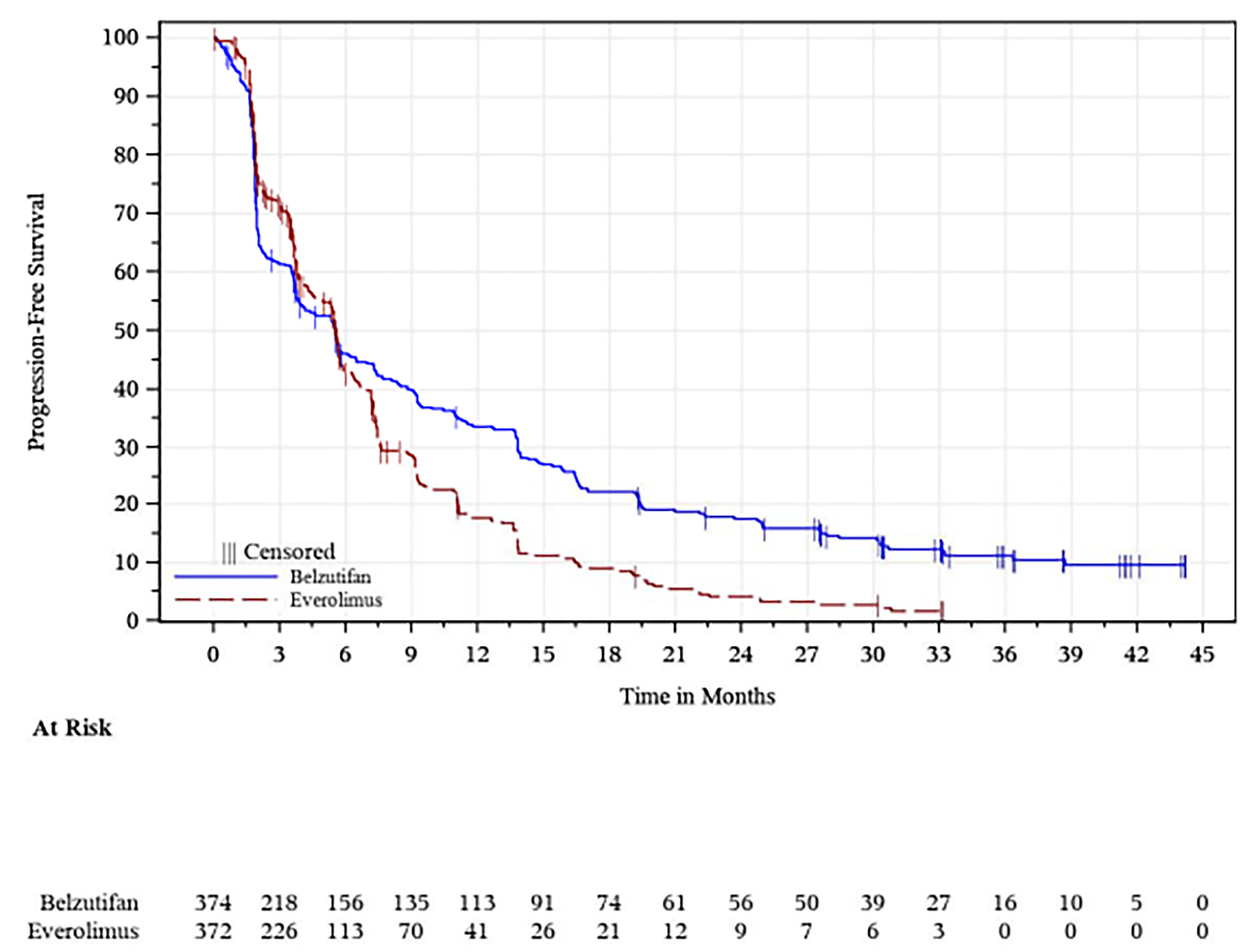
BICR = blinded independent central review; ITT = intention to treat; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: LITESPARK-005 Statistical Report final analysis.27
Overall Survival
The corresponding statistical hypothesis-testing P value boundary for OS was not met at any of the prespecified analyses (interim and final analyses) of the trial. A summary of results for OS at the final analysis of the LITESPARK-005 trial is provided in Table 18.
At the time of the final analysis (data cut-off: April 15, 2024), the median duration of follow-up was 21.4 months (range, 0.2 to 47.6 months) in the belzutifan group and 18.3 months (range, 0.8 to 49.2 months) in the everolimus group.27 At the time of the final analysis, 513 OS events were observed (approximately 106% of the 483 events planned for the final analysis; 254 occurring in the belzutifan group and 259 occurring the everolimus group).27 The corresponding HR for OS was 0.92 (95% CI, 0.77 to 1.10; one-sided P = 0.17644), which was not statistically significant.27 Median OS was 21.4 months (95% CI, 18.2 to 24.3 months) in the belzutifan group and 18.2 months (95% CI, 15.8 to 21.8 months) in the everolimus group.27 The OS rates based on KM estimation were numerically higher in the belzutifan group than in the everolimus group at 12 months, 18 months, 24 months, 30 months, and 36 months; the estimated OS rate at 36 months was █████ ████ ███ ████ ██ █████ in the belzutifan group compared with 28.0% (95% CI, 23.1% to 33.1%) in the everolimus group (between-group difference for belzutifan compared with everolimus, ████ ████ ███ ████ ██ █████).27 The KM curves for OS are shown in Figure 6.
Subgroup analyses of OS in the final analysis were consistent with results from the primary analysis in all prespecified subgroups. Full details of the subgroup analyses for OS are presented in Appendix 1.
Table 18: Summary of Results for OS in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
Variable | Belzutifan (n = 374) | Everolimus (n = 372) |
|---|---|---|
Median follow-up time, months (range) | 21.4 (0.2 to 47.6) | 18.3 (0.8 to 49.2) |
Number of events (%) | 254 (67.9) | 259 (69.6) |
Kaplan-Meier estimates, monthsa | ||
Median (95% CI) | 21.4 (18.2 to 24.3) | 18.2 (15.8 to 21.8) |
HR vs. everolimus (95% CI)b | 0.92 (0.77 to 1.10) | |
P valuec | 0.17644 | |
OS rate at month 6, % (95% CI) | 81.6 (77.2 to 85.1) | 81.1 (76.8 to 84.8) |
Difference at 6 months vs. everolimus (95% CI) | 0.5 (−5.1 to 6.1) | |
OS rate at month 12, % (95% CI) | 67.9 (62.9 to 72.4) | 65.8 (60.7 to 70.3) |
Difference at 12 months vs. everolimus (95% CI) | 2.1 (−4.7 to 8.9) | |
OS rate at month 18, % (95% CI) | ████ █████ ██ █████ | 50.7 (45.5 to 55.6) |
Difference at 18 months vs. everolimus (95% CI) | ███ █████ ██ █████ | |
OS rate at month 24, % (95% CI) | 45.2 (40.1 to 50.1) | 41.2 (36.2 to 46.2) |
Difference at 24 months vs. everolimus (95% CI) | 4.0 (−3.1 to 11.1) | |
OS rate at month 30, % (95% CI) | 35.9 (31.0 to 40.8) | 33.1 (28.3 to 38.0) |
Difference at 30 months vs. everolimus (95% CI) | 2.8 (−4.1 to 9.7) | |
OS rate at month 36, % (95% CI) | ████ █████ ██ █████ | 28.0 (23.1 to 33.1) |
Difference at 36 months vs. everolimus (95% CI) | ███ █████ ██ █████ | |
CI = confidence interval; HR = hazard ratio; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; ITT = intention to treat; OS = overall survival; RCC = renal cell carcinoma; vs. = versus.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with the Efron method of tie handling, with treatment as a covariate stratified by IMDC risk group (favourable vs. intermediate vs. poor) and number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 or 3).
cOne-sided P value based on log-rank test stratified by IMDC risk group (favourable vs. intermediate vs. poor) and number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 or 3).
Sources: LITESPARK-005 Statistical Report final analysis27 and addendum.30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Figure 6: Kaplan-Meier Analysis for OS — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
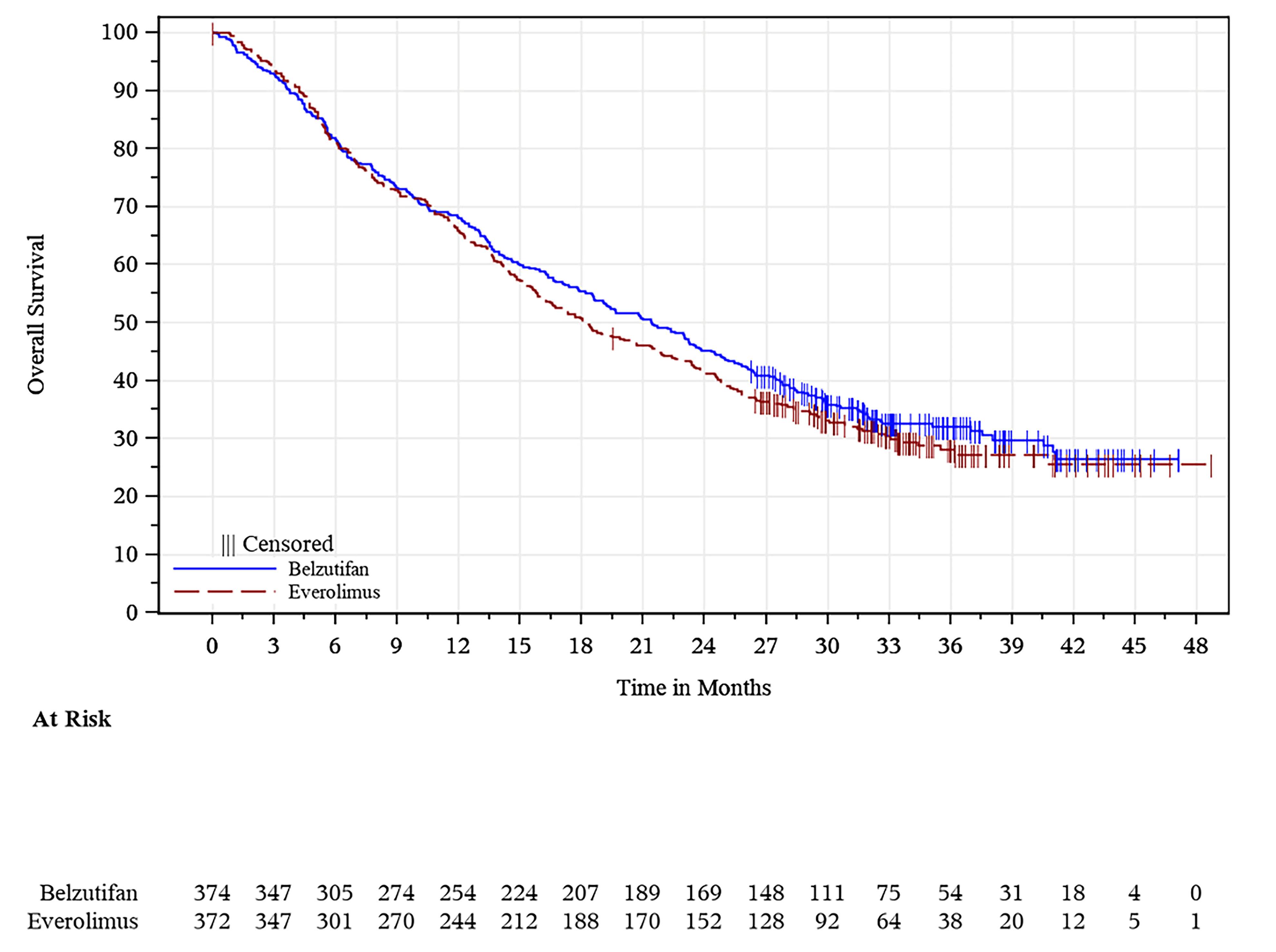
ITT = intention to treat; KM = Kaplan-Meier; OS = overall survival.
Source: LITESPARK-005 Statistical Report final analysis.27
ORR Based on BICR per RECIST 1.1
A summary of results for ORR based on BICR per RECIST 1.1 is provided in Table 19. At the time of the first interim analysis (data cut-off: November 1, 2022), the ORR per RECIST 1.1 by BICR in the belzutifan group was █████ ██████ ███ ████ ██ █████ compared to ████ ██████ ███ ███ ██ ████.26 The estimated difference in the percentage of patients with confirmed ORR for belzutifan versus everolimus was █████ ██████ ███ ████ ██ █████ ███████ ██████████.26 The P value crossed the prespecified boundary for statistical significance of 0.001 at the time of the first interim analysis.26 A higher proportion of patients had confirmed CR and PR in the belzutifan group (CR = 2.7%; PR = 19.3%) than in the everolimus group (CR = 0%;, PR = 3.5%).26
At the time of the second interim analysis (data cut-off: June 13, 2023), the ORR was █████ ██████ ███ ████ ██ █████ in the belzutifan group compared to 3.5% (99.9% CI, 1.2% to 7.8%) in the everolimus group.25 The estimated difference in the percentage of patients with confirmed ORR for belzutifan versus everolimus was █████ ██████ ███ ████ ██ █████ ███████ ███████ ██████████. At the time of the second interim analysis, a higher proportion of patients had confirmed CR and PR in the belzutifan group (CR = 3.5%; PR = 19.3%) than in the everolimus group (CR = 0%; PR = 3.5%).25
At the time of the final analysis (data cut-off: April 15, 2024), results of ORR for belzutifan compared to everolimus remained consistent with those of the second interim analysis.27
Duration of Response
A summary of results for DOR is provided in Table 19. At the time of the first interim analysis (data cut-off: November 1, 2022), the median DOR based on RECIST 1.1 by BICR was not yet reached (range, 1.7+ to 23.2+ months) in the belzutifan group and was 17.2 months (range, 3.8 to 18.0+ months) in the everolimus group.26 The proportion of responders was higher in the belzutifan group than the everolimus group at each response duration time point (i.e., 6 months through 21 months) based on KM estimates; 74.2% of responding patients in the belzutifan group and 68.4% of responding patients in the everolimus group had a DOR lasting 12 months or longer.26
At the time of the second interim analysis (data cut-off: June 13, 2023), the median DOR based on RECIST 1.1 by BICR was 19.5 months (range, 1.9+ to 31.6+ months) in the belzutifan group compared to 13.7 months (range, 3.8 to 21.2+ months) in the everolimus group.25 As was observed at the first interim analysis, the proportion of responders was higher in the belzutifan group than in the everolimus group at each response duration time point based on KM estimates; 73.2% of responding patients in the belzutifan group and 61.5% of responding patients in the everolimus group had a DOR lasting 12 months or longer.25
At the time of the final analysis (data cut-off: April 15, 2024), the median DOR was 19.3 months (range, 1.9+ to 40.1+ months) in the belzutifan group and 13.7 months (range, 3.8 to 29.5+ months) in the everolimus group; the proportion of responders remained higher in the belzutifan group than in the everolimus group at each response duration time point based on KM estimates.27
Time to Response
A summary of results for time to response is provided in Table 19. At the time of the first interim analysis (data cut-off: November 1, 2022), the median time to response was 3.7 months (range, 1.7 to 16.6) in the belzutifan group and 3.7 months (range, 1.8 to 5.4) in the everolimus group.26 At the time of the final analysis (data cut-off: April 15, 2024), the median time to response was 3.8 months (range, 1.7 to 22.0) in the belzutifan group and 3.7 months (range, 1.8 to 5.7) in the everolimus group.27
Table 19: Summary of Results for Response Outcomes in the LITESPARK-005 Trial — ITT Population (Interim Analysis 1 [Data Cut-Off of November 1, 2022] and Final Analysis [Data Cut-Off of April 15, 2024])
Variable | Interim analysis 1 (data cut-off of November 1, 2022) | Final analysis (data cut-off of April 15, 2024) | ||
|---|---|---|---|---|
Belzutifan (n = 374) | Everolimus (n = 372) | Belzutifan (n = 374) | Everolimus (n = 372) | |
ORR (by BICR per RECIST 1.1) | ||||
Number of patients with responsea | 82 | 13 | 85 | 13 |
ORR based on BICR per RECIST 1.1, % (99.9% CI) | ████ (████ ██ ████) | ███ ████ ██ ████ | 22.7 (████ ██ ████) | 3.5 (1.2 to 7.8) |
Difference vs. everolimus (%), | ||||
Estimate (99.9% CI)b,c | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
P valued | < 0.00001 | < 0.00001e | ||
Best confirmed objective response based on BICR per RECIST 1.1, n (%) | ||||
Complete response | 10 (2.7) | 0 (0.0) | 13 (3.5) | 0 (0.0) |
Partial response | 72 (19.3) | 13 (3.5) | 72 (19.3) | 13 (3.5) |
Stable disease | 147 (39.3) | 245 (65.9) | 143 (38.2) | 245 (65.9) |
Progressive disease | 126 (33.7) | 80 (21.5) | 127 (34.0) | 80 (21.5) |
Not evaluablef | 5 (1.3) | 8 (2.2) | 5 (1.3) | 9 (2.4) |
No assessmentg | 14 (3.7) | 26 (7.0) | 14 (3.7) | 26 (6.7) |
Time to response, months | ||||
Mean (SD) | 4.7 (3.3) | 3.5 (1.2) | 5.4 (4.2) | 3.6 (1.4) |
Median (range) | 3.7 (1.7 to 16.6) | 3.7 (1.8 to 5.4) | 3.8 (1.7 to 22.0) | 3.7 (1.8 to 5.7) |
Duration of response,h,i months | ||||
Median (range) | NR (1.7+ to 23.2+) | 17.2 (3.8 to 18.0+) | 19.3 (1.9+ to 40.1+) | 13.7 (3.8 to 29.5+) |
Patients with extended response duration, n (%) | ||||
≥ 6 months | 65 (93.1) | 10 (76.9) | 78 (94.0) | 10 (76.9) |
≥ 9 months | 42 (81.7) | 6 (68.4) | 67 (80.7) | 8 (61.5) |
≥ 12 months | 25 (74.2) | 6 (68.4) | 59 (71.1) | 8 (61.5) |
≥ 15 months | 17 (71.1) | 2 (51.3) | 50 (60.2) | 6 (46.2) |
≥ 18 months | 9 (61.9) | 0 (NR) | 39 (50.4) | 4 (30.8) |
≥ 21 months | 5 (61.9) | 0 (NR) | 33 (45.1) | 3 (23.1) |
BICR = blinded independent central review; CI = confidence interval; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; ITT = intention to treat; LS = least squares; NR = not reached; ORR = objective response rate; Q = quartile; QoL = quality of life; PFS = progression-free survival; PRO = patient-reported outcome; RCC = renal cell carcinoma; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SD = standard deviation; VAS = visual analogue scale; vs. = versus.
aIncludes patients with confirmed complete response or partial response.
bBased on the Miettinen and Nurminen method stratified by IMDC risk group (favourable vs. intermediate vs. poor) and number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 or 3).
cBased on the procedure for testing for multiplicity in the trial, the ultimate alpha used for testing ORR was determined to be 0.001. Thus, 99.9% CIs were reported alongside the point estimates for ORR.
dOne-sided P value for testing. H0: difference in % = 0 vs. H1: difference in % > 0.
eThis P value was from a supplemental analysis to the primary analysis and, thus, cannot be used to make inferences about PFS.
fIncludes patients with insufficient data for the assessment of response per RECIST 1.1.
gIncludes patients without postbaseline assessment on the data cut-off date.
hFrom product-limit (Kaplan-Meier) method for censored data.
iThe + sign indicates there was no progressive disease at the time of the last disease assessment.
Sources: LITESPARK-005 Clinical Study Report interim analysis 1,25 LITESPARK-005 Statistical Report final analysis,27 and addendum.30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Health-Related Quality of Life
The HRQoL outcomes of interest (i.e., EORTC QLQ-C30 and FKSI-DRS) were assessed at the first interim analysis (data cut-off: November 1, 2022)26 and second interim analysis (data cut-off: June 13, 2023).25 Results for HRQoL outcomes of interest were not assessed at the final analysis (data cut-off: April 15, 2024). The HRQoL outcomes were not controlled for multiplicity; thus, all reported P values and CIs are nominal and descriptive. Week 17 was selected as the time point for assessing change from baseline, based on the meeting of prespecified criteria for completion and compliance rates (i.e., a completion rate of 60% or greater and a compliance rate of 80% or greater) during the blinded data review before database lock. A summary of results for HRQoL in the LITESPARK-005 trial is provided in Table 20.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
At the time of the first interim analysis (data cut-off: November 1, 2022), the completion rate at baseline was 96.2% for patients in the belzutifan group and 94.1% for patients in the everolimus group.26 At week 17, the completion rate was 65.0% for patients in the belzutifan group and 56.7% in the everolimus group.26 At week 17, in the belzutifan group, there was no change in EORTC QLQ-C30 GHS/QoL scores from baseline █████ ██████ ████ ███ █████ ██ ██████, whereas in the everolimus group, there was a numerical decrease (worsening) in score from baseline (LS mean, −6.13; 95% CI, −8.51 to −3.75).26 At week 17, the difference in LS mean change in EORTC QLQ-C30 GHS/QoL scores from baseline between the belzutifan and everolimus groups was ████ ██████ ████ ███ ████ ██ █████ ███████ █████████.26 Median TTD in the EORTC QLQ-C30 GHS/QoL score was longer in the belzutifan group (median = not reached [95% CI, 11.99 months to not reached]) compared with the everolimus group (median = 10.15 months [95% CI, 6.47 to 12.98 months]), with a corresponding HR of 0.74 (95% CI, 0.58 to 0.95; nominal P < 0.0179).26
At the second interim analysis (data cut-off: June 13, 2023), similar results were noted for change in EORTC QLQ-C30 GHS/QoL from baseline between the 2 treatment groups (difference in LS means for belzutifan compared to everolimus = 6.38; 95% CI, 3.21 to 9.55).25 The TTD in the EORTC QLQ-C30 GHS/QoL score continued to be longer in the belzutifan group (median = 19.35 months; 95% CI, 11.89 months to not reached) compared with the everolimus group (median = 10.19 months; 95% CI, 6.47 to 12.98 months), with a corresponding HR of 0.75 (95% CI, 0.58 to 0.96; nominal P < 0.0185).25 Results for the EORTC QLQ-C30 instrument were not assessed at the final analysis (data cut-off: April 15, 2024).
Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms
At the time of the first interim analysis (data cut-off: November 1, 2022), the completion rate at baseline was 96.5% for patients in the belzutifan group and 94.4% for patients in the everolimus group.26 At week 17, the completion rate was 65.3% for patients in the belzutifan group and 56.7% for the everolimus group.26 At week 17, both groups reported a numerical decrease (worsening) in FKSI-DRS score from baseline, with a larger decrease in score noted for patients in the everolimus group (LS mean change = −1.61 points; 95% CI, −2.17 to −1.06 points) compared with the belzutifan group (LS mean change = −0.07 points; 95% CI, −0.59 to 0.45 points).26 The difference in LS mean change in FKSI-DRS from baseline between the belzutifan and everolimus groups was 1.54 points (95% CI, 0.81 to 2.28 points; nominal P < 0.0001).26 The TTD in the FKSI-DRS score was longer in the belzutifan group (median = not reached; 95% CI, not reached to not reached) compared with the everolimus group (median = 11.99 months; 95% CI, 6.44 to 15.70 months), with a corresponding HR of 0.53 (95% CI, 0.41 to 0.69; nominal P < 0.0001).26
At the second interim analysis, similar results were noted for change in FKSI-DRS from baseline between the 2 treatment groups (difference in LS means for belzutifan compared to everolimus = 1.45; 95% CI, 0.70 to 2.19).25 The TTD in the FKSI-DRS score continued to be longer in the belzutifan group (median = not reached; 95% CI, 21.26 months to not reached) compared with the everolimus group (median = 11.9 months; 95% CI, 5.55 to 15.38 months), with a corresponding HR of 0.53 (95% CI, 0.41 to 0.69; nominal P < 0.0001).25 Results for the FKSI-DRS instrument were not assessed at the final analysis (data cut-off: April 15, 2024).
Table 20: Summary of Results for HRQoL Outcomes in the LITESPARK-005 Trial — ITT Population (Interim Analysis 1 [Data Cut-Off of November 1, 2022] and Interim Analysis 2 [Data Cut-Off of June 13, 2023])
Variable | Interim analysis 1 (data cut-off of November 1, 2022) | Interim analysis 2 (data cut-off of June 13, 2023) | ||
|---|---|---|---|---|
Belzutifan (n = 374) | Everolimus (n = 372) | Belzutifan (n = 374) | Everolimus (n = 372) | |
EORTC QLQ-C30 GHS/QoL | ||||
Week 17 | ||||
Number of patients assessed | 240 | 202 | 234 | 200 |
Mean (SD) | 68.51 (19.58) | 60.68 (21.80) | 68.30 (19.75) | 60.54 (21.84) |
Change from baseline to week 17a | ||||
Number of patients assessed | 368 | 356 | 363 | 353 |
LS mean (95% CI) | ███ █████ █ █████ | −6.13 (−8.51 to −3.75) | 0.28 (−1.98 to 2.53) | −6.11 (−8.51 to −3.70) |
Difference in LS means | ████ █████ ██ █████ | 6.38 (3.21 to 9.55) | ||
P value | < 0.0001b,c | < 0.0001b,c | ||
TTDd | ||||
Number of patients assessed | 355 | 335 | 351 | 333 |
Median TTD, months (95% CI)e | NR (11.99 to NR) | 10.15 (6.47 to 12.98) | 19.35 (11.89 to NR) | 10.19 (6.47 to 12.98) |
HR (95% CI)f | 0.74 (0.58 to 0.95) | 0.75 (0.58 to 0.96) | ||
P value | 0.0179b,g | 0.0185b,g | ||
FKSI-DRS | ||||
Week 17 | ||||
Number of patients assessed | 241 | 202 | 235 | 200 |
Mean (SD) | 30.91 (4.51) | 29.20 (4.97) | 30.82 (4.52) | 29.15 (4.96) |
Change from baseline to week 17a | ||||
Number of patients assessed | 368 | 356 | 363 | 353 |
LS mean (95% CI) | −0.07 (−0.59 to 0.45) | −1.61 (−2.17 to −1.06) | −0.17 (−0.70 to 0.36) | −1.62 (−2.17 to −1.06) |
Difference in LS means | 1.54 (0.81 to 2.28) | 1.45 (0.70 to 2.19) | ||
P value | < 0.0001b,c | 0.0002b,c | ||
TTDh | ||||
Number of patients assessed | 356 | 336 | 352 | 334 |
Median TTD, months (95% CI)e | NR | 11.99 | NR (21.26 to NR) | 11.99 (5.55 to 15.38) |
HR (95% CI)f | 0.53 (0.41 to 0.69) | 0.53 (0.41 to 0.69) | ||
P value | < 0.0001b,g | < 0.0001b,g | ||
CI = confidence interval; cLDA = constrained longitudinal data analysis; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FKSI-DRS = Functional Assessment of Cancer Therapy–Kidney Symptom Index–Disease-Related Symptoms; GHS = global health status; HR = hazard ratio; HRQoL = health-related quality of life; IMDC = International Metastatic Renal Cell Carcinoma Database Consortium; LS = least squares; NR = not reached; PRO = patient-reported outcome; QoL = quality of life; SD = standard deviation; TTD = time to deterioration; VEGF = vascular endothelial growth factor.
aBased on a cLDA model, with PRO scores as the response variable, with covariates for the treatment-by-time interaction and stratification factors of IMDC prognostic scores (0 versus 1 to 2 versus 3 to 6) and the number of prior antiangiogenic therapies for advanced RCC (1 versus 2 or 3) as covariates.
bTwo-sided P value is based on a t test.
cCorresponds to outcome that was not controlled for multiple comparisons.
dDeterioration is defined as the time to first onset of a ≥ 10-point deterioration (i.e., decrease in score) from baseline, with confirmation at the subsequent visit of a ≥ 10-point deterioration from baseline under the right-censoring rule. If the first deterioration is at the last PRO assessment time point in the current database, no confirmation is required.
eFrom product-limit (Kaplan-Meier) method for censored data.
fBased on Cox regression model with the Efron method of tie handling, with treatment as a covariate, stratified by IMDC prognostic scores (0 versus 1 to 2 vs 3 to 6) and the number of prior VEGF- or VEGF receptor–targeted therapies for advanced RCC (1 versus 2 or 3).
gTwo-sided P value based on the log-rank test stratified by IMDC prognostic scores (0 vs 1 to 2 vs 3 to 6) and the number of prior VEGF- or VEGF receptor–targeted therapies for advanced RCC (1 versus 2 or 3).
hDeterioration is defined as the time to first onset of a ≥ 3-point deterioration (i.e., decrease in score) from baseline with confirmation at the subsequent visit of a ≥ 3-point deterioration from baseline under the right-censoring rule. If the first deterioration is at the last PRO assessment time point in the current database, no confirmation is required.
Sources: LITESPARK-005 Clinical Study Report interim analysis 126 and LITESPARK-005 Clinical Study Report interim analysis 2.25
Harms
The key harms results for the safety population (i.e., all patients as treated [APaT] population) are summarized in Table 21.
Adverse Events
Almost all patients in the LITESPARK-005 trial reported at least 1 AE (99.2% for both belzutifan and everolimus treatment groups).27 The most common AE in both treatment groups was anemia (83.1% in the belzutifan group versus 57.2% in the everolimus group), followed by fatigue (32.3%) and nausea (18.5%) in the belzutifan group and stomatitis (38.1%) and fatigue (25.8%) in the everolimus group.27
Serious Adverse Events
At least 1 SAE was reported in █████ of patients in the belzutifan group and 38.6% of patients in the everolimus group, with the most common SAEs reported being hypoxia (7.3% in the belzutifan group and none in the everolimus group) and anemia (5.4% in the belzutifan group and 2.2% in everolimus group).27
Discontinuation Due to Adverse Events
Discontinuation of the study treatment due to AEs was notably higher among patients in the everolimus group (15.3%) than those in the belzutifan group (████).27 The most common AEs that led to treatment discontinuation in the both of the treatment groups were related to respiratory, thoracic, and mediastinal disorders (1.9% for the belzutifan group compared with 6.9% for the everolimus group).27
Mortality
Death as a result of AEs was slightly higher among patients in the everolimus group (5.3%) than patients in the belzutifan group (3.8%).27 The most common AEs that led to death in both groups were related to infections and infestations (0.8% in the belzutifan group vs 3.1% for the everolimus group).27 The most common infections and infestations leading to death in the belzutifan group were abdominal sepsis (0.3%), pneumonia (0.3%), and sepsis (0.3%), whereas the most common infections and infestations leading to death in the everolimus group were COVID-19-related pneumonia (1.1%) and unspecified pneumonia (0.8%).27
Notable Harms
Anemia, hypoxia, and dyspnea were identified as clinical AEs of special interest, as they have been associated with treatment with belzutifan. At the final analysis, a higher rate of anemia-related events was reported in patients in the belzutifan group compared with patients in the everolimus group (83.3% for the belzutifan group and 57.5% for the everolimus group).27 In both groups, most events related to anemia were grade 2 (41.9% for the belzutifan group and 29.7% for the everolimus group), and treatment discontinuation related to anemia was low (0.3% for the belzutifan group and 0.6% for the everolimus group).27 Among the patients who reported anemia, 39.0% were treated with belzutifan and 48.3% reported the event resolved.27
Notably, a higher rate of hypoxia was reported in the belzutifan group compared with the everolimus group (14.2% for the belzutifan group and 1.1% for the everolimus group).27 In both groups, most events related to hypoxia were grade 3 (10.5% for the belzutifan group and 0.8% for the everolimus group), and treatment discontinuation related to hypoxia was low (0.8% for the belzutifan group and 0% for the everolimus group).27 Among the patients who reported hypoxia, 83.0% were treated with belzutifan and 75.0% treated with everolimus reported the event resolved.27
Similar rates of dyspnea were reported in the belzutifan (15.3%) and everolimus (14.4%) groups.27 In both groups, most events related to dyspnea were grade 1 (10.2% for belzutifan group and 8.1% for everolimus group), and treatment discontinuation related to dyspnea was low (0.3% for belzutifan group and 0% in everolimus group).27 Among patients who reported dyspnea, 57.9% were treated with belzutifan and 32.7% treated with everolimus reported the event resolved.27
Table 21: Summary of Harms Results in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024)
Adverse eventsa | Belzutifan (n = 372) | Everolimus (n = 360) |
|---|---|---|
Most common AEs (incidence of 10% or higher in either group), n (%)a-e | ||
≥ 1 AEs | 369 (99.2) | 357 (99.2) |
Anemia | 309 (83.1) | 206 (57.2) |
Fatigue | 120 (32.3) | 93 (25.8) |
Nausea | 69 (18.5) | 44 (12.2) |
Peripheral edema | 64 (17.2) | 65 (18.1) |
Constipation | 63 (16.9) | 30 (8.3) |
Back pain | 62 (16.7) | 32 (8.9) |
Arthralgia | 60 (16.1) | 29 (8.1) |
Dyspnea | 57 (15.3) | 52 (14.4) |
Asthenia | 56 (15.1) | 61 (16.9) |
Decreased appetite | 55 (14.8) | 57 (15.8) |
Hypoxia | 53 (14.2) | 4 (1.1) |
Dizziness | 50 (13.4) | 6 (1.7) |
Vomiting | 50 (13.4) | 32 (8.9) |
Diarrhea | 49 (13.2) | 72 (20.0) |
Headache | 49 (13.2) | 28 (7.8) |
Increased alanine aminotransferase | 48 (12.9) | 33 (9.2) |
Increased aspartate aminotransferase | 43 (11.6) | 33 (9.2) |
COVID-19 | 40 (10.8) | 34 (9.4) |
Increased blood creatinine | 33 (8.9) | 44 (12.2) |
Cough | 33 (8.9) | 76 (21.1) |
Pruritus | 31 (8.3) | 60 (16.7) |
Pyrexia | 23 (6.2) | 46 (12.8) |
Rash | 17 (4.6) | 69 (19.2) |
Hypertriglyceridemia | 15 (4.0) | 54 (15.0) |
Stomatitis | 13 (3.5) | 137 (38.1) |
Hyperglycemia | 12 (3.2) | 55 (15.3) |
Pneumonitis | 3 (0.8) | 53 (14.7) |
SAEs (incidence of 5% or higher in either group), n (%)c-f | ||
Patients with ≥ 1 SAE | ███ ██████ | 139 (38.6) |
Hypoxia | 27 (7.3) | 0 (0.0) |
Anemia | 20 (5.4) | 8 (2.2) |
Stopped treatment due to AEs (incidence of 1% or higher in either group), n (%)b-d | ||
Patients who stopped | ██ █████ | 55 (15.3) |
Cardiac disorders | 2 (0.5) | 4 (1.1) |
Gastrointestinal disorders | 3 (0.8) | 5 (1.4) |
General disorders and administration-site conditions | 2 (0.5) | 5 (1.4) |
Infections and infestations | 2 (0.5) | 7 (1.9) |
Respiratory, thoracic, and mediastinal disorders | 7 (1.9) | 23 (6.4) |
Pneumonitis | 0 (0.0) | 17 (4.7) |
Skin and subcutaneous tissue disorders | 0 (0.0) | 4 (1.1) |
Deaths (incidence higher than 0% in either group), n (%)b-d | ||
Patients who died | 14 (3.8) | 19 (5.3) |
Cardiac disorders | 2 (0.5) | 0 (0.0) |
Myocardial infarction | 1 (0.3) | 0 (0.0) |
Pericardial hemorrhage | 1 (0.3) | 0 (0.0) |
Gastrointestinal disorders | 1 (0.3) | 2 (0.6) |
Intra-abdominal hemorrhage | 0 (0.0) | 1 (0.3) |
Pancreatitis acute | 0 (0.0) | 1 (0.3) |
Upper gastrointestinal hemorrhage | 1 (0.3) | 0 (0.0) |
General disorders and administration-site conditions | 2 (0.5) | 0 (0.0) |
Death | 1 (0.3) | 0 (0.0) |
Multiple organ dysfunction syndrome | 1 (0.3) | 0 (0.0) |
Infections and infestations | 3 (0.8) | 11 (3.1) |
Abdominal sepsis | 1 (0.3) | 0 (0.0) |
COVID-19 | 0 (0.0) | 2 (0.6) |
COVID-19 pneumonia | 0 (0.0) | 4 (1.1) |
Clostridium difficile infection | 0 (0.0) | 1 (0.3) |
Pneumonia | 1 (0.3) | 3 (0.8) |
Sepsis | 1 (0.3) | 1 (0.3) |
Injury, poisoning, and procedural complications | 1 (0.3) | 0 (0.0) |
Road traffic accident | 1 (0.3) | 0 (0.0) |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 2 (0.5) | 0 (0.0) |
Gastric cancer | 1 (0.3) | 0 (0.0) |
Squamous cell carcinoma | 1 (0.3) | 0 (0.0) |
Nervous system disorders | 1 (0.3) | 0 (0.0) |
Seizure | 1 (0.3) | 0 (0.0) |
Renal and urinary disorders | 1 (0.3) | 1 (0.3) |
Acute kidney injury | 0 (0.0) | 1 (0.3) |
Nephrolithiasis | 1 (0.3) | 0 (0.0) |
Respiratory, thoracic, and mediastinal disorders | 1 (0.3) | 4 (1.1) |
Pleural effusion | 0 (0.0) | 1 (0.3) |
Pulmonary embolism | 0 (0.0) | 2 (0.6) |
Respiratory failure | 1 (0.3) | 1 (0.3) |
Surgical and medical procedures | 0 (0.0) | 1 (0.3) |
Assisted suicide | 0 (0.0) | 1 (0.3) |
AEs of special interest, n (%)b,d | ||
Anemia-related events | 310 (83.3) | 207 (57.5) |
Anemia | 309 (83.1) | 206 (57.2) |
Decreased hemoglobin | 2 (F0.5) | 2 (0.6) |
Dyspnea | 57 (15.3) | 52 (14.4) |
Hypoxia | 53 (14.2) | 4 (1.1) |
AE = adverse event; ITT = intention to treat; SAE = serious adverse event.
aGrades are based on National Cancer Institute Common Terminology Criteria for Adverse Events (Version 5.0).
bNonserious AEs up to 30 days after the last dose and SAEs up to 90 days after last dose are included.
cMedical Dictionary for Regulatory Activities (MedDRA) 27.0 preferred terms “Neoplasm progression,” “Malignant neoplasm progression,” and “Disease progression” not related to the drug are excluded.
dEvery participant is counted a single time for each applicable row and column.
eA specific AE appears on this table only if its incidence in 1 or more of the columns meets the incidence criterion in the table subhead, after rounding.
fSAEs up to 90 days after the last dose are included.
Sources: LITESPARK-005 Statistical Report final analysis.27 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Critical Appraisal
Internal Validity
LITESPARK-005 is a phase III, open-label, active-controlled randomized trial that evaluated the efficacy and safety of belzutifan compared to everolimus in patients with advanced RCC who were previously treated with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. Notable strengths of the trial included the use of an ITT analysis and the stratification of randomization according to IMDC prognostic scores and the number of prior VEGF-targeted therapies for advanced RCC. The randomization process of the trial was deemed to be appropriate, although there was limited detail on how randomization numbers allocated to patients were obtained. Moreover, the LITESPARK-005 trial had an open-label study design, indicating that treatments were not blinded to patients and investigators. Key efficacy outcomes PFS and ORR were assessed based on BICR, which may have mitigated the effects of assessment bias. However, the lack of blinding of patients may have contributed to performance bias in results for patient-reported outcomes.
The 2 treatment groups were balanced in terms of baseline patient and disease characteristics. Moreover, prior and concomitant non-oncologic medications were, overall, balanced in the 2 treatment groups. Although some imbalances in commonly reported categories of concomitant medications received were noted, the clinical experts consulted by CDA-AMC anticipated that these imbalances would have minimal impact on the treatment effect.
It was also noted that ████ patients in the everolimus group received ██████████ █████████ █████████ compared with patients in the belzutifan group (█████ ██████ █████). This difference could potentially introduce a confounding effect on OS, as the survival results might be partially attributable to treatments administered after disease progression, rather than the study treatment itself. Although this difference could favour the ██████████ group, the risk of bias due to deviations from the intended interventions was deemed to be low by the clinical experts consulted by CDA-AMC. The clinical experts consulted by CDA-AMC confirmed that the oncologic therapies used in the LITESPARK-005 trial were largely reflective of those used in Canadian clinical practice, although some differences in treatment sequence were observed. For instance, █████ of patients in the LITESPARK-005 trial received cabozantinib as a subsequent therapy after belzutifan or everolimus, whereas the clinical experts consulted by CDA-AMC anticipated that cabozantinib would be used first, followed by belzutifan. The clinical experts noted that if belzutifan were publicly reimbursed, most patients in clinical practice would be expected to receive belzutifan as third-line treatment or, to a lesser extent, as a fourth-line treatment, and cabozantinib would mainly remain as a second-line treatment. After treatment with belzutifan, the clinical experts anticipated there would be no further approved and publicly funded treatment options in clinical practice in Canada.
In the LITESPARK-005 trial, approximately 13%, 43%, 43%, and 1% of patients received 1, 2, 3, or 4 prior lines of therapy, respectively. Exploratory subgroup results for OS and PFS by line of therapy (i.e., 1, 2, or 3 prior lines of therapy) were consistent with those for the overall trial population. Although some uncertainty remains regarding the impact of differences in prior lines of therapy between the trial and expected clinical practice on the generalizability of results, the clinical experts consulted by CDA-AMC felt it would be reasonable to generalize the trial results to anticipated clinical practice, and did not raise concerns regarding the applicability of the LITESPARK-005 trial results in the Canadian clinical context.
Two interim analyses and 1 final analysis were prespecified in the statistical analysis plan and have been completed. A multiple testing procedure was employed to control the overall type I error for the primary end points of PFS and OS and the key secondary end point of ORR. Of note, the primary analysis for PFS and ORR was assessed at the first interim analysis, which could potentially have resulted in an overestimation of the treatment effect for belzutifan. However, 519 PFS events had occurred at the first interim analysis, which constituted 82.9% of the total expected events for PFS at the final analysis. Moreover, the results for PFS and ORR at the first interim analysis were, overall, consistent with those from the final analysis. Thus, the review team determined that the risk of overestimation was small.
In the ITT population of the LITESPARK-005 trial, a smaller proportion of patients in the belzutifan group discontinued study treatment (85.5%) compared with patients in the everolimus group (98.6%).27 The imbalance in study treatment discontinuation between the 2 groups was primarily driven by discontinuation due to AEs (████ for the belzutifan group compared with 15.3% for the everolimus group).27 For the assessment of PFS by BICR per RECIST 1.1, it was noted that a larger proportion of patients in the everolimus group (18.3%) were censored due to the initiation of new anticancer therapy before a PFS event than those in the belzutifan group (5.6%).26 However, the trial performed sensitivity analyses which counted the initiation of new anticancer therapy as a PFS event. The results of these analyses were consistent with those of the primary analysis for the ITT population, which suggested that the between-group imbalances in patients starting new anticancer therapy had little impact on results for PFS.26
A stratified Cox proportional hazards model, adjusted for stratification factors, was used to estimate the HRs and CIs for PFS. This model assumes proportional hazards across treatment arms. Visual inspection of the KM curves by the CDA-AMC review team revealed that for PFS, the curves for the intervention and comparator treatment arms crossed multiple times and did not separate until approximately 6 months. Although this suggests that the HRs may not accurately reflect the treatment effect over time, it is likely a result of a variation in effects between the treatment and an active control during the early stages of treatment initiation. The clinical experts consulted by CDA-AMC suggested that belzutifan may have a longer duration of efficacy compared to everolimus, which may explain why the benefit of belzutifan is observed at later time points beyond those corresponding to median PFS. The KM-estimated between-group differences in the probability of PFS at clinically relevant follow-up times, used herein to inform the certainty of evidence for this end point, were not affected by this limitation.
Key HRQoL outcomes of interest in the LITESPARK-005 trial included the EORTC QLQ-C30 and FKSI-DRS instruments. The FKSI-DRS has been validated in patients with RCC, with evidence of reliability, responsiveness, and MID (estimated to be between 2 and 3 points54). For the EORTC QLQ-C30 scale score, a change of 10 points in the scale score and the summary score is conventionally considered to be a MID.26 No literature was identified that estimated MIDs specifically in patients with advanced RCC. The clinical experts consulted by CDA-AMC agreed that the EORTC QLQ-C30 and FKSI-DRS were suitable for measuring HRQoL outcomes in patients with advanced RCC. However, they noted that the EORTC QLQ-C30 is a preferred measure of HRQoL; the FKSI-DRS instrument may be more applicable to patients with localized disease (i.e., outside of the target population). Of note, HRQoL was not adjusted for multiple testing in the LITESPARK-005 trial; therefore, the ability to draw conclusions from results for HRQoL are limited. Moreover, high attrition rates were noted in the analyses for change from baseline to week 17; at week 17, 61.0% and 57.4% of patients completed the EORTC QLQ-C30 and FKSI-DRS questionnaires, respectively.26 The impact of belzutifan on HRQoL was considered to be of low certainty by the CDA-AMC reviewers, owing to the potential for reporting and attrition bias. The belzutifan group had lower attrition rates at week 17 than the everolimus group. This may be attributed to the smaller proportion of patients in the belzutifan group than in the everolimus group discontinuing the study treatment, which was driven by treatment discontinuations due to AEs. However, the magnitude and direction of the potential bias arising from the imbalance in missing data remains unclear. Of note, the analysis for mean change in HRQoL score from baseline treated missing data as missing at random; however, the review team found that this assumption is not likely sufficient to account for missing data related to treatment discontinuation.
External Validity
The clinical experts consulted by CDA-AMC agreed that the eligibility criteria for the LITESPARK-005 trial were standard for clinical trials but were more restrictive than what is observed in patients in clinical practice. Nonetheless, they agreed that patients receiving belzutifan in clinical practice are expected to have a good performance status and an absence of hypoxia due to the safety profile of belzutifan. The experts consulted for this review agreed that the baseline characteristics in the LITESPARK-005 trial were generally representative of the heavily pretreated patient population with RCC.
The clinical experts consulted by CDA-AMC also agreed that prior and concomitant non-oncologic medications received by patients in the trial were representative of a heavy pretreated population of patients with RCC in clinical practice.
The LITESPARK-005 trial assessed the safety and efficacy of belzutifan compared with everolimus. Although once considered a standard treatment for pretreated advanced RCC, everolimus has been superseded by newer treatment options (e.g., cabozantinib and axitinib) and is currently under restricted access in most jurisdictions in Canada, except for Alberta, where it is listed under full benefit.10-18 The clinical experts consulted by CDA-AMC agreed that everolimus is a less relevant comparator for later-line advanced RCC compared to axitinib or cabozantinib, and is rarely used in Canadian clinical practice. Thus, the stand-alone results of the trial may not provide a full assessment of the efficacy and safety of belzutifan compared to existing treatments for advanced RCC in Canadian clinical practice. The clinical experts consulted by CDA-AMC suggested that everolimus may be similar to axitinib in terms of efficacy, based on how these drugs perform in clinical practice. Apart from AEs related to the mechanism of action of belzutifan (i.e., anemia and hypoxia), the clinical experts did not suggest any additional safety concerns with belzutifan compared to other key comparators for RCC in Canada. Of note, pERC previously discussed that everolimus was similar to axitinib in terms of efficacy and safety as part of the recommendation for funding cabozantinib for the treatment of advanced RCC.20 The clinical experts consulted by CDA-AMC anticipated that some patients who receive nivolumab-cabozantinib in the first line may still receive axitinib (rather than belzutifan) in the second line, followed by belzutifan in the third line, to avoid having to eliminate 1 line of treatment. The clinical experts also agreed that belzutifan should be reserved for the next line of therapy after cabozantinib, noting that there is currently a lack of head-to-head RCTs and no evidence to suggest that belzutifan performs substantially better than cabozantinib. The clinical experts consulted by CDA-AMC noted that treatment options in third-line or later-line settings for advanced RCC remain limited, with a lack of standard treatment options noted for fourth-line advanced RCC. The experts emphasized that a treatment’s ability to delay progression and achieve a response in these treatment settings would be highly valued in clinical practice. Thus, the clinical experts agreed that belzutifan would be of value to the Canadian treatment landscape for advanced RCC, as it introduces a treatment option for later lines of therapy for advanced RCC.
Several studies submitted by the sponsor supported the clinical utility of PFS as a surrogate outcome for OS in advanced or metastatic RCC.58-60 Two of these studies evaluated the surrogacy of PFS for OS in patients with metastatic RCC,59,60 whereas the remaining study evaluated surrogacy in patients with advanced RCC.58 Two studies assessed PFS as a surrogate for OS using correlation analyses based on published trials, which may have been subject to publication bias.58,59 These studies also cited limitations related to the confounding of OS due to crossover and the receipt of postprogression oncologic therapies in the included trials.58,59 The remaining study assessed the correlation between PFS and OS using individual patient-level data from 2 phase III RCTs that enrolled patients with first-line metastatic RCC; such results may not be generalizable to patients with later-line advanced RCC.60 The validity of a surrogate is likely both disease-specific and intervention-specific, and none of the studies evaluated belzutifan treatment. The CDA-AMC review team determined that the results for PFS and OS in the LITESPARK-005 trial are unlikely to suggest evidence of surrogacy. Thus, based on these studies, it remains unclear whether the belzutifan treatment effect on PFS could be interpreted as a surrogate outcome for OS for the target population of this review.
Last, the clinical experts consulted by CDA-AMC agreed that the management of anemia and hypoxia related to treatment with belzutifan in Canadian clinical practice were similar to methods for managing these AEs in the LITESPARK-005 trial.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.28,29
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word likely for evidence of moderate certainty (e.g., X intervention likely results in Y outcome).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word may for evidence of low certainty (e.g., X intervention may result in Y outcome).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as very uncertain.
Following the GRADE approach, evidence from RCTs starts as high-certainty evidence and can be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessments for PFS, OS, ORR, SAEs, and discontinuation due to AEs were set according to the presence or absence of an important effect, based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of evidence assessment for the EORT QLQ-C30 GHS/QoL score was sourced from the literature.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for belzutifan versus everolimus.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
No long-term extension studies were included in this submission.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
No ITCs were included in this submission.
Objectives and Methods for the Summary of Indirect Evidence
The sponsor conducted an assessment to determine the feasibility of ITCs estimating the comparative efficacy and safety of belzutifan and cabozantinib for the treatment of adult patients with advanced RCC, after prior treatment with an immune checkpoint inhibitor and an antiangiogenic therapy.61 The sponsor’s rationale underlying such ITCs is that belzutifan’s proposed place in therapy suggests that it would serve as an alternative to cabozantinib in patients with advanced RCC who are ineligible for the full dose of 60 mg. Given the absence of direct evidence comparing belzutifan to cabozantinib for advanced RCC, the sponsor conducted a feasibility assessment for ITCs comparing belzutifan and cabozantinib in advanced RCC, using data from their respective pivotal trials, LITESPARK-00525-27 and METEOR.62 In particular, the feasibility of a network meta-analysis and MAIC were assessed. The sponsor conducted a global systematic literature review to identify clinical trials investigating the efficacy and safety of second-line or later-line therapies for the treatment of adult patients with unresectable, locally advanced or metastatic RCC after prior treatment with an immune checkpoint inhibitor and an antiangiogenic therapy.61
Appraisal of the Feasibility Assessment
The systematic literature review identified 2 relevant RCTs for the feasibility assessment: LITESPARK-005 (belzutifan) and METEOR (cabozantinib). Both trials were phase III, open-label RCTs that evaluated patients with advanced, clear cell RCC and a minimum KPS score of 70%. Notably, both trials had everolimus as a common comparator to their respective interventions. Although the trials were comparable in terms of age, sex, race, and IMDC risk, the trials significantly differed in terms of ECOG PS and prior therapy. In particular, the LITESPARK-005 trial enrolled more patients who had an ECOG PS of 1 than the METEOR trial, and had more stringent criteria pertaining to prior treatment (patients in the LITESPARK-005 trial were required to have received prior treatment with an IO drug and a VEGF-TKI and received no more than 3 prior therapies, whereas patients in the METEOR trial were required to have received treatment with a VEGF-TKI but had no upper limit on number of prior therapies). Although the METEOR trial had subgroup data available for patients who had received an IO drug and a VEGF-TKI, it had a small sample size (n = 32 patients; 18 who received cabozantinib and 14 who received everolimus), which was expected to introduce significant uncertainty in the analyses. Moreover, despite the stratification of randomization, the subgroup of patients in the METEOR trial contained more patients who had been heavily pretreated in the cabozantinib group than in the everolimus group. However, patients in both arms of the LITESPARK-005 trial were balanced in terms of the number of prior VEGF-TKIs received.
To minimize bias from the heterogeneity of patient baseline characteristics, the feasibility of conducting alternative methods of ITCs was also assessed.61 Both unadjusted (i.e., Bucher method) and adjusted (i.e., MAIC) approaches were considered. The Bucher method was deemed to be infeasible due to compromised randomization in the METEOR trial’s subgroup of patients previously treated with IO drugs and VEGF-TKIs, whereas MAICs were infeasible due to the lack of reporting of key effect modifiers (i.e., IMDC risk classification and number of prior lines of therapy) for this subgroup. The small sample size of the subgroup was also a key limitation for both approaches, given its potential to introduce uncertainty in the analysis.
Based on the results of the feasibility assessment, the CDA-AMC review team agreed that neither a network meta-analysis (i.e., Bucher ITC) nor alternative methods of MAICs (i.e., anchored MAIC) were likely to provide unbiased treatment effect estimates for the comparison of belzutifan and cabozantinib for the treatment of adult patients with RCC.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
No studies addressing gaps in the evidence from the systematic review were included in this submission.
Discussion
Summary of Available Evidence
One pivotal phase III, open-label, active-controlled randomized trial (LITESPARK-005) submitted by the sponsor was summarized in this report. The objective of the LITESPARK-005 trial was to evaluate the efficacy and safety of belzutifan (n = 374) compared to everolimus (n = 372) in patients with advanced RCC who were previously treated with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The trial enrolled adult patients with unresectable, locally advanced, or metastatic clear RCC who were required to have had disease progression on or after having received treatment with both a PD-1 or PD-L1 inhibitor and a VEGF-TKI. Moreover, patients were required to have adequate organ function, a KPS score of at least 70%, and to have received no more than 3 prior systemic regimens for locally advanced or metastatic RCC. The randomization of patients in the trial was stratified according to IMDC prognostic score (0 versus 1 to 2 versus 3 to 6) and the number of prior VEGF- or VEGF receptor–targeted therapies for advanced RCC (1 versus 2 or 3). The primary outcomes of the LITESPARK-005 trial were PFS and OS. The key secondary outcome of the trial was ORR, and other secondary outcomes included DOR, HRQoL, and safety. The baseline demographic characteristics were balanced in the 2 treatment groups. The median age of patients in the trial was 63 years (range, 22 to 90 years).27 Most patients were male (77.9%) and white (78.8%).27 Most patients had a KPS score that ranged between 90% and 100% (64.1%) and an ECOG PS of 1 (55.1%).27 Most patients had an intermediate or poor IMDC prognostic risk score (78.3%) and stage IV RCC at diagnosis (57.0%).27 Most patients had 2 or more organs involved with disease at baseline (91.2%), with the lung being the most common site of metastatic disease (64.6%).27 Most patients had a prior nephrectomy (69.7%), received 2 (43.3%) or 3 (42.8%) prior lines of therapy, and received 1 prior VEGF- or VEGF receptor–targeted therapy (50.5%).27
In the absence of direct evidence comparing belzutifan to cabozantinib for the treatment of advanced RCC, the sponsor conducted a feasibility assessment for ITCs that compared belzutifan and cabozantinib in patients with advanced RCC who had received prior treatment with an immune checkpoint inhibitor and an antiangiogenic therapy.61
Based on the results of the feasibility assessment, the CDA-AMC review team agreed that neither a network meta-analysis (i.e., Bucher ITC) nor alternative methods of MAICs (i.e., anchored MAIC) were likely to provide unbiased treatment effect estimates for the comparison of belzutifan and cabozantinib for the treatment of adult patients with RCC. In light of these results, there remains an evidence gap pertaining to the efficacy and safety of belzutifan compared with cabozantinib for advanced RCC in Canadian clinical practice.
Interpretation of Results
Efficacy
Among patients with advanced RCC, there remains an unmet need for later-line treatments with novel mechanisms of action to improve survival, response, and HRQoL. The LITESPARK-005 trial assessed PFS and OS as primary end points, whereas response outcomes (i.e., ORR [key secondary outcome] and DOR) and HRQoL were assessed as secondary end points.
At the first interim analysis of the trial, the predefined success criterion for superiority based on PFS was met. Belzutifan was associated with a 25% reduction in the risk of progression or death compared to everolimus; this reduction was found to be statistically significant (HR = 0.75; 95% CI, 0.63 to 0.90; P = 0.00077).26 Results for PFS at the second interim analysis and the final analysis were consistent with those observed at the primary analysis. However, median PFS was similar for both the belzutifan (5.6 months; 95% CI, 3.9 to 7.0 months) and everolimus (5.6 months; 95% CI, 4.8 to 5.8 months) groups.26 Despite similarities in median PFS, the clinical experts consulted by CDA-AMC commented that the KM curves for belzutifan and everolimus eventually separate, at later time points, and a higher proportion of patients treated with belzutifan remain alive and progression-free at these time points. The clinical experts consulted by CDA-AMC suggested that belzutifan may have a longer duration of efficacy compared to everolimus, which may explain why the benefit of belzutifan is observed at time points beyond those corresponding to median PFS. The clinical experts consulted by CDA-AMC suggested a threshold of 10% as a clinically important between-group difference for the proportion of patients remaining alive and progression-free. Based on this threshold, belzutifan results in a clinically important increase in the probability of patients being alive and progression-free compared with everolimus. Of note, several studies submitted by the sponsor supported the clinical utility of PFS as a surrogate outcome for OS in patients with advanced or metastatic RCC.58-60 However, it is important to note that 2 of these studies evaluated the surrogacy of PFS for OS in patients with untreated, advanced RCC,58,60 whereas the remaining study evaluated surrogacy among patients with metastatic RCC.59 Moreover, the review team determined that the results for PFS and OS in the LITESPARK-005 trial are unlikely to suggest evidence of surrogacy. Thus, based on these studies, it remains unclear whether PFS can be interpreted as a surrogate outcome for OS in the target population of this review.
The corresponding statistical hypothesis-testing P value boundary for OS was not met at any of the prespecified analyses (interim or final) of the trial. At the final analysis, belzutifan was found to be associated with an 8% reduction in the risk of death compared to everolimus; however, this result was not found to be statistically significant (HR = 0.92; 95% CI, 0.77 to 1.10; one-sided P = 0.17644).27 Median OS was 21.4 months (95% CI, 18.2 to 24.3 months) in the belzutifan group and 18.1 months (95% CI, 15.8 to 21.8 months) in the everolimus group.27 Based on a threshold of 5% as a clinically important between-group difference for the proportion of patients remaining alive, suggested by the clinical experts, it is likely that belzutifan results in little to no difference in the probability of patients remaining alive at 18 months and 36 months compared with everolimus.
At the first interim analysis of the trial, the predefined success criterion for superiority based on ORR was met. The ORR among patients receiving belzutifan was significantly higher than that among patients receiving everolimus (between-group difference, █████ ████ ███ ████ ██ ██████ ███████ █████████).26 The clinical experts consulted by CDA-AMC suggested a threshold of 10% as a clinically important difference in ORR. Based on a threshold of 10%, belzutifan results in a clinically important increase in ORR. Moreover, belzutifan was associated with a longer DOR compared with everolimus. Notably, there were a higher proportion of patients with confirmed CR in the belzutifan group (2.7%) compared with the everolimus group (0%).26 The clinical experts consulted by CDA-AMC believed that this observation was impactful among patients receiving treatment for RCC in later-line settings. They also commented on the importance to patients of knowing whether a CR would be sustained, and for how long.
Key HRQoL outcomes of interest from the LITESPARK-005 trial included the EORTC QLQ-C30 and FKSI-DRS instruments. The between-group differences in LS mean change in the EORTC QLQ-C30 GHS/QoL score and the FKSI-DRS score from baseline to week 17 all favoured belzutifan over everolimus.26 The clinical experts consulted by CDA-AMC suggested that the EORTC QLQ-C30 instrument is a preferred measure of HRQoL; the FKSI-DRS instrument may be more applicable to patients with localized disease (i.e., outside of the target population).
A change of 10 points in the EORTC QLQ-C30 score and summary score is conventionally considered to be an MID.26 The sponsor’s Clinical Study Report26 defined a score of 10 or greater as clinically meaningful. No literature was identified that estimated MIDs specifically in patients with pretreated, advanced RCC. At a 10% threshold, belzutifan may result in little to no difference in LS mean change in the EORTC QLQ-C30 GHS/QoL score. The interpretation of results was limited by the open-label study design of the trial, the low completion rates at week 17, and an imbalance of attrition in the belzutifan and everolimus treatment groups.
This review highlighted a few issues that may affect the generalizability of the results of the LITESPARK-005 trial to clinical practice in Canada. The comparator treatment in the LITESPARK-005 trial has been superseded by newer treatment options (e.g., cabozantinib and axitinib) and is rarely used in Canadian clinical practice, according to the clinical experts consulted by CDA-AMC. Thus, the stand-alone results of the trial may not provide a full assessment of the efficacy and safety of belzutifan compared to existing treatments for advanced RCC in Canadian clinical practice. However, the clinical experts consulted by CDA-AMC suggested that everolimus may be similar to axitinib in terms of efficacy and safety, based on how these drugs perform in clinical practice. They also agreed that belzutifan should be reserved for the next line of therapy after cabozantinib (unless there is intolerance to cabozantinib), noting that there is currently a lack of head-to-head RCTs and no evidence to suggest that belzutifan performs substantially better than cabozantinib. Nonetheless, the clinical experts consulted by CDA-AMC noted that treatment options for third-line or later-line settings for advanced RCC remain limited, and there is a lack of standard treatment options for fourth-line advanced RCC. They emphasized that a treatment’s ability to delay progression and achieve response in a heavily pretreated patient population would be highly valued in clinical practice. Thus, the clinical experts consulted by CDA-AMC agreed that belzutifan would be of value to the Canadian treatment landscape for advanced RCC, as it introduces a treatment option for later lines of therapy for advanced RCC.
Harms
Safety was assessed as a secondary end point in the LITESPARK-005 trial. In the LITESPARK-005 trial, almost all patients experienced at least 1 AE in both of the belzutifan and everolimus groups.27 The frequency of AEs was the same for both groups, and the most common AE in both treatment groups was anemia.27 The incidence of SAEs was higher among patients receiving belzutifan compared with everolimus, with anemia as the most common SAE for both groups.27 Notably, the rate of discontinuations due to AEs was lower among patients receiving belzutifan compared with those receiving everolimus, with the most common AEs being related to respiratory, thoracic, and mediastinal disorders.27 Moreover, deaths resulting from AEs were lower in the belzutifan group compared with the everolimus group.27 Based on the Health Canada product monograph for belzutifan38 and validation from clinical experts consulted by CDA-AMC, clinical AEs of special interest related to belzutifan were anemia, hypoxia, and dyspnea. The incidences of all 3 AEs of special interest were higher in the belzutifan group compared with the everolimus group. However, both groups reported low rates of treatment discontinuation associated with these AEs.27 The clinical experts consulted by CDA-AMC agreed that the safety profile of belzutifan in the LITESPARK-005 trial is generally consistent with what they expect to observe with belzutifan treatment in clinical practice. Although the experts noted that anemia and hypoxia were not observed with other existing treatments for advanced RCC, they explained that these AEs were related to the mechanism of action of belzutifan, which is novel compared to existing treatment options for advanced RCC. Aside from this observation, the clinical experts did not note any additional safety concerns for belzutifan compared to other existing treatment options for advanced RCC in Canada. Due to incidences of hypoxia and anemia, the clinical experts consulted by CDA-AMC noted that treatment with belzutifan should be supervised by medical oncologists with expertise in managing these AEs. They also commented that these AEs are generally manageable in clinical practice and can be mitigated by dose reductions and interruptions. Given a paucity of head-to-head trials comparing belzutifan to other current treatment options for RCC, the safety profile of belzutifan compared with relevant comparators for RCC in Canadian clinical practice remains unclear.
Conclusion
According to patients with advanced RCC and to clinicians, there remains an unmet need for later-line treatments with novel mechanisms of action to improve survival, response, and HRQoL. One phase III, open-label, active-controlled RCT was included in this review. The LITESPARK-005 trial evaluated the efficacy and safety of belzutifan compared to everolimus in patients with advanced RCC following treatment with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. The LITESPARK-005 trial met the predefined success criterion for superiority based on PFS, one of the dual primary outcomes, and on ORR, the key secondary end point. According to the GRADE approach, the results demonstrated that, when compared with everolimus, belzutifan demonstrates a clinically important benefit for PFS and ORR. However, belzutifan likely demonstrates little to no difference in OS, the other primary outcome, compared with everolimus. Although belzutifan may result in little to no difference in HRQoL outcomes, the interpretation of these results is limited by the open-label design of the trial, the low completion rates at week 17, and an imbalance of attrition between the belzutifan and everolimus treatment groups. Overall, no new safety signals were observed with belzutifan in the LITESPARK-005 trial. Compared to everolimus, belzutifan likely results in a clinically important decrease in AEs leading to treatment discontinuation, but makes little to no difference in the incidence of SAEs. There was a lack of studies directly comparing the efficacy and safety of belzutifan to currently available treatment options, especially axitinib and cabozantinib. The clinical experts consulted by CDA-AMC suggested that everolimus may have similar efficacy as that of axitinib based on how these drugs perform in clinical practice. They also noted that there is currently no evidence to suggest that belzutifan performs substantially better than cabozantinib.
References
1.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada, and the Public Health Agency of Canada. Canadian Cancer Statistics 2024. Canadian Cancer Society; 2024. Accessed July 17, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2024-statistics/2024-cmaj/2024_cancer-specific-stats.pdf
2.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
3.Powles T, Albiges L, Bex A, et al. Renal cell carcinoma: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2024;35(8):692-706. doi: 10.1016/j.annonc.2024.05.537 PubMed
4.Iskandar AS, Zarrabi KK, Tester WJ. Renal cell carcinoma: entering the age of biomarkers. Can J Urol. 2024;31(4):11921-11930. PubMed
5.Osawa T, Takeuchi A, Kojima T, Shinohara N, Eto M, Nishiyama H. Overview of current and future systemic therapy for metastatic renal cell carcinoma. Jpn J Clin Oncol. 2019;49(5):395-403. doi: 10.1093/jjco/hyz013 PubMed
6.Atkins M. Post TW, ed. Clinical presentation, diagnosis, and staging of renal cell carcinoma. UpToDate; 2024. Accessed November 22, 2024. https://www.uptodate.com
7.CADTH Reimbursement Review Provisional Funding Algorithm Report for renal cell carcinoma. Accessed September 16, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/PH0037-Renal-Cell-Carcinoma.pdf
8.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines - Kidney Cancer Version 2.2025. Accessed December 6, 2024. https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf
9.Canil C, Kapoor A, Basappa NS, et al. Management of advanced kidney cancer: Kidney Cancer Research Network of Canada (KCRNC) consensus update 2021. Can Urol Assoc J. 2021;15(4):84-97. doi: 10.5489/cuaj.7245 PubMed
10.Government of New Brunswick. New Brunswick Drug Plans Formulary. Accessed November 20, 2024. https://www2.gnb.ca/content/dam/gnb/Departments/h-s/pdf/en/NBDrugPlan/NewBrunswickDrugPlansFormulary.pdf
11.Minister of Health and Wellness Province of Prince Edward Island. PEI Pharmacare Formulary. Accessed November 20, 2024. https://www.princeedwardisland.ca/sites/default/files/publications/pei_pharmacare_formulary.pdf
12.Newfoundland and Labrador Health and Community Services. Special authorization criteria. Accessed November 20, 2024. https://www.gov.nl.ca/hcs/files/Criteria-Oct-2024.pdf
13.Nova Scotia Pharmacare. Appendix III -Criteria for Coverage of Exception Status Drugs. Accessed November 20, 2024. https://novascotia.ca/dhw/pharmacare/documents/Criteria-for-Exception-Status-Coverage.pdf
14.Ontario Ministry of Health. Exceptional Access Program Reimbursement Criteria for Frequently Requested Drugs. Accessed November 20, 2024. https://files.ontario.ca/moh-frequently-requested-drugs.pdf
15.Saskatchewan Cancer Agency. Drug Formulary. Accessed November 20, 2024. https://saskcancer.ca/health-care-professionals/drug-formulary
16.B.C. Cancer. Protocol Summary for Therapy for Advanced Renal Cancer using Everolimus - Protocol GUEVER. Accessed October 31, 2024. http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Genitourinary/GUEVER_Protocol.pdf
17.Cancer Care Alberta. Renal Cell Carcinoma. Clinical Practice Guidelines GU-003-Version 11. 2023. Accessed September 17, 2024. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-gu003-renal-cell.pdf
18.Alberta Health Services. Outpatient Cancer Drug Benefit Program. Accessed November 20, 2024. https://www.albertahealthservices.ca/assets/programs/ps-1025651-drug-benefit-list.pdf
19.CADTH Reimbursement Review: Opdivo for Metastatic Renal Cell Carcinoma. Accessed September 18, 2024. https://www.cda-amc.ca/opdivo-metastatic-renal-cell-carcinoma-details
20.CADTH Reimbursement Review: Cabometyx for Renal Cell Carcinoma Resubmission. Accessed September 16, 2024. https://www.cda-amc.ca/cabometyx-renal-cell-carcinoma-resubmission-details
21.Kastrati K, Mathies V, Kipp AP, Huebner J. Patient-reported experiences with side effects of kidney cancer therapies and corresponding information flow. J Patient Rep Outcomes. 2022;6(1):126. doi: 10.1186/s41687-022-00533-z PubMed
22.Johnston H, Deal AM, Morgan KP, Patel B, Milowsky MI, Rose TL. Dose Intensity in Real-World Patients With Metastatic Renal Cell Carcinoma Taking Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin Genitourin Cancer. 2023;21(3):357-365. doi: 10.1016/j.clgc.2023.02.007 PubMed
23.Choueiri TK, Halabi S, Sanford BL, et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol. 2017;35(6):591-597. doi: 10.1200/JCO.2016.70.7398 PubMed
24.Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1814-23. doi: 10.1056/NEJMoa1510016 PubMed
25.Merck Canada Inc. P005V02MK6482. Clinical Study Report LITESPARK-005: Belzutifan (MK-6482) versus Everolimus in Participants with Renal Cell Carcinoma. Second Interim Analysis (IA2) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: WELIREG (belzutifan), tablets, 40 mg, oral. November 25, 2024.
26.Merck Canada Inc. P005V01MK6482. Clinical Study Report LITESPARK-005: Belzutifan (MK-6482) versus Everolimus in Participants with Renal Cell Carcinoma. First Interim Analysis (IA1) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: WELIREG (belzutifan), tablets, 40 mg, oral. November 25, 2024.
27.Merck Canada Inc. P005V03MK6482. Statistical Analysis Report LITESPARK-005: Belzutifan (MK-6482) versus Everolimus in Participants with Renal Cell Carcinoma. Final Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: WELIREG (belzutifan), tablets, 40 mg, oral. November 25, 2024.
28.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
29.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
30.BARDS HTA Statistics. Data on File. Protocol 005: Belzutifan (MK-6482) Versus Everolimus in Participants with Renal Cell Carcinoma. Final Analysis. HTA Report Addendum [sponsor supplied reference]. 2024.
31.Merck Canada Inc. Sponsor Summary of Clinical Evidence: WELIREG for the Treatment of Advanced Renal Cell Carcinoma After Prior Treatment with a PD-1 or PD-L1 Inhibitor and VEGF-TKI [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: WELIREG (belzutifan), 40 mg oral tablet. November 25, 2024.
32.Atkins B. Post TW, ed. Epidemiology, pathology, and pathogenesis of renal cell carcinoma. UpToDate; 2024. Accessed November 22, 2024. https://www.uptodate.com
33.Escudier B, Porta C, Schmidinger M, et al. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):706-720. doi: 10.1093/annonc/mdz056 PubMed
34.Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380(12):1116-1127. doi: 10.1056/NEJMoa1816714 PubMed
35.Motzer R, Alekseev B, Rha SY, et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N Engl J Med. 2021;384(14):1289-1300. doi: 10.1056/NEJMoa2035716 PubMed
36.Choueiri TK, Powles T, Burotto M, et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2021;384(9):829-841. doi: 10.1056/NEJMoa2026982 PubMed
37.Novartis Pharmaceuticals Canada, Inc. AFINITOR (everolimus): tablets, 2.5 mg, 5 mg, 7.5 mg and 10 mg [product monograph]. Updated November 16, 2017. Accessed October 9, 2024. https://pdf.hres.ca/dpd_pm/00042203.PDF
38.Merck Canada Inc. Welireg (belzutifan): Tablets, 40 mg, oral [draft product monograph]. July 11, 2022. Updated November 22, 2024.
39.Pfizer Products, U. L. C. INLYTA (axitinib): Tablets, 1 mg, 3 mg, 5 mg and 7 mg [product monograph]. 2021. Updated September 28, 2021. Accessed October 9, 2024. https://pdf.hres.ca/dpd_pm/00063084.PDF
40.Ipsen Biopharmaceuticals Canada Inc. CABOMETYX (cabozantinib): Tablets, 20 mg, 40 mg, 60 mg [product monograph]. Updated October 6, 2021. Accessed January 30, 2025. https://pdf.hres.ca/dpd_pm/00063172.PDF
41.Choueiri TK, Plimack ER, Bauer TM, et al. Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). J Clin Oncol. 2020;38(6_suppl):611-611. doi: 10.1200/JCO.2020.38.6_suppl.611
42.Albiges L, Rini BI, Peltola K, et al. LBA88 Belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC): Randomized open-label phase III LITESPARK-005 study. Ann Oncol. 2023;34:S1329-S1330. doi: 10.1016/j.annonc.2023.10.090
43.Albiges L, Choueiri TK, Peltola K, et al. Belzutifan Versus Everolimus for Previously Treated Advanced Clear Cell Renal Cell Carcinoma; Subgroup Analysis of the Phase 3 LITESPARK-005 Study [accessed by sponsor]. Presented at: Kidney Cancer Research Summit; 2024; Boston, Massachusetts.
44.Rini BI, Suarez Rodriguez C, Albiges L, et al. LBA74 Final analysis of the phase III LITESPARK-005 study of belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC) [accessed by sponsor]. Presented at: European Society for Medial Oncology Congress; 2024; Barcelona, Spain.
45.Rini BI, Suarez Rodriguez C, Albiges L, et al. LBA74 Final analysis of the phase III LITESPARK-005 study of belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC). Ann Oncol. 2024;35:S1262-S1263. doi: 10.1016/j.annonc.2024.08.2317
46.Powles T, Albiges L, Jalkanen KJ, et al. Belzutifan versus everolimus in participants (pts) with previously treated advanced renal cell carcinoma (RCC): Patient-reported outcomes (PROs) in the phase 3 LITESPARK-005 study. J Clin Oncol. 2024;42(4_suppl):361-361. doi: 10.1200/JCO.2024.42.4_suppl.361PubMed
47.Merck Sharp & Dohme LLC. NCT04195750: A Study of Belzutifan (MK-6482) Versus Everolimus in Participants With Advanced Renal Cell Carcinoma (MK-6482-005) [accessed by sponsor]. ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT04195750
48.ClinicalTrials.eu. EU Clinical Trials Register [accessed by sponsor]. https://www.clinicaltrialsregister.eu/
49.Novartis. Prescribing Information: Afinitor (everolimus) tablets for oral administration. U.S. Food and Drug Administration; 2022. Accessed February 28, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/203985s023,022334s051lbl.pdf
50.Choueiri TK, Powles T, Peltola K, et al. Belzutifan versus Everolimus for Advanced Renal-Cell Carcinoma. N Engl J Med. 2024;391(8):710-721. doi: 10.1056/NEJMoa2313906 PubMed
51.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
52.King MT. The interpretation of scores from the EORTC quality of life questionnaire QLQ-C30. Qual Life Res. 1996;5(6):555-67. doi: 10.1007/BF00439229 PubMed
53.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-44. doi: 10.1200/JCO.1998.16.1.139 PubMed
54.Cella D, Yount S, Brucker PS, et al. Development and validation of a scale to measure disease-related symptoms of kidney cancer. Value Health. 2007;10(4):285-93. doi: 10.1111/j.1524-4733.2007.00183.x PubMed
55.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-21. doi: 10.1016/j.ejca.2012.02.059 PubMed
56.Clopper C, Pearson E. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26(4):404-413.
57.Maurer W, Bretz F. Multiple Testing in Group Sequential Trials Using Graphical Approaches. Stat Biopharm Res. 2013;5(4):311-320. doi: 10.1080/19466315.2013.807748
58.Bria E, Massari F, Maines F, et al. Progression-free survival as primary endpoint in randomized clinical trials of targeted agents for advanced renal cell carcinoma. Correlation with overall survival, benchmarking and power analysis. Crit Rev Oncol Hematol. 2015;93(1):50-9. doi: 10.1016/j.critrevonc.2014.08.001 PubMed
59.Delea TE, Khuu A, Heng DY, Haas T, Soulieres D. Association between treatment effects on disease progression end points and overall survival in clinical studies of patients with metastatic renal cell carcinoma. Br J Cancer. 2012;107(7):1059-68. doi: 10.1038/bjc.2012.367 PubMed
60.Halabi S, Rini B, Escudier B, Stadler WM, Small EJ. Progression-free survival as a surrogate endpoint of overall survival in patients with metastatic renal cell carcinoma. Cancer. 2014;120(1):52-60. doi: 10.1002/cncr.28221 PubMed
61.BARDS HTA Statistics. Data on File. Statistical Report for Indirect Treatment Comparisons (ITCs) of Belzutifan (MK-6482) in LiteSpark-005 trial (Internal) An Open-label, Randomized Phase 3 Study of Belzutifan (MK-6482) Versus Everolimus in Participants with Advanced Renal Cell Carcinoma That Has Progressed After Prior PD-1/L1 and VEGF-Targeted Therapies vs. Cabozantinib in METERO trial (External) A Phase 3, Randomized, Controlled Study of Cabozantinib (XL184) vs Everolimus in Subjects With Metastatic Renal Cell Carcinoma That Has Progressed After Prior VEGFR Tyrosine Kinase Inhibitor Therapy [sponsor supplied reference]. 2024.
62.Motzer RJ, Escudier B, Powles T, Scheffold C, Choueiri TK. Long-term follow-up of overall survival for cabozantinib versus everolimus in advanced renal cell carcinoma. Br J Cancer. 2018;118(9):1176-1178. doi: 10.1038/s41416-018-0061-6 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Subgroup Analyses for Progression-Free Survival Based on BICR Assessment per RECIST 1.1
Figure 7: Subgroup Analyses for Progression-Free Survival in the LITESPARK-005 Trial — ITT Population (Interim Analysis 1; Data Cut-Off: November 1, 2022) (Part 1)
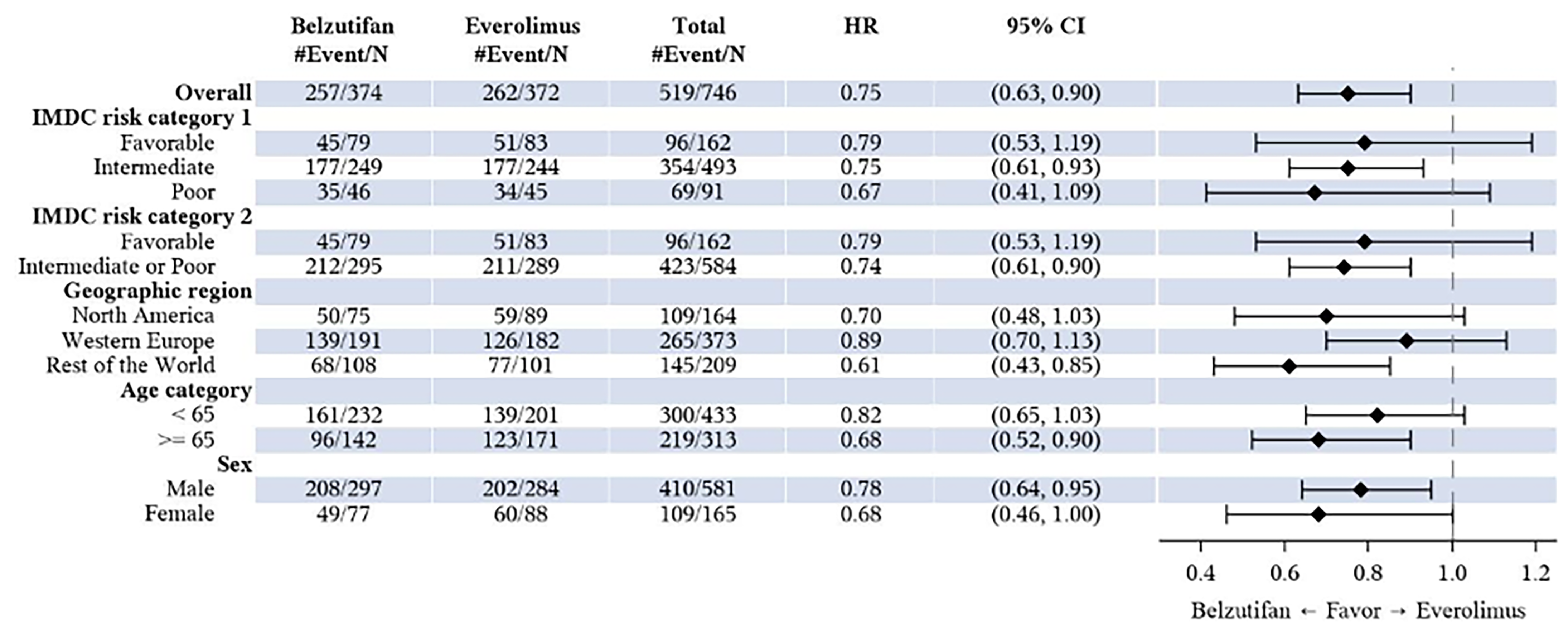
BICR = blinded independent central review; CI = confidence interval; IMDC = International Metastatic RCC Database Consortium; ITT = intention to treat; PFS = progression-free survival; VEGF/VEGFR = vascular endothelial growth factor/vascular endothelial growth factor receptor.
Source: LITESPARK-005 CSR IA1.26
Figure 8: Subgroup Analyses for Progression-Free Survival in the LITESPARK-005 Trial — ITT Population (Interim Analysis 1; Data Cut-Off November 1, 2022) (Part 2)
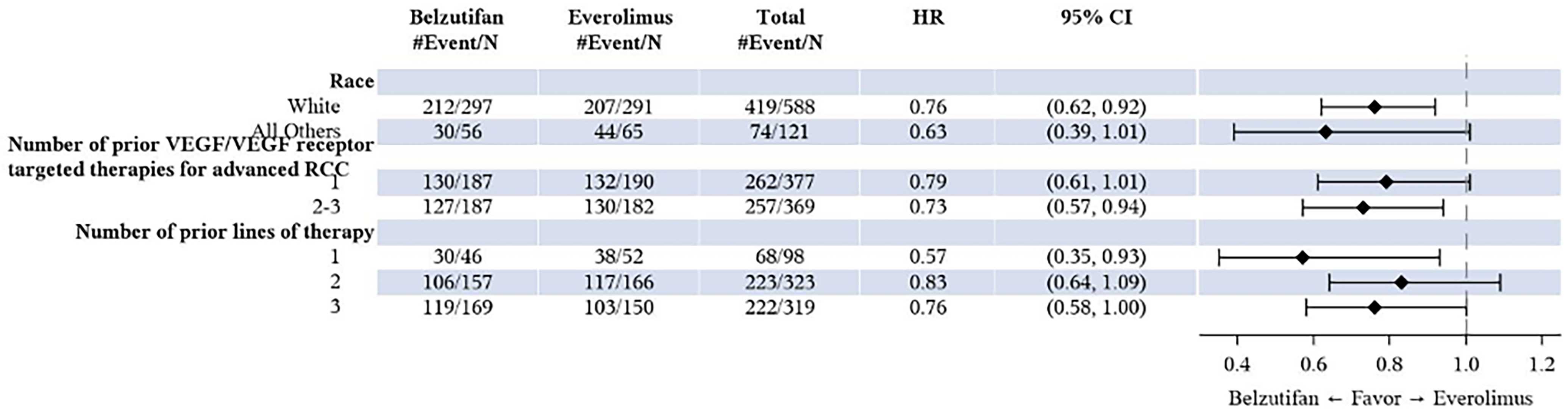
BICR = blinded independent central review; CI = confidence interval; IMDC = International Metastatic RCC Database Consortium; ITT = intention to treat; PFS = progression-free survival; RCC = renal cell carcinoma; VEGF = vascular endothelial growth factor.
Source: LITESPARK-005 CSR IA1.26
Supplemental Data From Interim Analysis 2 of the LITESPARK-005 Trial
Table 22: Progression-Free Survival and Key Response Outcomes in the LITESPARK-005 Trial — ITT Population (Interim Analysis 2; Data Cut-Off: June 13, 2023)
Variable | Belzutifan (N = 374) | Everolimus (N = 372) |
|---|---|---|
Progression-free survival (by BICR per RECIST 1.1) | ||
Median follow-up time, months (range) | 18.5 (0.2 to 37.5) | 17.2 (0.8 to 39.1) |
Number of events (%) | 289 (77.3) | 276 (74.2) |
Death | 24 (6.4) | 41 (11.0) |
Documented progression | 265 (70.9) | 235 (63.2) |
Number of censored (%) | 85 (22.7) | 96 (25.8) |
Last assessment before 2 or more consecutive missed assessments | 3 (0.8) | 6 (1.6) |
Last assessment showing no progression | 56 (15.0) | 7 (1.9) |
Last radiologic assessment before new anticancer therapy showing no progression | 22 (5.9) | 68 (18.3) |
Randomization | 4 (1.1) | 15 (4.0) |
KM estimates (months)a | ||
Median (95% CI) | 5.6 (3.8 to 6.5) | 5.6 (4.8 to 5.8) |
HR vs Everolimus (95% CI)b | 0.74 (0.63 to 0.88) | |
P valuec | < 0.00031 | |
PFS rate at month 6 (%) (95% CI) | 46.1 (40.9 to 51.2) | 42.8 (37.1 to 48.3) |
PFS rate at month 9 (%) (95% CI) | 39.9 (34.8 to 45.0) | 28.7 (23.5 to 34.2) |
PFS rate at month 12 (%) (95% CI) | 33.7 (28.8 to 38.7) | 17.6 (13.2 to 22.4) |
PFS rate at month 18 (%) (95% CI) | 22.5 (18.2 to 27.0) | 9.0 (5.8 to 13.0) |
PFS rate at month 24 (%) (95% CI) | 18.0 (14.0 to 22.5) | 3.7 (1.7 to 7.1) |
ORR (by BICR per RECIST 1.1) | ||
Number of patients with responsed | 85 | 13 |
ORR (%) (99.9% CI)e | █████ ██████ ███ ████ ██ █████ | 3.5 (1.2 to 7.8) |
Difference in % vs. everolimus | ||
Estimate (99.9% CI)e,f | █████ ██████ ███ ████ ██ █████ | |
P valueg | ██ ███ | |
Best Objective Response, n (%) | ||
CR | 13 (3.5) | 0 (0.0) |
PR | 72 (19.3) | 13 (3.5) |
SD | 143 (38.2) | 245 (65.9) |
PD | 127 (34.0) | 80 (21.5) |
Not evaluableh | 5 (1.3) | 8 (2.2) |
No assessmenti | 14 (3.7) | 26 (7.0) |
Time to Response (months) | ||
Mean (SD) | 5.4 (4.0) | 3.5 (1.2) |
Median (range) | 3.8 (1.7 to 22.0) | 3.7 (1.8 to 5.4) |
Duration of Responsej (months) | ||
Median (range) | 19.5 (1.9+ to 31.6+) | 13.7 (3.8 to 21.2+) |
Number (%) of patients with extended response duration | ||
≥ 6 months | 75 (93.9) | 10 (76.9) |
≥ 9 months | 65 (82.5) | 8 (61.5) |
≥ 12 months | 54 (73.2) | 8 (61.5) |
≥ 15 months | 40 (62.1) | 6 (46.2) |
≥ 18 months | 21 (51.1) | 4 (30.8) |
≥ 21 months | 18 (48.4) | 1 (15.4) |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; IMDC = International Metastatic RCC Database Consortium; LS = least square; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; RCC = renal cell carcinoma; SD = standard deviation.
Note: “+” indicates there is no progressive disease by the time of last disease assessment.
aFrom product-limit (Kaplan-Meier) method for censored data.
bBased on Cox regression model with Efron's method of tie handling with treatment as a covariate stratified by International Metastatic RCC Database Consortium (IMDC) risk group (favourable vs. intermediate vs. poor) and Number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 to 3).
cOne-sided P value based on log-rank test stratified by International Metastatic RCC Database Consortium (IMDC) risk group (favourable vs. intermediate vs. poor) and Number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 to 3).
dIncludes patients with confirmed complete response or partial response.
eBased on the procedure for testing for multiplicity in the trial, the ultimate alpha used for testing ORR was determined to be 0.001. Thus, 99.9% CIs were reported alongside the point estimates for ORR.
fBased on Miettinen and Nurminen method stratified by International Metastatic RCC Database Consortium (IMDC) risk group (favourable vs. intermediate vs. poor) and Number of prior antiangiogenic therapies for advanced RCC (1 vs. 2 to 3).
gOne-sided P value for testing. H0: difference in % = 0 vs. H1: difference in % > 0.
hNot Evaluable includes patients with insufficient data for assessment of response per RECIST 1.1.
iNo Assessment includes patients without postbaseline assessment on the data cut-off date.
jFrom product-limit (Kaplan-Meier) method for censored data.
Source: LITESPARK-005 CSR IA2.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.31
Subgroup Analyses for Overall Survival
Figure 9: Subgroup Analyses for Overall Survival in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off: April 15, 2024) (Part 1)
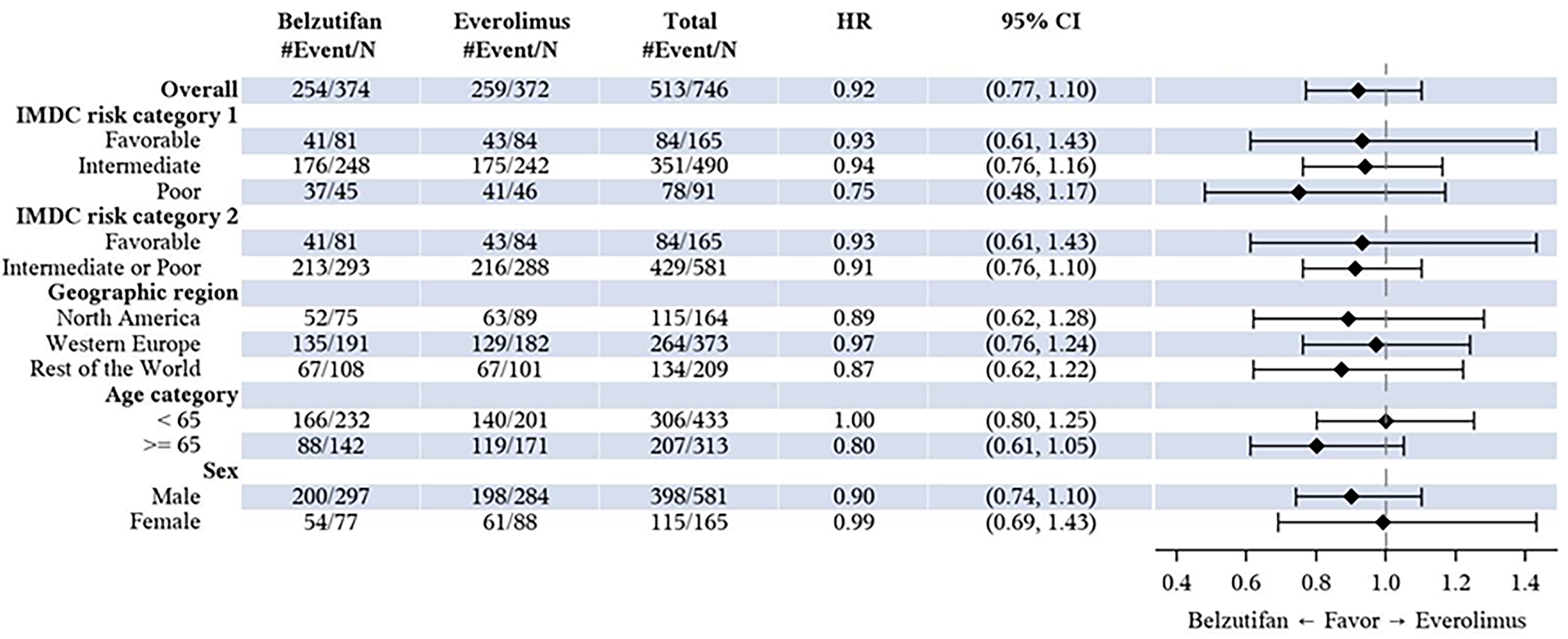
CI = confidence interval; HR = hazard ratio; IMDC = International Metastatic RCC Database Consortium; ITT = intention to treat; OS = overall survival.
Notes: Only patients with data in the specified subgroups are included.
Based on Cox model: stratified for overall population and unstratified for subgroups.
Subgroup factors of IMDC risk categories and number of prior VEGF/VEGF receptor–targeted therapies for advanced RCC are collected from eCRF.
Source: LITESPARK-005 Statistical Report FA.27
Figure 10: Subgroup Analyses for Overall Survival in the LITESPARK-005 Trial — ITT Population (Final Analysis; Data Cut-Off of April 15, 2024]) (Part 2)
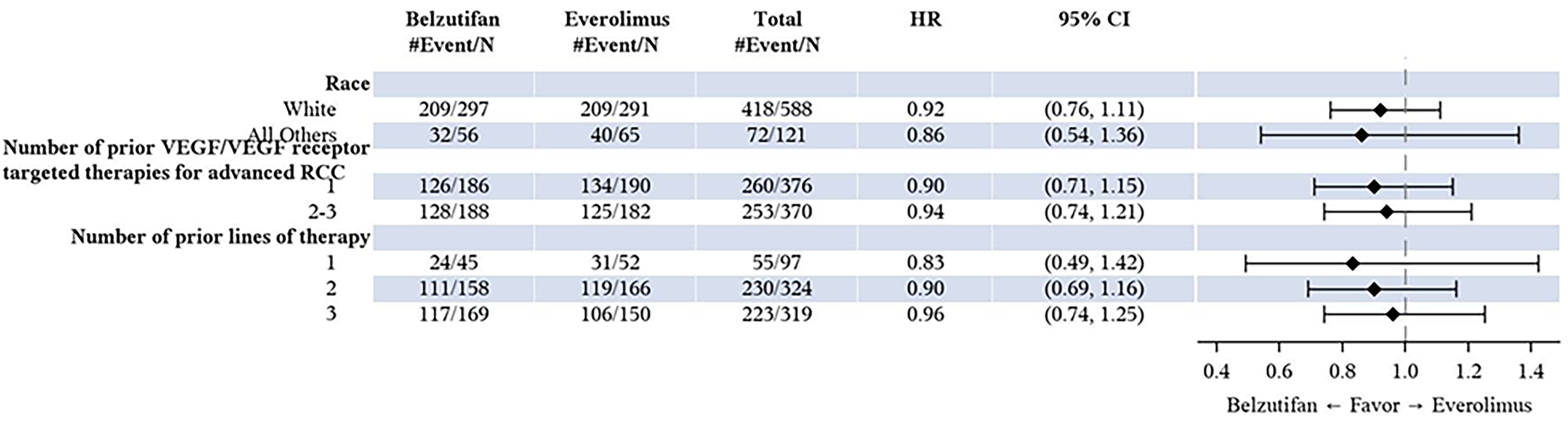
CI = confidence interval; IMDC = International Metastatic RCC Database Consortium; ITT = intention to treat; OS = overall survival; VEGF = vascular endothelial growth factor.
Notes: Only patients with data in the specified subgroups are included.
Based on Cox model: stratified for overall population and unstratified for subgroups
Subgroup factors of IMDC risk categories and number of prior VEGF/VEGF receptor–targeted therapies for advanced RCC are collected from eCRF.
Source: LITESPARK-005 Statistical Report FA.27
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
KM
Kaplan-Meier
LY
life-year
NICE
National Institute for Health and Care Excellence
NOC
notice of compliance
OS
overall survival
PD-1
programmed death receptor-1
PD-L1
programmed death-ligand 1
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RCC
renal cell carcinoma
RDI
relative dose intensity
TKI
tyrosine kinase inhibitor
ToT
time on treatment
VEGF
vascular endothelial growth factor
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Belzutifan (Welireg), 40 mg, tablet, oral |
Indication | For the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 17, 2024 |
Reimbursement request | As per indication |
Sponsor | Merck Canada Inc. |
Submission history | Previously reviewed: Yes Indication: For the treatment of adult patients with von Hippel-Lindau disease who require therapy for associated nonmetastatic RCC central nervous system hemangioblastomas and nonmetastatic pancreatic neuroendocrine tumours, not requiring immediate surgery Recommendation date: September 1, 2023 Recommendation: Recommended with clinical criteria and/or conditions |
NOC = Notice of Compliance; RCC = renal cell carcinoma; TKI = tyrosine kinase inhibitor; VEGF = vascular endothelial growth factor.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult patients with advanced RCC that has progressed after prior PD-1 or PD-L1 inhibitor and VEGF targeted therapies. |
Treatment | Belzutifan |
Dose regimen | 120 mg once daily until disease progression or unacceptable toxicity occurs. |
Submitted price | Belzutifan: $213.33 per 40 mg tablet |
Submitted treatment cost | The 28-day cost of belzutifan is $██████, as calculated by the sponsor (accounting for dose reductions and dose skipping). |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (38 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; KM = Kaplan-Meier; LY = life-year; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; RCC = renal cell carcinoma; VEGF = vascular endothelial growth factor; WTP = willingness to pay.
Conclusions
The LITESPARK-005 trial evaluated the efficacy and safety of belzutifan compared to everolimus in patients with advanced renal cell carcinoma (RCC) after previous treatment with a programmed death receptor-1 (PD-1) or programmed death-ligand (PD-L1) inhibitor and a vascular endothelial growth factor (VEGF)-tyrosine kinase inhibitor (TKI). Results from the LITESPARK-005 trial demonstrated that, compared to everolimus, belzutifan demonstrated clinically important benefit for objective response rate and progression-fee survival (PFS). However, belzutifan likely demonstrated no clinically relevant difference in survival compared to everolimus. Although belzutifan may have resulted in an important difference in health-related quality of life (HRQoL) outcomes, the interpretation of these results is limited by the open-label design of the trial, the low completion rates at week 17, and the imbalance of attrition between the belzutifan and everolimus treatment groups. However, because everolimus is rarely used to treat advanced RCC in Canada, the generalizability of the LITESPARK-005 trial results may be limited in Canadian clinical practice. There was a lack of studies directly comparing the efficacy and safety of belzutifan to currently available treatment options, especially cabozantinib and axitinib. The clinical experts consulted by Canada's Drug Agency (CDA-AMC) suggested that the efficacy of everolimus may be similar to that of axitinib based on how these drugs perform in clinical practice. They also noted that there is currently no evidence to suggest that belzutifan performs substantially better than cabozantinib. Nonetheless, the clinical experts noted that treatment options in third-line or later-line settings for advanced RCC remain limited and emphasized that a treatment’s ability to delay progression and achieve response in these treatment settings would be highly valued in clinical practice. Thus, the clinical experts agreed that belzutifan would be of value to the Canadian treatment landscape for advanced RCC because it would introduce a treatment option in later lines of therapy for advanced RCC.
In the base-case reanalysis, CDA-AMC adopted a one-piece extrapolation of PFS and assumed treatment waning begins at 35.8 months and ends at 72 months. The CDA-AMC base case results align with those of the sponsor’s submitted analysis, indicating that belzutifan is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained.
In the absence of comparative data on the efficacy of belzutifan and axitinib, along with the fact that everolimus is rarely used and is no longer considered a standard of care in Canadian clinical practice, the usefulness of sequential analysis is limited; therefore, pairwise comparisons between belzutifan and comparators were used in the CDA-AMC base-case reanalysis. In the CDA-AMC base case, belzutifan was associated with an incremental cost-effectiveness ratio (ICER) of $731,313 per QALY gained (incremental costs = $214,542; incremental QALYs = 0.29) compared to everolimus and an ICER of $664,048 per QALY gained (incremental costs = $194,792; incremental QALYs = 0.29) compared to axitinib. These results were driven by belzutifan drug acquisition costs, which accounted for nearly all of the total incremental costs incurred, and approximately 78% of the incremental benefit was estimated to be incurred after the trial period (i.e., extrapolations after 156 weeks). To be considered cost-effective at a WTP threshold of $50,000 per QALY gained, belzutifan would require a price reduction of 82% relative to everolimus and a price reduction of 74% relative to axitinib. The CDA-AMC reanalyses remain associated with uncertainty, as the predicted survival benefit with belzutifan was not supported by the LITESPARK-005 trial results, which indicated no clinically relevant difference in overall survival (OS) compared with everolimus.
CDA-AMC was unable to address limitations related to the lack of long-term clinical evidence and the absence of comparative efficacy data for axitinib versus belzutifan, and the costs of subsequent treatments were not appropriately modelled to align with Canadian clinical practice. According to clinical expert feedback obtained by CDA-AMC, belzutifan is most likely to be used as third-line or fourth-line treatment, after cabozantinib, and they anticipate that there would be no further approved subsequent treatment options. When subsequent treatments were excluded from a scenario analysis, the ICERs for belzutifan increased further. CDA-AMC notes that the estimates of the cost-effectiveness of belzutifan versus axitinib are based entirely on assumptions of equal efficacy between axitinib and everolimus. Given these limitations, there remains considerable uncertainty about the cost-effectiveness results and further price reductions may be required.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from Kidney Cancer Canada from an international online survey of patients done in affiliation with the International Kidney Cancer Coalition. The survey included 2,213 respondents, 139 of whom were from Canada. Of the 3 patients with experience with belzutifan, 2 were interviewed. Kidney Cancer Canada reported that although kidney cancer survival rates have improved in recent years as a result of new treatments, survival is still poor in those with stage IV disease. The group emphasized the importance of new treatments to improve disease stability and treatment tolerability to improve outcomes and quality of life. Patient feedback indicated a desire for novel drugs and new mechanisms of disease control beyond existing VEGF-TKI drugs and checkpoint inhibitors. One respondent who was previously treated with sunitinib and pazopanib was currently receiving belzutifan through the LITESPARK-005 study. Belzutifan reportedly resulted in substantial shrinkage of this patient’s tumour and the patient remained on treatment after 4 years. Another respondent had a diagnosis of von Hippel-Lindau disease and was previously treated with sunitinib and axitinib for RCC; after approximately 3 years of treatment with belzutifan, the patient reported positive outcomes. Both interviewed patients with belzutifan experience reported tolerable side effects with treatment, including anemia, low oxygen, fatigue, nausea, neuropathy (reported to have started with previous treatments), rash, elevated blood pressure, and chest tightening.
Input was received from 2 clinician groups: the Ontario Health (Cancer Care Ontario) (OH [CCO]) Genitourinary Cancer Drug Advisory Committee; and the Kidney Cancer Research Network of Canada. Clinician input stated that first-line systemic treatment for advanced RCC includes pembrolizumab-axitinib, pembrolizumab-lenvatinib, nivolumab-cabozantinib, nivolumab-ipilimumab, sunitinib, and pazopanib. Second-line treatment options include sunitinib, pazopanib, cabozantinib, axitinib, and nivolumab. Clinician input noted that belzutifan’s mechanism of action differs from current treatments, highlighting the need for more therapies because there are limited treatment options and there is a need for novel therapies that target different pathways in advanced RCC. Clinician input stated that belzutifan would be recommended for use in later-line settings (second, third, and fourth) as monotherapy.
Drug plan input noted that the comparator in the pivotal LITESPARK-005 trial was everolimus, which is no longer routinely used in Canada and is not considered a relevant comparator. Drug plan input stated that, based on the CDA-AMC provisional funding algorithm for metastatic RCC, patients are eligible for 1 immune checkpoint inhibitor and 2 TKI therapies; depending on the choice of first-line therapy, 2 or 3 lines of therapy may be funded. These were determined by drug plans to be more relevant comparators than everolimus. Drug plan input expressed concern around the discontinuation criteria for belzutifan and the potential large budget impact given the volume of patients with metastatic RCC. Drug plans also highlighted the reproductive risk to patients of all genders (i.e., belzutifan can render some hormonal contraceptives ineffective and exposure during pregnancy can cause embryo-fetal harm). Last, drug plan input highlighted the existence of confidential pan-Canadian Pharmaceutical Alliance (pCPA) pricing for nivolumab, pembrolizumab, and many TKIs, as well as the availability of generic everolimus.
Several of these concerns were addressed in the sponsor’s model:
Costs and utility decrements related to adverse events (AEs) were included in the model.
Subsequent treatment with sunitinib, pazopanib, cabozantinib, axitinib, or nivolumab as third-line therapy was included in the model based on transformations of the trial data.
In addition, CDA-AMC addressed some of these concerns as follows:
CDA-AMC removed subsequent therapies, after belzutifan and everolimus, in a scenario analysis due to their lack of generalizability to the provisional funding algorithm for metastatic RCC.
CDA-AMC was unable to address the following concerns raised in the input received:
CDA-AMC could not address the reproductive risk to patients of all sexes due to belzutifan exposure.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of belzutifan compared with everolimus and axitinib.1 The modelled population is consistent with the Health Canada indication and the reimbursement request, and these populations are aligned with the sponsor’s LITESPARK-005 trial population.
Belzutifan is available as 40 mg oral tablets. The recommended dose of belzutifan is 120 mg (3 tablets) administered orally once daily until disease progression or unacceptable toxicity occurs.2 The cost of belzutifan is $213.33 per 40 mg tablet, leading to an estimated 28-day cost of $██████ per patient, as calculated by the sponsor.1 Comparators in the analysis included everolimus (10 mg once daily) and axitinib (5 mg twice daily). The estimated 28-day cost of everolimus was $█████ and the 28-day cost of axitinib was $5,857, as calculated by the sponsor.1 The sponsor’s estimated costs considered discontinuation, dose reductions, and dose skipping, which were observed in the LITESPARK-005 trial for belzutifan and everolimus.3 Data informing relative dose intensity (RDI) for axitinib was sourced from National Institute for Health and Care Excellence (NICE) technology appraisal guidance TA333 and TA417.1
The clinical outcomes of interest were QALYs and life-years over a lifetime horizon (38 years). Discounting (1.5% per annum) was applied to both costs and outcomes, and a cycle length of 1 week was used, along with a half-cycle correction. The base case perspective was that of the Canadian publicly funded health care payer.
Model Structure
The sponsor submitted a partitioned survival model (PSM) consisting of 3 mutually exclusive health states: progression-free, progressive disease, and death.1 All patients entered the model in the progression-free state and received belzutifan or a comparator. The allocation of patients to health states is based on treatment-specific PFS and OS functions. The proportions of patients in the progression-free health state were derived from the respective PFS curves from the LITESPARK-005 trial (for patients treated with belzutifan and everolimus). The sponsor assumed equal efficacy between everolimus and axitinib. The proportion of patients in the progressive disease health state was equal to the difference between the OS and the PFS curves, and patients discontinued first-line treatment upon disease progression. Patients in the progressed disease state can receive a subsequent line of therapy, consisting of axitinib, cabozantinib, nivolumab, pazopanib, or sunitinib. Patients transitioning to the death state remained there until the end of the model time horizon. A figure of the sponsor’s model structure is available in Appendix 3 (Figure 1).
Model Inputs
The target population of the economic evaluation was based on the phase III, open-label LITESPARK-005 trial, which evaluated belzutifan versus everolimus for advanced RCC that has progressed after prior treatment with PD-1 or PD-/L1 inhibitor and a VEGF-targeted therapy.3 The mean age of the population was 62.4 years, mean weight was 78.8 kg, and 22.1% of patients were female (77.9% male).3
The key clinical inputs (i.e., PFS, OS, and treatment discontinuation) for belzutifan and everolimus were obtained from the April 15, 2024, LITESPARK-005 trial data cut-off, with a median follow-up of 35.8 months.3 Kaplan-Meier (KM) curves were generated and used to estimate median time-to-event estimates for each treatment group. Extrapolations of the PFS, OS, and time-on-treatment (ToT) data were performed. For PFS, a piecewise approach was used in which hazard rates of PFS failure were based on the observed KM data up to week 27, followed by extrapolation fitted to the post–27 week data, using a log-logistic distribution for belzutifan and everolimus. For OS, a log-logistic distribution was selected for belzutifan and everolimus. ToT data were fit with a gamma distribution for belzutifan and a log-normal distribution for everolimus. The sponsor assumed equal efficacy between everolimus and axitinib, based on the previous CDA-AMC appraisal of cabozantinib in patients with previously treated, advanced RCC.4
The submitted model included grade 3, 4, or 5 AEs occurring in 5% or more of patients in the LITESPARK-005 trial.3 AE risks for axitinib were assumed equivalent to those of everolimus. AEs included anemia, hyperglycemia, hypertriglyceridemia, hypoxia, and pneumonia.
Health state utility values in the model were informed by the EQ-5D-5L data collected in the LITESPARK-005 trial, calculated based on the average of all patient visits in each health state and scored using the EQ-5D-5L value set for Canada.5 The estimated values were █████ for progression-free disease and █████ for progressed disease.1 Disutility due to grade 3 or higher AEs was applied to each treatment arm at model entry (██████).1
Costs included were related to drug acquisition, subsequent treatment, AEs, disease management by health state, and terminal care. Drug acquisition costs were calculated by the sponsor as a function of unit drug costs, RDI, and the proportion of patients on treatment. The sponsor included RDI to account for dose reductions or interruptions in the administration of belzutifan (██████ and everolimus (█████), based on the pivotal trial3 and based on NICE TA333 and TA417 for axitinib (102%).6,7 Drug administration costs were assumed to be zero for belzutifan and all orally administered therapies. Costs of subsequent treatment were modelled based on the LITESPARK-005 trial data informing the proportion of patients who discontinued study treatment and received at least 1 subsequent systemic treatment, and their respective ToT, for the following: axitinib, cabozantinib, nivolumab, pazopanib, and sunitinib.3 All drug costs were sourced from IQVIA DeltaPA, and administration costs were included for any treatments administered as IV infusions.8 Disease management costs consisted of the costs for outpatient visits, hospitalizations and inpatient stays, emergency department visits, laboratory tests, diagnostic tests, and imaging, which were obtained from the Ontario Schedule of Benefits for Physician and Laboratory Services9,10 and the Alberta Interactive Health Data Application.11 Resource utilization frequency was assumed to be the same for all treatments, but differed for patients in the progression-free and progressed disease health states. Costs for AEs were derived from the Canadian Institute for Health Information (CIHI) Patient Cost Estimator.12 A one-off terminal care cost was applied based on a published study.13
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented here.
Base-Case Results
In the sponsor’s base case, belzutifan was associated with an estimated cost of $513,618 and 2.78 QALYs over a lifetime horizon. In sequential analysis, belzutifan was associated with an ICER of $563,740 compared to everolimus (incremental costs = $254,506; incremental QALYs = 0.45). Belzutifan had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. The key cost driver among patients receiving belzutifan was drug acquisition, which accounted for 99% of the total incremental costs incurred. The sponsor’s analysis predicted that approximately 85% of the incremental benefit was obtained after the trial period based on extrapolations (i.e., after the trial period of 156 weeks). Axitinib was extendedly dominated (i.e., more costly with equivalent benefits to everolimus). Additional results from the sponsor’s submitted economic evaluation base case are available in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Everolimus | 259,111 | 2.33 | Reference |
Belzutifan | 513,618 | 2.78 | 563,740 vs. everolimus |
Dominated treatments | |||
Axitinib | 280,439 | 2.33 | Extendedly dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analysis is based on publicly available prices of comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including alternate discount rates, various time horizons, alternate distributions for PFS and OS, alternate assumptions for ToT, treatment waning, and alternate health state utility values. Across all scenario analyses, the ICER comparing belzutifan to everolimus ranged from $485,138 to $964,574 per QALY gained, and it was most sensitive to shortening the model time horizon and the inclusion of treatment waning.
The sponsor conducted a scenario analysis from a societal perspective that included additional costs associated with work productivity impairment. In this analysis, the ICER per QALY gained was $560,631 relative to everolimus, lower than the sponsor’s base-case analysis using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Uncertainty regarding the PFS as modelled: The sponsor used a piecewise approach to model PFS, which entailed using KM data for 27 weeks directly from the LITESPARK-005 trial (median follow-up = 35.8 months; maximum follow-up = 49.2 months), and parametric extrapolation thereafter, fitted to the post–27 week data (using a log-logistic distribution), instead of using a more robust parametric distribution for the entirety of the model time horizon (38 years) or a piecewise model (i.e., different parametric distributions fitted to the different trial periods). In the LITESPARK-005 trial, the KM curves for PFS crossed at approximately 25 weeks and was subsequently higher for belzutifan than everolimus. It is generally more accepted to use the extrapolated parametric distributions over the entire model time horizon because it results in more consistent hazard estimates and avoids limitations related to a small number of events at later times points in observed data, the stepped-nature of the intervals for assessment in trials, and restricted mean estimates using incomplete data.14 Therefore, there remains uncertainty with the sponsor’s approach because it does not adequately capture the full uncertainty surrounding PFS (e.g., for the initial 27 weeks of the model).
In the CDA-AMC reanalysis, CDA-AMC adopted a one-piece extrapolation of PFS for all comparators that maintained the sponsor’s original choice of parametric distribution (log-logistics).
Uncertainty in the comparative efficacy of belzutifan versus comparators and the surrogacy of PFS and OS: Based on the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach outlined in the CDA-AMC clinical review, belzutifan demonstrates a clinically important PFS benefit, but likely results in no clinically relevant difference in OS compared with everolimus. Evidence provided by the sponsor, including the results of the LITESPARK-005 trial, was deemed to be insufficient to suggest surrogacy between PFS and OS for the indicated population. Thus, it remains unclear whether PFS can be interpreted as a surrogate outcome for OS in the modelled population.
Of note, the parametric distributions chosen by the sponsor to extrapolate PFS and OS (in addition to other modelling assumptions such as excluding treatment waning; refer to the following limitation) resulted in an incremental gain of 0.58 QALYs in the PFS health state in patients treated with belzutifan, and predicted an overall incremental gain of 0.53 life-years associated with belzutifan versus comparators. Therefore, the model’s predicted OS benefit with belzutifan remains highly uncertain and is likely due to extrapolation (i.e., the predicted benefit occurs after the observed trial period).
Furthermore, in the absence of randomized evidence for belzutifan versus axitinib (deemed to be the most relevant comparator for this patient population), the sponsor assumed that the efficacy of axitinib would be equivalent to that of everolimus. Although this assumption appeared to meet face validity, according to clinical expert feedback obtained by CDA-AMC, uncertainty remains because of the lack of supporting data. The lack of comparative evidence limits the usefulness of sequential analysis; therefore, pairwise comparisons between belzutifan and comparators were presented in the CDA-AMC base-case reanalysis.
CDA-AMC could not address the limitations surrounding the clinical evidence or the uncertainty of the OS benefit. The cost-effectiveness of belzutifan is uncertain, especially compared to axitinib.
Uncertainty in the long-term efficacy of belzutifan: The sponsor assumed no treatment-waning effect with belzutifan, meaning that the treatment effect of belzutifan was experienced indefinitely in the model. This assumption implies that the treatment effect of belzutifan will persist for approximately 30 years after treatment discontinuation. Importantly, 50% of the total incremental QALYs were accrued by patients from the period beyond which there are no observed clinical trial data (i.e., extrapolated period), highlighting uncertainty in the long-term efficacy of belzutifan. The clinical experts consulted by CDA-AMC for this review indicated that they are unaware of evidence that may support this assumption and that it is plausible for treatment effect waning to occur. Due to high censoring and the loss to follow-up that occurred after 24 months in the LITESPARK-005 trial, the clinical experts noted additional uncertainty in the long-term efficacy of belzutifan versus everolimus after this time point. Therefore, in the absence of evidence to support the long-term effectiveness of belzutifan, CDA-AMC assumed that treatment waning would begin at the median follow-up time of the LITESPARK-005 trial (i.e., 35.8 months) and would last until year 6. That is, the treatment effect of belzutifan would continue to be experienced for approximately 3 years after treatment discontinuation. High uncertainty remains and, according to the CDA-AMC Methods and Guidelines for Extrapolating Clinical Evidence Within Economic Evaluations,15 the inclusion of a treatment-waning effect is appropriate in this context when long-term clinical data are lacking.
In the CDA-AMC reanalysis, treatment waning begins at 35.8 months and ends at 72 months.
Structural uncertainty due to the use of a PSM. The sponsor used a PSM to calculate long-term costs and outcomes for each treatment. Although the PSM approach is commonly used in health technology assessments of oncology treatments, the model introduces structural assumptions about the relationship between PFS and OS that likely do not accurately reflect causal relationships within the disease pathway (for example, that PFS and OS are assumed to be independent), and it does not explicitly model progression or response to subsequent treatments, which could lead to inaccurate estimates of the long-term treatment effect of the modelled intervention.
CDA-AMC was unable to assess the concerns identified with this limitation.
Prior and subsequent exposure to therapies in the model was not aligned with clinical practice in Canada: The submitted analysis included everolimus and axitinib as comparators for belzutifan, and modelled subsequent treatment based on assumptions and transformations from the LITESPARK-005 trial data. Notably, the sponsor excluded cabozantinib from the analysis, citing a lack of comparative efficacy data and a lack of feasibility to conduct an indirect treatment comparison, although it is a widely used treatment in clinical practice.
Based on the comparator status table and treatment paradigm for RCC, “only one of axitinib or cabozantinib is funded in the eligible second or third-line treatment settings; third-line treatment of any kind is not funded in patients who received cabozantinib plus nivolumab followed by second-line axitinib” and “for patients treated with nivolumab + ipilimumab first-line and VEGF TKI (sunitinib or pazopanib) second line, either cabozantinib or axitinib may be used as third-line therapy.” Clinical expert feedback received by CDA-AMC confirmed that cabozantinib alone is generally used as second-line or third-line treatment (if not used in prior lines) and that belzutifan is expected to be used as third-line or fourth-line treatment, after cabozantinib. According to this feedback, belzutifan is unlikely to displace cabozantinib in the second line. This is reflected in the sponsor’s own budget impact analysis (BIA) of market shares, which indicated cabozantinib occupies the majority of market shares in this patient population, with minimum displacement by belzutifan. The clinical experts noted there is currently a lack of head-to-head randomized controlled trials and no evidence to suggest that belzutifan performs substantially better than cabozantinib. Although feedback supports that cabozantinib would likely be used before belzutifan, the exclusion of this comparator means that the cost-effectiveness of cabozantinib versus belzutifan remains unknown.
Clinical expert feedback received by CDA-AMC and reiterated by the drug plans indicates that, although available and occasionally used, everolimus is not included in the CDA-AMC provisional funding algorithm because it is no longer considered a standard of care in patients with previously treated RCC. This leaves axitinib as the most relevant comparator for belzutifan as third-line and fourth-line treatments, for which there is no direct or indirect comparative evidence available.
Based on LITESPARK-005 trial data, ████% of patients receiving belzutifan and ████% of patients receiving everolimus (total = ████%) did not receive subsequent systemic treatment, although the sponsor assumes these numbers to be ████% and ████%, respectively, in the economic model. In the LITESPARK-005 full intention-to-treat population, the remaining ████% of total patients were documented to have received many different subsequent treatments, including but not limited to monoclonal antibodies, protein kinase inhibitors, PD-1 or PDL-1 inhibitors, platinum compounds, poly(ADP-ribose) and polymerase (PARP) inhibitors, pyrimidine analogues, serotonin antagonists, antineoplastic agents, VEGF receptor TKIs, and VEGF receptor inhibitors. However, the submitted model only considered subsequent treatment options that are recommended for reimbursement as a subsequent treatment of advanced RCC in Canada and have an observed utilization of at least 3% among all patients. Based on the sponsor’s naive adjustment of subsequent treatment usage, of the patients who received at least 1 subsequent systemic treatment, approximately ██% received axitinib, ██% received cabozantinib, ██% received nivolumab or sunitinib, and ██% received pazopanib. Therefore, the sponsor assumed that, based on the recorded use of protein kinase inhibitors as subsequent treatment in the trial (n = ███ patients receiving cabozantinib, lenvatinib, pazopanib, sorafenib, or sunitinib, or ██% of a total ███ patients receiving subsequent therapy), this would represent the number of patients expected to receive cabozantinib as subsequent therapy in the model. In the model, it was assumed that ████% of patients receiving belzutifan and ████% of patients receiving everolimus would receive cabozantinib as subsequent systemic treatment, which is neither aligned with the clinical trial nor with expectations in clinical practice according to clinical expert feedback received by CDA-AMC. Based on the treatment paradigm for RCC previously described and belzutifan’s anticipated place in therapy, patients are not expected to receive further subsequent treatment in the economic model after belzutifan or everolimus. Clinical expert feedback received by CDA-AMC confirmed that patients would not receive cabozantinib as a subsequent treatment because they would have likely received it before the use of belzutifan (i.e., in third line or fourth line). Therefore, the sponsor’s assumptions regarding patient exposure to treatments (prior or subsequent treatments) are not generalizable to clinical practice in Canada.
Subsequent therapy was excluded from the analysis in a scenario analysis.
CDA-AMC could not address the exclusion of cabozantinib from the analysis. The cost-effectiveness of belzutifan versus cabozantinib is unknown.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The sponsor applied RDI to calculate drug acquisition costs of belzutifan and comparators based on trial evidence from LITESPARK-005 for belzutifan and everolimus and sourced from the literature for axitinib. | Uncertain. CDA-AMC notes that it is uncertain whether the trial-based proportion of doses skipped may reflect real-world clinical practice, as the doses received by patients may be different from the planned dosing schedule for several reasons (i.e., expected vs. observed doses). However, it seems to have been a conservative approach, given the way the sponsor modelled costs for everolimus (i.e., only included the 10 mg strength tablet for everolimus) and the flat pricing of everolimus across variable strengths, which would have increased the drug costs of everolimus. |
CDA-AMC = Canada’s Drug Agency; RDI = relative dose intensity.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes to model parameter values and assumptions, in consultation with clinical experts. The reanalyses were derived by adopting a one-piece extrapolation of PFS and assuming that treatment waning would begin at 35.8 months and end at 72 months.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. PFS extrapolation | Piecewise (KM data + log-logistics) | One-piece (log-logistics) |
2. Treatment waning | No treatment waning | Treatment waning beginning at 35.8 months and ending at 72 months |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada's Drug Agency; KM = Kaplan-Meier; PFS = progression-free survival.
In the absence of comparative efficacy data for axitinib in the analysis, the usefulness of sequential analysis is limited; therefore, CDA-AMC presented a pairwise comparison between belzutifan and comparators in the CDA-AMC base-case reanalysis. In the CDA-AMC base case, belzutifan was associated with an estimated cost of $472,978 and 2.62 QALYs over a lifetime horizon. In the pairwise comparison to everolimus, belzutifan was associated with an ICER of $731,313 per QALY gained (incremental costs = $214,542; incremental QALYs = 0.29). Compared to axitinib, belzutifan was associated with an ICER of $664,048 per QALY gained (incremental costs: $194,792; incremental QALYs: 0.29). Belzutifan had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained against either comparator. The key cost driver for patients receiving belzutifan was drug acquisition, which accounted for nearly 100% of the total incremental costs incurred. Similar to the sponsor’s base-case analysis, the CDA-AMC reanalysis predicted that approximately 78% of the incremental benefit was obtained after the trial period based on extrapolations (i.e., after the trial period of 156 weeks). A detailed breakdown of the disaggregated results and stepped reanalyses is available in Appendix 4.
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | Pairwise ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case (probabilistic) | |||
Belzutifan | 513,618 | 2.78 | Reference |
Everolimus | 259,111 | 2.33 | 563,740 vs. belzutifan |
Axitinib | 280,439 | 2.33 | 516,518 vs. belzutifan |
CDA-AMC base case (probabilistic) | |||
Belzutifan | 472,978 | 2.62 | Reference |
Everolimus | 258,436 | 2.33 | 731,313 vs. belzutifan |
Axitinib | 278,186 | 2.33 | 664,048 vs. belzutifan |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; vs. = versus.
Note: The reanalysis is based on publicly available prices of comparator treatments.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s results and the CDA-AMC base case.
The CDA-AMC reanalysis suggests that belzutifan would require a price reduction of 82% to be considered cost-effective at a WTP threshold of $50,000 per QALY gained, relative to everolimus. With this price reduction, the per-patient, 28-day, drug acquisition costs for belzutifan (administered 120 mg once daily) would be $3,192 (instead of ██████) at a unit price of $38 per 40 mg tablet.
The CDA-AMC reanalysis suggests that belzutifan would require a price reduction of 74% to be considered cost-effective at a WTP threshold of $50,000 per QALY gained, relative to axitinib. With this price reduction, the per-patient, 28-day, drug acquisition costs for belzutifan (administered 120 mg once daily) would be $4,620 (instead of ██████) at a unit price of $55 per 40 mg tablet.
Table 7: CDA-AMC Price Reduction Analyses
Analysis Price reduction | Unit drug cost ($) | Pairwise ICERs for belzutifan vs. comparators ($/QALY) | |||
|---|---|---|---|---|---|
Sponsor’s base case vs. everolimus | Sponsor’s base case vs. axitinib | CDA-AMC reanalysis vs. everolimus | CDA-AMC reanalysis vs. axitinib | ||
No price reduction | 213 | 563,740 | 516,499 | 731,313 | 664,048 |
10% | 192 | 501,068 | 453,828 | 648,295 | 581,022 |
20% | 171 | 438,397 | 391,156 | 565,276 | 497,996 |
30% | 149 | 375,725 | 328,485 | 482,258 | 414,971 |
40% | 128 | 313,054 | 265,813 | 399,239 | 331,945 |
50% | 107 | 250,382 | 203,142 | 316,221 | 248,920 |
60% | 85 | 187,711 | 140,470 | 233,202 | 165,894 |
70% | 64 | 125,039 | 77,799 | 150,184 | 82,868 |
80% | 43 | 62,368 | 15,127 | 67,165 | Dominant |
90% | 38 | Dominant | Dominant | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CDA-AMC conducted scenario analyses to evaluate the impact of excluding subsequent treatment from the analysis due to the lack of face validity surrounding the availability of subsequent treatment options that patients received in the LITESPARK-005 trial. When subsequent treatments were excluded, the ICER increased slightly to $751,204 per QALY gained versus everolimus and to $683,740 per QALY gained versus axitinib.
Issues for Consideration
Belzutifan has been previously reviewed by CDA-AMC for the treatment of adult patients with von Hippel-Lindau disease who require therapy for associated nonmetastatic RCC, central nervous system hemangioblastomas, or nonmetastatic pancreatic neuroendocrine tumours, not requiring immediate surgery.16 Belzutifan received a recommendation of reimburse with clinical criteria and/or conditions on September 1, 2023.16 The conditions described were if prescribed by a specialist with expertise in von Hippel-Lindau disease–associated tumours and if the cost of belzutifan is reduced. The cost of belzutifan in the previous submission is aligned with the current review at $213.33 per 40 mg tablet.
Overall Conclusions
The LITESPARK-005 trial evaluated the efficacy and safety of belzutifan compared to everolimus in patients with advanced RCC who were previously treated with a PD-1 or PD-L1 inhibitor and a VEGF-TKI. Results from the LITESPARK-005 trial demonstrated that, compared with everolimus, belzutifan demonstrated a clinically important benefit in objective response rate and PFS. However, belzutifan likely demonstrated no clinically relevant difference in survival compared to everolimus. Although belzutifan may have resulted in an important difference in HRQoL outcomes, the interpretation of these results is limited by the open-label design of the trial, the low completion rates at week 17, and an imbalance of attrition between the belzutifan and everolimus treatment groups. However, because everolimus is rarely used to treat advanced RCC in Canada, the generalizability of the results of the LITESPARK-005 trial to Canadian clinical practice may be limited. There is a lack of studies directly comparing the efficacy and safety of belzutifan to currently available treatment options, especially cabozantinib and axitinib. The clinical experts consulted by CDA-AMC suggested that everolimus and axitinib may have similar efficacy, based on how these drugs perform in clinical practice. They also noted that there is currently no evidence to suggest that belzutifan performs substantially better than cabozantinib. Nonetheless, the clinical experts noted that treatment options in third-line or later-line settings for advanced RCC remain limited and emphasized that a treatment’s ability to delay progression and achieve a response in these treatment settings would be highly valued in clinical practice. Thus, the clinical experts agreed that belzutifan would be of value in the Canadian treatment landscape for advanced RCC because it would introduce a treatment option for later lines of therapy for advanced RCC.
In the base-case reanalysis, CDA-AMC adopted a one-piece extrapolation of PFS and assumed treatment waning begins at 35.8 months and ends at 72 months. The CDA-AMC base-case results align with those of the sponsor’s submitted analysis, indicating that belzutifan is not cost-effective at a WTP threshold of $50,000 per QALY gained.
In the absence of comparative efficacy data informing the efficacy of belzutifan and axitinib, along with the fact that everolimus is rarely used and is no longer considered a standard of care in Canadian clinical practice, the usefulness of sequential analysis is limited; therefore, pairwise comparisons between belzutifan and comparators were used in the CDA-AMC base-case reanalysis. In the CDA-AMC base case, belzutifan was associated with an ICER of $731,313 per QALY gained (incremental costs = $214,542; incremental QALYs = 0.29) compared to everolimus and an ICER of $664,048 per QALY gained (incremental costs = $194,792; incremental QALYs = 0.29) compared to axitinib. These results were driven by belzutifan’s drug acquisition costs, which accounted for nearly all the total incremental costs incurred, and approximately 78% of the incremental benefit was estimated to be incurred after the trial period (i.e., extrapolations after 156 weeks). To be considered cost-effective at a WTP threshold of $50,000 per QALY gained, belzutifan would require a price reduction of 82% relative to everolimus and a price reduction of 74% relative to axitinib. The CDA-AMC reanalyses remain associated with uncertainty because the predicted survival benefit with belzutifan was not supported by the LITESPARK-005 trial results, which indicated no clinically relevant difference in OS compared with everolimus.
CDA-AMC was unable to address limitations with the lack of long-term clinical evidence and the absence of comparative efficacy data for axitinib versus belzutifan, and the costs of subsequent treatments were not appropriately modelled to align with Canadian clinical practice. According to clinical expert feedback obtained by CDA-AMC, belzutifan is most likely to be used as a third-line or fourth-line treatment, after cabozantinib, and they anticipate there would be no further approved subsequent treatment options. When subsequent treatments were excluded in a scenario analysis, the ICERs of belzutifan increased further. CDA-AMC notes that the estimates of the cost-effectiveness of belzutifan versus axitinib are based entirely on the assumption that axitinib and everolimus have equal efficacy. Given these limitations, there remains considerable uncertainty in the cost-effectiveness results and further price reductions may be required.
References
1.Merck Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Welireg (belzutifan), tablets, 40 mg, oral. November 25, 2024.
2.Merck Canada Inc. Welireg (belzutifan): tablets, 40 mg, oral [product monograph]. November 22, 2024.
3.Merck Canada Inc. P005V02MK6482. Clinical Study Report LITESPARK-005: Belzutifan (MK-6482) versus Everolimus in Participants with Renal Cell Carcinoma. Second Interim Analysis (IA2) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: WELIREG (belzutifan), tablets, 40 mg, oral. November 25, 2024.
4.pan-Canadian Oncology Drug Review. pCODR Expert Review Committee (PERC) final recommendation: Cabozantinib (Cabometyx) for the treatment of patients with advanced renal cell carcinoma (RCC) who have received prior therapy [accessed by sponsor]. 2019. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10163CabozantinibRCCResub_FnRec_2019-02-20_ApprovedByChair_Post_20Feb2019_final.pdf
5.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. doi: 10.1097/MLR.0000000000000447 PubMed
6.National Institute for Health and Care Excellence. Nivolumab for previously treated advanced renal cell carcinoma (NICE technology appraisal TA417) [accessed by sponsor]. 2016. https://www.nice.org.uk/guidance/ta417
7.National Institute for Health and Care Excellence. Axitinib for treating advanced renal cell carcinoma after failure of prior systemic treatment (NICE technology appraisal TA333) [accessed by sponsor]. 2015. https://www.nice.org.uk/guidance/ta333
8.Ontario Ministry of Health and Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index [accessed by sponsor]. 2024. https://www.formulary.health.gov.on.ca/formulary/
9.Ontario Health Insurance Plan. Schedule of Benefits: Physician Services Under the Health Insurance Act (Feb 20, 2024 (effective Apr 1, 2024)) [accessed by sponsor]. 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
10.Ontario Health Insurance Plan. Schedule of Benefits for Laboratory Services (July 5, 2023 (effective July 24, 2023)) [accessed by sponsor]. 2024. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf
11.Government of Alberta. Alberta Interactive Health Data Application [accessed by sponsor]. https://www.alberta.ca/interactive-health-data
12.Canadian Institute for Health Information. Patient Cost Estimator, 2020-2021 [accessed by sponsor]. https://www.cihi.ca/en/patient-cost-estimator
13.De Oliveira C, Pataky R, Bremner KE, et al. Estimating the Cost of Cancer Care in British Columbia and Ontario: A Canadian Inter-Provincial Comparison. Healthc Policy. 2017;12(3):95-108. doi: 10.12927/hcpol.2017.25024 PubMed
14.Latimer MR. Survival analysis for economic evaluations alongside clinical trials - extrapolation with patient-level data. (NICE DSU Technical Support Document 14). Decision Support Unit, ScHARR, University of Sheffield; 2013. Accessed March 5, 2025. https://www.sheffield.ac.uk/media/34225/download
15.CADTH Methods and Guidelines: Extrapolating Clinical Evidence Within Economic Evaluations. 2023. Accessed February 18, 2025. https://www.cda-amc.ca/sites/default/files/attachments/2023-05/MH0011-Extrapolating%20Clinical%20Evidence%20Within%20Economic%20Evaluations_0.pdf
16.pan-Canadian Oncology Drug Review. pCODR Expert Review Committee (PERC) final recommendation: Belzutifan. 2023. Accessed February 18, 2025. https://www.cda-amc.ca/sites/default/files/DRR/2023/PC0309%20Welireg%20-%20CADTH%20Final%20Recommendation%20Final.pdf
17.IQVIA. DeltaPA. 2023. Accessed January 17, 2025. https://www.iqvia.com/
18.Merck Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Welireg (belzutifan), tablets, 40 mg, oral. November 25, 2024.
19.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
20.Cardenas LM, Ghosh S, Finelli A, et al. Trends of Utilization of Systemic Therapies for Metastatic Renal Cell Carcinoma in the Canadian Health Care System. JCO Glob Oncol. 2023;9:e2300271. doi: 10.1200/GO.23.00271 PubMed
21.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2018. Canadian Cancer Society; 2018. Accessed June 12, 2024. https://www.cancer.ca/Canadian-Cancer-Statistics-2018-EN.pdf
22.Sutherland G, Dinh T. Understanding the gap: A pan-Canadian analysis of prescription drug insurance coverage [accessed by sponsor]. Canadian Alliance for Sustainable Health Care; 2017. https://innovativemedicines.ca/wp-content/uploads/2017/12/20170712-understanding-the-gap.pdf
23.Drug Intelligence, Inc. ONCO-CAPPS Patient Treatment Dynamics Metastatic Renal Cell Carcinoma (mRCC). 2024 Q2 Update and Trends [accessed by sponsor]. 2024.
24.Drug Intelligence, Inc. ONCO-CAPPS Metastatic Renal Cell Carcinoma (mRCC) - MAT Dec 2017 [accessed by sponsor]. 2018.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison for Advanced RCC
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Average 28-day cost ($)a |
|---|---|---|---|---|---|---|
Belzutifan | 40 mg | Oral tablet | 213.3300a | 120 mg once daily until disease progression or unacceptable toxicity occurs | 639.99 | 17,920 |
Rapamycin (mTOR) Inhibitor | ||||||
Everolimus (generics) | 2.5 mg 5 mg 10 mg | Oral tablet | 50.6637b | 10 mg once daily until disease progression or unacceptable toxicity occurs | 50.66 | 1,419 |
Tyrosine Kinase Inhibitor | ||||||
Axitinib | 1 mg 5 mg | Oral tablet | 20.5073 102.5387 | 5 mg twice daily until disease progression or unacceptable toxicity occurs | 205.08 | 5,742 |
Cabozantinib | 20 mg 40 mg 60 mg | Oral tablet | 293.3333 | 60 mg once daily until disease progression or unacceptable toxicity occurs | 293.33 | 8,213 |
Pazopanib | 200 mg | Oral tablet | 27.3225 | 800 mg orally once daily until disease progression or unacceptable toxicity occurs | 109.29 | 3,060 |
Sunitinib | 25 mg 50 mg | Oral tablet | 32.5618 65.1238 | 50 mg orally once daily on a schedule of 4 weeks on treatment followed by 2 weeks off | 65.12 | 1,823 |
Immune Checkpoint Inhibitor | ||||||
Nivolumab | 10 mg/mL | 4 mL 10 mL Vial for IV infusion | 782.2200 1,955.5600 | 240 mg every 2 weeks or 480 mg every 4 weeks | 335.24 | 9,386 |
Note: All prices are from IQVIA Delta PA (accessed January 2025),17 unless otherwise indicated, and do not include dispensing fees.
aSponsor’s submitted price.1
bWholesale price reported by IQVIA DeltaPA (January 2025) for generic everolimus in Manitoba, New Brunswick, Nova Scotia, Prince Edward Island, and Newfoundland and Labrador. In other jurisdictions where generic prices are higher, up to $172.2559 per tablet, the 28-day cost would be 4,823.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CDA-AMC key limitation “Limited generalizability of the trial data due to population’s exposure to prior therapies, subsequent therapy use, and misalignment with clinical practice in Canada” |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to CDA-AMC key limitation “Structural uncertainty due to the use of a PSM.” |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CDA-AMC key limitation “Uncertainty regarding the progression-free survival as modelled” |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CDA-AMC key limitation “Uncertainty regarding the progression-free survival as modelled” |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
PFS = progression-free survival; PSM = partitioned survival model; vs. = versus.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Belzutifan | Everolimus | Axitinib |
|---|---|---|---|
Discounted LYs | |||
Total | 3.47 | 2.94 | 2.94 |
Progression-free | 1.43 | 0.73 | 0.73 |
Progressive disease | 2.04 | 2.20 | 2.20 |
Discounted QALYs | |||
Total | 2.78 | 2.33 | 2.33 |
Progression-free | 1.20 | 0.62 | 0.62 |
Progressive disease | 1.60 | 1.73 | 1.73 |
Adverse events | −0.02 | −0.01 | −0.01 |
Discounted costs ($) | |||
Total | 513,618 | 259,111 | 280,439 |
Acquisition | 283,562 | 30,343 | 51,604 |
Administration | 0 | 0 | 0 |
Subsequent treatment costs | 61,412 | 67,359 | 67,430 |
Adverse event costs | 5,650 | 3,508 | 3,503 |
Disease management costs | 85,572 | 79,802 | 79,802 |
Terminal care costs | 77,421 | 78,099 | 78,099 |
LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Pairwise ICER ($/QALY)a |
|---|---|---|---|---|
Sponsor base case | Belzutifan | 512,821 | 2.77 | Reference |
Everolimus | 259,282 | 2.33 | 571,038 vs. belzutifan | |
Axitinib | 280,471 | 2.33 | 523,315 vs. belzutifan | |
1. CDA-AMC reanalysis 1 – one-piece model | Belzutifan | 514,133 | 2.77 | Reference |
Everolimus | 260,039 | 2.33 | 574,725 vs. belzutifan | |
Axitinib | 281,228 | 2.33 | 526,800 vs. belzutifan | |
2. CDA-AMC reanalysis 2 – treatment waning | Belzutifan | 469,169 | 2.64 | Reference |
Everolimus | 257,276 | 2.33 | 649,616 vs. belzutifan | |
Axitinib | 277,058 | 2.33 | 629,770 vs. belzutifan | |
CDA-AMC base case (deterministic) (reanalysis 1 + 2) | Belzutifan | 472,375 | 2.63 | Reference |
Everolimus | 258,033 | 2.33 | 721,529 vs. belzutifan | |
Axitinib | 277,814 | 2.33 | 654,940 vs. belzutifan | |
CDA-AMC base case (probabilistic) (reanalysis 1 + 2) | Belzutifan | 472,978 | 2.62 | Reference |
Everolimus | 258,436 | 2.33 | 731,313 vs belzutifan | |
Axitinib | 278,186 | 2.33 | 664,048 vs belzutifan |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; vs. = versus.
aNote that the sponsor base case results were recalculated to present pairwise ICERs to align with the presentation of the CDA-AMC base case results.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Table 12: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Belzutifan | Everolimus | Axitinib |
|---|---|---|---|
Discounted LYs | |||
Total | 3.28 | 2.94 | 2.94 |
Progression-free | 1.17 | 0.69 | 0.69 |
Progressive disease | 2.11 | 2.25 | 2.25 |
Discounted QALYs | |||
Total | 2.62 | 2.33 | 2.33 |
Progression-free | 0.98 | 0.58 | 0.58 |
Progressive disease | 1.66 | 1.77 | 1.77 |
Adverse events | −0.02 | −0.01 | −0.01 |
Discounted costs ($) | |||
Total | 472,978 | 258,436 | 278,186 |
Acquisition | 243,752 | 28,266 | 48,090 |
Administration | 0 | 0 | 0 |
Subsequent treatment costs | 61,744 | 67,579 | 67,521 |
Adverse event costs | 5,638 | 3,516 | 3,501 |
Disease management costs | 83,364 | 80,162 | 80,162 |
Terminal care costs | 78,480 | 78,912 | 78,912 |
LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 13: Scenario Analyses Conducted on the CDA-AMC Base Case (Probabilistic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Belzutifan | 513,618 | 2.78 | Reference |
Everolimus | 259,111 | 2.33 | 563,740 vs. belzutifan | |
Axitinib | 280,439 | 2.33 | 516,518 vs. belzutifan | |
CDA-AMC base case | Belzutifan | 472,978 | 2.62 | Reference |
Everolimus | 258,436 | 2.33 | 731,313 vs. belzutifan | |
Axitinib | 278,186 | 2.33 | 664,048 vs. belzutifan | |
CDA-AMC Scenario 1: Subsequent treatments excluded | Belzutifan | 411,234 | 2.623 | Reference |
Everolimus | 190,857 | 2.330 | 751,204 vs. belzutifan | |
Axitinib | 210,666 | 2.330 | 683,740 vs. belzutifan |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted BIA assessed the introduction of belzutifan for the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI.18 The analysis was taken from the perspective of the Canadian public drug plans using an epidemiology-based approach, with drug acquisition included in the base case. A 3-year time horizon was used, from 2026 to 2028, with 2025 as a base year. The population size was estimated starting with incident kidney cancer cases followed by a series of attritions (Figure 2). Key inputs to the BIA are documented in Table 15.
The reference case scenario included axitinib, cabozantinib, and everolimus. The new drug scenario included the same comparators with the addition of belzutifan. The market share estimates were derived from market research data and input from Canadian clinical experts.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
1L = first line; 2L = second line; 3L = third line; 4L = fourth line; IO = immunotherapy; TKI = tyrosine kinase inhibitor.
aCanadian Cancer Society (2024).19
bCardenas et al. (2023).20
cCanadian Cancer Society (2018).21
dDinh T and Sutherland G (2017).22
fExpert Opinion.
Source: Sponsor’s budget impact submission.18
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Estimated eligible target population in the base year | 333 |
Number of patients eligible for drug under review | 349 / 366 / 384 |
Market uptake (3 years) | |
Uptake (reference scenario) Axitinib Cabozantinib Everolimus | ███████████ ████████ ████████████ |
Uptake (new drug scenario) Belzutifan Axitinib Cabozantinib Everolimus | ███████ ██████ ████ ██████ |
Cost of treatment (per patient, per week)a | |
Belzutifan Axitinib Cabozantinib Everolimus Sunitinibb Pazopanibb | $4,480 $1,436 $2,053 $1,038 $303.91 $765.03 |
aWeekly costs were applied to parametric distributions fitted to Kaplan-Meier time-on-treatment (ToT) data from the LS-005 trial.
bSunitinib and pazopanib were included as treatment options in the third-line and fourth-line settings only.
Summary of the Sponsor’s BIA Results
The estimated budget impact of funding belzutifan for the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI was $679,425 in Year 1, $4,026,792 in Year 2, and $8,821,194 in Year 3, for a 3-year total of $13,527,411.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty regarding the anticipated market shares of comparators. The sponsor assumed market uptake for belzutifan of ||%, ██% and ██% in years 1, 2, and 3 of the BIA, respectively, based on █████████ market research. Clinical expert feedback obtained by CDA-AMC and input from clinician groups suggest that the estimated market uptake of belzutifan appeared to be reasonable. The market shares for axitinib and everolimus were estimated to be ██% and ||%, respectively, in the base year. However, clinical expert feedback obtained by CDA-AMC indicated that the market shares for axitinib and everolimus did not meet face validity, as axitinib is anticipated to be the most used comparator in third and fourth-line treatment whereas everolimus is rarely used in clinical practice in these patients. Feedback indicated that a lower market share for everolimus of 1% is more reflective of clinical practice in Canada, corresponding to a ██% market share for axitinib.
CDA-AMC adjusted market shares in the reference scenario to reflect ██% and 1% for axitinib and everolimus, respectively.
In the new drug scenario, CDA-AMC adjusted market shares to reflect the expected higher use of axitinib over everolimus, resulting in ██%, ||%, ||% in years 1, 2, and 3 for axitinib and 1%, 1% 0% for everolimus.
The number of eligible patients is uncertain. The sponsor derived the population for the BIA using an epidemiology-based approach with a series of attritions based on published literature and clinical assumptions, eventually resulting in approximately 349 incident patients that would potentially be eligible for belzutifan in Year 1. Clinical expert feedback obtained by CDA-AMC indicated that the literature used by the sponsor appeared to meet face validity, however noted uncertainty in the parameters based on clinical assumption and market research. These parameters included the proportion of patients who received IO and TKI in first-line treatment and the fourth-line re-treatment rate. Although the final estimated number of eligible patients appeared to be reasonable, CDA-AMC notes that there remains uncertainty in the number of eligible patients due to a lack of data to inform assumptions regarding these parameters in the analysis. If the number of eligible patients is higher than estimated by the sponsor, the expected budget impact of reimbursing belzutifan would also be higher than estimated by the sponsor.
CDA-AMC could not address this limitation in reanalysis.
Patient prior and subsequent exposure to therapies in the model was not found to be aligned with clinical practice in Canada: As detailed in the limitations section of the cost-effectiveness analysis, patients’ modelled exposure to previous therapies and subsequent treatments received are not aligned with Canadian clinical practice according to clinical experts consulted by CDA-AMC. Clinical expert feedback received by CDA-AMC indicated that cabozantinib alone is generally used in second or third-line treatment (if not used in prior lines) and that belzutifan is expected to be used in third-line or fourth-line treatment. According to this feedback, cabozantinib is the most widely used treatment in second-line and belzutifan is unlikely to displace cabozantinib in second-line. This is reflected in the sponsor’s own BIA market shares that indicate cabozantinib occupies the majority of the market share in the patient population.
Clinical expert feedback received by CDA-AMC indicated that the use of subsequent therapy as assumed by the sponsor did not reflect clinical practice in Canada. CDA-AMC notes that the distribution of use was based on LITESPARK-005 but was naively adjusted by the sponsor to reflect only treatments used by more than 3% of participants in each arm and those that are reimbursed in Canada. Given their prior exposure to therapies, a notable proportion of patients in the LITESPARK-005 trial had yet to receive cabozantinib, and therefore the sponsor assumes a high proportion of cabozantinib use in subsequent lines of treatment in the model (i.e., approximately ██% of patients after receiving belzutifan or everolimus, and ██% of patients after receiving axitinib). Based on the treatment paradigm for RCC (previously described) and belzutifan’s anticipated place in the treatment paradigm, it is not expected for patients to receive further subsequent treatment in the economic model, after belzutifan or everolimus, due to the lack of further approved treatment options at this time. Clinical expert feedback received by CDA-AMC confirmed that patients would not receive cabozantinib as subsequent treatment since they would have likely received it before using belzutifan (in third-line or fourth-line).
Subsequent treatment was excluded from the analysis in a scenario analysis.
In a scenario analysis, CDA-AMC assumed that belzutifan is only available in third or fourth-line treatment and assumed that uptake would reach 100% by year 3 to reflect clinical expert feedback obtained by CDA-AMC that belzutifan will likely completely displace axitinib and everolimus. Note that this scenario assumes cabozantinib is only used in second-line treatment, and therefore no subsequent treatment options will be available in or after third or fourth-line treatment.
Drug prices paid by public drug plans are uncertain. Both the sponsor’s and CDA-AMC’s analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
CDA-AMC could not address this limitation in reanalysis.
CDA-AMC Reanalyses of the BIA
Table 16: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Market shares of axitinib and everolimus | Reference Scenario Base year/ year 1/ year 2/ year 3 Axitinib: █████████ Cabozantinib: ████████ Everolimus: ███████ New Drug Scenario Base year/ year 1/ year 2/ year 3 Belzutifan: ████████ Axitinib: ███████ Cabozantinib: ████████ Everolimus: ████████ | Reference Scenario Base year/ year 1/ year 2/ year 3 Axitinib: █████████ Cabozantinib: ███████ Everolimus: 1% / 1% / 1% /1% New Drug Scenario Base year/ year 1/ year 2/ year 3 Belzutifan: ███████ Axitinib: ████████ Cabozantinib: ████████ Everolimus: 1% / 1% / 0% / 0% |
CDA-AMC base-case | Reanalysis 1 | |
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18.
In the CDA-AMC reanalysis, the estimated budget impact of funding belzutifan for the treatment of adult patients with advanced RCC following a PD-1 or PD-L1 inhibitor and a VEGF-TKI was $629,470 in Year 1, $3,752,798 in Year 2, and $8,253,600 in Year 3, for a 3-year total of $12,635,867.
Table 17: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 13,527,411 |
CDA-AMC base case (reanalysis 1) | 12,635,867 |
BIA = budget impact analysis
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 18):
Assuming no subsequent treatment, aligned with the scenario analysis conducted in the cost-effectiveness analysis
Assuming that belzutifan is only available in third or fourth-line treatment and that uptake will reach 100% by year 3. This scenario also assumes cabozantinib is only used in the second-line and that no subsequent treatment options are available in or after third or fourth-line treatment.
Table 18: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 11,950,671 | 24,286,530 | 29,509,618 | 32,420,477 | 86,216,625 |
New drug | 11,950,671 | 24,965,955 | 33,536,409 | 41,241,672 | 99,744,036 | |
Budget impact | 0 | 679,425 | 4,026,792 | 8,821,194 | 13,527,411 | |
CDA-AMC base case | Reference | 12,027,848 | 24,416,173 | 29,667,247 | 32,599,398 | 86,682,819 |
New drug | 12,027,848 | 25,045,643 | 33,420,045 | 40,852,998 | 99,318,686 | |
Budget impact | 0 | 629,470 | 3,752,798 | 8,253,600 | 12,635,867 | |
CDA-AMC scenario analysis 1: no subsequent therapy | Reference | 10,237,025 | 17,917,708 | 20,988,044 | 23,006,181 | 61,911,933 |
New drug | 10,237,025 | 18,548,632 | 24,867,752 | 31,673,105 | 75,089,489 | |
Budget impact | 0 | 630,924 | 3,879,709 | 8,666,924 | 13,177,556 | |
CDA-AMC scenario analysis 2: use in 3L and 4L | Reference | 4,567,531 | 7,996,864 | 9,367,624 | 10,268,559 | 27,633,046 |
New drug | 4,567,531 | 10,188,391 | 22,793,895 | 40,192,131 | 73,174,416 | |
Budget impact | 0 | 2,191,527 | 13,426,271 | 29,923,572 | 45,541,370 |
BIA = budget impact analysis; 3L = third line; 4L = fourth line
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.