Drugs, Health Technologies, Health Systems
Reimbursement Review
Daratumumab (Darzalex SC)
Sponsor: Janssen Inc.
Therapeutic area: Multiple myeloma, eligible for autologous stem cell transplant
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ASCT
autologous stem cell transplant
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CMRG
Canadian Myeloma Research Group
CR
complete response
CyBorD
cyclophosphamide-bortezomib-dexamethasone (also known as VCd)
DAC
Drug Advisory Committee
DOR
duration of response
DVRd
daratumumab-bortezomib-lenalidomide-dexamethasone
DVTd
daratumumab-bortezomib-thalidomide-dexamethasone
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-MY20
European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire for Myeloma
ESS
effective sample size
FLC
free light chain
GRADE
Grading of Recommendations, Assessment, Development and Evaluation
HDT
high-dose therapy
HR
hazard ratio
HRQoL
health-related quality of life
IMWG
International Myeloma Working Group
IPD
individual patient data
ISS
International Staging System
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MM
multiple myeloma
MRD
minimal residual disease
NDMM
newly diagnosed multiple myeloma
NGF
next-generation flow cytometry
NGS
next-generation sequencing
NMA
network meta-analysis
OH-CCO
Ontario Health (Cancer Care Ontario)
OR
odds ratio
OS
overall survival
PFS
progression-free survival
PR
partial response
QoL
quality of life
RCT
randomized controlled trial
RMST
restricted mean survival time
SAE
serious adverse event
SC
subcutaneous
sCR
stringent complete response
SD
standard deviation
SE
standard error
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TE
transplant-eligible
VCd
cyclophosphamide-bortezomib-dexamethasone (also known as CyBorD)
VGPR
very good partial response
VRd
bortezomib-lenalidomide-dexamethasone
VTd
bortezomib-thalidomide-dexamethasone
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Daratumumab (Darzalex SC), 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for subcutaneous injection |
Sponsor | Janssen Inc. |
Indication | Daratumumab in combination with bortezomib, lenalidomide, and dexamethasone, followed by maintenance treatment in combination with lenalidomide, for the treatment of adult patients with newly diagnosed multiple myeloma who are eligible for autologous stem cell transplant |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | November 27, 2024 |
Recommended dose | 1,800 mg administered subcutaneously, over approximately 3 to 5 minutes |
NOC = Notice of Compliance.
Introduction
Multiple myeloma (MM) is a hematological malignancy defined by the proliferation of plasma cells and excessive production of the abnormal immunoglobulin monoclonal M protein.1,2 Patients commonly experience fatigue and bone pain,3 as well as renal or nervous system problems, recurring infections, and fever.4 In Canada, an estimated 4,100 individuals had newly diagnosed multiple myeloma (NDMM), approximately 1,750 deaths due to MM occurred in 2024, and an estimated 1,895 patients with transplant-eligible (TE) NDMM were living in Canada as of 2024.5-7 Despite treatment advances in recent years, MM remains incurable8,9 and patients face a poor prognosis, with a 5-year survival rate of approximately 50%.10 Moreover, most patients with MM relapse and many develop refractoriness to commonly used treatments.11 However, the use of an autologous stem cell transplant (ASCT) to treat patients with NDMM is associated with significantly improved clinical outcomes and is considered the standard of care for TE NDMM.9,12 According to the clinical experts consulted for this review, for patients with TE NDMM, the current treatment consists of a multiphase approach that includes induction therapy and ASCT (with high-dose chemotherapy) with or without consolidation therapy, followed by maintenance therapy. The clinical experts noted that the induction therapy currently consists of 4 to 6 cycles of a multidrug regimen with either cyclophosphamide-bortezomib-dexamethasone (CyBorD) or bortezomib-lenalidomide-dexamethasone (VRd), with VRd being the most commonly used regimen in Canada. Following recovery from ASCT, post-transplant therapies include consolidation and maintenance therapies. As consolidation therapy is not publicly funded in all jurisdictions in Canada, currently, less than one-half (< 50%) of patients would receive a brief consolidation therapy with VRd (2 to 4 cycles) followed by maintenance therapy with lenalidomide alone, or bortezomib plus lenalidomide, or a proteasome inhibitor (e.g., ixazomib) plus lenalidomide for the 10% to 25% of patients with high-risk cytogenetics. Although maintenance therapy with lenalidomide is an option under the provisional funding algorithm of Canada’s Drug Agency (CDA-AMC), the clinical experts indicated that almost all patients will receive maintenance therapy with lenalidomide, other than the small percentage (2% to 5%) of patients who may be intolerant to lenalidomide. Because patients are not cured and will eventually relapse, there is a need for new treatments that will result in deepened responses, higher levels of minimal residual disease (MRD) negativity, and longer remissions. In addition, it is well established that patients with high-risk cytogenetics have lower response rates and shorter durations of response. Deeper responses to treatments are documented with complete responses that are also MRD-negative, as determined by next-generation flow cytometry (NGF) or next-generation sequencing (NGS). However, MRD testing in MM is currently not part of the standard of care, and neither NGF nor NGS testing is available in most of the clinical centres in Canada, potentially presenting some barriers if daratumumab becomes funded.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of daratumumab administered as an 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution by subcutaneous (SC) injection for the treatment of adult patients with NDMM who are eligible for ASCT.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from 2 clinical experts consulted by for the purpose of this review.
Patient Input
CDA-AMC received 1 patient group input from Myeloma Canada, which conducted a patient and caregiver survey regarding the use of daratumumab-bortezomib-lenalidomide-dexamethasone (DVRd) for the treatment of patients with NDMM receiving an ASCT in Canada. The survey, which was conducted between September 26 and October 10, 2024, was shared via email and social media by Myeloma Canada and the Leukemia & Lymphoma Society of Canada. Survey eligibility was determined by self-reports from patients and caregivers regarding their experience with MM and whether they (or the person they care for) were eligible for ASCT at the time of diagnosis and received an ASCT or were waiting to receive a transplant as part of their first line of therapy. The survey received a total of 84 responses; of these, 39 were deemed complete and eligible.
Survey respondents emphasized the importance of controlling MM-related symptoms, such as bone issues, kidney problems, mobility, pain, and infections. Respondents most frequently noted that MM-related symptoms had an extreme impact on their ability to work, travel, and conduct volunteer activities.
The results of the survey also highlighted several financial implications related to treatment for MM, such as loss of income or pension funds due to absence from work, disability or early retirement, and costs associated with parking, drugs, and travel. The results of the survey also noted negative psychosocial impacts associated with treatment for MM, with the interruption of life goals and accomplishments (e.g., career and retirement) having an extreme impact on quality of life (QoL).
The most important factors related to MM treatment among patients and caregivers surveyed were the effectiveness of treatment and achieving a long remission, maintenance of QoL and mental health, management of side effects, portability of treatment to reduce the number of visits to treatment centres and mitigate impacts on day-to-day activities, and the cost and accessibility of treatment.
Respondents with experience with DVRd frequently noted DVRd-related impacts on QoL such as treatment side effects, frequency of trips to receive treatment, and tolerability of the mode of drug administration. However, most respondents reported that treatment with DVRd improved their overall QoL, had side effects that were mostly manageable, was effective in controlling MM, and met their expectations for treatment. Few respondents described daratumumab as costly or emphasized the need for financial coverage to access treatment. Myeloma Canada’s input noted the importance of ensuring that patients in Canada have equal access to DVRd, regardless of socioeconomic status. Myeloma Canada emphasized the importance of proactively informing patients about potential vision problems related to DVRd, given that past surveys found that this side effect was of significant concern to patients.
Clinician Input
Input From Clinical Experts Consulted for This Review
The 2 clinical experts consulted by CDA-AMC agreed that a substantial number of patients with MM do not respond to current first-line treatment options, creating a need for new treatments that will result in deepened responses, higher levels of MRD negativity, and longer remissions among these patients. The clinical experts indicated that daratumumab would be administered in combination with VRd as first-line therapy for MM and would be given wherever VRd is currently used to treat patients with NDMM. The clinical experts indicated that DVRd should be administered as a first-line therapy to patients with NDMM who are eligible for ASCT. They also indicated that the assessment of response to treatment for MM consists of regular monitoring of monoclonal protein via serum protein electrophoresis, serum free light chain (FLC) assays, and monitoring of standard disease parameters for MM. The experts agreed that assessment of response to treatment is performed monthly in clinical practice, although they noted that it may be reduced to every 2 to 3 months for patients exhibiting a stable disease response. The clinical experts stated that treatment with daratumumab would be discontinued in the event of disease progression or toxicity. One expert indicated that daratumumab can be prescribed and administered by any physician with experience treating MM in a variety of treatment settings, including rural and community settings. However, the other expert noted that SC daratumumab should be administered in established chemotherapy units by specialized hematologists or oncologists.
Clinician Group Input
Clinician group input for this review was received from 2 clinician groups: the Canadian Myeloma Research Group (CMRG) and the Ontario Health (Cancer Care Ontario) (OH-CCO) Hematology Cancer Drug Advisory Committee (DAC). A total of 33 clinicians (25 from the CMRG and 7 from the DAC) provided input for this submission.
Both the CMRG and DAC agreed that goals for the treatment of MM include the achievement of an antimyeloma response, long-term control of MM-related disease and symptoms, and prolonging of survival. Similar to the clinical experts consulted by CDA-AMC, the clinician groups emphasized an unmet need related to available first-line treatment options for MM. The DAC indicated that some patients with MM do not respond adequately to first-line treatment. The CMRG highlighted the importance of first-line treatment, given that MM remains incurable. The DAC stated that DVRd could become the new standard of care for TE NDMM. However, the CMRG noted that the addition of daratumumab to maintenance therapy may result in increased visits to cancer centres to receive injections. The CMRG also emphasized the importance of increasing the capacity for MRD testing in Canada to minimize long-term toxicity and financial and patient QoL burdens related to daratumumab.
Similar to the clinical experts consulted by CDA-AMC, both clinician groups agreed that patients with NDMM who are eligible for a transplant would be best suited for treatment with DVRd. The DAC indicated that daratumumab can be delivered in any treatment setting with staff experienced in administering the drug, whereas the CMRG noted that daratumumab is appropriate for administration in outpatient settings and that consideration for funding the drug in inpatient settings may be required.
Both clinician groups agreed that standard myeloma and organ-response criteria are used to assess responses to treatment in clinical practice. The CMRG elaborated that assessments of response are based on the tests for the monoclonal protein in the serum and urine, bone marrow biopsies, and imaging studies. In addition to these tests, MRD testing was described as an emerging parameter of response assessment in MM. The CMRG also indicated that responses, in the context of MM, are assessed every 1 to 3 months depending on clinical stability and the regimen used for treatment. Similar to the clinical experts consulted by CDA-AMC, the CMRG indicated that clinically meaningful responses correlate with a partial response (PR) or better according to the International Myeloma Working Group (IMWG) Consensus Criteria. The clinical experts consulted by CDA-AMC and both clinician groups agreed that treatment with daratumumab should be discontinued upon the occurrence of disease progression, unacceptable toxicity, and/or intolerance.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for DVRd: relevant comparators, consideration for initiation of therapy, considerations for discontinuation of therapy, generalizability, funding algorithm, care provision issues, and systemic and economic issues. The clinical experts consulted for the purpose of this review provided advice on the potential implementation issues raised by the drug programs (Table 5).
Clinical Evidence
Systematic Review
Description of Studies
One phase III, open-label, active-controlled randomized controlled trial (RCT) (PERSEUS, N = 709) was designed to evaluate whether the addition of daratumumab to VRd followed by maintenance therapy with daratumumab and lenalidomide prolongs progression-free survival (PFS) compared to VRd as induction and consolidation therapy followed by maintenance therapy with lenalidomide in patients with NDMM who are eligible for ASCT. The demographic characteristics were balanced between treatment groups. The median age of all patients was 60.0 years, with a range of 31 to 70 years. Most patients were male (58.7%; female: 41.3%) and white (92.1%); while 1.4% of patients were Asian, 1.3% were Black, 0.6% were Native Hawaiian or other Pacific Islander, and 0.4% were American Indian or Alaska Native. At baseline, most patients (63.6%) had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0. More than one-half (51.4%) of patients had disease that was International Staging System (ISS) stage I, and approximately one-fifth (21.7%) had high-risk cytogenetics such as del(17p), t(4;14), and t(14;16) chromosomal anomalies. The primary objective of the PERSEUS trial was to evaluate the efficacy of DVRd compared to VRd in patients with TE NDMM in prolonging PFS. The secondary outcomes included the rate of a complete response (CR) or better (key secondary), MRD negativity rate (key secondary), overall survival (OS) (key secondary), rate of a very good partial response (VGPR) or better, duration of response (DOR) (for CR or better), and health-related quality of life (HRQoL) assessments. The study was funded by the European Myeloma Network in collaboration with Janssen Research & Development.
Efficacy Results
Only those efficacy outcomes identified as important for this review are reported. Efficacy and safety data were evaluated at a planned interim analysis with a data cut-off date of August 1, 2023.
Progression-Free Survival
At the time of the first interim analysis, the median duration of follow-up for PFS was ████ ██████ months (range, ████ to ████) in the DVRd group, and ████ months in the VRd group (range, ████ to ████). Fifty patients (14.1%) in the DVRd treatment group and 103 patients (29.1%) in the VRd group experienced a PFS event; among them, ██ ██████ had disease progression and ██ ████% died in the DVRd group, and ██ ███████ had disease progression and ██ ██████ died in the VRd group. The median PFS was not reached (95% confidence interval [CI], not estimable) for both the DVRd and VRd groups. The Kaplan-Meier (KM) estimates of the probability of PFS at 48 months were 84.3% (95% CI, 79.5% to 88.1%) for the DVRd group and 67.7% (95% CI, 62.2% to 72.6%) for the VRd group, and the between-group difference was █████ (95% CI, ████ to █████). The PFS results were consistent across all prespecified and additional sensitivity analyses and subgroup results, except for patients aged 65 years or older.
Very Good Partial Response or Better Rate
The VGPR or better rate was 95.2% (95% CI, 92.4% to 97.2%) in the DVRd group and 89.3% (95% CI, 85.6% to 92.3%) in the VRd group, with a between-group difference of ████ (95% CI, ████ to █████) █ The stratified Cochran-Mantel-Haenszel (CMH) estimate of the odds ratio (OR) was 2.40 (95% CI, 1.33 to 4.35; nominal P = 0.0029).
Overall MRD Negativity Rate
The proportions of patients reported to have negative overall MRD in bone marrow by NGS (threshold of 10-5) and a CR or better were 75.2% (95% CI, █████ to █████) in the DVRd group and 47.5% (95% CI, ████% to ████%) in the VRd group, with a between-group difference of ████% (95% CI █ █████ to ███████ The CMH estimate of OR was 3.40 (95% CI, 2.47 to 4.69; P < 0.0001).
Overall Survival
The median durations of follow-up for OS were ████ months (range, ████ to ████) in the DVRd group and ████ months in the VRd group (range, ████ to ████). Thirty-four patients (9.6%) in the DVRd treatment group and 44 patients (12.4%) in the VRd group had died. The median OS was not reached (95% CI, not estimable) for either the DVRd or VRd group. The KM estimates of OS probability at 48 months were 89.4% (95% CI, 85.4% to 92.4%) for the DVRd group and 87.5% (95% CI, 83.5% to 90.6%) for the VRd group, with a between-group difference of ████ (95% CI, ████ % to ███%).
Duration of Response (For Complete Response or Better)
The median DOR (for CR or better response) was not reached in either the DVRd or VRd group. Among patients who had a CR or better (312 versus 248 for DVRd versus VRd, respectively), ██ patients (███%) in the DVRd group and ██ patients (████%) in the VRd group achieved a CR or better but developed disease progression or died due to disease progression █ ███ patients (████%) in the DVRd group and ███ patients (████%) in the VRd group were censored. The KM estimates of event-free probability at 42 months were ████% (95% CI, ████% to ████%) in the DVRd group and ████% (95% CI, ████% to ████%) in the VRd group, and the between-group difference was ████% (95% CI, ███% to ███%).
Change From Baseline in EQ-5D-5L Utility Score
At baseline, the mean EQ-5D-5L utility score was ███ (standard deviation [SD] = ████) in the DVRd group and ███ (SD = ████) in the VRd group. At maintenance cycle 34 (approximately 40 months of treatment), patients in the DVRd group reported a least squares mean increase (an improvement) from baseline in the EQ-5D-5L utility score of ███ (standard error [SE] = ████) compared to ███ (SE = ████) in patients in the VRd group, with a between-group difference of █ (95% CI █ █ to █; nominal P = ██████).
Harms Results
At the time of the first interim analysis (data cut-off: August 1, 2023), 349 of 351 patients (99.4%) in the DVRd group and 344 of 347 patients (99.1%) in the VRd group experienced at least 1 treatment-emergent adverse event (TEAE). The most common TEAEs were infections and infestations (86.9% versus 76.7% for DVRd versus VRd, respectively), blood and lymphatic system disorders (83.2% versus 73.2%), including neutropenia (69.2% versus 58.8%), thrombocytopenia (48.4% versus 34.3%), anemia (22.2% versus 20.7%), and gastrointestinal disorders (81.8% versus 77.2%). Serious adverse events (SAEs) were reported among 57.0% of patients in the DVRd group and 49.3% of patients in the VRd group; infections and infestations (35.0% versus 27.4%), including pneumonia (11.4% versus 6.1%), were the most reported SAEs. Withdrawals due to TEAEs were reported among 116 patients (33.0%) in the DVRd group and 104 patients (30.0%) in the VRd group. Thirty-four patients (9.7%) in the DVRd group and 43 patients (12.4%) in the VRd group had died at the time of the first interim analysis. The most reported cause of death was disease progression (4.6% versus 5.5%). Notable harms identified by the clinical experts included cytopenia, systemic administration-related reactions, and infections and infestations. Infections and infestations were observed in 305 patients (86.9%) in the DVRd group and 266 (76.7%) in the VRd group. Cytopenia (comprising neutropenia, anemia, thrombocytopenia, and lymphopenia group terms) was reported in ███ patients (████%) in the DVRd group and ███ patients (████%) in the VRd group. Systemic administration-related reactions, defined as systemic reactions related to daratumumab subcutaneous administration, were reported in ██ patients (███%) in the DVRd group, and the majority were grade 1 or 2 events.
Critical Appraisal
The choice of VRd as the comparator in the PERSEUS trial was clinically relevant, according to the clinical experts consulted for this review. The methods of randomization involved stratification using ISS at screening (I versus II versus III) and cytogenetics (standard risk versus high risk) that were considered appropriate. There was generally no notable imbalance in the baseline patient demographic and disease characteristics between treatment groups, except for the involved FLC in serum, which was not a prognostic factor according to the clinical experts, and the impact of the imbalance in FLC levels was assumed to be minimal. As the PERSEUS trial is ongoing, only results from an interim analysis were available for this review. At the time of the interim analysis, the median PFS and median OS were not reached in either treatment group. While a benefit and a trend toward an improved OS was observed with DVRd treatment and this was supported by improvements in MRD negativity, the longer-term assessment of treatment effect in terms of both median survival time and hazard ratios (HRs) is unknown. All patients in the DVRd group received preadministration medications (e.g., antihistamines, corticosteroids, analgesics, and drugs for obstructive airway diseases) before receiving daratumumab to prevent infusion-related reactions, whereas no patients in the VRd group received preinjections. Although the clinical experts indicated that the use of preinjections would not have an impact on the study results, given the adverse event prophylaxis effects of the preinjected medications, the review team noted that the higher frequency of the use of preinjections in the DVRd group may bias the safety results in favour of DVRd. Additionally, a larger proportion of patients in the DVRd group used immune sera and immunoglobulins compared with the VRd group, and this may bias the safety results in favour of the DVRd group, given immune sera and immunoglobulins could reduce the frequency of adverse events, such as infections, according to the clinical experts. Fewer patients received subsequent treatment in the DVRd group compared with the VRd group, and this may be a potential source of bias for OS results against the DVRd group. The benefit of DVRd in terms of the overall MRD negativity rate was likely overestimated as a higher proportion of patients in the VRd group compared with the DVRd group discontinued treatment due to disease progression, and patients who did not have MRD negativity at a given time point were considered MRD-positive in the analysis. In the analysis of HRQoL, which was measured using change from baseline in EQ-5D-5L utility score at maintenance cycle 34, a notably smaller proportion of patients in the DVRd group (████%) compared with the VRd group (████%) was lost to follow-up at maintenance cycle 34 day 1 (approximately 40 months of treatment). Given adverse events and disease progression were the common reasons for treatment discontinuation, the disproportionate number of missing data between treatment groups would introduce bias in favour of the VRd group. Many of the outcomes used in the PERSEUS trial (PFS, MRD negativity rate, OS, VGPR or better rate, DOR, and HRQoL) were identified as clinically important by patients and/or clinicians; however, because VGPR or better rate, DOR, and HRQoL were not part of the statistical testing strategy and were not adjusted for multiple testing, the ability to draw conclusions from these results may be limited.
The eligibility criteria of the PERSEUS trial were standard but stricter than what is found in clinical practice, according to the clinical experts. For example, patients aged 70 years or older were excluded; those patients could be candidates for daratumumab-lenalidomide in clinical practice. The baseline characteristics of the PERSEUS trial may be indicative of the overrepresentation of patients who were white (92.1%), as the clinical experts indicated there is a more diversified patient population, including patients of other ethnic groups, in their clinical practice. The proportion of patients who received consolidation therapy (75%) in the trial also did not appear to be reflective of clinical practice, where, according to the clinical experts consulted for this review, less than one-half (< 50%) of patients would receive brief consolidation in clinical practice, given that consolidation therapy is not currently funded in all jurisdictions in Canada. These limitations may restrict the generalizability of the study results to clinical practice in Canada.
GRADE Summary of Findings and Certainty of Evidence
For the pivotal PERSEUS trial identified in the sponsor’s systematic review, the Grading of Recommendations, Assessment, Development and Evaluation (GRADE) approach was used to assess the certainty of the evidence for outcomes considered most relevant to expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.13,14 In the GRADE approach, evidence from RCTs starts as high-certainty evidence and can be rated down because of study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for PFS, rate of a VGPR or better, overall MRD-negative rate (at 10-5), OS, DOR (defined as the duration of CR or better), and harms were set according to the presence of an important effect based on thresholds agreed upon by clinical experts consulted for this review. For safety and HRQoL, as measured by the EQ-5D-5L utility score, no minimal important difference (MID) was established, and the clinical experts could not provide a threshold of important difference. As a result, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect.
The selection of GRADE outcomes was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
clinical outcomes (PFS, VGRP or better rate, overall MRD negativity rate, OS, and duration of CR or better)
HRQoL
safety.
Table 2: Summary of Findings for DVRd Versus VRd for Patients With TE NDMM
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
VRd | DVRd | Difference | |||||
Progression-free survival | |||||||
Probability of being alive and progression-free at 48 months Follow-up (median): DVRd: █ █████ months VRd: █████ months | 709 (1 RCT) | HR = 0.42 (0.30 to 0.59) | 677 per 1,000 | 843 per 1,000 (795 to 881 per 1,000) | ███ per █████ (██ to ███ per 1,000) | Higha | DVRd results in a clinically important increase in the probability of patients being alive and progression-free at 48 months compared with VRd. |
Very good partial response rate | |||||||
The proportion of patients who achieved a VGPR or better (CR, sCR, or VGPR) Follow-up (median): DVRd: █████ months VRd: █████ months | 709 (1 RCT) | OR = 2.40 (1.33 to 4.35) | 893 per 1,000 | 952 per 1,000 (924 to 972 per 1,000) | ██ per 1,000 (██ to ███ per 1,000) | Moderateb,c | DVRd likely results in a clinically important increase in VGPR or better rate at 48 months compared with VRd. |
Overall MRD negativity rate at 10-5 in bone marrow | |||||||
The proportion of patients who achieved overall MRD-negative status (at 10-5) Follow-up (median): DVRd: █████ months VRd: █████ months | 709 (1 RCT) | OR = 3.40 (2.47 to 4.69) | 475 per 1,000 | 752 per 1,000 (███ to ███ per 1,000) | ███ per 1,000 (███ to ███ per 1,000) | Highd | DVRd results in a clinically important increase in overall MRD negativity rate at 48 months compared with VRd. |
Overall survival | |||||||
Probability of being alive at 48 months Follow-up (median): DVRd: █████ months VRd: █████ months | 709 (1 RCT) | HR = 0.73 (0.47 to 1.14) | 875 per 1,000 | 894 per 1,000 (854 to 924 per 1,000) | ██ per 1,000 (██ █████ ██ ██ ████ per 1,000) | Moderatee | DVRd likely results in little to no difference in the probability of being alive at 48 months compared with VRd. |
Duration of response (for CR or better) | |||||||
Probability of remaining in response of CR or sCR at 42 months Follow-up (median): DVRd: █████ months VRd: █████ months | 675 (1 RCT) | HR = ████ (████ ██ █████ | ███ per 1,000 | ███ ███ █████ ████ ██ ███ ███ █████) | ███ ███ █████ ███ ██ ███ ███ █████) | Moderatec,f | DVRd likely results in a clinically important increase in the probability of remaining in response of CR or sCR at 42 months compared with VRd. |
Health-related quality of life | |||||||
Least squares mean change from baseline in EQ-5D-5L utility score at maintenance cycle 34 (approximately 40 months of treatment) Follow-up (median): DVRd: █████ months VRd█ █████ months | 296 (1 RCT) | NR | ███ (NR) | ███ (NR) | | ██ ██ ██ | Highc,g | DVRd results in little to no difference in the change from baseline in EQ-5D-5L utility score at maintenance cycle 34 (approximately 40 months of treatment) compared with VRd. |
Harms | |||||||
Incidence infections and infestations at 48 months Follow-up (median): DVRd: █████ months VRd: █████ months | 698 (1 RCT) | NR | ███ per 1,000 | ███ per 1,000 (NR) | 102 per 1,000 (██ ██ ███ ███ ██████ | Moderatec,h | DVRd likely results in a clinically important increase in the incidence infections and infestations at 48 months compared with VRd. |
Incidence of cytopenia at 48 months Follow-up (median): DVRd: █████ months VRd: █████ months | 698 (1 RCT) | NR | ███ per 1,000 | ███ per 1,000 (NR) | ██ ███ █████ ███ ██ ███ ███ █████) | Highc,i | DVRd results in little to no difference in the incidence of cytopenia at 48 months compared with VRd. |
Incidence of systemic administration-related reactions at 48 months Follow-up (median): DVRd: █████ months VRd: █████ months | 698 (1 RCT) | NR | NA | ██ per 1,000 (NR) | NA | NA | NA |
CI = confidence interval; CR = complete response; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; HR = hazard ratio; MID = minimal important difference; MRD = minimal residual disease; NA = not applicable; NDMM = newly diagnosed multiple myeloma; NR = not reported; OR = odds ratio; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; sCR = stringent complete response; TE = transplant-eligible; VGPR = very good partial response; VRd = bortezomib-lenalidomide-dexamethasone.
Note: Study limitations (internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aImprecision was not rated down. There is no established between-group MID for PFS at 48 months, but the clinical experts considered a 5% difference between groups in the probability of patients being alive and progression-free a possible threshold of clinical importance. The point estimate and the upper and lower bounds of the 95% CI for the between-group difference suggested a clinically important difference for DVRd versus VRd based on a 5% threshold.
bRated down 1 level for serious imprecision. There is no established between-group MID for VGPR or better rate at 48 months, but the clinical experts considered a 10% difference between groups in the proportion of patients who achieved a VGPR or better (CR, sCR, or VGPR) a possible threshold of clinical importance. The point estimate and the lower bound of the 95% CI for the between-group difference suggested no clinically important difference between the 2 groups while the upper bound of the 95% CI suggested a clinically important difference for DVRd versus VRd based on a 10% threshold. The statistical testing for VGPR or better rate was not adjusted for multiplicity in the PERSEUS trial and should be considered supportive evidence.
cThe statistical testing for this end point was not adjusted for multiplicity in analysis in the PERSEUS trial and should be considered supportive evidence.
dImprecision was not rated down. There is no established between-group MID for the overall MRD negativity rate (at 10-5) at 48 months, but the clinical experts considered a 10% difference between groups in overall MRD negativity rate (at 10-5) a possible threshold of clinical importance. The point estimate and the upper and lower bounds of the 95% CI for the between-group difference suggested a clinically important difference for DVRd versus VRd based on a 10% threshold.
eRated down 1 level for serious imprecision. There is no established between-group MID for OS at 48 months, but the clinical experts considered a 5% difference between groups in the probability of patients being alive at 48 months a possible threshold of clinical importance. The point estimate and the lower bound of the 95% CI for the between-group difference suggested no clinically important difference between the 2 groups while the upper bound of the 95% CI suggested a clinically important difference for DVRd versus VRd based on a 5% threshold.
fRated down 1 level for serious imprecision. There is no established between-group MID for duration of CR or better at 42 months, but the clinical experts considered a 10% difference between groups in the probability of patients remaining in response (CR or sCR) at 42 months a possible threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested a clinically important difference between the 2 groups while the lower bound of the 95% CI suggested no clinically important difference for DVRd versus VRd based on a 10% threshold.
gImprecision was not rated down. There is no established MID for change from baseline in EQ-5D-5L utility score and the clinical experts could not provide a threshold of important difference, so target of certainty appraisal was any effect for the change from baseline in EQ-5D-5L utility score at 48 months. The review team judged that the point estimate and 95% CI suggested no important difference between the 2 groups.
hRated down 1 level for serious imprecision. There is no established between-group MID for the incidence of infections and infestations at 48 months, but the clinical experts considered a 10% difference between groups in the incidence of infections and infestations at 48 months a possible threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested a clinically important difference between the 2 groups while the lower bound of the 95% CI suggested no clinically important difference for DVRd versus VRd based on a 10% threshold.
iImprecision was not rated down. There is no established MID for the incidence of cytopenia at 48 months, but the clinical experts considered a 20% difference between groups in the incidence of cytopenia at 48 months a possible threshold of clinical importance. The point estimate and the upper and lower bounds of the 95% CI for the between-group difference suggested no clinically important difference for DVRd versus VRd based on a 20% threshold.
Sources: Clinical Study Report for PERSEUS (2024),15 sponsor-provided additional information,16 and the sponsor’s Summary of Clinical Evidence.17
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparisons
Description of Studies
In the absence of head-to-head evidence to compare DVRd with all relevant comparators, 5 sponsor-conducted indirect treatment comparisons (ITCs) in patients with TE NDMM were included in this review.18 Based on the results of a feasibility study, the sponsor noted that a lack of available studies with a common comparator to form a connected network made fitting a random-effects model, such as a network meta-analysis (NMA), infeasible. To compare DVRd against CyBorD, the sponsor conducted 2 unanchored matching-adjusted indirect comparisons (MAICs), the PERSEUS trial versus the GMMG-MM5 trial and the PERSEUS trial versus the VCAT trial, to indirectly compare patient cohorts who received DVRd with those who received CyBorD; in each comparison, the former study assessed patients who received the full treatment sequence (i.e., induction through maintenance treatments), while the latter study assessed patients who received induction through consolidation treatments. Three unanchored MAICs that indirectly compared daratumumab-lenalidomide versus lenalidomide as maintenance treatments for patients with TE NDMM (the PERSEUS trial versus the Myeloma XI trial, PERSEUS versus the IFM 2005-02 trial, and PERSEUS versus the CALGB 100104 trial) were also submitted. These 3 MAICs were conducted by the sponsor to assess the incremental benefit of adding daratumumab to a maintenance regimen consisting of lenalidomide alone, given that, in the PERSEUS trial, no rerandomization occurred upon initiation of maintenance treatment to minimize confounding. The maintenance studies included patients regardless of MRD response to induction through consolidation treatment. Individual patient data (IPD) from the DVRd versus daratumumab-lenalidomide cohort of the PERSEUS trial were matching-adjusted, based on relevant covariates identified in the literature or thorough expert opinion, to aggregate data from the comparator trials identified by a systematic literature review (SLR). OS and PFS were outcomes of interest. Balance between the populations in each comparison, after weighting, was assessed using the effective sample size (ESS) and the distribution of weights.
Efficacy Results
The ESS for the DVRd group after match adjustment was █████ █████% of the original sample size from the PERSEUS trial in the PERSEUS versus GMMG-MM5 trial comparison, and █████ ██████ of the original sample size from the PERSEUS trial in the PERSEUS versus VCAT studies. The ESS for the daratumumab-lenalidomide group after match adjustment was ██████ █████, and █████ ███████ ██████ ███ ████% of the original sample size from the PERSEUS trial, respectively, in the comparison of the PERSEUS versus Myeloma XI trials, PERSEUS versus IFM 2005-02 trials, and PERSEUS versus CALGB 100104 studies, respectively.
DVRd Versus CyBorD: Full Treatment Sequence
Following weighting, results of the comparison of the PERSEUS versus GMMG-MM5 studies were in favour of DVRd compared with CyBorD with respect to OS (HR = █████ ███ ███ ████ ██ █████ ██████) and PFS (HR = 0████ ███ ██ ████ ██ █████ ████████).
DVRd Versus CyBorD: Induction Through Consolidation
Following weighting, PFS results of the comparison of the PERSEUS versus VCAT studies were in favour of DVRd compared with CyBorD (HR = █████ ███ ███ ████ ██ █████ ████████). Results of the sensitivity analysis for the PERSEUS versus VCAT trials were consistent with the base case. OS was not assessed in this ITC.
Daratumumab-Lenalidomide Versus Lenalidomide Maintenance Treatment
Following weighting, the difference in restricted mean survival time (RMST) between daratumumab-lenalidomide and lenalidomide alone with respect to OS at 3 years of maintenance therapy was ███ ██████ ████ ███ █████ ██████ ██ ████ ███████ in the PERSEUS versus Myeloma XI trials, ████ ██████ ████ ███ ████ ██████ ██ ████ ██████) in the PERSEUS versus IFM 2005-02 trials, and ████ ██████ ████ ███ █████ ██████ ██ ████ ██████) in the PERSEUS versus CALGB 100104 trials. The difference in RMST between daratumumab-lenalidomide and lenalidomide alone with respect to PFS at 3 years of maintenance therapy was ███ ██████ ████ ███ ████ ██████ ██ ████ ███████ in the PERSEUS versus Myeloma XI trials, ███ ██████ ████ ███ ████ ██████ ██ ████ ██████) in the PERSEUS versus IFM 2005-02 trials, and ████ months (███ ███ ████ ██████ ██ ████ ██████) in the PERSEUS versus CALGB 100104 trials.
Harms Results
Harms were not assessed in the ITCs.
Critical Appraisal
Studies included in the ITCs were identified by a sponsor-conducted SLR using appropriate methods. A feasibility assessment for a comprehensive ITC was subsequently conducted to inform study selection; however, the reasons for study exclusion were not documented, creating a potential risk of selection bias, although the extent of such bias is unclear. Other important limitations of the MAICs included an inability to adjust for potential prognostic factors (e.g., ECOG PS [not adjusted for in MAICs, except the PERSEUS versus VCAT studies] and the presence of extramedullary plasmacytomas). As well, there were temporal discordances in the study period between the included studies, during which major changes in subsequent treatment patterns occurred and, as a result, this could be a potential source of bias for the OS results. The duration of follow-up differed between studies, which may bias the comparisons of the HR. Additional limitations of the 3 MAICs assessing maintenance treatments included heterogeneity in induction and consolidation regimens between studies and a lack of adjustment for MRD negativity rates at the baseline of maintenance therapy (not adjusted for in the PERSEUS versus IFM 2005-02 trials and PERSEUS versus CALGB 100104 trials), which was identified as an important prognostic factor for maintenance treatment. A sizable reduction in ESS (█████ ██ █████) of the PERSEUS trial cohort after the match-adjustment process was observed in the comparisons versus the VCAT, Myeloma XI, IFM 2005-02, and CALGB 100104 studies, suggesting that there was a poor population overlap between studies and that the results may be heavily influenced by a subset of the sample in the PERSEUS trial that may not be representative of the full sample. MRD negativity rate, HRQoL, and harms outcomes, which are important to patients and clinicians, were not assessed in the analyses, representing a gap in evidence.
Studies Addressing Gaps in the Evidence From the Systematic Review
This section summarizes 1 sponsor-submitted RCT (AURIGA) to address a gap in comparative evidence and focusing on the use of daratumumab-lenalidomide versus lenalidomide monotherapy, as maintenance therapy, for NDMM after ASCT.
Description of Studies
The AURIGA trial (NCT03901963) is a phase III, open-label, active-controlled, multicentre RCT that evaluated the clinical benefit of adding daratumumab to maintenance treatment with lenalidomide among adult patients with TE NDMM who are MRD-positive after induction therapy and ASCT.19,20 The AURIGA trial randomized 200 patients across 52 sites in the US and Canada to receive either daratumumab-lenalidomide or lenalidomide as maintenance therapy for after induction and ASCT for TE NDMM.19,21 Eligible patients had NDMM, were treated with a minimum of 4 cycles of induction therapy, and had received high-dose therapy (HDT) and ASCT within 12 months of the start of induction therapy, with patients being within 6 months of ASCT on the date of randomization.19 Patients were also required to have a response of VGPR or better (assessed according to the IMWG 2016 criteria22) at the time of randomization, residual disease as defined by detectable MRD, and an ECOG PS score of 0, 1, or 2.19
Efficacy Results
Primary End Point
At the clinical cut-off date of April 4, 2024, the MRD conversion rate from MRD positivity to MRD negativity (10-5) from baseline to 12 months since the initiation of maintenance therapy was 50.5% in the daratumumab-lenalidomide treatment group compared with 18.8% in the lenalidomide treatment group.23,24 The corresponding OR (daratumumab-lenalidomide versus lenalidomide) was 4.51 (95% CI, 2.37 to 8.57; P < 0.0001), which was statistically significant at the prespecified 2-sided alpha level of 0.05.23,24
Secondary End Points
At a median study follow-up time of 32.3 months, 45 PFS events were observed. Of the 45 events, 19 were in the daratumumab-lenalidomide treatment group and 26 were in the lenalidomide treatment group.23,24 The corresponding HR was 0.53 (95% CI, 0.29 to 0.97), demonstrating a 47% reduction in the risk of disease progression or death in patients receiving daratumumab-lenalidomide compared to those receiving lenalidomide.23,24 The estimated 30-month PFS rates were 82.7% and 66.4% for the daratumumab-lenalidomide and lenalidomide treatment groups, respectively.23,24
The overall MRD (10-5) negativity conversion rate from baseline throughout the study treatment period was higher in the daratumumab-lenalidomide treatment group compared to the lenalidomide treatment group (60.6% versus 27.7%, respectively), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 4.12 (95% CI, 2.26 to 7.52; P < 0.0001).23,24
The sustained MRD negativity rate at 6 months or later was higher in the daratumumab-lenalidomide treatment group compared with the lenalidomide treatment group (35.4% versus 13.9%), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 3.40 (95% CI, 1.69 to 6.83) and a P value of 0.0005.23,24 Similarly, the sustained MRD negativity rate at 12 months or later was higher in the daratumumab-lenalidomide treatment group compared with the lenalidomide treatment group (17.2% versus 5.0%, respectively), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 4.08 (95% CI, 1.43 to 11.62; P = 0.0065).23,24
At a median study follow-up time of 32.3 months, a total of 15 OS events were observed.23,24 Of the 15 events, 5 were in the daratumumab-lenalidomide treatment group and 9 were in the lenalidomide treatment group.23,24 The median OS was not reached for either treatment group. The estimated 30-month OS rates were 94.6% and 91% for the daratumumab-lenalidomide and lenalidomide treatment groups, respectively.23,24
The overall rate of a CR or better response according to IMWG criteria was higher in the daratumumab-lenalidomide treatment group compared with the lenalidomide treatment group (75.8%; 95% CI, 66.1% to 83.8% versus 61.4%; 95% CI, 51.2% to 70.9%), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 2.00 (95% CI, 1.08 to 3.69; P = 0.0255).23,24
Health-related quality of life, functioning, and symptoms were assessed using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30), European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire for Myeloma Module (EORTC QLQ-MY20), and the EQ-5D-5L questionnaire. Overall, there was no difference in HRQoL, symptoms, and functioning between the daratumumab-lenalidomide and lenalidomide treatment groups and no detriment of HRQoL with the addition of daratumumab to maintenance therapy with lenalidomide.
Harms Results
The incidence of TEAEs in the AURIGA trial was 99% for both the daratumumab-lenalidomide and lenalidomide treatment groups.23,24 The most frequently reported TEAEs (an incidence of 30% or higher in either arm) were neutropenia (daratumumab-lenalidomide: 64.6%; lenalidomide: 61.2%), diarrhea (daratumumab-lenalidomide: 61.5%; lenalidomide: 55.1%), and fatigue (daratumumab-lenalidomide: 45.8%; lenalidomide: 46.9%). Injection-related reactions were reported in 13.5% of patients in the daratumumab-lenalidomide treatment group.23,24 Compared with the lenalidomide treatment group, patients in the daratumumab-lenalidomide treatment group experienced higher incidences of grade 3 or 4 TEAEs (74.0% versus 67.3%, respectively) and serious TEAEs (30.2% versus 22.4%, respectively).23,24 Rates of discontinuation due to TEAEs were also higher in the daratumumab-lenalidomide treatment group compared to the lenalidomide treatment group (12.5% versus 7.1%, respectively).23,24 Two deaths related to TEAEs occurred in the daratumumab-lenalidomide treatment group and a single TEAE-related death occurred in the lenalidomide treatment group.23,24
Critical Appraisal
Strengths of the AURIGA trial included the stratification of patients by cytogenetic risk before randomization and the use of an intention-to-treat (ITT) analysis to account for all randomized patients.19 A key limitation of the AURIGA trial was its open-label study design, which may have contributed to performance bias in results for patient-reported outcomes. Moreover, a larger proportion of patients in the daratumumab-lenalidomide treatment group had high cytogenetic risk according to available local cytogenetic risk data at diagnosis (daratumumab-lenalidomide: 23.9%; lenalidomide: 16.9%).19 The sponsor study report noted that any potential treatment effect due to this imbalance would have favoured the lenalidomide treatment group. Finally, subjects with missing or unevaluable MRD status were considered to have MRD-positive status for the analysis of the primary end point. Given that a larger proportion of patients in the lenalidomide treatment group dropped out of the study, the imputation of all missing subjects as having an MRD-positive status would have likely biased the results in favour of the daratumumab-lenalidomide treatment group.
Although the AURIGA trial recruited patients living in the US and Canada, the trial results do not explicitly state the proportion of patients living in Canada nor do they provide a subgroup analysis of these patients. Although it may be argued that there are similarities between patients with MM living in Canada and those living in the US, it is unclear how representative the findings of the trial are to patients with MM living in Canada and being managed in clinical practice. Moreover, the trial only included patients who were MRD-positive and who achieved a VGPR or better response after transplant.19 Clinical experts consulted by CDA-AMC indicated that patients who achieve a PR or better are able to proceed with maintenance therapy as long as they do not show signs of progressive disease. The applicability of findings from the trial would therefore be limited for this subset of patients. The AURIGA trial excluded patients who received daratumumab or other anti-CD38 therapies.19 The generalizability of the trial results may therefore be limited for patients in the PERSEUS trial. This is important because the AURIGA trial was submitted to address the lack of evidence pertaining to the efficacy of daratumumab-lenalidomide as maintenance therapy from the PERSEUS trial. The results of the PERSEUS and AURIGA trials showed similar trends in terms of improvement in PFS, MRD negativity, and response associated with the addition of daratumumab to their respective regimens.
Conclusions
One phase III, ongoing open-label, active-controlled RCT (PERSEUS) comparing DVRd with VRd in treating patients with NDMM who are eligible for ASCT were included in this review. Although not currently funded for first-line treatment in patients with TE NDMM, data from both patients whose disease is TE or transplant-noneligible increasingly indicates that daratumumab-based induction and maintenance approaches may yield higher and deeper clinical responses. Results from the PERSEUS trial demonstrated that when compared to VRd, the addition of daratumumab to VRd results in a clinically significant benefit in PFS. The treatment benefit with DVRd was also consistently presented in the rate of a VGPR or better and the overall MRD-negative rate. However, treatment benefits in OS and DOR (for CR or better) were uncertain as the median OS and median DOR were not reached in either study group in the interim analysis. The treatment benefit of DVRd was not observed in HRQoL. The pivotal trial showed that, compared with VRd, DVRd results in little to no difference in the change from baseline in EQ-5D-5L utility scores.
Compared with VRd, there is moderate-certainty to high-certainty evidence that DVRd likely results in an increase in the incidence of infections and infestations, and results in little to no difference in the incidence of cytopenia. Overall, no new safety signals were identified in the PERSEUS trial, and the observed safety profile of DVRd is similar to that of VRd.
No direct evidence between DVRd versus CyBorD was submitted. Indirect evidence from 2 sponsor-conducted MAICs suggested that DVRd, compared with CyBorD, was associated with improved PFS in patients with TE NDMM; however, the magnitude of benefit was uncertain due to limitations of the analyses, including different durations of follow-up and the lack of adjustment for potential prognostic factors. Potential heterogeneity in subsequent treatment use further increased the uncertainty in the OS findings, and as such, no definitive conclusions could be drawn on the relative effect of DVRd versus CyBorD on OS.
A phase III, ongoing open-label RCT (AURIGA) and 3 sponsor-conducted MAICs were submitted to address a gap in comparative evidence regarding daratumumab-lenalidomide versus lenalidomide alone as maintenance therapy for NDMM after ASCT. The AURIGA trial found that, when compared to lenalidomide alone, the addition of daratumumab to lenalidomide as maintenance therapy in patients who had MRD-positive disease at baseline resulted in a clinically significant benefit in the MRD conversion rate from baseline to 12 months. Moreover, treatment benefits were observed for PFS, overall MRD (10-5) negativity conversion rate from baseline, sustained MRD negativity rates at 6 and 12 months, and the overall CR or better response rate. There was no difference in HRQoL, symptoms, and functioning between the daratumumab-lenalidomide and lenalidomide treatment groups. However, a few limitations of the trial were noted, which included its open-label study design, imbalance in proportions of patients having high cytogenetic risks according to available local cytogenetic risk data at diagnosis, and handling of patients with missing or unevaluable data on MRD status in the primary end-point analysis. No definitive conclusions could be drawn from the MAICs conducted in patients with TE NDMM (regardless of MRD status at maintenance baseline) with respect to the relative effects of daratumumab-lenalidomide versus lenalidomide due to important methodological limitations of the analyses.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of daratumumab, as an 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for SC injection, in combination with VRd, followed by maintenance treatment in combination with lenalidomide, in the treatment of adult patients with NDMM who are eligible for ASCT.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the review team.
MM is a hematological malignancy defined by plasma cell proliferation and excessive production of abnormal immunoglobulin monoclonal M protein.1,2 Most cases of MM are preceded by monoclonal gammopathy of undetermined significance, which is an asymptomatic, premalignant plasma cell condition characterized by the presence of abnormal M protein in the blood.25 Another precursor is smouldering MM, which is a more advanced premalignant condition that develops into MM more rapidly than monoclonal gammopathy of undetermined significance.25 Regardless of the preceding condition, once MM develops, patients commonly experience fatigue and bone pain,3 as well as renal or nervous system problems, recurring infections, and fever.4
Despite treatment advances in recent years, MM remains incurable8,9 and patients face a poor prognosis with a 5-year survival rate of approximately 50%.10 Moreover, most patients with MM relapse and many develop refractoriness to commonly used treatments.11 However, treatment with ASCT among patients with NDMM is associated with significantly improved clinical outcomes and is considered the standard of care for TE NDMM.9,12 Eligibility for ASCT is determined by a variety of factors, including age, performance status, comorbidities, frailty, disability, and availability of social and family support.26-28
In Canada, an estimated 4,100 individuals had NDMM and approximately 1,750 deaths due to MM occurred in 2024.5 Although no studies in Canada have directly estimated the prevalence of TE NDMM, the number of patients with TE NDMM and attrition rates after first-line ASCT can be derived from recent observational studies focused on the patient population with MM in Canada,6,7 from which the estimated prevalence of TE NDMM can be imputed. Assuming that 49.7% of patients with NDMM are eligible for ASCT and end up receiving ASCT7 and a 7% attrition rate in first-line ASCT therapy,6 an estimated 1,895 patients with TE NDMM were living in Canada as of 2024.
MM is typically diagnosed by a primary care physician, based on criteria developed by the IMWG.25,29 According to these criteria, MM is diagnosed based on the presence of either at least 10% clonal bone marrow plasma cells or a biopsy-proven plasmacytoma and the presence of a myeloma-defining event. Such events may include end-organ damage defined as part of the “CRAB” criteria (i.e., hypercalcemia, renal insufficiency, anemia, and bone lesions), a clonal bone marrow plasma cell percentage of 60% or greater, an involved-uninvolved serum FLC ratio equal to or greater than 100 (the involved FLC level must be ≥ 100 mg/L), and more than 1 focal lesion on MRI studies.25
Several laboratory tests are recommended for the diagnosis of MM, which include complete blood count, peripheral blood smear, serum protein analysis, including protein electrophoresis, immunofixation electrophoresis, serum FLC assay, and urine analysis.30 In addition, bone marrow aspiration and core biopsy are used to measure the percentage of plasma cells to measure the percentage of plasma cells relative to total nucleated cells, as well as in situ hybridization for kappa and lambda immunoglobulin light chains to determine clonality.31 Chromosomal aberrations are typically assessed using fluorescence in situ hybridization targeted to myeloma cells following either plasma cell staining or cell sorting.30 Bone lesions in MM have historically been detected by radiographic skeletal surveys; however, the IMWG currently recommends that bone disease be assessed with newer imaging techniques if available.30,32 These may include whole-body low-dose CT, MRI, and/or 18F-fluorodeoxyglucose PET or CT.30
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the review team.
Patients with NDMM are typically categorized into 2 groups defined by their disease’s suitability for ASCT: TE NDMM or transplant-ineligible NDMM. The clinical experts consulted for this review indicated that the goals of treatment are to improve survival, prolong the time to relapse, and reduce cancer-related complications.
According to the clinical experts, for patients with TE NDMM (the patient population relevant to this submission), the current treatment consists of a multiphase approach, including induction therapy, ASCT (with high-dose chemotherapy) with or without consolidation therapy, followed by maintenance therapy (Figure 1). The clinical experts noted that the induction therapy currently consists of 4 to 6 cycles of a multidrug regimen of either CyBorD or VRd, with VRd being the most commonly used regimen in Canada. Most patients (95%) respond to induction therapy and will proceed to stem cell collection using growth factors (granulocyte colony-stimulating factor with or without plerixafor) on their own or following chemotherapy, such as cyclophosphamide. ASCT consists of a single administration of high-dose melphalan followed a day later by stem cell infusion. The clinical experts indicated that a small number (5% to 10%) of patients will not proceed to ASCT due to other health issues, patient choice, and failure to mobilize stem cells, and a fraction (10%) of patients who do not have a good response to the initial regimen may be switched to a second-line regimen that contains daratumumab or isatuximab to produce a better response. A small number (about 6%) of patients undergo ASCT and then go into an extended remission, according to the clinical experts. Following recovery from ASCT, post-transplant therapies include consolidation and maintenance therapies. As consolidation therapy is not publicly funded in all jurisdictions in Canada, currently, less than one-half (< 50%) of patients would receive a brief consolidation therapy with VRd (2 to 4 cycles) followed by maintenance therapy with lenalidomide alone, or maintenance with bortezomib plus lenalidomide or a proteasome inhibitor (e.g., ixazomib) plus lenalidomide for patients with high-risk cytogenetics (about 10% to 25% of patients), according to the clinical experts. Although the CDA-AMC provisional funding algorithm for MM describes maintenance therapy with lenalidomide as optional, the clinical experts indicated that almost all patients will receive maintenance therapy with lenalidomide, other than a very small percentage (2% to 5%) of patients who may be intolerant to lenalidomide. Additionally, the clinical experts mentioned that a tandem transplant is considered for patients with high-risk cytogenetics, such as del(17p), t(4;14),and t(14;16) chromosomal anomalies, in some centres; the tandem transplant consists of a second transplant around 3 months after the first, followed by maintenance therapy as described previously.
Figure 1: Treatment Pathway for Patients With TE NDMM
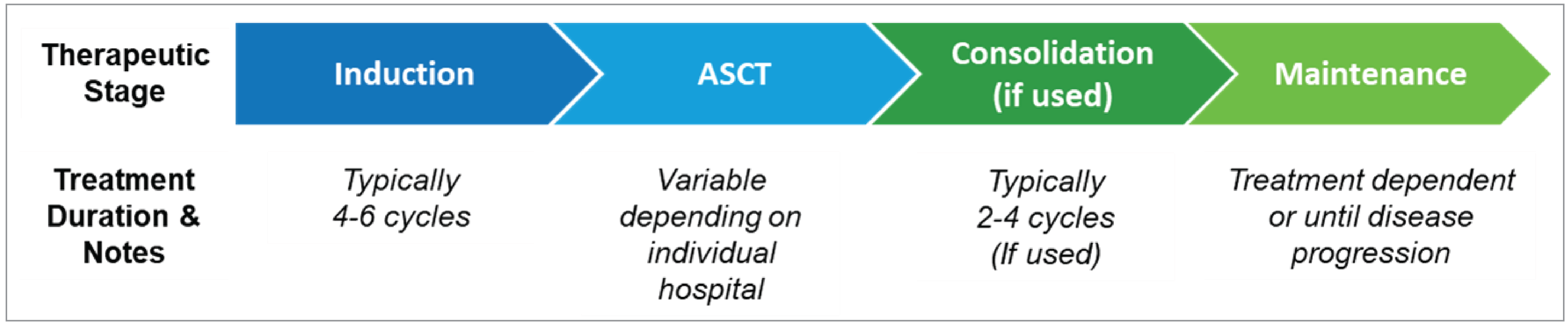
ASCT = autologous stem cell transplant; NDMM = newly diagnosed multiple myeloma; TE = treatment-eligible.
Source: Sponsor’s Summary of Clinical Evidence.17
Drug Under Review
Key characteristics of daratumumab, along with other treatments available for patients with NDMM who are eligible for ASCT, are summarized in Table 3.
Daratumumab is a fully human immunoglobulin G1 monoclonal antibody that targets CD38, a transmembrane glycoprotein expressed at high levels on the surface of MM tumour cells. Based on findings from preclinical studies, daratumumab binds to the CD38 protein and induces immune-mediated tumour cell death through Fc-mediated cross-linking.33
Daratumumab is available in IV or SC forms of administration. Daratumumab in IV form has previously received positive recommendations for reimbursement from the pan-Canadian Oncology Drug Review in several combination regimens indicated for NDMM and relapsed and/or refractory MM.34-36 Daratumumab SC, which is the main focus of the current submission, contains recombinant human hyaluronidase PH20, which increases tissue dispersion and the absorption of coadministered daratumumab.37,38
Daratumumab SC received a Notice of Compliance from Health Canada on November 27, 2024, for use in combination with VRd, followed by maintenance treatment in combination with lenalidomide, for the treatment of adult patients with newly diagnosed MM who are eligible for ASCT. The reimbursement request aligns with the Health Canada indication. Daratumumab SC, in combination with VRd, was recently approved in the US and received a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use for the treatment of adult patients with NDMM who are eligible for ASCT.39,40 These recommendations were supported by data for the phase III PERSEUS trial, which evaluated the efficacy and safety of daratumumab SC added to VRd (DVRd) compared to VRd alone among adult patients with NDMM who are eligible for ASCT.41
Daratumumab was previously reviewed by CDA-AMC for the following indications in the MM and light-chain amyloidosis settings, which were different from the current reimbursement request.
On January 28, 2022, a recommendation for reimbursement was issued for daratumumab in combination with CyBorD for the treatment of adult patients with newly diagnosed light-chain amyloidosis.42
On March 5, 2020, a recommendation for reimbursement was issued for daratumumab in combination with lenalidomide and dexamethasone for the treatment of patients with NDMM who are ineligible for ASCT.43
On August 29, 2019, a recommendation for reimbursement was issued for daratumumab in combination with bortezomib, melphalan, and prednisone, for the treatment of patients with NDMM who are not suitable for ASCT.44
On October 5, 2017, a recommendation for reimbursement was issued for daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with MM who have received at least 1 prior therapy.45
On December 1, 2016, daratumumab monotherapy was not recommended for reimbursement for the treatment of “patients with MM who 1) have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory drug, or 2) have failed or are intolerant to a proteasome inhibitor and who have failed or are intolerant to an immunomodulatory drug.”46
Daratumumab SC is supplied as an 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for injection. According to the draft product monograph for daratumumab SC, the recommended SC dose is 1,800 mg administered over approximately 3 to 5 minutes. During the induction treatment phase, the recommended SC dose is administered weekly (for a total of 8 doses) in weeks 1 to 8 and every 2 weeks (for a total of 4 doses) in weeks 9 to 16. After HDT and ASCT, the recommended SC dose is administered every 2 weeks (for a total of 4 doses) in weeks 17 to 24 during the consolidation treatment phase and every 4 weeks from week 25 onward until disease progression during the maintenance treatment phase.37
Testing Procedure Considerations
MRD refers to the persistence of a small amount of cancer cells after treatment, leading to relapse even after achieving a CR.52 In MM, the absence of MRD, known as MRD negativity, has been associated with significant improvements in PFS and OS, making it a strong prognostic factor.53-55 In addition, emerging evidence from clinical trials and other studies have demonstrated the use of MRD status in informing clinical decision-making (e.g., de-escalation, discontinuation, or change in treatment) related to various therapies.56-59
MRD status can be assessed using various methods, commonly NGF and NGS. Both methods use a bone marrow aspiration sample. NGF-based MRD testing, developed by EuroFlow,60 has been validated in real-world patients with MM as well as in clinical trials.52 NGS-based MRD testing (e.g., Adaptive Biotechnologies’ clonoSEQ) is considered the gold standard and requires a smaller sample than NGF does.61 NGS has high sensitivity that can be generalizable across institutions but requires a baseline sample for screening and identifying the predominant clonotype specific to each patient to monitor.61 NGF-based testing can detect the presence of residual cancer cells at sensitivity thresholds up to 10-5 cells (i.e., 1 cancer cell among 100,000 bone marrow cells) and NGS-based testing is sensitive up to 10-6 cells (i.e., 1 cancer cell in 1 million bone marrow cells).61 Emerging technologies, such as mass spectrometry–based MRD testing using peripheral blood samples, are also being evaluated in Canada.62
Table 3: Key Characteristics of Daratumumab, Bortezomib, and Lenalidomide
Characteristic | Darzalex SC | Revlimid | ||
|---|---|---|---|---|
Mechanism of action | Daratumumab is a fully human IgG1 mAb that targets the CD38 protein expressed on the surface of MM cells. Based on in vitro studies, by binding to CD38, daratumumab induces immune-mediated tumour cell death or apoptosis through Fc-mediated cross-linking. Darzalex SC contains rHuPH20, which increases tissue dispersion and the absorption of coadministered daratumumab. | Daratumumab is a fully human IgG1 mAb that targets the CD38 protein expressed on the surface of MM cells. Based on in vitro studies, by binding to CD38, daratumumab induces immune-mediated tumour cell death or apoptosis through Fc-mediated cross-linking. | Bortezomib is a reversible inhibitor of the 26S proteasome, a large protein complex that degrades ubiquitinated proteins. Inhibition of the 26S proteasome affects multiple cell signalling cascades and can lead to cell death. | The mechanism of lenalidomide is not fully characterized, but it is immunomodulatory with anti-inflammatory activity and antiangiogenic effects. It inhibits TNF-alpha production, stimulates thymus and natural killer cells, reduces serum levels of VEGF, and promotes G1 cell cycle arrest and apoptosis of malignant cells. |
Indicationa |
|
|
|
|
Route of administration | SC | IV | IV | PO |
Recommended dose | Combination therapy with VRd
Combination therapy with Rd: 1,800 mg/kg SC over approximately 3 to 5 minute, weekly for weeks 1 through 8, every 2 weeks for weeks 9 through 24, and every 4 weeks for week 25 onward until disease progression Combination therapy with VMP: 1,800 mg/kg SC over approximately 3 to 5 minute, weekly for weeks 1 through 6, every 3 weeks for weeks 7 through 54, and every 4 weeks for weeks 55 onward until disease progression | Combination therapy with VTd
Combination therapy with Rd: 16 mg/kg BW IV weekly for weeks 1 to 8, every 2 weeks for weeks 9 through 24, and every 4 weeks for weeks 25 onward until disease progression Combination therapy with VMP: 16mg/kg BW IV weekly for weeks 1 to 6, every 3 weeks for weeks 7 through 54, and every 4 weeks for week 55 onward until disease progression | Suitable for SCT: 1.3 mg/m2 BSA IV twice weekly on days 1, 4, 8, and 11, followed by a rest period of up to 20 days, which is considered a treatment cycle Unsuitable for SCT
| 25 mg/day PO on days 1 through 21 per 28-day cycle |
Serious adverse effects or safety issues |
|
|
|
|
Other |
|
|
|
|
ASCT = autologous stem cell transplant; BSA = body surface area; BW = body weight; CBC = complete blood count; DVT = deep-vein thrombosis; HBV = hepatitis B virus; IgG = immunoglobulin G; IgG1 = immunoglobulin G1; IRR = infusion-related reaction; mAb = monoclonal antibody; min = minute; MM = multiple myeloma; NDMM = newly diagnosed multiple myeloma; PO = oral; Rd = lenalidomide-dexamethasone; rHuPH20 = recombinant human hyaluronidase PH20; SC = subcutaneous; SCT = stem cell transplant; TNF = tumour necrosis factor; VEGF = vascular endothelial growth factor; VMP = bortezomib-melphalan-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone; VTd = bortezomib-thalidomide-dexamethasone.
aHealth Canada–approved indication.
Sources: Darzalex SC product monograph,37 Darzalex product monograph,33,47 Velcade product monograph,48,49 and Revlimid product monograph.50,51
In the sponsor-submitted PERSEUS trial included in this review,15 patients who achieved and sustained MRD negativity for 12 months and had been on maintenance for at least 24 months discontinued treatment with daratumumab. The clinical experts consulted for this review indicated that it would be reasonable to apply the same discontinuation criteria (i.e., MRD negativity that is sustained for 12 months) to clinical practice if daratumumab is funded. However, because most clinical centres in Canada do not have MRD testing available or funded for MM, the clinical experts noted that there would be barriers to implementing MRD-based treatment-discontinuation criteria. Alternative options for treatment-discontinuation criteria include less-sensitive testing methods (e.g., immunofixation electrophoresis or serum FLC assays), an arbitrary time point (e.g., 2 years) without any testing, or achievement of CR. However, the clinical experts noted that none of these can be considered a surrogate for MRD testing, and that there is no evidence in the sponsor-submitted trial to support these alternative criteria. If daratumumab is funded, but MRD testing is not available, 1 clinical expert indicated that they would want to continue treatment, rather than using less-sensitive testing methods to determine treatment discontinuation, as the latter would mean that patients who are not truly MRD-negative may be undertreated. The clinical experts suggested that continuing maintenance treatment with daratumumab until disease progression or toxicity is another option that is consistent with other trials.63,64
We considered the potential impacts of MRD testing to determine eligibility for treatment discontinuation with daratumumab in adult patients with NDMM who are eligible for ASCT if daratumumab becomes funded, including those on health systems, patients (and their families and caregivers), and costs. MRD testing in MM is currently not part of the standard of care, and NGF or NGS testing is not available in most clinical centres in Canada, potentially presenting some barriers if daratumumab is funded. Key considerations and relevant information available from materials submitted by the sponsor,17 input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Table 4.
Table 4: Considerations for MRD Testing for Establishing Treatment Discontinuation With Daratumumab in Patients With NDMM Eligible for Autologous Stem Cell Transplant
Consideration | Criterion | Available information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | The number of patients eligible for MRD testing would be the subset of incident NDMM patients who are transplant-eligible and would be eligible to receive daratumumab in the first-line setting for induction, consolidation, and maintenance if daratumumab becomes funded. Assuming approximately 4,000 patients were diagnosed with MM in 2024,10,65 and about one-half of them were eligible for autologous stem cell transplant, the sponsor estimated that █████ patients would be eligible to receive daratumumab in Canada per year.17 According to the clinical experts, this number may be an overestimate. Each patient would likely need to undergo MRD testing multiple times every year, adding to the total number of tests required. The total number of MRD tests expected to be conducted is dependent on the daratumumab uptake rate as well as the frequency and duration of testing. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | NGS and NGF platforms and capabilities are available in some jurisdictions in Canada.66 However, MRD testing for MM is not publicly funded, nor is MRD testing for MM routinely conducted in all jurisdictions in Canada. According to the sponsor, MRD testing for MM may be carried out at some sites on an as-needed basis. The sponsor estimated that 16 centres across the country (in Alberta, British Columbia, Nova Scotia, Ontario, Quebec, and Saskatchewan,) can conduct MRD testing for MM.17 It is unclear whether NGS, NGF, or both technologies are available in these centres. | |
Testing procedure as part of routine care | At the time of writing this report, MRD testing for MM is not routinely conducted in Canada. | |
Repeat testing requirements | According to the clinical experts, MRD testing should ideally be initiated before treatment initiation with daratumumab and, once a CR is achieved, repeated every 3 months during treatment with daratumumab. The experts noted the possibility of continued MRD testing even after daratumumab is discontinued, to detect relapses. Guidance from a working group in Canada in 2025 suggested MRD testing at 12 months after therapy, but noted that the ideal time points for MRD testing in patients with MM who are transplant-eligible were still being refined.67 The overall time for and frequency at which a patient needs to be monitored is therefore uncertain. The experts also highlighted the practical and patient preference–based limitations to frequent (i.e., more than 1 to 2 times per year) testing even though more frequent testing would provide a more precise real-time assessment of MRD status. | |
Impacts on human and other health care resources by provision of the testing procedure | Based on input from the clinical experts, provision of MRD testing for daratumumab discontinuation in patients with NDMM if daratumumab becomes funded is anticipated to affect human and other health care resources. Because testing is not routinely conducted currently and is only offered at a limited number of institutions on an ad hoc basis, initial resources may be required for most institutions to establish testing (e.g., infrastructure, equipment) or to establish protocol or procedures for out-of-jurisdiction testing. Impacts on human health care resources, such as staffing needs and training of pathologists and other staff, are also possible. Ontario Health (Cancer Care Ontario) has identified several barriers to implementing MRD testing in other hematological cancers. These include financial impacts, staffing, access to validated testing, and awareness in the community.66 These might be applicable to MRD testing in NDMM as well. Cancer Care Ontario has also identified development of a structured reporting format and continued clinical evaluation for addressing some of the health care and patient-oriented considerations.66 | |
Patient related | Accessibility of the testing procedure in jurisdictions across Canada | Currently, MRD testing for MM is not publicly funded in Canada. According to the sponsor, some institutions may offer and fund ad hoc MRD testing.17 Even so, the clinical experts pointed out that the testing is not accessible to a large number of patients across the country. If daratumumab is funded, patients are likely to face barriers related to access to testing. |
Expected turnaround times for the testing procedure | According to the sponsor, for NGS-based MRD testing using Adaptive Biotechnologies’ clonoSEQ offered by commercial laboratories outside of Canada, the reported US laboratory standard of the turnaround time from sample receipt and reconciliation to result delivery is 7 days for fresh specimens and 14 days for stored specimens.68 NGF-based MRD testing has a shorter turnaround time, with results made available in a “few days.”55 It is uncertain if these turnaround times would apply to clinical settings in Canada. The clinical experts noted that a 2-week to 4-week turnaround time would be acceptable for clinical decision-making. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | NGS-based and NGF-based MRD testing methods both require collecting bone marrow samples, using a relatively invasive and time-consuming procedure known as aspiration. Because multiple tests over and after the duration of treatment with daratumumab would be required, with new samples taken each time, patients would be required to visit the testing centre multiple times a year for sample collection. Patients may experience a psychological burden as they await their MRD testing results. In a small survey of prospective patients, those with an MRD-positive result were disappointed and concerned about their prognosis, while those who were MRD-negative were more confident and optimistic.69 If daratumumab is funded but MRD testing is not publicly funded or accessible, or if the institution requires patients to pay out of pocket, patients may experience a financial burden. | |
Clinical | Clinical use and validity of the testing procedure | Evidencea demonstrating the diagnostic accuracy and clinical use of NGS-based and NGF-based MRD testing methods in MM is available.52,55 Furthermore, there is evidencea to suggest the use of MRD negativity, determined by these testing methods, as a tool to guide discontinuation of maintenance therapy in patients with MM.59,70 Multiple studies have shown improved outcomes in patients who have achieved MRD negativity as assessed by 1 of these methods.9,55 In a large systematic review and meta-analysis encompassing 29 studies that assessed the MRD status by NGF and 9 studies by NGS, patients who tested MRD-negative had significantly improved PFS and OS, compared with those who were MRD-positive. These results were independent from the method of MRD evaluation.71 |
Risks of harm associated with the testing procedure | To test for the MRD status using NGS or NGF, samples are collected each time through bone marrow aspiration. Based on how often MRD testing is required to determine the potential for treatment discontinuation, patients might need to undergo bone marrow aspiration multiple times during and after treatment with daratumumab. While rare, patients may experience procedure-related adverse effects, such as pain, excessive bleeding, infections, or neurologic damage due to nerve injury.72 | |
Cost | Projected cost of the testing procedure | Based on publicly available information, the cost of NGS using Adaptive Biotechnologies’ clonoSEQ is approximately CA$2,500.73 The cost of NGF ranges from US$300 to US$400.62 The clinical experts identified the cost of the tests as a main barrier to implementation. |
CR = complete response; MM = multiple myeloma; MRD = minimal residual disease; NDMM = newly diagnosed multiple myeloma; NGF = next-generation flow cytometry; NGS = next-generation sequencing; OS = overall survival; PFS = progression-free survival.
aCDA-AMC has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by Myeloma Canada.
CDA-AMC received 1 patient group input from Myeloma Canada, which conducted a patient and caregiver survey regarding DVRd for the treatment of patients with NDMM receiving an ASCT in Canada. The survey was available from September 26 to October 10, 2024, and was shared via email and social media by Myeloma Canada and the Leukemia & Lymphoma Society of Canada. Survey eligibility was determined by asking patients (and caregivers) to self-report that they (or the person they care for) were eligible for ASCT at the time of diagnosis and received an ASCT or were waiting to receive a transplant as part of their first line of therapy. Upon verifying their eligibility for, or experience with, DVRd, respondents were divided into 3 subsets, and correspondingly given different questions. These subsets were as follows: patients who would be eligible for treatment with DVRd and their caregivers (i.e., newly diagnosed and have not yet received treatment), patients who received first-line treatment with ASCT and their caregivers, and patients who had experience with DVRd and their caregivers. The survey received 84 responses. Of these, 18 were incomplete (i.e., a respondent did not finish answering survey questions), and 27 ineligible responses were removed from the dataset, leaving 39 complete and eligible responses in the survey.
When asked to rate the importance of controlling various MM-related symptoms, respondents most frequently rated bone issues (i.e., fractures, breaks, and bone pain) as “extremely important to control,” followed by kidney problems, mobility, pain, and infections. Respondents also most frequently noted that MM-related symptoms had an extreme impact on their ability to work, travel, and conduct volunteer activities. Of the 34 patients with MM who responded to the survey, 17 required the support of a caregiver to help manage MM-related and treatment-related symptoms. Of these 17 patients, 3 indicated that they were unable to access the support they needed.
The results of the survey highlighted several financial implications related to treatment for MM. Surveyed patients and caregivers most frequently noted the loss of income or pension funds due to absence from work, disability, or early retirement as a significant financial implication related to MM treatment. Other common significant financial implications were costs associated with parking, drugs, and travel. The results of the survey also noted negative psychosocial impacts associated with treatment for MM. Of the various psychological and social difficulties related to MM, patients and caregivers most frequently rated the interruption of life goals and accomplishments (e.g., career and retirement) as having an extreme impact on their QoL. Other psychological and social difficulties related to MM that were frequently noted to have significant to extreme impacts on QoL were anxiety, difficulty sleeping, and loss of sexual desire.
Patients and caregivers completing the survey were asked to identify the factors that they consider to be the most important to MM treatment. The results of the survey found that the key factors were the effectiveness of treatment and achieving a long remission, maintenance of QoL and mental health, management of side effects, portability of treatment to reduce the number of visits to treatment centres and mitigate impact on day-to-day activities, and the cost and accessibility of treatment.
Among the subset of respondents who received first-line treatment with ASCT combined with a drug regimen other than DVRd (n = 11), drug regimens received included: CyBorD, VRd, ixazomib plus lenalidomide-dexamethasone, lenalidomide monotherapy, lenalidomide-dexamethasone, and a sequence of CyBorD, VRd, and thalidomide. When asked questions regarding the safety profile of DVRd, respondents in this subset most frequently noted that they felt that it was “slightly worrisome” compared to the safety profiles of other treatment options available to them or the person that they cared for. When asked about their perceptions of the advantages and disadvantages of DVRd compared to past treatments received, respondents noted that they believed that DVRd would result in increased control of MM and its symptoms and improve their QoL. However, they also expected that treatment would increase the frequency of trips to treatment centres. When asked similar questions about the safety profile of DVRd, respondents who were deemed eligible for treatment with DVRd (n = 6) most frequently noted that it was “somewhat worrisome” compared to other available treatments. Respondents also noted treatment side effects, frequency of trips to receive treatment, and tolerability of the mode of administration as factors that would affect their QoL. Regarding the most common side effects for DVRd, respondents from both subsets most frequently rated infections as “not at all bearable” or “slightly bearable,” followed by nausea, fever, and diarrhea. Nonetheless, based on their knowledge at the time of the survey, respondents from both subsets most frequently indicated that they would have been interested in receiving DVRd as first-line treatment for themselves or the person they care for.
Twenty-two respondents had prior treatment experience with DVRd. When asked to indicate and rate their experienced side effects while receiving DVRd, respondents most frequently rated diarrhea as “not at all bearable” or “slightly bearable,” followed by infections and neuropathy. One patient reported stopping treatment with DVRd due to rapid deterioration of vision. Respondents also noted that the supportive care they received was effective to some degree in managing side effects related to DVRd. Respondents who had prior treatment experience with DVRd most frequently reported that treatment side effects significantly affected their QoL, whereas the frequency of trips to receive treatment and tolerability of the mode of administration only somewhat affected their QoL. Most respondents indicated that treatment with DVRd improved their overall QoL, and the side effects of DVRd were mostly manageable. Most respondents also noted that treatment with DVRd was effective in controlling MM and met their expectations in treating MM. Respondents had access to DVRd treatment through various avenues, which included compassionate access, clinical trials, insurance coverage (e.g., private or public provincial), through a doctor, and self-funding. However, few respondents acknowledged that daratumumab was costly and emphasized the need for financial coverage to access treatment.
Myeloma Canada noted that the number of respondents to the survey who had experience with DVRd was greater than that of previous surveys for other treatments, which suggests that DVRd is already widely used in Canada. The results of the survey suggest that patients view DVRd as an optimal treatment choice but also acknowledge that the regimen can be expensive due to the cost of daratumumab. Patients who do not have access to private insurance or those unable to self-fund treatment may therefore face barriers to accessing DVRd. Myeloma Canada highlighted reimbursement of DVRd as an equity issue and emphasized the importance of ensuring that patients in Canada have equal access to this treatment, regardless of socioeconomic status. Myeloma Canada emphasized the importance of proactively informing patients about potential vision problems related to DVRd, given that past surveys found that this side effect was of significant concern to patients. Proactively informing patients about potential vision problems and other side effects related to DVRd would allow them to weigh their options and make an informed choice regarding treatment for MM.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of MM.
Unmet Needs
The clinical experts consulted by CDA-AMC agreed that there is a substantial number of patients with MM who have suboptimal (incomplete or transient) responses to current first-line treatment options. Both experts agreed that these patients will need new treatments that result in deepened responses, higher levels of MRD negativity, and longer remissions. One expert cited concerns of eventual drug resistance and emergence of drug-resistant variants of disease related to continuous therapy. One expert also emphasized the need for patient education regarding incoming treatments for MM.
Place in Therapy
Daratumumab targets the underlying disease process for MM and, according to the clinical experts consulted by CDA-AMC, daratumumab would be administered in combination with VRd as first-line therapy for MM and would be given wherever VRd is currently used to treat patients with NDMM. One expert noted that DVRd would be given as an induction therapy before ASCT and as consolidation and maintenance therapy after ASCT. The experts did not recommend any alternative first-line regimens for patients with MM before initiating treatment with DVRd.
Patient Population
According to the clinical experts consulted by CDA-AMC, DVRd should be administered as a first-line therapy to patients with NDMM who are eligible for ASCT. One expert noted that age and health status were the most important factors in determining eligibility for ASCT, and that less than 5% of patients aged 70 years or older would be eligible for transplant. It was also noted that eligibility for ASCT rarely changes after receipt of induction therapy. The clinical experts indicated that patients with MM would be identified by physicians with experience in treating MM. No issues related to the diagnosis or misdiagnosis of MM were identified. The clinical experts indicated that all eligible patients would benefit from treatment with DVRd. They did not identify a subgroup of patients who would receive more or less treatment benefit from DVRd compared to other subgroups. The clinical experts also agreed that it was not possible to identify which patients would receive more or less treatment benefit from DVRd.
The PERSEUS trial listed several exclusion criteria, including (but not limited to) the presence of specified comorbidities (e.g., asthma and cardiac conditions), peripheral neuropathy, prior or concurrent non–MM-related malignancy, meningeal involvement, and recent treatment with plasmapheresis or radiation therapy. It was agreed that the PERSEUS trial enrolled a more restrictive patient population compared to what is observed in clinical practice, given that patients would not be excluded from receiving treatment in clinical practice. However, 1 clinical expert indicated that patients excluded from the PERSEUS trial were less likely overall to be eligible for ASCT or receive treatment with DVRd in clinical practice.
Assessing the Response to Treatment
The clinical experts indicated that the assessment of response to treatment for MM consists of regular monitoring of monoclonal M protein via serum protein electrophoresis, serum FLC assays, and standard disease parameters for MM. The experts agreed that assessment of response to treatment is performed monthly in clinical practice, although it was noted that this may be reduced to every 2 to 3 months for patients exhibiting stable disease response. They also agreed that bone marrow biopsy would be used to assess depth of response and would be performed at diagnosis, before ASCT and post-ASCT.
One clinical expert indicated that the definition of a clinically meaningful response to treatment depended on when it was assessed relative to the receipt of ASCT. For instance, a response of PR or greater achieved before ASCT was considered to be clinically meaningful but a patient would be expected to achieve a greater than PR after receipt of ASCT. The other expert indicated that clinically meaningful responses to treatment also included reduction in MM-related symptoms (e.g., pain), improvements in key hematological outcomes (e.g., reduction or normalization of light chains and paraproteins, improvements in peripheral blood chemistry) and stabilization for improvement in bone imaging.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC agreed that treatment with daratumumab would be discontinued in the event of disease progression or toxicity, although they noted that adverse events related to daratumumab were rare in clinical practice. These criteria were largely aligned with the discontinuation criteria for daratumumab in the PERSEUS trial. Patients who achieved a CR or better in the PERSEUS trial were only able to discontinue treatment with daratumumab if MRD negativity was sustained for a minimum of 12 months and after receipt of a minimum of 24 months of maintenance therapy. Although 1 clinical expert suggested that sustained MRD negativity can be a criterion for discontinuation of daratumumab, the expert noted that this criterion is not formally used in clinical practice due to a lack of MRD testing across jurisdictions in Canada.
Prescribing Considerations
One clinical expert indicated that daratumumab can be prescribed and administered by any physician with experience treating MM in a variety of treatment settings, including rural and community settings. However, the other expert noted that SC daratumumab should be administered in established chemotherapy units by specialized hematologists or oncologists, adding that some infusion centres also have the expertise to administer this treatment. The second expert also noted that chemotherapy units should be able to manage injection-site reactions, although these were described as rare. One of the experts emphasized that access to qualifying specialists may vary across jurisdictions in Canada. Consequently, travel may be required of some patients, particularly those living in remote areas, to receive treatment.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Clinician group input for this review was received from 2 clinician groups: the CMRG and OH-CCO’s DAC. A total of 33 clinicians (25 from the CMRG and 7 from the DAC) provided input for this submission.
Both the CMRG and DAC agreed that goals for the treatment of MM include the achievement of an antimyeloma response, long-term control of MM-related disease and symptoms, and prolonging of survival. The CMRG also emphasized the importance of minimizing TEAEs and the improvement of QoL among patients with MM. Both clinician groups noted an unmet need related to current first-line treatment options for MM. Similar to the clinical experts consulted by CDA-AMC, the OH-CCO DAC agreed that some patients with MM do not respond adequately to first-line treatment. The CMRG highlighted the importance of first-line treatment, given that MM remains incurable. The CMRG added that the majority of patients with MM experience their longest period of disease control during first-line treatment, and that much of the improvements observed for long-term survival is dependent on maximizing disease control within this line.
The DAC stated that DVRd could become the new standard of care for TE NDMM. The CMRG noted that the addition of daratumumab to maintenance therapy may result in increased visits to cancer centres to receive injections. The CMRG also emphasized the importance of increasing the capacity for MRD testing in Canada to minimize long-term toxicity and financial and patient QoL burdens related to daratumumab. The group considered MRD testing a valuable prognostic tool, as it can be used to identify patients who are expected to have very long-term disease control, which will likely result in decreased MM-related morbidity and long-term health care utilization. The CMRG acknowledged that implementation of widespread MRD testing would result in costs incurred to provincial jurisdictions. However, the group noted that implementation will result in long-term cost savings for the health system, largely due to the de-escalation of daratumumab in eligible patients, as confirmed by MRD testing.
Similar to the clinical experts consulted by CDA-AMC, both clinician groups agreed that patients with NDMM who are eligible for a transplant would be best suited for treatment with DVRd. The OH-CCO DAC indicated that daratumumab can be delivered in any treatment setting with experience administering the drug; this includes community oncology clinics and medical facilities with expertise in administering cellular therapies for hematologic malignancies. The CMRG noted that daratumumab is appropriate for administration in outpatient settings, although consideration for funding the drug in inpatient settings may be required.
Both clinician groups agreed that standard myeloma and organ-response criteria are used to assess responses to treatment in clinical practice. The CMRG elaborated that the assessment of response is based on tests for the monoclonal M protein in the serum and urine, bone marrow biopsies, and imaging studies. In addition to these tests, MRD testing was noted as an emerging parameter of response assessment in MM. Similar to the clinical experts consulted by CDA-AMC, the CMRG indicated that clinically meaningful responses correlate with a PR or better according to the IMWG Consensus Criteria, which would include improvement in MM-related symptoms and improvements in energy and ability to perform activities of daily living. The CMRG also indicated that response, in the context of MM, is assessed every 1 to 3 months, depending on clinical stability and the regimen used for treatment. Similar to the clinical experts consulted by CDA-AMC, the OH-CCO DAC and the CMRG agreed that treatment with daratumumab should be discontinued upon the occurrence of disease progression, unacceptable toxicity, and/or intolerance.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The standard arm in the phase III PERSEUS trial used VRd induction (4 cycles) followed by ASCT and VRd consolidation (2 cycles), then lenalidomide maintenance therapy alone. In Canada, the standard of care is usually VRd or CyBorD induction (up to 4 cycles) followed by ASCT and 1 of either lenalidomide (more common) or bortezomib (less common) maintenance. VRd consolidation for 2 cycles after transplant is not commonly used. The choice of maintenance therapy is sometimes determined based on cytogenetics (e.g., some hematologists favour bortezomib maintenance in myeloma with del17p). Rarely, a second tandem transplant may be used as consolidation. | Comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Can daratumumab (or isatuximab) and/or lenalidomide be given to patients who relapse after maintenance therapy is discontinued? If so, what would be the appropriate progression-free interval for re-treatment? Should re-treatment with daratumumab (or use of isatuximab) be dependent on MRD status at time of discontinuation? Could daratumumab be restarted if the myeloma becomes MRD-positive after previous MRD negativity, but no other signs of “classic” disease progression (definitions follow)? Note: Most jurisdictions follow clinical trial definitions for determining whether refractory to treatment for drug funding decisions (i.e., disease progression within 60 days of stopping treatment or progression on any dose, including progression while on maintenance therapy). These definitions include:
| It would be advisable to restart daratumumab or isatuximab for patients who stopped maintenance therapy due to reasons other than relapse (e.g., toxicity). The clinical experts indicated that patients who stopped treatment due to relapse or disease progression or have received the drug but stopped due to other reasons recently (< 3 months) would not be eligible for re-treatment with daratumumab. For re-treatment with lenalidomide, 1 clinical expert mentioned having had success re-treating with lenalidomide in patients who were previously treated with lenalidomide but stopped the treatment for reasons other than relapse (e.g., side effects). Ninety days would be the appropriate progression-free interval for re-treatment for patients who stopped daratumumab for reasons other than relapse. However, 1 clinical expert indicated that treatment could be restarted almost at any time if patients had stopped daratumumab because of toxicity. Re-treatment with daratumumab (or isatuximab) should be dependent on whether patients’ disease respond to the drug. According to 1 clinical expert, patients who stop treatment because they are MRD-negative would be eligible for re-treatment with daratumumab or isatuximab if the treatment needs to be restarted for relapse; patients who relapse while receiving daratumumab would not be eligible for daratumumab or isatuximab. One clinical expert stated that patients who are not MRD-negative would presumably continue daratumumab or isatuximab past the 2-year mark. However, if the funding decision is such that all patients stop daratumumab maintenance at 2 years, then all patients would be potential candidates for re-treatment at relapse regardless of MRD status. The clinical experts agreed that daratumumab can be restarted if the myeloma becomes MRD-positive after previous MRD negativity, with no other signs of “classic” disease progression.” One clinical expert commented that patients with very early MRD changes without signs of “classic” disease progression may be more responsive to re-treatment. |
Would patients with high-risk cytogenetics such as del17p, t(4;14), t(14;16) benefit equally? | Patients with high-risk cytogenetics, such as del17p, t(4;14), and t(14;16), would benefit equally. |
Should this regimen be available for patients with amyloidosis who would be considered eligible for ASCT? | This regimen should be available for patients with amyloidosis who would be considered eligible for ASCT. |
Considerations for continuation or renewal of therapy | |
NA | NA |
Considerations for discontinuation of therapy | |
In the PERSEUS phase III trial, daratumumab maintenance was discontinued after a minimum of 24 months if the patient was MRD-negative for at least 12 months. MRD testing in myeloma is not standard of care in Canada. Should the same discontinuation criteria apply in standard practice? What criteria should be used to assess response or to discontinue daratumumab if MRD testing is not available? If MRD testing is available and daratumumab is discontinued due to MRD negativity, can it be restarted if myeloma becomes MRD-positive without other classical signs of disease progression (i.e., biochemical, clinical, or radiological)? And if so, would daratumumab need to be reloaded (weekly for 8 weeks, then biweekly for 4 months, then monthly), or would it be restarted at day 1 of an every-28-days cycle? This may have an impact on the budget impact analysis. | The availability of MRD testing is a key consideration when deciding to discontinue maintenance treatment with daratumumab. One clinical expert indicated that it is reasonable to apply the same daratumumab discontinuation criteria used in the PERSEUS trial to clinical practice only if centres have access to MRD testing that is as sensitive as the test used in the trial, while the other suggested that the use of MRD testing as daratumumab discontinuation criteria may not be feasible, given that the test is not available to most centres. If daratumumab is funded, 1 clinical expert stated that, in the absence of an appropriate assay for MRD testing, patients are anticipated to be kept on maintenance treatment with daratumumab until disease progression or unacceptable toxicity. Both clinical experts agreed that treatment with daratumumab should be discontinued in the event of disease progression or toxicity. The clinical experts agreed that, if MRD testing is available and daratumumab is discontinued due to MRD negativity, treatment can be restarted if patients’ multiple myeloma becomes MRD-positive without other classical signs of disease progression (i.e., biochemical, clinical, or radiological). One clinical expert noted that re-treatment with daratumumab would need to meet the therapeutic level in serum with weekly administrations before spacing out the treatments. |
Lenalidomide was continued in the study until disease progression irrespective of MRD status. Should lenalidomide continue until disease progression in standard practice? | Lenalidomide should be continued until disease progression in standard practice. |
Considerations for prescribing of therapy | |
NA | NA |
Generalizability | |
Some patients with newly diagnosed plasma cell leukemia or amyloidosis are treated similarly to those with myeloma and receive an autologous stem cell transplant. Should this regimen be given to patients with newly diagnosed plasma cell leukemia or amyloidosis planned for autologous stem cell transplant? | The clinical experts agreed that patients with newly diagnosed plasma cell leukemia or amyloidosis planned for autologous stem cell transplant would be eligible for this regimen. |
Should daratumumab be added to induction or maintenance therapy for patients who are on alternate induction or maintenance regimens? If so, what is the time frame to consider adding daratumumab to either induction or maintenance treatment? | One clinical expert indicated that, for patients who have started an alternative induction therapy regimen, daratumumab could be added any time once approved. However, for patients who are on maintenance, the time frame to consider adding daratumumab is rather arbitrary; for example, within the first 6 months or even a year. Results from the AURIGA trial demonstrated that, when compared to lenalidomide alone, the addition of daratumumab to lenalidomide as maintenance therapy in patients who had MRD-positive disease at maintenance baseline resulted in benefits in the following study outcomes: MRD conversion rate from baseline to 12 months, progression-free survival, overall survival, overall MRD (10-5) negativity conversion rate from baseline, sustained MRD negativity rates at 6 and 12 months, and the overall rate of a CR or better. However, no definitive conclusions could be drawn from the MAICs conducted in patients with transplant-eligible NDMM (regardless of MRD status at maintenance baseline) with respect to the relative effects of daratumumab plus lenalidomide versus lenalidomide alone due to important methodological limitations of the analyses. |
For patients who started VRd induction at time of implementation, would daratumumab be recommended to be added to induction, and if so, is there a maximum number of cycles to consider before not adding daratumumab? | Daratumumab should be added for patients who started VRd induction at time of implementation. The number of cycles would need to be individualized based on patients’ response to treatment and timing of stem cell collection. It would be best for patients to receive at least 2 cycles of DVRd pretransplant, even if that means adding cycles before stem cell collection or transplant. |
Funding algorithm | |
Request an initiation of a rapid provisional funding algorithm. | Comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
MRD testing in multiple myeloma is not standard of care in Canada and is not available in all jurisdictions. | Comment from the drug programs to inform pERC deliberations. |
Red blood cell genotyping is required for daratumumab and is available in jurisdictions. | Comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
PAG is concerned about the potential large budget impact of daratumumab if recommended for newly diagnosed transplant-eligible myeloma. | Comment from the drug programs to inform pERC deliberations. |
Confidential pCPA pricing exists for daratumumab for indications in the transplant-ineligible myeloma population. Generics are available for lenalidomide and bortezomib. | Comment from the drug programs to inform pERC deliberations. |
ASCT = autologous stem cell transplant; CyBorD = cyclophosphamide-bortezomib-dexamethasone; CR = complete response; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; MRD = minimal residual disease; NA = not applicable; NDMM = newly diagnosed multiple myeloma; PAG = Provincial Advisory Group; pCPA = pan-Canadian Pharmaceutical Alliance; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; VRd = bortezomib-lenalidomide-dexamethasone.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of daratumumab in the form of an 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for SC injection in the treatment of adult patients with NDMM who are eligible for ASCT. The focus will be placed on comparing DVRd to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of daratumumab is presented in 3 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The assessment of the certainty of the evidence in this first section using the GRADE approach follows a critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. The third section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
One phase III, open-label, active-controlled RCT identified in systematic review
Five ITCs using MAICs
One additional study addressing gaps in evidence.
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | PERSEUS trial |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, active-controlled randomized controlled trial |
Locations | 115 centres in 14 countries (Australia, Belgium, Czechia, Denmark, France, Germany, Greece, Italy, Netherlands, Norway, Poland, Spain, Switzerland, and Türkiye) |
Patient enrolment dates | Start date: January 19, 2019 End date: August 1, 2023 |
Randomized (N) | A total of 709 participants were randomized: 355 to the DVRd group and 354 to the VRd group |
Inclusion criteria | Participants who were aged 18 to 70 years with monoclonal plasma cells levels in the bone marrow of ≥ 10% or the presence of a biopsy-proven plasmacytoma and documented MM satisfying at least 1 of the CRAB criteria or biomarkers of malignancy criteria:
|
Exclusion criteria |
|
Drugs | |
Intervention | Participants assigned to the DVRd treatment group received daratumumab SC in the form of 1,800 mg once every week on days 1, 8, 15, and 22 for cycles 1 to 2, then every 2 weeks on days 1 and 15 for cycles 3 to 6. For maintenance cycles 7 and onward, participants received daratumumab once every 4 weeks on day 1 until documented disease progression or unacceptable toxicity. Participants in the DVRd treatment group received bortezomib, lenalidomide, and dexamethasone:
|
Comparator(s) | Participants in the VRd treatment group received bortezomib, lenalidomide, and dexamethasone in a manner similar to the treatment received by the DVRd group |
Study duration | |
Screening phase | Up to 28 days before randomization |
Treatment phase | From cycle 1 day 1 to discontinuation of all study treatment, included 4 cycles of induction, followed by ASCT, then 2 cycles of consolidation, followed by maintenance therapy until disease progression or unacceptable toxicity. |
Follow-up phase | Participants entered the follow-up phase after documented disease progression or unacceptable toxicity leading to all study treatment discontinuation or if a response of PR or better had not been achieved by cycle 7 day 1. |
Outcomes | |
Primary end point | PFS measured from the time from the date of randomization to the date of disease progression (assessed by IMWG 2011 criteria)22 or death |
Secondary and exploratory end points | Secondary
Exploratory
|
Publication status | |
Publications | Sonneveld et al. (2024)41 ClinicalTrials.gov identifier: NCT03710603 |
ASCT = autologous stem cell transplant; CR = complete response; CRAB = calcium, renal, anemia, bone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-MY20 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Multiple Myeloma; FEV1 = forced expiratory volume in 1 second; FLC = free light chain; IMWG = International Myeloma Working Group; MM = multiple myeloma; MRD = minimal residual disease; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival on the next line of therapy; PO = orally; PR = partial response; PRO = patient-reported outcome; rHuPH20 = recombinant human hyaluronidase PH20; SC = subcutaneous; sCR = stringent complete response; VGPR = very good partial response; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Clinical Study Report for PERSEUS (2024)15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
One pivotal trial (PERSEUS) was included in the systematic review. The PERSEUS trial is a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of DVRd compared to VRd in patients with TE NDMM. The primary objective of the PERSEUS trial is to evaluate the efficacy of DVRd compared to VRd in patients with TE NDMM in prolonging PFS. Secondary objectives are to evaluate the efficacy of DVRd compared to VRd in patients with NDMM who were eligible for ASCT with respect to response rates according to the IMWG criteria, MRD negativity rate, OS, time to response, DOR, PFS on the next line of therapy, and safety. The primary end point was PFS according to according to the IMWG 2011 criteria. Secondary end points included the rate of CR or better (key secondary), MRD negativity rate (key secondary), OS (key secondary), overall response rate, rate of VGPR or better, stringent complete response (sCR) rate, time to response, and DOR. HRQoL was measured using the EORTC QLC-C30, EORTC QLQ-MY20, and the EQ-5D-5L. The PERSEUS trial was funded by the European Myeloma Network in collaboration with Janssen Research & Development. The PERSEUS trial enrolled adults who had NDMM and were eligible for ASCT.
Patient enrolment for the PERSEUS trial began on December 14, 2018. The first patient was screened on January 3, 2019. Two interim analyses and 1 final analysis for PFS were planned, and were to be performed at approximately 143 PFS events (50% information fraction), 185 PFS events (65% information fraction), and 285 PFS events, respectively. Given the required accumulation of PFS events for the second interim and final analyses had not yet occurred at the time of this report, the review team focused on the results from the first interim analysis with the data cut-off of August 1, 2023. A total of 709 patients were randomized at a 1:1 ratio to receive either DVRd as induction and consolidation therapy followed by maintenance therapy with daratumumab and lenalidomide (n = 355) or VRd as induction and consolidation therapy followed by maintenance therapy with lenalidomide (n = 354). Randomization was conducted using an interactive web response system and stratified by the ISS at screening (I versus II versus III) and cytogenetics (standard risk versus high risk as defined by the presence of del17p, t[4;14] or t[14;16] chromosomal anomalies). Eligible patients were recruited at 115 centres across Europe (13 countries: Belgium, Czechia, Denmark, France, Germany, Greece, Italy, Netherlands, Norway, Poland, Spain, Switzerland, Türkiye) and Australia. The study had no sites in Canada.
The PERSEUS trial consists of 3 phases: screening, treatment, and follow-up (Figure 2). In the screening phase, patients provided written consent for study participation and were screened for eligibility within 28 days before randomization. The treatment phase consisted of 28-day cycles, including 4 cycles of induction, followed by ASCT, then 2 cycles of consolidation, followed by maintenance therapy until disease progression or unacceptable toxicity. Patients in the DVRd group received DVRd induction and consolidation therapy and daratumumab and lenalidomide maintenance therapy; patients in the VRd group received VRd induction and consolidation therapy and lenalidomide maintenance therapy alone. After a minimum of 24 months of maintenance therapy, patients in the DVRd group who had a CR or better discontinued therapy with daratumumab when sustained MRD negativity (at the threshold of 10-5) was established for a minimum of 12 months. These patients continued lenalidomide maintenance therapy until disease progression or unacceptable toxicity. After discontinuing daratumumab therapy due to sustained MRD negativity, patients restarted maintenance therapy with daratumumab if there was a recurrence of MRD (at ≥ 10-4) confirmed by loss of CR without IMWG-defined disease progression, which was measured by relevant central laboratory assessment (serum or urine M protein or ≥ 5% plasma cells in bone marrow). After reinitiating daratumumab, patients continued maintenance with daratumumab and lenalidomide therapy until disease progression or unacceptable toxicity. Patients entered the follow-up phase following documented disease progression (analyzed by computerized algorithm and concordance confirmed by investigator assessment) or unacceptable toxicity leading to discontinuation of all study treatment or if a PR or better had not been achieved by cycle 7 day 1. Patients were monitored for long-term safety, PFS on the next line of therapy, new malignancies, subsequent therapy, and HRQoL during the follow-up phase.
Figure 2: PERSEUS Study Design Diagram
CR = complete Iresponse; Dara = daratumumab; D-VRd = daratumumab-bortezomib-lenalidomide-dexamethasone; MRD = minimal residual disease; PD = progressive disease; PFS2 = progression-free survival on the next line of therapy; PRO = patient-reported outcome; R = lenalidomide; VRd = bortezomib-lenalidomide-dexamethasone.
Note: Arm A refers to the VRd group and arm B refers to the D-VRd group.
Source: Clinical Study Report for PERSEUS (2024).15
Populations
Inclusion and Exclusion Criteria
Patients were eligible for inclusion in the trial if they were aged 18 to 70 years and had NDMM with monoclonal plasma cells in the bone marrow (≥ 10%) or if the presence of a biopsy-proven plasmacytoma indicated they were eligible for HDT and ASCT, and had an ECOG PS score of 0 to 2 (on a scale from 0 to 5, with higher scores indicating greater disability). Patients were excluded if they had received prior systemic therapy or a stem cell transplant for any plasma cell dyscrasia, grade 2 or higher peripheral neuropathy or neuropathic pain, prior or concurrent invasive malignancy within 5 years of randomization, radiation therapy for treatment of plasmacytoma within 14 days of randomization, plasmapheresis within 28 days of randomization, clinical signs of meningeal involvement of MM, pulmonary disease, or moderate or severe persistent asthma within the past 2 years, or uncontrolled asthma.
Interventions
Patients in the DVRd group received DVRd as induction and consolidation therapy followed by maintenance therapy of daratumumab and lenalidomide. Treatment consisted of 28-day (4 week) cycles. All injections were administered in outpatient settings.
Daratumumab SC was administered at 1,800 mg once every week on days 1, 8, 15, and 22 for cycles 1 to 2, then every 2 weeks on days 1 and 15 for cycles 3 to 6. For maintenance (cycles 7 and onward), patients received daratumumab once every 4 weeks on day 1 until documented disease progression or unacceptable toxicity. Patients with a CR or better were allowed to discontinue daratumumab maintenance if they demonstrated sustained MRD negativity for 12 months and had received a minimum of 24 months of maintenance therapy.
Bortezomib was administered subcutaneously at 1.3 mg/m2 twice per week (days 1, 4, 8, and 11) for cycles 1 to 6 (i.e., 4 induction cycles [cycles 1 to 4] and 2 consolidation cycles [cycles 5 and 6]). Patients did not receive bortezomib after cycle 6.
Lenalidomide was administered orally at 25 mg on days 1 to 21 in cycles 1 to 6 (i.e., 4 induction cycles and 2 consolidation cycles). Patients started maintenance therapy (cycle 7 and onward) by receiving lenalidomide orally at 10 mg daily on days 1 to 28 (continuously) of each cycle until disease progression or unacceptable toxicity. After 3 cycles of maintenance therapy, if well tolerated, the lenalidomide dose could be increased to 15 mg daily, at the discretion of the investigator.
Dexamethasone was administered at 40 mg daily on days 1 to 4 and days 9 to 12 of each cycle during induction and consolidation (cycles 1 to 6). Patients did not receive dexamethasone after cycle 6.
Patients who had a post-ASCT recovery period that required more than 12 weeks off daratumumab skipped consolidation therapy and received a dose of daratumumab every 2 weeks for 4 doses (cycles 7 to 8 of maintenance therapy) then continued every 4 weeks (for cycle 9 and onward).
Patients in the VRd group received VRd as induction and consolidation therapy followed by maintenance therapy with lenalidomide. Patients received VRd in the same manner as the DVRd group.
Throughout the study, any concomitant medications or treatments deemed necessary to provide adequate supportive care (including treatment and/or management of adverse events) were permitted. Concomitant administration of strong CYP3A4 inducers was prohibited with the use of bortezomib. Concomitant use of other drugs that target CD38, medications used for other indications that have antimyeloma properties (e.g., interferon and clarithromycin), approved or investigational treatments for MM, and investigational agents were prohibited during the study.
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered the most relevant to expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the PERSEUS Trial
Outcome measure | Time point | PERSEUS trial |
|---|---|---|
Efficacy outcomes | ||
PFS | At 48 months | Primarya |
VGPR or better rate | NR | Secondary |
MRD negativity rate | NR | Secondarya |
OS | At 48 months | Secondarya |
DOR (CR or better) | NR | Secondary |
Health-related quality of Life | ||
Change from baseline in EQ-5D-5L utility score | At maintenance cycle 34 day 1 | Secondary |
Safety outcomes | ||
Cytopenia | NR | Secondary |
Systemic administration-related reactions | ||
Infections and infestations | ||
CR = complete response; DOR = duration of response; MRD = minimal residual disease; NR = not reported; OS = overall survival; PFS = progression-free survival; VGPR = very good partial response.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: Clinical Study Report for PERSEUS (2024)15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Efficacy Outcomes
Progression-Free Survival
The primary end point in the PERSEUS trial was PFS, which was defined as the time from the date of randomization to the date of first documented disease progression (based on a validated computer algorithm) or death due to any cause, whichever occurred first. Patients with no postbaseline disease assessment, those who experienced disease progression or death immediately after 2 or more missed consecutive disease evaluations, those who withdrew consent for study participation, and those who were lost to follow-up or started subsequent antimyeloma therapy were censored. Patients who died after the end of study (for example, those who died after consent withdrawal, with the death information obtained from public information) were censored at the date of the last disease assessment before the end of study for PFS analysis. Intercurrent events included subsequent antimyeloma therapy, 2 or more missing consecutive disease evaluations immediately after disease progression or death, and death due to COVID-19. PFS was considered important to patients and clinicians, according to the clinical experts and patient and clinician groups. In addition, PFS was used to inform the pharmacoeconomic model submitted to the drug agency.
Patients were considered to have disease progression if they met the IMWG response criteria22 of an increase of 25% from lowest confirmed response value in 1 or more of the following criteria:
The absolute increase in serum M protein must be at least 0.5 g/dL.
The absolute increase in urine M protein must be at least 200 mg per 24 hours.
In patients without measurable serum and urine M protein levels, the difference between involved and uninvolved FLC levels (absolute increase) must be greater than 10 mg/dL.
In patients without measurable serum and urine M protein levels and without measurable involved FLC levels, bone marrow plasma cell percentage irrespective of baseline status (absolute increase) must be at least 10%.
Definite development of new bone lesions or soft tissue plasmacytomas or definite increase in the size of existing bone lesions or soft tissue plasmacytomas must be evident.
Development of hypercalcemia (corrected serum calcium > 11.5 mg/dL or 2.65 mmol/L) that can be attributed solely to the plasma cell proliferative disorder must be evident.
Rate of Complete Response or Better
The rate of CR or better, a key secondary end point in the PERSEUS trial, was defined as the proportion of patients who achieve a CR or better (i.e., CR or sCR). The definitions of CR and sCR were based on the IMWG response criteria:22
CR: Negative immunofixation on the serum and urine and disappearance of any soft tissue plasmacytomas and less than 5% plasma cells in bone marrow.
sCR: CR as defined previously plus a normal FLC ratio and the absence of clonal cells in a bone marrow biopsy by immunohistochemistry (kappa:lambda ratio ≤ 4:1 or ≥ 1:2 for patients with kappa and/or lambda light chains, respectively, after counting ≥ 100 plasma cells).
Rate of Very Good Partial Response or Better
The rate of a VGPR or better, a secondary end point in the PERSEUS trial, was defined as the proportion of patients who achieved a VGPR or better (i.e., sCR, CR, or VGPR). Response to treatment was analyzed by a validated computer algorithm. The rate of a VGPR or better was selected for the GRADE assessment as it was considered important by clinical experts. The definition of a VGPR was based on the IMWG response criteria:22 serum and urine M protein detectable by immunofixation but not on electrophoresis, or at least 90% reduction in serum M protein plus urine M protein level to less than 100 mg per 24 hours.
Overall Rate of Minimal Residual Disease Negativity
Overall rate of MRD negativity was a key secondary end point in the PERSEUS trial, which was defined as the proportion of patients who achieved MRD negativity (at 10-5) as determined by a bone marrow aspirate and a CR or better at any time after the date of randomization during the study (and before progressive disease, subsequent therapy, or both). MRD status results were reported based on NGS. For patients in the DVRd group whose MRD negativity at the 10-5 threshold could not be determined by NGS (due to the lack of an MRD index clone or nonunique clone sequence), MRD by NGF could be used to guide daratumumab stopping and restarting. Overall MRD negativity rate was considered important for informing expert committee deliberations by clinical experts.
Overall Survival
OS was assessed as a key secondary end point in the PERSEUS trial and was defined as the time from the date of randomization to the date of the patient’s death due to any cause. Patients who were lost to follow-up were censored at the time of loss to follow-up. Patients who died after withdrawing consent were considered to have had an OS event. Patients who were still alive at the clinical cut-off date for the analysis were censored at the last known alive date, which was determined by the maximum collection or assessment date from among selected data domains within the clinical database. PFS was selected for GRADE assessment as it was considered important to patients and clinicians according to the clinical experts, and patient and clinician group inputs. In addition, OS was used to inform the pharmacoeconomic model submitted to the drug agency.
Duration of Response (For Complete Response or Better)
Duration of CR or better was defined as the time from the date of first documentation of a confirmed response of CR or better to the date of first documentation of a confirmed disease progression, or death due to disease progression, whichever occurred first, for patients for whom a CR or better was their best response. Patients who experience a CR or better response without disease progression were censored at the last disease evaluation before subsequent therapy. The duration of the CR or better was considered important for informing expert committee deliberations by the clinical experts.
Duration of Response (For Partial Response or Better)
Duration of PR or better was defined as the date of the initial documentation of a PR or better to the date of the first documented evidence of disease progression, or death due to disease progression, whichever occurred first, for patients who had a PR or better as their best response. Patients who experienced a PR or better response without disease progression were censored at the last disease evaluation before subsequent therapy. The definition of VGPR was based on the IMWG response criteria:22
greater than 50% reduction of serum M protein and reduction in 24 hours of urinary M protein by greater than 90% or to less than 200 mg per 24 hours. If the serum and urine M protein were not measurable, a greater than 50% decrease in the difference between involved and uninvolved FLC levels was required in place of the M protein criteria. If serum and urine M protein were not measurable, and a serum FLC assay was also not measurable, a reduction of greater than 50% in plasma cells was required in place of M protein, provided the baseline bone marrow plasma cell percentage was greater than 30%. In addition to these criteria, if present at baseline, a reduction in the size of soft tissue plasmacytomas of less than 50% was also required.
Health-Related Quality of Life
Change From Baseline in EQ-5D-5L Utility Score
In the PERSEUS trial, HRQoL was measured using the EQ-5D-5L, a 5-item questionnaire that assesses 5 domains, including mobility, self-care, usual activities, pain or discomfort, and anxiety or depression, plus a visual analogue scale rating “health today” that uses anchors of 0 (worst imaginable health state) and 100 (best imaginable health state). The scores for the 5 separate questions are categorical and cannot be analyzed as cardinal numbers. However, the scores for the 5 dimensions are used to compute a single utility score (0.0 to 1.0), representing the general health status of the individual (values of less than 0 are allowed according to the UK scoring algorithm). HRQoL was selected for GRADE assessment as it was considered important to patients and was used to the inform the pharmacoeconomic model submitted to the drug agency. Table 8 provides details of the EQ-5D-5L.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Rate of CR or better | In the context of the PERSEUS trial, the rate of CR or better is defined as the proportion of patients who achieve a response of CR or better (i.e., CR or sCR). The definitions of CR and sCR are based on the IMWG response criteria:22
|
| Not identified for patients with MM |
Rate of VGPR or better | In the context of the PERSEUS trial, the rate of a VGPR or better is defined as the proportion of patients who achieved a VGPR or better (i.e., sCR, CR, or VGPR). The definition of VGPR was based on the IMWG response criteria:22
|
| Not identified for patients with MM |
MRD negativity rate | In the context of the PERSEUS trial, the MRD negativity rate is defined as the proportion of patients who achieve MRD negativity (at 10-5) by bone marrow aspirate and achieve a CR or better at any time after the date of randomization during the study (and before progressive disease, subsequent therapy, or both). |
| Not identified for patients with MM |
EQ-5D-5L | THE EQ-5D-5L is a 2-part, generic instrument used to describe and value health status.79,80 The descriptive system assesses health status based on 5 dimensions: mobility, self-care, usual activities, pain or discomfort, and anxiety or depression.79,80 Each dimension has increasing levels of severity (i.e., no problems, slight problems, moderate problems, severe problems, and unable to or extreme problems).79,80 Scores for the 5 dimensions are used to generate a unique health state, which can be converted to a summary index score based on societal (countries or regions) preference weights for the health state.79,80 Index scores range from less than 0 (negative values represent worse than dead, which is represented by 0) to 1 (full health), with higher scores representing higher health utility.79,80 Alongside the descriptive system, overall health at the time of the survey is self-rated using the EQ-5D visual analogue scale, with anchors ranging from 0 (worst imaginable health state) to 100 (best imaginable health state).79,80 | Validity: Not estimated in patients with MM Reliability: Not estimated in patients with MM Responsiveness: Not estimated in patients with MM | Not identified for patients with MM |
CR = complete response; EFS = event-free survival; FLC = free light chain; HDT = high-dose therapy; HR = hazard ratio; IFM = Intergroupe Francophone du Myélome; IMWG = International Myeloma Working Group; ISS = International Staging System; MM = multiple myeloma; MRD = minimal residual disease; NDMM = newly diagnosed multiple myeloma; NGS = next-generation sequencing; PR = partial response; RRMM = relapsed or refractory multiple myeloma; OS = overall survival; PFS = progression-free survival; sCR = stringent complete response; SCT = stem cell transplant; TTNT = time to next treatment; VGPR = very good partial response; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
Safety Outcomes
In the PERSEUS trial, adverse events were coded using the Medical Dictionary for Regulatory Activities and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0. TEAEs were defined as adverse events that occur or worsen after administration of the first dose during that stage and through 30 days after the last dose of study drug in that stage and before the next phase of treatment begins. Notable harms included systemic administration-related reactions, local injection-site reactions, cytopenia, infections and infestations, new malignancies, and treatment-emergent interferences for blood typing. Among them, systemic administration-related reactions, cytopenia, and infections and infestations were selected for GRADE assessment as they were considered important to clinical experts.
Statistical Analysis
A summary of statistical analysis of efficacy end points is provided in Table 9.
Sample Size and Power Calculation
It was assumed that median PFS is 63 months for the VRd group, and the addition of daratumumab would decrease the risk of progression or death by 31% (HR = 0.69; estimated median PFS of 91 months for DVRd group). To achieve 85% power with a 2-sided alpha of 0.05, it was determined that 285 PFS events were needed. Assuming a 12-month accrual and 64 months of additional follow-up, approximately 690 patients (345 per group) would be required. Long-term follow-up for survival would continue until approximately 310 deaths had been observed or 9 years had elapsed after the last patient is randomized, whichever occurred earlier. Following this protocol would provide approximately 70% power to detect a 25% reduction in the risk of death (HR = 0.75) with a log-rank test at a 2-sided alpha of 0.05. However, an HR of 0.71 or less would provide at least 80% power. A large phase III trial (CASSIOPEIA [MMY3006]) comparing daratumumab-bortezomib-thalidomide-dexamethasone (DVTd) versus bortezomib-thalidomide-dexamethasone (VTd) in a similar study population (N = 1,085) observed an HR of 0.66 from first randomization. By censoring the events from patients in the VTd group who crossed over after part 1 of the study, with a median follow-up of 18.8 months, such an HR would provide approximately 93.5% power for this study with 310 deaths.81 After the significance of PFS is established, testing of OS may continue as planned until a definitive conclusion on OS is reached.
Statistical Test or Model
Analysis of PFS was based on the ITT analysis set. For PFS, the primary analysis consisted of a stratified log-rank test for the comparison of the PFS distribution between the 2 treatment groups. The P value from a stratified log-rank test was reported. The KM method was used to estimate the distribution of overall PFS for each treatment group. The treatment effect, measured by HR for (DVRd versus VRd), and its 2-sided 95% CI were estimated using a stratified Cox regression model with treatment as the sole explanatory variable. Stratification factors used in the analyses included ISS staging (I, II, and III), and cytogenetic risk (standard and high). The median PFS with a 95% CI was provided. The KM PFS curve was also plotted by treatment group. In addition, PFS rates with 95% CIs estimated by the KM method at landmarks (e.g., 12 months and 18 months) were reported for each treatment group. The number and percentage of patients who had a PFS event or were censored was reported.
Multiple Testing Procedure
Two interim analyses and 1 final analysis for the primary end point (PFS) were planned, which was intended to be performed at approximately 143 PFS events (50% information fraction), 185 PFS events (65% information fraction), and 285 PFS events, respectively. If the superiority of DVRd over VRd alone with respect to PFS could be established at the first or second interim analysis, the interim PFS analysis would serve as the primary PFS analysis, which otherwise was to occur when approximately 285 PFS events had been observed. The significance level at each interim analyses for PFS would be determined based on the observed number of PFS events at each interim analysis, using the Hwang-Shih-DeCani alpha spending function. A total of 153 PFS events were observed at this first interim PFS analysis, representing an approximately 54% PFS information fraction, with a corresponding stopping boundary of a 2-sided P value of 0.0126 for the analysis of PFS.
A hierarchical test proposed by Tang and Geller (1999)82 was used for the primary superiority hypothesis and 3 secondary superiority hypotheses. Only if the primary end point of PFS was statistically significant would the following secondary end points be sequentially tested, each with an overall 2-sided alpha of 0.05, by using the hierarchical testing approach that strongly controls the type I error rate. The secondary end points were tested in the following order:
overall CR or better rate
overall MRD negativity rate (threshold of 10-5)
overall survival.
The overall rate of CR or better and MRD negativity rate (10-5) were only tested at the interim or final PFS analysis when PFS was statistically significant. For OS, a descriptive analysis was performed at the time of the first PFS interim analyses; few OS events were expected. If PFS significance was established before the second interim or final PFS analysis, OS analysis was to continue to be performed at approximately the same time the second interim and final PFS analysis would occur (with 185 and 285 PFS events, respectively). Because the gap between the final PFS and final OS analysis was expected to be lengthy, additional periodic updates may be added until a definitive conclusion on OS is reached.
Data Imputation Methods
No data imputation methods were reported for the primary analysis of PFS in the PERSEUS trial.
In the analysis of overall MRD negativity rate, patients whose tested samples were found to be MRD-positive or ambiguous, and patients who were not tested were considered not to have achieved MRD negativity. Patients who did not have MRD negativity at a given time point were considered MRD-positive in the analysis.
Subgroup Analyses
Subgroup analyses were performed on the primary efficacy end point of PFS in the ITT analysis set. Subgroups analyses did not take multiplicity into account. Two subgroups were considered relevant based on input from the clinical experts consulted by CDA-AMC: age (< 65 years versus ≥ 65 years), and cytogenetic risk (standard versus high [more than 1 high-risk del17p, t(4;14), or t(14;16) chromosomal anomaly]).
Additionally, prespecified subgroups based on sex (male versus female), race (white versus other), ISS stage (I versus II versus III), type of MM (immunoglobin G versus non–immunoglobin G), and baseline ECOG PS (0 versus ≥ 1) were also conducted.
Sensitivity Analyses
In the PERSEUS trial, the following sensitivity and supplementary analyses were performed to evaluate the robustness of the primary analysis of PFS:
Sensitivity analyses:
progressive disease based on investigator assessment according to the IMWG response criteria
using unstratified log-rank test and unstratified Cox regression model
not censored for death or disease progression after missing more than 1 disease evaluation.
Supplementary analyses:
censoring for treatment discontinuation due to COVID-19
censoring for death due to COVID-19 for patients who had not developed a confirmed progressive disease
not censoring data due to start of subsequent antimyeloma therapies for patients who had not developed a confirmed progressive disease
per-protocol population that contains less than 95% of the ITT population
time-dependent Cox regression model to test the treatment effect adjusting for the possible confounding from transplant.
Secondary Outcomes
The overall rates of a CR or better and MRD negativity based on the computerized algorithm and corresponding exact 2-sided 95% CI from the Clopper-Pearson method were calculated for each treatment group based on the ITT population. A comparison of the 2 treatment groups was made using the stratified CMH test. The Mantel-Haenszel OR (DVRd versus VRd) was used to measure the treatment effect, with its 2-sided 95% CI and P value reported.
Analysis of OS was based on the ITT analysis set. For OS, the primary analysis consisted of a stratified log-rank test for the comparison of the OS distribution between the 2 treatment groups. The P value from a stratified log-rank test was reported. The KM method was used to estimate the distribution of overall OS for each treatment group. The treatment effect, measured by HR (DVRd versus VRd), and its 2-sided 95% CI were estimated using a stratified Cox regression model with treatment as the sole explanatory variable. The median OS with 95% CI was provided. The KM OS curve was also plotted by treatment group. In addition, OS rates with 95% CIs were estimated by the KM method at landmarks (e.g., 12 months and 18 months) and reported for each treatment group. The number and percentage of patients who had an OS event or were censored were reported. However, descriptive analysis without formal comparison was performed at the time of the first PFS interim analysis due to the small number of events expected.
The analyses of DOR (CR or better, or PR or better) were based on patients in the ITT analysis set who achieved a CR or better or a PR or better. The distribution of DOR was estimated using the KM method. Given that the DOR was calculated using a subset of patents who had a response of a PR or better, which means the ITT principle was not strictly followed, no formal statistical comparison of DOR between the treatment groups was made.
Summary descriptive statistics were calculated for the EQ-5D-5L baseline values (screening visit) by treatment group. The descriptive statistics and change from baseline to the end of maintenance cycle 34 (approximately 40 months of treatment) was calculated by treatment group. A mixed-effects model with repeated-measures analysis was created to estimate the change from baseline at each time point between the 2 treatments. Patients in the ITT analysis set who had a baseline value and at least 1 postbaseline value were included in the analysis. Change from baseline were fitted to a mixed-effects model, using patients as a random effect, and baseline value, treatment group, time in week, treatment-by-time interaction, and stratification factors as fixed effects.
Safety Outcomes
All safety analyses were based on the safety analysis set, unless otherwise specified. Descriptive statistics were calculated for continuous safety variables and frequency counts and percentages were tabulated for categorical safety variables.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS | Stratified log-rank test and KM method | ISS staging (I vs. II vs. III) and cytogenetic risk (standard vs. high) | NR |
|
Overall CR or better | Clopper-Pearson method for exact 2-sided 95% CI and the rate and corresponding 2-sided 95% CI. Comparison of the 2 treatment groups: 1. Mantel-Haenszel estimate of the common odds ratio for stratified tables is used 2. P value from the stratified Cochran-Mantel-Haenszel chi-square test. | ISS staging (I vs. II vs. III) and cytogenetic risk (standard vs. high) | NR | Overall response based on investigator assessment according to the IMWG response criteria |
Overall MRD negativity rate | Exact 2-sided 95% CI from the Clopper-Pearson method | NR |
| NR |
OS | KM method | ISS staging (I vs. II vs. III) and cytogenetic risk (standard vs. high) |
| NR |
DOR | Descriptively using the KM estimates | NR |
| NR |
EQ-5D-5L | Descriptive statistics | NR | NR |
CI = confidence interval; CR = complete response; DOR = duration of response; IMWG = International Myeloma Working Group; ISS = International Staging System; KM = Kaplan-Meier; MRD = minimal residual disease; NR = not reported; OS = overall survival; PFS = progression-free survival; vs. = versus.
Sources: Statistical Analysis Plan for PERSEUS.83 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Analysis Populations
A summary of analysis populations used in the PERSEUS trial and relevant to this review is provided in Table 10.
Table 10: Analysis Populations of the PERSEUS Trial
Population | Definition | Application |
|---|---|---|
ITT analysis set | Patients who had been randomly assigned to the DVRd group or VRd group | All efficacy analyses |
PP analysis set | All patients who were randomized who did not have a major protocol deviation due to not meeting all entry criteria; if the PP analysis set comprises > 95% of the ITT analysis set, no analysis by PP will be performed | Supplementary analysis |
Safety analysis set | All patients who had received at least 1 dose of study treatment (partial or complete) in the study | Safety analysis |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ITT = intention to treat; PP = per protocol; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Statistical Analysis Plan for the PERSEUS trial.83 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Results
Patient Disposition
At the time of the first interim analysis (data cut-off: August 1, 2023), a total of 709 patients (DVRd: 355; VRd: 354) were randomized. Of the 709 randomized participants, 351 patients in the DVRd group and 347 patients in the VRd group received study treatment (Table 11). Generally, higher proportions of patients in the DVRd group than in the VRd group completed induction (95.2% for DVRd versus 90.7% for VRd), mobilization (91.8% versus 87.0%, respectively), ASCT (87.0% versus 83.1%), consolidation (76.9% versus 73.2%), and received maintenance (90.7% versus 84.7%). Overall, more patients in the DVRd group (73.2%) remained on treatment than in the VRd group (44.9%), and fewer patients in the DVRd group (25.6%) discontinued all treatment compared with the VRd group (53.1%). The majority of discontinuations occurred during induction and maintenance. The most common reasons for discontinuation of all components of study treatment (> 10% of total in either group) were adverse events (9.0% versus 22.0% for DVRd versus VRd groups, respectively) and progressive disease (8.2% versus 20.3%), which were reported less frequently in the DVRd group than in the VRd group.
Table 11: Summary of Patient Disposition From the PERSEUS Trial (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
Patient disposition | DVRd (N = 355) | VRd (N = 354) |
|---|---|---|
Screened, N | 824 | |
Randomized, N (%) | 355 | 354 |
Not treated | 4 (1.1) | 7 (2.0) |
Treated | 351 (98.9) | 347 (98.0) |
Completed induction, n (%) | 338 (95.2) | 321(90.7) |
Completed mobilization, n (%) | 326 (91.8) | 308 (87.0) |
Completed ASCT, n (%) | 309 (87.0) | 294 (83.1) |
Completed consolidation, n (%) | 273 (76.9) | 259 (73.2) |
Received maintenance, n (%) | 322 (90.7) | 300 (84.7) |
Ongoing | 260 (73.2) | 159 (44.9) |
Stopped daratumumab | 207 (58.3) | NA |
Still on treatment, n (%) | 260 (73.2) | 159 (44.9) |
Discontinued all treatment, n (%) | 91 (25.6) | 188 (53.1) |
Reason for discontinuation, n (%) | ||
Adverse events | 32 (9.0) | 78 (22.0) |
Progressive disease | 29 (8.2) | 72 (20.3) |
Patient refused further treatment | 10 (2.8) | 14 (4.0) |
Death | 9 (2.5) | 11 (3.1) |
Death by COVID-19 | 3 (0.8) | 0 |
Physician decision | 8 (2.3) | 9 (2.5) |
Lost to follow-up | 3 (0.8) | 2 (0.6) |
COVID-19 | 1 (0.3) | 1 (0.3) |
Noncompliance with study drug | 0 | 1 (0.3) |
Other | 0 | 1 (0.3) |
ITT analysis set, N | 355 | 354 |
PP analysis set, N | 348 | 350 |
Safety analysis set, N | 351 | 347 |
ASCT = autologous stem cell transplant; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ITT = intention to treat; PP = per protocol; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Clinical Study Report for PERSEUS (2024).15
Baseline Characteristics
At baseline, overall, demographic characteristics were well balanced between treatment groups. The median age of all study patients was 60.0 years, with a range of 31 to 70 years. Most of the study patients were male (58.7%; female: 41.3%) and white (92.1%); 1.4% patients were Asian, 1.3% of patients were Black, 0.6% of patients were Native Hawaiian or other Pacific Islander, and 0.4% of patients were American Indian or Alaska Native. At baseline, most of the study patients (63.6%) had an ECOG PS of 0.
Overall, the baseline disease characteristics were balanced between treatment groups, except for the involved FLC in serum, which was higher in the DVRd group than in the VRD group (median: 581.0 mg/L for DVRd, 777.0 mg/L for VRd). More than one-half (51.4%) of patients had disease that was ISS stage I, and approximately one-fifth (21.7%) had high-risk cytogenetics such as del(17p), t(4;14), and t(14;16) chromosomal anomalies. The median time from diagnosis to randomization was 1.15 months (range, 0.0 to 184.6). Generally, the baseline disease history was balanced across treatment groups, except for hypertension (35.2% versus 30.2% for DVRd versus VRd, respectively).
The baseline characteristics outlined in Table 12 are limited to those most relevant to this review or those the review team concluded may affect the outcomes or interpretation of the study results.
Table 12: Summary of Baseline Characteristics From the PERSEUS Trial (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
Characteristic | DVRd (N = 355) | VRd (N = 354) |
|---|---|---|
Demographic | ||
Age (years) | ||
Mean (SD) | 58.7 (7.8) | 58.1 (8.1) |
Median (range) | 61.0 (32 to 70) | 59.0 (31 to 70) |
Sex, n (%) | ||
Female | 144 (40.6) | 149 (42.1) |
Male | 211 (59.4) | 205 (57.9) |
Race, n (%) | ||
American Indian or Alaska Native | 2 (0.6) | 1 (0.3) |
Asian | 4 (1.1) | 6 (1.7) |
Black or African American | 5 (1.4) | 4 (1.1) |
Native Hawaiian or other Pacific Islander | 2 (0.6) | 2 (0.6) |
White | 330 (93.0) | 323 (91.2) |
Not reported | 12 (3.4) | 18 (5.1) |
Baseline ECOG PS score,a n (%) | ||
0 | 221 (62.3) | 230 (65.0) |
1 | 114 (32.1) | 108 (30.5) |
2 | 19 (5.4) | 16 (4.5) |
3 | 1 (0.3) | 0 (0) |
Disease characteristics | ||
Type of measurable disease, n (%) | ||
Serum | 282 (79.4) | 281 (79.4) |
IgG | 204 (57.5) | 185 (52.3) |
IgA | 65 (18.3) | 85 (24.0) |
Other (IgD, IgM, IgE, and biclonal) | 13 (3.7) | 11 (3.1) |
Urine only | 43 (12.1) | 46 (13.0) |
Serum FLC only | 29 (8.2) | 27 (7.6) |
Not evaluable | 1 (0.3) | 0 (0) |
Serum M protein (g/dL) | ||
Patients with measurable serum M protein at baseline | 219 | 212 |
Mean (SD) | 3.5 (1.8) | 3.5 (1.6) |
Median (range) | 3.2 (1 to 11) | 3.3 (1 to 9) |
Urine M protein (g/dL) | ||
Patients with measurable urine M protein at baseline | 43 | 46 |
Mean (SD) | 1,560.3 (1,149.3) | 1,830.5 (2,419.2) |
Median (range) | 1,321.3 (217 to 4,396) | 1,242.3 (254 to 14,679) |
Serum FLC involved (mg/L) | ||
Patients with measurable serum FLC at baseline | 29 | 27 |
Mean (SD) | 1,864.7 (4,740.6) | 3,928.0 (13,686.6) |
Median (range) | 581.0 (113 to 25,700) | 777.0 (201 to 72,200) |
Serum FLC uninvolved (mg/L) | ||
Patients with measurable serum FLC at baseline | 29 | 27 |
Mean (SD) | 9.4 (6.43) | 8.2 (4.48) |
Median (range) | 8.9 (1 to 27) | 8.1 (1 to 19) |
ISS staging,b n (%) | ||
Number of patients contributing to the analysis | 355 | 353 |
I | 186 (52.4) | 178 (50.4) |
II | 114 (32.1) | 125 (35.4) |
III | 55 (15.5) | 50 (14.2) |
Number of extramedullary plasmacytomas, n (%) | ||
0 | 340 (95.8) | 338 (95.5) |
≥ 1 | 15 (4.2) | 16 (4.5) |
Bone marrow plasma cells (biopsy or aspirate), n (%) | ||
< 10% | 14 (4.0) | 14 (4.0) |
10% to 30% | 138 (39.1) | 132 (37.3) |
> 30% | 201 (56.9) | 208 (58.8) |
Cytogenetic risk, n (%) | ||
Standard risk | 264 (74.4) | 266 (75.1) |
High risk | 76 (21.4) | 78 (22.0) |
del(17p) | 36 (10.1) | 34 (9.6) |
t(4;14) | 33 (9.3) | 38 (10.7) |
t(14;16) | 11 (3.1) | 14 (4.0) |
Indeterminate | 15 (4.2) | 10 (2.8) |
Time from diagnosis of multiple myeloma to randomization (months) | ||
Median (range) | 1.2 (0.0 to 46.5) | 1.1 (0.1 to 184.6) |
Medical history | ||
Patients with 1 or more medical history, n (%) | 340 (95.8) | 327 (92.4) |
Commonly reported medical history (reported in ≥ 10% in either treatment group), n (%) | ||
Hypertension | 125 (35.2) | 107 (30.2) |
Anemia | 53 (14.9) | 51 (14.4) |
Hepatitis B immunization | 49 (13.8) | 52 (14.7) |
Back pain | 42 (11.8) | 43 (12.1) |
Bone pain | 36 (10.1) | 29 (8.2) |
Hepatitis B | 32 (9.0) | 43 (12.1) |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FLC = free light chain; IgA = immunoglobulin A; IgD = Immunoglobulin D; IgE = Immunoglobulin E; IgG = immunoglobulin G; IgM = immunoglobulin M; ISS = International Staging System; ITT = intention to treat; SD = standard deviation; VRd = bortezomib-lenalidomide-dexamethasone.
aOne patient had an ECOG PS of 0 at randomization that worsened to an ECOG PS of 3 at baseline.
bISS staging was derived based on the combination of serum beta-2 microglobulin and albumin.
Sources: Clinical Study Report for PERSEUS (2024).15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Major Protocol Deviations
A summary of major protocol deviations in the ITT population of the PERSEUS trial at the data cut-off of August 1, 2023, is provided in Table 13.
Twenty participants (5.6%) in the DVRd group and 26 (7.3%) in the VRd group reported major protocol deviations. Commonly reported major protocol deviations included: received wrong treatment or incorrect dose (received ASCT at time points not permitted) (2.3% versus 0.6% for DVRd versus VRd, respectively) and entered the trial but did not satisfy inclusion or exclusion criteria (2.0% versus 1.1%), which were reported more frequently in the DVRd group than the VRd group; and received a disallowed concomitant treatment (1.4% versus 4.2%), which was reported less frequently in the DVRd group than the VRd group.
Exposure to Study Treatments
At the time of the first interim analysis (data cut-off: August 1, 2023), patients in the DVRd group had slightly longer median durations of treatment and received more cycles of treatment than did patients in the VRd group (duration = 45.7 months versus 42.2 months for DVRd versus VRd; treatment cycle [median] = 43.0 versus 38.0, respectively) (Table 14). Overall, total dose and dose intensity were balanced between treatment groups, except for the median dose intensity of lenalidomide, which was lower in the DVRd group (█████ ████████) than the VRd group (█████ ████████). Cycle delays were reported by 89.5% patients in the DVRd group and 80.7% patients in the VRd group. Generally, dose delay, reduction, discontinuation, and skipped doses were balanced between treatment groups except for lenalidomide. Compared to the VRd group, higher proportions of patients in the DVRd group reported lenalidomide dose reduction (63.8% versus 51.3% for DVRd versus VRd, respectively), discontinuation (26.5% versus 19.6%), and skipped doses (84.3% versus 76.7%). Adverse events were the most commonly reported reasons for lenalidomide dose reduction (61.3% versus 49.9%), discontinuation (22.5% versus 12.4%), and skipped doses (74.4% versus 62.8%), which were reported by more patients in the DVRd group than in the VRd group.
Table 13: Summary of Major Protocol Deviations From the PERSEUS Trial (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
Major protocol deviations | DVRd (N = 355) | VRd (N = 354) |
|---|---|---|
Patients with major protocol deviations, n (%) | 20 (5.6) | 26 (7.3) |
Received wrong treatment or incorrect dose (received ASCT at time points not permitted) | 8 (2.3) | 2 (0.6) |
Entered but did not satisfy inclusion or exclusion criteria | 7 (2.0) | 4 (1.1) |
Received a disallowed concomitant treatment | 5 (1.4) | 15 (4.2) |
COVID-19 related | 3 (0.8) | 0 |
Developed withdrawal criteria but not withdrawn | 0 | 2 (0.6) |
Other | 1 (0.3) | 4 (1.1) |
ASCT = autologous stem cell transplant; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ITT = intention to treat; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Clinical Study Report for PERSEUS (2024).15
Table 14: Summary of Patient Exposure From the PERSEUS Trial (Safety Analysis Set; Data Cut-Off Date: August 1, 2023)
Exposure | DVRd (N = 351) | VRd (N = 347) | |||||
|---|---|---|---|---|---|---|---|
Total number of treatment cycles received | |||||||
Mean (SD) | ████ ██████ | ████ ██████ | |||||
Median (range) | ████ ██ ██ ███ | ████ ██ ██ ███ | |||||
Duration (months) | |||||||
Mean (SD) | 40.0 (13.5) | 34.1 (16.4) | |||||
Median (range) | 45.7 (0.5 to 54.3) | 42.2 (0.1 to 53.9) | |||||
Total dosea | D (mg/cycle) | V (mg/m2) | R (mg) | d (mg) | V (mg/m2) | R (mg) | d (mg) |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Median (range) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Dose intensitya (mg/cycle) | D | V | R | d | V | R | d |
Mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Median (range) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Cycle delays, n (%) | 314 (89.5) | 280 (80.7) | |||||
Treatment | D | V | R | d | V | R | d |
Injection or dose delay, n (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
AEs | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Dose reduction, n (%) | ████ | ████ | 224 (63.8) | ████ | ████ | 178 (51.3) | ████ |
AEs | ████ | ████ | 215 (61.3) | ████ | ████ | 173 (49.9) | ████ |
Injection or dose skipped, n (%) | ████ | ████ | 296 (84.3) | ████ | ████ | 266 (76.7) | ████ |
AEs | ████ | ████ | 261 (74.4) | ████ | ████ | 218 (62.8) | ████ |
Discontinuation, n (%) | ████ | ████ | 93 (26.5) | ████ | ████ | 68 (19.6) | ████ |
AEs | ████ | ████ | 79 (22.5) | ████ | ████ | 43 (12.4) | ████ |
AE = adverse event; D = daratumumab; d = dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; NA = not applicable; NR = not reported; R = lenalidomide; SD = standard deviation; V = bortezomib; VRd = bortezomib-lenalidomide-dexamethasone.
aAll treatment cycles.
bDaratumumab dose reduction was not permitted per protocol.
Sources: Clinical Study Report for PERSEUS (2024).15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Prior Therapy
A summary of prior therapy from the PERSEUS trial in the ITT population at the data cut-off of August 1, 2023, is provided in Table 15. ███ patients in the DVRd group received preadministration medications before receiving daratumumab to prevent infusion-related reactions compared to ██ patients in the VRd group. Antihistamines for systemic use was the most commonly reported preinjection medication in the DVRd group (100%) followed by corticosteroids for systemic use (████%), analgesics (███████ and drugs for obstructive airway diseases ████████.
Table 15: Summary of Preinjection Medications in the PERSEUS Trial (Safety Analysis Set; Data Cut-Off Date: August 1, 2023)
Prior therapy | DVRd (N = 351) | VRd (N = 347) |
|---|---|---|
Patients with ≥ 1 preinjection medications | ███ ███████ | █ |
Commonly reported preinjection medications (received by ≥ 50% of patients), n (%) | ||
Antihistamines for systemic use | ███ ███████ | █ |
Corticosteroids for systemic use | ███ ██████ | █ |
Corticosteroids, plain | ███ ██████ | █ |
Dexamethasone | ███ ██████ | █ |
Analgesics | ███ ██████ | █ |
Other analgesics and antipyretics | ███ ██████ | █ |
Paracetamol | ███ ██████ | █ |
Drugs for obstructive airway diseases | ███ ██████ | █ |
Other systemic drugs for obstructive airways diseases | ███ ██████ | █ |
Montelukast | ███ ██████ | █ |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Clinical Study Report for PERSEUS (2024).15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Concomitant Medication
At the time of the first interim analysis (data cut-off: August 1, 2023), the reported use of concomitant medications was similar between the DVRd (████) and VRd (████) groups (Table 16). Generally, the commonly reported concomitant medications were balanced between treatment groups, except for vaccines (█████ ███████% for DVRd versus VRd, respectively) and immunostimulants (████% versus████%), which were more frequently reported in the DVRd group than in the VRd group. Other concomitant medications that were reported by a higher proportion of patients in the DVRd group than in the VRd group by a difference of more than 10% were drugs for obstructive airway diseases (█████ ███ ███████ and immune sera and immunoglobulins (█████ ███ ████).
Table 16: Concomitant Medication Use From the PERSEUS Trial (Safety Analysis Set; Data Cut-Off Date: August 1, 2023)
Concomitant medication | DVRd (N = 351) | VRd (N = 347) |
|---|---|---|
Subjects with ≥ 1 concomitant medications | ███ ███████ | ███ ███████ |
Commonly reported concomitant medication (received by ≥ 50% of patients in either group), n (%) | ||
Antibacterials for systemic use | ███ ██████ | ███ ██████ |
Sulfonamides and trimethoprim | ███ ██████ | ███ ██████ |
Beta-lactam and penicillin | ███ ██████ | ███ ██████ |
Antivirals for systemic use | ███ ██████ | ███ ██████ |
Analgesics | ███ ██████ | ███ ██████ |
Acetylsalicylic acid | ███ ██████ | ███ ██████ |
Paracetamol | ███ ██████ | ███ ██████ |
Drugs for acid-related disorders | ███ ██████ | ███ ██████ |
Vaccines | ███ ██████ | ███ ██████ |
Viral vaccines | ███ ██████ | ███ ██████ |
Antithrombotic agents | ███ ██████ | ███ ██████ |
Drugs for treatment of bone diseases | ███ ██████ | ███ ██████ |
Immunostimulants | ███ ██████ | ███ ██████ |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Clinical Study Report for PERSEUS (2024).15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Subsequent Treatment
At the time of the first interim analysis (data cut-off: August 1, 2023), the use of subsequent treatments was reported by a smaller proportion of the DVRd group (9.4%) compared with the VRd group (26.8%) (Table 17). The most commonly reported subsequent treatment was systemic therapy (9.4% versus 26.8%), which was reported less frequently in the DVRd group than in the VRd group.
Table 17: Summary of Subsequent Treatment From the PERSEUS Trial (Safety Analysis Set; Data Cut-Off Date: August 1, 2023)
Subsequent treatment | DVRd (N = 351) | VRd (N = 347) |
|---|---|---|
Patients with 1 or more antimyeloma subsequent therapies, n (%) | 33 (9.4) | 93 (26.8) |
Subsequent surgery | | █████ | | █████ |
Subsequent radiotherapy | | █████ | ██ █████ |
Subsequent allogenic transplant | | ███ | | █████ |
Subsequent autologous transplant | 10 (2.8) | 15 (4.3) |
Subsequent systemic therapy | 33 (9.4) | 93 (26.8) |
Antineoplastic agents | 31 (8.8) | 89 (25.6) |
Other antineoplastic agents | 27 (7.7) | 65 (18.7) |
Monoclonal antibodies and conjugates | 16 (4.6) | 73 (21.0) |
Alkylating agents | 17(4.8) | 32 (9.2) |
Cytotoxic antibiotics and related substance | 8 (2.3) | 7 (2.0) |
Plant alkaloids and other natural products | 5 (1.4) | 7 (2.0) |
Antimetabolites | 1 (0.3) | 4 (1.2) |
Corticosteroids for systemic use | ██ █████ | ██ ██████ |
Immunosuppressants | ██ █████ | ██ ██████ |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Clinical Study Report for PERSEUS (2024).15 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Efficacy
Key efficacy results from the PERSEUS trial in the ITT population at the data cut-off of August 1, 2023, are provided in Table 18.
Progression-Free Survival
At the time of the first interim analysis (data cut-off: August 1, 2023), the median duration of follow-up for PFS was ████ months (range, ████ to ████) in the DVRd group, and ████ months in the VRd group (range, ████ to ████) (Table 18). Fifty patients (14.1%) in the DVRd treatment group and 103 patients (29.1%) in the VRd group experienced a PFS event; among them, ██ ██████ had disease progression and ██ ██████ died in the DVRd group, and ██ ███████ had disease progression and 19 (5.4%) died in the VRd group. The median PFS was not reached (95% CI, not estimable) for either the DVRd or VRd group. The KM estimate of PFS probability at 48 months was 84.3% (95% CI, 79.5% to 88.1%) for the DVRd group, and 67.7% (95% CI, 62.2% to 72.6%) for the VRd group, with a between-group difference of █████ (95% CI, ████ to ███████ The interim analysis showed an improvement in PFS for patients receiving DVRd compared with VRD (HR = 0.42; 95% CI, 0.30 to 0.59; P < 0.0001). This means patients in the DVRd group were 58% less likely to experience death or progression compared with those in the VRD group. The KM estimate of the PFS distribution among the interim analysis population is depicted in Figure 3.
Results from planned sensitivity and supplementary analyses were consistent with those of the primary analysis. These included progressive disease based on investigator assessment (HR = 0.42; 95% CI, 0.30 to 0.59; nominal P < 0.0001), PFS using unstratified analysis (HR = █████ ███ ███ ████ ██ ████; nominal P < 0.0001), patients who were censored who discontinued treatment due to COVID-19 (HR = █████ ███ ███ ████ ██ ████; nominal P < 0.0001), patients who were censored who died due to COVID-19 (HR = █████ 95% CI █ ████ ██ ████; nominal P < 0.0001), patients who were not censored who started subsequent therapy (HR = █████ ███ ███ ████ ██ ████; nominal P < 0.0001), patients who were not censored with 2 missing consecutive disease evaluations (HR = █████ ███ ███ ████ ██ ████; nominal P < 0.0001), and PFS using transplant and maintenance as time varying covariate (HR = 0 ████ ███ ███ ████ ██ ████; nominal P < 0.0001). Subgroup analyses of PFS in the primary analysis were consistent with the primary analysis across all prespecified subgroups, except for subgroups of patients aged 65 years or older (HR = 0.97; 95% CI, 0.52 to 1.81). Figure 5 in Appendix 1 provides detailed subgroup analyses data.
Rate of Complete Response or Better
In the first interim analysis (data cut-off date: August 1, 2023), the CR or better (sCR plus CR) rates were 87.9% (95% CI, 84.0% to 91.1%) in the DVRd group and 70.1% (95% CI, 65.0% to 74.8%) in the VRd group (Table 18). Among patients who achieved a CR or better, 69.3% (95% CI, 64.2% to 74.1%) of patients in the DVRd group and 44.6% (95% CI, 39.4% to 50.0%) of patients in the VRd had an sCR, and 18.6% (95% CI, 14.7% to 23.0%) of patients in the DVRd group and 25.4% (95% CI, 21.0% to 30.3%) of patients in the VRd group had a CR. The stratified CMH estimate of OR was 3.13 (95% CI, 2.11 to 4.65; P < 0.0001). Similar results were observed for the sensitivity analysis, in which determination of response was based on investigator assessment.
Rate of Very Good Partial Response or Better
In the interim first analysis (data cut-off date: August 1, 2023), the rates of a VGPR or better (sCR, CR, and VGPR) were 95.2% (95% CI, 92.4% to 97.2%) in the DVRd group and 89.3% (95% CI, 85.6% to 92.3%) the VRd group, with a between-group difference of ████ ████ ███ ████ ██ ████%). Among patients who achieved a VGPR or better, 7.3% (95% CI, 4.8% to 10.5%) of patients in the DVRd group and 19.2% (95% CI, 15.2% to 23.7%) of patients in the VRd group had a VGPR. The stratified CMH estimate of OR was 2.40 (95% CI, 1.33 to 4.35; nominal P = 0.0029). Similar results were observed for the sensitivity analysis, in which determination of response was based on investigator assessment.
Overall Rate of Minimal Residual Disease Negativity
In the first interim analysis (data cut-off: August 1, 2023), the proportions of patients reported to have a negative overall MRD in bone marrow by NGS (threshold of 10-5) and a CR or better were 75.2% (95% CI, 70.4% to 79.6%) in the DVRd group and 47.5% (95% CI, 42.2% to 52.8%) in the VRd group, with a between-group difference of █████ (95% CI, █████ ██ ██████. The Mantel-Haenszel estimate of OR was 3.40 (95% CI, 2.47 to 4.69; P < 0.0001).
Figure 3: KM Plot for Progression-Free Survival (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
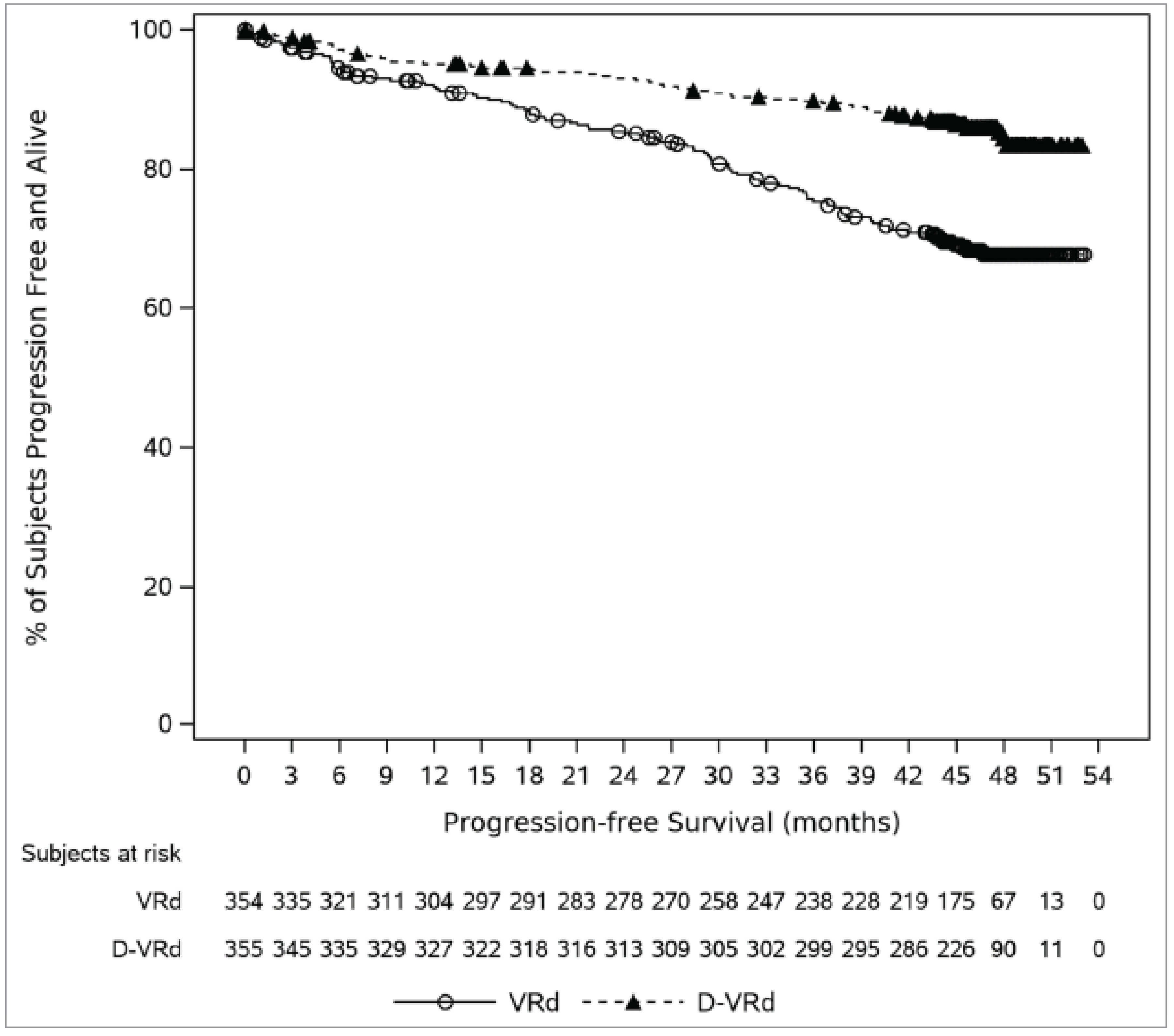
D-VRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ITT = intention to treat; KM = Kaplan-Meier; PFS = progression-free survival; VRd = bortezomib-lenalidomide-dexamethasoneI.
Source: Clinical Study Report for PERSEUS (2024).15
Overall Survival
At the time of the first interim analysis (data cut-off: August 1, 2023), the median durations of follow-up for OS were ████ months (range, ████ to ████) in the DVRd group, and ████ months in the VRd group (range, ████ to ████) (Table 18). Thirty-four patients (9.6%) in the DVRd treatment group and 44 patients (12.4%) in the VRd group had died. The median OS was not reached (95% CI, not estimable) for either the DVRd or VRd group. The KM estimates of OS probability at 48 months were 89.4% (95% CI, 85.4% to 92.4%) for the DVRd group and 87.5% (95% CI, 83.5% to 90.6%) for the VRd group, with a between-group difference of ████ (95% ███ █████ ██ ██████. There was an observed treatment benefit in OS for patients receiving DVRd compared with those receiving VRd (HR = 0.73; 95% CI, 0.47 to 1.14); however, no formal comparison was performed at the time of the first interim analysis due to the small number of events expected. The KM estimate of the OS distribution among the interim analysis population is depicted in Figure 4.
Figure 4: KM Plot for Overall Survival (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
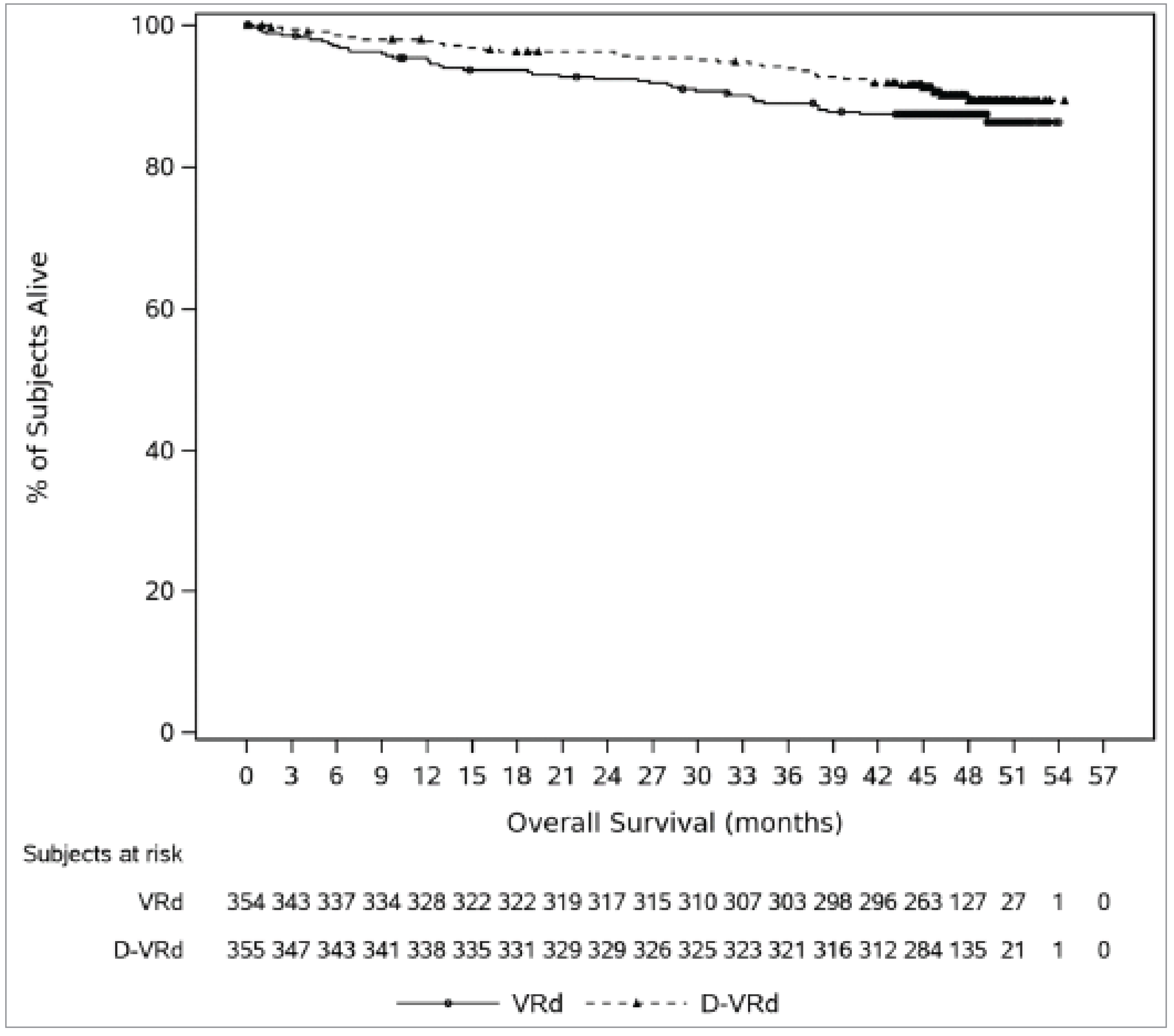
D-VRd = daratumumab-bortezomib-lenalidomide-dexamethasone; KM = Kaplan-Meier; ITT = intention to treat; OS = overall survival; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Clinical Study Report for PERSEUS (2024).15
Duration of Response (For Complete Response or Better)
At the time of the first interim analysis (data cut-off: August 1, 2023), with median follow-ups of ████ months (range, ████ to ████) in the DVRd group and ████ months in the VRd group (range, ████ to ████), the median DOR (for a CR or better) was not reached in either the DVRd or VRd group. Among patients who had a CR or better (312 versus 248 for DVRd versus VRd ██ ██ patients ████%) in the DVRd group and ██ patients (████%) in the VRd group achieved a CR or better but developed disease progression or died due to disease progression, ███ patients (████%) in the DVRd group and ███ patients (████%) in the VRd group were censored. The KM estimates of event-free probability at 42 months were ████% (95% CI, ████% to ████%) in the DVRd group and ████% (95% CI, █████ ██ ████%) in the VRd group, with a between-group difference of ████% (95% CI, ████ ██ ███%). The corresponding HR was 0.40 (95% CI, 0.24 to 0.67; nominal P = 0.0003).
Duration of Response (For Partial Response or Better)
At the time of the first interim analysis (data cut-off: August 1, 2023), with median follow-ups of ████ months (range, ████ to ████) in the DVRd group and ████ months in the VRd group (range, ████ to ████), the median DOR (for a PR or better) was not reached in either the DVRd or VRd group. Among patients who had a PR or better (343 versus 332 for DVRd versus VRd, respectively), ██ patients (███%) in the DVRd group and ██ patients (████%) in the VRd group achieved a PR or better but developed disease progression or died due to disease progression, ███ patients (████%) in the DVRd group and ███ patients (████%) in the VRd group were censored.
Health-Related Quality of Life
Change From Baseline in EQ-5D-5L Utility Score
At baseline, the mean EQ-5D-5L utility scores were ███ ███ █ █████ in the DVRd group and ███ ███ █ ████) in the VRd group. At maintenance cycle 34 (approximately 40 months of treatment), patients in the DVRd group reported a least squares mean increase (an improvement) from baseline in the EQ-5D-5L utility score of ███ ███ █ ████) compared to ██████ █ ████) in patients in the VRd group, with a between-group difference of █ (95% CI, █ ██ █; nominal P = ████████.
Table 18: Summary of Key Efficacy Results From the PERSEUS Trial (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
Variable | DVRd N = 355 | VRd N = 354 |
|---|---|---|
Progression-free survival | ||
Median follow-up time, months (range) | ██ █████ | ██ ██████ |
Patients with events, n (%) | 50 (14.1) | 103 (29.1) |
Disease progression | ██ █████ | ██ ██████ |
Death | ██ █████ | ██ █████ |
Censored, n (%) | 305 (85.9) | 251 (70.9) |
KM estimate of median PFS, months (95% CI) | NE (NE to NE) | NE (NE to NE) |
KM estimate of PFS probability at 48 months, % (95% CI) | 84.3 (79.5 to 88.1) | 67.7 (62.2 to 72.6) |
Absolute difference in PFS probability between study groups at 48 months, % (95% CI) | ████ ████ ██ █████ | |
HR (95% CI)a | 0.42 (0.30 to 0.59) | |
P valueb | < 0.0001 | |
Response rate | ||
Response rate by category, n (%) | ||
sCR | 246 (69.3) | 158 (44.6) |
95% CI | 64.2 to 74.1 | 39.4 to 50.0 |
CR | 66 (18.6) | 90 (25.4) |
95% CI | 14.7 to 23.0 | 21.0 to 30.3 |
VGPR | 26 (7.3) | 68 (19.2) |
95% CI | 4.8 to 10.5 | 15.2 to 23.7 |
PR | 5 (1.4) | 16 (4.5) |
95% CI | 0.5 to 3.3 | 2.6 to 7.2 |
CR or better (sCR + CR), n (%) | 312 (87.9) | 248 (70.1) |
95% CI | 84.0 to 91.1 | 65.0 to 74.8 |
OR (95% CI)c | 3.13 (2.11 to 4.65) | |
P valued | < 0.0001 | |
VGPR or better (sCR + CR + VGPR), n (%) | 338 (95.2) | 316 (89.3) |
95% CI | 92.4 to 97.2 | 85.6 to 92.3 |
Absolute between-group difference, % (95% CI) | ███ ████ ██ █████ | |
OR (95% CI)c | 2.40 (1.33 to 4.35) | |
Nominal P valued | 0.0029 | |
Overall minimal residual disease negativity rate | ||
Overall MRD negativity rate (10-5), n (%) | 267 (75.2) | 168 (47.5) |
95% CI | 70.4 to 79.6 | 42.2 to 52.8 |
Absolute between-group difference, % (95% CI) | ████ █████ ██ █████ | |
OR (95% CI)c | 3.40 (2.47 to 4.69) | |
P valued | < 0.0001 | |
Overall survival | ||
Median follow-up time, months (range) | ██ █████ | ██ ██████ |
Patients with events, n (%) | 34 (9.6) | 44 (12.4) |
Censored, n (%) | 321 (90.4) | 310 (87.6) |
KM estimate of median OS, months (95% CI) | NE (NE to NE) | NE (NE to NE) |
KM estimate of OS probability at 48 months, % (95% CI) | 89.4 (85.4 to 92.4) | 87.5 (83.5 to 90.6) |
Absolute difference in OS probability between study groups at 48 months, % (95% CI) | ███ █████ ██ ████ | |
HR (95% CI)a | 0.73 (0.47 to 1.14) | |
Duration of response | ||
Median follow-up time, months (range) | ██ █████ | ██ ██████ |
Patients who achieved a PR or better contributing to the analysis contributing to the analysis, n | 343 | 332 |
Duration of CR or better | ||
Patients with events,e n (%) | ██ █████ | ██ ██████ |
Censored, n (%) | ██ █████ | ██ ██████ |
KM estimate of median DOR (CR or better), months (95% CI) | ██ █████ | ██ ██████ |
KM estimate of CR or better probability at 42 months, % (95% CI) | ██ █████ | ██ ██████ |
Absolute difference in CR or better probability between study groups at 42 months, % (95% CI) | ██ █████ | |
HR (95% CI)a | ██ █████ | |
Nominal P valueb | ██ █████ | |
Duration of PR or better | ██ █████ | |
Patients with events,e n (%) | ██ █████ | ██ ██████ |
Censored, n (%) | ██ █████ | ██ ██████ |
KM estimate of median DOR (PR or better), months (95% CI) | ██ █████ | ██ ██████ |
Change from baseline in EQ-5D-5L utility score | ||
Patients contributing to the analysis at maintenance cycle 34 day 1, n (%) | ██ █████ | ██ ██████ |
Baseline, mean (SD) | ██ █████ | ██ ██████ |
Change from baseline at maintenance cycle 34 day 1, LS mean (SE) | ██ █████ | ██ ██████ |
Absolute between-group difference for DVRd vs. VRd (95% CI) | ██ █████ | |
Nominal P value | ██ █████ | |
CI = confidence interval; CR = complete response; DOR = duration of response; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; HR = hazard ratio; KM = Kaplan-Meier; LS = least squares; MRD = minimal residual disease; NE = not estimable; OR = odds ratio; OS = overall survival; PFS = progression-free survival; PR = partial response; sCR = stringent complete response; SD = standard deviation; SE = standard error; VGPR = very good partial response; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
aHazard ratio and 95% CI from a Cox proportional hazards model with treatment as the sole explanatory variable and stratified with ISS staging (I vs. II vs. III), and cytogenetic risk (high risk vs. standard risk or unknown). A hazard ratio less than 1 indicates an advantage for DVRd.
bP value was based on the log-rank test stratified with ISS staging (I, II, vs. III), and cytogenetic risk (high risk vs. standard risk or unknown).
cMantel-Haenszel estimate of the common odds ratio for stratified tables was used. The stratification factors were ISS staging (I vs. II vs. III) and cytogenetic risk (high risk vs. standard risk).
dP value from the stratified Cochran Mantel-Haenszel chi-square test.
eEvents referred to patients with CR or better (or PR or better for duration of PR or better) who developed disease progression or died due to disease progression.
Sources: Clinical Study Report for PERSEUS (2024),15 and sponsor-provided additional information.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Harms
Harms data in the PERSEUS trial first interim analysis safety analysis set (data cut-off: August 1, 2023) are summarized in Table 19.
Treatment-Emergent Adverse Events
At the time of the first interim analysis (data cut-off: August 1, 2023), 349 of 351 patients (99.4%) in the DVRd group and 344 of 347 patients (99.1%) in the VRd group had experienced at least 1 TEAE. Based on system organ class, the most common TEAEs were infections and infestations (86.9% versus 76.7% for DVRd versus VRd, respectively), blood and lymphatic system disorders (83.2% versus 73.2%), including neutropenia (69.2% versus 58.8%), thrombocytopenia (48.4% versus 34.3%), and anemia (22.2% versus 20.7%), and gastrointestinal disorders (81.8% versus 77.2%), including conditions such as nausea, diarrhea, constipation, and vomiting, which were reported more frequently in the DVRd group than in the VRd group.
Serious Adverse Events
In the same analysis, 200 patients (57.0%) in the DVRd group and 171 (49.3%) in the VRd group reported at least 1 SAE. Infections and infestations were reported in 35.0% and 27.4% of the DVRd and VRd groups, respectively. Pneumonia (11.4% versus 6.1%) was the most reported SAE.
Withdrawals Due to Adverse Events
Totals of 116 patients (33.0%) in the DVRd group and 104 patients (30.0%) in the VRd group stopped a component of treatment due to TEAEs. The most common TEAEs that caused treatment discontinuation were nervous system disorders (████% versus ███% for DVRd versus VRd, respectively) including peripheral sensory neuropathy (████ versus █████) and gastrointestinal disorders (███% versus ███%).
Mortality
Thirty-four patients (9.7%) in the DVRd group and 43 patients (12.4%) in the VRd group died at the time of the first interim analysis (data cut-off: August 1, 2023). The most reported cause of death was disease progression (4.6% versus 5.5% for DVRd versus VRd, respectively). TEAEs were reported as the primary cause of death for ██ ██████ patients in the DVRd group and ██ patients (███%) in VRd group, with 3 (0.9%) in the DVRd group and 4 (1.2%) in the VRd group dying of AEs related to the study drug. While the PERSEUS trial period overlapped with the COVID-19 pandemic, following the mitigation strategies via protocol amendment, COVID-19-related mortality was observed in 4 patients (1.1%) in the DVRd group and 1 (0.3%) in the VRd group.
Notable Harms
Notable harms identified by the clinical experts included cytopenia, systemic administration-related reactions, and infections and infestations. Infections and infestations were observed in 305 patients (86.9%) in the DVRd group and 266 patients (76.7%) in the VRd group. Cytopenia (comprising neutropenia, anemia, thrombocytopenia, and lymphopenia group terms) was reported in ███ ███████ patients in the DVRd group and ███ █████%) patients in the VRd group. Systemic administration-related reactions were defined as systemic reactions related to daratumumab SC administration and was reported in ██ ████%) patients in the DVRd group; the majority were grade 1 or 2 events.
Table 19: Summary of Harms Results From the PERSEUS Trial (Safety Analysis Set; Data Cut-Off Date: August 1, 2023)
Adverse events | DVRd (N = 351) | VRd (N = 347) |
|---|---|---|
Most common adverse events (≥ 70% of patients in either group), n (%) | ||
Patients ≥ 1 adverse event | 349 (99.4) | 344 (99.1) |
Infections and infestations | 305 (86.9) | 266 (76.7) |
Blood and lymphatic system disorders | 292 (83.2) | 254 (73.2) |
Neutropenia | 243 (69.2) | 204 (58.8) |
Thrombocytopenia | 170 (48.4) | 119 (34.3) |
Anemia | 78 (22.2) | 72 (20.7) |
Gastrointestinal disorders | 287 (81.8) | 268 (77.2) |
General disorders and administration site conditions | 265 (75.5) | 255 (73.5) |
Nervous system disorders | 262 (74.6) | 253 (72.9) |
Peripheral sensory neuropathy | 188 (53.6) | 179 (51.6) |
Serious adverse events (≥ 5% of patients in either group), n (%) | ||
Patients with ≥ 1 serious adverse event | 200 (57.0) | 171 (49.3) |
Infections and infestations | 123 (35.0) | 95 (27.4) |
Pneumonia | 40 (11.4) | 21 (6.1) |
Blood and lymphatic system disorders | 24 (6.8) | 18 (5.2) |
Gastrointestinal disorders | 23 (6.6) | 23 (6.6) |
Respiratory, thoracic, and mediastinal disorders | 23 (6.6) | 12 (3.5) |
Cardiac disorders | 21 (6.0) | 12 (3.5) |
Nervous system disorders | 19 (5.4) | 13 (3.7) |
Patients who stopped treatment due to adverse events (≥ 5% of patients in either group), n (%) | ||
Patients who stopped | 116 (33.0) | 104 (30.0) |
Nervous system disorders | ██ ██████ | ██ █████ |
Peripheral sensory neuropathy | ██ █████ | ██ █████ |
Gastrointestinal disorders | ██ █████ | ██ █████ |
Deaths, n (%) | ||
Patients who died | 34 (9.7) | 43 (12.4) |
Disease progression | 16 (4.6) | 19 (5.5) |
AEs | ██ █████ | ██ █████ |
AEs that were unrelated to study drug | ██ █████ | ██ █████ |
AEs that were related to study drug | 3 (0.9) | 4 (1.2) |
COVID-19 | 4 (1.1) | 1 (0.3) |
Other | | █████ | | █████ |
Adverse events of special interest, n (%) | ||
Infections and infestations | 305 (86.9) | 266 (76.7) |
Absolute difference in risk between study groups, % (95% CI) | ████ ████ ██ █████ | |
Cytopenia | ███ ██████ | ███ ██████ |
Absolute difference in risk between study groups, % (95% CI) | ███ ████ ██ █████ | |
Systemic administration-related reactions | ██ █████ | ██ |
AE = adverse event; CI = confidence interval; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; NA = not applicable; VRd = bortezomib-lenalidomide-dexamethasone.
Sources: Clinical Study Report for PERSEUS (2024),15 sponsor-provided additional information.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Critical Appraisal
Internal Validity
The PERSEUS trial is an ongoing phase III, open-label, active-control RCT to evaluate the efficacy and safety of DVRd compared to VRd in patients with TE NDMM. The choice of VRd as the comparator was clinically relevant, according to the clinical experts. The methods of randomization involved stratification using ISS at screening (I versus II versus III) and cytogenetics (standard risk versus high risk as defined by the presence of del17p, t[4;14] or t[14;16] chromosomal anomalies), which were considered appropriate. There was generally no notable imbalance in the baseline patient demographic and disease characteristics between treatment groups, except for the involved FLC in serum, which was higher in the DVRd group (581.0 mg/L) than in the VRD group (777.0 mg/L). The clinical experts pointed out that FLC level at baseline is not a prognostic factor, and, considering that other important factors, such as immunoglobin A, cytogenetics, and extramedullary disease were balanced between groups, the impact of the imbalance in FLC levels would be minimal.
In the ITT analysis set, a smaller proportion of patients in the DVRd group (9.0%) discontinued all treatments compared with the VRd group (22.0%) due to adverse events. Totals of ███ █████%) patients in the DVRd group and ███ █████%) patients in the VRd group stopped any component of treatment due to adverse events. In fact, the overall incidence of adverse events, despite being marginally higher in infection and infestations, blood and lymphatic disorders, and gastrointestinal disorders in DVRd versus VRd, was generally comparable between the 2 groups. Patients in the VRd group only received treatment with lenalidomide during maintenance therapy, and any discontinuation of lenalidomide led to discontinuation of all study drugs. In contrast, in the DVRd group, if patients discontinued lenalidomide during maintenance therapy, daratumumab study treatment was not discontinued. The lower proportion of patients who discontinued all treatments in the DVRd group compared with the VRd group is likely an artifact of the fact that those patients who were on daratumumab might otherwise have been counted as discontinuing treatments due to adverse events if they were in the VRd group.
The longer treatment duration in the DVRd group may contribute to the higher lenalidomide dose reduction, discontinuation, and skipped doses observed in the DVRd group compared with the VRd group, and, according to the clinical experts, the longer patients were on a treatment, the more likely they were to experience adverse events that would warrant a dose interruption or reduction. The clinical experts pointed out that treatment with daratumumab may be contributing to an increased risk of neutropenia that would cause lenalidomide dose reductions.
In the PERSEUS trial, all patients in the DVRd group received preadministration medications before receiving daratumumab, whereas no patients in the VRd group received preinjections. Antihistamines, corticosteroids (e.g., dexamethasone), analgesics, and drugs for obstructive airway diseases were the commonly reported preinjections used in the DVRd group. In particular, the clinical experts indicated that, among these preinjections, dexamethasone has an antimyeloma effect, and other preinjections were administered to prevent infusion-related reactions. Although the clinical experts indicated that the use of preinjections would not affect the study results, considering the adverse event prophylaxis effects of the preinjected medications, the review team noted that a higher frequency of the use of preinjections in the DVRd group could bias the safety results in favour of DVRd. Additionally, a larger proportion of patients in the DVRd group used immune sera and immunoglobulins compared with the VRd group, and this may bias the safety results in favour of the DVRd group, given immune sera and immunoglobulins can reduce the frequency of adverse events such as infections, according to the clinical experts. Fewer patients received subsequent treatment in the DVRd group than in the VRd group, which would bias the OS results against DVRd.
Data imputation was conducted in the analysis of overall MRD negativity rate, with patients who did not have MRD negativity at a given time point were considered MRD-positive. Given the notably higher proportion of patients who discontinued treatment in the VRd group (53.1%) than in the DVRd group (25.6%), and the fact that progressive disease was a common reason for treatment discontinuation, the review team noted that it was inappropriate to assume that patients for whom data for MRD negativity were missing were MRD-positive. The way missing data were handled may introduce bias and likely overestimate the benefit of DVRd for the overall MRD negativity rate. The attrition rate in the analysis of HRQoL measured using change from baseline in the EQ-5D-5L utility score at maintenance cycle 34 day 1 was notable, with only ███ ██ ███ █████%) patients contributing to the analysis. Specifically, a notably lower proportion of patients in the DVRd group (███ ██ ███ ████████ than the VRd group (███ ██ ███ ██████) were lost to follow-up at maintenance cycle 34 day 1 (approximately 40 months of treatment). Adverse events and disease progression were the common reasons for treatment discontinuation, these could contribute to missing data on HRQoL, and the disproportion of missing data between treatment groups would introduce bias in favour of the VRd group as the characteristics of patients who did not stay on treatment may differ from those who did. Moreover, no sensitivity analyses were conducted for the analysis of HRQoL.
As the PERSEUS trial is ongoing, results were only available from the interim analysis. At the time of the interim analysis, the median PFS and median OS were not reached in both treatment groups. While a treatment benefit and trend toward an improved OS with DVRd treatment was evident, an observation that is supported by improvements in MRD negativity, the longer-term treatment effect in terms of both median survival time and HR is unknown.
A multiple testing procedure was used to control the overall type I error for the primary end point of PFS and secondary end points of the rate of a CR or better, overall MRD negativity rate, and OS in the interim analysis. Many of the outcomes used in the PERSEUS trial (PFS, MRD negativity rate, OS, rate of a VGPR or better, DOR, and HRQoL) were identified as clinically important by patients and/or clinicians. However, because the rate of a VGPR or better, DOR, and HRQoL were not part of the statistical testing strategy and were not adjusted for multiple testing, the ability to draw conclusions from these results may be limited.
External Validity
The clinical experts commented that, in general, the trial eligibility criteria were standard but stricter than those in use in clinical practice. The trial included adults who were aged 70 years or younger, according to the clinical experts, and about 10% of patients who were aged between 70 and 75 years with good performance status (e.g., no major systemic illness) would be eligible for ASCT and therefore eligible for DVRd in clinical practice. However, a subgroup analysis found that there was no significant effect on PFS in patients aged 65 years or older, likely due to the small sample size. Further study is needed to assess the actual treatment effect among older patients. The clinical experts expressed a willingness to be flexible on ECOG PS scores given that patients with a poor ECOG PS at diagnosis would likely improve with treatment. Although meningeal involvement of MM is rare for patients with NDMM, the clinical experts stated they would still administer systemic therapy for those patients. Additionally, although patients with moderate or persistent asthma were excluded from the trial, the clinical experts would consider such patients eligible for DVRd in clinical practice. Overall, the patient population in the PERSEUS trial may not be representative of the patient profile in clinical practice.
The baseline characteristics of the PERSEUS trial may be indicative of the overrepresentation of patients with TE NEMM who were white (92.1%), and this represents an evidence gap in the study results’ generalizability. The clinical expert indicated that there is a more diversified patients’ population, including patients of other ethics groups, in their clinical practice compared to the patient population in the PERSEUS trial.
In the PERSEUS trial, consolidation therapy was part of the study treatment and 532 of 709 patients (75%) completed consolidation. This may not be reflective of clinical practice, given consolidation was not funded in all jurisdictions in Canada, according to the clinical experts. Because less than one-half of patients would receive brief consolidation in clinical practice, the study results may not be generalizable to clinical practice in Canada.
There were no study sites in Canada in the PERSEUS trial, which may also compromise the generalizability of the study results to clinical practice in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal PERSEUS trial identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:13,14
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The reference points for the certainty of evidence assessment for PFS, rate of a VGPR or better, overall MRD-negative rate (at 10-5), OS, DOR (CR or better), and harms were set according to the presence of an important effect based on thresholds agreed upon by the clinical experts consulted by the review team for this review. Safety and HRQoL were measured using EQ-5D-5L utility scores, and because there is no established MID and the clinical experts could not provide a threshold of important difference, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for DVRd versus VRd.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
Objectives for the Summary of Indirect Evidence
In the absence of head-to-head evidence comparing DVRd against other relevant comparator therapies used in the management of TE NDMM, the sponsor submitted 1 inverse probability of treatment-weighting analysis and 5 MAICs to indirectly compare the treatment effect of DVRd with other treatments in patients with TE NDMM.18
The inverse probability of treatment-weighting analysis assessed the efficacy of DVRd (the PERSEUS trial) with DVTd and VTd (CASSIOPEIA trial). The clinical experts consulted by CDA-AMC noted that DVTd and VTd regimens are not used in clinical practice in Canada and should not be considered relevant comparators of DVRd. This analysis was therefore not of interest to this review and will not be summarized.
The objective of this section is to summarize and critically appraise the methods and findings of the 5 MAICs.
Description of Indirect Treatment Comparison
In the absence of direct comparative evidence between DVRd and CyBorD, which is a relevant comparator, the sponsor submitted 2 unanchored MAICs of the efficacy of DVRd (in the form of IPD) and CyBorD (aggregate data) in patients with TE NDMM. This includes a comparison of the PERSEUS and GMMG-MM5 studies, which evaluated patients who received treatment from the induction phase through the maintenance phase (the full treatment sequence), and a comparison of the PERSEUS and VCAT studies, which evaluated patients who received induction treatment through consolidation treatment. Results from the PERSEUS versus GMMG-MM5 study were also used to inform the submitted pharmacoeconomic model. CyBorD was referred to as cyclophosphamide-bortezomib-dexamethasone (VCd) in the GMMG-MM5 and VCAT studies; these regimens were assumed by the sponsor to be therapeutically equivalent as they consist of identical components, although minor differences in dexamethasone dosing may occur between CyBorD and VCd across regions. This regimen will be referred to as CyBorD/VCd in the MAICs hereinafter.
The clinical experts consulted by CDA-AMC indicated that patients may benefit from the addition of daratumumab to lenalidomide (daratumumab-lenalidomide) regardless of the type of treatment regimen (DVRd, VRd, or CyBorD) received in the induction and consolidation phase. In the PERSEUS trial, no rerandomization occurred upon initiation of maintenance treatment, limiting the ability to assess the incremental benefit of adding daratumumab to the maintenance regimen consisting of lenalidomide alone. In an effort to address this gap in evidence, the sponsor submitted 3 unanchored MAICs (PERSEUS versus Myeloma XI; PERSEUS versus IFM 2005-02; PERSEUS versus CALGB 100104) that compared the efficacy of daratumumab-lenalidomide against lenalidomide as maintenance treatments based on IPD from the PERUSEUS trial and aggregate data from the comparator studies in patients with TE NDMM.
Both OS and PFS were assessed in the ITCs. A summary of the population, intervention, comparison, outcome of the MAICs is presented in Table 20.
Table 20: Overview of PICO for Indirect Treatment Comparisons
Study | Analysis method | Population | Intervention/comparator | Outcomes |
|---|---|---|---|---|
PERSEUS vs. GMMG-MM5 | Unanchored MAIC | TE NDMM | Full treatment sequence: DVRd vs. CyBorD/VCd | PFS OS |
PERSEUS vs. VCAT | Unanchored MAIC | TE NDMM | Induction and consolidation treatments: DVRd vs. CyBorD/VCd | PFS |
PERSEUS vs. Myeloma XI | Unanchored MAIC | TE NDMM | Maintenance treatment: daratumumab-lenalidomide vs. lenalidomide | PFS OS |
PERSEUS vs. IFM 2005-02 | Unanchored MAIC | TE NDMM | — | |
PERSEUS vs. CALGB 100104 | Unanchored MAIC | TE NDMM | — |
CyBorD/VCd = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; MAIC = matching-adjusted indirection comparison; NDMM = newly diagnosed multiple myeloma; OS = overall survival; PICO = population, intervention, comparison, outcome; PFS = progression-free survival; TE = transplant-eligible; vs. = versus
Source: MAIC Technical Report.18
Study Selection Methods
Study inclusion was informed by an SLR conducted by the sponsor to identify RCTs assessing the clinical efficacy and safety of drug regimens used in the induction through consolidation phases in addition to the maintenance phase for the treatment of TE NDMM. The study selection criteria and methods of the SLR are summarized in Table 21. The original SLR was conducted in 2018 using multiple literature databases (Embase, MEDLINE, and the Cochrane Library) and updated in November 2023. This SLR included studies published from January 1, 2000, to September 19, 2023. Study selection was conducted independently by 2 researchers. Data were extracted by a single reviewer and confirmed by a second reviewer, with a final quality check. A risk-of-bias assessment was conducted for all included comparator studies using the Cochrane Risk of Bias tool version 2.0. The reason for study exclusion of the SLR was documented.
Studies included in the SLR were grouped for feasibility assessment based on the phase of treatment. The 3 study groupings included full treatment sequence, induction through consolidation, and maintenance.
Indirect Treatment Comparison Analysis Methods
A summary of the analysis methods for the ITCs is shown in Table 22.
Feasibility Assessment
Studies were selected for inclusion in the ITCs based on results from a feasibility assessment, which included a review of study design, treatment characteristics (e.g., frequency, dosing, administration, and duration), population and patient characteristics (inclusion and exclusion criteria, baseline characteristics), comparability of characteristics that may be possible treatment-effect modifiers or prognostic variables, and outcome availability and outcome definitions.
Table 21: Study Selection Criteria and Methods for Indirect Treatment Comparisons
Characteristics | Indirect comparison |
|---|---|
Population | Patients with previously untreated MM who are eligible for transplant |
Intervention | Approved treatments, treatments used in routine care, or treatments under investigation provided as a single-drug or a combination treatment. These include but are not limited to the following:a
|
Comparator | Approved treatments, treatments used in routine care, or treatments under investigation provided as a single-drug or combination treatment. |
Outcome | Efficacy:
Safety:
Time point:
|
Study designs | RCTs (phase II and III) |
Exclusion criteriab | Population:
Intervention or comparator:
Time point:
Date limit:
|
Databases searched | Embase, MEDLINE, Cochrane Library (Cochrane Database of Systematic Reviews and Cochrane Central Register of Controlled Trials) |
Selection process | Articles screened independently by 2 researchers, with disagreements resolved by a third researcher |
Data extraction process | Data were extracted by a single reviewer and independently validated by a second reviewer, with disagreements resolved by a third researcher; a final quality check was conducted |
Quality assessment | Quality assessment (risk of bias) was performed using the Cochrane Risk of Bias tool version 2.0 |
AE = adverse event; ASCT = autologous stem cell transplant; CR = complete response; CyBorD/VCd = bortezomib-cyclophosphamide- dexamethasone; DOR = duration of response; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; DVTd = daratumumab- bortezomib-thalidomide-dexamethasone; IRd = ixazomib-lenalidomide-dexamethasone; Isa-VRd = isatuximab-bortezomib-lenalidomide-dexamethasone; KRd = carfilzomib-lenalidomide-dexamethasone; KTd = carfilzomib-thalidomide-dexamethasone; MM = multiple myeloma; MRD = minimal residual disease; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival on the next line of treatment; PR = partial response; RCT = randomized controlled trial; sCR = stringent complete response; SLR = systematic literature review; TTP = time to progression; VDd = bortezomib-doxorubicin-dexamethasone; VGPR = very good partial response; VRd = bortezomib-lenalidomide-dexamethasone; VTd = bortezomib-thalidomide-dexamethasone.
aThis SLR was conducted at the global level; as such, not all comparators denoted are relevant to the setting in Canada. Only those studies applicable to this review will be described herein.
bThis list does not include all exclusion criteria of the SLR protocol. Only key exclusion criteria of interest to this review are summarized in this table.
Sources: MAIC Technical Report,18 ITC Systematic Literature Review Report.85 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Analysis Methods
Based on the results from the feasibility assessment, the sponsor identified several limitations in the comparisons between the SLR-identified studies. The sponsor noted that the studies originated from various settings with a wide range of recruitment centres and that there were significant differences in inclusion and exclusion criteria, along with inadequate reporting of key treatment confounders, making comparisons difficult and potentially biased from the outset. Furthermore, there was considerable heterogeneity in baseline characteristics, including those that were identified by the sponsor as treatment-effect modifiers (ECOG PS, ISS, and types of myeloma). Differences were also identified in the treatment arms that could serve as anchors, particularly regarding the administration of lenalidomide, including variations in dosing schedules and dosages. Additionally, the sponsor noted that a lack of available studies comparing the same treatments or providing indirect evidence loops made fitting a random-effects model, such as an NMA, infeasible. Given these issues, the sponsor chose to use the unanchored MAIC method to compare DVRd with relevant comparators. A MAIC was conducted based on IPD from the index trial (PERSEUS) and aggregate data from the comparator studies (GMMG-MM5, VCAT, Myeloma XI, IFM 2005-02, and CALGB 100104). The covariates used for match adjustments were informed by a literature review and analyses of treatment-effect modifiers and prognostic factors using the data from the PERSEUS trial, along with input from the clinical experts.
Unanchored MAICs with RMST (PERSEUS versus GMMG-MM5 and PERSEUS versus VCAT):
Published KM curves were used to generate pseudo-IPD for the comparator study. To estimate the underlying IPD from the comparator’s published KM curves, the Guyot method,86 following the guidelines outlined by Aiello et al. (2023)87 on data preparation and analysis, was used. MAIC weights for the PERSEUS trial were constructed using the method proposed by Signorovitch et al. (2010).88 Balance between the 2 populations after weighting was assessed using the ESS and the distribution of weights.
In the base-case scenario comparing PFS (both analyses) and OS (the PERSEUS versus GMMG-MM5 comparison only), the variables included for weighting were median age, sex, ISS, and myeloma type (immunoglobin G) in the analysis of the PERSEUS versus GMMG-MM5 studies; and median age, aged 65 years and older, sex, ECOG PS, ISS, and cytogenetic risk in the analysis of the PERSEUS versus VCAT studies. No sensitivity analysis was conducted for the analysis of the PERSEUS versus GMMG-MM5 studies. A sensitivity analysis was conducted accounting for race as a covariate, in addition to the main analysis covariates for the PERSEUS trial versus the VCAT study analysis.
A dataset combining weighted IPD from the PERSEUS trial and the constructed IPD (setting weights for constructed observations equal to 1), was then used to fit a weighted Cox proportional hazards model with a binary treatment indicator (i.e., DVRd versus CyBorD/VCd). The estimated regression coefficient for the treatment indicator was used to represent the log of the HR for DVRd versus CyBorD/VCd, and its corresponding 95% CI was derived. Robust standard errors were estimated using the sandwich estimator.
A population-adjusted indirect comparison RMST difference analysis was conducted to investigate the comparative survival effectiveness at given survival milestones.
Unanchored MAICs with RMST (PERSEUS versus Myeloma XI; PERSEUS versus IFM 2005-02; PERSEUS versus CALGB 100104):
The covariates that were matched included median age, sex, cytogenetic risk, myeloma type, and MRD negativity at the beginning of the maintenance phase in the analysis of the PERSEUS versus Myeloma XI trials; and median age (of the comparator trials), sex, and myeloma type in the comparisons of PERSEUS versus IFM 2005-02 and PERSEUS versus CALGB 100104 trials. No sensitivity analysis was conducted for all studies. A MAIC restricted to data from the maintenance phase data for the PERSEUS trial arm of interest was used. Only baseline characteristics available at the start of maintenance of the PERSEUS trial were used in the MAIC. Given the nature of the data, the sponsor deemed the RMST to be the most appropriate method to summarize the differences in the survival curves.
Assessment of Model Fit
A validation of the reconstructed KM curves (using the Guyot method) and the original KM plots was conducted both visually and statistically by comparing the original and reconstructed curves, numbers at risk, and corresponding HRs in Cox models. The proportional hazards assumption was assessed for OS and PFS by inspecting the log-cumulative hazards curves and Schoenfeld residuals; in the MAICs, the Grambsch-Therneau test was also used.
Results
Summary of Included Studies
Fifteen, 25, and 23 unique studies assessing full treatment sequence, induction through consolidation treatment, and maintenance treatment, respectively, were identified by the SLR and were considered for inclusion in the ITCs. A feasibility assessment determined that 6 studies were used to inform the ITCs, including the 1 study of DVRd (PERSEUS), 2 studies of CyBorD/VCd (GMMG-MM5, full sequence treatment; and VCAT, induction through consolidation), and 3 studies of lenalidomide maintenance therapy (Myeloma XI, IFM 2005-02, and CALGB 100104). Reasons for study exclusion were not reported. Risk-of-bias assessments conducted by the sponsor suggested that the included studies were associated with a low risk of bias.
A summary of the homogeneity assessment for the MAICs is presented in Table 23 (DVRd versus CyBorD/VCd) and Table 24 (daratumumab-lenalidomide versus lenalidomide maintenance treatment).
Table 22: Indirect Treatment Comparison Analysis Methods
Methods | DVRd vs. CyBorD/VCd | Daratumumab-lenalidomide vs. lenalidomide maintenance | |||
|---|---|---|---|---|---|
PERSEUS trial vs. GMMG-MM5 trial | PERSEUS trial vs. VCAT trial | PERSEUS trial vs. Myeloma XI trial | PERSEUS trial vs. IFM 2005-02 trial | PERSEUS trial vs. CALGB 100104 trial | |
Analysis methods | Unanchored MAIC | ||||
Data sources | Main analysis:
| Main analysis:
| Main analysis:
| Main Analysis:
| Main analysis:
|
Assessment of model fit | A validation of the reconstructed KM curves (Guyot method) and the original KM plots was conducted both visually and statistically by comparing the original and reconstructed curves, numbers at risk, and corresponding HRs by running Cox models. The proportional hazards assumption was assessed for OS and PFS by inspecting the log-cumulative hazards curves and Schoenfeld residuals; in the MAICs, the Grambsch-Therneau tests were also used. | ||||
Outcomes | PFS and OS | PFS (Note: The VCAT trial did not report OS) | PFS and OS — RMST at 1, 2, and 3 years of maintenance | ||
Covariates used for weighting or match adjustments | Main analysis:
| Main analysis:
| Main analysis:
| Main analysis:
| Main analysis:
|
Sensitivity analyses | NR | A sensitivity analysis was conducted accounting for race as a covariate, in addition to the main analysis covariates. | NR | NR | NR |
CyBorD/VCd = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; IgG = immunoglobin G; IPD = individual patient data; ISS = International Staging System; KM = Kaplan-Meier; MAIC = matching-adjusted indirection comparison; MRD = minimal residual disease; NR = not reported; OS = overall survival; PFS = progression-free survival; RMST = restricted mean survival time; vs. = versus.
Sources: MAIC Technical Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
DVRd Versus CyBorD/VCd Comparisons (PERSEUS Versus GMMG-MM5 and PERSEUS Versus VCAT Studies)
Inclusion and exclusion criteria were generally similar between the PERSEUS trial and the comparator studies (GMMG-MM5 and VCAT); the studies enrolled patients who had NDMM, were TE, and were naive to MM treatment. The PERSEUS trial differed from the GMMG-MM5 trial in that the PERSEUS trial enrolled patients with an ECOG PS of 0 to 2, while the GMMG-MM5 trial enrolled patients with a WHO performance status of 0 to 2. The age criterion differed between the PERSEUS trial (aged 18 to 70 years) and the VCAT trial (aged 18 years or older). In terms of study intervention, the same treatment regimen was used throughout induction and consolidation treatment in the PERSEUS trial cohort (DVTd regimen) and the GMMG-MM5 trial cohort (VCd regimen), whereas in the VCAT trial cohort, VCd was used in the induction phase, followed by either bortezomib-thalidomide-prednisolone, or thalidomide-prednisolone in the consolidation phase. The definitions of study outcomes were similar between studies. The median duration of follow-up in the PERSEUS study cohort (approximately 48 months) was shorter than that in the GMMG-MM5 trial cohort (approximately 60 months) and longer than that in the VCAT trial cohort (approximately 23 months). Study locations also differed between the PERSEUS trial (Europe and Australia), the GMMG-MM5 trial (Germany), and the VCAT (Australia, Korea, and China) study. All were phase III, multicentre, open-label RCTs. There was temporal discordance in the study period between the PERSEUS trial (2019 to 2023) versus the GMMG-MM5 trial (2010 to 2012) and VCAT (2012 to 2016) trial.
Daratumumab-Lenalidomide Versus Lenalidomide Maintenance Treatment Comparisons (PERSEUS Versus Myeloma XI, PERSEUS Versus IFM 2005-02, and PERSEUS Versus CALGB 100104 Studies)
Patients included in the PERSEUS trial and the comparator studies similarly had treatment-naive NDMM at study entry, and had subsequently received induction and consolidation regimens, in addition to ASCT, before maintenance treatment. The induction and consolidation regimens differed between studies; patients in the PERSEUS trial received DVRd induction or consolidation treatments; TE patients in the Myeloma XI trial received regimens consisting of cyclophosphamide-thalidomide-dexamethasone, cyclophosphamide-lenalidomide-dexamethasone, or carfilzomib-cyclophosphamide-lenalidomide-dexamethasone; and patients in the IFM 2005-02 and CALGB 1001104 studies received treatment based on physician’s choice (regimens not specified). At study entry, the age of patients differed between the PERSEUS (18 to 70 years), Myeloma XI (18 years or older), and IFM 2005-02 (18 to 65 years) trials. As well, the selection criterion for ECOG PS differed (0 to 2 in the PERSEUS trial; 0 to 1 in the CALGB 100104 trial; and was not specified in the Myeloma XI and IFM 2005-02 trials). The definitions of study outcomes were similar between studies. Study location also differed between trials: PERSEUS was conducted in Europe and Australia; Myeloma XI in the UK; IFM 2005-02 in France, Belgium, and Switzerland; and CALGB 100104 in the US. All were phase III, multicentre, RCTs; the PERSEUS and Myeloma XI trials used an open-label design, while the IFM 2005-02 and CALGB100104 trials were double-blinded. There was temporal discordance in the study period between the PERSEUS trial (2019 to 2023) versus the Myeloma XI (2011 to 2017), IFM 2005-02 (2006 to 2008), and CALGB 100104 (2005 to 2009) trials.
Table 23: Assessment of Homogeneity for MAICs — DVRd Versus CyBorD/VCd
Characteristics | PERSEUS trial vs. GMMG-MM5 trial (full treatment sequence) | PERSEUS trial vs. VCAT trial (induction through consolidation) |
|---|---|---|
Treatment history | Both studies enrolled patients who were naive to MM treatment | Both studies enrolled patients who were naive to MM treatment |
Trial eligibility criteria |
|
|
Dosing of comparators | PERSEUS trial, DVRd arm:
GMMG-MM5 trial, CyBorD/VCd arm:
| PERSEUS trial, DVRd arm: dosing specified in the second column VCAT trial Induction: 3 cycles of CyBorD/VCd consisting of:
After ASCT, patients were randomized to (1:1) TP or VTP consolidation therapy:
|
Definitions of end points | Definitions of OS and PFS were similar between studies; tumour assessment criteria was based on IMWG criteria in both trials. | Definition of PFS was similar between studies; tumour assessment criteria was based on IMWG criteria in both trials. |
Duration of follow-up | PFS analysis
OS analysis
| PFS analysis
|
Study location | PERSEUS trial: Europe and Australia; GMMG-MM5 trial: Germany only | PERSEUS trial: Europe and Australia; VCAT trial: Australia, Korea, and China |
Study design | Both were phase III, multicentre, open-label RCTs | Both were phase III, multicentre, open-label RCTs |
Study period | PERSEUS trial: January 2019 to August 2023 GMMG-MM5 trial: July 2010 to October 2012 | PERSEUS trial: January 2019 to August 2023 VCAT trial: January 2012 to January 2016 |
ASCT = autologous stem cell transplant; CR = complete response; CyBorD/VCd = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IMWG = International Myeloma Working Group; MAIC = matching-adjusted indirection comparison; MM = multiple myeloma; NDMM = newly diagnosed multiple myeloma; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PO = orally; SC = subcutaneous; TE = transplant-eligible; TP = thalidomide-prednisone; vs. = versus; VTP = bortezomib-thalidomide-prednisone.
aTreatment discontinued if the patient had achieved a CR or better and MRD negativity that was sustained for 12 months and had been treated on maintenance for at least 24 months.
Sources: MAIC Technical Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Table 24: Assessment of Homogeneity for MAICs — Daratumumab-Lenalidomide Versus Lenalidomide (Maintenance)
Characteristics | PERSEUS trial vs. Myeloma XI trial | PERSEUS trial vs. IFM 2005-02 trial | PERSEUS trial vs. CALGB 100104 trial |
|---|---|---|---|
Treatment history | |||
At trial entry | Both studies enrolled patients who were naive to MM treatment | Both studies enrolled patients who were naive to MM treatment | Both studies enrolled patients who were naive to MM treatment |
Treatment received in the induction or consolidation phases | PERSEUS trial: DVRd Myeloma XI (TE patients) trial: CTD, CRD, or KCRD In both studies, patients had received ASCT in addition to the mentioned therapies (in TE patients only in the Myeloma XI trial). | PERSEUS trial: DVRd IFM 2005-02 trial: choice of treatment given at physician’s discretion In both studies, patients had received ASCT in addition to the mentioned therapies. | PERSEUS trial: DVRd CALGB 100104 trial: choice of treatment given at physician’s discretion In both studies, patients had received ASCT in addition to the mentioned therapies. |
Eligibility criteria | |||
At trial entry | Both studies enrolled patients who had NDMM; the PERSEUS trial included patients whose disease was TE only, the Myeloma XI trial included patients whose disease was both TE and non-TE but for the comparison, only patients whose disease was TE were included. Aged 18 to 70 years in the PERSEUS trial; 18 years or older in the Myeloma XI trial The PERSEUS trial enrolled patients with ECOG PS of 0 to 2; performance status not specified in the inclusion or exclusion criteria of the Myeloma XI trial. | Both studies enrolled patients who had TE NDMM Aged 18 to 70 years in the PERSEUS trial; 18 to 65 years in the IFM 2005-02 trial The PERSEUS trial enrolled patients with ECOG PS of 0 to 2; performance status not specified in the IFM 2005-02 trial inclusion and exclusion criteria. | Both studies enrolled patients who had TE NDMM. Patients were aged between 18 and 70 years in both studies. ECOG PS of 0 to 2 in the PERSEUS trial and 0 to 1 in the CALGB 100104 trial |
At maintenance phase baseline | PERSEUS trial: completed assigned induction, ASCT, and consolidation therapy Myeloma XI trial: completed assigned induction/consolidation therapy, had achieved at least a minimal response and received at least 100 mg/m2 melphalan if assigned | PERSEUS trial: completed assigned induction, ASCT, and consolidation therapy IFM 2005-02 trial: ≤ 6 months after transplant; no signs of progression after the transplant | PERSEUS trial: completed assigned induction, ASCT, and consolidation therapy CALGB 100104 trial: were within 12 months of initiation of induction therapy and had received at most 2 induction regimens; no disease progression during induction therapy |
Dosing of comparators | PERSEUS trial, daratumumab-lenalidomide arm in the maintenance phase: daratumumab every 4 weeks on day 1 of each cycle, in addition to lenalidomide 10 mg/day on days 1 to 28 of each 28-day cycle, until disease progression or unacceptable toxicity.a After 3 cycles of maintenance therapy, if well tolerated, the lenalidomide dose may be increased to 15 mg daily. Myeloma XI trial lenalidomide maintenance arm: lenalidomide 10 mg orally on days 1 through 21 of each 28-day cycle until progressive disease in the absence of toxicity | PERSEUS trial, daratumumab-lenalidomide arm: dosing supplied in second column IFM 2005-02 trial: 10 g daily, after 3 months, could be increased to 15 mg daily | PERSEUS trial, daratumumab-lenalidomide arm: dosing supplied in second column CALGB 100104 trial: 10 g daily, after 3 months, could be increased to 15 mg daily |
Definitions of end points | Definition of OS and PFS were similar between studies; tumour assessment criteria was based on IMWG criteria in both studies | Definition of PFS was similar between studies; tumour assessment criteria was based on IMWG criteria in both trials | Definition of OS and PFS were similar between studies; tumour assessment criteria was based on IMWG criteria in both studies |
Study location | PERSEUS trial: Europe and Australia; Myeloma XI trial: UK | PERSEUS trial: Europe and Australia; IFM 2005-02 trial: France, Belgium, and Switzerland | PERSEUS trial: Europe and Australia; CALGB 100104 trial: US |
Study design | Both were phase III, multicentre, open-label RCTs | Both were phase III, multicentre, RCTs. The PERSEUS trial was open-label, the IFM 2005-02 trial was double-blind. | Both studies are phase III, multicentre RCTs. The PERSEUS trial is open-label; the CALGB trial is double-blind. |
Study period | PERSEUS trial: January 2019 to August 2023 Myeloma XI trial: January 2011 to August 2017 | PERSEUS trial: January 2019 to August 2023 IFM 2005-02 trial: January 2006 to August 2008 | PERSEUS trial: January 2019 to August 2023 CALGB100104 trial: April 2005 to July 2009 |
ASCT = autologous stem cell transplant; CR = complete response; CRD = cyclophosphamide-lenalidomide-dexamethasone; CTD = cyclophosphamide-thalidomide-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IMWG = International Myeloma Working Group; KCRD = carfilzomib-cyclophosphamide-lenalidomide-dexamethasone; MAIC = matching-adjusted indirection comparison; MM = multiple myeloma; MRD = minimal residual disease; NDMM = newly diagnosed multiple myeloma; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; TE = transplant-eligible; vs. = versus.
aDiscontinue treatment if patient had achieved a CR or better and MRD negativity that was sustained for 12 months and had been treated on maintenance for at least 24 months.
Sources: MAIC Technical Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Results
The ESS for the DVRd group after match adjustment was █████% of the original sample size from the PERSEUS trial) in the comparison of the PERSEUS versus GMMG-MM5 studies, and ██████% of the original sample size in the PERSEUS trial) in the comparison of the PERSEUS versus VCAT studies. The ESS for daratumumab-lenalidomide group after match adjustment was ████, and █████ %, and ████% of the original sample size from the PERSEUS trial, respectively) in the comparisons of the PERSEUS versus Myeloma XI, PERSEUS versus IFM 2005-02, and PERSEUS versus CALGB 100104 studies, respectively.
Overall Survival
DVRd versus CyBorD/VCd: Following weighting, OS results of the DVRd versus CyBorD/VCd comparison were in favour of DVRd in the comparison of the PERSEUS trial versus GMMG-MM5 study assessing the full treatment sequence (HR = █████ ███ ███ ████ ██ █████ ██████). This outcome was not assessed in the PERSEUS trial versus VCAT study assessing induction treatment through consolidation treatment (Table 25).
Daratumumab-lenalidomide versus lenalidomide maintenance treatment: Following weighting, the difference in RMST between daratumumab-lenalidomide and lenalidomide with respect to OS at 3 years of maintenance therapy was ███ ██████ ████ ███ █████ ██████ ██ ████ ███████ in the comparisons of the PERSEUS and Myeloma XI trials, ████ ██████ ████ ███ ████ ██████ ██ ████ ███████ in the PERSEUS trial versus IFM 2005-02 study comparison, and ████ ██████ ████ ███ █████ ██████ ██ ████ ███████ in the comparison of the PERSEUS and CALGB 100104 trials (Table 26).
Progression-Free Survival
DVRd versus CyBorD/VCd: Following weighting, PFS results of the DVRd versus CyBorD/VCd comparison were in favour of DVRd in the PERSEUS trial versus the GMMG-MM5 study assessing the full treatment sequence (██ █ █████ ███ ██ ████ ██ █████ ████████) and the comparison of the PERSEUS trial versus the VCAT study assessing induction treatment through consolidation treatment (██ █ █████ ███ ███ ████ ██ █████ ████████) (Table 25) Results of the sensitivity analysis for the PERSEUS versus VCAT trials were consistent with the base case (HR = █████ ███ ███ ████ ██ █████ ████████).
Daratumumab-lenalidomide versus lenalidomide maintenance treatment: Following weighting, the difference in RMST between daratumumab-lenalidomide and lenalidomide with respect to PFS at 3 years of maintenance therapy was ███ ██████ ████ ███ ████ ██████ ██ ████ ██████) in the comparisons of the PERSEUS and Myeloma XI trials, ███ ██████ ████ ███ ████ ██████ ██ ████ ██████) in the PERSEUS trial versus IFM 2005-02 study comparison, and ████ ██████ ████ ███ ████ ██████ ██ ████ ███████ in the comparison of the PERSEUS and CALGB 100104 trials (Table 26).
Table 25: MAIC Results — DVRd vs. CyBorD (Full Sequence Treatment; Induction Through Consolidation)
Parameter | Overall survival | Progression-free survival | ||
|---|---|---|---|---|
Unweighted | Weighted | Unweighted | Weighted | |
PERSEUS trial vs. GMMG-MM5 trial (full treatment sequence): DVRd vs. CyBorD | ||||
N or ESS | N = 355 | ESS = ██████ | N = 355 | ESS = ██████ |
HR (95% CI) | ████ ████ | ████ ████ | ████ ███ | ████ ███ |
P value | ██████ | █████ | ██████ | ██████ |
PERSEUS trial vs. VCAT trial (induction through consolidation): DVRd vs. CyBorD | ||||
N or ESS | Not assessed | Not assessed | N = 354 | ESS = ██████ |
HR (95% CI) | Not assessed | Not assessed | ████ ████ | ████ ████ |
P value | Not assessed | Not assessed | ██████ | ██████ |
RMST after induction and consolidation (4 cycle of 28 days) for DVRd (SE) | Not assessed | Not assessed | █████ █ | █████ █ |
CI = confidence interval; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide- dexamethasone; ESS = effective sample size; HR = hazard ratio; MAIC = matching-adjusted indirection comparison; RMST = restricted mean survival time; SE = standard error; vs. = versus.
Sources: MAIC Technical Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Critical Appraisal of Indirect Treatment Comparisons
Study inclusion for the ITCs was informed by a sponsor-conducted SLR that used appropriate methods to reduce the risk of bias and error in study selection and data extraction (e.g., a priori patient, intervention, comparison, outcome selection criteria, duplicate independent reviewers for article screening, and data extraction by a single reviewer with validation by another reviewer). A risk-of-bias assessment was conducted by the sponsor using the Cochrane Risk of Bias 2.0 tool, in which all included studies were reported to have a low concern for bias. Study inclusion and choice of ITC method were informed by a feasibility assessment as well as an assessment of the proportional hazards assumption in each of the studies of interest. Of the 15, 25, and 23 identified studies assessing full treatment sequence, induction through consolidation treatment, and maintenance treatment, respectively, 1, 1, and 3 studies were selected as the comparator studies for inclusion into the MAICs. The reasons for study exclusion were not provided and, as such, there is a potential risk of selection bias, although the extent of such bias is unclear.
Table 26: MAIC Results — Daratumumab-Lenalidomide Versus Lenalidomide (Maintenance Therapy)
Parameter | Overall survival | Progression-free survival | ||||
|---|---|---|---|---|---|---|
Lenalidomide | Daratumumab-lenalidomide | Difference in RMST | Lenalidomide | Daratumumab-lenalidomide | Difference in RMST | |
PERSEUS versus Myeloma XI studies | ||||||
N or ESS | █ █ ███ | ███ █ ██████ | █ █ ███ | █ █ ███ | ███ █ ██████ | █ █ ███ |
RMST at 1 year of maintenance (95% CI) (months) | ████ ████ ███████ | ████ ████ ███████ | █ █ ███ | ████ ████ ███████ | ████ ████ ███████ | █ █ ███ |
RMST at 2 years of maintenance (95% CI) (months) | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ |
RMST at 3 years of maintenance (95% CI) (months) | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ |
PERSEUS versus IFM 2005-02 studies | ||||||
N or ESS | █ █ ███ | ███ █ ██████ | █ █ ███ | █ █ ███ | ███ █ ██████ | █ █ ███ |
RMST at 1 year of maintenance (95% CI) (months) | ████ ████ ███████ | ████ ████ ███████ | █ █ ███ | ████ ████ ██████ | ████ ████ ███████ | █ █ ███ |
RMST at 2 years of maintenance (95% CI) (months) | ████ ████ █████ | ████ ████ ██████ | █ █ ███ | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ |
RMST at 3 years of maintenance (95% CI) (months) | ████ ████ ██████ | ████ ████ █████ | █ █ ███ | ████ ████ █████ | ████ ████ ██████ | █ █ ███ |
PERSEUS versus CALGB 100104 studies | ||||||
N or ESS | █ █ ███ | ███ █ █████ | █ █ ███ | █ █ ███ | ███ █ █████ | █ █ ███ |
RMST at 1 year of maintenance (95% CI) (months) | ████ ████ █████ | ██ ████ ███████ | █ █ ███ | ████ ████ ███████ | ████ ████ ███████ | █ █ ███ |
RMST at 2 years of maintenance (95% CI) (months) | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ | ████ ████ █████ | ████ ████ █████ | █ █ ███ |
RMST at 3 years of maintenance (95% CI) (months) | ████ ████ ██████ | ████ ████ ██████ | █ █ ███ | ████ ████ █████ | ████ ████ ██████ | █ █ ███ |
CI = confidence interval; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ESS = effective sample size; MAIC = matching-adjusted indirection comparison; NA = not applicable; RMST = restricted mean survival time.
Sources: MAIC Technical Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
With respect to the comparisons of DVRd versus CyBorD/VCd (the PERSEUS versus GMMG-MM5 trials and the PERSEUS versus VCAT trials), all studies were phase III, open-label RCTs that enrolled patients who had TE NDMM and were naive to MM treatment. Different performance status scales were used to assess patients’ eligibility for study enrolment between the PERSEUS study (ECOG PS of 0 to 2) and the GMMG-MM5 study (WHO performance status of 0 to 2), although the clinical experts consulted by CDA-AMC noted that such a difference is expected to be minor and unlikely to have affected the study results. There was temporal discordance in the study period between the PERSEUS trial (2019 to 2023) versus both the GMMG-MM5 (2010 to 2012) and VCAT (2012 to 2016) trials. The clinical experts indicated that, in the past decade, significant changes have been made to the treatment approach in the second-line or later-line settings, such as the introduction of monoclonal antibodies in the relapse and refractory setting and availability of newer-generation proteasome inhibitors. This suggests that subsequent treatment use may have differed between the PERSEUS and comparator studies. Subsequent treatment use was not reported in the comparator trials. Potential differences in subsequent treatments could introduce bias to the OS results. Additionally, compared to the PERSEUS trial, the duration of follow-up was longer for the GMMG-MM5 study and shorter for the VCAT study, which could lead to overestimates of the relative effect (HR) of DVRd versus VRd in the comparison of the PERSEUS versus GMMG-MM5 trials and underestimates of the relative effect (HR) of DVRd versus VRd in the comparison of the PERSEUS versus VCAT trials.
With respect to the comparisons of daratumumab-lenalidomide versus lenalidomide maintenance therapy (PERSEUS versus Myeloma XI; PERSEUS versus IFM 2005-02; PERSEUS versus CALGB 100104), the included studies similarly enrolled patients who had treatment-naive NDMM at study entry, and had subsequently received induction and consolidation regimens, in addition to ASCT, before maintenance treatment. However, differences in the induction and consolidation regimens between the index (DVRd) and comparator studies (cyclophosphamide-thalidomide-dexamethasone, cyclophosphamide-lenalidomide-dexamethasone, or carfilzomib-cyclophosphamide-lenalidomide-dexamethasone in the Myeloma XI trial; physician’s choice in the IFM 2005-02 and CALGB 100104 trials) were identified. Such difference in treatment history between studies could not be adjusted for in the match-adjustment process. Additionally, eligibility criteria for ECOG PS differed (0 to 2 in the PERSEUS trial; 0 to 1 in the CALGB 100104 trial; and not specified in the Myeloma XI and IFM 2005-02 trials). There is also a potential risk of bias to the OS results due to likely differences in subsequent treatment use, given the temporal discordance in the study period between the PERSEUS trial (2019 to 2023) and the comparator studies (Myeloma XI, 2011 to 2017; IFM 2005-02, 2006 to 2008; CALGB 100104, 2005 to 2009).
A feasibility assessment was conducted by the sponsor to explore the possibility of conducting an NMA. The review team considered the sponsor’s justification for not choosing to conduct an NMA, due to notable heterogeneity between studies identified, to be reasonable. The choice to conduct an unanchored MAIC was justified by the lack of a common comparator across the included trials. Adjustment for all prognostic factors and treatment-effect modifiers is necessary for valid inference of the results from an unanchored MAIC, although this assumption may be implausible, as noted in the National Institute for Health and Care Excellence decision support unit technical support document.89 The covariates used for match adjustments were informed by a literature review and analyses of treatment-effect modifiers and prognostic factors using data from the PERSEUS trial and clinical expert input. Austin (2014) suggested that identification of covariates that were potentially prognostically important should be based on clinical expertise or an SLR, rather than statistical testing in the study sample.90 This was also noted in the technical support document.89 Of the sponsor-identified covariates, age, ECOG PS, ISS, baseline cytogenetic risk, and the presence of extramedullary plasmacytomas were noted to be of important prognostic value, according to the clinical experts consulted by CDA-AMC. These factors were adjusted for in the MAICs, except for ECOG PS and baseline cytogenetic risk, which were not adjusted for in the comparisons against the GMMG-MM5, Myeloma XI, IFM 2005-02, and CALGB 100104 studies. The mean age of the PERSEUS trial cohort was matched with the median age of the comparator trial cohorts. Because the mean and median ages of a study cohort are unlikely to be identical, the weighted results may be biased; however, the impact is likely to be small. As well, the presence of extramedullary plasmacytomas was not adjusted for in any of the MAICs. No adjustments were made in the base case of all MAICs for the following covariates identified by the sponsor to have potential prognostic or treatment-effect modifying effects: levels of lactate dehydrogenase, creatine clearance category, satisfied MM diagnostic criteria, the presence of extramedullary plasmacytomas, serum calcium levels, number of bone lesions, and platelet levels. No sensitivity analysis was performed to assess the potential impact on study results if these factors were adjusted for. Reasons for the lack of adjustment for these factors were not documented. MRD negativity at the beginning of maintenance treatment was a prognostic factor, according to clinical expert input received by CDA-AMC, and was acknowledged by the sponsor to be a predictor of long-term survival outcomes. Among the 3 comparisons of maintenance treatments, this factor was not adjusted for in the comparison of the PERSEUS and IFM 2005-02 trials or the PERSEUS and CALGB 100104 trials. A lack of adjustment for potential prognostic factors or treatment-effect modifiers suggests that the study results were likely biased due to confounding.
Treatment with VCd was the intervention of interest in the GMMG-MM5 and VCAT studies. The sponsor assumed VCd to be therapeutically equivalent to CyBorD, which is currently used in clinical practice in Canada. This assumption was considered to be reasonable by the clinical experts consulted by CDA-AMC. The experts identified some differences in the dosing of the component drugs (e.g., dosing interval and dosage of lenalidomide maintenance treatment) between the included studies versus clinical practice, but they were generally minor and not expected to have any major impact on study results. Of note, the treatment regimen assessed in the VCAT study does not align with those in clinical practice. In the VCAT study, following VCd induction therapy and ASCT, patients received either bortezomib-thalidomide-prednisone or thalidomide-prednisone consolidation therapy. According to the clinical experts consulted by CDA-AMC, CyBorD is rarely used in current practice to treat TE NDMM and if, on rare occasions, CyBorD is chosen as the induction therapy, no consolidation therapy is typically required following ASCT (only lenalidomide maintenance therapy would be given subsequently).
After the weighting process, the ESS of the study population of the PERSEUS trial was reduced by █████ in the analysis comparing the PERSEUS and GMMG-MM5 trials. A notable reduction in the ESS of the PERSEUS trial cohort was observed in the analysis of the PERSEUS versus VCAT trials (by ███████) and the comparisons of the maintenance treatments (by █████ ██ █████), which suggested poor population overlap between the PERSEUS trial and comparator studies. A significant reduction in sample size could contribute to imprecision, increasing uncertainty of the results. Reductions in ESS also suggest that results may be influenced by a subset of the sample in the trials that may not be representative of the full sample. Given that the populations from the MAICs were weighted to the comparator studies, the results may be driven by a subset of patients in the daratumumab group, limiting generalizability to the full population presented by the PERSEUS study.
The studies assessed OS and PFS, which are outcomes important to patients and clinicians. The comparative effects of daratumumab and comparator treatments on other efficacy outcomes of interest, including the MRD negativity rate, HRQoL, and harms, were not investigated and are therefore unknown.
Overall, results from the MAICs suggested that DVRd may improve PFS compared to CyBorD/VCd, although the magnitude of benefit is uncertain due to study limitations (heterogeneity in the duration of follow-up between studies and the lack of adjustment for important prognostic factors). The comparative evidence for OS was less certain; no firm conclusions could be drawn from the OS analyses due to potential bias resulting from the differential use of subsequent treatments. Similar limitations were identified in the MAICs comparing daratumumab-lenalidomide versus lenalidomide maintenance therapy. Heterogeneity in study populations (e.g., history of induction and consolidation treatment regimen, ECOG PS) between studies also added to the uncertainty of the daratumumab-lenalidomide versus lenalidomide comparisons. Given these limitations, no definitive conclusions could be drawn on the relative effects of daratumumab-lenalidomide versus lenalidomide in the patient population of interest (TE NDMM regardless of MRD status at the baseline of maintenance treatment).
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the review team.
The sponsor indicated that the PERSEUS trial was designed to evaluate the incremental benefit of adding daratumumab across induction and consolidation treatment phases with the VRd regimen and the maintenance treatment phase with lenalidomide alone.91 However, the sponsor highlighted a gap in evidence pertaining to the comparative efficacy in the maintenance phase for daratumumab plus lenalidomide (daratumumab-lenalidomide) versus lenalidomide alone, as patients in the DVRd arm of the PERSEUS trial would need to be rerandomized to receive either daratumumab-lenalidomide or lenalidomide alone. The sponsor also highlighted the importance of maintenance therapy in prolonged disease and improved patient outcomes.92 To address this evidence gap, the sponsor conducted 1 RCT, the AURIGA trial, to provide evidence regarding the clinical benefit of daratumumab when included as part of maintenance therapy. An overview of the noted evidence gap and study description and results for the AURIGA trial are presented in Table 27.
Table 27: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Direct comparative evidence to demonstrate the clinical benefit of daratumumab when included as part of maintenance therapy | Phase III open-label, active-controlled, multicentre RCT with 200 participants |
|
MRD = minimal residual disease; PFS = progression-free survival; RCT = randomized controlled trial.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.17
Description of Studies
The AURIGA trial (NCT03901963) is a phase III, open-label, active-controlled, multicentre RCT that evaluated the clinical benefit of adding daratumumab to maintenance treatment with lenalidomide among adult patients with TE NDMM who are MRD-positive after induction therapy and ASCT.19,21 The AURIGA trial randomized 200 patients across 52 sites in the US and Canada to receive either daratumumab-lenalidomide or lenalidomide monotherapy as maintenance therapy for after induction and ASCT for TE NDMM.19,21 Randomization was performed according to a 1:1 ratio and stratified by cytogenetic risk (high risk versus standard or unknown risk) according to investigator assessment.19,21 Details of the AURIGA trial are summarized in Table 28.
Table 28: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | AURIGA trial |
|---|---|
Design and population | |
Study design | Phase III, active-controlled, multicentre RCT |
Enrolled, N | 200 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Treatment is administered in 28-day cycles Daratumumab: 1,800 mg SC administered weekly for cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks for cycle 7 onward up to 36 cycles or until disease progression, unacceptable toxicity, or patient withdrawal Lenalidomide: 10 mg POa administered on days 1 to 28 per cycle, for up to 36 cycles or until disease progression, unacceptable toxicity, or patient withdrawal |
Comparator | Lenalidomide: 10 mg POa administered on days 1 to 28 per cycle, for up to 36 cycles or until disease progression, unacceptable toxicity, or patient withdrawal |
Outcomes | |
Primary end point | Percentage of participants with MRD-negative status as determined by NGS |
Secondary end points | PFS, overall and sustained MRD (10-5) negativity, percentage of participants achieving a CR or sCR, OS, HRQoL (EORTC QLQ-C30, EORTC QLQ-MY20, EQ-5D-5L), TEAEs |
Notes | |
Publications | None |
anti-HBc = Hepatitis B core antibody; ASCT = autologous stem cell transplant; COPD = chronic obstructive pulmonary disease; CR = complete response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-MY20 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire for Multiple Myeloma; FEV1 = forced expiratory volume in 1 second; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; HDT = high-dose therapy; HRQoL = health-related quality of life; IMWG = International Myeloma Working Group; MM = multiple myeloma; MRD = minimal residual disease; NDMM = newly diagnosed multiple myeloma; NGS = next-generation sequencing; RCT = randomized controlled trial; PCR = polymerase chain reaction; PFS = progression-free survival; PO = oral; OS = overall survival; SC = subcutaneous; sCR = stringent complete response; TEAE = treatment-emergent adverse event; VGPR = very good partial response.
aDose increases to 15 mg after 3 cycles of maintenance therapy if tolerated.
Sources: AURIGA Clinical Study Report19 and AURIGA Clinical Protocol.93
Populations
Key inclusion and exclusion criteria for the AURIGA trial are summarized in Table 28. Briefly, patients who were eligible were aged 18 to 79 years and had NDMM.19 Patients were treated with a minimum of 4 cycles of induction therapy and had received HDT and ASCT within 12 months of the start of induction therapy, with patients being within 6 months of ASCT on the date of randomization.19 Patients were also required to have a VGPR or better (according to IMWG 2016 criteria22) at the time of randomization, residual disease as defined by detectable MRD, and an ECOG PS of 0, 1, or 2.19 Patients were required to not have progressed on treatment for MM before screening and could not have been treated with any of the following: anti-CD38 therapy (e.g., daratumumab) or focal radiation therapy within 14 days before randomization (with the exception of palliative radiotherapy for symptomatic management but not on measurable extramedullary plasmacytoma, or plasmapheresis within 28 days of randomization).19 Patients with a history of malignancy other than MM were not eligible for inclusion unless all treatment of that malignancy was completed at least 2 years before consent and there was no evidence of disease before the date of randomization.19
Interventions
Details on interventions administered for the AURIGA trial are summarized in Table 28. Patients were randomized 1:1 to receive either maintenance therapy consisting of either daratumumab-lenalidomide or lenalidomide alone.19 Maintenance therapy for both arms was administered in 28-day cycles.19 Patients in both treatment groups received oral lenalidomide administered daily during each cycle.19 If tolerated, the dose of lenalidomide was increased from 10 mg to 15 mg after 3 cycles of maintenance therapy.19 Patients in the daratumumab-lenalidomide treatment group received daratumumab administered in the form of an 1,800 mg SC injection once weekly for cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks for cycle 7 and onward.19 Maintenance therapy continued for up to 36 cycles or until disease progression, unacceptable toxicity, or patient withdrawal.19
To reduce the risk of reactions related to the administration of daratumumab, patients receiving daratumumab-lenalidomide were given 20 mg of dexamethasone (or equivalent), antipyretics (oral paracetamol or acetaminophen [650 mg to 1,000 mg]), and antihistamines (oral or IV diphenhydramine [25 to 50 mg] or equivalent) before every SC injection of daratumumab.19 Patients were also administered low-dose (≤ 20 mg) oral methylprednisolone (or equivalent) the day after every SC injection of daratumumab to reduce the risk of delated administration-related reactions.19 No concomitant medications and/or treatments were specified with the administration of lenalidomide.19
Outcomes
Outcomes of the AURIGA trial are summarized in Table 28. The primary end point was the MRD conversion rate from baseline to 12 months after maintenance therapy as determined by NGS.19 This outcome was defined as the proportion of patients who had achieved MRD-negative status (at a threshold of 10-5) by 12 months after maintenance therapy before progressive disease or subsequent antimyeloma therapy.19 Patients who had achieved MRD-negative status on or after progressive disease or switched to subsequent antimyeloma therapy before progressive disease were not considered MRD-negative in the primary end-point analysis.19
The AURIGA trial used 5 secondary end points.
PFS, defined as the duration from the date of randomization to either progressive disease or death, whichever occurred first. Disease progression was determined according to IMWG 2016 criteria and a computer algorithm. For subjects who have not progressed and were alive at the cut-off date for analysis, data were censored at the last disease evaluation before the start of any subsequent antimyeloma therapy. Patients who were lost to follow-up were censored at the last disease assessment before patients were lost to follow-up. Patients who withdrew consent from the study before disease progression were censored at the last disease assessment. Patients without any postbaseline disease assessment after randomization were censored at the date of randomization.19
The overall MRD negativity conversion rate at any time after the date of randomization, defined as the proportion of patients with overall MRD-negative status at any time after the date of randomization.19
Sustained MRD negativity rate at 6 months or later, defined as the proportion of patients who have achieved MRD negativity status (at 10-5) in 2 bone marrow aspirate examinations a minimum of 6 months apart, without any examination showing MRD-positive status between assessments.19
OS, measured from the date of randomization to the date of the patient’s death due to any cause. If the patient was alive and vital status was unknown at the time of analysis, the patient’s data were censored at the date the patient was last known to be alive.19
Response rates including rate of CR and sCR, defined as the proportion of patients who achieved the respective responses before the first subsequent antimyeloma therapy in accordance with the IMWG criteria, during or after the study treatment.19
Assessments of HRQoL, functioning, and symptoms used the EORTC QLQ-C30, EORTC QLQ-MY20, and EQ-5D-5L. These outcomes were measured at baseline, 6 months, 12 months, 24 months, and 36 months.
The EORTC QLQ-C30 includes 30 items resulting in 5 functional scales (physical, role, emotional, cognitive, and social), 1 Global Health Status scale, 3 symptom scales (fatigue, nausea and vomiting, and pain), and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties). The item and scale scores are transformed to a 0-to-100 scale, in which a higher score for Global Health Status and functional scales represents a greater HRQoL and better functioning. However, a higher score for symptom scales indicates more (worse) symptoms.
The EORTC QLQ-MY20 is used alongside the EORTC QLQ-C30 to address issues of more relevance to patients with MM. The EORTC QLQ-MY20 consists of 20 items that make up 4 scales: disease symptoms, side effects of treatment, future perspective, and body image. Recall, response options, and interpretation are similar to those of the EORTC QLQ-C30.
The EQ-5D-5L is a 5-item questionnaire that assesses 5 domains, including mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression plus a visual analogue scale rating “health today,” with anchors at 0 (worst imaginable health state) and 100 (best imaginable health state). The scores for the 5 separate questions are categorical and cannot be analyzed as cardinal numbers. However, the scores for the 5 dimensions are used to compute a single utility score ranging from 0.0 to 1.0, representing the general health status of the individual.
Safety assessments included the rate of TEAEs (defined as adverse events that onset during the treatment phase or are a consequence of a pre-existing condition that has worsened since baseline), serious TEAEs, TEAEs leading to the discontinuation of treatment, and TEAEs leading to death.19
Statistical Analysis
The efficacy analysis was performed based on the ITT population, which included all patients who were randomized.19,93 Outcomes related to HRQoL were performed on the full analysis set population. Safety was evaluated for all patients who received at least 1 dose of randomized therapy.19,93 Continuous variables were summarized using descriptive statistics (e.g., mean, SD, and range).19,93 Categorical variables were summarized using frequency tables. For time-to-event variables, the KM method was used to produce descriptive summaries.19,93
The primary end point of the study was the MRD negativity conversion rate from baseline to 12 months after maintenance therapy.19 No interim analysis was planned for the primary end point.19,93 The primary analysis was performed after all participants who were randomized had completed 12 months of maintenance treatment, had disease progression, died, or had been discontinued or withdrawn from study treatment.19,93 The primary analysis was conducted at a 2-sided alpha level of 0.05.19,93 A single interim analysis was conducted for PFS at the time of analysis for the primary end point.19,93 A final analysis for PFS was expected to occur at the end of the study (i.e., 36 months after the last patient was randomized).19,93
The study sample size for the AURIGA trial was based on the primary end point of MRD negativity conversion rate.19,93 It was estimated that a sample size of approximately 214 participants (i.e., 107 per treatment group) would be required to demonstrate a 20% treatment difference in the MRD negativity conversion rate by the end of 12 months of maintenance, with a power of at least 85% and a 2-sided alpha of 0.05 using a continuity-corrected chi-square test.19,93 However, due to recruitment challenges, enrolment in the study ended after 200 patients (93% of the target sample size) had been randomized.19,93
The primary end point was compared between the treatment groups using a stratified CMH test with baseline cytogenetic risk (high versus standard or unknown) as the stratification factor.19,93 A Mantel-Haenszel OR along with its 2-sided 95% CI was calculated.19,93 To account for the possibility of subjects not adhering to the protocol visit schedule and evaluate the MRD negativity status, the postbaseline MRD records from randomization to 12 months were selected. Subjects with a missing or unevaluable MRD status were considered MRD-positive. Subjects who achieved MRD-negative status on or after progressive disease or switched to subsequent antimyeloma therapy before progressive disease were not considered MRD-negative in the primary end-point analysis.
Time-to-event outcomes, including PFS and OS, were analyzed using a stratified log-rank test for the comparison of the distribution between the 2 treatment groups.19,93 The KM method was used to estimate the distribution for each treatment.19,93 The analyses of the overall MRD negativity conversion rate at any time after randomization, durable MRD negativity rate, and response rates were conducted similarly to that for the primary end point. For key HRQoL outcomes, descriptive statistics at each time point and the change in score from baseline were summarized by treatment group. A mixed-effects model with repeated-measures analysis was conducted to estimate the change in HRQoL from baseline at each time point between the 2 treatment groups. Time to worsening and time to improvement of HRQoL were also derived, with a distribution-based method used to define improvement or worsening in scores. Time to improvement was summarized using descriptive statistics. Time to worsening was estimated using the KM method and an HR for daratumumab-lenalidomide relative to lenalidomide, and its associated 95% CI was calculated based on the stratified Cox proportional hazards model using the stratification factor at randomization.
Verbatim terms used by investigators in the electronic case report forms to identify adverse events were coded using the Medical Dictionary for Regulatory Activities.19,93 All reported adverse events were included in the analysis. For each adverse event, the percentage of patients who experienced at least 1 occurrence of the given event was summarized by treatment group.19,93
Results
Patient Disposition
At the time of clinical cut-off (April 4, 2024), a total of 200 patients were randomized to receive maintenance therapy with either daratumumab-lenalidomide or lenalidomide; 99 patients were randomized to the daratumumab-lenalidomide treatment group and 101 patients to the lenalidomide treatment group.19,21 Of those, 194 patients were treated: 96 with daratumumab-lenalidomide and 98 with lenalidomide. Among the 194 treated patients, 27 in the daratumumab-lenalidomide treatment group (28.1%) and 47 (48.0%) in the lenalidomide treatment group discontinued all study treatments during the study.19,21 The most common reason for discontinuation of daratumumab in the daratumumab-lenalidomide treatment group was disease progression.19,21 A higher rate of lenalidomide discontinuation was recorded in the lenalidomide treatment group compared to the daratumumab-lenalidomide treatment group, with the most common reason for discontinuation being disease progression.19,21 Of the 200 randomized patients, 7 patients in the daratumumab-lenalidomide treatment group (7.1%) and 15 patients in the lenalidomide treatment group (14.9%) terminated study participation prematurely.19,21
Baseline Characteristics
Baseline demographic and disease characteristics were generally balanced between the daratumumab-lenalidomide and lenalidomide treatment groups.19,21 The median age was 63.0 years for the daratumumab-lenalidomide treatment group and 62.0 years for the lenalidomide treatment group.19,21 Most patients had an ECOG PS of 0 or 1.19,21 The median number of induction cycles received by patients before screening was 5.0 for both treatment groups. Most patients received induction therapy containing lenalidomide and bortezomib for at least 2 cycles.19,21
Study randomization was carried out by stratifying the cytogenetic risk status (high versus standard or unknown) as assessed by investigator, resulting in ████% ███ ████% of patients with a high cytogenetic risk in the daratumumab-lenalidomide and lenalidomide treatment groups, respectively.19,21 Based on available local cytogenetic risk data at diagnosis (i.e., fluorescence in situ hybridization and/or karyotype), more patients had a high cytogenetic risk in the daratumumab-lenalidomide treatment group compared to those in the lenalidomide treatment group (23.9% versus 16.9%, respectively).19,21 The imbalance was driven by the difference in occurrence of the del(17p) chromosomal anomaly at diagnosis (daratumumab-lenalidomide: 14.1%; lenalidomide: 3.4%).19,21 When including the t(14;20) and gain/amp 1q anomalies, following the revised IMWG cytogenetic risk definition, the percentage of participants with a high cytogenetic risk at diagnosis was balanced between the treatment groups (daratumumab-lenalidomide: 34.4%; lenalidomide: 33.7%).19,21 This was due to higher percentage of gain/amp 1q participants in the lenalidomide arm compared with the daratumumab-lenalidomide treatment group.19,21
Exposure to Study Treatments
At the time of clinical cut-off, 85 patients (88.5%) in the daratumumab-lenalidomide treatment group and 77 patients (78.6%) in the lenalidomide arm had received 12 or more cycles of treatment.19,21 The median number of treatment cycles received was 33.0 among patients in the daratumumab-lenalidomide treatment group and 21.5 among patients in the lenalidomide treatment group.19,21 The median duration of study treatment was 30.65 months (range, 0.7 to 37.5 months) among patients receiving daratumumab-lenalidomide and 20.55 months (range, 0.0 to 37.7 months) among patients receiving lenalidomide.19,21
Efficacy
Primary End Point — MRD Conversion Rate From Baseline to 12 Months
At the clinical cut-off date of April 4, 2024, the MRD conversion rate from MRD positivity to MRD negativity (10-5) from baseline to 12 months since the initiation of maintenance therapy was 50.5% in the daratumumab-lenalidomide treatment group compared with 18.8% in the lenalidomide treatment group.23,24 The corresponding OR (daratumumab-lenalidomide versus lenalidomide) was 4.51 (95% CI, 2.37 to 8.57; P < 0.0001), which was statistically significant at the prespecified 2-sided alpha level of 0.05.23,24
Secondary End Point — Progression-Free Survival
At a median study follow-up time of 32.3 months, a total of 45 PFS events were observed. Of these 45 events, 19 were observed among the daratumumab-lenalidomide treatment group and 26 were observed in the lenalidomide treatment group.23,24 The corresponding HR was 0.53 (95% CI, 0.29 to 0.97), demonstrating a 47% reduction in the risk of disease progression or death in patients receiving daratumumab-lenalidomide compared to those receiving lenalidomide.23,24 The corresponding P value of 0.0361 did not cross the stopping boundary of 0.015 for this interim analysis of PFS. A final analysis of PFS is expected to be conducted at the end of the study.23,24 The estimated 30-month PFS rates were 82.7% and 66.4% for the daratumumab-lenalidomide and lenalidomide treatment groups, respectively.23,24
Secondary End Point — Overall MRD (10-5) Negativity Conversion Rates at Any Time
The overall MRD (10-5) negativity conversion rate from baseline throughout the study treatment period was higher in the daratumumab-lenalidomide treatment group compared to the lenalidomide treatment group (60.6% versus 27.7%), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 4.12 (95% CI, 2.26 to 7.52) and a P value of less than 0.0001.23,24
Secondary End Point — Sustained MRD (10-5) Negativity Rates
The sustained MRD negativity rate at 6 months or later was greater in the daratumumab-lenalidomide treatment group compared with the lenalidomide treatment group (35.4% versus 13.9%, respectively), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 3.40 (95% CI, 1.69 to 6.83) and a P value of 0.0005.23,24 Similarly, the sustained MRD negativity rate at 12 months or later was higher in the daratumumab-lenalidomide treatment group compared with lenalidomide treatment group (17.2% versus 5.0%, respectively), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 4.08 (95% CI, 1.43 to 11.62) and a P value of 0.0065.23,24
Secondary End Point — Overall Survival
At a median study follow-up time of 32.3 months, 15 OS events were observed.23,24 Of these 15 events, 5 were observed in the daratumumab-lenalidomide treatment group and 9 were observed in the lenalidomide treatment group.23,24 The median OS was not reached for either treatment group.23,24 The estimated 30-month OS rates were 94.6% and 91% for the daratumumab-lenalidomide and lenalidomide treatment groups, respectively.23,24
Secondary End Point — Rates of CR or Better
The overall rate of a CR or better (i.e., CR plus sCR) under the IMWG criteria was higher in the daratumumab-lenalidomide treatment group compared with the lenalidomide treatment group (75.8%; 95% CI, 66.1% to 83.8% versus 61.4%; 95% CI, 51.2% to 70.9%), with a corresponding OR (daratumumab-lenalidomide versus lenalidomide) of 2.00 (95% CI, 1.08 to 3.69) and a P value of 0.0255.23,24
Secondary End Point — HRQoL
Overall, there was no difference in HRQoL, symptoms, and functioning between the daratumumab-lenalidomide and lenalidomide treatment groups and no decline in the HRQoL with the addition of daratumumab to maintenance therapy with lenalidomide.
████████ ██████ ███ ███ ███ ████████ ██ ███ █████ ███████ ████ ██████████ ███████ █████████ ██████ ███ ████ █████████ ████ ███ ████████ ██ ███ ██████ █████████ █████ ███ ██ ██████████ ██ ███ ██ ████ ██████ ██ ████ ████████ ████ ████████ ██ ███ ██████████ ██████ ████████ ██ ███████████ █████ ██ ███ █ ████ ███████ ██ █████ ████████ ██ ████████ ███ ████ ███████ ████ ████ ████ ███ ████ ██ █████ ██ █ ████ ███ █████ █████ ██████████ █████████ ██████ ███ ███ ██████████ ███ ███████ █████████ ██ ███ █████ ███████ ████ ████ ██████████ ███████ █████████ ███████ ████████ ██████ ██ ███ █████████ ████ ██████████ ███████ █████████ ███████ ███ ██ ████ ███████ ████ ████████ ██████ ██████ █████████ ██ ████ ██████ ████ ███ █████████ █████████ ████████ ███████████ ███ █████████ ████ ████████ ██ █████ ██ ███ █ █████ ███ ████ ███ █████ █████ ██ ███ ████ ███ █████ █████ ███ ██ ██ ████ ███████ ████ ████████ ██████ ██████ █████████ ██ ████ ███ ████ █████████ ██████ ███████ █████ ██ ███ █ █████ ███ ████ ███ █████ ██████ ██ ████ ████ ███ ███ ███ ██████████ ███ ███ ███████ █████████ ███ ██ ████ ███████ ████ ████████ ████ ██████ ████ █████████████ █████████ ██ █████ ███ ████ █████████ ██████ ████ ████████ ███████ █████ ██ ███ █ █████ ███ ████ ███ █████ ██████ ██ ███ ████ ███ █████ █████ ██████████ ██ ███████████ ███████████ ████ ████████ ███████ █████████ ██████ ██ ███ ████ ██ ███████████ ██ ██████████ █████████ ██████ ██ ███ █████ ████████ ███████ ███████ █████ ████ ██████████ ██████ █████████ ███████ ███ ██ ████ ██████ ████ ████████ ██ ███ █████ ████████ ███████ ███████ █████ ██████ █████████ ████████ ████ ████ ███ ████ ███████████ ███████ ██████ █████████ ██████ ██ ███ █ ██ █████ ██ █████ ████ ████ ███ █████ ████ ██ ██ ████ ███ █████ ███ ██████████ ██ ███████████ ████ ████████ ███████ █████████ ██████ ██ ███ ████ ██ ███████████ ██ ███████████████████ ██████ ██ ███ ████████ ███████ █████ ███ ████████ ███ ████ ██████████ ███████ █████████ ███████ ███ ██ ██ ████ ██████ ████ ████████ ███████ ███████████ █████ ██ ███ ███ ████████ ███████ █████ ███ ███ ████████ ███ ████ ██████ ███ ███ ██████ ████ ██████████ ██ ████████ ██████ █████████ ███████
Harms
The incidence of TEAEs was 99% for both of the daratumumab-lenalidomide and lenalidomide treatment groups.23,24 The most frequently reported TEAEs (those with an incidence of 30% or higher in either arm) were neutropenia (daratumumab-lenalidomide: 64.6%; lenalidomide: 61.2%), diarrhea (daratumumab-lenalidomide: 61.5%; lenalidomide: 55.1%), and fatigue (daratumumab-lenalidomide: 45.8%; lenalidomide: 46.9%). Injection-related reactions were reported in 13.5% of patients in the daratumumab-lenalidomide treatment group.23,24 Compared with the lenalidomide treatment group, patients in the daratumumab-lenalidomide treatment group experienced higher incidences of grade 3 or 4 TEAEs (74.0% versus 67.3%) and serious TEAEs (30.2% versus 22.4%).23,24 Rates of discontinuation due to TEAEs were also higher in the daratumumab-lenalidomide treatment group compared to the lenalidomide treatment group (12.5% versus 7.1%).23,24 Three deaths related to TEAEs were reported; 2 in the daratumumab-lenalidomide treatment group and 1 in the lenalidomide treatment group.23,24
Critical Appraisal
Internal Validity
The AURIGA trial demonstrated notable strengths, such as patient stratification by cytogenetic risk before randomization and the use of an ITT analysis to account for all patients who were randomized.19 However, several key limitations should be considered when interpreting the results. For example, the AURIGA trial was open-label, which indicates that patients and investigators were not blinded to treatment. The trial protocol suggests evidence of independent outcome assessment, such as the establishment of an independent data-monitoring committee to review safety data and use of computerized algorithms to measure key efficacy outcomes (e.g., MRD negativity conversion rate, PFS, and response rates). However, the lack of blinding of patients may have contributed to performance bias in results for patient-reported outcomes. Moreover, although baseline characteristics were generally balanced between the 2 treatment groups, a larger proportion of patients in the daratumumab-lenalidomide treatment group had a high cytogenetic risk according to available local cytogenetic risk data at diagnosis (daratumumab-lenalidomide: 23.9%; lenalidomide: 16.9%).19 The sponsor study report noted that any potential treatment effect due to this imbalance would have favoured the lenalidomide treatment group. Finally, for the analysis of the primary end point, subjects with missing or unevaluable MRD status were considered to be MRD-positive. Given that a larger proportion of patients in the lenalidomide treatment group dropped out of the study, the imputation of all missing subjects as having MRD-positive status would have likely biased results in favour of the daratumumab-lenalidomide treatment group.
External Validity
The AURIGA trial recruited patients across 52 treatment centres in the US and Canada.19 However, the trial results do not explicitly state the proportion of patients living in Canada or provide a subgroup analysis of these patients. Although patients with MM living in Canada and those living in the US may share some characteristics, it is unclear how representative the findings of the trial are to patients with MM living in Canada and being managed in clinical practice. Moreover, the trial only included patients who were MRD-positive and who achieved a VGPR or better after transplant.19 Clinical experts consulted by CDA-AMC agreed that, in clinical practice, most patients starting maintenance therapy exhibit a VGPR or better after transplant. However, the experts also indicated that patients who achieve a PR or better are able to proceed with maintenance therapy as long as they do not show signs of progressive disease. The applicability of findings from the trial are therefore limited for this subset of patients.
In addition, the AURIGA trial excluded patients who received daratumumab or other anti-CD38 therapies.19 The generalizability of the trial results may be limited for patients receiving daratumumab as a part of induction and consolidation therapy in clinical practice. Because patients receiving daratumumab-lenalidomide as maintenance therapy in the PERSEUS trial would have received daratumumab as a part of induction and consolidation therapy,91 the results of the AURIGA trial may not be fully generalizable to patients in the PERSEUS trial. This is important because the AURIGA trial was submitted to address the lack of evidence pertaining to efficacy of daratumumab-lenalidomide as maintenance therapy from the PERSEUS trial. The results of the PERSEUS and AURIGA trials showed similar trends in terms of improvement in PFS, MRD negativity, and response associated with the addition of daratumumab to their respective regimens.
Discussion
Summary of Available Evidence
One pivotal trial (PERSEUS) was included in the sponsor-submitted systematic review. The PERSEUS trial was a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of DVRd (n = 355) compared to VRd (n = 354) in patients with TE NDMM. The trial enrolled adults with newly diagnosed and documented MM who were eligible for HDT and ASCT and had an ECOG PS of 0 to 2. Randomization was conducted using an interactive web response system and stratified by ISS at screening (I versus II versus III) and cytogenetics (standard risk versus high risk). The demographic characteristics were balanced between treatment groups. The median age of all patients was 60.0 years with a range of 31 to 70 years. Most patients were male (58.7%) (female: 41.3%) and white (92.1%) (1.4% of patients were Asian, 1.3% of patients were Black, 0.6% of patients were Native Hawaiian or other Pacific Islander, and 0.4% of patients were American Indian or Alaska Native). At baseline, most patients (63.6%) had an ECOG PS of 0. About one-half (51.4%) of patients had disease that was ISS stage I, and approximately one-fifth (21.7%) had high-risk cytogenetics such as a del(17p), t(4;14), or t(14;16) chromosomal anomaly. The primary objective of the PERSEUS trial was to evaluate the efficacy of DVRd compared to VRd in patients with TE NDMM in prolonging PFS. Secondary and other outcomes included the rate of a VGPR or better, MRD negativity rate, OS, DOR (CR or better), and HRQoL assessments. The study was funded by the European Myeloma Network in collaboration with Janssen Research & Development.
In an effort to address the absence of direct comparative evidence between DVRd versus CyBorD, the sponsor conducted 2 unanchored MAICs (PERSEUS versus GMMG-MM5 and PERSEUS versus VCAT) that indirectly compared OS and PFS for DVRd with CyBorD in patients with TE NDMM.
The sponsor also noted that, in the PERSEUS trial, no rerandomization occurred upon initiation of maintenance treatment to minimize confounding, limiting the ability to assess the incremental benefit of adding daratumumab to the maintenance regimen of lenalidomide alone. To address this gap in evidence, the sponsor submitted 1 phase III, open-label, RCT (AURIGA) and 3 unanchored MAICs (PERSEUS versus Myeloma XI; PERSEUS versus IFM 2005-02; PERSEUS versus CALGB 100104) assessing the clinical benefits of daratumumab-lenalidomide versus lenalidomide maintenance therapy in adult patients with TE NDMM following completion of induction therapy and ASCT. The AURIGA trial enrolled patients who were MRD-positive at the baseline of maintenance therapy, whereas studies informing MAICs included patients regardless of MRD status at the baseline of maintenance therapy. Outcomes assessed in the AURIGA trial included the MRD conversion rate from baseline to 12 months after maintenance therapy with a primary end point of PFS, overall MRD negativity conversion rate at any time after the date of randomization, OS, rate of a CR or better, and HRQoL (secondary end points). OS and PFS were outcomes of interest in the MAICs.
Interpretation of Results
Efficacy
Based on results of the interim analysis of the PERSUS trial (data cut-off: August 1, 2023), treatment with DVRd resulted in a statistically significant and clinically meaningful improvement in the primary end point of PFS compared with VRd for patients with TE NDMM (HR = 0.42; 95% CI, 0.30 to 0.59; P < 0.0001), and the between-group difference in OS probability at 48 months was 16.6% (95% CI, 9.9% to 23.4%). Generally, the improvements observed in PFS, rate of VGPR or better, overall MRD negativity rate, and DOR (CR or better) were considered clinically meaningful according to the clinical experts consulted for this review. The median PFS was not reached in either of the treatment groups and could not be interpreted at the time of the interim analysis. The clinical experts stated that the durations of follow-up (median: █████ months for DVRd, █████ months for VRd) in the interim analysis were adequate for the assessment of PFS; however, longer-term disease progression and survival data are required to assess the overall OS and PFS benefits (i.e., both median survival and relative risk reduction) and absolute OS and PFS benefits beyond 48 months. Overall, there is high certainty that DVRd results in a clinically important increase in the probability of patients being alive and progression-free when compared with VRd at 48 months based on the 5% threshold suggested by the clinical experts.
Sensitivity analyses, including investigator assessments, were conducted for PFS and the rate of a CR or better to assess the robustness of the results. Overall, the results were consistent with the primary analysis. Subgroup analyses of PFS were consistent with the primary analysis across all prespecified subgroups, except for the subgroup of patients aged 65 years or older (HR = 0.97; 95% CI, 0.52 to 1.81). This subgroup represented approximately 25% of the study population in the primary analysis (181 patients of 709 total), with a low number of events (39), and an imbalance of patients with high cytogenetic risk between treatment groups (25.5% versus 19.5% for DVRd versus VRd, respectively). The clinical experts confirmed that an imbalance in patients’ cytogenetic profile could be an important contributor to the observed result. The review team noted that, although the subgroup analyses of the primary end point were prespecified, the PERSEUS trial was not powered to detect statistically significant differences between DVRd and VRd treatment groups, making it impossible to draw definitive conclusions from the subgroup analyses.
There were imbalances in treatment exposure, preinjections, concomitant medications, and subsequent treatments. Specifically, VRd exposures throughout the entire treatment period appeared to be slightly higher in the DVRd group than in the VRd group, particularly for lenalidomide (total dose: ████ mg versus ████ mg, respectively). This may be because, for unknown reasons, a consistently higher proportion of patients completed induction (95% versus 90% for DVRd versus VRd, respectively), mobilization (91% versus 87%), ASCT (87% versus 83%), and consolidation (76% versus 73%) and received maintenance treatment (91% versus 85%) in the DVRd group compared with the VRd group. Collectively, these could have biased the results in favour of DVRd. A total of ███ patients in the DVRd group received preadministration medications before receiving daratumumab, whereas ██ patients in the VRd group received preinjections, and the use of corticosteroids may have had an antimyeloma effect, according to clinical experts. Notable differences in the use of concomitant treatments were reported in the use of immunostimulants (█████ ███████%, DVRd versus VRd, respectively), which may not have had a significant impact on PFS or OS but may have affected the HRQoL outcomes.
More patients in the VRd group received subsequent antimyeloma treatments, such as systematic antineoplastic agents (25.6%), than in the DVRd group (8.8%). This imbalanced use of antimyeloma treatments between treatment groups would bias the treatment effect, such as on OS, against the DVRd group (daratumumab with lenalidomide versus lenalidomide alone in the maintenance phase of the trial), and this could help explain why OS differed significantly as measured by HR and 95% CI, despite a significant difference on PFS between the 2 arms.
The PERSUS trial showed a moderate to high certainty of evidence that, when compared with VRd, DVRd likely results in a clinically important increase in the rate of a VGPR or better and a clinically important increase in the overall MRD negativity rate. Studies have shown that a VGPR or better and MRD negativity are surrogate outcomes positively correlated with OS.75-78 Despite the reported improvements in the rate of a VGPR or better and MRD negativity rate, the degree to which the observed benefits in the trial in terms of these 2 outcomes could be translated to an improvement in OS remains uncertain, as the median OS was not reached in either treatment group and, therefore was not interpretable at the time of the interim analysis. In addition, because relatively few patients received subsequent antimyeloma treatment in the DVRd group compared with the VRd group, the OS results could be biased against the DVRd group by the subsequent antimyeloma treatments. Overall, there is moderate certainty that DVRd likely resulted in a little to no difference in OS when compared with VRd at 48 months, based on the 5% threshold suggested by the clinical experts.
In the PERSEUS trial, the median DOR (measured as a CR or better) was not reached in either treatment group and was not interpretable at the time of the interim analysis. Despite the high probabilities of remaining in response (CR or sCR) at 42 months, it remains uncertain how long the treatment response could be sustained. █████████ ██ ███ ████████ ███ ███ █████ ██████ ███ ███████████ ██ █████████ ██ ██ ██ ██████ ████████ ███ █████████████ ███ ██ ████ ███ ████ ██ ██ ███ ██ ██████ ███████ ██ █████ ████████ ███████████ ██ █████ ███ ██ ███████ ████████████ ████████ ██████ ████ ████ ████████ ███ ████████ █████████ ██ ███████ █████████ █████████ ███ ███ ███ ██████ ███ ███████████ █████ ███ ██ █████████ ██ █████ ██ ███████ ███ ██████ ██ ████████ ██ ████ █████████ ████ ██ ██ █████ ██ ██ █ ██ █████ ███ █████ ███████ ██ █ █████████████ ███████████ ██ █████ ███ ███ ███████ ████████ █████████ ██ ███ ███ ██ ███████ ██ ██ ██████ ███████ ██ ██ ██████. There was therefore high uncertainty on the sustainability of treatment benefit from DVRd over VRd given the limited evidence available beyond 42 months.
A treatment benefit of DVRd in comparison with VRd was not observed in HRQoL, measured as change from baseline in EQ-5D-5L utility score at maintenance cycle 34 (approximately 40 months of treatment). However, a high proportion of data were missing. The number of patients contributing to the analysis was ███ out of ███ patients in the DVRd group and ███ out of ███ patients in the VRd group at maintenance cycle 34.
Patients aged 70 years or older were excluded from the PERSEUS trial; however, the clinical experts noted that a small proportion (about 10%) of patients with TE NDMM who are aged between 70 and 75 years and have good performance status would be potential candidates for DVRd in clinical practice. Moreover, the PERSEUS trial included predominantly (more than 90%) patients who were white. In addition, the PERSEUS trial had no study site in Canada. These limitations may compromise the generalizability of the study results to the clinical practice in Canada.
Evidence from the AURIGA trial suggests that, when compared to lenalidomide alone, the addition of daratumumab to lenalidomide as maintenance therapy was associated with improvements in the MRD conversion rate from baseline to 12 months after maintenance therapy (primary end point), PFS, overall and sustained MRD negativity rates, and the rate of a CR or better. At a median study follow-up time of 32.3 months, median OS was not reached for either treatment group. Five OS events were observed in the daratumumab-lenalidomide treatment group and 9 were observed in the lenalidomide treatment group.23,24 However, the OS benefit remains unclear, given that the difference in OS between the 2 groups was small in magnitude.
Moreover, no differences in HRQoL, symptoms, and functioning between the daratumumab-lenalidomide and lenalidomide treatment groups and no detriment of HRQoL with the addition of daratumumab to maintenance therapy with lenalidomide were seen in the AURIGA trial. However, these results may need to be interpreted with caution due to limitations of the trial. These limitations included its open-label study design, an imbalance in the proportion of patients with a high cytogenetic risk according to available local cytogenetic risk data at diagnosis, and handling of patients with missing or unevaluable data regarding MRD status in the primary end-point analysis. Moreover, it is unclear how representative the findings are to patients with MM living in Canada and being managed in clinical practice. The generalizability of results of the AURIGA trial may be limited for patients who initiated maintenance therapy after achieving a PR and for patients who received daratumumab as a part of induction and consolidation therapy in clinical practice.
Indirect evidence from 3 MAICs (PERSEUS versus Myeloma XI; PERSEUS versus IFM 2005-02; PERSEUS versus CALGB 100104) suggested that the addition of daratumumab to lenalidomide maintenance therapy in patients with TE NDMM (regardless of MRD status at the baseline of maintenance therapy) may improve PFS but there was insufficient evidence to show a difference in OS. Several important study limitations were identified, including heterogeneity in study populations (e.g., history of induction and consolidation treatment regimen, ECOG PS), temporal discordance in study period (which was suggestive of potential differences in subsequent treatment use), and the lack of adjustment for important prognostic factors for maintenance treatment (e.g., MRD negativity rate at the baseline of maintenance therapy, cytogenetic risk). These limitations preclude a firm conclusion on the treatment effects of daratumumab-lenalidomide versus lenalidomide maintenance therapy in patients who had TE NDMM (regardless of MRD status at the baseline of maintenance therapy).
Indirect evidence from the PERSEUS versus GMMG-MM5 trial comparison suggested that DVRd, compared with CyBorD, was associated with improved PFS in patients with TE NDMM who received full treatment sequence (i.e., induction therapy through maintenance therapy). The PFS benefit of DVRd relative to CyBorD in the induction through consolidation treatment phases was similarly observed in the PERSEUS versus VCAT MAIC. However, the magnitude of PFS benefit is uncertain due to limitations of the studies, including differences in the duration of follow-up between studies and inability to adjust for potential prognostic factors (which may introduce unmeasurable confounding in the relative treatment-effect estimates). Furthermore, there was a risk of bias in the OS results due to temporal discordance in study period between included studies, during which major changes in subsequent treatment pattern occurred. As such, no firm conclusions could be made on the comparative OS benefit of DVRd versus CyBorD.
Harms
Generally, no new safety signals for DVRd were identified in the PERSEUS trial. Increased incidences of infections, gastrointestinal disorders, and cytopenia were observed in the DVRd group. The clinical experts commented that, although the safety profile of DVRd was similar to that of VRd, added toxicities due to the addition of daratumumab would be expected in clinical practice. Given the infections, gastrointestinal disorders, and cytopenia commonly reported in both the DVRd and VRd treatment groups, the clinical experts did not anticipate that there would be an increase in disease management burden. The review team noted that, because more patients in the DVRd group than the VRd group received immune sera and immunoglobulins (which could reduce the frequency of adverse events such as infections), the results of infections may be biased in favour of the DVRd group. Inputs from patient and clinician groups emphasized concerns on stem cell collection issues and vision problems due to DVRd. The clinical experts indicated that, although stem cell yields may be lower in patients who received daratumumab, the impact would be minimal as an adequate amount of stem cells can be collected from those patients without a significant difference in the need for plerixafor. The clinical experts noted that vision problems may be related to the use of dexamethasone. The comparative harms between DVRd and CyBorD were unknown as no such evidence was submitted.
Conclusion
One phase III, ongoing open-label, active-controlled RCT (PERSEUS) comparing DVRd with VRd in treating patients with NDMM who are eligible for ASCT were included in this review. Although not currently funded for first-line treatment of patients with TE NDMM, there are increasing data from both patients whose disease is TE or transplant-noneligible that daratumumab-based induction and maintenance approaches may yield higher and deeper clinical responses Results from the PERSEUS trial demonstrated that, when compared to VRd, the addition of daratumumab to VRd results in a clinically significant benefit in PFS. The treatment benefit with DVRd was also consistently present in the rate of a VGPR or better and overall MRD-negative rate. However, treatment benefits in OS and DOR (for a CR or better) were uncertain as the median OS and median DOR were not reached in either study group in the interim analysis. Treatment with DVRd did not produce an HRQoL benefit. The results from the pivotal trial showed that, compared with VRd, DVRd results in little to no difference in the change from baseline in EQ-5D-5L utility scores.
Compared with VRd, there is moderate- to high-certainty evidence that DVRd likely results in an increase in the incidence of infections and infestations, and results in little to no difference in the incidence of cytopenia. Overall, no new safety signals were identified in the PERSEUS trial, and the observed safety profile of DVRd is similar to that of VRd.
No direct evidence comparing DVRd against CyBorD was submitted. Indirect evidence from 2 sponsor-conducted MAICs suggests that DVRd, compared with CyBorD, was associated with improved PFS in patients with TE NDMM; however, the magnitude of benefit was uncertain due to limitations of the analyses, including differences in the duration of follow-up and the lack of adjustment for potential prognostic factors. Potential heterogeneity in subsequent treatment use further increased the uncertainty in the OS findings, and no definitive conclusions could be drawn on the relative effect of DVRd versus CyBorD on OS.
A phase III, ongoing open-label RCT (AURIGA) and 3 sponsor-conducted MAICs were submitted to address a gap in comparative evidence of daratumumab-lenalidomide versus lenalidomide alone as maintenance therapy for NDMM after ASCT. Results from the AURIGA trial demonstrated that, when compared to lenalidomide alone, the addition of daratumumab to lenalidomide as maintenance therapy in patients who had MRD-positive disease at baseline resulted in a clinically significant benefit in the MRD conversion rate from baseline to 12 months. Moreover, treatment benefits were observed for PFS, overall MRD negativity conversion rate from baseline (at a threshold of 10-5), sustained MRD negativity rates at 6 and 12 months, and the overall rate of a CR or better. There was no difference in HRQoL, symptoms, and functioning between the daratumumab-lenalidomide and lenalidomide treatment groups. However, a few limitations of the trial were noted, including its open-label design, imbalance in proportion of patients with a high cytogenetic risk according to available local cytogenetic risk data at diagnosis, and handling of patients with missing or unevaluable data regarding MRD status in the primary end point analysis. No definitive conclusions could be drawn from the MAICs conducted in patients with TE NDMM (regardless of MRD status at maintenance baseline) with respect to the relative effects of daratumumab-lenalidomide versus lenalidomide due to important methodological limitations of the analyses.
References
1.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. 2015;12(1):42-54. doi: 10.1038/nrclinonc.2014.200 PubMed
2.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9. doi: 10.1038/leu.2008.291 PubMed
3.Bird SA, Boyd K. Multiple myeloma: an overview of management. Palliat Care Soc Pract. 2019;13:1178224219868235. doi: 10.1177/1178224219868235 PubMed
4.Canadian Cancer Society. Symptoms of multiple myeloma. Accessed February 1, 2024. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/signs-and-symptoms
5.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. Cmaj. 2024;196(18):E615-E623. doi: 10.1503/cmaj.240095 PubMed
6.McCurdy A, Mian H, LeBlanc R, et al. Redefining attrition in multiple myeloma (MM): a Canadian Myeloma Research Group (CMRG) analysis. Blood Cancer J. 2023;13(1):111. doi: 10.1038/s41408-023-00883-x PubMed
7.Côté J, LeBlanc R, Mian H, et al. Real-world results of autologous stem cell transplantation in newly diagnosed multiple myeloma: a report from the Canadian Myeloma Research Group database. Blood Cancer J. 2023;13(1):137. PubMed
8.Podar K, Leleu X. Relapsed/Refractory Multiple Myeloma in 2020/2021 and Beyond. Cancers (Basel). 2021;13(20):5154. doi: 10.3390/cancers13205154 PubMed
9.Mikhael J, Ismaila N, Cheung MC, et al. Treatment of Multiple Myeloma: ASCO and CCO Joint Clinical Practice Guideline. J Clin Oncol. 2019;37(14):1228-1263. doi: 10.1200/JCO.18.02096 PubMed
10.Government of Canada. Canadian Cancer Statistics. 2023. Accessed April 1, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf
11.Kurtin SE. Relapsed or Relapsed/Refractory Multiple Myeloma. J Adv Pract Oncol. 2013;4(Suppl 1):5-14.
12.Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Hemasphere. 2021;5(2):e528. PubMed
13.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
14.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
15.Janssen Research Development. Clinical Study Report: MMY3014 (PERSEUS). A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (DVRd) vs VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Previously Untreated Multiple Myeloma who are Eligible for High-dose Therapy [internal sponsor's report]. January 9, 2024.
16.Janssen Inc. Janssen Inc. response to November 5, 2024 CDA-AMC request for additional information regarding daratumumab CDA-AMC review: Between-group difference [internal additional sponsor's information]. 2024.
17.Janssen Inc. Sponsor Summary of Clinical Evidence: DVRd for the Treatment of Adult Patients with Transplant-eligible Newly Diagnosed Multiple Myeloma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Daratumumab, 1800 mg/15 mL (120 mg/mL) single-dose vial solution for subcutaneous injection. 2024.
18.Janssen Inc. Technical Report for the Indirect Treatment Comparison (ITC) of PERSEUS DVRd vs Relevant Treatment Alternatives in Newly Diagnosed Transplant-Eligible Multiple Myeloma (NDMM TE) version 1.1 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: daratumumab injection, 1800 mg/15 mL (120 mg/mL) solution for subcutaneous injection. 2024 September 19.
19.Janssen Research Development. Clinical Study Report: MMY3021 (AURIGA). A Randomized Study of Daratumumab Plus Lenalidomide Versus Lenalidomide Alone as Maintenance Treatment in Patients with Newly Diagnosed Multiple Myeloma Who Are Minimal Residual Disease Positive After Frontline Autologous Stem Cell Transplant [internal sponsor's report]. September 19, 2024.
20.Janssen. AURIGA Topline Results. 2024 [sponsor submitted reference].
21.Janssen. AURIGA Topline Results. 2024 [sponsor submitted reference].
22.International Myeloma Foundation. International Myeloma Working Group (IMWG) Uniform Response Criteria for Multiple Myeloma. Accessed July 1, 2024. https://www.myeloma.org/resource-library/international-myeloma-working-group-imwg-uniform-response-criteria-multiple
23.Janssen (data on file). AURIGA Topline Results. 2024 [sponsor submitted reference].
24.Badros A, Foster L, Anderson LD, Jr., et al. Daratumumab with lenalidomide as maintenance after transplant in newly diagnosed multiple myeloma: the AURIGA study. Blood. 2025. doi: 10.1182/blood.2024025746 PubMed
25.Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97(8):1086-1107. doi: 10.1002/ajh.26590 PubMed
26.Quach H, Joshua D, Ho J, et al. Treatment of patients with multiple myeloma who are eligible for stem cell transplantation: position statement of the M yeloma F oundation of A ustralia M edical and S cientific A dvisory G roup. Intern Med J. 2015;45(1):94-105. PubMed
27.Guglielmelli T, Palumbo A. Incorporating novel agents in the management of elderly myeloma patients. Curr Hematol Malig Rep. 2013;8:261-269. doi: 10.1007/s11899-013-0177-y PubMed
28.Rosko A, Giralt S, Mateos M-V, Dispenzieri A. Myeloma in elderly patients: when less is more and more is more. Am Soc Clin Oncol Educ Book. 2017;37:575-585. PubMed
29.Rajkumar SV. Updated Diagnostic Criteria and Staging System for Multiple Myeloma. Am Soc Clin Oncol Educ Book. 2016;35:e418-23. doi: 10.1200/EDBK_159009 PubMed
30.Bergstrom DJ, Kotb R, Louzada ML, Sutherland HJ, Tavoularis S, Venner CP. Consensus Guidelines on the Diagnosis of Multiple Myeloma and Related Disorders: Recommendations of the Myeloma Canada Research Network Consensus Guideline Consortium. Clin Lymphoma Myeloma Leuk. 2020;20(7):e352-e367. doi: 10.1016/j.clml.2020.01.017 PubMed
31.LeBlanc R, Bergstrom DJ, Côté J, Kotb R, Louzada ML, Sutherland HJ. Management of Myeloma Manifestations and Complications: The Cornerstone of Supportive Care: Recommendation of the Canadian Myeloma Research Group (formerly Myeloma Canada Research Network) Consensus Guideline Consortium. Clin Lymphoma Myeloma Leuk. 2022;22(1):e41-e56. doi: 10.1016/j.clml.2021.07.028 PubMed
32.Hillengass J, Usmani S, Rajkumar SV, et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019;20(6):e302-e312. doi: 10.1016/S1470-2045(19)30309-2 PubMed
33.Cancer Care Ontario. Daratumumab Drug Formulary. Accessed October 1, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44446
34.CADTH. CADTH Drug Reimbursement Expert Review Committee final recommendation: Daratumumab in Combination with Lenalidomide and Dexamethasone, or Bortezomib and Dexamethasone (Darzalex - Janssen). 2017. Accessed October 9, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_daratumumab_darzalex_mm_2ndln_fn_rec.pdf
35.CADTH. CADTH Drug Reimbursement Expert Review Committee final recommendation: Daratumumab in Combination with Bortezomib, Melphalan and Prednisone (Darzalex - Janssen). 2019. Accessed October 9, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10148Daratumumab%2BVMPforMM_fnRec_REDACT_Post_29Aug2019_final.pdf
36.CADTH. CADTH Drug Reimbursement Expert Review Committee final recommendation: Daratumumab in Combination with Lenalidomide and Dexamethasone (Darzalex - Janssen). 2020. Accessed October 9, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10189DaratumumabNDMM_fnRec_2020-03-03_ApprovedbyChair_Post_05Mar2020_final.pdf
37.Janssen Pharmaceuticals. DARZALEX SC Health Canada Product Monograph [Draft]. 2024 [sponsor submitted reference].
38.Kang DW, Bittner B, Sugarman BJ, Zepeda ML, Printz MA. Dispersive effects and focused biodistribution of recombinant human hyaluronidase PH20: A locally acting and transiently active permeation enhancer. PLoS One. 2021;16(7):e0254765. doi: 10.1371/journal.pone.0254765 PubMed
39.European Medicines Agency. Summary of opinion (post authorisation): Daratumumab (Darzalex - Janssen). 2024. Accessed October 9, 2024. https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-darzalex-ii-72_en.pdf
40.FDA. FDA approves daratumumab and hyaluronidase-fihj with bortezomib, lenalidomide, and dexamethasone for multiple myeloma. 2024. Accessed October 9, 2024. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-daratumumab-and-hyaluronidase-fihj-bortezomib-lenalidomide-and-dexamethasone-multiple
41.Sonneveld P, Dimopoulos MA, Boccadoro M, et al. Daratumumab, Bortezomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2024;390(4):301-313. doi: 10.1056/NEJMoa2312054 PubMed
42.CDA-AMC Drug Reimbursement Expert Review Committee final recommendation: Daratumumab (Darzalex - Janssen Inc.). CDA-AMC; January 28, 2022. Accessed December 3, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0257%20Darzalex%20-%20CADTH%20Final%20Rec%20Final.pdf
43.CDA-AMC. CDA-AMC Drug Reimbursement Expert Review Committee final recommendation: Daratumumab (Darzalex - Janssen Inc.). March 5, 2020. Accessed December 3, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10189DaratumumabNDMM_fnRec_2020-03-03_ApprovedbyChair_Post_05Mar2020_final.pdf
44.CDA-AMC. CDA-AMC Drug Reimbursement Expert Review Committee final recommendation: Daratumumab (Darzalex - Janssen Inc.). August 29, 2019. Accessed December 3, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10148Daratumumab%2BVMPforMM_fnRec_REDACT_Post_29Aug2019_final.pdf
45.CDA-AMC. CDA-AMC Drug Reimbursement Expert Review Committee final recommendation: Daratumumab (Darzalex - Janssen Inc.). October 5, 2017. Accessed December 3, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_daratumumab_darzalex_mm_2ndln_fn_rec.pdf
46.CDA-AMC. CDA-AMC Drug Reimbursement Expert Review Committee final recommendation: Daratumumab (Darzalex - Janssen Canada Inc.). December 1, 2016. Accessed December 3, 2024. https://www.cda-amc.ca/sites/default/files/pcodr/pcodr_daratumumab_darzalex_mm_fn_rec.pdf
47.Janssen Inc. Darzalex (daratumumab): 20 mg/mL concentrate for solution for infusion [product monograph]. April 30, 2024. https://pdf.hres.ca/dpd_pm/00075445.PDF
48.Teva Canada Limited. Velcade (bortezomib): Lyophilized powder, 3.5 mg / vial bortezomib, as the mannitol boronic ester, Intravenous or subcutaneous injection [product monograph]. 2023. https://pdf.hres.ca/dpd_pm/00070960.PDF
49.Cancer Care Ontario. Bortezomib Drug Formulary. 2024. Accessed February 1, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44116
50.Bristol Meyers Squibb. Revlimid (lenalidomide): capsules, 2.5 mg, 5 mg, 10 mg, 15 mg, 20 mg and 25 mg, oral [product monograph]. February 9, 2024. https://pdf.hres.ca/dpd_pm/00074535.PDF
51.Cancer Care Ontario. Lenalidomide Drug Formulary. Accessed July 1, 2024. https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/43951
52.Medina-Herrera A, Sarasquete ME, Jiménez C, Puig N, García-Sanz R. Minimal residual disease in multiple myeloma: past, present, and future. Cancers. 2023;15(14):3687. PubMed
53.Committee ODA. Position on use of MRD as an endpoint in MM clinical trials: April 12, 2024 Meeting of the Oncologic Drugs Advisory Committee Meeting Summary. 2024 [sponsor submitted reference]. https://www.fda.gov/advisory-committees/advisory-committee-calendar/april-12-2024-meeting-oncologic-drugs-advisory-committee-meeting-announcement-04122024#event-materials
54.Munshi NC, Avet-Loiseau H, Anderson KC, et al. A large meta-analysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood advances. 2020;4(23):5988-5999. PubMed
55.Anderson KC, Auclair D, Adam SJ, et al. Minimal Residual Disease in Myeloma: Application for Clinical Care and New Drug Registration. Clin Cancer Res. 2021;27(19):5195-5212. doi: 10.1158/1078-0432.CCR-21-1059 PubMed
56.Rosiñol L, Oriol A, Ríos R, et al. Lenalidomide and dexamethasone maintenance with or without ixazomib, tailored by residual disease status in myeloma. Blood. 2023;142(18):1518-1528. doi: 10.1182/blood.2022019531 PubMed
57.Martinez-Lopez J, Alonso R, Wong SW, et al. Making clinical decisions based on measurable residual disease improves the outcome in multiple myeloma. J Hematol Oncol. 2021;14(1):126. doi: 10.1186/s13045-021-01135-w PubMed
58.Dytfeld D, Wróbel T, Jamroziak K, et al. Carfilzomib, lenalidomide, and dexamethasone or lenalidomide alone as maintenance therapy after autologous stem-cell transplantation in patients with multiple myeloma (ATLAS): interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 2023;24(2):139-150. doi: 10.1016/S1470-2045(22)00738-0 PubMed
59.Derman BA, Major A, Cooperrider J, et al. Discontinuation of maintenance therapy in multiple myeloma guided by multimodal measurable residual disease negativity (MRD2STOP). Blood Cancer Journal. 2024;14(1):170. doi: 10.1038/s41408-024-01156-x PubMed
60.The EuroFlow Consortium. About EuroFlow. Accessed September 6, 2024. https://euroflow.org/about/
61.Szalat R, Anderson K, Munshi N. Role of minimal residual disease assessment in multiple myeloma. Haematologica. 2024;109(7):2049-2059. doi: 10.3324/haematol.2023.284662 PubMed
62.Nichols M, Chin-Yee I, Rutledge A, Chin-Yee B, Louzada ML, Phua CW. Advancing Multiple Myeloma Care: Targeted Mass Spectrometry for Measurable Residual Disease (MRD) Detection in Multiple Myeloma at London Health Sciences Centre Advocate. 2024;31(3).
63.Facon T, Kumar S, Plesner T, et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N Engl J Med. 2019;380(22):2104-2115. doi: 10.1056/NEJMoa1817249 PubMed
64.Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936-945. PubMed
65.Canadian Cancer Society. Multiple myeloma statistics. 2024. Accessed December 4, 2024. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/statistics
66.Sabloff M, Feilotter H, Sivajohanathan D, Howlett C, Ross C, Schuh A ea. Minimal residual disease testing in acute leukemia. Ontario Health - Cancer Care Ontario; 2022. https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/63341
67.Mian HS, Visram A, Shih SC-M, et al. Minimal Residual Disease Testing Infrastructure in Multiple Myeloma: Guidance for Clinical Trial and Routine Practice Use in Canada. Clin Lymphoma Myeloma Leuk. 2025. doi: https://doi.org/10.1016/j.clml.2025.01.010 PubMed
68.Adaptive Biotechnologies. Ordering clonoSEQ. Updated 2024. Accessed August 27, 2024. https://www.clonoseq.com/ordering-clonoseq/
69.Correia N, Dowling E, Popat R, et al. Functional and psychological impact of minimal residual disease assessment on patients with multiple myeloma. Br J Haematol. 2023;202(5):e43-e45. doi: 10.1111/bjh.18948 PubMed
70.Azvolinsky A. Discontinuing Maintenance Therapy for Multiple Myeloma Is Feasible, New Study Shows. American Society of Hematology; 2024. Accessed January 3, 2025. https://ashpublications.org/ashclinicalnews/news/8395/Discontinuing-Maintenance-Therapy-for-Multiple
71.Bravo-Pérez C, Sola M, Teruel-Montoya R, et al. Minimal residual disease in multiple myeloma: something old, something new. Cancers. 2021;13(17):4332. PubMed
72.Bain BJ. Bone marrow biopsy morbidity and mortality. Br J Haematol. 2003;121(6):949-51. doi: 10.1046/j.1365-2141.2003.04329.x PubMed
73.Carlson JJ, Eckert B, Zimmerman M. Cost-effectiveness of next-generation sequencing minimal residual disease testing during maintenance treatment for multiple myeloma. 2019;37(15 suppl). doi: 10.1200/JCO.2019.37.15_suppl.e19529
74.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v5.0. 2018. Accessed February 1, 2024. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40
75.Chan EHL, De-Silva D, Lin AHF, et al. Clinical benefit of depth of response for relapsed/refractory multiple myeloma patients treated on clinical trials: retrospective analysis from two tertiary centres. Br J Haematol. 2019;186(1):162-165. doi: 10.1111/bjh.15743 PubMed
76.van de Velde HJ, Liu X, Chen G, Cakana A, Deraedt W, Bayssas M. Complete response correlates with long-term survival and progression-free survival in high-dose therapy in multiple myeloma. Haematologica. 2007;92(10):1399-406. doi: 10.3324/haematol.11534 PubMed
77.Mangal N, Salem AH, Menon RM, Freise KJ. Use of depth of response to predict progression-free survival in relapsed or refractory multiple myeloma: Evaluation of results from 102 clinical trials. Hematol Oncol. 2018. doi: 10.1002/hon.2514 PubMed
78.Perrot A, Lauwers-Cances V, Corre J, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132(23):2456-2464. doi: 10.1182/blood-2018-06-858613 PubMed
79.van Reenen M, Stolk E, Boye K, et al. EQ-5D-5L User Guide Version 3.0. 2019. Accessed November 19, 2024. https://euroqol.org/publications/user-guides
80.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-36. doi: 10.1007/s11136-011-9903-x PubMed
81.Johnson & Johnson Research & Development. Evaluation of the comparative efficacy of daratumumab, bortezomib, lenalidomide, and dexamethasone versus daratumumab, bortezomib, and dexamethasone in transplant-eligible patients with previously untreated multiple myeloma. 2024 [sponsor submitted reference].
82.Tang DI, Geller NL. Closed testing procedures for group sequential clinical trials with multiple endpoints. Biometrics. 1999;55(4):1188-92. doi: 10.1111/j.0006-341x.1999.01188.x PubMed
83.Janssen Research & Development. Statistical Analysis Plan: MMY3014. A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide and Dexamethasone (DVRd) vs. VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Previously Untreated Multiple Myeloma who are Eligible for High-dose Therapy [internal sponsor's report]. August 31, 2023.
84.Janssen Inc. Response to Canada's Drug Agency request for additional information regarding daratumumab CDA-AMC review: Between-group difference [internal additional sponsor's information]. November 5, 2024.
85.Janssen Inc. Systematic literature review to support HTA submissions for daratumumab + VRd for transplant-eligible patients with newly diagnosed multiple myeloma. Evidence for the induction, consolidation, and maintenance phases (ICM) Study Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: daratumumab injection, 1800 mg/15 mL (120 mg/mL) solution for subcutaneous injection. 2024.
86.Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9. doi: 10.1186/1471-2288-12-9 PubMed
87.Aiello E, Gulas I, Mackay E, Laliman-Khara V. MSR124 Improving the Efficiency of Using the Guyot Algorithm to Construct Pseudo Individual Patient Data From Kaplan-Meier Survival Curves: Standardized Guidance and Data Preparation. Value in Health. 2023;26(12):S417.
88.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials. Pharmacoeconomics. 2010;28(10):935-945. PubMed
89.Phillippo D, Ades, T., Dias, S., Palmer, S., Abrams, K. R., & Welton, N. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. (Technical Support Documents). NICE Decision Support Unit; 2016. Accessed January 10, 2025. https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf
90.Austin PC. The use of propensity score methods with survival or time-to-event outcomes: reporting measures of effect similar to those used in randomized experiments. Stat Med. 2014;33(7):1242-58. doi: 10.1002/sim.5984 PubMed
91.Janssen Research & Development. Clinical Study Report: A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (DVRd) vs VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Previously Untreated Multiple Myeloma who are Eligible for High-dose Therapy (January 9, 2024; EEDMS-RIM-973762, 1.0). 2024.
92.Dimopoulos MA, Jakubowiak AJ, McCarthy PL, et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020;10(2):17. PubMed
93.Janssen. Clinical Protocol A Randomized Study of Daratumumab Plus Lenalidomide Versus Lenalidomide Alone as Maintenance Treatment in Patients with Newly Diagnosed Multiple Myeloma Who Are Minimal Residual Disease Positive After Frontline Autologous Stem Cell Transplant. 2024 [sponsor submitted reference].
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 5: Forest Plot of Subgroup Analyses on PFS (ITT Analysis Set; Data Cut-Off Date: August 1, 2023)
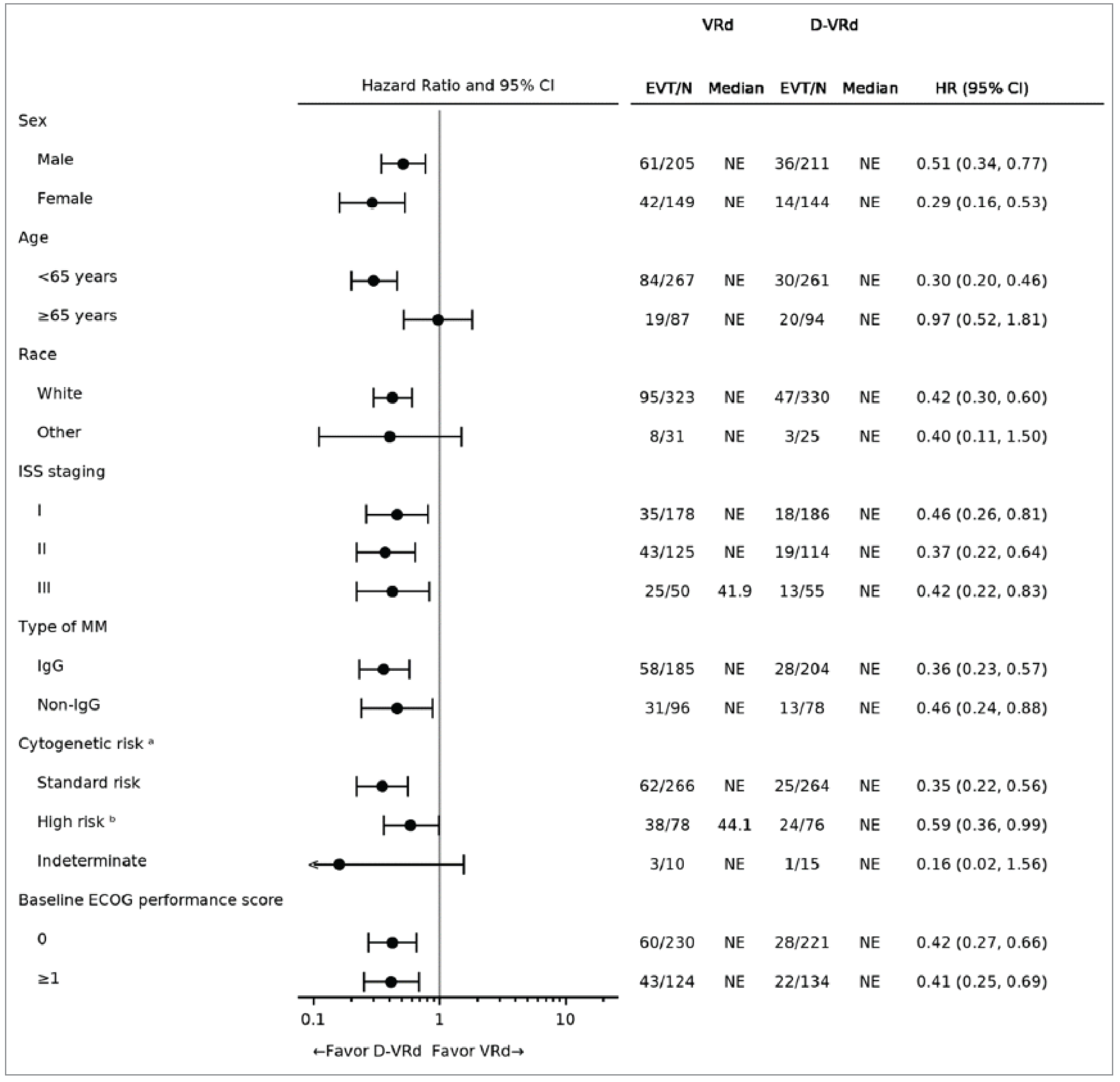
CI = confidence interval; D-VRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ECOG = Easter Cooperative Oncology Group; EVT = Number of subjects with an event in each subgroup; FISH = fluorescence in situ hybridization; HR = hazard ratio; IgG = Immunoglobulin G; ITT = intent-to-treat; ISS = International Staging System; MM = multiple myeloma; NE = not evaluable; PFS = progression-free survival; VRd = bortezomib-lenalidomide-dexamethasone.
Note: Hazard ratio and 95% CI from a Cox proportional hazards model with treatment as the sole explanatory variable. A hazard ratio of less than 1 indicates an advantage for DVRd.
aCytogenetic risk is based on FISH.
bPatient may have more than 1 high-risk abnormality [del17p, t(4;14) or t(14;16)].
Source: Clinical Study Report for PERSEUS (2024).15
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CyBorD
cyclophosphamide-bortezomib-dexamethasone
DVRd
daratumumab-bortezomib-lenalidomide-dexamethasone
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LY
life-year
MM
multiple myeloma
MRD
minimal residual disease
OS
overall survival
PFS
progression-free survival
PPS
postprogression survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RWE
real-world evidence
SC
subcutaneous
TTD
time to treatment discontinuation
VRd
bortezomib-lenalidomide-dexamethasone
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Daratumumab (Darzalex SC), 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for subcutaneous injection |
Indication | In combination with bortezomib, lenalidomide, and dexamethasone, followed by maintenance treatment in combination with lenalidomide, for the treatment of adult patients with newly diagnosed MM who are eligible for ASCT |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | November 27, 2024 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Yes Indication: Darzalex IV for the treatment of patients with MM who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an IMiD, or those who have failed or are intolerant to a proteasome inhibitor and who have failed to respond or are intolerant to an IMiD Recommendation date: December 16, 2016 Recommendation: Do not reimburse Indication: Darzalex IV in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for treatment of patients with MM who have received at least 1 prior therapy Recommendation date: October 17, 2017 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Darzalex IV in combination with bortezomib, melphalan, and prednisone, for the treatment of patients with newly diagnosed MM who are not suitable for ASCT Recommendation date: August 29, 2019 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Darzalex IV in combination with lenalidomide and dexamethasone for the treatment of patients with newly diagnosed MM who are ineligible for ASCT Recommendation date: March 5, 2020 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Darzalex SC in combination with bortezomib, cyclophosphamide, and dexamethasone, for the treatment of adult patients with newly diagnosed light-chain amyloidosis Recommendation date: January 28, 2022 Recommendation: Reimburse with clinical criteria and/or conditions |
ASCT = autologous stem cell transplant; IMiD = immunomodulatory drug; MM = multiple myeloma; NOC = Notice of Compliance; SC = subcutaneous.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adult patients with newly diagnosed MM who are transplant-eligible |
Treatment | Daratumumab in combination with DVRd, followed by daratumumab-lenalidomide in maintenance |
Dose regimen | For each 28-day cycle: Cycle 1 and 2: 1,800 mg on days 1, 8, 15, and 22 Cycle 3 to 6: 1,800 mg on days 1 and 15 Cycle 7 and onward: 1,800 mg on day 1 |
Submitted price | Daratumumab: $8,028 per single-dose vial |
Submitted treatment cost | $37,005 per cycle 1 and 2; $20,949 per cycle 3 to 6; $9,215 per cycle 7 and onward |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (40 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; HR = hazard ratio; ITC = indirect treatment comparison; LY = life-year; MM = multiple myeloma; MRD = minimal residual disease; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
Conclusions
Results of the PERSEUS trial demonstrated that, when compared to bortezomib-lenalidomide-dexamethasone (VRd), daratumumab-bortezomib-lenalidomide-dexamethasone (DVRd) results in a clinically significant benefit in progression-free survival (PFS) for patients with newly diagnosed multiple myeloma (MM) who are eligible for an autologous stem cell transplant (ASCT). However, treatment benefits in overall survival (OS) were uncertain due to the immaturity of data in the interim analysis. An indirect treatment comparison (ITC) of DVRd compared to cyclophosphamide-bortezomib-dexamethasone (CyBorD) was associated with numerous limitations. The exact magnitude of effect when comparing DVRd versus CyBorD therefore remains uncertain.
The CDA-AMC reanalysis addressed key limitations with respect to model structure, extrapolation of OS, and subsequent therapy costs. Results of the CDA-AMC reanalysis suggest the incremental cost-effectiveness ratio (ICER) for DVRd was $460,578 per quality-adjusted life-year (QALY) gained compared to VRd ($315,884 in incremental costs and 0.69 incremental QALYs). Incremental QALYs were driven by the mortality benefit associated with DVRd of an additional 0.88 life-years (LYs) relative to VRd. This mortality benefit was seen predominately in the extrapolated period, for which there are no trial data, with 92% of incremental QALYs being generated after 5 years. Conversely, more than 90% of the incremental costs were incurred in the first 5 years, driven primarily by the acquisition cost of daratumumab. Although some of the incremental costs of using DVRd in the first-line setting are offset by lower drug costs in subsequent treatment lines, these savings are uncertain and are much smaller than the initial upfront costs of using daratumumab in the first-line setting. Based on the CDA-AMC reanalysis, the unit cost of daratumumab as part of the DVRd regimen would need to be $2,649 (a 67% reduction in price) for daratumumab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY versus VRd.
In the CDA-AMC reanalysis, as per the PERSEUS trial, a proportion of patients were assumed to discontinue daratumumab maintenance therapy based on minimal residual disease (MRD) negativity status. If MRD testing is not routinely conducted in Canada, the reanalysis substantially underestimates daratumumab treatment costs. Where MRD testing is not used to inform maintenance therapy discontinuation, the cost of DVRd increases substantially, by $624,724 per patient lifetime. This causes the ICER to increase to beyond $1 million per QALY gained.
Finally, given the immaturity of the data in the trial, and that the analysis makes no use of any external evidence, the long-term outcomes for both costs and health impacts are uncertain. A more robust analysis would require a more complex model structure and real-world evidence (RWE) to inform subsequent treatment pathways, as well as achieve a better estimate of long-term survival.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from Myeloma Canada, which surveyed 39 patients and caregivers from September to October 2024. Respondents were adult patients or caregivers of adult patients with newly diagnosed MM who fell into 1 of 3 categories: those who were currently eligible for treatment with DVRd, those who had received first-line treatment with ASCT, or those with experience with DVRd. All respondents emphasized the importance of managing their myeloma symptoms, noting that MM affected the physical, social, and psychological aspects of their health-related quality of life (HRQoL). Among the subgroup of respondents (22 individuals) who had experience with DVRd, most reported a positive experience, stating that it improved their overall HRQoL with relatively manageable side effects. The patient group input commented that, while many patients in Canada are already receiving DVRd as first-line therapy, its availability is inconsistent across Canada.
Clinician input was received from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee. Clinician input noted that current treatments typically include CyBorD or VRd for induction, followed by ASCT. Consolidation is not publicly funded, and maintenance treatment with lenalidomide is possible. The primary goals of care are to prolong survival, achieve disease remission, improve symptoms, and prevent organ damage. Clinician input noted that some patients do not respond well to existing first-line options, and additional options are therefore needed. It was further noted that DVRd may become the standard of care for patients with transplant-eligible MM.
Drug plan input received for this review raised concerns with the choice of comparator in the submitted trials, as consolidation and maintenance therapy are not standard care in practices in Canada. The plans also questioned the feasibility of discontinuing daratumumab maintenance after 24 months among patients who achieve MRD negativity for at least 12 months, given that MRD testing is not routine in Canada. They asked if alternative criteria could be used to assess response or guide treatment discontinuation in the absence of MRD testing, and whether it would be appropriate to re-treat patients with daratumumab after discontinuing maintenance therapy.
Several of these concerns were addressed in the sponsor’s model:
Two outcomes valued by patients and clinicians, PFS and OS, were included in the model.
The impact of disease and treatment on a patient’s HRQoL was captured with utility values.
The sponsor considered the impact of adverse events (AEs).
VRd and CyBorD were included as comparators.
In addition, CDA-AMC addressed concerns as follows:
The feasibility of discontinuing daratumumab maintenance after 24 months among patients who achieve MRD negativity.
CDA-AMC was unable to address the concerns raised in input:
The misalignment with consolidation therapy between the trial and clinical practice in Canada.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of DVRd followed by daratumumab-lenalidomide maintenance against VRd followed by lenalidomide maintenance or CyBorD followed by lenalidomide maintenance, for the treatment of adult patients with transplant-eligible, newly diagnosed MM. This population aligns with the Health Canada indication and represents the sponsor’s reimbursement request.
Daratumumab (Darzalex) is available as 1,800 mg per 15 mL (120 mg/mL) single-use vials for subcutaneous (SC) injection.1 Daratumumab is recommended for use in combination with bortezomib, lenalidomide, and dexamethasone followed by maintenance treatment in combination with lenalidomide.1 The recommended dose is 1,800 mg weekly (on days 1, 8, 15, and 22) during cycles 1 and 2, 1,800 mg biweekly (on days 1 and 15) during cycles 3 through 6, and 1,800 mg every 4 weeks (on day 1) from cycle 7 onward.1 The submitted price for daratumumab is $8,028.00 per vial, with 28-day cycle costs ranging from $20,949.02 to $37,005.02 during induction, $20,949.02 during consolidation, and $9,215.20 during maintenance.2 The comparators in this analysis are VRd and CyBorD, as defined in the PERSEUS and GMMG-MM5 clinical trials, respectively.
The clinical outcomes modelled were OS, PFS, and time to treatment discontinuation (TTD). The model simulated QALYs, LYs, and costs for each treatment over a lifetime horizon of 40 years. Discounting at 1.5% per year was applied to both costs and outcomes, and a cycle length of 28 days was used with a half-cycle correction applied.2 The analysis was conducted from the perspective of the public health care payer in Canada.
Model Structure
The sponsor submitted a partitioned survival model (PSM) composed of 3 health states: “progression-free,” “postprogression,” and “death.” At any point in the model, the proportions of patients who were progression-free, had experienced progressed disease (as defined by International Myeloma Working Group criteria), or died were derived from non–mutually exclusive survival curves. Patients entered the model in the progression-free health state and may have remained stable, progress, or die. The proportions of patients in the progressed-disease or death state were estimated using the area under the extrapolated survival curves from the PERSEUS trial data. Specifically, OS was partitioned to estimate the proportion of patients in the death state, while the PFS curve was used to estimate the proportion of patients in the progression-free health state. The difference between the OS and PFS curves at each time point provided the proportion of patients in the postprogression state. Additionally, the submitted model includes an option to use a Markov model, which estimates direct transition probabilities between these 3 health states based on treatment-specific PFS and postprogression survival (PPS) curves. In this approach, OS was calculated by aggregating mortality rates from the progression-free and postprogression states.
Model Inputs
The target population reflected the population assessed in the PERSEUS trial, a phase III clinical trial comparing the efficacy of DVRd and VRd in adult patients with transplant-eligible, newly diagnosed MM. This population had a mean age of 58.4 years, was 58.7% male, and had a mean weight of 76 kg and a mean body surface area of 1.89 m2.2,3 These characteristics were used to inform the drug dosage regimens, as well as the age-specific and sex-specific distribution of the general population mortality risk, which the sponsor used to cap the lower bound for all-cause mortality in the model.
Efficacy data on PFS, OS, and TTD for DVRd and VRd were derived from the intention-to-treat population of the PERSEUS trial (data cut-off: August 1, 2023; median duration of PFS follow-up: 47.5 months; maximum duration of follow-up: 54.4 months). Parametric distributions were derived for DVRd and VRd using the Kaplan-Meier curves of PFS, OS, and TTD, enabling extrapolations of observed trial data to a lifetime model horizon. The sponsor explored dependent parametric models by assessing the proportional hazards assumption (i.e., constant treatment effect on the hazards).2 Extrapolation curves were selected based on clinical plausibility, visual inspection, and statistical fit to the trial’s Kaplan-Meier data. The sponsor applied a joint parametric distribution using treatment as a predictor in its base-case analysis. The Gompertz distribution was selected for the PFS curves of DVRd and VRd, and an exponential distribution was used to extrapolate OS for both DVRd and VRd.2 PFS and OS data for CyBorD were estimated by applying hazard ratios (HRs) from the sponsor-conducted matching-adjusted indirect comparison to the reference curves of DVRd. HRs of 4.00 (95% confidence interval]: 2.70 to 5.88) and 1.85 (95% confidence interval: 1.11 to 3.03) were used to inform the PFS and OS curves of CyBorD, respectively.2 Finally, the sponsor capped PFS at OS for all treatments, with OS further capped by general population mortality.
Induction and consolidation TTD data for DVRd and VRd were informed by the TTD observed in the PERSEUS trial. Induction and consolidation TTD data for CyBorD were modelled based on the planned median treatment duration in the GMMG-MM5 trial.2 Patients were assumed to discontinue treatment if they experienced progression, and in such instance, they were modelled to receive subsequent lines of therapy. For patients receiving daratumumab-lenalidomide maintenance, time on daratumumab treatment during the maintenance phase depended on achieving MRD negativity. In the PERSEUS trial, patients who received daratumumab-lenalidomide maintenance for at least 24 months, achieved a complete response or better, and maintained an MRD-negative response for at least 12 months were eligible to discontinue daratumumab treatment and continue with lenalidomide monotherapy. This was incorporated into the model, which assumed that 64.29% of patients starting daratumumab-lenalidomide maintenance met the criteria to discontinue daratumumab, as observed in the PERSEUS trial.3 The long-term extrapolation of maintenance treatment discontinuation for the following groups was based on extrapolated TTD data from the PERSEUS trial on daratumumab-lenalidomide maintenance for patients who did not achieve sustained MRD negativity, lenalidomide monotherapy maintenance for patients who achieved MRD negativity following daratumumab discontinuation at 24 months, and lenalidomide maintenance for patients who received lenalidomide monotherapy as maintenance. In the sponsor’s submitted base case, the exponential distribution was selected for all groups.2 The sponsor assumed that patients who discontinued daratumumab due to sustained MRD negativity could not restart daratumumab SC maintenance.
For subsequent therapies, patients were assumed to be treated until progression. The median PFS from respective clinical trials was used to generate a TTD curve, assuming an exponential distribution.
The analysis included AEs classified as grade 3 or higher that occurred in at least 5% of patients in either the DVRd or VRd arms of the PERSEUS trial.2 For DVRd and VRd, cumulative probabilities of AEs occurring during the induction and consolidation phases were informed by data from the PERSEUS trial. For CyBorD, rates of AE incidence during the induction phase were obtained from the GMMG-MM5 trials. AEs were applied as a single lump sum at the start of induction treatment.
Measures of HRQoL were captured in the model using utility values derived from EQ-5D-5L data obtained from the PERSEUS trial. Utility scores were derived using UK-specific utility weights. Health-state utility values increased progressively from the induction phase (0.721) through transplant (0.721) and consolidation (0.721), reaching their highest levels during the progression-free maintenance phase (0.721). For patients who experienced progression, the utility value in the postprogression phase (0.744) was slightly lower than during progression-free maintenance but remained higher than the values observed in earlier treatment stages. AE-related disutility values were included in the analysis, based on published literature and adjusted according to the mean duration of each AE, as reported in the National Institute for Health and Care Excellence technology appraisals for idecabtagene vicleucel (TA917) and ciltacabtagene autoleucel (TA765) for the treatment of relapsed or refractory MM.2,4-6
The analysis included costs related to drug acquisition and administration, comedications, and subsequent treatments, along with routine follow-up care, monitoring, AE management, and end-of-life care. Dosing schedules for induction, consolidation, and maintenance therapy were based on data from the PERSEUS trial for DVRd and VRd, and from the GMMG-MM5 trial for CyBorD. The acquisition cost of daratumumab was provided by the sponsor while the cost of other drugs was obtained from the Ontario Drug Benefit Formulary, Régie de l’assurance maladie du Québec, and previous CDA-AMC reviews.7-13 Second-line, third-line, and fourth-line subsequent therapy costs following progression were included in the model, consisting of acquisition, administration, and resource use.2 The sponsor assumed these costs would remain consistent, regardless of prior treatment.2 The sponsor assumed a cost of $75 per administration for both IV and SC treatments.14 Routine follow-up care and monitoring costs were acquired by health state. Weekly frequency and unit costs associated with each resource were informed by clinical expert feedback obtained by the sponsor and the Ontario Ministry of Health Schedule of Benefits for laboratory and physician services, respectively.2,14,15 The cost of MRD testing was informed by Carlson et al. (2019).16 A proportion of patients were assumed to undergo ASCT as informed by the PERSEUS or GMMG-MM5 trials. ASCT was assumed to cost $89,325 per patient based on published literature.17 Costs associated with AE management and end-of-life care were informed by published literature.2,18,19
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 1,000 iterations. The deterministic results were aligned with submitted probabilistic results. The probabilistic findings are presented in the following section.
Base-Case Results
As part of an additional information request, the sponsor resubmitted an economic model that resolved errors identified by the CDA-AMC review. In the sponsor’s updated submitted pairwise probabilistic comparisons, DVRd was more costly and more effective than VRd and CyBorD. Compared to VRd, DVRd was associated with 1.26 additional QALYs at an additional cost of $194,652, resulting in an ICER of $154,823 per QALY gained. Compared to CyBorD, DVRd was associated with 2.75 additional QALYs at an additional cost of $189,924, resulting in an ICER of $69,110 per QALY gained.
Given the duration of follow-up in the PERSEUS trial (median PFS follow-up: 47.5 months; maximum follow-up: 54.4 months) in contrast to the model’s lifetime horizon of 40 years, the majority of the incremental QALYs gained by patients receiving DVRd relative to VRd (98.7%) and CyBorD (99.0%) were derived from the period beyond which no observed trial data are available (i.e., the extrapolated period).
In the sponsor’s analysis, the probabilities of DVRd being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained were 0.4% and 19.4% when compared to VRd and CyBorD, respectively.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($ per QALY) |
|---|---|---|---|---|---|
DVRd vs. VRd | |||||
VRd | 824,693 | Reference | 11.30 | Reference | Reference |
DVRd | 1,019,346 | 194,652 | 12.55 | 1.26 | 154,823 |
DVRd vs. CyBorD | |||||
CyBorD | 829,421 | Reference | 9.81 | Reference | Reference |
DVRd | 1,019,346 | 189,924 | 12.55 | 2.75 | 69,110 |
CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
Alongside the original economic model, the sponsor conducted several deterministic scenario analyses that included the use of a Markov model structure, alternative discount rates and time horizon, excluding wastage, alternative dosing schedules, individual fitting for OS of DVRd and VRd, alternative proportions of patients who discontinue daratumumab maintenance due to sustained MRD negativity, excluding the cost of MRD testing and its impact on daratumumab maintenance therapy, and age-adjusted utility values.
However, the sponsor did not submit updated scenario analyses for the resubmitted model that resolved errors identified by CDA-AMC. As such, the impact of these scenarios on the sponsor-submitted cost-effectiveness of DVRd are based on an analysis with known errors and could be misleading.
Appraisal of the Sponsor’s Economic Evaluation
Several key limitations to the sponsor’s analysis were identified that have notable implications on the economic analysis:
The OS benefit of DVRd is uncertain. OS for patients receiving DVRd was estimated by conducting a survival analysis on the DVRd OS Kaplan-Meier curve observed from the PERSEUS trial (median follow-up of 47.6 months). A proportional hazards model with treatment as a predictor was used to estimate OS for patients receiving VRd. The sponsor’s base-case analysis estimates that DVRd would extend life by approximately 1.7 LYs (discounted) compared to VRd. Assuming a mean starting age of approximately 60 years, an estimated 77% and 59% of patients who received DVRd are expected to be alive at years 10 and 20, respectively. At 30 years, when the average patient age surpasses 90 years, an estimated 32% of patients are predicted to still be alive. To achieve this level of life expectancy, a substantial proportion of patients would need to be cured, meaning a patient’s MM does not affect their mortality. In the submitted analysis, at approximately 20 years, 60% of patients who received DVRd have a mortality rate that matches the general population. Likewise, the sponsor estimates that, at 21 years, 49% of patients who started on VRd have a mortality rate that matches the general population. This suggests that a substantial proportion of patients with MM will be cured even on VRd (current practice).
The sponsor’s base-case OS curves were considered inappropriate for several reasons. First, long-term RWE from Canada shows that median OS for patients in Canada with transplant-eligible, newly diagnosed MM is around 10 years.20 For patients receiving VRd, the sponsor estimates median OS to be around 20 years. Although data from this RWE study is older (patients were diagnosed between 2007 and 2018), clinical expert feedback for this review noted that median OS expectations will not have deviated from these estimates to the extent proposed by the sponsor, which suggests patients are currently living 10 years longer than what was presented in this data. Second, the sponsor assumes that the probability of death is constant over time. This would indicate that progression has no effect on OS. Over time, as more patients experience progression and treatment resistance, the mortality rate will likely start to increase. This was demonstrated in the PERSEUS trial, which showed OS to be worse in patients who progressed than in those who remained progression-free. This is also in line with RWE.21,22 Third, there is no evidence that a cure for MM is achievable in such a large percentage of patients. Clinical expert feedback noted that some patients may respond positively to a transplant, but treatment resistance is expected in most cases, and the cancer will eventually progress. Finally, clinical expert feedback noted that, even if residual risk directly related to MM was eliminated, the patient cohort would continue to have a higher risk of death compared with that of the general population due to comorbidities. When using the PSM, none of the extrapolations of OS were considered appropriate for this review, as all resulted in a substantial number of patients being cured.
Overall, only 10% of patients in the DVRd arm of the PERSEUS trial had died by the end of follow-up. The sponsor used this observation to predict the remaining 90% of OS events over 35 years without referencing any external data or accounting for future treatment pathways. By relying on highly immature data to extrapolate OS, the sponsor’s model generates long-term survival estimates that do not match clinical expectations and would suggest a fundamental shift in long-term, newly diagnosed MM prognosis for those even on current standard of care. A more appropriate model structure, such as a Markov model, can link survival outcomes to progression events, and eliminate the problematic assumption that PFS and OS are independent. This option was provided by the sponsor but was not presented as a base case.
The CDA-AMC base case used the Markov model structure submitted by the sponsor. However, this approach is still uncertain as data informing this approach come exclusively from the trial, which has immature data and does not cover the full MM treatment pathway.
The submitted Markov structure was problematic and associated with limitations. First, an assumption had to be made regarding what proportion of PFS events were preprogression deaths as opposed to progression events. The sponsor assumed the ratio of preprogression deaths to progression events was constant over time. For example, this means that, throughout the model, 18% of patients who received VRd would die before progression and 82% would progress. As part of an additional information request, CDA-AMC requested data on preprogression deaths and progression events in the PERSEUS trial at 1 year, 2 years, 3 years, and 4 years. The data showed the likelihood of dying before progression in both arms was highest in the first year (approximately 50%) and then fell substantially in subsequent years. This data were used to improve the accuracy of the Markov model. Second, an error in the model was identified that caused the ratio of deaths to progression events to be equal for both VRd and DVRd.
In the CDA-AMC base case, the proportion of PFS events that were deaths for year 1 was assumed to be 50% for both treatment arms. For subsequent years, rates of 7% and 25% were used for VRd and DVRd, respectively. These values were estimated by pooling the number of deaths and progression events in year 2 and onward (Table 13).
Assuming the mortality rate postprogression and the ratio of deaths to progression events are fixed over time, this leads to patients who receive DVRd having worse survival outcomes than those on VRd. This outcome is highly uncertain given the absence of long-term evidence. Assuming that the surviving patient cohorts between the DVRd and VRd treatment arms become more homogenous over time, an assumption was made that, beyond the trial data (5 years), mortality rates for those initially receiving DVRd would not exceed those of patients who initially received VRd.
A constant hazard rate over time was assumed. In the sponsor’s submitted analysis, the HR for PFS and OS was assumed to remain constant indefinitely (proportional hazards). This means the relative reduction in progression and mortality risk associated with receiving DVRd was assumed to remain the same throughout the patient’s lifetime. Evidence from the trial demonstrated that the proportional hazards assumption likely does not hold for OS, even within the trial data, based on visual inspection of the log-cumulative hazard plots and a significant P value for the Schoenfeld test. This further supports the use of a Markov model over a PSM, as a fixed treatment effect is assumed in a PSM. As noted in the Clinical Review report, median PFS and OS were not achieved in the first interim analysis. As such, there is substantial uncertainty regarding the long-term treatment effect of DVRd for patients with newly diagnosed MM who are transplant-eligible.
In general, the proportional hazards assumption is unlikely to hold over the long-term in most cases.23 As data from the PERSEUS trial are highly immature and cover such a short period relative to the model’s time horizon, it is difficult to predict how hazard rates will change over time. Clinical expert feedback for this review noted that it is unlikely that the HR will remain constant for the remainder of the full-time horizon (40 years); particularly as there are limited data for a long-term and durable treatment effect for those who discontinue daratumumab due to sustained MRD-negative status.
Additionally, clinical expert feedback noted that a patient with sustained MRD negativity will likely have a similar prognosis regardless of the treatment used to achieve MRD negativity; which is why new studies are exploring MRD negativity as a stopping rule.24 Numerous trials (POLLUX, CASTOR, ALCYONE, and MAIA) have demonstrated that PFS in those who achieve MRD negativity is similar regardless of whether MRD negativity was achieved using daratumumab or another regimen.25-28 As patients who are MRD-positive progress at a faster rate than those who are MRD-negative, the progression-free population of the DVRd and VRd will eventually consist predominately of those who are MRD-negative. The treatment benefit in a cohort of patients who are MRD-negative is likely to be smaller than it is in a population of patients who are both MRD-positive and MRD-negative. In general, the composition of the surviving cohort changes over time, creating a “survivor bias,” meaning those who remain progression-free for an extended period will have similar prognoses. This further supports the idea of a treatment waning effect.
CDA-AMC assumed the HR for PFS would approach 1 over time using the sponsor’s prebuild function, in which the HR would start trending toward 1 at year 5 and continue for 15 years, until hitting 1 at year 20. This means that the probability of progression at 20 years post-treatment initiation is the same regardless of whether VRd or DVRd was received initially.
Although there is strong rationale to indicate waning will occur, the period of time over which cessation of effect occurs is highly uncertain. Visual inspection of the PFS curves and waning during the trial period were considered when deriving a period over which the HR converges to 1. There was insufficient evidence to suggest PFS would change dramatically in a short time after the trial period, making the time frame of full treatment cessation under 5 years appear unlikely, but not impossible. Long-term evidence is required to establish the treatment benefit with daratumumab beyond 5 years, particularly in those who discontinue due to MRD negativity. To assess the uncertainty associated with treatment waning on the cost-effectiveness of DVRd, CDA-AMC conducted a scenario analysis that assumed 5 and 25 years of waning (Figure 2 in Appendix 4).
Given the known heterogeneity of PFS between those who achieve MRD negativity and those who do not, a more appropriate analysis would have stratified the survival analysis based on MRD status. This would lead to more accurate long-term survival estimates.
Discontinuing daratumumab maintenance therapy based on MRD status may not be reflective of clinical practice in Canada. In the sponsor’s submitted economic analysis, for those who receive DVRd, 26% have not met the criteria for discontinuation (they are either MRD-positive or have not been MRD-negative for long enough) at 2 years, while 60% are eligible for discontinuation based on MRD negativity status, and 14% have either progressed or died. At 4 years, nearly everyone who met the criteria for daratumumab discontinuation, based on MRD negativity, has come off treatment despite being progression-free. This was aligned with the PERSEUS clinical trial, in which patients could discontinue daratumumab maintenance (but remain on lenalidomide) if they had achieved a complete response or better and sustained MRD-negative status at and beyond 24 months after starting maintenance therapy.
Clinical expert feedback received by CDA-AMC noted that current clinical practice across Canada does not include MRD testing. Using MRD status to inform treatment discontinuation may therefore not be reflective of clinical practice in Canada. However, using sustained MRD-negative status to inform maintenance therapy discontinuation may become more frequent in clinical practice. Likewise, evidence that informs the economic analysis is based on the PERSEUS trial, and no evidence is available for patients who remain on daratumumab until progression.
CDA-AMC conducted a scenario analysis in which MRD testing is unavailable and therefore patients are treated until progression. In this scenario, anti-CD38 treatments (isatuximab and daratumumab) were assumed to not be used in subsequent treatment lines for those who progress on DVRd, as patients would not be re-treated with another anti-CD38 antibody immediately after progression. Likewise, costs associated with MRD testing were removed.
Subsequent therapy costs are associated with significant uncertainty. In the sponsor’s base-case analysis, a proportion of patients who experienced progression were assumed to receive subsequent treatments. Second-line, third-line, and fourth-line treatments were included in the model, with the sponsor using median treatment duration and median PFS from relevant clinical trials to estimate the cost of each subsequent therapy. Subsequent therapies were not used to estimate OS. It was also assumed that patients treated with DVRd were not eligible for another anti-CD38 antibody.
While CDA-AMC acknowledges the complexity of modelling multiple lines of therapy, the structure of the submitted economic analysis fails to accurately capture a patient’s treatment pathway. Subsequent therapy use will likely affect long-term OS, and without directly modelling this, the model relies heavily on assumptions that reduce the accuracy of the results (e.g., relying on median PFS to estimate drug costs and time to progression).
Additionally, the sponsor’s assumption that patients treated with DVRd would not receive an anti-CD38 antibody in later lines of therapy does not align with clinical expectations. Clinical expert feedback received by CDA-AMC noted that a patient who discontinues daratumumab due to MRD negativity and progresses later would be considered eligible for re-treatment with another anti-CD38. Uncertainty remains as to how much time would need to elapse before a patient would be considered eligible for re-treatment. In the PERSEUS trial, ███ of patients who received subsequent therapies in the DVRd arm received a monoclonal antibody as a subsequent line of therapy. This evidence was used to inform OS in the model. If these patients did not receive a monoclonal antibody, then the trial may misestimate OS gains for daratumumab. Likewise, the sponsor’s distribution for subsequent therapies in the VRd arm also does not align with the trial data. The sponsor assumes 100% of patients treated with VRd will go on to receive an anti-CD38. However, in the trial only 80% received a monoclonal antibody as subsequent therapy. This may be more reflective of clinical practice as some individuals may not receive an anti-CD38 due to patient preference regarding IV therapies. The clinical experts consulted for this review noted that the distribution of subsequent therapy use in the second line would change over time for those who received DVRd. The more time that passes between discontinuing daratumumab, based on MRD status, and progression, the more likely the individual would be considered for re-treatment with another anti-CD38. The CDA-AMC base case may therefore underestimate subsequent therapy costs for those receiving DVRd, as the proportion of patients who are re-treated with an anti-CD38 is fixed over the model’s time horizon.
Because subsequent therapies, such as ciltacabtagene autoleucel, teclistamab, and elranatamab, are currently undergoing active negotiations with the pan-Canadian Pharmaceutical Alliance, they are not currently available for public reimbursement in the fourth-line setting.29-31 In the model, subsequent therapies only affected costs and had no impact on OS. As a result, the impact of receiving ciltacabtagene autoleucel, teclistamab, and elranatamab on OS has not been accounted for.
In a base-case reanalysis, CDA-AMC adjusted the market share of second-line subsequent therapies to better align with the PERSEUS trial and expectations of clinical practice in Canada (Table 5).
Additionally, given the uncertainty associated with the availability of fourth-line subsequent therapies (i.e., ciltacabtagene autoleucel, teclistamab, and elranatamab) in clinical practice in Canada, CDA-AMC removed fourth-line therapies in a scenario analysis.
Due to the submitted model structure, any subsequent therapy costs estimated in the model are uncertain.
Uncertainty was inadequately captured in the probabilistic analysis. The results from the probabilistic analysis were considered unreliable for several reasons.
First, when the model is run probabilistically, parameters in the model are independently sampled. However, there are likely dependencies across many of the parameters used to inform the model. For example, when run probabilistically, progression and TTD are likely to be correlated, given that progression is the leading cause for discontinuing treatment. Likewise, changes in PPS will likely be linked to the distribution of subsequent therapies the patient receives. Without accounting for these correlations, the probabilistic sensitivity analysis introduces biases into the analysis.
Second, as the trial data from the PERSEUS trial are immature, and the model incorporates no long-term evidence from RWE, long-term uncertainty is not accurately incorporated. Trial data, which is the only evidence source for the model, are available for 4 years but the model’s time horizon is 40 years. A more appropriate model structure would have used external data to inform PFS and OS in subsequent treatment lines to more accurately capture long-term uncertainty.
Third, while the sponsor used reported standard errors for some inputs, such as body weight and body surface area, the standard error used to parametrize uncertainty for many parameters was arbitrarily set to 10% rather than based on underlying data. Uncertainty was therefore not appropriately captured in the probabilistic analysis.
Finally, when assessing different runs from the probabilistic analysis, the model frequently produced results that led to a substantial number of patients being “cured,” with OS exceeding general population mortality for many patients regardless of starting treatment. As a cure is not explicitly modelled, this result was considered inappropriate.
In a base-case reanalysis, CDA-AMC ran the model deterministically.
Generalizability of trial results to the clinical practice is uncertain. Primary efficacy (i.e., OS, PFS, and TTD) data in the sponsor’s submitted economic model was informed by the PERSEUS trial, which enrolled adult patients with newly diagnosed MM who are eligible for ASCT. In both treatment arms, patients in the PERSEUS trial received consolidation and maintenance. However, as consolidation is not funded in all jurisdictions and maintenance therapy is optional, there may be some generalizability concerns between results observed in the trial versus those seen in clinical practice in Canada. As noted in the Clinical Review report, there may be additional generalizability concerns with the trial population versus patients in clinical practice in Canada due to the overrepresentation of patients who were white (> 90%) and exclusion of patients aged 70 years and older who may be candidates for DVRd.
CDA-AMC was unable to address this limitation.
The comparative clinical evidence of DVRd versus CyBorD is uncertain. In the absence of direct head-to-head evidence comparing DVRd to CyBorD, the relative effectiveness of CyBorD in the model (i.e., OS and PFS) was informed by a sponsor-conducted ITC. Specifically, the sponsor submitted 2 unanchored matching-adjusted indirect comparisons that aimed to evaluate the comparative efficacy between DVRd (individual patient data) and CyBorD (aggregate data) in patients with transplant-eligible newly diagnosed MM. As noted in the Clinical Review report, significant limitations remain when assessing these indirect comparisons, including a risk of selection bias in study inclusion, potential differences in subsequent therapy use, sizable reductions in effective sample size, and a lack of adjustment for potential prognostic factors that may introduce unmeasurable confounding in the relative treatment-effect estimates. This renders the conclusion regarding the relative efficacy of DVRd versus CyBorD uncertain. Given the lack of direct evidence for DVRd versus CyBorD and limitations within the sponsor’s ITC, to what extent DVRd provides a net benefit compared to regimens outside what was used in the trial (i.e., VRd) remains uncertain.
In the CDA-AMC base case, direct head-to-head evidence from the PERSEUS trial was used to estimate the relative efficacy of DVRd versus VRd. In the absence of direct evidence and limitations with the sponsor’s submitted matching-adjusted indirect comparisons, the cost-effectiveness of DVRd relative to CyBorD is unknown.
Clinical experts consulted by CDA-AMC noted that CyBorD is most often used when lenalidomide is not a viable option. Given that daratumumab is given in combination with lenalidomide, DVRd is unlikely to displace CyBorD. Therefore, CyBorD was not considered highly relevant for this submission.
The key assumptions made by the sponsor and appraised by CDA-AMC are presented in Table 4.
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
VRd dosing schedule aligned with dosing schedule used in the PERSEUS clinical trial. | Reasonable. Clinical expert input noted that the dosing schedule of VRd used alone or in combination with daratumumab in the PERSEUS clinical trial is likely reflective of clinical practice in Canada. However, there may be jurisdictional differences, such as in the dosing schedule presented by Cancer Care Ontario, that are not expected to affect efficacy. This may have a small impact on costs but not enough to substantially influence cost-effectiveness. |
CDA-AMC = Canada’s Drug Agency; VRd = bortezomib-lenalidomide-dexamethasone.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base-case analyses were derived by making changes in the model parameter values and assumptions, in consultation with clinical experts. The changes, summarized in Table 5, include the selection of a Markov model with the rate of mortality varying by time, including treatment waning, and updating market-share values for second-line subsequent therapies. Due to limitations characterizing uncertainty in the analysis, results of the CDA-AMC base case are presented deterministically.
In the CDA-AMC reanalysis, DVRd was associated with an ICER of $460,578 per QALY gained compared to VRd ($315,884 in incremental costs and 0.69 incremental QALYs). Incremental costs were primarily due to the cost of daratumumab SC.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Overall survival | Model structure: PSM Key assumptions that influence overall survival: 1. Constant mortality risk assumed for DVRd and VRd 2. Mortality risk capped by the general population mortality risk | Model structure: Markov model Key assumptions that influence overall survival: 1. Time-varied ratio of preprogression death to progression events: VRd:
DVRd:
2. Mortality for DVRd assumed to not exceed VRd after 5 years. |
2. Treatment-waning | Excluded | Included for PFS Hazard ratio begins converging toward 1 at year 5 and reaches 1 after 15 years (year 20 in the model). |
3. Subsequent therapies in the second line | ||
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 |
CDA-AMC = Canada’s Drug Agency; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; PSM = partitioned survival model; VRd = bortezomib-lenalidomide-dexamethasone.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | VRd | 824,693 | 11.30 | Reference |
DVRd | 1,019,346 | 12.55 | 154,823 | |
CDA-AMC reanalysis 1 | VRd | 598,878 | 7.59 | Reference |
DVRd | 826,179 | 8.92 | 171,046 | |
CDA-AMC reanalysis 2 | VRd | 846,635 | 11.32 | Reference |
DVRd | 1,086,167 | 12.24 | 258,053 | |
CDA-AMC reanalysis 3 | VRd | 776,078 | 11.32 | Reference |
DVRd | 1,146,483 | 12.57 | 295,917 | |
CDA-AMC base case (reanalysis 1 + 2 + 3) | VRd | 559,227 | 7.59 | Reference |
DVRd | 875,111 | 8.28 | 460,578 |
CDA-AMC = Canada’s Drug Agency; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone.
Note: Deterministic results are reported, unless otherwise stated. The reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price-reduction analyses based on the sponsor’s results and the CDA-AMC reanalysis (Table 7). Results from the reanalysis suggest a price reduction of approximately 67% would be required for daratumumab SC (as part of DVRd) to achieve cost-effectiveness versus VRd at a willingness-to-pay threshold of $50,000 per QALY. The unit cost of daratumumab SC would be $2,649 at a price reduction of 67%. Price reductions of between 80% and 90% would be required to achieve cost-effectiveness at a threshold of $50,000 per QALY if daratumumab was administered until progression under the assumption MRD testing is unavailable (Table 16).
CDA-AMC conducted additional scenario analyses to explore the impact of an alternative waning assumption, exclusion of discontinuation of daratumumab maintenance therapy based on MRD negativity, and exclusion of fourth-line subsequent therapies.
Table 7: CDA-AMC Price-Reduction Analyses
Analysis | Unit drug cost ($) | ICERs for DVRd vs. VRd ($ per QALY) | |
|---|---|---|---|
Price reduction | $ | Sponsor base case | CDA-AMC reanalysis (deterministic) |
No price reduction | 8,028 | 154,823 | 460,578 |
10% | 7,225 | 126,265 | 399,281 |
20% | 6,422 | 93,771 | 337,984 |
30% | 5,620 | 61,278 | 276,687 |
40% | 4,817 | 28,784 | 215,390 |
50% | 4,014 | Dominant | 154,093 |
60% | 3,211 | Dominant | 92,796 |
70% | 2,408 | Dominant | 31,499 |
80% | 1,606 | Dominant | Dominant |
90% | 803 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
Figure 3 outlines how the ICER changes based on the different assumption regarding PFS treatment waning. In the figure, the HR for PFS from the trial (DVRd versus VRd) is applied for the first 5 years of the model. The HR then gradually decreases until it hits 1. The x-axis starts at 10 years, reflecting 5 years of full treatment benefit followed by 5 years of waning benefit, and then from year 10 onward the HR for PFS is 1. If a patient remained progression-free for 10 years after treatment initiation, then the risk of the patient progressing in following years would be the same regardless of whether they received upfront treatment with DVRd or VRd. In this analysis the ICER is $778,247 per QALY gained. Conversely, if it takes 25 years for full treatment waning to occur (i.e., 5 years of full benefit followed by 25 years of waning benefit, with an HR of 1 being applied to PFS from year 30 onward), the ICER decreases to $358,569 per QALY gained. There are limited data to indicate when additional treatment benefit will stop. If treatment waning occurs earlier than what is outlined in the CDA-AMC base case, this has a larger effect on the ICER than if treatment waning occurred later.
In an analysis in which patients were not allowed to discontinue daratumumab maintenance therapy based on MRD negativity (i.e., treated until progression) the ICER for DVRd compared to VRd was $1,327,480 per QALY gained. Price reductions of between 80% and 90% would be required to achieve cost-effectiveness at a threshold of $50,000 per QALY (Table 16).
When fourth-line subsequent therapies were excluded in the analysis, the incremental cost and incremental QALYs were $305,368 and 0.69, respectively. The ICER for DVRd compared to VRd was $445,246 per QALY gained.
Issues for Consideration
Clinical expert feedback obtained by CDA-AMC noted that MRD testing is not routinely conducted in clinical practice in Canada. As patients in the PERSEUS trial who were treated with DVRd, achieved a complete response or better, and sustained MRD-negative status at and beyond 24 months after starting maintenance therapy could discontinue daratumumab maintenance (but would remain on lenalidomide), there may be an incentive to increase testing. However, should the testing frequency increase with DVRd reimbursement, the true impact of MRD testing on health care resource use remains unknown.
Overall Conclusions
Results of the PERSEUS trial demonstrate that, compared to VRd, DVRd results in a clinically significant benefit in PFS for patients with newly diagnosed MM who are eligible for an ASCT. However, treatment benefits in OS were uncertain due to the immaturity of the data in the interim analysis. The evidence comparing DVRd and CyBorD was derived from an ITC that was associated with numerous limitations. The exact magnitude of any difference in effect between DVRd and CyBorD therefore remains uncertain.
The CDA-AMC reanalysis addressed key limitations with respect to model structure, extrapolation of OS, and subsequent therapy costs. Results of the reanalysis suggest the ICER for DVRd was $460,578 per QALY gained compared to VRd ($315,884 in incremental costs and 0.69 incremental QALYs). Incremental QALYs were driven by the mortality benefit associated with DVRd (an additional 0.88 LYs relative to VRd). This mortality benefit was seen predominately in the extrapolated period, for which there is no trial data, with 92% of incremental QALYs being generated after 5 years. Conversely more than 90% of the incremental costs were incurred in the first 5 years and driven primarily by the acquisition cost of daratumumab. Although some of the incremental costs of using DVRd in the first-line setting are offset by lower drug costs in subsequent treatment lines, these savings are uncertain and are much smaller than the initial upfront costs of using daratumumab in the first-line setting. Based on the CDA-AMC reanalysis, the unit cost of daratumumab as part of the DVRd regimen would need to be $2,649 (a 67% reduction in price) to be considered cost-effective at a threshold of $50,000 per QALY versus VRd.
In the reanalysis, as per the PERSEUS trial, a proportion of patients were assumed to discontinue daratumumab maintenance therapy based on MRD negativity status. If MRD testing is not routinely conducted in Canada, the reanalysis substantially underestimates daratumumab treatment costs. Where MRD testing is not used to inform maintenance therapy discontinuation, the cost of DVRd increases substantially by $624,724 per patient over their lifetime. This causes the ICER to increase beyond $1 million per QALY gained.
Finally, given the immaturity of data, long-term outcomes for both costs and health impacts are uncertain. As the model relies exclusively on trial data and does not use any external data to inform potential long-term mortality outcomes, the results of the analysis may not accurately reflect long-term outcomes. Uncertainty in long-term treatment benefit is reflected in the scenario analyses, with the ICER ranging from $358,569 to $778,247 per QALY gained, based on assumptions regarding the durability of PFS treatment benefit over 40 years. There is also outstanding uncertainty that could not be addressed by the scenario analyses, as none of these analyses could address the limitation associated with not directly modelling treatment benefits associated with subsequent lines of therapy.
References
1.Janssen Inc. Darzalex SC (daratumumab injection): 120 mg/mL solution for subcutaneous injection [product monograph]. July 29, 2020.
2.Janssen Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Darzalex SC (daratumumab injection), 120 mg/mL solution for subcutaneous injection. October 1, 2024.
3.Sonneveld P, Dimopoulos MA, Boccadoro M, et al. Daratumumab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2024;390(4):301-313. doi:10.1056/NEJMoa2312054 PubMed
4.National Institute for Health and Care Excellence. Daratumumab with lenalidomide and dexamethasone for untreated multiple myeloma when a stem cell transplant is unsuitable (ID4014: Committee Papers). 2022. Accessed June 10, 2024. https://www.nice.org.uk/guidance/ta917/documents/committee-papers
5.National Institute for Health and Care Excellence. Daratumumab in combination for untreated multiple myeloma when a stem cell transplant is suitable (NICE technology appraisal guidance TA763). 2022. Accessed June 10, 2024. https://www.nice.org.uk/guidance/ta763
6.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-4. doi:10.1177/0272989X11401031 PubMed
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed Nov 4, 2024. https://www.formulary.health.gov.on.ca/formulary/
8.CADTH Reimbursement Review: Isatuximab (Sarclisa). Can J Health Technol. 2022;2(4). doi:10.51731/cjht.2022.305
9.CADTH Reimbursement Review: Selinexor (Xpovio). Can J Health Technol. 2022;2(10). doi:10.51731/cjht.2022.467
10.CADTH Reimbursement Review: Ciltacabtagene Autoleucel (Carvykti). Can J Health Technol. 2023;3(8). doi:10.51731/cjht.2023.706
11.CADTH Reimbursement Recommendation: Elranatamab (Elrexfio). Can J Health Technol. 2024;4(6). doi:10.51731/cjht.2024.922
12.CADTH Reimbursement Recommendation: Teclistamab (Tecvayli). Can J Health Technol. 2024;4(4). doi:10.51731/cjht.2024.874
13.Ramq. List of Medications [accessed by sponsor]. 2024. Accessed September 2024. https://www.ramq.gouv.qc.ca/sites/default/files/documents/non_indexes/liste_med_2024-07-04_en.pdf
14.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). 2023. Accessed November 5, 2024. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
15.Ontario Ministry of Health. Schedule of benefits for laboratory services (Effective July 24, 2023). 2023. Accessed November 4, 2024. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/sob_lab_2023.pdf
16.Carlson JJ, Eckert B, Zimmerman M. Cost-effectiveness of next-generation sequencing minimal residual disease testing during maintenance treatment for multiple myeloma. J Clin Oncol. 2019;37(15_suppl):e19529-e19529. doi:10.1200/JCO.2019.37.15_suppl.e19529
17.Interprovincial Health Insurance Agreements Coordinating Committee. Interprovincial billing rates for designated high cost transplants [accessed by sponsor]. 2024. Accessed September 2024. https://www.ontario.ca/files/2024-04/moh-interprovincial-billing-rates-high-cost-transplants-en-2024-04-08.pdf
18.Janssen Research Development. Clinical Protocol A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (D-VRd) vs VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Previously Untreated Multiple Myeloma who are Eligible for High-dose Therapy [sponsor supplied reference]. 2022.
19.Zargar M, McFarlane T, Chan KKW, Wong WWL. Cost-Effectiveness of Nivolumab in Recurrent Metastatic Head and Neck Squamous Cell Carcinoma. Oncologist. 2018;23(2):225-233. doi:10.1634/theoncologist.2017-0277 PubMed
20.Mian H, Reece D, Masih-Khan E, et al. Survival and Outcomes of Newly Diagnosed Multiple Myeloma Patients Stratified by Transplant Status 2007-2018: Retrospective Analysis from the Canadian Myeloma Research Group Database. Clin Lymphoma Myeloma Leuk. 2022;22(8):608-617. doi:10.1016/j.clml.2022.03.002 PubMed
21.Bayani DB, Lin YC, Nagarajan C, et al. Modeling first-line daratumumab use for newly diagnosed, transplant-ineligible, multiple myeloma: A cost-effectiveness and risk analysis for healthcare payers. Pharmacoecon Open. 2024;8(5):651-664. doi:10.1007/s41669-024-00503-9 PubMed
22.CADTH Reimbursement Review: Optimal pharmacotherapy for transplant-ineligible multiple myeloma. Can J Health Technol. 2024;4(4). Accessed December 20, 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/TR0014-MM-ESHPM-Combined-Report.pdf
23.Coyle D, Haines A, Lee K. Extrapolating clinical evidence within economic evaluations. Can J Health Technol. 2023;3(5). doi:10.51731/cjht.2023.649
24.Derman BA, Major A, Cooperrider J, et al. Discontinuation of maintenance therapy in multiple myeloma guided by multimodal measurable residual disease negativity (MRD2STOP). Blood Cancer J. 2024;14(1):170. doi:10.1038/s41408-024-01156-x PubMed
25.Dimopoulos MA, San-Miguel J, Belch A, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica. 2018;103(12):2088-2096. doi:10.3324/haematol.2018.194282 PubMed
26.Spencer A, Lentzsch S, Weisel K, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica. 2018;103(12):2079-2087. doi:10.3324/haematol.2018.194118 PubMed
27.Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N Engl J Med. 2018;378(6):518-528. doi:10.1056/NEJMoa1714678 PubMed
28.Facon T, Kumar S, Plesner T, et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N Engl J Med. 2019;380(22):2104-2115. doi:10.1056/NEJMoa1817249 PubMed
29.pan-Canadian Pharmaceutical Alliance. Carvykti (ciltacabtagene autoleucel). 2024. Accessed December 13, 2024. https://www.pcpacanada.ca/negotiation/22916
30.pan-Canadian Pharmaceutical Alliance. Elrexfio (elranatamab). 2024. Accessed December 13, 2024. https://www.pcpacanada.ca/negotiation/22724
31.pan-Canadian Pharmaceutical Alliance. Tecvayli (teclistamab). 2024. Accessed December 13, 2024. https://www.pcpacanada.ca/negotiation/22654
32.IQVIA. DeltaPA. 2023. Accessed June 13, 2024. https://www.iqvia.com/
33.Cancer Care Ontario. CYBORD Regimen. 2019. Accessed Oct 25, 2024. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46446
34.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi:10.1503/cmaj.240095 PubMed
35.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. doi:10.1503/cmaj.191292 PubMed
36.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2019. Canadian Cancer Society; 2019. Accessed June 3, 2024. https://cancer.ca/Canadian-Cancer-Statistics-2019-EN
37.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian cancer statistics 2021. Canadian Cancer Society; 2021. Accessed June 3, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf
38.Brenner DR, Poirier A, Woods RR, et al. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194(17):E601-E607. doi:10.1503/cmaj.212097 PubMed
39.Cote J, LeBlanc R, Mian H, et al. Real-world results of autologous stem cell transplantation in newly diagnosed multiple myeloma: A report from the Canadian Myeloma Research Group database. Blood Cancer J. 2023;13(1):137. doi:10.1038/s41408-023-00905-8 PubMed
40.McCurdy A, Mian H, LeBlanc R, et al. Redefining attrition in multiple myeloma (MM): a Canadian Myeloma Research Group (CMRG) analysis. Blood Cancer J. 2023;13(1):111. doi:10.1038/s41408-023-00883-x’ PubMed
Appendix 1: Cost-Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost-Comparison Table for Transplant-Eligible Newly Diagnosed Multiple Myeloma
Treatment | Strength / concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | 28-day cycle cost ($)a |
|---|---|---|---|---|---|---|
Daratumumab (Darzalex SC) | 1,800 mg/15 mL | Single-dose vial | 8,028.0000b | Cycle 1 and 2: 1,800 mg on days 1, 8, 15, and 22 Cycle 3 to 6: 1,800 mg on days 1 and 15 Cycles 7+: 1,800 mg on day 1 | Cycle 1 and 2: 1,146.86 Cycle 3 to 6: 573.43 Cycles 7+: 286.71 | Cycle 1 and 2: 32,112 Cycle 3 to 6: 16,056 Cycles 7+: 8,028 |
Bortezomib | 3.5 mg | Powder in vial (for infusion) | 654.3132 | Cycle 1 to 6: 1.3 mg/m2 days 1, 4, 8, and 11 | Cycle 1 to 6: 93.47 | Cycle 1 to 6: 2,617 |
Lenalidomide | 2.5 mg 5 mg 10 mg 15 mg 20 mg 25 mg | Cap | 82.3750 85.0000 90.2500 95.5000 100.7500 106.0000 | Cycle 1 to 6: 25 mg on days 1 to 21 Cycle 7+: 10 mg days 1 to 28 | Cycle 1 to 6: 79.50 Cycle 7+: 90.25 | Cycle 1 to 6: 2,226 Cycle 7+: 2,527 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | Cycles 1 to 6: 40 mg on days 1 to 4, 9 to 12 | Cycle 1 to 6: 1.75 | Cycle 1 to 6: 49 |
DVRd | Cycle 1 and 2: 1,322 Cycle 3 to 6: 748 Cycles 7+:377 | Cycle 1 and 2: 37,004 Cycle 3 to 6: 20,948 Cycles 7+: 10,555 | ||||
VRd | ||||||
Bortezomib | 3.5 mg | Powder in vial (for infusion) | 654.3132 | Cycle 1 to 6: 1.3 mg/m2 days 1, 4, 8, and 11 | Cycle 1 to 6: 93.47 | Cycle 1 to 6: 2,617 |
Lenalidomide | 2.5 mg 5 mg 10 mg 15 mg 20 mg 25 mg | Cap | 82.3750 85.0000 90.2500 95.5000 100.7500 106.0000 | Cycle 1 to 6: 25 mg on days 1 to 21 Cycle 7+: 10 mg days 1 to 28 | Cycle 1 to 6: 79.50 Cycle 7+: 90.25 | Cycle 1 to 6: 2,226 Cycle 7+: 2,527 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | Cycle 1 to 6: 40 mg on days 1 to 4, 9 to 12 | Cycle 1 to 6: 1.75 | Cycle 1 to 6: 49 |
VRd | Cycle 1 to 6: 174.72 Cycle 7+: 90.25 | Cycle 1 to 6: 4,892 Cycle 7+: 2,527 | ||||
CyBorD33 | ||||||
Bortezomib | 3.5 mg | Powder in vial (for infusion) | 654.3132 | Cycle 1 to 3: 1.5 mg/m2 days 1, 8, 15, 22 | 93.47 | 2,617 |
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | Cycle 1 to 3: 300 mg/m2 days 1, 8, 15, 22 | 0.75 | 21 |
Dexamethasone | 0.5 mg 4 mg | Tablet | 0.1564 0.6112 | Cycle 1 and 2: 40 mg days 1 to 4, 9 to 12, 17 to 20 Cycle 3+: 40 mg days 1, 8, 15, 22 | Cycle 1 and 2: 2.62 Cycle 3+: 0.87 | Cycle 1 and 2: 73 Cycle 3+: 24 |
CyBorD | Cycle 1 and 2: 96.84 Cycle 3+: 95.10 Maintenance: 90.25 | Cycle 1 and 2: 2,712 Cycle 3+: 2,663 Maintenance: 2,527 | ||||
CDA-AMC = Canada’s Drug Agency; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed October 2024), unless otherwise indicated, and do not include dispensing fees.
aDosing schedules are aligned with those from the PERSEUS clinical trial, unless otherwise stated.
bSponsor-submitted price.2
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | Refer to CDA-AMC critical appraisal |
Model structure is adequate for decision problem | No | Refer to CDA-AMC critical appraisal |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CDA-AMC critical appraisal |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CDA-AMC critical appraisal |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.2
Table 10: What Treatments Patients Receive Upon Progression (Sponsor’s Base Case)
Treatment | Progression during induction | Progression during maintenance | ||
|---|---|---|---|---|
DVRd | VRd | DVRd | VRd | |
DRd | 0% | 3.75% | 0% | 3.75% |
DVRd | 0% | 0% | 0% | 6.25% |
IsaKd | 0% | 71.25% | 0% | 77.5% |
Kd | 50% | 0% | 50% | 0% |
Pd | 25% | 0% | 25% | 0% |
SVd | 25% | 0% | 25% | 0% |
IsaPd | 0% | 25% | 0% | 12.5% |
DRd = daratumumab-lenalidomide-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; IsaKd = isatuximab-carfilzomib-dexamethasone; IsaPd = isatuximab-pomalidamide-dexamethasone; Kd = carfilzomib-dexamethasone; Pd = pomalidomide-dexamethasone; SVd = selinexor-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Assumptions from sponsor’s submission
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | DVRd | VRd | CyBorD |
|---|---|---|---|
Discounted LYs | |||
Total | 17.89 | 16.22 | 14.06 |
Induction, ASCT, and consolidation | 0.64 | 0.63 | 0.48 |
Maintenance | 10.88 | 6.45 | 4.43 |
Postprogression | 6.36 | 9.14 | 9.15 |
Discounted QALYs | |||
Total | 12.55 | 11.30 | 9.81 |
Induction, ASCT, and consolidation | 0.47 | 0.46 | 0.35 |
Maintenance | 8.16 | 4.91 | 3.39 |
Postprogression | 4.28 | 6.24 | 6.32 |
Disutilities | −0.36 | −0.32 | −0.26 |
Discounted costs ($) | |||
Total | 1,019,346 | 824,693 | 829,421 |
Induction and consolidation treatment | 134,163 | 23,633 | 8,131 |
Drug acquisition | 2,498 | 1,450 | 676 |
Drug administration | 1,946 | 1,884 | 24 |
Concomitant medications | 3,011 | 2,338 | 1,552 |
Routine monitoring | 9,823 | 6,832 | 4,250 |
Adverse event management | 79,324 | 75,747 | 78,271 |
ASCT | 134,163 | 23,633 | 8,131 |
Maintenance treatment | |||
Drug acquisition | 410,287 | 66,910 | 61,190 |
Drug administration | 2,930 | 0 | 0 |
Concomitant medications | 43,145 | 29,751.26 | 27,208 |
Routine monitoring | 29,372 | 13,614 | 9,333 |
Adverse event management | 3,159 | 3,295 | 3,283 |
Postprogression | |||
Second-line treatment | |||
Drug acquisition | 88,223 | 458,659 | 488,382 |
Drug administration | 1,550 | 6,063 | 6,400 |
Routine monitoring | 2,277 | 4,691 | 5,130 |
Third-line treatment | |||
Drug acquisition | 21,069 | 18,446 | 19,393 |
Drug administration | 431.59 | 194 | 212 |
Routine monitoring | 967.48 | 1,116 | 1,135 |
Fourth-line treatment | |||
Drug acquisition | 129,026 | 46,304 | 49,318 |
Drug administration | 878 | 1,061 | 1,063 |
Routine monitoring | 15,748 | 21,557 | 1,193 |
Terminal care | 39,518 | 41,147 | 43,277 |
Indirect | 0 | 0 | 0 |
ASCT = autologous stem cell transplant; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; LY = life-year; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Disaggregated Summary of CDA-AMC Economic Evaluation Results (Deterministic)
Parameter | DVRd | VRd |
|---|---|---|
Discounted LYs | ||
Total | 11.57 | 10.69 |
Induction, ASCT, and consolidation | 0.64 | 0.63 |
Maintenance | 8.96 | 6.08 |
Postprogression | 1.97 | 3.98 |
Discounted QALYs | ||
Total | 8.28 | 7.59 |
Induction, ASCT, and consolidation | 0.47 | 0.46 |
Maintenance | 6.81 | 4.65 |
Postprogression | 1.36 | 2.80 |
Disutilities | −0.36 | −0.32 |
Discounted costs ($) | ||
Total | 875,111 | 559,227 |
Induction and consolidation treatment | ||
Drug acquisition | 134,178 | 23,637 |
Drug administration | 2,496 | 1,449 |
Concomitant medications | 1,946 | 1,884 |
Routine monitoring | 3,013 | 2,339 |
Adverse event management | 9,817 | 6,829 |
ASCT | 79,307 | 75,788 |
Maintenance treatment | ||
Drug acquisition | 408,960 | 68,031 |
Drug administration | 2,929 | 0 |
Concomitant medications | 42,447 | 30,250 |
Routine monitoring | 25,322 | 12,820 |
Adverse event management | 3,159 | 3,295 |
Postprogression | ||
Second-line treatment | ||
Drug acquisition | 58,855 | 231,116 |
Drug administration | 829 | 3,130 |
Routine monitoring | 908 | 2,608 |
Third-line treatment | ||
Drug acquisition | 7,412 | 11,514 |
Drug administration | 141 | 161 |
Routine monitoring | 327 | 644 |
Fourth-line treatment | ||
Drug acquisition | 41,922 | 27,491 |
Drug administration | 326 | 594 |
Routine monitoring | 4,659 | 8,729 |
Terminal care | 46,157 | 46,918 |
Indirect | 0 | 0 |
ASCT = autologous stem cell transplant; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; LY = life-year; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone.
Table 13: Breakdown of PFS Data Over Time
Year | VRd | DVRd | ||||
|---|---|---|---|---|---|---|
Progression | Preprogression death | Total PFS events | Progression | Preprogression death | Total PFS events | |
1 | 14 | 14 | 28 | 10 | 7 | 17 |
2 | 20 | 1 | 21 | 6 | 1 | 7 |
3 | 30 | 2 | 32 | 10 | 1 | 11 |
4 | 20 | 2 | 22 | 8 | 6 | 14 |
Total | 84 | 19 | 103 | 34 | 15 | 49 |
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; PFS = progression-free survival; VRd = bortezomib-lenalidomide-dexamethasone.
Source: PERSEUS trial.
Table 14: What Treatments Patients Receive Upon Progression (CDA-AMC Base Case)
Treatment | Progression during induction | Progression during maintenance | ||
|---|---|---|---|---|
DVRd | VRd | DVRd | VRd | |
DRd | 0% | 5% | 5% | 5% |
IsaKd | 0% | 65% | 35% | 65% |
Kd | 50% | 5% | 15% | 5% |
Pd | 25% | 5% | 15% | 5% |
SVd | 25% | 10% | 20% | 10% |
IsaPd | 0% | 10% | 10% | 10% |
DRd = daratumumab-lenalidomide-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; IsaKd = isatuximab-carfilzomib-dexamethasone; IsaPd = isatuximab-pomalidamide-dexamethasone; Kd = carfilzomib-dexamethasone; Pd = pomalidomide-dexamethasone; SVd = selinexor-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Based on clinical expert opinion.
Scenario Analyses
Table 15: Scenario Analyses Conducted on the CDA-AMC Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | VRd | 824,693 | 11.30 | Reference |
DVRd | 1,019,346 | 12.55 | 154,823 | |
CDA-AMC base case | VRd | 559,227 | 7.59 | Reference |
DVRd | 875,111 | 8.28 | 460,578 | |
CDA-AMC scenario 1: No 4L Subsequent Therapies | VRd | 523,365 | 7.59 | Reference |
DVRd | 828,734 | 8.28 | 445,246 | |
CDA-AMC scenario 2: No daratumumab maintenance discontinuation due to MRD status | VRd | 559,227 | 7.59 | Reference |
DVRd | 1,469,669 | 8.28 | 1,327,480 |
CDA-AMC = Canada’s Drug Agency; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; MRD = minimal residual disease; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone.
Note: Deterministic results are reported, unless otherwise stated.
Figure 2: Impact of Treatment Waning on D-VRD PFS
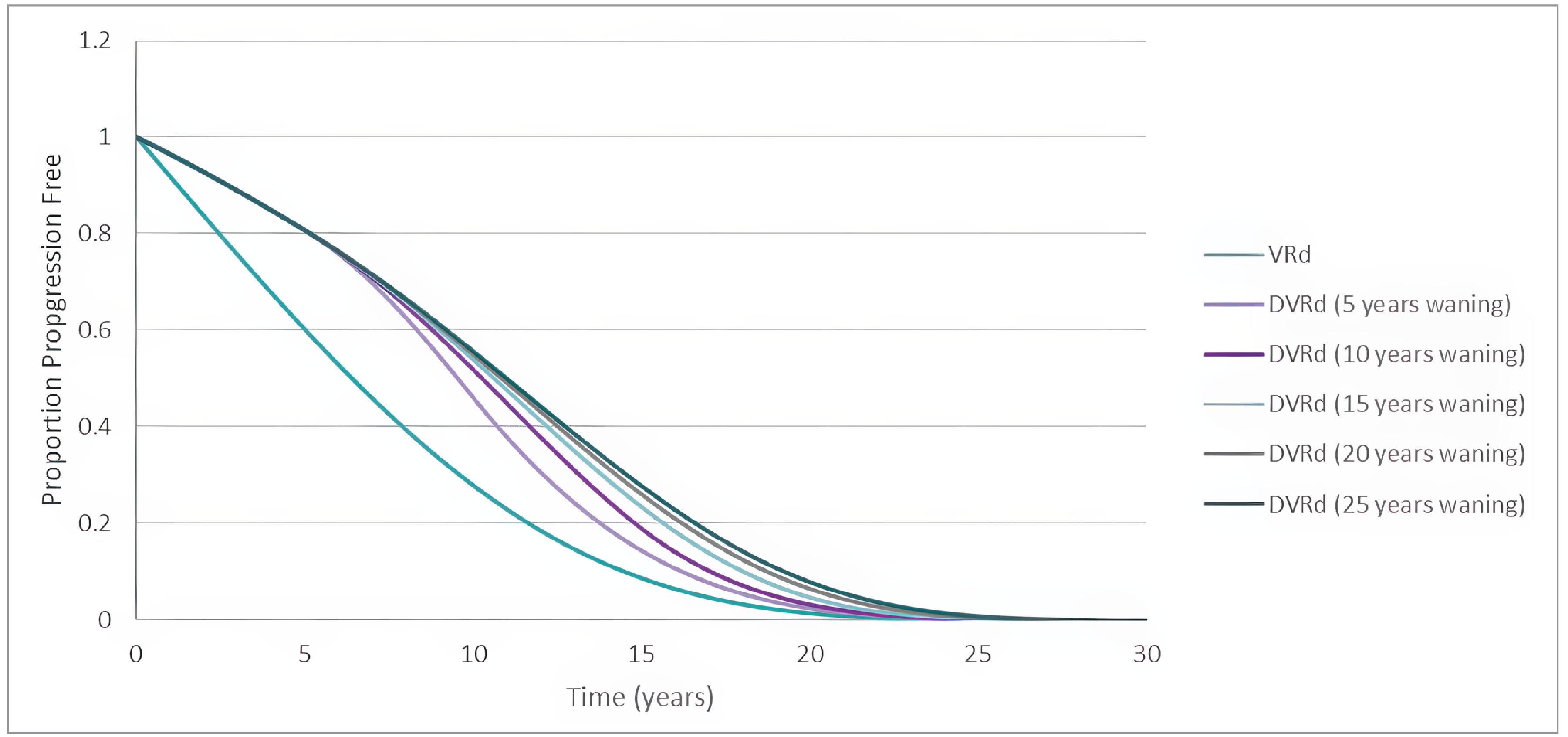
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; PFS = progression-free survival; QALY = quality-adjusted life-year. VRd = bortezomib-lenalidomide-dexamethasone.
Source: Output using the sponsor’s submitted model.
Figure 3: Impact of Treatment Waning on ICER
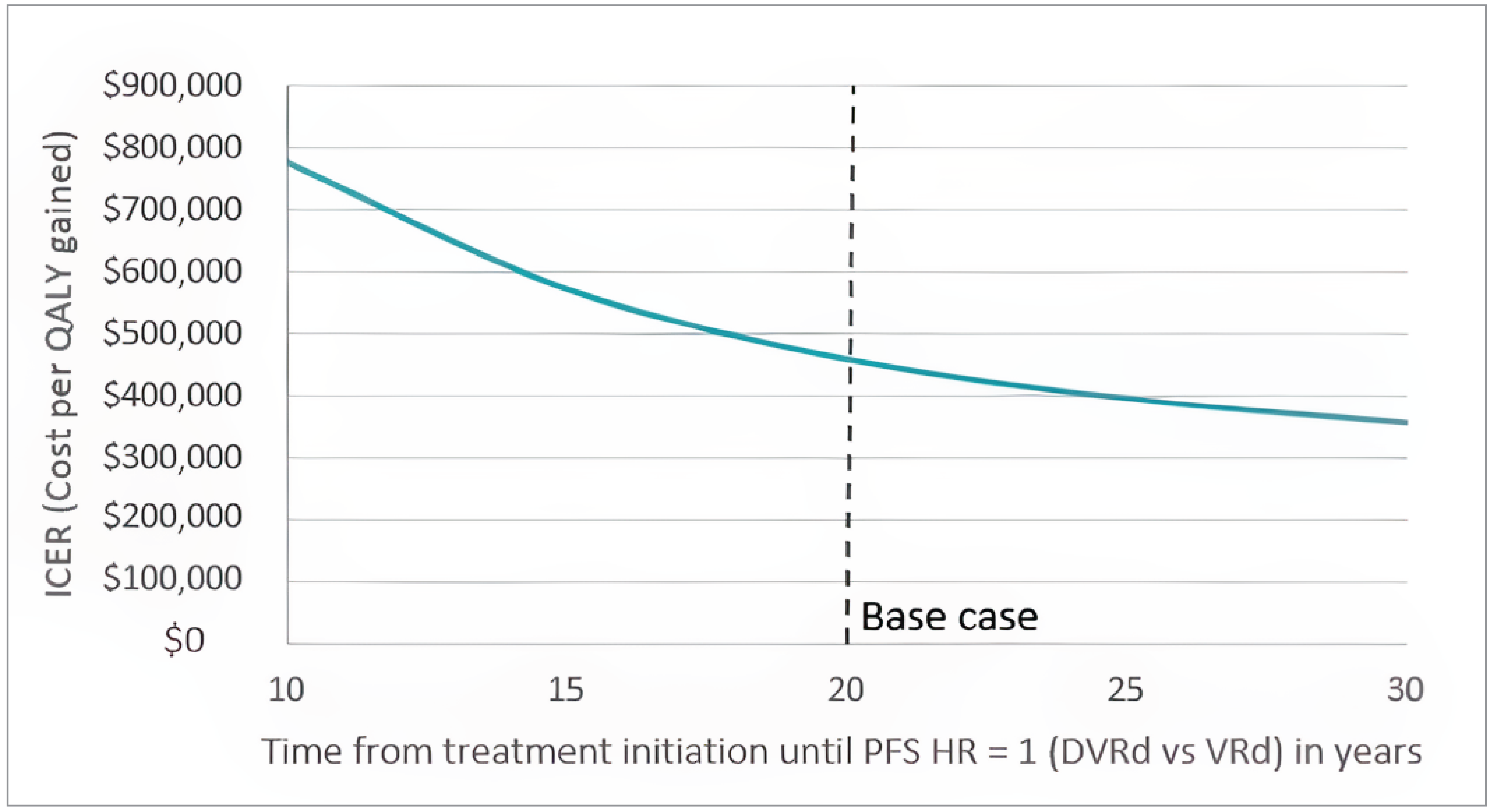
DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; PFS = progression-free survival; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone.
Source: Output using the sponsor’s submitted model.
Table 16: CDA-AMC Price-Reduction Analyses (No MRD Testing)
Analysis | Unit drug cost ($) | ICERs for DVRd vs. VRd ($ per QALY) | |
|---|---|---|---|
Price reduction | $ | Sponsor base case | CDA-AMC reanalysis (deterministic) |
No price reduction | 8,028 | 775,976 | 1,327,480 |
10% | 7,225 | 682,337 | 1,175,346 |
20% | 6,422 | 588,698 | 1,023,212 |
30% | 5,620 | 495,060 | 871,078 |
40% | 4,817 | 401,421 | 718,944 |
50% | 4,014 | 307,783 | 566,809 |
60% | 3,211 | 214,144 | 414,675 |
70% | 2,408 | 120,505 | 262,541 |
80% | 1,606 | 26,867 | 110,407 |
90% | 803 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; VRd = daratumumab-bortezomib-lenalidomide-dexamethasone; ICER = incremental cost-effectiveness ratio; MRD = minimal residual disease; QALY = quality-adjusted life-year; VRd = bortezomib-lenalidomide-dexamethasone; vs. = versus.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 17: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA to estimate the 3-year budget impact of reimbursing daratumumab SC for the treatment of adult patients with newly diagnosed MM who are transplant-eligible. The analysis was taken from the perspective of the public drug plan in Canada. A 3-year time horizon was used from 2025 to 2027 (with 2024 as the baseline year). The target population size was derived with an epidemiological approach based on the projected incidence of MM in 2024 published by Canadian Cancer Statistics Advisory Committee and other published literature.34-40 Key inputs to the BIA are documented in Table 18.
State the key assumptions:
Subsequent treatments were not considered in the analysis
Median PFS from the survival analysis conducted in the submitted pharmacoeconomic analysis was used to inform treatment duration for DVRd-DR and VRd. Median PFS for CyBorD was informed by the GMMG-MM5 trial.
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Projected incidence of MM in Canada (2024) | 4,10034 |
Average incidence growth rate (2019 to 2023) | |
% eligible for ASCT | 49.7%39 |
Attrition rate in first-line ASCT | 7.0%40 |
% pan-Canadian population | 78%34 |
Number of patients eligible for drug under review | 1,555 / 1,625 / 1,699 |
Market uptake (3 years) | |
Uptake (reference scenario) VRd CyBorD | 90% / 90% / 90% 10% / 10% / 10% |
Uptake (new drug scenario) DVRd VRd CyBorD | ██% / ██% / ██% ██% / ██% / ██% ██% / ██% / ██% |
Cost of treatment (per patient, per year) | |
DVRd VRd CyBorD | Year 1: $234,773; Year 2+: $142,948 Year 1: $50,129; Year 2+: $38,584 Year 1: $33,276; Year 2+: $38,584 |
ASCT = autologous stem cell transplant; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; MM = multiple myeloma; VRd = bortezomib-lenalidomide-dexamethasone.
Summary of the Sponsor’s BIA Results
In the sponsor’s base-case analysis, the estimated incremental budget impact of funding daratumumab SC for the treatment of adult patients with newly diagnosed MM who are transplant-eligible was $114,823,568 in year 1, $274,958,778 in year 2, and $436,019,907 in year 3. Therefore, the 3-year incremental budget impact was $825,802,253.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The market uptake of daratumumab is uncertain. In the sponsor’s submitted model, the market uptake of daratumumab was estimated to be ██%, ██%, and ██% in year 1, year 2, and year 3, respectively. These estimates were informed by sponsor internal projections and clinical expert feedback. However, clinical expert feedback received by CDA-AMC notes that daratumumab is expected to have substantially higher uptake upon reimbursement. As daratumumab has been frequently used in MM across multiple treatment lines clinicians have a lot of experience and comfort in using the treatment. Given the clinical efficacy demonstrated in the PERSEUS trial and familiarity with daratumumab clinical experts noted that uptake will be considerably higher than the estimates provided by the sponsor. Clinical experts consulted by CDA-AMC were unsure of any reason why patients eligible for daratumumab would not receive it if it were publicly funded.
CDA-AMC conducted a scenario analysis assuming 80%, 85%, and 90% of daratumumab uptake in year 1, year 2, and year 3, respectively.
Treatment duration used to inform drug costs is inaccurate. To inform time on treatment, the sponsor assumed patients would remain on treatment until median PFS was reached. As median PFS for DVRd was not reached in the PERSEUS trial, the sponsor used median PFS estimates from the conducted survival analyses. Based on this, it was assumed that the median duration was 142.44 months, 76.68 months, and 40.91 months for DVRd, VRd and CyBorD, respectively. As median duration of PFS was beyond 3-years, this assumption means that 100% of patients remain on treatment for 3 years with no treatment discontinuation during this period. This does not align with data or clinical expectations as this assumes there are no deaths within the first 3 years of treatment, for example.
Additionally, drug costs for daratumumab maintenance therapy are uncertain due to the potential implementation of treatment discontinuation due to a patient’s MRD status. If discontinuation in clinical practice in Canada is expected to align with protocol in the PERSEUS trial (i.e., patients who received DR maintenance for at least 24 months, achieved a complete response or better, and maintained an MRD-negative response for at least 12 months were eligible to discontinue daratumumab treatment and continue with lenalidomide monotherapy) then the budget impact of DVRd may be lower.
In the CDA-AMC reanalysis, drug costs were extracted from the base-case economic model (where the economic model was run with a 1, 2, and 3-year time horizon at a 0% discount rate) to estimate drug costs incurred for each incident case in the BIA. These costs are in Table 20. These costs are based on daratumumab discontinuation based on the PERSEUS trial protocol.
In a scenario analysis, CDA-AMC explored the impact of assuming treatment until progression. This is relevant if MRD testing is not conducted, and a patient does not discontinue daratumumab based on MRD status.
Impact of DVRd on subsequent therapy costs is uncertain. Subsequent therapy costs were not included in the sponsor’s submitted budget impact analysis. However, subsequent therapy will likely be influenced by DVRd’s place in therapy, as patients who progress while actively receiving daratumumab in the first-line setting would not be eligible for an additional anti-CD38 which would reduce the budget impact. CDA-AMC notes that there are limitations with estimating subsequent therapies in the sponsors submitted economic analysis as outlined in the critical appraisal of the economic analysis.
To assess the impact subsequent therapy costs may have on the budget impact, CDA-AMC conducted a scenario analysis using drug costs plus subsequent therapy costs extracted from the CDA-AMC base-case economic model (where the economic model was run with a 1, 2, and 3-year time horizon at a 0% discount rate) to estimate drug costs incurred for each incident case in the BIA.
The number of patients eligible for DVRd is uncertain. CDA-AMC identified several uncertainties with the estimation of the eligible patient population for DVRd. In the sponsor’s approach to estimating the eligible patient population parameter inputs were sourced from published literature. Of this, it was estimated that the average incidence growth rate for 2019 to 2023 was 4.5% (calculated as an average in the rate of change from 2019 and 2023 incidence population reported by the Brenner et al. (2020),35 Brenner et al. (2024),34 the Canadian Cancer Society, and Brenner et al. (2022).25-28,38 CDA-AMC acknowledges that this approach is reasonable; however, notes the variability in the rate of change per year (notable between 2019 to 2020 and 2021 to 2022) introduces some uncertainty in the incidence growth rate estimate. CDA-AMC further notes that the sponsor estimates there is a 7% attrition rate to first-line ASCT informed by McCurdy et al. (2023).40 However, the attrition rate reported in McCurdy et al. (2023)40 reports on the proportion of patients who “progress without treatment” and “death,” which is not equivalent to the population who does not receive first-line ASCT. That said, clinical expert feedback received by CDA-AMC noted that 7% of patients eligible for transplant not receiving it appears reasonable.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
Table 19: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. DVRd market share | ██% / ██% / ██% | 80% / 85% / 90% |
2. Drug costs | Annual costs informed from dosing schedules informed by product monographs | Costs extracted from the CUA model based on a 1-, 2-, or 3-year time horizon at a 0% discount rate (refer to Table 16) |
CDA-AMC base case | reanalysis 1 + 2 | |
CDA-AMC = Canada’s Drug Agency; CUA = cost-utility analysis; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone.
Table 20: Cost of Treatments in the CDA-AMC BIA Reanalysis
Treatment | Year 1 | Year 2 | Year 3 |
|---|---|---|---|
DVRd | $165,641 | $108,843 | $95,706 |
VRd | $27,551 | $12,638 | $10,387 |
CyBorD | $13,993 | $12,044 | $9,898 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; CyBorD = cyclophosphamide-bortezomib-dexamethasone; DVRd = daratumumab-bortezomib-lenalidomide-dexamethasone; VRd = bortezomib-lenalidomide-dexamethasone.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 21 and a more detailed breakdown is presented in Table 22. Results of the CDA-AMC reanalysis suggests that the estimated incremental budget impact of funding daratumumab SC in combination with VRd followed by maintenance treatment in combination with lenalidomide, for the treatment of adult patients with newly diagnosed MM was $171,746,567 in year 1, $310,413,347 in year 2, and $451,311,054 in year 3. Therefore, the 3-year incremental budget impact was $933,470,968.
Table 21: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 825,802,253 |
CDA-AMC reanalysis 1 | 1,172,250,699 |
CDA-AMC reanalysis 2 | 654,137,201 |
CDA-AMC base case | 933,470,968 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 22):
including cost of subsequent therapies
including treat until progression.
Table 22: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 72,044,505 | 132,694,865 | 196,097,099 | 262,376,066 | 591,168,030 |
New drug | 72,044,505 | 247,518,433 | 471,055,876 | 698,395,972 | 1,416,970,282 | |
Budget impact | 0 | 114,823,568 | 274,958,778 | 436,019,907 | 825,802,253 | |
CDA-AMC base case | Reference | 38,957,080 | 59,431,361 | 77,502,580 | 81,019,085 | 217,953,026 |
New drug | 38,957,080 | 231,177,927 | 387,915,928 | 532,330,140 | 1,151,423,995 | |
Budget impact | 0 | 171,746,567 | 310,413,347 | 451,311,054 | 933,470,968 | |
CDA-AMC scenario analysis 1: Subsequent therapies | Reference | 46,609,110 | 94,128,202 | 157,020,011 | 164,144,440 | 415,292,653 |
New drug | 46,609,110 | 261,002,450 | 445,622,553 | 564,167,967 | 1,270,792,969 | |
Budget impact | 0 | 166,874,248 | 288,602,541 | 400,023,527 | 855,500,316 | |
CDA-AMC scenario analysis 2: Treat until progression | Reference | 38,957,080 | 59,431,361 | 77,502,580 | 81,019,085 | 217,953,026 |
New drug | 38,957,080 | 231,369,462 | 392,950,605 | 552,057,607 | 1,176,377,673 | |
Budget impact | 0 | 171,938,101 | 315,448,024 | 471,038,522 | 958,424,647 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.