Drugs, Health Technologies, Health Systems
Reimbursement Review
Pemigatinib (Pemazyre)
Sponsor: Incyte Biosciences Canada Corporation
Therapeutic area: Cholangiocarcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
5-FU
fluorouracil
AE
adverse event
ASC
active symptom control
BTC
biliary tract cancers
CCA
cholangiocarcinoma
CCF
Cholangiocarcinoma Foundation
CDA-AMC
Canada's Drug Agency
CI
confidence interval
CR
complete response
DCR
disease control rate
DOR
duration of response
eCCA
extrahepatic cholangiocarcinoma
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-BIL21
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Cholangiocarcinomas and Gallbladder Cancer Module 21
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ESS
effective sample size
FOLFIRI
folinic acid, fluorouracil, and irinotecan hydrochloride
FOLFOX
folinic acid, fluorouracil, and oxaliplatin
HR
hazard ratio
HRQoL
health-related quality of life
iCCA
intrahepatic cholangiocarcinoma
IRC
independent review committee
ITC
indirect treatment comparison
KM
Kaplan-Meier
MAIC
matching-adjusted indirect comparison
mFOLFOX
modified folinic acid, fluorouracil, and oxaliplatin
MID
minimally important difference
NA
not applicable
NDA
New Drug Application
ORR
objective response rate
OS
overall survival
PD
progressive disease
pERC
pan-Canadian Oncology Review Expert Review Committee
PFS
progression-free survival
PR
partial response
QoL
quality of life
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
RWE
real-world evidence
SD
standard deviation
TEAE
treatment-emergent adverse events
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Pemigatinib (Pemazyre), tablets, 4.5 mg, 9 mg, and 13.5 mg, oral |
Indication | For the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 17, 2021 |
Sponsor | Incyte Biosciences Canada Corporation |
Resubmission notes | Pemigatinib was reviewed in 2021 and received a do not reimburse recommendation. This resubmission and associated Clinical Review Report includes the final data cut for the pivotal trial, FIGHT-202, and additional analyses within the indirect treatment comparison. These data were not included in the Clinical Review Report of the 2021 review but were considered during the reconsideration phase. Within the Studies Addressing Gaps in the Systematic Review section are 4 studies that are meant to support the results of the pivotal trial. These studies were not available at the time of the 2021 review or the reconsideration phase. |
CCA = cholangiocarcinoma; NOC = Notice of Compliance.
Sources: Application overview, product monograph.
Introduction
Gallbladder cancer and cholangiocarcinoma (CCA) are known as biliary tract cancers (BTCs) and account for 10% to 15% of all primary liver cancer.1,2 CCAs are most commonly adenocarcinomas and comprise 2 main subtypes: intrahepatic CCA (iCCA), which initiate from the biliary tree within the liver; and extrahepatic CCA (eCCA), which initiate outside the liver parenchyma.2,3 In Canada and the US, respectively, there are approximately 400 and 5,000 new cases of CCA diagnosed each year.4 The median age at diagnosis is 65 years in Western industrialized nations.5 The 5-year relative survival rates for iCCA and eCCA, respectively, are 9% and 10%.6 Diagnosis of CCA is most commonly made in the advanced stages (70% of patients are diagnosed with unresectable, locally advanced or metastatic disease),7 owing to an absence of symptoms until later in the course of the disease.8 The rate of recurrence is high in the minority of patients who are able to undergo potentially curative surgery.9 Symptoms commonly appear when a bile duct is blocked, and include jaundice; itching; light-coloured, greasy stools; dark urine; abdominal pain; loss of appetite and/or weight loss; fever; and nausea and vomiting.8
One of the most frequent genetic alterations in patients with iCCA involves FGFR2.7 FGFR2 fusions or rearrangements are found in 10% to 20%10 of patients with iCCA, whereas they rarely occur in patients with eCCA. Alterations involving other FGFRs are rare, with an incidence below 0.5%.11 Although there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known if patients with FGFR2 alterations represent a distinct prognostic subgroup.11
For patients with advanced-stage or unresectable CCA and a good Eastern Cooperative Oncology Group Performance Status (ECOG PS) (0 or 1), standard-of-care, first-line treatment is gemcitabine plus platinum (cisplatin or carboplatin) in combination with immunotherapy (durvalumab or pembrolizumab [Pemazyre]).12,13 If there are concerns about a patient’s renal function, oxaliplatin may be substituted for cisplatin.2 For patients with an ECOG PS of 2, gemcitabine monotherapy may be considered as first-line therapy.2 The median overall survival (OS), median progression-fee survival (PFS), and objective response rate (ORR) in patients with BTCs treated with standard-care, first-line palliative treatment, consisting of gemcitabine plus platinum therapy in combination with immunotherapy, range from 12.7 to 12.8 months, 6.5 to 7.2 months, and 26.7% to 28.7%, respectively.12,13 The clinical experts consulted by Canada's Drug Agency (CDA-AMC) noted that second-line treatment options are limited for patients whose disease has progressed after first-line treatment.
The ABC-06 trial14 compared the efficacy and safety of a modified regimen of folinic acid, fluorouracil [5-FU], and oxaliplatin (mFOLFOX) plus active symptom control (ASC) with ASC alone in patients with locally advanced or metastatic BTC (including CCA and gallbladder or ampullary carcinoma) who had progressed on first-line cisplatin and gemcitabine therapy. At the median follow-up time of 21.7 months, median OS was 6.2 months in the mFOLFOX group and 5.3 months in the ASC-alone group (hazard ratio [HR] = 0.69; 95% confidence interval [CI], 0.50 to 0.97; P = 0.031); median PFS was 4 months in the mFOLFOX group; and an objective response was observed in 5% of patients in the mFOLFOX group.2 According to the clinical experts consulted by CDA-AMC, other second-line therapies used in clinical practice in Canada include folinic acid, 5-FU, and irinotecan hydrochloride (FOLFIRI); 5-FU (alone or in combination with cisplatin or oxaliplatin); and capecitabine (alone or in combination with cisplatin or oxaliplatin). However, second-line mFOLFOX is currently the only treatment backed up by phase III trial data in this setting.5,14 The clinical experts consulted by CDA-AMC agreed that there is an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain health-related quality of life (HRQoL), delay disease progression, and prolong survival. In the absence of effective treatment options in the second-line setting, participation in clinical trials is recommended, as is best supportive care that includes the alleviation of biliary obstruction and full access to palliative care and symptom management.2
Pemigatinib (Pemazyre) is a small molecule kinase inhibitor with antitumour activity that inhibits FGFRs. FGFRs are receptor tyrosine kinases that activate signalling pathways in tumour cells.15 On September 17, 2021, pemigatinib was approved by Health Canada for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement. The sponsor’s requested reimbursement criteria for pemigatinib are in line with the Health Canada–approved indication. Pemigatinib underwent review through the standard Health Canada review pathway. Oral pemigatinib is available as 4.5 mg, 9 mg, and 13.5 mg tablets. The recommended starting dose is 13.5 mg, administered orally, for 14 consecutive days, followed by 7 days off therapy, in 21-day cycles. The product monograph states that treatment is to be continued until disease progression or unacceptable toxicity. Furthermore, the initiation of a low-phosphate diet is recommended when the phosphate level is greater than 5.5 mg/dL, and the addition of a phosphate-lowering therapy should be considered when the level is greater than 7 mg/dL. The dose of phosphate-lowering therapy is to be adjusted until the phosphate level returns to less than 7mg/ dL. It is recommended that the discontinuation of phosphate-lowering therapy be considered during pemigatinib treatment breaks or if the phosphate level falls below normal.16
In 2022, pemigatinib was reviewed by CADTH for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement, and received a do not reimburse recommendation. The CDA-AMC pan-Canadian Oncology Review Expert Review Committee (pERC) deliberated on the evidence from the FIGHT-202 trial and from an indirect treatment comparison (ITC) of pemigatinib and mFOLFOX. Although pERC acknowledged the rarity of FGFR2-positive CCA, ultimately the uncertainty related to the noncomparative evidence provided by the FIGHT-202 trial led to the recommendation against reimbursing pemigatinib. As part of this resubmission, the sponsor has submitted 4 additional studies that provide real-world evidence (RWE) in support of the FIGHT-202 trial data for pemigatinib.
The objective of this CDA-AMC review is to perform a systematic review of the beneficial and harmful effects of pemigatinib for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient groups that responded to the CDA-AMC call for patient input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
Two patient group inputs were received for this submission. One was a joint input from 5 patient groups — Cholangio-Hepatocellular Carcinoma Canada, Colorectal Cancer Resource & Action Network, Canadian Cancer Survivor Network, Canadian Cholangiocarcinoma Collaborative, and Gastrointestinal Society — and a separate input was from the Cholangiocarcinoma Foundation (CCF). The joint input was based on telephone and Zoom interviews with a total of 12 respondents who had treatment experience with pemigatinib. Among them, 11 participants were in Canada (Alberta, British Columbia, and Ontario) and 1 was in Israel.
The joint patient input highlighted the absence of any Canadian-reimbursable, first-line targeted therapy for patients with CCA and a FGFR2 fusion mutation. The respondents interviewed for the joint input reported various symptoms associated with chemotherapy, including nausea; loss of train of thought; inability to move; hair loss; swelling of the feet, hands, and face; and shortness of breath on exertion. Respondents also indicated that their quality of life (QoL) had been impacted while they were on systemic chemotherapy. Respondents highlighted some aspects of their treatment that were difficult to control, such as complications while taking treatments, inability to access pemigatinib because of its high cost, and difficult-to-control side effects (i.e., nausea, shortness of breath, flu-like symptoms, fatigue, inability to move, drowsiness, constipation, poor QoL).
The CCF input highlighted that for patients with FGFR2 fusions or rearrangements, pemigatinib represents both an alternative and a chance at improved outcomes. The patients interviewed for the joint input emphasized that the side effects were worth the benefits with respect to their QoL while on the targeted drug. The input also pointed to the feasibility of pemigatinib and the convenience of its oral administration. The CCF input further noted that the inability to access pemigatinib places an undue burden on patients who are already going through a challenging phase.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts consulted by CDA-AMC indicated that there are currently no effective, standard, funded second-line treatment options. Palliative therapy (e.g., FOLFOX, FOLFIRI, 5-FU, and capecitabine) and best supportive care are recommended for patients in the CCA setting. The clinical experts identified an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. The clinical experts consulted by CDA-AMC stated that pemigatinib is to be used in adult patients with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other alterations, as in the FIGHT-202 trial. Among patients enrolled in cohort A of the FIGHT-202 trial, the clinical experts did not identify any patient subgroups that would potentially be best suited for or benefit the least from pemigatinib. The clinical experts consulted by CDA-AMC felt that it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy.
The clinical experts agreed that patients would be identified as possible candidates for pemigatinib if they had an FGFR2 alteration. Clinical assessment to evaluate the response to treatment with pemigatinib would include regular radiological imaging (i.e., CT and/or MRI) and a CA19-9 biomarker test every 2 to 3 months to determine if a patient is experiencing disease progression. In addition, patients would be seen by an oncologist every 3 to 4 weeks for clinical assessment (i.e., of disease symptoms and performance status). The clinical experts indicated that the most clinically meaningful responses to treatment include disease control (i.e., disease stability or response), improvement in disease-related symptoms, improved pain control, weight gain, the regaining of a more active lifestyle, maintenance of HRQoL, and prolonged PFS and OS. Acceptable drug-related toxicity was also noted as a clinically meaningful outcome.
In the opinion of the clinical experts consulted by CDA-AMC, treatment with pemigatinib should be discontinued if a patient experiences disease progression, has a worsening performance status, is intolerant to or experiences unacceptable toxicity from pemigatinib (which cannot be improved with dose delays or reductions), or is not interested in continuing treatment.
Clinician Group Input
Clinician group input was received from the Canadian Gastrointestinal Oncology Evidence Network and other cholangiocarcinoma-treating physicians for this review. The clinicians noted that treatment goals for the management of CCA are extending survival, delaying disease progression, and maintaining QoL while on therapy. In terms of unmet needs, the clinicians suggested that new second-line treatments with meaningful survival benefits are required for this patient population. The clinicians who contributed to this input anticipate that pemigatinib will offer patients improved efficacy in terms of survival, PFS, response rate, and disease control. The clinicians further suggested that it would be reasonable to consider pemigatinib upfront for patients deemed unsuitable for cisplatin or gemcitabine plus durvalumab or pembrolizumab as first-line therapy. The clinicians involved in this input emphasized that a clinically meaningful response to treatment would be the achievement of tumour control (response or disease stabilization) and the maintenance of or improvement in QoL.
A clinician submission was received from a single community oncologist with experience treating 2 patients with CCA with pemigatinib. The first patient, diagnosed in their 70s, responded well to first-line chemotherapy and radiation, and the disease was controlled for 3 years. When the tumour began to grow again, the patient received gemcitabine and cisplatin, but the disease progressed after a few months. Testing revealed FGFR2 fusion, and the patient was able to enrol in a Patient Support Program to receive pemigatinib. This patient has continued to respond to pemigatinib for 2 and a half years. The second patient treated with pemigatinib was a 26-year-old person who had recently given birth. The clinician noted that while the response was brief of 4 months, the improvement in quality of life and the time she was able to spend with her child was precious. The clinician stated that, based on RWE with pemigatinib and its status as standard second-line therapy for patients with CCA and FGFR2 fusion or other alterations in other parts of the world, pemigatinib should be reimbursed in Canada.
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators, consideration for initiation of therapy, care provision, system issues, and economic considerations. The clinical experts consulted by CDA-AMC weighed evidence from the FIGHT-202 trial and supportive RWE studies and other clinical considerations to provide responses to drug program implementation questions from the Provincial Advisory Group. Refer to Table 3 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The FIGHT-202 trial is a multicentre, open-label, single-arm, phase II trial that evaluated the efficacy and safety of pemigatinib in patients with advanced and/or metastatic or surgically unresectable CCA with FGFR2 alterations, other FGF-FGFR alterations, or no FGF-FGFR alterations who had failed previous therapy. Patients were assigned to 3 cohorts, depending on FGF or FGFR status (cohort A: FGFR2 fusions or rearrangements; cohort B: FGF-FGFR alterations other than FGFR2 fusions or rearrangements; or cohort C: no FGF-FGFR alterations). This CDA-AMC review focuses on cohort A; cohort B and cohort C were not part of the requested reimbursement criteria to CDA-AMC and were not submitted for approval to Health Canada and are, therefore, beyond the scope of this review. A total of 147 patients were enrolled to received oral pemigatinib (13.5 mg orally once daily on a 2-weeks-on and 1-week-off schedule for each 21-day cycle). The primary outcome was ORR in cohort A, and secondary outcomes included ORR in cohort B, cohort A plus B, and cohort C, and, in all 3 cohorts, PFS, duration of response (DOR), disease control rate (DCR), OS, and safety. Exploratory end points included HRQoL and symptom severity.
Adults diagnosed with advanced and/or metastatic or surgically unresectable CCA with FGFR2-positive disease who had documented disease progression after at least 1 line of systemic therapy were enrolled in cohort A of the FIGHT-202 trial. At baseline, 107 patients were identified as having FGFR2 fusions or rearrangements and were grouped into cohort A. Cohort B included 20 patients with FGF-FGFR alterations other than FGFR2, and cohort C included 18 patients with no identified FGF-FGFR alterations. One patient grouped into an “undetermined” group was not assigned to any of the 3 cohorts because the local FGF-FGFR status results could not be confirmed by the central genomics laboratory. For patients in cohort A, mean age was 55.3 years (standard deviation [SD], 12.02 years), most patients were female (60.7%), and most were enrolled in trial sites in North America (59.8%) or Europe (29.9%). Almost all patients (89.0% of patients overall and 98.1% of patients in cohort A) had iCCA. The majority of patients in cohort A had metastatic disease (82.2%), with the lung and lymph nodes being the most common metastatic sites (54.2% and 53.3%, respectively). Median time since diagnosis was 1.28 years (range, 0.03 to 11.1 years) for patients in cohort A. The majority of patients in cohort A had an ECOG PS of 1 (53.3%), and all patients had received at least 1 line of systemic therapy for advanced or metastatic disease (60.7%, 27.1%, and 12.1% of patients received 1, 2, and ≥ 3 prior lines, respectively). Renal impairment grades were normal or mild for most patients in cohort A (39.3% or 43.9%, respectively), as were hepatic impairment grades (44.9% or 48.6%, respectively).17
The futility analysis, which was performed on October 12, 2017,18 was prespecified a priori in the statistical analysis plan. The timing of the subsequent analysis (March 22, 2019), when the predetermined threshold (i.e., lower limit of the 95% CI for ORR > 15%) would be assessed, was not prespecified a priori in the statistical analysis plan; however, the sponsor’s proposed timing was agreed upon by the FDA during their review of pemigatinib.
Efficacy Results
At the July 8, 2021, data cut-off date, the median duration of follow-up was 45.4 months in cohort A. Median OS was 17.48 months (95% CI, 14.36 to 22.93 months). The probability of patients surviving to 6 months was 88.7% (95% CI, 81.0% to 93.4%) and to 12 months was 67.6% (95% CI, 57.7% to 75.6%). Median PFS was 7.03 months (95% CI, 6.08 to 10.48 months). The PFS probability at 6 months was 61.1% (95% CI, 51.0% to 69.8%) and at 12 months was 32.3% (95% CI%, 22.9 to 42.1%).
As of the July 8, 2021, data cut-off date, the proportion of patients who achieved an objective response was 37.0% (95% CI, 27.94% to 46.86%); of these 40 patients, 3 (2.8%) achieved a complete response (CR) and 37 (34.3%) achieved a partial response (PR). Among the 40 patients who achieved an objective response, median DOR was 9.13 months (95% CI, 6.01 to 14.49 months). The probability of maintaining a response for at least 6 months was 67.8% (95% CI, 50.4% to 80.3%) and for at least 12 months was 41.2% (95% CI, 24.8% to 56.8%).
The proportion of patients with a best response of CR, PR, or stable disease was 82.4% (95% CI, 73.9% to 89.1%); of these 89 patients, 3 (2.8%) achieved a CR, 37 (34.3%) achieved a PR, and 49 (45.5%) achieved stable disease for 39 or more days after the first pemigatinib dose. ███ ███ ███████ █████ ██ ████████████ ██████████ ██████ █████████ ██████████ ███████ ████ █████ █████ ██ ███████████ ██████ █████████ ██████ ███ ███ █████ ████ ███ ████ ██ ██████.
The descriptive summary statistics of observed scores for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Cholangiocarcinomas and Gallbladder Cancer Module 21 (EORTC QLQ-BIL21) from baseline to cycle 33 (March 22, 2019, data cut-off date) or to cycle 42 (April 7, 2020, data cut-off date) were reported to be variable with no consistent trend.17 A definition for what constituted a clinically meaningful change from baseline in the target population was not provided. A post hoc analysis assessed observed mean changes from baseline to week 16 by subgroups of patients (i.e., patients with CR or PR, stable disease, or progressive disease [PD]). Results suggested that changes from baseline appeared directionally more favourable in patients with CR, PR, or stable disease than in patients with PD.
Harms Results
All patients in cohort A (100.0%) experienced at least 1 treatment-emergent adverse event (TEAE). The most commonly reported TEAEs were alopecia (59.3%), hyperphosphatemia (55.6%), diarrhea (53.7%), fatigue (46.3%), and nausea (42.6%). The percentage of patients who experienced serious TEAEs was █████ in cohort A. The most common serious TEAEs were pyrexia and cholangitis (each occurring in 4.6% of patients), abdominal pain (occurring in 3.7% of patients), and cholangitis infection (occurring in 2.8% of patients). Adverse events (AEs) led to the discontinuation of study treatment in ████ of patients in cohort A. None of the patients who withdrew from the FIGHT-202 study had an AE as the primary reason. TEAEs leading to treatment discontinuation ████████ ██████████ ███████████ ███████ ████████████████ ██████████ ███████ ████ ████ ███████████ ███████ ██████████████████ ███████ ███████ █████ █████████ ███████ ███ ██████ ███████ TEAEs leading to death occurred relatively rarely in cohort A █████████ ███ ████████ ███████ ██ ██████ ███ ████ ████ ████████████.
The percentage of patients who experienced nail toxicity TEAEs was █████ in cohort A. The most commonly reported nail toxicities included █████████████ ████████ ████ ██████████████ ████████ ████ █████████ ████████ ███████████ ███████ ██████████ ███ █████████████ █████████ ██ ████ ██ ████████ ████. No serious nail toxicity TEAEs occurred in cohort A.17
The percentage of patients who experienced serous retinal detachment TEAEs in cohort A was ████. The most commonly reported serous retinal detachment was ███████ ██████████ ██████ ████████ ██ █████████████ █████ ███████ ██████████ ██ ███████ ███████ ██████████ ███████ ███ ███████████ ███████ ███████ ██████ ███████ ██████████ ████ ███ ███████████ ██ ███ ████████.17
The percentage of patients who experienced hyperphosphatemia TEAEs in cohort A was ██████. The most commonly reported hyperphosphatemia events were █████████████████ ███████ ███ █████ ██████████ █████████ ██████. No serious hyperphosphatemia TEAEs occurred in cohort A.17
The percentage of patients who experienced hypophosphatemia TEAEs in cohort A was █████. The most commonly reported hypophosphatemia events were ████████████████ ███████ ███ █████ ██████████ █████████ ██████. No serious hypophosphatemia TEAEs occurred in cohort A.17
Critical Appraisal
The primary objective of phase II (randomized or nonrandomized) trials is to document safety outcomes and to investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict the harm and/or effectiveness of treatments. The clinical experts consulted by CDA-AMC noted that, despite the high unmet need, it would not be feasible to conduct a randomized controlled trial (RCT) in this small patient population to compare a targeted therapy such as pemigatinib with currently available therapies in the second-line setting in clinical practice in Canada. The FIGHT-202 trial included no formal statistical significance, but hypotheses testing and point estimates with 95% CIs were reported to estimate the magnitude of the treatment effect. A greater than 95% probability of having a 95% CI for ORR in cohort A with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. The subgroup analyses were noninferential, wide CIs reflected uncertainty in the effect estimates, and small sample sizes limited generalizability to a broader population. Interpretation of time-to-event end points such as OS or PFS is limited in single-arm studies; because all patients in cohort A received the same treatment, the extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. Although there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether patients with FGFR2 alterations represent a distinct prognostic subgroup.11 The clinical experts agreed that progression on prior systematic therapy is a major prognostic factor in the target population, and did not anticipate that patients would derive any substantial benefit from their underlying disease biology at the time they enrolled in the FIGHT-202 trial. The results for patient-reported outcomes were inconclusive, given the noncomparative, open-label design of the trial; the lack of a prespecified analysis of the patient-reported outcomes data; the substantial decline in patients available for assessments over time; and the lack of a definition for what constituted a clinically meaningful change from baseline in the target population.
Indirect Comparisons
Description of Studies
Two studies — FIGHT-202 and ABC-06 — were included in the sponsor’s ITC. The sponsor submitted an ITC in the form of an unanchored matching-adjusted indirect comparison (MAIC) between cohort A of the FIGHT-202 study and each of the 2 treatment groups in the ABC-06 study. The ABC-06 study compared an mFOLFOX regimen (oxaliplatin, L-folinic acid, and 5-FU) plus ASC with ASC alone in patients with locally advanced or metastatic BTC. Cohort A of the FIGHT trial only included patients with unresectable, locally advanced or metastatic CCA who had the FGFR2 mutation.
Efficacy Results
Overall Survival Pemigatinib Versus mFOLFOX Plus ASC
The results of the ITC favoured pemigatinib for PFS and OS over mFOLFOX plus ASC and over ASC alone. Median OS was █████ ██████ ██████ █████ ██ ███ ██ █ █████ ███████ for the pemigatinib group versus ████ ████ ███ ████ ██ █████ months for the mFOLFOX plus ASC group, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.209 (95% CI, 0.127 to 0.313), the HR using the results from the April 7, 2020, data cut-off date was █████ ████ ███ █████ ██ ██████, and the HR using the results from the July 8, 2021, study-close date was █████ ████ ███ █████ ██ ██████. Supplemental OS analyses were based on the July 8, 2021, data cut-off date comparing pemigatinib to mFOLFOX plus ASC in patients who received only 1 prior therapy. The number and effective sample size (ESS) for this subgroup were ██ and ████, respectively, and resulted in an HR of █████ ████ ███ █████ ██ ██████.
Overall Survival Pemigatinib Versus ASC Alone
Median OS was █████ ██████ ██████ █████ ██ ███ ██ █ █████ ███████ for the pemigatinib group versus ████ ████ ███ ████ ██ █████ months for the ASC group, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.163 (95% CI, 0.099 to 0.249), the HR using the results from the April 7, 2020, data cut-off date was ██████████ ███ █████████████, and the HR using the results from the July 8, 2021, study-close date was █████ ████ ███ █████ ██ ██████. Supplemental OS analyses were based on the July 8, 2021, data cut-off date comparing pemigatinib to ASC alone in patients who received only 1 prior therapy. The number and ESS for this subgroup were ██ and ████, respectively, and resulted in an HR of █████ ████ ███ █████ ██ ██████.
PFS for Pemigatinib Versus mFOLFOX Plus ASC
Median PFS was █████████ ███ ███████ ██████ months versus ████ ████ ████████ ████████ months for the pemigatinib versus mFOLFOX plus ASC groups, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.436 (95% CI, 0.319 to 0.599), the HR using the results from the April 7, 2020, data cut-off date was ██████████ ███ ████████ ██████, and that remained unchanged at the July 8, 2021, study-close date. Supplemental PFS analyses were based on the July 8, 2021, data cut-off date comparing the pemigatinib to mFOLFOX plus ASC in patients who received only 1 prior therapy. The number and ESS for this subgroup were ██ and ████, respectively, and resulted in an HR of █████ ████ ███ █████ ██ ██████.
For PFS, pemigatinib versus ASC alone was not assessed.
Harms Results
No comparisons for harms or safety were incorporated in the sponsor’s ITC.
Critical Appraisal
There were potentially important underlying differences between the FIGHT-202 and ABC-06 studies. In particular, FGFR2 alterations were not reported in the ABC-06 trial. Given that FGFR2 alterations occur almost exclusively in patients with iCCA and that the prevalence of FGFR2 alterations is less than 20%8 in patients with iCCA, there is likely a large disparity in FGFR2 mutation status between the study populations. Although the FIGHT-202 study only included patients with CCA, the ABC-06 study included patients with BTC, which encompasses gallbladder cancer and ampullary cancer, in addition to CCA. In cohort A of the FIGHT-202 study, 98% of patients had iCCA, compared with 42% in the mFOLFOX plus ASC group and 47% in the ASC group. Because the disease type and FGFR2 status were more restricted in the FIGHT-202 study, these differences could not be addressed through the weighting of patients in the pemigatinib group.
The covariates chosen for adjustment were based on age, sex, ECOG PS, and serum albumin. The following baseline characteristics were also available for both studies but did not appear to be considered: disease stage, the percentage of patients with prior surgery for cancer, and the number of lines of prior systemic therapy for advanced or metastatic cancer. The clinical experts consulted by CDA-AMC for this review were of the opinion that the number of lines of previous therapy was of key importance in terms of prognosis. The clinical experts were not aware of any additional prognostic factors and/or effect modifiers that were not reported in both studies and should have been considered.
Although there are retrospective studies suggesting that the presence of FGFR2 mutations in patients with CCA may be associated with better prognosis,19,20 the clinical experts consulted by CDA-AMC were of the opinion that FGFR2 mutation status is not an important prognostic factor in the indicated patient population. The clinical experts considered the fact that patients in both the FIGHT-202 and ABC-06 trials had progressed on prior systemic therapy to be of greater importance in terms of prognosis. The clinical experts expected patients in the FIGHT-202 study to have more advanced disease than patients in the ABC-06 study, because the FIGHT-202 study population was more heavily pretreated overall. It is unclear whether the pemigatinib group was more or less similar to the ABC-06 groups in this respect after weighting, as the weighting process did not take the number of prior lines of systemic therapy into account. If substantial differences remained, these differences could have led to bias against pemigatinib in all of the comparisons.
The ESS of the pemigatinib group was reduced by approximately 50% after weighting to the mFOLFOX plus ASC and ASC-alone groups, and it is unclear how representative the postweighting pemigatinib groups are of cohort A of the FIGHT-202 study.
Comparisons of pemigatinib and other relevant comparators (FOLFIRI, 5-FU alone or in combination with cisplatin or oxaliplatin, and capecitabine alone or in combination with cisplatin or oxaliplatin) were not available. Given that mFOLFOX plus ASC is the only therapy beyond the first-line setting with RCT evidence of an OS benefit, the clinical experts consulted by CDA-AMC said they expect that mFOLFOX plus ASC will have the greatest efficacy of all the relevant comparators.
In summary, for the unanchored MAIC to produce unbiased treatment effect estimates, all effect modifiers and prognostic variables need to be adjusted in the analysis. Residual confounding remains the major limitation of the MAIC, despite adjustments for age, sex, ECOG PS, and serum albumin in the comparisons of pemigatinib with mFOLFOX plus ASC and ASC alone. Although any bias introduced by the differences between the FIGHT-202 and ABC-06 studies in the number of prior lines of systemic therapy may have been against pemigatinib, the substantial differences in FGFR2 mutation status and tumour site between trials introduce a high degree of uncertainty in the OS and PFS results. Furthermore, MAICs cannot account for unknown cross-trial differences; thus, the MAIC estimates are susceptible to bias from unknown sources of confounding. An evaluation of potential bias from residual confounding was not reported; therefore, the magnitude of this bias in the relative treatment effect estimates is unclear. Overall, uncertainty remains around the magnitude of the additional benefit that pemigatinib provides for OS or PFS compared with mFOLFOX plus ASC or ASC alone.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
Parisi et al.21 conducted a multicentre, observational, retrospective study that assessed the effectiveness and safety of pemigatinib in patients with previously treated, locally advanced or metastatic CCA with FGFR2 fusion or rearrangements. Patients referred to 14 Italian centres and 25 French centres from July 2020 to September 2022 were evaluated (N = 72). These patients were initially included in 2 separate cohort studies but were pooled into a single dataset for analysis. An exploratory analysis compared PFS among patients in the cohort who had received pemigatinib in the second line to those who had received chemotherapy in the second line (and pemigatinib in a later line).
The study by Saverno et al.22 was a retrospective, observational, multisite chart review study based in the US. Physicians in the Cardinal Health Oncology Provider Extended Network were instructed to randomly select up to 10 patients who met the eligibility criteria during the index period. Between February 3, 2021, and February 22, 2023, physicians abstracted details related to demographics, clinical characteristics, biomarker testing patterns, treatment patterns, and clinical outcomes (N = 120).
The Ding et al.23 study was a retrospective, multisite physician survey designed to assess demographics, clinical characteristics, FGFR2 testing, and real-world treatment patterns and outcomes of patients with unresectable, locally advanced or metastatic CCA treated with pemigatinib (N = ██).
Post hoc analyses were conducted to compare patients from the FIGHT-202 study who received pemigatinib as second-line therapy with patients from the FIGHT-202 who received second-line systemic therapy before enrolling in the FIGHT-202 study.24 In total, 65 patients received pemigatinib as second-line therapy in the FIGHT-202 study and 41 patients received second-line systemic therapy before enrolling in the FIGHT-202 study, 39 of whom were evaluable for PFS. Of the 41 patients who received second-line systemic therapy, 38 received chemotherapy (gemcitabine plus cisplatin, 5-FU plus leucovorin calcium plus oxaliplatin, or 5-FU plus oxaliplatin) and 3 received anti-PD1 immunotherapy.
Efficacy Results
In the Parisi et al. study,21 median follow-up for the overall cohort was 19.5 months (95% CI, 15.0 to 30.5 months). Of the overall cohort of 72 patients, 2 patients recorded a CR and 31 patients recorded a PR, for an ORR of 45.8%. The median DOR was 7.0 months (95% CI, 5.8 to 9.3 months). Patients who received pemigatinib in the second-line setting had a median PFS of 8.6 months (95% CI, 6.6 months to not applicable [NA]), whereas patients who received chemotherapy in the second-line setting (and received pemigatinib in a later line) had a median PFS of 3.4 months (95% CI, 2.1 months to NA), with an HR of 3.88 (95% CI, 1.81 to 8.31; P < 0.001).
In the Saverno et al.study,22 the median duration of treatment in the first-line setting was 4.9 months (95% CI, 4.4 to 5.7 months); of these patients, 94.7% received chemotherapy as their first-line treatment. Most patients received pemigatinib in the second-line setting (94.2%), whereas 5.8% received pemigatinib in the third-line setting. The median duration of treatment with pemigatinib was 7.4 months (95% CI, 6.2 to 8.8 months). The ORR for the 116 patients with disease response data available was 59.2% (95% CI, 50.0% to 68.4%). The proportion of patients reporting a best response of CR was 5.0%, a best response of PR was 54.2%, and a best response of stable disease was 27.5%, for a DCR of 86.7%. Median PFS was 7.4 months (95% CI, 6.4 to 8.6 months). The PFS probability was 95.8% (95% CI, 90.3 to 98.2%) at 3 months and 71.5% (95% CI, 61.4 to 79.4) at 6 months. Median OS was not reported; the OS probability was 95.8% (95% CI, 90.3 to 98.2%) at 3 months and 88.4% (95% CI, 80.3 to 93.3%) at 6 months.
In the Ding et al.study,23 the ORR in the ██ patients with survey responses was █████. No patients included in the survey results achieved a CR. The proportion of patients achieving a best response of PR was █████ and the proportion of patients achieving a best response of stable disease was █████, for a DCR of ███. Median PFS was ████ ██████ ████ ███ ███ ██ ███. The PFS probability was ███ ████ ███ ██ ██ █████ at 6 months and ███ ████ ███ ██ ██ ████ at 12 months.
In the Bibeau et al. (2022) study,24 median PFS in patients receiving second-line pemigatinib therapy was 7.0 months (95% CI, 4.9 to 11.1 months), whereas median PFS in patients who received second-line therapy before enrolling in the FIGHT-202 study was 4.2 months (95% CI, 3.0 to 5.3 months). Median PFS in the 102 patients with evaluable results for first-line systemic therapy was 5.5 months (95% CI, 4.0 to 8.0 months).
Harms Results
In the Parisi et al. study,21 the proportion of patients who reported at least 1 TEAE was 97.2%, with the most common events being fatigue (69.4%), nail toxicities (61.1%), and hyperphosphatemia (55.6%).
Harms were not reported in the Saverno et al. study.22
Harms were not reported in the Ding et al. study.23
Harms were not reported in the Bibeau et al. (2022) study.24
Critical Appraisal
The clinical experts consulted considered the reported baseline characteristics in all 3 RWE studies to be representative of the expected patient population of Canada. The quality and completeness of the real-world data source were not reported. All 3 RWE studies were observational studies with no comparator arm; as such, it is difficult to assign, with certainty, causation of the effects seen to the study drug. It is not possible to determine the extent to which observed effects can be attributed to pemigatinib, as opposed to placebo or the natural history of the disease, in the absence of a frame of reference for comparison. Because of the retrospective nature of the study designs, ORR and progression assessments were conducted by the treating physician, potentially introducing bias; in phase II and phase III trials, assessments are commonly conducted by central review. The timing of assessments in observational, retrospective studies can also make the interpretation of time-to-progression outcomes challenging if patients are not being assessed at standardized time points.
The patient selection methodology in the Saverno et al. study22 potentially introduced selection bias, as the physicians were instructed to select, at random, 10 patients who fit the inclusion criteria during the index period. Because there was no methodology reported that indicated that the selecting physicians were blinded to the clinical outcomes of patients when making selections, it is possible that selection bias was introduced. Additionally, patients required at least 4 months of follow-up to be included (unless they died). It is not clear how many patients were excluded because of a lack of adequate follow-up, nor whether these patients might have differed in an important way in their prognosis.
The patient selection methodology in the Ding et al. study23 potentially introduced selection bias, as only ██ of a total ██ potential patients were included in the analysis. It is unknown how representative the ██ selected patients were of the larger group, as there was no response from the associated physicians, although the sponsor did provide a supplemental analysis showing that the total ██ patient population had a slightly longer mean duration of treatment (████ ██████ ██ █████ ██████), suggesting that the reduced patient population was not biased toward a longer treatment duration.
The study by Parisi et al.21 attempted to provide a comparative assessment of PFS for patients who received pemigatinib as second-line therapy during the study and patients who received other systemic therapy as second-line therapy before their inclusion in the study. Similar analyses were conducted in the Bibeau et al. (2022) study,24 drawing from patients in the FIGHT-202 study. Unadjusted comparisons were presented with no attempt to balance prognostic and confounding variables across groups and no assessment of the extent nor direction of residual confounding. The comparison was also affected by selection bias; patients in the comparator group needed to survive long enough to have received pemigatinib in a later line of therapy (this particular bias would favour the comparator), and those following different treatment trajectories (i.e., no pemigatinib in a later line) were excluded. The small sample size in each group introduced further uncertainty.
Conclusions
One phase II, singe-arm, open-label trial (FIGHT-202) provided evidence on the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (cohort A) who failed previous therapy. The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 15%) in cohort A. The clinical experts consulted by CDA-AMC felt that the achieved ORR of 37% (July 8, 2021, data cut-off date) was clinically meaningful for the target population, and durable (median, 9.13 months; 95% CI, 6.01 to 14.49 months). In the opinion of the clinical experts, the observed responses appeared to be higher than what is seen with currently used therapies in the second line in this setting (for example, 5% from the ASC plus FOLFOX group in the ABC-06 trial). There was uncertainty around the magnitude of the clinical benefit, given the limitations in the evidence from the noncomparative phase II clinical trial and the supportive RWE studies, the consistency of the results, and the high unmet need. The clinical experts consulted by CDA-AMC noted that, despite the high unmet need, it would not be feasible to conduct an RCT comparing a targeted therapy such as pemigatinib with currently available therapies in second line in clinical practice in Canada. Although the secondary efficacy outcomes, OS and PFS, appeared to be of the observed ORR achievements, the nonrandomized design of the FIGHT-202 trial made attributing PFS and OS events to pemigatinib challenging. The 3 RWE studies submitted to address gaps in the evidence showed outcomes similar to those of the pivotal trial (ORR ranged from 45.8% to 59.2%). Despite similar (and some additional) uncertainties to that of the pivotal trial, consistent positive results across both the pivotal trial and the RWE studies increase the confidence that the outcomes of the pivotal trial may be replicable in real-world practice. In the absence of a direct comparison between pemigatinib and relevant treatment options, the sponsor submitted an ITC. However, the CDA-AMC critical assessment identified limitations of the sponsor’s submitted unanchored MAIC (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which contributed to uncertainty around the magnitude of benefit of pemigatinib over other treatments. Similarly, 1 RWE study presented exploratory comparative information, but the conclusions were limited by methodological limitations. The results for the HRQoL and symptom severity exploratory outcomes remained inconclusive, owing to a number of important limitations. The toxicity profile of pemigatinib was considered manageable by the clinical experts consulted by CDA-AMC and, based on results from both the pivotal trial and the RWE studies, appeared favourable compared with currently available chemotherapy options.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of pemigatinib (4.5 mg, 9 mg, and 13.5 mg oral tablets) in adults with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Hepatobiliary cancers are highly lethal cancers arising in the liver (hepatocellular carcinoma), gallbladder, and bile ducts (iCCA and eCCA). Gallbladder cancer and CCA are known as BTCs.3 The most common type of liver cancer is hepatocellular carcinoma, followed by BTC, which account for 70% to 85% and 10% to 15% of all primary liver cancers, respectively.1,2 Gallbladder cancer is the most common type of BTC.3
CCAs are most commonly adenocarcinomas3 and comprise 2 main subtypes: iCCA, which initiates from the biliary tree within the liver; and eCCA, which initiates outside the liver parenchyma. eCCA is subdivided into perihilar CCA (or Klatskin tumour) and distal CCA.2 eCCA accounts for 80% to 90% of all CCAs, and iCCA is the least frequently reported subtype.2 The incidence of CCA is generally low (0.3 to 3.5 per 100,000 people) in Australia, Europe, and the US, but is higher in other parts of the world were certain parasite infections are common (e.g., China, Korea, and Thailand); Northeast Thailand has the higher incidence (90 per 100,000 people) of CCA in the world.2 In Canada and the US, respectively, there are approximately 400 and 5,000 new cases of CCA diagnosed each year.4 The median age at diagnosis is 65 years in Western industrialized nations.5 The 5-year relative survival rates for iCCA and eCCA, respectively, are 9% and 10%. The 5-year relative survival rates broken down by localized, regional, and distant stages of disease are 25%, 8%, and 2% for iCCA, respectively, and 15%, 16%, and 2% for eCCA, respectively.6
Although most CCAs arise spontaneously, without any known risk factors7, established risk factors for CCAs include primary sclerosing cholangitis, chronic ulcerative colitis, cysts in the bile ducts, and infection with a Chinese liver fluke parasite.25 Additionally, in Western countries, hepatitis C and liver cirrhosis have been identified as risk factors for iCCA; obesity, diabetes mellitus, metabolic disease, and certain substances (alcohol, tobacco, oral contraceptive pills, dioxin, and asbestos) have also been suggested as risk factor for CCA.7
The diagnosis of CCA is most commonly made at the advanced stage (70% of patients are diagnosed with unresectable, locally advanced or metastatic disease) because of the absence of symptoms until later in the course of the disease.7 The rate of recurrence is high among the minority of patients who are able to undergo potentially curative surgery.9 Symptoms commonly appear when a bile duct is blocked, and include jaundice; itching; light-coloured, greasy stools; dark urine; abdominal pain; loss of appetite and/or weight loss; fever; and nausea and vomiting.8
Different genetic alterations in BTC with oncogenic properties have been identified in recent years. Nearly 40% of patients harbour genetic alterations (e.g., IDH1/2, FGFR2, BRAF, and HER2/neu);9 however, evaluation of targeted treatment options is hampered by the overall low patient numbers.11 FGFR2 alterations are one of the most frequent genetic alterations in patients with iCCA.7 The FGFR2 fusions or rearrangements are found in 10% to 20%10 of patients with iCCA, but they rarely occur in patients with eCCA.11 Alterations involving other members of the FGFR family are rare; the incidence is below 0.5%.11 Although there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known whether patients with FGFR2 alterations represent a distinct prognostic subgroup11 and/or respond differently to chemotherapy than patients with unselected CCA.26 Retrospective studies20,27-29 in the first-line setting suggest that patients with CCA and FGFR alterations appear to have a better prognosis than patients in an unselected CCA population and that FGFR alterations occur more frequently in young women,9 although limitations preclude the drawing of strong conclusions. A number of phase II studies have been published that report results for FGFR2-directed therapies. Notably, 2 noncomparative phase II trials have reported very similar results for patients with iCCA and FGFR2 fusions or rearrangements. For pemigatinib, the objective response was 35.5% (38 of 107 patients), with an estimated median PFS of 6.9 months, and for infigratinib, the objective response was 31.0% (22 of 71 patients), with a median PFS of 5.8 months.11 Pemigatinib was approved by the FDA in April 2020 for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement detected by an FDA-approved test.30 Since March 2021, pemigatinib has been authorized in the European Union31 as monotherapy and is indicated for the treatment of adults with locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement who have progressed after at least 1 line of systemic therapy.32 The FDA has granted accelerated approval to infigratinib for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement detected by an FDA-approved test.33 Health Canada approved infigratinib, but it was cancelled premarket. There are 3 phase III RCTs of patients with CCA who are positive for FGFR2 fusions or rearrangements: the FIGHT-302 trial34 (NCT03656536) compares first-line standard of care (gemcitabine plus cisplatin) with pemigatinib (administered at a different frequency than in the FIGHT-202 trial [i.e., not administered continuously]; the estimated completion date is June 28, 2026); the PROOF trial35 (NCT03773302) compares standard of care with infigratinib; and the FOENIX-CCA336 (NCT04093362) compares standard of care with futibatinib (the estimated study completion date is February 2026) with primary end point PFS.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Most patients with CCA have advanced-stage disease at the time of diagnosis and although surgery is the preferred treatment option, only 35% of patients are eligible for surgical resection with curative intent.5 For patients with advanced-stage or unresectable CCA and a good EGOC PS (0 or 1), standard-of-care first-line treatment is gemcitabine plus platinum therapy in combination with immunotherapy.12,13 For patients with an ECOG PS of 2, gemcitabine monotherapy may be considered as first-line therapy. In patients with BTCs treated with standard-care first-line palliative treatment with gemcitabine plus platinum therapy in combination with immunotherapy, median OS ranges from 12.7 to 12.8 months, median PFS ranges from 6.5 to 7.2 months, and the ORR ranges from 26.7% to 28.7%.12,13 According to expert opinion, when disease progresses on first-line therapy, fewer than 50% of patients will be able to tolerate any second-line therapies, but a recent retrospective analysis from within Canada reported that only 30% of patients go on to receive second-line chemotherapy.37,38 Patients with molecularly unselected iCCA treated with standard first-line therapy were shown to have a median PFS of 8.4 months and a median OS of 15.4 months.26 There are currently no standard funded treatment options for patients whose disease has progressed on first-line treatment.9 In the absence of proven treatment options in the second-line setting for patients with CCA, participation in clinical trials is recommended, as is best supportive care, which includes alleviating biliary obstruction and full access to palliative care and symptom management.2 According to the clinical experts consulted by CDA-AMC, the second-line therapies used in clinical practice in Canada include FOLFOX, FOLFIRI, 5-FU (alone or in combination with cisplatin or oxaliplatin), and capecitabine (alone or in combination with cisplatin or oxaliplatin). A systematic review39 that included 761 patients participating in case reports, retrospective analyses, or phase II trials treated with second-line therapies for advanced BTC reported a mean OS of 7.2 months (95% CI, 6.2 to 8.2 months), a PFS of 3.2 months (95% CI, 2.7 to 3.7 months), and a response rate of 7.7% (95% CI, 4.6% to 10.9%).39
Second-line treatment with FOLFOX is currently the only regimen based on phase III trial data in this setting.5 The ABC-06 trial14 compared the efficacy and safety of mFOLFOX plus ASC with ASC alone in patients with locally advanced or metastatic BTC (including CCA and gallbladder or ampullary carcinoma) who had progressed on first-line cisplatin and gemcitabine therapy. At a median follow-up of 21.7 months, median OS was 6.2 months in the mFOLFOX group and 5.3 months in the ASC-alone group (HR = 0.69; 95% CI, 0.50 to 0.97; P = 0.031); median PFS was 4 months in the mFOLFOX group; and objective response was observed in 5% of patients in the mFOLFOX group.14
In Canada, there are currently no standard funded targeted treatment options for patients with CCA who harbour generic alterations that have been identified for targeted therapeutics. Another common genetic alteration in patients with iCCA is an IDH1/2 mutation, which is found in 10% to 23% of patients with iCCA; a targeted treatment, ivosidentib,40 has received priority review by the FDA for the treatment of patients with previously treated IDH1-mutant CCA.41
There was consensus among the clinicians that there is an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. It was also mentioned by the clinical experts that there are currently no biomarker-directed regimens specific to patients with FGFR2-positive CCA. The experts said they anticipate more promising benefits with a targeted therapy option in later lines than with chemotherapy for a disease that is steadily growing more resistant.
Drug Under Review
Pemigatinib is a molecule kinase inhibitor with antitumour activity that inhibits FGFRs. FGFRs are receptor tyrosine kinases that activate signalling pathways in tumour cells.15
On September 17, 2021, pemigatinib was issued market authorization with conditions by Health Canada for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement. The sponsor’s requested reimbursement criteria for pemigatinib are per the Health Canada–approved indication. In addition, the Health Canada indication states that the “clinical effectiveness of pemigatinib is based on ORR and DOR from a single-arm phase II trial in patients with specific FGFR2 rearrangements. Treatment with pemigatinib should be initiated following confirmation of a susceptible genetic alteration using a validated test.”16 Pemigatinib underwent review by Health Canada through a standard review pathway. Pemigatinib has no other Health Canada–approved indication.
After being granted priority review, with breakthrough therapy and orphan drug designation, pemigatinib received accelerated approval by the FDA in April 2020 for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement detected by an FDA-approved test.30 Since March 2021, pemigatinib has been authorized in the European Union31 as monotherapy and is indicated for the treatment of adults with locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement who have progressed after at least 1 line of systemic therapy.32 Pemigatinib has also been approved in Japan42 and various other countries.
Oral pemigatinib is available as 4.5 mg, 9 mg, and 13.5 mg tablets. The recommended starting dosage is 13.5 mg administered orally for 14 consecutive days, followed by 7 days off therapy, in 21-day cycles. The product monograph states that treatment is to be continued until disease progression or unacceptable toxicity. Furthermore, it is recommended that a low-phosphate diet be initiated when a patient’s phosphate level is greater than 5.5 mg/dL and that phosphate-lowering therapy be added when the level is greater than 7 mg/dL. The dose of phosphate-lowering therapy is to be adjusted until the phosphate level returns to less than 7 mg/dL. It is recommended that the discontinuation of phosphate-lowering therapy be considered during pemigatinib treatment breaks or when the phosphate level falls below normal.16
Table 2: Key Characteristics of Pemigatinib
Characteristics | Pemigatinib |
|---|---|
Mechanism of action | Inhibits FGFRs (1 to 3) by blocking the signalling of FGFRs and reducing the cell capabilities of cancerous cell lines that lead to constitutive activation of FGFR signalling pathways.43 |
Indicationa | For the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement. |
Route of administration | Oral |
Recommended dose | 13.5 mg administered orally once daily for 14 consecutive days, followed by 7 days off treatment, every 21-day cycle. |
Serious adverse effects or safety issues | According to the product monograph, hyperphosphatemia was reported in 59% of all patients who received pemigatinib, so recommendations include dietary phosphate restriction, administration of phosphate-lowering therapy, and dose modification when required. Serous retinal detachment occurred in 7.5% of all patients treated with pemigatinib, so recommendations include an ophthalmological examination before the initiation of therapy, every 2 months for the first 6 months of treatment, every 3 months afterward, and urgently at any time for visual symptoms. Pemigatinib treatment may cause fetal harm and may impair fertility in females. |
aThe full Health Canada–approved indication states that pemigatinib is indicated for the treatment of adults with previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangement; the clinical effectiveness of pemigatinib is based on overall response rate and duration of response from a single-arm phase II trial of patients with specific FGFR2 rearrangements; and that treatment with pemigatinib should be initiated after confirmation of a susceptible genetic alteration using a validated test.16
Sources: Application overview,26 product monograph.16
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website: https://www.cda-amc.ca/pemigatinib-0.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Two patient group inputs were received for this review. One was a joint input from 5 patient groups — Cholangio-Hepatocellular Carcinoma Canada, Colorectal Cancer Resource & Action Network, Canadian Cancer Survivor Network, Canadian Cholangiocarcinoma Collaborative, and Gastrointestinal Society — and a separate input was from the CCF. The joint input was based on telephone and Zoom interviews with a total of 12 respondents who had treatment experience with pemigatinib. Among them, 11 participants were across Canada (Alberta, British Columbia, and Ontario) and 1 was from Israel.
The joint patient input highlighted the absence of any reimbursable, first-line targeted therapy in Canada for patients with CCA and the FGFR2 fusion mutation. All respondents except 1 in the joint input indicated receiving gemcitabine and cisplatin. Half of the respondents in the joint input had received chemotherapy as first-line treatment, and the other half had received different initial treatments. The respondents interviewed in the joint input reported various symptoms associated with chemotherapy, including nausea; loss of train of thought; inability to move; hair loss; swelling of the feet, hands, and face; and shortness of breath on exertion. Respondents also indicated that their QoL had been impacted while they were on systemic chemotherapy. Respondents highlighted some aspects of their treatment that were more difficult to control, such as complications while taking treatments, inability to access pemigatinib because of its high cost, and difficult-to-control side effects (i.e., nausea, shortness of breath, flu-like symptoms, fatigue, inability to move, drowsiness, constipation, poor QoL). The joint input noted that symptoms also impacted patients' daily living activities.
According to the joint patient input received, respondents reported the average rating for QoL on pemigatinib as 9 out of 10. They also indicated some side effects while taking pemigatinib, such as thinning of hair, fingernail and toenail issues, dry eyes, and longer eyelashes. The patients interviewed in the joint input emphasized that the side effects were worth the benefits with respect to their QoL while on the targeted drug.
The CCF input highlighted that for patients with FGFR2 fusions or rearrangements, treatment with pemigatinib represents both an alternative and a chance to improve outcomes. The input also pointed to the drug’s feasibility and convenience for patients because of its oral administration. The CCF input further noted that the inability to access pemigatinib places an undue burden on patients who are already going through a challenging phase. The input emphasized that this could be an opportunity to address a critical unmet need and demonstrate compassion and commitment to those who often feel left behind by the health care system in Canada.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of CCA.
Unmet Needs
The clinical experts consulted by CDA-AMC noted that there are currently no standard funded second-line treatment options for patients with unresectable, locally advanced or metastatic CCA with FGFR2 alterations. The clinical experts noted that currently mFOLFOX is the most commonly used therapy in the target population in clinical practice in Canada. The first phase III RCT of patients with CCA in the second-line setting compared the chemotherapy option mFOLFOX plus ASC with ASC alone after standard first-line chemotherapy. The minimal survival benefit (OS of 6.2 months with mFOLFOX and 5.3 months with ASC alone) highlights the lack of options for these patients. It was emphasized by the clinical experts that patients who have progressed on first-line chemotherapy often have a rapidly declining performance status. The clinical experts noted that mFOLFOX is a difficult treatment to tolerate for patients who are already quite ill. There was consensus among the clinicians that there is an unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, delay worsening of symptoms, maintain HRQoL, delay disease progression, and prolong survival. The option of oral therapy was also noted by the experts as preferable for patients in terms of access and comfort.
Place in Therapy
The clinical experts consulted by CDA-AMC stated that pemigatinib was to be used in adult patients with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement, as was used in the FIGHT-202 trial. It was agreed that oral pemigatinib would likely shift the current treatment paradigm. The FIGHT-202 trial excluded patients who were intolerant to standard first-line therapy but did not experience PD. The clinical experts consulted by CDA-AMC felt that it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy, given the favourable safety profile of oral pemigatinib. Furthermore, the clinical experts anticipated that the benefit of treatment with pemigatinib would be experienced regardless of the number of previous lines of systemic therapy received, as long as patients have the FGFR2 alteration. However, the clinical experts agreed that patients should not have previously been treated with an FGFR2-targeted therapy.
Patient Population
The clinical experts noted that the presence of FGFR2 fusions or other rearrangements should be determined before the initiation of treatment with pemigatinib. The clinical experts noted that it would be ideal to have molecular FGFR2 testing results available after the patient commences first-line therapy and before the disease progresses. A valid test would involve next-generation sequencing, which was used in the FIGHT-202 trial to enrol patients in cohort A. The sponsor noted that FGFR2 testing is currently available and funded in Alberta, Ontario, and Quebec, and is provided on a case-by-case basis in Nova Scotia and New Brunswick. It was emphasized that patients without the FGFR2 alteration would not be expected to derive any benefit from pemigatinib.
Assessing the Response to Treatment
The clinical experts agreed that a patient’s clinical condition and CT imaging are used to assess response to treatment. It was suggested that patients should be assessed clinically every 3 weeks and assessed radiographically with CT imaging every 2 to 3 months. One clinical expert noted that a clinically meaningful response would be a maintenance or improvement in QoL and a prolongation of survival.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC agreed that patients should discontinue treatment if there is clear evidence of disease progression based on imaging, poor tolerance of treatment that cannot be improved with dose delays or dose reductions, or the patient chooses to discontinue treatment.
Prescribing Considerations
Pemigatinib is an oral drug that is self-administered in a patient’s home. The clinical experts consulted by CDA-AMC agreed that patients should have regular access to outpatient oncology clinics to ensure that treatment tolerance is confirmed and that the disease has not progressed.
Additional Considerations
The clinical experts reiterated the rarity of this disease and the inability to conduct a phase III trial.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Clinician group input was received from the Canadian Gastrointestinal Oncology Evidence Network and other cholangiocarcinoma-treating physicians for this review. A total of 15 clinicians provided input for this submission.
The clinicians noted that the treatment goals for the management of CCA are extending survival, delaying disease progression, and maintaining QoL while on therapy. In terms of unmet needs, the clinicians suggested that new second-line treatments with a meaningful survival benefit are required for this patient population. The clinicians who contributed to this input said that they anticipate that pemigatinib will offer patients improved efficacy in terms of survival, PFS, response rate, and disease control. The clinicians also emphasized that the convenient oral route of administration of pemigatinib will likely contribute to improvements in QoL for patients, because it requires fewer visits to a cancer centre and less chair time than alternative treatment options. The clinicians further suggested that it would be reasonable to consider pemigatinib upfront for patients deemed unsuitable for first-line therapy with cisplatin or gemcitabine plus durvalumab or pembrolizumab. The clinicians noted that pemigatinib would be best suited for patients with CCA who harbour FGFR2 gene fusions or other rearrangements and have an ECOG PS of 0 to 2, after prescreening for FGF-FGFR status using DNA or RNA sequencing. The clinician group also noted that patients with biliary cancer but no FGFR2 fusions or rearrangements should not be treated with pemigatinib. The clinicians who contributed to this input emphasized that a clinically meaningful response to treatment would be to achieve tumour control (response or disease stabilization) and to maintain or improve QoL. In terms of treatment discontinuation, the clinicians explained that cancer progression on imaging, poor tolerance of the treatment which cannot be improved with dose delays or reductions, or patient decision to stop treatment are the deciding factors.
A clinician submission was received from a single community oncologist with experience treating 2 patients with CCA with pemigatinib. The first patient had been diagnosed in their 70s and responded well to first-line chemotherapy and radiation, which controlled the disease for 3 years. When the tumour began to grow again, the patient received gemcitabine and cisplatin, but the disease progressed after a few months. Testing revealed FGFR2 fusion, and the patient was able to enrol in the Patient Support Program to receive pemigatinib. This patient has continued to respond to pemigatinib for 2 and a half years. The second patient treated with pemigatinib was a 26-year-old person who had recently given birth. The community oncologist noted that although the response was brief — 4 months — the improvement in QoL and the time she was able to spend with her child was precious. The clinicians reiterated that, based on RWE and the status as standard second-line therapy elsewhere in the world, pemigatinib should be reimbursed in Canada.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There is no established standard of care. IV chemotherapy (e.g., mFOLFOX, FOLFIRI, capecitabine) or best supportive care may be used. New evidence submitted for this review is not comparative data. | The clinical experts noted that given the small number of eligible patients, a comparative study is not feasible. Results from the FIGHT-202 trial are consistent with observations from other studies that have evaluated drugs in small biomarker-selected patient populations. |
Considerations for initiation of therapy | |
Patients with an ECOG PS of 0 to 2 were eligible for pemigatinib the FIGHT-202 trial, and were included in the RWE submitted. Are patients with an ECOG PS greater than 2 eligible for treatment? | The clinical experts agreed that patients with an ECOG PS of 3 are not included in the available evidence for pemigatinib and are unlikely to be offered treatment with pemigatinib because they are too unwell. |
Standard first-line treatment is typically cisplatin and gemcitabine. Should patients who have experienced disease progression while on cisplatin and gemcitabine be eligible for pemigatinib? | The clinical experts agreed that patients who received cisplatin plus gemcitabine as first-line therapy whose progressed should be eligible for pemigatinib. |
Care provision issues | |
Oral pemigatinib is available in 4.5 mg, 9 mg, and 13.5 mg tablets in a 14-day blister pack (not unit dose format). The recommended starting dose is 13.5 mg administered orally once daily for 14 consecutive days, followed by 7 days off therapy, in 21-day cycles. Wastage is not considered in economic analyses, but is likely to occur when dose adjustment is needed or when patients are admitted to hospital. | Comment provided to inform pERC deliberation. |
Serous retinal detachment with symptoms of blurred vision, visual floaters, or photopsia (estimated in 11% of patients and in 1.3% of patients with grade 3 to 4 disease). Although not specified in the product monograph, other sponsor information suggests an comprehensive ophthalmological examination, which includes optical coherence tomography, before the initiation of pemigatinib, every 2 months for the first 6 months, and every 3 months thereafter. After the onset of visual symptoms, patient should be referred for ophthalmologic evaluation and then every 3 weeks until resolution or discontinuation of pemigatinib. The cost of ophthalmological exams should be considered in economic analyses. | Comment provided to inform pERC deliberation. |
A review of drug interactions is needed; pemigatinib interacts with CYP3A inhibitors and inducers. | Comment provided to inform pERC deliberation. |
Genetic testing (FGFR2) for CCA is not always funded routinely, but needs to be funded in conjunction with this treatment. | Comment provided to inform pERC deliberation. |
System and economic issues | |
Costs associated with FGFR2 testing should be considered and incorporated into economic analyses. | Comment provided to inform pERC deliberation. |
CCA = cholangiocarcinoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FOLFIRI = folinic acid, fluorouracil, and irinotecan; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; pERC = pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of pemigatinib (oral tablets of 4.5 mg, 9 mg, and 13.5 mg) for the treatment of adults with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or other rearrangement. The focus will be placed on comparing pemigatinib to relevant comparators and identifying gaps in the current evidence. As this is a resubmission (the original clinical review was conducted in 2021), the Clinical Evidence section consists of the evidence identified by the CDA-AMC (known at the time as CADTH) when the original systematic review was conducted. For the current review, the sponsor has submitted the final data cut from the pivotal trial; this will be presented in the first section. The second section includes indirect evidence from the sponsor that was originally provided in the 2021 review, additional analyses presented in the reconsideration process of the 2021 review, and another analysis based on the final data cut from the pivotal trial. The third section includes studies that were considered by the sponsor to address important gaps in the systematic review evidence. These 4 additional studies are meant to support the results of the pivotal trial. These studies were not available at the time of the 2021 review or the reconsideration phase.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
1 pivotal study or RCT identified in systematic review
1 ITC
4 additional studies addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by the literature search conducted by CADTH staff. Refer to the 2021 Pemigatinib Clinical Review Report for details of the systematic review. Focus is placed on the final data cut, which was not available during the 2021 review.
Description of Studies
Characteristics of the included study are summarized in Table 4.
Table 4: Details of the FIGHT-202 Study
Detail | FIGHT-202 |
|---|---|
Design and population | |
Study design | Phase II, multicentre, open-label, single-arm, multicohort trial |
Locations | Patients enrolled at 67 sites across 12 countries:
|
Patient enrolment dates | January 17, 2017, to March 22, 2019 |
Data cut-off dates | Futility analysis: October 3, 2017 Initial data cut: March 22, 2019 Updated data cut: August 30, 2019 Updated data cut: April 7, 2020 Study-close date: July 8, 2021 |
Enrolled (N) | 146 patientsa
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Pemigatinib: 13.5 mg orally once daily on a 2-weeks-on and 1-week-off schedule for each 21-day cycle Treatment should continue until radiological disease progression, unacceptable toxicity, withdrawal of consent, or physician choice |
Comparator(s) | Not applicabled |
Outcomes | |
Primary end point | ORR (cohort A) |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publicationsg | Publication: Abou-Alfa et al. (2020),44 primary analysis results (data cut-off date of March 22, 2019) ClinicalTrial.gov entry: ClinicalTrials.gov. [internet] Bethesda (Maryland): National Library of Medicine (US). February 23, 2023. Identifier: NCT02924376. A Study of Pemigatinib in Participants With Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma (FIGHT-202). Updated June 17, 2021. [cited December 23, 2024] Available from: https://clinicaltrials.gov/ct2/show/NCT02924376 |
AE = adverse event; CCA = cholangiocarcinoma; CNS = central nervous system; CYP3A4 = cytochrome P3A4; DCR = disease control rate; DOR = duration of response; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer QLQ-Cholangiocarcinomas and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PK = pharmacokinetic; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aOne patient was not assigned to any cohort because of an inadequate tissue sample. This patient was not included in the efficacy analyses.44
bTo be enrolled and for initial cohort assignment, patients had to have results from a certified local laboratory; however, final cohort assignment for statistical analyses was based on next-generation sequencing results (using the Foundation Medicine clinical trial assay) from the central genomics laboratory.26
cAbnormal laboratory parameters: total bilirubin ≥ 1.5 × upper limit of normal (ULN); ≥ 2.5 × ULN if Gilbert syndrome or disease involving liver); aspartate transaminase (AST) and alanine aminotransferase (ALT) > 2.5 ULN (AST and ALT > 5 × ULN in the presence of liver metastases); creatinine clearance ≤ 30 mL/min based on Cockroft-Gault; serum phosphate > institutional ULN; serum calcium outside of the institutional normal range or serum albumin-correct calcium outside of the institutional normal range when serum albumin is outside of the institutional normal range; potassium levels < institutional lower limit of normal, but supplementation can be used to correct potassium level during the screening.
dThe FIGHT-202 study was a noncomparative single-arm phase II trial.
eAs specified in the protocol of the FIGHT-202 trial, only patients from the US were allowed to enrol in cohort C.
fSpecific PK analyses were not specified a priori in the statistical analysis plan.26
gThe EORTC QLQ-BIL21 is only translated and validated in the primary languages of these countries.26
Sources: Abou-Alfa et al. (2020),44 Clinical Study Report,17 sponsor’s response.18
FIGHT-202 is a multicentre, open-label, single-arm phase II trial that evaluated the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations, alterations in another FGF-FGFR, or no FGF -FGFR alterations, who failed previous therapy. Patients were assigned to 3 cohorts, depending on their FGF-FGFR status (cohort A consisted of patients with FGFR2 fusions or rearrangements; cohort B consisted of patients with FGF-FGFR alterations other than FGFR2 fusions or rearrangements; and cohort C consisted of patients with negative for FGF-FGFR alterations). This CDA-AMC review focuses on cohort A; cohort B and cohort C were not part of the requested reimbursement criteria to CDA-AMC and not submitted for approval to Health Canada and are therefore beyond the scope of this review. The primary objective of the trial was to assess the efficacy of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (patients in cohort A) who failed at least 1 previous treatment. Patients in this international trial were enrolled at 67 sites across 12 countries, which are listed in Table 4. The majority of sites were in the US, followed by Europe; there were no sites in Canada. Enrolment started on January 12, 2017, and ended on March 22, 2019.17
A total of 146 patients were enrolled to received oral pemigatinib (13.5 mg orally once daily on a 2-weeks-on, 1-week-off schedule for each 21-day cycle). Best supportive care was administered as needed and included, but was not limited to, palliative radiotherapy for bone lesions, stent placement, or replacement for blocked bile ducts.18 Pemigatinib was administered until documented disease progression or unacceptable toxicity.26 The study design is depicted in Figure 1.
To be enrolled and for initial cohort assignment, patients had to have results from a certified local laboratory; however, final cohort assignment for statistical analyses was based on next-generation sequencing results (using the Foundation Medicine clinical trial assay) from the central genomics laboratory.26 The study consisted of 3 phases: the screening phase (lasting up to 28 days); the treatment phase; and the follow-up phase. During the follow-up phase, patients were followed for safety (final follow-up visit was 30 to 35 days after the end of treatment), for disease status every 9 weeks (for patients who discontinued pemigatinib for reasons other than disease progression), and for OS at least every 12 weeks.26 Response assessment, according to Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1),45 was based on radiologic imaging and performed by an independent review committee, and occurred every 2 cycles (every 6 weeks) for the first 4 cycles, every 3 cycles (every 9 weeks) thereafter, and at the treatment discontinuation visit.26 Tumour response was also evaluated by the investigator.26 Safety and tolerability were evaluated by monitoring the frequency, duration, and severity of AEs.26
At the March 22, 2019, data cut-off date, the predetermined threshold for a positive study outcome (lower limit of the 95% CI for ORR > 15%) was achieved.17 Although the timing of the March 22, 2019, data cut-off date was not prespecified a priori in the statistical analysis plan, the sponsor’s proposed timing was agreed to by the FDA during their review process of pemigatinib.5 Two additional updated analyses occurred at the August 2019 and April 2020 data cut-off dates; the former was a 4-month safety update required for the FDA New Drug Application (NDA), and latter was performed to support the safety data summaries for another indication outside of Canada.18 The final study-close data cut-off date was July 8, 2021. The FIGHT-202 trial was sponsored by the Incyte Corporation.44
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria used in the FIGHT-202 trial are described in Table 4. Briefly, the trial enrolled adults 18 years and older who were diagnosed with advanced, metastatic, or surgically unresectable CCA and who progressed after at least 1 line of systemic therapy (prior therapy with selective FGFR inhibitors was not permitted). Patients had to have documentation of FGF-FGFR gene alteration status and radiologically measurable disease according to RECIST 1.1. At screening, patients had to have an ECOG PS of 0 to 2, a life expectancy of at least 12 weeks, adequate hepatic and renal function, and no clinically significant corneal or retinal disorder confirmed by ophthalmologic examination.17
Interventions
Patients enrolled in the FIGHT-202 trial self-administered oral pemigatinib once daily on a 2-weeks-on, 1-week-off schedule. Treatment and dose are described in Table 5.
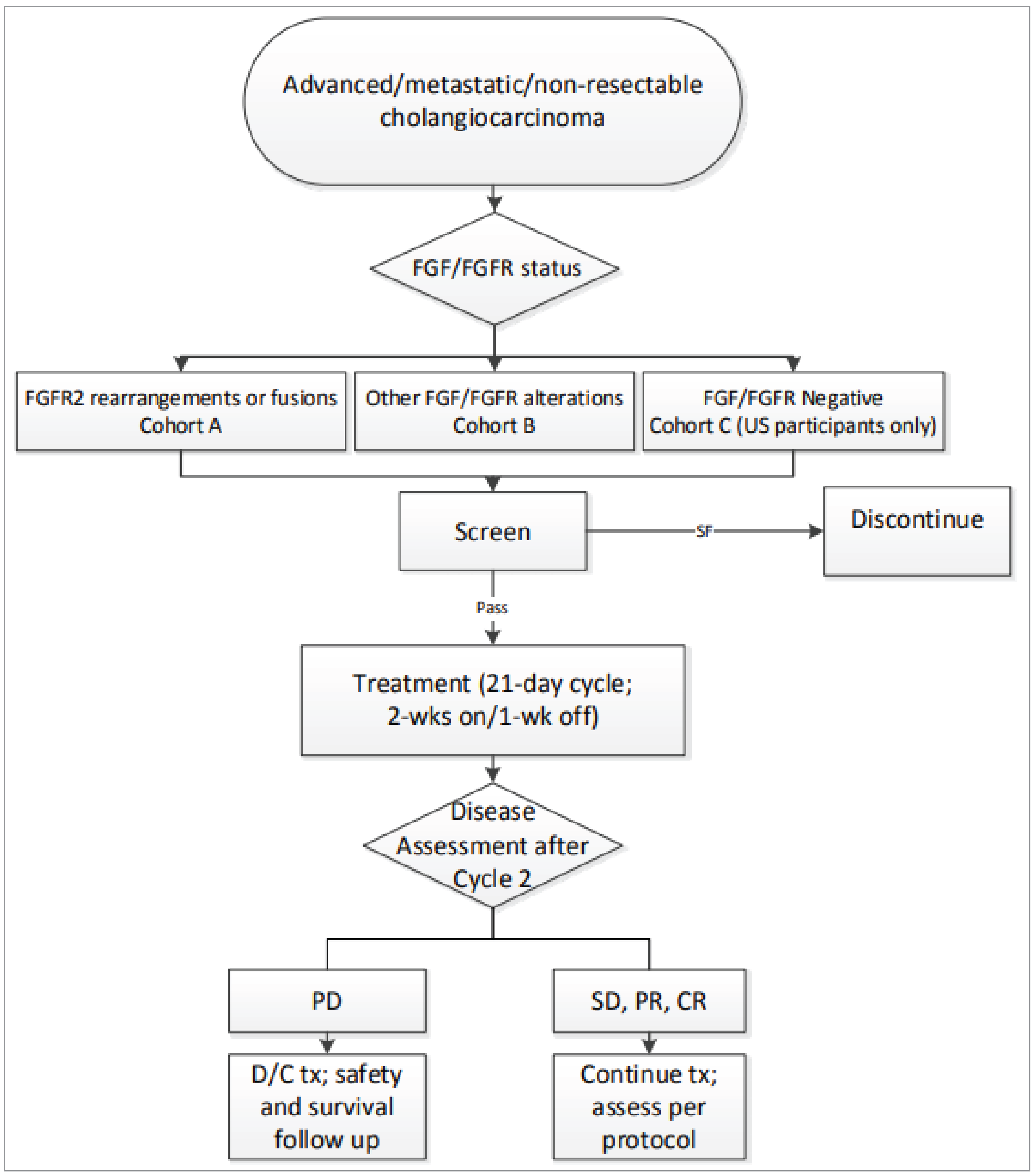
CR = complete response; D/C = discontinue; PD = progressive disease; PR = partial response; SD = stable disease; SF = screen fail; tx = therapy; wk = week.
Source: Clinical Study Report.17
Table 5: Treatment Regimen in the FIGHT-202 Trial
Detail | FIGHT-202 |
|---|---|
Dose | Pemigatinib: 13.5 mg administered orally (tablets) once daily for 2 weeks continuously (14 days) followed by a 1-week (7 days) pause for each 21-day treatment cycle. |
Treatment discontinuation | Patients were withdrawn from the study treatment when the following criteria were met:
Patients may be withdrawn from the study treatment when the following criteria are met:
|
AE = adverse event; ECG = electrocardiogram; IEC = independent ethics committee; IRB = institutional review board; QTc = corrected QT interval.
Source: Sponsor’s submission.26
Dose Modifications
Dose interruptions, delays, or modifications were permitted and were guided by the occurrence of toxicities (related or unrelated to the study drug). A maximum of 2 dose reductions (from a daily dose of 13.5 mg to 9 mg and further to 6 mg) was recommended. Patients could not receive a dose below 6 mg daily. No treatment schedule modification was allowed. At the occurrence of any grade 3 toxicity not manageable with supportive care or an aspartate transaminase and/or alanine transaminase level more than 5.0 × the upper limit of normal, treatment could be interrupted for up to 14 days until toxicity is resolved to grade 1 or lower. Once a dose interruption occurred, pemigatinib was restarted either at the same dose as before the dose interruption or at the next lower dose, and monitored as clinically indicated. Situations in which treatment was delayed for more than 14 days before it was restarted had to be discussed on case-by-case basis with the sponsor. In cases of recurrent grade 3 toxicity after 2 dose reductions or of any other grade 4 toxicity, pemigatinib administration had to be discontinued; exceptions to that required the sponsor’s approval.17
Concomitant Medication
Concomitant medications could be used to treat comorbidities or AEs, as long as they did not include potent CYP3A4 inhibitors and inducers, moderate CYP3A4 inducers (there was no restriction on topical ketoconazole), another selective FGFR inhibitor, an investigational study drug for any indication, or any anticancer medications other than the study drug.26
Outcomes
A list of efficacy end points identified in the CDA-AMC review protocol that were assessed in the clinical trial included in this review is provided in Table 6 and is subsequently summarized.
Table 6: Summary of Outcomes of Interest Identified in the CDA-AMC Review Protocol
Outcome measurea | FIGHT-202 end point |
|---|---|
OS | Secondary (cohort A, cohort B, and cohort C)b |
PFS | Secondary (cohort A, cohort B, and cohort C)b |
ORR | Primary (cohort A) |
Secondary (cohort B)b | |
Secondary (cohort A plus cohort B)b | |
Secondary (cohort C)b | |
DOR | Secondary (cohort A, cohort B, and cohort C)b |
DCR | Secondary (cohort A, cohort B, and cohort C)b |
TTP | Not measured in FIGHT-202 |
HRQoL | |
EORTC QLQ-C30 | Exploratory (cohort A, cohort B, and cohort C)b |
Symptom severity | |
EORTC QLQ-BIL21 | Exploratory (cohort A, cohort B, and cohort C)b |
Safety | |
Frequency, duration, and severity of AEs | Secondary (cohort A, cohort B, and cohort C)b |
AE = adverse events; DCR = disease control rate; DOR = duration of response; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer QLQ-Cholangiocarcinomas and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TTP = time to progression.
aOutcomes are presented in order of priority identified in the CDA-AMC review protocol.
bThis CDA-AMC review presents results just for cohort A; cohort B and cohort C were not part of the requested reimbursement criteria to CDA-AMC, were not submitted for approval to Health Canada, and are therefore beyond the scope of this review.
Source: Statistical analysis plan.26
Overall Survival
OS was a secondary outcome of the FIGHT-202 trial and was defined as the time from the date of the first dose until the date of death from any cause.26 The OS analyses were performed for patients in cohort A, cohort B, and cohort C.26
Progression-Free Survival
PFS was a secondary outcome of the FIGHT-202 trial and was measured as the time from the date of the first dose of study drug until the date PD was first recorded or death, whichever occurred first. The date of PD was the time point at which progression was first documented. Disease progression for the analysis of PFS was determined according to the RECIST 1.1 criteria assessed by an independent centralized radiological review committee. PFS was also analyzed based on investigator assessment. The PFS analyses were performed for patients in cohort A, cohort B, and cohort C.26
Objective Response Rate
ORR assessed in cohort A was the primary outcome of the FIGHT-202 trial. ORRs evaluated in cohort B, cohort A plus cohort B, and cohort C were secondary outcomes. ORR was defined as the proportion of patients who achieved a best overall response of CR (the disappearance of all target lesions) or a PR (a decrease of ≥ 30% in the sum of the longest diameters of target lesions) at any postbaseline visit before first PD. Best overall response was defined as the best response documented after baseline but before and including the first PD, in the following order: CR, PR, stable disease, PD, and not evaluable. Clinical response for the analysis of ORR was determined based on RECIST 1.1, assessed by an independent radiological review committee using central genomics laboratory results, and required confirmation of CR and PR at least 4 weeks after the initial assessment. The only clinical data transmitted by the site to the independent review committee (IRC) were radiation history, prior surgeries, and investigator-documented benign radiographic abnormalities. For the patient to be considered stable, the criteria for stable disease had to be met at least once after the date of the first dose, at a minimum interval of 39 days; if these criteria were not met, patients were recorded as having an overall response of PD, if the next available assessment indicated PD, or not evaluable, if there was no additional assessment available. ORR was also analyzed based on investigator assessment; however, confirmation of CR and PR was not required.26
Duration of Response
DOR was a secondary outcome of the FIGHT-202 trial and was defined as the interval from the date of CR or PR (i.e., an overall response contributing to an objective response) to the date of death or first overall response of PD, whichever occurred first.26 Clinical response for the analysis of DOR was determined based on RECIST 1.1, assessed by an independent radiological review committee. The DOR analyses were performed for patients in cohort A, cohort B, and cohort C. DOR was also analyzed based on investigator assessment.26
Disease Control Rate
The DCR was a secondary outcome of the FIGHT-202 trial and was defined as the proportion of patients with a best response of CR, PR, or stable disease. Clinical response for the analyses of DCR was determined based on RECIST 1.1, assessed by an independent radiological review committee using central genomics laboratory results, and required confirmation of CR and PR at least 4 weeks after the initial assessment.26 The DCR analyses were performed for patients in cohort A, cohort B, and cohort C. The DCR was also analyzed based on investigator assessment.26
Time to Progression
This outcome was not assessed in the FIGHT-202 trial.
Health Related Quality of Life
The HRQoL outcomes measured in the trial included the EORTC QLQ-C30. Neither an analysis plan or objective nor a minimally important difference (MID) for the EORTC QLQ-C30 instrument was specified a priori in the statistical analysis plan; it was noted, however, that the scores for each scale were to be calculated. According to the study’s protocol, scores for each scale and changes from baseline to each visit were to be measured and summarized descriptively.26 The EORTC QLQ-C30 was assessed at baseline, then at every 3 cycles, starting with cycle 3, until discontinuation of the study treatment, and then at the end-of-treatment visit.26
The EORTC QLQ-C30 is 1 of the most commonly used patient-reported outcomes measures in oncology clinical trials. It is a multidimensional, cancer-specific, evaluative measure of HRQoL. It was designed specifically to assess changes in participants’ HRQoL in response to treatment in clinical trials. The core questionnaire of the EORTC QLQ-C30 consists of 30 questions that are scored to create 5 multiitem functional scales, 3 multiitem symptom scales, 6 single-item symptom scales, and a 2-item QoL scale. Most questions have 4 response options (not at all, a little, quite a bit, very much), and scores on these items range from 1 to 4. For the 2 items that form the global QoL scale, however, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent). Version 3.0 of the questionnaire, used in the included trials in this report, is the most current version. Each raw scale score is converted to a standardized score that ranges from 0 to 100, using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better QoL (i.e., higher scores simply reflect higher levels of response on that scale). Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and QoL scales would reflect an improvement. It is available in 118 different languages on the EORTC website and is intended for use in adult populations only.
The reliability of the EORTC QLQ-C30 instrument was evaluated in an international study of patients with BTC.46 Internal consistency was assessed and was acceptable for all scales except for the physical function, cognitive function, and nausea/vomiting scales, which had mixed results. Results for test-retest reliability were also mixed, with the intraclass correlation coefficient for the scales ranging from 0.52 to 0.92. Construct validity and responsiveness were not assessed for the EORTC QLQ-C30.46 Estimates for MIDs in the literature were not found for the EORTC QLQ-C30 in patients with CCA or BTC.
Symptom Severity
Symptom severity was assessed in the trial using the EORTC QLQ-BIL21. The EORTC QLQ-BIL21 was an exploratory outcome in the FIGHT-202 trial. Neither an analysis plan or objective nor a MID for the EORTC QLQ-BIL21 instrument were specified a priori in the statistical analysis plan; it was noted, however, that the scores for each scale were to be calculated. According to the study’s protocol, scores for each scale and changes from baseline to each visit were to be measured and summarized descriptively.26 The EORTC QLQ-BIL21 was assessed at baseline and then every 3 cycles, starting with cycle 3, until discontinuation of the study treatment and at the end-of-treatment visit.26
The EORTC QLQ-BIL21 is a disease-specific module to be used in addition to the EORTC QLQ-C30 to assess HRQoL in patients with CCA and gallbladder cancer.47 It consists of 21 questions, with 18 of the items grouped into 5 scales: eating symptoms (4 items), jaundice symptoms (3 items), tiredness (3 items), pain symptoms (4 items), and anxiety symptoms (4 items).47 The remaining 3 items are single-item assessments of treatment side effects, difficulties with drainage bags and/or tubes, and concerns about weight loss.47 Patients complete the questionnaire based on a 1-week recall period by rating each item on a 4-point Likert scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much).47 The scores are then transformed linearly to a 0 to 100 scale to yield scale scores using EORTC guidelines, with higher scores indicating more severe symptoms.46,47 The questions have been translated, according to QoL group guidelines, into Dutch, German, Hindi, Italian, Mandarin Chinese, and Spanish.46,47
An international study was conducted to validate the EORTC QLQ-BIL21 in patients with BTC.46 The study included 172 adult patients with CCA and 91 patients with gallbladder cancer who had an expected minimum survival of 3 months and were undergoing treatment.46 Internal consistency was assessed and was acceptable for all multiitem scales of the EORTC QLQ-BIL21 instrument. Test-retest reliability was assessed using the intraclass correlation coefficient, which showed good reproducibility. Known group validity was assessed and was shown to distinguish groups based on Karnofsky performance status. There was some evidence of responsiveness to change on the eating, jaundice, tiredness, pain, treatment side effects, and anxiety scales. The single-item assessment of difficulties with drainage bags and tubes was considered irrelevant by 29 patients.46 The study authors noted that not all patients experience drains during their treatment and that perhaps there should be an NA option for responding to that item.46 Estimates for MIDs in the literature were not found for the EORTC QLQ-BIL21 in patients with CCA or BTC.
Safety
Safety was designated a secondary outcome in the FIGHT-202 trial and was defined as any untoward medial occurrence associated with the use of a drug in humans, whether or not the event was considered drug related, that occurred after a patient provided informed consent.26 Although data listings included all AEs, the analysis of AEs was limited to TEAEs.26 A TEAE included any AE reported for the first time or the worsening of a preexisting event after the first dose of pemigatinib.26 Abnormal laboratory values or test results observed in patients only constituted AEs if they were associated with clinical signs or symptoms, were considered clinically meaningful, required therapy (e.g., hematologic abnormality requiring transfusion), or required changes in the investigation study drug.26 Disease progression was recorded as an AE only if there were no other identifiable AEs or serious adverse events associated with the disease progression at the time of reporting.26
AEs were organized based on the Medical Dictionary for Regularity Activities (MedDRA) preferred term and system organ class. The severity of AEs was defined according to the US National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events version 4.03 (CTCAE v4.03). If a toxicity was not included in those criteria, it was graded on a scale of 1 to 4, where 1 = mild, 2 = moderate, 3 = severe, and 4 = life-threatening.26
All AEs were documented by the investigator from the date a patient gave informed consent to at least 30 to 35 days after the end-of-treatment visit (or the last dose of the study drug if the end-of-treatment visit did not occur).26
The FIGHT-202 trial included the following parameters for the analysis of AEs: the number of patients reporting TEAEs, serious adverse events, grade 3 or 4 TEAEs, fatal TEAEs, a temporary interruption of pemigatinib, or a permanent discontinuation of pemigatinib due to TEAEs; a summary of TEAEs and grade 3 or 4 TEAEs by system organ class, preferred term, decreasing order of frequency, and maximum severity; a summary of TEAEs leading to death, treatment-emergent serious adverse events, TEAEs leading to dose modifications (reductions, interruptions), and TEAEs leading to the discontinuation of pemigatinib by system organ class and preferred term.26
The FIGHT-202 trial monitored the following parameters: patients’ physical examination, changes in vital signs, electrocardiogram findings, and changes in clinical laboratory blood and urine sample evaluations.26
Statistical Analysis
Sample Size Determination
For the final analysis of the primary end point (ORR in cohort A), a sample size of approximately 100 patients with documentation of FGFR2 translocation from the central genomics laboratory was planned.26 A sample size of 100 patients was selected to guarantee an adequate population for robust response data and safety assessments.44 With the assumption that 33% of patients treated with pemigatinib would achieve an objective response, a sample size of 100 patients (assuming 10% of patients would be lost to follow-up) was estimated to provide a greater than 95% probability of having a 95% CI with a lower limit of greater than 15%.44 If the lower limit of the 95% CI for ORR exceeded 15%, it was predetermined that the trial results would be considered positive.17 The minimum clinically meaningful proportion of patients with an objective response was considered to be 15% on the basis of ORR results reported in previous studies48-50 of patients with CCA.44 For the analyses of cohort B and cohort C, a sample size of up to 20 patients was planned for each cohort, allowing a greater than 80% chance of observing at least 4 objective responders per cohort, if the underlying ORR was 30%.44
Interim Analysis
An interim analysis for futility for cohort A was planned after approximately 25 patients had enrolled and had at least 1 postbaseline tumour assessment or had permanently discontinued treatment.26 Enrolment in cohort A could have been terminated for futility if 2 or fewer of the 25 patients had achieved a response; there was a less than 10% probability that the proportion of patients with an objective response would be greater than 15% at the final analysis, based on a sample size of 60 patients in cohort A.26 Initially, the trial was designed to enrol 60 patients in cohort A; however, protocol Amendment 5, approved on October 3, 2017, increased the sample size of cohort A to approximately 100 patients.26 The futility analysis was conducted on October 3, 2017. Because the futility boundary was not crossed, the study proceeded as planned.18
The timing of the final analysis, when the predetermined threshold (i.e., the lower limit of the 95% CI for ORR was > 15%) would be assessed, was not prespecified a priori in the statistical analysis plan. According to the FDA report, the sponsor proposed a data cut-off date of March 22, 2019, during the pre-NDA meeting, held on August 8, 2019, during which the FDA acknowledged that the suggested data cut-off date would provide a minimum of 7 months of follow-up for all patients in the efficacy set and a minimum of 6 months of follow-up from the time of initial response for 92% of responders. The sponsor agreed further to provide additional follow-up data for DOR (no other efficacy outcome was analyzed at this time), corresponding to an August 30, 2019, data cut-off date, based on data with at least 6 months of follow-up from the time of initial response for all responders and a minimum of 12 months of follow-up for all patients in the efficacy set. The data cut-off date of August 30, 2019, also aligned with the 4-month safety update required for the NDA.18 According to the FDA, the proposed data analyses were sufficient to support the filing of an NDA under the provision of accelerated approval.5
An updated data analysis occurred at the April 7, 2020, data cut-off date to support safety data summaries for another indication outside of Canada.18 Because the April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date, some efficacy analyses (i.e., survival and response outcomes) were performed in addition to safety analyses and were provided to relevant regulatory authorities.18
Primary Outcome
The primary outcome in the FIGHT-202 trial was ORR in patients enrolled in cohort A. A brief overview of the statistical methods used for the primary outcome is provided in Table 7.
No statistical comparisons of cohorts were planned, and no formal hypothesis testing or inferential analyses were performed.44 The 95% CI for ORR was estimated using the exact method for binomial distribution.26 Patients with insufficient baseline or on-study response assessment data were considered nonresponders and were included in the denominators in the calculation of ORR.26 One sensitivity analysis for ORR was planned in the per-protocol population.26
Secondary Outcomes
A brief overview of the statistical methods used for the secondary outcomes is provided in Table 7.
Three secondary outcomes involved ORR in the FIGHT-202 trial: ORR in patients in cohort B; ORR in patients in cohort A plus cohort B; and ORR in patients in cohort C. For the 3 secondary ORR outcomes, the planned analyses were conducted in the same manner as the primary ORR outcome, and the 95% CI for ORR was estimated using the exact method for binomial distribution. No sensitivity analyses were planned for the 3 secondary ORR outcomes.26
PFS was a secondary outcome in the FIGHT-202 trial and was assessed for cohort A, cohort B, and cohort C; no statistical comparisons of cohorts were planned and no formal hypothesis testing or inferential analyses were performed. The number of patients whose disease progressed, who died, and who were censored were summarized. A Kaplan-Meier (KM) plot of PFS was presented with its 95% CI, which was estimated using the Brookmeyer and Crowley method,26,51 Censoring was based on the FDA Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics.26,52,53 Reasons for censoring included a lack of baseline tumour assessment, a lack of adequate postbaseline response assessment, a lack of progression, study discontinuation for undocumented progression or for toxicity or other reason, initiation of a new anticancer treatment (date of censoring was the last adequate response assessment before the initiation of the new anticancer treatment), and death or progression after more than 1 missed assessment. Outcomes of progression included progression documented between scheduled response assessments (date of progression was the date of first overall response of PD), death before first PD assessment (date of progression was date of death), and death between adequate assessment visits (date of progression was date of death). No sensitivity analysis was planned for PFS.26
DOR was a secondary outcome in the FIGHT-202 trial and was assessed for cohort A, cohort B, and cohort C. The number of patients who responded, who died or whose disease progressed, and who were censored were summarized. A Kaplan–Meier (KM) plot of DOR was presented with its 95% CI, which was estimated using the Brookmeyer and Crowley method.26,51 Censoring of DOR was done in the same manner as the censoring of PFS, as previously described. No sensitivity analysis was planned for DOR.26
The DCR was a secondary outcome in the FIGHT-202 trial and was assessed for cohort A, cohort B, and cohort C. The 95% CI for DCR was estimated using the exact method for binomial distribution.26 Patients with insufficient baseline or on-study response assessment data were considered nonresponders and were included in the denominators in the calculation of DCR.26 No sensitivity analysis for DCR was planned.26
OS was a secondary outcome in the FIGHT-202 trial and was assessed for cohort A, cohort B, and cohort C. The number of patients who died and the number who were censored were summarized. The KM plot of OS was presented with its 95% CI, which was estimated using the Brookmeyer and Crowley method.26,51 Reasons for censoring include being lost to follow-up or still being alive at the time of the analysis (censoring occurred at the date the patient was last known to be alive or the clinical data cut-off date for the analysis, whichever occurred first). The date the patient was last known to be alive was specified as the last study visit or the date the patient was last known to be alive from the survival follow-up, whichever occurred last.26 No sensitivity analysis for OS was planned.26
All analyses performed for the HRQoL outcome (EORTC QLQ-C30) and symptom severity outcome (EORTC QLQ-BIL21) were done in the efficacy-evaluable population (defined in the Analysis Populations section) for cohort A, cohort B, and cohort C. Analyses were considered descriptive (i.e., noninferential) in nature. No statistical comparisons of cohorts were planned. The standardized scores for each scale of the EORTC QLQ-C30 and the EORTC QLQ-BIL21 were calculated in accordance with the scoring guidance of the respective measure. A raw score was considered to be missing if the number of missing-item values totalled at least 50% of items that contributed to a scale.26 No MID was defined.26
In addition, results of a post hoc analysis were published in abstract format for a poster session at the 2021 Gastrointestinal Cancer Symposium.54 Additional information about this post hoc analysis and its results was provided by the sponsor in the Clinical Summary document.26 In their abstract, Valle et al. (2021)54 performed subgroup analyses in cohort A in which the changes from baseline to week 16 (cycle 6) in EORTC QLQ-C30 and EORTC QLQ-BIL21 scores were summarized for the following response subgroups: CR or PR; stable disease; and PD. Valle et al. (2021)54 noted that treatment-related changes in HRQoL would be expected to be apparent by week 16. Upon request, the sponsor explained that because of a rapid decline in the number of patients available to complete the questionnaires, a robust assessment of the data could only be conducted between baseline and cycle 6.18 Three key independent physicians, consulted by the sponsor on how the drop-off in patients after cycle 6 may have impacted interpretation of the data, noted that changes in HRQoL would be apparent 4 to 6 cycles after treatment initiation.18 The post hoc analyses were performed on the March 22, 2019, data cut-off date and were considered descriptive (i.e., noninferential) in nature. It was reported by the sponsor that nominal statistical significance was determined by the lack of overlap of the associated standard error bars around the point estimate.26 Graphs of the observed mean changes from baseline within each subgroup were generated and presented in the Clinical Summary document.26
Subgroup Analyses
Subgroup analyses of specific groups of patients were planned a priori in the statistical analysis plan. For each subgroup, a forest plot and the respective outcome’s 95% CI was provided. Subgroup analyses were conducted for the primary outcome, ORR in cohort A (subgroup analyses were conducted for all patient groups listed here), PFS in cohort A (subgroup analyses were performed for all groups listed here except for renal and/or hepatic impairment), and DOR for cohort A (subgroup analyses were only conducted for patients with renal and/or hepatic impairment):
age category (< 65 years versus 65 to < 75 years versus ≥ 75 years)
sex (female versus male)
region (North America versus Western Europe versus rest of the world)
baseline ECOG PS (0 versus 1 or 2)
metastatic disease present (yes versus no)
lines of prior therapy (1 line versus 2 lines versus ≥ 3 lines)
received previous platinum treatment (yes versus no)
renal impairment grade (normal versus mild versus moderate versus severe)
hepatic impairment grade (normal versus mild versus moderate versus severe).
The ECOG PS subgroup, planned a priori in the statistical analyses plan, aligned with the subgroup prespecified in the protocol for this CDA-AMC review. Only the subgroup identified in the CDA-AMC review protocol is reported in the Efficacy section.
Table 7: Statistical Analysis of Efficacy End Points
End pointa | Statistical model | Sensitivity analyses |
|---|---|---|
OS Definition: time from the start of the study drug (day 1) until the date of death from any cause. | The KM method was used to estimate median OS and 95% CIs. The 95% CI for OS was calculated using the Brookmeyer and Crowley method.51 | None |
PFS Definition: time from the start of the study drug (day 1) to the date of PD2 or death from any cause, whichever occurred first. | The KM method was used to estimate median PFS and 95% CIs. The 95% CI for PFS was calculated using the Brookmeyer and Crowley method.51 | None |
ORR (primary end point) Definition: best overall response is the best response recorded after baseline but before and including the first PD, in the order of CR, PR, stable disease, PD, and NE. A best overall response of CR or PR needs to be confirmed by independent review committee.26 | The 95% CI for ORR was calculated using the exact method for binomial distribution. | ORR was based on the PPb population for a sensitivity analysis. |
DOR Definition: time from the first overall response contributing to an objective responsec to the earlier of death from any cause or the first overall response of PD that occurred after the first overall response contributing to the objective response. | The KM method was used to estimate median DOR and 95% CIs. The 95% CI for DOR was calculated using the Brookmeyer and Crowley method.51 | None |
DCR Definition: the proportion of patients with a best response of CR, PR, or stable disease, based on RECIST 1.1, assessed by the IRC. | The 95% CI for DCR was calculated using the exact method for binomial distribution. | None |
EORTC QLQ-C30 | It was specified a priori in the statistical analysis plan26 that the scores for each scale would be calculated. No further analyses were specified. | None |
EORTC QLQ-BIL21 | It was specified a priori in the statistical analysis plan26 that the scores for each scale would be calculated. No further analyses were specified. | None |
CI = confidence interval; CR = complete response; DCR = disease control rate; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer QLQ-Cholangiocarcinomas and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; IRC = independent review committee; KM = Kaplan-Meier; NE = not evaluable; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PP = per protocol; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aOutcomes are presented in order of priority, as identified in the CDA-AMC review protocol.
bThe PP population includes patients in the efficacy-evaluable population, who are considered to be sufficiently compliant with the protocol.
cProgressive disease is based on RECIST 1.1, as assessed by the IRC.
Source: Sponsor’s submission.26
Table 8: Analysis Populations in the FIGHT-202 Trial
Analysis population | Description |
|---|---|
Efficacy-evaluable population | All patients who have FGF-FGFR alteration confirmed by the central genomics laboratory and who received at least 1 dose of pemigatinib, as well as all patients in the USa who have a negative FGF-FGFR alteration confirmed by the central genomics laboratory and who received at least 1 dose of pemigatinib |
PP population | Patients in the efficacy-evaluable population who were considered to be sufficiently compliant with the protocol (the decision to exclude a patient from the PP population was done by the clinical team before the database freeze) To identify potential patients for exclusion from the PP population, the following procedures were conducted:
|
Safety population | All enrolled patients who had received at least 1 dose of study drug |
PP = per protocol.
aAs specified in protocol Amendment 3 of the FIGHT-202 trial, only patients from the US were allowed to enrol in cohort C.
Source: Sponsor’s submission.26
Analysis Populations
All efficacy data were analyzed using the efficacy-evaluable population, as defined in Table 8. In sensitivity analyses, ORR in cohort A was analyzed using the per-protocol population, as defined in Table 8. Analyses of safety were performed using the safety population (Table 8).
Results
Patient Disposition
Details of patient disposition in cohort A of the FIGHT-202 trial are summarized in Table 9. Of the 171 patients screened, 146 patients were enrolled: 107 patients in cohort A, 20 patients in cohort B, and 18 patients in cohort C (the FGF-FGFR alteration for 1 patient was undetermined). The 25 patients who were not enrolled failed to meet the trial eligibility criteria. All patients enrolled in cohort A received pemigatinib. At study close, 106 of 108 patients (98.1%) in cohort A had discontinued treatment. The most common reason for treatment discontinuation was PD, which was reported by 77 of 108 patients (71.3%).17
At study close, 105 of 108 patients (97.2%) in cohort A had terminated the study, 72 (66.7%) of whom had died. Reasons for study discontinuation included withdrawal of consent (n = 7 [6.5%]), PD (n = 4 [3.7%]), and loss to follow-up (n = 3 [2.8%]).17
Table 9: Patient Disposition, Safety Population (Data Cut-Off Dates: March 22, 2019; April 7, 2020; and July 8, 2021)
Disposition | FIGHT-202 | ||
|---|---|---|---|
Screened, n | 171 | ||
Enrolled, na | 146 | ||
Cohort assignment,b n | |||
Cohort A | 107 | ||
Cohort B | 20 | ||
Cohort C | 18 | ||
Undetermined FGF or FGFR alterationc | 1 | ||
Cohort A (safety population) | |||
Data cut-off date | March 22, 2019 | April 7, 2020 | July 8, 2021 |
Treated, n | 107 | 108d | 108d |
With ongoing treatment, n (%) | 31 (29.0) | 10 (9.3) | █████ |
Discontinued from treatment phase, n (%) | 76 (71.0) | 98 (90.7) | 106 (98.1) |
Reason for discontinuation from treatment phase, n (%) | |||
Progressive disease | 57 (53.3) | 73 (67.6) | 77 (71.3) |
Withdrawal by participant | 5 (4.7) | 7 (6.5) | 8 (7.4) |
Adverse event | 4 (3.7) | 6 (5.6) | 6 (5.6) |
Physician decision | 4 (3.7) | 6 (5.6) | 7 (6.5) |
Othere | 5 (4.7) | 5 (4.6) | 5 (4.6) |
Death | 1 (0.9) | 1 (0.9) | 1 (0.9) |
Lost to follow-up | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Protocol violation | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Patients still in study, n (%) | 59 (55.1) | 35 (32.4) | 3 (2.8) |
Patients who discontinued the study, n (%) | 48 (44.9) | 73 (67.6) | 105 (97.2) |
Reason for discontinuation from the study n (%) | |||
Death | 38 (35.5) | 60 (55.6) | ██ ██████ |
Withdrew consent | 6 (5.6) | 7 (6.5) | █████ |
Progressive disease | 2 (1.9) | 3 (2.8) | █████ |
Lost to follow-up | 2 (1.9) | 3 (2.8) | █████ |
Efficacy-evaluable populationf | 145 | ||
Per-protocol populationg | 142 | ||
HRQoL population | |||
EORTC QLQ-C30 population | |||
Cohort A assignment | 107 | 108d | ███ |
Cohort B assignment | 20 | 20 | ██ |
Cohort C assignment | 18 | 17 | ██ |
Symptom severityh,k | |||
EORTC BIL-21 population | |||
Cohort A assignment | NRi | 83 | ██ |
Cohort B assignment | NR | 12 | ██ |
Cohort C assignment | NR | 17 | ██ |
Safety populationj | 146 | ||
EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer QLQ-Cholangiocarcinomas and Gallbladder Cancer Module 21; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; NR = not reported; PP = per protocol.
aEligibility criteria not met by patients were inclusion criteria (including, primarily, documentation of FGF or FGFR gene alteration status through central laboratory, ECOG PS of 0 to 2, and life expectancy ≥ 12 weeks), followed by exclusion criteria (including, primarily, abnormal laboratory parameters). In addition, 1 patients was not enrolled in any of the cohorts because of an inability of the central laboratory to confirm the FGFR gene alteration status.18
bCohort determination is based on tumour FGF or FGFR status determined by the central genomics laboratory.
cOne participant from the safety population was assigned to a group labelled undetermined and excluded from the efficacy-evaluable population because the local laboratory FGF or FGFR result could not be confirmed centrally, owing to technical issues with the tissue sample.
dThe April 7, 2020, and July 8, 2021, data cut-off dates included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.18
eOther reasons were consistent with progressive disease, but were not considered progressive disease, and included a clinical decline in the patient without growth of the tumour, a patient being considered to have progressive disease by the central radiology group but not by the investigator, receipt of anticancer therapy to treat brain metastasis, withdrawal of patient consent after being taken off treatment because of a suspicious lung lesion on CT.18
fOne participant from the safety population was assigned to a group labelled undetermined and excluded from the efficacy-evaluable population because the local laboratory FGF or FGFR result could not be confirmed centrally, owing to technical issues with the tissue sample.17
gThree participants in the efficacy-evaluable population were excluded from the PP population because of protocol deviations;17 1 patient had received prior therapy with another FGFR inhibitor (for fewer than 11 days and ending 30 days before first dose of pemigatinib); 1 patient underwent trisegmentectomy or extended right hepatectomy during the study; and 1 patient underwent surgical removal of pulmonary malignant lesions during the study.17
hIn Amendment 1 (September 14, 2016), language was added to denote that only patients at sites in Germany, Italy, Korea, UK, and US were administered the EORTC QLQ-BIL21, because the questionnaire is only translated and validated in the primary languages of those countries.26
iUpon request, the number of patients who contributed to the results for the April 7, 2020, data cut-off date was provided, but not the number for the March 22, 2019, data cut-off date.
jThe safety population included all 146 patients enrolled in the study as of the March 22, 2019, data cut-off date.17
kHRQoL and symptom severity were assessed in the efficacy-evaluable population. Patients who replied to at least 1 question on the patient-reported outcomes instruments were considered in the analyses.18
Sources: Clinical Study Report,17 and sponsor’s submission,18,26 sponsor’s response.18
Baseline Characteristics
The baseline characteristics of patients who comprised the safety population of the FIGHT-202 trial are summarized in Table 10. At baseline, 107 patients were identified as having FGFR2 fusions or rearrangements and were grouped into cohort A. Cohort B included 20 patients with FGF or FGFR alterations other than FGFR2, and cohort C included 18 patients with no identified FGF or FGFR alterations. One patient, grouped into an undetermined group, was not assigned to any of the 3 cohorts, as the local FGF or FGFR status results could not be confirmed by the central genomics laboratory. For patients in cohort A, the mean age was 55.3 years (SD =12.02 years), most patients were female (60.7%), and most were enrolled in trial sites in North America (59.8%) or Europe (29.9%). Almost all patients (89% of patients overall and 98.1% of patients in cohort A) had iCCA. The majority of patients in cohort A had metastatic disease (82.2%), with the lung and lymph nodes being the most common metastatic sites (54.2% and 53.3%, respectively). Median time since diagnosis was 1.28 years (range, 0.03 to 11.1 years) in patients in cohort A. The majority of patients in cohort A had an ECOG PS of 1 (53.3%) or 0 (42.1%), and all patients had received at least 1 line of prior systemic therapy for advanced and/or metastatic disease (60.7%, 27.1%, and 12.1% of patients received 1, 2, or ≥ 3 prior lines, respectively). Renal and hepatic impairment grades were normal or mild for most patients in cohort A (39.3% and 43.9% for normal and mild renal impairment grades, respectively; 44.9% and 48.6% for normal and mild hepatic grades, respectively).17
Table 10: Summary of Baseline Characteristics, Safety Population
Characteristic | FIGHT-202 | |||
|---|---|---|---|---|
Cohort A FGFR2 fusions or rearrangements N = 107 | Cohort B Other FGF-FGFR alterations N = 20 | Cohort C No FGF-FGFR alterations N = 18 | All patients N = 146a | |
Age, years | ||||
Mean (SD) | 55.3 (12.02) | 61.9 (10.99) | 63.7 (10.68) | 57.2 (12.08) |
Median (minimum to maximum) | 56.0 (26 to 77) | 63.0 (45 to 78) | 65.0 (31 to 78) | 59.0 (26 to 78) |
Age category, n (%) | ||||
< 65 years | 82 (76.6) | 10 (50.0) | 7 (38.9) | 100 (68.5) |
65 to < 75 years | 20 (18.7) | 7 (35.0) | 8 (44.4) | 35 (24.0) |
≥ 75 years | 5 (4.7) | 3 (15.0) | 3 (16.7) | 11 (7.5) |
Sex, n (%) | ||||
Male | 42 (39.3) | 9 (45.0) | 10 (55.6) | 62 (42.5) |
Female | 65 (60.7) | 11 (55.0) | 8 (44.4) | 84 (57.5) |
Geographical region, n (%) | ||||
North America | 64 (59.8) | 6 (30.0) | 18 (100.0) | 89 (61.0) |
Western Europe | 32 (29.9) | 3 (15.0) | 0 (0.0) | 35 (24.0) |
Rest of the worldb | 11 (10.3) | 11 (55.0) | 0 (0.0) | 22 (15.1) |
Race, n (%) | ||||
White | 79 (73.8) | 9 (45.0) | 15 (83.3) | 104 (71.2) |
Asian | 11 (10.3) | 11 (55.0) | 0 (0.0) | 22 (15.1) |
Black or African American | 7 (6.5) | 0 (0.0) | 1 (5.6) | 8 (5.5) |
American Indian or Alaska Native | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (0.7) |
Otherc | 4 (3.7) | 0 (0.0) | 1 (5.6) | 5 (3.4) |
Missing | 6 (5.6) | 0 (0.0) | 0 (0.0) | 6 (4.1) |
ECOG PS, (%) | ||||
0 | 45 (42.1) | 7 (35.0) | 7 (38.9) | 59 (40.4) |
1 | 57 (53.3) | 10 (50.0) | 8 (44.4) | 76 (52.1) |
2 | 5 (4.7) | 3 (15.0) | 3 (16.7) | 11 (7.5) |
≥ 3 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Renal impairment grade at baseline, n (%)d | ||||
Normal | 42 (39.3) | 6 (30.0) | 7 (38.9) | 55 (37.7) |
Mild | 47 (43.9) | 13 (65.0) | 7 (38.9) | 68 (46.6) |
Moderate | 18 (16.8) | 1 (5.0) | 3 (16.7) | 22 (15.1) |
Severe | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (0.7) |
Hepatic impairment grade at baseline, n (%)e | ||||
Normal | 48 (44.9) | 13 (65.0) | 13 (72.2) | 75 (51.4) |
Mild | 52 (48.6) | 7 (35.0) | 4 (22.2) | 63 (43.2) |
Moderate | 7 (6.5) | 0 (0.0) | 1 (5.6) | 8 (5.5) |
CCA location, n (%) | ||||
Intrahepatic | 105 (98.1) | 13 (65.0) | 11 (61.1) | 130 (89.0) |
Extrahepatic | 1 (0.9) | 4 (20.0) | 7 (38.9) | 12 (8.2) |
Other | 0 (0.0) | 3 (15.0)f | 0 (0.0) | 3 (2.1) |
Missing | 1 (0.9)g | 0 (0.0) | 0 (0.0) | 1 (0.7) |
Stage at initial diagnosis, n (%) | ||||
I | ██ ██████ | █████ | █████ | ██ █████ |
II | ██ ██████ | █████ | ██████ | ██ ███ |
III | █████ | ██████ | ██████ | ██ ██ |
IV | ██ ██████ | ██ ██████ | ██ ██████ | ██ ███ |
Missing | █████ | █████ | █████ | █████ |
Time since diagnosis (years) | ||||
Mean (SD) | 1.57 (1.619) | 1.01 (0.676) | 1.52 (1.240) | 1.49 (1.481) |
Median | 1.28 | 0.73 | 0.98 | 1.10 |
Minimum to maximum | 0.03h to 11.1 | 0.2 to 2.5 | 0.3 to 4.3 | 0.03 to 11.1 |
Metastatic disease, n (%) | ||||
Yes | 88 (82.2) | 20 (100.0) | 16 (88.9) | 125 (85.6) |
No | 16 (15.0) | 0 (0.0) | 2 (11.1) | 18 (12.3) |
Not evaluable | 1 (0.9) | 0 (0.0) | 0 (0.0) | 1 (0.7) |
Missing | 2 (1.9) | 0 (0.0) | 0 (0.0) | 2 (1.4) |
Number of previous systematic therapies for advanced or metastatic disease, n (%)i | ||||
1 | 65 (60.7) | 12 (60) | 12 (66.7) | 89 (61.0) |
2 | 29 (27.1) | 7 (35.0) | 2 (11.1) | 38 (26.0) |
≥ 3 | 13 (12.1) | 1 (5.0) | 4 (22.2) | 19 (13.0) |
CCA = cholangiocarcinoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; SD = standard deviation.
aOne participant from the safety population was assigned to a group labelled undetermined and was excluded from the efficacy-evaluable population because the local laboratory FGF-FGFR result could not be confirmed centrally, owing to technical issues with the tissue sample.
bRest of world consists of Israel, Japan, South Korea, Taiwan, and Thailand.
cIncludes Hispanic, Latino, or Spanish (n = 1) or not reported (n = 4).
dBaseline renal impairment grade (normal, mild, moderate, or severe) based on estimated glomerular filtration rate (eGFR) (calculated using the Modification of Diet in Renal Disease Study equation): normal renal function = eGFR ≥ 90 mL/min per 1.73 m2; mild renal impairment = eGFR ≥ 60 and < 90 mL/min per 1.73 m2; moderate renal impairment = eGFR ≥ 30 to < 60 mL/min per 1.73 m2; severe renal impairment = eGFR < 30 mL/min per 1.73 m2.
eDegree of hepatic impairment based on National Cancer Institute hepatic working group criteria.
fIncludes gallbladder (n = 2) and ampulla of Vater (n = 1).
gAt baseline, this participant had stage IV CCA (T3 N0 M1), presumed intrahepatic, with current sites of disease of the liver, omentum, and peritoneum.
hParticipant's date of diagnosis was entered incorrectly by the site. The time since diagnosis is 22.11 months, based on the correct date of diagnosis.
iMaximum number of 5 therapies in patients with FGFR2 fusions or rearrangements and 3 in the other patient cohorts.
Source: Clinical Study Report.17
All patients in cohort A received at least 1 prior systemic cancer therapy (refer to Table 11). The majority of patients received platinum-based chemotherapy regimens before study enrolment, most commonly gemcitabine, reported as gemcitabine (85.0%) or gemcitabine hydrochloride (7.5%) and cisplatin (75.7%). The second and third most commonly received pyrimidine analogues were 5-FU (29.0%) and capecitabine (14.0%); the second most frequently administered platinum compound was oxaliplatin (38.3%).17 Eight (7.5%) patients in cohort A received locoregional radioembolization before study entry.44
Table 11: Summary of Prior Systemic Cancer Therapy, Safety Population
Prior treatment, N (%) WHO drug class or WHO drug term | Pemigatinib |
|---|---|
Cohort A (N = 107) | |
Pyrimidine analogues | 106 (99.1) |
Gemcitabine | 91 (85.0) |
Fluorouracil | 31 (29.0) |
Capecitabine | ██ ██████ |
Gemcitabine hydrochloride | 8 (7.5) |
Tegafur | █████ |
Floxuridine | █████ |
Platinum compounds | 101 (94.4) |
Cisplatin | 81 (75.7) |
Oxaliplatin | 41 (38.3) |
Carboplatin | 6 (5.6) |
███████████ ██████ ███ ██████████████ | ██ ██████ |
███████ ████ | ██ ██████ |
███████ ████████ | █████ |
███████ ████████████ | █████ |
██████ ████████ | █████ |
█████ ██████████████ ██████ | ██ ██████ |
██████████ | ██ ██████ |
██████████ █████████████ | █████ |
███████ | █████ |
██████████ | █████ |
██████████ ███████ | █████ |
█████████ | █████ |
Investigational drug | 6 (5.6) |
Protein kinase inhibitors | 6 (5.6) |
Source: Clinical Study Report.17
Exposure to Study Treatments
Exposure to pemigatinib in cohort A at the March 22, 2019, April 7, 2020, and July 8, 2021, data cut-off dates is summarized in Table 12. The median duration of treatment with pemigatinib was 220 days (range, 7 to 1,554 days) at the July 8, 2021, study-close date. The median overall compliance rate was high as of the July 8, 2021, study-close date (100%; mean = 99.85%; SD =2.233%), indicating high treatment adherence in cohort A. Treatment compliance was evaluated by pill counts completed at the site.17
Table 12: Exposure to Pemigatinib, Safety Population
Exposure | Pemigatinib Cohort A | ||
|---|---|---|---|
Data cut-off date | March 22, 2019 | April 7, 2020 | July 8, 2021 |
Treated, n | 107 | 108a | 108a |
Duration of treatment, daysb | |||
Mean (SD) | 247.4 (170.25) | █████ █████ | █████ █████ |
Median (minimum to maximum) | 219.0 (7 to 730) | █████ ███ ██ | █████ ███ ██ |
Number of treatment cycles | |||
Mean (SD) | 11.8 (7.93) | █████ ██ | ████ ███ |
Median | 10.0 | ██████ | ████ |
Minimum to maximum | 1 to 34 | ██ ██ | ██ ██ |
Participants exposed, n (%) | |||
≤ 1 month | 3 (2.8) | █████ | ████████ |
> 1 to 3 months | 18 (16.8) | ██ ██████ | █████████ |
> 3 to 6 months | 21 (19.6) | ██ ██████ | ██████████ |
> 6 to 9 months | 26 (24.3) | ██ ██████ | ██████████ |
> 9 to 12 months | 18 (16.8) | ██ ██████ | █████████ |
> 12 to 15 months | 8 (7.5) | ██ █████ | ███████ |
> 15 to 18 months | 5 (4.7) | █████ | ████████ |
> 18 to 21 months | 5 (4.7) | █████ | ████████ |
> 21 to 24 months | 3 (2.8) | ██ █████ | ███████ |
> 24 months | 0 (0.0) | █████ | ███████ |
Overall compliance (%)c | |||
Mean (SD) | 100.37 (3.296) | █████ ████ | █████ ████ |
Median | 100.00 | ██████ | ██████ |
Minimum to maximum | 90.0 to 124.4 | █████ █████ | █████ █████ |
SD = standard deviation.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who was enrolled after the August 30, 2019, data cut-off date.18
bTreatment duration in days is defined as: (date of last dose – date of first dose + 1).
cThe compliance rate (%) for each patient was computed as: (total actual dose [mg]) / (total prescribed dose [mg]) * 100.
Sources: Clinical Study Report,17 sponsor’s response.18
Subsequent Treatments
Information on subsequent treatment received is available for some patients only, and is current as of the April 7, 2020, data cut-off date.18 According to the sponsor, the study sites of the FIGHT-202 trial were not required to provide information on posttreatment therapies once pemigatinib was discontinued; however, it was requested.18 For cohort A, information on subsequent treatments was available for 34 patients, the majority of whom received FOLFIRI (N = 11) as a first subsequent treatment.18 The next most commonly received subsequent treatments after pemigatinib included immunotherapies (2 patients received nivolumab and 2 patients received pembrolizumab) and targeted therapies (2 patients received TAS-120 and 1 patient received sulfatinib), and 3 patients received capecitabine and 2 patients received gemcitabine plus cisplatin. A range of chemotherapies were received by 1 patient each, including 5-FU plus gemcitabine, irinotecan plus leucovorin, epirubicin plus cisplatin, and gemcitabine plus oxaliplatin).18
Efficacy
Only efficacy outcomes and analyses of the subgroups identified in the review protocol are reported here. This CDA-AMC review focuses on cohort A; cohort B and cohort C were not part of the requested reimbursement criteria to CDA-AMC and were not submitted for approval to Health Canada so are beyond the scope of this review.
Overall Survival
The OS results of the FIGHT-202 trial for cohort A are summarized in Table 13 at the March 22, 2019, April 7, 2020, and July 8, 2021, data cut-off dates. As of the July 8, 2021, study-close date, with a median follow-up of 45.4 months, 76 (70.4%) death events occurred in cohort A. There were 32 (29.6%) patients censored. Median OS was 17.48 months (95% CI, 14.36 to 22.93 months). The KM curve is depicted in Figure 2. The survival probabilities of patients surviving to 6 months and to 12 months were 88.7% (95% CI, 81.0% to 93.4%) and 67.6% (95% CI, 57.7% to 75.6%), respectively.17
Table 13: Summary of Primary and Secondary End Points in the FIGHT-202 Study (Data Cut-Off Dates: March 22, 2019; April 7, 2020; and July 8, 2021)
Variable | Pemigatinib Cohort A | ||
|---|---|---|---|
N = 107 | N = 108a | N = 108a | |
Efficacy outcomes, efficacy-evaluable population | |||
Data cut-off date | March 22, 2019 | April 7, 2020 | July 8, 2021 |
Median follow-up time,b months (minimum to maximum) | 15.44 (7.0 to 24.7) | 27.9 (4.9 to 37.2) | 45.4 (19.9 to 53.7) |
Secondary outcome: OS | |||
Median OS, months (95% CI)c | 21.06 (14.82 to NE) | 17.48 (14.42 to 22.93) | 17.48 (14.36 to 22.93) |
Events (death), n (%) | 40 (37.4) | ██████████ | 76 (70.4) |
Censored, n (%) | 67 (62.6) | ██████████ | 32 (29.6) |
KM estimates of OS at: | |||
3 months (95% CI) | 96.2 (90.3 to 98.6) | ███████████ | ████ █████ █ |
6 months (95% CI) | 88.6 (80.8 to 93.4) | ███████████ | 88.7 (81.0 to 93.4) |
9 months (95% CI) | 77.4 (68.0 to 84.4) | 76.1 (66.7 to 83.2) | ████ █████ ██ |
12 months (95% CI) | 67.5 (56.4 to 76.3) | 67.3 (57.4 to 75.4) | 67.6 (57.7 to 75.6) |
Secondary outcome: PFS (IRC assessment) | |||
Median PFS, months (95% CI)c | 6.93 (6.18 to 9.59) | 7.03 (6.08 to 10.48) | 7.03 (6.08 to 10.48) |
Events (disease progression or death), n (%) | 71 (66.4) | ██ ██████ | 85 (78.7) |
Disease progression, n (%) | 63 (58.9) | ██ ██████ | ██ ██████ |
Death, n (%) | 8 (7.5) | █████ | ██ █████ |
Censored, n (%) | 36 (33.6) | ██ ██████ | 23 (21.3) |
KM estimates of PFS at: | |||
3 months (95% CI) | 78.9 (69.7 to 85.5) | ████ █████ ██ | ████ █████ █ |
6 months (95% CI) | 61.7 (51.5 to 70.4) | ████ █████ ██ | 61.1 (51.0 to 69.8) |
9 months (95% CI) | 45.3 (34.9 to 55.1) | ████ █████ ██ | ████ █████ ██ |
12 months (95% CI) | 29.2 (18.9 to 40.2) | ████ █████ ██ | 32.3 (22.9 to 42.1) |
Primary outcome: ORR (IRC assessment) | |||
Objective response,d n (%) | 38 (35.5) | 40 (37.0) | 40 (37.0) |
95% CIe | 26.50 to 45.35 | 27.94 to 46.86 | 27.94 to 46.86 |
Best overall response, n (%) | |||
Confirmed complete response, n (%) | 3 (2.8) | 4 (3.7) | 3 (2.8) |
Confirmed partial response, n (%) | 35 (32.7) | 36 (33.3) | 37 (34.3) |
Stable disease, n (%) | 50 (46.7) | 49 (45.4) | 49 (45.4) |
Progressive disease, n (%) | 16 (15.0) | 16 (14.8) | 16 (14.8) |
Not evaluable,f n (%) | 3 (2.8) | 3 (2.8) | 3 (2.8) |
Secondary outcome: DOR (IRC assessment)g | |||
Participants with confirmed objective responses, n (%) | 38 (35.5) | 40 (37.0) | 40 (37.0) |
Participants with events, n (%) | 21 (55.3) | ██ ██████ | 30 (75.0) |
Disease progression, n (%) | 20 (52.6) | ██ ██████ | ██ ██████ |
Death, n (%) | 1 (2.6) | █████ | █████ |
Participants censored, n (%) | 17 (44.7) | ██ ██████ | 10 (25.0) |
Median DOR, months (95% CI)c | 7.49 (5.65 to 14.49) | 8.08 (5.65 to 13.14) | 9.13 (6.01 to 14.49) |
KM estimates of DOR | |||
3 months (95% CI) | 100.0 (100.0 to 100.0) | █████ ███ | █████ ███ |
6 months (95% CI) | 68.5 (49.0 to 81.8) | ████ █████ █ | 67.8 (50.4 to 80.3) |
9 months (95% CI) | 47.4 (27.6 to 64.9) | ████ █████ ██ | ████ █████ █ |
12 months (95% CI) | 37.4 (18.6 to 56.2) | ████ █████ ██ | 41.2 (24.8 to 56.8) |
Secondary outcome: DCR (IRC assessment) | |||
Disease control,h n (%) | 88 (82.2) | Not availablei | 89 (82.4) |
95% CIe | 73.7 to 89.0 | 73.9 to 89.1 | |
Best response, n (%) | |||
Confirmed complete response, n (%) | 3 (2.8) | █████ | |
Confirmed partial response, n (%) | 35 (32.7) | ██ ██████ | |
Stable disease ≥ 39 days, n (%) | 50 (46.7) | ██ ██████ | |
CDA-AMC = Canada's Drug Agency; CI = confidence interval; CR = complete response; DCR = disease control rate; DOR = duration of response; IRC = independent review committee; ITT = intention to treat; KM = Kaplan-Meier; N/A = not applicable; NE = not evaluable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Note: Outcomes are presented in order of priority, as identified in the CDA-AMC review protocol.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who was enrolled after the August 30, 2019, data cut-off date.18
bFollow-up time for all patients in cohort A in the efficacy-evaluable population.
cThe 95% CI was calculated using the Brookmeyer and Crowley method (1982).
dParticipants who had a best overall response of complete response or partial response.
eThe CI was calculated based on the exact method for binomial distribution.
fPostbaseline tumour assessment was not performed because of study discontinuation (2 participants) or was performed before the minimum interval of 39 days for an assessment of stable disease (1 participant).
gComplete and partial responses are confirmed.
hParticipants who have a best overall response of CR, partial response, or stable disease with measurements that meet the stable disease criteria after the date of the first dose at a minimum interval of 39 days.
iThe DCR outcome was not generated for the April 7, 2020, data cut-off date because the primary focus of the analyses at that data cut-off date was for the integrated safety summary for a new regulatory submission.18
Sources: Clinical Study Report,17 sponsor’s response.18

Progression-Free Survival
The PFS results (based on IRC assessment) of the FIGHT-202 trial for cohort A are summarized in Table 13 for the March 22, 2019, April 7, 2020, and July 8, 2021 data cut-off dates. As of the analysis at the July 8, 2021, data cut-off date, with a median follow-up time of 45.4 months, ██ ███████ patients experienced disease progression, ██ ██████ patients had died, and 23 (21.3%) patients were censored. Median PFS was 7.03 months (95% CI, 6.08 to 10.48 months). The KM curve is depicted in Figure 3. The PFS probabilities at 6 months and 12 months were 61.1 (95% CI, 51.0 to 69.8) and 32.3 (95% CI, 22.9 to 42.1), respectively.

The PFS results by subgroup of interest were available for the March 22, 2019, data cut-off date. Subgroups presented are those specified a priori in the protocol for this CDA-AMC review (Table 14). The treatment effect on PFS was consistent with the PFS analysis for all patients in cohort A for patients with an ECOG PS of 0 and patients with an ECOG PS of 1 or 2. Of note, the sample sizes of these subgroups were small (45 patients with an ECOG PS of 0 and 62 patients with an ECOG PS of 1 or 2), and the relatively wide CIs in the subgroups reflected uncertainty in the effect estimates.
Table 14: PFS by ECOG PS in Cohort A, Efficacy-Evaluable Population (March 22, 2019, Data Cut-Off Date)
Subgroups | Pemigatinib cohort A (N = 107) | Median PFS, months (95% CI) | |
|---|---|---|---|
Number of patients | Number of patients with events | ||
ECOG PS | |||
0 | 45 | 25 | 9.59 (6.93 to 13.08) |
1 or 2 | 62 | 46 | 6.18 (4.73 to 9.00) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; PFS = progression-free survival.
Source: Clinical Study Report.17
Objective Response Rate
The ORR results (based on IRC assessment) for cohort A of the FIGHT-202 trial are summarized in Table 13 at the March 22, 2019, April 7, 2020, and July 8, 2021, data cut-off dates.
As of the analysis at the July 8, 2021, study-close date, the proportion of patients who achieved an objective response was 37.0% (N = 40) (95% CI, 27.94% to 46.86%), which included 3 (2.8%) patients with CRs and 37 (34.3%) patients with PRs.
The ORR results by subgroup of interest were available for the March 22, 2019, cut-off date. Subgroups presented are those specified a priori in the protocol for this CDA-AMC review (Table 15). The ORR results for the subgroup of interest suggested that the treatment effects on ORR for the subgroups with an ECOG PS of 0 and an ECOG PS of 1 or 2 were generally consistent with the overall population in cohort A. Of note, the sample sizes of these subgroups were small (45 patients with an ECOG PS of 0 and 62 patients with an ECOG PS of 1 or 2), and the relatively wide CIs in the subgroups reflected uncertainty in the effect estimates.
Table 15: ORR by ECOG PS in Cohort A, Efficacy-Evaluable Population (March 22, 2019, Data Cut-Off Date)
Subgroups | Pemigatinib cohort A (N = 107) | ORR (95% CI) |
|---|---|---|
Number of patients | ||
ECOG PS | ||
0 | 45 | 48.9 (33.70 to 64.23) |
1 or 2 | 62 | 25.8 (15.53 to 38.50) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ORR = objective response rate.
Note: Response assessed by independent reviewer and response was confirmed.
Source: Clinical Study Report.17
Duration of Response
The DOR results (based on IRC assessment) for cohort A of the FIGHT-202 trial for are summarized in Table 13 at the March 22, 2019, April 7, 2020, and July 8, 2021 data cut-off dates.
As of the July 8, 2021, data cut-off date, the median DOR was 9.13 months (95% CI, 6.01 to 14.49 months) among the 40 patients who achieved an objective response. The KM curve is depicted in Figure 4. The probabilities of maintaining a response for at least 6 months and for at least 12 months were 67.8% (95% CI, 50.4% to 80.3%) and 41.2% (95% CI, 24.8% to 56.8%), respectively.

Note: Data are from the independent centralized radiological review committee, per RECIST 1.1, and the response has been confirmed.
Source: Clinical Study Report.17
Disease Control Rate
The DCR results (based on IRC assessment) for cohort A of the FIGHT-202 trial are summarized in Table 13 for the March 22, 2019, and July 8, 2021, data cut-off dates. As of the July 8, 2021, data cut-off date, the proportion of patients with a best response of CR, PR, or stable disease was 82.4% (N = 89) (95% CI, 73.9% to 89.1%), including ██████ ████████ ████ ███ ██ ███████ ████████ ████ ███ ███ ██ ███████ ████████ ████ ██ ███ ██ ██ ████ ████ █████ ███ █████ ███████████ █████.
Patient-Reported Outcomes
Health-Related Quality of Life
EORTC QLQ-C30 Instrument
Completion rates for the EORTC QLQ-C30 declined over time. After week 16 (cycle 6), approximately 72% of patients were still available for completion in cohort A; at cycles 12 and 18 the percentage of available patients had dropped to approximately 56% and 36%, respectively.18 The descriptive summary statistics of observed mean scores and mean changes from baseline at each assessment point for the EORTC QLQ-C30 (Global Health Status/QoL scale) at the April 2, 2020, data cut-off date are summarized in Figure 5 and Figure 6, respectively. A definition of what constituted a clinically meaningful change from baseline in the target population was not provided. Overall observed scores from baseline to cycle 33 (March 22, 2019, data cut-off date) or to cycle 42 (April 7, 2020, data cut-off date) were reported to be variable, with no consistent trends.17


Post Hoc Analysis on HRQoL
Descriptive statistics of observed mean changes from baseline to week 16 (cycle 6) by subgroups of patients (i.e., patients with CR or PR, stable disease, or PD) were reported by Valle et al. (2021)54 (baseline to cycle 6) and in Figure 7 (baseline to cycle 39) for the March 22, 2019, data cut-off date.
The analysis population included 100 evaluable patients of the 107 patients in cohort A. A definition of the evaluable population was not provided.54 The 3 subgroups of CR or PR, stable disease, and PD included 36, 48, and 15 patients, respectively. The results suggested that the overall mean score change for the overall health status scale was maintained in patients with CR or PR (−0.3; SD = 1.3), and stable disease (−0.3; SD = 1.1), and declined in patients with PD (−1.2; SD = 0.8).54 Although similar results were observed for the emotional function scale, all patients appeared to show decline on the role and social functioning scales.54 The sponsor reported that the difference in mean change from baseline between patients with PD and those with CR or PR or with stable disease were driven by a reported increase in feelings of worry and tension in patients with PD at cycle 6.26 The sponsor additionally reported that differences in mean change from baseline were seen for the constipation scale between patients with PD and those with CR or PR or with stable disease; however, results were not reported and no definition of what constitutes a meaningful change was provided.26 Overall, Valle et al. (2021)54 concluded that changes in HRQoL appeared directionally more favourable in patients with CR or PR or with stable disease than in patients with PD.54

BL = baseline; CR/PR = complete response or partial response; SD = stable disease; PD = progressive disease; QoL = quality of life.
Source: Sponsor’s submission.26
Symptom Severity
EORTC QLQ-BIL21 Instrument
Results were available for the April 7, 2020, data cut-off date. Completion rates for the EORTC QLQ-BIL21 instrument declined over time. After week 16 (cycle 6), there were approximately ███ of patients still available for completion in cohort A; at cycle 12 and cycle 18, respectively, the percentage of available patients had dropped to approximately ███ and ███.18 The descriptive summary statistics of observed mean scores and mean changes from baseline at each assessment point for the EORTC QLQ-BIL21 (eating and pain scales) at the April 7, 2020, data cut-off date are summarized in Figure 8 (eating scale), Figure 9 (eating scale), Figure 10 (pain scale), and Figure 11 (pain scale). A definition for what constituted a clinically meaningful change from baseline in the target population was not provided. Overall observed scores from baseline to cycle 33 (March 22, 2019, data cut-off date) or to cycle 42 (April 7, 2020, data cut-off date) were reported to be variable, with no consistent trends.17




Post Hoc Analysis on HRQoL
Descriptive statistics of observed mean changes from baseline to week 16 (cycle 6) by subgroups of patients (i.e., patients with CR or PR, stable disease, or PD) were reported by Valle et al. (2021)54 (baseline to cycle 6) and in Figure 12 (baseline to cycle 39) for the March 22, 2019, data cut-off date. The analysis population included 100 evaluable patients of the total 107 patients in cohort A. A definition of the evaluable population was not provided.54 The 3 subgroups included 36, 48, and 15 patients with CR or PR, stable disease, and PD, respectively.54 The results suggested that the overall mean score change for the pain scales appeared to decline in patients with CR or PR (−5.7; SD = 20.0), and stable disease (−3.5; SD = 11.4), and increase in patients with PD (8.3; SD = 5.9).54 The sponsor reported that the difference in mean change from baseline between patients with PD and those with CR or PR or with stable disease were driven by a reported increase in pain at night in patients with PD at cycle 6.26 Similar results were observed for the anxiety scale, and the sponsor reported that the difference in mean change from baseline between patients with PD and those with CR or PR or with stable disease were driven by a reported increase in worry and decreased ability to enjoy oneself in patients with PD at cycle 6.26,54 All patients appeared to experience an increase in treatment side effects.54 Overall, Valle et al. (2021)54 concluded that changes in HRQoL appeared directionally more favourable in patients with CR or PD or with stable disease than in patients with PD.54

BL = baseline; CR/PR = complete response or partial response; SD = stable disease; PD = progressive disease; QoL = quality of life.
Source: Sponsor’s submission.26
Harms
Only harms identified in the review protocol are reported here. Refer to Table 16 for detailed harms data in cohort A at the March 22, 2019, April 7, 2020, and July 8, 2021 data cut-off dates. Results are similar at all data cut-off dates, with no new safety concerns identified at study close; therefore, the safety results of the March 22, 2019, analyses are described here.
Adverse Events
All patients in cohort A experienced at least 1 TEAE (100.0%). The most commonly reported TEAEs were alopecia (59.3%), hyperphosphatemia (55.6%), diarrhea (53.7%), fatigue (46.3%), and nausea (42.6%).17
Grade 3 or higher TEAEs occurred in 66.7% of patients in cohort A (Table 21). The most commonly reported grade 3 or higher TEAE was hypophosphatemia. The percentage of patients experiencing hypophosphatemia was 14.8%. Other grade 3 or higher TEAEs included stomatitis (9.3%), arthralgia (6.5%), palmar-plantar erythrodysesthesia syndrome (6.5%), abdominal pain (5.6%), and fatigue (4.6%).17
Serious Adverse Events
The percentage of patients experiencing serious TEAEs was █████ in cohort A. The most common serious TEAEs were ███████ ███ ████████████ ████ █████████ ██ ████ ██ █████████ █████████ █████ █████████ ██ ████ ██ █████████ ███ ███████████ ██████████ █████████ ██ ████ ██ █████████.17
Withdrawal Due to Adverse Events
None of the patients withdrew from the FIGHT-202 study because of an AE as primary reason. AEs led to discontinuation of the study treatment in 6.5% of patients in cohort A. TEAEs leading to treatment discontinuation were each experienced ██ ███ ███████ ████ ███ ████████ ██████████ ███████████ ███████ ████████████████ ██████████ ███████ ████ ████ ███████████ ███████ ██████████████████ ███████ ███████ █████ █████████ ███████ ███ ██████ ███████.17
Mortality
TEAEs leading to death occurred relatively rarely in cohort A █████████ and included failure to thrive and bile duct obstruction. None of the TEAEs leading to death were considered treatment related.17
Notable Harms
Notable harms specified in the protocol included nail toxicity, serous retinal detachment, hyperphosphatemia, and hypophosphatemia.17
Nail Toxicity
The percentage of patients experiencing nail toxicity TEAEs was █████ in patients in cohort A. The most commonly reported nail toxicity included █████████████ ████████ ████ ██████████████ ████████ ████ █████████ ████████ ███████████ ███████ ██████████ ███ █████████████ █████████ ██ ████ ██ ████████ ████. The percentage of patients experiencing nail toxicity TEAEs of grade 3 or higher was █████ █████████ ████ ██████████████ ████ █████████ █████████████ ███ ██████████. No serious nail toxicity TEAE occurred in cohort A.17
Serous Retinal Detachment
The percentage of patients experiencing serous retinal detachment TEAEs in cohort A was █████ The most commonly reported serous retinal detachment was ███████ ██████████ ██████ ████████ ██ █████████████ █████ ███████ ██████████ ██ ███████ ███████ ██████████ ███████ ███ ███████████ ███████ ██████ ███████ ██████████ ████ ██ █████ █ ██ ██████ ███ █ ███████ ██████ ███████ ██████████ ████ ███ ███████████ ██ ███ ███████ ████ ███ ████████ ██████ ███████ ██████████.17
Hyperphosphatemia
The percentage of patients experiencing hyperphosphatemia TEAEs in cohort A was █████. The most commonly reported hyperphosphatemia events were █████████████████ ███████ ███ █████ ██████████ █████████ ██████. No hyperphosphatemia TEAEs of grade 3 or higher or serious hyperphosphatemia TEAEs occurred in cohort A.17
Hypophosphatemia
The percentage of patients experiencing hypophosphatemia TEAEs in cohort A was █████. The most commonly reported hypophosphatemia events were ████████████████ ███████ ███ █████ ██████████ █████████ ██████. Hypophosphatemia TEAEs of grade 3 or higher occurred in █████ of patients, including only hypophosphatemia events. No serious hypophosphatemia TEAE occurred in cohort A.17
Table 16: Summary of Harms, Safety Population (Data Cut-Off Dates: March 22, 2019; April 7, 2020; and July 8, 2021)
Harms | Pemigatinib cohort A | ||
|---|---|---|---|
N = 107 | N = 108 | N = 108 | |
Data cut-off date | March 22, 2019 | April 7, 2020 | July 8, 2021 |
Patients with at least 1 TEAE | |||
n (%) | 107 (100.0) | ████ | 108a (100.0) |
Most common events,b n (%) | |||
Alopecia | 63 (58.9) | ████ | 64 (59.3) |
Hyperphosphatemia | 59 (55.1) | ████ | 60 (55.6) |
Diarrhea | 56 (52.3) | ████ | 58 (53.7) |
Dysgeusia | 51 (47.7) | ████ | 45 (41.7) |
Fatigue | 48 (44.9) | ████ | 50 (46.3) |
Nausea | 43 (40.2) | ████ | 46 (42.6) |
Constipation | 43 (40.2) | ████ | 46 (42.6) |
Stomatitis | 41 (38.3) | ████ | 46 (42.6) |
Dry mouth | 41 (38.3) | ████ | 42 (38.9) |
Dry eye | 34 (31.8) | ████ | 38 (35.2) |
Vomiting | 33 (30.8) | ████ | 36 (33.3) |
Decreased appetite | 32 (29.9) | ████ | 34 (31.5) |
Arthralgia | 31 (29.0) | ████ | 37 (34.3) |
Dry skin | 27 (25.2) | ████ | 30 (27.8) |
Hypophosphatemia | 26 (24.3) | ████ | 28 (25.9) |
Back pain | 24 (22.4) | ████ | 27 (25.0) |
Pain in extremity | 25 (23.4) | ████ | 26 (24.1) |
Abdominal pain | 24 (22.4) | ████ | 25 (23.1) |
Palmar-plantar erythrodysesthesia syndrome | 21 (19.6) | ████ | 23 (21.3) |
Urinary tract infection | 17 (15.9) | ████ | 21 (19.4) |
Decreased weight | 18 (16.8) | ████ | 20 (18.5) |
Headache | 20 (18.7) | ████ | 20 (18.5) |
Dizziness | 17 (15.9) | ████ | 19 (17.6) |
Epistaxis | 19 (17.8) | ████ | 19 (17.6) |
Hypercalcemia | 16 (15.0) | ████ | 17 (15.7) |
Dehydration | 17 (15.9) | ████ | 17 (15.7) |
Peripheral edema | 16 (15.0) | ████ | 16 (14.8) |
Anemia | 16 (15.0) | ████ | 16 (14.8) |
Pyrexia | 13 (12.1) | ████ | 15 (13.9) |
Asthenia | 14 (13.1) | ████ | 15 (13.9) |
Myalgia | 15 (14.0) | ████ | 15 (13.9) |
Gastroesophageal reflux disease | 13 (12.1) | ████ | 13 (12.0) |
Dyspepsia | 10 (9.3) | ████ | 14 (13.0) |
Upper abdominal pain | 10 (9.3) | ████ | 12 (11.1) |
Patients with at least 1 grade 3 or higher TEAE | |||
n (%) | 64 (59.8) | ████ | 72 (66.7) |
Most common events,c n (%) | |||
Hypophosphatemia | 13 (12.1) | ████ | 16 (14.8) |
Stomatitis | 8 (7.5) | ████ | 10 (9.3) |
Arthralgia | 7 (6.5) | ████ | 7 (6.5) |
Palmar-plantar erythrodysesthesia syndrome | 6 (5.6) | ████ | 7 (6.5) |
Abdominal pain | 5 (4.7) | ████ | 6 (5.6) |
Fatigue | 4 (3.7) | ████ | 5 (4.6) |
Diarrhea | 3 (2.8) | ████ | 4 (3.7) |
Hypotension | 4 (3.7) | ████ | 4 (3.7) |
Cholangitis | 3 (2.8) | ████ | 4 (3.7) |
Increased blood bilirubin | 2 (1.9) | ████ | 4 (3.7) |
Hyponatremia | 3 (2.8) | ████ | 3 (2.8) |
Anemia | 3 (2.8) | ████ | 3 (2.8) |
Increased blood alkaline phosphatase | 3 (2.8) | ████ | 3 (2.8) |
Dehydration | 3 (2.8) | ████ | 3 (2.8) |
Increased aspartate aminotransferase | 3 (2.8) | ████ | 3 (2.8) |
Hypertension | 3 (2.8) | ████ | 3 (2.8) |
Urinary tract infection | 3 (2.8) | ████ | 3 (2.8) |
Hyperbilirubinemia | 3 (2.8) | ████ | 3 (2.8) |
Hypokalemia | 2 (1.9) | ████ | 3 (2.8) |
Nausea | 3 (2.8) | ████ | 3 (2.8) |
Patients with at least 1 serious TEAE | |||
n (%) | 43 (40.2) | ████ | ████ |
Most common events,d n (%) | |||
Pyrexia | 5 (4.7) | ████ | ████ |
Cholangitis | 4 (3.7) | ████ | ████ |
Abdominal pain | 4 (3.7) | ████ | ████ |
Cholangitis infection | 3 (2.8) | ████ | ████ |
Small intestinal obstruction | 2 (1.9) | ████ | ████ |
Chills | 2 (1.9) | ████ | ████ |
Fatigue | 2 (1.9) | ████ | ████ |
Bile duct obstruction | 2 (1.9) | ████ | ████ |
Urinary tract infection | 2 (1.9) | ████ | ████ |
Sepsis | 1 (0.9) | ████ | ████ |
Bacteremia | 2 (1.9) | ████ | ████ |
Increased blood bilirubin | 0 (0.0) | ████ | ████ |
Failure to thrive | 2 (1.9) | ████ | ████ |
Hypercalcemia | 2 (1.9) | ████ | ████ |
Dehydration | 2 (1.9) | ████ | ████ |
Device occlusion | 2 (1.9) | ████ | ████ |
Acute kidney injury | 2 (1.9) | ████ | ████ |
Pleural effusion | 2 (1.9) | ████ | ████ |
Patients who stopped treatment because of TEAEs | |||
n (%) | 5 (4.7) | ████ | ████ |
Most common events, n (%) | |||
Intestinal obstruction | 1 (0.9) | ████ | ████ |
Gastrointestinal hemorrhage | 1 (0.9) | ████ | ████ |
Bile duct obstruction | 1 (0.9) | ████ | ████ |
Hyperbilirubinemia | 1 (0.9) | ████ | ████ |
Biliary tract infection | 0 (0.0) | ████ | ████ |
Sepsis | 0 (0.0) | ████ | ████ |
Paraplegia | 1 (0.9) | ████ | ████ |
Acute kidney injury | 1 (0.9) | ████ | ████ |
Deaths | |||
Due to TEAEs,e n (%) | 3 (2.8) | ████ | ████ |
Most common events, n (%) | |||
Bile duct obstruction | 1 (0.9) | ████ | ████ |
Failure to thrive | 2 (1.9) | ████ | ████ |
Notable harms | |||
Nail toxicity, n (%) | |||
Nail toxicity (any grade TEAEs) | 56 (52.3) | ████ | ████ |
Onychomadesis | 13 (12.1) | ████ | ████ |
Nail discoloration | 12 (11.2) | ████ | ████ |
Nail dystrophy | 10 (9.3) | ████ | ████ |
Onycholysis | 10 (9.3) | ████ | ████ |
Paronychia | 9 (8.4) | ████ | ████ |
Onychoclasis | 9 (8.4) | ████ | ████ |
Nail disorder | 5 (4.7) | ████ | ████ |
Onychomycosis | 4 (3.7) | ████ | ████ |
Nail infection | 1 (0.9) | ████ | ████ |
Nail ridging | 3 (2.8) | ████ | ████ |
Nail toxicity | 3 (2.8) | ████ | ████ |
Nail hypertrophy | 1 (0.9) | ████ | ████ |
Onychalgia | 1 (0.9) | ████ | ████ |
Nail toxicity (grade 3 or higher TEAEs) | 3 (2.8) | ████ | ████ |
Nail toxicity (serious TEAEs) | 0 (0.0) | ████ | ████ |
Serous retinal detachment, n (%) | |||
Serous retinal detachment (any grade TEAEs) | 4 (3.7) | ████ | ████ |
Retinal detachment | 2 (1.9) | ████ | ████ |
Chorioretinal folds | 1 (0.9) | ████ | ████ |
Detachment of retinal pigment epithelium | 0 (0.0) | ████ | ████ |
Maculopathy | 1 (0.9) | ████ | ████ |
Retinal thickening | 1 (0.9) | ████ | ████ |
Serous retinal detachment (grade 3 or higher TEAEs) | 1 (0.9) | ████ | ████ |
Serous retinal detachment (serious TEAEs) | 1 (0.9) | ████ | ████ |
Hyperphosphatemia, n (%) | |||
Hyperphosphatemia (any grade TEAEs) | 62 (57.9) | ████ | ████ |
Hyperphosphatemia | 59 (55.1) | ████ | ████ |
Increased blood phosphorus | 4 (3.7) | ████ | ████ |
Hyperphosphatemia (any grade 3 or higher TEAEs) | 0 (0.0) | ████ | ████ |
Hyperphosphatemia (serious TEAEs) | 0 (0.0) | ████ | ████ |
Hypophosphatemia, n (%) | |||
Hypophosphatemia (any grade TEAEs) | 27 (25.2) | ████ | ████ |
Hypophosphatemia | 26 (24.3) | ████ | ████ |
Decreased blood phosphorus | 1 (0.9) | ████ | ████ |
Hypophosphatemia (grade 3 or higher TEAEs) | 13 (12.1) | ████ | ████ |
Hypophosphatemia (serious TEAEs) | 0 (0.0) | ████ | ████ |
NR = not reported; TEAE = treatment-emergent adverse event.
Note: A patient is counted only once for multiple events within preferred term and/or system organ class.
aThe April 7, 2020, data cut-off date included 1 additional patient in cohort A who had been enrolled after the August 30, 2019, data cut-off date.18
bFrequency > 10% of patients at 1 or both of the 2 data cut-off dates.
cFrequency ≥ 2% of patients at 1 or both of the 2 data cut-off dates.
dFrequency ≥ 1% of patients at 1 or both of the 2 data cut-off dates.
eNone considered treatment related.
Sources: Clinical Study Report,17 sponsor’s response.18
Critical Appraisal
Internal Validity
Primary objective of phase II studies: The primary objective of phase II (randomized or nonrandomized) trials is to document the safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use in confirmatory phase III trials. It is unclear whether the results observed in this phase II trial will translate into positive phase III trials or into real-world clinical practice. There are currently no randomized phase III trials under way for this review’s target population. The clinical experts consulted by CDA-AMC noted that, despite the high unmet need, it would not be feasible to conduct an RCT comparing a targeted therapy such as pemigatinib with currently available therapies in the second-line setting in clinical practice in Canada. According to the clinical experts, the development of phase III RCTs is hindered by the overall low patient numbers with the current indication, and an equipoise between pemigatinib and other chemotherapy drugs does not exist.
Limited interpretation of time-to-event end points: The interpretation of time-to-event end points such as OS or PFS is limited in single-arm studies. Although ORR may be directly attributable to the drug effect, the nonrandomized design makes it a challenge to interpret PFS and OS events attributable to pemigatinib, because all patients in cohort A received the same treatment. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear.53 The FDA’s multidiscipline review of the assessment of pemigatinib for the present indication included the following reviewer comment: “In a single-arm trial, FDA considers time-to-event end points to be uninterpretable, and the results will not be described in this review.”5 Consequently, the efficacy outcomes contributing to the FDA’s accelerated approval of pemigatinib in the current setting included response outcomes and no survival end points.5
Prognostic value of FGFR2: Although there is strong genetic and functional evidence that FGFR genetic alterations can drive the formation of tumours,7 it is currently not known if patients with an FGFR2 alteration represent a distinct prognostic subgroup.11 Retrospective studies20,27-29 of patients with CCA have suggested that patients with FGFR alterations have a better prognosis than those in an unselected CCA population,9,20 although limitations preclude the ability to draw strong conclusions. Given the single-arm design of the FIGHT-202 trial, it could be a challenge to disentangle the extent to which the observed results are due to a potential prognostic effect of FGFR2 fusions or rearrangements or are due to drug-associated effects. The clinical experts consulted on this issue by CDA-AMC agreed that, although there is exploratory, preliminary data that FGFR2 genetic alterations may be associated with a more indolent disease progression,20 there is currently insufficient evidence to determine whether patients with an FGFR2 fusion represent a distinct prognostic subgroup. The clinical experts were of the opinion that FGFR2 mutation status was likely not an important prognostic factor in the indicated patient population. The clinical experts considered that the medium time from initial diagnosis to enrolment in cohort A of the FIGHT-202 trial was approximately 1 year, which is reflective of medium OS with standard first-line therapy in this setting. The duration of first-line therapy for patients in cohort A of the FIGHT-202 trial was also reflective of clinical practice, according to the clinical experts. The clinical experts further agreed that progression on prior systematic therapy is a key prognostic factor in these patients, and they stated that they anticipate that patients would not have derived any substantial benefit from their underlying disease biology at the time they enrolled into the FIGHT-202 trial.
Open-label design: The FIGHT-202 trial had an open-label design in which the investigator and the study participants were aware of their treatment status, which increases the risk of detection bias and performance bias. This has the potential to bias results and outcomes in favour of pemigatinib if the assessor (investigator or patient) believes the study drug is likely to provide a benefit. However, to mitigate the impact of this bias, the investigators relied on an IRC to evaluate responses using standardized criteria (i.e., based on RECIST 1.1, assessed by an independent radiological review committee using central genomics laboratory results26 with confirmation of CR and PR at least 4 weeks after the initial assessment).26 Results of the response assessments by the investigator and IRC generally consistent, suggesting that the potential confounding effects on response outcomes from the open-label design is likely not substantial. Furthermore, subjective outcomes (i.e., adverse outcomes and patient-reported outcomes) may be biased because of the open-label design. For example, if study personnel and patients knew that the treatment was pemigatinib (which is known to cause nail toxicity, hyperphosphatemia, and other AEs), this could have influenced the reporting of harms. Overall, the magnitude and direction of this bias remains unclear.
Statistical analyses: No formal statical significance or hypotheses testing were performed, so no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. In cohort A, a greater than 95% probability of having a 95% CI for ORR with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. Results for ORR appeared consistent with the sample size assumptions, and the study recruited the intended number of patients.
Subgroup analysis: Methodological issues limited the ability to interpret the results from subgroup analyses. The subgroup analyses were noninferential, wide CIs reflected uncertainty in the effect estimates, and small sample sizes limited the generalizability to a broader population.
Small sample size: There were a limited number of patients included in the efficacy-evaluable dataset (n = 107) of cohort A. The magnitude of the treatment effect estimates observed in a small study sample may not be replicable in a larger study sample or generalizable to the target population in real-world clinical practice.
HRQoL and symptom severity assessments: The interpretation of results for EORTC QLQ-C30 and EORTC QLQ-BIL21 (i.e., the ability to assess trends over time) at later cycles is limited by the substantial decline in patients available to provide assessment over time (after week 16 [cycle 6], approximately 72% of patients were still available for completion; at cycle 12 and cycle 18, respectively, the percentage of available patients had dropped to approximately 56% and 36%). In addition, selection bias over time should be considered when interpreting results, as the long-term survivors tend to be the healthier patients. Furthermore, given the lack of prespecified statistical analyses a priori in the statistical analysis plan for patient-reported outcomes, results from post hoc analyses (i.e., changes from baseline to week 16 summarized by disease response subgroup) are considered exploratory in nature. As well, patient-reported outcomes in the post hoc analysis were measured up to week 16, which may not represent an accurate picture of a patient’s experience with pemigatinib over a prolonged period of time. Given that the trial was nonrandomized, the impact of pemigatinib on patient-reported outcomes in relation to other therapies is unknown.
The reliability of the EORTC QLQ-C30 instrument was evaluated in an international study of patients with BTC,46 which showed that internal consistency was acceptable for most scales and that results for test-retest reliability were mixed.46 Estimates for MIDs in the literature were not found for the EORTC QLQ- QLQ-C30 in patients with CCA or BTC. The sponsor did not report an MID and did not define the magnitude in change from baseline that would constitute a clinically meaningful change in the target population. Therefore, it is unclear whether the changes from baseline experienced by patients in the FIGHT-202 trial are reflective of a clinically meaningful change in patients with unresectable, locally advanced or metastatic CCA and FGFR2 alterations.
The EORTC QLQ-BIL21 instrument was validated in an international study of patients with BTC, which showed acceptable internal consistency for all multiitem scales and good test-retest reliability. Known group validity was shown to distinguish between subgroups, and there was some evidence of responsiveness. Estimates for MIDs in the literature were not found for the EORTC QLQ-BIL21 in patients with CCA or BTC. The sponsor did not report an MID and did not define the magnitude in change from baseline that would constitute a clinically meaningful change in the target population. Therefore, it is unclear if the changes from baseline experienced by patients in the FIGHT-202 trial are reflective of a clinically meaningful change in patients with unresectable, locally advanced or metastatic CCA and FGFR2 alterations.
Overall, the methodological issues noted render results from the EORTC QLQ-C30 and EORTC QLQ-BIL21 instruments inconclusive.
External Validity
The clinical experts consulted by CDA-AMC, overall, agreed that the baseline patient characteristics of cohort A in the FIGHT-202 trial were reflective of the patients they care for in clinical practice in Canada for the present indication. Although the majority of patients in cohort A were enrolled at trial sites in Europe and the US, according to the clinical experts consulted by CDA-AMC, the population enrolled in the trial was consistent with the population expected to be treated in clinical practice in Canada; furthermore, no different treatment effect would be expected with the different disease management practices in countries. According to the clinical experts, as long as patients have the FGFR2 alteration, pemigatinib would be appropriate to administer after any of the prior therapies received by patients in the trial. However, the clinical experts agreed that patients should not have previously been treated with an FGFR2-targeted therapy. The majority of patients (61%) in cohort A of the FIGHT-202 trial had received 1 line of systemic therapy before trial enrolment, 27% of patients had received 2 lines, and 12% had received 3 or more lines. The clinical experts said they anticipate seeing a benefit from treatment with pemigatinib, regardless of the number of previous lines of systemic therapy received, as long as patients have the FGFR2 alteration. Furthermore, FGFR2 alterations occur rarely in patients with eCCA; there was 1 patient in cohort A of the FIGHT-202 trial with FGFR2-positive CCA, but all other patients in cohort A had iCCA. The clinical experts noted that patients with iCCA and eCCA are managed in a similar way in clinical practice, and that results observed in cohort A are generalizable to patients with FGFR2-positive eCCA, based on the fact that FGFR2 is the target of the mechanism of action of pemigatinib and there is no biological rationale to assume that pemigatinib’s safety profile would be different in patients with eCCA.
Concomitant medications received by patients in the trial appeared reflective of the medications that patients would receive in clinical practice in Canada, according to the clinical experts consulted by CDA-AMC.
Noncomparative design: The noncomparative design of the FIGHT-202 trial precludes the ability to compare the relative therapeutic benefit or safety of pemigatinib with currently available therapies in clinical practice in Canada. As noted previously, the clinical experts consulted by CDA-AMC agreed that direct randomized comparisons between pemigatinib and currently used therapies are not feasible in the setting of previously treated, unresectable, locally advanced or metastatic CCA in patients with an FGFR2 fusion or other rearrangement. In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC,55 in the form of an unanchored MAIC, comparing the efficacy of pemigatinib in cohort A in the FIGHT-202 trial with that in each of the 2 treatment groups in the ABC-06 study.
Relevance of trial efficacy outcomes: The primary outcome in in cohort A of the FIGHT-202 trial was ORR, and secondary outcomes included DOR, DCR, PFS, and OS. According to the clinical experts consulted by CDA-AMC, ORR, durability of response, and DCR are clinically meaningful end points for patients with unresectable, locally advanced or metastatic CCA who have progressed on prior therapy. Responses in this patient population are important because of the accompanying delay in the worsening of symptoms and the slower decline in ECOG PS. According to the clinical experts, the majority of patients will have stable disease, followed by PR and, rarely, CR in response to first-line treatment; therefore, the clinical experts were not concerned about the low number of patients who achieved a CR in the FIGHT-202 trial, but they emphasized the clinical relevance and importance of maintaining stable disease to prevent an otherwise fast decline in patients in this setting. The clinical experts suggested that it is reasonable to postulate that the 37% ORR in the FIGHT-202 trial may translate into clinical benefits in terms of PFS and OS. They also stated that durable responses could potentially delay tumour progression and result in a prolonged survival benefit in this patient population.
Excluded patient subgroups: The FIGHT-202 trial excluded patients who would be intolerant to standard first-line therapy without experiencing PD. The clinical experts consulted by CDA-AMC said that it would be reasonable to generalize the results from cohort A to patients with FGFR2 alterations who are intolerant to first-line therapy, given the favourable safety profile of oral pemigatinib. The clinical experts noted that because patients with an ECOG PS greater than 2 were excluded from the FIGHT-202 trial, there are no data to support the generalizability of treatment benefit in this patient population. The clinical experts were of the opinion that it would be reasonable to leave it to the discretion of the treating physician to apply some flexibility in terms of using pemigatinib with slightly lower lab parameters than those outlined in the trial. It was agreed by the clinical experts that because patients with untreated brain or central nervous system metastases or with brain or central nervous system metastases that have progressed were excluded from the trial, there are no data to support the generalizability of treatment benefit in this patient population.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Because FIGHT-202 was a noncomparative study, the systematic review does not provide any direct evidence for the relative efficacy of pemigatinib compared with a relevant comparator. A focused literature search for network meta-analyses dealing with pemigatinib or CCA was run in MEDLINE All (1946‒ ) via Ovid and in Embase (1974‒ ) via Ovid on July 16, 2021. No limits were applied, and conference abstracts were excluded from the search results. No relevant studies were identified that compared pemigatinib with mFOLFOX, FOLFIRI, 5-FU alone or in combination with cisplatin or oxaliplatin, capecitabine alone or in combination with cisplatin or oxaliplatin, or best supportive care in adults with previously treated, unresectable, locally advanced or metastatic CCA and FGFR2 fusions or other rearrangements.
The sponsor submitted an ITC55 in the form of a MAIC between cohort A of the FIGHT-202 study and each of the 2 treatment groups in the ABC-06 study. The ABC-06 study compared an mFOLFOX regimen plus ASC with ASC alone in patients with BTC. The results of the MAIC were used to inform the sponsor’s pharmacoeconomic submission.
Methods of Sponsor-Submitted MAIC
Objectives
Given the lack of an RCT comparing pemigatinib with a standard-of-care regimen, an ITC was conducted to provide evidence for the relative efficacy of pemigatinib compared with relevant comparators.
Study Selection Methods
There were 2 literature searches, performed on November 9, 2018 (original search) and April 21, 2020 (updated search), to identify available clinical efficacy, safety, and tolerability evidence related to the second-line treatment of patients with advanced and/or metastatic CCA and FGFR2 fusions or rearrangements. Both searches used the same English-language search strategy in multiple databases (MEDLINE In-Process, Embase, MEDLINE, and the Cochrane Library), and the date ranges were from database inception to November 9, 2018, for the original search and from October 1, 2018, to April 21, 2020, for the updated search. The following sources were also searched: conference proceedings from 6 oncology conferences starting from the year 2016; reference lists of relevant studies, systematic reviews published in the previous 2 years, and meta-analyses; and reference lists from relevant articles from 7 health technology assessment agencies. For the original search, 2 independent reviewers screened abstracts and a third reviewer assessed abstracts when there was disagreement or uncertainty. In these cases, the consensus of the majority was used to make the final decision. Full-text screening of relevant abstracts was performed in the same manner as for abstract screening. For the updated search, 2 independent reviewers screened abstracts and full-text articles and a third reviewer independently resolved uncertainties regarding study inclusion at each screening stage.
Studies were eligible if they included adults with advanced and/or metastatic or surgically unresectable CCA and FGFR2 fusions or rearrangements for whom at least 1 treatment had failed. Studies were excluded if they reported on pediatric patients, patients without metastatic and/or advanced-stage cancer or FGFR2 fusions or rearrangements, patients who were treatment naive, or patients with resectable CCA. Articles unclear about disease stage, FGFR2 status, or treatment line were included during the abstract screening. Studies had to be single-arm trials with a pharmacological intervention or had to compare a pharmacological intervention with placebo, best supportive care (defined by the study authors), or any other pharmacological intervention. Eligible study designs were RCTs, single-arm studies, observational studies, and systematic reviews, with the latter used only for bibliography searches. Preclinical studies, case reports, case series, pharmacokinetic studies, and economic studies were excluded. A list of relevant outcomes was provided, although it is unclear whether articles were screened based on available outcomes. These outcomes were response rate, OS, PFS, time to treatment discontinuation, DOR, mortality, HRQoL, incidence of AEs, study and/or treatment discontinuation, relationship between intermediate outcomes (PFS and response rate) and OS, DCR, stable disease, time on treatment, time to response, overall response rate, and patient-reported outcomes.
In the original search, 35 relevant results were identified, which reported on 8 noncomparative studies, 1 retrospective observational study, and 11 ongoing studies with no results available. In addition, 32 articles were flagged as having a patient population with BTC and 79 articles were flagged as having a patient population with unclear FGFR2 mutation status. From the updated search, 829 new articles were included at the full-text screening stage; in addition, the 111 flagged articles from the original search were included in the full-text screening. One of the most common reasons for excluding full-text publications was BTCs (n = 117). In response56 to a request for clarification by the CDA-AMC review team, the sponsor indicated that the following types of studies were excluded: studies that did not report a subgroup of patients with CCA, studies in which patients with CCA made up less than 80% of the study population, and studies in which the percentage of patients with each type of BTC was not reported.
After full-text screening in the updated search, 209 relevant publications were identified (of which 23 were identified from conference proceedings and 35 were the relevant results from the original search), reporting on 108 studies. Study quality for the 108 studies was assessed using the Downs and Black checklist, although it was unclear how, or if, these assessments were used.
For potential inclusion in the MAIC, additional criteria were applied to the 108 studies identified in the systematic literature searches. It was not clear at what point these criteria were established, and rationales were not provided for all of the criteria. KM plots of both OS and PFS were required so that pseudo patient-level data for these outcomes could be derived. The study had to include a treatment (if not pemigatinib) that was representative of standard of care, which seemed to be defined as chemotherapy. A minimum sample size of 20 patients was established, although a justification for this cut-off was not provided. After applying these additional criteria and eliminating a study of patients receiving fourth-line or later-line therapy, 8 studies remained. Although the proportion of patients with an ECOG PS of 0 or 1 and the proportion with iCCA had to be high to match the patient population in the FIGHT-202 study, these criteria do not appear to have been applied at this stage of the study selection process.
Of the 8 remaining studies, 2 were single-arm trials (including the FIGHT-202 study), 2 were RCTs, and 4 were retrospective studies. In the studies other than the FIGHT-202 study, publication dates ranged from 2012 to 2020, sample sizes for each treatment group ranged from 30 to 255, treatments included chemotherapy and ASC, median age ranged from 54 years to 65 years where reported, the percentage of male patients ranged from 43.0% to 66.7%, the percentage of patients with iCCA ranged from 16.7% to 94.6%, and the percentage of patients with an ECOG PS of 0 or 1 ranged from 64.0% to 100.0% where reported. FGFR2 mutation status was not reported for any of the studies, aside from the FIGHT-202 study.
Despite the sponsor’s explanation that studies were excluded during the full-text screening if they did not report on the percentage of patients with CCA or if the percentage of patients with CCA was less than 80%, CDA-AMC reviewers noted examples in the 8 considered studies that appear to contradict this. The types of biliary cancers included in 1 study were not reported in the cited conference abstract57 and the percentage of patients with CCA was less than 80% in 3 studies.14,58,59
The ITC authors focused on 2 studies — by Kim et al.59 and Lamarca et al. (2021)14 — based on sample size and recent date of publication (thought to reflect more accurately the current standard of care). The ITC was originally developed for the pemigatinib submission to the National Institute for Health and Care Excellence (NICE), and the study by Lamarca et al. (2021),14 also known as the ABC-06 study, was chosen because it recruited patients in the UK.
MAIC Analysis Methods
The MAIC approach was selected because of the noncomparative nature of the FIGHT-202 study. The choice of the ABC-06 study for comparison with the FIGHT-202 study does not appear to be based on its specific interventions; however, the mFOLFOX regimen was considered to be a relevant comparator in the second-line setting by the clinical experts consulted by the ITC authors. Both the ASC group and the mFOLFOX plus ASC group from the ABC-06 study were compared with the pemigatinib group from the FIGHT-202 study.
According to the ITC authors, the following baseline characteristics were available for patients in cohort A of the FIGHT-202 study and for patients in the ABC-06 study: median age, percentage of male patients, percentage of patients with iCCA, percentage of patients with an ECOG PS of 0 or 1, and percentage of patients with a serum albumin concentration of less than 35 g/L. Of these, the following covariates were chosen for adjustment: age, sex, proportion of patients with an ECOG PS of 0 or 1, and proportion of patients with a serum albumin concentration of 35 g/L or greater. The selection of covariates for adjustment does not appear to be preplanned, and no rationale was provided for the covariates selected. Additionally, the following baseline characteristics were available for both studies and did not appear to be considered: disease stage, percentage of patients with prior surgery for cancer, and number of lines of prior systemic therapy for advanced or metastatic cancer. In the FIGHT-202 study, 9 patients from cohort A were excluded from the MAIC because of missing serum albumin values.
For each arm in the ABC-06 study, a logistic regression model was estimated, using the method of moments based on individual patient data from the FIGHT-202 study and summary data from the relevant arm in the ABC-06 study. The model was used to approximate propensity scores for patients in the FIGHT-202 study, which for each patient reflected the odds of being included in the ABC-06 study versus the FIGHT-202 study, based on the distributions of the covariates included in the model. The baseline characteristics used to reweight the pemigatinib group, along with ESS, were presented for the pemigatinib group before weighting and after weighting to match the mFOLFOX plus ASC and ASC-alone groups. The PFS and OS outcomes were the only disease-related outcomes available for both studies and were generated on a pseudo patient level for the ABC-06 study by estimating times and survival probabilities from the published KM PFS and OS curves using a software tool. HRs for PFS and OS were determined using Cox proportional hazard models and bootstrapping was used to estimate standard errors and CIs.
Results of Sponsor-Submitted MAIC
Summary of Included Studies
The study selected for the MAIC with cohort A of the FIGHT-202 study (the pemigatinib group) — the ABC-06 study — was a phase III, open-label, multicentre RCT conducted in the UK with patients enrolled from 2014 to 2018. Adult patients with locally advanced or metastatic BTC and disease progression in the previous 6 weeks on first-line cisplatin plus gemcitabine chemotherapy were randomized (1:1) to receive ASC alone or mFOLFOX plus ASC. Randomization was stratified by platinum sensitivity (sensitive or refractory and/or resistant), serum albumin concentration (< 35 g/L or ≥ 35 g/L), and disease stage (locally advanced or metastatic).
Patients in the ABC-06 study had to have an ECOG PS of 0 to 1; a life expectancy longer than 3 months; adequate hematological, renal, and hepatic function; and no evidence of ongoing infection, inadequate biliary drainage, metastatic disease to the brain, or clinically significant cardiovascular disease.
All patients received ASC, which consisted of early identification and treatment of biliary-related complications and cancer-related symptom management. Interventions included biliary drainage, antibiotics, analgesia, steroids, antiemetics, other palliative treatment for symptom control, palliative radiotherapy (e.g., for painful bone metastases), and transfusion of blood products. Patients had study visits every 4 weeks for ASC, which included physical examination, assessment of ECOG PS, symptom monitoring, review of concomitant medication, and assessment of liver and renal function with full blood count. Patients in the mFOLFOX plus ASC group also received chemotherapy every 2 weeks for a maximum of 12 cycles. At each cycle, patients received oxaliplatin 85 mg/m2 and L-folinic acid 175 mg (or folinic acid 350 mg) through IV infusion over 2 hours and 5-FU 400 mg/m2 through a 5-minute to 10-minute bolus on day 1. 5-FU 2,400 mg/m2 was started as a continuous IV infusion on day 1 and was finished on day 2. A maximum of 2 dose-reduction levels for each drug was allowed, representing a 20% and 50% reduction from the initial dose. If treatment was delayed for more than 28 days because of toxicity, the patient permanently discontinued treatment. If oxaliplatin was discontinued because of toxicity, treatment with the other components of the regimen could continue with an increase in the 5-FU dose, according to local practice. Patients with disease progression in the mFOLFOX plus ASC group were subsequently treated at the clinician’s discretion. Patients in both groups could receive treatment with experimental therapies in phase I trials after disease progression.
Radiological assessment with CT (and optionally MRI) was performed in the mFOLFOX plus ASC group every 12 weeks until disease progression, and images were evaluated by investigators according to RECIST 1.1 criteria. Patients in the ASC-alone group only underwent radiological assessments when clinically indicated. The primary end point of the ABC-06 study was OS in the intention-to-treat population. Secondary end points included PFS, radiological response, and QoL, all in the ASC plus mFOLFOX group alone. Results for HRQoL outcomes were unavailable in Lamarca et al.14
Key baseline characteristics of patients in cohort A of the FIGHT-202 study and the ASC-alone and mFOLFOX plus ASC groups are presented in Table 17. Compared with both treatment groups in the ABC-06 study, patients in cohort A of the FIGHT-202 study were younger (median age, 56 years versus 65 years) and less likely to be male (39% male versus 53% and 46% male). Almost all patients in cohort A of the FIGHT-202 study had iCCA, whereas only 42% and 47% of patients in the mFOLFOX plus ASC and ASC-alone groups, respectively, had iCCA. Almost all patients in both studies had an ECOG PS of 0 or 1, with a greater proportion of patients in cohort A of the FIGHT-202 study having an ECOG status of 0 than in the mFOLFOX plus ASC and ASC-alone groups. FGFR2 mutation status was not reported in the ABC-06 study. Almost 40% of patients in cohort A of the FIGHT-202 study and no patients in the ABC-06 study had received more than 1 line of prior systemic therapy.
Characteristic | Cohort A of FIGHT-202, pemigatinib (preweighting) N = 107 | mFOLFOX plus ASC N = 81 | ASC alone N = 81 |
|---|---|---|---|
Median age,a years (range) | ██ ███ ██ ███ | ██ ███ ██ ███ | ██ ███ █ |
Male,a n (%) | ██ ████ | ██ ████ | ██ ████ |
Tumour site, n (%) | ██ ████ | ██ ████ | ██ ████ |
Intrahepatic | ██ ████ | ██ ████ | ██ ████ |
Extrahepatic | ██ ████ | ██ ████ | ██ ████ |
Gallbladder | ██ ████ | ██ ████ | ██ ████ |
Ampulla | ██ ████ | ██ ████ | ██ ████ |
Missing | ██ ████ | ██ ████ | ██ ████ |
Patients with FGFR2 fusions or rearrangements, n (%) | ██ ████ | ██ ████ | ██ ████ |
ECOG PS,a n (%) | ██ ████ | ██ ████ | ██ ████ |
0 | ██ ████ | ██ ████ | ██ ████ |
1 | ██ ████ | ██ ████ | ██ ████ |
2 | ██ ████ | ██ ████ | ██ ████ |
Missing | ██ ████ | ██ ████ | ██ ████ |
Serum albumin ≥ 35 g/L,a n (%) | ██ ████ | ██ ████ | ██ ████ |
Disease stage, n (%) | ██ ████ | ██ ████ | ██ ████ |
Locally advanced | ██ ████ | ██ ████ | ██ ████ |
Metastatic | ██ ████ | ██ ████ | ██ ████ |
Unknown and/or missing | ██ ████ | ██ ████ | ██ ████ |
Previous surgery, n (%) | ██ ████ | ██ ████ | ██ ████ |
Number of prior systemic therapies, n (%) | ██ ████ | ██ ████ | ██ ████ |
1 | ██ ████ | ██ ████ | ██ ████ |
2 | ██ ████ | ██ ████ | ██ ████ |
≥ 3 | ██ ████ | ██ ████ | ██ ████ |
ASC = active symptom control; ECOG PS = Eastern Cooperative Oncology Group Performance Status; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil; NA = not applicable; NR = not reported.
aMean age and percentages of male patients, patients with an ECOG PS ≤ 1, and patients with serum albumin ≥ 35 g/L were covariates in the logistic regression model used for adjustment.
Sources: Sponsor-submitted MAIC report,55 FIGHT-202 Clinical Study Report,17 and Lamarca et al. (2021).14
Results
The regression model covariates are presented in Table 18 for the pemigatinib group before weighting and after weighting to match the mFOLFOX plus ASC group and the ASC-alone group. After weighting, the mean age in the pemigatinib group matched the median age in the ABC-06 study groups, and the proportions of patients who were male, had an ECOG PS of 0 or 1, and had a serum albumin concentration of 35 g/L or greater were the same in the pemigatinib group and the ABC-06 study groups. The ESS of the pemigatinib group was reduced to ████ ███ ████ after weighting to match the mFOLFOX plus ASC and ASC-alone groups, respectively. Other baseline characteristics after weighting were not presented in the sponsor-submitted ITC report.
Table 18: Summary of Model Covariates Before and After Weighting
Model covariate | Pemigatinib, before weighting N = 98 | Pemigatinib, weighted to mFOLFOX plus ASC ESS = 48.2 | Pemigatinib, weighted to ASC alone ESS = 47.3 |
|---|---|---|---|
Mean age, years | ████ | ████ | ████ |
Male, % | ████ | ████ | ████ |
ECOG PS ≤ 1, % | ████ | █████ | █████ |
Serum albumin ≥ 35 g/L, n (%) | ████ | ████ | ████ |
ASC = active symptom control; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil.
Source: Sponsor-submitted MAIC report.55
The median follow-up duration in the ABC-06 study at the data cut-off date was 21.7 months (interquartile range, 17.2 to 30.8 months). There were 2 patients lost to follow-up in the ASC-alone group and 1 patient lost to follow-up in the mFOLFOX plus ASC group.
Overall Survival
Results for OS in the pemigatinib and mFOLFOX plus ASC groups are presented in Table 19. After weighting of the pemigatinib group to match the mFOLFOX plus ASC group, median OS was █████ ██████ ██████ █████ ██ ███ ██ █ █████ ███████ for the pemigatinib group versus ████ ██████ ████ ███ ████ ██████ ██ ████ ███████ for the mFOLFOX plus ASC group, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.209 (95% CI, 0.127 to 0.313), the HR using the results from the April 7, 2020, data cut-off date was █████ ████ ███ █████ ██ ██████, and the HR using the results from the July 8, 2021, study-close date was █████ ████ ███ █████ ██ ██████. KM curves for OS are shown in Figure 13 for the unweighted pemigatinib group, the weighted pemigatinib group, and the mFOLFOX plus ASC group. Supplemental OS analyses were provided from the July 8, 2021, data cut-off date comparing pemigatinib to mFOLFOX plus ASC in patients who had received only 1 prior therapy. The number and ESS for this subgroup was ███████ and resulted in an HR of █████ ████ ███ █████ ██ ██████.
Table 19: Overall Survival, Pemigatinib Versus mFOLFOX Plus ASC
Outcome | Pemigatinib, before weighting N = 98 | Pemigatinib, weighted to mFOLFOX plus ASC ESS = 48.2 | mFOLFOX plus ASC N = 81 |
|---|---|---|---|
OS events, n | ██ | ██ | ██ |
Median OS, months (95% CI),a March 22, 2019, data cut-off date | █████ ███ | █████ ███ | ████ ████ |
HR (95% CI),b March 22, 2019, data cut-off date | █████ ████ | 0.209 (0.127 to 0.313) | ███ |
HR (95% CI),b April 7, 2020, data cut-off date | █████ █████ | █████ █████ | ███ |
HR (95% CI),b July 8, 2021, study close | █████ █████ | █████ █████ | ███ |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil; HR = hazard ratio; NA = not applicable; OS = overall survival.
aKaplan-Meier estimates.
bHRs were determined using Cox proportional hazard models, with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.55

ASC = active symptom control; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil; Pemi = pemigatinib; SoC = standard of care.
Note: The results presented for the pemigatinib groups are from the March 22, 2019, data cut-off date.
Source: MAIC report.55
Results for OS for pemigatinib versus ASC-alone are presented in Table 20. After weighting of the pemigatinib group to match the ASC-alone group, median OS was 16.53 months (lower limit of 95% CI = 15.28 months) for the pemigatinib group versus ████ ██████ ████ ███ ████ ██████ ██ ████ ███████ for the ASC group, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.163 (95% CI, 0.099 to 0.249), the HR using the results from the April 7, 2020, data cut-off date was █████ ████ ███ █████ ██ ██████ and the HR using the results from the July 8, 2021, study-close date was █████ ████ ███ █████ ██ ██████. KM curves for OS are shown in Figure 14 for the unweighted pemigatinib group, the weighted pemigatinib group, and the ASC-alone group. Supplemental OS analyses were provided from the July 8, 2021, data cut-off date comparing pemigatinib to ASC alone in patients who had received only 1 prior therapy. The number and ESS for this subgroup was ███████ and resulted in an HR of █████ ████ ███ █████ ██ ██████.
Table 20: Overall Survival, Pemigatinib Versus ASC Alone
Outcome | Pemigatinib, before weighting N = 98 | Pemigatinib, weighted to ASC ESS = 47.3 | ASC N = 81 |
|---|---|---|---|
OS events, n | █████ ████ | █████ ████ | █████ █████ |
Median OS, months (95% CI)a March 22, 2019, data cut-off date | █████ ████ | █████ ████ | ████ █████ |
HR (95% CI),b March 22, 2019, data cut-off date | █████ █████ | 0.163 (0.099 to 0.249) | █████ ████ |
HR (95% CI),b April 7, 2020, data cut-off date | █████ ████ | █████ ████ | █████ ████ |
HR (95% CI),b July 8, 2021, study close | █████ ████ | █████ █████ | █████ ████ |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; HR = hazard ratio; NA = not applicable; OS = overall survival.
aKaplan-Meier estimates.
bHRs were determined using Cox proportional hazard models, with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.55

ASC = active symptom control; Pemi = pemigatinib; SoC = standard of care.
Note: The results presented for the pemigatinib groups are from the March 22, 2019, data cut-off date.
Source: Sponsor-submitted MAIC report.55
Progression-Free Survival
Results for PFS for pemigatinib versus mFOLFOX plus ASC are presented in Table 21. After weighting of the pemigatinib group to match the mFOLFOX plus ASC group, median PFS was ████ ██████ ████ ███ ████ ██████ ██ █████ ███████ versus ████ ██████ ████ ███ ████ ██████ ██ ████ ███████ for the pemigatinib versus mFOLFOX plus ASC groups, based on the March 22, 2019, data cut-off date for the FIGHT-202 study. The corresponding HR was 0.436 (95% CI, 0.319 to 0.599), the HR using the results from the April 7, 2020, data cut-off date was █████ ████ ███ █████ ██ ██████ and remained unchanged at the July 8, 2021, study-close date. KM curves for PFS are shown in Figure 15 for the unweighted pemigatinib group, the weighted pemigatinib group, and the mFOLFOX plus ASC group. Supplemental PFS analyses were provided from the July 8, 2021, data cut-off date comparing the pemigatinib to mFOLFOX plus ASC in patients who had received only 1 prior therapy. The number and ESS for this subgroup was ███████ and resulted in an HR of █████ ████ ███ █████ ██ ██████.
Table 21: Progression-Free Survival, Pemigatinib Versus mFOLFOX Plus ASC
Outcome | Pemigatinib, before weighting N = 98 | Pemigatinib, weighted to mFOLFOX plus ASC ESS = 48.2 | mFOLFOX plus ASC N = 81 |
|---|---|---|---|
PFS events, n | ███ ██████ | ███ ██████ | ███ ██████ |
Median PFS, months (95% CI),a March 22, 2019, data cut-off date | ████ █████ | ████ █████ | ████ ████ |
HR (95% CI),b March 22, 2019, data cut-off date | █████ █████ | 0.436 (0.319 to 0.599) | ███ ██████ |
HR (95% CI),b April 7, 2020, data cut-off date | █████ █████ | █████ ████ | ███ █████ |
HR (95% CI),b July 8, 2021, study close | █████ █████ | █████ █████ | ███ █████ |
ASC = active symptom control; CI = confidence interval; ESS = effective sample size; HR = hazard ratio; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil; NA = not applicable; PFS = progression-free survival.
aKaplan-Meier estimates.
bHRs were determined using Cox proportional hazard models, with bootstrapping to estimate CIs.
Source: Sponsor-submitted MAIC report.55

ASC = active symptom control; mFOLFOX = modified oxaliplatin, L-folinic acid, and fluorouracil; Pemi = pemigatinib; SoC = standard of care.
Note: The results presented for the pemigatinib groups are from the March 22, 2019, data cut-off date.
Source: Sponsor-submitted MAIC report.55
Critical Appraisal of Sponsor-Submitted MAIC
Because of the noncomparative design of the FIGHT-202 study, the use of an MAIC to compare pemigatinib with a relevant comparator was appropriate. The following limitations of the systematic literature search methods were identified: the final stage of study selection for the MAIC did not appear to use preplanned criteria; and the criteria for excluding publications during full-text screening for the reason BTCs (biliary tract cancers) may not have been consistently applied. However, the clinical experts consulted by CDA-AMC for this review agreed that the ABC-06 study was likely the most relevant trial in this setting, given that it represents the only RCT evidence for treating CCA beyond the first-line setting. The selection of an RCT rather than a retrospective study was an appropriate choice because patients who enrol in a clinical trial likely differ, overall, from those included in retrospective studies.
Although the statistical methods used to reweight the pemigatinib group (cohort A of the FIGHT-202 study) and estimate 95% CIs were appropriate, there were potentially important underlying differences between the FIGHT-202 and ABC-06 studies. In particular, all patients in cohort A of the FIGHT-202 study had FGFR2 fusions or rearrangements, whereas patients in the ABC-06 study were not selected based on FGFR2 mutation status, and FGFR2 mutation status was not reported. Given that FGFR2 fusions and rearrangements occur almost exclusively in patients with iCCA and that the prevalence of FGFR2 fusions and rearrangements is less than 20%60 in patients with iCCA, there is likely a large disparity in FGFR2 mutation status between the study populations. Although the FIGHT-202 study only included patients with CCA, the ABC-06 study included patients with BTC, which encompasses gallbladder cancer and ampullary cancer in addition to CCA. Because of this difference and the different distribution of FGFR2 fusions and rearrangements in the different types of CCA, 98.1% of patients in cohort A of the FIGHT-202 study had iCCA, compared with 42% and 47% of patients in the mFOLFOX plus ASC and ASC-alone groups, respectively. Because disease type and FGFR2 status were more restricted in the FIGHT-202 study, these differences could not be addressed with the weighting of patients in the pemigatinib group. The natural history among subtypes of bile duct cancer appears variable, with different prognoses.61
The covariates chosen for adjustment were based on age, sex, ECOG PS, and serum albumin. Although the ITC authors claimed that this selection was based on the availability of this information for both the FIGHT-202 and ABC-06 studies, the following baseline characteristics were also available for both studies and did not appear to be considered: disease stage, the percentage of patients with prior surgery for cancer, and the number of lines of prior systemic therapy for advanced or metastatic cancer. It was not clear if the selection of covariates for adjustment was based on the need to adjust for predetermined prognostic factors or effect modifiers. Baseline characteristics other than the chosen covariates were not reported for the pemigatinib group after weighting. The clinical experts consulted by CDA-AMC for this review were of the opinion that the number of lines of previous therapy was of key importance in terms of prognosis. The clinical experts were not aware of any additional prognostic factors and/or effect modifiers that were not reported in both studies and should have been considered.
Another difference noted was related to the median duration of follow-up, which was 21.7 months (interquartile ranges, 17.2 to 30.8 months) in the ABC-06 trial and 15.44 months (minimum to maximum, 7.0 to 24.7 months) and ████ ██████ █████ ████ ████ █████, respectively, at the March 22, 2019, and April 7, 2020, data cut-off dates in cohort A of the FIGHT-202 trial. Different lengths of follow-up between the trials was not adjusted for in the MAIC and may also contribute to heterogeneity, especially for survival analyses.
Although there are retrospective studies suggesting that the presence of FGFR2 mutations in CCA may be associated with better prognosis,19,20 the clinical experts consulted by CDA-AMC were of the opinion that FGFR2 mutation status is not an important prognostic factor in the indicated patient population. The clinical experts considered the fact that patients in both the FIGHT-202 and ABC-06 trials had progressed on prior systemic therapy to be of greater importance in terms of prognosis. The clinical experts expected patients in the FIGHT-202 study to have more advanced disease than patients in the ABC-06 study because the FIGHT-202 study population was more heavily pretreated overall. It is unclear whether the pemigatinib group was more or less similar to the ABC-06 groups in this respect after weighting, as the weighting process did not take the number of prior lines of systemic therapy into account. If substantial differences remained, these differences could have led to bias against pemigatinib in all of the comparisons.
The limitations with regard to external validity identified for cohort A in the FIGHT-202 study also apply to the MAIC results. In addition, the ESS of the pemigatinib group was reduced by approximately 50% after weighting to the mFOLFOX plus ASC and ASC-alone groups, and it is unclear how representative the postweighting pemigatinib groups are of cohort A in the FIGHT-202 study. It is apparent that patients from cohort A in the FIGHT-202 study with an ECOG PS of 2 were effectively excluded from the pemigatinib group during the weighting process. Other changes noted in the pemigatinib group after weighting were that mean age increased by approximately 10 years and the percentage of male patients increased from 39.8% to 53.0% and 46.0% for comparisons with mFOLFOX plus ASC and ASC alone, respectively. Although both male and female patients would have been adequately represented in the postweighting pemigatinib groups, it is not clear how the age distribution was affected.
After adjustment based on the 4 chosen covariates, there was a substantial loss in precision for the efficacy estimates, as the ESS was reduced by approximately 50%. Adding more covariates for adjustment may have further reduced the available precision.
Comparisons of pemigatinib with other relevant comparators (FOLFIRI, 5-FU alone or in combination with cisplatin or oxaliplatin, and capecitabine alone or in combination with cisplatin or oxaliplatin) were not available. Given that mFOLFOX plus ASC is the only therapy beyond the first-line setting with RCT evidence of an OS benefit, the clinical experts consulted by CDA-AMC noted that mFOLFOX plus ASC would likely have the greatest efficacy out of all the relevant comparators.
Summary
For the unanchored MAIC to produce unbiased treatment effect estimates, all effect modifiers and prognostic variables need to be adjusted in the analysis. Residual confounding remains the major limitation of the MAIC, despite adjustment for age, sex, ECOG PS, and serum albumin in the comparisons of pemigatinib with mFOLFOX plus ASC and ASC alone. Although any bias introduced by the differences between the FIGHT-202 and ABC-06 studies in the number of prior lines of systemic therapy may have been against pemigatinib, the substantial differences in FGFR2 mutation status and tumour site between trials introduce a high degree of uncertainty to the OS and PFS results. Furthermore, MAICs cannot account for unknown cross-trial differences; thus, the MAIC estimates are susceptible to bias from unknown confounding. An evaluation of potential bias from residual confounding was not reported; therefore, the magnitude of this bias in the relative treatment effect estimates is unclear. Overall, uncertainty remains around the magnitude of additional benefit that pemigatinib provides for OS or PFS versus mFOLFOX plus ASC or ASC alone.
Studies Addressing Gaps in the Systematic Review
Parisi et al. (2024)
Description of Studies
Parisi et al.21 conducted a multicentre, observational, retrospective study that assessed the effectiveness and safety of pemigatinib in patients with previously treated, locally advanced or metastatic CCA with FGFR2 fusion or rearrangements. Patients referred to 14 Italian centres and 25 French centres from July 2020 to September 2022 and treated with pemigatinib as a second line or later line of systemic treatment were evaluated (N = 72). These patients were initially included in 2 separate cohort studies but were pooled into a single dataset for analysis.
The primary end point of the study was ORR, according to RECIST 1.1, reported by the treating physician. Secondary end points included PFS, OS, and safety. PFS was defined as the time from the start of treatment to objective disease progression, determined by the investigator, or death, whichever occurred first. OS was defined as the time from the start of treatment to death; patients without an event at the end of follow-up were censored. An exploratory analysis was conducted to compare patients in the study receiving pemigatinib as second-line therapy to those that previously received second-line chemotherapy.
Statistical analyses conducted included descriptive patient and treatment characteristics. KM methods were used to estimate survival outcomes, with the reverse KM method used to calculate median follow-up time. Univariate analysis of OS and PFS, along with estimation of HR and 95% CI, was conducted using Cox proportional hazards regression. An exploratory analysis compared PFS among patients in the cohort who had received pemigatinib in the second line to those who had received chemotherapy in the second line (and pemigatinib in a later line).
Population
The mean age was 56.9 years (SD = 13.6 years), 55 patients (76%) were female and 17 patients (24%) were male, 69 patients (96%) had metastatic disease at treatment initiation, and 3 patients (4%) had locally advanced disease at treatment initiation. The proportion of patients with an ECOG PS of 0, 1, and 2 were 38%, 43%, and 16%, respectively. The proportion of patients who had received 1 prior systemic therapy was 59.7%, 2 prior systemic therapies was 20.8%, and 3 or more prior systemic therapies was 19.4%.21
Results
Median follow-up for the overall cohort was 19.5 months (95% CI, 15.0 to 30.5 months). Of the overall cohort of 72 patients, 2 patients recorded a CR and 31 patients recorded a PR, for an ORR of 45.8%. Median DOR was 7 months (95% CI, 5.8 to 9.3 months). Median OS was 17.1 months (95%CI; 12.7 months to NA, and median PFS was 8.7 months (95% CI, 7.3 to 11.8 months).21
In the overall cohort, 43 patients (59.7%) received pemigatinib as second-line therapy and 21 patients (29.1%) received chemotherapy (either 5-FU or cisplatin plus gemcitabine) as second-line therapy. Patients who received pemigatinib in the second-line setting had a median PFS of 8.6 months (95% CI, 6.6 months to NA), whereas patients who received chemotherapy in the second-line setting (and received pemigatinib in a later line) had a median PFS of 3.4 months (95% CI, 2.1 months to NA), with an HR of 3.88 (95% CI, 1.81 to 8.31; P < 0.001).21
Harms
The proportion of patients who reported at least 1 TEAE was 97.2%, with the most common event being fatigue (69.4%), nail toxicities (61.1%), and hyperphosphatemia (55.6%). The proportion of patients with at least 1 grade 3 or higher TEAE was 22.2%. All of the TEAEs were grade 3, and no patients had a grade 4 or grade 5 TEAE.21
Critical Appraisal of Parisi et al. (2024)
As has been noted in this report, CCA with FGFR2 alterations is a rare and serious disease, so conducting a phase III RCT is infeasible. As such, the pivotal trial was a single-arm, phase II trial, with inherent limitations that have been noted elsewhere in this report. To supplement the total body of evidence, the multicentre, observational, retrospective study conducted by Parisi et al.21 was submitted as RWE of the effectiveness of pemigatinib in patients with CCA and FGFR2 alterations. The following is a critical appraisal of the Parisi et al. 21 study in isolation. The totality of the evidence in the context of a rare disease is covered in the Discussion section.
The Parisi et al. 21 study included patients from French and Italian cancer centres; data for the analysis were pooled from 2 retrospective cohort studies. A rationale and discussion of the appropriateness of the pooling was not provided, and it is possible that the pooling obscured heterogeneity in findings across the studies. The quality and completeness of each of the sources was not reported. The inclusion criteria required receiving at least 1 cycle of pemigatinib (i.e., patients who died or who discontinued treatment during the first cycle because of progression or an AE were excluded); however, given the length of a treatment cycle (21 days), the impact of this selection bias may be small. Although there was no representation of patients from Canada, the clinical experts consulted suggested that patient care would not be meaningfully different, and noted that the reported baseline characteristics were representative of the expected patient population in Canada. As this was an observational study with no comparator arm, it is difficult to assign, with certainty, causation of the effects seen to the study. Because of the retrospective nature of the study design, ORR and progression assessments were conducted by the treating physician, potentially introducing bias; in contrast, assessments are commonly conducted by central review in phase II and phase III trials. The timing of assessments in observational, retrospective studies can also make interpretation of the time-to-progression outcomes a challenge if patients are not being assessed at standardized time points. Analysis of PFS can be sensitive to the censoring rules applied,62 and no censoring rules or reasons were provided in the publication. Complete information on baseline characteristics, treatment patterns, and outcomes was required for inclusion in the study. The impact of is not clear, as there was no information on the number of patients excluded for this reason or on the potential missingness mechanism.
The study attempts to provide a comparative assessment of PFS in patients who received pemigatinib as second-line therapy in the study and patients who received chemotherapy as second-line therapy before inclusion in the Parisi et al. study.21 This analysis should not be viewed as a replacement for randomized comparative evidence. Unadjusted comparisons are presented with no attempt to balance prognostic and confounding variables across groups and no assessment of the extent or the direction of bias due to confounding. There is a risk of selection bias because eligibility for the chemotherapy group required that patients survive long enough to subsequently receive pemigatinib in the third or later line (this particular bias would favour the comparator group). Additionally, patients who followed different treatment trajectories (i.e., did not receive pemigatinib in a later line) were excluded and may be prognostically different from the included patients (e.g., received another treatment in a later line based on patient or clinician preference or did not continue further treatment). Similar criteria were not applied to the pemigatinib group. The sample size has been reduced from the overall cohort; because of this, it is unclear whether there are important differences between the patients who received pemigatinib in the second line and those who received chemotherapy in second line, and it is unclear whether this post hoc subset of patients would be expected to have different outcomes. A predefined protocol for the study was not readily available, which increases the risk of selective outcome reporting based on the direction effect suggested by the results.
Saverno et al. (2025)
Description of Study
The study by Saverno et al.22 was a retrospective, observational, multisite chart review study based in the US, funded by Incyte Biosciences. Physicians in the Cardinal Health Oncology Provider Extended Network were instructed to randomly select up to 10 patients who met eligibility criteria during the index period. To be eligible for inclusion, patients had to be 18 years or older and had to have been prescribed pemigatinib for unresectable, locally advanced or metastatic CCA between February 3, 2021, and February 22, 2023. Patients needed to have at least 4 months of follow-up, but patients who died during this 4-month period were included. Physicians abstracted details related to demographics, clinical characteristics, biomarker testing patterns, treatment patterns, and clinical outcomes. The primary objective of the study was to describe patient demographics and clinical characteristics, FGFR2 testing patterns, treatment patterns, and pemigatinib use patterns. An exploratory objective was to describe the clinical effectiveness of pemigatinib.
Outcomes of interest included ORR, PFS, and OS. The ORR was defined as the proportion of patients with a CR or PR; no further criteria for determining response were provided. PFS was defined as the time from treatment initiation to physician-reported disease progression (which included treatment discontinuation if the reason for that was listed as progression) or death, whichever occurred first. The definition of progression was not reported. Patients without progression were censored at the start of new anticancer therapy if the reason for discontinuation was not disease progression, or at the date of last encounter if the patient was still on the study drug.
The overall response rate was summarized using descriptive statistics. The KM method was used for time-to-event outcomes, and point estimates for PFS and OS were reported at the 3-month and 6-month time points.
Population
In total, 120 patients were included in the study, identified by a total of 18 physicians. The median age was 65 years (range, 37.0 to 83.0), and 50.8% of patients were female. Most patients had an FGFR2 fusion rearrangement (91.9%). The majority of patients (78.3%) had an ECOG PS of 0 to 1 at baseline, 90.0% of patients had metastatic disease when pemigatinib was initially prescribed, and 70% had iCCA at time of initial diagnosis.22
Results
The median duration of treatment in the first-line setting was 4.9 months (95% CI, 4.4 to 5.7 months); of these patients, 94.7% received chemotherapy as their first-line treatment. Most patients received pemigatinib in the second-line setting (94.2%), and 5.8% received pemigatinib in the third-line setting. The median duration of treatment with pemigatinib was 7.4 months (95% CI, 6.2 to 8.8 months). At the time of data collection, 60 patients (50.0%) had discontinued treatment, with disease progression (confirmed with scan) being the most commonly reported reason. Pemigatinib dose reductions or dose-frequency reductions were reported by 12.5% of patients, and dose interruptions were reported by 2.5% of patients.22
ORR in the 116 patients with disease response data available was 59.2% (95% CI, 50.0% to 68.4%). The proportion of patients reporting a best response of CR was 5.0%, a best response of PR was 54.2%, and a best response of stable disease was 27.5%, for a DCR of 86.7%. Median PFS was 7.4 months (95% CI, 6.4 to 8.6 months). The PFS probability was 95.8% (95% CI, 90.3% to 98.2%) at 3 months and 71.5% (95% CI, 61.% to 79.4%) at 6 months. %Median OS was not reported; the OS probability was 95.8% (95% CI, 90.3% to 98.2%) at 3 months and 88.4% (95% CI, 80.3% to 93.3%) at 6 months.22
Harms
Harms were not reported by Saverno et al.22
Critical Appraisal
As has been noted in this report, CCA with FGFR2 alterations is a rare and serious disease, so conducting a phase III RCT is infeasible. As such, the pivotal trial was a single-arm, phase II trial, with inherent limitations that have been noted elsewhere in this report. To supplement the total body of evidence, a retrospective, observational, multisite study by Saverno et al.22 was submitted as RWE of the effectiveness of pemigatinib in patients with CCA and FGFR2 alterations. The following is a critical appraisal of the Saverno et al. study22 in isolation. The totality of evidence in the context of a rare disease is covered in the Discussion section.
The clinical experts consulted considered the reported baseline characteristics to be representative of the expected patient population in Canada. The quality and completeness of the real-world data source was not reported. The length of follow-up was short, so the study was not able to provide estimates of long-term effect. As this was an observational study with no comparison group, it is difficult to assign, with certainty, causation of the effects seen to the study drug. Because of the retrospective nature of the study design, ORR and progression assessments were conducted by the treating physician, potentially introducing bias; in contrast, assessments are commonly conducted by central review in phase II and phase III trials. The specific definition of progression was not provided, and it is not clear how this was assessed. The timing of assessments in observational, retrospective studies can also make interpretation of the time-to-progression outcomes a challenge if patients are not being assessed at standardized time points. The estimation of PFS is sensitive to the censoring assumptions used in the analysis.62 In this study, patients were censored on initiation of a subsequent treatment (if the reason was not progression), although it is unclear whether these patients had the same risk and timing of the outcome as those who did not initiate subsequent treatment. Alternative censoring rules could have provided different results, but these were not reported.
Last, the patient selection methodology in the Saverno et al. study22 potentially introduced selection bias, as the physicians were instructed to select, at random, 10 patients who fit the inclusion criteria during the index period. There was no methodology reported that indicated that selecting physicians were blinded to the clinical outcomes of patients when making selections. There is therefore a risk that patients with a better prognosis were preferentially included. Additionally, patients required at least 4 months of follow-up to be included (unless they died). It is not clear how many patients were excluded because of a lack of adequate follow-up, nor whether these patients might have differed in an important way in their prognosis. A predefined protocol for the study was not readily available, which increases the risk of selective outcome reporting based on the direction effect suggested by the results.
Ding et al. (2024)
Description of Studies
The Ding et al. study23 was a retrospective, multisite physician survey to assess demographics, clinical characteristics, FGFR2 testing, and real-world treatment patterns, and outcomes of patients with unresectable, locally advanced or metastatic CCA treated with pemigatinib. Of note, results from the Ding et al. study23 presented in this report are based on a poster presentation.
As part of the Incyte Solutions Patient Support Program, between September 2021 and January 2023, prescribing physicians in Canada were asked to identify patients who met the study criteria and to provide information on demographics, disease characteristics, treatment patterns, and outcome data. The study eligibility criteria were not reported.
Outcomes of interest included ORR, PFS, and OS. The definition of these outcomes and the method with which they were collected were not reported. Estimates of ORR were provided using descriptive statistics. Estimates of time-to-event outcomes were conducted using the KM method.
Of the ██ patients who received pemigatinib treatment in the Patient Support Program, survey responses were received for ██ █████ of them.
Population
The median (range) age at diagnosis was ██ █████ ███ ██ ████ ██ ████████ ███████ ████ ██████. The CCA location at initial diagnosis was unknown in ████████ ███████, and intrahepatic in ██ ████████ ██████████████ ███████ had an ECOG PS of 0 and ██ ████████ ███████ had an ECOG PS of 1.23
Results
The median duration of follow-up for patients on pemigatinib was ██ ███████ █ ██ ███ months. ORR in the ██ patients with survey responses was █████. No patients included in the survey results achieved a CR. The proportion of patients reporting a best response of PR was █████ and the proportion of patients achieving a best response of stable disease was █████, for a DCR of ███. Median PFS was ████ ██████ ████ ███ ███ ██ ███. The PFS probability was ███ ████ ███ ██ ██ █████ at 6 months and ███ ████ ███ ██ ██ ████ at 12 months.
Harms
Harms were not reported by Ding et al.23
Critical Appraisal
As has been noted in this report, CCA with FGFR2 alterations is a rare and serious disease, so conducting a phase III RCT is infeasible. As such, the pivotal trial was a single-arm, phase II trial, with inherent limitations that have been noted elsewhere in this report. To supplement the total body of evidence, a retrospective, observational, multisite study conducted by Ding et al.23 was submitted as RWE of the effectiveness of pemigatinib in patients with CCA and FGFR2 alterations. The following is a critical appraisal of the Ding et al. study23 in isolation. The totality of evidence in the context of a rare disease is covered in the Discussion section.
The study conducted by Ding et al.23 was a retrospective, multisite physician survey. It attempted to provide patient demographics and patient outcomes in all patients who received pemigatinib as part of the Incyte Solutions Patient Support Program in Canada. Of note, results from the Ding et al. study23 presented in this report are based on a poster presentation. There are several aspects of this study that make interpretation a challenge. Data quality and completeness were not reported. As this was an observational study with no comparison group, it is difficult to assign, with certainty, causation of the effects seen to the study drug. The small sample size of ██ patients is a limitation. With such a small number of patients, there is increased susceptibility to random variation, the results are likely to be unstable, and generalizability to a larger population may be reduced. Next is the potential bias related to the selection of the ██ patients compared to the ██ patients who made up the full Patient Support Program. It is unknown how representative the ██ selected patients are of the larger group, as there was no response from the associated physicians, although the sponsor did provide a supplemental analysis that showed that the total ██ patient population had a slightly longer mean duration of treatment (████ ██████ ██ █████,, which suggests that the reduced patient population was not biased toward longer treatment duration. Additionally, the inclusion and exclusion criteria were not described, so it is not clear whether application of these criteria would have introduced selection bias. Last, as this was a retrospective study, ORR and progression assessments were done by the treating physician; in contrast, central review is more common in controlled studies. Additionally, assessments would have been done at nonstandardized time points, so the reliability of time-to-progression end points is somewhat less reliable. The definition of each end point was not provided. Estimation of PFS, in particular, is sensitive to the censoring assumptions used in the analysis,62 but these were not reported. The proportion of patients censored and reasons for censoring were also unreported. A predefined protocol for the study was not readily available, which increases the risk of selective outcome reporting based on the direction effect suggested by the results.
Bibeau et al. (2022)
Description of Studies
Post hoc analyses were conducted to compare patients from the FIGHT-202 study who received pemigatinib as a second-line therapy with patients from the FIGHT-202 study who received second-line systemic therapy before enrolment in the FIGHT-202 study.24 In total, 65 patients received pemigatinib as second-line therapy in the FIGHT-202 study and 41 patients received second-line systemic therapy before enrolment in the FIGHT-202 study, 39 of whom were evaluable for PFS. Of the 41 patients who received second-line systemic therapy, 38 received chemotherapy (gemcitabine plus cisplatin, 5-FU plus leucovorin calcium plus oxaliplatin, or 5-FU plus oxaliplatin) and 3 received anti-PD-1 immunotherapy.
Population
In the subset of 39 patients who received second-line systemic therapy before enrolment in the FIGHT-202 study, median age was 51.5 years (range, 27 to 76 years), 26 patients (66.7%) were female, the proportion of patients with an ECOG PS of 0, 1, and 2 was 43.6%, 53.9%, and 2.6%, respectively. In addition, 53.9% of patients had received prior cancer surgery and 33.3% had received prior radiation. The CCA location was intrahepatic in 38 patients (97.4%) and other in 1 patient (2.6%).24
In the subset of 65 patients who received pemigatinib as second-line therapy during the FIGHT-202 study, median age was 54.7 years (range, 25 to 75 years), 39 patients (60.0%) were female, the proportion of patients with an ECOG PS of 0, 1, and 2 was 41.5%, 56.9%, and 1.5%, respectively. In addition, 24.6% of patients had received prior cancer surgery and 16.9% had received prior radiation. The CCA location was intrahepatic in 64 patients (98.5%) and extrahepatic in 1 patient (1.5%).24
Results
Median PFS in patients receiving second-line pemigatinib therapy was 7.0 months (95% CI, 4.9 to 11.1 months), whereas median PFS in patients who received second-line therapy before enrolment in the FIGHT-202 study was 4.2 months (95% CI, 3.0 to 5.3 months). Median PFS for the 102 patients with evaluable results for first-line systemic therapy was 5.5 months (95% CI, 4.0 to 8.0 months).24
Critical Appraisal
The comparative results for patients who received second-line therapy before enrolment in the FIGHT-202 study are uncertain. Some of the listed aggregate baseline characteristics appear to be fairly similar, but there were notable differences in region, prior cancer surgery, and prior radiation treatment. It is unclear if there are other important prognostic factors that differ between these 2 groups. Unadjusted comparisons are presented with no attempt to balance prognostic and confounding variables across groups, and no assessment of the extent or direction of bias due to confounding is presented. There is a risk of selection bias because eligibility for the comparator group required that patients survive long enough to be enrolled in the FIGHT-202 study (this particular bias favours the comparator group). Additionally, patients who followed different treatment trajectories (i.e., did not receive pemigatinib in a later line and/or did not enter the FIGHT-202 study) were excluded and may have been prognostically different from the included patients (e.g., received another treatment in a later line based on patient or clinician preference, did not wish to continue further treatment, were not eligible for or did not wish to enter the trial). Similar criteria were not applied to the pemigatinib group. Data for the comparison group were collected retrospectively from patient records (real-world data source). The quality and completeness of the real-world data source were not reported. Given the differences in the data sources across cohorts (i.e., retrospective chart review versus prospective trial), there are likely to be differences in the timing (i.e., fixed timing versus at the clinician’s discretion) and method of outcome measurement (i.e., independent centralized radiological review committee versus investigator) and in the background care, including concomitant treatments. These differences cannot be adjusted within the comparison, which increases the uncertainty of the findings. No formal statistical analyses were undertaken, and between-group differences and CIs were not provided to evaluate the potential magnitude and precision of differences across groups. The sample size in the comparison groups was small and the results might not be replicated in a larger study sample. The post hoc nature of the analysis increases the risk of selective outcome reporting based on the direction effect suggested by the results.
Discussion
Summary of Available Evidence
The CDA-AMC clinical review included 1 phase II trial of pemigatinib that evaluated the efficacy and safety of pemigatinib in patients with advanced and/or metastatic or surgically unresectable CCA with FGFR2 alterations, other FGF-FGFR alterations, or no FGF-FGFR alterations who failed previous therapy. FIGHT-202 was a multicentre, open-label, single-arm, phase II trial that assigned 146 patients to 1 of 3 cohorts, depending on the patient’s FGF-FGFR status (cohort A: FGFR2 fusions or rearrangements; cohort B: FGF-FGFR alterations other than FGFR2 fusions or rearrangements; or cohort C: negative for FGF-FGFR alterations). This CDA-AMC review focuses on cohort A, as cohort B and cohort C were not part of the requested reimbursement criteria to CDA-AMC and were not submitted for approval to Health Canada. All enrolled participates received oral pemigatinib (13.5 mg orally once daily on a 2-weeks-on and 1-week-off schedule for each 21-day cycle). The primary outcome was ORR in cohort A, and secondary outcomes included ORR in cohort B, cohort A plus cohort B, and cohort C, and PFS, DOR, DCR, OS, and safety assessed in all 3 cohorts. Exploratory end points included HRQoL and symptom severity.
Adults diagnosed with advanced and/or metastatic or surgically unresectable CCA with FGFR2-positive disease who had documented disease progression after at least 1 line of systemic therapy were enrolled in cohort A of the FIGHT-202 trial. The majority of patients had iCCA, an ECOG PS of 0 or 1, had received 1 or 2 previous lines of systemic therapy for advanced or metastatic disease, and were aged 56 years (range, 26 to 77 years).
Four studies addressed gaps in the systematic review: Parisi et al.21 was a multicentre, observational, retrospective study of 72 patients; Saverno et al.22 was a retrospective, observational, multisite chart review study of 120 patients; Ding et al.23 was a retrospective, multisite physician survey reporting on 18 patients; and Bibeau et al. (2022)24 was a post hoc chart review that analyzed previous lines of therapy outcomes in patients included in the FIGHT-202 study.
In addition, 1 sponsor-submitted ITC was summarized and appraised for this review.
Interpretation of Results
Efficacy
In 2022, pemigatinib was reviewed by CADTH for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement, and received a do not reimburse recommendation. pERC deliberated on the evidence available from the FIGHT-202 trial, as well as an ITC of pemigatinib compared to FOLFOX. Although pERC acknowledged the rarity of FGFR2-positive CCA, ultimately, uncertainty related to the noncomparative evidence provided by the FIGHT-202 trial led to the recommendation against reimbursing pemigatinib. As part of this resubmission, the sponsor has submitted 4 additional studies that provide RWE in support of the FIGHT-202 trial data for pemigatinib.
The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 15%) in cohort A. As of the July 8, 2021, study-close date, after a median follow-up time of 45.4 months, the proportion of patients with an objective response (the primary end point in cohort A) was 37.0% (95% CI, 27.94% to 46.86%). A total of 3 patients had achieved a CR, 37 patients had achieved a PR, and 49 patients had achieved stable disease as the best response; ███████ ███████ ████ ███ █████. Median DOR was 9.13 months (95% CI, 6.01 to 14.49 months). The FIGHT-202 trial included no formal statistical significance or hypotheses testing, and point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. A greater than 95% probability of having a 95% CI for ORR in cohort A with a lower limit larger than 15% was the basis for the sample size determination and was regarded as the threshold for a positive study outcome. Results for the subgroup of interest, as prespecified in the protocol for this CDA-AMC systemic literature review, suggested that the effect on ORR was similar across subgroups of patients with an ECOG PS of 0 and of 1 or 2. However, given that the trial was not designed to detect differences in treatment effects across subgroups, no conclusions can be drawn on the basis of subgroup results. OS and PFS were assessed as secondary outcomes in the FIGHT-202 trial; median OS and PFS, respectively, were 17.48 months (95% CI, 14.36 to 22.93 months) and 7.03 months (95% CI, 6.08 to 10.48 months) at the July 8, 2021, study-close date. Interpretation of time-to-event end points such as OS or PFS is limited in single-arm studies; because all patients in cohort A received the same treatment, the extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear.53 The primary objective of phase II (randomized or nonrandomized) trials is to document safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict harm and/or the effectiveness of treatments. The clinical experts consulted by CDA-AMC noted that, despite the high unmet need, it would not be feasible to conduct an RCT in this setting comparing a targeted therapy such as pemigatinib with currently available therapies in the second-line setting in clinical practice in Canada. According to the clinical experts, the development of phase III RCTs is hindered by the overall low patient numbers with the current indication, and equipoise between pemigatinib and other chemotherapy regimens does not exist.
The RWE studies submitted to address gaps in the evidence were all single arm and noncomparative; as such, they have many of the same difficulties with interpretation as the pivotal trial, with the additional limitations of retrospective RWE. However, results were supportive of the findings from the pivotal trial, with ORR and DCR of 45.8% and 84.7%, respectively, in the Parisi et al. study,21 59.2% and 86.7%, respectively, in the Saverno et al. study,22 and █████ and ███ respectively, in the Ding et al. study.23 Despite the limitations of the studies, the clinical experts consulted by CDA-AMC indicated that these results were consistent with those from the FIGHT-202, which increased their confidence in the outcomes produced in the pivotal trial. Particularly of note, according to the clinical experts, was the OS of 17.1 months (95% CI, 12.7 months to NA) in the Parisi et al. study.21 In the experience of the clinical experts, this level of OS is notable for this patient population in the context of an RWE study, and provides a signal consistent with that seen when naively comparing the median OS of █████ months reported in the FIGHT-202 study and the median OS of 5.18 months reported in the mFOLFOX plus ASC arm in the ABC-06 trial.14 Overall, although uncertainty related to the pivotal trial has not changed since the 2022 CADTH recommendation, the totality of the evidence and the consistency of that evidence addresses some of the uncertainty that was noted in the previous recommendation.
According to the clinical experts consulted by CDA-AMC and the registered clinician groups that provided input for this submission, the responses achieved with pemigatinib were clinically relevant, important to patients in this setting, and clearly higher than what would likely be observed with currently used therapies in this setting. The clinically experts consulted by CDA-AMC noted that durable responses in this patient population are important because of an accompanying delay in the worsening of symptoms and a slower decline in ECOG PS. The clinical experts emphasized the clinical relevance and importance of maintaining stable disease to the prevention of an otherwise fast decline in patients in this, oftentimes, last line of treatment. This view was echoed by the input provided by the patient advocacy group, which highlighted tumour response, maintenance of response, delay in disease progression, and QoL as important treatment goals for patients.
In the absence of a direct comparison of pemigatinib and relevant treatment options, the sponsor submitted an ITC55 in the form of an unanchored MAIC that compared the efficacy of pemigatinib (cohort A of the FIGHT-202 trial) with each of the 2 treatment groups in the ABC-06 study. For PFS and OS, the results of the ITC favoured pemigatinib over mFOLFOX plus ASC and over ASC alone. The CDA-AMC critical assessment identified several limitations of the sponsor’s submitted MAIC, including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables in the MAIC. The clinical experts agreed with the CDA-AMC clinical review team that, given the absence of robust comparative data on PFS and OS, the ability to interpret the relative treatment effects of pemigatinib, of mFOLFOX plus ASC, and of ASC alone was limited, and no firm conclusions could be drawn about how pemigatinib compared with other relevant treatment options. The clinical experts consulted by CDA-AMC stated, however, that based on the FIGHT-202 results and on the poor results with existing treatment options in clinical practice, pemigatinib appears to offer improved clinical benefits compared with current therapies, with better tolerability. The clinical experts consulted by CDA-AMC noted that there is currently insufficient evidence to determine whether patients with FGFR2 fusion-positive disease represent a distinct prognostic subgroup. The clinical experts agreed that progression on prior systematic therapy is a key prognostic factor in these patients, and noted that patients would be unlikely to have derived any substantial benefit from their underlying disease biology at the time they enrolled in the FIGHT-202 trial. Although a comparison of ORR was not included in the ITC provided by the sponsor, the 37% ORR from the FIGHT-202 study, as the clinical experts noted, is a stark increase over the 5% ORR reported in the mFOLFOX plus ASC arm of the ABC-06 trial.14 The clinical experts consulted by CDA-AMC suggested that based on a current understanding of the targeting biomarker-selected patient populations, it would be difficult to argue that the difference in ORR is not related to benefits from the targeting of FGFR2.
Input received by the patient advocacy group, the registered clinicians, as well as the clinical experts consulted by CDA-AMC, highlighted HRQoL as an important outcome and treatment goal for patients. Overall observed scores from baseline to cycle 42 were reported to be variable, with no consistent trend for EORTC QLQ-C30 and EORTC QLQ-BIL21 scores. However, given the noncomparative, open-label design of the trial, the lack of a prespecified analysis of the patient-reported outcomes data, the substantial decline in patients available for assessment over time, and the lack of a definition for what constituted a clinically meaningful change from baseline in the target population, the results were inconclusive. The clinical experts consulted by CDA-AMC noted that HRQoL in this setting is low and unstable. They stated that given the observed responses in the FIGHT-202 trial, pemigatinib would likely improve or at least maintain patients’ HRQoL.
Although patients recruited to the FIGHT-202 trial were considered to be representative of patients in clinical practice in Canada, the clinical experts consulted by CDA-AMC noted that it would be reasonable to generalize the results from cohort A of the FIGHT-202 trial to patients who are intolerant to first-line therapy, which was a group of patients excluded from the trial. As well, given the acceptable safety profile of pemigatinib, the clinical experts felt that it would be reasonable to leave it to the discretion of the treating physician to apply some flexibility in the terms of using pemigatinib in patients with slightly lower lab parameters than those outlined in the trial. The clinical experts said that they anticipate seeing the benefit of treatment with pemigatinib, regardless of the number of previous lines of systemic therapy received, as long as patients have the FGFR2 alteration. However, the clinical experts agreed that patients should not have previously been treated with an FGFR2-targeted therapy. Furthermore, the clinical experts noted that patients with iCCA and eCCA are managed in a similar way in clinical practice, and that the results observed in cohort A are generalizable to patients with FGFR2-positive eCCA, based on the fact that FGFR2 is the target of the mechanism of action of pemigatinib and there is no biological rationale to assume that pemigatinib’s safety profile would be different in patients with eCCA.
Harms
The single-arm, nonrandomized design of the FIGHT-202 trial, as well as the RWE studies, adds uncertainty to the interpretation of the safety events attributable to pemigatinib, because all patients in cohort A received the same treatment. All patients in cohort A experienced at least 1 TEAE. The most commonly reported TEAEs included alopecia, hyperphosphatemia, diarrhea, and dysgeusia. The most commonly reported TEAEs of grade 3 or higher and serious TEAEs, respectively, were ████████████████ ███ ███████. The clinical experts consulted by CDA-AMC noted that most TEAEs associated with pemigatinib could be managed with dose modifications and that treatment discontinuation due to TEAEs was relatively rare. From the review of notable harms, it appeared that toxicities from pemigatinib were mostly seen as █████████████████ ███ ████ ██████████. There were few deaths, overall, and no TEAE leading to death was considered treatment related. Overall, the clinical experts consulted by CDA-AMC agreed with the registered clinicians who provided input to this submission that the TEAEs observed with pemigatinib were, overall, acceptable and could be adequately managed in clinical practice. This was reflective of patients’ experience with pemigatinib reported in the patient input received, which stated that, overall, patients had little challenge dealing with the side effects of pemigatinib. Furthermore, it was emphasized by the clinical experts consulted by CDA-AMC that the toxicity of pemigatinib appears to be favourable compared with currently available chemotherapy options, based on the pivotal trial and the supportive RWE studies. Examples of side effects from chemotherapy that could be avoided with pemigatinib are neuropathy and neutropenia, according to the clinical experts.
Conclusions
One phase II, singe-arm, open-label trial (FIGHT-202) provided evidence on the efficacy and safety of pemigatinib in patients with advanced, metastatic, or surgically unresectable CCA with FGFR2 alterations (cohort A) whose previous therapy failed. The FIGHT-202 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 15%) in cohort A. The clinical experts consulted by CDA-AMC felt that the achieved ORR of 37% (July 8, 2021, data cut-off date) was clinically meaningful for the target population, and durable (median, 9.13 months; 95% CI, 6.01 to 14.49 months). In the opinion of the clinical experts, the observed responses appeared to be higher than what is seen with currently used therapies in the second line in this setting (for example, 5% from the mFOLFOX plus ASC group in the ABC-06 trial). There was uncertainty around the magnitude of the clinical benefit, given the limitations of the evidence from the noncomparative phase II clinical trial and the supportive RWE studies; however, the results were consistent and the unmet need is high. The clinical experts consulted by CDA-AMC noted that, despite the high unmet need, it would not be feasible to conduct an RCT comparing a targeted therapy such as pemigatinib with currently available therapies in the second-line setting in clinical practice in Canada. Although OS and PFS, both secondary efficacy outcomes, appeared to be supportive of the observed ORR achievements, the nonrandomized design of the FIGHT-202 trial made interpreting PFS and OS events attributable to pemigatinib a challenge. The 3 RWE studies submitted to address gaps in the evidence showed outcomes similar to those of the pivotal trial, with ORR ranging from 45.8% to 59.2%. Despite similar (and some additional) uncertainties to those of the pivotal trial, consistent positive results in both the pivotal trial and the RWE studies increase the confidence that the outcomes of the pivotal trial may be replicable in real-world practice. In the absence of a direct comparison of pemigatinib with relevant treatment options, the sponsor submitted an ITC. However, the CDA-AMC critical assessment identified limitations of the sponsor’s submitted unanchored MAIC (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which contributed to the uncertainty around the magnitude of benefit of pemigatinib over other treatments. Similarly, 1 RWE study presented exploratory comparative information, but the conclusions were limited by methodological limitations. Results for the HRQoL and symptom severity exploratory outcomes remained inconclusive because of a number of important limitations. The toxicity profile of pemigatinib was considered manageable by the clinical experts consulted by CDA-AMC, and appeared to be favourable compared with currently available chemotherapy options based on results from both the pivotal trial and the RWE studies.
References
1.Zhou X, Wang J, Tang M, et al. Hepatocellular carcinoma with hilar bile duct tumor thrombus versus hilar cholangiocarcinoma on enhanced computed tomography: a diagnostic challenge. BMC Cancer. 2020;20(1):54. doi:10.1186/s12885-020-6539-7 PubMed
2.Valle JW, Borbath I, Khan SA, et al. Biliary cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27:v28-v37. doi:10.1093/annonc/mdw324 PubMed
3.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN Guidelines): hepatobiliary cancers. Version 3.2021. 2021 June 15. Accessed August 10, 2021. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1438
4.Meza-Junco J, Montano-Loza AJ, Ma M, et al. Cholangiocarcinoma: has there been any progress? Can J Gastroenterol. 2010;24(1):52-57. doi:10.1155/2010/704759 PubMed
5.Center for Drug Evaluation Research. Multidiscipline review(s): Pemazyre (pemigatinib) administration. Company: Incyte Corporation. Application No.:213736Orig1s000. Approval date: 04/17/2020 (FDA approval package). U.S. Food and Drug Administration (FDA); 2020. Accessed July 8, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213736Orig1s000MultidisciplineR.pdf
6.American Cancer Society. Survival rates for bile duct cancer. 2021. Accessed July 25, 2021. https://www.cancer.org/cancer/bile-duct-cancer/detection-diagnosis-staging/survival-by-stage.html
7.National Institute for Health and Care Excellence. Single technology appraisal: pemigatinib for treating relapsed or refractory advanced cholangiocarcinoma with FGFR2 alterations [NICE ID3740]. Committee papers. 2021. Accessed October 19, 2021. https://www.nice.org.uk/guidance/ta722/documents/committee-papers-3
8.Amerian Cancer Society. Signs and symptoms of bile duct cancer. 2021. Accessed July 25, 2021. https://www.cancer.org/cancer/bile-duct-cancer/detection-diagnosis-staging/signs-symptoms.html
9.Committee for Medicinal Products for Human Use. Assessment report: pemazyre, international non-proprietary name: pemigatinib. European Medicines Agency; 2021. Accessed June 28, 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/pemazyre
10.Lamarca A, Barriuso J, McNamara MG, et al. Molecular targeted therapies: ready for “prime time” in biliary tract cancer. J Hepatol. 2020;73(1):170-185. doi:10.1016/j.jhep.2020.03.007 PubMed
11.Saborowski A, Lehmann U, Vogel A. FGFR inhibitors in cholangiocarcinoma: what's now and what's next? Ther Adv Med Oncol. 2020;12:1758835920953293. doi:10.1177/1758835920953293 PubMed
12.Kelley RK, Ueno M, Yoo C, et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;401(10391):1853-1865. doi:10.1016/S0140-6736(23)00727-4 PubMed
13.Oh DY, Ruth He A, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022;1(8):EVIDoa2200015. doi:10.1056/EVIDoa2200015
14.Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22(5):690-701. doi:10.1016/s1470-2045(21)00027-9 PubMed
15.National Library of Medicine. PubChem compound summary for CID 86705695, pemigatinib. 2021. Accessed September 8, 2021. https://pubchem.ncbi.nlm.nih.gov/compound/Pemigatinib
16.Incyte Corporation. Pemazyre (pemigatinib tablets): 4.5 mg, 9 mg, and 13.5 mg, oral [product monograph]. September 8, 2021.
17.Incyte Corporation. Clinical Study Report: INCB 54828-202. A phase 2, open-label, single-arm, multicenter study to evaluate the efficacy and safety of INCB054828 in subjects with advanced/metastatic or surgically unresectable cholangiocarcinoma including FGFR2 translocations who failed previous therapy (FIGHT-202) [internal sponsor's report]. August 12, 2019.
18.Incyte Biosciences Canada. Incyte response to September 1, 2021 DRR request for additional information regarding pemigatinib DRR review [internal additional sponsor's information]. September 10, 2021.
19.De Luca A, Esposito Abate R, Rachiglio AM, et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21(18)doi:10.3390/ijms21186856 PubMed
20.Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precis Oncol. 2018;(2):1-12. doi:10.1200/PO.17.00080 PubMed
21.Parisi A, Delaunay B, Pinterpe G, et al. Pemigatinib for patients with previously treated, locally advanced or metastatic cholangiocarcinoma harboring FGFR2 fusions or rearrangements: a joint analysis of the French PEMI-BIL and Italian PEMI-REAL cohort studies. Eur J Cancer. 2024;200:113587. doi:10.1016/j.ejca.2024.113587 PubMed
22.Saverno K, Zimmerman Savill KM, Brown-Bickerstaff C, et al. Real-world use of pemigatinib for the treatment of cholangiocarcinoma in the US. Oncologist. 2025;30(1):oyae204. doi:10.1093/oncolo/oyae204 PubMed
23.Ding P, Tam V, Ramjeesingh R, et al. Pemigatinib in the real-world management of cholangiocarcinoma (CCA) through a Canadian patient support program (PSP) [poster] Cholangiocarcinoma Foundation 2024 Annual Conference. Accessed January 10, 2025. https://www.incytemi.com/document/Poster/CCF%202024%20-%20Pemigatinib%20in%20RW%20Management%20of%20CCA%20Through%20Canadian%20Patient%20Support%20Program%20(poster).pdf
24.Bibeau K, Féliz L, Lihou CF, et al. Progression-free survival in patients with cholangiocarcinoma with or without FGF/FGFR alterations: a FIGHT-202 post hoc analysis of prior systemic therapy response. JCO Precis Oncol. 2022;6:e2100414. doi:10.1200/po.21.00414 PubMed
25.NIH National Cancer Institute. Bile duct cancer (cholangiocarcinoma) treatment (PDQ®) – patient version. 2021. Accessed July 10, 2021. https://www.cancer.gov/types/liver/patient/bile-duct-treatment-pdq#_1
26.Incyte Biosciences Canada Corporation. Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets [internal sponsor's package]. June 23, 2021.
27.Bibeau K, Féliz L, Barrett S, et al. Progression-free survival in patients with cholangiocarcinoma with FGFR2 fusions or rearrangements: an exploration of response to systemic therapy. J Clin Oncol. 2020;38(4_suppl):588-588. doi:10.1200/JCO.2020.38.4_suppl.588
28.Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9(12):e115383. doi:10.1371/journal.pone.0115383 PubMed
29.Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45(8):1630-8. doi:10.1016/j.humpath.2014.03.014 PubMed
30.U.S. Food & Drug Administration. FDA grants accelerated approval to pemigatinib for cholangiocarcinoma with an FGFR2 rearrangement or fusion. 2020. Accessed September 3, 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion
31.European Medicines Agency. EU/3/18/2066: orphan designation for the treatment of biliary tract cancer - pemigatinib. 2018. Accessed August 8, 2021. https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3182066
32.European Medicines Agency. Product information: Pemazyre (pemigatinib). Annex I - summary of product characteristics. 2021. Accessed June 28, 2021. https://www.ema.europa.eu/en/documents/product-information/pemazyre-epar-product-information_en.pdf
33.U.S. Food & Drug Administration. FDA grants accelerated approval to infigratinib for metastatic cholangiocarcinoma. May 28, 2021. Accessed September 3, 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-infigratinib-metastatic-cholangiocarcinoma
34.Incyte Corporation. NCT03656536: A study to evaluate the efficacy and safety of pemigatinib versus chemotherapy in unresectable or metastatic cholangiocarcinoma (FIGHT-302). ClinicalTrials.gov; 2021. Accessed August 20, 2021. https://www.clinicaltrials.gov/ct2/show/NCT03656536
35.QED Therapeutics Inc. NCT03773302: Phase 3 study of BGJ398 (oral infigratinib) in first line cholangiocarcinoma with FGFR2 gene fusions/translocations. ClinicalTrials.gov; 2021. Accessed September 3, 2021. https://clinicaltrials.gov/ct2/show/NCT03773302
36.Taiho Oncology Inc. NCT04093362: Futibatinib versus gemcitabine-cisplatin chemotherapy as first-line treatment of patients with advanced cholangiocarcinoma harboring FGFR2 gene rearrangements (FOENIX-CCA3). ClinicalTrials.gov; 2021. Accessed September 3, 2021. https://www.clinicaltrials.gov/ct2/show/NCT04093362
37.Mathers B, Abadi S, Davies JM, et al. Use, response and outcomes of second-line chemotherapy in patients with advanced biliary tract cancers. J Oncol Pharm Pract. 2023;29(6):1381-1386. doi:10.1177/10781552221122058 PubMed
38.Seung SJ, Saherawala H, Syed I, et al. Real-world treatment patterns and survival outcomes for treated biliary tract cancer patients using administrative databases in Ontario. J Gastrointest Oncol. 2023;14(4):1806-1816. doi:10.21037/jgo-23-155 PubMed
39.Lamarca A, Hubner RA, David Ryder W, et al. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol. 2014;25(12):2328-2338. doi:10.1093/annonc/mdu162 PubMed
40.Agios Pharmaceuticals Inc. NCT02989857: Study of AG-120 in previously treated advanced cholangiocarcinoma with IDH1 mutations (ClarIDHy) (ClarIDHy). ClinicalTrials.gov; 2021. Accessed September 3, 2021. https://clinicaltrials.gov/ct2/show/NCT02989857
41.Onclive.com. FDA grants priority review to ivosidenib in IDH1-mutant cholangiocarcinoma. May 5, 2021. Accessed September 8, 2021. https://www.onclive.com/view/fda-grants-priority-review-to-ivosidenib-in-idh1-mutant-cholangiocarcinoma
42.Businesswire. Incyte announces approval of Pemazyre® (pemigatinib) in Japan for the treatment of patients with unresectable biliary tract cancer (BTC) with a Fibroblast Growth Factor Receptor 2 (FGFR2) fusion gene, worsening after cancer chemotherapy. March 23, 2021. Accessed October 12, 2021. https://www.businesswire.com/news/home/20210323005530/en/Incyte-Announces-Approval-of-Pemazyre%C2%AE-pemigatinib-in-Japan-for-the-Treatment-of-Patients-with-Unresectable-Biliary-Tract-Cancer-BTC-with-a-Fibroblast-Growth-Factor-Receptor-2-FGFR2-Fusion-Gene-Worsening-After-Cancer-Chemotherapy
43.DrugBank. Pemigatinib. 2021. Accessed September 9, 2021. https://go.drugbank.com/drugs/DB15102
44.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671-684. doi:10.1016/S1470-2045(20)30109-1 PubMed
45.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-47. doi:10.1016/j.ejca.2008.10.026 PubMed
46.Kaupp-Roberts SD, Yadegarfar G, Friend E, et al. Validation of the EORTC QLQ-BIL21 questionnaire for measuring quality of life in patients with cholangiocarcinoma and cancer of the gallbladder. Br J Cancer. 2016;115(9):1032-1038. doi:10.1038/bjc.2016.284 PubMed
47.Friend E, Yadegarfar G, Byrne C, et al. Development of a questionnaire (EORTC module) to measure quality of life in patients with cholangiocarcinoma and gallbladder cancer, the EORTC QLQ-BIL21. Br J Cancer. 2011;104(4):587-92. doi:10.1038/sj.bjc.6606086 PubMed
48.Lamarca A, Palmer DH, Wasan HS, et al. ABC-06 | A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced / metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J Clin Oncol. 2019;37(15_suppl):4003-4003. doi:10.1200/JCO.2019.37.15_suppl.4003
49.Lowery MA, Goff LW, Keenan BP, et al. Second-line chemotherapy in advanced biliary cancers: a retrospective, multicenter analysis of outcomes. Cancer. 2019;125(24):4426-4434. doi:10.1002/cncr.32463 PubMed
50.Ying J, Chen J. Combination versus mono-therapy as salvage treatment for advanced biliary tract cancer: a comprehensive meta-analysis of published data. Crit Rev Oncol Hematol. 2019;139:134-142. doi:10.1016/j.critrevonc.2019.01.001 PubMed
51.Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. 1982;38(1):29-41.
52.U.S. Food & Drug Administration. Clinical trial endpoints for the approval of non-small cell lung cancer drugs and biologics: guidance for industry. 2015. Accessed September 3, 2021. https://www.fda.gov/downloads/drugs/guidances/ucm259421.pdf
53.U.S. Food & Drug Administration. Clinical trial endpoints for the approval of cancer drugs and biologics: guidance for industry. 2018. Accessed September 10, 2021. https://www.fda.gov/downloads/Drugs/Guidances/ucm071590.pdf.
54.Valle JW, Bibeau K, Cho Y, et al. Longitudinal evaluation of quality of life (QoL) in patients (Pts) with FGFR2-driven cholangiocarcinoma (CCA) treated with pemigatinib. J Clin Oncol. 2021;39(3_suppl):276-276. doi:10.1200/JCO.2021.39.3_suppl.276
55.Incyte Biosciences Canada Corporation. Pemigatinib 4.5 mg, 9 mg, 13.5 mg, oral tablets: indirect and mixed treatment comparison report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets. June 23, 2021.
56.Incyte Biosciences Canada Corporation. Incyte response to August 4, 2021 DRR request for additional information regarding pemigatinib DRR review [internal additional sponsor's information]. August 10, 2021.
57.Westin GFM, Alsidawi S, Chandrasekharan C, et al. Outcomes of second line treatment in patients with advanced and metastatic biliary cancers. J Clin Oncol. 2017;35(4_suppl):420-420. doi:10.1200/JCO.2017.35.4_suppl.420
58.Belkouz A, de Vos-Geelen J, Mathôt RAA, et al. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: an open-label, single arm, phase 2 trial. Br J Cancer. 2020;122(5):634-639. doi:10.1038/s41416-019-0698-9 PubMed
59.Kim BJ, Yoo C, Kim KP, et al. Efficacy of fluoropyrimidine-based chemotherapy in patients with advanced biliary tract cancer after failure of gemcitabine plus cisplatin: retrospective analysis of 321 patients. Br J Cancer. 2017;116(5):561-567. doi:10.1038/bjc.2016.446 PubMed
60.Bekaii-Saab TS, Bridgewater J, Normanno N. Practical considerations in screening for genetic alterations in cholangiocarcinoma. Ann Oncol. 2021;28:28. doi:10.1016/j.annonc.2021.04.012 PubMed
61.Ji JH, Song HN, Kim RB, et al. Natural history of metastatic biliary tract cancer (BTC) patients with good performance status (PS) who were treated with only best supportive care (BSC). Jpn J Clin Oncol. 2015;45(3):256-60. doi:10.1093/jjco/hyu210 PubMed
62.Lesan V, Olivier T, Prasad V. Progression-free survival estimates are shaped by specific censoring rules: Implications for PFS as an endpoint in cancer randomized trials. Eur J Cancer. 2024;202:114022. doi:10.1016/j.ejca.2024.114022 PubMed
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASC
active symptom control
CCA
cholangiocarcinoma
CDA-AMC
Canada's Drug Agency
FOLFIRI
folinic acid, fluorouracil, and irinotecan hydrochloride
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
mFOLFOX
modified folinic acid, fluorouracil, and oxaliplatin
NGS
next-generation sequencing
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
ToT
time on treatment
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pemigatinib (Pemazyre), tablet |
Submitted price | Pemigatinib, $830.30 per 4.5 mg, 9 mg, or 13.5 mg tablets |
Indication | Proposed: For the treatment of adults with previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor 2 (FGFR2 fusion or other rearrangement) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 8, 2021 |
Reimbursement request | As per indication |
Sponsor | Incyte Biosciences Canada |
Submission history | No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-effectiveness analysis Partitioned survival model |
Target population | Adult patients with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement, aligned with the proposed Health Canada indication |
Treatment | Pemigatinib |
Comparators | ASC alone (consisting of treatments that include biliary drainage, antibiotics, analgesia, steroids, and antiemetics, as well as palliative radiotherapy and blood transfusions) mFOLFOX plus ASC |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (20 years) |
Key data sources | FIGHT-202 trial, a phase II, open-label, single-arm, multinational trial (pemigatinib) and sponsor-conducted MAIC (mFOLFOX plus ASC and ASC alone) |
Submitted results | Sequential analyses:
|
Key limitations |
|
CDA-AMC reanalysis results |
|
ASC = active symptom control; CCA = cholangiocarcinoma; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Conclusions
The CDA-AMC clinical review found that, given the absence of robust comparative data on progression-free survival (PFS) and overall survival (OS), the ability to interpret the relative treatment effects observed between pemigatinib and modified folinic acid, fluorouracil, and oxaliplatin (mFOLFOX) plus active symptom control (ASC) versus ASC alone was limited. This was due to identified limitations with the sponsor’s submitted unanchored matching-adjusted indirect comparison (MAIC) (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which contributed to the uncertainty around the magnitude of benefit of pemigatinib over other treatments.
Given the high degree of uncertainty concerning the magnitude of clinical benefit, CDA-AMC was unable to perform a base-case analysis. The reanalysis performed by CDA-AMC utilizes more appropriate assumptions, but these estimates are highly uncertain.
CDA-AMC undertook reanalyses to address limitations relating to: the incorporation of MAIC-derived comparative efficacy estimates into the sponsor’s analysis, long-term extrapolations for pemigatinib PFS and OS, selecting comparator extrapolations for PFS and OS, the assumption that utility values vary by whether patients are on or off treatment, genetic testing costs, relative dose intensity (RDI), and mFOLFOX costs.
Based on the CDA-AMC reanalysis, the incremental cost-effectiveness ratio (ICER) for pemigatinib relative to ASC and mFOLFOX was estimated to be $252,718 and $261,226 per quality-adjusted life-year (QALY) gained, respectively. A sequential analysis could not be performed due to the efficacy of pemigatinib being contingent on whether data were matched to the ASC arm or ASC plus mFOLFOX arm of the ABC-06 trial. At these ICERs, at least a 95% to 100% reduction in the price of pemigatinib is required for pemigatinib to achieve an ICER of $50,000 per QALY gained compared with mFOLFOX and ASC, respectively. The reason a price reduction of 100% would be required is due to the high cost of testing, estimated to be $38,000 to identify a single patient eligible for treatment with pemigatinib. If testing costs were $0 then, to be cost-effective relative to ASC, a 77% price reduction is needed, or a 72% price reduction versus mFOLFOX.
The uncertainty in the comparative efficacy data for pemigatinib meant that the magnitude of benefit associated with pemigatinib compared with ASC and mFOLFOX could not be reliably determined. Consequently, CDA-AMC was unable to determine a base-case estimate regarding pemigatinib’s cost-effectiveness. Instead, CDA-AMC conducted an exploratory reanalysis on the sponsor’s base case. According to the clinical experts consulted by CDA-AMC for this review, pemigatinib could be equal to or better than alternative treatments currently received by patients. The price reductions noted by CDA-AMC assume substantially improved efficacy with pemigatinib, which is highly uncertain.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
Three groups collaborated for a single patient input submission: 2 patient groups in Canada, the Canadian Liver Foundation and the Canadian Organization for Rare Disorders, and 1 international organization, the Cholangiocarcinoma Foundation. Patient input was gathered through an online survey and through a virtual focus group with 3 participants in Canada that included patients and caregivers affected by bile duct cancers, including those with FGFR2 gene fusions or rearrangements. Twenty-seven respondents completed the entire survey, 12 of whom identified as Canadian. A total of 15% of respondents had been diagnosed with FGFR2 fusions. Patients reported that the experience of cholangiocarcinoma (CCA) influenced their overall quality of life, with fatigue being noted as the most problematic and common symptom, followed by anxiety. Other concerning symptoms reported included unintended weight loss, insomnia, and gastrointestinal problems. Among the 74% of patients who had received treatment, all had received chemotherapy and most indicated that the side effects were worth the benefits, which were noted to include reduced pain and increased OS. In terms of hopes for improved outcomes, patients noted that there is a lack of treatment options and that quality of life is valued as much or more than quantity. One survey respondent living in Canada and 2 focus group participants had received pemigatinib. Patients who received pemigatinib noted that dose adjustments were required due to side effects (including hair loss, headaches, diarrhea, and sore joints) but found that with these changes, the drug was tolerable. Patients receiving pemigatinib hoped for a reduction in nodule size, no new growth and stability in the tumour, and remission.
Registered clinician input was received from 2 groups, the Ontario Health Gastrointestinal Cancer Drug Advisory Committee and the Canadian Gastrointestinal Oncology Evidence Network, plus other CCA-treating physicians. The clinician input noted that the current care pathway for patients with unresectable disease includes cisplatin plus gemcitabine for first-line therapy. Second-line treatments include mFOLFOX; folinic acid, fluorouracil, and irinotecan hydrochloride (FOLFIRI); and capecitabine. Clinicians reported that prolonging survival, delaying disease progression and maintaining quality of life, reducing symptom severity, and minimizing AEs) are desired outcomes for a new treatment. Clinicians noted that patients would first need to be treated with a standard-of-care first-line therapy before receiving pemigatinib, in line with pemigatinib’s position in the FIGHT-202 trial, meaning that pemigatinib would be used as a later or last line of treatment. The Canadian Gastrointestinal Oncology Evidence Network noted that FGFR2 testing is required to identify eligible patients but that there is no current publicly funded mechanism for this testing in Canada, and such testing is not currently routine.
The drug plan input noted there is no standard of care for patients upon progression after first-line therapy, but that second-line options included mFOLFOX or FOLFIRI, capecitabine, or best supportive care. The drug plan input considered whether patients who were currently receiving second-line treatment (e.g., mFOLFOX) should switch to pemigatinib, or whether pemigatinib should be used for subsequent therapy. The drug plans also noted that genetic testing for FGFR2 is not routinely available and asked what the best timing would be for testing of mutation status.
Several of the drug plans’ concerns were addressed in the sponsor’s model:
OS and the health-state utilities capturing symptoms were included
AEs associated with pemigatinib and comparators were included in the pharmacoeconomic analysis
genetic testing was included in the sponsor’s analysis.
In addition, CDA-AMC addressed some of these concerns, as follows:
genetic testing was assumed not to have occurred in patients not receiving pemigatinib, given that genetic testing is not covered in a world without pemigatinib.
Economic Review
The current review is for pemigatinib for adult patients with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of pemigatinib compared with ASC alone and with mFOLFOX plus ASC. The model population comprised adult patients with previously treated, unresectable, locally advanced or metastatic CCA with an FGFR2 fusion or rearrangement, which was aligned with the proposed Health Canada indication.
Pemigatinib is available as a 4.5 mg, 9 mg, or 13.5 mg tablet. The recommended dose of pemigatinib is 13.5 mg orally once daily for 14 consecutive days followed by 7 days off therapy, in 21-day cycles. At the sponsor’s submitted price of $830.2987 per 13.5 mg tablet, the 28-day cycle cost of pemigatinib is $15,499, or $202,039 annually if patients remain on therapy for a full year (assuming a total of 17.4 21-day cycles annually). No drug-acquisition costs were modelled for ASC, which could consist of biliary drainage, antibiotics, analgesia, steroids, and antiemetics, as well as palliative radiotherapy and blood transfusions.2 The cost of mFOLFOX used by the sponsor in the model was $3,333 per 28-day cycle. A 24-week stopping rule was applied for mFOLFOX, in line with its use in the ABC-06 study. No wastage was assumed in the model.
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime (20-year) time horizon from the perspective of a public health care payer in Canada. Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
The sponsor submitted a partitioned survival model with 5 health states: progression free on treatment, progression free off treatment, progressed disease on treatment, progressed disease off treatment, and death (Figure 1). All patients entered the model in the progression-free on-treatment state. Because patients enter the model progression free after having received at least 1 line of prior therapy, “progression free” in the model refers to disease progression during or after receiving pemigatinib or a comparator. The proportion of people with progression-free and progressed disease was first determined by fitting survival curves to unadjusted PFS and OS data from cohort A of the FIGHT-202 study. The proportion of pemigatinib patients who remained on therapy over time was determined by fitting survival curves to time on treatment (ToT) data from cohort A of the FIGHT-202 trial. As ToT was always less than PFS, the default in the model was such that the proportion of patients with progressed disease on treatment was always 0.
Model Inputs
The model’s baseline population characteristics and clinical efficacy parameters for pemigatinib were characterized by the planned subgroup (cohort A) of the FIGHT-202 study. The FIGHT-202 study was a phase II, open-label, single-arm study to evaluate the efficacy and safety of pemigatinib in patients with previously treated, locally advanced, or metastatic CCA with and without FGFR2 fusions or rearrangements. Cohort A of the FIGHT-202 trial consisted of patients with FGFR2 fusions or rearrangements. The sponsor assumed that the baseline patient characteristics of cohort A of the FIGHT-202 study (mean age = 55 years; proportion males = 39%; body surface area = 1.88 m2) reflected the population in Canada. Mean age and sex distribution were used to adjust the general population mortality data, sourced from Statistics Canada, to match the demographics of cohort A.
PFS, OS, and ToT curves for pemigatinib were generated using unadjusted data from cohort A of the FIGHT-202 study. Extrapolation curves were selected based on clinical plausibility and visual and statistical fit to the trial’s Kaplan-Meier (KM) data. Figure 2 and Figure 4 present the observed and predicted OS and PFS for pemigatinib, respectively. Comparator survival data were informed by relative treatment effects derived from the sponsor-conducted MAIC study. The MAIC considered patient-level data from FIGHT-202 for pemigatinib matched to aggregate data from ABC-06, a randomized, phase III, open-label study comparing mFOLFOX plus ASC with ASC alone in patients with all types of biliary tract cancers. The resulting weighted hazard ratios (HRs) for OS for pemigatinib compared with mFOLFOX plus ASC and ASC alone were applied to the sponsor’s selected survival curve for pemigatinib OS to derive the comparator OS (Figure 3). As no PFS data for ASC alone were reported in ABC-06, an HR for PFS for ASC alone was not derived. Instead, it was assumed that PFS for ASC alone was equal to that of mFOLFOX plus ASC. Therefore, the MAIC-derived HR for PFS for mFOLFOX plus ASC was applied to the sponsor’s selected survival curve for pemigatinib and used to derive PFS for both mFOLFOX plus ASC and ASC alone (Figure 5). While ToT was modelled using a survival curve for pemigatinib (Figure 6), for comparators, ToT was assumed to be equal to PFS. Grade 3 or greater adverse events (AEs) were included if they occurred in 5% or more of patients for any comparator, and were naively derived from FIGHT-202 for pemigatinib and ABC-06 for mFOLFOX plus ASC and ASC alone.
Health-state utility values were derived from cohort A of the FIGHT-202 study. European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) data were mapped to EQ-5D-3L utilities, with a UK tariff applied.3 Utilities were age-adjusted by applying a multiplier to the health-state utility value. An IV medication administration disutility of 0.025 was applied to patients receiving mFOLFOX plus ASC while patients were on treatment.4 Disutility and durations of AEs sourced from the literature were applied based on their frequency. In the absence of data to inform AE disutility, assumptions based on clinical expert opinion were used to estimate the disutility associated with the AE.
Costs in the model included the cost of treatment acquisition, drug administration, health care resource use, and AE costs. Dose interruptions for pemigatinib were adjusted by calculating the percentage of doses received as a proportion of the expected number of doses without any interruptions. To adjust for patients taking pemigatinib for 2 weeks then not taking it for a week, in the model, pemigatinib weekly costs were adjusted by averaging the number of days per week they would be taking medication (4.67 days). Costs of subsequent therapies upon progression were not included in the model. Pain medication was included for patients in the progressed-disease health states. A cost per administration of mFOLFOX was derived based on chair time and a nurse visit to discontinue infusion. Health care resource use included medical oncologist visits, CT scans, and blood tests using costs from the Ontario Schedule of Benefits and Fees and the Schedule of Benefits for Laboratory Services.5,6 Frequency of health care resource use by health state is presented in Table 12. End-of-life costs were approximated based on the costs associated with pancreatic cancer from de Oliveira et al. (2016) and inflated to 2021 values.7 AE costs were sourced from the Ontario Case Costing Initiative database ambulatory and inpatient care codes.8 Costs for FGFR genetic testing were applied to 100% of pemigatinib patients and 75% of mFOLFOX plus ASC and ASC alone patients. These costs incorporated the costs of testing patients who would test negative by adjusting the cost of the test according to the prevalence of the mutation. The cost of the genetic test was based on a previous CADTH review where the cost of adding a gene to a panel was $750.9
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (2,500 iterations for the base-case and scenario analyses). The probabilistic findings are presented subsequently.
Base-Case Results
Pemigatinib was associated with a QALY gain of 1.23 at an additional cost of $177,324, resulting in an ICER of $143,604 compared with ASC alone. Compared with ASC alone, mFOLFOX plus ASC was dominated (i.e., less effective and more costly). In a pairwise comparison with pemigatinib, the ICERs for ASC and mFOLFOX versus pemigatinib were $143,604 and $127,359 per QALY gained, respectively. At the end of the 20-year time horizon, 1% of pemigatinib patients remained alive. Of the 1.65 QALYs accrued for pemigatinib, 0.92 (59%) occurred during the first 2 years of the model time horizon.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | Pairwise ICER ($/QALY) (pemigatinib vs. comparator) | Sequential ICER ($/QALY) |
|---|---|---|---|---|---|
ASC | 69,907 | 0.61 | 0.42 | 143,604 | Reference |
mFOLFOX + ASC | 89,316 | 0.67 | 0.41 | 127,359 | Dominated |
Pemigatinib | 247,231 | 2.56 | 1.65 | NA | 143,604 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted extensive probabilistic scenario analyses. When a shorter (10-year) time horizon was used, the ICER for pemigatinib compared with ASC increased to $159,040. Results were also sensitive to using the MAIC-adjusted survival analysis (rather than the sponsor’s unadjusted base case), increasing the ICER to $160,408 and $166,411 when mFOLFOX and ASC were adjusted, respectively. When the Weibull and generalized gamma curves were used to extrapolate pemigatinib OS, the ICER increased to $207,363 and $160,554 per QALY compared with ASC. Using the sponsor’s societal perspective, the ICER for pemigatinib increased to $179,274 compared with ASC.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative efficacy estimates derived from the MAIC are uncertain. As FIGHT-202 was a single-arm study and because no head-to-head studies were conducted comparing pemigatinib with ASC or mFOLFOX, the sponsor conducted an unanchored MAIC to derive comparator efficacy rates to be used in the model. The CDA-AMC Clinical Review Report identified several limitations with the sponsor’s MAIC, including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables in the MAIC. Not conducting a head-to-head trial means there are potentially known and unknown confounders that could influence the comparative efficacy estimates derived from the MAIC. Consequently, residual confounding remains the major limitation of the MAIC, despite adjusting for some baseline characteristics.
As the sponsor’s MAIC created 2 different patient populations — 1 that made cohort A of FIGHT-202 look similar to the ASC arm of ABC-06 and a second population that made cohort A of FIGHT-202 look similar to the mFOLFOX arm of ABC-06 — the model produced 2 different results for pemigatinib. One that should only be interpreted alongside the ASC arm of the ABC-06 trial and 1 that should only be interpreted alongside the mFOLFOX arm. Therefore, only pairwise comparisons between pemigatinib and each of the comparators are appropriate.
Additionally, the sponsor’s approach to deriving PFS and OS for comparators was to take unadjusted extrapolations from FIGHT-202 and apply MAIC-derived HRs to get ASC and mFOLFOX PFS and OS estimates. First, it is inappropriate to use the unadjusted FIGHT-202 KM data because it assumes that the MAIC HRs derived from an adjusted FIGHT-202 population will apply to the entire FIGHT-202 trial, which would negate the need for matching in the first place. Second, the approach of applying HRs to pemigatinib extrapolations assumes there are proportional hazards between pemigatinib and comparators. As there are no head-to-head data, there is no evidence that a proportional hazards assumption will hold; therefore, applying constant HRs is inappropriate.
Finally, AE rates were derived from a naive comparison rather than the MAIC, which further assumes that the baseline characteristics between FIGHT-202 and ABC-06 were the same which, based on the sponsor’s MAIC, was not found to be the case.
In the CDA-AMC reanalyses, data from the FIGHT-202 trial were matched to the ASC arm of the ABC-06 trial, as per the sponsor’s MAIC. Rather than deriving comparative efficacy from weighted HRs, CDA-AMC fitted independent survival curves to the ASC data and the ASC-matched pemigatinib data. A pairwise analysis was then performed between ASC and pemigatinib. Data from the FIGHT-202 trial was then matched to the mFOLFOX arm of the ABC-06 trial, as per the sponsor’s MAIC. CDA-AMC fitted independent survival curves to the FOLFOX data and the mFOLFOX-matched pemigatinib data. A pairwise analysis was then performed between mFOLFOX and pemigatinib.
CDA-AMC was unable to address the limitation regarding naively derived AE rates; however, it is not expected that this would have a large influence on cost-effectiveness estimates.
Sponsor’s selected parametric functions for long-term outcomes did not meet face validity. In the FIGHT-202 trial, after 36 months of exposure, approximately 5% of patients on pemigatinib remained progression-free. For OS, after 22 months, approximately 41% of patients remained alive. To extrapolate pemigatinib outcomes to the model time horizon, the sponsor used parametric survival functions. The sponsor selected a log-logistic distribution to extrapolate pemigatinib OS, which resulted in a sustained post-progression survival benefit. This meant that patients who progressed on pemigatinib would have substantially improved OS outcomes relative to those who progressed on another therapy. According to the clinical experts consulted by CDA-AMC for this review, this is neither expected nor proven. In the sponsor’s report, they also note: “One clinician consulted in the development of the economic model noted that they would expect approximately 10% of patients to be progression-free at 2 years.”2 The sponsor’s chosen log-logistic curve predicts 13% progression-free at 2 years, which is the furthest away from 10% compared with all other options. Although the sponsor argues the log-logistic has better statistical fit, this only relates to a curve’s ability to fit the known data and has little weight in determining long-term outcomes. For ASC and mFOLFOX, the data from the ABC-06 trial were very mature and most events, either progression or death, had occurred within the trial period.10 This meant that model outcomes for ASC and mFOLFOX were more influenced by the survival curve's ability to interpolate, rather than extrapolate, the data.
The CDA-AMC reanalyses selected alternative pemigatinib survival functions such that the post-progression survival benefit associated with pemigatinib was aligned with clinical expert expectations.
For mFOLFOX and ASC, the CDA-AMC chose curves that best interpolated the data while ensuring that post progression survival outcomes were not greater than for pemigatinib.
The sponsor’s approach to modelling ToT was uncertain. In the sponsor’s submission, ToT was assumed to equal PFS for mFOLFOX. The clinical experts consulted for this review deemed this to be inappropriate, as mFOLFOX has cumulative neurotoxicity such that some patients may not be able to tolerate sustained treatment until progression. Instead, ToT for some mFOLFOX patients may be less than PFS, as they could discontinue before progression. In the sponsor’s submission, as there is no ToT data available from ABC-06, this assumption could not be changed.
For pemigatinib, a separate ToT curve was fitted to the FIGHT-202 trial data to estimate pemigatinib treatment costs. The experts consulted by CDA-AMC for this review stated that while ToT for pemigatinib could be less than PFS, they would expect this to be rare in clinical practice and, if it were to occur, would expect progression in 1 to 2 months. The sponsor assumed that time to treatment discontinuation followed an exponential function, meaning that the rate of discontinuation was constant over time. However, they assumed a log-normal distribution for PFS, meaning that a considerable proportion of patients would discontinue therapy but remain progression-free for an extended period. Examining the pemigatinib PFS and ToT KM data demonstrated that these curves closely followed each other, indicating that PFS and ToT were similar for pemigatinib in FIGHT-202.
Finally, ToT was based on the full patient population from the FIGHT-202 trial, whereas the PFS data were based on an adjusted population to match the ASC and mFOLFOX arms of the ABC-06 trial. Given that ToT is inherently linked to PFS, any changes to PFS through matching should lead to a change in ToT.
To reflect the clinical experts’ expectations regarding ToT for pemigatinib and to ensure that modelled costs for pemigatinib reflected the approach for estimating costs for mFOLFOX, in the CDA-AMC reanalyses, ToT was assumed to be equal to PFS for pemigatinib.
Costs of genetic testing are uncertain. According to clinician feedback and the sponsor, genetic testing for FGFR2 fusions or rearrangements is not currently publicly funded.2 To estimate genetic testing costs, the sponsor applied a cost of $750, which was sourced from a previous CADTH review and was the cost associated with adding a genetic test onto an existing next-generation sequencing (NGS) panel.11 This approach is inappropriate, as the cost of the initial NGS panel is not accounted for, and NGS panels are not currently publicly funded. As a scenario analysis, the sponsor explored the costs associated with NGS panel testing from FoundationOne, a private laboratory, which was US$5,800, which was deemed to be a more appropriate approximation of NGS costs but was not converted to Canadian dollars. The sponsor’s base case also appropriately applied the prevalence of FGFR2 fusions and rearrangements to calculate testing costs, such that the cost of testing patients found to be negative was incorporated into the model.
In addition to 100% of pemigatinib patients receiving genetic testing, the sponsor assumed that 75% of patients receiving mFOLFOX and ASC would also undergo testing. While some patients in the current context may receive genetic testing either privately through out-of-pocket payments or through research studies, given that testing is not currently funded and will not inform treatment decisions if pemigatinib is not available, it is inappropriate to apply genetic testing costs to comparator arms.
In the CDA-AMC reanalyses, the cost of genetic testing was the cost associated with the NGS panel from FoundationOne converted to Canadian dollars using the average Bank of Canada exchange rate from August 30, 2020 to August 30, 2021 (1.2690).12 Additionally, CDA-AMC assumed 0% of ASC and mFOLFOX patients will receive publicly funded genetic testing.
Health-state utility values do not meet face validity. The sponsor derived utility values from FIGHT-202 for the following health states: progression-free on treatment, progression-free off treatment, progressed disease on treatment, progressed disease off treatment (Table 13). Patients with progression-free disease who were off treatment had a lower utility value (█████) applied than that of patients in any of the progressed-disease health states. According to the clinical experts consulted for this review, there is no reason for a patient with progression-free disease who is off treatment to have worse quality of life than someone with progressed disease. Additionally, results of the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30 questionnaire was assessed at baseline and then every 3 cycles starting with cycle 3 until discontinuation of the study treatment and at the end-of-treatment visit, meaning that the progression-free off-treatment utility value is taken at the time of discontinuation. If patients discontinue treatment due to intolerable AEs, their health-related quality of life will likely improve upon discontinuation. Finally, applying these utility values led to results that are not clinically expected; mFOLFOX plus ASC was found to predict greater life-years than ASC alone, but because no ASC patients fell into the progression-free off-treatment health state, treatment with ASC led to greater QALYs (Table 3).
To address this limitation, utilities in the CDA-AMC reanalyses did not differ depending on whether patients were on or off treatment.
The analysis did not include the costs of subsequent therapies. The sponsor’s model incorporated costs and outcomes of pemigatinib, ASC, and FOLFOX; however, upon progression, no additional treatments are incorporated. This is inappropriate as, according to the clinical experts consulted for this review, if patients progress on second-line treatment and have a good Eastern Cooperative Oncology Group Performance Status, they can receive subsequent chemotherapy. Treatment with subsequent therapies was observed in FIGHT-202, as discussed in the CDA-AMC Clinical Review Report. Further, subsequent therapy options are expected to differ depending on second-line treatments according to clinical experts; if patients received pemigatinib as second-line therapy, they would be expected to receive mFOLFOX upon progression, but if patients received second-line mFOLFOX, they would receive FOLFIRI or other drugs upon progression. As subsequent treatments differ by second-line therapy and, given there are differences in the costs of third-line regimens, this introduces some uncertainty in the cost-effectiveness estimates.
CDA-AMC was unable to address this limitation. The direction and magnitude of not incorporating subsequent therapies is unknown.
The following limitations were identified but were not deemed key limitations:
The incorporation of relative dose intensity (RDI) is inappropriate. The sponsor incorporated an RDI of ██████ which was calculated from FIGHT-202 based on doses received as a proportion of the expected number of doses without interruptions. CDA-AMC was unable to validate this value as a means of accounting for dose interruptions. RDI was not reported in the sponsor’s clinical study report; however, treatment compliance was reported and found to be high, as concluded in the CDA-AMC Clinical Review Report. Additionally, although patients may miss a dose of pemigatinib, this might not influence overall costs to public drug plans, as full drug claims will be dispensed. Finally, RDI was not applied to mFOLFOX therapy.
CDA-AMC reanalyses assumed an RDI of 1 for pemigatinib.
Costs used for the mFOLFOX regimen were uncertain. Costs for the components for the mFOLFOX regimen were sourced from a previous CADTH review.13 CDA-AMC found wholesale prices for mFOLFOX components from the IQVIA Delta PA database14 that were deemed to be more appropriate.
The CDA-AMC reanalysis used wholesale costs from IQVIA Delta PA.
Health care resource use estimates are uncertain. The sponsor incorporated health care resource use by assuming that the estimated clinician visits and bloodwork would occur once every 3 months for those with progression-free and progressed disease, and the CT scan would occur once every 3 and 12 months for those with progression-free and progressed disease, respectively.2 According to the clinical experts consulted for this review, patients are expected to visit clinicians and have bloodwork monitoring done more frequently when in the progressed-disease state versus the progression-free disease state. Additionally, while the study protocol for pemigatinib specifies that patients receiving pemigatinib will need to visit an eye specialist and undergo optic coherence tomography imaging before initiation and at regular intervals (every 3 cycles), or as clinically indicated thereafter, to monitor for serous retinal detachment, the costs of these exams for pemigatinib patients were not included.15
Given the relatively low overall costs of resource use compared with other model costs, assumptions regarding visit frequency are unlikely to change conclusions regarding the cost-effectiveness of pemigatinib. Not incorporating eye exam or optical coherence tomography costs for pemigatinib favours pemigatinib, as only patients receiving pemigatinib would require eye exams.
ASC costing assumption estimates are uncertain. According to the clinical experts consulted for this review, ASC may differ depending on a patient’s second-line therapy; however, the magnitude and direction are uncertain. Likewise, given that pemigatinib improves OS, this would lead to more ASC costs being incurred in the pemigatinib arm.
Exclusion of ASC costs is likely to favour pemigatinib but the magnitude of impact on cost-effectiveness is unlikely to be large.
The analysis does not include all relevant comparators. The sponsor’s analysis compared pemigatinib with ASC and mFOLFOX, as these were the only comparators indicated for CCA treatment. In addition to ASC and mFOLFOX, there are several other off-label medications for second-line treatment of CCA that are used in clinical practice in Canada, based on feedback from the clinical experts consulted for this review (per Appendix 1 and the comparator table provided by the sponsor).15 According to CDA-AMC economic guidelines, all interventions currently used and potentially displaced should be identified, and those that decision-makers are currently funding or that are commonly used should be included.16 While not indicated for CCA, some off-label treatments are listed as a full benefit in jurisdictions, meaning that clinicians can prescribe medications for any indication, including those that are off-label.15 Additionally, CDA-AMC economic guidelines note that comparator selection should not be limited by the availability of data.16 The exclusion of comparators that may be displaced if pemigatinib is publicly reimbursed may favour pemigatinib, as these comparators are associated with lower annual costs (refer to Appendix 1), although the comparative benefits are unknown.
CDA-AMC was unable to address this limitation and, as such, the cost-effectiveness of pemigatinib compared with off-label therapies that are currently reimbursed is unknown.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC reanalyses addressed several limitations within the economic model, summarized in Table 4. CDA-AMC was unable to address limitations regarding the uncertainty in comparative efficacy estimates due to FIGHT-202 being a single-arm trial and uncertainties arising in the MAIC, and not including costs of subsequent therapies. As such, the changes shown in Table 4 reflect a CDA-AMC reanalysis rather than a base-case estimate of the cost-effectiveness of pemigatinib compared with ASC and mFOLFOX. The CDA-AMC reanalysis was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 4: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption | ||
|---|---|---|---|---|
Correctionsa to sponsor’s base case | ||||
None | — | — | ||
Changes to derive the CDA-AMC base case | ||||
1. Comparative efficacy | Weighted hazard ratios | Extrapolated curves | ||
2. MAIC-adjusted survival analysis | Pemigatinib OS and PFS were unadjusted | Pemigatinib OS and PFS were adjusted to match the ASC and FOLFOX arms from the ABC-06 trial:
| ||
3. Time on treatment for pemigatinib | Not equal to PFS | Equal to PFS | ||
4. Pemigatinib PFS extrapolation | Log-normal | Weibull | ||
5. Pemigatinib OS extrapolation | Log-logistic | Weibull | ||
6. Comparator PFS extrapolation | Not applicable (based on HRs) | Log-normal (PFS assumed equal for ACS and FOLFOX due to absence of evidence for ASC PFS) | ||
7. Comparator OS extrapolation | Not applicable (based on HRs) | ASC: Log-normal mFOLFOX: Weibull | ||
8. Utility values | Assume treatment status effect (refer to Table 13). | Do not assume treatment status effect (█████ and █████ for progression-free and progressed disease, respectively) | ||
9. Genetic testing frequency and costs | $750 per test:
| $7,360 per test:
| ||
10. Relative dose intensity | █████% | 100% | ||
11. mFOLFOX costs | Total per week = $833.20:
| Total per week = $307.13: | ||
CDA-AMC reanalysis: ASC vs. pemigatinib | — | 1 + 2a + 3 + 4 + 5 + 6 + 7a + 8 + 9 + 10 | ||
CDA-AMC reanalysis: mFOLFOX vs. pemigatinib | — | 1 + 2b + 3 + 4 + 5 + 6 + 7b + 8 + 9 + 10 + 11 | ||
ASC = active symptom control; CDA-AMC = Canada's Drug Agency; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; OS = overall survival; PFS = progression-free survival; vs. = versus.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions, or standard errors in probabilistic analyses) that are not identified as limitations.
The results of CDA-AMC’s stepped analysis are presented in Table 14 and Table 15 for pemigatinib versus ASC and mFOLFOX, respectively. The efficacy of pemigatinib in the sponsor’s analysis is dependent on whether the population in FIGHT-202 is adjusted to the ASC cohort or mFOLFOX cohort from the ABC-06 trial. CDA-AMC notes that outcomes for pemigatinib were not substantially different when the population was adjusted to the ASC cohort relative to the FOLFOX population. Compared with ASC, pemigatinib was $209,585 more expensive and yielded 0.83 greater QALYs, leading to an ICER of $252,718 per QALY gained (Table 5). Compared with FOLFOX, pemigatinib was $198,154 more expensive and yielded 0.76 greater QALYs, leading to an ICER of $261,226 per QALY gained (Table 6). Changing the survival curve to extrapolate pemigatinib OS resulted in the largest change to the sponsor’s base case. At a willingness-to-pay threshold of $50,000, pemigatinib is 0% likely to be cost-effective compared with either ASC or mFOLFOX. Of the total costs for pemigatinib, 61% were drug costs (Table 16). The majority of costs for ASC and mFOLFOX came from terminal care costs. Of the 1.24 total QALYs for pemigatinib when compared with ASC, 0.62 are accrued during the first 2 years of the model’s time horizon. Of the 1.25 total QALYs for pemigatinib when compared with mFOLFOX, 0.53 are accrued during the first 2 years of the model’s time horizon.
Scenario Analysis Results
CDA-AMC undertook price-reduction analyses in its reanalysis (Table 7). These analyses demonstrated that a price reduction of 95% would be required for pemigatinib to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared with mFOLFOX. For ASC, a price reduction approaching 100% is required for pemigatinib to be considered cost-effective when compared with ASC. When testing costs were set to $0, the price reduction required for pemigatinib to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared with ASC and mFOLFOX was 77% and 72%, respectively.
Table 5: Summary of the CDA-AMC Reanalysis Results for Pemigatinib vs. ASC
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
ASC | 66,895 | 0.58 | 0.41 | Reference |
Pemigatinib (ASC adjusted) | 276,480 | 1.79 | 1.24 | 252,718 |
ASC = active symptom control; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Table 6: Summary of the CDA-AMC Reanalysis Results for Pemigatinib vs. mFOLFOX
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
mFOLFOX | 77,945 | 0.72 | 0.49 | Reference |
Pemigatinib (mFOLFOX adjusted) | 276,099 | 1.82 | 1.25 | 261,226 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; QALY = quality-adjusted life-year; vs. = versus.
Table 7: CDA-AMC Price-Reduction Analyses
Analysis | ICERs for pemigatinib vs. ASC and mFOLFOX | |||
|---|---|---|---|---|
Sponsor base case ($) | CDA-AMC reanalysis ($) | |||
Price reduction | ASC | mFOLFOX | ASC | mFOLFOX |
No price reduction | 143,604 | 127,359 | 252,718 | 261,226 |
10% | 128,897 | 112,792 | 232,758 | 238,427 |
20% | 115,506 | 99,314 | 211,560 | 214,927 |
30% | 101,650 | 85,533 | 190,917 | 194,114 |
40% | 87,167 | 71,079 | 171,020 | 170,806 |
50% | 72,816 | 56,824 | 149,908 | 148,989 |
60% | 58,475 | 42,566 | 129,915 | 126,879 |
70% | 44,407 | NA | 109,882 | 104,537 |
80% | NA | NA | 88,930 | 82,937 |
90% | NA | NA | 68,379 | 59,688 |
100% | NA | NA | 48,908 | 38,348 |
ASC = active symptom control; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; NA = not applicable; vs. = versus.
Issues for Consideration
The clinical experts consulted for this review would generalize the results from the FIGHT-202 trial to patients with CCA in the first line of treatment who do not tolerate standard of care for the first line, not just to patients whose disease has progressed. Because the inclusion criteria for the FIGHT-202 trial specified that patients’ disease must have progressed after at least 1 line of prior systemic therapy, the cost-effectiveness of pemigatinib for patients whose cancer does not progress is not known. Use of pemigatinib in the population with non–progressed disease would also lead to a higher budget impact than that estimated by the sponsor and CDA-AMC.
Treatment with pemigatinib will require testing to determine eligibility. Genetic testing for FGFR2 fusions or rearrangements to determine pemigatinib eligibility is not routinely available or funded by the public health care payer.
Overall Conclusions
The CDA-AMC clinical review found that, given the absence of robust comparative data on PFS and OS, the ability to interpret the relative treatment effects observed between pemigatinib and mFOLFOX plus ASC and ASC alone was limited. This was due to identified limitations with the sponsor’s submitted unanchored MAIC (including heterogeneity across study designs and populations and the inability to adjust for all potential confounders and prognostic variables), which contributed to the uncertainty around the magnitude of benefit of pemigatinib over other treatments.
Given the high degree of uncertainty concerning the magnitude of clinical benefit, CDA-AMC was unable to perform a base-case analysis. The reanalysis performed by CDA-AMC utilizes more appropriate assumptions but notes that these estimates are highly uncertain.
CDA-AMC undertook reanalyses to address limitations relating to: the incorporation of MAIC-derived comparative efficacy estimates into the sponsor’s analysis, long-term extrapolations for pemigatinib PFS and OS, selecting comparator extrapolations for PFS and OS, the assumption that utility values vary by whether patients are on or off treatment, genetic testing costs, RDI, and mFOLFOX costs.
Based on the CDA-AMC reanalysis, the ICER for pemigatinib relative to ASC and mFOLFOX was estimated to be $252,718 and $261,226 per QALY gained, respectively. A sequential analysis could not be performed due to the efficacy of pemigatinib being contingent on whether the MAIC matched the data from the ASC arm or mFOLFOX arm of the ABC-06 trial. At these ICERs, at least a 95% to 100% reduction in the price of pemigatinib is required for pemigatinib to achieve an ICER of $50,000 per QALY gained compared with mFOLFOX and ASC, respectively. The reason price reductions approach 100% is due to the high cost of testing, estimated to be $38,000, to identify a single patient eligible for treatment with pemigatinib. If testing costs were $0, then to be cost-effective relative to ASC, a 77% price reduction is needed, or 72% versus FOLFOX.
The uncertainty in the comparative efficacy data for pemigatinib meant that the magnitude of benefit associated with pemigatinib compared with ASC and mFOLFOX could not be reliably determined. Consequently, CDA-AMC was unable to determine a base-case estimate regarding pemigatinib’s cost-effectiveness. Instead, CDA-AMC conducted an exploratory reanalysis on the sponsor’s base case. According to the clinical experts consulted by CDA-AMC for this review, pemigatinib could be equal to or better than alternative treatments currently received by patients. The price reductions noted by CDA-AMC assume substantially improved efficacy with pemigatinib, which is highly uncertain.
References
1.CADTH. pemigatinib (Pemazyre). 2022. Accessed January 15, 2025 https://www.cda-amc.ca/pemigatinib
2.Incyte Biosciences Canada Corporation. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets. June 23, 2021.
3.Longworth L, Yang Y, Young T, et al. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. National Institute for Health Research (NIHR); 2014. Accessed October 18, 2021. https://www.ncbi.nlm.nih.gov/books/NBK261616/?report=classic
4.National Institute for Health and Care Excellence. Single technology appraisal: Pomalidomide with dexamethasone for treating relapsed and refractory multiple myeloma after at least two regimens including lenalidomide and bortezomib (review of TA338) [ID985] Committee papers. 2016. Accessed August 11, 2021. https://www.nice.org.uk/guidance/ta427/documents/committee-papers
5.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (effective April 1, 2020). 2020. Accessed September 23, 2021. http://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf
6.Ontario Ministry of Health. Schedule of benefits for laboratory services: (effective July 1, 2020). 2020. Accessed October 18, 2021. http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf
7.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. doi:10.1186/s12885-016-2835-7 PubMed
8.Ontario Health and Long-Term Care. Ontario Case Costing Initiative (OCCI). 2017. Accessed October 18, 2021. https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi
9.Canada’s Drug Agency. Larotrectinib for neurotrophic tyrosine receptor kinase (NTRK) locally advanced or metastatic solid tumours – details. 2019. Accessed August 11, 2021. https://cadth.ca/larotrectinib-neurotrophic-tyrosine-receptor-kinase-ntrk-locally-advanced-or-metastatic-solid
10.Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22(5):690-701. doi:10.1016/S1470-2045(21)00027-9 PubMed
11.CADTH. Pan-Canadian oncology drug review. Final economic guidance report: larotrectinib (Vitrakvi) for neurotrophic tyrosine receptor kinase (NTRK) positive solid tumours. 2019. Accessed September 3, 2021. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2019/10159LarotrectinibNTRK%2BSolidTumours_fnEGR_NOREDACT-ABBREV_Post_31Oct2019_final.pdf
12.Bank of Canada. Daily exchange rates lookup: currency converter. 2021. Accessed September 9, 2021. https://www.bankofcanada.ca/rates/exchange/daily-exchange-rates-lookup/
13.CADTH. Pan-Canadian oncology drug review. Final economic guidance report: panitmumab (Vectibix) for left-sided metastatic colorectal cancer. 2018. Accessed September 14, 2021. https://cadth.ca/sites/default/files/pcodr/pcodr_panitumumab_vectibix_ls_mcrc_fn_egr%20.pdf
14.IQVIA. DeltaPA. IQVIA; 2021. Accessed January 15, 2025. https://www.iqvia.com/
15.Incyte Biosciences Canada Corporation. Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets [internal sponsor's package]. June 23, 2021.
16.CADTH. Guidelines for the economic evaluation of health technologies: Canada. 4th ed. 2017. Accessed October 18, 2021. https://www.cadth.ca/dv/guidelines-economic-evaluation-health-technologies-canada-4th-edition
17.Cancer Care Ontario. FOLFIRI - regimen monograph. 2019. Accessed August 26, 2021. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/49306
18.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. 2024. Accessed January 15, 2025. https://www.formulary.health.gov.on.ca/formulary/
19.Nehls O, Oettle H, Hartmann JT, et al. Capecitabine plus oxaliplatin as first-line treatment in patients with advanced biliary system adenocarcinoma: a prospective multicentre phase II trial. Br J Cancer. 2008;98(2):309-315. doi:10.1038/sj.bjc.6604178 PubMed
20.Cancer Care Ontario. CAPECISP - regimen monograph. 2019. Accessed September 14, 2021. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46811
21.Flemming JA, Zhang‐Salomons J, Nanji S, et al. Increased incidence but improved median overall survival for biliary tract cancers diagnosed in Ontario from 1994 through 2012: a population‐based study. Cancer. 2016;122(16):2534-2543. doi:10.1002/cncr.30074 PubMed
22.Mukkamalla SKR, Naseri HM, Kim BM, et al. Trends in incidence and factors affecting survival of patients with cholangiocarcinoma in the United States. J Natl Compr Canc Netw. 2018;16(4):370-376. doi:10.6004/jnccn.2017.7056 PubMed
23.Bridgewater J, Lopes A, Wasan H, et al. Prognostic factors for progression-free and overall survival in advanced biliary tract cancer. Ann Oncol. 2016;27(1):134-140. doi:10.1093/annonc/mdv483 PubMed
24.Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precis Oncol. 2018;2:1-12. doi:10.1200/PO.17.00080 PubMed
25.Lowery MA, Goff LW, Jordan E, et al. Second-line chemotherapy (CTx) outcomes in advanced biliary cancers (ABC): a retrospective multicenter analysis. J Clin Oncol. 2016;34(4 suppl)
26.Incyte Biosciences Canada Corporation. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets June 23, 2021.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from the clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison for Previously Treated, Unresectable, Locally Advanced or Metastatic Cholangiocarcinoma
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
Pemigatinib | 4.5 mg 9 mg 13.5 mg | Tablet | 830.2987a | 13.5 mg orally once daily for 14 consecutive days followed by 7 days off therapy, in 21-day cycles | 830.30 | 15,499b |
mFOLFOX | ||||||
Oxaliplatin (generic) | 5 mg/mL | Solution for injection 50 mg/10 mL 100 mg/20 mL 200 mg/40 mL | 45.0000c 90.0000c 180.0000c | 85 mg/m2 every 14 days | 12.86 | 360 |
Leucovorin (generic) | 10 mg/mL | Vial for injection 50 mg/5 mL 500 mg/50 mL | 68.9000c | 350 mg/m2 every 14 days | 63.98 | 1,791 |
Fluorouracil (generic) | 50 mg/mL | Solution for injection 0.5 g/10 mL 5 g/100 mL | 16.0900c 160.9000c | 400 mg/m2 then 2,400 mg/m2 every 14 days | 2.30 11.49 | 64 322 |
mFOLFOX | 88.33 | 2,473 | ||||
CDA-AMC = Canada’s Drug Agency; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
aSponsor’s submitted price.
bThis cost represents the average 28-day cost. In a given 28-day period, due to patients being on therapy for 14 days followed by being off therapy for 7 days, patients may incur a minimum of 14 days of cost up to a maximum of 21 days depending on when the patient starts and pauses treatment. This was calculated by taking the average cost per day for a 21-day course ($830.2987 × 14/21 = $553.53) and then calculating the average cost per 28 days ($553.53 × 28 = $15,499).
cIQVIA Delta PA.14
Table 9: CDA-AMC Cost Comparison for Off-Label Treatments Used in Previously Treated, Unresectable, Locally Advanced or Metastatic Cholangiocarcinoma
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
FOLFIRIb | ||||||
Irinotecan (generic) | 20 mg/mL | Solution for injection 40 mg/2 mL 100 mg/5 mL 500 mg/25 mL | 208.3400a 520.8500a 2,604.2500a | 180 mg/m2 every 14 days | 126.49 | 3,542 |
Leucovorin (generic) | 10 mg/mL | Vial for injection 50 mg/5 mL 500 mg/50 mL | 68.9000a | 350 mg/m2 every 14 days | 63.98 | 1,791 |
Fluorouracil (generic) | 50 mg/mL | Solution for injection 0.5 g/10 mL 5 g/100 mL | 16.0900a 160.9000a | 400 mg/m2 then 2,400 mg/m2 every 14 days | 2.30 11.49 | 64 322 |
FOLFIRI | 204 | 5,719 | ||||
Capecitabine (may be used along or in combination with cisplatin or oxaliplatin)d | ||||||
Capecitabine alone | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesd | 11.59 | 216e |
XELOX (capecitabine plus oxaliplatin) | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesd | 11.59 | 216e |
Oxaliplatin | 5 mg/mL | Solution for injection 50 mg/10 mL 100 mg/20 mL 200 mg/40 mL | 45.0000a 90.0000a 180.0000a | 130 mg/m2 on day 1 in 21 day cycled | 10.71 | 300 |
XELOX (capecitabine plus oxaliplatin) | 22.30 | 516 | ||||
Capecitabine plus cisplatin | ||||||
Capecitabine | 150 mg 500 mg | Tablet | 0.4575c 1.5250c | 1,000 mg/m2 twice daily for 14 consecutive days followed by 7 days off therapy, in 21-day cyclesf | 11.59 | 216e |
Cisplatin | 1 mg/mL | Solution for injection 10 mg/10 mL 50 mg/50 mL 100 mg/100 mL | 27.0000a 135.0000a 270.0000a | 60 mg/m2 on day 1 in 21 day cyclef | 15.43 | 432 |
Capecitabine plus cisplatin | 27.02 | 648 | ||||
Paclitaxel | ||||||
Paclitaxel | 6 mg/mL | Solution for injection 30 mg/5 mL 100 mg/16.7 mL 300 mg/50 mL | 300.0000a 1,196.8000a 3,740.0000a | 180 mg/m2 every 21 daysg | 192.38 | 5,387 |
Note: All surface area–based dosing assumed a body surface area of 1.88 m2.
aIQVIA Delta PA.14
bDose obtained from Cancer Care Ontario and confirmed to be appropriate by the clinical experts consulted for this review.17
cOntario Drug Benefit formulary (accessed January 2025).18
dDose obtained from Nehls O, Oettle H, Hartmann JT, et al.19 and confirmed to be appropriate by the clinical experts consulted for this review.
eThis cost represents the average 28-day cost. In a given 28-day period, due to patients being on therapy for 14 days followed by being off therapy for 7 days, patients may incur a minimum of 14 days of cost up to a maximum of 21 days depending on when the patient starts and pauses treatment. This was calculated by taking the average cost per day for a 21-day course ($11.59 × 14/21 = $7.73) and then calculating the average cost per 28 days ($7.73 × 28 = $216).
fDose obtained from Cancer Care Ontario20 and confirmed to be appropriate by the clinical experts consulted for this review.
gDose informed by clinical expert feedback.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | As the clinical information was derived from a single-arm trial the population had to be restricted to match trial populations of other comparators. This restriction of trial data, although necessary to avoid an even more flawed naive comparison, means that the data being used in the economic model is not reflective of the full Health Canada population. |
Model has been adequately programmed and has sufficient face validity | Yes | Not applicable. |
Model structure is adequate for decision problem | No | Partition survival models assume no explicit relationship between progression-free survival and overall survival. In the review, this leads to the perverse conclusion that patients who progress can live substantially longer than those who do not progress. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | Not applicable. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to limitation regarding comparative efficacy assumptions. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | Not applicable. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 11: Sponsor’s Base-Case Distribution Choices and Survival Estimates for Pemigatinib
Extrapolation | Sponsor’s selected survival distribution | Survival estimates |
|---|---|---|
Overall survival | Log-logistic | 12% alive at 5 years |
Progression-free survival | Log-normal | 13% progression-free at 2 years |
Time on treatment | Exponential | 11% on treatment at 2 years |
Figure 2: Observed and Predicted Overall Survival Data for Pemigatinib
KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 3: ASC Alone OS-Informed MAIC HR Compared With Pemigatinib OS
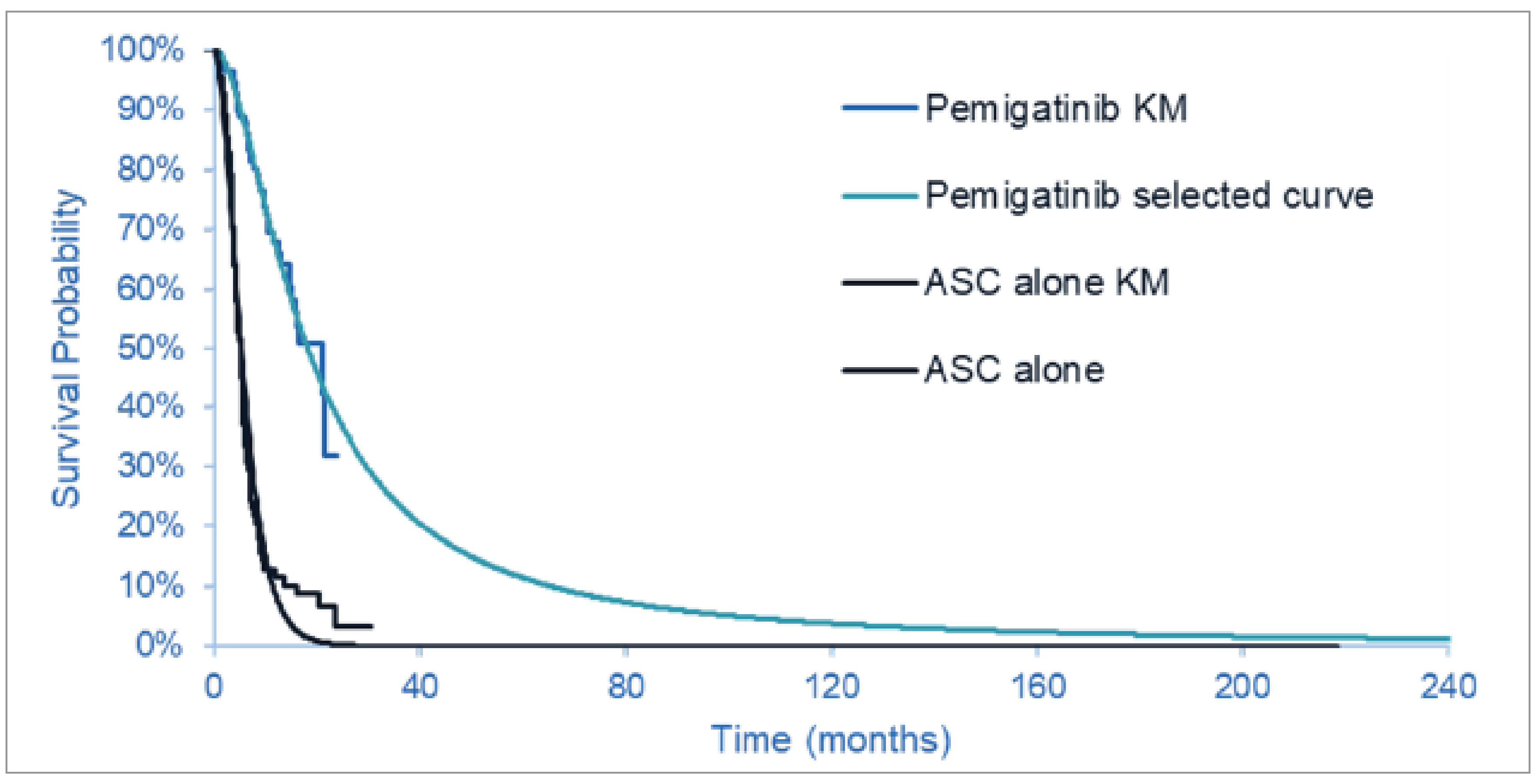
ASC = active symptom control; HR = hazard ratio; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; OS = overall survival.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 4: Observed and Predicted Progression-Free Survival Data for Pemigatinib
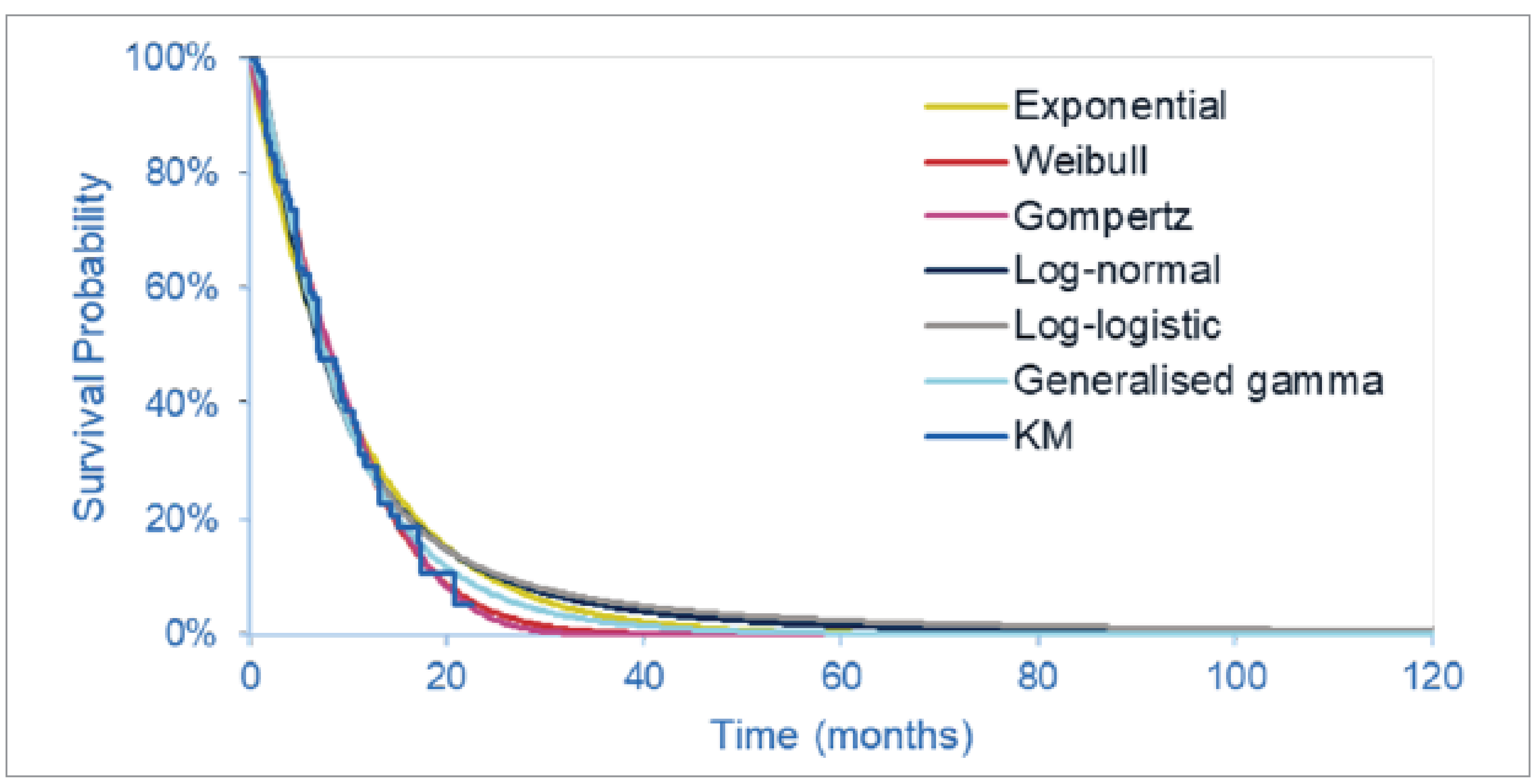
KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 5: mFOLFOX Plus ASC PFS-Informed MAIC HR, Compared With Pemigatinib PFS
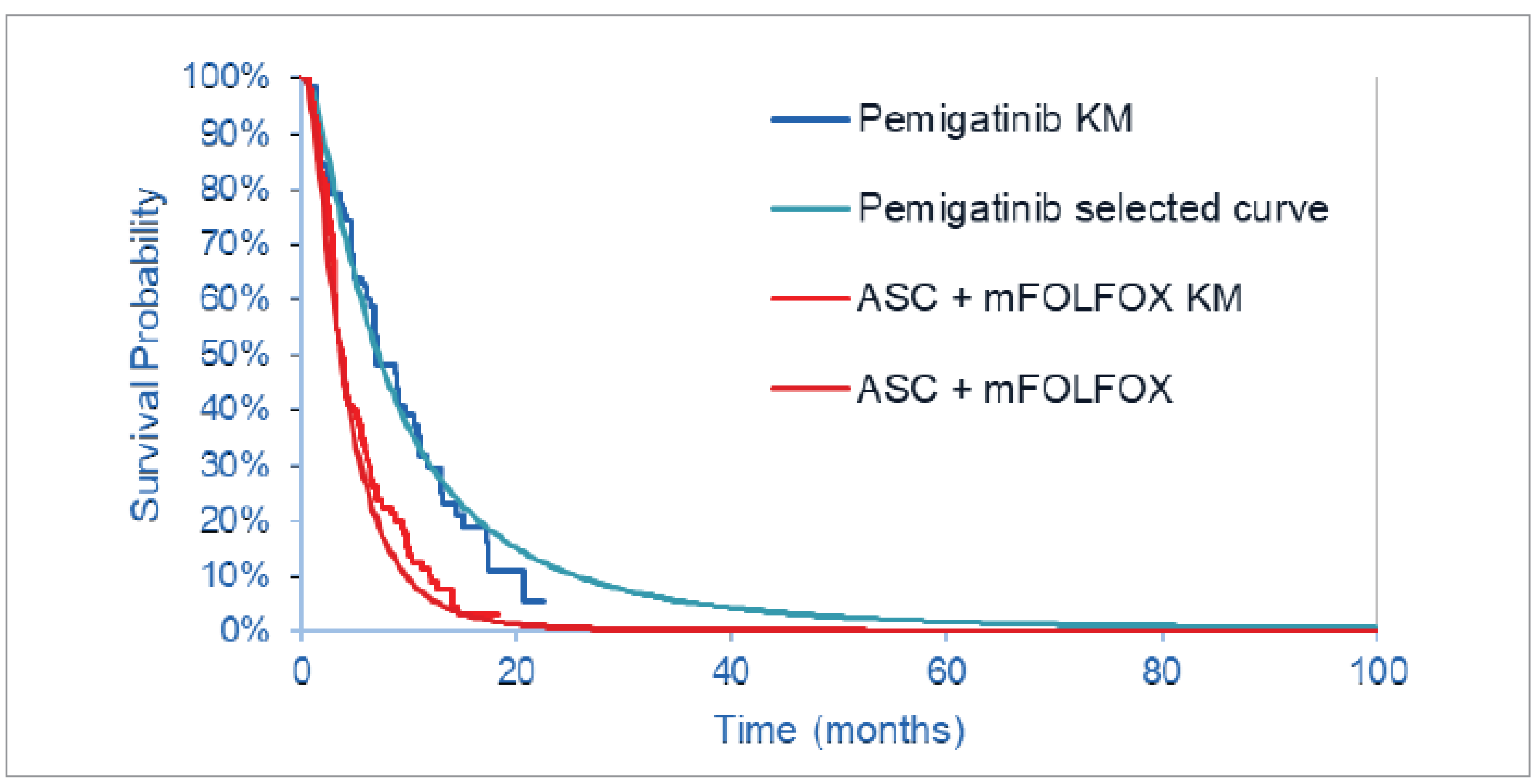
ASC = active symptom control; HR = hazard ratio; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.2
Figure 6: Observed and Predicted Time-on-Treatment Data for Pemigatinib
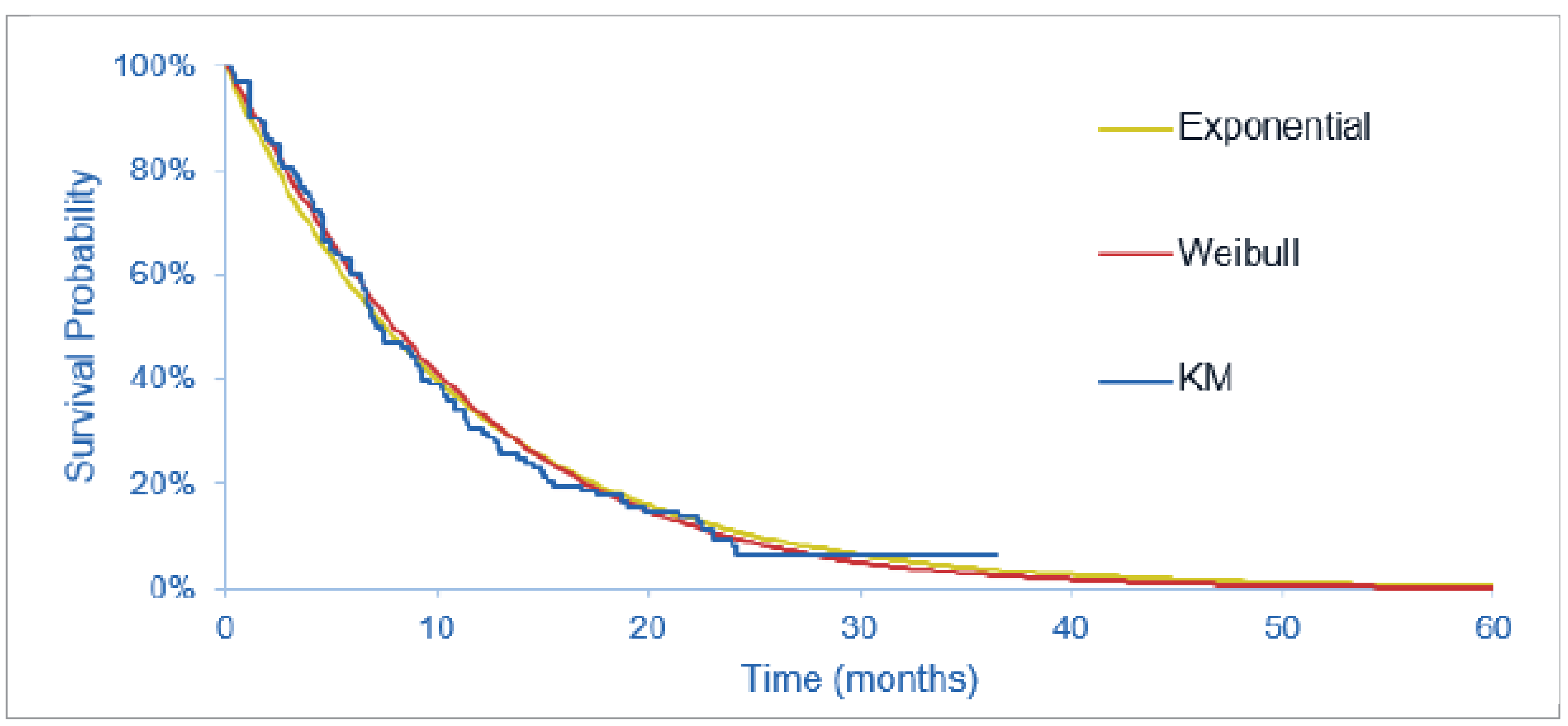
KM = Kaplan-Meier.
Source: Sponsor’s pharmacoeconomic submission.2
Table 12: Frequency of Health Care Resource Use
Resource | Monthly visit frequency | |
|---|---|---|
Progression-free | Progressed disease | |
Clinical exam | 0.333 | 0.333 |
CT scan | 0.333 | 0.083 |
Blood tests | 0.333 | 0.333 |
Source: Sponsor’s pharmacoeconomic submission.2
Table 13: Sponsor’s Health-State Utility Values
Health state | Mean utility value | Source |
|---|---|---|
Progression-free on treatment | █████ | FIGHT-202 utility analysis |
Progression-free off treatment | █████ | FIGHT-202 utility analysis |
Progressive disease on treatment | █████ | FIGHT-202 utility analysis |
Progressive disease off treatment | █████ | FIGHT-202 utility analysis |
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 14: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results for Pemigatinib vs. ASC
Stepped analysis | Drug | Total costs ($) | Total Lys | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case (deterministic) | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 148,488 | |
CDA-AMC reanalysis 1: Sponsor fitted survival curves to PFS and OS for patients receiving ASC | ASC | 69,281 | 0.57 | 0.40 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 146,327 | |
CDA-AMC reanalysis 2a: ASC adjusted survival analysis | ASC | 69,882 | 0.79 | 0.54 | Ref. |
Pemigatinib | 252,616 | 2.40 | 1.59 | 175,467 | |
CDA-AMC reanalysis 3: Pemigatinib ToT | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 278,023 | 2.53 | 1.69 | 163,867 | |
CDA-AMC reanalysis 4: Pemigatinib PFS | ASC | 69,794 | 0.60 | 0.42 | Ref. |
Pemigatinib | 244,091 | 2.53 | 1.67 | 139,321 | |
CDA-AMC reanalysis 5: Pemigatinib OS | ASC | 69,768 | 0.60 | 0.42 | Ref. |
Pemigatinib | 253,093 | 1.94 | 1.28 | 212,835 | |
CDA-AMC reanalysis 6: Reanalysis 1 + Sponsor fitted survival curve to PFS for patients receiving ASC | ASC | 69,821 | 0.57 | 0.4 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 146,327 | |
CDA-AMC reanalysis 7a: Reanalysis 1 + CDA-AMC fitted survival curve to OS for patients receiving ASC | ASC | 69,828 | 0.58 | 0.40 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 147,024 | |
CDA-AMC reanalysis 8: Utility values | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.73 | 139,844 | |
CDA-AMC reanalysis 9: Genetic testing | ASC | 66,853 | 0.60 | 0.42 | Ref. |
Pemigatinib | 286,890 | 2.53 | 1.65 | 178,771 | |
CDA-AMC reanalysis 10: Relative dose intensity | ASC | 69,776 | 0.60 | 0.42 | Ref. |
Pemigatinib | 256,835 | 2.53 | 1.65 | 151,978 | |
CDA-AMC reanalysis 1 + 2a + 3 + 4 + 5 + 6 + 7a+ 8 + 9 + 10 (deterministic) | ASC | 66,905 | 0.58 | 0.41 | Ref. |
Pemigatinib | 274,857 | 1.77 | 1.22 | 255,631 | |
CDA-AMC reanalysis 1 + 2a + 3 + 4 + 5 + 6 + 7a+ 8 + 9 + 10 (probabilistic) | ASC | 66,895 | 0.58 | 0.41 | Ref. |
Pemigatinib | 276,480 | 1.79 | 1.24 | 252,718 |
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; Ref. = reference; ToT = time on treatment; vs. = versus.
Note: Results of all steps are presented deterministically. The cumulative CDA-AMC base case is presented probabilistically, as well.
Table 15: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results for Pemigatinib vs. mFOLFOX
Stepped analysis | Drug | Total costs ($) | Total Lys | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case (deterministic) | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 132,099 | |
CDA-AMC reanalysis 1: Sponsor fitted survival curves to PFS and OS for patients receiving ASC | mFOLFOX | 90,613 | 0.82 | 0.50 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 141,451 | |
CDA-AMC reanalysis 2b: mFOLFOX adjusted survival analysis | mFOLFOX | 92,121 | 0.85 | 0.53 | Ref. |
Pemigatinib | 252,589 | 2.45 | 1.62 | 148,078 | |
CDA-AMC reanalysis 3: Pemigatinib ToT | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 278,023 | 2.53 | 1.69 | 147,931 | |
CDA-AMC reanalysis 4: Pemigatinib PFS | mFOLFOX | 89,276 | 0.66 | 0.41 | Ref. |
Pemigatinib | 244,091 | 2.53 | 1.67 | 122,779 | |
CDA-AMC reanalysis 5: Pemigatinib OS | mFOLFOX | 89,070 | 0.67 | 0.41 | Ref. |
Pemigatinib | 253,093 | 1.94 | 1.28 | 189,873 | |
CDA-AMC reanalysis 6: Reanalysis 1+ CDA-AMC fitted survival curve to PFS for patients receiving mFOLFOX | mFOLFOX | 90,613 | 0.82 | 0.50 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 141,451 | |
CDA-AMC reanalysis 7b: Reanalysis 1 + CDA-AMC fitted survival curve to OS for patients receiving mFOLFOX | mFOLFOX | 90,230 | 0.72 | 0.44 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 134,484 | |
CDA-AMC reanalysis 8: Utility values | mFOLFOX | 89,282 | 0.66 | 0.46 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.73 | 128,237 | |
CDA-AMC reanalysis 9: Genetic testing | mFOLFOX | 86,359 | 0.66 | 0.41 | Ref. |
Pemigatinib | 286,890 | 2.53 | 1.65 | 162,259 | |
CDA-AMC reanalysis 10: Relative dose intensity | mFOLFOX | 89,282 | 0.66 | 0.41 | Ref. |
Pemigatinib | 256,835 | 2.53 | 1.65 | 135,575 | |
CDA-AMC reanalysis 11: mFOLFOX costs | mFOLFOX | 80,516 | 0.66 | 0.41 | Ref. |
Pemigatinib | 252,540 | 2.53 | 1.65 | 139,192 | |
CDA-AMC reanalysis 1 + 2b + 3 + 4 + 5 + 6 + 7b+ 8 + 9 + 10 + 11 (deterministic) | mFOLFOX | 78,735 | 0.72 | 0.49 | Ref. |
Pemigatinib | 274,862 | 1.79 | 1.24 | 264,674 | |
CDA-AMC reanalysis 1 + 2b + 3 + 4 + 5 + 6 + 7b+ 8 + 9 + 10 + 11 (probabilistic) | mFOLFOX | 77,945 | 0.72 | 0.49 | Ref. |
Pemigatinib | 276,099 | 1.82 | 1.25 | 261,226 |
ICER = incremental cost-effectiveness ratio; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; Ref. = reference; ToT = time on treatment; vs. = versus.
Note: results of all steps are presented deterministically. The cumulative base case is presented probabilistically, as well.
Table 16: Disaggregated Summary of CDA-AMC’s Reanalysis Results (Pemigatinib vs. ASC)
Parameter | Pemigatinib | ASC | Incremental |
|---|---|---|---|
Discounted Lys | |||
Total | 1.79 | 0.58 | 1.21 |
Progression-free | 0.82 | 0.48 | 0.34 |
Progressed disease | 0.97 | 0.10 | 0.87 |
Discounted QALYs | |||
Total | 1.24 | 0.41 | 0.83 |
Progression-free, on treatment | 0.58 | 0.34 | 0.24 |
Progressed disease, off treatment | 0.66 | 0.07 | 0.59 |
Discounted costs ($) | |||
Total | 276,480 | 66,895 | 209,585 |
Acquisition | 169,439 | 0 | 169,439 |
Administration | 0 | 0 | 0 |
Adverse events | 1,360 | 270 | 1,090 |
Resource use | 41,231 | 982 | 40,249 |
Terminal care | 64,451 | 65,643 | −1,192 |
ICER ($/QALY) | 252,718 | ||
ASC = active symptom control; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Table 17: Disaggregated Summary of CDA-AMC’s Reanalysis Results (Pemigatinib vs. mFOLFOX)
Parameter | Pemigatinib | mFOLFOX | Incremental |
|---|---|---|---|
Discounted Lys | |||
Total | 1.82 | 0.72 | 1.09 |
Progression-free, on treatment | 0.82 | 0.33 | 0.48 |
Progression-free, off treatmenta | 0 | 0.14 | −0.14 |
Progressed disease | 1 | 0.25 | 0.75 |
Discounted QALYs | |||
Total | 1.25 | 0.49 | 0.76 |
Progression-free, on treatment | 0.58 | 0.23 | 0.35 |
Progression-free, off treatmenta | 0 | 0.10 | −0.10 |
Progressed disease | 0.68 | 0.17 | 0.51 |
Discounted costs ($) | |||
Total | 276,099 | 77,945 | 198,154 |
Acquisition | 169,439 | 5,339 | 163,868 |
Administration | 0 | 5,138 | −5,138 |
Adverse events | 1,359 | 953 | 406 |
Resource use | 41,237 | 1,143 | 40,095 |
Terminal care | 64,296 | 65,373 | −1,077 |
ICER ($/QALY) | 261,226 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; QALY = quality-adjusted life-year; vs. = versus.
aCDA-AMC notes that in the sponsor’s model, the maximum ToT for mFOLFOX is set to be 24 weeks or so; after that, patients can remain progression-free but they all switch to the “progression-free without treatment” state at 25 weeks.
Detailed Results of CDA-AMC Base Case
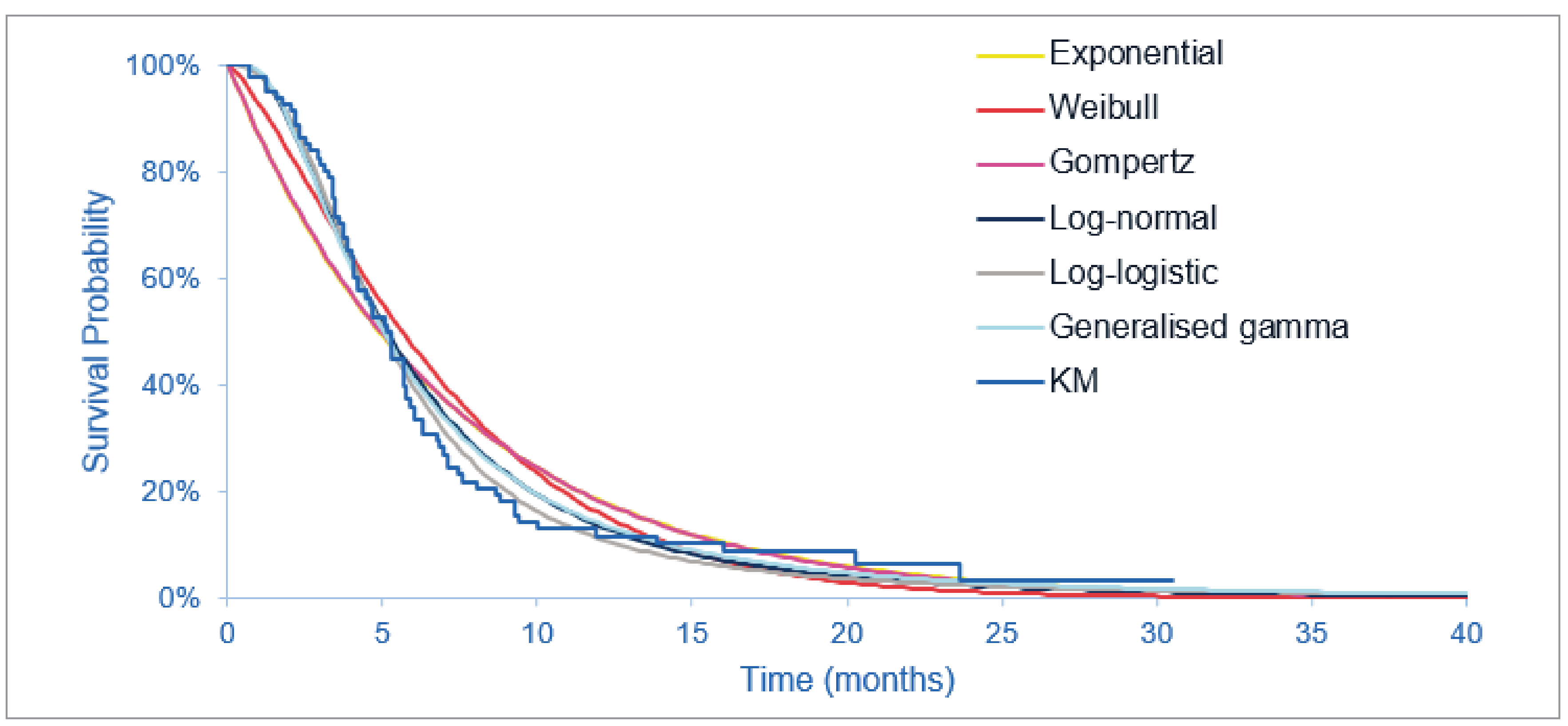
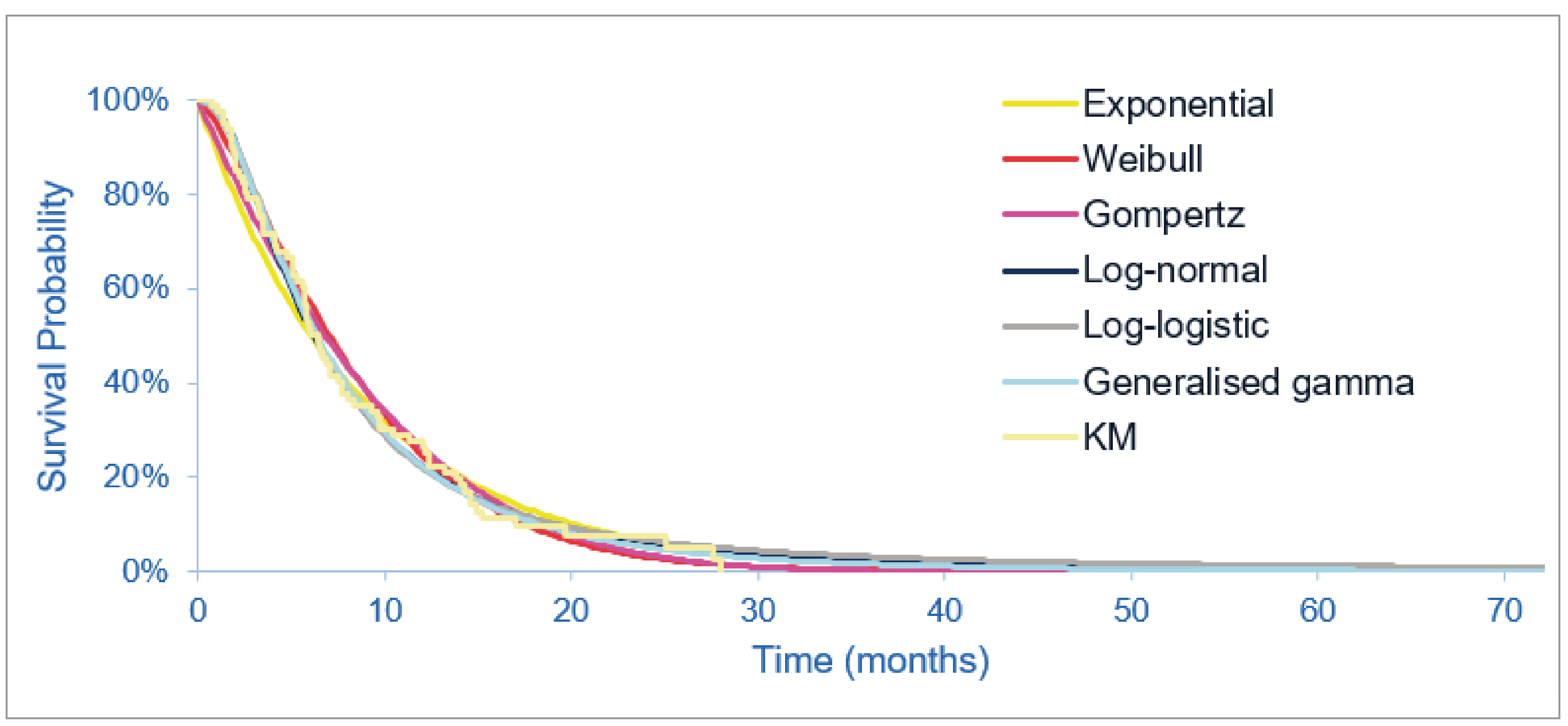
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 18: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; eCCA = extrahepatic cholangiocarcinoma; iCCA = intrahepatic cholangiocarcinoma.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA estimating the budget impact of reimbursing pemigatinib for the treatment of previously treated, unresectable, locally advanced or metastatic CCA patients with an FGFR2 fusion or rearrangement. The BIA base case was undertaken from a publicly funded drug plan perspective considering only drug costs over a 3-year time horizon. Pemigatinib costs were calculated by incorporating an RDI of ██████ observed in FIGHT-202. Stopping rules were applied for comparator treatments according to their respective clinical trials. Costs of subsequent therapies upon progression on second-line therapy were not included.
The analytic framework, which used an epidemiology-based approach, leveraged data from multiple sources in the literature and assumptions based on clinical expert input to determine the estimated population size (refer to Table 19). New patients were added to the BIA each year using a population growth rate of 4.4%.21 The sponsor compared a reference scenario where pemigatinib is not reimbursed as adjuvant therapy, with a new-drug scenario, where pemigatinib is funded as adjuvant therapy as per the Health Canada indication. Treatments available in the reference included mFOLFOX plus ASC, ASC alone, and clinical trial drugs. As it was assumed that all therapies will be taken with ASC, and ASC was assumed to not differ between comparators, costs for ASC were not assigned. A scenario analysis was conducted exploring drug costs along with costs of genetic testing and drug administration. Key inputs to the BIA are documented in Table 19.
Table 19: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Incidence of cholangiocarcinoma | 2.8 per 100,00022 |
Patients who are unresectable and eligible for first-line treatment with chemotherapy | 70%23 |
Percentage of patients completing genetic testing (including FGFR2) | 100%-Assumption |
Percentage FGFR2-positive | 19.24%24 (CCAs only) |
Percentage of patients treated with first line moving to second line | 100%-Assumption |
Percentage of eligible patients who move to third line | 52%25 |
Percentage of patients with public drug program coverage | 80%-Assumption |
Number of patients eligible for the drug under review | 143 / 149 / 154 |
Market uptake (3 years) | |
Uptake (reference scenario) ASC mFOLFOX + ASC Clinical trials | 45% / 45% / 45% 45% / 45% / 45% 10% / 10% / 10% |
Uptake (new-drug scenario) Pemigatinib ASC mFOLFOX + ASC Clinical trials | ███ █ ███ █ ███ ███ █ ███ █ ███ ███ █ ███ █ ███ 10% / 10% / 10% |
Cost of treatment (per patient) | |
Cost of treatment over 1 week Pemigatinib ASC mFOLFOX + ASC Clinical trials | $3,784.00 $0 $833.20 $0 |
ASC = active symptom control; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
Summary of the Sponsor’s BIA Results
The sponsor’s base case estimated the net budget impact of introducing pemigatinib for the treatment of previously treated, unresectable, locally advanced or metastatic CCA patients with an FGFR2 fusion or rearrangement to be $5,241,637 in year 1, $10,944,538 in year 2, and $14,282,622 in year 3, for a total budget impact over 3 years of $30,468,797.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
The incidence-based approach to estimating market size may underestimate pemigatinib costs. The sponsor used an incidence approach to estimate the number of patients eligible for pemigatinib, along with applying an annual incidence growth rate. The sponsor’s approach only captures the costs associated with newly diagnosed patients and does not capture pemigatinib costs incurred by patients remaining on pemigatinib beyond the first year of treatment. This is inappropriate as in both the sponsor’s and CDA-AMC’s pharmacoeconomic analysis, approximately 30% of patients receiving pemigatinib do so for more than 1 year.
As CDA-AMC was unable to address this limitation, the CDA-AMC base case likely underestimates the budget impact associated with reimbursing pemigatinib as costs are only captured for the first year of treatment with pemigatinib.
The uptake of pemigatinib is not aligned with clinical expert expectations. In the sponsor’s base case, it was assumed that 30% / 60% / 75% of eligible patients would uptake pemigatinib, should it become available. According to the clinical experts consulted by CDA-AMC for this review, approximately 80% of patients are expected to initiate treatment upon pemigatinib becoming available, and this is expected to reach 90% to 100% by year 3.
In the CDA-AMC reanalysis, the proportion of eligible patients who will use pemigatinib in year 1, 2, and 3 was changed to 80%, 85%, and 90%, respectively.
RDI implementation was inappropriate. The sponsor calculated an RDI of █████% based on the FIGHT-202 trial.26 CDA-AMC was unable to validate this value as a means of accounting for dose interruptions. RDI was not reported in the sponsor’s Clinical Study Report, however, treatment compliance was reported and found to be high, as concluded in the CDA-AMC Clinical Review Report. Additionally, though patients may miss a dose of pemigatinib, this might not influence overall costs to public drug plans, as full drug claims will be dispensed. Finally, an RDI was not applied to mFOLFOX therapy.
CDA-AMC reanalyses assumed an RDI of 100%.
The rate of growth of CCA is uncertain. The sponsor incorporated population growth in the model based on the growth in incidence of CCA.26 Two CCA growth rates were sourced from the literature: 1 for intrahepatic CCA (iCCA) (7%) and 1 for extrahepatic CCA (eCCA) (1.80%).21 The growth rate used in the sponsor’s base case was the mean of the iCCA and eCCA growth rates (4.40%).26 According to the CDA-AMC Clinical Review Report, 98% of cohort A in FIGHT-202 had intrahepatic CCA.
To align with the expected patient population receiving pemigatinib, in the CDA-AMC reanalyses, CCA growth rates were changed to those associated with intrahepatic CCA (7%).
Assigning clinical trials market shares is inappropriate. In the sponsor’s reference and new-drug scenario, 10% of eligible patients were assumed to be in clinical trials, which has no associated drug costs. This is inappropriate because it reduces the market size for the number of patients eligible for pemigatinib. Should pemigatinib become available and be deemed to be clinically effective, it is less likely that patients with FGFR2 fusions would enrol in clinical trials.
In CDA-AMC reanalysis, 0% market share was assigned to clinical trials in the reference and new-drug scenario.
The percentage of patients diagnosed who are unresectable and eligible for first-line treatment with chemotherapy is not aligned with clinical practice in Canada. In the sponsor’s base case, 70% of diagnosed patients are unresectable and eligible for 1L treatment with chemotherapy.23 This value was derived from a study examining prognostic factors in advanced biliary tract cancers in the UK, which reported that 30% and 70% of patients had locally advanced and metastatic disease, respectively.23 According to clinical experts consulted for this review, 80% to 90% of patients will be unresectable and eligible for first-line chemotherapy at diagnosis.
CDA-AMC reanalyses assumed that 85% of diagnosed patients are eligible for first-line chemotherapy to align with clinical practice in Canada.
Costs used for the mFOLFOX regimen were uncertain. Costs for the components for the mFOLFOX regimen were sourced from a previous CDA-AMC review.13 CDA-AMC found wholesale prices for mFOLFOX components from the IQVIA Delta PA database, which were deemed to be more appropriate. Additionally, the dose used for Leucovorin was inconsistent with that used in the ABC-06 trial.
The CDA-AMC reanalysis used wholesale costs from IQVIA.
Genetic testing costs are not transparently programmed into the sponsor’s BIA model. While the sponsor’s BIA model allows for exploration of broader health care system costs like administration and genetic testing, this is conducted through a visual basics programmed scenario analysis. As such, it is unclear whether the scenario considers genetic testing for just pemigatinib patients or if patients receiving mFOLFOX and ASC also have genetic testing costs applied. As the costs of FGFR2 testing are not currently publicly covered, it is not expected that genetic testing costs will be covered for patients in the reference scenario or for patients who receive ASC and mFOLFOX in the new-drug scenario.
CDA-AMC was unable to resolve this issue, but notes the cost of additional testing may be substantial.
The proportion of patients eligible for public coverage is uncertain. The sponsor’s base-case analysis assumed that 80% of eligible patients in each jurisdiction would have public coverage.26 IV oncology drugs are likely to be fully covered. Depending on the jurisdiction, oral oncology drugs may be fully reimbursed or may only be reimbursed by regular public drug plans. Neither of these scenarios is reflected in the sponsor’s base case.
To address uncertainty regarding the proportion eligible for public drug plan coverage, CDA-AMC conducted a scenario analysis exploring 100% coverage across jurisdictions.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by: increasing uptake of pemigatinib, changing the RDI to 100%, using the growth rate associated with iCCA, removing clinical trials market share from the reference and new-drug scenarios, assuming 85% of patients are diagnosed and unresectable, and changing mFOLFOX costs. Table 20 notes the assumptions used by the sponsor in comparison to those used by CDA-AMC in its reanalysis.
Applying these changes increased the total 3-year budget impact to $63,606,331. The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 21 and a more detailed breakdown is presented in Table 22.
Table 20: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case (none) | ||
Changes to derive the CDA-AMC base case | ||
1. Uptake | ███ █ ███ █ ███ | 80% / 85% / 90%a |
2. Relative dose intensity | █████% | 100% |
3. Growth rate | 4.40% | 7% |
4. Clinical trials market share | 10% | 0% |
5. Percent diagnosed and unresectable | 70% | 85% |
6. mFOLFOX prices | Fluorouracil: $0.003/mg Oxaliplatin: $10.20/mg Calcium folinate: $0.05/mg | Fluorouracil: $0.03218/mg14 Oxaliplatin: $0.7254/mg14 Calcium folinate $1.378/mg14 |
CDA-AMC base case | 1 + 2 + 3 + 4 + 5 + 6 | |
ASC = active symptom control; BIA = budget impact analysis; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin.
aThe market share for clinical trials was set to 0% in this step. The remaining market share was evenly distributed across ASC and mFOLFOX.
Table 21: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $30,468,797 |
CDA-AMC reanalysis 1: Uptake 80% / 85% / 90% | $47,063,969 |
CDA-AMC reanalysis 2: RDI 100% | $31,252,365 |
CDA-AMC reanalysis 3: Growth rate 7% | $32,245,142 |
CDA-AMC reanalysis 4: Removing clinical trials | $30,026,435 |
CDA-AMC reanalysis 5: 85% unresectable | $36,997,825 |
CDA-AMC reanalysis 6: mFOLFOX prices | $31,602,144 |
CDA-AMC base case | $63,606,331 |
BIA = budget impact analysis; mFOLFOX = modified folinic acid, fluorouracil, and oxaliplatin; RDI = relative dose intensity.
Note: CDA-AMC also conducted additional scenario analyses to address remaining uncertainty:
Reduced the price of pemigatinib to the value in which it would be cost-effective at a $50,000 per QALY threshold (95 and 100% compared with mFOLFOX and ASC, respectively).
100% of the population is eligible for public coverage.
Table 22: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|
Submitted base case | Reference | $1,270,369 | $1,326,265 | $1,384,621 | $3,981,255 |
New drug | $6,512,006 | $12,270,803 | $15,667,243 | $34,450,052 | |
Budget impact | $5,241,637 | $10,944,538 | $14,282,622 | $30,468,797 | |
CDA-AMC base case | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $19,417,451 | $22,018,662 | $24,888,897 | $66,325,010 | |
Budget impact | $18,571,801 | $21,113,817 | $23,920,712 | $63,606,331 | |
CDA-AMC scenario analysis 1a: 95% pemigatinib price reduction | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $1,131,546 | $1,229,874 | $1,336,422 | $3,697,842 | |
Budget impact | $285,896 | $325,028 | $368,238 | $979,163 | |
CDA-AMC scenario analysis 1b: 100% pemigatinib price reductiona | Reference | $845,650 | $904,845 | $968,184 | $2,718,679 |
New drug | $169,130 | $135,727 | $96,818 | $401,675 | |
Budget impact | -$676,520 | -$769,118 | -$871,366 | -$2,317,004 | |
CDA-AMC scenario analysis 2: 100% public drug coverage | Reference | $1,057,062 | $1,131,056 | $1,210,230 | $3,398,349 |
New drug | $24,271,814 | $27,523,328 | $31,111,121 | $82,906,263 | |
Budget impact | $23,214,752 | $26,392,271 | $29,900,891 | $79,507,913 |
BIA = budget impact analysis.
aAlthough cost saving from a drug budget perspective there would still be incremental costs to the health system due to testing costs which are not included here.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.