Drugs, Health Technologies, Health Systems
Reimbursement Review
Osimertinib (Tagrisso)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Unresectable (stage III) non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AJCC
American Joint Committee on Cancer
BICR
blinded independent central review
cCRT
concurrent chemoradiation therapy
CCSN
Canadian Cancer Survivor Network
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CNS
central nervous system
CRT
chemoradiation therapy
ctDNA
circulating tumour DNA
DCO
data cut-off
DoR
duration of response
eCRF
electronic case report form
EMA
European Medicines Agency
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
KM
Kaplan-Meier
LCC
Lung Cancer Canada
LCC MAC
Lung Cancer Canada Medical Advisory Committee
LHF
Lung Health Foundation
MID
minimal important difference
MTP
multiple testing procedure
NC
not calculable
NGS
next-generation sequencing
NSCLC
non–small cell lung cancer
OH-CCO
Ontario Health–Cancer Care Ontario
ORR
objective response rate
OS
overall survival
PCR
polymerase chain reaction
PFS
progression-free survival
PR
partial response
QoL
quality of life
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
SAE
serious adverse event
sCRT
sequential chemoradiation therapy
SD
standard deviation
TKI
tyrosine kinase inhibitor
TTD
time to treatment discontinuation or death
TTDM
time to death or distant metastases
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Osimertinib (Tagrisso), 40 mg and 80 mg daily, oral tablets |
Sponsor | AstraZeneca Canada Inc. |
Indication | For the treatment of patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based chemoradiation therapy. Osimertinib has been issued market authorization with conditions, pending the results of trials to verify its clinical benefit. The condition for authorization includes submission of the final Clinical Study Report of the LAURA trial. |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Priority review; Project ORBIS (Type C) |
NOC date | April 23, 2025 |
Recommended dose | 80 mg per day; can be reduced to 40 mg per day based on individual safety and tolerability |
NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions; NSCLC = non–small cell lung cancer.
Introduction
Lung cancer is the most commonly diagnosed cancer in Canada and the leading cause of cancer-related deaths.1,2 In 2024 alone, approximately 32,100 people living in Canada were expected to be diagnosed with lung cancer, and 20,700 annual deaths were expected to be attributable to lung cancer.3 Survival from lung cancer of all stages and histologies are poor, with an overall 5-year net survival of 22%.2 Lung cancer is classified into non–small cell lung cancer (NSCLC) or small cell lung cancer, with NSCLC accounting for approximately 88% of cases in Canada, excluding Quebec.1 One of the most prominent NSCLC molecular alterations are EGFR mutations which cause abnormal signalling pathways that drive tumour growth.4 EGFR mutations are more frequently observed in never smokers, people of Asian ethnicity, patients with adenocarcinoma, and females.5,6 The most common EGFR mutations are exon 19 deletions and the exon 21 codon 858 point mutation (L858R), accounting for 70% to 90% of EGFR mutations.7,8 NSCLC is often asymptomatic and due to its insidious nature patients may live for several years before presentation.9 The most common symptoms include unspecific cough, chest and shoulder pain, hemoptysis, weight loss, dyspnea, hoarseness, bone pain, fever, and recurring infections with bronchitis and pneumonia.9,10 Diagnostic procedures include imaging of the lungs, sputum cytology, and tissue biopsy.11
The treatment goals for locally advanced, unresectable stage III NSCLC are to cure the disease; prevent local recurrence, distant metastases, and disease progression; and improve quality of life (QoL).12-15 Before establishing a treatment plan, tumours should undergo testing for oncogenic drivers, including EGFR mutations. Testing for EGFR mutations, including EGFR exon 19 deletions and exon 21 (L858R) substitution mutations, is currently performed at the time of diagnosis as part of the standard of care for all nonsquamous NSCLC including locally advanced, unresectable NSCLC in Canada.
The current conventional treatment for patients presenting with unresectable stage III NSCLC, irrespective of EGFR mutation status, in the first-line setting is definitive concurrent chemoradiation therapy (cCRT).12,16-18 In patients who cannot tolerate cCRT, such as those with poor performance status or comorbid conditions, sequential chemoradiation therapy (sCRT) is recommended.12,19,20 In Canada, durvalumab, a PD-L1 inhibitor, is currently reimbursed for the treatment of patients with unresectable, stage III NSCLC whose disease has not progressed following concurrent platinum-based chemoradiation therapy (CRT). However, post hoc subgroup analysis of the PACIFIC trial suggested that durvalumab consolidation therapy has similar efficacy and more frequent adverse effects than placebo in patients with EGFR-mutated disease.21 As a result, in clinical practice, and based on a recent European Society for Medical Oncology consensus statement, durvalumab consolidation post-CRT is not used nor recommended for patients with unresectable NSCLC and EGFR mutations,22 leaving these patients with no effective reimbursed consolidation treatment. In the absence of an effective and available consolidation therapy (also referred to as maintenance therapy) for unresectable EGFR-mutated NSCLC in Canada, clinical experts consulted for this review with experience in the diagnosis and management of NSCLC indicated that active surveillance is the current standard of care for these patients. Active surveillance consists of regular imaging (e.g., CT scan) and clinical monitoring to detect early disease progression every 6 months for the first 2 years following completion of CRT and yearly thereafter for the next 3 years.23 Most patients with locally advanced, unresectable (stage III) EGFR-mutated NSCLC who remain progression-free during or post CRT are at risk of recurrence during the first year of the surveillance phase.24
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of osimertinib 80 mg taken orally once daily for the treatment of locally advanced, unresectable (stage III) NSCLC in patients whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT.
Osimertinib has been previously reviewed by Canada’s Drug Agency (CDA-AMC) for the treatment of locally advanced or metastatic NSCLC in patients whose tumours have EGFR exon 19 deletions or exon 21 (L858R) mutations and used as a monotherapy or in combination with pemetrexed and platinum-based chemotherapy; in patients with stage IB to stage IIIA NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations; and locally advanced or metastatic EGFR T790M mutation–positive NSCLC in patients who have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy. All final CDA-AMC recommendations were to reimburse with clinical criteria and/or conditions.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical expert(s) consulted for the purpose of this review.
Patient Input
CDA-AMC received 1 joint patient input submission from the Lung Health Foundation (LHF), Lung Cancer Canada (LCC), and the Canadian Cancer Survivor Network (CCSN). LHF is a registered charity that assists and empowers people living with or caring for others with lung disease. LCC is a registered national charitable organization that supports patients through providing education, research, and advocacy. CCSN is a national network that promotes the standard of care and provides support for patients with cancer, and issues related to survivorship or quality of end-of-life care.
The information was gathered through an online survey conducted from July 2024 to December 2024 with 23 respondents from Canada identifying as living with lung cancer, including 20 patients and 3 caregivers, all of whom had experience with osimertinib. Additionally, LFH conducted interviews with 6 patients (5 females and 1 male) who completed the online survey in January 2025. Interviewees ranged in age from 49 years to 72 years. Four interviewees were diagnosed with stage IV NSCLC with an EGFR exon 19 mutation. One interviewee was diagnosed with stage IV lung cancer (not specific as to which type) with the same mutation, and another was diagnosed with stage IIIC adenocarcinoma.
Based on the patient group input, fatigue was the most detrimental physical symptom of lung cancer, reported by 81% of survey respondents, followed by reduced appetite or weight loss (47.6%), cough (33.3%), pain (28.6%), and shortness of breath (23.8%). Additionally, 39.1% of survey respondents specifically cited fatigue and 26.1% cited shortness of breath as the primary reasons they were unable to perform daily activities. Fatigue as the most detrimental physical symptom was also echoed by interviewees, with 3 out of 6 interviewees noting fatigue and weakness as the reason they had to stop working.
As noted in the input, lung cancer impacted multiple aspects of patients’ daily life. The most impacted daily activity was the ability to work (57.1% of survey respondents and 67% of interviewees), followed by participation in sports (38.1% of respondents), travel (33.3% of respondents), leisure or hobbies (28.6% of respondents), and housework (23.8% of respondents). Survey respondents felt their experiences with lung cancer led to negative impacts on their emotional well-being (43.5%), feeling cold (43.5%), feeling isolated (34.8%), and impacted their family relationships (26.1%). The interviewees noted that the negative impacts of having lung cancer included emotional distress, mental health challenges, concerns regarding insurance coverage (e.g., travel and car insurance), changing medications and managing side effects, and job limitations due to infection risks (e.g., exposure to viruses).
The patient groups noted that concerns surrounding personal health, financial burdens, and concern for family members were some of the reasons for experiencing anxiety. These experiences highlight the profound and intertwined physical, emotional, and social toll of lung cancer on patients, their families, and their caregivers. Many respondents noted that both their symptoms and the time it took to manage their treatments negatively impacted their ability to continue working.
Survey respondents tried a range of treatments, included targeted therapy (47.6%), chemotherapies (33.3%; such as pemetrexed and carboplatin, double platinum-based chemotherapy, and others), surgery (4.8%; such as lobectomy), radiation (4.8%), immunotherapy (4.8%), and clinical trials (4.8%). Side effects were frequently associated with current treatment options, with 81.0% of survey respondents mentioning fatigue as a side effect associated with lung cancer treatments. Two interviewees who shared their experiences with lung cancer medications reported adverse effects including fatigue, anemia, bothersome mouth feel or altered taste, joint pain, edema in extremities, nausea, and constipation. Additionally, both interviewees noted that while additional medications could be taken to manage side effects, such medications were costly. The patient groups highlighted that while current lung cancer treatments extend patients’ lives, there are still concerns around health-related quality of life (HRQoL), with patients continuing to experience residual symptoms and debilitating side effects.
When considering new medication to treat lung cancer, survey respondents identified the most important outcomes as improved HRQoL (78.3%), reduced symptoms (60.9%), improved energy (43.5%), improved symptom management (39.1%), improved and/or prolonged efficacy (26.0%), reduced cost (21.7%), reduced travel time to obtain medication or treatment (8.7%), and zero cost (4.3%). The most important outcomes reported by 2 interviewees included treatment effectiveness (i.e., extending life), improved QoL, ability to effectively manage lung cancer without destroying healthy cells, ease of administration (daily oral pill is preferred over an IV infusion), manageable side effects, and affordability.
The patient groups stated that most respondents had positive experiences with osimertinib, with 90.8% preferring or strongly preferring it to other treatments or medications they had tried. Two interviewees reported a substantial change in QoL with osimertinib, stating that it gave them the ability to have a normal life and to return to work. The 3 most common side effects of osimertinib reported by survey respondents were negative changes in appearance (e.g., hair and nail issues) (73.9%), fatigue (69.6%), and diarrhea (69.6%); however, 95.7% of respondents felt their side effects were manageable. The top 10 benefits of osimertinib reported by survey respondents included reduced or eliminated pain (36.4%), stabilization of cancer or prolonged life (27.3%), ability to exercise (27.3%), increased energy (22.7%), reduced fatigue (18.2%), increased participation in daily activities (18.2%), reduced shortness of breath (18.2%), improved appetite or weight gain (18.2%), reduced cough (13.6%), and improved mood (13.6%).
Overall, the patient groups highlighted that there is an unmet need for lung cancer therapies that prolong progression-free survival (PFS) and improve HRQoL for patients. Patients also raised consideration about accessibility and needs for equity across all provinces in diagnoses and treatments. In addition, respondents expressed concerns about their continued ability to access medications due to costs, lack of availability, and the development of resistance to those medications that work for them.
Clinician Input
Input From Clinical Experts Consulted for This Review
Two clinical experts with expertise in the diagnosis and management of NSCLC provided input for this review.
Both experts felt the overarching goal of treatment for unresectable NSCLC in patients whose tumours have EGFR mutations is to reduce the risk of recurrence, increase the proportion of patients who remain cancer-free in the long term, improve survival, and in doing so, enhance HRQoL. The clinical experts noted that conventional treatments (e.g., CRT) often lead to high rates of disease recurrence, and relapsed stage III NSCLC is generally considered incurable. Additionally, the experts flagged that patients with EGFR mutations are more likely to develop central nervous system (CNS) metastases compared to those without driver mutations, increasing the risk of symptoms at recurrence. Therefore, reducing relapse risk in patients with unresectable NSCLC and EGFR mutations was identified as a critical unmet need by the clinical experts consulted for this review.
While patients with unresectable NSCLC are eligible for consolidation treatment with durvalumab after CRT, the clinical experts consulted for this review indicated that most oncologists in Canada would not administer durvalumab after CRT in patients with EGFR mutation due to its limited efficacy and increased toxicity.
The experts believe that using osimertinib after successful completion of platinum-based CRT would provide a new therapeutic option for patients with unresectable NSCLC with EGFR mutations. Because osimertinib is currently reimbursed for relapsing cases, the clinical experts noted that using it in the current setting would cause a shift in the current treatment paradigm. Currently, patients with locally advanced or metastatic disease are eligible for osimertinib as first-line treatment with palliative intent. If recurrence occurs on maintenance osimertinib or within 6 months of stopping therapy, these patients would no longer be eligible for palliative osimertinib.
The clinical experts noted that patients with unresectable stage III NSCLC whose tumours contain a common EGFR mutation (i.e., exon 19 deletion or exon 21 [L858R] substitution) and who have not progressed following platinum-based CRT would be best suited for maintenance treatment with osimertinib. The clinical experts felt that any patient who received nonplatinum-based CRT should not be routinely eligible for osimertinib treatment, but felt there should be mechanisms for individual patient requests. The experts noted that EGFR mutations are identified with molecular testing, either using polymerase chain reaction (PCR) or next-generation sequencing (NGS), and this testing should be performed at diagnosis or during CRT treatment. Following platinum-based CRT, a CT scan should be performed to evaluate disease response or progression before initiating treatment with osimertinib.
Outcome metrics used in clinical practice are generally aligned with those used in clinical trials. The clinical experts consulted for this review noted that the metrics most often used in clinical practice include progression, overall survival (OS), QoL (e.g., symptoms and the patients’ overall impression), and prevention of brain metastases. The experts noted that in current clinical practice, imaging is performed every 3 months to 6 months for the first 2 years following CRT and then annually thereafter. If osimertinib is reimbursed for patients with unresectable EGFR-mutated NSCLC who have not progressed during or following platinum-based CRT, the experts anticipated more frequent visits would be required when starting treatment (e.g., assessments approximately every 4 weeks for the first 3 months) and then every 3 months thereafter for the duration of treatment.
Clinical experts consulted for this review indicated that osimertinib should be discontinued in the event of disease progression or significant toxicities that cannot be safely managed with dose reductions or modifications. The clinical experts consulted for this review felt that in the event of disease progression that is not amenable to any other local therapies, patients should be able to continue treatment with osimertinib if there is evidence of ongoing clinical benefit. The clinical experts noted that if a patient discontinued maintenance osimertinib treatment in the absence of recurrence, they should be eligible for first-line palliative intent osimertinib treatment if at least 6 months have passed since discontinuation and recurrence.
The experts indicated that treatment with osimertinib should be prescribed and managed by a physician familiar with managing systemic lung cancers (i.e., medical oncologist or pulmonologist). In some regions, care may be supervised by another physician (e.g., a general practitioner) under the direction from a physician trained in managing systemic anticancer therapies.
Clinician Group Input
CDA-AMC received 2 clinician group input submissions from the LCC Medical Advisory Committee (LCC MAC) and Ontario Health–Cancer Care Ontario (OH-CCO) Lung Cancer Drug Advisory Committee. LCC is a national charity with the purpose of increasing awareness about lung cancer, providing support to patients with lung cancer, research, and advocacy. LCC MAC consists of clinicians in the field of lung cancer across the country. OH-CCO’s Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of Cancer Care Ontario’s mandate. In total, 27 clinicians contributed to this submission, 23 from LCC MAC and 4 from the OH-CCO Lung Cancer Drug Advisory Committee.
Both clinician groups and the clinical experts consulted for this review agreed that the current standard treatment for patients with unresectable, locally advanced NSCLC with EGFR mutations is cCRT. Clinician groups agreed that while consolidation immunotherapy with durvalumab is currently Health Canada approved and provincially funded for unresectable NSCLC agnostic of PD-L1 expression or EGFR mutation, it is not used in practice or recommended as conventional treatment for patients with EGFR mutations. Post hoc subgroup analyses of the PACIFIC trial revealed that patients with EGFR mutations receiving durvalumab had similar efficacy and increased toxicity compared to those receiving placebo. As such, both clinician groups agreed that currently there are no targetable treatments approved for patients with unresectable stage III NSCLC with EGFR mutations. Clinician groups noted that the goal of treatment is to cure the disease, as measured by OS, PFS, and disease-free survival, and LCC MAC added delaying disease progression in extrathoracic sites as a critical secondary goal.
The OH-CCO Lung Cancer Drug Advisory Committee explained patients with stage III (per standard Canadian staging techniques) unresectable NSCLC with EGFR exon 19 deletion or exon 21 (L858R) substation mutation who do not have disease progression by standard restaging techniques within 6 weeks of completion of platinum-based CRT (either concurrently or sequentially), with no contraindication to EGFR TKIs, no history of interstitial lung disease before CRT, and no evidence of symptomatic pneumonitis following definitive CRT, are best suited for treatment with osimertinib. The OH-CCO flagged that while one of the criteria is restaging within 6 weeks of completion of CRT, there should be some flexibility in this criterion as some centres have issues with CT scan wait times. While the OH-CCO Lung Cancer Drug Advisory Committee indicated that the reimbursement of molecular EGFR testing may not be universal across the country and additional funding may be needed for those provinces that do not currently have provincial reimbursement, LCC MAC indicated that they anticipated no increase in cost for testing for EGFR rearrangements to accommodate maintenance use of osimertinib.
LCC MAC stated that the outcomes to determine whether a patient is responding to treatment in clinical practice included PFS. LCC MAC suggested including the use of scans (CT of the body with or without brain imaging) and symptoms to monitor recurrence or progression with a frequency of every 3 months to 4 months while the OH-CCO Lung Cancer Drug Advisory Committee suggested restaging investigations every 3 months to 6 months.
Both clinician groups agreed that treatment should be discontinued if there is disease recurrence or progression, intolerance, or clinically important toxicity, and the OH-CCO Lung Cancer Drug Advisory Committee further added that treatment should be discontinued upon a patient’s request.
Clinician groups agreed that osimertinib can be delivered in a medical oncology outpatient setting. The OH-COO Lung Cancer Drug Advisory Committee further specified that osimertinib should be managed under the supervision of a specialist or medical oncologist with training in systemic therapy for thoracic malignancies.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. Key factors identified that could potentially impact the implementation of the CDA-AMC recommendation for using osimertinib following successful completion of platinum-based CRT in the first-line setting were the use in patients with a WHO performance status greater than 1 (i.e., beyond the pivotal trial’s inclusion criteria), initiation of treatment beyond 6 weeks following the completion of platinum-based CRT, and continuation of treatment following disease progression.
Clinical Evidence
Systematic Review
Description of Studies
The LAURA trial is an ongoing, multinational, phase III, double-blinded, placebo-controlled, randomized study that met the inclusion criteria for the systematic review conducted by the sponsor. This trial has no sites in Canada. The LAURA trial was aimed at evaluating the efficacy and safety of osimertinib as maintenance therapy in patients with locally advanced, unresectable (stage III) NSCLC with centrally confirmed EGFR mutations (exon 19 deletions or exon 21 [L858R] substitution) whose disease had not progressed during or following definitive platinum-based CRT. Following confirmation of eligibility, patients were randomized in a 2:1 ratio to receive either osimertinib 80 mg orally once daily or placebo until objective radiological disease progression (defined by Response Evaluation Criteria in Solid Tumours [RECIST] Version 1.1 and confirmed by blinded independent central review [BICR]) or another discontinuation criterion was met (e.g., patient decision or unacceptable toxicity). Randomization was stratified by prior chemoradiation strategy (cCRT versus sCRT), tumour stage before chemoradiation (stage IIIA versus stage IIIB or stage IIIC) and China cohort (enrolled at a Chinese site and patient declaring themselves of Chinese ethnicity versus enrolled at non-Chinese site or patient declaring themselves of non-Chinese ethnicity). Overall, 216 patients were randomized to either the osimertinib group (n = 143 patients) or the placebo group (n = 73 patients) and included in the full analysis set (FAS). The outcomes relevant to this review include OS, PFS, CNS PFS, response outcomes, time to death or distant metastases (TTDM), time to treatment discontinuation or death (TTD), patient-reported outcomes, and harms data. These were all collected at the planned January 5, 2024, data cut-off (DCO) date and OS was additionally collected at an unplanned, updated analysis using the November 29, 2024, DCO date.
In the FAS of the LAURA trial, slightly more patients were female (132 of 216 [61.1%]) than male (38.9%), the mean age of patients was 61.4 years (standard deviation [SD] = 10.95), and patients were primarily Asian (82.4%), followed by white (13.9%), American Indian or Alaska Native (1.4%), or another race (2.3%). At baseline, patients primarily had stage IIIA (35.2%) or stage IIIB (48.6%) NSCLC with just 16.2% of patients classified as having stage IIIC NSCLC. At screening, fewer patients in the osimertinib group were positive for exon 19 deletions (74 of 143 [51.7%]) and more were positive for exon 21 (L858R) substitutions (47.6%) compared to the placebo group (43 of 73 [58.9%] and 41.1%, respectively). Notable differences in baseline characteristics across treatment groups included fewer former smokers in the osimertinib group (25.9%) than in the placebo group (31.5%), more patients in the osimertinib group (55.9%) with a WHO performance status of 0 than in the placebo group (42.5%), and more patients in the osimertinib group receiving cCRT (91.6%) than in the placebo group (84.9%).
Efficacy Results
Efficacy outcomes presented in the following are from the most recent DCOs (November 29, 2024, for OS and January 5, 2024, for PFS, CNS PFS, TTDM, TTD, and HRQoL).
Overall Survival
At the January 5, 2024, DCO date, OS was tested and did not meet the prespecified boundary for declaring statistical significance.
At the November 29, 2024, DCO date, the median OS follow-up time for all patients was 39.36 months ███████ ████ ██ ██████ in the osimertinib group and 35.15 months ███████ ████ ██ ██████ in the placebo group at which point 40 deaths had occurred in the osimertinib group (of a total of 143 patients [28.0%]) and 26 deaths had occurred in the placebo group (of a total of 73 patients [35.6%]). The Kaplan-Meier (KM) estimate for median OS was 58.81 months (95% confidence interval [CI], 54.08 months to not calculable [NC]) in the osimertinib group and 53.98 months (95% CI, 42.05 months to NC) in the placebo group with a stratified hazard ratio (HR) of 0.67 (95% CI, 0.40 to 1.14). The between-group difference in the probability of survival for the osimertinib group versus the placebo group was ████ ████ ███ ████ ██ ████) at 36 months and █████ ████ ███ ███ ██ █████ at 48 months.
Progression-Free Survival
The median PFS follow-up time for all patients was 21.98 months (range, 0.03 months to 60.55 months) in the osimertinib group and 5.55 months (range, 0.03 months to 49.71 months) in the placebo group at which point 57 of 143 patients (39.9%) experienced progression events in the osimertinib group and 63 of 73 patients (86.3%) experienced progression events in the placebo group. The KM estimate for median PFS was 39.13 months (95% CI, 31.51 months to NC) in the osimertinib group and 5.55 months (95% CI, 3.71 months to 7.43 months) in the placebo group with a stratified HR of 0.16 (95% CI, 0.10 to 0.24) favouring the osimertinib group. The between-group difference in the probability of PFS for the osimertinib group versus the placebo group was █████ ████ ███ ████ ██ █████ at 12 months and █████ ████ ███ ████ ██ █████ at 36 months.
CNS Progression-Free Survival
At the January 5, 2024, DCO date, CNS PFS was not eligible for statistical testing (as OS was not statistically significant and therefore CNS PFS was not formally tested based on the multiple testing procedure [MTP]).
The median CNS PFS follow-up time for all patients was 24.64 months (██████ ████ ██ █████) in the osimertinib group and 5.72 months (██████ ████ ██ █████) in the placebo group at which point 29 CNS progression events had occurred in the osimertinib group (of a total of 143 patients [20.3%]) and 30 CNS progression events had occurred in the placebo group (of a total of 73 patients [41.1%]). The KM estimate for median CNS PFS was NC in the osimertinib group and 14.88 months (95% CI, 7.36 months to NC) in the placebo group with a stratified HR of 0.17 (95% CI, 0.09 to 0.32). The between-group difference in the probability of CNS PFS for the osimertinib group versus the placebo group was █████ ████ ███ ████ ██ █████ at 12 months and █████ ████ ███ ████ ██ █████ at 24 months.
Time to Death or Distant Metastases
TTDM was a secondary end point and not adjusted for multiplicity in the LAURA trial.
By the January 5, 2024, DCO date, 33 death or distant metastases events had occurred in the osimertinib group (of a total of 143 patients [23.1%]) and 31 death or distant metastases events had occurred in the placebo group (of a total of 73 patients [42.5%]) . The median TTDM was not reached (95% CI, 39.29 months to NC) in the osimertinib group and was 13.04 months (95% CI, 9.03 months to NC) in the placebo group with a stratified HR of 0.21 (95% CI, 0.11 to 0.38) favouring the osimertinib group.
Time to Treatment Discontinuation or Death
TTD was a secondary end point and not adjusted for multiplicity in the LAURA trial.
By the January 5, 2024, DCO date, 63 patients had a death or treatment discontinuation event in the osimertinib group (of a total of 143 patients [44.1%]) compared to 66 patients in the placebo group (of a total of 73 patients [90.4%]). The median TTD was 40.28 months (95% CI, 32.72 months to NC) in the osimertinib group compared to 8.31 months (95% CI, 6.14 months to 11.10 months) in the placebo group with a stratified HR of 0.21 (95% CI, 0.14 to 0.32) favouring the osimertinib group.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 Global Health Status/Quality of Life
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) questionnaire-based outcomes were secondary end points and not adjusted for multiplicity in the LAURA trial.
The between-group difference for the mean change from baseline across all visits to week 40 in the EORTC QLQ-C30 global health status/QoL did not meet the minimal important difference (MID) threshold of 4 points; however, the CI crosses the MID threshold of −4 points for deterioration (−1.9 points; 95% CI, −5.89 to 2.00 points).25
Harms Results
Safety outcomes presented in the following are from the January 5, 2024, DCO date and reported among the safety analysis set (i.e., all randomized patients who received at least 1 dose of study treatment).
Any Adverse Event
More patients in the osimertinib group experienced an adverse event (AE) (140 of 143 [97.9%]) than in the placebo group (64 of 73 [87.7%]). In both groups, the 3 most frequently reported AEs were radiation pneumonitis (47.6% in the osimertinib group and 38.4% in the placebo group), diarrhea (35.7% and 13.7%, respectively), and rash (23.8% and 13.7%, respectively).
Serious AEs
More patients in the osimertinib group experienced a serious AE (SAE) (55 of 143 [38.5%]) than in the placebo group (11 of 73 [15.1%]). In the osimertinib group, the most frequently reported SAEs were radiation pneumonitis (10.5%), pneumonia (4.9%), and gastroenteritis (1.4%) and pneumonitis (1.4%). In comparison, the most frequently reported SAEs in the placebo group were pneumonia (4.1%) and radiation pneumonitis (2.7%) with all other SAEs reported by only 1 patient.
Withdrawals Due to AEs
More patients in the osimertinib group prematurely stopped treatment due to an AE (██████ ████████ than in the placebo group (████ ██████). In the osimertinib group, the 3 most common AEs leading to treatment discontinuation were radiation pneumonitis (4.9%), █████████ ███████ ███ ███████████ ███████ ██ ███ ███████ █████ █ ████████ ██████ ███████████ █████████ ███████████ ████ ███ ██ █████████ ███████████████ █████ ███ █████ ███ ███████ ██ █████████ ███████████████ ████ ███████████ ██ ████ █ ████████
Death Due to AEs
A similar percentage of patients in both the osimertinib and placebo group experienced an AE with an outcome of death (█████ ██████ in the osimertinib group and ████ ██████ in the placebo group). In the osimertinib group, the fatal AEs were due to pneumonia (████), pneumonitis (████), and a road traffic accident (████). In the placebo group, the fatal AEs were due to myocardial infarction (████) and aortic aneurysm rupture (████).
Notable Harms
Generally, a higher percentage of patients in the osimertinib group tended to experience AEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review. In both treatment groups, radiation pneumonitis was the most common AE of special interest (experienced by 68 of 143 patients [47.6%] in the osimertinib group and 28 of 73 patients [38.5%] in the placebo group), followed by ███████ █████████ ██████ ███ █████ ████████████), and interstitial lung disease or pneumonitis (7.7% and 1.4%, respectively). Pneumonitis occurred in slightly more patients in the osimertinib group (████) than in the placebo group (████). Similarly, grade 3 or greater diarrhea occurred in ████ of patients in the osimertinib group and ████ in the placebo group.
Critical Appraisal
Despite the use of stratified randomization, differences in baseline characteristics across treatment groups poses questions about the comparability of the 2 groups. Notably, there was a higher percentage of patients with WHO performance status 0 at baseline in the osimertinib group (55.3%) versus the placebo group (42.5%) and fewer former smokers in the osimertinib group (25.9%) than the placebo group (31.5%).These differences may indicate that patients in the osimertinib group had a more favourable prognosis, which could introduce confounding and bias the results in favour of osimertinib. It is acknowledged that between-group differences in baseline characteristics may occur in trials with smaller sample sizes (e.g., < 100 patients per group) and multiple strata per covariates, which may apply to the LAURA trial.26,27 Subgroup analyses among current or former smokers were similar to the overall population. The sponsor conducted a post hoc subgroup analysis on baseline WHO performance status that was consistent with the PFS benefit for osimertinib in the primary analysis and the clinical experts consulted for this review did not anticipate the differences in WHO performance status to substantially impact the results. Nonetheless, the potential for residual confounding due to baseline imbalances remains a limitation in the internal validity of the trial.
Date of death, start and end dates for concomitant medication, radiotherapy, AEs, and subsequent anticancer treatment dates were imputed if missing. Instances where only partial information was available relied on assumptions of noninformative missingness (i.e., data missing completely at random or missing at random) which may introduce bias if missing not at random mechanisms are present. Additionally, this approach may not capture uncertainty in the imputed values resulting in an underestimation in the variability and narrowing the resulting CIs. A sensitivity analysis was conducted to assess the potential for evaluation time bias by imputing progression dates as the midpoint between the last progression-free and progression-confirming BICR assessments. Although the HR for the sensitivity analysis (████; 95% CI ████ ██ ████; refer to Table 19) ███ █████ to that for the primary PFS analysis (HR = 0.16; 95% CI, 0.10 to 0.24; refer to Table 15) it does not fully mitigate the risk of bias. It assumes progression occurs uniformly and does not account for differences in assessment schedules or missing data patterns between treatment groups. Importantly, the extent of missing data leading to imputation for these critical time-dependent variables was not reported, making it difficult to assess the overall impact of this approach on potential bias in the results.
BICR-assessed PFS, followed by OS, and BICR-assessed CNS PFS were addressed using a hierarchical MTP which controlled for type I error at the January 5, 2024, DCO date. At this time, PFS met the prespecified threshold for statistical significance. Per the testing procedure, the interim analysis of OS was also conducted at this DCO; however, OS failed to reach statistical significance. Consequently, the CNS PFS end point, which was next in the hierarchy, could not be formally tested. In this case, the reporting of a nominal P value for CNS PFS can be misleading and does not follow the statistical analysis plan; however, there is a substantial between-group difference that is unlikely attributable to chance or inflated type I error.
The OS analysis was based on the intention-to-treat approach, which assumes postprogression therapies are nondifferentially distributed between groups — a condition that may not hold given the observed disparities in postrelapse therapy used. A higher percentage of patients in the placebo group used subsequent anticancer treatments (█████) than in the osimertinib group (█████) by the November 29, 2024, DCO date. The largest discrepancy in postprogression therapies was for osimertinib (█████ in the placebo group versus █████ in the osimertinib group received subsequent osimertinib treatment at the November 29, 2024, DCO date). This imbalance challenges the intention-to-treat assumption and introduces potential bias in the OS analysis. The trial did not implement methods to adjust for treatment switching (e.g., rank-preserving structural failure time models). The crossover for patients on placebo who had progression potentially leads to an underestimation of the between-group difference in OS; however, the findings align with clinical practice, as osimertinib is the standard of care for disease that has progressed.
OS results from the LAURA trial are based on interim analyses, which may overestimate treatment effects.28 At the November 29, 2024, DCO date, the OS analysis included 66 events (in 40 of 143 patients [28.0%] in the osimertinib group and 26 of 73 patients [35.6%] in the placebo group), corresponding to approximately 30.6% of the total number of events required for the final analysis (120 events). This DCO date was not part of the prespecified analysis plan. Results from this time point may therefore be more susceptible to bias due to early data truncation and post hoc decision-making. This DCO date was conducted as part of regulatory marketing requirements.
Stratified Cox proportional hazards models were used to estimate the HRs and CIs for OS, PFS, and CNS PFS. The proportional hazards assumption is likely not met for OS based on the KM curves that cross several times over the duration of follow-up. While this suggests that the HRs may not reflect the treatment effect over time for OS, it may be partially explained by the frequent use of osimertinib in patients in the placebo group whose disease progressed. Clinical experts consulted for this review felt that the OS KM curves accurately reflected the natural history of EGFR-mutated NSCLC, with a typical median survival of 3 years to 4 years after recurrence. Alternative methods of analysis that do not rely on the proportional hazards assumption were not conducted for any of the time-to-event analyses, which makes it more difficult to assess the robustness of the results.
Visual inspection of KM plots indicated that fewer than 20% of patients in the placebo group remained at risk beyond 45 months for OS and beyond 12 months for PFS and CNS PFS. In the osimertinib group, fewer than 20% of patients were at risk beyond 51 months for OS and beyond 33 months for PFS and CNS PFS. Under such conditions, standard assumptions used in survival analysis (e.g., noninformative censoring) may be violated, which limits the reliability of survival estimates at later time points.29-31
The FDA and the EMA (European Medicines Agency) consider OS the gold standard primary outcome in trials, including NSCLC trials.32,33 However, PFS is considered to be an important cancer end point by the FDA in situations where survival may be prolonged, making an OS end point impractical.33,34 When observed differences in PFS are substantial in magnitude (viewed in the context of toxicities, the relatively short survival of patients with NSCLC, availability of alternative therapies, histologic or genetic subtype of NSCLC, and extent of prior treatment), the FDA and EMA consider PFS to provide clinical evidence to support approvals.32,33 While a correlation between PFS and OS in patients with locally advanced NSCLC has been shown,35,36 no correlation has been established for the current setting (i.e., unresectable EGFR-mutated NSCLC) and thus the validity of PFS as a surrogate for OS is unclear in the current context.
The completion rates for the EORTC QLQ-C30 and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module (EORTC QLQ-LC13) scales declined over time — particularly in the placebo group — reducing the certainty of the treatment effects at the later time points.
The clinical experts with expertise in the diagnosis and management of patients with NSCLC felt the inclusion and exclusion criteria used in the LAURA trial were generally aligned with those typically used in NSCLC trials and the sponsor’s reimbursement request. The clinical experts felt the reimbursement of osimertinib should include those with a WHO performance status of 0 to 2 (rather than only those with a WHO performance status of 0 and 1 as included in the trial).
The clinical experts consulted for this review indicated the baseline characteristics of the study population were representative of patients seen in clinical practice in Canada. Notably, a high percentage of patients enrolled in the LAURA trial identified as Asian (82.4%); however, this was partially expected as EGFR mutations are more common in patients of Asian ethnicity.5 The clinical experts consulted for this review did not expect racial differences to significantly impact treatment effects.
The clinical experts consulted for this review noted that the prior platinum-based CRT regimens used for treatment in the definitive setting for stage III disease in the LAURA trial were representative of those used to treat unresectable EGFR-mutated NSCLC in Canada. The same clinical experts also felt that watchful waiting was the most relevant comparator.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluations (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.37,38 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
OS (probabilities at 36 months and 48 months), PFS (probabilities at 12 months and 36 months), CNS PFS (probabilities at 12 months and 24 months)
HRQoL (average change in EORTC QLQ-C30 global health status/QoL score from baseline across all visits)
harms (pneumonitis, ≥ grade 3 diarrhea, withdrawal due to AEs, and fatal AEs).
Table 2 presents the GRADE summary of findings for osimertinib versus placebo for patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations and who have not progressed during or following platinum-based CRT.
Table 2: Summary of Findings for Osimertinib Versus Placebo for Patients With Locally Advanced, Unresectable (Stage III) NSCLC Whose Tumours Have EGFR Exon 19 Deletions or Exon 21 (L858R) Substitution Mutations and Have Not Progressed During or Following Platinum-Based CRT
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Osimertinib | Differencea | |||||
OS | |||||||
Probability of OS at 36 months Median follow-up: 39.36 months (range, ████ ██ █████) in the osimertinib group and 35.15 months (range, ████ ██ █████) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ██ ████ ███ █████ ███ ████ ███ █████ ██ ███ ████ ███ ██████ | Lowb | Osimertinib may result in a clinically important increase in the probability of being alive at 36 months compared to placebo. |
Probability of OS at 48 months Median follow-up: 39.36 months (range, ████ ██ █████) in the osimertinib group and 35.15 months (range, ████ ██ █████) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ████ ███ █████ ██ ██ ███ ████ ███ ██████ | Lowc | Osimertinib may result in a clinically important increased probability of being alive at 48 months compared to placebo. |
PFS | |||||||
Probability of PFS at 12 months Median follow-up: 22.18 months (range, 0.03 to 60.55 months) in the osimertinib group and 5.55 months (range, 0.03 to 49.71 months) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ████ ███ █████ ████ ██ ███ ████ ███ ██████ | Highd | Osimertinib results in a clinically important increase in the probability of being progression-free at 12 months compared to placebo. |
Probability of PFS at 36 months Median follow-up: 22.18 months (range, 0.03 to 60.55 months) in the osimertinib group and 5.55 months (range, 0.03 to 49.71 months) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ████ ███ ██████ | ███ ████ ███ █████ ████ ██ ███ ████ ███ ██████ | Moderatee | Osimertinib likely results in a clinically important increase in the probability of being progression-free at 36 months compared to placebo. |
CNS PFS | |||||||
Probability of CNS PFS at 12 months Median follow-up: 24.64 months (range, ████ ██ █████) in the osimertinib group and 5.72 months (range, ████ ██ █████) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ████ ███ █████ ████ ██ ███ ████ ███ ██████ | Highf | Osimertinib results in a clinically important increase in the probability of CNS PFS at 12 months compared to placebo. |
Probability of CNS PFS at 24 months Median follow-up: 24.64 months (range, ████ ██ █████) in the osimertinib group and 5.72 months (████ ██ █████) in the placebo group | 216 (1 RCT) | NR | ███ ███ █████ | ███ ███ █████ ████ ██ ███ ███ ██████ | ███ ████ ███ █████ ████ ██ ███ ████ ███ ██████ | Moderateg | Osimertinib likely results in a clinically important increase in the probability of CNS PFS at 24 months compared to placebo. |
EORTC QLQ-C30 global health status/quality of life | |||||||
Mean change from baseline over all visits (baseline to week 40) | 195 (1 RCT) | NR | −2.0 | −3.9 (−5.98 to −1.82) | −1.9 (−5.89 to 2.00) | Lowh | Osimertinib may result in little to no difference in quality of life compared to placebo. |
Harms at the January 5, 2024, DCO date | |||||||
Withdrawal due to AEs | 216 (1 RCT) | NR | ██ ███ █████ | ███ ███ █████ | ██ ████ ███ █████ ██ █████ ██ ███ ████ ███ ██████ | Moderatei | Osimertinib likely results in little to no difference in the frequency of withdrawal due to AEs compared to placebo. |
Fatal AEs | 216 (1 RCT) | NR | ██ ███ █████ | ██ ███ █████ | █████ ███ █████ ███ █████ ██ ██ ████ ███ ██████ | Lowj | Osimertinib may result in little to no difference in the frequency of fatal AEs compared to placebo. |
Pneumonitis | 216 (1 RCT) | NR | ██ ███ █████ | ██ ███ █████ | ██ ████ ███ █████ ██ █████ ███ █████ ██ ██ ████ ███ ██████ | Lowk | Osimertinib may result in little to no difference in the frequency of pneumonitis compared to placebo. |
Grade ≥ 3 diarrhea | 216 (1 RCT) | NR | ██ ███ █████ | ███ █████ | ██ ████ ███ █████ ███ █████ ███ █████ ██ ██ ████ ███ ██████ | Moderatel | Osimertinib likely results in little to no difference in the frequency of grade ≥ 3 diarrhea compared to placebo. |
AE = adverse event; CI = confidence interval; CNS = central nervous system; CRT = chemoradiation therapy; DCO = data cut-off; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; MID = minimally important difference; NR = not reported; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe between-group differences were requested from the sponsor to aid in the interpretation of the results for these end points.
bCertainty was rated down 1 level for risk of bias. While a planned interim analysis of OS was conducted at the January 5, 2024, DCO date and could have been used to avoid concerns related to post hoc decision-making, this analysis was based on a shorter duration of follow-up and fewer events (43 OS events), reducing its reliability for drawing robust conclusions. The later OS results, using the November 29, 2024, DCO date provide longer follow-up and more events (OS analysis included 66 events; 40 of 143 [28.0%] in the osimertinib group and 26 of 73 [35.6%] in the placebo group), but were from an unplanned, interim analysis, increasing the risk of post hoc decisions and the potential for inflated treatment effects. In both analyses, the presence of time-varying treatment effects, including a delayed separation and crossing of KM curves, limit the validity and reliability of the estimate of absolute difference between groups. Differences in postprogression anticancer therapies, including 74% of placebo group patients receiving osimertinib, also likely influence the between-group OS difference. No empirically derived and validated MID was identified for the between-group difference in the probability of OS. The clinical experts consulted for this review suggested that a 5% to 10% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 1 level for imprecision. Using the 5% between-group difference threshold, the point estimate suggests a clinically meaningful effect on OS at 36 months, while the lower bound of the 95% CI crossed the between-group difference threshold of 5%.
cCertainty was rated down 1 level for risk of bias. While a planned interim analysis of OS was conducted at the January 5, 2024, DCO date, and could have been used to avoid concerns related to post hoc decision-making, this analysis was based on a shorter duration of follow-up and fewer events (43 OS events), reducing its reliability for drawing robust conclusions. The later OS results, using the November 29, 2024, DCO date, provide longer follow-up and more events, but were from an unplanned analysis, increasing the risk of post hoc decisions and the potential for inflated treatment effects. In both analyses, the presence of time-varying treatment effects, including a higher probability of OS with placebo during the first 18 months and delayed separation and crossing of KM curves, limit the validity and reliability of the estimate of absolute difference between groups. Differences in postprogression anticancer therapies, including 74% of placebo group patients receiving osimertinib, also likely influence the between-group OS difference. In addition, at 48 months there was high attrition in the placebo group (n = 11), which may result in informative censoring and bias the between-group difference in OS at this time point. No empirically derived and validated MID was identified for the between-group difference in the probability of survival. The clinical experts consulted for this review suggested that a 5% to 10% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 1 level for imprecision. While the point estimate suggests a clinically meaningful effect on OS at 48 months, the lower bound of the 95% CI did not cross the threshold, suggesting little to no difference.
dCertainty was not rated down for indirectness for PFS as a surrogate outcome for OS. While direct validation of PFS as a surrogate for OS in the setting of unresectable EGFR-mutated NSCLC treated with maintenance therapy is lacking, the clinical experts considered PFS to be of clinical relevance for patients in delaying progression to metastatic disease and administration of more toxic treatment. Certainty was not rated down for imprecision. No empirically derived and validated MID was identified for the between-group difference in the probability of PFS. The clinical experts consulted for this review suggested that a 10% to 15% between-group difference would be clinically meaningful. The effect estimate as well as both lower and upper boundaries of the 95% CI of the between-group difference exceeded the threshold and suggested a benefit.
eCertainty was not rated down for indirectness for PFS as a surrogate outcome for OS. While direct validation of PFS as a surrogate for OS in the setting of unresectable EGFR-mutated NSCLC treated with maintenance therapy is lacking, PFS is a clinically relevant end point in oncology trials, including NSCLC, especially because patient input signalled value in therapies that prevent disease progression. Certainty was rated down 1 level for risk of bias due to high attrition at 36 months (n = 28 in the osimertinib group and n = 3 in the placebo group), which may result in informative censoring and bias the between-group difference in OS at this time point. No empirically derived and validated MID was identified for the between-group difference in the probability of PFS. The clinical experts consulted for this review suggested that a 10% to 15% between-group difference would be clinically meaningful and this value was used as the threshold. The effect estimate as well as both lower and upper boundaries of the 95% CI of the between-group difference exceeded the threshold and suggested a benefit.
fCNS PFS is not a validated surrogate for OS and does not capture extracranial progression. However, it is a clinically important outcome in the context of EGFR-mutated NSCLC, given the incidence and burden of CNS metastases in this population. The end point provides direct information on intracranial disease control that is relevant to patients and clinicians. No empirically derived and validated MID was identified for the between-group difference in the probability of CNS PFS. The clinical experts consulted for this review suggested that a 10% between-group difference would be clinically meaningful and this value was used as the threshold. The effect estimate as well as both lower and upper boundaries of the 95%CI of the between-group difference exceeded the threshold and suggested a benefit.
gCNS PFS is not a validated surrogate for OS and does not capture extracranial progression. However, it is a clinically important outcome in the context of EGFR-mutated NSCLC, given the incidence and burden of CNS metastases in this population. The end point provides direct information on intracranial disease control that is relevant to patients and clinicians. Certainty was rated down 1 level for risk of bias due to high attrition in the placebo group at 24 months (n = 8), which may result in informative censoring and bias the between-group difference in CNS PFS at this time point. No empirically derived and validated MID was identified for the between-group difference in the probability of CNS PFS. The clinical experts consulted for this review suggested that a 10% between-group difference would be clinically meaningful and this value was used as the threshold. The effect estimate as well as both lower and upper boundaries of the 95% CI of the between-group difference exceeded the threshold and suggested a benefit.
hCertainty was rated down 1 level for risk of bias. Beyond week 16 █████ ████ ███ of patients in the placebo group were available to contribute to the analysis. Certainty was rated down 1 level for imprecision. A MID for the EORTC QLQ-C30 global health status scale has not been definitively established, although a difference of 10 points is often cited. One recent review estimated the MID for the scale may be 4 points in patients with lung cancer, and 4 points was adopted as the MID for this assessment.25 Although the point estimate suggests little to no difference in HRQoL, the lower CI crosses the MID for deterioration and the possibility of a negative effect cannot be ruled out. EORTC QLQ-C30 was not adjusted for multiplicity in the trial and should be considered as supportive evidence.
iNo empirically derived and validated MID was identified for the between-group difference in the frequency of withdrawal due to AEs. The clinical experts consulted for this review suggested that a 10% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 1 level for imprecision. The between-group difference suggests no clinically meaningful effect on the frequency of withdrawal due to AEs at the January 5, 2024, DCO date while the upper bound of the 95% CI crosses the between-group difference threshold of 10%.
jNo empirically derived and validated MID was identified for the between-group difference in the frequency in fatal AEs. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 2 levels for imprecision. The between-group difference suggests no clinically meaningful effect on the frequency of fatal AEs at the January 5, 2024, DCO date while the lower bound of the 95% CI crosses the between-group difference threshold of 5%. Additionally, the absolute number of events was low in both groups, contributing to uncertainty.
kNo empirically derived and validated MID was identified for the between-group difference in the frequency of pneumonitis. The clinical experts consulted for this review suggested that a 5% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 2 levels for imprecision. The between-group difference suggests no clinically meaningful effect on the frequency of pneumonitis at the January 5, 2024, DCO date while the upper bound of the 95% CI crosses the between-group difference threshold of 5%. Additionally, the absolute number of events was low in both groups, contributing to uncertainty.
lNo empirically derived and validated MID was identified for the between-group difference in the frequency of grade ≥ 3 diarrhea. The clinical experts consulted for this review suggested that a 10% between-group difference would be clinically meaningful and this value was used as the threshold. Certainty was rated down 1 level for imprecision. The absolute number of events was low in both groups, contributing to uncertainty.
Source: The LAURA Clinical Study Report39 and LAURA Overall Survival Analysis Update.40 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
No indirect comparisons were submitted by the sponsor.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the evidence from the systematic review evidence were submitted by the sponsor.
Conclusions
The LAURA trial was a phase III, double-blinded, placebo-controlled, randomized, multinational trial comparing the efficacy and safety of maintenance osimertinib for patients with locally advanced, unresectable (stage III) NSCLC whose tumours harboured either EGFR exon 19 deletions or exon 21 (L858R) substitution mutations and whose disease has not progressed during or following platinum-based CRT. Compared to placebo, osimertinib results in a clinically important improvement in PFS and CNS PFS at 12 months (high-certainty evidence) and likely results in clinically important increases in PFS at 36 months and CNS PFS at 24 months (moderate certainty evidence). At the planned PFS interim analysis, relatively few OS events (43 events across both groups) had occurred and OS failed to achieve statistical significance. Results from a subsequent unplanned OS analysis showed osimertinib may result in a clinically important increase in OS at 36 months and 48 months compared to the placebo group (low certainty evidence at both time points). Results from the LAURA trial showed that osimertinib may result in little to no difference in HRQoL compared to placebo (low certainty of evidence). Clinical experts consulted for this review deemed the harms profile of osimertinib to be consistent with their expectations based on osimertinib’s known toxicity profile.
Clinical experts noted that conventional treatments (e.g., CRT) often lead to high rates of disease recurrence, and relapsing stage III NSCLC is generally considered incurable. Additionally, the experts flagged that patients with EGFR mutations are more likely to develop CNS metastasis compared to those with driver mutation–negative NSCLC, increasing the risk of symptoms at recurrence. Therefore, reducing relapse risk in patients with unresectable NSCLC and EGFR mutations was identified as a critical unmet need by the clinical experts consulted for this review.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of osimertinib, 80 mg oral tablets taken once daily in the treatment of locally advanced, unresectable (stage III) NSCLC in patients whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Lung cancer is the most commonly diagnosed cancer in Canada and the leading cause of cancer-related deaths.1,2 In 2024 alone, approximately 32,100 people living in Canada were expected to be diagnosed with lung cancer, and 20,700 annual deaths were expected to be attributable to lung cancer.3 Lung cancer predominantly affects males (53% of new lung cancer cases) and adults aged 50 years and older (98% of new lung cancer cases).2,3 Survival from lung cancer of all stages and histologies is poor, with an overall 5-year net survival of 22%.2 First Nations, Inuit, and Métis Peoples in Canada face significant lung cancer disparities. Adults of First Nations ancestry have a 35% lower 5-year survival rate despite similar incidence rates; adults of Métis ancestry are more likely to be diagnosed and have a 30% lower 5-year survival rate, and Inuit living in Inuit Nunangat are more than twice as likely to be diagnosed.41
Lung cancer is classified into NSCLC or small cell lung cancer, with NSCLC accounting for approximately 88% of cases in Canada, excluding Quebec.1 NSCLC is further classified into 3 main histologic subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.1 To date, several molecular alterations, defined as “driver mutations” have been identified in NSCLC, with the 2 prominent drivers being ALK rearrangements and EGFR mutations. ALK rearrangements occur through gene fusions, leading to abnormal cell proliferation.42 EGFR mutations are particularly common in adenocarcinomas, resulting in abnormal signalling pathways that drive tumour growth.4 Approximately 15% of Canadians with NSCLC have an EGFR-activating mutation in the region encoding the tyrosine kinase domain.6-8 EGFR mutations are more frequently observed in never smokers, people of Asian ethnicity, patients with adenocarcinoma, and females.5,6 The most common EGFR mutations are exon 19 deletions and the exon 21 codon 858 point mutation (L858R), accounting for 70% to 90% of EGFR mutations.43-47 A common feature of EGFR-mutated NSCLC is the development of CNS metastases, which are detected in approximately 25% of patients at diagnosis and can affect approximately 50% of all patients within 3 years from diagnosis.48 Brain metastases are associated with decreased QoL and poor prognosis and are a significant cause of cancer-related mortality.49,50
NSCLC diagnosis typically begins with a comprehensive medical history, smoking assessment, and standard tests, including evaluations of lung, renal, and liver function, along with blood and biochemistry tests.9 Imaging techniques such as X-rays, CT scans, PET scans of the chest and abdomen, and MRI of the CNS are then used before tissue biopsy is performed to confirm the diagnosis.11,51 To determine a patient’s prognosis and best course of treatment, NSCLC is staged using the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control TMN Staging System, which involves tumour, node, metastasis classification of the disease based on the size and spread of the primary tumour, lymph node involvement, and occurrence of metastasis.52 Once a confirmed diagnosis of NSCLC has taken place, patients undergo testing for oncogenic drivers, including EGFR mutations. Such testing is performed by molecular tissue testing on a sample obtained via biopsy.53
While resectability in stage III disease is not clearly defined and is dependent on a variety of factors including the size and location of the tumour, any lymph node metastases, and a surgical assessment, typically patients with stage III NSCLC without mediastinal nodal involvement (T3N1, T4N0 or T4N1) are considered resectable.14 An estimated 21% of patients diagnosed with NSCLC have stage III disease and among these patients, 87% are considered to have unresectable (nonoperable) disease.54
NSCLC is often asymptomatic and due to its insidious nature, patients may live for several years before presentation.9 The most common symptoms include unspecific cough, chest and shoulder pain, hemoptysis, weight loss, dyspnea, hoarseness, bone pain, fever, and recurring infections with bronchitis and pneumonia.9,10 Diagnostic procedures include imaging of the lungs, sputum cytology, and tissue biopsy.11
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
The treatment goals for locally advanced, unresectable stage III NSCLC are to prevent local recurrence, distant metastases, and progression as well as improve QoL.12-15
Before establishing a treatment plan, tumours should undergo testing for oncogenic drivers, including EGFR mutations. Such testing is performed by molecular tissue testing on a sample obtained via biopsy.53 The Canadian consensus for optimizing biomarker testing highlights the following recommendations for EGFR mutation testing.55
EGFR mutation testing should be performed as part of a comprehensive panel that includes the current standard of care biomarkers.
Comprehensive reflex biomarker testing, including EGFR, is recommended for all patients diagnosed with nonsquamous NSCLC regardless of disease stage and should be initiated by the pathologist at the time of initial diagnosis with targeted NGS.
When tissue biopsy harbours scarce tumour cells, when time for a tissue biopsy is too lengthy, or when invasive procedures for tissue procurement are contraindicated, liquid biopsy for the detection of activating EGFR mutations and other biomarkers is recommended at baseline, if available and publicly funded.
The current conventional treatment for patients presenting with unresectable stage III NSCLC, irrespective of EGFR mutation status, is definitive cCRT.12,16-18 The chemotherapy component typically consists of 2 to 4 cycles of preferred platinum-doublet options including cisplatin plus pemetrexed (nonsquamous only) or carboplatin plus pemetrexed, cisplatin plus etoposide, carboplatin plus paclitaxel, or cisplatin plus vinorelbine.12,19 For the radiotherapy portion, the current conventional radiation dose to be delivered with concurrent chemotherapy is 60 Gy given in 2 Gy fraction sizes delivered over 6 to 7 weeks; doses higher than 60 Gy and up to 70 Gy may be considered for select patients, with careful attention to doses to the heart, lungs, and esophagus.12,19 In patients with unresectable stage III NSCLC who cannot tolerate cCRT, sCRT is recommended12,19; typically sCRT is the preferred treatment in patients with poor performance status or comorbid conditions.20
While immunotherapy consolidation treatment after CRT has shown improved outcomes in patients with unresectable stage III NSCLC without EGFR mutations (e.g., durvalumab), subgroup analyses suggest it does not provide similar benefits in patients with EGFR mutations. In Canada, durvalumab, a PD-L1 inhibitor, is currently reimbursed for the treatment of patients with unresectable, stage III NSCLC whose disease has not progressed following concurrent platinum-based CRT. However, post hoc subgroup analysis of the PACIFIC trial suggested that durvalumab consolidation therapy has similar efficacy and more frequent adverse effects than placebo in patients with EGFR-mutated disease.21 As a result, in clinical practice, and based on a recent European Society for Medical Oncology consensus statement, the use of durvalumab consolidation post CRT is not recommended for patients with unresectable NSCLC and EGFR mutations,22 leaving these patients with no effective reimbursed immunotherapy consolidation treatment. In the absence of an effective and available consolidation therapy (also called maintenance therapy) for unresectable EGFR-mutated NSCLC in Canada, clinical experts consulted for this review with experience in the diagnosis and management of NSCLC indicated that active surveillance is the current standard of care for these patients. Active surveillance consists of regular imaging (e.g., CT scans) and clinical monitoring to detect early disease progression every 6 months for the first 2 years following completion of CRT and yearly thereafter for the next 3 years.23 Most patients with locally advanced, unresectable (stage III) EGFR-mutated NSCLC who remain progression-free during or post CRT are at risk of recurrence during the first year of the surveillance phase.24
Drug Under Review
Osimertinib, a TKI, is an oral, potent, and selective irreversible inhibitor of both EGFR-sensitizing mutations and the T790M resistance mutation that has limited activity against wild-type EGFR. Key characteristics of osimertinib are summarized in Table 3.
Osimertinib received conditional Health Canada authorization for the treatment of patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT. Health Canada’s authorization is conditional on the final OS results from the LAURA trial (at approximately 60% maturity) which will be used to confirm clinical benefit of osimertinib observed in the primary end point, PFS for the intention-to-treat target population. The final OS analysis is event driven and anticipated to occur in the first half of 2027.
The recommended dose of osimertinib is 80 mg tablet taken orally once a day.
The sponsor’s reimbursement request is the same as the Health Canada–approved indication. Osimertinib for the treatment of locally advanced, unresectable (stage III) NSCLC with EGFR exon 19 deletions or exon 21 (L858R) substitution mutations following completion of CRT without progression has been approved by FDA and EMA for this indication.56,57
In Canada, osimertinib is also Health Canada authorized for the following indications.
As adjuvant therapy after tumour resection in patients with stage IB to stage IIIA1 NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations.
For the first-line treatment of patients with locally advanced (not amenable to curative therapies), or metastatic NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations).
In combination with pemetrexed and platinum-based chemotherapy for the first-line treatment of patients with locally advanced (not amenable to curative therapies) or metastatic NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations.
For the treatment of patients with locally advanced or metastatic EGFR T790M mutation–positive NSCLC whose disease has progressed on or after EGFR TKI therapy.
Osimertinib has been previously reviewed by CDA-AMC and received recommendations for reimbursement with conditions:
in October 2024, in combination with pemetrexed and platinum-based chemotherapy for the first-line treatment of adult patients with locally advanced or metastatic NSCLC whose tumours have EGFR exon 19 deletions or exon 21 L858R mutations
in January 2022, as adjuvant therapy after tumour resection in patients with stage IB to stage IIIA NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations
in January 2019, for the first-line treatment of patients with locally advanced or metastatic NSCLC whose tumours have EGFR mutations
in May 2017, for the treatment of patients with locally advanced or metastatic EGFR T790M mutation–positive NSCLC who have progressed on or after EGFR TKI therapy.
Table 3: Key Characteristics of Osimertinib
Characteristic | Osimertinib |
|---|---|
Mechanism of action | A tyrosine kinase inhibitor; is an oral, potent, and selective irreversible inhibitor of both EGFR-sensitizing mutations and T790M resistance mutation that has limited activity against wild-type EGFR. |
Indicationa | For the treatment of patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based chemoradiation therapy. |
Route of administration | Oral |
Recommended dose | 80 mg tablet, orally, once a day |
Serious adverse effects or safety issues | Interstitial lung disease, QTcF interval prolongation, left ventricular dysfunction, and cardiomyopathy |
Other | Treatment with osimertinib should be initiated by a qualified physician experienced in the use of anticancer therapies. It is necessary that EGFR mutation–positive status (EGFR exon 19 deletions or exon 21 [L858R] substitution mutations) in tumour tissue specimens is determined using a validated test method by laboratories with demonstrated proficiency in the specific technology being used. |
NSCLC = non–small cell lung cancer; QTcF = QT interval corrected using the Fridericia formula.
aHealth Canada–approved indication.
Source: Osimertinib product monograph.58
Testing Procedure Considerations
With a growing landscape of targeted therapies available for the treatment of NSCLC, biomarker testing for oncogenic driver mutations is increasingly becoming part of a routine diagnostic work-up.55,59,60 Canadian consensus guidelines recommend testing for the presence of actionable biomarkers — at minimum EGFR, ALK, and ROS1 — to guide treatment.55,59,60 The clinical experts consulted for this review confirmed that EGFR testing, including testing for exon 19 deletions and exon 21 (L858R) substitution mutations, is currently conducted as part of routine care in patients with locally advanced, unresectable NSCLC, with testing performed at the time of initial diagnosis. It is estimated that 10% to 20% of patients with NSCLC have EGFR-sensitizing mutations, of which 70% to 90% are exon 19 deletions and exon 21 (L858R) substitution mutations55,59 EGFR mutations are more prevalent in people who are nonsmokers, females, and of Asian descent.55,59,60
EGFR mutations can be identified by either PCR or NGS testing methods, using biopsied tissue samples obtained as part of diagnostic work-up.18,55,60 NGS is preferred over single-gene PCR testing as parallel testing of multiple genes is more efficient overall in terms of turnaround times and tissue use.55,60,61 Liquid biopsy using circulating tumour DNA (ctDNA) from peripheral blood samples is also possible when insufficient tumour cells are available from tissue biopsy, patients are unable to undergo invasive tissue sampling procedures, or rapid biomarker testing results are required.18,55,60,62 However, according to the clinical experts, access to funded liquid biopsy testing in Canada is very limited and presents an ongoing challenge. Due to high false-negative rates, negative ctDNA results must be confirmed with tissue testing, but positive ctDNA results can be used to identify actionable biomarker targets.18,55,60,62
We considered the potential impacts of testing for EGFR mutations to ascertain eligibility for treatment with osimertinib in patients with locally advanced, unresectable (stage III) NSCLC, including those to health systems, patients (including families and caregivers), and costs. No new impacts are anticipated because EGFR testing is currently performed as the standard of care for patients with locally advanced, unresectable NSCLC across jurisdictions in Canada. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted for this review, and sources from the literature were validated by the review team when possible and are summarized in Table 4.
Table 4: Considerations for Testing for EGFR Exon 19 Deletions or Exon 21 (L858R) Substitution Mutations to Establish Treatment Eligibility With Osimertinib
Consideration | Criterion | Available information |
|---|---|---|
Health system related | Number of individuals in Canada expected to require the test (e.g., per year) | The sponsor estimates that in 2025, 16,107 patients with NSCLC will be tested for EGFR mutation status, which is around 77.4% of all patients with newly diagnosed cases of NSCLC. The clinical experts noted that, in Canadian clinical settings, almost all patients with NSCLC will be tested in cancer centres. Because testing for EGFR mutations is already part of the standard of care for locally advanced or metastatic NSCLC, there is no additional impact to health systems anticipated as part of establishing treatment eligibility for osimertinib. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | According to the clinical experts, testing for EGFR mutations, including exon 19 deletions and exon 21 (L858R) substitution mutations, is broadly available in all jurisdictions across Canada. | |
Testing procedure as part of routine care | According to the clinical experts, testing for EGFR mutations, including exon 19 deletions and exon 21 (L858R) substitution mutations, is currently performed during the diagnostic work-up as part of the standard of care for locally advanced, unresectable NSCLC.18,55,59,60 | |
Repeat testing requirements | According to the clinical experts, EGFR exon 19 deletions and exon 21 (L858R) substitution mutations are stable. Therefore, testing for these mutations does not need to be repeated. | |
Impacts on human and other health care resources by provision of the testing procedure | Because testing for EGFR mutations is publicly funded across jurisdictions and is currently part of the standard of care for locally advanced, unresectable NSCLC, use of the test result to establish treatment eligibility for osimertinib is not anticipated to impact human and other health care resources. | |
Patient related | Accessibility of the testing procedure in jurisdictions across Canada | Although biomarker testing is recommended for all patients with NSCLC, testing rates may be lower in rural or remote areas and among patients who are not treated by a medical oncologist.47 However, because EGFR mutation testing is part of the current standard of care, no additional impact on testing access is anticipated as part of establishing eligibility for osimertinib. |
Expected turnaround times for the testing procedure | Canadian consensus guidelines recommend turnaround times of no more than 21 calendar days for biomarker testing results.55 According to the clinical experts, the expected turnaround time for NGS is 2 weeks, although it may be closer to 4 to 6 weeks in certain cases (e.g., out of jurisdiction testing). However, the clinical experts indicated that testing is typically initiated at the time of initial diagnosis with NSCLC and results are expected to be available before initiation of osimertinib. Therefore, there is no additional impact of turnaround times to patients anticipated as part of establishing treatment eligibility for osimertinib. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | Because testing for EGFR mutations is currently part of the standard of care for locally advanced, unresectable NSCLC, there is no additional burden to patients, families, and/or caregivers anticipated from the testing as part of establishing treatment eligibility for osimertinib. | |
Clinical | Clinical utility and validity of the testing procedure | There is evidence to demonstrate the diagnostic accuracy and clinical utility of PCR and NGS testing for EGFR mutations.18,61,63,a The diagnostic accuracy of NGS-based genotyping of EGFR mutations is assumed equivalent to that of single-gene testing, and the estimated sensitivity and specificity of NGS is > 95% and 86% to 100%, respectively.63,64 According to the clinical experts, EGFR testing is relevant for patients with early-stage disease, locally advanced disease, and metastatic disease. Testing for actionable biomarkers allows clinicians to make treatment decisions regarding targeted therapies, which could improve overall survival of patients with NSCLC.18,61,63 |
Risks of harm associated with the testing procedure | The clinical experts indicated that tissue samples for EGFR testing are typically obtained through invasive procedures such as endobronchial ultrasound bronchoscopy, transthoracic biopsy, or biopsy of a metastatic site. Risks associated with these procedures include bleeding, infection, and injuries (e.g., pneumothorax).65 Because testing for EGFR mutations is currently part of the standard of care for locally advanced, unresectable NSCLC, there is no additional risk of harm associated with the testing as part of establishing treatment eligibility for osimertinib. | |
Cost | Projected cost of the testing procedure | The estimated cost of comprehensive genomic profiling using tissue testing is $1,400.66 However, because testing for EGFR mutations is currently part of the standard of care for locally advanced, unresectable NSCLC, there is no additional cost anticipated from the testing as part of establishing treatment eligibility. |
NGS = next-generation sequencing; NSCLC = non–small cell lung cancer; PCR = polymerase chain reaction.
aCanada’s Drug Agency has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 1 joint patient input submission from the LHF, LCC, and the CCSN. The LHF is a registered charity that assists and empowers people living with or caring for others with lung disease. LCC is a registered national charitable organization that supports patients through providing education, research, and advocacy. CCSN is a national network that promotes the standard of care and provides support for patients with cancer, and issues related to survivorship or quality of end-of-life care.
The information was gathered through an online survey conducted from July 2024 to December 2024 with 23 respondents identifying as living with lung cancer in Canada, including 20 patients and 3 caregivers, all of whom had experience with osimertinib. Additionally, LFH conducted interviews with 6 patients (5 females and 1 male) who completed the online survey in January 2025. Interviewees ranged in age from 49 years to 72 years. Four interviewees were diagnosed with stage IV NSCLC with an EGFR exon 19 mutation. One interviewee was diagnosed with stage IV lung cancer (not specific as to which type) with the same mutation, and another was diagnosed with stage IIIC adenocarcinoma.
Based on the patient group input, fatigue was the most detrimental physical symptom of lung cancer reported by 81% of survey respondents followed by reduced appetite or weight loss (47.6%), cough (33.3%), pain (28.6%), and shortness of breath (23.8%). Additionally, 39.1% of survey respondents specifically cited fatigue and 26.1% cited shortness of breath as the primary reasons they were unable to perform daily activities. Fatigue as the most detrimental physical symptom was also echoed by interviewees, with 3 out of 6 interviewees noting fatigue and weakness as the reason they had to stop working.
As noted in the input, lung cancer impacted multiple aspects of patients’ daily life. The most impacted daily activity was the ability to work (57.1% of survey respondents and 67% of interviewees), followed by participation in sports (38.1% of respondents), travel (33.3% of respondents), leisure or hobbies (28.6% of respondents), and housework (23.8% of respondents). Survey respondents felt their experiences with lung cancer led to negative impacts on their emotional well-being (43.5%), feeling cold (43.5%), feeling isolated (34.8%), and impacted their family relationships (26.1%). The interviewees noted that the negative impacts of having lung cancer included emotional distress, mental health challenges, concerns regarding insurance coverage (e.g., travel and car insurance), changing medications and managing side effects, and job limitations due to infection risks (e.g., exposure to viruses).
The patient groups noted that concerns surrounding personal health, financial burdens, and concern for family members were some of the reasons for experiencing anxiety. These experiences highlight the profound and intertwined physical, emotional, and social toll of lung cancer on patients, their families, and their caregivers. Many respondents noted that both their symptoms and the time it took to manage their treatments negatively impacted their ability to continue working.
Survey respondents tried a range of treatments, included targeted therapy (47.6%), chemotherapies (33.3%; such as pemetrexed and carboplatin, double platinum-based chemotherapy, and others), surgery (4.8%; such as lobectomy), radiation (4.8%), immunotherapy (4.8%), and clinical trials (4.8%). Side effects were frequently associated with current treatment options, with 81.0% of survey respondents mentioning fatigue as a side effect associated with lung cancer treatments. Two interviewees who shared their experiences with lung cancer medications reported adverse effects including fatigue, anemia, bothersome mouth feel or altered taste, joint pain, edema in extremities, nausea, and constipation. Additionally, both interviewees noted that while additional medications could be taken to manage side effects, such medications were costly. The patient groups highlighted that while current lung cancer treatments extend patients’ lives, there are still concerns around HRQoL, with patients continuing to experience residual symptoms and debilitating side effects.
When considering new medication to treat lung cancer, survey respondents identified the most important outcomes as improved HRQoL (78.3%), reduced symptoms (60.9%), improved energy (43.5%), improved symptom management (39.1%), improved and/or prolonged efficacy (26.0%), reduced cost (21.7%), reduced travel time to obtain medication or treatment (8.7%), and zero cost (4.3%). The most important outcomes reported by 2 interviewees included treatment effectiveness (i.e., extending life), improved QoL, ability to effectively manage lung cancer without destroying healthy cells, ease of administration (daily oral pill is preferred over an IV infusion), manageable side effects, and affordability.
The patient groups stated that most respondents had positive experiences with osimertinib, with 90.8% preferring or strongly preferring it to other treatments or medications they had tried. Two interviewees reported a substantial change in QoL with osimertinib, stating that it gave them the ability to have a normal life and to return to work. The 3 most common side effects of osimertinib reported by survey respondents were negative changes in appearance (e.g., hair and nail issues) (73.9%), fatigue (69.6%), and diarrhea (69.6%); however, 95.7% of respondents felt their side effects were manageable. The top 10 benefits of osimertinib reported by survey respondents included reduced or eliminated pain (36.4%), stabilization of cancer or prolonged life (27.3%), ability to exercise (27.3%), increased energy (22.7%), reduced fatigue (18.2%), increased participation in daily activities (18.2%), reduced shortness of breath (18.2%), improved appetite or weight gain (18.2%), reduced cough (13.6%), and improved mood (13.6%).
Overall, the patient groups highlighted that there is an unmet need for lung cancer therapies that prolong PFS and improve HRQoL for patients. Patients also raised consideration about accessibility and needs for equity across all provinces in diagnoses and treatments. In addition, respondents expressed concerns about their continued ability to access medications due to costs, lack of availability, and the development of resistance to those medications that work for them.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of lung cancer.
Unmet Needs
The clinical experts consulted for this review believed the overarching goal of treatment for unresectable NSCLC in patients whose tumours have EGFR mutations is to increase the proportion of patients who remain cancer-free in the long term, improve survival, reduce the risk of recurrence, and in doing so, enhance HRQoL. Clinical experts noted that conventional treatments (e.g., CRT) often lead to high rates of disease recurrence, and relapsing stage III NSCLC is generally considered incurable. Additionally, the experts flagged that patients with EGFR mutations are more likely to develop CNS metastasis compared to those with driver mutation–negative NSCLC, increasing the risk of symptoms at recurrence. Therefore, reducing relapse risk in patients with unresectable NSCLC and EGFR mutations was identified as a critical unmet need by the clinical experts consulted for this review.
Although consolidation treatment with durvalumab after CRT is currently reimbursed for patients in Canada with unresectable NSCLC, the clinical experts consulted for this review indicated that most oncologists in Canada would not administer durvalumab after CRT in patients who have an EGFR mutation as the current best evidence suggested a lack of efficacy and increased toxicity compared with placebo.
Place in Therapy
The experts felt maintenance treatment with osimertinib following the successful completion of platinum-based CRT would offer a new therapy for patients with unresectable NSCLC with EGFR mutations. Because osimertinib is currently reimbursed for relapsing cases, the clinical experts noted that using it in this setting would cause a shift in the current treatment paradigm. Currently, patients with locally advanced or metastatic disease are eligible for osimertinib as first-line treatment with palliative intent. If recurrence occurs on maintenance osimertinib or within 6 months of stopping therapy, these patients would no longer be eligible for palliative osimertinib.
Patient Population
The clinical experts noted that patients with unresectable stage III NSCLC whose tumours contain a common EGFR mutation (i.e., exon 19 deletion or exon 21 [L858R] point mutation) and who have not progressed following platinum-based CRT would be best suited for maintenance treatment with osimertinib. The clinical experts felt that any patient who received nonplatinum-based CRT should not be eligible for osimertinib treatment. The experts noted that EGFR mutations are identified with molecular testing, either using PCR or NGS, and this testing should be performed at diagnosis or during CRT treatment. Following platinum-based CRT, a CT scan must be performed to evaluate disease response or progression before initiating treatment with osimertinib. Patients who progress during or following platinum-based CRT would be eligible for osimertinib treatment as the initial therapy for recurrent disease (i.e., they would not be eligible for osimertinib under this reimbursement request).
Assessing the Response to Treatment
Outcome metrics used in clinical practice are generally aligned with those used in clinical trials. The clinical experts consulted for this review noted that the metrics most often used in clinical practice include response to treatment assessed by progression, OS, QoL (e.g., symptoms and the patients’ overall impression), and prevention of brain metastases. The experts noted that in current clinical practice, imaging is performed every 3 months to 6 months for the first 2 years following CRT and then annually thereafter. If osimertinib is reimbursed for patients with unresectable EGFR-mutated NSCLC who have not progressed during or following platinum-based CRT, the experts anticipated more frequent visits would be required when starting treatment (e.g., assessments approximately every 2 weeks for the first 3 months) and then every 3 months thereafter for the duration of treatment.
Discontinuing Treatment
Clinical experts consulted for this review indicated that osimertinib should be discontinued in the event of disease progression or significant toxicities that cannot be safely managed with dose reductions or modifications. The clinical experts consulted for this review felt that in the event of disease progression that is not amenable to any other local therapies, patients should be able to continue treatment with osimertinib if there is evidence of ongoing clinical benefit. The clinical experts noted that if a patient discontinued maintenance osimertinib treatment in the absence of recurrence, they should be eligible for first-line osimertinib treatment in the relapsed setting if at least 6 months have passed since discontinuation and recurrence.
Prescribing Considerations
Clinical experts indicated that treatment with osimertinib should be prescribed and managed by a physician familiar with managing systemic lung cancers (i.e., medical oncologist or pulmonologist). In some regions, care may be supervised by another physician (e.g., a general practitioner) under direction from a physician trained in managing systemic anticancer therapies.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
CDA-AMC received 2 clinician group input submissions, from LCC MAC and OH-CCO Lung Cancer Drug Advisory Committee. LCC is a national charity with the purpose of increasing awareness about lung cancer, providing support to patients with lung cancer, research, and advocacy. The LCC MAC consists of clinicians in the field of lung cancer across the country. The OH-CCO Cancer Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate. In total 27 clinicians contributed to this submission, 23 from LCC MAC and 4 from the OH-CCO Lung Cancer Drug Advisory Committee.
Both clinician groups and the clinical experts consulted for this review agreed that the current standard treatment for patients with unresectable, locally advanced NSCLC with EGFR mutations is cCRT. Clinician groups agreed that while consolidation immunotherapy with durvalumab is currently Health Canada approved and provincially funded for unresectable NSCLC agnostic of PD-L1 expression or EGFR mutation, it is not used in practice or recommended as conventional treatment for patients with EGFR mutations. Post hoc subgroup analyses of the PACIFIC trial revealed that patients with EGFR mutations receiving durvalumab had similar efficacy and increased toxicity compared to those receiving placebo. As such, both clinician groups agreed that currently there are no targetable treatments approved for patients with unresectable stage III NSCLC with EGFR mutations. Clinician groups noted that the goal of treatment is to cure the disease, as measured by OS, PFS, and disease-free survival, and LCC MAC added delaying disease progression in extrathoracic sites as a critical secondary goal.
The OH-CCO Lung Cancer Drug Advisory Committee explained patients with stage III (per standard Canadian staging techniques) unresectable NSCLC with EGFR exon 19 deletion or exon 21 (L858R) substation mutation who do not have disease progression by standard restaging techniques within 6 weeks of completion of platinum-based CRT (either concurrently or sequentially), with no contraindication to EGFR TKIs, no history of interstitial lung disease before CRT, and no evidence of symptomatic pneumonitis following definitive CRT are best suited for treatment with osimertinib. The OH-CCO flagged that while one of the criteria is restaging within 6 weeks of completion of CRT, there should be some flexibility in this criterion as some centres have issues with CT scan wait times. While the OH-CCO Lung Cancer Drug Advisory Committee indicated that the reimbursement of molecular EGFR testing may not be universal across the country and additional funding may be needed for those provinces that do not currently have provincial reimbursement, LCC MAC indicated that they anticipated no increase in cost for testing for EGFR rearrangements to accommodate maintenance use of osimertinib.
LCC MAC stated that the outcomes to determine whether a patient is responding to treatment in clinical practice included PFS. LCC MAC suggested including the use of scans (CT of the body with or without brain imaging) and symptoms to monitor recurrence or progression with a frequency of every 3 months to 4 months while the OH-CCO Lung Cancer Drug Advisory Committee suggested restaging investigations every 3 months to 6 months.
Both clinician groups agreed that treatment should be discontinued if there is disease recurrence or progression, intolerance, or clinically important toxicity, and the OH-CCO Lung Cancer Drug Advisory Committee further added that treatment should be discontinued upon a patient’s request.
Clinician groups agreed that osimertinib can be delivered in the medical oncology outpatient setting. The OH-COO Lung Cancer Drug Advisory Committee further specified that osimertinib should be managed under the supervision of a specialist or medical oncologist with training in systemic therapy for thoracic malignancies.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The LAURA trial randomized patients to osimertinib or placebo in a 2:1 ratio. Observation is an appropriate comparator for EGFR mutation–positive patients after CRT is complete for unresectable stage III NSCLC. | This was a comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
|
|
PAG is confirming that only patients with exon 19 and exon 21 EGFR-positive disease would be eligible for osimertinib and that other EGFR exon–positive patients would not be included (i.e., exon 20). | This was a comment from the drug programs to inform pERC deliberations. |
Considerations for discontinuation of therapy | |
In the trial, participants were able to continue osimertinib beyond disease progression if the investigator thinks there will be continued clinical benefit. What should be the discontinuation criteria for osimertinib? | The clinical experts consulted for this review felt in the event of disease progression that is not amenable to any other local therapies, patients should be able to continue treatment with osimertinib if there is evidence of ongoing clinical benefit. Patients in the LAURA trial were able to continue to receive osimertinib after blinded independent central review–confirmed progression, if in the opinion of the treating physician, they were continuing to derive clinical benefit. Therefore, the experts felt that osimertinib should be discontinued in the event of significant toxicities that cannot be safely managed with dose reductions or modifications or disease progression that is deemed by the treating physician to be amenable to other local therapies. |
Considerations for prescribing of therapy | |
Osimertinib is dosed at 80 mg orally once daily. Jurisdictions have familiarity with osimertinib as it is currently used in early-stage and metastatic EGFR-positive NSCLC. This is to enable implementation. | This was a comment from the drug programs to inform pERC deliberations. |
Generalizability | |
|
|
At time of implementation: For patients who have completed CRT and are now on observation, is there a maximum time frame that has to elapse between end of CRT and initiation of osimertinib? | The clinical experts consulted for this review felt osimertinib treatment should begin within 10 weeks of completing platinum-based CRT as this time frame would allow for sufficient time for patients to recover from CRT and receive reimaging scans while still allowing for maximum clinical benefit. |
Funding algorithm | |
Request an initiation of a rapid provisional funding algorithm. | This was a comment from the drug programs to inform pERC deliberations. |
Drug may change place in therapy of drugs reimbursed in subsequent lines. | This was a comment from the drug programs to inform pERC deliberations. |
PAG is informing pERC that if a patient has progressed while on osimertinib therapy or progressed within 6 months of stopping osimertinib for the indication under review, the patient will not be eligible for osimertinib in stage IV setting. | This was a comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Osimertinib requires monitoring of adverse events and ECG monitoring. | This was a comment from the drug programs to inform pERC deliberations. |
EGFR mutation testing is required to identify eligible patients for osimertinib. | This was a comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
PAG has identified osimertinib to have a high budget impact for the requested indication. | This was a comment from the drug programs to inform pERC deliberations. |
cCRT = concurrent chemoradiation therapy; CRT = chemoradiation therapy; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; NSCLC = non–small cell lung cancer; PAG = Provincial Advisory Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; sCRT = sequential chemoradiation therapy; TKI = tyrosine kinase inhibitor.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of osimertinib 80 mg oral tablets taken once daily in the treatment of locally advanced, unresectable (stage III) NSCLC in patients whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT. The focus will be placed on comparing osimertinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of osimertinib is presented in one section with our critical appraisal of the evidence included at the end of the section. The systematic review section includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this section using the GRADE approach follows the critical appraisal of the evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal study identified in a systematic review.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Characteristics of the included study are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | LAURA |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, double-blind, placebo-controlled, multicentre, international study |
Locations | A total of 74 study sites randomized at least 1 patient into the global cohort from 16 countries across Europe, Asia-Pacific, North America, and South America. There were no test sites in Canada. |
Patient enrolment dates | Start date: July 19, 2018 End date: July 25, 2022 |
Randomized (N) | Randomized (full analysis set) N = 216
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Osimertinib 80 mg orally once daily until objective radiological disease progression occurred as defined by RECIST 1.1 and as confirmed by BICR or until another discontinuation criterion was met. |
Comparator | Placebo tablets, oral, once daily until objective radiological disease progression occurred as defined by RECIST 1.1 and as confirmed by BICR or until another discontinuation criterion was met. |
Study duration | |
Screening phase | 28 days |
Treatment phase | Patients received randomized study treatment until objective radiological disease progression occurred as defined by RECIST 1.1 and as confirmed by BICR, or until another discontinuation criterion was met. |
Follow-up phase | Patients with BICR-confirmed progression per RECIST 1.1 entered survival follow-up and were contacted every 12 weeks to assess survival status. |
Outcomes | |
Primary end point | PFS using BICR assessment |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Lu S, Kato T, Dong X, et al. Osimertinib after Chemoradiotherapy in Stage III EGFR-Mutated NSCLC. N Engl J Med 2024;391(7):585-597. Lu S, Ahn MJ, Reungwetwattana T, et al. Osimertinib after definitive chemoradiotherapy in unresectable stage III epidermal growth factor receptor-mutated non-small-cell lung cancer: analyses of central nervous system efficacy and distant progression from the phase III LAURA study. Ann Oncol. Published online September 11, 2024. Lu S, Casarini I, Kato T, et al. Osimertinib Maintenance After Definitive Chemoradiation in Patients With Unresectable EGFR Mutation Positive Stage III Non-small-cell Lung Cancer: LAURA Trial in Progress. Clin Lung Cancer. 2021;22(4):371-375. NCT03521154 |
AE = adverse event; BICR = blinded independent central review; cCRT = concurrent chemoradiotherapy; CNS = central nervous system; CRT = chemoradiation therapy; CTCAE = Common Terminology Criteria for Adverse Events; CYP = cytochrome P450; DCR = disease control rate; DoR = duration of response; ECG = electrocardiogram; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; HRU = health resource use; IASLC = International Association for the Study of Lung Cancer; ILD = interstitial lung disease; NSCLC = non–small cell lung cancer; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS2 = time to second progression; PGI-S = Patient Global Impressions Scale–Severity; PRO = patient-reported outcomes; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; sCRT = sequential chemoradiation therapy; TFST = time to first subsequent therapy; TKI = tyrosine kinase inhibitor; TSST = time to subsequent treatment; TTD = time to treatment discontinuation; TTDM = time to death or distant metastases.
aPatients with a stage III tumour of squamous histology who had a pre-existing local positive test result (exon 19 deletion or L858R), irrespective of EGFR test used or lab accreditation, and confirmed by central testing during part I screening, were eligible for part II screening.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The LAURA trial is an ongoing, multinational, phase III, double-blinded, placebo-controlled, randomized study aimed at evaluating the efficacy and safety of osimertinib as maintenance therapy in patients with locally advanced, unresectable stage III NSCLC with centrally confirmed EGFR mutations (exon 19 deletions or exon 21 [L858R] substitutions) whose disease has not progressed during or following definitive platinum-based CRT. Patients were recruited from 74 international sites with none coming from Canada. Patients without a pre-existing approved local EGFR test result were required to undergo part I screening in which they provided a recent tumour tissue sample for central prospective EGFR mutation testing. All patients underwent part II screening to confirm eligibility, stage the disease, and confirm progression during or following platinum-based CRT. All patients who met the criteria for inclusion were randomized within 6 weeks of completion of CRT. Patients were randomized in a 2:1 ratio using an interactive voice or web response system to receive either osimertinib or placebo; no rationale for randomizing patients 2:1 was provided. Randomization was stratified by prior chemoradiation strategy (cCRT versus sCRT), tumour stage before chemoradiation (stage IIIA versus stage IIIB or stage IIIC) and China cohort (enrolled at a Chinese site and patient declaring themselves of Chinese ethnicity versus enrolled at non-Chinese site or patient declaring themselves of non-Chinese ethnicity) to allow separate randomization for China.
The primary objectives were to assess the PFS benefit of osimertinib compared to placebo. Key secondary objectives were to assess the OS and CNS PFS benefit of osimertinib treatment compared to placebo. All results presented here are from the January 5, 2024, DCO date, except for the OS outcome, for which data are based on the January 5, 2024, DCO date and an unplanned, updated analysis using the November 29, 2024, DCO date; the latter DCO date was requested as part of the regulatory marketing application.
Figure 1: Schematic of LAURA Trial Study Design
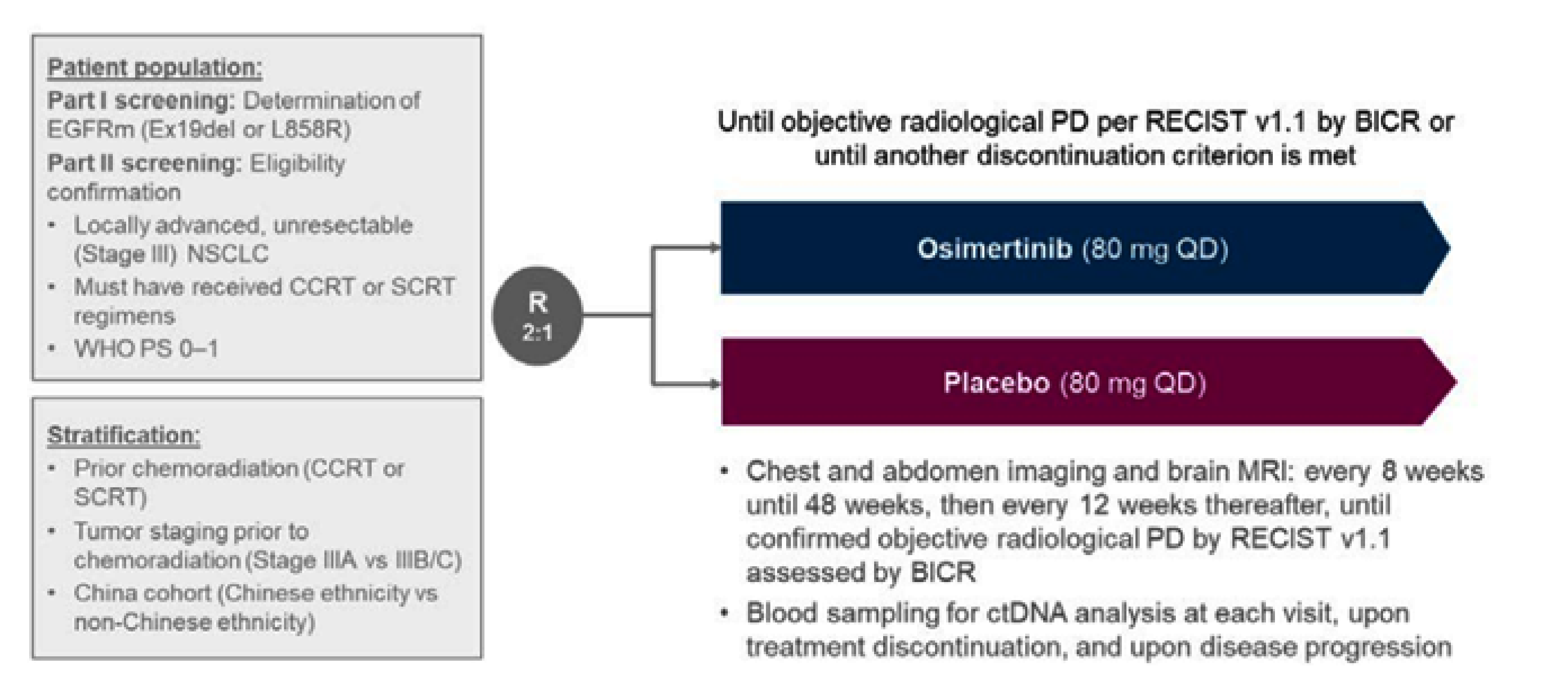
BICR = blinded independent central review; CRT = chemoradiation therapy; CCRT = concurrent chemoradiation therapy; ctDNA = circulating tumour DNA; EGFRm = epidermal growth factor receptor mutation; NSCLC = non–small cell lung cancer; PD = progressive disease; PS = performance status; QD = every day; RECIST = Response Evaluation Criteria in Solid Tumours; SCRT = sequential chemoradiation therapy; vs = versus.
Source: LAURA Clinical Study Report.39
Populations
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria are provided in Table 6. Briefly, the trial enrolled patients aged 18 years or older (aged ≥ 20 years in Japan) with histologically documented NSCLC of predominantly nonsquamous pathology who present with locally advanced, unresectable (stage III) disease (according to Version 8 of the International Association for the Study of Lung Cancer Staging Manual in Thoracic Oncology). Only patients with local or central laboratory confirmation that the tumour harboured 1 of the 2 common EGFR mutations known to be associated with EGFR TKI sensitivity (exon 19 deletion and/or exon 21 [L858R] substitutions), either alone or in combination with other EGFR mutations, and without disease progression during or following platinum-based cCRT or sCRT were eligible for randomization. Patients with mixed small cell and NSCLC histology, history of interstitial lung disease before CRT, symptomatic pneumonitis following chemoradiation, unresolved toxicity with a grade greater than 2, cardiac irregularities, inadequate bone marrow reserve or organ function, history of other malignancies, severe or uncontrolled systemic diseases, or any resection that would preclude adequate absorption of osimertinib were excluded from the study. In addition, patients with prior treatment for NSCLC outside of that received in the definitive setting for stage III disease, prior treatment with EGFR TKI therapy, major surgery within 4 weeks of the first dose of the study drug, and patients currently receiving and unable to stop medications or herbal supplements known to be strong inducers of cytochrome P450 3A4 were ineligible for randomization.
Interventions
In the LAURA trial, patients were randomized to 1 of 2 treatment groups to receive:
osimertinib 80 mg oral tablets, once daily, or
placebo oral tablets, once daily.
Placebo tablets were identical and presented in the same packaging as osimertinib to ensure blinding of the medication. Osimertinib or placebo was administered orally, once daily, commencing on day 1. Doses were to be taken approximately 24 hours apart at the same time each day without interruption. If a patient missed taking a scheduled dose, within a window of 12 hours, it was acceptable to take the dose. If it was more than 12 hours after the scheduled dose time, the missed dose was not to be taken, and patients were instructed to take the next dose at the next scheduled time. The initial dose of osimertinib and placebo was 80 mg every day. In the event of grade 3 or higher toxicity and/or unacceptable toxicity (any grade) not attributable to the disease or disease-related processes under investigation and deemed by the investigator to possibly be related to the study drug, the study drug could be interrupted and supportive therapy administered according to local practice or guidelines. If a toxicity resolved or reverted to less than grade 2 within 3 weeks of interruption, treatment with the study drug could be restarted at the same dose (i.e., 80 mg) or a lower dose (osimertinib 40 mg or matching placebo) with discussion and agreement with the AstraZeneca Study Team Physician, as needed. Once the dose of osimertinib or placebo was reduced to 40 mg every day, the patient was to remain on the reduced dose until termination from study treatment. If the toxicity did not resolve to grade 2 or less after 3 weeks, then the patient was withdrawn from the study drug and observed until resolution of the toxicity. In the event of any of the following, the patient was discontinued from the study treatment:
patient decision
patient experiencing any of the following AEs:
grade 3 or higher pneumonitis
grade 2 pneumonitis where symptoms did not resolve within 4 weeks after interrupting study treatment
recurrent symptomatic pneumonitis following prior dose interruption and study treatment rechallenge
QTc interval prolongation with signs or symptoms of serious arrhythmia
grade 3 or higher adverse reaction that did not improve to grade 0, 1, or 2, and treatment was not restarted within 3 weeks of interrupting study treatment
any AE that, in the opinion of the investigator or AstraZeneca, contraindicates further dosing
severe noncompliance with the Clinical Study Protocol as judged by the investigator and/or AstraZeneca representative
pregnancy
initiation of alternative anticancer therapy including another investigational agent
before primary PFS analysis: objective disease progression as per RECIST Version 1.1 assessed by BICR
postprimary PFS analysis: disease progression as assessed by investigator
patients who were incorrectly initiated on study treatment.
Patients who discontinued from the study treatment were to continue attending subsequent study visits and data collection continued according to protocol. If the patient did not agree to continue in-person study visits, a modified follow-up was arranged to ensure the collection of end points and safety information including new AEs and follow-up on any ongoing AEs and concomitant medications. Patients who agreed to modified follow-up were not considered to have withdrawn consent or to have withdrawn from the study. Before the primary PFS analysis, patients who discontinued the study drug for reasons other than RECIST Version 1.1–defined progression as assessed by BICR, continued assessments in accordance with the protocol. After the primary PFS analysis, patients who were in progression at follow-up continued to provide survival follow-up (every 12 weeks until death, withdrawal of consent, or the end of the study defined as at the time of final OS analysis), but had progression assessed in accordance with local clinical practice. Formal RECIST Version 1.1 measurements, electronic patient-reported outcomes, health resource use, WHO performance status, ctDNA, and blood borne biomarkers were no longer collected.
Following BICR-confirmed progression per RECIST Version 1.1, patients could receive open-label osimertinib if:
progression was confirmed by BICR, or, if progression occurred after the primary PFS analysis, investigator-assessed progression had been diagnosed
in the opinion of the treating physician, they were continuing to derive clinical benefit (for patients assigned to the osimertinib treatment group), or treatment was in accordance with local clinical practice at the judgment of their treating physician (for patients assigned to the placebo group)
they had not received any other anticancer therapy following the discontinuation of study treatment; palliative radiotherapy was allowed
disease extent had been characterized by CT or MRI scan at the time of progression.
All patients receiving open-label osimertinib postprogression entered survival follow-up data. Treatment with open-label osimertinib continued until the patient stopped deriving clinical benefit (as judged by the investigator).
Medication which is considered necessary for the patient’s safety and well-being could be given at the discretion of the investigator and recorded in the source document and appropriate sections of electronic case report form (eCRF). Once enrolled, all patients had to try to avoid concomitant use of medications, herbal supplements, and/or ingestion of foods that are strong inducers of cytochrome P450 3A4. Other anticancer therapies, investigational agents, and radiotherapy were not to be given while the patient initiated the study drug. Premedications were allowed after, but not before, the first dose of study drug. These included medications for the management of diarrhea, nausea, and vomiting, which were to be administered as directed by the investigator. Drugs that prolong the QT interval and have a known risk of torsades de pointes, even when taken as recommended, must have been discontinued before the start of administration of study treatment and were not to be coadministered with osimertinib or placebo for 2 weeks after discontinuing the study treatment.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
The following efficacy and HRQoL outcomes were included in the GRADE assessment: OS, PFS, CNS PFS, and the EORTC QLQ-C30. Select harms outcomes considered important for expert committee deliberations were also assessed using GRADE: pneumonitis, grade 3 or greater diarrhea, withdrawal due to AEs, and fatal AEs. These outcomes were deemed the most important for determining the comparative efficacy and safety of osimertinib compared to the current standard of care (i.e., watchful waiting).
Other outcomes were summarized in the review report to aid clinical decision-making but not appraised using GRADE, including: TTDM, TTD, response, and EORTC QLQ-LC13. While TTDM and TTD are informative for understanding the length of time to distant metastases, death, and treatment discontinuation, the CDA-AMC review team in consultation with the clinical experts consulted for this review, felt survival outcomes (i.e., OS, PFS, and CNS PFS) were more informative for clinical decision-making and captured outcomes most important to patients. The EORTC QLQ-C30 was appraised using GRADE as it is a generic measure that provides a comprehensive picture of patients’ HRQoL. The EORTC QLQ-LC13 focuses on lung cancer–specific symptoms and results were reported in this report.
Objective response rate (ORR) and duration of response (DoR) results from the LAURA trial are included in the appendix of this report as supportive evidence. The clinical experts cautioned that response outcomes may be confounded by prior chemoradiation and felt that for treatment selection PFS provides more relevant informative.
Time to second progression was deemed less clinically meaningful for decision-making and was not included in this report. Time to second progression was not identified as most important to guide treatment selection by the clinical experts consulted for this review and the relationship between this outcome and those deemed important by clinical experts is poor. Further, time to second progression may bias the intervention group due to the higher likelihood of remaining on study treatment.
While the Patient Global Impressions Scale–Severity and the EQ-5D-5L were assessed as exploratory objectives in the LAURA trial, the clinical experts consulted for this review felt the EORTC QLQ-C30 and EORTC QLQ-LC13 were the most informative HRQoL outcomes for decision-making and thus data on the Patient Global Impressions Scale–Severity and EQ-5D-5L are not reported in this Clinical Study Report. This is because the EORTC QLQ-C30 and EORTC QLQ-LC13 capture a comprehensive range of disease-specific symptoms, functioning impairments, and treatment-related effects that are particularly important for patients with locally advanced unresectable stage III NSCLC. In comparison, the Patient Global Impressions Scale–Severity and the EQ-5D-5L do not offer the same detailed, condition-specific insights into the HRQoL impacts of treatment and disease progression.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | End point | Outcome included in the CDA-AMC GRADE assessment? |
|---|---|---|---|
OS | At PFS IA (January 5, 2024), at OS nonplanned analysis (November 29, 2024), and at 12, 24, and 36 months | Key secondary | At 36 and 48 months (November 29, 2024, DCO date) |
PFS | At PFS IA (January 5, 2024) and at 6, 12, 18, 24, and 36 months | Primary | At 12 and 36 months |
CNS PFS | At PFS IA (January 5, 2024) and at 12 and 24 months | Key secondary | At 12 and 24 months |
TTDM | At PFS IA (January 5, 2024) | Secondary | No |
TTD | At PFS IA (January 5, 2024) | Secondary | No |
EORTC QLQ-C30 | At PFS IA (January 5, 2024) | Secondary | Mean change in global health status/QoL over all weeks |
EORTC QLQ-LC13 | At PFS IA (January 5, 2024) | Secondary | No |
Harms | At PFS IA (January 5, 2024) | Harms | Pneumonitis, grade ≥ 3 diarrhea, withdrawal due to AEs, and fatal AEs |
AE = adverse event; CDA-AMC = Canada's Drug Agency; CNS = central nervous system; DCO = data cut-off; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; GRADE = Grading of Recommendations, Assessment, Development and Evaluations; IA = interim analysis; OS = overall survival; PFS = progression-free survival; QoL = quality of life; TTD = time to treatment discontinuation or death; TTDM = time to death or distant metastases.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Tumour assessments included CT scan (preferred) or MRI with IV contrast of the chest and abdomen (including the entire liver and both adrenal glands). Tumour assessments were performed at screening (within 28 days before randomization), then after 8 weeks (± 1 week) relative to randomization for the first 48 weeks, and then every 12 weeks (± 1 week) until objective radiological disease progression per RECIST Version 1.1 as confirmed by BICR. Assessments occurred irrespective of whether the patient was receiving study treatment or had previously discontinued treatment for another discontinuation criterion and may have started alternative anticancer treatment. Additional scans were performed if clinically indicated (i.e., if disease progression was suspected).
EORTC QLQ-C30 was assessed at randomization, at week 4 and 8 (relative to randomization), thereafter every 8 weeks (± 3 days) until treatment discontinuation, and then at week 8 (± 3 days), week 16 (± 3 days), and week 32 (± 3 days) relative to the treatment discontinuation visit. EORTC QLQ-LC13 was assessed at randomization, weekly up to week 8 (relative to randomization), and thereafter every 4 weeks (± 3 days) until treatment discontinuation. For patients who discontinued study treatment before BICR-confirmed progression, patient-reported outcomes were collected at the study treatment discontinuation visit and continued to be collected at the same frequency as the treatment period during progression follow-up until BICR-confirmed disease progression, then at the disease progression visit and at week 8 (± 3 days), week 16 (± 3 days), and week 32 (± 3 days; relative to the disease progression visit).
AEs were assessed during the screening period, at randomization, and then at every visit including postprogression visits.
Overall Survival
OS was one of the key secondary outcomes of the LAURA trial and was defined as the time from randomization to death due to any cause.
Clinical experts consulted for this review advised an OS between-group difference of 5% to 10% is considered clinically meaningful.
Progression-Free Survival
PFS was the primary outcome of the LAURA trial based on BICR assessment according to RECIST Version 1.1 and was defined as the time from randomization until the date of objective disease progression or death (by any cause in the absence of progression), regardless of whether the patient withdrew from study treatment or received another anticancer therapy before progression.
Clinical experts consulted for this review advised a PFS between-group difference of 10% to15% is considered clinically meaningful.
CNS Progression-Free Survival
CNS PFS based on BICR assessment according to RECIST Version 1.1 was the second key secondary outcome in the LAURA trial and was defined as the time from randomization until the date of CNS objective disease progression or death (by any cause in the absence of CNS progression), regardless of whether the patient withdrew from randomized therapy or received another anticancer therapy before CNS progression.
Clinical experts consulted for this review advised a CNS PFS between-group difference of 10% is considered clinically meaningful.
Time to Death or Distant Metastases
TTDM was one of the secondary outcomes of the LAURA trial and was defined as the time from randomization until the first date of distant metastasis or date of death in the absence of distant metastasis. Distant metastasis was defined as any new lesion that was detected on a scan that was anywhere other than the lung or regional lymph node according to RECIST Version 1.1 or proven by biopsy.
Clinical experts consulted for this review advised a TTDM between-group difference of 3 months to 6 months is considered clinically meaningful.
Time to Treatment Discontinuation or Death
TTD was one of the secondary outcomes of the LAURA trial and was defined as the time from randomization to the earlier of the date of study treatment discontinuation (regardless of the reason for study treatment discontinuation) or death.
Clinical experts consulted for this review advised a TTD between-group difference of 3 months to 6 months is considered clinically meaningful.
HRQoL End Points
To compare disease-related symptoms and HRQoL, the EORTC QLQ-C30 and EORTC QLQ-LC13 were collected as other secondary outcomes in the LAURA trial.67,68 Descriptions of each measure are as follows (further details in Table 8).
The EORTC QLQ-C30 is a multidimensional, cancer-specific, self-administered, measure of HRQoL consisting of 30 questions across 5 multi-item functional scales (physical, role, emotional, cognitive, and social); 3 multi-item symptom scales (fatigue, pain, and nausea and vomiting); 5 single items assessing additional symptoms commonly reported by patients with cancer (dyspnea, loss of appetite, insomnia, constipation, and diarrhea); a 2-item global measure of health status/QoL; and a single item on the financial impact of the disease. Each item is evaluated using 4-point and 7-point Likert scales, raw scores for each scale are computed as the average of the items that contribute to a particular scale, and each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. Higher scores in symptom scales reflect more symptoms or problems, while higher scores in functional scales reflect healthier functioning.69 A higher score on the global scale reflects better HRQoL. The EORTC QLQ-C30 was scored according to the EORTC QLQ-C30 scoring manual.69
The EORTC QLQ-LC13 is a 13-item lung cancer–specific module used to assess lung cancer symptoms, treatment-related side effects, and pain medication. Except for a multi-item scale for dyspnea, all are single items. The dyspnea scale was only to be used if all 3 items were scored; otherwise, the items were treated as single-item measures.
The average change in scores from baseline across all visits were evaluated for the scales or items from EORTC QLQ-C30 and EORTC QLQ-LC13 and results are included in this Clinical Study Report.
A between-group MID in the EORTC QLQ-C30 global health status/QoL score has previously been estimated for patients with lung cancer as an increase or decrease of 4 points for improvement and deterioration, respectively.70 No MIDs for the EORTC QLQ-LC13 in patients with NSCLC were identified.
Harm End Points
To assess the safety and tolerability profile of osimertinib compared with placebo, AEs were collected throughout the study, from the date of informed consent until 28 days after the last dose of study treatment. The Medical Dictionary for Regulatory Activities Version 26.1 was used to code the AEs. AEs were also graded for severity according to the Common Terminology Criteria for Adverse Events Version 5.0. AE data were evaluated according to the following categories and results are included in this Clinical Study Report: all AEs, SAEs, withdrawals due to AEs, AEs with an outcome of death, and AEs of special interest. An AE of special interest is one of scientific and medical interest specific to understanding of the investigational product and may be serious or nonserious. The AEs of special interest in the LAURA trial included those listed in the product monograph and highlighted as important by clinical experts consulted for this review: lung disease including pneumonitis and radiation pneumonitis, cardiac effects (e.g., QT interval corrected using the Fridericia formula interval prolongation, left ventricular dysfunction, and cardiomyopathy), and grade 3 or greater diarrhea.
Clinical experts consulted for this review advised that a 5% between-group difference in the occurrence of pneumonitis and fatal AEs and a 10% between-group difference in grade 3 or greater diarrhea and withdrawal due to AEs is considered clinically meaningful.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | Cancer-specific self-reported measure of HRQoL. A 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, cognitive, and social); 3 multi-item symptom scales (fatigue, pain, and nausea and vomiting); and 7 single items (including dyspnea, appetite loss, sleep disturbance, constipation, and diarrhea); these are measured on a 4-point response scale. The instrument also contains an item assessing the perceived financial impact of the disease and treatment and two 7-point response scales pertaining to global health and QoL. A higher score for functional scales and for GHS represents better functioning ability or HRQoL. A higher score for symptom scales represents worse symptoms.67 | In studies with patients with lung cancer: Validity: Moderate to strong scale-to-scale correlations between the 5 EORTC QLQ-C30 functioning scales (r = 0.41 to 0.77); FACT-G global and physical well-being scale and EORTC QLQ-C30 scales (r = 0.40 to 0.64)71; HADS with all EORTC QLQ-C30 functioning scales (r = 0.28 to 0.75)72; and BPI scales with all EORTC QLQ-C30 scales except for nausea and vomiting (r = 0.20 to 0.72),72 supporting convergent validity. Known-groups approach: able to differentiate across different measures of cancer severity including cancer stages (effect size = 0.49); ECOG PS (effect size = 0.65); and self-reported health status (effect size = 1.39).71 Internal consistency reliability: Cronbach alpha ranging from 0.57 to 0.87 with 7 scales having acceptable internal consistency (alpha > 0.70).73 Responsiveness: Group differences (improved versus deteriorated based on ECOG PS) over 28 days between pretreatment and on-treatment periods showed a statistically significant difference in global QoL (P < 0.01) scale. No such difference was identified in patients whose ECOG PS remained unchanged.68 | A recent study25 analyzed MIDs for the EORTC QLQ-C30 using data from 21 phase III trials involving 13,015 patients across 9 cancer types (brain, colorectal, advanced breast, head/neck, lung, mesothelioma, melanoma, ovarian, and prostate). It found the following anchor-based MIDs for patients with lung cancer.
|
EORTC QLQ-LC13 | The EORTC QLQ-LC13 is a 13-item lung cancer–specific questionnaire. It is typically used in conjunction with the core questionnaire, the EORTC QLQ-C30. The module comprises a multi-item scale to assess dyspnea, and a series of single items assessing pain, coughing, sore mouth, dysphagia, peripheral neuropathy, alopecia, and hemoptysis. All of the scales and single-item measures range in score from 0 to 100. A high score for the scales and single items represents a high level of symptomatology or problems.67,68 | Validity: Construct validity has been established between pain score and disease type (P < 0.001). Also, based on ECOG PS, construct validity was confirmed in dyspnea, coughing, and pain (P < 0.001) scores.68 Correlation between spirometry result and dyspnea score was found to be weak (r = 0.24). BPI intensity score and EORTC QLQ-LC13 pain score were found to be modestly correlated (r > 0.4).72 Reliability: Good internal consistency reliability for the dyspnea multi-item scale (alpha = 0.81).68 Responsiveness: Dyspnea, coughing, and pain scores improved significantly over time between pretreatment and on-treatment period (P < 0.001 for all except for extrathoracic pain, which showed P < 0.05). Responsiveness of chest pain (P < 0.01), dyspnea (P < 0.001), and coughing (P < 0.001) to change in ECOG PS was also noted.68 | No relevant studies on MID in patients with NSCLC were identified. |
BPI = Brief Pain Inventory; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; FACT-G = Functional Assessment of Cancer Therapy–General; GHS = global health status; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; MID = minimal important difference; NA = not applicable; NSCLC = non–small cell lung cancer; QoL = quality of life.
Statistical Analysis
The statistical analyses for all study end points included in this review are presented in Table 9.
Sample Size and Power Calculation
The LAURA trial planned to enrol approximately 1,333 patients to randomize approximately 200 eligible patients. Of those it was planned that approximately 30 to 40 patients would be recruited in China to satisfy the China Regulatory Authority requirements. The primary analysis was to occur when approximately 120 PFS BICR events (approximately 60% maturity) had been observed in the global cohort. With 120 PFS BICR events, the study had 90% power to show a statistically significant difference in PFS at the 2-sided 5% level if the assumed true treatment effect HR was 0.53; this would translate to an approximate 7-month improvement from a median 8-month PFS on placebo. The smallest treatment difference that would be statistically significant is a PFS HR of 0.68 (translating to an approximate 4-month improvement).
Statistical Testing
The primary end point (PFS), key secondary end points (OS and CNS PFS), TTDM, and TTD were analyzed using a stratified log-rank test stratified by prior chemoradiation strategy (cCRT versus sCRT), disease stage before chemoradiation strategy (stage IIIA versus stage IIIB or stage IIIC), and China cohort (enrolled at a Chinese site and patient declaring themselves of Chinese ethnicity versus enrolled at non-Chinese site or patient declaring themselves as non-Chinese ethnicity) for generating the P value. The effect of treatment was estimated using a HR and 2-sided 95% CI. OS and CNS PFS were only analyzed if there were a sufficient number of events defined as more than 20 OS events (with at least 5 events per group) and at least 20 CNS PFS events across both treatment groups.
The assumption of proportionality in the primary analysis was assessed by examining the plots of complementary log–log (event times) versus log (time) and, if necessary, a time-dependent covariate was to be fitted to assess the extent to which random variation occurred. If a lack of proportionality was evident, the variation in treatment effect was described by presenting a piecewise HR calculated over distinct time periods. If lack of proportionality was found, which may be a result of a treatment-by-covariate interaction, it was investigated. If the resulting strata were too small (i.e., < 10 events) the strata were to be collapsed in the following predefined order to allow analysis: the China cohort strata were collapsed first, followed by prior chemoradiation strategy, and finally stage before chemoradiation. In the event of nonproportionality, the HR was to be interpreted as an average HR over the observed extent of follow-up.
Additionally, KM plots of OS, PFS, and CNS PFS were presented by treatment group. Summaries of the number and percentage of patients experiencing an event and the type of event were provided along with median time to event and associated 95% CIs for each treatment group. PFS rates at 6-month intervals, OS rates at set time intervals (e.g., 12 months and 24 months), and CNS PFS at 12 months and 24 months along with 95% CIs were estimated for each treatment group using the KM method.
Multiple Testing Procedure
To control for type I error rate at the 5% two-sided level, a sequential MTP was used for the primary end point (PFS) and key secondary end points (OS and CNS PFS). If any previous analysis in the sequence was not statistically significant, the alpha could not be transferred to subsequent analyses. Hypotheses were tested using a MTP with an alpha recycling strategy.74 With this approach, hypotheses were tested in the predefined order of PFS, OS, and CNS PFS.
The primary end point of BICR-assessed PFS and the key secondary efficacy end point of CNS PFS were planned to be analyzed only once, at the time of the primary analysis, when approximately 120 BICR-assessed PFS events had been observed in 200 patients (approximately 60% data maturity). Two analyses of the key secondary end point of OS were planned: 1 interim analysis at the time of the primary PFS analysis, and 1 final OS analysis when approximately 120 death events have been reported in 200 patients (approximately 60% data maturity). The alpha (2-sided 5%) level allocated to OS is controlled at the interim and final analyses by using the Lan-DeMets spending function that approximates an O’Brien-Fleming approach, where the alpha level applied at the interim and final analyses depends on the number of death events observed. This approach maintains an overall 2-sided 5% type I error across the 2 planned analyses of OS.
At the primary PFS analysis (January 5, 2024, DCO date), PFS was tested and declared statistically significant with a data maturity of 55.6% (120 PFS events in 216 patients). As per the MTP, OS was tested at this same DCO date and failed to meet the prespecified boundary for declaring statistical significance (P < 0.00036 was required to declare statistical significance at the interim analysis). As OS failed to reach statistical significance, CNS PFS statistical significance was not tested for drawing inferences.
All other clinical outcomes (e.g., TTDM and TTD), HRQoL outcomes, and harms data were not included in the MTP.
Data Imputation Methods
If a patient was known to have died where only a partial death date was available, then the date of death was imputed as the latest of the last date known to be alive plus 1 from the database and the death date using the available information provided as follows:
for missing day only, the first of the month was used
for missing day and month, January 1 was used.
If there is evidence of death but the date is entirely missing, it was treated as missing (i.e., censored at the last known alive date).
Missing safety data were generally not imputed; however, if the value was above or below the limit of quantification, the value was imputed to equal the limit. Missing start dates for concomitant medication, radiotherapy, and AE start dates were imputed as follows.
If the year was missing, the first dose date was imputed unless the end date suggested it could have started before this in which case January 1 of the same year was imputed as the end date .
If the year was present and the month and day were missing, then January 1 was imputed unless the year is the same as first dose date; in this situation, the first dose date was imputed .
If the year and month were present and the day was missing, the first of the month was imputed unless the month is the same month of the first dose of the study drug; in this situation, the first dose date was imputed.
For missing end concomitant medication, radiotherapy, and AE dates, the following was to be applied.
Missing day: the last day of the month was imputed unless the month is the same month of the last dose of the study drug; in this situation, the last dose date was imputed. For prior anticancer medications, the date of informed consent was imputed if the month is the same as the month informed consent was provided.
Missing day and month: December 31 was imputed unless the year is the same as the last dose date; in this situation, the last dose date was imputed. For prior anticancer medications, the date of informed consent was imputed if the month is the same as the month informed consent was provided.
Completely missing: consider when it started in relation to study drug. If the AE or medication was stopped and the start date is before the first dose date, then the first dose date was imputed. If it started on or after the first dose date, then a date that is after the last dose date (i.e., last dose + 1) was imputed, unless this is for a prior anticancer medication; in this situation, the date of informed consent was imputed.
For partial subsequent anticancer therapy dates, the following rules were applied for missing start dates:
Missing day: if the month is the same as the treatment end date, then the day after treatment was imputed, otherwise the first day of the month was used.
Missing day and month: if the year is the same as the treatment end date, then the day after treatment was imputed, otherwise January 1 of the same year as the anticancer therapy date was imputed.
Subgroup Analyses
Prespecified subgroup analyses were performed to assess the consistency of the treatment effect in the primary end point (PFS) across expected prognostic and/or predictive factors in the following subgroups:
age (aged < 65 years versus ≥ 65 years)
sex (female versus male)
smoking status (current or former smoker versus never smoker)
chemoradiotherapy (cCRT versus sCRT)
disease stage before chemoradiotherapy (stage IIIA versus stage IIIB or stage IIIC)
China cohort (Chinese patients enrolled at Chinese sites versus non-Chinese patients or patients enrolled at non-Chinese sites)
central plasma ctDNA EGFR mutation (exon 19 deletions or exon 21 [L858R] substitutions) status at screening (positive versus negative versus unknown)
tissue EGFR mutation at screening (exon 19 deletions versus exon 21 [L858R] substitutions)
race (Asian versus non-Asian)
response to prior chemoradiotherapy (complete response versus partial response [PR] versus stable disease versus nonevaluable response).
For each subgroup level of a factor, the HR and 95% CI was calculated from a Cox proportional hazards model that only contains a term for treatment, the factor, and treatment-by-factor interaction term. The Cox models were fitted using PROC PHREG with the Efron method to control for ties. If there were too few events available for a meaningful analysis of a particular subgroup comparison (i.e., < 20 events across both treatment groups), the relationship between that subgroup and the primary end point (PFS) was not formally analyzed. In this case, only descriptive summaries were provided. The clinical experts consulted for this review felt examining PFS by type of EGFR mutation at screening (exon 19 deletions or exon 21 [L858R] substitution mutations) would be relevant to investigate any differences in treatment effect. Differences in treatment effect across other subgroups were not expected based on clinical rationale.
Sensitivity Analyses
Sensitivity analyses on the primary efficacy analyses (PFS by BICR) were repeated to assess the following.
Ascertainment bias: This was determined with PFS based on the site investigator assessments according to RECIST Version 1.1. The stratified log-rank test was repeated, and a KM plot was presented with 95% CIs for PFS rated at 6-month intervals.
Stratification according to the eCRF: Stratified log-rank test was repeated where the stratification factors were recorded according to the eCRF.
Evaluation time bias: The stratified log-rank test was repeated using the midpoint between the time of progression and the previous nonmissing RECIST assessments. For patients who die in the absence of progression, the date of death was used to derive the PFS time used in the analysis. For those patients who did not experience disease progression during the study, the PFS time was right-censored at the date of the last nonmissing RECIST assessment. If the patient had no evaluable visits or did not have baseline data, they were to be censored at day 1. Note that midpoint values resulting in noninteger values were rounded down. To support this analysis, the means of patient-level average interassessment times were tabulated for each treatment. This approach used the BICR RECIST assessments.
Attrition bias: A stratified log-rank test and a reversed KM plot at the time of censoring, where the censoring indicator of the primary PFS analysis was reversed, were drafted where the actual PFS event times, rather than the censored times, of patients who progressed or died in the absence of progression immediately following 2 or more nonmissing tumour assessments, were included. In addition, and within the same sensitivity analysis, patients who took subsequent therapy before progression or death were censored at their last nonmissing assessment before taking the subsequent therapy.
The effect of COVID-19 deaths: If there were a sufficient number of patients with a confirmed or suspected COVID-19 death event (either ≥ 5 patients and/or ≥ 2% of the patient population), a sensitivity analysis was conducted to assess the potential impact of COVID-19 deaths on PFS. This was assessed by repeating the primary PFS analysis, but with patients whose disease did not progress before death and whose primary or secondary cause of death was due to COVID-19 infection, who had a COVID-19 infection reported as a fatal AE, or were censored at their last nonmissing assessment before their COVID-19 infection death date.
The primary analysis of PFS by BICR in the FAS was repeated using the evaluable for response analysis set as a sensitivity analyses. The evaluable for response analysis set includes all patients in the FAS who had measurable disease at baseline according to the BICR of baseline imaging data.
In the case that there was a sufficient number of patients with a confirmed or suspected COVID-19 death event (either at ≥ 5 patients and/or ≥ 2% of the patient population), a sensitivity analysis was planned to be conducted to assess the potential impact of COVID-19 deaths on OS. This was assessed by repeating the primary OS analysis, but for patients whose primary or secondary cause of death was due to COVID-19 infection, or who had a COVID-19 infection reported as a fatal AE, or who were censored at their last nonmissing assessment before their COVID-19 infection death date.
A sensitivity analysis of the primary CNS PFS analysis was conducted using the investigator-assessed CNS PFS using RECIST Version 1.1 assessments.
Table 9: Statistical Analysis of Efficacy End Points in the LAURA Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
OS | Provided there were sufficient events available for a meaningful analysis (≥ 20 deaths across both treatment groups with at least 5 events per group), the following models were used. Otherwise, descriptive summaries were to be provided.
| Stratified by adjustment factors employed in the randomization: prior platinum-based CRT (concurrent vs. sequential), disease stage before CRT (stage IIIA vs. stage IIIB or stage IIIC), and China cohort (enrolled at a site in China and patient declaring themselves of Chinese ethnicity vs. enrolled at site not in China or patient declaring themselves of non-Chinese ethnicity). | Any patient not known to have died at the time of analysis was censored based on the last recorded date on which the patient was known to be alive. | Not performed. |
PFS |
| Same for OS. | Patients who had not progressed or died at the time of analysis were censored at the time of the latest date of assessment from their last nonmissing RECIST assessment. If the patient progressed or died after ≥ 2 missed visits, the patient was censored at the time of the latest nonmissing RECIST Version 1.1 assessment before the 2 missed visits. If the patient had no postbaseline assessments or did not have baseline data, they were censored at day 1 unless they died within 2 visits of baseline. | Sensitivity analyses were performed for the following.
|
CNS PFS | Same methodology and model as for the primary analysis of PFS, provided sufficient events were available for a meaningful analysis (≥ 20 CNS PFS events across both treatment groups with at least 5 events per group); otherwise, descriptive summaries were to be provided. A competing risk analysis was also performed. | Same as for OS. | Patients who did not have CNS progression and had not died at the time of analysis were censored at the time of their last nonmissing RECIST assessment. However, if the patient experienced CNS progression or died after ≥ 2 missed visits, the patient was censored at the time of the latest nonmissing RECIST Version 1.1 assessment before the 2 missed visits. | A sensitivity analysis was conducted evaluating investigator-assessed CNS PFS using RECIST Version 1.1. |
TTDM | Analyzed using the same methodology and model as for the primary analysis of PFS; a competing risk analysis was also performed. | Same as for OS. | Patients who had not developed distant metastasis or died at the time of analysis were censored at the time of the latest date of assessment from their last evaluable RECIST Version 1.1 assessment. However, if the patient had distant metastasis or died after ≥ 2 missed visits, the patient was censored at the time of the latest evaluable RECIST Version 1.1 assessment before the 2 missed visits. If the patient had no evaluable visits or did not have baseline data, the patient was censored at day 1 (date of randomization) unless they died within 2 visits of baseline. | Not performed. |
TTD | Analyzed using the same methodology and model as for the primary analysis of PFS. | Same as for OS. | Any patient not known to have died at the time of analysis and not known to have discontinued study treatment was censored based on the last recorded date on which the patient was known to be alive. Patients who were randomized but had not received treatment were censored at the date of randomization. | Not performed. |
Change from baseline in EORTC QLQ-C30 and EORTC QLQ-LC13 | Change from baseline in each of the PRO symptom scores were analyzed using an MMRM analysis of the postbaseline scores for visits with the use of data from baseline up to 10 months or date of PD (whichever occurred earlier). | The model included patient, treatment, visit, and treatment-by-visit interaction as explanatory variables and the baseline score as a covariable along with the baseline score-by-visit interaction. Treatment, visit, and treatment-by-visit interaction were fixed effects in the model and patient was included as a random effect. Restricted maximum likelihood estimation was used. An overall adjusted mean estimate was derived to estimate the average treatment effect over visits giving each visit equal weight. | Visits with excessive missing data (≥ 75%) were excluded. If < 50% of the subscale items were missing, then the subscale score was divided by the number of nonmissing items and multiplied by the total number of items on the subscale. If at least 50% of the items are missing, that subscale was treated as missing.69 The dyspnea scale from the EORTC QLQ-LC13 was only used if all 3 items had been scored. Otherwise, the items were treated as single-item measures. | Not performed. |
BICR = blinded independent central review; CI = confidence interval; CNS = central nervous system; CRT = chemoradiotherapy; eCRF = electronic case report form; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; HR = hazard ratio; KM = Kaplan-Meier; MMRM = mixed effect model for repeated measures; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PRO = patient-reported outcome; RECIST = Response Evaluation Criteria in Solid Tumours; TTD = time to treatment discontinuation or death; TTDM = time to death or distant metastases; vs. = versus.
Source: LAURA Clinical Study Protocol,75 LAURA Statistical Analysis Plan,76 and LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The analysis population are defined in Table 10.
Table 10: Analysis Populations of the LAURA Trial
Study | Population | Definition | Application |
|---|---|---|---|
LAURA | FAS | Includes all randomized patients (i.e., the ITT population). | Demography and all efficacy analyses |
Evaluable for response analysis set | Includes all patients in the FAS who had measurable disease at baseline according to the BICR of baseline imaging data. | PFS and ORR sensitivity analyses | |
Safety analysis set | Includes all randomized patients who received at least 1 dose of study treatment. Safety data are not formally analyzed but summarized using the safety analysis set according to treatment received. Therefore, erroneously treated patients (e.g., those randomized to treatment A but actually given treatment B) are summarized according to the treatment actually received in the relevant treatment group. If a patient receives any active treatment, then they will be summarized in the active treatment group. | All safety analyses, laboratory measurements, vital signs, ECG, LVEF, and WHO performance status |
BICR = blinded independent central review; ECG = electrocardiogram; FAS = full analysis set; ITT = intention to treat; LVEF = left ventricular ejection fraction; ORR = objective response rate; PFS = progression-free survival.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition is summarized in Table 11. Of the 746 patients screened in the LAURA trial, 216 were ultimately randomized to either the osimertinib group (n = 143 patients) or the placebo group (n = 73 patients) and included in the FAS. All patients randomized received at least 1 dose of study treatment and were included in the safety analysis set. At the January 5, 2024, DCO date, 87 patients were ongoing on their randomized treatment (55.9% of patients in the osimertinib group and 9.6% of patients in the placebo group). In both groups, the primary reason for treatment discontinuation was due to progression (25.2% in the osimertinib group and 74.0% in the placebo group), followed by AEs (13.3% in the osimertinib group and 6.8% in the placebo group), and patient decision (2.8% in the osimertinib group and 4.1% in the placebo group). Following a treatment discontinuation event, a higher percentage of patients in the placebo group received open-label osimertinib (69.9%) compared to the osimertinib group (10.5%).
Table 11: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient Disposition | LAURA | |
|---|---|---|
Osimertinib n = 143 | Placebo n = 73 | |
Screened, N | 746 | |
Patients enrolled but not randomized, n | 530 | |
Reason for screening failure, n (%) | ||
Eligibility criteria not fulfilled | 519 (69.6) | |
Patient decision | 8 (1.1) | |
Death | 3 (0.4) | |
Randomized, n | 143 | 73 |
Patients who received at least 1 dose of study treatment, n (%) | 143 (100) | 73 (100) |
Ongoing study treatment at the DCO, n (%) | 80 (55.9) | 7 (9.6) |
Discontinued from study treatment, n (%) | 63 (44.1) | 66 (90.4) |
Patients with posttreatment open-label osimertinib use, n (%) | 15 (10.5) | 51 (69.9) |
Reason for study treatment discontinuation, n (%) | ||
RECIST disease progressiona | 36 (25.2) | 54 (74.0) |
AEb | 19 (13.3) | 5 (6.8) |
Patient decision | 4 (2.8) | 3 (4.1) |
Otherc | 3 (2.1) | 2 (2.7) |
Incorrect study drug initiatedd | 0 | 1 (1.4) |
Subsequent anticancer therapy | 1 (0.7) | 1 (1.4) |
FAS, n | 143 | 73 |
Safety analysis set, n | 143 | 73 |
Evaluable for response analysis set, n | 134 | 67 |
AE = adverse event; BICR = blinded independent central review; DCO = data cut-off; eCRF = electronic case report form; FAS = full analysis set; PD = progressive disease; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours.
Note: DCO date was January 5, 2024.
aBefore primary PFS analysis, assessed by BICR.
bIt is noted that the number of patients who discontinued study treatment due to AEs in this disposition summary differs from the number of patients with AEs leading to discontinuation of study treatment in the safety summaries. This discrepancy is due to AEs in 2 patients (1 patient in each treatment group) occurring outside of the safety data collection window.
cAny reason not specifically captured in earlier categories. Per the eCRF, other reasons for treatment discontinuation were captured as: “Other–Death” (2 patients) and “Other–PD by Investigator” (1 patient) in the osimertinib group; and “Other–Death” (1 patient) and “Other–PD by Investigator” (1 patient) in the placebo group (data on file).
dOne patient (1.4%) in the placebo group discontinued study treatment due to the reason of “incorrect study treatment initiation.” This patient was identified after the start of study treatment as having pre-existing brain metastases and was discontinued at the decision of the investigator.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The mean age in the osimertinib group (n = 143) was 60.9 years (SD = 10.38) compared to 62.4 years (SD = 12.01) in the placebo group (n = 73) and patients were primarily Asian (81.1% and 84.9%, respectively), had AJCC/Union for International Cancer Control disease stage IIIB (46.9% and 52.1%, respectively) or stage IIIC (16.8% and 15.1%, respectively), and had adenocarcinoma histology (97.2% and 94.5%, respectively). Notable differences were observed across the 2 treatment groups including proportionally more females than males in the osimertinib group (62.9% females and 37.1% males) than in the placebo group (57.5% females and 42.5% males), fewer former smokers in the osimertinib group (25.9%) than in the placebo group (31.5%), more patients in the osimertinib group (55.9%) with a WHO performance status of 0 at baseline than in the placebo group (42.5%), and more patients in the osimertinib group receiving cCRT (91.6%) than in the placebo group (84.9%). At screening, fewer patients in the osimertinib group were positive for exon 19 deletions (51.7%) and more were positive for exon 21 (L858R) substitutions (47.6%) compared to the placebo group (58.9% and 41.1%, respectively). Additionally, patients in the osimertinib group had a shorter mean time from unresectable stage III diagnosis to randomization than patients in the placebo group (117.6 days [SD = 37.71] versus 129.2 days [SD = 47.67], respectively) and more patients in the osimertinib group achieved a PR than in the placebo group (46.9% versus 37.0%, respectively).
Table 12: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristica | LAURA | |
|---|---|---|
Osimertinib n = 143 | Placebo n = 73 | |
Age (years) | ||
Mean (SD) | 60.9 (10.38) | 62.3 (12.01) |
Median (minimum to maximum) | 62.0 (36 to 84) | 64.0 (37 to 83) |
Age group (years), n (%) | ||
< 50 | 24 (16.8) | 12 (16.4) |
50 to < 65 | 57 (39.9) | 27 (37.0) |
65 to < 75 | 49 (34.3) | 20 (27.4) |
≥ 75 | 13 (9.1) | 14 (19.2) |
Sex, n (%) | ||
Female | 90 (62.9) | 42 (57.5) |
Male | 53 (37.1) | 31 (42.5) |
Race, n (%) | ||
American Indian or Alaska Native | 2 (1.4) | 1 (1.4) |
Asian | 116 (81.1) | 62 (84.9) |
Black or African American | 0 | 0 |
Native Hawaiian or other Pacific Islander | 0 | 0 |
White | 20 (14.0) | 10 (13.7) |
Other | 5 (3.5) | 0 |
Ethnic group, n (%) | ||
Hispanic or Latino | 11 (7.7) | 2 (2.7) |
Not Hispanic or Latino | 132 (92.3) | 71 (97.3) |
Smoking status, n (%) | ||
Never | 102 (71.3) | 49 (67.1) |
Smoker, current | 4 (2.8) | 1 (1.4) |
Smoker, former | 37 (25.9) | 23 (31.5) |
WHO performance status, n (%) | ||
0 (normal activity) | 80 (55.9) | 31 (42.5) |
1 (restricted activity) | 63 (44.1) | 42 (57.5) |
AJCC/UICC disease stage, n (%)b | ||
Stage IIIA | 52 (36.4) | 24 (32.9) |
Stage IIIB | 67 (46.9) | 38 (52.1) |
Stage IIIC | 24 (16.8) | 11 (15.1) |
Histology type, n (%) | ||
Adenocarcinoma | 139 (97.2) | 69 (94.5) |
Squamous cell carcinoma | 3 (2.1) | 2 (2.7) |
Otherc | 1 (0.7) | 2 (2.7) |
Time from unresectable stage III diagnosis to randomization (days) | ||
Mean (SD) | 117.6 (37.71) | 129.2 (47.67) |
Median (minimum to maximum) | 111.0 (55 to 265) | 121.0 (51 to 289) |
Prior CRT strategy, n (%) | ||
cCRT | 131 (91.6) | 62 (84.9) |
sCRT | 12 (8.4) | 11 (15.1) |
Response to prior CRT, n (%) | ||
Complete response | 4 (2.8) | 3 (4.1) |
Partial response | 67 (46.9) | 27 (37.0) |
Stable disease | 61 (42.7) | 37 (50.7) |
Progressive disease | 0 | 0 |
Nonevaluable | 11 (7.7) | 6 (8.2) |
Time from end date of radiotherapy to randomization date (weeks) | ||
Mean (SD) | 4.22 (1.39) | 4.05 (1.51) |
Median (minimum to maximum) | 4.29 (0.7 to 6.4) | 4.29 (0.4 to 6.0) |
Target lesion size by BICR | ||
Mean (SD), mm | 33.47 (17.65) | 35.93 (17.32) |
Median, mm | 29.40 | 30.50 |
No target lesions, n (%) | 9 (6.3) | 6 (8.2) |
Overall disease classification at randomization, n (%) | ||
Metastaticd | 0 | 1 (1.4) |
Locally advancede | 141 (98.6) | 72 (98.6) |
Missingf | 2 (1.4) | 0 |
Central plasma ctDNA EGFRm (exon 19 deletion or L858R) status at screening, n (%)g | ||
Positive | 10 (7.0) | 4 (5.5) |
Negative | 113 (79.0) | 58 (79.5) |
Unknownh | 20 (14.0) | 11 (15.1) |
Tissue EGFRm status at screening, n (%) | ||
Exon19 deletion positive | 74 (51.7) | 43 (58.9) |
L858R positive | 68 (47.6) | 30 (41.1) |
Missingi | 1 (0.7) | 0 |
AJCC = American Joint Committee on Cancer; BICR = blinded independent central review; cCRT = concurrent chemoradiotherapy; CRT = chemoradiotherapy; ctDNA = circulating tumour DNA; eCRF = electronic case report form; EGFRm = EGFR mutation; sCRT = sequential chemoradiation therapy; SD = standard deviation; UICC = Union for International Cancer Control.
Note: Data cut-off date was January 5, 2024.
aData reported in this table are based on values entered into the eCRF.
bDisease stage summarized based on values entered into the eCRF and categorized before CRT according to the TMN Classification of Malignant Tumours, 8th Edition.
cPer the eCRF, these patients had adenosquamous histology and were eligible for inclusion (data on file).
dPatient has any metastatic site of disease.
ePatient has only locally advanced sites of disease.
fThese patients had a complete response to prior CRT and therefore their disease was not detectable at randomization for classification (data on file).
gSummarized based on central cobas EGFR plasma testing results.
hStatus was unknown due to the unavailability or inadequacy of plasma samples for central testing.
iPatients randomized without an approved local positive EGFR test result or central EGFR test result.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Treatments
Study Treatments
At the January 5, 2024, DCO date, the mean total study treatment duration was longer in the osimertinib group (23.71 months [SD = 14.46]) than in the placebo group (11.77 months [SD = 10.88]) (refer to Table 13). Similar results were observed when excluding time related to dose interruptions indicating that the frequency of dose interruptions for any reason and their duration did not have a meaningful impact on the exposure to study treatments. More patients in the osimertinib group experienced a dose interruption (55.2%) or reduction (8.4%) than in the placebo group (28.8% and 1.4%, respectively).
Table 13: Summary of Patient Exposure to Study Treatment From Studies Included in the Systematic Review
Exposurea | LAURA | |
|---|---|---|
Osimertinib n = 143 | Placebo n = 73 | |
Total exposure (months)b | ||
Mean (SD) | 23.71 (14.46) | 11.77 (10.88) |
Median | 23.98 | 8.31 |
Minimum to maximum | 0.4 to 62.9 | 0.5 to 56.6 |
Total treatment-years | 282.5 | 71.6 |
Actual exposure (months)c | ||
Mean (SD) | 23.18 (14.44) | 11.53 (10.85) |
Median | 23.69 | 7.85 |
Minimum to maximum | 0.3 to 62.4 | 0.5 to 56.2 |
Total treatment-years | 276.3 | 70.2 |
Dose interruptions | ||
Patients with interruptions (any), n (%) | 79 (55.2) | 21 (28.8) |
Reason for interruption, n (%)d | ||
Adverse event | 76 (53.1) | 19 (26.0) |
Surgery | 1 (0.7) | 1 (1.4) |
Laboratory abnormality not reported as an adverse event | 1 (0.7) | 0 |
Othere | 8 (5.6) | 2 (2.7) |
Dose reductions | ||
Patients with dose reduction (any), n (%) | 12 (8.4) | 1 (1.4) |
Reason for dose reduction, n (%) | ||
Adverse event | 12 (8.4) | 1 (1.4) |
eCRF = electronic case report form; SD = standard deviation.
Note: Data cut-off date was January 5, 2024.
aExposure is calculated for on-study treatment only. Open-label osimertinib exposure is not included in this summary table. Total treatment-years = sum of treatment duration (months) for all patients per treatment.
bTotal treatment duration = (last dose date – first dose date + 1)/(365.25/12).
cActual treatment duration = total treatment duration, excluding dose interruptions.
dReasons for interruption are not mutually exclusive for patients with multiple interruptions although they were counted only once per category. Missed or forgotten doses have not been summarized as dose interruptions.
eNo specific pattern in the reasons for treatment interruption captured in the “Other” category was noted from a review of information captured in the eCRF.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
In both groups of the LAURA trial, the majority of patients received at least 1 concomitant medication (██████ of patients in the osimertinib group and █████ in the placebo group; Table 14). The most frequently used concomitant medication in the osimertinib group was ███████████████ (█████), followed by ██████ ████ ██████████ (█████) and ██████████ (█████). In the placebo group, the most commonly used concomitant medications were ██████ ████ ██████████ (█████), followed by ███████████████ (█████) and ██████████ (█████).
Table 14: Summary of Allowed Concomitant Medication in at Least 20% of Patients in Either Study Group of Studies Included in the Systematic Review
ATC classification generic terma | Patients, n (%) | |
|---|---|---|
Osimertinib n = 143 | Placebo n = 73 | |
Patients with allowed concomitant medication, n (%) | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
█████████████ | █████ | █████ |
ATC = anatomic therapeutic chemical; FAS = full analysis set; HMG-CoA = 3-hydroxy-3-methylglutaryl coenzyme A.
Note: Data cut-off date was January 5, 2024.
aA patient can have ≥ 1 generic terms reported under a given ATC code. Concomitant medications are those with a stop date on or after the first dose date of study treatment (and could have started before or during study treatment).
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Subsequent Treatments
By the January 5, 2024, DCO date, 63 of 143 patients (44.1%) in the osimertinib group had discontinued the study treatment and 42 patients (29.4%) went on to receive a subsequent anticancer treatment compared to 66 of 73 patients (90.4%) in the placebo group discontinuing the study treatment with 57 patients (78.1%) receiving a subsequent anticancer treatment (Table 15). In both treatment groups, patients tended to receive 1 or 2 subsequent lines of treatment (1 subsequent line: 19.6% of patients in the osimertinib group and 58.9% in the placebo group; 2 subsequent lines: 6.3% of patients in the osimertinib group and 15.1% in the placebo group), with relatively few patients receiving 3 or more lines of therapy. The most frequently reported category of subsequent anticancer treatment in both groups were EGFR TKIs (used by 19.6% of patients in the osimertinib group and 78.1% in the placebo group). Notably, 10.5% of patients in the osimertinib group and 69.9% of patients in the placebo group received osimertinib as subsequent anticancer treatment. The mean length of open-label osimertinib treatment was ████ ███ █ █████ months in the placebo group and ████ ███ █ █████ months in the osimertinib group.
Following a treatment discontinuation event, patients in the osimertinib group had a longer median time to first subsequent treatment (median = 43.83 months; 95% CI, 38.87 months to NC) compared to those in the placebo group (median = 9.46 months; 95% CI, 6.60 months to 11.53 months), favouring the osimertinib group (Table 15).
Similarly, at the unplanned November 29, 2024, OS analysis, more patients in the placebo group discontinued treatment (69 of 73 [94.5%]) and received a subsequent anticancer treatment (█████) than in the osimertinib group (74 of 143 [51.7%] and ██████ respectively; Table 15). Most patients in the placebo group received osimertinib as a subsequent anticancer treatment (█████) compared to █████ of patients in the osimertinib group. A higher percentage of patients in the osimertinib group received subsequent radiotherapy (19.6%) than in the placebo group (9.6%).
Table 15: Summary of Subsequent Treatments From Studies Included in the Systematic Review
Item | January 5, 2024, DCO date | November 29, 2024, DCO date | ||
|---|---|---|---|---|
Osimertinib n = 143 | Placebo n = 73 | Osimertinib n = 143 | Placebo n = 73 | |
Discontinued randomized study treatment, n (%)a | 63 (44.1) | 66 (90.4) | 74 (51.7) | 69 (94.5) |
Received any posttreatment anticancer therapya | 42 (29.4) | 57 (78.1) | █████ | █████ |
Received 1 line of therapy | 28 (19.6) | 43 (58.9) | █████ | █████ |
Received 2 lines of therapy | 9 (6.3) | 11 (15.1) | █████ | █████ |
Received 3 lines of therapy | 2 (1.4) | 3 (4.1) | █████ | █████ |
Received 4 lines of therapy | 2 (1.4) | 0 (0) | █████ | █████ |
Received ≥ 5 lines of therapy | 1 (0.7) | 0 (0) | █████ | █████ |
Did not receive posttreatment anticancer therapya | 21 (14.7) | 9 (12.3) | 20 (14.0) | 9 (12.3) |
Types of posttreatment disease-related anticancer therapy receivedb,c | ||||
EGFR TKI, n (%)a | 28 (19.6) | 57 (78.1) | 37 (25.9) | 60 (82.2) |
First-generation or second-generation EGFR TKI, n (%)a | 12 (8.4) | 7 (9.6) | █████ | █████ |
Third-generation EGFR TKI, n (%)a | 16 (11.2) | 52 (71.2) | █████ | █████ |
Osimertinib, n (%)a | 15 (10.5) | 51 (69.9) | █████ | █████ |
Open-label osimertinib exposure (months), mean (SD) | █████ | █████ | NR | NR |
Open-label osimertinib exposure (months), median (range) | █████ | █████ | NR | NR |
Aumolertinibd, n (%)a | 1 (0.7) | 1 (1.4) | █████ | █████ |
Furmonertinib, n (%)a | 0 (0) | 1 (1.4) | █████ | █████ |
Cytotoxic chemotherapy, n (%)a | 21 (14.7) | 11 (15.1) | 28 (19.6) | 15 (20.5) |
Platinum compounds | 19 (13.3) | 7 (9.6) | █████ | █████ |
Folic acid analogues (pemetrexed) | 13 (9.1) | 6 (8.2) | █████ | █████ |
Taxanes | 10 (7.0) | 5 (6.8) | █████ | █████ |
Other chemotherapye | 5 (3.5) | 2 (2.7) | █████ | █████ |
Radiotherapy, n (%)a | 21 (14.7) | 5 (6.8) | 28 (19.6) | 7 (9.6) |
VEGF inhibitor, monoclonal antibody, n (%)a | 8 (5.6) | 5 (6.8) | █████ | █████ |
PD-1 or PD-L1 inhibitor, immunotherapy, n (%)a | 5 (3.5) | 1 (1.4) | █████ | █████ |
EGFR and MET inhibitor, monoclonal antibody, n (%) | 0 (0) | 0 (0) | █████ | █████ |
Other, n (%)a | 2 (1.4) | 2 (2.7) | █████ | █████ |
Time to first subsequent treatment | ||||
Patients with an event, n (%)a | 53 (37.1) | 61 (83.6) | NR | NR |
Time to first subsequent treatment (months),f median (95% CI) | 43.83 (38.87 to NC) | 9.46 (6.60 to 11.53) | NR | NR |
DCO = data cut-off; FAS = full analysis set; NC = not calculable; NR = not reported; SD = standard deviation; TKI = tyrosine kinase inhibitor.
aThe number of patients is shown with percentages calculated as the proportion of patients in the FAS.
bA patient may be counted in multiple rows if they receive > 1 posttreatment anticancer therapy; includes anticancer therapies with a start date after the last dose date of study treatment.
cWHO Drug Dictionary version September 2022 format B3. A subsequent medical review has taken place to assign treatment classifications.
dNot available in Canada.
eIncludes pyrimidine analogues, vinca alkaloids and analogues, podophyllotoxin derivatives, and topoisomerase 1 inhibitors.
fCalculated using Kaplan-Meier techniques.
Source: LAURA Clinical Study Report39 and LAURA Overall Survival Analysis Update.40 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Efficacy outcomes presented in the following are from the most recent DCO dates (January 5, 2024, for all outcomes with the addition of the November 29, 2024, DCO date for OS).
Overall Survival
At the January 5, 2024, DCO date, OS was tested but did not meet the prespecified boundary for declaring statistical significance (P = 0. 530; P < 0.00036 was required to declare statistical significance at this interim analysis) (refer to Table 16 and Figure 5).
OS results at the January 5, 2024, DCO date and the November 29, 2024, DCO date are summarized in Table 16.
At the November 29, 2024, DCO date, the median OS follow-up time for patients was 39.36 months (range, ████ ██ █████) in the osimertinib group and 35.15 months (range, ████ ██ █████) in the placebo group. The OS stratified HR was 0.67 (95% CI, 0.40 to 1.14) favouring the osimertinib group. The between-group difference in survival probabilities for osimertinib versus placebo was ████ at 36 months and █████ at 48 months. The KM plot for OS in the FAS is presented in Figure 2.
Figure 2: Kaplan-Meier Plot of OS — FAS
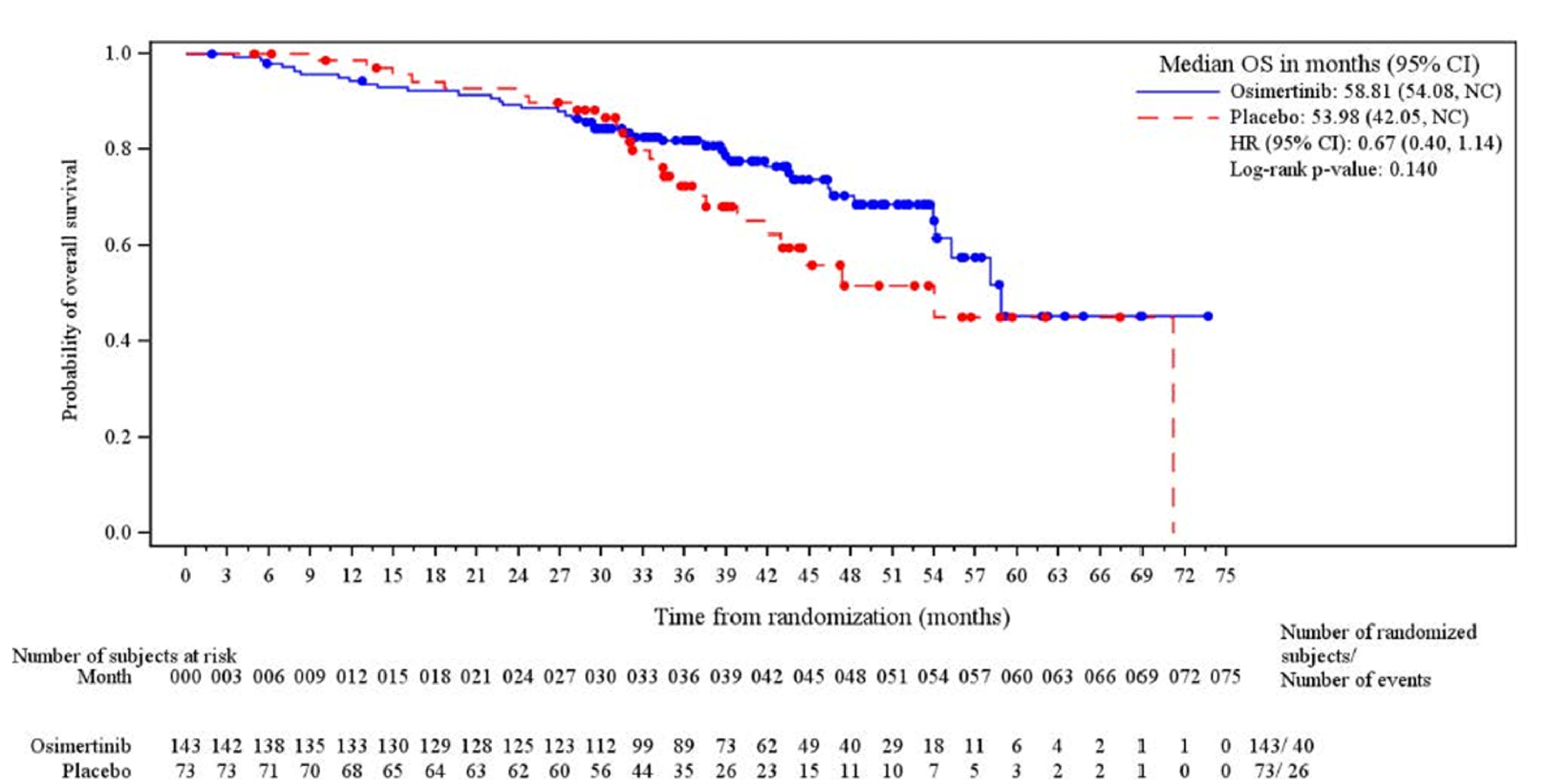
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NC = not calculable; OS = overall survival.
Notes: Circles indicate censored observations.
Data cut-off date was November 29, 2024.
Source: LAURA Overall Survival Analysis Update.40
Progression-Free Survival
PFS (based on BICR assessment according to RECIST Version 1.1) results at the January 5, 2024, DCO date are summarized in Table 16. The median PFS follow-up time for all patients was 21.98 months (range, 0.03 months to 60.55 months) in the osimertinib group and 5.55 months (range, 0.03 months to 49.71 months) in the placebo group. The PFS stratified HR was 0.16 (95% CI, 0.10 to 0.24), favouring the osimertinib group. The difference in PFS probabilities for osimertinib versus placebo was █████ at 12 months and █████ at 36 months. The KM plot for PFS in the FAS is presented in Figure 3.
With the exception of the sensitivity analysis to assess possible attrition bias which did not show a clear separation of the osimertinib and placebo curves (Figure 7), sensitivity analyses were generally consistent with those of the main analysis (Table 19, Figure 6, and Figure 8). Subgroup analyses are shown in Figure 9 and Table 21 and were generally consistent with the main analyses; however, some subgroups had relatively small sample sizes increasing the risk for imprecision.
Figure 3: Kaplan-Meier Plot of PFS by BICR Assessment — FAS
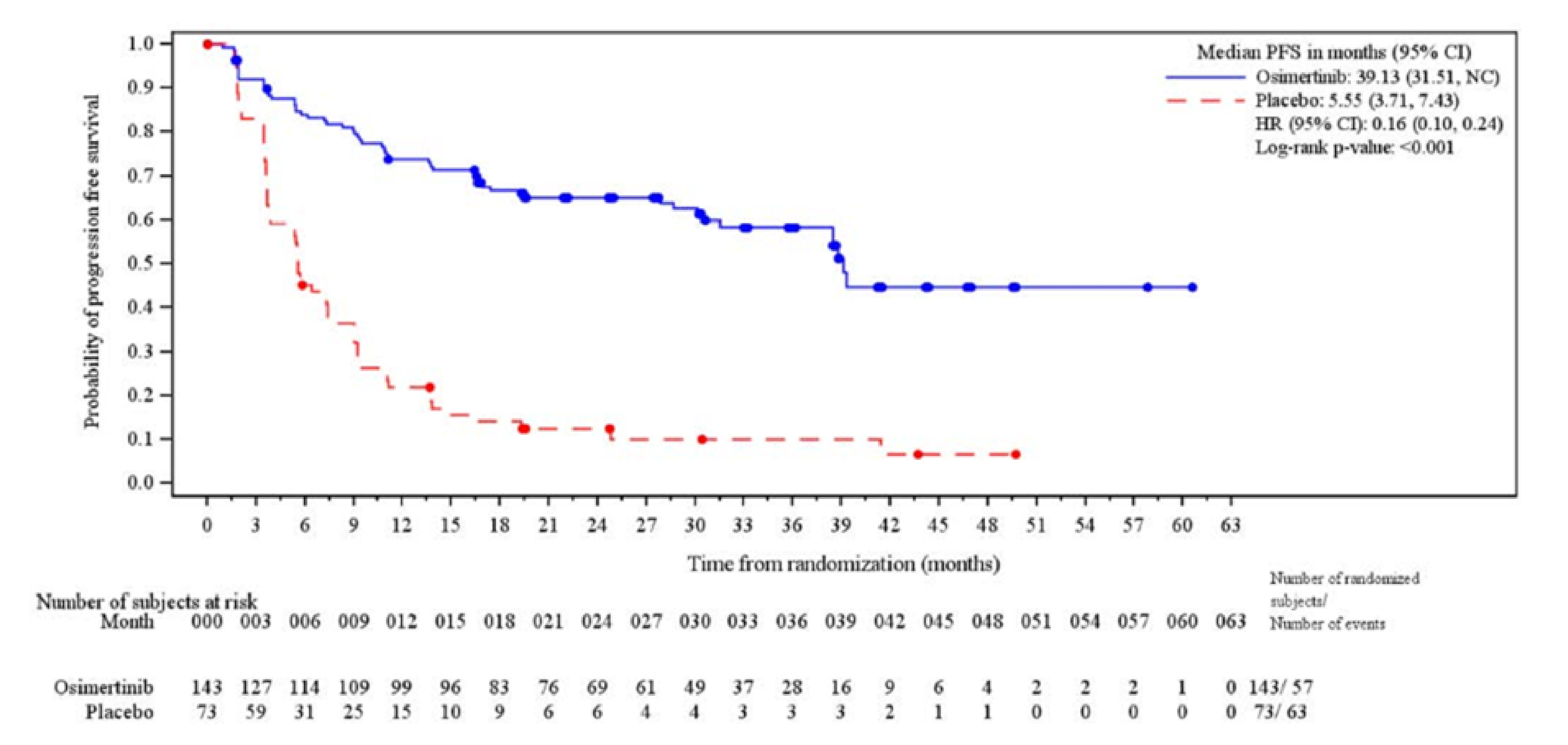
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NC = not calculable; PFS = progression-free survival.
Notes: Circles indicate censored observations.
Data cut-off date was January 5, 2024.
Source: LAURA Clinical Study Report.39
CNS Progression-Free Survival
CNS PFS (based on neuroradiologist BICR assessment according to RECIST Version 1.1) results at the January 5, 2024, DCO date are summarized in Table 16. The median CNS PFS follow-up time for all patients was 24.64 months (range, ████ ██ █████) in the osimertinib group and 5.72 months (range, ████ ██ █████) in the placebo group. The CNS PFS stratified HR was 0.17 (95% CI, 0.09 to 0.32) favouring the osimertinib group. The difference in CNS PFS probabilities for osimertinib versus placebo was █████ at 12 months and █████ at 24 months. The KM plot for CNS PFS in the FAS is presented in Figure 4.
The sensitivity analysis using investigator-assessed CNS PFS was consistent with the results of the main analysis (Table 20).
Figure 4: Kaplan-Meier Plot of CNS PFS by Neuroradiologist BICR Assessment — FAS
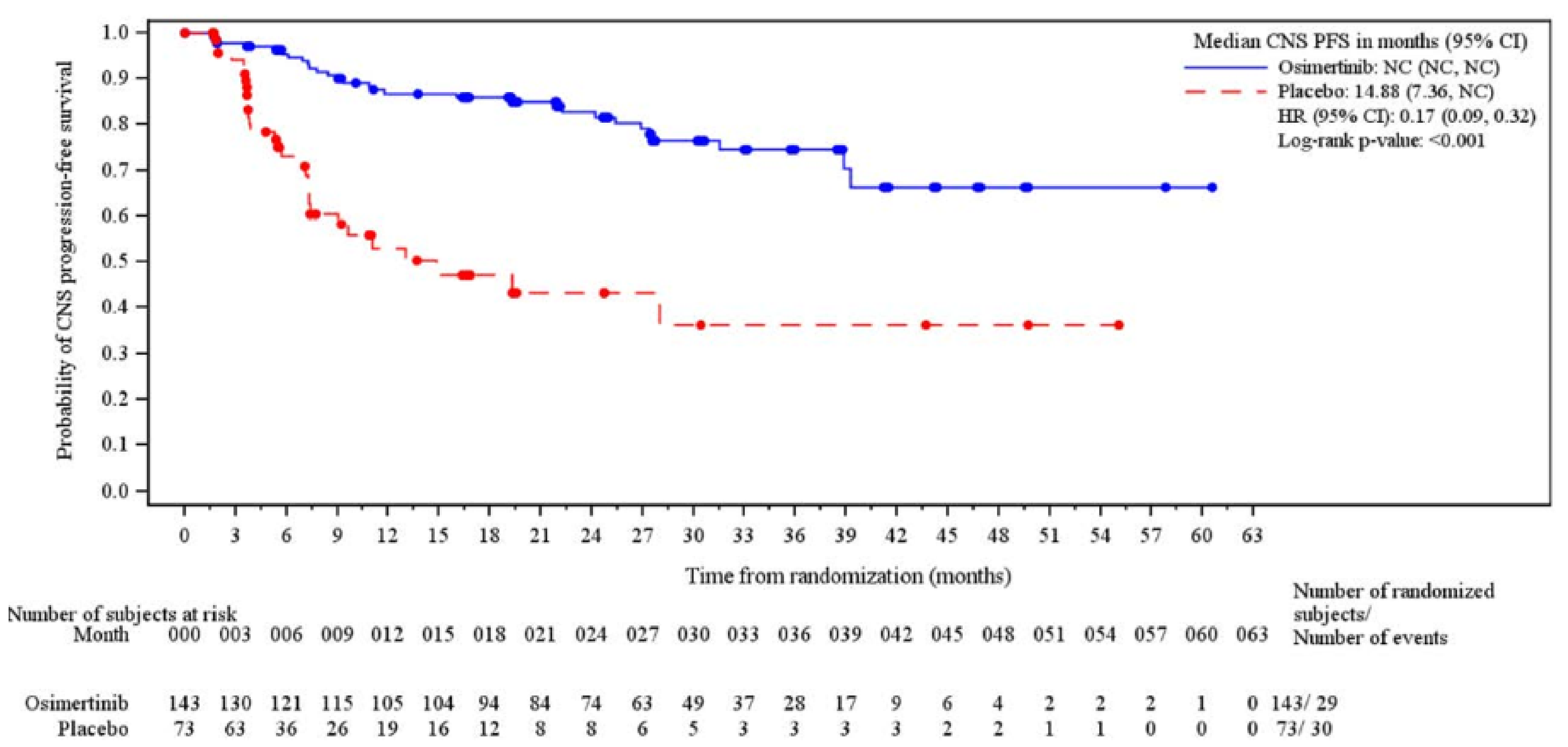
BICR = blinded independent central review; CI = confidence interval; CNS = central nervous system; FAS = full analysis set; HR = hazard ratio; NC = not calculable; PFS = progression-free survival.
Notes: Circles indicate censored observations.
Data cut-off date was January 5, 2024.
Source: LAURA Clinical Study Report.39
Time to Death or Distant Metastases
TTDM results at the January 5, 2024, DCO date are summarized in Table 16. A higher percentage of patients in the placebo group experienced a death or distant metastatic event than in the osimertinib group (42.5% versus 23.1%, respectively). The median TTDM was not reached (95% CI, 39.29 months to NC) in the osimertinib group and 13.04 months (95% CI, 9.03 months to NC) in the placebo group. The stratified HR was 0.21 (95% CI, 0.11 to 0.38), favouring the osimertinib group.
Time to Treatment Discontinuation or Death
TTD results at the January 5, 2024, DCO date are summarized in Table 16. A higher percentage of patients in the placebo group had a death or treatment discontinuation event than in the osimertinib group (90.4% versus 44.1%, respectively). The median TTD in the osimertinib group was 40.28 months (95% CI, 32.72 months to NC) compared to 8.31 months (95% CI, 6.14 months to 11.10 months) in the placebo group. The stratified HR was 0.21 (95% CI, 0.14 to 0.32), favouring the osimertinib group.
Table 16: Summary of Key Efficacy Results From Studies Included in the Systematic Review
Outcome | Osimertinib n = 143 | Placebo n = 73 | |
|---|---|---|---|
OS: January 5, 2024, DCO date | |||
Deaths, n (%) | 28 (19.6) | 15 (20.5) | |
Censored patients, n (%) | 115 (80.4) | 58 (79.5) | |
Still in survival at follow-upa | 111 (77.6) | 52 (71.2) | |
Terminated before deathb | 4 (2.8) | 6 (8.2) | |
Lost to follow-up | 0 (0) | 0 (0) | |
Withdrew consent | 4 (2.8) | 6 (8.2) | |
HR (95% CI)c | 0.81 (0.42 to 1.56) | ||
2-sided P valuec,d | 0.530 | ||
Median OS (months) (95% CI)e | 53.95 (46.49 to NC) | NC (42.05 to NC) | |
Difference (months) (95% CI) | NC (NR) | ||
OS rate at 12 months (%) (95% CI)e | 95.0 (89.8 to 97.6) | 98.6 (90.3 to 99.8) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 24 months (%) (95% CI)e | 90.3 (83.8 to 94.2) | 90.8 (80.5 to 95.8) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 36 months (%) (95% CI)e | 83.7 (75.3 to 89.4) | 73.7 (56.7 to 84.9) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 48 months (%) (95% CI)e | 65.1 (47.9 to 77.8) | 52.4 (26.6 to 72.9) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
Duration of follow-up in all patients (months), median (range) | 29.50 (1.87 to 62.88) | 28.09 (4.93 to 61.24) | |
Duration of follow-up in censored patients (months), median (range) | 30.92 (1.87 to 62.88) | 28.09 (4.93 to 61.24) | |
OS: November 29, 2024, DCO date | |||
Deaths, n (%) | 40 (28.0) | 26 (35.6) | |
HR (95% CI)c | 0.67 (0.40 to 1.14) | ||
2-sided P valuec,f | 0.140 | ||
Median OS (months) (95% CI)e | 58.81 (54.08 to NC) | 53.98 (42.05 to NC) | |
Difference (months) (95% CI) | ████ ████ | ||
OS rate at 12 months (%) (95% CI)e | ████ █████ ████ | ████ █████ ████ | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 24 months (%) (95% CI)e | ████ █████ ████ | ████ █████ ████ | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 36 months (%) (95% CI)e | 81.8 █████ ██ █████ | 72.5 █████ ██ █████ | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
OS rate at 48 months (%) (95% CI)e | 70.4 ███ ██ ████ | 51.5 ███ ██ ████ | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
Duration of follow-up in all patients (months), median (range) | 39.36 ██ ██ ██████ | 35.15 (███ ██ ████ | |
Duration of follow-up in censored patients (months), median (range) | ████ █████ ██ ██████ | ████ █████ ██ ██████ | |
PFS by BICR (per RECIST Version 1.1): January 5, 2024, DCO date | |||
PFS eventsg, n (%) | 57 (39.9) | 63 (86.3) | |
RECIST progression | 53 (37.1) | 62 (84.9) | |
Target lesionsh | 23 (16.1) | 16 (21.9) | |
Nontarget lesionsh | 6 (4.2) | 5 (6.8) | |
New lesionsh | 29 (20.3) | 45 (61.6) | |
Death in the absence of progression | 4 (2.8) | 1 (1.4) | |
Censored patients, n (%) | 86 (60.1) | 10 (13.7) | |
Censored RECIST progressioni | 1 (0.7) | 0 (0) | |
Censored death due to missing visitsi | 0 (0) | 1 (1.4) | |
Progression-free at time of analysis | 82 (57.3) | 6 (8.2) | |
Withdrawn consent | 3 (2.1) | 3 (4.1) | |
HR (95% CI)c | 0.16 (0.10 to 0.24) | ||
2-sided P valuec | < 0.001 | ||
Median PFS (months) (95% CI)e | 39.13 (31.51 to NC) | 5.55 (3.71 to 7.43) | |
Difference (months) (95% CI) | 33.58 (NR) | ||
PFS rate at 6 months (%) (95% CI)e | 84.0 (76.7 to 89.2) | 45.1 (33.3 to 56.1) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
PFS rate at 12 months (%) (95% CI)e | 73.7 (65.4 to 80.3) | 21.8 (13.0 to 32.1) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
PFS rate at 18 months (%) (95% CI)e | 66.8 (58.2 to 74.0) | 14.0 (7.0 to 23.4) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
PFS rate at 24 months (%) (95% CI)e | 65.1 (56.4 to 72.6) | 12.5 (5.9 to 21.6) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
PFS rate at 36 months (%) (95% CI)e | 58.4 (48.6 to 66.9) | 10.0 (4.0 to 19.2) | |
Difference in survival probability (%) (95% CI) | ████ █████ ████ | ||
Follow-up in all patients (months), median (range) | 21.98 (0.03 to 60.55) | 5.55 (0.03 to 49.71) | |
Follow-up in censored patients (months), median (range) | 27.71 (0.03 to 60.55) | 19.47 (0.03 to 49.71) | |
CNS PFS by neuroradiologist BICR assessment (per RECIST Version 1.1): January 5, 2024, DCO date | |||
CNS PFS events,j n (%) | 29 (20.3) | 30 (41.1) | |
CNS RECIST progression | 18 (12.6) | 26 (35.6) | |
CNS target lesionsh,k | 0 (0) | 0 (0) | |
CNS nontarget lesionsh,k | 1 (0.7) | 0 (0) | |
CNS new lesionsh | 17 (11.9) | 26 (35.6) | |
Death in the absence of progression | 11 (7.7) | 4 (5.5) | |
Censored patients, n (%) | 114 (79.7) | 43 (58.9) | |
Censored CNS RECIST progressioni | 1 (0.7) | 0 (0) | |
Censored deathi | 12 (8.4) | 7 (9.6) | |
No CNS event observed at time of analysis | 98 (68.5) | 31 (42.5) | |
CNS progression-free at time of analysis | 80 (55.9) | 6 (8.2) | |
Non-CNS progression observed | 18 (12.6) | 25 (34.2) | |
Withdrawn consent | 3 (2.1) | 5 (6.8) | |
HR (95% CI)c | 0.17 (0.09 to 0.32) | ||
2-sided P valuec,l | < 0.001 | ||
Median CNS PFS (months) (95% CI)e | NC (NC to NC) | 14.88 (7.36 to NC) | |
Difference (months) (95% CI) | NC (NR) | ||
CNS PFS rate at 12 months (%) (95% CI)e | 86.7 (79.4 to 91.5) | 53.0 (38.3 to 65.6) | |
Difference in CNS PFS rate (%) (95% CI) | ████ █████ ████ | ||
CNS PFS rate at 24 months (%) (95% CI)e | 82.7 (74.7 to 88.5) | 43.3 (28.0 to 57.7) | |
Difference in CNS PFS rate (%) (95% CI) | ████ █████ ████ | ||
Follow-up in all patients (months), median (range) | 24.64 ██ ██ ███ | 5.72 ███ ██ ███ | |
Follow-up in censored patients (months), median (range) | 27.40 (0.03 to 60.55) | 7.39 (0.03 to 55.06) | |
TTDM by BICR: January 5, 2024, DCO date | |||
Patients with events, n (%) | 33 (23.1) | 31 (42.5) | |
Median TTDM (months) (95% CI)e | Not reached (39.29 to NC) | 13.04 (9.03 to NC) | |
Difference (months) (95% CI) | NC (NR) | ||
HR, osimertinib versus placebo (95% CI)c | 0.21 (0.11 to 0.38) | ||
2-sided P valuec,m | < 0.001 | ||
TTD: January 5, 2024, DCO date | |||
Patients with events, n (%) | 63 (44.1) | 66 (90.4) | |
Median TTD (months) (95% CI)e | 40.28 (32.72 to NC) | 8.31 (6.14 to 11.10) | |
Difference (months) (95% CI) | 31.97 (NR) | ||
HR, osimertinib versus placebo (95% CI)c | 0.21 (0.14 to 0.32) | ||
2-sided P valuec,m | < 0.001 | ||
BICR = blinded independent central review; CI = confidence interval; CNS = central nervous system; DCO = data cut-off; HR = hazard ratio; IxRS = interactive voice and web response system; KM = Kaplan-Meier; NC = not calculable; NR = not reported; OS = overall survival; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours; SAP = statistical analysis plan; TTD = time to treatment discontinuation or death; TTDM = time to death or distant metastases.
aIncludes patients known to be alive at DCO.
bIncludes patients with unknown survival status or patients who were lost to follow-up.
cThe analysis was performed using a log-rank test stratified by disease stage before chemoradiation (stage IIIA versus stage IIIB or stage IIIC) based on values entered into the IxRS, after applying the SAP rule to collapse strata if there were < 10 events per stratum. A HR < 1 favours osimertinib to be associated with a longer PFS than placebo.
dHierarchical testing procedure to adjust for multiplicity stopped here as the P value was greater than the specified threshold of 0.00036.
eCalculated using the KM technique.
fNo threshold for significance was assigned to this analysis.
gPFS events that did not occur within 2 scheduled visits (plus visit window) of the last evaluable assessment (or randomization) were censored.
hTarget lesions, nontarget lesions, and new lesions are not necessarily mutually exclusive categories.
iOccurred after ≥ 2 consecutive missed visits following last nonmissing RECIST assessment (or randomization).
jCNS PFS events that did not occur within 2 scheduled visits (plus visit window) of the last nonmissing assessment (or randomization) are censored.
kIt is noted that while patients were free of CNS disease at baseline according to the investigator, the neuroradiologist BICR reader could independently assign target and nontarget lesions during CNS BICR assessment.
lP value should not be interpreted for rejecting the null hypothesis as a higher order comparison in the hierarchical procedure (OS at the DCO date of January 5, 2024) failed to reach statistical significance thereby preventing interpretation of the P value for CNS PFS comparisons.
mTested outside of the hierarchical testing procedure and not adjusted for multiplicity.
Source: LAURA Clinical Study Report39 and LAURA Overall Survival Analysis Update.40 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Health-Related Quality of Life
The percentage of patients who completed the EORTC QLQ-C30 at baseline was 92.9% in the osimertinib group and 93.2% in the placebo group. From baseline to week 40, more than 50% of patients in the osimertinib group completed the EORTC QLQ-C30 and the EORTC QLQ-LC13; however, in the placebo group by week 24 █████ ████ ███ of patients completed the assessments.
Baseline EORTC QLQ-C30 and EORTC QLQ-LC13 scores were similar across both treatment groups (Table 22).
While the MID of the EORTC QLQ-C30 varies by cancer type and subscale, global and subscale-specific changes did not exceed the MID in all instances.25 The point estimate for the between-group difference for the mean change in EORTC QLQ-C30 global health status/QoL score from baseline across all visits did not meet the MID threshold of 4 points; however, the CI crosses the MID threshold of −4 points for deterioration (−1.9 points; 95% CI, −5.89 to 2.00 points; Table 17). The only scores for which the CIs for the between-group difference for the mean change in scores from baseline across all visits did not include a value of 0 were for EORTC QLQ-C30 appetite loss (estimated difference = 8.1; 95% CI, 2.77 to 13.37; MID = 10) and for EORTC QLQ-C30 diarrhea (estimated difference = 7.9; 95% CI, 3.63 to 12.12; MID = not estimated).
Table 17: Mean Change From Baseline to Week 40 (Over All Visits) in EORTC QLQ-C30 and EORTC QLQ-LC13 Scores by Mixed Model for Repeated Measures Analysis — FAS
Outcome | Osimertinib n = 143 | Placebo n = 73 |
|---|---|---|
EORTC QLQ-C30 global measure of health status/quality of life | ||
Patients contributing to the analysis, n | 128 | 67 |
Adjusted mean change from baseline over all visits (95% CI)a | −3.9 (−5.98 to −1.82) | −2.0 (−5.30 to 1.40) |
Estimated difference (95% CI) | −1.9 (−5.89 to 2.00) | |
P valueb | █████ | |
EORTC QLQ-C30 function: Physical | ||
Patients contributing to the analysis, n | 128 | 67 |
Adjusted mean change from baseline over all visits (95% CI)a | −4.0 (−6.06 to −1.90) | −3.4 (−6.63 to −0.18) |
Estimated difference (95% CI) | −0.6 (−4.42 to 3.26) | |
P valueb | █████ | |
EORTC QLQ-C30 function: Role | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ ██████ | ████ ██████ ██ █████ |
Estimated difference (95% CI) | █████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 function: Emotional | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ █████ | ████ ██████ ██ █████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 function: Cognitive | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ ██████ | ████ ██████ ██ █████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 function: Social | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ ██████ | ████ ██████ ██ █████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Fatigue | ||
Patients contributing to the analysis, n | 128 | 67 |
Adjusted mean change from baseline over all visits (95% CI)a | 5.1 (2.49 to 7.68) | 3.4 (−0.62 to 7.50) |
Estimated difference (95% CI) | 1.6 (−3.18 to 6.46) | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Nausea and vomiting | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ █████ | ████ ██████ ██ ██████ |
Estimated difference (95% CI) | ███ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Pain | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ███ █████ ██ █████ | ███ █████ ██ █████ |
Estimated difference (95% CI) | ███ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Dyspnea | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ █████ ██ ██████ | ███ █████ ██ ██████ |
Estimated difference (95% CI) | ███ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Insomnia | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ ██████ ██ █████ | ███ ██████ ██ █████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Appetite loss | ||
Patients contributing to the analysis, n | 128 | 67 |
Adjusted mean change from baseline over all visits (95% CI)a | 3.3 (0.50 to 6.13) | −4.8 (−9.25 to −0.26) |
Estimated difference (95% CI) | 8.1 (2.77 to 13.37) | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Constipation | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ███ ██████ ██ █████ | ███ ██████ ██ █████ |
Estimated difference (95% CI) | ████ ██████ ██ █████ | |
P valueb | █████ | |
EORTC QLQ-C30 symptom: Diarrhea | ||
Patients contributing to the analysis, n | ███ | ██ |
Adjusted mean change from baseline over all visits (95% CI)a | ████ █████ ██ ██████ | ███ █ █████ ██ █████ |
Estimated difference (95% CI) | ███ █████ ██ ██████ | |
P valueb | ██████ | |
EORTC QLQ-LC13 symptom: Dyspnea | ||
Patients contributing to the analysis, n | 131 | 68 |
Adjusted mean change from baseline over all visits (95% CI)a | 7.6 (5.28 to 9.84) | 4.4 (1.04 to 7.83) |
Estimated difference (95% CI) | 3.3 (−0.98 to 7.24) | |
P valueb | █████ | |
EORTC QLQ-LC13 symptom: Coughing | ||
Patients contributing to the analysis, n | 131 | 68 |
Adjusted mean change from baseline over all visits (95% CI)a | 7.9 (5.00 to 10.71) | 8.0 (3.77 to 12.20) |
Estimated difference (95% CI) | −0.1 (−5.24 to 4.97) | |
P valueb | █████ | |
EORTC QLQ-LC13 symptom: Pain in chest | ||
Patients contributing to the analysis, n | 131 | 68 |
Adjusted mean change from baseline over all visits (95% CI)a | 4.2 (1.91 to 6.42) | 3.3 (−0.00 to 6.60) |
Estimated difference (95% CI) | 0.9 (−3.14 to 4.87) | |
P valueb | █████ | |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer Module; FAS = full analysis set; MMRM = mixed model for repeated measures.
Note: Data cut-off date was January 5, 2024.
aThe analysis was performed using an MMRM analysis of change from baseline score for all postbaseline assessments, with patient, treatment, visit, and treatment-by-visit interaction as explanatory variables and the baseline score as a covariable along with the baseline score-by-visit interaction. Treatment, visit, and treatment-by-visit interaction are fixed effects in the model and the patient is included as a random effect. Restricted maximum likelihood estimation is used. An overall adjusted mean estimate to estimate the average treatment effect over visits is derived giving each visit equal weight. The treatment-by-visit interaction is kept in the model regardless of significance. Data are summarized up to the earlier of the date of progression or 10 months of follow-up, excluding any visits with missing data from 75% of patients or more.
bNot adjusted for multiple testing.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Refer to Table 18 for harms data.
Adverse Events
More patients in the osimertinib group experienced an AE (140 of 143 [97.9%]) than in the placebo group (64 of 73 [87.7%]). In both groups, the 3 most frequently reported AEs were radiation pneumonitis (47.6% in the osimertinib group and 38.4% in the placebo group), diarrhea (35.7% and 13.7%, respectively), and rash (23.8% and 13.7%, respectively).
Serious Adverse Events
More patients in the osimertinib group experienced a SAE (55 of 143 [38.5%]) than in the placebo group (11 of 73 [15.1%]). In the osimertinib group, the most frequently reported SAEs were radiation pneumonitis (10.5%), pneumonia (4.9%), and gastroenteritis (1.4%) and pneumonitis (1.4%). In comparison, the most frequently reported SAEs in the placebo group were pneumonia (2.1%) and radiation pneumonitis (2.7%) with all other SAEs reported by only 1 patient.
Withdrawals Due to AEs
More patients in the osimertinib group prematurely stopped treatment due to an AE ███████ ████████ than in the placebo group █████ ███████. In the osimertinib group, the 3 most common AEs leading to treatment discontinuation were radiation pneumonitis (4.9%), █████████ ███████ ███ ███████████ ███████ ██ ███ ███████ █████ █ ████████ ██████ ██████████ █████████ ███████████ ████ ███ ██ █████████ ███████████████ █████ ███ █████ ███ ███████ ██ █████████ ███████████████ ████ ███████████ ██ ████ █ ████████
Mortality
A similar percentage of patients in both the osimertinib and placebo group experienced an AE with an outcome of death ██████ ██████ in the osimertinib group and ████ ██████ in the placebo group). In the osimertinib group, the fatal AEs were due to pneumonia ██████, pneumonitis (████), and a road traffic accident (████). In the placebo group, the fatal AEs were due to myocardial infarction (████) and aortic aneurysm rupture (████).
Notable Harms
Generally, a higher percentage of patients in the osimertinib group tended to experience AEs of special interest identified in the product monograph and highlighted as important by clinical experts consulted for this review. In both treatment groups, radiation pneumonitis was the most common AE of special interest (68 of 143 patients [47.6%] in the osimertinib group and 28 of 73 patients [38.5%] in the placebo group), followed by ███████ █████████ ██████ ███ █████ █████████████, and interstitial lung disease or pneumonitis (7.7% and 1.4%, respectively). Pneumonitis occurred in slightly more patients in the osimertinib group ██████ than in the placebo group ██████. Similarly, grade 3 or greater diarrhea occurred in ████ of patients in the osimertinib group and ████ in placebo group.
Table 18: Summary of Harms Results From Studies Included in the Systematic Review
AEa,b | Osimertinib n = 143 | Placebo n = 73 |
|---|---|---|
Most common AEs (reported in > 5% of patients in either treatment group), n (%) | ||
Patient with any AE | 140 (97.9) | 64 (87.7) |
Radiation pneumonitis | 68 (47.6) | 28 (38.4) |
Diarrhea | 51 (35.7) | 10 (13.7) |
Rash | 34 (23.8) | 10 (13.7) |
COVID-19 | 29 (20.3) | 6 (8.2) |
Paronychia | 24 (16.8) | 1 (1.4) |
Cough | 23 (16.1) | 7 (9.6) |
Decreased appetite | 21 (14.7) | 4 (5.5) |
Dry skin | 18 (12.6) | 4 (5.5) |
Pruritus | 18 (12.6) | 5 (6.8) |
Stomatitis | 17 (11.9) | 2 (2.7) |
White blood cell count decreased | 17 (11.9) | 2 (2.7) |
Pneumonia | 16 (11.2) | 6 (8.2) |
Anemia | 14 (9.8) | 3 (4.1) |
Herpes zoster | 13 (9.1) | 2 (2.7) |
Urinary tract infection | 11 (7.7) | 2 (2.7) |
Alanine aminotransferase increased | 10 (7.0) | 2 (2.7) |
Arthralgia | 10 (7.0) | 6 (8.2) |
Upper respiratory tract infection | 10 (7.0) | 1 (1.4) |
Dermatitis acneiform | 9 (6.3) | 2 (2.7) |
Dyspnea | 8 (5.6) | 5 (6.8) |
Aspartate aminotransferase increased | 8 (5.6) | 1 (1.4) |
Pneumonitis | 8 (5.6) | 1 (1.4) |
Sinus tachycardia | 8 (5.6) | 1 (1.4) |
Nasopharyngitis | 8 (5.6) | 0 |
Platelet count decreased | 8 (5.6) | 0 |
Productive cough | 7 (4.9) | 4 (5.5) |
Musculoskeletal chest pain | 5 (3.5) | 9 (12.3) |
Myalgia | 5 (3.5) | 6 (8.2) |
Headache | 2 (1.4) | 4 (5.5) |
SAEs (reported in ≥ 2 patients in either treatment group), n (%) | ||
Patient with any SAE | 55 (38.5) | 11 (15.1) |
Radiation pneumonitis | 15 (10.5) | 2 (2.7) |
Pneumonia | 7 (4.9) | 3 (4.1) |
Gastroenteritis | 2 (1.4) | 0 |
Pneumonitis | 2 (1.4) | 0 |
Patients who stopped treatment due to AEs (reported in ≥ 2 patients in either treatment group), n (%) | ||
Patient with any AE leading to treatment discontinuation | █████ | █████ |
Radiation pneumonitis | 7 (4.9) | █████ |
Pneumonia | █████ | █████ |
Pneumonitis | █████ | █████ |
Deaths, n (%) | ||
Patients with an AE with an outcome of death | █████ | █████ |
Pneumonia | █████ | █████ |
Myocardial infarction | █████ | █████ |
Aortic aneurysm rupture | █████ | █████ |
Pneumonitis | █████ | █████ |
Road traffic accident | █████ | █████ |
AEs of special interest, n (%) | ||
Any ILD/pneumonitisc | 11 (7.7) | 1 (1.4) |
ILD | 1 (0.7) | █████ |
Pneumonitis | █████ | █████ |
Pulmonary fibrosis | 3 (2.1) | █████ |
Radiation pneumonitis | 68 (47.6) | 28 (38.4) |
Cardiac effects | 1 (0.7) | 0 |
Cardiac disorders | █████ | █████ |
Left ventricular dysfunction | █████ | █████ |
Grade ≥ 3 diarrhea | █████ | █████ |
AE = adverse event; ILD = interstitial lung disease; PT = preferred term; SAE = serious adverse event.
Note: Data cut-off date was January 5, 2024.
aPatients with multiple events in the same PT are counted only once in that PT. Patients with events in > 1 PT are counted once in each of those PTs.
bIncludes AEs with an onset date on or after the date of first dose and up to and including the earlier of 28 days following the date of last dose of study medication and the day before the start of subsequent anticancer therapy.
cIncluded the following preferred terms: ILD, pneumonitis, acute interstitial pneumonitis, alveolitis, diffuse alveolar damage, idiopathic pulmonary fibrosis, lung disorder, organizing pneumonia, pulmonary toxicity, and pulmonary fibrosis.
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
While the LAURA trial planned to screen an estimated 1,333 patients to randomize approximately 200, only 746 were screened to enrol the 216 patients randomized. One possible explanation for the discrepancy between the planned and actual number of patients screened could be more patients than anticipated had EGFR testing performed before screening. However, as neither these data nor a rationale are provided, the exact reason for the difference remains unknown.
Allocation concealment methods used in the LAURA trial were deemed adequate by the CDA-AMC team. The LAURA trial randomized patients in a 2:1 ratio; however, no rationale for this ratio was provided. While such an approach is often used in oncology trials based on an established role of the investigational treatment and to improve data collection on the investigational treatment’s efficacy and harms, unequal allocation may reduce the precision of comparisons for the smaller control group and increase the potential for baseline imbalances, which could introduce bias or limit the interpretability of subgroup analyses. As will be described, there were notable differences in distribution of certain baseline characteristics between treatment groups, and the relatively smaller sample size in the placebo group potentially impacts the stability of the PFS estimates given the smaller number of patients at risk after 9 months (Figure 3).
Randomization in the LAURA trial was performed using an interactive voice or web response system and was stratified by prior platinum-based CRT strategy (cCRT versus sCRT), tumour stage before chemoradiation (stage IIIA versus stage IIIB or stage IIIC), and China cohort (enrolled at a Chinese site and patient declaring themselves of Chinese ethnicity versus enrolled at non-Chinese site or patient declaring themselves of non-Chinese ethnicity). These stratification factors were considered appropriate by the clinical experts consulted for this review and intended to balance known prognostic variables between treatment groups.
Despite randomization, differences in baseline characteristics between treatment groups were observed. Notable differences included a higher percentage of patients in the osimertinib group who had a WHO performance status of 0 (55.9%) at baseline than in the placebo group (42.5%), fewer former smokers in the osimertinib group (25.9%) than in the placebo group (31.5%), and a higher percentage of patients with PR in the osimertinib group (46.9%) than in the placebo group (37.0%) These differences may indicate that patients in the osimertinib group had a more favourable prognosis, which could introduce confounding and bias the results in favour of osimertinib. Another difference was observed in EGFR mutation types with 47.6% of patients in the osimertinib group and 41.1% in the placebo group having L858R-positive disease, while 51.7% and 58.9%, respectively, had exon 19 deletion–positive disease. It is acknowledged that between-group differences in baseline characteristics may occur in trials with smaller sample sizes (e.g., < 100 patients per group) and multiple strata per covariates, which may apply to the LAURA trial.26,27 Subgroup analyses among current or former smokers, by PR to prior CRT, and type of EGFR mutation were similar to the overall population findings. The sponsor conducted a post hoc subgroup analysis on baseline WHO performance status that was consistent overall with the PFS benefit for osimertinib in the primary analysis (refer to Table 21). While the results show some differences in the HRs between groups (HR = 0.17 for the WHO performance status of 0 subgroup and HR = 0.34 for the WHO performance status of 1 subgroup), these are inconclusive, due to the post hoc nature of the analyses and lack of test for interaction. Overall results were consistent with the PFS benefit for osimertinib in the primary analysis and the clinical experts consulted for this review did not anticipate the observed differences in WHO performance status at baseline to be clinically meaningful as to substantially impact the overall treatment effect. Nonetheless, the potential for residual confounding due to baseline imbalances remains a limitation in the internal validity of the trial.
While the LAURA trial was double-blinded, treatment unblinding was permitted for medical emergencies requiring knowledge of the treatment for appropriate management, and following BICR-confirmed progression of disease if the investigator deemed unblinding essential for the ongoing management of the patient. To mitigate potential bias related to subjective interpretation of disease progression and tumour response, a BICR committee reviewed all radiographic tumour assessments to determine response based on RECIST Version 1.1 criteria. As such, the risk of detection bias in tumour-related outcomes (e.g., PFS) due to selective unblinding is considered low. However, because unblinding was allowed for management decisions and may have occurred more frequently in one group (e.g., the placebo group upon progression), there remains a risk of bias for outcomes more susceptible to subjective influence or reporting bias, especially HRQoL and AEs. These domains rely more heavily on patient and investigator perception and were not centrally adjudicated, making them more vulnerable to differential reporting following unblinding.
While all patients in each treatment group received at least 1 dose of study treatment, the mean duration of treatment was longer for the osimertinib group (24 months [SD = 14]) than the placebo group (12 months [SD = 11]), reflecting longer PFS in the osimertinib group. Despite a higher percentage of patients in the osimertinib group experiencing dose interruptions (55.2%) than in the placebo group (28.8%), the mean treatment duration excluding time related to dose interruptions remained similar, indicating that the frequency and duration of interruptions for any reason did not have a meaningful impact on the cumulative exposure to study treatments.
Date of death, start and end dates for concomitant medication, radiotherapy, AEs, and subsequent anticancer treatment dates were imputed if missing. Instances where only partial information was available — dates with year only, or imputation based on first dose date — relied on assumptions of noninformative missingness (i.e., data missing completely at random or missing at random) which may introduce bias. If data were missing not at random, such as unrecorded events following specific treatments, these assumptions would be violated, potentially introducing bias in the estimation of treatment effects. Further, this imputation approach may not capture the uncertainty associated with the missing values, which could have led to underestimation in the variability and narrowing the resulting CIs. This undermines confidence in the precision of the effect estimates. A sensitivity analysis was conducted to assess the potential for evaluation time bias by imputing progression dates as the midpoint between the last progression-free and progression-confirming BICR assessments. This approach provides a standardized estimate of progression timing. Although the HR for the sensitivity (████; 95% CI ████ ██ ████; Table 19) was close to that for the primary PFS analysis (HR = 0.16; 95% CI, 0.10 to 0.24; Table 16) it does not fully mitigate the risk of bias. It assumes progression occurs uniformly and does not account for differences in assessment schedules or missing data patterns between treatment groups. Critically, the extent of missing data leading to imputation was not reported, making it difficult to assess the overall impact of this approach on potential bias in the results.
The LAURA trial used a hierarchical MTP to control the overall type I error rate across key efficacy end points, BICR-assessed PFS, followed by interim OS, and BICR-assessed CNS PFS. At the January 5, 2024, DCO date, PFS met the prespecified threshold for statistical significance. Per the testing procedure, the interim analysis of OS was also conducted at this DCO date, requiring a P value less than 0.00036 to declare statistical significance. The observed log-rank P value for OS was 0.530, which did not meet the statistical significance threshold. Consequently, the CNS PFS end point, which was next in the hierarchy, could not be formally tested. In this case, the reporting of a nominal P value for CNS PFS can be misleading and does not follow the statistical analysis plan; however, there is a substantial between-group difference that is unlikely attributable to chance or inflated type I error.
The OS analysis was based on the intention-to-treat approach, which assumes postprogression therapies are nondifferentially distributed between groups — a condition that may not hold given the observed disparities in postrelapse therapy used (Table 15). A higher percentage of patients in the placebo group had used subsequent anticancer treatments (█████) than in the osimertinib group (█████) by the November 29, 2024, DCO date. The largest discrepancy in postprogression therapies was for osimertinib (█████ in the placebo group versus █████ in the osimertinib group received subsequent osimertinib treatment at the November 29, 2024, DCO date). This imbalance challenges the intention-to-treat assumption and introduces potential bias in the OS analysis. The trial did not implement methods to adjust for treatment switching (e.g., rank-preserving structural failure time models). The crossover for patients on placebo who had progression potentially leads to an underestimation of the between-group difference in OS; however, the findings align with clinical practice, as osimertinib is the standard of care for disease that has progressed.
OS results from the LAURA trial are based on interim analyses, which may overestimate treatment effects.28 At the November 29, 2024, DCO date, the OS analysis included 66 events (40 of 143 patients [28.0%] in the osimertinib group and 26 of 73 patients [35.6%] in the placebo group), corresponding to approximately 30.6% of the total number of events required for the final analysis (120 events). This DCO date was not part of the prespecified analysis plan, nor was the number of events specified to trigger a second interim analysis. Results from this time point may therefore be more susceptible to bias due to early data truncation and post hoc decision-making. This DCO date was conducted as part of regulatory marketing requirements.
Cox proportional hazards models were used to estimate the HRs and CIs for OS, PFS, and CNS PFS. The proportional hazards assumption appeared to hold for PFS and CNS PFS based on visual inspection of KM plots. However, the early and rapid separation of the curves followed by the placebo curve plateauing could indicate either the hazard rates changed in this group leading to the hazards no longer being proportional, or the small (< 20%) proportion of patients remaining in the analysis led to less sensitive estimates, or both. Nevertheless, the visually apparent and sustained separation of the PFS and CNS PFS curves indicates there is likely little concern regarding the proportional hazards assumption. In contrast, the proportional hazards assumption is likely not met for OS. The placebo curve lies close to and slightly above that of osimertinib for the first 17 to 18 months. Following this, the curves overlap and remain close until around 30 months. From that point onward, the osimertinib curve shows higher survival compared to placebo, with a separation maintained through to approximately 58 months. While this suggests that the HRs may not reflect the treatment effect over time for OS, it may be partially explained by the frequent use of osimertinib in patients in the placebo group whose disease progressed. The clinical experts consulted for this review felt the OS curves were sufficiently close with no meaningful difference in the first 30 months. Clinical experts consulted for this review felt that the OS KM curves accurately reflected the natural history of EGFR-mutated NSCLC, with a typical median survival of 3 years to 4 years after recurrence. Alternative methods of analysis that do not rely on the proportional hazards assumption were not conducted for any of the time-to-event analyses, which makes it more difficult to assess the robustness of the results.
Visual inspection of KM plots indicated that fewer than 20% of patients in the placebo group remained at risk beyond 45 months for OS at the November 29, 2024, DCO date and beyond 12 months for PFS and CNS PFS at the January 5, 2024, DCO date. In the osimertinib group, fewer than 20% of patients were at risk beyond 51 months for OS at the November 29, 2024, DCO date and beyond 33 months for PFS and CNS PFS at the January 5, 2024, DCO date. Under such conditions, standard assumptions used in survival analysis (e.g., noninformative censoring) may be violated, which limits the reliability of survival estimates at later time points.29-31 Based on the available data, the clinical experts consulted for the review suggested 36 months and 48 months as clinically important time points for OS, 12 months and 36 months for PFS, and 12 months and 24 months for CNS PFS. Therefore, the certainty (potential risk of bias and imprecision) of the treatment effects of osimertinib versus placebo at the later time points for each of the end points may be reduced.
Due to too few events (i.e., < 10 events per strata), the stratified log-rank tests for PFS, OS, CNS PFS, TTDM, and TTD did not stratify by the China cohort or prior CRT strategy. This collapsing of stratification factors may introduce bias as it overlooks important differences such as geographic variations in clinical practice and variation in the efficacy of cCRT versus sCRT. It is acknowledged that to run the analysis, collapsing strata would be needed when the sample size is small. While the pooled analysis increases precision of PFS and OS estimates by combining strata, it sacrifices the accuracy of stratified analyses, which is most feasible when there are sufficient events per strata. Second, the proportional hazards assumption may no longer hold, potentially affecting the validity of the outcomes.
The FDA and the EMA consider OS the gold standard primary outcome in trials, including NSCLC trials.32,33 However, PFS is considered by the FDA to be an important cancer end point in situations where survival may be prolonged, making an OS end point impractical.33,34 When observed differences in PFS are substantial in magnitude (viewed in the context of toxicities, the relatively short survival of patients with NSCLC, availability of alternative therapies, histologic or genetic subtype of NSCLC, and extent of prior treatment), the FDA and EMA considered PFS to provide clinical evidence to support approvals.32,33 While a correlation between PFS and OS in patients with locally advanced NSCLC has been suggested,35,36 no correlation has been established for the current setting (i.e., unresectable EGFR-mutated NSCLC) and thus the validity of PFS as a surrogate for OS is unclear in the current context.
The primary PFS analysis and CNS PFS analysis censored patients who progressed or died after 2 or more consecutive missed visits. For both PFS and CNS PFS, censoring due to 2 or more consecutive missed visits was well balanced across both treatment groups. While results of the sensitivity analysis assessing attrition bias in PFS did not show a clear separation of the osimertinib and placebo curves which suggests possible attrition bias, the majority of PFS events in the placebo group occurred before 6 months (median PFS = ████ months in the placebo group) resulting in few patients censored beyond 6 months. Therefore, PFS attrition bias was deemed to be minimal.
Results from the EORTC QLQ-C30 and EORTC QLQ-LC13 showed that osimertinib did not meaningfully impact HRQoL compared to placebo. Across all items assessed, appetite loss and diarrhea (both assessed using the EORTC QLQ-C30) were the only items for which the CIs for the between-group difference in mean change in scores from baseline across all visits did not include the null value of 0. However, the completion rates for both scales declined over time reducing the certainty of the treatment effects at the later time points. By week 16 only ███ and ███ of patients in the placebo and osimertinib groups, respectively, were available to provide EORTC QLQ-C30 assessments; at week 32 these numbers had dropped to ███ and ████ respectively. For the EORTC QLQ-LC13, by week 16 only ███ and ███ of patients in the placebo and osimertinib groups, respectively, were available to provide assessments; at week 32 these numbers had dropped to ███ and ████ respectively.
External Validity
The clinical experts with expertise in the diagnosis and management of patients with NSCLC felt the inclusion and exclusion criteria used in the LAURA trial were generally aligned with those typically used in NSCLC trials and the sponsor’s reimbursement request. Additionally, the clinical experts felt the reimbursement of osimertinib should include patients with poorer performance status, that is, with a WHO performance status of 0 to 2 (rather than only those with a WHO performance status of 0 and 1 as included in the trial). The same clinical experts felt that the percentage of patients completing and discontinuing treatment throughout the trial aligned with clinical practice in Canada.
While none of the patients enrolled and randomized in the LAURA trial were from Canada, which may lower the generalizability of the results to the Canadian setting, the clinical experts consulted for this review indicated the baseline characteristics of the study population were generally representative of patients seen in Canadian clinical practice. Notably, a high percentage of patients enrolled in the LAURA trial identified as Asian (82.4%); however, this was partially expected as EGFR mutations are more common in patients of Asian ethnicity.5 The clinical experts consulted for this review did not anticipate the safety and efficacy of osimertinib to differ by race.
The outcomes measured in the LAURA trial addressed the key treatment goals identified by patient and clinician group inputs submitted to CDA-AMC and were deemed to be relevant by the consulted clinical experts.
The clinical experts consulted for this review noted that the prior platinum-based CRT regimens used for treatment in the definitive setting for stage III disease in the LAURA trial were representative of those used to treat unresectable EGFR-mutated NSCLC in Canada. The same clinical experts also felt that watchful waiting was the most relevant comparator. Additionally, osimertinib is currently reimbursed as first-line therapy in patients who progressed to metastatic disease (unresectable setting) and thus the clinical experts consulted for this review felt the high percentage of patients using postprogression osimertinib in the placebo group was representative of standard Canadian practice.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.37,38
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for osimertinib versus placebo.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Evidence
No indirect evidence was submitted.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies to address gaps within the systematic review evidence were submitted.
Discussion
Summary of Available Evidence
One ongoing phase III, double-blinded, placebo-controlled, randomized, multinational study, the LAURA trial (N = 216), met the inclusion criteria for the systematic review conducted by the sponsor. The LAURA trial enrolled adults with locally advanced, unresectable (stage III) NSCLC whose tumours harboured either EGFR exon 19 deletions or exon 21 (L858R) substitution mutations. None of the study sites were in Canada. Before randomization, patients received either concurrent or sequential platinum-based CRT. Only those whose disease did not progress during or following definitive platinum-based CRT, who had a WHO performance status of 0 or 1, and who did not have any unresolved toxicity greater than grade 2 were eligible for randomization. Patients were randomized 2:1 to receive either osimertinib 80 mg orally once daily or placebo until objective radiological disease progression (defined by RECIST Version 1.1 and confirmed by BICR) or another discontinuation criterion was met (e.g., patient decision or unacceptable toxicity). Patients assigned to osimertinib could continue receiving osimertinib (open label) after BICR-confirmed progression if, in the opinion of their treating physician, they were continuing to derive clinical benefit. The outcomes relevant to this review include the trial’s primary end point of PFS and select secondary end points of OS, CNS PFS, TTDM, TTD, response outcomes, and HRQoL assessed via EORTC QLC-C3 and EORTC QLC-LC13, and harms data. The trial population had a mean age of 61.4 years (SD = 10.95) and was primarily female (132 of 216 [61.1%]), never smokers (69.9%), and Asian (82.4%). Most patients were classified as having stage IIIA (35.2%) or stage IIIB (48.6%) NSCLC, received cCRT (89.4%), and were positive for exon 19 deletions (54.2%).
Interpretation of Results
The patient group input collected for this review highlighted the key treatment goals for patients are to prolong life, prolong PFS, reduce symptoms, and improve HRQoL. The effect of osimertinib following the successful completion of platinum-based CRT on achieving these goals was evaluated in the LAURA trial through the assessment of OS, PFS, CNS PFS, response outcomes, EORCT QLQ-C30 global health score/QoL, and AEs.
Efficacy
OS was determined to be the most clinically important outcome for this review. The clinical experts consulted for the review agreed with the importance of OS as an outcome and suggested a 5% to 10% between-group difference in the probability of OS to be clinically meaningful. Osimertinib compared to placebo may result in a clinically important increase in OS at 36 months and 48 months, based on the 5% threshold. However, the certainty of evidence for both time points was rated as low (per GRADE assessment; Table 2) due to risk of bias and imprecision. At a median OS follow-up of 39.36 months (range, ████ ██ █████) in the osimertinib group and 35.15 months (range, ████ ██ █████) in the placebo group, 66 deaths had occurred in the FAS and the KM estimate for median OS was 58.81 months (95% CI, 54.08 months to NC) in the osimertinib group and 53.98 months (95% CI, 42.05 months to NC) in the placebo group. Thus, a key limitation of the OS data is the insufficient sample size and duration of follow-up to adequately assess OS in the studied population. Further, a late divergence of the KM curves was observed (i.e., did not separate until approximately 30 months post randomization). The clinical experts consulted on this file agreed that this observation was expected. According to the experts, even in cases where patients experience disease progression, the median survival after recurrence is typically 3 years to 4 years. Additionally, they confirmed that the placebo curve followed the expected trajectory considering that the frequent use of osimertinib post progression in the placebo group is anticipated to reduce the observed treatment effect. Health Canada’s authorization of osimertinib is conditional on the final OS analysis. The final OS analyses is planned for when approximately 60% of OS events have occurred. However, uncertainties related to OS will likely remain due to the high level of cross over to the osimertinib group.
PFS was the primary outcome in the LAURA trial and considered to be a clinically important outcome for this review. The consulted clinical experts considered a 10% to 15% between-group difference to be clinically meaningful. At a median PFS follow-up of 21.98 months (range, 0.03 months to 60.55 months) in the osimertinib group and 5.55 months (range, 0.03 months to 49.71 months) in the placebo group, 120 progression events had occurred in the FAS and the median PFS was 39.13 months (95% CI, 31.51 months to NC) in the osimertinib group and 5.55 months (95% CI, 3.71 months to 7.43 months) in the placebo group. The between-group difference in PFS probabilities for osimertinib versus placebo were █████ ████ ███ ████ ██ █████ at 12 months and █████ ████ ███ ████ ██ █████ at 36 months. Using the thresholds suggested by the clinical experts, these point estimates suggest clinically meaningful increases in the probability of being progression-free at 12 months and 36 months. Certainty of evidence was rated as high at 12 months, but rated down at 36 months due to fewer than 20% of patients remaining at risk in both groups.29,31 While differences in baseline characteristics were observed across the treatment groups, including WHO performance status and smoking status, prespecified and post hoc PFS subgroup analyses were generally consistent with the primary result.
Given the uncertainty in the currently available OS results from the LAURA trial, the extent to which benefits observed in PFS translate into improvements in OS is a key consideration. While a correlation between PFS and OS in patients with locally advanced NSCLC treated with CRT has been suggested,35,36 no such correlation has been established for patients with locally advanced, unresectable EGFR-mutated NSCLC; thus, the validity of PFS as a surrogate for OS is unclear in the current context.
The between-group differences in CNS PFS rates at 12 months and 24 months were above the 10% to 15% threshold suggested by the clinical experts consulted for this review, indicating a clinically meaningful improvement in favour of osimertinib. However, the certainty of evidence was rated down at 24 months due to high attrition in the placebo group.
The antitumour effects of osimertinib compared with placebo were supported by the TTDM and TTD results. By the January 5, 2024, DCO date, the median TTDM was not reached in the osimertinib group and was 13.04 months in the placebo group. The HR and associated 95% CI favour the osimertinib group compared to placebo. The median TTD was approximately 5 times longer for the osimertinib group compared to the placebo group (40.28 months for the osimertinib group versus 8.31 months for the placebo group). The between-group difference in TTD was above the 6-month threshold suggested by clinical experts, demonstrating that osimertinib led to a clinically meaningful improvement in TTD compared to placebo. ORR and DoR results from the LAURA trial were also supportive of the observed PFS benefit (Table 23). However, these end points were outside of the MTP and were considered as supportive evidence.
Patient and clinician input highlighted that maintaining or improving HRQoL is an important treatment goal. HRQoL was measured in the LAURA trial using the well-established EORTC QLQ-C30, a cancer-specific HRQoL questionnaire. Published estimates for a between-group MID in the EORTC QLQ-C30 global health status/QoL score among patients with lung cancer are a 4-point increase or decrease, for improvement and deterioration, respectively.70 At the January 5, 2024, DCO date, the between-group difference in mean change across all visits until week 40 in EORTC QLQ-C30 global health status/QoL was −1.9 points (95% CI, −5.89 points to 2.00 points), indicating than osimertinib may result in little to no difference in HRQoL compared to placebo (low certainty per GRADE assessment).
Locally advanced, unresectable (stage III) EGFR-mutated NSCLC is a debilitating and life-threatening disease.2,77 While durvalumab, and alternative immunotherapy, is reimbursed in Canada after successful completion of platinum-based cCRT in patients with unresectable stage III NSCLC, post hoc subgroup analyses suggest it has similar efficacy and more frequent AEs compared to placebo in patients with EGFR-mutated NSCLC.21 Therefore, following the successful completion of CRT, there is currently no established treatment option for EGFR-mutated NSCLC, according to the clinical experts consulted for this review. Clinical experts noted that conventional treatments (e.g., CRT) followed by watchful waiting often lead to high rates of disease recurrence, as seen in the PFS data from the placebo group of the LAURA trial. The experts noted that relapsing stage III NSCLC is generally considered incurable. Additionally, the experts flagged that patients with EGFR mutations are more likely to develop CNS metastasis compared to those with driver mutation–negative NSCLC, increasing the risk of symptoms at recurrence. Therefore, reducing relapse risk in patients with unresectable NSCLC and EGFR mutations was identified as a critical unmet need by the clinical experts consulted for this review.
Harms
More patients in the osimertinib group experienced an AE (98%) and a SAE (39%) than in the placebo group (88% and 15%, respectively). Osimertinib likely results in little to no difference in withdrawals due to AEs (█████) compared to placebo (████) (moderate certainty of evidence per the GRADE assessment). A total of || patients in the safety analysis set experienced an AE with an outcome of death with ██████ occurring in the osimertinib group and ██████ occurring in the placebo group; based on the GRADE assessment, osimertinib may result in little to no difference in the incidence of fatal AEs compared to placebo. Pneumonitis and grade 3 or greater diarrhea were identified in the product monograph and highlighted as important by clinical experts consulted for this review. While a similar percentage of patients in the osimertinib group and placebo group experience pneumonitis (low certainty of evidence per GRADE assessment) and grade 3 or greater diarrhea (moderate certainty of evidence per GRADE assessment), the absolute number of events was low for both AEs.
The proposed product monograph further indicates that once osimertinib treatment has been initiated, patients should be monitored for interstitial lung disease (including pneumonitis), QT interval corrected using the Fridericia formula interval prolongation, left ventricular dysfunction, and cardiomyopathy.
For the LAURA trial, the clinical experts consulted for this review felt the data presented did not raise any new concerns about the safety profile of osimertinib, and the observed events were consistent with what is expected with the drug.
Conclusion
The LAURA trial was a phase III, double-blinded, placebo-controlled, randomized, multinational trial comparing the efficacy and safety of maintenance osimertinib for patients with locally advanced, unresectable (stage III) NSCLC whose tumours harboured either EGFR exon 19 deletions or exon 21 (L858R) substitution mutations and whose disease has not progressed during or following platinum-based CRT. Compared to placebo, osimertinib results in a clinically important improvement in PFS and CNS PFS at 12 months (high-certainty evidence) and likely results in clinically important increases in PFS at 36 months and CNS PFS at 24 months (moderate-certainty evidence). At the planned PFS interim analysis, relatively few OS events (43 events across both groups) had occurred and OS failed to achieve statistical significance. Results from a subsequent unplanned OS analysis showed osimertinib may result in a clinically important increase in OS at 36 months and 48 months compared to the placebo group (low-certainty evidence at both time points). Results from the LAURA trial showed that osimertinib may result in little to no difference in HRQoL compared to placebo (low certainty of evidence). Clinical experts consulted for this review deemed the harms profile of osimertinib to be consistent with their expectations based on osimertinib’s known toxicity profile.
Clinical experts noted that conventional treatments (e.g., CRT) often lead to high rates of disease recurrence, and relapsing stage III NSCLC is generally considered incurable. Additionally, the experts flagged that patients with EGFR mutations are more likely to develop CNS metastasis compared to those with driver mutation–negative NSCLC, increasing the risk of symptoms at recurrence. Therefore, reducing relapse risk in patients with unresectable NSCLC and EGFR mutations was identified as a critical unmet need by the clinical experts consulted for this review.
References
1.Canadian Cancer Statistics Advisory Committee: Canadian Cancer Statistics. Canadian Cancer Society, ed. A 2020 special report on lung cancer. 2020. Accessed March 13, 2025. cancer.ca/Canadian-Cancer-Statistics-2020-EN.
2.Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics 2023. Accessed December 3, 2024. https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_pdf_en.pdf?rev=7e0c86ef787d425081008ed22377754d&hash=DBD6818195657364D831AF0641C4B45C&_gl=1*1yvsjcf*_gcl_au*MjUzMzYyODg2LjE3Mzg2MTE0ODc
3.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-e623. doi: 10.1503/cmaj.240095 PubMed
4.O'Leary C, Gasper H, Sahin KB, et al. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals (Basel). 2020;13(10). doi: 10.3390/ph13100273 PubMed
5.Ellis PM, Verma S, Sehdev S, Younus J, Leighl NB. Challenges to implementation of an epidermal growth factor receptor testing strategy for non-small-cell lung cancer in a publicly funded health care system. J Thorac Oncol. 2013;8(9):1136-41. doi: 10.1097/JTO.0b013e31829f6a43 PubMed
6.Midha A, Dearden S, McCormack R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 2015;5(9):2892-911. PMC4633915, https://pmc.ncbi.nlm.nih.gov/articles/PMC4633915/ PubMed
7.Graham RP, Treece AL, Lindeman NI, et al. Worldwide Frequency of Commonly Detected EGFR Mutations. Arch Pathol Lab Med. 2018;142(2):163-167. doi: 10.5858/arpa.2016-0579-CP PubMed
8.Melosky B, Banerji S, Blais N, et al. Canadian consensus: a new systemic treatment algorithm for advanced EGFR-mutated non-small-cell lung cancer. Curr Oncol. 2020;27(2):e146-e155. doi: 10.3747/co.27.6007 PubMed
9.Canadian Cancer Society. Lung cancer. 2020. Updated May 2020. Accessed March 13, 2025. https://www.cancer.ca/en/cancer-information/cancer-type/lung/lung-cancer/?region=on
10.Birring SS, Peake MD. Symptoms and the early diagnosis of lung cancer. Thorax. 2005;60(4):268-9. doi: 10.1136/thx.2004.032698 PubMed
11.B.C. Cancer Agency. Lung. 2019. Updated August 6, 2019. Accessed April 1, 2025. http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-management-manual/lung/lung
12.Daly ME, Singh N, Ismaila N, et al. Management of Stage III Non-Small-Cell Lung Cancer: ASCO Guideline. J Clin Oncol. 2022;40(12):1356-1384. doi: 10.1200/jco.21.02528 PubMed
13.Swaminath A, Vella ET, Ramchandar K, et al. Surgery after Chemoradiotherapy in Patients with Stage III (n2 or N3, Excluding T4) Non-Small-Cell Lung Cancer: A Systematic Review. Curr Oncol. 2019;26(3):398-404. Accessed March 6, 2025. https://www.mdpi.com/1718-7729/26/3/4549 PubMed
14.Sathiyapalan A, Baloush Z, Ellis PM. Update on the Management of Stage III NSCLC: Navigating a Complex and Heterogeneous Stage of Disease. Curr Oncol. 2023;30(11):9514-9529. doi: 10.3390/curroncol30110689 PubMed
15.Shang S, Wang R, Wang F, Wu M, Chen D, Yu J. Treatment Patterns for Patients With Unresected Stage III NSCLC: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database. Front Oncol. 2022;12:874022. doi: 10.3389/fonc.2022.874022 PubMed
16.Remon J, Soria JC, Peters S. Early and locally advanced non-small-cell lung cancer: an update of the ESMO Clinical Practice Guidelines focusing on diagnosis, staging, systemic and local therapy. Ann Oncol. 2021;32(12):1637-1642. doi: 10.1016/j.annonc.2021.08.1994 PubMed
17.Robinson A, Vella ET, Ellis PM, et al. Recommendations for the Treatment of Patients with Clinical Stage III Non-Small Cell Lung Cancer: Endorsement of the 2019 National Institute for Health and Care Excellence Guidance and the 2018 Society for Immunotherapy of Cancer Guidance [sponsor submitted reference]. Vol. No.: 7-3 Version 4. 2020. Program in Evidence Based Care Guideline. https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/43311
18.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Non-Small Cell Lung Cancer Version 3.2025 — January 14, 2025. 2025. Accessed March 13, 2025. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf
19.Postmus PE, Kerr KM, Oudkerk M, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl_4):iv1-iv21. doi: 10.1093/annonc/mdx222 PubMed
20.Ramnath N, Dilling TJ, Harris LJ, et al. Treatment of stage III non-small cell lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e314S-e340S. doi: 10.1378/chest.12-2360
21.Naidoo J, Antonia S, Wu YL, et al. Brief Report: Durvalumab After Chemoradiotherapy in Unresectable Stage III EGFR-Mutant NSCLC: A Post Hoc Subgroup Analysis From PACIFIC. J Thorac Oncol. 2023;18(5):657-663. doi: 10.1016/j.jtho.2023.02.009 PubMed
22.Passaro A, Leighl N, Blackhall F, et al. ESMO expert consensus statements on the management of EGFR mutant non-small-cell lung cancer. Ann Oncol. 2022;33(5):466-487. doi: 10.1016/j.annonc.2022.02.003 PubMed
23.Eberhardt WE, De Ruysscher D, Weder W, et al. 2nd ESMO Consensus Conference in Lung Cancer: locally advanced stage III non-small-cell lung cancer. Ann Oncol. 2015;26(8):1573-88. doi: 10.1093/annonc/mdv187 PubMed
24.Lu S, Kato T, Dong X, et al. Osimertinib after Chemoradiotherapy in Stage III EGFR-Mutated NSCLC. N Engl J Med. 2024;391(7):585-597. doi: 10.1056/NEJMoa2402614 PubMed
25.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi: 10.1016/j.ejca.2023.04.027 PubMed
26.Nüesch E, Trelle S, Reichenbach S, et al. Small study effects in meta-analyses of osteoarthritis trials: meta-epidemiological study. BMJ. 2010;341. doi: 10.1136/bmj.c3515 PubMed
27.Zabor EC, Kaizer AM, Hobbs BP. Randomized controlled trials. Chest. 2020;158(1):S79-S87. doi: 10.1016/j.chest.2020.03.013 PubMed
28.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-7. doi: 10.1001/jama.2010.310 PubMed
29.Dudley WN, Wickham R, Coombs N. An Introduction to Survival Statistics: Kaplan-Meier Analysis. J Adv Pract Oncol. 2016;7(1):91-100. doi: 10.6004/jadpro.2016.7.1.8 PubMed
30.Gong Q, Fang L. Asymptotic properties of mean survival estimate based on the Kaplan-Meier curve with an extrapolated tail. Pharm Stat. 2012;11(2):135-40. doi: 10.1002/pst.514 PubMed
31.RoB2 Development Group. Higgins JP, Savović J, Page MJ, Sterne JA, eds. Revised Cochrane risk-of-bias tool for randomized trials (RoB 2). 2019. Accessed April 16, 2025. https://sites.google.com/site/riskofbiastool/welcome/rob-2-0-tool/current-version-of-rob-2
32.U.S. Department of Health and Human Services: Food and Drug Administration. Clinical Trial Endpoints for the Approval of Non-Small Cell Lung Cancer Drugs and Biologics - Guidance for Industry. 2015. Accessed March 21, 2025. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-non-small-cell-lung-cancer-drugs-and-biologics
33.European Medicines Agency. Guideline on the evaluation of anticancer medicinal product in man [sponsor submitted reference]. 2012.
34.FDA. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. 2018. Accessed 15 April 2025. https://www.fda.gov/media/71195/download
35.Hu C, Miccio JA, Dignam JJ, et al. Progression-Free Survival as a Surrogate Endpoint of Overall Survival in Patients with Locally Advanced Non-Small Cell Lung Cancer Treated with Chemoradiotherapy: Trial-Level Meta-Analysis and Individual-Level Analysis of NRG/RTOG 0617 and PROCLAIM. International Journal of Radiation Oncology*Biology*Physics. 2023;117(2, Supplement):S128. doi: 10.1016/j.ijrobp.2023.06.473
36.Mauguen A, Pignon JP, Burdett S, et al. Surrogate endpoints for overall survival in chemotherapy and radiotherapy trials in operable and locally advanced lung cancer: a re-analysis of meta-analyses of individual patients' data. Lancet Oncol. 2013;14(7):619-26. doi: 10.1016/s1470-2045(13)70158-x PubMed
37.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
38.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
39.AstraZeneca. Clinical Study Report: D5160C00048. A Phase III, Randomised, Double-Blind, Placebo-Controlled, Multicentre, International Study of Osimertinib as Maintenance Therapy in Patients with Locally Advanced, Unresectable EGFR Mutation-Positive Non-Small Cell Lung Cancer (Stage III) Whose Disease Has Not Progressed Following Definitive Platinum-Based Chemoradiation Therapy (LAURA)[internal sponsor's report]. 2024.
40.AstraZeneca. Overall Survival Analysis Update: AZD9291. TAGRISSO™ as Monotherapy for the Treatment of Adult Patients with Locally Advanced, Unresectable (Stage III) NSCLC whose Tumours have EGFR Exon 19 Deletions or Exon 21 (L858R) Mutations and whose Disease has not Progressed During or Following Platinum Based Chemoradiation Therapy - Overall Survival Analysis Update Report – LAURA [internal sponsor's report]. 2025.
41.Canadian Partnership Against Cancer. Lung Cancer and Equity: A Focus on Income and Geography. 2020. Accessed September 19, 2024. https://www.partnershipagainstcancer.ca/topics/lung-cancer-equity/
42.Yang X, Zhong J, Yu Z, et al. Genetic and treatment profiles of patients with concurrent Epidermal Growth Factor Receptor (EGFR) and Anaplastic Lymphoma Kinase (ALK) mutations. BMC Cancer. 2021;21(1):1107. doi: 10.1186/s12885-021-08824-2 PubMed
43.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129-39. doi: 10.1056/NEJMoa040938 PubMed
44.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497-500. doi: 10.1126/science.1099314 PubMed
45.Bhai P, Turowec J, Santos S, et al. Molecular profiling of solid tumors by next-generation sequencing: an experience from a clinical laboratory. Front Oncol. 2023;13:1208244. doi: 10.3389/fonc.2023.1208244 PubMed
46.Boyne DJ, Jarada T, Yusuf A, et al. 51P Testing patterns and outcomes of different EGFR-positive metastatic non-small cell lung cancer (NSCLC) patients in a Canadian real-world setting. Ann Oncol. 2022;33:S56. doi: 10.1016/j.annonc.2022.02.060
47.O'Sullivan DE, Jarada TN, Yusuf A, et al. Prevalence, Treatment Patterns, and Outcomes of Individuals with EGFR Positive Metastatic Non-Small Cell Lung Cancer in a Canadian Real-World Setting: A Comparison of Exon 19 Deletion, L858R, and Exon 20 Insertion EGFR Mutation Carriers. Curr Oncol. 2022;29(10):7198-7208. doi: 10.3390/curroncol29100567 PubMed
48.Rangachari D, Yamaguchi N, VanderLaan PA, et al. Brain metastases in patients with EGFR-mutated or ALK-rearranged non-small-cell lung cancers. Lung Cancer. 2015;88(1):108-11. doi: 10.1016/j.lungcan.2015.01.020 PubMed
49.Roughley A, Damonte E, Taylor-Stokes G, Rider A, Munk VC. Impact of brain metastases on quality of life and estimated life expectancy in patients with advanced non-small cell lung cancer. Value Health. 2014;17(7):A650. doi: 10.1016/j.jval.2014.08.2364 PubMed
50.Peters S, Bexelius C, Munk V, Leighl N. The impact of brain metastasis on quality of life, resource utilization and survival in patients with non-small-cell lung cancer. Cancer Treat Rev. 2016;45:139-62. doi: 10.1016/j.ctrv.2016.03.009 PubMed
51.Cancer Care Ontario. Lung Cancer Diagnosis Pathway Map. Accessed March 13, 2025. https://www.cancercareontario.ca/sites/ccocancercare/files/assets/LungCancerDiagnosisPathwayMap.pdf
52.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The eighth edition lung cancer stage classification. Chest. 2017;151(1):193-203. Accessed March 6, 2025. https://journal.chestnet.org/article/S0012-3692(16)60780-8/abstract
53.Cancer Care Ontario. Lung Cancer Tissue Pathway Map. Accessed April 7, 2025. https://www.cancercareontario.ca/sites/ccocancercare/files/assets/LungTissuePathwayMap.pdf
54.Seung SJ, Hurry M, Walton RN, Evans WK. Retrospective cohort study of unresectable stage III non-small-cell lung cancer in Canada. Curr Oncol. 2020;27(4):e354-e360. doi: 10.3747/co.27.6047 PubMed
55.Cheema PK, Gomes M, Banerji S, et al. Consensus recommendations for optimizing biomarker testing to identify and treat advanced EGFR-mutated non-small-cell lung cancer. Curr Oncol. 2020;27(6):321-329. doi: 10.3747/co.27.7297 PubMed
56.European Medicines Agency. Tagrisso (osimertinib). 2025. Accessed March 21, 2025. https://www.ema.europa.eu/en/medicines/human/EPAR/tagrisso
57.U.S. Food and Drug Administration. FDA approves osimertinib for locally advanced, unresectable (stage III) non-small cell lung cancer following chemoradiation therapy. 2024. Accessed March 21, 2025. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-osimertinib-locally-advanced-unresectable-stage-iii-non-small-cell-lung-cancer
58.TAGRISSO (osimertinib); 40 mg and 80 mg tablets. Product Monograph. AstraZeneca Canada Inc. (draft) [sponsor submitted reference].
59.Melosky B, Blais N, Cheema P, et al. Standardizing Biomarker Testing for Canadian Patients with Advanced Lung Cancer. Curr Oncol. 2018;25(1):73-82. PubMed
60.Ionescu DN, Stockley TL, Banerji S, et al. Consensus Recommendations to Optimize Testing for New Targetable Alterations in Non-Small Cell Lung Cancer. Curr Oncol. 2022;29(7):4981-4997. PubMed
61.Zheng Y, Helene V, X. LF, Barinder S, Sakshi S, and Sharda D. Diagnostic and Economic Value of Biomarker Testing for Targetable Mutations in Non-Small-Cell Lung Cancer: a Literature Review. Future Oncol. 2022;18(4):505-518. doi: 10.2217/fon-2021-1040 PubMed
62.Ontario Health. Plasma-Based Comprehensive Genomic Profiling DNA Assays for Non-Small Cell Lung Cancer: A Health Technology Assessment. Ont Health Technol Assess Ser. 2024;24(8):1-306. PMC11650780, Accessed March 6, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC11650780/ PubMed
63.Zou D, Ye W, Hess LM, et al. Diagnostic Value and Cost-Effectiveness of Next-Generation Sequencing–Based Testing for Treatment of Patients with Advanced/Metastatic Non-Squamous Non–Small-Cell Lung Cancer in the United States. J Mol Diagn. 2022;24(8):901-914. doi: 10.1016/j.jmoldx.2022.04.010 PubMed
64.Dagogo-Jack I, Azzolli CG, Fintelmann F, et al. Clinical Utility of Rapid EGFR Genotyping in Advanced Lung Cancer. JCO Precis Oncol. 2018;2018. doi: 10.1200/po.17.00299 PubMed
65.Ontario Health. Cell-Free Circulating Tumour DNA Blood Testing to Detect EGFR T790M Mutation in People With Advanced Non-Small Cell Lung Cancer: A Health Technology Assessment. Ont Health Technol Assess Ser. 2020;20(5):1-176. PMC7082730, Accessed March 6, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC7082730/ PubMed
66.Perdrizet K, Stockley TL, Law JH, et al. Integrating comprehensive genomic sequencing of non-small cell lung cancer into a public healthcare system. Cancer Treatment and Research Communications. 2022;31:100534. doi: 10.1016/j.ctarc.2022.100534 PubMed
67.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
68.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30a(5):635-42. doi: 10.1016/0959-8049(94)90535-5 PubMed
69.Fayers PM. The EORTC QLQ-C30 scoring manual. European Organisation for Research and Treatment of Cancer [sponsor submitted reference]. 2001.
70.Musoro JZ, Coens C, Fiteni F, et al. Minimally Important Differences for Interpreting EORTC QLQ-C30 Scores in Patients With Advanced Breast Cancer. JNCI Cancer Spectr. 2019;3(3):pkz037. doi: 10.1093/jncics/pkz037 PubMed
71.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. doi: 10.1186/1477-7525-9-106 PubMed
72.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi: 10.1007/s11136-007-9210-8 PubMed
73.Ozturk A, Sarihan S, Ercan I, Karadag M. Evaluating quality of life and pulmonary function of long-term survivors of non-small cell lung cancer treated with radical or postoperative radiotherapy. Am J Clin Oncol. 2009;32(1):65-72. doi: 10.1097/COC.0b013e31817e6ec2 PubMed
74.Burman CF, Sonesson C, Guilbaud O. A recycling framework for the construction of Bonferroni-based multiple tests. Stat Med. 2009;28(5):739-61. doi: 10.1002/sim.3513 PubMed
75.AstraZeneca. Clinical Study Protocol v5.0: D5160C00048. A Phase III, Randomised, Double-Blind, Placebo-Controlled, Multicentre, International Study of Osimertinib as Maintenance Therapy in Patients with Locally Advanced, Unresectable EGFR Mutation-Positive Non-Small Cell Lung Cancer (Stage III) Whose Disease Has Not Progressed Following Definitive Platinum-Based Chemoradiation Therapy (LAURA)[internal sponsor's protocol]. 2023.
76.AstraZeneca. Statistical Analysis Plan v3.0: D5160C00048. A Phase III, Randomised, Double-Blind, Placebo-Controlled, Multicentre, International Study of Osimertinib as Maintenance Therapy in Patients with Locally Advanced, Unresectable EGFR Mutation-Positive Non-Small Cell Lung Cancer (Stage III) Whose Disease Has Not Progressed Following Definitive Platinum-Based Chemoradiation Therapy (LAURA)[internal sponsor's SAP]. 2023.
77.Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48-51. PMC3256436, No potential conflict of interest. Accessed March 6, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC3256436/
Appendix 1: Overall Survival at the January 5, 2024, DCO
Please note that this appendix has not been copy-edited.
Figure 5: Kaplan-Meier Plot of Overall Survival at the January 5, 2024, DCO, FAS
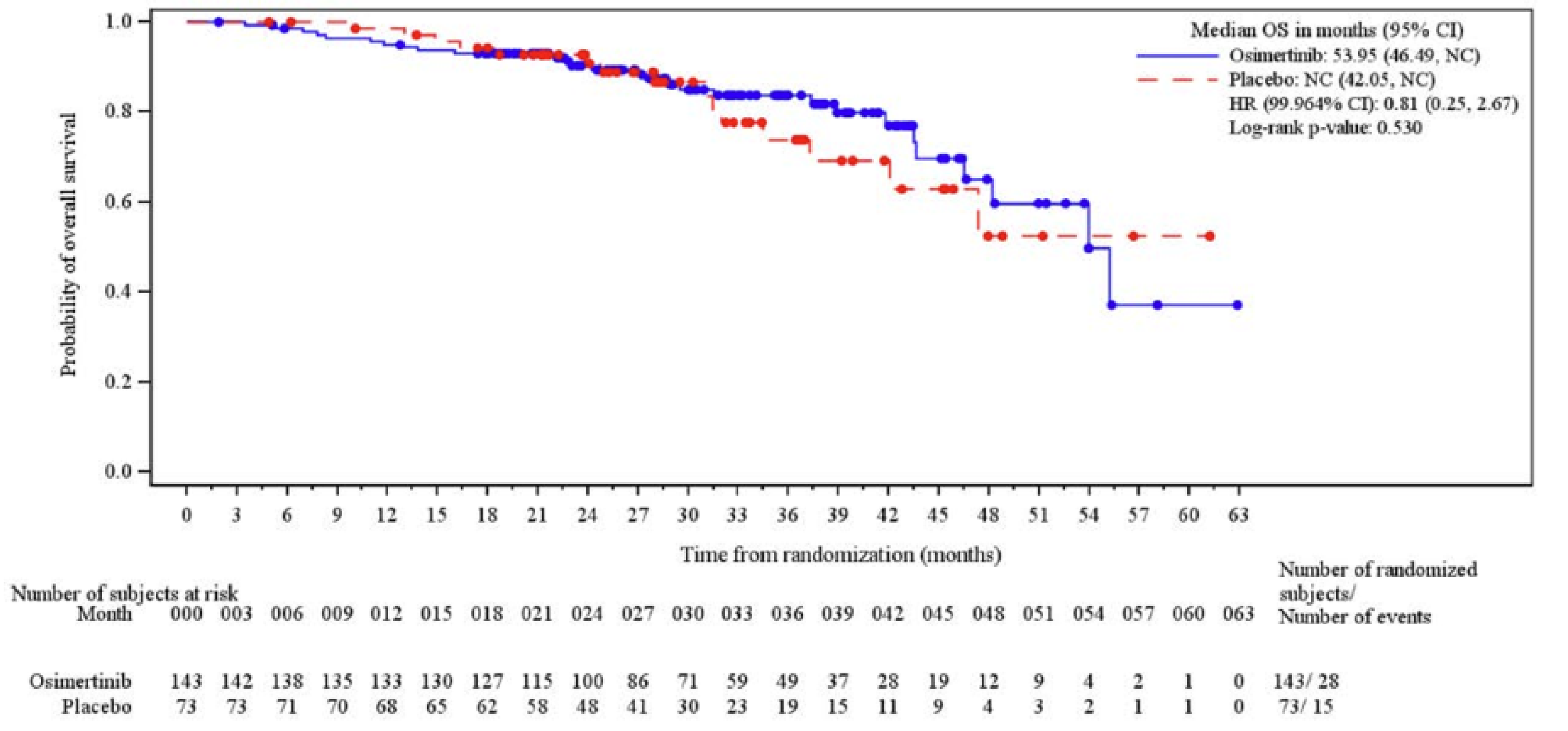
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; HR = hazard ratio; NC = not calculable; OS = overall survival.
Note: Circles indicate censored observations.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39
Appendix 2: The LAURA Trial Sensitivity Analyses
Please note that this appendix has not been copy-edited.
Table 19: Sensitivity Analysis: Progression-Free Survival
Outcome | Osimertinib N = 143 | Placebo N = 73 |
|---|---|---|
PFS by investigator assessment (ascertainment bias) according to RECIST v1.1 | ||
Total PFS events, n (%)a | 62 (43.4) | 63 (86.3) |
RECIST progression | █████ | █████ |
Target lesionsb | █████ | █████ |
Nontarget lesionsb | █████ | █████ |
New lesionsb | █████ | █████ |
Death in the absence of progression | █████ | █████ |
Censored patients, n (%) | █████ | █████ |
Censored deathc | █████ | █████ |
Progression-free at time of analysis | █████ | █████ |
Withdrawn consent | █████ | █████ |
HR (95% CI)d | 0.19 (0.12 to 0.29) | |
2-sided P valued | < 0.001 | |
Median PFS (months) (95% CI)e | 38.87 (26.71 to NC) | 7.29 (5.45 to 10.25) |
Difference in median PFS (months) (95% CI) | 31.58 (NR) | |
PFS rate at 6 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 12 months (%) (95% CI)e | 73.6 ███ █████ | 26.8 ███ █████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 18 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 24 months (%) (95% CI)e | 62.0 ██ █████ | 10.0 ██ █████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 30 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 36 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 42 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
PFS rate at 48 months (%) (95% CI)e | ███ ██ ████ | ███ ██ ████ |
Difference in survival probability (%) (95% CI) | ███ ██ ████ | |
Median (range) follow-up for PFS in all patients (months) | 22.18 (0.03 to 60.55) | 5.72 (0.03 to 55.06) |
Median (range) follow-up for PFS in censored patients (months) | 27.60 (0.03 to 60.55) | 22.14 (0.03 to 55.06) |
PFS by BICR according to RECIST v1.1 using the midpoint between time of progression and previous nonmissing RECIST assessment (evaluation time bias) | ||
Total PFS events, n (%) | ██ ████ | ██ ████ |
Median PFS (months) (95% CI)e | ██ ████ | ██ ████ |
HR (95% CI)d | ██ ████ | |
2-sided P valued | ██ ████ | |
PFS by BICR according to RECIST v1.1 including actual PFS event times of patient who progressed or died in the absence of progression immediately following 2 or more nonmissing tumour assessments (attrition bias) | ||
Total PFS events, n (%) | ██ ████ | ██ ████ |
Median PFS (months) (95% CI)e | ██ ████ | ██ ████ |
HR (95% CI)d | 0.16 (0.10 to 0.25) | |
2-sided P valued | ██ ████ | |
PFS by BICR according to RECIST v1.1 stratified according to the eCRF | ||
Total PFS events, n (%) | ██ ████ | ██ ████ |
Median PFS (months) (95% CI)e | ██ ████ | ██ ████ |
HR (95% CI)d | 0.16 (0.10 to 0.25) | |
2-sided P valued | ██ ████ | |
PFS by BICR according to RECIST v1.1 in the evaluable for response analysis set | ||
Number of patients | ██ ████ | ██ ████ |
Total PFS events, n (%) | ██ ████ | ██ ████ |
Median PFS (months) (95% CI)e | ██ ████ | ██ ████ |
HR (95% CI)d | 0.14 (0.09 to 0.22) | |
2-sided P valued | ██ ████ | |
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; IxRS = interactive voice/web response system; NC = not calculable; NR = not reported; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours; SAP = statistical analysis plan.
aPFS events that do not occur within 2 scheduled visits (plus visit window) of the last assessment (or randomization) are censored.
bTarget Lesions, Nontarget Lesions, and New Lesions are not necessarily mutually exclusive categories.
cOccurred after 2 or more consecutive missed visits following last nonmissing RECIST assessment (or randomization).
dThe analysis was performed using a log-rank test stratified by disease stage before chemoradiation (IIIA vs IIIB/IIIC) based on values entered into the IxRS, after applying the SAP rule to collapse strata if there are < 10 events per stratum. A HR < 1 favours osimertinib to be associated with a longer PFS than placebo.
eCalculated using the Kaplan-Meier technique.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 6: Sensitivity Analysis — Kaplan-Meier Plot of Progression-Free Survival According to Investigator Assessment, FAS
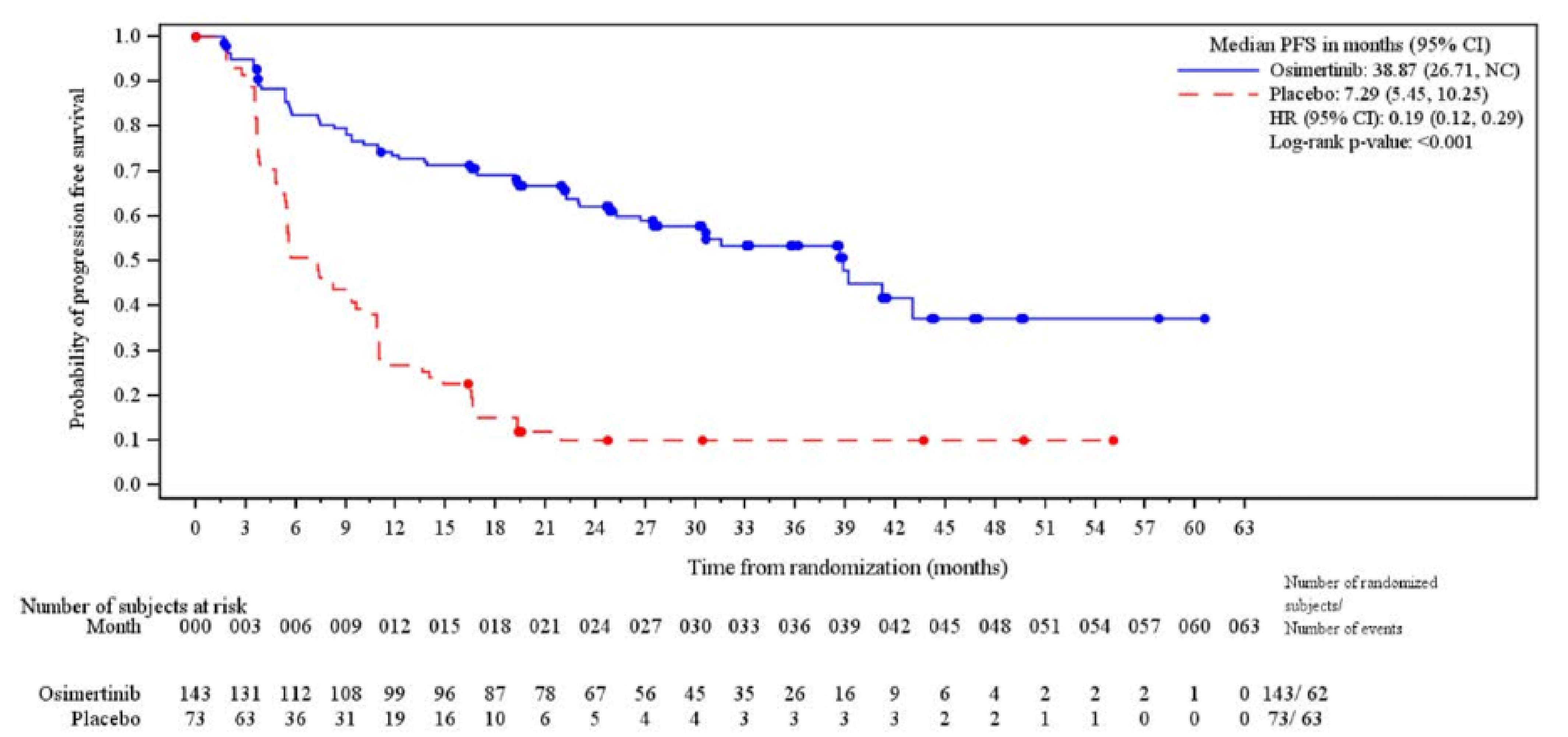
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NC = not calculable.
Note: Circles indicate censored observations.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39
Figure 7: Sensitivity Analysis — Kaplan-Meier Plot of Progression-Free Survival According to BICR With Censoring Flag Reversed to Assess Possible Attrition Bias, FAS
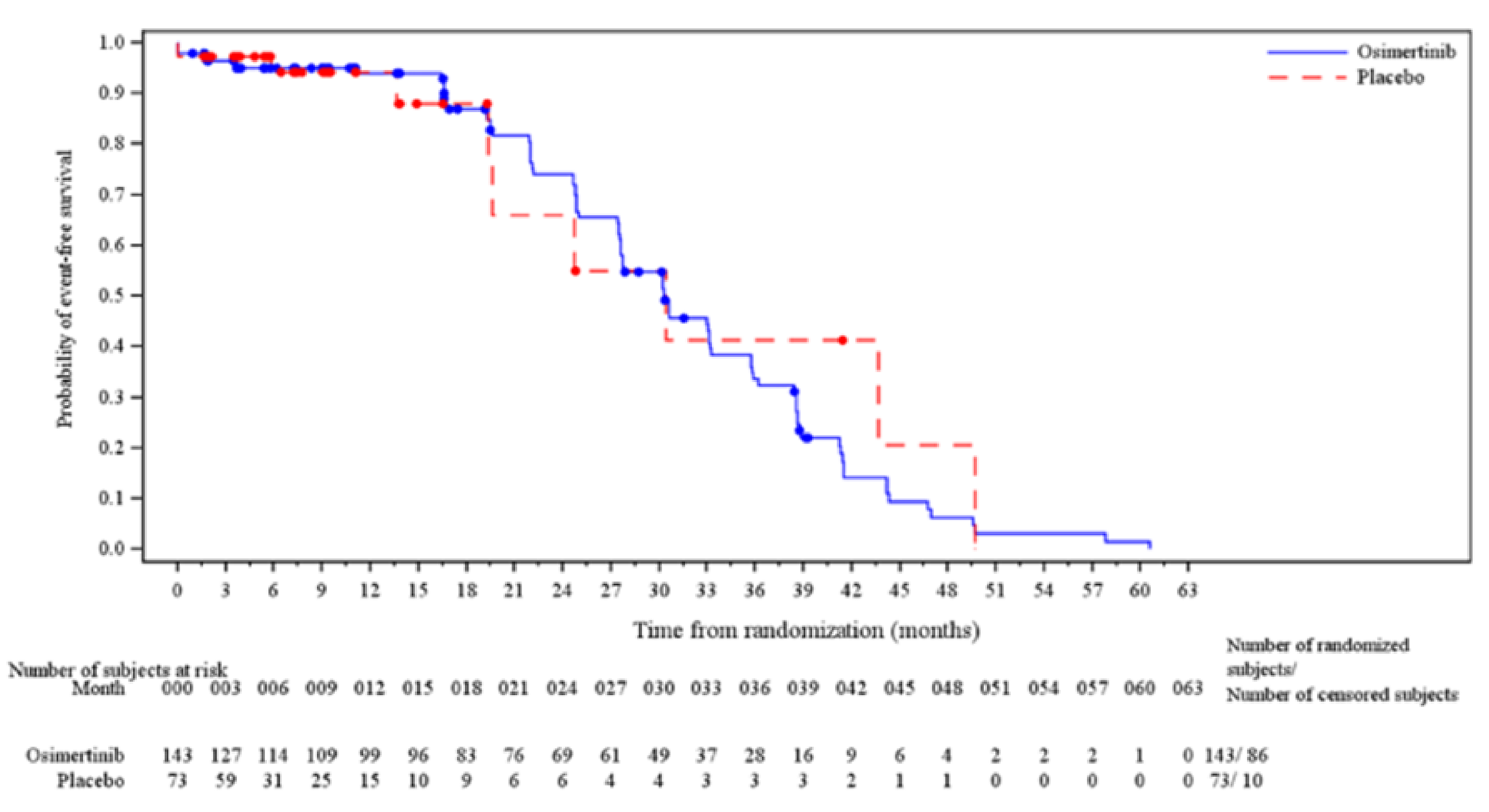
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NC = not calculable.
Note: Circles indicate censored observations.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39
Figure 8: Sensitivity Analysis — Kaplan-Meier Plot of Progression-Free Survival According to BICR, Evaluable for Response Set
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NC = not calculable.
Note: Circles indicate censored observations.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39
Table 20: Sensitivity Analysis: Central Nervous System Progression-Free Survival by Investigator Assessment, FAS
Outcome | Osimertinib N = 143 | Placebo N = 73 |
|---|---|---|
Total PFS events, n (%)a | 32 (22.4) | 28 (38.4) |
Median PFS (months) (95% CI)b | NC (39.16 to NC) | 16.39 (11.10 to NC) |
HR (95% CI)c | 0.19 (0.10 to 0.36) | |
2-sided P valuec | < 0.001 | |
CI = confidence interval; FAS = full analysis set; HR = hazard ratio; IxRS = interactive voice and web response system; NC = not calculable; PFS = progression-free survival.
aProgression determined by RECIST v1.1 assessments.
bThe analysis was performed using a log-rank test stratified by disease stage before chemoradiation (IIIA vs IIIB/IIIC) based on values entered into the IxRS, after applying the SAP rule to collapse strata if there are < 10 events per stratum. A HR < 1 favours osimertinib to be associated with a longer PFS than placebo.
cCalculated using the Kaplan-Meier technique.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 3: The LAURA Trial Subgroup Analyses
Please note that this appendix has not been copy-edited.
Figure 9: Subgroup Analysis — Kaplan-Meier Plot of Progression-Free Survival According to BICR, Evaluable for Response Set
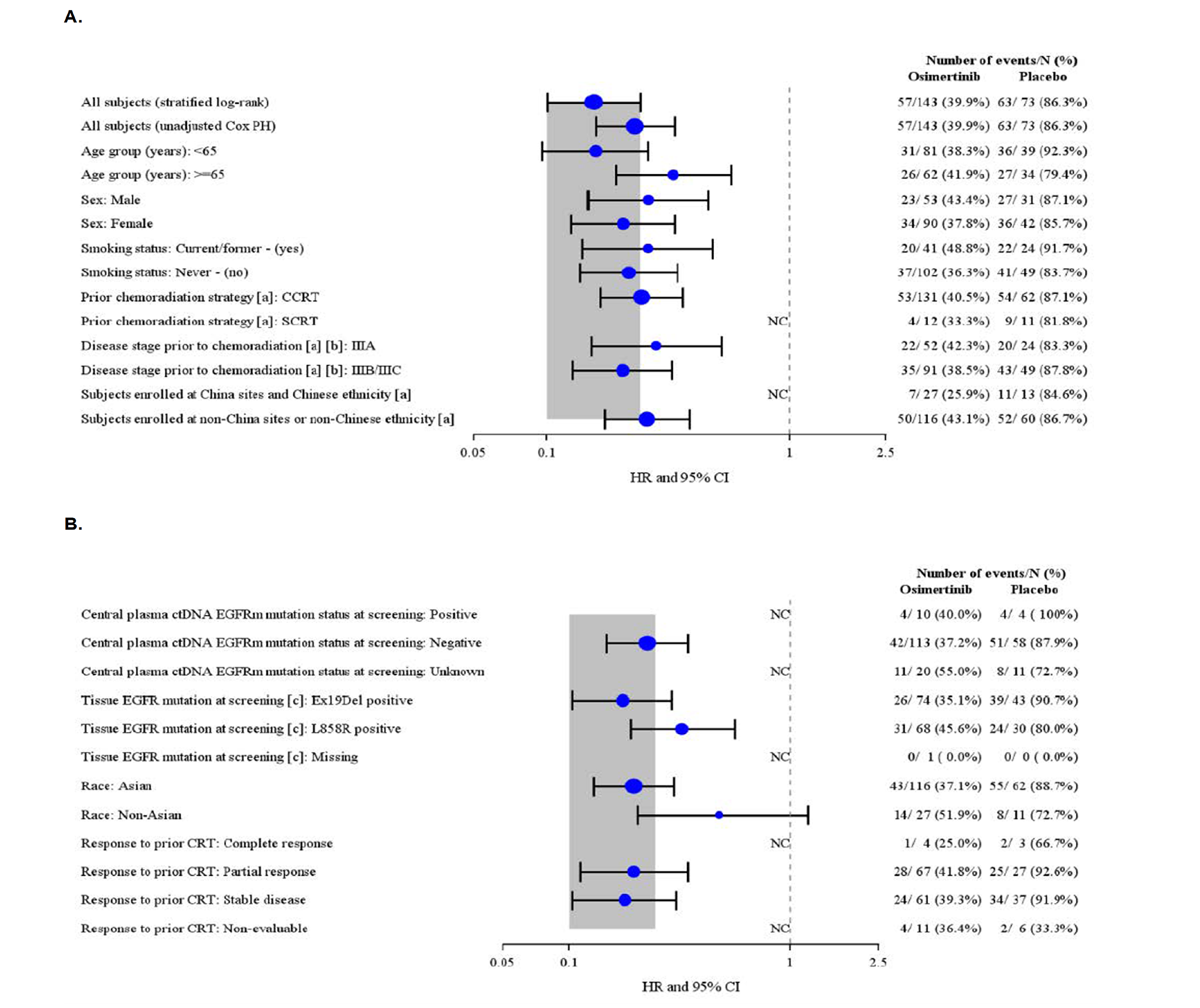
AJCC = American Joint Committee on Cancer; CI = confidence interval; CRT = chemoradiation; cCRT = concurrent chemoradiation; CRF = case report form; ctDNA = circulating tumour DNA; EGFR = epidermal growth factor receptor; FAS = full analysis set; HR = hazard ratio; NC = not calculable; sCRT = sequential chemoradiation therapy.
aSubgroup analyses for the stratification factors are based on the values entered into the CRF.
bTMN Classification of Malignant Tumours, 8th Edition.
cSummarized based on tissue-based central EGFR testing OR local pre-existing EGFR results.
Notes: Circles indicate censored observations.
Grey band represents the 95% CI for the overall HR (stratified log-rank test).
An HR < 1 implies a lower risk of progression on osimertinib than placebo. HRs and 95% CIs are presented where there are > = 20 events across both treatment groups only.
Data cut-off: January 5, 2024.
Source: LAURA Clinical Study Report.39
Table 21: Post Hoc Subgroup Analysis by WHO Performance Status
Subgroup | Hazard ratio (95% CI) |
|---|---|
WHO performance status 0 | 0.17 (0.10 to 0.28) |
WHO performance status 1 | 0.34 (0.20 to 0.56) |
CI = confidence interval.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39
Appendix 4: The LAURA Trial Baseline EORTC QLQ-C30 and EORTC QLQ-LC13 Scores
Please note that this appendix has not been copy-edited.
Table 22: Baseline EORTC QLQ-C30 and EORTC QLQ-LC13 Scores in the LAURA Trial
Functional/symptom scales/items | Score | Osimertinib N = 143 | Placebo N = 73 |
|---|---|---|---|
EORTC QLQ-C30 | n | 131 | 68 |
Global health status/QoL | Mean (SD) | 72.07 ███████ | 71.69 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Physical | Mean (SD) | 88.24 ███████ | 86.76 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Role | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Emotional | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Cognitive | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Social | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Fatigue | Mean (SD) | 18.83 ███████ | 22.39 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Nausea and Vomiting | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Pain | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Dyspnea | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Insomnia | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Appetite loss | Mean (SD) | 14.76 ███████ | 16.67 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Constipation | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Diarrhea | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Financial difficulties | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
EORTC QLQ-LC13 | n | 131 | 68 |
Dyspnea | Mean (SD) | 12.64 ███████ | 18.14 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Coughing | Mean (SD) | 20.61 ███████ | 24.51 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Hemoptysis | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Sore mouth | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Dysphagia | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Peripheral neuropathy | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Alopecia | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Pain in chest | Mean (SD) | 9.92 ███████ | 13.73 ███████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Pain in arm/shoulder | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ | |
Pain in other parts | Mean (SD) | █████ ██████ | █████ ██████ |
Median (min to max) | █████ ██████ | █████ ██████ |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer - Quality of Life Questionnaire 30 item v3; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer - 13 item Lung Cancer Quality of Life Questionnaire; max = maximum; min = minimum; SD = standard deviation.
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Appendix 5: The LAURA Trial ORR and DoR
Please note that this appendix has not been copy-edited.
Objective Response Rate
ORR was 1 of the secondary outcomes of the LAURA trial and was defined as the percentage of patients with at least 1 BICR-assessed visit response of CR (defined as the disappearance of all target lesions since baseline, any pathological lymph nodes selected as target lesions must have had a reduction in short axis to < 10mm) or PR (defined as at least a 30% decrease in the sum of the diameters of target lesions, taking as reference the baseline sum of diameters). A confirmed response of CR or PR meant that a response of CR or PR was recorded at 1 visit and confirmed by repeat imaging not less than 28 days after the visit when the response was first observed with no evidence of progression between the initial and CR or PR confirmation visit.
ORR by BICR was analyzed using a logistic regression stratified by chemoradiation (concurrent versus sequential), disease stage before chemoradiation (IIIA versus IIIB/IIIC) and China cohort (enrolled at a Chinese site and patient declaring themselves of Chinese ethnicity versus enrolled at non-Chinese site or patient declaring themselves of non-Chinese ethnicity). The results of the analysis were presented in terms of an odds ratio together with its associated 95% profile likelihood CI and 2-sided P value.
Duration of Response
DoR was 1 of the secondary outcomes of the LAURA trial and was defined as the time from the date of first documented response until date of documented progression or death in the absence of disease progression using BICR assessments. The end of response should coincide with the date of progression or death from any cause used for the RECIST v1.1 PFS end point. The time of the initial response was defined as the latest of the dates contributing toward the first visit response of PR or CR. If a patient did not progress following a response, then their DoR was the PFS censoring time. All BICR scans were used regardless of whether they were scheduled or not and both confirmed and unconfirmed responses were used. DoR was not defined for patients who did not have documented response. Descriptive data were provided for the DoR in patients responding to treatment based on the BICR RECIST v1.1 assessments in the FAS, including the associated KM curves and median DoR with associated 95% CI (without any formal comparison or P values attached).
Table 23: Summary of Objective Response Rate and Duration of Response in the LAURA Trial
Outcome | Osimertinib N = 143 | Placebo N = 73 |
|---|---|---|
ORR – 05 January 2024 DCOa | ||
Response, n (%) [95% CI]b | 82 (57.3) [48.8 to 65.6] | 24 (32.9) [22.3 to 44.9] |
Complete response | 3 (2.1) | 1 (1.4) |
Partial response | 79 (55.2) | 23 (31.5) |
Nonresponse | 61 (42.7) | 49 (67.1) |
Stable disease (≥ 8 weeks) | 45 (31.5) | 34 (46.6) |
Progression | 11 (7.7) | 12 (16.4) |
Not evaluable | 5 (3.5) | 3 (4.1) |
Odds ratio (95% CI)c | 2.77 (1.54, 5.08) | |
2-sided P valued, e | < 0.001 | |
DoR – 05 January 2024 DCO | ||
Number of respondersf | 82 | 24 |
Responders who subsequently progressed or died, n (%) | 28 (34.1) | 21 (87.5) |
Median DoR, months (95% CI)g | 36.9 (30.1 to NC) | 6.5 (3.6 to 8.3) |
Time to onset of responses, weeksf | ||
Mean (SD) | 25.90 (28.511) | 17.23 (10.203) |
Median (range) | 16.00 (6.9 to 180.3) | 15.79 (7.6 to 40.0) |
Number of confirmed respondersh | 77 | 19 |
Number of responders who subsequently progressed or died | 25 | 16 |
Median DoR, months (95% CI)g | 36.9 (31.6 to NC) | 7.4 (5.5 to 15.4) |
BICR = Blinded Independent Central Review; CI = confidence interval; DCO = data cut-off; DoR = duration of response; IxRS = interactive voice and web response system; KM = Kaplan-Meier; NC = not calculatable; ORR = objective response rate; RECIST = Response Evaluation Criteria in Solid Tumours; SD = standard deviation; vs. = versus.
aResponse includes both confirmed and unconfirmed BICR responses RECIST v1.1 assessments. All BICR scans are used regardless of whether they were scheduled or not. The analysis was performed using logistic regression stratified by disease stage before chemoradiation (IIIA vs. IIIB/IIIC) based on values entered into the IxRS, after applying the SAP rule to collapse strata to analyze the primary end point. An odds ratio > 1 favours osimertinib.
b95% CI calculated using the Clopper-Pearson exact method for binomial proportions.
cBased on the profile likelihood.
dBased on the likelihood ratio test.
eTested outside of the hierarchical testing procedure and were not adjusted for multiplicity.
fDerived from unconfirmed responses only.
gCalculated using KM technique.
hDerived from confirmed responses only (i.e., responses confirmed at least 4 weeks after the initial response).
Data cut-off: January 5, 2024
Source: LAURA Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CRT
chemoradiation therapy
ICER
incremental cost-effectiveness ratio
LY
life-year
NSCLC
non–small cell lung cancer
OS
overall survival
PFS
progression-free survival
PPS
postprogression survival
QALY
quality-adjusted life-year
TDT
time to treatment discontinuation
TTP
time to progression
Executive Summary
The Executive Summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Osimertinib (Tagrisso), 40 mg and 80 mg tablets, oral |
Indication | For the treatment of patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT. |
Submitted price | $322.13 per 40 mg or 80 mg tablet |
Health Canada approval status | NOC/c |
Health Canada review pathway | Priority review; Project ORBIS (Type C) |
NOC date | April 25, 2025 |
Reimbursement request | Per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Osimertinib (Tagrisso) has been reviewed for numerous indications by Canada’s Drug Agency. Indication: In combination with pemetrexed and platinum-based chemotherapy for the first-line treatment of patients with locally advanced (not amenable to curative therapies) or metastatic NSCLC whose tumours have EGFR exon 19 deletions or exon 21 L858R substitution mutations
Indication: Adjuvant therapy after tumour resection in patients with stage IB to stage IIIA NSCLC whose tumours have EGFR exon 19 deletions or L858R substitution mutations
Indication: NSCLC (first-line treatment)
Indication: NSCLC
|
CRT = chemoradiation therapy; NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions; NSCLC = non–small cell lung cancer.
Economic Review
The objective of the economic review is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of osimertinib compared to active surveillance for the treatment of patients with locally advanced, unresectable (stage III) non–small cell lung cancer (NSCLC) whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based chemoradiation therapy (CRT).
Key Messages
Osimertinib is available as an oral tablet (40 mg or 80 mg). At the submitted price of $322.13 per 40 mg or 80 mg tablet, the cost of osimertinib is expected to be $9,020 per 28-day cycle, based on the Health Canada–recommended dosage.
Clinical efficacy in the economic analysis was derived from the LAURA trial, which compared osimertinib with placebo in patients with locally advanced, unresectable NSCLC. Evidence submitted by the sponsor indicates that osimertinib is likely to improve progression-free survival (PFS) at 36 months and may improve overall survival (OS) at 48 months compared to placebo in this patient population.
The results of the Canada’s Drug Agency (CDA-AMC) base case suggest the following:
Osimertinib is predicted to be associated with higher costs to the health care system than active surveillance (incremental costs = $288,697), primarily driven by drug acquisition costs for osimertinib, although there will likely be some cost savings because of reduced subsequent treatment costs.
Osimertinib is predicted to be associated with a gain of 1.56 life-years (LYs) compared to active surveillance resulting in a gain of 1.42 quality-adjusted life-years (QALYs) compared to active surveillance.
The incremental cost-effectiveness ratio (ICER) of osimertinib compared to placebo is $203,741 per QALY gained in the CDA-AMC base case.
The degree of life extension associated with osimertinib versus active surveillance is dependent on the long-term efficacy of osimertinib. If the benefits provided by osimertinib wane over time, for example, due to treatment resistance, then the degree of life extension will be reduced and osimertinib becomes less cost-effective. There is no long-term evidence regarding the efficacy of osimertinib, meaning that long-term predictions of OS benefit are uncertain.
CDA-AMC estimates that the budget impact of reimbursing osimertinib for the indicated population will be approximately $19.6 million over the first 3 years of reimbursement compared to the amount currently spent on active surveillance, with an estimated expenditure of $45.1 million on osimertinib over this period. The budget impact of reimbursing osimertinib will depend on the number of people eligible for treatment, the uptake of osimertinib, and subsequent treatment costs.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of osimertinib from the perspective of a public health care payer in Canada over a lifetime horizon (25 years). The modelled population was adults with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations and whose disease has not progressed during or following platinum-based CRT. This aligned with the Health Canada–approved indication and participants in the LAURA trial. The sponsor’s base-case analysis included costs related to drug acquisition, disease management, adverse events, and terminal care.
In the sponsor’s base case, osimertinib was associated with incremental costs of $276,029 and 1.71 incremental QALYs relative to active surveillance. This resulted in an ICER of $161,884 per QALY gained. Of the incremental benefit compared to active surveillance, approximately 83% of the benefit of osimertinib was predicted to be accrued after the observation period of the LAURA trial. Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The sponsor assumed that the risk of disease progression decreased over time for patients receiving osimertinib. | In the LAURA trial, at 30 months postrandomization, the risk of progression increased in the osimertinib arm, though patient numbers are small. There are no data to suggest risk of disease progression for those who received osimertinib continues to decrease over time. | In the base-case analysis, CDA-AMC assumed a constant risk of disease progression over time for patients receiving osimertinib. | To explore uncertainty around this issue, CDA-AMC conducted a scenario analysis in which a treatment waning effect was assumed. |
The sponsor used a European value set to derive utility estimates rather than a Canadian one. | EQ-5D-5L was gathered in the LAURA trial. This can be turned into utilities using country-specific value sets of which a Canadian set is available. | In the base-case analysis, CDA-AMC used the Canadian EQ-5D-5L value set to derive utility estimates. | No scenario analysis was conducted. |
Long-term estimations of PPS are uncertain. | Given the absence of evidence, the sponsor’s projections of PPS may not accurately capture the long-term survival of patients who experience disease recurrence. | CDA-AMC did not address this limitation in the base-case analysis because there was insufficient evidence to suggest an alternative extrapolation of long-term PPS was more plausible. | To explore uncertainty around the issue, CDA-AMC conducted a scenario analysis in which alternative distributions were used to model PPS. |
Cost savings associated with subsequent treatment use are uncertain given the negotiated price of osimertinib for patients with progressed disease is unknown. | Osimertinib is currently used as a subsequent therapy for patients who progress. By using osimertinib in an earlier stage this will reduce its use in later lines. The cost savings associated with this will be influenced by any negotiated price arrangement currently in place. | CDA-AMC did not address this limitation in the base case because the negotiated price of osimertinib as subsequent treatment is unknown. | To explore uncertainty around the issue, CDA-AMC conducted a scenario analysis in which the price of osimertinib used as subsequent treatment in the progressed disease state was reduced. |
Correlation between events, for example, TTP and TDT, was not accounted for in the probabilistic analysis. | When assessing the uncertainty of PFS, TTP, and TDT in a probabilistic analysis, these events should be correlated. For example, if disease progression is expected to occur at a faster rate, patients will likely discontinue treatment sooner. | In the base-case analysis, CDA-AMC assumed that patients who progress discontinue treatment at their time of progression. The model was programmed such that TTP influences PFS, and TDT is equal to TTP. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; PFS = progression-free survival; PPS = postprogression survival; TDT = time to treatment discontinuation; TTP = time to progression.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 4.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the base case is provided in Appendix 4.
Impact on Health Care Costs
Osimertinib is predicted to be associated with additional health care costs compared to active surveillance (incremental costs = $288,697). The additional costs and cost savings associated with osimertinib relative to active surveillance can be derived from Figure 1. The increase in health care spending primarily results from drug acquisition costs with osimertinib in the indicated population ($526,351). There will be cost savings ($236,742) associated with reducing the use of osimertinib in later treatment lines as shown by the reduction in subsequent treatment costs. All other costs to the health care system are expected to be similar between osimertinib and active surveillance.
Figure 1: Impact of Osimertinib Versus Active Surveillance on Health Care Costs
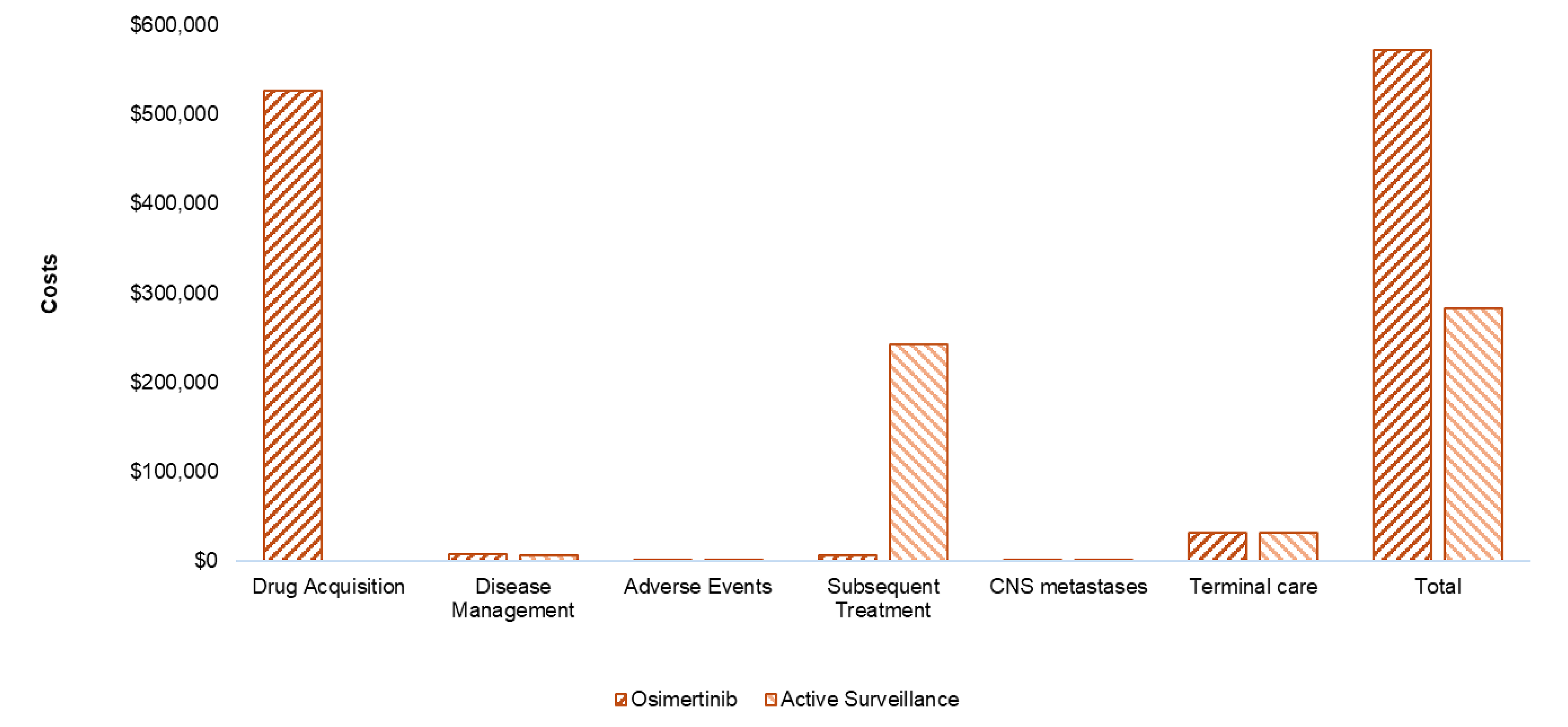
CNS = central nervous system.
Impact on Health
Relative to active surveillance, osimertinib is predicted to result in 1.42 additional QALYs per patient over the lifetime time horizon (refer to Figure 2). These QALY gains are associated with osimertinib prolonging time spent progression-free, relative to active surveillance. In the progression-free health state, patients have a better quality of life and a reduced risk of death. Approximately 83% of the predicted incremental benefit was accrued based on extrapolation where there are no trial data.
Figure 2: Impact of Osimertinib Versus Active Surveillance on Patient Health
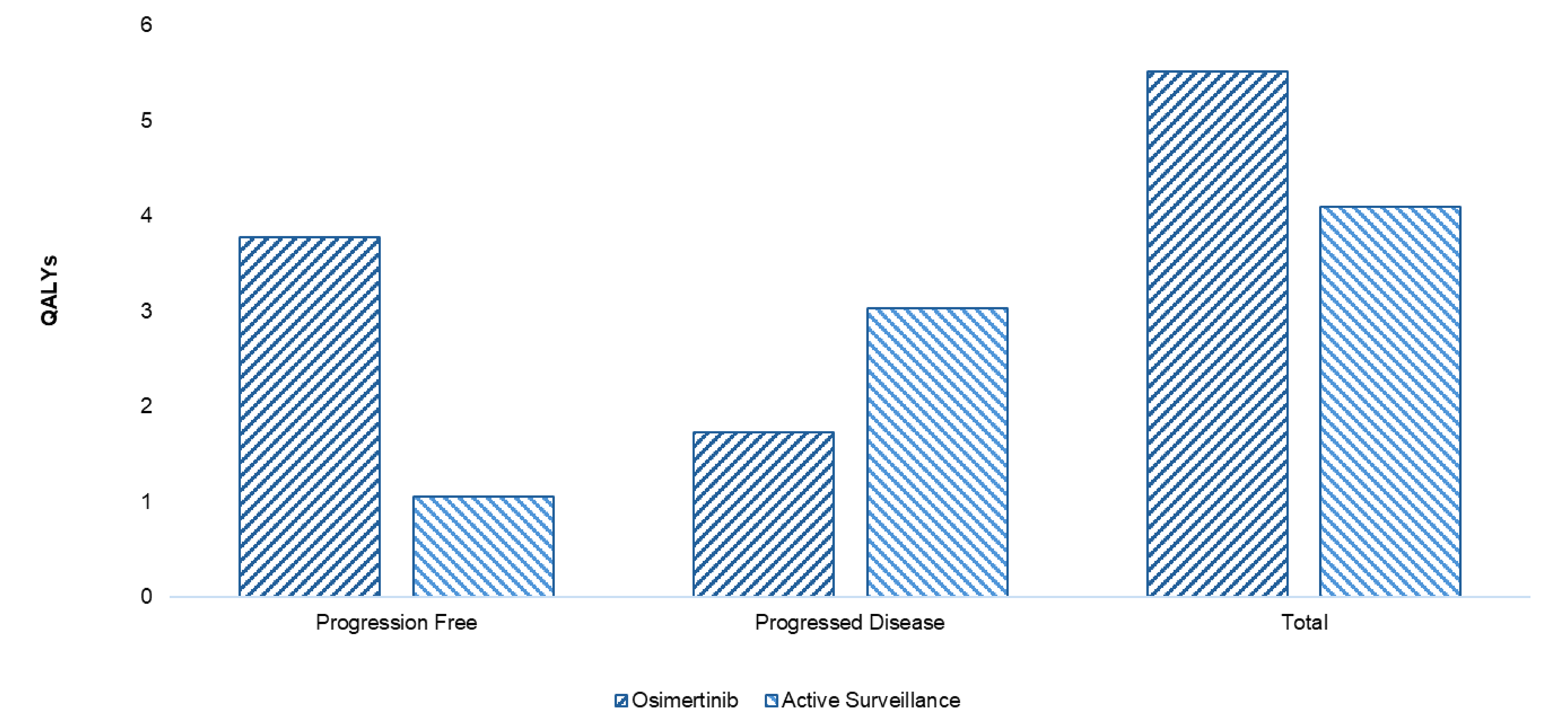
QALY = quality-adjusted life-year.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $203,741 per QALY gained for osimertinib compared to active surveillance (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Total LYs | ICER vs. active surveillance ($/QALY) |
|---|---|---|---|---|
Active surveillance | 282,982 | 4.10 | 4.95 | Reference |
Osimertinib | 571,679 | 5.51 | 6.51 | 203,741 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Uncertainty and Sensitivity
Uncertainty was explored through probabilistic analysis as well as through scenario analyses outlined in Table 2. In a scenario analysis that assumes a treatment waning effect, incremental QALYs decrease from 1.5 to 0.5 as life extension from osimertinib decreases. However, incremental costs decrease from $293,492 to $151,290 as patients spend less time on osimertinib given that they are treated until progression. Overall, the ICER increases from $195,354 to $290,971 per QALY gained. A scenario analysis which assumes a negotiated price for osimertinib exists in the later lines and this scenario increases the ICER by $15,323 per QALY for each 10% reduction in osimertinib’s list price. For example, if the negotiated price for osimertinib was 50% lower than the list price in the subsequent treatment line, the ICER only increases to $271,971 per QALY gained. This demonstrates that cost-effectiveness is contingent on the price paid for osimertinib in later lines. Finally, a scenario analysis that extended long-term survival for patients who experience progression increases the ICER to $290,971 per QALY gained. Long-term evidence is needed to confirm the magnitude of life extension osimertinib may provide relative to active surveillance.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing osimertinib for use in the Health Canada–indicated population.1 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of osimertinib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC estimated that 467 patients would be eligible for treatment with osimertinib, in the locally advanced or unresectable stage III setting, over a 3-year period (year 1 = 152; year 2 = 156; year 3 = 159), of whom 350 are expected to receive osimertinib (year 1 = 91; year 2 = 117; year 3 = 142). The estimated incremental budget impact of reimbursing osimertinib is predicted to be approximately $19.6 million over the first 3 years, with an expected expenditure of $45.1 million on osimertinib in the locally advanced or unresectable stage III setting.
Conclusion
Based on the CDA-AMC base case, osimertinib would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $203,741 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 9). There is uncertainty in the base-case analysis due to uncertainties in long-term health outcomes for those who receive osimertinib as well as the impact confidential price agreements have on cost savings in the subsequent treatment setting. The base-case analysis may therefore overestimate QALY gains and cost savings, meaning further price reductions than those presented may be required.
The budget impact of reimbursing osimertinib for the public drug plans in the first 3 years is estimated to be approximately $19.6 million. The 3-year expenditure on osimertinib in the locally advanced or unresectable stage III setting (i.e., not accounting for expenditure on osimertinib in later lines) is estimated to be $45.1 million.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
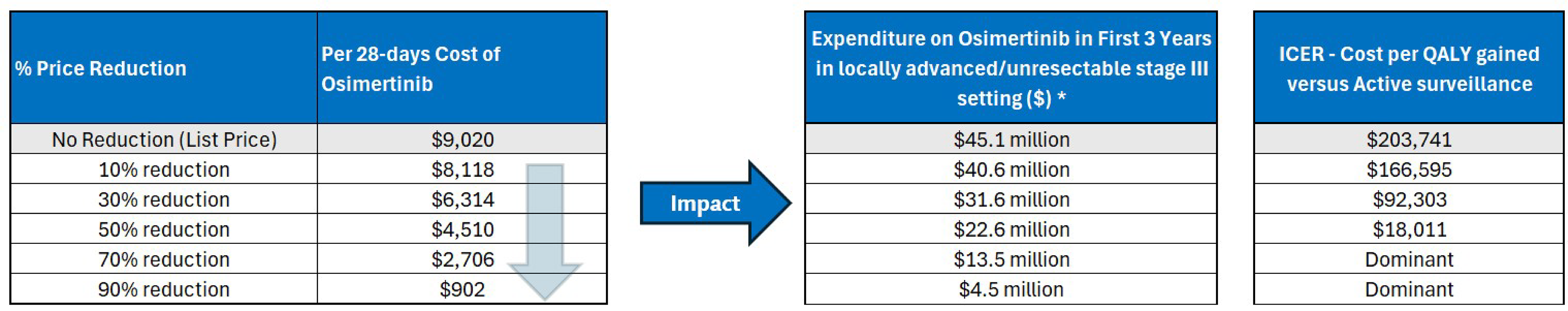
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: “Expenditure” includes only the drug cost of osimertinib. The term “dominant” indicates that a drug costs less and provides more QALYs than the comparator.
*This excludes expenditures on osimertinib in later-line settings.
References
1.AstraZeneca Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tagrisso (osimertinib tablets): tablets, 40 mg and 80 mg osimertinib (as osimertinib mesylate), oral. February 13, 2025.
2.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed February 13, 2025. https://www.formulary.health.gov.on.ca/formulary/
3.Ontario Ministry of Health. Exceptional Access Program (EAP). Accessed February 13, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
4.Iqvia Canada. IQVIA Delta Price Advisor. Accessed October 2023. [sponsor submitted reference].
5.Ontario Ministry of Health and Long-Term Care. Ontario Schedule of Benefits and Physician Services. [sponsor submitted reference]. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
6.Canadian Institute for Health Information. CIHI Patient Cost Estimator [sponsor submitted reference]. https://www.cihi.ca/en/patient-cost-estimator
7.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tagrisso (osimertinib tablets): tablets, 40 mg and 80 mg osimertinib (as osimertinib mesylate), oral. 2025 Feb 13.
8.Lu S, Kato T, Dong X, et al. Osimertinib after Chemoradiotherapy in Stage III EGFR-Mutated NSCLC. N Engl J Med. 2024;391(7):585-597. doi: 10.1056/NEJMoa2402614 PubMed
9.Planchard D, Jänne PA, Cheng Y, et al. Osimertinib with or without Chemotherapy in EGFR-Mutated Advanced NSCLC. N Engl J Med. 2023;389(21):1935-1948. doi: 10.1056/NEJMoa2306434 PubMed
10.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex [sponsor submitted reference]. https://doi.org/10.25318/1710000501-eng
11.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2018/19 [sponsor submitted reference]. https://www.sac-isc.gc.ca/DAM/DAM-ISC-SAC/DAM-HLTH/STAGING/texte-text/nihb-Annual_Report_2018-19_1589921777815_eng.pdf
12.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2020 to 2021 [sponsor submitted reference]. https://www.sac-isc.gc.ca/eng/1645718409378/1645718500555#chp2
13.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2021 to 2022 [sponsor submitted reference]. https://www.sac-isc.gc.ca/eng/1683039690813/1683039973755
14.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2019 to 2020 [sponsor submitted reference]. https://www.sac-isc.gc.ca/eng/1624462613080/1624462663098
15.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2022 to 2023 [sponsor submitted reference]. https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612
16.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-e623. doi: 10.1503/cmaj.240095 PubMed
17.Cancer Care Ontario. Ontario Cancer Facts: Lung cancer incidence higher in First nations people than other people in Ontario. [sponsor submitted reference]. https://www.cancercareontario.ca/en/cancer-facts/lung-cancer-incidence-higher-first-nations-people-other-people-ontario
18.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer [sponsor submitted reference]. www.cancer.ca/Canadian-Cancer-Statistics-2020-EN
19.Boyne DJ, Jarada T, Yusuf A, et al. 51P Testing patterns and outcomes of different EGFR-positive metastatic non-small cell lung cancer (NSCLC) patients in a Canadian real-world setting. Ann Oncol. 2022;33:S56. doi: 10.1016/j.annonc.2022.02.060
20.CADTH. CADTH Reimbursement Review Reports. Amivantamab (Rybrevant). Non-small cell lung cancer [sponsor submitted reference]. https://www.cadth.ca/sites/default/files/DRR/2023/PC0289-Rybrevant_combined.pdf
21.O'Sullivan DE, Jarada TN, Yusuf A, et al. Prevalence, Treatment Patterns, and Outcomes of Individuals with EGFR Positive Metastatic Non-Small Cell Lung Cancer in a Canadian Real-World Setting: A Comparison of Exon 19 Deletion, L858R, and Exon 20 Insertion EGFR Mutation Carriers. Curr Oncol. 2022;29(10):7198-7208. doi: 10.3390/curroncol29100567 PubMed
22.Shang S, Wang R, Wang F, Wu M, Chen D, Yu J. Treatment Patterns for Patients With Unresected Stage III NSCLC: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database. Front Oncol. 2022;12. doi: 10.3389/fonc.2022.874022 PubMed
23.Karacz CM, Yan J, Zhu H, Gerber DE. Timing, Sites, and Correlates of Lung Cancer Recurrence. Clin Lung Cancer. 2020;21(2):127-135.e3. doi: 10.1016/j.cllc.2019.12.001 PubMed
24.CADTH. CADTH Reimbursement Review. Osimertinib (Tagrisso) for NSCLC (PC0246) [sponsor submitted reference]. https://www.cda-amc.ca/sites/default/files/DRR/2022/PC0246-Tagrisso.pdf
25.Tsuboi M, Herbst RS, John T, et al. Overall Survival with Osimertinib in Resected EGFR-Mutated NSCLC. N Engl J Med. 2023;389(2):137-147. doi: 10.1056/NEJMoa2304594 PubMed
26.AstraZeneca Data on File. LAURA Clinical Study Report (data cut-off: January 5, 2024) [sponsor submitted reference].
27.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed February 13, 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
28.BC Cancer. Drug Funding. [sponsor submitted reference]. http://www.bccancer.bc.ca/health-professionals/clinical-resources/pharmacy/drug-funding
29.AB Health Services. Outpatient Cancer Drug Benefit Program. Programs & Services [sponsor submitted reference]. https://www.albertahealthservices.ca/findhealth/Service.aspx?id=1025651
30.Saskatchewan Cancer Agency. Frequently Asked Questions [sponsor submitted reference]. https://saskcancer.ca/health-care-professionals/drug-formulary#:~:text=Who%20pays%20for%20cancer%20drugs,cancer%20treatments%20taken%20at%20home
31.Cancer Care Manitoba. Manitoba Home Cancer Drug Program [sponsor submitted reference]. https://www.cancercare.mb.ca/Treatments/pharmacy/home-cancer-drug-program
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for the Treatment of Unresectable Stage III Non–Small Cell Lung Cancer
Treatment | Strength/concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 28-day cycle cost ($) |
|---|---|---|---|---|---|---|
Osimertinib (Tagrisso) | 40 mg 80 mg | Tablet | 322.1280a | 80 mg once daily | 322.13 | 9,020 |
aSponsor’s submitted price.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from a joint submission by the Lung Health Foundation, Lung Cancer Canada, and the Canadian Cancer Survivor Network. Patient input was gathered from an online survey of 20 patients and 3 caregivers conducted between July and December 2024 and from 6 telephone interviews conducted in January 2025. All respondents resided in Canada. Patients described fatigue as their most detrimental lung cancer symptom. Patients cited being unable to work, negative emotional well-being, financial cost burden, and concern for family members as key issues. Patients highlighted the importance of new treatment options that enhance quality of life and alleviate symptoms, given the side effects of current treatments. Most patients had a strong preference for osimertinib over other treatments, and those with experience with osimertinib described increased quality life. Patients who experienced side effects (e.g., changes in appearance, fatigue, and diarrhea) while taking osimertinib said they were largely manageable.
Clinician group input was received from the Medical Advisory Committee of Lung Cancer Canada and the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee. Clinician input indicated that consolidative immunotherapy with durvalumab is not used in clinical practice for patients with EGFR mutations. The clinician groups indicated that osimertinib would rapidly become the standard of care among patients with NSCLC with EGFR mutations without disease progression post concurrent CRT. Clinician input noted that the goal of treatment is to delay or prevent disease recurrence, and for patients to survive without cancer recurrence for 5 years. The clinician groups indicated that osimertinib could be used in both academic and nonacademic oncology clinics, including in outpatient settings.
Input from CDA-AMC–participating drug plans was not received.
Several of these concerns were addressed in the sponsor’s model:
The sponsor explored the quality of life and mortality impact associated with disease recurrence.
Side effects associated with treatment were considered.
Durvalumab was not considered a relevant comparator.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of osimertinib the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Osimertinib (Tagrisso) |
Submitted price of drug under review | $322.13 per 40 mg tablet $322.13 per 80 mg tablet |
Regimen | 80 mg per day |
Per-course cost of drug under review | $9,020 per patient every 28 days |
Model information | |
Type of economic evaluation | Cost-utility analysis Markov model |
Treatment | Osimertinib |
Included comparator | Placebo |
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (25 years) |
Cycle length | 30 days |
Modelled population | Patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations whose disease has not progressed during or following platinum-based CRT |
Characteristics of modelled population | Derived from the LAURA trial (61% female, 39% male, mean age = 61.4) |
Model health states |
For additional information, refer to Model Structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs included in the model |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis results | Results that differed from the sponsor’s base case by more than 20%
|
AE = adverse event; CIHI = Canadian Institute for Health Information; CRT = chemoradiation therapy; EAP = exceptional access program; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; TDT = time to treatment discontinuation; TTP = time to progression.
Model Structure
The sponsor submitted a Markov model with 3 health states: progression-free, progressed disease, and dead (Figure 4). Patients entered the model in the progression-free state. Over time patients will either progress and move to the progressed disease state or die. Upon progression the only remaining movement in the model is to the death state. Treatment duration in the progression-free state was derived from extrapolated time to treatment discontinuation (TDT) curves. As patients receiving osimertinib were treated until progression in the LAURA trial, the TDT curve was consistent with the PFS curve.
The movement of patients between health states over the model horizon was derived from parametric survival modelling. Transitions from the progression-free state to the progressed disease state (i.e., TP1) were derived from time to progression (TTP) curves seen in the LAURA trial. Upon entry into the progressed disease state, a proportion of patients receive subsequent treatment, which affects costs only. Transitions from the progression-free state to the dead state (i.e., TP2) were derived based on differences between PFS curves and TTP curves. Transitions from the progressed disease state to the dead state (i.e., TP3) were derived from postprogression survival (PPS) curves from the LAURA trial.
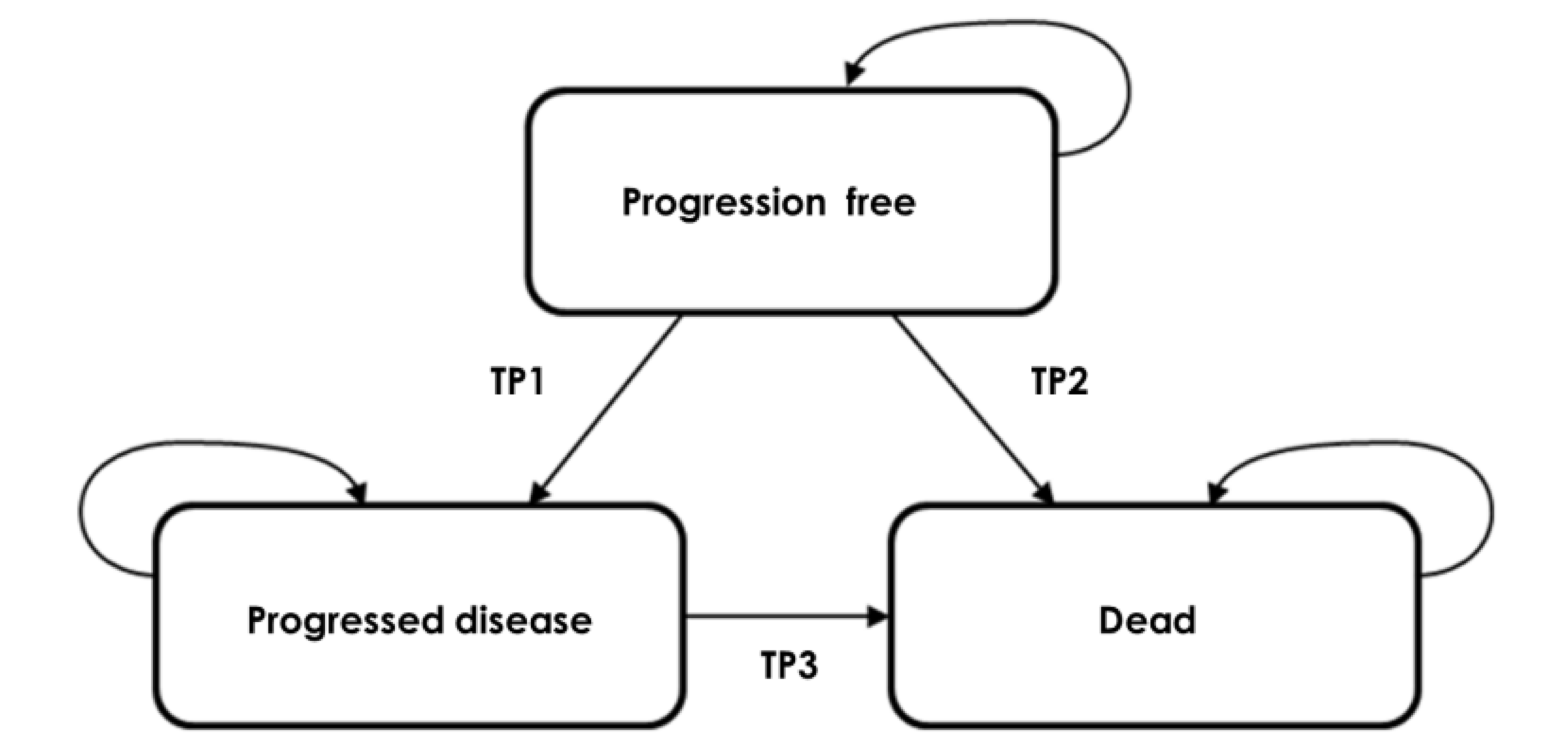
TP1 = transition probability 1, or the transition probability from progression-free to progressed disease; TP2 = transition probability 2, or the transition probability from progression-free to dead; TP3 = transition probability 3, or the transition probability from progressed disease to dead.
Source: Sponsor’s pharmacoeconomic submission.7
Appendix 4: Additional Details of the CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC clinical review found osimertinib results in clinically important improvement in PFS among patients with locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substation mutations and have not progressed during or following platinum-based CRT compared to placebo, based on evidence from the LAURA trial (follow-up of 36 months). Evidence from LAURA trial suggests that osimertinib may result in a clinically important increase in OS at 48 months compared to placebo. The CDA-AMC clinical review found that osimertinib may result in little to no difference in health-related quality of life.
Key clinical efficacy inputs for osimertinib were derived from the LAURA trial. The sponsor used survival models to estimate PFS, TTP, and PPS from patient-level data from the LAURA trial. Models were selected based on clinical validity and statistical fit. Data extrapolation for all efficacy inputs was required because some data from the trial were immature, and the sponsor’s economic model used a 25-year time horizon. The proportion of patients receiving subsequent treatments after discontinuation for each arm was based on data from the LAURA trial. Because of uncertainty in the clinical effectiveness data owing to immature data, the estimated gain predicted by the economic model in LYs, QALYs, and hence the overall cost-effectiveness, are uncertain.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The impact of osimertinib on long-term TTP is uncertain: In the submitted economic evaluation, the sponsor relied on parametric survival modelling to extrapolate the TTP and PFS beyond the observed time points in the LAURA trial. For the osimertinib arm, the TTP survival data from the LAURA trial were less than 40% mature. The sponsor fitted a gamma distribution to the observed osimertinib TTP Kaplan-Meier curve to determine the time spent in the progression-free health state over the 25-year time horizon. This assumes that the probability of progression will continue to decrease over time, indicating that over time osimertinib becomes more effective at preventing progression.
██████ ████ ████████ ███████████ ███████ ██ ███████████ ████ █████ ███ ██████ █ ████ ███████████ ████ ███████████████ ██████ ████████ ███████ ██████████ ████████ ███ ████████████ ████████████ ████████████ █████ ████████ █████████ ████████ ████████████ █████ ███████████ ████████ ██████ ████ ████ ███████████████ ████ █████ ████████████████ ████████████ █████ ██████ ███████ ███████████ ██████████████ █████████████ ██████████████ ██████████████ ██████ ████ ████████ (refer to Figure 5).
Figure 5: Hazards of TTP [Redacted]

TTP = time to progression.
Note: Smoothed hazards of TTP for osimertinib and placebo from the LAURA trial.8
Source: Sponsor’s pharmacoeconomic submission.7
██████ ████ ████████ ███████████ ███████ ██ ███████████ ████ █████ ███ ██████ ███████ ███████████ █████ ████████████ ████ ███████ █████████ ████████████████ ████ █████ █████ █████ ████████ █████████ ████████ ████████████ █████ ██████████ ████████ ██████ ████ ████ ███████████████ ████ █████ ████████████████ ██████████ █████ ██████ ███████ ███████████ ██████████████ █████████████ ██████████████ ████████████ ██████ ████ ████████ No evidence was presented which showed an improving treatment effect over time. None of the parametric distributions (i.e., exponential, Gompertz, log-normal, gamma, Weibull, log-logistic, and generalized gamma) considered by the sponsor captured an increasing hazard over time for those receiving osimertinib.
In consultation with clinical experts, an alternative parametric distribution with a waning treatment effect was considered plausible.
Given this limitation, and consistent with clinical expert opinion, the CDA-AMC base case used an exponential distribution for the extrapolation of the TTP curve for the osimertinib arm. This assumes that the treatment effect is constant over time.
As a scenario analysis, a treatment waning effect was explored. Figure 6 outlines what PFS and OS look like for patients who receive osimertinib in this scenario analysis.
Utilities do not reflect the population of Canada: In the base case of the submitted economic evaluation, health state utility values were derived using European Quality of Life 5-dimension 5 level questionnaire (EQ-5D-5L) from the LAURA trial. However, the sponsor did not use the Canadian value set to derive the utility values. The Canadian value set is derived using a representative sample of the population of people living in Canada and is therefore the most appropriate value set to derive utilities.
The CDA-AMC base case used the sponsor-calculated EQ-5D-5L Canadian value set for health state utility values.
TTP, PFS, and TDT are modelled independently: TTP was calculated from a reanalysis of PFS data from the LAURA trial. TDT was the time from randomization to the earliest date of study treatment discontinuation or death from the LAURA trial. When analyzed probabilistically, TDT and TTP are sampled independently. This assumes that there is no correlation between these 2 events. As progression is 1 of the main reasons for discontinuing treatment these 2 variables should be heavily correlated. Likewise, the sponsor also models PFS independently from TTP. PFS is made up of progression and preprogression deaths; therefore, TTP and PFS are directly correlated. Because of this discrepancy, results from the probabilistic analysis were inaccurate as they sample scenarios not considered clinically implausible, for example TTP decreases while time on treatment increases.
In the CDA-AMC base case, TDT was set to be equal to TTP in the osimertinib and placebo arms as data showed the 2 events were similar (refer to Figure 7).
Reviewing the data, PFS events are made up predominantly of progression events. Preprogression death is rare in this patient population and most patients die after progression occurs. Therefore, PFS and PPS are expected to be similar. CDA-AMC made PFS a function of TTP in the probabilistic analysis. For patients receiving osimertinib a hazard ratio of 1.075 was applied to TTP to derive the PFS curve. For patients receiving active surveillance a hazard ratio of 1.025 was applied to TTP to derive PFS. This ensured the PFS curves derived using this approach matched the curves derived from the survival analysis. Then when the model is run probabilistically the approach avoids the clinically implausible option of PFS exceeding TTP.
Long-term PPS is uncertain: In the submitted economic evaluation, the sponsor fitted Gompertz distributions to the observed PPS Kaplan-Meier curves for osimertinib and placebo to determine the time spent in the progressed disease state over the patient’s lifetime. Given the absence of long-term data, it is unclear what long-term PPS will look like for these patients. Clinical experts consulted for this review noted that patients who receive osimertinib in the progressed disease state may survive longer than what was presented by the sponsor.
Second, if osimertinib has a long-term treatment benefit in the first-line setting then this assumption may hold for later lines as well. Therefore, assumptions around treatment waning should be considered in both the first-line and subsequent settings of the model
In a CDA-AMC scenario analysis, and consistent with clinical expert opinion, generalized gamma distributions were used to extrapolate PPS Kaplan-Meier curves for osimertinib and placebo.
Time spent on subsequent therapies is uncertain: The duration of treatment in the progressed disease state for patients receiving osimertinib was informed by the FLAURA2 clinical trial.9 Participants of the FLAURA2 trial are not directly exchangeable with participants in the progressed disease state of the LAURA trial. For example, 97% of patients in the osimertinib monotherapy arm of the FLAURA2 trial had metastatic disease at trial entry and 40% had central nervous system metastases. In the placebo arm of the LAURA trial, distant metastases occurred in 37% of participants and new brain metastases occurred in 29% of participants.8 The length of time on treatment in the progressed disease state may therefore be misestimated in the sponsor’s model.
CDA-AMC was unable to address the limitations concerning the length of treatment in the progressed disease state. Given that patients who are metastatic may have a worse prognosis they may spend less time on treatment meaning the analysis may underestimate subsequent therapy costs and overestimate the ICER.
The progressed disease state is not homogenous: As assumption of Markov modelling is that all patients within a given health state are broadly homogenous, experiencing similar utilities, costs, and probabilities of death and progression. In the analysis, the progressed disease state is made up of patients who are experiencing a locally advanced recurrence as well as a distant metastatic recurrence. A distant metastatic recurrence is associated with a lower quality of life and a worse prognosis. By not accurately modelling this, the model derives less accurate predictions.
CDA-AMC was unable to address this limitation.
Additional issues were identified but were not considered to be key issues: The development of central nervous system metastases was assigned a one-off cost, but otherwise, the costs were the same for all patients in the progressed disease health state. The cost of osimertinib as a subsequent treatment is uncertain.
Figure 6: PFS and OS for the Treatment Waning Scenario
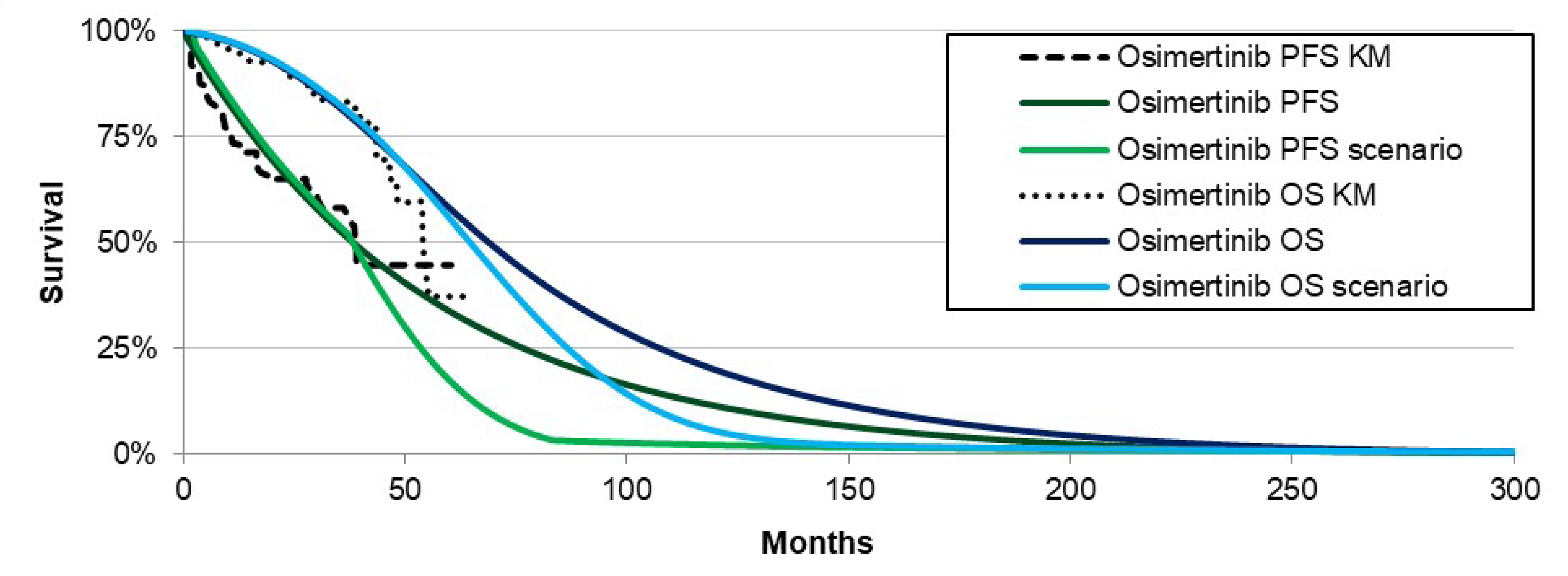
KM = Kaplan-Meier; OS = overall survival; PFS = progression-free survival.
Figure 7: KM Data on Time on Treatment Versus TTP [Redacted]

KM = Kaplan-Meier; TTP = time to progression.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 6). The base-case analysis was run probabilistically (1,000 iterations). The impact of these changes, individually and collectively, is presented in Table 7.
Table 6: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Alignment of TDT, TTP, and PFS | TDT, TTP, and PFS are sampled independently | Osimertinib:
Active surveillance:
|
2. Parametric extrapolation of TTP and TDT for the osimertinib arm | Parametric distribution of TTP:
Parametric distribution of TDT:
| Parametric distribution of TTP:
Parametric distribution of TDT:
|
3. Health utilities | Health state utilities ████ ████████ | Health state utilities PF: 0.89 PD: 0.83 |
CDA-AMC base case (health care payer perspective) | ― | 1 + 2 + 3 |
CDA-AMC = Canada’s Drug Agency; PD = progressed disease; PF = progression-free; PFS = progression-free survival; TDT = time to treatment discontinuation; TTP = time to progression.
Note: CDA-AMC was unable to resolve the issues with the duration of subsequent treatment and heterogeneity within the progressed disease state.
Table 7: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Active surveillance | 276,827 | 3.84 | Reference |
Osimertinib | 628,021 | 5.75 | 184,147 | |
CDA-AMC reanalysis 1 | Active surveillance | 276,719 | 3.85 | Reference |
Osimertinib | 640,921 | 5.82 | 184,819 | |
CDA-AMC reanalysis 2 | Active surveillance | 276,827 | 3.84 | Reference |
Osimertinib | 557,182 | 5.37 | 183,912 | |
CDA-AMC reanalysis 3 | Active surveillance | 276,827 | 3.94 | Reference |
Osimertinib | 628,021 | 5.83 | 185,450 | |
CDA-AMC base-case reanalysis 1 + 2 + 3 (deterministic) | Active surveillance | 276,719 | 3.95 | Reference |
Osimertinib | 570,211 | 5.45 | 195,354 | |
CDA-AMC base-case reanalysis 1 + 2 + 3 (probabilistic) | Active surveillance | 282,982 | 4.10 | Reference |
Osimertinib | 571,679 | 5.51 | 203,741 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 8: Disaggregated Results of the CDA-AMC Base Case
Parameter | Osimertinib | Active surveillance |
|---|---|---|
Undiscounted LYsa | ||
Total | 6.95 | 5.27 |
Progression-free | 4.59 | 1.31 |
Progressed disease | 2.35 | 3.96 |
Discounted QALYs | ||
Total | 5.52 | 4.14 |
Progression-free | 3.78 | 1.08 |
Progressed disease | 1.74 | 3.06 |
Discounted costs ($) | ||
Total | 571,994 | 274,150 |
Drug acquisition | 526,395 | 0 |
Disease management | 7,350 | 7,052 |
Adverse events | 11.39 | 2.27 |
Subsequent treatment | 6,119 | 233,779 |
CNS metastases | 225 | 752 |
Terminal care | 31,844 | 32,565 |
aDiscounted LYs were not provided in the model.
CDA-AMC = Canada’s Drug Agency; CNS = central nervous system; LY = life-year; QALY = quality-adjusted life-year.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 9). Note in these analyses, the price of osimertinib in subsequent lines remains at the full list price. Further price reductions will be required to achieve a given cost-effectiveness threshold based on any negotiated price reductions for osimertinib in subsequent treatment lines. Please refer to Figure 8 for the impact that price reductions of osimertinib in subsequent lines has on the ICER.
Table 9: Results of the Price Reduction Analysis (Holding Osimertinib Subsequent Therapy Costs Fixed)
Price reduction | Unit drug cost ($) | Cost per 28 days ($) | ICERs for osimertinib vs. active surveillance ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 322a | 9,020 | 161,884 | 203,741 |
10% | 290 | 8,118 | 127,373 | 166,595 |
20% | 258 | 7,216 | 93,326 | 129,449 |
30% | 225 | 6,314 | 59,278 | 92,303 |
40% | 193 | 5,412 | 25,231 | 55,157 |
50% | 161 | 4,510 | Dominant | 18,011 |
60% | 129 | 3,608 | Dominant | Dominant |
70% | 97 | 2,706 | Dominant | Dominant |
80% | 64 | 1,804 | Dominant | Dominant |
90% | 32 | 902 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The term dominant indicates that a drug costs less and provides more QALYs than the comparator.
aSponsor’s submitted price for osimertinib.7
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 10.
Using generalized gamma distributions to model long-term PPS in the osimertinib and placebo groups instead of Gompertz distributions.
Adding a treatment waning effect to TTP and PFS in the osimertinib group beginning at 3 years by increasing the hazard rates by 0.005 each 30-day cycle.
Reducing the price of osimertinib in the subsequent treatment line only.
Table 10: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | Active surveillance | 276,719 | 3.95 | Reference |
Osimertinib | 570,211 | 5.45 | 195,354 | |
CDA-AMC scenario 1: Generalized gamma distributions used to extrapolate PPS | Active surveillance | 277,201 | 4.36 | Reference |
Osimertinib | 570,107 | 5.36 | 290,971 | |
CDA-AMC scenario 2: Treatment waning effect beginning at 36 months | Active surveillance | 276,719 | 3.95 | Reference |
Osimertinib | 428,009 | 4.48 | 286,600 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; PPS = postprogression survival.
aDeterministic analyses.
Figure 8: CDA-AMC Scenario Analysis 3 — Impact of Reducing the Price of Osimertinib in the Subsequent Treatment Line Only on the ICER (Deterministic)
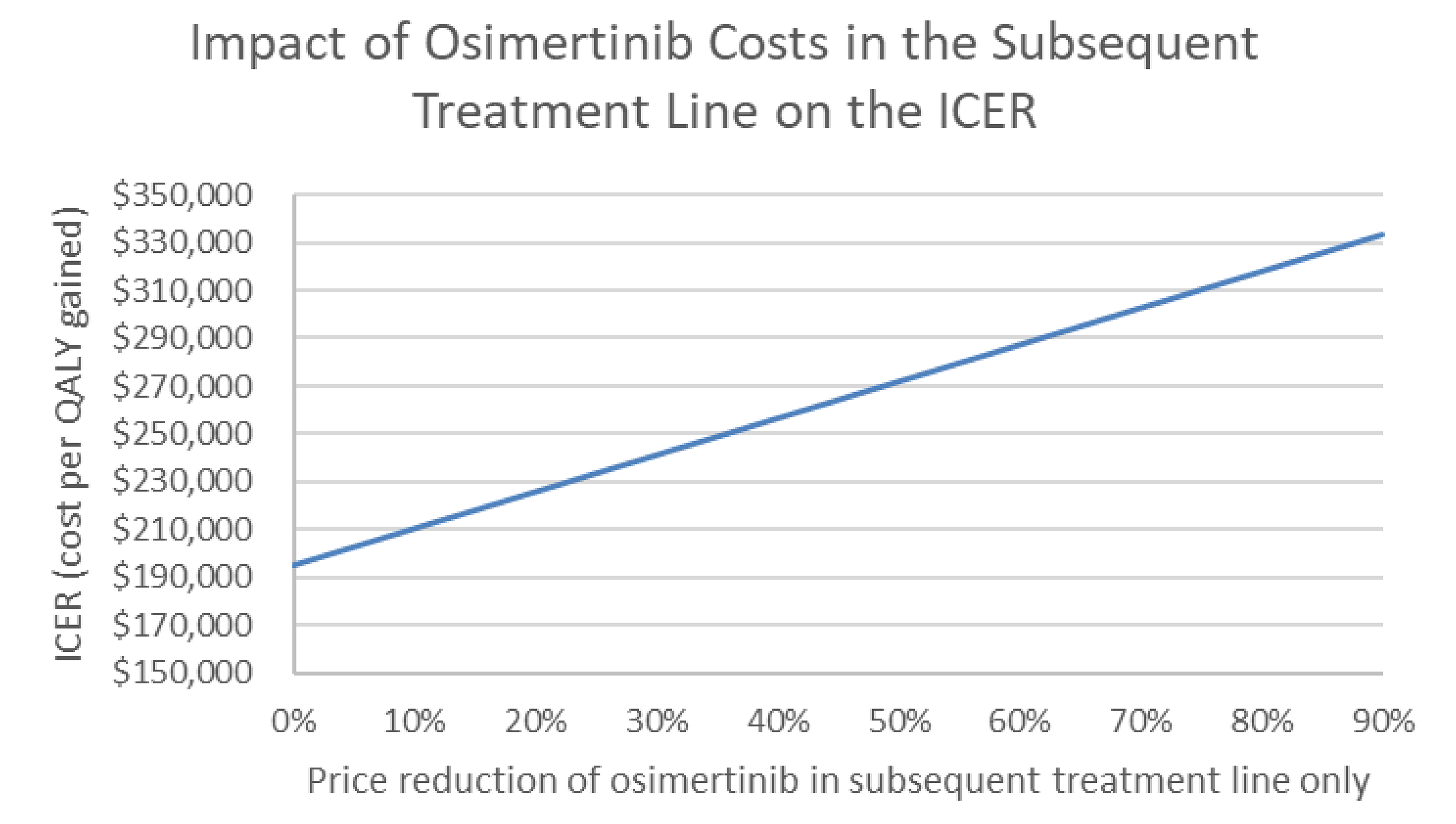
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Issues for Consideration
Osimertinib has been reviewed previously by CDA-AMC and has a letter of intent with the pan-Canadian Pharmaceutical Alliance (pCPA).
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing osimertinib for the treatment of locally advanced, unresectable (stage III) NSCLC whose tumours have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations (either alone or in combination with other EGFR mutations) and whose disease has not progressed during or following platinum-based CRT.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiologic approach. The sponsor’s base case included drug acquisition costs for osimertinib as well as for subsequent therapy. The market uptake for osimertinib was estimated using expert opinion obtained by the sponsor. The key inputs to the BIA are documented in Table 11.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing osimertinib would be $19,607,988 (year 1 = $3,988,099; year 2 = $7,555,692; year 3 = $8,064,197).
Table 11: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Adult population (18 years or older) | |
Lung cancer incidence | |
Percent with NSCLC | 88.0%18 |
Percent tested for EGFR mutation status | |
Percent with EGFR mutations | 15.2%21 |
Percent with exon 19 del or exon 21 L8588R mutations | 84.3%21 |
Population 1: Patients with unresectable stage III disease at diagnosis Percent stage III at diagnosis Percent unresectable disease | 22.7%22 56.8%22 |
Population 2: Patients with recurrent unresectable stage III disease Percent stage I-II/stage III at diagnosis Percent local/regional recurrence Tagrisso-naive or ≥ 6 months since completing adjuvant Tagrisso | 4.3%23 |
Percent receiving platinum-based CRTa | 80.0% |
Percent not progressed during or following platinum-based CRT | 80.0% |
Percent of patients covered by public drug plan | |
Number of incident patients eligible for drug under review | 152/156/159 |
Market shares (reference scenario) | |
Osimertinib | 0%/0%/0% |
Active surveillance | 100%/100%/100% |
Market shares (new drug scenario) | |
Osimertinib | █████████ |
Active surveillance | █████████ |
Cost of treatment (per patient per 28-day cycle) | |
Osimertinib | $9,020 |
Active surveillance | $0 |
CRT = chemoradiation therapy; NSCLC = non–small cell lung cancer.
aAmong patients with unresectable stage III disease at diagnosis or recurrent unresectable stage III disease.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Progression following platinum-based CRT is uncertain: Based on expert opinion, the sponsor assumed that 80% of patients did not progress during or following platinum-based CRT. According to clinical experts consulted by CDA-AMC for this review, 90% of patients do not progress during or following platinum-based CRT.
In a scenario analysis, CDA-AMC assumed 90% of patients did not progress during or following platinum-based CRT.
Subsequent therapy costs are uncertain: The sponsor uses data on secondary progression from the LAURA trial to inform time spent on subsequent therapies (approximately 48 months). However, secondary progression events are small in the trial and this is informed by 24 events out of 73 patients in the placebo arm. However, given the BIA time horizon is only 3 years this is unlikely to have a substantial impact unless time on subsequent therapy falls below 2 years. There is no data suggesting time on subsequent therapy to be this low.
In a scenario analysis, time spent on subsequent therapy was reduced to 28 months to align with the pharmacoeconomic analysis.
CDA-AMC Reanalyses of the BIA
No reanalysis was conducted on the sponsor’s base case.
CDA-AMC used the sponsor’s base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing osimertinib. The results are provided in Table 12.
Increasing the proportion of patients who do not progress during or following platinum-based CRT (to 90%) to align with clinical expectations.
Reduce median time spent on subsequent therapy to 28 months.
Table 12: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 1,936,710 | 11,461,931 | 24,858,587 | 39,533,462 | 75,853,980 |
Osimertinib | 0 | 0 | 0 | 0 | 0 | |
Active surveillance | 0 | 0 | 0 | 0 | 0 | |
Subsequent therapies | 1,936,710 | 11,461,931 | 24,858,587 | 39,533,462 | 75,853,980 | |
New drug total | 1,936,710 | 15,450,031 | 32,414,278 | 47,597,659 | 95,461,968 | |
Osimertinib | 0 | 5,176,790 | 14,894,671 | 25,411,086 | 45,482,548 | |
Active surveillance | 0 | 0 | 0 | 0 | 0 | |
Subsequent therapies | 1,936,710 | 10,308,227 | 17,648,205 | 22,408,344 | 52,301,486 | |
Budget Impact | 0 | 3,988,099 | 7,555,692 | 8,064,197 | 19,607,988 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: 90% of patients did not progress during or following platinum-based CRT | Reference total | 2,178,799 | 12,894,673 | 27,965,910 | 44,475,145 | 85,335,728 |
New drug total | 2,178,799 | 17,381,284 | 36,466,063 | 53,547,366 | 107,394,714 | |
Budget Impact | $0 | 4,486,612 | 8,500,153 | 9,072,222 | 22,058,986 | |
Scenario 2: time spent on subsequent therapy reduced to 28 months | Reference total | 1,937,160 | 11,464,592 | 23,681,442 | 30,089,651 | 65,235,685 |
New drug total | 1,937,160 | 15,452,415 | 31,235,430 | 38,875,857 | 85,563,702 | |
Budget Impact | 0 | 3,987,823 | 7,553,988 | 8,786,206 | 20,328,018 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; CRT = chemoradiation therapy.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.