Drugs, Health Technologies, Health Systems
Reimbursement Review
Tislelizumab (Tevimbra)
Sponsor: BeOne Medicines (Canada) ULC
Therapeutic area: Recurrent or metastatic nasopharyngeal carcinoma
Summary
What Is Nasopharyngeal Carcinoma?
Nasopharyngeal carcinoma (NPC) is a cancer that arises from the lining of the nasopharynx and causes a range of symptoms (e.g., nosebleeds; changes in vision, hearing, smell, or taste; sore throat; headaches; weight loss; and facial pain or paralysis) and can significantly impact health-related quality of life (HRQoL).
NPC is rare in North America but occurs more frequently in Southern China, Southeast Asia, North Africa, and the Arctic. The 5‑year prevalence in Canada (excluding Quebec) in 2018 was 840 cases, or 3 per 100,000 people.
What Are the Treatment Goals and Current Treatment Options for First-Line Treatment of Recurrent or Metastatic Nasopharyngeal Carcinoma?
Goals in the first-line treatment of recurrent or metastatic (R/M) NPC are to prolong survival, delay cancer progression, induce tumour shrinkage, maintain or improve quality of life, alleviate or control cancer-related symptoms, and minimize side effects from treatments.
Current first-line treatment for R/M NPC is palliative, with the preferred regimen consisting of platinum (cisplatin or carboplatin) and gemcitabine chemotherapy. Other chemotherapy agents may be considered for patients who are not candidates for this regimen.
Input from patients and clinicians emphasized that current treatments are associated with treatment resistance, cancer progression, and side effects, and that more effective and better-tolerated treatments are needed.
What Is Tevimbra and Why Did Canada’s Drug Agency Conduct This Review?
Tevimbra is a PD-1 inhibitor that is administered by IV infusion. At the time this review was conducted, Health Canada was reviewing Tevimbra in combination with gemcitabine and cisplatin (GC) for the first-line treatment of patients with R/M NPC, and then as a single agent after up to 6 cycles of combination therapy.
Canada’s Drug Agency (CDA-AMC) reviewed Tevimbra to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the indication under review by Health Canada.
The application was submitted by the sponsor before receiving a Notice of Compliance from Health Canada. The Health Canada indication changed during the review to clarify that the indication was for adults only. The approved indication for Tevimbra is in combination with GC for the first-line treatment of adult patients with R/M NPC. The Health Canada–approved product monograph incorporated a Serious Warnings and Precautions box outlining the risks of immune-mediated adverse reactions, infusion-related reactions, and complications in patients who receive an allogeneic hematopoietic stem cell transplant. The CDA-AMC review reflects the sponsor’s originally proposed indication and product monograph, which generally align with the final approved indication.
How Did CDA-AMC Evaluate Tevimbra?
CDA-AMC reviewed evidence submitted by the sponsor — clinical evidence on the beneficial and harmful effects, and also economic evidence — comparing Tevimbra with other treatments used in Canada for first-line treatment of R/M NPC. Gemcitabine plus cisplatin and gemcitabine plus carboplatin were considered relevant treatments to compare with Tevimbra when reviewing the evidence.
CDA-AMC identified equity and ethical considerations relevant to Tevimbra for the treatment of R/M NPC.
The review was also informed by separate submissions: a joint submission from 3 patient groups, 2 submissions from clinician groups, and input from participating public drug programs. The drug programs’ input covered issues that may affect their ability to implement a recommendation.
Three medical oncologists were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 phase III randomized controlled trial, the RATIONALE-309 trial, comparing Tevimbra plus GC with placebo plus GC in the first-line treatment of 263 patients with R/M NPC.
For the comparison of Tevimbra plus GC versus placebo plus GC:
Tevimbra plus GC demonstrated clinically important increases in the probability of progression-free survival compared to placebo plus GC at 9 months and 36 months, with findings of high and moderate certainty at these time points, respectively.
Tevimbra plus GC demonstrated increases in the probability of overall survival (OS) compared to placebo plus GC at 12 months and 36 months, with findings of moderate certainty at both time points.
HRQoL was measured by the change from baseline in the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 score for global health status and quality of life at cycles 4 and 8. The findings indicated little to no difference in HRQoL between Tevimbra plus GC and placebo plus GC, with the certainty in the results at these time points rated as moderate and low, respectively, due to study limitations.
Similar proportions of patients in both groups experienced serious adverse events. The evidence suggested that Tevimbra plus GC may result in little to no difference in the proportion of patients with 1 or more serious adverse events compared to placebo plus GC, with low certainty in the findings. The types of adverse events reported in the RATIONALE-309 trial were as expected for treatment with immunotherapy combined with GC.
Economic Evidence
Tevimbra is available as a 100 mg per 10 mL concentrate for solution for IV infusion. At the submitted price of $3,080.00 per vial, the 21-day cycle cost of Tevimbra is expected to be $6,160 per patient, based on the Health Canada–recommended dosage.1 When used in combination with chemotherapy, the total cost of Tevimbra plus chemotherapy is expected to be $7,645 in the first 6 cycles and $6,160 in subsequent cycles.
Clinical efficacy in the economic analysis was derived from the RATIONALE 309 trial, which compared Tevimbra plus GC with placebo plus GC. Evidence submitted by the sponsor indicated that Tevimbra plus GC likely improves progression-free survival and OS compared with placebo plus GC in the first-line treatment of patients with R/M NPC. However, the magnitude of the OS benefit remains uncertain, as many patients in the placebo plus GC arm crossed over to receive Tevimbra.
The results of the CDA-AMC base case suggest the following:
Tevimbra plus chemotherapy is predicted to be associated with higher costs to the health care system compared with chemotherapy alone (incremental costs = $107,597), primarily driven by increased costs associated with drug acquisition (incremental drug acquisition costs = $97,372). There are also higher costs associated with drug administration and disease management associated with Tevimbra use.
Tevimbra plus chemotherapy is predicted to be associated with a gain of 1.29 life-years compared to chemotherapy alone and may result in a gain of 0.87 quality-adjusted life-years compared to chemotherapy alone.
The incremental cost-effectiveness ratio of Tevimbra plus chemotherapy compared to chemotherapy alone was $124,171 per QALY gained in the CDA-AMC base case. However, the estimated incremental cost-effectiveness ratio is uncertain due to limitations in the long-term comparative efficacy data, long-term survival projections, and the submitted economic model structure. As a result, the economic analysis may not accurately assess the true impact of Tevimbra plus chemotherapy on patient health and health care resources. The cost-effectiveness of Tevimbra plus chemotherapy therefore remains uncertain and greater price reductions may be required to achieve a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing Tevimbra plus chemotherapy for the first-line treatment of recurrent or metastatic NPC will increase costs by approximately $26.0 million over the first 3 years of reimbursement compared to the amount currently spent on chemotherapy alone, with an estimated expenditure of $28.5 million on Tevimbra plus chemotherapy over this period (expenditure on Tevimbra alone = $26.3 million). The actual budget impact of reimbursing Tevimbra plus chemotherapy will depend on the number of patients eligible for treatment and the market uptake of Tevimbra plus chemotherapy.
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-H&N35
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck 35
GC
gemcitabine and cisplatin
GHS
global health status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ICI
immune checkpoint inhibitor
imAE
immune-mediated adverse event
ICER
incremental cost-effectiveness ratio
IRC
independent review committee
KM
Kaplan-Meier
LS
least squares
MID
minimal important difference
NCCN
National Comprehensive Cancer Network
NE
not evaluable
NPC
nasopharyngeal carcinoma
ORR
overall response rate
OS
overall survival
PFS
progression-free survival
PS
performance status
QALY
quality-adjusted life-year
QoL
quality of life
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
R/M
recurrent or metastatic
RPSFTM
rank-preserving structural failure time model
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tislelizumab, 100 mg/10 mL (10 mg/mL), IV infusion in the first-line treatment of patients with recurrent or metastatic (R/M) nasopharyngeal carcinoma (NPC). The focus will be placed on comparing tislelizumab to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, as outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost‑effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The application was submitted by the sponsor before receiving a Notice of Compliance from Health Canada. The CDA‑AMC review reflects the sponsor’s originally proposed indication and product monograph, which generally align with the final approved indication.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Tislelizumab (Tevimbra), 100 mg/10 mL (10 mg/mL), concentrate for solution, IV infusion |
Sponsor | BeOne Medicines (Canada) ULC |
Health Canada indication | Tislelizumab in combination with gemcitabine and cisplatin is indicated for the first‑line treatment of adult patients with recurrent or metastatic nasopharyngeal carcinoma (NPC). |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 18, 2025 |
Mechanism of action | PD-1 inhibitor |
Recommended dosage | 200 mg administered as an IV infusion once every 3 weeks in combination with cisplatin and gemcitabine chemotherapy for 4 to 6 cycles then continued as monotherapy until unacceptable toxicity or disease progressiona No dose reductions of tislelizumab are recommended; recommended treatment modifications to manage immune-related adverse reactions are provided in the product monograph |
Submission type | Initial submission |
Sponsor’s reimbursement request | Per indication |
Submitted price | $3,080.00 per 10 mL single-dose vial |
Information on the CDA-AMC review | |
Review type | Complex |
Clinical review focusb | Population: As defined in the Health Canada indication Intervention: Per recommended dosage Comparators: Gemcitabine plus cisplatin, gemcitabine plus carboplatin Outcomes: PFS, OS, ORR, DOR, HRQoL (EORTC QLQ-C30, EORTC QLQ‑H&N35), harms outcomes (AEs, SAEs, WDAEs, deaths, and notable harms [imAEs]) |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck 35; HRQoL = health-related quality of life; imAE = immune-mediated adverse event; PFS = progression-free survival; NOC = Notice of Compliance; NPC = nasopharyngeal carcinoma; ORR = objective response rate; OS = overall survival; SAE = serious adverse event; WDAE = withdrawal due to adverse event (from treatment).
aWhen tislelizumab is used in combination, refer to the full prescribing information of the combination therapy.1
bThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated. Outcomes of interest for the Economic Review were PFS, OS, treatment duration, and AEs.
Sources: Sponsor’s Summary of Clinical Evidence2 and product monograph for Tevimbra (tislelizumab).1
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed tislelizumab through the Reimbursement Review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts CDA-AMC consulted for this review.
Calls for patient group and clinician group input are issued for each Reimbursement Review. Three patient groups collectively submitted 1 patient group input: the Canadian Organization for Rare Disorders, the Canadian Cancer Survivor Network, and the Canadian Cancer Society. From May 29 to June 13, 2025, the 3 groups jointly conducted a survey to gather patient and caregiver perspectives on disease and treatment experience. Twelve patients with NPC and 3 caregivers completed this survey (i.e., a total of 15 patients and caregivers, with 14 from Canada and 1 patient from the US), of whom 3 patients were also interviewed. Clinician group submissions from Ad Hoc Treaters of NPC (7 clinicians contributed to the input) and the Ontario Health (Cancer Care Ontario) Head and Neck Cancer Drug Advisory Committee (4 clinicians contributed to the input) were received. The clinician group input was based on information gathered via emails and through expert review of clinical trial data from the RATIONALE-309 trial. The full submissions received are available on the project landing page for tislelizumab in the consolidated input document. The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may affect their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Three medical oncologists with expertise in the diagnosis and management of NPC participated as part of the review team.
Disease Background
NPC arises from the lining of the nasopharynx. It is a unique type of cancer that is differentiated from other cancers of the head and neck due to its epidemiology, etiology, clinical manifestation, and histopathology.3,4 NPC is rare in North America but occurs more frequently in the Arctic, North Africa, Southeast Asia, and Southern China.5,6 In Canada (excluding Quebec), according to data for 2019, a total of 235 new cases of NPC were diagnosed, representing a cancer incidence rate of 0.8 per 100,000.7 The 5-year prevalence in Canada (excluding Quebec) in 2018 was 840 cases, or 3.0 per 100,000 people.8 NPC is 25 to 40 times more frequent in Inuit than in white populations in Canada, Denmark, and the US, and represents 4% to 7% of all malignancies among Inuit.9
Signs of NPC include development of a neck mass and cervical lymphadenopathy with bilateral involvement.5 Symptoms commonly include epistaxis, nasal congestion, hearing loss, otitis media, headaches, sore throat that does not resolve, weight loss, facial numbness or pain or paralysis of the face, difficulty opening the jaw, and blurred or double vision.5,10 NPC is most often diagnosed when an individual seeks medical attention and presents with a lump in the neck. If NPC is suspected, a more thorough examination of the nasopharynx by a specialist and a tissue biopsy are required to confirm diagnosis. Recommended investigations at diagnosis for all patients with suspected or confirmed early-stage NPC include chest imaging, MRI with gadolinium of the nasopharynx and base of the skull to the clavicles, and/or CT with contrast to determine the local and regional extent of disease. A CT, PET-CT, and/or bone scan are also recommended to assess metastatic disease or advanced-stage disease, and examination under anesthesia with endoscopy may be recommended to determine the extent of disease.5
NPC is commonly staged according to the tumour-node-metastasis system. Stage I is an early localized stage of the disease, stage II refers to largely localized disease with some spread beyond the nasopharynx, stages III and IVa indicate advanced disease with more extensive involvement of nearby tissues or lymph nodes, and stage IVb denotes a metastatic stage in which the disease has spread to distant parts of the body.5,11 As NPC is difficult to detect due to variable symptoms and poor visualization by endoscopy, more than 70% of cases are detected at advanced stages (III to IV).12,13 Prognoses for patients with NPC are largely dependent on disease stage. Early-stage NPC (stage I or II) has a 5-year survival rate of approximately 70% to 90%, while patients with locally advanced NPC (stages III and IVa) have poorer 5-year survival rates ranging from 50% to 80%.14-17 Approximately 15% to 40% of patients with NPC initially diagnosed without metastasis will relapse or progress to developing metastases.18-20 At diagnosis, an estimated 7% of patients present with metastatic disease.14,21 Metastatic NPC has a poorer prognosis, with 3-year survival rates below 40%, and patients with recurrent disease have reported survival rates of approximately 30%.10,20,22-24
Patient Group Input
According to the patient group input, the most commonly reported symptoms among survey respondents were difficulty swallowing (16%), changes in vision, hearing, smell, or taste (14%), nasal congestion or blockage (11%), nosebleeds (11%), hearing loss or ringing in the ears (11%), headaches (8%), and swelling in the neck (8%). Approximately half of the survey respondents (55%) also stated that NPC had a moderate to severe impact on their overall health-related quality of life (HRQoL), with effects on physical health, emotional well-being, mental health, social interactions, work or daily activities, quality of sleep, nutritional intake, and pain and discomfort. The groups described how disease symptoms (e.g., fatigue, hearing loss, changes in visual appearance, breathing difficulties, and sleep disturbance) greatly impair patients’ social interactions and daily activities.
Current Management
Treatment Goals
Patient Group Input
The survey respondents emphasized the importance of effective first-line and second-line therapies for this patient population, for whom very few treatment options are available. They are looking for effective and safe therapies that can prolong their lives, relieve symptoms, improve quality of life (QoL), and bypass treatments with severe side effects, such as radiation. The patient groups stated that important outcomes for patients with R/M NPC include survival, improved HRQoL, symptom control, and minimizing side effects from treatments.
Clinician Input
According to the clinical experts consulted by CDA-AMC, the most important goals in the first-line treatment of R/M NPC are to improve survival, delay cancer progression, induce tumour shrinkage, alleviate cancer-related symptoms, and maintain or improve QoL. The clinician groups indicated that, because the treatment intent in the R/M NPC setting is not curative, the goals of treatment for these patients are to improve QoL (by improving cancer-related symptoms), control cancer, and prolong survival.
Current Treatment Options
No national Canadian guideline exists for the treatment of NPC; however, clinical practice guidelines are available from Alberta Health Services and internationally from the National Comprehensive Cancer Network (NCCN) and the European Society for Medical Oncology.5,25,26 The treatment approach is determined primarily by disease stage and not by histological subtype.5 According to Alberta Health Services guidelines, the preferred first-line treatment for patients with recurrent (not amenable to salvage surgery or radiation therapy) or metastatic NPC who have a good performance status is gemcitabine and cisplatin (GC) chemotherapy.5 Both the NCCN and European Society for Medical Oncology guidelines recommend adding a PD-1 inhibitor, including, in the case of NCCN, tislelizumab, to GC as a first-line option in the treatment of R/M NPC.25,26 Cisplatin may be substituted with carboplatin in the event of intolerance or contraindications.27 However, at the time of writing there is no Health Canada approval for the use of gemcitabine, cisplatin, or PD-1 inhibitors for the treatment of NPC in Canada.27-29 Radiotherapy may be used for patients with R/M NPC to palliate symptoms.5
Input received by clinical experts stated that the current first-line treatment for R/M NPC is doublet platinum and gemcitabine chemotherapy for up to 6 cycles, noting that platinum chemotherapy may comprise either cisplatin or carboplatin. Carboplatin may be substituted in clinical practice due to its more favourable toxicity profile or in cases of contraindication to cisplatin, and the 2 drugs are thought to be similar with regard to clinical activity in this setting. According to the clinical experts, for patients who are not candidates for this regimen, other chemotherapy agents (e.g., paclitaxel, docetaxel, 5-fluorouracil, capecitabine, cisplatin, or carboplatin) alone or in combination may be considered. Clinical experts also commented that palliative radiotherapy may be considered for oligometastatic cases. The clinician group input aligned with the clinical expert input, noting that the selection of chemotherapy agents may be based on patient comorbidities, performance status, and patient preferences.
According to the patient group input, the most common treatments received by the survey respondents included combined chemoradiation therapy, followed by radiation therapy, chemotherapy, and surgery. Patients’ experiences with currently available treatments varied from very positive to extremely poor. Some respondents said their treatments saved their lives, stabilized their conditions, and alleviated the symptoms, while others reported severe side effects from the treatments and subsequent impacts on their QoL. One patient with metastatic NPC, who was aged between 60 and 69 years and had experience with tislelizumab, was satisfied with this treatment, finding it effective in controlling their cancer with tolerable side effects. The patient and their caregiver stated that it was absolutely necessary to have tislelizumab available to appropriate patients as a first-line therapy.
Key characteristics of tislelizumab are summarized with other treatments available for first-line treatment of R/M NPC in the Supplemental Material document, in the Key Characteristics table in Appendix 1.
Unmet Needs and Existing Challenges
Patient Group Input
The patient groups identified a significant unmet need for additional treatment options that are more effective than the current standard of care. They indicated that some current treatments for R/M NPC, such as radiation and surgery, have severe side effects that can subsequently affect patients’ HRQoL. Side effects from current treatments reported by respondents included changes in smell or taste, loss of appetite, neuropathy, hearing loss, difficulty swallowing, radiation burns, weight loss, and fatigue. The patient groups emphasized the burden of NPC on various aspects of daily life, including physical health, emotional well-being and mental health, social interactions, work or daily activities, sleep, nutrition, and pain. Survey respondents also noted that NPC places a financial burden on patients and caregivers, with those with lower incomes often facing great financial difficulties. For example, 1 respondent noted having to travel long distances to access CT imaging, which resulted in additional expenses for food, gas, and accommodations. Patients also reported additional costs for meal replacements and medications to manage treatment side effects, as well as loss of income for patients and caregivers due to work absenteeism while receiving treatments and afterward.
Clinician Input
The clinical experts consulted by CDA-AMC commented that, although NPC is chemosensitive, not all patients respond and responses are not durable. According to the clinical experts, treatment with gemcitabine and platinum is associated with treatment resistance and cancer progression, as well as acute and long-term side effects, including nausea, acute kidney injury, ototoxicity, and renal toxicity. The clinical experts also pointed out that cumulative toxicity from continued cytotoxic therapy adversely affects QoL and indicated that more effective and better-tolerated treatments are needed. They noted that immunotherapy can provide durable disease control. The clinical experts identified subgroups of patients with NPC who experience challenges. These included older patients (who may have difficulty tolerating cytotoxic chemotherapy), patients with keratinizing histology (which is associated with poorer prognosis than nonkeratinizing histology), and patients with recurrent disease following definitive, curative-intent treatment and a short disease-free interval (which is associated with more aggressive disease). The clinical experts also commented that NPC disproportionately affects Inuit and some immigrant populations from regions with a higher risk of disease (the Arctic, East and Southeast Asia, and North Africa) who may already face barriers to diagnosis, access to care, and equitable outcomes.
The clinician groups indicated that the prognosis for patients with R/M NPC remains poor, noting that, while NPC tends to be initially chemosensitive, all patients invariably experience disease progression on chemotherapy. Therefore, there is a high unmet need for additional treatment options besides the existing first-line treatment with gemcitabine and platinum. The clinician groups noted the significant disparities in accessing immunotherapy as part of first-line therapy for R/M NPC, with Canada lagging behind the US and the European Union. The clinician groups suggested that, based on clinical trial data, treatment with tislelizumab in combination with gemcitabine and platinum will offer a meaningful benefit over chemotherapy alone, and they would recommend tislelizumab in the first-line treatment setting for patients with recurrent disease.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions and Implementation Considerations Proposed by the Sponsor table in Appendix 1). The implementation questions from the public drug programs and corresponding responses from the clinical experts CDA-AMC consulted for this review are summarized in the Supplemental Material document, in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1. The following has been summarized by the review team.
Place in Therapy
The clinical experts consulted by CDA-AMC expected that tislelizumab in combination with gemcitabine and platinum would become a new standard of care as first-line treatment for patients with R/M NPC and would cause a shift in the current treatment paradigm. The clinical experts noted that, as a PD-1 inhibitor, tislelizumab has a unique mechanism of action compared to cytotoxic chemotherapy, which addresses the underlying disease process and would be the first treatment to address the mechanisms responsible for immune evasion that are prevalent in NPC. The clinical experts stated that tislelizumab would complement gemcitabine and platinum treatment, resulting in improved outcomes over chemotherapy alone. According to the experts, the evidence supports first-line use of tislelizumab, and it would not be appropriate to require patients to first try or be intolerant of other treatments before qualifying for tislelizumab plus gemcitabine and platinum. According to the clinician group input, patients would benefit from tislelizumab as a first-line treatment in combination with platinum and gemcitabine, shifting the current treatment paradigm, for which there is no Health Canada–approved immunotherapy option.
Patient Population
According to the clinical experts consulted by CDA-AMC, patients with R/M NPC are in need of treatment with tislelizumab plus gemcitabine and platinum, particularly those with aggressive or symptomatic disease. The experts stated that patients best suited for treatment would be identified by an oncology specialist (radiation oncologist, surgeon, or medical oncologist) and that patients with earlier-stage NPC treated with curative intent would be routinely followed by an ear, nose, and throat specialist or radiation oncologist. The clinical experts commented that misdiagnosis or underdiagnosis is unlikely and noted that underdiagnosis may occur if patients are not under active surveillance following curative treatment. Clinical experts commented that a companion diagnostic is not required and there are currently no identified clinical prognostic tools and/or biomarkers to identify which patients are most likely to respond to treatment. According to the clinical experts, patients with R/M NPC are suited for treatment with tislelizumab, regardless of symptoms, disease burden, or demographic characteristics. According to the experts, patients least suited for treatment with tislelizumab would be those with a poor performance status, uncontrolled autoimmune conditions, or for whom life expectancy is not long enough to obtain a clinical benefit. The clinical experts noted that the initiation conditions proposed by the sponsor are appropriate for identifying the suitable patient population and that practising oncologists are familiar with these conditions, which could be readily implemented.
The clinician groups indicated that tislelizumab should be available to all fit patients (e.g., those having an Eastern Cooperative Oncology Group [ECOG] Performance Status (PS) score of 0 to 2 with no contraindication to immunotherapy) in the first-line R/M NPC setting. The clinician groups also suggested that the therapeutic benefit could continue even in later lines of therapy.
Assessing the Response to Treatment
The clinical experts consulted by CDA-AMC commented that a clinically meaningful response to treatment would include disease stabilization for at least 6 months or extended survival by at least 6 months, and that other considerations would include stabilization or improvement in disease-related symptoms (e.g., pain) and QoL scores. The experts stated that treatment response is assessed in clinical practice based on patient history and medical examination, including imaging, which is aligned with what is used in clinical trials, and that the timing of assessment for treatment response would range from every 9 weeks (i.e., 3 cycles) to every 3 months and possibly less frequently once a response is observed (depending on the practice and treating physician’s judgment). The clinical experts noted that the renewal conditions proposed by the sponsor are appropriate and could be readily implemented in clinical practice.
The clinician groups suggested that meaningful outcomes in the target population include stabilization or shrinkage of cancer seen on imaging, stabilization or improvement of clinical condition and cancer-related symptoms, stabilization or improvement of HRQoL, and prolongation of life or period without disease progression (i.e., overall survival [OS] and progression-free survival [PFS]).
The clinician groups also suggested conducting standard-of-care imaging (CT and/or MRI) every 3 to 4 months and regular clinical assessments to determine whether patients are responding or progressing on treatment.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC stated that any decision to discontinue treatment with tislelizumab should consider treatment response, tolerance, toxicities, and treatment alternatives. The experts noted that the discontinuation conditions proposed by the sponsor are appropriate and could be readily implemented.
According to the clinician groups, treatment with tislelizumab should be discontinued at disease progression (radiographic or symptomatic), occurrence of intolerance, or patient preference.
Prescribing Considerations
According to the clinical experts consulted by CDA-AMC, for treatment with tislelizumab, initial in-person diagnosis of R/M NPC, treatment, and monitoring should be carried out by a medical oncology specialist. Treatment can be administered and monitored in regional or community cancer centres with appropriate resources and supports in place. The clinical experts emphasized that long-distance travel for the required treatment and monitoring can be a significant challenge for patients living in remote areas of Canada, noting that this is true for all cancers and not unique to NPC. The experts noted that the prescribing conditions proposed by the sponsor are appropriate and may be readily implemented, acknowledging the previously mentioned challenges faced by patients in remote areas.
The clinician groups stated that medical oncologists with experience in treating cancers of the head and neck at regional cancer centres can prescribe tislelizumab to patients with R/M NPC.
Additional Considerations
According to the clinical experts consulted by CDA-AMC, the combination of an immune checkpoint inhibitor (ICI) with gemcitabine and platinum is a standard of care for the first-line treatment of patients with R/M NPC in other countries; however, currently there is no readily available access to ICIs for patients with NPC in Canada. The clinical experts commented that tislelizumab with gemcitabine and platinum would therefore likely be quickly adopted in Canada as a new treatment paradigm as this would be the first treatment to address the immune-evasion mechanisms that are prevalent in NPC. One clinical expert noted that, at their practice location, tislelizumab was adopted as the standard of care in 2022 and has been accessed through Health Canada’s Special Access Program. This clinical expert noted seeing firsthand the efficacy and tolerability of tislelizumab and expressed support for the drug to be readily available to all eligible patients with NPC in Canada.
Clinical Review
Methods
The review team considered evidence from the sponsor-submitted systematic review (pivotal studies and randomized controlled trials). Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and IV randomized controlled trials. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. The systematic literature review search was not limited by comparators.
The review team considered outcomes (and follow-up times) for review based on the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes, identified by patient and clinician groups and clinical experts as efficacy measures that address main treatment goals for R/M NPC, were assessed with GRADE: PFS, OS, and HRQoL (as measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 [EORTC QLQ-C30] global health status [GHS]/QoL domain). PFS and OS were also key inputs in the sponsor’s pharmacoeconomic model.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are provided in the Supplemental Material document, in Appendix 2.
Clinical Evidence
In this report, the following source of evidence submitted by the sponsor is reviewed and appraised:
1 pivotal phase III trial included in the systematic review, the RATIONALE-309 trial.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria and relevant outcome measures are in the Supplemental Material document in Appendix 3.
The RATIONALE-309 trial investigated the efficacy and safety of tislelizumab combined with gemcitabine and cisplatin (tislelizumab plus GC) compared with placebo combined with GC in the first-line treatment of patients with R/M NPC.30 The study was conducted at 42 sites across China, Taiwan, and Thailand (there were no sites in Canada).31 The first patient was randomized on April 17, 2019 (study start date), and the last patient visit occurred on December 8, 2023 (study end date).30 The study included a screening period (within 28 days before randomization to assess patient eligibility) and a treatment period. Patients were randomized 1:1 using interactive response technology to receive either tislelizumab plus GC or matched placebo plus GC, and randomization was stratified by sex and metastatic status (with or without liver metastases). Tumour imaging occurred approximately every 6 weeks for the first 6 months, every 9 weeks for the remainder of year 1, and every 12 weeks from year 2 onward based on Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1). Assessment of adverse events (AEs) occurred at each treatment visit, at the end‑of‑treatment visit, at the earlier of 30 days after the last dose of study treatment or initiation of new anticancer therapy (for AEs and serious adverse events [SAEs]), and until 90 days after the last dose of the study drug (for immune-mediated adverse events [imAEs]). Survival follow-up occurred approximately every 3 months after the safety follow-up visit that occurred 30 days after the last dose of study treatment.32 Patients in the tislelizumab plus GC arm who experienced progression as defined by RECIST 1.1 during chemotherapy or while receiving subsequent tislelizumab monotherapy were permitted to continue tislelizumab monotherapy if it was considered clinically beneficial by the investigator. Patients in the placebo plus GC arm who experienced progression according to RECIST 1.1 as confirmed by an independent review committee (IRC) had the option to cross over to receive tislelizumab monotherapy if it was considered clinically beneficial by the investigator. For both arms, once patients were receiving tislelizumab monotherapy, continuation of this treatment beyond investigator-assessed progression could be considered by the investigator, and tislelizumab could be continued until loss of clinical benefit, withdrawal of consent, study termination by the sponsor, start of a new anticancer therapy, or death (whichever occurred first).32,33
In this review report and accompanying appendices, results are presented from the interim analysis (data cut-off: March 26, 2021) and from approximately 17 additional months of follow-up as of the December 8, 2023, study end date (database lock: January 26, 2024).30,33
Table 2: Characteristics of Studies Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
RATIONALE-309 trial Double-blind, placebo-controlled, randomized, multicentre phase III study Total N = 263 |
|
| Intervention: Tislelizumaba (200 mg IV, day 1) + gemcitabine (1 g/m2 IV within 30 minutes, day 1 and day 8) + cisplatin (80 mg/m2 IV for 4 hours, day 1)b Comparator:
Both arms: Chemotherapy was administered q.3.w. for 4 to 6 cycles (at the investigator’s discretion) or until unacceptable toxicity or disease progressionc | Primary: PFS as assessed by the blinded IRCc Secondary:
|
DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-H&N35 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck 35; HRQoL = health-related quality of life; IRC = independent review committee; NPC = nasopharyngeal carcinoma; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PS = performance status; q.3.w. = every 3 weeks; R/M = recurrent or metastatic; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aThere was no dose reduction for tislelizumab or placebo. Patients could temporarily suspend study treatment if they experienced toxicity that is considered related to tislelizumab or placebo and required a dose to be withheld. Dose modification related to immune-mediated adverse events and infusion-related reactions were permitted.32
bDose modifications for chemotherapy were performed according to prescribing information, local practice, and the treating physician’s clinical judgment. Dose modifications were required if the patient’s body weight changed by greater than 10% from baseline (or the new reference body weight). Chemotherapy doses were not modified for any change in body weight of less than 10%.32
cAccording to RECIST 1.1.
dKey time points for both scales were cycle 4 and cycle 8, which were selected to measure the change in end points from baseline through the time point of completion of 3 cycles of chemotherapy, and from baseline through the time point of completion of 3 cycles of tislelizumab monotherapy, respectively.34
Sources: Sponsor’s Summary of Clinical Evidence,2 RATIONALE-309 Clinical Study Report (final analysis),30 and RATIONALE-309 Clinical Study Protocol.32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
The RATIONALE-309 trial’s sample-size calculation was based on the primary end point and assumed an exponential distribution for PFS. Assuming a median PFS of 7 months in the placebo plus GC arm, at a 1-sided alpha of 0.025, the study would have 82% power for a targeted hazard ratio (HR) of 0.65, which corresponded to an improvement in median PFS to 10.8 months. For the final analysis of PFS, assuming a PFS evaluation dropout rate of 5% per 12 months, a steady-state enrolment rate of 20 patients per month, and an enrolment ramp-up duration of 3 months, 181 PFS events were required. Assuming 256 patients were to be enrolled over a 14.3-month period, the final analysis would occur approximately 21.5 months after the first patient was randomized.35
Multiple testing of the primary end point of PFS was accounted for in the single planned interim efficacy analysis, which was to be conducted when approximately 127 PFS events (70% of the target number of 181 PFS events) were observed (estimated to occur after approximately 15.6 months). The interim boundary was based on the Lan-DeMets O’Brien-Fleming approximation spending function. At the interim analysis, the P value boundary was 0.007 and the approximate HR threshold was 0.649 for PFS; at the final analysis, they were 0.023 and 0.743, respectively. The actual boundaries were to be calculated based on the actual number of events observed at interim. No other multiplicity adjustment was implemented for other secondary end points.35
Efficacy analyses were performed in the intention-to-treat analysis set, which included all randomized patients, analyzed according to their randomized treatment arms. Safety analyses were performed in the safety analysis set, defined as all randomized patients who received any dose of any component of study drug, analyzed according to the actual treatment regimen received. The HRQoL analyses were performed in the HRQoL analysis set, which was defined as all randomized patients who received any dose of a study drug and completed at least 1 HRQoL assessment.35
Patient Disposition
Patient disposition for the RATIONALE-309 study is summarized in the Supplemental Material, Appendix 4.
In the RATIONALE-309 study, 326 patients were screened and 263 were randomized. At the end of the study (December 2023 data cut-off), all patients in both study arms had discontinued study treatment. Progressive disease was the primary reason for treatment discontinuation for 74 of 131 patients (56.5%) in the tislelizumab plus GC arm and for 91 of 132 patients (68.9%) in the placebo plus GC arm; withdrawal by patient occurred for 26 patients (19.8%) and 23 patients (17.4%), respectively. Also at the December 2023 data cut-off, all patients in both study arms had discontinued the study. Death was the primary reason for study discontinuation for 57 patients (43.5%) in the tislelizumab plus GC arm and for 66 patients (50.0%) in the placebo plus GC arm, and study terminated by sponsor was the primary reason for 51 patients (38.9%) and 41 patients (31.1%), respectively.2
Baseline Characteristics
Table 3: Summary of Baseline Characteristics From the RATIONALE-309 Trial (ITT Analysis Set)
Characteristic | Tislelizumab plus GC (N = 131) | Placebo plus GC (N = 132) |
|---|---|---|
Age (years), mean (SD) | 49.0 (10.62) | 49.5 (10.76) |
Age < 65 years, n (%) | 121 (92.4) | 120 (90.9) |
Age ≥ 65 years, n (%) | 10 (7.6) | 12 (9.1) |
Female, n (%) | 28 (21.4) | 29 (22.0) |
Male, n (%) | 103 (78.6) | 103 (78.0) |
Weight (kg), mean (SD) | 60.02 (10.248) | 60.46 (10.543) |
Race, n (%) | — | — |
Asian | 131 (100.0) | 132 (100.0) |
ECOG PS score, n (%) | — | — |
0 | 51 (38.9) | 46 (34.8) |
1 | 80 (61.1) | 86 (65.2) |
Histology, n (%) | — | — |
Undifferentiated nonkeratinized | 98 (74.8) | 96 (72.7) |
Differentiated nonkeratinized | 18 (13.7) | 21 (15.9) |
Keratinized squamous carcinoma | 8 (6.1) | 10 (7.6) |
Unclassified | 7 (5.3) | 5 (3.8) |
EBV DNA level, n (%) | — | — |
< 500 IU/mL | 26 (19.8) | 37 (28.0) |
≥ 500 IU/mL | 105 (80.2) | 95 (72.0) |
Patients with any prior anticancer drug therapy, n (%) | 83 (63.4) | 88 (66.7) |
Type of prior anticancer drug therapy, n (%) | — | — |
Adjuvant | 36 (43.4) | 32 (36.4) |
Neoadjuvant | 55 (66.3) | 59 (67.0) |
Concurrent with radiotherapy | 61 (73.5) | 67 (76.1) |
Other | 6 (7.2) | 4 (4.5) |
Patients with any prior anticancer surgeries, n (%) | 6 (4.6) | 4 (3.0) |
Patients with any prior anticancer radiotherapy, n (%) | 84 (64.1) | 91 (68.9) |
EBV = Epstein-Barr virus; ECOG = Eastern Cooperative Oncology Group; GC = gemcitabine and cisplatin; ITT = intention to treat; PS = performance status; SD = standard deviation.
Sources: Sponsor’s Summary of Clinical Evidence,2 RATIONALE-309 Clinical Study Report (interim analysis),33 and RATIONALE-309 Clinical Study Report (final analysis).30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure and use of concomitant medications and subsequent treatments in the included study are provided in the Supplemental Material document, in Appendix 4.
At the March 2021 interim analysis, for the primary end point of PFS as assessed by the IRC, the median follow-ups were 10.25 months (range, 0.3 to 23.3 months) in the tislelizumab plus GC arm and 9.77 months (range, 0.1 to 20.9 months) in the placebo plus GC arm.33 The mean duration of exposure to tislelizumab was 37.25 weeks (standard deviation [SD] = 20.650) in the tislelizumab plus GC arm and to placebo it was 30.52 weeks (SD = 14.927) in the placebo plus GC arm. The mean durations of exposure to gemcitabine were 16.35 weeks (SD = 5.174) in the tislelizumab plus GC arm and 16.04 weeks (SD = 4.628) in the placebo plus GC arm. The mean durations of exposure to cisplatin were 16.29 weeks (SD = 5.221) and 16.01 weeks (SD = 4.673) in the 2 study arms, respectively.2
At the December 2023 data cut-off, the median follow-ups for OS were 41.4 months (95% confidence interval [CI], 40.1 to 42.2) in the tislelizumab plus GC arm and 40.8 months (95% CI, 39.8 to 41.7) in the placebo plus GC arm; the median study follow-ups were 33.25 months (range, 0.3 to 52.4 months) and 25.00 months (range, 0.1 to 53.0 months) in the 2 groups, respectively.30 The mean durations of exposure to tislelizumab and placebo in the groups were 67.16 weeks (SD = 60.610) and 39.53 weeks (SD = 34.264), respectively. Exposure data for GC were not available for the December 2023 data cut-off because all patients had discontinued chemotherapy treatment at the time of the interim analysis.2
Regarding concomitant medications, at the December 2023 data cut-off, the most commonly received classes of medications in the tislelizumab plus GC and placebo plus GC groups were serotonin 5-HT3 antagonists (100.0% in each group), proton-pump inhibitors (90.2% and 92.3%, respectively), glucocorticoids (87.2% and 83.8%), colony-stimulating factors (83.5% and 90.0%), and solutions affecting the electrolyte balance (82.7% and 81.5%).30
As of the December 2023 data cut-off, 69 patients (52.3%) in the placebo plus GC arm had crossed over to tislelizumab monotherapy following initial disease progression and 13 patients (9.8%) in the tislelizumab plus GC arm had received tislelizumab monotherapy beyond progression. As of the December 2023 data cut-off, 78 patients (59.5%) in the tislelizumab plus GC arm and 32 patients (24.2%) in the placebo plus GC arm had received new subsequent anticancer systemic therapy.
Critical Appraisal
Internal Validity
The RATIONALE-309 study was a randomized, double-blind, placebo-controlled trial. Methods of randomization and treatment allocation by the interactive response technology system were adequate for limiting the risk of bias in the randomization process. Although the CDA-AMC Clinical Review team noted imbalances across treatment groups at baseline in some characteristics (e.g., Epstein-Barr virus DNA level, smoking status, and time since initial diagnosis), the clinical experts consulted by CDA-AMC for this review did not expect these imbalances to affect the interpretation of the efficacy or safety results. By the final data cut-off, all patients had discontinued both the treatments and the study. According to the clinical experts, the reasons for treatment discontinuation (progressive disease [primarily], withdrawal by patient, AEs, and physician decision) were generally aligned with clinical practice. However, 1 clinical expert noted that withdrawal by patient, which occurred in close to 20% of patients in each group, was less likely to occur than withdrawals due to AEs in practice. Although the proportion of patients who withdrew from treatment (i.e., withdrawal by patient) was similar in each group, the specific reasons for withdrawal were not provided. Therefore, the CDA-AMC review team could not assess whether any differences in circumstances leading to these withdrawals could have been a source of bias. Patients, investigators, and the IRC were all blinded to treatment allocation, and unblinding was performed once a patient had confirmed progression of disease by RECIST 1.1 as assessed by the IRC. Postprogression assessments (e.g., HRQoL and AEs) could be subject to bias due to patients’ and investigators’ knowledge of the treatments received; however, the extent of this potential bias is unknown.
The prespecified sample size was achieved and the trial was adequately powered for the primary end point of PFS. Multiple testing was accounted for in the interim and primary analyses for the primary end point using the Lan-DeMets O’Brien-Fleming approximation spending function, and these strategies were adhered to. No adjustments for multiplicity were made for other end points and these analyses were considered as supportive evidence; however, there are no significant concerns regarding type I errors because the results for these end points were directionally aligned but generally not statistically significant. Furthermore, results for overall response rate (ORR) and duration of response (DOR) were reported only for the interim analysis and should be interpreted with the understanding that treatment effects assessed at an interim analysis are prone to overestimation.36 In the assessment of ORR, patients without any postbaseline assessment were considered nonresponders; however, the risk of bias due to missing data was likely low.2 The DOR results do not accurately reflect the broader patient experience and are susceptible to selection bias, as DOR is measured only in patients who responded to treatment; these patients are a nonrandom subset of the study population that may differ biologically, including in genetic and immune profiles, which could influence prognosis.
The clinical experts consulted by CDA-AMC did not identify any differences in concomitant medications between treatment arms that would be expected to affect the treatment effect or tolerability of tislelizumab. The experts noted that some of the subsequent medications received by patients in the RATIONALE-309 trial differed from those currently available to patients with NPC in Canada, while some were representative of the treatments that would be expected in the Canadian context. Clinical experts provided differing input regarding whether the effect of subsequent therapies would be expected to affect interpretation of the OS results. In considering the differences in subsequent therapies received by trial participants (particularly immuno-oncology and targeted treatments) compared to those that would be received by patients in Canada, the CDA-AMC review team found that there is some uncertainty around whether the estimated magnitude of effect of tislelizumab on OS would reflect what would occur in clinical practice in Canada.
At the final analysis, important protocol deviations overall and by category were generally balanced between treatment groups and were unlikely to affect the results. The CDA-AMC review team did not identify any protocol amendments that would affect the interpretation of the results.
Two sensitivity analyses with different censoring rules (evaluating the impact of new anticancer treatment and the impact of 2 or more consecutive missing tumour assessments) were performed for PFS at the interim analysis. The stratified HR (and 95% CI) of these sensitivity analyses were consistent with the stratified HR (and 95% CI) of the primary analysis, supporting the robustness of the interim analysis PFS results.
Patients in the placebo plus GC arm who experienced disease progression had the option to cross over to receive tislelizumab monotherapy. According to the sponsor, this was permitted for ethical reasons, due to the rare and life-threatening nature of NPC and lack of treatment options.37 However, first-line GC followed by second-line tislelizumab monotherapy is not a relevant comparator in current practice. At the final analysis, 52.3% of patients in the placebo plus GC arm had crossed over to tislelizumab monotherapy. This introduces uncertainty regarding the magnitude of the treatment effect on OS due to indirectness, as patients in clinical practice are not treated with tislelizumab in the second line. Due to the protocol-allowable crossovers, the intention-to-treat–based OS effect estimate for tislelizumab could be underestimated; however, the magnitude of underestimation cannot be determined with the available data. Two supportive analyses using model-based approaches (the rank-preserving structural failure time model [RPSFTM] and the 2-stage method) were performed for OS to investigate the impact of the protocol-allowable crossovers. However, both methods are subject to unverifiable assumptions that are unlikely to be true. Specifically, the RPSFTM assumes that the treatment effect is equal for all patients no matter when the treatment is received (first-line or second-line) and the 2-stage method assumes that the model has adjusted for all potential confounders.38
Evidence was not available to validate PFS as a surrogate end point for OS in trials of tislelizumab or ICI treatment in patients with R/M NPC. The sponsor submitted 1 systematic review to support the validity of PFS as a surrogate for OS.39 Although this study suggested that PFS may be correlated with OS in patients with nonmetastatic NPC being treated with combined chemotherapy and radiotherapy, it did not provide evidence that the results are generalizable to patients with R/M NPC who are being treated with a PD-1 inhibitor. As such, whether PFS is a valid surrogate for patients with R/M NPC being treated with tislelizumab is uncertain.
For the analyses of both PFS and OS, there was a violation of the proportional hazards assumption as evidenced by the delay in separation of the curves for both outcomes and crossing of the curves at approximately 9 months for OS. As such, the ratio of the hazard functions is not constant over time and reliance on the HR alone to inform the effect of tislelizumab plus GC compared with placebo plus GC on PFS and OS may be misleading. Between-group differences in Kaplan-Meier (KM)–estimated probabilities of PFS and OS at clinically relevant follow-up times, which were used to judge the certainty of evidence for these end points, were not affected by this limitation. Censoring reasons for PFS did not present concerns for informative censoring. OS censoring results were generally balanced among groups and were as expected; however, as previously noted, it is uncertain whether any differences in circumstances leading to withdrawals could have been a source of bias.
The certainty of the point estimates for the between-group differences in KM-estimated probabilities of PFS at 36 months and the OS at 12 and 36 months and in the proportion of patients with at least 1 SAE were affected by imprecision. Although the PFS and OS estimates suggested a clinically important benefit with the addition of tislelizumab, the 95% CIs included the possibility of little to no clinically meaningful difference. The point estimate for SAEs suggested little to no difference between groups, but the 95% CI included the possibility of both a benefit and harm.
For the EORTC QLQ-C30 and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Head and Neck 35 (EORTC QLQ-H&N35) outcome measures, no evidence was available to support the psychometric properties (validity, reliability, and responsiveness) of these scales in patients with NPC; however, evidence exists to support the psychometric properties of these scales in other types of cancers and in head and neck cancers, respectively.40-46 A symptom index score from the EORTC QLQ-H&N35 was derived for use in the RATIONALE-309 trial but is not a validated score.34 Between-group anchor-based minimal important difference (MID) estimates from the literature were used as the thresholds for the EORTC QLQ-C30 GHS/QoL domain outcome in the GRADE assessment.47 These MID values were estimated for head and neck cancer; however, the clinical experts consulted by CDA-AMC noted that it is reasonable to extrapolate these MID estimates to NPC.
For EORTC QLQ-C30 and EORTC QLQ-H&N35 end points, the proportion of patients with missing data increased over time, with data missing for 17% of patients in the tislelizumab plus GC arm and 11% of patients in the placebo plus GC arm at cycle 4, and for 25% and 33%, respectively, at cycle 8. Missing data for these end points were imputed via a mixed-effects model for repeated measures;2 this method assumes that data are missing at random, which is unlikely to be true. As such, the HRQoL results are at risk of bias due to missing outcome data; the direction of bias is uncertain.
Because imAEs were reported up to 90 days after the last dose of tislelizumab or placebo, regardless of initiation of a new anticancer therapy after exiting the trial, there is the potential that the reported imAEs include those due to subsequent immuno-oncology treatments; however, the magnitude of the impact of subsequent treatments on imAE incidence could not be predicted. All other AE data include only AEs reported before patients started a new anticancer treatment (including crossovers). Although the efficacy results include placebo patients who crossed over to tislelizumab monotherapy, the harms results (with the exception of imAEs) for this group do not reflect AEs experienced following a crossover.
External Validity
The clinical experts consulted by CDA-AMC commented that, in general, the eligibility criteria used in the RATIONALE-309 trial appropriately captured patients who would be considered candidates for tislelizumab in clinical practice and are generally standard for a trial of this type of therapy, However, they noted that certain criteria may not apply in clinical practice. For example, patients with a glomerular filtration rate of less than 60 mL/min; archival tissue more than 2 years old; less than 6 months from prior curative treatment; or conditions such as diabetes, infections, autoimmune disease (e.g., rheumatoid arthritis), and cardiac risk factors may still be considered for treatment with tislelizumab. The RATIONALE-309 trial took place in Asia; all enrolled patients were of Asian race, and other populations affected by NPC in Canada (e.g., Inuit patients) were not represented in the trial.
The clinical experts consulted by CDA-AMC stated that the characteristics of randomized patients were generally reflective of the population of patients with NPC in Canada. According to the experts, the distribution of histology types is representative of what would be seen in practice in Canada and the results of the trial are generalizable to patients who are not of Asian race. One clinical expert noted that the typical disease biology of NPC in endemic Asian populations (undifferentiated, nonkeratinizing, Epstein-Barr virus–positive), is often seen in patients of Asian descent and Indigenous populations in Canada. This expert also commented that inclusion of patients with other disease subtypes (e.g., keratinizing histology) is representative of other populations of patients with NPC in Canada. The clinical experts agreed that patients with an ECOG PS score of 2 should be considered for treatment with tislelizumab when clinically appropriate. The small proportion of patients aged 65 years or older in the RATIONALE-309 trial suggested a potential limitation to the generalizability of the trial results to this population; however, 1 clinical expert noted that access to patients older than 65 years should not be restricted based on this finding.
The clinical experts consulted by CDA-AMC indicated that GC is a relevant comparator because current first-line treatment for R/M NPC in Canada is platinum (cisplatin or carboplatin) and gemcitabine doublet chemotherapy for up to 6 cycles. The experts acknowledged that the dosing regimens for tislelizumab and GC used in the RATIONALE-309 trial are aligned with what is currently used (or what would be expected) in practice in Canada, although there may be differences in chemotherapy infusion time at different settings. At the final analysis, 9.8% of patients in the tislelizumab plus GC arm received tislelizumab monotherapy beyond progression. The experts provided comments regarding different circumstances for continuation of tislelizumab beyond progression, including suspected pseudoprogression, insufficient time on immunotherapy (e.g., less than 3 months), or if there is a clinical benefit to the patient (e.g., appearance of small lesions but maintained symptomatic benefit). The clinical experts commented that, although certain concomitant medications used during the RATIONALE-309 trial are not commonly used in clinical practice in Canada, the results of the trial were still generalizable to patients with NPC in Canada.
According to input from the clinical experts consulted by CDA-AMC and patient and clinician groups, the most important goals in the first-line treatment of R/M NPC are to prolong survival, delay cancer progression, induce tumour shrinkage, maintain or improve QoL, alleviate or control cancer-related symptoms, and minimize side effects from treatments. As such, the main efficacy and harms outcomes assessed in the RATIONALE-309 trial align with the key outcomes of importance to patients and clinicians. The clinical experts consulted by CDA-AMC stated that treatment response is assessed in clinical practice by patient history, medical examination, and imaging, which align with what was used in clinical trials. The experts acknowledged that cycles 4 and 8 were clinically relevant and reasonable time points at which to study the effect of treatment on HRQoL end points.
Results
The key efficacy and harms results, including findings from the GRADE assessment, are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
Outcomes Assessed Using GRADE
PFS: At the final analysis (median follow-up durations of 33.3 months for the tislelizumab plus GC arm and 25.0 months for the placebo plus GC arm), the probabilities of PFS at 9 months were 52.7% (95% CI, 43.4 to 61.2) in the tislelizumab plus GC arm and 26.1% (95% CI, 18.3 to 34.5) in the placebo plus GC arm; the difference between groups was 26.6% (95% CI, 14.5 to 38.8). The probabilities of PFS at 36 months were 17.4% (95% CI, 10.7 to 25.5) and 6.3% (95% CI, 2.7 to 12.0), respectively, with a difference between groups of 11.1% (95% CI, 2.3 to 19.9).
OS: At the final analysis (median follow-up durations of 41.4 months in the tislelizumab plus GC arm and 40.8 months in the placebo plus GC arm), the probabilities of OS at 12 months were 89.6% (95% CI, 82.8 to 93.8) in the tislelizumab plus GC arm and 84.9% (95% CI, 77.1 to 90.2) in the placebo plus GC arm; the difference between groups was 4.7% (95% CI, −3.7 to 13.0). The probabilities of OS at 36 months were 55.6% (95% CI, 46.0 to 64.1) and 46.4% (95% CI, 36.9 to 55.3), respectively, with a difference between groups of 9.2% (95% CI, −3.8 to 22.2).
HRQoL: At baseline, the mean EORTC QLQ-C30 GHS/QoL scores were 62.9 (SD = 23.23) in the tislelizumab plus GC group and 67.6 (SD = 21.27) in the placebo plus GC group. At cycle 4, the changes from baseline in the least squares (LS) mean score were −2.48 (95% CI, −6.14 to 1.18) and −0.54 (95% CI, −4.11 to 3.03), respectively, with a difference in the LS mean of −1.94 (95% CI, −6.72 to 2.84). At cycle 8, the changes from baseline in the LS mean score were 2.72 (95% CI, −0.98 to 6.43) in the tislelizumab plus GC group and 3.03 (95% CI, −0.80 to 6.86) in the placebo plus GC group, with a difference in the LS mean of −0.31 (95% CI, −5.32 to 4.70).
Other Key Outcomes Not Assessed Using GRADE
PFS and OS were primary and secondary end points, respectively, in the RATIONALE-309 trial. The results for median PFS, median OS, and the HRs (95% CI) are presented here, but did not contribute to appraisals of the certainty of evidence.
PFS: At the time of the interim analysis (median follow-up durations of 10.3 months for the tislelizumab plus GC arm and 9.8 months for the placebo plus GC arm), tislelizumab plus GC was favoured over placebo plus GC for PFS by IRC (stratified HR = 0.52; 95% CI, 0.38 to 0.73; 1-sided stratified log-rank test P value < 0.0001). At the final analysis, the median PFS times as assessed by the IRC were 9.6 months (95% CI, 7.6 to 11.6) in the tislelizumab plus GC arm and 7.4 months (95% CI, 5.6 to 7.6) in the placebo plus GC arm; the stratified HR was 0.53 (95% CI, 0.39 to 0.71; 1-sided stratified log-rank test P value not reported).
Two sensitivity analyses with different censoring rules were performed for PFS at the interim analysis. For the first sensitivity analysis, in which PFS was derived without considering the occurrence of new anticancer treatment, the stratified HR was 0.53 (95% CI, 0.38 to 0.74). For the second sensitivity analysis, in which PFS was derived regardless of 2 or more missed tumour assessments, the stratified HR was 0.53 (95% CI, 0.38 to 0.73).33
OS: At the final analysis, median OS times were 45.3 months (95% CI, 33.4 to not evaluable [NE]) in the tislelizumab plus GC arm and 31.8 months (95% CI, 25.0 to NE) in the placebo plus GC arm; the stratified HR was 0.73 (95% CI, 0.51 to 1.05).2
Supplemental analyses intended to account for crossovers, using the RPSFTM and the 2-stage method, resulted in stratified HRs of 0.56 (95% CI, 0.27 to 1.19) and 0.62 (95% CI, 0.40 to 0.97), respectively. The probabilities of OS at 36 months were 55.6% (95% CI, 45.99 to 64.12) in the tislelizumab plus GC arm and 39.4% (95% CI, 28.52 to 49.99) in the placebo plus GC arm using the RPSFTM analysis and 55.6% (95% CI, 45.99 to 64.12) and 41.8% (95% CI, 31.77 to 51.45) in the 2 groups, respectively, following the 2-stage method.2
ORR: At the time of the interim analysis, the ORRs by IRC were 69.5% (95% CI, 60.8 to 77.2) in the tislelizumab plus GC arm and 55.3% (95% CI, 46.4 to 64.0) in the placebo plus GC arm, with an odds ratio of 1.85 (95% CI, 1.11 to 3.07). Results for ORR were not presented at the final analysis.2
DOR: At the time of the interim analysis, the median DORs by IRC were 8.5 months (95% CI, 6.5 to NE) in the tislelizumab plus GC arm and 6.1 months (95% CI, 4.7 to 6.2) in the placebo plus GC arm. Results for DOR were not presented at the final analysis.2
Harms
AEs were reported for up to 30 days after the last dose of the study drug or until initiation of another anticancer therapy (whichever occurred first). Assessment of imAEs occurred for up to 90 days after the last dose of tislelizumab or placebo, regardless of initiation of a new anticancer therapy after exiting the trial. For the placebo plus GC arm, only AEs reported before crossover to tislelizumab monotherapy are included in the safety analyses.30,48 Key results include the following for the final analysis (December 2023 data cut-off):
At least 1 treatment-emergent adverse event (TEAE) was reported for 133 of 133 patients (100%) in the tislelizumab plus GC arm and for 129 of 130 patients (99%) in the placebo plus GC arm. TEAEs that occurred in at least 30% of patients in either the tislelizumab plus GC or placebo plus GC group, respectively, were anemia (87% and 91%), decreased neutrophil count (62% and 59%), decreased white blood cell count (62% and 63%), nausea (59% and 72%), decreased platelet count (54% and 62%), decreased appetite (48% and 50%), vomiting (41% and 53%), neutropenia (35% and 29%), constipation (35% and 46%), leukopenia (35% and 35%), hyponatremia (31% and 29%), and hypothyroidism (31% and 15%).
At least 1 SAE was reported for 47 patients (35%) in the tislelizumab plus GC arm and for 46 patients (35%) in the placebo plus GC arm. SAEs that occurred in at least 2% of patients in either the tislelizumab plus GC or placebo plus GC group were decreased platelet count (6.8% and 10.8%, respectively), sepsis (3.0% and 0.0%), thrombocytopenia (3.0% and 2.3%), anemia (2.3% and 3.8%), decreased neutrophil count (2.3% and 4.6%), decreased white blood cell count (2.3% and 3.8%), pneumonia (1.5% and 3.1%), and leukopenia (0.0% and 3.1%).
At least 1 AE of grade 3 or higher was reported for 113 patients (85.0%) in the tislelizumab plus GC arm and for 111 patients (85.4%) in the placebo plus GC arm. AEs of grade 3 or higher that occurred in at least 30% of patients in either the tislelizumab plus GC or placebo plus GC group were decreased white blood cell count (30.1% and 39.2%, respectively), anemia (30.8% and 33.1%), and decreased neutrophil count (29.3% and 37.7%).
Any component of study treatment was discontinued due to AEs for 22 patients (17%) in the tislelizumab plus GC arm and 14 patients (11%) in the placebo plus GC arm. Tislelizumab was discontinued due to AEs for 12 patients (9%) in the tislelizumab plus GC arm and placebo was discontinued due to AEs for 6 patients (5%) in the placebo plus GC arm. Chemotherapy was discontinued due to AEs for 12 patients (9%) and 11 patients (8.5%) in the 2 treatment arms, respectively.
Death due to AEs was reported for 7 patients (5.3%) in the tislelizumab plus GC arm and for 2 patients (1.5%) in the placebo plus GC arm. TEAEs leading to death were reported for 6 patients (4.5%) and 2 patients (1.5%) in the 2 arms, respectively. In the tislelizumab plus GC arm, TEAEs in the tislelizumab plus GC arm comprised accidental death, death, depressed level of consciousness, hypoxia, myelodysplastic syndrome, pharyngeal hemorrhage, and pneumonia aspiration. TEAEs in the placebo plus GC arm comprised death, arrhythmia, and hypernatremia.30
At least 1 imAE was reported for 71 patients (53%) in the tislelizumab plus GC arm and 49 patients (38%) in the placebo plus GC arm. The most frequently reported imAEs by category that occurred in at least 5% of patients in either arm were immune-mediated skin adverse reactions (29% and 25%, respectively) and immune-mediated endocrinopathies (hypothyroidism) (29% and 15%).
Summary of Findings and Certainty of the Evidence
Literature-based MID estimates were used as the thresholds for the EORTC QLQ-C30 GHS/QoL domain outcome (between-group MID: 5 points for improvement and −7 points for deterioration).47 These MID values were estimated for head and neck cancer; however, the clinical experts consulted by CDA-AMC noted that it is reasonable to extrapolate these MID estimates to NPC. A summary of outcome measures is available in Appendix 3 of the Supplemental Material document. In the absence of literature-based MID estimates, the following thresholds for clinically meaningful between-group differences suggested by the clinical experts consulted by CDA-AMC were applied: 10 to 15 percentage points for probability of PFS at 9 months and 36 months, and 5 to 10 percentage points for probability of OS at 12 months and 36 months. In the absence of a known threshold, the certainty in the presence of a non-null effect was rated for serious AEs.
Table 4: Summary of Findings for Tislelizumab Plus GC vs. Placebo Plus GC for First-Line Treatment of Patients With R/M NPC (Final Analysis; December 8, 2023, Data Cut-off)
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo plus GC | Tislelizumab plus GC | Difference | |||||
PFS per RECIST 1.1 by blinded IRC | |||||||
Probability of PFS at 9 months Median follow-up: 25.0 months for placebo plus GC; 33.3 months for tislelizumab plus GC | 263 (1 RCT) | NA | 261 per 1,000 | 527 per 1,000 (434 to 612 per 1,000) | 266 more per 1,000 (145 to 388 more per 1,000)a | High | Tislelizumab plus GC results in a clinically important increase in the probability of PFS at 9 months compared to placebo plus GC. |
Probability of PFS at 36 months Median follow-up: 25.0 months for placebo plus GC; 33.3 months for tislelizumab plus GC | 263 (1 RCT) | NA | 63 per 1,000 | 174 per 1,000 (107 to 255 per 1,000) | 111 more per 1,000 (23 to 199 more per 1,000)a | Moderateb (Serious imprecision) | Tislelizumab plus GC likely results in a clinically important increase in the probability of PFS at 36 months compared to placebo plus GC. |
OS | |||||||
Probability of OS at 12 months Median follow-up: 40.8 months for placebo plus GC; 41.4 months for tislelizumab plus GC | 263 (1 RCT) | NA | 849 per 1,000 | 896 per 1,000 (828 to 938 per 1,000) | 47 more per 1,000c (37 less to 130 more per 1,000)a | Moderateb,d (Serious imprecision) | Tislelizumab plus GC likely results in an increase in the probability of OS at 12 months compared to placebo plus GC. The increase approached the threshold and may be considered clinically important. |
Probability of OS at 36 months Median follow-up: 40.8 months for placebo plus GC; 41.4 months for tislelizumab plus GC | 263 (1 RCT) | NA | 464 per 1,000 | 556 per 1,000 (460 to 641 per 1,000) | 92 more per 1,000 (38 less to 222 more per 1,000)a | Moderateb,d (Serious imprecision) | Tislelizumab plus GC likely results in a clinically important increase in the probability of OS at 36 months compared to placebo plus GC. |
HRQoL | |||||||
EORTC QLQ-C30 GHS/QoL domain LS mean change from baseline (0 [worst] to 100 [best]) Follow-up: cycle 4 | 226 (1 RCT) | NA | −0.54 (−4.11 to 3.03) | −2.48 (−6.14 to 1.18) | −1.94 (−6.72 to 2.84) | Moderatee (Serious risk of bias) | Tislelizumab plus GC likely results in little to no difference in HRQoL at cycle 4 compared to placebo plus GC. |
EORTC QLQ-C30 GHS/QoL domain LS mean change from baseline (0 [worst] to 100 [best]) Follow-up: cycle 8 | 186 (1 RCT) | NA | 3.03 (−0.80 to 6.86) | 2.72 (−0.98 to 6.43) | −0.31 (−5.32 to 4.70) | Lowf (Very serious risk of bias) | Tislelizumab plus GC may result in little to no difference in HRQoL at cycle 8 compared to placebo plus GC. |
Harms | |||||||
Patients with ≥ 1 SAE Median follow-up: 25.0 months for placebo plus GC; 33.3 months for tislelizumab plus GC | 263 (1 RCT) | NR | 354 per 1,000 | 353 per 1,000 | 1 less per 1,000 (116 less to 115 more per 1,000)a | Lowg (Very serious imprecision) | Tislelizumab plus GC may result in little to no difference in the proportion of patients with 1 or more SAE compared to placebo plus GC. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GC = gemcitabine and cisplatin; GHS = global health status; HRQoL = health-related quality of life; IRC = independent review committee; LS = least squares; NA = not applicable; NPC = nasopharyngeal carcinoma; NR = not reported; OS = overall survival; PFS = progression-free survival; QoL = quality of life; RCT = randomized controlled trial; R/M = recurrent or metastatic; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1; SAE = serious adverse event; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThis analysis was not part of the statistical analysis plan and was requested from the sponsor by CDA-AMC to facilitate the GRADE assessment.
bRated down 1 level for serious imprecision. The 95% CI for the difference between groups includes the possibility of little to no clinically meaningful difference, as informed by thresholds suggested by the clinical experts consulted by CDA-AMC.
cOf the clinical experts consulted by CDA-AMC, 2 considered this a clinically meaningful difference and 1 did not.
dPatients in the placebo plus GC group were permitted to cross over to tislelizumab monotherapy postprogression but usual care (i.e., first-line GC) followed by second-line tislelizumab monotherapy is not a relevant comparator in current practice. However, this was not rated down due to indirectness because the magnitude of the difference between groups is unlikely to be smaller.
eRated down 1 level for serious risk of bias due to missing outcome data.
fRated down 2 levels for very serious risk of bias due to missing outcome data.
gRated down 2 levels for very serious imprecision. No known minimal importance difference; the target of the certainty assessment was any effect. The 95% CI for the difference between groups includes the possibility of both benefit and harm.
Sources: Sponsor’s Summary of Clinical Evidence,2 RATIONALE-309 Clinical Study Report (final analysis),30 and sponsor’s response to CDA-AMC request for additional information.37 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
The sponsor did not submit any long-term extension studies for tislelizumab in the first-line treatment of R/M NPC.
Indirect Evidence
The sponsor did not submit any indirect treatment comparisons.
Studies Addressing Gaps in the Systematic Review Evidence
No additional studies were included in the sponsor’s submission.
Discussion
Efficacy
According to input from clinical experts consulted by CDA-AMC and patient and clinician groups, the most important goals in the first-line treatment of R/M NPC are to prolong survival, delay cancer progression, induce tumour shrinkage, maintain or improve QoL, alleviate or control cancer-related symptoms, and minimize side effects from treatments. As such, the main efficacy and harms outcomes assessed in the RATIONALE-309 trial align with the key outcomes of importance to patients and clinicians.
Input from the clinician groups regarding current management of NPC, unmet needs and existing challenges, and considerations for using tislelizumab generally aligned with input from clinical experts consulted by CDA-AMC for this review.
Evidence from the RATIONALE-309 trial demonstrated that first-line treatment for with tislelizumab plus GC resulted in improvement in the primary end point of median PFS in patients with R/M NPC compared to patients who received placebo plus GC. Clinically important increases in the probability of PFS were demonstrated for tislelizumab plus GC compared to placebo plus GC at 9 months and 36 months, with high and moderate certainty at these time points, respectively. The thresholds used to judge the clinical meaningfulness of the estimated effects are based on input from 3 clinical experts consulted by CDA-AMC. The KM plot for PFS showed separation of the curves between 6 and 9 months, with a higher proportion of patients in the tislelizumab plus GC arm experiencing PFS over time compared to the placebo plus GC arm. Of note, due to violation of the proportional hazards assumption, reliance on HR alone to interpret the effect of tislelizumab plus GC compared with placebo plus GC may be misleading; however, the between-group differences in KM-estimated probabilities at 9 and 36 months are not affected by this limitation. Sensitivity analyses demonstrated HR results consistent with the HR of the primary analysis, supporting the robustness of the PFS results. The validity of PFS as a surrogate end point for OS in the first-line treatment of R/M NPC with tislelizumab or ICI therapy is unknown. Evidence exists to support PFS as a surrogate end point for OS in trials of combined chemotherapy and radiotherapy to treat nonmetastatic NPC; however, these results may not be generalizable to treatments with different mechanisms of action.
For the secondary end point of OS at the final analysis, improvement in median OS was demonstrated for patients in the tislelizumab plus GC arm compared to patients in the placebo plus GC arm; this improvement was considered clinically meaningful by clinical experts consulted by CDA-AMC. In addition, the evidence suggests that, compared with placebo plus GC, tislelizumab plus GC likely results in increases in the probability of OS at 12 months and 36 months. All 3 clinical experts agreed that the improvement in the probability of OS at 36 months was clinically meaningful. The CDA-AMC review team rated the certainty in the probability of OS at both time points as moderate due to imprecision. The thresholds used to judge the clinical meaningfulness of the estimated effects are based on input from clinical experts consulted by CDA-AMC. The KM plot of OS showed initial crossing of the curves followed by small separation of the curves between 9 and 12 months; the OS benefit took time to accrue, with a higher proportion of patients in the tislelizumab plus GC arm experiencing OS over time compared to the placebo plus GC arm. Clinical experts commented that the shape of the KM curves of PFS and OS were as expected for this type of therapy, as the benefits of immunotherapy are expected to be delayed. According to the sponsor, for ethical reasons, crossover to tislelizumab monotherapy was permitted for patients in the placebo plus GC arm who experienced disease progression. The considerable proportion of patients who crossed over to tislelizumab monotherapy at the final analysis introduces uncertainty regarding the magnitude of treatment effect on OS. One clinical expert highlighted that the between-group difference in median OS was clinically meaningful despite crossovers being permitted. Supportive analyses performed for OS were subject to unverifiable assumptions. Because specific reasons for patient withdrawal from treatment were not available, it is uncertain whether any differences in circumstances leading to these withdrawals could have been a source of bias. There is uncertainty regarding whether OS results would be affected by subsequently received anticancer treatments, as well as the direction and magnitude of these potential effects.
At the interim analysis, ORR and median DOR were both greater in the tislelizumab plus GC arm compared to the placebo plus GC arm. Two of the clinical experts consulted by CDA-AMC considered the differences between groups for both end points to be clinically meaningful. The third clinical expert commented that ORR would only be clinically relevant if a patient is symptomatic, and did not consider the between-group difference in DOR to be clinically meaningful. Results for ORR and DOR end points are considered as supportive evidence and should be interpreted with the understanding that that treatment effects assessed at an interim analysis are prone to overestimation.
The clinical experts consulted by CDA-AMC identified the EORTC QLQ-C30 GHS/QoL domain as an HRQoL end point of key clinical relevance for patients with R/M NPC. At the final analysis, both treatment groups had a small decline in the LS mean score from baseline at cycle 4 and both groups had a small increase in the LS mean score from baseline at cycle 8. The evidence suggests little to no difference in the QLQ-C30 GHS/QoL domain between tislelizumab plus GC and placebo plus GC. The certainty in the evidence was rated as moderate and low at cycles 4 and 8, respectively, due to the risk of bias. The thresholds used to judge the target of certainty in the GRADE assessment are based on between-group MID estimates from the literature for head and neck cancer; however, the clinical experts consulted by CDA-AMC noted that it is reasonable to extrapolate these MID estimates to NPC. Limitations to the interpretation of the HRQoL results include missing outcome data and the potential for unblinding to treatment allocation upon confirmed progression to introduce bias.
Regarding the generalizability of the results of the RATIONALE-309 trial, the clinical experts consulted by CDA-AMC noted that the eligibility criteria in the RATIONALE-309 trial were generally expected for a clinical trial of immunotherapy, but added that, in clinical practice, some patients excluded from the trial, such as those with less than 6 months from prior curative treatment, an ECOG PS score of 2, or conditions such as diabetes, infections, autoimmune disease, and cardiac risk factors, may be considered for treatment with tislelizumab. According to the clinical experts, patients enrolled in the RATIONALE-309 trial were generally reflective of the population of patients with NPC in Canada. The experts stated that, although all patients in the trial were of Asian race, the results are considered generalizable to other patient populations affected by NPC in Canada (e.g., Inuit patients) due to the disease subtypes represented within the trial. There are potential generalizability limitations for patients aged 65 years and older as this cohort represented a small proportion of patients within the trial; however, 1 clinical expert noted that patients older than 65 years should not be restricted from accessing treatment based on this finding. One clinical expert commented that real-world data collected from patients with NPC in Canada and treated with tislelizumab plus platinum doublet chemotherapy would help address gaps in the study pertaining to efficacy and tolerability in certain groups of patients (e.g., older individuals, keratinizing histology, Inuit, and/or other endemic populations).
Harms
All but 1 patient in the RATIONALE-309 trial experienced at least 1 TEAE. The clinical experts consulted by CDA-AMC stated that the AEs reported in the trial are as expected for immunotherapy combined with GC. The experts noted that immunotherapy would be expected to introduce new toxicities. One clinical expert commented that, despite the possibility of increased incidence of SAEs with the introduction of a third agent, similar proportions of patients in both groups experienced AEs and SAEs of grade 3 or higher. The evidence suggested tislelizumab plus GC may result in little to no difference in the incidence of SAEs compared to placebo plus GC, with low certainty due to imprecision. A higher proportion of patients in the tislelizumab plus GC arm experienced imAEs compared with the placebo plus GC arm, a finding that is aligned with what would be expected with the addition of tislelizumab treatment, according to the clinical experts. It is also possible that reported imAEs included those due to subsequent immuno-oncology treatments; however, the magnitude of the impact of these subsequent treatments on imAE incidence could not be estimated. The tislelizumab product monograph includes a warning regarding the risk of immune-related adverse reactions, including pneumonitis, hepatitis, skin reactions, colitis, endocrinopathies (e.g., thyroid disorders and diabetes), and nephritis with renal dysfunction; as well as a warning regarding infusion-related reactions.1 The clinical experts commented that these reactions are known to occur with immuno-oncology therapy and are considered generally manageable in clinical practice. There is the possibility that unblinding could have affected the results for highly subjective AEs, and that imAEs from subsequently received anticancer treatments could have contributed to the number of imAEs reported in the RATIONALE-309 trial; however, the extent of these impacts is unknown.
Ethics and Equity Considerations
NPC is rare in North America but occurs more frequently in Southern China, Southeast Asia, North Africa, and the Arctic.5,6 NPC is 25 to 40 times more frequent in Inuit than in white populations in Canada, Denmark, and the US, and represents 4% to 7% of all malignancies in Inuit.9 According to the clinical experts consulted by CDA-AMC, in Canada, NPC disproportionately affects Inuit who may live in remote areas and some immigrant populations from regions with higher risk of the disease (namely, the Arctic, East and Southeast Asia, or North Africa). The RATIONALE-309 trial only included patients of Asian race; other populations disproportionately affected by NPC (e.g., Inuit patients) were not represented in the trial.
The clinician groups identified a high unmet need for additional treatment options and noted the significant disparities in accessing immunotherapy as part of first-line treatment for R/M NPC, with Canada lagging behind the US and the European Union. Similarly, clinical experts consulted by CDA-AMC commented that, in other countries, the combination of an ICI with gemcitabine and platinum is a standard of care as first-line treatment for patients with R/M NPC. Although tislelizumab may be available through an application to Health Canada’s Special Access Program, there is currently no readily available access to ICIs for patients with NPC in Canada. The clinical experts commented that patient populations typically affected by NPC may already face barriers to diagnosis, access to care, and equitable outcomes due to financial, language, or cultural challenges. For example, long-distance travel required for treatment and monitoring of NPC and being away from home and local support networks can be significant challenges for patients living in remote areas of Canada. The clinical experts added that some patients may encounter struggles with access, treatment, and supportive care due to language barriers, and that new immigrants may face financial challenges that affect, and are affected by, treatment. The clinical experts noted that the barriers for these populations are not unique to NPC. Inuit with NPC in Greenland and Denmark have been found to have lower OS rates, which may also be true of Inuit patients in Canada.27 The clinical experts indicated that there should be support for cultural sensitivities or language translation for Inuit and immigrant populations with NPC. In addition, patient group input indicated that NPC places a financial burden on patients and caregivers, and those with lower incomes often face great financial difficulties. Patient group input noted additional expenses related to travel for accessing imaging, costs for meal replacements and medications to manage side effects of treatments, and loss of income for patients and caregivers due to work absenteeism.
Conclusion
Evidence from the RATIONALE-309 trial demonstrated that, compared to placebo plus GC, first-line treatment with tislelizumab plus GC demonstrated a PFS benefit in adult patients with R/M NPC. Clinically important increases in the probability of PFS were demonstrated for tislelizumab plus GC compared to placebo plus GC at 9 months and 36 months, with high and moderate certainty in the findings at these time points, respectively. The trial evidence suggested that, compared with placebo plus GC, tislelizumab plus GC likely results in a benefit in OS, with increases in the probability of OS for tislelizumab plus GC compared to placebo plus GC at 12 months and 36 months, with moderate certainty in the findings at both time points. There is uncertainty in the magnitude of OS benefit as more than half of patients in the placebo plus GC group crossed over to receive tislelizumab in the second line, which is not reflective of current clinical practice in Canada. Little to no difference in HRQoL was shown between tislelizumab plus GC and placebo plus GC, as measured by the change from baseline in the EORTC QLQ-C30 GHS/QoL score at cycles 4 and 8, with moderate and low certainty in the results at these time points, respectively, due to study limitations.
Similar proportions of patients in both groups experienced SAEs, and the evidence suggested that tislelizumab plus GC may result in little to no difference in the proportion of patients with 1 or more SAEs compared to placebo plus GC, with low certainty in the findings. The types of AEs reported in the RATIONALE-309 trial were as expected for immunotherapy combined with GC.
Economic Review
Methods
The objective of the economic review undertaken by CDA-AMC is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of tislelizumab plus chemotherapy compared to chemotherapy alone for first-line treatment of patients with R/M NPC. The CDA-AMC appraisal was undertaken based on the submitted information, and the appraisal was not revised after the Notice of Compliance was received.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of tislelizumab plus chemotherapy for the first 6 cycles, followed by tislelizumab monotherapy, from the perspective of a public health care payer in Canada over a lifetime horizon of 30 years.49 The modelled population comprised adult patients with R/M NPC, which is aligned with the Health Canada proposed indication and was based on the participants from the RATIONALE 309 trial. The sponsor’s base-case analysis included costs related to drug acquisition, administration, premedication, monitoring, AEs, and subsequent treatments.49
In the sponsor’s base case, tislelizumab plus chemotherapy was associated with incremental costs of $102,166 and 2.27 incremental quality-adjusted life-years (QALYs) relative to chemotherapy alone, resulting in an incremental cost-effectiveness ratio (ICER) of $45,105 per QALY gained.49 Of the incremental benefit compared to chemotherapy alone (2.27 incremental QALYs), approximately 91% of the benefit was predicted to accrue beyond the trial follow-up period (median follow-up = 27 months). Additional information about the sponsor’s submission is summarized in the Supplemental Material document, in Appendix 10.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 5; full details are provided in the Supplemental Material document, in Appendix 11).
Table 5: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The long-term comparative efficacy of tislelizumab plus chemotherapy compared to chemotherapy alone is uncertain. | Evidence from the RATIONALE‑309 trial suggests that tislelizumab plus GC likely provides clinically meaningful improvements in PFS and OS compared to GC alone. Clinical expert feedback noted that the sponsor’s long-term survival estimates for both chemotherapy alone and tislelizumab plus chemotherapy may be overestimated within the submitted model. While the crossover to tislelizumab could have resulted in underestimation of the OS effect estimate, the high‑degree of crossover compounded with the absence of real-world survival data for both arms leave uncertainty in the magnitude of the OS benefit. | CDA-AMC selected the Weibull curves for PFS and OS of both tislelizumab plus chemotherapy and chemotherapy alone. | To explore uncertainty around this issue, CDA‑AMC conducted a scenario analysis in which the log-logistic approach was selected for all PFS and OS curves. |
The model fails to capture the causal disease pathway appropriately. | The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that may not accurately reflect the causal disease pathway. This approach could introduce postprogression survival bias, potentially biasing results in favour of tislelizumab plus chemotherapy. | This issue could not be addressed. | No scenario analysis was conducted. |
Exclusion of treatment waning is associated with uncertainty. | The sponsor assumed that the OS and PFS benefits of tislelizumab plus chemotherapy would continue indefinitely beyond the trial duration. In the absence of clinical evidence, both the magnitude and duration of treatment effect is uncertain, and the clinical experts noted that indefinite sustained treatment is unlikely. | CDA-AMC included treatment waning using the sponsor’s parameters, with the effect beginning to decline at 260 weeks and tapering over 104 toward the HR of chemotherapy alone. | CDA-AMC conducted a scenario analysis that excluded treatment waning. |
Inclusion of clinical trials as subsequent treatment is not reflective of clinical practice. | Clinical expert feedback received by CDA-AMC noted that, while patients requiring second-line and later therapy may be enrolled in clinical trials, such opportunities are rare, and therefore this is not common practice. | Subsequent treatment was assumed to be 100% chemotherapy. | No scenario analysis was conducted. |
The sponsor’s use of trial RDI for drug costs is inappropriate. | Inclusion of RDI may underestimate the total cost of treatment in clinical practice as the dose received by a patient may be different from the planned dose due to factors such as toxicity, treatment interruptions, or dose adjustments. Because tislelizumab is supplied in single-use vials, the full vial cost is incurred regardless of the administered dose. | CDA-AMC used an RDI of 100% for all treatments. | No scenario analysis was conducted. |
Tislelizumab treatment duration in clinical practice is uncertain. | The RATIONALE-309 trial reported 2 median treatment durations (by IRC and by investigator). The investigator-assessed duration included patients who continued tislelizumab beyond disease progression. | CDA-AMC explored uncertainty with this issue in a scenario analysis. | Due to the use of tislelizumab beyond progression in the RATIONALE‑309 trial, a scenario using the median treatment duration by investigator was conducted. |
CDA-AMC = Canada’s Drug Agency; GC = gemcitabine and cisplatin; IRC = independent review committee; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; RDI = relative dose intensity.
Note: Full details of the CDA-AMC identified issues are provided in the Supplemental Material document, in Appendix 11.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to the Supplemental Material document, Appendix 11, Table 19), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in the Supplemental Material document, in Appendix 11.
Impact on Health Care Costs
Tislelizumab plus chemotherapy is predicted to be associated with additional health care costs compared to chemotherapy alone (incremental costs = $107,597). This increase in health care spending results primarily from increased drug costs associated with tislelizumab (refer to Figure 1).
Figure 1: Impact of Tislelizumab Plus Chemotherapy vs. Chemotherapy Alone on Health Care Costs
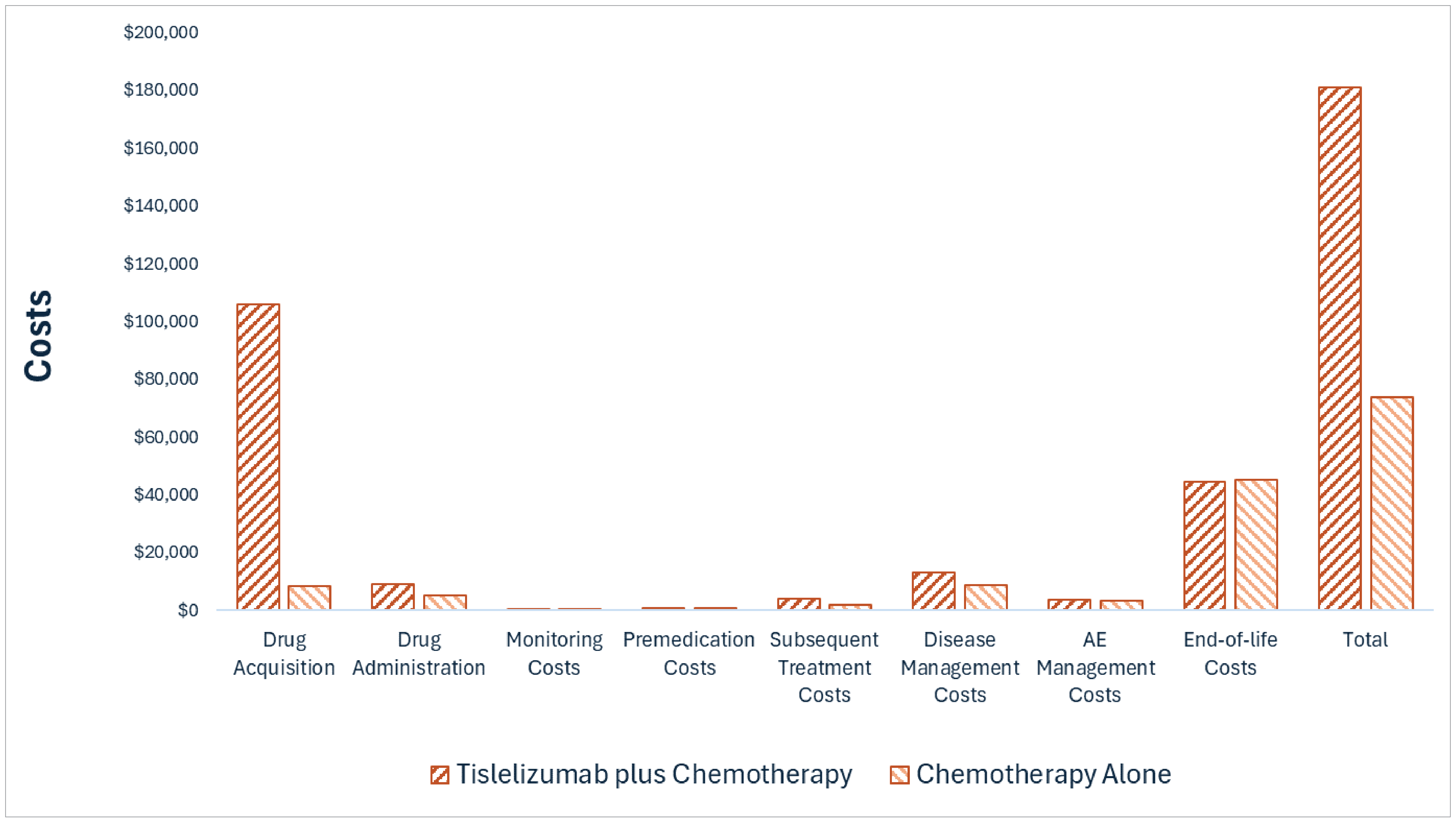
AE = adverse event; vs. = versus.
Impact on Health
Relative to chemotherapy alone, tislelizumab plus chemotherapy is predicted to increase the amount of time a patient remains in the progression-free health state by approximately 0.70 years and extend OS by approximately 1.29 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, tislelizumab plus chemotherapy is predicted to result in 0.87 additional QALYs per patient compared to chemotherapy alone. Approximately 81% of the predicted incremental benefit was accrued based on extrapolation.
Figure 2: Impact of Tislelizumab Plus Chemotherapy vs. Chemotherapy Alone on Patient Health
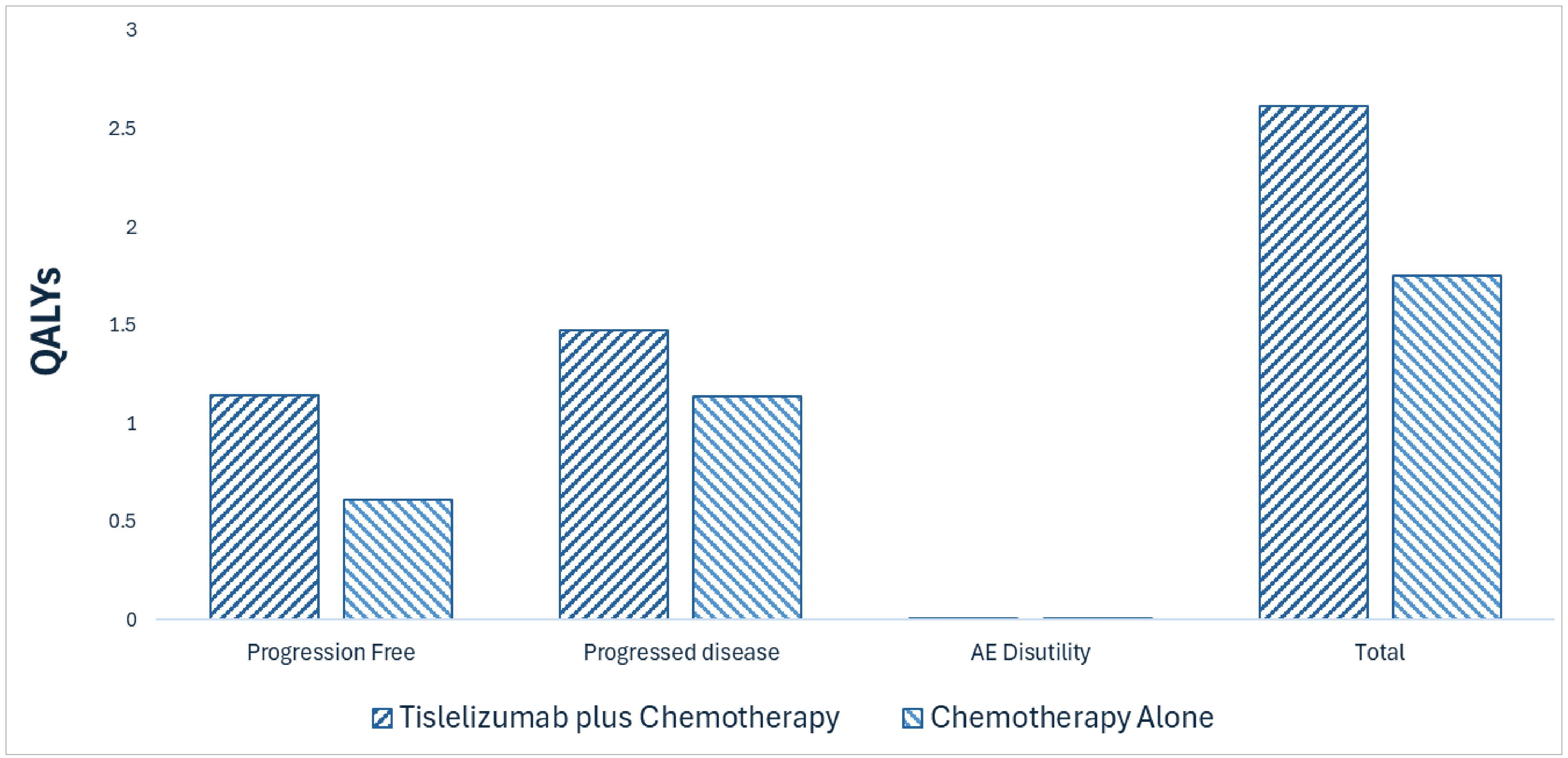
AE = adverse event; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $124,171 per QALY gained for tislelizumab plus chemotherapy compared to chemotherapy alone (refer to Table 6). Additional details on the CDA-AMC base case are available in Supplemental Material document, in Appendix 11.
Table 6: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | ICER vs. chemotherapy ($/QALY) |
|---|---|---|---|---|
Chemotherapy alone | 73,555 | 2.80 | 1.75 | Reference |
Tislelizumab plus chemotherapy | 181,153 | 4.09 | 2.61 | 124,171 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in the Supplemental Material document, in Appendix 11, Table 23. Uncertainty associated with efficacy had the largest impact on cost-effectiveness. Based on the results of these analyses, the ICER for tislelizumab plus chemotherapy may range from $78,151 to $133,016 per QALY gained compared with chemotherapy alone.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing tislelizumab plus chemotherapy for the first-line treatment of patients with recurrent or metastatic NPC, and then as a single agent after up to 6 cycles of combination therapy. The sponsor assumed that the cost would be borne by CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of tislelizumab was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in the Supplemental Material document, in Appendix 12.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (refer to Supplemental Material document, Appendix 12). CDA-AMC estimated that 419 patients would be eligible for treatment with tislelizumab plus chemotherapy over a 3-year period (year 1 = 135; year 2 = 140; year 3 = 144), of whom 391 are expected to receive tislelizumab plus chemotherapy (year 1 = 122; year 2 = 133; year 3 = 137). The estimated incremental budget impact of reimbursing tislelizumab plus chemotherapy is predicted to be approximately $25,979,875 over the first 3 years, with an expected expenditure of $28,501,534 on tislelizumab plus chemotherapy.
Conclusion
Based on the CDA-AMC base case, tislelizumab for use in combination with chemotherapy would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $124,171 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in the Supplemental Material document, in Appendix 11).
The budget impact of reimbursing tislelizumab for use in combination with chemotherapy to the public drug plans in the first 3 years is estimated to be approximately $26.0 million. The 3-year expenditure on tislelizumab for use in combination with chemotherapy (i.e., not accounting for current expenditure on comparators) is estimated to be $28.5 million.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
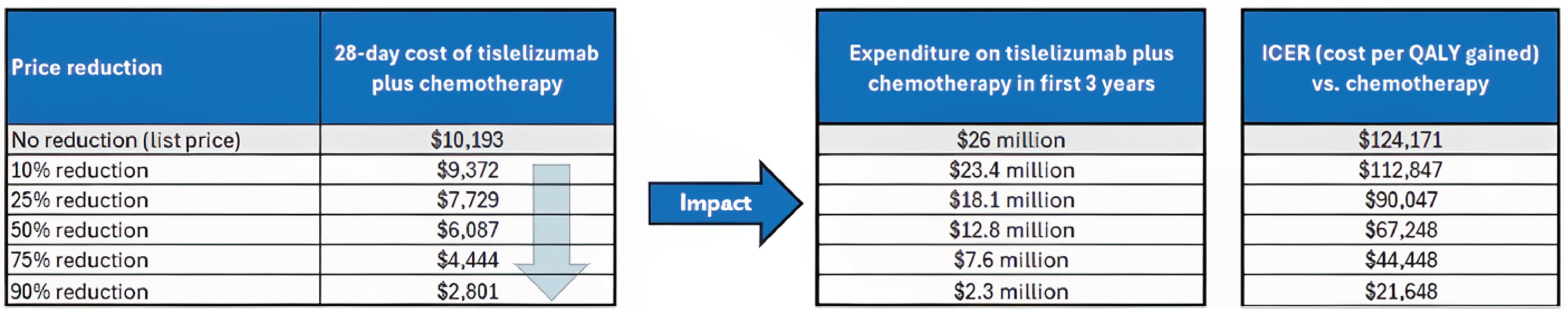
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Expenditure includes only the drug cost of tislelizumab plus chemotherapy. Price reductions only applied to tislelizumab.
References
1.BeOne Medicines I GmbH. Tevimbra (tislelizumab for injection): concentrate for solution for intravenous infusion, 100 mg/10 mL of tislelizumab [product monograph] [sponsor supplied reference].
2.BeiGene (Canada) ULC. Sponsor's clinical summary report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: tislelizumab, concentrate for solution for infusion, 100 mg/10 mL intravenous. June 2025.
3.Badoual C. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Oropharynx and Nasopharynx. Head Neck Pathol. 2022;16(1):19-30. doi:10.1007/s12105-022-01449-2
4.Cantu G. Nasopharyngeal carcinoma. A “different” head and neck tumour. Part B: treatment, prognostic factors, and outcomes. Acta Otorhinolaryngol Ital. 2023;43(3):155-169. doi:10.14639/0392-100X-N2223 PubMed
5.Alberta Health Services. Clinical Practice Guideline HN-003 – Version 2 Nasopharyngeal Cancer Treatment. March 2021. Accessed June 19, 2025. https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-hn003-nasopharyngeal.pdf
6.World Health Organization. GLOBOCAN 2022: Estimated cancer incidence, mortality and prevalence worldwide in 2022 [sponsor supplied reference]. 2022. Accessed September 2024. https://gco.iarc.who.int/media/globocan/factsheets/cancers/4-nasopharynx-fact-sheet.pdf
7.Statistics Canada. Table 13-10-0111-01 Number and rates of new cases of primary cancer, by cancer type, age group and sex [sponsor supplied reference]. 2021. Accessed September 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011101&pickMembers%5B0%5D=2.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.7&cubeTimeFrame.startYear=2017&cubeTimeFrame.endYear=2022&referencePeriods=20170101%2C20220101
8.Statistics Canada. Table 13-10-0751-01 Number of prevalent cases and prevalence proportions of primary cancer, by prevalence duration, cancer type, attained age group and sex [sponsor supplied reference]. 2022. Accessed January 2025. DOI: https://doi.org/10.25318/1310075101-eng
9.Friborg JT, Melbye M. Cancer patterns in Inuit populations. Lancet Oncol. 2008;9(9):892-900. doi:10.1016/S1470-2045(08)70231-6 PubMed
10.Howlett J, Hamilton S, Ye A, et al. Treatment and outcomes of nasopharyngeal carcinoma in a unique non-endemic population. Oral Oncol. 2021;114:105182. doi:10.1016/j.oraloncology.2021.105182 PubMed
11.Tang SQ, Mao YP, Xu C, et al. The evolution of the nasopharyngeal carcinoma staging system over a 10-year period: implications for future revisions. Chin Med J (Engl). 2020;133(17):2044-2053. doi:10.1097/CM9.0000000000000978 PubMed
12.Jiromaru R, Nakagawa T, Yasumatsu R. Advanced Nasopharyngeal Carcinoma: Current and Emerging Treatment Options. Cancer Manag Res. 2022;14:2681-2689. doi:10.2147/CMAR.S341472 PubMed
13.Wang KH, Austin SA, Chen SH, Sonne DC, Gurushanthaiah D. Nasopharyngeal Carcinoma Diagnostic Challenge in a Nonendemic Setting: Our Experience with 101 Patients. Perm J. 2017;21:16-180. doi:10.7812/TPP/16-180 PubMed
14.Blanchard P, Lee A, Marguet S, et al. Chemotherapy and radiotherapy in nasopharyngeal carcinoma: an update of the MAC-NPC meta-analysis. Lancet Oncol. 2015;16(6):645-55. doi:10.1016/S1470-2045(15)70126-9 PubMed
15.Guan H, He Y, Wei Z, et al. Assessment of induction chemotherapy regimen TPF vs GP followed by concurrent chemoradiotherapy in locally advanced nasopharyngeal carcinoma: A retrospective cohort study of 160 patients. Clin Otolaryngol. 2020;45(2):274-279. doi:10.1111/coa.13489 PubMed
16.Kazemian A, Ghalehtaki R, Farazmand B, et al. Long-term survival rates of patients with nasopharyngeal carcinoma treated by radiochemotherapy: a retrospective cohort study. Egypt J Otolaryngol. 2022;38(1):23. doi:10.1186/s43163-022-00212-2
17.Su SF, Han F, Zhao C, et al. Treatment outcomes for different subgroups of nasopharyngeal carcinoma patients treated with intensity-modulated radiation therapy. Chin J Cancer. 2011;30(8):565-73. doi:10.5732/cjc.010.10547 PubMed
18.Jiang Y, Chen C, Liu G, et al. Combination strategy exploration for prior treated recurrent or metastatic nasopharyngeal carcinoma in the era of immunotherapy. Sci Rep. 2024;14(1):1768. doi:10.1038/s41598-024-52326-7 PubMed
19.Lee AW, Ma BB, Ng WT, Chan AT. Management of Nasopharyngeal Carcinoma: Current Practice and Future Perspective. J Clin Oncol. 2015;33(29):3356-64. doi:10.1200/JCO.2015.60.9347 PubMed
20.Xu T, Tang J, Gu M, Liu L, Wei W, Yang H. Recurrent nasopharyngeal carcinoma: a clinical dilemma and challenge. Curr Oncol. 2013;20(5):e406-19. doi:10.3747/co.20.1456 PubMed
21.Tang LQ, Li CF, Li J, et al. Establishment and Validation of Prognostic Nomograms for Endemic Nasopharyngeal Carcinoma. J Natl Cancer Inst. 2016;108(1)doi:10.1093/jnci/djv291 PubMed
22.Huang H, Yao Y, Deng X, et al. Immunotherapy for nasopharyngeal carcinoma: Current status and prospects (Review). Int J Oncol. 2023;63(2):97. doi:10.3892/ijo.2023.5545 PubMed
23.Li JX, Huang SM, Wen BX, Lu TX. Prognostic factors on overall survival of newly diagnosed metastatic nasopharyngeal carcinoma. Asian Pac J Cancer Prev. 2014;15(7):3169-73. doi:10.7314/apjcp.2014.15.7.3169 PubMed
24.Toumi N, Ennouri S, Charfeddine I, Daoud J, Khanfir A. Prognostic factors in metastatic nasopharyngeal carcinoma. Braz J Otorhinolaryngol. 2022;88(2):212-219. doi:10.1016/j.bjorl.2020.05.022 PubMed
25.National Comprehensive Cancer Network. NCCN Guidelines Version 3.2025 Head and Neck Cancers. June 11, 2025. Accessed June 19, 2025. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1437
26.Bossi P, Chan AT, Even C, Machiels JP, ESMO Guidelines Committee. ESMO-EURACAN Clinical Practice Guideline update for nasopharyngeal carcinoma: adjuvant therapy and first-line treatment of recurrent/metastatic disease. Ann Oncol. 2023;34(3):247-250. doi:10.1016/j.annonc.2022.11.011 PubMed
27.Spreafico A, Winquist E, Ho C, et al. A Canadian Perspective on Systemic Therapy for Recurrent or Metastatic Nasopharyngeal Carcinoma. Curr Oncol. 2025;32(1):48. doi:10.3390/curroncol32010048 PubMed
28.Pfizer Canada ULC. Gemcitabine: sterile solution for injection (ready-to-use), 38 mg/mL gemcitabine (as gemcitabine hydrochloride), for intravenous use [product monograph]. February 27, 2013. Updated January 3, 2025. Accessed June 19, 2025. https://pdf.hres.ca/dpd_pm/00078192.PDF
29.Teva Canada Limited. Cisplatin: cisplatin injection solution, 1 mg / mL, intravenous [product monograph]. March 15, 2013. Updated June 25, 2024. Accessed June 19, 2025. https://pdf.hres.ca/dpd_pm/00076100.PDF
30.BeiGene Ltd. Final Abbreviated Clinical Study Report: BGB-A317-309. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study to Compare the Efficacy and Safety of Tislelizumab (BGB-A317) Combined With Gemcitabine Plus Cisplatin Versus Placebo Combined With Gemcitabine Plus Cisplatin as First-Line Treatment for Recurrent or Metastatic Nasopharyngeal Cancer [internal sponsor's report]. January 26, 2024.
31.Yang Y, Pan J, Wang H, et al. Tislelizumab plus chemotherapy as first-line treatment for recurrent or metastatic nasopharyngeal cancer: A multicenter phase 3 trial (RATIONALE-309). Cancer Cell. 2023;41(6):1061-1072 e4. doi:10.1016/j.ccell.2023.04.014 PubMed
32.BeiGene Ltd. Clinical Study Protocol (original, amendments 0.1, 1.0): BGB-A317-309. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study to Compare the Efficacy and Safety of Tislelizumab (BGB-A317) Combined With Gemcitabine Plus Cisplatin Versus Placebo Combined With Gemcitabine Plus Cisplatin as First-Line Treatment for Recurrent or Metastatic Nasopharyngeal Cancer [internal sponsor’s report]. March 26, 2021.
33.BeiGene Ltd. Clinical Study Report (interim analysis): BGB-A317-309. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study to Compare the Efficacy and Safety of Tislelizumab (BGB-A317) Combined With Gemcitabine Plus Cisplatin Versus Placebo Combined With Gemcitabine Plus Cisplatin as First-Line Treatment for Recurrent or Metastatic Nasopharyngeal Cancer [internal sponsor’s report]. July 15, 2021.
34.Yang Y, Pan J, Chen N, et al. Effects of tislelizumab on health-related quality of life in patients with recurrent or metastatic nasopharyngeal cancer. Head Neck. 2024;46(9):2301-2314. doi:10.1002/hed.27785 PubMed
35.BeiGene Ltd. Statistical Analysis Plan: BGB-A317-309. A Phase 3, Multicenter, Double-Blind, Randomized, Placebo-Controlled Study to Compare the Efficacy and Safety of Tislelizumab (BGB-A317) Combined With Gemcitabine Plus Cisplatin Versus Placebo Combined With Gemcitabine Plus Cisplatin as First-Line Treatment for Recurrent or Metastatic Nasopharyngeal Cancer [internal sponsor’s report]. January 13, 2021.
36.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-7. doi:10.1001/jama.2010.310 PubMed
37.BeiGene (Canada) ULC. BeiGene (Canada) ULC response to Canada’s Drug Agency request for additional information regarding tislelizumab review on July 11, 2025 [internal additional sponsor’s information]. July 23, 2025.
38.National Institute for Health and Care Excellence Decision Support Unit. NICE DSU Technical Support Document 16: Adjusting Survival Time Estimates in the Presence of Treatment Switching. July 2014 Accessed September 3, 2025. https://sheffield.ac.uk/sites/default/files/2022-02/TSD16_Treatment_Switching.pdf
39.Chen YP, Sun Y, Chen L, et al. Surrogate endpoints for overall survival in combined chemotherapy and radiotherapy trials in nasopharyngeal carcinoma: Meta-analysis of randomised controlled trials. Radiother Oncol. 2015;116(2):157-66. doi:10.1016/j.radonc.2015.07.030 PubMed
40.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. doi:10.1186/1477-7525-9-106 PubMed
41.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-28. doi:10.1007/s11136-007-9210-8 PubMed
42.Ahn MJ, Cho BC, Felip E, et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. N Engl J Med. 2023;389(22):2063-2075. doi:10.1056/NEJMoa2307980 PubMed
43.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi:10.1093/jnci/85.5.365 PubMed
44.Machingura A, Taye M, Musoro J, et al. Clustering of EORTC QLQ-C30 health-related quality of life scales across several cancer types: Validation study. Eur J Cancer. 2022;170:1-9. doi:10.1016/j.ejca.2022.03.039 PubMed
45.Parkar S, Sharma A. Validation of European Organization for Research and Treatment of Cancer Head and Neck Cancer Quality of Life Questionnaire (EORTC QLQ-H&N35) Across Languages: A Systematic Review. Indian J Otolaryngol Head Neck Surg. 2022;74(Suppl 3):6100-6107. doi:10.1007/s12070-021-02755-x PubMed
46.Bjordal K, Hammerlid E, Ahlner-Elmqvist M, et al. Quality of life in head and neck cancer patients: validation of the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-H&N35. J Clin Oncol. 1999;17(3):1008-19. doi:10.1200/JCO.1999.17.3.1008 PubMed
47.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi:10.1016/j.ejca.2023.04.027 PubMed
48.BeiGene (Canada) ULC. BeiGene (Canada) ULC response to Canada’s Drug Agency request for additional information regarding tislelizumab review on July 28, 2025 [internal additional sponsor’s information]. August 1, 2025.
49.BeiGene (Canada) ULC. Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: tislelizumab, concentrate for solution for infusion, 100 mg/10 mL intravenous. May 8, 2025.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.