Drugs, Health Technologies, Health Systems
Reimbursement Review
Vorasidenib (Voranigo)
Sponsor: Servier Canada Inc.
Therapeutic area: Astrocytoma or oligodendroglioma, IDH1 or IDH2 mutation
Summary
What Is Astrocytoma or Oligodendroglioma With IDH1 or IDH2 Mutation?
Gliomas are the most common type of central nervous system tumour. They arise from glial cells, the forerunners of brain cells such as astrocytes and oligodendrocytes. Approximately 24% of all brain and central nervous system tumours and 81% of malignant brain tumours are gliomas.
In 2017, age-standardized incidence rates for grade 2 glioma (according to 2016 WHO classification) in Canada were 0.30 per 100,000 for diffuse astrocytoma and 0.24 per 100,000 for oligodendroglioma.
IDH1 and IDH2 mutations are specific mutations that can be found in glioma cancers and can be identified using immunohistochemistry or next-generation sequencing. IDH testing using both immunohistochemistry and next-generation sequencing (where applicable) is performed as part of the standard of care for patients with glioma in Canada.
What Are the Treatment Goals and Current Treatment Options for Astrocytoma or Oligodendroglioma With IDH1 or IDH2 Mutation?
Treatment goals include living longer; delaying disease progression, including preservation of mental and nervous system function and slowing the disease from transforming into a high-grade glioma; as well as improving social and professional quality of life.
Delaying the need for radiotherapy and chemotherapy, as well as delaying disease progression and improving quality of life, were identified as important outcomes in the patient group input. Other important outcomes identified through clinician input included living longer and delaying the disease from changing into a higher-grade tumour.
After surgery, patients either receive treatment with radiotherapy and/or chemotherapy or receive no treatment (i.e., active surveillance) and are regularly followed up with MRI. Active surveillance is considered the alternative treatment option to Voranigo.
What Is Voranigo and Why Did Canada’s Drug Agency Conduct This Review?
Voranigo is a drug that is available as an oral tablet. Health Canada has approved Voranigo for the treatment of grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention.
Canada’s Drug Agency (CDA-AMC) reviewed Voranigo to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the treatment of grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention who are not in immediate need of radiotherapy or chemotherapy. The sponsor is seeking reimbursement for this patient population.
How Did CDA-AMC Evaluate Voranigo?
CDA-AMC reviewed the clinical and economic evidence regarding the beneficial and harmful effects of Voranigo for grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention who are not in immediate need of radiotherapy or chemotherapy.
CDA-AMC identified equity and ethical considerations relevant to Voranigo and grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation.
CDA-AMC considered the potential impacts of IDH mutation testing to ascertain eligibility for Voranigo for the treatment of grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention, including those related to health systems, patients (including families and caregivers), and costs.
The review was informed by materials submitted by the sponsor, including clinical and economic evidence.
The review was also informed by 2 patient group submissions and 2 clinician group submissions in response to a call for input, and by input from the participating public drug programs regarding issues that may affect their ability to implement a recommendation.
Two neuro-oncologists and 1 pediatric neuro-oncologist, representing the Atlantic provinces (Newfoundland and Labrador, Prince Edward Island, Nova Scotia, and New Brunswick) and Ontario, were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
One randomized controlled phase III trial (the INDIGO trial) comparing Voranigo with placebo in 331 patients with grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation.
For the comparison of Voranigo versus placebo:
Voranigo likely increased the proportion of patients whose disease remained progression-free at 24 months and the proportion of patients without a subsequent intervention at 24 months.
Voranigo also demonstrated a potential benefit on tumour growth rate. However, the impact of Voranigo on health-related quality of life, as measured by the change from baseline in Functional Assessment of Cancer Therapy – Brain total score, was very uncertain (due to risk of bias and imprecision).
The evidence was limited by a relatively short follow-up period because the trial was stopped early after an interim analysis demonstrated efficacy. Median progression-free survival in the Voranigo arm and median time to next intervention were not estimable, and the follow-up was not long enough to inform on potential longer-term benefits.
The generalizability of the evidence was limited by factors such as the study’s 1-year to 5-year postsurgery requirement, which may not be required in practice, and a lack of patients younger than 18 years.
It was not feasible to follow patients for long enough to assess the impact of Voranigo on overall survival; how well progression-free survival acts as a substitute for overall survival in this patient population and treatment context is uncertain.
Adverse events consisting of elevations in liver enzymes were reported in a numerically higher number of patients randomized to receive Voranigo than in patients randomized to receive placebo during the trial.
Economic Evidence
Voranigo is available as 10 mg and 40 mg tablets. At the submitted price of $399.17 per 10 mg tablet and $798.33 per 40 mg tablet, the cost of Voranigo per 28-day cycle is expected to be $22,353 per patient, based on the Health Canada–recommended dosage.
Clinical efficacy in the economic analysis was derived from the INDIGO trial, which compared Voranigo with active surveillance. Evidence submitted by the sponsor indicates that Voranigo likely increases the proportion of patients who are progression-free and the proportion of patients without a subsequent intervention at 24 months compared with active surveillance in patients aged 12 years and older with grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation following surgical intervention.
The results of the CDA-AMC base case suggest:
Voranigo is predicted to be associated with higher costs to the health care system than active surveillance (incremental costs = $1,190,165), driven primarily by increased costs associated with drug acquisition.
Voranigo is predicted to be associated with a gain of 2.97 life-years compared to active surveillance and may result in a gain of 2.08 quality-adjusted life-years (QALYs) compared to active surveillance.
The incremental cost-effectiveness ratio (ICER) of Voranigo compared to active surveillance was $571,629 per QALY gained in the CDA-AMC base case using a health care payer perspective. The estimated ICER was highly sensitive to the frequency of seizures and unplanned hospitalizations.
Given limitations with the estimation of productivity costs, an analysis using a societal perspective is only presented as a scenario analysis. In this analysis, the ICER increases to $583,257 per QALY gained.
CDA-AMC estimates that the budget impact of reimbursing Voranigo for the indicated population will be approximately $90 million over the first 3 years of reimbursement compared to the amount currently spent on active surveillance, driven by expenditures on Voranigo over this period. The actual budget impact of reimbursing Voranigo will depend on the number of people eligible for treatment and the uptake of Voranigo.
Abbreviations
AE
adverse event
ALT
alanine transaminase
AST
aspartate transaminase
BCC
Brain Cancer Canada
BIRC
blinded independent review committee
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CNS
central nervous system
DCO
data cut-off
DOR
duration of response
FACT-Br
Functional Assessment of Cancer Therapy – Brain
GGT
gamma-glutamyltransferase
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IHC
immunohistochemistry
LTE
long-term extension
MID
minimal important difference
NGS
next-generation sequencing
OH (CCO)
Ontario Health (Cancer Care Ontario)
OS
overall survival
PFS
progression-free survival
POGO
Pediatric Oncology Group of Canada
QALY
quality-adjusted life-year
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TGR
tumour growth rate
TTNI
time to next intervention
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of vorasidenib, 10 mg and 40 mg oral tablets, in the treatment of grade 2 (WHO 2016, 2021 grading system) astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in patients aged 12 years and older following surgical intervention. The focus will be placed on comparing vorasidenib to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, and this focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Vorasidenib (Voranigo), tablets, 10 mg and 40 mg, oral |
Sponsor | Servier Canada Inc. |
Health Canada indication | The treatment of Grade 2 (per WHO 2016, 2021 grading system) astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention. Treatment with vorasidenib should be initiated following confirmation of an IDH1 or IDH2 mutation through a validated test. |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | August 27, 2024 |
Mechanism of action | Small-molecule inhibitor of IDH1 and IDH2 enzymes, which decreases the production of 2-HG and may restore cellular differentiation. |
Recommended dosage | For patients weighing at least 40 kg, take 40 mg orally once daily. For patients weighing less than 40 kg, take 20 mg orally once daily. |
Submission type | Initial |
Sponsor’s reimbursement request | For the treatment of grade 2 (WHO 2016, 2021 grading system) astrocytoma or oligodendroglioma with a susceptible IDH1 mutation or IDH2 mutation in adults and pediatric patients aged 12 years and older who are not in immediate need of radiotherapy or chemotherapy following surgical intervention |
Submitted price | Vorasidenib: $399.17 per 10 mg tablet or $798.33 per 40 mg tablet |
Information on the CDA-AMC review | |
Review type | Complex |
Clinical review focusa | Population: As defined in the reimbursement request Subgroups: None identified for inclusion in the report Intervention: Per recommended dosage Comparators: Active surveillance Outcomes:
|
BIRC = blinded independent review committee; CDA-AMC = Canada’s Drug Agency; DOR = duration of response; FACT-Br = Functional Assessment of Cancer Therapy – Brain; NOC = Notice of Compliance; OS = overall survival; PFS = progression-free survival; TGR = tumour growth rate; TTNI = time to next intervention.
aThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed vorasidenib through the Reimbursement Review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each Reimbursement Review. Two patient group submissions from Brain Cancer Canada (BCC) and Heal Canada; and 2 clinician group submissions from the Pediatric Oncology Group of Canada (POGO) and Ontario Health (Cancer Care Ontario) (OH [CCO]) CNS Drug Advisory Committee were received. BCC summarized themes and experiences expressed by patients and caregivers, including those of 1 patient who had received vorasidenib for grade 2 oligodendroglioma. Heal Canada distributed an online survey in September 2024 consisting of 61 questions relating to treatment approaches in patients with glioma and domains of activity (e.g., working status, physical capability and productivity, and quality of life). The indication under review was discussed with members of the POGO submission panel (a collaboration of pediatric cancer clinicians) and input for this submission was sought from the POGO Technology and Therapeutic Advisory committee; 3 clinicians provided input. Information for the OH (CCO) submission was gathered by email and included input from 2 clinicians. The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may affect their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two neuro-oncologists and 1 pediatric neuro-oncologist with expertise in the diagnosis and management of astrocytoma or oligodendroglioma with susceptible IDH1 or IDH2 mutations participated as part of the review team, with representation from the Atlantic provinces (Newfoundland and Labrador, Prince Edward Island, Nova Scotia, and New Brunswick), and Ontario.
Disease Background
Gliomas are the most common form of central nervous system (CNS) neoplasm,1 accounting for approximately 24% of primary brain and other CNS tumours and approximately 81% of primary malignant brain tumours.2 Gliomas arise from glial cells or precursors of intrinsic brain cells (e.g., astrocytes and oligodendrocytes).2,3 Diffuse gliomas (e.g., IDH-mutant astrocytomas and oligodendrogliomas) are frequently characterized by continuous growth and aggressive transformation, leading to a poor prognosis and limited survival time.4 In general, the median overall survival (OS) for patients with minor IDH-mutant gliomas is approximately 10 years.5,6 The median survival for IDH-mutant grade 2 astrocytomas ranges from 5 to 8 years.7,8 Similarly, the median survival for IDH-mutant oligodendrogliomas is greater than 10 years.8,9 Symptoms of mutant IDH gliomas vary based on characteristics such as size, location, and degree of infiltration. Patients can present with focal or generalized symptoms over days to years depending on the speed of tumour growth and location.10 Onset of symptoms is usually experienced within 1 to 3 months of diagnosis,8,11 and symptoms can include headaches, nausea, vomiting, seizures, visual disturbance, speech and language problems, and changes in cognitive and/or functional ability, which vary depend on the tumour location.1 Patients may also be asymptomatic, without evident abnormalities on neurologic examination.12
According to the Brain Tumour Registry of Canada, between 2013 and 2017, the average annual age-standardized incidence rate for all primary CNS tumours (of which gliomas are a small subset) was 21.05 per 100,000 in Canada (excluding Quebec).13 The incidence of gliomas is higher in males than females.11 In 2017, age-standardized incidence rates for grade 2 gliomas (according to 2016 WHO Classification) in Canada were 0.30 per 100,000 for diffuse astrocytoma and 0.24 per 100,000 for oligodendroglioma.11,13 An updated incidence using the 2021 WHO classification is not yet available. Clinical experts consulted for this review noted that the disease does not differentially or disproportionately affect any systemically marginalized or equity-deserving groups, apart from indicating that their patients are generally young (approximately 40 years on average), and pediatric patients can also develop gliomas.
Patient groups highlighted that the disease and its treatments have a substantial impact on both patients and caregivers, significantly affecting their quality of life, financial stability, and productivity. Patients and caregivers place a high value on the quality of life (months and years of stable results between initial surgery and the next intervention). One patient group noted that it may take several days to weeks for results to come back for tumour samples from resection or biopsy surgery, and waiting for the tumour classification results may cause a delay in planning the next course of action.
Current Management
Treatment Goals
BCC highlighted the following outcomes as important when evaluating new treatment options: delay of disease progression (extended time to next intervention [TTNI]), improved quality of life including mental health, and avoidance of radiotherapy and chemotherapy. The clinical experts and clinician groups noted that the goals of treatment are prolonging survival, preserving neurologic and cognitive function as well as quality of life, delaying tumour progression, delaying transformation into high-grade glioma, and minimizing treatment-related toxicities. Both clinician groups added that effective treatment options at earlier stages of tumourigenesis are needed, and OH-CCO further noted improvement on the natural history of the disease is a goal of therapy.
Current Treatment Options
Treatment for low-grade astrocytoma and oligodendroglioma typically involves a combination of surgery, radiotherapy, and chemotherapy. The clinical experts indicated that the current treatment paradigm is based on stratification of patients based on their risk of progression. Treatment decisions depend on factors such as patient age, tumour size, location, presurgical neurologic symptoms, the extent of surgical resection carried out, and molecular biomarkers (e.g., 1p19q codeletion, CDKN2A homozygous deletion). Patients with gross total resection or who are asymptomatic and have favourable molecular biomarkers are usually followed with an active surveillance approach consisting of serial imaging (MRIs every 3 months initially, then every 6 months if the patient is clinically stable). This approach would also usually be applied if the surgery resulted in an incomplete resection. In all cases, if disease progression is found, patients would then undergo treatment with radiotherapy and chemotherapy. Patients deemed to be at high risk based on the same criteria would be treated with radiotherapy and chemotherapy (such as temozolomide for astrocytoma and procarbazine, lomustine, and vincristine for oligodendroglioma) post surgery.
The clinical experts consulted for this review also noted that, for both pediatric and adult patients, the standard of care is moving toward earlier biopsy or surgical resection, if possible, followed by confirmation of IDH mutation status and assessment of high-risk versus low-risk disease. The experts also noted that the standard of care is also shifting more toward the use of molecular biomarkers (e.g., CDKN2A homozygous deletion) to characterize the tumour, where the residual disease (if any) is located, and case-by-case assessment of the risk to the patient from treatment side effects versus disease progression.
Unmet Needs and Existing Challenges
Overall, patient groups highlighted a substantial impact of the disease and its treatment on both patients and caregivers, significantly affecting their quality of life, financial stability, and productivity. Specific challenges they highlighted from existing treatment options included side effects from chemotherapy (i.e., vomiting, fatigue, hair loss, changes in blood cell counts, and memory deficits) and radiotherapy (i.e., cognitive deficits, changes in brain function, hair loss, and fatigue). They noted barriers accessing specialized centres, which would be a particular concern to those who live in rural or remote locations and can both affect access to timely treatment and add travel time at the expense of work or other obligations. In addition, financial strain, long treatment sessions, significant impact on work and social life, missed or reduced work hours, and a burden on caregivers were also highlighted, which could have different impacts on patients depending on their socioeconomic status or the presence of family and/or friend support networks.
The clinical experts noted that, eventually, patients on active surveillance will progress, and the standard-of-care options are not curative. Eventually, patients’ cancer can develop resistance and recur aggressively or transform into a higher-grade glioma. In addition, radiotherapy can have a long-term neurocognitive impact, and both radiotherapy and chemotherapy are associated with debilitating side effects, such as fatigue and nausea. The experts noted that most patients with these cancers are young and often active in the workforce or raising families; therefore, the impacts on their quality of life can be considerable. They also highlighted access challenges for patients who live in rural settings. Radiotherapy, in particular, requires 6 weeks of daily treatment; therefore, additional time and cost may be incurred by patients in rural areas if they are required to travel to a cancer centre.
The clinician group input was consistent with the input from clinical experts, noting that current treatment options are limited and have many side effects (e.g., neurocognitive deficits, risk of secondary malignancy, risk of infertility, and hematologic toxicity).
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor table in Appendix 1). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Supplemental Material document (available from the project landing page), in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1. The following has been summarized by the review team.
Place in Therapy
The clinical experts indicated that in patients with grade 2 IDH-mutated gliomas, vorasidenib monotherapy would be used as a first-line treatment option for those who do not require immediate treatment after surgery. They estimated that approximately one-third of patients do not require immediate treatment following surgery (i.e., they move to active surveillance), while the remaining patients would require immediate treatment following surgery (i.e., they are considered to be at high risk). The experts also noted that patients who might not have been ideal for active surveillance but were not quite considered to have high-risk disease (and receive radiotherapy and chemotherapy), would also be considered candidates for vorasidenib. The experts noted that there is no evidence for the use of vorasidenib in recurrent gliomas.
Vorasidenib is the first treatment to target IDH mutations in gliomas and would represent a paradigm shift because fewer patients would undergo active surveillance. The clinical experts noted that patients who are young or of childbearing age may wish to discuss fertility preservation before initiating treatment with vorasidenib. One of the clinical experts also noted that vorasidenib is currently being studied in combination with other therapies, such as immune checkpoint inhibitors or temozolomide.
Patient Population
The clinical experts indicated that patients with low-grade gliomas (i.e., grade 2) who are young, in whom radiation needs to be delayed to prevent neurocognitive impacts, and patients with tumours in locations where a significant resection is not possible would be most in need of a treatment such as vorasidenib. Patients who have had a complete resection and who would otherwise undergo active surveillance would also be candidates for treatment. They noted that patients with gliomas are often young, may have young families, or may be more susceptible to experiencing financial burdens from treatment. The clinical experts emphasized that some subjectivity is involved in distinguishing between grade 2 and grade 3 gliomas, and that clarifying the meaning of “immediate need” for radiotherapy or chemotherapy would be important. The clinical expert with pediatric expertise noted that patients younger than 12 years should also be included if the indicated IDH mutations are present. The experts commented that, while a waiting period of 1 to 5 years after surgery was used in the pivotal trial, they would likely only wait a few months before starting to treat patients with vorasidenib. However, they also pointed out that, because the ideal duration of time after surgery as well as the impact of specific biomarkers were not hypotheses from the pivotal trial, the question of biomarkers or post surgery initiation would more likely be assessed on a case-by-case basis.
The experts indicated that, following surgery, patients with a predominantly nonenhancing IDH-mutated grade 2 glioma who have not received previous anticancer therapy would be most likely to respond to vorasidenib, although there is currently no biomarker that predicts response to vorasidenib. The experts noted that identifying eligible patients would be straightforward; postoperative MRIs are standard of care, as are the companion diagnostics (IDH immunostaining, followed by IDH sequencing if negative, 1p/19q codeletion status, and CDKN2A/B copy number review). Patients could be identified during a postoperative clinical consultation during which a review of pathology, imaging, and functional status, as well as neurologic symptoms, would be undertaken. Patients who were planning to have children in the near term, who had an enhancing glioma (i.e., a tumour that takes up contrast on a scan), and patients with unfavourable biomarkers (e.g., CDKN2AI deletion) would not be ideal candidates for vorasidenib.
The input from clinician groups broadly aligned with the input from the clinical experts; POGO indicated that patients whose characteristics align with the eligibility criteria of the INDIGO trial would be best suited for treatment with vorasidenib, and that eligible patients would be identified following surgical resection and/or biopsy. OH-CCO noted that patients who are symptomatic or have a significant burden of residual disease following surgery may be more appropriately treated with radiotherapy or chemotherapy.
Testing Procedure Considerations
All known oncogenic IDH mutations feature a missense mutation to the IDH1 or IDH2 gene, with the majority (greater than 85%) resulting from a missense mutation of arginine to histidine in codon 132 (R132H) of the IDH1 gene.14 Canadian and international guidelines recommend testing all patients with low-grade gliomas for IDH mutations as a diagnostic marker for tumour classification.15-17 As such, IDH testing is currently conducted as part of routine care, following surgical intervention (e.g., resection and biopsy). Immunohistochemistry (IHC) testing is available and publicly funded in all jurisdictions across Canada as part of standard of care. There is evidence to demonstrate the diagnostic accuracy of IHC testing for IDH mutation testing; however, CDA-AMC did not critically appraise that evidence. According to 1 study report, when compared to DNA-based sequencing, the sensitivity and specificity of IDH1 R132H IHC testing to detect R132H mutations were both 100%, and the sensitivity and specificity to detect all IDH1 R132 mutations were 94% and 100%, respectively.18 IDH mutation testing using IHC usually only needs to be performed once and does not need to be repeated. However, in patients with IHC-negative tumours who are younger than 55 years, expert consensus in Canada recommends sequencing of IDH1 R132 and IDH2 R172 genes using next-generation sequencing (NGS) to identify uncommon or noncanonical mutations.17,19 The expected turnaround time for IDH mutation testing is approximately 1 week (for IHC results) to 4 weeks (for NGS results). However, according to the clinical experts, turnaround times can vary depending on the region of Canada, particularly if the tissue must be sent out of jurisdiction for testing. Research into the use of liquid biopsy using circulating free DNA from cerebrospinal fluid — which contains circulating tumour DNA — is also under way, but this technique is not currently available for clinical use.20,21
The potential impacts of IDH testing were considered to ascertain eligibility for vorasidenib for the treatment of grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention, including those to health systems, patients (including families and caregivers), clinical use, and projected costs. If vorasidenib were to be reimbursed, no implementation barriers related to testing on health systems are anticipated because IDH mutation testing is currently the standard of care for patients with low-grade gliomas across jurisdictions in Canada.
Key considerations and relevant information available were validated by the review team when possible and are summarized in Appendix 1.
Assessing the Response to Treatment
The clinical experts consulted for this review noted that stabilization of the disease is the main criteria for treatment response, which would be assessed by regular MRIs and clinical evaluation. One clinical expert noted there is no consensus on the frequency of evaluation, but all the clinical experts noted that response to treatment could be assessed every 3 months initially or as needed if there is clinical functional deterioration. They noted that these assessments would not require modification of the current clinical approach to care. Monitoring would require in-person visits to obtain MRIs and to meet with the oncologist, but visits are unlikely to be more frequent than what is already done for active surveillance.
POGO noted that response to treatment should be assessed every 3 to 6 months using standard imaging techniques (i.e., MRI scans).
Discontinuing Treatment
The clinical experts and clinician groups indicated that a lack of response (as indicated by clinical or radiological progression), intolerability, or unacceptable toxicity would be reasons to discontinue treatment. Further, treatment should be stopped if there are signs of malignant transformation. The clinical experts also noted that family planning and patient decision would factor into discontinuation of treatment.
Prescribing Considerations
According to the clinical experts consulted for this review, a neuro-oncologist (e.g., a neurologist or medical oncologist with experience in treating patients with brain tumours) should prescribe vorasidenib. They did not anticipate that treatment with vorasidenib would differ from the current standard-of-care treatments as patients would still be followed up at a cancer centre with access to imaging and neuro-oncology resources. Both clinician groups agreed with the experts and added that a multidisciplinary team (trained in neurosurgery and neuropathology) would be required for diagnosis and to determine when the treatment should be started.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions (LTEs), indirect treatment comparisons, and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included pivotal studies and phase III RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Patients not in immediate need of radiotherapy or chemotherapy following surgical intervention were considered important for informing the reimbursement recommendation. Relevant comparators were drugs and nondrug treatments used in clinical practice in Canada to treat patients described in the reimbursement request under review and included watchful waiting. Radiotherapy and chemotherapy were not considered relevant comparators as they would be initiated in high-risk patients. LTEs of included pivotal studies and RCTs were included, regardless of whether there was a comparison group. Indirect treatment comparisons and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., missing comparator or longer follow-up time).
The review team selected outcomes (and follow-up times) for review considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes were those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes were included in the GRADE assessment: progression-free survival (PFS) by blinded independent review committee (BIRC) at 12 and 24 months; TTNI at 12 and 24 months; OS at 24 months; and change from baseline in Functional Assessment of Cancer Therapy – Brain (FACT-Br) total score at cycle 13, day 1, and at end of treatment. PFS, TTNI, and OS were selected as they address 3 of the main treatment goals for the disease (delaying disease progression, delaying transformation into high-grade glioma, and prolonging life). FACT-Br was included as it is a measure of health-related quality of life (HRQoL); the use of the FACT-Br subscale (accompanied by Functional Assessment of Cancer Therapy–General [FACT-G]) is validated in patients with brain metastases.
Additional outcomes and time points were selected for inclusion in the report but were not assessed using GRADE. Duration of response (DOR) results at 12 months were included to provide additional context. Tumour growth rate (TGR) as well as TGR before and after vorasidenib initiation was included as these are considered supportive outcomes, although the clinical experts noted that these are impractical to measure in clinical practice. These outcomes also form part of the input in the sponsor’s pharmacoeconomic model. EQ-5D-5L scores were included in Appendix 4 because they form part of the input in the sponsor’s pharmacoeconomic model. The proportion of patients with malignant transformation was included because it provides supportive data for a treatment goal (delay of disease transformation).
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are in the Supplemental Material document in Appendix 2.
Clinical Evidence
In this report, the following source of evidence submitted by the sponsor is reviewed and appraised:
1 pivotal study or RCT included in the systematic review, INDIGO.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Additional details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are in the Supplemental Material document in Appendix 3.
The INDIGO trial was a phase III, double-blind RCT evaluating the safety and efficacy of once-daily oral vorasidenib compared with once-daily placebo in patients aged 12 years and older and weighing 40 kg or more, for the treatment of grade 2 oligodendroglioma or astrocytoma with a susceptible IDH1 or IDH2 mutation. The trial included patients who were not considered by the investigator to be in immediate need of radiotherapy or chemotherapy; patients were excluded if they had high-risk features, including brainstem involvement either as primary location or by tumour extension, clinically relevant functional or neurocognitive deficits due to the tumour in the opinion of the investigator, or uncontrolled seizures. Patients had at least 1 prior surgery (biopsy, subtotal resection, or gross total resection), with the most recent surgery occurring at least 1 year and not more than 5 years before the date of randomization. Patients were randomized 1:1 to vorasidenib or placebo, and randomization was stratified based on 1p19q status (codeleted or not codeleted), and baseline tumour size (diameter ≥ 2 cm or < 2cm). Efficacy and safety assessments occurred according to a prespecified schedule, with tumour responses assessed by MRI every 12 weeks beginning at cycle 4, day 1. The study included a crossover option in which the investigator could request unblinding of a patient’s treatment assignment after radiographic progressive disease was centrally confirmed by the BIRC to determine eligibility to cross over to vorasidenib.
On February 24, 2023, the independent data monitoring committee recommended unblinding the study due to early demonstration of efficacy, as the study met its primary and key secondary end points. The study was unblinded on March 7, 2023, at which time patients receiving placebo were given the opportunity to cross over to vorasidenib provided certain eligibility criteria were met.
Table 2: Key Characteristics of the INDIGO Study
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
INDIGO Phase III, multicentre, randomized, double-blind, placebo-controlled RCT Total N = 331 |
|
| Intervention: vorasidenib 40 mg orally once daily on days 1 to 28 in 28-day cycles Comparator: placebo orally once daily on days 1 to 28 in 28-day cycles |
|
AE = adverse event; BIRC = blinded independent review committee; DOR = duration of response; FACT-Br = Functional Assessment of Cancer Therapy–Brain; PFS = progression-free survival; OS = overall survival; RCT = randomized controlled trial; SAE = serious adverse event; TGR = tumour growth rate; TTNI = time to next intervention; WDAE = withdrawal due to adverse event.
aThe INDIGO trial enrolled patients who had already been on active surveillance for at least 1 year, with the rationale that this ensured patients were clinically stable and to help homogenize the study population.
bConfirmed IDH1 (IDH1 R132H/C/G/S/L mutation variants tested) or IDH2 (IDH2 R172K/M/W/S/G mutation variants tested) gene mutation status disease by central laboratory testing during the prescreening period and available 1p19q status by local testing (e.g., fluorescence in situ hybridization, comparative genomic hybridization array, sequencing).
cHigh-risk features included brainstem involvement either as primary location or by tumour extension, clinically relevant functional or neurocognitive deficits due to the tumour in the opinion of the investigator (deficits resulting from surgery are allowed), or uncontrolled seizures (defined as persistent seizures interfering with activities of daily life and failure of 3 lines of antiepileptic drug regimens including at least 1 combination regimen).
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence,22 INDIGO Clinical Study Report,23 and INDIGO Study Protocol.24
Statistical Testing and Analysis Populations
The target sample size was 340 patients; a 10% dropout rate was 1 of the assumptions in the sample size calculation. For the primary end point (PFS), a total of 164 PFS events were required to have at least 90% power to detect a hazard ratio (HR) of 0.6 using a 1-sided log-rank test stratified by the randomization stratification factors at a significance level of 0.025, and a 3-look group sequential design with a gamma family (−24) alpha-spending function to determine the efficacy boundaries and a gamma family (−5) beta-spending function to determine the nonbinding futility boundary.
For TTNI, a total of 152 TTNI events were required to have approximately 80% power to detect an HR of 0.636 using a 1-sided log-rank test stratified by the randomization stratification factors at a significance level of 0.025, and a 2-look group sequential design with a gamma family (−22) alpha-spending function to determine the efficacy boundaries.
The study used a fixed-sequence testing procedure to control type I error at 2.5% (1-sided), for PFS (tested first) and TTNI (tested thereafter if PFS was significant). There was no multiplicity control for other end points. Multiplicity control across the interim analyses was achieved as previously described. Two interim analyses and a final analysis for PFS were planned.
Analysis populations included the full analysis set, the per-protocol set, and the safety analysis set. The full analysis set included all randomized patients and, in the analysis, patients were classified according to the intention-to-treat principle. The per-protocol set excluded patients who had not met the main inclusion criteria (no measurable lesions per modified Response Assessment in Neuro-Oncology [RANO] criteria for low-grade gliomas [LGGs], did not have a histopathological diagnosed grade 2 oligodendroglioma or astrocytoma, had prior anticancer therapy for glioma, or had not received any study drug). This dataset was used for PFS and TTNI analyses. The safety analysis set included all patients who had received at least 1 dose of study treatment; this was used for harms reporting. The study was unblinded on March 7, 2023, due to early demonstration of efficacy following the independent data monitoring committee recommendation. Two data cut-offs (DCOs) were included in the submission: September 6, 2022 (i.e., the planned interim analysis 2) and March 7, 2023 (representing an additional 6 months of study follow-up from the planned interim analysis 2 before unblinding of the study). Unless otherwise specified, data from the DCO on March 7, 2023, are included in the report and appraisal.
Patient Disposition
Patient disposition for the INDIGO trial is summarized in the Supplemental Material document, Appendix 4. Briefly, a total of 390 patients were screened and, of these, 331 were randomized (n = 163 in the placebo arm; n = 168 in the vorasidenib arm). As of the DCO on March 7, 2023, 55.8% of patients in the placebo arm had discontinued treatment compared to 26.2% in the vorasidenib arm. The most common reason for treatment discontinuation was centrally confirmed disease progression (47.2% of patients in the placebo arm, 18.5% of patients in the vorasidenib arm). The number of voluntary withdrawals and other reasons for discontinuing treatment (patient decision, adverse event [AE], and investigator decision) were broadly balanced between study arms, with each reason accounting for less than 5% of patients per study arm. Few patients (less than 5% in each arm) discontinued from the study.
A total of 17.8% of patients in the placebo arm and 21.4% of patients in the vorasidenib arm had at least 1 major protocol deviation. The frequency of protocol deviations was broadly balanced between study arms. The most common protocol deviation related to International Council for Harmonization or Good Clinical Practice deviations related to informed consent (12.3% in the placebo arm, 11.9% in the vorasidenib arm); the submission did not specify specific deviations. A total of 6.1% of patients in the placebo arm and 8.3% of patients in the vorasidenib arm reported deviations related to the protocol itself.
Baseline Characteristics
A summary of the baseline characteristics of patients in the INDIGO study is presented in Table 3. All but 1 of the patients were aged 18 years or older, with the 1 younger patient receiving placebo.
Table 3: Summary of Baseline Characteristics From the INDIGO Study (FAS)
Characteristic | INDIGO | |
|---|---|---|
Placebo (n = 163) | Vorasidenib (n = 168) | |
Demographic characteristics | ||
Age (years), mean (SD) | 39.8 (9.53) | 40.9 (10.51) |
Age category (years), n (%) | ||
< 16 | 0 | 0 |
16 to < 18 | 1 (0.6) | 0 |
18 to < 40 | 87 (53.4) | 76 (45.2) |
40 to < 65 | 74 (45.4) | 90 (53.6) |
≥ 65 | 1 (0.6) | 2 (1.2) |
Sex, n (%) | ||
Male | 86 (52.8) | 101 (60.1) |
Female | 77 (47.2) | 67 (39.9) |
Race,a n (%) | ||
American Indian or Alaska Native | 0 | 1 (0.6) |
Asian | 8 (4.9) | 5 (3.0) |
Black or African American | 1 (0.6) | 2 (1.2) |
Native Hawaiian or other Pacific Islander | 0 | 0 |
White | 132 (81.0) | 125 (74.4) |
Other | 1 (0.6) | 2 (1.2) |
Not reported | 21 (12.9) | 33 (19.6) |
BMI (kg/m2), mean (SD) | 26.52 (5.89) | 26.81 (5.75) |
Disease characteristics | ||
Histological subtype, n (%) | ||
Oligodendroglioma | 84 (51.5) | 88 (52.4) |
Astrocytoma | 79 (48.5) | 80 (47.6) |
Chromosome 1p19q codeletion status, n (%) | ||
Codeleted | 84 (51.5) | 88 (52.4) |
Not codeleted | 79 (48.5) | 80 (47.6) |
CDKN2A homozygous deletion, n (%) | 93 (57.1) | 109 (64.9) |
Present | 2 (1.2) | 0 |
Absent | 91 (55.8) | 109 (64.9) |
Karnofsky Performance Scale score, n (%)a | ||
100 | 87 (53.4) | 90 (53.6) |
90 to 80 | 76 (46.6) | 77 (45.8) |
70 to 60 | 0 | 1 (0.6) |
Tumour size at baseline (cm), n (%) | ||
Longest diameter of ≥ 2 cm | 137 (84.0) | 139 (82.7) |
Longest diameter of < 2 cm | 26 (16.0) | 29 (17.3) |
Pretreatment tumour growth (mm per year), mean (SD) | 2.79 (4.48) | 2.17 (2.98) |
Number of prior surgeries for glioma, n (%) | ||
1 | 134 (82.2) | 126 (75.0) |
≥ 2 | 29 (17.8) | 42 (25.0) |
Time from last glioma surgery to randomization (years), mean (SD) | 2.60 (1.285) | 2.66 (1.139) |
IDH1 positive | 152 (93.3) | 163 (97.0) |
R132H | 138 (84.7) | 146 (86.9) |
IDH2 positive | 11 (6.7) | 5 (3.0) |
BMI = body mass index; FAS = full analysis set; SD = standard deviation.
aRacial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.22
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure, use of concomitant medications and subsequent treatments in the included study are in the Supplemental Material document, Appendix 4. The mean duration of treatment was █████ █████████ █████████ █████ █████ months in the placebo arm and █████ ████ █████ months in the vorasidenib arm. The relative dose intensity (ratio of actual to planned dose intensity in milligrams per month) exceeded 90% in both treatment arms.
Treatment-emergent adverse events (TEAEs) leading to interruption of study treatment were reported less frequently in the placebo arm (25.2%) than the vorasidenib arm (32.9%). Fewer patients in the placebo arm (4.3%) also experienced dose reductions due to TEAEs compared to the vorasidenib arm (11.4%).
Concomitant medication use was reported only at the DCO on September 6, 2022. The majority of patients (99.4% in the placebo arm, 98.2% in the vorasidenib arm) reported at least 1 concomitant medication; the most common drug class in both treatment arms was antiepileptics (76.7% in the placebo arm, 73.7% in the vorasidenib arm), and other analgesics and antipyretics (46.0% and 36.5%, respectively). The numbers of patients reporting each medication class were broadly consistent between treatment arms, with the exception of other analgesics and antipyretics, anti-inflammatory and antirheumatic products (nonsteroidal), and antidepressants, with a numerically larger number of patients in the placebo arm (46.0%, 25.8%, and 22.7%, respectively) reporting using these classes of medications relative to the vorasidenib arm (36.5%, 18.0%, and 12.6%, respectively).
Six patients (3.7%) in the placebo arm and 19 patients (11.3%) in the vorasidenib arm received at least 1 subsequent antineoplastic therapy, most commonly temozolomide (3.1% and 8.3%, respectively). Post-progression, 70 (42.9%) of the patients in the placebo arm crossed over to receive vorasidenib. Six patients (3.7%) in the placebo arm and 14 patients (8.3%) in the vorasidenib arm received at least 1 subsequent anticancer radiotherapy. Five patients (3.1%) in the placebo arm and 14 patients (8.3%) in the vorasidenib arm received at least 1 subsequent anticancer surgery.
Critical Appraisal
Internal Validity
Overall, there was a low risk of bias arising from the randomization, blinding, and treatment allocation process. While unblinding is possible due to known harms, ascertainment of progressive disease by a BIRC mitigated the potential for bias in measurement of the primary end point (PFS). Although TTNI could be influenced by knowledge of treatment assignment, the decision to move a patient onto the next treatment was made based on radiographic progression determined before unblinding, which mitigated this potential. Baseline patient characteristics were generally balanced across treatment groups; however, there was a smaller proportion of male patients, a greater proportion of white patients, and a greater proportion of patients aged 18 to 40 years in the placebo arm relative to the vorasidenib arm. The relative treatment dose intensity was high in both arms and treatment discontinuations were primarily due to progressive disease and AEs. The use of concomitant treatments was generally similar across arms and not expected to affect efficacy.
Approximately twice as many patients in the placebo arm discontinued their randomized treatment relative to the vorasidenib arm, and the majority of discontinuations were due to disease progression. Post-progression, ██ ███████ of patients in the placebo arm crossed over to receive vorasidenib. Receiving vorasidenib post-progression does not reflect its likely use in practice nor its indication as a first-line therapy after surgery. The analysis of OS (which includes postcrossover data) may therefore not directly answer the clinical question of interest.25-27 While it is possible that this would result in OS estimates closer to the null, the available evidence does not allow the full impact of the crossover on OS to be determined with certainty, particularly given that the follow-up was short (as discussed in a following section). End points that consider only the on-treatment phase (e.g., TGR) or measured up to progression (i.e., PFS) are not affected. Because the analysis of TTNI considers crossover as an event, it is also not affected.
The INDIGO trial was stopped early for efficacy, and the submission transparently detailed the rationale for early unblinding following the recommendation of the independent data monitoring committee (i.e., the study met its primary and key secondary end points at the second interim analysis). As a result, the study recruitment period was shorter than planned at 25 months (42 months of nonuniform recruitment were assumed). The early study cut-off resulted in a lower number of events having accrued and the median PFS and TTNI not being reached: a total of 47.7% of PFS events, 32.0% of TTNI events and 0.3% of total OS events had occurred across arms by the DCO on March 7, 2023. The long-term results for these outcomes are therefore unclear, and, given that the findings are from an interim analysis, there is an increased risk that the treatment effect may be overestimated.28 The follow-up was also considered too short to collect meaningful OS information in this disease. PFS was used as a surrogate for OS, and the sponsor submitted evidence from a literature review of 91 studies,29 including 7,125 patients with glioblastoma, showing that PFS is a valid predictor of OS in that setting. However, the patient population differs from the population of interest (i.e., glioblastoma is more aggressive and OS expectations are different), none of the treatments were in the same class as vorasidenib, and PFS definitions in most studies differed from the definition used in the INDIGO trial. Due to these limitations, it remains uncertain whether the PFS benefit observed in the INDIGO trial will translate to future improvements in OS. The relatively short follow-up also means that long-term harms are unknown.
Only the primary and key secondary end points of PFS and TTNI were controlled for multiple comparisons. As such, the other outcomes are considered supportive of the impact of vorasidenib. Limited details were provided on the handling of missing data for TGR, time to malignant transformation, or seizure activity outcomes. There were few losses to follow-up from the trial, and there was no indication of informative censoring for the time-to-event end points. While censoring for subsequent treatments in the analysis of PFS can be informative, this occurred infrequently. Multiple sensitivity analyses, including those that did not censor for subsequent treatments and that counted all PFS events regardless of missing assessments, supported the robustness of the primary analysis. For the reporting of seizure activity, some patient outcome data were missing (n = 152 [93.3%] patients in the placebo arm and n = 162 [96.4%] patients in the vorasidenib arm reported at baseline, and only ███ ███████ in the placebo arm and ███ ███████ in the vorasidenib arm reported at cycle 13). HRQoL was measured by a validated tool for brain cancer (FACT-Br), and a literature-based minimum important difference (MID) was provided by the sponsor for patients with brain metastases,30,31 but not the specific tumour that is the subject of this review. In addition, the results for FACT-Br did not have data from the entire patient population (███ ███████ of patients in the placebo arm and ███ ███████ of patients reported results for FACT-Br at cycle 13, day 1; ██ ███████ in the placebo arm and ██ ███████ in the vorasidenib arm at end of treatment). This is possibly due to the early stopping of the trial but could also be due to patients being lost to follow-up or missing assessments. There were no details on how missing data were handled in the analysis. Given that the reasons for missing data are unknown and there are no sensitivity analyses with different plausible assumptions about the missing data, the findings are at risk of bias. DOR was only measured among patients with a response; the results are therefore at risk of bias because prognostic balance across treatment arms is not guaranteed in this subpopulation of patients. Similarly, time to malignant transformation was measured among only the patients who had surgery or biopsy as an intervention.
Some notable protocol changes occurred after recruitment began, including removing TGR as a key secondary outcome and readjusting the statistical analysis, including the power calculations, modifying TTNI to include death as an event, and changing the sponsor of the study. Although there were many major protocol violations, these deviations were balanced across the treatment arms and appear unlikely to have a serious impact on the efficacy results.
External Validity
According to the clinical experts consulted for this review, the baseline characteristics broadly represented the demographics and medical characteristics of the patients they might see in practice. Similarly, the frequency of visits that occurred during the study and the clinical investigations that occurred pre-enrolment (i.e., ascertainment of IDH status, MRI scan of the tumour, and determining whether it is enhancing or nonenhancing before enrolment) were generally similar to those seen in clinical practice, with some exceptions. Patients were required to have had surgery between 1 and 5 years before enrolment in the INDIGO trial, which is not reflective of real-world treatment with vorasidenib as the clinical experts indicated that they would initiate vorasidenib in patients a few months after surgery or as soon as the patients have recovered. The results from the INDIGO trial may not be generalizable to the population of patients aged 12 to 18 years, as no patients younger than 16 years were recruited into the study, and the majority (apart from 1 patient) were adults (aged ≥ 18 years). The product monograph for vorasidenib noted that the use of vorasidenib in pediatric patients is supported by additional population pharmacokinetic data demonstrating that age had no clinically meaningful effect on the pharmacokinetics of vorasidenib. The clinical experts consulted for this review further suggested that patients younger than the trial minimum (12 years) could be considered for vorasidenib. The study required that patients have MRI-visible, nonenhancing disease; the clinical experts noted that the nonenhancing disease is reasonable, but they would also consider patients who had had successful gross total resections, as these patients would likely also proceed to active surveillance. Of note, total resection was an example of a surgical procedure that was permitted for enrolment in the study. A Karnofsky Performance Status score of 80% or greater was required for enrolment in the study, and while the clinical experts noted that this functional status threshold covered most patients they would see in practice, they would consider patients with a lower score as well. Among the study exclusion criteria were patients taking steroids for the signs and symptoms of glioma, which the experts noted would not preclude them from considering these patients for vorasidenib. Patients with significant active cardiac disease or baseline renal, bone marrow, and hepatic function were also not included therefore, results may not be generalizable to these populations.
In addition to the patient population, it was noted in the submission that a modified version of the RANO-LGG criteria was used by the BIRC to assess disease progression. Specifically, the BIRC had reduced access to the patients’ clinical data, there was no measure for clinical deterioration used in the modified version (whereas the RANO-LGG contains this measure), and patients who were using steroids for symptom control were excluded from the study. In other words, the modified measure focused more on tumour volume for the purposes of blinded evaluation of disease progression. The clinical experts consulted for this review noted that, in low-grade gliomas, clinical progression is not often observed in the absence of radiographic progression. This was corroborated in the INDIGO study, in which only 1 patient in the placebo group discontinued treatment due to clinical progression in the absence of radiographic disease progression. However, the clinical experts confirmed that the RANO-LGG is used in practice alongside other clinical assessments but mentioned that it is not the only measure used to define disease progression. This may have some impact on the generalizability of the PFS, TTNI, TGR, and time to malignant transformation outcomes.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document. Results reflect the DCO on March 7, 2023, unless otherwise specified.
Efficacy
Key results include the following.
Progression-Free Survival
The median follow-up duration for PFS was 17.7 months (95% confidence interval [CI], █████ ████) and 16.7 months (95% CI, █████ ████) in the vorasidenib and placebo arms, respectively. A total of █████ of patients in the placebo arm and █████ of patients in the vorasidenib arm experienced a PFS event. Median PFS times were 11.4 months (95% CI, 11.1 to 13.9 months) in the placebo arm and not estimable (95% CI, 22.1 months to not estimable) in the vorasidenib arm. The HR was 0.35 (95% CI, 0.25 to 0.49; P = not reported) in favour of vorasidenib.
At 12 months, 47.3% (95% CI, █████ ██ █████) of patients in the placebo arm and 77.3% (95% CI, █████ ██ █████) of patients in the vorasidenib arm were progression-free. The difference in the proportion of patients whose disease remained progression-free at 12 months was 30.0% (95% CI, ██████ ██ ██████) in favour of vorasidenib. At 24 months, 26.2% (95% CI, 17.9% to 35.3%) of patients in the placebo arm and 58.8% (95% CI, 48.4% to 67.8%) of patients in the vorasidenib arm were progression-free. The difference in the proportion of patients whose disease remained progression-free at 24 months was 32.6% (95% CI, ██████ ██ ██████) in favour of vorasidenib.
Time to Next Intervention
A total of ██ ███████ patients in the placebo arm moved to a next intervention (██ ███████ patients reported crossing over to vorasidenib) and █████ of patients in the vorasidenib arm moved to a next intervention. The median TTNI was 20.1 months (95% CI, 17.5 to 27.1 months) in the placebo arm and was not estimable in the vorasidenib arm. The HR was 0.25 (95% CI, 0.16 to 0.40; P = not reported), in favour of vorasidenib.
At 12 months, 74.9% (95% CI, █████ ██ █████) of patients in the placebo arm and 90.3% (95% CI, █████ ██ █████) of patients in the vorasidenib arm were without a subsequent intervention. The difference in the proportion of patients without a subsequent intervention at 12 months was 15.4% (95% CI, █████ ██ ██████) in favour of vorasidenib. At 24 months, 41.4% (95% CI, █████ ██ █████) of patients in the placebo arm and 80.3% (95% CI, █████ ██ █████) of patients in the vorasidenib arm were without a subsequent intervention. The difference in the proportion of patients without a subsequent intervention at 24 months was 38.9% (95% CI, ██████ ██ ██████) in favour of vorasidenib.
Functional Assessment of Cancer Therapy – Brain
The mean FACT-Br total score at baseline was 158.8 points (standard deviation [SD] = 23.33 points) in the placebo arm and 158.2 points (SD = 26.40 points) in the vorasidenib arm. At cycle 13, day 1, the mean total score was █████ ██████ ████ ███ ███████ █████████████ in the placebo arm and █████ ██████ ████ ███ █████ ██ █████████ ████ in the vorasidenib arm (mean difference for vorasidenib versus placebo: ███ ██████ ████ ███ ████ ██ █████ representing mean changes from baseline of ████ ██████ ███ █ ██████ and ███ ██████ ███ █ █████ in the placebo and vorasidenib groups, respectively. The mean end of treatment total score was █████ ██████ ████ ███ █████ ██ ███████ █████ in the placebo arm and █████ ██████ ████ ███ █████ ██ █████████ ███ in the vorasidenib arm (mean difference for vorasidenib versus placebo: ███ █████ ████ ███ █████ ██ █████.
Overall Survival
The median OS follow-up was 20.2 ████ ███ █████ █████ months in the placebo arm and 19.8 ████ ███ █████ █████ months in the vorasidenib arm, respectively. In total, 0.0% of patients in the placebo arm and 0.6% of patients in the vorasidenib arm experienced an event. Median OS was not estimable in the placebo arm or vorasidenib arms. The HR was not estimable.
██ ██ ███████ ████ ████ ███ ███ ██████████ ██ ████████ ██ ███ ███████ ███ ███ ████ ████ ███ ███ ██████████ ██ ████████ ██ ███ ███████████ ███ ████ ██████ ███ ██████████ ██ ███ ██████████ ██ ████████ █████ ██ ██ ██████ ███ ███ ██ ██ ███████ ████ ████ ███ ███ ██████████ ██ ████████ ██ ███ ███████ ███ ███ █████ ████ ███ █████ ██ ██████ ██ ████████ ██ ███ ███████████ ███ ████ ██████ ███ ██████████ ██ ███ ██████████ ██ ████████ █████ ██ ██ ██████ ███ █████ ████ ███ ██████ ██ ███████.
Duration of Response
As of the DCO on September 6, 2022, 2.5% of patients in the placebo arm and 10.7% of patients in the vorasidenib arm reported a complete response, partial response, or minor response. There were 0 (0.0%) progressive disease events in the placebo arm and 3 (1.8%) progressive disease events in the vorasidenib arm.
At 12 months, the DOR rate was 100% (95% CI, not estimable) of patients in the placebo arm and 83.3% (95% CI, 48.2% to 95.6%) of patients in the vorasidenib arm; the difference was –16.7% (95% CI, –37.87% to 4.47%). At 24 months, the DOR rate was not estimable in either treatment arm. DOR was not assessed at the DCO on March 7, 2023.
Tumour Growth Rate
As of the DCO on March 7, 2023, the mean percent change in tumour volume over 6 months was 14.4% (95% CI, 12.0% to 16.8%) in the placebo arm and –1.3% (95% CI, –3.2% to 0.7%) in the vorasidenib arm. The treatment difference for placebo versus vorasidenib was 15.9% (95% CI, 12.6% to 19.3%).
In the placebo group, the mean percent change for every 6 months was 19.7% before treatment. The posttreatment mean percent change for every 6 months was 11.1%, representing a difference between pretreatment- and posttreatment TGR of █████ ████ ███ ██████ ██ ██████. In the vorasidenib group the mean percent change for every 6 months was 13.4% before treatment. The posttreatment mean percent change for every 6 months was –3.2%, representing a difference between pretreatment and posttreatment TGR of ██████ ████ ███ ██████ ██ ███████. The difference between placebo and vorasidenib was 8.7% (95% CI, 3.1% to 14.6%).
For TGR before and after vorasidenib crossover (conducted in the placebo arm only, among patients who crossed over), the mean percent change over 6 months was 23.9% (95% CI, 19.0% to 29.0%) in the precrossover placebo arm and 0.9% (95% CI, –3.6% to 5.6%) in the postcrossover placebo arm (n = 61). The difference in TGR precrossover versus postcrossover was –18.6% (95% CI, –23.4% to –13.5%).
Time to Malignant Transformation
A total of ██ patients in the placebo arm and ██ patients in the vorasidenib arm had histopathologically proven malignant transformation of their disease. The number of patients with malignant transformation was too small to perform an analysis.
Seizure Activity
A total of 20 patients in each study arm reported 1 or more seizures in the 30 days before baseline. The mean number of seizures in these patients was 13.20 (SD = 38.66) in the placebo arm and 8.40 (SD = 24.31) in the vorasidenib arm. As of the March 7, 2023, DCO, up to cycle 13, day 1, ██ patients in the placebo arm and ██ patients in the vorasidenib arm reported at least 1 seizure, corresponding to a mean of █████ ████████ ███ █ ██████ in the placebo arm and ███ ████████ ███ █ █████ in the vorasidenib arm, The mean change from baseline was ████ ████████ ███ █ ██████ and █████ ████████ ███ █ ████ in the placebo ██ █ ███ and vorasidenib ██ █ ████ arms, respectively.
A total of 69.9% of patients in the placebo arm and 61.3% of patients in the vorasidenib arm reported receiving antiseizure medication at baseline. In total, ██ patients in the placebo arm and ██ patients in the vorasidenib arm required the addition of at least 1 antiseizure medication (before crossover), and ██ and ██ patients in the placebo and vorasidenib arms who were not receiving any antiseizure medication at baseline required the addition of at least 1 antiseizure medication on-treatment, respectively.
Harms
Key results include the following.
Treatment-Emergent Adverse Events
The majority of patients in both study arms (95.1% in the placebo arm, 98.8% in the vorasidenib arm) reported TEAEs. A numerically greater proportion of patients in the vorasidenib arm reported grade 3 or higher TEAEs (26.9% versus 16.0% in the placebo arm), most commonly increases in levels of alanine transaminase (ALT) and aspartate transaminase (AST) (17 [10.2%] versus 2 [1.2%] and 8 [4.8%] versus 0, respectively).
The most common TEAEs in the vorasidenib arm were increased ALT (█████), fatigue (█████), COVID-19 (█████), increased AST (█████), headache (█████), diarrhea (█████), and nausea (█████). The most common TEAEs in the placebo arm were fatigue (█████), COVID-19 (█████), headache (█████), and nausea (█████).
A greater proportion of patients in the vorasidenib arm reported increased ALT (█████ ███ █████), increased AST (█████ ███ ████), increased gamma-glutamyltransferase (GGT) (█████ ███ ████), and diarrhea (█████ ███ █████) relative to placebo. The remaining TEAEs were broadly consistent between treatment arms.
Serious Adverse Events
More patients in the vorasidenib arm (█████) reported 1 or more SAEs relative to the placebo arm (████).
Apart from seizures, which were reported in ███████ patients in the placebo arm and ███████ patients in the vorasidenib arm, the remaining SAEs occurred in ███ █ ████████.
Other SAEs in the vorasidenib arm only occurred in ████████ ████. Most other SAEs in the placebo arm only occurred in ██ ███████ ████; ██ patients also reported suicidal ideation.
Withdrawals Due to Adverse Events
A total of ████ of patients in the placebo arm and ████ of patients in the vorasidenib arm stopped treatment due to AEs (based on the March 7, 2023, DCO). The reasons for stopping in the placebo arm reported at the September 6, 2022, DCO, were diarrhea (0.6%) and fatigue (0.6%); the reasons for stopping in the vorasidenib arm were increased ALT (3.0%), increased AST (1.8%), increased GGT (0.6%), and autoimmune hepatitis (0.6%).
Mortality
At the DCO on March 7, 2023, 1 patient in the vorasidenib group had died due to progressive disease.
Adverse Events of Special Interest
A total of █████ of patients in the vorasidenib arm reported increased ALT, relative to ████ in the placebo arm, and a total of ████ of patients in the vorasidenib arm reported increased AST, relative to ████ in the placebo arm.
Summary of Findings and Certainty of the Evidence
The sponsor did not identify any MIDs for the outcomes included in GRADE, and in the absence of literature-based MID estimates, a null target of assessment (i.e., certainty in the presence of a non-null effect) was used because thresholds for clinical importance could not be established.
Table 4: Summary of Findings for Vorasidenib vs. Active Surveillance for Patients Aged 12 Yearsa and Older With Grade 2 (WHO 2016, 2021 Grading System) Oligodendroglioma or Astrocytoma With Susceptible IDH1 or IDH2 Mutation — DCO on March 7, 2023
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Vorasidenib | Difference | |||||
Time-to-event outcomes | |||||||
Proportion of patients whose disease remains progression-free at 12 months Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NA | 473 per 1,000 | 773 per 1,000 (███ ██ ███ per 1,000) | 300 more per 1,000 (███ ██ ███ more per 1,000) | High | Vorasidenib results in an increase in the proportion of patients whose disease remains progression-free at 12 months when compared to placebo. The clinical importance of the increase is unclear. |
Proportion of patients whose disease remains progression-free at 24 months Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NA | 262 per 1,000 | 588 per 1,000 (484 to 678 per 1,000) | 326 more per 1,000 (███ ██ ███ more per 1,000) | Moderateb (serious study limitations) | Vorasidenib likely results in an increase in the proportion of patients whose disease remains progression-free at 24 months when compared to placebo. The clinical importance of the increase is unclear. |
Proportion of patients without a subsequent intervention at 12 months Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NA | 749 per 1,000 | 903 per 1,000 (███ ██ ███ per 1,000) | 154 more per 1,000 (██ ██ ███ more per 1,000) | High | Vorasidenib results in an increase in the proportion of patients without a subsequent intervention at 12 months when compared to placebo. The clinical importance of the increase is unclear. |
Proportion of patients without a subsequent intervention at 24 months Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NA | 414 per 1,000 | 803 per 1,000 (███ ██ ███ per 1,000) | 389 more per 1,000 (███ ██ ███ more per 1,000) | Moderateb (serious study limitations) | Vorasidenib likely results in an increase in the proportion of patients without a subsequent intervention at 24 months when compared to placebo. The clinical importance of the increase is unclear. |
Proportion of patients alive at 24 months Median follow-up: 19.8 months (vorasidenib), 20.2 months (placebo) | 331 (1 RCT) | NA | NE per 1,000 | NE per 1,000 (NE) | NE per 1,000 (NE) | Moderatec (serious study limitations) | Vorasidenib likely results in a nonestimable effect on the proportion of patients who are alive at 24 months. The clinical importance of the result is unclear. |
Health-related quality of life | |||||||
Change from baseline to cycle 13, day 1 in FACT-Br total score (184 [best] to 0 [worst]) Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NR | █████████ ██ █████ | ███ ███████ ██ █████ | █████████ ██ █████ | Very lowd,e (very serious study limitations and imprecision) | The evidence is very uncertain about the effect of vorasidenib on the change from baseline to cycle 13, day 1 in FACT-Br total scores when compared to placebo. |
Change from baseline to end of treatment in FACT-Br total score (184 [best] to 0 [worst]) Median follow-up: 17.7 months (vorasidenib), 16.7 months (placebo) | 331 (1 RCT) | NR | ████ ███████ ██ █████ | ████ ███████ ██ █████ | ███████████ ██ █████ | Very lowd,e (very serious study limitations and imprecision) | The evidence is very uncertain about the effect of vorasidenib on the change from baseline to end of treatment in FACT-Br total scores when compared to placebo. |
Harms | |||||||
Proportion of patients with ≥ 1 SAE Median follow-up: 19.8 months (vorasidenib), 20.2 months (placebo) | 331 (1 RCT) | NR | ██ ███ ████ | ███ ███ ████ ████ ██ ███ ███ █████ | ██ ████ ███ ████ ████ ██ ███ ████ ███ █████ | Moderatef (serious imprecision) | Vorasidenib likely results in an increase in the proportion of patients with at least 1 SAE when compared with placebo. The clinical importance of the increase is uncertain. |
Proportion of patients with increased ALT Median follow-up: 19.8 months (vorasidenib), 20.2 months (placebo) | 331 (1 RCT) | NR | ███ ███ ████ | ███ ███ ████ █████ ██ ███ ███ █████ | ███ ████ ███ ████ █████ ██ ███ ████ ███ █████ | High | Vorasidenib results in an increase in the proportion of patients with increased ALT when compared with placebo. The clinical importance of the increase is uncertain. |
Proportion of patients with increased AST Median follow-up: 19.8 months (vorasidenib), 20.2 months (placebo) | 331 (1 RCT) | NR | ██ ███ ████ | ███ ███ █████████ ██ ███ ███ █████ | ███ ████ ███ ████ █████ ██ ███ ████ ███ █████ | High | Vorasidenib results in an increase in the proportion of patients with increased AST when compared with placebo. The clinical importance of the increase is uncertain. |
ALT = alanine transaminase; AST = aspartate transaminase; CI = confidence interval; DCO = data cut-off; HRQoL = health-related quality of life; NA = not applicable; NR = not reported; FACT-Br = Functional Assessment of Cancer Therapy – Brain; RCT = randomized controlled trial; SAE = serious adverse event.
Note: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe INDIGO trial inclusion criteria were for patients aged 12 years or older; however, the INDIGO trial only had 1 patient aged between 16 and 18 years; the remaining participants were all adults. Therefore, the results in this table only directly apply to patients aged 18 years or older.
bRated down 1 level for serious study limitations. There is a large degree of censoring in the vorasidenib arm, and few patients remain at risk at this time point, which may reduce the reliability of the effect estimate.
cRated down 1 level for serious study limitations and indirectness. There is a large degree of censoring, which may reduce the reliability of the effect estimate, and due to short follow-up for this outcome only 1 event has accrued; the analysis includes patients in the placebo group who crossed over to vorasidenib, which is not reflective of clinical practice.
dRated down 2 levels for very serious study limitations. There is a large proportion of missing data, which results in high risk of bias.
eRated down 1 level for serious imprecision. Based on a non-null target of assessment, the CI around the point estimate contains the potential for a positive impact on HRQoL, no impact on HRQoL, or a negative impact on HRQoL.
fRated down 1 level for serious imprecision. Based on a non-null target of assessment, the CI around the point estimate contains the potential for no impact on SAEs as well as an increase in SAEs.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence,22 the INDIGO Clinical Study Report,23 the INDIGO Addendum Clinical Study Report,32 and additional information provided by the sponsor.33-35
Long-Term Extension Studies
The submission did not include any LTE studies.
Indirect Evidence
The submission did not include any indirect evidence.
Studies Addressing Gaps in the Systematic Review Evidence
The submission did not include any studies addressing gaps in the systematic review evidence.
Discussion
This report summarizes the evidence for vorasidenib compared to placebo for the treatment of grade 2 (WHO 2016, 2021 grading system) oligodendroglioma or astrocytoma with susceptible IDH1 or IDH2 mutation in patients aged 12 years or older who are not in immediate need of radiotherapy or chemotherapy after surgical intervention. The evidence appraisal was based on the interim analysis (6 months after additional follow-up) from a single multisite, phase III, double-blind, placebo-controlled trial (INDIGO; N = 331) in which patients were randomized to either vorasidenib once daily (n = 168) or placebo (n = 163).
Efficacy
The clinical experts indicated that patients on active surveillance will ultimately progress, and the standard-of-care pharmacological treatment options (i.e., surgery, radiotherapy, and chemotherapy) are not curative. The clinician groups also noted that current treatment options prolong survival but have many potential adverse effects. Patient input highlighted the impact this disease has on their lives and noted an unmet need for treatments that delay disease progression, improve quality of life, and avoid radiotherapy or chemotherapy. Many outcomes evaluated in the INDIGO trial were important and relevant to patients and clinicians, including TTNI, PFS, OS, and HRQoL.
The INDIGO trial was unblinded early at the recommendation of the independent data monitoring committee due to early demonstration of efficacy because the study met its primary end point and key secondary end point. As a result, the length of follow-up available is short in the context of this disease, and median PFS and TTNI in the vorasidenib arm were not estimable. Considerable differences were observed between patients randomized to placebo or to vorasidenib for the proportion of patients’ progression-free status and the proportion of patients without a subsequent intervention at 12 and 24 months favouring vorasidenib. The clinical experts consulted by the review team indicated that these findings were clinically significant. However, the longer-term efficacy is unknown. The follow-up of the INDIGO trial was too short to collect meaningful OS information, as patients may live for a long time with this cancer and therefore it is expected that events for analysis would require longer follow-up. Additionally, the OS analysis would be affected by the crossover of patients from the placebo group do vorasidenib after progression, which may not be reflective of current clinical practice. PFS was used as a surrogate for OS; however, information on the validity of PFS as a surrogate for OS in the population and treatment under review was unavailable. The sponsor provided multiple studies that suggested that PFS was a predictor of OS and could be an effective surrogate end point for OS. However, these studies were conducted in high-grade glioma and in adjuvant patients and are not aligned with the indication under review. It therefore remains unclear whether the association between PFS and OS is consistent in low-grade glioma. Nevertheless, given the slow-growing nature of diffuse glioma, the use of PFS as a primary end point to inform treatment efficacy is considered reasonable. In general, the clinical experts consulted by CDA-AMC indicated that the availability of a pharmacological option for patients on active surveillance would be an important change in the treatment paradigm for this disease, and they noted that the efficacy results to date were compelling. Of note, no MIDs were available for any outcomes, and no thresholds for clinical importance could be established. However, the clinical experts noted that in a disease setting where active surveillance is the default option, any improvement in current outcomes at a patient level would be meaningful to them.
The reimbursement request was narrower than the Health Canada indication as it the former requires patients to be in immediate need of radiotherapy or chemotherapy. As previously noted, the INDIGO trial enrolled patients who were at 1 to 5 years post surgery. The sponsor noted that this was done to homogenize the enrolled patient population, and to identify patients already undergoing active surveillance and characterize the TGR before treatment. However, according to the clinical experts consulted for this review, vorasidenib would be initiated relatively quickly in patients who would otherwise go to active surveillance, as opposed to an extended postsurgical duration. The impact of this delay in treatment on the efficacy of vorasidenib remains unclear. Furthermore, the Health Canada indication for vorasidenib is for patients aged 12 years and older. The study population for the INDIGO trial only contained 1 patient under the age of 18 years, who was in the placebo group. The product monograph notes that the use of vorasidenib in patients aged 12 years and older is supported by pharmacokinetic data. As such, the efficacy and safety of vorasidenib in patients aged less than 18 years is unknown, although none of the experts objected to using vorasidenib in a pediatric setting. Overall, the results from the study population enrolled in the INDIGO trial may not be fully generalizable to the patient population who may be considered candidates in clinical practice. The clinical experts consulted for this review also noted that low-grade tumours, good to excellent resection outcomes, and the absence of neurocognitive symptoms would suggest a patient could be considered to be at low risk and would be followed by active surveillance or considered for vorasidenib if available. They also indicated that the use of biomarkers and patient-specific clinical assessments in assessing whether to initiate chemotherapy or radiotherapy means that more patients may be considered for vorasidenib if there is ambiguity around the grade of tumour or concerns over the toxicities with chemotherapy or radiotherapy.
The clinical experts consulted for this review noted that all gliomas ultimately progress and can transform into higher-grade disease, which can take up to 5 years. In the INDIGO trial, only ████████ ██ ██ ███ ███████ ███ ███ █ ██ ███ ███████████ ████ had malignant transformation. Too few malignant transformations occurred during the study to conduct an analysis of the time to malignant transformation, and the short duration and early stoppage of the trial precludes drawing any conclusions on the effect of vorasidenib in preventing or slowing malignant transformation. Firm conclusions could also not be drawn for the exploratory end point of seizure activity, which is an outcome important to patients. The majority of patients in both arms were already taking antiseizure medication at baseline, irrespective of whether they had a seizure in the 30 days before baseline or not. During the treatment period, seizures (number and severity) were self-reported, and the SDs around the number of seizures were wide; thus, the impact of vorasidenib on seizure activity remains unknown.
HRQoL was highlighted as an outcome of importance to patients and was assessed in the INDIGO trial using 1 cancer-specific measure (FACT-Br). Results were very uncertain because they were affected by a risk of bias due to low numbers of patients reporting results, which could have been due to early study unblinding or missing assessments, and imprecision. The clinical experts consulted for the review noted that the trend of minimal impact on HRQoL was unsurprising because the patient population they would see for this indication in clinical practice are often younger and higher functioning.
Harms
In general, although nearly all patients in both arms of the INDIGO study reported TEAEs, there were few serious adverse events (SAEs) and no deaths were reported during the study. Fewer than 5% of patients in each study arm stopped treatment due to AEs. The TEAEs with the greatest difference between the vorasidenib and placebo arms were for increased ALT and increased AST, as well as increased GGT. The clinical experts noted that hepatic toxicity would be the main concern associated with vorasidenib treatment. The elevated ALT, AST, and GGT AEs observed with vorasidenib in the INDIGO trial are typically symptomatically inert; although the clinical experts noted that this would be monitored with regular bloodwork and would be manageable. The product monograph contains dosing adjustments due to hepatotoxicity and notes this plus fetal toxicity to be the main warnings associated with vorasidenib, which is a particular consideration for young patients who wish to start a family. Because the study has been stopped early, meaningful estimates of long-term harms are not available.
Ethics and Equity Considerations
The clinical experts commented that low-grade gliomas tend to affect people who are young, usually employed, and who might have young families or other people to provide for. The impact of this disease can ripple beyond the patient and affect their financial security and family if there are costs associated with obtaining treatment, taking time off work, or travelling to appointments, or if the toxicities of treatment require them to reduce or stop working. Because surgery is also a key part of the treatment paradigm, recovery from surgery is also a consideration that can affect patients and any caregivers. Patients noted that access to radiotherapy and chemotherapy often requires travel to a cancer centre. This may create inequities in access to treatment as patients who live closer to treatment centres will face fewer barriers compared to those who reside further away. A nonclinical challenge for all patients and caregivers includes the recovery from surgery, as well as potential toxicities from radiation or chemotherapy. Because vorasidenib is an oral tablet, patients can receive treatment at home and, assuming that the treatment is effective at prolonging the need for radiation or chemotherapy, vorasidenib may also provide nonclinical benefit if patients and caregivers are not required to manage potential toxicities as early in the disease course, potentially mitigating some access inequities.
Although patients aged 12 years and older are covered under the Health Canada indication, the pediatric clinical expert noted that they do sometimes see gliomas in patients younger than 12 years and hypothesized that they could receive a benefit from treatment. According to the experts, compassionate access is also possible for patients younger than 12 years as they tend to be exceptional cases (e.g., patients with specific syndromes or predisposition to tumours). However, the lack of evidence for patients younger than 18 years suggests that an unmet need remains for pediatric patients. An additional consideration raised by the experts was family planning and reproductive choice. Because vorasidenib is not safe to take during pregnancy, the clinical experts noted that some patients and their partners may need to consider whether to delay treatment until fertility preservation options can be explored or until they have decided about future pregnancies. This consideration may have resource impacts because gliomas tend to affect younger patients, and vorasidenib will be used as a first-line treatment in individuals young enough to have children or who may not have had children yet. In addition, the requirement for effective contraception while taking vorasidenib may not be acceptable to all patients and would have an impact on their reproductive choice. Finally, there may be access issues because neuro-oncologist follow-up and care would still be required for prescribing vorasidenib, and regular MRI imaging required to follow tumour progression.
Conclusion
Management of diffuse gliomas (astrocytoma or oligodendroglioma) after surgery currently involves either active surveillance, or treatment with chemotherapy and radiotherapy, which are associated with substantial morbidity and toxicity. Patient and clinician groups highlighted an important unmet need for new, additional treatment options that prevent or further delay the need for radiotherapy and chemotherapy. One phase III, double-blind, placebo-controlled, multicentre RCT (INDIGO) provided evidence regarding the efficacy and safety of vorasidenib as a first-line treatment in patients aged 12 years or older with oligodendroglioma or astrocytoma with susceptible IDH1 or IDH2 mutations and who are not in immediate need of radiotherapy or chemotherapy after surgical intervention.
The results of the INDIGO trial demonstrated a statistically significant improvement in median PFS and TTNI, as well as a likely increase in the proportion of patients whose disease remained progression-free, and the proportion of patients who did not require subsequent treatment at 24 months, both of which the clinical experts indicated were clinically important. However, the results were based on an interim analysis with fewer than one-third of patients in the vorasidenib group, and fewer than 50% of patients overall experiencing the primary end point at the time of study unblinding and early stoppage of the trial due to early demonstration of efficacy. As such, due to the relatively short follow-up, the results for the time-to-event outcomes of PFS, TTNI, OS, and DOR were not estimable in 1 or both treatment arms and were considered immature. The degree to which these results translate to long-term OS remain unclear and remains a gap in the evidence. The impact of vorasidenib on HRQoL was very uncertain due to missing data and imprecision. Harms results suggested an increase in ALT and AST levels with vorasidenib treatment. However, despite the elevated liver enzymes, the side-effect profile of vorasidenib was considered manageable according to the clinical experts consulted by CDA-AMC.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of vorasidenib compared to active surveillance for the treatment of grade 2 (WHO 2016, 2021 grading system8,36) astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation in adults and pediatric patients aged 12 years and older following surgical intervention. The sponsor submitted a cost-effectiveness analysis for a reimbursement population narrower than the Health Canada indication — specifically, patients not in immediate need of radiotherapy or chemotherapy. Voranigo is being reviewed by CDA-AMC through the complex review pathway; as such, CDA-AMC appraised 2 cost-effectiveness analyses submitted by the sponsor, 1 adopting a publicly funded health care payer perspective and 1 adopting a societal perspective. The sponsor-submitted budget impact analysis assesses the budgetary impact of reimbursing vorasidenib for the requested reimbursement population.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of vorasidenib from the perspective of a public health care payer in Canada and from a societal perspective over a lifetime horizon (60 years). The modelled population comprised patients with grade 2 astrocytoma or oligodendroglioma who were not in immediate need of radiotherapy or chemotherapy following surgical intervention, which is narrower than the Health Canada population, and was based on the participants in the INDIGO trial. The sponsor’s base-case analysis included costs related to drug acquisition, subsequent treatment, medical resource utilization, and terminal care. The sponsor’s societal base case included additional costs associated with patient and caregiver productivity loss.
In the sponsor’s base case, which adopted a health care payer perspective, vorasidenib was associated with incremental costs of $956,296 and 2.60 incremental quality-adjusted life-years (QALYs) relative to active surveillance. This resulted in an incremental cost-effectiveness ratio (ICER) of $367,366 per QALY gained. Of the incremental benefit compared to active surveillance (2.60 incremental QALYs), approximately 100% of the benefit was predicted to be accrued after the treatment duration of the INDIGO trial (median follow-up = 20 months). From a societal perspective, the ICER was $301,970 per QALY gained. Additional information about the sponsor’s submission is summarized in the Supplemental Material document, Appendix 10.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 5; full details are provided in the Supplemental Material document, Appendix 11).
Table 5: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The sponsor assumed that the protective effect of vorasidenib on seizure risk was maintained for a patient’s entire lifetime, thereby underestimating medical resource use for the vorasidenib group. | The sponsor assumed that the effect of vorasidenib on seizure risk was maintained for a patient’s entire lifetime, but at the time of disease progression, the average tumour size was similar in the 2 treatment groups of the INDIGO trial. | CDA-AMC assumed that the effect of vorasidenib on seizure risk did not persist once patients received subsequent treatment with radiotherapy or chemotherapy (i.e., entered the first-line radiotherapy or chemotherapy health state). | No scenario analysis was conducted. |
Long-term TTPD in the vorasidenib group was likely overestimated. | Based on the sponsor’s choice of TTPD extrapolation, the rate of disease progression or treatment discontinuation decreased over time. This assumption was considered clinically implausible, and no evidence was submitted to support it. | CDA-AMC used an exponential distribution that assumes the rate of TTPD is constant over time. | Given the lack of long-term data, the impact of vorasidenib on long-term TTPD is highly uncertain. CDA-AMC explored an alternative extrapolation of TTPD in a scenario analysis. |
Utility values lacked face validity and transparency. | The sponsor used an internal vignette study to adjust health state utility values sourced from the INDIGO trial and literature. The vignette study could not be validated, and the utilities sourced from the trial and literature lacked face clinical validity. | CDA-AMC used the unadjusted utility values sourced from the INDIGO trial and literature. | Given the limitations of the utility values presented by the sponsor, a scenario analysis was conducted with the vignette study utilities and a second scenario analysis was conducted with trial and literature-derived utilities adjusted by the vignette study utilities. |
The frequency and length of unplanned hospitalizations was overestimated. | The sponsor assumed that patients in the first-line radiotherapy or chemotherapy, second-line and later radiotherapy or chemotherapy, and BSC health states experienced 2 unplanned hospitalizations monthly, each lasting 7.2 days. An alternative evidence source from a Danish cohort study provided values that were seen as more clinically plausible, based on clinical expert opinion. | CDA-AMC used a Danish cohort study submitted by the sponsor to inform the frequency and length of hospitalizations. | No scenario analysis was conducted. |
The sponsor assumed that patients could return to the off-treatment PF health state after receiving surgery as first-line treatment. | According to clinical expert input, patients who receive surgery as first-line treatment in the next intervention period are not expected to have the same disease trajectory as patients whose disease did not progress. | CDA-AMC removed the transition from the first-line radiotherapy or chemotherapy health state to the off-treatment PD health state in the model. | No scenario analysis was conducted. |
Work productivity losses were uncertain in the societal perspective analysis. | The sponsor assumed that productivity losses persisted for the patients’ entire lifetime (i.e., used a human capital approach), and assumed that 100% patients were employed. | CDA-AMC did not present a societal perspective base case due to these issues. | A scenario analyses were conducted from a societal perspective. |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; PD = progressed disease; PF = progression-free; TTPD = time to progression or discontinuation.
Note: Full details of the CDA-AMC identified issues are provided in the Supplemental Material document, Appendix 11.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to the Supplemental Material document, Appendix 11, Table 17), in consultation with the clinical experts. Detailed information about the CDA-AMC base case is provided in the Supplemental Material document, Appendix 11.
Impact on Health Care Costs
Vorasidenib is predicted to be associated with additional health care costs compared to active surveillance (incremental costs = $1,190,165). This increase in health care spending results from drug acquisition costs associated with vorasidenib in the postsurgical resection period (refer to Figure 1). Vorasidenib is anticipated to reduce health care costs due to a reduction in medical resource use for treating seizures, for example (cost savings = $42,393); however, these are small relative to increased treatment costs ($1,232,557).
Figure 1: Impact of Vorasidenib vs. Active Surveillance on Health Care Costs
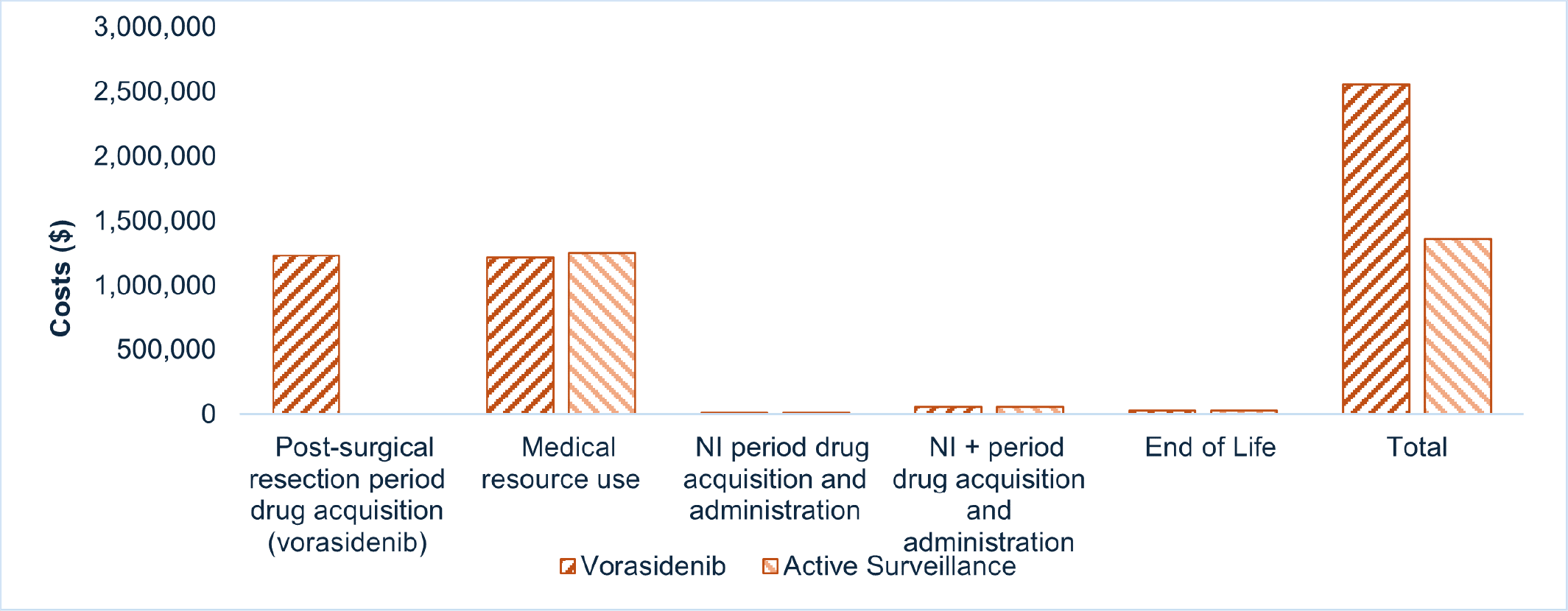
NI = next intervention; vs. = versus.
Note: Medical resource use costs include general practitioner, specialist, and hospital visits, tests and imaging, and disease management (i.e., platelet transfusion, seizure management).
Impact on Health
Relative to active surveillance, vorasidenib is predicted to increase the amount of time a patient remains in the postsurgical resection period (i.e., not receiving the next intervention) by approximately 3.31 years (refer to the Supplemental Material document, Appendix 11, Table 19), and extend OS by 2.97 years (refer to Figure 2). This occurs because the mortality risk is lower in the progression-free health states. Delaying time to progressed disease reduces the mortality risk, resulting in OS gains. Therefore, although no OS benefit was observed in the INDIGO trial, the analysis assumes that over time, survival gains will be realized as the risk of death remains higher in the active surveillance arm, where more disease progression has occurred.
Considering the impact of treatment on both quality and length of life, active surveillance is predicted to result in 2.08 additional QALYs per patient compared to active surveillance (refer to Figure 2 and the Supplemental Material document, Appendix 11, Table 19). Approximately 99% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 2: Impact of Vorasidenib vs. Active Surveillance on Patient Health
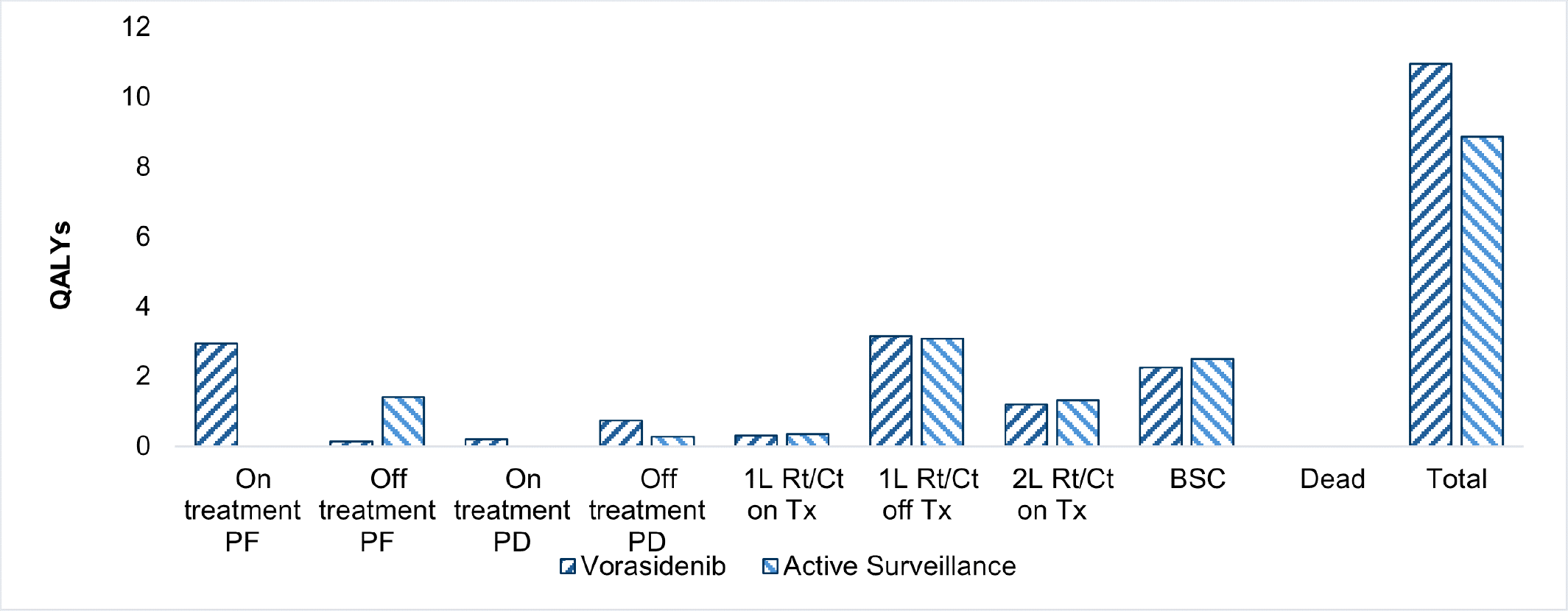
1L = first line; 2L = second line; BSC = best supportive care; Ct = chemotherapy; PD = progressed disease; PF = progression-free; Rt = radiotherapy; QALY = quality-adjusted life-year; Tx = treatment; vs. = versus.
Note: The postsurgical intervention period includes the on-treatment PF health state, the off-treatment PF health state, the on-treatment PD health state, and the off-treatment PD health state.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $571,629 per QALY gained for vorasidenib compared to active surveillance (refer to Table 6). Additional details on the CDA-AMC base case are available in the Supplemental Material document, Appendix 11.
Table 6: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | ICER vs. active surveillance ($/QALY) |
|---|---|---|---|---|
Publicly funded health care payer perspective | ||||
Active surveillance | 1,366,886 | 14.31 | 8.87 | Reference |
Vorasidenib | 2,557,051 | 17.28 | 10.95 | 571,629 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Uncertainty and Sensitivity
Uncertainty was explored in scenario analyses outlined in Table 5. Uncertainty surrounding health state utility values had the largest impact on cost-effectiveness (refer to the Supplemental Material document, Appendix 11, Table 21).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing vorasidenib for use in the reimbursement requested population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of vorasidenib was aligned with the price included in the sponsor’s economic evaluation. Additional information about the sponsor’s submission is provided in Supplemental Material document, Appendix 12.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Supplemental Material document, Appendix 12). CDA-AMC estimated that 277 patients would be eligible for treatment with vorasidenib over a 3-year period (year 1 = 109; year 2 = 89; year 3 = 79), of whom 208 would be expected to receive vorasidenib (year 1 = 77; year 2 = 67; year 3 = 64). The estimated incremental budget impact of reimbursing vorasidenib is predicted to be approximately $90 million over the first 3 years. The actual budget impact will depend on the number of people eligible for treatment and its uptake.
Conclusion
Based on the CDA-AMC base case, vorasidenib would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $571,629 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in the Supplemental Material document, Appendix 11, Table 20). The budget impact of reimbursing vorasidenib to the public drug plans in the first 3 years is estimated to be approximately $90 million.
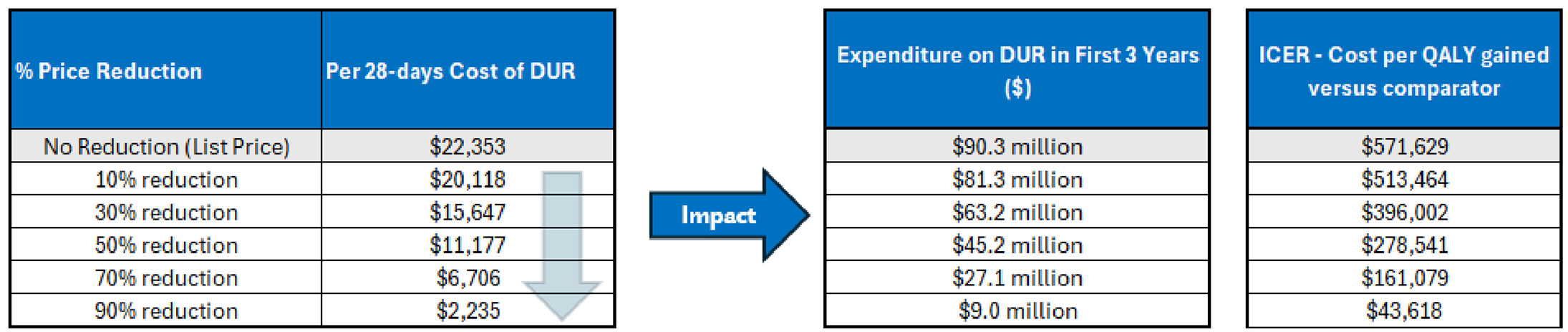
References
1.Servier Canada Inc. VORANIGO® (vorasidenib) Product Monograph. August 27, 2024 [sponsor supplied reference].
2.Mesfin Fb A-DMAG. [Updated 2023 May 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441874 [sponsor supplied reference].
3.Ostrom QT, Price M, Neff C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015-2019. Neuro Oncol. 2022;24(Suppl 5):v1-v95. doi:10.1093/neuonc/noac202 PubMed
4.Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Primers. 2015;1(1):15017. doi:10.1038/nrdp.2015.17 PubMed
5.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807-12. doi:10.1126/science.1164382 PubMed
6.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765-73. doi:10.1056/NEJMoa0808710 PubMed
7.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739-44. doi:10.1038/nature08617 PubMed
8.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-20. doi:10.1007/s00401-016-1545-1 PubMed
9.van den Bent MJ, French PJ, Brat D, et al. The biological significance of tumor grade, age, enhancement, and extent of resection in IDH-mutant gliomas: How should they inform treatment decisions in the era of IDH inhibitors? Neuro Oncol. 2024;26(10):1805-1822. doi:10.1093/neuonc/noae107 PubMed
10.Canadian Global Glioma Advisory Network Consultations. Report produced by Meducom Health Inc. for Servier Canada. September 2024 [sponsor supplied reference].
11.Byun YH, Park CK. Classification and Diagnosis of Adult Glioma: A Scoping Review. Brain Neurorehabil. 2022;15(3):e23. doi:10.12786/bn.2022.15.e23 PubMed
12.Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432-446. doi:10.1016/S0140-6736(18)30990-5 PubMed
13.Brain Tumour Registry. Table 16. Accessed at: https://braintumourregistry.ca/wp-content/uploads/2021/05/FINAL-Incidence-and-Mortality-Report-2021-logo-word-removed.pdf.
14.Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345. doi:10.1007/s11910-013-0345-4 PubMed
15.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Central Nervous System Cancers V.2.2024.© National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Available at NCCN.org [sponsor supplied reference].
16.Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170-186. doi:10.1038/s41571-020-00447-z PubMed
17.Mason WP, Harrison RA, Lapointe S, et al. Canadian Expert Consensus Recommendations for the Diagnosis and Management of Glioblastoma: Results of a Delphi Study. Current Oncology. 2025;32(4):207. PubMed
18.Capper D, Weißert S, Balss J, et al. Characterization of R132H Mutation-specific IDH1 Antibody Binding in Brain Tumors. Brain Pathol. 2010;20(1):245-254. doi:https://doi.org/10.1111/j.1750-3639.2009.00352.x PubMed
19.Lim-Fat MJ, Macdonald M, Lapointe S, et al. Molecular testing for adolescent and young adult central nervous system tumors: A Canadian guideline. Front Oncol. 2022;12:960509. doi:10.3389/fonc.2022.960509 PubMed
20.Jones JJ, Nguyen H, Wong SQ, et al. Plasma ctDNA liquid biopsy of IDH1, TERTp, and EGFRvIII mutations in glioma. Neuro-Oncology Advances. 2024;6(1)doi:10.1093/noajnl/vdae027 PubMed
21.Crucitta S, Pasqualetti F, Gonnelli A, et al. IDH1 mutation is detectable in plasma cell-free DNA and is associated with survival outcome in glioma patients. BMC Cancer. 2024;24(1):31. doi:10.1186/s12885-023-11726-0 PubMed
22.Servier Canada Inc. Sponsor Summary of Clinical Evidence Template: Voranigo (vorasidenib) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voranigo (vorasidenib tablets), 10 mg and 40 mg oral tablets. April 30, 2025.
23.Institut de Recherches Internationales Servier (I.R.I.S.). Clinical Study Report: AG881-C-004. A Phase 3, Multicenter, Randomized, Double-blind, Placebo-Controlled Study of AG-881 in Subjects With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation [internal sponsor's report]. September 29, 2023.
24.Institut de Recherches Internationales Servier (I.R.I.S.). Study Protocol: AG881-C-004. A Phase 3, Multicenter, Randomized, Double-blind, Placebo-Controlled Study of AG-881 in Subjects With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation [internal sponsor's report]. September 29, 2023.
25.Haslam A, Prasad V. When is crossover desirable in cancer drug trials and when is it problematic? Ann Oncol. 2018;29(5):1079-1081. doi:10.1093/annonc/mdy116 PubMed
26.Prasad V, Grady C. The misguided ethics of crossover trials. Contemp Clin Trials. 2014;37(2):167-9. doi:10.1016/j.cct.2013.12.003 PubMed
27.Manitz J, Kan-Dobrosky N, Buchner H, et al. Estimands for overall survival in clincial trials with treatment switching in oncology. Pharm Stat. 2022;21:150-162. PubMed
28.Bassler D, Briel M, Montori VM, et al. Stopping Randomized Trials Early for Benefit and Estimation of Treatment Effects: Systematic Review and Meta-regression Analysis. JAMA. 2010;303(12):1180-1187. doi:10.1001/jama.2010.310 PubMed
29.Han K, Ren M, Wick W, et al. Progression-free survival as a surrogate endpoint for overall survival in glioblastoma: a literature-based meta-analysis from 91 trials. Neuro Oncol. 2014;16(5):696-706. doi:10.1093/neuonc/not236 PubMed
30.Thavarajah N, Bedard G, Zhang L, et al. Psychometric validation of the functional assessment of cancer therapy--brain (FACT-Br) for assessing quality of life in patients with brain metastases. Support Care Cancer. 2014;22(4):1017-28. doi:10.1007/s00520-013-2060-8 PubMed
31.Lien K, Zeng L, Nguyen J, et al. FACT-Br for assessment of quality of life in patients receiving treatment for brain metastases: a literature review. Expert Rev Pharmacoecon Outcomes Res. 2011;11(6):701-8. doi:10.1586/erp.11.67 PubMed
32.Institut de Recherches Internationales Servier (I.R.I.S.). Clinical Study Report: AG881-C-004, Addendum 2. A Phase 3, Multicenter, Randomized, Double-blind, Placebo-Controlled Study of AG-881 in Subjects With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation [internal sponsor's report].
33.Servier. Servier response to Canada's Drug Agency request for additional information regarding vorasidenib review on May 23, 2025 [internal additional sponsor's information].
34.Servier. Servier response to Canada's Drug Agency request for additional information regarding vorasidenib review on July 29, 2025 [internal additional sponsor's information].
35.Servier. Servier response to Canada's Drug Agency request for additional information regarding vorasidenib review on August 13, 2025 [internal additional sponsor's information].
36.Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-1251. doi:10.1093/neuonc/noab106 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.