Drugs, Health Technologies, Health Systems
Reimbursement Review
Acalabrutinib (Calquence)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Mantle cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BR
bendamustine plus rituximab
BTK
Bruton tyrosine kinase
BTKi
Bruton tyrosine kinase inhibitor
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CR
complete response
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
FACT-Lym
Functional Assessment of Cancer Therapy–Lymphoma
FAS
full analysis set
FDG
fluorodeoxyglucose
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IRC
independent review committee
KM
Kaplan-Meier
LDi
longest transverse diameter
LLSC
Leukemia and Lymphoma Society of Canada
MCL
mantle cell lymphoma
MIPI
Mantle Cell Lymphoma International Prognostic Index
NHL
non-Hodgkin lymphoma
OH (CCO)
Ontario Health (Cancer Care Ontario)
ORR
overall response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PR
partial response
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
RCT
randomized controlled trial
R-CVP
rituximab, cyclophosphamide, vincristine, and prednisone
SAE
serious adverse event
SE
standard error
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Acalabrutinib (Calquence), 100 mg, oral tablets |
Sponsor | AstraZeneca Canada Inc. |
Indication | Acalabrutinib in combination with bendamustine and rituximab for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous stem cell transplant |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review and Project ORBIS (type A) |
NOC date | June 24, 2025 |
Recommended dose | 100 mg twice daily |
MCL = mantle cell lymphoma; NOC = Notice of Compliance.
Introduction
Mantle cell lymphoma (MCL) is an aggressive but rare form of non-Hodgkin lymphoma (NHL) that originates from a malignant transformation of B cells in the mantle zone of the lymph node. In 2024, there were an estimated 11,700 new cases of NHL diagnosed in Canada,1 of which 585 to 820 could be attributed to new MCL diagnoses.1,2 The overall 10-year prevalence of NHL is 141.0 cases per 100,000 persons, of which 7.1 to 9.9 cases per 100,000 would be attributed to MCL.3 MCL occurs 3 times more often in males than in females, and the median age at diagnosis is 67.5 years.4-7 Patients with MCL have poor survival: a 5-year survival of 65.9% compared with 73.3% for patients with any NHL.8
Approximately 90% of patients with MCL are classified as having aggressive disease, which requires upfront treatment.9-13 Patients with MCL often present with disseminated lymphadenopathy, splenomegaly, and bone marrow infiltration.4,5,14 Other common symptoms of MCL include B symptoms (weight loss, unexplained fever, night sweats), loss of appetite, nausea and/or vomiting, indigestion, abdominal pain, and bloating.15-17
Because the intent of current treatments for MCL is not curative, the main goal is to prolong survival and delay progression while minimizing toxicity, improving quality of life, and reducing the burden of the disease on both patients and their caregivers, according to the clinical experts consulted by Canada’s Drug Agency (CDA-AMC). Treatment pathways for MCL are generally based on suitability for autologous stem cell transplant (ASCT). Generally, patient eligibility for ASCT is limited by older age, declined physical function, comorbidities, and functional status, as assessed by the treating physician. An estimated 53.6% of patients with MCL are considered ineligible for ASCT, according to a study conducted in Ontario.18 According to clinical experts and input from clinician groups, the current standard of care for patients with MCL who are ineligible for ASCT is bendamustine plus rituximab (BR), followed by rituximab maintenance therapy every 3 months for up to 2 years. According to the clinical experts, for a small group of patients (approximately 5% to 10%) who have inconclusive disease pathology or who cannot tolerate intensive therapies, regimens other than BR (e.g., rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone [R-CHOP] or rituximab, cyclophosphamide, vincristine, and prednisone [R-CVP]) would be used. The clinical experts and clinician groups indicated that BR is the preferred option for patients with MCL who are ineligible for ASCT because it is associated with prolonged progression-free survival (PFS) and with less toxicity than R-CHOP. While ibrutinib in combination with BR is another potential treatment option, it does not have a reimbursement recommendation from CDA-AMC and has not been funded for the patient population under review by the jurisdictions. For patients who experience relapse after the initial therapy, the second-line therapy would often be monotherapy with a Bruton tyrosine kinase inhibitor (BTKi), such as ibrutinib, according to input from the clinical experts and clinician groups.
Acalabrutinib has been approved by Health Canada for use in combination with BR for the treatment of adult patients with previously untreated MCL who are ineligible for ASCT.19 The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of acalabrutinib 100 mg oral tablets in the treatment of MCL in adults who are ineligible for ASCT.
Acalabrutinib was previously reviewed by CADTH for chronic lymphocytic leukemia in the untreated and advanced or metastatic settings.20,21
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the CDA-AMC call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
CDA-AMC received input from 1 patient group, Lymphoma Canada, which conducted an anonymous patient survey between January 31 and April 6, 2025, shared via email and social media by Lymphoma Canada (102 responses from patients with MCL). The review team notes that the eligibility of these patients for ASCT and the advancement of their disease (newly diagnosed versus recurrent) are unknown, and thus their alignment with the current target population is unclear.
At the time of diagnosis of MCL, the respondents to the survey noted that they often experienced significant physical symptoms such as fatigue and/or lack of energy, abdominal issues, and enlarged lymph nodes. They also faced psychosocial impacts such as anxiety, stress, and fear of disease progression, which affected the emotional well-being of the patients and their families.
The results of the survey highlighted that patients with MCL often require immediate treatment (i.e., ASCT) and multiple lines of therapy. Common side effects with these treatments include fatigue, nausea, and hair loss, which have had negative impacts on the patients’ quality of life and were considered a significant issue by the survey respondents. Also, access to treatment can be challenging due to location and financial implications, impacting the survey respondents’ overall satisfaction and quality of life.
The surveyed patients prioritized longer disease remission, survival, improved quality of life, symptom control, and normalized blood counts in MCL treatments. They were willing to tolerate manageable side effects and emphasized the need for more therapy options to enhance their treatment outcomes and quality of life. Most surveyed patients believe it is very important to have choices in their treatment decision and a variety of treatment options to choose from.
From the survey responses, 8 patients indicated they had been treated with acalabrutinib in combination with BR with no prior treatment. These patients resided in Canada (5 patients) and the US (3 patients). These patients reported side effects with treatment with acalabrutinib plus BR such as fatigue, diarrhea, and neutropenia. Despite these issues, most patients rated their experience of treatment with acalabrutinib plus BR positively and would recommend the treatment to other patients with MCL.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts indicated that there is a significant unmet need for improved strategies for treating patients with MCL who are ineligible for ASCT. The treatment would ideally improve PFS without the addition of significant toxicity to an already older population at risk of complications. However, the clinical experts also highlighted the increased toxicity with acalabrutinib plus BR (e.g., diarrhea, infections, arrythmias) and noted that the overall survival (OS) was similar between treatment groups in the ECHO trial. The clinical experts identified patients who are most likely to respond to treatment with acalabrutinib would be aligned with the ECHO trial inclusion criteria. The clinical experts suggested that other meaningful treatment responses include complete response (CR) or partial response (PR) — assessed through blood work, physical exams, and end-of-treatment scans like CT or PET-CT — and improved quality of life. The clinical experts indicated that the most important factors in deciding to discontinue treatment with acalabrutinib include disease progression or unacceptable toxicity from acalabrutinib. The clinical experts indicated that acalabrutinib should be prescribed and monitored by a specialist in hematology and/or oncology. The clinical experts indicated that the appropriate setting for treatment with acalabrutinib plus BR would typically be the outpatient setting.
Clinician Group Input
Clinician group input for this review was received from 3 clinician groups: the Lymphoma Canada Scientific Advisory Board (Lymphoma Canada), the Leukemia and Lymphoma Society of Canada (LLSC) Pharmacist Network, and the Ontario Health (Cancer Care Ontario) (OH [CCO]) Hematology Cancer Drug Advisory Committee (Hem DAC). A total of 7 clinicians (4 from Lymphoma Canada,1 from LLSC Pharmacist Network, and 2 from OH [CCO] Hem DAC) provided input for this submission.
Input from the clinician groups aligned with that of the clinical experts consulted for this review with regard to treatment goals, the unmet needs of this patient population, assessment of treatment response, the drug’s place in therapy, decisions on discontinuing treatment, which specialists should manage these patients, and where patients should be treated with acalabrutinib. Clinicians from the OH (CCO) Hem DAC and the LLSC Pharmacist Network noted that BTKi therapy may be less suitable for patients at higher risk of bleeding disorders (e.g., cardiovascular bleeding) because BTKis can interfere with platelet function, increasing the risk of bleeding complications. Additionally, clinicians from the LLSC Pharmacist Network indicated that treatment with acalabrutinib would be least suitable for patients with uncontrolled infections or those on medications that may interact with acalabrutinib. Clinicians from the LLSC Pharmacist Network highlighted that when prescribing acalabrutinib, it is crucial to consider polypharmacy and to thoroughly review the patient’s current medications to manage potential interactions and minimize the risk of bleeding complications. Input from the LLSC Pharmacist Network suggested that treatment response is typically assessed every 2 to 3 months, and clinicians from Lymphoma Canada suggested serial imaging to assess treatment response could be performed at infrequent intervals (i.e., every 6 months). Input from the LLSC Pharmacist Network noted that when considering treatment sequencing, it is crucial to recognize that combining therapies with different mechanisms of action may exhaust multiple lines of treatment at once, especially in older patients with comorbidities who are ineligible for transplant, highlighting the need for careful patient selection for treatment with acalabrutinib due to a notable attrition rate following frontline treatment.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a recommendation for acalabrutinib: consideration for initiation of therapy, considerations for prescribing of therapy, and generalizability. The clinical experts consulted for the purpose of this review provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One ongoing phase III, double-blind, placebo-controlled randomized controlled trial (RCT) (ECHO trial; N = 598) evaluating the efficacy and safety of acalabrutinib plus BR compared to placebo plus BR in adult patients with previously untreated MCL who are ineligible for ASCT was included in the sponsor-submitted systematic review. The ECHO trial was not designed to demonstrate the efficacy and safety of acalabrutinib in induction (combined with BR) and maintenance (combined with rituximab) phases separately. The primary end point was PFS assessed by independent review committee (IRC) using the Lugano classification. Key secondary end points included overall response rate (ORR), assessed by IRC, and OS. Secondary end points outside the statistical hierarchy included PFS and ORR (investigator assessed), duration of response (DOR), health-related quality of life (HRQoL), and safety end points. Patients were randomized in a 1:1 ratio to receive either acalabrutinib plus BR (n = 299) or placebo plus BR (n = 299). Randomization was conducted using an interactive voice and/or web response system and stratified by geographic region (North America, Western Europe, other) and by simplified Mantle Cell Lymphoma International Prognostic Index (MIPI) score (low risk [0 to 3], intermediate risk [4 to 5], high risk [6 to 11]). Eligible patients were recruited in 189 study centres in 26 countries or regions, including 6 sites in Canada that enrolled a total of 25 patients.
Demographic and disease characteristics were well balanced between the treatment groups. The median age of all patients was 71.0 years (range, 65 to 86 years). Most patients were male (70.7%; female: 29.3%) and most were white (78.3%). The trial also included patients self-reporting their ethnicity as Asian, American Indian or Alaska Native, Black or African American, and multiple. Nearly half the patients (49.5%) had an Eastern Cooperative Oncology Group (ECOG) Performance Status score of 0. All patients (100%) had histologically documented MCL, and close to half the patients (42.3%) had a simplified MIPI score of 4 to 5 (intermediate risk). The most reported MCL type was classic (80.4%). Generally, the baseline disease history was balanced across treatment groups, ████████ ████████ ███████ was the most reported medical history in patients in the ECHO trial.
Efficacy Results
Only those efficacy outcomes identified as important for this review are reported. Efficacy and safety data were evaluated at a planned interim analysis, with a data cut-off date of February 15, 2024. The median duration of follow-up was 46.1 months in the acalabrutinib plus BR group and 44.4 months in the placebo plus BR group.
Progression-Free Survival
At the interim analysis, 110 patients (36.8%) in the acalabrutinib plus BR group and 137 patients (45.8%) in the placebo plus BR group experienced a PFS event. In the acalabrutinib plus BR group, 57 patients (19.1%) had disease progression based on IRC assessment and 53 (17.7%) died. In the placebo plus BR group, 99 patients (33.1%) had disease progression and 38 (12.7%) died. The median PFS was 66.4 months (95% confidence interval [CI], 55.1 months to not estimable) in the acalabrutinib plus BR group and 49.6 months (95% CI, 36.0 to 64.1 months) in the placebo plus BR group. There was a statistically significant improvement in PFS in the acalabrutinib plus BR group compared with the placebo plus BR group (hazard ratio [HR] = 0.73; 95% CI, 0.57 to 0.94; P = 0.0160). The Kaplan-Meier (KM) estimate of PFS probability at 48 months was █████ (95% CI, █████ to █████) for the acalabrutinib plus BR group and █████ (95% CI, █████ to █████) for the placebo plus BR group (Figure 3); the between-group difference was ████ (95% CI, ████ to █████). Similar results were observed in the analysis of PFS based on investigator assessment (HR = 0.68; 95% CI, 0.53 to 0.88; nominal P = 0.0028). The PFS results were consistent across most prespecified and additional sensitivity analyses and subgroups except for males (n = 425; HR = 0.91; 95% CI, 0.68 to 1.21) and females (n = 175; HR = 0.34; 95% CI, 0.19 to 0.58) (refer to Figure 5 in Appendix 1). The clinical experts consulted for this review advised that the treatment effects observed for the subgroup analysis by sex were likely attributable to chance.
Overall Survival
At the interim analysis, 97 patients (32.4%) in the acalabrutinib plus BR group and 106 patients (35.5%) in the placebo plus BR group had died. The median OS was not estimable at the time of the interim analysis for either treatment group, and there was no difference in the risk of death between acalabrutinib plus BR and placebo plus BR (HR = 0.86; 95% CI, 0.65 to 1.13; P = 0.2743). The KM estimate of OS probability at 48 months was █████ (95% CI, █████ to █████) for the acalabrutinib plus BR group and █████ (95% CI, █████ to █████) for the placebo plus BR group; the between-group difference was ████ (95% CI, ████ to █████). This analysis was not powered to detect a statistically significant difference in OS. The type I error rate in the OS analysis was not controlled for multiple comparisons, and the results should be considered as supportive evidence with limited interpretability.
Health-Related Quality of Life
At cycle 48 day 1 (approximately 48 months of treatment), ██ of 299 patients (█████) and ██ of 299 patients (█████) contributed to the analysis of Functional Assessment of Cancer Therapy–Lymphoma (FACT-Lym) total score. Patients in the acalabrutinib plus BR group reported an estimated least squares mean increase (improvement) from baseline in the FACT-Lym total score of ███ (standard error [SE] = ███) compared to ███ points (SE = ███) in patients in the placebo plus BR group. The between-group difference was ████ points (95% CI, ████ to ███; nominal P = █████). The type I error rate in the FACT-Lym analysis was not controlled for multiple comparisons, and the results should be considered as supportive evidence.
The results for the DOR for CR or PR and for EQ-5D-5L are reported in the Efficacy Outcomes section of this report.
Harms Results
At the time of the interim analysis, 296 of 297 patients (99.7%) in the acalabrutinib plus BR group and 294 of 297 patients (99.0%) in the placebo plus BR group had experienced at least 1 treatment-emergent adverse event (TEAE), with nausea being the most reported TEAE (42.8% versus 37.7% for the acalabrutinib plus BR group versus the placebo plus BR group). A total of 205 patients (69.0%) in the acalabrutinib plus BR group and 184 patients (62.0%) in the placebo plus BR group reported at least 1 serious adverse event (SAE). TEAEs leading to discontinuation of the study treatment were reported by 127 patients (42.8%) in the acalabrutinib plus BR group and 92 patients (31.0%) in the placebo plus BR group. The most common TEAE that caused treatment discontinuation was COVID-19 (4.7% versus 3.0%). Ninety-seven of the 299 patients (32.4%) in the acalabrutinib plus BR group and 106 of the 299 patients (35.5%) in the placebo plus BR group died during the study, including the crossover period. COVID-19 pneumonia was the most reported AE leading to death (5.1% versus 3.4% in the acalabrutinib plus BR group versus the placebo plus BR group). The clinical experts consulted by CDA-AMC identified the following AEs as notable harms for treatment with acalabrutinib: infections and infestations (78.1% versus 70.0% in the acalabrutinib plus BR group versus the placebo plus BR group), atrial fibrillation and/or flutter (6.7% versus 4.4%), and ventricular arrythmias (2.4% versus 2.4%).
Critical Appraisal
As the ECHO trial is ongoing, results were only available from the interim analysis for this review. The upper bound of the 95% CI for the median PFS in the acalabrutinib plus BR group was not estimable, and the median OS was not reached in either treatment group. The review team notes that the early reporting of the study results may lead to an overestimation of the treatment effect.22 Given that PFS is a surrogate outcome for OS, it remains uncertain whether these benefits will translate into an improvement in OS for this trial because the OS data were not interpretable at the time of the interim analysis. Furthermore, OS data were confounded by substantial crossover of patients from the placebo plus BR group to the acalabrutinib plus BR group upon disease progression. The study was not powered to detect a statistically significant difference in OS; moreover, DOR and HRQoL were not part of the statistical testing strategy and, thus, were not adjusted for multiple testing, limiting the credible conclusions that can be drawn from these results. There was a notable attrition rate observed at cycle 48 day 1 in the analysis of HRQoL measurements (i.e., FACT-Lym and EQ-5D-5L), with only approximately ███ of patients contributing to the analysis. Moreover, more patients in the acalabrutinib plus BR group (███) provided HRQoL data than in the placebo plus BR group (███). Withdrawal by patients was a common reason for study discontinuation; this could introduce bias because the characteristics of the patients who remained in the study may differ from those who did not. Generally, the clinical experts commented that the eligibility criteria of the ECHO trial were standard for clinical trials but were stricter than clinical practice in treating patients with MCL, indicating that the ECHO trial included a healthier patient population that may not be reflective of all patients with MCL who are ineligible for ASCT in clinical practice.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal ECHO trial identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.23,24 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The reference points for the certainty of evidence assessment for PFS, OS, HRQoL (measured using the FACT-Lym total score), and harms were set according to the presence of an important effect based on the established minimal important difference or thresholds agreed on by the clinical experts consulted for this review.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
PFS
OS
HRQoL (FACT-Lym)
safety.
Table 2: Summary of Findings for Acalabrutinib Plus BR vs. Placebo Plus BR for Patients With Untreated MCL
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo + BR | Acalabrutinib + BR | Difference | |||||
PFS | |||||||
Probability of being alive and progression-free at 48 months Follow-up (median): Acalabrutinib plus BR: 46.1 months Placebo plus BR: 44.4 months | 598 (1 RCT) | HR = 0.73 (0.57 to 0.94) | ███ per 1,000 | ███ per 1,000 (███ to ██ per 1,000) | ██ per 1,000 (| to ███ per 1,000) | Moderatea | Acalabrutinib plus BR likely results in a clinically important increase in the probability of being alive and progression-free at 48 months when compared with placebo plus BR. |
OS | |||||||
Probability of being alive at 48 months Follow-up (median): Acalabrutinib plus BR: 46.1 months Placebo plus BR: 44.4 months | 598 (1 RCT) | HR = 0.86 (0.65 to 1.13) | ███ per 1,000 | ███ per 1,000 (███ to ███ per 1,000) | ██ per 1,000 (███ ████ to ████ ████ per 1,000) | Moderateb,c | Acalabrutinib plus BR likely results in little to no clinically important difference in the probability of being alive at 48 months compared with placebo plus BR. |
HRQoL | |||||||
Least squares mean change from baseline in FACT-Lym total score at cycle 48 day 1 (0 [worse] to 168 [better]) Follow-up (median): Acalabrutinib plus BR: 46.1 months Placebo plus BR: 44.4 months | ███ (1 RCT) | NR | ███ ████ | ███ ████ ████ | ███ ████ (███ ████ to ███ ████) | Lowc,d | Acalabrutinib plus BR may result in little to no clinically important improvement in FACT-Lym total score at cycle 48 day 1 compared with placebo plus BR. |
Harms | |||||||
Incidence of infections and infestations Follow-up (median): NR | 594 (1 RCT) | NR | 710 per 1,000 | 781 per 1,000 (NR) | ██ █████ per 1,000 (███ to ███████ per 1,000) | Moderatec,e | Acalabrutinib plus BR likely results in little to no clinically important difference in the incidence of infections and infestations compared with placebo plus BR. |
Incidence of ventricular arrythmias Follow-up (median): NR | 594 (1 RCT) | NR | 24 per 1,000 | 24 per 1,000 | ██ per 1,000 (████████ to ██ ████ per 1,000) | Lowc,f | Acalabrutinib plus BR may result in little to no clinically important difference in the incidence of ventricular arrythmias compared with placebo plus BR. |
Incidence of atrial fibrillation and/or flutter Follow-up (median): NR | 594 (1 RCT) | NR | 44 per 1,000 | 67 per 1,000 | ██ █████ per 1,000 (██ █████ to ██ ████ per 1,000) | Moderatec,g | Acalabrutinib plus BR likely results in little to no clinically important difference in the incidence of atrial fibrillation and/or flutter compared with placebo plus BR. |
BR = bendamustine plus rituximab; CI = confidence interval; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HR = hazard ratio; HRQoL = health-related quality of life; MCL = mantle cell lymphoma; MID = minimal important difference; NR = not reported; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. There is no established between-group MID for PFS at 48 months, but the clinical experts suggested that a 7% difference between groups in the probabilities of PFS at 48 months could be considered a threshold of minimal clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested a minimal clinically important difference between the 2 groups, while the lower bound of the 95% CI suggested no clinically important difference for acalabrutinib plus BR vs. placebo plus BR based on this threshold. Additionally, PFS results were based on an interim analysis, and the treatment effect may be overestimated. Although PFS is a widely used surrogate outcome for OS in oncology trials, the correlation between PFS and OS is not well established in MCL.
bRated down 1 level for serious imprecision. There is no established between-group MID for OS at 48 months, but the clinical experts suggested that a 5% difference between groups in the probabilities of OS at 48 months could be considered a threshold of minimal clinical importance. The point estimate and the lower bound of the 95% CI for the between-group difference suggested no minimal clinically important difference between the 2 groups, while the upper bound of the 95% CI suggested a clinically important difference for acalabrutinib plus BR vs. placebo plus BR based on this threshold. Additionally, OS results were based on an interim analysis, and the treatment effect may be overestimated. The ECHO trial was not powered to evaluate OS. At the time of the data cut-off, data maturity was 34%. Given the trial design, the allowed crossover, and the data immaturity, the interpretability of these results is limited.
cThe statistical testing for this outcome was not adjusted for type I error rate for multiple comparisons in the trial and should be considered as supportive evidence.
dRated down 1 level for serious risk of bias due to missing data because the proportion of patients available for assessment diminished substantively over time. Rated down 1 level for serious imprecision. There is no established MID for between-group difference for FACT-Lym total score, but the estimated MID for the change from baseline is a 7-point increase. The point estimate and the upper bound of the 95% CI for the between-group difference suggested no clinically important increase, while the lower bound of the 95% CI suggested a clinically important difference between treatment groups based on a 7-point threshold, identified in the literature.
eRated down 1 level for serious imprecision. There is no established between-group MID for the incidence of infections and infestations, but the clinical experts considered that a 10% difference between groups at 48 months in the incidence of infections and infestations could be considered a threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested no clinically important difference, while the lower bound of the 95% CI suggested a clinically important difference between treatment groups based on a 10% threshold.
fRated down 2 levels for very serious imprecision. There is no established between-group MID for the incidence of ventricular arrythmias, but the clinical experts considered that a 1% difference between groups in the incidence of ventricular arrythmias could be considered a threshold of clinical importance. The point estimate for the between-group difference suggested no clinically important difference between the 2 groups, while the lower and upper bounds of the 95% CI suggested a clinically important difference in the incidence of ventricular arrythmias for acalabrutinib plus BR vs. placebo plus BR based on a 1% threshold, indicating a possible clinically important benefit and harm with acalabrutinib plus BR compared to placebo plus BR. Additionally, the incidence of ventricular arrythmias was based on a relatively low number of events in both treatment groups.
gRated down 1 level for serious imprecision. There is no established between-group MID for the incidence of atrial fibrillation and/or flutter, but the clinical experts considered that a 5% difference between groups in the incidence of atrial fibrillation and/or flutter could be considered a threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested no clinically important difference, while the lower bound of the 95% CI suggested a clinically important difference between treatment groups based on a 5% threshold.
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparisons
No indirect comparisons were submitted for this review.
Studies Addressing Gaps in the Evidence From the Systematic Review
The ECHO trial initially used acalabrutinib 100 mg capsules. As of February 27, 2023, Health Canada approved a 100 mg acalabrutinib maleate salt oral tablet formulation, which is now marketed to replace the capsule format; the capsule has not been supplied in Canada since July 31, 2024. The tablets reduce the impact of acid-reducing drugs (e.g., proton pump inhibitors) on acalabrutinib. The dosing is identical for the formulations, and the tablets are smaller in volume. The sponsor submitted 2 phase I, open-label, randomized, crossover studies (D8220C00018 and D8223C00013 trials) to assess the bioequivalence of the acalabrutinib maleate film-coated tablet and the acalabrutinib capsule. The results from the 2 studies concluded that acalabrutinib tablets and capsules are bioequivalent, indicating that the same efficacy and safety profile can be expected with the same dosing strength and schedule.27,28 Patients are able to co-administer tablets with acid-reducing drugs such as proton pump inhibitors, and this removes the need to stagger dosing with H2 receptor antagonists and antacids. The CDA-AMC review team considers the D8220C00018 and D8223C00013 trials out of scope for this review because the patients enrolled in the 2 trials were healthy patients without MCL. Therefore, the CDA-AMC review team acknowledges these studies but considers that they do not address gaps in the systematic review evidence for this review.
Conclusions
One ongoing phase III, double-blind, placebo-controlled RCT (ECHO trial) comparing acalabrutinib plus BR to placebo plus BR in adult patients with previously untreated MCL who are ineligible for ASCT was included in this review. The results from the ECHO trial demonstrated that the addition of acalabrutinib to BR likely results in a clinically important benefit in PFS. However, there was moderate certainty that acalabrutinib plus BR results in little to no clinically important difference in OS because the median OS was not reached in either study group in the interim analysis, and there were other limitations with the interpretation of the OS data, including crossover, lack of long-term follow-up, and an absence of adjustments for multiple comparisons. The results suggest that HRQoL may not be affected by the addition of acalabrutinib to BR because there was low certainty that acalabrutinib plus BR result in little to no clinically important difference in the FACT-Lym total score compared with placebo plus BR.
Compared with placebo plus BR, there is low to moderate certainty evidence that acalabrutinib plus BR results in little to no clinically important difference in the incidence of infections and infestations and of atrial fibrillation and/or flutter, and it may result in little to no clinically important difference in the incidence of ventricular arrythmias. Overall, no new safety signals were identified in the ECHO trial, and the observed safety profile of acalabrutinib plus BR is as expected but with added toxicities over standard treatment, according to feedback from the clinical experts.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the benefits and harms of acalabrutinib (100 mg, oral tablets) plus BR in the treatment of untreated MCL in patients who are ineligible for ASCT.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the review team.
MCL is an aggressive but rare form of NHL that originates from a malignant transformation of B cells in the mantle zone of the lymph node driven by the chromosomal translocation t(11;14)(q13;q32), resulting in an overexpression of cyclin D1 mRNA.2,29,30 MCL is a heterogeneous disease in terms of patient population, prognostic factors, disease stages, tumour burden, and molecular makeup. The majority of people with MCL are classified as having aggressive disease (about 90% of diagnoses); however, approximately 10% of people with MCL are classified as having indolent disease, which can transform to aggressive disease via mutations.9-13 MCL is commonly staged using the Lugano classification (an updated form of the Ann Arbor classification).31 Approximately 10% to 15% of people are diagnosed with stage I to II disease (localized disease) and have a better prognosis than those diagnosed at more advanced stages.17 Presentation at clinical stage III or IV (intermediate or advanced with extranodal spread) is more common and usually includes poor prognostic features (blastoid morphology, high Ki-67 proliferation index, and the presence of TP53 alterations) and a high tumour burden.5,17 People with MCL often present with disseminated lymphadenopathy, splenomegaly, and bone marrow infiltration.4,5,14 Other common symptoms of MCL include B symptoms (weight loss [> 10% of body weight in < 6 months], unexplained fever, and night sweats), loss of appetite, nausea and/or vomiting, indigestion, abdominal pain, and bloating.15-17 To establish a diagnosis of MCL, a tissue biopsy is performed to identify the morphology and molecular phenotype of the tumour cells. The standard diagnostic test to diagnose pathologically confirmed MCL includes testing for chromosome translocation t(11;14)(q13;q32) and/or overexpression of cyclin D1, with other relevant markers (e.g., CD5, CD19, CD20, and PAX5); this was confirmed to be the standard diagnostic process in Canada by the clinical experts consulted by CDA-AMC. Patients also typically undergo imaging with CT scans with contrast or PET-CT.32 As a rare form of NHL, MCL accounts for approximately 5% to 7% of all NHLs.2,5,33-35 In 2024, an estimated 11,700 new cases of NHL were diagnosed in Canada,1 of which 585 to 820 could be attributed to new MCL diagnoses.1,2 The overall 10-year prevalence of NHL is 141.0 cases per 100,000 persons, of which 7.1 to 9.9 cases per 100,000 would be MCL.3 MCL occurs 3 times more often in males than in females, and the median age at diagnosis is 67.5 years.4-7 Patients with MCL have poor survival: a 5-year survival of 65.9%, compared with 73.3% for patients with any NHL.8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Most patients with MCL require treatment following diagnosis due to the aggressive nature of the disease. Treatment pathways for MCL are generally based on suitability for ASCT, which is determined by oncologists on an individual patient basis, considering factors such as patient choice, younger age (generally < 65 years), physical fitness, frailty, and organ function. An estimated 53.6% of patients with MCL are considered ineligible for ASCT, according to a study conducted in Ontario.18 The clinical experts and clinician groups indicated that for patients with MCL who are ineligible for ASCT, the main goal of treatment is to prolong survival and PFS while minimizing toxicity from treatment in older patients, improving quality of life, and reducing the burden of disease on both patients and their caregivers. According to the clinical experts and the input from clinician groups, the current standard of care for patients with MCL who are ineligible for ASCT is BR, followed by rituximab maintenance therapy every 3 months for up to 2 years. For a small group of patients (approximately 5% to 10%) who have inconclusive disease pathology or who cannot tolerate intensive therapies, regimens other than BR (e.g., R-CHOP and R-CVP) would be used. The clinician groups indicated that a combination of bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone is another option for patients with MCL who are ineligible for ASCT, but the use of this regimen may be limited due to funding constraints. Generally, the clinical experts and clinician groups indicated that BR is the preferred option for patients with MCL who are ineligible for ASCT because it is associated with prolonged PFS and with less toxicity than R-CHOP. While ibrutinib in combination with BR is another potential treatment option, it does not have a reimbursement recommendation from CDA-AMC and was not funded for the patient population under review by the jurisdictions. For patients who experience relapse after the initial therapy, the second-line therapy would often be a BTKi, such as ibrutinib, according to input from the clinical experts and clinician groups.
Drug Under Review
The key characteristics of acalabrutinib are summarized in Table 3, along with those of other treatments available for patients with previously untreated MCL for whom ASCT is unsuitable.
Acalabrutinib is a second-generation, selective, covalent inhibitor of Bruton tyrosine kinase (BTK), which is a signalling molecule of the B-cell antigen receptor and cytokine receptor pathways; in B cells, BTK signalling results in survival and proliferation and is required for cellular adhesion, trafficking, and chemotaxis.26 Acalabrutinib forms a covalent bond with Cys481 in the BTK adenosine triphosphate pocket, permanently inactivating the enzyme and resulting in the inhibition of proliferation and survival signals in malignant B cells. Acalabrutinib is available in 100 mg oral tablets. The recommended dose of acalabrutinib for patients with MCL is 100 mg twice a day.19
Acalabrutinib is approved by Health Canada through the standard review and Project ORBIS (type A) pathways. The indication is for acalabrutinib in combination with BR for the treatment of adult patients with previously untreated MCL who are ineligible for ASCT.19 On January 16, 2025, the FDA approved acalabrutinib with BR for adults with previously untreated MCL who are ineligible for autologous hematopoietic stem cell transplant.36 The sponsor is seeking reimbursement for acalabrutinib as per the Health Canada indication.
Acalabrutinib was previously reviewed by CDA-AMC for chronic lymphocytic leukemia in the untreated and advanced or metastatic settings.20,21
Table 3: Key Characteristics of Acalabrutinib, Bendamustine, and Rituximab
Characteristic | Acalabrutinib | Bendamustine | Rituximab |
|---|---|---|---|
Mechanism of action | Acalabrutinib is a selective inhibitor of BTK, crucial for B-cell survival and proliferation, with minimal off-target effects. | Bendamustine is an alkylating drug that forms DNA crosslinks leading to cell death and is effective against both quiescent and dividing cells. | Rituximab binds specifically to the CD20 antigen on B lymphocytes, which is crucial for cell cycle initiation and differentiation, and is highly expressed in B-cell non-Hodgkin lymphomas. |
Indication | In combination with bendamustine and rituximab for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous stem cell transplant. | The combination regimen of BR is not specifically approved by Health Canada but is a well-accepted regimen for the first-line treatment of patients with MCL. | Rituximab given in combination with bendamustine or as part of R-CHOP is not specifically approved by Health Canada but is a well-accepted regimen for the first-line treatment of patients with MCL. |
Route of administration | Oral | IV | IV |
Recommended dose | When used in combination with BR: Start acalabrutinib 100 mg twice daily at cycle 1 (each cycle is 28 days). Start BR at cycle 1 for 6 cycles. For patients experiencing a response, treatment with rituximab continues for a maximum of an additional 12 doses every other cycle. Treatment with acalabrutinib should continue until disease progression or unacceptable toxicity. | Dosing regimen used in clinical practice, as sourced from Cancer Care Ontario Drug Formulary and BC Cancer protocols:37,38 BR are given every 28 days for a maximum of 6 cycles as follows: Rituximab: 375 mg/m2 by IV infusion on day 1. If the initial IV infusion is tolerated (no severe reactions requiring early termination), subsequent doses can be given at a dose of 1,400 mg as a slow subcutaneous injection. Bendamustine: 90 mg/m2 by IV infusion on days 1 and 2. | Dosing regimen used in clinical practice, as sourced from Cancer Care Ontario Drug Formulary and British Columbia Cancer Agency protocols:37,39 R-CHOP regimen is given every 21 days for a maximum of 6 cycles as follows: Doxorubicin: 50 mg/m2 by IV infusion (IV push) on day 1. Vincristine: 1.4 mg/m2 by IV infusion on day 1. Cyclophosphamide: 750 mg/m2 by IV infusion on day 1. Prednisone: 45 mg/m2 by IV infusion on days 1 to 5. Rituximab: 375 mg/m2 by IV infusion on day 1 or 2. If the initial IV infusion is tolerated (no severe reactions requiring early termination), subsequent doses can be given at a dose of 1,400 mg as a slow subcutaneous injection. |
Serious adverse effects or safety issues |
|
|
|
BR = bendamustine plus rituximab; BTK = Bruton tyrosine kinase; MCL = mantle cell lymphoma; R-CHOP = rituximab, cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone.
Source: Product monographs for acalabrutinib,19 bendamustine,40 and rituximab.41 Treatment protocols from Cancer Care Ontario Drug Formulary and British Columbia Cancer Agency.37-39
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by the patient groups.
CDA-AMC received input from 1 patient group, Lymphoma Canada, which conducted an anonymous patient survey between January 31 and April 6, 2025. The survey was shared via email and social media by Lymphoma Canada. The survey received 102 responses from patients who had MCL. The review team notes that the eligibility of these patients for ASCT and the advancement of their disease (newly diagnosed versus recurrent) are unknown, and thus their alignment with the current target population is unclear.
At the time of diagnosis of MCL, the respondents to the survey noted that they had often experienced significant physical symptoms like fatigue and/or lack of energy, abdominal issues, and enlarged lymph nodes. They also faced psychosocial impacts such as anxiety, stress, and fear of disease progression, which also affected the emotional well-being of the patients and their families.
The results of the survey highlighted that patients with MCL often require immediate treatment (i.e., ASCT) and multiple lines of therapy. Common side effects with these treatments include fatigue, nausea, and hair loss, which have negative impacts on the patients’ quality of life and were considered a significant issue for the survey respondents. Also, access to treatment can be challenging due to location and financial implications, impacting the survey respondents’ overall satisfaction and quality of life.
The survey respondents prioritized longer disease remission, survival, improved quality of life, symptom control, and normalized blood counts in MCL treatments. They were willing to tolerate manageable side effects and emphasized the need for more therapy options to enhance their treatment outcomes and quality of life. Most surveyed patients believe it is very important to have choice in their treatment decision and a variety of treatment options to choose from.
From the survey responses, 8 patients indicated they had been treated with acalabrutinib in combination with BR with no prior treatment. These patients resided in Canada (5 patients) and the US (3 patients). These patients reported side effects with treatment with acalabrutinib plus BR such as fatigue, diarrhea, and neutropenia. Despite these issues, most patients rated their experience of treatment with acalabrutinib plus BR positively and would recommend the treatment to other patients with MCL.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of MCL.
Unmet Needs
The clinical experts consulted by CDA-AMC noted that the patients’ disease inevitably relapses with current treatment strategies and that there is increased toxicity when novel drugs are added to current treatments. Therefore, the clinical experts emphasized the significant unmet needs for strategies that can improve PFS in patients with MCL who are ineligible for ASCT without the addition of significant toxicity to an older patient population who are at risk of complications. One clinical expert underscored the necessity for targeted strategies for patients with high-risk disease, such as those with early progression after frontline therapy, specific genetic markers like TP53 mutation, Ki-67 greater than 30%, a high-risk simplified MIPI score, and blastoid or pleiomorphic morphology.
Place in Therapy
The clinical experts indicated that acalabrutinib in combination with BR would be a good treatment option for patients with MCL who are ineligible for ASCT in the frontline setting. The clinical experts stated that the combination of acalabrutinib and BR targets multiple pathways, which may synergistically improve the response to treatment. However, the clinical experts also highlighted the increased toxicity with acalabrutinib plus BR (diarrhea, infections, and arrythmias) and noted that the OS was similar between the 2 treatment groups in the ECHO trial. Given that patients receive acalabrutinib until disease progression or unacceptable toxicity, 1 clinical expert mentioned that some patients may prefer a fixed-duration treatment (e.g., BR followed by maintenance rituximab) with fewer clinic visits and a shorter duration of treatment than with BR alone, even if they are eligible to receive the combination with acalabrutinib. This is because a treatment-free interval could potentially improve quality of life for patients.
Patient Population
The clinical experts indicated that people with MCL would be identified with a confirmed pathologic diagnosis with a characteristic chromosomal translocation and overexpression of cyclin D1 before any treatment. Currently, there is no consistent definition for patients’ eligibility for ASCT in clinical practice; the clinical experts stated that the decision on eligibility should be left to the discretion of the treating physician because it is a personalized assessment based on age, frailty, comorbidities, and functional status. According to the clinical experts, the patients who are most likely to experience response to treatment with acalabrutinib would be generally aligned with the ECHO trial inclusion criteria. The clinical experts noted that acalabrutinib in combination with BR would likely be used in patients with high-risk disease, for cases in which current treatment regimens provide less benefit (i.e., patients with a high-risk simplified MIPI score, blastoid histology, pleomorphic histology, Ki-67 ≥ 30%, and known TP53 mutation).
Assessing the Response to Treatment
The clinical experts indicated that a CR or PR and improvement in quality of life would be a meaningful response to treatment. According to the clinical experts, in clinical practice, the standard of care to assess treatment response remains blood work, physical examination of known sites of adenopathy, and typically either CT scans or, more commonly, a PET-CT scan. The assessment is most often done after the completion of chemoimmunotherapy. The clinical experts indicated that further scans in the absence of new signs or symptoms of MCL relapse are not typically required.
Discontinuing Treatment
The clinical experts indicated that the most important factor when deciding whether to discontinue treatment with acalabrutinib is disease progression. Intolerable toxicities such as severe bleeding or recurrent infections are other common reasons for discontinuing treatment with acalabrutinib in clinical practice.
Prescribing Considerations
The clinical experts indicated that acalabrutinib should be prescribed and monitored by a specialist in hematology and/or oncology. The appropriate setting for treatment with acalabrutinib would typically be the outpatient setting. Patients would typically be seen every 2 to 3 months while receiving acalabrutinib.
Clinician Group Input
This section was prepared by the review team based on the input provided by the clinician groups.
Clinician group input for this review was received from 3 clinician groups: the Lymphoma Canada Scientific Advisory Board (Lymphoma Canada), the LLSC Pharmacist Network, and the OH (CCO) Hem DAC. A total of 7 clinicians (4 from Lymphoma Canada,1 from the LLSC Pharmacist Network, and 2 from the OH [CCO] Hem DAC) provided input for this submission.
Input from the clinician groups aligned with that of the clinical experts consulted for this review with regard to treatment goals, the unmet needs of this patient population, assessment of treatment response, the drug’s place in therapy, decisions on discontinuing treatment, which specialists should manage these patients, and where patients should be treated with acalabrutinib. Clinicians from the OH (CCO) Hem DAC and the LLSC Pharmacist Network noted that BTKi therapy may be less suitable for patients at higher risk of bleeding disorders (e.g., cardiovascular bleeding) because BTKis can interfere with platelet function, increasing the risk of bleeding complications. Additionally, clinicians from the LLSC Pharmacist Network indicated that treatment with acalabrutinib would be least suitable for patients with uncontrolled infections or those receiving medications that may interact with acalabrutinib. Clinicians from the LLSC Pharmacist Network highlighted that when prescribing acalabrutinib, it is crucial to consider polypharmacy and to thoroughly review the patient’s current medications to manage potential interactions and minimize the risk of bleeding complications. Input from the LLSC Pharmacist Network suggested that treatment response is typically assessed every 2 to 3 months, as general follow-up, to monitor this medication. Clinicians from Lymphoma Canada suggested that serial imaging to assess treatment response could be performed at infrequent intervals (i.e., every 6 months). Input from the LLSC Pharmacist Network noted that when considering treatment sequencing, it is crucial to recognize that combining therapies with different mechanisms of action may exhaust multiple lines of treatment at once, especially in older patients with comorbidities who are ineligible for ASCT, highlighting the need for careful patient selection for treatment with acalabrutinib due to a notable attrition rate following frontline treatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions from the drug programs and the corresponding responses from the clinical experts consulted by for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
BR is an appropriate comparator for the first-line treatment of transplant-ineligible mantle cell lymphoma. Other regimens sometimes used are R-CVP and R-CHOP. | This is a comment from the drug programs to inform pERC deliberations. |
Considerations for initiation of therapy | |
Patients were enrolled in the ECHO trial if they were 65 years and older. Would patients who are younger than 65 years and not eligible for transplant be considered for acalabrutinib plus BR treatment? | The clinical experts indicated that patients younger than 65 years who are ineligible for transplant may be considered for acalabrutinib plus BR treatment, although this scenario would be relatively uncommon. |
Considerations for prescribing of therapy | |
During the maintenance rituximab phase, rituximab was administered every 2 months for 12 doses along with the acalabrutinib orally twice daily. In some jurisdictions, maintenance rituximab is administered every 3 months for 8 doses. | This is a comment from the drug programs to inform pERC deliberations. |
CDA-AMC had previously recommended brexucabtagene autoleucel for the treatment of adult patients with relapsed or refractory mantle cell lymphoma after 2 or more lines of systemic therapy. Prior therapy must have included an anthracycline or bendamustine-containing chemotherapy, an anti-CD20 monoclonal antibody therapy, and a BTK inhibitor.
| The clinical experts indicated that they would support the use of brexucabtagene autoleucel as a second-line treatment if patients had been exposed to an anthracycline or bendamustine, anti-CD20 monoclonal antibody, and BTK inhibitor in the first-line therapy. The clinical experts indicated that acalabrutinib and bendamustine can be administered with rituximab biosimilar or subcutaneous rituximab. The clinical experts indicated that if 1 drug of the combination of acalabrutinib, rituximab, and bendamustine needs to be discontinued, the remaining drugs can be continued. |
Generalizability | |
For patients who commenced on BR first-line treatment and are ineligible for transplant, should acalabrutinib be added to BR at the time of funding? If so, are there a maximum number of cycles of BR that would be given before considering not adding acalabrutinib? | The clinical experts consulted by CDA-AMC stated that there are no studies directly addressing the question. Considering the PFS benefit of BTK inhibitors in second-line treatment, the clinical experts stated it is unnecessary to offer acalabrutinib to patients who have already started BR as first-line therapy. Therefore, the clinical experts suggested that funding for acalabrutinib should be restricted to patients who have not yet initiated BR therapy. The review team notes that acalabrutinib is currently not funded in the second line for mantle cell lymphoma. Patients have access to acalabrutinib in the second-line setting only through private coverage or out-of-pocket payment. A patient support program is offered by the sponsor to help patients with private insurance navigate their coverage options. |
Funding algorithm (oncology only) | |
Request the initiation of a rapid provisional funding algorithm. | This is a comment from the drug programs to inform pERC deliberations. |
Drug may change the place in therapy of drugs reimbursed in subsequent lines. | This is a comment from the drug programs to inform pERC deliberations. |
Care provision issues | |
Acalabrutinib is an oral drug, which is an enabler for implementation. However, additional pharmacy resources will be required for drug-drug interaction monitoring and dispensing. | This is a comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
OWG is concerned with the budget impact from the addition of acalabrutinib to BR. | This is a comment from the drug programs to inform pERC deliberations. |
Rituximab biosimilars and rituximab subcutaneous have confidential negotiated prices. Bendamustine has multiple generics and confidential net prices. | This is a comment from the drug programs to inform pERC deliberations. |
BR = bendamustine plus rituximab; BTK = Bruton tyrosine kinase; CDA-AMC = Canada’s Drug Agency; OWG = Oncology Working Group; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; PFS = progression-free survival; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R-CVP = rituximab, cyclophosphamide, vincristine, and prednisone.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the benefits and harm effects of acalabrutinib 100 mg oral tablets plus BR for the treatment of previously untreated MCL in adult patients who are ineligible for ASCT. The focus will be placed on comparing acalabrutinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of acalabrutinib is presented in the systematic review section, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The systematic review includes the pivotal study and/or RCT that was selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. No long-term extension studies, indirect treatment comparisons, or studies addressing gaps were included in the review.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
one ongoing phase III, double-blind, placebo-controlled RCT (ECHO trial) identified in the systematic review.
Systematic Review
Contents within this section have been informed by the materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | ECHO trial |
|---|---|
Designs and populations | |
Study design | Ongoing phase III, double-blind, placebo-controlled RCT |
Locations | 189 study centres in 26 countries or regions: Argentina, Australia, Belgium, Brazil, Canada (25 patients across 6 sites), China, Czech Republic, France, Germany, Greece, Hungary, Israel, Italy, Japan, Mexico, New Zealand, Peru, Poland, Republic of Korea, Romania, Russia, Spain, Taiwan, Ukraine, US, Vietnam |
Patient enrolment dates | Start date: May 8, 2017 End date: March 27, 2023 |
Randomized (N) | A total of 635 patients were randomized (global cohort).a In the interim analysis from February 15, 2024, 598 patients were included: 299 patients were randomized to the acalabrutinib plus BR group and 299 patients were randomized to the placebo plus BR group. |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Acalabrutinib, 100 mg capsuleg administered twice daily until disease progression or toxicity. In combination with BR:
Dosing schedule:
|
Comparator(s) |
|
Study duration | |
Screening phase | 30 days before the first administration of the study drug. |
Treatment phase | Until disease progression. |
Follow-up phase | Until disease progression; following disease progression, survival status is assessed every 3 months until death, study withdrawal, loss to follow-up, or study termination. |
Outcomes | |
Primary end point | PFS Time frame: from randomization until disease progression (per the Lugano classification, based on IRC assessment) or death from any cause. |
Secondary and exploratory end points | Key secondary
Secondary
Exploratory
Harms: Incidence of AEs, SAEs, and AEs leading to study drug dose modification or treatment discontinuation |
Publication status | |
Publications | Wang et al. (2025)42 ClinicalTrials.gov identifier: NCT02972840 |
AE = adverse event; AIHA = autoimmune hemolytic anemia; ALT = alanine aminotransferase; ANC = absolute neutrophil count; aPTT = activated partial thromboplastin time; AST = aspartate aminotransferase; BR = bendamustine plus rituximab; CMV = cytomegalovirus; CNS = central nervous system; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; INR = international normalized ratio; IRC = independent review committee; ITP = idiopathic thrombocytopenic purpura; MCL = mantle cell lymphoma; ORR = overall response rate; OS = overall survival; PCR = polymerase chain reaction; PD = progressive disease; PFS = progression-free survival; PR = partial response; RCT = randomized controlled trial; SAE = serious adverse event; TTR = time to response; ULN = upper limit of normal.
aA total of 635 patients were randomized into a global cohort, including 85 patients from a cohort from China (81 patients from mainland China + 4 patients from Taiwan). However, for the analyses presented in this document (the interim analysis from February 15, 2024), only 598 (94.2%) of the 635 patients were included because 37 patients (all from China) were not included due to having < 2 years of follow-up at the study data cut-off. As such, 598 patients were randomized 1:1 into 2 treatment groups.
bProvided they met other eligibility criteria, patients who were receiving hormonal therapy alone were allowed to enrol in the study.
cIf a patient had major surgery, they must have recovered adequately from any toxicity and/or complications from the intervention before the first dose of the study drug.
dSuch as uncontrolled or untreated symptomatic arrhythmias, congestive heart failure, or myocardial infarction within 6 months of the first dose of the study drug, or any Class 3 or 4 cardiac disease as defined by the New York Heart Association Functional Classification, or corrected QT interval > 480 msec (calculated using the Fridericia formula: QT interval / [RR interval]0.33) at screening. Patients with controlled, asymptomatic atrial fibrillation during screening were allowed to enrol in the study.
ePatients may have used topical or inhaled corticosteroids or low-dose steroids (≤ 20 mg prednisone equivalent/day for ≤ 2 weeks) as a therapy for comorbid conditions. During study participation, patients may have also received systemic (e.g., IV or oral) corticosteroids as needed for treatment-emergent comorbid conditions.
fPatients with positive anti–hepatitis B core and negative surface antigen test results needed to have a negative PCR result before randomization. Those with positive hepatitis B surface antigen or positive hepatitis B PCR test results were not included. Patients with positive hepatitis C antibody test results needed to have a negative PCR result before randomization. Those with positive hepatitis C PCR test results were not included.
gThe ECHO trial was conducted using acalabrutinib 100 mg capsules because that was the only available format at the time of the trial. On February 27, 2023, a new 100 mg tablet formulation of acalabrutinib was approved by Health Canada that is bioequivalent to the existing capsule format. The tablet formulation of acalabrutinib is currently marketed.
Source: ECHO Clinical Study Report (2024);25 ECHO study protocol.43 Details included in the table are from the sponsor’s summary of clinical evidence.26
One pivotal trial (ECHO trial) was included in the systematic review (Table 5). The ECHO trial is an ongoing phase III, double-blind, placebo-controlled RCT to evaluate the efficacy and safety of acalabrutinib plus BR compared to placebo plus BR in adult patients with previously untreated MCL who are ineligible for ASCT. The ECHO trial was not designed to demonstrate the efficacy and safety of acalabrutinib in induction (combined with BR) and maintenance (combined with rituximab) phases separately. Randomization was conducted using an interactive voice and/or web response system and stratified by geographic region (North America, Western Europe, other) and simplified MIPI score (low risk [0 to 3], intermediate risk [4 to 5], high risk [6 to 11]). Eligible patients were recruited in 189 study centres in 26 countries or regions. The 6 sites in Canada enrolled a total of 25 patients. A total of 635 patients were randomized into a global cohort. However, 598 of the 635 patients (94.2%) were included in the interim analysis (data cut-off date: February 15, 2024), which is presented in this report. Patients were randomized at a 1:1 ratio to receive either acalabrutinib plus BR (n = 299) or placebo plus BR (n = 299).
The ECHO trial consisted of 3 phases: screening, treatment, and follow-up (Figure 1). In the screening phase, patients were screened for eligibility 30 days before the first administration of the study drug. The treatment phase consisted of 28-day treatment cycles until disease progression (assessed by IRC) or unacceptable toxicity. The primary end point was PFS, assessed by IRC using the Lugano classification. The key secondary end points were ORR (assessed by IRC) and OS. Secondary end points outside the statistical hierarchy included PFS and ORR (investigator assessed), DOR, HRQoL, and safety end points. Patients in the placebo plus BR group with disease progression at any time may have been eligible to cross over to receive acalabrutinib monotherapy until disease progression or unacceptable toxicity. After disease progression, all patients were followed up with approximately every 3 months (from last visit) by clinic visit or telephone to assess survival status until death, withdrawal of consent, loss to follow-up, or study termination by the sponsor, whichever came first.
One interim analysis and 1 final analysis for PFS were planned for the study when approximately 227 and 268 IRC-assessed PFS events were observed, respectively. The study will end at the time of the final analysis, which is anticipated to occur approximately 101 months after the first patient was randomized. Because of the potential impact of COVID-19 death on the primary IRC-assessed PFS analysis (i.e., interim analysis), the data cut-off date was determined so that approximately 10% more IRC-assessed PFS events would be accrued than the 227 IRC-assessed PFS events that the protocol had prespecified for the interim analysis. This report focuses on the results from the interim analysis, with a data cut-off date of February 15, 2024. The final analysis for IRC-assessed PFS events has an anticipated data cut-off date in ████████ ████ based on sponsor correspondence.44
Figure 1: ECHO Trial Study Design
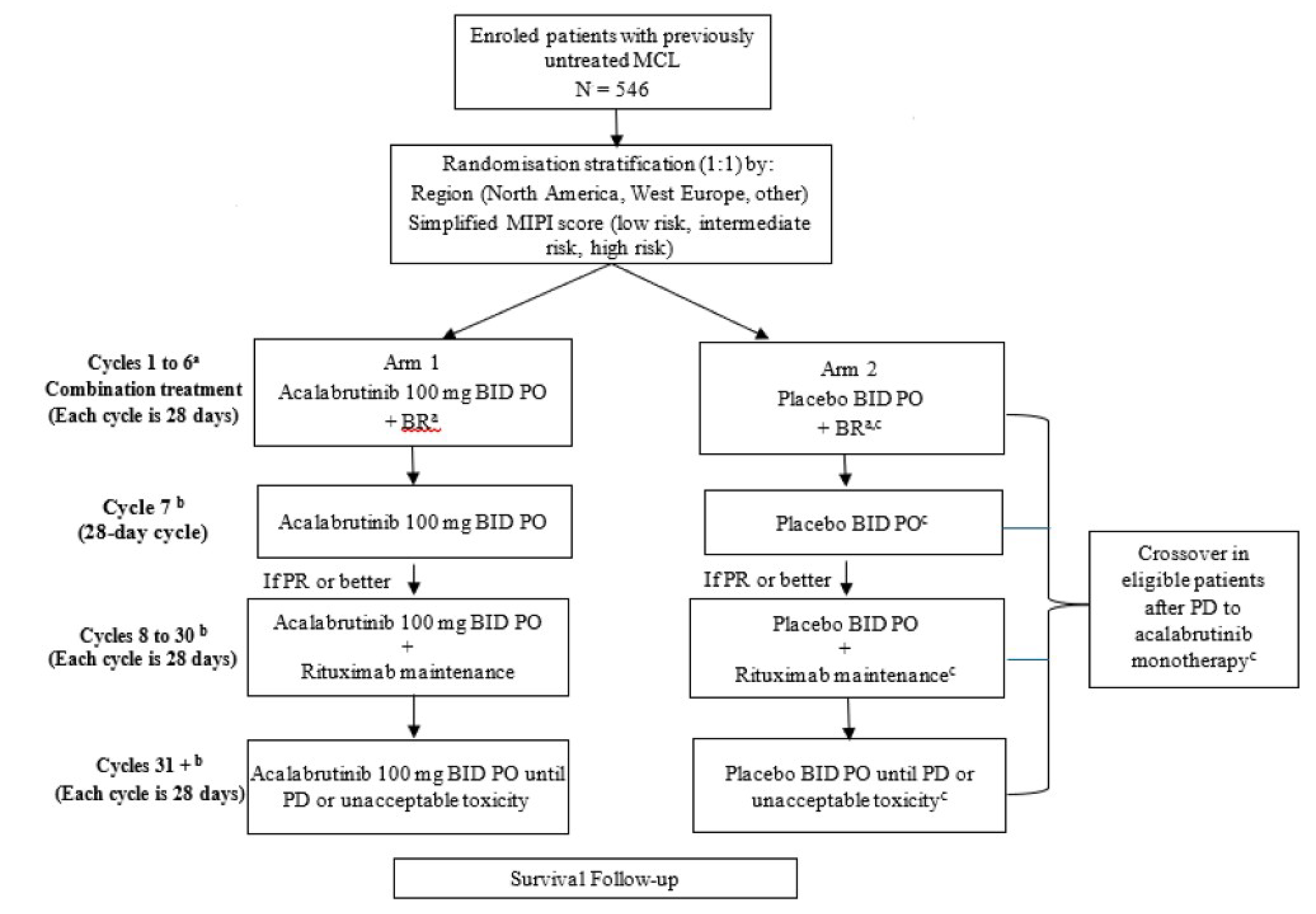
BID = twice daily; BR = bendamustine plus rituximab; MCL = mantle cell lymphoma; MIPI = Mantle Cell Lymphoma International Prognostic Index; PD = progressive disease; PO = orally; PR = partial response.
a Bendamustine 90 mg/m2 IV on days 1 and 2 plus rituximab 375 mg/m2 IV on day 1 of each 28-day cycle. Every attempt was to be made to complete 6 cycles of BR. Thus, if a dose delay of up to 28 days resulted in omission of BR for that cycle, BR may have been given for an additional cycle.
b After 6 cycles of acalabrutinib or placebo in combination with BR, patients who were tolerating treatment and whose disease was not progressing were to receive monotherapy acalabrutinib 100 mg twice a day or placebo twice a day. In addition, patients with a response (PR or greater) were to receive rituximab 375 mg/m2 on day 1 of every other cycle (starting on the next even-numbered cycle after the completion of 6 cycles of BR) for a maximum of 12 additional doses (through no later than cycle 30). Thereafter, patients continued to receive monotherapy acalabrutinib 100 mg twice a day (or last tolerated dose) or placebo twice a day until PD or unacceptable toxicity.
c Patients in the placebo plus BR arm who had PD at any time may have been eligible to cross over to receive acalabrutinib 100 mg twice a day as monotherapy until PD or unacceptable toxicity.
Source: ECHO Clinical Study Report (2024).25
Populations
Inclusion and Exclusion Criteria
Eligible patients were aged 65 years or older and had pathologically confirmed MCL with chromosome translocation t(11;14)(q13;q32), overexpressed cyclin D1, or both, and radiologically measurable disease. Patients also had to have MCL requiring treatment, no history of systemic anticancer therapies, and an ECOG Performance Status of 2 or less. At screening, MCL diagnoses were retrospectively confirmed by a central laboratory using tumour biopsies. The trial included patients with low-risk to high-risk features (e.g., high-risk simplified MIPI score, blastoid and pleomorphic variants, or Ki-67 index ≥ 30%). The trial did not include patients for whom the goal of therapy was tumour debulking before stem cell transplant.
Interventions
Patients were randomized 1:1 to receive acalabrutinib plus BR or placebo plus BR. Acalabrutinib (100 mg orally twice a day) or matching placebo was administered from cycle 1 until disease progression or unacceptable toxicity. Bendamustine (90 mg/m2 IV) was given on days 1 and 2 of each cycle for up to 6 cycles, and rituximab (375 mg/m2 IV) was given on day 1 of each cycle for 6 cycles. Each treatment cycle was 28 days. Combination treatment was to continue for up to 6 cycles, after which patients who were tolerating therapy and whose disease had not progressed could receive acalabrutinib (100 mg orally twice a day) monotherapy or placebo (twice a day) monotherapy. In addition, patients with a response of PR or better were to receive maintenance rituximab (375 mg/m2) on day 1 of every other cycle (starting on the next even-numbered cycle after the completion of 6 cycles of BR) for a maximum of 12 additional doses (through no later than cycle 30). Thereafter, patients continued to receive acalabrutinib (100 mg, or last tolerated dose, orally twice a day) monotherapy or placebo (twice a day) monotherapy until disease progression or unacceptable toxicity. Patients randomized to the placebo plus BR group who at any time during the study had disease progression assessed by the investigator and confirmed by an unblinded nonstudy team physician employed by the sponsor and who were eligible to cross over could have received treatment with acalabrutinib (100 mg orally twice a day) monotherapy until disease progression or unacceptable toxicity. Patients who are still receiving treatment at the end of the study and deriving clinical benefit from acalabrutinib treatment may be eligible to enrol in a separate safety extension study.
The ECHO trial was conducted using acalabrutinib 100 mg capsules, which was the format available at the time the trial was initiated (2017). Reimbursement is being requested for the tablet formulation of acalabrutinib, which is currently marketed and will be the format used in clinical practice. As of July 31, 2024, the capsule formulation is no longer supplied in Canada. The sponsor conducted 2 phase I, open-label, randomized, crossover studies (D8220C00018 and D8223C00013 trials) to evaluate bioequivalence between the capsule and tablet (i.e., acalabrutinib maleate salt) formulations of acalabrutinib.27,28
Patients were withdrawn from the study treatment because of disease progression, adverse events (AEs), start of alternative anticancer therapy, investigator decision, patient’s withdrawal of consent from the study, decision by the sponsor to terminate the study, patient being lost to follow-up, death, or other reasons.
Standard supportive care medications (e.g., antiemetics, antipyretics, antibiotics, transfusion of blood products) were permitted as per institutional standards. Prophylactic use of growth factors or administration in response to severe myelosuppression was permitted. During the study, a short course (e.g., ≤ 2 weeks) of high-dose corticosteroids (> 20 mg/day) was permitted for premedication to manage infusion-related reactions or to manage other inflammatory reactions. Corticosteroids to treat the underlying MCL were not allowed during the study. Any nonstudy anticancer therapies, including chemotherapy, anticancer immunotherapy, experimental therapy, or radiotherapy, were prohibited. Warfarin or equivalent vitamin K antagonists (e.g., phenprocoumon) were prohibited. Concomitant administration of drugs that are strong inhibitors or inducers of CYP3A were prohibited. Patients who required proton pump inhibitor therapy were required to switch to alternate acid-reducing drugs to participate in the study, although this is no longer a concern with the new tablet formulation.
Outcomes
A list of the efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted for this review and input from the patient and clinician groups and public drug plans. Using the same considerations, the review team selected the end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | Type of outcome | GRADE assessment |
|---|---|---|---|
Efficacy outcomes | |||
PFS by IRC | Event-driven analysis at 48 months | Primarya | Include |
OS | Event-driven analysis at 48 months | Key secondarya | Include |
DOR | Event-driven analysis at 48 months | Secondary | Do not include |
Health-related quality of life | |||
Change from baseline in EQ-5D-5L utility score | At cycle 48 day 1 (approximately 48 months of treatment) | Exploratory | Do not include |
Change from baseline in FACT-Lym total score | At cycle 48 day 1 (approximately 48 months of treatment) | Exploratory | Include |
Safety outcomes | |||
Infections and infestations | NR | Harms | Include |
Ventricular arrythmias | NR | Harms | Include |
Atrial fibrillation and/or flutter | NR | Harms | Include |
DOR = duration of response; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; GRADE = Grading of Recommendations Assessment, Development and Evaluation; IRC = independent review committee; NR = not reported; OS = overall survival; PFS = progression-free survival.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: ECHO Clinical Study Report (2024);25 ECHO study protocol.43 Details included in the table are from the sponsor’s summary of clinical evidence.26
Efficacy Outcomes
Assessments
In the ECHO trial, during treatment, CT scans were performed at week 12, then every 12 weeks until week 96 and every 24 weeks thereafter. PET-CT scans were performed at weeks 12 and 24 and then only to confirm CR. Patients who experienced confirmed CR did not need further PET-CT scans, but PET-CT could be used at the investigator’s discretion if disease progression was suspected but not proven with CT. Bone marrow aspiration and biopsy were performed at screening or before the first dose of the study drug.
Progression-Free Survival
PFS was the primary end point in the ECHO trial and was defined as the time from the date of randomization until disease progression (assessed by the IRC) or death from any cause (including COVID-19), whichever occurs first. PFS events include death or first disease progression that occurred on or before the earliest of the following: data cut-off date (February 15, 2024, for the interim analysis), subsequent start date of therapy for MCL, 2 or more consecutively missed response assessments, or study exit. Patients were censored if there was no disease progression or death at the time of data cut-off; if there was no disease progression or death before loss to follow-up, crossover, or study exit; if there was no response assessment postrandomization; if they missed 2 or more consecutive response assessments; or if they started subsequent therapy for MCL. The PFS outcome is considered clinically meaningful and important to patients and clinicians according to the clinical experts and the patient and clinician groups. In addition, PFS was used to inform the pharmacoeconomic model submitted to CDA-AMC.
Response assessments were based on the Lugano classification for NHL, which incorporates assessments via PET-CT and CT alone. Patients were considered to have disease progression if any 1 or more of the criteria were met based on the Lugano classification for NHL.31 Refer to Appendix 1 for the detailed response assessment criteria.
Overall Survival
OS was assessed as a key secondary end point in the ECHO trial and was defined as the time from the date of randomization to the date of death due to any cause, regardless of whether the patient withdrew from randomized therapy or received another therapy for MCL. Patients who were lost to follow-up immediately after randomization, not known to have died at or before the data cut-off date, or not known to have died at or before loss to follow-up or study exit were censored. OS was selected for GRADE assessment because it was considered to be clinically meaningful and important to patients and clinicians according to the clinical experts and the patient and clinician groups. In addition, OS was used to inform the pharmacoeconomic model submitted to CDA-AMC.
Duration of Response
DOR was a secondary end point in the ECHO trial and was defined as the time from the date of the first documented CR or PR until the date of the first documented disease progression (assessed by the IRC) or death due to any cause in the absence of disease progression, before starting any subsequent therapy for MCL. The definitions of CR and PR were based on the Lugano classification for NHL.31 Refer to Appendix 1 for the detailed response assessment criteria.
Change From Baseline in EQ-5D-5L
In the ECHO trial, the exploratory end point HRQoL was measured using the EQ-5D-5L. HRQoL, assessed using change from baseline in EQ-5D-5L utility score, was used to inform the pharmacoeconomic model submitted to CDA-AMC. EQ-5D-5L measures were assessed during the first week of the treatment phase (screening visit following randomization) and on day 1 of cycles 3, 5, and 8, then every 4 cycles. Refer to Table 7 for details of the EQ-5D-5L.
Change From Baseline in FACT-Lym Total Score
In the ECHO trial, FACT-Lym was an exploratory end point used to measure HRQoL. FACT-Lym was selected for GRADE assessment because it was considered to be clinically meaningful and important to patients and clinicians according to the clinical experts and the patient and clinician groups. Refer to Table 7 for details of the FACT-Lym.
Safety Outcomes
In the ECHO trial, AEs were coded using the Medical Dictionary for Regulatory Activities and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). TEAEs were defined as any event with an onset date on or after the date of the first dose of the study drug or any ongoing event that worsened in severity after the date of the first dose of the study drug and within 30 days after the date of the last dose of the study drug or the first date starting a new therapy for MCL. Notable harms in the ECHO trial included atrial fibrillation and/or flutter, ventricular arrhythmia, and infections and infestations. These outcomes were selected for GRADE assessment as they were considered important by the clinical experts consulted by CDA-AMC.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
PFS | Progression was defined according to the Lugano classification for NHL.31 PFS is not affected by crossover or confounded by later lines of therapy. | A moderate correlation between PFS and OS (R2 = 0.69; 95% CI, 0.40 to 0.91) in patients with MCL has been observed in a meta-analysis.45 However, this relationship was based on studies with chemotherapy, not targeted therapy, and is therefore uncertain in the context of acalabrutinib.46,47 | No established MID was reported in patients with untreated MCL. |
EQ-5D-5L | The EQ-5D-5L is a 2-part, generic instrument used to describe and value health status.48,49 It assesses health status based on 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression.48,49 Each dimension has 5 increasing levels of severity (no problems, slight problems, moderate problems, severe problems, and unable to or extreme problems).48,49 There is also a visual analogue scale rating “health today,” with anchors ranging from 0 (worst imaginable health state) to 100 (best imaginable health state). The scores for the 5 dimensions are used to compute a single utility score (ranging from 0 to 1) to represent the general health status of the individual, with higher scores representing higher health utility. | Validity: Not estimated in patients with untreated MCL. Reliability: Not estimated in patients with untreated MCL. Responsiveness: Not estimated in patients with untreated MCL. | No established MID was reported in patients with untreated MCL. In patients with various cancers, the estimated MID ranged from 0.01 to 0.09 for improvement and from –0.04 to –0.03 for deterioration, with a weighted value of 0.03 for improvement and deterioration.50,51 |
FACT-Lym | The FACT-Lym is designed to assess HRQoL in patients with lymphoma. The FACT-Lym total score ranges from 0 to 168. This score is based on responses to items across several subscales that measure physical, social and/or family, emotional, and functional well-being, as well as lymphoma-specific symptoms. Higher scores indicate better HRQoL.52,53 The FACT-Lym total score (ranging from 0 to 168) computed the sum of all 5 subscale domain scores. A higher score indicates a better HRQoL. The recall period for each question is “during the past 7 days.” | Validity: In patients with NHL, the FACT-Lym subscale demonstrated concurrent validity, with significant correlations to SF-36 physical (𝑟 = 0.62) and mental (𝑟 = 0.48) summary scores.53 The construct validity of the FACT-Lym subscale was tested in a single-arm, open-label study that enrolled 60 patients with relapsed or refractory MCL. The mean lymphoma subscale score showed a significant decline from baseline to the 30-day assessment (−4.8 points; P < 0.05). Additionally, changes in scores from baseline to discontinuation significantly differed between patients with an improved or stable ECOG PS (n = 20) and those with a worsened ECOG PS (n = 16), with the latter experiencing significantly greater declines in scores.52 Reliability: In patients with NHL, the FACT-Lym subscale exhibited good internal consistency across 3 time points (Cronbach alpha = 0.79, 0.85, and 0.84 at baseline, 3 to 7 days, and 8 to 12 weeks, respectively) and strong test-retest reliability (𝑟 = 0.84).53 Responsiveness: In patients with NHL, responsiveness exceeded established FACT-Lym subscale scores in detecting changes based on ECOG PS and treatment status. The subscale effectively distinguished between patients’ retrospective ratings of change (better, unchanged, worse; P < 0.001).53 | No established MID was reported in patients with untreated MCL. In patients with relapsed or refractory MCL, the estimated MID ranged from 6.5 points to 11.2 points for the FACT-Lym total score across the varying methodologies. A threshold of a 5-point decrease46 and a 7-point increase54 in mean FACT-Lym total score has been deemed clinically meaningful. |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; HRQoL = health-related quality of life; MCL = mantle cell lymphoma; MID = minimal important difference; NHL = non-Hodgkin lymphoma; OS = overall survival; PFS = progression-free survival; SF-36 = Short Form (36) Health Survey.
Statistical Analysis
A summary of the statistical analysis of efficacy end points is provided in Table 8.
Sample Size and Power Calculation
The study was sized to achieve approximately 90% power at final analysis to detect an HR of 0.67 in IRC-assessed PFS, which, under the model assumptions, translated to a 49% improvement in median PFS from 52.9 months in the placebo plus BR group to 79 months in the acalabrutinib plus BR group, with a 2-sided test at an alpha level of 0.05. One interim analysis (presented in this report) and 1 final analysis have been planned for the ECHO trial. The targeted number of IRC-assessed PFS events for the primary analysis was 227 events (85% information fraction; approximately 42% data maturity) for the interim analysis and 268 events (approximately 49% data maturity) for the final analysis. The interim analysis and final analysis for the primary end point of IRC-assessed PFS events were projected to occur at approximately 80 months and 101 months, respectively, after the first patient was randomized. Because of the potential impact of COVID-19 death on the primary IRC-assessed PFS analysis, the data cut-off date was determined to accrue approximately 10% more IRC-assessed PFS events than the 227 IRC-assessed PFS events prespecified in the protocol for the interim analysis. At the data cut-off date of the interim analysis, February 15, 2024, the actual number of IRC-assessed PFS events was 247.
Statistical Test or Model
The analysis of PFS was based on the full analysis set (FAS). For PFS, the primary analysis consisted of a stratified log-rank test for the comparison of the PFS distribution between the 2 treatment groups. The P value from a stratified log-rank test was reported. The treatment effect, measured by the HR for acalabrutinib plus BR versus placebo plus BR and its 2-sided 95% CI, was estimated using a stratified Cox regression model, with treatment as the sole explanatory variable. The KM method was used to estimate the distribution of overall PFS for each treatment group. Stratification factors used in the analyses included geographic region (North America, Western Europe, other) and simplified MIPI score (low risk [0 to 3], intermediate risk [4 to 5], high risk [6 to 11]). The median PFS with a 95% CI was provided. The KM PFS curve was also plotted by treatment group. The number and percentage of patients who had a PFS event or were censored (exited the study or were lost to follow-up, started subsequent therapy for MCL, missed 2 or more consecutive scheduled response assessments, or were in the study at the data cut-off date without prior documentation of disease progression or death) was reported.
Multiple Testing Procedure
To control the overall type I error at a 2-sided 0.05 level, the Lan-DeMets alpha-spending function based on the O’Brien-Fleming boundaries was used to split the alpha into alpha 1 and alpha 2 for the interim and final analyses, respectively, of IRC-assessed PFS. The nominal alpha 1 and alpha 2 levels were based on the actual number of IRC-assessed PFS events observed at the time of data cut-off. An alpha-exhaustive recycling strategy was used to adjust for multiple comparisons.55 With this approach, if the primary efficacy end point — IRC-assessed PFS in the acalabrutinib plus BR group versus the placebo plus BR group — achieved statistical significance at either the PFS interim analysis (data cut-off date: February 15, 2024) or the PFS final analysis (data cut-off date is anticipated to be in ████████ ████), then the 5% alpha will be recycled to test the following key secondary end points in the interim and final analyses in a fixed sequential hierarchical manner:
IRC-assessed ORR
OS.
The hypotheses were tested using alpha recycling, where the alpha that becomes available after each rejected hypothesis is recycled to the secondary hypotheses not yet rejected. This testing procedure stops when the entire alpha is allocated to nonrejected hypotheses. Implementation of this predefined ordered testing procedure, including recycling, can control type I error at 5% (2 sided) among all hypotheses included in the multiple testing procedure. A diagram of the decision notes for the trial data appears in Figure 2.
Figure 2: Multiplicity Adjustment [Redacted]

FA = final analysis; H = hypothesis; IA = interim analysis; Stat. Sig. = statistically significant.
███ ███████ ██ | | ███ ██ | | between the acalabrutinib plus BR group and the placebo plus BR group.████ ███████ ██████████ | | ███ ██████████ | | between the acalabrutinib plus BR group and the placebo plus BR group.████ ██ ██ | | ███ ██ | between the acalabrutinib plus BR group and the placebo plus BR group.██████ ███ ██ ████████ ██ ████ ████ ██ ██████ ████ █████ | | █████ ██ ██████ ███ ████ ███████ ███ ███████ ████████ ██ █████ ████████ ████████ ███ ███████ ████████ ███████ ██ ██ █████████████ ████████████ ██ ███ ███████ ███ █████ ██ ███ ███ ███ ██ ██ ██ ███ █████████████ ███████████ ██ ███ ████ ███████ ███ ███████ █████████ ███ ███████████ ██ ███ ████ ███████ ███ █████ █████████ ███ ██ ███████ ████████ ████ ████ ██ █████████ ██ ███ ████ ██ ███ ████ ███████ ███ █████ ████████ ████ ████ ██ ███ ██ ███ ████████████ ██ ████ █████ ███ ██ ████████ ███████ ██████████ ████ ██ ████████ ██ ██████ ███ ██████ ███ ███ ████ ███████ ███ ███████ ████████ ███ █████ █████████ █████████████
Source: ECHO trial statistical analysis plan.56
Data Imputation Methods
In general, missing data in the primary analysis of IRC-assessed PFS were not imputed and were treated as missing.
Subgroup Analyses
Subgroup analyses were performed on the primary efficacy end point of IRC-assessed PFS in the FAS. Subgroup analyses did not control for the type I error rate for multiple comparisons. The following subgroups were considered:
sex (male versus female)
age category (years) (< 70 versus ≥ 70; < 75 versus ≥ 75)
race (white versus nonwhite [groupings used in original source])
geographic region (North America, Western Europe, other)
baseline ECOG Performance Status (0, 1, 1 or 2, ≥ 2)
tumour bulk (largest diameter) (< 5 cm versus ≥ 5 cm; < 10 cm versus ≥ 10 cm)
Ann Arbor staging for lymphoma (I to III versus IV)
histologically documented MCL (yes versus no)
MCL type (classic type, blastoid variant and pleomorphic variant, other)
Ki-67 (%) (< 30% versus ≥ 30%; < 50% versus ≥ 50%)
bone marrow involvement (yes versus no)
extranodal disease (yes versus no)
gastrointestinal disease (yes versus no)
simplified MIPI score (low risk [0 to 3], intermediate risk [4 to 5], low or intermediate risk [0 to 5], high risk [6 to 11])
lactate dehydrogenase greater than upper limit of normal (yes versus no)
COVID-19 vaccine status (yes versus no).
Sensitivity Analyses
In the ECHO trial, sensitivity and supplementary analyses were performed to evaluate the robustness of the primary analysis of IRC-assessed PFS; these analyses are described in Table 8.
Secondary and Exploratory Outcomes
Overall Survival
A stratified log-rank test was used for the comparison of OS. Additionally, a stratified Cox regression model was used to provide the estimated OS HR and 2-sided 95% CI for acalabrutinib plus BR relative to placebo plus BR. KM estimates and 95% CIs were calculated for event time quartiles and event-free probabilities at selected times.
The study was not powered to detect statistically significant OS differences. The 7-year (84-month) OS rate was assumed to be 49% in the placebo plus BR group for the final analysis. At the time of the interim analysis, the number of OS events was projected to be approximately ███, with an information fraction of ████ and data maturity of ███. The trial will continue to assess OS as a key secondary end point, and it is estimated that ███ data maturity (corresponding to ███ death events) will be reached by ████████ █████ ██ ████ ███ █████ █ ███████████ ███████ ████████ ████████ ███ ██ ██████████████ ████ ██ █████ ███ ███████ ████ ██ ██████ ██ ███████ ██ ███ ████ ██ ███ █████ ████████ ███ ████████████ ███ ████ ███ █████████ ██████ ██ ██ ██████ ██ ███ ███ ███████████ ████████ ██ █████████████ ████ ████ ████ ████████ ██ ████.
Duration of Response
The results of IRC-assessed DOR were summarized with descriptive statistics. The analysis of DOR was conducted on a subset of patients in the FAS with CR or PR at the interim analysis.
Health-Related Quality of Life
Change from baseline, defined as postbaseline value minus baseline value, was calculated at each assessment. At each postbaseline assessment, the change in subscale scores from baseline was calculated for each scale or item.
Summary descriptive statistics were calculated for the EQ-5D-5L and FACT-Lym baseline values (screening visit) by treatment group. The descriptive statistics and change from baseline to cycle 48 day 1 (approximately 48 months of treatment) were calculated by treatment group.
Safety Outcomes
All safety analyses were based on the safety analysis set, unless otherwise specified. Descriptive statistics were calculated for continuous safety variables, and frequency counts and percentages were tabulated for categorical safety variables.
Table 8: Statistical Analysis of Efficacy End Points From the ECHO Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PFS (IRC assessed) |
|
| Patients who exited the study, were lost to follow-up, started a subsequent therapy for MCL, missed 2 or more consecutive scheduled response assessments, or were in the study at the data cut-off date without prior documentation of PD or death were censored. |
|
OS |
|
| Any patient not known to have died at the time of the analysis was censored based on the last recorded date on which the patient was known to be alive. Any patient recorded as alive or to have died after the data cut-off date was censored at the date of the data cut-off. |
|
DOR (IRC assessed and investigator assessed) |
| Not applicable | The same censoring rules for PFS were applied to DOR. | Not performed |
Change from baseline at cycle 48 day 1 in EQ-5D-5L utility score | Summarized with descriptive statistics | Not applicable | For patients who discontinued study treatment for any reason, patient-reported outcome assessments ≤ 1 day after the date of treatment discontinuation were included in the analysis. Any assessments beyond that were not consistent with the planned schedule of assessments and were not included in the analysis. | Not performed |
Change from baseline in FACT-Lym total score | Summarized with descriptive statistics | Not applicable | For patients who discontinued study treatment for any reason, patient-reported outcome assessments ≤ 1 day after the date of treatment discontinuation were included in the analysis. Any assessments beyond that were not consistent with the planned schedule of assessments and were not included in the analysis. | Not performed |
BR = bendamustine plus rituximab; DOR = duration of response; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; IRC = independent review committee; KM = Kaplan-Meier; MCL = mantle cell lymphoma; MIPI = Mantle Cell Lymphoma International Prognostic Index; OS = overall survival; PD = progressive disease; PFS = progression-free survival.
Source: ECHO Clinical Study Report (2024);25 ECHO statistical analysis plan.56 Details included in the table are from the sponsor’s summary of clinical evidence.26
Analysis Populations
A summary of the analysis populations used in the ECHO trial that are relevant to this review is provided in Table 9.
Table 9: Analysis Populations of the ECHO Trial
Population | Definition | Application |
|---|---|---|
Full analysis set | All patients randomized at least 24 months before the data cut-off date of the interim analysis. Includes patients who were randomized but did not subsequently receive treatment. | Used for interim and final analysis and to summarize demographics, baseline characteristics, and disease characteristics. |
Safety analysis set | All randomized patients who received at least 1 dose of the study drug. | Used for safety analyses; summarized for the main study period and crossover period separately. |
Per-protocol analysis population | Subset of the full analysis set, excluding patients:
| Used for sensitivity analysis to assess for deviation bias. |
Source: ECHO statistical analysis plan.56 Details included in the table are from the sponsor’s summary of clinical evidence.26
Results
This report focuses on the results from the interim analysis, with a data cut-off date of February 15, 2024.
Patient Disposition
Patient disposition in the interim analysis in the ECHO trial is summarized in Table 10. A total of 854 patients were screened, and 219 patients did not meet the screening criteria. Having measurable disease of less than 2 cm was the most reported reason for patients not meeting the screening criteria (██ of ███ ███████). In total, 635 patients were randomized; however, 37 patients (all from China) were not included due to having less than 2 years of follow-up at the data cut-off date of February 15, 2024. As a result, the FAS included 598 patients: 299 patients in the acalabrutinib plus BR group and 299 patients in the placebo plus BR group. Similar proportions of patients in both treatment groups were randomized and treated (99.3% versus 99.0% for acalabrutinib plus BR versus placebo plus BR). There were ███ ███████ patients in the acalabrutinib plus BR group and ███ ███████ patients in placebo plus BR group who discontinued all study treatment; the commonly reported reasons for treatment discontinuation included AEs (█████ versus █████), which was reported ████ ██████████ with acalabrutinib plus BR, and objective evidence of disease progression (█████ versus █████), which was reported ████ ██████████ with acalabrutinib plus BR.
Table 10: Summary of Patient Disposition From the ECHO Trial
Patient disposition | Acalabrutinib + BR (N = 299) | Placebo + BR (N = 299) |
|---|---|---|
Screened, N | 854 | |
Did not meet screening criteria, N | 219 | |
Common reasons (occurred in more than 10% of patients), n (%) | ||
Measurable disease < 2 cm | ██ ██████ | |
Estimated creatinine clearance of < 50 mL/min | ██ ██████ | |
Patient withdrew from study or PI decision to withdraw patient | ██ ██████ | |
Total randomized, N | 635 | |
Included in interim analysis, N | 598 | |
Randomized, N (%) | 299 (100) | 299 (100) |
Not treated, n (%) | 2 (0.7) | 2 (0.7) |
Treated, n (%) | 297 (99.3) | 296 (99.0)a |
Discontinued from study, n (%) | 142 (47.5) | 146 (48.8) |
Death | 96 (32.1) | 103 (34.4) |
Patient’s withdrawal of consent from study | 35 (11.7) | 30 (10.0) |
Patient lost to follow-up | █████ | █████ |
Other | █████ | ██ █████ |
Discontinued all study treatment,b n (%) | ███ ██████ | ███ ██████ |
Adverse event | ███ ██████ | ███ ██████ |
Objective evidence of PD | ██ ██████ | ██ ██████ |
Death | ██ █████ | ██ █████ |
Patient’s withdrawal of consent | ██ █████ | █████ |
Investigator’s decision | █████ | | █████ |
Clinical PD | █████ | | █████ |
Patient lost to follow-up | | █████ | | █████ |
Other | ██ █████ | ██ █████ |
Patients received acalabrutinib or placebo, n (%) | 95 (31.8) | 77 (25.8) |
Patients discontinued acalabrutinib or placebo, n (%) | 202 (67.6) | 219 (73.2) |
Adverse event | 129 (43.1) | 94 (31.4) |
Objective evidence of PD | 37 (12.4) | 86 (28.8) |
Death | 9 (3.0) | 12 (4.0) |
Patient’s withdrawal of consent from study | 8 (2.7) | 6 (2.0) |
Investigator’s decision | 5 (1.7) | 5 (1.7) |
Clinical PD | 2 (0.7) | 3 (1.0) |
Patient lost to follow-up | 1 (0.3) | 1 (0.3) |
Other | 11 (3.7) | 12 (4.0) |
Patients completed bendamustine per protocol, n (%) | ███ ██████ | ███ ██████ |
Patients discontinued bendamustine, n (%) | ██ ██████ | ██ ██████ |
Adverse event | ██ ██████ | ██ ██████ |
Objective evidence of PD | █████ | ██ █████ |
Investigator’s decision | █████ | | █████ |
Patient’s withdrawal of consent from study | █████ | | █████ |
Clinical PD | ██ | █████ |
Death | ██ | █████ |
Patient lost to follow-up | █████ | ██ |
Other | █████ | | █████ |
Patients completed rituximab per protocol, n (%) | 154 (51.5) | 137 (45.8) |
Patients received rituximab, n (%) | 1 (0.3) | 1 (0.3) |
Patients discontinued rituximab, n (%) | 142 (47.5) | 159 (53.2) |
Adverse event | ██ ██████ | ██ ██████ |
Objective evidence of PD | ██ ██████ | ██ ██████ |
Death | █████ | ██ █████ |
Investigator’s decision | █████ | ██ █████ |
Patient’s withdrawal of consent from study | █████ | █████ |
Clinical PD | █████ | █████ |
Patient lost to follow-up | █████ | ██ |
Other | ██ █████ | ██ █████ |
Patients ongoing any study treatment,c n (%) | 95 (31.8) | 77 (25.8) |
FAS, N | 299 | 299 |
SAS, N | 297 | 297 |
BR = bendamustine plus rituximab; FAS = full analysis set; PD = progressive disease; PI = principal investigator; SAS = safety analysis set.
██ ███████ ██ ███ ███████ ████ ██ █████ ███ █ ████ ██ █████████ ███ ████ ████████████ ███████ █████████ ███████ ██ █████████████
bDiscontinuation was based on the earlier discontinued drug of the regimen for each group. Discontinued all study treatment based on randomized treatment assignment.
cAt least 1 of the study drugs was ongoing (acalabrutinib, placebo, bendamustine, or rituximab).
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt by the review team to affect the outcomes or interpretation of the study results.
Overall, demographic characteristics, baseline disease characteristics, and medical history were balanced between the treatment groups. The median age of all study patients was 71.0 years, with a range of 65 to 86 years. The ECHO trial enrolled mostly males (70.7%), and the largest group of patients (49.5%) had an ECOG Performance Status of 0. Most patients were white (78.3%), and the study also included patients self-reporting their ethnicity as Asian, American Indian or Alaska Native, Black or African American, and multiple. All the patients (100%) had histologically documented MCL, and close to half the patients (42.3%) had a simplified MIPI score of 4 to 5 (intermediate risk). The most reported MCL type was classic (80.4%). The majority of patients (86.0%) had Ann Arbor stage IV disease, 90.5% had extranodal disease, and 71.7% had bone marrow involvement. ████████ █████████ ███████ was the most reported comorbidity in patients in the ECHO trial.
Table 11: Summary of Baseline Characteristics From the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
Characteristic | Acalabrutinib + BR (N = 299) | Placebo + BR (N = 299) |
|---|---|---|
Demographic | ||
Age (years) | ||
Mean (SD) | 71.6 (4.7) | 71.6 (4.6) |
Median (range) | 71.0 (65 to 85) | 71.0 (65 to 86) |
Sex, n (%) | ||
Female | 85 (28.4) | 90 (30.1) |
Male | 214 (71.6) | 209 (69.9) |
Race, n (%) | ||
American Indian or Alaska Native | 2 (0.7) | 2 (0.7) |
Asian | 44 (14.7) | 49 (16.4) |
Black or African American | 1 (0.3) | 2 (0.7) |
White | 233 (77.9) | 235 (78.6) |
Multiple | 5 (1.7) | 0 |
Not reported | 14 (4.7) | 11 (3.7) |
ECOG Performance Status, n (%) | ||
0 | 156 (52.2) | 140 (46.8) |
1 | 129 (43.1) | 132 (44.1) |
2 | 12 (4.0) | 23 (7.7) |
Disease characteristics | ||
Histologically documented MCL, n (%) | 299 (100.0) | 299 (100.0) |
MCL type, n (%) | ||
Classic type | 238 (79.6) | 243 (81.3) |
Blastoid variant | 26 (8.7) | 20 (6.7) |
Pleomorphic variant | 15 (5.0) | 18 (6.0) |
Other | 0 | 5 (1.7) |
Unknown | 19 (6.4) | 11 (3.7) |
Not done | 1 (0.3) | 2 (0.7) |
Ann Arbor staging for lymphoma, n (%) | ||
I | 2 (0.7) | 1 (0.3) |
II | 15 (5.0) | 11 (3.7) |
III | 31 (10.4) | 24 (8.0) |
IV | 251 (83.9) | 263 (88.0) |
Gastrointestinal disease, n (%) | ||
Yes | 74 (24.7) | 75 (25.1) |
No | 225 (75.3) | 224 (74.9) |
Extranodal disease, n (%) | ||
Yes | 264 (88.3) | 277 (92.6) |
No | 35 (11.7) | 22 (7.4) |
Tumour bulk,a n (%) | ||
< 5 cm | 187 (62.5) | 186 (62.2) |
≥ 5 cm and < 10 cm | 92 (30.8) | 92 (30.8) |
≥ 10 cm | 20 (6.7) | 21 (7.0) |
Bone marrow involvement (central lab), n (%) | ||
Involved | 211 (70.6) | 218 (72.9) |
Not involved | ██ ██████ | ██ ██████ |
Indeterminate | █████ | █████ |
Missing | █████ | █████ |
Ki-67, n (%) | ||
< 30% | 133 (44.5) | 126 (42.1) |
≥ 30% | 139 (46.5) | 147 (49.2) |
< 50% | 210 (70.2) | 199 (66.6) |
≥ 50% | 62 (20.7) | 74 (24.7) |
Indetermined | 4 (1.3) | 4 (1.3) |
Missing | 23 (7.7) | 22 (7.4) |
TP53 status,b n (%) | ||
Mutated | 22 (7.4) | 29 (9.7) |
Unmutated | 97 (32.4) | 83 (27.8) |
Unknown | 180 (60.2) | 187 (62.5) |
Simplified MIPI score, n (%) | ||
Low risk (0 to 3) | 99 (33.1) | 101 (33.8) |
Intermediate risk (4 to 5) | 128 (42.8) | 125 (41.8) |
High risk (6 to 11) | 72 (24.1) | 73 (24.4) |
LDH > upper limit of normal, n (%) | ||
Yes | 52 (17.4) | 54 (18.1) |
No | 245 (81.9) | 241 (80.6) |
Missing | 2 (0.7) | 4 (1.3) |
Medical history | ||
Commonly reported medical history (in ≥ 30% of patients in either group), n(%) | ||
████████ █████████ | ███ ██████ | ███ ██████ |
█████████ █████████ | ███ ██████ | ███ ██████ |
████████████████ █████████ | ███ ██████ | ███ ██████ |
███████ ██████████ | ███ ██████ | ███ ██████ |
███████████████ ███ ██████████ ██████ █████████ | ███ ██████ | ██ ██████ |
████████████ ████████ ███ ███████████ ████████ | ██ ██████ | ███ ██████ |
BR = bendamustine plus rituximab; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; LDH = lactate dehydrogenase; MCL = mantle cell lymphoma; MIPI = Mantle Cell Lymphoma International Prognostic Index; SD = standard deviation.
aFor target lesions at baseline, investigator assessment was used. Tumour bulk was defined as the largest diameter of a nodal or extranodal lesion. Tumour burden was defined as the sum of the product of diameters of all target lesions.
bTP53 status was determined post hoc.
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Important Protocol Deviations
A summary of important protocol deviations from the ECHO trial in the FAS population at the data cut-off of February 15, 2024, is provided in Table 12. Overall, the important protocol deviations were balanced between treatment groups. Forty-three patients (14.4%) in the acalabrutinib plus BR group and 37 patients (12.4%) in the placebo plus BR group reported important protocol deviations. Of the total important deviations, ████ of patients had deviations determined to be ████████████████, occurring primarily in the dosing and laboratory categories.
Table 12: Summary of Important Protocol Deviations From the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
Important protocol deviations | Acalabrutinib + BR (N = 299) | Placebo + BR (N = 299) |
|---|---|---|
Patients with any important protocol deviations, n (%) | 43 (14.4) | 37 (12.4) |
Common important protocol deviations (reported in ≥ 1.0% of patients in either group), n (%) | ||
Laboratory | 17 (5.7) | 15 (5.0) |
Dosing | 16 (5.4) | 12 (4.0) |
Required visit and/or procedure | 7 (2.3) | 6 (2.0) |
Noncompliance | 3 (1.0) | 2 (0.7) |
Other | 3 (1.0) | 3 (1.0) |
BR = bendamustine plus rituximab; FAS = full analysis set.
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Exposure to Study Treatments
At the time of the interim analysis (February 15, 2024, data cut-off), the median duration of exposure to acalabrutinib in the acalabrutinib plus BR group (28.6 months) was longer than the exposure to placebo in the placebo plus BR group (24.6 months) (Table 13). In total, 231 of 297 patients (77.8%) in the acalabrutinib plus BR group and 232 of 297 patients (78.1%) in the placebo plus BR group completed 6 cycles of BR per protocol, and 222 patients (74.7%) in the acalabrutinib plus BR group and 225 patients (75.8%) in the placebo plus BR group completed 6 cycles of acalabrutinib or placebo plus BR.
Table 13: Summary of Patient Exposure From the ECHO Trial — SAS (Data Cut-Off Date: February 15, 2024)
Exposure | Acalabrutinib + BR (N = 297) | Placebo + BR (N = 297) |
|---|---|---|
Acalabrutinib or placebo | ||
Patients contributing to the analysis, n | 297 | 296a |
Duration of exposure (months) | ||
Mean (SD) | 32.5 (23.5) | 29.1 (23.1) |
Median (range) | 28.6 (0.1 to 80.1) | 24.6 (0.03 to 76.4) |
Bendamustine | ||
Patients contributing to the analysis, n | 297 | 296a |
Duration of exposure (months) | ||
Mean (SD) | 5.28 (1.315) | 5.19 (1.193) |
Median (range) | 5.55 (0.9 to 11.1) | 5.55 (0.9 to 7.4) |
Rituximab | ||
Patients contributing to the analysis, n | 297 | 297 |
Duration of exposure (months) | ||
Mean (SD) | 20.30 (9.504) | 18.41 (10.192) |
Median (range) | 27.47 (0.9 to 28.6) | 23.92 (0.9 to 29.5) |
BR = bendamustine plus rituximab; SAS = safety analysis set; SD = standard deviation.
aOne patient did not have placebo exposure data.
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Concomitant Medications
At the time of the first interim analysis (data cut-off: February 15, 2024), the use of concomitant medications was reported █████████ between the acalabrutinib plus BR group ███████ and the placebo plus BR group (99.7%). Some common concomitant medications (reported in ≥ 30% of patients in either group) were reported by a ██████ proportion of patients in the acalabrutinib plus BR group than in the placebo plus BR group, with a difference of greater than ██ include: ██████████████ ███ ████████ ███ ██████ ███ ███████ █████████ ████████████ ██████ ███ ███████ █████ ███████████ ███ █████████ █████████ ██████ ███ ███████ ████████ ██████ ███ ███████ ███ ███████ ███████████ ██████ ███ ██████. All other concomitant therapies were reported at a frequency less than ███. Detailed results on concomitant therapies can be found in Table 17 in Appendix 1.
Subsequent Treatments
A smaller proportion of patients received at least 1 subsequent therapy for MCL in the acalabrutinib plus BR group (10.0%) than in the placebo plus BR group (29.4%) (Table 14). Among these patients, 13 of 30 patients in the acalabrutinib plus BR group and 76 of 88 patients in the placebo plus BR group received a subsequent BTKi-based therapy. Among the 76 patients in the placebo plus BR group, acalabrutinib was counted as the subsequent therapy for MCL for 55 patients, of which 51 crossed over to the acalabrutinib plus BR group and received acalabrutinib monotherapy after experiencing disease progression.
Table 14: Summary of Subsequent Treatments From the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
Subsequent treatment | Acalabrutinib + BR (N = 299) | Placebo + BR (N = 299) |
|---|---|---|
Patients with ≥ 1 subsequent therapy for MCL, n (%) | 30 (10.0) | 88 (29.4) |
Number of subsequent regimens, n (%) | ||
1 | ██ █████ | ██ ██████ |
2 | █████ | ██ █████ |
3 | █████ | █████ |
≥ 4 | ██ | █████ |
Patients with ≥ 1 BTKi as subsequent therapy for MCL, n (%) | 13 (4.3) | 76 (25.4) |
Acalabrutinib | 1 (0.3) | 55 (18.4) |
Ibrutinib | 7 (2.3) | 17 (5.7) |
Nemtabrutinib | 1 (0.3) | 0 |
Pirtobrutinib | 2 (0.7) | 4 (1.3) |
Zanubrutinib | 2 (0.7) | 5 (1.7) |
BR = bendamustine plus rituximab; BTKi = Bruton tyrosine kinase inhibitor; FAS = full analysis set; MCL = mantle cell lymphoma.
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Efficacy
Key efficacy results from the ECHO trial in the FAS population in the interim analysis are provided in Table 15. At the time of the interim analysis (data cut-off: February 15, 2024), the median duration of follow-up was 46.1 months in the acalabrutinib plus BR group and 44.4 months in the placebo plus BR group.
Progression-Free Survival
In total, 110 patients (36.8%) in the acalabrutinib plus BR group and 137 patients (45.8%) in the placebo plus BR group experienced a PFS event; among them, in the acalabrutinib plus BR group 57 patients, (19.1% of all patients in the group) had disease progression based on IRC assessment and 53 patients (17.7%) died, and in the placebo plus BR group, 99 patients (33.1% of all patients in the group) had disease progression and 38 patients (12.7%) died (Table 15). The median PFS was 66.4 months (95% CI, 55.1 months to not estimable) in the acalabrutinib plus BR group and 49.6 months (95% CI, 36.0 to 64.1 months) in the placebo plus BR group. There was an improvement in PFS in the acalabrutinib plus BR group compared with the placebo plus BR group (HR = 0.73; 95% CI, 0.57 to 0.94; P = 0.0160). The KM estimate of PFS probability at 48 months was █████ (95% CI, █████ to ██████ for the acalabrutinib plus BR group and █████ (95% CI, █████ to █████) for the placebo plus BR group (Figure 3). The between-group difference was ████ (95% CI, ████ to █████). Similar results were observed in the analysis of PFS based on investigator assessment (HR = 0.68; 95% CI, 0.53 to 0.88; nominal P = 0.0028).
The results from the planned sensitivity analyses were consistent with those of the primary analysis. Subgroup analyses of IRC-assessed PFS in the primary analysis were consistent with the primary analysis across most prespecified subgroups, including those based on the simplified MIPI score.57 However, the treatment effect was different between male patients (HR = 0.91; 95% CI, 0.68 to 1.21) and female patients (HR = 0.34; 95% CI, 0.19 to 0.58). Refer to Figure 5 in Appendix 1 for the detailed subgroup analyses data.
Figure 3: Kaplan-Meier Plot for Progression-Free Survival in the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
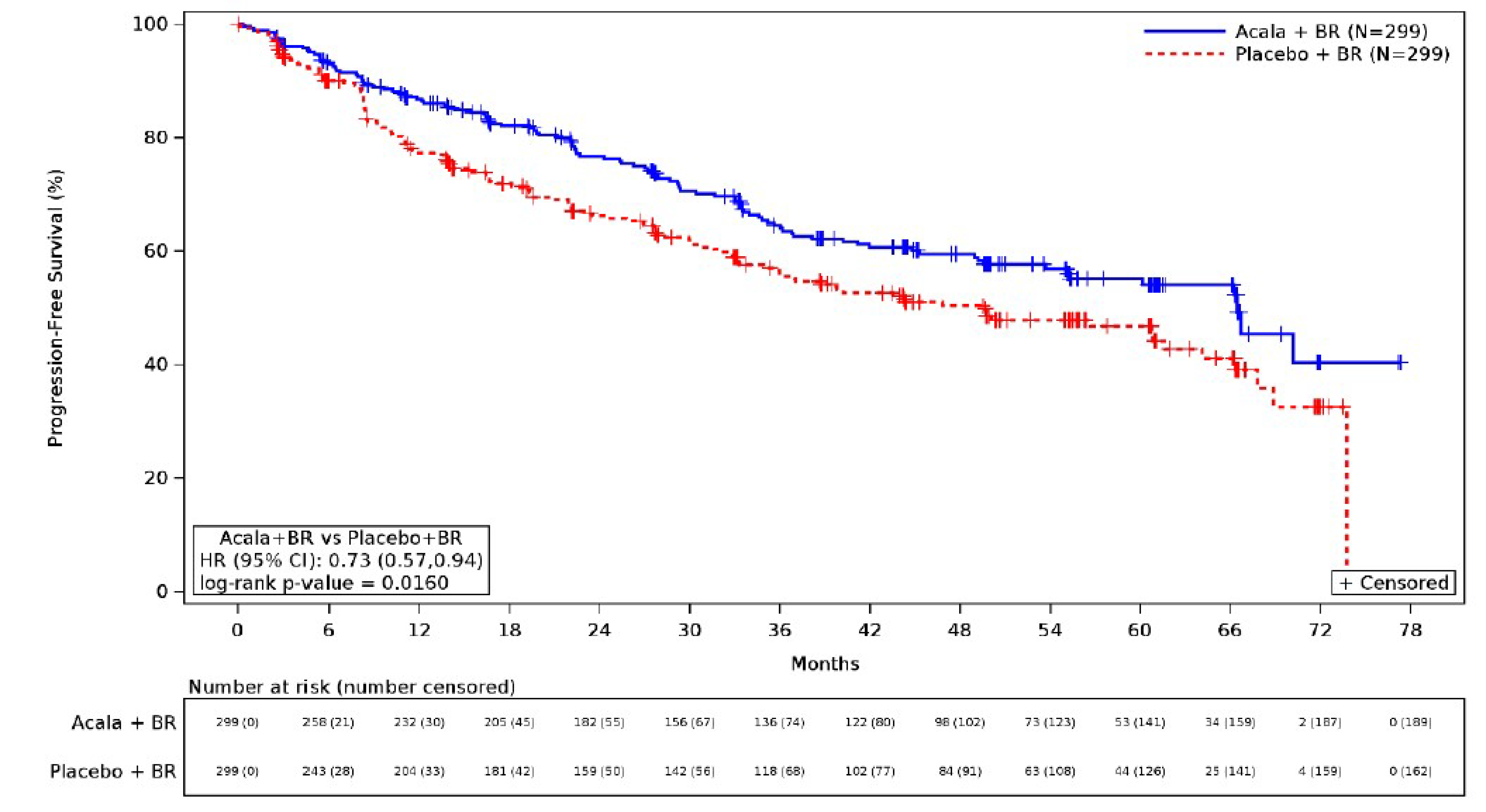
Acala = acalabrutinib; BR = bendamustine plus rituximab; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; vs = versus.
Source: ECHO Clinical Study Report (2024).25
Overall Survival
At the interim analysis, 97 patients (32.4%) in the acalabrutinib plus BR group and 106 patients (35.5%) in the placebo plus BR group had died. The median OS was not estimable at the time of the interim analysis for either treatment group, and the HR for risk of death for acalabrutinib plus BR compared with placebo plus BR was 0.86 (95% CI, 0.65 to 1.13; nominal P = 0.2743). The KM estimate of OS probability at 48 months was █████ (95% CI, █████ to █████) for the acalabrutinib plus BR group and █████ (95% CI, █████ to █████) for the placebo plus BR group. The between-group difference was ████ (95% CI, █████ to █████). This analysis was not powered to detect statistically significant differences in OS. Because the first key secondary end point, ORR, was not statistically significant, the testing stopped for the interim analysis; therefore, the type I error rate in the OS analysis was not controlled for multiple comparisons, and the results should be interpreted with caution because they were considered supportive evidence. The KM curve estimate of the OS event time distribution is depicted in Figure 4.
Figure 4: Kaplan-Meier Plot for Overall Survival in the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
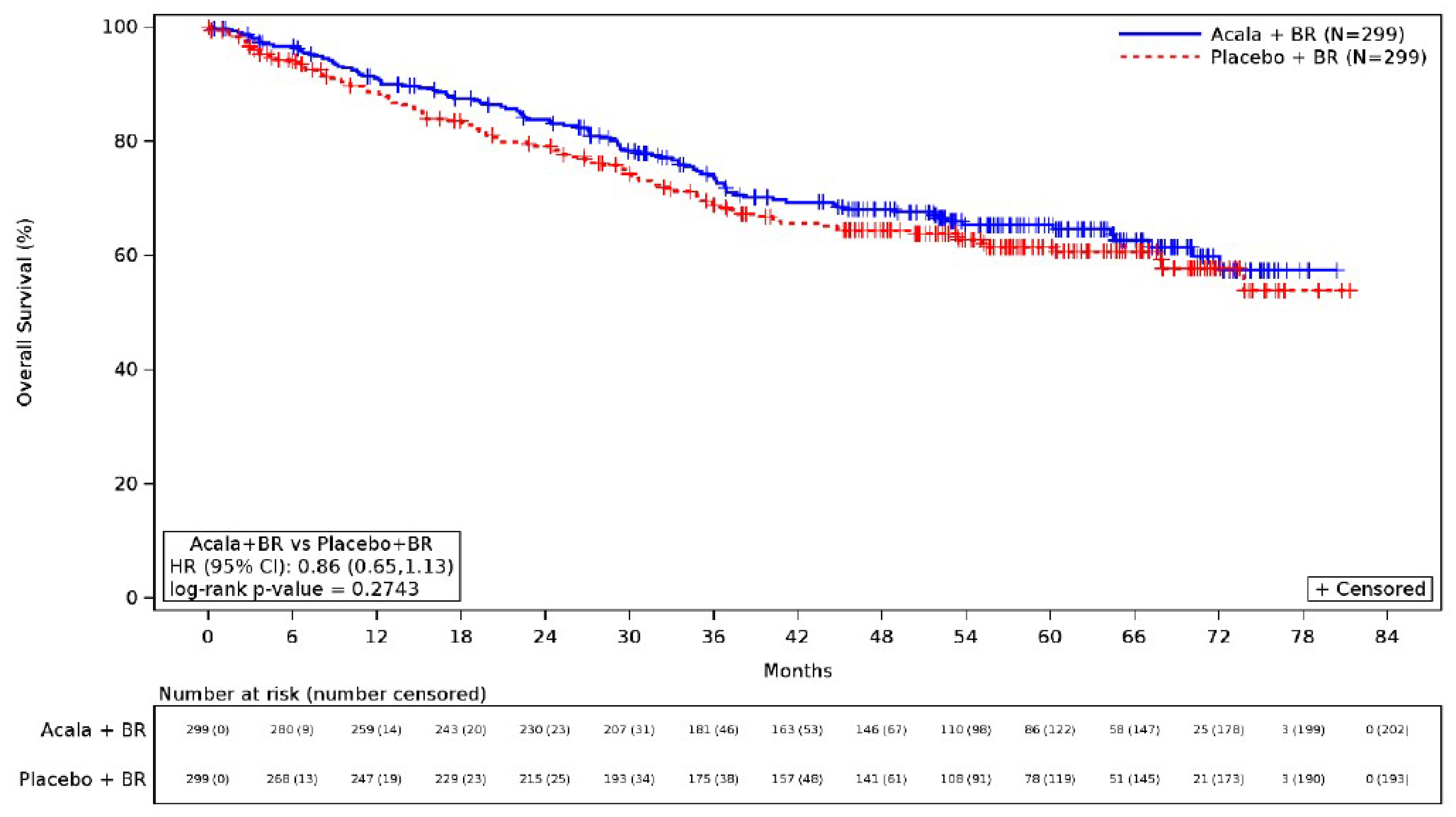
Acala = acalabrutinib; BR = bendamustine plus rituximab; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; vs = versus.
Source: ECHO Clinical Study Report (2024).25
Duration of Response
At the time of the interim analysis, among patients who had a CR or PR based on IRC assessment (272 versus 263 for acalabrutinib plus BR versus placebo plus BR), 99 patients (36.4%) in the acalabrutinib plus BR group and 117 patients (44.5%) in the placebo plus BR group developed disease progression or died, and 173 patients (63.3%) in the acalabrutinib plus BR group and 146 patients (55.5%) in the placebo plus BR group were censored. The median DOR (for PR or better based on IRC assessment) was 63.5 months (95% CI, 52.5 months to not estimable) in the acalabrutinib plus BR group and 53.8 months (95% CI, 37.6 to 66.1 months) in the placebo plus BR group. The KM estimate of DOR probability at 48 months was 59.9% (95% CI, 53.0% to 66.1%) for the acalabrutinib plus BR group and 51.1% (95% CI, 44.0% to 57.7%) for the placebo plus BR group. Similar results were observed in the analysis of DOR based on investigator assessment despite the median DOR not being reached in the acalabrutinib plus BR group. The type I error rate was not controlled for multiple comparisons in the DOR analysis, and the results should be considered supportive evidence.
Health-Related Quality of Life
Change From Baseline in EQ-5D-5L Utility (Index) Score at Cycle 48 Day 1
At the interim analysis, at cycle 48 day 1 (approximately 48 months of treatment), patients in the acalabrutinib plus BR group reported an estimated least squares mean change from baseline in the EQ-5D-5L utility (index) score of ████ (SE = ████) compared to ████ points (SE = ████) in patients in the placebo plus BR group. The between-group difference was █████ points (95% CI, █████ to ████; nominal P = █████). The type I error rate in the EQ-5D-5L analysis was not controlled for multiple comparisons, and the results should be considered supportive evidence.
FACT-Lym Total Score
At the interim analysis, at cycle 48 day 1 (approximately 48 months of treatment), patients in the acalabrutinib plus BR group reported an estimated least squares mean increase (improvement) from baseline in the FACT-Lym total score of ███ points (SE = ███) compared to ███ points (SE = ███) in patients in the placebo plus BR group. The between-group difference was ████ points (95% CI, ████ to ███; nominal P = █████). The FACT-Lym analysis did not control for type I error rate for multiple comparisons, and the results should be considered supportive evidence.
Table 15: Summary of Key Efficacy Results From the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
Variable | Acalabrutinib + BR N = 299 | Placebo + BR N = 299 |
|---|---|---|
PFS (IRC assessment) | ||
Patients with events, n (%) | 110 (36.8) | 137 (45.8) |
Disease progression | 57 (19.1) | 99 (33.1) |
Death | 53 (17.7) | 38 (12.7) |
Censored patients, n (%) | 189 (63.2) | 162 (54.2) |
PFS (months), median (95% CI) | 66.4 (55.1 to NE) | 49.6 (36.0 to 64.1) |
HR (95% CI)a | 0.73 (0.57 to 0.94) | |
P valueb | 0.0160 | |
PFS event-free probability at 48 months,c % (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ |
Absolute difference between study groups,c,d % (95% CI) | ███ ████ ██ █████ | |
OS | ||
Patients with events, n (%) | 97 (32.4) | 106 (35.5) |
Censored patients, n (%) | ███ ██████ | ███ ██████ |
OS (months), median (95% CI) | NE (72.1 to NE) | NE (73.8 to NE) |
HR (95% CI)a | 0.86 (0.65 to 1.13) | |
Nominal P valueb,c | 0.2743 | |
OS event-free probability at 48 months,c % (95% CI) | ████ (████ to ████) | ████ (████ to ████) |
Absolute difference between study groups,d,e % (95% CI) | ███ (████ to ████) | |
DOR (IRC assessment) | ||
Patients with PR or CR contributing to the analysis, n | 272 | 263 |
Patients with events, n (%) | 99 (36.4) | 117 (44.5) |
Censored patients, n (%) | 173 (63.6) | 146 (55.5) |
DOR (months), median (95% CI) | 63.5 (52.5 to NE) | 53.8 (37.6 to 66.1) |
DOR probability at 48 months,d % (95% CI) | 59.9 (53.0 to 66.1) | 51.1 (44.0 to 57.7) |
Change from baseline at cycle 48 day 1 in EQ-5D-5L utility (index) score | ||
Patients contributing to the analysis, n (%) | ██ | ██ |
Baseline (points), mean (SD) | ████ ██████ | ████ ██████ |
Change from baseline (points), LS mean (SE) | ████ ██████ | ████ ██████ |
Nominal P valuec | █████ | |
Absolute between-group difference for acalabrutinib plus BR vs. placebo plus BR, % (95% CI) | █████ ██████ ██ █████ | |
Change from baseline at cycle 48 day 1 in FACT-Lym total score | ||
Number of patients contributing to the analysis at cycle 48 day 1, n (%) | ██ | ██ |
Baseline (points), mean (SD) | █████ ██████ | █████ ██████ |
Change from baseline (points), LS mean (SE) | ███ █████ | ███ █████ |
Nominal P valuec | █████ | |
Absolute between-group difference for acalabrutinib plus BR vs. placebo plus BR, % (95% CI) | ████ █████ ██ ████ | |
BR = bendamustine plus rituximab; CI = confidence interval; CR = complete response; DOR = duration of response; FACT-Lym = Functional Assessment of Cancer Therapy–Lymphoma; FAS = full analysis set; HR = hazard ratio; IRC = independent review committee; LS = least squares; NE = not estimable; OS = overall survival; PFS = progression-free survival; PR = partial response; SD = standard deviation; SE = standard error; vs. = versus.
aEstimated based on stratified Cox proportional hazards model for HR (95% CI) using geographic region (North America, Western Europe, other) and simplified Mantle Cell Lymphoma International Prognostic Index score (low risk [0 to 3], intermediate risk [4 to 5], high risk [6 to 11]) as stratification factors.
bEstimated based on stratified or unstratified log-rank test for P value and adjusted for type I error for multiple comparisons.
cThis end point was not controlled for type I error rate for multiple comparisons and should be interpreted as supportive evidence.
dEstimated by Kaplan-Meier probabilities.
e95% CI estimated based on SE of PFS probability at each time point calculated using the Breslow method and a Satterthwaite approximation of the SE of the difference.
Source: ECHO Clinical Study Report (2024)25 and sponsor-provided additional data (April 30, 2025).58 Details included in the table are from the sponsor’s summary of clinical evidence.26
Harms
Refer to Table 16 for harms data in the ECHO trial at the interim analysis for the safety analysis set (data cut-off date: February 15, 2024).
Treatment-Emergent AEs
At the time of the interim analysis, 296 of 297 patients (99.7%) in the acalabrutinib plus BR group and 294 of 297 patients (99.0%) in the placebo plus BR group experienced at least 1 TEAE. The most commonly reported TEAEs (reported by ≥ 30% of patients in either group) were nausea (42.8% versus 37.7% for acalabrutinib plus BR versus for acalabrutinib plus BR ), neutropenia (40.1% versus 41.4%), diarrhea (37.4% versus 27.9%), COVID-19 (30.6% versus 20.9%), and headache (30.3% versus 14.1%). All other TEAEs were reported at a frequency less than 30%. Most of the TEAEs were grade 3 or higher, as reported by 264 patients (88.9%) in the acalabrutinib plus BR group and 262 patients (88.2%) in the placebo plus BR group. The most reported grade 3 or higher TEAE was neutropenia (35.4% in the acalabrutinib plus BR group versus 37.0% in the placebo plus BR group).
Serious AEs
In the same analysis, 205 patients (69.0%) in the acalabrutinib plus BR group and 184 patients (62.0%) in the placebo plus BR group reported at least 1 SAE. The commonly reported SAEs (reported by ≥ 5% of patients in either group) were COVID-19 pneumonia (13.8% versus 11.4% for acalabrutinib plus BR versus placebo plus BR), pneumonia (9.4% versus 7.1%), COVID-19 (8.8% versus 6.4%), and pyrexia (5.7% versus 5.1%). All other SAEs were reported at a frequency less than 5%. Grade 3 or higher SAEs were reported by 191 patients (64.3%) in the acalabrutinib plus BR group and 166 patients (55.9%) in the placebo plus BR group. Commonly reported SAEs of grade 3 or higher were COVID-19 pneumonia (13.1% versus 10.1% for acalabrutinib plus BR versus placebo plus BR), pneumonia (8.1% versus 6.1%), and COVID-19 (7.7% versus 6.1%).
Withdrawals Due to AEs
TEAEs leading to discontinuation of study treatment were reported by 127 patients (42.8%) in the acalabrutinib plus BR group and 92 patients (31.0%) in the placebo plus BR group. The most common TEAE that caused treatment discontinuation was COVID-19 (4.7% versus 3.0%).
Mortality
There were 97 of 299 patients (32.4%) in the acalabrutinib plus BR group and 106 of 299 patients (35.5%) in the placebo plus BR group who died. The most reported cause of death was AEs (15.4% versus 13.7% for acalabrutinib plus BR versus placebo plus BR). COVID-19 pneumonia (5.1% versus 3.4%) and COVID-19 (2.7% versus 2.0%) were the most reported AEs leading to death. Disease progression was the cause of death in 30 patients (10.0%) and 43 patients (14.4%) in the acalabrutinib plus BR and placebo plus BR groups, respectively.
Notable Harms
Based on input from clinician groups, expert committee members, and the clinical experts consulted by CDA-AMC, the review team identified infections and infestations, ventricular arrythmias, and atrial fibrillation and/or flutter as notable harms for treatment with acalabrutinib, and these were included in the GRADE table. Infections and infestations were observed in 232 patients (78.1%) in the acalabrutinib plus BR group and 211 patients (71.0%) in the placebo plus BR group. Ventricular arrythmias were reported similarly in the treatment groups (2.4% in both groups). Atrial fibrillation and/or flutter was observed in 20 patients (6.7%) in the acalabrutinib plus BR group and 13 patients (4.4%) in the placebo plus BR group. Hemorrhage (28.3% versus 17.2% for acalabrutinib plus BR versus placebo plus BR) was also an AE of clinical interest.
Table 16: Summary of Harms Results From the ECHO Trial — SAS (Data Cut-Off Date: February 15, 2024)
Adverse events | Acalabrutinib + BR (N = 297) | Placebo + BR (N = 297) |
|---|---|---|
Most common adverse events (reported in ≥ 30% of patients in either group), n (%) | ||
Patients with ≥ 1 adverse event, n (%) | ||
Any grade | 296 (99.7) | 294 (99.0) |
Nausea | 127 (42.8) | 112 (37.7) |
Neutropenia | 119 (40.1) | 123 (41.4) |
Diarrhea | 111 (37.4) | 83 (27.9) |
COVID-19 | 91 (30.6) | 62 (20.9) |
Headache | 90 (30.3) | 42 (14.1) |
Grade ≥ 3 | 264 (88.9) | 262 (88.2) |
Nausea | 4 (1.3) | 4 (1.3) |
Neutropenia | 105 (35.4) | 110 (37.0) |
Diarrhea | 9 (3.0) | 7 (2.4) |
COVID-19 | 26 (8.8) | 21 (7.1) |
Headache | 4 (1.3) | 2 (0.7) |
Serious adverse events (reported in ≥ 5% of patients in group), n (%) | ||
Patients with ≥ 1 SAE, n (%) | ||
Any grade | 205 (69.0) | 184 (62.0) |
COVID-19 pneumonia | 41 (13.8) | 34 (11.4) |
Pneumonia | 28 (9.4) | 21 (7.1) |
COVID-19 | 26 (8.8) | 19 (6.4) |
Pyrexia | 17 (5.7) | 15 (5.1) |
Grade ≥ 3 | 191 (64.3) | 166 (55.9) |
COVID-19 pneumonia | 39 (13.1) | 30 (10.1) |
Pneumonia | 24 (8.1) | 18 (6.1) |
COVID-19 | 23 (7.7) | 18 (6.1) |
Pyrexia | 7 (2.4) | 4 (1.3) |
Patients who stopped treatment due to adverse events (reported in ≥ 1% of patients in either group), n (%) | ||
Patients with ≥ 1 TEAE leading to discontinuation of acalabrutinib or placebo, n (%) | 127 (42.8) | 92 (31.0) |
COVID-19 | 14 (4.7) | 9 (3.0) |
COVID-19 pneumonia | 13 (4.4) | 8 (2.7) |
Neutropenia | 12 (4.0) | 10 (3.4) |
Pneumonia | 5 (1.7) | 1 (0.3) |
Hepatitis B reactivation | 4 (1.3) | 5 (1.7) |
Sepsis | 1 (0.3) | 3 (1.0) |
Deaths (reported in ≥ 1% of patients in either group), n (%) | ||
Patients contributing to the analysis, n | 299 | 299 |
Patients who died, n (%) | 97 (32.4) | 106 (35.5) |
Adverse event | 46 (15.4) | 41 (13.7) |
COVID-19 pneumonia | 15 (5.1) | 10 (3.4) |
COVID-19 | 8 (2.7) | 6 (2.0) |
Pneumonia | 3 (1.0) | 0 |
Dyspnea | 1 (0.3) | 0 |
Disease progression | 30 (10.0) | 43 (14.4) |
Other | 14 (4.7) | 16 (5.4) |
Unknown | 7 (2.3) | 6 (2.0) |
Notable harms, n (%) | ||
Infections and infestations | 232 (78.1) | 211 (71.0) |
Absolute difference in risk between study groups, % (95% CI) | ████ ██████ ██ █████ | |
Ventricular arrythmias | 7 (2.4) | 7 (2.4) |
Absolute difference in risk between study groups, % (95% CI) | █████ ██ ████ | |
Atrial fibrillation and/or flutter | 20 (6.7) | 13 (4.4) |
Absolute difference in risk between study groups, % (95% CI) | ████ █████ ██ ████ | |
Hemorrhage | 84 (28.3) | 51 (17.2) |
BR = bendamustine plus rituximab; CI = confidence interval; SAE = serious adverse event; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
Source: ECHO Clinical Study Report (2024)25 and sponsor-provided additional data (April 30, 2025).58 Details included in the table are from the sponsor’s summary of clinical evidence.26
Critical Appraisal
Internal Validity
The ECHO trial was well conducted overall, but it was not without limitations that could introduce bias and/or uncertainty. Patients were randomized via an interactive voice and/or web response system, which was adequate to conceal allocation until randomization; therefore, there is a low risk of bias arising from the randomization process. There was generally no notable imbalance in the baseline patient demographic and disease characteristics between treatment groups.
The choice of placebo plus BR as the comparator was clinically relevant as BR is the current standard of care for the target patient population in Canada according to the clinical experts consulted by the CDA-AMC review team. Patients in the placebo plus BR group were allowed to cross over to receive acalabrutinib monotherapy if they experienced disease progression. The clinical experts commented that the crossover was reasonable and reflective of current treatment sequencing in clinical practice in Canada. The review team notes that acalabrutinib is currently not funded in the second line for MCL. Patients have access to acalabrutinib in the second-line setting only through private coverage or out-of-pocket payment. A patient support program is offered by the sponsor to help patients with private insurance navigate their coverage options.
The ECHO trial was double blinded and used an IRC; there is therefore a low risk of bias in the measurement of objective outcomes such as OS, and bias in measuring PFS is reduced. There is some risk that patients could have become aware of the treatment group to which they were assigned due to differences in treatment-related AEs; if this occurred, there would be some concerns for risk of bias in subjective outcomes, such as HRQoL.
PFS and OS were reported in the ECHO trial as survival outcomes. PFS was the primary efficacy end point in the ECHO trial. The clinical experts consulted by the CDA-AMC review team noted that the selection of PFS as the primary efficacy end point was acceptable and informative. This is because with current treatments for MCL the intent of therapy is not curative, and PFS provides information on the relative estimates of treatment effect, compared to BR alone, on disease control and stabilization. The clinical experts confirmed that the definition of disease progression used in the trial (criteria based on the Lugano classification for NHL) was well accepted and reflective of current clinical practice in Canada. The analysis of PFS was based on blinded IRC assessment, which is appropriate to maintain trial integrity with interim analyses. PFS is often used as a surrogate end point for OS in oncology trials.59,60 A meta-analysis reported a moderate correlation between PFS and OS in NHL (including MCL). However, this correlation was not based on chemotherapy in combination with immunotherapy and is therefore uncertain in the context of acalabrutinib.45-47 The review team noted that a clear surrogate relationship between PFS and OS in untreated MCL is uncertain in the context of acalabrutinib use. Despite the reported improvements in PFS, it remains uncertain whether these benefits will translate into an improvement in OS.
The clinical experts acknowledged OS as a key clinically important outcome for patients with MCL. However, only results from the interim analysis were available for this review. The median OS was not reached in either treatment group; therefore, uncertainty remains in the OS evidence, and this finding is not interpretable at the time of the interim analysis. The final OS analysis, which is anticipated to occur in ████████ ████ when the projected number of OS events is ███, will provide further information. The OS results are further confounded by the crossover, when patients in the control arm (placebo plus BR) were allowed to switch to the experimental treatment (acalabrutinib monotherapy) upon disease progression, which may dilute any true survival benefit attributable to acalabrutinib in the first-line setting. Moreover, no statistical method was used to adjust for crossover. The ECHO study was not powered to detect statistically significant OS differences because the analysis of OS was not controlled for type I error rate for multiple comparisons; therefore, the OS results should be interpreted with caution because they were considered supportive evidence.
Because the upper bound of the 95% CI for median PFS in the acalabrutinib plus BR group was not estimable and the median OS was not reached in either treatment group, the early reporting of the study results in an interim analysis may lead to an overestimation of the treatment effect.22 The sponsor has planned final PFS and OS analyses with a longer follow-up time and more events (anticipated to happen in 2027); however, there is an increased risk of type I error because multiple analyses may raise the probability of falsely rejecting the null hypothesis. Although the sponsor used the Lan-DeMets alpha-spending function to control type I error for PFS, ORR, and OS, the testing hierarchy stopped at ORR because the P value exceeded 0.05. The OS analysis in the ECHO trial was not controlled for type I error because it was conducted after the ORR analysis, following the predefined sequence of end points. As a result, the findings related to OS may be subject to an increased risk of type I error, limiting credible conclusions about this end point. Many of the outcomes used in the ECHO trial (PFS, OS, DOR, and HRQoL) were identified as clinically important by patients and/or clinicians. However, DOR and HRQoL were not part of the statistical testing strategy and thus were not adjusted for multiple testing; therefore, these results should be considered supportive evidence only.
As is common in oncology trials, the results for OS are reflective of the effects of acalabrutinib plus BR versus placebo plus BR and of any subsequent non–protocol-specified treatment for MCL received in each group (rather than each treatment in isolation). Although the effects of acalabrutinib plus BR versus placebo plus BR are confounded by subsequent treatments (used more frequently in the placebo plus BR group), the comparison is relevant because these treatments are reflective of those used in practice in Canada according to the clinical experts consulted for this review.
Sensitivity analyses, including investigator assessment, were conducted for PFS to assess the robustness of the results. Overall, the results were consistent with the primary analysis. The results from the analysis of DOR for CR and PR generally aligned with the PFS analysis. Subgroup analyses of PFS indicated that the treatment effect was notably different between male and female patients. However, there were no statistical tests for treatment by subgroup interactions, and there were no adjustments for multiple testing, limiting credible conclusions about effect modification.
A higher proportion of patients in the acalabrutinib plus BR group than in the placebo plus BR group used concomitant antibacterials, and a lower proportion of patients in the acalabrutinib plus BR group than in the placebo plus BR group used concomitant antigout preparations; this may affect the safety of bendamustine because concomitant use of these medications may be associated with rash, according to the clinical experts and the product monograph for bendamustine.40,61 These imbalances may bias the safety results in favour of acalabrutinib.
Few patients were lost to follow-up; therefore, there is a low risk of bias due to missing outcome data for the time-to-event end points (OS and PFS).
A treatment benefit of acalabrutinib plus BR in comparison with placebo plus BR on HRQoL was not observed in the ECHO trial at cycle 48. There was notable attrition in the number of patients included in the analysis of HRQoL, with only approximately ███ of patients contributing to the analysis at cycle 48 day 1. Moreover, more patients in the acalabrutinib plus BR group (███) provided HRQoL data than in the placebo plus BR group (███). Therefore, there is a high risk of bias due to missing outcome data for HRQoL because the characteristics of patients who remained in the study may differ from those who did not. Missing data were not imputed in the HRQoL analyses. Moreover, no sensitivity analyses were conducted for the analysis of HRQoL. Statistical testing for this outcome was not adjusted for multiple testing in the trial and should be considered supportive evidence.
The certainty of evidence for the between-group effect estimates for PFS and OS was affected by imprecision. Although the point estimates for the between-group differences in KM-estimated probabilities of PFS at 48 months of follow-up suggested a clinically important benefit with acalabrutinib, the lower bound of the 95% CIs included the possibility of no benefit based on the minimal important difference of 7% as estimated by clinical experts. For OS, although the point estimate and upper bound of the 95% CI for between-group differences in KM-estimated probabilities at 48 months of follow-up suggested little to no clinically important difference, the lower bound of the 95% CIs included the potential for a clinically important benefit with acalabrutinib based on the minimal important difference of 5% as estimated by clinical experts. This uncertainty was also impacted by the interim analysis and the uncertain surrogate relationship between PFS and OS for untreated MCL.
External Validity
According to the clinical experts consulted, the characteristics of the patients enrolled in the ECHO trial were mostly reflective of patients in clinical practice in Canada with untreated MCL who would be eligible for treatment with acalabrutinib, with a few exceptions. Generally, the clinical experts commented that the eligibility criteria of the ECHO trial were standard for clinical trials but stricter than current clinical practice in Canada. The indication for acalabrutinib is for the treatment of patients with MCL who are ineligible for ASCT. In the ECHO trial, patients were required to be aged 65 years or older. According to the clinical experts, 65 years is a historical age cut-off for patients’ eligibility for ASCT, but a more contemporary cut-off of 70 years is commonly used in clinical practice; therefore, the clinical decision of whether to proceed with ASCT would be individualized based on the patient’s comorbidities and functional status. The clinical experts indicated that although it is very uncommon for patients to have no measurable disease (< 5% of cases), they still expect this treatment with acalabrutinib to be effective in such cases because some patients’ disease may only involve bone marrow and blood. The trial did not include patients whose absolute neutrophil count was less than 1.0 × 109/L or whose platelet count was less than 75 × 109/L and patients with disease involvement in the bone marrow whose absolute neutrophil count was less than 0.75 × 109/L or whose platelet count was less than 50 × 109/L. The clinical experts commented that this is a standard trial exclusion criterion but that in clinical practice those patients may be generally considered eligible for treatment with acalabrutinib. Additionally, patients whose estimated creatinine clearance was less than 50 mL/min were not eligible for the ECHO trial. The clinical experts indicated that a cut-off of 30 mL/min is generally used for acalabrutinib therapy in clinical practice.19
Overall, the patients in the ECHO trial may be somewhat healthier than in general clinical practice in Canada. Nearly half the patients (49.5%) in the ECHO trial had an ECOG Performance Status score of 0. The clinical experts commented that this percentage is higher than what they typically observe in clinical practice: they see a higher percentage of patients with an ECOG Performance Status score ranging from 0 to 2. Patients with Ann Arbor stage I to II disease were included in the trial; however, there were few patients with this stage disease (< 6%). The clinical experts indicated that patients with Ann Arbor stage I or II MCL generally have a better prognosis than those with more advanced stages, and typical systemic therapy regimens are not always used for them because they often get combined modality therapy. Therefore, the ECHO trial results may not be applicable to patients with Ann Arbor stage I to II disease in Canadian clinical practice. Overall, the review team notes that the study included a patient population that may be somewhat healthier than patients with MCL who are ineligible for ASCT in clinical practice. As a result, the absolute PFS and OS outcomes observed in the trial may be more favourable than would be expected in routine care.
The selected end points were considered relevant to patients and clinicians, who underscored the need for treatments that improve PFS, extend OS, and maintain HRQoL while limiting toxicity. Although maintaining HRQoL is important for patients, the results from the ECHO trial do not allow the review team to confidently draw conclusions about the effect of acalabrutinib plus BR versus placebo plus BR on this end point due to the risk of bias due to attrition.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal study and/or RCT identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:23,24
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The reference points for the certainty of evidence assessment for PFS, OS, and harms were set according to the presence of an important effect based on thresholds agreed on by the clinical experts consulted for this review.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for acalabrutinib plus BR versus placebo plus BR.
Acalabrutinib plus BR likely results in a clinically important increase in the probability of being alive and progression-free at 48 months when compared with placebo plus BR (moderate certainty due to serious imprecision).
Acalabrutinib plus BR likely results in little to no clinically important difference in the probability of being alive at 48 months compared with placebo plus BR (moderate certainty due to serious imprecision).
Acalabrutinib plus BR may result in little to no clinically important improvement in FACT-Lym total score at cycle 48 day 1 compared with placebo plus BR (low certainty due to very serious imprecision).
Acalabrutinib plus BR likely results in little to no clinically important difference in the incidence of infections and infestations compared with placebo plus BR (moderate certainty due to serious imprecision).
Acalabrutinib plus BR may result in little to no clinically important difference in the incidence of ventricular arrythmias compared with placebo plus BR (low certainty due to very serious imprecision).
Acalabrutinib plus BR likely results in little to no clinically important difference in the incidence of atrial fibrillation and/or flutter compared with placebo plus BR (moderate certainty due to serious imprecision).
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
No indirect comparisons were submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
The ECHO trial initially used acalabrutinib 100 mg capsules. As of February 27, 2023, Health Canada approved a 100 mg acalabrutinib maleate salt oral tablet formulation, which is now marketed to replace the capsule format; the capsule has not been supplied in Canada since July 31, 2024. The tablets reduce the impact of acid-reducing drugs (e.g., proton pump inhibitors) on acalabrutinib. The dosing is identical for the formulations, and the tablets are smaller in volume. The sponsor submitted 2 phase I, open-label, randomized, crossover studies (D8220C00018 and D8223C00013 trials) to assess the bioequivalence of the acalabrutinib maleate film-coated tablet and the acalabrutinib capsule. The results from the 2 studies concluded that acalabrutinib tablets and capsules are bioequivalent, indicating that the same efficacy and safety profile can be expected with the same dosing strength and schedule.27,28 Patients are able to co-administer tablets with acid-reducing drugs such as proton pump inhibitors, and this removes the need to stagger dosing with H2 receptor antagonists and antacids. The CDA-AMC review team considers the D8220C00018 and D8223C00013 trials out of scope for this review because the patients enrolled in the 2 trials were healthy patients without MCL. Therefore, the CDA-AMC review team acknowledges these studies but considers that they do not address gaps in the systematic review evidence for this review.
Discussion
Summary of Available Evidence
The ECHO trial is an ongoing phase III, double-blind, placebo-controlled RCT to evaluate the efficacy and safety of acalabrutinib plus BR (n = 299) compared to placebo plus BR (n = 299) in adult patients with previously untreated MCL who are ineligible for ASCT. The primary objective of the ECHO trial was to evaluate the efficacy of acalabrutinib plus BR compared to placebo plus BR in prolonging PFS in patients with previously untreated MCL who are ineligible for ASCT. Secondary outcomes included ORR, OS, HRQoL, and safety. The baseline demographic and disease characteristics were balanced between the 2 treatment groups. The median age of all patients was 71.0 years and ranged from 65 to 86 years. Most patients were male (70.7%; female: 29.3%), and most were white (78.3%). The trial also included patients self-reporting their ethnicity as Asian, American Indian or Alaska Native, Black or African American, and multiple. Nearly half the patients (49.5%) had an ECOG Performance Status of 0. All the patients (100%) had histologically documented MCL, and close to half the patients (42.3%) had a simplified MIPI score of 4 to 5 (intermediate risk). The most reported MCL type was classic (80.4%). The majority of patients (86.0%) had Ann Arbor stage IV disease, 90.5% had extranodal disease, and 71.7% had bone marrow involvement. Generally, the baseline disease history was balanced across treatment groups, ████████ ████████ ███████ was the most reported medical history in patients in the ECHO trial.
Interpretation of Results
Efficacy
The indication for acalabrutinib plus BR is for the treatment of adult patients with previously untreated MCL who are ineligible for ASCT. Currently, BR is the standard of care for the target patient population in clinical practice in Canada. The review team notes a limitation that there is no evidence describing the comparative effectiveness of acalabrutinib plus BR versus other funded regimens used in this population (e.g., R-CHOP and R-CVP). However, the clinical experts consulted by CDA-AMC stated that these regimens are only used rarely (estimated < 15% of patients in Canada) for the treatment of transplant-ineligible MCL, for example in patients for whom the pathology is inconclusive, who have severe skin reactions to BR, or who have compelling reasons to complete therapy sooner with R-CHOP than with BR.
According to the clinical experts consulted, the characteristics of the patients enrolled in the ECHO trial were mostly reflective of patients in clinical practice in Canada. However, the ECHO trial may have included a favourable patient population that may not be reflective of all patients with MCL who are ineligible for ASCT in clinical practice. For example, in clinical practice in Canada, the clinical experts observe a higher percentage of patients with a poorer ECOG Performance Status (ranging from 0 to 2) than that in the ECHO trial and fewer patients with a better prognosis (Ann Arbor stage I to II) than were included in the trial. Overall, these limitations may impact the generalizability of the study results to clinical practice.
Survival (OS and PFS) and HRQoL outcomes (EQ-5D-5L and FACT-Lym) were reported in the ECHO study. These efficacy end points were selected as being important based on the input from the patient and clinician groups as well as from the clinical experts consulted by the CDA-AMC review team. These efficacy outcomes aligned with the outcomes identified by patients as important: prolonging life, achieving longer disease remission, controlling disease symptoms, and improving quality of life. Because the intent of therapy with current treatments is not curative, the goals of treatment for patients with MCL include managing symptoms, maintaining quality of life, and prolonging PFS, rather than solely extending life. Additionally, the input from the clinician groups highlighted that crossover was employed in the ECHO trial, which makes it unlikely that an OS advantage would be demonstrated, particularly because the ECHO trial was not designed to evaluate OS in any meaningful way.
While acknowledging OS as the most clinically relevant efficacy end point for patients with MCL, the clinical experts consulted by the CDA-AMC review team also considered PFS an important end point. Based on the results of the interim analysis of the ECHO trial (data cut-off date: February 15, 2024), treatment with acalabrutinib plus BR resulted in an improvement in the primary end point of PFS compared with placebo plus BR for patients with MCL who are ineligible for ASCT (HR = 0.73; 95% CI, 0.57 to 0.94; P = 0.0160); the between-group difference in PFS probability at 48 months was ████ (95% CI, ████ to █████). There was no established minimal important difference for the PFS probability in this population; the clinical experts consulted by CDA-AMC suggested a 7% difference as a threshold of minimal clinical importance based on their clinical experience. Therefore, based on the GRADE approach, acalabrutinib plus BR likely results in a clinically meaningful benefit in PFS at 48 months compared to placebo plus BR, based on moderate certainty evidence. However, there was a potential risk of overestimation in treatment effect, as the evidence was based on the results from an interim analysis.22 Additionally, although some studies have shown that PFS is a surrogate outcome that is positively correlated with OS in NHL, this correlation was based on trials of chemotherapy and not targeted therapy and is therefore uncertain in the context of acalabrutinib.45-47 Despite the reported improvements in PFS, the degree to which the observed PFS benefits in the ECHO trial could be translated to an improvement in OS remains uncertain as the median OS was not reached in either treatment group and the data lacked maturity.
Sensitivity analyses including investigator assessment were conducted for PFS to assess the robustness of the results; overall, the results were consistent with those of the primary analysis. The results from the analysis of DOR for CR and PR appeared aligned with the PFS analysis. Subgroup analyses of PFS indicated that the treatment effect was notably different between males (HR = 0.91; 95% CI, 0.68 to 1.21) and females (HR = 0.34; 95% CI, 0.19 to 0.58). Because the female subgroup represented approximately 29.3% of the study population in the primary analysis — 175 patients of 598 total with a low number of events (60 events) — the review team notes that the results were very uncertain. Additionally, although the subgroup analyses of the primary end point were prespecified, 95% CIs were wide and generally overlapping across categories within each subgroup (except male versus female sex), there were no statistical tests for treatment by subgroup interactions, and there were no adjustments for multiple testing, limiting credible conclusions about effect modification.62,63 Although credible effect modification could not be inferred from these analyses, the results raise uncertainty as to whether one should expect the real-world magnitude of benefit to differ between males and females. However, the clinical experts consulted for this review advised that the difference in treatment effects observed for the subgroup analysis by male and female sex were likely attributable to chance.
The evidence regarding OS reviewed in this report was based on an interim analysis, and the median OS was not reached in either treatment group. At the time this report was prepared, the duration of follow-up in the interim analysis (median for acalabrutinib plus BR: 46.1 months; median for placebo plus BR: 44.4 months) was considered adequate for the assessment of the primary efficacy end point of PFS but inadequate for the assessment of OS, as per feedback from the clinical experts. Therefore, there remains a gap in the evidence regarding OS to be addressed by the final analysis (anticipated ████████ ████) with longer follow-up and more events. The point estimate of the interim OS analysis favoured the acalabrutinib group, but the results were not statistically significant due to imprecision (i.e., the 95% CI crossed the null). Based on the assessment of the certainty of evidence using the GRADE approach, there was moderate certainty that acalabrutinib plus BR results in little to no clinically important difference in the probability of being alive at 48 months, compared to placebo plus BR. Additionally, the ECHO trial was not designed to detect a statistically significant difference in OS. Because the first key secondary end point, ORR, did not reach statistical significance, testing was halted during the interim analysis. Consequently, the type I error rate in the OS analysis was not adjusted for multiple comparisons, and the results should be viewed as supportive evidence with limited interpretability.
Patient and clinician groups indicated that maintaining or improving HRQoL is an important goal of treatment for MCL. The results of the ECHO trial suggested that there may be little to no treatment benefit (and no detriment) of acalabrutinib plus BR in comparison with placebo plus BR for HRQoL measured as change from baseline in FACT-Lym total score at cycle 48 (approximately 48 months of treatment), based on a 7-point threshold for increase as identified in the literature.54 Similar findings were observed in the EQ-5D-5L utility score. However, there was a high proportion of missing data; therefore, these data are at risk of bias due to attrition. The number of patients contributing to the analysis was ██ out of 299 patients in the acalabrutinib plus BR group and ██ out of 299 patients in the placebo plus BR group at maintenance cycle 48 day 1. This high rate of attrition introduces uncertainty and difficulty in interpreting the effect of acalabrutinib plus BR on HRQoL. Additionally, the type I error rate for the HRQoL analysis was not adjusted for multiple comparisons, and the results should be viewed as supportive evidence.
Harms
Acalabrutinib has been available in Canada for patients with chronic lymphocytic leukemia since 2020, providing extensive clinical experience with the drug. Overall, no new safety signals for acalabrutinib plus BR were identified in the ECHO trial. Generally, increased AEs were observed, and more patients stopped treatment due to AEs in the acalabrutinib plus BR group than in the placebo plus BR group. For example, greater incidence of diarrhea and COVID-19 were observed in the acalabrutinib plus BR group than in the placebo plus BR group. The clinical experts commented that imbalances were expected because these are known AEs of acalabrutinib. B-cell suppressive drugs like acalabrutinib and rituximab may increase the risk of developing viral infections (e.g., COVID-19) according to the clinical experts. The incidence of diarrhea and COVID-19 in the acalabrutinib plus BR may be underestimated compared to the placebo plus BR group because higher proportions of patients in the acalabrutinib plus BR group received antidiarrheals and COVID-19 vaccination. The clinical experts and input from the clinician group highlighted increased hemorrhage and cardiovascular events (i.e., ventricular arrythmias and atrial fibrillation and/or flutter) with acalabrutinib. The clinical experts commented that these are AEs related to the class of BTK inhibitors (e.g., acalabrutinib and ibrutinib). The clinical experts indicated that they may consider prescribing acalabrutinib to more fit patients with controlled cardiovascular disease but would avoid prescribing the drug to patients with higher risk of cardiovascular events, given a lack of survival benefit in those patients in this trial. Overall, the increased diarrhea, infections, hemorrhage, and cardiovascular events were expected with acalabrutinib and would contribute to disease management burden, according to feedback from the clinical experts.
Conclusion
One ongoing phase III, double-blind, placebo-controlled RCT (ECHO trial) comparing acalabrutinib plus BR to placebo plus BR in adult patients with previously untreated MCL who are ineligible for ASCT was included in this review. The results from the ECHO trial demonstrated that the addition of acalabrutinib to BR likely results in a clinically important benefit in PFS. However, there was moderate certainty that acalabrutinib plus BR results in little to no clinically important difference in OS because the median OS was not reached in either study group in the interim analysis, and there were other limitations with the interpretation of the OS data, including crossover, lack of long-term follow-up, and an absence of adjustments for multiple comparisons. The results suggest that HRQoL may not be affected by the addition of acalabrutinib to BR because there was low certainty that acalabrutinib plus BR result in little to no clinically important difference in the FACT-Lym total score compared with placebo plus BR.
Compared with placebo plus BR, there is low to moderate certainty evidence that acalabrutinib plus BR results in little to no clinically important difference in the incidence of infections and infestations and of atrial fibrillation and/or flutter, and it may result in little to no clinically important difference in the incidence of ventricular arrythmias. Overall, no new safety signals were identified in the ECHO trial, and the observed safety profile of acalabrutinib plus BR is as expected but with added toxicities over standard treatment, according to feedback from the clinical experts.
References
1.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-e623. doi:10.1503/cmaj.240095 PubMed
2.Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol. 1998;16(8):2780-95. doi:10.1200/jco.1998.16.8.2780 PubMed
3.Statistics Canada. Table 13-10-0751-01 Number of prevalent cases and prevalence proportions of primary cancer, by prevalence duration, cancer type, attained age group and sex [sponsor supplied reference]. https://doi.org/10.25318/1310075101-eng
4.Epperla N, Hamadani M, Fenske TS, Costa LJ. Incidence and survival trends in mantle cell lymphoma. Br J Haematol. 2018;181(5):703-706. doi:10.1111/bjh.14699 PubMed
5.Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer. 2008;113(4):791-8. doi:10.1002/cncr.23608 PubMed
6.Velders GA, Kluin-Nelemans JC, De Boer CJ, et al. Mantle-cell lymphoma: a population-based clinical study. J Clin Oncol. 1996;14(4):1269-74. doi:10.1200/jco.1996.14.4.1269 PubMed
7.Radkiewicz C, Bruchfeld JB, Weibull CE, et al. Sex differences in lymphoma incidence and mortality by subtype: A population-based study. Am J Hematol. 2023;98(1):23-30. doi:10.1002/ajh.26744 PubMed
8.Xie S, Yu Z, Feng A, et al. Analysis and prediction of relative survival trends in patients with non-Hodgkin lymphoma in the United States using a model-based period analysis method. Front Oncol. 2022;12:942122. doi:10.3389/fonc.2022.942122 PubMed
9.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-90. doi:10.1182/blood-2016-01-643569 PubMed
10.Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748. doi:10.1038/s41375-022-01620-2
11.Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229-1253. doi:10.1182/blood.2022015851 PubMed
12.Dreyling M, Campo E, Hermine O, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl_4):iv62-iv71. doi:10.1093/annonc/mdx223 PubMed
13.Villa D, Kansara R, Lemieux C, Kuruvilla J. Updates in the treatment of mantle cell lymphoma: A Canadian expert framework. Canadian Hematology Today. 2022;1(S12):2-11. doi:10.58931/cht.2022.1S1227
14.Sandoval-Sus JD, Sotomayor EM, Shah BD. Mantle Cell Lymphoma: Contemporary Diagnostic and Treatment Perspectives in the Age of Personalized Medicine. Hematol Oncol Stem Cell Ther. 2017;10(3):99-115. doi:10.1016/j.hemonc.2017.02.003 PubMed
15.Doorduijn JK, Kluin-Nelemans HC. Management of mantle cell lymphoma in the elderly patient. Clin Interv Aging. 2013;8:1229-36. doi:10.2147/cia.s35082 PubMed
16.Cheah CY, Seymour JF, Wang ML. Mantle Cell Lymphoma. J Clin Oncol. 2016;34(11):1256-69. doi:10.1200/jco.2015.63.5904 PubMed
17.Inamdar AA, Goy A, Ayoub NM, et al. Mantle cell lymphoma in the era of precision medicine-diagnosis, biomarkers and therapeutic agents. Oncotarget. 2016;7(30):48692-48731. doi:10.18632/oncotarget.8961 PubMed
18.Suleman A, Ante Z, Liu N, et al. Outcomes of Transplant-Eligible and Transplant-Ineligible Patients with Mantle Cell Lymphoma in Ontario, Canada. Blood. 2023;142(Supplement 1):1665-1665. doi:10.1182/blood-2023-177788
19.AstraZeneca Canada Inc. Calquence (acalabrutinib as acalabrutinib maleate): 100 mg oral tablets [product monograph]. June 24, 2025.
20.CADTH. pCODR Expert Review Committee (pERC) final recommendation: Acalabrutinib (Calquence - AstraZeneca Canada Inc.) for previously untreated chronic lymphocytic leukemia. 2020. Accessed May 28, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10210AcalabrutinibCLL%28previously%20untreated%29_FnRec_pERC%20Chair%20Approved_REDACT_Post08Jan2021_final.pdf
21.CADTH. pCODR Expert Review Committee (pERC) final recommendation: Acalabrutinib (Calquence - AstraZeneca Canada Inc.) for previously treated chronic lymphocytic leukemia. 2021. Accessed May 28, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10211AcalabrutinibCLL_fnRec_REDACT_EC_Post17Nov2020_final.pdf
22.Ciolino JD, Kaizer AM, Bonner LB. Guidance on interim analysis methods in clinical trials. J Clin Transl Sci. 2023;7(1):e124. e124. doi:10.1017/cts.2023.552
23.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
24.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
25.AstraZeneca Canada Inc. Clinical Study Report (Data cut off: 15 February 2024): D8220C00004 (ACE-LY-308). A Phase 3, Randomized, Double-blind, Placebo-controlled, Multicenter Study of Bendamustine and Rituximab (BR) Alone Versus in Combination with Acalabrutinib (ACP-196) in Subjects with Previously Untreated Mantle Cell Lymphoma (ECHO) [internal sponsor's report]. July 18, 2024.
26.AstraZeneca Canada Inc. Acalabrutinib in combination with bendamustine and rituximab for the treatment of adult patients with previously untreated mantle cell lymphoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Calquence (acalabrutinib), 100 mg, oral tablets. March 2025.
27.AstraZeneca AB. Clinical Study Report (Version 1.0): Study D8220C00018 (ACE-HV-115). A 2-Part, Phase I, Open-label, Single-dose, Sequential Randomized Crossover Study of New Acalabrutinib Maleate Tablet in Healthy Subjects to Evaluate Relative Bioavailability, Proton Pump Inhibitor (Rabeprazole) Effect, Food Effect and Particle Size Effect [internal sponsor's report]. May 31, 2021.
28.AstraZeneca AB. Clinical Study Report: D8223C00013. A Phase I, Open-Label, Randomized, 2-Treatment, 2-Period, Crossover Study in Healthy Subjects to Assess the Bioequivalence of Acalabrutinib Tablet and Acalabrutinib Capsule [internal sponsor's report]. August 18, 2021.
29.Armitage JO, Longo DL. Mantle-Cell Lymphoma. N Engl J Med. 2022;386(26):2495-2506. doi:10.1056/NEJMra2202672 PubMed
30.Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750-62. doi:10.1038/nrc2230 PubMed
31.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-68. doi:10.1200/jco.2013.54.8800 PubMed
32.Jain P, Wang ML. Mantle cell lymphoma in 2022-A comprehensive update on molecular pathogenesis, risk stratification, clinical approach, and current and novel treatments. Am J Hematol. 2022;97(5):638-656. doi:10.1002/ajh.26523 PubMed
33.Anderson JR, Armitage JO, Weisenburger DD. Epidemiology of the non-Hodgkin's lymphomas: distributions of the major subtypes differ by geographic locations. Non-Hodgkin's Lymphoma Classification Project. Ann Oncol. 1998;9(7):717-20. doi:10.1023/a:1008265532487 PubMed
34.Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459. doi:10.3322/caac.21357 PubMed
35.Al-Hamadani M, Habermann TM, Cerhan JR, Macon WR, Maurer MJ, Go RS. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am J Hematol. 2015;90(9):790-5. doi:10.1002/ajh.24086 PubMed
36.U. S. Food and Drug Administration. FDA approves acalabrutinib with bendamustine and rituximab for previously untreated mantle cell lymphoma. January 16, 2025. Accessed May 28, 2025. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-acalabrutinib-bendamustine-and-rituximab-previously-untreated-mantle-cell-lymphoma
37.B. C. Cancer Agency. BC Cancer Protocol Summary for Treatment of Non-Hodgkin Lymphoma with Bendamustine and Rituximab (LYBENDR). 2024. Accessed June 10, 2025. https://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma-Myeloma/LYBENDR_Protocol.pdf
38.Ontario Health/Cancer Care Ontario. Bendamustine + rituximab Protocol (BEND+RITU Regimen) [sponsor submitted reference]. https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/46691
39.Ontario Health/Cancer Care Ontario. CHOP+R Regimen: Cyclophosphamide-Hydroxyldaunorubicin (DOXOrubicin)-ONCOVIN® (VinCRIStine)-Prednisone-riTUXimab [sponsor supplied reference]. https://www.cancercareontario.ca/en/drugformulary/regimens/45801
40.Teva Canada Limited. Treanda (bendamustine hydrochloride for injection): 25 mg/ vial and 100 mg/ vial for intravenous infusion [product monograph]. January 10, 2018. Accessed May 05, 2025. https://pdf.hres.ca/dpd_pm/00043152.PDF
41.Hoffmann-La Roche Ltd. Rituxan SC (rituximab): 120 mg/mL solution for subcutaneous injection [product monograph]. November 19, 2020. Accessed May 05, 2025. https://pdf.hres.ca/dpd_pm/00058908.PDF
42.Wang M, Salek D, Belada D, et al. Acalabrutinib Plus Bendamustine-Rituximab in Untreated Mantle Cell Lymphoma. J Clin Oncol. 2025:101200jco2500690. doi:10.1200/jco-25-00690
43.Acerta Pharma, BV. Clinical Study Protocol (Version 4.0): D8220C00004 (ACE-LY-308). Phase 3, Randomized, Double-blind, Placebo-controlled, Multicenter Study of Bendamustine and Rituximab (BR) Alone Versus in Combination with Acalabrutinib (ACP-196) in Subjects with Previously Untreated Mantle Cell Lymphoma [internal sponsor's report]. June 06, 2023. Accessed May 05, 2025.
44.AstraZeneca Canada Inc. AstraZeneca Canada Inc. response to Canada's Drug Agency request for additional information regarding Calquence(acalabrutinib) review on April 17, 2025: Long-term data [internal additional sponsor's information]. April 22, 2025.
45.Zhu R, Lu D, Chu YW, et al. Assessment of Correlation Between Early and Late Efficacy Endpoints to Identify Potential Surrogacy Relationships in Non-Hodgkin Lymphoma: a Literature-Based Meta-analysis of 108 Phase II and Phase III Studies. AAPS J. 2017;19(3):669-681. doi:10.1208/s12248-017-0056-x PubMed
46.Wang ML, Jurczak W, Jerkeman M, et al. Ibrutinib plus Bendamustine and Rituximab in Untreated Mantle-Cell Lymphoma. N Engl J Med. 2022;386(26):2482-2494. doi:10.1056/NEJMoa2201817 PubMed
47.Merino M, Kasamon Y, Theoret M, Pazdur R, Kluetz P, Gormley N. Irreconcilable Differences: The Divorce Between Response Rates, Progression-Free Survival, and Overall Survival. J Clin Oncol. 2023;41(15):2706-2712. doi:10.1200/jco.23.00225 PubMed
48.van Reenen M, Stolk E, Boye K, et al. EQ-5D-5L User Guide Version 3.0. 2019. Accessed June 01, 2025. https://euroqol.org/publications/user-guides
49.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-36. doi:10.1007/s11136-011-9903-x PubMed
50.Bourke S, Bennett B, Oluboyede Y, et al. Estimating the minimally important difference for the EQ-5D-5L and EORTC QLQ-C30 in cancer. Health Qual Life Outcomes. 2024;22(1):81. doi:10.1186/s12955-024-02294-3 PubMed
51.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. doi:10.1186/1477-7525-5-70 PubMed
52.Carter GC, Liepa AM, Zimmermann AH, Morschhauser F. Validation of the Functional Assessment of Cancer Therapy–Lymphoma (FACT-LYM) in Patients with Relapsed/Refractory Mantle Cell Lymphoma. Blood. 2008;112(11):2376. doi:10.1182/blood.V112.11.2376.2376
53.Hlubocky FJ, Webster K, Beaumont J, et al. A preliminary study of a health related quality of life assessment of priority symptoms in advanced lymphoma: the National Comprehensive Cancer Network-Functional Assessment of Cancer Therapy - Lymphoma Symptom Index. Leuk Lymphoma. 2013;54(9):1942-6. doi:10.3109/10428194.2012.762977 PubMed
54.Cheson BD, Trask PC, Gribben JG, et al. Health-related quality of life and symptoms in patients with rituximab-refractory indolent non-Hodgkin lymphoma treated in the phase III GADOLIN study with obinutuzumab plus bendamustine versus bendamustine alone. Ann Hematol. 2017;96(2):253-259. doi:10.1007/s00277-016-2878-5 PubMed
55.Burman CF, Sonesson C, Guilbaud O. A recycling framework for the construction of Bonferroni-based multiple tests. Stat Med. 2009;28(5):739-61. doi:10.1002/sim.3513 PubMed
56.AstraZeneca. Statistical Analysis Plan (Version 6.0): D8220C00004 (ACE-LY-308). A Phase 3, Randomized, Double blind, Placebo-controlled, Multicenter Study of Bendamustine and Rituximab (BR) Alone Versus in Combination with Acalabrutinib (ACP-196) in Subjects with Previously Untreated Mantle Cell Lymphoma [internal sponsor's report]. July 12, 2024.
57.Dreyling M, Mayer J, Belada D, et al. High-Risk Subgroups and MRD: An Updated Analysis of the Phase 3 ECHO Trial of Acalabrutinib with Bendamustine/Rituximab in Previously Untreated Mantle Cell Lymphoma. Blood. 2024;144(Supplement 1):1626-1626. doi:10.1182/blood-2024-200290
58.AstraZeneca Canada Inc. AstraZeneca Canada Inc. response to Canada's Drug Agency request for additional information regarding Calquence(acalabrutinib) review on April 30, 2025: Additional efficacy and safety data [internal additional sponsor's information]. May 7, 2025.
59.U.S. Food and Drug Administration. Clinical trial endpoints for the approval of cancer drugs and biologics. Guidance for Industry. December 2018. Accessed May 29, 2025. https://www.fda.gov/media/71195/download
60.Committee for Medicinal Products for Human Use. Guideline on the clinical evaluation of anticancer medicinal products. European Medicines Agency; November 18, 2018. Accessed May 29, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-evaluation-anticancer-medicinal-products-revision-6_en.pdf
61.B. C. Cancer Agency. BC Cancer Drug Manual for Bendamustine. 2018. Accessed June 10, 2025. https://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Bendamustine_monograph.pdf
62.Schandelmaier S, Briel M, Varadhan R, et al. Development of the Instrument to assess the Credibility of Effect Modification Analyses (ICEMAN) in randomized controlled trials and meta-analyses. CMAJ. 2020;192(32):E901-e906. doi:10.1503/cmaj.200077 PubMed
63.Committee for Medicinal Products for Human Use. Guideline on the investigation of subgroups in confirmatory clinical trials. European Medicines Agency; 2019. Accessed June 11, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-subgroups-confirmatory-clinical-trials_en.pdf
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Response Assessments
Response assessment criteria based on the Lugano Classification for NHL:31
Progressive Disease
Progressive metabolic disease based on PET-CT scans:
Score 4 or 5 with an increase in intensity of uptake from baseline and/or new fluorodeoxyglucose (FDG)-avid foci consistent with lymphoma at interim or end-of-treatment assessment.
New FDG-avid foci consistent with lymphoma rather than another etiology (e.g., infection, inflammation). If uncertain regarding etiology of new lesions, biopsy or interval scan may be considered.
New or recurrent FDG-avid foci in bone marrow.
At least 1 of the following based on CT scans:
Cross product of the longest transverse diameter (LDi) of a lesion and perpendicular diameter progression response:
An individual node and/or lesion must be abnormal with:
LDi greater than 1.5 cm and
Increase by at least 50% from the nadir of the perpendicular diameter or the nadir of the cross product of the LDi and the perpendicular diameter and
An increase in LDi or shortest axis perpendicular to the LDi from nadir:
0.5 cm for lesions at most 2 cm
1.0 cm for lesions greater than 2 cm
In the setting of splenomegaly, the splenic length must:
increase by greater than 50% of the extent of its prior increase
beyond baseline (e.g., a 15-cm spleen must increase to more than 16 cm). If no prior splenomegaly, must increase by at least 2 cm from baseline
New or recurrent splenomegaly.
New or clear progression of preexisting nonmeasured lesions.
Regrowth of previously resolved lesions.
A new node more than 1.5 cm in any axis.
A new extranodal site greater than 1.0 cm in any axis; if less than 1.0 cm in any axis, its presence must be unequivocal and must be attributable to lymphoma.
Assessable disease of any size unequivocally attributable to lymphoma.
New or recurrent involvement in bone marrow.
Complete Response
Complete metabolic response based on PET-CT scans:
Score 1, 2, or 3 with or without a residual mass on the 5-point scale
In Waldeyer’s ring or extranodal sites with high physiologic uptake or with activation within spleen or marrow (e.g., with chemotherapy or myeloid colony-stimulating factors), uptake may be greater than normal mediastinum and/or liver. In this circumstance, complete metabolic response may be inferred if uptake at sites of initial involvement is no greater than surrounding normal tissue even if the tissue has high physiologic uptake
No new lesions
No evidence of FDG-avid disease in bone marrow
Complete radiologic response (all of the following) based on CT scans:
Target nodes and/or nodal masses must regress to no more than 1.5 cm in LDi
No extralymphatic sites of disease
Absent of nonmeasured lesion
Organ enlargement(s) regress to normal
No new lesions
Normal by morphology; if indeterminate, immunohistochemistry negative in bone marrow
Partial Response
Partial metabolic response based on PET-CT scans:
Score 4 or 5 with reduced uptake compared with baseline and residual mass(es) of any size
No new lesions
Residual uptake higher than uptake in normal bone marrow but reduced compared with baseline (diffuse uptake compatible with reactive changes from chemotherapy allowed). If there are persistent focal changes in the bone marrow in the context of a nodal response, consideration should be given to further evaluation with MRI or biopsy or an interval scan
Partial remission (all of the following) based on CT scans:
At least 50% decrease in sum of the product of the perpendicular diameters for multiple lesions of up to 6 target measurable nodes and extranodal sites
When a lesion is too small to measure on CT, assign 5 mm by 5 mm as the default value
When a lesion is no longer visible, 0 by 0 mm
For a node greater than 5 mm by 5 mm, but smaller than normal, use actual measurement for calculation
Absent and/or normal, regressed, but no increase in nonmeasured lesions
Spleen must have regressed by more than 50% in length beyond normal
No new lesions
Figure 5: Forest Plot of Subgroup Analyses on PFS in the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
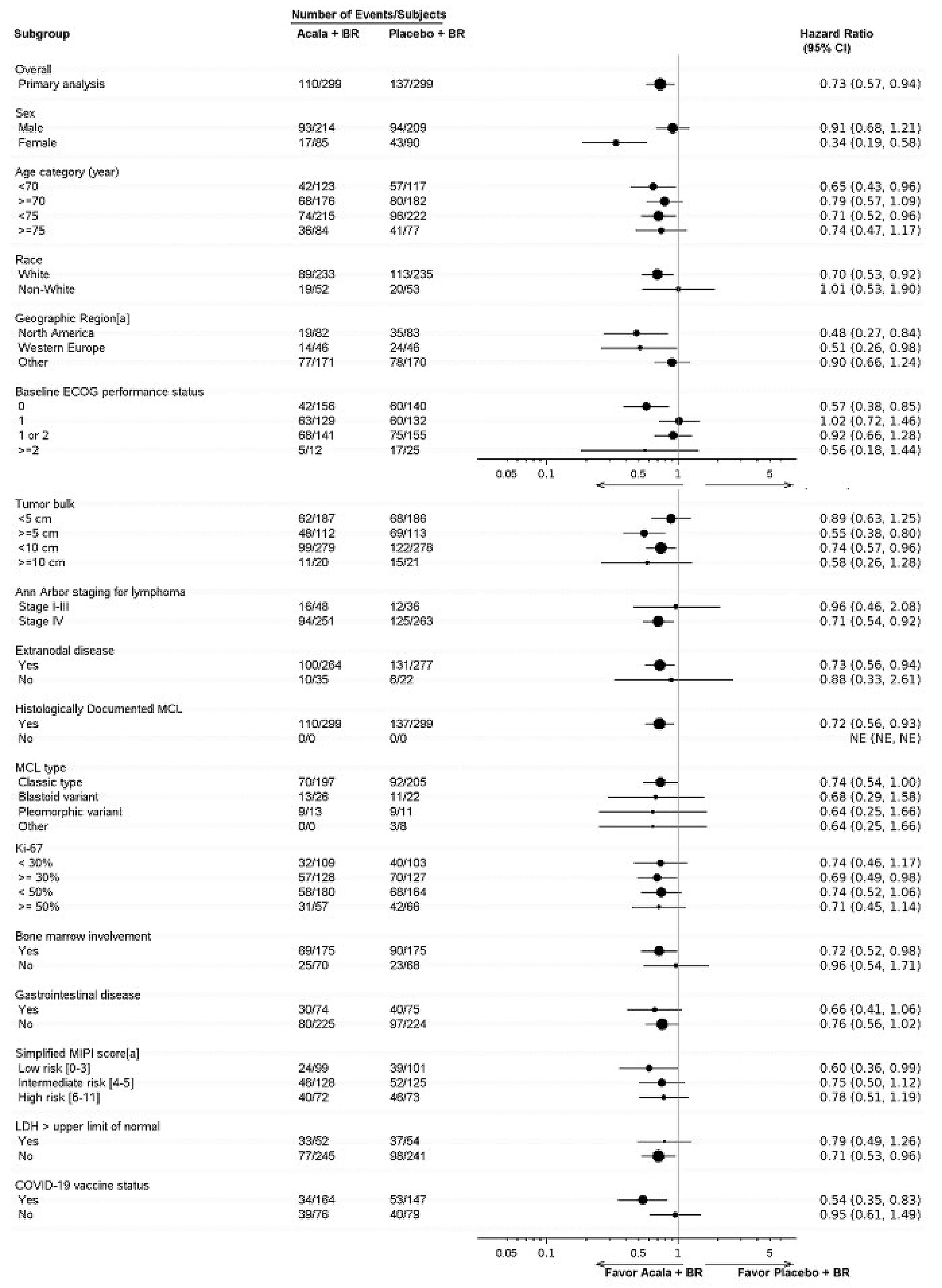
Source: ECHO Clinical Study Report (2024).25
Table 17: Summary of Concomitant Medications From the ECHO Trial — FAS (Data Cut-Off Date: February 15, 2024)
Concomitant medications | Acalabrutinib + BR (N = 299) | Acalabrutinib + BR (N = 299) |
|---|---|---|
Patients with at least 1 concomitant medication, n (%) | ███ ██████ | ███ ██████ |
██████ ███████████ ██████████ █████████ ██ █ ███ ██ ████████ ██ ██████ ███████ █ ███ | ||
██████████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ |
██████████████ ███ ████████ ███ | ███ ██████ | ███ ██████ |
███████████ ███ █████████████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ |
███████████████ ███ ████████ ███ | ███ ██████ | ███ ██████ |
█████████████ | ███ ██████ | ███ ██████ |
██████████████ ███ ████████ ███ | ███ ██████ | ███ ██████ |
██████████ ███ ████████ ███ | ███ ██████ | ███ ██████ |
█████████ | ██ ██████ | ██ ██████ |
█████ ███ ████████████ █████████ | ███ ██████ | ███ ██████ |
██████████████ ██████ | ███ ██████ | ███ ██████ |
█████████ ████████████ | ███ ██████ | ███ ██████ |
███████████ | ███ ██████ | ███ ██████ |
█████ ███████████ ███ █████████ █████████ | ███ ██████ | ███ ██████ |
████████ ███████████ | ███ ██████ | ███ ██████ |
████████ | ███ ██████ | ███ ██████ |
█████ █████ ██ ███ ████████████████ ██████ | ███ ██████ | ███ ██████ |
███████████████ ██████ | ███ ██████ | ███ ██████ |
█████████████ | ███ ██████ | ███ ██████ |
███████ ███████████ | ███ ██████ | ██ ██████ |
███████████████ ███ ███████████ ████████ | ██ ██████ | ██ ██████ |
█████████ | ██ ██████ | ███ ██████ |
█████ ███ ████████████ | ██ ██████ | ██ ██████ |
████ ████████ ██████ | ██ ██████ | ██ ██████ |
████████ | ██ ██████ | ██ ██████ |
Source: ECHO Clinical Study Report (2024).25 Details included in the table are from the sponsor’s summary of clinical evidence.26
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BIA
budget impact analysis
BR
bendamustine plus rituximab
BTK
Bruton tyrosine kinase
CDA-AMC
Canada’s Drug Agency
ICER
incremental cost-effectiveness ratio
MCL
mantle cell lymphoma
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
RDI
relative dose intensity
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of acalabrutinib in combination with bendamustine and rituximab (BR) compared to BR alone for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous stem cell transplant (ASCT).
Item | Description |
|---|---|
Drug product | Acalabrutinib (Calquence), 100 mg, oral tablets |
Indication | Acalabrutinib in combination with bendamustine and rituximab for the treatment of adult patients with previously untreated mantle cell lymphoma (MCL) who are ineligible for autologous stem cell transplant |
Submitted price | Acalabrutinib: $142.77 per 100 mg tablet |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review and Project ORBIS (type A) |
NOC date | June 24, 2025 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: Yes Indication: As monotherapy for the treatment of patients with chronic lymphocytic leukemia who have received at least 1 prior therapy Recommendation date: November 17, 2020 Recommendation: Reimburse with clinical criteria and/or conditions Indication: With or without obinutuzumab, for the treatment of patients with previously untreated chronic lymphocytic leukemia for whom a fludarabine-based regimen is inappropriate Recommendation date: January 8, 2021 Recommendation: Reimburse with clinical criteria and/or conditions |
MCL = mantle cell lymphoma; NOC = Notice of Compliance.
Summary
Acalabrutinib is available as 100 mg tablets.1 At the submitted price of $142.77 per tablet, the cost of acalabrutinib is expected to be $7,995 per patient per 28-day cycle, based on the Health Canada–recommended dosing.1,2 In combination with BR, the per-cycle cost of acalabrutinib is expected to be $13,574 per patient. The combination regimen may be administered for up to 6 cycles. Following this, acalabrutinib may be continued with maintenance rituximab, which is estimated to cost $9,035 per patient per 28-day cycle.
Clinical efficacy in the economic analysis was derived from the ECHO trial, which compared acalabrutinib plus BR to placebo plus BR.3 Evidence submitted by the sponsor indicates that acalabrutinib plus BR results in a benefit in progression-free survival (PFS); however, the treatment benefit in overall survival (OS) is uncertain because of immature data.
The results of the CDA-AMC base-case analysis suggest the following:
Acalabrutinib plus BR is predicted to be associated with higher costs to the health care system than BR alone (incremental costs = $339,591), primarily driven by increased drug acquisition costs associated with acalabrutinib.
Acalabrutinib plus BR is predicted to be associated with a gain of 0.74 life-years compared to BR alone. When the impact on health-related quality of life is also considered, acalabrutinib plus BR is predicted to be associated with a gain of 0.64 quality-adjusted life-years (QALYs) compared to BR alone.
The incremental cost-effectiveness ratio (ICER) of acalabrutinib plus BR compared to BR alone was $533,458 per QALY gained in the CDA-AMC base case. The estimated ICER is uncertain because of uncertainty in the long-term survival estimates, the comparative efficacy, and the subsequent therapy assumptions. Given the unresolved uncertainty in the long-term comparative efficacy, the economic analysis may not accurately assess the impact on patient health. As such, the cost-effectiveness estimates are uncertain and higher price reductions may be required to achieve a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing acalabrutinib for use in combination with BR for the treatment of adult patients with previously untreated MCL who are ineligible for ASCT will be approximately $25.6 million over the first 3 years of reimbursement compared to the amount currently spent on comparators. The expenditure on acalabrutinib over this period is expected to be $30.9 million (acalabrutinib plus BR = $40.9 million). The actual budget impact of reimbursing acalabrutinib will depend on the number of patients treated and the type and proportion of subsequent therapies received following progression.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of acalabrutinib plus BR from the perspective of a public drug plan payer in Canada over a lifetime horizon (30 years).2 The modelled population comprised adult patients with previously untreated MCL who are ineligible for ASCT and was based on the participants in the ECHO trial. The sponsor’s base-case analysis included costs related to drug acquisition (using the submitted price for acalabrutinib and public list prices for comparators), administration, subsequent therapy, disease management and monitoring, adverse events (AEs), and terminal care. In the sponsor’s base case, acalabrutinib plus BR was associated with incremental costs of $238,579 and with 0.72 incremental QALYs compared to BR. This resulted in an ICER of $333,097 per QALY gained. Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The long-term OS benefit predicted for acalabrutinib plus BR relative to BR alone is uncertain. | The sponsor predicted an OS benefit for acalabrutinib plus BR vs. BR alone based on immature data from the ECHO trial. The Clinical Review of the ECHO trial concluded, with moderate certainty due to serious imprecision, that there was little to no clinically important difference in the probability of being alive at 48 months. | This issue could not be addressed due to the absence of more mature data. | CDA-AMC conducted a scenario analysis that assumed equal OS between acalabrutinib plus BR and BR alone. |
The comparative efficacy of acalabrutinib plus BR vs. BR alone is uncertain. | The Clinical Review of the ECHO trial noted concerns with the generalizability of the trial results to the patient population expected to receive treatment with acalabrutinib plus BR in clinical practice and the potential overestimation of the effect of the treatment due to the use of an interim analysis. | This issue could not be addressed due to the absence of more robust data. | No scenario analysis was conducted owing to a lack of long-term comparative data and real-world evidence. |
The use of RDI to estimate treatment costs is inappropriate. | A reduction in RDI may be due to a delayed dose, a missed dose, or a reduction in dose. Each of these have different impacts on drug costs, which were not explicitly modelled. | RDI was assumed to be 100% in the CDA-AMC base case. | CDA-AMC conducted a scenario analysis that maintained the sponsor’s RDI assumptions. |
Subsequent therapy costs are associated with uncertainty. | The sponsor’s assumptions about the composition and distribution of subsequent therapies did not align with the actual treatments and proportions observed in the ECHO trial, resulting in a misalignment between benefits and costs. | CDA-AMC aligned the proportion of patients receiving BTK inhibitors with the ECHO trial. The costs of CAR T-cell therapy and rituximab monotherapy were excluded. | No scenario analysis was conducted due to the structural limitations of the model. |
The sponsor used inconsistent sources for utility values. | The sponsor used utility values for the progression-free health state from the ECHO trial but sourced a utility decrement associated with disease progression from the literature, despite available evidence from the ECHO trial. | CDA-AMC adopted health state utilities derived from the ECHO trial in reanalysis. | CDA-AMC conducted a scenario analysis using the sponsor’s estimated utility value for progressed disease. |
The sponsor’s analysis excluded R-CHOP, a relevant comparator for patients with MCL who are ineligible for ASCT. | According to clinical expert input and published literature, R-CHOP is a potential first-line therapy option for patients with MCL who are ineligible for SCT. The cost-effectiveness of acalabrutinib plus BR compared to R-CHOP is unknown. | This issue could not be addressed. | No scenario analysis was conducted due to the absence of comparative data. |
BR = bendamustine plus rituximab; BTK = Bruton tyrosine kinase; CAR = chimeric antigen receptor; CDA-AMC = Canada’s Drug Agency; MCL = mantle cell lymphoma; OS = overall survival; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; RDI = relative dose intensity; ASCT = autologous stem cell transplant.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 7), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
Acalabrutinib plus BR is expected to be associated with additional health care costs compared to BR alone (incremental costs = $339,591). This increase in health care spending primarily results from drug acquisition costs associated with acalabrutinib (refer to Figure 1). All other costs to the health system are expected to be similar, except for costs associated with subsequent therapies, where costs are $98,201 lower for those who receive acalabrutinib plus BR in the first-line setting. This is because patients who do not receive acalabrutinib plus BR upfront incur higher downstream costs due to the higher use of more costly subsequent therapy with a BTK inhibitor.
Impact on Health
Relative to BR alone, acalabrutinib plus BR is expected to increase the proportion of patients who remain in the progression-free state by approximately 1.69 years and to extend OS by 0.74 years. Because of the impact of treatment on both quality and length of life, acalabrutinib plus BR is expected to result in an additional 0.64 QALYs per patient compared to BR alone (refer to Figure 2).
Figure 1: Impact of Acalabrutinib Plus BR vs. BR Alone on Health Care Costs
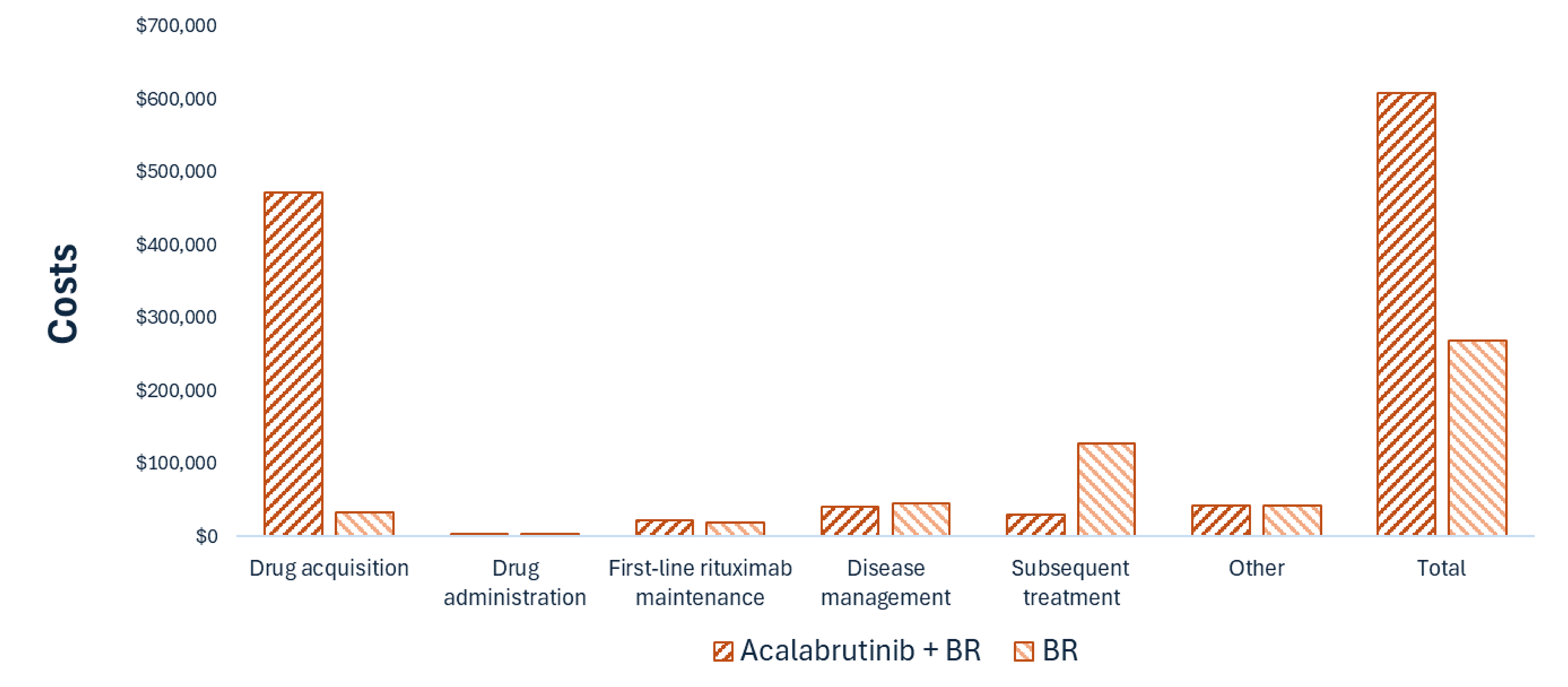
BR = bendamustine plus rituximab; vs. = versus.
Note: Other costs include costs related to adverse events and terminal care.
Figure 2: Impact of Acalabrutinib Plus BR vs. BR Alone on Patient Health
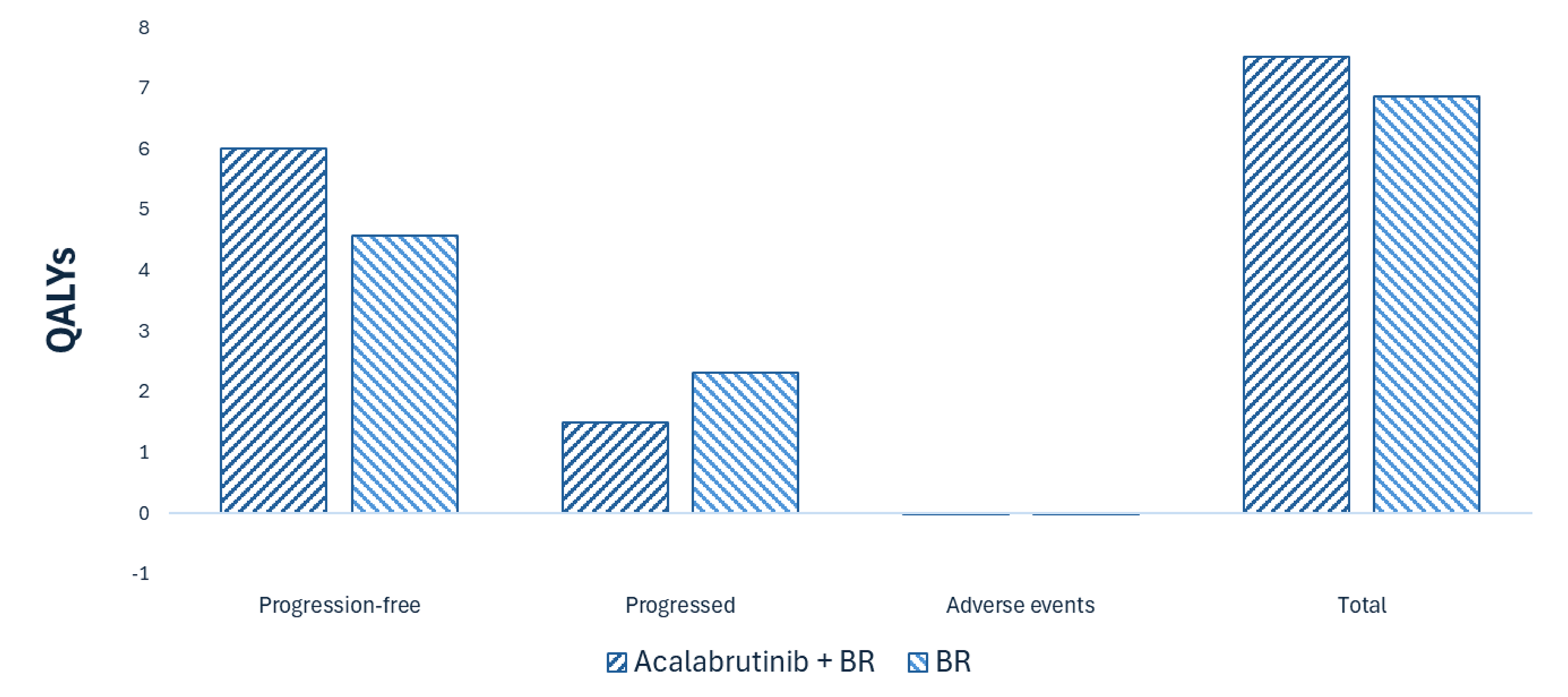
BR = bendamustine plus rituximab; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base-case analysis suggest an ICER of $533,458 per QALY gained for acalabrutinib plus BR compared to BR alone (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. BR alone ($/QALY) |
|---|---|---|---|
BR alone | 268,627 | 6.88 | Reference |
Acalabrutinib plus BR | 608,218 | 7.51 | 533,458 |
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
CDA-AMC notes that the sponsor’s assumptions on the long-term efficacy of acalabrutinib and the characterization of uncertainty result in a 16% probability that treatment with acalabrutinib plus BR results in fewer life-years than treatment with BR alone and, as such, a 16% probability that acalabrutinib plus BR is dominated by BR alone (i.e., acalabrutinib plus BR is more costly and less effective). Due to immature data and the lack of long-term evidence, extrapolation of OS was uncertain. The impact of uncertainty on long-term OS for patients receiving acalabrutinib plus BR was explored in a scenario analysis. The results of this analysis (refer to Table 11) show that adding acalabrutinib to BR in the first-line setting leads to a very small improvement in quality of life when using data on quality of life from the ECHO trial. Therefore, if acalabrutinib plus BR does not extend life, there are very small QALY gains associated with extending the time until disease progression, and a key benefit of treatment comes from reducing subsequent therapy costs. Finally, scenario analyses were conducted that focused on alternative quality-of-life estimations from the literature and on allowing for relative dose intensity (RDI). Of these, allowing for RDI had the largest impact, decreasing the ICER to $439,971 per QALY gained.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2029) budget impact of reimbursing acalabrutinib plus BR for use in the treatment of adult patients with previously untreated MCL who are ineligible for ASCT.4 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of acalabrutinib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 257 patients would be eligible for treatment with acalabrutinib plus BR over a 3-year period (year 1 = 253; year 2 = 255; year 3 = 257), of whom 128 are expected to receive acalabrutinib plus BR (year 1 = 38; year 2 = 77; year 3 = 128). The estimated incremental budget impact of reimbursing acalabrutinib plus BR is expected to be approximately $25.6 million over the first 3 years, with an expected expenditure of $30.9 million on acalabrutinib. The actual budget impact will depend on the number of patients treated with acalabrutinib and the type and proportion of subsequent therapies received following disease progression.
Conclusion
Based on the CDA-AMC base case, acalabrutinib plus BR would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $533,458 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 10). The estimated cost-effectiveness of acalabrutinib plus BR compared to BR alone is uncertain due to immature OS trial data and uncertainty in the long-term comparative clinical efficacy of treatment.
The budget impact of reimbursing acalabrutinib plus BR to the public drug plans in the first 3 years is estimated to be approximately $25.6 million. The 3-year expenditure on acalabrutinib (i.e., not accounting for current expenditure on comparators) is estimated to be $30.9 million. The estimated budget impact is uncertain due to uncertainty around subsequent therapy use in clinical practice.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
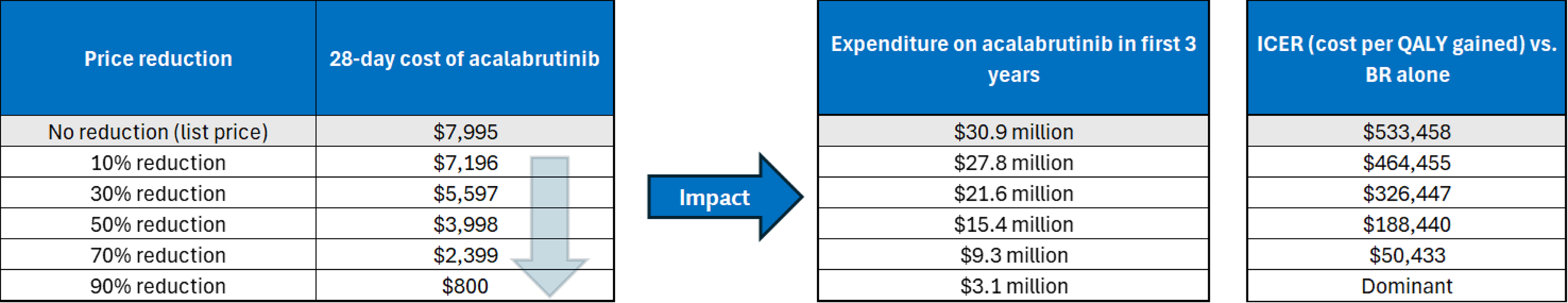
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Expenditure includes only the drug cost of acalabrutinib. The term dominant indicates that a drug costs less and provides more QALYs than the comparator.
References
1.AstraZeneca Canada Inc. Calquence (acalabrutinib): 100 mg acalabrutinib tablets (as acalabrutinib maleate), oral [product monograph]. June 24, 2025.
2.AstraZeneca Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Calquence (acalabrutinib), 100 mg acalabrutinib tablets (as acalabrutinib maleate), oral. March 27, 2025.
3.AstraZeneca Canada Inc. Drug Reimbursement Review sponsor submission: Calquence (acalabrutinib), 100 mg acalabrutinib tablets (as acalabrutinib maleate), oral [internal sponsor's package]. March 27, 2025.
4.AstraZeneca Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Calquence (acalabrutinib), 100 mg acalabrutinib tablets (as acalabrutinib maleate), oral. March 27, 2025.
5.Iqvia. DeltaPA. 2023. Accessed April 5, 2025. https://www.iqvia.com/
6.Cancer Care Ontario. Funded evidence-informed regimens. Accessed April 5, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens
7.Accord Healthcare Inc. Cyclophosphamide: 500 mg, 1000 mg and 2000 mg per vial, lyophilized powder for injection [product monograph]. September 14, 2022. Accessed May 1, 2025. https://pdf.hres.ca/dpd_pm/00067461.PDF
8.Omega Laboratories Ltd. Doxorubicin Hydrochloride: 2 mg/mL; 10 mg (5 mL), 50 mg (25 mL) and 200 mg (100 mL) vials [product monograph]. January 21, 2013. Accessed May 1, 2025. https://pdf.hres.ca/dpd_pm/00019011.PDF
9.Natco Pharma (Canada) Inc. Nat-Bendamustine (bendamustine hydrochloride for injection): 25 mg/ vial and 100 mg/ vial lyophilized powder for injection, for intravenous infusion [product monograph]. March 4, 2020. Accessed May 1, 2025. https://pdf.hres.ca/dpd_pm/00060095.PDF
10.Apotex Inc. Prednisone: 1 mg, 5 mg and 50 mg tablets, oral [product monograph]. May 28, 2015. Accessed May 1, 2025. https://pdf.hres.ca/dpd_pm/00030893.PDF
11.Hoffmann-La Roche Limited. Rituxan (rituximab for injection): 10 mg/mL intravenous infusion [product monograph]. June 2, 2023. Accessed May 1, 2025. https://assets.roche.com/f/173850/x/5af6aeabe9/rituxaniv_pm_e.pdf
12.Pfizer Canada ULC. Vincristine Sulfate: 1 mg/mL injection [product monograph]. July 26, 2021. Accessed May 1, 2025. https://pdf.hres.ca/dpd_pm/00062283.PDF
13.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed April 5, 2025. https://www.formulary.health.gov.on.ca/formulary/
14.AstraZeneca Canada Inc. Clinical Study Report: (Data cut-off: 15 February 2024): D8220C00004 (ACE-LY-308). A Phase 3, Randomized, Double-blind, Placebo-controlled, Multicenter Study of Bendamustine and Rituximab (BR) Alone Versus in Combination with Acalabrutinib (ACP-196) in Subjects with Previously Untreated Mantle Cell Lymphoma (ECHO) [internal sponsor's report]. July 18, 2024.
15.Statistics Canada. Life expectancy and other elements of the complete life table, single-year estimates, Canada, all provinces except Prince Edward Island [sponsor supplied reference]. Accessed February 2025. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310083701&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.3&pickMembers%5B2%5D=4.3&cubeTimeFrame.startYear=2023&cubeTimeFrame.endYear=2023&referencePeriods=20230101%2C20230101
16.National Institute for Health and Care Excellence. Bortezomib for previously untreated mantle cell lymphoma (Technology appraisal guidance TA370) [sponsor supplied reference]. 2015. https://www.nice.org.uk/guidance/ta370
17.National Institute for Health and Care Excellence. Ramucirumab for previously treated locally advanced or metastatic non-small-cell lung cancer (Technology appraisal guidance TA403) [sponsor supplied reference]. 2016. https://www.nice.org.uk/guidance/ta403
18.National Institute for Health and Care Excellence. Zanubrutinib for treating chronic lymphocytic leukaemia (Technology appraisal guidance TA931) [sponsor supplied reference]. 2023. https://www.nice.org.uk/guidance/ta931
19.Ontario Ministry of Health. Exceptional Access Program (EAP). Accessed January 15, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
20.Canada's Drug Agency. Reimbursement Review -- Brexucabtagene Autoleucel (Tecartus). November 2021 [sponsor supplied reference].
21.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)) [sponsor supplied reference]. 2023. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
22.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2)(2):e90-e106. doi:10.3747/co.20.1223
23.Ontario Ministry of Health. Schedule of benefits for laboratory services: (effective July 24, 2023) [sponsor supplied reference]. 2023. https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf
24.Lagerquist O, Poseluzny D, Werstiuk G, et al. The cost of transfusing a unit of red blood cells: a costing model for Canadian hospital use. ISBT Science Series. 2017;12(3):375-380. doi:10.1111/voxs.12355
25.Ontario Regional Blood Coordinating Network. Provincial Platelet Audit Report [sponsor supplied reference]. 2017. https://transfusionontario.org/wp-content/uploads/2020/06/Plt-Audit-Report_final-1-2.pdf
26.Canadian Institute for Health Information. Patient cost estimator. 2024. Accessed January 15, 2025. https://www.cihi.ca/en/patient-cost-estimator
27.Walker H, Anderson M, Farahati F, et al. Resource use and costs of end-of-Life/palliative care: Ontario adult cancer patients dying during 2002 and 2003. J Palliat Care. 2011;27(2):79-88. doi:10.1177/08258597110270020 PubMed
28.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2024;25(1):147-155. doi:10.1007/s10198-023-01570-1 PubMed
29.Medscape. Mantle Cell Lymphoma - Treatment. 2025. Accessed June 16, 2025. https://emedicine.medscape.com/article/203085-overview#a9
30.CADTH. pCODR Expert Review Committee (pERC) final recommendation: Acalabrutinib (Calquence - AstraZeneca Canada Inc.) for previously untreated chronic lymphocytic leukemia. 2021. Accessed June 16, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10210AcalabrutinibCLL%28previously%20untreated%29_FnRec_pERC%20Chair%20Approved_REDACT_Post08Jan2021_final.pdf
31.CADTH. pCODR Expert Review Committee (pERC) final recommendation: Acalabrutinib (Calquence - AstraZeneca Canada Inc.) for previously treated chronic lymphocytic leukemia. 2020. Accessed June 16, 2025. https://www.cda-amc.ca/sites/default/files/pcodr/Reviews2020/10211AcalabrutinibCLL_fnRec_REDACT_EC_Post17Nov2020_final.pdf
32.pan-Canadian Pharmaceutical Alliance. PCPA status: Calquence (acalabrutinib) for treated chronic lymphocytic leukemia. 2021. Accessed June 16, 2025. https://www.pcpacanada.ca/negotiation/21413
33.pan-Canadian Pharmaceutical Alliance. PCPA status: Calquence (acalabrutinib) for untreated chronic lymphocytic leukemia. 2021. Accessed June 16, 2025. https://www.pcpacanada.ca/negotiation/21416
34.Cancer Care Ontario. Funded evidence-informed regimens: ACAL. Accessed April 5, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/67571
35.Cancer Care Ontario. Funded evidence-informed regimens: ACAL(MNT). Accessed April 5, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/67581
36.Cancer Care Ontario. Funded evidence-informed regimens: ACAL+OBIN. Accessed April 5, 2025. https://www.cancercareontario.ca/en/drugformulary/regimens/67576
37.Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659-667. doi:10.1016/S0140-6736(17)33108-2 PubMed
38.AstraZeneca Canada Inc. Physician Interviews. Data on File (2025) [sponsor supplied reference].
39.Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol. 1998;16(8):2780-95. doi:10.1200/jco.1998.16.8.2780 PubMed
40.Anderson JR, Armitage JO, Weisenburger DD. Epidemiology of the non-Hodgkin's lymphomas: distributions of the major subtypes differ by geographic locations. Non-Hodgkin's Lymphoma Classification Project. Ann Oncol. 1998;9(7):717-20. doi:10.1023/a:1008265532487 PubMed
41.Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459. doi:10.3322/caac.21357 PubMed
42.Al-Hamadani M, Habermann TM, Cerhan JR, Macon WR, Maurer MJ, Go RS. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am J Hematol. 2015;90(9):790-5. doi:10.1002/ajh.24086 PubMed
43.Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer. 2008;113(4):791-8. doi:10.1002/cncr.23608 PubMed
44.Leukemia & Lymphoma Society of Canada. Mantle Cell Lymphoma [Factsheet; sponsor supplied reference]. 2020. https://www.bloodcancers.ca/sites/default/files/2021-11/2020%20LLSC%20Fact%20Sheet-Disease_MantleCellLymphoma-Final_EN.pdf
45.Armitage JO, Longo DL. Mantle-Cell Lymphoma. N Engl J Med. 2022;386(26):2495-2506. doi:10.1056/NEJMra2202672 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans
Table 4: Cost Comparison for Untreated Mantle Cell Lymphoma
Treatment | Strength and/or concentration | Form | Price | Recommended dosage | Average daily cost ($) | Average 28-day cost ($) |
|---|---|---|---|---|---|---|
Acalabrutinib (Calquence) | 100 mg | Tablet | 142.7738a | 100 mg twice daily until disease progression or unacceptable toxicity | 285.55 | 7,995 |
Acalabrutinib plus bendamustine and rituximab | — | — | — | — | 485 | 13,574 |
Acalabrutinib plus maintenance rituximab | — | — | — | — | 323 | 9,035 |
Chemoimmunotherapy | ||||||
Bendamustine (generics) | 25 mg 100 mg | Lyophilized powder in vial for injection and IV infusion | 250.0000 1,000.0000 | 90 mg/m2 on days 1 and 2 of each 28-day cycle for up to 6 cycles | 125.00 | 3,500 |
Cyclophosphamide | 500 mg 1,000 mg 2,000 mg | Lyophilized powder in vial for injection | 107.8100 115.0000 150.0000 | 750 mg/m2 on day 1 of 21-day cycle for up to 6 cycles | 7.14 | 200 |
Doxorubicin (Hydroxydaunorubicin) | 10 mg 50 mg 200 mg | 2 mg/mL vial for injection | 50.0000 252.2500 770.0000 | 50 mg/m2 on day 1 of 21-day cycle for up to 6 cycles | 21.43 | 600 |
Prednisone | 1 mg 5 mg 50 mg | Tablet | 0.1276b 0.0220b 0.1735b | 100 mg daily on days 1 to 5 of 21-day cycle for up to 6 cycles | 0.08 | 2 |
Rituximab (Ruxience, Riximyo, Truxima) | 100 mg 500 mg | 10 mL/mL vial for IV solution | 297.0000b 1,485.0000b | 375 mg/m2 on day 1 of each 28-day or 21-day cycle for up to 6 cycles Maintenance: 375 mg/m2 every other cycle (i.e., every 56 days) for up to 2 years | 74.25 to 99.00 Maintenance: 37.13 | 2,079 to 2,772 Maintenance: 1,040 |
Vincristine | 1 mg 2 mg 5 mg | 1 mg/mL vial for injection and IV infusion | 30.6000b 61.2000 153.0000 | 1.4 mg/m2 (maximum of 2 mg) on day 1 of 21-day cycle for up to 6 cycles | 2.91 | 82 |
Bendamustine and rituximab | — | — | — | — | 199.25 | 5,579 |
Rituximab maintenance | — | — | — | — | 37.13 | 1,040 |
R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) | — | — | — | — | 130.57 | 3,656 |
Note: All prices are from the IQVIA DeltaPA database (accessed July 15, 2025),5 and dosage was obtained from Cancer Care Ontario drug formulary,6 unless otherwise indicated. Formulation information was obtained from respective product monographs.7-12 Costs assume weight of 80 kg and body surface area of 1.8 m2. Drug wastage is included.
aSponsor’s submitted price.2
bOntario Drug Benefit formulary, accessed July 15, 2025.13
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Lymphoma Canada, which was collected by surveying patients with MCL in Canada. Patients with MCL reported symptoms such as fatigue or lack of energy, enlarged lymph nodes, night sweats, indigestion, abdominal pain or bloating, weight loss, low platelet counts, and leukocytosis. These challenges collectively had a negative impact on patients’ mental health, quality of life, and ability to carry out daily activities. Current first-line treatments for patients who are not eligible for transplant included BR and rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). Patients received treatment with Bruton tyrosine kinase (BTK) inhibitors (ibrutinib, acalabrutinib, zanubrutinib) in subsequent lines of therapy. Treatment goals identified by patient input included achieving a longer disease remission, improving OS, enhancing quality of life, maintaining the ability to perform daily activities, controlling disease-related symptoms, and normalizing blood counts on laboratory tests. There were 8 patients with previously untreated with MCL who reported having experience with acalabrutinib plus BR. Two of these patients were still undergoing treatments, 2 had been in remission for 1 to 2 years, 1 patient was in remission for less than 6 months, and 3 patients relapsed after treatment. Patient input also reported AEs such as fatigue, diarrhea, neutropenia, allergic reaction, and psychological impacts.
Clinician input was received from Lymphoma Canada, Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee Canadian and LLSC Pharmacist Network, which was gathered through interviews with clinical experts, published literature and pharmacists with experience in managing and supporting treatment of oncology patients. The current standard of treatment for MCL aims to improve PFS, maintain remission, extend OS, ensure a manageable safety profile, and enhance quality of life. The first-line therapy for MCL patients who are ineligible for ASCT included BR, followed by maintenance therapy with rituximab for up to 2 years typically, and a regimen of bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP). For patients with relapsed or refractory MCL, monotherapy with oral BTK inhibitors such as ibrutinib, acalabrutinib and zanubrutinib was reported to be the standard of care. Treatment response would be assessed using CT scans, PET scans, routine blood laboratory tests, and assessments of organ function. The clinician input noted that acalabrutinib treatment would be discontinued upon disease progression and AEs such as significant or recurrent infection, bleeding, atrial fibrillation, or uncontrollable hypertension. Acalabrutinib would be best suited for patients who are not eligible for transplant, particularly older individuals or those with significant comorbidities who cannot tolerate more aggressive treatments. Input received from pharmacists highlighted that, despite the convenience associated with oral administration compared to IV therapies, significant pharmacist involvement would be required for patient education, AE monitoring, and drug interaction monitoring.
Input from CDA-AMC–participating drug plans noted that BR is the common treatment option for adults with MCL who are previously untreated and are ineligible for ASCT, with R-CVP and R-CHOP used less frequently. The drug plan input highlighted that reimbursement of acalabrutinib in first-line therapy may impact the sequencing of therapies in subsequent lines of therapy. In addition, there were concerns with the use of additional pharmacy resources to manage potential drug interactions. The input also expressed concerns regarding the overall budget impact of adding acalabrutinib to a regimen of BR, and noted existence of confidential pricing for acalabrutinib, rituximab biosimilars, rituximab subcutaneous and bendamustine.
Several of these concerns were addressed in the sponsor’s model:
Clinical outcomes valued by patients and clinicians (i.e., PFS and OS) were included in the model.
CDA-AMC addressed some of these concerns as follows:
The impact of treatment on health-related quality of life was included by use of EQ-5D-L data collected in the ECHO trial.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of acalabrutinib plus BR, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Acalabrutinib (Calquence), oral tablets (100 mg)1 |
Submitted price of drug under review | $142.7738 per 100 mg tablet2 |
Regimen | 100 mg twice daily until progressive disease or toxicity, in combination with bendamustine 90 mg/m2 on days 1 and 2 of each 28-day cycle and rituximab 375 mg/m2 on day 1 of each 28-day cycle, both for up to 6 cycles followed by maintenance rituximab (every 2 cycles for up to 2 years)1 |
28-day cycle cost of drug under review | Acalabrutinib: $7,122 per patient2 Acalabrutinib plus BR: $12,201 per patient for up to 6 cycles2 Acalabrutinib plus maintenance rituximab: $8,904 per patient2 Drug costs accounted for relative dose intensity (89% for acalabrutinib) |
Model information | |
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Treatment | Acalabrutinib plus bendamustine and rituximab (BR) |
Included comparator | BR |
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (30 years) |
Cycle length | 28 days |
Modelled population | Patients aged 18 years and older with previously untreated MCL who are ineligible for ASCT |
Characteristics of modelled population | Derived from the ECHO trial (mean age: 71.60 years, mean weight: 77.04 kg, mean height: 168.38 cm, 71% men, 29% women)14 |
Model health states |
|
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs included in the model |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis resultsa |
|
AE = adverse event; BR = bendamustine plus rituximab; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-years; RDI = relative dose intensity; TTD = time-to-treatment discontinuation.
aResults of scenario analyses that had a meaningful impact on the estimated ICER compared to the sponsor’s base case. Additional scenarios were submitted that had no meaningful impact on the estimated ICER included adopting shorter time horizon, alternative discount rates, alternative survival distributions for OS, PFS, and TTD, sourcing utility values from the ECHO trial and assuming vial sharing.
Table 6: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
BR alone | 311,644 | Reference | 6.69 | Reference | Reference |
Acalabrutinib plus BR | 550,223 | 238,579 | 7.41 | 0.72 | 333,097 |
BR = bendamustine plus rituximab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The key clinical efficacy data in the model (OS, PFS, health-related quality of life, time-to-treatment discontinuation) were derived from the ECHO trial (data cut-off: February 15, 2024). Evidence from the ECHO trial suggests that, based on moderate certainty due to serious imprecision, acalabrutinib plus BR may result in a clinically significant benefit in PFS. However, the treatment benefit in OS was uncertain as the median OS was not reached in either study group in this interim analysis. The early reporting of the study results in an interim analysis may lead to an overestimation of the treatment effect. Evidence from the ECHO trial also suggests that there is likely little to no treatment benefit of acalabrutinib plus BR in comparison to BR alone for EQ-5D-5L from baseline. The observed safety profile of acalabrutinib plus BR was as expected but with added disease toxicities, as per feedback from the clinical experts.
Estimates of relative efficacy for the economic evaluation were obtained from the data on OS and PFS from the ECHO trial,14 and extrapolated over the time horizon based on sponsor’s assumptions. Approximately 74% of additional life-years lived and 72% of QALYs gained with acalabrutinib plus BR were accrued beyond the maximum trial follow-up (i.e., 84 months) and were based on the sponsor’s extrapolations of the trial data. Given the immaturity of the OS data, the sponsor’s extrapolation of OS is highly uncertain. Drug acquisition cost accounted for the majority of incremental costs for acalabrutinib plus BR, which were partially offset by reduced costs of subsequent treatment in second and third-line of therapy. The type and distribution of patients across subsequent therapy were based on sponsor’s assumptions.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The impact of acalabrutinib plus BR on long-term OS is uncertain: The sponsor submitted a partitioned survival model in which the long-term clinical efficacy of acalabrutinib plus BR was based on the sponsor’s extrapolations of OS using efficacy data from the ECHO trial.14 In the sponsor’s base-case analysis, OS was extrapolated by fitting an exponential distribution, which resulted in a survival benefit of approximately 9.14 months. However, the Clinical Review of the ECHO trial OS data concluded that, based on moderate certainty due to serious imprecision, there was no clinically important difference in the probability of being alive at 48 months. The Clinical Review noted that the treatment benefit in OS associated with acalabrutinib plus BR compared to BR alone was uncertain because the median OS was not reached in either study arms in the interim analysis. It was also noted that the ECHO trial was not powered to evaluate OS in any meaningful way, and that these results should be interpreted with caution given the trial design and OS data maturity (34%). The estimated hazard ratio (HR = 0.86; 95% confidence interval: 0.65, 1.13) was not statistically significant and crossed the null, indicating that a survival benefit cannot be confidently concluded.14 As such, the modelled impact of acalabrutinib plus BR on long-term OS is highly uncertain. This uncertainty is partially reflected in the sponsor’s probabilistic analysis, which found a 16% probability that acalabrutinib plus BR could result in fewer life-years compared to BR alone. This has important clinical implications: while the mean OS is higher with acalabrutinib plus BR in the base case, there remains a nontrivial likelihood that treatment could lead to lower survival.14 Given these limitations, the long-term OS benefit modelled in the sponsor’s base case is highly uncertain.
This issue could not be addressed by CDA-AMC due to the lack of long-term data on OS. To explore the impact of uncertainty in the modelled OS, a scenario analysis was conducted assuming an OS hazard ratio of 1 (i.e., no difference in OS between acalabrutinib plus BR and BR alone).
The comparative efficacy of acalabrutinib plus BR versus BR alone is uncertain: In the sponsor’s model, clinical efficacy was informed by the ECHO trial which was extrapolated beyond the trial period. The CDA-AMC clinical review highlighted limitations associated with the trial, including the reliance on result from an interim analysis which may lead to an overestimation of the treatment effect. Furthermore, the Clinical Review of the ECHO trial noted concerns with the generalizability of trial results to clinical practice. The ECHO trial enrolled patients who had more favourable baseline characteristics, including a better ECOG Performance Status and less severe disease than those anticipated to be treated in clinical practice in Canada. These characteristics are associated with better prognosis, raising uncertainty about whether the modelled benefits would be realized in clinical practice. The long-term efficacy of acalabrutinib plus BR versus BR alone is also uncertain due to a lack of long-term comparative evidence. Approximately 74% of additional life-years lived and 72% of QALYs gained with acalabrutinib plus BR were accrued beyond the maximum trial follow-up (i.e., 84 months) and were based on the sponsor’s extrapolations of the trial data.
This issue could not be addressed by CDA-AMC.
The use of RDI to estimate treatment cost is inappropriate: The sponsor adjusted the total treatment cost of first-line treatments by adopting RDI values of less than 100% estimated based on data from the ECHO trial28 This consideration of RDI is problematic as this parameter can be influenced by several factors. For example, the dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, and subsequent dose re-escalation; each of these have different impacts on drug costs. It is also unclear how the adopted RDI values interact with considerations about drug wastage, which was assumed to be included by the sponsor. Furthermore, prescriptions for acalabrutinib may be filled and reimbursed regardless of treatment adherence.
In CDA-AMC reanalysis, RDI values for acalabrutinib, BR were set to be 100%. The sponsor’s estimated RDI values were adopted in scenario analysis.
The costs associated with subsequent therapy are uncertain: In the pharmacoeconomic model, OS was informed by data from the ECHO trial;28 however, the costs of subsequent therapy were based on sponsor-sought expert opinion regarding the composition and distribution of second- and third-line treatments. While subsequent therapy use in clinical practice in Canada may differ from those observed in the trial, this approach introduced misalignment between the modelled efficacy and the costs associated with subsequent therapy.
Notably, the sponsor assumed that no patients on acalabrutinib plus BR received a BTK inhibitor as subsequent therapy in the model, however, there were 13 patients (43%) on acalabrutinib plus BR who received a BTK inhibitor as subsequent therapy, in the ECHO trial. The use of BTK inhibitors was captured in the OS estimates from the trial, but their associated costs were excluded. Conversely, the sponsor assumed that 50% of patients receiving a third- line therapy would receive chimeric antigen receptor T-cell therapy, whereas fewer than 5% did so in the trial, resulting in an overestimation of these costs. Moreover, given the duration of trial follow-up, it is uncertain whether the OS estimates fully capture the potential survival benefits of third-line therapies. These misalignments resulted in a disconnect between the modelled OS and cost estimates associated with subsequent therapy. As a result, the estimated cost offsets from subsequent therapy may be overstated and are subject to uncertainty.
Additionally, the proportion of patients who experience disease progression receiving subsequent therapy lacked face validity. In the trial, the proportion of patients receiving subsequent therapy differed between patients on acalabrutinib plus BR and BR alone. However, based on expert clinical input received for this review, this does not reflect the anticipated clinical practice in Canada. According to expert feedback, the majority of patients who experience disease progression are anticipated to be offered subsequent therapy regardless of the treatment they received in the first line.
To address this limitation, CDA-AMC aligned the proportion of patients receiving BTK inhibitors with the ECHO trial, retaining the sponsor’s assumption that the cost of BTK inhibitor is reflective of ibrutinib treatment. The sponsor’s assumption that the rest of patients incur the cost of R-CHOP was retained. Moreover, given the trial does not provide data on which treatments were used in which line of subsequent therapy, the treatments associated with third-line of therapy were excluded.
CDA-AMC maintained the sponsor’s assumption that more patients who experience disease progression following treatment with BR alone would be treated with subsequent therapy than those treated with acalabrutinib plus BR because it was observed in the trial, however, it is uncertain whether this would be observed in clinical practice in Canada.
Different sources for utilities were used: In the sponsor’s base case, utility values for the progression-free health state were derived from the ECHO trial data,14 however, utility for the progressed health state was estimated by applying a decrement associated with progressed disease that was sourced from the literature.16 This approach introduced inconsistency because utility values were obtained from different sources, despite the fact that trial-based utilities for both health states were available. As a result, the model applied a difference of 0.071 between the progression-free and progressed health states. In contrast, the ECHO trial data demonstrated a much smaller difference in health utilities between the 2 health states, and the CDA-AMC Clinical Review of the ECHO trial concluded there is likely little to no treatment benefit as measured using EQ-5D-5L for acalabrutinib plus BR compared to BR alone.
In reanalysis, CDA-AMC adopted the health utility derived from the ECHO trial data for progressed health state. The sponsor’s estimated utility values were considered in a scenario analysis.
Important AEs were excluded: The sponsor incorporated Grade 3 or higher AEs occurring in at least 5% of patients in the ECHO trial and modelled a one-time disutility and cost associated with these AEs that were applied during the first cycle. There are other important AEs, such as atrial fibrillation or flutter and hemorrhage, that occurred more frequently in the acalabrutinib plus BR arm of the ECHO trial but were not captured in the model. The exclusion of these AEs introduced uncertainty into the model, particularly with respect to the potential underestimation of both AE-related costs and the disutility experienced by patients receiving acalabrutinib plus BR. Given these AEs were excluded, the model may not fully reflect the impact of treatment-related AEs on costs and QALYs.
This issue could not be addressed by CDA-AMC because the model structure lacked flexibility to incorporate additional AEs or explore their impact through scenario analyses. The impact of excluding cardiac and hemorrhagic events on QALYs and costs are unknown.
Relevant comparator was excluded: The sponsor’s submitted base case compared acalabrutinib plus BR to BR alone, excluding R-CHOP in line with a deviation request accepted by CDA-AMC. However, according to clinical expert input received by CDA-AMC for this review and published guidelines,29 some patients with MCL who are ineligible for ASCT may be treated with R-CHOP as a first line of therapy (< 15% of patients). Given R-CHOP was excluded from the sponsor’s analysis, the cost-effectiveness of acalabrutinib plus BR compared to R-CHOP is unknown.
CDA-AMC could not address this limitation due to lack of comparative effectiveness data.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 7). The impact of these changes, individually and collectively, is presented in Table 8.
Table 7: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. RDI for first-line therapies | < 100% | 100% |
2. Distribution of patients across subsequent therapies | Acalabrutinib plus BR Ibrutinib: 0% R-CHOP: 100% BR alone Ibrutinib: 82.1% R-CHOP: 17.9% Third-line therapies were included | Acalabrutinib plus BR Ibrutinib: 43.3% R-CHOP: 56.7% BR alone Ibrutinib: 86.4% R-CHOP: 13.6% Third-line therapies were excluded |
3. Health state utilities | Utility for progression-free state was derived from ECHO trial data, and for progressed disease was derived from alternative source Progression free: █████ Progressed disease: █████ | Utilities for both health states were derived from ECHO trial data Progression free: █████ Progressed disease: █████ |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 + 3 |
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; RDI = relative dose intensity.
Note: CDA-AMC was unable to resolve the issues with lack of mature OS data, and remaining limitations of modelling subsequent therapy.
Table 8: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | BR alone | 311,644 | 6.69 | Reference |
Acalabrutinib plus BR | 550,223 | 7.41 | 333,097 | |
Sponsor’s base case (deterministic) | BR alone | 311,898 | 6.69 | Reference |
Acalabrutinib plus BR | 549,810 | 7.41 | 326,658 | |
CDA-AMC reanalysis 1 | BR alone | 320,434 | 6.69 | Reference |
Acalabrutinib plus BR | 606,890 | 7.41 | 393,311 | |
CDA-AMC reanalysis 2 | BR alone | 259,899 | 6.69 | Reference |
Acalabrutinib plus BR | 551,212 | 7.41 | 399,980 | |
CDA-AMC reanalysis 3 | BR alone | 311,898 | 6.87 | Reference |
Acalabrutinib plus BR | 549,810 | 7.53 | 359,504 | |
CDA-AMC base case: (Reanalysis 1 + 2 + 3) (deterministic) | BR alone | 268,436 | 6.87 | Reference |
Acalabrutinib plus BR | 608,293 | 7.53 | 513,553 | |
CDA-AMC base case (Reanalysis 1 + 2 + 3) (probabilistic) | BR alone | 268,627 | 6.88 | Reference |
Acalabrutinib plus BR | 608,218 | 7.51 | 533,458 |
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 9: Disaggregated Results of the CDA-AMC Base Case
Parameter | Acalabrutinib plus BR | BR alone |
|---|---|---|
Discounted LYs | ||
Total | 8.75 | 8.00 |
By health state or data source | ||
Progression free | 6.99 | 5.30 |
Progressed | 1.76 | 2.70 |
Discounted QALYs | ||
Total | 7.51 | 6.88 |
By health state or data source | ||
Progression free | 6.01 | 4.56 |
Progressed | 1.50 | 2.31 |
AEs | −0.002 | −0.002 |
Discounted costs ($) | ||
Total | 608,218 | 268,627 |
First-line treatment acquisition costs (Induction – BR only) | 32,310 | 31,920 |
First-line treatment administration costs (Induction – BR only) | 3,260 | 3,211 |
First-line treatment acquisition costs (Continuous – acalabrutinib only) | 439,248 | 0 |
First-line treatment administration costs (Continuous – acalabrutinib only) | 0 | 0 |
First-line rituximab maintenance cost | 21,279 | 18,916 |
Disease Management – progression-free state | 22,892 | 17,356 |
Disease Management – progressed state | 18,134 | 27,885 |
Subsequent therapy (fixed duration) | 3,689 | 2,168 |
Subsequent therapy (treat to progression) | 25,962 | 125,684 |
AEs | 2,345 | 1,848 |
End of life care | 39,100 | 39,639 |
AE = adverse event; BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 10).
Table 10: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Cost per 28 days ($) | ICERs for acalabrutinib plus BR vs. BR alone ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 143a | 7,995 | 333,097 | 533,458 |
10% | 129 | 7,196 | 274,375 | 464,455 |
20% | 114 | 6,396 | 226,175 | 395,451 |
30% | 100 | 5,597 | 164,506 | 326,447 |
40% | 86 | 4,797 | 115,656 | 257,444 |
50% | 71 | 3,998 | 60,396 | 188,440 |
60% | 57 | 3,198 | 3,271 | 119,437 |
70% | 43 | 2,399 | Dominant | 50,433 |
80% | 29 | 1,599 | Dominant | Dominant |
90% | 14 | 800 | Dominant | Dominant |
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; vs. = versus.
aSponsor’s submitted price for acalabrutinib.2
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 11.
Scenario 1: A hazard ratio of 1 was assumed for OS of acalabrutinib plus BR compared to BR alone.
Scenario 2: The sponsor’s estimated RDI values were adopted.
Scenario 3: Utility for progressed state was estimated by applying sponsor’s estimate of disutility associated with progressed disease.
Table 11: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | BR alone | 268,627 | 6.88 | Reference |
Acalabrutinib plus BR | 608,218 | 7.51 | 533,458 | |
CDA-AMC scenario 1: assuming HR of 1 for OS | BR alone | 266,943 | 6.87 | Reference |
Acalabrutinib plus BR | 612,645 | 6.88 | 117,730,007 | |
CDA-AMC scenario 2: adopt sponsor’s estimated RDI | BR alone | 261,071 | 6.87 | Reference |
Acalabrutinib plus BR | 551,842 | 7.53 | 439,971 | |
CDA-AMC scenario 3: adopt sponsor’s utilities | BR alone | 268,099 | 6.69 | Reference |
Acalabrutinib plus BR | 607,988 | 7.40 | 478,571 |
BR = bendamustine plus rituximab; CDA-AMC = Canada’s Drug Agency; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aProbabilistic analyses.
Issues for Consideration
CDA-AMC has previously recommended acalabrutinib for the treatment of patients with chronic lymphocytic leukemia in the untreated and advanced or metastatic settings.30,31 The cost-effectiveness results of these evaluations may not be directly comparable to those in the current review, owing to differences in target population, model structure, clinical effectiveness parameters, health state utility values, and cost inputs. CDA-AMC further notes that the pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for acalabrutinib for the aforementioned indications.32,33 As such, acalabrutinib has a confidential negotiated price, and is currently funded by jurisdictional cancer formularies.34-36 The drug plan input also noted that existence of confidential prices for rituximab (biosimilar and subcutaneous), and bendamustine. The CDA-AMC reanalyses in this review are based on the publicly available price of acalabrutinib and comparators, which may be different than the confidential price and may influence the results of the cost-utility analysis and BIA.
Acalabrutinib is also indicated as monotherapy for the treatment of patients with MCL who have received at least 1 prior therapy, however, this indication has not been reviewed by CDA-AMC and it is generally not publicly funded in this population in clinical practice in Canada.1,37 Acalabrutinib was not included as a subsequent therapy in refractory or relapsed setting in the cost-utility analysis and BIA.
Drug plan input raised concerns about the potential need for additional pharmacy resources related to drug-drug interaction monitoring and dispensing. The product monograph notes that dosing may need to be de-escalated and subsequently re-escalated in response to certain AEs.1 However, these potential resource implications and dose modifications were not incorporated in this cost-utility analysis and BIA owing to a lack of data.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing acalabrutinib in combination with BR for the treatment of adult patients with previously MCL who are ineligible for ASCT.4
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2029), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological based approach. The sponsor’s base case included drug acquisition costs with first-line and subsequent lines of treatments. To estimate the number of patients receiving subsequent therapies, the sponsor used Kaplan-Meier curve for PFS and data on subsequent therapy from the ECHO trial to derive the proportion of progressing patients receiving subsequent therapy at the end of each year of treatment. The market uptake for acalabrutinib plus BR was estimated using internal market research and sponsor-sought expert input.38 The key inputs to the BIA are documented in Table 12.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing acalabrutinib plus BR for the treatment of adult patients with previously MCL who are ineligible for ASCT would be $22,157,052 (year 1 = $2,521,720; year 2 = $6,738,085; year 3 = $12,897,247).
Table 12: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3) |
|---|---|
Target population | |
Starting number of people | |
Annual incidence of non-Hodgkin lymphoma | 0.033%41 |
Percentage with mantle cell lymphoma | 6.00%42 |
Percentage receiving treatment | 93.40%43 |
Percentage transplant ineligible | 53.60%44 |
Percentage of patients covered by public drug plan | 96.0%45 |
Number of patients eligible for drug under review | 253 / 255 / 257 |
Market shares (reference scenario) | |
Acalabrutinib plus BR | 0% / 0% / 0% |
BR | 90% / 90% / 90% |
R-CHOPa | 10% / 10% / 10% |
Market shares (new drug scenario) | |
Acalabrutinib plus BR | ███ █ ███ █ ███ |
BR | ███ █ ███ █ ███ |
R-CHOP | ███ █ ███ █ ███ |
Cost of treatment (per patient per 28-day cycle)b | |
Acalabrutinib | $7,122.24 |
Bendamustine | $2,950.78 |
Rituximab (induction) | $1,962.16 |
Rituximab (maintenance) | $811.34 |
Cyclophosphamide | $327.47 |
Doxorubicin | $591.04 |
Vincristine | $77.53 |
Prednisone | $2.05 |
Cost of subsequent treatment (per patient per 28-day cycle) | |
Ibrutinib | $11,181.52 |
R-CHOP | $3,886.29 |
BR | $5,531.01 |
BR = bendamustine plus rituximab; R-CHOP = rituximab (induction and maintenance), cyclophosphamide, doxorubicin, vincristine, and prednisone.
aR-CHOP represents R-CHOP plus other rituximab-based chemoimmunotherapies that the sponsor assumed have minimal utilization in clinical practice in Canada.
bCost estimates were based on relative dose intensity below 100%.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of RDI to estimate treatment cost is inappropriate: As noted in the “Key Issues of the Submitted Economic Evaluation,” the use of RDI to reflect deviations from planned dose due to dose delays, missed doses, dose reduction to manage toxicity, and subsequent dose re-escalation is problematic. Furthermore, prescriptions for acalabrutinib may be filled and reimbursed regardless of treatment adherence.
In the CDA-AMC reanalysis, RDI was set to 100% for all first-line drugs.
Subsequent therapies in the BIA are uncertain: The modelled subsequent therapy in the BIA is based on data from the ECHO trial and sponsor-sought expert input. Although the exclusion of BTK inhibitor rechallenge for patients receiving acalabrutinib plus BR aligns with clinical expert feedback received by CDA-AMC for this review, the observation that different proportions of patients in each arm would receive subsequent therapy does not meet face validity. According to expert feedback, the majority of patients who experience progression, regardless of treatment arm, would be offered subsequent therapy in clinical practice. In the BIA, the sponsor estimated a higher proportion of patients in the BR arm received subsequent therapy compared to the acalabrutinib plus BR arm based on data from the ECHO trial.14 The model also assumed that patients who progress incur the full cost of subsequent therapy immediately, which may overestimate cost offsets associated with reduced progression in the acalabrutinib plus BR arm. These assumptions introduce uncertainty regarding the estimated budget impact, particularly in terms of the magnitude of downstream treatment costs.
CDA-AMC could not address this issue due to absence of real-world evidence on subsequent therapy use. A scenario analysis was conducted in which the proportion of patients from the full ECHO trial population who received subsequent therapy (i.e., 47.8%) was applied to both comparators to test the impact of this assumption on the estimated budget impact.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 13.
Table 13: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. RDI | < 100% | 100% |
CDA-AMC base case | ― | Reanalysis 1 |
BR = bendamustine plus rituximab; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; RDI = relative dose intensity.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15. In the CDA-AMC base case, the 3-year budget impact of reimbursing acalabrutinib plus BR for the treatment of adult patients with previously MCL who are ineligible for ASCT was $25,581,166 (year 1 = $2,888,065; year 2 = $7,763,239; year 3 = $14,929,862).
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 22,157,052 |
CDA-AMC reanalysis 1 | 25,581,166 |
CDA-AMC base case: reanalysis 1 | 25,581,166 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing acalabrutinib. The results are provided in Table 15.
Assuming the same proportion of patients who experience disease progression (proportion of full ECHO trial population: 47.8%) receive subsequent therapy in acalabrutinib plus BR, BR alone and R-CHOP arms.
Table 15: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 10,811,852 | 16,662,458 | 22,096,397 | 27,234,441 | 65,993,296 |
Acalabrutinib plus BR | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 10,811,852 | 16,662,458 | 22,096,397 | 27,234,441 | 65,993,296 | |
New drug total | 10,811,852 | 19,184,179 | 28,834,482 | 40,131,687 | 88,150,349 | |
Acalabrutinib plus BR | 0 | 4,168,325 | 10,926,268 | 21,021,940 | 36,116,533 | |
All other comparators | 10,811,852 | 15,015,853 | 17,908,214 | 19,109,748 | 52,033,816 | |
Budget impact | 0 | 2,521,720 | 6,738,085 | 12,897,247 | 22,157,052 | |
CDA-AMC base case | Reference total | 11,912,123 | 18,208,365 | 23,754,247 | 28,903,836 | 70,866,447 |
Acalabrutinib plus BR | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 11,912,123 | 18,208,365 | 23,754,247 | 28,903,836 | 70,866,447 | |
New drug total | 11,912,123 | 21,096,430 | 31,517,486 | 43,833,698 | 96,447,613 | |
Acalabrutinib plus BR | 0 | 4,710,256 | 12,371,193 | 23,796,543 | 40,877,992 | |
All other comparators | 11,912,123 | 16,386,173 | 19,146,293 | 20,037,155 | 55,569,620 | |
Budget impact | 0 | 2,888,065 | 7,763,239 | 14,929,862 | 25,581,166 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Assume the same proportion of patients who experience disease progression receive subsequent therapy | Reference total | 11,014,585 | 16,195,112 | 20,449,808 | 24,299,169 | 60,944,089 |
New drug total | 11,014,585 | 19,242,678 | 28,720,051 | 40,357,248 | 88,319,977 | |
Budget impact | 0 | 3,047,566 | 8,270,243 | 16,058,079 | 27,375,888 | |
BR = bendamustine plus rituximab; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.