Drugs, Health Technologies, Health Systems
Reimbursement Review
Nivolumab Plus Ipilimumab (Opdivo Plus Yervoy)
Sponsor: Bristol Myers Squibb Canada Co.
Therapeutic area: Unresectable or advanced hepatocellular carcinoma
Summary
What Is Hepatocellular Carcinoma?
Hepatocellular carcinoma (HCC) is liver cancer that starts in the cells that make up the body of the liver. Patients identified emotional distress, loss of independence, disruption to employment and family life, and reduction in quality of life as the negative impact of HCC.
In Canada, the estimated incidence of liver and intrahepatic bile duct cancer was 4,700 new cases, and the estimated mortality was 3,500 deaths in 2023.
What Are the Treatment Goals and Current Treatment Options for HCC?
Treatment goals for HCC include improving survival, slowing cancer progression, maintaining quality of life, and minimizing adverse events (AEs).
The current treatment options for previously untreated, unresectable HCC are atezolizumab plus bevacizumab, durvalumab plus tremelimumab, lenvatinib, and sorafenib.
What Is Opdivo and Yervoy and Why Did Canada’s Drug Agency Conduct This Review?
Opdivo is a drug that is administered by IV infusion. Health Canada has approved Opdivo, in combination with Yervoy, for the first-line treatment of adult patients with unresectable or advanced HCC.
Canada’s Drug Agency (CDA-AMC) reviewed Opdivo, in combination with Yervoy, to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the first-line treatment of adult patients with unresectable or advanced HCC.
How Did CDA-AMC Evaluate Opdivo Plus Yervoy?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Opdivo plus Yervoy versus other treatments used in Canada for the first-line treatment of adult patients with unresectable or advanced HCC. Atezolizumab plus bevacizumab, durvalumab plus tremelimumab, lenvatinib, and sorafenib were considered as relevant treatments to compare with Opdivo plus Yervoy when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Opdivo and Yervoy and HCC.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission and 2 clinician group submissions in response to the call for input, and by input from the participating public drug programs around issues that may impact the ability to implement a recommendation.
Two medical oncologists with expertise in the diagnosis and management of unresectable or advanced HCC with representation from Ontario and British Columbia were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 phase III, open-label, randomized controlled trial (the CheckMate 9DW trial) comparing Opdivo plus Yervoy with sorafenib or lenvatinib in 668 patients with unresectable or advanced HCC who have not received any systemic therapy for the carcinoma
1 simulated treatment comparison (STC) of Opdivo plus Yervoy versus atezolizumab plus bevacizumab and durvalumab plus tremelimumab.
For the comparison of Opdivo plus Yervoy versus sorafenib or lenvatinib:
Opdivo plus Yervoy likely results in increased overall survival (OS) compared with sorafenib or lenvatinib, but this benefit appears delayed. At 6 months, OS is likely lower with Opdivo plus Yervoy (moderate certainty); by 24 months, OS is likely higher (moderate certainty). At 12 months, the evidence suggests little to no difference in OS (low certainty). There is uncertainty due to imprecision in the estimates (95% confidence intervals include the possibility of little to no clinically important difference), especially at 12 months. It is unclear which patient characteristics may influence the timing or magnitude of benefit. Also of note, most (85%) participants in the tyrosine kinase inhibitor group received lenvatinib.
Opdivo plus Yervoy may result in improvement in health-related quality of life, compared with sorafenib or lenvatinib, but the clinical importance of the effect is uncertain. There is uncertainty in the evidence due to risk of bias arising from study limitations.
Opdivo plus Yervoy results in increased any-grade serious AEs and likely results in increased any-grade AEs leading to withdrawal of treatment, compared with sorafenib or lenvatinib; but the clinical importance of the effect is uncertain. There is uncertainty due to imprecision in the estimates (95% CI includes the possibility of little to no clinically important difference).
Overall, the proportion of trial participants with AEs, serious AEs, AEs leading to withdrawal of treatment, and immune-mediated AEs — especially grade 3 or 4 AEs — was numerically greater in the group that received Opdivo plus Yervoy than the group that received sorafenib or lenvatinib. This is aligned with expectations for the treatment due to the relatively high dose of ipilimumab.
For the comparison of Opdivo plus Yervoy versus atezolizumab plus bevacizumab and versus durvalumab plus tremelimumab:
STCs suggest Opdivo plus Yervoy may result in delayed improvements in OS and progression-free survival (PFS) compared with durvalumab plus tremelimumab and atezolizumab plus bevacizumab but may also lead to lower survival (higher mortality) during the early months after the start of treatment. The certainty of this evidence is low due to methodological limitations, including violation of the proportional hazard assumption and lack of a common comparator across trials.
There remains a lack of evidence for the treatment effect of Opdivo plus Yervoy versus atezolizumab plus bevacizumab and versus durvalumab plus tremelimumab on outcomes important to patients and clinicians, including health-related quality of life and harms.
Economic Evidence
Opdivo is available as an IV solution at a strength of 10 mg/mL. At the submitted price of $782.22 per 40 mg/4 mL vial, the 28-day cycle cost of Opdivo is expected to be $2,086 per patient in the first 4 cycles (combination phase) and $9,387 in subsequent cycles (single-drug phase), based on the Health Canada–recommended dosage. Yervoy is available as an IV solution at a strength of 5 mg/mL.1 At the submitted price of $5,800.00 per 50 mg/10 mL vial, the 28-day cycle cost of Yervoy is expected to be $38,667 per patient in the combination phase, based on the Health Canada–recommended dosage. Therefore, the 28-day cycle cost of Opdivo plus Yervoy is expected to be $40,753 per patient in the first 4 cycles (combination phase) and then $9,387 in subsequent cycles (single-drug phase).
Clinical efficacy in the economic analysis for Opdivo plus Yervoy versus sorafenib and versus lenvatinib was derived from the pooled data of the CheckMate 9DW trial.2 Evidence submitted by the sponsor indicates that among patients with unresectable or advanced HCC, Opdivo plus Yervoy is likely to result in a clinically important decrease in the probability of OS and PFS compared with sorafenib and lenvatinib at 6 months; however, a clinically important increase in the probability of OS and PFS at 24 months was likely observed. The time-varying treatment effect and the inability to reliably identify patients who are considered to be at high risk of early mortality may limit the generalizability of the observed treatment effect to clinical practice.
Due to limitations associated with the sponsor’s submitted STC, the comparative effectiveness between Opdivo plus Yervoy versus durvalumab plus tremelimumab and versus atezolizumab plus bevacizumab was deemed of low certainty. Therefore, the evidence of clinical benefit is too uncertain to estimate the price reduction needed to ensure cost-effectiveness of Opdivo plus Yervoy in relation to durvalumab plus tremelimumab or atezolizumab plus bevacizumab.
CDA-AMC noted several limitations that add uncertainty to the results of the sponsor’s cost-utility analysis.3 These limitations could not be addressed through reanalysis due to the lack of alternative data (refer to Table 8 and Appendix 11 in the Supplemental Material). Consequently, no base-case reanalysis was performed. CDA-AMC notes that the predicted clinical benefit of Opdivo plus Yervoy remains uncertain given that more than 70% of the incremental quality-adjusted life-years with Opdivo plus Yervoy compared with all comparators were accrued beyond the trial period, during which time there is no clinical evidence.
CDA-AMC estimates that the cost savings of reimbursing Opdivo in combination with Yervoy for the treatment of unresectable or advanced HCC will be approximately $2,072,919 over the first 3 years of reimbursement compared with the amount currently spent on comparators. The expenditure on Opdivo plus Yervoy over this period is predicted to be $28.8 million. The actual budget impact of reimbursing Opdivo plus Yervoy will depend on the confidentially negotiated price of comparators.
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
ECOG PS
Eastern Cooperative Oncology Group Performance Status
FACT-Hep
Functional Assessment of Cancer Therapy–Hepatobiliary
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HCC
hepatocellular carcinoma
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IO
immuno-oncology
ITC
indirect treatment comparison
KM
Kaplan-Meier
MID
minimal important difference
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
SOC
standard of care
STC
simulated treatment comparison
TKI
tyrosine kinase inhibitor
TTD
time to treatment discontinuation
WDAE
withdrawal due to adverse event
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of nivolumab 10 mg/mL plus ipilimumab 5 mg/mL injections as a first-line treatment for adult patients with unresectable or advanced hepatocellular carcinoma (HCC). The review focuses on comparing nivolumab plus ipilimumab with relevant comparators in clinical practice in Canada (atezolizumab plus bevacizumab, durvalumab plus tremelimumab, lenvatinib, and sorafenib), and on identifying gaps in the current evidence. The scope of the review, including the intervention, comparators, and outcomes of interest, is summarized in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Nivolumab (Opdivo), 10 mg/mL, single-use vials, IV infusion Ipilimumab (Yervoy), 5 mg/mL, single-use vials, IV infusion |
Sponsor | Bristol Myers Squibb Canada Co. |
Health Canada indication | Opdivo (nivolumab), in combination with ipilimumab, is indicated for the first-line treatment of adult patients with unresectable or advanced hepatocellular carcinoma. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard, Project Orbis (Type B) |
NOC date | June 19, 2025 |
Mechanism of action | Nivolumab blocks PD-1 receptor interaction with its ligands, PD-L1 and PD-L2, enhancing T-cell function and antitumour response. Ipilimumab blocks CTLA-4 receptor interaction with CD80/CD86, enhancing T-cell function and antitumour response. |
Recommended dosage | Combination phase:
Single-drug phase:
|
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | Nivolumab: $782.22 per 40 mg/4 mL vial; $1,955.56 per 100 mg/10 mL vial Ipilimumab: $5,800.00 per 50 mg/10 mL vial; $23,200.00 per 200 mg/40 mL vial |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focus | Population: As defined in the Health Canada indication Subgroups: None Intervention: Per recommended dosage Comparators: Atezolizumab plus bevacizumab,a durvalumab plus tremelimumab,a lenvatinib,a and sorafenib Outcomes: OS, PFS, FACT-Hep, standard harms outcomes (AEs, SAEs, WDAEs, deaths), and notable harms outcomes (immune-mediated AEsb) |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; FACT-Hep = Functional Assessment of Cancer Therapy–Hepatobiliary; NOC = Notice of Compliance; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aCDA-AMC has previously issued a reimbursement recommendation for this drug for the same indication or a similar indication.
bImmune-mediated AEs are identified in the serious warnings and precautions box in the product monograph for Opdivo (nivolumab).4
Submission History for Nivolumab and Ipilimumab
CDA-AMC previously reviewed nivolumab and ipilimumab through the reimbursement review process in the following therapeutic areas: malignant pleural mesothelioma, non–small cell lung cancer, advanced or metastatic renal cell carcinoma, and metastatic melanoma.
At the time of this submission, the CDA-AMC review of nivolumab and ipilimumab in unresectable or metastatic microsatellite instability-high or mismatch repair deficient colorectal cancer is ongoing through the reimbursement review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted by CDA-AMC for this review (hereafter referred to as the clinical experts consulted for this review).
Calls for patient group and clinician group input are issued for each Reimbursement Review. One patient group submission was received from Liver Canada, and 2 clinician group submissions were received from the Canadian Gastrointestinal Oncology Evidence Network and the Ontario Health (Cancer Care Ontario) Gastrointestinal Cancer Drug Advisory Committee. Patient input was gathered from 14 survey respondents across Canada in May 2025, including patients with liver cancer and caregivers. Five specialists in gastrointestinal cancer across Canada and 4 clinician members of the Drug Advisory Committee provided input based on their review of relevant evidence. The full submissions received are available on the project landing page. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two medical oncologists with expertise in the diagnosis and management of unresectable or advanced HCC participated as part of the review team, with representation from Ontario and British Columbia.
Disease Background
HCC, defined as liver cancer that starts in the cells that make up the body of the liver (hepatocytes), accounts for more than 80% of primary liver cancer cases.5 Worldwide, liver cancer was the sixth most common cancer (ranked out of 33 other cancer types), with an estimated 866,136 new cases, and the third leading cause of cancer-related death, with an estimated 758,725 deaths, in 2022.6 Among people living in Canada, liver and intrahepatic bile duct cancer was the 14th most common cancer (ranked out of 23 other cancer types), with an estimated 4,700 new cases, and the sixth leading cause of cancer-related death, with an estimated 3,500 deaths, in 2023.7 The predicted 5-year net survival for cases of liver and intrahepatic bile duct cancer diagnosed in 2015 to 2017 was 18% in Canada.7 In addition, the 5-year person-based prevalence in Canada, defined as the number of individuals living with or beyond a diagnosis of liver cancer on January 1, 2018, was 4,265 individuals.8
Important risk factors of HCC include chronic hepatitis B and C viral infections, which account for approximately 80% of HCC cases worldwide, alcohol-related cirrhosis (alcohol consumption), metabolic dysfunction-associated steatotic liver disease, and diabetes mellitus.5
Liver cancer may not cause symptoms in its early stages and is often not diagnosed until it reaches an advanced stage, when it can no longer be surgically removed (unresectable).9 Signs and symptoms of liver cancer include, but are not limited to, pain in the upper-right abdomen, a lump on the right side below the ribs, swelling of the abdomen and/or legs and feet, easy bruising or bleeding, and jaundice.10
Patient Group Input
Patients identified the following debilitating symptoms of HCC: chronic pain, fatigue, significant weight loss, muscle wasting, and impaired mobility. Patients also reported emotional distress, loss of independence, disruption to employment and family life, and reduction in quality of life as the negative impact of HCC. The patient group indicated that many patients are forced to stop working, require caregiver support, and transition into supportive living environments.
Current Management
Treatment Goals
Patient Group Input
Patients identified living longer, maintaining quality of life, minimizing adverse events (AEs), slowing cancer progression, and ease of access as important treatment outcomes.
Clinical Expert Input
The clinical experts identified improving survival, maintaining quality of life, and achieving disease control as important goals of first-line treatment for adults with unresectable or advanced HCC. The clinical experts indicated that response to treatment (i.e., tumour shrinking) contributes to improvements in health-related quality of life (HRQoL) when tumours compromise anatomical structures (e.g., biliary system causing cholangitis or vascular obstruction), by reducing associated pain. Although bleeding-related AEs are an important consideration, the clinical experts indicated that this is less likely to be applicable to nivolumab plus ipilimumab.
Clinician Group Input
In addition to the treatment goals identified by the clinical experts consulted for this review, clinicians indicated that the ideal therapy should have a reasonable safety profile (i.e., should not significantly impact quality of life and should not increase the risk of bleeding due to varices or large tumours that may rupture).
Current Treatment Options
The Provisional Funding Algorithm for unresectable HCC, dated January 24, 2024, is available on the project landing page. The treatment options for previously untreated, unresectable HCC include atezolizumab plus bevacizumab, durvalumab plus tremelimumab, and tyrosine kinase inhibitors (TKIs).11 Durvalumab plus tremelimumab would be suitable for patients who are considered to be at high risk of bleeding (i.e., would not be eligible for atezolizumab plus bevacizumab).11 If immunotherapy is unavailable or not indicated for a patient, lenvatinib would be an alternative first-line treatment. In cases of intolerance or contraindication to lenvatinib, sorafenib can be offered.11 Note that durvalumab plus tremelimumab concluded negotiations with the pan-Canadian Pharmaceutical Alliance, with a letter of intent on February 28, 2024, for unresectable HCC in adult patients who require systemic therapy as a first-line treatment.12
Clinical Expert Input
Input from the clinical experts on first-line treatment for adults with unresectable or advanced HCC aligns with the 2024 Provisional Funding Algorithm. The clinical experts noted that, although these treatment options improve survival and control disease progression, immunotherapy combinations have demonstrated superiority over TKIs.
Clinician Group Input
Clinicians generally agreed with the clinical experts consulted for this review.
Key characteristics of nivolumab plus ipilimumab, along with other treatments available for unresectable or advanced HCC, are summarized in the Key Characteristics table in Appendix 1 in the Supplemental Material (available on the project landing page).
Unmet Needs and Existing Challenges
Patient Group Input
Patients identified a need for additional treatment options and access to treatments with improved tolerability.
The patient group indicated that people from equity-deserving groups, including Indigenous Peoples, people who have recently immigrated to Canada, and people experiencing low income, face barriers to diagnosis, care coordination, and access to novel therapies for HCC. These barriers were reported to contribute to disease progression and poorer health outcomes. As such, the patient group identified a need for systemic changes to improve early detection, referral pathways, equitable drug and testing access, and to avoid disparities in survival and quality of life, particularly in rural communities.
Clinical Expert Input
The clinical experts identified a need for additional treatment options that offer, for example, a higher likelihood of a complete response, an increased probability of downstaging to curative therapy, and the potential to allow patients to discontinue treatment.
The clinical experts noted that people with IV drug or alcohol dependency (associated with hepatitis C viral etiology), people who have immigrated to Canada from regions where hepatitis B is endemic, and patients with metabolic-associated liver cirrhosis are often underrepresented in clinical trials due to a lack of support. The clinical experts further noted that these equity-deserving groups often face barriers to accessing health care and lack support in navigating health care systems due to socioeconomic status and other structural inequities.
Clinician Group Input
In consideration of the increased risk of bleeding due to underlying liver conditions (e.g., cirrhosis) and varices in these patients, the clinicians indicated that therapies that do not further increase this risk are clinically important. Clinicians indicated that the single-dose tremelimumab in combination with regular-interval durvalumab (STRIDE) regimen is the only pure immunotherapy treatment funded to date that is associated with a lower risk of bleeding.
Considerations for Using the Drug Under Review
The content in this section is informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (presented in the Conditions Proposed by the Sponsor table in Appendix 1 in the Supplemental Material). Implementation questions from the public drug programs and the corresponding responses from the clinical experts consulted for this review are summarized in the Drug Program Input and Clinical Expert Response table in Appendix 1 in the Supplemental Material. The following information has been summarized by the review team.
Place in Therapy
Clinical Expert Input
The clinical experts indicated that the anticipated place in therapy for nivolumab plus ipilimumab would be as a first-line immunotherapy combination treatment option. The clinical experts noted that additional lines of therapy are TKI-based; however, funding is generally limited to second-line treatment, even though some therapies have been evaluated in the third-line setting (i.e., cabozantinib).
Clinician Group Input
Clinicians generally agreed with the clinical experts consulted for this review and indicated that the choice of therapy is based on patient characteristics and preferences. Clinicians suggested that nivolumab plus ipilimumab may be more practical for smaller centres that do not routinely use durvalumab plus tremelimumab, given that nivolumab plus ipilimumab is also used for other tumour sites.
Patient Population
Clinical Expert Input
The clinical experts identified patients with advanced (unresectable) or metastatic HCC and preserved Eastern Cooperative Oncology Group Performance Status (ECOG PS) and/or Child-Pugh status as those most in need of intervention. Although there are no known predicators to identify patients who would most likely respond to treatment with nivolumab plus ipilimumab, the clinical experts indicated that patients with prior treatment experience and poor ECOG PS scores and/or Child-Pugh status would be least suitable for treatment with this regimen.
Regarding the initiation conditions proposed by the sponsor, the clinical experts indicated that these conditions are standard and reasonable; however, some patients with borderline Child-Pugh B7 status could also be considered for treatment (e.g., those with borderline albumin levels). The 2023 CADTH Reimbursement Recommendation for tremelimumab in combination with durvalumab recommends that patients with severe autoimmune or inflammatory disorders are ineligible for treatment with tremelimumab in combination with durvalumab due to lack of evidence. The clinical experts suggested that this condition would be applicable to nivolumab plus ipilimumab and noted that assessment of the severity of autoimmune or inflammatory disorders in this context would be based on clinical judgment.
Clinician Group Input
Clinicians generally agreed with the clinical experts consulted for this review. Additionally, clinicians indicated that patients with a prior liver transplant would be least suitable for treatment with nivolumab plus ipilimumab. Clinicians defined preserved liver function as Child-Pugh class A and defined decompensated liver function as Child-Pugh class B and C.
Assessing Response to Treatment
Clinical Expert Input
The clinical experts indicated that clinical and/or biochemical response measures, along with on-treatment imaging, are used to assess response to treatment in practice. They noted that imaging is generally performed every 3 to 4 months, depending on access across Canada. The clinical experts indicated that stable disease or better, as defined by the Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST 1.1), as well as improvements in symptoms, quality of life, and survival, are considered clinically meaningful responses to treatment in practice.
The sponsor did not propose any renewal conditions. The clinical experts deferred to previous reimbursement recommendation conditions and funding algorithms in HCC for applicable renewal conditions. The 2020 CADTH Reimbursement Recommendation for atezolizumab and bevacizumab for HCC states: “The pan-Canadian Oncology Drug Review Expert Review Committee (pERC) agrees with the clinical guidance panel that re-treatment would be reasonable if the treatment was discontinued for reasons other than progression (e.g., treatment break, intolerance). Re-treatment would be reasonable if progression occurs more than 6 months after stopping treatment with atezolizumab plus bevacizumab.”13
Clinician Group Input
Clinicians generally agreed with the clinical experts consulted for this review. Additionally, clinicians suggested to assess response to treatment every 2 months at the beginning to detect early progression. They indicated that elevated alpha-fetoprotein levels may be used as a biomarker to assess treatment response and highlighted the importance of monitoring for grade 3 and grade 4 toxicities.
Discontinuing Treatment
Clinical Expert Input
The clinical experts identified toxicity, disease progression, and patient preference as factors that should be considered when deciding to discontinue treatment with nivolumab plus ipilimumab.
The clinical experts agreed with the discontinuation conditions proposed by the sponsor.
Clinician Group Input
In addition to the factors identified by the clinical experts consulted for this review, clinicians indicated that decompensation of liver cirrhosis should be considered when deciding to discontinue treatment with nivolumab plus ipilimumab.
Prescribing Considerations
Clinical Expert Input
The clinical experts indicated that the drug regimen under review belongs to a class that is already commonly used in practice by medical oncologists and physician extenders, including in resource-limited regions.
The clinical experts agreed with the prescribing conditions proposed by the sponsor.
Clinician Group Input
Clinicians elaborated that nonmedical oncologists (i.e., surgical oncologists and radiation oncologists), with general oversight of a medical oncologist, may be involved in prescribing in some jurisdictions. Additionally, clinicians suggested the use of outpatient infusion clinics, including satellite clinics.
Clinical Review
Methods
The review team considered trials in the sponsor’s systematic review (pivotal trials and randomized controlled trial [RCTs]), indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and phase IV RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included atezolizumab plus bevacizumab, durvalumab plus tremelimumab, lenvatinib, and sorafenib. ITCs and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., a missing comparator or longer follow-up time).
The review team selected outcomes and follow-up times for review considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes were those considered relevant to expert committee deliberations and were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes were assessed using GRADE because they address important treatment goals for advanced or unresectable HCC and are considered important to patients per patient and clinician input:
overall survival (OS)
progression-free survival (PFS)
Functional Assessment of Cancer Therapy–Hepatobiliary (FACT-Hep) total score
serious adverse events (SAEs)
withdrawal due to adverse events (WDAEs).
Although time on treatment and time to treatment discontinuation (TTD) are inputs in the sponsor’s pharmacoeconomic model, these outcomes were not included in the sponsor’s inclusion criteria for the systematic review and were not assessed as efficacy end points in the study included in the systematic review.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are summarized in Appendix 2 in the Supplemental Material.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal study included in the systematic review, the CheckMate 9DW study
1 ITC.
The sponsor also submitted evidence from CheckMate 040, a phase I/II, dose-escalation, open-label, noncomparative study of nivolumab alone or nivolumab in combination with ipilimumab in patients with advanced HCC, with or without chronic viral hepatitis. In addition, the sponsor also submitted evidence from a randomized, open-label study of nivolumab versus sorafenib in patients with advanced HCC who were naive to systemic therapy. Within the CheckMate 040 study, the sponsor identified cohort 4 as the most relevant for this review — the nivolumab plus ipilimumab combination cohort, consisting of 3 groups receiving different doses of nivolumab and ipilimumab. Notably, cohort 4 included patients whose disease was intolerant to or had progressed on sorafenib, reflecting a second-line treatment setting rather than the first-line setting specified in the indication under review. Additionally, the sponsor did not identify any evidence gaps in the submission. For these reasons, the CheckMate 040 study was not included in this review.
Systematic Review
Description of Study
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the intervention and comparators, including prohibited and permitted treatments and relevant outcome measures, are summarized in Appendix 3 in the Supplemental Material.
CheckMate 9DW is an ongoing study evaluating the efficacy and safety of nivolumab plus ipilimumab as first-line therapy compared with investigator’s choice of standard of care (SOC) with either sorafenib or lenvatinib, according to patient profile and local standards, in adults with unresectable or advanced HCC. The primary objective was to compare the OS of nivolumab plus ipilimumab with sorafenib or lenvatinib, measured as the time from randomization to the date of death due to any cause. The study is being conducted at 163 sites across 25 countries, including 1 site in Canada from which 10 patients were included in the randomization.
There are 3 phases in the study design: screening, treatment, and follow-up. From January 2020 to November 2021, patients were enrolled and randomized in a 1:1 ratio using interactive response technology, to receive open-label treatment with either nivolumab plus ipilimumab or investigator’s choice of sorafenib or lenvatinib. Randomization was stratified by etiology (hepatitis B virus, hepatitis C virus, or uninfected), macrovascular invasion and/or extrahepatic spread (present or absent), and alpha-fetoprotein baseline level (< 400 ng/mL or ≥ 400 ng/mL).
Tumour images were reviewed on a rolling basis by blinded independent central review, with reviewers blinded to treatment assignment, clinical data, and investigator assessments of the submitted images. Summaries of unblinded, aggregate efficacy and safety analyses were prepared by an external independent statistician for review by an independent data monitoring committee in closed meetings.
Table 2: Characteristics of the Study Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
CheckMate 9DW study: Global, phase III, open-label, sponsor-blind,a RCT Total N = 668 |
|
| Intervention:f,g,h
Comparator:h
| Primary:
Exploratory:
Safety:
|
AE = adverse event; BICR = blinded independent central review; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FACT-Hep = Functional Assessment of Cancer Therapy–Hepatobiliary; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; IPI = ipilimumab; NIV = nivolumab; OS = overall survival; PD = progressive disease; PFS = progression-free survival; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SAE = serious adverse event; SOC = standard of care; WDAE = withdrawal due to adverse event.
aThe sponsor was blinded to aggregate safety and efficacy data by treatment assignments, including comparisons between treatment groups, until the independent data monitoring committee reviewed the prespecified interim analysis results of OS on March 15, 2024.
bPrior neoadjuvant or adjuvant systemic therapy was permitted if recurrence occurred ≥ 12 months after treatment completion.
cAdequate hepatic function was defined by using the following screening laboratory values that met the following criteria: serum albumin ≥ 2.8 g/dL (transfusion to meet this level was not permitted), serum total bilirubin ≤ 3 mg/dL, serum aspartate aminotransferase ≤ 5 times the upper limit of normal, and alanine aminotransferase ≤ 5 times the upper limit of normal.
dInhaled or topical steroids and adrenal replacement steroid doses >10 mg/day prednisone equivalents were permitted in the absence of active autoimmune disease. Participants with type 1 diabetes mellitus, hypothyroidism only requiring hormone replacement, and skin disorders (e.g., vitiligo, psoriasis, and alopecia) not requiring systemic treatment, or conditions not expected to recur in the absence of an external trigger were permitted to enrol.
ePalliative radiotherapy for symptomatic control was acceptable if it was completed at least 2 weeks before randomization and no additional radiotherapy for the same lesion was planned.
fIV infusions were administered in an outpatient hospital or cancer centre clinic, including satellite clinics, by trained medical personnel at each site.
gA minimum of 1 dose of NIV plus IPI is required before proceeding to the nivolumab monotherapy dosing.
hPatients could be treated beyond disease progression according to protocol-defined conditions.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence14 and the Clinical Study Report for the CheckMate 9DW study.15
This report is based on the primary Clinical Study Report that provided results from the planned interim analysis of the primary efficacy end point of OS. As of the clinical cut-off date of January 31, 2024, and database lock date of February 28, 2024, a total of 422 OS events had occurred, representing 81% of the total number of events planned for the final OS analysis. On March 15, 2024, the independent data monitoring committee concluded a statistically significant improvement in the primary end point of OS with nivolumab plus ipilimumab compared with sorafenib or lenvatinib in the study population. Consequently, the interim analysis was considered the final analysis of the primary end point. The median follow-up was 35.2 months (range, 26.8 months to 48.9 months).
Statistical Testing and Analysis Populations
In a global protocol amendment dated August 2, 2021, the statistical assumptions on power calculation, including the number of expected events and the follow-up time to trigger the interim and final analyses, were modified based on the survival results from the CheckMate 459 study.2 In a subsequent global protocol amendment dated February 1, 2023, these statistical assumptions were further modified based on the survival results from external data from the LEAP-002 study.16
The sample size determination was based on the comparison of OS between nivolumab plus ipilimumab and sorafenib or lenvatinib. Based on a planned sample size of 650 patients who were randomized in a 1:1 ratio to each group, 87% power would be achieved for an average OS hazard ratio (HR) of 0.74 (nivolumab plus ipilimumab compared to sorafenib or lenvatinib), using an overall 2-sided type I error rate of 0.05. The key assumptions for sample size determination included a piecewise exponential distribution for OS in each group; a median OS of 19 months for sorafenib or lenvatinib, based on OS data for lenvatinib from the LEAP-002 study16; and a median OS of 23 months for nivolumab plus ipilimumab. The target average HR of 0.74 was modelled as a 2-piece HR, with a delayed effect corresponding to an HR of 1.0 in the first 15 months, followed by a constant HR of 0.6 thereafter.
The data were derived from the preplanned interim analysis of the primary end point of OS, conducted after at least 416 OS events had been observed, corresponding to 80% of the total 520 OS events planned for the final analysis. The alpha allocation for the interim and final analyses was based on the Lan-DeMets alpha spending function approach, using an O’Brien-Fleming stopping boundary to control the overall 2-sided type I error rate at 5%. The stopping boundary was dependent on the actual number of deaths at the time of the analysis. If the trial was stopped for superiority of OS at the interim analysis, the P value from the interim stratified log-rank test was considered the final primary analysis result.
Efficacy and HRQoL analyses were based on the randomized population, defined as all patients who were randomized to a treatment group. Safety analyses were based on the treated population, defined as all patients who received at least 1 dose of a study drug.
Patient Disposition
A total of 1,148 patients were enrolled, of whom 668 were randomized and 657 received treatment. Among patients who were treated, 21% (71 of 332 patients) in the nivolumab plus ipilimumab group and 16% (52 of 325 patients) in the sorafenib or lenvatinib group discontinued the study. The most frequently reported reason for discontinuing the study was death, occurring in 15% of patients in the nivolumab plus ipilimumab group and 8% of patients in the sorafenib or lenvatinib group; all other reasons for study discontinuation were reported in fewer than 10% of patients in each group. Among patients who were treated, all 332 patients in the nivolumab plus ipilimumab group and 95% of patients (310 of 325 patients) in the sorafenib or lenvatinib group discontinued study treatment. The most frequently reported reason for discontinuing treatment was disease progression, reported in 40% of patients in the nivolumab plus ipilimumab group and 69% of patients in the sorafenib or lenvatinib group, followed by study drug toxicity, reported in 22% and 11% of patients, respectively. All other reasons for treatment discontinuation were reported in fewer than 10% of patients in each group.
Important protocol deviations were reported in 67% of patients (225 of 335 patients who were randomized) in the nivolumab plus ipilimumab group and 70% of patients (232 of 333 patients who were randomized) in the sorafenib or lenvatinib group. Although protocol deviations were frequently reported, the reasons for protocol deviations were similar in both treatment groups. Details pertaining to the protocol deviations are summarized in Appendix 4 in the Supplemental Material.
Baseline Characteristics
A summary of baseline characteristics for the randomized analysis population in the CheckMate 9DW study is presented in Table 3.
Table 3: Summary of Baseline Characteristics From the Study Included in the Systematic Review (Randomized Population)
Characteristic | CheckMate 9DW | |
|---|---|---|
Nivolumab plus ipilimumab (N = 335) | Sorafenib or lenvatinib (N = 333) | |
Age (years), median (range) | 65 (20 to 86) | 66 (20 to 89) |
Age category, n (%) | ||
< 65 years | 162 (48.4) | 149 (44.7) |
≥ 65 years and < 75 years | 126 (37.6) | 123 (36.9) |
≥ 75 years | 47 (14.0) | 61 (18.3) |
Sex, n (%) | ||
Female | 64 (19.1) | 56 (16.8) |
Male | 271 (80.9) | 277 (83.2) |
Race,a n (%) | ||
Asian | 140 (41.8) | 152 (45.6) |
Black or African American | 11 (3.3) | 4 (1.2) |
Native Hawaiian or other Pacific Islander | 1 (0.3) | 0 |
White | 179 (53.4) | 174 (52.3) |
Other | 4 (1.2) | 3 (0.9) |
ECOG PS, n (%) | ||
0 | 233 (69.6) | 243 (73.0) |
1 | 102 (30.4) | 89 (26.7) |
Not reported | 0 | 1 (0.3) |
Weight (kg), median (range) | 71.20 (39.0 to 134.9) | 70.00 (36.5 to 160.0) |
Etiology per central lab, n (%) | ||
Hepatitis B virus | 99 (29.6) | 98 (29.4) |
Hepatitis C virus | 27 (8.1) | 27 (8.1) |
Uninfected (nonviral-related HCC)b | 209 (62.4) | 208 (62.5) |
Macrovascular invasion, n (%) | ||
Yes | 77 (23.0) | 92 (27.6) |
No | 258 (77.0) | 241 (72.4) |
Extrahepatic spread, n (%) | ||
Yes | 188 (56.1) | 172 (51.7) |
No | 147 (43.9) | 161 (48.3) |
Alpha-fetoprotein category, n (%) | ||
< 400 ng/mL | 227 (67.8) | 220 (66.1) |
≥ 400 ng/mL | 108 (32.2) | 113 (33.9) |
Child-Pugh score, n (%) | ||
5 | 254 (75.8) | 263 (79.0) |
6 | 72 (21.5) | 58 (17.4) |
7 | 8 (2.4) | 11 (3.3) |
8 | 1 (0.3) | 0 |
Not reported | 0 | 1 (0.3) |
BCLC staging, n (%) | ||
0 | 3 (0.9) | 3 (0.9) |
A | 25 (7.5) | 18 (5.4) |
B | 61 (18.2) | 67 (20.1) |
C | 246 (73.4) | 242 (72.7) |
D | 0 | 0 |
Unknown | 0 | 3 (0.9) |
Prior adjuvant systemic therapy, n (%) | 1 (0.3) | 1 (0.3) |
Prior surgery related to cancer, n (%) | 133 (39.7) | 133 (39.9) |
Prior radiotherapy, n (%) | 32 (9.6) | 24 (7.2) |
Prior nonsystemic cancer therapy related to HCC local only, n (%) | 142 (42.4) | 158 (47.4) |
Prior nonsystemic treatment for HCC, n (%) | ||
Transcatheter arterial chemoembolization | 118 (35.2) | 129 (38.7) |
Radiofrequency ablation | 51 (15.2) | 60 (18.0) |
Transcatheter arterial embolization | 12 (3.6) | 14 (4.2) |
Microwave ablation | 9 (2.7) | 16 (4.8) |
Yttrium-90 microspheres | 8 (2.4) | 8 (2.4) |
Hepatic arterial infusion chemotherapy | 5 (1.5) | 2 (0.6) |
Percutaneous ethanol injection | 4 (1.2) | 2 (0.6) |
Other | 5 (1.5) | 6 (1.8) |
BCLC = Barcelona Clinic Liver Cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HCC = hepatocellular carcinoma.
aCategories are as reported in the study.
bFor stratification purposes, the “uninfected” category includes patients with past or resolved hepatitis B or C virus infection.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence14 and the Clinical Study Report for the CheckMate 9DW study.15
Treatment Exposure, Concomitant Medications, and Subsequent Treatments
Among the 332 patients treated in the nivolumab plus ipilimumab group during the combination phase, patients received a median of 4 doses (range, 1 dose to 4 doses) of each drug. Of those who were treated in the nivolumab plus ipilimumab group, ███ ████████ ████████ █ ██████ ██ ██ █████ ███████ █ ██ ███ ██ █████████ ███ ██ ██ ███ ███████████ █████. A summary of treatment exposure by ipilimumab dose category is presented in Appendix 4 in the Supplemental Material. Among the 325 patients who were treated in the SOC group, 50 received sorafenib 400 mg, 62 received lenvatinib 8 mg, and 213 received lenvatinib 12 mg.
The median duration of treatment was 4.68 months (range, < 0.1 months to 24.4 months) in the nivolumab plus ipilimumab group and 6.93 months (range, < 0.1 months to 45.8 months) in the sorafenib or lenvatinib group.
A total of 62 patients in the nivolumab plus ipilimumab group (16 patients during the combination phase and 46 patients during the nivolumab monotherapy phase) and 157 patients in the sorafenib or lenvatinib group were treated beyond progression. Treatment beyond progression was defined as patients whose last available dose date was after the date of radiographic progression, as assessed by the investigator using RECIST 1.1. A summary of treatment beyond progression is presented in Appendix 4 in the Supplemental Material.
A total of █████ ██ ████████ ████ ██ ███ ███████ █████████ ██ ███ █████████ ████ ██████████ █████ ███ █████ ██ ████████ ████ ██ ███ ███████ █████████ ██ ███ █████████ ██ ██████████ █████ ████████ ███████████ ███████████. Notably, immune-modulating concomitant medications were administered for the management of AEs in █████ ██ ████████ ████ █████████ in the nivolumab plus ipilimumab group and █████ ██ ████████ ███ █████████ in the sorafenib or lenvatinib group. Dermatological drugs were the most common type of concomitant immune-modulating medication administered in both groups (█████ in the nivolumab plus ipilimumab group and █████ in the sorafenib or lenvatinib group).
A total of 45.1% of patients (151 of 335 patients who were randomized) in the nivolumab plus ipilimumab group and 55.6% of patients (185 of 333 patients who were randomized) in the sorafenib or lenvatinib group received subsequent anticancer therapy. Details regarding subsequent treatments are summarized in Appendix 4 in the Supplemental Material.
Critical Appraisal
Internal Validity
Immune-checkpoint inhibitors and TKIs belong to 2 separate drug classes with key differences in administration, dosing schedules, and safety profiles, making blinding impractical in the study. An open-label design was used to avoid placing participants at risk of AEs and to ensure appropriate identification and management of AEs according to the study drug received. The potential risk of bias due to the open-label design was likely mitigated for response-related end points, including PFS, by a blinded independent central review of tumour assessments.
Among patients randomized to receive SOC, 50 patients received sorafenib and 275 patients received lenvatinib. Regarding the use of a pooled comparator group, the REFLECT study concluded that “lenvatinib was noninferior to sorafenib in OS for patients with untreated advanced hepatocellular carcinoma.”17 However, a reanalysis using “a covariate adjustment of REFLECT suggested that the original noninferiority trial likely underestimated the true effect of lenvatinib on OS due to an imbalance in baseline prognostic covariates and the greater use of post-treatment therapies in the sorafenib group.”18 Given that there is evidence of between-study heterogeneity between the CheckMate 9DW study and the REFLECT study, as discussed in the Indirect Evidence section, there are concerns regarding the reliability of comparative effect estimates derived from pooled comparator group inputs. The magnitude and direction of any resulting bias would be difficult to determine in the absence of more in-depth information.
Randomization was achieved via interactive response technology, a method that is sufficient for concealing treatment allocation until the time of assignment. Randomization was stratified by etiology, the presence or absence of macrovascular invasion and/or extrahepatic spread, and baseline alpha-fetoprotein levels. These factors have been identified as prognostic, as discussed in the Indirect Evidence section, and were also identified as standard stratification factors by the clinical experts consulted for this review. Randomization appears to have adequately balanced baseline demographic and disease characteristics between groups, and the risk of selection bias related to the randomization process is likely low.
OS remains the gold standard for evaluating the efficacy of immunotherapy, as it captures both the effects of treatment and treatment-related toxicity on survival.19 Although immunotherapy clinical trials have demonstrated OS benefits, differences in PFS have not always been observed; as such, analyses of PFS may be limited in their ability to predict long-term survival.19 The clinical experts consulted for this review agreed that this limitation is particularly relevant in HCC. In addition, RECIST 1.1 does not capture pseudoprogression, defined as a radiographic tumour flare that occurs before a clinical response and may be observed with immunotherapy.19 In a systematic review and meta-analysis of 77 phase III clinical studies evaluating immunotherapy across 15 different types of tumours, including HCC (4 studies), a weak association was observed between the clinical end point of OS and the surrogate end point of PFS (i.e., the coefficient of determination [R2] and its associated 95% CI were less than 0.7).20 Although evidence from the literature suggests that PFS may not be a valid surrogate for OS, PFS remains a clinically relevant end point for patients who have identified slowing cancer progression as an important treatment outcome.
Evidence supporting the validity, reliability, and responsiveness of the FACT-Hep questionnaire as a measure of HRQoL, as well as the between-group minimal important difference (MID) of 8 to 9 points in patients with hepatobiliary carcinoma,21,22 is summarized in Appendix 3 in the Supplemental Material.
The potential risk of overestimation of treatment effect due to interim analysis was likely reduced because the type I error was controlled for and the interim analysis was conducted by an external independent data monitoring committee. In addition, a relatively high proportion of events (81% of the total number of events planned for the final OS analysis) had occurred at the time of the interim analysis.
In the analyses of OS and PFS (primary definition), censoring due to lost to follow-up was low, occurring in fewer than 2% of patients who were randomized to each group. In the analysis of PFS (primary definition), censoring due to the absence of on-study tumour assessment without death was also low, affecting fewer than 4% of patients who were randomized to each group. Although censoring due to receipt of subsequent anticancer therapy was relatively high (> 15% of patients who were randomized to each group) in the analysis of PFS (primary definition), the results were generally consistent with the results from the analysis of PFS (secondary definition, irrespective of subsequent therapy), thereby supporting the results using the primary definition, which accounted for subsequent therapy.
For the analyses of OS and PFS, there was a violation of the proportional hazards assumption, as evidenced by the early survival detriment and PFS detriment with nivolumab plus ipilimumab relative to sorafenib or lenvatinib. As such, the ratio of the hazard functions was not constant over time, and reliance on the HR alone to inform the OS and PFS benefits may be misleading. To address the challenge with interpreting the HR when there is violation of the proportional hazards assumption, 3 prespecified supportive analyses were performed to account for the delayed effect. The results from the supportive analyses — piecewise HR, unstratified Max-Combo test, and restricted mean survival time analyses — were generally aligned with the between-group differences in Kaplan-Meier (KM) estimated probabilities of OS and PFS at clinically relevant follow-up time points.
Between-group differences in KM-estimated probabilities of OS and PFS at clinically relevant follow-up time points, which were used herein to judge the certainty of evidence for these end points, are not affected by these limitations.
Less than 50% of patients in each group remained available for HRQoL assessments at week 25, and less than 30% of patients remained in each group at week 53. As such, there is a risk of attrition bias, although the direction of this bias cannot be predicted. In addition, the open-label study design likely introduced a risk of bias in the measurement of HRQoL using FACT-Hep, a patient-reported subjective outcome measure.
External Validity
Overall, the clinical experts consulted for this review suggested that the trial population was a fair representation of patients with unresectable or advanced HCC seen in practice in Canada. Although some patient subgroups were slightly underrepresented in the study, such as patients with borderline Child-Pugh B7 status (e.g., on the borderline for albumin) and patients with hepatitis C viral etiology, the clinical experts consulted for this review suggested that the study results are generalizable to these groups in practice, given that various aspects of a study (e.g., study design and enrolment) evolve over time.
With respect to continuing treatment with nivolumab plus ipilimumab beyond initial investigator-assessed disease progression, the clinical experts consulted for this review agreed that the criteria used in the CheckMate 9DW trial are standard across immuno-oncology (IO) studies and suggested that they are relevant to clinical practice. The criteria for treatment beyond progression used in the study are summarized in Appendix 3 in the Supplemental Material.
The Clinical Study Report indicated that, at the time of initiating the study, the standard first-line therapy for advanced HCC was either sorafenib or lenvatinib. The sponsor clarified that no first-line immunotherapy was available in Canada at the time of patient accrual, which occurred between January 6, 2020, and November 8, 2021. Although the clinical experts consulted for this review indicated that the use of TKIs in the study reflected clinical practice in Canada at that time, with greater use of lenvatinib than sorafenib, TKIs are not the most relevant comparators for this review. The Provisional Funding Algorithm for unresectable HCC dated January 24, 2024, indicated that lenvatinib and sorafenib are alternative first-line treatment options if immunotherapy is unavailable or not indicated for the patient. As such, there remains an evidence gap comparing nivolumab plus ipilimumab with IO regimens for unresectable or advanced HCC in the first-line setting.
The outcomes assessed in this review were selected to align with the treatment goals for advanced or unresectable HCC and with outcomes considered important to patients.
Results
The key efficacy and harms results, along with findings from the GRADE assessment, are presented in this section. Detailed efficacy and harms results, including sensitivity and supportive analyses of the primary end point of OS, are presented in Appendix 4 in the Supplemental Material.
Efficacy
Overall survival:
At the time of the clinical cut-off date, 57.9% of patients (194 of 335 patients) in the nivolumab plus ipilimumab group and 68.5% of patients (228 of 333 patients) in the sorafenib or lenvatinib group had experienced an event (death).
The KM curves crossed at 12 months and remained separated thereafter in favour of nivolumab plus ipilimumab.
The absolute difference in the KM estimate of the probability of OS for nivolumab plus ipilimumab compared with sorafenib or lenvatinib was █████ ████ ███ ██████ ██ ██████ ██ █ ██████, was –1.3% (95% confidence interval [CI], –8.4% to 5.8%) at 12 months, and was 10.2% (95% CI, 2.5% to 18.0%) at 24 months.
The median OS was 23.66 months (95% CI, 18.83 months to 29.44 months) in the nivolumab plus ipilimumab group and 20.63 months (95% CI, 17.48 months to 22.54 months) in the sorafenib or lenvatinib group. Nivolumab plus ipilimumab was favoured over sorafenib or lenvatinib (HR = 0.79; 97.43% CI, 0.64 to 0.99; P value = 0.0180); the boundary for statistical significance was 0.0257.
Three prespecified supportive analyses were performed to assess the treatment effect of nivolumab plus ipilimumab versus sorafenib or lenvatinib when taking delayed effect into consideration, including a piecewise HR model. The piecewise HR was 1.65 (95% CI, 1.12 to 2.43) at 6 months or earlier and 0.61 (95% CI, 0.48 to 0.77) after 6 months.
Based on the results from the exploratory prespecified subgroup analyses of OS, the point estimates for the HR generally favoured nivolumab plus ipilimumab over sorafenib or lenvatinib. Among patients with hepatitis C viral etiology per central lab (past and resolved hepatitis C viral infections were categorized as uninfected for stratification purposes), the point estimate favoured sorafenib or lenvatinib.
In a post hoc analysis, among patients with hepatitis C viral etiology per case report form (with past and resolved hepatitis C viral infections were categorized as hepatitis C virus for documentation of medical history), the point estimate for the HR favoured nivolumab plus ipilimumab.
Progression-free survival:
At the time of the clinical cut-off date, 65.4% of patients (219 of 335 patients) in the nivolumab plus ipilimumab group and 64.6% of patients (215 of 333 patients) in the sorafenib or lenvatinib group had experienced an event (disease progression or death).
The KM curves of PFS (primary definition) crossed between 9 and 12 months and remained separated in favour of nivolumab plus ipilimumab until 36 months, at which point fewer than 5 patients in each group remained at risk thereafter. Similarly, the KM curves for PFS (secondary definition) crossed at 12 months and remained separated in favour of nivolumab plus ipilimumab until almost 42 months, after which fewer than 5 patients in each group remained at risk thereafter.
The absolute difference in the KM estimate of the probability of PFS (primary definition) for nivolumab plus ipilimumab compared with sorafenib or lenvatinib was █████ ████ ███ ██████ ██ ██████ ██ █ ███████ ███ ████ ████ ███ █████ ██ ██████ ██ ██ ███████ ███ ███ █████ ████ ███ ████ ██ ██████ ██ ██ ██████.
The median PFS (primary definition) was 9.07 months (95% CI, 6.60 months to 10.51 months) in the nivolumab plus ipilimumab group and 9.20 months (95% CI, 7.89 months to 11.07 months) in the sorafenib or lenvatinib group. The HR for PFS (primary definition) was 0.87 (95% CI, 0.72 to 1.06) following treatment with nivolumab plus ipilimumab versus sorafenib or lenvatinib.
The median PFS (secondary definition) was 8.97 months (95% CI, 6.67 months to 10.51 months) in the nivolumab plus ipilimumab group and 9.33 months (95% CI, 8.31 months to 11.14 months) in the sorafenib or lenvatinib group. The HR for PFS (secondary definition) was 0.82 (95% CI, 0.69 to 0.98) following treatment with nivolumab plus ipilimumab versus sorafenib or lenvatinib.
Health-related quality of life:
At baseline, the mean FACT-Hep total score was 139.28 (standard deviation [SD] = 22.441) in the nivolumab plus ipilimumab group and 139.58 (SD = 22.053) in the sorafenib or lenvatinib group.
At week 13, FACT-Hep measurements were available for 53.4% of patients (179 of 335 patients) in the nivolumab plus ipilimumab group and 70.0% of patients (233 of 333 patients) in the sorafenib or lenvatinib group. The mean change from baseline in the FACT-Hep total score was –2.42 (SD = 21.878) in the nivolumab plus ipilimumab group and –8.25 (SD = 19.369) in the sorafenib or lenvatinib group.
At week 25, FACT-Hep measurements were available for 42.7% of patients (143 patients) in the nivolumab plus ipilimumab group and 49.2% of patients (164 patients) in the sorafenib or lenvatinib group. The mean change from baseline in the FACT-Hep total score was –0.99 (SD = 23.212) in the nivolumab plus ipilimumab group and –8.45 (SD = 17.757) in the sorafenib or lenvatinib group.
At week 53, FACT-Hep measurements were available for 27.8% of patients (93 patients) in the nivolumab plus ipilimumab group and 27.0% of patients (90 patients) in the sorafenib or lenvatinib group. The mean change from baseline in the FACT-Hep total score was 5.91 (SD = 20.015) in the nivolumab plus ipilimumab group and –6.52 (SD = 20.835) in the sorafenib or lenvatinib group.
Harms
Adverse Events
Events reported between first dose and 30 days after last dose of study therapy:
A total of 331 of 332 patients treated with a study drug (99.7%) in the nivolumab plus ipilimumab group and 320 of 325 patients treated with a study drug (98.5%) in the sorafenib or lenvatinib group had at least 1 any-grade AE.
The most common any-grade AEs (> 20%) reported in the nivolumab plus ipilimumab group were pruritus (114 patients; 34.3%), increased aspartate aminotransferase (98 patients; 29.5%), rash (81 patients; 24.4%), increased alanine aminotransferase (79 patients; 23.8%), and diarrhea (73 patients; 22.0%).
The most common any-grade AEs (> 20%) reported in the sorafenib or lenvatinib group were hypertension (145 patients; 44.6%), diarrhea (127 patients; 39.1%), palmar-plantar erythrodysesthesia syndrome (104 patients; 32.0%), decreased appetite (91 patients; 28.0%), hypothyroidism (88 patients; 27.1%), and proteinuria (71 patients; 21.8%).
A total of 230 patients (69.3%) in the nivolumab plus ipilimumab group and 203 patients (62.5%) in the sorafenib or lenvatinib group had at least 1 grade 3 or 4 AE.
The most common grade 3 or 4 AE (> 5%) reported in the nivolumab plus ipilimumab group was increased aspartate aminotransferase (25 patients; 7.5%).
The most common grade 3 or 4 AE (> 5%) reported in the sorafenib or lenvatinib group was hypertension (43 patients; 13.2%).
Serious AEs
Events reported between first dose and 30 days after last dose of study therapy:
A total of 176 patients (53.0%) in the nivolumab plus ipilimumab group and 143 patients (44.0%) in the sorafenib or lenvatinib group experienced at least 1 any-grade SAE.
A total of 154 patients (46.4%) in the nivolumab plus ipilimumab group and 115 patients (35.4%) in the sorafenib or lenvatinib group experienced at least 1 grade 3 or 4 SAE.
Malignant neoplasm progression was the most frequently reported any-grade SAE (24 patients; 7.2%) and grade 3 or 4 SAE (16 patients; 4.8%) in the nivolumab plus ipilimumab group.
Malignant neoplasm progression was also the most frequently reported any-grade SAE (32 patients; 9.8%) and grade 3 or 4 SAE (19 patients; 5.8%) in the sorafenib or lenvatinib group.
Events reported regardless of the dose date:
A total of 65.1% of patients in the nivolumab plus ipilimumab group and 55.7% of patients in the sorafenib or lenvatinib group had at least 1 any-grade SAE.
Withdrawal Due to AEs
Events reported between first dose and 30 days after last dose of study therapy:
A total of 88 patients (26.5%) in the nivolumab plus ipilimumab group and 75 patients (23.1%) in the sorafenib or lenvatinib group discontinued at least 1 study drug in the regimen due to any-grade AEs.
A total of 69 patients (20.8%) in the nivolumab plus ipilimumab group and 51 patients (15.7%) in the sorafenib or lenvatinib group discontinued at least 1 study drug in the regimen due to grade 3 or grade 4 AEs.
Malignant neoplasm progression was the most frequently reported any-grade AE and grade 3 or grade 4 AE leading to discontinuation in the nivolumab plus ipilimumab group (9 patients; 2.7% for each).
Malignant neoplasm progression was also the most frequently reported any-grade AE (20 patients; 6.2%) and grade 3 or 4 AE leading to discontinuation (14 patients; 4.3%) in the sorafenib or lenvatinib group.
Events reported regardless of the dose date:
A total of 35.5% of patients in the nivolumab plus ipilimumab group and 23.4% of patients in the sorafenib or lenvatinib group stopped at least 1 study drug in the regimen due to AEs.
Deaths
As of the clinical cut-off date:
A total of 192 patients (57.8%) in the nivolumab plus ipilimumab group and 224 patients (68.9%) in the sorafenib or lenvatinib group died.
A total of 19.7% of patients (66 of 335 patients who were randomized) in the nivolumab plus ipilimumab group and █████ of patients (██ of 333 patients who were randomized) in the sorafenib or lenvatinib group died within ████ months after randomization.23
The primary reasons for death reported in the nivolumab plus ipilimumab group included disease progression (139 patients; 41.9%), study drug toxicity per investigator assessment (12 patients; 3.6%), other causes unrelated to the study drug per investigator assessment (35 patients; 10.5%), and unknown causes (6 patients; 1.8%).
Among the 12 patients who died primarily due to study drug toxicity (per investigator assessment) in the nivolumab plus ipilimumab group, reported causes included immune-mediated hepatitis (4 patients; 1.2%), hepatic failure (3 patients; 0.9%), hepatic insufficiency, decompensated cirrhosis, diarrhea-colitis, autoimmune hemolytic anemia or hepatic failure, and dysautonomia (1 patient each; 0.3% each).
A total of 11 patients (3.3% of patients) in the nivolumab plus ipilimumab group died primarily due to study drug toxicity (per investigator assessment) within 5.84 months after randomization.23
The primary reasons for death reported in the sorafenib or lenvatinib group included disease progression (202 patients; 62.2%), study drug toxicity per investigator assessment (3 patients; 0.9%), other reasons unrelated to the study drug per investigator assessment (14 patients; 4.3%), and unknown causes (5 patients; 1.5%).
Among the 3 patients who died primarily due to study drug toxicity (per investigator assessment) in the sorafenib or lenvatinib group, reported causes included hepatorenal syndrome, ischemic stroke, and acute kidney injury (1 patient each; 0.3%).
One patient (0.3% of patients) in the sorafenib or lenvatinib group died primarily due to study drug toxicity (per investigator assessment) within 5.84 months after randomization.23
Immune-Mediated AEs
Events reported between the first dose and 100 days after the last dose of study therapy:
The most common immune-mediated any-grade AEs (> 10%) reported in the nivolumab plus ipilimumab group were hepatitis (63 patients who were treated with a study drug; 19.0%), rash (51 patients; 15.4%), and hypothyroidism (46 patients; 13.9%).
The most common immune-mediated grade 3 or 4 AEs (> 10%) reported in the nivolumab plus ipilimumab group was hepatitis (51 patients; 15.4%).
No immune-mediated AEs (any-grade or grade 3 or 4) were reported in greater than 10% of patients in the sorafenib or lenvatinib group.
Summary of Findings and Certainty of the Evidence
The literature-based MID estimate was used as the threshold for FACT-Hep (MID: 8 to 9 points). Details are provided in the summary of outcome measures in Appendix 3 in the Supplemental Material. In the absence of literature-based MID estimates, thresholds suggested by the clinical experts consulted for this review were applied for OS (threshold: 5%) and PFS (threshold: 5%). For outcomes without established thresholds, certainty in the presence of a nonnull effect was assessed for SAEs and WDAEs.
Table 4: Summary of Findings for Nivolumab Plus Ipilimumab vs. Sorafenib or Lenvatinib in Patients With Advanced or Unresectable HCC
Outcome and follow-up | Patients (study), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Sorafenib or lenvatinib | Nivolumab plus ipilimumab (95% CI) | Difference (95% CI) | |||||
OS | |||||||
Kaplan-Meier estimates of the probability that a patient is alive | |||||||
Probability of OS at 6 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | ███ ███ | ███ ███ | ███ ███ | Moderatea (Serious risk of imprecision) | Nivolumab plus ipilimumab likely results in a clinically important decrease in the probability of OS at 6 months compared with sorafenib or lenvatinib. |
Probability of OS at 12 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | 697 per 1,000 | 684 per 1,000 (631 to 732 per 1,000) | 13 less per 1,000 (84 less to 58 more per 1,000) | Lowb (Very serious risk of imprecision) | Nivolumab plus ipilimumab may result in little to no clinically important difference in the probability of OS at 12 months compared with sorafenib or lenvatinib. |
Probability of OS at 24 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | 392 per 1,000 | 494 per 1,000 (438 to 548 per 1,000) | 102 more per 1,000 (25 more to 180 more per 1,000) | Moderatea (Serious risk of imprecision) | Nivolumab plus ipilimumab likely results in a clinically important increase in the probability of OS at 24 months compared with sorafenib or lenvatinib. |
PFS | |||||||
Kaplan-Meier estimates of the probability that a patient has not progressed and is alive | |||||||
Probability of PFS at 6 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | ███ ███ | ███ ███ | ███ ███ | Moderatea (Serious risk of imprecision) | Nivolumab plus ipilimumab likely results in a clinically important decrease in the probability of PFS at 6 months compared with sorafenib or lenvatinib. |
Probability of PFS at 12 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | ███ ███ | ███ ███ | ███ ███ | Lowb (Very serious risk of imprecision) | Nivolumab plus ipilimumab may result in little to no clinically important difference in the probability of PFS at 12 months compared with sorafenib or lenvatinib. |
Probability of PFS at 24 months Median follow-up: 35.2 months | 668 (1 RCT) | NR | ███ ███ | ███ ███ | ███ ███ | High | Nivolumab plus ipilimumab results in a clinically important increase in the probability of PFS at 24 months compared with sorafenib or lenvatinib. |
HRQoL | |||||||
FACT-Hep questionnaire assesses the effects of HCC and its treatment on HRQoL. Each item is rated on a 5-point scale ranging from 0 (not at all) to 4 (very much); higher scores indicate better HRQoL. | |||||||
Mean change from baseline in the FACT-Hep total score at 13, 25, and 53 weeks Median follow-up: 35.2 months | 668 (1 RCT) | NR | At 13 weeks: Nivolumab plus ipilimumab: –2.42 Sorafenib or lenvatinib: –8.25 Between-group difference: NR At 25 weeks: Nivolumab plus ipilimumab: –0.99 Sorafenib or lenvatinib: –8.45 Between-group difference: NR At 53 weeks: Nivolumab plus ipilimumab: 5.91 Sorafenib or lenvatinib: –6.52 Between-group difference: NR | Lowc (Very serious risk of study limitation) | Nivolumab plus ipilimumab may improve HRQoL compared with sorafenib or lenvatinib, but the clinical importance of the effect is uncertain. | ||
Harmsd | |||||||
Patients with any-grade SAEs at clinical cut-off date Median follow-up: 35.2 months | 668 (1 RCT) | 1.20 (1.03 to 1.41) | 440 per 1,000 | 530 per 1,000 (475 to 585 per 1,000) | 90 more per 1,000 (14 more to 166 more per 1,000) | Highe | Nivolumab plus ipilimumab results in an increase in the proportion of patients with any-grade SAEs compared with sorafenib or lenvatinib. The clinical importance of the increase is uncertain. |
Patients with WDAEs (any-grade) at clinical cut-off date Median follow-up: 35.2 months | 668 (1 RCT) | 1.15 (0.88 to 1.50) | 231 per 1,000 | 265 per 1,000 (218 to 316 per 1,000) | 34 more per 1,000 (32 less to 100 more per 1,000) | Moderatef (Serious risk of imprecision) | Nivolumab plus ipilimumab likely results in an increase in the proportion of patients with WDAEs (any grade) compared with sorafenib or lenvatinib. The clinical importance of the increase is uncertain. |
CI = confidence interval; FACT-Hep = Functional Assessment of Cancer Therapy–Hepatobiliary; HCC = hepatocellular carcinoma; HRQoL = health-related quality of life; NR = not reported; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse events.
Notes: Clinical cut-off date: January 31, 2024.
Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. The 95% CI for the between-group difference includes the possibility of little to no clinically important difference based on a threshold of 5% (50 per 1,000), as informed by the clinical experts consulted for this review.
bRated down 2 levels for very serious imprecision. The 95% CI for the between-group difference includes the possibility of clinically important benefit and harm based on a threshold of 5% (50 per 1,000), as informed by the clinical experts consulted for this review.
cRated down 2 levels for very serious study limitations. Less than 50% of patients remained in each group to provide assessments at week 25 and less than 30% of patients remained in each group at week 53. The open-label study design likely introduced a risk of bias in the measurement of HRQoL using patient-reported outcomes such as FACT-Hep. There is no between-group estimate to judge the clinical significance versus the target threshold; hence, the clinical importance of the estimated effect is uncertain.
dIncludes events reported between first dose and 30 days after last dose of study therapy.
eThe null was used as the threshold to inform the target of the certainty rating and the precision of the effect; hence, the clinical importance of the estimated effect is uncertain.
fRated down 1 level for serious imprecision. The 95% CI for the between-group difference includes the possibility of clinically important benefit and harm. The null was used as the threshold to inform the target of the certainty rating and the precision of the effect; hence, the clinical importance of the estimated effect is uncertain.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence,14 the Clinical Study Report for the CheckMate 9DW study,15 and the sponsor response to the request for additional information by Canada’s Drug Agency regarding the Opdivo and Yervoy review on June 19, 2025; July 16, 2025; and July 28, 2025.24-26
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor for this review.
Indirect Evidence
Direct comparative evidence between nivolumab plus ipilimumab and sorafenib or lenvatinib was available in the CheckMate 9DW study; however, an evidence gap was identified for comparisons between nivolumab plus ipilimumab and IO regimens for unresectable or advanced HCC. An ITC was used to address this evidence gap.
Description of Indirect Comparisons
One ITC was submitted by the sponsor comparing nivolumab plus ipilimumab with durvalumab plus tremelimumab and with atezolizumab plus bevacizumab with respect to efficacy and safety outcomes in patients with unresectable or advanced HCC who had not received prior systemic therapy.
The primary objective of the ITC was to compare the efficacy of nivolumab plus ipilimumab versus durvalumab plus tremelimumab and versus atezolizumab plus bevacizumab, separately, to inform health economic modelling. This objective was addressed by performing standard and piecewise Bucher ITCs and anchored simulated treatment comparisons (STCs) for the primary outcomes of interest (PFS, OS, and TTD).
The secondary objective of the ITC was to assess the sensitivity of the primary analysis results to methodological assumptions. This was evaluated by performing unanchored matching-adjusted indirect comparisons for the primary outcomes of interest (PFS, OS, and objective response rate).
Study Selection and Review Methods
The ITC was informed by a systematic literature review (SLR) of evidence on first-line systemic therapies in adults with unresectable or advanced HCC. Relevant interventions included sorafenib (400 mg twice daily), lenvatinib (≥ 60 kg: 12 mg once daily; < 60 kg: 8 mg once daily), atezolizumab plus bevacizumab (1,200 mg IV and 15 mg/kg IV every 3 weeks), tremelimumab plus durvalumab (300 mg IV single dose and 1,500 mg IV every 4 weeks), and nivolumab plus ipilimumab (per recommended dosing regimen). There were no restrictions on the comparator for inclusion. Relevant outcomes included OS, PFS, objective response rate, duration of response, time to symptom deterioration, AEs, SAEs, treatment discontinuations, HRQoL scores, and utility values. Eligible study designs included RCTs, SLRs, and (network) meta-analyses. The bibliographies of SLRs and (network) meta-analyses of relevant studies were handsearched for additional primary studies.
The database search was conducted in August 2024, using MEDLINE, Embase, Cochrane Database of Systematic Reviews, Cochrane Central Register of Controlled Studies, and Database of Abstracts of Reviews of Effects. Additionally, WHO International Clinical Studies Registry Platform, Health Canada’s Clinical Study Database, ClinicalTrials.gov, and key conference proceedings from the past 2 years were handsearched in August 2024.
Two independent reviewers screened articles, with discrepancies resolved by consensus. Due to the high volume of literature identified at the full-text screening stage, only studies with a sample size of at least 350 patients were prioritized for extraction. One reviewer performed data extraction for each included study, with verification by a second independent reviewer. One reviewer completed the quality assessment of extracted studies using the Cochrane risk of bias 2 tool for RCTs,27 with verification by a second independent reviewer. Studies with critical flaws were excluded, and studies with significant biases were excluded or addressed through sensitivity analyses.
ITC Analysis Methods
The authors of the ITC conducted a feasibility assessment to inform the ITC methodology by first identifying treatment effect modifiers and prognostic factors, followed by evaluating heterogeneity across the included studies. The authors also assessed the assumption of proportional hazards for each time-to-event outcome of interest across the included studies.
The results of the feasibility assessment highlighted 3 key challenges that influenced the choice of ITC methodology. Heterogeneity in the study populations — █████████ █████████ ██ ████ ███ █████████████ █████████ ███ ████████████ ██████ — was addressed using STC population adjustment methodology to conduct the primary pairwise analyses. Violations of the proportional hazards assumption were addressed by using piecewise STC models as an alternative to standard proportional hazards models. Differences in the comparator groups were addressed by performing sensitivity analyses to assess how varying comparator groups could impact the results.
Although additional analyses were conducted, only the anchored STCs were used to inform the accompanying pharmacoeconomic submission and are therefore the focus of this review. Details of the sponsor’s anchored STC analysis methods are summarized in the Indirect Comparison Analysis Methods table in Appendix 6 in the Supplemental Material.
Summary of Included Studies
Overview
The SLR search yielded 4,986 records. After screening, 142 publications representing 55 unique studies were included in the SLR, of which 51 were excluded from the ITC because the studies did not report on relevant comparators. A total of 4 unique studies were included in the ITC: CheckMate 9DW (N = 668),28 IMbrave150 (N = 501),29 HIMALAYA (N = 1,171),28 and REFLECT (N = 954).17
Study Characteristics
CheckMate 9DW, HIMALAYA, IMbrave150, and REFLECT are phase III, global, open-label randomized studies. The CheckMate 9DW study is the only study with a maximum treatment duration, with nivolumab plus ipilimumab administered for up to 2 years. Criteria for treatment beyond progression were established in the CheckMate 9DW, HIMALAYA, and IMbrave150 studies, but were not reported in the REFLECT study.
Atezolizumab plus bevacizumab and durvalumab plus tremelimumab were compared with sorafenib using identical dosing schedules in the IMbrave150 and HIMALAYA studies, respectively. Hence, the REFLECT study, which evaluated lenvatinib versus sorafenib, was included in the ITC to facilitate anchored comparisons between the comparator studies and the CheckMate 9DW study, which compared nivolumab plus ipilimumab with investigator’s choice of sorafenib or lenvatinib. The dosing for the comparators was according to the approved dosing regimen for sorafenib (400 mg oral twice daily) and lenvatinib (< 60 kg: 8 mg oral once daily; ≥ 60 kg: 12 mg oral daily).
In all included studies, OS was defined as the time from the date of randomization until death by any cause; TTD was defined as the time from first dose of study therapy until treatment discontinuation, although this outcome was not reported in the REFLECT study. PFS was defined as the time from randomization to the first documented disease progression or death due to any cause, whichever occurred first; however, in all included studies, there was some variation in the end point definition (independent versus investigator assessment). Additionally, different tumour measurement criteria were used, with the CheckMate 9DW, HIMALAYA, and IMbrave150 studies using RECIST 1.1, while the REFLECT study used modified RECIST 1.1 criteria.
The median OS and PFS were longer in the sorafenib or lenvatinib group from the CheckMate 9DW study compared with the sorafenib groups in the IMbrave150 and HIMALAYA studies and with either group in the REFLECT study. Additionally, there was some variation in the median follow-up times across the comparator groups, ranging from 10.4 months in the IMbrave150 study to 35.2 months in the CheckMate 9DW study. Follow-up duration was comparable between the CheckMate 9DW and HIMALAYA studies, slightly shorter in the REFLECT study, and notably shorter in the IMbrave150 study.
Using the Cochrane risk of bias 2 tool for RCTs, the authors concluded that there were some concerns for the overall risk of bias in the CheckMate 9DW, HIMALAYA, and IMbrave150 studies, while there was a low risk for bias in the REFLECT study.
Patient Characteristics
All patients across the included studies had unresectable or advanced HCC and had not received prior systemic therapy. Across the included studies, patients had a Child-Pugh score of 5 or 6 and met screening laboratory criteria indicating adequate hematologic and organ function.
Patients with HCC occupying 50% or more of the liver and with clear bile duct invasion were excluded from the CheckMate 9DW and REFLECT trials, while such exclusions did not apply to the HIMALAYA and IMbrave150 trials. Additionally, patients with portal vein invasion at the main portal branch were included in the IMbrave150 trial, whereas they were excluded from the other 3 trials.
Across the included studies, patients were aged a median of 62 years to 66 years, and at least 80% of patients in each treatment group were male. There was some variation across studies in population distributions of potential treatment effect modifiers. The REFLECT study had a higher proportion of patients from Asia than the other studies. The IMbrave150 study had a higher proportion of patients with macrovascular invasion and extrahepatic spread compared with the CheckMate 9DW and HIMALAYA studies, in which these proportions were comparable. The REFLECT study had a higher proportion of patients with extrahepatic spread compared with the other studies; however, macroscopic portal vein invasion was not reported in the REFLECT study. The IMbrave150 and REFLECT studies had higher proportions of patients with hepatitis B virus–related HCC compared with the CheckMate 9DW and HIMALAYA studies. The CheckMate 9DW study had a higher proportion of patients with an ECOG PS score of 0 than the other studies. In addition, the CheckMate 9DW and REFLECT studies had higher proportions of patients with a Barcelona Clinic Liver Cancer staging of 0, A, or B, and a Child-Pugh score of 5 or 6 (Child-Pugh class A in the REFLECT study) compared with the HIMALAYA and IMbrave150 studies.
Critical Appraisal of ITCs
Studies included in the STC were selected from those identified in the SLR, which was conducted using standard methods with defined research questions specified a priori. Multiple databases were searched in August 2024. The interventions and comparators were refined for the inclusion of studies in the STC and were consistent with the objective and reflective of practice in the first-line treatment setting. The methods used for study selection and data extraction were sufficient to limit the risk of error and bias in these procedures.
The authors concluded there were some concerns for the overall risk of bias in the CheckMate 9DW, HIMALAYA, and IMbrave150 studies; however, insufficient information was provided to predict the direction of any potential bias. Although the risk of bias of included studies was assessed using a relevant tool, the assessment was conducted at the study level rather than at the level of the reported result (i.e., for each outcome). This method ignores the fact that risk of bias can vary depending on the effect estimate being evaluated, particularly for domains such as performance bias, detection bias, attrition bias, and reporting bias. As such, the risk of bias reported by the sponsor for each study may not universally apply to OS and PFS.
STCs are a population adjustment methodology used in ITC to estimate treatment effects when individual patient data are available and when there are differences in treatment effect modifiers (and prognostic factors in unanchored analyses) between 2 studies.30 Importantly, because baseline covariates included as predictors in regression models are adjusted for, STCs estimate a conditional treatment effect (i.e., a relative effect at the patient level for specific covariate values) instead of a marginal treatment effect (i.e., a relative effect at the population level), with the latter being relevant for decision-making in health technology assessment.30 Due to the noncollapsibility of certain effect measures, such as (log) HRs, conditional and marginal estimands may differ. This discrepancy arises because adjusting for covariates in nonlinear models can shift the estimated treatment effect away from the null.30,31 As a result, combining a conditional estimate from an STC model with an unadjusted (marginal) estimate from an aggregate comparator study may introduce bias.31 Similarly, Remiro-Azócar et al.30 caution that if a marginal estimand is targeted for decision-making, using a conditional estimate may misrepresent the true relative treatment effect in the target population.
Although an anchored Bucher (unadjusted) ITC, which produces marginal estimands, was performed, the results are not presented in this report. The main limitation of this approach is that the ITC estimates did not adjust for between-study differences in baseline patient characteristics. Because there were notable baseline differences between the included studies, as described in the feasibility assessment, definitive conclusions based on the Bucher results are not advised. As such, it could not be determined how well the conditional estimates from the STC applies to the marginal estimands.
An underlying assumption of anchored STCs is the conditional constancy of relative effects, whereby relative treatment effects are assumed to be constant between-study populations at any given level of effect modifiers.32 To reduce bias, all unbalanced effect modifiers should be adjusted for using regression-based population adjustment methods.32 While prognostic factors are typically assumed to be similarly distributed across trials in anchored STCs, this assumption is weakened when the common comparator differs across trials, as in this case (sorafenib or lenvatinib versus sorafenib only). In such scenarios, residual imbalances in prognostic factors may affect both the validity and precision of the estimated treatment effects, and adjustment for additional prognostic variables and relevant effect modifiers could improve precision. If important prognostic covariates are missed or effect modifiers are incorrectly omitted, the STC may yield imprecise or biased estimates.32,33 Although albumin-bilirubin score, number of target lesions, and target lesion size were identified as potential prognostic factors in the STC technical report, these variables were not adjusted for in the analyses. The clinical experts consulted for this review did not express major concern about their exclusion. PD-L1 tumour proportion score was identified as a treatment effect modifier in the ITC feasibility assessment but was not adjusted for due to insufficient data and uncertainty regarding assay type. The sponsor clarified that, because the STC was anchored, prognostic factors were assumed to be balanced and therefore not considered necessary for adjustment. However, given the heterogeneity in control groups and trial designs, this assumption may not hold, and the potential for residual bias reduces certainty in the results.
Importantly, STCs do not account for differences in study design.33 Heterogeneity in the end point definition of PFS across studies was addressed by using the independent assessment for comparisons between the CheckMate 9DW and IMbrave150 studies, while investigator-assessed PFS was used for comparisons between the CheckMate 9DW and HIMALAYA studies. The use of investigator-assessed PFS introduces a potential risk in the outcome measurement bias. Additionally, there are notable differences in the follow-up times across the comparator groups. The implications of including studies with differential follow-up durations in the STC are unknown but likely introduced bias in the results as a source of methodological heterogeneity.
There was violation of the proportional hazards assumption across studies. As such, the ratio of the hazard functions was not constant over time, and reliance on the HR alone to inform the effects of nivolumab plus ipilimumab compared with the immunotherapies on OS and PFS may be misleading. To address this issue, the authors fitted piecewise proportional hazards models as the STC regression model. The follow-up time was divided into multiple intervals, with separate Cox proportional hazards models fitted to each interval to allow for deviation from the proportional hazards assumption. Hence, interpretation of the results should be specific to the distinct time intervals and not the overall treatment effect.
The selection of cut points for the piecewise analyses was based on visual assessments of log cumulative hazard plots, complementary log-log survival plots, Schoenfeld residual plots, and the results of the Schoenfeld global test. Although the Schoenfeld residual plots suggested the inclusion of later cut points at 15 months to 18 months for some studies, the authors indicated there were insufficient data at the later time points to justify additional cut points. As such, a single cut point at 6 months was selected for the analysis of OS and PFS. The sponsor clarified that introducing additional cut points would increase the risk of overfitting and bias, particularly at later time points where sample sizes are limited and frailty effects may differ between groups, as noted by Bartlett et al.34 Additionally, the sponsor indicated that selection of a single cut point was also supported by input from the sponsor's clinical experts regarding the delayed effect of nivolumab plus ipilimumab compared with sorafenib or lenvatinib. However, based on visual assessment of the plots and test results by the review team, there appears to still be a violation of the proportional hazards assumption for OS and PFS within some of the segments using the 6-month cut point; this would introduce bias in the estimated treatment effects, limiting confidence in the results. Of note, the KM curves for OS in the CheckMate 9DW study crossed at 12 months. This limitation is also applicable to the STC-derived risk differences, but to a lesser extent, because the STC-derived HRs were applied only to the survival estimates for nivolumab plus ipilimumab.
The STC-derived differences in the PFS probabilities between durvalumab plus tremelimumab versus nivolumab plus ipilimumab appear generally consistent with the results of the piecewise survival analyses at 6 months and beyond. However, the corresponding results appear to be less consistent for comparisons between durvalumab plus tremelimumab versus nivolumab plus ipilimumab for OS, and for atezolizumab plus bevacizumab versus nivolumab plus ipilimumab for OS and PFS, at 6 months and beyond. These inconsistencies, combined with the absence of sensitivity analyses using alternative modelling approaches, introduce additional uncertainty regarding the robustness and reliability of the STC-derived estimates for certain treatment comparisons.
Bartlett et al.34 cautioned against interpreting changes in period-specific HRs as changes in treatment effect, as period-specific HRs are subject to selection bias. Specifically, period-specific HRs are estimated among patients who have survived to the start of the period of interest. Bartlett et al.34 suggested that despite randomization, there will generally be systematic differences in the distribution of “frailty factors”34 between survivors in each group at later time points. Hence, Bartlett et al.34 advised the use of improved interpretable estimands, such as comparisons of survival probabilities at specified time points, median survival, or differences or ratios of restricted mean survival time.
A population-adjusted anchored ITC may be considered when there is a common comparator that connects the evidence.32 In the primary analysis, pairwise anchoring was performed to sorafenib or lenvatinib in the CheckMate 9DW study, under the assumption that lenvatinib and sorafenib in the CheckMate 9DW study are equivalent to sorafenib in the HIMALAYA and IMbrave150 studies. This assumption was based on the REFLECT study, which concluded that “lenvatinib was non-inferior to sorafenib in OS for patients with untreated advanced hepatocellular carcinoma.”17 However, a reanalysis using “a covariate adjustment of REFLECT suggested that the original noninferiority trial likely underestimated the true effect of lenvatinib on OS due to an imbalance in baseline prognostic covariates and the greater use of post-treatment therapies in the sorafenib group.”18 Given this discrepancy, the authors performed 2 sensitivity analyses to assess how varying comparator groups could impact the results. However, these sensitivity analyses were limited by small sample size, inclusion of a subset of patients in the analysis (introducing a risk of bias due to the likely loss of prognostic balance between groups), and incorporation of HRs from the REFLECT study without population adjustment, which would introduce bias that cannot be predicted. These limitations contribute to uncertainty in the validity of the comparator assumptions, and by extension, the reliability of the treatment effect estimates derived from the anchored STC.
Current STC population-adjusted methodology is limited to producing estimates applicable to the comparator populations, assuming that the 2 different comparator populations (HIMALAYA and IMbrave150 study populations) represent the target population relevant to the Clinical Review.32,33 Although all studies included patients with advanced or unresectable HCC who had not received prior systemic therapy, it is likely that there are differences between each comparator and the target populations, raising concerns regarding the generalizability of the STC results.33 For example, the IMbrave150 study included a higher proportion of patients with macrovascular invasion compared with the CheckMate 9DW and HIMALAYA studies, in which the proportions were comparable. As a result, the IMbrave150 study population included patients with a relatively poorer prognosis,35 a point with which the clinical experts consulted for this review agreed.
Efficacy Results of ITC
Key results from the STCs are presented in Table 5, Table 6, and Table 7. Results from the sensitivity analyses are presented in Appendix 6 in the Supplemental Material.
TTD is an input in the sponsor’s pharmacoeconomic model. A summary of STC results for TTD is presented in Appendix 6 in the Supplemental Material.
Table 5: Summary of Simulated Treatment Comparison Results for PFS and OS (Primary Analysis)
Comparator | Simulated treatment comparison | ||
|---|---|---|---|
NIV + IPI vs. comparator, HR (95% CI)a | |||
Overall | 0 to 6 months | ≥ 6 months | |
OS | |||
DUR + TRE | ████████ | ████████ | ████████ |
ATE + BEV | ████████ | ████████ | ████████ |
PFSb | |||
DUR + TRE | ████████ | ████████ | ████████ |
ATE + BEV | ████████ | ████████ | ████████ |
ATE = atezolizumab; BEV = bevacizumab; CI = confidence interval; DUR = durvalumab; HR = hazard ratio; IPI = ipilimumab; NIV = nivolumab; OS = overall survival; PFS = progression-free survival; TRE = tremelimumab; vs. = versus.
Note: Results where the 95% CI excludes the null are indicated in bold text (redacted).
aThe primary analysis is a pairwise anchored analysis using the sorafenib or lenvatinib comparator group in the CheckMate 9DW study. This analysis was anchored on the sorafenib group in the HIMALAYA and IMbrave150 studies and in the lenvatinib and sorafenib group in the CheckMate 9DW study. Lenvatinib and sorafenib in the CheckMate 9DW study was assumed to be equivalent to sorafenib in the HIMALAYA and IMbrave150 studies for the purpose of the analysis to facilitate the indirect comparison of NIV plus IPI versus DUR plus TRE and ATE plus BEV.
bTo address the heterogeneity in the definition of PFS across studies, an independent assessment of PFS was used for comparisons between the CheckMate 9DW and the IMbrave150 studies, while investigator-assessed PFS was used for comparisons between the CheckMate 9DW study and the HIMALAYA study.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence14 and the sponsor’s indirect treatment comparison report.35
Efficacy
Overall Survival
STC-derived HRs:
At 0 to 6 months, durvalumab plus tremelimumab and atezolizumab plus bevacizumab were favoured over nivolumab plus ipilimumab.
At 6 months and beyond, nivolumab plus ipilimumab was favoured over durvalumab plus tremelimumab and atezolizumab plus bevacizumab; however, this is associated with some uncertainty due to imprecision (i.e., the 95% CIs included the null, suggesting the potential for little to no difference).
STC-derived risk differences:
At 12 months, nivolumab plus ipilimumab was favoured over durvalumab plus tremelimumab.
At 6 months and 24 months, no treatment was favoured between durvalumab plus tremelimumab versus nivolumab plus ipilimumab.
At 6 months, 12 months, and 24 months, no treatment was favoured between atezolizumab plus bevacizumab versus nivolumab plus ipilimumab.
Table 6: Summary of ITC Results for OS Probability and Risk Difference
Treatment | Median OS (95% CI) | KM-estimated OS probability (95% CI)a | STC-derived risk differenceb | ||||
|---|---|---|---|---|---|---|---|
Comparator vs. NIV + IPI (95% CI) | |||||||
6 months | 12 months | 24 months | 6 months | 12 months | 24 months | ||
NIV + IPI | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
DUR + TRE | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
ATE + BEVc | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
ATE = atezolizumab; BEV = bevacizumab; CI = confidence interval; DUR = durvalumab; HR = hazard ratio; IPD = individual patient data; IPI = ipilimumab; ITC = indirect treatment comparison; KM = Kaplan-Meier; NIV = nivolumab; OS = overall survival; STC = simulated treatment comparison; TRE = tremelimumab; vs. = versus.
Note: Results where the 95% CI excludes the null are indicated in bold text (redacted).
aKM survival curves were fitted to the IPD from the CheckMate 9DW study and the pseudo-IPD generated from published KM curves from the HIMALAYA and the IMbrave150 studies. Median OS was calculated at the point at which the OS probability crossed 0.5 and OS probability was calculated by taking the KM estimate at the relevant time point.
bTo produce risk differences, STC-derived HRs were applied to the survival estimates from the KM data for NIV plus IPI and the difference between the survival probabilities at the specified time points were taken.
cData were only reported for up to 30 months for ATE plus BEV.
Sources: Sponsor response to the request for additional information by Canada's Drug Agency regarding the Opdivo and Yervoy review on June 19, 2025, and July 16, 2025.25,26
Progression-Free Survival
STC-derived HRs:
At 0 months to 6 months, no treatment was favoured between nivolumab plus ipilimumab versus durvalumab plus tremelimumab, while atezolizumab plus bevacizumab was favoured over nivolumab plus ipilimumab.
At 6 months and beyond, nivolumab plus ipilimumab was favoured over durvalumab plus tremelimumab and atezolizumab plus bevacizumab.
STC-derived risk differences:
At 6 months, 12 months, and 24 months, nivolumab plus ipilimumab was favoured over durvalumab plus tremelimumab.
At 12 months, nivolumab plus ipilimumab was favoured over atezolizumab plus bevacizumab.
At 6 months and 24 months, no treatment was favoured between atezolizumab plus bevacizumab versus nivolumab plus ipilimumab.
Harms
Harms were not assessed in the STC analyses.
Table 7: Summary of ITC Results for PFS Probability and Risk Difference
Treatment | Median PFS (95% CI) | KM-estimated PFS probability (95% CI)a | STC-derived risk differenceb | ||||
|---|---|---|---|---|---|---|---|
Comparator vs. NIV + IPI (95% CI) | |||||||
6 months | 12 months | 24 months | 6 months | 12 months | 24 months | ||
Investigator-assessed PFS | |||||||
NIV + IPI | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
DUR + TRE | ███ | ███ | ███ | ███ | |||
BICR-assessed PFS | |||||||
NIV + IPI | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
ATE + BEV | ███ | ███ | ███ | ███ | |||
ATE = atezolizumab; BEV = bevacizumab; BICR = blinded independent central review; CI = confidence interval; DUR = durvalumab; HR = hazard ratio; IPD = individual patient data; IPI = ipilimumab; ITC = indirect treatment comparison; KM = Kaplan-Meier; NIV = nivolumab; PFS = progression-free survival; STC = simulated treatment comparison; TRE = tremelimumab; vs. = versus.
Note: Results where the 95% CI excludes the null are indicated in bold text (redacted).
aKM survival curves were fitted to the IPD from the CheckMate 9DW study and the pseudo-IPD generated from published KM curves from the HIMALAYA and the IMbrave150 studies. Median PFS was calculated at the point at which the PFS probability crossed 0.5 and PFS probability was calculated by taking the KM estimate at the relevant time point.
bTo produce risk differences, STC-derived HRs were applied to the survival estimates from the KM data for NIV plus IPI and the difference between the survival probabilities at the specified time points were taken.
Sources: Sponsor response to the request for additional information by Canada's Drug Agency regarding the Opdivo and Yervoy review on June 19, 2025, and July 16, 2025.25,26
Studies Addressing Gaps in the Systematic Review Evidence
No other studies are included in this review. The sponsor did not identify a gap in the systematic review evidence.
Discussion
Efficacy
Patients and clinicians have identified a need for additional treatment options with improved survival, tolerability, and safety. These needs align with the treatment goals identified for first-line treatment in adults with unresectable or advanced HCC: improving survival, slowing disease progression, maintaining HRQoL, and minimizing AEs.
In the CheckMate 9DW study, there was an apparent early OS detriment with nivolumab plus ipilimumab compared with sorafenib or lenvatinib in adults with unresectable or advanced HCC who had not received any prior systemic therapy for the carcinoma. Notably, there were more deaths that occurred within 6 months after randomization in the nivolumab plus ipilimumab group compared with the sorafenib or lenvatinib group. At 6 months, the study suggested that there was likely a clinically important decrease in the probability of OS with nivolumab plus ipilimumab compared with sorafenib or lenvatinib. ███████████ ███████████ █████ █████████ ██ ███ ████████ ███ ███ ██████ ████████ ████████ ██████████████ ████ ████████ ████ █████ ████ ██ █████ ███████ ████ █████████ ████ ██████████ ███ ████ █ ██████ ████ ██ █████ ████████ ██ █████ ███████ ████ █████████ ██ ██████████. The reviewer report indicated that the analyses were confounded by uncontrolled poor prognostic factors and a limited number of events (deaths).23 The reviewer report also indicated that the sponsor did not identify any specific risk factors associated with early death in patients with HCC.23 Accordingly, the product monograph for nivolumab advises physicians to consider this risk before prescribing treatment with nivolumab plus ipilimumab in patients with poor prognostic factors.4
The KM curves of OS crossed at 12 months and remained separated in favour of nivolumab plus ipilimumab thereafter, suggesting a delayed benefit for OS. At 24 months, the CheckMate 9DW study suggested that there was likely a clinically important increase in the probability of OS with nivolumab plus ipilimumab compared with sorafenib or lenvatinib. However, the clinical experts consulted for this review indicated that the observed delayed benefit was not aligned with their expectations for the treatment. Given the time-varying treatment effect and the limitations in identifying patients who are considered to be at high risk of early mortality, the extent of uncertainty in the evidence is likely underestimated by the CIs for the between-group differences in OS at 24 months. Specifically, the OS benefit is conditional on early survival, and there are no known predictors to identify patients most likely to benefit from treatment with nivolumab plus ipilimumab, per clinical expert input. As a result, the generalizability of the observed study results for OS (and PFS benefit, as discussed later in this section) at a fixed time point of 24 months remains unclear based on the available data.
As is common in oncology studies,36 the results for OS are reflective of the combined effects of nivolumab plus ipilimumab versus sorafenib or lenvatinib and any subsequent anticancer therapies received in each group, rather than the effects of each treatment in isolation. Although the comparative effects are confounded by subsequent immunotherapy-containing regimens (used more frequently in the sorafenib or lenvatinib group), the comparison is of direct relevance because these subsequent treatments generally reflect those used in practice, according to the clinical experts consulted for this review. An exception is further treatment with immunotherapy, which may occur depending on the funding algorithm used in each jurisdiction across Canada or participation in a clinical trial, but is not reflective of general practice, according to the clinical experts consulted for this review.
Similar to the OS results, there was an early PFS detriment with nivolumab plus ipilimumab relative to sorafenib or lenvatinib. At 6 months, the study suggested a likely clinically important decrease in the probability of PFS with nivolumab plus ipilimumab compared with sorafenib or lenvatinib. The KM curves of PFS crossed between 9 and 12 months and remained separated in favour of nivolumab plus ipilimumab thereafter, suggesting a delayed benefit for PFS. At 24 months, the CheckMate 9DW study suggested a clinically important increase in the probability of PFS with nivolumab plus ipilimumab compared with sorafenib or lenvatinib.
The CheckMate 9DW study suggested that nivolumab plus ipilimumab may improve HRQoL compared with sorafenib or lenvatinib; however, the clinical importance of this effect is uncertain. There is low certainty in the assessment of HRQoL due to attrition and the use of a patient-reported outcome measure in an open-label study design. Furthermore, there was no between-group estimate to assess the clinical significance versus the target threshold.
The sponsor-submitted STC suggested that nivolumab plus ipilimumab may result in an early OS detriment and, relative to atezolizumab plus bevacizumab only, an early PFS detriment, followed by a delayed effect on OS and PFS, in adults with unresectable or advanced HCC who had not received prior systemic therapy, compared with durvalumab plus tremelimumab and atezolizumab plus bevacizumab. There is low certainty in the STC results, primarily due to violations of the proportional hazards assumption within some of the segments using the 6-month cut point and heterogeneity that could not be adequately addressed due to the lack of a common comparator. In particular, the results from the STC-derived differences in the OS and PFS probabilities between atezolizumab plus bevacizumab versus nivolumab plus ipilimumab appeared to be less consistent with the results from the piecewise survival analyses at 6 months and beyond. While exploring areas of uncertainty in the reliability of the STC-derived estimates, notable differences were identified in the follow-up times across the comparator groups with a notably shorter median follow-up for the IMbrave150 study compared with the other included studies. The implications of including studies with differential follow-up in the STC are unknown based on the submitted information but likely introduced bias in the results as a source of important methodological heterogeneity. Additionally, there are generalizability concerns because treatment effects were estimated in the comparator populations. Importantly, there remains a lack of evidence for the treatment effect of nivolumab plus ipilimumab versus other relevant immunotherapies on outcomes important to patients and clinicians, including HRQoL and harms.
Given the limitations of the comparative evidence from the STC, the clinical experts consulted for the review were asked to provide their opinions on the place in therapy for nivolumab plus ipilimumab. They indicated that immunotherapy, including nivolumab plus ipilimumab, would be preferred over sorafenib or lenvatinib in most cases, consistent with the funding algorithm. The clinical experts consulted for this review further noted that:
nivolumab plus ipilimumab would be preferred over atezolizumab plus bevacizumab for patients with a higher risk of bleeding and those with larger tumour size
the use of nivolumab plus ipilimumab compared with durvalumab plus tremelimumab remains unclear until higher quality comparative evidence emerges and clinicians gain experience with both regimens in practice.
Harms
Overall, patients in the nivolumab plus ipilimumab group experienced numerically more AEs, SAEs, WDAEs, and immune-mediated AEs, particularly grade 3 or grade 4 AEs, than patients in the sorafenib or lenvatinib group. The clinical experts consulted for this review indicated that the greater toxicity observed with nivolumab plus ipilimumab was consistent with expectations for the treatment, given the relatively high dose of ipilimumab. The most frequently reported any-grade AE in the nivolumab plus ipilimumab group was pruritus, and the most frequently reported grade 3 or 4 AE was increased aspartate aminotransferase. The most frequently reported any-grade and grade 3 or 4 SAE and WDAE in the nivolumab plus ipilimumab group was malignant neoplasm progression. The Health Canada reviewer report23 highlighted an increased risk of SAEs and WDAEs in patients aged 75 years or older receiving nivolumab plus ipilimumab, which is reflected in the product monograph warning.4 The most frequently reported any-grade and grade 3 or 4 immune-mediated AE in the nivolumab plus ipilimumab group was hepatitis.
The clinical experts consulted for this review identified a clinically important difference in the number of deaths primarily due to study drug toxicity (i.e., deaths considered related to the study drug per investigator), with more deaths observed in the nivolumab plus ipilimumab group compared with the sorafenib or lenvatinib group. Although most of these deaths were attributed to liver-related AEs in the nivolumab plus ipilimumab group, there is a risk of bias due to the measurement of outcome (assessed by investigator) because the study used an open-label design. The clinical experts anticipated that this observation may influence prescribing decisions, emphasizing the importance of careful selection of patients based on adequate liver function to mitigate hepatic risks, consistent with the Health Canada reviewer report.23
The product monograph of nivolumab4 warns that monotherapy and combination therapy with ipilimumab can cause severe and potentially fatal immune-mediated AEs, including pneumonitis, interstitial lung disease, encephalitis, myocarditis, Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune hemolytic anemia. The product monograph further warns that while most of the immune-mediated AEs occur during treatment, onset after the last dose has been reported.4 The product monograph advises early diagnosis and appropriate management to minimize potentially life-threatening complications. Of note, immune-modulating concomitant medications for the management of AEs were administered more frequently in the nivolumab plus ipilimumab group than in the sorafenib or lenvatinib group, with dermatologic drugs being the most common.
No new safety signals were identified in the 5-year follow-up results from the CheckMate 040 study, which evaluated nivolumab plus ipilimumab combination therapy in patients with advanced HCC previously treated with sorafenib.37 The Health Canada reviewer report23 indicated that the risks associated with nivolumab plus ipilimumab can be properly mitigated through close monitoring and adherence to the risk mitigation measures described in the respective product monographs.
Ethics and Equity Considerations
The clinical experts consulted for this review noted that people with IV drug or alcohol dependency (associated with hepatitis C viral etiology), people who have immigrated to Canada from regions where hepatitis B is endemic, and patients with metabolic-associated liver cirrhosis often face barriers to accessing health care and lack support in navigating health care systems due to socioeconomic status and other structural inequities. Additionally, the patient group indicated that people from equity-deserving groups, including Indigenous Peoples, people who have recently immigrated to Canada, and people experiencing low income, face barriers to diagnosis, care coordination, and access to novel therapies for HCC, contributing to disease progression and poorer health outcomes. As such, the patient group identified a need for systemic changes to improve early detection, referral pathways, and equitable drug and testing access, to avoid disparities in survival and quality of life, particularly in rural communities.
The clinical experts consulted for this review noted that these equity-deserving groups are often underrepresented in clinical trials due to a lack of support. Although patients with hepatitis C viral etiology of HCC were slightly underrepresented in the CheckMate 9DW study, the clinical experts consulted for this review suggested that the study results are generalizable to this patient group in clinical practice. However, the lower representation of this patient group in the study introduces uncertainty regarding the generalizability of the observed magnitude of benefit and harm to patients with hepatitis C viral etiology, with implications for clinical decision-making, informed consent and patient decision-making, and health system decision-making.
Conclusion
One ongoing, phase III, open-label RCT (the CheckMate 9DW study) provided evidence on the efficacy and safety of nivolumab plus ipilimumab as first-line therapy compared with investigator’s choice of SOC with either sorafenib or lenvatinib in adults with unresectable or advanced HCC. The results of the CheckMate 9DW study indicate that treatment with nivolumab plus ipilimumab likely results in increased OS compared with sorafenib or lenvatinib; however, this benefit is delayed. At 6 months of treatment, nivolumab plus ipilimumab likely resulted in a clinically important decrease in the probability of OS compared with sorafenib or lenvatinib. The KM curves of OS crossed at 12 months and remained separated thereafter, and nivolumab plus ipilimumab was likely associated with a clinically important increase in OS compared with sorafenib or lenvatinib at 24 months. The time-varying treatment effect and the inability to reliably identify patients who are considered to be at high risk of early mortality potentially limit the generalizability of the observed treatment effect to clinical practice. The study suggested that nivolumab plus ipilimumab may improve HRQoL compared with sorafenib or lenvatinib, but the clinical importance of the effect is uncertain. Compared with patients in the sorafenib or lenvatinib group, those in the nivolumab plus ipilimumab group experienced numerically more AEs, SAEs, WDAEs, and immune-mediated AEs, particularly grade 3 or grade 4 AEs.
An STC submitted by the sponsor provided indirect evidence comparing nivolumab plus ipilimumab with other IO regimens, which informed the evaluation of nivolumab plus ipilimumab as an alternative first-line treatment option. The STC suggested that nivolumab plus ipilimumab may result in an early OS detriment and delayed benefit for OS in adults with unresectable or advanced HCC who have not received prior systemic therapy, compared with durvalumab plus tremelimumab and atezolizumab plus bevacizumab. There is low certainty in the STC results, primarily due to violations of the proportional hazards assumption within some segments using the 6-month cut point and heterogeneity that could not be accounted for because of the lack of a common comparator across studies. Additionally, there are generalizability concerns related to estimating treatment effects in the comparator populations. Importantly, there remains a lack of evidence for the treatment effect of nivolumab plus ipilimumab versus other relevant immunotherapies on outcomes important to patients and clinicians, including HRQoL and harms.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of nivolumab plus ipilimumab compared to sorafenib, lenvatinib, durvalumab plus tremelimumab, and atezolizumab plus bevacizumab for the first-line treatment of adult patients with HCC.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of nivolumab plus ipilimumab from the perspective of a public health care payer in Canada over a 10-year horizon. The modelled population was comprised of patients with unresectable or advanced HCC, which aligns with the Health Canada indication. The sponsor’s base-case analysis included costs related to drug acquisition, administration, subsequent treatment, resource use, AEs, and end-of-life care.
The sponsor’s base case consisted of 4 pairwise cost-effectiveness results:
Compared with sorafenib: The incremental cost-effectiveness ratio (ICER) was $112,133 per quality-adjusted life-year (QALY) gained (incremental costs = $79,034; incremental QALYs = 0.70); approximately 74% of the benefit was predicted to be accrued after the treatment duration of the CheckMate 9DW trial (maximum follow-up period = 48.3 months).
Compared with lenvatinib: The ICER was $180,098 per QALY gained (incremental costs = $127,629; incremental QALYs = 0.71); approximately 74% of the benefit was predicted to be accrued after the treatment duration of the CheckMate 9DW trial.
Compared with durvalumab plus tremelimumab: Nivolumab plus ipilimumab was dominant (incremental cost savings = $70,052; incremental QALYs = 0.32); approximately 80% of the benefit was predicted to be accrued after the treatment duration of the CheckMate 9DW trial.
Compared with atezolizumab plus bevacizumab: Nivolumab plus ipilimumab is dominant (incremental cost savings = $107,677; incremental QALYs = 0.41); approximately 81% of the benefit was predicted to be accrued after the treatment duration of the CheckMate 9DW trial.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 8; full details are provided in Appendix 11 in the Supplemental Material).
Table 8: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative effectiveness of nivolumab plus ipilimumab relative to IOs is uncertain. | The STC used to inform the comparative efficacy of nivolumab plus ipilimumab relative to durvalumab plus tremelimumab and to atezolizumab plus bevacizumab has several limitations, such as the violation of the proportional hazards assumption within some of the segments using the 6‑month cut point and heterogeneity that could not be accounted for due to the lack of a common comparator across studies. There are also generalizability concerns due to estimating treatment effects in the comparator populations. | CDA-AMC could not address this issue due to uncertainty in the comparative clinical information. | No scenario analysis was conducted given the lack of available evidence. |
The long-term treatment effect of nivolumab plus ipilimumab is uncertain. | The long-term comparative effectiveness of nivolumab plus ipilimumab relative to all comparators is uncertain due to a lack of long-term clinical data and the low certainty of the STC results. | CDA-AMC could not address this issue due to uncertainty associated with the lack of long-term clinical information. | No scenario analysis was conducted given the lack of available evidence. |
The assumption for TOT is uncertain. | The sponsor applied different approaches to model TOT for nivolumab plus ipilimumab and the comparators: observed data from the CheckMate 9DW study were used for nivolumab plus ipilimumab, and the sponsor assumed that TOT for all other comparators was noninferior to extrapolated PFS. This approach creates a gap in which patients remain in PFS without associated treatment costs and leads to an underestimation of treatment costs for nivolumab plus ipilimumab, introducing potential bias in favour of nivolumab plus ipilimumab. | CDA-AMC could not resolve this issue due to the absence of evidence to support an alternative assumption for TOT. | CDA-AMC conducted a scenario analysis in which TOT for nivolumab plus ipilimumab was equal to extrapolated PFS curves. |
The approach used to model the efficacy of TKI comparators is uncertain. | The sponsor used pooled data from the comparator arm of the CheckMate 9DW trial, consisting of 85% lenvatinib and 15% sorafenib, to inform survival for both sorafenib and lenvatinib in the economic model, assuming equal clinical efficacy between sorafenib and lenvatinib. However, clinical expert input noted that this distribution does not reflect clinical practice in Canada (approximately 95% lenvatinib and 5% sorafenib). Additionally, the sponsor’s submission indicated that the clinical benefits of lenvatinib and sorafenib may differ. Therefore, the sponsor's approach may not accurately represent real-world outcomes. | CDA-AMC could not address the inaccuracy in the sorafenib and lenvatinib distribution used in the model due to the lack of clinical data reflecting the alternative ratio observed in clinical practice in Canada. | CDA-AMC conducted a scenario analysis using the respective data from the CheckMate 9DW study to inform survival and TOT for sorafenib and lenvatinib separately. |
CDA-AMC = Canada’s Drug Agency; IO = immuno-oncology; KM = Kaplan-Meier; PFS = progression-free survival; QALY = quality-adjusted life-year; STC = simulated treatment comparison; TKI = tyrosine kinase inhibitor; TOT = time on treatment.
Note: Full details of the identified issues by CDA-AMC are provided in Appendix 10 in the Supplemental Material.
CDA-AMC Assessment of Cost-Effectiveness
CDA-AMC noted several limitations that add uncertainty to the sponsor’s base case, including uncertainty in comparative effectiveness, uncertainty in assumption for time on treatment, and uncertainty in assumptions related to the efficacy and distribution of TKIs, sorafenib and lenvatinib. There limitations could not be addressed through reanalysis due to the lack of supporting clinical evidence. As such, no reanalyses were performed.
Impact on Health Care Costs
In the sponsor’s base case, nivolumab plus ipilimumab is predicted to be associated with cost savings compared with durvalumab plus tremelimumab (cost savings = $70,052) and with additional health care costs compared with lenvatinib (incremental cost = $127,629). In both comparisons, the differences in health care spending results from differences in drug acquisition costs (refer to Figure 1).
Nivolumab plus ipilimumab is stopped after 2 years, whereas treatment with other IO combinations continues until progression. Consequently, drug acquisition costs (and total costs) were lower for nivolumab plus ipilimumab than for the other IO combinations.
Figure 1: Impact of Nivolumab Plus Ipilimumab Versus Comparators on Health Care Costs
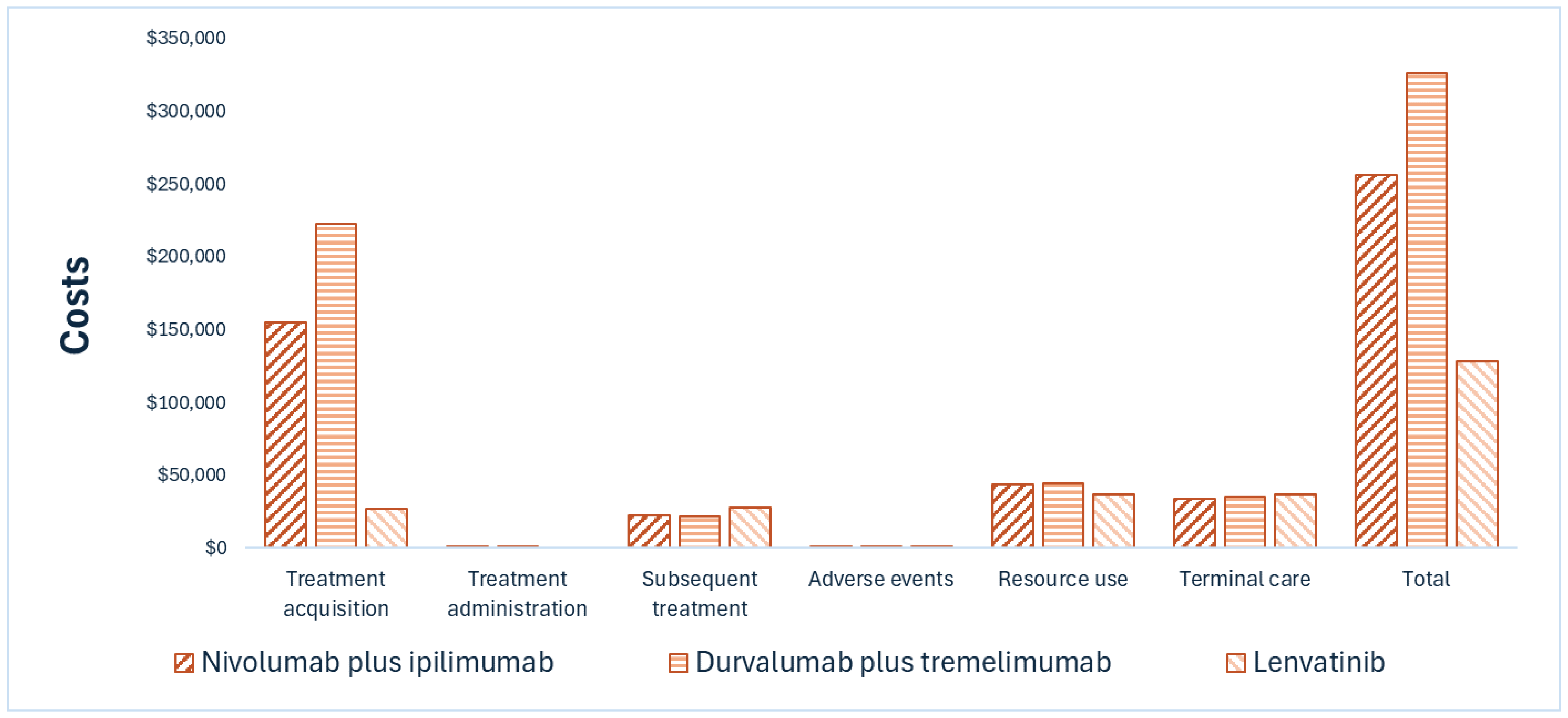
Note: Results for atezolizumab plus bevacizumab and sorafenib are not presented in this figure. For detailed results, refer to Appendix 11 in the Supplemental Material.
Impact on Health
In the sponsor’s base case, nivolumab plus ipilimumab is predicted to result in 0.71 additional QALYs per patient compared with lenvatinib over a 10-year time horizon. However, the efficacy data analysis used pooled data from sorafenib and lenvatinib, which adds uncertainty to the estimated health benefit. Clinical expert input indicated that TKIs are generally reserved for patients who are not eligible for IO therapy. As such, comparisons with TKIs are of uncertain rigour or relevance.
The sponsor’s submitted base case suggested that nivolumab plus ipilimumab was associated with more QALYs (2.63) than durvalumab plus tremelimumab (2.25) or atezolizumab plus bevacizumab (2.15). Due to the low certainty in the results of the STC, there is insufficient evidence to draw robust conclusions on the comparative effectiveness of nivolumab plus ipilimumab versus durvalumab plus tremelimumab or versus atezolizumab plus bevacizumab in this population.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2025 to 2027) budget impact of reimbursing nivolumab in combination with ipilimumab for use in the first-line treatment of adult patients with unresectable or advanced HCC. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of nivolumab was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 in the Supplemental Material.
Although the sponsor estimated that reimbursing nivolumab in combination with ipilimumab for use in the first-line treatment of adult patients with unresectable or advanced HCC would result in cost savings for the public drug plan, this result is highly uncertain and dependent on the confidentially negotiated prices of each comparator.
Conclusion
Based on the CDA-AMC Clinical Review, nivolumab plus ipilimumab is likely to result in a clinically important increase in OS compared with sorafenib and lenvatinib at 24 months in adult patients with unresectable or advanced HCC. Results of the sponsor-submitted STC suggested, with low certainty, that nivolumab plus ipilimumab may be associated with an early survival detriment and a delayed effect on OS compared with durvalumab plus tremelimumab and atezolizumab plus bevacizumab in adult patients with unresectable or advanced HCC. The limitations identified by CDA-AMC could not be addressed through reanalysis, mainly due to the lack of direct comparative evidence against IO comparators and the absence of long-term clinical data for nivolumab plus ipilimumab relative to all comparators. Therefore, no reanalysis by CDA-AMC was performed. Clinical expert input sought by CDA-AMC for this review noted that adult patients who receive treatment with lenvatinib or sorafenib do so because IO therapies are contraindicated. As such, these patients would also not be eligible to receive nivolumab plus ipilimumab, complicating the interpretation of ICER estimates by the CDA-AMC and corresponding price reductions relative to lenvatinib or sorafenib.
According to the sponsor’s base case, nivolumab plus ipilimumab was dominant over durvalumab plus tremelimumab and atezolizumab plus bevacizumab (i.e., less costly and more effective). However, the estimated ICERs compared to IOs remain uncertain due to the lack of direct comparative evidence and the absence of long-term clinical data. The evidence is therefore insufficient to support a higher price for nivolumab plus ipilimumab than for the other IOs indicated in this population.
The estimated budget impact of reimbursing nivolumab in combination with ipilimumab to the public drug plans over the first 3 years is cost savings of approximately $2,072,919. The 3-year expenditure for nivolumab plus ipilimumab (i.e., not accounting for current expenditure on comparators) is estimated to be $28.8 million.
Figure 2: Summary of the Sponsor’s Economic Analysis and Price Reduction

Note: Expenditure includes only the drug cost for nivolumab plus ipilimumab. The term dominant indicates that a drug costs less and provides more QALYs than the comparator. Full results are presented in Appendix 11 in the Supplemental Material.
References
1.Bristole Myers Squibb. Anticipated Nivolumab + ipilimumab List Price [sponsor supplied reference]. 2025.
2.Yau T, Park JW, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022;23(1):77-90. doi: 10.1016/s1470-2045(21)00604-5 PubMed
3.Bristol Myers Squibb. Pharmacoeconomic Report: Cost-effectiveness Analysis of OPDIVO® (nivolumab) in Combination with YERVOY® (ipilimumab) for the First-line Treatment of Adult Patients with Unresectable or Advanced Hepatocellular Carcinoma (HCC) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opdivo (nivolumab) in combination with Yervoy (ipilimumab). May 22, 2025.
4.Bristol-Myers Squibb Canada. Opdivo (nivolumab for injection): Intravenous Infusion, 10 mg nivolumab /mL, 40 mg and 100 mg single-use vials [product monograph]. 2025. Updated June 19, 2025.
5.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589-604. doi: 10.1038/s41575-019-0186-y PubMed
6.Ferlay J EM, Lam F, Laversanne M, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F. Global Cancer Observatory: Cancer Today. International Agency for Research on Cancer; 2024. Accessed June 19, 2025. https://gco.iarc.who.int/today
7.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian cancer statistics 2023. Canadian Cancer Society; 2023. Accessed August 12, 2025. https://cancer.ca/en/cancer-information/resources/publications/canadian-cancer-statistics-2023
8.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian Cancer Statistics: A 2022 special report on cancer prevalence. Canadian Cancer Society; 2022. Accessed August 12, 2025. cancer.ca/Canadian-Cancer-Statistics-2022-EN.
9.Canadian Cancer Society. Survival statistics for liver cancer [sponsor supplied reference]. Updated September 2019. Accessed April 2025. https://cancer.ca/en/cancer-information/cancer-types/liver/prognosis-and-survival/survival-statistics
10.Canadian Cancer Society. Symptoms of liver cancer. Updated September 2019. Accessed June 19, 2025. https://cancer.ca/en/cancer-information/cancer-types/liver/signs-and-symptoms
11.CADTH. Provisional Funding Algorithm: Unresectable hepatocellular carcinoma. 2024. https://www.cda-amc.ca/unresectable-hepatocellular-carcinoma-2
12.pan-Canadian Pharmaceutical Alliance. Concluded with a Letter of Intent: Imfinzi and Imjudo (durvalumab and tremelimumab). Updated February 28, 2024. Accessed June 19, 2025. https://www.pcpacanada.ca/negotiation/22493
13.CADTH. Drug Reimbursement Expert Review Committee final recommendation: Atezolizumab (Tecentriq) - Hoffman-La Roche Limited). November 17, 2020. Accessed August 18, 2025. https://www.cda-amc.ca/tecentriq-avastin-hepatocellular-carcinoma-details
14.Bristol Myers Squibb. Summary of clinical evidence for Opdivo® (nivolumab) plus Yervoy® (ipilimumab), for the first-line treatment of adult patients with unresectable or advanced hepatocellular carcinoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opdivo (nivolumab) in combination with Yervoy (ipilimumab). May 22, 2025.
15.Bristol Myers Squibb. Clinical Study Report: CA2099DW. A Randomized, Multi-Center, Phase 3 Study Of Nivolumab In Combination With Ipilimumab Compared To Sorafenib Or Lenvatinib As First-Line Treatment In Participants With Advanced Hepatocellular Carcinoma (Check Mate 9DW: CHECKpoint pathway and nivoluMAb clinical Trial Evaluation 9DW) [internal sponsor's report]. April 25, 2024.
16.Llovet JM, Kudo M, Merle P, et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(12):1399-1410. doi: 10.1016/s1470-2045(23)00469-2 PubMed
17.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163-1173. doi: 10.1016/S0140-6736(18)30207-1 PubMed
18.Briggs A, Daniele B, Dick K, et al. Covariate-adjusted analysis of the Phase 3 REFLECT study of lenvatinib versus sorafenib in the treatment of unresectable hepatocellular carcinoma. Br J Cancer. 2020;122(12):1754-1759. doi: 10.1038/s41416-020-0817-7 PubMed
19.Anagnostou V, Yarchoan M, Hansen AR, et al. Immuno-oncology Trial Endpoints: Capturing Clinically Meaningful Activity. Clin Cancer Res. 2017;23(17):4959-4969. doi: 10.1158/1078-0432.Ccr-16-3065 PubMed
20.Zhang Z, Pan Q, Lu M, Zhao B. Intermediate endpoints as surrogates for outcomes in cancer immunotherapy: a systematic review and meta-analysis of phase 3 trials. EClinicalMedicine. 2023;63:102156. doi: 10.1016/j.eclinm.2023.102156 PubMed
21.Steel JL, Eton DT, Cella D, Olek MC, Carr BI. Clinically meaningful changes in health-related quality of life in patients diagnosed with hepatobiliary carcinoma. Ann Oncol. 2006;17(2):304-12. doi: 10.1093/annonc/mdj072 PubMed
22.Heffernan N, Cella D, Webster K, et al. Measuring health-related quality of life in patients with hepatobiliary cancers: the functional assessment of cancer therapy-hepatobiliary questionnaire. J Clin Oncol. 2002;20(9):2229-39. doi: 10.1200/jco.2002.07.093 PubMed
23.Bristol Myers Squibb. Health Canada reviewer's report: Nivolumab (Opdivo), in combination with ipilimumab for the first-line treatment of adult patients with unresectable hepatocellular carcinoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Nivolumab (Opdivo): Intravenous Infusion, 10 mg nivolumab /mL, 40 mg and 100 mg single-use vials. June 17, 2025.
24.Bristol Myers Squibb. Bristol Myers Squibb response to Canada's Drug Agency request for additional information regarding Opdivo and Yervoy review on July 28, 2025. [internal additional sponsor's information]. August 1, 2025.
25.Bristol Myers Squibb. Bristol Myers Squibb response to Canada's Drug Agency request for additional information regarding Opdivo and Yervoy review on June 19, 2025. [internal additional sponsor's information]. June 26, 2025.
26.Bristol Myers Squibb. Bristol Myers Squibb response to Canada's Drug Agency request for additional information regarding Opdivo and Yervoy review on July 16, 2025. [internal additional sponsor's information]. July 23, 2025.
27.Cochrane Methods. Risk of Bias 2 (RoB 2) tool [sponsor supplied reference]. 2019. https://methods.cochrane.org/risk-bias-2
28.Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022;1(8):EVIDoa2100070. doi: 10.1056/EVIDoa2100070
29.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894-1905. doi: 10.1056/NEJMoa1915745 PubMed
30.Remiro-Azócar A, Heath A, Baio G. Methods for population adjustment with limited access to individual patient data: A review and simulation study. Res Synth Methods. 2021;12(6):750-775. doi: 10.1002/jrsm.1511 PubMed
31.Phillippo DM, Dias S, Ades AE, Welton NJ. Assessing the performance of population adjustment methods for anchored indirect comparisons: A simulation study. Stat Med. 2020;39(30):4885-4911. doi: 10.1002/sim.8759 PubMed
32.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Med Decis Making. 2018;38(2):200-211. doi: 10.1177/0272989x17725740 PubMed
33.Macabeo B, Quenéchdu A, Aballéa S, François C, Boyer L, Laramée P. Methods for Indirect Treatment Comparison: Results from a Systematic Literature Review. J Mark Access Health Policy. 2024;12(2):58-80. doi: 10.3390/jmahp12020006 PubMed
34.Bartlett JW, Morris TP, Stensrud MJ, Daniel RM, Vansteelandt SK, Burman CF. The Hazards of Period Specific and Weighted Hazard Ratios. Stat Biopharm Res. 2020;12(4):518-519. doi: 10.1080/19466315.2020.1755722 PubMed
35.Bristol Myers Squibb. Indirect Treatment Comparisons for Nivolumab + Ipilimumab in Advanced Unresectable Hepatocellular Carcinoma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opdivo (nivolumab) in combination with Yervoy (ipilimumab). July 11, 2025.
36.U.S. Department of Health and Human Services, Food and Drug Administration, Oncology Center of Excellence, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry. 2018. Accessed August 15, 2025. https://www.fda.gov/media/71195/download
37.Melero I, Yau T, Kang YK, et al. Nivolumab plus ipilimumab combination therapy in patients with advanced hepatocellular carcinoma previously treated with sorafenib: 5-year results from CheckMate 040. Ann Oncol. 2024;35(6):537-548. doi: 10.1016/j.annonc.2024.03.005 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.