Drugs, Health Technologies, Health Systems
Reimbursement Review
Daratumumab (Darzalex SC)
Sponsor: Janssen Inc.
Therapeutic area: Multiple myeloma in patients ineligible for autologous stem cell transplant
Summary
What Is Multiple Myeloma?
Multiple myeloma is a cancer of plasma cells (the white blood cells that make immunoglobulins) found in the bone marrow. In 2025, approximately 4,285 people in Canada were newly diagnosed with multiple myeloma (NDMM). Among them, 2,125 people were not suitable for treatment with autologous stem cell transplant (ASCT) or did not plan to receive ASCT initially. The prognosis for these patients is poorer than for those who receive ASCT (median overall survival [OS] is 4.5 years compared to 8 to 10 years).
What Are the Treatment Goals and Current Treatment Options for NDMM?
Input from the patient group and the clinicians indicated that treatment goals for NDMM are to alleviate symptoms, achieve long-term remission, preserve health-related quality of life, minimize treatment side effects, prevent new complications from the treatment, and prolong survival.
Current treatments for patients with NDMM who are not undergoing ASCT include daratumumab in combination with lenalidomide and dexamethasone (DRd); the combination of bortezomib, lenalidomide, and dexamethasone (VRd); daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone; daratumumab in combination with bortezomib, melphalan, and prednisone (DVMp); and isatuximab in combination with VRd (Isa-VRd). Among these, DRd is the standard of care in Canada, according to the clinical experts consulted by Canada’s Drug Agency (CDA-AMC).
What Is Darzalex SC and Why Did CDA-AMC Conduct This Review?
Darzalex SC is a monoclonal antibody targeting the CD38 protein, which is expressed on the surface of cells in a variety of blood cancers, including multiple myeloma. Health Canada has approved Darzalex SC (a subcutaneous formulation of daratumumab) in combination with other drugs (bortezomib, lenalidomide, and dexamethasone), referred to as D-VRd hereinafter, for the treatment of adult patients with NDMM who are not undergoing ASCT as initial therapy.
CDA-AMC reviewed Darzalex SC to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the indication under review by Health Canada.
How Did CDA-AMC Evaluate Darzalex SC?
CDA-AMC reviewed evidence submitted by the sponsor, including clinical evidence on the beneficial and harmful effects, as well as economic evidence, of Darzalex SC versus other treatments used in Canada for adult patients with NDMM who are not undergoing ASCT as initial therapy. DRd, VRd, Isa-VRd, DVMp, and daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone were considered relevant treatments to compare with Darzalex SC when reviewing the evidence.
CDA-AMC identified equity and ethical considerations relevant to treatment with Darzalex SC in patients with NDMM for whom ASCT is not planned as the initial therapy.
The review was also informed by separate submissions from 1 patient group, 1 clinician group, and the participating public drug programs. The drug programs’ input covered issues that may impact their ability to implement a recommendation.
Two hematologists with representation from the Prairies (Manitoba and Alberta) were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 randomized controlled phase III trial (the CEPHEUS study) comparing D-VRd with VRd in 395 patients with NDMM who are not undergoing ASCT as initial therapy
2 propensity score–based analyses and 1 anchored matching-adjusted indirect comparison of D-VRd versus DRd, DVMp, and Isa-VRd.
For the comparison of D-VRd versus VRd:
D-VRd demonstrated a clinically important increase in the probability of progression-free survival (PFS) compared to VRd at 54 months, with high certainty in the findings.
D-VRd demonstrated increases in the overall minimal residual disease (MRD) negativity rate and very good partial response or better rate compared to VRd at a median follow-up of 58.7 months, with high and moderate certainty in the findings, respectively.
There was no statistically significant difference between D-VRd and VRd in the OS at 54 months; the evidence was of moderate certainty.
D-VRd may not result in any clinically important difference in patients’ health-related quality of life compared to VRd; the evidence was of low certainty because of a large amount of missing data.
D-VRd likely results in an increase in the incidence of serious adverse events, infections and infestations, and cytopenia compared to VRd; the evidence was of moderate to high certainty.
For the comparison of D-VRd versus other active treatments (DRd, DVMp, and Isa-VRd):
Results from the propensity-based methods suggested that treatment with D-VRd may be related to longer PFS and a higher MRD negativity rate when compared to DRd and DVMp, although the magnitude of benefit is uncertain. This uncertainty was likely related to the cross-trial heterogeneity (e.g., different patients’ characteristics at baseline). Additionally, it is possible that the comparisons were not adjusted for all effect modifiers. There was no statistically significant difference in OS between D-VRd and DRd or DVMp.
Results from a matching-adjusted indirect comparison analysis suggested that evidence is insufficient to conclude whether D-VRd or Isa-VRd is favoured for prolonging PFS or OS, or improving the MRD negativity rate, in patients with NDMM who are not undergoing ASCT as initial therapy. The estimates were affected by serious imprecision (i.e., wide 95% confidence intervals) that included the null, suggesting that either treatment could be favoured.
Economic Evidence
Darzalex SC is available as single-dose vials (1,800 mg per 15 mL).1 At the submitted price of $8,028.00 per 1,800 mg vial,2 the 21-day cycle cost of Darzalex SC is expected to be $24,084 per patient in cycles 1 and 2, $8,028 per patient in cycles 3 to 8, and the 28-day cycle cost of Darzalex SC is expected to be $8,028 per patient in cycle 9 and onwards, based on the Health Canada–recommended dosage. The regimen cost of D-VRd is expected to be $28,210 per patient in cycles 1 and 2 (21 days), $12,154 per patient in cycles 3 to 8 (21 days), and $10,278 per patient in cycles 9 and beyond (28 days).
Key clinical efficacy outcomes in the economic analysis for D-VRd versus VRd were derived from the CEPHEUS trial.3 Evidence submitted by the sponsor indicates that D-VRd is likely to improve PFS compared with VRd among patients 18 years and older with NDMM who are not undergoing ASCT as initial therapy. However, treatment benefit in OS was uncertain because median OS was not reached in either treatment group at the latest data cut-off for analyses, and the between-group difference in the 54-month OS rate was not statistically significant. For D-VRd versus all other comparators, clinical efficacy was informed by sponsor-submitted indirect treatment comparisons, which suggest that D-VRd may improve PFS when compared to DRd and DVMp, although the magnitude of benefit is uncertain because of study limitations.4 There was no statistically significant difference in OS between D-VRd and DRd or DVMp.4,5 When compared to Isa-VRd, there was insufficient evidence to conclude whether D-VRd or Isa-VRd is favoured for prolonging PFS or OS.6 There remain several critical limitations to these analyses, including heterogeneity in the included trials and some patients’ characteristics at baseline, and the possibility that not all confounding factors were adjusted for. Consequently, the estimates of PFS and OS used in the model are associated with uncertainty.
The results of the CDA-AMC base case suggest:
D-VRd is predicted to be associated with higher costs to the health care system than VRd (incremental costs = $1,072,043) and DRd (incremental costs = $311,182). These cost increases are driven by drug acquisition costs associated with D‑VRd.
D-VRd is predicted to result in no additional life-years compared to VRd and DRd. D-VRd may result in a gain of 0.10 quality-adjusted life-years (QALYs) compared to VRd, and 0.04 QALYs gained compared to DRd.
The incremental cost-effectiveness ratio of D-VRd compared to VRd was $10,623,156 per QALY gained and $7,029,398 per QALY gained compared to DRd in the CDA-AMC base case.
The estimated incremental cost-effectiveness ratio was highly sensitive to the predicted OS benefit and the drug acquisition costs of Darzalex SC. Approximately 82% and 89% of the incremental benefit relative to VRd and DRd, respectively, was gained in the extrapolated period (i.e., after 5.3 years). In the absence of comparative evidence beyond this time point and uncertainty in the comparative clinical evidence, the QALYs gained remain highly uncertain and may be overestimated. Higher price reductions than those presented in this report may therefore be required to achieve cost-effectiveness at a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing D-VRd for the indicated population will result in savings of approximately $39 million over the first 3 years of reimbursement compared to the amount currently spent on comparators, with an estimated expenditure of $369 million on D-VRd over this period. The actual budget impact of reimbursing D-VRd will depend on the market uptake of D-VRd, the negotiated prices of reimbursed comparators, and the distribution of therapies that are likely to be displaced by D-VRd. The magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption given the difference between the sponsor’s estimate and the CDA-AMC estimate.
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BIA
budget impact analysis
CI
confidence interval
Cloglog
complimentary log-log
CMRG
Canadian Myeloma Research Group
CR
complete response
DCyBorD
daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone
DRd
daratumumab in combination with lenalidomide, and dexamethasone
DVMp
daratumumab in combination with bortezomib, melphalan, and prednisone
D-VRd
daratumumab in combination with bortezomib, lenalidomide, and dexamethasone
ECOG PS
Eastern Cooperative Oncology Group Performance Status
eGFR
estimated glomerular filtration rate
EMD
extramedullary disease
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-MY20
European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Multiple Myeloma Module 20
ESS
effective sample size
FMEC
Formulary Management Expert Committee
GHS
global health status
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IMWG
International Myeloma Working Group
IPD
individual patient data
IPTW
inverse probability of treatment weighting
Isa-VRd
isatuximab in combination with bortezomib, lenalidomide, and dexamethasone
ISS
International Staging System
ITC
indirect treatment comparison
ITT
intention to treat
KM
Kaplan-Meier
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MM
multiple myeloma
MRD
minimal residual disease
NDMM
newly diagnosed multiple myeloma
OR
odds ratio
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PH
proportional hazard
PRO
patient-reported outcome
QALY
quality-adjusted life-year
RCT
randomized controlled trial
Rd
lenalidomide and dexamethasone
R-ISS
revised International Staging System
ROBINS-I
Risk of Bias In Nonrandomized Studies of Interventions
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SLR
systematic literature review
SMD
standardized mean difference
TEAE
treatment-emergent adverse event
VGPR
very good partial response
VRd
bortezomib, lenalidomide, and dexamethasone
Background
Introduction
This report reviews and critically appraises the evidence submitted by the sponsor on the beneficial and harmful effects of daratumumab subcutaneous (SC), 1,800 mg per 15 mL (120 mg/mL) single-dose vial solution for SC injection, in combination with bortezomib, lenalidomide, and dexamethasone (referred to as D-VRd hereinafter), for the treatment of adult patients with newly diagnosed multiple myeloma (NDMM) who are not undergoing autologous stem cell transplant (ASCT) as initial therapy. The focus is placed on comparing daratumumab to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence; this focus is outlined in Table 1.
This report also reviews and critically appraises the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis (BIA). The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a CDA-AMC base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The application was submitted by the sponsor before receiving a Notice of Compliance from Health Canada. This report reflects the anticipated indication and recommended dosage for daratumumab during the initial CDA-AMC review period.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Daratumumab (Darzalex SC), 1800 mg/15 mL (120 mg/mL) single-dose vial solution for subcutaneous injection. |
Sponsor | Janssen Inc. |
Health Canada indication | Daratumumab in combination with bortezomib, lenalidomide, and dexamethasone for the treatment of adult patients with newly diagnosed multiple myeloma who are not candidates for autologous stem cell transplant. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 18, 2025 |
Mechanism of action | An IgG1κ human monoclonal antibody (mAb) that targets the CD38 protein expressed on the surface of cells in a variety of hematological malignancies. |
Recommended dosage | 1800 mg administered subcutaneously, over approximately 3 to 5 minutes |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $8,028.00 per 1,800 mg vial |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: as defined in the Health Canada indication Intervention: per recommended dosage Comparators: DRdb, VRdb, DVMpb, DCyBorD, Rdb, Isa-VRdb Outcomes: PFS, OS, response rate, HRQoL, SAEs, cytopenia |
CDA-AMC = Canada’s Drug Agency; DCyBorD = daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone; DRd = daratumumab in combination with lenalidomide and dexamethasone; DVMp = daratumumab in combination with bortezomib, melphalan, and prednisone; HRQoL = health-related quality of life; Isa-VRd = isatuximab in combination with bortezomib, lenalidomide, and dexamethasone; mAb = monoclonal antibody; NOC = Notice of Compliance; OS = overall survival; PFS = progression-free survival; Rd = lenalidomide and dexamethasone; SAE = serious adverse event; VRd = bortezomib, lenalidomide, and dexamethasone.
aThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated.
bCDA-AMC has previously issued a reimbursement recommendation for this drug for the same indication or a similar indication.
Submission History for the Drug Under Review
CDA-AMC previously reviewed daratumumab in combination with other drugs (e.g., daratumumab in combination with lenalidomide and dexamethasone [DRd], and daratumumab in combination with bortezomib, melphalan, and prednisone [DVMp]) through the reimbursement review process for adult patients with NDMM who are ineligible for ASCT, and CDA-AMC issued a conditional recommendation of reimbursement for the use of DRd or DVMp in these patients.
CDA-AMC also previously reviewed D-VRd for patients with NDMM who are eligible for ASCT, and reviewed DVMp for adult patients with newly diagnosed light chain amyloidosis, and issued a conditional recommendations of reimbursement for these patients.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), input from clinical experts consulted for this review; they were also informed by a literature search.
Calls for patient group and clinician group input are issued for each reimbursement review. One patient group submission from Myeloma Canada and 1 clinician group submission from the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee were received. For the patient group input, a patient and caregiver survey was used and distributed via email and social media to eligible patients. Survey eligibility was determined by patient and caregiver self-report of experience with multiple myeloma (MM), that they (or the person they care for) had MM and were ineligible for ASCT at the time of diagnosis, and/or did not receive an ASCT as their first-line therapy. There were 101 responses in total, with 60 complete responses. Respondents were divided into 3 subsets: patients newly diagnosed who have yet to receive any treatment; patients who had MM who were ineligible for transplant or did not receive first-line treatment with ASCT, and their caregivers; and patients who received treatment with D-VRd as first-line therapy in place of ASCT. For the clinician group input, information was gathered by email. The full submissions are available on the project landing page in the consolidated input document Patient and Clinician Group Input for Darzalex SC. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two hematologists with expertise in the diagnosis and management of MM participated as part of the review team, with representation from the Prairies (Manitoba and Alberta).
Disease Background
MM is a rare hematological cancer characterized by the proliferation of plasma cells and an overproduction of abnormal immunoglobulin monoclonal protein (M-protein).7,8 Common clinical manifestations of MM include symptoms of fatigue and bone pain, as well as fever, frequent infections, kidney problems, and nervous system impairment.9,10 The diagnosis of MM is usually made by a family physician and can be prompted by noticeable signs and symptoms or discovered incidentally during the review of lab results ordered for a different condition.11 Moreover, the International Myeloma Working Group (IMWG) diagnostic criteria can be used for MM, and diagnosis is made when at least 1 MM-defining event is present and either at least 10% clonal bone marrow plasma cells or a biopsy-proven plasmacytoma are present.12 Approximately 4,100 cases of MM were diagnosed in Canada in 2024, with an estimated 1,750 MM-related deaths.13 The estimated projected prevalence in Canada of NDMM in 2025 is 4,285 patients, and the projected prevalence of patients not undergoing ASCT as initial therapy is 2,125 patients.14-16 Based on data regarding the MM burden in Canada, lower incidence rates of MM are observed in large metropolitan areas and high-latitude regions of the country, whereas high incidence rates were observed in smaller municipalities and rural areas. In addition, increased density of crop farms and agricultural industries correlate with a higher incidence of MM.17 Other risk factors for MM include age (most people diagnosed with MM are aged 65 years or older), sex (male patients are slightly more likely to develop MM than female patients), race (e.g., MM is more than twice as common in Black people than in white people), family history of MM, a weakened immune system, history of monoclonal gammopathy of undetermined significance, excess body weight, and exposure to radiation or to certain chemicals.18
Numerous factors influence the prognosis of MM, including disease stage, LDH levels, beta2-microglobulin levels, serum albumin levels, and chromosomal changes.19 Among patients with NDMM who are not undergoing ASCT as initial therapy, the prognosis can be particularly poor, with a median overall survival (OS) of 4.5 years compared to the median OS of approximately 8 to 10 years among those who receive ASCT.20,21
Input from the patient group identified that NDMM greatly impacted their quality of life, limiting their ability to travel, exercise, and work. Patients shared that NDMM disrupted their life goals and achievements and was associated with loss of sexual desire, difficulty sleeping, and increased anxiety and worry. They also reported financial strain largely because of hospital parking costs, travel expenses, and drug costs.
Current Management
Treatment Goals
According to Myeloma Canada, which submitted input to CDA-AMC, an ideal treatment should be accessible and effective in achieving long-term remission while preserving quality of life and minimizing disruptions to daily activities. Patients also highlighted the importance of limiting side effects of the treatments, ensuring continuity of care, and being supported by a proactive and responsive care team. Therefore, achieving long-term remission, preserving patients’ health-related quality of life (HRQoL) and autonomy, minimizing side effects of the treatment, and ensuring continuity of care are deemed important outcomes by the patient group.
According to the clinical experts consulted for this review, important treatment goals for patients with NDMM who are not undergoing ASCT as initial therapy are to alleviate symptoms, improve treatment response, extend the duration of remission, improve HRQoL, prevent new complications from the treatment, and prolong survival. The clinical experts and the clinician group considered the following outcomes clinically important in the target population: treatment response measured with standard response criteria adopted in clinical practice, improvement in symptoms related to MM, and survival.
Current Treatment Options
Following an MM diagnosis, patients’ eligibility for receiving an ASCT as initial therapy is determined by their ability to tolerate the treatment, which is based on clinical perception and depends on various factors, such as age, performance status, comorbidities, frailty status, the presence of disabilities, and the availability of social and family support.22-24
For patients with NDMM who are ineligible for ASCT as initial therapy, the Canadian Myeloma Research Group (CMRG) recommends that all patients undergo a frailty status assessment (via an assessment of performance status and comorbidities or using a validated tool such as the IMWG frailty index) to assess their overall fitness.25 Real-world data from 2007 to 2021 showed that the combinations of cyclophosphamide, bortezomib, and dexamethasone (47%); lenalidomide and dexamethasone (Rd) (24%); bortezomib, melphalan, and prednisone (23%); and bortezomib, lenalidomide, and dexamethasone (VRd) (6%) were commonly used initial regimens in patients with NDMM who were ineligible for transplant before daratumumab became available.25 At present, regimens that do not contain daratumumab — such as VRd; the combination of cyclophosphamide, bortezomib, and dexamethasone; and Rd (for patients unable to tolerate triplet regimens or living in rural settings) — remain potential alternatives, but not preferred options, according to CMRG.25 Currently, for patients deemed fit enough to tolerate treatment with an anti-CD38 monoclonal antibody, CMRG and the Formulary Management Expert Committee (FMEC) recommend that first-line therapy for patients with NDMM who are ineligible for transplant includes an anti-CD38 monoclonal antibody, with daratumumab in combination with lenalidomide and dexamethasone (DRd) representing the first-choice regimen.25,26 For patients who have frailty, high-risk disease, or renal insufficiency, the use of a daratumumab-containing regimen is also recommended, but additional considerations are necessary to ensure tolerability and selection of the most appropriate combination therapy.25 With respect to the full population of interest for this submission (patients who are not undergoing ASCT as initial therapy), FMEC recommends that treatment options for patients who choose not to undergo ASCT are consistent with the options offered to patients with NDMM who are ineligible for transplant.26 The clinical experts consulted for this review echoed that for patients with NDMM who are not undergoing ASCT as initial therapy, a regimen containing daratumumab is generally given as first-line treatment.
Beyond DRd, other daratumumab-containing regimens include daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone (DCyBorD) and DVMp.25 FMEC no longer considers DVMp clinically relevant in Canada for patients with NDMM who are ineligible for transplant because of the increased toxicity associated with melphalan.26 In addition, clinical experts consulted by CDA-AMC noted that both DCyBorD and DVMp are not commonly used in the first-line setting because of their safety profile and clinical inferiority compared with DRd. Furthermore, DCyBorD is only explicitly mentioned in recommendations for subpopulations of patients with high-risk disease or severe and/or rapidly progressing acute renal insufficiency related to MM,25 which is considered a highly restricted population compared to the overall population of patients with NDMM who are not undergoing ASCT as initial therapy.
In addition, isatuximab in combination with VRd (Isa-VRd) was recently reviewed, and received a positive reimbursement recommendation from CDA-AMC in June 2025 for patients with NDMM who are not eligible for ASCT.27
Key characteristics of daratumumab are summarized with other treatments available for patients with NDMM who are not undergoing ASCT in Table 1 in Appendix 1 in the Supplemental Material document (available on the project landing page).
Unmet Needs and Existing Challenges
Patients with MM may experience disease-related symptoms (e.g., fatigue, anemia, bone pain) and unwanted side effects from the treatments that create substantial burdens in their daily living, physical well-being, and psychosocial well-being. The disease also impairs caregivers’ overall well-being.28 According to the input submitted by Myeloma Canada, the patient group emphasized the need for treatments accessible to those who are ineligible for or choose not to undergo ASCT as initial therapy. Given the financial and quality of life burdens associated with ASCT, patients highlighted the importance of having greater autonomy in treatment decisions. They expressed a need for therapies that achieve long-term remission, minimize side effects, and preserve quality of life. Patients also noted barriers to care, including delayed treatments, inaccessibility of treatments in rural or physician-short areas, unavailability in certain provinces, and not being comprehensively covered by insurance.
The clinical experts consulted by CDA-AMC and the clinician group that provided input to this review identified the challenges with the current first-line therapies for patients with NDMM who are not undergoing ASCT as initial therapy. They noted that even though the current standard treatment (e.g., DRd) is highly effective, not all patients respond adequately: a small portion of these patients do not respond initially, and some patients eventually become refractory to the treatment. The clinician group also emphasized the need for an alternative option that could serve as the first-line treatment for patients who are either ineligible for or choose not to undergo ASCT.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to Table 2 in Appendix 1 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in Table 3 in Appendix 1 in the Supplemental Material document. The following has been summarized by the review team.
Place in Therapy
The clinical experts consulted by CDA-AMC expected that daratumumab in combination with bortezomib, lenalidomide, and dexamethasone (D-VRd) would increase the response rates with the available standard of care for NDMM, such as VRd or DRd, because of the synergistic effects after combining these active drugs for MM. If adopted, D-VRd would become 1 of the first-line regimens for appropriate patients with NDMM who are ineligible for transplant. Subsequently, it would slightly change the current treatment paradigm, in that this 4-drug combination would be the first-line therapy instead of the existing 3-drug regimen. Because this is a first-line therapy for patients with NDMM who are ineligible for transplant, patients are not recommended to try other treatments before initiating D-VRd, unless D-VRd is deemed not suitable according to certain clinical criteria.
The clinician group also suggested that D-VRd be used as first-line therapy for patients with NDMM who are ineligible for or choose not to undergo ASCT.
Patient Population
According to the clinical experts consulted by CDA-AMC, D-VRd should be prescribed as first-line therapy to all patients with NDMM who are ineligible for transplant if they are physically fit. Because the addition of bortezomib to the current standard of treatment (e.g., DRd) adds both drug toxicity and patient inconvenience (because of the additional SC injections), treatment with D-VRd should only be considered in patients with good performance status and manageable comorbidities. The clinical experts noted that patients who are older (e.g., aged older than 80 years), have frailty, have poor performance status, or have existing neuropathy may not be candidates for treatment with D-VRd. Instead, treatment with DRd would be preferred if the patients are older or have poorer performance status.
The clinician group also stated that D-VRd therapy is appropriate for patients with NDMM who are ineligible for transplant or have deferred transplant.
Testing Procedure Considerations
Minimal residual disease (MRD) refers to the small number of cancer cells that persist after treatment, which can cause relapses despite achievement of a complete response (CR).29 The IMWG defines MRD negativity as no more than 1 cancer cell detected in 100,000 bone marrow cells (10–5) and sustained MRD negativity as 2 consecutive MRD-negative results for at least 12 months.30 MRD status can be assessed using bone marrow aspirates by next-generation flow cytometry or next-generation sequencing.30 Multiple studies reported that in NDMM, achieving MRD negativity was associated with improved survival outcomes in patients with NDMM who are either eligible or ineligible for transplant.31-34
In the sponsor-submitted CEPHEUS trial for this review, sustained MRD negativity was a primary end point but did not guide treatment adjustment in patients with NDMM not undergoing ASCT as initial therapy.35 Consistent with the trial, the clinical experts consulted for this review confirmed that sustained MRD negativity would not be used to guide treatment decisions in the indicated population.
While guidelines in Canada recommend that MRD testing should be available and accessible as a routine standard of care assay for all patients with MM (i.e., those who are eligible or ineligible for transplant), according to the clinical experts, testing capacity is limited and MRD testing is not routinely available or funded for this patient population. The clinical experts also noted that in clinical practice, there is no strong interest in having MRD testing available for patients who are ineligible for transplant because MRD status currently does not inform treatment decisions in them.36 Therefore, its limited availability and funding are not expected to affect the implementation and adoption of daratumumab therapy.
CDA-AMC considered the potential impacts of MRD testing to informing treatment adjustment with daratumumab, including those to health systems, patients (including families and caregivers), and costs. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Table 4 in Appendix 1 in the Supplemental Material document.
Assessing the Response to Treatment
The clinical experts consulted by CDA-AMC indicated that clinically meaningful outcomes to determine a patient’s response to treatment in the target population include treatment response rate, change in MM symptoms, and survival in the long-term. In terms of treatment response, various measurements are available. The experts considered a “partial response or better” (defined by IMWG response criteria) as the most clinically relevant treatment response measurement, and they suggested that this outcome be assessed every 4 weeks by blood tests. Improvements in MM symptoms can also be examined every 4 weeks in clinical practice.
The clinician group agreed with the clinical experts consulted by CDA-AMC that the treatment response can be examined by standard myeloma and organ response criteria adopted in clinical practice.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC stated that the decision to discontinue treatment with daratumumab should consider disease progression and toxicities related to the treatment (especially neuropathy). The experts also suggested that bortezomib in this regimen be used for a limited number of cycles.
The clinician group also suggested that treatment with D-VRd should be discontinued if disease progression, relapse, or significant intolerance occurs.
Prescribing Considerations
According to clinical experts consulted by CDA-AMC, for treatment with D-VRd, a specialist (i.e., a hematologist, or in some cases a medical oncologist) would be required to diagnose myeloma and create a treatment plan. Thereafter, the treatment can be delivered through a community oncology site under the supervision of a specialist.
The clinician group agreed that D-VRd therapy can be an outpatient treatment, while the consideration for inpatient funding may be needed.
Additional Considerations
The clinical experts consulted by CDA-AMC indicated that if the D-VRd regimen becomes available in Canada, it is unlikely to be prescribed the same way as in the CEPHEUS trial. In clinical practice, most centres treat patients with 28-day cycles, which appears to be less toxic. In the 28-day cycle regimen with VRd or D-VRd, patients receive bortezomib once per week, dexamethasone once per week, and lenalidomide from day 1 to day 21.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions, indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III clinical trials that reported results for D-VRd in the population of interest. Relevant patients and interventions were defined by the indication or reimbursement request and the recommended dosage in the product monograph. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included VRd, DRd, and Isa-VRd. Long-term extensions of included pivotal studies and RCTs were included, regardless of whether there was a comparison group. ITCs and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time).
The review team selected outcomes (and follow-up times) for review considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes, identified by patient and clinician groups and by clinical experts as efficacy measures that address the main treatment goals for NDMM, were assessed with GRADE: progression-free survival (PFS), OS, MRD negativity rate, response rate (very good partial response [VGPR] or better), and HRQoL (as measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 [EORTC QLQ-C30] Global Health Status (GHS) and Quality of Life domain, and the EORTC QLQ Multiple Myeloma Module 20 [EORTC QLQ-MY20]). PFS was also a key input in the sponsor’s pharmacoeconomic model.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are provided in Appendix 2 in the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal RCT included in the systematic review, the CEPHEUS study3
3 ITCs comparing:
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are in Appendix 3 in the Supplemental Material document.
The CEPHEUS study is a phase III, open-label, randomized, multicentre study that evaluated daratumumab SC in combination with VRd (D-VRd) versus VRd in patients with NDMM who were not undergoing ASCT as initial therapy (including both patients with NDMM who are ineligible for transplant and patients in whom transplant was not planned as initial therapy).3 The primary objective of this study was to determine if the addition of daratumumab to VRd improves overall MRD negativity (≥ CR) compared with VRd alone. The study was initiated on November 15, 2019, and the final PFS analysis clinical cut-off date was May 7, 2024. Patients from 98 centres across 13 countries (Brazil, Canada, Czechia, France, Germany, Israel, Japan, the Netherlands, Poland, Spain, Türkiye, the UK, and the US) were included in the study.
The CEPHEUS trial evaluated daratumumab SC combined with bortezomib, lenalidomide, and dexamethasone (hereinafter referred to as the D-VRd cohort), compared with bortezomib, lenalidomide, and dexamethasone (hereafter referred to as the VRd cohort). In total, 395 patients were randomly assigned (in a 1:1 ratio) to receive either VRd or D-VRd, using an interactive web response system. Eligible patients were stratified before randomization by International Staging System (ISS) stage I, stage II, or stage III (based on central laboratory results for beta2-microglobulin and albumin) and age or transplant eligibility (aged younger than 70 years and ineligible; aged younger than 70 years and transplant not planned as initial therapy; or aged 70 years or older). Because of the timing and administration route of daratumumab and the lack of a placebo control group, study intervention allocation could not be blinded.
The CEPHEUS trial consisted of 3 phases: a screening phase, a treatment phase, and a follow-up phase. The screening phase commenced up to 28 days before randomization (cycle 1, day 1). The treatment phase extended from cycle 1, day 1 until treatment discontinuation because of progressive disease (PD) or unacceptable toxicity. During the treatment phase, patients were to receive treatment with either VRd or D-VRd for 8 cycles. Bortezomib was planned as a fixed-dose treatment of 8 cycles in both groups. After completing 8 cycles of therapy, participants were to continue with Rd or D-Rd until disease progression or unacceptable toxicity. Participants who discontinued treatment with any component of study treatment (bortezomib, lenalidomide, dexamethasone, or daratumumab) could continue to receive treatment with the other components of study treatment, as assigned. Patients entered the follow-up phase once they had documented disease progression or unacceptable toxicity and all treatment was discontinued.
Patients who discontinued before disease progression were to continue to have disease evaluations until confirmed disease progression, death, withdrawal of consent, loss to follow-up, or the end of data collection. After disease progression was documented, follow-up evaluations continued at least every 16 weeks until the final PFS analysis.
In the CEPHEUS trial, 3 analyses were planned: a primary MRD analysis (date: █████ ██ ████), an interim PFS analysis (date: █████████ ██ ████), and a final PFS analysis (date: May 7, 2024). Results of all efficacy and safety outcomes are available at the data cut-off date of May 7, 2024.
Intervention: Treatment was administered as a 3-week (21-day) regimen for cycles 1 to 8 and as a 4-week (28-day) regimen for cycle 9 onward, as follows:1,37
daratumumab administered SC at 1,800 mg weekly for cycles 1 and 2, every 3 weeks for cycles 3 to 8 (with the first dose of the every-3-week dosing schedule given at week 7), and every 4 weeks for cycle 9 and beyond (with the first dose of the every-4-week dosing schedule given at week 25) until disease progression or unacceptable toxicity occurs
bortezomib administered SC at 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1 to 8
lenalidomide administered orally at 25 mg daily on day 1 to day 14 of cycles 1 to 8 and on day 1 to day 21 of cycle 9 and beyond until disease progression or unacceptable toxicity occurs
dexamethasone administered orally at 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 of cycles 1 to 8; in cycles 9 and beyond, dexamethasone is administered orally at 40 mg on days 1, 8, 15, and 22 of each cycle, until disease progression or unacceptable toxicity occurs.
For all patients, the following preinjection drugs had to be administered approximately 1 to 3 hours before every dose of daratumumab SC to reduce the risk of administration-related reactions:1 a corticosteroid (20 mg dexamethasone or equivalent), an antihistamine (25 mg to 50 mg administered orally or IV diphenhydramine or equivalent), and an antipyretic (650 mg to 1,000 mg acetaminophen). To reduce the risk of administration-related reactions, administering low-dose oral methylprednisolone (≤ 20 mg) or equivalent the day after daratumumab SC injection was recommended.1 No predrug or postdrug requirements are specified in the Health Canada product monographs for bortezomib, lenalidomide, or dexamethasone.38-40
In the CEPHEUS trial, the primary efficacy outcome was the overall MRD negativity (≥ CR) rate, and the secondary efficacy outcomes included PFS, OS, sustained MRD negativity (≥ CR), other MRD analyses, response rate, or time to subsequent antimyeloma therapy.3 Additionally, safety outcomes such as AEs monitoring were reported. Patient-reported outcomes (PROs) such as EORTC QLQ-C30, EORTC QLQ‑MY20, and EQ-5D-5L scores were included to assess participants’ symptoms, functioning, and general well-being (Table 2).3
Table 2: Characteristics of Studies Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
The CEPHEUS study Multicentre, phase III, open-label RCT N = 395 |
|
| Intervention Daratumumab administered as a SC injection combined with bortezomib (SC), lenalidomide (p.o.), and dexamethasone (p.o.) Comparator Bortezomib (SC), lenalidomide (p.o.), and dexamethasone (p.o.) | Primary
Secondary
|
ASCT = autologous stem cell transplant; CR = complete response; DoR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-MY20 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Myeloma 20; HRQoL = health-related quality of life; MM = multiple myeloma; MRD = minimal residual disease; NCI-CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; NDMM = newly diagnosed multiple myeloma; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PFS2 = second progression-free survival; p.o. = orally; RCT = randomized controlled trial; SC = subcutaneous; TTSAT = time to subsequent antimyeloma therapy; VGPR = very good partial response.
Sources: Clinical Protocol42 and Clinical Study Report for CEPHEUS.3 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
The CEPHEUS trial sample size calculation was based on the primary end point. The anticipated overall MRD negativity (≥ CR) rate (10–5) for the control group in this trial was estimated to be at most ███.43 The CEPHEUS trial assumed that the addition of daratumumab to VRd would lead to a ███ absolute increase in the overall MRD negativity (≥ CR) rate (███ D-VRd versus ███ VRd alone). Therefore, a sample size of 360 patients (180 in each group) was needed to achieve 80% power to detect such a treatment difference at a 2-sided alpha of 0.05. This sample size would provide approximately 80% power to detect a 37% reduction in the risk of progression or death (hazard ratio [HR] = 0.63, translating to an improvement in median PFS from 43 months to 68 months) with a log-rank test at a 2-sided alpha of 0.05. The event size for the final analysis of PFS was not decreased from 162.
Three analyses were planned: a primary MRD analysis (date: █████ ██ ████), an interim PFS analysis (date: █████████ ██ ████), and a final PFS analysis (date: May 7, 2024).
In general, continuous variables were summarized using descriptive statistics such as mean, standard deviation (SD), median, and range. Categorical variables were summarized using frequency and percentage. For the efficacy assessment of response and disease progression, a validated computerized algorithm was utilized to determine response and disease progression for each patient. This computerized algorithm is based on the IMWG response criteria44,45 and has been used and validated by an independent review committee. As a sensitivity analysis, investigator assessments of treatment response and disease progression per the IMWG response criteria were also performed. All statistical hypothesis tests and 95% confidence intervals (CIs) presented were 2-sided.
Clinical Trial End Points
Progression-free survival: Analysis of PFS was based on the intention-to-treat (ITT) analysis set. The Kaplan-Meier (KM) method was used to estimate the distribution of overall PFS for each treatment group. The median PFS and corresponding 95% CIs are also provided. In addition, the numbers and percentages of patients who had a PFS event or were censored were reported. The reasons for PFS censoring were summarized accordingly. The KM PFS curve was also plotted by treatment groups.
If a subsequent antimyeloma therapy started before disease progression or death, the PFS date was censored at the date of the last disease assessment before the start of subsequent antimyeloma therapy. The treatment comparison of the distribution of overall PFS was based on a stratified log-rank test. The P value from a stratified log-rank test was also reported. HRs and their 95% CIs were estimated based on a stratified Cox regression model with treatment as the sole explanatory variable. Stratification factors used in the analyses include ISS staging (I, II, and III) and age or transplant eligibility (aged younger than 70 years and ineligible, aged younger than 70 years and transplant not planned as initial therapy, or aged 70 years or older).
Overall MRD negativity (≥ CR) rate: The overall MRD negativity (≥ CR) rate was the primary efficacy end point in the CEPHEUS trial. It was defined as the proportion of participants in the ITT population who achieved both MRD negativity by next-generation sequencing per clonoSEQ test (at or lower than a sensitivity threshold of 10–5) in bone marrow aspirate and a CR or better response at any time after the date of randomization (and before disease progression, receipt of subsequent therapy, or both). It was calculated for each treatment group based on the ITT analysis set. The corresponding 95% exact CI was provided. Reasons for missing or unevaluable MRD status were tabulated by treatment group. Stratified Cochran-Mantel-Haenszel estimates of odds ratio (ORs) and their 95% CIs and P values from the Fisher exact test were used to test if the MRD negativity (≥ CR) rate is the same between the 2 treatment groups. Stratification factors used in the analysis include ISS staging (I, II, and III) and age or transplant eligibility (aged younger than 70 years and ineligible for transplant, aged younger than 70 years and transplant not planned as initial therapy, or aged 70 years or older).
Other efficacy variables (OS, VGPR or better rate): The analysis of OS was performed in a similar manner as described in the preceding PFS section. At the primary MRD analysis, the OS analysis was exploratory and descriptive. The KM curves of OS were also provided by treatment groups.
The VGPR or better rate was defined as a VGPR, CR, or stringent CR according to the IMWG criteria. It was calculated for each treatment group based on the ITT analysis set. The corresponding 95% exact CIs were provided. The stratified Cochran-Mantel-Haenszel estimate of OR and its 95% CI and P value for testing treatment difference were reported. Stratification factors used in the analysis include ISS staging (I, II, and III), and age or transplant eligibility (aged younger than 70 years and ineligible, aged younger than 70 years and transplant not planned as initial therapy, or aged 70 years or older).
Safety outcomes: Safety analyses were based on the safety analysis set and presented by treatment group. Descriptive statistics (e.g., mean, SD, median, range) were calculated for continuous safety variables. Frequency counts and percentages were tabulated for categorical safety variables.
Patient-reported outcomes: Analysis of PRO data was performed on the ITT analysis set. For patients with multiple records at the same visit, the closest one to the visit date was selected as the scheduled assessment. Completion rates of the EORTC QLQ-C30, the EORTC QLQ-MY20, and the EQ-5D-5L at each time point were generated based on the actual number of PRO assessments received over the number of expected assessments. The PRO end points are secondary and not part of the statistical hierarchy. Type I error control was not applied to the PRO data. Summary descriptive statistics (n, mean, SD, median, and range) at each time point and the change from baseline were summarized by treatment group.
Details of statistical analyses of efficacy and safety end points for patients in the CEPHEUS trial are provided in Appendix 3 in the Supplemental Material document.
Multiple testing procedure: The primary hypothesis was tested at the 0.05 significance level (overall). If the primary end point of overall MRD negativity (≥ CR) rate was statistically significant, the key secondary end points (i.e., CR or better rate, PFS, and durable MRD negativity [≥ CR] rate) would be sequentially tested, each with an overall 2-sided alpha of 0.05, by using a hierarchical testing approach as proposed by Tang and Geller (1999)46 that controls family-wise type I error rate. Because of the short follow-up time at the primary MRD data cut-off, PFS and durable MRD negativity (≥ CR) data were premature, and the hierarchical test was performed on them starting at the interim PFS data cut-off (██ events occurred). The significance level at each data cut-off was determined by the alpha spending function specific to end points:
For the CR or better rate, the information fraction was expected to be 80% at the primary MRD data cut-off. The O’Brien-Fleming alpha spending function, as implemented by the Lan-DeMets method, was used for alpha spending: ██████ (2-sided) at the primary MRD data cut-off and ██████ (2-sided) at the interim PFS data cut-off.
For PFS, the exact significance level at the interim analysis and final PFS analysis was to be determined by the observed number of events per the O’Brien-Fleming alpha spending function. Assuming ██ PFS events are observed at the interim analysis, the alpha to be spent was ██████ (2-sided) for the interim analysis and ██████ (2-sided) for the final PFS analysis (162 PFS events occur).
To control the overall type I error rate, the inverse normal test with the same fixed weights (i.e., information fractions of interim and final analyses) as originally planned was used to combine the log-rank statistics before and after the interim analysis.
For durable MRD negativity (≥ CR) rate, the information fraction was expected to be 80% at the interim PFS data cut-off. The O’Brien-Fleming alpha spending function, as implemented by the Lan-DeMets method, was used for alpha spending: ██████ (2-sided) at the interim PFS data cut-off and ██████ (2-sided) at the final PFS cut-off.
If the null hypothesis for any of these end points failed to be rejected at the interim analysis, then any subsequent end point(s) listed in the statistical hierarchy were not to be tested until the next analysis time point (e.g., final PFS analysis), if applicable. If the null hypothesis for any end point was rejected at any interim analysis, it remained rejected and was not retested at any subsequent time points. The PRO end points are secondary and not part of the statistical hierarchy. Type I error control was not applied to PRO data.
The following prespecified subgroup analyses were performed for the efficacy and/or safety end points: sex, age, baseline renal function, region, weight, baseline Eastern Cooperative Oncology Group Performance Status (ECOG PS), baseline ISS, and cytogenetic risk.
Analysis sets: Efficacy analyses were performed in the ITT analysis set, which included all randomized patients, analyzed according to their randomized treatment groups. Safety analyses were performed in the safety analysis set, defined as all randomized patients who received at least 1 dose of study drug (partial or complete) in the study.
Patient Disposition
Patient disposition for the CEPHEUS trial is summarized in Appendix 4 in the Supplemental Material document.
Of the 395 randomized participants, 392 participants were treated (197 participants with D-VRd and 195 participants with VRd). Three patients were randomized to the VRd group but were not treated (1 patient each because of an AE, loss to follow-up, and withdrawal by the patient).
In the 197 patients who were allocated to D-VRd treatment, all patients (100%) received treatment. In total, 95 patients (48.2%) discontinued the treatment; common reasons for treatment discontinuation included PD (13.7%) and AEs (8.1%). A total of 198 patients were allocated to VRd treatment, with 3 patients (1.5%) not receiving treatment. The remaining 195 patients (98.5%) received VRd. In total, 128 patients (65.6%) discontinued treatment; common reasons for study discontinuation included PD (26.2%) and AEs (16.4%).
Sixty-two patients (31.5%) and 69 patients (35.4%) in the D-VRd and VRd groups, respectively, discontinued the study. The most common reason for discontinuation of the study was death (50 patients [25.4%] in the D-VRd group and 54 patients [27.7%] in the VRd group).
Baseline Characteristics
In the study population, the median age was 70.0 years (range, 31 years to 80 years). Approximately half of the patients were female (49.9%) and half were male (50.1%). The proportions of patients with ECOG PS scores of 0, 1, and 2 were 39.2%, 51.4%, and 9.4%, respectively.
The median time from initial diagnosis of MM to randomization was 1.18 months. Of the 395 patients who had evaluable baseline cytogenetic data reported, 52 patients (13.2%) had a high-risk cytogenetic abnormality, 298 patients (75.4%) had a standard-risk cytogenetic abnormality, and this information was unevaluable or missing for 45 patients (11.4%). Overall, 34.4% of patients were reported as ISS stage I, 37.5% as stage II, and 28.1% as stage III. The proportions of patients aged younger than 70 years with MM who were ineligible for transplant, aged younger than 70 years who did not choose transplant, or aged 70 years or older were 17.7%, 26.8%, and 55.4%, respectively.
Additional patient baseline characteristics from the CEPHEUS trial are presented in Appendix 4 in the Supplemental Material document.
Table 3: Summary of Patient Baseline Characteristics From the CEPHEUS Trial (ITT Analysis Set; Final PFS Analysis, May 7, 2024)
Characteristic | The CEPHEUS trial | ||
|---|---|---|---|
D-VRd (N = 197) | VRd (N = 198) | Total (N = 395) | |
Age (years) | |||
N | 197 | 198 | 395 |
Mean (SD) | 69.0 (7.01) | 68.9 (7.31) | 69.0 (7.15) |
Median | 70.0 | 70.0 | 70.0 |
Range | (42 to 79) | (31 to 80) | (31 to 80) |
Sex, n (%) | |||
N | 197 | 198 | 395 |
Female | 110 (55.8) | 87 (43.9) | 197 (49.9) |
Male | 87 (44.2) | 111 (56.1) | 198 (50.1) |
Race, n (%) | |||
N | 197 | 198 | 395 |
Asian | 11 (5.6) | 14 (7.1) | 25 (6.3) |
Black or African American | 10 (5.1) | 9 (4.5) | 19 (4.8) |
Native Hawaiian or Other Pacific Islander | 0 | 1 (0.5) | 1 (0.3) |
White | 162 (82.2) | 156 (78.8) | 318 (80.5) |
Not reported | 13 (6.6) | 16 (8.1) | 29 (7.3) |
Other | 1 (0.5) | 2 (1.0) | 3 (0.8) |
Baseline ECOG PS score, n (%) | |||
N | 197 | 198 | 395 |
0 | 71 (36.0) | 84 (42.4) | 155 (39.2) |
1 | 103 (52.3) | 100 (50.5) | 203 (51.4) |
2 | 23 (11.7) | 14 (7.1) | 37 (9.4) |
ISS stage,a n (%) | |||
N | 197 | 198 | 395 |
I | 68 (34.5%) | 68 (34.3%) | 136 (34.4%) |
II | 73 (37.1%) | 75 (37.9%) | 148 (37.5%) |
III | 56 (28.4%) | 55 (27.8%) | 111 (28.1%) |
Time since initial MM diagnosis (months) | |||
N | 197 | 198 | 395 |
Mean (SD) | 1.49 (1.028) | 1.60 (1.100) | 1.54 (1.065) |
Median | 1.15 | 1.26 | 1.18 |
Range | (0.4 to 5.8) | (0.3 to 8.0) | (0.3 to 8.0) |
Cytogenetic risk, n (%) | |||
N | 197 | 198 | 395 |
Standard riskb | 149 (75.6) | 149 (75.3) | 298 (75.4) |
High riskb | 25 (12.7) | 27 (13.6) | 52 (13.2) |
del(17p) | 12 (6.1) | 14 (7.1) | 26 (6.6) |
t(4;14) | 12 (6.1) | 13 (6.6) | 25 (6.3) |
t(14;16) | 4 (2.0) | 2 (1.0) | 6 (1.5) |
Unevaluable or missing | 23 (11.7) | 22 (11.1) | 45 (11.4) |
Transplant eligibility, n (%) | |||
N | 197 | 198 | 395 |
Transplant ineligible | 144 (73.1) | 145 (73.2) | 289 (73.2) |
Transplant deferredc | 53 (26.9) | 53 (26.8) | 106 (26.8) |
BSA = body surface area; del = deletion; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FISH = fluorescence in situ hybridization; ISS = International Staging System; ITT = intention to treat; MM = multiple myeloma; p = p-arm; PFS = progression-free survival; SD = standard deviation; t = translocation; VRd = bortezomib, lenalidomide, and dexamethasone.
Notes: Percentages are calculated with the number of patients in each cohort with available data as the denominator.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aISS stage is derived based on the combination of serum beta2-microglobulin and albumin.
bStandard risk includes patients who tested negative for del(17p), t(14;16), or t(4;14) by FISH. High risk includes patients who tested positive for any of del(17p), t(14;16), or t(4;14) by FISH.
cTransplant not planned as initial therapy.
Sources: Clinical Study Report for CEPHEUS.3 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure, use of concomitant drugs, and subsequent treatments in the CEPHEUS trial are in Appendix 4 in the Supplemental Material document.
Overall, the median duration of study treatment was 56.28 months (range, 0.1 months to 64.6 months) in the D-VRd group compared to 34.33 months (range, 0.5 months to 63.8 months) in the VRd group. The median number of treatment cycles was 59.0 cycles (range, 1 cycle to 71 cycles) in the D-VRd group compared with 37.0 cycles (range, 1 cycle to 70 cycles) in the VRd group.
Regarding concomitant drugs, at the May 7, 2024, data cut-off, all study participants received concomitant drugs, and the use of concomitant therapies was similar between treatment groups.
As of the May 7, 2024, data cut-off, fewer patients in the D-VRd group (22 patients [11.2%]) received subsequent antimyeloma therapies compared with the VRd group (65 patients [33.3%]). Of the patients receiving subsequent therapies, the 3 most common subsequent antineoplastic drugs were daratumumab (D-VRd: █████; VRd: ███████), bortezomib (D-VRd: ██████; VRd: ███████), and carfilzomib (D-VRd: ██████; VRd: ███████).
Critical Appraisal
Internal Validity
In the CEPHEUS trial, appropriate methods of randomization were employed. An interactive web response system was used in treatment allocation. The randomization was stratified by ISS (stage I, II, or III, assessed by the central laboratory) and age or transplant eligibility. There were some imbalances between the 2 treatment groups in patients’ baseline characteristics, such as type of myeloma by immunofixation, number of lytic bone lesions, percentage of plasma cells, and bone marrow cellularity. According to the clinical experts consulted by CDA-AMC, these imbalances are less likely to have an impact on the treatment effect of the study drug. The CEPHEUS trial is an open-label RCT. This study design may introduce bias because of the awareness of the treatment assignment and may potentially affect the assessment of patient- or clinician-reported outcomes, such as HRQoL or some safety outcomes. It is possible that patients would tend to report more AEs that are known for a particular drug in a regimen. The use of concomitant therapies during the study was generally balanced between the 2 treatment groups, whereas more patients in the VRd group required subsequent antimyeloma therapy compared to those in the D-VRd group. The clinical experts did not express concern about the imbalance between the treatment groups in this regard.
The MRD negativity rate was a primary outcome in the CEPHEUS trial. This is a biomarker that is commonly used for the assessment of treatment response in patients with MM. Although the clinical experts consulted by CDA-AMC agreed that this is a reasonable clinical trial end point in the study population, they noted that MRD testing is not widely adopted by clinicians in practice to monitor disease progression or to determine a patient’s response to treatment. Furthermore, a minimal important difference (MID) for the MRD negativity rate has not been established yet. Studies have demonstrated the correlations between the MRD negativity rate and survival outcomes, especially PFS, in patients with MM.47-49 In 1 meta-analysis evaluating the utility of MRD detection in patients with NDMM, MRD-negative status was associated with significantly better PFS (HR = 0.41; 95% CI, 0.36 to 0.48; P < 0.001) and OS (HR = 0.57; 95% CI, 0.46 to 0.71; P < 0.001) compared to MRD-positive status.47
In the CEPHEUS trial, multiplicity adjustment was performed in the analysis of the primary end point (MRD negativity rate) and the key secondary end points (CR or better rate, PFS, and durable MRD negativity [≥ CR] rate). There was no control for multiplicity for other efficacy or safety outcomes in the study, including OS and HRQoL; thus, all other secondary end points were considered supportive. However, type I error was not of concern given that these additional end points did not reach a conventional level of statistical significance.
In the analysis of PFS, the sponsor’s Statistical Analysis Plan noted that the proportional hazard (PH) assumption of the PFS analysis would be examined graphically. However, it was unclear whether the PH assumption was violated because the results were not provided.
Predefined sensitivity analyses were conducted to adjust for the impact of COVID-19 on the survival data and examine the robustness of the results from the primary analysis. In general, there was no imputation planned for missing efficacy end points. Missing data in the CEPHEUS trial was handled using different approaches. For patients with missing MRD samples, failure to calibrate baseline MRD, or otherwise unevaluable samples, MRD status was considered MRD-positive. Missing or partial dates in AE, concomitant therapies, MM diagnosis date, and onset date of subsequent antimyeloma therapy were imputed. It is unknown what the impact of these missing data would be on the efficacy or safety assessments.
Prespecified subgroup analyses were conducted to examine the treatment effect according to various patient baseline characteristics. Results of these subgroups were generally consistent with the overall direction of the effect of D-VRd compared to VRd; however, some subgroups had small sample sizes, resulting in wide CIs (i.e., patients with high-risk cytogenetic status [PFS: HR = 0.88; 95% CI, 0.47 to 1.84]).
In the CEPHEUS trial, HRQoL was assessed using cancer-specific questionnaires: EORTC QLQ-C30 and EORTC QLQ-MY20. The latter was specifically designed for patients with MM and has been validated in patients with this condition; in addition, an MID used to determine the clinical relevance of the study findings was identified from the literature. On the other hand, at cycle 36 of the CEPHEUS trial, the completion rates of these 2 questionnaires were low and imbalanced in the ITT population (for EORTC QLQ-C30, █████ versus ██████ and for EORTC QLQ-MY20, ████% versus ████%, in the D-VRd group and VRd group, respectively). The analysis methods assumed that data were missing at random, but this assumption may not hold true. As such, the risk of bias because of missing outcomes data and its impact on study findings are uncertain.
External Validity
Based on the feedback from the clinical experts consulted by CDA-AMC, the eligibility criteria in the CEPHEUS trial were standard in clinical trials of MM but stricter than clinical practice. Patients in the real world are likely a few years older than those recruited in the CEPHEUS trial. In clinical practice, patients with poor performance status (e.g., an ECOG PS score of 3) may receive D-VRd. The trial population represented relatively healthier patients (by excluding those patients with a frailty index ≥ 2) and those with a better ECOG PS score of 0 or 1 (by excluding those patients with an ECOG PS score > 2). Therefore, it remains unknown if the efficacy and safety profiles observed in this trial could be extrapolated to patients not studied in this trial.
According to the baseline of patients in the CEPHEUS study, the experts noted that the proportion of patients who deferred transplant (26.8%) was higher than the clinical practice, where the number of patients who need to defer ASCT is very limited. Overall, the findings of the CEPHEUS study can be generalized to patients with NDMM who are not undergoing ASCT as initial therapy.
There were study sites in Canada in the CEPHEUS trial, which may support the generalizability of the study findings to clinical practice in Canada.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
At the primary MRD analysis cut-off, the primary end point (MRD negativity rate at 10–5) and the key secondary efficacy end point (CR or better rate) were formally tested for statistical significance; updated results for these end points are also provided for the final PFS analysis. At the interim PFS analysis cut-off, the other 2 major secondary end points (PFS and sustained MRD negativity rate) were formally tested for statistical significance; updated results of PFS and sustained MRD negativity rate are also provided for the final PFS analysis. The remaining secondary efficacy end points (including OS) are provided for the final PFS analysis. Safety data are presented for the final PFS analysis.
Efficacy
Progression-free survival: This was a key secondary outcome in the CEPHEUS trial.
At the final PFS analysis cut-off date (May 7, 2024), with a median follow-up of 58.71 months, a total of 154 PFS events were observed (D-VRd: 63 events [32.0%]; VRd: 91 events [46.0%]). The addition of daratumumab to VRd resulted in a PFS with a HR (D-VRd versus VRd) of 0.57 (95% CI, 0.41 to 0.79; P = 0.0005). The median PFS was not reached in the D-VRd group and was 52.63 months in the VRd group. Additionally, data from 68.0% of participants in the D-VRd group and from 54.0% of participants in the VRd group were censored at the time of the clinical cut-off of the final PFS analysis. Most of these participants were censored because of the clinical cut-off (D-VRd: 120 of 134 [89.6%]; VRd: 79 of 107 [73.8%]). The 54-month PFS rate was 68.1% (95% CI, 60.8% to 74.3%) in the D-VRd group and 49.5% (95% CI, 41.8% to 56.8%) in the VRd group, with a between-group difference of ████% (95% CI, ████ to ████%).
Prespecified sensitivity and supplementary analyses were consistent with the final PFS result per the computerized algorithm (refer to Appendix 4 in the Supplemental Material document).
At the final PFS analysis, the treatment effect on PFS (D-VRd versus VRd) consistently favoured D-VRd across all prespecified subgroups, including age, ISS stage, cytogenetic risk, and baseline ECOG PS score (refer to Appendix 4 in the Supplemental Material document).
Overall MRD negativity (≥ CR) rate at 10–5: This was a primary efficacy outcome in the CEPHEUS trial.
At the primary MRD analysis cut-off (April 8, 2021) with a median follow-up of █████ months, the overall MRD negativity rate at 10–5 was ████% (95% CI, ████% to ████%) in the D-VRd group and ████% (95% CI, ████% to ████%) in the VRd group. At the final PFS analysis cut-off (May 7, 2024), with a median follow-up of 58.71 months, the overall MRD negativity rate at 10–5 was 60.9% (95% CI, 53.7% to 67.8%) in the D-VRd group and 39.4% (95% CI, 32.5% to 46.6%) in the VRd group; the OR was 2.37 (95% CI, 1.58 to 3.55). The between-group difference in overall MRD negativity rate at 10–5 was ████% (95% CI, ████% to ████%).
The treatment effect on the overall MRD negativity (≥ CR) rate (D-VRd compared with VRd) was generally consistent across the prespecified subgroups.
VGPR or better rate: At the final PFS analysis cut-off (May 7, 2024), with a median follow-up of 58.71 months, the VGPR or better rate was 92.9% (95% CI, 88.4% to 96.1%) in the D-VRd group and 86.9% (95% CI, 81.4% to 91.2%) in the VRd group (OR = 1.97; 95% CI, 0.99 to 3.90; P = 0.0495).
Overall survival: At the final PFS analysis clinical cut-off (May 7, 2024), with a median follow-up of 58.7 months, OS data were not mature, with a total of 111 deaths: 51 of 197 patients (25.9%) in the D-VRd group versus 60 of 198 patients (30.3%) in the VRd group. The median OS was not reached for either treatment group. The HR (D-VRd versus VRd) was 0.85 (95% CI, 0.58 to 1.24). The 54-month OS rate was 74.2% (95% CI, 67.3% to 79.8%) in the D-VRd group and 69.6% (95% CI, 62.5% to 75.7%) in the VRd group, with a between-group difference of ███% (95% CI, ████% to ████%).
Two sensitivity analyses of OS to adjust for the impact of deaths because of COVID-19 were performed and demonstrated an improved HR and its 95% CI, which supported a positive OS trend favouring D-VRd. Censoring for death because of COVID-19 resulted in an HR of 0.69 (95% CI, 0.45 to 1.05).
Health-Related Quality of Life
EORTC QLQ-C30 GHS subscale: Baseline scores on the GHS subscale were comparable between treatment groups. The least squares mean (LSM) of the change from baseline scores showed numerical improvement in GHS from cycle 7 onward, with similar improvements across the 2 treatment groups (in cycle 36, the LSM was 8.2 points [95% CI, ███ ██ ████] in the DVR-d group versus 8.5 points [95% CI ██ ████ P = █████] in the VRd group).
EORTC QLQ-MY20 Disease Symptom Scale: Baseline scores on the EORTC QLQ-MY20 Disease Symptom Scale were comparable across treatment groups. The LSMs of the change from baseline in the Disease Symptom Scale scores showed improvement at every cycle (cycle 2, day 1 through cycle 36) and were numerically similar across treatment groups, with both treatment groups achieving more than a 10-point reduction, indicating an improvement in MM-related symptoms. The largest improvements were observed in cycle 9 (cycle 9, day 1: ██████ █████ ████ ███ █████ ██ ██████ ████ █████ ████ ███ █████ ██ ███████; cycle 36, day 1: ██████ ████ ████ ███ █████ ██ ██████ ████ ██ ████ ███ ███ ██ ██████████).
Harms
Safety was evaluated using the safety analysis set from the CEPHEUS trial, composed of 392 patients who were randomized and received at least 1 dose of study intervention (partial or complete) in the study.
Adverse events: All participants in each treatment group experienced treatment-emergent adverse events (TEAEs) (100% in the D-VRd group and 100% in the VRd group). The incidence of grade 3 or grade 4 TEAEs was higher in the D-VRd group (92.4%) versus in the VRd group (85.6%). The most commonly reported AEs were upper respiratory tract infection (reported by 39.6% of patients in the D-VRd group versus 32.8% of patients in the VRd group), COVID-19 infection (38.1% versus 24.6%, respectively), neutropenia (55.8% versus 39.0%, respectively), thrombocytopenia (46.7% versus 33.8%, respectively), anemia (37.1% versus 31.8%, respectively), peripheral sensory neuropathy (55.8% versus 61.0%, respectively), peripheral edema (42.1% versus 39.0%, respectively), fatigue (32.0% versus 30.8%, respectively), diarrhea (56.9% versus 59.0%, respectively), constipation (38.1% versus 42.1%, respectively), insomnia (32.0% versus 32.3%, respectively), vascular disorders (45.7% versus 37.4%, respectively), and renal and urinary disorders (37.6% versus 30.8%, respectively).
Serious AEs: The incidence of treatment-emergent serious AEs (SAEs) was slightly higher in the D-VRd group (72.1%) compared with the VRd group (67.2%). The treatment-emergent SAEs that occurred with a frequency of 5% or higher in either treatment group were pneumonia (13.7% in the D-VRd group versus 12.8% in the VRd group), COVID-19 (11.2% versus 8.2%, respectively), pulmonary embolism (5.6% versus 2.6%, respectively), and diarrhea (5.1% versus 3.1%, respectively).
Withdrawal because of AEs: The proportion of participants with TEAEs leading to discontinuation of all study treatment was lower in the D-VRd group (7.6%) compared with the VRd group (15.9%). TEAEs leading to discontinuation of all study treatments that occurred in more than 1 participant in either treatment group were pneumonia (2 participants [1.0%] in the D-VRd group versus 0 participants [0%] in the VRd group), peripheral sensory neuropathy (1 participant [0.5%] versus 5 [2.6%], respectively), peripheral sensorimotor neuropathy (0 participants [0%] versus 3 participants [1.5%], respectively), diarrhea (0 participants [0%] versus 3 participants [1.5%], respectively), and rash (0 participants [0%] versus 2 participants [1.0%], respectively). The proportions of participants with TEAEs leading to discontinuation of any component of study treatment (i.e., daratumumab, bortezomib, lenalidomide, or dexamethasone) were similar in the 2 treatment groups (D-VRd: ██████ VRd: ███████).
Deaths: As of the final PFS analysis (May 7, 2024), 25.9% of patients in the D-VRd group and 30.3% of patients in the VRd group of the safety analysis set had died. The primary causes of death were either AEs (37 patients [18.8%] in the D-VRd group versus 25 patients [12.8%] in the VRd group) or disease progression (8 patients [4.1%] in the D-VRd group versus 16 patients [8.2%] in the VRd group).
The overall incidence of grade 5 TEAEs was higher in the D-VRd group (16.8%) compared with the VRd group (10.8%). Of the grade 5 TEAEs (fatal AEs), grade 5 non–COVID-19 TEAEs were reported in 10.7% of participants in the D-VRd group and 7.7% in the VRd group, and grade 5 COVID-19 TEAEs were reported in 6.1% of participants in the D-VRd group and 3.1% of participants in the VRd group.
Notable Harms
Cytopenia: Treatment-emergent cytopenia (consisting of neutropenia, anemia, thrombocytopenia, and lymphopenia group terms) of any grade was reported in a higher proportion of participants in the D-VRd group (79.2%) compared with the VRd group (63.6%).
Infections and infestations: The proportions of patients with infections and infestations — including overall (91.9% in the D-VRd group versus 85.6% in the VRd group), grade 3 or grade 4 (40.1% versus 31.8%, respectively), and SAEs (39.6% versus 35.4%, respectively) — were all higher in the D-VRd group compared with the VRd group.
The most common grade 3 or grade 4 infections and infestations (frequency ≥ 5% in either treatment group) were pneumonia (14.2% in the D-VRd group versus 12.8% in the VRd group) and COVID-19 (11.2% in the D-VRd group versus 4.6% in the VRd group). The proportions of participants with TEAEs of infections and infestations leading to discontinuation of all study treatment were low and similar in both groups (2.0% in the D-VRd group versus 1.0% in the VRd group).
Summary of Findings and Certainty of the Evidence
Literature-based MID estimates were used as the thresholds for the EORTC QLQ-MY20 (MID: 10 points for the Disease Symptom Scale; details are provided in the Summary of Outcome Measures in Appendix 3 in the Supplemental Material document).50 Note that this MID value was derived from a mixed population of patients, including those with NDMM and relapsed or refractory MM. In the absence of literature-based MID estimates, thresholds suggested by the clinical experts were used for the following outcomes: risk of SAE (threshold of 10%), risk of cytopenia (threshold of 10%), and risk of infection or infestation (threshold of 10%). In the absence of a known threshold, the certainty in the presence of a nonnull effect was rated for the probability of PFS at 54 months, the probability of OS at 54 months, the proportion of patients who achieved MRD negativity, and the mean change from baseline in the EORTC QLQ-C30 GHS scale. For these outcomes, even though the clinical experts were unable to suggest a specific threshold for a clinically important effect, with their input, the review team judged whether the point estimates and relevant bounds of the 95% CIs represented clinically important effects.
Table 4: Summary of Findings for D-VRd vs. VRd for Patients With NDMM Who Are Not Undergoing ASCT
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
VRd | D-VRd | Difference | |||||
Survival | |||||||
Probability of PFS at 54 months Median follow-up: 58.71 months | 395 (1 RCT) | NA | 495 per 1,000 | 681 per 1,000 (608 per 1,000 to 743 per 1,000) | 185 more per 1,000 (██ █ per 1,000)a | High | D-VRd results in an increase in the probability of PFS compared to VRd. |
Probability of OS at 54 months Median follow-up: 58.71 months | 395 (1 RCT) | NA | 696 per 1,000 | 742 per 1,000 (██ █ per 1,000) | ██ more per 1,000 (██ █ per 1,000)a | Moderateb | D-VRd likely results in an increase in the probability of OS compared to VRd. The clinical importance of the increase is uncertain. |
Response rates | |||||||
Proportion of patients with MRD negativity (≥ CR) status at 10–5 Median follow-up: 58.71 months | 395 (1 RCT) | OR = 2.37 (1.58 to 3.55) | 394 per 1,000 | 609 per 1,000 (██ █ per 1,000) | 215 more per 1,000 (██ █ more per 1,000)a | High | D-VRd results in an increase in the proportion of patients with MRD negativity (≥ CR) status ≥ 10–5 when compared with VRd. |
Proportion of patients with VGPR Median follow-up: 58.71 months | 395 (1 RCT) | OR = █ █ to██ ███) | ███ per 1,000 | ███ per 1,000 (███ █ per 1,000) | ██ more per 1,000 (██ █ more per 1,000)a | Moderateb | D-VRd likely results in an increase in the proportion of patients with VGPR when compared to VRd. The clinical importance of the increase is uncertain. |
Health-related quality of life | |||||||
EORTC QLQ-C30 GHS and QoL domain, LS mean change from baseline (0 [worst] to 100 [best]) Follow-up: ████ █ | 395 (1 RCT) | NA | █████ | █████ | █████ | Lowc | D-VRd may result in little to no difference in HRQoL at cycle 36 compared to VRd. |
EORTC QLQ-MY20 Disease Symptom Scale, LS mean change from baseline (higher score = worse symptoms) Follow-up: ████ █ | 395 (1 RCT) | NA | █████ | █████ | █████ | Lowc | D-VRd may result in little to no difference in HRQoL at cycle 36 compared to VRd. |
Harms | |||||||
Proportion of patients with ≥ 1 SAE Median follow-up: 58.71 months | 395 (1 RCT) | NA | 672 per 1,000 | 721 per 1,000 (95% CI NR) | ██ more per 1,000 (██ █ more per 1,000)a | Moderateb | D-VRd likely results in an increase in the proportion of patients with 1 or more SAE compared to VRd. The clinical importance of the increase is uncertain. |
Proportion of patients with cytopenia Median follow-up: 58.71 months | 395 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (95% CI NR) | ███ more per 1,000 (██ █ more per 1,000)a | High | D-VRd results in an increase in the proportion of patients with 1 or more cytopenia compared to VRd. The clinical importance of the increase is uncertain. |
Proportion of patients with infections and infestations Median follow-up: 58.71 months | 395 (1 RCT) | NA | ███ per 1,000 | ███ per 1,000 (95% CI NR) | ██ more per 1,000 (██ █ more per 1,000)a | Moderateb | D-VRd likely results in an increase in the proportion of patients with 1 or more infection or infestation compared to VRd. The clinical importance of the increase is uncertain. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; CR = complete response; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-MY20 = European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Multiple Myeloma Module 20; GHS = Global Health Status; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HRQoL = health-related quality of life; LS = least square; MRD = minimal residual disease; NA = not applicable; NR = not reported; OR = odds ratio; OS = overall survival; PFS = progression-free survival; QoL = quality of life; RCT = randomized controlled trial; SAE = serious adverse event; VGPR = very good partial response; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThis analysis was not part of the Statistical Analysis Plan and was requested from the sponsor by CDA-AMC to facilitate the GRADE assessment.
bRated down 1 level for serious imprecision: no known MID so the target of certainty appraisal was any effect; the 95% CI for the difference between groups includes the possibility of no difference.
cRated down 2 levels for serious imprecision and serious risk of bias: the 95% CI for the difference between groups includes the possibility of no difference; in addition, there were large amount of missing HRQoL data at cycle 36.
Sources: Clinical study report for the CEPHEUS study3 and additional information provided by the sponsor.51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
At the time of this submission, there was no direct evidence comparing D-VRd against other relevant comparators (except for VRd in the CEPHEUS trial) used in the treatment of adult patients with NDMM who were not undergoing ASCT as initial therapy. The sponsor submitted 3 ITC reports, including 2 inverse probability of treatment weighting (IPTW)4,5 analyses and 1 matching-adjusted indirect comparison (MAIC),6 to indirectly compare the treatment effect of D-VRd to those of other active treatments in the target population.
The objective of this section is to summarize and critically appraise the sponsor-submitted ITCs, and to inform the pharmacoeconomic model.
Description of ITCs
The objectives of the submitted ITC reports were to derive estimates of relative efficacy of D-VRd versus existing therapies for patients with NDMM who are not undergoing ASCT as initial therapy, using various statistical methods. The IPTW analyses assessed the efficacy of D-VRd versus DRd, Rd, and DVMp, whereas the MAIC analysis assessed the efficacy of D-VRd versus Isa-VRd.
Study Selection and Review Methods
The CEPHEUS trial is the key data source of the ITCs. All other studies considered for use in ITCs were identified via an internally sponsored systematic literature review (SLR) of clinical trials, as well as a previously published SLR on RCTs including patients with NDMM who are ineligible for transplant.52 Studies in the internally sponsored SLR were screened and selected based on criteria for population, intervention, comparison, outcomes, and study design presented in Table 15 in Appendix 6 in the Supplemental Material document. Treatments of interest included D-VRd and any licensed treatments, treatments used in routine care, or treatments under investigation provided as a single drug or a combination treatment. Multiple databases and trial registries were searched for potentially relevant articles. The ITC reports did not specify the time frame of the literature search.
Two reviewers independently conducted study selections. A third reviewer was involved in resolving any discrepancies between the first 2 reviewers during the process. Data extraction was conducted by 1 reviewer and verified by a second reviewer. The quality of each included primary, full-text RCT publication was assessed using the National Institute for Health and Care Excellence risk of bias criteria. A risk of bias assessment was completed for the CEPHEUS study using the Risk of Bias In Nonrandomized Studies of Interventions (ROBINS-I) assessment.53
A summary of the study selection criteria and methodology used to conduct the SLR contributing to the ITC is described in Table 15 in Appendix 6 in the Supplemental Material document.
ITC Analysis Methods
Feasibility assessments were performed to compare the study, population, and outcome characteristics of the relevant RCTs and informed the selection of the most appropriate ITC methods, such as network meta-analysis, population-adjusted indirect comparisons (e.g., MAIC), or other adjusted treatment comparisons (e.g., propensity score methods). Based on results of feasibility assessments, 2 different ITC methods were deemed most appropriate by the sponsor for deriving robust comparative efficacy estimates of D-VRd versus relevant comparators based on data availability, whether it is possible to form a connected network, and the existence of different distributions of treatment effect modifiers and prognostic variables. These methods include IPTW and MAIC.
These methods were performed using individual patient data (IPD) from the CEPHEUS study and either IPD (in IPTW analyses) or summary-level data (in MAIC analyses) from the comparator trials.
Additional information on the analysis methods for the ITCs is presented in Appendix 6 in the Supplemental Material document.
Inverse Probability of Treatment Weighting
D-VRd (the CEPHEUS study) versus DRd (the MAIA study): A feasibility assessment5,54 was conducted to determine if an ITC was suitable and which method was most appropriate to generate comparative efficacy estimates for D-VRd versus DRd, leveraging IPD from the CEPHEUS3 and MAIA trials.5 Trials were evaluated based on study design, treatment characteristics, inclusion and exclusion criteria, baseline patient characteristics, and outcomes (definitions, assessment criteria and frequency, availability, and maturity of data). Heterogeneity existed between these 2 trials in eligibility criteria and patient demographic or disease characteristics at baseline, whereas the definitions of OS, PFS, MRD negativity (≥ CR) rate and response rates, and follow-up durations used in the MAIA study were aligned with the CEPHEUS study. Selection of baseline covariates used for the ITC was informed by literature reviews and an empirical assessment of the role of baseline characteristics as prognostic variables in 3 clinical trials (the CEPHEUS, MAIA, and ALCYONE studies). Initially, age, ECOG PS, ISS, type of myeloma, cytogenetic risk, hemoglobin, albumin, LDH, AST, serum calcium, frailty, extramedullary disease (EMD), sex, renal function, and liver function were identified as prognostic for PFS and OS. The final selection of baseline covariates used for adjustment in the ITC was determined by clinicians consulted by the sponsor, who reviewed the results of the 2 literature reviews and the regression analyses. Based on their input, a base-case covariate set that included the baseline patient characteristics identified as the most important prognostic variables and/or treatment effect modifiers was used for the primary analyses. To account for differences in trial populations, IPTW using average treatment effect weights derived from propensity scores was applied. In addition, to assess the validity of the PHs assumption, a multistep approach was followed.54 To measure the estimate use and develop an interpretation of the comparison between the 2 trials, HRs were reported for time-to-event outcomes (i.e., PFS, OS, second PFS, and COVID-19–adjusted OS), whereas ORs were calculated for binary outcomes (i.e., overall response rate, ≥ CR rate, stringent CR rate, ≥ VGPR rate, and MRD [≥ CR]).
Sensitivity analyses to evaluate the impact of alternative statistical methods and variable adjustment were conducted using multivariate regression and doubly robust methods.
D-VRd (the CEPHEUS study) versus Rd (the MAIA study): The ITC of D-VRd versus Rd followed the same methods used for the ITC of D-VRd versus DRd.5 Results of this comparison are not presented in this review because the Rd regimen is rarely used in clinical practice, according to the clinical experts consulted by CDA-AMC.
D-VRd (the CEPHEUS study) versus DVMp (the ALCYONE study): A feasibility assessment4,54 was conducted to determine if an ITC was suitable and what method was most appropriate to generate comparative efficacy estimates for D-VRd versus DVMp, leveraging IPD from the CEPHEUS3 and ALCYONE4 trials. The CEPHEUS and ALCYONE trials had overlapping but distinct eligibility criteria; therefore, heterogeneity existed between the 2 studies in terms of patients’ demographic and disease characteristics at baseline. The definitions of PFS, OS, and MRD negativity status used in the ALCYONE study were aligned with the CEPHEUS study. With similar follow-up durations, both trials provide comparable long-term data for assessing efficacy and safety outcomes. The method for selecting baseline covariates used for adjustment in the ITC aligns with the approach described in the comparison between the CEPHEUS and MAIA studies presented earlier in this section, and the final list of covariates is presented in Table 17 in Appendix 6 in the Supplemental Material document. Furthermore, similar to the IPTW conducted for the CEPHEUS and MAIA studies, an IPTW with average treatment effects in the treated patients derived from propensity scores was performed for the CEPHEUS and ALCYONE studies with the purpose of balancing the patient characteristics at baseline in both groups.
Note that the population in the CEPHEUS study was first restricted to only patients with NDMM who are ineligible for transplant, excluding those who were eligible for transplant (but for whom transplant was not planned as initial therapy), to ensure comparability with the population in the ALCYONE study. The comparison was considered unanchored because there is no common comparator that serves as a connecting bridge between the treatments of interest.
In the base-case analysis, variables including ISS stage, cytogenetic risk, age, ECOG PS, type of MM, EMD, frailty (based on simplified frailty score), sex, estimated glomerular filtration rate (eGFR), anemia, and LDH greater than 280 U/L were adjusted across trials. After adjusting, baseline characteristics showed standardized mean differences (SMDs) less than 0.1. The full model included all available adjustment variables, including those from the base-case analysis plus calcium, race, and time of diagnosis, whereas the unadjusted analysis did not include any adjustments. The adequacy of the weighting procedure was evaluated through balance diagnostics, which were conducted on the weighted sample using SMDs, in which values less than 0.1 were considered to indicate sufficient balance achievement.
Similar to the preceding section, to measure the treatment effects and develop an interpretation of the comparison between the 2 trials, HRs were reported for time-to-event outcomes (i.e., PFS, OS, and COVID-19–adjusted OS), whereas ORs were calculated for binary outcomes (i.e., overall response rate, ≥ CR rate, stringent CR rate, ≥ VGPR rate, and MRD [≥ CR]).
Sensitivity analyses evaluating the impact of alternative statistical methods and variable adjustment were conducted using multivariate regression and doubly robust methods, as described for the IPTW conducted for the CEPHEUS and MAIA studies.4
Matching-Adjusted Indirect Comparison
D-VRd (the CEPHEUS study) versus Isa-VRd (the IMROZ study): A feasibility assessment6,54 was conducted to determine if an ITC was suitable and what method was most appropriate to generate comparative efficacy estimates for D-VRd versus Isa-VRd, leveraging IPD from the CEPHEUS trial3 and published summary-level data from the IMROZ trial.6 The CEPHEUS and IMROZ trials were evaluated based on study design, treatment characteristics, inclusion and exclusion criteria, baseline patient characteristics, and outcomes (definitions, assessment criteria, and the frequency, availability, and maturity of data).6 Heterogeneity existed between the 2 trials in their eligibility criteria and patients’ demographic and disease characteristics at baseline, whereas the outcome definitions for PFS, OS, and MRD negativity rate were comparable. An anchored MAIC is deemed appropriate for comparing D-VRd from the CEPHEUS trial with Isa-VRd from the IMROZ trial (VRd was the common comparator for both studies). This MAIC between D-VRd and Isa-VRd in patients with NDMM who are ineligible for transplant investigated multiple end points, including PFS and OS (through HRs), and response outcomes (including MRD negativity [≥ CR] rate and ≥ CR rate, through ORs).
The method is considered anchored when both trials share a common control group that serves as a connecting bridge between the treatments of interest. The base-case model included revised ISS (R-ISS) stage, cytogenetic risk, age, ECOG PS, and frailty status. Additional variables including myeloma type, EMD, sex, and renal function were incorporated into the full model for sensitivity analyses. The final selection of covariates is presented in Table 17 in Appendix 6 in the Supplemental Material document. As reported in the preceding section, to assess the validity of the PHA, a multistep approach was followed.54
A sensitivity analysis was conducted by applying a closer match to the IMROZ study inclusion criteria to the population in the CEPHEUS trial. The population in the CEPHEUS study was restricted to include only patients who were either aged65 years or older (and therefore considered ineligible for transplant) or patients aged younger than 65 years who were determined to be ineligible for transplant based on their age or transplant ineligibility randomization stratification factor.
Summary of Included Studies
In total, 4 RCTs were included in the ITC analyses: the CEPHEUS, MAIA, ALCYONE, and IMROZ studies, in which D-VRd was compared to DRd, Rd, DVMp, and Isa-VRd. The IPTW approach was used in the comparisons between the CEPHEUS, MAIA, and ALCYONE studies, while MAIC analysis was used in the comparison between the CEPHEUS and IMROZ studies. Details in the comparisons between studies in the trial characteristics and patient characteristics are provided in Appendix 6 in the Supplemental Material document.
Assessment of Homogeneity
There was overlap between trials in their eligibility criteria, such as age and performance status; however, discrepancy in some trial and patient characteristics were found. The CEPHEUS trial enrolled patients who were ineligible for transplant , as well as patients for whom transplant was not planned as initial therapy; however, the MAIA, ALCYONE, and IMROZ studies only enrolled patients who were ineligible for transplant. Patients in all 4 trials were required to have an ECOG PS score of 2 or lower, whereas the CEPHEUS trial additionally excluded patients with a frailty score of 2 or higher. Definitions of PFS, OS, and MRD negativity rate aligned with each other. Dose and treatment regimens in the comparator trials were not consistent across the trials. The median follow-up was 58.7 months in the CEPHEUS trial, 40.1 months in the ALCYONE trial, 64.5 months in the MAIA trial, and 59.7 months in the IMROZ trial.
Sources of similarities and differences across the 4 RCTs are summarized in Table 18 in Appendix 6 in the Supplemental Material document.
Effect Modifiers
The authors of the ITC report identified several potential important effect modifiers for PFS and OS for patients with NDMM: ISS stage, cytogenetic risk, age, ECOG PS score, MM subtype, EMD, frailty, sex, renal function, anemia, and LDH levels. These factors were identified through published subgroup data of RCTs conducted in the NDMM setting. Regression analyses assessing prognostic association of baseline covariates with PFS and OS were performed using pooled individual-level patient data from the CEPHEUS, MAIA, and ALCYONE studies. Final selection of baseline covariates used for adjustment in the ITC was determined by clinicians with expertise in MM who were consulted by the sponsor. Based on their input, a base-case covariate set that included the baseline patient characteristics identified as the most important prognostic variables and/or treatment effect modifiers was used for the primary analyses. Sensitivity analyses that adjusted for additional baseline covariates were also performed.
Selected covariates used for adjustment in the ITCs are summarized in Table 17 in Appendix 6 in the Supplemental Material document.
Critical Appraisal of ITCs
Research protocols for the ITC analyses were provided by the sponsor. In these ITC reports, relevant studies were identified by searching multiple databases based on prespecified inclusion and exclusion criteria. Appropriate approaches were also used to reduce the risk of bias and error in data extraction. The risk of bias of the included studies was assessed using the ROBINS-I tool. All included trials were considered to not have major issues in the risk of bias based on the ROBINS-I tool. However, this is a tool for nonrandomized observational studies, of which at least 3 of 7 domains (e.g., confounding, misclassification of intervention, or differential selection of participants into the study) are not applicable to randomized controlled studies. Moreover, those issues specifically related to RCTs including, for example, blinding, allocation concealment, and deviation from intended treatments were not assessed or not assessed appropriately. The results of the risk of bias assessment were not used in the ITC, such as a sensitivity analysis (i.e., excluding trial with a high risk of bias), to examine the robustness of study findings based on the quality of the clinical evidence.
Feasibility assessments were conducted by the sponsor to explore the appropriateness of different indirect analysis methods. Based on the reported information, analysis methods of the ITC (such as statistical method selection, model fit assessment, or assessment of homogeneity) were considered appropriate.
Age, ECOG PS score, ISS, type of myeloma, cytogenetic risk, hemoglobin, albumin, LDH, serum calcium, frailty, EMD, sex, renal function, and race were identified by the sponsor as important effect modifiers for the treatments in the study population. These variables were identified through a literature search, previous subgroup analyses, and through communications with the clinicians consulted by the sponsor. According to the clinical experts consulted by CDA-AMC, all relevant effect modifiers have been included in the ITC analyses, which helps adequately address the issues of heterogeneity.
The ITC reports noted clinical and methodological heterogeneity between the CEPHEUS study and the comparator trials. All trials were phase III, open-label RCTs. PFS, OS, and MRD negativity rate were consistently defined in all the included trials. In terms of patients’ baseline characteristics, there were variations in age, type of MM, ECOG PS score, or race.
The eligibility criteria of patient enrolment in these trials were largely similar, although some differences were noted. For example, the CEPHEUS trial enrolled patients with NDMM who were not undergoing ASCT, including those who were ineligible for transplant, as well as those who preferred not having ASCT as initial therapy; however, the MAIA, ALCYONE, and IMROZ trials all enrolled only patients who were ineligible for transplant. The median follow-up times were different between the trials; therefore, the comparisons of treatment outcomes were based on different time points of the trials. These heterogeneities between the trials included in the ITC could compromise the validity of the ITC results.
In the 2 IPTW analyses, after matching, the effective sample size (ESS) was ██% of the original sample size in the ALCYONE study for DVMp, ██% of the original sample size in the MAIA study for DRd, and ███ ██ ██% of the original sample size in the CEPHEUS study for D-VRd. In the MAIC analysis, the sponsor noted that the retention of the ESS was ██ ██% of the original sample size in IMROZ, indicating successful matching and that the key prognostic factors and effect modifiers were well balanced in these comparisons. Even though the matching process was generally successful, some residual imbalances remained for variables such as age, anemia, or type of MM, which could have an impact on result interpretation. Furthermore, the potential for unidentified or unmeasured confounding factors remains a concern, and the extent of their impact cannot be determined.
In addition to base-case analyses, sensitivity analyses that adjusted for additional baseline patient characteristics were performed to examine the robustness of the findings.
In these sponsor-submitted ITC reports, clinical outcomes that are important to patients and clinicians, such as HRQoL and safety outcomes, were not assessed. Therefore, the effect of D-VRd relative to other active treatments for patients with NDMM who are not undergoing ASCT as initial therapy was unknown.
Efficacy Results of ITCs
Key results of the ITCs are presented in Table 5.
IPTW: D-VRd Versus DRd
For the comparison between D-VRd and DRd, after base-case IPTW, the ESS decreased from 321 to ██████ for DRd and from 144 to ██████ for D-VRd. All SMDs were reduced to less than 0.1 in the base-case model, with the largest remaining imbalances observed in the variables of being aged 75 years or older (−0.03), having the immunoglobulin G subtype of MM (−0.03), and having hemoglobin levels lower than 100 mg/L (−0.03). In the sensitivity model (i.e., full model), the ESS for D-VRd was estimated at 124.35 patients. The sample size was not normally distributed, and more than 25% were weighted greater than 2. Based on the weighted and unweighted ESS, the median weight was estimated at 1.54 for the base-case model.
Progression-free survival: In the base-case analysis, after incorporating adjustments for the type of MM, EMD, frailty, sex, eGFR at baseline, anemia status, and LDH categories, treatment with D-VRd was statistically favoured versus treatment with DRd for PFS (HR = 0.62; 95% CI, 0.44 to 0.88).
In the full model that included all available adjustment variables, including calcium, race, and time of diagnosis, treatment with D‑VRd was statistically favoured versus treatment with DRd for PFS (HR = 0.62; 95% CI, 0.43 to 0.88). Doubly robust and multivariate regression analysis showed similar results across all adjustment sets.
The KM curves demonstrated overlapping survival probabilities throughout the follow-up period, with separation becoming apparent after approximately 30 months.
Overall survival: In the base-case analysis, after incorporating adjustments for the type of MM, EMD, frailty, sex, eGFR status, anemia status, and LDH status, treatment with D-VRd was not statistically significantly different from treatment with DRd in terms of OS (HR = 0.80; 95% CI, 0.53 to 1.19).
In the full model that included all available adjustment variables, including calcium, race, and time of diagnosis, treatment with D‑VRd was not statistically significantly different from treatment with DRd in terms of OS (HR = 0.81; 95% CI, 0.54 to1.22). Doubly robust and multivariate regression analysis showed similar results across all adjustment sets.
The KM curves demonstrated overlapping survival probabilities throughout the follow-up period. The log-cumulative hazards and complimentary log-log (cloglog) plots showed that the PH assumption was met across different scenarios.
MRD negativity (≥ CR) rate: In the base-case analysis, after incorporating adjustments for the type of MM, EMD, frailty, sex, eGFR, anemia status, and LDH status, treatment with D-VRd was statistically favoured versus treatment with DRd in terms of MRD negativity rate (HR = 3.04; 95% CI, 2.01 to 4.61).
In the full model that included all available adjustment variables including calcium, race, and time of diagnosis, treatment with D-VRd was statistically favoured versus treatment with DRd in terms of MRD negativity rate (OR = 3.07; 95% CI, 2.01 to 4.69). Doubly robust and multivariate regression analysis showed similar results across all adjustment sets.
IPTW: D-VRd Versus DVMp
For the comparison between D-VRd and DVMp, after base-case IPTW-adjustment, the ESS decreased from 322 to ██████ for DVMp and from 144 to █████ for D-VRd. All SMDs were reduced to less than 0.1 in the base-case model, with the largest remaining imbalances observed in patients aged 70 to 75 years (0.05) and eGFR (0.06). The sample size was not log-normally distributed, with most of the distribution being below 2 (75% quantile being 1.58 for the base-case model and 1.54 for the full model). Based on the weighted and unweighted ESS, the median weight was estimated at 1.36 for the base-case model and 1.25 in the full model.
Progression-free survival: In the base-case analysis, after incorporating adjustments for LDH, treatment with D-VRd was statistically favoured versus treatment with DVMp for PFS (HR = █████ █████ ████ ██ ████).
In the full model, in which additional matching for sex, eGFR, anemia, LDH level, calcium, ethnicity, and time to diagnosis was performed, treatment with D-VRd was statistically favoured versus treatment with DVMp for PFS (HR = █████ █████ ████ ██ ████).
The KM curves demonstrated nonoverlapping survival probabilities throughout the follow-up period. The log-cumulative hazards and cloglog plots showed that the PH assumption was met across different scenarios. However, the Schoenfeld test showed nonsignificant P values for the base case but statistically significant values for the sensitivity analysis (███████ for base-case and ███████ for sensitivity), suggesting that the PH assumption for the sensitivity analysis was violated.
Overall survival: In the base-case analysis, after incorporating adjustments for LDH levels, treatment with D-VRd was not statistically significantly different from treatment with DVMp in terms of OS (HR = █████ █████ ████ ██ ████).
In the full model that included all available adjusted variables, including calcium, race, and time of diagnosis, treatment with D-VRd was not statistically significantly different from treatment with DVMp in terms of OS (HR = █████ ███ ██ ████ ██ ████). Doubly robust and multivariate regression analysis showed similar results across all adjustment sets. The KM curves demonstrated overlapping survival probabilities throughout the follow-up period. The log-cumulative hazards and cloglog plots showed overlap across different scenarios. The Schoenfeld test also showed significant P values for all the scenarios (0.006483 for base case and 0.00053 for sensitivity).
MRD negativity (≥ CR) rate: In the base-case analysis, after incorporating adjustments through ISS up to frailty, treatment with D-VRd was statistically favoured versus treatment with DVMp in terms of MRD negativity rate (OR = █████ ███ ██ ████ ██ ██████).
In the full model that included all available adjusted variables, including calcium, race, and time of diagnosis, treatment with D-VRd was statistically favoured versus treatment with DVMp in terms of MRD negativity rate (OR = █████ ███ ██ ████ ██ ████). Doubly robust and multivariate regression analysis showed similar results across all adjustment sets.
MAIC: D-VRd Versus Isa-VRd
For the comparison between D-VRd and Isa-VRd, the ESS for D-VRd was estimated at ██████ patients in the base case and ██████ patients in the sensitivity model (i.e., full model). The sample size was not normally distributed and, as expected, only a few patients had an MAIC weight greater than 2, which contributed only marginally to the ESS. Based on the weighted and unweighted ESS, the median weight was estimated at 0.86. The retention of ESS after matching was 84%, indicating successful balancing of patient characteristics while maintaining statistical power.
Progression-free survival: After matching for key baseline characteristics including R-ISS stage, cytogenetic risk, age, ECOG PS score, myeloma type, EMD, frailty status, sex, and eGFR status at baseline, there was no statistically significant difference between treatment with D-VRd or treatment with Isa-VRd in PFS (HR █████ ███ ██ ████ ██ ██████).
In a sensitivity analysis including years since diagnostic and ethnicity in addition to the previous baseline characteristics, treatment with D-VRd was not statistically significantly different from treatment with Isa-VRd in PFS (HR = █████ ███ ██ ████ ██ ████).
Overall survival: After matching for key baseline characteristics including R-ISS stage, cytogenetic risk, age, ECOG PS score, myeloma type, EMD, frailty status, sex, and eGFR status at baseline, there was no statistically significant difference between treatment with D-VRd and treatment with Isa-VRd in OS (HR = █████ █████ ████ ██ ████).
In a sensitivity analysis including years since diagnosis and ethnicity in addition to the previous baseline characteristics, treatment with D-VRd was not statistically significantly different from treatment with Isa-VRd in OS (HR = █████ ███ ██ ████ █ ████).
MRD negativity (≥ CR) rate: In the base-case analysis, after including adjustments for the eGFR, treatment with D-VRd was not statistically significantly different from treatment with Isa-VRd in MRD negativity rate (OR = █████ ███ ██ ████ ██ ████).
In the full model, after incorporating all matching variables including race, treatment with D-VRd was not statistically significantly different from treatment with Isa-VRd in MRD negativity rate (OR = █████ ███ ██ ████ ██ ████).
Table 5: Summary of Key Outcomes Across Adjustment Sets (D-VRd vs. Comparators)
Comparisons | PFS, HR (95% CI) | OS, HR (95% CI) | MRD negativity (≥ CR), OR (95%CI) |
|---|---|---|---|
D-VRd vs. DRd | |||
Unadjusted | 0.66 (0.47 to 0.93) | 0.78 (0.52 to 1.16) | ████████ |
Base case | 0.62 (0.44 to 0.88) | 0.80 (0.53 to 1.19) | ████████ |
Full model | ████████ | ████████ | ████████ |
D-VRd vs. DVMp | |||
Unadjusted | ████████ | ████████ | ████████ |
Base case | ████████ | ████████ | ████████ |
Full model | ████████ | ████████ | ████████ |
D-VRd vs. Isa-VRd | |||
Unadjusted | ████████ | ████████ | ████████ |
Base case | ████████ | ████████ | ████████ |
Full model | ████████ | ████████ | ████████ |
CI = confidence interval; CR = complete response; DRd = daratumumab in combination with lenalidomide and dexamethasone; DVMp = daratumumab in combination with bortezomib, melphalan, and prednisone; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; HR = hazard ratio; IPTW = inverse probability of treatment weighting; Isa-VRd = isatuximab in combination with bortezomib, lenalidomide, and dexamethasone; MRD = minimal residual disease; OR = odds ratio; OS = overall survival; PFS = progression-free survival; Rd = lenalidomide and dexamethasone; VGPR = very good partial response; vs. = versus.
Sources: The IPTW report (the CEPHEUS vs. MAIA trials),5 the IPTW report (the CEPHEUS vs. ALCYONE trials),4 and the MAIC report (the CEPHEUS vs. IMROZ trials).6 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence for adults with NDMM who are not undergoing ASCT as initial therapy were submitted by the sponsor.
Discussion
One pivotal trial (the CEPHEUS study) was included in the sponsor-submitted systematic review. The CEPHEUS trial was a phase III, open-label, active-controlled RCT to evaluate the efficacy and safety of D-VRd (n = 197) compared to VRd (n = 198) in patients with NDMM who were not undergoing ASCT as initial therapy. Study participants were required to have a diagnosis of MM per the IMWG criteria and an ECOG PS score of 0 to 2. Eligible patients were stratified before randomization by ISS stage I, II, or III (based on central laboratory results for beta2-microglobulin and albumin) and age and transplant eligibility (aged younger than 70 years and ineligible for transplant, aged younger than 70 years and chose not to undergo transplant, or aged 70 years or older). In this trial, patients’ demographic characteristics were generally balanced between treatment groups. In the study population, the median age was 70.0 years (range, 31 years to 80 years). Approximately half of the patients were female (49.9%) and half were male (50.1%). The proportions of patients with ECOG PS scores of 0, 1, and 2 were 39.2%, 51.4%, and 9.4%, respectively. The median time from initial diagnosis of MM to randomization was 1.18 months. Of the 395 patients who had evaluable baseline cytogenetic data reported, 52 patients (13.2%) had a high-risk cytogenetic abnormality, 298 patients (75.4%) had a standard-risk cytogenetic abnormality, and this information was unevaluable or missing for 45 patients (11.4%). Overall, 34.4% of patients were reported as ISS stage I, 37.5% as stage II, and 28.1% as stage III. The proportions of patients aged younger than 70 years with MM who were ineligible for transplant, those aged younger than 70 years who chose not to undergo transplant (meaning transplant was not planned as initial therapy), and patients aged 70 years or older were 17.7%, 26.8%, and 55.4%, respectively. The primary objective of the CEPHEUS trial was to determine if the addition of daratumumab to VRd improves overall MRD negativity (≥ CR) compared with VRd alone. Other outcomes included PFS, VGPR or better rate, OS, HRQoL assessments, and safety.
Three ITC reports were submitted by the sponsor to compare the treatment efficacy of D-VRd with other active treatments in patients with NDMM who were not undergoing ASCT as initial therapy. The comparative efficacy of D-VRd versus DRd, DVMp, and Isa-VRd was evaluated based on evidence from 4 phase III RCTs: the CEPHEUS, MAIA, ALCYONE, and IMROZ studies.
Efficacy
In the CEPHEUS trial, overall MRD negativity rate was the primary efficacy outcome. Although this is an outcome commonly used in clinical trials of MM, input from patient groups, clinical experts consulted for this review, and the clinician groups indicated that improved survival is regarded as the most important treatment outcome. However, thresholds of clinically important survival have not yet been established in the target population. Based on the results in the CEPHEUS trial (data cut-off: May 7, 2024; median follow-up of 58.71 months), the median PFS was not reached in the D-VRd group and was 52.6 months in the VRd group. The corresponding HR was 0.57 (95% CI, 0.41 to 0.79; P = 0.0005), which met the prespecified efficacy boundary for a statistically significant PFS benefit for D-VRd according to the multiplicity scheme. The KM curves of PFS showed separation at approximately 30 months, and this was maintained thereafter. At 54 months, the proportions of patients with no disease progression were 68.1% in the D-VRd group and 49.5% in the VRd group. The clinical experts consulted by CDA-AMC confirmed that the difference of 18.5% between D-VRd and VRd was clinically meaningful, and the improvement in PFS benefit in patients treated with D-VRd was promising. Overall, there is high certainty that D-VRd results in a clinically important increase in the probability of PFS when compared with VRd at 54 months. Sensitivity analyses including the impact of COVID-19 were conducted for PFS to assess the robustness of the results, and overall, the results were consistent with the primary analysis. Subgroup analyses of PFS were generally consistent with the primary analysis across nearly all prespecified subgroups, despite the subgroups with wide 95% CIs that included the null; for example, for patients with high-risk cytogenetics, the HR for PFS was 0.88 (95% CI, 0.47 to 1.84). The review team notes that although the subgroup analyses of PFS were prespecified, the CEPHEUS trial was not powered to detect statistically significant differences between the D-VRd and VRd treatment groups; thus, no definitive conclusions can be drawn from the subgroup analyses.
OS is 1 of the important clinical outcomes in cancer treatment. At the latest cut-off date of May 7, 2024, the median OS was not reached in either group, suggesting that the OS data were still immature after approximately 5 years of follow-up. The corresponding HR was 0.85 (95% CI, 0.58 to 1.24; P = 0.3950). The KM curves of OS rates at various time points showed no statistically significant difference in OS between the 2 groups up to 54 months. According to the GRADE assessment, there is moderate certainty that D-VRd likely results in an increase in the probability of OS when compared with VRd at 54 months; however, the clinical importance of the increase is uncertain. In the CEPHEUS trial, the median duration of study treatment was longer in the D-VRd group (56.3 months) compared to the VRd group (34.3 months). After disease progression, ████% of patients in the D-VRd group received subsequent antimyeloma treatment (████% of them received antineoplastic drugs, ████% received systemic corticosteroids, and ████% received immunosuppressants), compared to ████% of such patients in the VRd group (████% of them received antineoplastic drugs [including ████% received daratumumab], ████% received systemic corticosteroids, and ████% received immunosuppressants). This imbalance in the use of subsequent antimyeloma treatments between treatment groups could have biased the treatment effect on OS against the D-VRd group. Because rescue therapy could impact OS but not PFS provided that these drugs were administered after a patient’s disease progression, this could be a potential reason why OS did not show a significant difference between treatment groups despite a significant difference observed for PFS.
The overall MRD negativity rate was the primary efficacy outcome in the CEPHEUS trial. Findings from the study suggested that treatment with D-VRd resulted in a statistically significantly higher overall MRD negativity rate compared with VRd for patients with NDMM who were not undergoing ASCT (OR = 2.37; 95% CI, 1.58 to 3.55) (between-group difference = 21.5%; 95% CI, 10.9% to 31.1%). This also met the protocol-defined threshold for clinically meaningful benefit. The clinical experts consulted by CDA-AMC indicated that this surrogate outcome is not routinely used in clinical practice because of the availability and accessibility across Canada and its value in treatment selection; however, previous studies have demonstrated the correlation between MRD negativity status and PFS benefits in patients with MM. Patients’ response to treatment was also measured with the VGPR and better rate. Results of this measure showed that patients in the D-VRd group had a superior response to those in the VRd group, which was consistent with the results of overall MRD negativity rate.
HRQoL measured by the EORTC QLQ-C30 and EORTC QLQ-MY20 questionnaires were secondary outcomes in the CEPHEUS trial. At cycle 36, the proportions of patients who completed the 2 questionnaires and contributed to the analysis were slightly lower in the VRd group: 61.5% of patients in the D-VRd group versus 50.1% in the VRd group completed the EORTC QLQ-C30, and 61.5% of patients in the D-VRd group versus 51.0% in the VRd group completed the EORTC QLQ-MY20. Based on the analyses of the HRQoL data, a trend of improved GHS and decreased disease symptoms was observed for both D-VRd and VRd treatment groups, whereas the between-group differences for these outcomes were not statistically significant. The results suggested that treatment with D-VRd may result in little to no difference in HRQoL compared to treatment with VRd. Clinically important between-group differences in the change in the subscale scores for GHS in the EORTC QLQ-C30 and the subscale scores for Disease Symptoms in EORTC QLQ-MY20 were not demonstrated. The results are at risk of bias because of missing outcomes data.
In the sponsor-submitted ITCs, the comparative efficacy of D-VRd versus DRd, DVMp, and Isa-VRd was evaluated based on evidence from 4 phase III RCTs. Results from the propensity-based methods suggested that treatment with D-VRd may be related to longer PFS and a higher MRD negativity rate when compared to DRd and DVMp, although the magnitude of benefit is uncertain. This uncertainty was likely related to the heterogeneity between studies (e.g., different patient characteristics at baseline). Additionally, it is possible that the comparisons were not adjusted for all effect modifiers. There was insufficient evidence to show a difference in OS between D-VRd and DRd or DVMp. Results from an anchored MAIC analysis suggested that evidence is insufficient to conclude whether D-VRd or Isa-VRd is favoured for prolonging PFS or OS, or improving the MRD negativity rate, in patients with NDMM who are not undergoing ASCT as initial therapy. The estimates were affected by serious imprecision (i.e., wide 95% CIs) that included the null, suggesting that either treatment could be favoured.
Harms
Safety analyses were conducted for all patients who received at least 1 dose of study drug in the CEPHEUS trial. Study findings from this trial suggested that no new safety signals for D-VRd were identified. Increased incidences of infections, cytopenia, vascular disorders, and renal and urinary disorders were observed in the D-VRd group (all grades or grade 3 and grade 4), whereas slightly higher incidences of gastrointestinal disorders and peripheral sensory neuropathy were observed in the VRd group. The clinical experts commented that although the safety profile of D-VRd was similar to that of VRd, added toxicities because of the addition of daratumumab would be expected in clinical practice. The experts also noted that the safety data for daratumumab given in combination with VRd were consistent with the known safety profiles of daratumumab and VRd.
Harm outcomes were not evaluated in the sponsor-submitted ITC reports.
Ethics and Equity Considerations
Patient groups identified the negative impact of MM on patients and their caregivers’ HRQoL. Patients with MM may experience disease-related symptoms (e.g., fatigue, anemia, bone pain) and unwanted side effects from the treatments that create substantial burdens in their daily living, physical well-being, and psychosocial well-being. The disease also impairs caregivers’ overall well-being.28 Many treatments for MM are administered via IV injections or infusions and are administered frequently. Routine laboratory checkups, repeated blood sample collections that may require painful needlesticks, and bone marrow aspiration and biopsy are frequently required for diagnosis and monitoring of the disease response and recurrence.28
The formulation of daratumumab for SC administration has been developed to avoid the long infusion time that frequently requires hospitalization with IV administration of daratumumab and to lessen the rate and severity of infusion-related reactions observed with IV daratumumab. In the CEPHEUS trial, daratumumab SC was administered in an outpatient setting. According to the clinical experts consulted by CDA-AMC, daratumumab SC can be delivered through a community oncology site under the supervision of a specialist, which may decrease the burden on patients because it eliminates the need to travel to cancer centres and also reduces chair time.
Conclusion
Patients and clinicians highlighted the need for new treatment options that alleviate symptoms, achieve long-term remission, preserve HRQoL, reduce side effects compared to the current treatments, prevent new complications from the treatment, and prolong life. Results from the CEPHEUS trial demonstrated that when compared to VRd, the addition of daratumumab to VRd results in a clinically important benefit in PFS with high certainty. The treatment benefit with D-VRd was also consistently presented in the overall MRD-negative rate (with high certainty) and the VGPR or better rate (with moderate certainty). However, the treatment effect of the D-VRd regimen on OS was uncertain because the median OS was not reached in either treatment group at the latest data cut-off for analyses. Evidence from the CEPHEUS trial suggested that adding daratumumab to VRd may not result in a clinically important difference in patients’ HRQoL, compared to VRd; however, the evidence was with low certainty because of a large amount of missing data. Compared with VRd, there is moderate- to high-certainty evidence that D-VRd likely results in an increase in the incidence of SAEs, infections and infestations, and cytopenia.
Indirect evidence from 2 sponsor-conducted propensity score–based analyses and 1 anchored MAIC analysis assessed the comparative efficacy of D-VRd versus DRd, DVMp, and Isa-VRd in patients with NDMM who are not undergoing ASCT as initial therapy. The results of these ITCs suggested that D-VRd may be related to longer PFS and a higher MRD negativity rate when compared to DRd and DVMp, although the magnitude of benefit is uncertain because of study limitations. There was no statistically significant difference in OS between D-VRd and DRd or DVMp. When compared to Isa-VRd, there was insufficient evidence to conclude whether D-VRd or Isa-VRd is favoured for prolonging PFS or OS, or improving MRD negativity rate.
Economic Review
Methods
The review team appraised the submitted pharmacoeconomic evidence, which included an economic evaluation comparing the cost-effectiveness of daratumumab in combination with bortezomib, lenalidomide, and dexamethasone (D-VRd) to comparators for the requested reimbursement population (i.e., patients aged 18 years or older with NDMM) who are ineligible for ASCT, which is narrower than the Health Canada indication.2 The Health Canada indication is for patients aged 18 years or older with NDMM who are not undergoing ASCT as initial therapy, including those who have deferred transplant. The sponsor additionally submitted a BIA assessing the budgetary impact of reimbursing D-VRd for the requested reimbursement population.55
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of D-VRd from the perspective of a public health care payer in Canada over a lifetime horizon (28 years). The modelled population consisted of patients aged 18 years or older with NDMM who are ineligible for ASCT, which is narrower than the Health Canada indication and was based on the participants in the CEPHEUS trial. The sponsor’s base-case analysis included costs related to drug acquisition, administration, subsequent treatment, AEs, health care resource use, and end-of-life care.
The sponsor’s base case consisted of 5 pairwise cost-effectiveness results compared with D-VRd:
the incremental cost-effectiveness ratio (ICER) versus VRd was $578,965 per quality-adjusted life-year (QALY) gained (incremental costs = $773,900; incremental QALYs gained = 1.34)
the ICER versus DRd was $204,735 per QALY gained (incremental costs = $146,505; incremental QALYs gained = 0.72)
the ICER versus DVMp was $472,146 per QALY gained (incremental costs = $389,487; incremental QALYs gained = 0.82)
D-VRd is dominant when compared to Isa-VRd (incremental cost savings = $21,843; incremental QALYs gained = 0.33)
the ICER versus Rd was $383,296 per QALY gained (incremental costs = $832,318; incremental QALYs gained = 2.17).
Additional information about the sponsor’s submission is summarized in Appendix 10 in the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 6; full details are provided in Appendix 11 in the Supplemental Material document).
Table 6: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative clinical efficacy of D-VRd vs. DRd, DVMp, Isa-VRd, and Rd is uncertain. | There is a lack of head-to-head evidence between D-VRd and nontrial comparators, and the CDA‑AMC Clinical Review identified several limitations with the sponsor’s ITCs. | CDA-AMC could not address this issue in the base case because of a lack of robust clinical evidence. | No scenario analysis — the best available evidence was used in the CDA-AMC base case. |
The OS benefit predicted for D-VRd relative to other comparators is likely overestimated in the sponsor’s base case. | Based on the sponsor’s choice of OS extrapolation, a curative effect is suggested with D-VRd that is not supported by evidence. | CDA-AMC assumed equivalent OS between D-VRd and comparators in the base case to reflect the uncertainty in the long-term survival benefit expected with D-VRd. | No scenario analysis was conducted. |
Subsequent therapy costs are highly uncertain. | The sponsor used a single one-time cost for all patients transitioning to the postprogression health state, failing to account for the relationship between time in the progressed health state and time on treatment. Cilta-cel was included as a second-line treatment option, which is not accurate.56 | CDA-AMC excluded subsequent therapy costs from the base-case analysis. | To explore uncertainty around this issue, CDA‑AMC conducted a scenario analysis which included subsequent therapy costs, with the exception of cilta-cel. |
The approach to modelling TTD was inappropriate for comparators. | The use of unadjusted HRs from the ITC does not account for important differences between trial populations. | CDA-AMC equated TTD to PFS for both D-VRd and its comparators. | To explore uncertainty around this issue, CDA‑AMC conducted a scenario analysis which included the sponsor’s base-case TTD assumptions. |
The sponsor excluded DCyBorD and CyBorD from the analysis. | DCyBorD and CyBorD are relevant comparators for this indication. | This issue could not be addressed. The cost-effectiveness of D-VRd vs. these comparators remains unknown. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; cilta-cel = ciltacabtagene autoleucel; CyBorD = cyclophosphamide, bortezomib, and dexamethasone; DCyBorD = daratumumab in combination with cyclophosphamide, bortezomib, and dexamethasone; DRd = daratumumab in combination with lenalidomide and dexamethasone; DVMP = daratumumab in combination with melphalan, prednisone, and bortezomib; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; HR = hazard ratio; Isa-VRd = isatuximab in combination with bortezomib, lenalidomide, and dexamethasone; ITC = indirect treatment comparison; OS = overall survival; PFS = progression-free survival; Rd = lenalidomide and dexamethasone; TTD = time to treatment discontinuation; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 in the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 22 in Appendix 11 in the Supplemental Material document), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 11 in the Supplemental Material document.
Impact on Health Care Costs
D-VRd is predicted to be associated with additional health care costs compared to VRd (incremental costs = $1,072,043) and DRd (incremental costs = $311,182). This increase in health care spending results from drug acquisition costs associated with D-VRd (refer to Figure 1).
Figure 1: Impact of D-VRd vs. Comparators on Health Care Costs
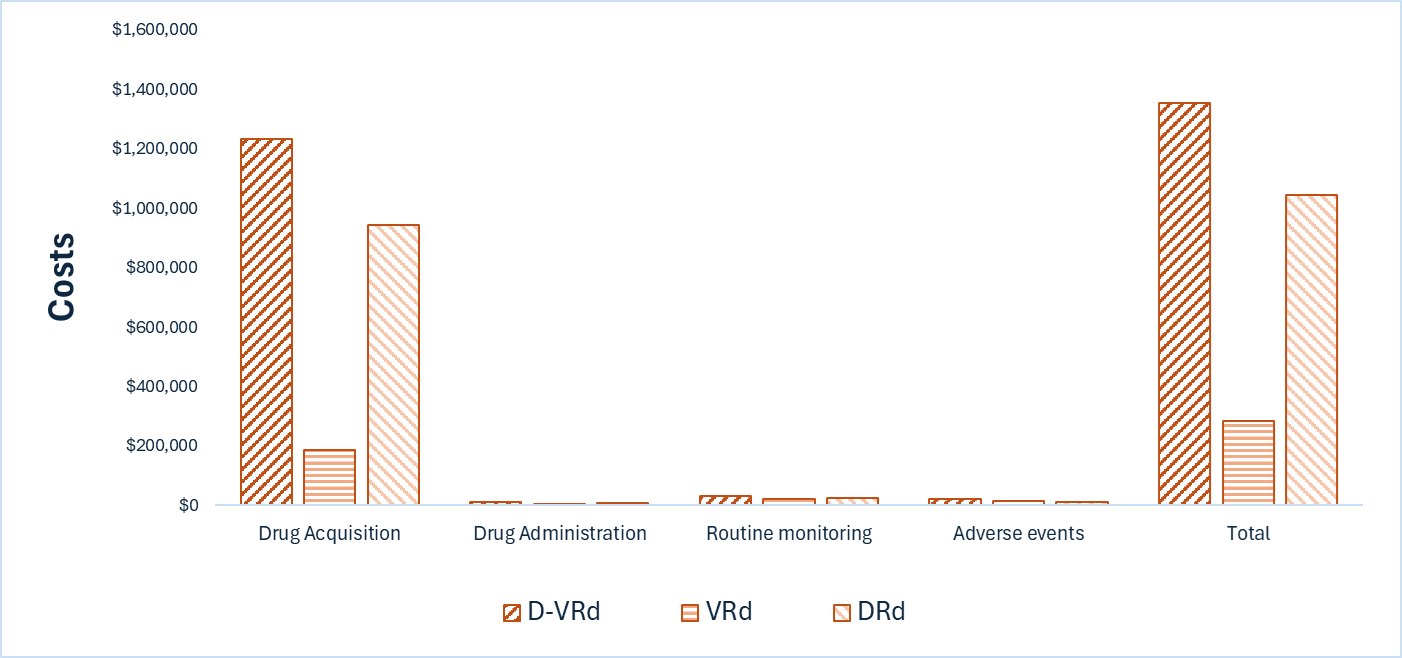
DRd = daratumumab in combination with lenalidomide and dexamethasone; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Impact on Health
Treatment with D-VRd is predicted to increase the amount of time a patient remains in the PFS health state by approximately 2.9 years versus VRd and 2.2 years versus DRd (refer to Figure 2). No survival benefit was predicted with D-VRd. When considering the impact of treatment on both quality and length of life, D-VRd is predicted to result in 0.10 additional QALYs per patient compared to VRd and 0.04 additional QALYs per patient compared to DRd. Approximately 82% and 92% of the predicted incremental benefit versus VRd and DRd, respectively, was accrued on the basis of extrapolation.
Figure 2: Impact of D-VRd vs. Comparators on Patient Health
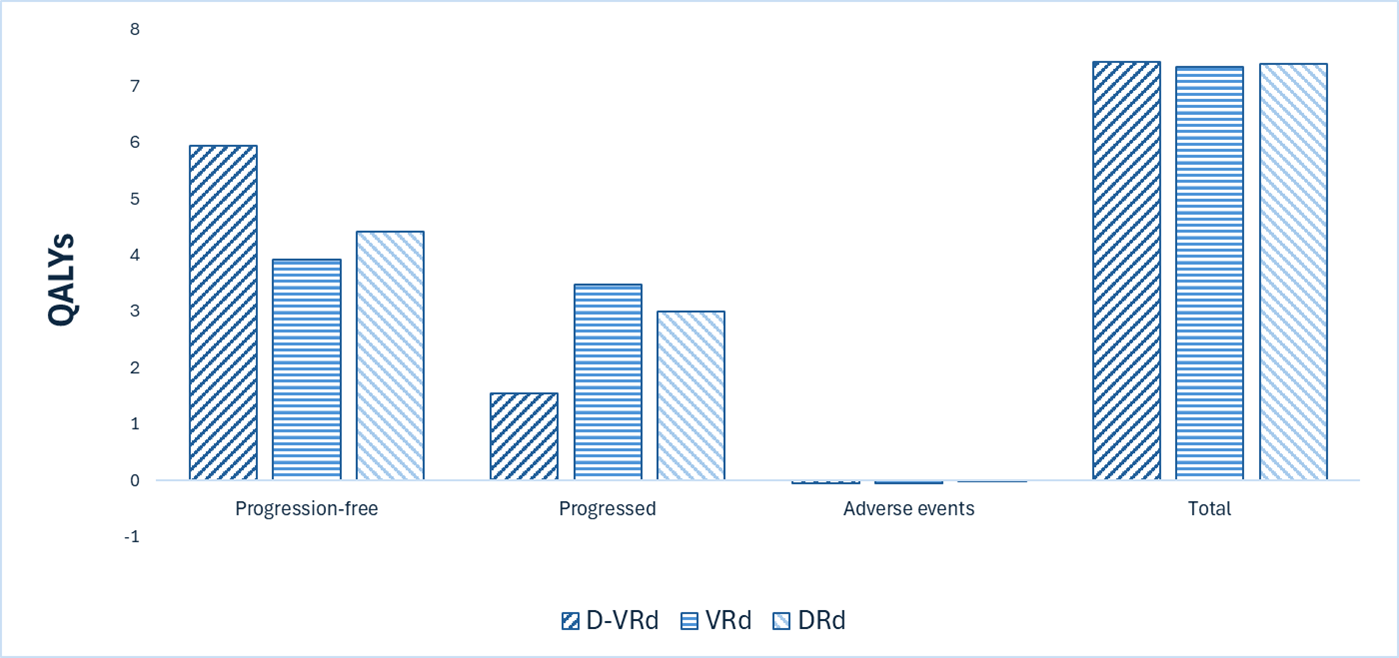
DRd = daratumumab in combination with lenalidomide and dexamethasone; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; QALY = quality-adjusted life-year; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Overall Results
The results of the CDA-AMC base-case analysis suggest an ICER of $10,623,156 per QALY gained for D-VRd compared to VRd and an ICER of $7,029,398 per QALY gained for D-VRd compared to DRd (refer to Table 7). Additional details on the CDA-AMC base-case analysis are available in Appendix 11 in the Supplemental Material document.
Table 7: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total LYs gained | Total QALYs gained | Pairwise ICER ($ per QALY gained) |
|---|---|---|---|---|
D-VRd vs. VRd | ||||
VRd | 283,327 | 10.84 | 7.34 | Reference |
D-VRd | 1,355,370 | 10.83 | 7.44 | 10,623,156 |
D-VRd vs. DRd | ||||
DRd | 1,044,188 | 10.84 | 7.39 | Reference |
D-VRd | 1,355,370 | 10.83 | 7.44 | 7,029,398 |
CDA-AMC = Canada’s Drug Agency; DRd = daratumumab in combination with lenalidomide and dexamethasone; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality‑adjusted life-year; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in Table 2. The use of alternative time to treatment discontinuation modelling approaches had the largest impact on the cost-effectiveness conclusions (refer to Table 26 in Appendix 11 in the Supplemental Material document). Based on the results of these analyses, the ICER for D-VRd compared with VRd may decrease to $7,029,948 per QALY gained, and the ICER for D-VRd versus DRd may decrease to $2,671,109 per QALY gained. CDA-AMC could not address the remaining uncertainty surrounding the lack of long-term clinical data to inform efficacy of D-VRd versus comparators.
Summary of the Budget Impact
The sponsor submitted a BIA to estimate the 3-year (2026 to 2028) budget impact of reimbursing D-VRd in the reimbursement-requested population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of daratumumab was aligned with the price included in the sponsor’s economic evaluation, whereas the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 in the Supplemental Material document. CDA-AMC identified issues with the sponsor’s estimated budget impact and made changes to model assumptions in consultation with clinical experts to derive the CDA-AMC base case (refer to Appendix 12 in the Supplemental Material document). CDA-AMC estimated that 6,866 patients would be eligible for D-VRd over a 3-year period (year 1 = 2,188 patients; year 2 = 2,287 patients; year 3 = 2,391 patients), of whom 1,214 patients are expected to receive D-VRd (year 1 = 223 patients; year 2 = 485 patients; year 3 = 507 patients). The estimated incremental budgetary savings of reimbursing D-VRd is predicted to be approximately $39 million over the first 3 years, with an expected expenditure of $369 million on D-VRd. The actual budgetary savings of reimbursing D-VRd will depend on the market uptake of D-VRd, the negotiated price of reimbursed comparators, and market displacement.
Conclusion
Based on the CDA-AMC base case, D-VRd would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $10,623,156 for each additional QALY gained in comparison to VRd, and at least $7,029,398 for each additional QALY gained in comparison to DRd. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 25 in Appendix 11 in the Supplemental Material document). The cost-effectiveness of D-VRd compared with VRd and DRd is highly uncertain because of limited long-term evidence. Notably, 82% and 89% of the incremental QALYs gained relative to VRd and DRd, respectively, accrue beyond 5.3 years in the absence of comparative data, likely leading to an overestimation of benefit. The budgetary savings of reimbursing D-VRd to the public drug plans in the first 3 years are estimated to be approximately $39 million. The 3-year expenditure on D-VRd (i.e., not accounting for current expenditure on comparators) is estimated to be $369 million. The estimated budgetary savings are uncertain because of uncertainty in the market uptake of D-VRd and uncertainty about market displacement.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction

CDA-AMC = Canada’s Drug Agency; DRd = daratumumab in combination with lenalidomide and dexamethasone; D-VRd = daratumumab in combination with bortezomib, lenalidomide, and dexamethasone; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VRd = bortezomib, lenalidomide, and dexamethasone; vs. = versus.
Notes: Expenditure includes only the drug cost of daratumumab.
The term dominant indicates that a drug costs less and provides more QALYs than the comparator.
References
1.Janssen Pharmaceuticals. DARZALEX SC Health Canada Product Monograph [Draft; EDMS-RIM-1448453 v.4.0 Darzalex SC] [sponsor supplied reference]. 2024.
2.Janssen Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: DARZALEX (daratumumab), 1800 mg/15 mL (120 mg/mL) solution for subcutaneous injection. August 18, 2025.
3.Janssen Inc. Clinical Study Report: 54767414MMY3019. A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (D-VRd) with VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Untreated Multiple Myeloma and for Whom Hematopoietic Stem Cell Transplant is Not Planned as Initial Therapy [internal sponsor's report]. August 12, 2024.
4.Janssen Research and Development. A Propensity Score Based Indirect Treatment Comparison Comparing DVRd vs. DVMP and VMP of ALCYONE in Newly Diagnosed Multiple Myeloma (NDMM) Patients Who Were Ineligible to Undergo Transplantation (v3.2) [sponsor supplied reference]. 2025.
5.Janssen Research and Development. An Inverse Probability of Treatment Weighting (IPTW)-Based Indirect Treatment Comparison Study of DVRd vs. DRd and Rd in Newly Diagnosed Multiple Myeloma (NDMM) Patients Who Were Ineligible to Undergo Transplantation (v5.4) [sponsor supplied reference]. 2025.
6.Janssen Research and Development. An Anchored Based Matching Adjusted Indirect Treatment Comparison (MAIC) Study Comparing DVRd vs. IsaVRd in Newly Diagnosed Multiple Myeloma (NDMM) Patients Who Were Ineligible to Undergo Transplantation. Version 4.2 [sponsor supplied reference]. 2025.
7.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. Jan 2015;12(1):42-54. doi:10.1038/nrclinonc.2014.200 PubMed
8.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. Jan 2009;23(1):3-9. doi:10.1038/leu.2008.291 PubMed
9.Bird SA, Boyd K. Multiple myeloma: an overview of management. Palliat Care Soc Pract. 2019;13:1178224219868235. doi:10.1177/1178224219868235 PubMed
10.Canadian Cancer Society. Symptoms of multiple myeloma [sponsor supplied reference]. Accessed February 2024, https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/signs-and-symptoms
11.Mayo Clinic. Multiple myeloma [sponsor supplied reference]. Accessed February 2024, https://www.mayoclinic.org/diseases-conditions/multiple-myeloma/diagnosis-treatment/drc-20353383
12.Rajkumar SV. Multiple myeloma: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. Sep 2024;99(9):1802-1824. doi:10.1002/ajh.27422 PubMed
13.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. May 12 2024;196(18):E615-E623. doi:10.1503/cmaj.240095 PubMed
14.McCurdy A, Mian H, LeBlanc R, et al. Redefining attrition in multiple myeloma (MM): a Canadian Myeloma Research Group (CMRG) analysis. Blood Cancer J. Jul 20 2023;13(1):111. doi:10.1038/s41408-023-00883-x PubMed
15.Janssen Data on File. Market Share Estimates for 1L TIE NDMM (ONCO-CAPPS data) [sponsor supplied reference]. 2025.
16.Janssen Pharmaceuticals. Data on file - DVRd Clinical Expert Consultation Meeting Minutes. 2025 [sponsor supplied reference].
17.Tsang M, Le M, Ghazawi FM, et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: A population study. Cancer. Jul 15 2019;125(14):2435-2444. doi:10.1002/cncr.32128 PubMed
18.Canadian Cancer Society. Risk factors for multiple myeloma. Accessed November 7, 2025, 2025. https://cancer.ca/en/cancer-information/cancer-types/multiple-myeloma/risks
19.Myeloma Canada. Multiple Myeloma Patient Handbook [sponsor supplied reference]. 2023. Accessed February 2024. https://myeloma.ca/wp-content/uploads/2023/08/web_mc_patient_hanbook_en_2023_final-1.pdf
20.Mian H, Reece D, Masih-Khan E, et al. Survival and Outcomes of Newly Diagnosed Multiple Myeloma Patients Stratified by Transplant Status 2007-2018: Retrospective Analysis from the Canadian Myeloma Research Group Database. Clin Lymphoma Myeloma Leuk. Aug 2022;22(8):608-617. doi:10.1016/j.clml.2022.03.002 PubMed
21.Kurtin SE. Relapsed or Relapsed/Refractory Multiple Myeloma. Journal of the Advanced Practitioner of Oncology. 2013;4(Suppl 1):5-14.
22.Quach H, Joshua D, Ho J, et al. Treatment of patients with multiple myeloma who are eligible for stem cell transplantation: position statement of the Myeloma Foundation of Australia Medical and Scientific Advisory Group. Intern Med J. 2015;45(1):94-105. PubMed
23.Guglielmelli T, Palumbo A. Incorporating novel agents in the management of elderly myeloma patients. Curr Hematol Malig Rep. Dec 2013;8(4):261-9. doi:10.1007/s11899-013-0177-y PubMed
24.Rosko A, Giralt S, Mateos M-V, Dispenzieri A. Myeloma in elderly patients: when less is more and more is more. American Society of Clinical Oncology Educational Book. 2017;37:575-585. PubMed
25.Côté J, Kotb R, Bergstrom DJ, et al. First Line Treatment of Newly Diagnosed Transplant Ineligible Multiple Myeloma: Recommendations from the Canadian Myeloma Research Group Consensus Guideline Consortium. Clin Lymphoma Myeloma Leuk. May 2023;23(5):340-354. doi:10.1016/j.clml.2023.01.016 PubMed
26.CADTH Reimbursement Recommendation: Optimal Pharmacotherapy for Transplant-Ineligible Multiple Myeloma; Therapeutic Review [sponsor supplied reference]. 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/TR0014%20Final%20Recommendation%20and%20Reasons.pdf
27.Reimbursement Recommendation: Isatuximab (Sarclisa) [sponsor supplied reference]. CDA-AMC; 2025. Accessed July 2, 2025. https://www.cda-amc.ca/isatuximab-0
28.Fragola M. Patient-Reported Outcomes and Assessment of Quality of Life: A Focus on Multiple Myeloma. J Adv Pract Oncol. Jul 2020;11(5):513-520. doi:10.6004/jadpro.2020.11.5.7 PubMed
29.Medina-Herrera A, Sarasquete ME, Jimenez C, Puig N, Garcia-Sanz R. Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future. Cancers (Basel). Jul 20 2023;15(14):3687. doi:10.3390/cancers15143687 PubMed
30.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. Aug 2016;17(8):e328-e346. doi:10.1016/S1470-2045(16)30206-6 PubMed
31.CDA-AMC Reimbursement Recommendation: Daratumumab (Darzalex SC) [sponsor supplied reference]. https://www.cda-amc.ca/sites/default/files/DRR/2025/PC0388_Darzalex_DRAFT_Recommendation.pdf
32.Anderson KC, Auclair D, Adam SJ, et al. Minimal Residual Disease in Myeloma: Application for Clinical Care and New Drug Registration. Clin Cancer Res. 10 01 2021;27(19):5195-5212. doi:10.1158/1078-0432.Ccr-21-1059
33.Cavo M, San-Miguel J, Usmani SZ, et al. Prognostic value of minimal residual disease negativity in myeloma: combined analysis of POLLUX, CASTOR, ALCYONE, and MAIA. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov't. Blood. 02 10 2022;139(6):835-844. doi:10.1182/blood.2021011101
34.Derman BA, Major A, Cooperrider J, et al. Discontinuation of maintenance therapy in multiple myeloma guided by multimodal measurable residual disease negativity (MRD2STOP). Blood Cancer J. 10 07 2024;14(1):170. doi:10.1038/s41408-024-01156-x
35.Janssen Inc. Sponsor's clinical summary report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: DARZALEX (daratumumab), 1800 mg/15 mL (120 mg/mL) solution for subcutaneous injection. August 18, 2025.
36.Mian HS, Visram A, Shih SC, et al. Minimal Residual Disease Testing Infrastructure in Multiple Myeloma: Guidance for Clinical Trial and Routine Practice Use in Canada. Clin Lymphoma Myeloma Leuk. Jan 23 2025;doi:10.1016/j.clml.2025.01.010 PubMed
37.Usmani SZ, Facon T, Hungria V, et al. Daratumumab plus bortezomib, lenalidomide and dexamethasone for transplant-ineligible or transplant-deferred newly diagnosed multiple myeloma: the randomized phase 3 CEPHEUS trial. Nat Med. Apr 2025;31(4):1195-1202. doi:10.1038/s41591-024-03485-7 PubMed
38.Health Canada. APO-DEXAMETHASONE Product Monograph [sponsor supplied reference]. Accessed July 2024. https://pdf.hres.ca/dpd_pm/00065302.PDF
39.Health Canada. ACT BORTEZOMIB Product Monograph [sponsor supplied reference]. Accessed May 2, 2023. https://pdf.hres.ca/dpd_pm/00070960.PDF
40.Health Canada. REVLIMID Product Monograph [sponsor supplied reference]. Accessed Feb 9, 2024. https://pdf.hres.ca/dpd_pm/00074535.PDF
41.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03 [sponsor supplied reference]. Accessed February 2024, https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_40
42.Janssen Research and Development. Clinical Protocol: A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (D-VRd) with VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Untreated Multiple Myeloma and for Whom Hematopoietic Stem Cell Transplant is Not Planned as Initial Therapy (21 February 2025; EDMS-ERI-167846090, 9.0) [sponsor supplied reference]. 2025.
43.Janssen Research and Development. Statistical Analysis Plan: A Phase 3 Study Comparing Daratumumab, VELCADE (bortezomib), Lenalidomide, and Dexamethasone (D-VRd) with VELCADE, Lenalidomide, and Dexamethasone (VRd) in Subjects with Untreated Multiple Myeloma and for Whom Hematopoietic Stem Cell Transplant is Not Planned as Initial Therapy (28 May 2021; EDMS-ERI-182800649, 3.0) [sponsor supplied reference]. 2021.
44.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. Sep 2006;20(9):1467-73. doi:10.1038/sj.leu.2404284 PubMed
45.Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. May 5 2011;117(18):4691-5. doi:10.1182/blood-2010-10-299487 PubMed
46.Tang DI, Geller NL. Closed testing procedures for group sequential clinical trials with multiple endpoints. Biometrics. Dec 1999;55(4):1188-92. doi:10.1111/j.0006-341x.1999.01188.x PubMed
47.Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncol. Jan 1 2017;3(1):28-35. doi:10.1001/jamaoncol.2016.3160 PubMed
48.Landgren O, Giralt S. MRD-driven treatment paradigm for newly diagnosed transplant eligible multiple myeloma patients. Bone Marrow Transplant. Jul 2016;51(7):913-4. doi:10.1038/bmt.2016.24 PubMed
49.Lahuerta JJ, Mateos MV, Martínez-López J, et al. Influence of pre- and post-transplantation responses on outcome of patients with multiple myeloma: sequential improvement of response and achievement of complete response are associated with longer survival. J Clin Oncol. Dec 10 2008;26(35):5775-82. doi:10.1200/jco.2008.17.9721 PubMed
50.Sully K, Trigg A, Bonner N, et al. Estimation of minimally important differences and responder definitions for EORTC QLQ-MY20 scores in multiple myeloma patients. Eur J Haematol. Nov 2019;103(5):500-509. doi:10.1111/ejh.13316 PubMed
51.Janssen Inc. Janssen Inc. response to CDA-AMC request for additional information regarding Darzalex SC (daratumumab) review on October 6, 2025: between-group differences for efficacy outcomes [internal additional sponsor's information]. 2025.
52.Facon T, San-Miguel J, Dimopoulos MA, et al. Treatment Regimens for Transplant-Ineligible Patients With Newly Diagnosed Multiple Myeloma: A Systematic Literature Review and Network Meta-analysis. Adv Ther. May 2022;39(5):1976-1992. doi:10.1007/s12325-022-02083-8 PubMed
53.Sterne JA, Hernan MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. Oct 12 2016;355:i4919. doi:10.1136/bmj.i4919 PubMed
54.Janssen Research and Development. Data on file - Protocol: Evaluation of the comparative efficacy of daratumumab in combination with bortezomib, lenalidomide and dexamethasone (D-VRd) in CEPHEUS versus approved comparator treatments in transplant-not-intended/transplant-ineligible (TNI/TIE) patients with newly diagnosed multiple myeloma (NDMM) [sponsor supplied reference]. 2024.
55.Janssen Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: DARZALEX (daratumumab), 1800 mg/15 mL (120 mg/mL) solution for subcutaneous injection. August 18, 2025.
56.pan-Canadian Pharmaceutical Alliance (pCPA). Carvykti (ciltacabtagene autoleucel). Accessed 2025 November 3. https://www.pcpacanada.ca/index.php/negotiation/22916
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.