Drugs, Health Technologies, Health Systems
Reimbursement Review
Encorafenib (Braftovi)
Sponsor: Pfizer, Inc.
Therapeutic area: Metastatic colorectal cancer with a BRAF V600E mutation
Summary
What Is Colorectal Cancer?
Colorectal cancer (CRC) is caused by the abnormal growth of mucosal cells in the inner lining of the colon or rectum. In most cases, CRC starts as a polyp on the intestinal mucosa, eventually developing into a carcinoma.
CRC is the fourth most common cancer in Canada, with an estimated 25,200 cases diagnosed in 2024. It is the third leading cause of cancer-related mortality, with an estimated 9,400 deaths in 2024. Approximately 55% of patients present with metastatic colorectal cancer (mCRC) at diagnosis; 5-year survival rates for these patients are poor. The BRAF V600E mutation is the most common type of BRAF mutation affecting patients with mCRC. It occurs in 8% to 12.5% of cases and is a well-established poor prognostic factor.
BRAF V600E mutation testing using next-generation sequencing is performed as part of the standard of care (SOC) for patients with mCRC in Canada.
What Are the Treatment Goals and Current Treatment Options for CRC?
Treatment goals for patients with BRAF V600E-mutant mCRC include extending overall survival, improving disease control and patient quality of life, and reducing toxicities related to treatment.
Among patients with mCRC and microsatellite-low, microsatellite-stable, or proficient mismatch repair status, the recommended first-line treatment option is a multidrug chemotherapy regimen, such as 1 of the following: 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI); modified 5-fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6); 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI); or capecitabine in combination with oxaliplatin (CAPOX) (with or without bevacizumab in all cases).
What Is Braftovi and Why Did Canada’s Drug Agency Conduct This Review?
Braftovi is available as an oral capsule. Health Canada has approved Braftovi in combination with cetuximab plus mFOLFOX6 for the treatment of patients with mCRC who have a BRAF V600E mutation, as detected by a validated test.
Canada’s Drug Agency (CDA-AMC) reviewed Braftovi to inform a recommendation to the participating public drug programs on whether this drug should be reimbursed for patients with mCRC who have a BRAF V600E mutation, as detected by a validated test.
How Did CDA-AMC Evaluate Braftovi?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Braftovi in combination with cetuximab plus mFOLFOX6 versus other treatments used in Canada for patients with mCRC who have a BRAF V600E mutation, as detected by a validated test. mFOLFOX6, FOLFOXIRI, FOLFIRI, and CAPOX (all regimens with or without bevacizumab) were considered relevant treatments to compare with Braftovi in combination with cetuximab plus mFOLFOX6 when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Braftovi and mCRC.
CDA-AMC considered the potential impacts of BRAF V600E mutation testing, as detected by a validated test, to ascertain eligibility for Braftovi in combination with cetuximab plus mFOLFOX6 in patients with mCRC, including impacts related to health systems, patients (including families and caregivers), and costs.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence as well as literature searches.
The review was also informed by 1 patient group submission and 2 clinician group submissions in response to our call for input, and by input from the participating public drug programs around issues that may affect their ability to implement a recommendation.
Two oncologists in Ontario with expertise in the diagnosis and management of CRC were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 phase III randomized controlled trial (the BREAKWATER trial) comparing Braftovi in combination with cetuximab plus mFOLFOX6 versus SOC chemotherapy (i.e., 1 of mFOLFOX6, FOLFOXIRI, or CAPOX, with or without bevacizumab) in 489 patients with previously untreated BRAF V600E-mutant mCRC
an assessment of the feasibility of indirect treatment comparisons comparing Braftovi in combination with cetuximab plus mFOLFOX6 versus relevant treatments for untreated mCRC in Canada.
For the comparison of Braftovi in combination with cetuximab plus mFOLFOX6 with SOC chemotherapy, based on Interim Analysis 2 of the BREAKWATER trial:
Treatment with Braftovi in combination with cetuximab plus mFOLFOX6 resulted in a clinically important increase in the probability of patients being alive and progression-free at 12 months and 15 months.
Treatment with Braftovi in combination with cetuximab plus mFOLFOX6 resulted in a clinically important increase in the probability of patients being alive at 12 months and 24 months compared with SOC chemotherapy.
Treatment with Braftovi in combination with cetuximab plus mFOLFOX6 likely resulted in a clinically important increase in the probability of patients being alive and progression-free after the next line of therapy at 21 months.
The evidence was very uncertain about the effect of Braftovi in combination with cetuximab plus mFOLFOX6 versus SOC chemotherapy on mean change from baseline at weeks 24 and 48 in European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 Global Health Status score.
Treatment with Braftovi in combination with cetuximab plus mFOLFOX6 likely resulted in little to no difference in the proportion of patients who experienced 1 or more serious adverse events.
There was no evidence to inform how Braftovi in combination with cetuximab plus mFOLFOX6 compares with the individual SOC chemotherapy regimens included in the SOC chemotherapy arm of the BREAKWATER trial. Given that FOLFIRI was not included in the SOC chemotherapy arm, there was also no evidence for the comparison of Braftovi in combination with cetuximab plus mFOLFOX6 with this regimen.
Economic Evidence
Braftovi is available as a 75 mg capsule. At the submitted price of $52.76 per 75 mg capsule, the 28-day cycle cost of Braftovi is expected to be $211 per patient, based on the Health Canada–recommended dosage. When Braftovi is used in combination with cetuximab and mFOLFOX6, the expected 28-day cycle cost is $18,843.
Key clinical efficacy data used to inform the economic model were derived from the BREAKWATER trial, which compared Braftovi in combination with cetuximab plus mFOLFOX6 to SOC chemotherapy. Evidence submitted by the sponsor indicates that Braftovi in combination with cetuximab and mFOLFOX6 is likely to improve overall survival, progression-free survival, and progression-free survival after next line of therapy compared with SOC chemotherapy among patients with mCRC who have a BRAF V600E mutation.
The results of the CDA-AMC base case suggest that:
Braftovi in combination with cetuximab and mFOLFOX6 is predicted to be associated with higher costs to the health care system than SOC chemotherapy (incremental cost = $367,543), with the higher cost driven primarily by drug acquisition.
Braftovi in combination with cetuximab and mFOLFOX6 is predicted to be associated with a gain of 1.04 life-years compared to SOC chemotherapy and may result in a gain of 0.85 quality-adjusted life-years compared to SOC chemotherapy.
The incremental cost-effectiveness ratio of Braftovi in combination with cetuximab and mFOLFOX6 compared to SOC chemotherapy was $432,400 per quality-adjusted life-year gained in the CDA-AMC base case. The estimated incremental cost-effectiveness ratio was sensitive to the distribution of subsequent treatment.
CDA-AMC estimates that the budget impact of reimbursing Braftovi in combination with cetuximab and mFOLFOX6 for the indicated population will be approximately $175 million over the first 3 years of reimbursement compared to the amount currently spent on SOC chemotherapy, with an estimated expenditure of $70 million on Braftovi over this period. The actual budget impact of reimbursing Braftovi in combination with cetuximab and mFOLFOX6 will depend on its uptake. The incremental budget impact of reimbursing Braftovi in combination with cetuximab and mFOLFOX6 is predicted to be greater than $40 million in year 2 and year 3, and the economic feasibility of adoption must be addressed.
Abbreviations
AE
adverse event
BICR
blinded independent central review
CAPOX
capecitabine and oxaliplatin
CCRAN
Colorectal Cancer Resource & Action Network
CDA-AMC
Canada’s Drug Agency
CGOEN
Canadian Gastrointestinal Oncology Evidence Network
CI
confidence interval
CRC
colorectal cancer
dMMR
deficient mismatch repair
DOR
duration of response
EC
encorafenib and cetuximab
ECOG
Eastern Cooperative Oncology Group
EGFR
epidermal growth factor receptor
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FAS
full analysis set
FOLFIRI
5-fluorouracil, leucovorin, and irinotecan
FOLFOX
5-fluorouracil, leucovorin, and oxaliplatin
FOLFOXIRI
5-fluorouracil, leucovorin, oxaliplatin, and irinotecan
GHS
Global Health Status
GI DAC
Gastrointestinal Cancer Drug Advisory Committee
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IA1
Interim Analysis 1
IA2
Interim Analysis 2
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
KM
Kaplan-Meier
MAB
Medical Advisory Board
mCRC
metastatic colorectal cancer
mFOLFOX6
modified 5-fluorouracil, leucovorin, and oxaliplatin
MID
minimal important difference
MMR
mismatch repair
MSI
microsatellite instability
MSI-L
microsatellite instability-low
MSS
microsatellite stable
NGS
next-generation sequencing
OH (CCO)
Ontario Health (Cancer Care Ontario)
ORR
objective response rate
OS
overall survival
PCR
polymerase chain reaction
PFS
progression-free survival
PFS2
progression-free survival after next line of treatment
pMMR
proficient mismatch repair
QALY
quality-adjusted life-year
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SOC
standard of care
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of Braftovi (encorafenib) (75 mg oral capsules) in combination with cetuximab (EC) plus modified 5-fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) for the treatment of patients with metastatic colorectal cancer (mCRC) with a BRAF V600E mutation, as detected by a validated test. The focus will be on comparing EC plus mFOLFOX6 to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence. This focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the economic review is aligned with the scope of the clinical review unless otherwise stated. For most reviews, Canada’s Drug Agency (CDA-AMC) develops a base case that is informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Encorafenib (Braftovi), 75 mg capsules taken orally |
Sponsor | Pfizer Canada ULC |
Health Canada indication | Encorafenib, in combination with cetuximab and mFOLFOX6, for the treatment of patients with mCRC with a BRAF V600E mutation, as detected by a validated test |
Health Canada approval status | NOC/c |
Health Canada review pathway | Priority review |
NOC date | July 25, 2025 |
Mechanism of action | Encorafenib is a selective BRAF kinase inhibitor that targets and inhibits the activity of mutant BRAF V600E, thereby blocking the MAPK-ERK signalling pathway responsible for promoting tumour cell proliferation. |
Recommended dosage | The recommended dose of encorafenib is 300 mg (four 75 mg capsules) orally q.d. in combination with cetuximab and mFOLFOX6 until disease progression or unacceptable toxicity. Encorafenib may be taken with or without food. |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $52.76 per capsule |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusb | Population: As defined in the Health Canada indication Subgroups: None Intervention: Per recommended dosage Comparators:
Outcomes: ORR, PFS, OS, DOR, PFS2, HRQoL (measured through the EORTC QLQ-C30), safety |
CAPOX = capecitabine and oxaliplatin; CDA-AMC = Canada’s Drug Agency; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FOLFIRI = 5-fluorouracil, leucovorin, and irinotecan; FOLFOXIRI = 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan; HRQoL = health-related quality of life; mCRC = metastatic colorectal cancer; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival after next line of treatment; q.d. = once daily.
aCDA-AMC has previously issued a reimbursement recommendation for this drug for the same indication or a similar indication.
bThe economic review aligns with the scope of the clinical review unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC has previously reviewed EC for the treatment of patients with mCRC who have a BRAF V600E mutation (as detected by a validated test) after prior therapy. The agency issued a recommendation to reimburse with clinical criteria and/or conditions. CDA-AMC has also previously reviewed encorafenib in combination with binimetinib for the treatment of patients with unresectable or metastatic melanoma who have a BRAF V600 mutation, as detected by a validated test. The agency issued a recommendation to reimburse with clinical criteria and/or conditions.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (i.e., patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
CDA-AMC received 1 patient group submission from the Colorectal Cancer Resource & Action Network (CCRAN) in collaboration with the Canadian Cancer Survivor Network. According to the patient group input, CCRAN and the network conducted outreach campaigns through social media and email to recruit patients with BRAF V600E-mutant mCRC. CCRAN also reached out to US-based colorectal cancer (CRC) patient groups, CRC medical oncologists (16 in Canada and 5 in US), and trial investigators to help identify patients with mCRC who had treatment experience with encorafenib. The survey was completed by 77 patients with mCRC or their respective caregivers. Outreach efforts resulted in 5 telephone interviews with 4 patients with BRAF V600E-mutant mCRC and 1 caregiver in Canada who had either accessed the BREAKWATER trial or encorafenib in combination with an epidermal growth factor receptor (EGFR) inhibitor with or without chemotherapy.
CDA-AMC received 2 clinician group submissions. One joint submission was received from the Canadian Gastrointestinal Oncology Evidence Network (CGOEN), the Medical Advisory Board (MAB) of Colorectal Cancer Canada, and other clinicians with expertise in treating gastrointestinal cancers; this joint submission included 22 clinicians. Information was gathered through literature review and virtual discussion among experts. Another submission was received from the Ontario Health (Cancer Care Ontario) (OH [CCO]) Gastrointestinal Cancer Drug Advisory Committee (GI DAC), which gathered information from 3 clinicians through email.
The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may affect the programs’ ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group inputs are summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two oncologists in Ontario with expertise in the diagnosis and management of CRC participated in this review.
Disease Background
CRC is caused by the abnormal growth of mucosal cells in the inner lining of the colon or rectum. In most cases, CRC starts as a polyp on the intestinal mucosa, eventually developing into a carcinoma.1,2 In Canada, CRC is the fourth most common cancer (with an estimated 25,200 cases diagnosed in 2024) and the third leading cause of cancer-related mortality (with an estimated 9,400 deaths in 2024).3 Approximately 55% of patients present with mCRC at diagnosis; 5-year survival rates for these patients are poor.4 In Canada, stage IV colon cancer is associated with a 5-year survival rate of 11% to 12%, while stage IV rectal cancer is associated with a 5-year survival rate of 13%.13
CRC is often characterized by several genetic mutations, including those affecting the BRAF gene. BRAF is a protein kinase in the mitogen-activated protein kinase pathway that regulates cell proliferation and growth.6,7 Mutations in the BRAF gene can result in a constitutively active MAPK pathway, leading to tumour cell growth.7 The BRAF V600E mutation is the most common type of BRAF mutation affecting patients with mCRC; it occurs in 8% to 12.5% of patients with mCRC and is a well-established poor prognostic factor.5-8 Notably, patients with BRAF V600E-mutant mCRC have a significantly worse prognosis and lower quality of life (QoL) than those with wild-type mCRC, given that they are more likely to experience rapid disease progression and shorter life expectancy.14-17 In Canada, testing for the BRAF V600E mutation and other genomic biomarkers is recommended by guidelines to inform selection of appropriate therapy for patients with mCRC; it is a routine part of the diagnostic work-up.18 The clinical experts consulted by CDA-AMC did not identify any specific marginalized or equity-deserving populations who would be disproportionately affected by CRC.
According to the patient group input, living with mCRC comes with a unique set of physical and psychological challenges. Interviewed and surveyed patients with mCRC reported symptoms that included but were not limited to abdominal pain, changes in bowel habits (e.g., constipation, diarrhea), bloody stools, nausea, fatigue, and disease complications (e.g., fistula formation). Survey respondents indicated that CRC significantly affected patients’ QoL by limiting their ability to work, engage in exercise, and fulfill family obligations.
Current Management
Treatment Goals
According to the patient group input, interviewed and surveyed patients and caregivers reported that it was very important that new drug therapies improve their physical condition and QoL. Interview respondents also valued personalized and targeted therapies for mCRC, a holistic and multidisciplinary approach to disease management, the ability to remain asymptomatic, the ability to extend survival, and reduced toxicity from treatment. Patients emphasized the need for faster and equitable access to funded drug therapies and clinical trials in Canada.
The clinical experts consulted by CDA-AMC indicated that the goals of treatment for patients with BRAF V600E-mutant mCRC include extending overall survival (OS) and improving disease control and patients’ QoL. Similarly, both clinician group inputs indicated that improvements in OS and maintenance of QoL were 2 crucial goals of treatment for mCRC; the groups noted that patients with BRAF V600E-mutant mCRC often experience poor clinical outcomes.
Current Treatment Options
According to the CDA-AMC provisional funding algorithm for mCRC and treatment guidelines for CRC, treatment pathways for mCRC are guided by microsatellite instability (MSI) status, mismatch repair (MMR) status, tumour location, and the presence of genetic markers (e.g., BRAF, RAS).9-12
Among patients with mCRC who have a BRAF V600E mutation and microsatellite instability-low (MSI-L), microsatellite-stable (MSS), or proficient mismatch repair (pMMR) status, the recommended first-line treatment option is a multidrug chemotherapy regimen (e.g., 5-fluorouracil, leucovorin, and irinotecan [FOLFIRI]; mFOLFOX6; 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan [FOLFOXIRI]; or capecitabine and oxaliplatin [CAPOX]) administered with or without bevacizumab.9-12 The clinical experts consulted by CDA-AMC agreed that FOLFIRI and 5-fluorouracil, leucovorin, and oxaliplatin (FOLFOX) with or without bevacizumab are the most common first-line chemotherapy regimens administered among such patients in clinical practice, while FOLFOXIRI and CAPOX were less common.
After a patient with mCRC who has a BRAF V600E mutation and MSI-L, MSS, or pMMR status has experienced disease progression while receiving first-line treatment, the recommended second-line treatment (regardless of tumour location) is an alternate chemotherapy regimen; the choice depends on the regimen administered in the first-line treatment setting. However, the clinical experts consulted by CDA-AMC and the treatment guidelines for CRC recommend encorafenib in combination with an EGFR inhibitor (e.g., cetuximab or panitumumab).9-12 The clinical experts consulted by CDA-AMC noted that treatment with an EGFR monoclonal antibody is not recommended without the use of a BRAF inhibitor among patients with BRAF mutations. Among patients with a BRAF V600E mutation, recommended third-line treatments include an alternate chemotherapy regimen or encorafenib in combination with an EGFR inhibitor (if an EGFR inhibitor was not received in the previous line).9-12 Fourth-line and later-line treatments may include trifluridine-tipiracil in combination with bevacizumab, followed by fruquintinib.9-12
Key characteristics of encorafenib are summarized along with those of other treatments available for mCRC in the Supplemental Material document (available on the project landing page), Appendix 1, Table 1.
Unmet Needs and Existing Challenges
Based on the patient group input submitted to CDA-AMC, due to the aggressive nature of the mutational status, patients with BRAF-mutant mCRC are often treated with an aggressive, cytotoxic chemotherapeutic combination regimen (e.g., FOLFOX, FOLFIRI, or FOLFOXIRI [with or without bevacizumab]) in the first-line setting. The patient group input noted that certain regimens (e.g., FOLFOXIRI) can be toxic and difficult to tolerate, can significantly impair QoL, and often have limited efficacy in patient populations with BRAF mutation. Some of the patient respondents expressed frustration with respect to managing the required logistics, travel, and frequency of appointments and the difficulty of fitting treatments into their busy lives. Survey respondents also noted a variety of side effects from various systemic treatments, such as fatigue (reported by 86% of respondents), peripheral neuropathy (72%), hair loss (71%), diarrhea (67%), nausea (67%), cognitive problems (60%), mouth sores (55%), and low white blood cell count (48%). Moreover, 69% of survey respondents required additional prescription medicines to help manage treatment-induced side effects; of this proportion, 40% reported incurring out-of-pocket expenses for these additional medications. The patient group input highlighted that there is a significant unmet clinical need for efficacious and tolerable therapeutics for patients with BRAF-mutant mCRC, who often experience poor clinical outcomes.
Clinical experts consulted by CDA-AMC noted that patients with BRAF-mutant mCRC need effective treatment options. The experts consulted by CDA-AMC emphasized that, despite the addition of bevacizumab to standard of care (SOC) chemotherapy, patients with BRAF-mutant mCRC experience more aggressive disease, less response to treatment, and much shorter survival durations. Further, the clinical experts consulted for this review noted that these patients often experience failure of most SOC treatment options as well as earlier and extensive recurrence of cancer compared to patients without a BRAF mutation. One clinical expert noted that patients who received bevacizumab in the first-line setting cannot receive it as a second-line treatment in some jurisdictions (e.g., Ontario).
One clinician group who submitted input to CDA-AMC (CGOEN and the MAB of Colorectal Cancer Canada) stated that current therapies are limited by the rapid development of resistance, leading to disease progression within a relatively short time frame. Thus, novel therapies are urgently needed to improve OS, provide more durable disease control, minimize symptom burden, and prevent the development of symptoms due to rapid growth of these cancers. The OH (CCO) GI DAC clinician group input reported that the prognosis for patients with BRAF-mutant mCRC remains very poor, with many patients dying within the first year of diagnosis. Thus, improving access to more effective first-line regimens for these patients is critical.
Considerations for Using the Drug Under Review
The contents of this section have been informed by input from the clinical experts consulted for the purpose of this review, input from clinician groups, and the reimbursement conditions proposed by the sponsor (refer to Appendix 1, Table 2 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized (refer to Appendix 1, Table 3 in the Supplemental Material document). The following information has been summarized by the review team.
Place in Therapy
The clinical experts consulted by CDA-AMC indicated that EC plus mFOLFOX6 should be administered as a first-line treatment for patients with mCRC who have a BRAF V600E mutation. They agreed that it would not be appropriate for these patients to try other treatments before initiating treatment with encorafenib. However, 1 clinical expert noted that it would be reasonable for patients whose disease progresses while receiving SOC chemotherapy to switch to treatment with encorafenib. Both clinical experts consulted by CDA-AMC agreed that the introduction of EC plus mFOLFOX6 would shift the treatment paradigm for mCRC, and that this regimen would be considered as the SOC for first-line mCRC.
Both clinician groups that submitted input to CDA-AMC agreed that EC plus mFOLFOX6 would be best suited as a first-line regimen for patients with BRAF V600E-mutant mCRC and pMMR status. The CGOEN and MAB of Colorectal Cancer Canada added that patients should have mCRC that is MSS. The groups further noted that the introduction of encorafenib as a first-line treatment would displace the drug from the second-line setting and ensure that a greater proportion of patients can access it earlier in their disease course.
Patient Population
The clinical experts consulted by CDA-AMC indicated that patients with BRAF-mutant mCRC are most in need of an intervention. They further agreed that all patients with BRAF-mutant mCRC should be eligible to receive EC plus mFOLFOX6, provided they have reasonable Eastern Cooperative Oncology Group (ECOG) Performance Status and RAS wild-type, MSS disease. The clinical experts consulted by CDA-AMC noted that patients who have deficient mismatch repair (dMMR) mutation status would be least suitable for treatment with encorafenib and should receive immunotherapy as a first-line treatment for mCRC. The clinical experts consulted by CDA-AMC did not identify any specific marginalized or equity-deserving populations that would be disproportionately affected by CRC.
The input from the clinician groups was consistent with the clinical experts’ identification of the patient population suitable for treatment with EC plus mFOLFOX6. The CGOEN and MAB of Colorectal Cancer Canada indicated that companion testing for BRAF V600E mutations and MMR status is already part of the SOC for CRC, is already reimbursed, and can be deployed easily at centres without concerns about test quality. They also noted that misdiagnosis of a BRAF mutation is unlikely, given that testing for the mutation is performed using a method based on either next-generation sequencing (NGS) or polymerase chain reaction (PCR).
The sponsor outlined several criteria pertaining to the initiation of treatment with EC and chemotherapy among patients aged 16 years or older. These included good ECOG Performance Status, the presence of histologically or cytologically confirmed stage IV mCRC, the presence of measurable disease per Response Evaluation Criteria in Solid Tumours Version 1.1 criteria, and the presence of a BRAF V600E mutation in tumour tissue or blood (as detected by a validated test). Moreover, the sponsor outlined criteria that would exclude patients from receiving encorafenib. These included prior receipt of systemic treatment for metastatic disease, prior treatment with any BRAF or EGFR inhibitor, the presence of symptomatic brain metastases, MSI-high and/or dMMR disease, and disease with a RAS mutation. The clinical experts consulted by CDA-AMC agreed with the proposed conditions.
Testing Procedure Considerations
BRAF V600 mutation status is used by clinicians to inform mCRC prognosis and treatment planning in patients.19 Canadian and international guidelines recommend testing for a minimum set of biomarkers — including MMR and/or MSI status, BRAF V600, and extended RAS (i.e., KRAS and NRAS genes) — in patients with mCRC before initial treatment.11,19-23 According to the clinical experts consulted for this review, the testing for this minimum set of biomarkers is broadly available and publicly funded across Canada as part of the SOC in patients with mCRC. However, they did mention that, for various reasons, reflex testing of biopsy samples from patients with mCRC is not always done (e.g., the pathologist is unaware of the staging when the biopsy sample is from the primary tumour). In these situations, testing would need to be specifically requested by the treating oncologist. BRAF mutations, including the V600E variant, can be identified using PCR or NGS methods on formalin-fixed, paraffin-embedded samples of primary, recurrent, or metastatic tumour tissue.19 Commercial PCR and NGS panels are highly sensitive and specific (> 98%) in detecting BRAF V600E mutations in multiple cancer types, including CRC.24 According to the Canadian guidelines, multigene NGS testing is preferred in order to optimize turnaround time and use of tissue; it also provides the option to add new biomarkers in the future.19 The clinical experts noted that NGS is the most common method used across jurisdictions in Canada to identify BRAF V600E mutations. The Canadian guidelines on biomarker testing in patients with mCRC also recommend that molecular testing results be available to the treating oncologist by the time of the first consultation to determine first-line therapy options.19 The clinical experts mentioned that the turnaround time for NGS results is 4 to 6 weeks at many centres. This could mean a delay in initiating targeted therapy (e.g., EC) in the first-line setting, especially if the testing was not performed reflexively during routine work-up. The clinical experts also confirmed that testing for BRAF V600E mutations in this patient population needs to be performed only once; it does not need to be repeated. One patient group mentioned that, due to the poor prognosis associated with BRAF V600E mutations, patients, families, and caregivers may experience anxiety and a greater emotional burden when learning that a tumour expresses this mutation.25
We considered the potential impacts of BRAF V600E mutation testing to ascertain eligibility for EC plus mFOLFOX6 in patients with mCRC, including those to health systems, patients (including families and caregivers), and costs. No new impacts are anticipated because BRAF V600E mutation testing is currently performed as SOC for patients with mCRC across jurisdictions in Canada. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team, when possible, and are summarized in Appendix 1, Table 4 in the Supplemental Material document.
Assessing the Response to Treatment
The clinical experts consulted by CDA-AMC agreed that response to treatment for mCRC is assessed by CT scans performed every 2 to 3 months. They indicated that a clinically meaningful response to treatment for mCRC would be stable disease (i.e., lack of disease progression) or shrinkage without any disease shown on CT scan. They noted that clinically meaningful responses to treatment for mCRC also include improvement of disease-related symptoms and tolerance to treatment.
The CGOEN and the MAB of Colorectal Cancer Canada also agreed that response to treatment for mCRC is assessed by CT scans performed every 2 to 3 months. The OH (CCO) GI DAC clinician group recommended that CT scans be performed every 3 to 4 months, or more frequently if clinically indicated. The OH (CCO) GI DAC noted that response to treatment would be assessed using tumour markers, regular imaging, and assessment of clinical symptoms. The CGOEN and the MAB of Colorectal Cancer Canada indicated that clinically meaningful responses to treatment include radiographic response, clinical stability, maintenance or improvement in ECOG Performance Status, improvement in cancer-related symptoms (e.g., pain), and the absence of rapid clinical deterioration. They also noted that laboratory markers (e.g., carcinoembryonic antigen levels) may be supportive indicators of disease activity; however, these alone are not considered sufficient to guide treatment decisions.
The sponsor proposed renewal conditions for the reimbursement of encorafenib. These conditions required ongoing clinical and radiographic evidence of response and tolerance to therapy as well as clinical assessments every 2 to 4 weeks, with radiological assessments every 8 to 12 weeks. The clinical experts consulted by CDA-AMC agreed that these conditions aligned with assessment of response to treatment for mCRC and could be readily implemented. Of note, 1 clinical expert consulted by CDA-AMC noted that in clinical practice, CT scans may be obtained only every 3 to 4 months, due to resource limitations.
Discontinuing Treatment
The clinical experts consulted by CDA-AMC indicated that treatment with encorafenib should be discontinued in the event of disease progression, intolerance to treatment not managed by dose modifications (e.g., reductions or delays), or decline in ECOG Performance Status. In the BREAKWATER trial, patients with disease progression who continued to derive clinical benefit from the study intervention per the investigator were eligible to continue, provided the risk-benefit profile for doing so was favourable. The clinical experts consulted noted that these situations would be rare in clinical practice, but possible. Cases of oglio-progression may be managed by a local modality before discontinuation, according to the clinical experts.
Both clinician groups that submitted input to CDA-AMC agreed that treatment with encorafenib should be discontinued upon disease progression or unacceptable toxicity. The sponsor proposed conditions for the discontinuation of EC plus mFOLFOX6, which stated that treatment should be discontinued in the case of disease progression based on Response Evaluation Criteria in Solid Tumours Version 1.1 criteria, unacceptable toxicity, pregnancy, or breastfeeding. The clinical experts consulted by CDA-AMC agreed with the proposed conditions and indicated that these could be readily implemented.
Prescribing Considerations
The clinical experts consulted by CDA-AMC agreed that treatment should be prescribed and administered by a specialist in medical oncology. They further indicated that patients living in remote areas without proximity to a medical oncologist can be assessed at cancer centres and receive treatment at local hospitals that can administer oncologic treatments. They also noted that FOLFOX and FOLFIRI (in combination with cetuximab or panitumumab) have been administered as first-line therapies for RAS wild-type, left-sided mCRC for several years, and that the addition of oral encorafenib is not expected to negatively affect patients’ ability to access treatment.
Both clinician groups that submitted input to CDA-AMC agreed that patients with BRAF-mutant mCRC would be treated and monitored by medical oncologists. The CGOEN and the MAB of Colorectal Cancer Canada clinician group added that oncologists are familiar with the management of adverse events (AEs) related to encorafenib, given that the drug is already indicated for the treatment of mCRC in the second-line setting.
The sponsor proposed conditions for prescribing EC and chemotherapy. These stated that treatment should be offered only by medical oncologists and at centres with experience in treating CRC, and that IV cetuximab and chemotherapy should be administered in approved oncology infusion clinics in outpatient settings. The clinical experts consulted by CDA-AMC agreed that these proposed conditions aligned with the prescribing of encorafenib in clinical practice. However, they noted that there are some areas in Canada where nonmedical oncologists administer treatment under the guidance of medical oncologists at cancer centres.
Additional Considerations
The clinical experts consulted by CDA-AMC noted that the use of cetuximab in the pivotal study should likely be considered interchangeable with panitumumab.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (i.e., pivotal studies and randomized controlled trials [RCTs]) and an indirect treatment comparison (ITC) feasibility assessment report in the evidence for inclusion. The sponsor did not submit any long-term extension studies, ITCs, or studies addressing gaps in evidence. Studies eligible for the systematic review included published and unpublished pivotal studies and phase III and IV RCTs. Relevant patients and interventions were defined by the reimbursement request and the dosage recommended in the product monograph. Relevant comparators were drugs used in clinical practice in Canada to treat patients described in the indication under review, which included chemotherapeutic drugs (e.g., mFOLFOX6, FOLFOXIRI, FOLFIRI, and CAPOX) administered with or without bevacizumab.
The review team selected outcomes (and follow-up times) for review considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations; these were selected in consultation with committee members (Table 2). Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.
Although objective response rate (ORR) and duration of response (DOR) were considered important outcomes for patients with mCRC, the clinical experts consulted by CDA-AMC indicated that response outcomes were considered as supportive outcomes to OS and progression-free survival (PFS). Thus, ORR and DOR were not formally included in the GRADE assessment. However, the results for these outcomes are discussed in this review.
Table 2: List of Outcomes Included for GRADE Assessment
Outcome | Rationale |
|---|---|
PFS | PFS, a measure of disease control, was identified as an important treatment goal by clinical experts consulted by CDA-AMC as well as by clinician and patient groups. Moreover, PFS was an outcome used to inform the pharmacoeconomic model. |
OS | Improvement in OS was noted to be an important treatment goal by clinical experts consulted by CDA-AMC as well as by clinician and patient groups. Moreover, OS was an outcome used to inform the pharmacoeconomic model. |
PFS2 | PFS2 was an outcome used to inform the pharmacoeconomic model. |
HRQoL (measured using the EORTC QLQ-C30) | The EORTC QLQ-C30 is a validated measure of HRQoL among patients with CRC. Improvement in HRQoL was noted to be an important treatment goal by clinical experts consulted by CDA-AMC as well as by clinician and patient groups. |
Proportion of patients with 1 or more SAEs | Reduction in adverse effects related to treatment for mCRC was noted to be an important treatment goal by patient groups. |
CDA-AMC = Canada’s Drug Agency; CRC = colorectal cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HRQoL = health-related quality of life; mCRC = metastatic colorectal cancer; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival after next line of treatment; SAE = serious adverse event.
The methods used for data extraction, risk of bias appraisal, and certainty of evidence assessment are described in Appendix 2 of the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal RCT included in the systematic review (the BREAKWATER trial)
1 feasibility assessment of ITCs.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the BREAKWATER trial are summarized in Table 3. Details pertaining to relevant outcome measures are in the Supplemental Material document (Appendix 3).
At the time of this review, the BREAKWATER trial is an ongoing, phase III, open-label, multicentre RCT evaluating the efficacy and safety of EC alone or in combination with chemotherapy (i.e., EC plus mFOLFOX6) compared with SOC chemotherapy (i.e., 1 of mFOLFOX6, FOLFOXIRI, or CAPOX; all regimens with or without bevacizumab) among patients with previously untreated, BRAF V600E-mutant mCRC. Before the phase III portion of the trial, a 2-cohort safety lead-in part was conducted to evaluate the tolerability and pharmacokinetics of EC plus mFOLFOX6 or EC plus FOLFIRI in patients with BRAF V600E-mutant mCRC with 1 or 0 prior systemic treatments for metastatic disease. An ongoing, open-label cohort 3 involving 2 randomized treatment arms is being conducted to assess EC plus FOLFIRI versus FOLFIRI with or without bevacizumab in patients with untreated BRAF V600E-mutant mCRC.26 Cohort 3 was added to the study after the approval of Protocol Amendment 5 and commenced after enrolment into phase III was completed.27 The focus of this review is the phase III portion of the BREAKWATER trial. The results of the safety lead-in portion (or cohort 3 of the BREAKWATER trial) will not be presented in further detail.
Although the BREAKWATER trial was originally designed to include EC alone as a treatment arm (arm A), enrolment into this arm was discontinued after the approval of Protocol Amendment 5. The report will not discuss the results pertaining to arm A in further detail, given that it is out of scope for the review.
The BREAKWATER trial is ongoing at 258 sites across 29 countries, including 5 centres in Canada. The phase III portion consisted of a 28-day screening period to assess the eligibility of patients for inclusion in the study. Using interactive response technology, a total of 479 patients (arms B and C) were randomized in a 1:1 ratio to receive either EC plus mFOLFOX6 (n = 236) or SOC chemotherapy (n = 243).
Randomization was stratified by ECOG Performance Status score (0 versus 1) and region (US and Canada versus Europe versus rest of world). Patients were required to receive their first dose of study drug within 5 days of randomization. Patients received their respective study intervention until disease progression (confirmed by blinded independent central review [BICR]), withdrawal of consent, loss to follow-up, unacceptable toxicity, or death. Patients with disease progression who continued to derive clinical benefit from the study intervention per the investigator were eligible to continue, provided the risk-benefit profile for doing so was favourable. Following treatment discontinuation, patients were contacted approximately every 3 months to assess survival status and the initiation of subsequent anticancer therapies or date of disease progression. Patients were then followed up until withdrawal of consent, loss to follow-up, death, or the final OS analysis, whichever occurred first. The dual primary end points of the BREAKWATER trial were PFS and ORR (both assessed by BICR); OS was measured as a key secondary end point. Other secondary end points measured in the BREAKWATER trial and of interest to this review included progression-free survival after next line of treatment (PFS2), DOR, health-related quality of life (HRQoL) as measured by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) instrument, and safety outcomes.
At the time of this review, 2 interim analyses of the phase III portion of the BREAKWATER trial had reported the efficacy and safety results of EC plus mFOLFOX6 compared with SOC chemotherapy. Interim Analysis 1 (IA1) (data cut-off date: December 22, 2023) reported the primary analysis for ORR, while Interim Analysis 2 (IA2) (data cut-off date: January 6, 2025) reported the primary analysis for PFS. Results for IA1 and IA2 are presented in this report.
Table 3: Characteristics of the Study Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
BREAKWATER trial Open-label, phase III, multicentre, 3-arm RCT Total N = 479a |
|
| Intervention: EC + mFOLFOX6
Comparators: SOC, based on investigator’s choice of the following regimens:
| Dual primary end points:
Key secondary end point:
Secondary end points:
|
AE = adverse event; BICR = blinded independent central review; CAPOX = capecitabine and oxaliplatin; dMMR = deficient mismatch repair; DOR = duration of response; EC = encorafenib and cetuximab; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FOLFOXIRI = 5-fluorouracil, leucovorin, oxaliplatin, and irinotecan; IA1 = Interim Analysis 1; IA2 = Interim Analysis 2; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; MMR = mismatch repair; MSI = microsatellite instability; MSI-H = microsatellite instability-high; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival after next line of treatment; q.2.w. = every 2 weeks; q.3.w. = every 3 weeks; q.d. = once daily; RCT = randomized controlled trial; SOC = standard of care.
aFour hundred and seventy-nine patients were randomized to receive either EC plus mFOLFOX6 (n = 236) or SOC chemotherapy (n = 243).
bAlternatively, 200 mg/m2 levo-leucovorin (through a 120-minute IV infusion) could be administered.
cLeucovorin could be coadministered with oxaliplatin or irinotecan infusion.
Sources: Kopetz et al. (2025);28 Elez et al. (2025);29 BREAKWATER Trial Protocol;27 BREAKWATER trial, IA1;30 BREAKWATER trial, IA2.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Testing and Analysis Populations
Approximately 620 patients were planned to be randomized, initially in a 1:1:1 ratio to receive 1 of EC alone, EC plus mFOLFOX6, or SOC chemotherapy.31 After the approval of Protocol Amendment 5, enrolment in the EC-alone arm was terminated, and patients were randomized 1:1 to receive either EC plus mFOLFOX6 or SOC chemotherapy. A total of 235 patients were planned for each of these arms.31
Approximately 250 PFS events by BICR were required to achieve at least 85% power to detect a hazard ratio (HR) of 0.67 using a 1-sided stratified log-rank test at a significance level of 0.023.31 A sample size of 220 patients per arm provided 90% power to detect a statistically significant benefit of ORR using a 1-sided chi-square test at a significance level of 0.001, assuming ORRs by BICR of 35% and 65% for the SOC arm and EC plus mFOLFOX6 arm, respectively.31 The sponsor estimated that 297 OS events would achieve 85% power to detect an HR of 0.70 using a 1-sided stratified log-rank test at a significance level of 0.023.
A hierarchical testing procedure was used to control the family-wise type I error rates for ORR, OS, and PFS. A summary of the multiple testing procedure used in the BREAKWATER trial is presented in the Supplemental Material document (Appendix 3, Figure 1).
The full analysis set (FAS) consisted of all patients who were randomized in the phase III portion of the trial. The primary and secondary end point analyses conducted at the time of the analysis for PFS were based on the FAS. The primary analysis for ORR and DOR was performed using a subset of the FAS, which comprised the first 110 patients randomized in each of the EC plus mFOLFOX6 and SOC chemotherapy arms (i.e., FAS including only the ORR subset). The safety analysis set consisted of all patients who received at least 1 dose of study intervention.
The censoring rules for PFS and OS are described in the Supplemental Material document (Appendix 3, tables 6, 7, and 8).
Patient Disposition
Patient disposition in the BREAKWATER trial is summarized in the Supplemental Material document (Appendix 4, Table 9).
A total of 1,047 patients were screened for participation, with 637 randomized to receive study intervention. Of these, 236 patients received EC plus mFOLFOX6, and 243 patients received SOC chemotherapy. At IA1, 137 patients who had received EC plus mFOLFOX6 (58.1%) and 82 patients who had received SOC chemotherapy (33.7%) continued to receive treatment in the study. At IA2, 67 patients who had received EC plus mFOLFOX6 (28.4%) and 16 patients who had received SOC chemotherapy (6.6%) continued to receive treatment in the study.
In both analyses, the most common reason for treatment discontinuation among both arms was progressive disease (IA1: 20.8% of patients receiving EC plus mFOLFOX6 versus 31.7% of patients receiving SOC chemotherapy; IA2: 42.8% versus 48.6%, respectively). Other reasons included AEs (IA1: 4.7% versus 9.1%; IA2: 7.2% versus 10.7%), death (IA1: 3.4% versus 4.1%; IA2: 3.8% versus 4.1%), withdrawal by patient (IA1: 5.5% versus 11.5%; IA2: 7.2% versus 15.2%), global deterioration in health status (IA1: 3.4% versus 1.6%; IA2: 4.2% versus 3.7%), and other (IA1: 4.2% versus 8.2%; IA2: 6.4% versus 11.1%).
Baseline Characteristics
The baseline demographic and disease characteristics of patients in the BREAKWATER trial are presented in Table 4.
Treatment Exposure, Concomitant Medications, and Subsequent Treatments
Details of patients’ treatment exposure and use of concomitant medications and subsequent treatments in the BREAKWATER trial are in the Supplemental Material document (Appendix 4, tables 10 to 13).
At the time of IA2, the median durations of treatment were 49.8 weeks (range, 1.3 to 161.9 weeks) in the EC plus mFOLFOX6 arm and 25.9 weeks (range, 2.0 to 150.0 weeks) in the SOC chemotherapy arm;26 41.4% of patients in the EC plus mFOLFOX6 arm and 10.9% of patients in the SOC chemotherapy arm received 60 or more weeks of study treatment.
The most common regimens administered in the SOC chemotherapy arm were mFOLFOX6 (39.9%), FOLFOXIRI (24.3%), and CAPOX (16.9%) (all 3 regimens administered with bevacizumab). Others included mFOLFOX6 (7.4%), FOLFOXIRI (3.3%), and CAPOX (2.5%) (all 3 regimens administered without bevacizumab). Lastly, 14 patients (5.8%) in the SOC chemotherapy arm did not receive treatment.
All patients in the EC plus mFOLFOX6 and SOC chemotherapy arms used concomitant medications. In both arms, the most frequently used concomitant medications were dexamethasone (65.1% for EC plus mFOLFOX6 compared with 61.6% for SOC chemotherapy), paracetamol (50.9% for EC plus mFOLFOX6 compared with 47.6% for SOC chemotherapy), and ondansetron (40.9% for EC plus mFOLFOX6 compared with 41.5% for SOC chemotherapy).
At the time of IA2, 45.8% of patients in the EC plus mFOLFOX6 arm and 57.2% of patients in the SOC chemotherapy arm had received subsequent systemic anticancer treatments. In both arms, the most frequently administered subsequent therapies were FOLFIRI-based regimens (24.2% for the EC plus mFOLFOX6 arm compared with 16.5% for the SOC chemotherapy arm), BRAF inhibitor–based regimens (8.1% for the EC plus mFOLFOX6 arm compared with 41.2% for the SOC chemotherapy arm), and single-drug chemotherapy–based regimens (7.2% for the EC plus mFOLFOX6 arm compared with 7.8% for the SOC chemotherapy arm).
Table 4: Summary of Baseline Characteristics in the BREAKWATER Trial
Characteristic | EC + mFOLFOX6 (n = 236) | SOC chemotherapy (n = 243) |
|---|---|---|
Demographic characteristics | ||
Age, years, n (%) | ||
Median (range) | 60.0 (24 to 81) | 62.0 (28 to 84) |
< 18 years | 0 | 0 |
18 to 64 years | 150 (63.6) | 139 (57.2) |
≥ 65 years | 86 (36.4) | 104 (42.8) |
≥ 75 years | 16 (6.8) | 24 (9.9) |
Sex, n (%) | ||
Female | 113 (47.9) | 124 (51.0) |
Male | 123 (52.1) | 119 (49.0) |
Race, n (%) | ||
Asian | 88 (37.3) | 91 (37.4) |
Black or African American | 0 (0.0) | 1 (0.4) |
Multiracial | 0 (0.0) | 2 (0.8) |
White | 141 (59.7) | 144 (59.3) |
Not reported | 7 (3.0) | 5 (2.1) |
Disease characteristics | ||
Body site, n (%) | ||
Colon, ascending | 78 (33.1) | 72 (29.6) |
Colon, descending | 18 (7.6) | 16 (6.6) |
Colon, rectosigmoid | 17 (7.2) | 16 (6.6) |
Colon, sigmoid | 30 (12.7) | 32 (13.2) |
Colon, transverse | 39 (16.5) | 35 (14.4) |
Colon, hepatic flexure | 9 (3.8) | 14 (5.8) |
Colon, splenic flexure | 1 (0.4) | 7 (2.9) |
Rectum | 24 (10.2) | 27 (11.1) |
Cecum | 20 (8.5) | 24 (9.9) |
Side of tumour, n (%) | ||
Left | 90 (38.1) | 98 (40.3) |
Right | 146 (61.9) | 145 (59.7) |
Stage at initial diagnosis, n (%) | ||
I | 3 (1.3) | 2 (0.8) |
II | 13 (5.5) | 10 (4.1) |
III | 38 (16.1) | 45 (18.5) |
IV | 182 (77.1) | 186 (76.5) |
Primary tumour resection, n (%) | ||
Complete | 116 (49.2) | 110 (45.3) |
Partial | 14 (5.9) | 11 (4.5) |
None | 106 (44.9) | 122 (50.2) |
Number of organs involved, n (%) | ||
≤ 2 | 119 (50.4) | 127 (52.3) |
≥ 3 | 117 (49.6) | 116 (47.7) |
Liver metastases, n (%)a | 147 (62.3) | 160 (65.8) |
ECOG Performance Status | ||
0 | 128 (54.2) | 131 (53.9) |
1 | 104 (44.1) | 98 (40.3) |
Missing | 4 (1.7) | 14 (5.8) |
Central BRAF V600E status | ||
Detected | 226 (95.8) | 224 (92.2) |
Indeterminate | 0 (0.0) | 1 (0.4) |
Not detected | 4 (1.7) | 2 (0.8) |
Not available | 6 (2.5) | 16 (6.6) |
Local MSI and/or dMMR status, n (%) | ||
MSI-H and/or dMMR | 1 (0.4) | 0 (0.0) |
MSS and/or pMMR | 229 (97.0) | 227 (93.4) |
Not available | 6 (2.5) | 16 (6.6) |
Carcinoembryonic antigen at baseline, n (%) | ||
≤ 5 mcg/litre | 64 (27.1) | 63 (25.9) |
> 5 mcg/litre | 167 (70.8) | 163 (67.1) |
Missing | 5 (2.1) | 17 (7.0) |
C-reactive protein at baseline, n (%) | ||
≤ 10 mcg/litre | 125 (53.0) | 118 (48.6) |
> 10 mcg/litre | 105 (44.5) | 108 (44.4) |
Missing | 6 (2.5) | 17 (7.0) |
dMMR = mismatch repair deficiency; EC = encorafenib and cetuximab; ECOG = Eastern Cooperative Oncology Group; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; MSI = microsatellite instability; MSI-H = microsatellite instability-high; MSS = microsatellite stable; pMMR = proficient mismatch repair; SOC = standard of care.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: BREAKWATER Trial IA2 Clinical Study Report.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
There were no concerns identified with respect to the randomization process conducted in the BREAKWATER trial. The clinical experts consulted by CDA-AMC indicated that the stratification of randomization according to ECOG Performance Status and geographical region was appropriate. The BREAKWATER trial had an open-label design and indicated that patients and investigators were not blinded to treatment. The trial assessed several efficacy end points (e.g., PFS, ORR, DOR) using BICR; therefore, there is low risk of bias in the measurement of these end points. However, the open-label design contributes to risk of bias in the measurement of HRQoL (which was self-reported) and PFS2 (which was assessed by investigators) — likely in favour of EC plus mFOLFOX6 — and in the reporting of subjective harms. Given that OS is an objective end point, the risk of bias in the measurement of this end point is low.
Due to the open-label nature of the trial, there are some concerns about risk of bias due to deviations from the intended interventions. Notably, at both interim analyses, more patients in the SOC chemotherapy arm discontinued study treatment due to withdrawal by patient (IA1: 5.5% in the EC plus mFOLFOX6 arm compared with 11.5% in the SOC chemotherapy arm; IA2: 7.2% in the EC plus mFOLFOX6 arm compared with 15.2% in the SOC chemotherapy arm) or to other reasons (IA1: 4.2% in the EC plus mFOLFOX6 arm compared with 8.2% in the SOC chemotherapy arm; IA2: 6.4% in the EC plus mFOLFOX6 arm compared with 11.1% in the SOC chemotherapy arm). The rationales for classifying patients in these categories were not clearly elaborated in the submitted materials. Thus, it is uncertain whether these reasons for treatment discontinuation could have influenced the interpretations of the outcomes.
The analyses of most efficacy end points in the BREAKWATER trial were performed using the FAS, which consisted of all patients who were randomized in the phase III portion of the trial. This is appropriate for measuring the effect of assignment to the intended interventions. The primary analysis of ORR was conducted on a subset of the FAS, which consisted of the first 110 patients randomized in each of the EC plus mFOLFOX6 and the SOC chemotherapy arms (i.e., the FAS including only the ORR subset). It is unclear whether the baseline characteristics of this subset of patients was balanced for important prognostic factors; however, the results from IA2, which were analyzed on the FAS, were consistent with those of the primary analysis. Of note, DOR was measured, as is typical, among patients who demonstrated a response to treatment. Thus, it should be considered that prognostic balance may not have been maintained within this subpopulation of patients. Differences in response duration could be driven by prognostic differences between the arms.
Within the SOC chemotherapy arm, 5.8% of patients did not receive treatment. The reasons for this were not clearly elaborated in the submitted materials; as a result, there is uncertainty about how this may have affected the interpretation of the results. However, given the small proportion, the CDA-AMC team did not anticipate a significant impact.
All patients used 1 or more concomitant medications during the trial. However, several imbalances in the type of concomitant medications used were noted between the 2 treatment arms. The clinical experts consulted by CDA-AMC noted that several imbalances are likely attributed to the use of comedications for prophylaxis measures and management of cetuximab-related side effects in the EC plus mFOLFOX6 arm. Such imbalances included larger proportions of patients in the EC plus mFOLFOX6 arm receiving doxycycline (17.2% versus 0.4% for the SOC chemotherapy arm) and hydrocortisone (14.7% versus 6.1% for the SOC chemotherapy arm). The clinical experts consulted by CDA-AMC did not expect these imbalances to affect the interpretation of the trial results.
A larger proportion of patients in the SOC chemotherapy arm received subsequent oncologic treatments (57.2%) compared with the EC plus mFOLFOX6 arm (45.8%). However, there were no protocol-related deviations related to subsequent oncologic treatment. Moreover, the clinical experts consulted by CDA-AMC indicated that the use of subsequent therapies in the BREAKWATER trial was largely aligned with those used among patients in clinical practice. Thus, the risk of bias related to the imbalance in subsequent oncologic treatments between arms was deemed low by the CDA-AMC review team.
At the time of this review, the BREAKWATER trial is ongoing; 2 interim analyses have been completed. The dual primary end point of ORR was met during IA1, and the dual primary end point of PFS and key secondary end point of OS were met during IA2. At the time of IA2, approximately 81.5% of the events required for the final analysis of OS had occurred. Thus, the CDA-AMC review team did not identify serious concerns about overestimation of the treatment effect.
For the assessment of PFS based on BICR, a large proportion of patients were censored due to the initiation of new anticancer therapy in both the EC plus mFOLFOX6 (22.5%) arm and the SOC chemotherapy arm (25.9%). However, the trial conducted a supplementary analysis based on a treatment policy strategy, which included observations that occur after the intercurrent event of initiating new anticancer therapy. The results of this supplementary analysis were consistent with those of the primary analysis for PFS by BICR, which suggested that censoring due to the initiation of anticancer therapy had little impact on the results for PFS. The CDA-AMC review team did not identify any concerns for informative censoring for the other survival end points (i.e., OS and PFS2).
A stratified Cox proportional hazards model, adjusted for stratification factors, was used to estimate the HRs for PFS. This model assumes proportional hazards across treatment arms. The Schoenfeld’s residual test indicated departure from the proportional hazards assumption for the primary analysis of PFS by BICR. However, the BREAKWATER trial performed an analysis based on restricted mean survival time differences for PFS by BICR, the results of which supported an efficacy benefit for EC plus mFOLFOX6 compared with SOC chemotherapy. The Kaplan-Meier (KM)–estimated probabilities of PFS at clinically relevant follow-up times are not subject to the proportional hazards assumption. For OS, there were no major concerns regarding departure from the proportional hazards assumption based on Schoenfeld’s residual test and visual inspection of the KM curves. Although Schoenfeld’s residual test was not performed for PFS2, the CDA-AMC review team did not identify any major concerns with respect to departure from the proportional hazards assumption based on visual inspection of the KM curves for this end point.
In both interim analyses of the BREAKWATER trial, a larger proportion of patients in the SOC chemotherapy arm versus the EC plus mFOLFOX6 arm were not evaluable for confirmed ORR (IA1: 5.5% in the EC plus mFOLFOX6 arm compared with 17.3% in the SOC chemotherapy arm; IA2: 7.3% in the EC plus mFOLFOX6 arm compared with 15.2% in the SOC chemotherapy arm). The imbalance in the proportions of patients who were not evaluable across the treatment arms was attributed to higher proportions of patients receiving SOC chemotherapy having no postbaseline assessment due to early death (IA1: 1.8% in the EC plus mFOLFOX6 arm compared with 4.5% in the SOC chemotherapy arm; IA2: 1.3% in the EC plus mFOLFOX6 arm compared with 4.5% in the SOC chemotherapy arm) or due to other reasons (IA1: 3.6% in the EC plus mFOLFOX6 arm compared with 8.2% in the SOC chemotherapy arm; IA2: 3.0% in the EC plus mFOLFOX6 arm compared with 6.6% in the SOC chemotherapy arm). The submitted evidence did not elaborate further on the underlying causes for patients having no postbaseline assessment due other reasons. Thus, it is unclear how the between-group imbalance pertaining to these patients could have influenced interpretations of the outcome. However, other possible reasons for patients who were not evaluable for ORR (e.g., no adequate baseline assessment, early stable disease, initiation of new anticancer therapy before the first postbaseline assessment, or late progressive disease) were low and balanced between the 2 treatment arms and were not expected to affect the interpretation of ORR.
HRQoL was measured in the BREAKWATER trial using the EORTC QLQ-C30. The clinical experts consulted by CDA-AMC agreed that the key time points for measuring HRQoL among patients with mCRC were 3 months (12 weeks), 6 months (24 weeks), and 12 months (48 weeks). Although the EORTC QLQ-C30 has not yet been validated in patients with BRAF V600E-mutant mCRC, it was previously validated in patients across several other cancer types, including CRC.32-34 According to a systematic review by Musoro et al. (2023),33 the between-group minimal important difference (MID) for clinically important improvement in EORTC QLQ-C30 Global Health Status (GHS)/QoL score is estimated as a difference of 6 points among patients with CRC. The same review also estimated a difference of 8 points as a between-group MID for clinically important deterioration in EORTC QLQ-C30 GHS/QoL score among the same patient population.33
Conclusions regarding change in mean EORTC QLQ-C30 score were further limited by the potential for bias in the measurement of the outcome owing to the open-label design of the trial (likely favouring the EC plus mFOLFOX6 arm) and to high rates of missing data at key time points. At 12, 24, and 48 weeks, 79%, 69%, and 48% of patients, respectively, in the EC plus mFOLFOX6 arm and 69%, 50%, and 16% of patients, respectively, in the SOC chemotherapy arm completed the assessments. The analysis of EORTC QLQ-C30 data in the BREAKWATER trial was conducted using a complete case analysis. Within this analysis, there were no imputations of missing outcome data, and data were assumed to be missing completely at random. Given that this assumption is likely unrealistic, there is risk of bias due to missing outcome data at the key time points assessed.
External Validity
The clinical experts consulted by CDA-AMC agreed that the eligibility criteria of the BREAKWATER trial were generally representative of patients with BRAF V600E-mutant mCRC in clinical practice. The clinical experts consulted by CDA-AMC added that the BRAF V600E mutation could be detected based on tissue or blood samples; however, they noted that tumour-based testing was more common in Canada. The clinical experts consulted by CDA-AMC noted that patients who have dMMR mutation status would be least suitable for treatment with encorafenib and should receive immunotherapy as a first-line treatment for mCRC. The clinical experts further noted that these patients would be treated with immunotherapy (e.g., pembrolizumab, nivolumab, and ipilimumab) in the first-line setting, whereas EC plus mFOLFOX6 may be offered as a second-line treatment after disease progression in patients receiving immunotherapy. The clinical experts consulted by CDA-AMC also noted that patients with appendiceal cancer are treated in a similar manner to patients with CRC and would be offered the same treatments used for CRC in clinical practice. The clinical experts consulted by CDA-AMC noted that the baseline characteristics of patients in the BREAKWATER trial were representative of patients with mCRC in clinical practice. However, they agreed that younger patients were more common in clinical practice (i.e., those younger than 61 years, which was the median age of patients in the trial). Of note, patients who identified as Black or African American, or as multiracial, were also underrepresented in the trial.
The BREAKWATER trial compared the efficacy and safety of EC plus mFOLFOX6 to SOC chemotherapy, which consisted of the study investigator’s choice of mFOLFOX6 with or without bevacizumab, FOLFOXIRI with or without bevacizumab, or CAPOX with or without bevacizumab. The dosing of EC plus mFOLFOX6 and regimens in the SOC chemotherapy arm administered in the BREAKWATER trial were aligned with their respective Health Canada product monographs. The chemotherapy regimens administered in the SOC chemotherapy arm are relevant first-line treatments for BRAF V600E-mutant mCRC. However, the exclusion of FOLFIRI as a chemotherapy regimen in the SOC chemotherapy arm is not aligned with clinical practice in Canada. The clinical experts consulted by CDA-AMC indicated that FOLFIRI is a common chemotherapy regimen administered for first-line mCRC. However, the experts agreed that doublet combination therapies, such as mFOLFOX6, FOLFIRI, and CAPOX, could be considered similar in terms of efficacy; safety and tolerability across regimens may vary. Furthermore, the clinical experts consulted by CDA-AMC agreed that the results pertaining to EC plus mFOLFOX6 in the BREAKWATER trial could be generalized to the use of EC in combination with other chemotherapy regimens (e.g., FOLFIRI or CAPOX) in clinical practice.
The clinical experts consulted by CDA-AMC noted that FOLFIRI and mFOLFOX6 (with or without bevacizumab) are the most common chemotherapy regimens administered for first-line BRAF V600E-mutant mCRC in clinical practice, whereas FOLFOXIRI and CAPOX are less common. The most commonly administered regimens in the SOC chemotherapy arm of the BREAKWATER trial were mFOLFOX6 plus bevacizumab, FOLFOXIRI plus bevacizumab, and CAPOX plus bevacizumab. Apart from the previously noted exclusion of FOLFIRI in the trial, the distribution of chemotherapy regimens among patients in the SOC chemotherapy arm is generally aligned with what is observed in clinical practice in Canada.
Limited details were provided regarding the rationale for the investigator’s choice of regimen in the SOC arm in the BREAKWATER trial. Thus, it is unclear whether the rationale for choice of regimen administered as SOC in the trial is aligned with the rationale used in clinical practice in Canada. The clinical experts consulted by CDA-AMC indicated that the choice of chemotherapy regimen for patients with mCRC is influenced by several factors, such as patient preference and the presence of comorbidities. For example, the clinical experts noted that FOLFIRI may be preferred over FOLFOX regimens among patients with peripheral neuropathy related to diabetes.
The clinical experts consulted by CDA-AMC agreed that the use of concomitant medications and subsequent anticancer therapies among patients in the BREAKWATER trial was representative of how patients with mCRC are treated in clinical practice. Notably, a larger proportion of patients in the SOC arm (41.2%) received a BRAF inhibitor–based combination therapy compared with those in the EC plus mFOLFOX6 arm (8.1%). The clinical experts consulted by CDA-AMC indicated that this observation aligned with the treatment paradigm for mCRC in clinical practice, given that BRAF inhibitor–based combination therapy would be a second-line treatment option among patients who received multidrug chemotherapy regimens as a first-line treatment for BRAF V600E-mutant mCRC. The clinical experts suggested that FOLFIRI-based chemotherapy would be a subsequent line of treatment after disease progression while receiving EC plus mFOLFOX6. Among patients in the EC plus mFOLFOX6 arm, 24.2% received FOLFIRI-based regimens. Although the clinical experts suggested that this estimate may be lower than expected in clinical practice, the experts indicated that the estimate reflects the fact that patients whose disease progresses while they receive therapy often experience rapid disease progression and may not be well enough to receive further therapy.
The BREAKWATER trial measured outcomes that were considered relevant to clinicians and patients with mCRC, including PFS, OS, and HRQoL. The clinical experts noted that ORR was a relevant clinical outcome in terms of measuring disease control; however, they considered ORR to be a supportive outcome to OS and PFS. Of note, the evidence underlying HRQoL (measured by EORTC QLQ-C30) was of very low certainty due to several limitations noted previously. Thus, although the BREAKWATER trial evaluated HRQoL among patients with BRAF V600E-mutant mCRC, the results of the trial do not present any conclusive evidence for this end point.
The BREAKWATER trial permitted the enrolment of patients aged 16 years and older. However, based on the reported baseline characteristics, no participants younger than 18 years were enrolled. According to the Health Canada product monograph for encorafenib, “The safety and effectiveness of Braftovi [encorafenib] have not been established in pediatric patients; therefore, Health Canada has not authorized an indication for pediatric use.”35
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in in Appendix 4 of the Supplemental Material document.
Efficacy
ORR by BICR
Full details of the results for ORR in the BREAKWATER trial are provided in the Supplemental Material document (Appendix 4, Table 14).
An assessment of the primary end point of ORR by BICR was performed for the first 110 patients randomized to each treatment arm (i.e., FAS including only the ORR subset) and was met at IA1. At the time of the data cut-off for this analysis, the ORR was 60.9% (95% confidence interval [CI], 51.6% to 69.5%) in the EC plus mFOLFOX6 arm compared with 40.0% (95% CI, 31.3% to 49.3%) in the SOC chemotherapy arm. The corresponding odds ratio for ORR was 2.443 (95% CI, 1.348 to 4.380; P = 0.0008).30 An updated analysis of ORR was performed at the time of IA2. The results were consistent with those of the primary analysis for ORR.26
Duration of Response
Results for DOR are presented in the Supplemental Material document (Appendix 4, Table 15).
PFS by BICR
Full details of the results for PFS in the BREAKWATER trial are provided in the Supplemental Material document (Appendix 4, Table 16). The KM curve for PFS2 is presented in Appendix 4, Figure 2).
PFS by BICR was met at IA2. At the time of the data cut-off for this analysis, the median durations of follow-up were 16.8 months (95% CI, 15.1 to 18.4 months) for patients in the EC plus mFOLFOX6 arm and 9.8 months (95% CI, 8.5 to 13.0 months) for patients in the SOC chemotherapy arm.
At the time of IA2, 254 PFS events had occurred (122 events [51.7%] in the EC plus mFOLFOX6 arm and 132 events [54.3%] in the SOC chemotherapy arm). The median PFS periods by BICR were 12.8 months (95% CI, 11.2 to 15.9 months) for patients in the EC plus mFOLFOX6 arm and 7.1 months (95% CI, 6.8 to 8.5 months) for patients in the SOC chemotherapy arm. The stratified HR for PFS by BICR for the EC plus mFOLFOX6 arm versus the SOC chemotherapy arm was 0.53 (95% CI, 0.407 to 0.677; P < 0.0001). The KM-estimated probabilities of PFS at 12 months were 54.3% (95% CI, 46.9% to 61.0%) for patients in the EC plus mFOLFOX6 arm and 29.3% (95% CI, 22.0% to 36.8%) for patients in the SOC chemotherapy arm ██████████████ ███████████ █████ ████ ███ ████ ██ ███████.26 The KM-estimated probabilities of PFS at 15 months were 44.4% (95% CI, 37.1% to 51.5%) for patients in the EC plus mFOLFOX6 arm and 20.2% (95% CI, 13.6% to 27.8%) for patients in the SOC chemotherapy arm (█████████████ ███████████ █████ ████ ███ ████ ██ ███████.26
Results of the planned sensitivity and supplementary analyses for PFS were consistent with those of the primary analysis.
Overall Survival
Full details of the OS results in the BREAKWATER trial are provided in the Supplemental Material document (Appendix 4, Table 17). The KM curve for PFS2 is presented in the Appendix 4, Figure 3.
At the time of IA2, the median follow-up durations for OS were 21.8 months (95% CI, 20.4 to 23.4 months) in the EC plus mFOLFOX6 arm and 22.2 months (95% CI, 18.9 to 23.5 months) in the SOC chemotherapy arm. A total of 242 OS events occurred (with 81.5% planned events required for the final analysis); these involved 94 patients (39.8%) in the EC plus mFOLFOX6 arm and 148 patients (60.9%) in the SOC chemotherapy arm.26 The median OS was 30.3 months (95% CI, 21.7 months to not estimable) for patients in the EC plus mFOLFOX6 arm compared with 15.1 months (95% CI, 13.7 to 17.7 months) for patients in the SOC chemotherapy arm.26 The corresponding HR for OS was 0.49 (95% CI, 0.375 to 0.632; P < 0.0001). The KM-estimated probabilities of OS at 12 months were 80.1% (95% CI, 74.4% to 84.7%) for patients in the EC plus mFOLFOX6 arm and 66.0% (95% CI, 59.4% to 71.7%) for patients in the SOC chemotherapy arm ██████████████ ███████████ █████ ████ ███ ███ ██ ██████.26 The KM-estimated probabilities of OS at 24 months were 52.0% (95% CI, 43.9% to 59.4%) for patients in the EC plus mFOLFOX6 arm and 29.0% (95% CI, 22.1% to 36.3%) for patients in the SOC chemotherapy arm ██████████████ ███████████ █████ ████ ███ ████ ██ ███████.26
PFS After Next Line of Treatment
Full details of the results for PFS2 in the BREAKWATER trial are provided in Supplemental Material document (Appendix 4, Table 18). The KM curve for PFS2 is presented in Appendix 4, Figure 4.
At the time of IA2, the median PFS2 durations measured by the investigators were 20.7 months (95% CI, 19.0 to 23.9 months) for patients in the EC plus mFOLFOX6 arm and 12.7 months (95% CI, 11.2 to 13.7 months) for patients in the SOC chemotherapy arm. The KM-estimated probabilities of PFS2 at 12 months were 74.5% (95% CI, 68.2% to 79.7%) for patients in the EC plus mFOLFOX6 arm and 51.7% (95% CI, 44.7% to 58.3%) for patients in the SOC chemotherapy arm ██████████████ ███████████ █████ ████ ███ ████ ██ ███████.26 The KM-estimated probabilities of PFS2 at 21 months were 48.6% (95% CI, 40.9% to 55.8%) for patients in the EC plus mFOLFOX6 arm and 20.4% (95% CI, 14.7% to 26.8%) for patients in the SOC chemotherapy arm ██████████████ ███████████ █████ ████ ███ ████ ██ ███████.26
EORTC QLQ-C30 GHS/QoL Scores
Full details of the results for mean change in EORTC QLQ-C30 GHS/QoL score from baseline in the BREAKWATER trial are provided in the Supplemental Material document (Appendix 4, Table 19).
Mean change in EORTC QLQ-C30 GHS/QoL score from baseline was reported at IA2. An increase indicates improvement in HRQoL. The between-group differences in mean change in EORTC QLQ-C30 GHS/QoL score from baseline for EC plus mFOLFOX6 compared with SOC chemotherapy were █████ ██████ ████ ███ ████ ██ ████ at week 12, █████ ██████ ████ ███ ████ ██ ████ at week 24, and ████ ██████ ████ ███ ████ ██ █████ at week 48. Of note, the between-group differences at clinically relevant follow-up times were not tested statistically.
Harms
Full details of the harms results in the BREAKWATER trial are presented in the Supplemental Material document (Appendix 4, Table 20). Key results include the following:
All patients in the EC plus mFOLFOX6 arm (100%) and almost all patients in the SOC chemotherapy arm (99.1%) experienced at least 1 AE. The most common AEs in the EC plus mFOLFOX6 arm were nausea (53.9% for EC plus mFOLFOX6 versus 49.8% for SOC chemotherapy), anemia (46.1% for EC plus mFOLFOX6 versus 25.3% for SOC chemotherapy), and diarrhea (41.8% for EC plus mFOLFOX6 versus 50.2% for SOC chemotherapy).
Serious adverse events (SAEs) occurred in 46.1% of patients who received EC plus mFOLFOX6 and 38.9% of patients who received SOC chemotherapy. No specific SAE occurred in more than 5% of patients in either arm. The most common SAEs in the EC plus mFOLFOX6 arm were intestinal obstruction (4.7% for EC plus mFOLFOX6 versus 2.2% for SOC chemotherapy), pyrexia (3.9% for EC plus mFOLFOX6 versus 1.3% for SOC chemotherapy), febrile neutropenia (0.9% for EC plus mFOLFOX6 versus 3.9% for SOC chemotherapy), and abdominal pain (2.6% for EC plus mFOLFOX6 versus 3.1% for SOC chemotherapy).
AEs leading to dose reductions of any study intervention occurred in 65.5% of patients in the EC plus mFOLFOX6 arm and in 54.1% of patients in the SOC chemotherapy arm. AEs leading to dose interruptions of any study intervention occurred in 65.5% of patients in the EC plus mFOLFOX6 arm and in 54.1% of patients in the SOC chemotherapy arm.
AEs leading to treatment discontinuation occurred in 26.7% of patients in the EC plus mFOLFOX6 arm and in 17.5% of patients in the SOC chemotherapy arm. No specific AE leading to treatment discontinuation occurred in more than 3% of patients in either arm. The most common AEs leading to treatment discontinuation were asthenia (3.0% for EC plus mFOLFOX6 versus 0.4% for SOC chemotherapy), anemia (2.6% for EC plus mFOLFOX6 versus 0% for SOC chemotherapy), lipase increase (2.2% for EC plus mFOLFOX6 versus 0.4% for SOC chemotherapy), and intestinal obstruction (0.9% for EC plus mFOLFOX6 versus 2.2% for SOC chemotherapy arm).
Rates of all-cause grade 5 AEs (i.e., those leading to death) were similar between the EC plus mFOLFOX6 arm (4.3%) and the SOC chemotherapy arm (4.4%).
Compared with the SOC chemotherapy arm, more patients in the EC plus mFOLFOX6 arm reported 1 or more AEs of special interest for encorafenib (79.3% for EC plus mFOLFOX6 compared with 65.9% for SOC chemotherapy). These AEs included:
cutaneous nonsquamous cell carcinoma (1.3% for EC plus mFOLFOX6 compared with 0.4% for SOC chemotherapy)
hepatic failure (1.7% for EC plus mFOLFOX6 compared with 1.3% for SOC chemotherapy)
myelosuppression (77.6% for EC plus mFOLFOX6 compared with 64.6% for SOC chemotherapy)
QT prolongation (6.9% for EC plus mFOLFOX6 compared with 2.2% for SOC chemotherapy)
hypomagnesemia (16.4% for EC plus mFOLFOX6 compared with 4.8% for SOC chemotherapy)
skin and subcutaneous tissue disorders (81.0% for EC plus mFOLFOX6 compared with 34.5% for SOC chemotherapy).
Summary of Findings and Certainty of the Evidence
A summary of findings for EC plus mFOLFOX6 compared with SOC chemotherapy among patients with mCRC is presented in Table 5. Literature-based MID estimates were used as the thresholds for clinically important between-group differences for the EORTC QLQ-C30 (MID = difference of 6 points for improvement or 8 points for deterioration).33 Refer to the summary of outcome measures in Appendix 3 of the Supplemental Material document. In the absence of literature-based MID estimates, thresholds for clinically important between-group differences suggested by the clinical experts were used for PFS (threshold: 10%), OS (threshold: 5%) PFS2 (threshold: 15%), and SAEs (threshold: 10%).
Table 5: Summary of Findings for EC Plus mFOLFOX6 vs. SOC Chemotherapy for Patients With mCRC
Outcome and follow-up | Patients (studies), N | Relative effects (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
SOC chemotherapy | EC + mFOLFOX6 (95% CI) | Difference (95% CI) | |||||
PFS in the FAS population, IA2 (data cut-off date: January 6, 2025) | |||||||
Probability of being alive and progression-free at 12 months Follow-up (median): EC + mFOLFOX6: 16.8 months SOC chemotherapy: 9.8 months | 479 (1 RCT) | NR | 293 per 1,000 | 543 per 1,000 (469 per 1,000 to 610 per 1,000) | ███ ████ ███ █████ ████ ████ ██ ███ ████ ███ ██████ | Higha,b | EC + mFOLFOX6 results in a clinically important increase in the probability of patients being alive and progression-free at 12 months compared with SOC chemotherapy. |
Probability of being alive and progression-free at 15 months Follow-up (median): EC + mFOLFOX6: 16.8 months SOC chemotherapy: 9.8 months | 479 (1 RCT) | NR | 202 per 1,000 | 444 per 1,000 (371 per 1,000 to 515 per 1,000) | ███ ████ ███ █████ ████ ████ ██ ███ ████ ███ ██████ | Higha,b | EC + mFOLFOX6 results in a clinically important increase in the probability of patients being alive and progression-free at 15 months compared with SOC chemotherapy. |
OS in the FAS population, IA2 (data cut-off date: January 6, 2025) | |||||||
Probability of being alive at 12 months Follow-up (median): EC + mFOLFOX6: 21.8 months SOC chemotherapy: 22.2 months | 479 (1 RCT) | NR | 660 per 1,000 | 801 per 1,000 (744 per 1,000 to 847 per 1,000) | ███ ████ ███ █████ ███ ████ ██ ███ ████ ███ ██████ | Higha,c | EC + mFOLFOX6 results in a clinically important increase in the probability of patients being alive at 12 months compared with SOC chemotherapy. |
Probability of being alive at 24 months Follow-up (median): EC + mFOLFOX6: 21.8 months SOC chemotherapy: 22.2 months | 479 (1 RCT) | NR | 290 per 1,000 | 520 per 1,000 (439 per 1,000 to 594 per 1,000) | ███ ████ ███ █████ ████ ████ ██ ███ ████ ███ ██████ | Higha,c | EC + mFOLFOX6 results in a clinically important increase in the probability of patients being alive at 24 months compared with SOC chemotherapy. |
PFS2 in the FAS population, IA2 (data cut-off date: January 6, 2025) | |||||||
Probability of being alive and progression-free after next line of therapy at 12 months Follow-up (median): EC + mFOLFOX6: ████ ██████ SOC chemotherapy: ████ ██████ | 479 (1 RCT) | NR | 517 per 1,000 | 745 per 1,000 (682 per 1,000 to 797 per 1,000) | ███ ████ ███ █████ ████ ████ ██ ███ ████ ███ ██████ | Lowa,d,e,f | EC + mFOLFOX6 may result in a clinically important increase in the probability of patients being alive and progression-free after the next line of therapy at 12 months compared with SOC chemotherapy. |
Probability of being alive and progression-free after next line of therapy at 21 months Follow-up (median): EC + mFOLFOX6: ████ ██████ SOC chemotherapy: ████ ██████ | 479 (1 RCT) | NR | 204 per 1,000 | 486 per 1,000 (409 per 1,000 to 558 per 1,000) | ███ ████ ███ █████ ████ ████ ██ ███ ████ ███ ██████ | Moderatea,d,e | EC + mFOLFOX6 likely results in a clinically important increase in the probability of patients being alive and progression-free after the next line of therapy at 21 months compared with SOC chemotherapy. |
Health-related quality of life, safety population, IA2 (data cut-off date: January 6, 2025)g | |||||||
Mean change from baseline in EORTC QLQ-C30 GHS/QoL score at week 24 | 254 (1 RCT) | NR | −0.1 | −3.3 (−7.1 to 0.5) | █████ █████ ██ ████ | Very lowa,d,h,i | The evidence is very uncertain about the effect of EC + mFOLFOX6 on the mean change from baseline in EORTC QLQ-C30 GHS/QoL score when compared with SOC chemotherapy at week 24. |
Mean change from baseline in EORTC QLQ-C30 GHS/QoL score at week 48 | 131 (1 RCT) | NR | −1.9 | 0.7 (−3.3 to 4.8) | ████ █████ ██ █████ | Very lowa,d,h,j | The evidence is very uncertain about the effect of EC + mFOLFOX6 on the mean change from baseline in EORTC QLQ-C30 GHS/QoL score when compared with SOC chemotherapy at week 48. |
Harms, safety population, IA2 (data cut-off date: January 6, 2025) | |||||||
Proportion of patients with 1 or more SAEs Follow-up (median): EC + mFOLFOX6: NR SOC chemotherapy: NR | 461 (1 RCT) | NR | 389 per 1,000 | 461 per 1,000 | 73 more per 1,000 (17 less per 1,000 to 163 more per 1,000) | Moderatea,d,k | EC + mFOLFOX6 likely results in little to no clinically important difference in the proportion of patients with ≥ 1 SAEs compared with SOC chemotherapy. |
CI = confidence interval; EC = encorafenib and cetuximab; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FAS = full analyses set; GHS = Global Health Status; GRADE = Grading of Recommendations Assessment, Development and Evaluation; IA2 = Interim Analysis 2; mCRC = metastatic colorectal cancer; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; MID = minimal important difference; NR = not reported; OS = overall survival; PFS = progression-free survival; PFS2 = progression-free survival after next line of treatment; QoL = quality of life; RCT = randomized controlled trial; SAE = serious adverse event; SOC = standard of care; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe values for the GRADE assessment of this outcome were sourced from an interim analysis. Although limitations regarding internal validity were identified regarding results from interim analyses, certainty was not rated down for study limitations because the CDA-AMC review team thought it was unlikely to have an important impact on the results.
bThere is no established between-group MID for the probability of PFS at 12 months or 15 months; however, the clinical experts consulted by CDA-AMC considered that a 10% difference between arms at these time points could be considered a threshold of clinical importance.
cThere is no established between-group MID for the probability of OS at 12 months or 24 months; however, the clinical experts consulted by CDA-AMC considered that a 5% difference between arms at these time points could be considered a threshold of clinical importance.
dThis end point was not statistically tested in the BREAKWATER trial. It should be considered as supportive evidence.
eRated down by 1 level for risk of bias because this outcome was measured by the study investigator, and its measurement is affected by the open-label study design.
fRated down by 1 level for serious imprecision. There is no established between-group MID for the PFS2 rate at 12 months or 21 months. However, the clinical experts consulted by CDA-AMC considered that a 15% difference between groups in the probability of patients who were alive and progression-free after next line of therapy at this time point could be considered a threshold of clinical importance. The point estimate and upper bound of the 95% CI for the between-group difference suggested a clinically important difference between the EC plus mFOLFOX6 and SOC chemotherapy arms; however, the lower bound of the 95% CI did not suggest a clinically important difference between the 2 arms.
gThe clinical experts consulted by CDA-AMC suggested that clinically important time points for assessing HRQoL for patients with mCRC were 12 weeks, 24 weeks, and 48 weeks. To assess the impact of treatment on long-term HRQoL, changes in mean EORTC QLQ-C30 score measured at 24 weeks and 48 weeks were included in the GRADE assessment. Although not formally included in the GRADE assessment, change in mean EORTC QLQ-C30 score measured at 12 weeks is described in the clinical report.
hRated down by 2 levels for risk of bias due to the reporting of the outcome being affected by the open-label study design and low number of patients eligible for analysis at the specified time point.
iRated down by 1 level for serious imprecision. According to a systematic review by Musoro et al. (2023),33 the between-group MID for clinically important improvement in EORTC QLQ-C30 GHS/QoL score is 6 points among patients with CRC. The same review also identified −8 points as the between-group MID for clinically important deterioration in EORTC QLQ-C30 GHS/QoL score among in the same patient population. Based on these thresholds, the point estimate and upper bound of the 95% CI suggested no clinically important difference between the EC plus mFOLFOX6 and SOC chemotherapy arms, whereas the lower bound of the 95% CI suggested a clinically important deterioration in HRQoL between the 2 arms.
jRated down by 1 level for serious imprecision. According to a systematic review by Musoro et al. (2023),33 the between-group MID for clinically important improvement in EORTC QLQ-C30 GHS/QoL score is 6 points among patients with CRC. The same review also identified −8 points as the between-group MID for clinically important deterioration in EORTC QLQ-C30 GHS/QoL score among the same patient population. Based on these thresholds, the point estimate and lower bound of the 95% CI suggested no clinically important difference between the EC plus mFOLFOX6 and SOC chemotherapy arms, whereas the upper bound of the 95% CI suggested a clinically important improvement in HRQoL between the 2 arms.
kRated down by 1 level for serious imprecision. There is no established between-group MID for the proportion of patients with 1 or more SAEs; however, the clinical experts consulted by CDA-AMC considered that a 10% difference between arms could be considered a threshold of clinical importance. The upper bound of the 95% CI for the between-group difference suggested a clinically important difference between the EC plus mFOLFOX6 arm and the SOC chemotherapy arm; however, the point estimate and lower bound of the 95% CI did not suggest a clinically important difference between the 2 arms.
Sources: BREAKWATER Trial Clinical Study Report, IA2.26 The between-group differences in the probabilities of PFS, OS, and PFS2, mean change in EORTC QLQ-C30 GHS/QoL score from baseline to week 24 and week 48, and proportion of patients with SAEs in this table were not part of the statistical analysis plan and were requested from the sponsor.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
No ITCs were included in this submission.
Appraisal of the Feasibility Assessment
The sponsor conducted an assessment to determine the feasibility of using ITCs to estimate the comparative efficacy of EC plus mFOLFOX6 versus relevant comparators for the treatment of adult patients with BRAF V600E-mutant mCRC. Full details of this assessment are presented in the Supplemental Material document (Appendix 6).
The CDA-AMC review team agreed that, based on the evidence informing the assessment, network meta-analyses were infeasible due to the identified biases (e.g., differences in study characteristics, eligibility criteria, treatment characteristics, and outcome assessment) and the lack of a common comparator available to compare EC plus mFOLFOX6 with FOLFIRI plus bevacizumab. The sponsor also proposed the assumption that FOLFIRI can be considered clinically equivalent to mFOLFOX6, FOLFOXIRI, and CAPOX. Under this assumption, the sponsor stated that unanchored ITCs were not required due to the availability of direct comparative evidence for EC plus mFOLFOX6 versus mFOLFOX6, FOLFOXIRI, and CAPOX (with or without bevacizumab) in the BREAKWATER trial. Although the CDA-AMC review team acknowledged the plausibility of this assumption, the team disagreed that it could be used to justify the use of BREAKWATER trial evidence in place of ITCs. In the BREAKWATER trial, there were no subgroups comparing EC plus mFOLFOX6 to each of the chemotherapy regimens administered in the SOC chemotherapy arm. Moreover, the sponsor did not submit additional evidence comparing efficacy and safety between the chemotherapy regimens administered in the SOC chemotherapy arm. Thus, considering the paucity of evidence supporting the sponsor’s assumption, there remains uncertainty as to whether these regimens could be considered equivalent in terms of efficacy and harms.
The clinical experts consulted by CDA-AMC agreed that doublet combination therapies (i.e., mFOLFOX6, FOLFIRI, and CAPOX) could be considered relatively similar in terms of efficacy. However, the clinical experts noted that there may be some differences between the regimens in terms of safety and tolerability.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in evidence were submitted by the sponsor.
Discussion
Efficacy
Compared with patients with wild-type mCRC, patients harbouring BRAF V600E-mutant mCRC tend to experience poorer clinical outcomes, such as reduced OS, PFS, and HRQoL. These patients also experience high attrition rates across subsequent lines of therapy and are less likely to receive treatment for mCRC in later-line settings, as per both the clinical experts consulted for this review and the input from clinician groups. Thus, there remains an unmet need for first-line treatments with novel mechanisms of action to improve clinical outcomes and HRQoL in this patient population. The BREAKWATER trial assessed the efficacy and safety of EC plus mFOLFOX6 compared with SOC chemotherapy as a first-line treatment among patients with BRAF V600E-mutant mCRC.
The Health Canada–approved indication for this review is as follows: Encorafenib, in combination with cetuximab and mFOLFOX6, for the treatment of patients with mCRC with a BRAF V600E mutation, as detected by a validated test. The clinical experts consulted by CDA-AMC noted that it would be reasonable to allow treating physicians discretion to combine EC with alternative doublet chemotherapy regimens, such as FOLFIRI or CAPOX, in clinical practice. The clinical experts consulted by CDA-AMC agreed that doublet combination therapies (i.e., mFOLFOX6, FOLFIRI, and CAPOX) could be considered relatively similar in terms of efficacy. However, the clinical experts noted that there may be some differences between the regimens in terms of safety and tolerability. According to the experts consulted, patients with early recurrence (i.e., within 6 months of the start of adjuvant treatment with mFOLFOX6) may preferably receive EC plus FOLFIRI, given that these patients are considered resistant to mFOLFOX6. Patients’ preferences or comorbidities may also guide the choice of chemotherapy backbone. For instance, mFOLFOX6 requires a central catheter, which may be challenging for some patients, while other patients may have diabetes-related peripheral neuropathy, making FOLFIRI a more suitable option.
The BREAKWATER trial assessed PFS by BICR as a dual primary outcome at IA2. Based on the clinical experts’ identified threshold of 10%, EC plus mFOLFOX6 resulted in a clinically important increase in the probability of patients being alive and progression-free at 12 months and 15 months compared with SOC chemotherapy. However, there was greater uncertainty in the KM estimates of the probability of PFS at time points beyond 15 months due to the small number of patients at risk. The results of the prespecified sensitivity and supplementary analyses were consistent with those of the primary assessment.
The BREAKWATER trial assessed OS as a key secondary outcome at IA2. Based on the clinical experts’ identified threshold of 5%, EC plus mFOLFOX6 resulted in a clinically important increase in the probability of patients being alive at 12 months and 24 months compared with SOC chemotherapy. There was greater uncertainty in the KM estimates of the OS rate at time points beyond 24 months due to the small number of patients at risk.
The BREAKWATER trial assessed ORR among patients in the FAS including only the ORR subset (i.e., the first 110 patients randomized to each arm) as a dual primary outcome at IA1. At IA2, an updated analysis of ORR was conducted using the FAS, and it was not adjusted for multiplicity. The results of this analysis demonstrated that patients treated with EC plus mFOLFOX6 continued to experience increased ORR compared with those receiving SOC chemotherapy. Of note, ORR was not formally included in the GRADE assessment because the clinical experts consulted by CDA-AMC noted that ORR was considered a supportive outcome to OS and PFS. However, the clinical experts consulted by CDA-AMC agreed that the benefit in ORR observed with EC plus mFOLFOX6 compared with SOC chemotherapy in the BREAKWATER trial was clinically meaningful.
PFS2 was assessed as a secondary outcome in the BREAKWATER trial. According to the threshold of 15% identified by the clinical expert, low-certainty evidence suggests that EC plus mFOLFOX6 may result in a clinically important increase in the probability of patients being alive and progression-free after the next line of therapy at 12 months. According to the same threshold, moderate-certainty evidence suggests that EC plus mFOLFOX6 likely results in a clinically important increase in the probability of patients being alive and progression-free after the next line of therapy at 21 months. The evidence informing the PFS2 rate at both time points was rated down because this outcome was assessed by the study investigator (reporting may have been affected by the open-label design of the trial). Evidence informing the PFS2 rate at 12 months was further rated down due to imprecision in the estimates.
Patients and clinicians identified improvement in HRQoL as an important goal of treatment for patients with mCRC. HRQoL as measured using the EORTC QLQ-C30 was a key end point of interest measured in the BREAKWATER trial. No literature that identified MIDs pertaining to change in EORTC QLQ-C30 score over time among patients with BRAF-mutant mCRC was identified. However, according to a systematic review by Musoro et al. (2023),33 the between-group MID for clinically important improvement in EORTC QLQ-C30 GHS/QoL score is 6 points in patients with CRC. The same review also identified −8 points as the between-group MID for clinically important deterioration in EORTC QLQ-C30 GHS/QoL score in the same patient population.33 According to these thresholds, the evidence is very uncertain about the effect of EC plus mFOLFOX6 on the mean change in EORTC QLQ-C30 GHS/QoL score from baseline to weeks 24 and 48 compared with SOC chemotherapy. At both time points, the evidence informing mean change in EORTC QLQ-C30 GHS/QoL score from baseline was rated down due to imprecision in the estimates. The interpretation of these results was further limited by the open-label study design and low numbers of patients eligible to complete the questionnaires, particularly at longer follow-up time points, in both arms.
The sponsor also conducted a feasibility assessment of ITCs comparing the efficacy (e.g., ORR, PFS, OS) of EC plus mFOLFOX6 compared to relevant treatments for BRAF V600E-mutant mCRC. The conclusion of this assessment was that the ITCs were not feasible due to sources of bias (e.g., differences in study characteristics, eligibility criteria, treatment characteristics, and outcome assessment) that could not be adjusted for using traditional ITC methods, as well as the lack of common comparator arms comparing EC plus mFOLFOX6 with FOLFIRI plus bevacizumab. Of note, the sponsor stated that unanchored ITCs were not required because FOLFIRI can be considered clinically equivalent to other relevant first-line chemotherapy regimens for mCRC (i.e., mFOLFOX6, FOLFOXIRI, and CAPOX) and because direct evidence is available in the BREAKWATER trial comparing EC plus mFOLFOX6 to SOC chemotherapy. However, the lack of data measuring the efficacy A compared with each regimen in the SOC chemotherapy arm (e.g., subgroup data stratified by SOC chemotherapy regimen in the BREAKWATER trial) contributes to uncertainty in the sponsor’s assumption. The sponsor did not submit any evidence to support the assertion that the first-line chemotherapy regimens are equivalent in benefits and harms.
The clinical experts consulted by CDA-AMC agreed that doublet combination therapies (i.e., mFOLFOX6, FOLFIRI, and CAPOX) could be considered relatively similar in terms of efficacy. However, the clinical experts noted that there may be some differences between these regimens in terms of safety and tolerability.
This review highlighted a few considerations pertaining to the generalizability of the results of the BREAKWATER trial to clinical practice in Canada. For instance, the clinical experts consulted for this review noted that patients who have MSI-high or dMMR mutation status would be least suitable for treatment with encorafenib and should receive immunotherapy as first-line treatment for mCRC. Moreover, those who identified as Black or multiracial were underrepresented in the BREAKWATER trial. The chemotherapy regimens administered as a part of the SOC chemotherapy arm are relevant first-line treatments for BRAF V600E-mutant mCRC. However, the exclusion of FOLFIRI as a regimen from the SOC chemotherapy arm is not aligned with clinical practice in Canada, given that the clinical experts consulted by CDA-AMC indicated that FOLFIRI is a common chemotherapy regimen administered for first-line mCRC. Due to this difference, the overall distribution of chemotherapy regimens administered in the BREAKWATER trial differs from that observed in clinical practice in Canada.
Harms
In the BREAKWATER trial, all patients who received EC plus mFOLFOX6 and 99.1% of patients who received SOC chemotherapy experienced at least 1 AE. The most common AEs in the EC plus mFOLFOX6 arm were nausea, anemia, and diarrhea, whereas the most common AEs in the SOC chemotherapy arm were diarrhea, nausea, and fatigue. The incidence of SAEs was numerically higher in the EC plus mFOLFOX6 arm compared with SOC chemotherapy, with intestinal obstruction being the most common SAE among the EC plus mFOLFOX6 arm and febrile neutropenia being the most common SAE among the SOC chemotherapy arm. Moderate-certainty evidence suggests that EC plus mFOLFOX6 likely results in little to no difference in the proportion of patients with SAEs compared with SOC chemotherapy. Evidence was rated down due to imprecision in the estimates based on a 10% threshold for a clinically important difference suggested by the clinical experts consulted for the review. Incidences of dose reduction, dose interruption, and discontinuation due to AEs were higher among patients who received EC plus mFOLFOX6 than among those who received SOC chemotherapy. Similar rates of all-cause grade 5 AEs (i.e., those leading to death) were observed among patients who received EC plus mFOLFOX6 compared to those who received SOC chemotherapy.
Based on the Health Canada product monograph for encorafenib and validation from the clinical experts consulted by CDA-AMC, the key AEs of special interest related to encorafenib included myelosuppression events, QT prolongation, hepatic failure, cutaneous nonsquamous cell carcinoma, and facial paresis. The clinical experts noted that febrile neutropenia is an AE of special interest because patients with this AE are more likely to be hospitalized. Higher rates of overall myelosuppression events and QT prolongation were observed among patients treated with EC plus mFOLFOX6 compared with SOC chemotherapy. However, rates of febrile neutropenia were lower among patients treated with EC plus mFOLFOX6 compared with SOC chemotherapy. Rates of hepatic failure, nonsquamous cell carcinoma, and facial paresis were low (i.e., less than 2%) and similar between the treatment arms. The clinical experts consulted by CDA-AMC indicated that hypomagnesemia and skin toxicities should be considered AEs of special interest related to EC plus mFOLFOX6, given that these are common AEs related to cetuximab. As expected, rates of hypomagnesemia and common skin toxicities (e.g., rash, alopecia, and dermatitis acneiform) were higher among the EC plus mFOLFOX6 arm than among the SOC chemotherapy arm. The clinical experts consulted by CDA-AMC agreed that the safety profile of EC plus mFOLFOX6 demonstrated in the BREAKWATER trial is consistent with what is generally observed with the individual components of this regimen in clinical practice. Despite noting higher incidences of anemia in the EC plus mFOLFOX6 arm compared with SOC chemotherapy, the clinical experts consulted by CDA-AMC agreed that the AEs related to treatment with EC plus mFOLFOX6 were generally manageable in clinical practice. The clinical experts agreed that there are differences in safety and tolerability among SOC chemotherapy regimens. Because there is a lack of safety data pertaining to EC plus mFOLFOX6 compared to each of the individual regimens in the SOC chemotherapy arm, the safety profile of EC plus mFOLFOX6 compared with each chemotherapy regimen remains unclear.
Ethics and Equity Considerations
According to the patient group input submitted to CDA-AMC, interviewed patients and caregivers highlighted the need for better, quicker, equitable access to funded drug therapies and clinical trials in Canada. The clinical experts consulted by CDA-AMC indicated that treatment with EC plus mFOLFOX6 should be administered by a specialist in medical oncology. They noted that patients living in remote areas may not have access to such a specialist, but suggested these patients may receive treatment at a local hospital, with ongoing follow-up every few months at a cancer centre. The clinical experts consulted by CDA-AMC agreed that all patients with BRAF V600E-mutant mCRC would be eligible for treatment. Of note, patients who identified as Black or multiracial were underrepresented in the BREAKWATER trial, with only 1 patient (who received SOC chemotherapy) in the trial. In Canada, patients who identify as Black often face barriers to several aspects of cancer care, including screening, access to targeted therapies, and enrolment in clinical trials.36
Conclusion
There remains an unmet need for first-line treatments with novel mechanisms of action to improve clinical outcomes and HRQoL among patients with BRAF V600E-mutant mCRC. One phase III, open-label RCT was included in this review. The BREAKWATER trial evaluated the efficacy and safety of EC plus mFOLFOX6 compared with SOC chemotherapy among patients with previously untreated BRAF V600E-mutant mCRC. The dual primary end points of the BREAKWATER trial were ORR and PFS, while OS was evaluated as a key secondary end point. Results from the GRADE assessment indicated that EC plus mFOLFOX6 resulted in clinically important benefits in PFS and OS compared to SOC chemotherapy. Although ORR was not formally included in the GRADE assessment, the clinical experts consulted by CDA-AMC indicated that treatment with EC plus mFOLFOX6 was associated with clinically meaningful improvements in this outcome compared with SOC chemotherapy. Low- to moderate-certainty evidence suggests that EC plus mFOLFOX6 may result in clinically important benefits in PFS2 compared to SOC chemotherapy. The evidence is very uncertain about the effect of EC plus mFOLFOX6 on HRQoL compared with SOC chemotherapy. The interpretation of these results is limited by the open-label design of the trial and the low number of patients who were eligible to complete the EORTC QLQ-C30 at all clinically relevant follow-up times in both arms. Overall, no new safety signals were observed with EC plus mFOLFOX6 in the BREAKWATER trial; in addition, treatment with EC plus mFOLFOX6 likely resulted in little to no difference in the incidence of SAEs. The distribution of chemotherapy regimens administered in the SOC chemotherapy arm of the BREAKWATER trial differs from that seen in clinical practice in Canada, given that the SOC chemotherapy arm did not include the FOLFIRI regimen. The clinical experts consulted by CDA-AMC agreed that the efficacy of FOLFIRI and other regimens administered in the SOC chemotherapy arm were similar; however, they noted differences in the regimens’ safety and tolerability. The lack of subgroup data stratified by SOC chemotherapy regimen in the BREAKWATER trial limits the ability to compare the efficacy and safety of EC plus mFOLFOX6 with each individual regimen used in the SOC arm.
Economic Review
Methods
The CDA-AMC review team reviewed and critically appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of Braftovi in combination with cetuximab and mFOLFOX6 compared to chemotherapy with or without bevacizumab for the treatment of patients with mCRC who have a BRAF V600E mutation.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of EC plus mFOLFOX6 from the perspective of a public health care payer over a 40-year time horizon. The modelled population comprised patients with mCRC who have a BRAF V600E mutation; this aligns with the Health Canada population and was based on the participants in the BREAKWATER trial. The sponsor’s base-case analysis included costs related to drug acquisition and administration, medical resource use, AEs, and terminal care.
In the sponsor’s base case, EC plus mFOLFOX6 was associated with an incremental cost of $226,722 and 0.85 incremental quality-adjusted life-years (QALYs) relative to SOC chemotherapy. This resulted in an incremental cost-effectiveness ratio (ICER) of $265,036 per QALY gained. In a scenario analysis in which FOLFIRI was used with EC instead of mFOLFOX6, the incremental cost increased to $277,963, resulting in an ICER of $326,596 per QALY gained.
Approximately 43.5% of the incremental benefit compared to SOC chemotherapy (0.85 incremental QALYs) was predicted to be accrued after the observation period of the BREAKWATER trial (average observation period = 3.2 years). Additional information about the sponsor’s submission is summarized in Appendix 10 of the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 6; full details are provided in Appendix 11 of the Supplemental Material document).
Table 6: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The costs of leucovorin and bevacizumab were incorrect in the sponsor’s model. | The sponsor used the list price per 5 mg tablet of leucovorin instead of the price of the IV form. The IV form of leucovorin is included in the mFOLFOX6 and FOLFOXIRI chemotherapy regimens. The list price of bevacizumab decreased in January 2025; however, the sponsor used the previous list price. | CDA-AMC updated the cost of leucovorin based on the 5 mL vial and updated the cost of bevacizumab in the economic model and budget impact analysis. | CDA-AMC explored the impact of using the 50 mL vial of leucovorin instead of the 5 mL vial. |
The sponsor applied RDIs to treatment costs. | Applying RDIs to the treatment costs based on the BREAKWATER trial likely underestimates the cost of treatment to the health system. | CDA-AMC applied 100% RDIs to treatment costs. | CDA-AMC applied mean RDIs from the BREAKWATER trial to treatment costs. |
The distribution of subsequent treatments was informed by clinical expert opinion. | Efficacy estimates informing the economic model were from the BREAKWATER trial, whereas the distribution of subsequent treatment was informed by clinical expert opinion. This leads to a misalignment between treatment efficacy outcomes and subsequent treatment costs. | CDA-AMC used the distribution of subsequent treatment from the BREAKWATER trial. | CDA-AMC used the distribution of subsequent treatment informed by clinical expert opinion sourced by the sponsor and validated by clinical experts consulted by CDA-AMC. |
CDA-AMC = Canada’s Drug Agency; RDI = relative dose intensity.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 of the Supplefmental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6) in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 11 of the Supplemental Material document.
Impact on Health Care Costs
EC plus mFOLFOX6 is predicted to be associated with additional health care costs compared to SOC chemotherapy (incremental cost = $367,543). This increase in health care spending results from increased drug acquisition costs in all treatment lines associated with EC plus mFOLFOX6 (refer to Figure 1).
Figure 1: Impact of EC Plus mFOLFOX6 vs. SOC Chemotherapy on Health Care Costs
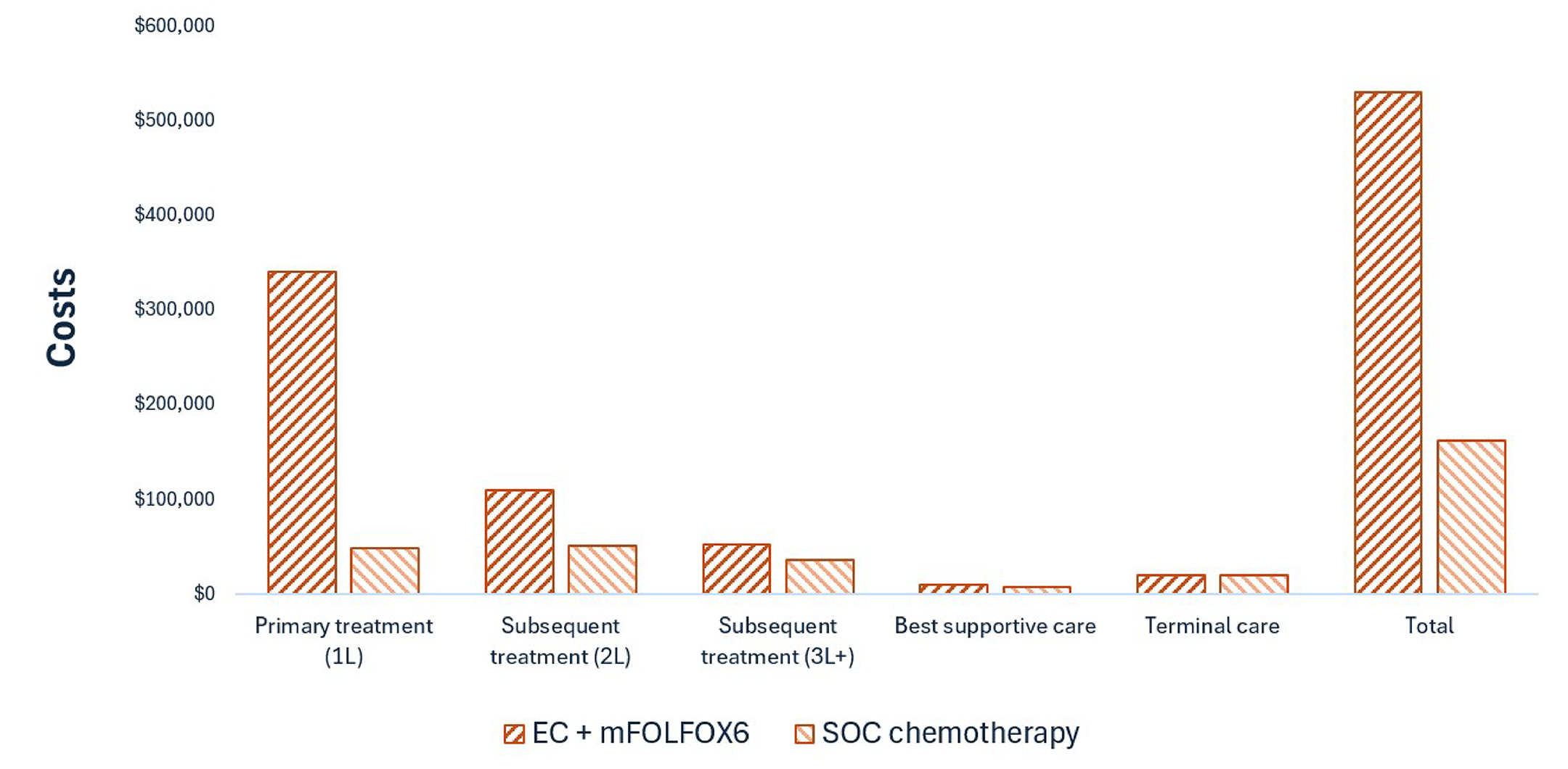
1L = first line; 2L = second line; 3L+ = third line and beyond; EC = encorafenib and cetuximab; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; SOC = standard of care; vs. = versus.
Note: Treatment costs include those associated with drug acquisition, drug administration, routine management, and adverse events.
Impact on Health
Relative to SOC chemotherapy, EC plus mFOLFOX6 is predicted to increase the amount of time a patient remains in the progression-free health state by approximately 7.5 months (refer to Figure 2) and to extend OS by 12 months. Considering the impact of treatment on both quality and length of life, EC plus mFOLFOX7 is predicted to result in 0.85 additional QALYs per patient compared to SOC chemotherapy. Approximately 43% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 2: Impact of EC Plus mFOLFOX6 vs. SOC Chemotherapy on Patient Health
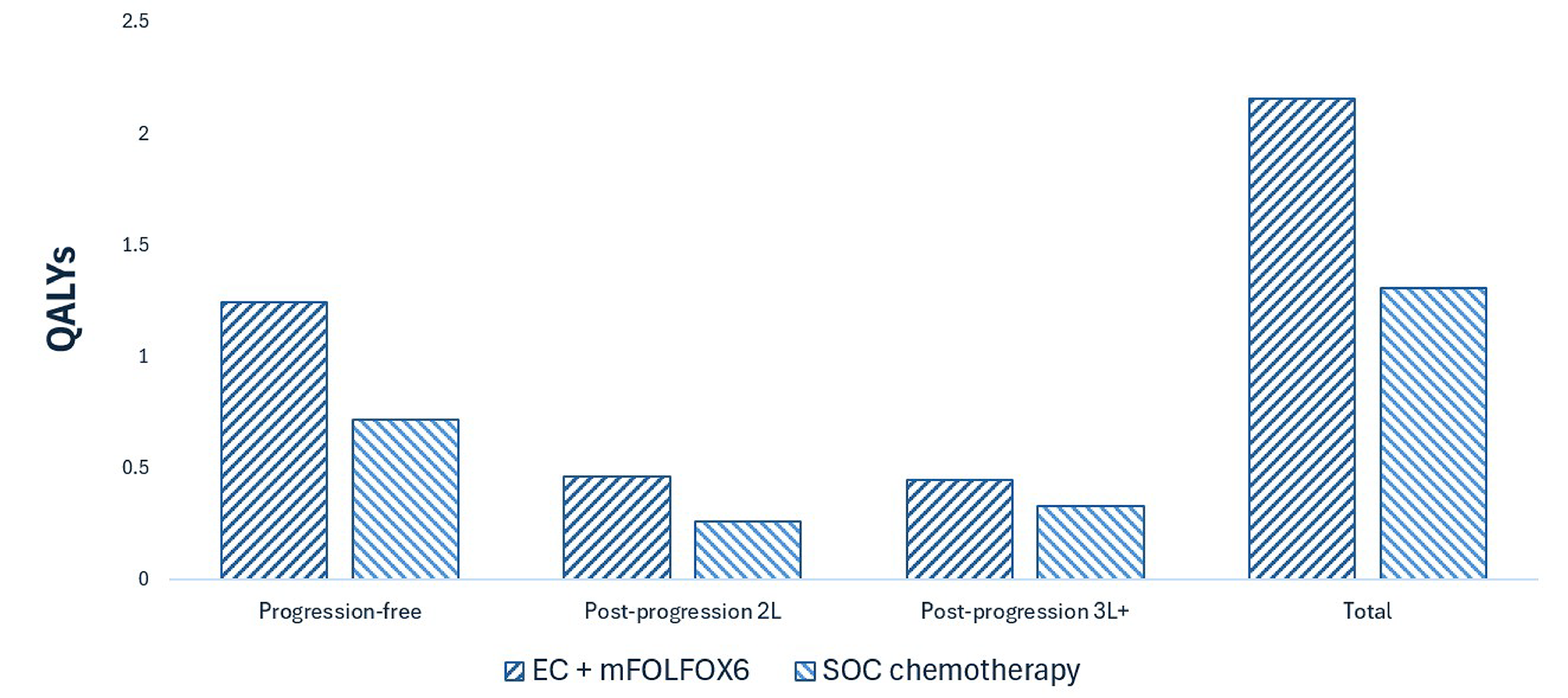
2L = second line; 3L+ = third line and beyond; EC = encorafenib and cetuximab; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; SOC = standard of care; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $432,400 per QALY gained for EC plus mFOLFOX6 compared to SOC chemotherapy (refer to Table 7). Additional details on the CDA-AMC base case are available in Appendix 11 of the Supplemental Material document.
Table 7: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Total LYs | ICER vs. SOC chemotherapy ($/QALY) |
|---|---|---|---|---|
SOC chemotherapy | 162,196 | 1.31 | 1.61 | Reference |
EC + mFOLFOX6 | 529,739 | 2.16 | 2.65 | 432,400 |
CDA-AMC = Canada’s Drug Agency; EC = encorafenib and cetuximab; ICER = incremental cost-effectiveness ratio; LY = life-year; mFOLFOX6 = modified 5-fluorouracil, leucovorin, and oxaliplatin; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored in the scenario analyses outlined in Table 6. Uncertainty around the distribution of subsequent treatment had the largest impact on cost-effectiveness (refer to Table 28 in the Supplemental Material document).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing EC plus mFOLFOX6 for use in the Health Canada–indicated population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of encorafenib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 of the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 2,012 patients would be eligible for EC plus mFOLFOX6 over a 3-year period (year 1 = 659; year 2 = 671; year 3 = 683), of whom 822 patients could be expected to receive EC plus mFOLFOX6 (year 1 = 21; year 2 = 228; year 3 = 573). The estimated incremental budget impact of reimbursing encorafenib in combination with cetuximab and mFOLFOX6 is predicted to be approximately $175 million over the first 3 years, with an expected expenditure of $70 million on encorafenib. The actual budget impact of reimbursing encorafenib in combination with cetuximab and mFOLFOX6 will depend on its uptake, which is likely underestimated in the CDA-AMC base case.
Conclusion
Based on the CDA-AMC base case, EC plus mFOLFOX6 would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $432,400 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 27 in the Supplemental Material document).
The budget impact to the public drug plans of reimbursing encorafenib for use in this context in the first 3 years is estimated to be approximately $175 million. The 3-year expenditure on encorafenib (i.e., not accounting for current expenditure on comparators) is estimated to be $70 million. The estimated budget impact is highly uncertain due to uncertainty regarding the uptake of EC plus mFOLFOX6.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
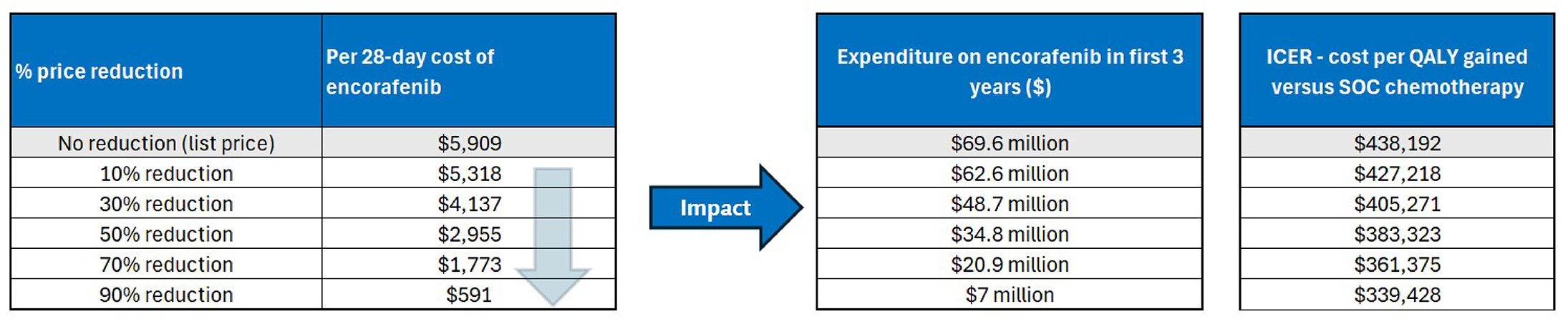
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: Expenditure includes only the drug cost of encorafenib. Given that encorafenib is provided as part of EC plus mFOLFOX6, the expenditure for the full combination is higher than what is presented here. Deterministic results are presented.
References
1.Duan B, Zhao Y, Bai J, et al. Colorectal Cancer: An Overview. In: Morgado-Diaz JA, ed. Gastrointestinal Cancers [Internet]. Brisbane (AU): Exon Publications; 2022:1-12:chap 1.
2.American Cancer Society. What Is Colorectal Cancer? 2024. Accessed 10 April. https://www.cancer.org/cancer/types/colon-rectal-cancer/about/what-is-colorectal-cancer.html
3.Brenner DR, Gillis J, Demers AA, et al. Projected estimates of cancer in Canada in 2024. CMAJ. 2024;196(18):E615-E623. doi:10.1503/cmaj.240095PubMed
4.CADTH. CADTH Reimbursement Review: Encorafenib (Braftovi) in metastatic colorectal cancer [2L]. Canadian Journal of Health Technologies. 2021;1(9):1-170.
5.Tabernero J, Ros J, Elez E. The Evolving Treatment Landscape in BRAF-V600E-Mutated Metastatic Colorectal Cancer. Am Soc Clin Oncol Educ Book. 2022;42:1-10. doi:10.1200/EDBK_349561 PubMed
6.Sutherland RL, Boyne DJ, Brenner DR, Cheung WY. The Impact of BRAF Mutation Status on Survival Outcomes and Treatment Patterns among Metastatic Colorectal Cancer Patients in Alberta, Canada. Cancers (Basel). 2023;15(24):1-11. doi:10.3390/cancers15245748 PubMed
7.Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol. 2015;6(6):660-7. doi:10.3978/j.issn.2078-6891.2015.077 PubMed
8.Chu JE, Johnson B, Kugathasan L, et al. Population-based Screening for BRAF (V600E) in Metastatic Colorectal Cancer Reveals Increased Prevalence and Poor Prognosis. Clin Cancer Res. 2020;26(17):4599-4605. doi:10.1158/1078-0432.CCR-20-1024 PubMed
9.Benson AB, Venook AP, Adam M, et al. Colon Cancer, Version 3.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024;22(2 D)doi:10.6004/jnccn.2024.0029
10.CDA-AMC. Provisional Funding Algorithm for Metastatic Colorectal Cancer (February 2025) [sponsor submitted reference]. 2025. Accessed 01 May, 2025. https://www.cda-amc.ca/sites/default/files/2025-01/ph0062-mcrc_draft_algorithm_report.pdf
11.Cervantes A, Adam R, Rosello S, et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(1):10-32. doi:10.1016/j.annonc.2022.10.003 PubMed
12.Morris VK, Kennedy EB, Baxter NN, et al. Treatment of Metastatic Colorectal Cancer: ASCO Guideline. J Clin Oncol. 2023;41(3):678-700. doi:10.1200/JCO.22.01690 PubMed
13.Ellison LF, Saint-Jacques N. Five-year cancer survival by stage at diagnosis in Canada. Health Rep. 2023;34(1):3-15. doi:10.25318/82-003-x202300100001-eng PubMed
14.Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306-15. doi:10.1016/S1470-2045(15)00122-9 PubMed
15.Thomsen M, Guren MG, Skovlund E, et al. Health-related quality of life in patients with metastatic colorectal cancer, association with systemic inflammatory response and RAS and BRAF mutation status. Eur J Cancer. 2017;81:26-35. doi:10.1016/j.ejca.2017.04.026 PubMed
16.Innocenti F, Ou F-S, Zemla T, et al. Somatic DNA mutations, MSI status, mutational load (ML): Association with overall survival (OS) in patients (pts) with metastatic colorectal cancer (mCRC) of CALGB/SWOG 80405 (Alliance). Journal of Clinical Oncology. 2017;35(15_suppl):3504-3504. doi:10.1200/JCO.2017.35.15_suppl.3504
17.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386-422. doi:10.1093/annonc/mdw235 PubMed
18.Yu IS, Aubin F, Goodwin R, et al. Tumor Biomarker Testing for Metastatic Colorectal Cancer: a Canadian Consensus Practice Guideline. Ther Adv Med Oncol. 2022;14:1-29. doi:10.1177/17588359221111705 PubMed
19.Yu IS, Aubin F, Goodwin R, et al. Tumor Biomarker Testing for Metastatic Colorectal Cancer: a Canadian Consensus Practice Guideline. Ther Adv Med Oncol. 2022;14:17588359221111705. doi:10.1177/17588359221111705 PubMed
20.National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology: Colon Cancer, Version 4.2025. 2025. Accessed October 01, 2025. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf
21.National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology: Rectal Cancer, Version 3.2025. 2025. Accessed October 01, 2025. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf
22.Cervantes A, Candia Montero L, Pentheroudakis G, Martins-Branco D, Ducreux M, Cremolini C. ESMO Metastatic Colorectal Cancer Living Guideline v1.3. 2025;
23.NICE. Colorectal cancer. 2021. Accessed October 01, 2025. https://www.nice.org.uk/guidance/ng151
24.Hummel M, Hegewisch-Becker S, Neumann JHL, Vogel A. BRAF testing in metastatic colorectal carcinoma and novel, chemotherapy-free therapeutic options. Pathologe. 2021;42(Suppl 1):98-109. doi:10.1007/s00292-021-00946-5 PubMed
25.Phillips WJ, Marginean H, Alrehaili M, et al. Real-world evaluation of treatment patterns and clinical outcomes in patients with BRAF-V600E metastatic colorectal cancer (mCRC) in Canada. Cancer Treat Res Commun. 2025;43:100896. doi:10.1016/j.ctarc.2025.100896 PubMed
26.Pfizer Inc. An Open-Label, Multicenter, Randomized Phase 3 Study of First-Line Encorafenib Plus Cetuximab With or Without Chemotherapy Versus Standard of Care Therapy With a Safety Lead-In of Encorafenib and Cetuximab Plus Chemotherapy in Participants With Metastatic BRAF V600E-Mutant Colorectal Cancer [Interim CSR; Phase 3 PFS PCD 06 Jan 2025; Version 1.0]. 2025.
27.Pfizer Inc. An Open-Label, Multicenter, Randomized Phase 3 Study of First-Line Encorafenib Plus Cetuximab With or Without Chemotherapy Versus Standard of Care Therapy With a Safety Lead-In of Encorafenib and Cetuximab Plus Chemotherapy in Participants With Metastatic BRAF V600E-Mutant Colorectal Cancer [Protocol; 31 May 2024; Amendment 7]. 2024.
28.Kopetz S, Yoshino T, Van Cutsem E, et al. Encorafenib, cetuximab and chemotherapy in BRAF-mutant colorectal cancer: a randomized phase 3 trial. Nat Med. 2025;31(3):901-908. doi:10.1038/s41591-024-03443-3 PubMed
29.Elez E, Yoshino T, Shen L, et al. Encorafenib, Cetuximab, and mFOLFOX6 in BRAF-Mutated Colorectal Cancer. N Engl J Med. 2025;392(24):2425-2437. doi:10.1056/NEJMoa2501912 PubMed
30.Pfizer Inc. An Open-Label, Multicenter, Randomized Phase 3 Study of First-Line Encorafenib Plus Cetuximab With or Without Chemotherapy Versus Standard of Care Therapy With a Safety Lead-In of Encorafenib and Cetuximab Plus Chemotherapy in Participants With Metastatic BRAF V600E-Mutant Colorectal Cancer [Interim CSR; Phase 3 ORR PCD 22 Dec 2023; Version 1.0]. 2024.
31.Pfizer Inc. An Open-Label, Multicenter, Randomized Phase 3 Study of First-Line Encorafenib Plus Cetuximab With or Without Chemotherapy Versus Standard of Care Therapy With a Safety Lead-In of Encorafenib and Cetuximab Plus Chemotherapy in Participants With Metastatic BRAF V600E-Mutant Colorectal Cancer [SAP; 07 May 2024; Version 7.0]. 2024.
32.Cocks K, Wells JR, Johnson C, et al. Content validity of the EORTC quality of life questionnaire QLQ-C30 for use in cancer. Eur J Cancer. 2023;178:128-138. doi:10.1016/j.ejca.2022.10.026 PubMed
33.Musoro JZ, Coens C, Sprangers MAG, et al. Minimally important differences for interpreting EORTC QLQ-C30 change scores over time: A synthesis across 21 clinical trials involving nine different cancer types. Eur J Cancer. 2023;188:171-182. doi:10.1016/j.ejca.2023.04.027 PubMed
34.Musoro JZ, Sodergren SC, Coens C, et al. Minimally important differences for interpreting the EORTC QLQ-C30 in patients with advanced colorectal cancer treated with chemotherapy. Colorectal Dis. 2020;22(12):2278-2287. doi:10.1111/codi.15295 PubMed
35.Pfizer Canada ULC. BRAFTOVI (encorafenib) – Product Monograph [sponsor submitted reference]. 2025. https://pdf.hres.ca/dpd_pm/00081199.PDF
36.Ezeife DA, Padmore G, Vaska M, Truong TH. Ensuring equitable access to cancer care for Black patients in Canada. CMAJ. 2022;194(41):E1416-E1419. doi:10.1503/cmaj.212076 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.