Drugs, Health Technologies, Health Systems
Health Technology Review
Botulinum Toxin A Product Comparison for Upper Limb Spasticity, Lower Limb Spasticity, and Cervical Dystonia
Summary
Main Take-Aways
Botulinum neurotoxin type A (BoNT-A) products (onabotulinumtoxin A [onaBoNT-A], abobotulinumtoxin A [aboBoNT‑A], and incobotulinumtoxin A [incoBoNT-A]) are comparably effective and safe for the treatment of limb spasticity in children and adults and for the treatment of cervical dystonia (CD) in adults.
Switching between products due to previous treatment failure is possible.
Key Messages
What Is the Issue?
BoNT-A has been used to treat excessive muscle contractility in patients with movement disorders, including CD and spastic conditions, as well as for cosmetic purposes. Health Canada has approved 3 BoNT-A products for various therapeutic uses: onaBoNT-A, aboBoNT-A, and incoBoNT-A.
Decision-makers are interested in understanding the comparative clinical efficacy and safety of these products and the clinical effectiveness and safety of switching from 1 product to another in pediatric and adult patients with limb spasticity or CD. They are also interested in recommendations from evidence-based guidelines on the use of BoNT‑A products for these indications in children and adults.
What Did We Do?
We searched key resources, including journal citation databases, and conducted a focused internet search for relevant evidence published since 2013.
What Did We Find?
Clinical evidence suggests that onaBoNT-A and aboBoNT-A are comparatively effective and safe for the treatment of limb spasticity in children. OnaBoNT-A, aboBoNT-A, and incoBoNT-A are comparable in terms of efficacy and safety for the treatment of CD in adults.
Switching from onaBoNT-A or aboBoNT-A due to treatment failure to incoBoNT-A in adult patients with CD is possible.
Evidence-based guidelines recommend onaBoNT-A, aboBoNT-A, and incoBoNT-A as treatment options for upper limb spasticity, lower limb spasticity, and CD in adults.
What Does This Mean?
OnaBoNT-A, aboBoNT-A, and incoBoNT-A can be used to treat upper and lower limb spasticity in children and adults and CD in adults. Switching between products is possible, provided that the recommended conversion ratios are applied.
Abbreviations
AAN
American Academy of Neurology
aboBoNT-A
abobotulinumtoxin A
AE
adverse event
BoNT-A
botulinum neurotoxin type A
CD
cervical dystonia
CDQ-24
24-item Craniocervical Dystonia Questionnaire
CGI-I
Clinical Global Impressions of Illness
COST
European Cooperation in Science and Technology
GAS
Goal Attainment Scale
GCA
global clinical assessment
HRQoL
health-related quality of life
HTA
health technology assessment
incoBoNT-A
incobotulinumtoxin A
MAS
Modified Ashworth Scale
NAB
neutralizing antibody
NMA
network meta-analysis
onaBoNT-A
onabotulinumtoxin A
RCP
Royal College of Physicians
RCT
randomized controlled trial
SAE
serious adverse event
SR
systematic review
TWSTRS
Toronto Western Spasmodic Torticollis Rating Scale
Context and Policy Issues
What Are Limb Spasticity and Cervical Dystonia?
Limb spasticity is characterized by involuntary muscle tightness and stiffness in the limbs, leading to increased muscle tone and resistance to stretching.1 Symptoms include muscle stiffness, muscle spasm, uncontrolled movements, impaired coordination, pain, and in some severe cases muscle deformities.1 Limb spasticity is caused by damage to the parts of the brain and spinal cord that control muscle movement.1 Common neurologic conditions that can lead to limb spasticity include stroke, cerebral palsy, multiple sclerosis, and injuries to the brain or spinal cord.1
Cervical dystonia (CD), also known as spasmodic torticollis, is characterized by involuntary contraction of the neck muscles, causing the head to twist and turn and leading to neck pain, disability, abnormal posture, and social withdrawal.2 Symptoms include involuntary muscle contractions, abnormal head and neck postures, spasms, tremors, and pain.2 Although the cause of CD is often unknown, it is considered a neurologic disorder involving a miscommunication between the brain and muscles.2 Risk factors for CD include hereditary factors, use of dopamine antagonists or antipsychotics, and brain injury.2
What Are the Medications for Treatment of Limb Spasticity and CD?
Local injections of botulinum neurotoxin type A (BoNT-A) are currently the preferred treatment for limb spasticity and CD.3 When injected into the muscle, it causes muscle weakness and makes the muscle relax by inhibiting the release of acetylcholine from nerve terminals into the neuromuscular junction, thus preventing muscle contraction. Three BoNT-A products have been approved by Health Canada for various therapeutic uses:
onabotulinumtoxin A (onaBoNT-A [Botox]), the original and most widely known brand, approved for a broad range of therapeutic applications including blepharospasm (uncontrollable twitching or closing of the eyelids), strabismus (misaligned eyes), and spasticity (upper and/or lower limb) in children and adults as well as hyperhidrosis (excessive sweating), chronic migraine, bladder dysfunction, and CD in adults4
abobotulinumtoxin A (aboBoNT-A [Dysport], approved for therapeutic purposes such as spasticity in adults and children (upper and/or lower limb) and CD in adults5
incobotulinumtoxin A (incoBoNT-A [Xeomin]), approved for therapeutic use in adults for conditions such as CD, blepharospasm, upper limb spasticity, and chronic sialorrhea.6
Why Is It Important to Do This Review?
BoNT-A products have different complex protein structures and differ in molecular weight, onset of action, dosing, potency, and pharmacologic properties.7,8 For instance, aboBoNT-A and onaBoNT-A are toxins associated with high molecular weight complex proteins, while in incoBoNT-A the complex proteins have been removed through a purification process.8 Repeated BoNT-A injection therapy can lead to reduced responsiveness to treatment (partial secondary treatment failure) and to the development of neutralizing antibodies (NABs).7,8 Higher content of bacteria proteins, adjuvant activity of complex proteins, and the size and amount of complex proteins may contribute to NAB development and secondary nonresponsiveness.7,8
A CADTH review9 published in 2018 found that BoNT-A products were comparable in terms of clinical efficacy and safety for the treatment of adults with CD following a switch between 3 different products. Studies included in the review involved patients who had begun treatment with 1 BoNT-A product and then switched to a different product for the purpose of assessing the comparative clinical efficacy and safety of the 2 products at set conversion ratios. However, evidence assessing the switching between BoNT-A products due to clinical reasons such as secondary therapeutic failure or side effects is still lacking. In addition, head-to-head comparisons of the clinical efficacy and safety of different BoNT-A products were not the objective of the 2018 CADTH review, and the review did not include the pediatric population.9
Decision-makers are interested in understanding the comparative efficacy and safety of alternative BoNT-A products for managing upper and lower limb spasticity and CD in children and adults. They also want to know whether patients can recapture benefit when switching between BoNT-A products for medical (e.g., disease resistance to previous treatment) or nonmedical reasons.
Research Questions
What are the comparative clinical efficacy and safety of BoNT-A products in pediatric and adult patients with upper limb spasticity, lower limb spasticity, or CD?
What are the clinical effectiveness and safety of switching between BoNT-A products in pediatric and adult patients with upper limb spasticity, lower limb spasticity, or CD?
What are the evidence-based guidelines regarding the use and administration (including switching guidance) of BoNT-A products for pediatric and adult patients with upper limb spasticity, lower limb spasticity, or CD?
Methods
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE, the Cochrane Database of Systematic Reviews, the International HTA Database, the websites of health technology assessment (HTA) agencies in Canada and of major international HTA agencies, as well as a focused internet search. We also searched Embase starting from October 1, 2025. The search approach was customized to retrieve a limited set of results, balancing comprehensiveness with relevance. The search strategy comprised both focused controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. Search concepts were developed based on elements of the research questions and selection criteria. The main search concepts were botulinum toxin A products, cervical dystonia, and upper limb or lower limb spasticity. Search filters were applied to limit retrieval to HTAs, systematic reviews (SRs), meta-analyses, or indirect treatment comparisons, any types of clinical trials or observational studies. The search was completed on October 14, 2025, and was limited to English-language documents published since January 1, 2013 (the start date of the search conducted for the 2018 CADTH review).9 Internet links were provided, where available. The search strategy is available on request.
Selection Criteria and Methods
One reviewer screened citations and selected studies. In the first level of screening, titles and abstracts were reviewed and potentially relevant articles were retrieved and assessed for inclusion. The final selection of full-text articles was based on the inclusion criteria presented in Table 1.
Criteria | Description |
|---|---|
Population | Pediatric and adult patients with upper limb spasticity, lower limb spasticity, or cervical dystonia |
Intervention |
|
Comparator |
|
Outcomes |
|
Study designs | Systematic reviews, meta-analyses, randomized controlled trials, health technology assessment reports, nonrandomized studies,a evidence-based guidelines |
AE = adverse event; BoNT-A = botulinum neurotoxin type A; SAE = serious adverse event; SF-36 = Short Form 36 Health Survey; TWSTRS = Toronto Western Spasmodic Torticollis Rating Scale; WDAE = withdrawal due to adverse event.
aWe took a hierarchical approach to identifying evidence. If no health technology assessments, systematic reviews, meta-analyses, or randomized controlled trials were identified, we proceeded to identify and include nonrandomized studies.
Exclusion Criteria
Articles were excluded if they did not meet the selection criteria outlined in Table 1 or if they were published before 2013. Studies included in the 2018 CADTH review9 were excluded. Studies involving BoNT-A products that have not been approved by Health Canada or that have been approved by Health Canada but for cosmetic use only were excluded. Guidelines with unclear methodology were also excluded.
Critical Appraisal of Individual Studies
The included publications were critically appraised by 1 reviewer using the following tools as a guide: A MeaSurement Tool to Assess systematic Reviews 2 (AMSTAR 2)10 for SRs, the questionnaire to assess the relevance and credibility of a network meta-analysis (NMA)11 for network meta-analyses, the Downs and Black checklist12 for randomized controlled trials (RCTs) and nonrandomized studies, and the Appraisal of Guidelines for Research and Evaluation (AGREE) II instrument13 for guidelines. Summary scores were not calculated for the included studies; rather, the strengths and limitations of each included publication were described narratively.
Summary of Evidence
Quantity of Research Available
A total of 882 citations were identified in the literature search. Following the screening of titles and abstracts, 857 citations were excluded and 25 potentially relevant reports from the electronic search were retrieved for full-text review. Two potentially relevant publications were retrieved from the grey literature search for full-text review. Of these potentially relevant articles, 17 publications were excluded for various reasons, and 10 publications met the inclusion criteria and were included in this report. These comprised 2 SRs, 1 RCT, 4 nonrandomized studies, and 3 evidence-based guidelines. Appendix 1 presents the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA)14 flow chart of the study selection (Figure 1).
Summary of Study Characteristics
Appendix 2 provides details regarding the characteristics of the included SRs15,16 (Table 2), the included primary studies17-21 (Table 3), and the included evidence-based guidelines22-24 (Table 4).
Included Studies for Research Question 1: What Are the Comparative Clinical Efficacy and Safety of BoNT-A Products in Pediatric and Adult Patients With Upper Limb Spasticity, Lower Limb Spasticity, or CD?
Study Design
Two SRs with Bayesian NMA15,16 and 3 primary studies17-19 were included to answer research question 1.
The SR by Danchenko et al. (2021)15 included 6 RCTs (8 publications) to examine the comparative efficacy, safety, and cost-effectiveness of aboBoNT-A and onaBoNT-A in children with upper limb spasticity. The authors used the Bayesian NMA with noninformative prior distributions for all outcomes.
The SR by Guyot et al. (2019)16 included 10 RCTs to examine the comparative efficacy and safety of aboBoNT-A and onaBoNT-A in children with lower limb spasticity. The authors used the Bayesian NMA with noninformative prior distributions for all outcomes.
The 3 primary studies were a double-blind, multicentre, noninferiority, 2-period crossover RCT by Yun et al. (2015),17 a retrospective cohort study by Petracca et al. (2023),18 and a retrospective cohort study by Jochim et al. (2019).19
Country of Origin
One SR15 was conducted by authors in France, Canada, and the UK, while the other16 was conducted by authors in France and the Netherlands.
The included primary studies were conducted by authors in the Republic of Korea,17 Italy18 and Germany.19
Patient Population
The patient population included in the SR by Danchenko et al. (2021)15 was children and adolescents (aged 4 to 16 years) receiving BoNT-A for the treatment of upper limb spasticity, with the proportion of female participants ranging from 9% to 50% and the proportion of male participants ranging from 50% to 91%.
The patient population included in the SR by Guyot et al. (2019)16 was children and adolescents (aged 2 to 17 years) receiving BoNT-A for the treatment of lower limb spasticity, with the proportion of female participants ranging from 20% to 62% and the proportion of male participants ranging from 38% to 80%.
The patient population in the RCT by Yun et al. (2015)17 was adults with CD. The mean age of participants was 53.3 years, with fewer male (39%) than female (61%) participants.
Patients in both retrospective cohort studies18,19 were adults with acquired CD. The study by Petracca et al. (2023)18 included 23 patients (56% female, 44% male) but did not report the age of participants. Patients in the study by Jochim et al. (2019)19 had a mean age of approximately 45 years.
Interventions and Comparators
In the SR by Danchenko et al. (2021),15 all analyses were based on a network of 4 interventions: aboBoNT-A 8 unit (U), aboBoNT-A 16 U, onaBoNT-A (doses pooled within the approved range), or placebo or no treatment. Follow-up periods in the included primary studies varied between 6 and 16 weeks.
In the SR by Guyot et al. (2019),16 the interventions investigated included aboBoNT-A and onaBoNT-A at different dosages (aboBoNT-A: 10, 15, or 30 U/kg/leg; onaBoNT-A: 4 U/kg, 8 U/kg, 3 U/kg, 0.5 to 4 U/kg/leg, 4 U/kg/leg, 4 U/kg/leg + casting, or 12 U/kg distributed among left and right gastrocnemius muscle). Follow-up periods in the included primary studies varied between 12 and 52 weeks.
The crossover RCT by Yun et al. (2015)17 compared the efficacy and safety of aboBoNT-A (n = 48) versus onaBoNT-A (n = 48) at a 2.5:1 ratio. Follow-up was 16 weeks in the first period followed by a 4-week washout before switching to the other formulation, with a follow-up up of 16 weeks in the second period.
The retrospective cohort study by Petracca et al. (2023)18 included 23 patients who received a total of 739 treatment injections (235 with aboBoNT-A, 72 with incoBoNT-A, and 432 with onaBoNT-A). The follow-up duration was up to 25 years. Mean doses of treatment with aboBoNT-A, incoBoNT-A, and onaBoNT-A were 737 U, 138 U, and 158 U, respectively.
The retrospective cohort study by Jochim et al. (2019)19 compared the efficacy and safety of aboBoNT-A (N = 209) versus onaBoNT-A (N = 135), with a mean total dose per treatment of 663 U and 138 U, respectively. The observation period was up to 27 years for aboBoNT-A and 17 years for onaBoNT-A.
Outcomes
The efficacy outcomes in all included studies (2 SRs,15,16 1 RCT,17 and 2 retrospective cohort studies18,19) included the Modified Ashworth Scale (MAS), the Goal Attainment Scale (GAS), the Tardieu Scale, the Tsui Scale, the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), the Clinical Global Impression of Illness (CGI-I) scale, global clinical assessment (GCA), latency, total duration of improvement, and peak effect duration. Brief descriptions of these outcome measures are as follows:
MAS is a clinical tool used to measure spasticity (increased muscle tone) in patients with central nervous system lesions. MAS uses a scale from 0 to 4, where 0 means no increased tone and 4 indicates rigidity.
The Tardieu Scale is a way to measure spasticity that considers resistance to passive movement at both slow and fast speeds. The Tardieu Scale grades spasticity from 0 to 4 based on the quality of the muscle's reaction to a fast passive stretch. The grades are 0 for no resistance, 1 for slight resistance throughout, 2 for a clear catch at a specific angle, 3 for fatigable clonus (less than 10 seconds), and 4 for unfatigable clonus (more than 10 seconds). A higher grade indicates a stronger spastic reflex.
The Tsui Scale is a clinical rating scale used to assess the severity of CD. It is a 4-item scale with a maximum score of 25. A higher score on the scale indicates greater severity of the condition.
TWSTRS is a tool used to measure the severity of CD (spasmodic torticollis) and to evaluate how well treatments are working. It is a composite scale that assesses 3 main areas: severity (physical signs; max = 35), disability (impact on daily life; max = 30), and pain (max = 20). A higher score on the scale indicates greater severity of the condition.
CGI-I is expressed as the proportions of patients with CGI of illness score of 1 = normal or not at all ill, 2 = borderline mildly ill, or 3 = mildly ill.
GCA was based on the perceptions of patients and/or their next of kin of improvement, expressed as 0% to 100% compared with the baseline condition. GCA was rated from 0 to 6 (0 = no effect, 1 = < 20%, 2 = 20% to 40%, 3 = 40% to 60%, 4 = 60% to 80%, 5 = 80% to 90%, 6 = complete resolution).
Latency was defined as the interval between injection and the first sign of improvement noticed by the patient.
Total duration of improvement was defined as the interval between the first day of improvement and the last day of reported benefit.
Peak effect duration was the number of days the patient experienced the best clinical effect.
Safety outcomes reported in all included studies included overall adverse events (AEs), serious AEs (SAEs), treatment-related AEs, and common AEs.
Included Studies for Research Question 2: What Is the Clinical Effectiveness and Safety of Switching Between BoNT-A Products for Pediatric and Adult Patients With Upper Limb Spasticity, Lower Limb Spasticity, or CD?
The previous CADTH review9 included 1 SR, 1 RCT, and 2 nonrandomized studies and assessed the clinical effectiveness of switching between BoNT-A products for adult patients with CD. This review identified and included 2 additional primary studies20,21 to answer research question 2.
Study Design
One retrospective cross-sectional study20 and 1 prospective cohort study21 were included to answer research question 2.
Country of Origin
Both studies were conducted by authors in Germany.20,21
Patient Population
Both studies included adult patients with CD.20,21
In the cross-sectional study,20 59 patients with secondary treatment failure after aboBoNT-A or onaBoNT-A therapy who were switched to incoBoNT-A (switched group) were compared with 34 patients with CD who were treated exclusively with incoBoNT-A (incoBoNT-A group).20 Secondary treatment failure in botulinum toxin therapy is when a patient’s disease stops responding to treatment after an initial successful response, most often due to the development of NABs. The mean ages in the switched group and the incoBoNT-A group were 47 years and 51 years, respectively. Slightly more females (63% in the switched group and 56% in the incoBoNT-A group) than males (37% in the switched group and 44% in the incoBoNT-A group,) participated in the study.
In the prospective cohort study,21 patients were those with partial secondary treatment failure under previous long-term BoNT‑A treatment. Partial secondary treatment failure in botulinum toxin therapy occurs when a patient’s disease initially responds well to treatment but the treatment later has a gradual, incomplete loss of effectiveness, often due to the development of NABs or due to progression of the underlying disease. Details of patient characteristics were not reported.
Interventions and Comparators
In the cross-sectional study,20 the efficacy of changing the switched group to incoBoNT-A was compared with the treatment efficacy measured in the group treated exclusively with incoBoNT-A. The follow-up period was 2,700 days (30 treatment cycles). A conversion ratio of 3:1 between aboBoNT-A and onaBoNT-A and a conversion ratio of 1:1 between onaBoNT-A and incoBoNT-A were used.
In the prospective cohort study,21 the efficacy of changing the switched group to incoBoNT-A was assessed based on comparisons with baseline measures. The follow-up period was 48 weeks. Two hundred units of incoBoNT-A were administered every 12 weeks with 4 injection cycles over a total of 48 weeks.
Outcomes
The efficacy outcomes in the cross-sectional study20 for the switched group were initial clinical severity of CD before aboBoNT-A or onaBoNT-A therapy, best outcome during aboBoNT-A or onaBoNT-A therapy, severity at the time of switching to incoBoNT-A due to secondary treatment failure, and severity at recruitment (i.e., during incoBoNT-A therapy). For the comparator group (i.e., the group treated exclusively with incoBoNT-A), outcomes were initial clinical severity of CD before incoBoNT-A therapy, best outcome during incoBoNT-A therapy, and severity at recruitment (i.e., during incoBoNT-A therapy). All outcomes were assessed using the Tsui Scale.
The efficacy outcomes in the prospective cohort study21 included TWSTRS, the 24-item Craniocervical Dystonia Questionnaire (CDQ-24), and NAB titres.
Neither study reported safety outcomes.
The Tsui Scale and TWSTRS have already been defined. The CDQ-24 is a 24-item disease-specific instrument measuring patients’ health-related quality of life (HRQoL) based on 5 subscales: stigma, emotional well-being, pain, activities of daily living, and social or family life. Patients with more than 20% improvement in CDQ-24 scores were classified as having a disease response, and those with a worsening of more than 20% were classified as not having a disease response.
Included Studies for Research Question 3: What Are the Evidence-Based Guidelines Regarding Use and Administration (Including Switching Guidance) of BoNT-A Products for Pediatric and Adult Patients With Upper Limb Spasticity, Lower Limb Spasticity, or CD?
Study Design
Three evidence-based guidelines22-24 were included to answer research question 3.
The Royal College of Physicians (RCP) (UK) 2018 guideline22 was developed by its Guideline Development Group, whose members first identified the key priority areas for the guidelines to address and set 1 or more questions for each priority area. The group then considered their recommendation for each question according to a list of 11 questions related to benefits and harms, resource use, equity, acceptability, and feasibility. The group conducted a systematic literature search for each question. They graded the quality of evidence, which ranged from high to very low based on the level of confidence in the estimate of effect. The group graded the strength of recommendations based on consensus.
The levels of evidence related to benefits were classified as follows:
high (Research Grade A)
moderate (Research Grade B)
low (Research Grade C)
user or professional opinion.
Harms and costs were classified as no, probably no, uncertain, probably yes, yes, and varies.
The strength of recommendations was classified as follows:
very strong (high-level research evidence [Grade A] with minimal harms and costs)
strong (lower-level evidence but minimal harms and costs)
moderate (lower-level evidence and potentially significant harms and/or costs)
weak (lower-level evidence and potentially significant harms and costs).
Authors of the European Cooperation in Science and Technology (COST) guideline23 conducted systematic literature searches on the topic of botulinum neurotoxin treatment for CD. The levels of evidence and strength of recommendations were classified according to the American Academy of Neurology (AAN) guideline. However, the guideline did not report how the recommendations were developed.
The levels of evidence were classified as follows:
Class I: An RCT of the intervention of interest with masked or objective outcome assessment conducted in a representative population. Relevant baseline characteristics are presented and substantially equivalent among treatment groups, or there is appropriate statistical adjustment for differences. The following are also required:
concealed allocation
clearly defined primary outcome(s)
clearly defined exclusion and inclusion criteria
adequate accounting for participants who drop out (with at least 80% of enrolled participants completing the study) and crossovers, with numbers sufficiently low to have minimal potential for bias
for noninferiority or equivalence trials claiming to prove efficacy for 1 or both drugs, the following are also required:
The standard treatment used in the study is substantially similar to that used in previous studies establishing efficacy of the standard treatment (e.g., for a drug, the mode of administration, dose, and dosage adjustments are similar to those previously shown to be effective).
The inclusion and exclusion criteria for patient selection and the outcomes of patients on the standard treatment are substantially equivalent to those shown in previous studies establishing efficacy of the standard treatment.
The interpretation of the results of the study is based on an observed cases analysis.
Class II: An RCT of the intervention of interest conducted in a representative population with masked or objective outcome assessment that lacks 1 of the 5 criteria listed as sub-bullets under class I or a prospective matched cohort study with masked or objective outcome assessment in a representative population that meets the second through fifth sub-bullets in class I. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
Class III: All other controlled trials (including well-defined natural history controls or patients serving as their own controls) conducted in a representative population, in which the outcome is independently assessed or independently derived by objective outcome measurement.
Class IV: Studies not meeting class I, II, or III criteria, including those based on consensus or expert opinion. Recommendation strength was classified as follows:
A = Established as effective, ineffective, or harmful for patients with the given condition in the specified population. Requires 2 consistent class I studies.
B = Probably effective, ineffective, or harmful for patients with the given condition in the specified population. Requires 1 class I study or 2 consistent class II studies.
C = Possibly effective, ineffective, or harmful for patients with the given condition in the specified population. Requires 1 class II study or 2 consistent class III studies.
U = Data are inadequate or conflicting; given current knowledge, the treatment is unproven. Studies do not meet criteria for class I through class III.
The AAN guideline24 was developed by guideline panel members who voted on their levels of agreement with each recommendation, which helped determine its strength. A formal process of reviewing and analyzing the existing literature was conducted. Studies were evaluated for their quality and the strength of the overall evidence was determined. The evidence and other factors were used to formulate specific recommendations.
The levels of evidence and strength of recommendations of the AAN guideline have already been described.
Country of Origin
The authors of the RCP guideline, the COST guideline, and the AAN guideline were from the UK,22 Europe,23 and the US,24 respectively.
Target Population and Intended Users
The patient population of the RCP guideline22 was adults with spasticity due to neurologic illness or injury. The intended users were health professionals involved in the treatment of patients with spasticity as well as providers and purchasers of rehabilitation services.
The patient population of the COST guideline23 was adult patients with CD. The intended users were clinicians involved in the treatment of patients with CD.
The patient populations of the AAN guideline24 were adult patients with blepharospasm, CD, spasticity, or migraine headache. The intended users were clinicians involved in the treatment of patients with those conditions. Only patients with CD and spasticity were relevant to this review.
Interventions and Practice Considered
The RCP guideline22 provides recommendations to promote the appropriate use of BoNT-A in the treatment of patients with spasticity, and it gives guidance on its administration and the wider principles of management.
The COST guideline23 provides recommendations about the comparison of different formulations of BoNT-A in improving motor symptoms, pain, and HRQoL and in relation to the dosage conversion ratio.
The AAN guideline24 provides recommendations on the use of botulinum toxin for the treatment of blepharospasm, CD, spasticity, and migraine headache.
Outcomes
All included guidelines22-24 considered all relevant outcomes related to the clinical effectiveness, HRQoL, and safety outcomes.
Summary of Critical Appraisal
SRs With NMA
Both SRs15,16 with NMA had similar strengths and weaknesses in methodology. Both SRs15,16 used the same NMA method and approach (Bayesian). The authors of both SRs15,16 clearly presented the literature search methods, search terms, search dates, search strategy, and criteria for the SRs and attempted to identify and include all relevant RCTs. All included RCTs were assessed for risk of bias using the Cochrane Risk of Bias tool, and results related to risk of bias and heterogeneity were presented and discussed. However, a published protocol was not established before the conduct of the review. As such, there was a risk of bias due to selective reporting.
The authors of both SRs15,16 performed pairwise meta-analysis using random-effects model where more than 1 study was available. In the analysis and synthesis of evidence, the authors of both SRs15,16 used a Bayesian NMA approach, which was based on noninformative prior distributions and a normal approximation of the treatment response. The Bayesian NMA approach was used due to sparse data (few studies). However, the authors did not provide raw data by study and treatment as used in the analysis or model that could have a risk of error in the analysis and limit the reproducibility of the findings.
Assessment of inconsistency could not be conducted in either SR15,16 due to a lack of closed loops in the network of evidence, thus limiting the assessment of incoherence, and the statistical manifestation of intransitivity. The between-study heterogeneity (e.g., in age, gender, medication status, comorbidities) and study designs (e.g., length of follow-up, drug dosage) did not uphold the transitivity assumption, thus compromising the accuracy of the indirect estimates. Reporting of potential treatment effect modifiers across studies was often incomplete, which precludes a comprehensive appraisal of similarity. The robustness of the findings could not be determined as the sensitivity analyses were not planned and conducted for all treatment effect modifiers. Overall, both SRs15,16 with NMA had major weaknesses in the statistical methods for analysis and synthesis of evidence, which limit the certainty of the findings.
Randomized Controlled Trial
For reporting, the authors of the included RCT17 clearly described the objective of the study, the intervention of interest, the main outcomes, and the main findings of the study. However, the demographic characteristics of the randomized participants were not clearly reported. As such there is a risk of potential differences in baseline characteristics between groups. Actual P values and AEs of the intervention were reported in the RCT. In terms of external validity, the patients included in the RCT may represent the entire eligible population, as they were recruited from multiple centres. The treatment settings (i.e., hospitals) appeared to be representative of the settings where most of the patients accessed care in the Republic of Korea. However, the strict patient inclusion and exclusion criteria applied in the studies may limit generalizability to other populations. As the RCT was conducted in the Republic of Korea, the applicability of the findings to health care settings in Canada may be limited.
For internal validity related to bias, the RCT was registered with ClinicalTrials.gov. As such, the risk of bias in the selection of the reported results was low. Statistical tests were used appropriately and the main outcome measures were accurate and reliable. Adherence to the intervention was reliable, as more than 90% of patients completed the study. The primary efficacy end points were accurately measured using appropriate outcome measures. For internal validity related to confounding, patients in both intervention groups appeared to have been recruited from the same population and over the same period of time, and methods of randomization and allocation concealment were described, thus ensuring comparability and minimizing selection bias. Sample size calculation was performed and provided enough statistical power to detect a real effect. Efficacy evaluation was conducted for the modified intention-to-treat set population that provides the most realistic assessment of a treatment's effectiveness in a real-world setting. However, adjustment for multiplicity in the analysis of secondary outcomes was not performed. As such, there was increased risk for false-positive results (type I errors). For a crossover design, a carryover effect may potentially exist despite a 4-week washout period before starting a new treatment. Therefore, the effect of the second treatment may have been incorrectly assessed. Overall, the included RCT17 had certain weaknesses in external validity and internal validity related to confounding, which should be considered in the interpretation of its findings.
Nonrandomized Studies
In term of reporting, none of the 4 nonrandomized studies18-21 clearly reported the demographic characteristics of the participants, which could indicate a risk of potential differences in baseline characteristics between groups. AEs were not reported in 2 studies.20,21 For external validity, the strict patient inclusion and exclusion criteria applied in all 4 studies18-21 may limit generalizability to the other populations. As the studies were conducted in Germany and Italy, the applicability of findings to health care settings in Canada may be limited. For internal validity related to bias, 3 studies18-20 were of retrospective design, which may have potential biases in selection (nonrepresentative sample), information (errors in data collection or measurement), and loss to follow-up.
The prospective cohort study by Hefter et al. (2021)21 had several potential biases such as confounding bias, attrition bias (loss to follow-up), and information bias (such as ascertainment bias and observer bias). The open-label design of this study (participants, investigators, and outcome assessors were aware of the treatment) may have led to a potential bias in efficacy outcomes, which may have resulted in the overreporting of known harms. For internal validity related to confounding, none of the 4 studies18-21 identified or adjusted the confounding factors in the analyses. As such, the unmeasured factors may have influenced outcomes. A sample size calculation was not performed in any of the nonrandomized studies,18-21 which means they may not have had enough statistical power to detect real effects. Adjustment for multiplicity in the analysis of secondary outcomes was not performed in any of the 4 studies.18-21 This may have increased the risk of false-positive results (type I errors). Some of the strengths in reporting and internal validity related to bias of the 4 nonrandomized studies18-21 are presented in the Table 6 of Appendix 3. Overall, all 4 nonrandomized studies18-21 had major weaknesses in external validity, internal validity related to bias, and internal validity related to confounding. These shortcomings limit the certainty of the findings.
Guidelines
For reporting, all 3 included guidelines22-24 were explicit in terms of scope and purpose (i.e., objectives, health questions, and populations) and presented recommendations clearly (i.e., they were specific and unambiguous, and it was easy to find key recommendations, with options for managing different conditions or health issues provided). In terms of the involvement of interested parties, the authors of 2 included guidelines22,24 clearly defined target users and the guideline development groups, and they reported that the views and preferences of patients were sought. The COST guideline authors23 also defined target users and reported that the views and preferences of patients were sought, but they did not define the guideline development groups involved. The methodology for the development of the 3 guidelines22-24 was robust. The authors of all 3 guidelines22-24 clearly reported methods for evidence collection, criteria for selection, and methods for evidence synthesis. There were explicit links between the recommendations and supporting evidence and the methods used for formulating the recommendations. Also, the authors of the 3 guidelines22-24 considered health benefits and risks of side effects in formulating the recommendations. Plans for updating the guidelines22-24 were not indicated.
The 3 guidelines22-24 were reviewed by relevant professional experts and published in peer-reviewed journals. However, there were some limitations related to guideline implementation and applicability in the 3 guidelines.22-24 Specifically, facilitators and barriers to application were not specified, advice or tools related to how the recommendations can be put into practice were lacking, and monitoring or auditing criteria were unclear. The authors of the included guidelines22-24 declared the competing interests of all guideline development group members and disclosed that the views of the funding body had no influence on the content of the guidelines. Overall, the included guidelines22-24 were robust in terms of scope and purpose, the involvement of interested parties, rigour of development, clarity of presentation, and editorial independence.
Additional details regarding the strengths and limitations of included publications are provided in Appendix 3.
Summary of Findings
Appendix 4 presents the main study findings.
Comparative Clinical Efficacy and Safety of BoNT-A Products in Pediatric Patients With Upper Limb Spasticity or Lower Limb Spasticity
Key results of the NMA for relevant outcomes for the comparative clinical efficacy of BoNT-A products are presented in Table 8 and those for safety are presented in Table 9.
Upper Limb Spasticity
In terms of MAS score changes from baseline at 6 weeks to 16 weeks’ follow-up, patients treated with aboBoNT‑A (8 U or 16 U) and onaBoNT-A (1 U to 6.6 U combined) were likely to have comparable results in terms of improvement in elbow and wrist spasticity.15
A sensitivity analysis on MAS scores for elbows and wrists at weeks 6 and 16 suggested that aboBoNT-A was likely more efficacious than onaBoNT-A in the improvement of elbow spasticity, but not wrist spasticity, across doses and time points. This analysis excluded 1 study that introduced a large amount of heterogeneity into the network of evidence.15
AboBoNT-A was likely associated with a higher median percentage of patients whose elbow or wrist spasticity responded to treatment compared to onaBoNT-A at 6 weeks and 16 weeks. However, the difference was uncertain due to imprecision.15
For safety, patients who received aboBoNT-A and onaBoNT-A were likely to have comparable results in terms of overall AEs, SAEs, or treatment-related AEs at 12 to 16 weeks’ follow-up.15
Lower Limb Spasticity
In terms of MAS score changes from baseline to 12 weeks’ follow-up, aboBoNT-A (15 U/kg/leg) likely resulted in improvement in spasticity compared with onaBoNT-A (4U/kg/leg, 4 U/kg/leg + casting, 4 U/kg, or 8 U/kg). AboBoNT-A (10 U/kg/leg) was also likely better than onaBoNT-A (4 U/kg/leg, 4 U/kg/leg + casting) but comparable to onaBoNT-A (4 U/kg, or 8 U/kg).16
In terms of GAS scores at 12 weeks’ follow-up, both aboBoNT-A 10 U/kg/leg and aboBoNT-A 15 U/kg/leg likely resulted in better outcomes compared with onaBoNT-A 12 U/kg.16
In terms of Tardieu Scale spasticity scores at 12 weeks’ follow-up, aboBoNT-A (10 U/kg/leg or 15 U/kg/leg) and onaBoNT-A (3 U/kg) were likely to have comparable results in resistance to passive movement at both slow and fast speeds.16
Regarding safety, aboBoNT-A and onaBoNT-A were likely to have comparable results in terms of overall AEs at 12 weeks follow-up.16
Comparative Clinical Efficacy and Safety of BoNT-A Products in Adult Patients With CD
Three studies (1 RCT17 and 2 retrospective cohort studies18,19) provided evidence for the comparative clinical efficacy and safety of BoNT-A products in adult patients with CD. Details of efficacy results are provided in Table 10 and safety results are provided in Table 11.
In the RCT by Yun et al. (2015):17
In terms of changes in Tsui Scale scores from baseline to 4 weeks’ follow-up, there were no differences between aboBoNT-A and onaBoNT-A groups in the improvement of severity of CD.
In terms of changes in TWSTRS scores from baseline to 4 weeks’ follow-up, there were no differences between aboBoNT-A and onaBoNT-A groups in the total TWSTRS score, TWSTRS Severity subscore, TWSTRS Disability subscore, or TWSTRS Pain subscore.
The aboBoNT-A and onaBoNT-A groups had similar proportions of patients with improvement in CGI-I scores and Patient Global Impression of Improvement scores.
Regarding safety, there were no differences between aboBoNT-A and onaBoNT-A groups in terms of overall AEs, severe AEs, treatment-related AEs, AEs leading to discontinuation of treatment, or common AEs such as muscle weakness and dysphagia.
In the retrospective cohort study by Petracca et al. (2023):18
For efficacy, the comparisons between incoBoNT-A, onaBoNT-A, and aboBoNT-A treatment resulted in little to no difference due to small to very small effects sizes in terms of latency (i.e., the interval between injection and the first sign of improvement noticed by the patient), maximum duration of improvement, total duration of improvement, and the patient’s perception of improvement (GCA score).
For safety, participants who received incoBoNT-A appeared to have fewer cases of AEs than those who received onaBoNT-A or aboBoNT-A , with muscle weakness and dysphagia reported as common AEs. However, statistical comparisons between groups were not performed.
In the retrospective cohort study by Jochim et al. (2019):19
In terms of satisfactory effect, the proportions of patients in the aboBoNT-A and onaBoNT-A groups who reported favourable results after at least 80% of their individual treatments were 68.0% and 72.0%, respectively. Whether this result differed significantly between groups was not determined.
For safety, overall AEs (15% of participants in the aboBoNT-A group versus 10% in the onaBoNT-A group), muscle weakness (6% of participants in the aboBoNT-A group versus 2% in the onaBoNT-A group), and dysphagia (6% of participants in the aboBoNT-A group versus 2% in the onaBoNT-A group) were reported, without performing statistical comparisons between groups.
Clinical Effectiveness and Safety of Switching Between BoNT-A Products for CD
Two studies (1 retrospective cross-sectional study20 and 1 prospective cohort study21) provided evidence regarding switching between BoNT-A products for adults with CD (Table 12).
In the retrospective cross-sectional study by Hefter et al. (2022),20 Tsui Scale scores were used to measure clinical severity:
Before aboBoNT-A or onaBoNT-A treatment, the mean Tsui Scale score of the switched group was 8.77 (standard deviation [SD] = 3.44). Before incoBoNT-A treatment, the mean Tsui Scale score of the group treated exclusively with incoBoNT-A was 7.83 (SD = 3.25).
During treatment with aboBoNT-A or onaBoNT-A, the mean Tsui Scale score for best outcome among the switched group was 4.04 (SD = 3.25) and occurred after a mean treatment duration of 3.7 years (SD = 3.6 years). During treatment with incoBoNT-A), the mean Tsui Scale score for best outcome of the group treated exclusively with incoBoNT-A was 1.71 (SD = 1.77) and occurred after a mean treatment duration of 2.52 years (SD = 1.86 years). The difference between groups was statistically significant (P < 0.005).
After approximately 7.7 years of treatment on average with aboBoNT-A or onaBoNT-A, patients in the switched group developed secondary treatment failure, with a mean Tsui Scale score of 8.32 (SD = 3.43).
After being treated with incoBoNT-A, both the switched group and the group treated exclusively with incoBoNT-A had lower Tsui Scale scores compared to before treatment. The mean Tsui Scale scores of the switched group and the group treated exclusively with incoBoNT-A were 5.40 (SD = 2.92) and 3.27 (SD = 2.35), respectively. The difference between groups was statistically significant (P < 0.001). This suggests that although the switched group regained benefit with incoBoNT-A therapy after development of secondary treatment failure with previous aboBoNT-A or onaBoNT-A treatment, such benefit in the switched group was still inferior to that of the group treated exclusively with incoBoNT-A.
In the prospective cohort study by Hefter et al. (2021):21
Patients for whom previous botulinum toxin treatment (aboBoNT-A, onaBoNT-A, or rimabotulinumtoxin B) had partially failed had improvement in the TWSTRS Severity subscore after switching to incoBoNT-A treatment. The difference between scores after and before treatment was statistically significant (P < 0.01).
There were no differences between after and before treatment in TWSTRS total score, Disability subscore, or Pain subscore.
For HRQoL assessed with CDQ-24, the authors reported that median CDQ-24 scores improved between baseline and week 48, but this difference failed to reach the statistical significance level of 5% (data not shown).
During incoBoNT-A treatment, NAB titres decreased in 3.22% of patients, did not change in 45.2% of patients, and increased in 22.6% of patients.
Evidence-Based Guidelines Regarding Use and Administration (Including Switching Guidance) of BoNT-A Products for Adult Patients With Upper Limb Spasticity, Lower Limb Spasticity, or CD
A summary of recommendations from the included guidelines22-24 is presented in Table 13.
The RCP guideline22 recommends that:
BoNT-A is a safe and effective for the treatment of both upper and lower limb spasticity in adults (strength of recommendation: strong).
Maximum doses per treatment session are 200 U to 240 U (arm), and 300 U (leg) for onaBoNT-A (Botox), 1,500 U (total body dose of arm and leg) for aboBoNT-A (Dysport), and 500 U (arm) for incoBoNT-A (Xeomin) (strength of recommendation: very strong). The recommended doses for individual muscles are presented in Appendix 2 of the guideline.
The COST guideline23 recommends that:
AboBoNT-A (strength of recommendation: A), incoBoNT-A (strength of recommendation: B), and onaBoNT-A (strength of recommendation: A) are effective in improving CD in adults.
BoNT-A treatment improves quality of life (strength of recommendation: B) and reduces pain (strength of recommendation: A) associated with CD.
The conversion ratio of onaBoNT-A to aboBoNT-A is 1:3 (strength of recommendation: A) or 1:2.5 (strength of recommendation: B) and the conversion ratio of onaBoNT-A to incoBoNT-A is 1:1 (strength of recommendation: B).
An initial dose of 500 U of incoBoNT-A is effective in improving CD in adults (strength of recommendation: A).
There are no clear recommendations on the optimal starting dose of onaBoNT-A (strength of recommendation: U).
The AAN guideline24 recommends that:
For CD in adults, aboBoNT-A (strength of recommendation: A) and incoBoNT-A and onaBoNT-A (strength of recommendation: B) should be used as treatment options.
For upper limb spasticity in adults, aboBoNT-A (strength of recommendation: A), incoBoNT-A (strength of recommendation: B), and onaBoNT-A (strength of recommendation: B) should be used as treatment options.
For lower limb spasticity in adults, onaBoNT-A and aboBoNT-A should be used as treatment options (strength of recommendation: A). There is insufficient evidence to support or refute a benefit of incoBoNT-A.
Limitations
Evidence Gaps
Evidence on the comparative clinical efficacy and safety of BoNT-A products for the treatment of CD in children and for the treatment of limb spasticity in adults was not identified. There is also a lack of evidence on the clinical effectiveness of switching between BoNT-A products for the treatment of limb spasticity in both children and adults. The included guidelines22-24 provided recommendations on the use of BoNT-A products as treatment options for limb spasticity and CD in adults, but not in children.
Generalizability
The findings of the included clinical studies, including SRs,15,16 RCTs,17,25 and nonrandomized studies18-21 may have limited generalizability to health care settings in Canada due to limitations in external validity. However, the recommendations of the included guidelines22-24 should be applicable to the Canadian context.
Certainty of Evidence
The 2 SRs15,16 with NMA had several limitations. First, the evidence base was limited, with only 6 RCTs included in 1 SR15 and 10 RCTs included in the other.16 Many of those RCTs had small sample sizes. Second, the certainty of evidence was low due to the lack of covariate analysis to adjust for potential treatment effect modifiers or for the risk of bias assessment. Third, outcome measures for efficacy and tolerability outcomes were limited and inconsistently reported in the included studies. Fourth, there was a lack of data to explore the clinically relevant efficacy outcomes such as those assessed by clinicians.
The included RCT17 had several limitations. First, total duration of treatment was relatively short (maximum 16 weeks) to obtain long-term follow-up data. Second, the relatively small sample size may reduce statistical power, making it difficult to detect true effects and limiting generalizability. Third, the findings can be confounded by carryover effects, although a 4-week washout period was applied. Fourth, the study did not compare onset, potency, or duration of action between products; thus, it was unclear which products had a faster or slower onset, are more potent, or had more prologued effect or shorter effect.
The main limitations of the retrospective studies18-20 and prospective cohort study21 related to the design and the analysis of nonhomogeneous populations in terms of causes and severity of CD. Given small sample sizes and different types of CD, definitive conclusions could not be drawn about treatment outcomes associated with various conditions that cause CD. The findings in those studies should be considered as exploratory that require further research with well-controlled studies.
Although the included guidelines22-24 were generally methodologically robust, they were not without limitations that affect providing up-to-date recommendations. The most recently published guideline identified in this review was the RCP guideline22 published in 2018, followed by the COST23 and ANN24 guidelines published in 2017 and 2016, respectively. It is apparent that new evidence published thereafter would have been missed, and therefore guidelines should be updated to include the latest evidence in their recommendations.
Conclusions and Implications for Decision- or Policy-Making
This review included 2 SRs with NMA,15,16 1 RCT,17 4 nonrandomized studies,18-21 and 3 guidelines.22-24
Comparative Clinical Efficacy and Safety of BoNT-A Products in Pediatric Patients With Upper or Lower Limb Spasticity
In children with upper or lower limb spasticity, evidence from 2 SRs15,16 with NMA suggests that treatment with aboBoNT-A may result in better improvement in tone (MAS score) and functional outcomes (GAS score) compared with onaBoNT-A. AboBoNT-A and onaBoNT-A were comparable in terms of Tardieu Scale spasticity grade. Both treatments appear to have comparable safety profiles in terms of overall AEs, SAEs, and treatment-related AEs. Limitations of the NMA included the potential for intransitivity (differences in patient and study characteristics across comparisons in the network), which would bias the effect estimates and study-level risks of bias and lead to imprecision in the effect estimates. As such, the results should be interpreted in the context of heterogeneity and sparsity of the evidence.
Comparative Clinical Efficacy and Safety of BoNT-A Products in Adult Patients With CD
Evidence from 1 crossover RCT17 suggests that aboBoNT-A and onaBoNT-A, when applied at a 2.5:1 ratio, have similar efficacy (assessed by Tsui Scale, TSWTRS, CGI-I, and Patient Global Impression of Improvement scores) for the treatment of CD in adults. Both treatments appear to have comparable safety profiles in terms overall AEs, severe AEs, treatment-related AEs, AEs leading to discontinuation of treatment, and common AEs such as muscle weakness and dysphagia. Similar findings in efficacy and tolerability between aboBoNT-A and onaBoNT-A were reported in the 2 retrospective cohort studies.18,19 One retrospective cohort study18 also included incoBoNT-A in the comparison with aboBoNT-A and onaBoNT-A and found little to no difference in efficacy among treatments.
Clinical Effectiveness and Safety of Switching Between BoNT-A Products for CD
Evidence from 1 retrospective cross-sectional study20 and 1 prospective cohort study21 suggests that adult patients with CD who had developed secondary treatment failure with previous BoNT-A products and switched to incoBoNT-A treatment may experience improvement in long-term functional outcomes (assessed by Tsui Scale and TWSTRS Severity subscale scores) and HRQoL (assessed by CDQ-24). Due to limitations of the included studies, these findings should be explored further with randomized studies that carefully control for the type of CD, side effects, and outcomes.
Recommendations From Included Evidence-Based Guidelines
The 3 included guidelines22-24 recommend that aboBoNT-A, onaBoNT-A, and incoBoNT-A should be used as treatment options for upper or lower limb spasticity and CD in adults as they are safe and effective and they improve quality of life and pain. The conversion ratio of onaBoNT-A to aboBoNT-A is 1:3 or 1:2.5, and the conversion ratio of onaBoNT-A to incoBoNT-A is 1:1.
Considerations for Future Research
Future clinical research is needed in the areas of the use of BoNT-A products for the treatment of CD in children, switching between BoNT-A products for the treatment of limb spasticity in both children and adults, and comparisons between products in terms of potency and duration of action. New guidelines should be developed to reflect new evidence and provide recommendations on the use of BoNT-A products as treatment options for limb spasticity and CD in children.
Implications for Clinical Practice
Collective evidence in this review suggests that BoNT-A products such as aboBoNT-A, onaBoNT-A, and incoBoNT-A are effective and safe for the treatment of limb spasticity in children and adults and for the treatment of CD in adults. Switching between products is possible, provided that the recommended conversion ratios are applied.
References
1.Johns Hopkins Medicine. Spasticity. 2025. Accessed 30 October. https://www.hopkinsmedicine.org/health/conditions-and-diseases/spasticity
2.Cleveland Clinic. Cervical Dystonia. 2023. Accessed 30 October. https://my.clevelandclinic.org/health/diseases/25228-cervical-dystonia
3.Spasticity Treatment with Botulinum Toxins. 2025. Accessed 30 October. https://www.sralab.org/lifecenter/resources/spasticity-treatment-botulinum-toxins#:~:text=Muscle%20relaxation%20begins%20one%20to,causes%20pain%20or%20functional%20problems.
4.Product monograph. BOTOX. 2025. Accessed 30 November 2025. chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/BOTOX_PM_EN.pdf
5.Product monograph. DYSPORT THERAPEUTIC. 2021. Accessed 30 November 2025. chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://pdf.hres.ca/dpd_pm/00060067.PDF
6.Product monograph. XEOMIN. 2020. Accessed 30 October 2025. chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://pdf.hres.ca/dpd_pm/00058883.PDF
7.Aoki KR, Guyer B. Botulinum toxin type A and other botulinum toxin serotypes: a comparative review of biochemical and pharmacological actions. Eur J Neurol. 2001;8 Suppl 5:21-9. doi: 10.1046/j.1468-1331.2001.00035.x PubMed
8.Frevert J. Pharmaceutical, biological, and clinical properties of botulinum neurotoxin type A products. Drugs R D. 2015;15(1):1-9. doi: 10.1007/s40268-014-0077-1 PubMed
9.CADTH. Switching Botulinum Toxin A Products for Patients with Upper Limb Spasticity or Cervical Dystonia: A Review of Clinical Effectiveness. 2018. Accessed 23 October. https://www.ncbi.nlm.nih.gov/books/NBK531716/pdf/Bookshelf_NBK531716.pdf
10.Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. doi: 10.1136/bmj.j4008 PubMed
11.Jansen JP, Trikalinos T, Cappelleri JC, et al. Supplementary material to: Indirect treatment comparison/network meta-analysis study questionnaire to assess relevance and credibility to inform health care decision making: an ISPOR-AMCP-NPC Good Practice Task Force report. Value Health. 2014;17(2):157-73. doi: 10.1016/j.jval.2014.01.004. PubMed
12.Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. doi: 10.1136/jech.52.6.377 PubMed
13.Agree Next Steps Consortium. The AGREE II Instrument. AGREE Enterprise; 2017. Accessed January 1, 1800. https://www.agreetrust.org/wp-content/uploads/2017/12/AGREE-II-Users-Manual-and-23-item-Instrument-2009-Update-2017.pdf
14.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1-e34. doi: 10.1016/j.jclinepi.2009.06.006 PubMed
15.Danchenko N, Johnston KM, Haeussler K, Whalen J. Comparative efficacy, safety, and cost-effectiveness of abobotulinumtoxinA and onabotulinumtoxinA in children with upper limb spasticity: a systematic literature review, indirect treatment comparison, and economic evaluation. J Med Econ. 2021;24(1):949-961. doi: 10.1080/13696998.2021.1957582 PubMed
16.Guyot P, Kalyvas C, Mamane C, Danchenko N. Botulinum Toxins Type A (Bont-A) in the Management of Lower Limb Spasticity in Children: A Systematic Literature Review and Bayesian Network Meta-analysis. J Child Neurol. 2019;34(7):371-381. doi: 10.1177/0883073819830579 PubMed
17.Yun JY, Kim JW, Kim HT, et al. Dysport and Botox at a ratio of 2.5:1 units in cervical dystonia: a double-blind, randomized study. Mov Disord. 2015;30(2):206-13. doi: 10.1002/mds.26085 PubMed
18.Petracca M, Lo Monaco MR, Ialongo T, et al. Efficacy and safety of long-term botulinum toxin treatment for acquired cervical dystonia: a 25-year follow-up. J Neurol. 2023;270(1):340-347. doi: 10.1007/s00415-022-11343-0 PubMed
19.Jochim A, Meindl T, Mantel T, et al. Treatment of cervical dystonia with abo- and onabotulinumtoxinA: long-term safety and efficacy in daily clinical practice. J Neurol. 2019;266(8):1879-1886. doi: 10.1007/s00415-019-09349-2 PubMed
20.Hefter H, Urer B, Brauns R, et al. Significant Long-Lasting Improvement after Switch to Incobotulinum Toxin in Cervical Dystonia Patients with Secondary Treatment Failure. Toxins (Basel). 2022;14(1):06. doi: 10.3390/toxins14010044
21.Hefter H, Hartmann CJ, Kahlen U, Samadzadeh S, Rosenthal D, Moll M. Clinical Improvement After Treatment With IncobotulinumtoxinA (XEOMIN(R)) in Patients With Cervical Dystonia Resistant to Botulinum Toxin Preparations Containing Complexing Proteins. Front Neurol. 2021;12:636590. doi: 10.3389/fneur.2021.636590 PubMed
22.Royal College of Physicians. Spasticity in adults: management using botulinum toxin National guidelines 2018. 2018. Accessed 20 October. https://www.rcp.ac.uk/improving-care/resources/spasticity-in-adults-management-using-botulinum-toxin/
23.Contarino MF, Van Den Dool J, Balash Y, et al. Clinical Practice: Evidence-Based Recommendations for the Treatment of Cervical Dystonia with Botulinum Toxin. Front Neurol. 2017;8:35. doi: 10.3389/fneur.2017.00035 PubMed
24.Simpson DM, Hallett M, Ashman EJ, et al. Practice guideline update summary: Botulinum neurotoxin for the treatment of blepharospasm, cervical dystonia, adult spasticity, and headache [RETIRED]: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2016;86(19):1818-26. doi: 10.1212/WNL.0000000000002560 PubMed
25.Do KH, Chun MH, Paik NJ, et al. Safety and efficacy of letibotulinumtoxinA(BOTULAX(R)) in treatment of post stroke upper limb spasticity: a randomized, double blind, multi-center, phase III clinical trial. Clin Rehabil. 2017;31(9):1179-1188. doi: 10.1177/0269215516689331 PubMed
Appendix 1: Selection of Included Studies
Figure 1: PRISMA Flow Chart of Study Selection
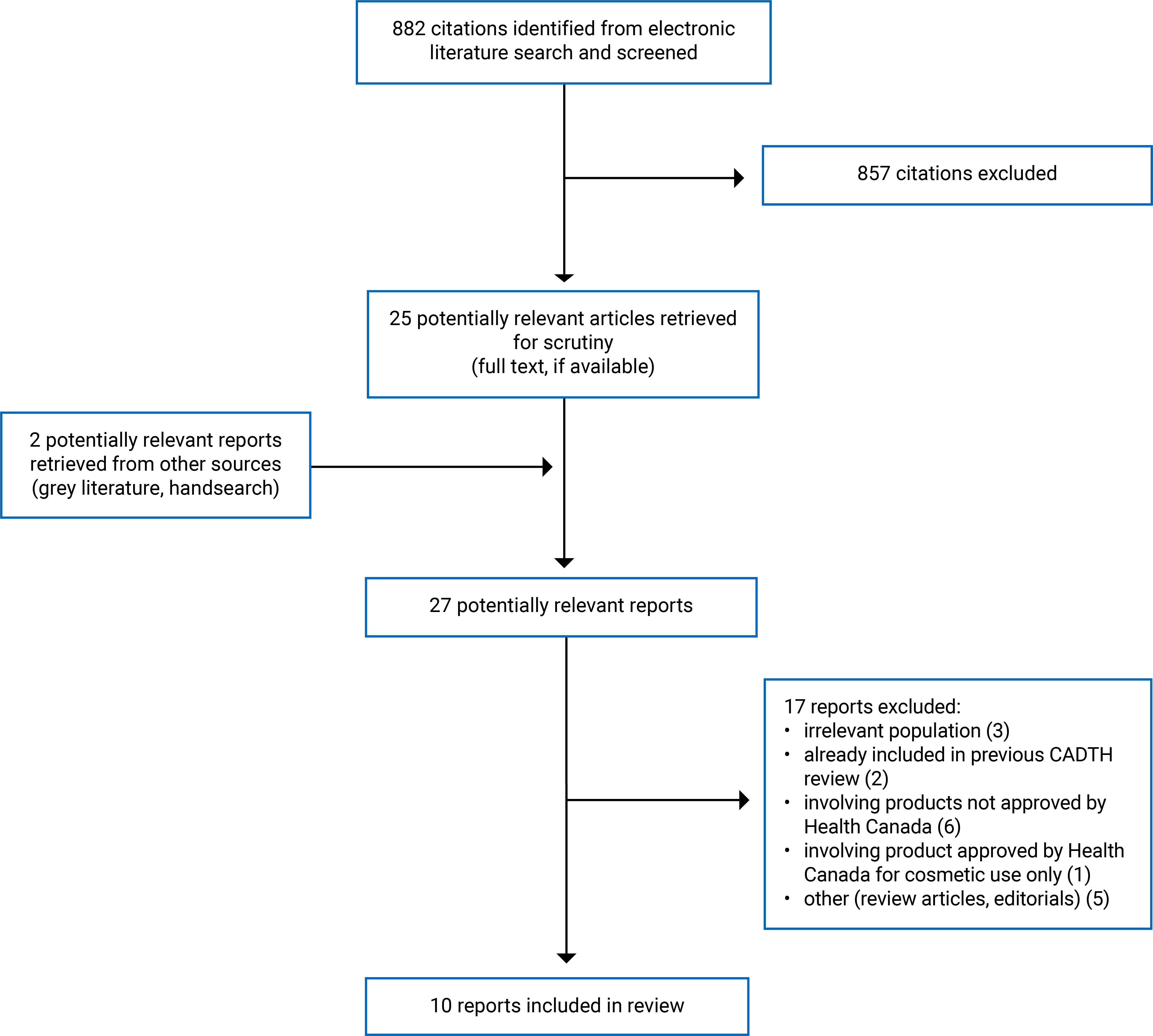
PRISMA = Preferred Reporting Items for Systematic reviews and Meta-Analyses.14
Appendix 2: Characteristics of Included Publications
Please note that this appendix has not been copy-edited.
Table 2: Characteristics of Included Systematic Reviews
Study citation, country, funding source | Study designs and numbers of primary studies included | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
Danchenko et al. (2021)15 France, Canada, UK Funding source: Ipsen | SR with NMA (Bayesian) 6 RCTs (8 publications) 1 RCT with aboBoNT-A 5 RCTs with onaBoNT-A | Children and adolescents receiving BoNT-A for the treatment of upper limb spasticity Sex:
Age, range: 4 to 16 years | Intervention: AboBoNT-A (Dysport) 2 U/kg to 16 U/kg Comparator: OnaBoNT-A (Botox) 1 U/kg to 6.6 U/kg | Outcomes:
Follow-up: 6 weeks to 16 weeks |
Guyot et al. (2019)16 France, the Netherlands Funding source: Ipsen | SR with NMA (Bayesian) 10 RCTs 3 RCTs with aboBoNT-A 7 RCTs with onaBoNT-A | Children and adolescents receiving BoNT-A for the treatment of lower limb spasticity Sex:
Age, range: 2 to 17 years | Intervention: AboBoNT-A (Dysport) 10 U/kg to 30 U/kg Comparator: OnaBoNT-A (Botox) 0.5 U/kg to 12 U/kg | Outcomes:
Follow-up: 12 weeks to 52 weeks |
aboBoNT-A = abobotulinumtoxin A; AE = adverse event; BoNT-A = botulinum neurotoxin type A; GAS = Goal Attainment Scale; MAS = Modified Ashworth Scale; NMA = network meta-analysis; onaBoNT-A = onabotulinumtoxin A; RCT = randomized controlled trial; SAE = serious adverse event; SR = systematic review.
aThe MAS is a clinical tool used to measure spasticity (increased muscle tone) in patients with central nervous system lesions. The MAS uses a 0 to 4 scale, with 0 meaning no increased tone and 4 indicating rigidity.
bThe GAS is a client-centred, individualized method used in health care and rehabilitation to measure progress toward personal goals. Each goal is evaluated on a 5-point scale, typically ranging from −2 to + 2: −2 = worse than expected outcome; −1 = baseline, or not quite as expected; 0 = expected outcome; +1 = better than expected outcome; +2 = much better than expected outcome.
cThe Tardieu Scale is a scale for measuring spasticity that considers resistance to passive movement at both slow and fast speed. The Tardieu Scale grades spasticity from 0 to 4 based on the quality of the muscle's reaction to a fast passive stretch. The grades are: 0 for no resistance, 1 for slight resistance throughout, 2 for a clear catch at a specific angle, 3 for fatigable clonus (less than 10 seconds), and 4 for unfatigable clonus (more than 10 seconds). A higher grade indicates a stronger spastic reflex.
Table 3: Characteristics of Included Primary Clinical Studies
Study citation, country, funding source | Study design | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
Randomized controlled trial (comparison) | ||||
Yun et al. (2015)17 Republic of Korea Funding source: Ipsen Ltd., and Ministry of Education, Science and Technology | Randomized, double-blind, multicentre, noninferiority, 2-period crossover study | Adult patients with CD Mean age (SD), years: 53.30 (10.76) Sex:
Mean time (SD) since onset of CD, years: 10.46 (8.62) | Intervention: AboBoNT-A (Dysport) (N = 48) Comparator: OnaBoNT-A (Botox) (N = 48) Conversion ratio: 2.5:1 First period with aboBoNT-A or onaBoNT-A, then switched to onaBoNT-A or aboBoNT-A in the second period | Outcomes:
Follow-up: Once per month for 16 weeks in the first period, followed by 4-week washout; switched to other formulation and then followed up once per month for 16 weeks in the second period |
Nonrandomized studies (comparison) | ||||
Petracca et al. (2023)18 Italy Funding source: University grant | Retrospective cohort study | Adult patients with acquired CD (N = 23) Age: NR Sex:
Mean disease duration (SD), years: 16.6 (17.5) |
| Outcomes:
Follow-up: up to 25 years |
Jochim et al. (2019)19 Germany Funding source: The authors declared that there was no funding | Retrospective cohort study | Adult patients with acquired CD Mean age (SD), years: aboBoNT-A: 43.7 (15.8) onaBoNT-A: 46.3 (15.3) Sex: NR |
| Outcomes:
Follow-up: up to 17 years for onaBoNT-A; up to 27 years for aboBoNT-A On average, patients had been treated for 5.9 (5.0) years with onaBoNT-A and 11.0 (7.7) years with aboBoNT-A |
Nonrandomized studies (switching) | ||||
Hefter et al. (2022)20 Germany Funding source: The authors declared that there was no external funding | Cross-sectional study | Adult patients with CD, with secondary treatment failure after aboBoNT-A or onaBoNT-A therapyh Mean age (SD), years:
Sex:
| Intervention: Switched group to incoBoNT-A (Xeomin) (N = 59) Comparator: Exclusively treated group with incoBoNT‑A (Xeomin) (N = 34) | Outcomes:
Follow-up: 2,700 days (30 treatment cycles) |
Hefter et al. (2021)21 Germany Funding source: NR | Single-centre, prospective, cohort study | Adult patients with CD, with partial secondary treatment failure under previous long-term BoNT-A (aboBoNT-A, onaBoNT-A, or RIMA) treatmenti | Intervention: Switch to incoBoNT-A (Xeomin) (N = 33); 200 U every 12 weeks (4 injection cycles = 48 weeks) Comparator: Baseline | Outcomes:
Follow-up: 48 weeks |
aboBoNT-A = abobotulinumtoxin A; AE = adverse event; ATSUI = Tsui at recruitment; BoNT-A = botulinum neurotoxin type A; BTSUI = best Tsui; CD = cervical dystonia; CDQ-24 = Craniocervical Dystonia Questionnaire (24-item); CGI-I, Clinical Global Impressions of Illness; GCA = global clinical assessment; incoBoNT-A= incobotulinumtoxin A; ITSUI = Tsui at onset of BoNT-A therapy; NAB = neutralizing antibodies; NR = not reported; onaBoNT-A = onabotulinumtoxin A; RIMA = rimabotulinumtoxin B; SD = standard deviation; STSUI = Tsui score at switch to incoBoNT-A; TWSTRS = Toronto Western Spasmodic Torticollis Rating Scale; U = unit.
aThe Tsui Scale is a clinical rating scale used to assess the severity of CD, a movement disorder affecting the neck and head. It is a 4-item scale with a maximum score of 25. A higher score on the scale indicates greater severity of the condition.
bTWSTRS is a tool used to measure the severity of CD (spasmodic torticollis) and evaluate how well treatments are working. It is a composite scale that assesses 3 main areas: severity (physical signs) (max = 35), disability (impact on daily life) (max = 30), and pain (max = 20). A higher score on the scale indicates greater severity of the condition.
cCGI-I is expressed as the proportions of patients with CGI of illness score of “1 = normal or not at all ill” or “2 = borderline mildly ill,” or “3 = mildly ill.”
dLatency was defined as the interval between injection and the first sign of improvement noticed by the patient.
eTotal duration of improvement was defined as the interval between the first day of improvement and the last day of reported benefit
fPeak effect duration was the number of days the patients experienced the best clinical effect.
gThe GCA was based on the patient’s and/or of next of kin’s perception of improvement expressed as 0% to 100% compared with the baseline condition. GCA was rated from 0 to 6 (0 = no effect, 1 = < 20%, 2 = 20% to 40%, 3 = 40% to 60%, 4 = 60% to 80%, 5 = 80% to 90%, 6 = complete resolution).
hSecondary treatment failure of botulinum toxin therapy is when a patient stops responding to treatment after an initial successful response, most often due to the development of NABs.
iPartial secondary treatment failure in botulinum toxin therapy occurs when a patient initially responds well to treatment but later experiences a gradual, incomplete loss of effectiveness of treatment, often due to the development of NABs or progression of the underlying disease. Criteria for partial secondary treatment failure were: the patient had previously had a good response by at least 3 Tsui Scale points; the patient presents with a systematic worsening of CD despite dose increase and/or change of botulinum neurotoxin preparations containing complexing proteins (systematic worsening was defined as an increase by at least 2 points over 3 consecutive Tsui Scale scores each determined about 3 months after injection); the patient reports reduced efficacy for these last 3 consecutive injections in comparison to previous injections.
jThe CDQ-24 is a 24-item disease-specific instrument measuring patients’ quality of life based on 5 subscales: stigma, emotional well-being, pain, activities of daily living, and social/family life. Patients with more than 20% improvement of CDQ24 were classified as having disease response, while those with a worsening of more than 20% were classified as not having disease response.
Table 4: Characteristics of Included Guidelines
Intended users, target population | Intervention and practice considered | Major outcomes considered | Evidence collection, selection, and synthesis | Evidence quality assessment | Recommendations development and evaluation | Guideline validation |
|---|---|---|---|---|---|---|
RCP (2018)22 | ||||||
Intended users: Health professionals involved in the management of spasticity and providers and purchasers of rehabilitation services Target population: Adults with spasticity due to neurologic illness or injury | To promote the appropriate use of BoNT-A in the management of spasticity, and give guidance on its administration and the wider principles of management | Effectiveness, HRQoL, economic, and safety outcomes | Systematic literature searches were conducted on each PICO question. | The quality of evidence was graded, ranging from high to very low, based on the confidence in the estimate of effect.a The strength of recommendations was graded based on consensus.b | The GDG first identified the key priority areas for the guidelines to address and set 1 or more PICO questions for each priority area. The GDG then considered its recommendation in respect of that PICO question according to a list of 11 questions relating to benefits and harms, resource use, equity, acceptability, and feasibility. | The final draft was circulated to all relevant parties and their comments incorporated. |
COST, Contarino et al. (2017)23 | ||||||
Intended users: Clinicians involved in the management of patients with CD Target population: Adult patients with CD | Comparison of different formulations of BoNT in improving motor symptoms, pain, and HRQoL, also in relation to the dosage conversion ratio | Effectiveness, HRQoL, and safety outcomes | Systematic literature searches were conducted on topic of BoNT treatment for CD (3 databases) | Level of evidencec and strength of recommendationd were classified according to the AAN guidelines | NR | Published in peer-reviewed journal |
AAN, Simpson et al. (2016)24 | ||||||
Intended users: Clinicians involved in the management of patients with blepharospasm, CD, spasticity, and migraine headache Target population: Adult patients with blepharospasm, CD, spasticity, and migraine headache | The use of BoNT for treatment of blepharospasm, cervical dystonia, spasticity, and migraine headache | Effectiveness, HRQoL, and safety outcomes | A formal process of reviewing and analyzing the existing literature | Studies were evaluated for their quality, and the strength of the overall evidence was determined.c The evidence and other factors were used to formulate specific recommendations.d | All guideline panel members vote on their level of agreement with each recommendation, which helped to determine its strength. | Published in peer-reviewed journal |
AAN = American Academy of Neurology; BoNT-A = botulinum neurotoxin type A; CD = cervical dystonia; COST = European Cooperation in Science and Technology; GDG = Guideline Development Group; HRQoL = health-related quality of life; NR = not reported; PICO = Population, Intervention, Comparison, and Outcome; RCP = Royal College of Physicians.
aLevel of evidence related to benefits: High (Research Grade A), Moderate (Research Grade B), Low (Research Grade C), User/professional opinion.
bStrength of recommendation: very strong = high-level research evidence (Grade A) with minimal harms and costs; strong= lower-level evidence but minimal harms and costs; moderate = lower-level evidence and potentially significant harms and/or costs; weak = lower-level evidence and potentially significant harms and costs.
cLevel of evidence: class I = a randomized, controlled clinical trial of the intervention of interest with masked or objective outcome assessment, in a representative population (relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences); class II = a randomized controlled clinical trial of the intervention of interest in a representative population with masked or objective outcome assessment that lacks 1 criteria a–e above or a prospective matched cohort study with masked or objective outcome assessment in a representative population that meets b–e above (relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences); class III = all other controlled trials (including well-defined natural history controls or patients serving as their own controls) in a representative population, where outcome is independently assessed, or independently derived by objective outcome measurement; class IV = studies not meeting class I, II, or III criteria, including studies based on consensus or expert opinion.
dStrength of recommendation: A = established as effective, ineffective, or harmful for the given condition in the specified population (requires 2 consistent class I studies); B = probably effective, ineffective, or harmful for the given condition in the specified population (requires 1 class I study or 2 consistent class II studies); C = possibly effective, ineffective, or harmful for the given condition in the specified population (requires 1 class II study or 2 consistent class III studies); U = data are inadequate or conflicting; given current knowledge, treatment is unproven (studies not meeting criteria for class I through class II).
Appendix 3: Critical Appraisal of Included Publications
Please note that this appendix has not been copy-edited.
Table 5: Strengths and Limitations of Systematic Reviews and Network Meta-Analyses Using AMSTAR 2 and the ISPOR Questionnaire
Strengths | Weaknesses |
|---|---|
Danchenko et al. (2021)15 | |
Objectives:
Search strategies:
Data collection:
Methods for analysis/synthesis of evidence:
Reporting:
Discussion:
| Search strategies:
Methods for analysis/synthesis of evidence:
Reporting:
|
Guyot et al. (2019)16 | |
Objectives:
Search strategies:
Data collection:
Methods for analysis/synthesis of evidence:
Reporting:
Discussion:
| Search strategies:
Methods for analysis/synthesis of evidence:
Reporting:
|
AMSTAR 2 = A MeaSurement Tool to Assess systematic Reviews 2;10 CrI = credible interval; ISPOR = International Society for Pharmacoeconomics and Outcomes Research;11 ITC = indirect treatment comparison; MA = meta-analysis; NA = not applicable; NMA = network meta-analysis; NR = not reported; RCT = randomized controlled trial.
Table 6: Strengths and Limitations of Clinical Studies Using the Downs and Black Checklist12
Strengths | Weaknesses |
|---|---|
Yun et al. (2015)17 | |
Reporting:
External validity:
Internal validity – bias:
Internal validity – confounding:
| Reporting:
External validity:
Internal validity – confounding:
|
Petracca et al. (2023)18 | |
Reporting:
Internal validity – bias:
| Reporting:
External validity:
Internal validity – bias:
Internal validity – confounding:
|
Jochim et al. (2019)19 | |
Reporting:
Internal validity – bias:
| Reporting:
External validity:
Internal validity – bias:
Internal validity – confounding:
|
Hefter et al. (2022)20 | |
Reporting:
Internal validity – bias:
| Reporting:
External validity:
Internal validity – bias:
Internal validity – confounding:
|
Hefter et al. (2021)21 | |
Reporting:
Internal validity – bias:
| Reporting:
External validity:
Internal validity – bias:
Internal validity – confounding:
|
Table 7: Strengths and Limitations of Guidelines Using AGREE II
Item | RCP (2018)22 | COST, Contarino et al. (2017)23 | AAN, Simpson et al. (2016)24 |
|---|---|---|---|
Domain 1: scope and purpose | |||
1. The overall objective(s) of the guideline is (are) specifically described. | Yes | Yes | Yes |
2. The health question(s) covered by the guideline is (are) specifically described. | Yes | Yes | Yes |
3. The population (e.g., patients, public) to whom the guideline is meant to apply is specifically described. | Yes | Yes | Yes |
Domain 2: stakeholdera involvement | |||
4. The guideline development group includes individuals from all relevant professional groups. | Yes | Yes | Yes |
5. The views and preferences of the target population (e.g., patients, public) have been sought. | Yes | Yes | Yes |
6. The target users of the guideline are clearly defined. | Yes | Yes | Yes |
Domain 3: rigour of development | |||
7. Systematic methods were used to search for evidence. | Yes | Yes | Yes |
8. The criteria for selecting the evidence are clearly described. | Yes | Yes | Yes |
9. The strengths and limitations of the body of evidence are clearly described. | Yes | Yes | Yes |
10. The methods for formulating the recommendations are clearly described. | Yes | Yes | Yes |
11. The health benefits, side effects, and risks have been considered in formulating the recommendations. | Yes | Yes | Yes |
12. There is an explicit link between the recommendations and the supporting evidence. | Yes | Yes | Yes |
13. The guideline has been externally reviewed by experts before its publication. | Yes | Yes | Yes |
14. A procedure for updating the guideline is provided. | Unclear | Unclear | Unclear |
Domain 4: clarity of presentation | |||
15. The recommendations are specific and unambiguous. | Yes | Yes | Yes |
16. The different options for management of the condition or health issue are clearly presented. | Yes | Yes | Yes |
17. Key recommendations are easily identifiable. | Yes | Yes | Yes |
Domain 5: applicability | |||
18. The guideline describes facilitators and barriers to its application. | Unclear | Unclear | Unclear |
19. The guideline provides advice and/or tools on how the recommendations can be put into practice. | Unclear | Unclear | Unclear |
20. The potential resource implications of applying the recommendations have been considered. | Yes | Unclear | Unclear |
21. The guideline presents monitoring and/or auditing criteria. | Unclear | Unclear | Unclear |
Domain 6: editorial independence | |||
22. The views of the funding body have not influenced the content of the guideline. | Yes | Yes | Yes |
23. Competing interests of guideline development group members have been recorded and addressed. | Yes | Yes | Yes |
AAN = American Academy of Neurology; AGREE II = Appraisal of Guidelines for Research and Evaluation II;13 COST = European Cooperation in Science and Technology; RCP = the Royal College of Physicians.
aWherever possible, Canada’s Drug Agency no longer uses the term stakeholder because of its harmful association with colonialism; in this case, the word appears in the original phrasing of AGREE II.
Appendix 4: Main Study Findings
Please note that this appendix has not been copy-edited.
Table 8: Summary of Comparative Clinical Efficacy Findings – Limb Spasticity
Study citation, study design, intervention vs. comparator | Outcomes | Results |
|---|---|---|
Danchenko et al. (2021)15 SR with NMA (Bayesian) 6 RCTs (8 publications) 1 RCT with aboBoNT-A 5 RCTs with onaBoNT-A AboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for upper limb spasticity in children | MAS (elbow) | Difference in mean change from baseline (95% CrI); P value
|
Sensitivity analysis (excluding 1 study with large heterogeneity) Difference of mean change from baseline (95% CrI); P value
| ||
MAS (wrist) | Difference of mean change from baseline (95% CrI); P value
| |
Sensitivity analysis (excluding 1 study with large heterogeneity) Difference of mean change from baseline (95% CrI); P value
| ||
MAS responder rates for elbow or wrist | Median % of responders (95% CrI)
| |
Guyot et al. (2019)16 SR with NMA (Bayesian) 10 RCTs 3 RCTs with aboBoNT-A 7 RCTs with onaBoNT-A aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for lower limb spasticity in children | MAS at 12 weeks | Difference of mean change from baseline (95% CrI)
|
GAS at 12 weeks | Difference of mean change from baseline (95% CrI)
| |
Tardieu Scale at 12 weeks | Difference of mean change from baseline (95% CrI)
|
aboBoNT-A = abobotulinumtoxin A; AE = adverse event; CrI = credible interval; GAS = Goal Attainment Scale; MAS = Modified Ashworth Scale; NMA = network meta-analysis; NR = not reported; onaBoNT-A = onabotulinumtoxin A; RCT = randomized controlled trial; SAE = serious adverse event; SR = systematic review; U = unit; vs. = versus.
Table 9: Summary of Comparative Safety Findings – Limb Spasticity
Study citation, study design, intervention vs. comparator | Outcomes | Results |
|---|---|---|
Danchenko et al. (2021)15 SR with NMA (Bayesian) 6 RCTs (8 publications) 1 RCT with aboBoNT-A 5 RCTs with onaBoNT-A aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for upper limb spasticity in children | Overall AEs at 16 and 52 weeks | RR (95% CrI); P value
|
SAEs at 12 to 16 weeks | RR (95% CrI); P value
| |
Treatment-related AEs at 12 to 16 weeks | RR (95% CrI); P value
| |
Guyot et al. (2019)16 SR with NMA (Bayesian) 10 RCTs 3 RCTs with aboBoNT-A 7 RCTs with onaBoNT-A aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for lower limb spasticity in children | Overall AEs at 12 weeks | OR (95% CrI)
|
aboBoNT-A = abobotulinumtoxin A; AE = adverse event; CrI = credible interval; NMA = network meta-analysis; onaBoNT-A = onabotulinumtoxin A; OR = odds ratio; RCT = randomized controlled trial; RR = relative risk; SAEs = serious adverse event; SD = standard deviation; SR = systematic review; U = unit; vs. = versus.
Table 10: Summary of Comparative Clinical Efficacy Findings – Cervical Dystonia
Study citation, study design, intervention vs. comparator | Outcomes | Results |
|---|---|---|
Yun et al. (2015)17 Randomized, double-blind, multicentre, noninferiority, 2‑period crossover study aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for CD in adults | Tsui Scale score |
|
Total TWSTRS score |
| |
TWSTRS Severity subscore |
| |
TWSTRS Disability subscore |
| |
TWSTRS Pain subscore |
| |
Patients scoring 1, 2, or 3 on CGI-I scale |
| |
Patients scoring 1, 2, or 3 on PGI-I scale |
| |
Petracca et al. (2023)18 Retrospective cohort study IncoBoNT-A (Xeomin) vs. onaBoNT-A (Botox) vs. aboBoNT-A (Dysport) for CD in adults | Latency |
|
Duration of maximum improvement |
| |
Total duration of improvement |
| |
GCA score |
| |
Jochim et al. (2019)19 Retrospective cohort study aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for CD in adults | Satisfactory effect | Patients reported favourable results after at least 80% of their individual treatments, n of N (%):
|
aboBoNT-A = abobotulinumtoxin A; CD = cervical dystonia; CGI-I = Clinical Global Impression of Illness; CI = confidence interval; GCA = global clinical assessment; incoBoNT-A = incobotulinumtoxin A; onaBoNT-A = onabotulinumtoxin A; PGI-I = Patient Global Impression of Improvement; SD = standard deviation; TWSTRS = Toronto Western Spasmodic Torticollis Rating Scale; vs. = versus.
Table 11: Summary of Comparative Safety Findings – Cervical Dystonia
Study citation, study design, intervention vs. comparator | Outcomes | Results |
|---|---|---|
Yun et al. (2015)17 Randomized, double-blind, multicentre, noninferiority, 2‑period crossover study aboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for CD in adults | Overall AEs | 14.9% (14 of 94) vs. 20.2% (19 of 94); P = 0.332 |
Severe AEs | 0 vs. 0; P = 1.00 | |
Treatment-related AEs | 14.9% (14 of 94) vs. 20.2% (19 of 94); P = 0.332 | |
AEs leading to discontinuation | 0 vs. 0; P = 1.00 | |
Muscle weakness | 9.6% (9 of 94) vs. 13.8% (13 of 94); P = 0.388 | |
Dysphagia | 6.4% (6 of 94) vs. 12.8% (12 of 94); P = 0.070 | |
Petracca et al. (2023)18 Retrospective cohort study IncoBoNT-A (Xeomin) vs. onaBoNT-A (Botox) vs. aboBoNT-A (Dysport) for CD in adults | Overall AEs | 0.8% vs. 4.7% vs. 3.5% |
Muscle weakness | 0 vs. 3.7% vs. 2.5% | |
Dysphagia | 0 vs. 0.4% vs. 0.8% | |
Allergic reactions | None | |
Jochim et al. (2019)19 Retrospective cohort study AboBoNT-A (Dysport) vs. onaBoNT-A (Botox) for CD in adults | Overall AEs | 15% (31 of 209) vs. 10% (14 of 135) |
Muscle weakness | 6% (13 of 209) vs. 2% (3 of 135) | |
Dysphagia | 6% (13 of 209) vs. 2% (3 of 135) |
aboBoNT-A = abobotulinumtoxin A; AE = adverse event; CD = cervical dystonia; onaBoNT-A = onabotulinumtoxin A; vs. = versus.
Table 12: Summary of Clinical Effectiveness Findings of Switching Between BoNT-A Products – Cervical Dystonia
Study citation, study design, intervention vs. comparator | Outcomes | Results |
|---|---|---|
Hefter et al. (2022)20 Cross-sectional study Group switched to incoBoNT-A (Xeomin) vs. the group treated exclusively with incoBoNT-A (Xeomin) for CD in adults | Initial clinical severity of CD before treatment (ITSUI score) | Mean score (SD):
|
Best outcome during therapy (BTSUI score) | Mean score (SD):
| |
Severity at the time of switching to incoBoNT-A (STSUI score) | Mean score (SD):
| |
Severity at recruitment (ATSUI score); i.e., after being treated with incoBoNT-A | Mean score (SD):
| |
Hefter et al. (2021)21 Single-centre, prospective, cohort study Switch to incoBoNT-A after partial secondary treatment failure under previous long-term BoNT-A treatment for CD in adults | TWSTRS | Mean score (SD):
|
CDQ-24 | Median CDQ-24 scores improved from baseline to week 48, but failed to reach the significant level of 5% (data not shown) | |
Development of NAB titres during incoBoNT‑A treatment | NAB titres:
|
aboBoNT-A = abobotulinumtoxin A; ATSUI = Tsui at recruitment; BoNT-A = botulinum neurotoxin type A; BTSUI = best Tsui; CD = cervical dystonia; CDQ-24 = Craniocervical Dystonia Questionnaire (24-item); incoBoNT-A = incobotulinumtoxin A; ITSUI = initial Tsui at onset of BoNT-A therapy; NAB = neutralizing antibodies; NS = not statistically significant; onaBoNT-A = onabotulinumtoxin A; SD = standard deviation; STSUI = Tsui at switch to incoBoNT-A; TWSTRS = Toronto Western Spasmodic Torticollis Rating Scale; vs. = versus.
Table 13: Summary of Recommendations in Included Guidelines
Recommendations and supporting evidence | Level of evidence and strength of recommendations |
|---|---|
RCP (2018)22 | |
“Botulinum toxin type A (BoNT-A) is a safe and effective treatment for upper and lower limb spasticity, resulting in both passive and active functional gains.” p. IX Supporting evidence: 1 RCT and 4 observational studies provided evidence to address the recommendation. | Level of evidence: RA, user/professional opinion Strength of recommendation: Strong |
“Clinicians must be aware that different BoNT-A products have different dosage schedules. “The current recommended maximum doses per treatment session within licensed usage for spasticity are:
“Clinicians should refer to Appendix 2 for the recommended doses for individual muscles.” p. X Supporting evidence: Not reported | Level of evidence: A Strength of recommendation: Very strong |
COST, Contarino et al. (2017)23 | |
Supporting evidence: 12 RCTs provided evidence to address these 3 recommendations. | Level of evidence: Class I Strength of recommendations: A for aboBoNT-A, B for incoBoNT-A, and onaBoNT-A) |
BoNT-A treatment improves QoL. Supporting evidence: 1 RCT provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: B |
BoNT-A reduces pain associated with CD. Supporting evidence: 5 RCTs provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: A |
Conversion ratio of onabotulinumtoxin A to abobotulinumtoxin A: 1 IU to 3 IU. Supporting evidence: 3 RCTs provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: A |
Conversion ratio of onabotulinumtoxin A to abobotulinumtoxin A: 1 IU to 2.5 IU. Supporting evidence: 1 RCT provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: B |
Conversion ratio of onabotulinumtoxin A to incobotulinumtoxin A: 1 IU to 1 IU. Supporting evidence: 1 open-label prospective crossover study and 1 RCT provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: B |
An initial total dose of 500 IU abobotulinumtoxin A is effective, although other dosages might be used. Supporting evidence: 2 RCTs provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: A |
An initial total dose of 120 IU incobotulinumtoxin A is probably effective. Supporting evidence: 1 RCT provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: B |
No clear recommendations can be given on the optimal starting doses of onabotulinumtoxin A (level U). Supporting evidence: None | Level of evidence: Not applicable Strength of recommendation: U |
AAN, Simpson et al. (2016)24 | |
For CD: “AboBoNT-A…should be offered (Level A), and onaBoNT-A and incoBoNT-A should be considered (Level B), as options for the treatment of CD.” p. 1821 Supporting evidence: Studies reviewed in the previous 2008 AAN guidelines | Level of evidence: Class I Strength of recommendations: A for aboBoNT-A, B for onaBoNT-A and incoBoNT-A) |
For upper limb spasticity in adults: “AboBoNT-A, incoBoNT-A, and onaBoNT-A are established as safe and effective for the reduction of adult upper limb spasticity and improvement of passive function.” p. 1822 Supporting evidence: Multiple class I studies for all preparations provided evidence to address the recommendation. | Level of evidence: Class I Strength of recommendation: A |
For lower limb spasticity in adults: “For focal manifestations of adult spasticity involving the lower limb that warrant treatment, onaBoNT-A and aboBoNT-A should be offered (Level A) as treatment options. “There is insufficient evidence to support or refute a benefit of incoBoNT-A…for treatment of adult lower limb spasticity.” p. 1822 Supporting evidence: Studies reviewed in the previous 2008 AAN guidelines | Level of evidence: Class I Strength of recommendation: A |
AAN = American Academy of Neurology; aboBoNT-A = abobotulinumtoxin A; BoNT-A = botulinum neurotoxin type A; CD = cervical dystonia; COST = European Cooperation in Science and Technology; incoBoNT-A = incobotulinumtoxin A; onaBoNT-A = onabotulinumtoxin A; RCP = Royal College of Physicians; RCT = randomized controlled trial.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.