CADTH Reimbursement Review
Abrocitinib (Cibinqo)
Sponsor: Pfizer Canada ULC
Therapeutic area: Atopic dermatitis, moderate to severe
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAD
American Academy of Dermatology
AD
atopic dermatitis
AE
adverse event
ALT
alanine transaminase
ANCOVA
analysis of covariance
AST
aspartate transaminase
BSA
body surface area
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CPK
creatine phosphokinase
CSPA
Canadian Skin Patient Alliance
DFI
Dermatitis Family Impact
DLQI
Dermatology Life Quality Index
DVT
deep vein thrombosis
EASI
Eczema Area and Severity Index
EASI-50
improvement of 50% or greater in the Eczema Area and Severity Index total score
EASI-75
improvement of 75% or greater in the Eczema Area and Severity Index total score
EASI-90
improvement of 90% or greater in the Eczema Area and Severity Index total score
EASI-100
improvement of 100% in the Eczema Area and Severity Index total score
eGFR
estimated glomerular filtration rate
EQ-5D-5L
EQ-5D 5-Levels questionnaire
EQ-5D-Y
EQ-5D Youth Scale
EQ VAS
EQ-5D Visual Analogue Scale
ESC
Eczema Society of Canada
ESS
effective sample size
FACIT-F
Functional Assessment of Chronic Illness Therapy–Fatigue
FAS
full analysis set
HADS
Hospital Anxiety and Depression Scale
HR
hazard ratio
ICER
Institute for Clinical and Economic Review
IGA
Investigator’s Global Assessment
IQR
interquartile range
ITC
indirect treatment comparison
JAK
Janus kinase
JAK1
Janus kinase-1
LSMD
least squares mean difference
MAIC
matching-adjusted indirect comparison
MACE
major adverse cardiovascular event
MAR
missing at random
MID
minimal important difference
NICE
National Institute for Health and Care Excellence
MNAR
missing not at random
NMA
network meta-analysis
NRS
numeric rating scale
PE
pulmonary embolism
Peds-FACIT-F
Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue
POEM
Patient-Oriented Eczema Measure
PP-NRS
peak pruritus numerical rating scale
PP-NRS4
improvement of 4 or greater from baseline on peak pruritus numerical rating scale
PSAAD
Pruritus and Symptoms Assessment for Atopic Dermatitis
PtGA
Patient Global Assessment
RCT
randomized controlled trial
SAE
serious adverse event
SCORAD
Scoring Atopic Dermatitis
SCORAD-50
improvement of 50% or greater in Scoring Atopic Dermatitis
SCORAD-75
improvement of 75% or greater in Scoring Atopic Dermatitis
SD
standard deviation
SF-36
Short Form (36) Health Survey
SF-36v2
Short Form (36) Health Survey Version 2
TCI
topical calcineurin inhibitor
TCS
topical corticosteroids
TP
tipping point
TEAE
treatment-emergent adverse events
ULN
upper limit of normal
VAS
visual analogue scale
VTE
venous thromboembolism
WDAE
withdrawal due to adverse event
WPAI-AD
Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Abrocitinib (Cibinqo), 200 mg, 100 mg, 50 mg, oral tablets |
Indication | Cibinqo (abrocitinib) is indicated for the treatment of patients 12 years and older with refractory moderate-to-severe atopic dermatitis, including pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advised |
Reimbursement request | As per indication |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard |
NOC date | June 29, 2022 |
Sponsor | Pfizer Canada ULC |
NOC = Notice of Compliance.
Introduction
Atopic dermatitis (AD) is the most common type of eczema. It is a chronic, relapsing, inflammatory skin condition characterized by severely itchy skin (pruritus) that results in red and swollen skin (rash). Lesions may appear as fluid-filled vesicles that ooze, crack, and crust. Pruritus of the skin can cause frequent scratching and may result in lichenification (thickening of the skin) and secondary skin infections. Atopic dermatitis typically involves the popliteal (skin folds behind the knees) and the antecubital (skin folds in front of the elbows) areas. It may also appear on the face, neck, and hands. Individuals with AD have skin with impaired barrier function and reduced water-holding capacity, resulting in dry skin that requires treatment with specific bathing, cleansing, and moisturizing practices.
The goals of AD management are to prevent flares (episodes of worsening of symptoms typically requiring escalation of treatment), and effectively manage flares when they occur by preventing disease progression. While there is no cure for AD, several therapeutic options are available to patients to manage the condition. The majority of patients treat AD by avoiding skin irritants and using general skin care methods and topical anti-inflammatory therapy. If these common methods fail to improve AD, patients may use off-label systemic therapy (i.e., immunosuppressant therapy) or other therapies such as phototherapy.
The most common pharmaceutical topical therapies include topical corticosteroids (TCS) and topical calcineurin inhibitors (TCIs). Topical corticosteroids act as anti-inflammatory therapy and are considered to be the first-line treatment for AD. Topical calcineurin inhibitors are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used long-term. In Canada, the 2 available second-line drugs are pimecrolimus and tacrolimus. Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available in Canada, although it is not recommended for reimbursement by CADTH. Phototherapy is another second-line therapy that is commonly used after failure of TCS, TCIs, and crisaborole.
Systemic therapy for the treatment of AD typically involves the use of antimicrobials, antihistamines, or immunomodulators. Immunomodulatory drugs, including methotrexate, cyclosporine, mycophenolate mofetil, azathioprine (listed in order of frequency of use in Canada), can be used in patients who are not responsive to other treatments. Dupilumab (Dupixent) is an interleukin-4 and interleukin-13 inhibitor indicated for use in adults and pediatrics with moderate to-severe AD whose disease is not adequately controlled with topical prescription therapies or for whom those therapies are not advisable. CADTH recommended that dupilumab be reimbursed with conditions and it is currently reimbursed by the participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure on or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine.
Abrocitinib is a selective Janus kinase-1 (JAK1) inhibitor indicated for the treatment of patients 12 years of age and older with refractory moderate-to-severe AD, including pruritus, who have had an inadequate response to other systemic drugs (steroid or biologic) or for whom these treatments are not advisable. The product monograph states that abrocitinib can be used with or without medicated topical therapies for AD. Abrocitinib is available as 50 mg, 100 mg, and 200 mg oral tablets. The dosage recommended in the product monograph is 100 mg or 200 mg orally once daily, based on individual goals of therapy and the potential risk for adverse reactions. For patients using the 200 mg once daily dosage, a reduction in the dosage to 100 mg once daily can be considered after symptom control is achieved at week 12. Relative to patients who maintained the 200 mg dose, the risk of occurrence of serious adverse reactions was lower in patients who reduced their dose to 100 mg beyond 12 weeks. If symptom control is lost after dose reduction, the dose can be increased to 200 mg. In patients with moderate renal impairment (an estimated glomerular filtration rate [eGFR] of 30 mL/min to < 60 mL/min) or severe renal impairment (an eGFR < 30 mL/min), the recommended dose of abrocitinib should be reduced by 50%.
The objective of this review is to evaluate the beneficial and harmful effects of oral abrocitinib 100 mg and 200 mg once daily for the treatment of patients 12 years of age and older with moderate-to-severe AD, including pruritus, who have had an inadequate response to prescribed topical therapy or for whom these treatments are not advisable.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups responded to CADTH’s call for patient input: the Canadian Skin Patient Alliance (CSPA), Eczéma Québec, and the Eczema Society of Canada (ESC). Eczéma Québec and the CSPA developed and circulated a web-based survey through both organizations’ newsletters and other channels. The survey drew 56 respondents. The ESC gathered survey data from more than 3,000 Canadians who live with AD on topics including quality-of-life impact, experience with systemic treatments, the AD patient journey, and experience with itch related to AD.
The patient groups reported that AD negatively affects mood and the ability to work, attend school, and participate in social interactions, and can cause patients to experience psychological distress. Itch is frequently experienced by patients and is considered the most burdensome symptom of AD, often affecting the ability of patients to sleep. The patient groups are seeking treatments that will reduce itch, decrease the occurrence of flares, reduce inflammation and rashes, and improve their ability to sleep and overall quality of life. Patients, particularly those who are adolescents, want to be able to have the confidence to be more outgoing and social, and patients with skin of colour want to avoid the visible changes in skin pigmentation that can result from scratching, flares, and scarring associated with AD.
Patients affected by AD must often try multiple treatments to find the best option for their circumstances, and these circumstances can change over time. The patient groups emphasized the importance of multiple treatment options to ensure that the specific circumstances of each patient can be addressed.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical expert consulted by CADTH noted that abrocitinib is potentially a useful addition to the currently available therapeutic options for AD. Abrocitinib may be particularly useful for patients who have contraindications to, experience adverse effects from, or who are unresponsive to the use off-label immunosuppressive drugs. Abrocitinib could also provide another treatment option for patients who have been treated with dupilumab but have demonstrated a suboptimal response, developed severe conjunctivitis or other ocular side effects from dupilumab, are intolerant to injections (e.g., due to severe injection-site reactions), and/or would prefer an orally administered treatment.
The clinical expert noted that abrocitinib should be used as an add-on therapy and that all patients should continue regimens involving emollients, TCS, and/or TCIs. Abrocitinib should not be used in combination with off-label immunosuppressives or dupilumab. The clinical expert was of the opinion that many specialists would consider a trial of methotrexate and cyclosporine before initiating treatment with abrocitinib.
The clinical expert suggested that patients less suitable for treatment with abrocitinib would be those with AD who are well controlled with topical therapy, phototherapy, and/or intermittent off-label immunosuppressive therapy, as well as those who are currently well controlled with dupilumab. Abrocitinib should be avoided in patients with potential contraindications to Janus kinase (JAK) inhibitors. Such contraindications include severe active infections, malignancies, ongoing treatment with chemotherapy such as checkpoint inhibitors, severe hepatic disease, severe renal disease, pregnancy and/or lactation, a history of thromboembolic events, and pre-existing hematologic disease.
In general, the outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials of AD treatments. Of these outcome measurements, an improvement of 75% of greater in the Eczema Area and Severity Index total score (EASI-75) after 16 weeks of treatment is a reasonable measure of response. In the opinion of the clinical expert, patients who initiate treatment with abrocitinib would be re-evaluated after 16 weeks (depending on the ability to arrange appointments). Those judged to be responders at this visit would be seen subsequently at 6-month intervals. Those who do not reach response targets at 16 weeks could be re-evaluated after 20 weeks following initiation of drug.
The factors anticipated by the clinical expert to be used as criteria for discontinuation included failure to achieve a clinically meaningful response at 16 to 20 weeks; failure to maintain an adequate response on long-term maintenance; development of a hypersensitivity response judged to be due to abrocitinib; treatment-emergent adverse effects (TEAEs) such as lymphopenia, neutropenia, arterial thrombosis, or venous thromboembolism (VTE); and treatment-emergent severe infections or malignancies.
Administration of the drug involves no special challenges. However, a specialist would still be required to diagnose, treat, and monitor patients taking abrocitinib. Appropriate specialists include pediatric dermatologists, general dermatologists, or pediatricians with experience and interest in AD.
Clinician Group Input
No clinician groups responded to the call for input for the review of abrocitinib.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review processes. The following were identified as key factors that could affect the implementation of a CADTH recommendation for abrocitinib.
Access to phototherapy may be limited in some areas of Canada. The clinical expert consulted by CADTH noted that phototherapy is typically accessible in urban areas, but access may be limited in rural areas. The expert noted that this barrier to phototherapy access should be considered in the reimbursement review decision-making process.
Could abrocitinib be initiated in patients who have failed previous treatment with a biologic drug? The clinical expert noted that patients who have failed dupilumab (with or without prior exposure to an immunomodulator) could be candidates to receive abrocitinib. The clinical expert noted that there is limited evidence supporting the sequential use of abrocitinib after an adequate trial of dupilumab in patients with moderate-to-severe AD.
Should patients be required to have a previous trial of (or be ineligible for) cyclosporine, methotrexate, and phototherapy before initiating treatment with abrocitinib? The clinical expert consulted by CADTH noted that a trial of 2 of the 4 immunomodulators (methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine) should be considered before initiating abrocitinib.
Could the reimbursement criteria that were recommended for dupilumab (e.g., initiation and renewal criteria) be applicable to abrocitinib? The clinical expert consulted by CADTH noted that the criteria for dupilumab could be applicable for abrocitinib and could be implemented in clinical practice.
Should patients be required to undergo an adequate trial with dupilumab before being eligible for treatment with abrocitinib? The clinical expert consulted by CADTH noted that prior therapy with dupilumab should not be required for a patient to be eligible for treatment with abrocitinib, as the 2 drugs have the same indication and potential place in therapy.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The evidence for this review was derived from the results of a systematic literature review of pivotal and phase III studies that was supplemented with additional studies to address important gaps in the evidence from randomized controlled trials (RCTs). The systematic review included 6 double-blind, phase III RCTs: a pair of 12-week placebo-controlled trials conducted with abrocitinib as monotherapy for AD (JADE MONO-1 [N = 387] and JADE MONO-2 [N = 391]); 2 placebo-controlled trials conducted with abrocitinib as combination therapy for AD (JADE COMPARE [N = 838 adults] and JADE TEEN [N = 287 adolescents]); 1 26-week active-controlled trial comparing abrocitinib and dupilumab as combination therapy (JADE DARE [N = 727]); and 1 placebo-controlled, responder-enriched, withdrawal trial (JADE REGIMEN [N = 789]). The evidence from these studies was supplemented with the interim results from 1 long-term extension-phase study (JADE EXTEND) and 3 indirect treatment comparisons (ITCs).
The included studies evaluated a range of outcomes that are important in the management of AD, including overall severity of AD (e.g., the Eczema Area and Severity Index [EASI] and Investigator’s Global Assessment [IGA]), severity of itching (e.g., peak pruritus numerical rating scale [PP-NRS]), symptoms (e.g., Patient-Oriented Eczema Measure [POEM] and Pruritus and Symptoms Assessment for Atopic Dermatitis [PSAAD]), health-related quality of life (e.g., Dermatology Life Quality Index [DLQI] and Children’s Dermatology Life Quality Index [CDLQI]), fatigue (e.g., Functional Assessment of Chronic Illness Therapy–Fatigue [FACIT-F] scale), patient-reported anxiety and depression, and the need for additional AD medications (e.g., corticosteroid-free days). In addition, the JADE REGIMEN study investigated the use of abrocitinib (100 mg once daily or 200 mg once daily) as a maintenance therapy for patients who achieved an initial response to the abrocitinib 200 mg once daily dosage regimen by evaluating the time to acute worsening of the patient’s condition (i.e., development of a disease flare in accordance with standardized criteria).
The eligibility criteria for the included RCTs were similar except for the differences in the age ranges for the combination-therapy studies (i.e., the JADE COMPARE and JADE DARE trials were limited to adults and the JADE TEEN trial was limited to adolescents) and the need to establish a response to abrocitinib 200 mg once daily to be randomized in the JADE REGIMEN trial. All of the trials enrolled patients with moderate-to-severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., those with refractory moderate-to-severe AD and an inadequate response to other systemic drugs). The proportions of patients with prior exposure to at least 1 systemic therapy for AD in the included trials were: 48.3% for JADE MONO-1, 41.4% for JADE MONO-2, 43.2% for JADE COMPARE, 47.9% for JADE DARE, 25.6% for JADE TEEN, and 59.5% for JADE REGIMEN (in both the open-label induction phase and the double-blind treatment phase).
Efficacy Results
In the active-controlled, combination-therapy trial (JADE DARE), treatment with abrocitinib 200 mg once daily was superior to dupilumab every 2 weeks in demonstrating an improvement of 90% or greater in the Eczema Area and Severity Index total score (EASI-90) and IGA responses in the initial 20 weeks after starting treatment, but there were no statistically significant differences between the 2 drugs at 26 weeks.1
When used as monotherapy and combination therapy, abrocitinib 100 mg once daily and 200 mg once daily resulted in statistically significant increases in the proportion of patients who demonstrated an EASI-75 and IGA response at 12 weeks compared with placebo (i.e., the co-primary end points). The adjusted differences for abrocitinib 100 mg once daily and 200 mg once daily (respectively) compared with placebo for an EASI-75 response in each study were: 27.9% (95% confidence interval [CI], 17.4 to 38.3; P < 0.0001) and 51.0% (95% CI, 40.5 to 61.5; P < 0.0001) for the JADE MONO-1 trial; 33.9% (95% CI, 23.3 to 44.4; P < 0.0001) and 50.5% (95% CI, 40.0 to 60.9; P < 0.0001) for the JADE MONO-2 trial; 31.9% (95% CI, 22.2 to 41.6; P < 0.0001) and 43.2% (95% CI, 33.7 to 52.7; P < 0.0001) for the JADE COMPARE trial; and 26.5% (95% CI, 13.1 to 39.8; P = 0.0002) and 29.4% (95% CI, 16.3 to 42.5; P < 0.0001) for the JADE TEEN trial. Similar results were demonstrated for IGA responses at 12 weeks compared with placebo: 15.8% (95% CI, 6.8 to 24.8; P = 0.0037) and 36.0% (95% CI, 26.2 to 45.7; P < 0.0001) for the JADE MONO-1 trial; 19.3% (95% CI, 9.6 to 29.0; P = 0.0008) and 28.7% (95% CI, 18.6 to 38.8; P < 0.0001 for the JADE MONO-2 trial; 23.1% (95% CI, 14.7 to 31.4; P < 0.0001) and 34.8% (95% CI, 26.1 to 43.5; P < 0.0001) for the JADE COMPARE trial; and 16.7% (95% CI, 3.5 to 29.9; P = 0.0147) and 20.6% (95% CI, 7.3 to 33.9; P = 0.0030) for the JADE TEEN trial. The clinical expert consulted by CADTH noted that the results for EASI-75 and IGA responses compared with placebo are clinically meaningful.
In the subgroup of patients with prior use of a systemic immunosuppressant for AD, the adjusted differences for abrocitinib 100 mg once daily and 200 mg once daily (respectively) compared with placebo for an IGA response were: 9.1% (95% CI, −1.2 to 19.4) and 36.2% (95% CI, 22.7 to 49.7) for the JADE MONO-1 trial; 20.4% (95% CI, 6.7 to 34.1) and 26.9% (95% CI, 12.1 to 41.6) for the JADE MONO-2 trial; 27.5% (95% CI, 14.4 to 40.6) and 43.9% (95% CI, 30.7 to 57.1) for the JADE COMPARE trial; and 18.6% (95% CI, −1.7 to 38.9) and 41.7% (95% CI, 18.0 to 65.3) for the JADE TEEN trial. For EASI-75 response, the adjusted differences for abrocitinib 100 mg once daily and 200 mg once daily (respectively) compared with placebo for IGA response were: 17.0% (95% CI, 2.6 to 31.4) and 49.3% (95% CI, 33.8 to 64.7) for the JADE MONO-1 trial; 30.9% (95% CI, 16.4 to 45.3) and 54.6% (95% CI, 39.4 to 69.7) for the JADE MONO-2; and 49.1% (95% CI, 35.5 to 62.7) and 63.0% (95% CI, 50.3 to 75.7) for the JADE COMPARE trial; and 24.7% (95% CI, −1.7 to 51.1) and 39.0% (95% CI, 12.4 to 65.7) for the JADE TEEN trial.
A statistically significantly greater proportion of patients in both the abrocitinib groups demonstrated an EASI-90 response at 12 weeks in the JADE MONO-1, JADE MONO-2, and JADE TEEN trials, and at 16 weeks in the JADE COMPARE trial. Similarly, a statistically significantly greater proportion of patients in both the abrocitinib groups demonstrated an improvement of 100% in the Eczema Area and Severity Index total score (EASI-100) response at 12 weeks in the JADE MONO-1 and JADE MONO-2 trials, and at 16 weeks in the JADE COMPARE trial. There was no statistically significant difference between the abrocitinib and placebo groups for EASI-100 response in the JADE TEEN trial.
Patient groups and the clinical expert consulted by CADTH identified itch as the most burdensome symptom of AD. In both the monotherapy and combination-therapy trials, both doses of abrocitinib resulted in a greater proportion of patients achieving a improvement of 4 or greater from baseline on the peak pruritus numerical rating scale (PP-NRS4). The adjusted differences for abrocitinib 100 mg once daily and 200 mg once daily (respectively) compared with placebo for a PP-NRS4 response in terms of least squares mean difference [LSMD] in each study were 22.5% (95% CI, 10.3 to 34.8; P = 0.0003) and 41.7% (95% CI, 29.6 to 53.9; P < 0.0001) for the JADE MONO-1 trial; 33.7% (95% CI, 22.8 to 44.7; P < 0.0001) and 43.9 (95% CI, 32.9 to 55.0; P < 0.0001) for the JADE MONO-2 trial; 18.1% (95% CI, 6.2 to 30.0; P = 0.0045) and 32.7% (95% CI, 21.0 to 44.4; P < 0.0001) for the JADE COMPARE trial; and 22.8% (95% CI, 8.0 to 37.7; P = 0.0035) and 25.6% (95% CI, 10.6 to 40.6; P = 0.0013) at 12 weeks for the JADE TEEN trial at 16 weeks. The results were statistically significant for all comparisons with the exception of the JADE TEEN trial (due to failure of the statistical testing hierarchy at a higher-order end point of PP-NRS4 at 4 weeks for the abrocitinib 100 mg group) and were considered to be clinically meaningful by the expert consulted by CADTH. No subgroup analyses were performed for PP-NRS4 in the placebo-controlled trials. In the JADE DARE trial, for the co-primary end point of PP-NRS4 at week 2, abrocitinib 200 mg once daily was superior to dupilumab 300 mg every 2 weeks (48.2% versus 25.5%, for a difference of 22.6% [95% CI, 15.8 to 29.5; P < 0.0001]). The difference between the groups that received abrocitinib 200 mg once daily and dupilumab every 2 weeks decreased over time and was similar between the 2 groups from week 12 onward.
Those living with moderate-to-severe AD can experience sleep disruption due to the symptoms of their condition, particularly persistent itch. Both 100 mg once daily and 200 mg once daily dosages of abrocitinib resulted in statistically significant improvements in FACIT-F compared with placebo in the JADE MONO-1 trial (LSMD = 3.6 [95% CI, 0.9 to 6.4; P = 0.0102] and 4.5 [95% CI, 1.8 to 7.3; P = 0.0013], respectively) and the JADE MONO-2 trial (LSMD = 3.3 [95% CI, 0.8 to 5.9; P = 0.0107] and 4.3 [95% CI, 1.8 to 6.9; P = 0.0010], respectively); there was no statistically significant difference between either abrocitinib group and placebo for the smaller subset of adolescent patients who completed the Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue (Peds-FACIT-F). In the combination-therapy trials, the FACIT-F scale was not evaluated in the JADE COMPARE trial and there was no statistically significant difference between either dose of abrocitinib and placebo in the Peds-FACIT-F in the JADE TEEN study. No subgroup analyses were performed for FACIT-F and Peds-FACIT-F.
Patient groups and the clinical expert consulted by CADTH reported that AD can have a profound negative impact on the mental well-being of those living with the condition, and these patients are at risk of experiencing depression. The monotherapy studies and the combination-therapy study in adults demonstrated that both 100 mg once daily and 200 mg once daily dosages of abrocitinib resulted in statistically significant improvements in Hospital Anxiety and Depression Scale (HADS) anxiety scores and depression scores compared with placebo. There was no statistically significant difference in HADS scores between the abrocitinib and placebo groups in the JADE TEEN trial or the abrocitinib and dupilumab groups in the JADE DARE trial. No subgroup analyses were performed for the HADS.
Patient groups noted the importance of treatments that can improve quality of life for those living with moderate-to-severe AD. The included trials evaluated health-related quality of life using the DLQI and CDLQI instruments for adults and adolescents, respectively. Treatment with both abrocitinib 100 mg once daily and 200 mg once daily (respectively) was associated with a statistically significantly greater improvement (i.e., lower scores) in DLQI scores compared with placebo in the JADE MONO-1 trial (LSMD = −2.8 [95% CI, −4.8 to −0.8; P = 0.0072] and −4.9 [95% CI, −6.9 to −2.9; P < 0.0001] at 12 weeks), the JADE MONO-2 trial (LSMD = −4.4 [95% CI, −6.2 to −2.7; P < 0.0001] and −5.9 [95% CI, −7.7 to −4.2; P < 0.0001] at 12 weeks), and the JADE COMPARE trial (LSMD = −2.8 [95% CI, −3.9 to −1.7; P < 0.0001] and −5.6 [95% CI, −6.7 to −4.5; P < 0.0001] at 16 weeks). Similarly, treatment with both abrocitinib 100 mg once daily and 200 mg once daily was associated with a statistically significantly greater improvement in CDLQI scores compared with placebo in the JADE TEEN trial (LSMD = −2.3 [95% CI, −3.7 to −0.8; P = 0.0026] and −2.3 [95% CI, −3.8 to −0.9; P = 0.0018], respectively). For the adolescent subgroup of patients in the monotherapies, only the 200 mg once daily group demonstrated a statistically significant improvement in CDLQI scores compared with placebo. In the JADE DARE trial, the change from baseline in DLQI scores was greater in the abrocitinib 200 mg group compared with the dupilumab treatment group from week 2 to week 20; however, the difference between the abrocitinib and dupilumab groups decreased over time and was no longer statistically significant at 26 weeks. No subgroup analyses were performed for the DLQI and CDLQI.
As shown in Table 2 and Table 3, treatment with both doses of abrocitinib typically resulted in statistically significant improvements in the additional secondary end points compared with placebo, including PSAAD, Scoring Atopic Dermatitis (SCORAD), POEM, Short Form (36) Health Survey Version 2 (SF-36v2), and Patient Global Assessment (PtGA), although most of these end points were analyzed outside of the statistically testing hierarchy. The JADE DARE trial demonstrated that abrocitinib was superior to dupilumab for improving SCORAD and POEM results in the initial weeks after treatment initiation, but there were no statistically significant differences at week 26. No subgroup analyses were performed for these end points.
Exploratory analyses demonstrated that initiating treatment with the abrocitinib 200 mg once daily regimen was generally more efficacious than the 100 mg once daily regimen for establishing a response to treatment in the 12- to 16-week time frame that was used in the phase III clinical trials. The clinical expert consulted by CADTH noted that specialists are likely to initiate treatment with the higher dosage for most patients and then may consider reducing the dosage based on the patient’s response to therapy and/or tolerability. This approach for reducing the 200 mg dosage is aligned with the dosing recommendations in the product monograph (i.e., for patients using the 200 mg once daily dosage, a reduction of the dosage to 100 mg once daily can be considered after symptom control is achieved).
Harms Results
Abrocitinib 100 mg once daily and 200 mg once daily were generally well tolerated, with few serious adverse events (SAEs) or withdrawals due to adverse events (WDAEs) for up to 16 weeks in the phase III trials and 48 weeks in the interim analysis of the long-term extension-phase study (JADE EXTEND). No subgroup analyses based on prior exposure to at least 1 systemic therapy for AD were performed for adverse events (AEs).
In the monotherapy studies (JADE MONO-1 and JADE MONO-2), the proportions of patients who had at least 1 AE were greater in the abrocitinib 100 mg once daily (69.2% and 62.7%, respectively) and 200 mg once daily (77.9% and 65.8%, respectively) groups compared with the placebo groups (57.1% and 53.8%, respectively). Nausea, headaches, and acne occurred in at least 5% more abrocitinib-treated patients compared with the placebo group. The proportions of patients with at least 1 SAE were similar between abrocitinib groups (3.2% in both) and the placebo group (3.9%) in the JADE MONO-1 trial. In the JADE MONO-2 trial, the proportions with at least 1 SAE were 3.2% in the abrocitinib 100 mg once daily group, 1.3% in the abrocitinib 200 mg once daily group, and 1.3% in the placebo group. In the JADE MONO-1 trial, the proportions of patients who withdrew because of AEs were 9.1% in the placebo group, 5.8% in the abrocitinib 100 mg once daily group, and 5.8% in the abrocitinib 200 mg once daily group. In the JADE MONO-2 trial, the proportions of patients who withdrew because of AEs were 12.8% in the placebo group, 3.8% in the abrocitinib 100 mg once daily group, and 3.2% in the abrocitinib 200 mg once daily group. Withdrawals due to AEs included events categorized as worsening AD, which contributed to the high proportion of WDAEs within the placebo groups of the monotherapy studies. Serious infections and opportunistic infections were rare in the monotherapy studies. Elevated blood creatine phosphokinase (CPK) was reported for numerically more patients in abrocitinib groups compared with placebo. No malignancies, major adverse cardiovascular events (MACEs), or VTE events were reported during the trials.
When used as combination therapy in adults, the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 200 mg group (61.9%) compared to the abrocitinib 100 mg (50.8%), dupilumab 300 mg every 2 weeks (50.0%), and placebo (53.4%) groups in the JADE COMPARE trial. In the JADE DARE trial, the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 200 mg group (74.0%) compared to the dupilumab 300 mg every 2 weeks group (65.5%). Most events were mild or moderate in severity in both the JADE COMPARE and JADE DARE trials. Nausea, headaches, and acne were the most reported AEs in the abrocitinib groups, and conjunctivitis was the most frequently reported in the dupilumab group. The proportions of patients with at least 1 SAE were 3.8% in the placebo group, 2.5% in the abrocitinib 100 mg once daily group, 0.9% in the abrocitinib 200 mg group, and 0.8% in the dupilumab group of the JADE COMPARE trial and 1.7% in the abrocitinib 200 mg group and 1.6% in the dupilumab every 2 weeks group of the JADE DARE trial. The proportions of patients who withdrew because of AEs were 3.8% in the placebo group, 2.5% in the abrocitinib 100 mg once daily group, 4.4% in the abrocitinib 200 mg once daily group, and 3.3% in the dupilumab group of the JADE COMPARE trial and 3.3% in the abrocitinib 200 mg once daily group and 2.5% in the dupilumab group of the JADE DARE trial.
When used as combination therapy in adolescents (in the JADE TEEN trial), the proportion of patients who had at least 1 AE was greater in the abrocitinib 200 mg once daily group (62.8%) compared to the abrocitinib 100 mg once daily (56.8%) and placebo (52.1%) groups. Nausea and acne were more commonly reported with abrocitinib compared with placebo. Two SAEs were reported in the placebo group and 1 SAE was reported in the abrocitinib 200 mg once daily group. The proportions of patients who withdrew because of AEs were 2.1% in the placebo group, 1.1% in the abrocitinib 100 mg once daily group, and 2.1% in the abrocitinib 200 mg once daily group.
Serious infections and opportunistic infections were rare in the combination-therapy studies. Herpes zoster and elevated blood CPK were reported for numerically more patients in the abrocitinib groups compared with the placebo group in both the JADE COMPARE and JADE TEEN trials. No malignancies, MACEs, or VTE events were reported during the trials for patients treated with abrocitinib patients (a malignancy was reported for 1 patient treated with dupilumab in the JADE COMPARE trial).
Table 2: Summary of Key Results From Monotherapy Studies
Results | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
IGA response at week 12 (subgroup with prior exposure to a systemic therapy)a | ||||||
Patients in subgroup analysis | 40 | 78 | 68 | 31 | 67 | 60 |
Responders, n (%) | 2 (5.0) | 11 (14.1) | 28 (41.2) | 2 (6.5) | 18 (26.9) | 20 (33.3) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 9.1 (−1.2 to 19.4) | 36.2 (22.7 to 49.7) | Reference | 20.4 (6.7 to 34.1) | 26.9 (12.1 to 41.6) |
P value | Reference | NR | NR | Reference | NR | NR |
IGA response at week 12 primary end point) (full analysis set)a | ||||||
Responders, n (%) | 6 (7.9) | 37 (23.7) | 67 (43.8) | 7 (9.1) | 44 (28.4) | 59 (38.1) |
Difference in responders (%) (95% CI) Active vs. placebo | Reference | 15.8 (6.8 to 24.8) | 36.0 (26.2 to 45.7) | Reference | 19.3 (9.6 to 29.0) | 28.7 (18.6 to 38.8) |
2-sided P value | Reference | 0.0037 | < 0.0001 | Reference | 0.0008 | < 0.0001 |
EASI-75 response at week 12 (subgroup with prior exposure to a systemic therapy)a | ||||||
Patients in subgroup analysis | 40 | 78 | 68 | 31 | 67 | 59 |
Responders, n (%) | 5 (12.5) | 23 (29.5) | 42 (61.8) | 2 (6.5) | 25 (37.3) | 36 (61.0) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 17.0 (2.6 to 31.4) | 49.3 (33.8 to 64.7) | Reference | 30.9 (16.4 to 45.3) | 54.6 (39.4 to 69.7) |
P value | Reference | NR | NR | Reference | NR | NR |
EASI-75 response at week 12 (primary end point) (full analysis set)a | ||||||
Responders, n (%) | 9 (11.8) | 62 (39.7) | 96 (62.7) | 8 (10.4) | 69 (44.5) | 94 (61.0) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 27.9 (17.4 to 38.3) | 51.0 (40.5 to 61.5) | Reference | 33.9 (23.3 to 44.4) | 50.5 (40.0 to 60.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
PP-NRS4 response at week 12 (key secondary end point) (full analysis set)a | ||||||
Estimated response rate | 15.3 | 37.7 | 57.2 | 11.5 (4.1 to 19.0) | 45.2 (37.1 to 53.3) | 55.3 (47.2 to 63.5) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 22.5 (10.3 to 34.8) | 41.7 (29.6 to 53.9) | Reference | 33.7 (22.8 to 44.7) | 43.9 (32.9 to 55.0) |
2-sided P value | Reference | 0.0003 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
Change from baseline in PSAAD at week 12 (key secondary end point) (full analysis set)b | ||||||
Baseline, mean (SD) | 5.5 (2.0) | 5.3 (2.3) | 5.4 (2.1) | 5.1 (2.1) | 5.4 (2.1) | 5.2 (2.0) |
LSM (95% CI) | −1.1 (−1.7 to −0.6) | −2.2 (−2.6 to −1.9) | −3.2 (−3.6 to −2.8) | −0.8 (−1.3 to −0.3) | −2.4 (−2.8 to −2.1) | −3.0 (−3.3 to −2.7) |
LSMD (95% CI) | Reference | −1.1 (−1.7 to −0.4) | −2.1 (−2.7 to −1.4) | Reference | −1.7 (−2.3 to −1.1) | −2.2 (−2.8 to −1.6) |
2-sided P value | Reference | 0.0010 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
SCORAD-75 response at week 12 (FAS)a | ||||||
Responders, n (%) | 3 (4.1) | 18 (12.4) | 45 (30.8) | 2 (2.6) | 29 (18.7) | 47 (30.3) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 8.2 (1.0 to 15.3) | 26.4 (17.6 to 35.3) | Reference | 16.2 (8.8 to 23.6) | 27.6 (19.3 to 35.8) |
2-sided P value | Reference | 0.0528 | < 0.0001 | Reference | 0.0005 | < 0.0001 |
Change from baseline in BSA at week 12 (%) (full analysis set)b | ||||||
Baseline, mean (SD) | 47.4 (22.7) | 50.8 (23.4) | 49.9 (24.4) | 48.2 (20.8) | 48.7 (21.4) | 47.7 (22.3) |
LSM (95% CI) | −11.4 (−16.0 to −6.8) | −25.1 (−28.3 to −22.0) | −33.4 (−36.6 to −30.3) | −10.0 (−14.8 to −5.1) | −26.9 (−30.2 to −23.6) | −30.6 (−33.8 to −27.3) |
LSMD (95% CI) | Reference | −13.8 (−19.3 to −8.2) | −22.0 (−27.6 to −16.5) | Reference | −16.9 (−22.8 to −11.1) | −20.6 (−26.5 to −14.8) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
Change from baseline in DLQI at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 13.9 (7.3) | 14.6 (6.5) | 14.6 (6.8) | 15.0 (7.1) | 15.4 (7.3) | 14.8 (6.0) |
LSM (95% CI) | −4.2 (−5.9 to −2.5) | −7.0 (−8.1 to −5.8) | −9.1 (−10.3 to −8.0) | −3.9 (−5.3 to −2.4) | −8.3 (−9.3 to −7.3) | −9.8 (−10.7 to −8.8) |
LSMD (95% CI) | Reference | −2.8 (−4.8 to −0.8) | −4.9 (−6.9 to −2.9) | Reference | −4.4 (−6.2 to −2.7) | −5.9 (−7.7 to −4.2) |
2-sided P value | Reference | 0.0072 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
Change from baseline in HADS anxiety component at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 6.0 (4.0) | 5.9 (4.1) | 5.6 (4.0) | 6.0 (3.7) | 5.5 (4.2) | 5.9 (3.9) |
LSM (95% CI) | −1.0 (−1.7 to −0.4) | −1.6 (−2.0 to −1.1) | −2.1 (−2.5 to −1.6) | −0.6 (−1.3 to 0.2) | −1.6 (−2.1 to −1.1) | −1.7 (−2.2 to −1.2) |
LSMD (95% CI) | Reference | −0.5 (−1.3 to 0.2) | −1.0 (−1.8 to −0.3) | Reference | −1.0 (−1.9 to −0.1) | −1.1 (−2.0 to −0.2) |
2-sided P value | Reference | 0.1675 | 0.0085 | Reference | 0.0240 | 0.0138 |
Change from baseline in HADS depression component at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 3.9 (3.5) | 4.1 (3.7) | 4.2 (3.7) | 4.4 (3.3) | 4.1 (4.0) | 4.0 (3.7) |
LSM (95% CI) | −0.2 (−0.8 to 0.4) | −1.4 (−1.8 to −0.9) | −1.8 (−2.2 to −1.4) | 0.3 (−0.3 to 0.9) | −1.0 (−1.5 to −0.6) | −1.4 (−1.8 to −1.0) |
LSMD (95% CI) | Reference | −1.1 (−1.9 to −0.4) | −1.6 (−2.3 to −0.9) | Reference | −1.3 (−2.1 to −0.6) | −1.7 (−2.5 to −0.9) |
2-sided P value | Reference | 0.0028 | < 0.0001 | Reference | 0.0008 | < 0.0001 |
Change from baseline in POEM at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 19.9 (6.1) | 19.5 (6.5) | 19.6 (5.9) | 19.2 (5.5) | 20.9 (5.7) | 19.7 (5.7) |
LSM (95% CI) | −3.7 (−5.5 to −1.9) | −6.8 (−8.0 to −5.6) | −10.6 (−11.8 to −9.4) | 3.6 (−5.3 to −1.9) | −8.7(−9.9 to −7.5) | −11.0 (−12.1 to −9.8) |
LSMD (95% CI) | Reference | −3.1 (−5.2 to −0.9) | −6.9 (−9.0 to −4.7) | Reference | −5.1 (−7.2 to −3.1) | −7.4 (−9.5 to −5.3) |
2-sided P value | Reference | 0.0049 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
PtGA responder at week 12 (FAS)a | ||||||
Responders, n (%) | 5 (6.8) | 32 (21.1) | 54 (36.0) | 3 (3.9) | 25 (16.2) | 45 (29.2) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 14.2 (5.3 to 23.2) | 29.3 (19.6 to 38.9) | Reference | 12.2 (4.5 to 19.9) | 25.2 (16.4 to 33.9) |
2-sided P value | Reference | 0.0075 | < 0.0001 | Reference | 0.0077 | < 0.0001 |
Change from baseline in SF-36 physical component summary at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 45.3 (9.2) | 44.2 (8.5) | 45.2 (8.2) | 46.7 (6.9) | 46.1 (9.3) | 46.6 (7.7) |
LSM (95% CI) | 0.5 (−1.4 to 2.4) | 4.3 (3.0 to 5.6) | 5.2 (3.9 to 6.5) | 1.2 (−0.5 to 2.9) | 4.0 (3.0 to 5.1) | 5.0 (3.9 to 6.0) |
LSMD (95% CI) | Reference | 3.8 (1.5 to 6.1) | 4.7 (2.4 to 7.0) | Reference | 2.9 (0.9 to 4.9) | 3.8 (1.8 to 5.8) |
2-sided P value | Reference | 0.0013 | < 0.0001 | Reference | 0.0052 | 0.0002 |
Change from baseline in SF-36 mental component summary at week 12 (full analysis set)b | ||||||
Baseline, mean (SD) | 50.2 (8.7) | 48.2 (11.1) | 48.8 (11.0) | 47.3 (9.4) | 48.4 (10.5) | 47.1 (10.3) |
LSM (95% CI) | −0.2 (−2.5 to 2.0) | 1.5 (−0.1 to 3.0) | 2.8 (1.3 to 4.3) | 0.4 (−1.9 to 2.7) | 2.2 (0.8 to 3.7) | 3.9 (2.5 to 5.3) |
LSMD (95% CI) | Reference | 1.7 (−1.0 to 4.4) | 3.0 (0.3 to 5.8) | Reference | 1.8 (−0.9 to 4.6) | 3.5 (0.8 to 6.2) |
2-sided P value | Reference | 0.2256 | 0.0275 | Reference | 0.1866 | 0.0113 |
Summary of adverse events, n (%) (safety analysis set) | ||||||
AEs | 44 (57.1) | 108 (69.2) | 120 (77.9) | 42 (53.8) | 99 (62.7) | 102 (65.8) |
SAEs | 3 (3.9) | 5 (3.2) | 5 (3.2) | 1 (1.3) | 5 (3.2) | 2 (1.3) |
Severe adverse events | 9 (11.7) | 8 (5.1) | 5 (3.2) | 5 (6.4) | 7 (4.4) | 6 (3.9) |
Study discontinuation due to AE | 7 (9.1) | 9 (5.8) | 9 (5.8) | 10 (12.8) | 6 (3.8) | 5 (3.2) |
Drug discontinuation due to AE | 1 (1.3) | 2 (1.3) | 0 | 0 | 2 (1.3) | 0 |
Interruption due to AE | 2 (2.6) | 4 (2.6) | 9 (5.8) | 2 (2.6) | 8 (5.1) | 5 (3.2) |
AE = adverse event; BSA = body surface area; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; LSM = least squares mean; LSMD = least squares mean difference; NR = not reported; POEM = Patient-Oriented Eczema Measure; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; SAE = serious adverse event; SCORAD-75 = improvement of 75% or greater in Scoring Atopic Dermatitis; SD = standard deviation; SF-36 = Short Form (36) Health Survey; vs. = versus.
Note: The full analysis set was used for all efficacy end points and the safety analysis set was used for all AE end points.
aThe estimate and CI for difference were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. P values were calculated using the Cochran-Mantel-Haenszel method adjusted by randomization strata (baseline disease severity and age category).
bThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, stratification factors, baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.2,3
Table 3: Summary of Key Results From Placebo-Controlled Combination-Therapy Studies
End point | JADE COMPARE | JADE TEEN | |||||
|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
IGA response at week 12 (subgroup with prior exposure to a systemic therapy)a | |||||||
Patients in subgroup analysis | 47 | 97 | 99 | 112 | 24 | 26 | 22 |
Responders, n (%) | 5 (10.6) | 37 (38.1) | 54 (54.5) | 41 (36.6) | 2 (8.3) | 7 (26.9) | 11 (50.0) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 27.5 (14.4 to 40.6) | 43.9 (30.7 to 57.1) | 26.0 (13.4 to 38.5) | Reference | 18.6 (−1.7 to 38.9) | 41.7 (18.0 to 65.3) |
2-sided P value | Reference | NR | NR | NR | Reference | NR | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | 1.5 (−11.6 to 14.7) | 17.9 (4.7 to 31.2) | Reference | NA | NA | NA |
IGA response at week 12 (primary end point) (full analysis set)b | |||||||
Responders, n (%) | 18 (14.0) | 86 (36.6) | 106 (48.4) | 88 (36.5) | 23 (24.5) | 37 (41.6) | 43 (46.2) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 23.1 (14.7 to 31.4) | 34.8 (26.1 to 43.5) | 22.5 (14.2 to 30.9) | Reference | 16.7 (3.5 to 29.9) | 20.6 (7.3 to 33.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0147 | 0.0030 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | 0.5 (−8.0 to 9.1) | 12.4 (3.5 to 21.3) | Reference | NA | NA | NA |
EASI-75 response at week 12 (subgroup with prior exposure to a systemic therapy)a | |||||||
Patients in analysis | 47 | 97 | 99 | 112 | 24 | 26 | 22 |
Responders, n (%) | 6 (12.8) | 60 (61.9) | 75 (75.8) | 68 (60.7) | 7 (29.2) | 14 (53.8) | 15 (68.2) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 49.1 (35.5 to 62.7) | 63.0 (50.3 to 75.7) | 47.9 (34.8 to 61.1) | Reference | 24.7 (−1.7 to 51.1) | 39.0 (12.4 to 65.7) |
2-sided P value | Reference | NR | NR | NR | Reference | NR | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | 1.1 (−12.1 to 14.4) | 15.0 (2.7 to 27.4) | Reference | NA | NA | NA |
EASI-75 response at week 12 (primary end point) (full analysis set)b | |||||||
Responders, n (%) | 35 (27.1) | 138 (58.7) | 154 (70.3) | 140 (58.1) | 39 (41.5) | 61 (68.5) | 67 (72.0) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 31.9 (22.2 to 41.6) | 43.2 (33.7 to 52.7) | 30.9 (21.2 to 40.6) | Reference | 26.5 (13.1 to 39.8) | 29.4 (16.3 to 42.5) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0002 | < 0.0001 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | 0.8 (−8.1 to 9.6) | 12.0 (3.3 to 20.7) | Reference | NA | NA | NA |
PP-NRS4 response at week 16 (JADE COMPARE) and week 12 (JADE TEEN) (key secondary end point) (full analysis set)b | |||||||
Responders, n (%) | 27 (28.7) | 79 (47.0) | 108 (62.8) | 108 (57.1) | 25 (29.8) | 40 (52.6) | 41 (55.4) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 18.1 (6.2 to 30.0) | 32.7 (21.0 to 44.4) | 28.3 (16.8 to 39.9) | Reference | 22.8 (8.0 to 37.7) | 25.6 (10.6 to 40.6) |
2-sided P value | Reference | 0.0045 | < 0.0001 | NR | Reference | 0.0035 | 0.0013 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | −10.2 (−20.5 to 0.1) | 5.2 (−4.8 to 15.2) | Reference | NA | NA | NA |
SCORAD-75 response at week 16 (JADE COMPARE) and week 12 (JADE TEEN) (full analysis set)b | |||||||
Responders, n (%) | 13 (10.6) | 61 (26.8) | 89 (40.3) | 68 (29.4) | 12 (12.9) | 33 (36.7) | 32 (34.8) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 16.2 (8.4 to 24.1) | 29.6 (21.2 to 37.9) | 18.8 (10.8 to 26.8) | Reference | 23.7 (11.7 to 35.8) | 21.7 (9.7 to 33.7) |
2-sided P value | Reference | 0.0004 | < 0.0001 | NR | Reference | 0.0002 | 0.0006 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumaba | NA | −2.6 (−10.9 to 5.6) | 10.6 (1.9 to 19.3) | Reference | NA | NA | NA |
Change from baseline in BSA at week 16 (JADE COMPARE) and week 12 (JADE TEEN) (%) (full analysis set)c | |||||||
Baseline, mean (SD) | 48.9 (24.9) | 48.1 (23.1) | 50.8 (23.0) | 46.5 (22.1) | 45.8 (22.4) | 51.2 (21.7) | 48.7 (21.7) |
LSM (95% CI) | −19.6 (−22.6 to −16.6) | −32.9 (−35.1 to −30.7) | −39.0 (−41.3 to −36.8) | −34.4 (−36.6 to −32.2) | −24.2 (−27.8 to −20.7) | −34.4 (−38.0 to −30.8) | −35.2 (−38.8 to −31.6) |
LSMD (95% CI) Active vs. placebo | Reference | −13.2 (−17.0 to −9.5) | −19.4 (−23.1 to −15.7) | −14.7 (−18.5 to −11.0) | Reference | −10.2 (−15.2 to −5.1) | −11.0 (−16.0 to −5.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0001 | < 0.0001 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 1.5 (−1.6 to 4.6) | −4.6 (−7.8 to −1.5) | Reference | NA | NA | NA |
Change from baseline in PtGA at week 16 (JADE COMPARE) and week 12 (JADE TEEN) (full analysis set)c | |||||||
Baseline, mean (SD) | NR | NR | NR | NR | |||
LSM (95% CI) | −0.7 (−0.9 to −0.6) | −1.2 (−1.3 to −1.0) | −1.6 (−1.7 to −1.5) | −1.4 (−1.5 to −1.2) | −0.9 (−1.1 to −0.7) | −1.4 (−1.6 to −1.2) | −1.6 (−1.8 to −1.4) |
LSMD (95% CI) Active vs. placebo | Reference | −0.4 (−0.6 to −0.2) | −0.9 (−1.1 to −0.6) | −0.6 (−0.8 to −0.4) | Reference | −0.5 (−0.8 to −0.2) | −0.7 (−0.9 to −0.4) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0008 | < 0.0001 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 0.2 (0.0 to 0.4) | −0.2 (−0.4 to 0.0) | Reference | NA | NA | NA |
Change from baseline in DLQI (JADE COMPARE) or CLQI (JADE TEEN) (full analysis set)c | |||||||
Baseline, mean | 15.2 (6.9) | 15.5 (6.4) | 16.3 (6.6) | 15.6 (6.7) | 14.0 (6.7) | 14.3 (6.1) | 13.6 (7.0) |
LSM (95% CI) | −6.2 (−7.1 to −5.3) | −9.0 (−9.7 to −8.4) | −11.7(−12.4 to −11.1) | −10.8 (−11.4 to −10.1) | −6.3 (−7.4 to −5.3) | −8.6 (−9.6 to −7.5) | −8.7 (−9.7 to −7.6) |
LSMD (95% CI) Active vs. placebo | Reference | −2.8(−3.9 to −1.7) | −5.6 (−6.7 to −4.5) | −4.6 (−5.7 to −3.5) | Reference | −2.3 (−3.7 to −0.8) | −2.3 (−3.8 to −0.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0026 | 0.0018 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 1.7 (0.8 to 2.7) | −1.0 (−1.9 to −0.1) | Reference | NA | NA | NA |
Change from baseline in HADS anxiety component (full analysis set)c | |||||||
Baseline, mean | 5.3 (3.9) | 5.3 (3.9) | 5.5 (3.8) | 5.1 (3.8) | 5.7 (3.7) | 5.7 (4.1) | 5.2 (4.3) |
LSM (95% CI) | −0.4 (−0.9 to 0.1) | −1.2 (−1.6 to −0.8) | −2.0 (−2.4 to −1.6) | −1.5 (−1.9 to −1.1) | −2.1 (−2.7 to −1.5) | −2.0 (−2.6 to −1.4) | −2.4 (−3.0 to −1.8) |
LSMD (95% CI) Active vs. placebo | Reference | −0.8 (−1.5 to −0.1) | −1.6 (−2.2 to −0.9) | −1.1 (−1.7 to −0.4) | Reference | 0.1 (−0.8 to 1.0) | −0.3 (−1.2 to 0.6) |
2-sided P value | Reference | 0.0175 | < 0.0001 | NR | Reference | 0.8603 | 0.4961 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 0.3 (−0.3 to 0.8) | −0.5 (−1.0 to 0.1) | Reference | NA | NA | NA |
Change from baseline in HADS depression component (full analysis set)c | |||||||
Baseline, mean | 4.1 (3.7) | 4.0 (3.3) | 3.9 (3.4) | 3.7 (3.7) | 3.8 (3.4) | 3.7 (3.3) | 3.3 (2.8) |
LSM (95% CI) | −0.3 (−0.8 to 0.2) | −1.0 (−1.4 to −0.7) | −1.6 (−1.9 to −1.2) | −1.2 (−1.5 to −0.8) | −1.0 (−1.5 to −0.5) | −1.4 (−1.9 to −0.8) | −1.2 (−1.7 to −0.6) |
LSMD (95% CI) Active vs. placebo | Reference | −0.7 (−1.3 to −0.1) | −1.3 (−1.9 to −0.7) | −0.9 (−1.5 to −0.3) | Reference | −0.4 (−1.1 to 0.4) | −0.2 (−0.9 to 0.6) |
2-sided P value | Reference | 0.0181 | < 0.0001 | NR | Reference | 0.3364 | 0.6632 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 0.1 (−0.4 to 0.6) | −0.4 (−0.9 to 0.1) | Reference | NA | NA | NA |
Change from baseline in POEM (full analysis set)c | |||||||
Baseline, mean | 20.4 (6.1) | 20.9 (5.5) | 21.5 (5.3) | 21.2 (5.5) | 19.8 (5.9) | 19.5 (6.4) | 19.2 (6.2) |
LSM (95% CI) | −5.0 (−6.3 to −3.8) | −9.2 (−10.1 to −8.2) | −12.5 (−13.4 to −11.6) | −10.8 (−11.8 to −9.9) | −6.9 (−8.3 to −5.6) | −11.1 (−12.5 to −9.7) | −10.9 (−12.2 to −9.5) |
LSMD (95% CI) Active vs. placebo | Reference | −4.1 (−5.7 to −2.6) | −7.5 (−9.0 to −5.9) | −5.8 (−7.4 to −4.2) | Reference | −4.1 (−6.1 to −2.2) | −3.9 (−5.9 to −2.0) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | < 0.0001 | < 0.0001 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 1.7 (0.4 to 3.0) | −1.7 (−3.0 to −0.4) | Reference | NA | NA | NA |
Change from baseline in PSAAD (full analysis set)c | |||||||
Baseline, mean | 5.3 (2.2) | 5.3 (2.1) | 5.6 (2.0) | 5.3 (1.9) | 5.0 (2.4) | 4.9 (2.1) | 4.8 (2.3) |
LSM (95% CI) | −1.7 (−2.0 to −1.3) | −2.8 (−3.1 to −2.6) | −3.6 (−3.8 to −3.4) | −3.4 (−3.6 to −3.2) | −2.0 (−2.4 to −1.6) | −2.5 (−2.9 to −2.1) | −2.7 (−3.1 to −2.3) |
LSMD (95% CI) Active vs. placebo | Reference | −1.2 (−1.6 to −0.8) | −1.9 (−2.3 to −1.5) | −1.7 (−2.1 to −1.3) | Reference | −0.5 (−1.1 to 0.0) | −0.7 (−1.3 to −0.1) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0664 | 0.0142 |
LSMD (95% CI) Abrocitinib vs. dupilumaba | NA | 0.5 (0.2 to 0.9) | −0.2 (−0.6 to 0.1) | Reference | NA | NA | NA |
Summary of adverse events, n (%) (safety analysis set) | |||||||
AE | 70 (53.4) | 121 (50.8) | 140 (61.9) | 121 (50.0) | 50 (52.1) | 54 (56.8) | 59 (62.8) |
SAE | 5 (3.8) | 6 (2.5) | 2 (0.9) | 2 (0.8) | 2 (2.1) | 0 | 1 (1.1) |
Severe adverse events | 3 (2.3) | 5 (2.1) | 4 (1.8) | 2 (0.8) | 2 (2.1) | 0 | 2 (2.1) |
Discontinued study due to AE | 5 (3.8) | 6 (2.5) | 10 (4.4) | 8 (3.3) | 2 (2.1) | 1 (1.1) | 2 (2.1) |
Discontinued drug due to AE | 2 (1.5) | 2 (0.8) | 1 (0.4) | 0 | 0 | 0 | 0 |
Interruption due to AE | 9 (6.9) | 15 (6.3) | 12 (5.3) | 9 (3.7) | 4 (4.2) | 4 (4.2) | 4 (4.3) |
AE = adverse event; BSA = body surface area; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported;; POEM = Patient-Oriented Eczema Measure; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily; SAE = serious adverse event; SCORAD-75 = improvement of 75% or greater in Scoring Atopic Dermatitis; vs. = versus.
Note: The full analysis set was used for all efficacy end points and the safety analysis set was used for all AE end points.
aDifferences between abrocitinib and dupilumab were calculated, but no statistical comparisons were made between the groups.
bThe estimate and CI for difference were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. P values were calculated using the Cochran-Mantel-Haenszel method adjusted by randomization strata (baseline disease severity and age category).
cThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, stratification factors, baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.4,5
Table 4: Summary of Key Results From Active-Controlled Combination-Therapy Study
Analyses | JADE DARE (full analysis set population) | JADE DARE (prior systemic immunosuppressant for AD) | ||
|---|---|---|---|---|
Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) | Abrocitinib 200 mg q.d. (N = 171) | Dupilumab 300 mg q.2.w. (N = 176) | |
EASI-90 response at week 4 (co-primary end point) | ||||
Patients in analysis | 354 | 364 | 171 | 176 |
Responders, n (%) | 101 (28.5) | 53 (14.6) | 45 (26.3) | 24 (13.6) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 14.1 (8.2 to 20.0) | 12.7 (4.4 to 21.0) | ||
2-sided P value | < 0.0001 | NA | ||
EASI-90 response at week 16 (key secondary end point) | ||||
Patients in analysis | 357 | 360 | 171 | 175 |
Responders, n (%) | 194 (54.3) | 151 (41.9) | 96 (56.1) | 73 (41.7) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 12.5 (5.3 to 19.7) | 14.4 (4.0 to 24.9) | ||
2-sided P value | 0.0008 | NA | ||
EASI-90 response at week 26 (secondary end point) | ||||
Patients in analysis | 348 | 361 | NA | |
Responders, n (%) | 190 (54.6) | 172 (47.6) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 6.9 (−0.4 to 14.3) | |||
2-sided P value | 0.0647 | |||
IGA response at week 26 (secondary end point) | ||||
Patients in analysis | 347 | 362 | NA | |
Responders, n (%) | 193 (55.6) | 185 (51.1) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 4.5 (−2.8 to 11.8) | |||
2-sided P value | 0.2293 | |||
PP-NRS4 at week 2 (co-primary end point) | ||||
Patients in analysis | 357 | 364 | 170 | 175 |
Responders, n (%) | 172 (48.2) | 93 (25.5) | 79 (46.5) | 39 (22.3) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 22.6 (15.8 to 29.5) | 24.2 (14.5 to 33.9) | ||
2-sided P value | < 0.0001 | NA | ||
PP-NRS4 at week 26 (secondary) | ||||
Patients in analysis | 354 | 363 | NA | |
Responders, n (%) | 241 (68.1) | 229 (63.1) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 5.0 (−1.9 to 11.9) | |||
2-sided P value | 0.1601 | |||
Change from baseline in BSA (%) at week 26 | ||||
Patients in analysis | 362 | 365 | NA | |
Baseline, mean (SD) | 42.5 (19.9) | 42.6 (21.3) | ||
LSM (95% CI) | −82.3 | −79.0 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | −3.4 (−7.1 to 0.4) | |||
2-sided P value | 0.0793 | |||
Change from baseline in SCORAD at week 26 | ||||
Patients in analysis | 362 | 365 | NA | |
Baseline, mean (SD) | 67.8 (12.8) | 66.8 (12.7) | ||
LSM (95% CI) | −71.5 (−73.9 to −69.1) | −68.2 (−70.6 to −65.9) | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | −3.3 (−6.6 to 0.1) | |||
2-sided P value | 0.0578 | |||
Change from baseline in DLQI at week 26 | ||||
Patients in analysis | 361 | 363 | NA | |
Baseline, mean (SD) | 14.0 (6.8) | 14.2 (6.3) | ||
LSM (95% CI) | −10.3 (−10.8 to −9.9) | −10.0 (−10.5 to −9.6) | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | −0.3 (−1.0 to 0.4) | |||
2-sided P value | 0.3814 | |||
Change from baseline in HADS depression component at week 26 | ||||
Patients in analysis | 362 | 365 | NA | |
BASELINE, mean (SD) | 3.3 (3.2) | 3.3 (3.0) | ||
LSM (95% CI) | −0.8 (−1.0 to −0.5) | −1.0 (−1.3 to −0.8) | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | 0.2 (−0.1 to 0.6) | |||
2-sided P value | 0.2132 | |||
Change from baseline in HADS anxiety component at week 26 | ||||
Patients in analysis | 362 | 365 | NA | |
Baseline, mean (SD) | 5.1 (3.7) | 5.2 (3.6) | ||
LSM (95% CI) | −1.1 (−1.4 to −0.7) | −1.2 (−1.5 to −0.9) | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | 0.1 (−0.3 to 0.6) | |||
2-sided P value | 0.4991 | |||
Change from baseline in POEM at week 26 | ||||
Patients in analysis | 362 | 365 | NA | |
Baseline, mean (SD) | 20.4 (5.8) | 20.9 (5.3) | ||
LSM (95% CI) | −13.8 (−14.5 to −13.1) | −13.4 (−14.0 to −12.7) | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | −0.4 (−1.3 to 0.5) | |||
2-sided P value | 0.3684 | |||
Summary of adverse events, n (%) (safety analysis set) | ||||
AE | 268 (74.0) | 239 (65.5) | 268 (74.0) | 239 (65.5) |
SAE | 6 (1.7) | 6 (1.6) | 6 (1.7) | 6 (1.6) |
Severe adverse events | 12 (3.3) | 9 (2.5) | 11 (3.0) | 8 (2.2) |
Discontinued study due to AE | 0 | 1 (0.3) | 12 (3.3) | 9 (2.5) |
Discontinued drug due to AE | 39 (10.8) | 27 (7.4) | 0 | 1 (0.3) |
Interruption due to AE | 268 (74.0) | 239 (65.5) | 39 (10.8) | 27 (7.4) |
AE = adverse event; CI = confidence interval; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; HADS = Hospital Anxiety and Depression Scale; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.d. = once daily; SAE = serious adverse event; vs. = versus.
Note: The full analysis set was used for all efficacy end points and the safety analysis set was used for all AE end points.
Source: Clinical Study Reports.1
Critical Appraisal
Randomization was stratified based on relevant prognostic factors in the JADE MONO-1, JADE MONO-2, and JADE TEEN trials (i.e., baseline AD severity [moderate or severe] in all 3 studies and age [< 18 years or ≥ 18 years] in the JADE MONO-1 and JADE MONO-2 trials). There was no stratification at the time of randomization in the JADE COMPARE trial, stratification was based only on age (< 18 years or ≥ 18 years) in the JADE REGIMEN trial, and stratification was based on disease baseline AD severity in the JADE DARE trial. The baseline and demographic characteristics were generally well balanced across the treatments of each of the studies. The study treatments were administered in a double-blind manner, and a double-dummy design was used to maintain blinding in the JADE COMPARE and JADE DARE trials to account for the oral administration of abrocitinib and the subcutaneous injection of dupilumab. The AE profile of abrocitinib and the comparators (placebo or dupilumab) was unlikely to compromise blinding in any of the included trials. As the trials were placebo-controlled, it is possible that some patients could have inferred their allocated treatment assignment due to improvement or lack of improvement in AD over the study period and the use of rescue medication, which occurred in a higher proportion of patients in the placebo groups of the included studies. Withdrawals due to AEs included events categorized as worsening AD, which contributed to the high proportion of WDAEs within the placebo groups of the monotherapy studies. Adherence to the study treatments was evaluated by counting the number of study drugs at each visit. Median compliance was 100% across all treatment groups. Few patients discontinued from the combination-therapy trials (completion rates ranged from 89.3% to 96.8% across the treatment groups), but the completion rates were considerably lower in the placebo groups of the monotherapy trials (79.2% and 66.7% in JADE MONO-1 and JADE MONO-2 trials, respectively) compared with the abrocitinib groups (range = 86.5% to 91.0%). True intention-to-treat analyses were not performed; however, the full analysis set (FAS) included nearly all randomized patients, and sensitivity analyses were performed to investigate the impact of missing data. Data were more commonly missing in the placebo arms of the studies, and this may have biased the results in favour of the active treatments as analysis approaches and imputation of missing data assumed the data were missing at random (MAR) (e.g., missing data were imputed as nonresponders); however, numerous sensitivities analysis were performed to investigate the impact of missing data and the results remained robust.
Hierarchies were statistically significant at all end points in the statistical testing in the JADE MONO-1, JADE MONO-2, JADE COMPARE, and JADE DARE trials. The statistical testing hierarchy was stopped at the first key secondary end point of the JADE TEEN trial (i.e., PP-NRS4 response); however, the sponsor continued to calculate and report P values for the remaining key secondary end point (i.e., nominal P values were considered to be descriptive). Subgroup analyses, secondary end points, and exploratory end points were tested without adjustment for multiple comparisons, and all P values are considered nominal. Subgroup analyses for patients with prior exposure to at least 1 systemic therapy for AD were limited to the primary and key secondary end points (e.g., IGA and EASI-75 responses). Imbalances in baseline disease severity were evident across the treatment groups in the subgroup analyses based on prior exposure to at least 1 systemic therapy for AD. The clinical expert consulted by CADTH indicated that, overall, these analyses suggest that the response to abrocitinib would likely be similar for those with and those without prior exposure to a systemic therapy for AD.
The diagnostic criteria used in the screening process for the included studies were consistent with Canadian clinical practice for identifying patients with moderate-to-severe AD. Overall, the clinical expert consulted by CADTH indicated that the populations enrolled in the included trials were a reasonable reflection of the target population in Canada. The clinical expert consulted by CADTH noted that the co-primary end points (EASI and IGA) are clinically relevant and can be evaluated in routine Canadian practice for determining response to treatment with abrocitinib (i.e., for the purposes of establishing renewal criteria for reimbursement by the public drug programs).
As AD is a chronic disease, abrocitinib would likely be used as a long-term treatment for patients who require systemic therapy. The placebo-controlled trials were short-term (12 and 16 weeks) with only limited data available from the longer-term studies (JADE EXTEND and JADE REGIMEN) at the time of this review. Complete reporting of the longer-term studies will help characterize the longer-term efficacy and safety of abrocitinib in the treatment of AD.
Indirect Comparisons
Description of Studies
CADTH summarized and appraised 3 ITCs: 2 unpublished comparisons submitted by the sponsor (1 network meta-analysis [NMA] and 1 matched-adjusted indirect comparison [MAIC]) and a published ITC by the Institute for Clinical and Economic Review (ICER). The NMAs compared abrocitinib against dupilumab (the only drug approved for use in the treatment of AD at the time of this review), upadacitinib and tralokinumab (currently under review by Health Canada and CADTH for use in the treatment of AD), and several drugs that were not listed as under review by Health Canada or CADTH at the time of this review (e.g., nemolizumab, lebrikizumab, and baricitinib). The MAIC compared abrocitinib 100 mg once daily and 200 mg once daily against cyclosporine and methotrexate (2 drugs that are not approved by Health Canada for use as systemic treatments for AD but are commonly used in Canada).
Efficacy Results
Population With Prior Exposure to a Systemic Therapy for AD (Subgroup Analysis From Sponsor’s Network Meta-Analysis
Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy studies. Comparisons could only be conducted for abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, dupilumab 200 or 300 mg every 2 weeks, and placebo. The odds ratios for IGA response were: ||||| |||| |||| |||| || |||||| for abrocitinib 200 mg once daily versus placebo, |||| |||| |||| |||| || |||||) for abrocitinib 200 mg once daily versus dupilumab 200 mg or 300 mg every 2 weeks, and |||| |||| |||| |||| || ||||| for abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily. The odds ratios for EASI-75 response were: ||||| |||| |||| |||| || |||||| for abrocitinib 200 mg once daily versus placebo, ||||| |||| |||| |||| || ||||||) for abrocitinib 200 mg once daily versus dupilumab 200 mg or 300 mg every 2 weeks, |||| |||| |||| |||| || |||||| for abrocitinib 200 mg once daily versus dupilumab 300 mg every 2 weeks, and |||| |||| |||| |||| || ||||| for abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily.
Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to a single composite end point (improvement of 50% or greater in the Eczema Area and Severity Index total score [EASI-50] plus DLQI improvement of ≥ 4 points) in the combination-therapy NMA. Comparisons could only be conducted for abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, dupilumab 300 mg every 2 weeks, and placebo. The odds ratios for achieving an EASI-50 response and a DLQI improvement of 4 or more points were: |||| |||| |||| |||| || |||||| for abrocitinib 200 mg once daily versus placebo, |||| |||| |||| |||| || ||||| for abrocitinib 200 mg once daily versus dupilumab 300 mg every 2 weeks, and |||| |||| |||| |||| || ||||| for abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily.
Overall Population
The sponsor’s NMA reported that ||||||||||| ||| || |||| |||||| |||||||||||| || || ||| || || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| were consistently the most efficacious treatments across the efficacy outcomes evaluated in the NMA. Based on improvements in the EASI, abrocitinib 200 mg once daily was superior to |||||||| ||||||||||| ||| || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| |||| ||||| |||||||||| |||| |||| || |||||||||||. When used in combination with topical therapies, abrocitinib 200 mg once daily was |||||||| || |||||||| ||||||||||| ||| || |||| |||||| ||| |||||||||||| ||| || ||||| | |||||. The results of the NMA conducted by the ICER were generally similar to those reported by the sponsor with respect to the comparative efficacy of abrocitinib 200 mg once daily. The sponsor’s NMA did not compare abrocitinib 100 mg once daily against all of the comparators (only placebo). However, the ICER’s NMA reported that, for most efficacy outcomes, abrocitinib 100 mg was either inferior or occasionally comparable to upadacitinib (30 mg and 15 mg once daily), abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks, while it was superior (or occasionally comparable) to both tralokinumab 300 mg every 2 weeks and placebo.
The sponsor-submitted MAIC reported that abrocitinib at both 100 mg once daily and 200 mg once daily dosages ||| || |||| ||||||||||| |||| |||||||||||| ||| ||||||||||||.
Harms Results
In the NMAs, the TEAEs and discontinuations due to AEs were similar across abrocitinib and the comparators. The sponsor-submitted MAIC reported that abrocitinib at both 100 mg once daily and 200 mg once daily dosages ||| |||| | ||||||| || |||||| |||||| ||||||| |||| |||||||| || |||||||||||| ||| ||||||||||||. No subgroup analyses were conducted for the AE end points.
Critical Appraisal
Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy NMAs and a single composite end point (EASI-50 plus DLQI improvement of ≥ 4 points) in the combination-therapy NMAs. Due to the small number of patients in the LIBERTY AD ADOL trial with prior exposure to at least 1 systemic therapy for AD (n = 11 for the dupilumab 200 mg every 2 weeks or 300 mg every 2 weeks group and n = 9 for the placebo group), there was considerable uncertainty in the estimates of effect for the monotherapy NMA for IGA response. Similar to the primary NMA analyses, abrocitinib 200 mg once daily was |||||||| || ||||||||| ||| || ||||| |||||| ||| ||||||| ||||||||. In the combination-therapy NMA, abrocitinib 200 mg once daily was ||||||| || ||||||||| ||| || ||| ||| ||||||||||||| | |||||||| ||||||| |||||||| ||| || ||||||||||| || || ||||| | |||||| || ||||.
The sponsor-submitted NMA did not report on the relative efficacy and safety of abrocitinib 100 mg when compared with other treatments. Most importantly, no conclusions regarding the long-term efficacy of abrocitinib compared to the active comparators relevant to this review can be drawn as the NMA used study results collected over a relatively short duration compared to the chronic nature of AD. The inherent heterogeneity across trials in the networks also introduces uncertain to interpretation of the results of the trials. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and results from the sponsor-submitted ITC must be interpreted with caution. The conclusion for the MAIC must be weighed against the highly unstable nature of unanchored indirect comparisons which, while being improvements on naive comparisons, are still highly prone to potential biases. Until direct evidence is available, the efficacy and safety differences between abrocitinib and cyclosporine-methotrexate will remain inconclusive.
Other Relevant Evidence
Description of Studies
The JADE EXTEND trial is an ongoing multi-centre, quadruple-masked, randomized phase III study of the long-term efficacy and safety of abrocitinib with or without topical medications in patients aged 12 years and older with moderate-to-severe AD. Patients who complete the JADE MONO-1, JADE MONO-2, JADE COMPARE, JADE TEEN, or JADE REGIMEN studies are eligible for enrolment in the JADE EXTEND trial. Only limited data for patients from the JADE MONO-1 and JADE MONO-2 trials were available at the time of the CADTH review. Patients in the JADE EXTEND trial remained on the same dose of abrocitinib that they received in the parent study, and patients in the placebo groups of the parent study were re-randomized to treatment with abrocitinib 100 mg once daily or 200 mg once daily. The end points reported for the JADE EXTEND trial included IGA, EASI-75, and PP-NRS4 response.
At the data cut-off date of April 22, 2020, for the interim analysis, 520 eligible patients who participated in the JADE MONO-1 and JADE MONO-2 trials were included in the JADE EXTEND trial. Abrocitinib monotherapy was maintained in 361 of 520 patients in the JADE EXTEND trial, while 159 patients received combination therapy of abrocitinib and topical medication. Approximately 25% of patients in both the 100 mg once daily and 200 mg once daily abrocitinib groups had discontinued from the JADE EXTEND trial by week 48.
Efficacy Results
The sponsor reported interim results for 48 weeks of treatment for patients who completed the JADE MONO-1 or JADE MONO-2 trials. The IGA response rate increased from 26.0% to 45.2% in the abrocitinib 100 mg once daily group and from 40.9% to 60.5% in the abrocitinib 200 mg once daily group between week 12 and week 48 of treatment. The EASI-75 response rate increased from 42.1% to 68.0% in the abrocitinib 100 mg once daily group and from 61.9% to 87.2% in the abrocitinib 200 mg once daily group between week 12 and week 48 of treatment. The PP-NRS4 response rate increased from 41.6% to 52.0% in the abrocitinib 100 mg once daily group and from 56.3% to 72.5% in the abrocitinib 200 mg once daily group between week 12 and week 48 of treatment.
The clinical expert consulted by CADTH noted that an important gap in the phase III evidence base is the use of abrocitinib in patients who experienced an inadequate response or whose condition is no longer controlled by treatment with dupilumab. As such, CADTH included the information available for this subgroup of patients from JADE EXTEND. The sponsor reported exploratory analyses to evaluate the efficacy of 12 weeks of abrocitinib treatment in patients who were previously treated with dupilumab for 16 weeks in the JADE COMPARE trial and failed to demonstrate IGA, EASI-75, and PP-NRS4 responses. Further subgroup analyses were conducted for primary nonresponders (defined as patients who did not achieve a response at any visit through week 16 of the JADE COMPARE trial) and secondary nonresponders (defined as patients who had achieved a response at any time before week 16 but were nonresponders at week 16). Responses for the IGA were reported for 34.3% and 47.2% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively. Responses of an EASI-75 were reported for 67.7% and 80.0% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively. Responses of a PP-NRS4 were reported for 37.8% and 81.0% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively.
Harms Results
No harms data were reported for JADE EXTEND at the time of the submission to CADTH.
Critical Appraisal
The JADE EXTEND trial is an ongoing, double-blind extension study that enrolled patients from the phase III RCTs. Only interim data were available at the time of the submission to CADTH, and reporting was limited to an interim analysis with partial reporting (i.e., a clinical study report was not available to enable a thorough appraisal). Extension studies are often limited by selection bias, as only patients who are tolerant to treatment and complete the parent studies are eligible to enrol. At the time of interim analysis, a large proportion of patients had withdrawn from both the abrocitinib 100 mg once daily (22.9%) and abrocitinib 200 mg once daily (20.0%) groups at 48 weeks.6 Issues with the generalizability of these data are the same as for the parent double-blind studies. Patients were considered to be dupilumab nonresponders if they failed to demonstrate an IGA, EASI 75, and PP-NRS4 response after 16 weeks of treatment, which was likely insufficient time to fully realize the maximal treatment effects for dupilumab. The CADTH reimbursement recommendation for dupilumab for patients aged 12 years and older with moderate-to-severe AD advises evaluating the response to treatment after 6 months of treatment.
Conclusions
Four double-blind RCTs demonstrated that, compared with placebo, 12 or 16 weeks of treatment with abrocitinib was associated with statistically significant and clinically meaningful improvements in a range of outcomes that are important to the management of AD, including overall severity of AD (EASI and IGA response), severity of itching (PP-NRS4 response), symptoms (POEM and PSAAD), health-related quality of life (DLQI and CDLQI), fatigue (FACIT-F), and patient-reported anxiety and depression. These trials included the use of abrocitinib as monotherapy (JADE MONO-1 [N = 387] and JADE MONO-2 [N = 391]) and as combination therapy (JADE COMPARE [N = 838 adults] and JADE TEEN [N = 287 adolescents]). One active-controlled trial demonstrated that abrocitinib 200 mg once daily was superior to dupilumab for improving symptoms in the initial weeks after starting treatment, but no significant differences were seen between the 2 drugs at 26 weeks. All of the trials enrolled patients with moderate-to-severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., those with refractory moderate-to-severe AD and an inadequate response to other systemic drugs). The sponsor conducted pre-specified subgroup analyses based on prior exposure to at least 1 systemic immunosuppressant for AD for the co-primary end points of each trial (i.e., EASI-75 and IGA response). The clinical expert consulted by CADTH indicated that the subgroup analyses suggest that the response to abrocitinib would likely be similar for those with and those without prior exposure to a systemic therapy for AD.
All of the included studies suggest that initiating treatment with abrocitinib using the 200 mg once daily regimen was generally more efficacious than the 100 mg once daily regimen for establishing a response to treatment in the 12- to 16-week time frame that was studied in the trials. In addition, the JADE REGIMEN study demonstrated that responders who continue to receive 200 mg once daily as maintenance treatment were less likely to experience a disease flare than those who received 100 mg once daily or placebo.
The product monograph states that there is a risk of serious infections, malignancies, and thrombosis with abrocitinib and other JAK inhibitors. Serious AEs and WDAEs were rare in the included studies. As AD is a chronic disease, abrocitinib would likely be used as a long-term treatment for patients who require systemic therapy. Abrocitinib was well tolerated in the target patient population (i.e., at least 12 years of age with moderate-to-severe AD) in the short term 12- and 16-week phase III studies. No safety data were reported for the interim analysis of the long-term extension study (JADE EXTEND) and only limited data were available from the 52-week JADE REGIMEN trial. Data on AEs in the JADE REGIMEN trial were generally consistent with those observed during the parent studies, but with a numerical increase in the incidence of SAEs per 100 person-years with abrocitinib 200 mg once daily (7.77; 95% CI, 4.25 to 13.04) compared with the abrocitinib 100 mg once daily (2.69; 95% CI, 0.73 to 6.88) and placebo (3.18; 95% CI, 0.39 to 11.49). The ongoing JADE EXTEND study will help better characterize the longer-term efficacy and safety of abrocitinib in the treatment of AD.
Network meta-analyses from the sponsor suggest that ||||||||||| ||| || |||| |||||| |||||||||||| || || ||| || || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| |||| ||||||||| ||||||||||| across the outcomes that were evaluated. Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy studies and a single composite end point (EASI-50 response and a DLQI improvement of ≥ 4 points) in the combination-therapy NMA. There was considerable uncertainty in the estimates of effect for the monotherapy NMA for IGA response; however, similar to the primary NMA analyses, abrocitinib 200 mg once daily ||| |||||||| || ||||||||| ||| || ||||| |||||| ||| ||||||| ||||||||. The NMA from the ICER suggests that abrocitinib 100 mg was either inferior or occasionally comparable to upadacitinib 30 mg and 15 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks. The sponsor-submitted MAIC reported that abrocitinib at dosages of both 100 mg once daily and 200 mg once daily ||| || |||| ||||||||||| |||| |||||||||||| ||| ||||||||||||. No subgroup analyses were reported for the MAIC, and the ICER’s NMA did not report a subgroup analysis based on prior exposure to at least 1 systemic therapy for AD. No conclusions regarding the long-term efficacy of abrocitinib compared to the active comparators relevant to this review can be drawn as the NMA used study results collected over a relatively short duration compared to the chronic nature of AD. The inherent heterogeneity across trials in the networks also introduces uncertainty to interpretation of the results of the trials. The robustness of the comparative efficacy was further compromised by a lack of precision in some of the findings, and results from the indirect comparisons must be interpreted with caution.
Introduction
Disease Background
Atopic dermatitis is the most common type of eczema. It is a chronic, relapsing, inflammatory skin condition characterized by severely itchy skin (pruritus) that results in red and swollen skin (rash). Lesions due to AD may appear as fluid-filled vesicles that ooze, crack, and crust. Pruritus of the skin can cause frequent scratching and may result in lichenification (thickening of the skin) and secondary skin infections. The disease typically involves skin folds in the popliteal (behind the knees) and antecubital (front of the elbows) areas. It may also appear on the face, neck, and hands. Individuals with AD have skin with impaired barrier function and reduced water-holding capacity, resulting in dry skin that requires treatment with specific bathing, cleansing, and moisturizing practices.
Atopic dermatitis is a hereditary form of eczema that generally presents in infancy with most cases beginning before the age of 5 years. The majority of these children will outgrow the condition by adolescence. It is common for children with AD to develop asthma and/or hay fever. This process is referred to as the “atopic march,” and AD is often the first step in the sequential development of these other atopic conditions. The clinical manifestations of AD vary with age, with infants showing AD on the extensor surfaces of extremities, face, neck, scalp, and trunk. Children are typically affected on the flexural surfaces of the extremities, neck, wrists, and ankles, while adolescents and adults are generally affected on the flexural surfaces of the extremities and the hands and feet.
The Canadian Dermatology Association reports that the lifetime prevalence of AD is up to 17% in the Canadian population, and evidence suggests that the prevalence has increased over the past 30 years. Patients often experience worsening itching symptoms throughout the night, and this may result in sleep loss, which may result in detrimental effects pertaining to school or work. Individuals with AD may also suffer from the social stigma of having a highly visible condition. Overall, these patient experiences describe a physically and mentally exhausting condition that can result in anxiety, depression, and a decrease in the quality of life.
The goals of AD management are to prevent flares (episodes of worsening of symptoms typically requiring escalation of treatment), and effectively manage flares when they occur by preventing disease progression. While there is no cure for AD, several therapeutic options are available to patients to manage the condition. The majority of patients treat AD by avoiding skin irritants and using general skin care methods and topical anti-inflammatory therapy. If these common methods fail to improve AD, patients may use off-label systemic therapy (i.e., immunosuppressant therapy) or other approaches, such as phototherapy.
Standards of Therapy
General Skin Care
General skin care practices for patients with AD include irritant avoidance and managing dry skin. The symptoms of AD may be reduced or prevented through the avoidance of known skin irritants or triggers.7-9 Some common irritants include temperature, humidity, dust, pets (animal dander), smoke, and grass. Using mild detergents with no bleach or fabric softener to wash clothing and double-rinsing clothing has been recommended to those with AD. Dry skin associated with AD can be countered through specific bathing, cleansing, and moisturizing practices. Baths using lukewarm water and emulsifying oils followed by the use of moisturizers area recommended. Limiting the use of soap and fragranced products may also help reduce symptoms.7,8
Topical Therapy
While a number of nonpharmacological topical therapies exist for treating the symptoms of AD, the most common therapy is the use of moisturizers. The use of moisturizers is important to combat dry skin through hydration and the prevention of trans-epidermal water loss. Moisturizers, which are routinely used to provide some barrier protection for the skin from irritants or allergens, can soften skin, reduce itching, and minimize cracking, fissuring, and lichenification. Moisturizers are routinely used frequently throughout the day, preferably after bathing. Moisturizers can contain a combination of emollients, humectants, and occlusive drugs.7,9,10 Emollients (e.g., glycol and glyceryl stearate and soy sterols) lubricate and soften the skin by smoothing out the surface of the skin and filling the spaces with droplets. Humectants (e.g., glycerol, lactic acid, and urea) attract water and increase the skin’s water-holding capacity. Humectants sting open skin and are not useful in children with AD. Occlusive drugs (e.g., petrolatum, dimethicone, and mineral oil) provide a layer of oil on the surface of the skin to slow trans-epidermal water loss, prevent water loss though evapouration, and increase the moisture content of the skin. The choice of moisturizer depends on the area of the body and the degree of dryness of the skin.7,9,10
The most common pharmaceutical topical therapies include the use of TCS and TCIs. The former act as anti-inflammatory therapy and are considered to be the first-line treatment for AD.7 The more than 30 different types of TCS come in the form of lotions, creams, oily creams, ointments, or gels, and they can be combined with other drugs such as antibiotics. Topical corticosteroids vary in potency. In Canada, low-potency (1%) hydrocortisone is the most commonly prescribed type of TCS for the face. For the body, triamcinolone or betamethasone valerate (moderate potency) are most commonly prescribed. Topical corticosteroids are applied directly to the area of affected skin before the use of emollients, and a response is typically seen within 10 to 14 days. Side effects associated with the long-term use of TCS include striae (stretch marks), petechiae (small red and/or purple spots), telangiectasia (small, dilated blood vessels on the surface of the skin), skin thinning, atrophy, and acne.7,9-11 Topical corticosteroids are also recommended for use in children according to the American Academy of Dermatology (AAD), with cautions regarding dosing, as children have a larger ratio of surface area to body mass, and there are mixed results from various studies suggesting that systemic absorption may have an impact on growth.7,10
Topical calcineurin inhibitors are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used long-term. In Canada, the 2 available second-line drugs are pimecrolimus and tacrolimus. Pimecrolimus 1% cream can be used for short-term and intermittent long-term therapy for mild-to-moderate AD and is effective in controlling pruritus.10,11 Topical tacrolimus, an ointment that can be used for short-term and intermittent long-term therapy of moderate-to-severe AD, demonstrates rapid and sustained AD symptom control. The most common AE associated with TCIs is application site–specific burning and irritation. A black-box warning remains for TCIs regarding lymphoma; however long-term 10-year surveillance studies have not found an increased risk of lymphoma over that of the general pediatric population.
Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available in Canada, although it is not recommended for reimbursement by CADTH.12 The advantage of TCIs and crisaborole is that both can be safely applied to the face and creases, whereas TCS that are more potent than 1% hydrocortisone are inappropriate. Other topical therapies for AD include treatments with diluted bleach baths, which can help reduce the occurrence of secondary skin infections.7,9
Systemic Therapy
Systemic therapy for the treatment of AD typically involves the use of antimicrobials, antihistamines, or immunomodulators.7,11 Systemic antibiotic treatment can be used to counter widespread secondary bacterial infection. Many patients encounter infection with Staphylococcus aureus, and this may cause new inflammation and exacerbate AD symptoms. The choice of systemic antibiotic drug depends upon the skin culture and sensitivity profile. Sedating antihistamines have been used in cases in which patients are not achieving adequate sleep due to itching.10
Immunomodulatory drugs, including (in order of frequency of use in Canada) methotrexate, cyclosporine, mycophenolate mofetil, azathioprine, can be used in patients who are not responsive to other treatments.7,10,11 However, these commonly used off-label treatments are used at the lowest dose for the shortest possible duration due to side effects.13,14 According to the AAD, cyclosporine is an effective treatment in pediatrics. The AAD noted the evidence for the use of methotrexate in pediatric AD is limited; however, a recent 12-week study showed it had a slower onset than low-dose cyclosporine but an increased time before relapse after discontinuation. Regarding azathioprine, the AAD noted there was evidence of efficacy in children; however, its use should be reserved for recalcitrant AD, or cases in which AD has a significant psychosocial impact.15 The AAD noted that mycophenolate mofetil was a relatively safe systemic therapy in pediatric AD, although its long-term (> 24 months) efficacy and safety in pediatrics have not been studied.11 With respect to corticosteroids, there is a longstanding understanding that chronic use can affect growth in children. The AAD does not recommend corticosteroid use in children with AD except as part of a short-term transition to systemic immunomodulators.
Dupilumab (Dupixent) is an interleukin-4 and interleukin-13 inhibitor indicated for use in adults and pediatrics with moderate to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. CADTH recommended that dupilumab be reimbursed with conditions and it is currently reimbursed by the participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure on or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine.16
Other Therapy
Phototherapy is another second-line therapy that is commonly used after failure of TCS, TCIs, and crisaborole. This therapy includes several sessions and is guided by a number of factors, including patient skin type and skin cancer history. According to AAD guidelines, phototherapy is considered to be a safe and effective treatment for AD in children. There are no studies of the long-term consequences of phototherapy use in pediatric AD patients; however, an increased risk of nonmelanoma skin cancer has been reported in children receiving psoralen and UV A radiation for psoriasis.17
Drug
Abrocitinib is a selective JAK1 inhibitor. All JAKs are intracellular enzymes that transmit signals arising from cytokine or growth factor receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Abrocitinib is indicated for the treatment of patients 12 years of age and older with refractory moderate-to-severe AD, including pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advisable. The product monograph states that abrocitinib can be used with or without medicated topical therapies for AD. The sponsor has requested that abrocitinib be reimbursed in accordance with the indication approved by Health Canada.
Abrocitinib is available as 50 mg, 100 mg, and 200 mg oral tablets. The dosage recommended in the product monograph is 100 mg or 200 mg orally once daily, based on the individual goals of therapy and potential risks of adverse reactions. The product monograph recommends patients using the 200 mg once daily dosage consider reducing the dosage to 100 mg once daily after symptom control is achieved at week 12. Relative to patients who maintained the 200 mg dose, the risk of occurrence of serious adverse reactions decreased in patients who reduced their dose to 100 mg beyond 12 weeks. If symptom control is lost after dose reduction, the dose can be increased to 200 mg.
Recommended dosage adjustments for patients with renal impairment are summarized in Table 5. No adjustment is required in patients with mild renal impairment (i.e., an eGFR of 60 mL/min to < 90 mL/min). In patients with moderate renal impairment (eGFR of 30 mL/min to < 60 mL/min) or severe renal impairment (eGFR < 30 mL/min), the recommended dose of abrocitinib is to be reduced by 50%, as shown in Table 5. Abrocitinib has not been studied in patients with end-stage renal disease on renal replacement therapy. No dosage adjustment is recommended in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. The product monograph states that abrocitinib has not been studied in patients with severe hepatic impairment (Child Pugh C). For patients receiving concomitant treatment with a strong inhibitor of cytochrome P450 2C19 (e.g., fluconazole, fluvoxamine, or fluoxetine), the use of abrocitinib is not recommended concomitantly with strong inducers of CYP enzymes (e.g., rifampin). The product monograph also states that treatment with abrocitinib should not be initiated in patients with a platelet count less than 150 × 103/mm3, an absolute lymphocyte count less than 0.5 × 103/mm3, or an absolute neutrophil count less than 1 × 103/mm3, or in those who have a hemoglobin value less than 8 g/dL.18
Table 5: Dosage Adjustments for Renal Impairment
Renal impairment stage | eGFR | Dose adjustment | |
|---|---|---|---|
Indicated dosage: 100 mg q.d. | Indicated dosage: 200 mg q.d. | ||
Mild | 60 to < 90 mL/min | None | None |
Moderate | 30 to < 60 mL/min | 50 mg q.d. | 100 mg q.d. |
Severe | < 30 mL/min | 50 mg q.d. | 100 mg q.d. |
eGFR = estimated glomerular filtration rate; q.d. = once daily.
Source: Product Monograph.18
Table 6: Key Characteristics of Systemic Therapies for Atopic Dermatitis
Detail | Abrocitinib | Dupilumab | Azathioprine | Mycophenolate mofetil | Cyclosporine | Methotrexate |
|---|---|---|---|---|---|---|
Mechanism of action | JAK1 inhibitor | IL-4 and IL-13 inhibitor | Immune suppressant | Immune suppressant | Immune suppressant | Immune suppressant |
Indication | Patients ≥ 12 years old with refractory moderate-to-severe AD, including pruritus, who have had an inadequate response to other systemic (steroid or biologic) drugs or for whom these treatments are not advisable | Patients ≥ 6 years old with moderate to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
|
|
|
|
Route of administration | Oral | Subcutaneous | Oral |
| Oral |
|
Recommended dosage | ≥ 12 years old: 100 mg or 200 mg q.d.; for patients using 200 mg q.d., after symptom control is achieved at week 12, consider dose reduction to 100 mg q.d.; relative to patients who maintain the 200 mg dose, the risk of serious adverse reactions decreased in patients who reduced their dose to 100 mg beyond 12 weeks; if symptom control is lost after dose reduction, the dose can be increased to 200 mg | ≥ 18 years old: 600 mg, followed by 300 mg q.2.w. 6 to 17 years
|
|
| Psoriasis:
| Varies with indication and clinical use |
Serious adverse effects or safety issues | Product monograph for abrocitinib contains black-box warnings regarding the risk of serious infections, malignancies, and thrombosis |
|
|
|
|
|
AD = atopic dermatitis; b.i.d. = twice a day; IL-13 = interleukin 13; IL-4 = interleukin 4; ITT = intention-to-treat; JAK1 = Janus kinase-1; q.2.w. = every 2 weeks; q.4.w = every 4 weeks; q.d. = once daily.
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
Three patient groups responded to CADTH’s call for patient input: the CSPA, Eczéma Québec, and the ESC.
The CSPA is a national nonprofit organization advocating, educating, and supporting Canadians affected by skin, hair, and nail disorders. Their mission is to promote skin health and improve the quality of life of Canadians living with skin disorders through advocacy, education, and awareness, supporting research, and working with affiliate member organizations that serve specific patient communities such as those with eczema, melanoma, or psoriasis.
Eczéma Québec was created as a branch of the McGill University Hospital Network Centre of Excellence for Atopic Dermatitis. Eczéma Québec is a Patient Advisory Committee and registered nonprofit organization. It established a network of adult AD patients and health care practitioners in the field of AD (encompassing specialist clinician dermatologists, general practitioners, nurse practitioners, and others), with a goal of building resources based on international best-practice guidelines. Eczéma Québec works with the Centre of Excellence to build knowledge translation tools featuring validated information to improve education, experience of care, and promote awareness and the health outcomes of this population.
Eczéma Québec and the CSPA developed and circulated a web-based survey in English and French using the Survey Monkey platform. The survey was distributed through both organizations’ newsletters and other channels. The survey drew 56 respondents. Of the respondents, 91% resided in Québec, 3.6% in Ontario, 3.6% in New Brunswick, and 1.8% in Manitoba. About 3-quarters (43 or 76.8%) of the respondents were patients while 7 (12.5%) were a parent of a patient. Most (80.4%) of the surveyed patients were female (gender at birth) and 19.6% were male. Of these respondents, 11 identified as male, 43 as female, and 2 as non-binary. The age groups (numbers of respondents, percentage of each) were: 18 to 24 (2, 3.6%), 25 to 34 (13, 23.2%), 35 to 44 (15, 26.8%), 45 to 54 (12, 21.4%), 55 to 64 (3, 5.4%), and over 65 years of age (9, 16.1%).
The ESC is a registered Canadian charity dedicated to improving the lives of Canadians living with eczema with a mission of providing support, education, awareness, and research. The ESC gathered survey data from more than 3,000 Canadians who live with AD on topics including quality-of-life impact, experience with systemic treatments, the AD patient journey, and experience with itch related to AD. Respondents included adults living with AD and the caregivers of children living with AD. Information for this submission was also gathered via questionnaires and 1-on-1 interviews. Patients and caregivers who shared their experiences using abrocitinib accessed the drug through a clinical trial.
Disease Experience
Based on patients’ experiences shared by the 3 patient groups for this review, AD negatively affects the individual and their family and can lead to psychological distress. The disease is not only referred to as the most common chronic inflammatory skin disease, but also as 1 of the highest-ranking disorders that causes disability-adjusted life-years in patients worldwide. Patients frequently report that itch is the most burdensome symptom of AD. Some also experience pain or a burning sensation from their AD. The severity of AD correlates with impacts on health-related quality of life as well as lost productivity at school and burdens on health systems. All respondents experienced itching because of their condition. Some patients shared challenges due to the pain they are experiencing. For example, with respect to their clothing: “The symptoms come and go between seasons. My biggest outbreaks are on my right heel, and it makes wearing shoes (even the comfiest shoes I own) very uncomfortable.”
Other reported experiences included: “At my worst, I had to be hospitalized. My AD was seeping, oozing, and infected. I was only sleeping a couple of hours a night. It was a very tough time for me” and “It felt like my whole body was burning, especially on my neck and chest. […] Aside from that, the itchiness is uncontrollable and would wake me up at night.”
Nearly half (48.5%) of the joint survey respondents had symptoms for more than 10 years; 5 (15.2%) had lived with symptoms for 1 to 2 years, 4 (12.1%) for 3 to 5 years, and 4 (12.1%) for 5 to 10 years. These patients and their caregivers also reported on the severity of the condition: 3 (9.1%) reported having a mild form of the disease, 16 (48.5%) from a moderate form, and 14 (42.4%) were living with a severe form of the disease. One patient reported: “All my life, I have struggled with itch. The constant, debilitating itch that would never leave me alone.” Atopic dermatitis also has significant impacts in terms of the psychosocial burden of symptoms, as 1 respondent reflected: “If flaring, [it is] hard to do some things physically and [I’m] self-conscious so tend to stay home.”
Mood, work, school, and social interactions can all be affected by AD. In the ESC survey, 32% of adult respondents with moderate or severe AD had missed work events due to their condition, and 30% had to change careers or give up certain activities. A respondent to the CSPA–Eczéma Québec questionnaire shared their experience of AD by noting: “Work stoppage, Repeated Depression, Lack of Sleep. It’s hard to participate in social or seasonal activities.” Adult respondents of the ESC survey reported feeling itchy multiple times each day (reported by 72% of respondents with moderate AD and by 95% of respondents with severe AD); 71% of adult survey respondents with moderate or severe AD rated their overall itch as 7 out of 10 or greater, and at its worst, 42% of survey respondents rated it as 10 out of 10 — the worst itch imaginable.
From the joint survey, 6 (18.2%) of the respondents noted they would miss 1 to 2 days per month and 2 (6.1%) would miss more than 7 days each month to care for their condition. Some compared the sensation of itch to being bitten by thousands of mosquitoes at once. The respondents noted that the most prevalent areas where they experienced AD were the backs of their hands (63.64%) and their thighs and/or legs (54.55%), neck (51.52%), the inside the arms and/or the elbow folds (51.52%), the outside of the arms and/or the exterior part of the elbows (51.52%), scalp (48.48%), face (45.45%), ears (45.45%), abdomen (45.45%), the area around the eyes (39.39%), breasts, under breasts and/or nipples (39.39%), back (39.39%), backs of the knees (36.36%), the top of the feet (30.30%), the palms of their hands (30.30%), groin area and/or genitalia (24.24%), buttocks (21.21%), front of the knees (21.21%), soles of the feet (21.21%), and armpits (18.18%). Other symptoms included redness of the skin (87.88%), repeated rashes (84.85%), frequent scratching (84.85%), cracked skin (84.85%), dry and rough skin (78.79%), disrupted sleep (75.76%), bleeding (69.70%), flaking of the skin (69.70%), pain (69.70%), thickening of the skin (60.61%), oozing (48.48%), swelling (42.42%), lichenification (39.39%), and blistering (36.36%).
Experience With Currently Available Treatment
For many patients living with AD, frequent moisturizing, trigger avoidance, and the use of topical treatments work well to control their AD flares, but others are left suffering.
Patients affected by AD often have to try multiple treatments to find the right option for their circumstances, and these circumstances can change over time. Respondents considered it important that AD patients have multiple treatment options available for their specific circumstances: “I think that my problem is that I no longer have treatment options left” (translation). Another reported: “I have been a prisoner of my creams and ointments. My family has had to sit by powerlessly as I scratched my skin until [I was] bloody.”
Patients and caregivers can feel extremely frustrated when they follow their doctors’ instructions closely and still continue to experience treatment failure. Parents often feel it is their fault that the treatment is failing. In addition to experiencing debilitating and life-altering symptoms, patients and caregivers alike report often having difficulty accessing timely and appropriate care when they experience flares of their disease: “Accessibility to a competent health professional with regard to eczema is one of the biggest challenges” (translation).
Most patients expressed their dissatisfaction with the treatment options available to them and how these treatments addressed the most important symptom of their disease. One respondent stated: “Nothing works” while another shared that: “Nothing has stopped the itch.” Another source of frustration for these participants was that they did not see these treatments as long-term options but rather “temporary” (translation).
Although most respondents of the joint survey did not have specific experience with targeted treatments for AD (only 8 [14.3%] respondents had experience with dupilumab, 8 [14.3%] had experience with TCIs, and 16 [28.6%] had experience with cyclosporine), few of the other treatment options stood out as very effective or somewhat effective, with the treatment perceived as the most generally effective being TCS (66.7%), followed by the use of a moisturizer, emollient and/or ointments (47.8%), and topical phosphodiesterase type 4 inhibitors (30.0%).
According to the ESC survey on the use of systemic treatments for AD, oral corticosteroids were the most frequently used systemic treatments, but they also rated highest in safety concerns for patients. Patients also reported that the rebound flares experienced after taking oral corticosteroids can be devastating. Phototherapy is also sometimes used; however, some patients reported that it does not adequately control their AD in the long-term. In addition, a lack of access to phototherapy clinics is a significant barrier for many patients depending where they are located in the country. Furthermore, the COVID-19 pandemic significantly affected patients’ access to phototherapy clinics across the country.
Improved Outcomes
Overall, patients expressed the strongest desire for improvements in their ability to manage the itch and reduce flares, inflammation, and/or rashes and improve quality of life and sleep, and they a placed a slightly lower value on the importance of the mode of administration or lichenification.
In the joint survey, patients agreed that new treatments should be able to manage itch (28 of 28 strongly or somewhat agreed), reduce flares (26 of 27), manage redness and inflammation (26 of 27), give rapid results (26 of 28), address lichenification (thickening) of the skin (25 of 27), be easy to use (25 of 28), be covered by insurance or be affordable (23 of 27), allow patients to stop using topical treatments (23 of 28), and not require injections by the patient or someone else (19 of 28). No respondent strongly or somewhat disagreed that these outcomes were important in a new treatment. When referring to the preferred mode of administration: 67.9% (19) preferred daily pills taken by mouth, 50% (14) preferred daily topical medications, and 42.9% (9) preferred injections every other week that they could do themselves or with help. On the subject of topical medications, 1 patient commented: “If you knew how many layers of creams, I had to slather on my body with help.... It was just inhumane” (translation).
Patients were generally unwilling to accept serious side effects from a new treatment. However, they also commented that they are living with the serious effects of their disease, and the negative impacts of their disease would be weighed against the side effects of a treatment option. Across the spectrum of AD severity, patients and caregivers consistently reported carefully weighing the risks and benefits of any medication, ranging from topical medications to systemic medications. For those living with uncontrolled moderate or severe AD, they expressed a willingness to accept some acceptable level of side effects associated with a new treatment and a clinical trial if it meant it would bring them relief from their symptoms.
Patients, particularly those who are adolescents, want to be able to have the confidence to be more outgoing and social, and patients with skin of colour want to avoid the visible changes in skin pigmentation that can result from scratching, flares, and scarring.
Experience With Drug Under Review
The ESC interviewed Canadian patients about their experiences with abrocitinib, which was accessed through the clinical trial. The treatment was reported as being extremely effective at controlling itch, while also reducing flares and subsequently, the cycle of open sores, and skin infections. One patient indicated that, after starting abrocitinib, they no longer had skin infections from open sores that would ooze and then lead to hospitalization and the need for IV antibiotics. It was also reported that, in addition to itch relief and skin improvement, abrocitinib improved stress, sleep, mood, and concentration at work.
Medication delivery was also noted as a benefit of this treatment, compared to experiences patients have reported with complex and often uncomfortable skin care routines and topical treatments.
At the time that the responses from the joint survey responses were received, between March 29 and April 23 of 2021, none of the individuals who took part had direct experience with abrocitinib.
Additional Information
According to the ESC, patients who live with moderate-to-severe uncontrolled AD may never experience periods of clear skin despite adherent use of their treatments, such as topical medications. For this patient population, there is a significant gap in treatments, and more options are desperately needed. The burden of disease, quality-of-life impact, and suffering associated with moderate or severe AD can be debilitating, as the itch, discomfort, and pain can disrupt sleep, affect mental health, and lead to absenteeism from work and/or school. Abrocitinib offers hope to patients with uncontrolled moderate or severe AD by providing rapid improvement of itch and skin lesions, which in turn improves their quality of life.
According to the CSPA and Eczéma Québec, skin disorders are often diminished, disregarded, and dismissed. They are more than “just a rash.” The development of better-tailored treatment options for skin disorders on the horizon provides new hope that treatments will address the underlying pathology of skin disorders, rather than only treating the symptoms. Those living with skin disorders deserve to be treated with respect and dignity by the health system, which includes its embrace of new and tailored treatment options.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by a clinical specialist with expertise in the diagnosis and management of AD.
Unmet Needs
Presently, patients achieving suboptimal disease control with appropriate disease-specific skin care measures (irritant avoidance, emollients, and bleach baths), TCS and/or TCIs, crisaborole, and phototherapy are offered treatments with off-label immunosuppressive drugs. In Canada, the most commonly chosen immunosuppressive drug is methotrexate, followed by cyclosporine, azathioprine, and mycophenolate mofetil. Because of their potential toxicities, these drugs are generally prescribed as intermittent courses in the treatment of AD. There are patients for whom some or all of these drugs are contraindicated or for whom toxicities limit their use. There are also patients who do not respond to these drugs. Dupilumab is offered as second-line systemic therapy to the immunosuppressives, but reimbursement for dupilumab in Canada remains problematic.
Place in Therapy
Abrocitinib, a small-molecule reversible JAK1 inhibitor, is a potentially useful addition to the currently available therapeutic options for AD. It will, in the specialist’s opinion, be a useful drug in patients who have contraindications to, experience adverse effects from, or who are unresponsive to the off-label immunosuppressive drugs. It will also be useful in that subset of patients who respond to off-label immunosuppressive drugs but who require continuous long-term therapy to control their disease.
Abrocitinib will also potentially be of value in patients with AD who have been treated with dupilumab and had a suboptimal response, who develop severe conjunctivitis or other ocular side effects from dupilumab or are intolerant of injections and prefer an oral drug, and/or those who have severe injection-site reactions to dupilumab.
All patients with AD treated with abrocitinib would be expected to continue on with emollients, TCS, TCIs, and/or crisaborole. It is expected that abrocitinib would never be combined with off-label immunosuppressants or dupilumab (or the new biologics such as tralokinumab that are emerging treatments for AD).
Abrocitinib is unlikely to cause a significant shift in the current treatment paradigm for AD beyond its inclusion as another effective drug. It would be appropriate to recommend trials of methotrexate and cyclosporine before initiating treatment with abrocitinib. These older drugs are cost-effective and efficacious, and dermatologists are well versed in appropriate dosing, duration of therapy, and monitoring of patients for potential toxicities. Many patients can be managed with intermittent use of immunosuppressants. The clinical expert consulted by CADTH noted that immunosuppressants have likely been underutilized in clinical practice, partly due to the paucity of literature. As these are older drugs with low commercial value, adequate clinical trials are rare. This leads to a “low evidence” designation in reviews and treatment guidelines.
Patient Population
Although AD is not a diagnostic challenge for a dermatologist, the differential diagnosis includes psoriasis, ichthyoses, allergic contact dermatitis, irritant contact dermatitis, and cutaneous T-cell lymphoma. The barrier dysfunction of AD predisposes patients to superimposed allergic contact dermatitis and also dermatophytosis, and therefore patch tests and skin scrapings for potassium, oxygen, and hydrogen, and fungal culture may be of benefit in selected cases. A biopsy would usually be reserved for patients recalcitrant to all therapy for whom cutaneous T-cell lymphoma is a consideration, or occasionally, to differentiate AD from psoriasis. Abrocitinib would never be considered for pre-symptomatic patients.
All patients with moderate-to-severe AD could respond to treatment with abrocitinib. It is unclear whether abrocitinib can effectively treat those patients who have failed methotrexate, cyclosporine, and/or dupilumab. It is not currently possible to identify those patients who are most likely to exhibit a response to abrocitinib.
The patients with AD who could be considered least suited for treatment with abrocitinib include:
those who are well controlled with topical therapy, phototherapy, and/or intermittent off-label immunosuppressive therapy
those who are well controlled with dupilumab
those with potential contraindications to JAK inhibitors such as: severe active infections acute or chronic including latent tuberculosis, deep fungal infections and opportunistic infections; potentially malignancy, including ongoing treatment with chemotherapy such as checkpoint inhibitors; severe hepatic disease; severe renal disease; pregnancy and lactation; a history of thromboembolic events, and pre-existing hematologic disease, including lymphopenia and neutropenia.
Assessing Response to Treatment
In general, the outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials. The clinical expert consulted by CADTH anticipated that the EASI score will be chosen as the benchmark for reimbursement. As such, this will be calculated and recorded at each patient visit. Many clinicians will also record a DLQI score, although this value may not be required for reimbursement. Reduction in pruritus will also be noted but not formally scored using a scale. The patient’s impression of their overall improvement will also be recorded.
The benchmark response will be an EASI-75 at 16 weeks. However, EASI score reductions of 50% to 75% would be anticipated to be clinically meaningful, particularly to those who have had severe disease recalcitrant to all previous therapies. It is anticipated that patients who initiate treatment with abrocitinib would be re-evaluated after 16 weeks. Those who are judged to be responders at this visit would be seen subsequently once every 6 months. Those who have not reached response targets at 16 weeks would be re-evaluated at 20 weeks following initiation of drug. A decision about whether to stop or continue would be made at the 20-week visit. Bloodwork, including complete blood count and differential liver-function tests, creatinine, lipids, and CPK, would be analyzed monthly before the first follow-up visit and, if there are no concerns, every 3 months thereafter.
Discontinuing Treatment
The following factors would be considered when deciding to discontinue treatment with abrocitinib:
failure to achieve clinically meaningful response at 16 to 20 weeks
failure to maintain an adequate response on long-term maintenance
development of a hypersensitivity response judged to be due to abrocitinib
TEAEs, such as lymphopenia, neutropenia, arterial thrombosis, or VTE
treatment-emergent severe infection
treatment-emergent malignancy.
Prescribing Conditions
A specialist would be required to diagnose, treat, and monitor patients taking abrocitinib. Appropriate specialists would include a pediatric dermatologist, a general dermatologist, or a pediatrician with an interest in AD.
Clinician Group Input
No input was received from clinician groups.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 7.
Table 7: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Access to phototherapy seems to be limited across Canada. Is this factual or perceived among clinicians and dermatologists? | Phototherapy is mostly accessible in urban areas but not in rural areas. It is important to consider this barrier in the decision-making process. |
Would abrocitinib be initiated in patients who have failed previous treatment with a biologic drug? | Patients who have failed dupilumab plus 1 of the immunomodulators would be candidates to receive abrocitinib. This also would apply in those who have failed dupilumab alone, although there is high uncertainty due to lack of evidence for this clinical recommendation. |
Should it be required that patients had an adequate trial of (or be ineligible for) cyclosporine, methotrexate, and phototherapy before initiating abrocitinib? | A trial of 2 of the 4 immunomodulators (methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine) should be considered before initiating abrocitinib. |
The initiation criteria that were recommended by CDEC for dupilumab are:
| The initiation criteria for dupilumab are feasible to implement in clinical practice and could be applied to abrocitinib. It would also be practical to consider earlier than 6 months for the duration of the initial authorization (i.e., 16 to 20 weeks instead of 24 weeks) and proceed to assess the continuation/renewal of the indication. |
Will dupilumab (or other biologics approved for AD) be among the prior therapies required in the eligibility criteria for initiation of therapy with abrocitinib? Will prior therapies required for eligibility include dupilumab (or biologics approved for AD)? | The use of dupilumab as a prior therapy before initiating treatment with abrocitinib should not be an initiation criterion. Both drugs would have the same place of therapy in the population for this indication. |
CDEC renewal criteria for dupilumab are as follows:
| The renewal criteria are feasible to apply to abrocitinib, although the timing of 6 months (24 weeks) could be considered to earlier dates (e.g., 16 to 20 weeks). |
The included trials had a duration of 12 to 16 weeks, with the longest follow-up in the studies assessing up to 48 weeks. Based on the available evidence, have the long-term safety data been established with certainty? | The currently available evidence is not sufficient to establish the long-term safety profile of abrocitinib in the treatment of AD. |
The CDEC recommendation for dupilumab included the following 3 implementation considerations:
Should these 3 implementation considerations be also considered for abrocitinib? | These implementation considerations are relevant for the reimbursement of abrocitinib and should be noted in the recommendation. |
Can abrocitinib be used in combination with other JAK inhibitors, biologic DMARDs, phototherapy or immunosuppressants? | Abrocitinib should not be used in combination with other systemic treatments for AD (there is no evidence regarding the safety and efficacy of such combinations). |
Should abrocitinib be prescribed in consultation with a dermatologist and/or specialist? | A specialist would be required to diagnose, treat, and monitor patients taking abrocitinib. Appropriate specialists would include a pediatric dermatologist, a general dermatologist, or a pediatrician with an interest in AD. |
How would an “adequate trial” be defined in clinical practice for patients with AD who undergo therapy with phototherapy (where available), methotrexate, and cyclosporine? |
|
How would “ineligible” be defined in clinical practice for patients with AD who are ineligible to receive therapy with methotrexate or cyclosporine? | Risk factors or potential adverse reactions from the interventions would make patients ineligible. |
AD = atopic dermatitis; CDEC = CADTH Canadian Drug Expert Committee; DMARD = disease-modifying antirheumatic drug; EASI = Eczema Area and Severity Index; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; JAK = Janus kinase.
Clinical Evidence
The clinical evidence included in the review of abrocitinib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of oral abrocitinib 100 mg once daily and 200 mg once daily for the treatment of patients 12 years and older with moderate-to-severe AD, including pruritus, who have had an inadequate response to prescribed topical therapy or for whom these treatments are not advisable.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria are presented in Table 8. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 8: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients 12 years of age and older with moderate-to-severe AD, including pruritus, who have had an inadequate response to prescribed topical therapy or for whom these treatments are not advisable Subgroups:
|
Interventions | Abrocitinib 100 mg q.d. or 200 mg q.d. as monotherapy for AD Abrocitinib 100 mg q.d. or 200 mg q.d. in combination with topical therapies for AD |
Comparator | When used alone or in combination with topical therapy:
|
Outcomes | Efficacy outcomes
Harms outcomes
|
Study designs | Published and unpublished phase III and 4 RCTs |
AD = atopic dermatitis; AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; CPK = creatine phosphokinase; DFI = Dermatitis Family Impact; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; MACE = major adverse cardiovascular event; q.d. = once daily; RCT = randomized controlled trial; SAE = serious adverse events; SCORAD = Scoring Atopic Dermatitis; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.23
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Cibinqo (abrocitinib). Clinical trials registries searched included the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on May 25, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 15, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.24 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
A focused literature search for NMAs dealing with Cibinqo (abrocitinib) and AD was run-in MEDLINE All (1946–) on May 18, 2021. No limits were applied.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
Five studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 9. None of the potentially relevant studies were excluded.
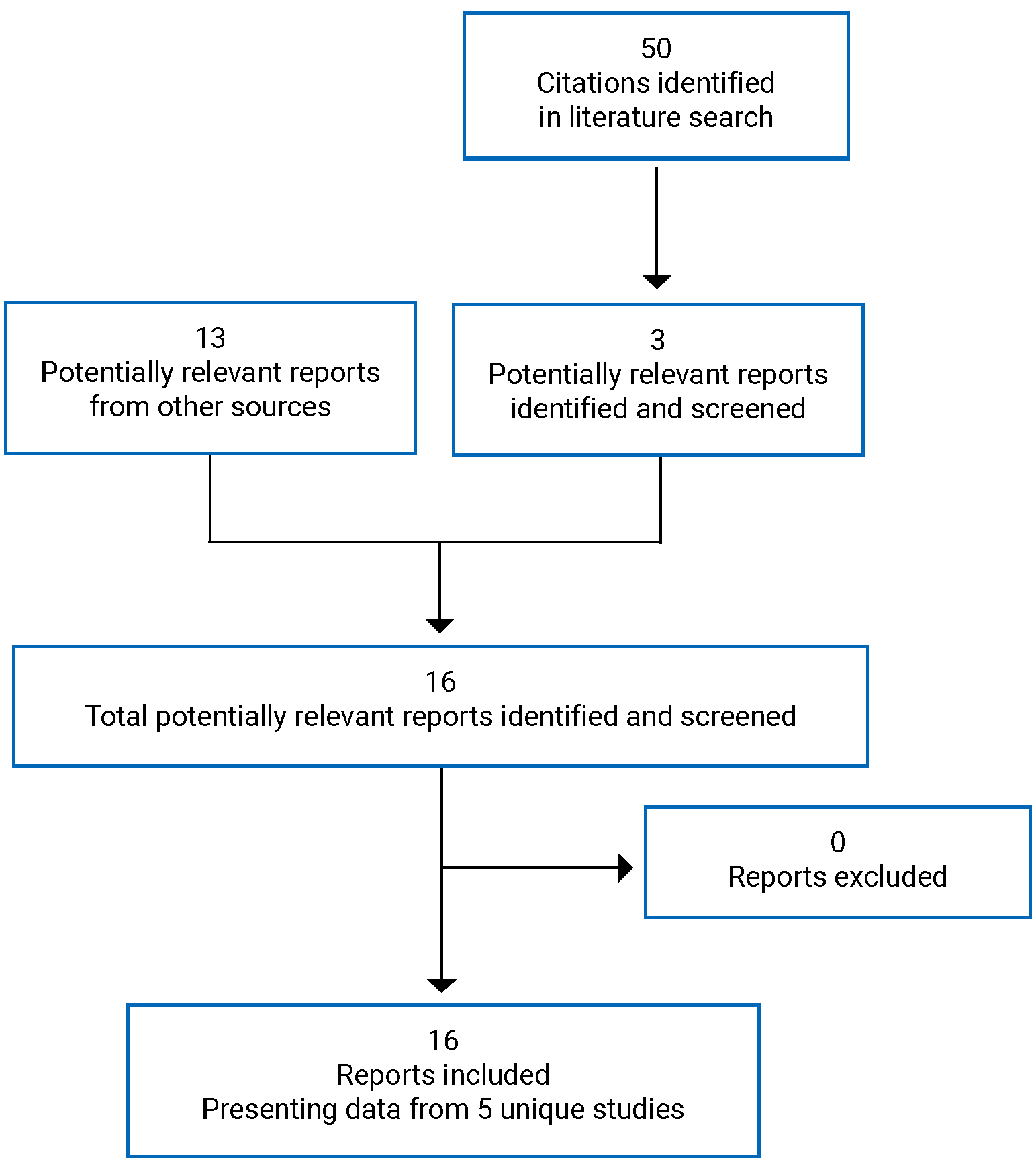
Table 9: Details of Included Monotherapy Studies
Detail | JADE MONO-1 | JADE MONO-2 |
|---|---|---|
Designs and populations | ||
Study design | Phase III, double-blind, placebo-controlled, parallel-group, RCT | |
Locations | 69 sites in 8 countries: the US, Canada, Germany, Australia, Poland, Czech Republic, the UK, and Hungary | 102 sites in 13 countries: the US, Poland, Republic of Korea, Japan, Australia, Bulgaria, Canada, Germany, the UK, China, Latvia, Hungary, and Czech Republic |
Patient enrolment dates | First visit: December 7, 2017 Last visit: March 26, 2019 | First visit: June 29, 2018 Last visit: August 13, 2019 |
Randomized (N) | 387
| 391
|
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention |
|
|
Comparator(s) |
|
|
Duration | ||
Phase | ||
Screening | 28 days | 28 days |
Double-blind | 12 weeks | 12 weeks |
Follow-up | 4 weeks or open-label extension | 4 weeks or open-label extension |
Outcomes | ||
Primary end point |
| |
Secondary and exploratory end points |
|
|
Notes | ||
Publications | ||
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CFB = change from baseline; DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; JAK = Janus kinase; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PP-NRS = severity of pruritus numerical rating scale; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = an improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = an improvement of 75% or greater in Scoring Atopic Dermatitis; SF-36v2 = Short Form (36) Health Survey Version 2; WPAI-AD = Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis.
Source: Clinical Study Reports.2,3
Table 10: Details of Included Combination-Therapy Studies
Detail | JADE COMPARE | JADE TEEN | JADE DARE |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind, double-dummy, placebo-controlled, parallel group RCT | Phase III, double-blind, placebo-controlled, parallel group RCT | Phase IIIb, double-blind, double-dummy, active-controlled, parallel group RCT |
Locations | 194 sites in 18 countries: the US, Poland, Republic of Korea, Japan, Australia, Bulgaria, Canada, Germany, the UK, Latvia, Hungary, Czech Republic, Chile, Spain, Italy, Mexico, Slovakia, and Taiwan | 99 sites in 13 countries: Australia, China, Czech Republic, Germany, Hungary, Italy, Japan, Latvia, Mexico, Poland, Spain, Taiwan, and the US | 151 sites in Australia, Bulgaria, Canada, Chile, Finland, Germany, Hungary, Italy, Latvia, Poland, Republic of Korea, Slovakia, Spain, Taiwan, and the US |
Patient enrolment dates | First visit: October 29, 2018 Last visit: March 6, 2020 | First visit: February 18. 2019 Last visit: April 8, 2020 | First visit: June 11, 2020 Last visit: July 13, 2021 |
Randomized (N) | 838
| 287
| 727
|
Inclusion criteria |
|
|
|
Exclusion criteria |
| ||
Drugs | |||
Intervention |
|
|
|
Comparator(s) |
|
|
|
Duration | |||
Phase | |||
Screening | 28 days | 28 days | 28 days |
Double-blind | 20 weeks | 12 weeks | 26 weeks |
Follow-up | 4 weeks or entry into open-label extension | 4 weeks or entry into open-label extension | 4 weeks or entry into open-label extension |
Outcomes | |||
Primary end point |
|
|
|
Secondary and exploratory end points |
|
|
|
Notes | |||
Publications |
| ||
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CFB = change from baseline; DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; JAK = Janus kinase; MOS = Medical Outcomes Study; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; PP-NRS = severity of pruritus numerical rating scale; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = an improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = an improvement of 75% or greater in Scoring Atopic Dermatitis.
Source: Clinical Study Reports.4,5
Table 11: Details of Included Withdrawal Study
Detail | JADE REGIMEN |
|---|---|
Designs and populations | |
Study design | Phase III multi-centre, randomized, responder-enriched, double-blind, placebo-controlled withdrawal study |
Locations | 21 countries/regions (the US; Argentina; Belgium; Brazil; Bulgaria; Canada; Chile; China; Germany; Israel; Italy; Latvia; Mexico; Netherlands; Poland; Romania; Russia; Serbia; Slovakia; Spain; Taiwan) |
Patient enrolment dates | Study start: June 11, 2018 Study end: October 7, 2020 |
Randomized (N) | 1,235 patients enrolled in open-label induction Period
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Open-label induction phase | Abrocitinib 200 mg q.d. |
Double-blind phase interventions |
|
Double-blind phase comparator | Placebo |
Open-label rescue therapy | Abrocitinib 200 mg q.d. plus topical therapy |
Duration | |
Phase | |
Screening | 28 days |
Run-in | 12 weeks open-label run-in with abrocitinib 200 mg q.d. |
Double-blind |
|
Follow-up | 4 weeks or entry into open-label extension |
Outcomes | |
Primary end point | A flare requiring rescue treatment: defined as a loss of at least 50% of the EASI response at week 12 and an IGA score of 2 or higher |
Secondary and exploratory end points |
|
Notes | |
Publications | |
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CFB = change from baseline; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; JAK = Janus kinase; NRS = numeric rating scale; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = an improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = an improvement of 75% or greater in Scoring Atopic Dermatitis.
Source: Clinicaltrials.gov,33 Gubelin et al. (2021),34 Sponsor’s clinical summary.35
Description of Studies
Table 12 provides an overview of the studies that were summarized and appraised by CADTH for the current review of abrocitinib. Five double-blind, phase III RCTs were included in the CADTH systematic review: 2 placebo-controlled trials conducted with abrocitinib as monotherapy for AD (JADE MONO-1 and JADE MONO-2)2,3; 2 placebo-controlled trials conducted with abrocitinib as combination therapy for AD (JADE COMPARE and JADE TEEN)4,5; and 1 placebo-controlled withdrawal trial (JADE REGIMEN).34,35 CADTH also reviewed additional studies that did not meet the eligibility criteria of the systematic review but may address important gaps in the evidence from the pivotal and supportive RCTs. These included the interim analysis from 1 long-term extension-phase study (JADE EXTEND)35,36 and 3 indirect comparisons (2 filed by the sponsor and 1 from the ICER).37-39
Table 12: Summary of Evidence Included in Review
Regimen | Study name | Design | Duration | Status |
|---|---|---|---|---|
Studies included in systematic review | ||||
Monotherapy | JADE MONO-1 | Phase III, double-blind, placebo-controlled, RCT | 12 weeks | Complete |
JADE MONO-2 | Phase III, double-blind, placebo-controlled, RCT | 12 weeks | Complete | |
Combination therapy | JADE COMPARE | Phase III, double-blind, double-dummy, RCT | 16 weeks | Complete |
JADE TEEN | Phase III, double-blind, placebo-controlled, RCT | 12 weeks | Complete | |
JADE DARE | Phase IIIb, double-blind, active-controlled, RCT | 26 weeks | Complete | |
Withdrawal | JADE REGIMEN | Phase III responder-enriched, double-blind, placebo-controlled withdrawal study | 52 weeks | Complete |
Long-term extension studies | ||||
Monotherapy combination therapy | JADE EXTEND | Open-label extension-phase study | Up to 5 years | Ongoing |
Indirect comparisons | ||||
Monotherapy Combination therapy | Sponsor NMA | Bayesian network meta-analysis | Variable | Final |
Sponsor MAIC | Matching-adjusted indirect comparison | Variable | Final | |
ICER NMA | Bayesian network meta-analysis | Variable | Final | |
ICER = Institute for Clinical and Economic Review; MAIC = matching-adjusted indirect comparison; NMA = network meta-analysis; RCT = randomized controlled trial.
Monotherapy Studies
The JADE MONO-1 and JADE MONO-2 trials were phase III, double-blind, placebo-controlled, parallel-group RCTs conducted to evaluate the efficacy and safety of abrocitinib monotherapy in patients aged 12 years and older with moderate-to-severe AD and a body weight of 40 kg or more. The studies consisted of a 28-day screening period, a 12-week double-blind treatment phase, and a 4-week safety follow-up period (or entry into the long-term extension study [EXTEND]).2,3 During the screening period, treatments for AD will be washed out, as applicable, according to eligibility requirements. Eligible patients were randomized in a 2:2:1 ratio to receive 200 mg of abrocitinib once daily, 100 mg of abrocitinib once daily, or matching placebo. Randomization was stratified by baseline disease severity (moderate [IGA = 3] or severe [IGA = 4] AD), and age (< 18 years or ≥ 18 years).2,3 The 2 trials were identically designed, except for an additional exclusion criterion in JADE MONO-2 (i.e., patients who had topical treatments for AD within 72 hours of the first dose of study medication) and the inclusion of the Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis (WPAI-AD) as an additional end point.35
The JADE MONO-1 trial was conducted at 69 sites in 8 countries: the US (n = 114), Canada (n = 64), Germany (n = 64), Australia (n = 51), Poland (n = 49), the Czech Republic (n = 19), the UK (n = 14), and Hungary (n = 12).2 The JADE MONO-2 trial was conducted at 102 sites in 13 countries: the US (n = 19), Poland (n = 14), Republic of Korea (n = 10), Japan (n = 8), Australia (n = 7), Bulgaria (n = 7), Canada (n = 7), Germany (n = 7), the UK (n = 6), China (n = 5), Latvia (n = 5), Hungary, (n = 4), and the Czech Republic (n = 3).3
Figure 2: Schematic of JADE MONO-1 and JADE MONO-2 Design
PF-04965842 = abrocitinib.
Source: Clinical Study Report.2
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
JADE COMPARE was a phase III, double-blind, double-dummy, placebo-controlled, parallel group, multi-centre study investigating the efficacy and safety of abrocitinib and dupilumab in comparison with placebo in adult subjects on background topical therapy, with moderate-to-severe AD. The study consisted of a 28-day screening period, a 20-week double-blind treatment phase, and a 4-week safety follow-up period (or entry into the long-term extension study [EXTEND]). During the screening period, treatments for AD will be washed out, as applicable, according to eligibility requirements. Eligible patients were randomized in a 4:4:4:1:1 ratio to receive 100 mg or 200 mg of abrocitinib once daily with dupilumab-matching placebo every 2 weeks, dupilumab 300 mg every 2 weeks (with a loading dose of 600 mg at baseline) with abrocitinib-matching placebo once daily, or 1 of 2 sequences of abrocitinib-matching placebo administered once daily with dupilumab-matching placebo administered every 2 weeks from day 1 for 16 weeks followed by either 100 mg or 200 mg of abrocitinib once daily. The 20-week double-blind treatment phase consisted of 2 parts: 1) a 16-week randomized, double-blind, placebo-controlled, double-dummy treatment period with patients receiving both injectable and oral investigational product; and 2) a 4-week phase during which patients only received the oral investigational product. At week 16, all patients were to cease administering injectable dupilumab or the matching placebo. This 4-week phase was included in the study to facilitate the washout of dupilumab before eligible patients entering the long-term extension study. Following week 16, patients who received placebo during part 1 of the study were to cross over to receive abrocitinib at a 100 mg once daily or 200 mg once daily (in accordance with how they were randomized) and those who received abrocitinib in part 1 continued on their randomized treatment. Those who received dupilumab in part 1 continued to take the oral placebo to maintain blinding. Randomization was not stratified by any baseline characteristics.4
The JADE COMPARE trial was conducted at 194 sites in 18 countries, including sites in the US (n = 46), Poland (n = 36), Republic of Korea (n = 7), Japan (n = 12), Australia (n = 10), Bulgaria (n = 5), Canada (n = 11), Germany (n = 13), the US (n = 11), Latvia (n = 5), Hungary (n = 5), the Czech Republic (n = 7), Chile (n = 4), Spain (n = 5), Italy (n = 2), Mexico (n = 4), Slovakia (n = 5), and Taiwan (n = 6).4
Figure 3: Schematic of JADE COMPARE Design
PF-04965842 = abrocitinib.
aAt week 2 and week 16, key secondary end points are measured.
bAt week 12, primary end points are measured.
cAt week 20, eligible subjects will enter the long-term extension study; ineligible subjects will instead enter the 4-week off-treatment follow-up period.
Source: Clinical Study Report.4
Active-Controlled Trial in Adults
The JADE DARE trial was a phase IIIb, double-blind, double-dummy, active-controlled, parallel group, multi-centre study investigating the efficacy and safety of abrocitinib compared with dupilumab in adult patients on background topical therapy, with moderate-to-severe AD. The study consisted of a 28-day screening period, a 26-week double-blind treatment phase, and a 4-week safety follow-up period (or entry into the long-term extension study [EXTEND]). During the screening period, treatments for AD will be washed out, as applicable, according to eligibility requirements. Eligible patients were randomized in a 1:1 ratio to receive 200 mg of abrocitinib once daily with dupilumab-matching placebo every 2 weeks or dupilumab 300 mg every 2 weeks (with a loading dose of 600 mg at baseline) with abrocitinib-matching placebo once daily. Randomization was stratified by baseline AD severity (moderate or severe).1
Figure 4: Schematic of JADE DARE Design
LTE = long-term extension study; PF-04965842 = abrocitinib; Q2W = every 2 weeks; QD = once daily; TCS = topical corticosteroids.
Source: Clinical Study Report.1
Adolescents
The JADE TEEN trial was a randomized, double-blind, placebo-controlled, parallel-group, phase III RCT to evaluate the efficacy and safety of abrocitinib in adolescent patients aged 12 to 18 years of age with moderate-to-severe AD. The study consisted of a 28-day screening phase, a 12-week double-blind treatment phase, and a 4-week follow-up period or entry into the open-label extension study. Eligible patients were randomized at a 1:1:1 ratio to receive abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, or placebo for 12 weeks. Randomization was stratified by baseline disease severity (moderate [IGA = 3] versus severe [IGA = 4] AD).5
The JADE TEEN trial was conducted at 99 sites in 13 countries, including sites in the Australia (n = 4), China (n = 10), Czech Republic (n = 3), Germany (n = 4), Hungary (n = 6), Italy (n = 1), Japan (n = 7), Latvia (n = 2), Mexico (n = 8), Poland (n = 13), Spain (n = 6), Taiwan (n = 3), and the US (n = 32).5
Figure 5: Schematic of JADE TEEN Design
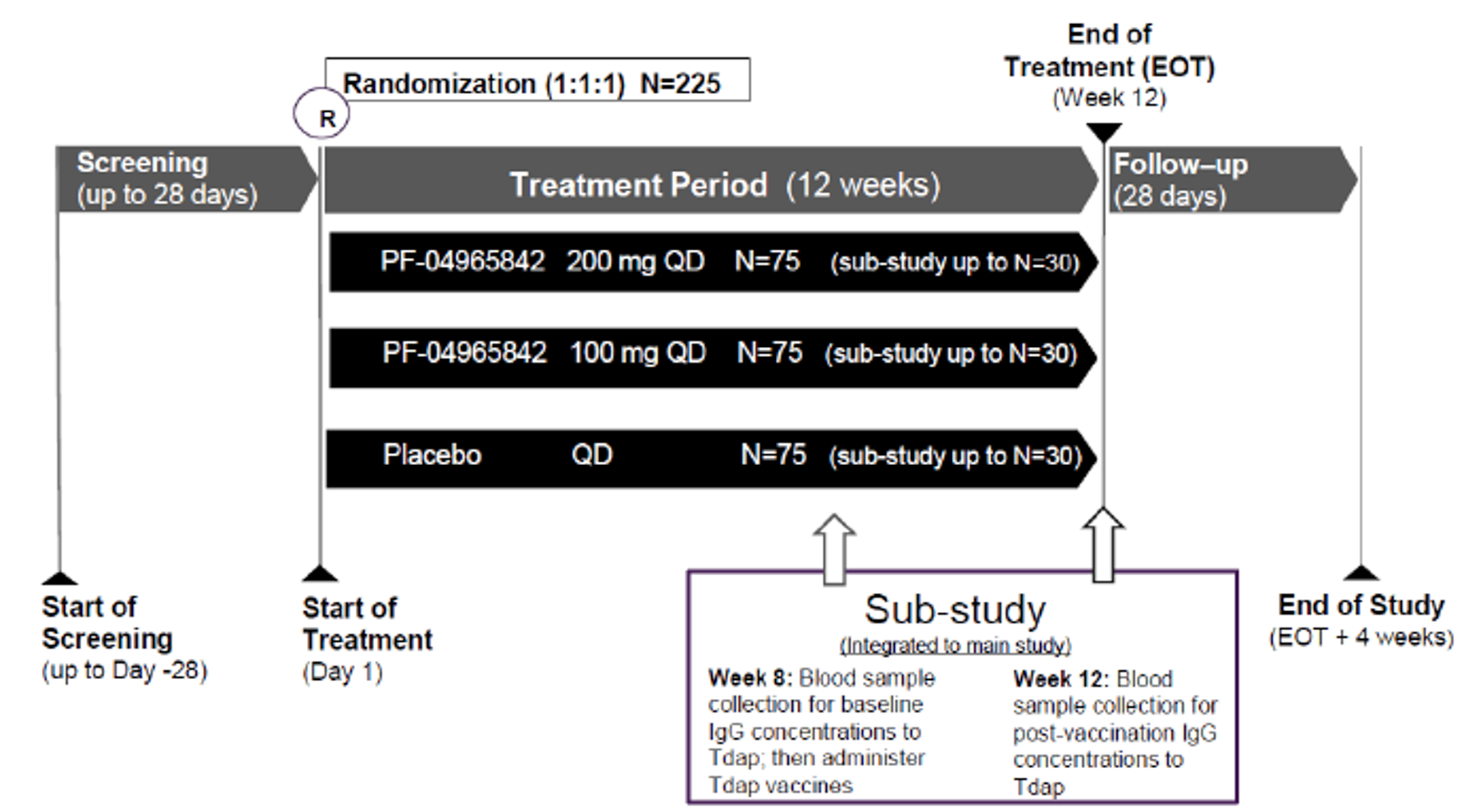
EOT = end of treatment; IgG = immunoglobin G; PF-04965842 = abrocitinib; QD = once daily.
Source: Clinical Study Report.5
Withdrawal Study
The JADE REGIMEN trial was a phase III multi-centre, randomized, responder-enriched, double-blind, placebo-controlled withdrawal study that evaluated the efficacy and safety of abrocitinib monotherapy in subjects aged 12 years and older with moderate-to-severe AD. The JADE REGIMEN trial consisted of an initial open-label induction treatment with abrocitinib 200 mg once daily for 12 weeks. Responders to treatment were subsequently randomized (1:1:1) ratio to receive abrocitinib 200 mg once daily, abrocitinib 100 mg once daily, or matching placebo in a double-blinded maintenance treatment period for 40 weeks. Responders were identified as patients that achieved an IGA score of clear (0) or almost clear (1), a reduction of 2 or more points from the baseline IGA score, and an EASI-75 response compared to baseline. Patients who experienced disease flares during the double-blind treatment period were provided with open-label rescue therapy with abrocitinib 200 mg once daily and topical medication for 12 weeks and assessed for disease improvement throughout the rescue therapy period. A patient was considered to experience a disease flare if a loss of response was observed. This loss of response was defined as a decrease of at least 50% of the EASI response compared to randomization and an IGA score of 2 or higher. Following the 40-week double-blind treatment period, patients could enter a 4-week untreated follow-up period or the JADE EXTEND extension study (if eligible).6,35
The JADE REGIMEN trial was conducted at 235 sites in 21 countries: the US, Argentina, Belgium, Brazil, Bulgaria, Canada; Chile, China, Germany, Israel, Italy, Latvia, Mexico, Netherlands, Poland, Romania, Russia, Serbia, Slovakia; Spain, and Taiwan.33
Figure 6: Schematic of JADE REGIMEN Design
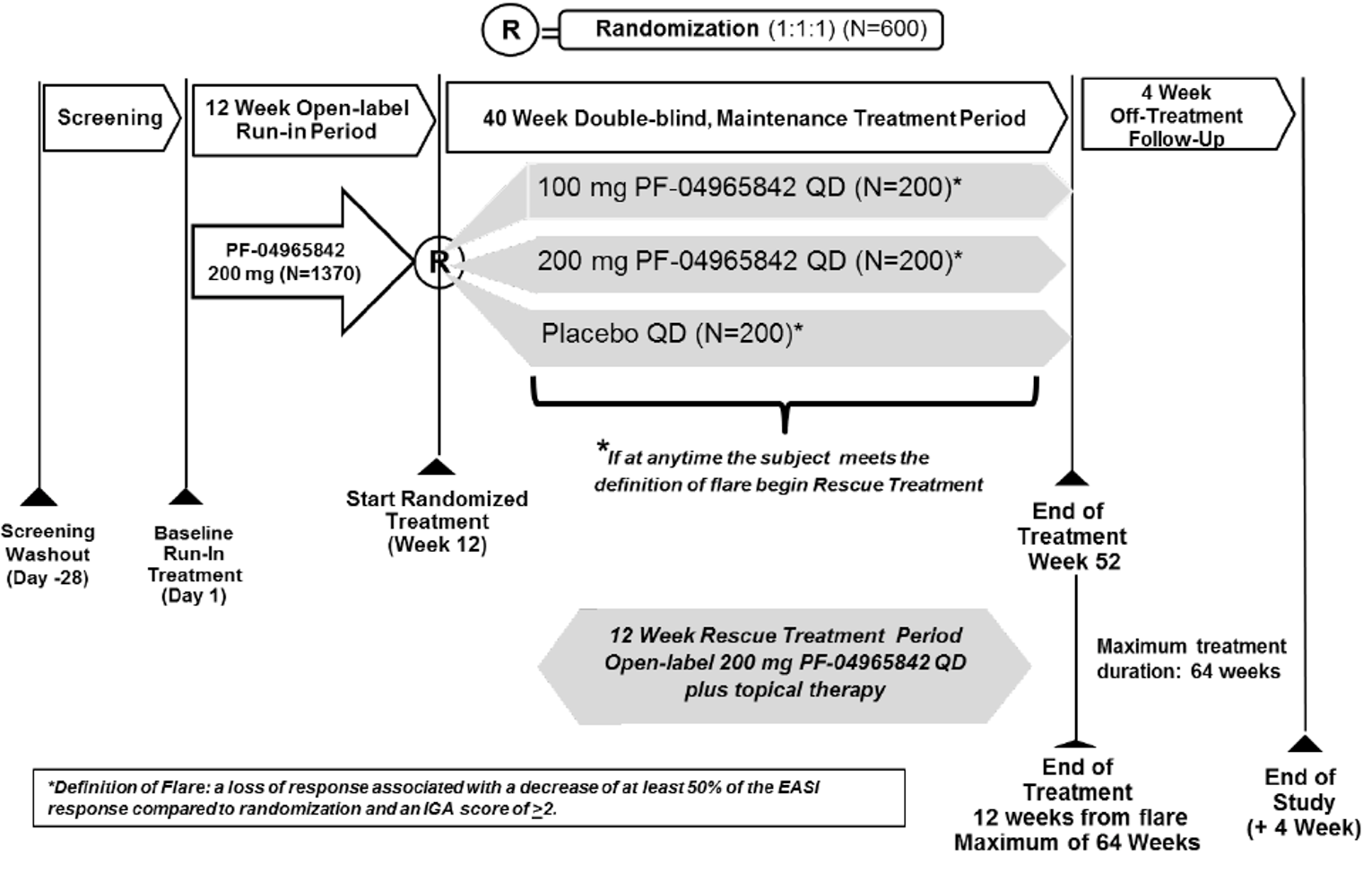
PF-04965842 = abrocitinib; QD = once daily.
Note: A flare is defined as a loss of response associated with a decrease of at least 50% of the EASI response compared to randomization and an IGA score of 2 or higher.
Source: Common Technical Document Module 2.7.3.6
Populations
Inclusion and Exclusion Criteria
Monotherapy Studies
Patients were eligible for the JADE MONO-1 and JADE MONO-2 trials if they met the following criteria: a diagnosis of chronic AD for at least 1 year before day 1 and had confirmed AD using the Hanifin and Rajka criteria at the screening and baseline visits; documented recent history (within 6 months before the screening visit) of an inadequate response to topical medications for at least 4 weeks, or for whom topical treatments are otherwise medically inadvisable (e.g., important side effects or safety risks), or who have required systemic therapies for control of their disease. Enrolment was limited to patients with moderate-to-severe AD, defined as an affected body surface area (BSA) of 10% or greater, an IGA score of 3 or higher, an EASI score of 16 or higher, and a pruritus numeric rating scale (NRS) score of 4 or higher at the baseline visit. Patients were to agree to avoid prolonged exposure to the sun and not use tanning booths, sun lamps or other UV light sources during the study. Any patients receiving concomitant medications for any reason other than AD were required to have been receiving a stable therapeutic regimen, defined as not starting a new drug or changing dosage within 7 days or 5 half-lives (whichever was longer) before day 1, and remain on a stable regimen throughout the duration of the studies.2,3
Patients were excluded if that had active forms of other inflammatory skin diseases or evidence of skin conditions at the day 1 visit that would have interfered with the evaluation of AD or the patient’s response to the study treatment, or if they had received prior treatment with any JAK inhibitors. Patients were also excluded if they had any psychiatric condition, including recent or active suicidal ideation or behaviour that met any of the following criteria: suicidal ideation associated with actual intent and a method or plan in the past year or previous history of suicidal behaviours in the past 5 years (both assessed using the Columbia Suicide Severity Rating Scale); any lifetime history of serious or recurrent suicidal behaviour; a total score of 8 or higher on the Suicidal Behaviours Questionnaire – Revised; or clinically significant depression, defined as a total score of 15 or higher on the Patient Health Questionnaire – 8 items. The JADE MONO-2 trial also excluded patients if they had topical treatments for AD within 72 hours of the first dose of study medication.35
Combination-Therapy Studies
Adults
Patients aged 18 years and older were eligible for the JADE COMPARE trial if they met the following criteria: a diagnosis of chronic AD for at least 1 year before day 1 and had confirmed AD using the Hanifin and Rajka criteria at the screening visit; documented recent history (within 6 months before the screening visit) of an inadequate response to topical medications for at least 4 weeks, or who have required systemic therapies for control of their disease. Enrolment was limited to patients with moderate-to-severe AD, defined as an affected BSA of 10% or greater, an IGA score of 3 or higher, an EASI score of 16 or higher, and a pruritus NRS score of 4 of higher at the baseline visit). During the last 7 days before day 1, patients could only use non-medicated topical therapy (i.e., emollients) for the treatment of AD without other active ingredients indicated to treat AD, or other additives that could affect AD (e.g., hyaluronic acid, urea, ceramide, or filaggrin degradation products) at least twice daily, with response to treatment remaining inadequate at baseline. Patients were to agree to avoid prolonged exposure to the sun and not use tanning booths, sun lamps, or other UV light sources during the study. Any patients receiving concomitant medications for any reason other than AD were required to have been receiving a stable therapeutic regimen, defined as not starting a new drug or changing dosage within 7 days or 5 half-lives (whichever was longer) before day 1, and remain on a stable regimen throughout the duration of the studies.4
Patients were excluded if that had active forms of other inflammatory skin diseases or evidence of skin conditions at the day 1 visit that would have interfered with the evaluation of AD (e.g., psoriasis, seborrheic dermatitis, or lupus) or the patient’s response to the study treatment. Patients could not have had received prior treatment with any systemic JAK inhibitor or dupilumab. Patients were also excluded if they had any psychiatric condition, including recent or active suicidal ideation or behaviour that met any of the following criteria: suicidal ideation associated with actual intent and a method or plan in the past year or previous history of suicidal behaviours in the past 5 years (both assessed using the Columbia Suicide Severity Rating Scale); any lifetime history of serious or recurrent suicidal behaviour; total score of 8 or higher on the Suicidal Behaviours Questionnaire – Revised; clinically significant depression, defined as a total score of 15 or higher on the Patient Health Questionnaire – 8 items.4
Adolescents
Patients aged 12 to 18 years of age were eligible for the JADE TEEN trial if they had a body weight of at least 25 kg and met the following criteria: a diagnosis of chronic AD for at least 1 year before day 1 and had confirmed AD using the Hanifin and Rajka criteria at the screening and baseline visits, documented recent history (within 6 months before the screening visit) of inadequate response to topical medications for at least 4 weeks, or for whom topical treatments are otherwise medically inadvisable (e.g., important side effects or safety risks), or who have required systemic therapies for control of their disease. Enrolment was limited to patients with moderate-to-severe AD, defined as an affected BSA of 10% or greater, an IGA score of 3 or higher, an EASI score of 16 or higher, and a pruritus NRS of 4 or higher at the baseline visit). Patients were to agree to avoid prolonged exposure to the sun and not use tanning booths, sun lamps, or other UV light sources during the study. During the last 7 days before day 1, the patient must have used only non-medicated topical therapy for the treatment of AD (i.e., emollients) at least twice daily, without other active ingredients indicated to treat AD, or other additives that could have affected AD (e.g., hyaluronic acid, urea, ceramide, or filaggrin degradation products), with response to treatment remaining inadequate at baseline. Any patients receiving concomitant medications for any reason other than AD were required to have been receiving a stable therapeutic regimen, defined as not starting a new drug or changing dosage within 7 days or 5 half-lives (whichever was longer) before day 1, and remain on a stable regimen throughout the duration of the studies.5
Patients were excluded if that had active forms of other inflammatory skin diseases or had evidence of skin conditions at the day 1 visit that would have interfered with the evaluation of AD or the patient’s response to the study treatment or had received prior treatment with any JAK inhibitors. Patients were also excluded if they had any psychiatric condition, including recent or active suicidal ideation or behaviour that met any of the following criteria: suicidal ideation associated with actual intent and a method or plan in the past year or previous history of suicidal behaviours in the past 5 years, both assessed using the Columbia Suicide Severity Rating Scale; any lifetime history of serious or recurrent suicidal behaviour; total score of 8 or higher on the Suicidal Behaviours Questionnaire – Revised; or clinically significant depression, defined as a total score of 15 or higher on the Patient Health Questionnaire – 8 items. The JADE MONO-2 trial also excluded patients if they had used topical treatments for AD within 72 hours of the first dose of study medication.35
Withdrawal Study
Patients were eligible for the JADE REGIMEN trial if they had a diagnosis of moderate-to-severe AD for at least 1 year and were aged 12 years and older. Patients had moderate-to-severe AD, a BSA of 10% or greater, an IGA score of 3 or higher, an EASI score of 16 or higher and a PP-NRS score of 4 or higher. Within 6 months of screening patients had to have documented inadequate response to treatment with medicated topic therapy for 4 weeks or longer or required use of a systemic therapy to control their AD.34,35
Patients were excluded if they met any of the following criteria: were unwilling to discontinue current AD medications before the study or required treatment with prohibited medications during the study; had experienced prior treatment with JAK inhibitors; had other active non-AD inflammatory skin diseases or conditions affecting skin; had a medical history including thrombocytopenia, coagulopathy or platelet dysfunction, Q-wave interval abnormalities, current or history of certain infections, cancer, lymphoproliferative disorders or other medical conditions (at the discretion of the investigator); or were pregnant, breastfeeding, or a woman of childbearing potential who was unwilling to use contraception.33
Baseline Characteristics
Monotherapy Studies
Baseline characteristics for the JADE MONO-1 and JADE MONO-2 trials are summarized in Table 13 and Table 14, respectively. The majority of patients in the JADE MONO-1 trial were men (57%) and the mean age of patients in the study was 32.5 years (standard deviation [SD] = 16.0). The proportion of patients with moderate disease (59%) was higher than the proportion of patients with severe disease (41%) as measured by IGA scores.35 The characteristics were well balanced across the 3 treatment groups in the JADE MONO-1 trial, with the exception of differences in the proportion of females (which ranged from 36.4% in the placebo group to 47.4% in the abrocitinib 200 mg once daily group), the proportion of White patients (which ranged from 67.5% in the abrocitinib 200 mg once daily group to 80.5% in the placebo group), and the median EASI score at baseline, which ranged from 22.9 in the placebo group to 27.3 in the abrocitinib 100 mg once daily group. For the subgroup of adolescent patients, the median EQ-5D Youth (EQ-5D-Y) Visual Analogue Scale (EQ VAS) scores were lower in the placebo group (45.0; interquartile range [IQR] = 32.0 to 77.5) compared with the abrocitinib 100 mg once daily (70.0; IQR = 48.0 to 87.5) and abrocitinib 200 mg once daily groups (66.0; IQR = 40.0 to 82.5]). Prior treatments for AD are summarized in Table 15, and 48% of those in the JADE MONO-1 trial had received topical drugs only (48.3% had prior exposure to at least 1 systemic therapy). Prior exposure to dupilumab was reported by 7.8% of the patient population (exposure to other biologics was not reported). Patients with prior exposure to at least 1 systemic immunosuppressant for AD had more severe disease at baseline relative to the overall study population within the abrocitinib groups of the JADE MONO-1 trial (e.g., those with severe disease based on baseline IGA ranged from 54.3% to 57.1% across the abrocitinib groups in the subgroup and from 40.9% to 41.% in the overall study population). The proportion of patients with severe disease at baseline was considerably lower in the placebo group of the subgroup analysis (32.1%) compared with overall study population (40.3%).40
Table 13: Summary of Baseline Characteristics for JADE MONO-1 (Safety Analysis Set)
Characteristic | Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) |
|---|---|---|---|
Overall study population | |||
Age in years < 18, n (%) | 17 (22.1) | 34 (21.8) | 33 (21.4) |
Age in years, median (IQR) | 29.0 (18.0 to 42.0) | 30.5 (19.0 to 43.0) | 27.0 (19.0 to 45.0) |
Female, n (%) | 28 (36.4) | 66 (42.3) | 73 (47.4) |
White, n (%) | 62 (80.5) | 113 (72.4) | 104 (67.5) |
Hispanic or Latino, n (%) | 6 (7.8) | 10 (6.4) | 4 (2.6) |
Height in cm, median (IQR) | 171.5 (165.5 to 178.0) | 170.2 (162.6 to 177.8) | 168.5 (162.5, 177.8) |
Weight in kg, median (IQR) | 73.0 (62.4 to 82.7) | 74.0 (65.2 to 89.3) | 75.0 (61.0 to 89.0) |
BMI in kg/m2, median (IQR) | 24.8 (21.6 to 28.1) | 25.9 (22.9 to 29.9) | 25.9 (22.6 to 29.2) |
AD duration in years, median (IQR) | 18.8 (13.8 to 29.8) | 21.3 (12.6 to 37.7) | 18.9 (12.8 to 31.6) |
IGA, % moderate/severe | 59.7/40.3 | 59.0/41.0 | 59.1/40.9 |
EASI, median (IQR) | 22.9 (19.2 to 37.6) | 27.3 (20.1 to 40.3) | 25.2 (19.2 to 41.7) |
BSA %, median (IQR) | 43.0 (28.0 to 63.0) | 47.0 (31.0 to 67.0) | 42.0 (30.0 to 69.0) |
Pruritus NRS severity, median (IQR) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) |
PSAAD, median (IQR) | 5.2 (4.1 to 6.8) | 5.4 (3.1 to 7.0) | 5.5 (3.7 to 6.9) |
SCORAD, median (IQR) | 64.5 (53.9 to 73.1) | 65.1 (56.3 to 77.2) | 62.8 (54.0 to 73.1) |
DLQI, median (IQR) | 13.0 (10.0 to 16.0) | 14.0 (10.0 to 18.0) | 14.0 (9.0 to 20.0) |
CDLQI, median (IQR) | 14.0 (7.5 to 18.0) | 11.0 (7.5 to 17.0) | 14.0 (10.5 to 17.0) |
POEM, median (IQR) | 21.0 (17.0 to 24.0) | 20.0 (15.0 to 26.0) | 21.0 (16.0 to 24.0) |
PtGA, % moderate/severe | 50.6/45.5 | 46.2/45.5 | 46.8/45.5 |
HADS Anxiety, median (IQR) | 6.0 (3.0 to 8.0) | 5.0 (3.0 to 8.0) | 5.0 (2.0 to 8.0) |
HADS Depression, median (IQR) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 6.0) |
EQ-5D-Y VAS, median (IQR) | 45.0 (32.0 to 77.5) | 70.0 (48.0 to 87.5) | 66.0 (40.0 to 82.5) |
EQ-5D-5L VAS, median (IQR) | 70.5 (52.0 to 84.0) | 70.0 (59.0 to 80.0) | 71.0 (57.0 to 80.0) |
EQ-5D-Y Index, median (IQR) | 0.7 (0.0 to 0.8) | 0.8 (0.5 to 0.8) | 0.7 (0.4 to 0.8) |
EQ-5D-5L Index, median (IQR) | 0.8 (0.7 to 0.8) | 0.8 (0.7 to 0.9) | 0.8 (0.8 to 0.9) |
Peds-FACIT-F, median (IQR) | 34.0 (27.5 to 41.0) | 38.0 (33.0 to 41.0) | 38.0 (34.5 to 39.0) |
FACIT-F, median (IQR) | 40.0 (34.0 to 48.0) | 40.0 (27.0 to 47.0) | 42.0 (33.0 to 46.0) |
SF-36 mental component, median (IQR) | 52.9 (43.5 to 56.9) | 51.0 (43.6 to 56.4) | 51.7 (42.3 to 57.4) |
SF36 physical component, median (IQR) | 46.0 (40.9 to 52.3) | 44.3 (39.8 to 50.3) | 46.7 (39.5 to 52.2) |
Subgroup of patients with prior exposure to systemic immunosuppressant | |||
Number of patients in subgroup | 28 | 46 | 42 |
Duration of disease in years, median (IQR) | 28.6 (14.8 to 38.8) | 26.2 (16.5 to 43.0) | 22.7 (13.7 to 37.1) |
Investigator Global Assessment | |||
Moderate | 19 (67.9) | 21 (45.7) | 18 (42.9) |
Severe | 9 (32.1) | 25 (54.3) | 24 (57.1) |
EASI, median (IQR) | 22.6 (19.1 to 33.4) | 26.9 (21.5 to 43.2) | 34.3 (22.4 to 45.0) |
BSA, median (IQR) | 37.0 (27.0 to 62.0) | 53.5 (31.0 to 73.0) | 56.0 (37.0 to 84.0) |
PP-NRS severity, median (IQR) | 7.0 (6.0 to 7.5) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) |
PSAAD, median (IQR) | 4.9 (3.6 to 7.2) | 4.5 (3.5 to 6.2) | 5.4 (3.8 to 6.8) |
SCORAD, median (IQR) | 60.3 (53.9 to 67.7) | 64.0 (55.5 to 76.8) | 66.4 (59.8 to 75.8) |
DLQI, median (IQR) | 13.0 (8.0 to 14.0) | 16.0 (11.0 to 20.0) | 12.0 (10.5 to 19.0) |
CDLQI, median (IQR) | 17.5 (14.0 to 23.0) | 8.5 (6.5 to 11.5) | 12.5 (6.0 to 15.0) |
POEM, median (IQR) | 21.5 (17.0 to 25.0) | 20.0 (15.0 to 25.0) | 22.0 (16.0 to 25.0) |
HADS Anxiety, median (IQR) | 7.0 (3.0 to 11.0) | 5.0 (3.0 to 7.0) | 5.0 (3.0 to 8.0) |
HADS Depression, median (IQR) | 3.0 (1.0 to 6.0) | 4.0 (2.0 to 7.0) | 5.0 (2.0 to 6.0) |
FACIT-F, median (IQR) | 38.0 (30.0 to 48.0) | 40.5 (31.0 to 45.0) | 38.0 (31.0 to 45.0) |
Peds-FACIT-F, median (IQR) | 27.5 (24.0 to 35.0) | 39.5 (35.0 to 42.0) | 38.5 (34.0 to 39.0) |
SF-36 physical component, median (IQR) | 47.0 (40.9 to 54.1) | 43.9 (40.4 to 50.4) | 45.9 (38.6 to 50.3) |
SF-36 mental component, median (IQR) | 49.9 (44.0 to 56.2) | 49.8 (40.7 to 56.6) | 48.2 (35.9 to 55.6) |
AD = atopic dermatitis; BMI = body mass index; BSA = body surface area; CDLQI = Children's Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator's Global Assessment; IQR = interquartile range; NRS = numeric rating scale; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PP-NRS = peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; SF-36 = Short Form (36) Health Survey; VAS = Visual Analogue Scale.
Source: Clinical Study Report2 and additional data provided by sponsor.40
The majority of patients in the JADE MONO-2 trial were men (59%) and the median age of patients in the study was 31.0 years. The proportion of patients with moderate disease (67.8%) was higher than the proportion of patients with severe disease (32.2%) as measured by IGA scores.35 Characteristics were well balanced across the 3 treatment groups in the JADE MONO-2 trial, with the exception of differences in race (e.g., the proportion of White patients ranged from 51.3% in the placebo group to 63.9% in the abrocitinib 100 mg once daily group). Prior treatments for AD are summarized in Table 15, and 57.8% of those in the JADE MONO-2 trial had received topical drugs only (41.4% had prior exposure to at least 1 systemic therapy). Prior exposure to dupilumab was reported in 3.6% of the patient population, and 2.8% had exposure to a biologic other than dupilumab. Patients with prior exposure to at least 1 systemic immunosuppressant for AD had more severe disease at baseline relative to the overall study population in the JADE MONO-2 trial (e.g., those with severe disease based on baseline IGA ranged from 43.3% to 48.8% across the 3 treatment groups in the subgroup and from 31.6% to 33.3% in the overall study population).40
Table 14: Summary of Baseline Characteristics for JADE MONO-2 (Safety Analysis Set)
Characteristic | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) |
|---|---|---|---|
Overall study population | |||
Age < 18 years, n (%) | 8 (10.3) | 17 (10.8) | 15 (9.7) |
Age ≥ 18 to < 65 years, n (%) | 69 (88.5) | 130 (82.3) | 133 (85.8) |
Age ≥ 65 years, n (%) | 1 (1.3) | 11 (7.0) | 7 (4.5) |
Age in years, median (IQR) | 29.0 (23.0 to 43.0) | 35.0 (25.0 to 48.0) | 29.0 (23.0 to 42.0) |
Male, n (%) | 47 (60.3) | 94 (59.5) | 88 (56.8) |
Female, n (%) | 31 (39.7) | 64 (40.5) | 67 (43.2) |
White, n (%) | 40 (51.3) | 101 (63.9) | 91 (58.7) |
African-American, n (%) | 6 (7.7) | 9 (5.7) | 6 (3.9) |
Asian, n (%) | 29 (37.2) | 46 (29.1) | 54 (34.8) |
Multiracial, n (%) | 1 (1.3) | 1 (0.6) | 2 (1.3) |
Hispanic or Latino, n (%) | 2 (2.6) | 3 (1.9) | 4 (2.6) |
Not Hispanic or Latino, n (%) | 73 (93.6) | 154 (97.5) | 150 (96.8) |
Height in cm, median (IQR) | 170.0 (162.5 to 176.0) | 169.8 (163.0 to 177.4) | 170.0 (164.0 to 176.0) |
Weight in kg, median (IQR) | 71.4 (62.0 to 81.5) | 72.6 (61.2 to 88.0) | 72.0 (60.5 to 82.8) |
BMI in kg/m2, median (IQR) | 23.8 (22.3 to 27.1) | 25.1 (22.1 to 28.3) | 24.2 (21.8 to 28.3) |
AD duration in years, median (IQR) | 19.9 (11.2 to 28.2) | 20.2 (9.1 to 30.9) | 18.9 (8.2 to 29.1) |
IGA, % moderate/severe | 66.7/33.3 | 67.7/32.3 | 68.4/31.6 |
EASI, median (IQR) | 25.9 (20.0 to 33.2) | 25.2 (19.7 to 33.7) | 24.9 (19.3 to 36.0) |
BSA %, median (IQR) | 45.0 (33.0 to 67.0) | 45.0 (32.0 to 64.0) | 44.0 (29.0 to 67.0) |
Pruritus NRS severity, median (IQR) | 7.0 (5.0 to 8.0) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) |
PSAAD, median (IQR) | 5.0 (3.4 to 6.8) | 5.4 (3.8 to 6.9) | 5.3 (3.7 to 6.7) |
SCORAD, median (IQR) | 64.9 (55.9 to 72.6) | 63.4 (55.8 to 71.1) | 62.5 (53.7 to 73.2) |
DLQI, median (IQR) | 14.5 (10.0 to 19.0) | 14.0 (10.0 to 21.0) | 14.0 (11.0 to 19.0) |
CDLQI, median (IQR) | 8.5 (8.0 to 12.0) | 12.0 (9.5 to 19.0) | 13.0 (10.0 to 16.0) |
POEM, median (IQR) | 19.5 (15.0 to 24.0) | 22.0 (18.0 to 25.5) | 20.0 (16.0 to 24.0) |
PtGA, % moderate/severe | 52.6/42.3 | 35.4/58.2 | 51.0/43.2 |
HADS Anxiety, median (IQR) | 5.5 (3.0 to 9.0) | 5.0 (2.0 to 8.0) | 5.0 (3.0 to 9.0) |
HADS Depression, median (IQR) | 4.0 (2.0 to 6.0) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 6.0) |
EQ-5D-Y VAS, median (IQR) | 68.5 (39.0 to 83.5) | 72.5 (57.0 to 80.0) | 68.0 (49.0 to 89.0) |
EQ-5D-5L VAS, median (IQR) | 70.0 (52.0 to 80.0) | 70.0 (51.0 to 80.5) | 70.0 (54.0 to 84.0) |
EQ-5D-Y Index Value, median (IQR) | 0.861 (0.812 to 0.861) | 0.796 (0.736 to 0.819) | 0.790 (0.731 to 0.861) |
EQ-5D-5L Index Value, median (IQR) | 0.818 (0.765 to 0.861) | 0.819 (0.688 to 0.861) | 0.820 (0.765 to 0.861) |
PEDS-FACIT-F, median (IQR) | 39.5 (35.5 to 45.0) | 41.5 (33.0 to 46.5) | 44.0 (34.0 to 46.0) |
FACIT-F, median (IQR) | 40.0 (28.0 to 46.0) | 41.5 (30.5 to 48.0) | 42.0 (31.0 to 46.0) |
SF-36 mental component, median (IQR) | 49.5 (40.7 to 55.1) | 51.4 (42.8 to 56.5) | 49.5 (39.8 to 55.6) |
SF-36 physical component, median (IQR) | 46.8 (41.5 to 52.4) | 47.5 (39.9 to 53.7) | 47.7 (42.4 to 52.4) |
Subgroup of patients with prior exposure to systemic immunosuppressant | |||
Number of patients in subgroup | 15 | 41 | 30 |
Duration of disease in years, median (IQR) | 17.2 (8.3 to 23.2) | 23.2 (15.0 to 33.2) | 18.2 (7.2 to 29.1) |
Investigator Global Assessment | |||
Moderate | 8 (53.3) | 21 (51.2) | 17 (56.7) |
Severe | 7 (46.7) | 20 (48.8) | 13 (43.3) |
EASI, median (IQR) | 27.2 (20.0 to 35.0) | 29.8 (24.8 to 44.8) | 28.5 (19.8 to 40.5) |
BSA, median (IQR) | 38.0 (23.0 to 69.0) | 57.0 (42.0 to 79.0) | 51.0 (27.5 to 74.0) |
PP-NRS severity, median (IQR) | 8.0 (6.0 to 9.0) | 7.0 (6.0 to 8.0) | 8.0 (7.0 to 9.0) |
PSAAD, median (IQR) | 7.0 (4.5 to 7.8) | 6.1 (4.4 to 7.0) | 5.8 (4.9 to 7.2) |
SCORAD, median (IQR) | 65.8 (54.9 to 74.2) | 69.0 (60.2 to 76.8) | 65.6 (57.2 to 82.1) |
DLQI, median (IQR) | 23.0 (15.0 to 23.0) | 18.5 (12.5 to 24.5) | 20.0 (12.0 to 24.0) |
CDLQI, median (IQR) | 13.0 (8.0 to 18.0) | 8.0 (8.0 to 8.0) | 15.0 (14.0 to 24.0) |
POEM, median (IQR) | 21.0 (16.0 to 26.0) | 23.0 (21.0 to 27.0) | 20.5 (16.0 to 24.0) |
HADS Anxiety, median (IQR) | 6.0 (3.0 to 10.0) | 5.0 (2.0 to 9.0) | 6.5 (4.0 to 9.0) |
HADS Depression, median (IQR) | 5.0 (2.0 to 7.0) | 5.0 (2.0 to 7.0) | 5.5 (3.0 to 9.0) |
FACIT-F, median (IQR) | 35.0 (35.0 to 41.0) | 33.5 (24.5 to 45.5) | 35.0 (23.0 to 43.0) |
Peds-FACIT-F, median (IQR) | 40.5 (29.0 to 52.0) | 45.0 (45.0 to 45.0) | 30.0 (18.0 to 42.0) |
SF-36 physical component, median (IQR) | 44.1 (38.5 to 46.8) | 39.9 (34.1 to 49.4) | 44.1 (35.4 to 50.6) |
SF-36 mental component, median (IQR) | 50.3 (45.3 to 55.1) | 46.3 (37.0 to 56.4) | 41.8 (38.8 to 53.8) |
AD = atopic dermatitis; BMI = body mass index; BSA = body surface area; CDLQI = Children's Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator's Global Assessment; IQR = interquartile range; NRS = numeric rating scale; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PP-NRS = peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; SF-36 = Short Form (36) Health Survey; SCORAD = Scoring Atopic Dermatitis.
Source: Clinical Study Report3 and additional data provided by sponsor.40
Table 15: Summary of Prior Medications for JADE MONO-1 and JADE MONO-2 (SAS)
Prior medications, n (%) | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
No prior medication | 0 | 1 (0.6) | 0 | 0 | 1 (0.6) | 2 (1.3) |
Topical drugs only | 34 (44.2) | 69 (44.2) | 82 (53.2) | 46 (59.0) | 87 (55.1) | 93 (60.0) |
High potency CS | 3 (3.9) | 2 (1.3) | 8 (5.2) | 3 (3.8) | 5 (3.2) | 3 (1.9) |
Medium- to low-potency CS | 3 (3.9) | 7 (4.5) | 17 (11.0) | 4 (5.1) | 5 (3.2) | 5 (3.2) |
Unknown strength CS | 27 (35.1) | 57 (36.5) | 55 (35.7) | 38 (7) | 74 (46.8) | 83 (53.5) |
TCIs | 8 (10.4) | 17 (10.9) | 25 (16.2) | 16 (20.5) | 29 (18.4) | 29 (18.7) |
Crisaborole | 0 | 5 (3.2) | 3 (1.9) | 0 | 1 (0.6) | 4 (2.6) |
Systemic drugs | 41 (53.2) | 78 (50.0) | 68 (44.2) | 32 (41.0) | 70 (44.3) | 60 (38.7) |
Systemic, non-biologic drugs | 33 (42.9) | 65 (41.7) | 59 (38.3) | 27 (34.6) | 57 (36.1) | 53 (34.2) |
Mycophenolate mofetil | 2 (2.6) | 2 (1.3) | 2 (1.3) | 0 | 2 (1.3) | 0 |
Methotrexate | 8 (10.4) | 10 (6.4) | 8 (5.2) | 2 (2.6) | 15 (9.5) | 4 (2.6) |
Azathioprine | 4 (5.2) | 1 (0.6) | 6 (3.9) | 1 (1.3) | 3 (1.9) | 2 (1.3) |
CS | 23 (29.9) | 49 (31.4) | 42 (27.3) | 22 (28.2) | 46 (29.1) | 45 (29.0) |
Ciclosporin | 10 (13.0) | 23 (14.7) | 19 (12.3) | 10 (12.8) | 24 (15.2) | 20 (12.9) |
Biologic | 5 (6.4) | 13 (8.2) | 7 (4.5) | |||
Dupilumab | 8 (10.4) | 13 (8.3) | 9 (5.8) | 2 (2.6) | 7 (4.4) | 5 (3.2) |
Other biologics | NR | NR | NR | 3 (3.8) | 6 (3.8) | 2 (1.3) |
CS = corticosteroids; NR = not reported; q.d. = once daily; SAS = safety analysis set; TCIs = topical calcineurin inhibitors.
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Baseline characteristics for the JADE COMPARE trial are summarized in Table 16. The overall proportion of female patients was 51.1%, a majority (64.6%) of the patients had moderate AD, and the median age of patients in the study was 34.0 years.35 Characteristics were well balanced across the 4 treatment groups in the JADE COMPARE trial, with the exception of differences in sex (ranging from 41.2% female in the placebo group to 55.4% in the dupilumab group), race (e.g., the proportion of White patients ranged from 66.4% in the placebo group to 76.5% in the abrocitinib 100 mg once daily group), and PtGA (ranging from 42.5% in the abrocitinib 200 mg once daily group to 51.9% in the placebo group).4 Prior treatments for AD are summarized in Table 17, and 56.5% of patients in the JADE COMPARE trial had received topical drugs only (43.2% had prior exposure to at least 1 systemic therapy). Only a minority of patients had prior exposure to a biologic (no patients had prior exposure to dupilumab and 2.3% of patients had prior exposure to a biologic other than dupilumab).35 Patients with prior exposure to at least 1 systemic immunosuppressant for AD had more severe disease at baseline relative to the overall study population in the JADE COMPARE trial (e.g., those with severe disease based on baseline IGA ranged from 41.7% to 69.0% across the treatment groups in the subgroup analysis and from 31.8% to 38.9% in the overall study population). The proportion of patients with severe disease at baseline was greater in the abrocitinib 200 mg group (69.0%) of the subgroup analysis compared with the other treatment groups (range = 41.7% to 47.3%).
Table 16: Summary of Baseline Characteristics for JADE COMPARE (Safety Analysis Set)
Characteristics | Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) |
|---|---|---|---|---|
Overall study population | ||||
Age 18 to < 65 years, n (%) | 121 (92.4) | 224 (94.1) | 211 (93.4) | 227 (93.8) |
Age ≥ 65 years, n (%) | 10 (7.6) | 14 (5.9) | 15 (6.6) | 15 (6.2) |
Age in years, median (IQR) | 34.0 (25.0 to 46.0) | 33.0 (25.0 to 46.0) | 36.0 (28.0 to 48.0) | 34.0 (25.0 to 47.0) |
Male, n (%) | 77 (58.8) | 120 (50.4) | 104 (46.0) | 108 (44.6) |
Female, n (%) | 54 (41.2) | 118 (49.6) | 122 (54.0) | 134 (55.4) |
White, n (%) | 87 (66.4) | 182 (76.5) | 161 (71.2) | 176 (72.7) |
Black or African-American, n (%) | 6 (4.6) | 6 (2.5) | 9 (4.0) | 14 (5.8) |
Asian, n (%) | 31 (23.7) | 48 (20.2) | 53 (23.5) | 46 (19.0) |
American Indian or Alaska Native, n (%) | 2 (1.5) | 1 (0.4) | 0 | 2 (0.8) |
Native Hawaiian or other Pacific Islander, n (%) | 1 (0.8) | 0 | 1 (0.4) | 0 |
Multiracial, n (%) | 1 (0.8) | 1 (0.4) | 1 (0.4) | 2 (0.8) |
Hispanic or Latino, n (%) | 16 (12.2) | 35 (14.7) | 36 (15.9) | 37 (15.3) |
Not Hispanic or Latino, n (%) | 113 (86.3) | 200 (84.0) | 187 (82.7) | 201 (83.1) |
Height in cm, median (IQR) | 170.2 (164.0 to 177.9) | 170.0 (162.9 to 176.1) | 168.0 (161.0 to 176.0) | 167.6 (162.2 to 174.5) |
Weight in kg, median (IQR) | 74.9 (63.0 to 84.3) | 73.0 (64.0 to 83.2) | 72.3 (62.7 to 84.0) | 72.8 (61.1 to 89.0) |
BMI in kg/m2, median (IQR) | 25.3 (21.9 to 29.1) | 25.1 (22.5 to 29.0) | 25.6 (22.9 to 29.1) | 25.6 (22.2 to 30.5) |
AD duration in years, median (IQR) | 21.3 (9.6 to 30.4) | 21.5 (8.6 to 30.6) | 23.3 (8.6 to 34.5) | 22.5 (9.6 to 33.2) |
IGA, % moderate/severe | 67.2/32.8 | 64.3/35.7 | 61.1/38.9 | 66.9/33.1 |
EASI, median (IQR) | 26.0 (20.8 to 41.4) | 25.3 (19.2 to 38.4) | 29.8 (21.6 to 39.4) | 26.8 (20.3 to 37.6) |
BSA %, median (IQR) | 42.9 (30.2 to 69.0) | 44.3 (28.3 to 65.5) | 48.1 (32.1 to 67.1) | 44.5 (28.0 to 62.0) |
Pruritus NRS severity, median (IQR) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) | 8.0 (7.0 to 9.0) | 7.0 (6.0 to 8.0) |
PSAAD, median (IQR) | 5.2 (3.4 to 6.9) | 5.2 (3.8 to 6.8) | 5.6 (4.2 to 7.0) | 5.2 (3.9 to 6.6) |
SCORAD, median (IQR) | 67.1 (58.7 to 76.6) | 66.2 (56.4 to 77.2) | 68.2 (60.6 to 77.4) | 67.8 (59.3 to 74.7) |
DLQI, median (IQR) | 15.0 (10.0 to 20.0) | 15.0 (10.0 to 20.0) | 16.0 (12.0 to 21.0) | 15.0 (11.0 to 21.0) |
POEM, median (IQR) | 21.0 (16.0 to 26.0) | 21.0 (18.0 to 25.0) | 22.0 (18.0 to 26.0) | 22.0 (18.0 to 26.0) |
PtGA, % moderate/severe | 51.9/44.3 | 47.5/47.1 | 42.5/54.4 | 48.3/48.3 |
HADS anxiety component, median (IQR) | 4.0 (3.0 to 8.0) | 4.5 (2.0 to 8.0) | 5.0 (2.0 to 8.0) | 4.0 (2.0 to 7.0) |
HADS depression component, median (IQR) | 3.0 (1.0 to 7.0) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 6.0) |
EQ-5D-5L VAS, median (IQR) | 71.0 (48.0 to 84.0) | 70.0 (51.0 to 83.0) | 69.5 (49.0 to 82.0) | 70.0 (50.0 to 85.0) |
EQ-5D-5L index, median (IQR) | 0.818 (0.716 to 0.861) | 0.808 (0.729 to 0.861) | 0.816 (0.692 to 0.846) | 0.818 (0.725 to 0.861) |
Subgroup of patients with prior exposure to systemic immunosuppressant | ||||
Number of patients in subgroup | 24 | 40 | 42 | 55 |
IGA n (%) | ||||
Moderate | 14 (58.3) | 23 (57.5) | 13 (31.0) | 29 (52.7) |
Severe | 10 (41.7) | 17 (42.5) | 29 (69.0) | 26 (47.3) |
EASI, median (IQR) | 33.4 (23.5 to 44.4) | 31.9 (22.2 to 44.2) | 40.5 (27.2 to 52.6) | 34.9 (24.0 to 46.0) |
BSA %, median (IQR) | 56.1 (39.7 to 79.5) | 51.9 (38.5 to 67.0) | 61.3 (43.8 to 83.7) | 54.1 (36.0 to 77.0) |
Pruritus NRS severity, median (IQR) | 8.0 (6.0 to 9.0) | 8.0 (7.0 to 8.0) | 8.0 (7.0 to 9.0) | 7.0 (6.0 to 9.0) |
PSAAD, median (IQR) | 5.0 (3.8 to 7.8) | 5.8 (4.3 to 6.8) | 6.6 (5.5 to 7.6) | 5.5 (4.6 to 6.9) |
SCORAD, median (IQR) | 69.9 (64.2 to 80.1) | 68.2 (57.6 to 79.2) | 76.1 (64.5 to 85.6) | 69.0 (61.8 to 79.5) |
DLQI, median (IQR) | 15.0 (12.0 to 20.5) | 16.5 (13.0 to 21.0) | 17.5 (13.0 to 21.0) | 16.0 (11.0 to 21.0) |
HADS anxiety, component median (IQR) | 3.5 (2.0 to 7.0) | 6.0 (3.0 to 8.5) | 5.5 (2.0 to 9.0) | 5.0 (3.0 to 8.0) |
HADS depression component, median (IQR) | 5.0 (2.0 to 7.5) | 5.0 (2.5 to 7.5) | 4.0 (1.0 to 7.0) | 3.0 (1.0 to 7.0) |
POEM, median (IQR) | 22.0 (18.0 to 25.5) | 22.0 (18.0 to 26.0) | 25.0 (20.0 to 28.0) | 21.0 (19.0 to 26.0) |
BMI = body mass index; BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator's Global Assessment; IQR = interquartile range; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; VAS = Visual Analogue Scale.
Source: Clinical Study Report4 and additional data provided by sponsor.40
Active-Controlled Trial in Adults
Baseline characteristics for the JADE DARE trial are summarized in Table 17. The overall proportion of female patients was 45.4%, a majority of the patients had moderate AD (60.0%), and the median age of patients in the study was 33.0 years. Characteristics were well balanced across the 2 treatment groups in the JADE DARE trial. Prior treatments for AD are summarized in Table 18, and 51.9% of those in the JADE COMPARE trial had received topical drugs only (47.9% had prior exposure to at least 1 systemic therapy). Only a minority of patients had prior exposure to a biologic (no patients had prior exposure to dupilumab and 2.2% in each treatment group of patients had prior exposure to a biologic other than dupilumab).1
Table 17: Summary of Baseline Characteristics for JADE DARE (Safety Analysis Set)
Characteristics | Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) |
|---|---|---|
Age in years, median (IQR) | 33.0 (24.0 to 47.0) | 32.0 (25.0 to 43.0) |
Age in years < 65, n (%) | 341 (94.2) | 354 (97.0) |
Age in years ≥ 65, n (%) | 21 (5.8) | 11 (3.0) |
Male, n (%) | 193 (53.3) | 204 (55.9) |
Female, n (%) | 169 (46.7) | 161 (44.1) |
White, n (%) | 269 (74.3) | 248 (67.9) |
Black or African-American, n (%) | 25 (6.9) | 26 (7.1) |
Asian, n (%) | 62 (17.1) | 83 (22.7) |
American Indian or Alaska Native, n (%) | 1 (0.3) | 0 |
Native Hawaiian or other Pacific Islander, n (%) | 1 (0.3) | 0 |
Multiracial, n (%) | 0 | 3 (0.8) |
Hispanic or Latino, n (%) | 30 (8.3) | 27 (7.4) |
Not Hispanic or Latino, n (%) | 331 (91.4) | 337 (92.3) |
Height in cm, median (IQR) | 170.0 (163.0 to 177.0) | 170.0 (162.6 to 177.8) |
Weight in kg, median (IQR) | 76.3 (65.0 to 88.1) | 74.5 (63.5 to 88.0) |
BMI in kg/m2, median (IQR) | 26.1 (22.4 to 29.4) | 25.6 (22.3 to 29.4) |
IGA, % moderate/severe | 59.7/40.3 | 60.3/39.7 |
IGA – hand, % moderate/severe | 36.7/20.4 | 39.7/19.2 |
EASI, median (IQR) | 24.5 (19.4 to 33.6) | 24.5 (19.2 to 33.5) |
BSA (%), median (IQR) | 39.0 (27.0 to 55.0) | 36.0 (26.0 to 55.0) |
Pain at its worst, median (IQR) | 7.0 (5.0 to 8.0) | 7.0 (5.0 to 8.0) |
PP-NRS, median (IQR) | 8.0 (7.0 to 8.0) | 7.0 (6.0 to 9.0) |
SCORAD, median (IQR) | 66.4 (58.9 to 76.8) | 65.2 (58.0 to 75.1) |
DLQI, median (IQR) | 14.0 (9.0 to 19.0) | 14.0 (9.0 to 19.0) |
HADS anxiety component, median (IQR) | 5.0 (2.0 to 8.0) | 5.0 (2.0 to 8.0) |
HADS depression component, median (IQR) | 2.0 (1.0 to 5.0) | 3.0 (1.0 to 5.0) |
POEM, median (IQR) | 21.0 (17.0 to 25.0) | 21.0 (18.0 to 25.0) |
EQ-5D-5L VAS score, median (IQR) | 72.0 (59.0 to 80.0) | 70.0 (58.5 to 80.0) |
EQ-5D-5L index value, median (IQR) | 0.8 (0.7 to 0.9) | 0.8 (0.7 to 0.9) |
BMI = body mass index; BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; IQR = interquartile range; PP-NRS = severity of peak pruritus numerical rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; VAS = Visual Analogue Scale.
Source: Clinical Study Report.1
Adolescents
Baseline characteristics for the JADE TEEN trial are summarized in Table 18. The overall proportion of females was 49.1%, the median age of patients was 15.0 years, and the proportion with moderate AD was 61.4% based on IGA scores.35 Characteristics were well balanced across the 3 treatment groups in the JADE TEEN trial, with the exception of differences in sex (ranging from 40.4% female in the abrocitinib 200 mg group to 54.2% in the placebo group), and percentage BSA (ranging from 38.2% in the placebo group to 48.0% in the abrocitinib 100 mg once daily group). Prior treatments for AD are summarized in Table 19, and 73.3% of those in the JADE TEEN trial had received topical drugs only (25.6% had prior exposure to at least 1 systemic therapy). Only a minority of patients had prior exposure to a biologic (1 patient in each group had prior exposure to dupilumab and 1 patient in the 200 mg abrocitinib group had prior exposure to a biologic other than dupilumab).5 Patients with prior exposure to at least 1 systemic immunosuppressant for AD had more severe disease at baseline relative to the overall study population in the JADE TEEN trial (e.g., those with severe disease based on baseline IGA ranged from 45.5% to 59.3% across the treatment groups in the subgroup analysis and from 35.1% to 40.6% in the overall study population). The proportion of patients with severe disease at baseline was greater in the abrocitinib 100 mg group (59.3%) of the subgroup analysis compared with the other treatment groups (range: 54.2% to 45.5%).
Table 18: Summary of Baseline Characteristics for JADE TEEN (Safety Analysis Set)
Characteristics | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) |
|---|---|---|---|
Overall study population | |||
Age < 18 years, n (%) | 95 (99.0) | 95 (100) | 94 (100) |
Age in years, median (IQR) | 14.0 (13.5 to 16.5) | 16.0 (14.0 to 17.0) | 15.0 (13.0 to 16.0) |
Male, n (%) | 44 (45.8) | 45 (47.4) | 56 (59.6) |
Female, n (%) | 52 (54.2) | 50 (52.6) | 38 (40.4) |
White, n (%) | 56 (58.3) | 52 (54.7) | 52 (55.3) |
Black or African-American, n (%) | 3 (3.1) | 9 (9.5) | 5 (5.3) |
Asian, n (%) | 32 (33.3) | 31 (32.6) | 31 (33.0) |
American Indian or Alaska Native, n (%) | 1 (1.0) | 3 (3.2) | 4 (4.3) |
Native Hawaiian or other Pacific Islander, n (%) | 1 (1.0) | 0 | 1 (1.1) |
Multiracial, n (%) | 1 (1.0) | 0 | 1 (1.1) |
Hispanic or Latino, n (%) | 25 (26.0) | 26 (27.4) | 25 (26.6) |
Not Hispanic or Latino, n (%) | 65 (67.7) | 63 (66.3) | 69 (73.4) |
Height in cm, median (IQR) | 163.8 (157.5 to 169.5) | 163.0 (156.0 to 170.0) | 163.3 (157.0 to 170.5) |
Weight in kg, median (IQR) | 55.3 (49.0 to 64.5) | 59.0 (49.5 to 69.8) | 57.5 (51.0 to 67.5) |
BMI in kg/m2, median (IQR) | 20.8 (19.0 to 22.8) | 21.5 (19.6 to 25.8) | 21.3 (19.3 to 24.8) |
AD duration in years, median (IQR) | 11.7 (6.8 to 14.1) | 10.9 (4.4 to 14.8) | 11.7 (3.6 to 14.1) |
IGA, % moderate/severe | 59.4/40.6 | 60.0/40.0 | 64.9/35.1 |
EASI, median (IQR) | 24.5 (19.5 to 34.9) | 26.5 (20.6 to 38.6) | 25.4 (19.8 to 37.7) |
BSA (%), median (IQR) | 38.2 (28.9 to 61.0) | 48.0 (36.0 to 67.1) | 47.0 (31.5 to 69.5) |
Pruritus NRS severity, median (IQR) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 8.0) |
PSAAD, median (IQR) | 4.5 (2.8 to 6.5) | 5.0 (3.0 to 6.2) | 4.8 (2.7 to 6.4) |
SCORAD, median (IQR) | 68.3 (57.9 to 78.7) | 67.2 (57.4 to 77.4) | 66.1 (56.4 to 76.4) |
CDLQI, median (IQR) | 14.0 (9.0 to 19.0) | 14.0 (10.0 to 19.0) | 13.0 (8.0 to 19.0) |
POEM, median (IQR) | 21.0 (16.0 to 24.0) | 21.0 (16.0 to 24.0) | 20.0 (15.0 to 24.0) |
PtGA, % moderate/severe | 43.8/50.0 | 43.2/54.7 | 51.1/40.4 |
HADS anxiety component, median (IQR) | 5.0 (3.0 to 8.0) | 5.0 (2.0 to 8.0) | 4.0 (2.0 to 7.0) |
HADS depression component, median (IQR) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 5.0) | 3.0 (1.0 to 5.0) |
EQ-5D-Y VAS, median (IQR) | 65.5 (48.0 to 85.0) | 66.0 (49.0 to 81.0) | 65.0 (50.0 to 82.0) |
EQ-5D-Y index value, median (IQR) | 0.727 (0.410 to 0.796) | 0.725 (0.585 to 0.796) | 0.727 (0.585 to 0.796) |
Peds-FACIT-F, median (IQR) | 41.5 (33.0 to 47.0) | 43.0 (34.0 to 46.0) | 40.0 (34.0 to 47.0) |
Subgroup of patients with prior exposure to systemic immunosuppressant | |||
Number of patients in subgroup | 24 | 27 | 22 |
IGA, n (%) | |||
Moderate | 11 (45.8) | 11 (40.7) | 12 (54.5) |
Severe | 13 (54.2) | 16 (59.3) | 10 (45.5) |
EASI | |||
median (IQR) | 29.1 (20.8 to 49.9) | 38.3 (27.0 to 47.8) | 26.5 (20.2 to 38.6) |
BSA %, median (IQR) | 49.0 (33.2 to 75.5) | 65.0 (41.3 to 78.0) | 47.2 (32.4 to 71.0) |
Pruritus NRS severity, median (IQR) | 7.0 (6.0 to 8.0) | 7.0 (6.0 to 9.0) | 6.0 (5.0 to 8.0) |
Night Time Itch Scale severity, median (IQR) | 7.0 (5.5 to 8.0) | 7.0 (5.0 to 8.0) | 6.0 (4.0 to 8.0) |
PSAAD, median (IQR) | 4.4 (3.7 to 6.2) | 5.6 (3.0 to 7.5) | 5.0 (2.1 to 6.6) |
SCORAD, median (IQR) | 64.4 (57.4 to 81.3) | 70.5 (63.1 to 83.6) | 65.9 (57.2 to 78.9) |
CDLQI, median (IQR) | 16.5 (9.5 to 25.0) | 14.0 (10.0 to 21.0) | 14.0 (8.0 to 19.0) |
HADS anxiety component, median (IQR) | 7.0 (3.0 to 9.0) | 5.0 (3.0 to 9.0) | 5.0 (3.0 to 9.0) |
HADS depression component, median (IQR) | 3.5 (2.0 to 6.0) | 3.0 (1.0 to 6.0) | 3.0 (1.0 to 5.0) |
POEM, median (IQR) | 22.0 (19.5 to 27.0) | 22.0 (18.0 to 26.0) | 19.0 (13.0 to 21.0) |
Peds-FACIT-F, median (IQR) | 40.0 (30.5 to 45.0) | 43.0 (34.0 to 48.0) | 38.5 (32.0 to 42.0) |
AD = atopic dermatitis; BMI = body mass index; BSA = body surface area; CDLQI = Children's Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator's Global Assessment; IQR = interquartile range; NRS = numeric rating scale; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis.
Source: Clinical Study Report5 and additional data provided by sponsor.40
Withdrawal Study
Baseline characteristics for the JADE REGIMEN trial are summarized in Figure 7. In the responder group (i.e., those who were randomized) the overall proportion of females was 45.0%, the mean age of patients was 32.1 years (SD = 14.8), and the proportion with moderate AD was 63.7% based on IGA scores.34 Characteristics were well balanced across the 3 treatment groups in the randomized maintenance phase. The proportion of patients with prior exposure to at least 1 systemic therapy for AD was 60% in both the open-label induction and the double-blind treatment phases. The proportion of patients with prior exposure to a biologic for AD was 7.0% (5.3% with dupilumab) and 5.5% (4.0% with dupilumab) in the open-label and double-blind phases, respectively.32
Table 19: Summary of Prior Medications for JADE COMPARE, JADE DARE, and JADE TEEN (SAS)
Prior medications, n (%) | JADE COMPARE | JADE DARE | JADE TEEN | ||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | DUP 300 mg q.2.w. (N = 243) | Abrocitinib 200 mg q.d. (N = 362) | DUP 300 mg q.2.w. (N = 365) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
No prior medication | 0 | 0 | 1 (0.4) | 1 (0.4) | 2 (0.6) | 0 (0.0) | 1 (1.0) | 0 | 2 (2.1) |
Topical drugs only | 83 (63.4) | 139 (58.4) | 122 (54.0) | 129 (53.3) | 188 (51.9) | 189 (51.8) | 71 (74.0) | 68 (71.6) | 70 (74.5) |
Systemic drugs | 48 (36.6) | 99 (41.6) | 103 (45.6) | 112 (46.3) | 172 (47.5) | 176 (48.2) | 24 (25.0) | 27 (28.4) | 22 (23.4) |
Non-biologic | 43 (32.8) | 96 (40.3) | 96 (42.5) | 108 (44.6) | 164 (45.3) | 168 (46.0) | 23 (24.0) | 26 (27.4) | 20 (21.3) |
Biologic (not DUP) | 5 (3.8) | 3 (1.3) | 7 (3.1) | 4 (1.7) | 8 (2.2) | 8 (2.2) | 0 | 0 | 1 (1.1) |
DUP | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) | 1 (1.1) | 1 (1.1) |
DUP = dupilumab; q.2.w. = every 2 weeks; q.d. = once daily; SAS = safety analysis set.
Source: Clinical Study Reports.1,4,5
Figure 7: Summary of Baseline Characteristics for JADE REGIMEN
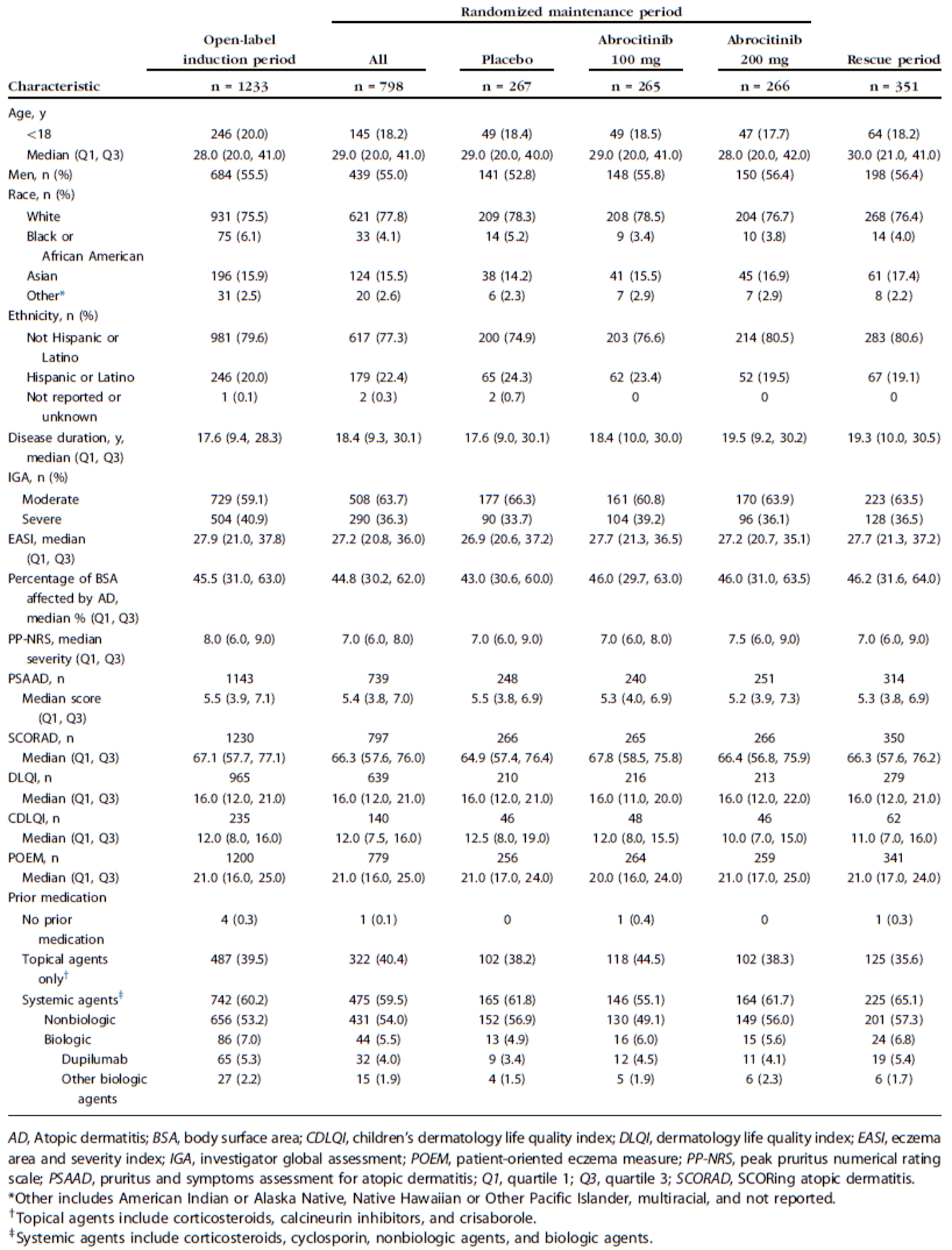
Source: Blauvelt A et al. J Am Acad Dermatol. 2022; 86(1):104 to 112. Copyright 2021 American Academy of Dermatology, Inc. Reprinted in accordance with Creative Commons Attribution 4.0 International Public Licence CC BY 4.0.32
Interventions
Study Treatments
Table 20 summarizes the randomized study treatments in the JADE MONO-1, JADE MONO-2, JADE COMPARE, JADE DARE, JADE TEEN, and JADE REGIMEN trials. Study investigators could temporarily interrupt the dosing of the study treatments for up to 14 consecutive days for safety reasons or while monitoring abnormal laboratory tests if the investigator judged that it was necessary. The investigators were instructed to use their judgment regarding unscheduled visits, laboratory tests, and clinical assessments required to monitor the patient during the time the treatment had been interrupted. If within the 14-day time frame, and the investigator judged that it was safe to restart dosing, the patient could resume receiving the study treatments. If the investigator judged that it was not safe to restart dosing, the patient was to be permanently discontinued from treatment and enter the 4-week follow-up period.2-5
Patients were to be permanently discontinued from the study treatments if they met any of the following criteria at any point in the studies:
marked prolongation of the QTcF interval to greater than 500 ms or greater than a 60 ms change from screening electrocardiogram
serious infection
any bleeding event judged by the investigator to be associated with a platelet count reduction
An AE that, in the judgment of the investigator, required discontinuation from treatment
Any AE or laboratory abnormality that, in the investigator’s judgment, required withholding the investigational product for more than 14 days
Two sequential laboratory results for any of the following: platelet counts below 50,000/mm3; neutrophil counts below 500/mm3; lymphocyte counts below 500/mm3; hemoglobin assessments below 8.0 g/dL or less than 30% from baseline value; aspartate aminotransferase (AST) or alanine transaminase (ALT) elevations of greater than 3 times the upper limit of normal (ULN) with at least 1 total bilirubin value greater than 2 times the ULN; AST or ALT elevations greater than 3 times the ULN with an abnormal international normalized ratio; AST or ALT elevations greater than 3 times the ULN accompanied by symptoms consistent with hepatic injury; and AST or ALT elevations greater than 5 times the ULN, regardless of total bilirubin or accompanying symptoms; increases in serum creatinine that are more than 50% above the average of screening and baseline values and an absolute increase in serum creatinine of 0.5 mg/dL or more.2-5
Table 20: Study Treatments in the Included Studies
Studies | Study drugs |
|---|---|
Monotherapy studies | |
JADE MONO-1 and JADE MONO-2 |
|
Combination-therapy studies | |
JADE COMPARE |
|
JADE DARE |
|
JADE TEEN |
|
Withdrawal study | |
JADE REGIMEN | Induction phase (open-label)
Maintenance phase (double-blind)
Rescue therapy (open-label)
|
q.2.w. = every 2 weeks; q.d. = once daily; SC = subcutaneous.
Source: Clinical Study Reports1-5 and Common Technical Document Module 2.7.36
Background Topical Therapy
Patients in JADE COMPARE, JADE DARE, and JADE TEEN were to comply with the standardized background topical therapy described in Table 21. Background topical therapy was not to be applied before attending a study visit on the day of the study visit. Background topical therapy instead should be applied after the visit, on study visit days.
Table 21: Background Therapy for JADE COMPARE, JADE DARE, and JADE TEEN
Category | Guidance |
|---|---|
Non-medicated topical therapy | Non-medicated topical emollient without other active ingredients indicated to treat AD, or other additives that could affect AD (e.g., hyaluronic acid, urea, ceramide, or filaggrin degradation products): must be applied at least twice daily to all body areas affected with AD, throughout at least the final 7 days before day 1 and throughout the remainder of the study |
Medicated topical therapy |
|
Other concomitant AD therapies | Oral antihistamines |
AD = atopic dermatitis; PDE-4 = phosphodiesterase type 4; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
Source: Clinical Study Reports.4,5
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 22. These end points are further summarized in the following section. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 22: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Time point | JADE MONO-1 | JADE MONO-2 | JADE COMPARE | JADE DARE | JADE TEEN | JADE REGIMEN |
|---|---|---|---|---|---|---|---|
EASI-75 response | At 12 weeks | Co-primary (12 weeks) | Co-primary (12 weeks) | Co-primary (12 weeks) | Secondary | Co-primary (12 weeks) | Secondary |
At 16 weeks | NA | NA | Key Secondary | Secondary | NA | Secondary | |
Other time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary | |
IGA response | At 12 weeks | Co-primary | Co-primary | Co-primary | Secondary | Co-primary | Secondary |
At 16 weeks | NA | NA | Key Secondary | Secondary | NA | Secondary | |
Other time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary | |
PP-NRS4 response | At 2 weeks | Key Secondary | Key Secondary | Key Secondary | Co-primary | Key secondary | Secondary |
At 4 weeks | Key Secondary | Key Secondary | Secondary | Secondary | Key secondary | Secondary | |
At 12 weeks | Key Secondary | Key Secondary | Secondary | Secondary | Key secondary | Secondary | |
Other time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary | |
Loss of response (loss ≥ 50% of EASI response and IGA score ≥ 2) | NA | NA | NA | NA | NA | Primary | |
Loss of response (IGA score ≥ 2) | NA | NA | NA | NA | NA | Key secondary | |
CFB in PP-NRS | All time points | NA | NA | Secondary | NA | Secondary | NR |
CFB in PSAAD score | At 12 weeks | Key Secondary | Key Secondary | NA | NA | Key secondary | Secondary |
Other time points | Other | Other | Other | NA | NA | Secondary | |
EASI-50 response | All time points | Secondary | Secondary | Secondary | NA | Secondary | NR |
EASI-90 response | At 4 weeks | Secondary | Secondary | Secondary | Co-primary | Secondary | NR |
At 16 weeks | NA | NA | Secondary | Key Secondary | NA | NR | |
Other time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary | |
EASI-100 response | All time points | Secondary | Secondary | Secondary | Secondary | Secondary | NR |
CFB in EASI | All time points | Secondary | Secondary | Secondary | Secondary | Secondary | NR |
CFB in BSA (%) | All time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary |
BSA response (5%) | All time points | Secondary | Secondary | NA | Secondary | Secondary | NA |
Steroid-free days | All time points | NA | NA | Secondary | NA | Secondary | NA |
SCORAD-50 | All time points | Secondary | Secondary | Secondary | NA | Secondary | Secondary |
SCORAD-75 | All time points | Secondary | Secondary | Secondary | NA | Secondary | Secondary |
CFB in SCORAD | All time points | Secondary | Secondary | Secondary | Secondary | Secondary | NR |
CFB in SCORAD VAS for sleep loss | All time points | Secondary | Secondary | Secondary | NA | Secondary | NR |
Time to ≥ 4-point improvement in NRS for severity | Secondary | Secondary | Secondary | Secondary | Secondary | NA | |
PSAAD response | All time points | Secondary | Secondary | Secondary | NA | NA | NR |
CFB DLQI | All time points | Secondary | Secondary | Secondary | Secondary | NA | Secondary |
CFB CDLQI | All time points | Secondary | Secondary | NA | NA | Secondary | Secondary |
DLQI response (≥ 2 points) | All time points | Secondary | Secondary | NA | Secondary | NA | NR |
DLQI response (≥ 4 points) | All time points | Secondary | Secondary | NA | Secondary | NA | NR |
CDLQI response (≥ 2.5 points) | All time points | Secondary | NA | NA | NA | Secondary | NR |
CFB in each HADS component | All time points | Secondary | Secondary | Secondary | Secondary | Secondary | Secondary |
HADS response (< 8) | All time points | Secondary | NA | NA | Secondary | NA | NR |
CFB in POEM | All time points | Secondary | Secondary | NA | Secondary | Secondary | Secondary |
PtGA response | All time points | Secondary | Secondary | NA | NA | Secondary | Secondary |
CFB in PtGA | All time points | Secondary | Secondary | Secondary | NA | Secondary | NA |
CFB in EQ-5D-5L | All time points | Secondary | Secondary | Secondary | Secondary | NA | Secondary |
CFB in EQ-5D-Y | All time points | Secondary | Secondary | NA | NA | Secondary | Secondary |
CFB in FACIT-F | All time points | Secondary | Secondary | NA | NA | NA | Secondary |
CFB in Peds-FACIT-F | All time points | Secondary | Secondary | NA | NA | Secondary | Secondary |
CFB in SF-36v2 | All time points | Secondary | Secondary | NA | NA | NA | Secondary |
CFI in DFI | At 12 weeks | NA | NA | NA | NA | Secondary | NR |
CFB in WPAI-AD | At 12 weeks | NA | Secondary | NA | NA | NA | NR |
MOS sleep scale | All time points | NA | NA | NA | Secondary | NA | NA |
Night Time Itch Scale response | All time points | NA | Exploratory | NA | NA | Exploratory | NR |
Skin pain NRS | All time points | NA | NA | NA | Secondary | NA | NA |
Time to achieve ≥ 4-point improvement from baseline in the Night Time Itch Scale | NA | Exploratory | NA | NA | Exploratory | NR | |
BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CFB = change from baseline; DFI = Dermatitis Family Impact; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; MOS = Medical Outcomes Study; NA = not applicable; NR = not reported; NRS = numeric rating scale; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PP-NRS = severity of pruritus numerical rating scale; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = improvement of 75% or greater in Scoring Atopic Dermatitis; SF-36v2 = Short Form (36) Health Survey Version 2; VAS = Visual Analogue Scale; WPAI-AD = Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis.
Source: Clinical Study Reports.2-4,6
Eczema Area and Severity Index
The EASI is a scale used in clinical trials to assess the severity and extent of AD.13,14,41,42 In the EASI, 4 disease characteristics of AD (erythema, infiltration and/or papulation, excoriations, and lichenification) are assessed for severity by the investigator on a scale of “0” (absent) to “3” (severe). The scores are added up for each of the 4 body regions (head, arms, trunk, and legs). The assigned percentages of BSA for each section of the body are 10% for head, 20% for arms, 30% for trunk, and 40% for legs. Each subtotal score is multiplied by the BSA represented by that region. In addition, the affected area of AD assessed as a percentage by each body region is converted to a score of 0 to 6, with the area expressed as 0 (none), 1 (1% to 9%), 2 (10% to 29%), 3 (30% to 49%), 4 (50% to 69%), 5 (70% to 89%), or 6 (90% to 100%). Each of the body area scores are multiplied by the area affected. The total EASI score therefore ranges from 0 to 72 points, with the highest score indicating worse severity of AD.13 It is suggested that the severity of AD based on EASI be categorized as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 2l.1 to 50.0 = severe; and 50.1 to 72.0 = very severe.43 The EASI-50, EASI-75, EASI-90, and EASI-100 end points indicate improvements of 50% or greater, 75% or greater, 90% or greater, and 100% improvement from baseline, respectively. The validity and reliability of the EASI was examined in several studies and was shown to be adequate.13,14,41,44 The overall minimal important difference (MID) is 6.6, based on results from 1 study.14
Investigator’s Global Assessment
The IGA is a 5-point scale that provides a global clinical assessment of AD severity ranging from 0 to 4, where “0” indicates clear, and “4” indicates severe AD.2-4,6 A decrease in score relates to an improvement in signs and symptoms. No information was found on what would constitute an MID in patients with AD. The clinical evaluator performed the assessment of the overall severity of AD and assigned an IGA score and category as described in Table 23.
Table 23: Investigator’s Global Assessment Score
Score | Category | Description |
|---|---|---|
0 | Clear | AD is cleared, except for any residual discoloration (post-inflammatory hyperpigmentation and/or hypopigmentation) |
1 | Almost clear | Overall, the AD is not entirely cleared, and remaining lesions are light pink (not including post-inflammatory hyperpigmentation), and/or have barely palpable hard thickened skin and/or papules, and/or have barely perceptible lichenification; excoriation and oozing/crusting are absent |
2 | Mild | Overall, the AD consists of lesions that are light red with slight, but definite hard thickened skin and/or papules, with slight but definite linear or picked scratch marks or penetrating surface injury, with slight but definite thickened skin, fine skin markings, and lichenoid scale; oozing and/or crusting is absent |
3 | Moderate | Overall, the AD consists of lesions that are red with easily palpable moderate hard thickened skin and/or papules; with moderate linear or picked scratch marks or penetrating surface injury, with moderate thickened skin, coarse skin markings, and coarse lichenoid scale with slight oozing and/or crusting |
4 | Severe | Overall, the AD consists of lesions that are deep, dark red with severe hard thickened skin and/or papules, with severe linear or picked scratch marks or penetrating surface injury, with severe thickened skin with very coarse skin markings and lichenoid scale, with moderate-to-severe oozing and/or crusting |
AD = atopic dermatitis.
Source: Clinical Study Reports.2-4,6
Body Surface Area
Measurements of BSA was derived from the sum of the BSA in handprints across 4 body regions as part of the EASI assessment. A handprint refers to that of each individual patient for their own measurement. The BSA efficacy ranges from 0 to 100%, with higher values representing greater severity of AD. Because the scalp, palms, and soles were excluded from the BSA assessment, the maximum possible value was less than 100%.2-4,6
Dermatology Life Quality Index
The DLQI is a dermatology-specific quality of life instrument. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life: symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment.45-47 The maximum score per aspect is either 3 (with a single question) or 6 (with 2 questions) and the scores for each can be expressed as a percentage of either 3 or 6. Each of the 10 questions is scored from 0 (not at all) to 3 (very much) and the overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30).46,47 The higher the score, the more quality of life is impaired. The meaning of the DLQI scores on a patient’s life is as follows: 0 to 1 indicates no effect; 2 to 5 a small effect; 6 to 10 a moderate effect; 11 to 20 a very large effect; and 21 to 30 an extremely large effect. Estimates of the MID have ranged from 2.2 to 6.9.46,47
Children’s Dermatology Life Quality Index
The CDLQI is a 10-item widely used and validated questionnaire used in clinical practice and clinical trials to measure the impact of skin disease on the quality of life in children.48,49 The CDLQI can be completed by the child alone and/or with help from the parents or guardian.48 It covers 6 areas of daily activities, including symptoms and feelings, leisure, school or holidays, personal relationships, sleep, and treatment. The questions are answered using a 4-point Likert scale (scored from 0 to 3 for each question) based on recall for the past week. Total scores therefore range from 0 to 30. A higher CDLQI score indicates a greater degree of quality-of-life impairment.48 No minimal clinically important difference was identified in the literature.
Functional Assessment of Chronic Illness Therapy–Fatigue
The Functional Assessment of Chronic Illness Therapy–Fatigue is a validated subject-completed questionnaire consisting of 13 items that assess fatigue. Instrument scoring yields a range from 0 to 52, with higher scores representing better overall health status (less fatigue).2,3 An MID of 3 to 4 of the total score was established in patients with rheumatoid arthritis,50 and 5.9 in patients with systemic lupus erythematosus,51 but no MID has been established in patients with AD.
Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue
The Peds-FACIT-F is a version of the FACIT-F for adolescents aged 12 to 17 years.5 The MID of the Peds-FACIT-F was calculated using anemia and the functional performance status as clinical anchors, and a difference greater than 4.7 points was considered clinically important.52 However, no MID for patients with AD has been established.
Hospital Anxiety and Depression Scale
The HADS is a patient-reported questionnaire designed to identify anxiety disorders and depression in patients at nonpsychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.53-55 The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, among which 7 items are related to anxiety and 7 items are related to depression. Patients provide responses to each item based on a 4-point Likert scale, from 0 (the best) to 3 (the worst); a person can score between 0 and 21 for each subscale (anxiety and depression). A high score is indicative of a poor state. Scores of 11 or more on either subscale are considered to be a “definite case” of psychological morbidity, while scores of 8 to 10 represented “probable case” and 0 to 7 “not a case.”55 No information on MID was found in the literature.
Severity of Pruritus Numerical Rating Scale
The PP-NRS is a tool that patients used to report the intensity of their itch during a daily recall period using an interactive voice response system. Patients were asked to rate their worst itching due to AD over the past 24 hours on an NRS anchored by the terms “no itch” (0) and “worst itch imaginable” (10). The proportion of patients with improvement (reduction of ≥ 4 points) of weekly average of peak daily pruritus NRS from baseline was reported in the included studies.2-4,6 The most appropriate definition of a responder on the pruritus NRS has been reported to be in the range of 3 to 4 points.56
Pruritus and Symptoms Assessment for Atopic Dermatitis
The PSAAD is a daily patient-reported symptom diary. The preliminary version of the PSAAD involved a 15-item questionnaire that included 11 items developed to measure the symptoms of AD, capturing those identified by patients to be most important, based on a 24-hour recall. The 11 items related to symptoms (itch, dryness, redness, flaking, discoloration, pain, bleeding, cracking, bumps, swelling, and weeping and/or oozing) are measured using a scale, ranging from 0 (none) to 10 (extreme). The total score is the average of the responses to each of the 11 items, with higher scores indicating worse symptoms. Four items were added for exploratory and psychometric validation purposes: sleep and usual-activities questions, patient global impression of severity, and patient global impression of change questions.2-4,6 The MID has been estimated to be a change of 0.63 of the total score.57
Patient Global Assessment
The PtGA required patients to evaluate their overall cutaneous disease at that point in time on a single-item, 5-point scale. The same category labels used in the IGA were used for the PtGA (severe [4], moderate [3], mild [2], almost clear [1], and clear [0]), with higher scores indicating worse AD.2-4,6 A literature search by CADTH did not identify an MID for the PtGA.
Scoring Atopic Dermatitis
The SCORAD tool was developed to standardize the evaluation of the extent and severity of AD.58 It assesses 3 components of AD: the affected BSA, severity of clinical signs, and symptoms. The severity of 6 specific symptoms of AD (redness, swelling, oozing and/or crusting, excoriation, skin thickening and/or lichenification, and dryness) is assessed using a 4-point scale (i.e., none = 0, mild = 1, moderate = 2 and or severe = 3) with a maximum of 18 total points. The symptoms (itch and sleeplessness) are recorded by the patient or relative on a VAS, where 0 is no symptom and 10 is the worst imaginable symptom, with a maximum possible score of 20. The SCORAD results are calculated based on the 3 components of the AD listed previously. The maximum possible total score of SCORAD is 103, with a higher score indicating poorer or a more severe condition.2-4,6 A difference of 8.7 points in SCORAD was estimated as the MID for the patients with atopic eczema (also known as AD).14
Patient-Oriented Eczema Measure
The POEM is a 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults.59 Based on frequency of occurrence during the past week, the 7 items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question were: “0” for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” for every day. The maximum total score was 28; a high score is indicative of poor quality of life (0 to 2 indicates clear or almost clear, 3 to 7 mild eczema, 8 to 16 moderate eczema, 17 to 24 severe eczema, and 25 to 28 very severe eczema).59 One study14 reported that the overall mean MID of the POEM was 3.4 points (SD = 4.8), when an IGA improving by 1 point was used as anchor. In 2018, the MID of the POEM in children (N = 300) with moderate-to-severe AD was calculated in 1 study.60 The authors recommended the following thresholds be used to interpret changes in POEM scores in children: a score of 3 to 3.9 indicates a probably clinically important change and a score of 4 or higher indicates a very likely clinically important change.60
Short Form (36) Health Survey Version 2
The SF-36v2 is a validated generic health status measure. It measures 8 general health domains: physical functioning, role limitations due to physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations due to emotional problems, and mental health. These domains can also be summarized as physical and mental component summary scores. The use of this scale was restricted to adult subjects and not for adolescents to complete. The acute version uses a recall period of 1 week. Eight scaled scores are converted to weighted sums of the questions in their section. Each scale is directly transformed into a 0-to-100 scale on the assumption that each question carries equal weight. Lower scores mean more disability. (i.e., a score of 0 indicates maximum disability and a score of 100 indicates no disability).61,62 The user’s manual for the SF-36v2 proposes MIDs of a change of 2 points on the physical component summary and 3 points for the mental component summary.
Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis
The WPAI is an instrument used to measure loss of productivity at work and impairment in daily activities over the past 7 days.63 The questionnaire includes 4 items (absenteeism, presenteeism, overall work impairment, and activity impairment) that range from 0% to 100%, with higher values indicating greater impairment. While absenteeism represents the percentage of work-time-missed due to AD, presenteeism represents the percentage of impairment while at work due to AD. Overall work impairment represents the total percentage of work time missed due to either absenteeism or presenteeism (as those are mutually exclusive). Activity impairment represents the percentage of impairment during daily activities other than work. The 4 items are all evaluated using an 11-point Likert-type scale from 0 (no effect) to 10 (completely prevented), and the scores are multiplied by 10 to produce a percentage. CADTH did not identify an MID for the WPAI-AD; however, the MID has been estimated to be 20% for the Work Productivity and Activity Impairment Questionnaire for use in patients with psoriasis.64 The questionnaire was only completed by adult patients and included as an outcome only in the JADE MONO-2 trial.
Dermatitis Family Impact Questionnaire
The Dermatitis Family Impact (DFI) is a validated 10-item measure filled out by the parent or caregiver of the patient to assess the impact of the patient’s eczema on the family.6 Each item is scored using a 4-point scale (3 [very much], 2 [a lot], 1 [a little], or 0 [not at all]).5 Total scores range from 0 to 30, with higher scores indicating a greater impact on the family and/or caregivers. A literature search by CADTH did not identify an MID for the DFI.
EQ-5D
The EQ-5D is a generic quality-of-life instrument that has been applied to a wide range of health conditions and treatments including AD.65,66 The first of 2 parts of the EQ-5D constitute a descriptive system that classifies respondents (aged ≥ 12 years) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression. Each dimension has 3 possible levels (1, 2, or 3) representing “no problems,” “some problems,” and “extreme problems,” respectively. Respondents are asked to choose a level that reflects their own health state for each of the 5 dimensions. A scoring function can be used to assign a value to self-reported health states from a set of population-based preference weights.65,66 The second part is a 20 cm Visual Analogue Scale (EQ VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state,” respectively. Respondents are asked to rate their own health by drawing a line from an anchor box to the point on the EQ VAS that best represents their own health on that day. The third part is the EQ-5D index score, which is generated by applying a multi-attribute utility function to the descriptive system. Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). The EQ-5D therefore produces 3 types of data for each respondent.
The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores of less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The MID for the EQ-5D ranges from 0.033 to 0.074. No additional validity and MID information were found in literature search for EQ-5D in patients with AD.
Rescue Therapy due to Protocol-Defined Flare
The primary end point of JADE REGIMEN was the proportion of patients entering rescue during the maintenance period due to a protocol-defined flare. A flare requiring rescue treatment was defined as a loss of at least 50% of the EASI response at week 12 and an IGA score of 2 or higher.6,35
Statistical Analysis
Primary Outcomes of the Studies
The co-primary end points of 4 of the included studies (JADE MONO-1, JADE MONO-2, JADE COMPARE, and JADE TEEN) were: response based on an IGA score of clear or almost clear and a reduction from baseline of 2 or more points at 12 weeks; and response based on an EASI-75 response at 12 weeks. The co-primary end points were analyzed using the Cochran-Mantel-Haenszel (CMH) test adjusted by baseline disease severity group (moderate and severe). The differences between each active group and the placebo group in the proportion of patients achieving an IGA response or an EASI-75 along with its 95% CI (using the normal approximation for the difference in binomial proportions) were reported by the sponsor. Both end points were required to achieve statistical significance for a given dose to meet the primary objective of the studies. Patients who withdrew from the trials were counted as nonresponders for end points after the time of withdrawal.
Sensitivity analyses were performed using MAR and missing not at random (MNAR) approaches. Missing observations were imputed using a tipping point analysis to estimate the treatment effect under the assumption that the missing data mechanism is MAR or, more generally, is MNAR. A longitudinal logit-normal mixed model was fit using only the observed data. Under the MAR framework, imputations were based on the posterior predictive probability of response obtained from the posterior distribution under the mixed model. Under an MNAR framework, imputations for the active treatment groups were based on a linear combination of the posterior predictive probability of response for the active group and the placebo group. For each such completed dataset, the estimates of the proportions and CMH-weighted difference of proportions between each active dose group and placebo will be obtained and Rubin’s rule will be used to combine the multiple estimates and standard errors across the imputed datasets and provide P values.
The co-primary end points of the JADE DARE trial were: PP-NRS4 from baseline at week 2 and EASI-90 at week 4. The co-primary end points were analyzed using the CMH test adjusted by baseline disease severity group (moderate and severe). Patients who withdrew from the trials were counted as nonresponders for end points after the time of withdrawal. The protocol first tested the superiority of PP-NRS4 responses at week 2 and then EASI-90 at week 4 between abrocitinib and dupilumab. If both hypotheses were rejected, the procedure continued to test the noninferiority of EASI-90 at week 16 between abrocitinib and dupilumab. Noninferiority would be declared if the lower bound of the CI for the response difference (abrocitinib minus dupilumab) was greater than −10%. If noninferiority was achieved, the procedure continued to test the superiority of EASI-90 at week 16 between abrocitinib and dupilumab.
Key Secondary and Other Outcomes of the Studies
Binary secondary outcomes were analyzed in the same manner as the co-primary end points. Continuous secondary end points were analyzed using a mixed model for repeated measures approach that included fixed factors of treatment, week, treatment-by-week interaction, randomization strata (baseline disease severity and/or age category depending on the individual trial), baseline value, and an unstructured covariance matrix. The time-to-event end point (i.e., PP-NRS4) was summarized using the Kaplan–Meier method to estimate median, quartiles, and probabilities of an event. A log-rank test was used to compare times to event data between treatment groups.
Statistical Testing Hierarchy
Statistical testing for the co-primary and key secondary end points was adjusted for multiple comparisons using a sequential Bonferroni-based iterative multiple-testing procedure to control the familywise type I error at 5% for each of the 2 abrocitinib dosages (200 mg once daily and 100 mg once daily) versus placebo (Table 24).
In the JADE COMPARE trial, the sponsor used a series of hypotheses related to the PP-NRS4 outcome at week 2 that were assessed at a 2.5% significance level in the order specified in sequence A of Table 24. If all hypotheses in sequence A were rejected, then the unused alpha level of 2.5% was passed on to the assessments for the week-16 end points in sequence B at a 5% significance level. The statistical significance for each hypothesis in sequence B could not be claimed unless the prior hypothesis in the sequence was statistically significant. In sequence B, if 1 hypothesis was not rejected at an alpha level of 5% then no statistical significance was claimed for any subsequent hypotheses in the sequence. In sequence A, if 1 hypothesis was not rejected at an alpha level of 2.5% then no statistical significance was claimed for any subsequent hypotheses in the sequence. In this case, the assessing for statistical significance in sequence B was at the 2.5% significance level. If all hypotheses in sequence B were rejected, then the unused alpha level of 2.5% was passed back for assessing the hypotheses in sequence A at the 5% level. In sequence B, if a hypothesis was not rejected at an alpha level of 2.5% then no statistical significance was claimed for any subsequent hypotheses in the sequence. Failure to demonstrate statistical significance for abrocitinib 100 mg once daily versus dupilumab for PP-NRS4 meant the sequence B end points were evaluated at a 2.5% significance level.
Table 24: Statistical Testing Hierarchies
Study | Statistical testing hierarchy |
|---|---|
JADE MONO-1 JADE MONO-2 JADE TEEN | 1. IGA and EASI-75 at week 12 (200 mg q.d. vs. placebo) 2. PP-NRS4 response at 2 weeks (200 mg q.d. vs. placebo) Sequence A 3. PP-NRS4 response at 4 weeks (200 mg q.d. vs. placebo) 4. PP-NRS4 response at 12 weeks (200 mg q.d. vs. placebo) 5. IGA and EASI-75 at week 12 (100 mg q.d. vs. placebo) 6. PP-NRS4 response at 2 weeks (100 mg q.d. vs. placebo) 7. PP-NRS4 response at 4 weeks (100 mg q.d. vs. placebo) 8. PP-NRS4 response at 12 weeks (100 mg q.d. vs. placebo) 9. CFB in PSAAD at 12 weeks (200 mg q.d. vs. placebo) 10. CFB in PSAAD at 12 weeks (100 mg q.d. vs. placebo) |
JADE COMPARE | 1. IGA and EASI-75 at week 12 (200 mg q.d. vs. placebo) 2. IGA and EASI-75 at week 12 (100 mg q.d. vs. placebo) Sequence A 3. PP-NRS4 response at 2 weeks (200 mg q.d. vs. placebo) 4. PP-NRS4 response at 2 weeks (100 mg q.d. vs. placebo) 5. PP-NRS4 response at 2 weeks (200 mg q.d. vs. dupilumab) 6. PP-NRS4 response at 2 weeks (100 mg q.d. vs. dupilumab) Sequence B 7. IGA at week 16 (200 mg q.d. vs. placebo) 8. EASI-75 at week 16 (200 mg q.d. vs. placebo) 9. IGA at week 16 (100 mg q.d. vs. placebo) 10. EASI-75 at week 16 (100 mg q.d. vs. placebo) |
JADE DARE | 1. PP-NRS4 response at week 2 (200 mg q.d. vs. dupilumab) 2. EASI-90 at week 4 (superiority; 200 mg q.d. vs. dupilumab) 3. EASI-90 at week 16 (noninferiority; 200 mg q.d. vs. dupilumab) 4. EASI-90 at week 16 (superiority; 200 mg q.d. vs. dupilumab) |
CFB = change from baseline; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; IGA = Investigator’s Global Assessment; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; q.d. = once daily.
Source: Clinical Study Reports.1-4,6
Power Calculation
Monotherapy Studies
The JADE MONO-1 and JADE MONO-2 trial were planned to have a total sample size of 375 patients with 2:2:1 randomization to 1 of the following groups: abrocitinib 200 mg once daily (n = 150), abrocitinib 100 mg once daily (n = 150), or placebo (n = 75). The planned sample size would provide at least 95% power to detect a difference in the IGA response rate of at least 20% between either dose of abrocitinib and placebo, assuming the placebo response rate was 6% at 12 weeks. This would also provide at least 99% power to detect a difference in the EASI-75 response rate of at least 30% between either dose of abrocitinib and placebo, assuming the placebo response rate was 15% at 12 weeks.2,3
Combination-Therapy Studies
Adults
The JADE COMPARE trial was planned to have a total sample size of 700 patients with 4:4:4:1:1 randomization to 1 of the following groups: abrocitinib 200 mg once daily (n = 200), abrocitinib 100 mg once daily (n = 200), dupilumab (n = 200), matching placebo for 16 weeks followed by abrocitinib 100 mg once daily (n = 50), and matching placebo for 16 weeks followed by abrocitinib 200 mg once daily (n = 50). The 2 placebo sequences were combined for the purposes of analyses at all visits up to and including week 16, essentially resulting in a 2:2:2:1 randomization scheme. The planned sample size would provide at least 96% power to detect a difference of at least 20% in the IGA response rate between either dose of abrocitinib and placebo, assuming the placebo response rate is 12% at 12 weeks. This would also provide at least 99% power to detect a difference of at least 30% in the EASI-75 response rate between either dose of abrocitinib and placebo, assuming the placebo response rate was 23% at 12 weeks.
The JADE DARE trial was planned to have a total sample size of 600 patients with 1:1 randomization to abrocitinib 200 mg once daily (n = 300) or dupilumab (n = 300). The proposed sample size would provide adequate power for all superiority hypotheses for the primary end points (PP-NRS4 at week 2 and EASI-90 at week 4) and the key secondary end point (EASI-90 at week 16).1
Adolescents
The JADE TEEN trial was planned to have a total sample size of 225 patients (75 patients in each of the 3 treatment groups). The planned sample size would provide at least 80% power to detect a difference of at least 20% in the IGA response rate between either dose of abrocitinib and placebo, assuming the placebo response rate was 12% at 12 weeks. This would also provide at least 96% power to detect a difference of at least 30% in the EASI-75 response rate between either dose of abrocitinib and placebo, assuming the placebo response rate was 23% at 12 weeks.5
Subgroup Analyses
Monotherapy Studies
In the JADE MONO-1 and JADE MONO-2 trials, subgroup analyses were performed for co-primary end points for the following: age group (< 18 years, ≥ 18 years; < 40 years, ≥ 40 years; < 65 years, or ≥ 65 years); sex (male or female); race (White, Black or African-American, Asian, or other); region of enrolment (US, Canada, or Australia; Europe; Asia; or Latin America); body weight (less than or equal to the median value in the FAS or above the median value); AD duration group (< 26 years or ≥ 26 years); baseline disease severity (moderate or severe); baseline EASI group (16 to 25 or > 25); baseline percentage BSA group (10 to 30, > 30 to 50, or > 50); and previous use of a systemic immunosuppressant for AD (yes or no).2,3 Of these, the subgroups of interest for CADTH’s review were: age group (< 18 years or ≥ 18 years), baseline disease severity (moderate or severe), and previous use of a systemic immunosuppressant for AD (yes or no).
Combination-Therapy Studies
Adults
In the JADE COMPARE trial, subgroup analyses were performed for co-primary end points for the following: age group (< 40 years, ≥ 40 years; < 65 years, or ≥ 65 years); sex (male or female); race (White, Black or African-American, Asian, or other); region of enrolment (US, Canada, or Australia; Europe; Asia; or Latin America); body weight (less than or equal to the median value in the FAS or above the median value); AD duration group (< 26 years or ≥ 26 years); baseline disease severity (moderate or severe); baseline EASI group (16 to 25 or > 25); baseline percentage BSA group (10 to 30, > 30 to 50, or > 50); and previous use of systemic immunosuppressants for AD (yes or no).4 Of these, the subgroups of interest for CADTH’s review were: baseline disease severity (moderate or severe) and previous use of systemic immunosuppressants for AD (yes or no).
In the JADE DARE trial, subgroup analyses were performed for co-primary end points for the following: age group (< 40 years or ≥ 40 years; < 65 years or ≥ 65 years); sex (male or female); race (White, Black or African-American, Asian, or other); region of enrolment (US, Canada, or Australia; Europe; Asia; or Latin America); body weight (< 70 kg, ≥ 70 kg to ≤ 100 kg, > 100 kg); AD duration group (< 26 years or ≥ 26 years); baseline disease severity (moderate or severe); baseline EASI group (16 to 25 or > 25); baseline percentage BSA group (10 to 30, > 30 to 50, or > 50); previous cyclosporine exposure (exposed or naive); prior AD medications (systemic drugs or topical drugs only); and baseline PP-NRS group (4 to 6 or ≥ 7). Of these, the subgroups of interest for CADTH’s review were baseline disease severity (moderate or severe) and previous use of systemic immunosuppressant for AD (yes or no).
Adolescents
In the JADE TEEN trial, subgroup analyses were performed for co-primary end points for the following: age group less than or equal to the median value in the FAS or above the median value); sex (male or female); race (White, Black or African-American, Asian, or other); region of enrolment (US, Canada, or Australia; Europe; Asia; or Latin America); AD duration group (less than or equal to the median value in the FAS or above the median value); baseline disease severity (moderate or severe); baseline EASI group (16 to 25 or > 25); baseline percentage BSA group (10 to 30, > 30 to 50, or > 50); and previous use of a systemic immunosuppressant for AD (yes or no).6 Of these, the subgroups of interest for CADTH’s review were: baseline disease severity (moderate or severe) and previous use of systemic immunosuppressant for AD (yes or no).
Analysis Populations
The analysis sets that were used to evaluate the safety and efficacy end point in the included studies are summarized in Table 25.
Data | Definition | Use |
|---|---|---|
JADE MONO-1, JADE MONO-2, JADE COMPARE, and JADE TEEN | ||
FAS | All randomized patients who received ≥ 1 dose of study treatment; analyses for binary end points that were defined based on a threshold of change from baseline were also required the baseline value to be equal to or greater than the threshold (e.g., baseline values for PP-NRS4 had to be ≥ 4) | Efficacy and PRO end points |
Per-protocol analysis set | Subset of FAS who had no major protocol violations before week 12 | Primary end points |
Safety analysis set | All patients who received ≥ 1 dose of study drug classified according to actual treatment received | Safety |
JADE DARE | ||
FAS | All randomized participants who received at least 1 dose of study intervention; participants were analyzed according to the intervention to which they were randomized | Efficacy and PRO end points |
Per-protocol analysis set | All randomized participants who received at least 1 dose of study intervention who had no major protocol violations and met the following criteria:
| Supportive analysis for key secondary end point |
Safety analysis set | All patients who received ≥ 1 dose of study drug classified according to actual treatment received | Safety |
FAS = full analysis set; EASI = Eczema Area and Severity Index; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; PRO = patient-reported outcome.
Source: Clinical Study Reports.1-4
Results
Patient Disposition
Monotherapy Studies
Patient disposition in the JADE MONO-1 and JADE MONO-2 trials is summarized in Table 26. The proportion of patients who completed the trials ranged from 86.5% to 91% in the abrocitinib groups compared with 66.7% to 79.2% in the placebo groups. Adverse events were the most reported reason for early discontinuations in both the JADE MONO-1 and JADE MONO-2 trials. The proportion of patients who withdrew because of AEs was identical in the abrocitinib 100 mg once daily and 200 mg once daily groups in both the JADE MONO-1 trial (5.8% in both groups) and the JADE MONO-2 trial (3.2% in both groups). In both studies, a numerical higher proportion of patients withdrew from the placebo groups because of AEs (9.1% and 10.3% in JADE MONO-1 and JADE MONO-2, respectively), with worsening AD the most cited reason.
Table 26: Patient Disposition in JADE MONO-1 and JADE MONO-2
Disposition, n (%) | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
Screened | 553 | 554 | ||||
Screen failure | 150 | 163 | ||||
Not randomized | 16 | 0 | ||||
Randomized | 77 (100) | 156 (100) | 154 (100) | 78 (100) | 158 (100) | 155 (100) |
Treated | 77 (100) | 156 (100) | 154 (100) | 78 (100) | 158 (100) | 155 (100) |
Not treated | 0 | 0 | 0 | 0 | 0 | 0 |
Safety analysis set | 77 (100) | 156 (100) | 154 (100) | 78 (100) | 158 (100) | 155 (100) |
Full analysis set | 77 (100) | 156 (100) | 154 (100) | 78 (100) | 158 (100) | 155 (100) |
Per-protocol analysis set | 57 (74.0) | 132 (84.6) | 132 (85.7) | 52 (66.7) | 128 (81.0) | 130 (83.9) |
Discontinued | 16 (20.8) | 21 (13.5) | 17 (11.0) | 26 (33.3) | 21 (13.3) | 14 (9.0) |
AE | 7 (9.1) | 9 (5.8) | 9 (5.8) | 8 (10.3) | 5 (3.2) | 5 (3.2) |
Death | 0 | 0 | 0 | 0 | 1 (0.6) | 0 |
Lack of efficacy | 2 (2.6) | 1 (0.6) | 0 | 7 (9.0) | 5 (3.2) | 4 (2.6) |
Lost to follow-up | 1 (1.3) | 2 (1.3) | 1 (0.6) | 1 (1.3) | 1 (0.6) | 1 (0.6) |
Protocol deviation | 1 (1.3) | 2 (1.3) | 2 (1.3) | 1 (1.3) | 1 (0.6) | 1 (0.6) |
Withdrawal by patient | 4 (5.2) | 5 (3.2) | 3 (1.9) | 9 (11.5) | 6 (3.8) | 1 (0.6) |
Medication error without AE | 1 (1.3) | 0 | 0 | 0 | 0 | 0 |
Withdrawal by parent or guardian | 0 | 0 | 1 (0.6) | 0 | 0 | 0 |
Other | 0 | 2 (1.3) | 1 (0.6) | 0 | 2 (1.3) | 2 (1.3) |
Completed | 61 (79.2) | 135 (86.5) | 137 (89.0) | 52 (66.7) | 137 (86.7) | 141 (91.0) |
AE = adverse event; q.d. = once daily
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Patient disposition in the JADE COMPARE and JADE TEEN trials is summarized in Table 27.
Adults
A total of 1,234 patients were screened for enrolment in the JADE COMPARE trial and 838 were randomized. The proportions of patients who completed the trial were 89.3% in the placebo group, 91.2% in the abrocitinib 100 mg once daily group, 92.0% in the abrocitinib 200 mg once daily group, and 92.1% in the dupilumab group. Adverse events and withdrawals by the patients were the most common reasons for discontinuation from the JADE COMPARE trial.4 A total of 940 patients were screened for enrolment in the JADE DARE trial and 727 were randomized. The proportion of patients who completed the trial was 90.3% in the abrocitinib 200 mg once daily group and 91.5% in the dupilumab group. Adverse events and withdrawals by the patients were the most common reasons for discontinuation from JADE DARE.4
Adolescents
A total of 408 patients were screened for enrolment in the JADE TEEN trial and 287 were randomized. The proportions of patients who completed the trials were 93.8% in the placebo group, 96.8% in the abrocitinib 100 mg once daily group, and 96.8% in the abrocitinib 200 mg once daily group. Adverse events and losses to follow-up were the most common reasons for discontinuation from the JADE TEEN trial.5
Table 27: Patient Disposition in JADE COMPARE, JADE DARE, and JADE TEEN
Disposition, n (%) | JADE COMPARE | JADE DARE | JADE TEEN | ||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | DUP 300 mg q.2.w. (N = 243) | Abrocitinib 200 mg q.d. (N = 362) | DUP 300 mg q.2.w. (N = 365) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
Screened | 1,234 | 940 | 408 | ||||||
Screen Failure | 394 | 213 | 121 | ||||||
Not Randomized | 2 | 0 | 0 | ||||||
Randomized | 131 (100) | 238 (100) | 226 (100) | 243 (100) | 362 (100.0) | 365 (100.0) | 96 (100) | 95 (100) | 96 (100) |
Treated | 131 (100) | 238 (100) | 226 (100) | 242 (99.6) | 362 (100.0) | 365 (100.0) | 96 (100) | 95 (100) | 94 (97.9) |
Not treated | 0 | 0 | 0 | 1 (0.4) | 0 (0.0) | 0 (0.0) | 0 | 0 | 2 (2.1) |
Safety analysis set | 131 (100) | 238 (100) | 226 (100) | 242 (99.6) | 362 (100.0) | 365 (100.0) | 96 (100) | 95 (100) | 94 (97.9) |
Full analysis set | 131 (100) | 238 (100) | 226 (100) | 242 (99.6) | 362 (100.0) | 365 (100.0) | 96 (100) | 95 (100) | 94 (97.9) |
PP analysis set | 93 (71.0) | 174 (73.1) | 161 (71.2) | 172 (70.8) | 320 (88.4) | 337 (92.3) | 70 (72.9) | 73 (76.8) | 78 (81.3) |
Discontinued | 14 (10.7) | 21 (8.8) | 18 (8.0) | 19 (7.9) | 35 (9.7) | 31 (8.5) | 6 (6.3) | 3 (3.2) | 3 (3.2) |
AE | 5 (3.8) | 5 (2.1) | 8 (3.5) | 6 (2.5) | 10 (2.8) | 9 (2.5) | 2 (2.1) | 1 (1.1) | 2 (2.1) |
Death | 0 | 0 | 0 | 0 | 2 (0.6) | 0 (0.0) | 0 | 0 | 0 |
Lack of efficacy | 0 | 1 (0.4) | 0 | 1 (0.4) | 2 (0.6) | 0 (0.0) | 0 | 0 | 0 |
Lost to follow-up | 1 (0.8) | 2 (0.8) | 1 (0.4) | 2 (0.8) | 2 (0.6) | 4 (1.1) | 2 (2.1) | 1 (1.1) | 0 |
Pregnancy | 0 | 0 | 1 (0.4) | 1 (0.4) | 0 | 0 | 0 | 0 | 0 |
Protocol deviation | 2 (1.5) | 2 (0.8) | 2 (0.9) | 1 (0.4) | 4 (1.1) | 3 (0.8) | 0 | 0 | 1 (1.1) |
Withdrawal by patient | 5 (3.8) | 9 (3.8) | 3 (1.3) | 6 (2.5) | 11 (3.0) | 11 (3.0) | 0 | 0 | 0 |
Withdrawal by parent or guardian | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) | 1 (1.1) | 0 |
Medication error without AE | 0 | 1 (0.4) | 1 (0.4) | 0 | 1 (0.3) | 0 | 0 | 0 | 0 |
Other | 1 (0.8) | 1 (0.4) | 2 (0.9) | 2 (0.8) | 3 (0.8) | 4 (1.1) | 1 (1.0) | 0 | 0 |
Ongoing or completed | 117 (89.3) | 217 (91.2) | 208 (92.0) | 223 (92.1) | 327 (90.3) | 334 (91.5) | 90 (93.8) | 92 (96.8) | 91 (96.8) |
AE = adverse event; DUP = dupilumab; PP = per-protocol; q.2.w. = every 2 weeks; q.d. = once daily.
Source: Clinical Study Reports.1,4,5
Withdrawal Study
Patient disposition in the JADE REGIMEN trials is summarized in Figure 8. A total of 1,797 patients were screened for eligibility and 1,233 (68.6%) received at least 1 dose of 200 mg abrocitinib in the open-label induction phase. A total of 435 patients were discontinued from the open-label phase of the study (35.2%), with 315 patients (25.6%) failing to demonstrate a response at week 12. A total of 798 patients were randomized in the maintenance phase (64.7% of those who were treated in the open-label phase).
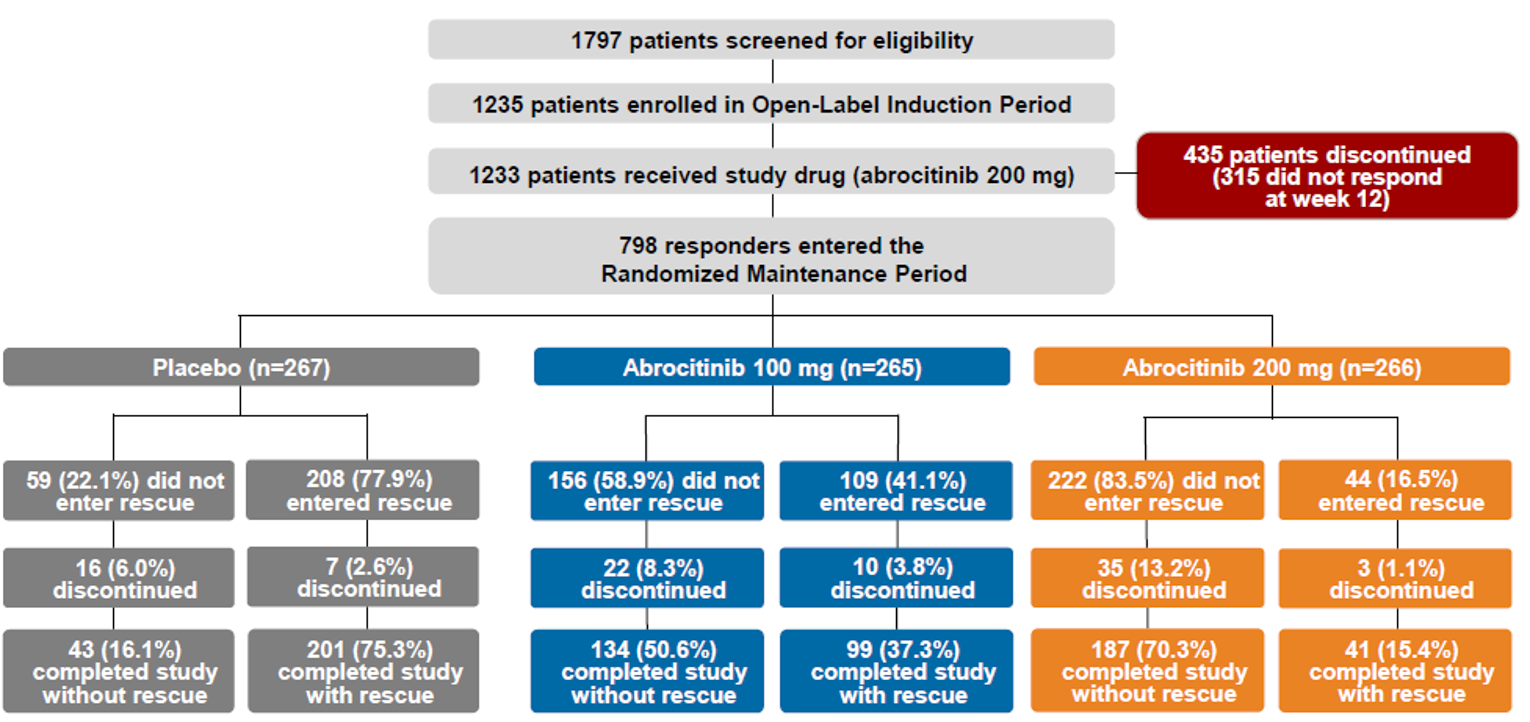
Exposure to Study Treatments
Exposure to the study treatments is summarized in Table 28 for the monotherapy studies (JADE MONO-1 and JADE MONO-2) and in Table 29 for the combination-therapy studies (JADE COMPARE, JADE DARE, and JADE TEEN).
Monotherapy Studies
The median exposure time to the study drugs was similar across the treatment groups of the JADE MONO-1 trial (range = 84 to 85 days) and the JADE MONO-2 trial (range = 82 to 84 days).2,3 The mean exposure was lower in the placebo groups of both trials (71.7 days and 66.3 days in JADE MONO-1 and JADE MONO-2, respectively) compared with the abrocitinib 100 mg once daily groups (77.7 days and 77.3 days) and abrocitinib 200 mg once daily groups (78.2 days and 80.1 days).2,3 The sponsor noted that the difference was due to fewer placebo-treated patients completing the full 12-week double-blind treatment period.2
Table 28: Summary of Exposure in JADE MONO-1 and JADE MONO-2 (Safety Analysis Set)
Exposure | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
Duration of treatment (days) | ||||||
Median (range) | 84 (6, 100) | 85 (1, 96) | 85 (1, 91) | 82 (5, 92) | 84 (7, 93) | 84 (2, 93) |
Mean (SD) | 71.7 (24.5) | 77.7 (19.6) | 78.2 (18.7) | 66.3 (27.8) | 77.3 (18.8) | 80.1 (15.2) |
Exposure category, n (%) | ||||||
< 1 week | 1 (1.3) | 3 (1.9) | 2 (1.3) | 2 (2.6) | 0 | 1 (0.6) |
≥ 1 to < 4 weeks | 7 (9.1) | 6 (3.8) | 6 (3.9) | 11 (14.1) | 8 (5.1) | 5 (3.2) |
≥ 4 to < 8 weeks | 8 (10.4) | 7 (4.5) | 7 (4.5) | 11 (14.1) | 9 (5.7) | 4 (2.6) |
≥ 8 to < 10 weeks | NR | NR | NR | 1 (1.3) | 2 (1.3) | 3 (1.9) |
≥ 8 to < 12 weeks | 22 (28.6) | 37 (23.7) | 40 (26.0) | NR | NR | NR |
≥ 10 weeks | NR | NR | NR | 53 (67.9) | 139 (88.0) | 142 (91.6) |
≥ 12 weeks | 39 (50.6) | 103 (66.0) | 99 (64.3) | NR | NR | NR |
NR = not reported; q.d. = once daily; SD = standard deviation.
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
In the JADE COMPARE trial, the median exposure time to the study drugs was similar across all 4 treatment groups (range = 111 to 112 days).4 In the JADE TEEN trial, the median exposure time to the study treatments was similar across all 3 treatment groups (range = 84 to 85 days).5 In the JADE DARE trial, the median exposure time to the study treatments was similar across the abrocitinib and 200 mg once gaily and dupilumab every 2 weeks groups (167 and 171 days, respectively).
Table 29: Summary of Exposure in JADE COMPARE, JADE DARE, and JADE TEEN (Safety Analysis Set)
Exposure | JADE COMPARE | JADE DARE | JADE TEEN | ||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 242) | Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
Duration of treatment (days) | |||||||||
Median (range) | 111 (1 to 119) | 111 (1 to 128) | 112 (5 to 130) | 112 (5 to 121) | 166.6 (39.5) | 171.2 (27.9) | 84 (43 to 105) | 85 (28 to 105) | 84 (8 to 113) |
Mean (SD) | 103.0 (23.8) | 104.9 (21.0) | 106.3 (18.5) | 104.6 (21.4) | 180 (2 to 230) | 180 (2 to 194) | 82.2 (8.3) | 82.4 (10.7) | 81.3 (13.7) |
Exposure category, n (%) | |||||||||
< 1 week | 1 (0.8) | 3 (1.3) | 2 (0.9) | 1 (0.4) | 3 (0.8) | 1 (0.3) | 0 | 0 | 0 |
≥ 1 to < 4 weeks | 4 (3.1) | 4 (1.7) | 3 (1.3) | 8 (3.3) | 9 (2.5) | 3 (0.8) | 0 | 0 | 3 (3.2) |
≥ 4 to < 8 weeks | 5 (3.8) | 4 (1.7) | 3 (1.3) | 4 (1.7) | 9 (2.5) | 4 (1.1) | 2 (2.1) | 3 (3.2) | 0 |
≥ 8 to < 10 weeks | 0 | 4 (1.7) | 3 (1.3) | 3 (1.2) | NR | NR | 5 (5.2) | 4 (4.2) | 2 (2.1) |
≥ 8 to < 12 weeks | NR | NR | NR | NR | 4 (1.1) | 2 (0.5) | NR | NR | NR |
≥ 10 weeks | NR | NR | NR | NR | NR | NR | 89 (92.7) | 88 (92.6) | 89 (94.7) |
≥ 10 to < 12 weeks | 2 (1.5) | 2 (0.8) | 3 (1.3) | 4 (1.7) | NR | NR | NR | NR | NR |
≥ 12 to < 14 weeks | 6 (4.6) | 9 (3.8) | 4 (1.8) | 8 (3.3) | NR | NR | NA | ||
≥ 12 to < 16 weeks | NR | NR | NR | NR | 2 (0.6) | 9 (2.5) | |||
≥ 14 weeks | 113 (86.3) | 212 (89.1) | 208 (92.0) | 214 (88.4) | NR | NR | |||
≥ 16 to < 20 weeks | NR | NR | NR | NR | 7 (1.9) | 4 (1.1) | |||
≥ 20 to < 24 weeks | NR | NR | NR | NR | 34 (9.4) | 41 (11.2) | |||
≥ 24 weeks | NR | NR | NR | NR | 294 (81.2) | 301 (82.5) | |||
NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.d. = once daily; SD = standard deviation.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Appendix 3 provides detailed efficacy data.
IGA Response
Monotherapy Studies
Table 30 provides a summary of IGA response in the JADE MONO-1 and JADE MONO-2 trials. A statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an IGA response at week 12 compared with placebo in the JADE MONO-1 trial (15.8% [95% CI, 6.8 to 24.8] and 36.0% [95% CI, 26.2 to 45.7], respectively) and in the JADE MONO-2 trial (19.3% [95% CI, 9.6 to 29.0] and 28.7% [95% CI, 18.6 to 38.8], respectively).2,3 Results in the analyses performed using the per-protocol analysis set demonstrated a statistically significantly greater proportion of patients with an IGA response in the abrocitinib 100 mg group (19.9% [95% CI, 9.9 to 29.9] and 18.8% [95% CI, 6.8 to 30.8]) and abrocitinib 200 mg once daily group (39.8% [95% CI, 29.2 to 50.4] and 26.7% [95% CI, 14.4 to 38.9]) for the JADE MONO-1 and JADE MONO-2 trials, respectively. The results in the tipping point (TP) sensitivity analysis were similar to those of the primary analyses.
The analyses conducted at earlier time points demonstrated that a statistically significantly greater proportion of patients in the abrocitinib 200 mg once daily groups achieved an IGA response compared with the placebo group at 2 weeks, 4 weeks, and 8 weeks in both the JADE MONO-1 and JADE MONO-2 trials. The proportion of patients who demonstrated an IGA response in the abrocitinib 100 mg once daily group was statistically significantly greater compared with placebo at all time points in the JADE MONO-2 trial and at week 8 in the JADE MONO-1 trial.2,3
Subgroup analyses are summarized in Table 95. For abrocitinib 100 mg once daily, subgroup analyses found IGA responses based on baseline disease severity were 15.0 (95% CI, 2.1 to 27.8) and 18.4 (95% CI, 6.0 to 30.9) for patients with moderate AD and 17.1% (95% CI, 5.4 to 28.7) and 20.6% (95% CI, 6.5 to 34.8) for severe AD in the JADE MONO-1 and JADE MONO-2 trials, respectively. For abrocitinib 200 mg once daily, subgroup analyses found IGA responses based on baseline disease severity were 41.6% (95% CI, 27.9 to 55.4) and 30.7% (95% CI, 17.8 to 43.6) for patients with moderate AD and 27.4% (95% CI, 14.4 to 40.5) and 24.7% (95% CI, 10.1 to 39.4) for those with severe AD in the JADE MONO-1 and JADE MONO-2 trials, respectively. Response rates for patients with prior use of a systemic immunosuppressant for AD were 9.1% (95% CI, −1.2 to 19.4) and 20.4% (95% CI, 6.7 to 34.1) for abrocitinib 100 mg once daily and 36.2% (95% CI, 22.7 to 49.7) and 26.9% (95% CI, 12.1 to 41.6) for abrocitinib 200 mg once daily in the JADE MONO-1 and JADE MONO-2 trials, respectively. For those without prior use of a systemic immunosuppressant for AD, the response rates were 22.2% (95% CI, 7.6 to 36.9) and 18.7% (95% CI, 5.6 to 31.8) with 100 mg once daily and 34.8% (95% CI, 20.0 to 49.5) and 30.2% (95% CI, 16.8 to 43.6) for abrocitinib 200 mg once daily in the JADE MONO-1 and JADE MONO-2 trials, respectively.2,3
Compared with the abrocitinib 100 mg once daily dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an IGA response at 2 weeks (5.9; 95% CI, 0.2 to 11.6), 4 weeks (16.4; 95% CI, 8.0 to 24.9), 8 weeks (15.4; 95% CI, 5.7 to 25.1), and 12 weeks (20.0; 95% CI, 9.9 to 30.1) in the JADE MONO-1 trial and at 2 weeks (9.3; 95% CI, 2.7 to 15.8), 4 weeks (19.0; 95% CI, 9.9 to 28.0), and 8 weeks (15.2; 95% CI, 5.6 to 24.9) in the JADE MONO-2 trial.2,3
Table 30: IGA Response in JADE MONO-1 and JADE MONO-2 (Full Analysis Set)
Response | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
IGA response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | ||||||
Patients in analysis | 40 | 78 | 68 | 31 | 67 | 60 |
Responders, n (%) | 2 (5.0) | 11 (14.1) | 28 (41.2) | 2 (6.5) | 18 (26.9) | 20 (33.3) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 9.1 (−1.2 to 19.4) | 36.2 (22.7 to 49.7) | Reference | 20.4 (6.7 to 34.1) | 26.9 (12.1 to 41.6) |
2-sided P value | Reference | NR | NR | Reference | NR | NR |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 27.1 (13.1 to 41.1) | NA | 6.5 (−9.5 to 22.4) | ||
IGA response at week 12 (primary end point) | ||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 155 |
Responders, n (%) | 6 (7.9) | 37 (23.7) | 67 (43.8) | 7 (9.1) | 44 (28.4) | 59 (38.1) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 15.8 (6.8 to 24.8) | 36.0 (26.2 to 45.7) | Reference | 19.3 (9.6 to 29.0) | 28.7 (18.6 to 38.8) |
2-sided P value | Reference | 0.0037 | < 0.0001 | Reference | 0.0008 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 20.0 (9.9 to 30.1) | NA | 9.7 (−0.7 to 20.0) | ||
IGA response at week 2 (secondary end point) | ||||||
Patients in analysis | 77 | 155 | 154 | 76 | 157 | 152 |
Responders, n (%) | 0 | 6 (3.9) | 15 (9.7) | 0 | 8 (5.1) | 22 (14.5) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 3.9 (−0.7 to 8.5) | 9.8 (4.0 to 15.7) | Reference | 5.1 (0.2 to 10.0) | 14.2 (7.8 to 20.5) |
2-sided P value | Reference | 0.0802 | 0.0045 | Reference | 0.0459 | 0.0005 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 5.9 (0.2 to 11.6) | NA | 9.3 (2.7 to 15.8) | ||
IGA response at week 4 (secondary end point) | ||||||
Patients in analysis | 76 | 152 | 152 | 77 | 155 | 153 |
Responders, n (%) | 4 (5.3) | 16 (10.5) | 41 (27.0) | 1 (1.3) | 22 (14.2) | 51 (33.3) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 5.2 (−1.9 to 12.4) | 21.7 (13.0 to 30.5) | Reference | 12.9 (6.3 to 19.4) | 31.8 (23.6 to 39.9) |
2-sided P value | Reference | 0.1888 | < 0.0001 | Reference | 0.0019 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 16.4 (8.0 to 24.9) | NA | 19.0 (9.9 to 28.0) | ||
IGA response at week 8 (secondary end point) | ||||||
Patients in analysis | 75 | 153 | 154 | 78 | 157 | 154 |
Responders, n (%) | 5 (6.7) | 31 (20.3) | 55 (35.7) | 8 (10.3) | 35 (22.3) | 58 (37.7) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 13.8 (5.2 to 22.4) | 29.3 (19.8 to 38.7) | Reference | 11.9 (2.4 to 21.4) | 26.9 (17.0 to 36.9) |
2-sided P value | Reference | 0.0071 | < 0.0001 | Reference | 0.0246 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 15.4 (5.7 to 25.1) | NA | 15.2 (5.6 to 24.9) | ||
CI = confidence interval; NA = not applicable; NR = not reported; IGA = Investigator’s Global Assessment; q.d. = once daily; vs. = versus.
Note: The estimate and CI for differences were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. P values were calculated using the Cochran-Mantel-Haenszel method adjusted by randomization strata (baseline disease severity and age category).
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 31 provides a summary of IGA response in the JADE COMPARE trial. For the primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an IGA response at week 12 compared with the placebo group (23.1% [95% CI, 14.7 to 31.4] and 34.8% [95% CI, 26.1 to 43.5], respectively). Results were similar in the analyses performed using the per-protocol analysis set (24.2% [95% CI, 14.1 to 34.2] and 33.1% [95% CI, 22.7 to 43.4]) for the abrocitinib 100 mg once daily and 200 mg once daily groups, respectively) and in the TP sensitivity analysis. Compared with placebo, the proportion of patients with an IGA response was greater in both abrocitinib groups at all other time points (2 weeks, 4 weeks, 8 weeks, and 16 weeks). Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an IGA response at 8 weeks (15.3% [95% CI, 6.5 to 24.2]), 12 weeks (12.1% [95% CI, 3.2 to 21.1]), and 16 weeks (13.1% [95% CI, 4.2 to 22.1]).4
Subgroup analyses are summarized in Table 95. The analyses stratified based on baseline disease severity demonstrated results similar to those of the primary analysis for abrocitinib 100 mg once daily for patients with moderate AD (23.3% [95% CI, 11.7 to 34.8]) and severe AD (22.7% [95% CI, 12.4 to 33.0]). For abrocitinib 200 mg once daily, subgroup analyses found IGA responses based on baseline disease were 23.3% (95% CI, 11.7 to 34.8) for patients with moderate AD and 22.7% (95% CI, 12.4 to 33.0) for those with severe AD. Response rates for patients with prior use of a systemic immunosuppressant for AD were 27.5% (95% CI, 14.4 to 40.6) for abrocitinib 100 mg once daily and 43.9 (95% CI, 30.7 to 57.1) for abrocitinib 200 mg once daily. For those without prior therapy prior use of a systemic immunosuppressant for AD, the response rates were 19.7% (95% CI, 8.4 to 30.9) and 27.5% (95% CI, 15.6 to 39.4) with 100 mg once daily and 200 mg once daily, respectively.
A greater proportion of dupilumab-treated patients demonstrated an IGA response at week 12 compared to placebo (22.5%; 95% CI, 14.2 to 30.9). The sponsor’s exploratory comparisons demonstrated that a similar proportion of patients achieved an IGA response with abrocitinib 100 mg once daily versus dupilumab (0.5%; 95% CI, −8.0 to 9.1) and a greater proportion of patients treated with abrocitinib 200 mg once daily achieved an IGA response versus dupilumab (12.4%; 95% CI, 3.5 to 21.3).4
Table 31: IGA Response in JADE COMPARE (Full Analysis Set)
Response | JADE COMPARE | |||
|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | |
IGA response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | ||||
Patients in analysis | 47 | 97 | 99 | 112 |
Responders, n (%) | 5 (10.6) | 37 (38.1) | 54 (54.5) | 41 (36.6) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 27.5 (14.4 to 40.6) | 43.9 (30.7 to 57.1) | 26.0 (13.4 to 38.5) |
2-sided P value | Reference | NR | NR | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 1.5 (−11.6 to 14.7) | 17.9 (4.7 to 31.2) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 16.4 (2.6 to 30.2) | NA | |
IGA response at week 12 (primary end point) | ||||
Patients in analysis | 129 | 235 | 219 | 241 |
Responders, n (%) | 18 (14.0) | 86 (36.6) | 106 (48.4) | 88 (36.5) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 23.1 (14.7 to 31.4) | 34.8 (26.1 to 43.5) | 22.5 (14.2 to 30.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 0.5 (−8.0 to 9.1) | 12.4 (3.5 to 21.3) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 12.1 (3.2 to 21.1) | NA | |
IGA response at week 16 (key secondary end point) | ||||
Patients in analysis | 124 | 230 | 221 | 232 |
Responders, n (%) | 16 (12.9) | 80 (34.8) | 105 (47.5) | 90 (38.8) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 22.1 (13.7 to 30.5) | 35.0 (26.3 to 43.7) | 25.6 (17.1 to 34.1) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −3.5 (−12.2 to 5.2) | 9.4 (0.4 to 18.5) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 13.1 (4.2 to 22.1) | NA | |
IGA response at week 2 (secondary end point) | ||||
Patients in analysis | 128 | 230 | 223 | 236 |
Responders, n (%) | 8 (6.3) | 35 (15.2) | 41 (18.4) | 11 (4.7) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 9.3 (3.0 to 15.6) | 13.0 (6.4 to 19.6) | −1.6 (−6.7 to 3.5) |
2-sided P value | Reference | 0.0093 | 0.0007 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 10.9 (5.5 to 16.2) | 14.5 (8.8 to 20.2) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 3.5 (−3.3 to 10.3) | NA | |
IGA response at week 4 (secondary end point) | ||||
Patients in analysis | 129 | 234 | 223 | 238 |
Responders, n (%) | 8 (6.2) | 59 (25.2) | 70 (31.4) | 45 (18.9) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 19.4 (12.6 to 26.3) | 25.7 (18.3 to 33.0) | 12.7 (6.3 to 19.1) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 6.8 (−0.5 to 14.1) | 13.2 (5.4 to 20.9) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 6.6 (−1.5 to 14.7) | NA | |
IGA response at week 8 (secondary end point) | ||||
Patients in analysis | 129 | 232 | 225 | 239 |
Responders, n (%) | 13 (10.1) | 83 (35.8) | 114 (50.7) | 68 (28.5) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 26.2 (18.3 to 34.1) | 41.1 (32.9 to 49.4) | 18.3 (10.7 to 25.9) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 8.0 (−0.3 to 16.2) | 23.0 (14.4 to 31.6) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 15.3 (6.5 to 24.2) | NA | |
CI = confidence interval; IGA = Investigator’s Global Assessment; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
Note: The difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. The CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). The P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
Source: Clinical Study Report.4
Active-Controlled Trial in Adults
Table 32 shows the results for IGA response at 26 weeks in the JADE DARE trial. There was no statistically significantly difference between the abrocitinib 200 mg once daily and dupilumab every 2 weeks groups (4.5%; 95% CI, −2.8 to 11.8).1 There were no subgroup data analyses based on prior exposure to systemic AD therapy.
Table 32: IGA Response in JADE DARE (Full Analysis Set)
Response | JADE DARE | |
|---|---|---|
Abrocitinib 200 mg q.d. (N = 362) | DUP 300 mg q.2.w. (N = 365) | |
IGA response at week 26 | ||
Patients in analysis | 347 | 362 |
Responders, n (%) | 193 (55.6) | 185 (51.1) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 4.5 (−2.8 to 11.8) | |
2-sided P value | 0.2293 | |
CI = confidence interval; IGA = Investigator’s Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily
Note: The difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. The CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). The P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
Source: Clinical Study Report.1
Adolescents
Table 33 provides a summary of IGA response in the JADE TEEN trial. For the primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an IGA response at week 12 compared with the placebo group (16.7% [95% CI, 3.5 to 29.9]) and 20.6% [95% CI, 7.3 to 33.9]), respectively). Results in the analyses performed using the per-protocol analysis set demonstrated a statistically significantly greater proportion of patients with an IGA response in the abrocitinib 200 mg group (17.8%; 95% CI, 2.8 to 32.9; P = 0.0238), but not in the abrocitinib 100 mg once daily group (15.1%; 95% CI, −0.3 to 30.5; P = 0.0586). The results of the TP sensitivity analysis were similar to those of the primary analyses. Compared with placebo, the proportion of patients with an IGA response was greater in the abrocitinib 200 mg group at all other time points (2 weeks, 4 weeks, and 8 weeks) and was greater in the abrocitinib 100 mg group at 4 weeks and 8 weeks. Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an IGA response at 4 weeks (18.2%; 95% CI, 5.8 to 30.7) and 8 weeks (17.7%; 95% CI, 3.8 to 31.6).5
Subgroup analyses are summarized in Table 95. For abrocitinib 100 mg once daily, subgroup analyses found IGA responses based on baseline disease severity were 15.4% (95% CI, −2.8 to 33.6) for patients with moderate AD and 18.6% (95% CI, 0.0 to 37.2) for severe AD. For abrocitinib 200 mg once daily, subgroup analyses based on baseline disease severity found IGA responses were 18.1% (95% CI, 0.5 to 35.7) for patients with moderate AD and 24.7% (95% CI, 4.9 to 44.5) for those with severe AD. Response rates for patients with prior use of a systemic immunosuppressant for AD were 18.6 (95% CI, −1.7 to 38.9) for abrocitinib 100 mg once daily and 41.7% (95% CI, 18.0 to 65.3) for abrocitinib 200 mg once daily. For those without prior use of a systemic immunosuppressant for AD, the response rates were 17.6% (95% CI, 1.3 to 34.0) and 15.1% (95% CI, −0.7 to 30.9) with 100 mg once daily and 200 mg once daily, respectively.5
Table 33: IGA Response in JADE TEEN (Full Analysis Set)
Response | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) |
|---|---|---|---|
IGA response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | |||
Patients in analysis | 24 | 26 | 22 |
Responders, n (%) | 2 (8.3) | 7 (26.9) | 11 (50.0) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 18.6 (−1.7 to 38.9) | 41.7 (18.0 to 65.3) |
2-sided P value | Reference | NR | NR |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 23.1 (−3.9 to 50.0) | |
IGA response at week 12 (primary end point) | |||
Patients in analysis | 94 | 89 | 93 |
Responders, n (%) | 23 (24.5) | 37 (41.6) | 43 (46.2) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 16.7 (3.5 to 29.9) | 20.6 (7.3 to 33.9) |
2-sided P value | Reference | 0.0147 | 0.0030 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 3.9 (−10.4 to 18.2) | |
IGA response at week 2 (secondary end point) | |||
Patients in analysis | 91 | 92 | 94 |
Responders, n (%) | 1 (1.1) | 6 (6.5) | 12 (12.8) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 5.4 (−0.3 to 11.0) | 11.3 (4.2 to 18.3) |
2-sided P value | Reference | 0.0586 | 0.0027 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 5.8 (−2.5 to 14.1) | |
IGA response at week 4 (secondary end point) | |||
Patients in analysis | 96 | 92 | 94 |
Responders, n (%) | 3 (3.1) | 18 (19.6) | 36 (38.3) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 16.3 (7.4 to 25.1) | 34.3 (24.2 to 44.5) |
2-sided P value | Reference | 0.0004 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 18.2 (5.8 to 30.7) | |
IGA response at week 8 (secondary end point) | |||
Patients in analysis | 94 | 91 | 92 |
Responders, n (%) | 15 (16.0) | 28 (30.8) | 45 (48.9) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 14.8 (3.0 to 26.7) | 32.6 (20.0 to 45.1) |
2-sided P value | Reference | 0.0161 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 17.7 (3.8 to 31.6) | |
CI = confidence interval; IGA = Investigator’s Global Assessment; NA = not applicable; NR = not reported; q.d. = once daily; vs. = versus.
Source: Clinical Study Report.5
Eczema Area and Severity Index Score
Monotherapy Studies
Table 34 provides a summary of response for EASI-50, EASI-75, EASI-90, and EASI-100 in JADE MONO-1 and JADE MONO-2. For the co-primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an EASI-75 response at week 12 compared with the placebo group in the JADE MONO-1 trial (27.9% [95% CI, 17.4 to 38.3] and 51.0% [95% CI, 40.5 to 61.5], respectively) and in the JADE MONO-2 trial (33.9% [95% CI, 23.3 to 44.4] and 50.5% [95% CI, 40.0 to 60.9], respectively).2,3 Results of the analyses performed using the per-protocol analysis set demonstrated a statistically significantly greater proportion of patients with an EASI-75 response in the abrocitinib 100 mg group (34.2% [95% CI, 22.9 to 45.5] and 35.6% [95% CI, 22.6 to 48.5]) and abrocitinib 200 mg once daily group (59.2% [95% CI, 48.1 to 70.3] and 48.7% [95% CI, 35.9 to 61.4]) for the JADE MONO-1 and JADE MONO-2 trials, respectively. The results of the TP sensitivity analysis were similar to those of the primary analyses.
The analyses conducted at earlier time points demonstrated that a statistically significantly greater proportion of patients in the abrocitinib 200 mg once daily groups achieved an EASI-75 response compared with the placebo group at 2 weeks, 4 weeks, and 8 weeks in both the JADE MONO-1 and JADE MONO-2 trials. The proportion of patients who demonstrated an EASI-75 response in the abrocitinib 100 mg once daily group was statistically significantly greater compared with placebo at all time points in the JADE MONO-2 trial and at week 8 in the JADE MONO-1 trial.2,3 Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an EASI-75 response at 2 weeks, 4 weeks, 8 weeks, and 12 weeks in both the JADE MONO-1 and JADE MONO-2 trials.2,3
Subgroup analyses are summarized in Table 95. Response rates for patients with prior use of a systemic immunosuppressant for AD were 17.0% (95% CI, 2.6 to 31.4) and 30.9% (95% CI, 16.4 to 45.3) for abrocitinib 100 mg once daily and 49.3% (95% CI, 33.8 to 64.7) and 54.6% (95% CI, 39.4 to 69.7) for abrocitinib 200 mg once daily in the JADE MONO-1 and JADE MONO-2 trials, respectively. For those without prior use of a systemic immunosuppressant for AD, the response rates were 38.9% (95% CI, 23.8 to 54.0) and 37.0% (95% CI, 22.7 to 51.2) with 100 mg once daily and 52.4% (95% CI, 37.9 to 66.9) and 48.0% (95% CI, 34.2 to 61.8) 200 mg once daily in the JADE MONO-1 and JADE MONO-2 trials, respectively.2,3
In both the JADE MONO-1 and JADE MONO-2 trials, a statistically significantly greater proportion of patients in both the abrocitinib groups demonstrated an EASI-50, EASI-90, or EASI-100 response at 12 weeks.2,3
Table 34: EASI-50, EASI-75, EASI-90, and EASI-100 in JADE MONO-1 and JADE MONO-2 (FAS)
Response | JADE MONO-1 | JADE MONO-2 | |||||
|---|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | ||
EASI-75 response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | |||||||
Patients in analysis | 40 | 78 | 68 | 31 | 67 | 59 | |
Responders, n (%) | 5 (12.5) | 23 (29.5) | 42 (61.8) | 2 (6.5) | 25 (37.3) | 36 (61.0) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 17.0 (2.6 to 31.4) | 49.3 (33.8 to 64.7) | Reference | 30.9 (16.4 to 45.3) | 54.6 (39.4 to 69.7) | |
2-sided P value | Reference | NR | NR | Reference | NR | NR | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 32.3 (16.9 to 47.6) | NA | 23.7 (6.7 to 40.7) | |||
EASI-75 response at week 12 (primary end point) | |||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 154 | |
Responders, n (%) | 9 (11.8) | 62 (39.7) | 96 (62.7) | 8 (10.4) | 69 (44.5) | 94 (61.0) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 27.9 (17.4 to 38.3) | 51.0 (40.5 to 61.5) | Reference | 33.9 (23.3 to 44.4) | 50.5 (40.0 to 60.9) | |
2-sided P value | Reference | < 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 23.0 (12.3 to 33.7) | NA | 16.5 (5.6 to 27.4) | |||
EASI-75 response at week 2 (secondary end point) | |||||||
Patients in analysis | 77 | 155 | 154 | 76 | 157 | 152 | |
Responders, n (%) | 3 (3.9) | 16 (10.3) | 37 (24.0) | 1 (1.3) | 16 (10.2) | 37 (24.3) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 6.5 (−0.3 to 13.3) | 20.3 (12.0 to 28.6) | Reference | 8.8 (2.8 to 14.9) | 22.7 (15.0 to 30.3) | |
2-sided P value | Reference | 0.0869 | 0.0001 | Reference | 0.0150 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 13.8 (5.6 to 22.0) | NA | 14.0 (5.8 to 22.2) | |||
EASI-75 response at week 4 (secondary end point) | |||||||
Patients in analysis | 76 | 152 | 152 | 77 | 155 | 153 | |
Responders, n (%) | 11 (14.5) | 42 (27.6) | 72 (47.4) | 5 (6.5) | 41 (26.5) | 78 (51.0) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 13.1 (2.6 to 23.6) | 33.0 (21.7 to 44.2) | Reference | 20.0 (10.9 to 29.0) | 44.3 (34.8 to 53.8) | |
2-sided P value | Reference | 0.0259 | < 0.0001 | Reference | 0.0004 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 19.8 (9.4 to 30.3) | NA | 24.3 (14.2 to 34.5) | |||
EASI-75 response at week 8 (secondary end point) | |||||||
Patients in analysis | 75 | 154 | 154 | 78 | 157 | 154 | |
Responders, n (%) | 10 (13.3) | 59 (38.3) | 89 (57.8) | 10 (12.8) | 68 (43.3) | 93 (60.4) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 25.0 (14.2 to 35.8) | 44.6 (33.6 to 55.6) | Reference | 30.4 (19.7 to 41.2) | 47.4 (36.8 to 58.0) | |
2-sided P value | Reference | 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 19.6 (8.8 to 30.4) | NA | 17.1 (6.3 to 27.8) | |||
EASI-50 response at week 12 | |||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 154 | |
Responders, n (%) | 17 (22.4) | 90 (57.7) | 116 (75.8) | 15 (19.5) | 106 (68.4) | 123 (79.9) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 35.3 (23.3 to 47.4) | 53.5 (42.0 to 65.0) | Reference | 48.7 (37.2 to 60.1) | 60.1 (49.1 to 71.0) | |
2-sided P value | Reference | < 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 18.1 (7.9 to 28.4) | NA | 11.5 (2.0 to 21.0) | |||
EASI-90 response at week 12 | |||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 154 | |
Estimated response rate | 4 (5.3) | 29 (18.6) | 59 (38.6) | 3 (3.9) | 37 (23.9) | 58 (37.7) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 13.3 (5.4 to 21.2) | 33.4 (24.3 to 42.5) | Reference | 20.1 (11.9 to 28.3) | 33.5 (24.6 to 42.5) | |
2-sided P value | Reference | 0.0066 | < 0.0001 | Reference | 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 20.1 (10.3 to 29.8) | NA | 13.7 (3.6 to 23.7) | |||
EASI-100 response at week 12 | |||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 154 | |
Estimated response rate | 0 | 10 (6.4) | 20 (13.1) | 0 | 8 (5.2) | 11 (7.1) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 6.4 (1.2 to 11.6) | 13.1 (6.7 to 19.4) | Reference | 5.2 (0.3 to 10.1) | 7.0 (1.8 to 12.2) | |
2-sided P value | Reference | 0.0255 | 0.0010 | Reference | 0.0419 | 0.0180 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 6.6 (0.1 to 13.2) | NA | 1.9 (−3.6 to 7.4) | |||
CI = confidence interval; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; FAS = full analysis set; NA = not applicable; NR = not reported; q.d. = once daily; vs. = versus
Note: The difference and CI were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. The P values were calculated using the Cochran-Mantel-Haenszel method adjusted by randomization strata (baseline disease severity and age category).
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 35 provides a summary of response for EASI-50, EASI-75, EASI-90, and EASI-100 in the JADE COMPARE trial. For the co-primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an EASI-75 response at week 12 compared with the placebo group (31.9% [95% CI, 22.2 to 41.6] and 43.2% [95% CI, 33.7 to 52.7], respectively). Results were similar in the analyses performed using the per-protocol analysis set (33.1% [95% CI, 21.8 to 44.4] and 43.5% [95% CI, 32.5 to 54.6] for the abrocitinib 100 mg once daily and 200 mg once daily groups, respectively) and in the TP sensitivity analysis. Compared with placebo, the proportion of patients with an EASI-75 response was greater in both abrocitinib groups at all other time points (2 weeks, 4 weeks, 8 weeks, and 16 weeks). Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an EASI-75 response at 4 weeks (13.0%; 95% CI, 4.0 to 22.1), 8 weeks (12.4%; 95% CI, 3.6 to 21.3), 12 weeks (11.5%; 95% CI, 2.8 to 20.2), and 16 weeks (10.7%; 95% CI, 2.0 to 19.4).4
Subgroup analyses are summarized in Table 95. For abrocitinib 100 mg once daily, subgroup analyses found EASI-75 responses based on baseline disease severity were 26.0% (95% CI, 13.3 to 38.8) for patients with moderate AD and 43.1% (95% CI, 28.8 to 57.5) for severe AD. For abrocitinib 200 mg once daily, subgroup analyses based on baseline disease severity found EASI-75 responses were 30.5% (95% CI, 17.6 to 43.4) for patients with moderate AD and 66.3% (95% CI, 53.3 to 79.3) for those with severe AD. Response rates for patients with prior use of a systemic immunosuppressant for AD were 49.1% (95% CI, 35.5 to 62.7) for abrocitinib 100 mg once daily and 63.0% (95% CI, 50.3 to 75.7) for abrocitinib 200 mg once daily. For those without prior use of a systemic immunosuppressant for AD, the response rates were 21.2% (95% CI, 7.9 to 34.4) and 30.5% (95% CI, 17.1 to 43.9) with 100 mg once daily and 200 mg once daily to respectively.
A greater proportion of dupilumab-treated patients demonstrated an EASI-75 response at week 12 compared with placebo (30.9%; 95% CI, 21.2 to 40.6). The sponsor’s exploratory comparisons demonstrated that a similar proportion of patients achieved an EASI-75 response with abrocitinib 100 mg once daily versus dupilumab at 12 weeks (0.8%; 95% CI, −8.1 to 9.6) and at 16 weeks (−5.1; 95% CI, −13.9 to 3.7). A greater proportion of patients treated with abrocitinib 200 mg once daily achieved an EASI-75 response versus dupilumab at 12 weeks (12.0%; 95% CI, 3.3 to 20), but not at 16 weeks (5.5%; 95% CI, −3.1 to 14.1).4 As shown in Figure 9, the EASI response rate in the dupilumab group was greater at each time point in the JADE COMPARE trial; whereas those in the abrocitinib groups appeared to have plateaued by 16 weeks.
A statistically significantly greater proportion of patients in both the abrocitinib groups demonstrated an EASI-50, EASI-90, or EASI-100 response at 16 weeks.4
Table 35: EASI-50, EASI-75, EASI-90, and EASI-100 Response in JADE COMPARE (FAS)
Response | JADE COMPARE | |||
|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | |
EASI-75 response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | ||||
Patients in analysis | 47 | 97 | 99 | 112 |
Responders, n (%) | 6 (12.8) | 60 (61.9) | 75 (75.8) | 68 (60.7) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 49.1 (35.5 to 62.7) | 63.0 (50.3 to 75.7) | 47.9 (34.8 to 61.1) |
2-sided P value | Reference | NR | NR | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 1.1 (−12.1 to 14.4) | 15.0 (2.7 to 27.4) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 13.9 (1.1 to 26.7) | NA | |
EASI-75 response at week 12 (primary end point) | ||||
Patients in analysis | 129 | 235 | 219 | 241 |
Responders, n (%) | 35 (27.1) | 138 (58.7) | 154 (70.3) | 140 (58.1) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 31.9 (22.2 to 41.6) | 43.2 (33.7 to 52.7) | 30.9 (21.2 to 40.6) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 0.8 (−8.1 to 9.6) | 12.0 (3.3 to 20.7) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 11.5 (2.8 to 20.2) | NA | |
EASI-75 response at week 16 (key secondary end point) | ||||
Patients in analysis | 124 | 229 | 221 | 232 |
Responders, n (%) | 38 (30.6) | 138 (60.3) | 157 (71.0) | 152 (65.5) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 29.7 (19.5 to 39.9) | 40.4 (30.4 to 50.4) | 34.7 (24.6 to 44.8) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −5.1 (−13.9 to 3.7) | 5.5 (−3.1 to 14.1) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 10.7 (2.0 to 19.4) | NA | |
EASI-75 response at week 2 (secondary end point) | ||||
Patients in analysis | 128 | 228 | 223 | 235 |
Responders, n (%) | 14 (10.9) | 58 (25.4) | 67 (30.0) | 33 (14.0) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 15.1 (7.4 to 22.8) | 19.8 (11.9 to 27.8) | 3.1 (−3.8 to 10.0) |
2-sided P value | Reference | 0.0006 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 11.8 (4.7 to 18.9) | 16.5 (9.1 to 24.0) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 4.9 (−3.2 to 13.1) | NA | |
EASI-75 response at week 4 (secondary end point) | ||||
Patients in analysis | 128 | 233 | 223 | 238 |
Responders, n (%) | 20 (15.6) | 104 (44.6) | 128 (57.4) | 91 (38.2) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 29.4 (20.6 to 38.3) | 42.0 (33.0 to 51.0) | 22.6 (13.8 to 31.4) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. DUP | NA | 6.7 (−2.2 to 15.5) | 19.2 (10.3 to 28.2) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 13.0 (4.0 to 22.1) | NA | |
EASI-75 response at week 8 (secondary end point) | ||||
Patients in analysis | 129 | 232 | 224 | 239 |
Responders, n (%) | 24 (18.6) | 129 (55.6) | 152 (67.9) | 126 (52.7) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 37.3 (28.1 to 46.5) | 49.3 (40.2 to 58.4) | 34.1 (24.9 to 43.3) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 3.1 (−5.8 to 12.1) | 15.0 (6.2 to 23.8) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 12.4 (3.6 to 21.3) | NA | |
EASI-50 response at week 16 (secondary end point) | ||||
Patients in analysis | 124 | 229 | 221 | 232 |
Responders, n (%) | 71 (57.3) | 186 (81.2) | 193 (87.3) | 195 (84.1) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 24.1 (14.0 to 34.1) | 30.1 (20.3 to 39.8) | 26.7 (16.9 to 36.6) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −2.7 (−9.6 to 4.2) | 3.1 (−3.3 to 9.6) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 6.1 (−0.6 to 12.9) | NA | |
EASI-90 response at week 16 (secondary end point) | ||||
Patients in analysis | 124 | 229 | 221 | 232 |
Responders, n (%) | 14 (11.3) | 87 (38.0) | 108 (48.9) | 90 (38.8) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 26.8 (18.5 to 35.2) | 37.5 (28.9 to 46.0) | 27.3 (19.0 to 35.7) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −0.5 (−9.3 to 8.4) | 10.1 (1.0 to 19.2) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 10.9 (1.8 to 20.0) | NA | |
EASI-100 response at week 16 (secondary end point) | ||||
Patients in analysis | 124 | 229 | 221 | 232 |
Responders, n (%) | 5 (4.0) | 29 (12.7) | 30 (13.6) | 12 (5.2) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 8.7 (3.2 to 14.2) | 9.3 (3.7 to 15.0) | 1.1 (−3.4 to 5.6) |
2-sided P value | Reference | 0.0082 | 0.0059 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 7.7 (2.5 to 12.9) | 8.3 (3.1 to 13.5) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 1.0 (−5.2 to 7.1) | NA | |
CI = confidence interval; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; FAS = full analysis set; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
Note: The difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). P value was calculated using the CMH method adjusted by baseline disease severity.
Source: Clinical Study Report.4
Figure 9: IGA and EASI-75 Responses in JADE COMPARE
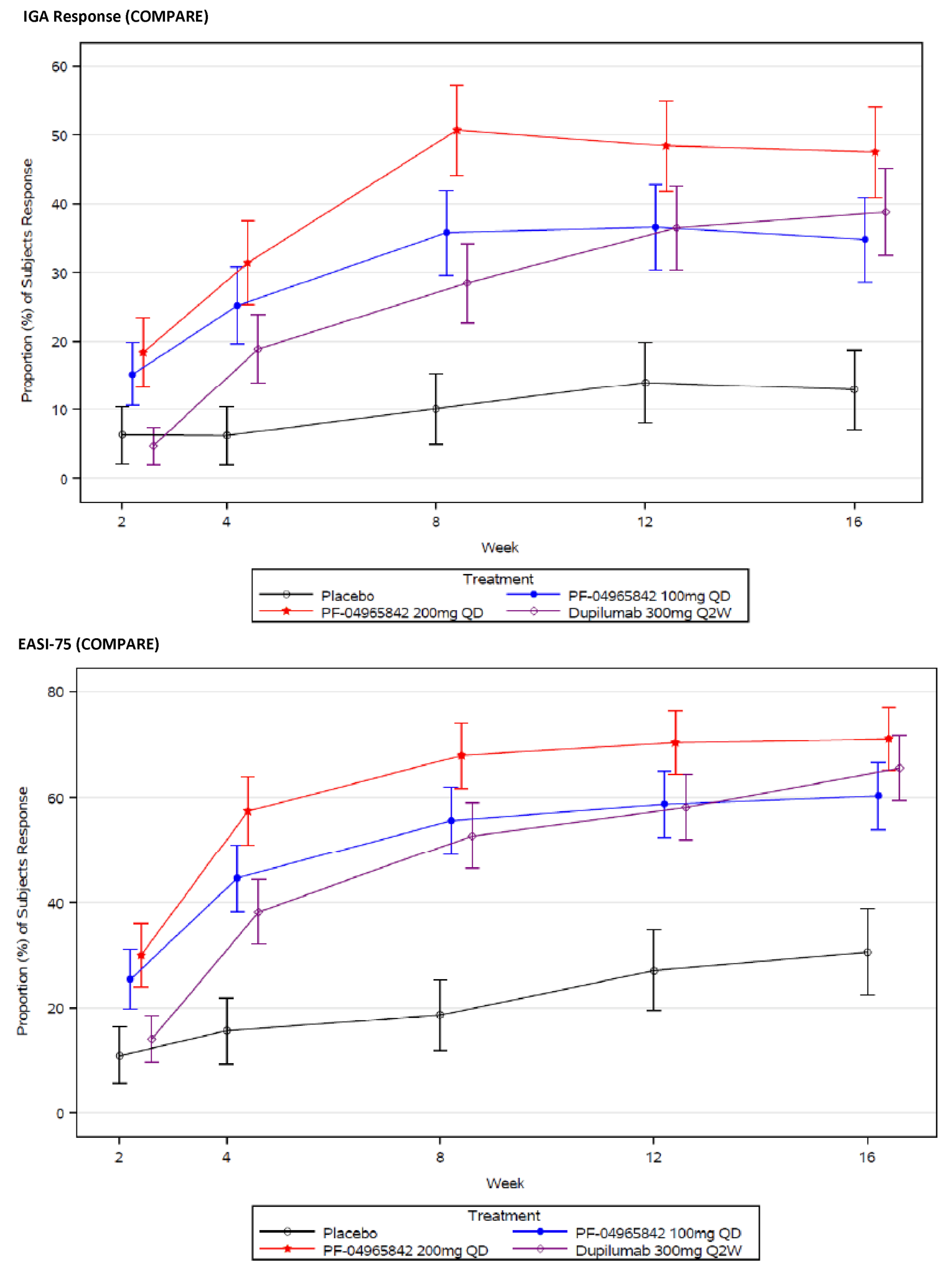
PF-04965842 = abrocitinib; Q2W = every 2 weeks; QD = once daily.
Source: Clinical Study Report.4
Active-Controlled Trial in Adults
Table 36 provides a summary of response for EASI-75, EASI-90, and EASI-100 in the JADE DARE trial. For the co-primary end point of EASI-90 response at 4 weeks, abrocitinib 300 mg once daily was superior to dupilumab every 2 weeks (28.5% versus 14.6% for a difference of 14.1% [95% CI, 8.2 to 20.0]; P < 0.0001). Response rates for patients with prior use of a systemic immunosuppressant for AD were similar to the overall patient population (26.3% versus 13.6%, for a difference of 12.7% [95% CI, 4.4 to 21.0]).1
For the key secondary end point of EASI-90 at week 16, abrocitinib 200 mg once daily was both noninferior (the lower bound of the 95% CI for the response difference was greater than −10%) and superior (2-sided P value for response difference was < 0.05) compared with dupilumab 300 mg every 2 weeks. A significantly greater proportion of patients from the abrocitinib 200 mg once daily group compared with the dupilumab group demonstrated an EASI-90 response at 16 weeks (54.3% versus 41.9%, for a difference of 12.5 [95% CI, 5.3 to 19.7]). Results were similar in the analyses performed using the per-protocol analysis set (15.2%; 95% CI, 7.6 to 22.7) and in the sensitivity analysis using multiple imputation. Response rates for patients with prior use of a systemic immunosuppressant for AD were similar to those of the overall patient population (56.1% versus 41.7%, for a difference of 14.4% [95% CI, 4.0 to 24.9]).1 As shown in Figure 10, the EASI-90 response rate was greater in the abrocitinib group compared with the dupilumab group from week 2 to week 20 but was no longer different at week 26.1
There was no statistically significant difference in the EASI-75 response between abrocitinib 200 mg once daily and dupilumab at 26 weeks (0.7%; 95% CI, −5.9 to 7.2). However, a greater proportion of abrocitinib-treated patients demonstrated an EASI-100 response at week 26 compared with dupilumab (22.7% versus 13.9%, for a difference of 8.8% [95% CI, 3.2 to 14.5]).1
Table 36: EASI-75, EASI-90, and EASI-100 Response in JADE DARE (Full Analysis Set)
Response | JADE DARE (full analysis set population) | JADE DARE (prior systemic immunosuppressant for AD) | ||
|---|---|---|---|---|
Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) | Abrocitinib 200 mg q.d. (N = 171) | Dupilumab 300 mg q.2.w. (N = 176) | |
EASI-90 response at week 4 (co-primary end point) | ||||
Patients in analysis | 354 | 364 | 171 | 176 |
Responders, n (%) | 101 (28.5) | 53 (14.6) | 45 (26.3) | 24 (13.6) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 14.1 (8.2 to 20.0) | 12.7 (4.4 to 21.0) | ||
2-sided P value | < 0.0001 | NA | ||
EASI-90 response at week 16 (key secondary end point) | ||||
Patients in analysis | 357 | 360 | 171 | 175 |
Responders, n (%) | 194 (54.3) | 151 (41.9) | 96 (56.1) | 73 (41.7) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 12.5 (5.3 to 19.7) | 14.4 (4.0 to 24.9) | ||
2-sided P value | 0.0008 | NA | ||
EASI-90 response at week 26 (secondary end point) | ||||
Patients in analysis | 348 | 361 | NA | |
Responders, n (%) | 190 (54.6) | 172 (47.6) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 6.9 (−0.4 to 14.3) | |||
2-sided P value | 0.0647 | |||
EASI-75 response at week 26 (secondary end point) | ||||
Patients in analysis | 348 | 361 | NA | |
Responders, n (%) | 254 (73.0) | 261 (72.3) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 0.7 (−5.9 to 7.2) | |||
2-sided P value | 0.8395 | |||
EASI-100 response at week 26 (secondary end point) | ||||
Patients in analysis | 348 | 361 | NA | |
Responders, n (%) | 79 (22.7) | 50 (13.9) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 8.8 (3.2 to 14.5) | |||
2-sided P value | 0.0023 | |||
CI = confidence interval; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; NA = not applicable; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
Note: The difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. The CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
Source: Clinical Study Report.1
Figure 10: EASI-90 and PP-NRS4 Responses in JADE DARE
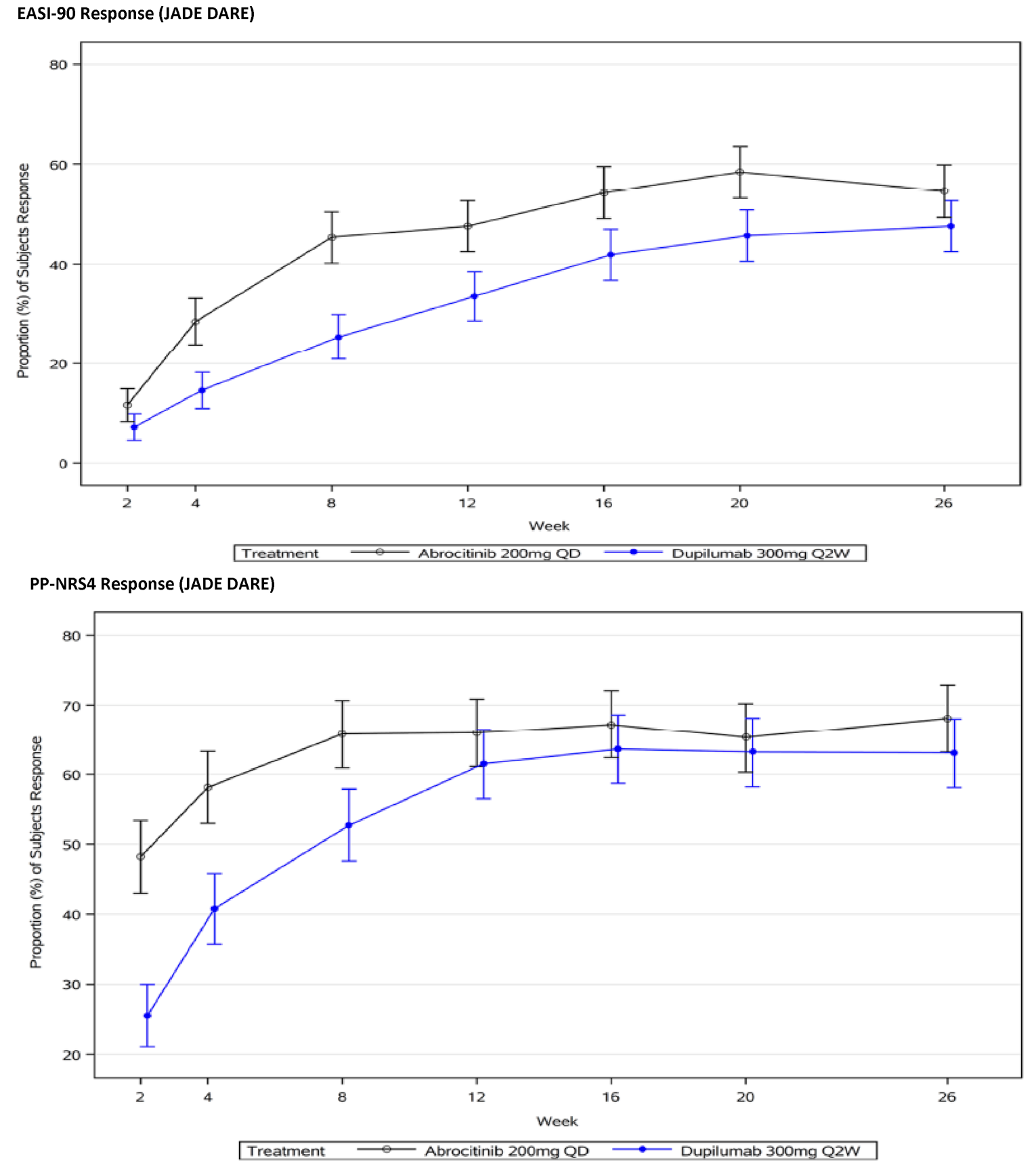
Q2W = every 2 weeks; QD = once daily.
Source: Clinical Study Report.1
Adolescents
Table 37 provides a summary of EASI-50, EASI-75, EASI-90, and EASI-100 responses in the JADE TEEN trial. For the primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated an EASI-75 response at week 12 compared with the placebo group (26.5% [95% CI, 13.1 to 39.8] and 29.4% [95% CI, 16.3 to 42.5]), respectively). Results of the analyses performed using the per-protocol analysis set demonstrated a statistically significantly greater proportion of patients with an EASI-75 response in the abrocitinib 100 mg group (29.8%; 95% CI, 15.1 to 44.5; P = 0.0002) and abrocitinib 200 mg once daily group (31.3%; 95% CI, 17.0 to 45.7; P < 0.0001). The results in the TP sensitivity analysis were similar to those of the primary analyses. Compared with placebo, the proportion of patients with an EASI-75 response was greater in the abrocitinib groups at all other time points (2 weeks, 4 weeks, and 8 weeks). Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an EASI-75 response at 4 weeks (22.3%; 95% CI, 8.3, 36.2), but not at 2 weeks, 8 weeks, or 12 weeks.5
For abrocitinib 100 mg once daily, subgroup analyses found EASI-75 responses based on baseline disease severity were 19.6% (95% CI, 2.2 to 36.9) for patients with moderate AD and 14.1% (95% CI, −3.3 to 31.5) for severe AD. For abrocitinib 200 mg once daily, subgroup analyses based on baseline disease severity were 36.6% (95% CI, 15.9 to 57.4) for patients with moderate AD and 54.5% (95% CI, 34.8 to 74.1) for those with severe AD. Response rates for patients with prior use of a systemic immunosuppressant for AD were 24.7% (95% CI, −1.7 to 51.1) for abrocitinib 100 mg once daily and 39.0% (95% CI, 12.4 to 65.7) for abrocitinib 200 mg once daily. For those without prior use of a systemic immunosuppressant for AD, the response rates were 28.9% (95% CI, 13.0 to 44.8) and 27.5% (95% CI, 12.0 to 43.1) with 100 mg once daily and 200 mg once daily, respectively.5
A statistically significantly greater proportion of patients in both the abrocitinib groups demonstrated an EASI-50 or EASI-90 response at 12 weeks. There was no statistically significant difference between abrocitinib 100 mg once daily or abrocitinib 200 mg once daily and placebo for the proportion of patients with an EASI-100 response at 12 weeks.5
Table 37: EASI-50, EASI-75, EASI-90, and EASI-100 Response in JADE TEEN (FAS)
Response | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) |
|---|---|---|---|
EASI-75 response at week 12 (subgroup of patients with prior use of systemic immunosuppressant) | |||
Patients in analysis | 24 | 26 | 22 |
Responders, n (%) | 7 (29.2) | 14 (53.8) | 15 (68.2) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 24.7 (−1.7 to 51.1) | 39.0 (12.4 to 65.7) |
2-sided P value | Reference | NR | NR |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 14.3 (−13.0 to 41.6) | |
EASI-75 response at week 12 (primary end point) | |||
Patients in analysis | 94 | 89 | 93 |
Responders, n (%) | 39 (41.5) | 61 (68.5) | 67 (72.0) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 26.5 (13.1 to 39.8) | 29.4 (16.3 to 42.5) |
2-sided P value | Reference | 0.0002 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 3.1 (−9.9 to 16.2) | |
EASI-75 response at week 2 (secondary end point) | |||
Patients in analysis | 91 | 92 | 94 |
Responders, n (%) | 4 (4.4) | 18 (19.6) | 24 (25.5) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 15.0 (5.9 to 24.2) | 20.5 (10.7 to 30.3) |
2-sided P value | Reference | 0.0017 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 5.4 (−6.5 to 17.3) | |
EASI-75 response at week 4 (secondary end point) | |||
Patients in analysis | 96 | 92 | 94 |
Responders, n (%) | 14 (14.6) | 38 (41.3) | 60 (63.8) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 26.5 (14.3 to 38.8) | 48.8 (36.7 to 60.8) |
2-sided P value | Reference | < 0.0001 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 22.3 (8.3 to 36.2) | |
EASI-75 response at week 8 (secondary end point) | |||
Patients in analysis | 93 | 91 | 92 |
Responders, n (%) | 31 (33.3) | 55 (60.4) | 63 (68.5) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 27.1 (13.3 to 40.9) | 35.0 (21.5 to 48.4) |
2-sided P value | Reference | 0.0002 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 8.1 (−5.7 to 21.9) | |
EASI-50 response at week 12 (secondary end point) | |||
Patients in analysis | 94 | 89 | 93 |
Responders, n (%) | 65 (69.1) | 78 (87.6) | 81 (87.1) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 18.2 (6.9 to 29.4) | 16.8 (5.6 to 28.0) |
2-sided P value | Reference | 0.0026 | 0.0048 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | −0.7 (−10.4 to 8.9) | |
EASI-90 response at week 12 (secondary end point) | |||
Patients in analysis | 94 | 89 | 93 |
Responders, n (%) | 17 (18.1) | 37 (41.6) | 46 (49.5) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 23.4 (10.5 to 36.2) | 30.9 (18.0 to 43.8) |
2-sided P value | Reference | 0.0006 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 7.7 (−6.7 to 22.2) | |
EASI-100 response at week 12 (secondary end point) | |||
Patients in analysis | 94 | 89 | 93 |
Responders, n (%) | 2 (2.1) | 2 (2.2) | 8 (8.6) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 0.0 (−4.7 to 4.7) | 6.1 (−0.4 to 12.5) |
2-sided P value | Reference | 0.9852 | 0.0653 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 6.1 (−0.5 to 12.6) | |
CI = confidence interval; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; EASI-100 = improvement of 100% in the Eczema Area and Severity Index total score; FAS = full analysis set; NA = not applicable; NR = not reported; q.d. = once daily; vs. = versus.
Note: The difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. The CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
Source: Clinical Study Report.5
Severity of Pruritus Numerical Rating Scale
Monotherapy Studies
Table 38 provides a summary of PP-NRS4 response and time to PP-NRS4 response in the JADE MONO-1 and JADE MONO-2 trials. For the key secondary primary end point, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated a PP-NRS4 response at week 12 compared with the placebo group in the JADE MONO-1 trial (22.5% [95% CI, 10.3 to 34.8] and 41.7% [95% CI, 29.6 to 53.9], respectively) and in the JADE MONO-2 trial (33.7% [95% CI, 22.8 to 44.7] and 43.9 [95% CI, 32.9 to 55.0], respectively).2,3 The analyses conducted at earlier time points demonstrated that a statistically significantly greater proportion of patients in both abrocitinib groups achieved an PP-NRS4 response compared with the placebo group at 2 weeks, 4 weeks, and 8 weeks in both the JADE MONO-1 and JADE MONO-2 trials. Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an EASI-75 response at 2 weeks, 4 weeks, 8 weeks, and 12 weeks in both the JADE MONO-1 and JADE MONO-2 trials.2,3 The Kaplan–Meier analyses to estimate the time to first PP-NRS4 showed the time to response was shorter in both the abrocitinib groups compared with placebo in the JADE MONO-1 and JADE MONO-2 trials. Kaplan–Meier curves showing the time to PP-NRS4 response are provided in Table 38.
Table 38: PP-NRS4 Response in JADE MONO-1 and JADE MONO-2 (Full Analysis Set)
Response | JADE MONO-1 | JADE MONO-2 | |||||
|---|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | ||
PP-NRS4 response at week 2 (key secondary end point) | |||||||
Patients in analysis | 74 | 147 | 147 | 76 | 156 | 153 | |
Estimated response rate, % (95% CI) | 2.7 (NR) | 20.4 (NR) | 45.6 (NR) | 3.9 (0.0 to 8.3) | 23.1 (16.5 to 29.7) | 35.3 (27.7 to 42.9) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 18.0 (10.2 to 25.8) | 42.5 (33.6 to 51.4) | Reference | 19.2 (11.0 to 27.4) | 31.2 (22.3 to 40.2) | |
2-sided P value | Reference | 0.0004 | < 0.0001 | Reference | 0.0002 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 24.9 (14.8 to 35.0) | NA | 12.1 (2.2 to 22.1) | |||
PP-NRS4 response at week 4 (key secondary end point) | |||||||
Patients in analysis | 74 | 147 | 147 | 76 | 156 | 153 | |
Estimated response rate, % (95% CI) | 17.2 (NR) | 32.2 (NR) | 58.8 (NR) | 4.0 (0.0 to 8.4) | 33.4 | 52.8 (44.7 to 60.8) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 15.0 (1.9 to 28.0) | 41.1 (27.8 to 54.4) | Reference | 29.5 (20.5 to 38.4) | 48.8 (39.5 to 58.2) | |
2-sided P value | Reference | 0.0251 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 26.5 (13.7 to 39.2) | NA | 19.4 (8.4 to 30.4) | |||
PP-NRS4 response at week 8 (secondary end point) | |||||||
Patients in analysis | 74 | 147 | 147 | 76 | 156 | 153 | |
Estimated response rate, % (95% CI) | 14.4 (NR) | 34.3 (NR) | 59.9 (NR) | 12.0 (4.6 to 19.4) | 40.4 (32.6 to 48.2) | 54.4 (46.4 to 62.4) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 20.0 (7.4 to 32.7) | 45.3 (32.7 to 57.8) | Reference | 28.5 (17.8 to 39.3) | 42.4 (31.4 to 53.4) | |
2-sided P value | Reference | 0.0019 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 25.5 (13.5 to 37.6) | NA | 14.0 (2.9 to 25.1) | |||
PP-NRS4 response at week 12 (key secondary end point) | |||||||
Patients in analysis | 74 | 147 | 147 | 76 | 156 | 153 | |
Estimated response rate, % (95% CI) | 15.3 (NR) | 37.7 (NR) | 57.2 (NR) | 11.5 (4.1 to 19.0) | 45.2 (37.1 to 53.3) | 55.3 (47.2 to 63.5) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 22.5 (10.3 to 34.8) | 41.7 (29.6 to 53.9) | Reference | 33.7 (22.8 to 44.7) | 43.9 (32.9 to 55.0) | |
2-sided P value | Reference | 0.0003 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 19.3 (7.3 to 31.2) | NA | 10.2 (−1.1 to 21.5) | |||
Time to PP-NRS4 response through 12 weeks | |||||||
Responders, n (%) | 23 (31.1) | 73 (49.7) | 106 (72.1) | 20 (026.3) | 90 (57.7) | 110 (71.9) | |
Nonresponders, n (%) | 51 (68.9) | 74 (50.3) | 41 (27.9) | 56 (73.7) | 66 (42.3) | 43 (28.1) | |
Median time to event in days, (interquartile range) | 92.0 (85.0 to NE) | 84.0 (56.0 to NE) | 14.0 (11.0 to 29.0) | 112.0 (112.0 to NE) | 58.0 (56.0 to 83.0) | 29.0 (16.0 to 31.0) | |
Log-rank test (P value) | Reference | 0.0071 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
CI = confidence interval; NE = not evaluable; NA = not applicable; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.d. = once daily; vs. = versus.
Note: Each complete imputed dataset was analyzed using the Cochran-Mantel-Haenszel risk difference method adjusting by randomization strata, separately for each week. Results from multiply imputed datasets were combined using Rubin's rules to obtain treatment difference, 95% CI, and P value. Missing responses after permanent discontinuation were defined as nonresponders. Any intermittent missing responses were imputed 500 times using random Bernoulli draws and a posterior probability of response at each visit. Posterior probabilities were estimated under a Bayesian framework from a logit-normal generalized linear mixed model with treatment, visit, treatment by visit interaction as fixed effects and a latent subject-level, zero mean, normally distributed random effect, with a logit link function.
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 39 provides a summary of PP-NRS4 response and time to PP-NRS4 response in the JADE COMPARE trial. For the key secondary end points of PP-NRS4 response at week 2 and week 16, a statistically significantly greater proportion of patients in the abrocitinib 200 mg once daily groups demonstrated a PP-NRS4 response compared with the placebo group at both time points (34.9% [95% CI, 26.0 to 43.7] and 32.7% [95% CI, 21.0 to 44.4], respectively). For the abrocitinib 100 mg once daily group, a statistically significantly greater proportion of patients in the abrocitinib 100 mg once daily group demonstrated a PP-NRS4 response compared with the placebo group at week 16 (18.1%; 95% CI, 6.2 to 30.0), but not at week 2 (5.2%; 95% CI, −2.9 to 13.4). Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an PP-NRS4 response at 2 weeks (17.2%; 95% CI, 8.4 to 26.0) and 16 weeks (15.7%; 95% CI, 5.4 to 26.1).
The Kaplan–Meier analyses to estimate the time to first PP-NRS4 showed the time to response was shorter in both the abrocitinib groups compared with placebo in the JADE COMPARE trial. Kaplan–Meier curves showing the time to PP-NRS4 response are provided in Table 40.
Table 39: PP-NRS4 Response in JADE COMPARE (Full Analysis Set)
Response | Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) |
|---|---|---|---|---|
PP-NRS4 response at week 2 (key secondary end point)a | ||||
Patients in analysis | 130 | 236 | 226 | 239 |
Responders, n (%) | 18 (13.8) | 75 (31.8) | 111 (49.1) | 63 (26.4) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 17.9 (9.5 to 26.3) | 34.9 (26.0 to 43.7) | 12.5 (4.4 to 20.7) |
2-sided P value | Reference | 0.0002 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | 5.2 (−2.9 to 13.4) | 22.1 (13.5 to 30.7) | Reference |
2-sided P value | NA | 0.2084 | < 0.0001 | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 17.2 (8.4 to 26.0) | NA | |
PP-NRS4 response at week 16 (key secondary end point)a | ||||
Patients in analysis | 94 | 168 | 172 | 189 |
Responders, n (%) | 27 (28.7) | 79 (47.0) | 108 (62.8) | 108 (57.1) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 18.1 (6.2 to 30.0) | 32.7 (21.0 to 44.4) | 28.3 (16.8 to 39.9) |
2-sided P value | Reference | 0.0045 | < 0.0001 | NR |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −10.2 (−20.5 to 0.1) | 5.2 (−4.8 to 15.2) | Reference |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 15.7 (5.4 to 26.1) | NA | |
Time to PP-NRS4 responseb through 16 weeks | ||||
Responders, n (%) | 57 (43.8) | 155 (65.7) | 169 (74.8) | 178 (74.2) |
Nonresponders, n (%) | 73 (56.2) | 81 (34.3) | 57 (25.2) | 62 (25.8) |
Median time to event, days (IQR) | NE (84.0 to NE) | 29.0 (16.0 to 56.0) | 13.0 (10.0 to 16.0) | 31.0 (29.0 to 57.0) |
P value (Abrocitinib vs. placebo) | Reference | < 0.0001 | < 0.0001 | NA |
CI = confidence interval; NA = not applicable; NE = not evaluable; NR = not reported; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
aThe difference and CI were calculated based on the weighted average of difference by disease severity group using the normal approximation of binomial proportions. CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were 0 or 100% responders). P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
bKaplan–Meier estimates based on the Brookmeyer and Crowley method. Log-rank test was adjusted for baseline disease severity.
Source: Clinical Study Report.4
Active-Controlled Trial in Adults
For the co-primary end point of PP-NRS4 at week 2, abrocitinib 200 mg once daily was superior to dupilumab 300 mg every 2 weeks (48.2% versus 25.5%, for a difference of 22.6%; 95% CI, 15.8 to 29.5; P < 0.0001). Results were similar in the sensitivity analysis using multiple imputation. Response rates for patients with prior use of a systemic treatments for AD were similar to the overall patient population (46.5% versus 22.3%, for a difference of 24.2%; 95% CI, 14.5 to 33.9).1 As shown in Table 39, the difference between abrocitinib 200 mg once daily and dupilumab every 2 weeks groups decreased over time and was similar between the 2 groups from week 12 onward.1
The Kaplan–Meier analyses to estimate the time to first PP-NRS4 showed the time to response was shorter in the abrocitinib 200 mg group compared with the dupilumab every 2 weeks group in the JADE DARE trial. Kaplan–Meier curves showing the time to PP-NRS4 response are provided in Table 40.
Table 40: PP-NRS4 Response in JADE DARE (Full Analysis Set)
Response | JADE DARE (full analysis set population) | JADE DARE (prior systemic immunosuppressant for AD) | ||
|---|---|---|---|---|
Abrocitinib 200 mg q.d. (N = 362) | Dupilumab (N = 365) | Abrocitinib (N = 171) | Dupilumab (N = 176) | |
PP-NRS4 at week 2 (co-primary end point) | ||||
Patients in analysis | 357 | 364 | 170 | 175 |
Responders, n (%) | 172 (48.2) | 93 (25.5) | 79 (46.5) | 39 (22.3) |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 22.6 (15.8 to 29.5) | 24.2 (14.5 to 33.9) | ||
2-sided P value | < 0.0001 | NA | ||
PP-NRS4 at week 26 | ||||
Patients in analysis | 354 | 363 | NA | |
Responders, n (%) | 241 (68.1) | 229 (63.1) | ||
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | 5.0 (−1.9 to 11.9) | |||
2-sided P value | 0.1601 | |||
Time to PP-NRS4 responsea through 16 weeks | ||||
Responders, n (%) | 313 (87.7) | 303 (83.2) | NA | |
Nonresponders, n (%) | 44 (12.3) | 61 (16.8) | ||
Median time to event (days) (IQR) | 11.0 (9.0 to 14.0) | 25.0 (21.0 to 30.0) | ||
P value (abrocitinib vs. dupilumab) | < 0.0001 | |||
CI = confidence interval; FAS = full analysis set; IQR = interquartile range; NA = not applicable; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
aKaplan–Meier estimates based on the Brookmeyer and Crowley method. Log-rank test was adjusted for baseline disease severity.
Source: Clinical Study Report.4
Adolescents
Table 41 provides a summary of PP-NRS4 response and time to PP-NRS4 response in the JADE TEEN trial. For the key secondary end points of PP-NRS4 response at week 2, week 4, and week 12, a statistically significantly greater proportion of patients in the abrocitinib 200 mg once daily groups demonstrated a PP-NRS4 response compared with the placebo group at all time points (26.1% [95% CI, 13.9 to 38.3], 29.4% [95% CI, 16.0 to 42.9], and 25.6% [95% CI to 10.6, 40.6] at weeks 2, 4, and 12 respectively). For the abrocitinib 100 mg once daily group, a statistically significantly greater proportion of patients demonstrated a PP-NRS4 response compared with the placebo group at week 2 (14.7%; 95% CI, 3.5 to 25.9), but not at week 4 (10.9; 95% CI, −1.8 to 23.6). Failure to demonstrate statistical significance for abrocitinib 100 mg once daily versus placebo at week 4 stopped the statistical testing hierarchy; the results for PP-NRS4 response at week 12 were therefore not considered to be statistically significant.5
Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated an PP-NRS4 response at 4 weeks (18.4%; 95% CI, 4.1 to 32.7), but not at the week 2 or week 12 time points.
The Kaplan–Meier analyses to estimate the time to first PP-NRS4 showed the time to response was shorter in both the abrocitinib groups compared with placebo in JADE TEEN. Kaplan–Meier curves showing the time to PP-NRS4 response are provided in Figure 11.
Table 41: PP-NRS4 Response in JADE TEEN (Full Analysis Set)
Response | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) |
|---|---|---|---|
PP-NRS4 response at week 2 (key secondary end point)a | |||
Patients in analysis | 95 | 92 | 88 |
Responders, n (%) | 12 (12.6) | 25 (27.2) | 34 (38.6) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 14.7 (3.5 to 25.9) | 26.1 (13.9 to 38.3) |
2-sided P value | Reference | 0.0119 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 11.7 (−1.8 to 25.2) | |
PP-NRS4 response at week 4 (key secondary end point)a | |||
Patients in analysis | 92 | 89 | 84 |
Responders, n (%) | 19 (20.7) | 28 (31.5) | 42 (50.0) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 10.9 (−1.8 to 23.6) | 29.4 (16.0 to 42.9) |
2-sided P value | Reference | 0.0971 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 18.4 (4.1 to 32.7) | |
PP-NRS4 response at week 12 (key secondary end point)a | |||
Patients in analysis | 84 | 76 | 74 |
Responders, n (%) | 25 (29.8) | 40 (52.6) | 41 (55.4) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 22.8 (8.0 to 37.7) | 25.6 (10.6 to 40.6) |
2-sided P value | Reference | 0.0035 | 0.0013 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 2.6 (−13.4 to 18.7) | |
Time to PP-NRS4 response through 16 weeksb | |||
Responders, n (%) | 41 (42.7) | 55 (59.1) | 59 (65.6) |
Nonresponders, n (%) | 55 (57.3) | 38 (40.9) | 31 (34.4) |
Median time to event (days) (IQR) | 90.0 (62.0 to NE) | 70.0 (30.0 to 85.0) | 29.0 (15.0 to 61.0) |
P value (abrocitinib vs. placebo) | Reference | 0.0159 | 0.0003 |
CI = confidence interval; NA = not applicable; NE = not evaluable; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.d. = once daily; vs. = versus.
aThe difference and CI were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. The CI for the response rate was based on normal approximation (or the Clopper-Pearson exact method when there were no or if all were responders). The P value was calculated using the Cochran-Mantel-Haenszel method adjusted by baseline disease severity.
bKaplan–Meier estimates based on the Brookmeyer and Crowley method. Log-rank test was adjusted for baseline disease severity.
Source: Clinical Study Report.5
Protocol-Defined Flare
Figure 11 summarizes the time to protocol-defined flare during the randomized maintenance period of the JADE REGIMEN trial. Compared with the placebo group, the risk of a protocol-defined flare during the maintenance period was statistically significantly reduced in the abrocitinib 100 mg once daily group (hazard ratio [HR] = 0.27; 95% CI, 0.211 to 0.341; P < 0.0001) and abrocitinib 200 mg once daily group (HR = 0.10; 95% CI, 0.070 to 0.136; P < 0.0001).34 The sponsor reported that subgroup analyses based on prior exposure to at least 1 systemic therapy for AD for the JADE REGIMEN study have not yet been conducted and therefore were not available at the time of CADTH’s review.32
Figure 11: Time to Protocol-Defined Flare During the Randomized Maintenance Period
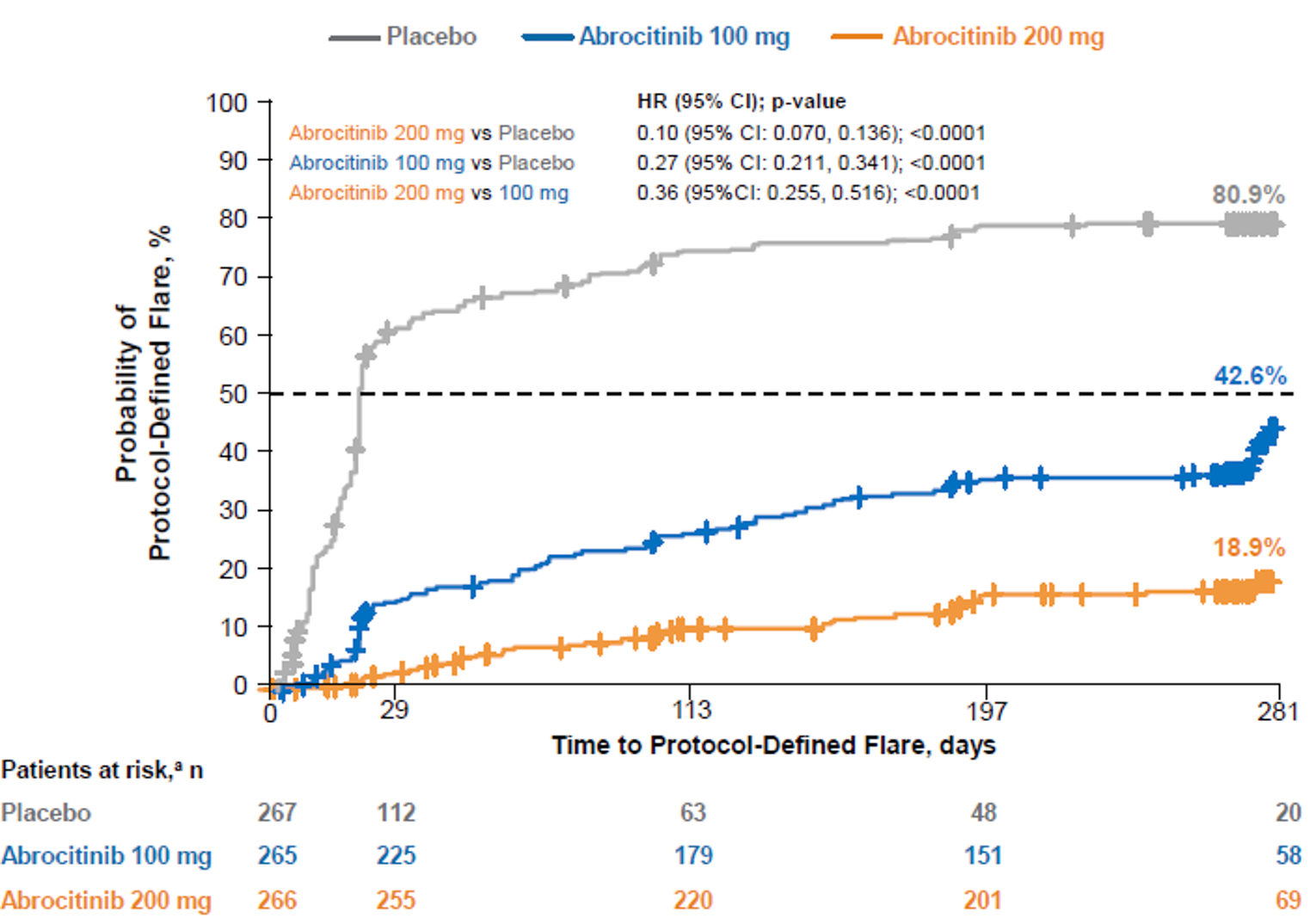
CI = confidence interval; HR = hazard ratio.
Source: Gubelin et al. (2021).34
Pruritus and Symptoms Assessment for Atopic Dermatitis
Monotherapy Studies
Table 42 provides a summary of change from baseline in PSAAD in the JADE MONO-1 and JADE MONO-2 trials. For the key secondary primary end point, treatment with abrocitinib 100 mg once daily or abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in change from baseline in PSAAD in both the JADE MONO-1 (LSMD = −1.1 [95% CI, −1.7 to −0.4] and −2.1 [95% CI, −2.7 to −1.4]) and JADE MONO-2 studies (LSMD = −1.7 [95% CI, −2.3 to −1.1] and −2.2 [95% CI, −2.8 to −1.6]).2,3 Compared with the 100 mg once daily abrocitinib dosage regimen, treatment with abrocitinib 200 mg once daily demonstrated a greater reduction in PSAAD at 12 weeks in both the JADE MONO-1 and JADE MONO-2 trials (LSMD = −1.0 [95% CI, −1.5 to −0.5] and −0.6 [95% CI, −1.0 to −0.1]).2,3
Table 42: Change From Baseline in PSAAD in JADE MONO-1 and JADE MONO-2 (FAS)
Response | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
Change from baseline in PSAAD at week 12 (key secondary end point) | ||||||
Patients in analysis | 68 | 137 | 138 | 77 | 156 | 155 |
Baseline, mean (SD) | 5.5 (2.0) | 5.3 (2.3) | 5.4 (2.1) | 5.1 (2.1) | 5.4 (2.1) | 5.2 (2.0) |
LSM (95% CI) | −1.1 (−1.7 to −0.6) | −2.2 (−2.6 to −1.9) | −3.2 (−3.6 to −2.8) | −0.8 (−1.3 to −0.3) | −2.4 (−2.8 to −2.1) | −3.0 (−3.3 to −2.7) |
Active vs. placebo, LSMD (95% CI) | Reference | −1.1 (−1.7 to −0.4) | −2.1 (−2.7 to −1.4) | Reference | −1.7 (−2.3 to −1.1) | −2.2 (−2.8 to −1.6) |
2-sided P value | Reference | 0.0010 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
200 mg q.d. vs. 100 mg q.d., LSMD (95% CI) | NA | −1.0 (−1.5 to −0.5) | NA | −0.6 (−1.0 to −0.1) | ||
CI = confidence interval; FAS = full analysis set; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; q.d. = once daily; vs. = versus.
Note: The mixed model for repeated measures contained fixed factors of treatment, week, treatment-by-week interaction, randomization strata (baseline disease severity and age category), baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Adults
Table 50 summarizes change from baseline in PSAAD in the JADE COMPARE trial. Compared with placebo, treatment with abrocitinib 100 mg once daily or abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in change from baseline in PSAAD at week 16 (LSMD = −1.2 [95% CI, −1.6 to −0.8] and −1.9 [95% CI, −2.3 to −1.5], respectively). Similarly, treatment with dupilumab resulted in a greater reduction from baseline in PSAAD compared with placebo (LSMD = −1.7; 95% CI, −2.1 to −1.3). The sponsor’s exploratory comparisons demonstrated a greater reduction from baseline with dupilumab compared with abrocitinib 100 mg once daily (LSMD = 0.5; 95% CI, 0.2 to 0.9) and a similar reduction compared with abrocitinib 200 mg once daily group (LSMD = −0.2; 95% CI, −0.6 to 0.1) at 16 weeks. The reduction in PSAAD from baseline was greater in the abrocitinib 200 mg once daily group compared with the abrocitinib 100 mg once daily group at 16 weeks (LSMD = −0.8; 95% CI, −1.1 to −0.4).
Adolescents
Table 50 summarizes change from baseline in PSAAD in the JADE TEEN trial. The statistical testing hierarchy was stopped at a higher-level end point (i.e., PP-NRS4 response for abrocitinib 100 mg once daily versus placebo at week 4); the results for abrocitinib 100 once daily and abrocitinib 200 mg once daily versus placebo for change from baseline in the total PSAAD score at week 12 are therefore not considered statistically significant.5
Scoring Atopic Dermatitis
Monotherapy Studies
Table 44 summarizes the responder analyses for patients who demonstrated an improvement of 50% or greater in Scoring Atopic Dermatitis (SCORAD-50) or an improvement of 75% or greater in Scoring Atopic Dermatitis (SCORAD-75) and change from baseline in SCORAD VAS for sleep loss in the JADE MONO-1 and JADE MONO-2 trials. Compared with placebo, a greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated SCORAD-50 and SCORAD-75 responses at 12 weeks in both the JADE MONO-1 and JADE MONO-2 trials. Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received abrocitinib 200 mg once daily demonstrated SCORAD-50 and SCORAD-75 responses at 12 weeks in both the JADE MONO-1 trial (20.2% [95% CI, 9.1 to 31.3] and 18.4% [95% CI, 9.3 to 27.5], respectively) and the JADE MONO-2 trial (13.4% [95% CI, 2.5 to 24.3] and 11.3% [95% CI, 1.9 to 20.7], respectively).2,3
For change from baseline in SCORAD VAS at 12 weeks, treatment with abrocitinib 100 mg once daily was associated with a statistically significantly greater reduction in SCORAD VAS compared with placebo in the JADE MONO-1 trial (LSMD = −1.3 [95% CI, −2.1 to −0.6]) and the JADE MONO-2 trial (LSMD = −0.9 [95% CI, −1.7 to −0.2], respectively). Treatment with abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction from baseline in SCORAD VAS compared with placebo in the JADE MONO-1 trial (LSMD = −2.1; 95% CI, −2.9 to −1.4) and the JADE MONO-2 trial (LSMD = −1.7; 95% CI, −2.5 to −1.0).2,3
Table 43: Change From Baseline and Responder Analyses for SCORAD in JADE MONO-1 and JADE MONO-2 (Full Analysis Set)
Response and change from baseline | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
SCORAD-50 response at week 12 | ||||||
Patients in analysis | 73 | 145 | 146 | 78 | 155 | 155 |
Responders, n (%) | 12 (16.4) | 53 (36.6) | 83 (56.8) | 10 (12.8) | 76 (49.0) | 97 (62.6) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 19.6 (8.1 to 31.1) | 40.0 (28.3 to 51.7) | Reference | 36.2 (25.4 to 47.1) | 49.6 (38.9 to 60.3) |
2-sided P value | Reference | 0.0026 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 20.2 (9.1 to 31.3) | NA | 13.4 (2.5 to 24.3) | ||
SCORAD-75 response at week 12 | ||||||
Patients in analysis | 73 | 145 | 146 | 78 | 155 | 155 |
Responders, n (%) | 3 (4.1) | 18 (12.4) | 45 (30.8) | 2 (2.6) | 29 (18.7) | 47 (30.3) |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 8.2 (1.0 to 15.3) | 26.4 (17.6 to 35.3) | Reference | 16.2 (8.8 to 23.6) | 27.6 (19.3 to 35.8) |
2-sided P value | Reference | 0.0528 | < 0.0001 | Reference | 0.0005 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 18.4 (9.3 to 27.5) | NA | 11.3 (1.9 to 20.7) | ||
Change from baseline in SCORAD visual analogue scale at week 12 | ||||||
Patients in analysis | 77 | 154 | 153 | 78 | 158 | 155 |
LSM (95% CI) | −1.6 (−2.2 to−1.0) | −2.9 (−3.4 to −2.5) | −3.7 (−4.2 to −3.3) | −2.1 (−2.7 to −1.5) | −3.0 (−3.4 to −2.6) | −3.8 (−4.2 to −3.4) |
LSMD (95% CI) Abrocitinib vs. placebo | Reference | −1.3 (−2.1 to −0.6) | −2.1 (−2.9 to −1.4) | Reference | −0.9 (−1.7 to −0.2) | −1.7 (−2.5 to −1.0) |
2-sided P value | Reference | 0.0005 | < 0.0001 | Reference | 0.0164 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.8 (−1.4 to −0.2) | NA | −0.8 (−1.4 to −0.2) | ||
CI = confidence interval; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = improvement of 75% or greater in Scoring Atopic Dermatitis; vs. = versus.
Note: The mixed model for repeated measures contained fixed factors of treatment, week, treatment-by-week interaction, randomization strata (baseline disease severity and age category), baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 45 provides a summary of change from baseline in SCORAD and responder analyses (SCORAD-50 and SCORAD-75) in the JADE COMPARE trial. Compared with placebo, a greater proportion of patients in both the abrocitinib groups demonstrated SCORAD-50 and SCORAD-75 responses at 16 weeks. Compared with the 100 mg once daily abrocitinib dosage regimen, a greater proportion of patients who received 200 mg once daily demonstrated SCORAD-50 and SCORAD-75 responses at 16 weeks (12.6% [95% CI, 3.8 to 21.5] and 13.4% [95% CI, 4.8 to 22.0], respectively).4
For change from baseline in SCORAD VAS at 16 weeks, treatment with both abrocitinib 100 mg once daily and abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in SCORAD VAS compared with placebo at 12 weeks (LSMD = −1.1 [95% CI, −1.6 to −0.6] and −2.1 [95% CI, −2.6 to −1.6], respectively).4
Active-Controlled Trial in Adults
For SCORAD total score, the least squares mean in percent change from baseline in percent SCORAD was greater in the abrocitinib 200 mg group compared with the dupilumab treatment group from week 2 to week 20. The difference between the abrocitinib and dupilumab groups decreased over time and was no longer statistically significant at 26 weeks (LSMD = −3.3 [95% CI, −6.6 to 0.1]; P = 0.0578).1
Adolescents
Table 45 summarizes change from baseline and responder analyses for SCORAD in the JADE TEEN trial. Compared with placebo, a greater proportion of patients in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated SCORAD-50 responses at 12 weeks (37.8% [95% CI, 24.8 to 50.7] and 35.2 [95% CI, 22.0 to 48.4], respectively) and SCORAD-75 responses at 12 weeks (23.7% [95% CI, 11.7, to 35.8] and 21.7% [95% CI, 9.7 to 33.7], respectively). There was no statistically significant difference between the abrocitinib 100 mg once daily group and the abrocitinib 200 mg once daily group for the proportion of patients who demonstrated SCORAD-50 and SCORAD-75 responses at 12 weeks.5
For change from baseline in SCORAD VAS at 12 weeks, treatment with both abrocitinib 100 mg once daily and abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in SCORAD VAS compared with placebo at 12 weeks (LSMD = −0.7 [95% CI, −1.4 to −0.1] and −1.2 [95% CI, −1.9 to −0.5], respectively).5
Table 44: Change From Baseline and Responder Analyses for SCORAD in JADE COMPARE and JADE TEEN (FAS)
Response and change from baseline | JADE COMPARE | JADE TEEN | |||||
|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
SCORAD-50 response at week 16 (JADE COMPARE) and week 12 (JADE TEEN) | |||||||
Patients in analysis | 123 | 228 | 221 | 231 | 93 | 90 | 92 |
Responders, n (%) | 41 (33.3) | 128 (56.1) | 152 (68.8) | 156 (67.5) | 35 (37.6) | 68 (75.6) | 68 (73.9) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 23.0 (12.6 to 33.4) | 35.4 (25.2 to 45.6) | 34.1 (23.9 to 44.3) | Reference | 37.8 (24.8 to 50.7) | 35.2 (22.0 to 48.4) |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | < 0.0001 | < 0.0001 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −11.3 (−20.1 to −2.5) | 0.9 (−7.7 to 9.5) | Reference | NA | NA | NA |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 12.6 (3.8 to 21.5) | NA | NA | −2.1 (−14.6 to 10.4) | ||
SCORAD-75 response at week 16 (JADE COMPARE) and week 12 (JADE TEEN) | |||||||
Patients in analysis | 123 | 228 | 221 | 231 | 93 | 90 | 92 |
Responders, n (%) | 13 (10.6) | 61 (26.8) | 89 (40.3) | 68 (29.4) | 12 (12.9) | 33 (36.7) | 32 (34.8) |
Difference in responders, % (95% CI) Active vs. placebo | Reference | 16.2 (8.4 to 24.1) | 29.6 (21.2 to 37.9) | 18.8 (10.8 to 26.8) | Reference | 23.7 (11.7 to 35.8) | 21.7 (9.7 to 33.7) |
2-sided P value | Reference | 0.0004 | < 0.0001 | NR | Reference | 0.0002 | 0.0006 |
Difference in responders, % (95% CI) Abrocitinib vs. dupilumab | NA | −2.6 (−10.9 to 5.6) | 10.6 (1.9 to 19.3) | Reference | NA | NA | NA |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 13.4 (4.8 to 22.0) | NA | NA | −1.6 (−15.6 to 12.4) | ||
Change from baseline in SCORAD visual analogue score at week 16 (JADE COMPARE) and week 12 (JADE TEEN) | |||||||
Patients in analysis | 129 | 237 | 225 | 241 | 96 | 95 | 93 |
LSM (95% CI) | −2.6 (−3.0 to −2.2) | −3.7 (−4.0 to −3.4) | −4.8 (−5.1 to −4.5) | −4.3 (−4.6 to −4.0) | −2.7 (−3.2 to −2.2) | −3.5 (−3.9 to −3.0) | −3.9 (−4.4 to −3.4) |
LSMD (95% CI) Active vs. placebo | Reference | −1.1 (−1.6 to −0.6) | −2.1 (−2.6 to −1.6) | −1.6 (−2.1 to −1.1) | −0.7 (−1.4 to −0.1) | −1.2 (−1.9 to −0.5) | |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | 0.0270 | 0.0004 | |
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 0.6 (0.2 to 1.0) | −0.5 (−0.9 to −0.1) | Reference | NA | NA | |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −1.1 (−1.5 to −0.6) | NA | 0.0578 | |||
CI = confidence interval; FAS = full analysis set; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis; SCORAD-75 = improvement of 75% or greater in Scoring Atopic Dermatitis; vs. = versus.
Source: Clinical Study Reports.4,5
Percentage Body Surface Area
Monotherapy Studies
Compared with placebo, treatment with abrocitinib 100 mg once daily or abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in change from baseline in BSA at week 12 in both the JADE MONO-1 trial (LSMD = −13.8 [95% CI, −19.3 to −8.2] and −22.0 [95% CI, −27.6 to −16.5], respectively) and the JADE MONO-2 trial (LSMD = −16.9 [95% CI, −22.8 to −11.1] and −20.6 [95% CI, −26.5 to −14.8]). The LSMD between the abrocitinib 200 mg once daily group and the abrocitinib 100 mg once daily group was −8.3 (95% CI, −12.7 to −3.8).
Table 45: Change From Baseline and Responder Analyses for Body Surface Area in JADE MONO-1 and JADE MONO-2 (Full Analysis Set)
Change from baseline and response | JADE MONO-1 | JADE MONO-2 | |||||
|---|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | ||
Change from baseline in percentage BSA at week 12a | |||||||
Patients in analysis | 77 | 156 | 154 | 78 | 158 | 155 | |
Baseline, mean (SD) | 47.4 (22.7) | 50.8 (23.4) | 49.9 (24.4) | 48.2 (20.8) | 48.7 (21.4) | 47.7 (22.3) | |
LSM (95% CI) | −11.4 (−16.0 to −6.8) | −25.1 (−28.3 to −22.0) | −33.4 (−36.6 to −30.3) | −10.0 (−14.8 to −5.1) | −26.9 (−30.2 to −23.6) | −30.6 (−33.8 to −27.3) | |
LSMD (95% CI) Active vs. placebo | Reference | −13.8 (−19.3 to −8.2) | −22.0 (−27.6 to −16.5) | Reference | −16.9 (−22.8 to −11.1) | −20.6 (v26.5 to −14.8) | |
2-sided P value | Reference | < 0.0001 | < 0.0001 | Reference | < 0.0001 | < 0.0001 | |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −8.3 (−12.7 to −3.8) | NA | −3.7 (−8.3 to 0.9) | |||
Patients achieving percentage BSA < 5% at week 12b | |||||||
Patients in analysis | 76 | 156 | 153 | 77 | 155 | 154 | |
Estimated response rate | 4 (5.3) | 33 (21.2) | 59 (38.6) | 3 (3.9) | 35 (22.6) | 53 (34.4) | |
Difference in responders, % (95% CI) Abrocitinib vs. placebo | Reference | 15.8 (7.5 to 24.0) | 33.3 (24.0 to 42.7) | Reference | 18.5 (10.5 to 26.5) | 30.2 (21.4 to 39.0) | |
2-sided P value | Reference | 0.0019 | < 0.0001 | Reference | 0.0003 | < 0.0001 | |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 17.4 (7.5 to 27.3) | NA | 11.8 (2.0 to 21.6) | |||
BSA = body surface area; CI = confidence interval; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; q.d. = once daily; vs. = versus.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
bThe difference and CI were calculated based on the weighted average of difference for each randomization stratum using the normal approximation of binomial proportions. P value was calculated using the Cochran-Mantel-Haenszel method adjusted by randomization strata (baseline disease severity and age group).
Source: Clinical Study Reports.2,3
Combination-Therapy Studies
Table 46 provides a summary of change from baseline in BSA at week 16 in the JADE COMPARE trial and week 12 in the JADE TEEN trial.
Placebo-Controlled Trial in Adults
Compared with placebo, treatment with abrocitinib 100 mg once daily or abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in change from baseline in BSA at week 16 (LSMD = −13.2 [95% CI, −17.0, to −9.5] and −19.4 [95% CI, −23.1 to −15.7], respectively). Similarly, treatment with dupilumab resulted in a greater reduction from baseline in BSA compared with placebo (LSMD = −14.7; 95% CI, −18.5 to −11.0). The sponsor’s exploratory comparisons demonstrated a greater reduction from baseline in BSA with abrocitinib 200 mg once daily compared with dupilumab (LSMD = −4.6; 95% CI, −7.8 to −1.5). The reduction in BSA from baseline was greater in the abrocitinib 200 mg once daily group compared with the abrocitinib 100 mg once daily group at 16 weeks (LSMD = −6.2; 95% CI, −9.3 to −3.0).
Active-Controlled Trial in Adults
The least squares mean in percent change from baseline in percent BSA was greater in the abrocitinib 200 mg group compared with the dupilumab treatment group from week 2 to week 20. The difference between the abrocitinib and dupilumab groups decreased over time and was no longer statistically significant at 26 weeks (LSMD = −3.4; 95% CI, −7.1 to 0.4; P = 0.0793).
Adolescents
Compared with placebo, treatment with abrocitinib 100 mg once daily or abrocitinib 200 mg once daily was associated with a statistically significantly greater reduction in change from baseline in BSA at week 12 (LSMD = −10.2 [95% CI, −15.2 to −5.1] and −11.0 [95% CI, −16.0 to −5.9], respectively). The LSMD between the abrocitinib 200 mg once daily group and the abrocitinib 100 mg once daily group was −0.8 (95% CI, −5.9 to 4.3).
Table 46: Change From Baseline for BSA in JADE COMPARE AND JADE TEEN (Full Analysis Set)
Change from baseline | JADE COMPARE (16 weeks) | JADE DARE | JADE TEEN (12 weeks) | ||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | DUP 300 mg q.2.w. (N = 243) | Abrocitinib 200 mg q.d. (N = 362) | DUP 300 mg q.2.w. (N = 365) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
Change from baseline in BSA (%)a | |||||||||
Patients in analysis | 131 | 238 | 226 | 242 | 362 | 365 | 96 | 95 | 94 |
Baseline, mean (SD) | 48.9 (24.9) | 48.1 (23.1) | 50.8 (23.0) | 46.5 (22.1) | 42.5 (19.9) | 42.6 (21.3) | 45.8 (22.4) | 51.2 (21.7) | 48.7 (21.7) |
LSM (95% CI) | −19.6 (−22.6 to −16.6) | −32.9 (−35.1 to −30.7) | −39.0 (−41.3 to −36.8) | −34.4 (−36.6 to −32.2) | −82.3 | −79.0 | −24.2 (−27.8 to −20.7) | −34.4 (−38.0 to −30.8) | −35.2 (−38.8 to −31.6) |
LSMD (95% CI) Active vs. placebo | Reference | −13.2 (−17.0 to −9.5) | −19.4 (−23.1 to −15.7) | −14.7 (−18.5 to −11.0) | NA | Reference | −10.2 (−15.2 to −5.1) | −11.0 (−16.0 to −5.9) | |
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | 0.0793 | Reference | 0.0001 | < 0.0001 | |
LSMD (95% CI) Abrocitinib vs. DUP | NA | 1.5 (−1.6 to 4.6) | −4.6 (−7.8 to −1.5) | Reference | −3.4 (−7.1 to 0.4) | NA | NA | NA | |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −6.2 (−9.3 to −3.0) | NA | NA | NA | −0.8 (−5.9 to 4.3) | |||
BSA = body surface area; CI = confidence interval; DUP = dupilumab; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
Corticosteroid-Free Days
Combination-Therapy Studies
Adults
Table 47 summarizes the results for corticosteroid-free days up to week 16 in JADE COMPARE. Compared with placebo, patients in the abrocitinib 200 mg once daily group had a greater number of corticosteroid-free days up (LSMD = 11.8; 95% CI, 3.1 to 20.5).
Table 47: Corticosteroid-Free Days in JADE COMPARE (Full Analysis Set)
Characteristic | JADE COMPARE | |||
|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | |
Corticosteroid-free days up to week 16a | ||||
Patients in analysis | 131 | 238 | 226 | 242 |
LSM (95% CI) | 21.8 (14.9 to 28.8) | 30.2 (25.1 to 35.4) | 33.6 (28.3 to 38.9) | 28.1 (23.0 to 33.2) |
Active vs. placebo, LSMD (95% CI) | Reference | 8.4 (−0.3 to 17.0) | 11.8 (3.1 to 20.5) | 6.3 (−2.4 to 14.9) |
2-sided P value | Reference | 0.0577 | 0.0082 | NR |
Abrocitinib vs. dupilumab, LSMD (95% CI) | NA | 2.1 (−5.1 to 9.4) | 5.5 (−1.8 to 12.9) | Reference |
200 mg q.d. vs. 100 mg q.d., LSMD (95% CI) | NA | 3.4 (−4.0 to 10.8) | NA | |
CI = confidence interval; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported; q.d. = once daily; vs. = versus.
aAn analysis of covariance model was used including treatment as a main effect and baseline disease severity as covariates.
Source: Clinical Study Report.4
Adolescents
Table 48 summarizes the results for corticosteroid-free days up to week 12 and days without background therapy up to week 12 in JADE TEEN. Compared with placebo, patients in the abrocitinib 200 mg once daily group had a greater number of corticosteroid-free days up (LSMD = 8.3; 95% CI, 1.5 to 5.1) and a greater number of days when medicated background therapy was not used (LSMD = 9.0 days; 95% CI, 3.6 to 14.4).5
Table 48: Days Without Corticosteroids or Background Medication in JADE TEEN (FAS)
Characteristic | JADE TEEN | ||
|---|---|---|---|
Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
Corticosteroid-free days up to week 12a | |||
Patients in analysis | 83 | 89 | 82 |
LSM (95% CI) | 6.8 (2.0 to 11.6) | 10.9 (6.2 to 15.5) | 15.1 (10.2 to 19.9) |
Active vs. placebo, LSMD (95% CI) | Reference | 4.1 (−2.6 to 10.7) | 8.3 (1.5 to 15.1) |
2-sided P value | Reference | 0.2309 | 0.0176 |
200 mg q.d. vs. 100 mg q.d., LSMD (95% CI) | NA | 4.2 (2.5 to 10.9) | |
Days when medicated background therapy not used up to week 12a | |||
Patients in analysis | 91 | 91 | 88 |
LSM (95% CI) | 4.0 (0.2, 7.8) | 7.6 (3.8 to 11.4) | 13.0 (9.1 to 16.9) |
Active vs. placebo, LSMD (95% CI) | Reference | 3.6 (−1.8, 9.0) | 9.0 (3.6 to 14.4) |
2-sided P value | Reference | 0.1877 | 0.0013 |
200 mg q.d. vs. 100 mg q.d., LSMD (95% CI) | NA | 5.4 (0.0 to 10.8) | |
CI = confidence interval; FAS = full analysis set; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; q.d. = once daily; vs. = versus.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
Source: Clinical Study Report.5
Dermatology Life Quality Index and Children's Dermatology Life Quality Index
Monotherapy Studies
Table 50 provides a summary of results for change from baseline in DLQI and CDLQI. Treatment with both abrocitinib 100 mg once daily and 200 mg once daily was associated with a statistically significantly greater improvement in DLQI compared with placebo in the JADE MONO-1 trial (LSMD = −2.8 [95% CI, −4.8 to −0.8] and −4.9 [95% CI, −6.9 to −2.9], respectively) and the JADE MONO-2 trial (LSMD = −4.4 [95% CI, −6.2 to −2.7] and −5.9 [95% CI, −7.7 to −4.2], respectively).2,3 For the subset of patients who completed the CDLQI questionnaire, treatment with abrocitinib 200 mg once daily was associated with a statistically significantly greater improvement in CDLQI compared with placebo in both the JADE MONO-1 trial (LSMD = −3.6 [95% CI, −6.2 to −0.9]) and the JADE MONO-2 trial (LSMD = −7.1; 95% CI, −11.2 to −2.9); there was no significant difference between the abrocitinib 100 mg once daily group and placebo in either study.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in DLQI. Compared with placebo, the abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks groups demonstrated a statistically significantly greater reduction from baseline in DLQI at 16 weeks (LSMD = −2.8 [95% CI, −3.9 to −1.7] to −5.6 [95% CI, −6.7 to −4.5], and −4.6 [95% CI, −5.7 to −3.5], respectively).4 The sponsor’s exploratory comparisons demonstrated a greater reduction from baseline with dupilumab compared with abrocitinib 100 mg once daily (LSMD = 1.7; 95% CI, 0.8 to 2.7) and a greater reduction with abrocitinib 200 mg once daily compared with both dupilumab (LSMD = −1.0; 95% CI, −1.9 to −0.1) and abrocitinib 100 mg once daily group at 16 weeks (LSMD = −2.7; 95% CI, −3.7 to −1.8).
Active-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in DLQI. The change from baseline in DLQI was greater in the abrocitinib 200 mg group compared with the dupilumab treatment group from week 2 to week 20. The difference between the abrocitinib and dupilumab groups decreased over time and was no longer statistically significant at 26 weeks (LSMD = −0.3; 95% CI, −1.0 to 0.4).1
Adolescents
Table 50 provides a summary of results for change from baseline in CDLQI. Treatment with both abrocitinib 100 mg once daily and 200 mg once daily was associated with a statistically significantly greater improvement in CDLQI compared with placebo in JADE TEEN (LSMD = −2.3 [95% CI, −3.7 to −0.8] and −2.3 [95% CI, −3.8 to −0.9], respectively).5
Hospital Anxiety and Depression Scale
Monotherapy Studies
Table 50 provides a summary of results for change from baseline in HADS scores. For the HADS anxiety component score at 12 weeks, treatment with abrocitinib 100 mg once daily was associated with a statistically significantly greater improvement from baseline compared with placebo in the JADE MONO-2 trial (LSMD = −1.0; 95% CI, −1.9 to −0.1), but not in the JADE MONO-1 trial (LSMD = −0.5; 95% CI, −1.3 to 0.2); treatment with abrocitinib 200 mg once daily was associated with a statistically significantly greater improvement compared with placebo in both the JADE MONO-1 and JADE MONO-2 trials (LSMD = −1.0 [95% CI, −1.8 to −0.3] and −1.1 [95% CI, −2.0 to −0.2], respectively). For the HADS depression component score at 12 weeks, both the100 mg once daily and 200 mg once daily dosages resulted in statistically significant improvements compared with placebo in the JADE MONO-1 and JADE MONO-2 trials.2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in HADS scores. Compared with placebo, the abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks groups demonstrated a statistically significantly greater reduction from baseline in HADS anxiety component score (LSMD = −0.8 [95% CI, −1.5 to −0.1] to −1.6 [95% CI, −2.2 to −0.9], and −1.1 [95% CI, −1.7 to −0.4], respectively) and HADS depression component score at 16 weeks (LSMD = −0.7 [95% CI, −1.3 to −0.1] to −1.3 [95% CI, −1.9 to −0.7] and −0.9 [95% CI, −1.5 to −0.3], respectively).4 The sponsor’s exploratory comparisons did not demonstrate a difference between dupilumab and abrocitinib 100 mg once daily or abrocitinib 200 mg once daily for change from baseline HADS anxiety component scores or HADS depression component scores at 16 weeks. The reduction from baseline was greater in the abrocitinib 200 mg once daily group compared with the abrocitinib 100 mg once daily group at 16 weeks for both the HADS anxiety component score (LSMD = −0.7; 95% CI, −1.3 to −0.2) and depression component scores (LSMD = −0.5; 95% CI, −1.0 to 0.0).4
Active-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in HADS at 26 weeks. There were no differences between the abrocitinib and dupilumab groups at any of the time points that were assessed for both the HADS depression component and HADS anxiety component (LSMD = 0.2 [95% CI, −0.1 to 0.6] and 0.1 [95% CI, −0.3 to 0.6], respectively).1
Adolescents
Table 50 provides a summary of results for change from baseline in HADS. There was no statistically significant difference between the abrocitinib 100 mg once daily and 200 mg once daily groups compared with placebo for change from baseline in the HADS anxiety component score at 12 weeks (LSMD = 0.1 [95% CI, −0.8 to 1.0] and −0.3 [95% CI, −1.2 to 0.6], respectively) or the change from baseline in the HADS depression component at week 12 (LSMD = −0.4 [95% CI, −1.1 to 0.4] and −0.2 [95% CI, −0.9 to 0.6], respectively).5
Patient-Oriented Eczema Measure
Monotherapy Studies
Table 50 provides a summary of results for POEM scores. For change from baseline in POEM scores at 12 weeks, both 100 mg once daily and 200 mg once daily dosages resulted in statistically significant improvements compared with placebo in the JADE MONO-1 trial (LSMD = −3.1 [95% CI, −5.2 to −0.9] and −6.9 [95% CI, −9.0 to −4.7], respectively) and the JADE MONO-2 trial (LSMD = −5.1 [95% CI, −7.2 to −3.1] and −7.4 [95% CI, −9.5 to −5.3], respectively).2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in the POEM. Compared with placebo, the abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks groups demonstrated a statistically significantly greater reduction from baseline in the POEM (LSMD = −4.1 [95% CI, −5.7 to −2.6] to −7.5 [95% CI, −9.0 to −5.9], and −5.8 [95% CI, −7.4 to −4.2] respectively).4 The sponsor’s exploratory comparisons demonstrated a greater reduction from baseline with dupilumab compared with abrocitinib 100 mg once daily (LSMD = 1.7; 95% CI, 0.4 to 3.0) and a greater reduction with abrocitinib 200 mg once daily compared with both dupilumab (LSMD = −1.7; 95% CI, −3.0 to −0.4) and abrocitinib 100 mg once daily group at 16 weeks (LSMD = −3.3; 95% CI, −4.7 to −2.0).4
Active-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in POEM. Changes in POEM were assessed at week 12, week 16, and week 26. The change from baseline POEM was greater in the abrocitinib 200 mg group compared with the dupilumab treatment group at week 12 and week 16; however, the difference between the abrocitinib and dupilumab groups decreased over time and was no longer statistically significant at 26 weeks (LSMD = −0.4; 95% CI, −1.3 to 0.5).1
Adolescents
Table 50 provides a summary of results for change from baseline in POEM. Treatment with both abrocitinib 100 mg once daily and 200 mg once daily was associated with a statistically significantly greater reduction from baseline in POEM at 12 weeks compared with placebo in JADE TEEN (LSMD-4.1 [95% CI, −6.1 to −2.2] and −3.9 [95% CI, −5.9 to −2.0], respectively).5
Patient Global Assessment
Monotherapy Studies
Table 50 provides a summary of results for PtGA. For change from baseline in PtGA score at 12 weeks, both 100 mg once daily and 200 mg once daily doses resulted in statistically significant improvements compared with placebo in the JADE MONO-1 trial (LSMD = −0.5 [95% CI, −0.8 to −0.2] and −0.9 [95% CI, −1.3 to −0.6], respectively) and the JADE MONO-2 trial (LSMD = −0.6 [95% CI, −0.9 to −0.3] and −1.0 [95% CI, −1.3 to −0.7], respectively).2,3 Similarly, both 100 mg once daily and 200 mg once daily doses resulted in statistically significantly greater proportion of patients with PtGA response at 12 weeks in both the JADE MONO-1 trial (14.2% [95% CI, 5.3 to 23.2] and 29.3% [95% CI, 19.6 to 38.9], respectively) and the JADE MONO-2 trial (12.2% [95% CI, 4.5 to 19.9] and 25.2% [95% CI, 16.4 to 33.9], respectively).2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in PtGA. Treatment with abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks demonstrated statistically significantly greater reductions from baseline in PtGA compared with placebo in the JADE COMPARE trial (LSMD = −0.4 [95% CI, −0.6 to −0.2] to −0.9 [95% CI, −1.1 to −0.6], and −0.6 [95% CI, −0.8 to −0.4], respectively).4 The sponsor’s exploratory comparisons demonstrated a greater reduction from baseline with dupilumab compared with abrocitinib 100 mg once daily (LSMD = 0.2; 95% CI, 0.0 to 0.4) and a similar reduction compared with abrocitinib 200 mg once daily group (LSMD = −0.2; 95% CI, −0.4 to 0.0) at 16 weeks. The reduction from baseline was greater in the abrocitinib 200 mg once daily group compared with the abrocitinib 100 mg once daily group at 16 weeks (LSMD = −0.4; 95% CI, −0.6 to −0.3).4
Adolescents
Table 50 provides a summary of results for change from baseline in PtGA. Both abrocitinib 100 mg once daily and 200 mg once daily demonstrated a statistically significantly greater reduction from baseline in PtGA compared with placebo in the JADE TEEN trial (LSMD = −0.5 [95% CI, −0.8 to −0.2] and −0.7 [95% CI, −0.9 to −0.4], respectively).5
Functional Assessment of Chronic Illness Therapy–Fatigue
Monotherapy Studies
Table 50 provides a summary of results for change from baseline in FACIT-F and Peds-FACIT-F. Both abrocitinib 100 mg once daily and 200 mg once daily demonstrated statistically significant improvements in FACIT-F compared with placebo in the JADE MONO-1 trial (LSMD = 3.6 [95% CI, 0.9 to 6.4] and 4.5 [95% CI, 1.8 to 7.3], respectively) and the JADE MONO-2 trial (3.3 [95% CI, 0.8 to 5.9] and 4.3 [95% CI, 1.8 to 6.9], respectively); there was no statistically significant difference between abrocitinib and placebo for the smaller subset of adolescent patients who completed the Peds-FACIT-F.2,3
Combination-Therapy Studies
Adolescents
Table 50 provides a summary of results for change from baseline in Peds-FACIT-F. There was no statistically significant difference between the abrocitinib 100 mg once daily and 200 mg once daily groups compared with placebo for change from baseline in Peds-FACIT-F at week 12 in the JADE TEEN trial (LSMD = 2.0 [95% CI, −0.1 to 4.0] and 1.8 [95% CI, −0.2 to 3.8], respectively).5
Short Form (36) Health Survey
Monotherapy Studies
Table 50 provides a summary of results for change from baseline in the SF-36. Both abrocitinib 100 mg once daily and 200 mg once daily dosage groups demonstrated statistically significant improvements in the SF-36 physical component score compared with placebo in the JADE MONO-1 trial (LSMD = 3.6 [95% CI, 0.9 to 6.4] and 4.5 [95% CI, 1.8 to 7.3], respectively) and the JADE MONO-2 trial (2.9 [95% CI, 0.9 to 4.9] and 3.8 [95% CI, 1.8 to 5.8], respectively). For the SF-36 mental component score, only the 200 mg once daily dosage resulted in a statistically significantly improvement compared with placebo in the JADE MONO-1 and JADE MONO-2 trials (LSMD = 3.0 [95% CI, 0.3 to 5.8] and 3.5 [95% CI, 0.8 to 6.2], respectively).2,3
EQ-5D 5-Levels Questionnaire
Monotherapy Studies
Table 50 provides a summary of results for change from baseline in the EQ-5D 5-Levels questionnaire (EQ-5D-5L) and EQ-5D-Y. Both the abrocitinib 100 mg once daily and 200 mg once daily dosage groups demonstrated statistically significant improvements in the EQ-5D-5L index scores and EQ VAS compared with placebo in the JADE MONO-1 and JADE MONO-2 trials. There was no statistically significant difference between abrocitinib 100 mg once daily and placebo for change from baseline in the EQ-5D-Y index score and EQ VAS in either study and the results were mixed for the abrocitinib 200 mg once daily group (i.e., statistically significant for the EQ-5D-Y VAS only in the JADE MONO-1 trial and the EQ-5D-Y index score only in the JADE MONO-2 trial).2,3
Combination-Therapy Studies
Placebo-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in the EQ-5D-5L. The abrocitinib 200 mg once daily and dupilumab 300 mg every 2 weeks groups demonstrated statistically significant improvements in the EQ-5D-5L VAS compared with the placebo group; there was no statistically significant difference between the abrocitinib 100 mg once daily and placebo group. Treatment with abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks demonstrated statistically significantly greater reduction from baseline in EQ-5D index score compared with placebo.4
Active-Controlled Trial in Adults
Table 50 provides a summary of results for change from baseline in EQ-5D-5L index and EQ VAS scores at 26 weeks. There were no differences between the abrocitinib and dupilumab groups for either the index score or the EQ VAS at 26 weeks (LSMD = 0.007 [95% CI −0.008 to 0.022] and −0.816 [95% CI, −2.914 to 1.281], respectively).1
Adolescents
Table 50 provides a summary of results for change from baseline in the EQ-5D-Y. Both doses of abrocitinib resulted in a statistically significant increase in EQ-5D-Y index score at week 12 compared with placebo. For change from baseline in EQ-5D-Y VAS at week 12, the abrocitinib 200 mg once daily demonstrated a statistically significant increase compared with placebo; there was no statistically significant difference between the abrocitinib 200 mg once daily compared with placebo.5
Dermatitis Family Impact
Combination-Therapy Studies
Adolescents
Table 50 provides a summary of results for change from baseline in DFI in the JADE TEEN trial. Both abrocitinib 100 mg once daily and 200 mg once daily resulted in statistically significant improvements in the DFI questionnaire compared with placebo in the JADE TEEN trial (LSMD = −1.5 [95% CI, −3.3 to 0.3] and −2.1 [95% CI, −3.9 to −0.3], respectively).5
Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis
Monotherapy Studies
Change from baseline in the WPAI-AD was only included as an outcome in the JADE MONO-2 trials. Results are summarized in Table 50. For the WPAI-AD percent work-time-missed component, there was no statistically significant difference between the abrocitinib 100 mg once daily or abrocitinib 200 mg once daily groups compared with placebo at 12 weeks (LSMD = 1.6; [95% CI, −4.5 to 7.7] and −1.0 [95% CI, −7.3 to 5.3], respectively). For the remaining components of the WPAI-AD, both the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups demonstrated statistically significant improvements compared with the placebo group in percent impairment while working (LSMD = −13.8 [95% CI, −22.8 to −4.9] and −17.9 [95% CI, −27.2 to −8.7], respectively), percent overall work impairment (LSMD = −13.7 [95% CI, −22.8 to −4.6] and −17.9 [95% CI, −27.3 to −8.5], respectively), and percent activity impairment (LSMD = −16.1 [95% CI, −23.9 to −8.3] and −18.2 [95% CI, −26.0 to −10.5], respectively).3
Table 49: Patient-Reported Outcomes in JADE MONO-1 and JADE MONO-2 (Full Analysis Set)
Outcomes | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
Change from baseline in DLQI at week 12a | ||||||
Patients in analysis | 60 | 121 | 119 | 70 | 140 | 139 |
Baseline, mean (SD) | 13.9 (7.3) | 14.6 (6.5) | 14.6 (6.8) | 15.0 (7.1) | 15.4 (7.3) | 14.8 (6.0) |
LSM (95% CI) | −4.2 (−5.9 to −2.5) | −7.0 (−8.1 to −5.8) | −9.1 (−10.3 to −8.0) | −3.9 (−5.3 to −2.4) | −8.3 (−9.3 to −7.3) | −9.8 (−10.7 to −8.8) |
LSMD (95% CI) Active vs. placebo | Reference | −2.8 (−4.8 to −0.8) | −4.9 (−6.9 to −2.9) | Reference | −4.4 (−6.2 to −2.7) | −5.9 (−7.7 to −4.2) |
2-sided P value | Reference | 0.0072 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −2.1 (−3.8 to −0.5) | NA | −1.5 (−2.9 to −0.1) | ||
Change from baseline in CDLQI at week 12a | ||||||
Patients in analysis | 16 | 32 | 32 | 8 | 16 | 15 |
Baseline, mean (SD) | 13.6 (7.0) | 11.7 (6.6) | 13.2 (5.5) | 10.1 (3.8) | 13.8 (5.8) | 12.9 (5.7) |
LSM (95% CI) | −3.9 (−6.1 to−1.7) | −6.4 (−7.9 to −5.0) | −7.5 (−8.9 to −6.0) | −2.7 (−6.1 to 0.8) | −4.8 (−7.2 to −2.5) | −9.7 (−12.1 to −7.4) |
LSMD (95% CI) Active vs. placebo | Reference | −2.5 (−5.2 to 0.1) | −3.6 (−6.2 to −0.9) | Reference | −2.2 (−6.3 to 2.0) | −7.1 (−11.2 to −2.9) |
2-sided P value | Reference | 0.0629 | 0.0100 | Reference | 0.3031 | 0.0015 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −1.0 (−3.1 to 1.1) | NA | −4.9 (−8.2 to −1.6) | ||
Change from baseline in HADS anxiety component at week 12a | ||||||
Patients in analysis | 76 | 152 | 152 | 78 | 156 | 153 |
Baseline, mean (SD) | 6.0 (4.0) | 5.9 (4.1) | 5.6 (4.0) | 6.0 (3.7) | 5.5 (4.2) | 5.9 (3.9) |
LSM (95% CI) | −1.0 (−1.7 to −0.4) | −1.6 (−2.0 to −1.1) | −2.1 (−2.5 to −1.6) | −0.6 (−1.3 to 0.2) | −1.6 (−2.1 to −1.1) | −1.7 (−2.2 to −1.2) |
LSMD (95% CI) Active vs. placebo | Reference | −0.5 (−1.3 to 0.2) | −1.0 (−1.8 to −0.3) | Reference | −1.0 (−1.9 to −0.1) | −1.1 (−2.0 to −0.2) |
2-sided P value | Reference | 0.1675 | 0.0085 | Reference | 0.0240 | 0.0138 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.5 (−1.1 to 0.1) | NA | −0.1 (−0.8 to 0.6) | ||
Change from baseline in HADS depression component at week 12a | ||||||
Patients in analysis | 76 | 152 | 152 | 78 | 156 | 153 |
Baseline, mean (SD) | 3.9 (3.5) | 4.1 (3.7) | 4.2 (3.7) | 4.4 (3.3) | 4.1 (4.0) | 4.0 (3.7) |
LSM (95% CI) | −0.2 (−0.8 to 0.4) | −1.4 (−1.8 to −0.9) | −1.8 (−2.2 to −1.4) | 0.3 (−0.3 to 0.9) | −1.0 (−1.5 to −0.6) | −1.4 (−1.8 to −1.0) |
LSMD (95% CI) Active vs. placebo | Reference | −1.1 (−1.9 to −0.4) | −1.6 (−2.3 to −0.9) | Reference | −1.3 (−2.1 to −0.6) | −1.7 (−2.5 to −0.9) |
2-sided P value | Reference | 0.0028 | < 0.0001 | Reference | 0.0008 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.5 (−1.1 to 0.1) | NA | −0.4 (−1.0 to 0.2) | ||
Change from baseline in POEM at week 12a | ||||||
Patients in analysis | 77 | 153 | 153 | 78 | 156 | 154 |
Baseline, mean (SD) | 19.9 (6.1) | 19.5 (6.5) | 19.6 (5.9) | 19.2 (5.5) | 20.9 (5.7) | 19.7 (5.7) |
LSM (95% CI) | −3.7 (−5.5 to −1.9) | −6.8 (−8.0 to −5.6) | −10.6 (−11.8 to −9.4) | 3.6 (−5.3 to −1.9) | −8.7 (−9.9 to −7.5) | −11.0 (−12.1 to −9.8) |
LSMD (95% CI) Active vs. placebo | Reference | −3.1 (−5.2 to −0.9) | −6.9 (−9.0 to −4.7) | Reference | −5.1 (−7.2 to −3.1) | −7.4 (−9.5 to −5.3) |
2-sided P value | Reference | 0.0049 | < 0.0001 | Reference | < 0.0001 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −3.8 (−5.5 to −2.1) | NA | −2.3 (−3.9 to −0.6) | ||
Change from baseline PtGA at week 12a | ||||||
Patients in analysis | 77 | 154 | 153 | 78 | 157 | 154 |
Baseline, mean (SD) | NR | NR | NR | NR | NR | NR |
LSM (95% CI) | −0.5 (−0.8 to −0.3) | −1.0 (−1.2 to −0.9) | −1.5 (−1.7 to −1.3) | −0.4 (−0.7 to −0.1) | −1.0 (−1.2 to −0.8) | −1.4 (−1.6 to −1.2) |
LSMD (95% CI) Active vs. placebo | Reference | −0.5 (−0.8 to −0.2) | −0.9 (−1.3 to −0.6) | Reference | −0.6 (−0.9 to −0.3) | −1.0 (−1.3 to −0.7) |
2-sided P value | Reference | 0.0014 | < 0.0001 | Reference | 0.0001 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.4 (−0.7 to −0.2) | NA | −0.4 (−0.6 to −0.1) | ||
PtGA responder at week 12a | ||||||
Patients in analysis | 73 | 152 | 150 | 76 | 154 | 154 |
Estimated response rate | 5 (6.8) | 32 (21.1) | 54 (36.0) | 3 (3.9) | 25 (16.2) | 45 (29.2) |
Difference in responders, Abrocitinib vs. placebo | Reference | 14.2 (5.3 to 23.2) | 29.3 (19.6 to 38.9) | Reference | 12.2 (4.5 to 19.9) | 25.2 (16.4 to 33.9) |
2-sided P value | Reference | 0.0075 | < 0.0001 | Reference | 0.0077 | < 0.0001 |
Difference in responders, % (95% CI) 200 mg vs. 100 mg | NA | 15.0 (5.2 to 24.9) | NA | 13.0 (3.9 to 22.2) | ||
Change from baseline in EQ-5D-5L VAS at week 12a | ||||||
Patients in analysis | 60 | 121 | 119 | 70 | 140 | 138 |
Baseline, mean (SD) | 67.9 (21.4) | 65.9 (19.1) | 66.5 (19.9) | 66.3 (19.7) | 65.9 (22.4) | 67.1 (20.9) |
LSM (95% CI) | 1.035 (−3.451 to 5.520) | 8.604 (5.509 to 11.699) | 10.409 (7.328 to 13.489) | 1.511 (−2.969 to 5.991) | 7.470 (4.513 to 10.427) | 12.392 (9.469 to 15.315) |
LSMD (95% CI) Active vs. placebo | Reference | 7.569 (2.119 to 13.019) | 9.374 (3.933 to 14.815) | Reference | 5.959 (0.591 to 11.328) | 10.881 (5.532 to 16.230) |
2-sided P value | Reference | 0.0067 | 0.0008 | Reference | 0.0297 | 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 1.805 (−2.562 to 6.172) | NA | 4.922 (0.764 to 9.080) | ||
Change from baseline in EQ-5D-5L index score at week 12a | ||||||
Patients in analysis | 60 | 121 | 119 | 70 | 140 | 138 |
Baseline, mean (SD) | 0.8 (0.1) | 0.8 (0.1) | 0.8 (0.1) | 0.792 (0.151) | 0.789 (0.163) | 0.803 (0.132) |
LSM (95% CI) | 0.014 (−0.021 to 0.050) | 0.058 (0.034 to 0.083) | 0.078 (0.054 to 0.103) | 0.000 (−0.030 to 0.030) | 0.075 (0.056 to 0.095) | 0.098 (0.078 to 0.117) |
LSMD (95% CI) Active vs. placebo | Reference | 0.044 (0.001 to 0.087) | 0.064 (0.021 to 0.107) | Reference | 0.075 (0.039 to 0.111) | 0.097 (0.061 to 0.133) |
2-sided P value | Reference | 0.0461 | 0.0037 | Reference | 0.0001 | < 0.0001 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 0.020 (−0.014 to 0.055) | NA | 0.022 (−0.006 to 0.050) | ||
Change from baseline in EQ-5D-Y VAS at week 12a | ||||||
Patients in analysis | 16 | 32 | 32 | 8 | 16 | 15 |
Baseline, mean (SD) | 54.3 (25.1) | 65.0 (25.0) | 63.0 (24.0) | 62.3 (25.5) | 68.2 (16.5) | 63.9 (23.9) |
LSM (95% CI) | 4.276 (−3.397 to 11.948) | 10.347 (5.347 to 15.347) | 17.224 (12.151 to 22.297) | 2.670 (−10.715 to 16.055) | 3.927 (−5.418 to 13.271) | 16.622 (6.989 to 26.254) |
LSMD (95% CI) Active vs. placebo | Reference | 6.071 (−3.107 to 15.249) | 12.948 (3.754 to 22.143) | Reference | 1.257 (−15.090 to 17.603) | 13.952 (−2.530 to 30.434) |
2-sided P value | Reference | 0.1915 | 0.0064 | Reference | 0.8767 | 0.0944 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 6.877 (−0.246 to 14.000) | NA | 12.695 (−0.739 to 26.129) | ||
Change from baseline in EQ-5D-Y index score at week 12a | ||||||
Patients in analysis | 16 | 31 | 32 | 8 | 16 | 15 |
Baseline, mean (SD) | 0.5 (0.4) | 0.6 (0.4) | 0.6 (0.4) | 0.852 (0.072) | 0.786 (0.084) | 0.809 (0.103) |
LSM (95% CI) | 0.153 (−0.007 to 0.314) | 0.160 (0.056 to 0.265) | 0.215 (0.109 to 0.322) | 0.033 (−0.027 to 0.093) | 0.035 (−0.005 to 0.076) | 0.162 (0.121 to 0.203) |
LSMD (95% CI) Active vs. placebo | Reference | 0.007 (−0.184 to 0.198) | 0.062 (−0.130 to 0.254) | Reference | 0.003 (−0.071 to 0.076) | 0.129 (0.056 to 0.202) |
2-sided P value | Reference | 0.9429 | 0.5212 | Reference | 0.9421 | 0.0010 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 0.055 (−0.094 to 0.204) | NA | 0.127 (0.069 to 0.184) | ||
Change from baseline in FACIT-F at week 12a | ||||||
Patients in analysis | 49 | 106 | 108 | 70 | 140 | 138 |
Baseline, mean (SD) | 38.9 (9.5) | 36.5 (11.9) | 38.7 (10.0) | 36.8 (10.8) | 38.1 (11.7) | 37.9 (11.1) |
LSM (95% CI) | −1.3 (−3.6 to 1.0) | 2.4 (0.8 to 3.9) | 3.3 (1.7 to 4.8) | 0.0 (−2.2 to 2.2) | 3.4 (2.0 to 4.7) | 4.3 (3.0 to 5.7) |
LSMD (95% CI) Active vs. placebo | Reference | 3.6 (0.9 to 6.4) | 4.5 (1.8 to 7.3) | Reference | 3.3 (0.8 to 5.9) | 4.3 (1.8 to 6.9) |
2-sided P value | Reference | 0.0102 | 0.0013 | Reference | 0.0107 | 0.0010 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 0.9 (−1.3 to 3.1) | NA | 1.0 (−0.9 to 2.9) | ||
Change from baseline in Peds-FACIT-F at week 12a | ||||||
Patients in analysis | 13 | 31 | 30 | 8 | 16 | 15 |
Baseline, mean (SD) | 33.7 (7.4) | 35.2 (8.2) | 35.8 (6.8) | 40.1 (7.3) | 38.1 (10.4) | 39.7 (9.0) |
LSM (95% CI) | 1.2 (−1.4 to 3.9) | 2.2 (0.5 to 3.9) | 2.1 (0.3 to 3.8) | 2.2 (−3.3 to 7.8) | −0.3 (−4.1 to 3.5) | 5.1 (1.2 to 9.0) |
LSMD (95% CI) Active vs. placebo | Reference | 1.0 (−2.1 to 4.2) | 0.9 (−2.3 to 4.1) | Reference | −2.5 (−9.3 to 4.2) | 2.8 (−4.0 to 9.6) |
2-sided P value | Reference | 0.5241 | 0.5821 | Reference | 0.4510 | 0.4023 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.1 (−2.6 to 2.3) | NA | 5.4 (−0.1 to 10.8) | ||
Change from baseline in SF-36 physical component summary at week 12 a | ||||||
Patients in analysis | 50 | 106 | 108 | 70 | 140 | 138 |
Baseline, mean (SD) | 45.3 (9.2) | 44.2 (8.5) | 45.2 (8.2) | 47.3 (9.4) | 48.4 (10.5) | 47.1 (10.3) |
LSM (95% CI) | 0.5 (−1.4 to 2.4) | 4.3 (3.0 to 5.6) | 5.2 (3.9 to 6.5) | 1.2 (−0.5 to 2.9) | 4.0 (3.0 to 5.1) | 5.0 (3.9 to 6.0) |
LSMD (95% CI) Active vs. placebo | Reference | 3.8 (1.5 to 6.1) | 4.7 (2.4 to 7.0) | Reference | 2.9 (0.9 to 4.9) | 3.8 (1.8 to 5.8) |
2-sided P value | Reference | 0.0013 | < 0.0001 | Reference | 0.0052 | 0.0002 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 0.9 (−0.9 to 2.7) | NA | 0.9 (−0.6 to 2.4) | ||
Change from baseline in SF-36 mental component summary at week 12a | ||||||
Patients in analysis | 50 | 106 | 108 | 70 | 140 | 138 |
Baseline, mean (SD) | 50.2 (8.7) | 48.2 (11.1) | 48.8 (11.0) | 46.7 (6.9) | 46.1 (9.3) | 46.6 (7.7) |
LSM (95% CI) | −0.2 (−2.5 to 2.0) | 1.5 (−0.1 to 3.0) | 2.8 (1.3 to 4.3) | 0.4 (−1.9 to 2.7) | 2.2 (0.8 to 3.7) | 3.9 (2.5 to 5.3) |
LSMD (95% CI) Active vs. placebo | Reference | 1.7 (−1.0 to 4.4) | 3.0 (0.3 to 5.8) | Reference | 1.8 (−0.9 to 4.6) | 3.5 (0.8 to 6.2) |
2-sided P value | Reference | 0.2256 | 0.0275 | Reference | 0.1866 | 0.0113 |
LSMD (95% CI) 200 mg vs. 100 mg | NA | 1.4 (−0.8 to 3.5) | NA | 1.7 (−0.4 to 3.7) | ||
Change from baseline in WPAI-AD (percent work time missed) at week 12a | ||||||
Patients in analysis | NA | 42 | 93 | 79 | ||
Baseline, mean (SD) | NR | NR | NR | |||
LSM (95% CI) | −1.7 (−7.0 to 3.5) | −0.1 (−3.3 to 3.0) | −2.7 (−6.2 to 0.8) | |||
LSMD (95% CI) Active vs. placebo | Reference | 1.6 (−4.5 to 7.7) | −1.0 (−7.3 to 5.3) | |||
2-sided P value | Reference | 0.6102 | 0.7605 | |||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −2.6 (−7.2 to 2.1) | ||||
Change from baseline in WPAI-AD (percent impairment while working) at week 12a | ||||||
Patients in analysis | NA | 42 | 92 | 76 | ||
Baseline, mean (SD) | NR | NR | NR | |||
LSM (95% CI) | −4.7(−12.4 to 2.9) | −18.5 (−23.2 to −13.9) | −22.7 (−27.8 to −17.5) | |||
LSMD (95% CI) Active vs. placebo | Reference | −13.8 (−22.8 to −4.9) | −17.9 (−27.2 to −8.7) | |||
2-sided P value | Reference | 0.0027 | 0.0002 | |||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −4.1 (−11.0 to 2.8) | ||||
Change from baseline in WPAI-AD (percent overall work impairment) at week 12a | ||||||
Patients in analysis | NA | 42 | 92 | 76 | ||
Baseline, mean (SD) | NR | NR | NR | |||
LSM (95% CI) | −5.0 (−12.8 to 2.8) | −18.7 (−23.4 to −14.0) | −22.9 (−28.2 to −17.6) | |||
LSMD (95% CI) Active vs. placebo | Reference | −13.7 (−22.8 to −4.6) | −17.9 (−27.3 to −8.5) | |||
2-sided P value | Reference | 0.0035 | 0.0002 | |||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −4.2 (−11.3 to 2.9) | ||||
Change from baseline in WPAI-AD (percent activity impairment) at week 12a | ||||||
Patients in analysis | NA | 70 | 139 | 138 | ||
Baseline, mean (SD) | NR | NR | NR | |||
LSM (95% CI) | −3.3 (−9.8 to 3.3) | −19.4 (−23.5 to −15.2) | −21.5 (−25.6 to −17.4) | |||
LSMD (95% CI) Active vs. placebo | Reference | −16.1 (−23.9 to −8.3) | −18.2 (−26.0 to −10.5) | |||
2-sided P value | Reference | 0.0001 | < 0.0001 | |||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −2.1 (−7.9 to 3.7) | ||||
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HADS = Hospital Anxiety and Depression Scale; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily; SF-36 = Short Form (36) Health Survey; vs. = versus; WPAI-AD = Work Productivity and Activity Impairment Questionnaire–Atopic Dermatitis.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.2,3
Table 50: Patient-Reported Outcomes in JADE COMPARE and JADE TEEN (Full Analysis Set)
End points (week 16 JADE COMPARE and week 12 JADE TEEN) | JADE COMPARE (16 weeks) | JADE TEEN (12 weeks) | |||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | DUP 300 mg q.2.w. (N = 243) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |||
Change from baseline in PtGAa | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | NR | NR | NR | NR | NR | NR | NR | ||
LSM (95% CI) | −0.7 (−0.9 to −0.6) | −1.2 (−1.3 to −1.0) | −1.6 (−1.7 to −1.5) | −1.4 (−1.5 to −1.2) | −0.9 (−1.1 to −0.7) | −1.4 (−1.6 to −1.2) | −1.6 (−1.8 to −1.4) | ||
LSMD (95% CI) Active vs. placebo | Reference | −0.4 (−0.6 to −0.2) | −0.9 (−1.1 to −0.6) | −0.6 (−0.8 to −0.4) | Reference | −0.5 (−0.8 to −0.2) | −0.7 (−0.9 to −0.4) | ||
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0008 | < 0.0001 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 0.2 (0.0 to 0.4) | −0.2 (−0.4 to 0.0) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.4 (−0.6 to −0.3) | NA | NA | −0.2 (−0.4 to 0.1) | ||||
Change from baseline in DLQI (JADE COMPARE) and CDLQI (JADE TEEN)a | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | 15.2 (6.9) | 15.5 (6.4) | 16.3 (6.6) | 15.6 (6.7) | 14.0 (6.7) | 14.3 (6.1) | 13.6 (7.0) | ||
LSM (95% CI) | −6.2 (−7.1 to −5.3) | −9.0 (−9.7 to −8.4) | −11.7 (−12.4 to −11.1) | −10.8 (−11.4 to −10.1) | −6.3 (−7.4 to −5.3) | −8.6 (−9.6 to −7.5) | −8.7 (−9.7 to −7.6) | ||
LSMD (95% CI) Active vs. placebo | Reference | −2.8 (−3.9 to −1.7) | −5.6 (−6.7 to −4.5) | −4.6 (−5.7 to −3.5) | Reference | −2.3 (−3.7 to −0.8) | −2.3 (−3.8 to −0.9) | ||
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0026 | 0.0018 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 1.7 (0.8 to 2.7) | −1.0 (−1.9 to −0.1) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −2.7 (−3.7 to −1.8) | NA | NA | −0.1 (−1.6 to 1.4) | ||||
Change from baseline in HADS anxiety componenta | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | 5.3 (3.9) | 5.3 (3.9) | 5.5 (3.8) | 5.1 (3.8) | 5.7 (3.7) | 5.7 (4.1) | 5.2 (4.3) | ||
LSM (95% CI) | −0.4 (−0.9 to 0.1) | −1.2 (−1.6 to −0.8) | −2.0 (−2.4 to −1.6) | −1.5 (−1.9 to −1.1) | −2.1 (−2.7 to −1.5) | −2.0 (−2.6 to −1.4) | −2.4 (−3.0 to −1.8) | ||
LSMD (95% CI) Active vs. placebo | Reference | −0.8 (−1.5 to −0.1) | −1.6 (−2.2 to −0.9) | −1.1 (−1.7 to −0.4) | Reference | 0.1 (−0.8 to 1.0) | −0.3 (−1.2 to 0.6) | ||
2-sided P value | Reference | 0.0175 | < 0.0001 | NR | Reference | 0.8603 | 0.4961 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 0.3 (−0.3 to 0.8) | −0.5 (−1.0 to 0.1) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.7 (−1.3 to −0.2) | NA | NA | −0.4 (−1.3 to 0.5) | ||||
Change from baseline in HADS depression componenta | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | 4.1 (3.7) | 4.0 (3.3) | 3.9 (3.4) | 3.7 (3.7) | 3.8 (3.4) | 3.7 (3.3) | 3.3 (2.8) | ||
LSM (95% CI) | −0.3 (−0.8 to 0.2) | −1.0 (−1.4 to −0.7) | −1.6 (−1.9 to −1.2) | −1.2 (−1.5 to −0.8) | −1.0 (−1.5 to −0.5) | −1.4 (−1.9 to −0.8) | −1.2 (−1.7 to −0.6) | ||
LSMD (95% CI) Active vs. placebo | Reference | −0.7 (−1.3 to −0.1) | −1.3 (−1.9 to −0.7) | −0.9 (−1.5 to −0.3) | Reference | −0.4 (−1.1 to 0.4) | −0.2 (−0.9 to 0.6) | ||
2-sided P value | Reference | 0.0181 | < 0.0001 | NR | Reference | 0.3364 | 0.6632 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 0.1 (−0.4 to 0.6) | −0.4 (−0.9 to 0.1) | — | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.5 (−1.0 to 0.0) | NA | NA | 0.2 (−0.6 to 0.9) | ||||
Change from baseline in POEM a | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 95 | 95 | 94 | ||
Baseline, mean (SD) | 20.4 (6.1) | 20.9 (5.5) | 21.5 (5.3) | 21.2 (5.5) | 19.8 (5.9) | 19.5 (6.4) | 19.2 (6.2) | ||
LSM (95% CI) | −5.0 (−6.3 to −3.8) | −9.2 (−10.1 to −8.2) | −12.5 (−13.4 to −11.6) | −10.8 (−11.8 to −9.9) | −6.9 (−8.3 to −5.6) | −11.1 (−12.5 to −9.7) | −10.9 (−12.2 to −9.5) | ||
LSMD (95% CI) Active vs. placebo | Reference | −4.1 (−5.7 to −2.6) | −7.5 (−9.0 to −5.9) | −5.8 (−7.4 to −4.2) | Reference | −4.1 (−6.1 to −2.2) | −3.9 (−5.9 to −2.0) | ||
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | < 0.0001 | < 0.0001 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 1.7 (0.4 to 3.0) | −1.7 (−3.0 to −0.4) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −3.3 (−4.7 to −2.0) | NA | NA | 0.2 (−1.7 to 2.2) | ||||
Change from baseline in PSAAD a | |||||||||
Patients in analysis | 130 | 237 | 225 | 241 | 95 | 95 | 93 | ||
Baseline, mean (SD) | 5.3 (2.2) | 5.3 (2.1) | 5.6 (2.0) | 5.3 (1.9) | 5.0 (2.4) | 4.9 (2.1) | 4.8 (2.3) | ||
LSM (95% CI) | −1.7 (−2.0 to −1.3) | −2.8 (−3.1 to −2.6) | −3.6 (−3.8 to −3.4) | −3.4 (−3.6 to −3.2) | −2.0 (−2.4 to −1.6) | −2.5 (−2.9 to −2.1) | −2.7 (−3.1 to −2.3) | ||
LSMD (95% CI) Active vs. placebo | Reference | −1.2 (−1.6 to −0.8) | −1.9 (−2.3 to −1.5) | −1.7 (−2.1 to −1.3) | Reference | −0.5 (−1.1 to 0.0) | −0.7 (−1.3 to −0.1) | ||
2-sided P value | Reference | < 0.0001 | < 0.0001 | NR | Reference | 0.0664 | 0.0142 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | 0.5 (0.2 to 0.9) | −0.2 (−0.6 to 0.1) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.8 (−1.1 to −0.4) | NA | NA | −0.2 (−0.8 to 0.4) | ||||
Change from baseline in EQ-5D-5L VAS (JADE COMPARE) and EQ-5D-Y VAS (JADE TEEN) a | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | 64.5 (22.9) | 66.0 (22.2) | 63.7 (23.7) | 65.1 (23.5) | 63.5 (24.8) | 63.2 (22.2) | 64.9 (21.6) | ||
LSM (95% CI) | 7.840 (4.952 to 10.727) | 11.223 (9.129 to 13.318) | 16.711 (14.581 to 18.841) | 14.405 (12.315 to 16.496) | 9.944 (6.373 to 13.515) | 14.226 (10.624 to 17.828) | 15.756 (12.153 to 19.360) | ||
LSMD (95% CI) Active vs. placebo | Reference | 3.383 (−0.185 to 6.951) | 8.871 (5.285 to 12.458) | 6.565 (3.000 to 10.130) | Reference | 4.282 (−0.790 to 9.354) | 5.812 (0.738 to 10.887) | ||
2-sided P value | Reference | 0.0630 | < 0.0001 | NR | Reference | 0.0976 | 0.0249 | ||
LSMD (95% CI) Abrocitinib vs. dupilumab | NA | −3.182 (−6.141 to −0.223) | 2.306 (−0.681 to 5.292) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | 5.488 (2.499 to 8.476) | NA | NA | 1.530 (−3.565 to 6.626) | ||||
Change from baseline in EQ-5D-5L index score (JADE COMPARE) and EQ-5D-Y index score (JADE TEEN) a | |||||||||
Patients in analysis | 131 | 238 | 226 | 241 | 96 | 95 | 94 | ||
Baseline, mean (SD) | 0.789 (0.145) | 0.787 (0.133) | 0.767 (0.150) | 0.778 (0.160) | 0.617 (0.342) | 0.633 (0.312) | 0.635 (0.297) | ||
LSM (95% CI) | 0.067 (0.047 to 0.087) | 0.093 (0.079 to 0.107) | 0.133 (0.119 to 0.148) | 0.113 (0.099 to 0.127) | 0.146 (0.101 to 0.192) | 0.228 (0.182 to 0.274) | 0.253 (0.207 to 0.299) | ||
LSMD (95% CI) Active vs. placebo | Reference | 0.026 (0.002 to 0.051) | 0.066 (0.042 to 0.091) | 0.046 (0.022 to 0.071) | Reference | 0.082 (0.017 to 0.146) | 0.106 (0.042 to 0.171) | ||
2-sided P value | Reference | 0.0342 | < 0.0001 | NR | Reference | 0.0132 | 0.0013 | ||
LSMD (95% CI) Abrocitinib vs. DUP | NA | −0.020 (−0.040 to 0.000) | 0.020 (0.000 to 0.040) | Reference | NA | NA | NA | ||
LSMD (95% CI) 200 mg vs. 100 mg | NA | 0.040 (0.020 to 0.060) | NA | NA | 0.025 (−0.040 to 0.089) | ||||
Change from baseline in Peds-FACIT-F (JADE TEEN)a | |||||||||
Patients in analysis | NA | 96 | 95 | 94 | |||||
Baseline, mean (SD) | 38.4 (11.2) | 39.3 (9.9) | 39.2 (9.0) | ||||||
LSM (95% CI) | 2.5 (1.1 to 3.9) | 4.5 (3.0 to 5.9) | 4.3 (2.9 to 5.7) | ||||||
LSMD (95% CI) Active vs. placebo | Reference | 2.0 (−0.1 to 4.0) | 1.8 (−0.2 to 3.8) | ||||||
2-sided P value | Reference | 0.0576 | 0.0804 | ||||||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.2 (−2.2 to 1.8) | |||||||
Change from baseline in Dermatitis Family Impact (JADE TEEN)a | |||||||||
Patients in analysis | NA | 92 | 95 | 93 | |||||
LSM (95% CI) | −5.2 (−6.5 to −3.9) | −6.7 (−7.9 to −5.4) | −7.3 (−8.6 to −6.0) | ||||||
LSMD (95% CI) Active vs. placebo | Reference | −1.5 (−3.3 to 0.3) | −2.1 (−3.9 to −0.3) | ||||||
2-sided P value | Reference | 0.1045 | 0.0211 | ||||||
LSMD (95% CI) 200 mg vs. 100 mg | NA | −0.6 (−2.4 to 1.2) | |||||||
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; EQ-5D-Y = EQ-5D Youth Scale; HADS = Hospital Anxiety and Depression Scale; LSM = least squares mean; LSMD = least squares mean difference; NA = not applicable; NR = not reported; Peds-FACIT-F = Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue; POEM = Patient-Oriented Eczema Measure; PSAAD = Pruritus and Symptoms Assessment for Atopic Dermatitis; PtGA = Patient Global Assessment; q.2.w. = every 2 weeks; q.d. = once daily; vs. = versus.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.4,5
Table 51: Patient-Reported Outcomes in JADE DARE (Full Analysis Set)
Analyses | Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) |
|---|---|---|
Change from baseline in DLQI at week 26a | ||
Patients in analysis | 361 | 363 |
Baseline, mean (SD) | 14.0 (6.8) | 14.2 (6.3) |
LSM (95% CI) | −10.3 (−10.8 to −9.9) | −10.0 (−10.5 to −9.6) |
LSMD (95% CI); abrocitinib vs. dupilumab | −0.3 (−1.0 to 0.4) | |
2-sided P value | 0.3814 | |
Change from baseline in HADS depression component at week 26a | ||
Patients in analysis | 362 | 365 |
Baseline, mean (SD) | 3.3 (3.2) | 3.3 (3.0) |
LSM (95% CI) | −0.8 (−1.0 to −0.5) | −1.0 (−1.3 to −0.8) |
LSMD (95% CI); abrocitinib vs. dupilumab | 0.2 (−0.1 to 0.6) | |
2-sided P value | 0.2132 | |
Change from baseline in HADS anxiety component at week 26a | ||
Patients in analysis | 362 | 365 |
Baseline, mean (SD) | 5.1 (3.7) | 5.2 (3.6) |
LSM (95% CI) | −1.1 (−1.4 to −0.7) | −1.2 (−1.5 to −0.9) |
LSMD (95% CI); Abrocitinib vs. dupilumab | 0.1 (−0.3 to 0.6) | |
2-sided P value | 0.4991 | |
Change from baseline in POEM at week 26a | ||
Patients in analysis | 362 | 365 |
Baseline, mean (SD) | 20.4 (5.8) | 20.9 (5.3) |
LSM (95% CI) | −13.8 (−14.5 to −13.1) | −13.4 (−14.0 to −12.7) |
LSMD (95% CI); Abrocitinib vs. dupilumab | −0.4 (−1.3 to 0.5) | |
2-sided P value | 0.3684 | |
Change from baseline in EQ-5D-5L index score at week 26a | ||
Patients in analysis | 362 | 364 |
Baseline, mean (SD) | 0.776 (0.154) | 0.787 (0.134) |
LSM (95% CI) | 0.128 (0.117 to 0.139) | 0.121 (0.111 to 0.132) |
LSMD (95% CI); abrocitinib vs. dupilumab | 0.007 (−0.008 to 0.022) | |
2-sided P value | 0.3646 | |
Change from baseline in EQ-5D-5L VAS at week 26a | ||
Patients in analysis | 362 | 364 |
Baseline, mean (SD) | 68.4 (19.5) | 67.7 (18.3) |
LSM (95% CI) | 13.484 (11.982 to 14.985) | 14.300 (12.836 to 15.764) |
LSMD (95% CI); abrocitinib vs. dupilumab | −0.816 (−2.914 to 1.281) | |
2-sided P value | 0.4450 | |
CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ-5D-5L = EQ-5D 5-Levels questionnaire; HADS = Hospital Anxiety and Depression Scale; LSM = least squares mean; LSMD = least squares mean difference; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.d. = once daily; VAS = Visual Analogue Scale; vs. = versus.
aThe mixed model for repeated measures contained fixed factors of treatment, visit, treatment by visit interaction, baseline disease severity, baseline value and an unstructured covariance matrix.
Source: Clinical Study Reports.1
Harms
Only those harms identified in the review protocol are reported here. Table 53 provides a summary of the AEs in the JADE MONO-1 and JADE MONO-2 trials, Table 54 provides a summary of the AEs that were reported in the JADE COMPARE, JADE DARE, and JADE TEEN trials, and Table 55 provides a summary of the aggregate AEs that were reported in the JADE REGIMEN trial. Because limited data were available for the JADE REGIMEN trial, the events are reported as incidence rates.
Adverse Events
Monotherapy Studies
In both the JADE MONO-1 and JADE MONO-2 trials, the sponsor reported that the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 100 mg once daily (69.2% and 62.7%, respectively) and 200 mg groups (77.9% and 65.8%, respectively) compared with the placebo groups (57.1% and 53.8%, respectively). Most events were mild or moderate in severity. Nausea was the most reported AE to occur at a greater frequency in the abrocitinib group than in the placebo group. Other commonly reported AEs that occurred in at least 5% more patients treated with abrocitinib compared with patients treated with placebo were headaches and acne.
Combination-Therapy Studies
In the JADE COMPARE trial, the sponsor reported that the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 200 mg group (61.9%) compared to the abrocitinib 100 mg (50.8%), dupilumab 300 mg every 2 weeks (50.0%), and placebo (53.4%) groups. In the JADE DARE trial, the sponsor reported that the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 200 mg group (74.0%) compared to the dupilumab 300 mg every 2 weeks group (65.5%). Most events were mild or moderate in severity in both the JADE COMPARE and JADE DARE trials. Nausea, headaches, and acne were the most reported AEs in the abrocitinib groups and conjunctivitis was the most frequently reported AE in the dupilumab group.1,4
In the JADE TEEN trial, the sponsor reported that the proportion of patients who had at least 1 TEAE was greater in the abrocitinib 200 mg group (62.8%) compared to the abrocitinib 100 mg (56.8%) and placebo (52.1%) groups. Most events were mild or moderate in severity. Nausea and acne were more commonly reported with abrocitinib compared with placebo.5
Withdrawal Study
Table 55 provides a summary of the TEAEs that were reported at an incidence rate of at least 4 per 100 patient-years in any of the 3 treatment groups in the JADE REGIMEN trial (excluding events that were classified as AD. The incidence of nausea was greater and the incidence of herpes zoster was numerically greater in the abrocitinib 200 mg once daily group compared with the abrocitinib 100 mg once daily and placebo groups. The incidence of increased blood CPK and acne were numerically greater in the abrocitinib 200 mg once daily group (7.85 [95% CI, 4.29 to 13.18] and 4.43 [95% CI, 1.91 to 8.73], respectively) and the abrocitinib 100 mg once daily group (4.08 [95% CI, 1.50 to 8.89] and 3.42 [95% CI, 1.11 to 7.97], respectively) compared with placebo group (1.59 [95% CI, 0.04 to 8.84]) and 0.00 [95% CI, 0.00 to 5.85], respectively).34
Serious Adverse Events
Monotherapy Studies
The proportion of patients with at least 1 SAE was similar between abrocitinib groups (3.2% in both) and the placebo group (3.9%) in the JADE MONO-1 trial. In the JADE MONO-2 trial, the proportions with at least 1 SAE were 3.2% in the abrocitinib 100 mg once daily group, 1.3% in the abrocitinib 200 mg once daily group, and 1.3% in the placebo group.2,3
Combination-Therapy Studies
In the JADE COMPARE trial, the proportions of patients with at least 1 SAE were 3.8% in the placebo group, 2.5% in the abrocitinib 100 mg once daily group, 0.9% in the abrocitinib 200 mg group, and 0.8% in dupilumab group.4 In the JADE DARE trial, the proportions of patients with at least 1 SAE were 1.7% in the abrocitinib 200 mg group and 1.6% in dupilumab every 2 weeks group. In the JADE TEEN trial, 2 SAEs were reported in the placebo group and 1 SAE was reported in the abrocitinib 200 mg group.5
Withdrawal Study
In the JADE REGIMEN trial, incidence rates for SAEs per 100 person-years were higher in the abrocitinib 200 mg group (7.77; 95% CI, 4.25 to 13.04) compared with the abrocitinib 100 mg group (2.69; 95% CI, 0.73 to 6.88) and placebo (3.18; 95% CI, 0.39 to 11.49).34
Withdrawal Due to Adverse Events
Monotherapy Studies
In the JADE MONO-1 trial, the proportions of patients who withdrew because of AEs were 9.1% in the placebo group, 5.8% in the abrocitinib 100 mg once daily group, and 5.8% in the abrocitinib 200 mg once daily group. In the JADE MONO-2 trial, the proportions of patients who withdrew because of AEs were 12.8% in the placebo group, 3.8% in the abrocitinib 100 mg once daily group, and 3.2% in the abrocitinib 200 mg once daily group.2,3
Combination-Therapy Studies
In the JADE COMPARE trial, the proportions of patients who withdrew because of AEs were 3.8% in the placebo group, 2.5% in the abrocitinib 100 mg once daily group, 4.4% in the abrocitinib 200 mg once daily group, and 3.3% in the dupilumab group.4 In the JADE DARE trial, the proportions of patients who withdrew because of AEs were 3.3% in the abrocitinib 200 mg once daily group and 2.5% in the dupilumab group.1 In the JADE TEEN trial, the proportions of patients who withdrew because of AEs were 2.1% in the placebo group, 1.1% in the abrocitinib 100 mg once daily group, and 2.1% in the abrocitinib 200 mg once daily group.5
Notable Harms
Monotherapy Studies
As shown in Table 53, serious infections and opportunistic infections were rare in the monotherapy studies. Elevated blood CPK was reported for numerically more patients in abrocitinib groups compared with placebo. No malignancies, MACEs, or VTE events were reported during the trials.2,3
Combination-Therapy Studies
As shown in Table 54, serious infections and opportunistic infections were rare in the combination -studies. Herpes zoster and elevated blood CPK were reported for numerically more patients in the abrocitinib groups compared with placebo in both the JADE COMPARE and JADE TEEN trials.4,5 No malignancies, MACEs, or VTE events were reported during the trials for abrocitinib-treated patients (a malignancy was reported for 1 patients treated with dupilumab in the JADE COMPARE trial).4
Mortality
No patients died in the JADE MONO-1, JADE COMPARE, or JADE TEEN studies.2,4,5 One 73-year-old patient in the abrocitinib 100 mg once daily group in the JADE MONO-2 trial died from a cardiovascular event. (The patient had a history of cardiovascular disease and the event was not considered related to the study treatment.3) Two patients died in the JADE DARE trial (both in the abrocitinib 200 mg once daily): 1 from COVID-19 and 1 from cardiorespiratory arrest or intracranial hemorrhage.1
Table 52: Summary of Harms in JADE MONO-1 and JADE MONO-2
Adverse events, n (%) | JADE MONO-1 | JADE MONO-2 | ||||
|---|---|---|---|---|---|---|
Placebo (N = 77) | Abrocitinib 100 mg q.d. (N = 156) | Abrocitinib 200 mg q.d. (N = 154) | Placebo (N = 78) | Abrocitinib 100 mg q.d. (N = 158) | Abrocitinib 200 mg q.d. (N = 155) | |
Patients with ≥ 1 adverse event | ||||||
≥ 1 AE | 44 (57.1) | 108 (69.2) | 120 (77.9) | 42 (53.8) | 99 (62.7) | 102 (65.8) |
Most common AEsa | ||||||
Nausea | 2 (2.6) | 14 (9.0) | 31 (20.1) | 2 (2.6) | 12 (7.6) | 22 (14.2) |
Vomiting | 1 (1.3) | 4 (2.6) | 6 (3.9) | 1 (1.3) | 2 (1.3) | 8 (5.2) |
Thrombocytopenia | — | — | — | 0 | 0 | 5 (3.2) |
Nasopharyngitis | 8 (10.4) | 23 (14.7) | 18 (11.7) | 5 (6.4) | 20 (12.7) | 12 (7.7) |
Upper respiratory tract infection | 5 (6.5) | 11 (7.1) | 11 (7.1) | 3 (3.8) | 14 (8.9) | 5 (3.2) |
Headache | 2 (2.6) | 12 (7.7) | 15 (9.7) | 2 (2.6) | 9 (5.7) | 12 (7.7) |
Acne | 0 | 1 (0.6) | 4 (2.6) | 0 | 2 (1.3) | 9 (5.8) |
Atopic dermatitis | 13 (16.9) | 22 (14.1) | 8 (5.2) | 12 (15.4) | 9 (5.7) | 6 (3.9) |
Patients with ≥ 1 SAE | ||||||
≥ 1 SAE | 3 (3.9) | 5 (3.2) | 5 (3.2) | 1 (1.3) | 5 (3.2) | 2 (1.3) |
Patients who stopped or interrupted treatment due to adverse events | ||||||
Discontinued | 1 (1.3) | 2 (1.3) | 0 | 0 | 2 (1.3) | 0 |
Interruption | 2 (2.6) | 4 (2.6) | 9 (5.8) | 2 (2.6) | 8 (5.1) | 5 (3.2) |
Patients who discontinued study due to adverse events | ||||||
Discontinued | 7 (9.1) | 9 (5.8) | 9 (5.8) | 10 (12.8) | 6 (3.8) | 5 (3.2) |
Notable harms | ||||||
Serious infections | 1 (1.3) | 1 (0.9) | 1 (0.6) | 1 (1.3) | 3 (1.9) | 0 |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 0 | 0 | 0 | 0 | 1 (0.6) |
Herpes zoster | 0 | 1 (0.9) | 2 (1.3) | 0 | 0 | 2 (1.3) |
Active tuberculosis | 0 | 0 | 0 | 0 | 0 | 0 |
Malignancy (excluding NMSC) | 0 | 0 | 0 | 0 | 0 | 0 |
NMSC | 0 | 0 | 0 | 0 | 0 | 0 |
Hepatic disorder | ||||||
Anemia | 0 | 0 | 2 (1.3) | 0 | 0 | 1 (0.6) |
Neutropenia | 0 | 0 | 1 (0.6) | 0 | 0 | 1 (0.6) |
Lymphopenia | 0 | 0 | 0 | 0 | 0 | 0 |
Increased blood CPK | 0 | 3 (1.9) | 5 (3.2) | 2 (2.6) | 3 (1.9) | 5 (3.2) |
MACE | 0 | 0 | 0 | 0 | 0 | 0 |
AE = adverse event; CPK = creatine phosphokinase; MACE = major adverse cardiovascular event; NMSC = nonmelanoma skin cancer; SAE = serious adverse event; VTE = venous thromboembolism; q.d. = once daily.
Source: Clinical Study Reports.2,3
Table 53: Summary of Harms in JADE COMPARE, JADE DARE, and JADE TEEN
Adverse events, n (%) | JADE COMPARE | JADE DARE | JADE TEEN | ||||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 131) | Abrocitinib 100 mg q.d. (N = 238) | Abrocitinib 200 mg q.d. (N = 226) | Dupilumab 300 mg q.2.w. (N = 243) | Abrocitinib 200 mg q.d. (N = 362) | Dupilumab 300 mg q.2.w. (N = 365) | Placebo (N = 96) | Abrocitinib 100 mg q.d. (N = 95) | Abrocitinib 200 mg q.d. (N = 94) | |
Patients with ≥ 1 adverse event | |||||||||
≥ 1 AE | 70 (53.4) | 121 (50.8) | 140 (61.9) | 121 (50.0) | 268 (74.0) | 239 (65.5) | 50 (52.1) | 54 (56.8) | 59 (62.8) |
Most common AEsa | |||||||||
Nausea | 2 (1.5) | 10 (4.2) | 25 (11.1) | 7 (2.9) | 70 (19.3) | 8 (2.2) | 1 (1.0) | 7 (7.4) | 17 (18.1) |
Conjunctivitis | 3 (2.3) | 2 (0.8) | 3 (1.3) | 15 (6.2) | 8 (2.2) | 35 (9.6) | |||
Nasopharyngitis | 9 (6.9) | 22 (9.2) | 15 (6.6) | 23 (9.5) | 14 (3.9) | 12 (3.3) | 9 (9.4) | 8 (8.4) | 8 (8.5) |
Upper respiratory tract infection | 6 (4.6) | 12 (5.0) | 9 (4.0) | 9 (3.7) | 10 (2.8) | 9 (2.5) | 10 (10.4) | 9 (9.5) | 10 (10.6) |
Headache | 6 (4.6) | 10 (4.2) | 15 (6.6) | 13 (5.4) | 47 (13.0) | 24 (6.6) | 7 (7.3) | 5 (5.3) | 8 (8.5) |
Acne | 0 | 7 (2.9) | 15 (6.6) | 3 (1.2) | 46 (12.7) | 10 (2.7) | 1 (1.0) | 3 (3.2) | 5 (5.3) |
Vomiting | < 5% | 11 (3.0) | 6 (1.6) | 0 | 4 (4.2) | 5 (5.3) | |||
Folliculitis | < 5% | 12 (3.3) | 3 (0.8) | 1 (1.0) | 7 (7.4) | 2 (2.1) | |||
Pharyngitis | < 5% | < 2% | 3 (3.1) | 5 (5.3) | 3 (3.2) | ||||
Dizziness | < 5% | 10 (2.8) | 4 (1.1) | 1 (1.0) | 0 | 6 (6.4) | |||
Patients with ≥ 1 SAE | |||||||||
≥ 1 SAEb | 5 (3.8) | 6 (2.5) | 2 (0.9) | 2 (0.8) | 6 (1.7) | 6 (1.6) | 2 (2.1) | 0 | 1 (1.1) |
Patients who stopped or interrupted treatment due to adverse events | |||||||||
Discontinued | 2 (1.5) | 2 (0.8) | 1 (0.4) | 0 | 0 | 1 (0.3) | 0 | 0 | 0 |
Interruption | 9 (6.9) | 15 (6.3) | 12 (5.3) | 9 (3.7) | 39 (10.8) | 27 (7.4) | 4 (4.2) | 4 (4.2) | 4 (4.3) |
Patients who discontinued study due to adverse events | |||||||||
Discontinued | 5 (3.8) | 6 (2.5) | 10 (4.4) | 8 (3.3) | 12 (3.3) | 9 (2.5) | 2 (2.1) | 1 (1.1) | 2 (2.1) |
Notable harms | |||||||||
Serious infections | 0 | 2 (0.8) | 0 | NR | 3 (0.8) | 0 | 0 | 0 | 0 |
Herpes zoster | 0 | 2 (0.8) | 4 (1.8) | NR | 9 (2.5) | 2 (0.5) | 0 | 1 (1.1) | 0 |
Active tuberculosis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
Malignancy (excluding nonmelanoma skin cancer) | 0 | 0 | 0 | 1 (0.4) | 0 | 0 | 0 | 0 | 0 |
Nonmelanoma skin cancer | 0 | 0 | 1 (0.4) | 0 | 0 | 0 | 0 | 0 | 0 |
Anemia | 0 | 1 (0.4) | 0 | 0 | 5 (1.4) | 0 | 0 | 1 (1.1) | 0 |
Neutropenia | 0 | 0 | 0 | 0 | 2 (0.6) | 0 | 0 | 0 | 0 |
Lymphopenia | 0 | 1 (0.4) | 1 (0.4) | 0 | 4 (1.1) | 1 (0.3) | 0 | 0 | 0 |
Increased blood CPK | 3 (2.3) | 7 (2.9) | 6 (2.7) | 2 (0.8) | 14 (3.9) | 13 (3.6) | 0 | 4 (4.2) | 4 (4.3) |
MACE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
VTE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
AE = adverse event; CPK = creatine phosphokinase; MACE = major adverse cardiovascular event; SAE = serious adverse event; VTE = venous thromboembolism; q.2.w. = every 2 weeks; q.d. = once daily.
Table 54: Summary of Harms in JADE REGIMEN
Incidence rate per 100 patient-years (95% CI) | JADE REGIMEN | ||
|---|---|---|---|
Placebo (N = 267) | Abrocitinib 100 mg q.d. (N = 265) | Abrocitinib 200 mg q.d. (N = 266) | |
TEAEs with incidence rate ≥ 4 per 100 patient-years (excluding AD) | |||
Nausea | 1.61 (0.04 to 8.95) | 1.35 (0.16 to 4.87) | 4.43 (1.91 to 8.72) |
Bronchitis | 4.78 (0.99 to 13.97) | 2.03 (0.42 to 5.92) | 0.54 (0.01 to 3.03) |
Conjunctivitis | 4.84 (1.00 to 14.14) | 2.02 (0.42 to 5.91) | 1.09 (0.13 to 3.92) |
Herpes zoster | 1.59 (0.04 to 8.84) | 1.34 (0.16 to 4.85) | 4.40 (1.90 to 8.67) |
Nasopharyngitis | 8.01 (2.60 to 18.70) | 6.98 (3.35 to 12.84) | 10.17 (6.03 to 16.07) |
Upper respiratory tract infection | 9.80 (3.60 to 21.34) | 5.48 (2.37 to 10.80) | 4.42 (1.91 to 8.71) |
Increased blood CPK | 1.59 (0.04 to 8.84) | 4.08 (1.50 to 8.89) | 7.85 (4.29 to 13.18) |
Asthma | 4.77 (0.98 to 13.95) | 0.67 (0.02 to 3.74) | 2.19 (0.60 to 5.60) |
Acne | 0.00 (0.00 to 5.85) | 3.42 (1.11 to 7.97) | 4.43 (1.91 to 8.73) |
Pruritus | 6.44 (1.76 to 16.49) | 2.72 (0.74 to 6.97) | 1.64 (0.34 to 4.78) |
SAEs | |||
SAE (excluding events of AD) | 3.18 (0.39 to 11.49) | 2.69 (0.73 to 6.88) | 7.77 (4.25 to 13.04) |
Patients who discontinued study due to adverse events | |||
Discontinuations due to AEs | 6.38 (1.74 to 16.34) | 3.36 (1.09 to 7.85) | 8.76 (5.01 to 14.23) |
AD = atopic dermatitis; AE = adverse event; CPK = creatine phosphokinase; SAE = serious adverse event; q.d. = once daily; TEAE = treatment-emergent adverse event.
Note: Includes all patients who were randomly assigned at week 12 and received at least 1 dose of study medication during the maintenance phase.
Source: Gubelin et al. (2021).34
Critical Appraisal
Internal Validity
Randomization was performed using an appropriate methodology with adequate allocation concealment (i.e., interactive response technology system). Patients were stratified based on relevant prognostic factors in the JADE MONO-1, JADE MONO-2, and JADE TEEN trials (i.e., baseline AD severity [moderate or severe] in all 3 studies and age [< 18 years or ≥ 18 years] in the JADE MONO-1 and JADE MONO-2 trials). In contrast, there was no stratification at the time of randomization in the JADE COMPARE trial and stratification was based only on age (< 18 years or ≥ 18 years) in the JADE REGIMEN trial. Despite these differences, baseline and demographic characteristics were generally well balanced across the treatments of each of the studies. In the JADE MONO-1 trial, the median EASI scores were lower in the placebo group (22.9; IQR = 19.2 to 37.6) compared with the abrocitinib 100 mg once daily (27.3; IQR = 20.1 to 40.3) and abrocitinib 100 mg once daily groups (25.2; IQR = 19.2 to 41.). Given the lower baseline scores in the placebo group, the patients in the placebo group would have less room to demonstrate improvements in EASI scores compared with the abrocitinib groups. These characteristics were well balanced in the JADE MONO-2 trial. There were imbalances in baseline disease severity across the treatment groups in the subgroup analyses based on prior exposure to at least 1 systemic therapy for AD. In the JADE MONO-1 trial, the proportion of patients with severe disease at baseline was 32.1% in the placebo group compared with 54.3% and 57.1% in the abrocitinib 100 mg once daily and 200 mg once daily treatment groups, respectively. In the JADE COMPARE trial, the proportion of patients with severe disease at baseline was greater in the abrocitinib 200 mg group (69.0%) of the subgroup analysis compared with the other treatment groups (range = 41.7% to 47.3%). In the JADE TEEN trial, the proportion of patients with severe disease at baseline was greater in the abrocitinib 100 mg group (59.3%) of the subgroup analysis compared with the other treatment groups (range = 54.2% to 45.5%).
The study treatments were administered in a double-blind manner and a double-dummy design was used to maintain blinding in the JADE COMPARE and JADE DARE trials to account for the oral administration of abrocitinib and the subcutaneous injection of dupilumab. The abrocitinib tablets and the dupilumab injections were identical in appearance to the corresponding placebo formats. Treatment with abrocitinib is associated with an increased risk of gastrointestinal AEs, but the clinical expert consulted by CADTH noted that the event profile of abrocitinib was unlikely to compromise blinding cross the studies. In addition, the sponsor did not report an increase in injection-site reactions for patients who received dupilumab in the JADE COMPARE or JADE DARE trial. As all of the trials except JADE DARE were placebo-controlled, it is possible that some patients could have inferred their allocated treatment assignment due to improvement or lack of improvement in AD over the study period and the use of rescue medication, which occurred in a higher proportion of patients in the placebo groups of the included studies. Although objective measures evaluated by the investigators would not likely be affected, patient-reported outcomes could have been influenced by inferring the allocated treatment; the direction and magnitude of the bias is unknown. This may have been most noticeable for patients in the withdrawal study (JADE REGIMEN) as all patients were responders, and a subsequent switch to placebo and loss of response would likely have unblinded patients to their new treatment assignment.
Patient disposition was thoroughly documented and well reported by the sponsor in its application to CADTH. Few patients discontinued from the 3 combination-therapy trials (completion rates ranged from 89.3% to 96.8% across the treatment groups), but the completion rates were considerably lower in the placebo groups of the monotherapy trials (79.2% and 66.7% in the JADE MONO-1 and JADE MONO-2 trials, respectively) compared with the abrocitinib groups (range = 86.5% to 91.0%). True intention-to-treat analyses were not performed; however, each FAS included nearly all randomized patients, and sensitivity analyses were performed to investigate the impact of missing data.
A greater proportion of patients who discontinued the study treatments were in the placebo groups in the JADE MONO-1 and JADE MONO-2 trials (ranging from 20.8% to 33.3% in the placebo group, 13.3% to 13.5% with abrocitinib 100 mg once daily, and from 11.0% to 9.0% with abrocitinib 200 mg once daily, respectively). This introduces the potential for bias against the null (i.e., toward an inflated efficacy of abrocitinib) as more placebo patients would have been imputed as nonresponders. The statistical analysis protocol for the co-primary end points assumed that data were MAR, which is not supported by the differential losses to follow-up and reasons for discontinuations between the placebo and abrocitinib groups. This assumption is strong and unverifiable and may, in some situations, increase the bias in the observed results, particularly where patients discontinue therapy due to lack of efficacy as observed in the JADE MONO-1 and JADE MONO-2 trial. However, sensitivity analyses were conducted based on per-protocol populations, and a completed TP analysis also supports the robustness of the conclusions of the primary analyses.
Adherence to the study treatments was evaluated by counting the number of study drugs at each visit, and median compliance was 100% across all treatment groups. In accordance with the study protocols, the use of concomitant medications was documented and reported throughout all of the included studies. The use of concomitant medications for AD (including corticosteroids) was greater in the placebo group of the JADE MONO-1 trial, potentially bia87sing some end points against abrocitinib,
The JADE COMPARE trial included dupilumab as an active comparator, although statistical analyses were limited to a single end point of PP-NRS4 at 2 weeks. This was a 16-week trial, which the clinical expert consulted by CADTH noted was likely insufficient to fully realize the maximal treatment effects for dupilumab, potentially biasing the results in favour of abrocitinib in both the JADE COMPARE trials and the indirect comparisons filed by the sponsor. The sponsor reported that the 16-week end point was selected to be consistent with the LIBERTY trials, which used 16 weeks as the primary end point in a study of dupilumab. In addition, the sponsor correctly noted the results of the 52-week LIBERTY AD CHRONOS study, in which EASI-75 responses for dupilumab every 2 weeks were 69% at week 16 and 65% at week 52.67 However, the results from the 26-week JADE DARE trial demonstrate that there is an earlier onset of response with abrocitinib once daily treatment compared with dupilumab every 2 weeks, but no significant differences between the 2 treatments were observed at 26 weeks.1,68
The proportions of adolescent patients in the monotherapy studies were relatively small at 84 of 387 (22%) and 40 of 391 (10%) in the JADE MONO-1 and JADE MONO-2 trials, respectively). Subgroup analyses for the adolescent patients in these trials found that a greater proportion of patients demonstrated an EASI-75 response for both abrocitinib doses, but greater uncertainty was associated with the estimated effect (as shown by the wide CIs in Table 95).
Statistical power calculations were reported for all of the included studies, and a sufficient number of patients were enrolled and completed the studies. The co-primary and key secondary end points of the included studies were tested using a hierarchical approach to limit the overall type I error rate to 0.05. All end points within the statistical testing hierarchies were statistically significant in the JADE MONO-1 and JADE MONO-2 trials. The statistical testing hierarchy was stopped at the first key secondary end point (PP-NRS4) of the JADE TEEN trial; however, the sponsor continued to calculate and report P values for the remaining key secondary end point (i.e., nominal P values were considered to be descriptive). In the JADE COMPARE trial, failure to demonstrate statistical significance for abrocitinib 100 mg once daily versus dupilumab for PP-NRS4 meant the sequence B end points were evaluated at a 2.5% significance level (Table 25); all of the end points in the sequence were statistically significant. Subgroup analyses and secondary and exploratory end points were tested without adjustment for multiple comparisons, and all P values were considered nominal. The majority of subgroups in the randomization scheme, beyond age and baseline severity, were not included as stratification variables, and differences between groups that may introduce bias in the observed subgroups would therefore be expected.
The critical appraisal of the JADE REGIMEN trial was limited by the small amount of information available regarding this study. In response to a request from CADTH, the sponsor noted that the Clinical Study Report was not available at the time of submission to Health Canada. To ensure consistency with the data filed to and reviewed by Health Canada, the sponsor elected not to include the Clinical Study Report in the submission to CADTH. Information available for the CADTH review, limited to an oral presentation at the AAD Conference, is summarized in the clinical summary of the CADTH submission and is provided with the submission (as it is public information).
External Validity
The diagnostic criteria used in the screening process for all of the included studies were consistent with Canadian clinical practice for identifying patients with moderate-to-severe AD. These criteria are similar to those used in other phase III trials for drugs used in the treatment of patients with moderate-to-severe AD. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., those with refractory moderate-to-severe AD and an inadequate response to other systemic drugs). The clinical expert consulted by CADTH indicated that the populations enrolled in the included trials were a reasonable reflection of the target population in Canada and that the response to abrocitinib would likely be similar for those with and those without prior exposure to at least 1 systemic therapy for AD. Black and African patients, members of First Nations, and people of Asian descent may be under-represented in the included studies in comparison with White patients.
The included RCTs were conducted at dermatology clinics. The clinical expert consulted by CADTH indicated that this is consistent with how abrocitinib would likely be used in Canadian clinical practice (i.e., prescribed and evaluated by specialists with experience in the diagnosis and management of AD). The same clinical assessor performed the evaluation of AD for any individual patient throughout the study, except in exceptional circumstances.
The dosage recommended in the product monograph is 100 mg or 200 mg orally once daily, based on individual goals of therapy and the potential risks of adverse reactions. Patients using the 200 mg once daily dosage could consider reducing the dosage to 100 mg once daily after symptom control is achieved at week 12. Relative to patients who maintained the 200 mg dose, the risk of occurrence of serious adverse reactions decreased in patients who reduced their dose to 100 mg beyond 12 weeks. If symptom control was lost after dose reduction, the dose could be increased to 200 mg. All the included studies investigated the use of abrocitinib at the starting dosage recommended in the product monograph (i.e., either 100 mg once daily or 200 mg once daily). The JADE REGIMEN trial was the only study that investigated dose reduction from 200 mg once daily to 100 mg once daily. In the JADE COMPARE trial, dupilumab was administered at the dosage recommended in the Canadian product monograph (i.e., 300 mg administered by IV injection in the morning and 150 mg in the evening every 2 weeks). Due to the need to ensure blinding, patients in the JADE COMPARE and JADE DARE trials underwent a more complicated dosage regimen than would be required for typical administration of abrocitinib or dupilumab (i.e., they received both orally administered tablets daily and subcutaneous injections once every 2 weeks). Nevertheless, as noted above, adherence with study treatments was high throughout the run-in and double-blind treatment periods. The JADE COMPARE, JADE DARE, and JADE TEEN trials compared the addition of abrocitinib to standardized AD management therapies. The clinical expert consulted by CADTH noted that these therapies are reasonably consistent with those used in Canadian clinical practice.
The included studies evaluated a range of outcomes that are important in the management of AD, including overall severity of AD (e.g., EASI and IGA), severity of itching (e.g., PP-NRS), symptoms (e.g., POEM and PSAAD), health-related quality of life (e.g., DLQI and CDLQI), fatigue (e.g., FACIT-F and Peds-FACIT-F), patient-reported anxiety and depression, and need for additional AD medications (e.g., corticosteroid-free days). In addition, the JADE REGIMEN study investigated the use of abrocitinib (100 mg once daily or 200 mg once daily) as maintenance therapy for patients who achieved an initial response to the 200 mg once daily dosage regimen by evaluating the time to acute worsening of the patient’s condition (i.e., development of a disease flare in accordance with standardized criteria). The clinical expert consulted by CADTH noted that the EASI and IGA are clinically relevant and can be used in routine Canadian practice to evaluate the response to treatment with abrocitinib for patients with moderate-to-severe AD (i.e., for the purposes of establishing renewal criteria for reimbursement by the public drug programs). Subgroup analyses for patients with prior exposure to at least 1 systemic therapy for AD were limited to IGA and EASI-75 responses (i.e., the co-primary end points). The clinical expert consulted by CADTH indicated that these analyses suggested that the response to abrocitinib would likely be similar for those with and those without prior exposure to a systemic therapy for AD.
As AD is a chronic disease, abrocitinib would likely be used as a long-term treatment for patients who require systemic therapy. The placebo-controlled trials were short-term (12 and 16 weeks) with only limited data available from the longer-term JADE EXTEND and JADE REGIMEN trials at the time this review. Complete reporting of the longer-term studies will help characterize the longer-term efficacy and safety of abrocitinib in the treatment of AD.
Patients enrolled in the included RCTs received extensive contact with health professionals over the study periods (e.g., 10 clinic visits over the 20-week study period of the JADE COMPARE trial). Although common in clinical trial settings, this level of contact is not reflective of routine care for patients with moderate-to-severe AD in Canada. The clinical expert consulted by CADTH noted that patients would likely been seen 4 months after initiating treatment with a therapy such as abrocitinib (depending on the ability to arrange appointments). This level of contact in the clinical trials is typical for AD studies and would not be expected to significantly affect the generalizability of the results to the target population in Canada. Adherence with study treatments was high throughout the treatment periods of all studies (i.e., median adherence of 100%). The clinical expert consulted by CADTH noted that the level of adherence observed in the included studies is not reflective of typical adherence in Canada, particularly for adolescent patients, for whom adherence to treatments, including orally administered treatments, is considerably lower.
Table 55: Assessment of Generalizability of Evidence
Domain | Factor | Evidence | CADTH's assessment of generalizability |
|---|---|---|---|
Population | Diagnostic criteria | Patients were required to have confirmed AD using the Hanifin and Rajka criteria. | The diagnostic criteria used in the screening process for all of the included studies were consistent with Canadian clinical practice for identifying patients with moderate-to-severe AD. |
Age | The included trials enrolled patients who were at least 12 years of age at screening. | This is reflective of the indication under review by CADTH. | |
Severity of AD | The trials enrolled patients with moderate-to-severe AD (defined as an affected BSA ≥ 10%, IGA ≥ 3, EASI ≥ 16, and pruritus NRS ≥ 4). | The clinical expert consulted by CADTH indicated that, overall, the populations enrolled in the included trials were a reasonable reflection of the target population in Canada. | |
Intervention | Abrocitinib | All the included studies investigated the use of abrocitinib at the starting dosage recommended in the product monograph (i.e., 100 mg q.d. and 200 mg q.d. with or without TCS). | The clinical expert consulted by CADTH noted that the 200 mg dosage may be preferred as the initial treatment and the reduced dosage may be used in younger patients or others where a reduce dosage is warranted. |
Concomitant AD therapies | The JADE COMPARE, JADE DARE, and JADE TEEN trials used a standardized regimen for combination therapy. | The clinical expert consulted by CADTH noted that the standardized regimen was a reasonable representation of combination therapy in Canada. | |
Comparator | Dupilumab | The JADE COMPARE and JADE DARE trials used dupilumab at the dosage recommended in the Canadian product monograph (i.e., 300 mg q.2.w.) | The 16-week JADE COMPARE trial was likely insufficient to fully realize the maximal treatment effects for dupilumab, potentially biasing the results in favour of abrocitinib in both the JADE COMPARE trial and the ITCs filed by the sponsor. This is supported by results from the 26-week JADE DARE trial, which showed an earlier onset of response for abrocitinib compared with dupilumab, but no significant differences between the 2 treatments at 26 weeks. |
Outcomes | Described in detail in Table 23 | The included studies evaluated outcomes that are important in the management of AD, including overall response, severity of itching, symptoms, health-related quality of life, fatigue, patient-reported anxiety, and depression, and need for additional AD medications. | The clinical expert consulted by CADTH noted that these are clinically relevant end points for evaluating response to treatment for AD. |
Setting | Contact with health care professionals | Patients enrolled in the included RCTs received extensive contact with health professionals over the study periods (e.g., 10 clinic visits over the 20-week study period of the JADE COMPARE trial). | The clinical expert consulted by CADTH noted that patients would likely be seen 4 months after initiating treatment with a therapy such as abrocitinib (depending on the ability to arrange appointments). This level of contact in the clinical trials is typical for AD studies and would not be expected to significantly impact the generalizability of the results to the target population in Canada. |
Outpatient setting | The included RCTs were conducted at specialized dermatology clinics. | The clinical expert consulted by CADTH indicated that this is consistent with how abrocitinib would be used in clinical practice (i.e., prescribed and evaluated by specialists with experience in the diagnosis and management of AD). |
AD = atopic dermatitis; BSA = body surface area; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; ITC = indirect treatment comparison; NRS = numeric rating scale; q.2.w. = every 2 weeks; q.d. = once daily; RCT = randomized controlled trial; TCS = topical corticosteroids.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Several treatments are under development for moderate-to-severe AD. Three JAK inhibitors (abrocitinib, upadacitinib, and baricitinib) and monoclonal antibodies such as lebrikizumab, tralokinumab, and nemolizumab are all being developed. Other than the JADE COMPARE trial, there is no other RCT that compares the efficacy and safety of abrocitinib with other active comparators, and only indirect evidence can provide informative data. The aim of this section is to summarize and critically appraise any ITCs that compare abrocitinib with other treatments for the management of moderate-to-severe AD in both monotherapy and combination regimes.
Patients with moderate-to-severe AD were evaluated in this review. Two unpublished ITCs submitted by the sponsor (1 NMA37 and 1 MAIC39) and a published ITC by the ICER38 identified in a separate literature search were summarized and critically appraised.
Description of Indirect Treatment Comparisons Identified
The sponsor submitted an NMA to evaluate the clinical efficacy and safety of investigational and approved systemic treatments of AD based on evidence from RCTs. The sponsor performed a systematic review to identify relevant studies for inclusion in the NMA. The included studies compared abrocitinib, baricitinib, dupilumab, nemolizumab, and placebo in both monotherapy and combination therapy. In addition, upadacitinib, lebrikizumab, and tralokinumab were compared in combination regimens. While a long list of outcomes was planned, the NMA only included EASI, IGA, SCORAD, PP-NRS, DLQI, HADS, POEM, TEAEs, and discontinuation due to AEs.
The ICER performed an NMA to compare the efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib with each other, dupilumab, and placebo in patients with moderate-to-severe AD in both monotherapy and combination therapy with TCS. While a second population of mild-to-moderate AD was also analyzed, it will not be summarized in this section as this target population is not of interest to the indication under review. While many outcomes were planned for inclusion, only EASI-50, EASI-75, EASI-90, IGA 0 and 1 and PP-NRS4 were reported in the NMA.
In addition, the sponsor also submitted an unanchored MAIC to supplement the NMA. The purpose of this analysis was to compare abrocitinib with cyclosporine, methotrexate, and azathioprine through the use of 2 trials (METHODA and NTR1916) that were not included in the previous NMA due to a lack of connectivity to the network. An unanchored MAIC between these trials and the JADE COMPARE trial was conducted by adjusting the weights of patients in the latter trial (for which individual patient data were available) to make the comparison. It was eventually determined that the NTR1916 trial was too clinically dissimilar to the JADE COMPARE trial (mainly due to the former's use of rescue medication) to perform the MAIC, and NTR1916 (as well as the azathioprine intervention) was dropped from the analysis. The final analyses therefore contained only the METHODA and JADE COMPARE studies and included comparisons between abrocitinib and cyclosporine as well as between abrocitinib and methotrexate. Outcomes examined were EASI, SCORAD, DLQI, and safety.
Table 56: Overview of Included Indirect Treatment Comparisons
Detail | Sponsor-submitted NMA | ICER NMA | Sponsor-submitted MAIC |
|---|---|---|---|
Population | Adults and children (≥ 12 years old) with moderate-to-severe AD | Adults with moderate-to-severe AD | Adults (≥ 18 years old) with moderate-to-severe AD |
Intervention |
|
|
|
Comparators |
|
|
|
Outcomes |
|
|
|
Study design | RCTs | RCTs | RCTs |
Publication characteristics | English only; 2-year limitation on conference abstracts | English only; included abstracts from conference proceedings | NA |
Exclusion criteria | Studies evaluating topical therapies, phototherapy alone, treatments intended primarily for infections, and systemic corticosteroids or antihistamines | Articles indexed as guidelines, letters, editorials, narrative reviews, case reports, or news items | NA |
Databases searched | MEDLINE, MEDLINE In-process, Embase, Cochrane Library (CDSR and CENTRAL), and Database of Abstracts of Reviews of Effects and health technology assessments database as well as hand searching of various grey literature sources; no limit was placed on publication date | MEDLINE, Embase, Cochrane Library (CDSR and CENTRAL) searches conducted from 1996 to present | NA |
Selection process | Abstracts screened independently by 2 reviewers with discrepancies resolved by third reviewer; full-text articles reviewed by single investigator and validated by second independent investigator with discrepancies resolved by a third party | Full-text articles screened by single reviewer, providing justification for exclusions | NA |
Data-extraction process | One investigator extracted full articles, with independent validation from a second investigator; extracted data logic reviewed and validated for additional quality assurance | Not specified | Not specified |
Quality assessment | Cochrane risk-of-bias assessment | Criteria published by the US Preventive Services Task Forces | Not specified |
AD = atopic dermatitis; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; HADS = Hospital Anxiety and Depression Scale; ICER = Institute for Clinical and Economic Review; IGA = Investigator's Global Assessment; ITC = indirect treatment comparison; MAIC = matched-adjusted indirect comparison; NA = not applicable; POEM = Patient-Oriented Eczema Measure; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; RCT = randomized controlled trial; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Source: Sponsor-submitted ITCs37,39 and ITC performed by ICER.38
Sponsor-Submitted Network Meta-Analysis
Objectives
While the rationale behind conducting the NMA was not explicitly stated, it can be presumed to be a lack of head-to-head evidence comparing abrocitinib with treatments other than placebo.
Study Eligibility, Selection Process, and Data Extraction
Multiple electronic databases, including MEDLINE, Embase, the Cochrane Central Register and Database of Systematic Reviews, Database of Abstracts of Reviews of Effects, and Health Technology Assessment database were searched, as was the grey literature for the past 2 years from the following meetings: World Congress of Dermatology, British Academy of Dermatologists, American Academy of Dermatology, and European Academy of Dermatology and Venereology. Clinical trials indexed on Clinicaltrials.gov, the European Medicines Agency European public assessment reports, product labels indexed on Drugs@FDA, and reference lists of eligible systematic literature reviews were also searched.
Studies were included if they were RCTs that reported the population and outcomes of interest. Studies were excluded if they were not randomized, had patients with only mild AD (or healthy volunteers), had all patients younger than 12 years of age, or less than 80% of patients met inclusion criteria. Also excluded were studies that looked solely at topical therapies, phototherapies, treatments intended primarily for infections, systemic corticosteroids, and systemic antihistamines; although studies that looked at these interventions in combination with systemic therapies were included.
Title and abstracts identified from searches were reviewed independently by 2 separate reviewers to determine if they should be included or excluded, with any discrepancies resolved by a third reviewer. Full-text articles and conference abstracts identified during initial screening by a single investigator were validated by a second reviewer; any discrepancies were resolved by a third party.
Studies that met eligibility criteria were extracted into a data-extraction template independently by 1 investigator and validated by a second. Extracted data logic was reviewed and validated for additional quality assurance.
Comparators
The authors planned to compare all JAK inhibitors, biologics, immunomodulators, and retinoids with each other as well as topical or phototherapies and placebo. The full list of used comparators is provided in Table 57.
Outcomes
The outcomes investigated in the NMA included clinical end points, quality-of-life measurements and AEs (Table 57). for the complete list of outcomes.
Quality Assessment of Included Studies
Included studies were checked for quality using the Cochrane risk-of-bias tool for RCTs and checked for potential bias according to 5 criteria: randomization, deviations from intended interventions, missing outcome data, measurement of outcome, and selection of the reported result. Risk of bias was assessed in each of these domains by a single reviewer and quality checked by an independent reviewer.
Evidence Networks
The evidence networks for the NMAs are presented in Figure 12 for the subgroup analyses of patients with prior exposure to at least 1 systemic therapy for AD and in Figure 13 for the base-case analysis. Networks for other outcomes were similar and based on reporting of the outcomes in each trial.
Figure 12: Evidence Network Diagrams for Prior Immunosuppressant Failure Subgroup
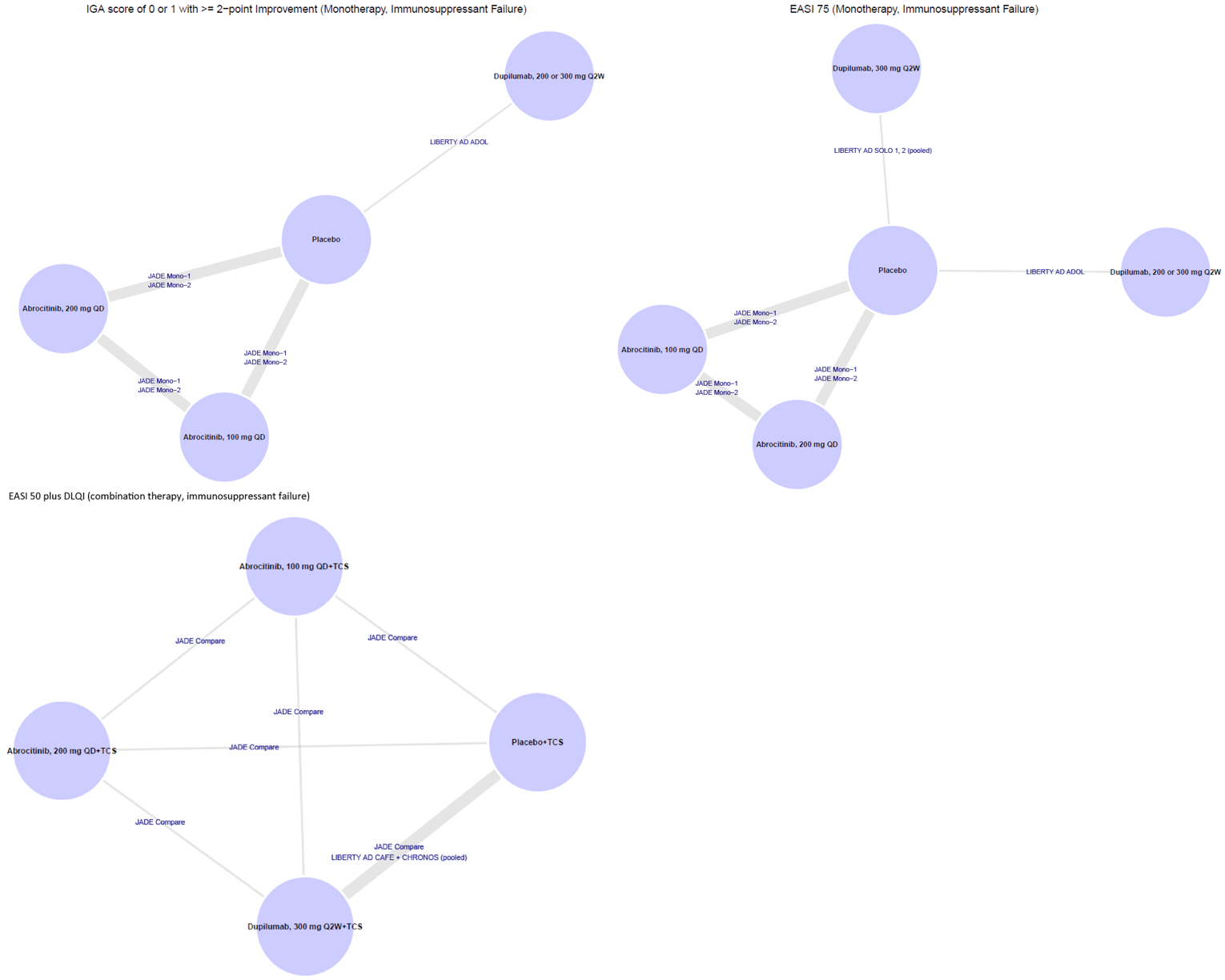
Source: Sponsor-submitted network meta-analysis.37
Figure 13: Evidence Network Diagrams for Base-Case Analysis
AE = adverse event; EASI = Eczema Area and Severity Index; QD = daily; Q2W = every 2 weeks; Q4W = every 4 weeks; TCS = topical corticosteroids.
Source: Sponsor-submitted network meta-analysis.37
Indirect Treatment Comparison Methods
Bayesian multinomial analyses were performed across the EASI-50, EASI-75, and EASI-90 response thresholds; standard Bayesian univariate analyses were carried out for all other outcomes. Results of the included studies were synthesized using Bayesian NMA models to obtain the relative treatment effects for abrocitinib versus each comparator. The analysis was conducted in OpenBUGS 3.2.3 using code and methods referenced in the National Institute for Health and Care Excellence Decision Support Unit's series of Technical Support Documents.69-71
The multinomial analyses were conducted using a probit link and the proportional odds assumption. The latter assumes identical pattern of response at each EASI level, allowing for borrowed strength across levels. In addition, 2 enhanced models were also conducted. The first was an adjustment for baseline risk using a regression component; this has been suggested to adjust for trials that have similar treatment response rates, but differing placebo response rates. A class-effects component was also proposed but not used due to sparseness of data, making the plan untenable. The second adjustment removed the assumption of equal treatment effects across thresholds and added a random-effects component to the difference between levels. All analyses were run using both the fixed and random effects for a total of 6 models. A seventh using fixed effects and incorporating both adjustments was also run; the equivalent random-effects model was not run due to sparse data that were deemed insufficient to estimate all variance components.
For all other outcomes, a standard Bayesian NMA was conducted using both fixed- and random-effects models. Noninformative priors (distributions not specified) were used for baseline and treatment effects. Markov chain Monte Carlo simulations were performed using 100,000 iterations as a discarded burn-in followed by another 100,000 simulations to estimate the posterior distributions. Convergence was checked through inspection of the ratio of Monte Carlo error to the SDs of the posteriors and confirmed by evaluating 3-chain Brooks-Gelman-Rubin plots; if convergence was not achieved, the run-in was increased. Model fit was assessed by comparing the deviance information criteria of the fixed- and random-effects models; a difference of 5 points was considered a substantially better-fitting model. Parameters were estimated from the median and the 2.5th and 97.5th percentiles of the posterior distributions. In choosing between fixed- and random-effects models when the deviance information criteria were similar, fixed effects would be preferred when data were sparse (i.e., no more than 2 studies looking at each comparison) while random-effects models would be preferred otherwise.
Statistical heterogeneity was assessed by computing the I2 statistic on pairwise comparisons with at least 2 trials reporting. High heterogeneity was explored through a review of patient characteristics from a clinical perspective. Consistency was not assessed as there were no instances of independent direct and indirect evidence to consider.
Table 57: Indirect Treatment Comparison Analysis Methods for Sponsor-Submitted NMA
Indirect treatment comparison methods | Network meta-analysis |
|---|---|
Priors | Noninformative (otherwise not specified) |
Assessment of model fit | Chosen based on deviance information criterion and other pre-specified factors |
Assessment of consistency | Not performed |
Assessment of convergence | Ratio of Monte Carlo error to the standard deviation of the posterior and confirmation by a Brooks-Gelman-Rubin plot |
Follow-up time points | 12 to 16 weeks |
Sensitivity analyses | Fixed and random effects; adjustment for baseline risk |
Subgroup analyses |
|
AD = atopic dermatitis; IGA = Investigator’s Global Assessment; NMA = network meta-analysis.
Source: Sponsor-submitted NMA.37
Results
Out of 25 trials identified for potential inclusion in the NMA, 19 were eventually included. Some trials included treatment arms that were not of interest (e.g., baricitinib 1 mg in BREEZE AD 1) and these arms were excluded from the NMA. One trial (TREBLE) included 3 arms of lebrikizumab but 2 treatment arms in which participants received a single administration of lebrikizumab; these arms were excluded from the NMA as this regimen differs significantly from common comparators received over a minimum of 12 weeks. Two trials were excluded because they evaluated dupilumab in dosages that differed from the approved dosage. Other trials were excluded because they did not have placebo arms and did not connect to the rest of the network. In total 7,073 patients were included in the NMA.
All 19 trials recommended for inclusion in the NMA were phase II or III RCTs that included a placebo arm. All were conducted over 12 to 16 weeks and were similar in terms of age (with 1 exception), geographic region, race, and duration of AD. While there was some variation in baseline disease severity, this did not result in exclusion from the NMA. In terms of outcome definitions, EASI was measured consistently across the trials in terms of the proportion of patients reporting an EASI-50, EASI-75, or EASI-90. While IGA scales differed slightly (0 to 4 versus 0 to 5), response scores were based on achieving a score of 0 or 1, which was defined similarly across scales. All but 3 of the trials were considered to be at low risk of bias.
Table 58 shows the characteristics and baseline demographics of the included studies.
Table 58: Summary of Baseline Characteristics
Trial | Sample size | Mean/median age | % male | % White | Average duration of AD (years) | % patients IGA 4 | Average EASI score at baseline | Overall risk of bias |
|---|---|---|---|---|---|---|---|---|
LIBERTY AD ADOL | 251 | 14.5 | 57.5 | 61.1 | 12.4 | 53.3 | 35.4 | Low |
JADE MONO-2 | 391 | 31.4 | 58.6 | 49.2 | 19.6 | 32.2 | 25.2 | Low |
JADE MONO-1 | 387 | 32.5 | 56.8 | 72.1 | NR | NR | NR | Low |
JADE COMPARE | 837 | 34.3 | 48.8 | 72.4 | 22.2 | 35.4 | 27.1 | Low |
LIBERTY AD SOLO 2 | 708 | 34.5 | 57.5 | 68.5 | 25.3 | 49 | 29.6 | Low |
BREEZE-AD2 | 615 | 35 | 61.5 | 68.5 | NR | 50.3 | 31.7 | Low |
XCIMA | 211 | 35.3 | 44 | NR | NR | 44 | 28.8 | Some |
BREEZE-AD1 | 624 | 35.5 | 62.7 | 59.2 | NR | 41.7 | 33.5 | Low |
Guttman-Yassky | 104 | 36.3 | 54.7 | 49.1 | 21.6 | NR | 21.3 | Low |
Thaci | 379 | 37 | 63 | NR | 28 | 48 | 32.3 | Low |
LIBERTY AD CHRONOS | 740 | 37.4 | 60.6 | 67 | 26.5 | 47.8 | 32.9 | Low |
Wollenberg | 103 | 37.5 | 33.6 | 57.3 | NR | NR | NR | Low |
TREBLE | 104 | 37.7 | 68.2 | 68.5 | NR | 21.5 | 25.2 | Low |
LIBERTY AD CAFE | 215 | 37.7 | 61.9 | 96.7 | 28.7 | 47.4 | 31.7 | Low |
LIIBERTY AD SOLO 1 | 671 | 38.5 | 55.5 | 67 | 27 | 48.5 | 31.1 | Low |
Silverberg | 114 | 40.5 | 52.6 | 74.6 | NR | 32.5 | 26.4 | Low |
Gooderham | 167 | 40.8 | 47.9 | 70 | 23 | 40.9 | 25.6 | Low |
BREEZE AD 7 | 329 | NR | NR | NR | NR | NR | NR | Some |
Reich | 123 | NR | NR | NR | NR | NR | NR | Some |
AD = atopic dermatitis, EASI = Eczema Area Severity Index, IGA = Investigator’s Global Assessment, NR = not reported.
Source: Sponsor-submitted network meta-analysis.37
As can be seen in Figure 14 the included trials almost universally compared a single treatment (usually in multiple doses) with a placebo arm. The lone exception is the JADE COMPARE trial, which had arms for abrocitinib, dupilumab, and placebo (all in combination with topical therapy). Table 60 outlines the dosages and regimens for all the treatments of interest.
Table 59: Doses and Regimens of Treatments of Interest
Treatment | Number of trials | Route |
|---|---|---|
Monotherapy regimens | ||
Abrocitinib 100 mg q.d. | 3 | Oral |
Abrocitinib 200 mg q.d. | 2 | Oral |
Baricitinib 2 mg q.d. | 2 | Oral |
Baricitinib 4 mg q.d. | 2 | Oral |
Dupilumab 200 or 300 mg q.2.w. | 2 | Subcutaneous |
Dupilumab 300 mg q.2.w. | 2 | Subcutaneous |
Nemolizumab 0.5 mg/kg q.4. w. | 1 | Subcutaneous |
Upadacitinib 15 mg q.d. | 1 | Oral |
Upadacitinib 30 mg q.d. | 1 | Oral |
Combination regimens | ||
Abrocitinib 100 mg q.d. | 1 | Oral |
Abrocitinib 200 mg q.d. | 1 | Oral |
Baricitinib 2 mg q.d. | 2 | Oral |
Baricitinib 4 mg q.d. | 2 | Oral |
Dupilumab 300 mg q.2.w. | 3 | Subcutaneous |
Lebrikizumab 125 mg q.4.w. | 1 | Subcutaneous |
Nemolizumab 30 mg q.4.w | 1 | Subcutaneous |
Tralokinumab 300 mg q.2.w. | 1 | Subcutaneous |
q.2.w. = every 2 weeks; q.4.w = every 4 weeks; q.d. = once daily.
Source: Sponsor-submitted network meta-analysis.37
Population With Prior Exposure to a Systemic Therapy for Atopic Dermatitis (Subgroup Analysis)
Monotherapy
Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy studies. Comparisons could only be conducted for abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, dupilumab 200 mg or 300 mg every 2 weeks and placebo. Results are summarized in Figure 14 for each comparator versus placebo and versus abrocitinib 200 mg once daily.
The odds ratios for IGA response were: abrocitinib 200 mg once daily versus placebo (||||||||||||||||||||||||||||||||||||]), abrocitinib 200 mg once daily versus dupilumab 200 or 300 mg every 2 weeks ||||||||||||||||||||||||||||||), and abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily (||||||||||||||||||||||||||).
The odds ratios for EASI-75 response were: abrocitinib 200 mg once daily versus placebo (||||||||||||||||||||||||||||||||||||), abrocitinib 200 mg once daily versus dupilumab 200 or 300 mg every 2 weeks (||||||||||||||||||||||||||||||||), abrocitinib 200 mg once daily versus dupilumab 300 mg every 2 weeks. (|||| |||| |||| |||| || ||||||), and abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily (|||| |||| |||| |||| || |||||).
Combination Therapy
Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to a single composite end point (EASI-50 plus DLQI improvement of ≥ 4 points) in the combination-therapy NMA. Comparisons could only be conducted for abrocitinib 100 mg once daily, abrocitinib 200 mg once daily, dupilumab 300 mg every 2 weeks, and placebo. Results are summarized in Figure 14 for each comparator versus placebo and versus abrocitinib 200 mg once daily.
The odds ratios for achieving an EASI-50 response and a DLQI improvement of 4 or more points were: abrocitinib 200 mg once daily versus placebo (|||| |||| |||| |||| || ||||||), abrocitinib 200 mg once daily versus dupilumab 300 mg every 2 weeks (|||| |||| |||| |||| || |||||), and abrocitinib 200 mg once daily versus abrocitinib 100 mg once daily (|||| |||| |||| |||| || |||||).
Figure 14: Subgroup Analyses for IGA Response, EASI-75 Response, and EASI-50 Plus DLQI improvement of 4 or More Points

CrI = credible interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; q.d. = daily; q.2.w. = every 2 weeks; q.4.w = every 4 weeks; TT = topical therapy.
Source: Sponsor-submitted NMA.37
Overall Population (Base Case)
The results of the NMA showed that abrocitinib 200 mg was |||||||| || ||||||||| ||| ||| ||||||||||| || || ||| | |||| ||||||||||| |||| |||||| ||| ||||||||||| ||| || |||| ||||||| || ||||||||| |||| |||||||| |||||| || || || || |||||| || |||||||||| || |||| |||||||| ||| |||||||| |||| ||||||||||| ||| || ||| |||||||| || |||||||||||| ||| || ||| || ||| ||| ||||||||| ||| || |||| ||| ||| ||| ||||||||| ||||||||||| ||| || ||| |||||||| || ||||||||||| || || ||| | ||| || |||| || ||||||||||| ||| ||| ||| ||||| ||| || |||||||||| |||| ||||||||||| ||| || ||| |||||||| || ||||||||| (Table 61). Other comparators (upadacitinib and nemolizumab) were not available to compare IGA responses. Subgroup analyses were conducted by patients reporting AD treatment failure, IGA score (3 versus 4), age (adolescent versus adult), and patients from Asia for the outcomes of EASI-75 and IGA. Results from all subgroup analyses conducted were consistent with the base-case analyses Examining other areas of efficacy (SCORAD-50, PP-NRS, DLQI, HADS, and POEM) showed no differences between abrocitinib and other active comparators, although many treatments are absent from these comparisons because their studies did not report the outcomes.
For combination therapies, abrocitinib 200 mg was |||||||| || ||||||||||| ||| ||| ||||||||||| || || ||| | ||| ||| |||||||||||| ||||| ||| || ||||| || ||||||||| |||| ||||||||| ||| ||| |||||||| ||||| ||| || |||||||||| |||| ||||||||||| ||| || ||| |||||||| || ||||| |||||||||| (Table 64). Upadacitinib was not included in this analysis. Among other outcomes, ||||||||||| ||| || ||| |||||||| || ||||||||||| || || ||| | ||| ||| |||| ||||||| ||| || ||||| ||||||||||| |||| |||||||||.
Analysis of safety outcomes (TEAEs and discontinuation due to AEs) |||||| || ||||||||||| ||||| ||||||, but as the credible intervals were wide and many interventions were missing from the analyses, ||||| || || |||||||||| |||||||| || |||||||||||.
Table 61 to Table 66 summarize the main results for the important outcomes in the network meta-analyses.
Table 60: Network Meta-Analysis Results for Binary and Ordinal Outcomes in Monotherapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments | |||
|---|---|---|---|---|
EASI-50/75/90 probit difference (95% CrI) | IGA response OR (95% CrI) | PP-NRS response OR (95% CrI) | SCORAD-50 OR (95% CrI) | |
Placebo | ||||| ||||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| |
Baricitinib 2 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||||| | |||||| |
Baricitinib 4 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||||| | |||||| |
Dupilumab 200 mg | |||||| | |||| |||||| ||||| | |||||| | |||||| |
Dupilumab 200 or 300 mg | |||||| |||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||||| |
Dupilumab 300 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||||| |
Nemolizumab 0.5 mg/kg | ||||| ||||||| |||||| | |||||| | |||||| | |||||| |
Upadacitinib 15 mg | ||||| |||||||| |||||| | |||||| | |||||| | |||| |||||| ||||| |
Upadacitinib 30 mg | |||||| |||||||| |||||| | |||||| | |||||| | |||| |||||| ||||| |
Abrocitinib 100 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||| |||||| ||||| |
Model fit parameters | ||||
Model used | |||||||||| || | |||||||| || | |||||||||| || | |||||||||| || |
Unadjusted fixed-effects DIC | |||||| | ||||| | ||||| | ||||| |
Unadjusted random-effects DIC | |||||| | ||||| | ||||| | ||||| |
Adjusted fixed-effects DIC | |||||| | ||||| | ||||| | ||||| |
Adjusted random-effects DIC | |||||| | ||||| | ||||| | ||||| |
CrI = credible interval; DIC = deviance information criterion; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; IGA = Investigator's Global Assessment; NA = not available; PP-NRS = peak pruritis numerical rating scale; OR = odds ratio; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Source: Sponsor-submitted network meta-analysis.37
Table 61: Network Meta-Analysis Results for Continuous Efficacy Outcomes in Monotherapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments | ||
|---|---|---|---|
CFB in DLQI MD (95% CrI) | CFB in POEM MD (95% CrI) | HADS total score MD (95% CrI) | |
Placebo | ||||| ||||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
Baricitinib 2 mg | |||||| | |||||| | |||||| |
Baricitinib 4 mg | |||||| | |||||| | |||||| |
Dupilumab 200 mg | |||| ||||||| ||||| | |||||| | |||||| |
Dupilumab 200 or 300 mg | |||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
Dupilumab 300 mg | ||||| ||||||| ||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
Nemolizumab 0.5 mg/kg | |||||| | |||||| | |||||| |
Upadacitinib 15 mg | |||||| | ||||| ||||||| ||||| | |||||| |
Upadacitinib 30 mg | |||||| | |||| ||||||| ||||| | |||||| |
Abrocitinib 100 mg | ||||| ||||||| ||||| | ||||| ||||||| |||||| | ||||| ||||||| ||||| |
Model fit parameters | |||
Model used | Unadjusted random effects | Unadjusted random effects | Unadjusted random effects |
Unadjusted fixed-effects DIC | 35.49 | 45.93 | 28.68 |
Unadjusted random-effects DIC | 35.49 | 47.35 | 29.31 |
Adjusted fixed-effects DIC | NA | NA | NA |
Adjusted random-effects DIC | NA | NA | NA |
CFB = change from baseline; CrI = credible interval; DIC = deviance information criterion; DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; NA = not available; NMA = network meta-analysis; POEM = Patient-Oriented Eczema Measure.
Source: Sponsor-submitted NMA.37
Table 62: NMA Results for Binary Safety Outcomes in Monotherapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments | |
|---|---|---|
Treatment-emergent AE OR (95% CrI) | Discontinuation due to AE OR (95% CrI) | |
Placebo | |||| |||||| ||||| | |||| |||||| ||||| |
Baricitinib 2 mg | |||||| | |||||| |
Baricitinib 4 mg | |||||| | |||||| |
Dupilumab 200 mg | |||||| | |||||| |
Dupilumab 200 or 300 mg | |||| |||||| ||||| | |||| |||||| |||||||| |
Dupilumab 300 mg | |||| |||||| ||||| | |||| |||||| ||||| |
Nemolizumab 0.5 mg/kg | |||||| | |||| |||||| ||||| |
Upadacitinib 15 mg | |||||| | |||||| |
Upadacitinib 30 mg | |||||| | |||||| |
Abrocitinib 100 mg | |||| |||||| ||||| | |||| |||||| ||||| |
Model fit parameters | ||
Model used | Unadjusted random effects | Unadjusted random effects |
Unadjusted fixed-effects DIC | 88.17 | 85.24 |
Unadjusted random-effects DIC | 86.81 | 85.85 |
Adjusted fixed-effects DIC | NA | NA |
Adjusted random-effects DIC | NA | NA |
AE = adverse event; CrI = credible interval; NA = not available, NMA = network meta-analysis; OR = odds ratio.
Source: Sponsor-submitted NMA.37
Table 63: NMA Results for Binary and Ordinal Outcomes in Combination Therapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments (all with topical therapy) | |||
|---|---|---|---|---|
EASI-50/75/90 probit difference (95% CrI) | IGA response OR (95% CrI) | PP-NRS response OR (95% CrI) | SCORAD-50 OR (95% CrI) | |
Placebo | ||||| ||||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| ||||| |
Baricitinib 2 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||||| |
Baricitinib 4 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||||| |
Dupilumab 300 mg | ||||| |||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||| |||||| ||||| |
Lebrikizumab 125 mg | ||||| |||||||| |||||| | |||| |||||| |||||| | |||||| | |||| |||||| ||||| |
Nemolizumab 30 mg | ||||| |||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||||| |
Tralokinumab 300 mg | ||||| ||||||| |||||| | |||||| | |||||| | |||| |||||| ||||| |
Abrocitinib 100 mg | ||||| ||||||| |||||| | |||| |||||| ||||| | |||| |||||| ||||| | |||| |||||| ||||| |
Model fit parameters | ||||
Model used | Adjusted fixed effects | Unadjusted random effects | Unadjusted random effects | Unadjusted fixed effects |
Unadjusted fixed-effects DIC | 101.21 | 123.6 | 88.52 | 68.02 |
Unadjusted random-effects DIC | 102.37 | 125.5 | 89.11 | 68.86 |
Adjusted fixed-effects DIC | 100.43 | 124.4 | 89.62 | NA |
Adjusted random-effects DIC | 102.22 | 126.1 | 89.48 | NA |
CrI = credible interval; DIC = deviance information criterion; EASI-50/75/90 = improvement of 50%/75%/90% or greater in the Eczema Area and Severity Index total score; IGA = Investigator's Global Assessment; NA = not available; OR = odds ratio; PP-NRS = peak pruritis numerical rating scale; NMA = network meta-analysis; OR = odds ratio; q.d. = once daily; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Source: Sponsor-submitted NMA.37
Table 64: NMA Results for Continuous Outcomes in Combination Therapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments (all with topical therapy) | ||
|---|---|---|---|
CFB in DLQI MD (95% CrI) | CFB in POEM MD (95% CrI) | HADS total score MD (95% CrI) | |
Placebo | ||||| ||||||| |||||| | ||||| |||||||| |||||| | ||||| ||||||| ||||| |
Baricitinib 2 mg | ||||| ||||||| |||||| | ||||| |||||||| ||||| | |||||| |
Baricitinib 4 mg | ||||| ||||||| |||||| | ||||| |||||||| ||||| | |||||| |
Dupilumab 300 mg | ||||| ||||||| ||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
Lebrikizumab 125 mg | |||||| | |||||| | |||||| |
Nemolizumab 30 mg | |||||| | |||||| | |||||| |
Tralokinumab 300 mg | ||||| ||||||| ||||| | |||||| | |||||| |
Abrocitinib 100 mg | ||||| ||||||| |||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
Model fit parameters | |||
Model used | Unadjusted random effects | Unadjusted random effects | Unadjusted random effects |
Unadjusted fixed-effects DIC | 35.99 | 29.81 | 19.05 |
Unadjusted random-effects DIC | 37.82 | 29.84 | 17.04 |
Adjusted fixed-effects DIC | NA | NA | NA |
Adjusted random-effects DIC | NA | NA | NA |
CFB = change from baseline; DIC = deviance information criterion; DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; MD = mean difference; NMA = network meta-analysis; NA = not applicable; POEM = Patient-Oriented Eczema Measure.
Source: Sponsor-submitted NMA.37
Table 65: NMA Results for Binary Safety Outcomes in Combination Therapy
Treatment | Abrocitinib 200 mg q.d. vs. all other treatments (all with topical therapy) | |
|---|---|---|
Treatment-emergent AE OR (95% CrI) | Discontinuation due to AE OR (95% CrI) | |
Placebo | |||| |||||| ||||| | |||| |||||| ||||| |
Baricitinib 2 mg | |||| |||||| ||||| | |||| |||||| ||||||| |
Baricitinib 4 mg | |||| |||||| ||||| | |||| |||||| ||||| |
Dupilumab 300 mg | |||| |||||| ||||| | |||| |||||| ||||| |
Lebrikizumab 125 mg | |||||| | |||||| |
Nemolizumab 30 mg | |||||| | |||||| |
Tralokinumab 300 mg | |||||| | |||||| |
Abrocitinib 100 mg | |||| |||||| ||||| | |||| |||||| ||||| |
Model fit parameters | ||
Model used | Unadjusted random effects | Unadjusted fixed effects |
Unadjusted fixed-effects DIC | 83.17 | 44.06 |
Unadjusted random-effects DIC | 83.57 | 44.28 |
Adjusted fixed-effects DIC | NA | NA |
Adjusted random-effects DIC | NA | NA |
AE = adverse event; CrI = credible interval; DIC = deviance information criterion; OR = odds ratio; NA = not available; NMA = network meta-analysis.
Source: Sponsor-submitted NMA.37
Critical Appraisal of Sponsor-Submitted Network Meta-Analysis
The protocol for the systematic review conducted by the sponsors was comprehensive, capturing relevant comparators and clinically relevant end points, according to the clinical expert consulted by CADTH for this review. The authors performed relevant subgroup analyses to explore potential heterogeneity across base-case analyses. They also performed sensitivity analyses that compared fixed- and random-effects methods as well as adjustments for baseline risk, comparing these analyses to see which had the better model fit. Their decision to analyze all the efficacy variables as continuous or ordinal was considered to be appropriate by the clinical expert consulted by CADTH.
The use of the Cochrane risk-of-bias tool to assess study quality was appropriate; most studies were found to be of low risk of bias and none to be at high risk of bias. Only 2 domains (selection of reported results and missing outcome data) were reported to be at high risk by any studies (1 case for each domain).
Some critical appraisal points can be made regarding the sponsor review:
A lack of reporting on statistical heterogeneity, statistical consistency, and publication bias was evident. In their methods, the authors report that they will analyze pairwise heterogeneity using the I2 statistic, but this is never reported on. While no pairwise comparisons involved more than 3 studies, I2 values would have been useful in determining the amount of statistical heterogeneity present in these analyses. Furthermore, while the authors did not conduct a consistency analysis to explore transitivity due to the absence of any closed loops that contained independent direct and indirect evidence, there was 1 closed loop (from the JADE COMPARE trial) in the combination therapy (placebo-dupilumab-abrocitinib) that could have been explored for consistency. In the absence of other measures of transitivity in the analysis, this loop should have been explored.
There was a lack of reporting regarding prior distributions: For the multinomial model, the authors are explicit about the priors they use, and the sensitivity analysis conducted around them. However, for the standard NMAs there is no mention of what distributions are used for the priors (only that they are noninformative) nor is there any mention of sensitivity analyses of the choices of these priors. In small NMAs (as many of those conducted were) the prior for the between-study variance in a random-effects model can often be influential on the final results,72 and more information regarding this issue would have strengthened the robustness of the findings.
While most of the trials had an end point of 16 weeks, all the abrocitinib trials had an end point of 12 weeks. The authors suggest this may underestimate the effects of abrocitinib, but the true effect this point of clinical heterogeneity has on the final results is uncertain. One trial (LIBERTY AD ADOL) was performed on adolescents, and this may have added heterogeneity to the analysis.
The authors focused their reporting on abrocitinib 200 mg and did not give the same attention to abrocitinib 100 mg (only comparing it with placebo). No league tables were given to allow for comparisons across all interventions.
Many of the networks, particularly in the combination therapies, were relatively sparse, creating wide credible intervals for many comparisons, particularly in the safety analyses. High uncertainty remains in many of the estimates.
The LIBERTY AD ADOL trial involved adolescents and used a treatment dose based on weight (dupilumab 200 mg or 300 mg). These differences from the other trials in the NMA warrant a cautious approach to the comparisons with the dupilumab 200 mg or 300 mg dose due to increased clinical heterogeneity.
The choice of model was based on the statistical significance parameter. The authors made an a priori decision when selecting which model to use as the primary analysis. If differences in the deviance information criteria were less than 2 points, they would choose the unadjusted random-effects model when data were substantive, and the unadjusted fixed-effects model when data were sparse. However, in the monotherapy analysis of IGA score they chose the adjusted random-effects analysis even though the deviance information criteria were similar (145.5 compared to 144.5) because the adjusted model had a statistically significant difference in baseline risk. The authors indicated that the unadjusted results are available in an appendix, but this does not seem to be the case as only the adjusted results are reported. Because these adjusted results present data that favour abrocitinib, it would be important to see what the difference the adjustment makes on the robustness of the findings.
The probit differences provided as the main output in the multinomial analysis are not clinically meaningful and can only be evaluated in terms of relative efficacy. Converting the probit differences to odds ratios (for example) would have given them more clinical meaning and interpretability.
Network Meta-Analysis Conducted by Institute for Clinical and Economic Review
Objectives and Rationale
The objectives of the ITC were to assess the relative efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib, as compared to each other, dupilumab, and placebo in populations with moderate-to-severe AD. A separate analysis that examined mild-to-moderate AD will not be considered for the purposes of this evaluation.
Study Eligibility, Selection Process, and Data Extraction
MEDLINE, Embase, and the Cochrane Library (both the Cochrane Database of Systematic Reviews and Central Register of Controlled Trials) were searched. Specific study design filters to identify RCTs for each database were applied.
Studies were included if they were RCTs that reported the outcomes of interest, included a treatment of interest, were done on the population of interest, were published since 1996, and were reported in English. Abstracts and conference proceedings were included. Articles indexed as guidelines, letters, editorials, narrative reviews, case reports, or news items were excluded. Screening was performed by a single investigator, as was inclusion and exclusion. How data extraction was conducted and whether more than 1 reviewer was involved were not reported.
Comparators
The comparators of interest were abrocitinib, baricitinib, tralokinumab, and upadacitinib compared to each other, topical therapies, and dupilumab (Table 57).
Outcomes
Efficacy outcomes identified to be assessed were patient-reported pruritus or itching, EASI-50 to EASI-75, and EASI-90 or relative change from baseline, IGA, sleep, SCORAD, POEM, DLQI, CDLQI, anxiety and depression (using the HADS), EQ-5D, measures of productivity (e.g., WPAI), and other patient-reported symptom and quality-of-life measures. Safety outcomes intended to be assessed were AEs, TEAEs, SAEs, discontinuation due to AEs, thrombotic events, infections, hematological abnormalities, malignancy, and all-cause mortality.
After data collection, NMAs were performed only on EASI, IGA, and PP-NRS (Table 57). Other outcomes were not reported due to inconsistent or limited data reporting and only described narratively.
Quality Assessment of Included Studies
Quality assessment was performed using criteria published by the US Preventive Services Task Force, rating each study as good, fair, or poor. Criteria used in the ratings were comparability of groups, reliability and validity of measurements instruments, intervention clarity, outcomes, attention to confounders, and performance of intention-to-treat analysis. Publication bias for the review was assessed by searching the clinicaltrials.gov database of trials to identify trials completed more than 2 years ago that would have met the inclusion criteria but for which no findings have been published.
Evidence Networks
The evidence networks for both monotherapy and combination therapies are shown below (Figure 15 and Figure 16).
Figure 15: Network Diagram for Monotherapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks, TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Source: ICER network meta-analysis.38
Figure 16: Network Diagram for Combination-Therapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks, TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Source: ICER network meta-analysis.38
Indirect Treatment Comparison Methods
The NMA was conducted using a Bayesian framework. The IGA and PP-NRS variables were analyzed as dichotomous outcomes using a binomial likelihood and log link. The EASI outcomes were analyzed as ordinal data with 4 distinct groups (EASI < 50, EASI-50, EASI-75, and EASI-90). Mutually exclusive groups were created by reclassifying the data as less than 50, 50 to 74, 75 to 89, and 90 or greater. A multinomial likelihood model with a probit link used methods from the National Institute for Health and Clinical Excellence Decision Support Unit.71 Unspecified noninformative priors were used for all model parameters. A Markov chain Monte Carlo simulation was performed, discarding the first 50,000 iterations as burn-in and basing inferences on an additional 50,000 iterations using 3 chains. Convergence was determined through visual examinations of the Brook-Gelman-Rubin diagnostic and historical plots. Models were run for both fixed and random effects as well as both unadjusted and adjusted for differences in placebo response. Models with the lowest deviance information criterion were considered to have the best fit between fixed and random effects. The model with placebo adjustment was considered a better fit if the regression coefficient was statistically significant and there was a reduction in between-trial heterogeneity. Only the analysis for the primary model was reported.
No information was given on details of the assessment of statistical heterogeneity, statistical consistency, or transitivity. Many trials had multiple arms with multiple doses of the same drug; these were treated as separate nodes within the NMA, and there was no pooling of arms to create nodes.
The models performed are summarized in Table 67.
Table 66: Network Meta-Analysis Reported by Institute for Clinical and Economic Review
Outcome | Trial type | Model | Number of trials |
|---|---|---|---|
EASI | a) Monotherapy only b) Combination only | Multinomial with probit link | a) 15 b) 6 |
IGA | a) Monotherapy only b) Combination only | Binomial with log link | a) 14 b) 6 |
PP-NRS4 | a) Monotherapy only b) Combination only | Binomial with log link | a) 14 b) 5 |
EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale.
Source: Institute for Clinical and Economic Review network meta-analysis.38
Table 67: Analysis Methods Used for ICER Network Meta-Analysis
ITC methods | NMA |
|---|---|
Priors | Noninformative |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | Not reported |
Assessment of convergence | Brook-Gelman-Rubin diagnostic and historical plots |
Follow-up time points | 12 to 16 weeks |
Sensitivity analyses | Not reported in NMA (only cost-effectiveness analysis) |
Subgroup analyses (descriptive only; not included in NMA) | Age (children/adolescents/adults) Disease severity (moderate/severe) |
ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis.
Source: ICER NMA.38
Results
Out of 58 trials identified for potential inclusion in the NMA, 21 were eventually included: 15 in the monotherapy analysis and 6 in the combination-therapy analysis. Reasons for exclusion were not specified. All 21 trials enrolled adults and only 2 trials (Heads Up and JADE COMPARE) included active comparator groups (i.e., dupilumab 300 mg every 2 weeks). All trials were conducted over 12 to 16 weeks, used stable doses, and were similar in terms of age, duration of AD, and disease diversity. Although dupilumab was tested at different doses, only the FDA-approved dosage of 300 mg once every 2 weeks was included in the NMA. All studies were of parallel design and assessed to be of “good” quality according to the US Preventive Services Task Force rating scale. All studies used some form of imputation, but the methods varied across studies. Multiple imputation, last observation carried forward, and nonresponse imputation were used in various combinations to account for missing data. Table 69 lists the important characteristics and baseline demographics of the included studies.
Table 68: Baseline Characteristics of Included Studies
Trial | Monotherapy or combination therapy | Doses | Sample size (N) | EASI (mean) | Mean age, years | Mean disease duration, years | IGA score of 4 (%) |
|---|---|---|---|---|---|---|---|
Abrocitinib trials | |||||||
JADE MONO-1a | Monotherapy | 100 mg 200 mg | 387 | 30.2 | 32.4 | 23.4 | 40.7 |
JADE MONO-2a | Monotherapy | 100 mg 200 mg | 391 | 28.5 | 35.1 | 21.0 | 32.2 |
JADE COMPARE | Combination | 100 mg 200 mg Dupilumab 300 mg | 837 | 30.9 | 37.7 | 22.7 | 35.4 |
Gooderham (2019) | Monotherapy | 100 mg 200 mg | 167 | 25.6 | 40.8 | 23.0 | 40.8 |
Baricitinib trials | |||||||
BREEZE-AD 1 | Monotherapy | 1 mg 2 mg 4 mg | 624 | 31.0 | 35.7 | 25.7 | 41.8 |
BREEZE-AD 2 | Monotherapy | 1 mg 2 mg 4 mg | 615 | 33.5 | 34.5 | 24.0 | 50.5 |
BREEZE-AD 5 | Monotherapy | 1 mg 2 mg | 440 | 27.1 | 39.7 | 23.7 | 41.7 |
BREEZE-AD 7 | Combination | 2 mg | 329 | 29.57 | 33.8 | 24.03 | 45.0 |
Guttman-Yassky (2018) | Combination | 2 mg 4 mg | 104 | 21.23 | 36.5 | 22.03 | NR |
Tralokinumab trials | |||||||
ECZTRA 1 | Monotherapy | 300 mg | 802 | 29.3 | 37.0 | 27.5 | 50.9 |
ECZTRA 2 | Monotherapy | 300 mg | 794 | 28.9 | 32.0 | 25.3 | 49.2 |
ECZTRA 3 | Combination | 300 mg | 380 | 25.5 | 36.0 | 26.0 | 46.3 |
Upadacitinib trials | |||||||
MEASURE UP 1a | Monotherapy | 15 mg 30 mg | 847 | 29.5 | 34.0 | 20.7 | 45.2 |
MEASURE UP 2a | Monotherapy | 15 mg 30 mg | 836 | 29.1 | 33.6 | 24.3 | 54.9 |
AD-UP | Combination | 15 mg 30 mg | 907 | 29.6 | 34.1 | 23.4 | 52.9 |
Heads Up | Monotherapy | Upadacitinib 30 mg Dupilumab 300 mg | 692 | 29.8 | 36.7 | 24.2 | 50.1 |
Guttman-Yassky 2018 | Monotherapy | 7.5 mg 15 mg 30 mg | 167 | 25.6 | 40.8 | 23.0 | 40.8 |
Dupilumab trials | |||||||
LIBERTY AD SOLO 1 | Monotherapy | 300 mg | 671 | 30.7 | 38.7 | 26.7 | 48.3 |
LIBERTY AD SOLO 2 | Monotherapy | 300 mg | 708 | 29.4 | 34.7 | 24.8 | 48.3 |
LIBERTY AD CHRONOS | Combination | 300 mg | 740 | 29.8 | 31.2 | 26.7 | 47.7 |
Thaci 2016 | Monotherapy | 100 mg 200 mg 300 mg | 379 | 31.9 | 37.0 | 28.0 | 47.3 |
EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment.
Note: All time points at 16 weeks, except JADE MONO-1, JADE MONO-2, (12 weeks) and COMPARE (12 or 16 weeks).
aPooled estimates from this trial were in patients 12 years of age and older.
Source: ICER network meta-analysis.38
Monotherapy
Table 69: Network Meta-Analysis Inputs for Monotherapy Outcomes
Trial | Arm | IGA | PP-NRS4 | EASI scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Response | Response | 50 | 75 | 90 | |||||||
N | n | N | n | N | n | N | n | N | n | ||
JADE MONO-1 | Abrocitinib 200 mg | 120 | 58 | 121 | 68 | RD | RD | 120 | 78 | RD | RD |
Abrocitinib 100 mg | 122 | 28 | 122 | 44 | RD | RD | 122 | 47 | RD | RD | |
Placebo | 60 | 4 | 60 | 11 | RD | RD | 60 | 7 | RD | RD | |
JADE MONO-2 | Abrocitinib 200 mg | 140 | 53 | 140 | 75 | RD | RD | 139 | 85 | RD | RD |
Abrocitinib 100 mg | 139 | 42 | 141 | 67 | RD | RD | 139 | 62 | RD | RD | |
Placebo | 70 | 7 | 70 | 8 | RD | RD | 70 | 8 | RD | RD | |
Goderham (2019) | Abrocitinib 200 mg | 48 | 21 | 44 | 28 | 48 | 38 | 48 | 31 | 48 | 21 |
Abrocitinib 100 mg | 54 | 16 | 50 | 25 | 54 | 30 | 54 | 22 | 54 | 14 | |
Placebo | 52 | 3 | 51 | 13 | 52 | 14 | 52 | 8 | 52 | 5 | |
ECZTRA 1 | Tralokinumab 300 mg | 601 | 95 | 594 | 119 | 601 | 250 | 601 | 150 | 601 | 87 |
Placebo | 197 | 14 | 194 | 20 | 197 | 42 | 197 | 25 | 197 | 8 | |
ECZTRA 2 | Tralokinumab 300 mg | 591 | 131 | 575 | 144 | 591 | 295 | 591 | 196 | 591 | 108 |
Placebo | 201 | 22 | 200 | 19 | 201 | 41 | 201 | 23 | 201 | 11 | |
MEASURE UP 1 | Upadacitinib 30 mg | 243 | 148 | 238 | 145 | RD | RD | 243 | 192 | RD | RD |
Upadacitinib 15 mg | 239 | 119 | 234 | 125 | RD | RD | 239 | 166 | RD | RD | |
Placebo | 241 | 21 | 233 | 26 | RD | RD | 241 | 43 | RD | RD | |
MEASURE UP 2 | Upadacitinib 30 mg | 247 | 125 | 246 | 150 | RD | RD | 247 | 180 | RD | RD |
Upadacitinib 15 mg | 243 | 93 | 240 | 103 | RD | RD | 243 | 144 | RD | RD | |
Placebo | 242 | 12 | 238 | 24 | RD | RD | 242 | 32 | RD | RD | |
Heads Up | Upadacitinib 30 mg | NR | NR | 336 | 120 | RD | RD | 348 | 247 | 348 | 211 |
Dupilumab 300 mg q.2.w. | NR | NR | 340 | 188 | RD | RD | 344 | 210 | 344 | 133 | |
Guttman-Yassky (2020) | Upadacitinib 30 mg | 42 | 21 | 36 | 19 | 42 | 35 | 42 | 29 | 42 | 21 |
Upadacitinib 15 mg | 42 | 13 | 32 | 19 | 42 | 30 | 42 | 22 | 42 | 11 | |
Placebo | 41 | 1 | 35 | 2 | 41 | 9 | 41 | 4 | 41 | 1 | |
BREEZE-AD 1 | Baricitinib 2 mg | 123 | 14 | 100 | 12 | 123 | 37 | 123 | 23 | 123 | 13 |
Baricitinib 1 mg | 127 | 15 | 105 | 11 | 127 | 32 | 127 | 22 | 127 | 11 | |
Placebo | 249 | 12 | 222 | 16 | 249 | 38 | 249 | 22 | 249 | 12 | |
BREEZE-AD 2 | Baricitinib 2 mg | 123 | 13 | 106 | 16 | 123 | 34 | 123 | 22 | 123 | 11 |
Baricitinib 1 mg | 125 | 11 | 100 | 6 | 125 | 23 | 125 | 16 | 128 | 8 | |
Placebo | 244 | 11 | 213 | 10 | 244 | 30 | 244 | 15 | 244 | 6 | |
BREEZE-AD 5 | Baricitinib 2 mg | 146 | 35 | 131 | 33 | 146 | 51 | 146 | 43 | 146 | 30 |
Baricitinib 1 mg | 147 | 19 | 132 | 21 | 147 | 29 | 147 | 19 | 147 | 11 | |
Placebo | 147 | 8 | 123 | 7 | 147 | 19 | 147 | 12 | 147 | 5 | |
SOLO 1 | Dupilumab 300 mg q.2.w. | 244 | 85 | 213 | 87 | 224 | 154 | 224 | 115 | 224 | 80 |
Placebo | 224 | 23 | 212 | 26 | 224 | 55 | 224 | 33 | 224 | 17 | |
SOLO 2 | DUP 300 mg q.2.w. | 233 | 84 | 225 | 81 | 233 | 152 | 233 | 103 | 233 | 70 |
Placebo | 236 | 20 | 221 | 21 | 236 | 52 | 236 | 28 | 236 | 17 | |
THACI (2016) | Dupilumab 300 mg q.2.w. | 64 | 19 | NR | NR | 64 | 50 | 64 | 34 | 64 | 1R9 |
Placebo | 61 | 1 | NR | NR | 61 | 18 | 61 | 7 | 61 | 2 | |
IGA = Investigator’s Global Assessment; NR = not reported; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.2.w. = every 2 weeks; RD = data redacted.
Source: ICER network meta-analysis.38
Figure 17 through Figure 21 show the NMA league tables comparing all outcomes in monotherapy across all included outcomes. All analyses were done using the random-effects, unadjusted models. Values greater than 1 favour the treatment in the column.
Figure 17: Network Meta-Analysis Results of EASI-75 in Monotherapy Trials in Adults
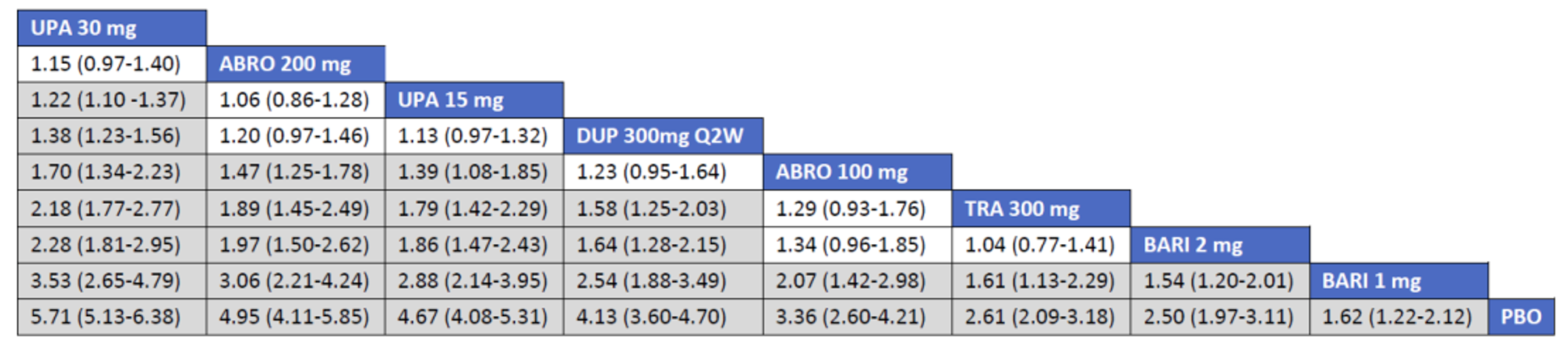
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib; Q2W = every 2 weeks.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source: ICER network meta-analysis.38
Figure18: Network Meta-Analysis Results of EASI-90 in Monotherapy Trials in Adults
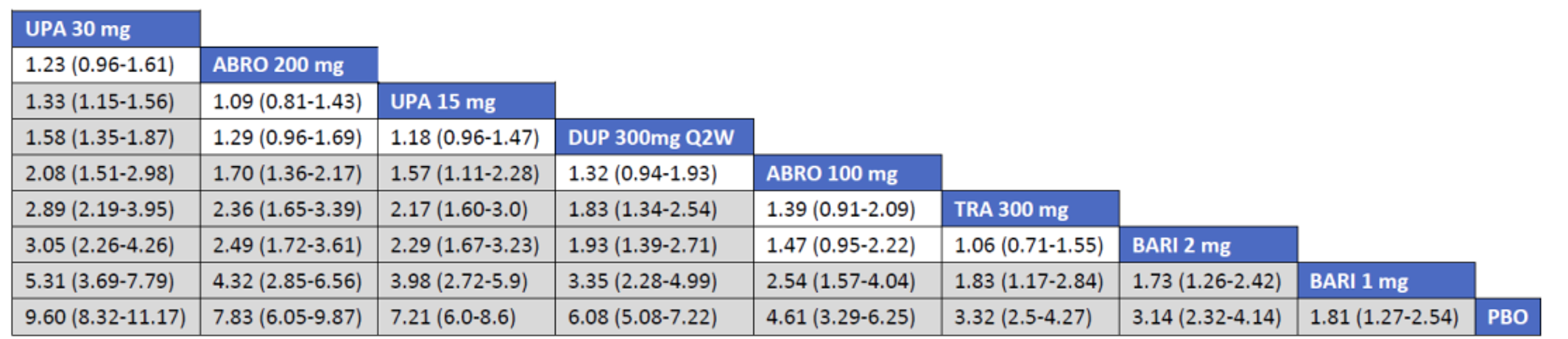
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib; Q2W = every 2 weeks.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 19: Network Meta-Analysis Results of EASI-50 in Monotherapy Trials in Adults
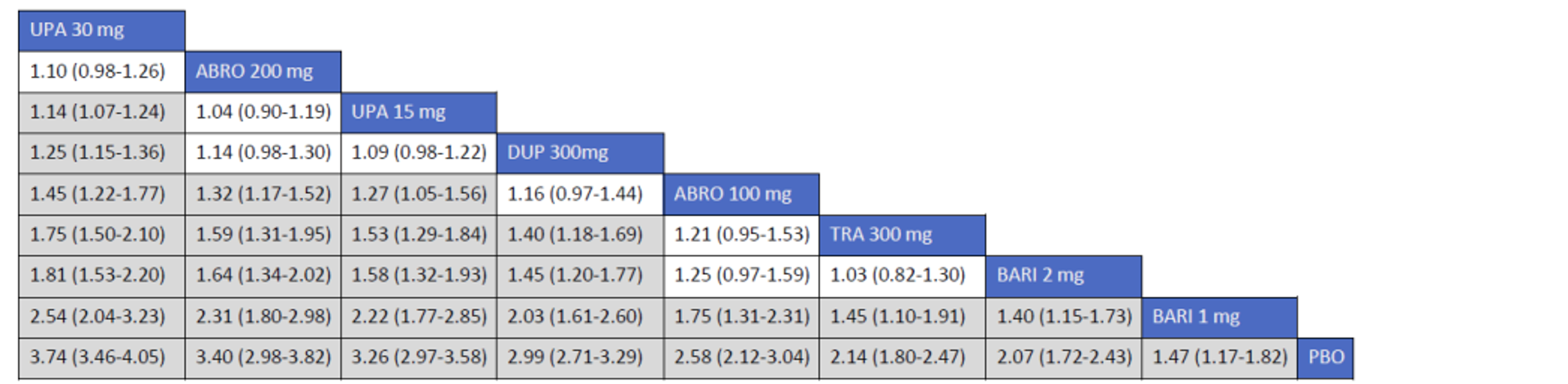
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 20: Network Meta-Analysis Results of IGA in Monotherapy Trials in Adults
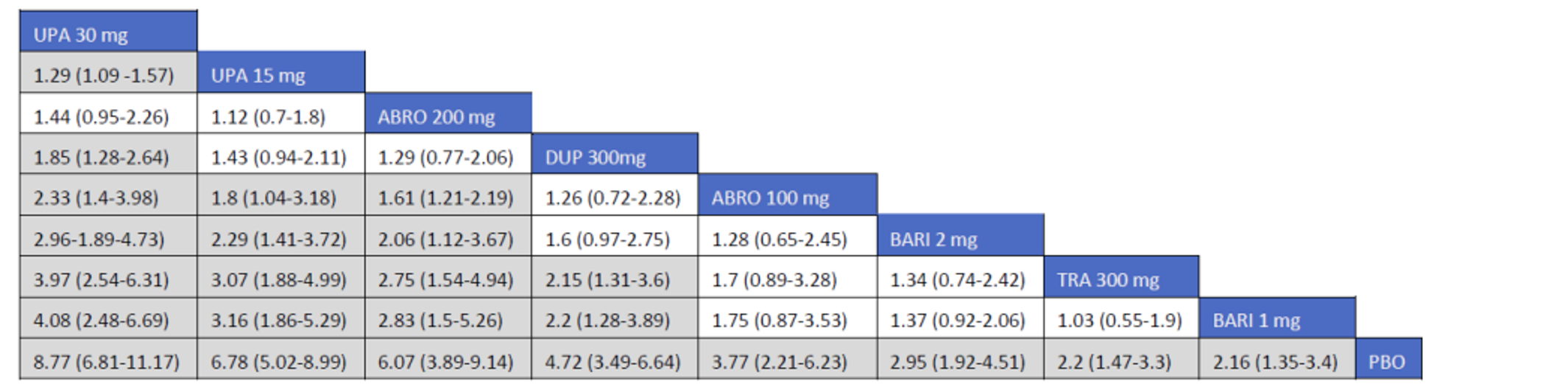
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; IGA = Investigator’s Global Assessment; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 21: Network Meta-Analysis Results of PP-NRS4 in Monotherapy Trials in Adults
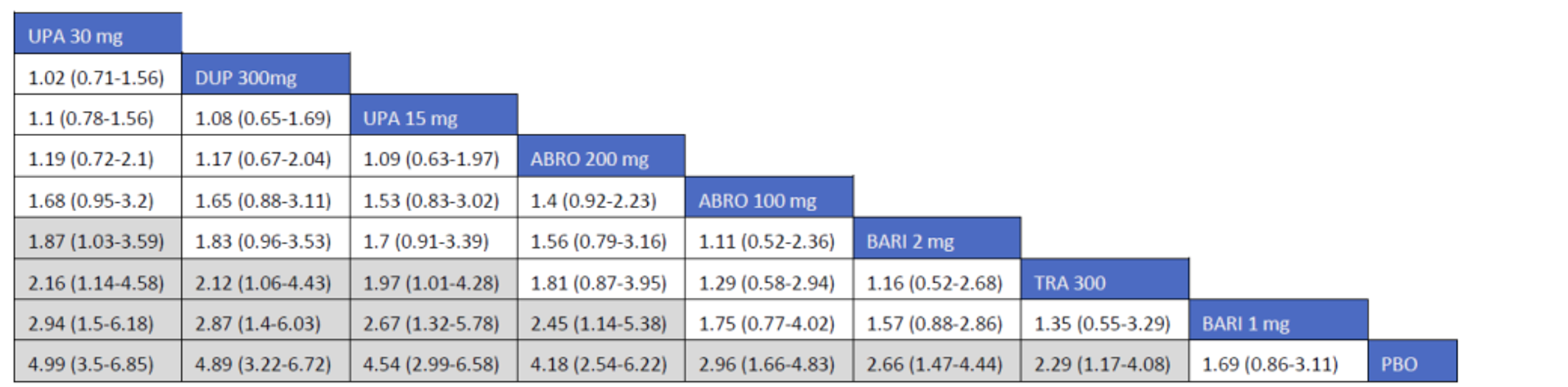
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source: ICER network meta-analysis.38
The results for monotherapy show that for EASI-50, EASI-75, and EASI-90 as well as IGA, abrocitinib 200 mg was superior to all other treatments except for upadacitinib (both 30 mg and 15 mg) and dupilumab 300 mg, which were comparable. In terms of PP-NRS, abrocitinib 200 mg showed superiority to baricitinib 1 mg and placebo, but was comparable to all other interventions.
Abrocitinib 100 mg was superior to placebo and baricitinib 1 mg for EASI-50, EASI-75, and EASI-90, but had inferior results when compared with upadacitinib (30 mg and 15 mg) and abrocitinib 200 mg; it was comparable with dupilumab, tralokinumab, and baricitinib 2 mg. In terms of IGA response, abrocitinib 100 mg only showed superiority to placebo it was inferior to upadacitinib (30 mg and 15 mg) and abrocitinib 200 mg, while being comparable to the other interventions. In terms of PP-NRS, abrocitinib 100 mg was superior to only placebo, while being comparable to all other interventions.
Combination Therapy
Table 70: Network Meta-Analysis Inputs for Combination-Therapy Outcomes
Trial | Arm | IGA | PP-NRS4 | EASI scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Response | Response | 50 | 75 | 90 | |||||||
N | n | N | n | N | n | N | n | N | n | ||
JADE COMPARE | Abrocitinib 200 mg | 221 | 105 | 172 | 108 | 221 | 193 | 221 | 157 | 221 | 108 |
Abrocitinib 100 mg | 230 | 80 | 168 | 79 | 229 | 186 | 229 | 138 | 229 | 87 | |
Dupilumab 300 mg q.2.w. | 232 | 90 | 189 | 108 | 232 | 195 | 232 | 152 | 232 | 90 | |
Placebo | 124 | 16 | 94 | 27 | 124 | 71 | 124 | 38 | 124 | 14 | |
ECZTRA 3 | Tralokinumab 300 mg | 252 | 98 | 249 | 113 | 252 | 200 | 252 | 141 | 252 | 83 |
Placebo | 126 | 33 | 126 | 43 | 126 | 73 | 126 | 45 | 126 | 27 | |
AD-UP | Upadacitinib 30 mg | 260 | 150 | 258 | 168 | RD | RD | 260 | 201 | RD | RD |
Upadacitinib 15 mg | 261 | 107 | 252 | 134 | RD | RD | 261 | 172 | RD | RD | |
Placebo | 264 | 30 | 256 | 39 | RD | RD | 264 | 68 | RD | RD | |
BREEZE-AD7 | Baricitinib 2 mg | 109 | 26 | 97 | 37 | 109 | 70 | 109 | 47 | 109 | 18 |
Placebo | 109 | 16 | 104 | 21 | 109 | 45 | 109 | 25 | 109 | 15 | |
Guttman-Yassky (2018) | Baricitinib 2 mg | 37 | 8 | NR | NR | 37 | 21 | 37 | 11 | 37 | 7 |
Placebo | 49 | 4 | NR | NR | 49 | 18 | 49 | 10 | 49 | 3 | |
LIBERTY AD CHRONOS | Dupilumab 300 mg q.2.w. | 106 | 41 | 102 | 60 | 106 | 85 | 106 | 73 | 106 | 42 |
Placebo | 315 | 39 | 299 | 59 | 315 | 118 | 315 | 73 | 315 | 35 | |
IGA = Investigator’s Global Assessment; NR = not reported; q.2.w. = every 2 weeks; RD = data redacted.
Note: All interventions (including placebo) were administered with topical corticosteroids.
Source: ICER network meta-analysis.38
Table 77 through Table 81 show the NMA league tables comparing all outcomes in combination therapy. All analyses were done using the random-effects, unadjusted models. Values greater than 1 favour the treatment in the column.
Figure 22: Network Meta-Analysis Results of EASI-75 in Combination-Therapy Trials in Adults
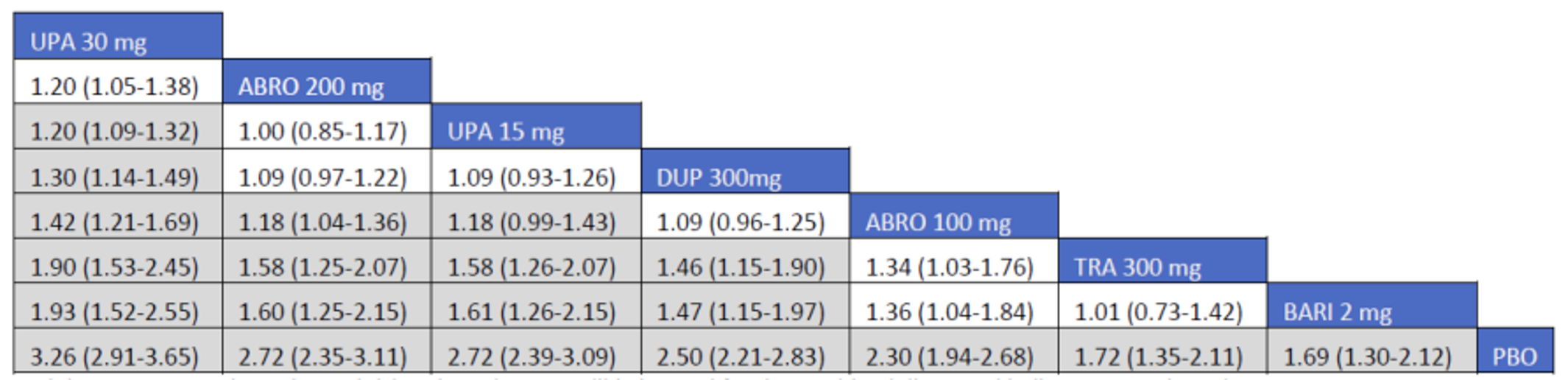
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 23: Network Meta-Analysis Results of EASI-90 in Combination-Therapy Trials in Adults
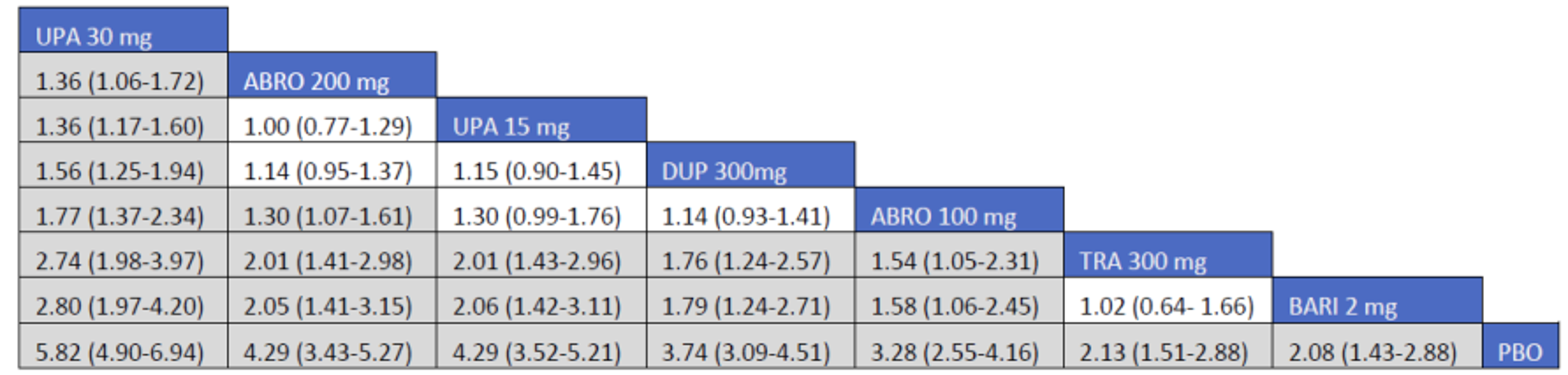
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-90 = improvement of 90% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 24: Network Meta-Analysis Results of EASI-50 in Combination-Therapy Trials in Adults
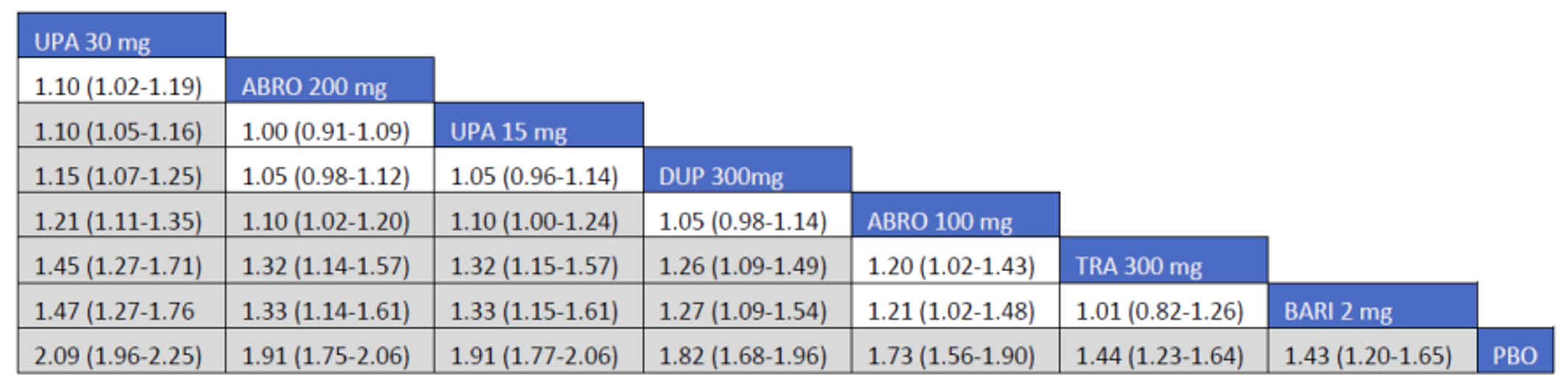
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain 1.
Source = ICER network meta-analysis.38
Figure 25: Network Meta-Analysis Results of IGA in Combination-Therapy Trials in Adults
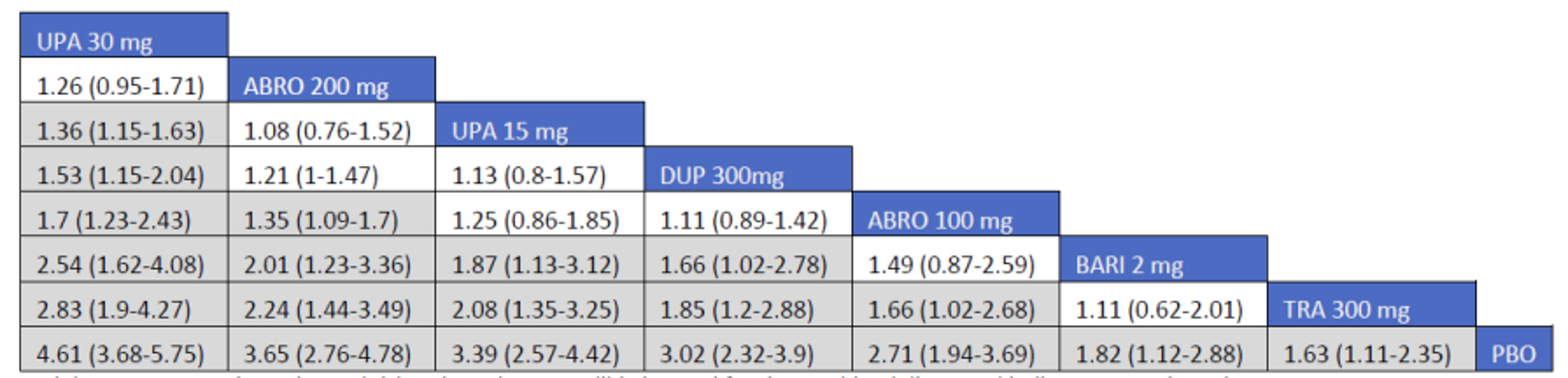
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; IGA = Investigator’s Global Assessment; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source = ICER network meta-analysis.38
Figure 26: Network Meta-Analysis Results of PP-NRS4 in Combination-Therapy Trials in Adults
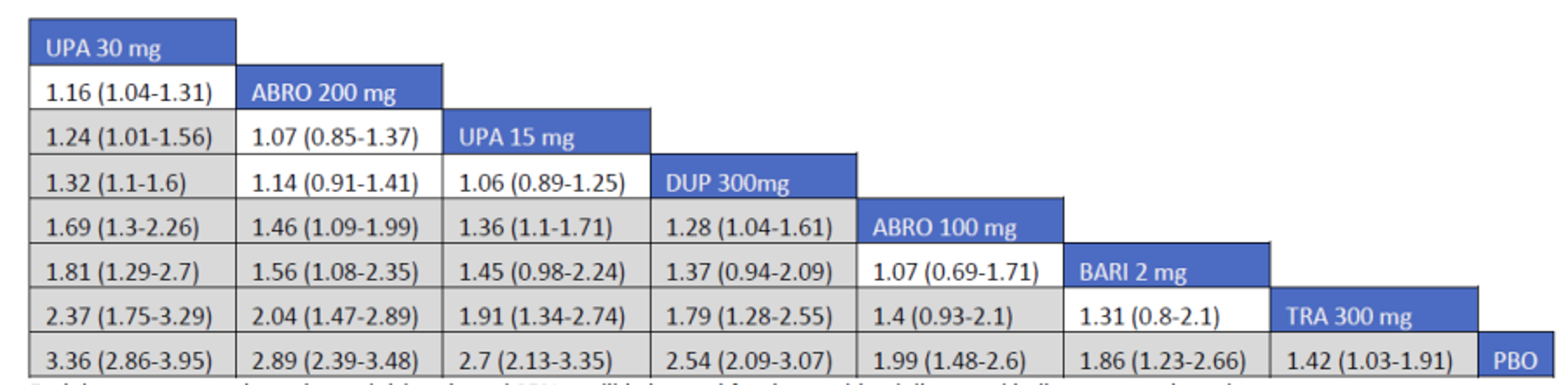
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source = ICER network meta-analysis.38
With respect to combination therapy for the EASI, IGA, and PP-NRS outcomes, abrocitinib 200 mg showed superiority to abrocitinib 100 mg, baricitinib 2 mg, tralokinumab 300 mg, and placebo. It was comparable with dupilumab 300 mg and upadacitinib 15 mg. Comparing abrocitinib 200 mg with upadacitinib 30 mg, the 2 were comparable for EASI-50, EASI-75, IGA, and PP-NRS while abrocitinib 200 mg was inferior for EASI-90.
Abrocitinib 100 mg in combination therapy showed inferiority to upadacitinib (30 mg and 15 mg) and abrocitinib 100 mg, comparability to dupilumab 300 mg, tralokinumab 300 mg, and baricitinib 2 mg, and superiority to placebo in EASI-50 and EASI-75. For EASI-90, it was superior to tralokinumab 300 mg, baricitinib 2 mg, and placebo, comparable to upadacitinib 15 mg and dupilumab 300 mg, and inferior to upadacitinib 30 mg and abrocitinib 200 mg. For IGA response, abrocitinib 100 mg showed superiority to tralokinumab and placebo, comparability to baricitinib 2 mg, dupilumab 300 mg, and upadacitinib 15 mg, and inferiority to upadacitinib 30 mg and abrocitinib 200 mg. For PP-NRS, abrocitinib 100 mg showed superiority to tralokinumab 300 mg and placebo, comparability with baricitinib 2 mg, and inferiority to all other treatments.
Harms
An NMA of harms data was not performed. Narrative summaries of the reported safety data indicated that nausea, conjunctivitis, and herpetic infection were more common in treatments than in placebo. Treatment-emergent AEs, SAEs, and discontinuation due to AEs were low and generally similar among treatments.
While no subgroup analyses were performed within the NMA, age and disease severity were examined using in-confidence data provided to the ICER by manufacturers. Results in adolescents were deemed to be similar to adults in abrocitinib, while abrocitinib showed greater efficacy among patients with severe disease compared to those with moderate disease.
Critical Appraisal of ICER Network Meta-Analysis
The eligibility criteria, PICOT process (patient, intervention, comparison, outcome, and time), and search strategy were comprehensive, and the authors reported that they followed Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines in the conduct of the review. Quality assessment of included studies was performed using a referenced tool. End point data were collected at 16 weeks for all trials except for the monotherapy abrocitinib trials, for which data were collected at 12 weeks. The pivotal trials for abrocitinib and upadacitinib enrolled adolescents, but the majority of patients were still 18 years of age or older, and the reviewers attempted to stratify by age. The trials were similar in terms of key baseline characteristics age (31 to 41 years), duration of disease (21 to 28 years) and disease severity (32% to 55% IGA score of 4). No unpublished studies meeting the inclusion criteria were identified in the clincaltrials.gov database, which was an indication of no evidence of publication bias.
Many critical appraisal points can be made regarding the ICER NMA:
Study screening was not verified by a second party, as all studies were screened by a single reviewer. The recommended practice is for screening by 2 or more independent reviewers to reduce the risk of selection bias.
For data extraction, there is no mention of duplicate data-extraction or data-validation methods.
While most of the trials had an end point of 16 weeks, all the monotherapy abrocitinib trials had an end point of 12 weeks. The true effect of this point of clinical heterogeneity on the final results is uncertain.
The results from the combination therapy were based on only 6 trials; despite this, credible intervals were relatively precise, and were in general more precise than the results of monotherapy (for which there were 15 trials). The reason for this apparent discrepancy is unclear.
The authors made no mention of the transitivity issue and the testing for consistency. While the lack of head-to-head comparisons among active treatments would make tests for consistency difficult, closed loops were present within the network that could have been tested.
There appears to have been no sensitivity analysis done within the NMA to explore any possible assumptions made by the reviewers. There is also no indication of an adjustment made in the model to account for the correlation in the 3 arm trials.
NMA results are presented only for EASI, IGA, and PP-NRS outcomes; other planned outcomes were not explored due to inconsistent or limited data reporting. Tables are presented giving narrative information on safety data. The lack of reporting for these additional outcomes of interest increases the likelihood of reporting bias.
All trials included in the review used imputation to adjust for missing data (combinations of multiple imputation, nonresponder imputation, or last observation carried forward), although there was no systematic difference in imputation methods across end points. It is unknown what kind of effect this may have had on final results.
Because there was insufficient information to perform any NMA on the populations of adolescents and children, information in those areas was restricted to descriptions of trial specific results.
Sponsor-Submitted Matching-Adjusted Indirect Comparison
Objectives and Rationale
The objective of the MAIC was to compare the efficacy and safety of abrocitinib to cyclosporin, methotrexate, and azathioprine in patients with moderate-to-severe AD.
Study Selection Methods
The 3 studies selected to potentially take part in the MAIC (JADE COMPARE, METHODA, and NTR1916) were chosen from the results of the literature search carried out for the sponsor-submitted NMA. The choice was based on the similarities in trial design among the 3 trials and the availability of the individual patient data for the JADE COMPARE study. While not specifically stated, the selections do not appear to have been systematic.
How data extraction and quality assessment were performed is not described. Table 57 provides further details.
Feasibility Assessment Methods
A compatibility assessment was performed through a comparative review to assess the similarities and differences among the JADE COMPARE, METHODA, and NTR1916 trials to determine whether they could be adequately adjusted in the MAIC analysis. Some of the important characteristics for the 3 studies are presented in Table 71.
Table 71: Comparative Characteristics of JADE COMPARE, METHODA, and NTR1916
Characteristic | JADE COMPARE | METHODA | NTR1916 | Comments |
|---|---|---|---|---|
Interventions | Abrocitinib 100 mg Abrocitinib 200 mg | Cyclosporine Methotrexate | Azathioprine Methotrexate | — |
Study period | 2018 to 2020 | 2008 to 2012 | 2009 to 2010 | JADE COMPARE more recent |
Blinding | Double-blind | Open-label | Single-blind | Different |
Treatment period | 16 weeks | 24 weeks | 24 weeks | JADE COMPARE shorter |
Number randomized | 838 | 97 | 43 | JADE COMPARE larger |
Topical therapy | Per-protocol use required | Allowed in first 4 weeks | Allowed | Lone exposure to topical therapy in JADE COMPARE |
Rescue therapy | None | None | Allowed in first 8 weeks | NTR1916 different |
Prohibited medications during trial | Systemic corticosteroids, immunosuppressant treatment, live vaccine, phototherapy, herbal medications | Systemic corticosteroids, immunosuppressant treatment, live vaccine, phototherapy | Not reported | Similar for JADE COMPARE and METHODA |
AD diagnosis | Moderate to severe | Moderate to severe | Severe | Different for NTR 1916 |
Age (mean) | 38.0 | 32.5 | 39.9 | METHODA younger |
% male | 48.3 | 60.8 | 52.4 | |
Mean weight (kg) | 75.7 | 71.4 | NR | |
Mean/median disease duration (years) | 23.0 | 23.5 | 36.3 | NTR1916 longer duration |
Mean baseline EASI | 31.2 | 18.5 | 29.2 | Much lower for METHODA |
Mean baseline SCORAD | 68.0 | 55.4 | 57.8 | Higher for JADE COMPARE |
Mean baseline DLQI | 15.9 | 12.5 | NR | METHODA lower. |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; SCORAD = Scoring Atopic Dermatitis.
Source: Sponsor-submitted matching-adjusted indirect comparison.39
Potential differences that could potentially not be adjusted for in a MAIC are highlighted below:
Trial design: JADE COMPARE was a large global study, while METHODA and NTR1916 were smaller single-country studies. The 3 trials also differed in blinding methods, time period, use of topical therapy, length of washout period, and use of rescue medication. Because these factors were consistent within trials, they could not be adjusted for in the MAIC.
Inclusion criteria: The JADE COMPARE and METHODA trials included patients with moderate-to-severe AD compared to NTR1916, which included only patients with severe AD. This difference could be partially, but not fully, offset through characteristic balancing in the MAIC.
Baseline characteristics: Most baseline characteristics were of reasonable similarity, with the exception of SCORAD (higher in the JADE COMPARE trial than in the METHODA trial), disease duration (shorter in the JADE COMPARE trial than in the NTR1916 trial) and proportion of patients with asthma and/or allergic rhinitis (smaller proportion in the JADE COMPARE trial than in the NTR1916 trial). These differences could reduce the effective sample size of the JADE COMPARE trial in the MAIC.
Instruments: Two of the main outcomes were measured differently in the JADE COMPARE and NTR1916 trials: SCORAD (103 maximum versus 108 maximum) and IGA (5-point scale versus 6-point scale). While SCORAD discrepancy was suspected to be a typographical error, the JADE COMPARE trial had much a lower IGA of less than 2 compared with the NTR1916 trial.
Outcomes: Based on availability and comparability with the JADE COMPARE, trial, outcomes of EASI-50, SCORAD-50, and DLQI of 5 or lower were chosen for the METHODA trial, and an IGA score of less than 2, SCORAD-50, and POEM score were chosen for the NTR1916 trial. The selection of AEs was selected based those that were commonly compared.
The final feasibility assessment determined that only the comparison between the JADE COMPARE and METHODA trials would be feasible. The potential MAIC between the JADE COMPARE and NTR1916 trials was not undertaken due to the allowance for rescue medication in the NTR1916 trial. This difference could not be adjusted for in the MAIC and was deemed too significant to leave unadjusted. The clinical expert consulted by CADTH for this review agreed with this assessment.
Indirect Treatment Comparison Analysis Methods
The authors conducted the analysis using 2 approaches. The first used observed data only; in this approach, patients with missing data in the METHODA trial were not used in the analysis. The second approach used nonresponse imputation, in which patients missing from the final results were assumed to be nonresponders. This second approach was considered the primary analysis.
Selection of matching variables is important in an unanchored MAIC. Both prognostic variables and treatment effect modifiers that are unbalanced between studies should be matched. The authors carried out ad hoc univariate and multivariate analyses to identify predictors for each of the examined outcomes (EASI-50, SCORAD-50, DLQI of 5 or lower, and proportion of patients with AEs). The analyses were run on the pooled abrocitinib arms from the JADE COMPARE trial (excluding patients who received prior cyclosporine or methotrexate to match with the exclusion criteria of the METHODA trial). The following potential predictors were entered into the model:
baseline SCORAD index
baseline SCORAD pruritus VAS
weight
sex
baseline DLQI
baseline BSA(%)
age
baseline EASI of 19 or greater
disease duration (years)
baseline EASI index.
Univariate predictors with a P value of less than 0.2 were entered into a multivariate model. Sex was also added to the multivariate model even though it did not reach the 0.2 threshold due to its association with all outcome variables. The following variables were used for the matching in the base-case as well as sensitivity analyses.
EASI-50
baseline DLQI score
baseline BSA
sex
baseline EASI index (sensitivity analysis only).
SCORAD-50
baseline SCORAD index
sex.
DLQI of 5 or lower
baseline DLQI score
weight
sex.
Safety outcomes
disease duration
baseline BSA
baseline DLQI score
age
baseline SCORAD Index (removed for a sensitivity analysis).
The MAIC analysis was conducted using a method described by Signorovitch et al. (2012) and guidelines from the National Institute for Health and Care Excellence (NICE),73,74 whereby a logistic propensity-score model that included all baseline characteristics in the matching was created. The weights were calculated as wi = exp(α+ xi′β), where xi′ is the vector of baseline variables included for matching. The β coefficients were estimated by the method of moments rather than the maximum likelihood (as is usually the case) because only aggregate data for the x’s were available for the competitor populations.74,75
Once the β coefficients were estimated, the equation was applied to the patients from the JADE COMPARE trial to calculate the individual patient weights. The weights were then used to calculate the effective sample size (ESS) achieved after weighting patients. The ESS was calculated by (Σwi)2 (Σwi2). If the populations were perfectly balanced before adjustment, wi for all patients would equal 1, and the ESS would be the original size of the index population. Adjustment for population differences assigns patients uneven weights, leading to an inevitable reduction in ESS. A small ESS indicates an irregular distribution of weights across patients, meaning that only a small fraction of patients drives the treatment effect. To account for an increased uncertainty caused by the reduction in ESS in the analyses, the weights were further normalized by dividing each wi by their sum (Σwi) and then multiplying it by the ESS. The sum of the normalized weights for all patients is guaranteed to equal 1.
For the purpose of visually examining the distribution of the weights, they were scaled relative to the original unit weight for each individual as recommended by NICE.73 This was done by dividing each wi by the sum (Σwi) and then multiplying it by the initial sample size, N. The distribution of the rescaled weights was assessed using a histogram for each MAIC analysis. A rescaled weight of greater than 1 would mean that an individual carries more weight in the reweighted population than in the JADE COMPARE trial, and a rescaled weight of less than 1 would mean that an individual carries less weight. The mean of rescaled weights is guaranteed to equal 1. The weights were derived in R version 3.5.1 using the code published by NICE.73
Once the weights were obtained, all dichotomous outcomes for the abrocitinib arms were recomputed using a weighted frequency approach. Relative treatment effects between abrocitinib and cyclosporine as well as between abrocitinib and methotrexate were computed as odds ratios for EASI-50, SCORAD-50, a DLQI score of 5 or lower and obtained using weighted logistic regression, and a robust sandwich estimator to compute standard errors.74 For AE outcomes, adjusted risk differences were obtained using a weighted binomial model with identity link function and a robust sandwich estimator for computation of standard errors. Analyses were conducted using SAS 9.4 and/or R version 3.5.1.
Results
The characteristics of the JADE COMPARE and METHODA trials can be seen in Table 82. Efficacy and safety results for each of the 4 comparisons were considered. For each comparison all 3 efficacy outcomes (SCORAD-50, EASI-50, and a DLQI score of 5 or lower) as well as 3 important safety outcomes (at least 1 AE, TEAE, and SAE) were examined. For each outcome, the base-case analysis, the sensitivity analysis (when performed), and the naive analysis (a computation carried out without any matching adjustment) were presented. For the efficacy outcomes, both the efficacy analyses done with nonresponse imputation as well as the comparison using the original data were presented. The nonresponse imputation was considered as the primary analysis in these cases.
Abrocitinib 100 mg Versus Cyclosporine
When comparing abrocitinib 100 mg with cyclosporine, the original sample size of 238 was reduced in the various analyses to a low ESS of 31 (in the EASI-50 sensitivity analysis) to a high ESS of 145 (in the base-case safety analyses). Table 72 lists the comparisons between abrocitinib 100 mg and cyclosporine. For efficacy outcomes, abrocitinib had a ||||||||||||| |||||| |||||||||| || |||||||| ||||||||| ||||||| |||||||| || |||||||||||| || ||| |||||||| ||||| ||||| ||||||||| ||| |||||| |||| ||| ||||||||||||| ||||||||| ||||||| ||||||||||. The MAIC adjustment did not change greatly from the naive analyses, nor did the sensitivity analyses. ||||||||||| ||||||| ||| ||| || ||| |||||| |||| ||| ||| ||||||| ||||| |||||||||||| |||| |||||||||| || |||||||||||.
For safety outcomes, ||||||||||| |||||| ||||||||||||| ||||| |||||| ||| ||| ||||| ||| |||| |||||||||| ||||||| |||| ||| ||||||||||||||||| |||||||||| |||| ||||||| ||| ||||| |||||| |||||||| |||| |||||||||||||||. Results were also similar across the base case, the sensitivity analysis, and the naive comparisons.
Table 72: Abrocitinib 100 mg Versus Cyclosporine
Outcome | Base case | Sensitivity analysis | Naive comparison |
|---|---|---|---|
SCORAD-50 (NRI) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| ||||| |
SCORAD-50 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
EASI-50 (NRI) | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |||| ||||||| |||||| |
EASI-50 (OD) | ||||| ||||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
DLQI ≤ 5 (NRI) | |||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
DLQI ≤ 5 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
At least 1 AE | |||||| ||||||| ||||| | |||||| ||||||| |||||| | |||||| ||||||| |||||| |
Treatment-related AE | |||||| ||||||| |||||| | |||||| ||||||| |||||| | |||||| ||||||| |||||| |
SAE | ||||| |||||| |||| | ||||| |||||| |||| | |||| |||||| |||| |
AE = adverse event; DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; NA = not applicable; NRI = nonresponse imputation; OD = original data; SAE = serious adverse event; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Note: SCORAD-50, EASI-50, and DLQI values are odds ratios (with 95% confidence intervals); safety outcomes are risk differences (with 95% confidence intervals). For odds ratios, numbers larger than 1 favour abrocitinib, for risk differences. numbers less than 0 favour abrocitinib.
Source: Sponsor-submitted matching-adjusted indirect comparison.39
Abrocitinib 100 mg Versus Methotrexate
When comparing abrocitinib 100 mg with methotrexate, the original sample size of 238 was reduced in the various analyses to a low ESS of 38 (in the EASI-50 sensitivity analysis) to a high ESS of 155 (in the base-case EASI-50 analysis).
Table 73 lists the comparisons between abrocitinib 100 mg and methotrexate. For efficacy outcomes, abrocitinib ||| | ||||||||||||| |||||| |||||||||| || |||||||| ||||||||| ||||||| ||| ||||||||| |||||||| || |||||||||||| || ||| |||||||| ||||| ||||| |||||| ||| ||| ||||||||||||| ||||||||| ||||||| ||||||||||| ||| |||| |||||||||| ||| ||| |||||| ||||||| |||| ||| ||||| ||||||||| ||| ||| ||| ||||||||||| ||||||||| ||||||||||| ||||||| ||| ||| || ||||||| ||| |||||| |||| ||| ||| ||||||| ||||| |||||||||||| |||| |||||||||| || |||||||||||.
For safety outcomes, abrocitinib |||||| ||||||||||||| ||||| |||||| ||| ||| ||||| ||| |||| ||||||||| ||||||| ||| |||| ||| ||||||||||| ||| ||| |||| |||| ||||||||| ||| |||||||| |||||||||||| || ||| ||||||||||| |||||||| |||| || |||||||| |||||| |||||| |||| ||| ||||| ||| ||||| |||||| |||||||| |||| ||||||||||||||| ||| ||| |||| ||||. Results were also similar across the base case, the sensitivity analysis, and the naive comparisons for all safety outcomes.
Table 73: Abrocitinib 100 mg Versus Methotrexate
Outcome | Base case | Sensitivity analysis | Naive comparison |
|---|---|---|---|
SCORAD-50 (NRI) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
SCORAD-50 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
EASI-50 (NRI) | ||||| ||||||| ||||||| | ||||| ||||||| ||||||| | ||||| ||||||| ||||||| |
EASI-50 (OD) | ||||| |||||| ||||||| | ||||| |||||| ||||||| | ||||| ||||||| |||||| |
DLQI ≤ 5 (NRI) | ||||| ||||||| |||||| | |||||| | ||||| |||||| |||||| |
DLQI ≤ 5 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
At least 1 adverse event | |||||| ||||||| ||||| | |||||| ||||||| |||||| | |||||| ||||||| |||||| |
Treatment-related adverse event | |||||| ||||||| |||| | |||||| ||||||| ||||| | |||||| ||||||| ||||| |
Serious adverse event | |||| ||| ||| |||||||||| | |||| ||| ||| |||||||||| | |||| ||| ||| |||||||||| |
DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; NA = not applicable; NRI = nonresponse imputation; OD = original data; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Note: SCORAD-50, EASI-50, and DLQI values are odd ratios (with 95% confidence intervals); Safety outcomes are risk differences (with 95% confidence intervals) For odds ratios, numbers larger than 1 favour abrocitinib; for risk differences, numbers less than 0 favour abrocitinib.
Source: Sponsor-submitted matching-adjusted indirect comparison.39
Abrocitinib 200 mg Versus Cyclosporine
When comparing abrocitinib 200 mg with cyclosporine the original sample size of 226 was reduced in the various analyses to a low ESS of 16 (in the EASI-50 sensitivity analysis) to a high ESS of 104 (in the base-case DLQI ≤ 5 analysis).
Table 74 lists the comparisons between abrocitinib 200 mg and cyclosporine. For efficacy outcomes, ||||||||||| ||| | ||||||||||||| |||||| |||||||||| || |||||||| ||||||||| ||||||| |||||||| || |||||||||||| || ||| |||||||| ||||| ||||| ||| ||||||||| ||| |||||| |||| ||| ||||||||||||| ||||||||| ||||||| ||||||||||| ||| |||||||||| ||| |||| |||||||||| ||| |||| || |||||| |||||||| || ||| ||||| |||||||| |||||||| |||| |||| |||||||||| ||| ||||||||||| ||| |||||, but the 2 were more similar for EASI-50 and a DLQI of 5 or lower. The EASI-50 responses showed a significantly larger effect in the sensitivity analysis compared to the base case. ||||||||||| ||||||| ||| ||| || |||||| |||| |||||||||| ||||||| || ||||||||||| |||| ||| ||| ||||||||| ||| ||||||||||| |||| |||| |||| |||| ||||||||||| |||| ||.
For safety outcomes, while abrocitinib had ||||||| |||||| || |||||| ||| ||| ||||| ||| |||| |||||||||| ||||||| |||| |||||||| |||| ||| ||||||||||||| ||||||||||| ||| ||| |||||||| |||| || ||||||||||| ||||||||| ||| ||||| |||||| |||||||| |||| |||||||||||||||. Results were similar across the base case, the sensitivity analysis, and the naive comparisons for most safety outcomes, although there were some potential differences.
Table 74: Abrocitinib 200 mg Versus Cyclosporine
Outcome | Base case | Sensitivity analysis | Naive comparison |
|---|---|---|---|
SCORAD-50 (NRI) | ||||| ||||||| |||| | |||||| | ||||| ||||||| ||||| |
SCORAD-50 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
EASI-50 (NRI) | ||||| ||||||| |||||| | ||||| ||||||| ||||||| | ||||| ||||||| ||||| |
EASI-50 (OD) | ||||| ||||||| ||||||| | ||||| |||||| |||||| | ||||| ||||||| |||||| |
DLQI ≤ 5 (NRI) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
DLQI ≤ 5 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| |||||| |
At least 1 adverse event | |||||| ||||||| |||| | |||||| ||||||| |||| | |||||| ||||||| ||||| |
Treatment-related adverse event | |||||| ||||||| |||| | |||||| ||||||| |||| | ||||||| ||||||| ||||| |
Serious adverse event | |||| |||||| |||| | |||| |||||| |||| | ||||| |||||| |||| |
DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; NA = not applicable; NRI = nonresponse imputation; OD = original data; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Note: SCORAD-50, EASI-50, and DLQI values are odd ratios (with 95% confidence intervals); Safety outcomes are risk differences (with 95% confidence intervals). For odds ratios, numbers larger than 1 favour abrocitinib; for risk differences number less than 0 favour abrocitinib.
Source: Sponsor-submitted matching-adjusted indirect comparison.39
Abrocitinib 200 mg Versus Methotrexate
When comparing abrocitinib 200 mg with methotrexate the original sample size of 238 was reduced in the various analyses to a low ESS of 19 (in the EASI-50 sensitivity analysis) to a high ESS of 124 (in the base case DLQI ≤ 5 analysis).
Table 75 lists the comparisons between abrocitinib 200 mg and methotrexate. For the EASI-50 and DLQI scores of 5 or lower outcomes, ||||||||||| ||| | ||||||||||||| |||||| |||||||||| || |||||||| ||||||||| |||||||||| |||||||| || |||||||||||| || ||| |||||||| |||| ||| ||||||||||| |||||||| ||| |||||||| ||||||||| ||| ||| ||||||||||||| ||||||||| ||||||| ||||||||||. The MAIC adjustment did not change greatly from the naive analyses for EASI-50 or DLQI score of 5 or lower but was different for SCORAD-50 (|||||||| |||||||| |||||| | ||||||| ||||||| ||| ||||||||||||| ||| ||||||||||| |||||||| ||| ||||||| |||||| | |||||| ||||||| ||| |||||||||||| ||||||||||| ||||||| ||| ||| || ||||||| |||| |||| ||||||||| |||| ||| ||| ||||||| ||||| |||||||||||| |||| |||||||||| || |||||||||||.
For safety outcomes, ||||||||||| |||||| ||||||||||||| ||||| |||||| ||| ||| ||||| ||| ||| || |||| |||| |||| ||| ||||||||||| ||||||||| ||| ||||| |||||| |||||||| |||| ||||||||||||||| ||| ||| |||| ||||| ||||||| |||| |||| ||||||| |||||| ||| |||| |||| ||| ||||||||||| ||||||||| ||| |||||||| |||||||| ||| ||| ||||| |||||||||| ||||| |||| ||||||| |||||||| |||||| |||||| ||| ||||||||||| || ||| ||||| |||||||||||.
Table 75: Abrocitinib 200 mg Versus Methotrexate
Outcome | Base case | Sensitivity analysis | Naive comparison |
|---|---|---|---|
SCORAD-50 (NRI) | ||||| ||||||| ||||| | |||||| | ||||| ||||||| ||||||| |
SCORAD-50 (OD) | ||||| ||||||| |||||| | |||||| | ||||| ||||||| ||||| |
EASI-50 (NRI) | |||||| ||||||| ||||||| | |||||| ||||||| ||||||| | |||||| ||||||| ||||||| |
EASI-50 (OD) | ||||| ||||||| ||||||| | |||||| ||||||| ||||||| | ||||| ||||||| ||||||| |
DLQI ≤ 5 (NRI) | ||||| |||||| |||||| | |||||| | ||||| |||||| ||||| |
DLQI ≤ 5 (OD) | ||||| |||||| |||||| | |||||| | ||||| ||||||| ||||| | |
At least 1 adverse event | |||||| ||||||| ||||| | |||||| ||||||| ||||| | ||||| ||||||| ||||| |
Treatment-related adverse event | ||||| ||||||| ||||| | ||||| ||||||| ||||| | |||| ||||||| |||| |
Serious adverse event | |||| ||| ||| |||||||||| | |||| ||| ||| |||||||||| | ||| ||| ||| |||||||||| |
DLQI = Dermatology Life Quality Index; EASI-50 = improvement of 50% or greater in the Eczema Area and Severity Index total score; NA = not applicable; NRI = nonresponse imputation; OD = original data; SCORAD-50 = improvement of 50% or greater in Scoring Atopic Dermatitis.
Note: SCORAD-50, EASI-50, and DLQI values are odd ratios (with 95% confidence intervals); Safety outcomes are risk differences (with 95% confidence intervals).
Source: Sponsor-submitted matching-adjusted indirect comparison.39
Critical Appraisal of Sponsor-Submitted Matching-Adjusted Indirect Comparison
The sponsor submitted an unanchored MAIC comparing abrocitinib with both cyclosporine and methotrexate for patients with moderate-to-severe AD between the JADE COMPARE and METHODA trials. A comparison using a MAIC was necessitated by the lack of a placebo arm in the METHODA trial, which prevented it from being included in the NMA. Guidelines from NICE were followed, and a thorough analysis of matching variables was conducted to perform the indirect comparison.
However, many critical appraisal points can be made with respect to the unanchored MAIC:
By its very nature an unanchored MAIC indirect analysis is subject to limitations. Although based on RCT data, the use of adjusted comparisons of arms from each of the trials without a common comparator or anchor makes it more akin to comparisons in observational studies (as they are not using comparisons between randomized groups within the trials). It is on the level with a propensity-score analysis, which attempts to control for confounding through regression in observational studies. The potential for confounding due to unused or unreported covariates is unknown but greatly increased over anchored comparisons. The MAIC results provided by the sponsor are therefore associated with substantial uncertainty.
No justification was given for the method of selecting the matching variables for each of the outcomes; they were described as ad-hoc. In an unanchored MAIC, the assumption of conditional constancy of absolute effects requires that adjustment be made for all effect modifiers and prognostic variables. There is no way to be certain important matching variables that could bias results were not considered in the ad hoc analysis, particularly when some of the analyses had so few matching variables.
While the original data analyses may bias comparisons in favour of cyclosporin and methotrexate, the nonresponder imputation performed in the primary analyses could bias results in favour of abrocitinib. Because the METHODA trial had more missing data compared with the JADE COMPARE trial, there were more imputed nonresponders in the METHODA trial. It is unknown how many of them are true nonresponders.
Because individual patient data were only available for the JADE COMPARE study, the matching had to carried out at an aggregate level, with weights adjusted in the JADE COMPARE trial to make it as similar as possible to the METHODA trial with respect to the matching variables. Clinical differences between the JADE COMPARE and NTR1916 trials (most prominently NTR1916’s use of rescue medication) prevented the inclusion of the NTR1916 trial in the analysis. But there were many differences between the JADE COMPARE and METHODA trials that could also not be adjusted using the MAIC. These include study period (the 2 were a decade apart), randomization method (double-blind versus open-label), treatment period (16 versus 24 weeks), and use of topical therapy. The effect these differences may have had on the comparisons is unknown. Due to the unanchored nature of the comparison, these concerns are more serious than they would be in a standard anchored indirect comparison. Unanchored MAICs make the much stronger assumption of “conditional constancy of absolute effects,” i.e., that the absolute treatment effects are assumed to be constant at any given level of the effect modifiers and prognostic variables, and all effect modifiers and prognostic variables are required to be known. This is unlikely to be true in these analyses.
All results from the MAIC were based on only 2 studies that were compared indirectly. The METHODA study had only 97 patients and, while the JADE COMPARE study had 838 patients, more than half were not comparable with the METHODA trial, and, after adjusting for matching variables, this was further reduced in the ESS computation. Because each efficacy comparison used only half the patients in each study, all the comparisons compared about 50 patients in the METHODA trial indirectly with an ESS of 50 to 150 patients (or even fewer in sensitivity analyses) in the JADE COMPARE study. The small ESS indicates that the weights were highly variable due to a lack of population overlap, and the resulting estimate may be unstable. Not only would some selection bias be inherent in this approach, but the imprecision is reflected in the wide confidence intervals of the estimates, and this should be considered when interpreting the results.
For the EASI-50 and safety outcomes, sensitivity analysis was performed based on perceived clinical importance of baseline EASI (for EASI) and lack of perceived clinical importance of baseline SCORAD index (for AEs). For the most part these analyses did not change from the base case but there were some exceptions (e.g., abrocitinib 200 mg versus cyclosporin showed a larger odds ratio in the sensitivity analysis). This shows that with such a small ESS, the choices of matching variables can have an impact on the result. Whether important variables have been left out of the matching process cannot be ascertained from the given data.
Despite the statistically significant differences observed between abrocitinib and both cyclosporin and methotrexate in many of the outcomes, these results need to be interpreted with extreme caution, as the unanchored nature of the MAIC will always be susceptible to an unknown amount of potential confounding. If any of the clinical heterogeneity between the JADE COMPARE and METHODA trials that could not be adjusted in the MAIC are correlated with the outcome, the estimates are likely to be biased in the absence of an anchor variable.
Summary of Indirect Evidence
The 2 NMAs (by the sponsor and the ICER) were conducted more than a year apart (search dates: October 2019 versus January 2021, respectively). Population characteristics for inclusion in the NMAs were similar. While the ICER’s NMA restricted interventions to abrocitinib, baricitinib, tralokinumab, upadacitinib, dupilumab, and placebo, the sponsor-submitted analysis was more inclusive, also examining the biologics lebrikizumab and nemolizumab as well as immunomodulators (none of which were included in the NMA but were examined in the later MAIC). The list of outcomes examined were similar between the 2 studies; the ICER looked at more outcomes initially than did the sponsor, but in the end, both analyzed the EASI and IGA. While the sponsor’s NMA performed formal ITCs on safety outcomes, the ICER’s review presented safety outcomes only in the form of descriptive summaries. While many studies were included in both NMAs, some studies included in 1 review were omitted from the other. In the case of studies included by the ICER that were omitted from the sponsor’s analysis, most had been unavailable at the time of the sponsor’s review. It is less clear why some of the studies in the sponsor’s analysis (e.g., Wallenberg, Reich) were omitted from the ICER’s analysis.
Both ITCs used Bayesian NMA methods to conduct their analyses. Although both NMAs chose to use a multinomial probit model to analyze the EASI score thresholds together, the sponsor presented the results as probit differences while ICER presented results separately for EASI-50, EASI-75, and EASI-90 as risk ratios. For all other variables the sponsors used a binomial model with an estimated odds ratio for binary efficacy outcomes and an estimated risk difference for safety outcomes. The ICER used a risk ratio to model all variables. Both NMAs used an adjustment for baseline risk model when the model fit was superior.
The use of different metrics (odds ratios versus risk ratios) makes it difficult to determine how similar the results of the 2 ITCs were. Furthermore, the probit differences presented for the EASI in the sponsor's analysis are not clinically meaningful. A comparison of results for the EASI for monotherapy are listed in Table 76.
Table 76: Comparison of Sponsor and ICER NMAs for EASI in Monotherapy
Intervention | Active vs. placebo | |
|---|---|---|
Sponsor NMA: EASI improvement probit difference (95% CrI) | ICER NMA: EASI-75 risk ratio (95% CrI) | |
Upadacitinib 30 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Abrocitinib 200 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Upadacitinib 15 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Dupilumab 200 or 300 mg q.2.w. | ||||| ||||||| |||||| | |||||| |
Dupilumab 300 mg q.2.w. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Abrocitinib 100 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Baricitinib 4 mg q.d. | ||||| ||||||| |||||| | |||||| |
Tralokinumab 300 mg q.2.w. | |||||| | |||| |||||| ||||| |
Baricitinib 2 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Baricitinib 1 mg q.d. | |||||| | |||| |||||| ||||| |
Nemolizumab 0.5 mg/kg q.4. w. | ||||| |||||||| |||||| | |||||| |
CrI = credible interval; EASI = Eczema Area and Severity Index; ICER = Institute for Clinical and Economic Review; NA = not available; NMA = network meta-analysis; NA = not available; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.d. = once daily.
Note: All estimates are relative to placebo. Numbers for the sponsor’s analysis are probit differences (with 95% CrIs) from a multinomial model (higher numbers are more favourable to treatment; numbers higher than 0 indicate superiority to placebo). Numbers for the ICER’s analysis are risk ratios (with 95% CrIs) from a binomial model for achievement of an improvement of 75% or greater in the EASI total score (higher risk ratio is more favourable to treatment; numbers higher than 1 indicate superiority to placebo).
Source: Sponsor-submitted NMA and ICER NMA.37,38
While it was not possible to compare the results from the 2 NMAs due to the different natures of the chosen methods, the 2 analyses do agree on the relative order of efficacy for the treatments they had in common. || |||| ||||||| ||| |||||||||| |||| |||||||| || ||||||| ||||||| ||| ||||||||||| || ||| ||||||| ||||||| |||| |||||||||||| || || ||||||| ||| |||| ||||||||. Table 77 lists similar results for combination therapy.
Table 77: Comparison of Sponsor and ICER NMAs for EASI in Combination Therapy
Intervention | Active vs. placebo | |
|---|---|---|
Sponsor NMA: EASI improvement probit difference (95% CrI) | ICER NMA: EASI-75 risk ratio (95% CrI) | |
Upadacitinib 30 mg q.d. | |||||| | |||| ||||| ||||| |
Abrocitinib 200 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Upadacitinib 15 mg q.d. | |||||| | |||| |||||| ||||| |
Dupilumab 300 mg q.2.w. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Abrocitinib 100 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Nemolizumab 30 mg q.4.w. | ||||| ||||||| |||||| | |||||| |
Lebrikizumab 125 mg q.4.w. | ||||| ||||||| |||||| | |||||| |
Tralokinumab 300 mg q.2.w. | ||||| ||||||| |||||| | |||| |||||| ||||| |
Baricitinib 4 mg q.d. | ||||| ||||||| |||||| | |||||| |
Baricitinib 2 mg q.d. | ||||| ||||||| |||||| | |||| |||||| ||||| |
CrI = credible interval; EASI = Eczema Area and Severity Index; ICER = Institute for Clinical and Economic Review; NA = not available; NMA = network meta-analysis; NA = not available; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.d. = once daily.
Note: All estimates are relative to placebo. Numbers for the sponsor’s analysis are probit differences (with 95% CrIs) from a multinomial model (higher numbers are more favourable to treatment; numbers higher than 0 indicate superiority to placebo). Numbers for the ICER’s analysis are risk ratios (with 95% CrIs) from a binomial model for achievement of an improvement of 75% or greater in the EASI total score (higher risk ratio is more favourable to treatment; numbers higher than 1 indicate superiority to placebo).
Source: Sponsor-submitted NMA and ICER NMA.37,38
For combination therapy, the relative order also remained unchanged for the treatments reported in both NMAs. All treatments in both reviews showed superiority to placebo. The other outcomes where it was possible to compare the 2 NMAs were for IGA 0 or 1 and PP-NRS4 in monotherapy. Side-by-side results are presented in Table 78 and Table 79:
Table 78: Comparison of Sponsor and ICER NMAs for IGA 0 or 1 in Monotherapy
Intervention | Active vs. placebo | |
|---|---|---|
Sponsor NMA OR (95% CrI) | ICER NMA risk ratio (95% CrI) | |
Upadacitinib 30 mg q.d. | |||||| | |||| |||||| |||||| |
Upadacitinib 15 mg q.d. | |||||| | |||| |||||| ||||| |
Abrocitinib 200 mg q.d. | |||| |||||| |||||| | |||| |||||| ||||| |
Dupilumab 300 mg q.2.w. | |||| |||||| |||||| | |||| |||||| ||||| |
Dupilumab 200 mg q.2.w. | |||| |||||| |||||| | |||||| |
Dupilumab 200 or 300 mg q.2.w. | |||| |||||| |||||| | |||||| |
Abrocitinib 100 mg q.d. | |||| |||||| ||||| | |||| |||||| ||||| |
Baricitinib 4 mg q.d. | |||| |||||| ||||| | |||||| |
Baricitinib 2 mg q.d. | |||| |||||| ||||| | |||| |||||| ||||| |
Tralokinumab 300 mg q.2.w. | |||||| | |||| |||||| ||||| |
Baricitinib 1 mg q.d. | |||||| | |||| |||||| ||||| |
CrI = credible interval; ICER = Institute for Clinical and Economic Review; NA = not available; NMA = network meta-analysis; OR = odds ratio; q.2.w. = every 2 weeks; q.d. = once daily.
Note: All estimates are relative to placebo. Numbers for the sponsor’s analysis are odds ratios (with 95% CrIs) from a binomial model (higher numbers are more favourable to treatment; numbers higher than 1 indicate superiority to placebo). Numbers for the ICER’s analysis are risk ratios (with 95% CrIs) from a binomial model (higher risk ratio is more favourable to treatment; numbers higher than 1 indicate superiority to placebo).
Source: Sponsor-submitted NMA and ICER NMA.37,38
Table 79: Comparison of Sponsor and ICER NMAs for PP-NRS4 in Monotherapy
Intervention | Active vs. placebo | |
|---|---|---|
Sponsor NMA OR (95% CrI) | ICER NMA risk ratio (95% CrI) | |
Upadacitinib 30 mg q.d. | |||||| | |||| |||||| ||||| |
Upadacitinib 15 mg q.d. | |||||| | |||| |||||| ||||| |
Dupilumab 200 or 300 mg q.2.w. | ||||| |||||| |||||| | |||||| |
Dupilumab 300 mg q.2.w. | |||| |||||| ||||| | |||| |||||| ||||| |
Abrocitinib 200 mg q.d. | |||| |||||| |||||| | |||| |||||| ||||| |
Abrocitinib 100 mg q.d. | |||| |||||| ||||| | |||| |||||| ||||| |
Baricitinib 2 mg q.d. | |||||| | |||| |||||| ||||| |
Tralokinumab 300 mg q.2.w. | |||||| | |||| |||||| ||||| |
Baricitinib 1 mg q.d. | |||||| | |||| |||||| ||||| |
CrI = credible interval; ICER = Institute for Clinical and Economic Review; NA = not available; NMA = network meta-analysis; OR = odds ratio; PP-NRS4 = improvement of 4 or greater from baseline on peak pruritus numerical rating scale; q.2.w. = every 2 weeks; q.d. = once daily.
Note: All estimates are relative to placebo. Numbers for the sponsor’s analysis are odds ratios (with 95% CrIs) from a binomial model (higher numbers are more favourable to treatment; numbers higher than 1 indicate superiority to placebo). Numbers for the ICER’s analysis are risk ratios (with 95% CrIs) from a binomial model (higher risk ratio is more favourable to treatment; numbers higher than 1 indicate superiority to placebo).
Source: Sponsor-submitted NMA and ICER NMA.37,38
For the IGA and PP-NRS outcomes in monotherapy, |||| |||| |||||| |||| ||||||||||| ||| ||||||||| |||| |||| ||||||||||| |||| ||||||||||| ||| ||||||||||||. The 2 NMAs did show some disagreements, particularly with the relative efficacy of abrocitinib 200 mg versus dupilumab 300 mg in terms of PP-NRS4, the ||||||| ||||||||| |||||| ||| |||||| |||||||| ||| |||||||||||, while the ICER’s analysis found dupilumab had more efficacy.
The sponsor also submitted an unanchored MAIC to compare abrocitinib (100 mg and 200 mg once daily) with cyclosporine and methotrexate, 2 immunomodulators that were unable to be included in the NMA due to a lack of connectivity to the network. While the authors concluded |||| ||||||||||| ||| |||| |||||| ||| |||| |||||| |||||||| ||| | ||||||| || |||||| |||||| |||||||, these conclusions are uncertain due to the potential confounding nature of the ITC. The risk factors and prognostic factors that could not be adjusted between the studies as well as the absence of a common comparator are likely to lead to substantial uncertainty in the MAIC results.76
Conclusion
Three ITCs were identified, reviewed, and critically appraised. Two were submitted by the sponsor (1 NMA and 1 unanchored MAIC) and the other was published by ICER. The 2 NMAs approached the ITC, similarly, using Bayesian methods to conduct their analyses on similar populations. The 2 NMAs differed by about 15 months in the time of last search and the inclusion of more treatments in the sponsor-submitted review.
The sponsor-submitted NMA reported that the EASI score for abrocitinib 200 mg was |||||||| || |||||||| ||||||||||| ||| ||| ||||||||||| | ||| | ||| ||||||||||| ||| |||||| ||| ||||||||| ||| || when these treatments were used as monotherapy. In combination with topical therapies it was reported that abrocitinib 200 mg was |||||||| || |||||||| ||||||||||| ||| ||| ||||||||||| | ||| | ||| ||| |||||||||||| ||| ||| || |||||||| |||||||||||| || || ||| || ||| ||||||||||| ||| ||| ||| ||||||||| ||| || |||| |||||||||||| ||| |||| ||||||||||| |||||||||| |||||| ||| ||||| |||||||| ||||||||| ||||||||| |||||||| ||||||| |||||| ||| |||||||||||||||| ||| || ||||||| |||||| |||| ||||||| |||||| ||| |||||||||||||. The sponsor-submitted NMA did not report on the efficacy and safety of abrocitinib 100 mg when compared with other treatments. Most importantly, no conclusions regarding the long-term efficacy of abrocitinib relative to the active comparators relevant to this review can be drawn as the NMA used study results collected over a relatively short duration compared to the chronic nature of AD. There is also uncertainty due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and results from the sponsor-submitted ITC must be interpreted with caution. Results from the ICER NMA generally agreed with the sponsor's NMA in terms of abrocitinib 200 mg once daily. Reported results for abrocitinib 100 mg showed that for most outcomes it was inferior (or occasionally comparable) to upadacitinib (30 mg and 15 mg once daily), abrocitinib 200 mg, and dupilumab 300 mg, while it was superior (or occasionally comparable) to baricitinib (2 mg or 1 mg), tralokinumab 300 mg, and placebo.
The sponsor-submitted MAIC showed that abrocitinib at both 100 mg and 200 mg once daily may be |||| ||||||||||| |||| ||| |||| | ||||||| |||||| ||||||| || |||||||||||| ||| ||||||||||||. However, these conclusions must be weighed against the highly unstable nature of unanchored indirect comparisons which, while improvements on naive comparisons, are still highly prone to potential biases. Until direct evidence is available, the efficacy and safety differences between abrocitinib and cyclosporine-methotrexate will remain inconclusive.
Other Relevant Evidence
This section includes submitted long-term extension studies and additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Long-Term Extension Study
Methods
The JADE EXTEND trial is an ongoing multi-centre, quadruple-masked, randomized phase III study for evaluating the long-term efficacy and safety of abrocitinib with or without topical medications in patients aged 12 years and older with moderate-to-severe AD.35 Patients who complete the JADE MONO-1, JADE MONO-2, JADE COMPARE, JADE TEEN, or JADE REGIMEN studies are eligible for enrolment in the JADE EXTEND trial. Only data for patients from the JADE MONO-1 and JADE MONO-2 trials that were filed for review with CADTH were available at the time of the data cut-off (April 22, 2020).
Populations
Eligible patients must have completed the full treatment period of the parent study, the full rescue treatment period of a qualifying parent study (if applicable), or the full open-label run-in period of the JADE REGIMEN study. However, no efficacy requirements have to be met to be eligible for the JADE EXTEND trial. Patients were excluded if they met any of the exclusion criteria outlined for their parent study. Patients were also excluded if they discontinued treatment early in a qualifying parent study or had experienced or were currently experiencing an AE in the parent study that may be considered an ongoing safety concern. Eligible patients who met the screening criteria had to agree to avoid prolonged exposure to the sun and UV light sources and the use of prohibited medications throughout the JADE EXTEND study. The demographic and baseline characteristics for the patients in the JADE EXTEND study are summarized Table 91. The characteristics were well balanced across the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily groups for age, gender, and duration of AD, and IGA, EASI, and PP-NRS scores.
Table 80: Demographic and Baseline Characteristics of JADE EXTEND
Characteristic | Abrocitinib 100 mg q.d. (n = 181) | Abrocitinib 200 mg q.d. (n = 180) |
|---|---|---|
Age in years, mean (SD) | 34.1 (16.2) | 33.8 (16.3) |
Male sex, n (%) | 103 (56.9) | 95 (52.8) |
Disease duration in years, mean (SD) | 22.2 (15.2) | 21.7 (15.2) |
IGA, n (%) Moderate Severe | 120 (66.3) 61 (33.7) | 121 (67.2) 59 (32.8) |
EASI, mean (SD) | 28.6 (11.6) | 28.1 (12.0) |
PP-NRS, mean (SD) | 7.1 (1.8) | 7.1 (1.9) |
EASI = Eczema Activity and Severity Index; IGA = Investigator’s Global Assessment; PP-NRS = peak pruritus numerical rating scale; q.d. = once daily; SD = standard deviation.
Source: Reich et al. (2020).36
Interventions
Eligible patients enrolled in the JADE EXTEND trial remain on the same dose of abrocitinib to which they were randomized in their qualifying parent study and are treated with or without TCS. Additionally, eligible patients in the placebo groups of their qualifying parent study are re-randomized to treatment with abrocitinib 100 mg once daily or 200 mg once daily.
Outcomes
The end points reported for the JADE EXTEND trial included IGA, EASI-75, and PP-NRS4 responses. All 3 end points were as described in the JADE MONO-1 study. The EASI-90 and EASI-100 responses are also being evaluating in the JADE EXTEND trial, but were not reported in the interim analysis filed with CADTH.
Statistical Analysis
Details regarding the statistical methodology applied in the interim analyses of the JADE EXTEND trial were not reported.
Patient Disposition
At data cut-off (April 22, 2020), 520 eligible patients who participated in the JADE MONO-1 and JADE MONO-2 trials were included in the JADE EXTEND trial. Of the 520 patients, 519 were enrolled from the 550 patients who completed the JADE MONO-1 or JADE MONO-2 trial and 1 patient had been previously discontinued from their parent study.6 Abrocitinib monotherapy was maintained in 361 of the 520 patients in the JADE EXTEND trial, while 159 patients received combination therapy of abrocitinib and topical medication. Interim results at 48 weeks reported treatment discontinuation in patients who received abrocitinib monotherapy. Specifically, 24.9% of the abrocitinib 100 mg once daily group and 25.0% of the abrocitinib 200 mg once daily group discontinued treatment, with the main reasons for discontinuation being an AE, study withdrawal, and lack of efficacy.
Table 81: Patient Disposition in JADE EXTEND (Interim Analysis)
Disposition, n (%) | Abrocitinib 100 mg q.d. with or without concomitant topical | Abrocitinib 200 mg q.d. with or without concomitant topical |
|---|---|---|
Randomized | 595 | 521 |
Treated | 595 | 521 |
Monotherapy subcohort | 388 | 335 |
Discontinued | 136 (22.9) | 104 (20.0) |
Adverse event | 37 (6.2) | 45 (8.6) |
Death | 0 | 1 (0.2) |
Lack of efficacy | 35 (5.9) | 13 (2.5) |
Lost to follow-up | 15 (2.5) | 7 (1.3) |
Pregnancy | 1 (0.2) | 0 |
Protocol deviation | 1 (0.2) | 5 (1.0) |
Study terminated by sponsor | 0 | 1 (0.2) |
Withdrawal by patient or parent/guardian | 47 (7.9) | 29 (5.6) |
Other | 0 | 3 (0.6) |
q.d. = once daily.
Source: Common Technical Document.6
Efficacy (Monotherapy)
The sponsor reported interim results for 48 weeks of treatment for patients who completed the JADE MONO-1 or JADE MONO-2 trial. The IGA response rate increased from 26.0% to 45.2% in the abrocitinib 100 mg once daily group and from 40.9% to 60.5% in the abrocitinib 200 mg once daily group between week 12 and 48 of treatment (Figure 27). The EASI-75 response rate increased from 42.1% to 68.0% in the abrocitinib 100 mg once daily group and from 61.9% to 87.2% in the abrocitinib 200 mg once daily group between week 12 and 48 of treatment. Consistent with the changes in EASI-75 response rates between weeks 12 and 48, the EASI-90 and EASI-100 response rates also increased in this time period (Figure 28). The PP-NRS4 response rate increased from 41.6% to 52.0% in the abrocitinib 100 mg once daily group and from 56.3% to 72.5% in the abrocitinib 200 mg once daily group between week 12 and 48 of treatment (Figure 29).
The sponsor evaluated patients who were nonresponders after 12 weeks of abrocitinib treatment in the parent studies to determine if continued treatment with abrocitinib in the JADE EXTEND trial could improve their condition. The sponsor reported IGA response rates of 22.4% and 26.7% were achieved by nonresponders in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily treatment groups at week 24, respectively. Similarly, nonresponders in the abrocitinib 100 mg once daily and abrocitinib 200 mg once daily treatment groups achieved EASI-75 response rates of 44.8% and 54.0% and PP-NRS response rates of 32.3% and 41.3% by week 24, respectively.
Figure 27: IGA Response in JADE EXTEND Over 48 Weeks (Monotherapy)
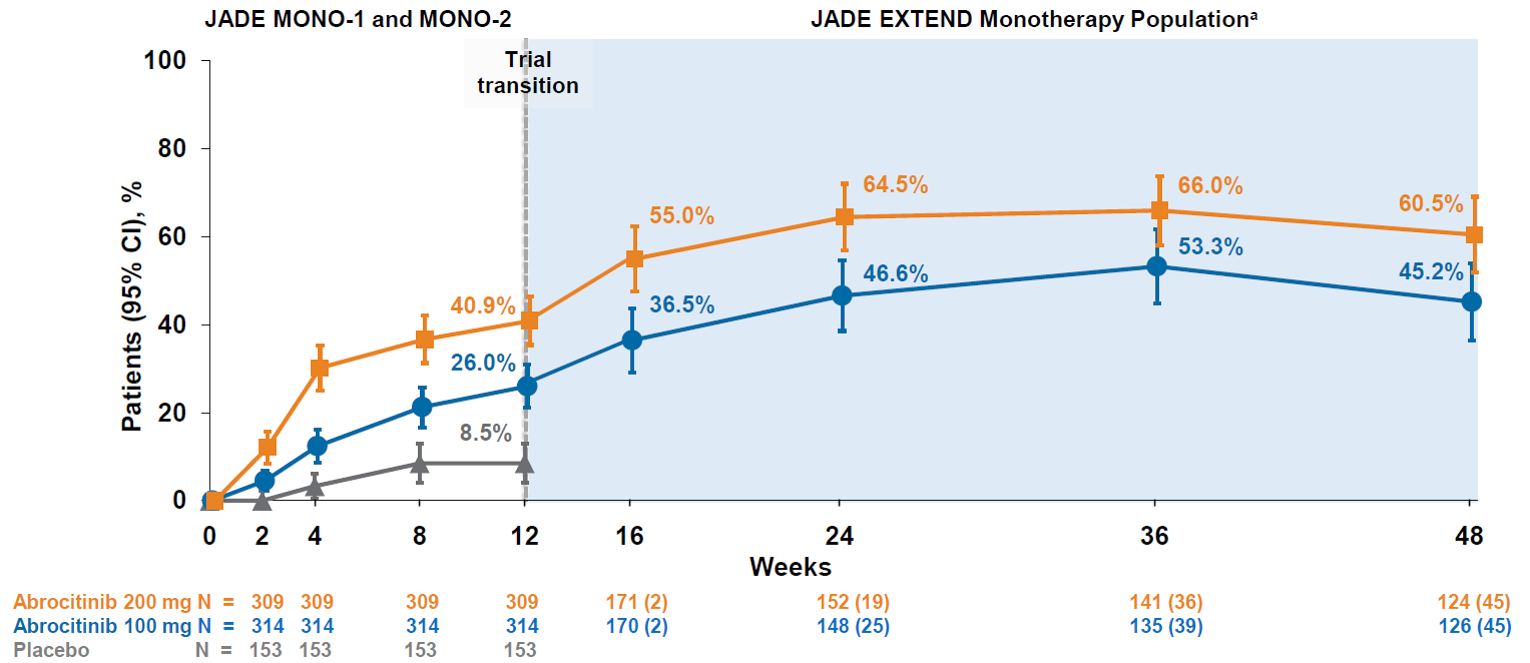
Note: Patients who withdrew from the JADE EXTEND study are shown in parentheses from weeks 16 to 24.
aParticipant numbers vary between time points assessed due to some participants not yet reaching a particular time point or if assessments for participants were missing.
Source: Reich et al. (2020).36
Figure 28: EASI-75 Response With Abrocitinib Monotherapy in JADE EXTEND Over 48 Weeks
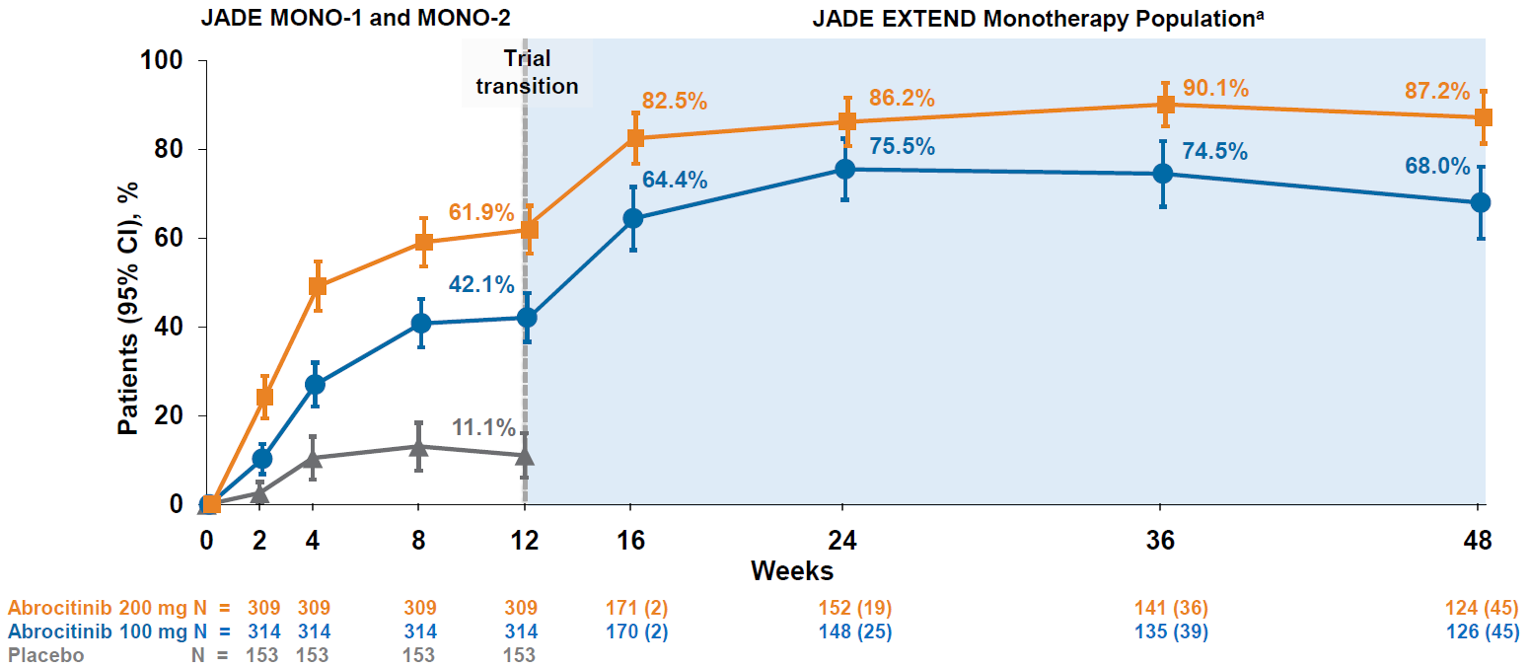
Note: Patients who withdrew from the JADE EXTEND study are shown in parentheses from weeks 16 to 24.
aParticipant numbers vary between time points assessed due to some participants not yet reaching a particular time point or if assessments for participants were missing.
Source: Reich et al. (2020).36
Figure 29: PP-NRS Response With Abrocitinib Monotherapy in JADE EXTEND Over 48 Weeks
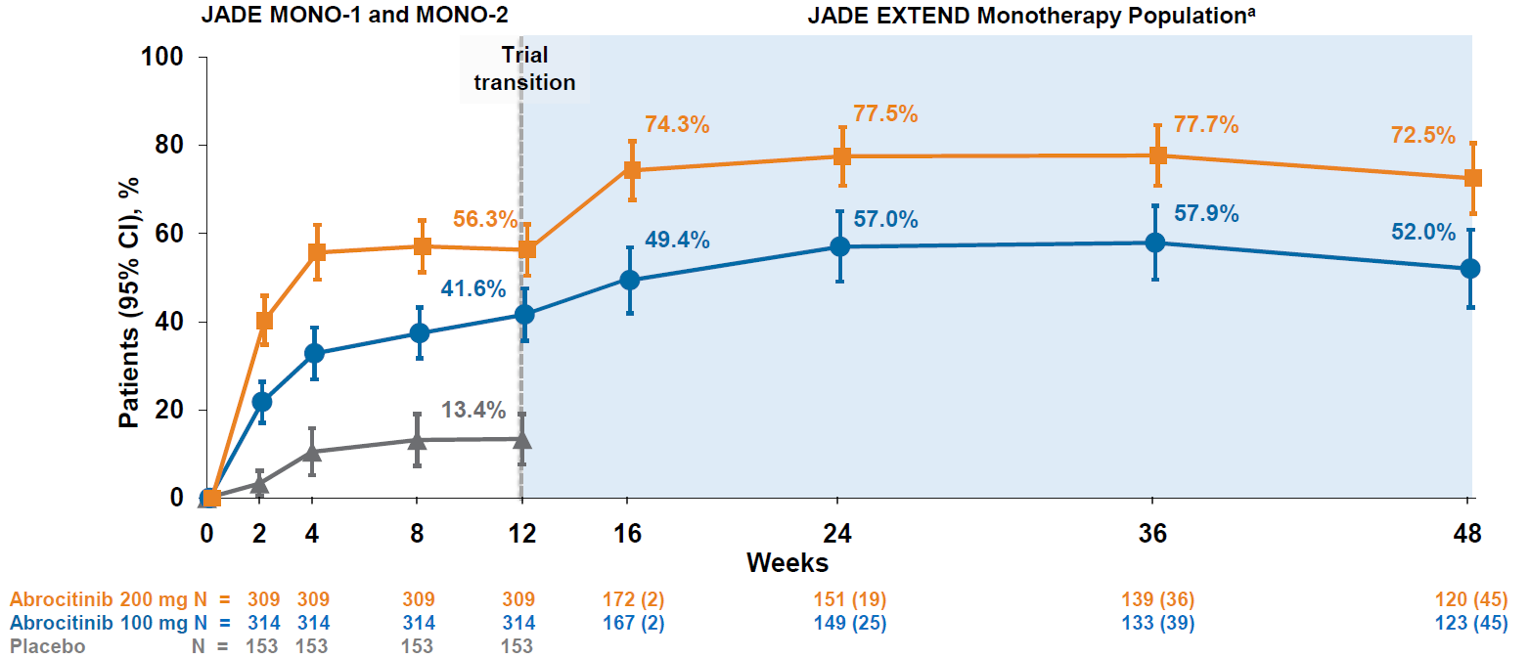
Note: Patients who withdrew from the JADE EXTEND study are shown in parentheses from weeks 16 to 24.
aParticipant numbers vary between time points assessed due to some participants not yet reaching a particular time point or if assessments for participants were missing.
Source: Reich et al. (2020).36
Efficacy (Combination Therapy)
The sponsor reported exploratory analyses to evaluate the efficacy of 12 weeks of abrocitinib treatment in patients who were previously treated with dupilumab for 16 weeks in the JADE COMPARE trial and failed to demonstrate IGA, EASI-75, and PP-NRS4 responses (Table 93). Further subgroup analyses were conducted for primary nonresponders (defined as patients who did not achieve a response at any visit through week 16 of the JADE COMPARE trial) and secondary nonresponders (defined as patients who had achieved a response at any time before week 16 but were nonresponders at week 16).
Responses from the IGA were reported for 34.3% and 47.2% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively. Responses of an EASI-75 were reported for 67.7% and 80.0% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively. Responses of a PP-NRS4 were reported for 37.8% and 81.0% of dupilumab nonresponders who received 12 weeks of abrocitinib 100 once daily and abrocitinib 200 once daily, respectively.
Table 82: Dupilumab-Treated Patients With IGA and EASI-75 Response at Week 12 in JADE EXTEND After Switching to Abrocitinib
Response in JADE COMPARE | Abrocitinib 100 mg q.d. | Abrocitinib 200 mg q.d. | |
|---|---|---|---|
IGA response at week 12 for patients treated with dupilumab in JADE COMPARE | |||
Dupilumab responders | N | 51 | 29 |
n (%) | 39 (76.5) | 25 (86.2) | |
95% CI | (64.8 to 88.1) | (73.7 to 98.8) | |
Dupilumab nonresponders | N | 70 | 36 |
n (%) | 24 (34.3) | 17 (47.2) | |
95% CI | (23.2 to 45.4) | (30.9 to 63.5) | |
Dupilumab primary nonresponders | N | 57 | 30 |
n (%) | 19 (33.3) | 14 (46.7) | |
95% CI | (21.1 to 45.6) | (28.8 to 64.5) | |
Dupilumab secondary nonresponders | N | 13 | 6 |
n (%) | 5 (38.5) | 3 (50.0) | |
95% CI | (12.0 to 64.9) | (10.0 to 90.0) | |
EASI-75 response at week 12 for patients treated with dupilumab in JADE COMPARE | |||
Dupilumab responders | N | 90 | 45 |
n (%) | 81 (90.0) | 43 (95.6) | |
95% CI | (83.8 to 96.2) | (89.5 to 100.0) | |
Dupilumab nonresponders | N | 31 | 20 |
n (%) | 21 (67.7) | 16 (80.0) | |
95% CI | (51.3 to 84.2) | (62.5 to 97.5) | |
Dupilumab primary nonresponders | N | 22 | 15 |
n (%) | 12 (54.5) | 11 (73.3) | |
95% CI | (33.7. 75.4) | (51.0 to 95.7) | |
Dupilumab secondary nonresponders | N | 9 | 5 |
n (%) | 9 (100.0) | 5 (100.0) | |
95% CI | (66.4 to 100.0) | (47.8 to 100.0) | |
CI = confidence interval; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; IGA = Investigator’s Global Assessment; N = number of subjects who met criteria at week 16 and were evaluable at week 12 of the JADE EXTEND trial; n (%) = number of subjects who met criteria (percentage based on N); q.d. = once daily.
Note: Dupilumab was administered at 300 mg once every 2 weeks.
Source: Clinical Summary.35
Harms
No harms data were reported specifically for the JADE EXTEND trial.
Critical Appraisal
Internal Validity
JADE EXTEND is an ongoing, double-blind extension study that enrolled patients from the phase III RCTs. The extension study maintained blinding and those who crossed over from placebo were randomized to 1 of the 2 abrocitinib dosage regimens. Only interim data were available at the time of the submission to CADTH, and reporting was limited to an interim analysis with partial reporting (i.e., a clinical study report was not available to enable a thorough appraisal). Extension studies are often limited by selection bias, as only patients who are tolerant to treatment and complete the parent studies are eligible to enrol. At the time of interim analysis, a large proportion of patients had withdrawn from both the abrocitinib 100 mg once daily (24.9%) and abrocitinib 200 mg once daily (25.0%) groups at 48 weeks.
External Validity
Issues with the generalizability of these data are the same as for the parent double-blind studies. The Systematic Review Critical Appraisal section discusses the external validity of the JADE MONO-1, JADE MONO-2, and JADE COMPARE trials. Patients were considered to be dupilumab nonresponders if they failed to demonstrate IGA, EASI-75, and PP-NRS4 responses after 16 weeks of treatment — a time period that was likely insufficient to fully realize the maximal treatment effects for dupilumab. The CADTH reimbursement recommendation for dupilumab for patients aged 12 years and older with moderate-to-severe AD recommends that the response to be treatment be evaluated after 6 months of treatment.
Discussion
Summary of Available Evidence
The evidence for this review was derived from a systematic literature review of pivotal and phase III studies that was supplemented with additional studies to address important gaps in the RCT evidence. The systematic review included 6 double-blind, phase III, RCTs: 2 12-week placebo-controlled trials conducted with abrocitinib as monotherapy for AD (JADE MONO-1 [N = 387] and JADE MONO-2 [N = 391]); 3 trials conducted with abrocitinib as combination therapy for AD (2 placebo-controlled trials [JADE COMPARE; N = 838 adults] and JADE TEEN; N = 287 adolescents] and 1 active-controlled trial comparing abrocitinib with dupilumab [JADE DARE; N = 727]); and 1 placebo-controlled, responder-enriched, withdrawal trial (JADE REGIMEN [N = 789]). The evidence from these studies was supplemented with the interim results from 1 long-term extension-phase study (JADE EXTEND) and 3 indirect comparisons (2 filed by the sponsor and 1 from the ICER).
The included studies evaluated a range of outcomes that are important in the management of AD, including overall severity of AD (e.g., EASI and IGA), severity of itching (e.g., PP-NRS), symptoms (e.g., POEM and PSAAD), health-related quality of life (e.g., DLQI and CDLQI), fatigue (e.g., FACIT-F and Peds-FACIT-F), patient-reported anxiety and depression (e.g., HADS), and need for additional AD medications (e.g., corticosteroid-free days). In addition, the JADE REGIMEN study investigated the use of abrocitinib (100 mg once daily or 200 mg once daily) as a maintenance therapy for patients who achieved an initial response to the 200 mg once daily dosage regimen by evaluating the time to acute worsening of the patient’s condition (i.e., development of a disease flare in accordance with standardized criteria).
The eligibility criteria for the included RCTs were similar except for the differences in the age ranges for the combination-therapy studies (i.e., the JADE COMPARE and JADE DARE trials were limited to adults and the JADE TEEN trial was limited to adolescents) and the need to establish a response to abrocitinib 200 mg once daily to be randomized in the JADE REGIMEN trial. All of the trials enrolled patients with moderate-to-severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., those with refractory moderate-to-severe AD and an inadequate response to other systemic drugs). The sponsor conducted pre-specified subgroup analyses based on prior exposure to at least 1 systemic immunosuppressant for AD for the co-primary end points of each trial (i.e., EASI-75 and IGA response). Subgroup analyses based on prior exposure to a systemic therapy were not reported for any of the secondary or exploratory end points included in the trials. The proportions of patients with prior exposure to at least 1 systemic therapy for AD in the included trials were 48.3% for the JADE MONO-1 trial, 41.4% for the JADE MONO-2 trial, 43.2% for the JADE COMPARE trial, 47.9% for the JADE DARE trial, 25.6% for the JADE TEEN trial, and 59.5% for the JADE REGIMEN trial in both the open-label induction phase and the double-blind treatment phase. The clinical expert consulted by CADTH noted that the criteria resulted in study populations that are reasonable reflections of the target population in Canada for systemically administered AD therapies, such as abrocitinib.
Interpretation of Results
Efficacy
With the exception of the JADE REGIMEN trial, all of the included studies used EASI-75 and IGA responses as co-primary end points. The sponsor reported that co-primary end points were required in the development program to address different preferences from international regulatory agencies. In both the monotherapy and combination-therapy trials, treatment with abrocitinib resulted in a statistically significant and clinically relevant increase in the proportion of patients with EASI-75 and IGA responses compared with placebo in the overall study populations. In the subgroup of patients with prior exposure to at least 1 systemic therapy for AD, the 200 mg dose of abrocitinib consistently demonstrated a benefit compared with placebo, with results that were similar to the primary analyses; however, there was greater uncertainty with the 100 mg once daily dosage. The clinical expert consulted by CADTH noted that these end points are clinically relevant and can be used in routine Canadian practice to evaluate responses to treatment with abrocitinib for patients with moderate-to-severe AD (i.e., for the purposes of establishing renewal criteria for reimbursement by the public drug programs). The clinical expert consulted by CADTH indicated that the subgroup analyses suggests that the response to abrocitinib would likely be similar for those with and without prior exposure to a systemic therapy for AD.
In their input to CADTH, patient groups and the clinical expert identified itch as the most burdensome symptom of AD. All of the included trials evaluated improvement in patient-reported itch severity using the PP-NRS instrument (a 10-point scale ranging from 0 [no itch] to 10 [worst itch imaginable]). The trials used a responder analysis based on the proportion of patients who achieved an improvement from baseline in PP-NRS of at least 4 units. In both the monotherapy and combination-therapy, placebo-controlled trials, both doses of abrocitinib demonstrated that a statistically significantly greater proportion of patients achieved a PP-NRS4 response, results that were considered to be clinically relevant by the expert consulted by CADTH. In the JADE DARE trial, abrocitinib 200 mg once daily was shown to be superior to dupilumab every 2 weeks for achieving a PP-NRS4 response in the initial weeks after treatment initiation (i.e., weeks 2, 4, and 8); however, there was no statistically significant difference from week 12 onward.
Patients with moderate-to-severe AD can experience sleep disruption due to the symptoms of their condition, particularly a persistent itch. The included monotherapy trials evaluated change from baseline in fatigue using validated scales for use in adults (FACIT-F) and adolescents (Peds-FACIT-F). Both doses of abrocitinib resulted in statistically significant improvements in FACIT-F compared with placebo in the JADE MONO-1 and JADE MONO-2 trials; there was no statistically significant difference between abrocitinib and placebo for the smaller subset of adolescent patients who completed the Peds-FACIT-F. The FACIT-F scale was not evaluated in the JADE COMPARE trial and there was no statistically significant difference between either dose of abrocitinib and placebo in the Peds-FACIT-F in the JADE TEEN study. CADTH did not identify an MID for the FACIT-F and Peds-FACIT-F specifically for patients with AD; however, the MID has been estimated to range from 3 to 5 in other inflammatory conditions (i.e., rheumatoid arthritis and systemic lupus erythematosus). The results for mean change from baseline in FACIT-F exceeded 3 in the JADE MONO-1 and JADE MONO-2 trials (LSMD = 3.3 and 3.6 with 100 mg once daily and LSMD = 4.3 to 4.5 with 200 mg once daily).
As reported by the patient groups who responded to the call for input for this review, AD can have a profound negative impact on the mental well-being of patients. Similarly, the clinical expert consulted by CADTH noted that those living with moderate-to-severe AD are at risk of experiencing depression as result of their condition. The phase III RCTs investigated the efficacy of abrocitinib in improving the symptoms of anxiety and depression using the HADS instrument. The monotherapy studies and the combination-therapy study in adults demonstrated that both 100 mg once daily and 200 mg once daily dosages of abrocitinib resulted in statistically significant improvements in HADS anxiety scores and HADS depression scores compared with placebo. There was no statistically significant difference between the abrocitinib and placebo groups in the JADE TEEN trial and no significant differences between abrocitinib and dupilumab in the JADE DARE trial. A literature search by CADTH did not identify an accepted MID for change from baseline in HADS scores. The clinical expert consulted by CADTH suggested that the results may be clinically relevant for short-term improvements in HADS scores. All of the phase III RCTs excluded patients who had any psychiatric condition, including clinically relevant depression and/or any history of suicidal ideation or behaviour. Patients were screened for these criteria using a variety of instruments that are not routinely applied in Canadian dermatology clinicals (i.e., the Columbia Suicide Severity Rating Scale, Suicidal Behaviours Questionnaire – Revised, and Patient Health Questionnaire – 8 items). As such, the results for the HADS end points may not be generalizable to AD patients who may be having more severe psychiatric conditions.
The clinical expert consulted by CADTH noted that the use of abrocitinib in patients who experienced an inadequate response or whose condition is no longer controlled by treatment with dupilumab represents an important gap in the phase III evidence base. As such, CADTH included the information available for this subgroup of patients from the JADE EXTEND trial. The sponsor reported exploratory analyses to evaluate the efficacy of 12 weeks of abrocitinib treatment in patients who were previously treated with dupilumab for 16 weeks in the JADE COMPARE trial and failed to demonstrate IGA, EASI-75, and PP-NRS4 responses. The 16-week time frame was likely insufficient to accurately evaluate if a patient would respond to dupilumab. For example, the CADTH reimbursement recommendation for dupilumab for patients aged 12 years and older with moderate-to-severe AD advises evaluating the response to treatment after 6 months of treatment.
The dosage recommended in the product monograph for abrocitinib is 100 mg or 200 mg orally once daily, based on individual goals of therapy and potential risks of adverse reactions. Exploratory analyses demonstrated that initiating treatment with the abrocitinib 200 mg once daily regimen was generally more efficacious than the 100 mg once daily regimen for establishing a response to treatment in the 12- to 16-week time frame that was studied in the phase III clinical trials. In addition, the JADE REGIMEN study demonstrated that responders (i.e., patients with an induction response after 12 weeks of abrocitinib 200 mg once daily) who continued to receive 200 mg once daily as maintenance treatment were less likely to experience a disease flare than those who received 100 mg once daily or placebo. The clinical expert consulted by CADTH noted that specialists are likely to initiate treatment with the higher dosage for most patients and then may consider reducing the dosage based on the patient’s response to therapy and/or tolerability.
CADTH reviewed and appraised the results of 3 indirect comparisons (2 unpublished analyses filed by the sponsor [1 NMA and 1 MAIC] and 1 published NMA from the ICER). The NMAs compared abrocitinib against dupilumab (the only drug approved for use in the treatment of AD at the time of this review), upadacitinib, tralokinumab (currently under review by Health Canada and CADTH for use in the treatment of AD), and several drugs that were not listed as under review by Health Canada or CADTH at the time of this review (e.g., nemolizumab and baricitinib). Comparisons with subgroup analyses based on prior exposure to at least 1 systemic therapy for AD were limited to abrocitinib versus dupilumab and placebo. The MAIC compared abrocitinib 100 mg once daily and 200 mg once daily against cyclosporine and methotrexate (2 drugs that are not approved by Health Canada for use as systemic treatments for AD but are commonly used in Canada). No subgroup analyses was reported for the MAIC, and the ICER NMA did not report a subgroup analysis based on prior exposure to at least 1 systemic therapy for AD.
The sponsor NMA reported that ||||||||||| ||| || |||| |||||| |||||||||||| || || ||| || || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| |||| |||||||||||| ||| |||| ||||||||||| |||||||||| |||||| ||| |||||||| |||||||| ||||||||| || ||| |||. Based on improvements in EASI, ||||||||||| ||| || |||| ||||| ||| |||||||| || |||||||| ||||||||||| ||| || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| |||| ||||| |||||||||| |||| |||| || |||||||||||| |||| |||| || ||||||||||| |||| ||||||| |||||||||| ||||||||||| ||| || |||| ||||| ||| |||||||| || |||||||| ||||||||||| ||| || |||| |||||| ||| |||||||||||| ||| || ||||| | |||||. Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy studies and a single composite end point (EASI-50 response and a DLQI improvement of ≥ 4 points) in the combination-therapy NMA. Due to the small number of patients in the LIBERTY AD ADOL trial with prior exposure to at least 1 systemic therapy for AD (n = 11 for the dupilumab 200 mg every 2 weeks or 300 mg every 2 weeks group, and n = 9 the placebo group), there was considerable uncertainty in the estimates of effect for the monotherapy NMA for IGA response. Similar to the primary NMA analyses, ||||||||||| ||| || |||| ||||| ||| |||||||| || ||||||||| ||| || ||||| |||||| ||| ||||||| ||||||||| || ||| ||||||||||| ||||||| ||| ||||||||||| ||| || |||| ||||| ||| ||||||| || ||||||||| ||| || ||| ||| ||||||||||||| | |||||||| ||||||| |||||||| ||| || ||||||||||| || || ||||| | |||||| || ||||.
The ICER’s NMA results were generally similar to those reported by the sponsor with respect to the comparative efficacy of abrocitinib 200 mg once daily. The sponsor’s NMA did not compare abrocitinib 100 mg once daily against all of the comparators (only placebo). However, the ICER NMA reported that, for most efficacy outcomes, abrocitinib 100 mg was either inferior or occasionally comparable to upadacitinib (30 mg and 15 mg once daily), abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks, while it was superior (or occasionally comparable) to tralokinumab 300 mg every 2 weeks and placebo.
The sponsor-submitted MAIC reported that abrocitinib at both 100 mg once daily and 200 mg once daily dosages ||| || |||| ||||||||||| |||| ||| |||| | ||||||| || |||||| |||||| ||||||| |||| |||||||| || |||||||||||| ||| ||||||||||||. However, these conclusions must be weighed against the highly unstable nature of unanchored indirect comparisons which, while being improvements on naive comparisons, are still highly prone to potential biases. Until direct evidence is available, the efficacy and safety differences between abrocitinib and cyclosporine-methotrexate will remain inconclusive.
Conclusions regarding the long-term efficacy of abrocitinib compared to the active comparators relevant to this review cannot be drawn as the NMA used study results collected over a relatively short duration compared to the chronic nature of AD. The inherent heterogeneity across trials in the networks also introduces uncertainty. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and the results of the sponsor-submitted ITCs must be interpreted with caution.
Harms
In 2021, the FDA announced that it had concluded that there may be an increased risk of serious heart-related events such as heart attack or stroke, cancer, blood clots, and death with tofacitinib (Xeljanz and Xeljanz extended release), which is another JAK inhibitor. The FDA stated that these findings were based on a review of a large safety clinical trial that compared tofacitinib against 2 tumour necrosis factor inhibitors (adalimumab and etanercept) in patients with rheumatoid arthritis who were 50 years of age or older and had at least 1 additional cardiovascular risk factor (N = 4,372).77,78 As result of this study, the FDA has mandated that updated warnings be included in the labels of all JAK inhibitors currently marketed in the US (i.e., tofacitinib, upadacitinib [Rinvoq], and baracitinib [Olumiant]).78 Health Canada cited the findings of the tofacitinib safety study as the rationale for restricting the indication for abrocitinib to those patients who have failed at least 1 systemic therapy for the treatment of AD.
The product monograph for abrocitinib contains black-box warnings regarding the risk of serious infections, malignancies, and thrombosis. It is recommended that treatment with abrocitinib should be interrupted if a patient develops a serious infection, sepsis, or opportunistic infection, until the infection is controlled.18 Similar warnings are currently included in all Canadian product monographs for JAK inhibitors (i.e., tofacitinib [Xeljanz], upadacitinib [Rinvoq], and baricitinib [Olumiant]).79-81 In contrast, the product monograph for dupilumab did not contain any black-box warnings at the time of this review.20 The clinical expert consulted by CADTH noted that specialists may demonstrate a preference for dupilumab based on the perception that the treatment may be associated with a reduced risk of SAEs in comparison with JAK inhibitors.
Serious adverse events and WDAEs were rare in the included studies. The most reported AE associated with abrocitinib in the clinical trials was nausea. The increase in nausea relative to placebo was dose-dependent (i.e., it was more commonly reported in those who initiated treatment with the 200 mg once daily dosage). The sponsor reported that nausea was typically mild to moderate in severity and resolved with continued treatment (median duration of the events was 15 days).
The included trials excluded patients with a history of coagulopathy or platelet dysfunction. The product monograph recommendations that abrocitinib be used with caution in patients at high risk for deep vein thrombosis (DVT) and pulmonary embolism (PE), noting that the following risk factors should be considered in determining an individual’s risk for DVT and/or PE: older age, obesity, medical history of DVT and/or PE, prothrombotic disorder, use of combined hormonal contraceptives or hormone replacement therapy, patients undergoing major surgery, or prolonged immobilization. The sponsor reported that, among all patients who were treated with abrocitinib, including in the interim analysis of the JADE EXTEND long-term extension study, PE was reported in 3 patients (0.18 per 100 patient-years) and DVT was reported in 2 patients (0.09 per 100 patient-years).
In the sponsor’s pooled safety analysis, overall infections were reported in 26.3% of patients who were treated with placebo and 35.2% and 34.6% of patients treated with abrocitinib 100 mg once daily and 200 mg once daily, respectively. Most infections were mild or moderate in severity. The most reported serious infections with abrocitinib were herpes simplex, herpes zoster, and pneumonia. Among all patients treated with abrocitinib, including the long-term extension study, serious infections were reported in 17 patients (2.65 per 100 patient-years) treated with abrocitinib 100 mg once daily and 24 patients (2.33 per 100 patient-years) treated with abrocitinib 200 mg once daily.
As AD is a chronic disease, abrocitinib would likely be used as a long-term treatment for patients who require systemic therapy. Abrocitinib was well tolerated in the target patient population (i.e., at least 12 years of age with moderate-to-severe AD) in the short term 12- and 16-week phase III studies. No safety data were reported for the interim analysis of the long-term extension study (JADE EXTEND) and only limited data were available from the 52-week JADE REGIMEN trial. Data on AEs in the JADE REGIMEN trial were generally consistent with those observed during the parent studies, but with a numerical increase in the incidence of SAEs per 100 person-years with abrocitinib 200 mg once daily (7.77; 95% CI, 4.25 to 13.04) compared with abrocitinib 100 mg once daily (2.69; 95% CI, 0.73 to 6.88) and placebo (3.18; 95% CI, 0.39 to 11.49). The ongoing JADE EXTEND study will help better characterize the longer-term efficacy and safety of abrocitinib in the treatment of AD.
In the indirect comparisons, |||||||||||||||||| ||| ||| |||||||||||||||| ||| || ||| |||| ||||||| |||||| ||| || ||| |||||||||||||.
Other Considerations
Abrocitinib is administered as a once daily oral treatment (irrespective of the dosage) and dupilumab is administered with subcutaneous injection once every 2 weeks. The patient groups who provided input to CADTH through the CSPA and Eczéma Québec survey of their preferences for the route of administration noted an overall preference for orally administered treatments. The clinical expert consulted by CADTH similarly noted that individual patient preferences are likely to vary with respect to oral versus subcutaneous administration. With respect to treatment adherence, the clinical expert noted that overall adherence may be greater with dupilumab (i.e., a single injection once every 2 weeks) compared with daily oral administration, particularly for adolescent patients who are typically less adherent to a daily treatment regimen in comparison with adults. The use of a double-dummy design in the JADE COMPARE and JADE DARE trials prevented any comparisons of adherence to the oral versus subcutaneous treatment regimens.
Conclusions
Four double-blind RCTs demonstrated that, compared with placebo, 12 or 16 weeks of treatment with abrocitinib was associated with statistically significant and clinically meaningful improvements in a range of outcomes that are important in the management of AD, including overall severity of AD (EASI and IGA response), severity of itching (PP-NRS4 response), symptoms (POEM and PSAAD), health-related quality of life (DLQI and CDLQI), fatigue (FACIT-F), and patient-reported anxiety and depression (HADS). These trials included the use of abrocitinib as monotherapy (JADE MONO-1 [N = 387] and JADE MONO-2 [N = 391]) and as combination therapy (JADE COMPARE [N = 838 adults] and JADE TEEN [N = 287 adolescents]). One active-controlled trial demonstrated that abrocitinib 200 mg once daily was superior to dupilumab for improving symptoms in the initial weeks after starting treatment, but that no significant differences were evident between the 2 drugs at 26 weeks. All of the trials enrolled patients with moderate-to-severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., those with refractory moderate-to-severe AD and an inadequate response to other systemic drugs). The sponsor conducted pre-specified subgroup analyses based on prior exposure to at least 1 systemic immunosuppressant for AD for the co-primary end points of each trial (i.e., EASI-75 and IGA response). The clinical expert consulted by CADTH indicated that the subgroup analyses suggest that the response to abrocitinib would likely be similar for those with and those without prior exposure to a systemic therapy for AD.
All the included studies suggested that initiating treatment with abrocitinib with the 200 mg once daily regimen was generally more efficacious than the 100 mg once daily regimen for establishing a response to treatment in the 12- to 16-week time frame studied in the trials. In addition, the JADE REGIMEN study demonstrated that responders who continue to receive 200 mg once daily as maintenance treatment were less likely to experience a disease flare than those who received 100 mg once daily or placebo.
The product monograph states that there is a risk of serious infections, malignancies, and thrombosis with abrocitinib and other JAK inhibitors. Serious AEs and WDAEs were rare in the included studies. As AD is a chronic disease, abrocitinib would likely be used as a long-term treatment for patients who require systemic therapy. Abrocitinib was well tolerated in the target patient population (i.e., at least 12 years of age with moderate-to-severe AD) in the short term 12- and 16-week phase III studies. No safety data were reported for the interim analysis of the long-term extension study (JADE EXTEND) and only limited data were available from the 52-week JADE REGIMEN trial. Data for AEs in the JADE REGIMEN trial were generally consistent with those observed during the parent studies, but with a numerical increase in the incidence of SAEs per 100 person-years with abrocitinib 200 mg once daily (7.77; 95% CI, 4.25 to 13.04) compared with the abrocitinib 100 mg once daily (2.69; 95% CI, 0.73 to 6.88) and placebo (3.18; 95% CI, 0.39 to 11.49). The ongoing JADE EXTEND study will help better characterize the longer-term efficacy and safety of abrocitinib in the treatment of AD.
The NMAs from the sponsor and ICER suggest |||| ||||||||||| ||| || |||| |||||| |||||||||||| || || ||| || || |||| |||||| ||| ||||||||| ||| || ||||| | ||||| |||| ||||||||| ||||||||||| |||||| ||| |||||||| |||||||| |||||||||. Subgroup analyses for patients reporting AD treatment failure with systemic immunosuppressants before study enrolment were limited to IGA response and EASI-75 for the monotherapy studies and a single composite end point (EASI-50 response and a DLQI improvement of ≥ 4 points) in the combination-therapy NMA. The effect estimates for the monotherapy NMA for IGA response were subject to considerable uncertainty; however, similar to the primary NMA analyses, ||||||||||| ||| || |||| ||||| ||| |||||||| || ||||||||| ||| || ||||| |||||| ||| ||||||| ||||||||. The NMA from the ICER suggested that abrocitinib 100 mg was either inferior or occasionally comparable to upadacitinib 30 mg and 15 mg once daily, abrocitinib 200 mg once daily, and dupilumab 300 mg every 2 weeks. The sponsor-submitted MAIC reported that abrocitinib at both 100 mg once daily and 200 mg once daily doses ||| || |||| ||||||||||| |||| |||||||||||| ||| ||||||||||||. No subgroup analyses were reported for the MAIC, and the ICER NMA did not report a subgroup analysis based on prior exposure to at least 1 systemic therapy for AD. No conclusions regarding the long-term efficacy of abrocitinib compared to the active comparators relevant to this review can be drawn as the NMA used study results collected over a relatively short duration compared to the chronic nature of AD. The inherent heterogeneity across trials in the networks also introduces uncertainty to interpretation of the results of the trials. The robustness of the comparative efficacy was further compromised by the lack of precision in some of the findings, and results from the indirect comparisons must be interpreted with caution.
References
1.Clinical Study Report: B7451050 (JADE DARE). A phase 3b randomized, double-blind, double-dummy, active-controlled multi-center study assessing the efficacy and safety of abrocitinib compared with dupilumab in adult participants on background topical therapy with moderate to severe atopic dermatitis [internal sponsor's report]. New York City (NY): Pfizer Inc; 2021 Dec 9.
2.Clinical Study Report: B7451012 (JADE MONO-1). A phase 3 randomized, double-blind, placebo-controlled, parallel group, multi-center study to evaluate the efficacy and safety of PF-04965842 monotherapy in subjects aged 12 years and older, with moderate to severe atopic dermatitis [internal sponsor's report]. New York City (NY): Pfizer Inc; 2019 Aug 27.
3.Clinical Study Report: B7451013 (JADE MONO-2). A phase 3 randomized, double-blind, placebo-controlled, parallel group, multi-center study to evaluate the efficacy and safety of PF-04965842 monotherapy in subjects aged 12 years and older, with moderate to severe atopic dermatitis [internal sponsor's report]. New York City (NY): Pfizer Inc; 2020 Jan 13.
4.Clinical Study Report: B7451029 (JADE COMPARE). A phase 3 randomized, double-blind, double-dummy, placebo-controlled, parallel group, multi-center study investigating the efficacy and safety of PF-04965842 and dupilumab in comparison with placebo in adult subjects on background topical therapy, with moderate to severe atopic dermatitis [internal sponsor's report]. New York City (NY): Pfizer Inc; 2020 May 28.
5.Clinical Study Report: B7451036 (JADE TEEN). A phase 3, randomized, double-blind, placebo-controlled, multi-center study investigating the efficacy and safety of PF-04965842 co-administered with background medicated topical therapy in adolescent participants 12 to <18 years of age with moderate-to-severe atopic dermatitis [internal sponsor's report].. New York City (NY): Pfizer Inc; 2020 Sep 10.
6.Common Technical Document: Module 2.7.3 summary of clinical efficacy [internal sponsor's report]. New York City (NY): Pfizer Inc; 2021.
7.Lynde C, Barber K, Claveau J, et al. Canadian practical guide for the treatment and management of atopic dermatitis. J Cutan Med Surg. 2005;8 Suppl 5:1-9. PubMed
8.Spergel JM. Epidemiology of atopic dermatitis and atopic march in children. Immunol Allergy Clin North Am. 2010;30(3):269-280. PubMed
9.Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657-682. PubMed
10.Stander S. Atopic dermatitis. N Engl J Med. 2021;384(12):1136-1143. PubMed
11.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
12.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: crisaborole (Eucrisa - Pfizer Canada Inc). Ottawa (ON): CADTH; 2019 Mar 27: https://www.cadth.ca/sites/default/files/cdr/complete/SR0570-Eucrisa-cdec-rec-april-2-2019.pdf. Accessed 2021 Jul 12.
13.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10(1):11-18. PubMed
14.Schram ME, Spuls PI, Leeflang MM, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. PubMed
15.Meggitt SJ, Gray JC, Reynolds NJ. Azathioprine dosed by thiopurine methyltransferase activity for moderate-to-severe atopic eczema: a double-blind, randomised controlled trial. Lancet. 2006;367(9513):839-846. PubMed
16.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent - sanofi-aventis Canada Inc). Ottawa (ON): CADTH; 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2021 Jul 12.
17.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
18.Cibinqo (abrocitinib): 50 mg, 100 mg, 200 mg oral tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021 Dec 17.
19.CellCept (mycophenolate mofetil): 250 mg capsules, 500 mg film-coated tablets, 200 mg/mL (when reconstituted) powder for oral suspension; CellCept i.v. (mycophenolate mofetil): 500 mg/vial injection [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2022 Jan 28: https://www.rochecanada.com/PMs/CellCept/CellCept_PM_CIE.pdf. Accessed 2022 Jan 31.
20.Dupixent (dupilumab): 300 mg single-use syringe (300 mg/2 mL), 300 mg single-use pen (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL), solution for subcutaneous injection [product monograph]. Laval (QC): Sanofi-aventis Canada Inc; 2021 Feb 22: https://pdf.hres.ca/dpd_pm/00060069.PDF. Accessed 2021 Jul 12.
21.Methotrexate, USP (methotrexate): 10 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019 Sep 12: https://www.pfizer.ca/sites/default/files/201910/Methotrexate_Tablets_PM_224778_12Sep2019_E.pdf. Accessed 2021 Jul 12.
22.Azathioprine: 50 mg tablets [product monograph]. Toronto (ON): Apotex Inc; 2020 Oct 28: https://pdf.hres.ca/dpd_pm/00059993.PDF. Accessed 2021 Jul 12.
23.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
24.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 May 25.
25.Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255-266. PubMed
26.Pfizer. NCT03349060: Study to evaluate efficacy and safety of PF-04965842 in subjects aged 12 years and older with moderate to severe atopic dermatitis (JADE Mono-1). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2019: https://clinicaltrials.gov/ct2/show/NCT03349060. Accessed 2021 May 14.
27.Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863-873. PubMed
28.Pfizer. NCT03575871: Study evaluating efficacy and safety of PF-04965842 in subjects aged 12 years and older with moderate to severe atopic dermatitis (JADE Mono-2). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2020: https://clinicaltrials.gov/ct2/show/NCT03575871. Accessed 2021 May 14.
29.Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101-1112. PubMed
30.Pfizer. NCT03796676: JAK1 inhibitor with medicated topical therapy in adolescents with atopic dermatitis (JADE TEEN). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT03796676. Accessed 2021 May 14.
31.Pfizer Inc. NCT04345367: Study of Abrocitinib Compared With Dupilumab in Adults With Moderate to Severe Atopic Dermatitis on Background Topical Therapy. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT04345367. Accessed May 14, 2021.
32.Blauvelt A, Silverberg JI, Lynde CW, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: Results from the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) REGIMEN phase 3 trial. J Am Acad Dermatol. 2022;86(1):104-112. PubMed
33.Pfizer. NCT03627767: Study to investigate efficacy and safety of PF-04965842 in subjects aged 12 years and over with moderate to severe atopic dermatitis with the option of rescue treatment in flaring subjects. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://www.clinicaltrials.gov/ct2/show/NCT03627767. Accessed 2021 May 14.
34.Gubelin W, Simpson EL, Blauvelt A, et al. Abrocitinib induction, randomized withdrawal, and response recapture with rescue therapy in patients with moderate-to-severe atopic dermatitis: results from the JADE REGIMEN phase 3 trial [slides] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets. Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
35.Clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50 mg, 100 mg, 200 mg oral tablets Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
36.Reich K, De Bruin-Weller M, Sondergaard Deleuran M, et al. Abrocitinib efficacy and safety as monotherapy over 48 weeks: results from a long-term extension study [slides] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets. Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
37.Systemic therapies for moderate-to-severe atopic dermatitis: a systematic literature review and meta-analysis (Final report) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50 mg, 100 mg, 200 mg oral tablets Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
38.Atlas SJ, Brouwer E, Fox G, et al. JAK inhibitors and monoclonal antibodies for the treatment of atopic dermatitis: effectiveness and value. (Evidence report). Boston (MA): Institute for Clinical and Economic Review (ICER); 2021: https://icer.org/wp-content/uploads/2020/12/Atopic-Dermatitis_Final-Evidence-Report_081721.pdf. Accessed 2021 Jul 12.
39.Matching adjusted indirect comparison (MAIC) for abrocitinib versus cyclosporine and methotrexate in patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50 mg, 100 mg, 200 mg oral tablets Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
40.Pfizer Canada ULC response to October 4, 2021 request for additional information regarding abrocitinib review [internal additional sponsor's information]. Kirkland (QC): Pfizer Canada ULC; 2021 Oct 5.
41.Badia-Tahull MB, Cobo-Sacristán S, Leiva-Badosa E, et al. Use of Subjective Global Assessment, Patient-Generated Subjective Global Assessment and Nutritional Risk Screening 2002 to evaluate the nutritional status of non-critically ill patients on parenteral nutrition. Nutr Hosp. 2014;29(2):411-419. PubMed
42.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
43.Leshem YA, Hajar T, Hanifin JM, Simpson EL. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172(5):1353-1357. PubMed
44.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. PubMed
45.Apfelbacher CJ, Ofenloch R. Good evidence on measurement properties of the Dermatology Life Quality Index, Itchy Quality of Life and 5-dimensions itch scales in atopic eczema, but problems with structural validity remain. Br J Dermatol. 2019;180(5):979-980. PubMed
46.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-216. PubMed
47.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. PubMed
48.Lewis-Jones MS, Finlay AY. The Children's Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-949. PubMed
49.Salek MS, Jung S, Brincat-Ruffini LA, et al. Clinical experience and psychometric properties of the Children's Dermatology Life Quality Index (CDLQI), 1995-2012. Br J Dermatol. 2013;169(4):734-759. PubMed
50.Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811-819. PubMed
51.Goligher EC, Pouchot J, Brant R, et al. Minimal clinically important difference for 7 measures of fatigue in patients with systemic lupus erythematosus. J Rheumatol. 2008;35(4):635-642. PubMed
52.Lai JS, Cella D, Kupst MJ, et al. Measuring fatigue for children with cancer: development and validation of the pediatric Functional Assessment of Chronic Illness Therapy-Fatigue (pedsFACIT-F). J Pediatr Hematol Oncol. 2007;29(7):471-479. PubMed
53.Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res. 2002;52(2):69-77. PubMed
54.Herrmann C. International experiences with the Hospital Anxiety and Depression Scale--a review of validation data and clinical results. J Psychosom Res. 1997;42(1):17-41. PubMed
55.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361-370. PubMed
56.Simpson E BL, Gadkari A, Eckert L, Reaney M. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate-to-severe atopic dermatitis: detailed analysis from randomized trials of dupilumab. J Am Acad Dermatol. 2017;76(6):AB93.
57.Hall R, Lebwohl MG, Bushmakin AG, et al. Development and Content Validation of Pruritus and Symptoms Assessment for Atopic Dermatitis (PSAAD) in adolescents and adults with moderate-to-severe AD. Dermatol Ther (Heidelb). 2021;11(1):221-233. PubMed
58.Severity scoring of atopic dermatitis: the SCORAD index. Consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1993;186(1):23-31. PubMed
59.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Arch Dermatol. 2004;140(12):1513-1519. PubMed
60.Howells L, Ratib S, Chalmers JR, Bradshaw L, Thomas KS. How should minimally important change scores for the Patient-Oriented Eczema Measure be interpreted? A validation using varied methods. Br J Dermatol. 2018;178(5):1135-1142. PubMed
61.Bronsard V, Paul C, Prey S, et al. What are the best outcome measures for assessing quality of life in plaque type psoriasis? A systematic review of the literature. J Eur Acad Dermatol Venereol. 2010;24 Suppl 2:17-22. PubMed
62.Ware JKM, Bjorner J. Determining important differences in scores. User's manual for the SF-36v2®. 2nd ed. London Quality Metric; 2007:125-133.
63.Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353-365. PubMed
64.Wu JJ, Lin C, Sun L, et al. Minimal clinically important difference (MCID) for work productivity and activity impairment (WPAI) questionnaire in psoriasis patients. J Eur Acad Dermatol Venereol. 2019;33(2):318-324. PubMed
65.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
66.EuroQol G. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
67.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. PubMed
68.Pfizer. NCT04345367: Study of abrocitinib compared with dupilumab in adults with moderate to severe atopic dermatitis on background topical therapy. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT04345367. Accessed 2021 May 14.
69.Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: Heterogeneity: subgroups, meta-regression, bias and bias-adjustment. London (GB): National Institute for Health and Care Excellence (NICE); 2012: https://www.ncbi.nlm.nih.gov/books/NBK395886/. Accessed 2021 Jul 12.
70.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 5: Evidence synthesis in the baseline natural history model. London (GB): National Institute for Health and Care Excellence (NICE); 2012: https://www.ncbi.nlm.nih.gov/books/NBK310368/. Accessed 2021 Jul 12.
71.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. London (GB): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/books/NBK310366/. Accessed 2021 Jul 12.
72.Lambert PC, Sutton AJ, Burton PR, Abrams KR, Jones DR. How vague is vague? A simulation study of the impact of the use of vague prior distributions in MCMC using WinBUGS. Stat Med. 2005;24(15):2401-2428. PubMed
73.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
74.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947. PubMed
75.Ishak KJ, Proskorovsky I, Benedict A. Simulation and matching-based approaches for indirect comparison of treatments. Pharmacoeconomics. 2015;33(6):537-549. PubMed
76.Hatswell AJ, Freemantle N, Baio G. The effects of model misspecification in unanchored matching-adjusted indirect comparison: results of a simulation study. Value Health. 2020;23(6):751-759. PubMed
77.Pfizer. NCT02092467: Safety study of tofacitinib versus tumor necrosis factor (TNF) inhibitor in subjects with rheumatoid arthritis. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT02092467. Accessed 2021 Oct 4.
78.U.S. Food and Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. 2021; https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death. Accessed 2021 Sep 9.
79.Olumiant (baricitinib): 2 mg tablets [product monograph]. Toronto (ON): Eli Lilly Canada Inc; 2020 Apr 9: https://pdf.hres.ca/dpd_pm/00055893.PDF. Accessed 2021 Jul 12.
80.Rinvoq (upadacitinib): 15 mg extended-release tablets [product monograph]. St-Laurent (QC): AbbVie Corporation; 2021 Jun 2: https://pdf.hres.ca/dpd_pm/00061353.PDF. Accessed 2021 Jul 12.
81.Xeljanz (tofacitinib citrate): 5 mg and 10 mg tablets; Xeljanz XR (tofacitinib citrate): 11 mg extended-release tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019 Oct 24: https://pdf.hres.ca/dpd_pm/00053735.PDF. Accessed 2021 Jul 12.
82.Landis JR, and Gary G. Koch. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159-174. PubMed
83.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
84.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
85.Rehal B, Armstrong AW. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality-of-life instruments 1985-2010. PLoS One. 2011;6(4):e17520. PubMed
86.Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets [internal sponsor's package]. Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
87.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. PubMed
88.Futamura M, Leshem YA, Thomas KS, Nankervis H, Williams HC, Simpson EL. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: many options, no standards. J Am Acad Dermatol. 2016;74(2):288-294. PubMed
89.Simpson E, Bissonnette R, Eichenfield LF, et al. The Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD): the development and reliability testing of a novel clinical outcome measurement instrument for the severity of atopic dermatitis. J Am Acad Dermatol. 2020;83(3):839-846. PubMed
90.Alexander H, Patton T, Jabbar-Lopez ZK, Manca A, Flohr C. Novel systemic therapies in atopic dermatitis: what do we need to fulfil the promise of a treatment revolution? F1000Res. 2019;8. PubMed
91.Chalmers JR, Schmitt J, Apfelbacher C, et al. Report from the third international consensus meeting to harmonise core outcome measures for atopic eczema/dermatitis clinical trials (HOME). Br J Dermatol. 2014;171(6):1318-1325. PubMed
92.Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed. New York (NY): Lawrence Erlbaum Associates; 1988.
93.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs Eczema Area and Severity Index and other measures of atopic dermatitis: a validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. PubMed
94.Grinich EE, Pawlitschek T, Simpson EL. Reporting of patient global assessments in atopic dermatitis randomized controlled trials. Dermatitis. 2020;31(1):e7-e9. PubMed
95.Chandran V, Bhella S, Schentag C, Gladman DD. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66(7):936-939. PubMed
96.Hewlett S, Dures E, Almeida C. Measures of fatigue: Bristol Rheumatoid Arthritis Fatigue Multi-Dimensional Questionnaire (BRAF MDQ), Bristol Rheumatoid Arthritis Fatigue Numerical Rating Scales (BRAF NRS) for severity, effect, and coping, Chalder Fatigue Questionnaire (CFQ), Checklist Individual Strength (CIS20R and CIS8R), Fatigue Severity Scale (FSS), Functional Assessment Chronic Illness Therapy (Fatigue) (FACIT-F), Multi-Dimensional Assessment of Fatigue (MAF), Multi-Dimensional Fatigue Inventory (MFI), Pediatric Quality Of Life (PedsQL) Multi-Dimensional Fatigue Scale, Profile of Fatigue (ProF), Short Form 36 Vitality Subscale (SF-36 VT), and Visual Analog Scales (VAS). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S263-286. PubMed
97.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
98.Reilly MC, Lavin PT, Kahler KH, Pariser DM. Validation of the Dermatology Life Quality Index and the Work Productivity and Activity Impairment-Chronic Hand Dermatitis questionnaire in chronic hand dermatitis. J Am Acad Dermatol. 2003;48(1):128-130. PubMed
99.Wallenhammar LM, Nyfjall M, Lindberg M, Meding B. Health-related quality of life and hand eczema--a comparison of two instruments, including factor analysis. J Invest Dermatol. 2004;122(6):1381-1389. PubMed
100.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
101.Fernandez-Penas P, Jones-Caballero M, Espallardo O, Garcia-Diez A. Comparison of Skindex-29, Dermatology Life Quality Index, Psoriasis Disability Index and Medical Outcome Study Short Form 36 in patients with mild to severe psoriasis. Br J Dermatol. 2012;166(4):884-887. PubMed
102.Heinl D, Prinsen CA, Deckert S, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: a systematic review. Allergy. 2016;71(3):358-370. PubMed
103.Gabes M, Tischer C, Apfelbacher C, quality of life working group of the Harmonising Outcome Measures for Eczema (HOME) initiative. Measurement properties of quality-of-life outcome measures for children and adults with eczema: an updated systematic review. Pediatr Allergy Immunol. 2020;31(1):66-77. PubMed
104.Silverberg JI, Gelfand JM, Margolis DJ, et al. Measurement properties of the Hospital Anxiety and Depression Scale used in atopic dermatitis in adults. J Invest Dermatol. 2019;139(6):1388-1391. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 25, 2021
Alerts: Weekly search updates until project completion
Study types: No filters were applied to limit the retrieval by study type
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(abrocitinib* or CIBINQO* or pf04965842 or “pf 04965842” or pf4965842 or pf 4965842 or 73SM5SF3OR).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*abrocitinib/ or (abrocitinib* or CIBINQO* or pf04965842 or “pf 04965842” or pf4965842 or pf 4965842).ti,ab,kw,dq.
3 use oemezd
(conference review or conference abstract).pt.
4 not 5
2 or 6
remove duplicates from 7
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms – abrocitinib; atopic dermatitis]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – abrocitinib; atopic dermatitis]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – abrocitinib; atopic dermatitis]
Grey Literature
Search dates: May 11– May 25, 2021
Keywords: abrocitinib; atopic dermatitis
Limits: Publication years: none
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals.
Appendix 2: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 84: Subgroup Data for IGA Response and EASI-75 Response (FAS)
Study | End point | Population | Difference in responders (%) abrocitinib vs. placebo (95% CI) | |
|---|---|---|---|---|
100 mg q.d. | 200 mg q.d. | |||
Monotherapy studies | ||||
JADE MONO-1 | IGA response | < 18 years | 14.0 (−8.0 to 35.9) | 14.8 (−7.4 to 37.0) |
≥ 18 years | 16.3 (6.5 to 26.1) | 41.7 (30.7 to 52.6) | ||
Moderate AD | 15.0 (2.1 to 27.8) | 41.6 (27.9 to 55.4) | ||
Severe AD | 17.1 (5.4 to 28.7) | 27.4 (14.4 to 40.5) | ||
Prior systemic immunosuppressant for AD | 9.1 (−1.2 to 19.4) | 36.2 (22.7 to 49.7) | ||
No prior systemic immunosuppressant for AD | 22.2 (7.6 to 36.9) | 34.8 (20.0 to 49.5) | ||
EASI-75 response | < 18 years | 31.6 (8.4 to 54.9) | 42.0 (18.6 to 65.5) | |
≥ 18 years | 26.9 (15.0 to 38.7) | 53.3 (41.6 to 65.1) | ||
Moderate AD | NR | NR | ||
Severe AD | NR | NR | ||
Prior systemic immunosuppressant for AD | 17.0 (2.6 to 31.4) | 49.3 (33.8 to 64.7) | ||
No prior systemic immunosuppressant for AD | 38.9 (23.8 to 54.0) | 52.4 (37.9 to 66.9) | ||
JADE MONO-2 | IGA response | < 18 years | 12.5 (−11.7 to 36.7) | 40.0 (9.4 to 70.6) |
≥ 18 years | 20.2 (9.8 to 30.6) | 27.9 (17.2 to 38.5) | ||
Moderate AD | 18.4 (6.0 to 30.9) | 30.7 (17.8 to 43.6) | ||
Severe AD | 20.6 (6.5 to 34.8) | 24.7 (10.1 to 39.4) | ||
Prior systemic immunosuppressant for AD | 20.4 (6.7 to 34.1) | 26.9 (12.1 to 41.6) | ||
No prior systemic immunosuppressant for AD | 18.7 (5.6 to 31.8) | 30.2 (16.8 to 43.6) | ||
EASI-75 response | < 18 years | 43.8 (13.5 to 74.0) | 60.0 (29.4 to 90.6) | |
≥ 18 years | 33.2 (22.0 to 44.3) | 49.7 (38.7 to 60.7) | ||
Moderate AD | NR | NR | ||
Severe AD | NR | NR | ||
Prior systemic immunosuppressant for AD | 30.9 (16.4 to 45.3) | 54.6 (39.4 to 69.7) | ||
No prior systemic immunosuppressant for AD | 37.0 (22.7 to 51.2) | 48.0 (34.2 to 61.8) | ||
Combination-therapy studies | ||||
JADE COMPARE | IGA response | Moderate AD | 23.3 (11.7 to 34.8) | 23.3 (11.7 to 34.8) |
Severe AD | 22.7 (12.4 to 33.0) | 22.7 (12.4 to 33.0) | ||
Prior systemic immunosuppressant for AD | 27.5 (14.4 to 40.6) | 43.9 (30.7 to 57.1) | ||
No prior systemic immunosuppressant for AD | 19.7 (8.4 to 30.9) | 27.5 (15.6 to 39.4) | ||
EASI-75 response | Moderate AD | 26.0 (13.3 to 38.8) | 30.5 (17.6 to 43.4) | |
Severe AD | 43.1 (28.8 to 57.5) | 66.3 (53.3 to 79.3) | ||
Prior systemic immunosuppressant for AD | 49.1 (35.5 to 62.7) | 63.0 (50.3 to 75.7) | ||
No prior systemic immunosuppressant for AD | 21.2 (7.9 to 34.4) | 30.5 (17.1 to 43.9) | ||
JADE TEEN | IGA response | Moderate AD | 15.4 (−2.8 to 33.6) | 18.1 (0.5 to 35.7) |
Severe AD | 18.6 (0.0 to 37.2) | 24.7 (4.9 to 44.5) | ||
Prior systemic immunosuppressant for AD | 18.6 (−1.7 to 38.9) | 41.7 (18.0 to 65.3) | ||
No prior systemic immunosuppressant for AD | 17.6 (1.3 to 34.0) | 15.1 (–0.7 to 30.9) | ||
EASI-75 response | Moderate AD | 19.6 (2.2 to 36.9) | 14.1 (–3.3 to 31.5) | |
Severe AD | 36.6 (15.9 to 57.4) | 54.5 (34.8 to 74.1) | ||
Prior systemic immunosuppressant for AD | 24.7 (−1.7 to 51.1) | 39.0 (12.4 to 65.7) | ||
No prior systemic immunosuppressant for AD | 28.9 (13.0 to 44.8) | 27.5 (12.0 to 43.1) | ||
AD = atopic dermatitis; CI = confidence interval; EASI-75 = ≥ 75% improvement in the EASI total score; FAS = full analysis set; IGA = Investigator’s Global Assessment; NA = not applicable; NR = not reported
Source: Clinical Study Reports2-5
Figure 30: Kaplan–Meier Plot of Time to PP-NRS4 Response in Monotherapy Studies
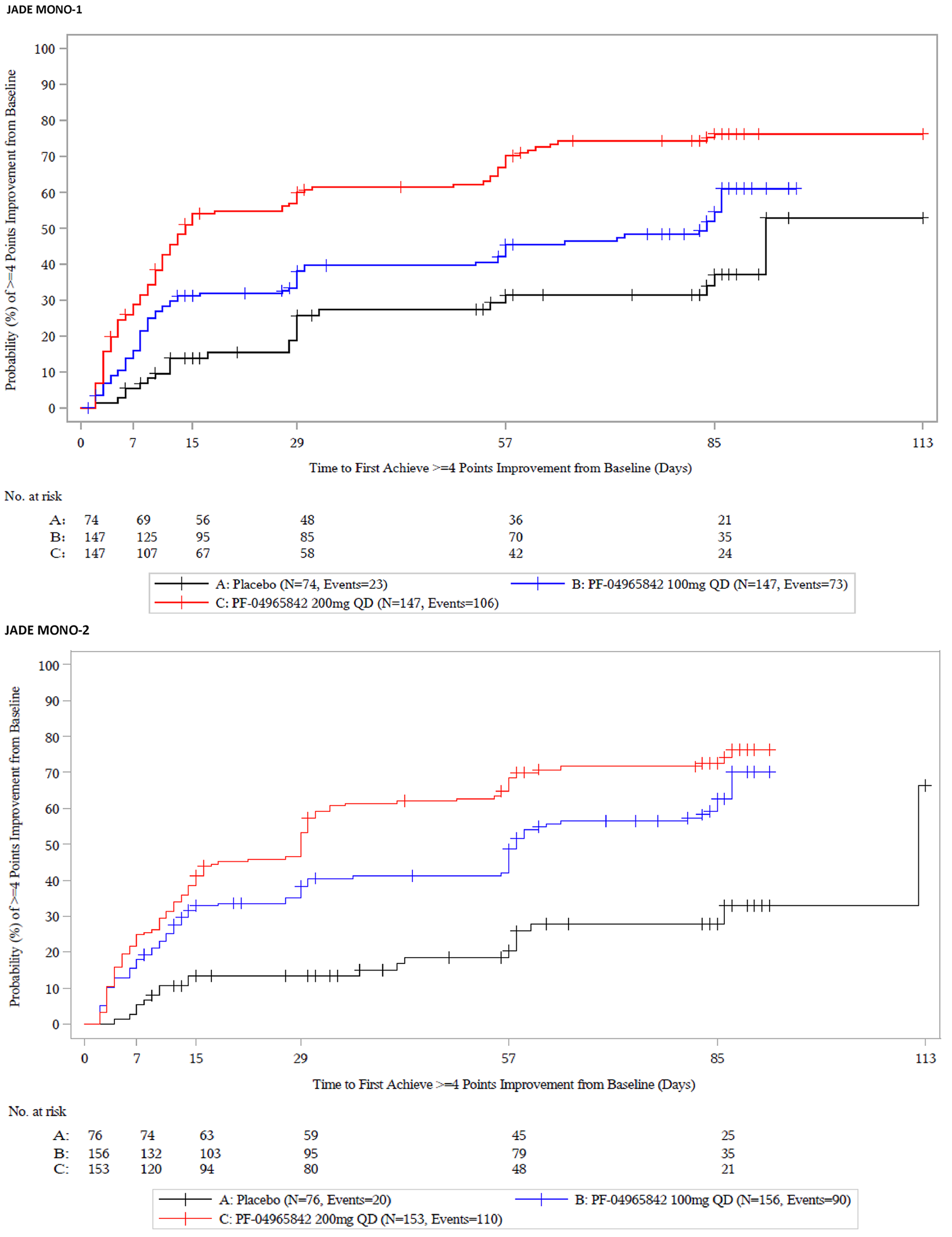
PF-04965842 = abrocitinib; q.d. = once daily
Figure 31: Kaplan–Meier Plot of Time to PP-NRS4 Response in Adult Combination-Therapy Studies
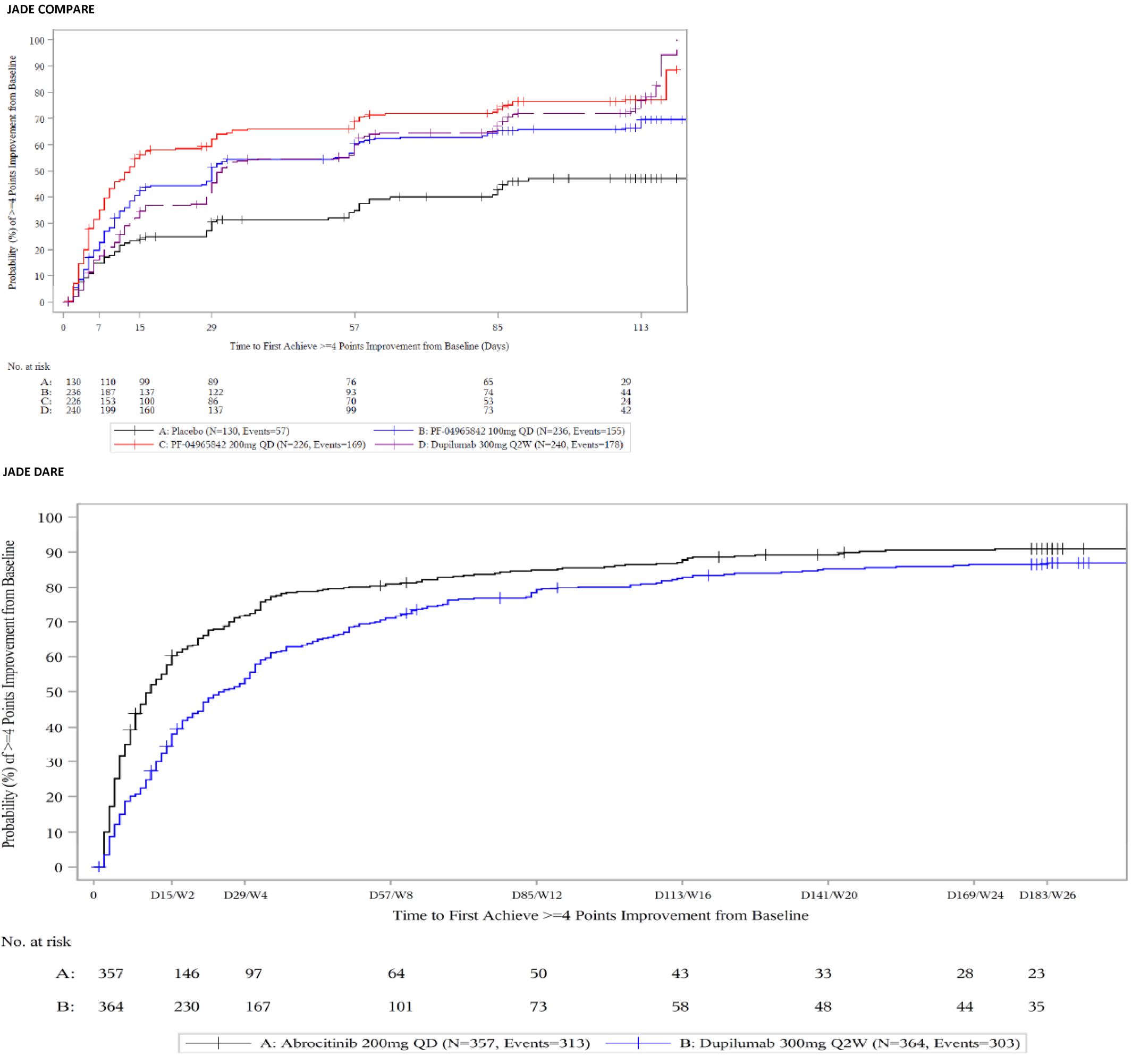
JADE COMPARE
PF-04965842 = abrocitinib; QD = once daily
JADE DARE
Q2W = every 2 weeks; QD = once daily
JADE COMPARE Source: Clinical Study Report4
JADE DARE Source: Clinical Study Report1
Figure 32: Kaplan–Meier Plot of Time to PP-NRS4 Response in Adolescent Combination-Therapy Study
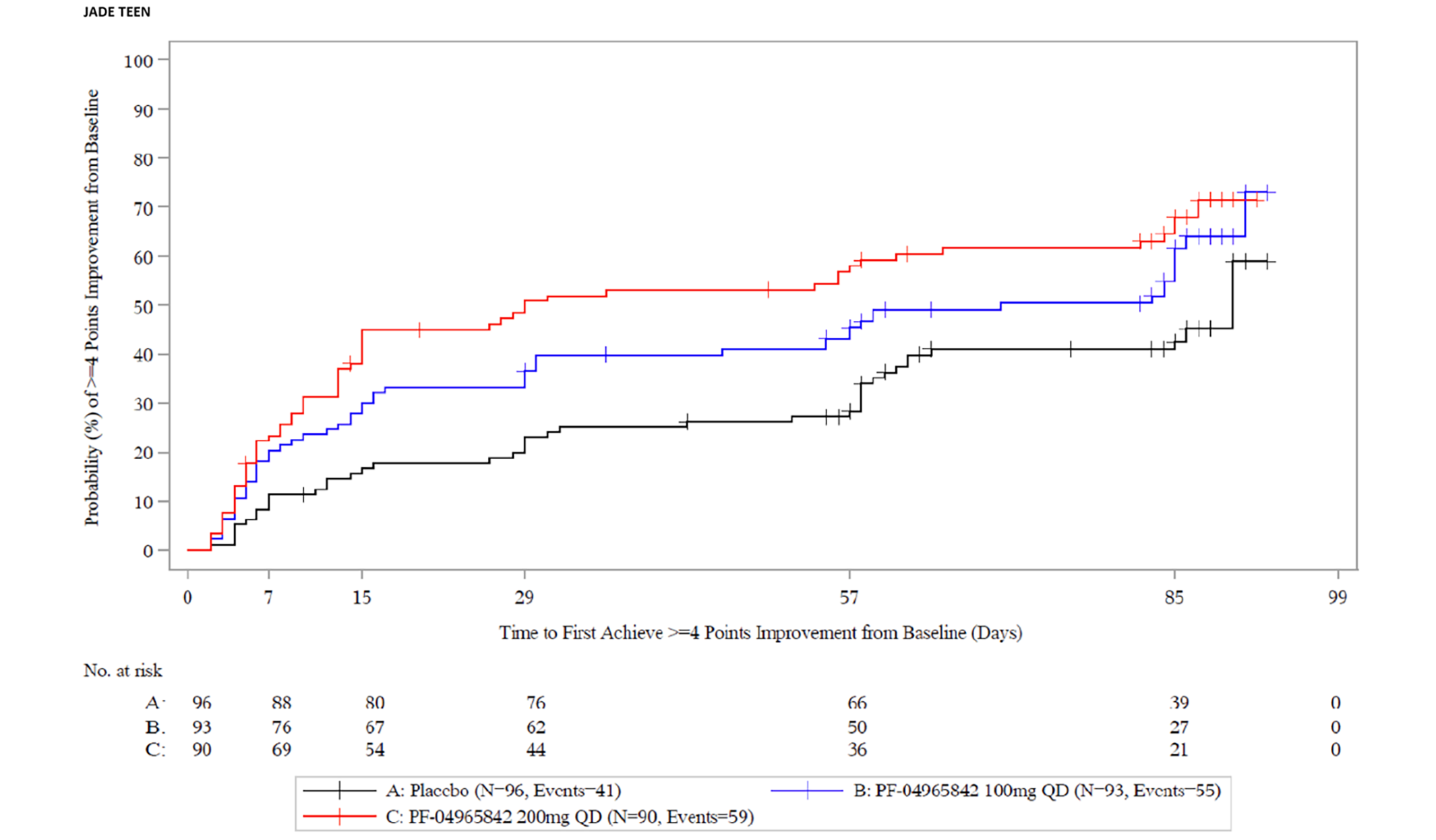
PF-04965842 = abrocitinib; QD = once daily
Source: Clinical Study Report5
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
To summarize the validity of the following end point measures:
Eczema Area and Severity Index (EASI)
Investigator Global Assessment (IGA)
Scoring Atopic Dermatitis (SCORAD)
Pruritus numerical rating score (NRS)
Patient-Oriented Eczema Measure (POEM)
Patient Global Assessment (PtGA)
Pruritus and Symptoms Assessment for Atopic Dermatitis (PSAAD)
FACIT Fatigue Scale (FACIT-F) and Pediatric FACIT-F (Peds-FACIT-F)
Dermatology life quality index (DLQI)
European quality of life-5 dimensions (EQ-5D)
Short Form-36 Health Survey, Version 2, Acute (SF-36v2, Acute)
Hospital anxiety and depression (HADS)
Findings
A focused literature search was conducted to identify the psychometric properties and MID of each of the stated outcome measures.
Interpretation of the reliability and validity metrics were based on the following criteria:
Inter-rater reliability, kappa statistics (level of agreement)82:
< 0 = poor agreement
0.00 to 0.21 = slight agreement
0.21 to 0.40 = fair agreement
0.41 to 0.60 = moderate agreement
0.61 to 0.8 = substantial
0.81 to 1.00 = almost perfect agreement
Internal consistency (Cronbach alpha) and test-retest reliability: ≥ 0.7 is considered acceptable.83
Validity; i.e., between-scale comparison (correlation coefficient, r)84:
≤ 0.3 = weak
0.3 to ≤ 0.5 = moderate
> 0.5 = strong
The findings about validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 85.
Table 85: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EASI | A scale used in clinical trials to assess the severity and extent of AD. The total EASI score ranges from 0 to 72 points, with the highest score indicating worse severity of AD. EASI-75 indicates ≥ 75% improvement from baseline. | Adequate construct and content validity, estimated between EASI and SCORAD, reports of moderate to high correlation (r = 0.84 to 0.93) between these 2 tools.14,13,41,42 Internal consistency of EASI is also adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94 respectively.42 Intra- and Inter-rater reliability has kappa values of test-retest reliability of 0.76.42 Responsiveness (sensitivity to change) was judged as adequate.14 | |
IGA | A scale that provides a global clinical assessment of AD by investigator. IGA is a 5-point scale that provides a global clinical assessment of AD severity (ranging from 0 to 4). “0” indicates clear, and “4” indicates severe AD. | No information on the validity and MID of the IGA scale in patients with AD were identified. | None identified. |
SCORAD | A tool used in clinical research to standardize the evaluation of the extent and severity of AD. The maximum possible total score of SCORAD is 103, in which, the higher score indicates poorer or a more severe condition. | Two systematic reviews found excellent agreement with global assessments of disease severity.42,85 Content validity was deemed adequate, good construct validity (Spearman r values ranging from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability were also adequate; the latter with several measurements of ICC from 0.84 to 0.99. Intra-observer reliability (test-retest), however, was unclear.42 | 8.7 points using IGA as anchor42 |
Pruritus NRS | A tool for patients with AD used to report the intensity of their itch. Patients rate average and maximum intensity of itch in past 24 hours based on a scale of 0 to 10 (0 = “no itch” and 10 = “worst itch imaginable.”) | The most appropriate definition of a responder on the pruritus NRS has been reported to be in the range of 3 to 4 points. | 3 to 4 points |
POEM | A 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults with eczema. Seven items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question were: “0” indicates for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” indicates for every day. The maximum total score is 28; a high score is indicative of severity (0 to 2 indicates for clear or almost clear; 3 to 7 for mild eczema; 8 to 16 for moderate eczema; 17 to 24 for severe eczema; 25 to 28 for very severe eczema). | Moderate concurrent validity (Spearman = 0.56). Good convergent validity when compared to DLQI, but moderate to weak when compared to EASI and NRS. Poor discriminant validity in predicting self-reported global severity. Moderate responsiveness. Good reliability (ICC = 0.90) | MID of 5 points change from baseline using global severity as anchor. |
PtGA | Defined as a patient-/parent-reported assessment that instructed patients to report the overall state of their condition in a holistic fashion integrating 1 or more disease domains including, but not limited to, signs, symptoms, quality of life, and perception of disease control. | For the studies included in this submission, the PtGA asks the subject to evaluate the overall cutaneous disease at that point in time on a single-item, 5-point scale. The same category labels used in the Investigator’s Global Assessment will be used for the Patient Global Assessment, i.e., “severe (4)”, “moderate (3)”, “mild (2)”, “almost clear (1)”, and “clear (0)” No data on validity, reliability, or responsiveness. | Not available. |
PSAAD | A daily patient reported symptom diary for the assessment of pruritus and symptoms in adolescents and adults with mild to severe AD, based on a 24-hour recall. PSAAD is based on 11 symptoms (itch, dryness, redness, flaking, discoloration, pain, bleeding, cracking, bumps, swelling, and weeping/oozing) on an 11-point scale, ranging from 0 (none) to 10 (extreme). The total score is the average of the responses to each of the 11 items. Total score range of 0 (none) to 10 (extreme). | The instrument has acceptable test-retest reliability with ICC of 0.81 and good internal consistency with Cronbach alpha > 0.9, as well as good responsiveness as assessed with the change from baseline in the PSAAD and compared to a patient global impression of change (PGIC). | 0.63 of the total estimated score. |
FACIT-F | A patient self-completed questionnaire to assess the intensity of fatigue (and its impact on daily life) at an individual’s level during their usual daily activities over the past week. It consists of a 13-item questionnaire that assesses self-reported tiredness, weakness, and difficulty conducting usual activities due to fatigue. The level of fatigue is measured on a 4-point Likert scale (4 = not at all fatigued to 0 = very much fatigued). The instrument scoring yields a range from 0 to 52, with higher scores representing better overall health status (less fatigue). | The instrument has good criterion validity and reliability in other conditions (rheumatoid arthritis and psoriatic arthritis), but no values were found in patients with AD. | A MID of 3 to 4 of the total score was established in patients with rheumatoid arthritis, and 5.9 in patients with and systemic lupus erythematosus. No MID is available for AD. |
Pediatric FACIT-F | Developed with some unique areas relevant to children, the tool has 11 items evaluated on a 5-point Likert scale (from 0 = none of the time, to 4 = all of the time) for patients 8 to 18 years old with a recall period of 7 days. The maximum score is 44 and higher scores representing better overall health status (less fatigue). | The instrument has good validity and reliability. However, no values were found from AD patients. | A difference > 4.7 points was considered of clinical importance in patients with cancer, but no information is available for AD. |
DLQI | A questionnaire used to assess 6 different aspects that may affect quality of life of patients in dermatology. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life. The overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30). The higher the score, the more quality of life is impaired. | The DLQI has shown good test-retest reliability, internal consistency reliability, construct validity and responsiveness in patients with psoriasis. In patients with AD, internal consistency could not be determined. Reliability was moderate (0.77). Other validity measures and MID information were not found. | 2.2 to 6.9 (psoriasis) Not available for AD |
CDLQI | Child-completed questionnaire (ages 3 to 16 years), designed to measure the impact of any skin disease on the quality of life with a recall period of 7 days. It comprises 10 questions asking about the impact of a skin disease on the life of the affected child, including symptoms, embarrassment, friendships, clothes, playing, sports, bullying, sleep, and impact of treatment. Each question on a 4-point Likert scale (from 0 to 3). Minimum of 0 and maximum of 30. Higher CDLQI scores indicate greater degree of impairment in HRQoL. | Adequate concurrent validity vs. Cardiff Acne Disability Index (CADI) and the Childhood Atopic Dermatitis Impact Scale (CADIS). Good convergent construct validity and divergent construct validity. Good internal consistency of the CDLQI (examined in 6 studies) with Cronbach alpha values ranging from 0.82 to 0.92. Test-retest reliability is adequate, with Spearman’s rank order correlation coefficient calculated in 4 studies (range 0.74 to 0.97). One study examined the ICC, finding 0.80. Good responsiveness to change was found in studies using Wilcoxon signed rank test and repeated measures ANOVA. | One study conducted in the US and Canada with 202 participants using a distribution- based approach, determined the MCID of the CDLQI in psoriasis to be 2.5. However, no MID is available for AD. |
EQ-5D-5L | Generic, preference based HRQoL instrument, consisting of an index score and VAS scale score that has been applied to a wide range of health conditions and treatments. | EQ-5D includes 3 parts: a descriptive system that classifies respondents (aged ≥ 12 years) into one of 243 distinct health states. The second part is a 20 cm visual analogue scale (EQ VAS) that has end points labelled 0 and 100. The third part is the EQ-5D index score which is generated by applying a multi-attribute utility function to the descriptive system. No information is found from literature search for EQ-5D in AD. | 0.033 to 0.074, Not available for AD |
SF-36 | The SF-36v2 -acute is a validated generic health status measure. It measures 8 general health domains: physical functioning, role limitations due to physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations due to emotional problems, and mental health. These domains can also be summarized as physical and mental component summary scores. The acute version uses one week recall. Eight scaled scores are converted to weighted sums of the questions in their section. Each scale is directly transformed into a 0 to 100 scale (these are t-scores with mean of 50 and standard deviation of 10). Lower scores mean more disability. (i.e., a score of 0 = maximum disability and a score of 100 = no disability). | The tool has been extensively validated for measuring quality of life in different clinical conditions. However, no specific values were found in patients with AD. Also, internal consistency coefficients for the 8 scales have been reported from many studies in other clinical conditions; for instance, intraclass correlation of 0.85 have been reported for patients with musculoskeletal problems and item-total correlations typically are in the mid-0.70s, but no specific numbers in AD. | No specific MID for patients with AD was available The User’s Manual proposed MIDs of a change of 2 points on the physical component summary and 3 points on the mental component summary. The MIDs for the SF-36v2 are based on clinical and other non–patient-reported anchors. |
HADS | A patient-reported questionnaire designed to identify anxiety disorders and depression in patients at non-psychiatric medical institutions. | The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, A person can score between 0 and 21 for each subscale (anxiety and depression). A high score was indicative of a poor state. No additional validity and MID information regarding HADS was found from the literature search for AD. | Not available |
AD = atopic dermatitis; DLQI = Dermatology life quality index; EASI = Eczema area and severity index; EQ-5D = European quality of life-5 dimensions; HADS = Hospital anxiety and depression scale; IGA = Investigator global assessment; MID = minimal important difference; NRS = numerical rating score; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring atopic dermatitis.
Eczema Area and Severity Index
The EASI is a scale used in clinical trials to assess the severity and extent of AD that has been recommended as the core outcome measure for the clinical signs of eczema.14,13,41,42 In EASI, 4 disease characteristics of AD (erythema, infiltration/papulation, excoriations, and lichenification) are assessed for severity by the investigator on a scale from “0” (absent) to “3” (severe). The scores are added up for each of the 4 body regions (head, arms, trunk, and legs). The assigned percentages of BSA for each section of the body are 10% for head, 20% for arms, 30% for trunk, and 40% for legs respectively. Each subtotal score is multiplied by the BSA represented by that region. In addition, the affected area of AD assessed as a percentage by each body region is converted to a score of 0 to 6, where the area is expressed as 0 (none), 1 (1% to 9%), 2 (10% to 29%), 3 (30% to 49%), 4 (50% to 69%), 5 (70% to 89%), or 6 (90% to 100%). Each of the body area scores are multiplied by the area affected. Therefore, the total EASI score ranges from 0 to 72 points, with the highest score indicating worse severity of AD. It is suggested that the severity of AD based on EASI are categorized as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 21.1 to 50.0 = severe; and 50.1 to 72.0 = very severe.43 EASI-75 indicates ≥ 75% improvement from baseline.86
The validity and reliability of the EASI has been examined in several studies and a systematic review of the literature.14,13,41,42 The correlation coefficients for assessing content and construct validity were estimated between EASI and SCORAD42 with reports of moderate to high correlation (r = 0.84 to 0.93) between these 2 tools. Internal consistency of EASI is adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94 respectively.42 Intra- and Inter-rater reliability has also been examined with adequate values of test-retest reliability and kappa values of 0.76.42 Responsiveness (sensitivity to change) was also judged as adequate by the systematic review authors. Overall, EASI has been considered a validated scale and can be used reliably in the assessment of severity and extent of AD.13,87
One study using anchor-based methods reported an overall MID of 6.6 points using IGA improving 1 point as anchor.14
Investigator’s Global Assessment
The IGA is an easy and rapid administered tool that provides a global assessment of AD severity. This tool has been widely used in many AD clinical trials and required by regulatory agencies for drug approval trials in AD and psoriasis in an effort to provide an understandable and meaningful end point measure for patients and clinicians.88 However, the instrument has had many issues with variable content validity, and variable definitions and implementations. For instance, the tool has had over 20 different names, it has been used with various numbers of scale categories (from 4- to 7-point scales), as well as variability in the content of the scales.88
The IGA used in the pivotal trials included in this submission to CADTH uses a 5-point scale, ranging from 0 to 4, reflecting a global consideration of erythema, induration, and scaling, where “0” indicates clear, and “4” indicates severe AD. A decrease in score relates to an improvement in signs and symptoms.87
A 2016 systematic review of the literature found no information on the validity and reliability of the IGA instrument in patients with AD as well as no information on what would constitute a MID in patients with AD.88,89
Scoring Atopic Dermatitis
The SCORAD tool used is in clinical research to standardize the evaluation of the extent and severity of AD.90 SCORAD was considered as a valid and reliable tool for the objective assessment of eczema clinical signs.91 The instrument assesses 3 components of AD: the extent of affected body surface area (0 to 100), severity (0 to 18), and symptoms (0 to 20). The extent of AD is assessed as a percentage of each defined body area and reported as the sum of all areas. The score ranges from 0 to 100. The severity of 6 specific signs of AD (redness, swelling, oozing/crusting, excoriation, skin thickening/lichenification, dryness) is assessed using a 4-point scale (i.e., none = 0, mild = 1, moderate = 2 and or severe = 3) with a minimum score of 0 and a maximum of 18. The subjective symptoms (itch and sleeplessness) are recorded by the patient or relative on a visual analogue scale, with scores ranging from 0 (no symptoms) to 10 (worst imaginable symptom) with a maximum possible score of 20. The total SCORAD is calculated based on the 3 components with a maximum possible total score of 103, in which, the higher score indicates poorer or a more severe condition.
Based on 2 systematic reviews, SCORAD has been found to be valid and reliable, with excellent agreement with global assessments of disease severity.42,85 Content validity was deemed adequate, with good construct validity (Spearman R’s ranging from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability were also adequate; the latter with several measurements of ICC from 0.84 to 0.99. Intra-observer reliability (test-retest), however, was unclear.42
Based on the analysis of the data from 3 RCTs in patients with atopic eczema, the MID was estimated using mean change scores of SCORAD of patients that showed a relevant improvement based on IGA, defined as an “improvement” or “decline” of ≥ 1 point in PGA and IGA; thus, a difference of 8.7 points in SCORAD was estimated as the MID for the patients with AD.14
Pruritus Numerical Rating Scale
The PP-NRS is a tool that patients used to report the intensity of their itch during a daily recall period using an interactive voice response system. Patients rate their overall (average) and maximum intensity of itch experienced during the past 24 hours based on a scale of 0 to 10 (0 = “no itch” and 10 = “worst itch imaginable”). The validity and reliability of the NRS is based on 3 phase III and 1 phase IIb RCTs in patients with AD.56 The pooled ICC from the 3 RCTs was 0.96, and the ICC from the phase IIb study ranged from 0.95 to 0.97. The ICC values indicated that the NRS scores were stable over a period of time when the patients’ disease was stable. To assess the validity of the NRS, a priori hypotheses were evaluated using correlational analyses and 3 known-groups analysis of variance (ANOVA) models (“absent/mild group based on the Pruritus Categorical Scale [PCS]; “poor” disease group based on the Patient Global Assessment of Disease and “no impact” on skin-related quality-of-life group based on DLQI total score).56 Results for all 3 known groups were in the anticipated direction and were statistically significant, and the effect sizes for the differences between the extreme categories for each known group were all above a Cohen threshold of 0.80 for large effect sizes.92 Based on the data from the phase IIb study, using EASI, IGA as anchors, NRS responder reportedly ranged between 2.2 and 4.2, with the highest estimates based on the most stringent clinical criteria (EASI 90 to 100 and IGA 0/1). Using PCS as an anchor, the responder was estimated as 2.6 points. These analyses suggested that the most appropriate definition of a responder on the pruritus NRS is in the range of 3 to 4 points.56
Patient-Oriented Eczema Measure
The POEM is a 7-item, questionnaire used in clinical trials to assess disease symptoms in children and adults. Based on frequency of occurrence during the past week, the 7 items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question were: “0” indicates for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” indicates for every day. The maximum total score is 28; a high score is indicative of poor quality of life (0 to 2 indicates for clear or almost clear; 3 to 7 for mild eczema; 8 to 16 for moderate eczema; 17 to 24 for severe eczema; 25 to 28 for very severe eczema).59
In 1 study,14 it was reported that the overall mean MID of the POEM was 3.4 points (SD = 4.8), using IGA as anchor. In 2018, the minimally important change (MIC) of POEM in children (N = 300) with moderate-to-severe atopic eczema was calculated in 1 study.60 Based on distribution-based methods, the estimated MIC were 1.07 (using 0.2 SD of baseline POEM scores) and 2.68 (using 0.5 SD of baseline POEM scores); The estimated MIC were 3.09 to 6.13 and 3.23 to 5.38 based on patient-/parent-reported anchor-based methods and investigator-reported anchor-based methods respectively. The authors recommended the following thresholds be used to interpret changes in POEM scores in children: a score of 3 to 3.9 indicates a probably clinically important change; ≥ 4, indicates a very likely clinically important change.60
The tool has been tested in its validity, reliability, and responsiveness. When compared to the PO-SCORAD and DLQI93 a moderate concurrent validity (Spearman = 0.56) was detected in adults. Good convergent validity when compared to DLQI, but moderate to weak when compared to EASI and NRS. Poor discriminant validity in predicting self-reported global severity. In other studies including children, content validity was poor to moderate as a measurement of clinical signs of AD.42,59 The same studies have revealed moderate responsiveness and good reliability (ICC = 0.90).93
The MID has been previously stated as 3.4 points in adults and from 3.0 to 3.9 in children based on a pooled cohort of 3 clinical trials. A recent prospective study established– a change of 5.0 points as a clinically meaningful threshold for adults using global severity of AD as anchor.93
Patient Global Assessment
The PtGA is defined as a patient-/parent-reported assessment that instructs patients to report the overall state of their condition in a holistic fashion integrating 1 or more disease domains including, but not limited to, signs, symptoms, quality of life, and perception of disease control.94 For the pivotal studies included in this submission to CADTH, the PtGA asks the patient to evaluate the overall cutaneous disease at a certain point in time on a single-item, 5-point scale. The same category labels used in the Investigator’s Global Assessment will be used for the Patient Global Assessment, i.e., “severe (4)”, “moderate (3)”, “mild (2)”, “almost clear (1)”, and “clear (0)”.94
There are several versions of the PtGA instrument. Although it has been cited as being of high importance to investigators, clinicians, and patients the PtGA has been used differently in several AD studies (34 instruments found in 40 AD studies in a recent systematic review)94 as well as the scale used for patients between studies (scales varying from 4 to 9 points). No individual studies were found to have examined the scope, validity, reliability, responsiveness, or MID for the PtGA.
Pruritus and Symptoms Assessment for Atopic Dermatitis
The PSAAD is a daily patient reported symptom diary for the assessment of pruritus and symptoms in atopic dermatitis in adolescents and adults with mild to severe AD.57 The preliminary version is a 15-item questionnaire that includes 11 items developed to measure symptoms of atopic dermatitis, capturing those identified by patients to be the most important, based on a 24-hour recall. Analysis of the PSAAD in the pivotal studies for this submission to CADTH was based solely on these 11 items, or 11 symptoms (itch, dryness, redness, flaking, discoloration, pain, bleeding, cracking, bumps, swelling, and weeping/oozing). Each item of the PSAAD assesses the severity of a single symptom on an 11-point scale, ranging from 0 (none) to 10 (extreme). The PSAAD total score is calculated as the average of the responses to each of the 11 items, for a PSAAD total score range of 0 (none) to 10 (extreme). One recent study assessed the convergent and known-group validity with other PRO measures ranged from low to high estimates (r = 0.24 to 0.91).57 The instrument has acceptable test-retest reliability with ICC of 0.81 and good internal consistency with Cronbach alpha > 0.9, as well as good responsiveness as assessed with the change from baseline in the PSAAD and compared to a patient global impression of change (PGIC). The MID was assessed in the same study using the PGIC and the patient global impression of severity as anchors, where a change in PSAAD total score of 0.63 considered as clinically important.57
Functional Assessment of Chronic Illness Therapy–Fatigue
The FACIT-F is a patient self-completed questionnaire to assess fatigue. The instrument asks about the intensity of fatigue (and its impact on daily life) at an individual’s level during their usual daily activities over the past week. This instrument has been evaluated in rheumatoid arthritis (RA) and psoriatic arthritis (PsA), primary Sjogren’s syndrome, osteoarthritis (OA), and systemic lupus erythematosus (SLE), as well as many other long-term conditions (e.g., multiple sclerosis, cancer, neurologic disorders).50,95,96 However, no MID for patients with MID has been established.
It consists of a 13-item questionnaire that assesses self-reported tiredness, weakness, and difficulty conducting usual activities due to fatigue. The level of fatigue is measured on a 4-point Likert scale (4 = not at all fatigued to 0 = very much fatigued). The instrument scoring yields a range from 0 to 52, with higher scores representing better overall health status (less fatigue).95,96
The instrument has good criterion validity with correlation between the FACIT and Fatigue Severity Scale (FSS) of −0.79 tool comparison. In a 2007 study in patients with psoriatic arthritis,95 the FACIT Fatigue Scale was found to have high internal consistency (Cronbach alpha = 0.96). Similarly, a study assessing patients with RA the Cronbach alpha was 0.86 to 0.87 at 3 time points.96 The instrument also showed good responsiveness after 24 weeks of antirheumatic treatments in patients with RA (n = 631), showing a mean change of 2.1 in patients who did not achieve American College of Rheumatology 20% criteria for improvement in disease activity (ACR20; effect size 0.19), compared to 12.4 in those who achieved ACR70 (effect size 1.13).96
A MID of 3 to 4 of the total score was established in patients with RA,50 and 5.9 in patients with SLE,51 but no MID has been established in patients with AD.
Pediatric Functional Assessment of Chronic Illness Therapy–Fatigue
The 11-item Peds-FACIT-F was developed via literature review, feedback from patient/parent/clinician, face-to-face consensus meeting and using Rasch Analysis.52 Some of the Peds-FACIT-F items are unique to children, whereas others share the same concepts captured in a parallel adult version. The tool has 11 items evaluated on a 5-point Likert-type scale (from 0 = none of the time, to 4 = all of the time) for patients 8 to 18 years old with a recall period of 7 days. The maximum score is 44 and higher scores representing better overall health status (less fatigue).
Concurrent validity of the Peds-FACIT-F has been examined in 1 study in children with cancer52 using Spearman r between scores on the Peds-FACIT-F and Multidimensional Fatigue Scale. Additionally, analysis of variance was used to determine whether the Peds-FACIT-F differentiated patients with different functional performance levels, anemic/nonanemic status, and risk levels. The concurrent validity was confirmed with Spearman r = 0.86, 0.71, and 0.57 for general fatigue, sleep, and cognition, respectively. Acceptable internal consistency was found when all patients were analyzed as a whole (Cronbach a = 0.89), and also when patients were analyzed separately by age group (a = 0.85 and 0.91 for children and adolescents, respectively).
The MID of the Peds-FACIT-F was calculated by using anemia and functional performance status as clinical anchors. Effect sizes (ES), defined as mean difference divided by the SD for each clinical anchor were calculated. An ES > 0.5 was considered moderate to large. A difference > 4.7 points was considered of clinical importance.52 However, no MID for patients with AD has been established.
Dermatology Life Quality Index
The DLQI is a widely used dermatology-specific quality of life instrument. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life.46,47 These aspects are symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment. The maximum score per aspect is either 3 (with a single questions) or 6 (with 2 questions) and the scores for each can be expressed as a percentage of either 3 or 6. Each of the 10 questions is scored from 0 (not at all) to 3 (very much) and the overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30).46,47 The higher the score, the more quality of life is impaired. The meaning of the DLQI scores on a patient’s life is as follows97:
0 to 1 = no effect.
Two to 5 = small effect.
Six to 10 = moderate effect.
Eleven to 20 = very large effect.
Twenty-one to 30 = extremely large effect.
The validity of the DLQI has been assessed in patients with eczema.85,98-100 The DLQI has shown good test-retest reliability (correlation between overall DLQI scores was 0.99, P < 0.0001 and of individual question scores was 0.95 to 0.98, P < 0.001),46 internal consistency reliability (with Cronbach alpha coefficients ranging from 0.75 to 0.92 when assessed in 12 international studies),97 construct validity,(as 37 separate studies have mentioned a significant correlation of the DLQI with either generic or dermatology-specific and disease-specific measures),97 and responsiveness (the DLQI being able to detect changes before and after treatment in patients with psoriasis in 17 different studies).97-99
Estimates of the MID have ranged from 2.2 to 6.9.47,97 It should be noted that some of the anchors that were used to obtain the DLQI MID were not patient-based (i.e., Basra et al.97 derived estimates from PASI and physician global assessment anchors, as well as a distribution-based approach).
Limitations associated with the DLQI include concerns regarding unidimensionality and the behaviour of items of the DLQI in different psoriatic patient populations with respect to their cross-cultural equivalence and age and gender; however, these concerns were only identified in 2 citations out of 12 international studies identified.97
The patient’s emotional aspects may be under-represented. To overcome this, it is suggested that the DLQI be combined with more emotionally-oriented measures such as the mental component of the SF-36 scales or Hospital anxiety and depression scale (HADS).97
There are no adequate benchmarks for the MID of DLQI scores in general dermatological conditions, although there have been some attempts to determine these in conditions such as psoriasis.97,101,102 No validity and MID information were found for the patients with AD.102
Children’s Dermatology Life Quality Index
The CDLQI is based on the adult version (DLQI). This is a child-completed questionnaire to be applied to children from 3 to 16 years of age, designed to measure the impact of any skin disease on the quality of life with a recall period of 7 days. It is 1 of the most commonly used instruments for measuring health-related quality of life in children with skin conditions.48,49,103 The instrument has 10 questions asking about the impact of a skin disease on the life of the affected child, including symptoms, embarrassment, friendships, clothes, playing, sports, bullying, sleep, and impact of treatment. Each question is answered on a 4-point Likert scale scored from 0 to 3,The 10 individual question scores are summed to provide a total score, with a maximum possible of 30, indicating maximum impact on QoL.
A 2013 systematic review did not identified studies demonstrating content validity.49 In the same review 3 studies demonstrated concurrent validity, 2 between CDLQI and Cardiff Acne Disability Index (CADI) and 1 between CDLQI and Childhood Atopic Dermatitis Impact Scale (CADIS). The CDLQI was correlated in 10 studies with SCORAD, the primarily sign-based severity scoring system for AD. Forty-five studies demonstrated convergent construct validity and 6 studies demonstrated divergent construct validity. The same review showed good internal consistency of the CDLQI (examined in 6 studies) with Cronbach alpha values ranging from 0.82 to 0.92. Similarly, test-retest reliability was adequate, with Spearman’s rank order correlation coefficient calculated in 4 studies (range 0.74 to 0.97). One study examined the ICC, finding 0.80. Good responsiveness to change was found in studies using Wilcoxon signed rank test and repeated measures ANOVA.
One study conducted in the US and Canada with 202 participants using a distribution- based approach, determined the MCID of the CDLQI in psoriasis to be 2.5. No specific MID has been determined for patients with AD.
EQ-5D Questionnaire
The EQ-5D is a generic quality-of-life instrument that has been applied to a wide range of health conditions and treatments including AD.65,66 The first of 2 parts of the EQ-5D is a descriptive system that classifies respondents (aged ≥ 12 years) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 3 possible levels (1, 2, or 3) representing “no problems,” “some problems,” and “extreme problems,” respectively. Respondents are asked to choose 1 level that reflects their own health state for each of the 5 dimensions. A scoring function can be used to assign a value (EQ-5D index score) to self-reported health states from a set of population-based preference weights.65,66 The second part is a 20 cm visual analogue scale (EQ VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state,” respectively. Respondents are asked to rate their own health by drawing a line from an anchor box to the point on the EQ VAS which best represents their own health on that day. The third part is the EQ-5D index score, which is generated by applying a multi-attribute utility function to the descriptive system. Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Hence, the EQ-5D produces 3 types of data for each respondent:
A profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121 or 33211.
A population preference-weighted health index score based on the descriptive system.
A self-reported assessment of health status based on the EQ VAS.
The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively.
The MID for the EQ-5D ranges from 0.033 to 0.074. However, no additional validity information was found from literature search for EQ-5Dand no MID has been established in AD.
Short Form (36) Health Survey Version 2 – Acute
The SF-36v2 is a validated generic health status measure. It measures 8 general health domains: physical functioning, role limitations due to physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations due to emotional problems, and mental health. These domains can also be summarized as physical and mental component summary scores. The use of this scale was restricted to adult subjects and not for adolescents to complete. The acute version uses 1 week recall. Eight scaled scores are converted to weighted sums of the questions in their section. Each scale is directly transformed into a 0 to 100 scale on the assumption that each question carries equal weight. Lower scores mean more disability. (i.e., a score of 0 = maximum disability and a score of 100 = no disability).61,62
The SF-36 acute has been extensively validated for measuring quality of life in different clinical conditions.61 However, no specific values were found in patients with AD. Alpha internal consistency coefficients for the 8 scales have been reported from many studies in other clinical conditions; for instance, intraclass correlation of 0.85 have been reported for patients with musculoskeletal problems and item-total correlations typically are in the mid-0.70s, but no specific numbers in AD.
The User’s Manual proposed the following MIDs: a change of 2 points on the physical component summary and 3 points on the mental component summary. The manual also proposes the following minimal mean group differences, in terms of t score points, for SF-36v2 individual dimension scores: physical functioning, 3; role physical, 3; bodily pain, 3; general health, 2; vitality, 2; social functioning, 3; role emotional, 4; and mental health, 3. It should be noted that these MID values were determined as appropriate for groups with mean t score ranges of 30 to 40; for higher t score ranges, values may be higher. MID values do not represent patient-derived scores. The MIDs for the SF-36v2 are based on clinical and other non–patient-reported anchors.61,62 No specific MID for patients with AD was available.
Hospital Anxiety and Depression Scale
The HADS is a widely used patient-reported questionnaire designed to identify anxiety disorders and depression in patients at non-psychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.53-55 The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, among which, 7 items are related to anxiety and 7 items are related to depression. Patients provided responses to each item based on a 4-point Likert scale. Each item is scored from 0 (the best) to 3 (the worst); thus, a person can score between 0 and 21 for each subscale (anxiety and depression). A high score was indicative of a poor state. Scores of 11 or more on either subscale were considered to be a 'definite case' of psychological morbidity, while scores of 8 to 10 represented 'probable case” and 0 to 7 'not a case'.55 One study104 indicated that HADS have good construct validity, with no overall floor or ceiling effects. HADS may be useful for the assessment of AD patients in clinical trials and practice. The author concluded that additional research is needed to confirm the construct validity and to assess content validity and feasibility in research and clinical practice.104 No additional validity and MID information regarding HADS was found from the literature search for AD.
Pharmacoeconomic Review
Abbreviations
AD
atopic dermatitis
BIA
budget impact analysis
EASI
Eczema Area and Severity Index
EASI-75
improvement of 75% or greater in the Eczema Area and Severity Index total score
EQ-5D-3L
EQ-5D 3-Levels questionnaire
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
NMA
network meta-analysis
QALY
quality-adjusted life-year
SoC
standard of care
TCI
topical calcineurin inhibitor
TCS
topical corticosteroids
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Abrocitinib (Cibinqo), oral tablets |
Submitted price | Abrocitinib, 50 mg, 100 mg: $48.67 per tablet Abrocitinib, 200 mg: $54.47 per tablet |
Indication | For the treatment of patients 12 years and older with refractory moderate-to-severe atopic dermatitis, including the relief of pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advisable |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 29, 2022 |
Reimbursement request | As per indication |
Sponsor | Pfizer Canada ULC |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree–Markov model hybrid |
Target population | Adults and adolescents (≥ 12 years of age) with moderate-to-severe AD who have had an inadequate response to prescribed topical therapies or for whom these treatments are not advisable; patients are assumed to have had no prior use of immunosuppressants |
Treatments | Abrocitinib 100 mg plus SoC Abrocitinib 200 mg plus SoC |
Comparators | SoC (a basket of topical corticosteroids, topical calcineurin inhibitors, phosphodiesterase type 4 inhibitors, oral antihistamines) Dupilumab plus SoC Cyclosporine plus SoC Methotrexate plus SoC |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | Lifetime (up to patient age of 110 years) |
Key data source | Treatment inputs for abrocitinib were informed by the JADE COMPARE, JADE EXTEND, JADE MONO-1, JADE MONO-2, JADE TEEN, and JADE DARE trials. A network meta-analysis was used to compare the effectiveness of abrocitinib vs. dupilumab and SoC; comparative effectiveness for methotrexate and cyclosporine was based on an unanchored MAIC |
Submitted results | Sequential base case: eligible for systemic therapy
|
Key limitations |
|
CADTH reanalysis results |
|
AD = atopic dermatitis; ICER = incremental cost-effectiveness ratio; MAIC = matching-adjusted indirect comparison; QALY = quality-adjusted life-year; SoC = standard of care.
Conclusions
Abrocitinib reduces the symptoms of atopic dermatitis (AD) (i.e., increases the likelihood of an improvement of 75% or greater in the Eczema Area and Severity Index total score [EASI-75]) among patients with moderate-to-severe AD compared to standard of care (SoC). The comparative effects of abrocitinib relative to dupilumab and other treatments for AD are uncertain and may depend in part on the timing of outcome assessment. Notably, the use of 16-week effectiveness data in the pharmacoeconomic model may bias the findings in favour of abrocitinib relative to dupilumab. The CADTH base case may therefore overestimate the incremental effectiveness of abrocitinib relative to dupilumab, biasing the cost-effectiveness results in favour of abrocitinib.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including assuming that abrocitinib will be used by patients who are refractory to or ineligible for systemic immunosuppressants; correcting the price of dupilumab; removing methotrexate and cyclosporine as comparators due to the high level of uncertainty with the comparative effectiveness data; assuming that health-state utility values are equal regardless of which treatment is received; assuming that responders to all treatments would receive the utility benefit starting at 8 weeks of treatment; and assuming that treatment effectiveness may wane over the entire analysis horizon.
In the CADTH reanalysis for patients refractory to or ineligible for systemic immunosuppressants, the incremental cost-effectiveness ratio (ICER) for abrocitinib 100 mg plus SoC compared with SoC is $156,735 per quality-adjusted life-year (QALY) gained, and the ICER for abrocitinib 200 mg plus SoC compared with abrocitinib 100 mg plus SoC is $231,013, while dupilumab plus SoC was dominated by (less effective and more costly than) abrocitinib 100 mg plus SoC. The pairwise ICER for abrocitinib 200 mg plus SoC in this population was $177,248 per QALY compared to SoC. In all analyses, price reductions of between 52% and 56% were needed for abrocitinib 100 mg and abrocitinib 200 mg, respectively, to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY. These estimates should be interpreted with the described limitations in mind, particularly the potential for bias in comparisons between abrocitinib and dupilumab due to the timing of assessment.
CADTH was unable to address several other limitations in the sponsor’s submission, including the anticipated preference of clinicians to prescribe abrocitinib 200 mg as a starting dose for most patients with a potential step-down to abrocitinib 100 mg depending on treatment response or adverse events. CADTH was also unable to address the lack of comparative clinical effectiveness data for some relevant treatment comparators, the impact of treatment adherence and adverse events, and the lack of long-term effectiveness data beyond 52 weeks. CADTH was also unable to assess the cost-effectiveness of abrocitinib among patients who have had an inadequate response to biologics. The inability to estimate the influence of these limitations means that the cost-effectiveness of abrocitinib is highly uncertain.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Patient input from caregivers and patients with AD was received from the Eczema Society of Canada, the Canadian Skin Patient Alliance, and Eczéma Québec, which collected input via online surveys, questionnaires, focus groups and 1-on-1 interviews. Patients and caregivers described how living with AD affects their quality of life, mental health, ability to work, social lives, and daily routines. Symptoms that affect quality of life include itch, redness of the skin, repeated rashes, frequent scratching, cracked skin, dry and rough skin, sleep disruption, bleeding from intense scratching, flaking of the skin, pain, thickening of the skin, oozing, swelling, lichenification, blistering, and impact on mobility. Patients reported that the mental health effects of AD include stress, distress, feelings of depression and anxiety, poor self-esteem, low energy, and suicidal thoughts. Patients also described limited accessibility to AD treatments and specialists. Patients described experience with current treatments including, but not limited to, frequent moisturizing, trigger avoidance, topical creams, immunosuppressants (e.g., methotrexate and cyclosporine), oral corticosteroids (e.g., prednisone), and phototherapy. Patients who had experience with abrocitinib through clinical trials reported reduced itch, flares, sores, and skin infections. Generally, patients were unwilling to accept serious side effects; however, some patients noted that they were willing to accept some side effects if treatment relieved their symptoms. Patients also reported frustration and financial strain from the trial-and-error nature of current treatments. Patients expressed a desire for a treatment that would reduce itch, flares, and rashes, improve quality of life and sleep, and reduce pain. Some patients noted that daily oral treatments would be preferred over injections; others noted that less-frequent injections would be preferred over daily treatments.
No clinician group input was received for this review.
Drug plan input received for this review noted that the recommended dose of abrocitinib (100 mg versus 200 mg daily) is based on individual goals of therapy and the potential risk for adverse reactions, although the optimal dose of abrocitinib is unclear. The plans questioned whether the initiation and renewal criteria for abrocitinib should be aligned with that for dupilumab, and whether a trial of dupilumab would be required before reimbursement of abrocitinib. However, the plans noted that dupilumab is not currently reimbursed in all jurisdictions. The plans also noted that the long-term safety of abrocitinib is not established, that patients taking abrocitinib are at increased risk of serious infections, and that treatment interruptions may be required to manage the adverse events associated with abrocitinib
Several of these concerns were addressed in the sponsor’s model:
Treatment effectiveness is modelled in the sponsor’s submission in terms of the Eczema Area and Severity Index (EASI) score, which considers the extent of redness, thickness, scratching, and lichenification.
The cost-effectiveness of abrocitinib compared to that of dupilumab was considered.
Quality of life was incorporated in the sponsor’s model using EQ-5D data captured in dupilumab clinical trials. However, the EQ-5D questionnaire is unlikely to capture all symptoms of AD that were noted by patients to affect quality of life.
Costs associated with some adverse events were included in the model; however, the impact of adverse events on quality of life may not be captured.
CADTH addressed some of these concerns in a base case that assumes abrocitinib will be used by patients who are refractory to or ineligible for systemic immunosuppressants.
CADTH was unable to address the following concerns raised from stakeholder input:
The structure of the sponsor’s model did not permit the assessment of the cost-effectiveness of abrocitinib after a trial of dupilumab.
The cost-effectiveness of abrocitinib relative to some relevant comparators (e.g., phototherapy and systemic immunosuppressants) could not be assessed due to a lack of data.
Economic Review
The current review is for abrocitinib (Cibinqo) for the treatment of moderate-to-severe AD. Abrocitinib is indicated “for the treatment of patients 12 years and older with refractory moderate-to-severe AD, including the relief of pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advisable.”1
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The submitted cost-utility analysis assessed the cost-effectiveness of 2 doses of abrocitinib 100 mg and 200 mg plus SoC compared with SoC alone, dupilumab plus SoC, methotrexate plus SoC, and cyclosporine plus SoC in patients aged 12 years and older whose symptoms are not adequately controlled by topical therapies and who had no prior use of immunosuppressants.2 Scenario analyses were provided for patients refractory to or ineligible for systemic immunosuppressants and for adolescents (aged 12 to 18 years), which is consistent with the Health Canada indication. The modelled population is based on patients in the phase III JADE trials (JADE MONO-1, JADE MONO-2, JADE COMPARE, JADE TEEN).
Abrocitinib is available as 50 mg, 100 mg, or 200 mg tablets. The recommended dosage is 100 mg or 200 mg orally once daily “based on individual goal of therapy and potential risk for adverse reactions.”1 The submitted price of abrocitinib is $48.67 per 50 mg or 100 mg tablet and $54.47 per 200 mg tablet. The annual per-patient cost for the recommended dosages is $17,777 for 100 mg abrocitinib taken daily (assuming that a single 100 mg tablet would be taken) and $19,896 for 200 mg daily (assuming that a single 200 mg tablet would be taken). In the sponsor’s model, the annual cost of dupilumab was assumed to be $24,988 per patient, while the annual per-patient costs of methotrexate and cyclosporine were assumed to be $1,816 and $99, respectively. The SoC was a basket of treatments, including mild- to medium-potency topical corticosteroids (TCS), topical calcineurin inhibitors (TCIs), phosphodiesterase type 4 inhibitors, and oral antihistamines; the proportion of each treatment within the basket was based on clinical expert opinion. No cost was associated with the use of SoC in the sponsor’s submission.
The clinical outcomes of interest were QALYs. The economic analysis was undertaken from the perspective of the publicly funded health care payer over a lifetime horizon (maximum patient age of 110 years). Discounting at 1.5% per year was applied to both costs and outcomes.
Model Structure
The model structure included a short-term (1-year) phase and a long-term maintenance phase. The short-term phase was based on a 1-year decision tree, with treatment response assessed at 16 and 52 weeks (Figure 1). Patients with moderate-to-severe AD entered the decision tree on abrocitinib 100 mg plus SoC, abrocitinib 200 mg plus SoC, dupilumab plus SoC, methotrexate plus SoC, cyclosporine plus SoC, or SoC alone. After 16 weeks of treatment, treatment response was assessed based on the EASI, with response defined as an EASI-75. Patients with a treatment response at 16 weeks stayed on their current treatment until 52 weeks. Those with a less than an EASI-75 (i.e., no treatment response) at 16 weeks discontinued active treatment (abrocitinib plus SoC, dupilumab plus SoC, methotrexate plus SoC, or cyclosporine plus SoC) and received subsequent therapy consisting of a basket of treatments (methotrexate, cyclosporine, TCIs, and phosphodiesterase type 4 inhibitors). Response was assessed for a second time at 52 weeks. Those with a sustained response at 52 weeks entered a Markov model on their current treatment. Those with a treatment response at 16 weeks but not at 52 weeks were assumed to have lost the response at 32 weeks and entered the Markov model on subsequent treatment only.
All patients entered the decision tree with the same baseline utility value. For patients with a treatment response at 16 weeks, the responder utility was assumed to be incurred at 8 weeks for abrocitinib plus SoC, methotrexate plus SoC, and cyclosporine plus SoC, and at 16 weeks for dupilumab and SoC. Those with an initial treatment response at 16 weeks but not at 52 weeks were assumed to accrue the responder utility benefit until the 16-week assessment, after which they were assumed to receive an average of the responder and nonresponder utility until the 52-week assessment. Patients with an initial response at 16 weeks but who discontinued treatment before 52 weeks were assumed to receive the average of the utility weight for treatment responders and nonresponders to SoC.
The Markov model consisted of 3 health states: maintenance treatment, subsequent treatment, and death (Figure 2). Patients entered the Markov model from the decision tree in either the maintenance treatment or subsequent treatment state, depending on treatment response at 52 weeks. Upon entering the Markov model, utilities were assigned based on whether patients were receiving active treatment or SoC and whether a patient was classified as a treatment responder or nonresponder. Those who entered the model in the maintenance treatment state remained on treatment until a loss of treatment response or treatment discontinuation, at which time they moved to the subsequent treatment state or death. Patients remained in the subsequent treatment state until death.
Model Inputs
The baseline characteristics in the model were based on pooled data from the JADE clinical trials (29 years, 74.3 kg).2 The sponsor assumed that patients independently administered all treatments, although a 1-time training session was assumed for dupilumab administration. Treatment adherence was not considered in the model.
The clinical efficacy (probability of achieving an EASI-75) for abrocitinib plus SoC, dupilumab plus SoC, and SoC at 16 weeks was derived from a network meta-analyses (NMA), while the efficacy of methotrexate plus SoC and cyclosporine plus SoC were derived from an unanchored matching-adjusted indirect comparison (MAIC).3 At 52 weeks, the probability of a treatment response was based on a naive comparison of clinical trial data for abrocitinib (ad hoc analysis of 48-week data from the JADE EXTEND trial) and dupilumab (from the LIBERTY AD CHRONOS trial), while the 52-week treatment response for cyclosporine and methotrexate was assumed to be equivalent to the average response of abrocitinib, dupilumab, and SoC. Treatment effectiveness was assumed to wane at a constant rate between 52 weeks and 5 years, and a response floor of 62% was assumed for all treatments except SoC (the response floor for SoC was 3%); waning of treatment effect was not modelled beyond the first 5 years of treatment. A discontinuation rate of 6.9% was applied to abrocitinib 100 mg plus SoC, abrocitinib 200 mg plus SoC, and dupilumab plus SoC in the first year of treatment based on the rate of discontinuation among treatment responders who discontinued abrocitinib 200 mg in the JADE EXTEND trial, with an annual discontinuation rate of 6.3% in year 2 and onward based on the results of the LIBERTY AD SOLO trial reported in the 2018 CADTH review of dupilumab.4 For methotrexate plus SoC and cyclosporine plus SoC, discontinuation was assumed to be 84.5% and 57.7% between 16 to 52 weeks, respectively, and 93.2% and 71.1%, respectively, annually thereafter.
The sponsor assumed that treatment did not affect mortality risk. Age- and sex-specific mortality rates were based on general population life tables from Statistics Canada.5
Health-state utilities were based on published EQ-5D 3-Levels questionnaire (EQ-5D-3L) data for dupilumab from the LIBERTY AD SOLO 1 and LIBERTY AD SOLO 2 trials6 and were assumed to vary by whether active treatment (abrocitinib, dupilumab, methotrexate, or cyclosporine) or SoC was received. The health-state utility values were age-adjusted using population norms from the Health Quality Council of Alberta.7 Disutilities related to adverse events were assumed to be captured as part of health-state utility values.
Adverse events, including acne, asthma, blepharitis, conjunctivitis allergic, AD, headaches, influenza, injection-site reactions, nasopharyngitis, nausea, oral herpes, sinusitis, upper respiratory tract infections, urinary tract infections, vomiting, and folliculitis, were based on those reported in clinical trials (JADE COMPARE for abrocitinib and SoC; LIBERTY AD CHRONOS for dupilumab8; and METHODA for methotrexate and cyclosporine9). Headaches, injection-site reactions, and nausea were assumed to be experienced only in the first treatment cycle, while all other adverse events could occur at any time over the analysis horizon, based on the proportion of patients with an event in the clinical trials, most of which had a treatment duration of 16 weeks.
The economic model included costs related to drugs (acquisition and administration), adverse events, disease management (i.e., visits to primary care providers, dermatologists, emergency room, and hospitalizations), monitoring, and subsequent treatment. Drug acquisition costs for abrocitinib 100 mg and 200 mg were based on the sponsor’s submitted price,3 while the cost of comparators was obtained from IQVIA for dupilumab, and the Ontario Drug Benefit Formulary10 for methotrexate and cyclosporine). For dupilumab, dosing was assumed to vary by patient age,11 and a 1-time administration cost was incorporated to reflect the cost of training patients to self-administer. No cost was associated with SoC in the model. Adverse events were assumed to be managed by 1 visit to a family physician visit. Disease-management costs varied by treatment response and were obtained from the CADTH pharmacoeconomic analysis of dupilumab.12 Treatment monitoring costs (renal function tests, urinalysis, lipid profile, complete count, liver function test, and hepatitis B and C) were included, with the frequency of testing based on clinical expert opinion for abrocitinib or on product monographs11 or guidelines13 for other comparators. Costs associated with subsequent treatment (methotrexate, cyclosporine, TCIs, and phosphodiesterase type 4 inhibitors) were incorporated after treatment discontinuation or lost response.
Summary of Sponsor’s Economic Evaluation Results
The sponsor’s submitted probabilistic and deterministic cost-utility analyses produced similar results; the results of the probabilistic analyses (with 1,500 iterations) are presented in the following section. The submitted analyses were based on the publicly available prices for SoC, methotrexate, and cyclosporine, and on the wholesale prices of dupilumab.
Base-Case Results
The sponsor’s base case assessed the cost-effectiveness of abrocitinib among patients eligible for systemic therapy (i.e., those who have had an inadequate response to prescribed topical therapy or for whom these treatments are not advisable). In the sponsor’s base-case analysis, both strengths of abrocitinib plus SoC were more costly and produced more QALYs than did treatment with SoC alone (Table 3). Based on sequential analyses, SoC alone would be the preferred treatment option if a decision-maker’s WTP threshold is below $67,246 per QALY. Abrocitinib 100 mg plus SoC would be the preferred treatment option for WTP thresholds between $67,246 and $96,122, while abrocitinib 200 mg plus SoC would be the preferred option above $96,122 per QALY. Methotrexate plus SoC and cyclosporine plus SoC were dominated by abrocitinib 100 mg plus SoC, indicating that they were both more costly and produced fewer QALYs compared to abrocitinib 100 mg plus SoC, while dupilumab plus SoC was dominated by abrocitinib 200 mg plus SoC. At a WTP of $50,000 per QALY, the probability of abrocitinib 100 mg plus SoC being considered cost-effective relative to SoC was 37%, while the probability of abrocitinib 200 mg plus SoC being optimal compared to SoC was 30% (The sponsor’s model did not permit the sequential consideration of the probability of cost-effectiveness). Compared to dupilumab, the pairwise probability of abrocitinib 100 mg plus SoC being considered optimal was 98%, while the probability of abrocitinib 200 mg plus SoC being optimal was 91%.
The drug costs associated with abrocitinib are key drivers of the ICER (Appendix 3, Table 12), and the majority of QALYs gained with abrocitinib plus SoC were accrued in the extrapolated period after the first year of treatment in the sponsor’s base case for both abrocitinib doses compared with SoC alone (90%). Additional details pertaining to the sponsor’s submission are provided in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Patients Eligible for Systemic Therapy
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Standard of care | 177,444 | 21.91 | Reference |
Abrocitinib 100 mg plus SoC | 249,053 | 22.98 | 67,246 |
Abrocitinib 200 mg plus SoC | 277,969 | 23.28 | 96,122 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the efficiency frontier are reported in the main body. Full results are reported in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor provided several scenario and sensitivity analyses, including scenarios involving patients refractory to or ineligible for systemic immunosuppressants; however, sequential analyses comparing abrocitinib 100 mg plus SoC and abrocitinib 200 mg plus SoC to the other comparators were not provided (i.e., abrocitinib 100 mg plus SoC and abrocitinib 200 mg plus SoC were compared to each other treatment in a pairwise fashion), limiting the interpretation of the findings.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The pharmacoeconomic evaluation does not reflect the intended clinical use of abrocitinib. The sponsor’s base case assesses the cost-effectiveness of abrocitinib among patients with moderate-to-severe AD who have had an inadequate response to prescribed topical therapies or for whom these treatments are not advisable, which is inconsistent with the Health Canada indication.1 The sponsor’s base-case analysis assumes that patients have had no prior exposure to systemic immunosuppressants; however, the clinical expert consulted by CADTH for this review indicated that, in clinical practice, abrocitinib will be most likely used after a trial of systemic immunosuppressants, rather than after a trial of prescribed topical therapy. The clinical expert noted that the expected place of abrocitinib in therapy is similar to that of dupilumab, which is recommended for patients who have had an adequate trial or who are ineligible for phototherapy (where available), methotrexate, and cyclosporine.14
The abrocitinib product monograph states that, for adult patients, the choice between the 200 mg and 100 mg abrocitinib doses should be “based on individual goal of therapy and potential risk for adverse reactions.”1 The clinical expert consulted by CADTH indicated that the majority of patients would be expected to start treatment on abrocitinib 200 mg and potentially step down to abrocitinib 100 mg depending on treatment response and/or adverse events. The sponsor’s pharmacoeconomic analysis assessed the cost-effectiveness of abrocitinib separately for the 200 mg and 100 mg doses, and patients were unable to transition between doses. Considering the cost-effectiveness of each dose separately does not reflect the expected use of abrocitinib in clinical practice.
The CADTH base case assumes that abrocitinib will be used by patients refractory to or ineligible for systemic immunosuppressants. CADTH was unable to assess the impact of switching between abrocitinib 200 mg and abrocitinib 100 mg. CADTH was also unable to address the cost-effectiveness of abrocitinib among patients who have had an inadequate response to biologics.
The comparative effectiveness of abrocitinib is uncertain. In the model, different sources of comparative effectiveness data were used depending on the treatment, and different assumptions were applied at the 16- and 52-week assessment time points in the decision model. At 16 weeks, effectiveness was based on the sponsor’s NMA, which included abrocitinib plus SoC, dupilumab plus SoC, and SoC, while the comparative effectiveness of methotrexate plus SoC and cyclosporine plus SoC were based on the sponsor’s unanchored MAIC for an improvement of 50% or greater in the EASI total score. The sponsor assumed that, for methotrexate and cyclosporine, the ratio of the response between an improvement of 50% and an improvement of 75% in the EASI would be equal to that observed in the NMA for all other treatments.
At 52 weeks, the effectiveness of abrocitinib plus SoC, dupilumab plus SoC, and SoC was based on a naive comparison of the proportion of initial responders who maintained an EASI-75 treatment response in the JADE EXTEND trial (for abrocitinib plus SoC) and the LIBERTY AD CHRONOS trial (for dupilumab plus SoC and SoC). The use of multiple sources of effectiveness data and different assumptions between time points increases the uncertainty associated with the comparative effectiveness of abrocitinib relative to other model comparators.
The clinical expert consulted by CADTH for this review indicated that the assessment of treatment response at 16 weeks may bias the findings against dupilumab due to a potentially longer time to achieve full effectiveness with dupilumab compared to abrocitinib. Currently available head-to-head studies of abrocitinib and dupilumab (the JADE DARE and JADE COMPARE trials) assessed outcomes at 16 weeks, which may bias the findings in favour of abrocitinib.
CADTH was unable to address the limitations associated with the comparative effectiveness of abrocitinib to methotrexate and cyclosporine. Given the high level of uncertainty associated with the use of an unanchored MAIC to inform the effectiveness of these treatments, methotrexate and cyclosporine were removed from the CADTH base case. The cost-effectiveness of abrocitinib versus methotrexate and cyclosporine was assessed in scenario analyses. CADTH was unable to address the potential bias in the effectiveness data for abrocitinib and dupilumab introduced by the use of effectiveness data ascertained after 16 weeks of treatment. CADTH conducted a scenario analysis in which the effectiveness of abrocitinib 100 mg plus SoC, abrocitinib 200 mg plus SoC, and dupilumab plus SoC was assumed to be equivalent.
Relevant comparators were omitted. The clinical expert consulted by CADTH for this review indicated that AD is treated with numerous off-label treatments, including immunosuppressants (e.g., azathioprine and mycophenolate mofetil), retinoids (e.g., acitretin and alitretinoin), and phototherapy. The cost-effectiveness of abrocitinib relative to these treatments is unknown.
CADTH was unable to address this limitation due to a lack of comparative clinical effectiveness data for most immunosuppressants, retinoids, and phototherapy. The cost-effectiveness of abrocitinib relative to other relevant comparators is unknown. Methotrexate and cyclosporine were removed from the CADTH base case due to the high level of uncertainty associated with the sponsor’s MAIC.
The utility values associated with the model health states are uncertain for several reasons. First, the sponsor adopted health-state utility values from a pooled analysis of EQ-5D-3L data from 2 dupilumab trials (LIBERTY AD SOLO-1 and LIBERTY AD SOLO-2),6 stating that data on health-related quality of life (HRQoL) collected in the dupilumab trials are more applicable to patients with AD than those collected in the abrocitinib JADE COMPARE and JADE MONO trials. The utility values from the SOLO trials are considerably different from those collected as part of the JADE trials, including the baseline utility value (0.6156 for SOLO1 and 2; 0.7584 for JADE MONO; and 0.7840 for JADE COMPARE) and the utility benefit gained with a response to active treatment (abrocitinib, dupilumab, methotrexate, or cyclosporine) ( + 0.26 for SOLO; + 0.13 for JADE MONO; and + 0.12 for JADE COMPARE), and the model is highly sensitive to the choice of utilities. The sponsor justified the use of the LIBERTY trial utilities on the basis of the exclusion of patients with “[a] psychiatric condition including recent (within the past year) or active suicidal ideation or behavior” from the JADE trials. The clinical expert consulted by CADTH indicated that the exclusion criteria for the 2 trials were not substantially different such that there would be an expected difference in HRQoL between trial populations.
Second, the sponsor’s submission inappropriately incorporated treatment-dependent utilities, such that patients who responded to active treatment received a greater increase in utility from baseline compared to those who responded to SoC. According to CADTH guidelines for the conduct of economic evaluations,15 utilities should reflect the health states within the model and should not be specific to treatment.
Third, the utilities adopted for nonresponders to SoC in the sponsor’s base case were assumed to have a lower utility (0.6084) compared with the baseline (0.6516). The nonresponder utilities (active treatment: 0.7777; SoC: 0.6084) were derived by the sponsor from published6 utility values for treatment responders. The resulting nonresponder utilities for patients who received SoC lack face validity; the clinical expert consulted by CADTH indicated that patients who do not respond to treatment will likely still have an HRQoL higher than at baseline due to contact with a health care professional.
Finally, for patients with a treatment response at 16 weeks, the corresponding increase in HRQoL (modelled in terms of the responder utility weight) was assumed to be incurred at 8 weeks for abrocitinib plus SoC, methotrexate plus SoC, and cyclosporine plus SoC, and at 16 weeks for dupilumab and SoC. This differential timing of HRQoL changes for dupilumab and SoC relative to abrocitinib and other model comparators were justified by the sponsor on the basis of findings from the JADE COMPARE trial, in which the proportion of patients with a treatment response to dupilumab appeared to change more gradually than with abrocitinib. However, this does not address the timing of HRQoL effects. As noted in the CADTH review of dupilumab,12 patients taking dupilumab are likely to develop AD-related changes soon after treatment onset, which may affect HRQoL.
CADTH explored the impact of adopting alternative health-state utility values in scenario analyses. In the CADTH base case, the responder utility was applied at 8 weeks for all treatments.
The long-term effectiveness of abrocitinib is uncertain. As noted in the CADTH clinical review, the 52-week JADE EXTEND trial is ongoing. While effectiveness in the sponsor’s model was based on data from the JADE EXTEND trial, the clinical study report for this trial was not available at the time of the review by CADTH. The sponsor assumed that the annual probability of treatment discontinuation after the first year would be equivalent between abrocitinib and dupilumab based on data reported for dupilumab.4 The sponsor similarly assumed without justification that effectiveness-waning would be equivalent between abrocitinib and dupilumab. The clinical expert consulted by CADTH for this review indicated that the validity of these assumptions is highly uncertain due to the lack of long-term data for abrocitinib.
CADTH explored alternative assumptions about the timing of effectiveness-waning in scenario analyses, although whether effectiveness-waning is equivalent between abrocitinib and dupilumab remains uncertain. CADTH was unable to address treatment discontinuation beyond 52 weeks due to the lack of long-term data past 52 weeks.
Treatment adherence is not considered. The sponsor’s model does not account for nonadherence to treatment. The clinical expert consulted by CADTH for this review indicated that, in practice, adherence would be less than 100% and may differ between adults and adolescents. The expert noted that adherence to treatments administered as daily tablets, such as abrocitinib, will likely be lower than for treatments administered less frequently via injection (i.e., dupilumab). Treatment adherence would be expected to affect patient outcomes (i.e., HRQoL and treatment response) as well as costs. In the submitted budget impact analysis (BIA),16 the sponsor estimated adherence to abrocitinib to be 62.7%, on the basis of percentage of patients with rheumatoid arthritis remaining on tofacitinib after 1 year of treatment.17 Whether adherence is similar between patients with AD and those with rheumatoid arthritis, and whether adherence is similar between abrocitinib and tofacitinib, is unknown.
CADTH was unable to explore the impact of reduced adherence in the model due to the structure of the model and a lack of clinical data about the rate of adherence and the impact on health outcomes.
The impact of adverse events is uncertain. The impact of adverse events on the cost-effectiveness of abrocitinib is uncertain for several reasons. First, the rates of adverse events in the sponsor’s assessment were applied annually in the model, based on the proportion of patients who experienced an event in trials with a 16-week treatment duration. This duration may be insufficient to capture the true risk of some events (e.g., malignancy). Although the pivotal abrocitinib AD trials showed few serious adverse events, the abrocitinib product monograph1 includes a serious warnings-and-precautions box for serious infections, malignancies, and clotting disorders. Second, the sponsor incorporated costs related to adverse events as a 1-time cost within the first model cycle (headaches, nausea, and injection-site reactions) or on an annual basis (all other adverse events), based on the proportion of patients with an event in the JADE COMPARE trial (for abrocitinib plus SoC and SoC), LIBERTY AD CHRONOS and LIBERTY AD ADOL trials (for dupilumab plus SoC), or METHODA9 trial (for methotrexate plus SoC and cyclosporine plus SoC). For dupilumab and abrocitinib, the proportion of patients with adverse events was obtained from different sources, despite the availability of a head-to-head trial including both treatments (JADE COMPARE). No adjustment or accounting for differences in patient characteristics or treatment durations was considered, and the proportion of patients who experienced adverse events in short-term clinical trials was assumed to be consistent over time, which is uncertain. Third, the sponsor assumed that disutilities related to adverse events would be captured as part of health-state utility values, which were based on EQ-5D-3L data collected in the dupilumab LIBERTY SOLO trials. It is unlikely that the impact of adverse events on quality of life would be adequately captured by EQ-5D-3L values. The EQ-5D lacks specific domains that might be affected by adverse events, and the EQ-5D was administered at set times during the trial and has a 1-day recall period, which is problematic in assessing the impact of adverse events in clinical trials.18 Additionally, quality-of-life measurements in clinical trials are often missing not at random. Further, applying dupilumab utility weights to other treatments fails to account for differences in their respective safety profiles. Finally, the adverse events included in the model do not capture the range of adverse events deemed to be of special interest to clinicians (e.g., herpes simplex, hepatitis B, and anemia).
CADTH was unable to address this limitation due to a lack of data and the structure of the sponsor’s model. The impact of adverse events on the ICER is therefore highly uncertain and may bias the ICER in favour of abrocitinib.
The cost-effectiveness of abrocitinib among adolescents is uncertain. Abrocitinib is indicated for use by patients 12 years of age and older. However, the modelled cohort in the sponsor’s base case was assumed to have a starting age of 29 years. Effectiveness at 16 weeks in the sponsor’s model was based on the sponsor’s NMA, which included the JADE MONO-1, JADE MONO-2, and JADE COMPARE trials. As noted in the CADTH clinical review, the JADE COMPARE trial enrolled patients aged 18 years and older, with a mean age of 34 years. While the JADE MONO-1 and MONO-2 trials enrolled patients aged 12 years and older, these trials enrolled relatively few participants younger than 18 years (JADE MONO-1: 21.7%; JADE MONO-2: 10.2%). To derive effective estimates at 16 weeks for patients aged 12 to 18 years, the sponsor adjusted the NMA findings by applying the ratio of overall response from the JADE COMPARE trial (age 18 and older) to that observed in the JADE TEEN trial (12 to 18 years). At 52 weeks, the probability of sustaining a treatment response was based on the JADE EXTEND trial, which is a long-term extension study. The clinical study report for the JADE EXTEND trial was not available at the time of the review by CADTH, and the relevance of findings from the JADE EXTEND trial to the adolescent subgroup is uncertain. The clinical expert consulted by CADTH also noted that adherence to treatment may be lower among adolescents than among adults, which would affect both costs and clinical outcomes.
CADTH explored the cost-effectiveness of abrocitinib in adolescents in scenario analyses. CADTH was unable to consider differential adherence estimates for adolescents due to the structure of the sponsor’s model.
Poor modelling practices were employed. The model includes numerous IFERROR statements, which can lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impracticable, as it remains unclear whether the model is running inappropriately by overriding errors. For some model parameters, the sponsor arbitrarily incorporated uncertainty as ± 15% of the mean value (e.g., for treatment response, discontinuation rate in the first year), ± 20% of the mean value (e.g., health-state utility values), or ± 50% of the mean value (costs of treating adverse events), which does not reflect the true uncertainty around the model’s parameters possible values.
CADTH was unable to fully validate the model and notes that the results presented should be treated with a degree of caution as the validity of the model calculations could not be thoroughly appraised.
One other limitation was identified, but it was not considered to be a key limitation: the price of dupilumab in the sponsor’s submission was based on that in the dupilumab CADTH submission12 and on the IQVIA10 wholesale price ($959), which is lower than the price on the Ontario Exceptional Access Program19 ($979).
The price of dupilumab in the CADTH base case is based on that in the Ontario Exceptional Access Program.
Additionally, key assumptions made by the sponsor were appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients enrolled in the JADE clinical trials were assumed to be representative of patients in Canada who would be eligible for abrocitinib. | Reasonable, although the clinical expert consulted by CADTH for this review noted that patients in the pivotal abrocitinib trials may have less-severe AD than those included in the dupilumab trials. |
Treatment with abrocitinib was assumed to be on a background of concomitant topical therapy. | Reasonable as abrocitinib can be used with or without topical corticosteroids. The clinical expert consulted by CADTH indicated that at least 80% of patients would be expected to use concomitant topical corticosteroids. |
Patients who discontinue active treatment (abrocitinib, dupilumab, methotrexate, and cyclosporine) were assumed to receive subsequent treatment, which was assumed to consist of a basket of treatments, including methotrexate, cyclosporine, TCIs, and phosphodiesterase type 4 inhibitors. | Uncertain. The clinical expert consulted by CADTH indicated that, a patient who starts on systemic treatments such as abrocitinib or dupilumab would be expected to remain on systemic treatment indefinitely; however, CADTH was unable to model treatment-specific sequences due to the structure of the sponsor’s model. |
A 75% reduction in EASI score from baseline was assumed to represent a treatment response. | Reasonable. The clinical expert consulted by CADTH indicated that an EASI-75 score would likely represent a clinically meaningful reduction. CADTH notes that EASI-75 has been used in previous submissions in this clinical area. Treatment decisions in practice not made based on the EASI score, although the EASI score is routinely used due to reimbursement requirements. |
No cost was incorporated for SoC, reflecting the assumption that the use of SoC would not vary between treatments. | Uncertain, but unlikely to have an important effect on the ICER. The clinical expert consulted by CADTH indicated that patients whose AD responds favourably to treatment may be able to reduce the amount of topical treatments used. |
Medical resource utilization was assumed to include general practitioner or specialist visits, outpatient visit to the clinic, and hospital admissions. | Reasonable. The sponsor adopted medical resource utilization costs from the previous CADTH review of dupilumab, with separate costs incorporated for treatment responders ($173.19 per patient annually) and nonresponders ($4,193.49 per patient annually).12 |
AD = atopic dermatitis; EASI = Eczema Area Severity Index; EASI-75 = improvement of 75% or greater in the Eczema Area and Severity Index total score; ICER = incremental cost-effectiveness ratio; SoC = standard of care; TCIs = topical calcineurin inhibitors.
CADTH Reanalyses of the Economic Evaluation
Several limitations with the sponsor’s submission could not be adequately addressed. Notably, CADTH could not address the anticipated preferential use of abrocitinib 200 mg as a starting dose for most patients, with a step-down to abrocitinib 100 mg depending on treatment response or adverse events.
CADTH was also unable to address the lack of comparative clinical effectiveness data for some relevant treatment comparators; the impact of treatment adherence or adverse events, and the lack of long-term effectiveness data beyond 52 weeks. CADTH was unable to address the potential bias stemming from the assessment of treatment outcomes at 16 weeks, and the incremental effectiveness between abrocitinib and dupilumab in the CADTH base case may be overestimated. CADTH was additionally unable to address the cost-effectiveness of abrocitinib among patients who have had an inadequate response to biologics.
Aligned with the Health Canada indication for abrocitinib, the CADTH base case considers the use of abrocitinib among patients refractory to or ineligible for systemic immunosuppressants. Abrocitinib was assumed to be used in combination with TCS.
CADTH undertook reanalyses that addressed limitations within the model, as summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Dupilumab costs | $959.595 per syringe based on IQVIA and the 2018 CADTH assessment of dupilumab4 | $978.70 per syringe19 |
Changes to derive the CADTH base case | ||
1. Target population | Patients eligible for systemic therapy | Patients refractory or ineligible for systemic immunosuppressants |
2. Comparators | Dupilumab plus SoC Methotrexate plus SoC Cyclosporine plus SoC SoC | Dupilumab plus SoC SoC |
3. Health-state utility values | Treatment-specific utility values were applied, such that the utility benefit for a treatment response (or nonresponse) was greater patients who received active treatment than for those who received SoC: Active treatmenta plus SoC:
SoC:
| Utility benefits were assumed to be equal for all treatments:
|
4. Timing of response onset | Abrocitinib: 8 weeks Dupilumab: 16 weeks SoC: 16 weeks | Abrocitinib: 8 weeks Dupilumab: 8 weeks SoC: 8 weeks |
5. Treatment effectiveness-waning | Effectiveness was assumed to wane in a linear manner over the first 5 years of treatment | Effectiveness was assumed to wane in a linear manner over the first 40 years of active treatmentb |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 + 5 | |
SoC = standard of care.
aActive treatment = abrocitinib, dupilumab, methotrexate, or cyclosporine.
bThe structure of the sponsor’s model permitted effectiveness-waning to be applied only for the first 40 years of treatment, despite a lifetime horizon. Effectiveness of SoC was assumed to wane over the first 5 years of treatment.
Base-Case Results
CADTH undertook a stepped analysis, incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 6; disaggregated results are presented in Appendix 4, Table 14).
In the CADTH base case, abrocitinib 100 mg plus SoC and abrocitinib 200 mg plus SoC were associated with more QALYs gained over a lifetime horizon compared with dupilumab plus SoC (incremental QALYs versus dupilumab plus SoC for abrocitinib 100 mg plus SoC: 0.11; for abrocitinib 200 mg plus SoC: 0.17), with lower costs (incremental cost versus dupilumab plus SoC for abrocitinib 100 mg plus SoC: −$60,734; for abrocitinib 200 mg plus SoC: −$23,000). In sequential analyses, abrocitinib 100 mg plus SoC was associated with an ICER of $156,735 per QALY gained compared to SoC alone, while abrocitinib 200 mg plus SoC was associated with an ICER of $231,013 compared with abrocitinib 100 mg plus SoC. Dupilumab was dominated by abrocitinib 200 mg plus SoC, although it is likely that the sponsor’s model overestimates the effectiveness of abrocitinib relative to dupilumab.
In pairwise comparison, the probability that abrocitinib 100 mg plus SoC would be considered optimal compared to SoC at a WTP threshold of $50,000 per QALY was 18%, while there is a 14% probability that abrocitinib 200 mg plus SoC would be optimal compared to SoC. (While the probability that a treatment is optimal should be assessed in sequential analyses, the sponsor’s model was not programmed for this assessment.) Compared to dupilumab plus SoC, the probability that abrocitinib 100 mg plus SoC or abrocitinib 200 mg plus SoC would be considered optimal is 98% and 85%, respectively.
In the first year of treatment, there were minimal differences in QALYS gained across treatments (SoC: 0.76; abrocitinib 100 mg plus SoC: 0.79; abrocitinib 200 mg plus SoC: 0.80; dupilumab plus SoC: 0.80), indicating that the majority of the incremental benefits (97%) for both abrocitinib doses were derived from extrapolated findings rather than observed benefit (Table 14). Drug acquisition costs for abrocitinib are key drivers of the ICER, representing 45% and 55% of the total costs associated with abrocitinib 100 mg plus SoC and abrocitinib 200 mg plus SoC, respectively.
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER vs. SoC | Sequential ICER |
|---|---|---|---|---|
Sponsor corrected base case (patients eligible for systemic therapy) | ||||
SoC | 177,356 | 21.95 | Reference | Reference |
Methotrexate plus SoC | 180,701 | 21.81 | Dominated | Dominated |
Cyclosporine plus SoC | 181,617 | 21.81 | Dominated | Dominated |
Abrocitinib 100 mg plus SoC | 249,386 | 23.01 | 68,321 | 68,321 vs.. SoC |
Abrocitinib 200 mg plus SoC | 277,239 | 23.29 | 74,541 | 97,685 vs. abrocitinib 100 |
Dupilumab plus SoC | 287,238 | 22.94 | 111,829 | Dominated |
CADTH base case (patients refractory to or ineligible for systemic immunosuppressants) | ||||
SoC | 181,688 | 27.57 | Reference | Reference |
Abrocitinib 100 mg plus SoC | 248,791 | 28.00 | 156,735 | 156,735 vs. SoC |
Abrocitinib 200 mg plus SoC | 286,525 | 28.17 | 177,248 | 231,013 vs. abrocitinib 100 |
Dupilumab plus SoC | 309,525 | 28.11 | 237,330 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care; vs. = versus.
Scenario Analysis Results
A number of scenario analyses were performed using the CADTH base case (Table 15). These analyses were performed to investigate the impact that critical assumptions had on cost-effectiveness. These scenarios analyses explored the impact of the following model parameters and assumptions on the ICER: assuming that the treatment effectiveness of abrocitinib and dupilumab is equivalent at 16 weeks; adopting alternative health-state utility values; including cyclosporine and methotrexate as a comparators; adopting alternative estimates of treatment effectiveness-waning, and adopting a societal perspective. Scenarios were also analyzed to explore the cost-effectiveness of abrocitinib in patient subgroups (by disease severity, among adolescents).
The cost-effectiveness estimates for abrocitinib 100 mg plus SoC and abrocitinib 200 mg plus SoC were highly sensitive to the health-state utility values chosen (Appendix 4, Table 16). When the sponsor-provided utility values from the abrocitinib trials were adopted, the ICER for abrocitinib 100 mg plus SoC was $536,944 per QALY gained compared to SoC, and the ICER for abrocitinib 200 mg plus SoC was $760,737 per QALY compared to abrocitinib 100 mg plus SoC.
Price-Reduction Analyses
CADTH undertook price-reduction analyses for the sponsor’s base case and CADTH’s base case (Table 7). For the sponsor’s base case, methotrexate and cyclosporine were removed as comparators for comparability to the CADTH reanalysis. A proportional price reduction was assumed for both abrocitinib 100 mg and abrocitinib 200 mg, and the price reduction of abrocitinib 100 mg was held constant when the ICER for abrocitinib 100 mg plus SoC versus SoC alone was at or lower than a $50,000 WTP threshold, while the price of abrocitinib 200 mg continued to be reduced.
For patients refractory to or ineligible for systemic immunosuppressants, a price reduction of 52% would be needed for abrocitinib 100 mg plus SoC to be considered optimal at a WTP of $50,000 compared with SoC in the CADTH base case (Table 7). For abrocitinib 200, a 56% price reduction would be required for abrocitinib 200 mg plus SoC to be considered optimal at a WTP of $50,000.
Table 7: CADTH Price-Reduction Analyses — Patients Refractory to or Ineligible for Systemic Immunosuppressants
Analysis | ICERs for abrocitinib vs. dupilumab plus SoC and SoC ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base casea,b (patients eligible for systemic immunosuppressants) | CADTH reanalysisa,b (patients refractory to or ineligible for systemic immunosuppressants) |
No price reduction | WTP < 56,915: SoC WTP 56,915 to 85,083: ABRO 100 mg plus SoC WTP ≥ 85,083: ABRO 200 mg plus SoC | WTP < 156,735: SoC WTP 156,735 to 231,013: ABRO 100 mg plus SoC WTP ≥ 231,013: ABRO 200 mg plus SoC |
10% | WTP < 49,538: SoC WTP 49,538 to 74,745: ABRO 100 mg plus SoC WTP ≥ 74,745: ABRO 200 mg plus SoC | WTP < 131,519: SoC WTP 131,519 to 188,271: ABRO 100 mg plus SoC WTP ≥ 188,271: ABRO 200 mg plus SoC |
20% | WTP < 47,846: SoC WTP ≥ 47,846: ABRO 200 mg plus SoC | WTP < 111,743: SoC WTP 111,743 to 162,165: ABRO 100 mg plus SoC WTP ≥ 162,165: ABRO 200 mg plus SoC |
30% | NA | WTP < 91,968: SoC WTP 91,968 to 136,058: ABRO 100 mg plus SoC WTP ≥ 136,058: ABRO 200 mg plus SoC |
40% | NA | WTP < 72,192: SoC WTP 72,192 to 109,952: ABRO 100 mg plus SoC WTP ≥ 109,952: ABRO 200 mg plus SoC |
50% | NA | WTP < 52,416: SoC WTP 52,416 to 83,846: ABRO 100 mg plus SoC WTP ≥ 83,846: ABRO 200 mg plus SoC |
52% | NA | WTP < 48,461: SoC WTP 48,461 to 78,625: ABRO 100 mg plus SoC WTP ≥ 78,625: ABRO 200 mg plus SoC |
56% | NA | WTP < 48,081: SoC WTP ≥ 48,081: ABRO 200 mg plus SoC |
ABRO = abrocitinib; ICER = incremental cost-effectiveness ratio; NA = not applicable; SoC = standard of care; vs. = versus; WTP = willingness-to-pay.
Note: The corrected price of dupilumab was used in all price-reduction scenarios.
aThe price of abrocitinib 100 mg was reduced until the ICER for abrocitinib 100 mg vs. SoC was less than $50,000, after which point the price of abrocitinib 100 mg was assumed to remain constant while the price of abrocitinib 200 mg continued to be reduced.
bDupilumab was dominated in all analyses.
Issues for Consideration
Additional treatments, including upadacitinib (a Janus kinase inhibitor) and tralokinumab (an interleukin-13 monoclonal antibody), are currently under consideration by Health Canada for the treatment of moderate-to-severe AD. The cost-effectiveness of abrocitinib relative to these other potential treatments is unknown.
A recent pharmacoeconomic analysis by ICER found that abrocitinib was more costly and more effective than dupilumab (i.e., it produced more QALYs).20 CADTH notes that there were methodological differences between the ICER analysis and the sponsor’s submission (e.g., model structure, clinical inputs, and drug prices) that preclude direct comparison of the findings. These differences highlight the uncertainty associated with both the sponsor’s submission and the CADTH reanalysis.
As noted in the patient and clinician input received for this review, some patients may prefer treatment that can be administered less frequently, while others may prefer daily treatments over injections. Abrocitinib is taken daily, compared with biweekly injections of dupilumab.
The clinical expert consulted by CADTH for this review noted that access to dermatological treatment is difficult for patients who already face barriers to health care, particularly members of racially and economically marginalized communities and those who live in remote areas. The analysis described in this report does not consider the differential impacts that may be experienced by patients receiving treatment with a tablet versus a syringe versus other methods of drug administration. Consequently, any differences in cost-effectiveness due to these factors is not reflected within this analysis.
Overall Conclusions
The CADTH clinical review found that abrocitinib reduces the symptoms of AD among patients with moderate-to-severe AD, although the comparative efficacy and safety of abrocitinib relative to other treatments for AD are uncertain. As noted by the clinical expert consulted by CADTH for this review, the use of 16-week effectiveness data in the pharmacoeconomic model may bias the findings in favour of abrocitinib relative to dupilumab. CADTH was unable to address this limitation in its reanalysis. Consequently, CADTH’s calculated ICERs and the price reductions necessary to reach a given cost-effectiveness threshold are likely underestimated.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including assuming that abrocitinib will be used by patients refractory or ineligible to systemic immunosuppressants; correcting the price of dupilumab; removing methotrexate and cyclosporine as comparators due to the high level of uncertainty with the comparative effectiveness data; assuming that health-state utility values are equal regardless of which treatment is received; assuming that treatment responders would receive the utility benefit starting at 8 weeks for all treatments; and assuming that treatment effectiveness may wane over the entire analysis horizon. Abrocitinib 100 mg plus SoC was more costly and more effective than SoC (ICER: $156,735 per QALY gained), abrocitinib 200 mg plus SoC was more costly and effective than abrocitinib 100 mg plus SoC (ICER: $231,013), and dupilumab plus SoC was dominated by (less effective and more costly than) abrocitinib 200 mg plus SoC. The cost-effectiveness results were highly sensitive to assumptions about health-state utility values and the price of both abrocitinib and dupilumab.
A scenario analysis was conducted to reflect the most likely clinical use of abrocitinib based on clinical expert feedback (i.e., abrocitinib 200 mg plus SoC versus dupilumab plus SoC versus SoC in patients refractory or ineligible for systemic immunosuppressants). In this analysis, abrocitinib 200 mg plus SoC was associated with an ICER of $177,248 per QALY compared with SoC among patients refractory to or ineligible for systemic immunosuppressants, while dupilumab plus SoC was dominated. In the CADTH base case and all scenarios, price reductions of between 52% and 56% were needed for abrocitinib 100 mg and abrocitinib 200 mg to be considered cost-effective at a WTP threshold of $50,000 per QALY.
CADTH was unable to address several potentially influential limitations within the sponsor’s submission. In addition to the potential bias in favour of abrocitinib versus dupilumab, the CADTH base case could not reflect the anticipated preference of clinicians to step down dosing from abrocitinib 200 mg, the lack of comparative clinical effectiveness data for some relevant comparators, the impact of treatment adherence and adverse events, and the lack of long-term clinical data beyond 52 weeks. CADTH was also unable to address the cost-effectiveness of abrocitinib among patients who have had an inadequate response to biologics. The impact of these limitations is unknown. Consequently, all estimates of the ICER and price reduction are highly uncertain and should be interpreted with these limitations in mind, particularly the potential for bias in comparisons between abrocitinib and dupilumab due to the timing of the assessment.
As noted in the CADTH clinical review, exploratory analyses demonstrated that initiating treatment with abrocitinib 200 mg was generally more efficacious than abrocitinib 100 mg for establishing a response to treatment in the 12- to 16-week time frame that was studied in the phase III clinical trials. The long-term comparative efficacy of abrocitinib 200 mg, abrocitinib 100 mg, and dupilumab is unknown. Based on the submitted evidence, the CADTH pharmacoeconomic reanalysis finds that both abrocitinib 100 mg and abrocitinib 200 mg are not cost-effective at a WTP threshold of $50,000. The long-term comparative efficacy and therefore cost-effectiveness of all comparators remains highly uncertain.
References
1.Cibinqo (abrocitinib): 50 mg, 100 mg, 200 mg oral tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022 June 29.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
3.Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets [internal sponsor's package]. Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
4.CADTH Common Drug Review pharmacoeconomic review report: dupilumab (Dupixent) for moderate-to-severe atopic dermatitis (AD). Ottawa (ON): CADTH; 2018 Jul: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0533_Dupixent_PE_Report.pdf?bcs-agent-scanner=b4d8fb92-0f36-5c46-a0a7-68cf0875f55a. Accessed 2021 Jul 8.
5.Statistics Canada. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2021; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2021 Jun 3.
6.Simpson EL. Dupilumab improves general health-related quality-of-life in patients with moderate-to-severe atopic dermatitis: pooled results from two randomized, controlled phase 3 clinical trials. Dermatol Ther. 2017;7(2):243-248. PubMed
7.Alberta provincial norms for EQ-5D-3L. Calgary (AB): Health Quality Council of Alberta; 2013: https://www.hqca.ca/wp-content/uploads/2018/05/Alberta_Provincial_Norms_for_EQ_5D_3L_1_.pdf. Accessed 2021 Jul 8.
8.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. PubMed
9.Goujon C, Viguier M, Staumont-Salle D, et al. Methotrexate versus cyclosporine in adults with moderate-to-severe atopic dermatitis: a phase III randomized noninferiority trial. J Allergy Clin Immunol Pract. 2018;6(2):562-569 e563. PubMed
10.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Jul 20.
11.Dupixent (dupilumab): 300 mg single-use syringe (300 mg/2 mL); 300 mg single-use pen (300 mg/2 mL); 200 mg single-use syringe (200 mg/1.14 mL) subcutaneous injection [product monograph]. Laval (QC): Sanofi-aventis Canada Inc; 2021 Aug 17: https://pdf.hres.ca/dpd_pm/00062540.PDF. Accessed 2021 Aug 19.
12.CADTH Drug Reimbursement Review pharmacoeconomic report: dupilumab (Dupixent) for the treatment of patients aged 12 years and older with moderate-to-severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Ottawa (ON): CADTH; 2020 Jun: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0636-dupixent-pharmacoeconomic-review-report.pdf. Accessed 2021 Jun 9.
13.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: Section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
14.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent - sanofi-aventis Canada Inc). Ottawa (ON): CADTH; 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2021 Jun 15.
15.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/pdf/guidelines_for_the_economic_evaluation_of_health_technologies_canada_4th_ed.pdf. Accessed 2021 Aug 23.
16.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cibinqo (abrocitinib) for moderate to severe atopic dermatitis, 50mg, 100mg, 200mg oral tablets. Kirkland (QC): Pfizer Canada ULC; 2021 Apr 26.
17.Pope J, Bessette L, Jones N, et al. Experience with tofacitinib in Canada: patient characteristics and treatment patterns in rheumatoid arthritis over 3 years. Rheumatology (Oxford). 2020;59(3):568-574. PubMed
18.Bansback N, Sun H, Guh DP, et al. Impact of the recall period on measuring health utilities for acute events. Health Econ. 2008;17(12):1413-1419. PubMed
19.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Jul 20.
20.Atlas SJ, Brouwer E, Fox G, et al. JAK inhibitors and monoclonal antibodies for the treatment of atopic dermatitis: effectiveness and value. (Evidence report). Boston (MA): Institute for Clinical and Economic Review (ICER); 2021: https://icer.org/wp-content/uploads/2020/12/Atopic-Dermatitis_Revised-Evidence-Report_070921.pdf?bcs-agent-scanner=74ce3d04-bf93-d641-9167-3c8727fa7d3d. Accessed 2021 Jul 12.
21.Ratio-Amcinonide (amcinonide USP): 0.1% w/w cream, ointment, lotion [product monograph] Toronto (ON): Teva Canada Limited; 2017 Jun 26: https://pdf.hres.ca/dpd_pm/00039966.PDF. Accessed 2021 Jul 20.
22.Diprosone (betamethasone dipropionate): 0.05% w/w cream (Merck standard), ointment (Merck standard), lotion (USP) [product monograph]. Kirkland (QC): Merck Canada Inc; 2020 May 19: https://pdf.hres.ca/dpd_pm/00057419.PDF. Accessed 2021 Jul 20.
23.Teva-Clobetasol (clobetasol 17-propionate): 0.05% cream, ointment, and scalp lotion [product monograph]. Toronto (CA): Teva Canada Limited; 2017 Dec 18: https://pdf.hres.ca/dpd_pm/00042697.PDF. Accessed 2021 Jul 20.
24.pdp-Desonide (desonide): 0.05% w/w cream & ointment [product monograph]. Montreal (QC): PENDOPHARM, Division of Pharmascience Inc; 2014 Jul 29: https://pdf.hres.ca/dpd_pm/00026056.PDF. Accessed 2021 Aug 19.
25.Topicort (desoximetasone): 0.25% cream (USP), 0.05% cream (mild), 0.05% gel, 0.25% ointment [product monograph]. Montreal (QC): Valeant Canada LP/Valeant Canada S.E.C; 2012 Feb 20: https://pdf.hres.ca/dpd_pm/00015890.PDF. Accessed 2021 Jul 20.
26.Lyderm (fluocinonide USP): 0.05% ointment, gel, cream [product monograph]. Brampton (ON): Taropharma, A Division of Taro Pharmaceuticals Inc; 2003 Sep 02: https://pdf.hres.ca/dpd_pm/00004233.PDF. Accessed 2021 Jul 20.
27.Tiamol (fluocinonide, USP): cream emollient base [product monograph]. TaroPharma, A Division of Taro Pharmaceuticals Inc: Brampton (ON); 2003 Sep 23: https://pdf.hres.ca/dpd_pm/00001696.PDF. Accessed 2021 Jul 20.
28.Ultravate (halobetasol propionate): 0.05% w/w topical cream, ointment [product monograph]. Laval (QC): Bausch Health, Canada Inc; 2019 Oct 15: https://pdf.hres.ca/dpd_pm/00053569.PDF. Accessed 2021 Jul 20.
29.HydroVal (hydrocortisone valerate USP): 0.2% cream & ointment [product monograph]. Brampton (ON): TaroPharma; 2008 Jul 25: https://pdf.hres.ca/dpd_pm/00006434.PDF. Accessed 2021 Jul 20.
30.Mometasone (mometasone furoate): 0.1% ointment USP [product monograph]. Toronto (ON): Teva Canada Limited; 2018 Jan 17: https://pdf.hres.ca/dpd_pm/00043401.PDF. Accessed 2021 Jul 20.
31.Government of Saskatchewan. Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2021 Jul 20.
32.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020: https://open.alberta.ca/dataset/30add047-29c2-4fc7-83b5-a8ab78605cdd/resource/48d0dd0b-bb33-478d-a85c-85a86d4555ad/download/health-somb-medical-price-list-2020-03.pdf. Accessed 2021 Jul 20.
33.Government B.C. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2021 Jul 20.
34.Elidel (pimecrolimus): 1% cream [product monograph]. Laval (QC): Valeant Canada LP; 2014 Sep 4: https://pdf.hres.ca/dpd_pm/00030576.PDF. Accessed 2021 Jul 20.
35.CADTH Common Drug Review pharmacoeconomic review report: crisaborole ointment, 2% (Eucrisa) for topical treatment of mild-to-moderate atopic dermatitis in patients two years of age and older. Ottawa (ON): CADTH; 2019 Apr: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0570-eucrisa-pharmacoeconomic-report.pdf. Accessed 2021 Jul 20.
36.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: crisaborole (Eucrisa - Pfizer Canada Inc). Ottawa (ON): CADTH; 2019 Mar 27: https://cadth.ca/sites/default/files/cdr/complete/SR0570-Eucrisa-cdec-rec-april-2-2019.pdf. Accessed 2021 Jul 20.
37.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020.
38.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
39.Dupilumab for treating moderate to severe atopic dermatitis. (Final appraisal document). London (GB): National Institute for Health and Care Excellence (NICE); 2018: https://www.nice.org.uk/guidance/ta534/documents/final-appraisal-determination-document?bcs-agent-scanner=0846a587-872b-4b48-9ea1-7fe22aaa4759. Accessed 2021 Jul 8.
40.Silverberg JI, Barbarot S, Gadkari A, et al. Atopic dermatitis in the pediatric population. Ann Allergy Asthma Immunol. 2021;126(4):417-428. PubMed
41.Burgess JA, Dharmage SC, Byrnes GB, et al. Childhood eczema and asthma incidence and persistence: a cohort study from childhood to middle age. J Allergy Clin Immunol. 2008;122(2):280-285. PubMed
42.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
43.Sutherland G, Dinh T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: http://innovativemedicines.ca/wp-content/uploads/2017/12/20170712-understanding-the-gap.pdf. Accessed 2021 Jul 20.
44.Prescribed drug spending in Canada. Ottawa (ON): Canadian Institute for Health Information (CIHI); 2020: https://www.cihi.ca/sites/default/files/document/prescribed-drug-spending-in-canada-2020-report-en.pdf. Accessed 2021 Jul 20.
45.Lyakhovitsky A, Barzilai A, Baum S, et al. Low-dose methotrexate treatment for moderate-to-severe atopic dermatitis in adults. J Eur Acad Dermatol Venereol. 2010;24(1):43-49. PubMed
46.Megna M, Napolitano M, Patruno C, et al. Systemic treatment of adult atopic dermatitis: a review. Dermatol Ther (Heidelb). 2017;7:1-23. PubMed
47.List of medications. Quebec (QC): Régie de l'assurance maladie du Québec (RAMQ); 2021: https://www.ramq.gouv.qc.ca/en/about-us/list-medications. Accessed 2021 Apr 30.
48.Eichenfield LF, DiBonaventura M, Xenakis J, et al. Costs and treatment patterns among patients with atopic dermatitis using advanced therapies in the United States: analysis of a retrospective claims database. Dermatol Ther (Heidelb). 2020;10(4):791-806. PubMed
49.Statistics Canada. Population estimates on July 1, by age and sex. Table: 17-10-0005-01 (formerly CANSIM 051-0001). 2021; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2021 Jul 20.
Appendix 1: Cost-Comparison Tables
Note that this appendix has not been copy-edited.
The comparators presented in Table 8 have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Comparators are not restricted to drugs or drug regimens and may be devices or procedures. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost-Comparison Table for Systemic Therapy of Moderate-to-Severe AD
Drug/ comparator | Strength/ concentration | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Average annual drug cost ($) |
|---|---|---|---|---|---|---|
Abrocitnib (Cibinqo) | 50 mg 100 mg 200 mg | Tablet | 48.6667a 54.4667a | 100 mg or 200 mg once daily | 48.67 54.47 | 17,765 19,882 |
Biologics | ||||||
Dupilumab (Dupixent) | 200 mg/ 1.14 mL 300 mg/ 2 mL | Pre-filled syringe | 978.7000b | Adolescents < 60 kg: 400 mg as an initial dose, followed by 200 mg every 2 weeks Adolescents ≥ 60 kg: 600 mg as an initial dose, followed by 300 mg every 2 weeks Adults: 600 mg as an initial dose, followed by 300 mg every 2 weeks | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,425 Year 2+: 25,446 |
aSponsor’s submitted price for each dosage.
bCost obtained from the Ontario Exceptional Access Program Formulary (July 2021).19
Note: Annual period assumes 52 weeks or 365 days for all comparators.
Table 9: CADTH Cost-Comparison Table for Systemic Therapy of Moderate-to-Severe Atopic Dermatitis (Not Indicated for AD)
Drug/ comparator | Strength/ concentration | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Average 24-week treatment course cost ($) |
|---|---|---|---|---|---|---|
Immunosuppressants a | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.2405 | Pediatric: 1.0 to 4.0 mg/kg per day Adult: 1.0 to 3.0 mg/kg per day | Pediatric: 0.24 to 0.96c Adult: 0.48 to 1.20 d | Pediatric: 41 to 162 Adult: 81 to 203 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.6700 0.9952 1.9400 3.8815 | Pediatric: 3.0 to 6.0 mg/kg per day Adult: 150 to 300 mg per day | Pediatric: 5.55 to 11.04c Adult: 5.82 to 11.64d | Pediatric: 932 to 1,855 Adult: 981 to 1,962 |
Methotrexate (generic) | 2.5 mg | Tablet | 0.6325 | Pediatric: 0.2 to 0.7 mg/kg per week Adult: 7.5 to 25 mg per week | Pediatric: 2.53 to 8.22c Adult: 1.90 to 6.33 per | Pediatric: 61 to 197 Adult: 46 to 152 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | Pediatric: 30.0 to 50.0 mg/kg per day Adult: 2,000 to 13,000 mg daily | Pediatric: 2.23 to 3.34c Adult: 2.97 to 4.45 | Pediatric: 375 to 563 Adult: 500 to 750 |
Retinoidsb | ||||||
Acitretin (Soriatane) | 10 mg 25 mg | Capsule | 1.2965 2.2770 | 10 to 50 mg once daily, max 75 mg daily | 1.30 to 6.83 | 218 to 1,148 |
Alitretinoin (Toctino) | 10 mg 30 mg | Capsule | 22.6490 | 30 mg once daily, dose may be reduced to 10 mg if unacceptable side effects | 22.65 | 3,815 |
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary10 (accessed July 2021), unless otherwise indicated, and do not include dispensing fees. Recommended doses from respective product monographs, unless otherwise indicated. Annual period assumes 52 weeks or 365 days for all comparators.
aRecommended dosage based on the American Atopic Dermatology Guidelines.13
bRecommended dosage aligned with the previous CADTH Pharmacoeconomic Review of dupilumab.12 According the clinical expert consulted by CADTH for a previous review,12 retinoids are primarily used to treat dermatitis on the hands of adults, not adolescents.
cAssumes child weight of 45 kg.
dAssumes adult weight of 70 kg.
According to the clinical expert consulted by CADTH for this review, the following treatments may also be used to treat moderate-to-severe AD in adolescents and adults (Table 10).
Table 10: CADTH Cost-Comparison Table of Topical Treatments for AD
Drug/comparator | Strength | Dosage form | Price | Price per gram or mL ($) | Recommended dosage |
|---|---|---|---|---|---|
Topical corticosteroids | |||||
Amcinonide (generics) | 0.1% | Cream 15 g 30 g 60 g | 2.9325 5.8650 11.7300 | 0.1955 | Thin amount to affected area twice daily, max 5 days on face, axillae, scrotum or scalp, 2 to 3 weeks elsewhere.21 |
Ointment 15 g 30 g 60 g | 5.9640 11.9280 23.8560 | 0.3609a | |||
Lotion 20 mL 60 mL | 5.9940 17.9820 | 0.2997a | |||
Betamethasone dipropionate (generic) | 0.05% | Cream 15 g 50 g 45 g 120 g | 3.0720 10.2400 9.2160 24.5760 | 0.2048 | Thin film to affected area twice daily, duration of therapy varies.22 |
Ointment 15 g 50 g 450 g | 3.2280 10.7600 96.8400 | 0.2152 | |||
7.7790 25.9300 24.5760 | 0.5186 | ||||
Lotion 30 mL 75 mL | 5.9400 14.8500 | 0.1980 | |||
Betamethasone valerate (generic) | 0.05% | Cream 454 g 450 g | 27.0584 26.8200 | 0.0596 | No recommended daily dose. Use as directed by clinicians.12 |
Ointment 454 g 450 g | 27.0584 26.8200 | ||||
0.1% | Cream 454 g 450 g | 40.3606 40.0050 | 0.0889 | ||
Ointment 454 g 450 g | |||||
Lotion 30 mL 60 mL 75 mL | 9.3750 18.7500 23.4375 | 0.3125 | |||
Clobetasol propionate (generic) | 0.05% | Cream 15 g 50 g 450 g 454 g | 3.4185 11.3950 102.5550 103.4666 | 0.2279 | Thin amount to affected area twice daily. Weekly application should not exceed 50 g, and limited to 2 consecutive weeks.23 |
Ointment 15 g 50 g 450 g | |||||
Lotion 20 mL 60 mL | 3.9800 11.9400 | 0.1990 | |||
Desonide (generic) | 0.05% | Cream 15 g 60 g | 3.9750 15.9000 | 0.2650 | Thin amount to affected area twice daily, may be increased in refractory cases.24 |
Ointment 60 g | 15.8820 | 0.2647 | |||
Desoximetasone (Topicort) | 0.05% 0.25% | Cream 20 g 60 g | 10.4300 31.2900 | 0.5215a | Thin amount to affected area twice daily.25 |
Cream 20 g 60 g | 14.6700 44.0100 | 0.7335a | |||
0.25% | Ointment 60 g | 14.6700 44.0100 | 0.7335a | ||
0.05% | Gel 15 g 60 g | 8.5605 34.2420 | 0.5707a | ||
Fluocinonide (Lidemol, Lyderm, Lidex) | 0.05% | Cream 15 g 60 g 400 g | 3.5670 14.2680 95.1200 | 0.2378 | Thin amount to affected area twice daily. Weekly application should not exceed 45 g, and limited to 2 weeks.26 |
Emollient Cream 30 g 100 g | 5.9400 19.8000 | 0.1980 | |||
Ointment 60 g | 18.2100 | 0.3035 | |||
Gel 60 g | 18.4560 | 0.3076 | |||
Fluocinonide (Tiamol) | 0.05% | Emollient Cream 25 g 100 g | 4.9500 19.8000 | 0.1980 | Thin amount 2 to 4 times daily.27 |
Halobetasol propionate (Ultravate) | 0.01% | Lotion 100 g | N/A | N/A | Thin amount to affected area twice daily, limited to 50 g weekly and 2 weeks without re-evaluation.28 |
0.05% | Cream 15 g 50 g | 17.1975 57.3250 | 1.1465c | ||
Ointment 50 g | 55.6750 | 1.1135c | |||
Hydrocortisone (various) | 1.0% | Cream 15 g 30 g 45 g 454 g | 2.577 5.1540 7.7310 77.9972 | 0.1718 | No recommended daily dose. Use as directed by clinicians.12 |
1.0% | Lotion 60 mL | 9.5220 7.1460 | 0.1587 0.1191a | ||
0.5% 1.0% | Ointment 15 g 454 g | 2.1000 17.7060 | 0.1400 0.0390 | ||
Hydrocortisone acetate | 1% | Cream 15 g 30 g | 3.0840 6.1680 | 0.2056 | Twice-daily application is generally recommended initially; intermittent use 1 to 2 times per week on areas that commonly flare for maintenance therapy. |
0.5% 1.0% | Ointment 28.4 g | 11.8087 | 0.4158c | ||
Hydrocortisone valerate (Hydroval) | 0.2% | Cream 15 g 45 g 60 g | 2.5005 7.5015 10.0020 | 0.1667 | Small amount to affected area twice daily. Discontinue as soon as lesions heal or if no response.29 |
Ointment 15 g 60 g | 2.5005 10.0020 | ||||
Mometasone furoate (generic) | 0.1% | Cream 15 g 50 g | 8.3130 27.7100 | 0.5542 | Thin film to affected areas twice daily.30 |
Ointment 15 g 50 g | 3.3780 11.2600 | 0.2252 | |||
Lotion 30 mL 60 mL | 10.0740 20.1480 | 0.3358 | |||
Triamcinolone acetonide (various) | 0.1% | Cream 15 g 30 g 500 g | 0.7995 1.5990 26.6500 | 0.0533 | No recommended daily dose. Use as directed by clinicians. |
Ointment 30 g | 4.5690 | 0.1523 | |||
0.5% | Cream 15 g 50 g | 18.84 62.80 37.68 | 1.2560b | ||
Ointment 30 g | |||||
Topical calcineurin inhibitors | |||||
Pimecrolimus (Elidel)d | 1% | Cream 10 g 30 g | 24.8800 74.6400 | 2.4880 | Thin layer to affected area twice daily, discontinue when resolved or after 3 weeks if no improvement or exacerbation. |
Tacrolimus | 0.03% 0.10% | Ointment 30 g | 78.5190 84.0000 | 2.6173 2.8000 | Thin layer to affected area twice daily. Discontinue after 6 weeks if no improvement or exacerbation. |
Phosphodiesterase type 4 inhibitor | |||||
Crisaborole (Eucrisa)e | 2% | Ointment 60 g | 2.3000f | 138.0000 | Thin layer to affected area twice daily. |
Phototherapy | |||||
UV light therapy | NA | NA | 1130.4000 to 1884.0000 | 7.85 per treatmentg | Administered 3 to 5 times per weekh |
NA = not applicable.
Note: Ontario Drug Benefit Formulary list prices10 unless otherwise indicated; recommended doses from respective product monographs unless otherwise indicated.
aSaskatchewan Formulary list price31 (July 2021).
bAlberta Formulary list price32 (July 2021).
cBritish Columbia Formulary list price (July 2021).33
dPimecrolimus is indicated for treatment of mild to moderate AD in patients 2 years of age and older.34
eCrisaborole received a do not reimburse recommendation from CDEC in March 2019 for treatment of mild to moderate AD in patients 2 years of age and older who have failed or are intolerant to a topical corticosteroid treatment.35,36
fCost obtained from IQVIA DELTA PA database (accessed August 2021)
gOntario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy.”37
hMinimum frequency of phototherapy sessions required per week for successful maintenance as well as length of maintenance period varies between individuals.13,38
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The modelled population is reflecting patients with no prior exposure to systemic immunosuppressants (i.e., post-topical therapy), which is inconsistent with the indication. The model allows for alternative populations and subgroups to be considered. |
Model has been adequately programmed and has sufficient face validity | No | Poor modelling practices were employed (see main text). In the sponsor’s submission, SoC alone was associated with higher total QALYs than either MTX + SoC or CYC + SoC, which lacks face validity. |
Model structure is adequate for decision problem | Yes | — |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | For some model parameters, the sponsor arbitrarily incorporated uncertainty as +/−15% of the mean value (e.g., for treatment response, discontinuation rate in the first year), +/− 20% of the mean value (e.g., health-state utility values), or +/−50% of the mean value (costs of treating adverse events), which does not reflect the true uncertainty around the model’s parameters possible values. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | — |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | — |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure – Decision Tree
AD = atopic dermatitis; M = Markov model; SoC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.2
Table 12: Disaggregated Summary of the Sponsor’s Base Case
Treatment | Component | Value | Incremental (vs. SoC) | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
SoC | Total | 21.91 | NA | NA |
Decision tree (Year 1) | 0.65 | NA | NA | |
Markov model (Year 2+) | 21.26 | NA | NA | |
MTX + SoC | Total | 21.77 | −0.14 | NA |
Decision tree (Year 1) | 0.68 | 0.03 | NA | |
Markov model (Year 2+) | 21.09 | −0.17 | NA | |
CYC + SoC | Total | 21.77 | −0.14 | 0.00 |
Decision tree (Year 1) | 0.69 | 0.04 | 0.01 | |
Markov model (Year 2+) | 21.08 | −0.18 | −0.01 | |
abrocitinib 100 mg plus SoC | Total | 22.98 | 1.07 | 1.21 |
Decision tree (Year 1) | 0.76 | 0.11 | 0.07 | |
Markov model (Year 2+) | 22.22 | 0.96 | 1.14 | |
abrocitinib 200 mg plus SoC | Total | 23.28 | 1.37 | 0.30 |
Decision tree (Year 1) | 0.78 | 0.13 | 0.02 | |
Markov model (Year 2+) | 22.50 | 1.24 | 0.28 | |
DUP + SoC | Total | 22.91 | 1.00 | −0.37 |
Decision tree (Year 1) | 0.73 | 0.08 | −0.05 | |
Markov model (Year 2+) | 22.19 | 0.93 | −0.31 | |
Discounted costs ($) | ||||
SoC | Total | 177,444 | ||
Drug costsa | 26,970 | NA | NA | |
Administration | 0 | NA | NA | |
Adverse events | 1,785 | NA | NA | |
Medical resource use | 148,689 | NA | NA | |
Treatment monitoring | 0 | NA | NA | |
MTX + SoC | Total | 180,859 | 3,415 | NA |
Drug costsa | 27,599 | 629 | NA | |
Administration | 0 | 0 | NA | |
Adverse events | 1,759 | −26 | NA | |
Medical resource use | 151,433 | 2,744 | NA | |
Treatment monitoring | 68 | 68 | NA | |
CYC + SoC | Total | 181,776 | 4,332 | 917 |
Drug costsa | 28,486 | 1,516 | 887 | |
Administration | 0 | 0 | 0 | |
Adverse events | 1,768 | −17 | 9 | |
Medical resource use | 151,399 | 2,710 | −34 | |
Treatment monitoring | 123 | 123 | 55 | |
ABRO 100 mg plus SoC | Total | 249,053 | 71,609 | 67,277 |
Drug costsa | 112,980 | 86,010 | 84,494 | |
Administration | 0 | 0 | 0 | |
Adverse events | 1,842 | 57 | 74 | |
Medical resource use | 133,435 | −15,254 | −17,964 | |
Treatment monitoring | 796 | 796 | 673 | |
ABRO 200 mg plus SoC | Total | 277,969 | 100,525 | 28,916 |
Drug costsa | 146,639 | 119,669 | 33,659 | |
Administration | 0 | 0 | 0 | |
Adverse events | 1,857 | 72 | 15 | |
Medical resource use | 128,484 | −20,205 | −4,951 | |
Treatment monitoring | 989 | 989 | 193 | |
DUP plus SoC | Total | 284,131 | 106,687 | 6,162 |
Drug costsa | 148,141 | 121,171 | 1,502 | |
Administration | 39 | 39 | 39 | |
Adverse events | 1,776 | −9 | −81 | |
Medical resource use | 134,176 | −14,513 | 5,692 | |
Treatment monitoring | 0 | 0 | −989 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
SoC | Ref. | Ref. | ||
MTX plus SoC | Dominated by SoC | NA | ||
CYC plus SoC | Dominated by SoC | NA | ||
ABRO 100 mg plus SoC | 67,246 | 67,246 v. SoC | ||
ABRO 200 mg plus SoC | 73,606 | 96,122 v. ABRO 100 | ||
DUP plus SoC | Dominated by ABRO 100 | NA | ||
ABRO = abrocitinib; CYC = cyclosporine; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; MTX = methotrexate; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; SoC = standard of care; v. = vs..
aIncludes the cost of subsequent treatment after discontinuation from or failure of (loss of response) treatment.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 13: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | SoC | 177,444 | 21.91 | Ref. |
CYC + SoC | 181,776 | 21.77 | NA | |
MTX + SoC | 180,859 | 21.77 | NA | |
ABRO 100 mg plus SoC | 249,053 | 22.98 | 67,246 v. SoC | |
ABRO 200 mg plus SoC | 277,969 | 23.28 | 96,122 v. ABRO 100 | |
DUP + SoC | 284,131 | 22.91 | NA | |
Sponsor’s corrected base case | SoC | 177,356 | 21.95 | Ref. |
CYC + SoC | 181,617 | 21.81 | Dominated | |
MTX + SoC | 180,701 | 21.81 | Dominated | |
ABRO 100 mg plus SoC | 249,386 | 23.01 | 68,321 v. SoC | |
ABRO 200 mg plus SoC | 277,239 | 23.29 | 97,685 v. ABRO 100 | |
DUP + SoC | 287,238 | 22.94 | Dominated | |
CADTH reanalysis 1: Post-systemic population | SoC | $181,734 | 21.66 | Ref. |
MTX + SoC | $181,872 | 21.69 | 5,875 v. SoC | |
CYC + SoC | $182,563 | 21.69 | Extended dominance | |
ABRO 100 mg plus SoC | $235,886 | 22.62 | 57,864 v. MTX + SoC | |
ABRO 200 mg plus SoC | $263,950 | 22.97 | 80,619 v. ABRO 100 mg plus SoC | |
DUP + SoC | $283,507 | 22.79 | Dominated | |
CADTH reanalysis 2: Comparators | SoC | 177,356 | 21.95 | Ref. |
ABRO 100 mg plus SoC | 249,386 | 23.01 | 68,321 v. SoC | |
ABRO 200 mg plus SoC | 277,239 | 23.29 | 97,685 v. ABRO 100 | |
DUP + SoC | 287,238 | 22.94 | Dominated | |
CADTH reanalysis 3: Health-state utility values | SoC | 177,356 | 27.82 | Ref. |
CYC + SoC | 181,617 | 27.78 | Dominated | |
MTX + SoC | 180,701 | 27.78 | Dominated | |
ABRO 100 mg plus SoC | 249,386 | 28.22 | 181,617 v. SoC | |
ABRO 200 mg plus SoC | 277,239 | 28.31 | 180,701 v. ABRO 100 | |
DUP + SoC | 287,238 | 28.16 | Dominated | |
CADTH reanalysis 4: Timing of response onset | SoC | 177,166 | 21.97 | Ref. |
CYC + SoC | 181,617 | 21.81 | Dominated | |
MTX + SoC | 180,701 | 21.81 | Dominated | |
ABRO 100 mg plus SoC | 249,386 | 23.01 | 69,270 v. SoC | |
ABRO 200 mg plus SoC | 277,239 | 23.29 | 97,685 v. ABRO 100 | |
DUP + SoC | 286,838 | 22.97 | Dominated | |
CADTH reanalysis 5: Treatment effectiveness-waning | SoC | 178,084 | 21.80 | Ref. |
CYC + SoC | 182,418 | 21.65 | Dominated | |
MTX + SoC | 181,485 | 21.66 | Dominated | |
ABRO 100 mg plus SoC | 265,040 | 23.14 | 64,702 v. SoC | |
ABRO 200 mg plus SoC | 302,569 | 23.56 | 89,516 v. ABRO 100 | |
DUP + SoC | 312,393 | 23.10 | Dominated | |
CADTH base case (1 + 2 + 3 + 4 + 5) | SoC | 181,688 | 27.57 | Ref. |
ABRO 100 mg plus SoC | 248,791 | 28.00 | 156,735 v. SoC | |
ABRO 200 mg plus SoC | 286,525 | 28.17 | 231,013 v. ABRO 100 | |
DUP + SoC | 309,525 | 28.11 | Dominated |
ABRO = abrocitinib; CYC = cyclosporine; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; MTX = methotrexate; QALY = quality-adjusted life-year; Ref. = reference; SoC = standard of care.
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. SoC) | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
SoC | Total | 27.57 | NA | NA |
Decision tree (Year 1) | 0.76 | NA | NA | |
Markov model (Year 2+) | 26.82 | NA | NA | |
ABRO 100 mg plus SoC | Total | 28.00 | 0.43 | NA |
Decision tree (Year 1) | 0.79 | 0.03 | NA | |
Markov model (Year 2+) | 27.21 | 0.39 | NA | |
ABRO 200 mg plus SoC | Total | 28.17 | 0.59 | 0.16 |
Decision tree (Year 1) | 0.80 | 0.04 | 0.01 | |
Markov model (Year 2+) | 27.37 | 0.55 | 0.15 | |
DUP + SoC | Total | 28.11 | 0.54 | −0.05 |
Decision tree (Year 1) | 0.80 | 0.04 | 0 | |
Markov model (Year 2+) | 27.31 | 0.49 | −0.05 | |
Discounted costs ($) | ||||
SoC | Total | 181,688 | NA | NA |
Drug Costsa | 27,568 | NA | NA | |
Administration | 0 | NA | NA | |
Adverse Events | 1,780 | NA | NA | |
Medical Resource Use | 152,340 | NA | NA | |
Treatment Monitoring | 0 | NA | NA | |
ABRO 100 mg plus SoC | Total | 248,791 | 67,103 | NA |
Drug Costsa | 111,905 | 84,337 | NA | |
Administration | 0 | 0 | NA | |
Adverse Events | 1,838 | 59 | NA | |
Medical Resource Use | 134,261 | −18,080 | NA | |
Treatment Monitoring | 787 | 787 | NA | |
ABRO 200 mg plus SoC | Total | 286,525 | 104,837 | 37,734 |
Drug Costsa | 156,629 | 129,060 | 44,724 | |
Administration | 0 | 0 | 0 | |
Adverse Events | 1,857 | 78 | 19 | |
Medical Resource Use | 130,016 | −25,373 | −7294 | |
Treatment Monitoring | 1,073 | 1,073 | 286 | |
DUP + SoC | Total | 309,257 | 127,568 | 22,731 |
Drug Costsa | 177,432 | 149,864 | 20,803 | |
Administration | 39 | 39 | 39 | |
Adverse Events | 1,769 | −10 | −88 | |
Medical Resource Use | 130,016 | −22,324 | 3,049 | |
Treatment Monitoring | 0 | 0 | −1,073 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
SoC | Ref. | Ref. | ||
ABRO 100 mg plus SoC | 156,735 | 156,735 v. SoC | ||
ABRO 200 mg plus SoC | 177,248 | 231,013 v. ABRO 100 | ||
DUP + SoC | 237,330 | Dominated | ||
ABRO = abrocitinib; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; SoC = standard of care; v. = vs..
aIncludes the cost of subsequent treatment after discontinuation from or failure of (loss of response) treatment.
Scenario Analyses
Table 15: CADTH Scenario Analyses
Detail | CADTH base case | CADTH scenario |
|---|---|---|
Scenario analyses | ||
1. Age | All patients (aged 12+) | Adolescents (aged 12 to 18 years)a |
2. Treatment effectiveness at 16 weeks (% EASI-75 responders) | Based on indirect evidence from the sponsor’s NMA; EASI score assessed at 16 weeksb: ABRO 100 mg plus SoC: 49.7% ABRO 200 mg plus SoC: 62.4% DUP + SoC: 63.6% SoC: 6.1% | The % of EASI-75 responders was assumed to be equivalent between ABRO + SoC and DUP + SoC: ABRO 100 mg plus SoC: 62.4% ABRO 200 mg plus SoC: 62.4% DUP + SoC: 62.4% SoC: 6.1% |
3. Health-state utility values | Sponsor-provided values (derived from the dupilumab LIBERTY SOLO1 and SOLO2 trials6), assuming that “active treatment” utilities apply to all treatments including SoC: Baseline: 0.6156 Treatment responders: 0.8772 Nonresponders: 0.7777 | Sponsor-provided values (derived from ABRO JADE COMPARE trial), assuming that “active treatment utilities apply to all treatments, including SoC”: Baseline: 0.7840 Treatment responders: 0.9020 Nonresponders: 0.8739 |
4. Health-state utility values | As above | Derived from the dupilumab LIBERTY SOLO1 and SOLO2 trials, nonresponders assumed to revert to baseline utility: Baseline: 0.6156 Treatment responders: 0.8723c Nonresponders: 0.6156 |
5. Health-state utility values | Age-adjusted | Age adjustment disabled |
6. Timing of response onset | ABRO: 8 weeks DUP: 8 weeks SoC: 8 weeks | ABRO: 8 weeks DUP: 16 weeks SoC: 16 weeks |
7. Comparators | DUP + SoC SoC | DUP + SoC MTX + SoCd CYC + SoCd SoC |
8. Treatment effectiveness-waning | Effectiveness was assumed to wane in a linear manner over the first 40 years of active treatmente | Time-dependent waning, assuming that the rate of effectiveness-waning for ABRO is equivalent to that reported for DUP from the CHRONOS trial; the rate of waning for SoC was assumed to be equivalent to that reported for BSC in the CHRONOS trial.39 |
9. Dupilumab price reduction | $978.70 per syringe.19 | 54% price reduction from a base price of $959.9350 based on the 2020 CADTH assessment of DUP.12 |
10. Analysis perspective | Health care payer | Societal (i.e., productivity costs included) |
ABRO = abrocitinib; AD = atopic dermatitis CYC = cyclosporine; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; IGA = Investigator’s Global Assessment; MTX = methotrexate; SoC = standard of care.
aLifetime horizon was assumed to be 91.5 years for the adolescent subgroup.
bOverall patient population.
cResponder utilities were derived using the weighted average of utilities for the placebo and DUP arms in the SOLO 1 and SOLO 2 trials.6
dComparative treatment effectiveness estimates for MTX and CYC were based on an unanchored MAIC; while estimates for other model comparators were based on a network meta-analysis.
eEffectiveness of SoC was assumed to wane over the first 5 years of treatment.
Table 16: CADTH Scenario Analyses Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
CADTH base case | |||
SoC | 181,688 | 27.57 | Ref. |
ABRO 100 mg plus SoC | 248,791 | 28.00 | 156,735v. SoC |
ABRO 200 mg plus SoC | 286,525 | 28.17 | 231,031 v. ABRO 100 mg plus SoC |
DUP + SoC | 309,525 | 28.11 | Dominated |
Scenario 1: Adolescents (aged 12 to 18 years) | |||
SoC | 211,704 | 33.27 | Reference |
ABRO 100 mg plus SoC | 304,677 | 33.81 | 171,849 v. SoC |
ABRO 200 mg plus SoC | 334,001 | 33.91 | 281,645 v. ABRO 100 mg plus SoC |
DUP + SoC | 345,274 | 33.76 | Dominated |
Scenario 2: Treatment effectiveness of ABRO and DUP assumed to be equal at 16 weeks | |||
SoC | 181,703 | 27.63 | Ref. |
ABRO 100 mg plus SoC | 264,274 | 28.19 | 147,051 v. SoC |
ABRO 200 mg plus SoC | 284,390 | 28.24 | 417,886 v. ABRO 100 mg plus SoC |
DUP + SoC | 305,393 | 28.16 | Dominated |
Scenario 3: Health-state utility values – JADE COMPARE | |||
SoC | 181,697 | 31.04 | Reference |
ABRO 100 mg plus SoC | 248,761 | 31.17 | 536,944 v. SoC |
ABRO 200 mg plus SoC | 284,359 | 31.21 | 760,737 v. ABRO 100 mg plus SoC |
DUP + SoC | 307,631 | 31.19 | Dominated |
Scenario 4: Health-state utility values – Simpson 2017,6 nonresponders assumed to receive baseline utility value | |||
SoC | 181,697 | 21.92 | Ref. |
ABRO 100 mg plus SoC | 248,761 | 23.06 | 58,672 v. SoC |
ABRO 200 mg plus SoC | 284,359 | 23.49 | 83,136 v. ABRO 100 mg plus SoC |
DUP + SoC | 307,631 | 23.31 | Dominated |
Scenario 5: Health-state utility values – age adjustment disabled | |||
SoC | 181,697 | 28.39 | Ref. |
ABRO 100 mg plus SoC | 248,761 | 28.83 | 149,916 v. SoC |
ABRO 200 mg plus SoC | 284,359 | 29.00 | 212,411 v. ABRO 100 mg plus SoC |
DUP + SoC | 307,631 | 28.93 | Dominated |
Scenario 6: Timing of response onset | |||
SoC | 181,734 | 27.60 | Ref |
ABRO 100 mg plus SoC | 248,761 | 28.07 | 142,897 v. SoC |
ABRO 200 mg plus SoC | 284,359 | 28.23 | 214,377 v. ABRO 100 mg plus SoC |
DUP + SoC | 308,023 | 28.13 | Dominated |
Scenario 7: Comparators | |||
SoC | 181,697 | 27.63 | Ref. |
CYC + SoC | 182,562 | 27.63 | Extended dominance |
MTX + SoC | 181,859 | 27.62 | Dominated |
ABRO 100 mg plus SoC | 248,761 | 28.07 | 151,294 v. SoC |
ABRO 200 mg plus SoC | 284,359 | 28.23 | 214,377 v. ABRO 100 mg plus SoC |
DUP + SoC | 307,631 | 28.17 | Dominated |
Scenario 8: Treatment effectiveness-waning | |||
SoC | 180,169 | 27.66 | Ref. |
ABRO 100 mg plus SoC | 233,016 | 27.95 | 179,713 v. SoC |
ABRO 200 mg plus SoC | 259,401 | 28.07 | 214,845 v. ABRO 200 mg plus SoC |
DUP + SoC | 277,651 | 28.02 | Dominated |
Scenario 9: DUP price reduction (54%)a | |||
SoC | 181,697 | 27.63 | Ref. |
DUP + SoC | 225,568 | 28.17 | 81,142 |
ABRO 100 mg plus SoC | 248,761 | 28.07 | Dominated |
ABRO 200 mg plus SoC | 284,359 | 28.23 | 856,402 |
Scenario 10: Societal perspective (productivity costs included) | |||
SoC | 417,637 | 27.63 | Ref. |
ABRO 100 mg plus SoC | 454,882 | 28.07 | 84.023 v. SoC |
ABRO 200 mg plus SoC | 492,653 | 28.23 | 227,458 v. ABRO 100 mg plus SoC |
DUP + SoC | 519,618 | 28.17 | Dominated |
ABRO = abrocitinib; CYC = cyclosporine; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; MTX = methotrexate; QALY = quality-adjusted life-year; Ref. = reference; SoC = standard of care.
aIn this analysis, the price of DUP was assumed to be $959.9350 per syringe based on the 2020 CADTH assessment of DUP.12 In all other scenarios, the price of DUP was assumed to be $978.70 based on the Ontario Exceptional Access Program price.
Exploratory Scenario
The clinical expert consulted by CADTH for this review indicated that the majority of patients will initiate treatment on the 200 mg ABRO dose, potentially reducing the dosage to 100 mg daily depending on treatment response and adverse events. CADTH was unable to explore the cost-effectiveness of this treatment approach owing to the structure of the sponsor’s model. CADTH conducted exploratory scenario analyses assuming that all patients taking ABRO received the 200 mg dose; that is, the cost-effectiveness of ABRO 200 mg plus SoC was compared with DUP + SoC and SoC among patients with prior exposure to systemic immunosuppressants.
Among patients refractory or ineligible for systemic immunosuppressants, ABRO 200 mg plus SoC was more effective and more costly than SoC alone, with an ICER of $177,248 per QALY gained compared to SoC over a lifetime horizon (Table 17), while DUP + SoC was dominated by ABRO 200 mg plus SoC.
Table 17: CADTH Exploratory Scenario – ABRO 200 mg plus SoC vs. DUP + SoC and SoC
Drug | Total costs ($) | Total QALYs | ICER vs. SoC ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Patients refractory or ineligible for systemic immunosuppressants | ||||
SoC | 181,688 | 27.57 | Ref. | Ref. |
ABRO 200 mg plus SoC | 286,525 | 28.17 | 177,248 | 177,248 v. SoC |
DUP + SoC | 309,257 | 28.11 | 237,330 | Dominated |
ABRO = abrocitinib; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; SoC = standard of care.
An additional exploratory price-reduction analysis was performed for this scenario (Table 18).
In this scenario, a price reduction of 56% would be required for ABRO 200 mg plus SoC to be considered at a WTP threshold of $50,000 per QALY.
In reality, the kind of ABRO dose reduction described by the clinical expert would result in a reduction in the cost of treatment but not necessarily a reduction in the effectiveness of treatment. Due to structural limitations of the model and a lack of evidence to inform the proportion of patients who would switch to a lower dose, CADTH was unable to estimate the cost-effectiveness of this approach.
Table 18: Exploratory Price-Reduction Analyses
Analysis | ICERs for ABRO 200 mg plus SoC vs. DUP + SoC and SoC ($/QALY) |
|---|---|
Price reduction | Patients refractory or ineligible for systemic immunosuppressants |
No price reduction | 177,248 vs. SoC |
10% | 146,985 vs. SoC |
20% | 125,484 vs. SoC |
30% | 103,983 vs. SoC |
40% | 82,483 vs. SoC |
50% | 60,982 vs. SoC |
56% | 48,081 vs. SoC |
ABRO = abrocitinib; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care; vs. = versus.
Note: The corrected price of dupilumab was used in all price-reduction scenarios. a Dupilumab was dominated in all price-reduction scenarios.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 19: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
CADTH identified the following key limitations with the sponsor’s analysis:
CADTH reanalysis included using the proportion of patients eligible for coverage to calculate market size. Based on CADTH reanalyses, the budget impact to the public drug plans of introducing ABRO for patients with moderate-to-severe AD is expected to be cost savings of $790,027 in year 1, $9,693,656 in year 2, and $39,556,691 in year 3, for a 3-year total budget impact of $50,040,374. The estimated budget impact is sensitive to the proportion of patients who are eligible for public drug plan coverage, assumptions around market share distribution, adherence rates and the proportion of patients receiving a systemic immunosuppressant. |
Summary of Sponsor’s BIA
The submitted BIA16 assessed the expected budgetary impact resulting from reimbursing ABRO for the treatment of moderate-to-severe AD for patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when systemic therapies are not advisable. The BIA was conducted from the perspective of the Canadian public drugs over a 3-year time horizon and included drug acquisition costs, markup, and dispensing fees. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. The analysis was performed using jurisdiction-specific values and results were consolidated by summing up individual provincial results. Key inputs to the BIA are documented in Table 21.
The sponsor estimated the current eligible population using an epidemiologic approach. The sponsor adopted an estimated diagnosed AD prevalence of 15.8%40 and incidence of 0.74% among Canadian adolescents41 (aged 12 to < 18 years), and a prevalence of 3.5%42 and incidence of 0.44%41 among adults (≥ 18 years). Approximately 40.2% of adolescents40 and 52.0% of adults42 were categorized as having moderate-to-severe disease. The sponsor assumed that 25% of patients are not adequately controlled with topical prescription therapies and that 30% of these patients receive a systemic immunosuppressant.16 The sponsor also assumed that 27.9% of patients between ages 12 to 64 years and 91.2% of patients aged ≥ 65 years will be covered by the public drug plans.43,44
The sponsor’s submission considered a reference scenario in which patients received DUP, MTX, and CYC and a new-drug scenario in which ABRO was reimbursed. The sponsor excluded azathioprine and mycophenolate as comparators, assuming these treatment options are not commonly used.
The cost of ABRO was based on the sponsors submitted price ($48.68 per 100 mg tablet, $54.47 per 200 mg tablet). The sponsor estimated an annual treatment cost of $12,906 for ABRO, assuming 35% of patients would be prescribed 100 mg daily and 65% would be prescribed 200 mg daily. The sponsor included an average annual treatment cost of $18,485 for DUP (first year: $19,194), $1,910 for CYC (average dose of 200 mg), and $133 MTX (15 mg weekly). The dosing regimens and costs were based on product monographs3,11 and published literature,16,45,46 and the RAMQ database analysis,47 and drug costs were adjusted for anticipated adherence.
The following key assumptions were made by the sponsor:
The sponsor assumed that 35% of patients will initiate treatment on ABRO 100 and 65% patients will initiate treatment on ABRO 200.
The sponsor assumed an adherence of 62.7% for oral treatments (ABRO, MTX, CYC)17 and 68.7% for subcutaneous injections (DUP).48
The sponsor assumed a market expansion of 13.3% for biologic treatment. ABRO was assumed to have a market share of 0.25% in year 1, 2.8% in year 2 and 10.5% in year 3 and capture market share from DUP, CYC and MTX.
The sponsor assumed that ABRO captured market share from DUP at an increasing rate of 20%, 30% and 40% in year 1, 2 and 3, respectively.
The sponsor assumed that 30% of patients not controlled by topical therapies will receive a systemic immunosuppressant.
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population for the Baseline Year

Note: General population data are sourced from Statistics Canada (Table 17 to 10 to 0005 to 01)49 and population growth is projected using annual growth rate of 1.58% for adolescents (12 to < 18 years) and 1.51% for adults (≥ 18 years) for the period of 2016 to 2020.
AD = atopic dermatitis.
Source: Sponsor’s Budget Impact Analysis.16
Table 20: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Prevalence of diagnosed AD | |
Adolescents | 15.8%a |
Adults | 3.5%b |
Incidence of diagnosed AD | |
Adolescents | 0.74%c |
Adults | 0.44%c |
Proportion of moderate-to-severe AD | |
Adolescents | 40.2%a |
Adults | 52.0%b |
Not adequately controlled with topical therapy | 25%d |
Receiving a systemic immunosuppressant | 30% d |
Percentage of public coverage according to agee | |
12 to 64 years | 27.9% |
≥ 65 years | 91.2% |
Annual population growth rate (2016 to 2020)f | |
Adolescents | 1.58% |
Adults | 1.51% |
Number of patients eligible for drug under review (Year 1 / Year 2 / Year 3) | 18,573 / 20,412 / 22,274 |
Market uptake (Year 1 / Year 2 / Year 3)d | |
Uptake (reference scenario) DUP CYC MTX | 15.6% / 17.2% / 18.9% 30.4% / 30.1% / 29.7% 54.0% / 52.7% / 51.4% |
Uptake (new drug scenario) ABRO DUP CYC MTX | 0.25% / 2.8% / 10.5% 15.6% / 16.3% / 14.7% 30.4% / 29.4% / 27.4% 53.8% / 51.5% / 47.4% |
Annual cost of treatment (per patient)g | |
ABRO DUP CYC MTX | $12,907h $18,485i $1,911j $133k |
ABRO = abrocitinib; AD = atopic dermatitis; CYC = cyclosporine; DUP = dupilumab; MTX = methotrexate.
aSilverberg, 2020.40
bBarbarot, 2018.42
bBurgess, 2008.41
dSponsor’s Budget Impact Analysis.16
eSutherland, 201743 and CIHI, 2020.44
fStatistics Canada, 2020 (Table: 17 to 10 to 0005 to 01).49
hAssuming a utilization proportion of 35% taking 100 mg daily and 65% taking 200 mg daily.
iAssuming average dosage of 300 mg every 2 weeks.
jAssuming average dosage of 200 mg daily.
kAssuming average dosage of 15 mg weekly.
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor estimated the net budget impact to the public drug plans of introducing ABRO for moderate-to-severe AD to be $357,180 in year 1, $3,484,285 in year 2, and $10,525,648 in year 3. The 3-year budget impact to the public drug plans was projected to be cost increase of $14,367,113.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The BIA may not reflect the clinical use of ABRO: The sponsor’s submitted budget impact assumed that ABRO will be used by patients with no prior exposure to systemic immunosuppressants and that ABRO captures market share from both DUP and systemic immunosuppressants (CYC, MTX). The HC indication notes ABRO use in patients with inadequate response to other systemic drugs, including biologics. According to the clinical expert consulted by CADTH for this review, the uptake of ABRO will depend on the relative efficacy and safety of treatments, and there is uncertainty as to which treatments are most likely to be displaced by ABRO. The clinical expert noted there are safety concerns with the off-label use of systemic immunosuppressants and that the clinical efficacy of ABRO would need to be established before physicians are comfortable prescribing ABRO. The budget impact is very sensitive to assumptions about the market share distribution of ABRO and the displacement of current comparators.
In CADTH reanalysis, ABRO was assumed to be used by patients with prior exposure to systemic immunosuppressants. In this scenario analysis, CADTH assumed 100% of market share of ABRO will be captured from DUP and there will be no change in the market share distribution of MTX and CYC with the introduction of ABRO.
The number of patients covered by public drug plans is underestimated: The sponsor estimated the proportion of patients eligible for public drug plan coverage by use of the number of patients enrolled in public plans for each jurisdiction.43 It is more appropriate to use the proportion of patients eligible, rather than enrolled, as the market size will be determined by all eligible for public coverage, and the BIA should consider all patients eligible regardless of whether they are presently enrolled. Should ABRO be reimbursed by public plans, it is assumed that all eligible patients for this treatment would enrol for public coverage.
In CADTH reanalysis, the proportion of patients eligible for public drug plan coverage was used to determine the market size for ABRO.43
Uncertainty in the proportion of patients prescribed ABRO 100 versus ABRO 200: The sponsor assumed that 35% of patients would be prescribed 100 mg ABRO daily and that 65% would be prescribed 200 mg ABRO daily, based on internal market research. In the draft product monograph,1 the recommended dose is based on the individual goal of therapy and potential risk for adverse reactions. According to the clinical expert consulted by CADTH for this review, the most likely clinical use of ABRO would be for patients to initiate treatment on the 200 mg dose, and potentially stepping down to ABRO 100 depending on treatment response and adverse events. The sponsor’s model did not explicitly consider the possibility of all patients treated on low dose or high dose.
CADTH conducted 2 scenario analyses: 1 in which all patients receive ABRO 100, and 1 where all patients receive ABRO 200.
Uncertainty regarding treatment adherence: The sponsor assumed that adherence would be similar for oral treatments (ABRO, CYC, MTX; 62.7%)17 and subcutaneous injections (DUP; 68.7%)48 based on published literature on other JAK inhibitors and DUP. There is no clinical evidence to support estimates around treatment adherence rates for AD patients. However, treatment adherence is a major driver of BIA results. According to the clinical expert consulted by CADTH for this review, adherence is expected to be lower for ABRO compared to DUP. Adherence may also vary by age group, with lower adherence expected for adolescents. The clinical expert indicated that, in clinical practice, treatment adherence may be in the range of 90% for DUP and 70% for ABRO.
In CADTH reanalysis, an adherence rate of 70% adherence was assumed for ABRO, MTX, and CYC, and 90% for DUP based on feedback received from clinical experts for this review. The sponsor’s BIA model was not flexible to readily test different adherence assumptions across age groups.
High uncertainty regarding the incidence of AD: The sponsor assumed an incidence rate of 7.41 per 1,000 person-years for adolescents and 4.37 per 1,000 person-years for adults.41 The sponsor obtained this data from a study examining the incidence rate of eczema using results from the Tasmanian Longitudinal Health Study. The evidence on incidence rates is outdated, non-Canadian and does not represent the demographics of the reimbursement population, producing high uncertainty in the number of new AD cases, size of eligible population and the budget impact. Moreover, the incidence rates are far higher than the average annual Canadian population growth and it is unlikely that new cases of AD are growing faster than the general population, overestimating the budget impact.
This limitation could not be addressed by CADTH owing to a lack of available data on incidence rates of AD in Canada.
The number of patients who receive treatment is overestimated: The sponsor assumes that all patients diagnosed with moderate-to-severe AD will receive treatment. However, some patients may have limited access to health care services, such as those living in remote or rural areas, and may not be able to access treatment for AD. According to clinical expert consulted by CADTH for this review, it is unlikely that 100% of the diagnosed population with moderate-to-severe AD will be treated. It is likely that estimated budgetary impact of reimbursing ABRO is overestimated under the sponsor’s assumption.
This limitation could not be addressed by CADTH owing to the structure of the sponsor’s model.
Additional limitations were identified, but were not considered to be key limitations. These limitations include: assuming same adherence for responders and nonresponders to treatment, excluding costs of TCS, assuming no differential use of TCS among different therapies, and excluding general discontinuation rates.
CADTH Reanalyses of the Budget Impact Analysis
Table 21: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Eligible population | NIHB beneficiaries were not subtracted from respective provinces, and annual population growth rate was 1.58% for adolescents and 1.51% for adults. | NIHB beneficiaries were removed from the Canadian population estimates for each age group (adults, adolescents) and jurisdiction, and corrected annual population growth rates were 1.57% for adolescents and 1.50% for adults were applied. |
2. DUP price change | $959.5950 per pre-filled syringe. | $978.7000 per pre-filled syringe.19 |
Revisions to sponsor’s base case | ||
1. Eligibility criteria | Patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when systemic therapies are not advisable. | Patients aged 12 years and older with moderate-to-severe atopic dermatitis “with refractory moderate-to-severe atopic dermatitis, including the relief of pruritus, who have had an inadequate response to other systemic drugs, (e.g., steroid or biologic), or for whom these treatments are not advisable.1” |
Changes to derive the CADTH base case | ||
1. Percentage of patients covered by public drug plans | Determined by the percentage of patients enrolled (a weighted average of 27.9% for individuals aged < 65 years and 91.2% for individuals aged ≥ 65 years), using Canadian population estimates for year 2020.43,44 | Determined by the percentage of patients eligible for enrollment43 (a weighted average of 63.6% for individuals aged < 65 years and 99.0% for individuals aged ≥ 65 years), using Canadian population estimates for year 2020.43,44 |
2. Adherence across treatments | The sponsor assumed 62.7% adherence for ABRO, CYC, MTX17 and 68.7% for DUP48 | Based on clinical expert’s feedback, an adherence rate of 70% was assumed for oral treatment and 90% was assumed for subcutaneous injections. |
CADTH base case | Reanalysis 1 + 2 | |
AD = atopic dermatitis; DUP = dupilumab, NIHB = Non-insured Health Benefits.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 22 and a more detailed breakdown is presented in Table 23. CADTH corrected the sponsor’s base case by removing the number of NIHB beneficiaries from the Canadian population estimates and updating the unit cost of DUP using the price listed on Ontario Exceptional Access Program Formulary.19 CADTH revised the sponsor’s corrected base case by updating eligibility criteria to patients with prior exposure to systemic immunosuppressants, using the number of patients eligible for public coverage, rather than enrolled, to estimate the percentage of patients who would be covered in each jurisdiction, and adopting an adherence of 90% for treatment with subcutaneous injection.
After applying these changes, the total 3-year budget impact of reimbursing ABRO for use by patients with prior exposure to systemic immunosuppressants was a cost savings of $50,040,374. The estimate of budget impact was highly sensitive to the percentage of patients covered by public drug plans and treatment adherence.
Table 22: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 14,367,113 |
CADTH correction 1 | 14,367,113 |
CADTH correction 2 | 13,998,294 |
Sponsor’s base case, corrected | 13,998,294 |
CADTH revision 1 | −15,952,094 |
Stepped analysis | |
CADTH reanalysis 1 | −28,919,481 |
CADTH reanalysis 2 | −27,571,415 |
CADTH base case | -50,040,374 |
BIA = budget impact analysis.
Note: CADTH revisions and reanalyses are carried out on sponsor’s corrected base case.
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 23.
All patients taking ABRO assumed to receive ABRO 200 in all years (proportion of patients receiving ABRO 100 = 0%).
All patients taking ABRO receive ABRO 100 in all years (proportion of patients receiving ABRO 200 = 0%).
Assuming 40% of patients receive systemic immunosuppressants.
Price reduction of 52% for ABRO 100.
Price reduction of 52% for ABRO 100 and of 56% for ABRO 200.
DUP price is $959.9350 per pre-filled syringe, based on the 2020 CADTH assessment of DUP.12
Price reduction of DUP (54%) from a base price of $959.9350 based on the 2020 CADTH assessment of DUP.12
Table 23: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 ($) (current situation) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 47,573,156 | 60,550,248 | 71,695,612 | 84,411,871 | 216,657,731 |
New drug | 47,573,156 | 60,907,428 | 5,179,897 | 94,937,520 | 231,024,845 | |
Budget impact | 0 | 357,180 | 3,484,285 | 10,525,648 | 14,367,113 | |
Submitted base case (corrected) | Reference | 48,293,326 | 61,507,976 | 72,853,171 | 85,800,992 | 220,162,139 |
New drug | 48,293,326 | 61,862,087 | 76,280,792 | 96,017,554 | 234,160,433 | |
Budget impact | 0 | 354,111 | 3,427,621 | 10,216,562 | 13,998,294 | |
Submitted base case (revised) | Reference | 48,293,326 | 61,507,976 | 72,853,171 | 85,800,992 | 220,162,139 |
New drug | 48,293,326 | 61,257,113 | 69,767,833 | 73,185,099 | 204,210,045 | |
Budget impact | 0 | −250,863 | −3,085,338 | −12,615,893 | −15,952,094 | |
CADTH base case | Reference | 111,699,806 | 142,618,690 | 168,936,272 | 199,015,062 | 510,570,023 |
New drug | 111,699,806 | 141,828,663 | 159,242,616 | 159,458,371 | 460,529,650 | |
Budget impact | 0 | −790,027 | −9,693,656 | −39,556,691 | −50,040,374 | |
CADTH scenario analysis: 100% of patients receive ABRO 200 | Reference | 111,699,806 | 142,618,690 | 168,936,272 | 199,015,062 | 510,570,023 |
New drug | 111,699,806 | 141,870,724 | 159,759,075 | 161,567,151 | 463,196,950 | |
Budget impact | 0 | −747,966 | −9,177,197 | −37,447,911 | −47,373,074 | |
CADTH scenario analysis: 100% of patients receive ABRO 100 | Reference | 111,699,806 | 142,618,690 | 168,936,272 | 199,015,062 | 510,570,023 |
New drug | 111,699,806 | 141,750,549 | 158,283,477 | 155,542,067 | 455,576,093 | |
Budget impact | 0 | −868,140 | −10,652,795 | −43,472,995 | −54,993,931 | |
CADTH scenario analysis: 40% of patients receive systemic immunosuppressants | Reference | 148,933,075 | 190,158,253 | 225,248,363 | 265,353,416 | 680,760,031 |
New drug | 148,933,075 | 189,104,884 | 212,323,488 | 212,611,162 | 614,039,533 | |
Budget impact | 0 | −1,053,369 | −12,924,875 | −52,742,254 | −66,720,498 | |
Price-reduction scenario: 52% for ABRO 100 | Reference | 111,699,806 | 142,618,690 | 168,936,272 | 199,015,062 | 510,570,023 |
New drug | 111,699,806 | 141,644,292 | 156,978,791 | 150,214,926 | 448,838,008 | |
Budget impact | 0 | −974,397 | −11,957,481 | −48,800,136 | −61,732,015 | |
Price-reduction scenario: 52% for ABRO 100, 56% for ABRO 200 | Reference | 111,699,806 | 142,618,690 | 168,936,272 | 199,015,062 | 510,570,023 |
New drug | 111,699,806 | 141,232,989 | 151,928,810 | 129,596,263 | 422,758,062 | |
Budget impact | 0 | −1,385,700 | −17,007,462 | −69,418,799 | −87,811,961 | |
Price-reduction scenario: DUP price ($959.9350)a | Reference | 110,011,656 | 140,380,502 | 166,238,069 | 195,784,319 | 502,402,890 |
New drug | 110,011,656 | 139,626,344 | 156,984,679 | 158,024,768 | 454,635,790 | |
Budget impact | 0 | −754,158 | −9,253,390 | −37,759,551 | −47,767,100 | |
Price-reduction scenario: DUP price reduced by 54%a | Reference | 51,469,243 | 63,333,716 | 73,667,116 | 85,282,276 | 222,283,109 |
New drug | 51,469,243 | 63,658,319 | 77,654,780 | 101,571,103 | 242,884,203 | |
Budget impact | 0 | 324,603 | 3,987,664 | 16,288,827 | 20,601,094 |
BIA = budget impact analysis; DUP = dupilumab.
Note: The scenario analyses are carried out on CADTH base case. Reanalyses are based on publicly available prices of the comparator treatments, unless otherwise noted.
aIn this analysis, the price of DUP was assumed to be $959.9350 per syringe based on the 2020 CADTH assessment of DUP.12 In all other scenarios, the price of DUP was assumed to be $978.70 based on the Ontario Exceptional Access Program price.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.