CADTH Reimbursement Review
Sodium phenylbutyrate-ursodoxicoltaurine (Albrioza)
Sponsor: Amylyx Canada
Therapeutic area: Amyotrophic lateral sclerosis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AA
active treatment to active treatment
AE
adverse event
ALS
amyotrophic lateral sclerosis
ALSFRS
Amyotrophic Lateral Sclerosis Functional Rating Scale
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
ATLIS
Accurate Test of Limb Isometric Strength
CALS
Canadian ALS Research Network
CI
confidence interval
DB
double-blind
del-FS
delta-functional scale
FVC
forced vital capacity
HR
hazard ratio
HRQoL
health-related quality of life
ICC
intraclass correlation coefficient
ITT
intention-to-treat
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
mITT
modified intention-to-treat
MMRM
mixed model of repeated measures
NEALS
Northeast Amyotrophic Lateral Sclerosis Consortium
NOC/c
Notice of Compliance with Conditions
OLE
open-label extension
PA
placebo to active treatment
PB-TURSO
sodium phenylbutyrate-ursodoxicoltaurine
PP
per protocol
PPN
percent predicted normal
RA
randomized to active treatment
RCT
randomized controlled trial
RP
randomized to placebo
RPSFT
rank preserving structural failure time
SAE
serious adverse event
SD
standard deviation
SE
standard error
SOC
standard of care
SVC
slow vital capacity
TEAE
treatment-emergent adverse event
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Sodium phenylbutyrate-ursodoxicoltaurine (Albrioza), 3 g/1 g per sachet, powder for oral suspension |
Indication | Proposed: For the treatment of amyotrophic lateral sclerosis |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard |
NOC/c date | June 10, 2022 |
Sponsor | Amylyx Canada |
NOC/c = Notice of Compliance with Conditions; TBD = to be determined.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig disease, is a rare, incurable, neurodegenerative disease.1 It primarily affects the nerve cells (neurons) that control voluntary muscles. As motor neurons in the brain (upper motor neurons) and spinal cord (lower motor neurons) deteriorate, they stop sending messages to the muscles, causing muscle weakness, muscle twitching (fasciculations), muscle tightness (spasticity), and muscle atrophy (shrinkage of muscle).2-4 Early symptoms include muscle twitching and cramping, especially in the hands and feet; loss of motor control in the hands and arms; impaired use of the arms and legs; weakness and fatigue; tripping and falling; dropping things; and slurred or thick speech, and difficulty in projecting the voice. Later symptoms include shortness of breath, difficulty breathing, difficulty swallowing (dysphagia), and paralysis.3 Respiratory failure is the most common cause of death.5,6
It is estimated that there are 3,000 Canadians currently living with ALS4 and approximately 1,000 patients die from ALS each year while a similar number is diagnosed every year in Canada.7 A review of Canadian data has estimated the annual incidence of ALS to be between 1.63 and 2.4 per 100,000 persons and prevalence to be 4.9 per 100,000 persons.8 The average age at the time of diagnosis is 55 years. While most patients develop ALS between 40 years of age and 70 years of age, it can occur in people as young as 20 and as old as 90 years of age.9 The median survival time from symptom onset to death ranges from 20 months to 48 months,10 with 25%11 to 30%12 of patients surviving 5 years and 10%11 to 20%12 of patients being alive at 10 years or more.
An ALS diagnosis is made by reviewing a patient’s symptoms, evidence of disease progression, and full medical history, as well as performing tests to rule out other diseases since there is no single diagnostic test or biomarker that can confirm or completely eliminate other causes of illnesses.13 Delays in diagnosis are common due to the overlap of initial symptoms with other conditions, and the estimated mean time between symptom onset and diagnosis can range from 15.1 months to 27.0 months in Canada.14 Patients with symptoms and signs suggestive of ALS are diagnosed based on the El Escorial criteria, which were originally designed to improve diagnostic certainty in clinical trials.13,15
Two disease-modifying therapies are currently available: riluzole and edaravone. “Canadian best practice recommendations for the management of amyotrophic lateral sclerosis,” published in 2020 in the Canadian Medical Association Journal, noted that specialty clinics for treating ALS that offer multidisciplinary care are important for the treatment of ALS and health care professionals from different specialties can help with challenges related to communication, nutrition, activities of daily living, and end-of-life care.4
The clinician group input for the review of sodium phenylbutyrate-ursodoxicoltaurine (PB-TURSO) highlighted that currently available treatments for ALS offer limited clinical benefit to patients. They stated that there is a clear need for new treatments that slow or reverse disease progression and preserve function. Since no current monotherapies stop disease progression, there is no rationale for patients to be required to fail or progress on a medication before starting another and multiple therapies may be used concurrently. According to the clinicians, the most important goals of treatment are to slow disease progression and prolong survival as well as preserve respiratory function, reduce symptoms, slow cognitive decline, promote health-related quality of life (HRQoL), maintain patient independence, and reduce caregiver burden.
Health Canada issued a Notice of Compliance with Conditions (NOC/c) for PB-TURSO on June 10, 2022, pending the results of trials to verify its clinical benefit. PB-TURSO is available as individual sachets, each containing 10 g of powder (3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine) to be reconstituted in 250 mL of room-temperature water and taken orally or administered via feeding tube within 1 hour of preparation. The recommended dosage is 1 sachet daily for the first 3 weeks and, if tolerated, 1 sachet twice daily thereafter. PB-TURSO can be taken alone or with riluzole and/or edaravone. The mechanism of action of PB-TURSO is unknown in patients with ALS, though it is hypothesized that the combination of sodium phenylbutyrate and ursodoxicoltaurine reduces neuronal death.16 Sodium phenylbutyrate is a pan-histone deacetylase inhibitor that ameliorates endoplasmic reticulum stress by upregulating chaperone proteins. Ursodoxicoltaurine ameliorates mitochondrial stress by reducing mitochondrial permeability and increasing the apoptotic threshold of the cell.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of sodium phenylbutyrate-ursodoxicoltaurine (3 g/1 g sachet, oral administration) for the treatment of patients with ALS.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 2 patient group submissions for the review of PB-TURSO. The ALS Society of Canada (ALS Canada) conducted an online survey of 629 patients and caregivers from Canada, the US, the UK, Israel, and the Netherlands between November 10 and November 24, 2021. A second patient group, ALS Action Canada, collected patients’ experiences and opinions through email communications and Zoom meetings from members of its organization living in British Columbia, Alberta, Ontario, and Nova Scotia.
Respondents felt that ALS had the most impact on mobility, motor function, fatigue, breathing, speech, and swallowing. It was also emphasized how ALS negatively impacted patients’ independence with performing activities of daily living, quality of life, and social activities. For caregivers, the aspects of life that were most negatively affected were travel options, family life, and emotional and psychological well-being. Patients also spent several hours each day on exercises for legs and arms and breathing, receiving treatments (drugs and supplements), and attending medical appointments. As ALS progresses and patients are faced with new challenges, the caregiver becomes increasingly important; however, finding qualified full-time support can be challenging.
Some patients felt that currently available medications (i.e., edaravone and riluzole) appeared to slow disease progression, help maintain motor function, and increase survival. Key challenges with these medications included side effects, limited access, affordability, and administration. A limited number of patients who had access to PB-TURSO (through clinical trials, Health Canada’s Special Access Program, or the sponsor’s compassionate program) felt that slowed disease progression and maintained motor function were the main benefits, although some said it was too soon to comment on the impact. A few respondents reported side effects that are mostly gastrointestinal-related as well as taste disturbances.
Patients and caregivers emphasized the importance of treatments that are simple to administer, significantly slow progression, help patients maintain their independence, reverse symptoms, and increase survival.
Clinician Input
Input From Clinical Expert Consulted by CADTH
The clinical expert highlighted that there are no available treatments that stop or reverse disease progression, reduce symptom severity (including neurologic decline), minimize adverse events (AEs), and improve HRQoL and survival. It was emphasized that current treatments demonstrate very modest benefits with slowing progression; however, as ALS is a terminal disease, all patients progress despite available medications.
Due to the progressive nature of ALS and low survival beyond 5 years after diagnosis, the clinical expert indicated that it is reasonable to target all potential pathological pathways as early as possible. Riluzole, edaravone, and PB-TURSO act on different pathways and targets in the body and may be used simultaneously with a 1-week to 2-week interval between starting a new treatment to assess tolerance and side effects. The clinical expert explained that it is important to start treatment early to spare healthy neurons and preserve muscle function in affected areas of the body.
The clinical expert noted that all patients diagnosed with ALS would be suitable for treatment with PB-TURSO. As there is no single diagnostic biomarker for the disease, neurologists confirm through medical history, a physical exam, and electromyography testing, and by excluding other diagnoses. The expert suggested that patients who are most suitable for receiving treatment be decided based on clinician judgment rather than functional rating scores or pulmonary function tests since patients may have difficulties accessing ALS specialty clinics to perform these tests.
Per input from the clinical expert, ALS is clinically heterogenous and the rate of progression is individual, which complicates monitoring outcomes and defining a response to therapy. According to “Canadian best practice recommendations for the management of amyotrophic lateral sclerosis,” patients should be routinely monitored every 3 months to 4 months.
The clinical expert indicated that patients with advanced disease may not derive benefit from treatment with PB-TURSO, but also noted that the definition of advanced disease may vary among clinicians and differ between clinicians and patients. It was suggested that treatment discontinuation could be considered for patients with advanced disease who are fully dependent for their activities of daily living, walking, transfers, and feeding. Changes to a patient’s goals of care or a desire to discontinue a medication should also be considered.
A neurologist or physiatrist with experience caring for patients with ALS is the most appropriate health care provider to prescribe PB-TURSO, though all patients should have a multidisciplinary care team and be followed by an ALS clinic.
Clinician Group Input
Input was received from the Canadian ALS Research Network (CALS), which consisted of 10 CALS members from across Canada.
The clinician group input was similar to that given by the clinical expert consulted by CADTH.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for PB-TURSO:
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability of trial populations to the broader populations in the jurisdictions
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The CENTAUR trial was a phase II, multi-centre, double-blind (DB), randomized, placebo-controlled trial to assess the safety, tolerability, and efficacy of PB-TURSO in adult patients with ALS. One sachet of medication was taken orally or via feeding tube once daily for the first 3 weeks and, if tolerated, 1 sachet twice daily thereafter. The CENTAUR trial was conducted at 25 Northeast Amyotrophic Lateral Sclerosis Consortium (NEALS) centres in the US and included 137 patients. Patients were randomized in a 2:1 ratio to receive either PB-TURSO and standard of care (SOC) (n = 89) or matching placebo and SOC (n = 48) for the duration of the 24-week DB treatment period. SOC included concomitant riluzole and/or edaravone. The study design consisted of (1) a screening period of up to 42 days, (2) a DB treatment period of 24 weeks with study evaluations taking place every 3 weeks, and (3) a follow-up period for up to 4 weeks. Patients who discontinued early from the study were asked to return to the study site for final safety assessments. After completing the DB trial, patients could enrol in the 132-week open-label extension (OLE) phase.
The primary safety outcome of the CENTAUR study was to confirm the safety and tolerability of PB-TURSO while the primary efficacy outcome was the rate of change (slope) of disease progression as measured by the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). Secondary outcomes included in the CADTH review protocol were the Accurate Test of Limb Isometric Strength (ATLIS) for measuring isometric muscle strength, slow vital capacity (SVC) percent predicted normal (PPN) for respiratory function, and survival (defined as death, tracheostomy, or permanent assisted ventilation) outcomes.
Patients had to have a diagnosis of definite ALS, be within 18 months of symptom onset, and have greater than 60% predicted normal SVC. The mean age of patients in the CENTAUR trial was 57.5 years (standard deviation [SD] = 9.50 years). Most patients were male (68.9%) and most patients were White (94.8%). The mean rate of disease progression (del-FS) before study entry was 0.95 points per month (SD = 0.43 points per month) in the PB-TURSO group and 0.93 points per month (SD = 0.60 points per month) in the placebo group. In general, mean ALSFRS-R total and domain scores were similar between the treatment groups as were mean SVC measurements. Mean ATLIS scores were numerically higher for those in the PB-TURSO group compared to the placebo group. The use of riluzole and/or edaravone at or before study entry was overall more common in the placebo group. Most (77.1%) patients in the placebo group had experience with riluzole compared to 67.8% of patients in the PB-TURSO group. Similarly, 50% of the placebo group and 25.3% of the PB-TURSO group had experience with edaravone. Most patients had experience with 1 of the treatments (87.5% in the placebo group and 71.3% in the PB-TURSO group), and more patients in the placebo group (39.6%) had experience with both medications compared to the PB-TURSO group (21.8%).
Efficacy Results
A summary of key efficacy results from the CENTAUR trial is provided in Table 2.
The primary efficacy outcome was the rate of change (slope) of disease progression as measured by the ALSFRS-R total score in the modified intention-to-treat (mITT) population. The slope for the ALSFRS-R total score was –1.24 points per month for the PB-TURSO group and –1.66 points per month for the placebo group. The treatment difference was 0.42 points per month (95% confidence interval [CI], 0.03 points per month to 0.81 points per month; P = 0.03) comparing the PB-TURSO versus placebo groups. The mean change from baseline to week 24 was –6.86 points (standard error [SE] = 0.66 points) and –9.18 points (SE = 0.88 points) in the PB-TURSO and placebo groups, respectively. Overall, the difference in change from baseline to week 24 between the PB-TURSO and placebo groups was 2.32 points (95% CI, 0.18 points to 4.47 points; P = 0.03). For the per-protocol (PP) population, the difference comparing the PB-TURSO group to the placebo group was 2.54 points (95% CI, 0.28 points to 4.81 points; P = 0.03). A pre-specified sensitivity analysis was conducted for the missing at random assumption, which resulted in a difference of 1.87 points (95% CI, 0.06 points to 3.69 points) for PB-TURSO versus placebo. According to the clinical expert consulted for this review, a difference of at least 2 points over a period of 6 months for most patients with ALS would be considered clinically meaningful if found to be reproducible through additional studies. Additionally, a change of 20% to 25% in the slope of ALSFRS-R was considered “at least somewhat clinically meaningful,” according to surveyed clinical experts.17
The first outcome in the testing hierarchy was the ATLIS total score. The mean changes from baseline to week 24 were –16.72% (SE = 1.05%) and –19.54% (SE = 1.45%) for the PB-TURSO and placebo treatment groups, respectively. Overall, the difference comparing PB-TURSO versus placebo was 2.82% (95% CI, –0.67% to 6.31%; P = 0.11) at week 24. Since the result was not statistically significant, statistical testing was stopped at the first outcome in the testing hierarchy and all subsequent P values were considered nominal (i.e., not adjusted for multiple testing). The differences from baseline to week 24 for SVC PPN were –17.11% (SE = 1.70%) and –22.22% (SE = 2.32%) for the PB-TURSO and placebo groups, respectively. Overall, the difference comparing PB-TURSO versus placebo was 5.11% (95% CI, –0.54% to 10.76%) at week 24. A total of 6 death or death equivalent events occurred based on the mITT population, resulting in a hazard ratio (HR) of 0.63 (95% CI, 0.11 to 3.92) for the PB-TURSO versus placebo treatment groups.
Harms Results
A summary of key harms results from the CENTAUR trial is provided in Table 2.
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Result | CENTAUR study | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
Primary efficacy end point: ALSFRS-R total score, week 24 (mITT population) | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 35.68 (5.78) | 36.67 (5.08) |
LSMa (week 24), mean (SE) | 29.06 (0.78) | 26.73 (0.98) |
Change from baseline,a mean (SE) | –6.86 (0.66) | –9.18 (0.88) |
Treatment group difference vs. controla (95% CI) | 2.32 (0.18 to 4.47) | Reference |
Rate of change per month,a mean (SE) | –1.24 (0.12) | –1.66 (0.16) |
Treatment group difference vs. controla (95% CI) | 0.42 (0.03 to 0.81) | Reference |
P valueb | 0.03 | Reference |
Secondary efficacy end point: ATLIS total score (percent of normal strength), week 24 (mITT population) | ||
Number of patients contributing to the analysis at week 24 | 55 | 32 |
Baseline, mean (SD) | 56.83 (20.08) | 53.92 (20.94) |
LSMa (week 24), mean (SE) | 39.08 (1.99) | 36.26 (2.22) |
Change from baseline,c mean (SE) | –16.72 (1.05) | –19.54 (1.45) |
Treatment group difference vs. controlc (95% CI) | 2.82 (–0.67 to 6.31) | Reference |
P valued | 0.11 | Reference |
Secondary efficacy end point: SVC (percent predicted normal), week 24 (mITT population) | ||
Number of patients contributing to the analysis at week 24 | 62 | 34 |
Baseline, mean (SD) | 83.62 (18.17) | 83.88 (15.92) |
LSMa (week 24), mean (SE) | 66.17 (2.33) | 61.06 (2.81) |
Change from baseline,a mean (SE) | –17.11 (1.70) | –22.22 (2.32) |
Treatment group difference vs. controla (95% CI) | 5.11 (–0.54 to 10.76) | Reference |
P valued | 0.08 | Reference |
Secondary efficacy end point: Survival (death or death equivalente, f) (mITT population) | ||
Number of events | 6 | |
Estimated percentage of risk event (SE) | 2.8 (1.69) | 4.4 (3.02) |
HR, PB-TURSO vs. placebo (95% CI) | 0.63 (0.11 to 3.92) | Reference |
P valued | 0.60 | Reference |
Harms, n (%) (safety population) | ||
AEsg | 86 (96.6) | 46 (95.8) |
SAEs | 11 (12.4) | 8 (16.7) |
WDAEs (from study treatment) | 18 (20.2) | 5 (10.4) |
Deaths | 5 (5.6) | 2 (2.2) |
Notable harms | ||
Gastrointestinal AEsg | ||
Diarrhea | 19 (21.3) | 8 (16.7) |
Nausea | 16 (18.0) | 6 (12.5) |
Constipation | 12 (13.5) | 12 (25.0) |
Salivary hypersecretion | 10 (11.2) | 1 (2.1) |
Abdominal pain | 7 (7.9) | 4 (8.3) |
Abdominal discomfort | 5 (5.6) | 0 |
Dry mouth | 3 (3.4) | 4 (8.3) |
Dysphagia | 3 (3.4) | 4 (8.3) |
Neurologic AEsg, h | ||
Headache | 13 (14.6) | 11 (22.9) |
Dizziness | 9 (10.1) | 2 (4.2) |
Insomnia | 3 (3.4) | 3 (6.3) |
Anxiety | 2 (2.2) | 3 (6.3) |
Respiratory AEsg, i | ||
Dyspnea | 9 (10.1) | 4 (8.3) |
Cough | 5 (5.6) | 3 (6.3) |
Respiratory failure | 4 (4.5) | 4 (8.3) |
Taste disturbancej | 3 (3.4) | 1 (2.1) |
AE = adverse event; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; CI = confidence interval; del-FS = delta-functional scale; HR = hazard ratio; LSM = least squares mean; MedDRA = Medical Dictionary for Regulatory Activities; mITT = modified intention-to-treat; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SAE = serious adverse event; SD = standard deviation; SE = standard error; SOC = standard of care; SVC = slow vital capacity; vs. = versus; WDAE = withdrawal due to adverse event.
aShared-baseline MMRM analysis with covariates of age and del-FS before the study was used in the model to compare treatment groups. The patient’s rate of decline before the study = (48 – ALSFRS-R at baseline) ÷ time in months from symptom onset to baseline. The maximum score on the ALSFRS-R is 48 points.
bP value has been adjusted for multiple testing (i.e., the type I error rate has been controlled).
cShared-baseline MMRM analysis with covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time was used in the model to compare treatment groups. The patient’s rate of decline before study = (ceiling maximum efficacy score – efficacy score at baseline) ÷ time in months from symptom onset to baseline. Where there was no defined maximum value (i.e., SVC and ATLIS), the observed maximum among all enrolled patients (PB-TURSO or placebo treatment) was used.
dP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
eDeath equivalent was defined as time to death, permanent assisted ventilation (noninvasive mechanical ventilation for more than 22 hours per day for more than 7 days), or tracheostomy.
fCox proportional hazards model with covariates of del-FS and age at baseline.
gFrequency of at least 5% of patients.
hNeurologic AEs include those listed under MedDRA’s Nervous System Disorders and Psychiatric Disorders headings.
iRespiratory AEs include those listed under MedDRA’s Respiratory, Thoracic, and Mediastinal Disorders heading.
jData here are those reported for MedDRA’s term for “dysgeusia.” It was noted in the CENTAUR Clinical Study Report that investigators were instructed to not capture the bad taste of medication as an AE.
Source: CENTAUR Clinical Study Report (2021).18
The primary safety outcome of the CENTAUR study was to confirm the safety and tolerability of PB-TURSO. Overall, 86 (96.6%) patients in the PB-TURSO group and 46 (95.8%) patients in the placebo group experienced at least 1 treatment-emergent adverse event (TEAE) during the CENTAUR trial. The 3 most frequently reported TEAEs in the PB-TURSO group were falls (28.1%), diarrhea (21.3%), and muscular weakness (20.2%). Among patients in the placebo group, the 3 most frequently reported TEAEs were falls (37.5%), constipation (25.0%), and headache (22.9%). The TEAEs that occurred more frequently (5% or greater) in patients who received PB-TURSO compared to placebo included nausea (18.0% versus 12.5%), salivary hypersecretion (11.2% versus 2.1%), viral upper respiratory tract infection (11.2% versus 4.2%), dizziness (10.1% versus 4.2%), abdominal discomfort (5.6% versus 0%), and asthenia (5.6% versus 0%).
In total, 23 serious adverse events (SAEs) were reported in 11 (12.4%) patients from the PB-TURSO group and 8 (16.7%) patients from the placebo group. Most SAEs were reported in single patients aside from respiratory failure (5 patients), bacteremia (2 patients), and nephrolithiasis (2 patients). Overall, 18 (20.2%) patients from the PB-TURSO group and 5 (10.4%) patients from the placebo group withdrew from the study medication due to a TEAE. The most frequently reported reasons were due to diarrhea (5.6%) in the active treatment group (versus 0 in the placebo group) and respiratory failure (6.3%) in the placebo group (versus 0 in the PB-TURSO group). There were 7 deaths reported during the CENTAUR trial in the safety population: 5 (5.6%) patients in the PB-TURSO group due to respiratory failure or respiratory arrest (3 patients), subdural hematoma (secondary to a fall; 1 patient), and diverticular perforation (1 patient) compared to 2 (2.2%) patients in the placebo group, both due to respiratory failure or respiratory arrest. The safety population included 2 additional patient deaths that were excluded from the mITT population.
Notable harms considered relevant to this review included gastrointestinal AEs, neurologic AEs, respiratory AEs, and taste disturbances. Frequently reported gastrointestinal AEs (in at least 5% of patients) that occurred more often among patients who received PB-TURSO compared with those who received placebo included diarrhea (21.3% versus 16.7%), nausea (18.0% versus 12.5%), salivary hypersecretion (11.2% versus 2.1%), and abdominal discomfort (5.6% versus 0%). Neurologic AEs occurring in at least 5% of patients such as dizziness were more frequent among patients who received PB-TURSO compared with those who received placebo (10.1% versus 4.2%). Headache (22.9% in the placebo group versus 14.6% in the PB-TURSO group), insomnia (6.3% in the placebo group versus 3.4% in the PB-TURSO group), and anxiety (6.3% in the placebo group versus 2.2% in the PB-TURSO group) were more frequent among patients who received placebo. Respiratory AEs that were reported in at least 5% of patients and were more common in patients who received PB-TURSO included dyspnea (10.1% in the PB-TURSO group versus 8.3% in the placebo group). Respiratory failure (8.3% in the placebo group versus 4.5% in the PB-TURSO group) and cough (6.3% in the placebo group versus 5.6% in the PB-TURSO group) were more frequently reported among patients who received placebo compared to those who received PB-TURSO. Dysgeusia, or taste disturbance, occurred in 3.4% of patients who received active treatment compared to 2.1% of patients who received placebo. It should be noted that investigators were instructed not to capture the bad taste of medication as an AE (as it was not classified as an AE per the Common Terminology Criteria for Adverse Events Version 5.0) and instead, to record issues with oral administration if there was a clinically untoward effect such as burning, vomiting, or anxiety.
Critical Appraisal
The key limitation was that the CENTAUR trial was a phase II trial with a small number of patients and a short duration. There were large proportions of patients who discontinued from the study and the number of patients available for the analysis at 24 weeks varied largely from the number of patients randomized at baseline. All efficacy outcomes had missing data at week 24, including the ALSFRS-R total score, due to patients discontinuing from the study (23% of the randomized population), leading to uncertainty in the results. For the ALSFRS-R instrument, information on its responsiveness to change and minimal important difference (MID) estimates were not found in the literature, though estimates of what would be clinically meaningful based on clinical expert opinion are available. Survival data were limited by the small number of patients and were immature at the end of 6 months due to few events having occurred. The low frequencies for SAEs, withdrawals due to adverse events (WDAEs), and deaths, due to small sample size and short duration, as well as the missing data available for analysis at 24 weeks compared to baseline, made it difficult to draw any firm conclusions from these results.
All the study centres were in the US, which limits the generalizability to Canadian practice on the basis of differences in health care provisions. The criteria of a definite ALS diagnosis within 18 months of symptom onset were very restrictive since they require patients to have a high level of symptoms that many patients would not meet within 18 months. These limitations prevented the capture of information for patients who are progressing slowly and may benefit from treatment with PB-TURSO. The clinical expert identified specific exclusion criteria that may also be restrictive: poorly controlled arterial hypertension (systolic blood pressure greater than 160 mm Hg or diastolic blood pressure greater than 100 mm Hg) at screening, a history of cholecystectomy, and exposure to antacids containing aluminum hydroxide or aluminum oxide within 2 hours of administration of PB-TURSO. Overall, the safety and efficacy of PB-TURSO are unknown outside the CENTAUR study population. Patient-reported outcomes such as those measuring ALS symptoms or HRQoL were not included in the CENTAUR trial and little is known about the impact of PB-TURSO from the patient perspective.
Indirect Comparisons
Description of Studies
Due to the lack of head-to-head comparisons between PB-TURSO and other ALS medications, the sponsor submitted a feasibility assessment for conducting a matching-adjusted indirect comparison (MAIC) to compare the relative efficacy of PB-TURSO to edaravone. In total, 1 randomized controlled trial (RCT) for PB-TURSO (the CENTAUR trial) and 2 RCTs for edaravone (Study 16 and Study 19) were included in the assessment. The sponsor stated that Study 16 and Study 19 were “sufficiently homogenous” for the data to be pooled using standard methods.19 The primary end point of the analysis was the difference between PB-TURSO and edaravone for the mean change in the ALSFRS-R total score from baseline to week 24.
There were notable differences between the studies in terms of study design (e.g., location, pre-baseline observation period, eligibility criteria) and baseline characteristics for sex, site of onset, duration of disease, ALSFRS-R baseline score, del-FS at baseline, proportion of patients with a definite ALS diagnosis, FVC or SVC at baseline, and use of riluzole or edaravone at baseline.
Efficacy Results
Adjustments were made for the following covariates: del-FS, baseline ALSFRS-R score, duration of disease, baseline FVC or SVC, and concomitant riluzole use at baseline. After matching to the pooled Study 16 and Study 19 data, the effective sample size of the CENTAUR trial was reduced from an original 135 patients to 24.8 patients. After adjusting for baseline edaravone use, the effective sample size was further reduced to 3.4 patients. The large reduction in effective sample size indicated that the study populations were substantially different from 1 another and that it was not reasonable to conduct an MAIC using these populations. The sponsor’s analysis identified del-FS to be the main reason for the reduction in effective sample size; del-FS was also considered to be the most clinically important covariate.
Critical Appraisal
The CADTH review team agreed that the reduction in effective sample size after adjustments indicated that there were substantial differences between the populations and that it would not be reasonable to compare the treatments between the CENTAUR trial and the pooled Study 16 and Study 19 data.
Other Relevant Evidence
Description of Studies
The OLE of the pivotal CENTAUR trial provided long-term safety and efficacy evidence for PB-TURSO. In total, 90 patients enrolled in the OLE and those who were randomized to placebo in the DB phase received PB-TURSO in the OLE. The primary end point was long-term safety and patients were analyzed based on whether they received PB-TURSO during both DB and OLE phases or placebo and PB-TURSO during the DB and OLE phases, respectively. The secondary end points were for survival (hospitalization, tracheostomy, permanent assisted ventilation, death), ALSFRS-R, ATLIS, and SVC. Patients were analyzed based on whether they were randomized to PB-TURSO or to placebo in the CENTAUR study. The survival analysis was expanded following database lock and unblinding to treatment assignment to include information on death events obtained via a vital status sweep for all patients randomized in the main trial with data cut-offs of February 29, 2020, and July 20, 2020.
A post-hoc analysis of the CENTAUR trial was conducted to compare the relative efficacy of PB-TURSO to edaravone to support the sponsor’s pharmacoeconomic model. The main subgroups of interest were patients who received PB-TURSO without edaravone and placebo with edaravone. The primary end point was the rate of change (slope) in the ALSFRS-R total score from baseline to week 24 using a shared-baseline mixed-effects model; the secondary end point was similar but used a change from baseline approach.
A second post-hoc analysis was performed on the survival data from patients in the CENTAUR trial up to 35 months post-baseline comparing the effect of switching treatments (i.e., placebo to PB-TURSO). In total, 34 of the 48 (71%) patients who received placebo during the DB phase enrolled in the CENTAUR-OLE and began receiving PB-TURSO. The sponsor noted that any beneficial effect on overall survival of PB-TURSO over placebo in the absence of a switch will be underestimated in the pre-specified intention-to-treat (ITT) analysis. The overall survival end point was defined as all-cause mortality. The main objective of the analysis was to model what the overall survival of patients in the CENTAUR trial may have been if patients from the placebo group had not switched treatments and received PB-TURSO. A rank preserving structural failure time (RPSFT) model was used and it was assumed that the treatment effect was consistent regardless of when it was given during the study (i.e., at randomization or upon enrolment to the OLE).
Efficacy Results
In the CENTAUR-OLE trial, the median survival for the ITT population was 25.0 months (95% CI lower bound = 20.8 months; upper bound not reached) and 18.5 months (95% CI lower bound = 14.9 months; upper bound not reached) for the PB-TURSO and placebo groups, respectively, yielding an HR of 0.56 (95% CI, 0.34 to 0.93) for death events at the July 20, 2020, data cut-off date. For death or death equivalent events, the median survival was 23.2 months (95% CI lower bound = 19.5 months; upper bound not reached) and 18.2 months (95% CI, 14.9 months to 23.1 months) for the PB-TURSO and placebo groups, respectively, yielding an HR of 0.57 (95% CI, 0.35 to 0.93) at the July 20, 2020, data cut-off date. Based on the Clinical Study Report with a data cut-off for March 1, 2021, which contains updated survival data, the median survival was 23.5 months and 18.7 months for patients randomized to PB-TURSO and placebo, respectively, resulting in an HR of 0.64 (95% CI, 0.42 to 1.00; P = 0.0475). At this data cut-off date, 94 death events were reported (69% of the ITT population), with 1 patient lost to follow-up. The FDA noted in the Combined FDA and Applicant Briefing Document for the March 30, 2022, meeting of the Peripheral and Central Nervous System Drugs Advisory Committee that, using the likelihood ratio test specified in the survival statistical analysis plan, the HR is 0.64 with a P value of 0.0518. Further, the FDA also noted that with the inclusion of 5 additional death events captured following the March 1, 2021, data cut-off date, the HR is 0.70 with a P value of 0.1109. However, it was unclear how this was determined, and the analysis would not have been from a planned data cut-off.
The treatment group differences (patients randomized to PB-TURSO versus placebo) were 4.23 points (95% CI, 0.56 points to 7.90 points) for the ALSFRS-R total score, 6.20% (95% CI, 0.01% to 12.39%) for the ATLIS total score, and 10.66% (95% CI, 0.63% to 20.69%) for SVC.
In the first post-hoc analysis, the estimated effect sizes between patients in the PB-TURSO without edaravone group and patients in the placebo with edaravone group varied from 2.62 points to 3.22 points using a shared-baseline approach. The secondary end point results (using a change from baseline approach) varied from 3.61 points to 4.41 points.
For the second post-hoc assessment’s primary and sensitivity analyses (with and without recensoring), the acceleration factor estimates were all less than 1, indicating that PB-TURSO had a beneficial effect on overall survival. Using the RPSFT model without recensoring, the median overall survival was approximately 13.5 months for the placebo group with an HR of 0.34 (95% CI, 0.13 to 0.87). When recensoring was applied, the median overall survival was approximately 15.2 months for the placebo group with an HR of 0.40 (95% CI, 0.18 to 0.88).
Harms Results
The proportion of patients who reported the most common AEs (5% or greater) was higher in the group that received placebo in the DB phase (placebo to active treatment [PA] group = 82.4%) compared with the group that received PB-TURSO in the DB phase (active treatment to active treatment [AA] group = 73.2%). The most common AEs were falls, nausea, and diarrhea. The incidence of SAEs was also higher in the PA group (20.6% versus 14.3%). The percentage of patients withdrawing from the study due to AEs was higher in the PA group (29.4%) than the AA group (10.7%). Five (14.7%) patients in the PA group and 2 (3.6%) patients in the AA group died before week 24 in the OLE study. The reasons were respiratory failure, disease progression or ALS, and cardiac arrest. The most common notable harms were nausea (17.6% in the PA group versus 12.5% in the AA group) and diarrhea (20.6% in the PA group versus 8.9% in the AA group). Dysgeusia was reported only in the PA group (2.9%). At the latest data cut-off date, the numbers of patients reporting at least 1 AE were 32 (94.1%) patients in the PA group and 49 (87.5%) patients in the AA group, with notable increases in the percentage of patients reporting respiratory failure, dyspnea, constipation, and pneumonia. Further, 13 (38.2%) patients and 18 (32.1%) patients experienced at least 1 SAE in the PA and AA groups, respectively.
Harms were not assessed in either of the 2 post-hoc analyses.
Critical Appraisal
Given the nature of OLE studies, there is bias that impacts how the results are interpreted such as the lack of blinding during the OLE phase, the lack of a control group, and the selection bias for patients who successfully completed the main trial. Also, there were large proportions of study discontinuations, and it is possible that treatment assignment from the main trial was deduced for some patients based on the differences in gastrointestinal AEs between groups. All efficacy end points are secondary outcomes, and it is not possible to make definitive conclusions based on the available data. Although vital status was available for all but 2 patients from the main trial ITT population, death equivalent events outside the study were not captured, contributing uncertainty to the death or death equivalent composite end point. Due to the crossover in the OLE, assuming any effect of PB-TURSO on survival is beneficial, bias from treatment switching would be against PB-TURSO. The generalizability issues identified for the DB phase regarding patient characteristics and outcome measures also apply to the OLE.
A key limitation to the post-hoc analyses was that neither assessment was pre-specified, and therefore they should be viewed as hypothesis-generating. Specific to the first post-hoc analysis, defining treatment groups by whether a patient received edaravone meant that the benefits of randomization were lost for these comparisons. Additionally, the groups included only a subset of the mITT population and sample sizes were small as a result. Given the serious limitations, it is not possible to make any conclusions on how treatment with PB-TURSO compared to edaravone. In the second post-hoc analysis, the overall survival assessment relied on the assumption of constant treatment effect associated with the RPSFT model to accommodate crossovers from placebo to PB-TURSO in the OLE. The validity of the main assumption of the RPSFT method is unknown and no conclusions can be drawn from the RPSFT model results.
Conclusions
The CENTAUR trial results indicated a statistically significant difference in favour of PB-TURSO over placebo for the primary outcome of slowing disease progression as measured by the rate of change of the ALSFRS-R total score in adults who have a diagnosis of definite ALS, have an SVC greater than 60% of the predicted value, and are within 18 months of symptom onset. The clinical relevance of the treatment effect is unclear due to uncertainty introduced by the amount of missing data. There were no other statistically significant findings, though results for the ATLIS, SVC, and the ALSFRS-R domain scores supported the primary end point result. Survival analyses conducted during the OLE study suggested a survival benefit for PB-TURSO over placebo, but variation in the results from different data cut-offs, lack of adjustment for analyses at multiple time points, missing data for death equivalent events, and treatment switching during the extension mean that the finding may not be robust, and the magnitude of the treatment effect is uncertain. Conclusions regarding efficacy outcomes, other than survival, beyond 24 weeks of treatment could not be drawn. Outcomes for HRQoL and caregiver burden, both identified by patients as being important, were not included in the CENTAUR trial. It should also be noted that the narrow eligibility criteria for the CENTAUR trial resulted in a trial population that was representative of only a subpopulation of patients with ALS. The comparative efficacy of PB-TURSO versus edaravone or riluzole is unknown as the only evidence available was a post-hoc analysis of the CENTAUR trial that had serious limitations. Firm conclusions regarding the safety of PB-TURSO could not be drawn due to the limited sample size of the CENTAUR trial, though the results suggest that gastrointestinal AEs associated with PB-TURSO contribute to treatment discontinuations. Overall, a major limitation of the CENTAUR trial is the fact that it is a phase II trial; it is important that the efficacy and safety findings be confirmed in phase III trials.
Introduction
Disease Background
ALS, also known as Lou Gehrig disease, is a rare, incurable, neurodegenerative disease.1 It primarily affects the nerve cells (neurons) that control voluntary muscles. Motor neurons innervate from the brain to the spinal cord and to muscles.2 As motor neurons in the brain (upper motor neurons) and spinal cord (lower motor neurons) deteriorate, they stop sending messages to the muscles, causing muscle weakness, muscle twitching (fasciculations), muscle tightness (spasticity), and muscle atrophy (shrinkage of muscle).2-4
Symptoms of ALS develop gradually and may be overlooked in the early stages. Symptoms typically start and spread within the body segment where onset occurs, then spread to other regions in subsequent months to years.5 Early symptoms include muscle twitching and cramping, especially in the hands and feet; loss of motor control in the hands and arms; impaired use of the arms and legs; weakness and fatigue; tripping and falling; dropping things; and slurred or thick speech, and difficulty in projecting the voice. Later symptoms include shortness of breath, difficulty breathing, difficulty swallowing (dysphagia), and paralysis.3 The loss of motor neurons leads to life-threatening complications such as dysphagia and respiratory failure.5 Dysphagia can cause aspiration while eating or drinking, resulting in issues such as pneumonia, malnutrition, and dehydration. Respiratory failure is the most common cause of death.5,6 Varying degrees of cognitive impairment occur in about 30% to 50% of patients with ALS (e.g., changes related to executive function and fluency, behavioural changes such as apathy and disinhibition).1 ALS does not commonly harm a person’s senses or sexual, bowel, or bladder functions.3 Disease progression is believed to be constant over time, though the rate of progression varies among patients.5 The ALSFRS-R is a 12-question score (refer to Appendix 3) to assess the severity and progression of the disease. Specifically, ALSFRS-R scores can be compared over time to determine the rate of progression.
It is estimated that there are 3,000 Canadians currently living with ALS.4 According to ALS Canada, approximately 1,000 patients die from ALS each year and a similar number is diagnosed every year in Canada.7 A review of Canadian data has estimated the annual incidence of ALS to be between 1.63 and 2.4 per 100,000 persons8 based on data from 3 provinces; this is consistent with an estimated worldwide incidence of 1 to 3 per 100,000 persons.5 The prevalence of ALS has been estimated to be 4.9 per 100,000 persons, though it is worth noting that this figure was based on Ontario data collected between 1978 and 1982 before the El Escorial criteria became the standard diagnostic criteria.8 In the US, annual ALS prevalence and incidence, respectively, are estimated to be 5.2 cases and 1.6 cases per 100,000 persons.1 The average age of patients at the time of diagnosis is 55 years, with most people developing ALS between the ages of 40 years and 70 years. However, the disease does occur in people as young as 20 and as old as 90 years of age.9 The median survival time from symptom onset to death ranges from 20 months to 48 months,10 with 25%11 to 30%12 of patients surviving 5 years and 10%11 to 20%12 of patients surviving to 10 years or more. There is a general consensus that older age and bulbar onset of ALS are associated with a worse prognosis.10
Limb onset is the most common ALS phenotype (occurring in approximately 70% of patients) and bulbar onset is less common (occurring in approximately 25% of patients).1 Apart from upper and lower motor neuron disease, some patients present with additional symptoms or signs (i.e., dementia, extrapyramidal, autonomic dysfunction, ocular motility disturbance, and/or sensory loss) and are considered to have ALS-plus syndrome.1
While most cases of ALS arise spontaneously (sporadic ALS), without any known risk factors, 5% to 10% of cases are associated with a family history of ALS (familial ALS).2 Sporadic and familial ALS have the same general signs and symptoms. Different genetic mutations have been identified in recent years and those most commonly associated with ALS are in the SOD1, C9orf72, TARDBP, and FUS genes.20 The SOD1 mutation is associated with an extremely aggressive course, with survival in many cases of only 11 months to 12 months.21
There is no single diagnostic test or biomarker that can confirm or completely rule out other causes of illness.13 The diagnosis of ALS is made primarily by a review of the symptoms and subsequent evidence of disease progression observed by the treating physician in addition to a patient’s full medical history and a series of tests to rule out other diseases. Delays in diagnosis are common due to the overlap of initial symptoms with other conditions, and the estimated mean time between symptom onset and diagnosis can range from 15.1 months to 27.0 months in Canada.14 Electromyography and a nerve conduction study may assist in an ALS diagnosis. MRI, blood and urine tests, and a muscle biopsy may be used to exclude the possibility of other diseases.
Patients with symptoms and signs suggestive of ALS are assigned a different level of diagnostic certainty based on the El Escorial criteria.15 The original criteria included 5 levels (suspected, possible, laboratory-supported probable, probable, and definite ALS), but were simplified to 3 levels (possible, probable, and definite ALS) in the revised El Escorial criteria in 1997. The classes of the criteria depend on the extent of involvement in different segments of the central nervous system: bulbar (cranial nerve segment), cervical spinal cord, thoracic spinal cord, and lumbosacral spinal cord. Possible, probable, and definite ALS diagnoses are defined as follows1,22:
Possible ALS — There is a presence of upper and lower motor neuron signs in 1 body segment, or upper motor neuron signs in at least 2 segments, or lower motor neuron signs in a body segment above upper motor neuron signs.
Probable ALS — There is a presence of upper and lower motor neuron signs in at least 2 body segments with upper motor neuron signs in a body segment above lower motor neuron signs.
Definite ALS — There is a presence of upper and lower motor neuron signs in at least 3 body segments.
The revised El Escorial World Federation of Neurology criteria or Airlie House criteria were originally designed for research purposes to improve the diagnostic certainty of patients in clinical trials and have been validated pathologically.13 The criteria were further updated to include electromyography information to improve diagnostic sensitivity and have been called the Awaji criteria. Most recently, the Gold Coast criteria were created to simplify and improve the diagnosis process. These criteria remove the uncertainty that is often associated with the labels of possible or probable ALS. A confirmed diagnosis of ALS requires13,23:
progressive motor impairment recorded by medical history or repeated clinical assessments that was preceded by normal motor function and
upper and lower motor neuron symptoms and signs in at least 1 limb or body region (bulbar, cervical, thoracic, and lumbosacral), or lower motor neuron symptoms and signs in at least 2 body segments and
investigations (e.g., electrophysiologic, neuroimaging, pathologic evidence of other processes) that exclude other causes of neuron degeneration.
“Canadian best practice recommendations for the management of amyotrophic lateral sclerosis” was published in the Canadian Medical Association Journal in 2020.4 The recommendations have been gathered and presented based on the best available evidence and expert consensus on best practices among clinicians treating ALS in Canadian practices. The guidance emphasizes that care and management should be focused on the patient and consider holistic and emotional well-being. It is recommended that an initial diagnosis of ALS be confirmed by a neurologist or physiatrist with expertise in ALS and patients should attend an ALS specialty clinic within 4 weeks.
When considering disease-modifying therapies, prescriptions should be made by clinicians experienced in the treatment of patients with ALS.4 Two medications are currently available: riluzole and edaravone. The clinician group input for the review of PB-TURSO highlighted that these 2 medications for ALS offer limited clinical benefit to patients and there is a clear need for new treatments that slow or reverse disease progression and preserve function. The group also indicated that since no current monotherapies stop disease progression, there is no rationale for patients to be required to fail or progress on a medication before starting another and multiple therapies may be used concurrently. It was also noted that the criteria for accessing edaravone are restrictive and many patients are ineligible based on poor baseline forced vital capacity (FVC) measures and ALSFRS-R scores.
Specialty clinics for treating ALS that offer multidisciplinary care have shown benefits to survival, fewer and shorter hospitalizations, increased use of adaptive equipment and supportive care, and improved quality of life.4 A team of health care professionals from different specialties can help with challenges related to communication, nutrition, activities of daily living, and end-of-life care. The Canadian guidelines state that a patient’s individual needs and rate of progression may determine the frequency of care visits, though it is recommended that regular monitoring be conducted at least every 3 months for respiratory function and nutritional status.4 For patients with respiratory insufficiency, noninvasive ventilation is the SOC and should be initiated within 4 weeks for patients who meet the outlined criteria. Invasive ventilation may be an option for patients who cannot be managed with noninvasive ventilation and should be considered with overall goals of care. Nutritional management may include dietary changes and a review of medications, as well as considerations for enteral feeding tube insertion. Recommendations are also outlined for the management of symptoms, dysarthria, exercise, cognition and behaviour, and caregiver support. While discussing disease management and expectations with a patient, palliative care and end-of-life care should also be considered, particularly if there is evidence of advanced disease.
According to the clinicians who provided written input, the most important goals of treatment are to slow disease progression and prolong survival as well as preserve respiratory function, reduce symptoms, slow cognitive decline, promote HRQoL, maintain patient independence, and reduce caregiver burden.
Drug
The key characteristics of PB-TURSO, edaravone, and riluzole have been summarized in Table 3.
PB-TURSO is indicated for the treatment of patients with ALS.16 It is available as individual sachets, each containing 10 g of powder (3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine). The contents of 1 sachet are to be reconstituted in 250 mL of room-temperature water and taken orally or administered via feeding tube within 1 hour of preparation. The product monograph states that patients may consume a snack, meal, honey, or milk with the medication to reduce the bitter flavour of the medication, but to avoid consuming the medication with fruit juice. The recommended dosage is 1 sachet daily for the first 3 weeks and 1 sachet twice daily thereafter. PB-TURSO can be taken alone or with edaravone and/or riluzole.
The mechanism of action of PB-TURSO is unknown in patients with ALS, though it is hypothesized that the combination of sodium phenylbutyrate and ursodoxicoltaurine reduces neuronal death based on in-vitro studies.16 Sodium phenylbutyrate is a pan-histone deacetylase inhibitor that ameliorates endoplasmic reticulum stress by upregulating chaperone proteins. Ursodoxicoltaurine ameliorates mitochondrial stress by reducing mitochondrial permeability and increasing the apoptotic threshold of the cell.
Health Canada issued a Notice of Compliance with Conditions (NOC/c) for PB-TURSO on June 10, 2022, pending the results of trials to verify its clinical benefit. The sponsor has requested reimbursement as per the approved Health Canada indication. PB-TURSO has not been previously reviewed by CADTH.
Table 3: Key Characteristics of PB-TURSO, Edaravone, and Riluzole
Characteristic | PB-TURSO | Edaravone | Riluzole |
|---|---|---|---|
Mechanism of action | The mechanism of action in patients with ALS is unknown. In-vitro, the combination of sodium phenylbutyrate and ursodoxicoltaurine may reduce neuronal cell death. Activity could be attributed to sodium phenylbutyrate, ursodoxicoltaurine, their metabolites, or derivatives. | The mechanism of action in patients with ALS is unknown. | The mechanism of action in patients with ALS is unknown. Its pharmacological properties include the following, some of which may be related to its effect: • an inhibitory effect on glutamate release • inactivation of voltage-dependent sodium channels • ability to interfere with intracellular events that follow transmitter binding at an excitatory amino acid receptor. |
Indicationa | For the treatment for patients with ALS | To slow the loss of function in patients with ALS, as measured by the ALSFRS-R | May extend survival and/or time to tracheostomy in some patients with ALS |
Route of administration | Oral (or via feeding tube) | IV infusion | Oral |
Recommended dosage | 1 sachet daily for the first 3 weeks, then 1 sachet twice daily thereafter One sachet contains 3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine. | 60 mg administered via IV infusion over a 60-minute period according to the following schedule:
Each infusion bag contains 30 mg/100 mL (0.3 mg/mL) of edaravone. | One 50 mg tablet every 12 hours. To be taken at least 1 hour before or 2 hours after a meal to avoid food-related decrease in bioavailability. |
Serious adverse events or safety issues |
| General: Hypersensitivity reactions, sulfite allergic reactions. |
|
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
aHealth Canada–approved indication.
Source: Health Canada product monographs for PB-TURSO,16 Radicava (2021),24 and Mylan-Riluzole (2010).25
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full patient group submissions are included in the Stakeholder Input section at the end of this report.
CADTH received 2 patient group submissions for the review of PB-TURSO. ALS Canada conducted an online survey of 629 patients and caregivers from Canada, the US, the UK, Israel, and the Netherlands between November 10 and November 24, 2021. A second patient group, ALS Action Canada, collected patients’ experiences and opinions through email communications and Zoom meetings from members of its organization living in British Columbia, Alberta, Ontario, and Nova Scotia.
Respondents felt that ALS had the most impact on mobility, motor function, fatigue, breathing, speech, and swallowing. It was also emphasized how ALS negatively impacted patients’ independence with performing activities of daily living, quality of life, and social activities. For caregivers, the aspects of life that were most negatively affected were travel options, family life, and emotional and psychological well-being. Patients also spent several hours each day on exercises for legs and arms and breathing, receiving treatments (drugs and supplements), and attending medical appointments. As ALS progresses and patients are faced with new challenges, the caregiver becomes increasingly important; however, finding qualified full-time support can be challenging.
Some patients felt that currently available medications (i.e., edaravone and riluzole) appeared to slow disease progression, help maintain motor function, and increase survival. Key challenges with these medications included side effects, limited access, affordability, and administration. A limited number of patients who had access to PB-TURSO (through clinical trials, Health Canada’s Special Access Program, or the sponsor’s compassionate program) felt that slowed disease progression and maintained motor function were the main benefits, although some said it was too soon to comment on the impact. A few respondents reported side effects that are mostly gastrointestinal-related as well as taste disturbances.
Patients and caregivers emphasized the importance of treatments that are simple to administer, significantly slow progression, help patients maintain their independence, reverse symptoms, and increase survival.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of ALS.
Unmet Needs
The clinical expert CADTH consulted for this review highlighted that there are no available treatments that stop or reverse disease progression, including neurologic decline, and that current treatments demonstrate very modest benefits with slowing ALS progression. It was emphasized that ALS is a terminal disease, and all patients will progress despite available medications. Strict criteria for the reimbursement of edaravone have limited the number of patients who can use the medication and it was noted that at the time of diagnosis, most patients are ineligible due to poor FVC measurements or ALSFRS-R scores.
Place in Therapy
The clinical expert indicated that due to the progressive nature of ALS and low survival beyond 5 years after diagnosis, it is reasonable to target all potential pathological pathways as early as possible. Riluzole, edaravone, and PB-TURSO act on different pathways and targets in the body, thus supporting the possibility of using all 3 medications simultaneously. Typically, riluzole is the first treatment prescribed to patients with ALS and it is expected that clinicians would discuss the use of edaravone or PB-TURSO if available and if the patient was eligible for reimbursement. When using multiple therapies, there is usually a 1-week to 2-week interval between starting a treatment and adding another to assess side effects. The clinical expert explained that due to the nonhomogeneous loss of motor neurons with ALS, it is important to start treatment early to spare healthy neurons and preserve muscle function.
Patient Population
The clinical expert noted that all patients diagnosed with ALS (either by the El Escorial criteria or Gold Coast criteria) would be suitable for treatment with PB-TURSO. It was clarified that the Gold Coast criteria are a better fit with how clinicians confirm a diagnosis of ALS, being both sensitive and specific, compared to the El Escorial criteria. The latter method uses categories of “possible” and “probable” which, although they are confirmed diagnoses of ALS, may suggest doubt due to the connotations associated with the words.
One of the challenges with diagnosing ALS is that there is no single diagnostic biomarker for the disease. Typically, neurologists confirm diagnosis through medical history, a physical exam, and electromyography testing, and by excluding other diagnoses.
Assessing Response to Treatment
Per input from the clinical expert, response to treatment may not be strictly defined since the goal of treatment is to delay or prevent disease progression by slowing the degeneration of motor neurons. Additionally, progression is individual to each patient, which makes monitoring outcomes difficult due to disease heterogeneity. There are no tools or biomarkers available to clinicians that can be used to definitively measure whether a patient is responding to a medication since all patients experience disease progression regardless of treatment. Per Canadian guidelines, patients should be routinely monitored every 3 months to 4 months.
Discontinuing Treatment
The clinical expert was of the opinion that patients with advanced disease may not derive benefit from treatment with PB-TURSO, though how “advanced disease” is defined may vary among clinicians and differ between clinicians and patients. For instance, the clinical expert suggested that patients with advanced disease would include those who are fully dependent for their activities of daily living. Furthermore, it was indicated that treatment discontinuation could be considered for patients with full dependency for walking, transfers, and nutrition. Changes to a patient’s goals of care or a desire to discontinue a medication should also be considered.
Prescribing Conditions
A neurologist or physiatrist with experience caring for patients with ALS would be the most appropriate to prescribe PB-TURSO, though all patients should have a multidisciplinary care team and be followed by an ALS clinic.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by a clinician group. The full clinician group submission is included in the Stakeholder Input section at the end of this report.
Input was received from CALS, which consisted of 10 CALS members from across Canada. The clinician group input was similar to that given by the clinical expert consulted by CADTH.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation question | Clinical expert response |
|---|---|
Relevant comparators | |
Patients were randomized 2:1 to PB-TURSO or matching placebo. Patients could remain on riluzole and/or initiate or remain on edaravone. Placebo is likely an appropriate comparator in this treatment space, as PB-TURSO could be considered early on in the disease course and could be considered as an add-on to riluzole or edaravone. Can CDEC comment on the relevant comparator? | The clinical expert agreed that placebo was the appropriate comparator since PB-TURSO is expected to be an add-on treatment to the current standard of care (riluzole and/or edaravone). |
In CENTAUR, a group of patients was allowed to initiate edaravone as well as PB-TURSO. For those patients who were newly initiated on edaravone, it is unclear what benefit was derived from edaravone vs. PB-TURSO. | For CDEC consideration. |
Considerations for initiation | |
Clinical trials used the revised El Escorial criteria; it is noted that these are relevant for clinical trials, but are not widely used in clinical practice. The CENTAUR study enrolled patients with definite ALS, based on the El Escorial criteria. The sponsor’s reimbursement request does not restrict reimbursement only to patients with definite ALS as per the El Escorial criteria. | According to the clinical expert, the use of definite ALS by the El Escorial criteria was based on clinical trial design decisions (to determine a rapid answer as to whether the drug was potentially effective). By restricting the inclusion criteria to a definite diagnosis of ALS by the El Escorial criteria and to being 18 months or less from ALS symptom onset, the trial would be restricted to patients who progress relatively rapidly. Restricting a trial to rapidly progressive patients would mean that less time would be required to demonstrate a change in rate of progression, as compared to a wider population of patients with ALS. There is no physiologic or pharmacological reason to predict that patients at other levels of the El Escorial diagnostic criteria (i.e., “probable” or “possible”) would not respond to the treatment. |
The sponsor notes that earlier access results in better treatment outcomes, and patients should not have to try alternative therapies before accessing PB-TURSO. All jurisdictions list edaravone with criteria. Riluzole is the other marketed ALS treatment. Some jurisdictions list it with criteria, some do not list it, and still others list it as a full benefit. Five of the 6 jurisdictions listing with criteria require an FVC of more than 60%. | According to the clinical expert, it is expected that PB-TURSO would be offered as an add-on therapy in addition to riluzole and/or edaravone. |
The CENTAUR trial looked at a measure called SVC. How does SVC relate to FVC? Is SVC used in clinical practice? | The clinical expert stated that it is not standard practice to measure SVC in ALS clinics in Canada, though its use may vary among clinicians. According to the expert, it is more common to measure FVC to assess respiratory function in clinical practice. It was also noted that SVC is felt to be generally equivalent to FVC. |
The reimbursement criteria for edaravone and riluzole require FVC scores above a certain threshold at the time of initiation. The edaravone reimbursement criteria require patients to have probable or definite ALS. The CENTAUR trial for PB-TURSO enrolled patients with definite ALS. | For CDEC consideration. |
Considerations for continuation or renewal | |
ALS disease progression is measured using the ALSFRS-R, which consists of 4 domains measuring 12 different activities of daily living in each domain. Each item score ranges from 0 to 4 with a maximum score of 48, which indicates normal function. As the disease is progressive in nature, what would be a reasonable reduction in the ALSFRS-R score that signifies benefit with treatment at 6 months? At the end of the 6-month randomized study period, there was a difference of 2.32 points between groups in the ALSFRS-R estimate; however, both groups experienced decline in the ALSFRS-R estimate. It will be difficult to withdraw therapy if patients are deteriorating despite therapy. | Due to the progressive nature of ALS, all patients are expected to decline over the course of the disease. Currently, there are no available therapies to stop or reverse disease progression and available treatments only show modest effects in slowing progression. The average decline among patients with ALS is approximately 1 point per month during the course of the disease, although decline may slow toward the end stages of the disease. It was noted that patients will decline faster or slower than this average without treatment, which makes comparisons to the average rate of decline challenging. The clinical expert explained that clinicians are unlikely to have an accurate reading of a patient’s rate of decline based on ALSFRS-R scores before treatment and while on treatment to compare on an individual patient level. |
Considerations for discontinuation | |
Clearly defined discontinuation criteria would be helpful. Given the progressive nature of the disease, it will be difficult to discontinue coverage of therapy. | Per “Canadian best practice recommendations for the management of amyotrophic lateral sclerosis,” patients with ALS should be routinely monitored every 3 months to 4 months.4 PB-TURSO could be continued until the goals of treatment change and become more palliative, or the patient requires total care or near-continuous ventilation as this indicates there are few surviving motor neurons and the drug no longer provides benefit. Specific examples for discontinuation could include: • BiPAP support for > 20 hours per day • wheelchair dependency and the need to be fed by a caregiver (i.e., dependent for all activities of daily living). |
In the CENTAUR trial, patients were treated within 13.5 months of diagnosis. Is it necessary to start therapy early for patients to demonstrate a beneficial response? | Early intervention is important to prevent motor neuron death toward the goals of slowing progression of ALS and preserving muscle function. |
Consider consistency with discontinuation criteria associated with other drugs reviewed by CADTH in the same therapeutic space. See previous comment regarding renewal criteria. | For CDEC consideration. |
Considerations for prescribing | |
The dosing regimen for PB-TURSO may be preferred to that for edaravone, which requires an IV infusion to be given over a 60-minute period according to the following schedule:
| The clinical expert agreed with this statement. |
PB-TURSO can be administered orally or per feeding tube, which is an important consideration in patients with ALS. | The clinical expert agreed with this statement. |
Some jurisdictions may have limited access to specialists experienced in diagnosing ALS and this may lead to delay in treatment initiation. In the CENTAUR trial, patients had an average of 13.5 months since symptom onset and 6 months since diagnosis before they were enrolled in the study. In some jurisdictions, it is likely that access to specialists and diagnosis exceeds this time frame. | The clinical expert stated that most jurisdictions in Canada should be able to confirm a diagnosis of ALS within 12 months to 18 months. Access to specialist diagnosis should not be an issue, particularly since “Canadian best practice recommendations for the management of amyotrophic lateral sclerosis” has required an appointment for confirmation of a diagnosis of ALS within 4 weeks of a referral to a specialist.4 |
In the CENTAUR trial, patients could remain on therapy with riluzole and/or edaravone. | It is expected that PB-TURSO would be offered as add-on therapy in addition to riluzole and/or edaravone. |
Edaravone criteria specify that patients should have probable or definite ALS. The CENTAUR study enrolled patients with definite ALS. Conditions for coverage of edaravone require patients to have scores of at least 2 points on each item of the ALSFRS-R, have ALS symptoms for 2 years or less, and have an FVC of greater than or equal to 80% predicted. In addition, patients must not require permanent noninvasive or invasive ventilation. The consistency of criteria depends on what the place in therapy for this product is determined to be. | The clinical expert consulted on this review emphasized that there are no available treatments that have a proven ability to stop disease progression. Those that are available only modestly slow disease progression. Patients should not have to demonstrate treatment failure before being able to access another therapy. |
Generalizability | |
The clinical trial was relatively small given the incidence of the disease. Approximately 1 in 50,000 people in Canada is diagnosed with ALS each year. A total of 137 patients were enrolled in the CENTAUR study. The study was conducted across a number of states in the US and enrolled patients with definite ALS based on the revised El Escorial criteria. Patients with probable and possible ALS may also seek treatment given the limited treatment options that exist. The RCT did not show any survival benefit. Survival was assessed in the open-label extension. It would be helpful if CDEC could comment on the methodology. The RCT is a 6-month, phase II trial, with a primary outcome of assessing the safety and tolerability of PB-TURSO. The primary efficacy end point was ALSFRS-R total score. | The clinical expert explained that it is highly unlikely that a 6-month trial could demonstrate survival benefit due to the natural history of the disease. The clinical expert added that it is much more likely that a survival benefit would be seen over a 12-month or 18-month trial. |
Patients will advocate for early access to the drug as 80% of patients with ALS die within 2 years to 5 years of being diagnosed. | The clinical expert agreed with this statement. |
System and economic issues | |
The duration of treatment was assumed by the sponsor to be the same as edaravone in the budget impact analysis. The sponsor is claiming that PB-TURSO has a survival benefit, whereas edaravone does not. Therefore, treatment duration should not be equivalent. The sponsor’s budget impact analysis assumed that all patients would be receiving riluzole. This is unlikely to be the case. | The clinical expert indicated that the majority of patients will be on riluzole. Some patients who do not tolerate riluzole could be tried on PB-TURSO. However, it would be extremely unusual to have a patient refuse riluzole or have a medical contraindication to riluzole and be prescribed PB-TURSO. |
PB-TURSO will likely be used as add-on therapy (in addition to riluzole and edaravone), thus increasing the overall treatment cost of ALS significantly. | The clinical expert agreed with this statement. |
There may be some patients who are on the product through the SAP at the time of Health Canada approval. These patients may have to transition to another method of coverage once the drug is approved by Health Canada. | The clinical expert agreed with this statement. |
Generic versions of riluzole are now available. These are listed in at least 1 jurisdiction at 35% of the price of the brand-name drug. | For CDEC consideration. |
What is an appropriate time horizon to assess cost-effectiveness of this drug? The sponsor used a Markov state-transition model to describe the progression of ALS over a lifetime horizon (10 years). Is 10 years an appropriate time frame? | The clinical expert felt 10 years would be a reasonable time frame. |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; BiPAP = bilevel positive airway pressure; CDEC = CADTH Canadian Drug Expert Committee; FVC = forced vital capacity; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RCT = randomized controlled trial; SAP = Special Access Program; SVC = slow vital capacity; vs. = versus.
Clinical Evidence
The clinical evidence included in the review of PB-TURSO (Albrioza) is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of PB-TURSO (3 g sodium phenylbutyrate/1 g ursodoxicoltaurine) sachet, oral administration, for the treatment of patients with ALS
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented in Table 5 was established before the granting of an NOC from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients with ALS Subgroups:
|
Interventiona | Sodium phenylbutyrate-ursodoxicoltaurine orally or via feeding tube as 1 sachet once daily for the first 3 weeks, then 1 sachet twice daily thereafter (1 sachet = 3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine) |
Comparatora |
|
Outcomes | Efficacy outcomes
Harms outcomes
|
Study designs | Published and unpublished phase II, phase III, and phase IV RCTs |
AE = adverse event; ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; FVC = forced vital capacity; HRQoL = health-related quality of life; RCT = randomized controlled trial; SAE = serious adverse event; SVC = slow vital capacity; WDAE = withdrawal due to adverse event; vs. = versus.
aStandard of care may include a background of best supportive care with or without edaravone and/or riluzole.
bThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.26
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were amx0035 (sodium phenylbutyrate and ursodoxicoltaurine). Clinical trials registries were searched: the US National Institutes of Health’s ClinicalTrials.gov, the WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on December 17, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on April 27, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.27 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Findings from the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study has been summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Included Studies — CENTAUR Trial
Factor | CENTAUR trial |
|---|---|
Designs and populations | |
Study design | Phase II, DB, RCT with placebo control and parallel groups |
Locations | 25 centres in the US |
Patient enrolment dates | June 22, 2017, to September 25, 2019 |
Randomized (N) | N = 137
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Interventionc | PB-TURSO orally or via feeding tube as 1 sachet once daily for the first 3 weeks, then 1 sachet twice daily thereafter (1 sachet = 3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine) |
Comparator(s)c | Matching placebo (designed to match in size, colour, presentation, and taste) |
Duration | |
Screening phase | Up to 42 days |
Double-blind phase | 24 weeks |
Follow-up phase | 28 days |
Open-label extensiond | Up to 132 weeks |
Outcomes | |
Primary end points | Rate of change in ALSFRS-R total score |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; DB = double-blind; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RCT = randomized controlled trial; SVC = slow vital capacity.
aRevised El Escorial criteria: Clinical evidence alone by the presence of upper motor neuron, as well as lower motor neuron, signs of neurodegeneration in at least 3 of 4 regions (i.e., brainstem [bulbar cranial motor neurons], cervical, thoracic, and lumbosacral spinal cord [anterior horn motor neurons]) of the central nervous system.
bExceptions include basal cell carcinoma or successfully treated squamous cell carcinoma of the skin, cervical carcinoma in situ, prostatic carcinoma in situ, or other malignancies curatively treated and with no evidence of disease recurrence for at least 3 years.
cWith standard of care (e.g., riluzole, edaravone).
dResults from the open-label extension are presented in the Other Relevant Evidence section of the CADTH review.
Source: CENTAUR Clinical Study Report (2021),18 Paganoni et al. (2020),28 and Paganoni et al. (2021).29
Description of Studies
One pivotal study was included in the CADTH review for PB-TURSO. The CENTAUR trial was a phase II, multi-centre, DB, randomized, placebo-controlled trial to assess the safety, tolerability, and efficacy of PB-TURSO in adult patients with ALS. One sachet of medication was taken orally or via feeding tube once daily for the first 3 weeks and, if tolerated, 1 sachet was taken twice daily thereafter. The CENTAUR trial was conducted at 25 NEALS centres in the US and included 137 patients. Patients were randomized in a 2:1 ratio to receive either PB-TURSO and SOC (n = 89) or matching placebo and SOC (n = 48) for the duration of the 24-week DB treatment period. The computer-generated randomization scheme was independently developed by an unblinded study statistician and managed by PCI Pharma Services.28
The study design consisted of (1) a screening period of up to 42 days, (2) a DB treatment period of 24 weeks with study evaluations taking place every 3 weeks, and (3) a follow-up period for up to 4 weeks. Patients who discontinued early from the study were asked to return to the study site for final safety assessments. The protocol was amended to add the OLE phase and after completing the DB trial, patients could enrol in the 132-week CENTAUR-OLE study, which has been summarized in the Other Relevant Evidence section of the CADTH report.
Populations
Inclusion and Exclusion Criteria
Patients were eligible to participate in the CENTAUR trial if they were aged 18 years to 80 years (inclusive) and had definite ALS (sporadic or familial) as defined by the World Federation of Neurology–revised El Escorial criteria. Patients must have also had ALS symptom onset in the past 18 months and an SVC of greater than 60% of predicted value for their sex, height, and age. Continued SOC treatment during the trial was acceptable and patients must have been on a stable dose of riluzole for at least 30 days before screening while those on edaravone or planning to initiate edaravone were also eligible (Note: edaravone was approved for use in the US after enrolment to the CENTAUR trial had begun). Patients were excluded if they had a tracheostomy, abnormal liver function, renal insufficiency, uncontrolled hypertension, biliary disease, or a history of cholecystectomy or heart failure.
Baseline Characteristics
Patient baseline characteristics are summarized in Table 7. Overall, the mean age of patients in the CENTAUR trial was 57.5 years (SD = 9.50 years). Most patients were male (68.9%) and most patients were White (94.8%).
The del-FS before study entry was calculated as the difference resulting from the subtraction of the patient’s ALSFRS-R score at baseline from 48 points (the maximum ALSFRS-R score, which corresponds to no functional impairment), divided by the number of months from symptom onset to baseline. For the overall study population, the mean rate was 0.94 points per month (SD = 0.49 points per month) and was numerically greater in the PB-TURSO group versus the placebo group. Patients who received PB-TURSO had a shorter mean time since ALS diagnosis of 5.9 months (SD = 3.33 months) compared to patients in the placebo group at 6.3 months (SD = 3.22 months), although the mean time since onset of ALS symptoms was similar between the groups (13.5 months and 13.6 months, respectively). The proportion of patients who experienced disease onset in the limb region was lower in the PB-TURSO group (67.8%) than the placebo group (79.2%). Conversely, a greater proportion of patients in the PB-TURSO group had bulbar onset (29.9%) compared to the placebo group (20.8%). In general, mean ALSFRS-R total and domain scores were similar between the treatment groups as were mean SVC measurements. Mean ATLIS scores were numerically higher for those in the PB-TURSO group compared to the placebo group.
The use of riluzole and/or edaravone at or before study entry was overall more common in the placebo group. Most patients in the placebo group (77.1%) had experience with riluzole compared to 67.8% of patients in the PB-TURSO group. Similarly, 50% of the placebo group and 25.3% of the PB-TURSO group had experience with edaravone. Most patients had experience with 1 of the treatments (87.5% in the placebo group and 71.3% in the PB-TURSO group), and more patients in the placebo group (39.6%) had experience with both medications compared to the PB-TURSO group (21.8%).
Table 7: Summary of Baseline Characteristics — CENTAUR Trial, Modified ITT Population
Characteristic | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
Demographics | ||
Age (years) | ||
Mean (SD) | 57.6 (10.45) | 57.3 (7.56) |
Median (range) | 59.0 (31.0 to 79.0) | 57.5 (36.0 to 79.0) |
Sex, n (%) | ||
Male | 61 (70.1) | 32 (66.7) |
Female | 26 (29.9) | 16 (33.3) |
Weight (pounds), mean (SD) | 177.2 (38.66) | 175.8 (43.67) |
Race, n (%) | ||
White | 82 (94.3) | 46 (95.8) |
Asian | 2 (2.3) | 1 (2.1) |
Black | 2 (2.3) | 1 (2.1) |
Unknown | 1 (1.1) | 0 |
Baseline disease characteristics and medical history | ||
Del-FS before studya | ||
Mean (SD) | 0.95 (0.43) | 0.93 (0.60) |
Median (range) | 0.89 (0.12 to 1.94) | 0.76 (0.13 to 3.15) |
Time since ALS diagnosis (months) | ||
Mean (SD) | 5.9 (3.33) | 6.3 (3.22) |
Median (range) | 5.3 (1.3 to 15.7) | 6.0 (1.3 to 17.6) |
Time since onset of ALS symptoms (months) | ||
Mean (SD) | 13.5 (3.83) | 13.6 (3.64) |
Median (range) | 13.9 (3.0 to 19.9) | 13.3 (4.8 to 19.6) |
Concomitant medication at or before study entry, n (%) | ||
Either edaravone or riluzole | 62 (71.3) | 42 (87.5) |
Edaravone only | 3 (3.4) | 5 (10.4) |
Riluzole only | 40 (46.0) | 18 (37.5) |
Edaravone, any experience | 22 (25.3) | 24 (50.0) |
Riluzole, any experience | 59 (67.8) | 37 (77.1) |
Both edaravone and riluzole | 19 (21.8) | 19 (39.6) |
Family history of ALS, n (%) | ||
Yes | 9 (10.3) | 7 (14.6) |
No | 76 (87.4) | 38 (79.2) |
Unknown | 2 (2.3) | 3 (6.3) |
Site of onset, n (%) | ||
Limb | 59 (67.8) | 38 (79.2) |
Bulbar | 26 (29.9) | 10 (20.8) |
Other | 2 (2.3) | 0 |
ALSFRS-R total score, mean (SD) | 35.7 (5.78) | 36.7 (5.08) |
ALSFRS-R breathing | 10.6 (1.92) | 11.0 (1.80) |
ALSFRS-R bulbar | 9.5 (2.40) | 10.0 (2.60) |
ALSFRS-R fine motor | 8.0 (2.69) | 8.0 (2.63) |
ALSFRS-R gross motor | 7.5 (2.84) | 7.6 (2.62) |
ATLIS total score, mean (SD) | 56.83 (20.08) | 53.92 (20.94) |
ATLIS upper extremities | 54.76 (24.40) | 51.44 (25.22) |
ATLIS lower extremities | 57.65 (24.89) | 57.10 (25.81) |
SVC (% predicted) | ||
Mean (SD) | 83.6 (18.17) | 83.9 (15.92) |
Median (range) | 82.0 (46.0 to 142.0) | 84.0 (52.0 to 125.0) |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; del-FS = delta-functional scale; ITT = intention to treat; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SOC = standard of care; SVC = slow vital capacity.
aPatient’s rate of decline before study = (48 – ALSFRS-R at baseline) ÷ time in months from symptom onset to baseline. The maximum score on the ALSFRS-R is 48 points.
Source: CENTAUR Clinical Study Report (2021).18
Interventions
In the CENTAUR trial, patients were randomized 2:1 to receive either PB-TURSO or matching placebo. PB-TURSO was supplied as a powder in individually packaged sachets, each containing 3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine. The contents of 1 sachet were to be reconstituted in 250 mL of room-temperature water, stirred vigorously, and taken orally or administered via feeding tube within 1 hour of preparation. Patients were made aware of the strong bitter flavour of the medication and were able to mask the taste by taking flavoured breath fresheners, consuming a snack or meal, or drinking milk. Patients were to consume 1 sachet daily (e.g., morning) for the first 3 weeks and, if tolerated, 1 sachet twice daily thereafter (e.g., morning and evening) for the duration of the study. The placebo sachets and contents were designed to match the active treatment in size, colour, presentation, and taste.
Concomitant medications, supplements, and assistive devices were recorded in the electronic case report form. Patients were able to continue SOC therapies for ALS if they were on a stable dose of riluzole and/or had started or were planning to start edaravone during the trial. Antacids containing aluminum hydroxide or aluminum oxide were not permitted within 2 hours of taking the study medication as they could inhibit absorption of ursodoxicoltaurine. Other prohibited medications included histone deacetylase inhibitors (e.g., valproate, vorinostat, romidepsin, chidamide, panobinostat, lithium, butyrate, suramin), probenecid, and bile acid sequestrants (i.e., cholestyramine, Questran, Welchol, Colestid, or Prevalite).
It was noted that early in the study, there was an error in kit distribution where the planned shipping sequence impacted the first 26 patients enrolled in the study (the first 17 patients received PB-TURSO and the next 9 patients received placebo).28 It was then corrected and a sensitivity analysis was performed using the mITT population wherein 25 patients were removed (1 patient did not have a secondary efficacy assessment and was not part of the mITT population). The sensitivity analysis assessed the effect on the primary efficacy end point. Patients, study site personnel, and the sponsor remained blinded to the treatment assignments until after the study database was locked.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized as follows. A detailed description and critical appraisal of the outcome measures are provided in Appendix 3.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | CENTAUR study |
|---|---|
Overall survival | Secondary |
Assessment of motor function using a validated scale (e.g., ALSFRS-R, ATLIS) | Primary/secondary/exploratory |
Mobility | Primary/exploratory |
Muscle strength | Secondary |
Use of feeding tube | Primary/exploratory |
Assessment of respiratory function | Primary/secondary/exploratory |
Determination of vital capacity | Secondary |
Use of respirator | Secondary |
Time to tracheostomy | Secondary |
Tracheostomy-free survival | Not reporteda |
HRQoL | Not reporteda |
Caregiver burden | Not reporteda |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; HRQoL = health-related quality of life.
aOutcome measure was not reported as an outcome in the CENTAUR trial.
Efficacy Outcomes
The ALSFRS-R is a questionnaire-based scale designed to allow clinicians to quickly measure physical function regarding activities of daily living for patients with ALS.17,30,31 Its use is supported by the FDA as a measure of treatment efficacy in ALS studies.32 The ALSFRS-R was revised from the original Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) questionnaire to include the assessment of respiratory dysfunction (e.g., dyspnea, orthopnea, respiratory insufficiency). Answers are rated on a 5-point scale (from 0 = total loss of function to 4 = no loss of function) to assess a patient’s ability and independence in 12 areas. Higher scores indicate better performance with a maximum overall score of 48 points. The 12 areas are grouped into 4 domains, each with 3 questions: bulbar (speech, salivation, swallowing), fine motor (handwriting, using utensils, dressing, and hygiene), gross motor (turning in bed, walking, climbing stairs), and breathing (dyspnea, orthopnea, respiratory insufficiency). There is also a single question (answered “yes” or “no”) assessing a patient’s use of a feeding tube for more than 50% of their daily nutrition. Whenever feasible, the same NEALS-certified evaluator administered the ALSFRS-R questionnaire for the duration of the study. Acceptable internal consistency and test-retest reliability have been demonstrated for the total score.31 The domain scores did not meet the threshold for acceptable internal consistency and not all had acceptable test-retest reliability.31 No evidence of responsiveness to change was identified from the literature search. Construct validity of the total score has been demonstrated with the Sickness Impact Profile (a general assessment of health), but not for the breathing domain score when compared with FVC.31 The clinical expert consulted for this review considered a 2-point difference between groups in mean change over a 6-month period to be clinically meaningful. Experts in the treatment of ALS from the NEALS consortium suggested that a change of 20% to 25% in the ALSFRS-R slope was clinically meaningful.17 From a patient perspective, a 9-point decrease in ALSFRS-R score was considered clinically meaningful, though this was based on only 30 individuals and over a 6-month time frame.33
The ATLIS device was used to measure a patient’s isometric muscle strength. In total, 12 muscle areas are assessed of which half are upper (right and left grip strength, elbow extension, and elbow flexion) and half are lower (right and left ankle dorsiflexion, knee extension, and knee flexion). The device uses a fixed load cell and wireless dynamometer with standard positions to improve reproducibility and reliability between tests. In the CENTAUR study, each maneuver was performed twice, with a third attempt if the first 2 differed by more than 15%. Measurements were standardized to PPN strength based on sex, age, weight, and height, and values were presented as upper, lower, and total ATLIS PPN values. If a patient had no movement in a limb or was too weak to perform a test, the score was recorded as 0. If a patient could move a limb but was unable to complete testing for another reason, the data were considered missing. The highest score from each attempt of a man oeuvre was used for the analysis. Upper extremity ATLIS scores were the average of the 6 standardized measures, but only if at least 4 of the 6 items were available. Lower extremity ATLIS scores were calculated in the same manner. The total ATLIS score was the average of the upper and lower extremity ATLIS scores and required that both subscores were available. It has been noted that there is a training effect for this test. Whenever feasible, the same NEALS-certified evaluator administered the ATLIS test for the duration of the study. Validity was demonstrated when comparing the ATLIS and Tufts Quantitative Neuromuscular Exam (the gold standard test for assessing strength) among healthy adults.32 Test-retest reliability and interrater reliability were acceptable for ATLIS measurements among patients with ALS.32 The longitudinal responsiveness of the ATLIS as ALS progresses is under investigation.32 No MID estimates were identified from the literature search.
Respiratory function was measured using SVC (PPN) in the upright position. Vital capacity was measured using conventional spirometers. Each patient performed at least 3 trials; however, if the variability between the 2 highest attempts was 10% or greater, up to 5 attempts could be made. The 3 best attempts were recorded, and the highest SVC measure was used for analysis. Values were standardized to PPN based on sex, age, weight, and height. Whenever feasible, the same NEALS-certified evaluator administered the SVC tests for the duration of the study. Patients with upper motor neuron dysfunction and bulbar weakness may not be able to perform the FVC maneuver since it can cause glottal closure resulting in inaccurate FVC measures; this does not typically occur with SVC manoeuvres.34 For this reason and to improve reproducibility, the FDA supports the use of SVC measures in ALS trials.34 Conversely, the clinical expert consulted for this review stated that SVC measures can be harder for patients to perform and it can be difficult to get reproducible results. The clinical expert also indicated that FVC measures are more commonly used in Canadian clinical practice than SVC measures, though the latter may be used in research settings. No evidence for the assessment of the psychometric properties or MID were identified from the literature search.
Additional details on the ALSFRS-R and ATLIS are provided in Appendix 3.
Survival outcomes were assessed as a secondary end point as individual events of death, tracheostomy, or permanent assisted ventilation. Permanent assisted ventilation was defined as noninvasive mechanical ventilation for more than 22 hours per day for more than 7 days. Analyses were performed for death or death equivalent (time to death, tracheostomy, or permanent assisted ventilation) as individual events, then as a composite of the events where any 1 of the events was considered a failure.
Harms Outcomes
The incidence and seriousness of AEs, WDAEs, and deaths were reported for the safety population during the DB phase and follow-up period. AEs, SAEs, and protocol-defined notable harms were described based on the Medical Dictionary for Regulatory Activities’ Preferred Term and associated System Organ Class version 16.1. Notable harms from the CADTH review protocol included gastrointestinal AEs, neurologic AEs, respiratory AEs, and taste disturbance. Symptoms of ALS progression were recorded as AEs. Investigational sites were instructed to not capture the bad taste of medication as an AE, and instead, record challenges with oral delivery of the study drug if they had a clinically untoward effect (e.g., burning, vomiting, anxiety).
Statistical Analysis
The statistical analysis of efficacy end points conducted in the CENTAUR trial is summarized in Table 9.
Table 9: Statistical Analysis of Efficacy End Points — CENTAUR Trial
End point | Statistical model | Adjustment factor | Sensitivity analysis |
|---|---|---|---|
CENTAUR trial | |||
Rate of change in the ALSFRS-R total score | MMRM | Age and del-FS before the study |
|
Rate of change in isometric muscle strength as measured by the ATLIS total score | MMRM | Age, del-FS before the study, and the change in ATLIS total score interacting with time | None |
Rate of change in SVC in the upright position | MMRM | Age, del-FS before the study, and the change in SVC interacting with time | None |
Rates of survival (defined as death, tracheostomy, and permanent assisted ventilation) | Cox proportional hazards ANCOVA | Del-FS and age at baseline for survival | None |
Rate of change in the subscale scores of the ALSFRS-R (i.e., bulbar, fine motor, gross motor, and breathing) | MMRM | Age, del-FS before the study, and the change in ALSFRS-R subscale scores interacting with time | None |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ANCOVA = analysis of covariance; ATLIS = Accurate Test of Limb Isometric Strength; del-FS = delta-functional scale; MMRM = mixed model of repeated measures; SVC = slow vital capacity.
Source: CENTAUR Clinical Study Report (2021).18
Primary Outcome Analysis
The primary efficacy outcome for the CENTAUR trial was the rate of change (slope) of the ALSFRS-R total score from baseline to week 24. Continuous variables were presented using descriptive statistics (e.g., number of patients, mean, SD, median, range) while categorical variables were reported as frequencies and percentages. The least squares mean and SE were estimated for the treatment groups at each scheduled time point. The difference between active treatment and placebo slopes was calculated along with the 95% CI and P value. Post-baseline assessments occurred approximately every 3 weeks in the clinic or by phone and were included in the efficacy analysis, though only results from week 24 were summarized in the CADTH review. Efficacy analyses were performed on the mITT population while analyses of harms was performed on the safety population. Post-hoc analyses of efficacy outcomes were performed using the ITT population, which was the same as the safety population.
The primary safety end point was to confirm the safety and tolerability of the fixed-dose combination of PB-TURSO. TEAEs that occurred on or after the start of dosing, including those consistent with disease progression, were reported based on System Organ Class and Preferred Term using the Medical Dictionary for Regulatory Activities version 16.1 for the safety population. SAEs, AEs leading to patient withdrawal, and deaths have been summarized in the CADTH report. If the proportion of treatment failures (the percentage of patients who discontinued medication due to an AE) was less than 40% with 80% confidence, 1-sided, the dose of PB-TURSO was considered tolerable. PB-TURSO was considered tolerable at a threshold of less than 40% discontinuations due to AEs or if fewer than 30 of 88 PB-TURSO–treated patients discontinued during the 24 weeks of treatment.
Power Calculation and Determination of Sample Size
For the efficacy analyses, the investigators based the study power calculations on patient criteria from the Pooled Resource Open-Access ALS Clinical Trials database35 and data from the first 6 months of a study on ceftriaxone (an ALS trial).28 Using a shared-baseline, mixed-effects analysis, and 2:1 randomization scheme, it was estimated that enrolling 131 patients for 6 months would provide 80% power to detect a 30% treatment effect in the rate of change in the ALSFRS-R total score when tested at a 2-sided alpha of 0.1.
Statistical Test or Model
A shared-baseline, mixed model of repeated measures (MMRM) analysis with covariates of age and del-FS before the study was used to compare PB-TURSO treatment and placebo groups (Table 9). The mixed-effects model accounts for variance between patients and deviation within patients from their average rate of decline.
It was noted in the sponsor’s submission that analyses have shown ALS has a linear disease progression; thus, the del-FS before study entry is a strong predictor of future disease progression. A patient’s rate of decline since symptom onset was included as a covariate in the statistical model and was calculated based on the ALSFRS-R score and time since ALS symptom onset (i.e., rate = [48 – ALSFRS-R at baseline] ÷ time in months from symptom onset to baseline; where 48 is the maximum ALSFRS-R total score). Time was a quantitative measure with the baseline or randomization visit representing day 0 and subsequent visits measured as days since randomization.
Although ALS appears to have a linear progression over time, due to the unknown effects of PB-TURSO, linearity was not immediately assumed in this study. To confirm linearity, a modified version of the statistical model that included quadratic terms for time was used. If the quadratic terms for time were nonsignificant (P > 0.10), linearity was assumed, and the linear primary model was used for the analysis. If any interaction term was significant (P < 0.10), the modified quadratic version of the model was used for the analysis.
Missing Data
In the MMRM model, it was assumed that missing data (other than death or death equivalent events) were missing at random. Sensitivity analyses were conducted for the efficacy end points to address missing data and are described as follows.
Subgroup Analyses
No subgroup analyses were performed for the CENTAUR trial.
Post-Hoc Analyses and Sensitivity Analyses
The primary analysis assumed a shared baseline between the PB-TURSO and placebo groups. An analysis using a change from baseline approach was performed on continuous outcomes for the mITT population.
To assess the impact of missing data, sensitivity analyses were conducted for the ALSFRS-R, ATLIS, and SVC measures. To explore the missing at random assumption, a sensitivity analysis (a multiple imputation model for missing not at random) imputed data for patients who discontinued for any reason. Patients in the placebo group had values imputed in a linear trajectory while those in the PB-TURSO group had values imputed in a linear trajectory after subtracting the difference in average slope between the treatment groups. In a second set of analyses (left censoring), values were censored by an intercurrent event of death. It was assumed that these censored values were lower than all observed values and the contribution to the likelihood for each patient was the product of the density of all the observed outcomes and of the conditional distribution of the censored outcomes. Fixed variable starting values were the point estimates from the primary analysis and variance parameters had a lower bound of 0.
Two patients were excluded from the mITT population for not having post-baseline efficacy assessments. An analysis of the ITT population included these 2 patients, who were in the PB-TURSO group.
The effect of concomitant medications on rate of progression was evaluated in a post-hoc analysis. The main efficacy model was used to compare efficacy outcome scores over time and included terms for maximum time on concomitant medication. Subgroups of interest included patients who had experience with edaravone only, riluzole only, edaravone or riluzole, and edaravone and riluzole. For the edaravone or riluzole group, the maximum time on either treatment was used for the analysis. A P value of less than 0.10 for the 3-way interaction term after correcting for all other factors was to be considered significant, indicating a significant interaction between time on concomitant medication and treatment over time.
Hierarchical Testing Strategy
Type I error was controlled using the hierarchical testing strategy. This was used for the primary and secondary end points and performed at a 2-sided 5% significance level. Once testing for statistical significance failed, all subsequent results were reported with nominal P values. Testing was conducted in the following order:
the rate of change of isometric muscle strength, as measured by the ATLIS device
the rate of change in plasma concentration of phosphorylated axonal neurofilament-H, a potential marker of neuronal death
the rate of change in SVC in the upright position, a measure of lung function
the rates of survival (defined as death, tracheostomy, or permanent assisted ventilation [noninvasive mechanical ventilation for more than 22 hours per day for more than 7 days]), and hospitalization
a concentration-response model of PB-TURSO at steady state after administration of PB-TURSO 4 g twice daily
the rate of 18 kDa translocator protein uptake, as measured by a PET scan (Magnetic Resonance-PET substudy).
Secondary and Exploratory Outcome Analyses
The same MMRM model used for the primary efficacy analysis was used for continuous secondary and exploratory efficacy outcomes. Covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time were used in the model to compare treatment groups (Table 9). A calculation similar to that used for estimating the del-FS before the study was used for the secondary and exploratory efficacy outcomes (i.e., rate = [ceiling maximum efficacy score – efficacy score at baseline] ÷ time in months from symptom onset to baseline). Where there was no defined maximum value (e.g., SVC and ATLIS scores), the observed maximum among all enrolled patients was used.
Survival analyses used a Cox proportional hazards model with baseline age and del-FS as covariates. Three survival events were considered: death, tracheostomy (irrespective of reason for tracheostomy), and permanent assisted ventilation (noninvasive mechanical ventilation for more than 22 hours per day for more than 7 days). An event must have occurred during the 24 weeks of treatment or 4 weeks of follow-up to be considered for the analysis. A combined survival analysis for time to “death or equivalent” was performed in which any 1 of the 3 events was an event and time to the first occurrence of an event was analyzed. This analysis included patients who withdrew from the study but whose death occurred during the 24-week study. An HR of greater than 1 indicated a covariate was positively associated with the event probability and negatively associated with the length of survival while an HR of less than 1 indicated a reduction in the hazard. To assess if a covariate had a statistically significant impact on the hazard of an event, a likelihood ratio test was used. Where a P value of less than 0.05 was found, the HR was examined to assess which treatment increased the hazard.
Analysis Populations
The mITT population (N = 135) included all patients who received at least 1 dose of study medication and had at least 1 post-baseline total ALSFRS-R score. Two patients who did not have post-baseline assessments were excluded from the mITT population. Patients were analyzed based on the medication they received. The mITT population was the primary population for efficacy analysis.
The safety population (N = 137) included all patients who received at least 1 dose of study medication and patients were analyzed based on the medication received. The safety population was the primary population for safety analysis, medical history, and concomitant medication use. The safety and ITT populations were equivalent.
The PP population (N = 134) included all patients in the mITT population who took the assigned medication per the study protocol and who did not have major protocol deviations that would exclude them from the PP analysis as determined by committee before database lock. Inclusion in the PP population was assessed by visit and patients remained in this population until either a major protocol deviation occurred or they had not taken the study medication for at least 30 days. Clinically major protocol deviations were decided on a case-by-case basis by the principal investigator and the sponsor’s medical director without knowing the treatment assignment and before unblinding.
Results
Patient Disposition
Patient disposition has been summarized in Table 10. In total, 177 individuals were screened for participation in the study, of whom 137 were randomized in a 2:1 ratio to receive PB-TURSO (n = 89) or matching placebo (n = 48). The major reasons for screening failures included not meeting entry criteria (30 [75%] patients), patient withdrawing consent during screening (4 [10%] patients), and other reasons (6 [15%] patients). The proportion of patients who completed the 24-week treatment was similar between the groups: 75.3% of those who received PB-TURSO and 79.2% of those who received placebo. The most common reason for discontinuation was due to AEs (12.4% of patients from the PB-TURSO group and 6.3% of patients from the placebo group) followed by disease progression (5.6% of patients from the PB-TURSO group and 4.2% of patients from the placebo group). Death as a reason for discontinuation was reported for 3 (3.4%) patients in the PB-TURSO group and 2 (4.2%) patients in the placebo group during the 24-week trial. Physician decision, patient decision, and loss to follow-up were the other reasons for discontinuation and were less commonly reported.
Table 10: Patient Disposition — CENTAUR Trial
Patient disposition | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC | Placebo + SOC | |
Screened, N | 177 | |
Randomized, N (%) | 89 | 48 |
Completed study, n (%)a | 67 (75.3) | 38 (79.2) |
Discontinued from study, n (%)a | 22 (24.7) | 10 (20.8) |
Reason for discontinuation, n (%)a | ||
Withdrawal by patient | 17 (19.1) | 6 (12.5) |
Adverse event | 11 (12.4) | 3 (6.3) |
Disease progression | 5 (5.6) | 2 (4.2) |
Patient decision | 1 (1.1) | 1 (2.1) |
Deathb or death equivalentc | 3 (3.4) | 2 (4.2) |
Physician decision | 2 (2.2) | 1 (2.1) |
Lost to follow-up | 0 | 1 (2.1) |
Discontinued study treatment before study completion, n (%)a | 7 (7.9) | 1 (2.1) |
ITT, n (%) | 89 (100) | 48 (100) |
mITT, n (%) | 87 (97.8) | 48 (100) |
Safety, n (%) | 89 (100) | 48 (100) |
PP, n (%) | 86 (96.6) | 48 (100) |
ITT = intention-to-treat; mITT = modified intention-to-treat; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; PP = per protocol; SOC = standard of care.
aBased on safety population.
bA total of 7 patients died during the 24-week DB study (5 patients who received PB-TURSO and 2 patients who received placebo). However, 3 patients discontinued participation before death (2 patients who received PB-TURSO and 1 patient who received placebo); therefore, death was not listed as the reason for discontinuation.
c“Death equivalent” was defined as requiring tracheostomy or permanent assisted ventilation.
Source: CENTAUR Clinical Study Report (2021).18
Exposure to Study Treatments
Based on the safety population (N = 137), the mean duration of exposure to study medication was 19.7 weeks (SD = 7.89 weeks) for the PB-TURSO group and 21.5 weeks (SD = 5.82 weeks) for the placebo group. The median time on treatment was 23.9 weeks (range = 0.6 weeks to 31.6 weeks) and 23.9 weeks (range = 1.0 weeks to 25.9 weeks) for the PB-TURSO and placebo groups, respectively.
Most patients in either group increased the study drug dose to 2 sachets during the trial: 79 (88.8%) patients in the PB-TURSO group and 45 (93.8%) patients in the placebo group.
Adherence was determined based on the number of sachets consumed divided by the number of sachets required for consumption according to the protocol plan. Nonadherence was defined as taking less than 80% of study medication or more than 125% of study medication based on sachet counts. All but 1 of the 137 patients in the safety population were started on a dose of 1 sachet. The single exception was a patient who was started on a 2-sachet dose by mistake. Overall, compliance for the groups was 90.1% for the PB-TURSO group and 90.2% for the placebo group.
Protocol Deviations
Protocol deviations have been summarized in Table 11. Protocol deviations were reported for |||||||||||||||||||| with |||||| and |||||| of patients in the PB-TURSO and placebo groups, respectively, having at least 1 protocol deviation. The most common reason was |||||||||||||||||||||||||||||||||||| of PB-TURSO patients and |||||| of placebo patients. |||||||||||| were less common at |||||| and |||||| for the PB-TURSO and placebo groups, respectively. Other reasons included ||||||||||||||||||||||||||||||, ||||||||||||||||||||||||||||||, ||||||||||||||||||||||||, ||||||||||||||||||||||||||||||||||||||||||, and ||||||||||||||||||. There was a total of |||||||||||||||||||||||||||||||||||||||||| (|||||| of PB-TURSO patients and |||||| of placebo patients), all others being minor deviations. Of those considered major deviations, |||||| patients had ||||||||||||||||||||||||||||||||||||, |||||| patients had ||||||||||||||||||||||||||||||||||||, |||||| patients had ||||||||||||||||||||||||||||||, and |||||||||||| patient had ||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Table 11: Protocol Deviations — CENTAUR Trial, All Randomized Patients
Protocol deviation | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 89) | Placebo + SOC (N = 48) | |
Protocol deviations, n (%) | |||||||||||||||||| | |||||||||||||||||| |
Total number of protocol deviations | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SOC = standard of care.
Source: CENTAUR Clinical Study Report (2021).18
Efficacy
Only the efficacy outcomes and analyses of subgroups identified in the review protocol are reported as follows.
Overall Survival
Survival analysis results are summarized in Table 12. For the composite secondary end point of death or death equivalent, 6 events occurred resulting in an HR of 0.63 (95% CI, 0.11 to 3.92). Five deaths occurred for an HR of 1.02 (95% CI, 0.15 to 9.75). Only 1 event was reported for each of permanent assisted ventilation and tracheostomy.
Table 12: Secondary Efficacy Outcomes for Survival at Week 24 — CENTAUR Trial, mITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
Death events onlya | ||
Number of events | 5 | |
Estimated percentage of risk event (SE) | 2.6 (1.65) | 2.6 (2.28) |
HR, PB-TURSO vs. placebo (95% CI) | 1.02 (0.15 to 9.75) | Reference |
P valueb | 0.99 | Reference |
Death or death equivalenta, c | ||
Number of events | 6 | |
Estimated percentage of risk event (SE) | 2.8 (1.69) | 4.4 (3.02) |
HR, PB-TURSO vs. placebo (95% CI) | 0.63 (0.11 to 3.92) | Reference |
P valueb | 0.60 | Reference |
CI = confidence interval; del-FS = delta-functional scale; HR = hazard ratio; mITT = modified intention to treat; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SE = standard error; SOC = standard of care; vs. = versus.
aCox proportional hazards model with covariates of del-FS and age at baseline.
bP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
c“Death equivalent” was defined as time to death, permanent assisted ventilation (noninvasive mechanical ventilation for more than 22 hours per day for more than 7 days), or tracheostomy.
Source: CENTAUR Clinical Study Report (2021).18
Assessment of Motor Function Using a Validated Scale Including Mobility, Muscle Strength, and Use of Feeding Tube
The results for the primary efficacy outcome of ALSFRS-R total score during the DB phase have been summarized in Table 13. The rate of change in the ALSFRS-R total score was –1.24 points per month for the PB-TURSO group and –1.66 points per month for the placebo group. The treatment difference was 0.42 points per month (95% CI, 0.03 points per month to 0.81 points per month; P = 0.03) comparing the active treatment versus placebo groups. The mean change from baseline to week 24 was –6.86 points (SE = 0.66 points) and –9.18 points (SE = 0.88 points) in the PB-TURSO and placebo groups, respectively. Overall, the difference between the PB-TURSO and placebo groups was 2.32 points (95% CI, 0.18 points to 4.47 points; P = 0.03). For the PP population, the difference comparing the PB-TURSO group to the placebo group was 2.54 points (95% CI, 0.28 points to 4.81 points; P = 0.03).
The result of the sensitivity analysis exploring the data missing at random assumption for the change in ALSFRS-R total score was 1.87 points (95% CI, 0.06 points to 3.69 points) comparing PB-TURSO versus placebo.
A sensitivity analysis using a left-censored model with ALSFRS-R, ATLIS, and SVC values was conducted to assess the risk of death biasing the results. The difference between the treatment and placebo groups for mean change in ALSFRS-R total score from baseline to week 24 was 2.33 points (95% CI, 0.18 points to 4.47 points).
To account for the drug shipping error at the beginning of the study that impacted the first 25 patients enrolled in the study and who were included in the mITT population, a post-hoc sensitivity analysis removed all 25 patients from the primary efficacy analysis. The difference between the PB-TURSO and placebo groups for the mean change in ALSFRS-R score from baseline to week 24 was 2.51 points (95% CI, 0.09 points to 4.94 points).
Table 13: Primary Efficacy Outcome at Week 24 — CENTAUR Trial, Modified ITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
Primary efficacy end point: ALSFRS-R total score | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 35.68 (5.78) | 36.67 (5.08) |
LSMa (week 24), mean (SE) | 29.06 (0.78) | 26.73 (0.98) |
Change from baseline,a mean (SE) | –6.86 (0.66) | –9.18 (0.88) |
Treatment group difference vs. controla (95% CI) | 2.32 (0.18 to 4.47) | Reference |
Rate of change per month,a mean (SE) | –1.24 (0.12) | –1.66 (0.16) |
Treatment group difference vs. controla (95% CI) | 0.42 (0.03 to 0.81) | Reference |
P valueb | 0.03 | Reference |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; del-FS = delta-functional scale; ITT = intention to treat; LSM = least squares mean; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SE = standard error; SOC = standard of care; vs. = versus.
aShared-baseline MMRM analysis with covariates of age and del-FS before the study. The patient’s rate of decline before study = (48 – ALSFRS-R at baseline) ÷ time in months from symptom onset to baseline. The maximum score on the ALSFRS-R is 48 points.
bP value has been adjusted for multiple testing (i.e., the type I error rate has been controlled).
Source: CENTAUR Clinical Study Report (2021).18
The results for the secondary outcome of the ATLIS PPN scores during the DB phase have been summarized in Table 14. For the ATLIS total score, the mean change from baseline to week 24 was –16.72% (SE = 1.05%) and –19.54% (SE = 1.45%) for the PB-TURSO and placebo treatment groups, respectively. Overall, the difference in the PB-TURSO group versus the placebo group was 2.82% (95% CI, –0.67% to 6.31%; P = 0.11). Since the result was not statistically significant, statistical testing was stopped at the first outcome in the hierarchical analysis and all subsequent P values were considered nominal. The difference in the PB-TURSO group versus the placebo group was 4.27% (95% CI, 0.16% to 8.38%) for the ATLIS upper extremity score and 2.09% (95% CI, –2.23% to 6.41%) for the ATLIS lower extremity score.
Results for the exploratory outcomes of the ALSFRS-R bulbar, fine motor, and gross motor domain scores during the DB phase have been summarized in Table 15. The fine motor domain covers handwriting, using utensils, dressing, and hygiene while the gross motor domain covers turning in bed, walking, and climbing stairs. At week 24, the difference in change from baseline to week 24 comparing the PB-TURSO group to the placebo group for the fine motor domain was 1.04 points (95% CI, 0.20 points to 1.87 points) and for the gross motor domain was 0.51 points (95% CI, –0.31 points to 1.34 points).
Table 14: Secondary Efficacy Outcomes for the ATLIS Scores at Week 24 — CENTAUR Trial, Modified ITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
ATLIS upper extremities score | ||
Number of patients contributing to the analysis at week 24 | 55 | 32 |
Baseline, mean (SD) | 54.76 (24.40) | 51.44 (25.22) |
LSMa (week 24), mean (SE) | 36.63 (2.32) | 32.36 (2.59) |
Change from baseline,a mean (SE) | –16.79 (1.25) | –21.06 (1.71) |
Treatment group difference vs. controla (95% CI) | 4.27 (0.16 to 8.38) | Reference |
P valueb | 0.04 | Reference |
ATLIS lower extremities score | ||
Number of patients contributing to the analysis at week 24 | 56 | 33 |
Baseline, mean (SD) | 57.65 (24.89) | 57.10 (25.81) |
LSMa (week 24), mean (SE) | 41.17 (2.37) | 39.09 (2.66) |
Change from baseline,a mean (SE) | –16.47 (1.32) | –18.56 (1.80) |
Treatment group difference vs. controla (95% CI) | 2.09 (–2.23 to 6.41) | Reference |
P valueb | 0.34 | Reference |
ATLIS total score (percent of normal strength) | ||
Number of patients contributing to the analysis at week 24 | 55 | 32 |
Baseline, mean (SD) | 56.83 (20.08) | 53.92 (20.94) |
LSMa (week 24), mean (SE) | 39.08 (1.99) | 36.26 (2.22) |
Change from baseline,a mean (SE) | –16.72 (1.05) | –19.54 (1.45) |
Treatment group difference vs. controla (95% CI) | 2.82 (–0.67 to 6.31) | Reference |
P valueb | 0.11 | Reference |
ATLIS = Accurate Test of Limb Isometric Strength; CI = confidence interval; del-FS = delta-functional scale; ITT = intention to treat; LSM = least squares mean; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SE = standard error; SOC = standard of care; SVC = slow vital capacity; vs. = versus.
aShared-baseline MMRM analysis with covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time were used in the model to compare treatment groups. The patient’s rate of decline before study = (ceiling maximum efficacy score – efficacy score at baseline) ÷ time in months from symptom onset to baseline. Where there was no defined maximum value (i.e., SVC), the observed maximum among all enrolled patients (PB-TURSO or placebo treatment) was used.
bP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: CENTAUR Clinical Study Report (2021).18
Table 15: Exploratory Efficacy Outcomes for the ALSFRS-R Domains at Week 24 — CENTAUR Trial, Modified ITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
ALSFRS-R bulbar domain score | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 9.51 (2.396) | 9.98 (2.597) |
LSMa (week 24), mean (SE) | 8.20 (0.320) | 7.68 (0.366) |
Change from baseline,a mean (SE) | –1.50 (0.201) | –2.02 (0.268) |
Treatment group difference vs. controla (95% CI) | 0.52 (–0.13 to 1.17) | Reference |
P valueb | 0.1188 | Reference |
ALSFRS-R fine motor domain score | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 7.99 (2.69) | 8.04 (2.63) |
LSMa (week 24), mean (SE) | 5.84 (0.31) | 4.80 (0.38) |
Change from baseline,a mean (SE) | –2.14 (0.27) | –3.18 (0.35) |
Treatment group difference vs. controla (95% CI) | 1.04 (0.20 to 1.87) | Reference |
P valueb | 0.01 | Reference |
ALSFRS-R gross motor domain score | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 7.54 (2.84) | 7.60 (2.62) |
LSMa (week 24), mean (SE) | 5.57 (0.34) | 5.05 (0.41) |
Change from baseline,a mean (SE) | –1.90 (0.25) | –2.42 (0.34) |
Treatment group difference vs. controla (95% CI) | 0.51 (–0.31 to 1.34) | Reference |
P valueb | 0.22 | Reference |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; del-FS = delta-functional scale; ITT = intention to treat; LSM = least squares mean; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SE = standard error; SOC = standard of care; vs. = versus.
aShared-baseline MMRM analysis with covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time were used in the model to compare treatment groups. The patient’s rate of decline before study = (48 – ALSFRS-R at baseline) ÷ time in months from symptom onset to baseline. The maximum score on the ALSFRS-R is 48 points.
bP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: CENTAUR Clinical Study Report (2021).18
Assessment of Respiratory Function Including Determination of Vital Capacity, Use of Respirator, Time to Tracheostomy, and Tracheostomy-Free Survival
There was 1 tracheostomy event in a patient who received placebo during the CENTAUR trial.
The results for the exploratory outcome of the ALSFRS-R breathing domain scores during the DB phase have been summarized in Table 16. The breathing domain covers dyspnea, orthopnea, and respiratory insufficiency. At week 24, the difference in mean change from baseline to week 24 comparing the PB-TURSO versus placebo groups for the breathing domain score was 0.36 points (95% CI, –0.53 points to 1.25 points).
Table 16: Exploratory Efficacy Outcomes for the ALSFRS-R Breathing Domain at Week 24 — CENTAUR Trial, Modified ITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
ALSFRS-R breathing domain score | ||
Number of patients contributing to the analysis at week 24 | 64 | 37 |
Baseline, mean (SD) | 10.64 (1.92) | 11.04 (1.80) |
LSMa (week 24), mean (SE) | 9.49 (0.29) | 9.13 (0.37) |
Change from baseline,a mean (SE) | –1.28 (0.28) | –1.64 (0.37) |
Treatment group difference vs. controla (95% CI) | 0.36 (–0.53 to 1.25) | Reference |
P valueb | 0.43 | Reference |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; del-FS = delta-functional scale; ITT = intention to treat; LSM = least squares mean; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SE = standard error; SOC = standard of care; vs. = versus.
aShared-baseline MMRM analysis with covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time were used in the model to compare treatment groups. The patient’s rate of decline before the study = (48 – ALSFRS-R at baseline) ÷ time in months from symptom onset to baseline. The maximum score on the ALSFRS-R is 48 points.
bP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: CENTAUR Clinical Study Report (2021).18
The results for the secondary outcome of SVC PPN during the DB phase have been summarized in Table 17. The differences from baseline to week 24 were –17.11% (SE = 1.70%) and –22.22% (SE = 2.32%) for the active and placebo treatment groups, respectively. Overall, the difference comparing the PB-TURSO versus placebo groups was 5.11% (95% CI, –0.54% to 10.76%).
Health-Related Quality of Life
HRQoL was not assessed in the CENTAUR trial.
Caregiver Burden
Caregiver burden was not assessed in the CENTAUR trial.
Harms
Only the harms identified in the review protocol are reported as follows. Detailed harms data are in Table 18.
Table 17: Secondary Efficacy Outcomes for SVC at Week 24 — CENTAUR Trial, Modified ITT Population
Outcome | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 87) | Placebo + SOC (N = 48) | |
SVC (percent predicted normal) | ||
Number of patients contributing to the analysis at week 24 | 62 | 34 |
Baseline, mean (SD) | 83.62 (18.17) | 83.88 (15.92) |
LSMa (week 24), mean (SE) | 66.17 (2.33) | 61.06 (2.81) |
Change from baseline,a mean (SE) | –17.11 (1.70) | –22.22 (2.32) |
Treatment group difference vs. controla (95% CI) | 5.11 (–0.54 to 10.76) | Reference |
P valueb | 0.08 | Reference |
CI = confidence interval; del-FS = delta-functional scale; ITT = intention to treat; LSM = least squares mean; MMRM = mixed model of repeated measures; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SD = standard deviation; SE = standard error; SOC = standard of care; SVC = slow vital capacity; vs. = versus.
aShared-baseline MMRM analysis with covariates of age, del-FS before the study, and the change in the efficacy outcome measure interacting with time were used in the model to compare treatment groups. The patient’s rate of decline before study = (ceiling maximum efficacy score – efficacy score at baseline) ÷ time in months from symptom onset to baseline. Where there was no defined maximum value (i.e., SVC), the observed maximum among all enrolled patients (PB-TURSO or placebo treatment) was used.
bP value is nominal and has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: CENTAUR Clinical Study Report (2021).18
Table 18: Summary of Harms — CENTAUR Trial, Safety Population
Harm | CENTAUR trial | |
|---|---|---|
PB-TURSO + SOC (N = 89) | Placebo + SOC (N = 48) | |
Patients with ≥ 1 TEAE by MedDRA System Organ Class and Preferred Term, n (%) | 86 (96.6) | 46 (95.8) |
Most common events,a n (%) | ||
Gastrointestinal disorders | 59 (66.3) | 30 (62.5) |
Diarrhea | 19 (21.3) | 8 (16.7) |
Nausea | 16 (18.0) | 6 (12.5) |
Constipation | 12 (13.5) | 12 (25.0) |
Salivary hypersecretion | 10 (11.2) | 1 (2.1) |
Abdominal pain | 7 (7.9) | 4 (8.3) |
Abdominal discomfort | 5 (5.6) | 0 |
Dry mouth | 3 (3.4) | 4 (8.3) |
Dysphagia | 3 (3.4) | 4 (8.3) |
Musculoskeletal and connective tissue disorders | 37 (41.6) | 19 (39.6) |
Muscular weakness | 18 (20.2) | 9 (18.8) |
Back pain | 5 (5.6) | 4 (8.3) |
Muscle spasms | 5 (5.6) | 3 (6.3) |
Arthralgia | 5 (5.6) | 2 (4.2) |
Musculoskeletal pain | 5 (5.6) | 2 (4.2) |
Neck pain | 2 (2.2) | 5 (10.4) |
Injury, poisoning, and procedural complications | 31 (34.8) | 22 (45.8) |
Fall | 25 (28.1) | 18 (37.5) |
Laceration | 6 (6.7) | 3 (6.3) |
Contusion | 5 (5.6) | 4 (8.3) |
Nervous system disorders | 35 (39.3) | 20 (41.7) |
Headache | 13 (14.6) | 11 (22.9) |
Dizziness | 9 (10.1) | 2 (4.2) |
Infections and infestations | 28 (31.5) | 19 (39.6) |
Viral upper respiratory tract infection | 10 (11.2) | 2 (4.2) |
Urinary tract infection | 5 (5.6) | 3 (6.3) |
Upper respiratory tract infection | 4 (4.5) | 3 (6.3) |
Respiratory, thoracic, and mediastinal disorders | 27 (30.3) | 12 (25.0) |
Dyspnea | 9 (10.1) | 4 (8.3) |
Cough | 5 (5.6) | 3 (6.3) |
Respiratory failure | 4 (4.5) | 4 (8.3) |
Investigations | 23 (25.8) | 10 (20.8) |
Weight, decreased | 5 (5.6) | 1 (2.1) |
Alanine aminotransferase, increased | 4 (4.5) | 3 (6.3) |
Aspartate aminotransferase, increased | 4 (4.5) | 3 (6.3) |
General disorders and administration site conditions | 20 (22.5) | 13 (27.1) |
Fatigue | 7 (7.9) | 3 (6.3) |
Asthenia | 5 (5.6) | 0 |
Edema peripheral | 2 (2.2) | 3 (6.3) |
Skin and subcutaneous tissue disorders | 17 (19.1) | 7 (14.6) |
Rash | 5 (5.6) | 4 (8.3) |
Psychiatric disorders | 14 (15.7) | 10 (20.8) |
Insomnia | 3 (3.4) | 3 (6.3) |
Anxiety | 2 (2.2) | 3 (6.3) |
Renal and urinary disorders | 11 (12.4) | 8 (16.7) |
Proteinuria | 6 (6.7) | 2 (4.2) |
Metabolism and nutrition disorders | 10 (11.2) | 4 (8.3) |
Decreased appetite | 7 (7.9) | 2 (4.2) |
Patients with ≥ 1 SAE by MedDRA System Organ Class and Preferred Term, n (%) | 11 (12.4) | 8 (16.7) |
Respiratory, thoracic, and mediastinal disorders | 3 (3.4) | 4 (8.3) |
Respiratory failure | 2 (2.2) | 3 (6.3) |
Respiratory arrest | 1 (1.1) | 0 |
Pulmonary embolism | 0 | 1 (2.1) |
Infections and infestations | 3 (3.4) | 1 (2.1) |
Bacteremia | 1 (1.1) | 1 (2.1) |
Implant site cellulitis | 1 (1.1) | 0 |
Pneumonia | 1 (1.1) | 0 |
Pneumonia, respiratory syncytial viral | 1 (1.1) | 0 |
Catheter site infection | 0 | 1 (2.1) |
Injury, poisoning, and procedural complications | 3 (3.4) | 1 (2.1) |
Skull fracture | 1 (1.1) | 0 |
Stoma site hemorrhage | 1 (1.1) | 0 |
Subdural hematoma | 1 (1.1) | 0 |
Pubic bone fracture | 0 | 1 (2.1) |
Gastrointestinal disorders | 2 (2.2) | 0 |
Diverticular perforation | 1 (1.1) | 0 |
Pneumoperitoneum | 1 (1.1) | 0 |
Renal and urinary disorders | 1 (1.1) | 1 (2.1) |
Nephrolithiasis | 1 (1.1) | 1 (2.1) |
Eye disorders | 1 (1.1) | 0 |
Vision blurred | 1 (1.1) | 0 |
Product issues | 0 | 1 (2.1) |
Device dislocation | 0 | 1 (2.1) |
WDAE, n (%)b | 18 (20.2) | 5 (10.4) |
Diarrhea | 5 (5.6) | 0 |
Abdominal pain, upper | 2 (2.2) | 0 |
Dysgeusia | 2 (2.2) | 0 |
Nausea | 1 (1.1) | 1 (2.1) |
Subdural hematoma | 1 (1.1) | 0 |
Diverticulitis | 1 (1.1) | 0 |
Adjustment disorder with depressed mood | 1 (1.1) | 0 |
Fatigue | 1 (1.1) | 0 |
Asthenia | 1 (1.1) | 0 |
Atrial fibrillation | 1 (1.1) | 0 |
Respiratory arrest | 1 (1.1) | 0 |
Rash | 1 (1.1) | 0 |
Edema peripheral | 1 (1.1) | 0 |
Joint swelling | 1 (1.1) | 0 |
Arthralgia | 1 (1.1) | 0 |
Abdominal discomfort | 1 (1.1) | 0 |
Weight, decreased | 1 (1.1) | 0 |
Decreased appetite | 1 (1.1) | 0 |
Lethargy | 1 (1.1) | 0 |
Sinusitis | 1 (1.1) | 0 |
Respiratory failure | 0 | 3 (6.3) |
Hypoxia | 0 | 1 (2.1) |
Neck pain | 0 | 1 (2.1) |
Pubic bone fracture | 0 | 1 (2.1) |
Deaths, n (%) | 5 (5.6) | 2 (2.2) |
Respiratory failure or respiratory arrest | 3 (3.4) | 2 (2.2) |
Subdural hematoma | 1 (1.1) | 0 |
Diverticular perforation | 1 (1.1) | 0 |
Notable harms, n (%) | ||
Gastrointestinal AEsa | ||
Diarrhea | 19 (21.3) | 8 (16.7) |
Nausea | 16 (18.0) | 6 (12.5) |
Constipation | 12 (13.5) | 12 (25.0) |
Salivary hypersecretion | 10 (11.2) | 1 (2.1) |
Abdominal pain | 7 (7.9) | 4 (8.3) |
Abdominal discomfort | 5 (5.6) | 0 |
Dry mouth | 3 (3.4) | 4 (8.3) |
Dysphagia | 3 (3.4) | 4 (8.3) |
Neurologic AEsa, c | ||
Headache | 13 (14.6) | 11 (22.9) |
Dizziness | 9 (10.1) | 2 (4.2) |
Insomnia | 3 (3.4) | 3 (6.3) |
Anxiety | 2 (2.2) | 3 (6.3) |
Respiratory AEsa, d | ||
Dyspnea | 9 (10.1) | 4 (8.3) |
Cough | 5 (5.6) | 3 (6.3) |
Respiratory failure | 4 (4.5) | 4 (8.3) |
Taste disturbancee | 3 (3.4) | 1 (2.1) |
AE = adverse event; MedDRA = Medical Dictionary for Regulatory Activities; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SAE = serious adverse event; SOC = standard of care; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
aFrequency of at least 5% of patients.
bAE leading to withdrawal of study medication.
cNeurologic AEs include those listed under MedDRA’s Nervous System Disorders and Psychiatric Disorders headings.
dRespiratory AEs include those listed under MedDRA’s Respiratory, Thoracic, and Mediastinal Disorders heading.
eData here are those reported for MedDRA’s term for “dysgeusia.” It was noted in the CENTAUR Clinical Study Report that investigators were instructed to not capture the bad taste of medication as an AE.
Source: CENTAUR Clinical Study Report (2021).18
Adverse Events
Overall, 86 (96.6%) patients in the PB-TURSO group and 46 (95.8%) patients in the placebo group experienced at least 1 TEAE during the 24-week DB phase. The TEAEs reported in at least 20% of patients in either group were falls (28.1% and 37.5%), diarrhea (21.3% and 16.7%), muscular weakness (20.2% and 18.8%), headache (14.6% and 22.9%), and constipation (13.5% and 25%) in the PB-TURSO and placebo groups, respectively.
The TEAEs that occurred more frequently (by more than 5%) in patients who received PB-TURSO compared to placebo included nausea (18.0% versus 12.5%), salivary hypersecretion (11.2% versus 2.1%), viral upper respiratory tract infection (11.2% versus 4.2%), dizziness (10.1% versus 4.2%), abdominal discomfort (5.6% versus 0%), and asthenia (5.6% versus 0%) in the PB-TURSO and placebo groups, respectively.
Most TEAEs (794 of 915) did not result in a dose change during the trial. TEAEs leading to medication interruption were more frequent in the PB-TURSO group (31 events) compared to the placebo group (5 events). The same trend was observed for dose reductions (8 events for the PB-TURSO group compared to 0 events for the placebo group) and withdrawal of study medication (26 events for the PB-TURSO group versus 7 events for the placebo group).
Serious Adverse Events
In total, 23 SAEs were reported in 11 (12.4%) patients from the PB-TURSO group and 8 (16.7%) patients from the placebo group. Most SAEs were reported in single patients aside from respiratory failure (5 patients), bacteremia (2 patients), and nephrolithiasis (2 patients).
Withdrawal Due to Adverse Events
Overall, 18 (20.2%) patients from the PB-TURSO group and 5 (10.4%) patients from the placebo group withdrew from the study medication due to a TEAE. The most frequently reported reasons were due to diarrhea among 5 (5.6%) patients in the active treatment group (versus 0 patients in the placebo group) and respiratory failure among 3 (6.3%) patients in the placebo group (versus 0 patients in the PB-TURSO group).
Mortality
There were 7 deaths reported in the safety population during the CENTAUR trial: 5 (5.6%) patients in the PB-TURSO group due to respiratory failure or respiratory arrest (3 patients), subdural hematoma (secondary to a fall; 1 patient), and diverticular perforation (1 patient) compared to 2 (2.2%) patients in the placebo group, both due to respiratory failure or respiratory arrest.
Notable Harms
Gastrointestinal Adverse Event
Frequently reported gastrointestinal AEs (in 5% of patients or greater) that occurred more commonly among patients who received PB-TURSO than those who received placebo included diarrhea (21.3% versus 16.7%), nausea (18.0% versus 12.5%), salivary hypersecretion (11.2% versus 2.1%), and abdominal discomfort (5.6% versus 0%) in the PB-TURSO and placebo groups, respectively. Patients who received PB-TURSO had gastrointestinal AEs more frequently that resulted in dose reductions (3%) or dose interruptions (9%) compared to patients who received placebo (0% and 2%, respectively).28
Neurologic Adverse Event
Neurologic AEs occurring in at least 5% of patients included dizziness (10.1% versus 4.2%), headache (14.6% versus 22.9%), insomnia (3.4% versus 6.3%), and anxiety (2.2% versus 6.3%) in the PB-TURSO and placebo groups, respectively.
Respiratory Adverse Event
Respiratory AEs occurring in at least 5% of patients included dyspnea (10.1% versus 8.3%), respiratory failure (6.3% versus 5.6%), and cough (5.6% versus 6.3%) in the PB-TURSO and placebo groups, respectively.
Taste Disturbance
Dysgeusia, or taste disturbance, occurred in 3.4% of patients who received PB-TURSO compared to 2.1% of patients who received placebo. It was noted that investigators were instructed not to capture the bad taste of medication as an AE and instead, to record issues with oral administration if there was a clinically untoward effect such as burning, vomiting, or anxiety.
Critical Appraisal
Internal Validity
The CENTAUR trial was a small phase II trial and evidence produced from such trials is considered exploratory36 rather than confirmatory.37 It is important for findings from a phase II trial to be confirmed in a phase III trial as the magnitude of treatment effect is subject to greater uncertainty in a phase II trial with relatively small sample size and short treatment duration.
Randomization appeared to adequately balance most of the baseline characteristics. A greater proportion of patients in the placebo group had experience with concomitant ALS medications (riluzole and/or edaravone), except for riluzole only (where a greater proportion of patients in the PB-TURSO group had experience). The clinical expert indicated the imbalances were not concerning and could be a result of the randomization of a small study population, and previous experience with these medications was not expected to impact the interpretation of the result. The site of onset was also imbalanced between the groups with a greater percentage of patients in the PB-TURSO group reporting bulbar onset and, conversely, a greater percentage of patients in the placebo group reporting limb onset. Onset in the bulbar region has been associated with faster decline in the ALSFRS-R score38; thus, the imbalance in site of disease onset would likely bias against PB-TURSO.
There were large proportions of patients who discontinued from the study (24.7% and 20.8% in the PB-TURSO and placebo groups, respectively) and only a small number were due to death or death equivalent events (3.4% and 4.2% in the PB-TURSO and placebo groups, respectively). Withdrawals from treatment due to AEs were more frequent in the PB-TURSO group than the placebo group (20.2% versus 10.4%, respectively) and diarrhea was the most common reason for withdrawal (5.6% in the PB-TURSO group versus 0% in the placebo group), with most other reasons being single-patient events. Consequently, the number of patients available for the analysis at 24 weeks varied largely from the number of patients randomized at baseline. This affected the assessment of all the outcomes, including the ALSFRS-R. Scores that were missing for the ALSFRS-R, ATLIS, and SVC were not imputed, and missing data were handled using a missing at random assumption. Since the data missing at random assumption cannot be tested, it has been recommended that sensitivity analyses be performed in ALS studies.34 The results of the sensitivity analysis for data missing at random supported the main analysis for the primary end point, though the treatment effect was smaller for the sensitivity analysis than for the main analysis. It is not possible to determine whether the sensitivity analysis accounted for the full potential impact of missing data.
The treatment duration for the CENTAUR trial was 24 weeks (or 6 months), which is considered the shortest acceptable duration for measuring a treatment effect by both the clinical expert consulted for this review as well as guidance provided by the FDA on ALS trials.34 The ALSFRS-R total score has demonstrated acceptable validity and reliability, but information on its responsiveness to change and MID estimates were not found in the literature. The clinical expert consulted for this review was of the opinion that a 2-point difference in change over 24 weeks between treatment groups is meaningful while experts from the NEALS consortium suggested a change of 20% to 25% in the ALSFRS-R slope was “at least somewhat clinically meaningful.”17 Estimates of the MID for the ATLIS and SVC were also not identified in the literature. Acceptable validity and reliability have been demonstrated for the ATLIS and information on its responsiveness to change is being assessed but is yet to be published. Evidence for the reliability, validity, and responsiveness of SVC was not found in the literature. According to the clinical expert consulted on this review, it can be difficult for patients to perform SVC manoeuvres and obtain reproducible results. Survival was a secondary outcome and data were immature at the end of 6 months due to few events having occurred. The small sample size, the relatively short duration of the trial, and the low frequencies for SAEs, WDAEs, and deaths made it difficult to draw any firm conclusions from these results.
Most patients reported at least 1 protocol deviation during the CENTAUR study and 19 patients had 21 major protocol deviations. The reasons for this included the study drug not being dispensed per protocol (n = 9), issues with the collection of informed consent (n = 7), and issues with eligibility criteria deviations (n = 4). Although the sponsor stated that, overall, the deviations were not believed to have an impact on the study’s integrity or conclusions, it is uncertain if or what effect these may have had on the results. However, the PP analysis for the primary end point, which excluded data from patients with a major protocol deviation from the time the deviation occurred onwards, was consistent with the main analysis.
External Validity
The clinical expert confirmed that patients in the CENTAUR trial were similar to those treated in a Canadian setting with a few exceptions. The mean age of patients in the study was younger, a greater proportion of patients was female, and a greater proportion of patients had limb onset compared with patients in Canadian clinical practice. The expert noted the possibility that the differences in age and proportion of females were at least partly due to the eligibility criteria requiring that patients have a definite ALS diagnosis within 18 months of symptom onset. The clinical expert explained that it is more likely that patients with definite ALS within 18 months have limb onset than bulbar onset, males are more likely to have limb onset, and males typically present at a younger age than females.
All the study centres were in the US, which limits the generalizability of the findings since general health care in the US, and particularly the SOC for ALS, may differ from that in Canada.8 According to the clinical expert consulted by CADTH, the diagnosis of definite ALS according to the El Escorial criteria (having 3 zones affected) and patients having 18 months or less between symptom onset and enrolment are very restrictive criteria. The expert explained that these criteria required patients to have a high level of symptoms that many patients would not meet within 18 months. The clinical expert stated that most patients would have a diagnosis of possible or probable ALS in 18 months and therefore would have been excluded from this study. Based on the requirement for a definite ALS diagnosis within 18 months, it can be inferred that the patients in the CENTAUR study had more rapidly progressive disease than most patients with ALS. The short time frame and inclusion of patients with rapidly progressing disease allowed the investigators to observe a measurable treatment effect in only 24 weeks, but these limitations prevented the capture of information for patients who are more slowly progressing. The clinical expert identified specific exclusion criteria that may also be restrictive: poorly controlled arterial hypertension at screening (systolic blood pressure greater than 160 mm Hg or diastolic blood pressure greater than 100 mm Hg), a history of cholecystectomy, and exposure to antacids containing aluminum hydroxide or aluminum oxide within 2 hours of administration of PB-TURSO.
The clinical expert felt that the proportions of patients accessing riluzole and edaravone in the CENTAUR study were similar to what is seen in Canadian ALS clinics. As is the case with most drug trials, clinical visits were more frequent than what is typical of regular practice. Study visits were much more frequent at every 3 weeks in the CENTAUR trial compared to the recommended monitoring at 3-month intervals per “Canadian best practice recommendations for the management of amyotrophic lateral sclerosis.”4
Use of the ALSFRS-R as a measure of efficacy was supported by both the clinical expert, since it is commonly used in Canadian practice, and the FDA.39 The clinical expert noted that the ATLIS and SVC are not typically used in Canadian ALS clinics, but instead for research purposes, though both were considered acceptable by the clinical expert and the FDA if a clinically meaningful change due to treatment could be demonstrated.39
Patient groups that submitted input for the CADTH review listed issues with motor function, mobility, fatigue, breathing, speech, and swallowing as being important. These were mostly addressed by the ALSFRS-R domains (bulbar, fine motor, gross motor, and breathing functions), the ATLIS (measure of muscle strength), and SVC (measure of respiratory function). The patient groups were also interested in a medication that is easy to administer, helps them maintain their current function and independence, slows progression, improves symptoms, and improves survival outcomes. There was a notable lack of patient-reported and HRQoL outcomes for measuring symptom burden in the CENTAUR trial. Therefore, it is uncertain what benefits there are for patient-centred outcomes.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
A focused literature search for network meta-analyses dealing with PB-TURSO (sodium phenylbutyrate and ursodoxicoltaurine) and ALS was run in MEDLINE All (1946–) on December 17, 2021. No limits were applied to the search. Indirect treatment evidence for PB-TURSO was not identified from the literature for this review.
A sponsor-submitted feasibility assessment for an MAIC comparing PB-TURSO to edaravone in the CENTAUR trial has been summarized and appraised as follows.
Description of Indirect Comparison
Due to the lack of head-to-head comparisons between PB-TURSO and other ALS medications, the sponsor submitted a feasibility assessment for conducting an MAIC to compare the relative efficacy of PB-TURSO to edaravone.
Methods
Objectives
The objective of the feasibility assessment was to determine if it was reasonable to conduct an MAIC comparing the relative efficacy of PB-TURSO to edaravone.
Study Selection Methods
One RCT for PB-TURSO was identified (the CENTAUR trial) and 4 RCTs for edaravone were identified (Study 16, Study 17, Study 18, and Study 19) based on the 2019 CADTH review of Radicava.40 Of the edaravone studies, 2 were considered appropriate for the purposes of the analysis: Study 16 and Study 19.
Indirect Treatment Comparison Analysis Methods
The analysis sets used from each of the studies were the mITT population from the CENTAUR study, and the full analysis sets from Study 16 and Study 19. The sponsor felt that the edaravone studies were “sufficiently homogenous” and their data were pooled using standard meta-analysis methods. The primary end point of the analysis was the difference between PB-TURSO and edaravone in the mean change in the ALSFRS-R total score from baseline to week 24.
An anchored comparison using a common placebo group was planned to evaluate the relative treatment effects of the 2 drugs. To conduct an anchored MAIC, it was assumed that all effect modifiers were accounted for as covariates in the matching process or otherwise unmeasured and balanced between studies. Covariates used in the analysis included baseline ALSFRS-R score, del-FS, duration of disease, baseline FVC or SVC, and concomitant riluzole use. For the CENTAUR trial data, an MMRM analysis was used with the MAIC weights applied.
Results
Summary of Included Studies
A summary of the patient characteristics for the CENTAUR trial, Study 16, and Study 19 is available in Table 19. There were notable differences between the studies in terms of study design (e.g., location, pre-baseline observation period, eligibility criteria, proportion of patients with definite ALS) and imbalances between the CENTAUR study and pooled Study 16 and Study 19 data for sex, site of onset, duration of disease, ALSFRS-R baseline score, del-FS at baseline, proportion of patients with a definite ALS diagnosis, FVC or SVC at baseline, and use of riluzole and/or edaravone at baseline. The del-FS was reported for patients in the CENTAUR trial but not in the edaravone studies; thus, the del-FS was estimated for those studies based on available data and a similar calculation to that used in the CENTAUR trial.
Table 19: Summary of Patient Characteristics for the CENTAUR Trial, Study 16, and Study 19
Characteristic | CENTAUR PB-TURSO | CENTAUR Placebo | Study 16 Edaravone | Study 16 Placebo | Study 19 Edaravone | Study 19 Placebo |
|---|---|---|---|---|---|---|
Patients, N | 87 | 48 | 101 | 104 | 69 | 68 |
Age (years), mean | 57.6 | 57.3 | 57.9 | 57.7 | 60.5 | 60.1 |
Male (%) | 70 | 67 | 62 | 66 | 55 | 60 |
Site of onset (%) | NA | NA | NA | NA | NA | NA |
Bulbar | 30 | 21 | 19 | 18 | 23 | 21 |
Limb | 68 | 79 | 81 | 82 | 77 | 79 |
Other | 2 | 0 | 0 | 0 | 0 | 0 |
Duration of disease (months), mean | 13.5 | 13.6 | 17.3 | 15.6 | 13.6 | 12.7 |
ALSFRS-R baseline total score, mean | 35.7 | 36.7 | 40.6 | 41.2 | 41.9 | 41.8 |
ALSFRS-R total score at start of pre-baseline period, mean | NA | NA | 42.5 | 43.3 | 43.6 | 43.5 |
Pre-baseline period, weeks | NA | NA | 12 | 12 | 12 | 12 |
Change in ALSFRS-R score in pre-baseline perioda (%) | NA | NA | NA | NA | NA | NA |
–4 | NA | NA | 8 | 11 | 7 | 4 |
–3 | NA | NA | 21 | 20 | 10 | 12 |
–2 | NA | NA | 39 | 38 | 30 | 37 |
–1 | NA | NA | 40 | 32 | 52 | 47 |
Del-FS at baseline, mean | 0.95 | 0.93 | 0.43b | 0.44b | 0.45b | 0.49b |
El Escorial Definite diagnosis (%) | 100 | 100 | 29 | 20 | 41 | 40 |
FVC or SVC at baseline, mean | 83.62 | 83.88 | 95.53 | 95.78 | 100.50 | 97.37 |
Riluzole use at baseline (%) | 68 | 77 | 89 | 89 | 91 | 91 |
Edaravone use at baseline (%) | 25 | 50 | NA | NA | NA | NA |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; del-FS = delta-functional scale; FVC = forced vital capacity; NA = not applicable; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SVC = slow vital capacity.
aPatients in Study 16 and Study 19 were required to progress or decline by 1 to 4 points on the ALSFRS-R during the pre-baseline period to be enrolled.
bDel-FS was estimated for Study 16 and Study 19 using this equation: (48 – mean ALSFRS-R baseline score) ÷ mean duration of disease in months. For consistency with the conversion factors used in the CENTAUR analyses, 1 year = 365.25 days ÷ 30.417 days, or 12.008 months.
Source: Sponsor’s Pharmacoeconomic Report submitted for the review of PB-TURSO.41
Results
After matching for the covariates, the effective sample size for the CENTAUR trial was reduced from an original 135 patients to 24.8 patients. By including baseline edaravone as a covariate, the matched CENTAUR trial population was further reduced to 3.4 patients. The large reduction in effective sample size after matching indicated that the study populations were substantially different from 1 another and that it was not reasonable to conduct an MAIC using these populations. The sponsor’s analysis identified del-FS as the main reason for the reduction in effective sample size; del-FS was also considered to be the most clinically important covariate.
Critical Appraisal
Eligibility criteria40 were narrower for Study 19 than Study 16 and baseline characteristics differed between the studies, indicating that the studies and populations likely were not similar enough for the data to be pooled and making it difficult to compare them effectively. The ALS diagnosis could not be adjusted for in the analysis since all patients in the CENTAUR trial had a definite ALS diagnosis, which the sponsor acknowledged could violate the assumption that all effect modifiers were accounted for. Furthermore, the del-FS in Study 16 and Study 19 was not reported and had to be estimated, which introduced uncertainty into the analysis. The CADTH review team noted that baseline edaravone use could not be excluded as an effect modifier and when included, greatly reduced the effective sample size. The CADTH review team agreed that the reduction in effective sample size after adjustments indicated that there were substantial differences between the populations and that it would not be reasonable to compare the treatments between the CENTAUR trial and the pooled Study 16 and Study 19 data.
Summary
Based on the sponsor-submitted assessment of the data presented from 1 RCT of PB-TURSO and 2 RCTs of edaravone, the CADTH reviewers agreed it was not reasonable to conduct an MAIC comparing the relative efficacy of PB-TURSO to edaravone.
Other Relevant Evidence
This section includes 1 long-term extension study and 2 post-hoc analyses for the pivotal CENTAUR trial included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
The AMX3500-OLE (CENTAUR-OLE) Trial
This study is an OLE to the main, 24-week, DB CENTAUR trial to evaluate the long-term safety and efficacy of PB-TURSO. The OLE was added in an amendment (dated October 20, 2017) to the CENTAUR trial protocol dated after the enrolment of the first patient in the CENTAUR trial. The primary objective of the OLE was to determine the long-term safety of PB-TURSO in patients with ALS. Efficacy outcomes were assessed as secondary outcomes. This report summarizes interim data (cut-off date of April 2, 2020), which include at least 24 weeks of data from the OLE and 48 weeks overall for all patients. This report also includes death data from a cut-off date of July 20, 2020. While an OLE duration of 52 weeks was initially planned, the treatment duration was extended to 132 weeks post-OLE enrolment in a subsequent protocol amendment.
Methods
The CENTAUR-OLE trial is a multi-centre OLE of the main CENTAUR trial. The OLE study was unblinded and all patients (except for 1 patient in the placebo group whose dose was reduced due to an AE [i.e., diarrhea]) received PB-TURSO twice daily; however, investigators, evaluators, and patients remained blinded to the randomized treatment assignment from the main DB study. A total of 97 patients completed the main DB CENTAUR study and 90 of the patients continued into the OLE to assess the long-term safety (primary outcome) and efficacy (secondary outcomes) of PB-TURSO.
Patients had to have their baseline visit within 28 days of the week 24 visit in the main trial. Study visits occurred at week 6, week 12, week 24, week 36, and every 16 weeks thereafter.
Populations
Inclusion Criteria
Patients must have completed all visits in the CENTAUR study. Patients who received a tracheostomy or permanent assisted ventilation while in the DB trial could elect to enrol in the OLE so long as they completed all visits in the main study.
Patients had to enrol in the OLE within 28 days of the week 24 visit of the main study.
Exclusion Criteria
Discontinued study drug prematurely in the DB phase for reasons other than a tracheostomy or permanent assisted ventilation.
Had exposure to or anticipated requirement for any disallowed medications (e.g., histone deacetylase inhibitors, probenecid, bile acid sequestrants, antacids containing aluminum hydroxide or aluminum oxide within 2 hours of administration of study drug).
Had unstable cardiac or other life-threatening disease emergent during the DB study.
Had any ongoing AEs that are clear contraindications to the study drug
Had any major medical conditions that would interfere with the study and place the patients at increased risk
Patients were analyzed according to the treatment to which they were randomized in the main trial. Baseline characteristics were comparable between the 2 groups in the safety, mITT, and PP analysis populations without any major differences in demographics, disease characteristics, and concomitant medications. However, a notable difference was observed in the safety and mITT populations, in which time since the first exposure to edaravone was longer in the group of patients who were randomized to PB-TURSO compared to those randomized to placebo in the main trial. In all 3 analysis populations, for both groups, most patients were male (> 70%), most were White (> 90%), and the mean age ranged between 57.7 years and 58.5 years (SD = 6.69 years and 10.22 years). The ranges of mean time since ALS diagnosis and since onset of symptoms were from 11.7 months to 11.8 months (SD = from 3.6 months to 3.73 months) and from 19.2 months to 19.3 months (SD = from 4.07 months to 4.15 months), respectively. Most patients (77.5% to 77.9%) were receiving either edaravone or riluzole and 18.8% to 19.8% of patients were on both, at or before OLE entry. The majority of patients (75.0% to 75.6%) were using riluzole compared to 21.1% to 22.1% of patients using edaravone. Scores for ALSFRS-R, ATLIS, and SVC were also similar between the 2 groups in the 3 populations. For example, the ranges of mean scores for ALSFRS-R total score, ATLIS upper and lower extremities, and SVC were 30.1 points to 30.6 points (SD = 8.69 points to 8.74 points), 44.98% to 46.18% (SD = 19.24% to 19.63%), and 71.0% to 71.7% (SD = 22.18% to 22.43%), respectively. Lastly, the mean OLE baseline del-FS scores (pre-randomization progression rates) were similar between groups among the 3 populations — namely, 0.95 points per month to 0.99 points per month (SD = 0.57 points per month to 0.58 points per month).
Interventions
Study drug administration was consistent with the CENTAUR trial; to maintain blinding from the main study, none of the patients went through the titration stage (i.e., initial 3 weeks of 1 sachet daily dosing).
Outcomes
The primary objective of the study was to assess the long-term safety of oral (or feeding tube) administration of PB-TURSO. Safety and tolerability were assessed using standard AE (including SAE) reporting.
The secondary objectives of the study were the:
rate of key study events (i.e., hospitalization, tracheostomy, permanent assisted ventilation, death)
rate of progression (motor function) as measured by ALSFRS-R scale and ATLIS
rate of progression (respiratory function) as measured by SVC (PPN).
All efficacy analyses were based on the 48 weeks of treatment (i.e., 24 weeks during each of the DB phase and the OLE phase), except for the survival analyses outlined in a separate statistical analysis plan, which were based on a data cut-off date of February 29, 2020. An additional survival analysis, outlined in an addendum to the survival statistical analysis plan, was performed with a data cut-off date of July 20, 2020. In the original statistical analysis plan for the OLE, only events occurring during the OLE and patients enrolled in the OLE were to be included in the survival analyses. Also, survival was to be analyzed using the same Cox proportional hazards model as was used in the main trial (del-FS and age at baseline as covariates).
The survival analyses based on the February 29, 2020, and July 20, 2020, cut-off dates were specified following unblinding to treatment assignment in the main trial. The following were additional methods specified in the survival statistical analysis plan:
All patients regardless of OLE entry were included in the survival analysis using a vital status sweep conducted by Omnitrace, a professional search firm, to supplement the main study and OLE data. It was assumed that death equivalent events did not occur in patients who were lost to follow-up or who did not enter the OLE.
Analysis was conducted in the ITT population.
The baseline ALSFRS-R total score was added as a covariate in the Cox proportional hazards model.
Vital status and date of death for all but 2 patients in the ITT population were obtained as of the July 20, 2020, data cut-off date.
Statistical Analysis
Overall, the efficacy evaluation for the OLE phase followed the same methods as outlined for the main study. Outcomes were compared between 2 groups — namely, patients randomized to PB-TURSO (active treatment) in the main trial (the AA group) and patients randomized to placebo in the main trial (the PA group).
Two sets of analyses were performed. The first analysis was performed using the original baseline and following patients through to the end of the OLE cut-off dates, and a second analysis was performed using only data from the OLE phase corrected for the new baseline at the beginning of the OLE phase of the study. All continuous efficacy measures used the same statistical models as the main study and followed the hierarchical order of the ALSFRS-R total score, key events (tracheostomy, hospitalization, and death), the ATLIS upper score, the ATLIS lower score, SVC PPN, 4 ALSFRS-R domains, and ATLIS total scores. The main analyses in the OLE were conducted in the mITT population.
Analysis Populations
The ITT population included all patients who received at least 1 dose of study medication in the main study. Patients in the ITT population were analyzed based on the study medication that they received in the main study. The mITT population included all patients who received at least 1 dose of study medication in the main study and had at least 1 post-baseline total ALSFRS-R score available. Patients were analyzed based on the treatment they were assigned to in the main study. The safety population included all patients who received at least 1 dose of study medication in the OLE. Patients were analyzed based on the actual study medication they received.
Patient Disposition
A total of 97 patients completed the CENTAUR study and were eligible to enrol in the OLE. In addition, a patient who had a brief drug disruption (approximately 1 week) at the very end of the main study was also permitted to enter the OLE. Of these, 90 (93%) patients continued to the OLE: 34 patients who had been originally randomized to placebo and 56 patients who had been originally randomized to PB-TURSO. A smaller percentage of patients discontinued (42.9% versus 52.9%) in the AA group compared to the PA group during the OLE. The most common reasons for study discontinuation were patient decision and death (Table 20).
Table 20: Patient Disposition — CENTAUR-OLE Trial
Patient disposition | PA groupa | AA groupb |
|---|---|---|
Completed RCT | 37 | 60 |
Enrolled in OLE | 34 | 56 |
Ongoing, n (%) | 16 (47.1) | 32 (57.1) |
Discontinued follow-up, n (%) | 18 (52.9) | 24 (42.9) |
Patient decision | 12 | 18 |
Death | 5 | 2 |
Physician decision | 1 | 2 |
Lost to follow-up | 0 | 2 |
Safety population | 34 | 56 |
mITT population | 32 | 54 |
Per-protocol population | 27 | 53 |
AA = active treatment to active treatment; mITT = modified intention-to-treat; OLE = open-label extension; PA = placebo to active treatment; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RCT = randomized clinical trial.
aPA group: This comprised patients randomized to placebo in the main 24-week randomized study and who entered the OLE and received PB-TURSO.
bAA group: This comprised patients randomized to active treatment (PB-TURSO) in the main 24-week randomized study and who entered the OLE and received PB-TURSO.
Source: CENTAUR-OLE Clinical Study Report (2021).42
Exposure to Study Treatments
The exposure data included all data from the baseline of the main study through the OLE. The mean duration of exposure in the safety population was 43.7 weeks (SD = 36.68 weeks) in the AA group and 30.9 weeks (SD = 33.36 weeks) in the PA group. The mean duration of exposure in the mITT population was 45.0 weeks (SD = 36.71 weeks) in the AA group and 32.7 weeks (SD = 33.58 weeks) in the PA group.
The mean compliance (derived by dividing the total number of sachets consumed by the total number of sachets required for consumption per the protocol plan) with PB-TURSO administration throughout the study period of 48 weeks in the safety population was 82.6% (SD = 26.54%) in the PA group and 87.9% (SD = 25.03%) in the AA group. In the mITT population, the mean compliance was 84.8% (SD = 23.73%) in the PA group and 88.3% (SD = 25.03%) in the AA group. Compliance rates were similar between the 2 groups in the safety and mITT populations.
All the patients enrolled in the OLE received at least 1 concomitant medication. In the safety population, the most common medications that patients received were riluzole and edaravone. In the PA group, 28 (82.4%) patients received riluzole and 12 (35.3%) patients received edaravone. In the AA group, 42 (75.0%) patients received riluzole and 20 (35.7%) patients received edaravone.
Efficacy
Efficacy analyses were performed from the initiation of the DB period through the data cut-off date in the OLE. Efficacy data analyses were also performed for the ITT and PP populations and the results were consistent with those from the mITT population (data not included in this report). Data analyses were also performed for the OLE-only portion of the study; however, the data have not been included in this report.
In the ITT population, the median survival was 25.0 months (95% CI lower bound = 20.8 months; upper bound not reached) and 18.5 months (95% CI, 14.9 months to 25.0 months) for the PB-TURSO and placebo groups, respectively, yielding an HR of 0.56 (95% CI, 0.34 to 0.93) for death events at the July 20, 2020, data cut-off date (Table 21). For death or death equivalent events, the median survival was 23.2 months (95% CI lower bound = 19.5 months; upper bound not reached) and 18.2 months (95% CI, 14.9 months to 23.1 months) for the PB-TURSO and placebo groups, respectively, yielding an HR of 0.57 (95% CI, 0.35 to 0.93) at the July 20, 2020, data cut-off date. Figure 2 and Figure 3 show the Kaplan–Meier curves for death events and death or death equivalent events, respectively, with a July 20, 2020, data cut-off date.
A Clinical Study Report with a data cut-off date of March 1, 2021, was provided by the sponsor and contained mature survival data (Table 21).42 The median survival was 23.5 months for the patients randomized to PB-TURSO and 18.7 months for the patients randomized to placebo, resulting in an HR of 0.64 (95% CI, 0.42 to 1.00; P = 0.0475) for death events only. For death or death equivalent events, the median survival was 23.2 months for patients randomized to PB-TURSO and 17.9 months for patients randomized to placebo, yielding an HR of 0.62 (95% CI, 0.40 to 0.96; P = 0.0308). The Combined FDA and Applicant Briefing Document for the March 30, 2022, meeting of the Peripheral and Central Nervous System Drugs Advisory Committee contains additional survival data and Figure 4 shows the Kaplan–Meier curve for death events in the ITT population based on the same March 1, 2021, data cut-off date.43 At this cut-off date, 94 death events were reported (69% of the ITT population), with 1 patient lost to follow-up.43 The FDA noted that, using the likelihood ratio test specified in the survival statistical analysis plan, the HR is 0.64 with a P value of 0.0518.43 The FDA also noted that, with the inclusion of 5 additional death events captured following the March 1, 2021, cut-off date, the HR is 0.70 with a P value of 0.1109.43 However, it was unclear how this was determined and the analysis would not have been from a planned data cut-off date.
Table 21: Secondary Outcomes for Death and Death Equivalent Events — CENTAUR-OLE Trial, ITT Populationa
NA, Event | CENTAUR-OLE | |
|---|---|---|
RAb + SOC N = 89 | RPc + SOC N = 48 | |
Death events only | ||
February 29, 2020, cut-off | ||
Number of events, n (%) | 36 (40.4) | 22 (45.8) |
Median survival,d months (95% CI) | 25.0 (20.3 to NR) | 18.5 (14.8 to NR) |
HR, PB-TURSO vs. placebo (95% CI) | 0.60 (0.35 to 1.04) | Reference |
P valuee | 0.07 | Reference |
July 20, 2020, cut-off | ||
Number of events, n (%) | 42 (47.2) | 28 (58.3) |
Median survival,d months (95% CI) | 25.0 (20.8 to NR) | 18.5 (14.9 to 25.0) |
HR, PB-TURSO vs. placebo (95% CI) | 0.56 (0.34 to 0.93) | Reference |
P valuee | 0.02 | Reference |
March 1, 2021, cut-off | ||
Number of events, n (%) | Not reported | Not reported |
Median survival,d months (95% CI) | 23.5 (not reported) | 18.7 (not reported) |
HR, PB-TURSO vs. placebo (95% CI) | 0.64 (0.42 to 1.00) | Reference |
P valuee | 0.05 | Reference |
Death or death equivalent eventsf | ||
February 29, 2020, cut-off | ||
Number of events, n (%) | 38 (42.7) | 23 (47.9) |
Median survival,d months (95% CI) | 23.8 (18.8 to NR) | 17.6 (14.8 to 25.0) |
HR, PB-TURSO vs. placebo (95% CI) | 0.56 (0.33 to 0.97) | Reference |
P valuee | 0.04 | Reference |
July 20, 2020, cut-off | ||
Number of events, n (%) | 47 (52.8) | 29 (60.4) |
Median survival,d months (95% CI) | 23.2 (19.5 to NR) | 18.2 (14.9 to 23.1) |
HR, PB-TURSO vs. placebo (95% CI) | 0.57 (0.35 to 0.93) | Reference |
P valuee | 0.02 | Reference |
March 1, 2021, cut-off | ||
Number of events, n (%) | Not reported | Not reported |
Median survival,d months (95% CI) | 23.2 (not reported) | 17.9 (not reported) |
HR, PB-TURSO vs. placebo (95% CI) | 0.62 (0.40 to 0.96) | Reference |
P valuee | 0.03 | Reference |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; del-FS = delta-functional scale; HR = hazard ratio; ITT = intention to treat; NR = not reached; OLE = open-label extension; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RA = randomized to active treatment; RP = randomized to placebo; SOC = standard of care; vs. = versus.
aITT population includes those in the ITT population from the main CENTAUR trial.
bRA group: Patients randomized to active treatment (PB-TURSO) in the CENTAUR trial, patients who continued in the OLE received PB-TURSO in the OLE.
cRP group: Patients randomized to placebo in the CENTAUR trial, patients who continued in the OLE received PB-TURSO in the OLE.
dSurvival was analyzed using a Cox proportional hazards model with covariates for age, baseline ALSFRS-R, and pre-randomization progression rate (del-FS score).
eP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
f“Death or death equivalent” is defined as death, tracheostomy, or permanent assisted ventilation (defined as more than 22 hours daily of noninvasive mechanical ventilation for more than 1 week or 7 days).
Source: CENTAUR-OLE Clinical Study Reports (2021).42,44
Figure 2: Kaplan–Meier Curves for Death Events — CENTAUR-OLE Trial, ITT Population (July 20, 2020, Data Cut-Off)
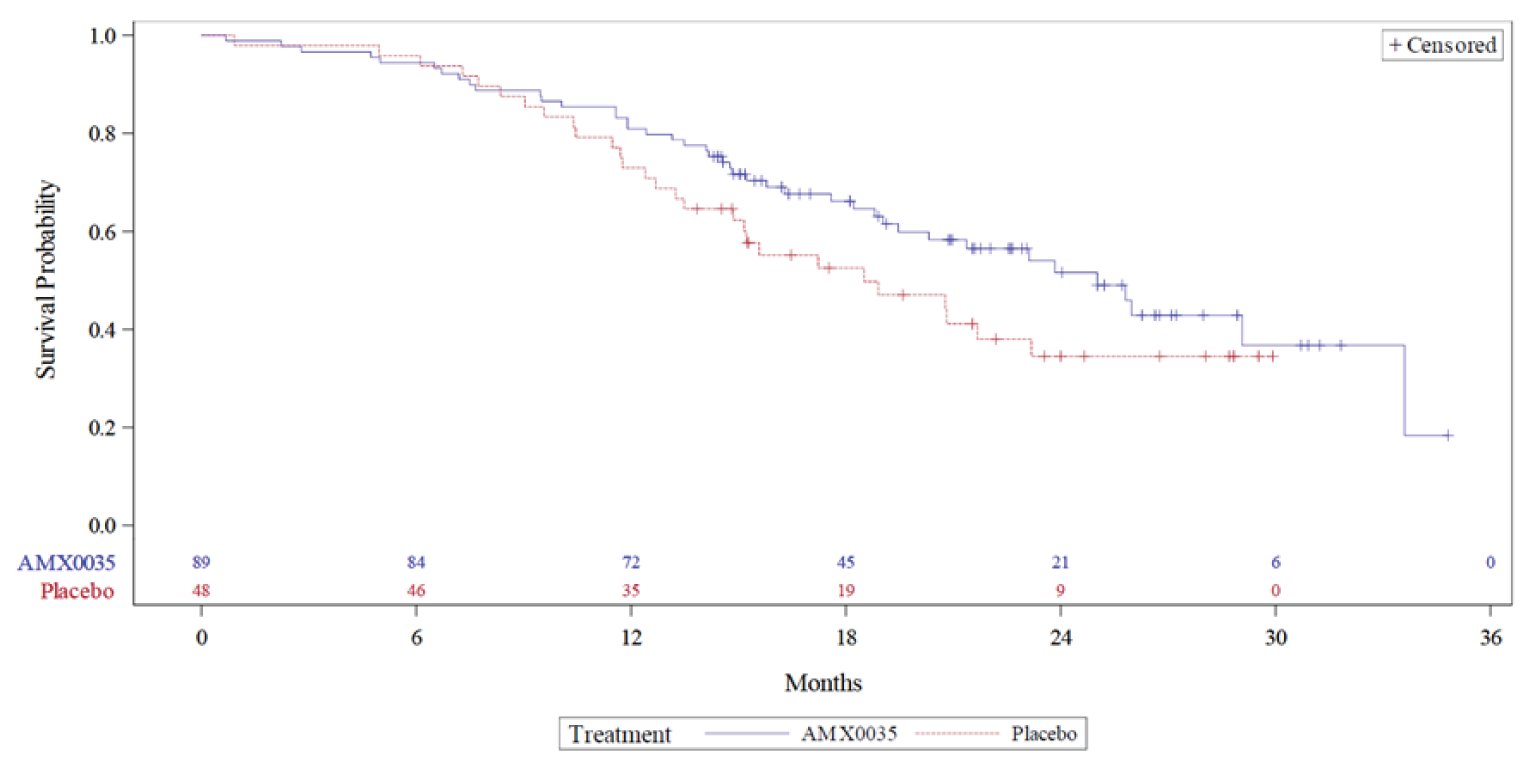
AMX0035 = sodium phenylbutyrate-ursodoxicoltaurine; ITT = intention to treat.
Source: CENTAUR-OLE Clinical Study Reports, with data cut-off dates of July 20, 2020, and March 1, 2021 (2021 for both reports).42,44
Figure 3: Kaplan–Meier Curves for Death or Death Equivalent Events — CENTAUR-OLE Trial, ITT Population (July 20, 2020, Data Cut-Off)
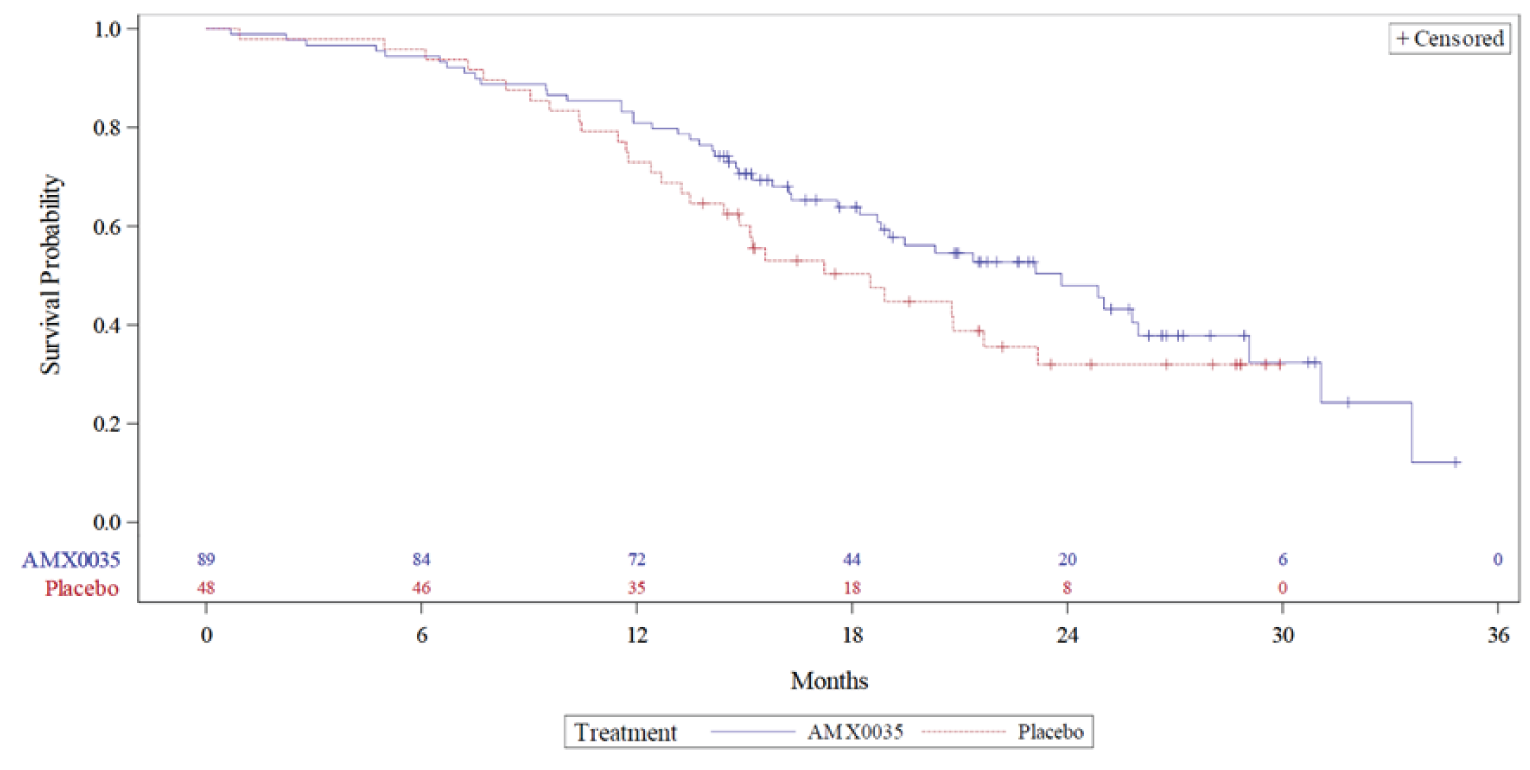
AMX0035 = sodium phenylbutyrate-ursodoxicoltaurine; ITT = intention to treat.
Source: CENTAUR-OLE Clinical Study Report (2021).42
Figure 4: Kaplan–Meier Curves for Death Events — CENTAUR-OLE Trial, ITT Population (March 1, 2021, Data Cut-Off)
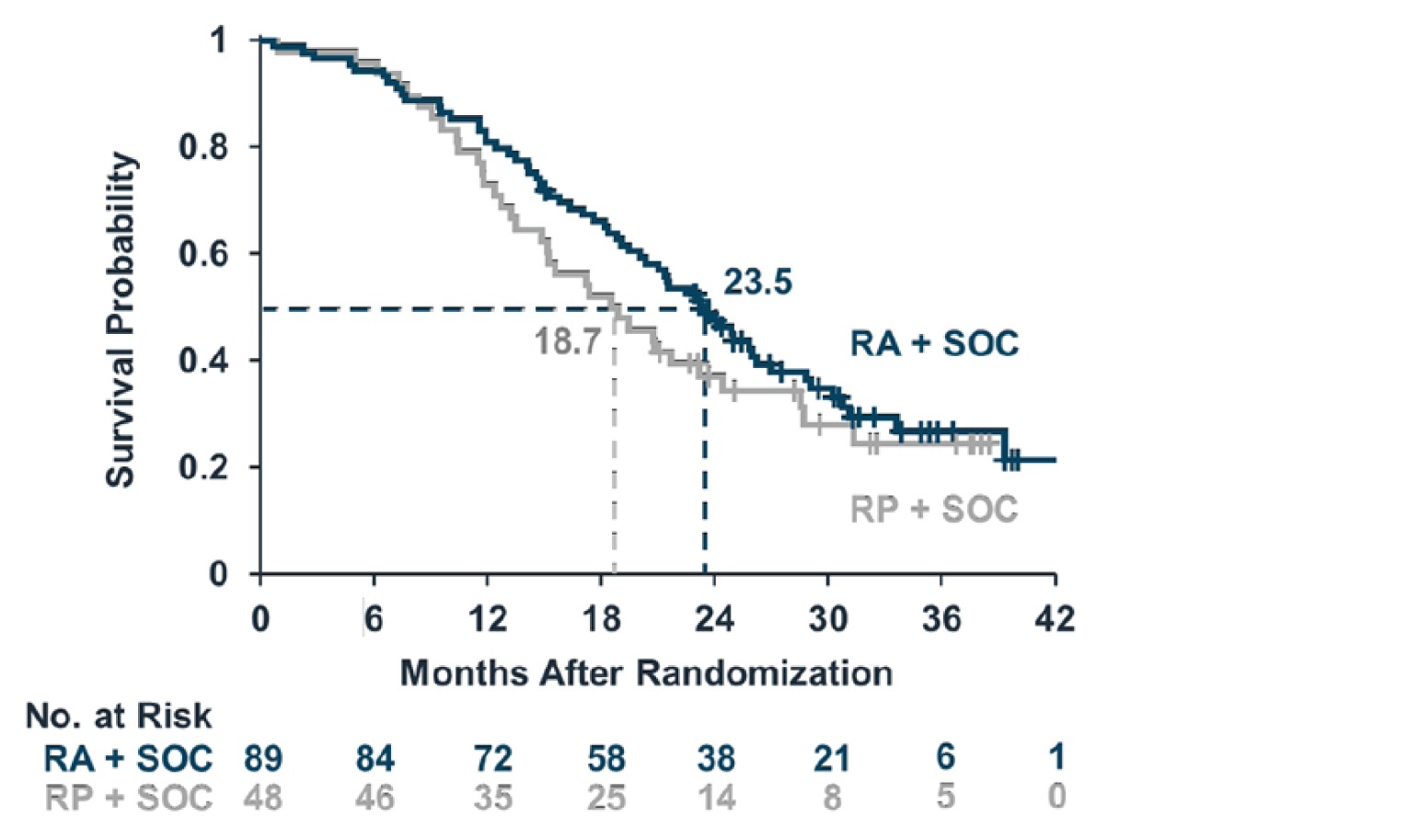
ITT = intention to treat; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RA = randomized to active treatment; RP = randomized to placebo; SOC = standard of care.
Note: The RA group comprised patients who were randomized to active treatment (PB-TURSO) in the CENTAUR trial, while the RP group comprised patients who were randomized to placebo in the CENTAUR trial.
Source: FDA report (2022).43
Patients who were randomized to active treatment (RA) of PB-TURSO in the CENTAUR trial were collectively called the RA group, while patients who were randomized to placebo (RP) in the CENTAUR trial were called the RP group. An analysis for change in the ALSFRS-R total score comparing the difference between the 2 groups (the RA group versus the RP group) from the main study baseline to week 48 was 4.23 points (95% CI, 0.56 points to 7.90 points) in favour of the RA group (Table 22). For ALSFRS-R domain scores, the difference between the RA group and the RP group in change of fine motor function from baseline was 1.70 points (95% CI, 0.27 points to 3.13 points) in favour of the RA group (Table 22). Results for the sensitivity analysis exploring the missing at random assumption for missing data were similar to the results for the main analysis (a difference of 4.28 points [95% CI, 1.69 points to 6.87 points]). For ATLIS scores, the difference between the RA group and the RP group in change of total muscle strength from baseline was 6.20% (95% CI, 0.01% to 12.39%) and change of upper extremities function was 7.83% (95% CI, 0.85% to 14.80%), both in favour of the RA group (Table 22). Lastly, the difference for SVC results from the main study baseline through week 48 overall between the 2 treatment groups (the RA group versus the RP group) was 10.66% (95% CI, 0.63% to 20.69%) in favour of the RA group (Table 22).
Table 22: Secondary Outcomes for ALSFRS-R, ATLIS, and SVC at Week 48 — CENTAUR-OLE Trial, Modified ITT Populationa
NA, Outcome | CENTAUR-OLE trial | |
|---|---|---|
RAb + SOC N = 87 | RPc + SOC N = 48 | |
ALSFRS-R total score | ||
Number of patients contributing to the analysis at week 48 | 36 | 19 |
Baseline, mean (SD) | 35.68 (5.78) | 36.67 (5.08) |
End of treatment (week 48), mean (SD) | 24.82 (10.08) | 23.74 (11.01) |
Change from baseline, mean (SE) | –14.44 (1.13) | –18.67 (1.51) |
Treatment group difference vs. control (95% CI) | 4.23 (0.56 to 7.90) | Reference |
P value | 0.02 | Reference |
ALSFRS-R bulbar domain score | ||
Number of patients contributing to the analysis at week 48 | 36 | 19 |
Baseline, mean (SD) | 9.51 (2.40) | 9.98 (2.60) |
End of treatment (week 48), mean (SD) | 7.15 (3.86) | 6.63 (3.95) |
Change from baseline, mean (SE) | –3.37 (0.36) | –4.03 (0.49) |
Treatment group difference vs. control (95% CI) | 0.67 (–0.53 to 1.86) | Reference |
P value | 0.27 | Reference |
ALSFRS-R fine motor domain score | ||
Number of patients contributing to the analysis at week 48 | 36 | 19 |
Baseline, mean (SD) | 7.99 (2.69) | 8.04 (2.63) |
End of treatment (week 48), mean (SD) | 5.18 (3.42) | 4.21 (3.85) |
Change from baseline, mean (SE) | –4.39 (0.45) | –6.09 (0.60) |
Treatment group difference vs. control (95% CI) | 1.70 (0.27 to 3.13) | Reference |
P value | 0.02 | Reference |
ALSFRS-R gross motor domain score | ||
Number of patients contributing to the analysis at week 48 | 36 | 19 |
Baseline, mean (SD) | 7.54 (2.84) | 7.60 (2.62) |
End of treatment (week 48), mean (SD) | 5.06 (3.07) | 4.58 (4.03) |
Change from baseline, mean (SE) | –3.71 (0.38) | –4.78 (0.51) |
Treatment group difference vs. control (95% CI) | 1.07 (–0.19 to 2.32) | Reference |
P value | 0.10 | Reference |
ALSFRS-R breathing domain score | ||
Number of patients contributing to the analysis at week 48 | 36 | 19 |
Baseline, mean (SD) | 10.64 (1.92) | 11.01 (1.80) |
End of treatment (week 48), mean (SD) | 7.43 (3.49) | 8.32 (2.93) |
Change from baseline, mean (SE) | –2.76 (0.42) | –3.71 (0.57) |
Treatment group difference vs. control (95% CI) | 0.95 (–0.44 to 2.33) | Reference |
P value | 0.18 | Reference |
ATLIS total score | ||
Number of patients contributing to the analysis at week 48 | 28 | 13 |
Baseline, mean (SD) | 56.83 (20.08) | 53.92 (20.94) |
End of treatment (week 48), mean (SD) | 40.41 (17.29) | 37.92 (18.26) |
Change from baseline, mean (SE) | –32.94 (1.89) | –39.14 (2.61) |
Treatment group difference vs. control (95% CI) | 6.20 (0.01 to 12.39) | Reference |
P value | 0.05 | Reference |
ATLIS upper extremities score | ||
Number of patients contributing to the analysis at week 48 | 29 | 13 |
Baseline, mean (SD) | 54.76 (24.40) | 51.44 (25.22) |
End of treatment (week 48), mean (SD) | 36.35 (20.78) | 31.36 (19.22) |
Change from baseline, mean (SE) | –33.38 (2.19) | –41.21 (2.99) |
Treatment group difference vs. control (95% CI) | 7.83 (0.85 to 14.80) | Reference |
P value | 0.03 | Reference |
ATLIS lower extremities score | ||
Number of patients contributing to the analysis at week 48 | 28 | 14 |
Baseline, mean (SD) | 57.65 (24.89) | 57.10 (25.81) |
End of treatment (week 48), mean (SD) | 43.58 (21.88) | 43.04 (28.29) |
Change from baseline, mean (SE) | –32.20 (2.37) | –36.94 (3.27) |
Treatment group difference vs. control (95% CI) | 4.74 (–3.0 to 12.48) | Reference |
P value | 0.23 | Reference |
SVC (percentage predicted) | ||
Number of patients contributing to the analysis at week 48 | 31 | 15 |
Baseline, mean (SD) | 83.62 (18.17) | 83.88 (15.92) |
End of treatment (week 48), mean (SD) | 64.23 (23.70) | 60.33 (21.43) |
Change from baseline, mean (SE) | –35.05 (2.99) | –45.72 (4.17) |
Treatment group difference vs. control (95% CI) | 10.66 (0.63 to 20.69) | Reference |
P value | 0.04 | Reference |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; CI = confidence interval; ITT = intention to treat; OLE = open-label extension; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RA = randomized to active treatment; RP = randomized to placebo; SD = standard deviation; SE = standard error; SOC = standard of care; SVC = slow vital capacity; vs. = versus.
amITT population includes those in the mITT population from the main CENTAUR trial.
bRA group: Patients randomized to active treatment (PB-TURSO) in the CENTAUR trial, patients who continued in OLE received PB-TURSO in the OLE.
cRP group: Patients randomized to placebo in the CENTAUR trial, patients who continued in OLE received PB-TURSO in the OLE.
Source: CENTAUR-OLE Clinical Study Report (2021).42
Harms
The safety data (Table 23) are from the baseline of the OLE study through week 24 of the OLE. The percentage of patients who reported at least 1 AE was higher in the PA group (82.4%) compared with the AA group (73.2%). The most common AEs (with a frequency of at least 5% of patients) included falls (|||||||||| in the PA group vs. |||||||||| in the AA group), nausea (17.6% in the PA group vs. 12.5% in the AA group), and diarrhea (20.6% in the PA group vs. 8.9% in the AA group). Of note, |||||||||||||||||||||||||||||| occurred more frequently during the first 3 weeks of the OLE in the PA group. According to the latest Clinical Study Report with a data cut-off date of March 1, 2021, the number of patients reporting at least 1 AE was 32 (94.1%) patients in the PA group and 49 (87.5%) patients in the AA group.42 Types of AEs were generally similar to those reported at the earlier data cut-off date, with notable increases in the percentage of patients reporting respiratory failure (|||||| in the PA group vs. |||||| in the AA group), dyspnea (|||||| in the PA group vs. |||||| in the AA group), constipation (|||||| in the PA group vs. |||||| in the AA group), and pneumonia (|||||| in the PA group vs. |||||| in the AA group).
The incidence of SAEs was also higher in the PA group (20.6% in the PA group and 14.3% in the AA group). The most common SAEs were |||||||||||||||||||||||||||||| (|||||| in the PA group versus |||||| in the AA group), |||||||||||||||||||||||| (|||||| in the PA group versus |||||| in the AA group), and |||||||||||||||||||||||||||||| (|||||| in the PA group versus |||||| in the AA group). Based on the March 1, 2021, data cut-off, 13 (38.2%) patients and 18 (32.1%) patients experienced at least 1 SAE in the PA and AA groups, respectively.42 |||||||||||||||||||||||||||||| (|||||| in the PA group versus |||||| in the AA group) and |||||||||||||||||||||||| (||||||| in the PA group versus |||||| in the AA group) were SAEs ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
The percentage of patients withdrawing from the study due to AEs was higher in the PA group (29.4%) than the AA group (10.7%) and was mostly due to |||||||||||||||||||||||| (|||||| in the PA group versus |||||| in the AA group).
Five (14.7%) patients in the PA group and 2 (3.6%) patients in the AA group died before week 24 in the OLE study. The reasons were respiratory failure (|||||| in the PA group versus |||||| in the AA group), disease progression or ALS (|||||| in the PA group and |||||| in the AA group), and cardiac arrest (|||||| in the PA group and |||||| in the AA group).
The most common notable harms were nausea (17.6% in the PA group and 12.5% in the AA group) and diarrhea (20.6% in the PA group and 8.9% in the AA group). Dysgeusia was reported only in the PA group (2.9% in the PA group and 0% in the AA group).
Table 23: Primary Outcome: Summary of Harms — CENTAUR-OLE Trial, Safety Population
Safety outcome | PAa + SOC N = 34 | AAb + SOC N = 56 |
|---|---|---|
Patients with ≥ 1 AE | 28 (82.4) | 41 (73.2) |
Most common events (frequency ≥ 5% of patients), n (%) | ||
Fall | |||||||||||| | |||||||||||| |
Nausea | 6 (17.6) | 7 (12.5) |
Diarrhea | 7 (20.6) | 5 (8.9) |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
Patients with ≥ 1 SAE | ||
n (%) | 7 (20.6) | 8 (14.3) |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
Patients who stopped treatment due to AEs | ||
n (%) | 10 (29.4) | 6 (10.7) |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
Deaths | ||
n (%) | 5 (14.7) | 2 (3.6) |
Respiratory failure | |||||||||||| | |||||||||||| |
Disease progression | |||||||||||| | |||||||||||| |
ALS | |||||||||||| | |||||||||||| |
Cardiac arrest | |||||||||||| | |||||||||||| |
Notable harms | ||
|||||||||||| | |||||||||||| | |||||||||||| |
Dysgeusia | 1 (2.9) | 0 (0) |
|||||||||||| | |||||||||||| | |||||||||||| |
Nausea | 6 (17.6) | 7 (12.5) |
Diarrhea | 7 (20.6) | 5 (8.9) |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| |
AA = active treatment to active treatment; AE = adverse event; ALS = amyotrophic lateral sclerosis; OLE = open-label extension; PA = placebo to active treatment; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; SAE = serious adverse event; SOC = standard of care.
aPA group: This comprised patients randomized to placebo in the main 24-week randomized study and who entered the OLE and received PB-TURSO.
bAA group: This comprised patients randomized to active treatment (PB-TURSO) in the main 24-week randomized study and who entered the OLE and received PB-TURSO.
cRespiratory AEs include those listed under MedDRA’s Respiratory, Thoracic, and Mediastinal Disorders heading.
dNeurologic AEs include those listed under MedDRA’s Nervous System Disorders and Psychiatric Disorders headings.
eInvestigational sites were instructed to not capture the bad taste of medication as an AE, but instead to capture emergent AEs relating to challenges of the oral delivery of study medication if they had a clinically untoward effect (e.g., burning, vomiting, anxiety).
Note: Redacted rows have been deleted.
Source: CENTAUR-OLE Clinical Study Report (2021).42
Critical Appraisal
Internal Validity
Given the nature of open-label studies, there may be bias that impacts how the results are interpreted such as the lack of blinding during the OLE phase and the lack of a control group (all patients received PB-TURSO in the OLE). Also, it is possible that treatment assignment from the main trial was deduced for some patients based on the differences in gastrointestinal AEs between groups.
There are many limitations impacting the ability to interpret the efficacy results. There is the possibility of selection bias since patients must have successfully completed the pivotal study and chosen to continue in the extension. In addition, there were large proportions of study discontinuations during the OLE (52.9% in the PA group and 42.9% in the AA group) and only a small proportion of these were due to death. The same limitations identified in the main trial regarding the properties of the ALSFRS-R, ATLIS, and SVC also apply to the OLE phase. Finally, all the efficacy end points were secondary end points. Therefore, it is not possible to make definitive conclusions about efficacy and/or durability of efficacy for the ALSFRS-R, ATLIS, and SVC end points.
Although vital status was available for all but 2 patients from the main trial ITT population, death equivalent events outside the main trial and OLE were not captured, contributing uncertainty to the death or death equivalent composite end point given the proportion of study discontinuations. It is important to capture death equivalent events since noninvasive and invasive ventilation can alter the disease trajectory and extend survival.4 Another source of uncertainty in the survival analyses is the fact that patients who received placebo in the main trial crossed over to receive PB-TURSO in the OLE. Assuming any effect of PB-TURSO on survival is beneficial, bias from treatment switching would be against PB-TURSO.
External Validity
As the CENTAUR-OLE trial is the extension study of the main CENTAUR trial, the generalizability issues identified for the main trial regarding patient characteristics and outcome measures also apply in the OLE. However, the short duration of the main trial for assessing survival was addressed by the longer follow-up period in the OLE.
Post-Hoc Analysis of PB-TURSO Versus Edaravone in the CENTAUR Study
The sponsor concluded that an MAIC was infeasible; therefore, a post-hoc analysis of the CENTAUR trial was conducted to compare the relative efficacy of PB-TURSO to edaravone to support the sponsor’s pharmacoeconomic model.
Methods
Details of the CENTAUR trial have been described previously in this CADTH report. Edaravone was approved for the treatment of ALS in the US after the study began, resulting in a protocol amendment that allowed patients to enrol if they had started or were planning to start edaravone treatment. To inform the inputs of the economic model, a post-hoc analysis comparing PB-TURSO and edaravone was performed. The sponsor noted that to best estimate the effect of PB-TURSO versus edaravone, the subgroups of interest included patients who received PB-TURSO without edaravone and patients who received placebo with edaravone.
Populations and Interventions
Separate analyses were conducted based on edaravone use defined as use at or before study entry, or use during or before the study.
Outcomes
The primary end point was the rate of change (slope) in ALSFRS-R total score from baseline to week 24 using a shared-baseline mixed-effects model. The secondary end point was the ALSFRS-R total score change from baseline to week 24 without using a shared-baseline approach.
Statistical Analysis
The mITT population (N = 135) was used for the post-hoc analysis. Covariates for the primary end point included age and pre-baseline del-FS (interacting with time). Covariates for the secondary end point were the same as those for the primary end point, with the addition of the baseline ALSFRS-R score. The sponsor indicated that age, sex, time since diagnosis, site of onset, and concomitant use of riluzole were possibly imbalanced between the groups; therefore, additional adjustments were made for sex, time since diagnosis, site of onset, and concomitant use of riluzole (interacting with time). The site of disease onset and riluzole use were considered to be the most important prognostic factors to adjust for.
Efficacy
For the primary end point using a shared-baseline approach, the estimated effect sizes between patients in the PB-TURSO without edaravone group and patients in the placebo with edaravone group varied from 2.62 points to 3.22 points in favour of PB-TURSO. For the secondary end point using a change from baseline approach, the estimated effect sizes between patients in the PB-TURSO without edaravone group and patients in the placebo with edaravone group varied from 3.61 points to 4.41 points in favour of PB-TURSO. The results for the secondary end point supported those of the primary outcome and were in the same direction, regardless of adjusting for the additional covariates.
Critical Appraisal
A key limitation was that the comparative efficacy results were based on post-hoc analyses, not pre-hypothesized; therefore, they should be viewed as hypothesis-generating. Defining treatment groups by whether a patient received edaravone meant that the benefits of randomization were lost for these comparisons. Additionally, the groups included only a subset of the mITT population and sample sizes were small as a result. Given these serious limitations, it is not possible to make any firm conclusions based on the data available for how treatment with PB-TURSO compared to edaravone.
Post-Hoc Analysis of Overall Survival Accounting for Treatment Switching
The sponsor conducted a post-hoc analysis of the survival data from patients in the CENTAUR trial and its extension up to 35 months post-baseline (with a data cut-off date of July 20, 2020) to account for the potential effects on overall survival of switching treatments (i.e., placebo to PB-TURSO).
Populations and Interventions
In total, 34 of the 48 (71%) patients who received placebo during the DB phase enrolled in the CENTAUR-OLE study and began receiving PB-TURSO. The sponsor noted that any beneficial effect on overall survival of PB-TURSO over placebo in the absence of a switch will be underestimated in the pre-specified ITT analysis.
Outcomes
The overall survival end point was defined as all-cause mortality. The main objective of the analysis was to model what the overall survival of patients in the CENTAUR trial may have been if the patients from the placebo group had not switched treatments and received PB-TURSO.
Statistical Analysis
The post-hoc analysis included all randomized patients (N = 137). Censoring dates were 30 days before the date of each patient’s survival check. Survival data were missing for 2 individuals and as a result, were censored at their last known date of survival. The primary analysis (“on treatment”) considered the duration of switch treatment effect to include only the days a patient received PB-TURSO and did not include time after a patient discontinued the drug.
Due to the large proportion of patients who switched, the inverse probability of censoring weighting and 2-stage models were ruled out in favour of a RPSFT model that assumed that the treatment effect was consistent regardless of when it was given during the study (i.e., at randomization or upon enrolment to the OLE). The model estimated the counterfactual survival time without PB-TURSO as well as the effect of PB-TURSO to extend survival while receiving the treatment. The acceleration factor was estimated using G-estimation and the Cox statistic with covariates for baseline age, pre-baseline ALSFRS-R slope, and baseline ALSFRS-R total score. Analyses were conducted with and without recensoring. For patients who received only placebo and not PB-TURSO, the adjusted survival time was the counterfactual survival time based on the acceleration factor estimate.
Kaplan–Meier curves were constructed for the PB-TURSO group based on observed data and for the placebo (without switching) group based on adjusted survival data. HRs were calculated using a Cox analysis and covariates for baseline age, pre-baseline ALSFRS-R slope, and baseline ALSFRS-R total score. Symmetric CIs were constructed for the log HR using the ITT P value.
Efficacy
Efficacy results using the RPSFT model for the on-treatment approach have been summarized in Table 24. Overall, the median overall survival was approximately 25.0 months for the PB-TURSO group (N = 89) versus 18.5 months for the placebo group (N = 48). This indicated that patients who received PB-TURSO had a survival benefit over those who received placebo, with an HR of 0.56 (95% CI, 0.34 to 0.93). Using the RPSFT model without recensoring, the median overall survival was approximately 13.5 months for the placebo group with an HR of 0.34 (95% CI, 0.13 to 0.87). When recensoring was applied, the median overall survival was between the results for the ITT and RPSFT without recensoring analyses — 15.2 months (HR = 0.40; 95% CI, 0.18 to 0.88).
Table 24: Summary of Results From the RPSFT Model, On-Treatment Approach — Intention-to-Treat Population
Modela | Median OS (months), placebo | Median OS (months), PB-TURSO | HR (95% CI) |
|---|---|---|---|
ITT | 18.5 | 25.1 | 0.56 (0.34 to 0.93) |
RPSFT model, no recensoring | 13.5 | 25.1 | 0.34 (0.13 to 0.87) |
RPSFT model, recensoring AF only | 15.2 | 25.1 | 0.40 (0.18 to 0.88) |
RPSFT model, full recensoring | NR | 25.1 | 0.44 (0.22 to 0.90) |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; AF = acceleration factor; CI = confidence interval; del-FS = delta-functional scale; HR = hazard ratio; NR = not reached; OS = overall survival; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; RPSFT = rank preserving structural failure time.
aOn-treatment approach used for the RPSFT model. Cox proportional hazards models include covariates for age, del-FS, and ALSFRS-R score at baseline.
Source: Sponsor’s Statistical Report for Modelling Overall Survival submitted for the review of PB-TURSO.41
Critical Appraisal
The aforementioned limitations regarding the ITT survival analyses performed during the OLE also apply to the RPSFT model results. While the methodology was intended to correct for bias introduced by treatment switching for patients randomized to placebo continuing in the OLE, the assessment relied on the assumption that treatment effect was consistent regardless of when it was given during the study. A treatment that is expected to be neuroprotective may not have the same treatment effect at later disease stages when there are fewer surviving motor neurons. The validity of the main assumption of the RPSFT method is unknown and no conclusions can be drawn from the RPSFT model results.
Discussion
Summary of Available Evidence
One pivotal trial for PB-TURSO, the CENTAUR trial (N = 137), met the inclusion criteria for the CADTH systematic review. The CENTAUR trial was a multi-centre, phase II, DB, randomized, placebo-controlled study. Patients were adults between 18 and 80 years of age, had a diagnosis of definite ALS (sporadic or familial) as defined by the World Federation of Neurology–revised El Escorial criteria, were within 18 months from the onset of ALS symptoms, had an SVC of greater than 60% of predicted value, and were on a stable dose of concomitant medication (i.e., riluzole and/or edaravone). Patients were randomized 2:1 to receive either PB-TURSO or matching placebo that was administered orally or via feeding tube as 1 sachet once daily for the first 3 weeks, then 1 sachet twice daily thereafter for up to 24 weeks. The primary outcomes of the CENTAUR study were to confirm the safety and tolerability of PB-TURSO as well as to assess the impact of PB-TURSO on the rate of change of the ALSFRS-R. Key secondary objectives included the rate of change of isometric muscle strength using the ATLIS, the rate of change of SVC, and rates of survival (defined as death, tracheostomy, or permanent assisted ventilation).
Patients who completed the DB CENTAUR trial could enrol in the CENTAUR-OLE study (N = 90), which evaluated the long-term safety and efficacy of PB-TURSO. All patients, regardless of which treatment they were randomized to in the DB phase, could receive PB-TURSO for up to an additional 132 weeks of treatment. The primary objective was to assess the long-term safety of PB-TURSO. Key secondary outcomes included the rate of key events (tracheostomy, permanent assisted ventilation, death) and the rate of change for the ALSFRS-R, ATLIS, and SVC.
In addition to the CENTAUR and CENTAUR-OLE studies, a sponsor-submitted feasibility assessment for an MAIC and 2 post-hoc analyses comparing the relative efficacy of PB-TURSO to edaravone and estimating overall survival while accounting for treatment crossover in the OLE study were summarized and appraised.
The key limitations with the CENTAUR trial include its phase II design, small number of patients, narrow eligibility criteria, and amount of missing data.
Interpretation of Results
Efficacy
The primary efficacy end point of the CENTAUR trial was to assess the rate of change of progression as measured by the ALSFRS-R. Using the ALSFRS-R as a primary efficacy outcome aligns with FDA guidance39 and the instrument is typically used in ALS clinics in Canada, as confirmed by the clinical expert consulted for this review. Over the 24-week study, the PB-TURSO group had a mean change from baseline in ALSFRS-R total score of –6.86 points (SE = 0.66 points) and the placebo group had a mean change of –9.18 points (SE = 0.88 points). Treatment with PB-TURSO showed a slowing of disease progression as indicated by the 2.32-point difference (95% CI, 0.18 points to 4.47 points; P = 0.03) from the placebo group, which was statistically significant, and the primary end point was met. According to the clinical expert consulted for this review, a difference of at least 2 points over a period of 6 months for most patients with ALS would be considered clinically meaningful if found to be reproducible through additional studies. Additionally, a change of 20% to 25% in the slope of ALSFRS-R was considered “at least somewhat clinically meaningful,” according to surveyed clinical experts.17 Therefore, a difference of 2.32 points between the treatment groups from the main analysis of the primary end point was both statistically significant and clinically meaningful, supporting the superiority of PB-TURSO over placebo for slowing progression as measured by the ALSFRS-R for patients in this particular study population. However, there was substantial missing data by the end of the 24-week treatment period and a sensitivity analysis exploring the missing at random assumption yielded a difference of 1.87 points between treatment groups, which may not be considered clinically meaningful. It should be noted that the threshold for clinical meaningfulness was based solely on expert opinion as an MID estimate was not found in the literature. While the ALSFRS-R total score results from the CENTAUR-OLE suggested continued benefit, study discontinuations during the OLE were even more extensive than in the main trial and firm conclusions could not be drawn.
Since the results of the secondary outcome for the ATLIS scores were not statistically significant, P values were nominal for all other secondary and exploratory outcomes. Results for the ATLIS, SVC, and ALSFRS-R domains were in the same direction and supported the results of the primary ALSFRS-R outcome. While the ATLIS and SVC are recommended outcome measures for clinical trials,34 the clinical expert consulted for this review indicated that they are not commonly used in Canadian ALS clinics and the generalizability of the study results for these outcomes may be limited.
Overall survival results from the first 24 weeks of the DB CENTAUR trial were immature as few events had occurred (6 events were considered death or death equivalent). Long-term survival results were assessed for up to 35 months post-randomization and all patients who enrolled in the CENTAUR-OLE study received PB-TURSO. Survival analyses were conducted using vital status sweeps to supplement data obtained in the main trial and OLE. For the outcome of death events only (July 2020 data cut-off), patients in the ITT population who were randomized to PB-TURSO in the main trial had a median survival of 25.0 months (95% CI lower bound = 20.8 months; upper bound not reached) and patients randomized to placebo had a median survival of 18.5 months (95% CI, 14.9 months to 25.0 months). This yielded an HR of 0.56 (95% CI, 0.34 to 0.93) for PB-TURSO versus placebo. Data available from the sponsor’s Clinical Study Report contained a later cut-off date of March 1, 2021.42,43 The reports showed median survivals of approximately 23.5 months and 18.7 months for patients randomized to PB-TURSO and placebo, respectively, resulting in an HR of 0.64 (95% CI, 0.42 to 1.00; P = 0.0453) for death events only. In the Combined FDA and Applicant Briefing Document for the March 30, 2022, meeting of the Peripheral and Central Nervous System Drugs Advisory Committee, the FDA also noted that an additional 5 deaths after March 1, 2021, were administratively censored and had the deaths been included in the analysis, the HR would have been 0.70 (P = 0.11) for the ITT population.43 However, it was unclear how this was determined and the analysis would not have been from a planned data cut-off. Additionally, there is uncertainty in the magnitude of the survival benefit given that information on death equivalent events was not available for patients who discontinued the main trial or OLE and patients randomized to placebo switched to treatment with PB-TURSO during the OLE. An RPSFT model was used to adjust the results of the survival analysis for death events to account for potential bias against PB-TURSO introduced by treatment switching, but the RPSFT model results were not interpretable due to uncertainty in the assumption of constant treatment effect underpinning the approach.
At week 48 post-randomization, results for the change in ALSFRS-R total score, ATLIS, and SVC from baseline to week 48 numerically favoured PB-TURSO. Due to the small number of patients, large proportions of missing data, selection bias for patients who successfully completed the DB phase, and efficacy outcomes being secondary end points, no firm conclusions about long-term efficacy for these outcomes can be made.
Riluzole and edaravone were identified as relevant comparators, but no published direct or indirect evidence comparing PB-TURSO with either treatment was found. To inform the submitted pharmacoeconomic model, the sponsor conducted a post-hoc analysis comparing patients who received PB-TURSO without edaravone to those who received placebo with edaravone in the CENTAUR trial (as an MAIC was deemed infeasible). The results of the analysis suggested that patients who received PB-TURSO without edaravone had less decline in the ALSFRS-R total score compared to patients who received placebo with edaravone in this study. Aside from the post-hoc nature of the analysis, the major limitations were the loss of randomization, the small number of patients included in the analysis, and not being sufficiently powered to find a treatment difference. Therefore, conclusions could not be drawn on the efficacy of PB-TURSO versus edaravone.
The mean rate of change on the ALSFRS-R was –1.24 points per month for the PB-TURSO group and –1.66 points per month for the placebo group. It was noted in both the FDA guidance34 as well as by the clinical expert consulted for this review that patients with ALS have an average decline of 1 point per month on the ALSFRS-R. It is important to also consider that the CENTAUR study population was more rapidly progressing than patients with ALS on average, as per the opinion of the clinical expert. Also, the safety and efficacy of PB-TURSO are unknown outside this study population, which was restricted to patients with a definite ALS diagnosis and within 18 months of symptom onset. The clinical expert noted that there is no physiologic or pharmacological reason indicating that patients at other levels of the El Escorial diagnostic criteria (i.e., “probable” or “possible”) would not respond to the treatment.
Since the CENTAUR trial was a phase II study, it is important to confirm the findings in a phase III study. The PHOENIX trial (NCT05021536) is an ongoing randomized, DB, phase III study evaluating the safety and efficacy of PB-TURSO versus placebo for 48 weeks in adults with ALS.45 The PHOENIX study population is broader than that of the CENTAUR trial, allowing the enrolment of patients with a diagnosis of definite ALS or clinically probable ALS and who are 24 months or less from symptom onset.
The FDA guidance recommends the use of patient-reported outcomes to support the primary analysis, though there were no outcomes specific to HRQoL in the CENTAUR study.34 It is uncertain what benefits treatment with PB-TURSO provides beyond the outcomes captured in the trial, particularly for symptom management, patients being able to maintain their independence, and caregiver burden, which were identified as being important to the patient groups that provided input for the review process.
Harms
During the CENTAUR trial, nearly all patients experienced at least 1 TEAE, with 86 (96.6%) patients in the PB-TURSO group and 46 (95.8%) patients in the placebo group reporting a TEAE. The TEAEs reported in more than 15% of patients in the PB-TURSO group were falls, diarrhea, muscular weakness, and nausea. In the placebo group, the TEAEs reported in more than 15% of patients were falls, constipation, headache, muscular weakness, and diarrhea. Gastrointestinal AEs (e.g., diarrhea, nausea, salivary hypersecretion, abdominal discomfort) were more frequently reported in the PB-TURSO group compared to the placebo group.
In total, 23 SAEs were reported among 19 (13.9%) patients during the CENTAUR trial, consisting of 11 (12.4%) patients from the PB-TURSO group and 8 (16.7%) patients from the placebo group. Overall, 18 (20.2%) patients from the PB-TURSO group and 5 (10.4%) patients from the placebo group withdrew from the study medication due to a TEAE. There were 7 deaths reported during the CENTAUR trial in the safety population: 5 (5.6%) patients in the PB-TURSO group due to respiratory failure or respiratory arrest (3 patients), subdural hematoma (1 patient), and diverticular perforation (1 patient) compared to 2 (2.2%) patients in the placebo group, both due to respiratory failure or respiratory arrest. According to the clinical expert, the reasons for AEs leading to withdrawal or death, aside from gastrointestinal AEs, were consistent with events that typically occur in individuals with ALS of this age group or were otherwise too infrequent (single-patient events) to draw any firm conclusions about associations with the treatment.
For notable harms, gastrointestinal AEs reported by more than 10% of patients for a single treatment group included diarrhea, nausea, constipation, and salivary hypersecretion. Neurologic AEs reported by more than 10% of patients overall included headache and dizziness. Dyspnea was the only respiratory AE reported by more than 10% of patients during the CENTAUR trial. The clinical expert highlighted medication taste as being a particular concern with PB-TURSO. Although dysgeusia was reported for 4 patients overall, the study investigators were instructed not to report the bad taste of medication as an AE as it was not classified as an AE per the Common Terminology Criteria for Adverse Events Version 5.0. Thus, taste disturbance was likely not captured (as it was intended in the CADTH review protocol) and it is unknown what impact this would have had on the results (e.g., harms, withdrawals). A patient group that provided input for the CADTH review indicated the bitter taste of the medication was a drawback but described it as being tolerable over time.
Overall, the clinical expert consulted for this review did not consider there to be any major concerns with the harms recorded or the imbalances between the treatment groups. The clinical expert noted that, although the medication’s disagreeable taste was not captured as a harm, this would be important information to both clinicians and patients. There did not appear to be any new safety concerns raised in the CENTAUR-OLE study. As with the main trial, safety data were limited by sample size. WDAEs, and particularly gastrointestinal-related WDAEs, occurred more frequently in the PB-TURSO group and this may impact some patients’ ability to continue with treatment.
Conclusions
The CENTAUR trial results indicated a statistically significant difference in favour of PB-TURSO over placebo for the primary outcome of slowing disease progression as measured by the rate of change of the ALSFRS-R total score in adults who have a diagnosis of definite ALS, have an SVC greater than 60% of the predicted value, and are within 18 months of symptom onset. The clinical relevance of the treatment effect is unclear due to uncertainty introduced by the amount of missing data. There were no other statistically significant findings, though results for the ATLIS, SVC, and the ALSFRS-R domain scores supported the primary end point result. Survival analyses conducted during the OLE study suggested a survival benefit for PB-TURSO over placebo, but variation in the results from different data cut-off dates, lack of adjustment for analyses at multiple time points, missing data for death equivalent events, and treatment switching during the extension mean that the finding may not be robust, and the magnitude of the treatment effect is uncertain. Conclusions regarding efficacy outcomes, other than survival, beyond 24 weeks of treatment could not be drawn. Outcomes for HRQoL and caregiver burden, both identified by patients as being important, were not included in the CENTAUR trial. It should also be noted that the narrow eligibility criteria for the CENTAUR trial resulted in a trial population that was representative of only a subpopulation of patients with ALS. The comparative efficacy of PB-TURSO versus edaravone or riluzole is unknown as the only evidence available was a post-hoc analysis of the CENTAUR trial that had serious limitations. Firm conclusions regarding the safety of PB-TURSO could not be drawn due to the limited sample size of the CENTAUR trial, though the results suggest that gastrointestinal AEs associated with PB-TURSO contribute to treatment discontinuations. Overall, a major limitation of the CENTAUR trial is the fact that it is a phase II trial; it is important that the efficacy and safety findings be confirmed in phase III trials.
References
1.Brotman RG, Moreno-Escobar MC, Joseph J, Pawar G. Amyotrophic lateral sclerosis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022: https://www.ncbi.nlm.nih.gov/books/NBK556151/. Accessed 2022 Feb 11.
2.National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. 2022; https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet#definition. Accessed 2022 Feb 11.
3.Johns Hopkins Medicine. ALS - amyotrophic lateral sclerosis. 2022; https://www.hopkinsmedicine.org/neurology_neurosurgery/centers_clinics/als/conditions/als_amyotrophic_lateral_sclerosis.html. Accessed 2022 Feb 11.
4.Shoesmith C, Abrahao A, Benstead T, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ. 2020;192(46):E1453-e1468. PubMed
5.Elman LB, McCluskey L. Clinical features of amyotrophic lateral sclerosis andother forms of motor neuron disease. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Dec 9.
6.Mayo Clinic. Amyotrophic lateral sclerosis (ALS). 2022; https://www.mayoclinic.org/diseases-conditions/amyotrophic-lateral-sclerosis/symptoms-causes/syc-20354022. Accessed 2022 Feb 11.
7.ALS Society of Canada. About ALS. 2018; https://als.ca/what-is-als/about-als/. Accessed 2022 Feb 11.
8.Wolfson C, Kilborn S, Oskoui M, Genge A. Incidence and prevalence of amyotrophic lateral sclerosis in Canada: a systematic review of the literature. Neuroepidemiology. 2009;33(2):79-88. PubMed
9.ALS Association. Who gets ALS? 2022; https://www.als.org/understanding-als/who-gets-als. Accessed 2022 Feb 11.
10.Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10(5-6):310-323. PubMed
11.ALS News Today. Prognosis of ALS. 2022; https://alsnewstoday.com/prognosis-of-als/. Accessed 2022 Feb 11.
12.Galvez-Jimenez N. Symptom-based management of amyotrophic lateralsclerosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Dec 12.
13.Elman LB, McCluskey L. Diagnosis of amyotrophic lateral sclerosis and otherforms of motor neuron disease. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Dec 12.
14.Hodgkinson VL, Lounsberry J, Mirian A, et al. Provincial differences in the diagnosis and care of amyotrophic lateral sclerosis. Can J Neurol Sci. 2018;45(6):652-659. PubMed
15.Agosta F, Al-Chalabi A, Filippi M, et al. The El Escorial criteria: strengths and weaknesses. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(1-2):1-7. PubMed
16.Albrioza (sodium phenylbutyrate and ursodoxicoltaurine): powder for suspension, 3 g/1 g sachet, oral [product monograph]. Milton (ON): Innomar Strategies; 2022 Jun 1.
17.Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler. 2010;11(1-2):178-180. PubMed
18.Clinical Study Report: AMX3500 (CENTAUR). Evaluation of the safety, tolerability, efficacy and activity of AMX0035, a fixed combination of phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA), for treatment of amyotrophic lateral sclerosis (ALS) [internal sponsor's report]. Cambridge (MA): Amylyx Pharmaceuticals, Inc; 2021 May 11.
19.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: AMX0035 (sodium phenylbutyrate and ursodoxicoltaurine), 3 mg sachet sodium phenylbutyrate sachet and 1 mg sachet ursodoxicoltaurine powder for suspension, oral. Calgary (AB): Amylyx Canada; 2021 Nov 24.
20.ALS and genetics. Toronto (ON): ALS Society of Canada; 2020: https://www.als.ca/wp-content/uploads/2020/12/Fact-Sheet-Genetics_FINAL.pdf. Accessed 2022 Feb 11.
21.Ghasemi M, Brown RH, Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med. 2018;8(5). PubMed
22.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124 Suppl:96-107. PubMed
23.Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131(8):1975-1978. PubMed
24.Radicava (edaravone): solution, 30mg/100mL (0.3mg/mL), intravenous administration [product monograph]. Jersey City (NJ): Mitsubishi Tanabe Pharma America, Inc., a US subsidiary of Mitsubishi Tanabe Pharma Corporation; 2021 Mar 25: https://pdf.hres.ca/dpd_pm/00060555.PDF. Accessed 2022 Feb 11.
25.Rilutek (riluzole): film-coated tablets, 50 mg [product monograph]. Laval (QC): sanofi-aventis Canada Inc; 2010 May 11: https://pdf.hres.ca/dpd_pm/00010806.PDF. Accessed 2022 Feb 11.
26.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
27.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Dec 10.
28.Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919-930. PubMed
29.Paganoni S, Hendrix S, Dickson SP, et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021;63(1):31-39. PubMed
30.Cedarbaum JM, Stambler N. Performance of the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) in multicenter clinical trials. J Neurol Sci. 1997;152 Suppl 1:S1-9. PubMed
31.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169(1-2):13-21. PubMed
32.Andres PL, Skerry LM, Munsat TL, et al. Validation of a new strength measurement device for amyotrophic lateral sclerosis clinical trials. Muscle Nerve. 2012;45(1):81-85. PubMed
33.Gordon PH, Cheng B, Montes J, Doorish C, Albert SM, Mitsumoto H. Outcome measures for early phase clinical trials. Amyotroph Lateral Scler. 2007;8(5):270-273. PubMed
34.Drug development for amyotrophic lateral sclerosis: guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration; 2017: https://www.als.org/sites/default/files/2020-07/Drug-Development-for-Amyotrophic-Lateral-Sclerosis-Guidance-for-Industry-August-2017.pdf. Accessed 2022 Feb 11.
35.Atassi N, Berry J, Shui A, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology. 2014;83(19):1719-1725. PubMed
36.Gray R, Manola J, Saxman S, et al. Phase II clinical trial design: methods in translational research from the Genitourinary Committee at the Eastern Cooperative Oncology Group. Clin Cancer Res. 2006;12(7 Pt 1):1966-1969. PubMed
37.The drug development process. Step 3: clinical research. Silver Spring (MD): U.S. Food and Drug Administration; 2018: https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. Accessed 2022 Feb 10.
38.Daghlas I, Lever TE, Leary E. A retrospective investigation of the relationship between baseline covariates and rate of ALSFRS-R decline in ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(3-4):206-211. PubMed
39.Center for Drug Evaluation and Research. Amyotrophic lateral sclerosis: developing drugs for treatment guidance for industry. Rockville (MD): U.S. Food and Drug Administration; 2019: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/amyotrophic-lateral-sclerosis-developing-drugs-treatment-guidance-industry. Accessed 2022 Feb 11.
40.Common Drug Review clinical review report: Edaravone (radicava) for the treatment of amyotrophic lateral sclerosis. Ottawa (ON): CADTH; 2019 Apr: https://cadth.ca/sites/default/files/cdr/clinical/sr0573-radicava-clinical-review-report.pdf. Accessed 2022 Feb 11.
41.Drug Reimbursement Review pharmacoeconomic report: economic evaluation of Albrioza (sodium phenylbutyrate and ursodoxicoltaurine) for the treatment of amyotrophic lateral sclerosis. Ottawa (ON): CADTH; [forthcoming 2022]: https://www.cadth.ca/sodium-phenylbutyrate-and-ursodoxicoltaurine. Accessed 2022 Apr 4.
42.Clinical Study Report: AMX3500-OLE (CENTAUR-OLE). Evaluation of the safety, tolerability, efficacy, and activity of AMX0035, a fixed combination of phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA), for treatment of amyotrophic lateral sclerosis: open-label extension [internal sponsor's report]. Cambridge (MA): Amylyx Pharmaceuticals, Inc; 2021 Oct 23.
43.Peripheral and Central Nervous System Drugs Advisory Committee. Combined FDA and Amylyx briefing document. Rockville (MD): U.S. Food and Drug Administration; 2022 Mar 30: https://www.fda.gov/media/157186/download. Accessed 2022 Mar 30.
44.Clinical Study Report: AMX3500-OLE (CENTAUR-OLE). Evaluation of the safety, tolerability, efficacy, and activity of AMX0035, a fixed combination of phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA), for treatment of amyotrophic lateral sclerosis: open-label extension [internal sponsor's report]. Cambridge (MA): Amylyx Pharmaceuticals, Inc; 2021 May 14.
45.Amylyx Pharmaceuticals Inc. NCT05021536: Phase III trial of AMX0035 for amyotrophic lateral sclerosis treatment (Phoenix). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT05021536. Accessed 2022 Feb 24.
46.McElhiney M, Rabkin JG, Goetz R, et al. Seeking a measure of clinically meaningful change in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5-6):398-405. PubMed
47.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
48.Shefner JM. Designing clinical trials in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):495-508, ix. PubMed
49.Gordon PH, Miller RG, Moore DH. ALSFRS-R. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5 (Suppl 1):90-93. PubMed
50.Andres PL, English R, Mendoza M, et al. Developing normalized strength scores for neuromuscular research. Muscle Nerve. 2013;47(2):177-182. PubMed
51.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
Appendix 1: Literature Search Strategy
Note that this table has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: December 17, 2021
Alerts: Weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Humans
Language limit: none
Conference abstracts: excluded.
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Relyvrio* or amx-0035 or amx0035 or PB-TURSO).ti,ab,kf,ot,hw,nm,rn.
Phenylbutyrates/ or (Buphenyl* or phenylbutyrate* or phenylbutanoate* or phenylbutanoic acid or TriButyrate* or ammonaps* or phenylbutyric acid* or pheburane* or lunaphen* or satisma* or nsc 657802 or nsc657802 or "acer 001" or acer001 or cmk 304 or cmk304 or lu 901 or lu901 or NT6K61736T or 7WY7YBI87E).ti,ab,kf,ot,hw,nm,rn.
Taurochenodeoxycholic Acid/ or (ursodoxicoltaurin* or tauroursodeoxycholic acid* or ursodeoxycholic acid* or ursodeoxycholyltaurine* or taurolite* or taurursodiol* or TUDCA or tauroursodeoxycholate* or taurochenodeoxycholic acid* or chenodeoxycholyltaurine* or taurochenodeoxycholate* or taurine chenodeoxycholate* or chenyl taurine sodium or ursodiol* or tauro* or ur 906 or ur906 or WHO 11388 or WHO11388 or 60EUX8MN5X or U7XRV7RZ1I).ti,ab,kf,ot,hw,nm,rn.
2 and 3
1 or 4
5 use medall
*"sodium phenylbutyrate plus taurursodiol"/ or (Relyvrio* or amx-0035 or amx0035 or PB-TURSO).ti,ab,kf,dq.
*"4 phenylbutyric acid"/ or (Buphenyl* or phenylbutyrate* or phenylbutanoate* or phenylbutanoic acid or TriButyrate* or ammonaps* or phenylbutyric acid* or pheburane* or lunaphen* or satisma* or nsc 657802 or nsc657802 or "acer 001" or acer001 or cmk 304 or cmk304 or lu 901 or lu901).ti,ab,kf,dq.
*taurursodiol/ or (ursodoxicoltaurin* or tauroursodeoxycholic acid* or ursodeoxycholic acid* or ursodeoxycholyltaurine* or taurolite* or taurursodiol* or TUDCA or tauroursodeoxycholate* or taurochenodeoxycholic acid* or chenodeoxycholyltaurine* or taurochenodeoxycholate* or taurine chenodeoxycholate* or chenyl taurine sodium or ursodiol* or tauro* or ur 906 or ur906 or WHO 11388 or WHO11388).ti,ab,kf,dq.
8 and 9
7 or 10
11 use oemezd
12 not (conference review or conference abstract).pt.
6 or 13
exp animals/
exp animal experimentation/ or exp animal experiment/
exp models animal/
nonhuman/
exp vertebrate/ or exp vertebrates/
or/15-19
exp humans/
exp human experimentation/ or exp human experiment/
or/21-22
20 not 23
14 not 24
remove duplicates from 25
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
Search: amx0035/sodium phenylbutyrate and ursodoxicoltaurine, amyotrophic lateral sclerosis
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search: amx0035/sodium phenylbutyrate and ursodoxicoltaurine, amyotrophic lateral sclerosis
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search: amx0035/sodium phenylbutyrate and ursodoxicoltaurine, amyotrophic lateral sclerosis
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search: amx0035/sodium phenylbutyrate and ursodoxicoltaurine, amyotrophic lateral sclerosis
Grey Literature
Search dates: December 13-21, 2021
Keywords: Search: amx0035/sodium phenylbutyrate and ursodoxicoltaurine, amyotrophic lateral sclerosis
Limits: Publication years: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Paganoni, S. and M. E. Cudkowicz (2020). "Sodium Phenylbutyrate-Taurursodiol for ALS. Reply." New England Journal of Medicine 383(23): 2294. | Editorial |
ALS = amyotrophic lateral sclerosis.
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
ALSFRS-R was the primary outcome of the CENTAUR study and a secondary outcome in the CENTAUR-OLE study.
ATLIS was a secondary outcome for the CENTAUR and CENTAUR-OLE studies.
Findings
Table 27: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusion about measurement properties | MID |
|---|---|---|---|
ALSFRS-R | A questionnaire-based, ordinal scale designed for use by clinicians to measure activities of daily living functionality of patients living with ALS. Composed of 12 items that cover 4 domains (gross motor activity, fine motor activity, respiratory function, and nutrition). An ordinal rating scale from 0 (absent function) to 4 (no impairment) and each score is summed for an overall score ranging from 0 to 48. | Reliability: For ALSFRS, ICCs for the total score were determined based on data collected from 3 trials and ranged from 0.94-0.96.30 Test-retest reliability for each item of the ALSFRS, Cohen’s kappa was greater than 0.76, except for “breathing” in 1 study where kappa = 0.59.30 Validity: For ALSFRS-R, internal consistency (Cronbach alpha) was greater than 0.67 for each domain (alpha > 0.70 is acceptable) and was 0.73 for the total score.31 ALSFRS-R was highly correlated with Sickness Impact Profile (Pearson correlation coefficient = −0.72), but the respiratory subscale was poorly correlated with FVC (Pearson correlation coefficient = 0.40).31 Responsiveness: No evidence has been identified. | Not identified in patients with ALS. A change of 20% to 25% in the slope of ALSFRS-R is considered clinically meaningful, according to clinical experts.17 A 1-unit change in clinical function corresponded to a 9-point decrease in the ALSFRS-R (95% CI, 8 points to 10 points; P = 0.025), according to patients.33 |
ATLIS | A muscle strength measuring device (and protocol) that produces interval-level data to assess weakening of muscles as a proxy for neuron loss in ALS. A certified evaluator is required. Assesses 12 muscle groups (right and left elbow and knee flexion and extension, ankle dorsiflexion, and grip). Raw scores are converted to PPN. A lower PPN value indicates weaker muscle strength. | Reliability: Test-retest reliability was acceptable for both healthy adults (average ICC = 0.92) and patients with ALS (average ICC = 0.97).32 Interrater reliability has been demonstrated in healthy adults (average ICC = 0.89) and patients with ALS (average ICC = 0.97).32 A significant difference in test scores attributable to the evaluator was observed patients with ALS (P = 0.009), but not in healthy adults (P = 0.179).32 Validity: The mean ATLIS score and Tufts Quantitative Neuromuscular Exam (‘gold standard’) score for all muscles were highly correlated with a Pearson correlation coefficient of 0.90.32 Responsiveness: Under investigation (NCT01911130) and results are not yet published.32 | Not identified. |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ATLIS = Accurate Test of Limb Isometric Strength; CI = confidence interval; FVC = forced vital capacity; ICC = intraclass correlation coefficient; MID = minimal important difference; PPN = percent predicted normal.
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
Description
The ALSFRS-R is a questionnaire-based scale designed to allow clinicians to quickly measure functionality or physical function regarding activities of daily living for patients living with ALS.17,30,31 The ALSFRS-R is composed of 12 items that cover 4 domains: gross motor activity, fine motor activity, respiratory function, and nutrition. More specifically, the topics that are addressed include speech, salivation, swallowing, handwriting, cutting food and handling utensils, dressing, and hygiene, turning in bed and adjusting bed clothes, walking, climbing stairs, as well as dyspnea, orthopnea, and respiratory insufficiency.31 The last 3 topics concerning respiratory function were an addition to the original ALSFRS, thus resulting in the revised version. Further, an alternative scale for patients with a gastrostomy tube is provided for the question concerning cutting food and handling utensils. Each question is scored on a 5-point scale from 0 to 4, where 0 = absent function and 4 = no impairment. The score for each question is summed for an overall score ranging from 0 to 48. ALSFRS-R is widely used in clinical trials and other patient-oriented research.46
Reliability and validity
The ALSFRS demonstrated test-retest reliability and internal consistency using data collected from 3 trials: 1) the study that originally validated the ALSFRS; 2) a 9-month placebo-controlled therapeutic RCT for ALS conducted in 36 centres in the US and Canada; and 3) a phase I and II study that evaluated the biological effect of a treatment for ALS in 279 patients at 21 sites over a 6-month period.30 For the total score, an intraclass correlation coefficient (ICC) of 0.96, 0.95, and 0.94 was determined for each of the studies, respectively.30 Acceptable test-retest reliability for each item of the ALSFRS was determined using Cohen’s kappa, which was greater than 0.76 for all items, except for “breathing” for 1 study where kappa = 0.59.
The ALSFRS-R was assessed for validity and internal consistency using data from a clinical trial for brain-derived neurotrophic factor for ALS, which included 387 placebo-treated patients who were evaluated monthly using the ALSFRS-R for 9 months.31 Internal consistency was assessed using a Cronbach alpha, which was greater than 0.67 for each individual domain of the scale; however, reliability should be 0.70 or higher.47 The total ALSFRS-R score met the 0.70 threshold with a Cronbach alpha of 0.73. This study also evaluated the construct validity of the ALSFRS-R by comparing it to the Sickness Impact Profile, a general assessment of health, as well as the FVC percentage for the respiratory subscale. The ALSFRS-R was well correlated with the Sickness Impact Profile (Pearson correlation coefficient = −0.72), but the correlation with FVC was poor (Pearson correlation coefficient = 0.40). The author suggested this may be attributed to the subjectivity of the ALSFRS-R compared with an objective FVC measure.31
MID
The determination of a clinically meaningful change in the ALSFRS-R was carried out in 2 studies. The first was a survey of members of the NEALS (ALS clinical experts), to determine whether a clinically significant change in the slope of ALSFRS-R decline could be agreed upon.17 A simple survey was sent to 65 experts, who were asked to rate the level of clinical meaningfulness of changes from 10% to 50% (time period not specified) in the ALSFRS-R slope (score versus time) from 1 to 7, where 1 = not very clinically meaningful, 4 = somewhat clinically meaningful, and 7 = very clinically meaningful. Forty-two (65%) surveys were returned. Briefly, a change of 20% to 25% in the slope (time period not specified) of ALSFRS-R was deemed clinically meaningful, as per expert opinion (93% and 100% of ALS experts rated a 20% and 25% decrease, respectively, as at least somewhat clinically meaningful).17
Gordon et al.33 also analyzed the performance of outcome measures used in early clinical trials for ALS based on a short duration (6 months), small sample size (N = 30) trial, to determine if the end points perform as they do in large trials, which end points have the least variability over 6 months, and whether any could act as surrogates for survival.33 The smallest clinically meaningful change according to patients was also explored. This was done by asking patients to rate their change from their last visit in terms of physical condition, emotional state, ability to enjoy social life, and overall quality of life. Each question was rated using a visual analogue scale from 1 to 7, where 1 = very much worse, 4 = about the same, and 7 = very much better. The response for each of the 4 questions was summed for a clinical meaningfulness score. The clinical meaningfulness score was used to reflect patient-perceived clinical change and was also determined to be associated with the ALSFRS-R using a linear mixed-effects model (P = 0.025). Based on this association, the authors reported a 1-unit change in the clinical meaningfulness score — namely, in patient-perceived clinical function, corresponding to a 9-point decrease in the ALSFRS-R (95% CI, 8 points to 10 points).
Other considerations and limitations
Results of the respiratory subscale do not correspond well with FVC, which is commonly used to measure respiratory status for patients with ALS. Also, the MID was not derived using 1 or more formal statistical approaches; rather, it was based on expert opinion and therefore, does not necessarily reflect what is clinically meaningful to patients.
Although there was a study that investigated the MID from the patient and caregiver perspectives, the results were mainly inconclusive or based on a small sample size. Also of note, the original validation studies were carried out nearly 20 years ago, which may affect the generalizability of the results when applied today, as standards of care have changed. Despite this, the FDA supports the use of the ALSFRS-R as a measure of efficacy for ALS treatment and as a demonstration of treatment effect on function in daily living.39
Lastly, there is no evidence of responsiveness to change for the ALSFRS-R scale. Also, it has been noted that the ALSFRS-R is relatively insensitive to change, provides only indirect evidence of motor neuron loss, and requires a large sample size due to large variability between patients.48,49 Therefore, it would be difficult to assess responders — namely, those who have been responding to treatment, compared to nonresponders or responses from the variabilities in measurements.
Accurate Test of Limb Isometric Strength
Description
The ATLIS was developed to overcome limitations of existing outcome protocols for ALS (e.g., Tufts Quantitative Neuromuscular Exam, hand-held dynamometry) such as space requirements, excessive physical burdens placed on patients and evaluators, and inter-patient variances. The ATLIS tests 12 selected muscle groups (left and right elbow and knee flexion and extension, ankle dorsiflexion, and grip) in a standardized, gravity-neutral position. The patient remains seated in the portable chair for all muscle tests and no position changes or stabilization are needed. The patient exerts maximal isometric force against a fixed load cell that transmits data wirelessly to a computer. Grip strength is measured using a wireless grip dynamometer. ATLIS tests both very weak muscle groups by using gravity-eliminated positions and very strong muscle groups by using a fixed load cell. Because progressive muscle weakness is the major clinical feature of ALS, and is highly correlated with motor neuron loss, measurement of maximal voluntary isometric strength produces interval data and is a widely used surrogate measure in ALS that demonstrates a linear decline over time. The ATLIS testing protocol takes approximately 15 minutes to administer. Administering the ATLIS requires training and quality assurance to assure that all evaluators follow the same testing procedures. If possible, the same evaluator should repeat measures of the same patient.32
Scoring
Andres et al.50 analyzed ATLIS data from 432 healthy adults to predict normal scores for individuals based on biometric factors such as sex, age, and size. Raw strength values in normal adults can vary by 2- or 3-fold, depending on biometric factors. Thus, the relative strength of specific muscles within and between individual patients using raw scores is difficult to interpret. Also, percent differences of raw score changes can be grossly distorted when raw values are small. Therefore, ATLIS raw data were collected from 192 men and 240 women and converted to PPN values using regression equations. The PPN can be calculated for 12 muscle groups as predicted by age, sex, weight, and height. As normalized data that have been controlled for differences in muscle sizes and biometric differences between individuals, PPN has been shown to be less variable and more intuitive to understand than raw data. Furthermore, the normalized scores allow individual muscle scores to be combined and allow meaningful comparisons between individuals whose normal strength is expected to be very divergent. The ATLIS PPN scores can be clinically useful in identifying relatively weak muscle groups in individual patients.
The sponsor for CENTAUR and CENTAUR-OLE trials used modified coefficients and intercepts from the originally published paper50 to calculate PPN and used ATLIS version 2 (modified equation tables are not included in this report). In the Clinical Study Report, the upper extremity ATLIS score represents an average of the 6 standardized upper muscle groups (left and right grip, elbow flexion and extension) and lower extremity ATLIS score represents an average of the 6 standardized lower muscle groups (left and right knee extension and flexion, ankle dorsiflexion). The total ATLIS score is an average of the upper and lower ATLIS scores.18,42
Reliability
Twenty healthy adults and 10 patients with ALS were tested twice by the same evaluator to determine test-retest reliability. Test sessions were separated by at least 1 hour and by no more than 1 week. The average difference between test and retest using the same evaluator for all muscle groups was 8.2% for the group of 20 healthy adults (average ICC = 0.92) and 8.6% for the 10 patients with ALS (average ICC = 0.97).32 Test-retest reliability has been demonstrated to be acceptable (ICC > 0.7).47
Interrater reliability was determined by testing 20 healthy adults and 10 patients with ALS by each of 2 different evaluators. Interrater reliability tests demonstrated a mean difference between tests using 2 different evaluators of 8.9% for the 20 healthy adults (average ICC = 0.89) and 8.3% for the 10 patients with ALS (average ICC = 0.97). There were no testing order effects in either the healthy adult or ALS groups. The ATLIS scores did not differ significantly between evaluators performing the tests in healthy patients (P = 0.179); however, there was a significant difference in test scores for patients with ALS attributed to the evaluator performing the test (difference = 0.716 ± 0.21, P = 0.009).32
Validity
Twenty healthy adults were tested using both ATLIS and a well-validated strength testing protocol (i.e., Tufts Quantitative Neuromuscular Exam) to assess criterion-based validity.32 The mean ATLIS score and Tufts Quantitative Neuromuscular Exam (‘gold standard’) score for all muscles for the 20 healthy adults were highly correlated,51 with a Pearson correlation coefficient of 0.90, although ATLIS scores were generally lower than the Tufts Quantitative Neuromuscular Exam scores.
Responsiveness to Change
The characteristics of the longitudinal responsiveness of the ATLIS as the disease progresses are under investigation (NCT01911130).32
Other Considerations and Limitations
The sponsor modified the regression equations to calculate PPN values from raw ATLIS data and it is unclear what impact this may have had on the reliability and validity of the ATLIS scores.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALS
amyotrophic lateral sclerosis
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
BIA
budget impact analysis
BSC
best supportive care
FT9
Fine’til 9
GT
gastric tube
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
NIV
noninvasive ventilation
PB-TURSO
sodium phenylbutyrate-ursodoxicoltaurine
QALY
quality-adjusted life-year
WTP
willingness-to-pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Sodium phenylbutyrate-ursodoxicoltaurine (Albrioza), powder for oral suspension |
Submitted price | Powder-filled sachet containing sodium phenylbutyrate (3 g) and ursodoxicoltaurine (1 g): $306.71 per sachet |
Indication | For the treatment of amyotrophic lateral sclerosis |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard review |
NOC date | June 10, 2022 |
Reimbursement request | As per indication |
Sponsor | Amylyx Canada |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients with ALS |
Treatment | PB-TURSO |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (10 years) |
Key data source |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
AE = adverse event; ALS = amyotrophic lateral sclerosis; BSC = best supportive care; FT9 = Fine’til 9; ICER = incremental cost-effectiveness ratio; LY = life-year; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year; vs. = versus.
Conclusions
The CADTH clinical review noted that results of the phase II, placebo-controlled CENTAUR trial demonstrated the superiority of sodium phenylbutyrate-ursodoxicoltaurine (PB-TURSO) over placebo for the primary outcome of slowing disease progression as measured by the slope of the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) in adults with a definite diagnosis of amyotrophic lateral sclerosis (ALS) within 18 months of symptom onset. However, the clinical relevance of the improvements seen in ALSFRS-R is uncertain. Evidence also indicates a potential survival benefit, as seen in the open-label extension of the trial, though the magnitude of this benefit is uncertain. Outcomes for health-related quality of life (HRQoL) were not included in the CENTAUR study and it is uncertain what benefits or drawbacks treatment with PB-TURSO provides beyond the outcomes captured in the trial. Long-term evidence for treatment efficacy and harms is limited as are studies comparing PB-TURSO to other approved ALS treatments.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness results. First, the sponsor included the clinical benefit of riluzole for patients on PB-TURSO but not the associated costs. Furthermore, patients were assumed to receive benefits from riluzole indefinitely after treatment discontinuation. In addition, the clinical expert consulted by CADTH for this review noted that the Fine’til 9 (FT9) staging system is not used in clinical practice and may not accurately represent the natural history of ALS. CADTH also noted limitations with treatment duration, discontinuation rates, adverse events (AEs), and the proportion of patients who receive no benefit with PB-TURSO. CADTH made various changes to address these limitations. The CADTH reanalysis resulted in an incremental cost-effectiveness ratio (ICER) of $2,086,658 per quality-adjusted life-year (QALY) for PB-TURSO plus riluzole compared to riluzole alone (incremental costs: $285,060; incremental QALYs: 0.137; incremental life-years: 0.218). Price reductions of approximately 98% would be required to achieve cost-effectiveness at a $50,000 per QALY threshold. This would reduce treatment costs to $4,478 per patient per year (assuming patients take 2 sachets daily). As PB-TURSO is not expected to displace any current therapies (but is added on to existing therapies), this would represent an incremental cost to the system. This price reduction would reduce the budget impact from $489 million to $10 million over 3 years.
CADTH notes the sponsor made several assumptions that implied PB-TURSO would only provide short-term benefits to a subpopulation of the Health Canada indication by implementing an 11-month treatment stopping rule and assuming 10% of patients with ALS would receive no benefit from PB-TURSO treatment. The CADTH reanalysis removed these assumptions as they were not reflective of clinical expert opinion regarding how this drug would be used in practice. Therefore, the 0.137 incremental QALYs associated with PB-TURSO treatment in the CADTH reanalysis are based on more favourable assumptions regarding PB-TURSO efficacy than suggested by the sponsor. CADTH notes nearly all (99%) of the incremental costs associated with PB-TURSO are attributable to treatment acquisition costs; this is the main driver behind the cost-effectiveness conclusions.
As the trial did not assess HRQoL outcomes, it is difficult to validate the benefit experienced by patients from delaying disease progression as calculated by the sponsor. The model structure the sponsor uses to extrapolate ALSFRS-R benefits into a QALY estimate is highly uncertain as it relies largely on data collected outside the trial. While a reduction in ALSFRS-R score always indicates progression, the impact of the progression on patient well-being is highly variable. The same absolute reduction in ALSFRS-R score in 2 different patients could translate to vastly different functional outcomes depending on the domain in which such reductions are experienced. Therefore, the absence of utility and HRQoL estimates from the trial to indicate how the delay in disease progression impacts patient’s lives makes validation of the sponsor’s approach challenging.
Overall, given the potential flaws with the model structure and difficulty with validating the model results, there is a high degree of uncertainty regarding the extent of incremental benefit associated with PB-TURSO using the sponsor-submitted approach. This contrasts to the high degree of certainty associated with the incremental costs and, therefore, opportunity costs to the health care system.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic review).
As part of the call for patient input, CADTH received feedback from the ALS Society of Canada (ALS Canada), a registered charity that advocates for government support and health care access for people living with ALS. ALS Canada collected data from 629 patients and caregivers through online surveys, for which patients were recruited via email, social media, and other online platforms. Almost all respondents lived in Canada, with a small number participating from the US, UK, Israel, and the Netherlands. Many patients being treated with riluzole and edaravone experienced increased survival and slower progression, and retained ability on these therapies, all of which were rated as the most important benefits of treatment. Few patients reported side effects with riluzole, but among those who did, the most difficult to manage were tiredness, weakness, muscle stiffness, and gastrointestinal problems. Additionally, some patients have difficulty swallowing pills, which makes riluzole administration challenging. For patients on edaravone, difficulties related to the IV administration were reported such as the scheduling of appointments and injection site pain. Ten patients and 10 caregivers of patients had experience with PB-TURSO. Some patients believed the drug was delaying their disease progression and preserving their speaking and breathing functions. In addition to the survey responses, 2 patients and 1 caregiver were interviewed regarding their experiences with the drug. One was from Canada and 2 were from the US. The interviewees reported a reduction in disease progression and increased independence with minimal side effects.
CADTH received clinician input from the Canadian ALS Research Network, a national network of clinicians at academic health care centres across Canada. The only disease-modifying therapies currently available for patients with ALS are riluzole and edaravone, which have shown limited benefit in slowing disease progression. Clinicians expect to try PB-TURSO in all patients, regardless of prior treatment status, as the drug has a novel mechanism of action. Clinicians noted that there is sound clinical rationale to introduce all 3 therapies concurrently in patients without contraindication.
Feedback from the drug plans was received. The drug plans noted that, in the CENTAUR trial, some patients were allowed to initiate edaravone along with PB-TURSO, making it unclear what benefit was derived from which drug. The plans noted that PB-TURSO is compatible with feeding tube administration while edaravone is not. Furthermore, the oral dosing of PB-TURSO may make it preferable to the IV dosing regimen of edaravone. Regarding the budget impact analysis (BIA), the drug plans felt that the market shares for PB-TURSO were underestimated, as was the duration of treatment when compared to edaravone. The drug plans noted the presence of a confidential negotiated price for edaravone.
Several of these concerns were addressed in the sponsor’s model:
the choice of comparators aligned with clinician and drug plan feedback
a disutility for edaravone administration was applied.
In addition, CADTH addressed some of these concerns as follows:
CADTH aligned the sponsor’s model inputs such that all patients were assumed to receive riluzole.
CADTH was unable to address the following concerns raised from stakeholder input:
CADTH reanalyses are based on publicly available prices and do not incorporate the presence of confidential negotiated prices.
CADTH did not consider the swallowing difficulties that some patients experience with riluzole.
Economic Review
The current review is for PB-TURSO for patients with ALS.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing PB-TURSO compared with edaravone or riluzole for the treatment of patients with ALS. The modelled population aligned with the CENTAUR trial1 on which the Health Canada indication was based and represents the reimbursement request.2
PB-TURSO is available in powder-filled sachets containing 3 g sodium phenylbutyrate and 1 g ursodoxicoltaurine for oral suspension. The recommended dose is 1 sachet daily for the first 3 weeks, and then 1 sachet twice daily thereafter if tolerated.2 The cost for PB-TURSO is $306.7123 per sachet;3 the cost per 1-month cycle was $11,964 in cycle 1 with once-daily dosing, and $17,812 in each cycle thereafter.3 Based on the CENTAUR trial, 90.8% of patients titrated to 2 sachets per day; the rest were assumed to remain on 1 sachet per day throughout the model.1 The annual cost of PB-TURSO, as calculated by CADTH, was $217,459 in the first year of treatment (Table 8).
The comparators for this analysis are edaravone, riluzole, and best supportive care (BSC) alone. Edaravone, an IV medication, has daily dosing for the first 14 days, followed by a 14-day washout period. Thereafter, the dosing is daily for 10 out of 14 days, followed by 14-day washout periods.4 The per cycle drug acquisition costs for edaravone were $12,880 in the first month, and $9,967 in each month thereafter. Riluzole is administered twice daily as an oral tablet and has a per month cost of $448.3 The annual costs of these comparators, as calculated by CADTH, was $124,254 for edaravone and $2,508 for riluzole (Table 8). Wastage was not relevant for any drug in this analysis due to the medication sizes aligning with the recommended doses. BSC was considered symptomatic disease management and did not have any cost or effectiveness associated with it; all patients were assumed to receive BSC alongside the other comparators.
Outcomes of the model included QALYs and life-years over a lifetime horizon of 10 years. Discounting (1.5% per annum) was applied to both costs and outcomes and a monthly cycle length with half-cycle correction was used.
Model Structure
The sponsor submitted a Markov model consisting of 5 mutually exclusive health states along with a death state. The model is based on the FT9 staging system, developed by Thakore et al.5 The FT9 staging system consists of 5 stages, defined by the number of ALSFRS-R domains impacted by ALS progression. The 5 states, labelled stage 0 through stage IV, correspond to the number of ALSFRS-R subscores that are 9 or less (of a normal 12); stage IV indicates that 4 ALSFRS-R subscores are 9 or less.5 The baseline distribution of patients across the 5 stages was determined by applying the staging criteria of the FT9 system to individual patient-level data of the randomized controlled trial phase of the CENTAUR trial.1,6 As ALS is a progressive disease, patients could only move forward or remain in state; they could not move backward. Non-sequential forward transitions were also possible, and patients were at risk of death from any state. A figure of the sponsor’s model structure is available in Figure 1, Appendix 3.
Model Inputs
The target population was based on the phase II, placebo-controlled CENTAUR trial, which enrolled patients with ALS (N = 137) expected to be at a high risk of progression based on baseline characteristics.1 The study was conducted in 2 phases: a randomized phase of 24 weeks and an open-label extension phase of up to 132 weeks. Based on the trial, the mean starting age of patients in the model was 57.5 years, and 69% of patients were male.1
As noted earlier, the baseline distribution of patients in FT9 states was based on individual patient data from the CENTAUR trial as follows: stage 0, 4.44%; stage I, ||||||||||%; stage II, ||||||||||%; stage III, ||||||||||%; and stage IV, ||||||||||%.3 However, due to a lack of relevant data from CENTAUR to inform transition probabilities between stages, the sponsor conducted a feasibility assessment for an indirect treatment comparison. The sponsor deemed an indirect treatment comparison to be unfeasible to compare the various treatments due to heterogeneity among included studies that could not be overcome by matching. Therefore, comparative efficacy data were derived from the published literature. Thakore et al. (2020) reported transition probabilities by FT9 stage for both BSC and riluzole (Table 10, Appendix 3).6 Data for PB-TURSO and edaravone were taken from a post-hoc analysis of the CENTAUR trial.3 While this trial initially randomized patients to PB-TURSO and placebo, the protocol was amended to allow patients who were taking edaravone to also be included and randomized to either arm. The sponsor used individual patient data from the modified intention-to-treat population from the CENTAUR trial to compare patients who received PB-TURSO without edaravone to the group that received placebo with edaravone to inform the comparison between PB-TURSO and edaravone. The sponsor created mixed-effects models comparing PB-TURSO to edaravone for change at 24 weeks from baseline in ALSFRS-R score in the modified intention-to-treat population from CENTAUR.3 These models were adjusted for age, gender, disease progression rate, time since diagnosis, site of onset, and concomitant use of riluzole. Ratios of the least squares mean versus placebo at week 24 from the mixed-effects model were performed, resulting in a rate ratio of |||||||||| for PB-TURSO and |||||||||| for edaravone, versus placebo.3 The sponsor set the rate ratio for edaravone to 1.0, under the assumption that edaravone could not be worse than placebo. The resulting rate ratios of |||||||||| for PB-TURSO and 1.0 for edaravone were applied to the transition probabilities for riluzole reported in Table 10. In addition to these transition probabilities, transitions to death due to background mortality were applied to all health states using Canadian life tables with age- and sex-adjusted mortality estimates.
Patients in the model were assumed to remain on treatment for 11 months, at which point 100% discontinued active therapy and moved to BSC. The 11-month value was based on the mean treatment exposure for PB-TURSO from the CENTAUR trial.1 An assumption was made that treatment duration would be equal for all treatments. In addition, efficacy persistence for 3 months after the end of treatment was assumed for all treatments. Patients could also discontinue treatment at a rate of 3.07% per cycle for PB-TURSO, and 1.86% per cycle for edaravone and riluzole, based on data from CENTAUR.1
Utility values were derived from Thakore et al. (2020), who collected HRQoL data from patients using the EQ-5D Three-Level tool.6 As these values were based on the US valuation of EQ-5D Three-Level, Canadian preference-based weights were used to calculate the final scores.7 The final utility values by FT9 stage were as follows: stage 0, 0.75; stage I, 0.70; stage II, 0.61; stage III, 0.54; and stage IV, 0.48.6 In addition, utility decrements were applied for AEs and edaravone use, given the IV administration. Rates of AEs were derived from CENTAUR, and utility decrements were sourced from the published literature.1,3 A utility decrement of 0.02 was applied for patients on edaravone once, in the first cycle, based on the literature.8
The doses and drug acquisition costs used in the model were all as described previously. In addition, the sponsor included administration costs for edaravone. The sponsor assumed that 50% of patients receiving edaravone would require a peripherally inserted central catheter, which was associated with insertion, removal, and maintenance costs derived from a Canadian study in pediatric patients.9 Edaravone administration was assumed to occur 50% of the time at home and 50% of the time in hospital, with additional nursing costs associated with each administration. Recurring and 1-time costs were included in the model to account for health care resource use. Recurring costs for gastric tubes (GTs) and noninvasive ventilation (NIV) were incorporated into the model on a monthly basis, the rates of which were derived from CENTAUR.1 The annual costs for a GT and NIV were $16,360 and $52,107, respectively, calculated from the published literature and converted to 2021 Canadian dollars.3 Additional monthly costs for disease management were incorporated and included physician visits, outpatient facilities, home health care, and dietary supplements and supplies.6 These costs ranged from $240 in stage 0 to $7,130 in stage IV.3 One-time costs were included related to powered wheelchairs for all patients, communication aids for 90% of patients, and hospitalizations for serious AEs. Finally, costs for AEs were applied as one-off costs based on the unit costs of a standard hospital stay and typical resource intensity weights.3 The AEs included in the sponsor’s model were those of a serious nature, occurring in at least 2% of patients in the CENTAUR trial, and consisted of respiratory disorders, speech disorders, pneumonia aspiration, and catheter-site infection.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented as follows.
Base-Case Results
The results of the sponsor’s analysis demonstrated that 3 comparators remained on the cost-effectiveness frontier: BSC alone, riluzole, and PB-TURSO (Table 3). Edaravone was subject to extended dominance (Table 11). Compared to riluzole, PB-TURSO was associated with incremental costs of $155,283 and QALYs of 0.21, resulting in an ICER of $735,528 per QALY. The probability of cost-effectiveness of PB-TURSO at a $50,000 per QALY willingness-to-pay (WTP) threshold was 0%. Additional results from the sponsor’s submitted economic evaluation base case are available in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 122,513 | 0.85 | Reference |
Riluzole | 137,894 | 0.91 | 220,625 vs. BSC |
PB-TURSO | 293,177 | 1.13 | 735,528 vs. riluzole |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted analyses are based on the publicly available prices of comparators and may not reflect confidential negotiated prices. Only treatments on the cost-effectiveness frontier are reported in this table.
Source: Sponsor’s Pharmacoeconomic Report (2021).3
Note that the submitted analyses are based on the publicly available prices of comparators and may not reflect confidential, negotiated prices. Only treatments on the cost-effectiveness frontier are reported in this table.
Sensitivity and Scenario Analysis Results
The sponsor conducted 2 scenario analyses involving shortening the time horizon and removing the persistence of efficacy for the active comparators. Results in these scenario analyses were similar to those in the base case, with edaravone being subjected to extended dominance in both. In sensitivity analyses in which clinical, cost, and utility parameters were varied by 20%, the parameters that had the largest impact on the results were the unit drug costs, treatment duration, and proportion titrating to twice daily for PB-TURSO.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
Rate ratios for the transition probabilities for PB-TURSO and edaravone are applied to riluzole transition probabilities. The sponsor calculated rate ratios of |||||||||| and 1.000 from the post-hoc analysis of the CENTAUR trial for PB-TURSO and edaravone, respectively, which were applied to the transition probabilities between FT9 stages for riluzole derived from Thakore et al.6 The rationale for this assumption was that both trial arms in CENTAUR included high proportions of riluzole use. There are 2 issues with this assumption.
First, by applying transition probabilities for PB-TURSO and edaravone to the transition probabilities observed for riluzole, the sponsor has assumed an additive effect of these drugs. For example, the transition probabilities for riluzole include a lower probability of death relative to BSC. The same lower probabilities of death are also used for PB-TURSO in the model (i.e., the benefits of a lower mortality rate with riluzole are also included for PB-TURSO). The result of this assumption is that the comparators in the pharmacoeconomic analysis should be PB-TURSO plus riluzole, compared to edaravone plus riluzole, compared to riluzole alone. This assumption does have some validity according to clinical experts consulted by CADTH, who stated that patients would be expected to receive these drugs concurrently, and that, due to their different mechanisms of action, there is some expectation of an additive effect. However, it is clear the sponsor intended to model each comparator as a monotherapy as this is stated explicitly in its pharmacoeconomic report. As such, the treatment costs for PB-TURSO and edaravone do not include the cost of riluzole, yet the sponsor has attributed the added effectiveness of riluzole to these treatments.
Second, after patients discontinue their primary therapy (i.e., PB-TURSO, edaravone, and/or riluzole), it is assumed that all treatment efficacy is lost after 3 months. However, because of the approach taken, patients discontinuing PB-TURSO and edaravone retain the riluzole transition probabilities post-discontinuation (i.e., patients who discontinue PB-TURSO and edaravone are expected to stay on riluzole indefinitely). Theoretically, the sponsor may be assuming that being treated with riluzole would lead to an indefinite benefit of reducing the rate of ALS progression, an assumption for which there is no evidence and is also not applied to patients receiving riluzole alone. Given the omission of riluzole acquisition costs for those receiving PB-TURSO and edaravone, it seems more likely that the implications of this assumption were not considered by the sponsor.
CADTH made 2 changes to the base case as a result of this assumption. First, drug acquisition costs for riluzole were included in the PB-TURSO and edaravone treatment costs. These costs last until treatment discontinuation, thus assuming patients discontinue all active therapies at the same time. Second, patients discontinuing any therapy were assumed to experience the transition probabilities associated with BSC (no active therapy).
FT9 staging system may not be appropriate for economic modelling. The sponsor’s model is based on a published ALS model used to estimate the cost-effectiveness of riluzole.5 The original publication compares the model’s predictive survival outcomes to those seen in a real-world setting to ensure the model is well calibrated and valid. However, unlike the published model, the sponsor assumes no regression through health states (i.e., patients cannot improve). Removing the assumption is valid but the remaining transition probabilities would need to be re-calibrated to ensure survival outcomes are supported by real-world evidence. As it stands, the sponsor’s model underestimates the survival associated with riluzole relative to the original publication by Thakore et al. and no further evidence is presented to indicate that this change is valid.6 This means either the choice of FT9 is inappropriate, or the survival outputs of the sponsor’s model are not valid.
Furthermore, the clinical expert engaged by CADTH for the review stated that the FT9 staging system is not used by Canadian clinicians in managing ALS. The expert noted that it is a novel staging system and is not expected to adequately represent the natural history of ALS. In addition, some issues were noted with the clinical plausibility of the staging system in the context of economic modelling. For example, the clinical expert noted that a patient with serious respiratory issues has a high probability of death; were this patient to have high scores in the other domains, they would only be classified as stage I with a low probability of death. Likewise, the healthiest stage IV patient with scores of 9 in all domains could still be entirely independent — able to clothe, bathe, eat, and climb stairs without assistance — yet receive a very low utility score in this model.10 These examples highlight the limitations of the FT9 system to capture the nuance of ALS.
From a methodological perspective, a health state in an economic model should represent a homogenous group of patients who have similar expected costs and quality-of-life considerations. This is not the case in the FT9 staging system. The healthiest stage IV patient described previously is very dissimilar to a stage IV patient with such severely progressed disease that they are unable to eat, speak, write, walk, or breath independently.10 While these 2 patients are considered the same in the sponsor’s model, they will have different costs, quality of life, and survival outcomes. Likewise, in later stages, some states include patients who require NIV and those who do not. This level of heterogeneity within a single health state is problematic as the sponsor assumes patients requiring NIV have the same survival outcomes as those who do not, all other factors remaining equal. The implications of heterogeneity in health states have been well documented in the literature.11 The model applies average health care costs to average survival in a very heterogenous cohort where it is likely that those with high health care costs have a shorter life expectancy. Therefore, the model structure likely overestimates total health care costs across the cohort’s lifetime.
CADTH was unable to address this limitation in reanalysis and notes that the model structure potentially lacks the required nuance to evaluate the benefit of delaying progression. This also makes validation of the model outputs difficult to validate as the staging system has limited clinical use. The current modelling approach appears to underestimate riluzole survival outcomes relative to evidence cited by the sponsor.
Treatment duration is underestimated. The sponsor assumed a maximum duration of therapy of 11 months for all comparators based on the observed maximum therapy of PB-TURSO in the CENTAUR trial.1 However, the product monograph for PB-TURSO does not stipulate any stopping criteria for this therapy, nor are any stopping criteria noted in the monographs for edaravone or riluzole.2,4,12 Given the progressive and ultimately fatal nature of ALS, there is no clinically plausible reason to implement an arbitrary maximum duration of treatment if a patient still has the potential to benefit from therapy. The clinical expert consulted for this review did not support a maximum treatment duration of PB-TURSO, but noted that patients may eventually discontinue if they progress to a very severe state (i.e., full dependency or continual respiratory support). However, CADTH notes that the sponsor has already considered discontinuation elsewhere in the model; thus, the implementation of a maximum treatment duration may double-count this element of the analysis. Finally, CADTH notes that a post-hoc analysis of the open-label extension phase of the CENTAUR trial considered a mean exposure to PB-TURSO of 14.7 months, further supporting the fact that the sponsor has underestimated treatment duration in its base case.
As part of the base case, CADTH removed the maximum treatment duration of 11 months for all comparators and assumed that patients would only discontinue per the discontinuation rates and due to death.
Differential discontinuation rates. The sponsor assumed discontinuation rates per cycle of 3.07% and 1.86% for PB-TURSO and other comparators, respectively, based on the CENTAUR trial.1 Details of how these rates were calculated were limited, but the sponsor stated discontinuation would be due to AEs or disease progression. The sponsor’s use of a rate ratio of |||||||||| for PB-TURSO necessarily assumes disease progression to be slower with PB-TURSO. As the rates of AEs included in the sponsor’s model are lower for PB-TURSO compared to edaravone and riluzole, it is unclear why the discontinuation rate for PB-TURSO would be higher. Data from the CENTAUR publication stated that the most common AEs leading to discontinuation of the trial regimen were diarrhea and respiratory failure, both of which were higher in the PB-TURSO group compared to the placebo group.1 However, diarrhea was not included in the sponsor’s model, and the rates of respiratory disorder differed from those reported in the publication. Therefore, there is uncertainty in the discontinuation rates for the active treatments. If there is a higher rate of discontinuation for PB-TURSO relative to current therapies, that would indicate that patients either progress faster on PB-TURSO or PB-TURSO has a worse safety profile.
As part of the base case, CADTH assumed the same discontinuation rate for all active therapies as that of PB-TURSO. If PB-TURSO does have a higher discontinuation rate, this would indicate that a poorer safety signal and disutility associated with these AEs would need to be included in the analysis.
AEs included may be related to disease progression. The sponsor included the following AEs in its model: respiratory disorder, speech disorder, pneumonia aspiration, and, for edaravone only, catheter-site infection. Other than catheter-site infection, all these AEs may be associated with the progression of ALS and are not treatment-related. As the sponsor has already included health state–specific utility values for the various disease stages, the inclusion of such AEs with their associated cost and disutility potentially double-counts this element of the analysis.
As part of the base case, CADTH excluded all AEs and their specified costs and disutility except for the disutility associated with catheter-site injections. The CADTH base case, therefore, has no differential AEs between treatments except for catheter-site injection that is associated with edaravone. This is also to align with the assumption of equivalent discontinuation rates among therapies.
Patients in FT9 stage IV are assumed to incur no benefit of treatment with PB-TURSO. The sponsor’s methodology leads to the consequence that patients in FT9 stage IV do not incur benefit with PB-TURSO while still incurring drug acquisition and health care costs. This assumption is implicit; as the rate ratio of 0.675 is only applied to the transition probabilities between health states and not to the probability of death, the probability of death from stage IV is equal across all active treatments. In effect, this assumes that patients beginning the model in stage IV do not have the capacity to benefit from treatment with PB-TURSO as they cannot progress to a more severe state. In the sponsor’s base case, the proportion of patients starting in stage IV is |||||||%; that is, |||||||% of patients will incur drug acquisition costs but will not derive benefit. This choice by the sponsor suggests that eligibility criteria for PB-TURSO should be restricted to patients with disease stage 0 to stage III. This assumption was not supported by the clinical expert, who noted that while patients with early-stage disease have the capacity to benefit more from treatment, clinical evidence for restricting treatment to these patients is lacking. Again, this raises concerns regarding the validity of the sponsor’s chosen model structure.
As part of the base case, CADTH assumed that the 10% of patients with stage IV disease at baseline would have stage III disease instead. This assumption improves the efficacy of PB-TURSO, as it allows these patients to still benefit from a delay in progression from stage III to stage IV. CADTH notes that there may be some patients who receive no benefit from PB-TURSO, though these have not been explicitly identified.
Uncertainty associated with disease management and 1-time costs. The sponsor included 1-time costs for ALS of a powered wheelchair, communication aid, and hospitalization, along with disease management costs, which included costs associated with GT and NIV. Regarding the 1-time costs for a wheelchair and communication aid, it was noted by the clinical expert that most or all patients with ALS would eventually require these devices, an assumption that the sponsor also supports. However, the sponsor’s methods of incorporating these 1-time costs into its analysis is associated with some uncertainty as there is the potential for double-counting due to these costs being incurred in both stage III and stage IV. As all patients with ALS are expected to incur these costs regardless of treatment, the only differential costs between treatments pertain to when these costs are incurred (economic analyses value future costs less than costs that occur closer to the present through discounting). Given the short time horizon, the impact of discounting is likely to be minimal. Furthermore, the clinical expert noted that patients with ALS do not frequently require hospitalizations, while the sponsor has included substantial costs associated with hospitalization in stage IV. The recurring costs for GT and NIV are also associated with uncertainty, as the clinical expert noted that while NIV is associated with high upfront costs for the purchase of equipment, the maintenance costs are relatively inexpensive.
CADTH performed scenario analyses in which all 1-time costs were excluded and, alternatively, recurring costs for GT and NIV were excluded.
Rate ratios used in the model are uncertain. The rate ratios used in the model were based on a post-hoc analysis of the CENTAUR trial. The sponsor split the trial cohort into those who received PB-TURSO alone, those who received PB-TURSO plus edaravone, those who received placebo alone, and those who received edaravone plus placebo. Given that patients were not randomized at baseline based on concomitant drug use, the sponsor attempted to control for confounders using a mixed-effects model. The outcomes of this post-hoc analysis are highly limited and produce results inconsistent with clinical expectation. For example, the results show that edaravone use leads to a higher rate of progression than placebo. These results cast doubt on the validity of the sponsor’s modelling approach. Indeed, the sponsor’s pharmacoeconomic report suggests that “the model is not estimating covariate effects accurately due to the model structure or study size.”3 Evidence from the CENTAUR trial indicates that the change in ALSFRS-R baseline score in patients who received PB-TURSO is –6.86 versus –9.18 in the placebo plus standard-of-care arm. The post-hoc analysis used in the economic model suggests that for patients receiving PB-TURSO alone, the reduction in ALSFRS-R is –|||||||||| relative to –|||||||||| for those who receive riluzole but not edaravone. Therefore, results from the post-hoc analysis are more favourable than what was demonstrated in the trial.
Notwithstanding the methodological limitations of the post-hoc analysis, results for edaravone do not meet face validity. In the sponsor’s base case, the only benefit from treatment with edaravone came from the fact that patients who are discontinuing experience riluzole transition probabilities rather than those for BSC. As CADTH removed this assumption, given it is not clinically plausible and contradicts other assumptions imposed by the sponsor, the only clinical difference remaining between edaravone and riluzole was the disutility of IV administration and potential catheter-site injections.
CADTH used the rate ratio from the post-hoc analysis but notes this gives a slightly more optimistic view of the evidence. CADTH notes, however, that the cost-effectiveness of PB-TURSO relative to edaravone is highly uncertain. CADTH did include edaravone in the base case but results pertaining to this comparator are largely based on flawed assumptions. It should be noted that PB-TURSO is not expected to displace edaravone but be used alongside it, given their different mechanisms of action. Therefore, the cost-effectiveness of PB-TURSO monotherapy relative to edaravone as a monotherapy is limited.
The majority of patients will be on active therapy. All patients in the model were assumed to receive BSC, consisting of symptomatic disease management, alongside their active therapies. The sponsor also considered BSC alone to be a relevant comparator, which was not associated with cost or effectiveness other than health care resource utilization costs. The clinical expert stated that essentially every patient with ALS would be offered riluzole first, given its demonstrated clinical benefit and favourable safety profile. The only patients who are expected to not receive active therapy are those for whom financial considerations present an obstacle; however, given the availability of generic riluzole, this was thought to make up a minority of patients. Furthermore, for those patients not on active therapy due to financial considerations, the introduction of PB-TURSO is unlikely to alleviate that concern given the list price is substantially higher than riluzole. As such, it is unlikely there are a group of patients currently receiving BSC alone who would receive PB-TURSO if it were funded. CADTH notes that the sponsor’s own assumptions support the exclusion of BSC as a comparator. In the BIA, 100% of patients are assumed to receive riluzole concomitantly with PB-TURSO or edaravone, and BSC alone is not a comparator.
As part of the base case, CADTH assumed that PB-TURSO would only displace active therapies. This change did not require any modification to the sponsor’s model, only the omission of BSC alone in the results. CADTH does provide BSC results in Appendix 4, but these are not used to draw conclusions regarding the cost-effectiveness of PB-TURSO in the Health Canada–approved indication.
One additional limitation was identified but was not considered to be a major limitation. The sponsor’s base case included a minor error in the transition probabilities for edaravone, in which the transition probability from stage II to death was adjusted by the discontinuation rate twice.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The proportion of patients with ALS titrating to 2 sachets of PB-TURSO daily was 90.8% based on the CENTAUR trial. | Likely appropriate, though rates may differ with larger patient numbers and in a real-world setting. |
In total, 50% of patients on edaravone are assumed to require use of a PICC. | Uncertain, but unlikely to affect the results. According to the clinical expert, the choice of whether to use a PICC is made at the hospital level and will vary by jurisdiction. |
Costs associated with the insertion, removal, and maintenance of the PICC were taken from a pediatric population. | Uncertain, but unlikely to affect the results. Infants may require anesthetic to have the PICC inserted, which would be associated with additional costs not incurred by an adult population. |
ALS = amyotrophic lateral sclerosis; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; PICC = peripherally inserted central catheter.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes, summarized in Table 5, included the addition of riluzole acquisition costs to the PB-TURSO and edaravone treatments, the use of BSC transition probabilities off-treatment, no specified maximum treatment duration, equal discontinuation rates, the exclusion of disease-related AEs, and the assumption that those patients with baseline stage IV would be classified as stage III.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Minor programming error | Transition probability for edaravone | Fixed the transition probability from stage II to death, as it had been adjusted by the discontinuation rate twice |
2. BSC as comparator | Included | Excluded |
Changes to derive the CADTH base case | ||
1. Riluzole acquisition costs in the PB-TURSO and edaravone treatments | Excluded | Included |
2. Transition probabilities for patients discontinuing PB-TURSO and edaravone | Transition probabilities for riluzole were used off-treatment. | Transition probabilities for BSC were used off-treatment. |
3. Maximum treatment duration of active comparators | 11 months | No specified maximum |
4. Discontinuation probability each month | PB-TURSO = 3.07% Edaravone = 1.86% Riluzole = 1.86% | PB-TURSO = 3.07% Edaravone = 3.07% Riluzole = 3.07% |
5. Adverse events included | Respiratory disorder Speech disorder Pneumonia aspiration Catheter-site injection | Catheter-site injection |
6. Proportion of patients beginning in stage IV | |||||||% | 0% (moved to stage III) |
CADTH reanalysis | — | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
BSC = best supportive care; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
In the CADTH base case, PB-TURSO plus riluzole was associated with estimated total costs and QALYs of $431,923 and 1.095, compared with total costs and QALYs of $146,863 and 0.958 for patients receiving riluzole alone. The ICER for PB-TURSO plus riluzole compared to riluzole alone was $2,086,658 per QALY, and the probability of cost-effectiveness at a $50,000 per QALY WTP threshold was 0%. In the CADTH base case, edaravone was dominated. Results of the stepped reanalysis are available in Table 6 with full disaggregated results for edaravone and BSC available in Table 13, Appendix 4.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER |
|---|---|---|---|---|---|
Sponsor’s base case (corrected) | Riluzole alone | 138,276 | 1.701 | 0.916 | Reference |
PB-TURSO + riluzole | 298,507 | 2.084 | 1.127 | 757,459 | |
CADTH reanalysis 1: Drug costs for riluzole | Riluzole alone | 138,276 | 1.701 | 0.916 | Reference |
PB-TURSO + riluzole | 302,287 | 2.084 | 1.127 | 775,328 | |
CADTH reanalysis 2: Off-treatment transition probabilities | Riluzole alone | 138,276 | 1.701 | 0.916 | Reference |
PB-TURSO + riluzole | 278,912 | 1.849 | 1.014 | 1,441,275 | |
CADTH reanalysis 3: No specified maximum treatment duration | Riluzole alone | 148,325 | 1.780 | 0.953 | Reference |
PB-TURSO + riluzole | 426,375 | 2.104 | 1.141 | 1,472,315 | |
CADTH reanalysis 4: Discontinuation rates | Riluzole alone | 136,995 | 1.692 | 0.912 | Reference |
PB-TURSO + riluzole | 298,507 | 2.084 | 1.127 | 749,907 | |
CADTH reanalysis 5: Adverse events | Riluzole alone | 137,043 | 1.701 | 0.917 | Reference |
PB-TURSO + riluzole | 298,447 | 2.084 | 1.128 | 765,040 | |
CADTH reanalysis 6: Stage IV proportions | Riluzole alone | 142,315 | 1.736 | 0.936 | Reference |
PB-TURSO + riluzole | 305,905 | 2.135 | 1.156 | 743,432 | |
CADTH base case: Reanalysis 1 + 2 + 3 + 4 + 5 + 6 (deterministic) | Riluzole alone | 146,954 | 1.782 | 0.958 | Reference |
PB-TURSO + riluzole | 431,727 | 2.000 | 1.095 | 2,083,058 | |
CADTH base case: Reanalysis 1 + 2 + 3 + 4 + 5 + 6 (probabilistic) | Riluzole alone | 146,863 | 1.781 | 0.958 | Reference |
PB-TURSO + riluzole | 431,923 | 1.999 | 1.095 | 2,086,658 |
ICER = incremental cost-effectiveness ratio; LY = life-year; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year.
Note: In all stepwise analyses, edaravone was dominated or extendedly dominated and, as such, does not appear on the efficiency frontier. Full results with edaravone are available in Appendix 4.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s base case and CADTH’s base case. The CADTH base case suggested a price reduction in excess of 98% would be necessary to achieve cost-effectiveness of PB-TURSO at a WTP threshold of $50,000 per QALY (Table 7).
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for PB-TURSO vs. riluzole | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 735,528 | 2,086,658 |
10% | 668,843 | 1,879,507 |
20% | 602,158 | 1,672,356 |
30% | 535,473 | 1,465,205 |
40% | 468,788 | 1,258,054 |
50% | 402,103 | 1,050,903 |
60% | 335,418 | 843,752 |
70% | 268,733 | 636,601 |
80% | 202,048 | 429,450 |
90% | 135,363 | 222,299 |
95% | 102,021 | 118,724 |
98% | 68,678 | 56,578 |
ICER = incremental cost-effectiveness ratio; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; vs. = versus.
CADTH undertook several scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of PB-TURSO plus riluzole in the base case, which are outlined as follows:
excluded all 1-time costs under the assumption that all patients would eventually require a powered wheelchair and communication aid
excluded costs for GT and NIV.
The results of these analyses are presented in Table 14, Appendix 4. The results of both these scenario analyses were very similar to the CADTH base case, indicating that disease management costs are not a driver of the model when balanced between treatments.
Issues for Consideration
An oral form of edaravone is anticipated to be reviewed for patients with ALS. This may offer an alternative to IV edaravone with a different cost and QALY profile.
Overall Conclusions
The CADTH clinical review noted that CENTAUR trial results demonstrated the superiority of PB-TURSO over placebo for the primary outcome of slowing disease progression as measured by the slope of the ALSFRS-R in adults with a definite diagnosis of ALS within 18 months of symptom onset. However, the clinical relevance of the improvements seen in ALSFRS-R is uncertain. Evidence also indicates a potential survival benefit, as seen in the open-label extension of the trial, though the magnitude of this benefit is uncertain. Outcomes for HRQoL were not included in the CENTAUR study and it is uncertain what benefits or drawbacks treatment with PB-TURSO provides beyond the outcomes captured in the trial. Long-term evidence for treatment efficacy and harms is limited as are studies comparing PB-TURSO to other approved ALS treatments.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness results. First, the sponsor included the clinical benefit of riluzole for patients on PB-TURSO but not the associated costs. Furthermore, patients were assumed to receive benefits from riluzole indefinitely after treatment discontinuation. In addition, the clinical expert noted that the FT9 staging system is not used in clinical practice and may not accurately represent the natural history of ALS. CADTH also noted limitations with treatment duration, discontinuation rates, AEs, and the proportion of patients who receive no benefit with PB-TURSO. CADTH made various changes to address these limitations. The CADTH reanalysis resulted in an ICER of $2,086,658 per QALY for PB-TURSO plus riluzole compared to riluzole alone (incremental costs: $285,060; incremental QALYs: 0.137). Price reductions of approximately 98% would be required to achieve cost-effectiveness at a $50,000 per QALY threshold. This would reduce treatment costs to $4,478 per patient per year (assuming patients take 2 sachets daily). As PB-TURSO is not expected to displace any current therapies (but is added on to existing therapies), this would represent an incremental cost to the system. This price reduction would reduce the budget impact from $489 million to $10 million over 3 years.
CADTH notes the sponsor made several assumptions that implied PB-TURSO would only provide short-term benefits to a subpopulation of the Health Canada indication, by implementing an 11-month treatment stopping rule and assuming 10% of patients with ALS would receive no benefit from PB-TURSO treatment. The CADTH reanalysis removed these assumptions as they were not reflective of clinical expert opinion regarding how this drug would be used in practice. Therefore, the 0.137 incremental QALYs associated with PB-TURSO treatment in the CADTH reanalysis are based on more favourable assumptions regarding PB-TURSO efficacy than suggested by the sponsor. CADTH notes nearly all (99%) of the incremental costs associated with PB-TURSO are attributable to treatment acquisition costs; this is the main driver behind the cost-effectiveness conclusions.
As the trial did not assess HRQoL outcomes, it is difficult to validate the benefit experienced by patients from delaying disease progression as calculated by the sponsor. The model structure that the sponsor uses to extrapolate ALSFRS-R benefits into a QALY estimate is highly uncertain as it relies largely on data collected outside of the trial. While a reduction in ALSFRS-R score always indicates progression, the impact of the progression on patient well-being is highly variable. The same absolute reduction in ALSFRS-R score in 2 different patients could translate to vastly different functional outcomes depending on the domain in which such reductions are experienced. Therefore, the absence of utility and HRQoL estimates from the trial indicating how the delay in disease progression impacts patients’ lives makes validation of the sponsor’s approach challenging.
Overall, given the potential flaws with the model structure and difficulty with validating the model results, there is a high degree of uncertainty regarding the extent of incremental benefit associated with PB-TURSO using the sponsor-submitted approach. This contrasts with the high degree of certainty associated with the incremental costs and, therefore, opportunity costs to the health care system.
References
1.Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919-930. PubMed
2.Albrioza (sodium phenylbutyrate and ursodoxicoltaurine): powder for suspension, 3 g/1 g sachet, oral [product monograph]. Milton (ON): Innomar Strategies; 2022 Jun 1.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Albrioza (sodium phenylbutyrate and ursodoxicoltaurine), 3 mg sachet sodium phenylbutyrate sachet and 1 mg sachet ursodoxicoltaurine powder for suspension, oral. Calgary (AB): Amylyx Canada; 2021 Nov 24.
4.Radicava (edaravone): solution, 30 mg/100mL (0.3mg/mL), intravenous administration [product monograph]. Jresey City (NJ): Mitsubishi Tanabe Pharma America Inc., a US subsidiary of Mitsubishi Tanabe Pharma Corporation; 2018 Oct 2: https://pdf.hres.ca/dpd_pm/00047612.PDF. Accessed 2022 Ja 11.
5.Thakore N, Lapin B, Kinzy T, Pioro E. Deconstructing progression of amyotrophic lateral sclerosis in stages: a Markov modeling approach. Amyotroph Lateral Scler Frontotemporal Degener. 2018:1-11. PubMed
6.Thakore N, Pioro E, Udeh B, Lapin B, Katzan I. A cost-effectiveness framework for amyotrophic lateral sclerosis, applied to riluzole. Value Health. 2020:1-9. PubMed
7.Bansback N, Tsuchiya A, Brazier J, Anis A. Canadian valuation of EQ-5D health states: preliminary value set and considerations for future valuation studies. PLoS One. 2012;7(2):e31115. PubMed
8.Matza L, Deger K, Vo P, Maniyar F, Goadsby P. Health state utilities associated with attributes of migraine preventive treatments based on patient and general population preferences. Qual Life Res. 2019;28(9):2359-2372. PubMed
9.Dong Z, Connolly B, Ungar W, Coyte P. Cost analysis of peripherally inserted central catheter in pediatric patients. Int J Technol Assess Health Care. 2018;34(1):38-45. PubMed
10.Cedarbaum J, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169:13-21. PubMed
11.Zaric G. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-396. PubMed
12.Riluzole (riluzole): 50 mg tablets [product monograph]. Toronto (ON): Apotex Inc.; 2012 Jul 9: https://pdf.hres.ca/dpd_pm/00017738.PDF. Accessed 2022 Jan 11.
13.Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Jan 11.
14.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Albrioza (sodium phenylbutyrate and ursodoxicoltaurine), 3 mg sachet sodium phenylbutyrate sachet and 1 mg sachet ursodoxicoltaurine powder for suspension, oral. Calgary (AB): Amylyx Canada; 2021 Nov 24.
15.Sutherland G, Dinh T. Understanding the gap. A pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: http://innovativemedicines.ca/wp-content/uploads/2017/12/20170712-understanding-the-gap.pdf. Accessed 2022 Feb 11.
16.Hodgkinson V, Lounsberry J, Mirian A, et al. Provincial differences in the diagnosis and care of amyotrophic lateral sclerosis. Can J Neurol Sci. 2018;45:652-659. PubMed
17.Clinical Study Report: AMX3500-OLE (CENTAUR-OLE). Evaluation of the safety, tolerability, efficacy, and activity of AMX0035, a fixed combination of phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA), for treatment of amyotrophic lateral sclerosis: open-label extension [internal sponsor's report]. Cambridge (MA): Amylyx Pharmaceuticals, Inc; 2021 May 14.
Appendix 1: Cost Comparison Table
Note that this table has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Amyotrophic Lateral Sclerosis
Treatment | Strength / concentration | Form (vial size if single use) | Price | Recommended dosagea | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
PB-TURSO | 3 g sodium phenylbutyrate 1 g ursodoxicoltaurine | Powder for suspension (sachet) | $306.7123b | 1 sachet daily for the first 3 weeks, followed by 2 sachets daily thereafter | First 3 weeks: $306.71 After 3 weeks: $613.42 | First year: $217,459 Subsequent years: $223,900 |
Active treatments | ||||||
Edaravone | 0.3 mg/mL | 100 mL | $460.2000 | Cycle 1: 60 mg daily for 14 days, followed by a 14-day drug-free period Cycle 2+: 60 mg daily for 10 out of 14 days, followed by 14-day drug-free periods | First 4 weeks: $460.20 After 4 weeks: $328.71 | First year: $124,254 Subsequent years: $119,652 |
Riluzole | 50 mg | Tablet | $3.4361 | 50 mg twice daily | $6.87 | $2,508 |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
Note: All prices are from the Saskatchewan Drug Plan Formulary (accessed January 2022),13 unless otherwise indicated, and do not include dispensing fees.
aRecommended doses as per the respective product monographs.2,4,12
bSponsor-submitted price.3
Appendix 2: Submission Quality
Note that this table has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The sponsor’s base case included patients who were expected never to benefit from treatment with PB-TURSO. |
Model has been adequately programmed and has sufficient face validity | No | CADTH identified a programming error. Parameters in both the PE model and BIA were found in multiple places within the model, complicating the validation process. |
Model structure is adequate for decision problem | No | The FT9 staging system is not used in clinical practice and does not adequately capture treatment benefits important to patients and clinicians, namely the slowing of disease progression. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Some uncertainty remains in the PE analysis due to issues with the FT9 staging system. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Details regarding some pharmacoeconomic and budget impact assumptions were lacking. |
BIA = budget impact analysis; FT9 = Fine’til 9; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; PE = pharmacoeconomic.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
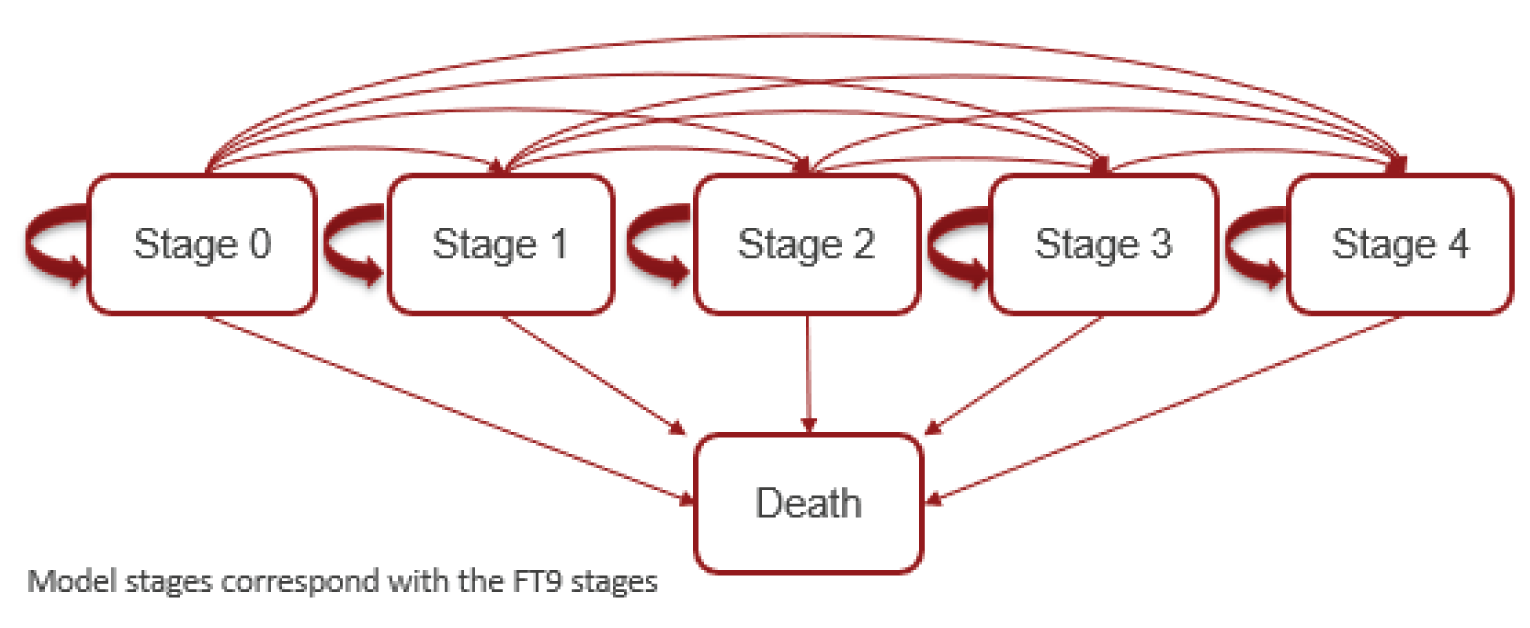
Table 10: Transition Matrix for Health State Transition Probabilities
FT9 stage | 0 | 1 | 2 | 3 | 4 | Death | |
|---|---|---|---|---|---|---|---|
Best supportive care | |||||||
0 | 0.69 | 0.27 | 0.04 | 0.00 | 0.00 | 0.00 | |
1 | 0.00 | 0.77 | 0.21 | 0.02 | 0.00 | 0.00 | |
2 | 0.00 | 0.00 | 0.86 | 0.12 | 0.01 | 0.01 | |
3 | 0.00 | 0.00 | 0.00 | 0.82 | 0.16 | 0.02 | |
4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.90 | 0.10 | |
Riluzole | |||||||
0 | 0.70 | 0.27 | 0.03 | 0.00 | 0.00 | 0.00 | |
1 | 0.00 | 0.81 | 0.18 | 0.02 | 0.00 | 0.00 | |
2 | 0.00 | 0.00 | 0.84 | 0.14 | 0.01 | 0.00 | |
3 | 0.00 | 0.00 | 0.00 | 0.84 | 0.14 | 0.02 | |
4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.07 | |
FT9 = Fine’til 9.
Source: Sponsor’s Pharmacoeconomic Report (2021).3
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Results of the Sponsor’s Base Case
Parameter | PB-TURSO | Edaravone | Riluzole | BSC |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 2.082 | 1.919 | 1.699 | 1.562 |
Stage 0 | 0.013 | 0.010 | 0.010 | 0.010 |
Stage I | 0.170 | 0.125 | 0.122 | 0.100 |
Stage II | 0.426 | 0.339 | 0.350 | 0.372 |
Stage III | 0.488 | 0.428 | 0.407 | 0.360 |
Stage IV | 0.986 | 1.018 | 0.811 | 0.721 |
Discounted QALYs | ||||
Total | 1.126 | 1.001 | 0.915 | 0.845 |
Stage 0 | 0.010 | 0.007 | 0.007 | 0.007 |
Stage I | 0.119 | 0.085 | 0.085 | 0.070 |
Stage II | 0.260 | 0.201 | 0.213 | 0.227 |
Stage III | 0.264 | 0.226 | 0.220 | 0.194 |
Stage IV | 0.474 | 0.483 | 0.389 | 0.346 |
Discounted costs ($) | ||||
Total | 293,177 | 258,757 | 137,894 | 122,513 |
Drug acquisition and administration | 139,229 | 107,403 | 3,827 | 0 |
Health state costs | 153,889 | 151,256 | 132,836 | 122,513 |
Adverse event costs | 59 | 97 | 1,231 | 0 |
Pairwise ICER vs. BSC | 607,704 | 873,257 | 220,625 | Reference |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year; vs. = versus.
Table 12: Probabilistic Cost-Effectiveness Sequential Analysis from Sponsor’s Base Case
Treatment | Cost | QALYs | Incremental cost | Incremental QALYs | ICER |
|---|---|---|---|---|---|
BSC | $122,513 | 0.85 | Reference | Reference | Reference |
Riluzole | $137,894 | 0.91 | $15,381 | 0.07 | $220,615 vs. BSC |
Edaravone | $258,757 | 1.00 | — | — | Extendedly dominated by PB-TURSO |
PB-TURSO | $293,177 | 1.13 | $155,283 | 0.21 | $735,528 vs. riluzole |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year; vs. = versus.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 13: Disaggregated Results of the CADTH Base Case
Parameter | PB-TURSO | Edaravone | Riluzole | BSC |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 1.999 | 1.781 | 1.781 | 1.600 |
Stage 0 | 0.013 | 0.009 | 0.009 | 0.010 |
Stage I | 0.168 | 0.122 | 0.122 | 0.100 |
Stage II | 0.455 | 0.346 | 0.346 | 0.371 |
Stage III | 0.559 | 0.453 | 0.453 | 0.401 |
Stage IV | 0.803 | 0.851 | 0.851 | 0.718 |
Discounted QALYs | ||||
Total | 1.095 | 0.929 | 0.958 | 0.866 |
Stage 0 | 0.010 | 0.007 | 0.007 | 0.007 |
Stage I | 0.118 | 0.083 | 0.085 | 0.070 |
Stage II | 0.278 | 0.205 | 0.211 | 0.227 |
Stage III | 0.303 | 0.238 | 0.246 | 0.217 |
Stage IV | 0.386 | 0.397 | 0.409 | 0.345 |
Discounted costs ($) | ||||
Total | 431,923 | 327,771 | 146,863 | 126,975 |
Drug acquisition and administration | 287,438 | 187,407 | 6,519 | 0 |
Health state costs | 144,485 | 140,343 | 140,343 | 126,975 |
Adverse event costs | 0 | 21 | 0 | 0 |
Pairwise ICER vs. riluzole ($/QALY) | 2,086,658 | Dominated | Reference | NA |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year; vs. = versus.
Scenario Analyses
Table 14: Summary of Scenario Analyses Conducted on CADTH Base Case
Scenario | Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER |
|---|---|---|---|---|---|
CADTH base case | Riluzole alone | 146,863 | 1.781 | 0.958 | Reference |
PB-TURSO + riluzole | 431,923 | 1.999 | 1.095 | 2,086,658 | |
1. Exclusion of 1-time costs | Riluzole alone | 114,180 | 1.781 | 0.958 | Reference |
PB-TURSO + riluzole | 400,291 | 1.998 | 1.094 | 2,096,061 | |
2. Exclusion of GT and NIV costs | Riluzole alone | 109,872 | 1.781 | 0.958 | Reference |
PB-TURSO + riluzole | 393,978 | 1.999 | 1.094 | 2,079,808 |
GT = gastric tube; ICER = incremental cost-effectiveness ratio; LY = life-year; NIV = noninvasive ventilation; PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this table has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
Summary of Sponsor’s Budget Impact Analysis
The submitted BIA assessed the introduction of PB-TURSO for the treatment of patients with ALS. The analysis was taken from the perspective of the Canadian public drug plans using an epidemiology-based approach, with drug acquisition, mark-ups, dispensing fees, and co-payments included. A 3-year time horizon was used, from 2022 to 2024, with 2021 as a base year. The population size was derived using a prevalence estimate from ALS Canada and a summary of the derivation of the population size is available in Figure 2.
The reference case scenario included edaravone while the new drug scenario included edaravone and PB-TURSO. All patients in both scenarios were assumed to receive concomitant riluzole and therefore, it was not associated with a cost. PB-TURSO was assumed to displace edaravone use in the market. Key inputs to the BIA are documented in Table 16.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
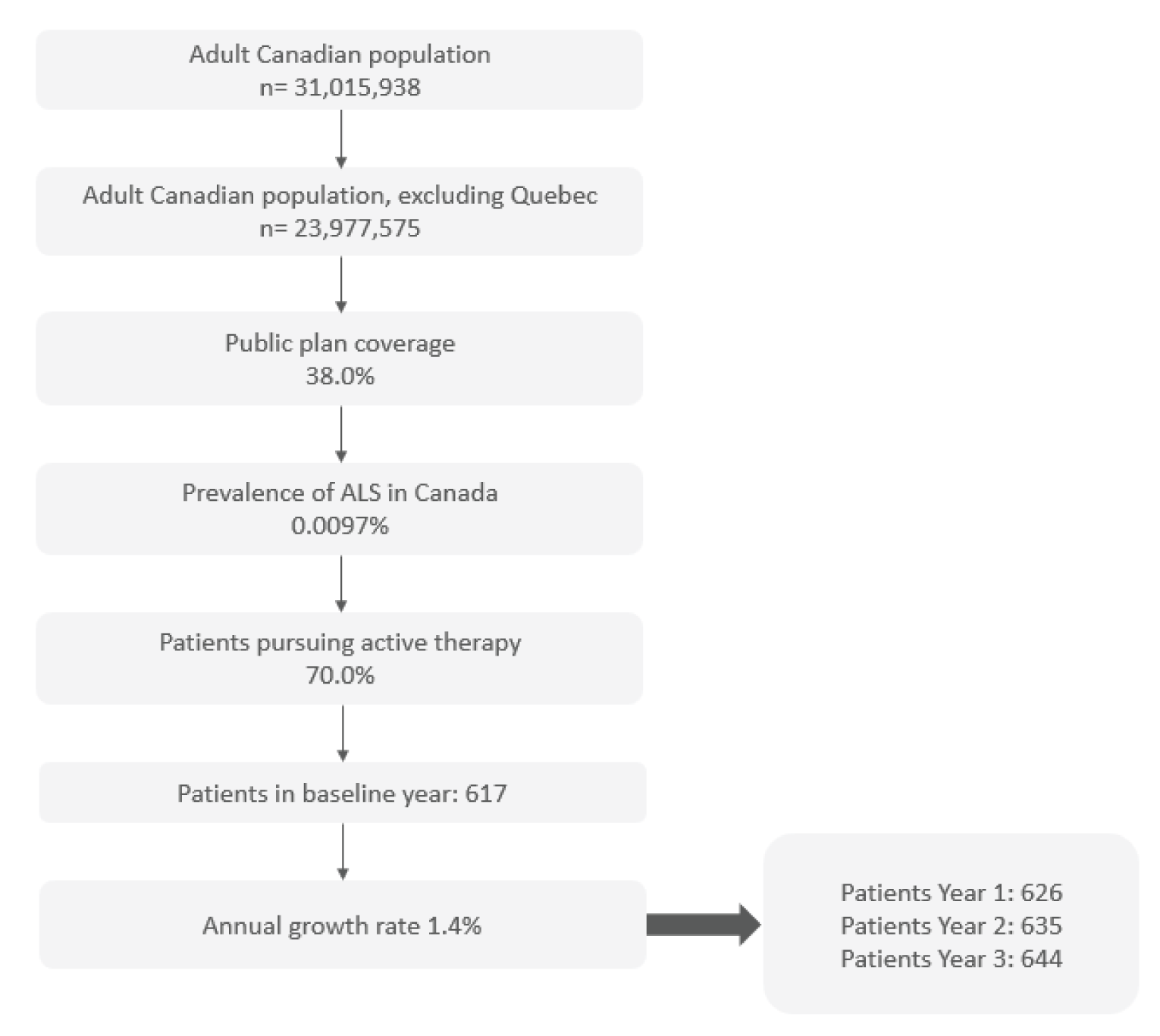
ALS = amyotrophic lateral sclerosis.
Source: Sponsor’s Pharmacoeconomic Report (2021).3
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Number of patients eligible for drug under review | 626/635/644 |
Market uptake (3 years) | |
Uptake (reference scenario) PB-TURSO + riluzole Edaravone + riluzole Riluzole alone | 0%/0%/0% 19.53%/25.75%/30.41% 80.47%/74.25%/69.59% |
Uptake (new scenario) PB-TURSO + riluzole Edaravone + riluzole Riluzole alone | 30%/43%/45% 12.03%/10.70%/10.16% 57.97%/46.30%/44.84% |
Cost of treatment (per patient) | |
Cost of treatment annually PB-TURSO + riluzole Edaravone + riluzole Riluzole alone | $190,086 $112,547 $0a |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
aAs all patients are assumed to receive riluzole concomitantly, it does not have an associated cost.
Summary of the Sponsor’s Budget Impact Analysis Results
The estimated budget impact of funding PB-TURSO for the treatment of patients with ALS was $29,270,156, $39,554,824, and $38,805,028 for year 1, year 2, and year 3, respectively, for a cumulative incremental impact of $107,630,008 over the 3-year time horizon.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Proportion of patients pursuing active therapy underestimated: The sponsor assumed that 70% of patients would pursue active therapy based on claims data for riluzole. The clinical expert consulted by CADTH felt that value was an underestimate, noting that essentially every patient with ALS would receive riluzole first, given its demonstrated clinical benefit and favourable safety profile. The only patients who are truly expected to not receive active therapy are those for whom financial considerations present an obstacle. However, as the sponsor has also restricted the population to those receiving public coverage, this issue of financial considerations does not apply, and therefore overestimates the proportion of patients who would not pursue active therapy.
As part of the base case, CADTH increased the proportion of patients expected to pursue active therapy to 85% (midway between 70% and 100%). The sponsor’s original assumptions were tested in scenario analysis.
Proportion of patients with public drug coverage underestimated: The sponsor generated an estimate of the proportion of patients with public drug coverage of 38.0%. This was based on work done by a consultancy using Statistics Canada data, details of which were lacking in the provided technical report.14 Given the mean age of 57.5 years assumed in the sponsor’s model, a public drug coverage rate of 38.0% was deemed to be underestimated, especially given the relatively low market shares of PB-TURSO. Market shares estimates encompass a variety of unknown assumptions including patient preference, prescribing patterns, ease of administration, and so forth. Without the means to validate either the market shares or the public drug coverage, uncertainty remains in the analysis. As such, CADTH sought a public source to estimate public drug coverage rates, the 2017 Understanding the Gap report.15 Data from this report were available by jurisdiction, and were used to estimate public drug coverage rates for an oral/self-administered product by calculating the proportion enrolled and proportion eligible for public drug coverage in Canada over the age of 25. While market shares remained unchanged, this approach mitigates some of the uncertainty and potential double-counting by the sponsor as regards the proportion of patients who would not receive PB-TURSO due to public coverage. CADTH also notes a paper published using data from the Canadian Neuromuscular Disease Registry suggests the mean age of diagnosis of patients with ALS is 61.8 with a standard deviation of 11.9.16 Given public coverage is close to 100% for patients over the age of 65 in most provinces and PB-TURSO is also indicated for patients after the point of diagnosis this further suggests the 38% public coverage estimate provided by the sponsor is an underestimate. Thus, CADTH sought to determine a more appropriate estimate of the proportion of patients receiving public coverage, noting various factors:
Using data from Hodgkinson et al.,16 it could be approximated that 39% of patients with ALS may be diagnosed over the age of 65. According to the Understanding the Gap report 90% of patients over the age of 65 are enrolled in public drug plans and 99% are eligible.
Again, using data from Hodgkinson et al.,16 it could be approximated that 61% of patients with ALS would be diagnosed before the age of 65. According to the Understanding the Gap report 25% of patients between 25 and 65 years of age are enrolled in public drug plans and 55% are eligible.
Taking all of the aforementioned into account, CADTH calculated Canadian public drug coverage rates to be between 50 and 73% at a national level, depending on what proportion of patients eligible for public coverage are enrolled on a public plan. Given there is uncertainty regarding what proportion of patients with ALS are over the age of 65 and what proportion of eligible patients are covered CADTH chose a value of 61% in the base case. Scenario analyses were performed in which higher and lower values for public drug coverage were assumed.
PB-TURSO is expected to be used concomitantly with edaravone: According to the clinician input received for this review, there is clinical rationale for patients to receive PB-TURSO concurrently with edaravone and riluzole. This fact was confirmed by the clinical expert consulted by CADTH. While the sponsor has assumed PB-TURSO and edaravone to be separate treatments with separate market shares, the clinical expert stated that PB-TURSO would not displace edaravone but would be used additively, leading to additional costs to the health care payer and no cost savings that arise from replacing 1 therapy with another.
As part of the base case, CADTH assumed that edaravone would not be displaced by PB-TURSO. CADTH assumed the decision to place a patient on edaravone would be made independently of whether access to PB-TURSO was available as they have separate mechanisms of actions and would therefore not be considered mutually exclusive treatment options. Therefore, the budget associated with edaravone would be equal in both the reference and new drug scenario. An exploratory scenario analysis was conducted to measure the impact of all patients on edaravone also receiving PB-TURSO.
Treatment duration is underestimated: As in the pharmacoeconomic analysis, treatment with PB-TURSO and edaravone was capped at 11 months. And as described previously, this treatment duration is likely underestimated in a real-world setting, as the product monograph does not stipulate stopping rules for PB-TURSO.2 A post-hoc analysis of the open-label extension phase of the CENTAUR trial suggests that more than half of enrolled patients received PB-TURSO for longer, specifically 14.7 months.17 The drug plan input received for this review also supported a higher duration of therapy with PB-TURSO.
As part of the base case, CADTH assumed a treatment duration of 14.7 months for both PB-TURSO and edaravone.
Uncertainty regarding patient co-payments: The sponsor included patient co-payments as part of their base case. There is uncertainty in the impact of this assumption due to the variability between types of programs/provinces and type of co-payment system implemented (e.g., income based, fixed ceiling, variable stepped rate). Further, co-payments are typically inclusive of all treatments received by the patient and may already reach the maximum amount to be paid prior to starting the new drug.
As part of the base case, CADTH excluded patient co-payments and tested in a scenario analysis.
CADTH Reanalyses of the Budget Impact Analysis
Based on the identified limitations, CADTH’s base-case analysis included an increase to the proportion of patients expected to receive active therapy, the proportion with public drug coverage, and the treatment duration, along with assuming that PB-TURSO would not displace edaravone (Table 17).
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Proportion of patients receiving active therapy | 70% | 85% |
2. Proportion of patients with public drug coverage | 38.0% | 61% |
3. Displacement of edaravone by PB‑TURSO | Assumed | Not assumed |
4. Treatment duration of PB-TURSO and edaravone | 11 months | 14.7 months |
5. Patient co-payments | Included | Excluded |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 + 5 | |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. Based on the CADTH base case, the budget impact of the reimbursement of PB-TURSO for the treatment of patients with ALS is expected to be $122,345,734 in year 1, $177,817,289in year 2, and $188,693,091 in year 3, with a 3-year total of $488,856,114. CADTH emphasizes that the sponsor’s base-case analysis has vastly underestimated the budget impact.
Scenario analyses were conducted on the proportion of patients expected to receive active therapy and/or public drug coverage. The 3-year budget impact totals resulting from these analyses ranged from $312,354,634 to $802,501,394, indicating that, in all scenarios tested, the sponsor’s budget impact estimate was still underestimated. Even with a 98% price reduction from the pharmacoeconomic analysis the 3-year budget impact is still expected to be greater than $10 million.
Table 18: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $107,630,008 |
CADTH reanalysis 1: Active therapy | $130,693,581 |
CADTH reanalysis 2: Public drug coverage | $175,763,614 |
CADTH reanalysis 3: PB-TURSO does not displace edaravone | $137,480,294 |
CADTH reanalysis 4: Increased treatment duration | $194,446,239 |
CADTH reanalysis 5: Excluded co-payments | $107,242,568 |
CADTH base case | $488,856,114 |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
Table 19: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $11,787,170 | $16,212,439 | $20,759,307 | $24,331,694 | $61,303,440 |
New drug | $11,787,170 | $45,482,595 | $60,314,130 | $63,136,722 | $168,933,448 | |
Budget impact | $0 | $29,270,156 | $39,554,824 | $38,805,028 | $107,630,008 | |
CADTH base case | Reference | $36,588,786 | $51,986,857 | $67,808,902 | $80,225,084 | $200,020,843 |
New drug | $36,588,786 | $174,332,591 | $245,626,191 | $268,918,175 | $688,876,957 | |
Budget impact | $0 | $122,345,734 | $177,817,289 | $188,693,091 | $488,856,114 | |
CADTH scenario analysis 1: 80% public drug coverage | Reference | $47,580,428 | $67,577,101 | $88,124,356 | $104,248,813 | $259,950,270 |
New drug | $47,580,428 | $226,445,269 | $319,023,350 | $349,270,234 | $894,738,853 | |
Budget impact | $0 | $158,868,167 | $230,898,994 | $245,021,421 | $634,788,583 | |
CADTH scenario analysis 2: 38% public drug coverage | Reference | $23,294,685 | $33,130,866 | $43,237,895 | $51,169,017 | $127,537,778 |
New drug | $23,294,685 | $111,303,678 | $156,854,259 | $171,734,475 | $439,892,412 | |
Budget impact | $0 | $78,172,812 | $113,616,364 | $120,565,458 | $312,354,634 | |
CADTH scenario analysis 3: 70% receive active therapy | Reference | $30,680,514 | $43,447,484 | $56,565,915 | $66,861,622 | $166,875,022 |
New drug | $30,680,514 | $144,552,401 | $203,511,802 | $222,795,129 | $570,859,332 | |
Budget impact | $0 | $101,104,917 | $146,945,887 | $155,933,507 | $403,984,311 | |
CADTH scenario analysis 4: included patient co-payments | Reference | $37,254,910 | $52,757,659 | $68,687,183 | $81,189,113 | $202,633,955 |
New drug | $37,254,910 | $175,527,915 | $247,121,473 | $270,536,943 | $693,186,332 | |
Budget impact | $0 | $122,770,256 | $178,434,291 | $189,347,830 | $490,552,377 | |
CADTH scenario analysis 5: 98% price reduction | Reference | $36,588,786 | $51,986,857 | $67,808,902 | $80,225,084 | $200,020,843 |
New drug | $36,588,786 | $54,545,061 | $71,526,995 | $84,170,587 | $210,242,642 | |
Budget impact | $0 | $2,558,204 | $3,718,093 | $3,945,502 | $10,221,800 | |
CADTH scenario analysis 6: all patients on edaravone would also receive PB‑TURSO | Reference | $36,588,786 | $51,986,857 | $67,808,902 | $80,225,084 | $200,020,843 |
New drug | $36,588,786 | $253,979,663 | $352,109,801 | $396,432,773 | $1,002,522,237 | |
Budget impact | $0 | $201,992,806 | $284,300,899 | $316,207,689 | $802,501,394 |
PB-TURSO = sodium phenylbutyrate-ursodoxicoltaurine.
Stakeholder Input
Patient Input
ALS Society of Canada
About ALS Society of Canada
Founded in 1977, the ALS Society of Canada (ALS Canada) is dedicated to supporting Canadians living with ALS and investing in research to achieve a future without ALS. We are a registered charity that receives no core government funding – all our services and research are funded through the generosity of donors. ALS Canada advocates federally, provincially, and locally for better government support and access within the healthcare system for people touched by ALS.
Information Gathering
The information contained in this submission was gathered by ALS Canada through an online survey and telephone interviews. All the data was contributed anonymously.
ALS Canada developed and administered a 20-minute online survey that was disseminated in English and French. Survey respondents were recruited by ALS Canada through promotion via email, social media, blog posts, e-newsletters and other online platforms, with the following populations invited to take part: people living with ALS (“patients”) and their caregivers and family members (“caregivers”). The online survey was open between November 10 and 24, 2021.
A total of 629 patients and caregivers responded to the English (558) and French (71) online surveys. Almost all were from Canada (primarily Ontario and Quebec), with a small number from the U.S., the U.K., Israel, and the Netherlands. Approximately 70% of respondents are, or were, caregivers to someone diagnosed with ALS. The remainder are currently living with the disease. Among the respondents living with the disease, approximately 60% indicated they had been diagnosed with ALS between six months and two years ago, with some having received their diagnosis more than three years ago. More than half of respondents were 55 years of age or older.
In November 2021, ALS Canada supplemented the information gathered through the surveys by conducting telephone interviews with two patients and one caregiver, all of whom had experience with AMX0035.
Disease Experience
ALS is a terminal disease that gradually paralyzes people because the brain is no longer able to communicate with the muscles of the body that we are typically able to move at will. As the connection with muscles of the body breaks down, someone living with ALS will lose the ability to walk, talk, eat, swallow, and eventually breathe. Of those who receive an ALS diagnosis, 80% will die within two to five years of diagnosis. Every year, approximately 1,000 Canadians die of ALS and a similar number are diagnosed. There are currently about 3,000 people living with ALS in Canada.
A diagnosis of ALS and the realities of living with the disease have a profound and pervasive effect on the lives of not only those who are struck by this devastating disease, but also anyone who loves and cares for them. The following is a summary of how the respondents of this survey – people living with ALS, caregivers and family of those who are living or have lived with the disease – describe its impact on their lives.
With respect to the wide range of symptoms patients experienced due to ALS, among the most severe are decreased muscle tone, and related difficulties with mobility (including walking and standing), gripping/holding things, muscle cramping/twitching and fatigue caused by muscle exhaustion. These symptoms were also among the most important to control for people living with ALS, in addition to difficulties breathing, speaking, and choking episodes.
When asked how living with ALS has negatively affected their quality of life, patients indicated that their social life, travel/hobbies, and family life suffered the most as seen in the figure below.

The impact of ALS was said to be very pervasive, especially the loss of independence which touched all aspects of patients’ lives. Among patients who need help performing daily tasks, such as feeding themselves, drinking, bathing and toileting, approximately half said they require support for up to five hours per week.
People living with ALS also indicated that they rely on a range of assistive devices with a walker, specialized bathroom equipment, non-invasive ventilation support, a lift chair, and standard and power wheelchairs identified as the most commonly used.
When asked to describe in their own words how their day-to-day life and quality of life have been impacted by ALS, patients said:
“I am unable to live with my wife, go walking, participate in family activities, enjoy my grandkids, or go driving. It has forced me to rely on caregivers to manage my day-to-day life.”
“My sense of self has taken a huge hit and continues to be chipped away at with each small decline in my physical abilities. I am finding it very difficult to maintain a positive outlook.”
“I have become very depressed and don't want to talk to my loved ones as I have difficulty talking, eating and breathing.”
“I miss going hiking in the countryside, being surrounded by nature, traveling with friends, and seeing the stars at night. I will now be living the rest of my life in a 10' x 20' room, with little exposure to the outside world.”
« Beaucoup de solitude, vie sociale pratiquement nulle, ne peux plus sortir seule, fini les restaurants, je ne peux plus venir en aide à mes enfants, m'occuper de mon petit-fils comme je le faisais avant, congédiement de mon employeur après 33 ans de bons services, projets de retraite irréalisables, extrêmement difficile émotionnellement, loisirs personnels plus rien ne va. Épuisement juste à essayer de communiquer. Fatigue continuelle. Savoir qu'un médicament pourrait aider à ralentir la maladie et ne pouvoir y avoir accès est inhumain. Je suis passé de totalement autonome, emploi professionnel à devoir demander de l'aide et confiner à la maison et ça n'a pas fini de s'aggraver. »
The results in the following figure clearly illustrate the degree to which ALS has negatively impacted the lives of the caregivers who responded. Family life, emotional/psychological well- being and travel options were the aspects of caregivers’ lives most impacted; with 44% of respondents saying that their family life had “completely changed.” They specifically mentioned pervasive feelings of overwhelming grief and struggles with mental health, including stress, anxiety, and helplessness/hopelessness as they watched their loved one’s body wither away and die. One person said: “This disease has eaten away at our family emotionally and financially and has stripped us of all that we have.”
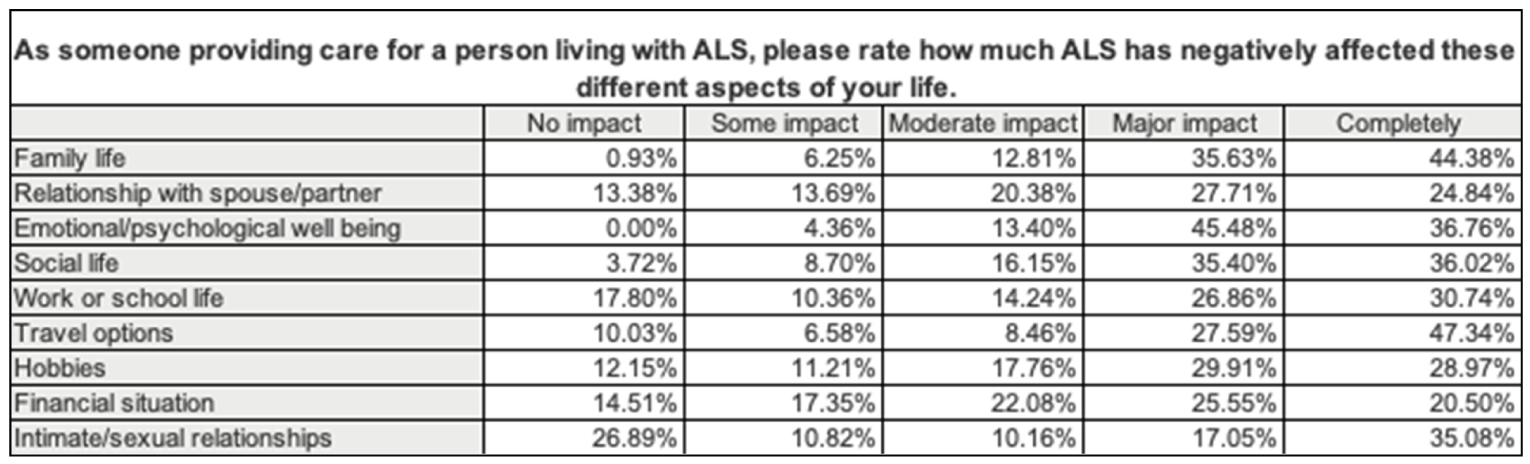
The day-to-day lives of caregivers providing assistance to people living with ALS are dramatically impacted by the disease. Needing to assist the patient under their care with aspects of daily living such as exercising (including physical therapy), bathing, walking, and using the toilet, were found to have “completely changed” the lives of approximately half the respondents.
Assistive devices and medical interventions are critical to the care of people living with ALS. Of the caregivers surveyed, with approximately 70% noting the need for specialized bathroom equipment, followed by hospital bed/mattress, walker, non-invasion ventilation support and standard/power wheelchairs.
When asked to describe in their own words how their day-to-day life and quality of life have been affected by providing care for someone living with ALS, caregivers said:
“The emotional toll on our family’s life is immeasurable. The fact that this disease holds out NO hope makes day-to-day demands almost impossible to carry out.”
“This horrible nature of disease is not easy to describe in words. It literally steals the life bit by bit – day-by-day with tremendous cruelty. The hardship to the patient and family is worse than any other affliction I have ever witnessed.”
“Knowing your loved one is not going to get better but will eventually expire is a daunting daily emotional struggle. Physically having to care for a loved one with every aspect of their care is draining, stressful, emotional, and physically exhausting.
“As a caregiver there really isn’t much quality of life. Your ALS person is your focus every moment of every day. Your eating habits change, your sleeping habits change. There really isn’t much else you have the energy for.”
« L’imprévisibilité de l’évolution de la maladie, la peur de ne pas avoir les moyens financiers nécessaires pour accompagner jusqu’au bout, la conciliation travail/être présent pour aider, pouvoir continuer mes propres projets de vie est un facteur de stress élevé au quotidien. La peine engendrée par la dégénération constante de la personne et la peur qu’elle ne trouve plus de bonheur à vivre ainsi est un poids au quotidien. »
« Ma santé mentale a été la première chose atteinte. L'effet de choc reste constant avec chaque petit changement chez la personne atteinte (ma mère). Vu la rapidité de la maladie, j'ai dû faire un choix sur ma carrière en diminuent mes heures pour les redonner comme proche aidant. Ce qui diminue mon revenu mensuel. De plus, mon moral au travail diminue de beaucoup, mon efficacité et mon rendement. Après avoir passé une journée avec ma mère malade, j'ai perdu tout désir, plaisir, et joie de vive. Ma tête reste sans arrêt dans l'inquiétude de n'être pas présente pour elle. »
Experiences with Currently Available Treatments
Rilutek (riluzole)
When it comes to approved treatments for ALS, 101 patients said they are taking/have taken Rilutek (riluzole). As seen in the figure below, among the benefits some patients believe they have seen from this therapy, all were identified as being the most important. That said, several patients commented that they are unsure if the drug is having an impact on their ALS, and others said it’s too early to tell.
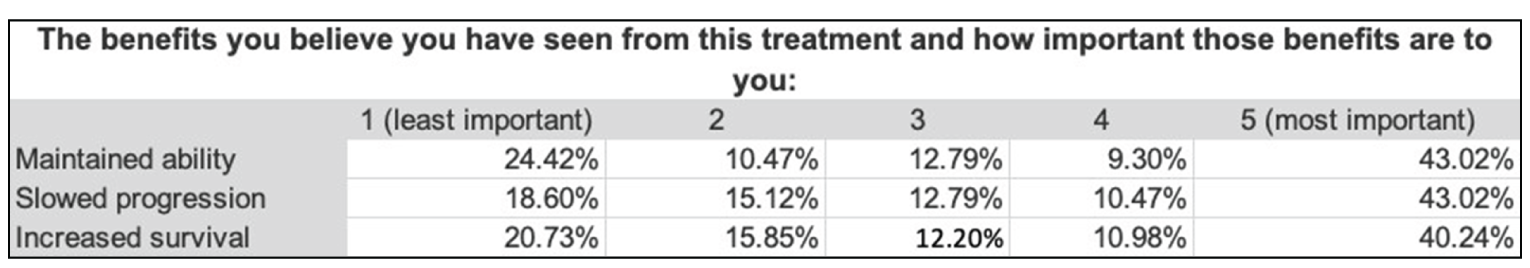
Most patients said they didn’t experience any side effects while taking riluzole, but among those who did, the most difficult to manage were tiredness, weakness, muscle stiffness and gastrointestinal problems.
And while many patients haven’t had any problems accessing riluzole, some mentioned difficulty due to partial or no private coverage, strict funding criteria, out-of-pocket costs, and a shortage of supply due to COVID-19. As an oral treatment, taking riluzole wasn’t a problem for most patients, but some mentioned having difficulty swallowing it, and others opted to dissolve it in water or take it through their feeding tube.
Among the caregiver respondents, 156 supported a person living with ALS who used/had used Rilutek (riluzole). Similar to the patient responses, the benefits observed by some of these caregivers – slowing disease progression, maintained ability and increased survival – were all identified as important, as seen in the figure below. However, there were several caregivers who stated that they could not tell or didn’t know if riluzole had helped the person living with ALS.
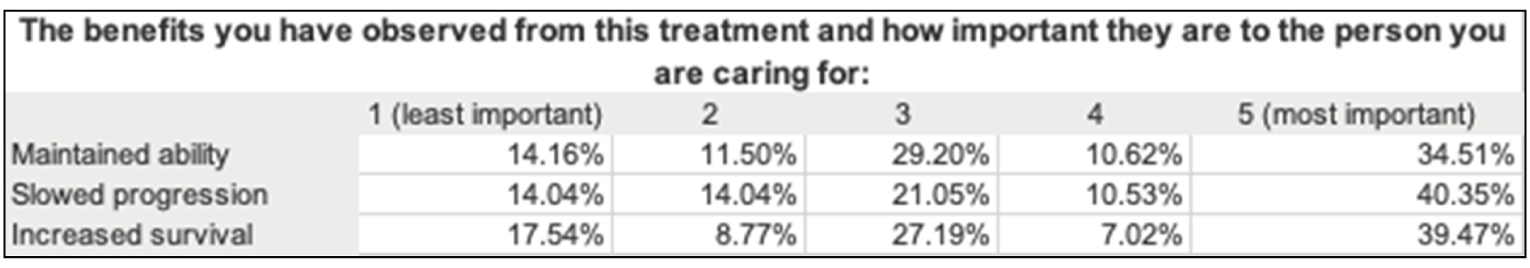
Varied side effects of riluzole were observed by caregivers, with most seemingly quite manageable including vomiting, runny nose, spinning sensation and nausea. Other caregivers noted that it can be difficult to tell the difference between the symptoms of ALS and the potential side effects of riluzole.
Almost two-thirds of respondents reported no difficulty with patients accessing riluzole, although just under 15% encountered “cost implications” as a challenge. Approximately 55% of respondents reported patients having difficulty swallowing riluzole and several caregivers added that the person living with ALS had no difficulties receiving riluzole.
When asked about any benefits caregivers had experienced as a result of taking riluzole, approximately 45% reported having more time with the person under their care because of delayed progression of ALS. A smaller number reported “independence for the person I am providing care for” and being “able to maintain my daily schedule with less interruptions due to care requirements” as benefits, while several others reported that there were no benefits at all.
Radicava (edaravone)
With respect to Radicava (edaravone), the other approved treatment for ALS, 43 patients said they have/had experience with the medication. Among the benefits some patients believe they have seen from this therapy, slowing disease progression, maintained ability and increased survival were all identified as important, as in the figure below.
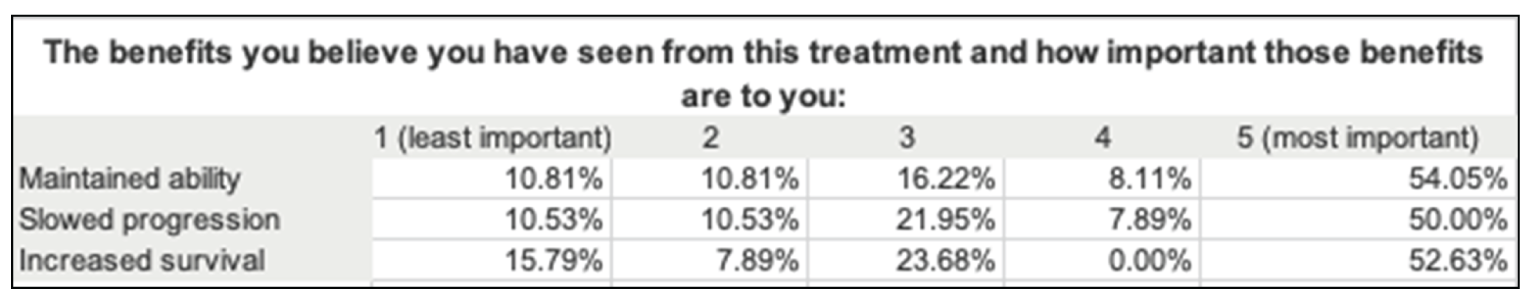
Most patients said they didn’t experience any side effects while taking edaravone, but some had difficulty managing changes to their normal walking gait, skin irritation, pressure or pain at the injection site and headaches. And while many patients haven’t had any problems accessing edaravone, some mentioned difficulty with out-of-pocket costs, travel to infusion clinics, and a lack of home care nurses available for infusions. Any difficulties with taking edaravone were related to the IV administration, including patients having to schedule activities of daily living around their infusion schedule and needing to have a port catheter implanted.
Of the caregiver respondents who supported a person living with ALS, 57 use/had used Radicava (edaravone). Interestingly, as seen in the following figure, maintained ability, slowed progression, and increased survival ranked equally as important as benefits observed by these caregivers. However, there were also several caregivers who stated that they could not tell or didn’t know if edaravone had helped the person living with ALS.
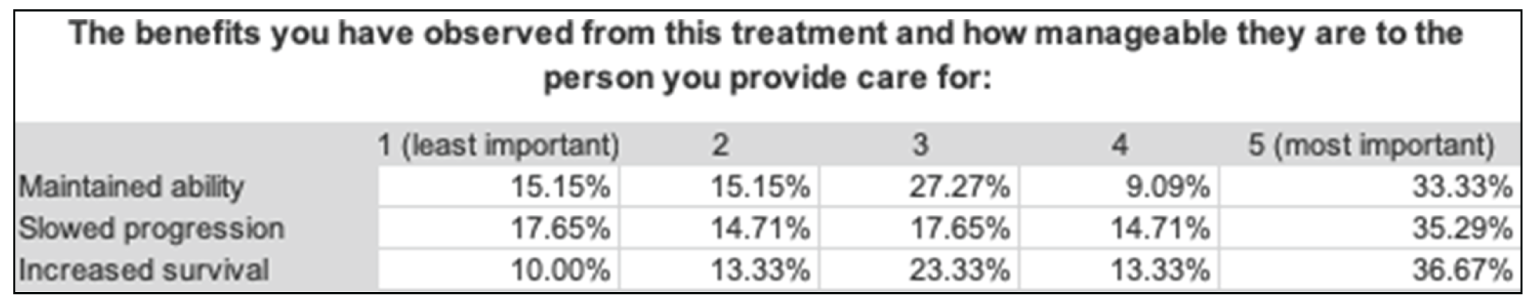
Varied side effects of edaravone observed by caregivers, with most seemingly quite manageable including eczema, fungal infection, hypoxia, glycosuria and skin inflammation or rash. However, almost 18% noted changes from normal walking gait as a “most severe” side effect.
Caregiver respondents reported a number of difficulties with patients accessing edaravone, including travel to clinic/hospital/outpatient clinic for infusions, a lack of home care nurses available to do daily infusions, and out-of-pocket costs.
The difficulties caregivers noted with patients receiving edaravone are significant. They include having to schedule activities of daily living around infusion schedules, being unable to self- administer edaravone, and the inability to get their IV started. Similarly, most challenges encountered by caregivers (78%) were related to administering and scheduling around infusions, with 30% reporting challenges related to costs, including a lack of private/public coverage and restrictive coverage criteria.
And finally, approximately 55% of respondents reported “more time with the person I am providing care for because of delayed progression” as a benefit to taking Radicava; however, many said they had experienced no benefits at all.
Other Treatments
When patients and caregivers were asked what treatments other than Rilutek (riluzole), Radicava (edaravone) and AMX0035 they were using to treat ALS or its symptoms, they mentioned a variety of natural supplements including antioxidants, as well as cannabis/cannabis products and Chinese medicine. They also listed several prescription and OTC products intended for symptomatic relief including Fentanyl patch, Hydromorphone, Lorazepam, Gabapentin, Keppra, Amantadine, Baclofen, Domperidone, Prucalopride, Prevacid, Restoralax, Lactulose, Nuedexta, Quinidex, Botox injections, Atropine, Abreva, Tylenol/Aleve, etc., stem cell therapy, human growth hormone, anti-depressants, and diuretics. And, some patients and caregivers mentioned participating in clinical trials for investigational therapies other than AMX0035.
Improved Outcomes
When asked what improvements they would like to see in a new treatment for ALS that are not achieved in currently available treatments, most patients mentioned maintaining current function and independence, delayed progression, symptom reversal and increased survival.
Some patients wanted to see specific improvements to their speech, mobility and energy levels, and others hoped for a cure for ALS and increased survival only if their symptoms improved.
If the desired improvements they hoped for in a new treatment were achieved, patients said the result would be:
“Not to be continually afraid of tomorrow, at least to stop the progression of the disease, to be able to live a little and not only to survive.”
“I could be independent again, enjoy life, and spend time with my grandchildren and children. I could speak again and eat again and talk again. I used to sing with my grandsons – we had so much fun together, now I can't even talk with them.”
“Not having to worry about running out of time and having to make judgements as to priorities. I would have more time with family and be able to dance at my daughters’ weddings.”
« Garder mon autonomie le plus longtemps possible est très important pour moi pour ne pas trop surcharger ma conjointe car nous avons 2 jeunes enfants à s'occuper. Prolonger ma vie est également très important pour moi pour pouvoir voir mes enfants grandir je souhaite au moins jusqu' à l’âge adulte. »
When asked about the improvements that caregivers would like to see in a new treatment for ALS, it is not surprising that they ranked all of the choices high – maintain current function and independence, delayed progression, symptom reversal and increased survival. Some caregivers expressed their hopes for a cure for ALS, as well as earlier diagnosis.
If the desired improvements they hoped for in a new treatment were achieved, caregivers said the result would be:
“Hope. We needed hope that there might be another outcome other than death. We never had that. You don't battle ALS. You live with ALS knowing that there is no hope of survival.”
“Increased possibility of having a “normal” relationship, rather than one focused on dealing with constant losses, and no hope. All we do as caregivers is prepare for the inevitable. That is very difficult.”
“There was no time to adjust and adapt, just jump in and sink or swim. Improved treatment would have allowed more time for mom to function independently with a lower level of support, allowing caregivers like myself to maintain our own lives simultaneously. You basically give up your own life to care for ALS patients.”
« Nous aurions plus de temps d'imparti pour effectuer des activités, des voyages et passé du temps précieux en famille. Maintenir l'autonomie de mon mari le plus longtemps possible et surtout de lui permettre de s'exprimer de façon normale. »
« Perte de l'autonomie de la personne et perte de jouissance de la vie car tout est concentré à aider et donner des soins, ça chambarde la vie du couple. Malgré que la progression de la maladie a été plus ou moins lente nous aurions certainement améliorer notre qualité de vie psychologique face à cette menace de perdre son conjoint. »
Experience With Drug Under Review – AMX0035
Of the 10 patients who have had the opportunity to try AMX0035 and the 10 caregivers whose loved ones had the opportunity to try AMX0035, most said they received it through a clinical trial, with a few having accessed it through compassionate use from the manufacturer or Health Canada’s Special Access Program.
Compared to other treatments they had taken, some patients on AMX0035 said they believed it was delaying their disease progression and most likely preserving speaking and breathing functions. While some patients and caregivers said it was too soon to see any impact from AMX0035, one patient said: “It helps me continue my everyday activities – I am still able to go to work and my doctors are amazed about my ability to do things.” One caregiver said AMX0035 slowed the progression of the disease, with the least amount of side effects. Other caregivers commented that AMX0035 has made them “feel at ease” that their loved one’s disease progression is slowing down, and that “it’s given hope to our family, and some hope is better than no hope.”
When asked about any negative impacts experienced on AMX0035 compared to other ALS treatments, a few patients and caregivers mentioned the “horrible taste.” While most patients didn’t experience any side effects while taking AMX0035, a few people (patients/caregivers) mentioned upset stomach/diarrhea, nausea, and loss of appetite which they managed by eating bland foods and drinking water. One caregiver said that the medication caused a “possible adverse reaction and progression actually sped up!?”
Most patients taking AMX0035 (80%) and caregivers (89%) of people on the treatment would recommend that it be made accessible to people living with ALS. One patient noted that, “even if it is not the cure, it can extend life, keep the quality of life longer, and in combination with other future therapies, make ALS livable or extend the life until there is a cure.” Another patient responded: “WHY NOT! No negative effects and trials prove (and my own experience) that it slows progression. The longer I can maintain a level of independence and efficiency, the better.” One caregiver of a patient on AMX0035 said: “I think whatever drug might work should be offered to ALS patients. They are fighting for their lives – every minute counts and a fast approval process might mean extra time with loved ones. I urge the government to fast track the approval process for AMX0035, a promising therapy. ALS patients deserve your help.”
In addition to the survey responses summarized above, two patients and one caregiver were interviewed regarding their experiences with AMX0035 and its impact on their lives. One was from Canada and two were from the U.S., and all three have been able to access AMX0035 through clinical trials.
The spouse of the caregiver interviewed was among the first patient to be dosed in an AMX0035 clinical trial (in 2017) and, as such, has considerable experience on the drug and has seen many benefits. In fact, during a two-month gap before this patient began the AMX0035 extension trial, their caregiver (spouse) noticed some signs of disease progression, which slowed down again once the extension trial began. Unlike with Radicava which they had to discontinue due to severe headaches, this patient hasn’t experienced any disadvantages or side effects from AMX0035. In fact, their caregiver believes that AMX0035 has allowed their family to have an extra year with her “in an able-bodied state.” They also credit AMX0035 for allowing their young children to “enjoy hearing their mom’s voice” for two years longer than might otherwise have been the case, before she lost the ability to speak nine months ago. This caregiver feels that AMX0035 delayed the progression of the disease and allowed his wife more time to do things herself. He also noted that their youngest son, who loves to sit on his mother’s lap, was able to do so for an additional two years due to AMX0035. He added that, “ALS doesn’t care if you’re rich or poor, and every family – the whole family – is going to be impacted the same.” Because of this, he feels that “it’s government’s responsibility to provide this treatment for everyone.”
One patient interviewed found out about the AMX0035 clinical trial through online research and has been taking the drug for more than three years. He believes he may have been part of the placebo group initially, because when he was switched to the open-label trial, he began to notice a positive difference in how he was feeling. Before AMX0035, he felt his right arm was getting weaker and he felt nerve twitching and now he says he can still use his right arm and hand. He believes that AMX0035 has “slowed the progression in my body” and even though AMX0035 didn’t reverse his disease, it has extended his quality of life to this day. He says that he feels more energetic and is still able to drive and walk. His wife helps him with things like doing up buttons, but otherwise is able to live independently. Even though he is also taking Radicava (he was on Rilutek but discontinued it due to side effects), he believes that AMX0035 is “definitely what’s saving me” and is the reason he can still play with his grandchildren and travel with this wife. He knows others who were diagnosed around the same time as he was and did not receive treatment with AMX0035, and many of them are confined to wheelchairs. This patient strongly believes that AMX0035 should be publicly funded: “If the government wants to spend less money on palliative care, this drug is a good way to do that and to give people a chance to live independently for longer. I would like to see everyone in the ALS community get it.”
The third patient interviewed was diagnosed with ALS in late December 2017 and was enrolled in a clinical trial for AMX0035 by the following March. The year of his ALS diagnosis, he recalled taking a trip to Europe with his family and having to be pushed around in a wheelchair. This patient, who is also taking Radicava and Rilutek, says that being on AMX0035 has meant “a complete turn-around of my ALS.” In fact, he has done so well that his physician has put him in his “ALS reversal group.” Prior to treatment with AMX0035 in March/April 2018, his wife had to cut up his food for him and help him button up his shirt and he also had to wear a leg brace. By August 2018, he felt so good that he built a 100- square foot deck for his home, including digging holes and mixing concrete and was back to work full-time by the end of that year. He owns a liquor store and prior to AMX0035 he wasn’t even able to hold a bottle of wine in his hand – now he’s stocking shelves! After his ALS diagnosis, he came home and got all his affairs in order, but now with AMX0035 he says, “I don’t have the outlook now that I’ll be dead in five years.” He hopes that AMX0335 “will continue to affect my ALS and make it so that I can continue to live a somewhat normal life without being in a wheelchair, or having people look after me. It’s not a drug I want to stop.”
Companion Diagnostic Test
N/A
Anything Else?
When asked if there was anything else they wanted to share about living with ALS or about their experience with AMX0025, this is what patients and caregivers said:
“Since ALS is currently without treatment to do anything except possibly slow progression by a number of months, any new therapies should be given an expedited review given the high unmet medical need.”
“I have given up many things and am losing more abilities each month. Anything that can slow down this progression is needed immediately! I am running out of time, and the future losses – paralysis, eating, talking, and breathing – are terrifying.”
“Remember, people are dying while you are doing this review. People with ALS have literally nothing to lose. Any advancements are crucial.”
“Living with ALS, time is not on our side. Any new treatments give patients and caregivers hope. Right now, there is not much hope, and it is much needed.”
“Approve this new drug and give all ALS patients access to it NOW!! This is a terminal disease. If the drug causes headaches, that’s manageable. We have to use every tool and drug to stop the progression of the disease, to stop the deaths and suffering.”
“Most ALS patients are looking for any glimmer of hope that a potential drug can provide. We don't expect or need a drug to solve all our ALS woes but to ‘buy more time’ for us. We live in constant hope and anticipation that one or more drugs will be available in our lifetime. Many patients before us have died because time ‘ran out’ for them. You must understand that delaying the availability of potential drugs can be a death sentence for us.”
« Il y a tellement d'espoir en ce médicament pour les familles dont un proche est atteint de la SLA il faut leur permettre d'y avoir accès le plus rapidement possible. »
« J’aimerais que toutes les personnes qui ont la SLA puissent avoir accès dès maintenant à AMX0035 car nous avons droit à un accès prioritaire. Avec une espérance de vie de 2 à 5 ans c’est maintenant qu’il faut agir et Santé Canada se doit de prioriser ce médicament. »
« Tout traitement qui peut ralentir la maladie est plus que la bienvenue dans un contexte où la maladie évoluée très rapidement. Ceci améliorera ma qualité de vie et celle de toute ma famille. »
Patient Group Conflict of Interest Declaration — The ALS Society of Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
ALS Canada completed the submission independently and with external support from a public affairs agency who was hired by ALS Canada on a fee-for-service basis.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
As noted above, a public affairs agency supported data collection and analysis along with internal resources.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
The companies listed below have sponsored ALS Canada signature events, the ALS Canada Research Forum scientific conference, and provided general donations.
Table 1: Conflict of Interest Declaration for ALS Society of Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AB Science Pharmaceutical | X | — | — | — |
Alexion Pharma Canada Corp. | — | — | X | — |
Amylyx Pharmaceuticals | — | — | X | — |
Apellis Pharmaceuticals | — | — | X | — |
Biogen Canada | — | — | X | — |
Cytokinetics Inc. | X | — | — | — |
DuPont Canada Inc. | X | — | — | — |
Hoffmann-La Roche Ltd. | X | — | — | — |
Impres Pharma Inc. | X | — | — | — |
Ingredion Canada Corporation | X | — | — | — |
Innomar Strategies | X | — | — | — |
Innovative Medicines Canada | X | — | — | — |
Ionis Pharmaceuticals Inc. | X | — | — | — |
Johnson & Johnson Family of Companies | X | — | — | — |
Mitsubishi Tanabe Pharma Canada | — | — | — | X |
Novartis Pharma Canada Inc. | — | X | — | — |
Pharma Consultants Inc. | X | — | — | — |
ALS Action Canada
About ALS Action Canada
ALS Action Canada is a patient led organization aimed at advocating for urgent access to promising therapies for Canadians living with Amyotrophic Lateral Sclerosis.
Information Gathering
CADTH is interested in hearing from a wide range of patients and caregivers in this patient input submission. Describe how you gathered the perspectives: for example, by interviews, focus groups, or survey; personal experience; or a combination of these. Where possible, include when the data were gathered; if data were gathered in Canada or elsewhere; demographics of the respondents; and how many patients, caregivers, and individuals with experience with the drug in review contributed insights. We will use this background to better understand the context of the perspectives shared.
Experiences and opinions were gathered through email and Zoom meetings with members of ALS Action Canada.
All members are Canadian Citizens living all across Canada. Specifically in the provinces of BC, Alberta, Manitoba, Ontario & Nova Scotia.
The only person in our group, as well as we believe in Canada, to have access to AMX0035 in the clinical trial is |||||||||||||||||||| participated in the trial in Sept 2018 out of Seattle, Washington and after 6 months he was moved to compassionate use and has continued on that ever since.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
ALS affects all aspects of a patients life as well as the lives of the patients caregivers and family. The disease is progressive meaning that new challenges occur daily that the patient has to adapt to in order to continue their independence. ALS also requires several hours a day for treatments, exercising for legs/arms, breathing exercises, constant regiment of drugs & supplements and appointments.
As the disease progresses motor function, speech and swallowing is most impacted. This makes every daily task increasingly difficult and eventually impossible. Caregivers take on a much greater role in the household as the disease progresses. Everything from getting dressed in the morning to eating meals has to be consistently adapted.
ALS profoundly impacts patients families as they live in crisis mode watching their loved one weaken losing simple independence and risking injury daily. The stress level on a family is extremely high.
When full time care is needed for the patient, finding qualified and available regular caregiving is a challenge that every family faces. Modifications to bedroom and bathroom are very expensive and require using contractors that are not knowledgeable in ALS accessibility requirements.
The main objective of ALS patients when starting a new drug is to slow or stop the progression of the disease. Managing symptoms is necessary but the goal for patients remains to stop the progression of ALS.
Experiences With Currently Available Treatments
CADTH examines the clinical benefit and cost-effectiveness of new drugs compared with currently available treatments. We can use this information to evaluate how well the drug under review might address gaps if current therapies fall short for patients and caregivers.
Describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
Rilizule
Has a small effect on prolonging survival. Clinical trial data shows that it may add 2 - 3 months to the life of an ALS patient.
The recommended dosage for Rilizule is 50 mg taken orally twice daily. Rilizule needs to be taken at least 1 hour before or 2 hours after a meal making it restrictive to a patients life.
These prescription directions are challenging to follow as ALS patients need to make sure to consume a higher than average amount of calories daily in order to keep on weight. As the disease progresses eating becomes challenging and rapid weight loss occurs as the patients muscles weaken and appetite decreases.
It is in pill form and as patients begin to have difficulty swallowing taking pills can become very challenging.
Many patients have side effects from Rilizule including nausea, frequent and/or painful urination, trouble sleeping, headache, increased cough.
Patients often report that Rilizule makes them very drowsy. Doctors advice to solve this side effect is to cut back to 1 pill a day when this occurs.
Liver enzymes can be affected therefore regular blood testing is required (Every 3 months) to continue using the medication.
Edaravone
Edaravone is administered by IV with dosage being one hour infusions every day for 10 days in a row with a 2 week break in between infusion cycles.
Patients have to travel to a hospital each day that they are getting the infusions to have it administered. This is very time consuming and challenging as the patient disease progresses limiting mobility.
Some patients have chosen to have a port access implanted so their Caregivers are able to administer the infusions at home. This option eliminates the daily trips to the hospital although the patient will still need to make two trips to the hospital every cycle to get access to the port to allow administration of the drug at home. As well the patient and caregiver have to keep a personal stock of IV supplies which for many patients are paid out of pocket.
Although a port limits the trips needed to the hospital it's not without risk. The port access of several patients in our group has become infected requiring antibiotics or removal of the port.
As the patients disease progresses they become at risk to not meeting the criteria for getting access to Edaravone. The criteria could be when you no longer can cut your food or walk independently up a flight of stairs. When the patient no longer meets this criteria they will get refused access to the treatment. This is a constant worry for ALS patients as Edaravone is currently just 1 of 2 treatments approved to slow disease progression.
Patients report that a difference while using Edaravone is hard to notice as it only slows progression marginally.
*There is a clear sign that neither of these drug options are adequate when ALS Clinics across Canada discourage newly diagnosed patients from using Rilizule and/or Edaravone as they don't have a significant effect and are very burdensome on the patient and caregiver. More and better treatment options are desperately needed for ALS patients.
Improved Outcomes
CADTH is interested in patients’ views on what outcomes we should consider when evaluating new therapies. What improvements would patients and caregivers like to see in a new treatment that is not achieved in currently available treatments? How might daily life and quality of life for patients, caregivers, and families be different if the new treatment provided those desired improvements?
What trade-offs do patients, families, and caregivers consider when choosing therapy?
A treatment option that is easily administered and does not have restrictive directions to take would be very beneficial to the patient and caregivers. Treatments that are quick to administer free up much needed time daily allowing the patient and caregiver to focus on other critical areas of care. As well not needing equipment to administer will save the patient funds and time travelling to and from the hospital for appointments. These are huge benefits to an ALS patient and their care givers.
ALS patients want to see a new treatment that significantly slows progression of the disease. Slowing progression of ALS can give the patient and their families time to adapt and adjust to each new loss of independence as well gives the patient time to wait for a future cure.
ALS takes over every aspect of the patient and their families lives. The uncertainty of not knowing what each new day will bring, could you fall, lose more function, choke on something you're eating. The worries are endless for the patient and their families. A treatment that can slow the progression of ALS in a significant way can ease some of worries for them.
Experience With Drug Under Review
CADTH will carefully review the relevant scientific literature and clinical studies. We would like to hear from patients about their individual experiences with the new drug. This can help reviewers better understand how the drug under review meets the needs and preferences of patients, caregivers, and families.
How did patients have access to the drug under review (for example, clinical trials, private insurance)? Compared to any previous therapies patients have used, what were the benefits experienced? What were the disadvantages? How did the benefits and disadvantages impact the lives of patients, caregivers, and families? Consider side effects and if they were tolerated or how they were managed. Was the drug easier to use than previous therapies? If so, how? Are there subgroups of patients within this disease state for whom this drug is particularly helpful? In what ways? If applicable, please provide the sequencing of therapies that patients would have used prior to and after in relation to the new drug under review. Please also include a summary statement of the key values that are important to patients and caregivers with respect to the drug under review.
AMX0035 is a repurposed combination of two currently approved drugs. It comes in a powder form that is taken twice daily dissolved in 8 ounces of warm water. Once dissolved this treatment is taken orally and because it is in liquid form this treatment option is very desirable for patients with swallowing issues or feeding tubes. The packages are stored at room temperature, making it easy to transport if needed. The treatment requires no equipment to be administered as well does not need to be taken with food or on an empty stomach. Although there is a bitter taste to the patients palate this becomes very tolerable once taken for an extended period of time.
|||||||||| participated in the clinical trial and experienced no side effects during the trial or on the continued compassionate use. He has been on AMX0035 for just over three years now. He started the trial one year after onset of symptoms and one month after being diagnosed with ALS. Since starting the compassionate use after the placebo controlled trial he has seen a remarkable slowing in the progression of his symptoms. ||||||’s weakness first started in his right hand and today, over 4 years from symptom onset, he is still able to eat with his right hand using an adaptive utensil and continues to get around independently because of AMX0035. His progression is slower than any other patient in the ALS community that we've come across with a similar diagnosis timeline which is evidence that AMX0035 has a positive impact on slowing progression.
The only other treatments to slow disease progression are Rilizule which offers very minimal effect on the progression of the disease, it's stated on average a slowing of progression of just 10%, and Edaravone which slows progression on average up to 30%. Rilizule is in pill form making it difficult for patients with swallowing issues as well is it restrictive as it needs to be taken at least 1 hour before or 2 hours after a meal twice a day.
Edaravone is much more invasive as it is administered by IV with dosage being one hour infusions every day for 10 days in a row with a 2 week break in between infusion cycles. Some patients goto routine appointments at the hospital for these infusions while others have to hire a private nurse at home or if a Port access is placed in the patient they may have their caregiver administer in the home, in most cases at their own expense for supplies and equipment needed.
Top line data from the trial showed a 44% average slowing of progression for patients on AMX0035 vs the placebo. This is significantly better than the current two approved treatment options offered to patients.
AMX0035 is by far the most effective, easily administered and accessible treatments available for ALS patients today.
Companion Diagnostic Test
There is no required companion diagnostic test to use AMX0035.
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
The criteria for access to AMX0035 needs to be much more broad. Only a small amount of patients in Canada would qualify to access it under the current SAP coverage guidelines. Access should be open to every patient that wants to try given what little options are available to the ALS community today and the aggressive nature of the disease.
For Canadians with ALS there is no risk-benefit assessment to be done because dying from ALS is a 100% certainty. We want CADTH to understand that the term “ALS” applies to a heterogenous subset of specific other motor neurone disease, some of which are genetically triggered, some which aren’t. It’s critical for CADTH to know that ALS is a “messy” disease because its causes are not yet clearly understood and different types of treatments are shown to work for different types of ALS. Science is working hard at untangling the “mess” that is ALS.
Patient Group Conflict of Interest Declaration — ALS Action Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Yes, we received clarity on the Diagnostic testing needed from |||||||||| at Amylyx.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No, all data was collected and analyzed entirely by ALS Action Canada membership.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Conflict of Interest Declaration for ALS Action Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Clinician Input
The Canadian ALS Research Network
About The Canadian ALS Research Network
The Canadian ALS Research Network (CALS) is a national network of clinicians at academic health care centres across Canada that specialize in ALS research and clinical care. Our mission is to connect Canadian ALS clinical research centres and improve both patient and clinic participation in clinical research. The network was established in 2008 and since then has built a strong reputation within Canada and internationally for rapid and effective study recruitment and completion.
Information Gathering
All members of CALS were invited to participate in Zoom meeting to discuss key questions related to this submission. This Zoom meeting took place on Monday, November 8, 2021 and included 10 CALS members from across Canada. Each question was discussed until consensus was reached regarding wording of responses to template questions. A freelance medical writer (||||||||||||||||||||||||||||||||||||||||) was engaged to work with |||||||||||||||||||||||||||||| to prepare a draft for circulation to all members of CALS. Where appropriate, revisions based on feedback were incorporated into the final draft. In addition, the following key references were used for background information and to describe the current standard of care for ALS:
Shoesmith C, Abrahao A, Benstead T, Chum M, Dupre N, Izenberg A, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ. 2020;192(46):E1453–68.
Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. New Engl J Med. 2017;377:162–72.
Product monograph for (RELYVRIOTM) Amylyx Pharmaceuticals Inc., 43 Thorndike Street, Cambridge, MA 02141
Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131:1975–8.
Hannaford A, Pavey N, van den Bos M, Geevasinga N, Menon P, Shefner JM, et al. Diagnostic utility of Gol d Coast Criteria in amyotrophic lateral sclerosis. Ann Neurol. 2021;89:979–86.
Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of sodium phenylbutyrate- taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383:919–30.
Paganoni S, Hendrix S, Dickson SP, Knowlton N, Macklin EA, Berry JD, et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021;63:31–9.
Current Treatments
Describe the current treatment paradigm for the disease.
Amyotrophic lateral sclerosis (ALS) is a debilitating, progressive disease characterized by the degeneration of motor neurons in the brain and spinal cord (1). Over the course of the disease, patients develop increasingly severe weakness in limb, bulbar, and respiratory muscles. This often leads to requirements for wheelchair use, gastrostomy feeding tube, noninvasive ventilatory support, and loss of autonomy. Death due to respiratory failure typically occurs within 5 years of diagnosis. For the estimated 3000 Canadians with ALS, treatment options for their disease remain extremely limited.
The only disease-modifying treatments for ALS are riluzole and edaravone. Neither offers substantial clinical benefit. Riluzole, which acts by suppressing excessive motor neuron firing through inhibition of glutamate, prolongs survival by a median duration of 3 months. Intravenous edaravone, which reduces oxidative stress, has been shown to slow the rate of clinical decline by 33% in a select group patients with preserved respiratory function and disease duration < 2 years. Given the limited benefit of these treatments, there is a desperate need for new therapeutic options with the aim of slowing or reversing neurologic decline.
Symptom management and quality-of-life optimization are priorities in patient care (as emphasized in the Canadian best practice recommendations [1]). This submission will not address these supportive therapies. Instead, we focus on disease-modifying treatments, aimed at slowing disease progression.
Treatment Goals
What are the most important goals that an ideal treatment would address?
An ideal treatment would delay or prevent disease progression (i.e., motor neuron degeneration), slow decline in lung capacity, reduce severity of symptoms, minimize adverse events, reduce loss of cognition, improve health-related quality of life, and ultimately increase patients’ ability to continue to work, reduce burden on caregivers, and prolong life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Currently, the mainstay of care for patients diagnosed with ALS consists of interventions and supports to manage symptoms (e.g., nasogastric feeding, prevention of aspiration, and ventilatory support). There are no available treatments to reverse the disease or to arrest the progression of neurological decline.
Currently approved treatments have only shown very modest benefits in slowing disease progression. Riluzole confers a survival benefit of roughly 3 months, and edaravone slows disease progression by roughly 33%. It is clear to clinicians who treat ALS patients that development of new neuroprotective treatments is required in order to provide a multimodal attack to address the various mechanisms of action of the disease.
Which patients have the greatest unmet need for an intervention such as the drug under review?
All patients diagnosed with ALS have an unmet need for a therapy that slows the progression of disease.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The cause of ALS remains unknown and multiple pathogenic mechanisms are thought to be involved. Importantly, these processes result in substantial neuronal death before the manifestation of clinical symptoms. Given the progressive nature of the disease and the low probability of survival beyond 5 years from diagnosis, targeting all potential pathological pathways early is appropriate. Riluzole acts by suppressing excessive motor neuron firing, while edaravone suppresses oxidative stress. RELYVRIO (sodium phenylbutyrate and ursodoxicoltaurine) exerts its therapeutic effects by reducing neuronal death, hypothesized to occur through simultaneous inhibition of endoplasmic reticulum (ER) and mitochondrial stress (3). RELYVRIO thus offers treatment via a novel pathway and could be initiated at any time.
Initiation earlier in the disease course is preferred, as this would theoretically pre-date the widespread loss of motor neurons (and therefore preserve function). Administration of the other approved ALS therapies (riluzole, edaravone) should not preclude the introduction of RELYVRIO and there is sound clinical rationale to introduce all three therapies concurrently in patients without contraindication.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
There is no rationale for first-, second- or third-line treatments in ALS, because there is no chance that current monotherapies will be successful; nor are there any reliable biomarkers to judge such success. Patients should not be required to demonstrate “failure” or decline on one agent before introduction of a subsequent agent. The concept of “failure” is not applicable in ALS treatments, as ultimately all agents fail – ALS is, at this time, always a terminal disease. Moreover, currently available agents are not sufficiently potent to justify waiting for “failure”, as there is always worsening/progression, even in patients who receive both available therapies. Note, however, that good clinical practice would dictate allowing sufficient time after initiation of one agent until addition of another agent, in order to monitor for tolerance and side effects.
How would this drug affect the sequencing of therapies for the target condition?
Refer to the previous response.
Which patients would be best suited for treatment with the drug under review?
Anyone with a diagnosis of ALS according to Gold Coast criteria (4), made by a neurologist, physiatrist, or neuromuscular specialist could be considered for treatment with this drug. Use of the use Gold Coast criteria has been shown to increase sensitivity for the diagnosis of ALS while maintaining high specificity when compared with the El Escorial criteria (5).
How would patients best suited for treatment with the drug under review be identified?
ALS is challenging to diagnose because initial symptoms can be similar to other diseases. No specific biomarker exists. A neurologist makes a clinical diagnosis of ALS based on history, physical examination, electromyography, and exclusion of alternative diagnoses. The typical delay from the first symptom of ALS to diagnosis is approximately 6 to 12 months (2), during which time progression occurs. Early intervention to prevent motor neuron death and slow progression is a key treatment objective in ALS. Therefore, patients should be considered for the drug under review immediately after diagnosis.
Which patients would be least suitable for treatment with the drug under review?
The mechanism of action of this agent is hypothesized to reduce neuronal death. As such, there must be neurons available to protect. Therefore, in theory, patients with advanced ALS/severe loss of motor neurons would not benefit from the drug under review. However, our expert recommendation is that this be based on the judgement of the clinician, rather than on arbitrary measures such as functional rating scores or pulmonary function test (PFT) results. Some patients may be profoundly affected in one body region, but have well-preserved function in another body region (e.g., a patient who is wheelchair-bound due to severe leg weakness, but exhibits minimal upper limb and/or bulbar weakness). In these patients, slowing of disease progression in the more well-preserved body region is certainly desirable.
Trial evidence (6,7) used strict eligibility criteria to ensure that the intervention with the drug would demonstrate an effect. However, there is no evidence to show that it would NOT demonstrate an effect in other patients. In Canada, access to specialist ALS clinics and pulmonary function testing is uneven depending on geography, and could pose obstacles to treatment if, for example, strict PFT criteria were to be applied.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
“Treatment response” for ALS cannot be strictly defined, as the goals of treatment are to slow the degeneration of motor neurons. Progression is individual and monitoring for outcomes (slowing of disease progression) is not feasible on an individual patient basis due to disease heterogeneity (hence, the dependence on clinical trial results). As such, a treatment strategy would more likely consist of starting the drug and following the patient at regular intervals until the point where the patient’s goals of care change (to a less interventional/more palliative approach), or the patient wishes to discontinue the drug.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Refer to the previous response.
What would be considered a clinically meaningful response to treatment?
Refer to the previous response.
How often should treatment response be assessed?
Although it is not possible to define “treatment response” in the case of this specific drug (due to the relentlessly progressive nature of ALS), patients’ clinical status is routinely monitored every 3 to 4 months, as per the Canadian best practice guidelines (1). At these visits, patients’ tolerance for the drug under review, as well as goals of care, should be explored.
What factors should be considered when deciding to discontinue treatment?
It is important for patients to have regularly scheduled visits with their ALS team to review benefits and goals of treatment. It is reasonable to expect that goals of treatment will change as the disease progresses (once the patient becomes severely disabled), however, there is no evidence to suggest that the drug will NOT provide benefit beyond a specific endpoint. Therefore, the drug could be continued until the focus of care becomes more palliative. When the focus of treatment becomes supportive/palliative and the patient requires total care and near-continuous ventilation, this would suggest that there are few surviving motor neurons. At this point, it would be logical to stop the drug, as it would not provide further benefit.
What settings are appropriate for treatment with the drug under review?
The drug under review should be prescribed by a neurologist or physiatrist with experience in the care of patients with ALS.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Patients with ALS should be regularly followed by a multidisciplinary ALS clinic that delivers team-based care (neurology, physiatry, respirology, and allied health professionals) and addresses issues including communication, nutrition, swallowing, mobility, activities of daily living, respiratory care, cognition, psychosocial issues, medical management and end-of-life care (1). As noted in response 6.12, patients receiving the drug under review should, at minimum, be followed by a neurologist or physiatrist experienced in the care of ALS patients.
Additional Information
Is there any additional information you feel is pertinent to this review?
The authors of this submission acknowledge the pressures on our publicly funded healthcare system, and the need to rationalize resources. However, we are confident that physicians treating patients with ALS, and indeed the patients themselves, can and will make informed judgements to ensure that the drug under review is used in those who can benefit the most. We therefore believe that RELYVRIO should be recommended for public reimbursement in Canada.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
A medical writer (|||||||||||||||||||||||||| from Halifax, Nova Scotia) assisted with the preparation of the first draft of this submission and the incorporation of revisions from the reviewing physicians.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Ari Breiner MD MSc FRCPC
Position: Neurologist (The Ottawa Hospital), Assistant Professor (University of Ottawa)
Date: 18-11-2021
Table 3: Conflict of Interest Declaration for CALS — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Angela Genge
Position: Neurologist, McGill University
Date: 06-12-2021
Table 4: Conflict of Interest Declaration for CALS — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Amylyx | X | — | — | — |
Declaration for Clinician 3
Name: Sylvie Gosselin
Position: MD, Neurologist (neuromuscular diseases), associate professor, faculty of Medecine, Sherbrooke University, Qc
Date: 06-12-2021
Table 5: Conflict of Interest Declaration for CALS — Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Ian Grant, MD FRCPC
Position: Neurologist, QEII Health Sciences Centre, Halifax NS
Date: 07-12-2021
Table 6: Conflict of Interest Declaration for CALS — Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Sandrine Larue, MD FRCPC
Position: Neurologist (Charles-Lemoyne Hospital), Professeur Adjoint (Université de Sherbrooke)
Date: 07-12-2021
Table 7: Conflict of Interest Declaration for CALS — Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Mitsubishi: oral presentation to a meeting | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Ahmad (Amer) Alavian Ghavanini
Position: Neurologist and Division Head of Neurology, Trillium Health Partners, Assistant Professor (University of Toronto)
Date: 08-12-2021
Table 8: Conflict of Interest Declaration for CALS — Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr Nicolas Dupré, MD MSc (bis) FRCP FAAN
Position: Neurologist (CHU de Québec), Full Professor (Université Laval)
Date: 09-12-2021
Table 9: Conflict of Interest Declaration for CALS — Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 8
Name: Dr. Theodore Mobach
Position: Calgary ALS Clinic Director
Date: 13-12-2021
Table 10: Conflict of Interest Declaration for CALS — Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 9
Name: Dr. Collin Luk MD, PhD, FRCPC
Position: ALS Clinical Research Fellow (University of Alberta)
Date: 13-12-2021
Table 11: Conflict of Interest Declaration for CALS — Clinician 9
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 10
Name: Dr. Agessandro Abrahao Junior
Position: Staff ALS Neurologist (Sunnybrook Health Sciences Centre), Assistant Professor (University of Toronto)
Date: 13-12-2021
Table 12: Conflict of Interest Declaration for CALS — Clinician 10
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 11
Name: Colleen O’Connell
Position: Physiatrist, Medical Director, Stan Cassidy Centre for Rehabilitation
Date: 14-12-2021
Table 13: Conflict of Interest Declaration for CALS — Clinician 11
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
MT Pharma | X | — | — | — |
Biogen, Calico, Annexon, Alexion, Cytokinetics | X | — | — | — |
Amylyx | X | — | — | — |
Declaration for Clinician 12
Name: Dr. Gordon Jewett, MD FRCPC
Position: Neurologist, neuromuscular fellow (University of Calgary
Date: 13-12-2021
Table 14: Conflict of Interest Declaration for CALS — Clinician 12
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Amylyx (advisory board participation) | X | — | — | — |
Declaration for Clinician 13
Name: Christen Shoesmith
Position: Neurologist and MND Clinic Director (London Health Sciences Centre) and Associate Professor in Clinical Neurological Sciences (Western University)
Date: 10-12-2021
Table 15: Conflict of Interest Declaration for CALS — Clinician 13
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Mitsubishi Tanabe Pharma Canada | X | — | — | — |
Biogen | X | — | — | — |
Amylyx | X | — | — | — |
Declaration for Clinician 14
Name: Dr Genevieve Matte, MDCM, FRCPC
Position: Neurologist (Centre Hospitalier de l’Université de Montréal), Assistant Professor (Université de Montréal)
Date: 10-12-2021
Table 16: Conflict of Interest Declaration for CALS — Clinician 14
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Amylyx Pharmaceuticals | X | — | — | — |
Mitsubishi-Tanabe Pharma Canada | — | — | X | — |
Declaration for Clinician 15
Name: Dr. Lorne Zinman, MD, M.Sc., FRCPC
Position: Neurologist (Sunnybrook Health Sciences Centre), Associate Professor (University of Toronto), Associate Scientist (Sunnybrook Research Institute)
Date: 14-12-2021
Table 17: Conflict of Interest Declaration for CALS — Clinician 15
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Amylyx | — | X | — | — |
Biogen | — | X | — | — |
Mitsubishi Tanabe Pharma Canada | — | X | — | — |
Cytokinetics | — | X | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.