CADTH Reimbursement Review
Ravulizumab (Ultomiris)
Sponsor: Alexion Pharma GmBH
Therapeutic area: Atypical hemolytic uremic syndrome
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ADAMTS13
a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13
aHUS
atypical hemolytic uremic syndrome
C3
complement component 3
C5
complement component 5
C5b-9
complement component 5b-9
CFH
complement factor H
CI
confidence interval
CKD
chronic kidney disease
CSR
Clinical Study Report
eGFR
estimated glomerular filtration rate
ESKD
end-stage kidney disease
FAS
full analysis set
FACIT-F
Functional Assessment of Chronic Illness Therapy – Fatigue
HRQoL
health-related quality of life
HUS
hemolytic uremic syndrome
ITC
indirect treatment comparison
LDH
lactodehydrogenase
MAC
membrane attack complex
MID
minimally important difference
MMRM
mixed model for repeated measures
PP
per protocol
RBC
red blood cell
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TMA
thrombotic microangiopathy
TTP
thrombotic thrombocytopenic purpura
ULN
upper limit of normal
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Ravulizumab (Ultomiris) 10 mg/mL solution for IV infusion |
Indication | For the treatment of adults and pediatric patients with aHUS to inhibit complement-mediated TMA |
Reimbursement request | Per indication |
Health Canada approval status | Pre-NOC |
Health Canada review pathway | Not specified (standard) |
NOC date | November 1, 2022 |
Sponsor | Alexion Pharma GmBH |
aHUS = atypical hemolytic uremic syndrome; NOC = Notice of Compliance; TMA = thrombotic microangiopathy.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a life-threatening, ultra-rare disease in which patients are susceptible to sudden and progressive episodes of complement-mediated thrombotic microangiopathy (TMA) that most commonly damage the kidneys and include extrarenal, multiorgan involvement.1,2 Patients typically present with signs and symptoms of the triad of thrombocytopenia, hemolysis, and acute kidney injury.3 The disease is primarily caused by an inherited or acquired dysregulation of complement-regulatory proteins, resulting in uncontrolled complement activation.2-4 Over the past few years, there has been an increasing consensus that, in the majority of patients, aHUS may involve both genetic predisposition (e.g., pathogenic variants, autoantibodies, or at-risk polymorphisms in complement genes) and a triggering condition in order for the clinical event of a TMA to occur.5-7 aHUS biomarkers include complement component 3 (C3), complement component 5a (C5a), complement component 5b-9 (C5b-9), factor B, complement factors B, H, and I, CH50, AH50, d-dimer, and anticomplement factor H (CFH) antibodies.8,9 Low levels of C3, CH50, AH50, and CFB—along with increased levels of C5a, C5b-9, Bb, anti-CFH autoantibodies, and d-dimer—are usually found in patients with aHUS.8 aHUS can occur at any age, but onset during childhood is more common than in adulthood (60% versus 40%, respectively). Diagnosis is currently based on excluding other causes of TMA.10,11 Therefore, the risk of misdiagnosis of aHUS may exist in clinical practice.11,12 Although a positive genetic test can help to confirm a previously clinically diagnosed case of aHUS, it is not required to make the diagnosis of aHUS or to commence treatment. A clinical differential diagnosis remains the primary method of establishing a diagnosis of aHUS.6,11 According to the clinical experts consulted by CADTH for this review, 30% to 40% of patients with aHUS may have no known genetic predisposition. According to the clinical experts, patients with aHUS who have DGKE mutations are unlikely to benefit from treatment with C5 inhibitors (e.g., eculizumab and ravulizumab).
The incidence and prevalence of aHUS vary widely.3,13 A 2020 systematic literature review of the global epidemiology of aHUS reported that, for all ages, the annual incidence ranged from 0.23 per million population to 1.9 per million population.13 It was also reported that, for all age groups, the annual incidence was 4.9 per million population.13 There is limited published prevalence data for aHUS specific to Canada and the US.13 A Canadian study published in 2004 reported an incidence of aHUS in children of 2 cases per million over a 4-year period.14 Most recently, a 2020 analysis of 37 patients in Canada (15 pediatric and 22 adult) enrolled in the aHUS Global Registry (an observational, noninterventional, multicentre study that prospectively and retrospectively collects data on patients who have a clinical diagnosis of aHUS, irrespective of treatment)15 estimated that there are potentially 74 patients with aHUS in Canada.16
Prior to the approval of ravulizumab, eculizumab, a terminal complement inhibitor, was considered the standard of care for the treatment of patients with aHUS in most jurisdictions for over a decade.17 Eculizumab is the only Health Canada–approved drug indicated for the treatment of aHUS.18 However, it is not reimbursed in all Canadian jurisdictions (refer to Appendix 1). Furthermore, eculizumab imposes a substantial treatment burden on patients due to its shorter half-life and requirement for biweekly doses.18 The frequent dosing schedule of eculizumab is burdensome for patients, potentially affecting their health-related quality of life (HRQoL). It is also health care resource–intensive, which also drives infusion-related costs with eculizumab.19 The clinical experts consulted by CADTH for this review indicated that there is an unmet need for alternative effective therapies with acceptable toxicity profiles that can help patients achieve TMA remission and improve HRQoL. The appropriate duration of treatment with anticomplement therapy is unknown.
Ravulizumab (10 mg/mL concentrate for solution for infusion) is a terminal complement inhibitor that specifically binds to C5, inhibiting its cleavage to C5a and C5b, preventing the generation of the terminal complement complex membrane attack complex (MAC) or C5b9. Health Canada has previously issued market authorization for ravulizumab for paroxysmal nocturnal hemoglobinuria.18,20 The Health Canada–proposed indication of interest for this review is for the treatment of adult patients with aHUS. The Health Canada–recommended dosing regimen consists of a single loading dose followed 2 weeks later by the first maintenance dose, with subsequent maintenance doses administered every 8 weeks for patients weighting greater than or equal to 20 kg or every 4 weeks for those weighing less than 20kg (Table 3). The sponsor’s reimbursement request is identical to the Health Canada–approved indication.
The objective of this clinical report is to review the beneficial and harmful effects of ravulizumab for the treatment of adult and pediatric patients with aHUS by inhibiting complement-mediated TMA.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group, aHUS Canada, provided input for the treatment of aHUS. This group gathered information from 19 caregivers and 41 patients from inside and outside Canada through an online survey conducted in June 2022. Of these 60 respondents, 19 had experience with the drug under review.
Respondents identified anemia, low platelet count, and acute renal failure as the most difficult primary symptoms to control. Lack of quality of life, helplessness, post-traumatic stress disorder, fatigue, headache, high blood pressure, inability to travel, frequent hospital visits, kidney issues and dialysis are some of the experiences that respondents have while living with aHUS. According to aHUS Canada, aHUS patients with dialysis who need a kidney transplant are not eligible for transplant in Canada unless they receive eculizumab infusions during the transplant. Caregivers to patients with aHUS also face emotional and financial challenges because the process to access eculizumab or alternatives differs from province to province. Respondents described financial struggles, anxiety about access to treatment, the need to protect organs, exhaustion, memory loss, and brain fog as aspects of the disease that are among the hardest to control.
Respondents identified plasma therapy (fresh frozen plasma or plasmapheresis), eculizumab infusions, and long-term dialysis as the currently available treatments for patients with aHUS. Reported side effects included nausea, headache, fatigue, anaphylactic reaction to plasma, vein collapse, infection, anxiety, kidney failure, uncontrolled blood pressure, migraines, exhaustion, memory loss, brain fog, central line issues, muscle crumps, insomnia, abdominal pain, fever and chills, weight gain or loss, and being refractory to plasma therapy, among others.
When discussing their expectations for new drugs, patients reported that access to treatment and freedom of choice were critical components in managing the disease. However, quality of life was the most commonly cited desired outcome, and it was affected by factors like choice in care, frequency of appointments, and drug affordability. The abilities to travel, focus on family, and have more time between appointments were also described as critical to patients’ mental health. Moreover, frequent blood tests and IV therapies or ports were reported to be significant problems for many patients. While 1 caregiver pointed out the importance of maintaining “venous access for continuous access to eculizumab,” other patients shared their ineligibility for ports due to damaged veins from the disease. Patient also expressed the importance of less frequent treatments.
When discussing their experiences with ravulizumab, patients listed benefits that included more energy, less vein damage, fewer treatments, fewer symptom fluctuations, greater freedom of choice, and less anxiety. However, they also reported experiencing headache, nausea, and body aches right after their infusion or during the month after the infusion.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Unmet Needs
The clinical experts consulted by CADTH for this review indicated that administering eculizumab every 2 weeks interferes with a patient’s quality of life by consuming time that could be spent working, travelling, or with friends and family. It can also be an issue when it comes to venous access fatigue and comes with the societal cost of nursing and allied health care support. The biggest limitation to the current treatment is prohibitive cost: most centres will fund an initial treatment or a few treatments, but very few centres have the resources to fund lifelong treatment. Inclusion in provincial formularies is inconsistent across provinces, and private insurance coverage is not common. Patients or their health teams advocate for subsidies or payment in full, but are not always successful. With respect to venous access fatigue, most patients would be candidates for portacaths or central lines, which are normally offered to chemotherapy patients.
The clinical experts indicated that the mechanism of action of ravulizumab is the same as that of eculizumab. Ravulizumab would not be added to other treatments: it would replace eculizumab as the treatment of choice for aHUS. The clinical experts indicated that they believe that ravulizumab would have likely similar or equivalent efficacy to eculizumab, with the potential for a better therapeutic profile and reduced therapeutic burden. These experts believed that ravulizumab would become the first-line treatment of choice because it offers improved patient quality of life and cost-effectiveness compared to eculizumab. The clinical experts mentioned that, theoretically—as CADTH has found with other biologics that use the same target molecule—tachyphylaxis to 1 medication may open up options to treat with the second, so acquired nonresponse may be a consideration to switch therapies. Improvements in patient HRQoL are expected to be significant after switching from eculizumab to ravulizumab
The clinical experts indicated that the patients most suitable for treatment with ravulizumab are those who have been diagnosed with aHUS. The patients least suitable are those with TMA that is clearly due to a secondary cause, such as malignant hypertension, malignancy, or infection. There may be some benefit in using eculizumab in some patients with autoimmune disease with histological evidence of TMA and evidence of complement dysregulation (e.g., some variants of lupus). According to the clinical experts, the patients with aHUS who are most in need of intervention are those with severe TMA with associated end organ damage, such as acute kidney injury or brain ischemia. The clinical experts indicated that patients who qualify for treatment would be identified by physicians with expertise in TMAs, such as nephrologists, hematologists, and internal medicine physicians. These physicians would make the diagnosis based on clinical examination, lab investigations, and genetic testing for complement dysregulation, and by excluding other causes of TMA.
To diagnose aHUS, there needs to be evidence of TMA, such as schistocytes, elevated lactate dehydrogenase, decreased haptoglobin, decreased hemoglobin, and thrombocytopenia. These lab abnormalities should also coincide with 1 or more of the following: neurological symptoms, acute renal failure, or gastrointestinal symptoms, although any organ system can be involved (e.g., pancreas, heart). Diagnosing aHUS can be very challenging because there is no single diagnostic test that can confirm it. In many situations, it is a diagnosis of exclusion. For this reason, misdiagnosis of this condition is a risk. One clinical expert indicated that testing has improved while the difficulties of diagnosis have decreased, suggesting that these diagnostic challenges may have been a greater issue 10 years or 15 years ago, when genetic and biochemical assessments of complement pathways were less accessible; however, these tests are now more available, often on a quick turnaround, even when sent out of province. One clinical expert indicated that haptoglobin is not the most reliable diagnostic indicator; lactodehydrogenase (LDH) level is more reliable.
The clinical experts indicated that etiologies that mimic TMA need be excluded, including infections, medications, malignancy, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, malignant hypertension, disseminated intravascular coagulation, preeclampsia, and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome. Thrombotic thrombocytopenia purpura (TTP) can be distinguished from aHUS by measuring ADAMTS13 level. If ADAMTS13 is higher than 5% and the patient is resistant to plasma exchange, then the diagnosis is more likely to be aHUS than TTP. Screening for complement mutations and antibodies should be performed. More sophisticated testing is available as well, including soluble C5b-9 levels: these levels are elevated during aHUS and reduced with treatment because C5b-9 is generated as a product of complement activation. If it is initially low, most centres will monitor C3 and complement component 4 levels for recovery.
The clinical experts indicated that early initiation of plasmapheresis until the diagnosis is confirmed is critical, given the high mortality risk of untreated TTP. One clinical expert indicated that most centres have access to ADAMTS13 activity testing with a turnaround time of 24 hours to 48 hours. The approach to treatment in adults, particularly older adults, may include plasmapheresis before the result is known. One clinical expert specializing in pediatric nephrology indicated that, if feasible, it is best to wait for the results for pediatric patients, because plasmapheresis is not recommended in this population; however, local resources also dictate its use and whether centres can procure C5 inhibitors quickly. In pediatrics, where TTP is less common, clinicians would likely not initiate plasmapheresis first, but agree it would be prudent to do so for adult patients, particularly older adults. The clinical experts emphasized that once aHUS has been diagnosed, C5 inhibition may be used as first-line therapy.
The clinical experts indicated that the treatment goals for aHUS are resolution of the TMA with normal platelet and LDH counts as well as resolution of acute kidney injury, neurological sequelae and stabilization of end organ damage. The required duration of treatment with C5 inhibition is unknown. Based on available data, if there are no high-risk complement genetic variants, then termination of treatment could be considered after 6 months to 12 months. However, according to the clinical experts, it is possible to discontinue treatment with ravulizumab in patients with aHUS without a genetic mutation in complement 3 months to 6 months after remission is achieved. Lifelong treatment may be considered for patients with high-risk complement genetic variations. The clinical experts mentioned that 30% to 40% of patients may have no known genetic disposition. As noted previously, patients with aHUS who have DGKE mutations are unlikely to benefit from treatment with C5 inhibitors (e.g., eculizumab and ravulizumab). Clinical experts highlighted that patients with DGKE mutations can safely come off C5 inhibitors if no response to treatment has been observed, because it is unlikely to help. The outcomes indicating a favourable response include resolution of TMA (i.e., normalization of LDH and platelet count), stabilization of end organ damage (such as acute kidney injury and brain ischemia), transplant graft survival (in susceptible individuals), and dialysis avoidance (in patients who have not yet developed end-stage kidney disease [ESKD]).
Close monitoring of the patient for 1 year after discontinuing therapy is recommended for monitoring relapse. Treatment discontinuation in patients with a high-risk mutation in complement is associated with a 50% relapse rate; therefore, discontinuing treatment in these patients is more challenging. Treatment discontinuation also needs to be considered in the setting of severe infections. However, 1 clinical expert indicated that this would entail restarting the medication, either with a reduced dose or with prophylactic anti-infectives.
The clinical experts indicated that ravulizumab can be given at home with nursing support or at an infusion centre. A specialist, such as a nephrologist or hematologist with expertise in TMA, needs to monitor the patient.
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
The drug plan identified the following jurisdictional implementation issues: considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for prescribing of therapy, and system and economic issues. The clinical experts consulted by CADTH provided responses to the drug program implementation questions. For details, refer to Table 4: Summary of Drug Plan Input and Clinical Expert Response.
Clinical Evidence
Pivotal and Protocol-Selected Studies
Description of Studies
Two manufacturer-sponsored studies were included in this review: Study 31121 and Study 312.22
Study 311 is an ongoing, phase III, prospective, multicentre, single-arm, open-label trial that includes adult patients with aHUS.21 Its key objective is to evaluate the safety and efficacy of ravulizumab (IV infusion) in adult patients (aged 18 years and older) with aHUS who are complement inhibitor treatment–naive. The study consists of a screening period (up to 7 days), a 26-week initial evaluation period, and an extension period until the product is registered or approved (in accordance with country-specific regulations) or for up to 4.5 years. Enrolment started on March 18, 2017, and is ongoing.21 The cut-off date for the data reported herein was July 2, 2019. As of the cut-off date, a total of 58 adult patients were included, and 56 patients had received at least 1 dose of ravulizumab. The primary outcome was complete TMA response during the initial 26-week evaluation period, which was defined as normalization of hematologic parameters (platelet count and LDH) and an improvement of at least 25% in serum creatinine from baseline. The secondary outcomes were hematologic normalization (platelet count and LDH), hematologic TMA parameters (platelet count, LDH, and hemoglobin), hemoglobin response (more than 2% increase), dialysis requirement status, estimated glomerular filtration rate (eGFR), chronic kidney disease (CKD) stage, fatigue (measured using the Functional Assessment of Chronic Illness Therapy – Fatigue scale [FACIT-F]), HRQoL (measured using the 3-Level EQ-5D), and safety. Health care resource utilization, patient-reported aHUS symptoms, and extrarenal signs and symptoms of aHUS were reported as exploratory outcomes on a by-patient basis (no summary data provided).
Study 312 is an ongoing, phase III, prospective, multicentre, single-arm, open-label trial conducted in pediatric patients (younger than 18 years) with aHUS.22 The study includes 2 cohorts (i.e., cohort 1 and cohort 2). Cohort 1 includes 21 children with aHUS who are complement inhibitor–naive. The key objective for cohort 1 is to evaluate the safety and efficacy of ravulizumab (IV infusion) in this group. Cohort 2 includes 10 children with aHUS treated with eculizumab. The key objective for cohort 2 is to evaluate the safety and efficacy of ravulizumab (IV infusion) in children with aHUS with stable TMA parameters before a switch to ravulizumab. The study consists of a screening period (up to 7 days), a 26-week initial evaluation period, and an extension period that runs until the product is registered or approved (in accordance with country-specific regulations) or for up to 4.5 years. Enrolment for this study started on September 1, 2017, and is ongoing.22 The cut-off date for the data reported herein was December 3, 2019. As of the cut-off date, a total of 21 pediatric patients were included in cohort 1, and 18 patients had received at least 1 dose of ravulizumab. In cohort 2, a total of 10 pediatric patients were included, and all 10 patients received at least 1 dose of ravulizumab. The primary outcome was complete TMA response during the initial 26-week evaluation period, which was defined as normalization of hematologic parameters (platelet count and LDH) and at least a 25% improvement in serum creatinine from baseline (for cohort 1 only). The secondary outcomes were hematologic normalization (platelet count and LDH), hematologic TMA parameters (platelet count, LDH, and hemoglobin, cohort 1 only), hemoglobin response (great than 2% increase, cohort 1 only), dialysis requirement status, eGFR, CKD stage, fatigue (measured using FACIT-F), and safety. Health care resource utilization, patient-reported aHUS symptoms, and extrarenal signs and symptoms of aHUS were reported as exploratory outcomes on a by-patient basis (no summary data provided).
Efficacy Results
Complete TMA Response
At week 26 of Study 311, complete TMA response was observed in 30 patients of the 56 patients in the full analysis set (FAS) (53.6%; 95% confidence interval [CI], 39.6% to 67.5%). At the data cut-off date (median follow-up time = 75.57 weeks), complete TMA response was observed in 34 patients of the 56 patients in the FAS (60.7%; 95% CI, 47.0% to 74.4%). In Study 312, cohort 1, at week 26, complete TMA response was observed in 14 patients of the 18 patients in the FAS (77.8%; 95% CI, 52.4% to 93.6%). At the data cut-off date (median follow-up time: 82.43 weeks), complete TMA response was observed in 17 patients of the 18 patients in the FAS (94.4%; 95% CI, 72.7% to 99.9%).
Hematologic Normalization
In Study 311, hematologic normalization was defined as normalization of platelets and LDH. At week 26, hematologic normalization was observed in 41 patients of 56 patients in the FAS (73.2%; 95% CI, 60.7% to 85.7%). As of the data cut-off date, hematologic normalization was observed in 45 patients of the 56 patients in the FAS (80.4%; 95% CI, 69.1% to 91.7%). In Study 312, cohort 1, at week 26, hematologic normalization was observed in 16 patients of the 18 patients (88.9%; 95% CI, 65.3% to 98.6%). As of the data cut-off date, hematologic normalization was observed in 17 patients of the 18 patients in the FAS (94.4%; 95% CI, 72.7% to 99.9%).
Individual Hematologic Parameters
In Study 311, the mean (standard deviation [SD]) platelet count improved to a normal value after the initiation of ravulizumab treatment and remained stable during the extension period at the data cut-off date. Similarly, the mean LDH value decreased from baseline to within a normal range at week 26 and was sustained during the extension period at the data cut-off date. The mean hemoglobin value increased more gradually over time. The mean hemoglobin value was 120.27 g/L (normal value = 130 g/L to 175 g/L) at week 26 and remained above 120 g/L during the extension period at the data cut-off date. At week 26, 40 patients of the 56 patients (71.4%; 95% CI, 58.7% to 84.2%) in the FAS achieved a hemoglobin response. As of the data cut-off date, 45 patients of the 56 patients (80.4%; 95% CI, 69.1% to 91.7%) in the FAS achieved a hemoglobin response. In Study 312, cohort 1, similar improvements were observed in platelet count, LDH, and hemoglobin at week 26 and at the data cut-off date. In Study 312, cohort 2, hematologic parameters (platelet count, LDH, and hemoglobin) remained stable during the initial 26 weeks as well as through the data cut-off date.
Time to Complete TMA Response
In Study 311, as of the data cut-off date, complete TMA response was achieved at a median time of 86 (range, 7 days to 401 days). In Study 312, for pediatric patients, the median time to complete TMA response was 30 days (range, 15 days to 351 days).
Fatigue (FACIT-F)
In Study 311, an improvement of at least 3 points in FACIT-F score, which is considered a clinically meaningful improvement,23 was observed in 37 patients of 44 patients (84.1%) with available data at week 26. During the extension period, 33 patients of 40 patients (82.5%) with available data had at least a 3-point improvement from baseline at the day 351 visit. In cohort 1 of Study 312, 3 patients of 9 patients (33.3%) had at least a 3-point improvement in the FACIT-F total score from baseline at week 26. All 9 patients had at least a 3-point improvement from baseline at day 351. In Study 312, cohort 2, there was no notable improvement or worsening compared to baseline in the pediatric FACIT-F scores for all 8 patients during the initial 26 weeks through day 351 of the extension period.
HRQoL (Measured Using the 3-Level EQ-5D)
In Study 311, patients in the FAS showed improved 3-Level EQ-5D scores at week 26, and this improvement continued to day 351 of the extension period.
Renal Function (eGFR, CKD Stage Shifting, Dialysis Status)
Estimated Glomerular Filtration Rate
In Study 311, the mean eGFR gradually improved during the initial 26 weeks. During the extension period, the mean eGFR remained stable above 50 mL/min/1.73 m2 for the 43 patients who reached the day 407 visit. Overall, the mean eGFR value at baseline was 15.86 mL/min/1.73 m2. The mean eGFRs were 51.83 mL/min/1.73 m2 at week 26 and 50.30 mL/min/1.73 m2 at day 407. In Study 312, cohort 1, the mean eGFR value at baseline was 26.4 mL/min/1.73m2 (SD = 21.17 mL/min/1.73m2). The eGFR was 108.5 mL/min/1.73 m2 (SD = 56.87 mL/min/1.73 m2) at week 26 and remained above 100 mL/min/1.73 m2 for the 14 patients who reached the day 407 visit. In Study 312, cohort 2, the eGFR remained generally stable for all 10 patients from week 26 through the data cut-off date.
CKD Stage
In Study 311, in patients with available baseline and week 26 data, 32 of 47 patients (68.1%) in the FAS had improvement in CKD stage compared to baseline; 2 patients experienced a worsening of their CKD stage. During the extension period, for the 42 patients with available baseline data and day 407 data, 29 patients (69.0%) had improvement in CKD stage compared to baseline; the 2 patients who experienced worsening CKD stage at week 26 remained at stage 5 at the last available visit during the extension period. In Study 312, all but 2 patients in cohort 1 had an improved CKD stage at week 26; the shift was substantial, with 14 patients improving by 2 or more stages. None of the patients worsened in CKD stage at week 26 or during the extension period. In Study 312, 8 patients of 10 patients in cohort 2 began at CKD stage 1 and remained stable; 2 patients worsened during the initial 26 weeks. During the extension period, all 10 patients had no change in CKD stage by day 351 compared to baseline (refer to Table 2).
Dialysis Requirement Status
In Study 311, at baseline or within 5 days before the first dose of the study drug, 29 patients (51.8%) in the FAS had received renal dialysis (Table 2, Table 23). During the initial 26 weeks, 17 patients of these 29 patients (58.6%) discontinued dialysis. As of the data cut-off date, 18 patients of 29 patients (62.1%) had discontinued dialysis. Of the 27 patients who were not on dialysis at baseline, 7 patients (25.9%) initiated dialysis during the initial 26 weeks. As of the data cut-off date, 4 patients (14.8%) remained or started on dialysis. In Study 312, cohort 1, of the 6 patients in the FAS who were receiving kidney dialysis at baseline, 4 patients discontinued dialysis within the first 36 days of exposure to ravulizumab (Table 2, Table 23). All 6 patients had discontinued dialysis by day 193. Among patients who were not on dialysis at baseline, 0 patients initiated dialysis after starting treatment with ravulizumab. In Study 312, as of the data cut-off date, 0 of the 10 patients in cohort 2 initiated dialysis after starting treatment with the study drug.
Plasma Therapy–Free Status
Plasma therapy was prohibited during the trials; therefore, it was not an outcome assessed in the pivotal studies. However, plasma therapy was reported in the section on the concomitant therapy. In Study 311, 3 patients (5.2%) received plasma therapy; this was considered a protocol violation. No patients received plasma therapy in either cohort of Study 312.
Other Outcomes
Mortality, bleeding, packed red blood cell (RBC) transfusions, and soluble MAC levels were not assessed as efficacy outcomes in the 2 pivotal studies (Study 311 and Study 312). Symptoms (aside from fatigue) and hospitalization were reported on a by-patient basis in the 2 Clinical Study Reports (CSRs) submitted by the sponsor; there were no summary data submitted. Therefore, symptom reduction and hospitalization are not reported herein.
Harms Results
The key harm findings of Study 311 and Study 312 are shown in Table 2. In both studies, as of the data cut-off date, all patients experienced at least 1 treatment-emergent adverse event (TEAE). In Study 311, the most common adverse events (occurring in at least 30% of patients) were headache (n = 22; 37.9%), diarrhea (n = 19; 32.8%), and vomiting (n = 18; 31.0%). In Study 312, cohort 1, the most common adverse events (occurring in at least 30% of patients) were pyrexia (n = 10, 47.6%) and headache, diarrhea, vomiting, and nasopharyngitis (each occurring in 7 patients [33.3%]). In Study 312, cohort 2, the most common adverse event (occurring in at least 30% of patients) was oropharyngeal pain (n = 3; 30%). In Study 311, a total of 33 patients (56.9%) experienced a serious adverse event (SAE). Each SAE was reported in 1 patient. Exceptions were pneumonia and hypertension, each of which occurred in 3 patients (5.2%), and septic shock, urinary tract infection, aHUS, and malignant hypertension, each of which occurred in 2 patients (3.4%). In Study 312, cohort 1, the SAEs that occurred in greater than or equal to 2 patients were gastroenteritis viral infection and abdominal pain; each occurred in 2 patients (9.5%). In Study 312, cohort 2, no SAE was reported in more than 1 patient. In Study 311, a total of 3 patients (5.2%) experienced adverse events leading to discontinuation of the study drug. In Study 312, cohort 1, a total of 1 patient (4.8%) experienced adverse events leading to discontinuation of the study drug. In Study 312, cohort 2, 0 patients experienced adverse events leading to discontinuation of the study drug.
In Study 311, 4 patients died during the initial 26-week evaluation period. One of these 4 patients died due a pretreatment SAE (cerebral arterial thrombosis); 3 patients (5.2%) died due to treatment-emergent SAEs that were not considered to be related to the study drug. In Study 312, cohort 1 and cohort 2, no patients had died due to adverse events as of the data cut-off date. Regarding notable harms, as identified in the review protocol, no meningococcal disease was reported in either study. In Study 311, sepsis, hypersensitivity to the drug, and antidrug antibodies were each reported in 1 patient (1.7%). Infusion-related reactions were not reported. In Study 312, cohort 1, 1 patient (5.6%) reported hypersensitivity; no other notable harms were reported. In Study 312, cohort 2, no notable harms were reported.
Table 2: Summary of Key Results From the Pivotal and Protocol-Selected Studies
Outcomes | Study 311 (N = 56) | Study 312 | ||||
|---|---|---|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | |||||
Week 26 | Extension period (median time = 75.6 weeks) | Week 26 | Extension period (median time = 82.4 weeks) | Week 26 | Extension period (median time = 50.9 weeks) | |
Complete TMA responsea | ||||||
N | 56 | 56 | 18 | 18 | NA | NA |
n, patients with response | 30 | 34 | 14 | 17 | NA | NA |
% (95% CI) | 53.6 (39.6 to 67.5) | 60.7 (47.0 to 74.4) | 77.8 (52.4 to 93.6) | 94.4 (72.7 to 99.9) | NA | NA |
Components of complete TMA responseb | ||||||
Hematologic normalization (platelet count and LDH) | ||||||
N | 56 | 56 | 18 | 18 | NA | NA |
n, patients with response | 41 | 45 | 16 | 17 | NA | NA |
% (95% CI) | 73.2 (60.7 to 85.7) | 80.4 (69.1 to 91.7) | 88.9 (65.3 to 98.6) | 94.4 (72.7 to 99.9) | NA | NA |
Platelet count normalization | ||||||
N | 56 | 56 | 18 | 18 | 10 | 10 |
n, patients with response | 47 | 48 | 17 | 17 | NA, remained stable | NA, remained stable |
% (95% CI) | 83.9 (73.4 to 94.4) | 85.7 (75.7 to 95.8) | 94.4 (72.7 to 99.9) | 94.4 (72.7 to 99.9) | NA | NA |
LDH normalization | ||||||
N | 56 | 56 | 18 | 18 | 10 | 10 |
n, patients with response | 43 | 47 | 16 | 17 | NA, remained stable | NA, remained stable |
% (95% CI) | 76.8 (64.8 to 88.7) | 0.839 (73.4 to 94.4) | 88.9 (65.3 to 98.6) | 94.4 (72.7 to 99.9) | NA | NA |
≥ 25% improvement in serum creatinine from baseline | ||||||
N | 56 | 56 | 18 | 18 | 10 | 10 |
n, patients with responder | 33 | 35 | 15 | 17 | NA, remained stable | NA, remained stable |
% (95% CI) | 58.9 (45.2 to 72.7) | 62.5 (48.9 to 76.1) | 83.3 (58.6 to 96.4) | 94.4 (72.7 to 99.9) | NA | NA |
Time to complete TMA response (days) | ||||||
Median (minimum, maximum), days | 86 (7, 401) | 30 (15, 351) | NA | NA | ||
Fatigue | ||||||
Number of patients with ≥ 3 improvement on FACIT-F | ||||||
N, patients with data | 44 | 40 | 9 | 9 | NR | NR |
Patients with ≥ 3 improvement, n (%) | 37 (84.1) | 33 (82.5%) | 3 (33.3%) | 9 (100%) | NR | NR |
Baseline, n | 51 | 9 | 8 | |||
Mean (SD) | 24.03 (15.279) | 31.44 (13.648) | 48.88 (5.410) | |||
Week 26 and extension period (day 351), n | 48 | 40 | 9 | 9 | 8 | 8 |
Mean (SD) | 42.85 (8.796) | 42.52 (9.802) | 48.22 (5.848) | 48.11 (5.968) | 48.88 (5.410) | 47.63 (4.470) |
Change from baseline, n | 44 | 40 | 9 | 9 | 8 | 8 |
Mean (SD) | 19.15 (16.212) | 19.29 (17.520) | 16.78 (14.704) | 16.67 (15.297) | 0.00 (2.268) | –1.25 (2.712) |
HRQoL (3-Level EQ-5D) | ||||||
Baseline, n | 53 | 53 | NR | NR | NR | NR |
Mean (SD) | 0.48 (0.271) | 0.48 (0.271) | NR | NR | NR | NR |
Week 26 and extension period (day 351), n | 48 | 44 | NR | NR | NR | NR |
Mean (SD) | 0.71 (0.085) | 0.71 (0.057) | NR | NR | NR | NR |
Change from baseline, n | 46 | 42 | NR | NR | NR | NR |
Mean (SD) | 0.22 (0.247) | 0.25 (0.256) | NR | NR | NR | NR |
CKD stage changes | ||||||
N | 47 | 42 | 17 | 17 | 10 | 10 |
CKD stage improved by at least 1 stage from baseline, n (%) | 32 (68.1%) | 29 (69.0%) | 14 (77.8%) | 14 (77.8%) | 0 | 0 |
CKD stage worsened by at least 1 stage from baseline, n (%) | 2 (4.3%) | 2 (4.8%) | 0 | 0 | 3 of 10 (30%) | 0 |
Dialysis status | ||||||
For patients with dialysis at baseline, but discontinued during the trial, n of N (%) | 17 of 29 (58.6%) | 18 of 29 (62.1%) | 4 of 6 (66.7%) | 6 of 6 (100%) | NA | NA |
For patients without dialysis at baseline, but started dialysis during the trial, n of N (%) | 7 of 27 (25.9%) | 4 of 27 (14.8%) | 0 | 0 | 0 | 0 |
Harms (safety analysis population) | ||||||
Patients with at least 1 AE, n (%) | 58 (100.0) | 21 (100.0) | 10 (100.0%) | |||
Patients with at least 1 SAE, n (%) | 33 (56.9) | 14 (66.7) | 1 (10.0) | |||
Patients with an AE leading to DC from the treatment, n (%) | 3 (5.2) | 1 (4.8) | 0 | |||
Deaths | 3 (5.2) | 0 | 0 | |||
Notable harms | ||||||
Meningococcal disease | 0 | 0 | 0 | |||
Sepsis | 1 (1.7) | NR | NR | |||
Infusion-related reaction | NR | NR | NR | |||
Hypersensitivity to the drug | 1 (1.7) | 1 (4.8) | NR | |||
Antidrug antibodies | 1 | 0 | 0 | |||
AE = adverse event; CKD = chronic kidney disease; CI = confidence interval; DC = discontinuation; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; LDH = lactate dehydrogenase; NA = not assessed; NR = not reported; SAE = serious adverse event; SD = standard deviation; TMA = thrombotic microangiopathy.
a95% CIs for the proportion were based on the asymptotic Gaussian approximation method with a continuity correction.
b95% CIs for the proportion were based on exact confidence limits using the Clopper-Pearson method.
Source: Clinical Study Reports.21,22
Critical Appraisal
The main limitation of the 2 included pivotal studies (Study 311 and Study 312) is the single-arm design, which does not include a comparator arm. Due to the rare and severe nature of aHUS, a randomized control group was not likely to be feasible. Such a design, in addition to a lack of consideration of confounding variables, precludes causal inferences (i.e., the outcomes cannot be directly attributed to ravulizumab). Without an active comparator, standard of care, or statistical hypothesis testing, it is not possible to confirm the relative therapeutic benefit or safety of ravulizumab versus other available treatments (such as eculizumab in this population) or standard of care. In addition, both Study 311 and 312 were open-label trials; the study investigators and patients were aware of their treatment status, which increases the risk of detection and performance biases that have the potential to influence outcome reporting. However, the primary and most secondary outcomes (aside from fatigue and HRQoL) are objective end points for which the risk of bias due to open-label design is low. The potential for bias is a greater concern for the subjective end points, such as safety, fatigue (measured using the FACIT-F), and HRQoL (measured using the 3-Level EQ-5D). The direction of anticipated bias related to these outcomes is unclear. It is possible that known harms and anticipated benefits would be overreported.
For the longer-term subjective end points (HRQoL and fatigue), there is a potential risk of bias because complete measures were lacking for a large number of patients (especially for the extension period), leading to substantial missing data on certain outcomes. There may have been differential recall bias, and/or those patients remaining in the study may have differed in some systematic way compared to those who remained in the study and provided responses. Overall, the magnitude and direction of the impact of these missing data and of recall bias on the patient-reported and HRQoL outcomes is unknown. No minimally important difference (MID) was identified for HRQoL measures in the aHUS population. The overall the findings for HRQoL should be viewed as supportive evidence only.
One more potential limitation was that the efficacy assessment was not based on the intention-to-treat population (for Study 311 and Study 312, cohort 1); instead, it included patients who received at least 1 dose of the study drug. A total of 2 patients (3.4%) in Study 311 and 3 patients (14.3%) in Study 312, cohort 1 were excluded from the primary FAS analysis. In addition, it is also noted that 43 patients (76.79%) in Study 311 and 14 patients (66.7%) in Study 312, cohort 1 experienced major protocol violations (N = 25, 43.1% in Study 311 and N = 9, 42.9% in Study 312), the majority of which were related to the eligibility criteria. Although a per-protocol (PP) analysis was performed (N = 44, 75.9% for Study 311 and N = 18, 85.7% for Study 312, cohort 1) and showed results that were consistent with the FAS analysis, not all patients with the major protocol violation, especially those related to eligibility criteria, were excluded from the analysis. Therefore, there is a potential impact on the results (although the direction of the impact is not clear). The main limitation of Study 312, cohort 2 (pediatric patients with aHUS who switched from eculizumab to ravulizumab) was that the sample size (N = 10) was small, which meant the overall dataset was more sensitive to outliers and skewed distribution.
Overall, according to the clinical experts consulted by CADTH, the inclusion and exclusion criteria of the 2 pivotal studies (Study 311 and Study 312) were reasonable and the baseline patient characteristics, concomitant medications, and prohibited medications were reflective of patients observed in clinical practice for the indication under review. Finally, it is not clear whether the magnitude of the treatment effect estimates observed in the relatively small study sample will be replicable in a larger study sample or generalizable to the target population in real-world clinical practice.
Indirect Treatment Comparisons
Direct comparisons between ravulizumab and eculizumab are likely to be infeasible due to the rare and severe nature of aHUS. Therefore, for this submission, a systematic literature review was conducted to identify any sources of indirect treatment comparisons (ITCs) between ravulizumab and eculizumab, or between ravulizumab and best supportive care. No ITCs were identified in the CADTH search.
Description of Studies
Overall, 1 study, a sponsor-submitted ITC, was available to assess the relative efficacy of ravulizumab versus eculizumab using a patient-level, propensity-based primary analysis.
Efficacy Results
Among adult patients with aHUS who had not had a kidney transplant, the sponsor did not note any statistically significant differences between ravulizumab and eculizumab with respect to mortality, complete TMA response, LDH, platelets, EQ-5D visual analogue scale (VAS), FACIT subscales, renal function, or dialysis status at 6 months when using a stabilized weights model. Sensitivity analyses exploring pediatric patients without kidney transplant, adult patients with kidney transplant, and adult patients without kidney transplant using propensity matching were broadly concordant with the primary analysis.
No data were available with respect to the presence of severe bleeding, hemoglobin concentration change over time, plasma therapy–free status, packed RBC transfusion, hospitalizations, or soluble MAC.
Harms Results
No evidence for relative safety or harms was presented for review.
Critical Appraisal
Overall, the submitted ITC was subject to several limitations that add uncertainty to the conclusions presented. Principally, it is unclear whether all clinically meaningful covariates were accounted for within the sponsor’s ITC; residual confounding may occur from these characteristics not being accounted for in the primary analysis. Similarly, there remain potentially important unmeasured confounding characteristics, such as a 10-year gap between the studies of eculizumab and ravulizumab. During this period, there may have been changes to standard of care, increased awareness or capacity to diagnose disease, and changes in health care system capacity. These are all confounding factors that cannot be excluded from the current analysis. Finally, a few reporting characteristics were absent, such as rationale of exclusion for studies, specification of the estimands used in the analysis, units of outcomes, and baseline covariates of interest.
Other Relevant Evidence
No other relevant evidence was identified.
Conclusions
The evidence for the clinical benefits and harms of ravulizumab in the treatment of aHUS was based on the 2 sponsor-submitted, pivotal, multinational, single-arm, open-label, prospective phase III trials (Study 311 for adults with aHUS and Study 312 for pediatric patients with aHUS). The majority of pediatric and adult patients who were complement inhibitor treatment–naive experienced hematological normalization, improved renal function, and improved HRQoL with ravulizumab treatment. Despite uncertainty around the magnitude of the clinical benefit attributable to ravulizumab (given the limitations inherent in the single-arm trial design), the lack of formal hypothesis testing, and the relatively small sample size, the clinical experts indicated that the benefits observed in the 2 trials appeared clinically meaningful, considering that aHUS is an extremely rare and life-threatening disease. For adult patients who were complement inhibitor–experienced, no evidence was identified to inform the switch from eculizumab to ravulizumab. The expected benefit of switching lies in the reduced number of infusions required (because of the longer half-life of ravulizumab versus eculizumab). Although the 10 patients who switched from eculizumab to ravulizumab in Study 312 appeared to have a maintained TMA response, due to the small sample size, it remains unclear whether these findings are reflective of what would be observed in the larger population of patients with aHUS. The sponsor also submitted a propensity score–weighted analysis comparing ravulizumab with eculizumab; however, due to several methodological limitations, no robust conclusion could be drawn on the comparative efficacy and safety of ravulizumab versus eculizumab. The safety profile of ravulizumab observed in the 2 trials appeared consistent with the known safety profile of ravulizumab, and no additional safety signals were identified.
Introduction
Disease Background
aHUS is a life-threatening, ultra-rare disease in which patients are susceptible to sudden and progressive episodes of complement-mediated TMA. These episodes most commonly affect the kidneys, but can also include extrarenal, multiorgan involvement.1,2 An acute aHUS episode requires emergency care; however, patients with aHUS are also at ongoing risk of systemic, life-threatening, and multisystem complications over the long-term. Extrarenal manifestations are common, especially in newly diagnosed patients (i.e., ≤ 6 months from diagnosis).24 aHUS is primarily caused by inherited or acquired dysregulation of complement-regulatory proteins, resulting in uncontrolled complement activation.2-4 Historically, kidney failure and death were common outcomes; however, improved understanding of the condition has led to the discovery of novel therapies (i.e., complement inhibitors, including eculizumab and ravulizumab) that may reduce the risk of these complications.25 The uncontrolled complement activation of aHUS causes inflammation, endothelial activation and damage, and a prothrombotic and/or procoagulant state, resulting in systemic TMA.2,26-28 Over the past few years, there has been an increasing consensus that, in the majority of patients, for the clinical event of a TMA to occur, aHUS may involve both genetic predisposition (e.g., pathogenic variants, autoantibodies, or at-risk polymorphisms in complement genes) and a triggering condition.5-7 Atypical HUS can occur at any age, but childhood onset is more common than adult onset (60% versus 40%, respectively). When onset occurs in childhood, the disease affects males and females equally, whereas in adulthood, the disease affects women more frequently. Most children affected by aHUS (70%) will have the disease before the age of 2 years. aHUS biomarkers include C3, C5a, C5b-9, factor B, complement factors B, H, and I, CH50, AH50, d-dimer, as well as anti-CFH antibodies.8,9 Low levels of C3, CH50, AH50, and CFB—along with increased levels of C5a, C5b-9, Bb, anti-CFH autoantibodies, and d-dimer—are usually noted in patients with aHUS.8
Diagnosis of aHUS is currently based on exclusion of other causes of TMA.10,11 Therefore, the potential risk of misdiagnosis of aHUS may exist in clinical practice.11 Although a positive genetic test can help to confirm a clinically diagnosed case of aHUS, complement gene mutations are identified in only 50% to 60% of patients with aHUS6,29,30 and are not required to make the diagnosis or commence treatment. A clinical differential diagnosis remains the primary method of establishing a diagnosis of aHUS.6,11 The clinical experts consulted by CADTH for this review indicated that 30% to 40% patients with aHUS may have no known genetic disposition.
The incidence and prevalence of aHUS vary widely.3,13 This is attributed to the heterogeneity of patients with aHUS, ambiguity surrounding its clinical presentation, and difficulties with diagnosing aHUS.13 A 2020 systematic literature review of the global epidemiology of aHUS reported that the annual incidence of aHUS ranged from 0.26 per million population to 0.75 per million population among people aged 20 years and younger. For all ages, the annual incidence ranged from 0.23 per million population to 1.9 per million population.13 These estimates are in line with the estimate reported in the US of 1 case per million to 2 cases per million in the general population.14 The 2020 systematic literature review also reported that the prevalence of aHUS ranged from 2.2 per million population to 9.4 per million population in people aged 20 years and younger, whereas the prevalence in all age groups (based on only 1 study) was 4.9 per million population.13 Most studies providing these data were from Europe and Oceania, given that there are limited published prevalence estimates for aHUS from countries such as Canada and the US.13 A Canadian study published in 2004 reported an incidence of aHUS in children of 2 cases per million over a 4-year period.14 It has also been reported that aHUS occurs in approximately 1 in 1 million births and affects 60 patients to 90 patients in Canada.31,32 Most recently, a 2020 analysis of 37 patients in Canada (15 pediatric and 22 adult) enrolled in the aHUS Global Registry (an observational, noninterventional, multicentre study that prospectively and retrospectively collects data on patients with a clinical diagnosis of aHUS, irrespective of treatment)15 estimated that there are potentially 74 patients with aHUS in Canada.16
Standards of Therapy
Prior to the approval of ravulizumab, the terminal complement inhibitor eculizumab was considered the standard of care in most jurisdictions for the treatment of patients with aHUS for more than a decade.17 Eculizumab is the only Health Canada–approved drug indicated for the treatment of aHUS.18 However, eculizumab is not reimbursed across all Canadian jurisdictions (refer to Appendix 1). Furthermore, eculizumab imposes a substantial treatment burden on patients, due to its shorter half-life (compared to ravulizumab) and requirement for biweekly dosing.18 This results in missed days of work or school to accommodate visits to an infusion centre and requires careful scheduling of travel and other life events to accommodate biweekly treatment. Frequent infusions also make venous access ports necessary for some patients, especially children, which puts them at risk of port-related complications (e.g., infection and thrombosis).19 The frequent dosing schedule of eculizumab is health care resource–intensive, which also drives infusion-related costs with eculizumab.19 The clinical experts consulted by CADTH for this review indicated that there is an unmet need for alternative effective therapies with acceptable toxicity profiles that help patients with aHUS achieve TMA remission and improved HRQoL. The clinical experts anticipated that ravulizumab would have similar or equivalent efficacy as eculizumab, with the potential of a better therapeutic profile and/or reduced therapeutic burden. The clinical experts indicated that patients with aHUS who have DGKE mutations are unlikely to benefit from treatment with C5 inhibitors (e.g., eculizumab and ravulizumab).
The appropriate duration of treatment with anticomplement therapy is unknown. Both eculizumab and ravulizumab are C5 inhibitors, the major difference between these drugs is duration of action; ravulizumab has a longer duration. Compared with eculizumab, ravulizumab may have a similar clinical benefit, but be less burdensome for patients and the health care system. The choice between them is individualized.9
Drug
Ravulizumab (10 mg/mL concentrate for solution for infusion) is a terminal complement inhibitor that specifically binds to C5, inhibiting its cleavage to C5a and C5b and preventing the generation of MAC or C5b9. Health Canada has previously issued market authorization for ravulizumab for paroxysmal nocturnal hemoglobinuria.18
The Health Canada–proposed indication of interest for this review is the treatment of adult patients with aHUS. The Health Canada–recommended dosing regimen consists of a single weight-based IV loading dose followed 2 weeks later by the first IV maintenance dose; IV maintenance doses are then administered every 8 weeks (or every 4 weeks for children weighing ˂ 20 kg) (Table 3). The sponsor’s reimbursement request is identical to the Health Canada–approved indication.
Table 3: Key Characteristics of Ravulizumab and Eculizumab
Characteristics | Ravulizumab | Eculizumab |
|---|---|---|
Mechanism of action | Ravulizumab is a terminal complement inhibitor that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a (the proinflammatory anaphylatoxin) and C5b (the initiating subunit of the membrane attack complex [or C5b-9]), preventing the generation of the terminal complement complex C5b9. | Eculizumab, the active ingredient in Soliris, is a monoclonal antibody that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a and C5b and preventing the generation of the terminal complement complex C5b-9 and free C5a. Eculizumab inhibits complement-mediated TMA in patients with aHUS. |
Indicationa | For the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA | For the treatment of patients with aHUS to reduce complement-mediated TMA |
Route of administration | IV infusion | IV infusion |
Recommended dose | For adult and pediatric patients with aHUS with a body weight ≥ 5 kg, dosage is based on the patient’s body weight. Frequency is q.4.w. for body weight < 20 kg and q.8.w. for body weight ≥ 20kg. The first maintenance dose is given 2 weeks after the loading dose, with doses as follows: ≥ 5 kg to < 10 kg (q.4.w.) Loading dose: 600 mg Maintenance: 300 mg ≥ 10 kg to < 20 kg (q.4.w.) Loading dose: 600 mg Maintenance: 600 mg ≥ 20 kg to < 30 kg (q.8.w.) Loading dose: 900 mg Maintenance: 2,100 mg ≥ 30 kg to < 40 kg (q.8.w.) Loading dose: 1,200 mg Maintenance: 2,700 mg ≥ 40 kg to < 60 kg (q.8.w.) Loading dose: 2,400 mg Maintenance: 3,000 mg ≥ 60 kg to < 100 kg (q.8.w.) Loading dose: 2,700 mg Maintenance: 3,300 mg ≥ 100 kg (q.8.w.) Loading dose: 3,000 mg Maintenance: 3,600 mg | For adult patients (aged ≥ 18 years), the dosage is 900 mg weekly for the first 4 weeks, followed by 1,200 mg for the fifth dose 1 week later, followed by 1,200 mg every 2 weeks thereafter. For patients with atypical HUS and aged ˂ 18 years, weight-based dosing regimens are as follows: ≥ 40 kg Induction: 900 mg weekly × 4 doses Maintenance: 1,200 mg at week 5, then 1,200 mg every 2 weeks 30 kg to < 40 kg Induction: 600 mg weekly × 2 doses Maintenance: 900 mg at week 3, then 900 mg every 2 weeks 20 kg to < 30 kg Induction: 600 mg weekly × 2 doses Maintenance: 600 mg at week 3, then 600 mg every 2 weeks 10 kg to < 20 kg Induction: 600 mg weekly × 1 dose Maintenance: 300 mg at week 2, then 300 mg every 2 weeks 5 kg to < 10 kg Induction: 300 mg weekly × 1 dose Maintenance:300 mg at week 2, then 300 mg every 3 weeks |
Serious adverse effects or safety issues |
|
|
aHUS = atypical hemolytic uremic syndrome; C5 = complement component 5; q.4.w. = once every 4 weeks; q.8.w. = once every 8 weeks; TMA = thrombotic microangiopathy.
aHealth Canada–approved indication.
Source: Product monographs for Ultomiris and Soliris.17,18,20
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient advocacy group, aHUS Canada, provided input on the treatment of aHUS. This group gathered information from 19 caregivers and 41 patients from inside and outside Canada through an online survey conducted in June 2022. Of these 60 respondents, 19 had experience with the drug under review.
Respondents identified anemia, low platelet count, and acute renal failure as the most difficult primary symptoms to control. Diminished quality of life, helplessness, post-traumatic stress disorder, fatigue and/or exhaustion, headache, high blood pressure, inability to travel, frequent hospital visits, and kidney issues and/or dialysis are some of the experiences that respondents have while living with aHUS. According to aHUS Canada, aHUS patients who require dialysis and need a kidney transplant are not eligible for a transplant in Canada unless they receive eculizumab infusions at the time of the transplant.
Caregivers to patients with aHUS also face emotional and financial challenges because the process to access eculizumab and alternatives differs from province to province. Respondents described financial struggles, anxiety about access to treatment, the need to protect organs, exhaustion, memory loss and/or brain fog as aspects of the disease that are hardest to control.
Respondents identified plasma therapy (fresh frozen plasma or plasmapheresis), eculizumab infusions, and long-term dialysis as the currently available treatments for patients with aHUS. Side effects reported by the respondents included nausea, headache, fatigue, anaphylactic reaction to plasma, vein collapse, infection, anxiety, kidney failure, uncontrolled blood pressure, migraines, exhaustion, memory loss and/or brain fog, central line issues, muscle crumps, insomnia, abdominal pain, fever and chills, weight gain or loss, and being refractory to plasma therapy, among others.
While discussing their expectations for new drugs, patients reported that access to treatment and freedom of choice were critical components in managing the disease. However, quality of life was the most commonly cited desired outcome, and it was affected by factors like choice in care, frequency of appointments, and drug affordability. The abilities to travel, focus on family, and have more time between appointments were also described as critical to patients’ mental health. Moreover, frequent blood tests and IV therapies or ports were reported to be significant problems for many patients. While 1 caregiver pointed out the importance of maintaining “venous access for continuous access to eculizumab,” other patients shared their ineligibility for ports due to damaged veins from the disease. Patients also expressed the importance of requiring less frequent treatments.
While discussing their experiences with ravulizumab, patients listed benefits that included more energy, less vein damage, fewer treatments, fewer symptom fluctuations, greater freedom of choice, and less anxiety. However, they also reported experiencing headache, nausea, and body aches right after their infusion or during the month after the infusion.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adult and pediatric patients with aHUS.
Unmet Needs
The clinical experts consulted by CADTH for this review indicated that administration of eculizumab every 2 weeks interferes with a patient’s quality of life by consuming time that could be spent working, travelling, or with friends and family. Administration of eculizumab every 2 weeks can also be an issue when it comes to venous access fatigue and comes with the societal cost of nursing and allied health care support. The biggest limitation to the current treatment is prohibitive cost: most centres will fund an initial treatment or a few treatments, but very few have the resources to fund lifelong treatment. Inclusion in provincial formularies is inconsistent across provinces. and private insurance coverage is not common. Often, patients or their health teams advocate for subsidies or payment in full, but they are not always successful. With respect to venous access fatigue, most patients would be candidates for portacaths or central lines, which are normally offered to chemotherapy patients.
Place in Therapy
The mechanism of action of ravulizumab is the same as that of eculizumab, which is the only other approved treatment for aHUS. Ravulizumab would not be added to other treatments. Instead, the clinical experts believed it would replace eculizumab as the treatment of choice for aHUS. The clinical experts anticipated that ravulizumab would have similar or equivalent efficacy as eculizumab, with the potential of a better therapeutic profile and/or reduced therapeutic burden. The reasons clinical experts believed ravulizumab could become the first-line treatment of choice included the potential for improved patient quality of life and better cost-effectiveness because of the fewer infusions required.
Theoretically, as with other biologics that use the same target molecule, tachyphylaxis to 1 medication may open up options to treat with the second; therefore, acquired nonresponse may be a consideration to switch therapies.
Patient Population
The patients most suitable for treatment with ravulizumab are those diagnosed with aHUS. The patients most in need of intervention are those with severe TMA with associated end organ damage, such as acute kidney injury or brain ischemia.
The patients who are least suitable for treatment with ravulizumab are those with TMA that is clearly due to a secondary cause, such as malignant hypertension, malignancy, or infection. There may be some benefit to using eculizumab in patients with certain autoimmune diseases in which there is histological evidence of TMA as well as evidence of complement dysregulation (e.g., some variants of lupus).
Patients who qualify for treatment would be identified by physicians with expertise in TMAs. These include nephrologists, hematologists, and internal medicine physicians, who would make the diagnosis based on clinical examination, lab investigations, and genetic testing for complement dysregulation, and by excluding other causes of TMA.
Diagnosing aHUS can be very challenging because no single diagnostic test can confirm the disease. In many situations, it is a diagnosis of exclusion. For this reason, misdiagnosis is a risk. One clinical expert indicated that testing has improved and the difficulties of diagnosis have decreased, suggesting that these diagnostic challenges may have been a greater issue 10 or 15 years ago, when genetic and biochemical assessments of complement pathways were less accessible; however, these tests are now more available, often on a quick turnaround, even when sent out of province. The diagnosis of aHUS requires evidence of TMA, such as schistocytes, elevated lactate dehydrogenase, decreased haptoglobin, decreased hemoglobin, and thrombocytopenia. These lab abnormalities should also coincide with 1 or more of the following: neurological symptoms, acute renal failure, or gastrointestinal symptoms, although any organ system can be involved (e.g., pancreas, heart). One clinical expert indicated that the haptoglobin is not the most reliable diagnostic indicator, and that LDH level is a better test. In patients with aHUS, early initiation of plasmapheresis until the diagnosis is confirmed is critical, given the increased mortality of untreated TTP. Most centres have access to ADAMTS13 activity testing with a turnaround of 24 hours to 48 hours. Adult patients may be offered treatment with plasmapheresis before the results are known; however, for pediatric patients, physicians would prefer to wait for the results, if feasible.
Etiologies that mimic TMA need be excluded during diagnosis, including infections, medications, malignancy, scleroderma, antiphospholipid antibody syndrome, systemic lupus erythematosus, malignant hypertension, disseminated intravascular coagulation, preeclampsia, and HELLP syndrome. TTP can be distinguished from aHUS by measuring ADAMTS13 level. If ADAMTS13 is greater than 5% and the patient is resistant to plasma exchange, then the diagnosis is more likely to be aHUS than TTP. Screening for complement mutations and antibodies should be performed. In pediatric populations, where TTP is less common, clinicians would likely not initiate plasmapheresis first, but agree this would be prudent to do for older patients, particularly older adults. Once aHUS has been diagnosed, C5 inhibition may be used as first-line therapy.
Treatment goals include the resolution of TMA, with normal platelet and LDH counts, as well as the resolution of acute kidney injury and/or neurological sequelae and the stabilization of end organ damage.
More sophisticated testing is available as well, including levels of soluble C5b-9. Levels of C5b-9 are elevated during aHUS and subside with treatment because C5b-9 is generated as a product of complement activation. If levels are initially low, most centres may follow C3 and complement component 4 levels to monitor for recovery. Duration of treatment with C5 inhibition is unknown. Based on the available data, if there are no high-risk complement genetic variants, then termination of treatment could be considered after 6 months to 12 months. Lifelong treatment may be considered for patients with high-risk complement genetic variations; however, 30% to 40% of patients may have no known genetic disposition. As noted previously, patients with aHUS who have DGKE mutations are unlikely to benefit from treatment with C5 inhibitors (e.g., eculizumab and ravulizumab). Clinical experts highlighted that patients with DGKE mutations can safely come off of C5 inhibitors because these are unlikely to help if no response to treatment has been observed.
Assessing Response to Treatment
The outcomes indicating a favourable response include the resolution of TMA (i.e., normalization of LDH and platelet count), the stabilization of end organ damage (such as acute kidney injury and brain ischemia), transplant graft survival (in susceptible individuals), and dialysis avoidance (in patients who are pre-ESKD).
Discontinuing Treatment
It is possible to discontinue treatment with ravulizumab in patients with aHUS who do not have a genetic complement mutation 3 months to 6 months after they achieve remission. Close monitoring of the patient for 1 year after they discontinue therapy is recommended to monitor for relapse. One clinical expert indicated that 30% to 40% of patients do not have a genetic diagnosis. Treatment discontinuation in patients with a high-risk mutation in complement is associated with a 50% relapse rate; therefore, discontinuing treatment in these patients is more challenging.
Treatment discontinuation also needs to be considered in the setting of severe infections. However, 1 clinical expert indicated that this would entail restarting the medication with reduced dose or prophylactic anti-infectives.
Prescribing Conditions
Ravulizumab can be given at home with nursing support or at an infusion centre. A specialist, such as a nephrologist or hematologist with expertise in TMA, needs to monitor the patient.
Additional Information
The clinical experts expressed that despite the cost savings associated with ravulizumab’s less frequent administration, the drug’s cost still needs to be reasonable. The experts highlighted that if the cost is much higher than that of eculizumab, most health systems would constrain use to eculizumab.
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
Drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical experts’ responses |
|---|---|
Considerations for initiation of therapy | |
Would patients who do not respond (or who stop responding) to treatment with eculizumab benefit from ravulizumab treatment? | There is no evidence of this. However, ravulizumab does give an immediate, complete, more sustained C5 inhibition compared to eculizumab; this may be a consideration in individual cases. There is also evidence that some patients who develop tachyphylaxis to specific biologics retain some responsiveness to biosimilars. |
Can a patient restart ravulizumab if they responded to previous treatment? If so, under what clinical conditions? | If a patient redevelops a TMA related to aHUS, ravulizumab needs to be restarted to prevent end organ damage. Note that discontinuation of C5 inhibitors, when these have been maintaining remission and withdrawal has subsequently caused relapse, may cause irreversible damage, resulting in progression of organ damage. Therefore, if a patient were in this situation and progressed to end-stage kidney disease with no history of other organ involvement, it may be futile to restart the medication because the patient would remain on dialysis. Restarting the medication post-transplant would be necessary if the patient were deemed a suitable transplant candidate. |
Consider alignment with current Canadian public drug plan initiation criteria for eculizumab. | No response required. For CDEC consideration. |
Considerations for continuation or renewal of therapy | |
Consider alignment with renewal criteria for eculizumab. | No response required. For CDEC consideration. |
Considerations for prescribing of therapy | |
Ravulizumab (Ultomiris) is dosed by weight and given by IV every 4 weeks (≥ 5 kg to < 20 kg) or every 8 weeks (≥ 20 kg). | No response required. For CDEC consideration. |
Consider alignment with prescribing criteria for eculizumab (Soliris): i.e., prescribed by or in consultation with a pediatric nephrologist, nephrologist, pediatric hematologist, or hematologist. | No response required. For CDEC consideration. |
System and economic issues | |
Consider adding a similar discussion point to ravulizumab for PNH. There is a risk of ravulizumab not being cost-effective vs. a biosimilar of eculizumab in the future. | No response required. For CDEC consideration. |
C5 = complement component 5; CDEC = CADTH Canadian Drug Expert Committee; PNH = paroxysmal nocturnal hemoglobinuria; TMA = thrombotic microangiopathy.
Clinical Evidence
The clinical evidence included in the review of ravulizumab is presented in 3 sections. The Systematic Review section includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review (when available). The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review (if submitted).
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of ravulizumab for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult and pediatric patients with aHUS Subgroups:
|
Intervention | Ravulizumab IV infusion at recommended doses (body weight-based), with maintenance doses administered every 4 weeks or every 8 weeks, starting 2 weeks after the loading dose. |
Comparators | Eculizumab IV infusion at recommended doses Supportive care (e.g., plasma exchange or infusion, plasmapheresis) |
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality due to AE, notable harms, harms of special interest (e.g., serious infections [including meningococcal, sepsis], infusion-related reactions, antidrug antibodies) |
Study designs | Published and unpublished clinical phase III interventional trials |
AE = adverse event; aHUS = atypical hemolytic uremic syndrome; CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; ESRD = end-stage renal disease; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; SAE = serious adverse event; TMA = thrombotic microangiopathy; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.33
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Ultomiris (ravulizumab). Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on July 07, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on October 26, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.)34 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and contacting appropriate experts. In addition, the sponsor was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies21,22 were identified from the literature for inclusion in the systematic review (Figure 1). The 2 included studies are presented in 7 documents12,21,22,35-38 and summarized in Table 6. A list of excluded studies is presented in Appendix 3.
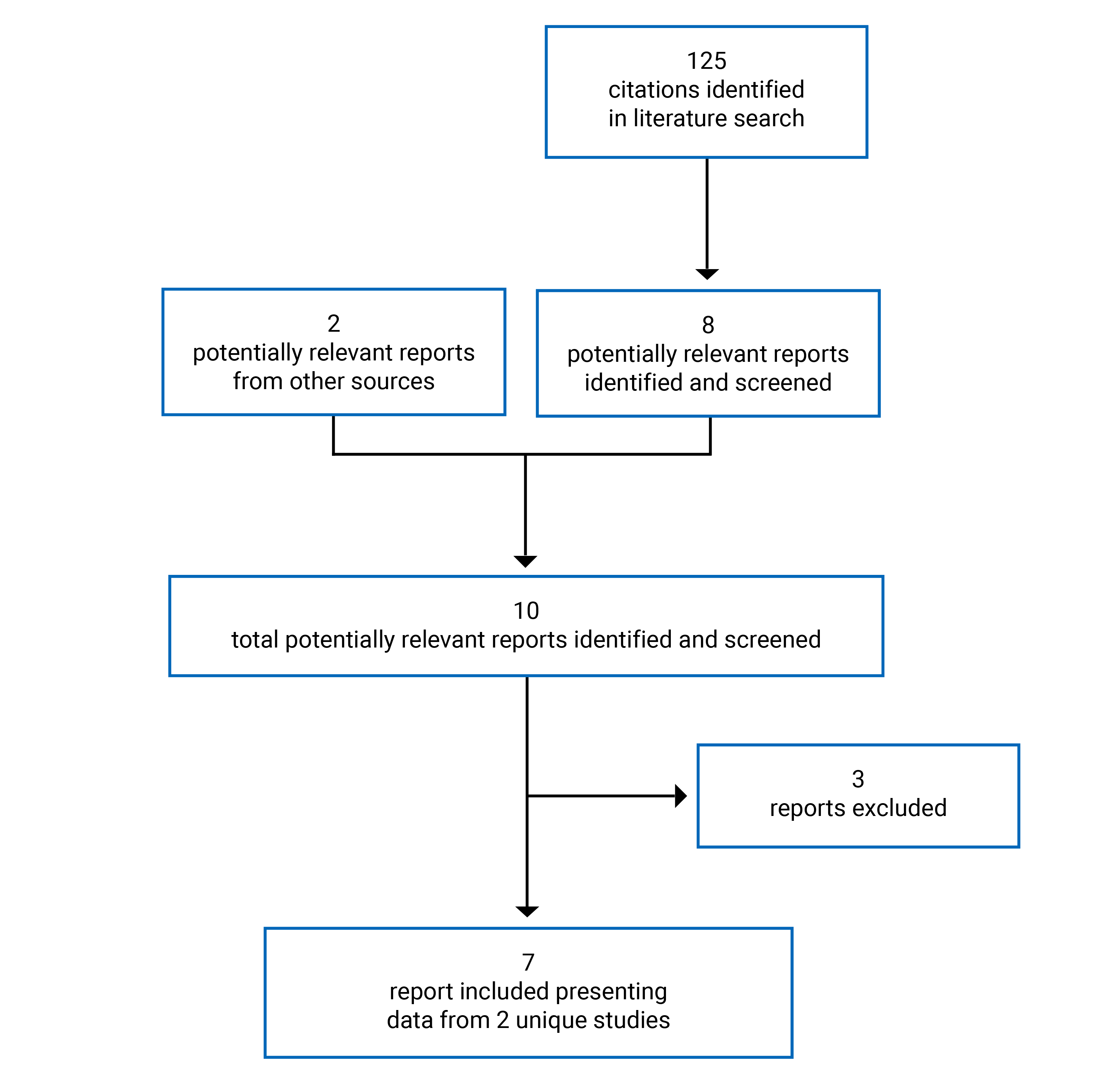
Table 6: Details of Study 311 (Adults)
Characteristics | Study 311 |
|---|---|
Designs and populations | |
Study design | Ongoing, phase III, multicentre, OL, single-arm trial |
Locations | 41 sites in 14 countries (Australia, Austria, Belgium, Canada, France, Germany, Italy, Japan, Korea, Russia, Spain, Taiwan, the UK, and the US). |
Patient enrolment date | Date first patient treated: March 18, 2017 |
Date of extension period data cut-off | July 2, 2019 |
Sample size (N) | 58 |
Inclusion criteria | Patients were eligible for enrolment in the study only if they met all the following criteria and none of the exclusion criteria:
|
Exclusion criteria | Patients were excluded if they met any of the following criteria:
|
Drugs | |
Intervention | During the 26-week initial evaluation period, patients received a weight-based loading dose of ravulizumab IV on day 1, followed by q.8.w. body weight–based maintenance doses on days 15, 71, and 127. After the 26-week initial evaluation period, all patients rolled into an extension period during which they continued to receive the ravulizumab q.8.w. weight-based IV maintenance dose. A patient who discontinued and restarted ravulizumab on a scheduled study visit received a loading dose, a supplemental maintenance dose 2 weeks later, and a maintenance dose 6 weeks later, resuming a q.8.w. regimen thereafter. If the decision to re-treat with ravulizumab occurred between scheduled study visits, the dosing regimen was to be determined by the Alexion medical monitor and the investigator. |
Comparator(s) | None |
Duration | |
Phase | |
Screening period | Up to 7 days |
Initial open-label evaluation period | 26 weeks |
Open-label extension period | Up to 4.5 years |
Outcomes | |
Primary end point | Complete TMA response during the 26-week initial evaluation period, as evidenced by normalization of hematologic parameters (i.e., platelet count and LDH) and ≥ 25% improvement in serum creatinine from baseline) |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | |
AMR = antibody-mediated rejection; CKD = chronic kidney disease; CNI = calcineurin inhibitor; eGFR = estimated glomerular filtration rate; ESKD = end-stage kidney disease; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; Hb = hemoglobin; HRQoL = health-related quality of life; HUS = hemolytic uremic syndrome; LDH = lactate dehydrogenase; LLN = lower limit of normal; mTORi = mammalian target of rapamycin inhibitor; OL = open label; q.8.w. = every 8 weeks; TMA = thrombotic microangiopathy; ULN = upper limit of normal.
Source: Study 311 Clinical Study Report.21
Table 7: Details of Study 312 (Children)
Characteristics | Study 312 (cohort 1) | Study 312 (cohort 2) |
|---|---|---|
Designs and populations | ||
Study design | Ongoing, phase III, multicentre, OL, single-arm trial | |
Locations | 20 sites in 8 countries (Belgium, Germany, Italy, Japan, Korea, Spain, UK, and the US) | |
Patient enrolment date | Date first patient treated: September 1, 2017 | |
Data cut-off date | December 3, 2019 | |
Sample size (N) | 21 | 10 |
Inclusion criteria | Patients were eligible for enrolment in the study only if they met all the following criteria (and none of the exclusion criteria):
| Patients were eligible for enrolment in the study only if they met all the following criteria (and none of the exclusion criteria):
|
Exclusion criteria | Patients were excluded if they met any of the following criteria:
| Patients were excluded if they met any of the following criteria:
|
Intervention | Patients received a weight-based loading dose of ravulizumab IV on day 1 followed by maintenance treatment with ravulizumab on day 15 and q.8.w. thereafter for patients weighing ≥ 20 kg or q.4.w. thereafter for patients weighing < 20 kg. The loading and maintenance doses were based on the patient’s body weight recorded on dosing regimen decision days. For cohort 2, the first dose was administered 14 days after the last dose of eculizumab. | |
Comparator(s) | None | |
Duration | ||
Phases | ||
Screening period | Up to 7 days | Up to 28 days |
Initial open-label evaluation period | 26 weeks | |
Open-label extension period | Up to 4.5 years | |
Outcomes | ||
Primary end point | Complete TMA response during the 26-week initial evaluation period | NA |
Secondary and exploratory end points | Secondary:
| Secondary:
|
| ||
Publications | Ariceta (2021)35 | Tanaka (2021)38 |
aHUS = atypical hemolytic uremic syndrome; AMR = antibody-mediated rejection; CKD = chronic kidney disease; CNI = calcineurin inhibitor; eGFR = estimated glomerular filtration rate; ESKD = end-stage kidney disease; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; HUS = hemolytic uremic syndrome; LDH = lactate dehydrogenase, LLN = lower limit of normal; mTORi = mammalian target of rapamycin inhibitor; NA = not applicable; OL = open label; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; TMA = thrombotic microangiopathy; ULN = upper limit of normal.
Source: Study 312 Clinical Study Report.22
Description of Studies
Two manufacturer-sponsored studies were included in this review: Study 31121 and Study 312.22
Study 311
Study 311 is an ongoing, phase III, prospective, multicentre, single-arm, open-label trial that includes adult patients with aHUS.21 The key objective of the Study 311 is to evaluate the safety and efficacy of ravulizumab (IV infusion) in complement inhibitor treatment–naive adult patients (aged ≥ 18 years) with aHUS. The trial was conducted at 41 sites in 14 countries (including Canada, the US, Australia, and 11 countries in Europe and Asia). The key characteristics of the study design are summarized in Table 6. The study consists of a screening period (≤ 7 days), a 26-week initial evaluation period, and an extension period until the product is registered or approved (in accordance with country-specific regulations) or for up to 4.5 years, whichever occurs first, as illustrated in Figure 2.
Enrolment started on March 18, 2017, and is ongoing.21 The cut-off date for the data presented herein was July 2, 2019. As of the cut-off date, a total of 58 patients were included and 56 patients had received at least 1 weight-based dose of IV ravulizumab. The primary outcome was complete TMA response during the initial 26-week evaluation period, which was defined as the normalization of hematologic parameters (platelet count and LDH) and an improvement of at least 25% in serum creatinine from baseline. The secondary outcomes were hematologic normalization (platelet count and LDH), hematologic TMA parameters (platelet count, LDH, and hemoglobin), hemoglobin response (> 2% increase), dialysis requirement status, eGFR, CKD stage, fatigue, HRQoL, and safety outcomes. Health care resource utilization, patient-reported aHUS symptoms, and extrarenal signs and symptoms of aHUS were reported as exploratory outcomes.
Major protocol deviations were reported in 43 patients (74.1%) in Study 311. Among those with protocol violations, 25 patients (43.1%) were in the eligibility and entry criteria and 15 patients (25.9%) were in the category of SAE reporting criteria (Table 39).
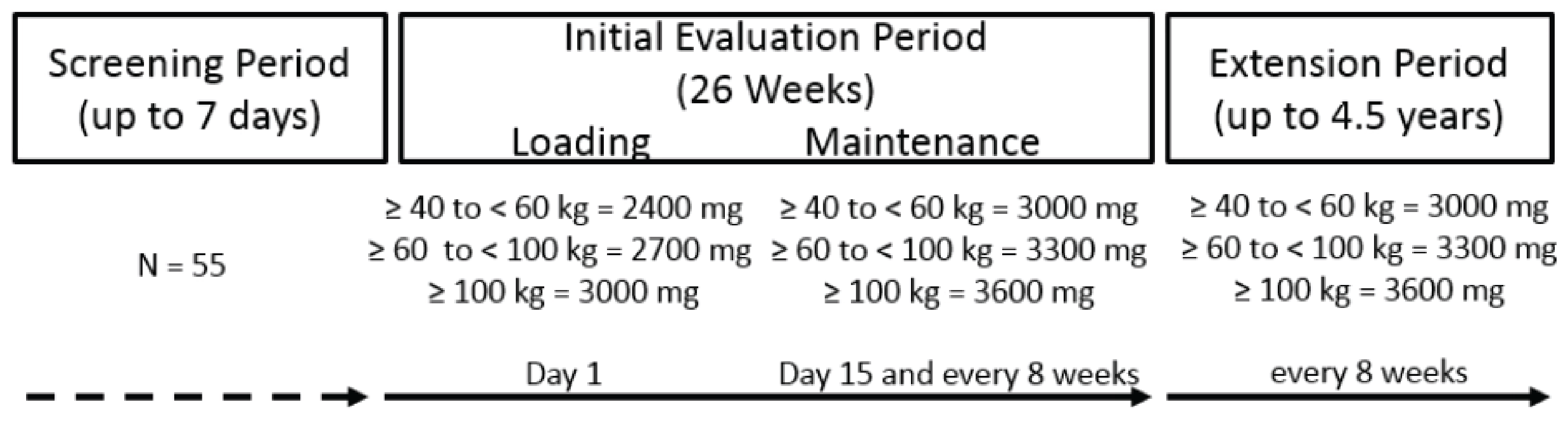
Study 312
Study 312 is an ongoing, phase III, prospective, multicentre, single-arm, open-label trial conducted in pediatric patients (aged < 18 years) with aHUS.22 Study 312 included 2 cohorts (cohort 1 and cohort 2). Cohort 1 included 21 complement inhibitor–naive children (aged < 18 years) with aHUS. The key objective for cohort 1 was to evaluate the safety and efficacy of ravulizumab (IV infusion) in complement inhibitor–naive children (aged < 18 years) with aHUS. Cohort 2 included 10 children (aged < 18 years) with aHUS who had been treated with eculizumab. The key objective for cohort 2 was to evaluate the safety and efficacy of ravulizumab (IV infusion) in children (aged < 18 years) with aHUS with stable TMA parameters following a switch from eculizumab to ravulizumab treatment. Study 312 was conducted at 20 sites in 8 countries (including the US and 7 countries in Europe and Asia; there were no sites in Canada). The key characteristics of the study design are summarized in Table 7 The study consists of a screening period (≤ 7 days), a 26-week initial evaluation period, and an extension period lasting until the product is registered or approved (in accordance with country-specific regulations) or for up to 4.5 years, whichever occurs first, as illustrated in Figure 3.
The enrolment for this study started on September 1, 2017, and is ongoing.22 The cut-off date for the data presented herein was December 3, 2019. As of the cut-off date, a total of 21 patients were included in Study 312, cohort 1, and 18 patients had received at least 1 weight-based dose of IV ravulizumab. A total of 10 patients were included in cohort 2, and all received at least 1 dose of ravulizumab. The primary outcome was complete TMA response during the initial 26-week evaluation period among patients in cohort 1; response was defined as the normalization of hematologic parameters (platelet count and LDH) and an improvement of greater than or equal to 25% in serum creatinine from baseline. The secondary outcomes were hematologic normalization (platelet count and LDH), hematologic TMA parameters (platelet count, LDH, and hemoglobin for cohort 1 only), hemoglobin response (> 2% increase; cohort 1 only), dialysis requirement status, eGFR, CKD stage, fatigue, and safety outcomes. Health care resource utilization, patient-reported aHUS symptoms, and extrarenal signs and symptoms of aHUS were reported as exploratory outcomes.
Major protocol deviations were reported in 14 patients (66.7%) in cohort 1. Among those with protocol violations, 9 patients (42.9%) were in the eligibility and entry criteria category and 7 patients (33.3%) were in the category of SAE reporting criteria (Table 39). No major protocol violations were reported in cohort 2.
Figure 3: Study Design Schematic for Study 312 (Cohort 1 and Cohort 2)
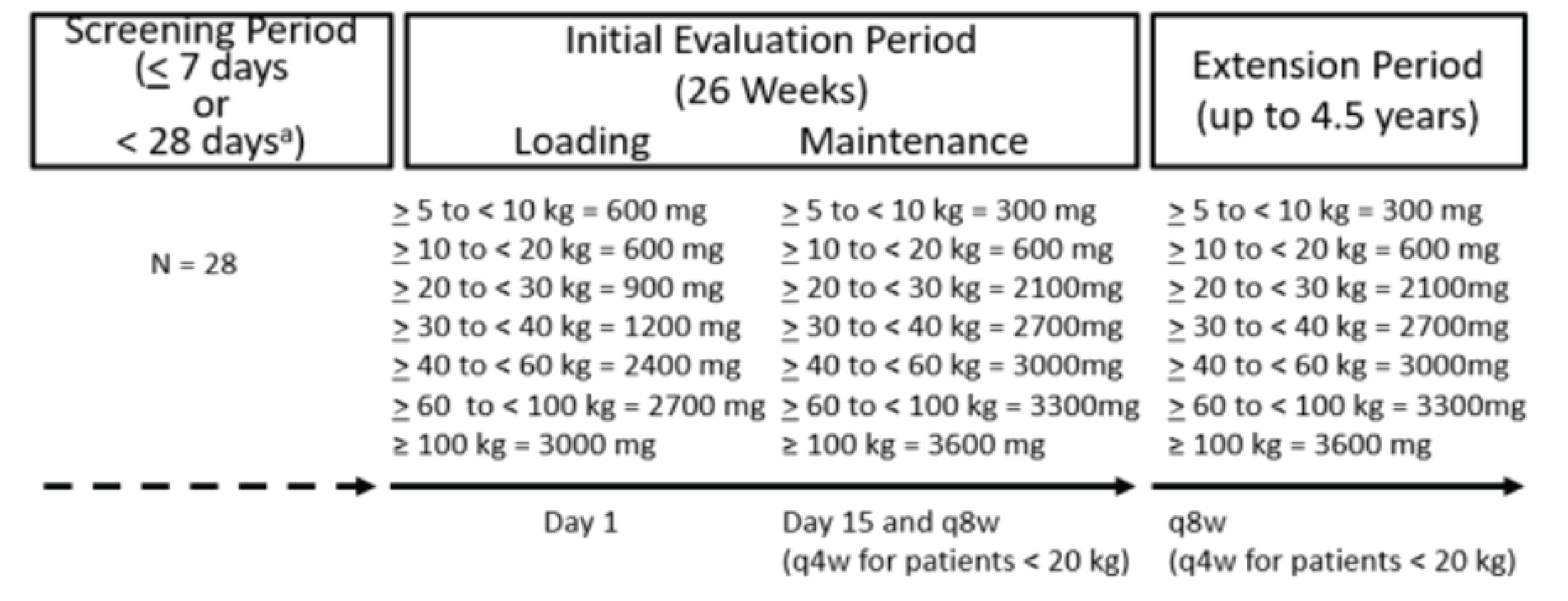
q.4.w. = every 4 weeks; q.8.w. = every 8 weeks.
Source: Study 312 Clinical Study Report.22
Populations
Inclusion and Exclusion Criteria
Study 311
Eligible patients were adults (aged 18 years and older) weighing at least 40 kg with evidence of active TMA during the screening period or within 28 days before the start of the screening period, defined as: platelet count less than 150,000/μL; lactate dehydrogenase greater than or equal to 1.5 times the upper limit of normal (ULN) and hemoglobin less than or equal to the lower limit of normal for age and gender; and serum creatinine level greater than or equal to the ULN during the screening period. (Patients who required dialysis for acute kidney injury were also eligible.) Patients with renal transplant were permitted, but must have had either a prior history of aHUS or persistent evidence of TMA in the 4 days after modifying the dose of calcineurin inhibitors or mammalian target of rapamycin inhibitors. Postpartum patients were permitted, but must have had either a prior history of aHUS or persistent evidence of TMA for more than 3 days after childbirth. Patients must have received meningococcal vaccination at the time of starting ravulizumab and were required to receive treatment doses of antibiotics from the time of the first dose of ravulizumab until at least 2 weeks after vaccination.
Key exclusion criteria were: a deficiency of ADAMTS13 (activity < 5%, suggestive of TTP); known Shiga toxin–related HUS; and other HUSs, such as drug exposure–related HUS with a positive direct Coombs test. Patients receiving immunosuppressive therapies were excluded unless they were part of an established post-transplant antirejection regimen, had confirmed anticomplement antibodies, or were using steroids for a different condition. Patients receiving plasma exchange and/or plasma infusion for a period of 28 days or longer before screening, or who were on chronic dialysis at screening, were excluded. Among patients without a kidney transplant, history of kidney disease suggesting an underlying disease other than aHUS were excluded.
Study 312
Study 312 included pediatric patients (aged < 18 years with > 5 kg body weight). For cohort 1, the key inclusion and exclusion criteria were the same as those for Study 311. Cohort 2 included children with a documented diagnosis of aHUS who were treated with eculizumab for at least 90 days before screening and showed clinical evidence of response, indicated by stable TMA parameters at screening, including LDH lower than 1.5 times ULN; platelet count 150,000/μL or higher; and eGFR greater than 30 mL/min/1.73 m2 using the Schwartz formula. The key exclusion criteria for cohort 2 were the same as those for cohort 1 (Table 7).
Baseline Characteristics
Study 311
The main baseline demographics and disease characteristics of the 56 adult patients (for the FAS population) in the trial are summarized in Table 8 and Table 9. In the FAS, the median age (years) at time of first aHUS symptoms was 40.1 years (range, 9.3 years to 76.6 years). The median age at the time of first infusion was 40.1 years (range, 19.5 years to 76.6 years). Thirty-seven patients (66.1%) were female. A total of 51.8% of patients were white and 26.8% were Asian. At baseline, 30 patients (53.6%) met the protocol-specified TMA criteria at day 1, based on central laboratory results. Genetic mutations were present in 2 patients (3.6%), while 52 patients (92.9%) presented without gene mutations (2 patients had unknown status). The median time from the first aHUS symptom to the first dose of ravulizumab was 0.28 months (range, 0 months to 215.0 months).
The baseline median platelet level was 95.25 × 109/L. The baseline median serum LDH level was 508.00 U/L. The baseline median eGFR was 10.00 mL/min/1.73 m2. At baseline, 39 patients (72.2%) presented with CKD stage 5, and 9 patients (16.7%) presented with CKD stage 4. At baseline, 29 patients (51.8%) were on dialysis.
Eight patients (14.3%) had received a kidney transplant before entering the study, but none of these transplants were related to aHUS. Prior to the study, the numbers (proportions) of patients who had received plasma exchange and/or plasma infusion (related to the current TMA), packed RBC transfusions, and platelet transfusions were 48 patients (82.8%), 17 patients (29.3%), and 6 patients (10.3%), respectively. Three patients (5.2%) received selective immunosuppressants (not eculizumab) before the study.
The majority of patients (92.9%) had pretreatment extrarenal signs or symptoms of aHUS. At the time of the first dose of the study drug, 48 of 56 patients (85.7%) were hospitalized due to aHUS (refer to Table 36). Fifty-three patients (94.6%) in the FAS had previous hospitalizations and/or emergency room visits due to aHUS. Twenty-seven patients (50.9%) had received intensive care, with a mean duration of stay in the intensive care unit of 10.1 days (SD = 10.0 days).
Study 312, Cohort 1
The main baseline demographics and disease characteristics of the 18 pediatric patients in cohort 1 (for the FAS population) are summarized in Table 8 and Table 9. The median age (years) at the time of first aHUS symptoms was 4.75 years (range, 0.8 years to 14.7 years). The median age at the time of first infusion was 5.2 years (range, 0.9 years to 17.3 years). Ten patients (55.6%) were female. A total of 50% of patients were white, and 27.8% were Asian. All 18 patients (100%) presented without gene mutation. The median time from the first aHUS symptom to the first dose of ravulizumab was not reported.
The baseline median platelet level was 51.25 × 109/L. The baseline median serum LDH level was 1963.00 U/L. The baseline median eGFR was 22.0 mL/min/1.73 m2. At baseline, a total of 6 patients (33.3%) presented with CKD stage 5 and 8 patients (44.4%) presented with CKD stage 4. At baseline, patients 6 (33.3%) were on dialysis.
One patient had received a kidney transplant (related to aHUS) before entering the study. Prior to the study, no patients received plasma exchange and/or plasma infusion related to the current TMA. The numbers (proportions) of patients who received packed RBC transfusions and platelet transfusions were 12 patients (57.1%) and 4 patients (19%), respectively. One patient (4.8%) received selective immunosuppressants (not eculizumab) before the study.
Thirteen patients (72.2%) had pretreatment extrarenal signs or symptoms of aHUS at baseline (refer to Table 36). All 18 patients had experienced a hospitalization and/or emergency room visit due to aHUS before the start of screening. Prior to screening, 7 patients (38.9%) had received intensive care during their hospitalizations due to aHUS, with a mean duration of stay in the intensive care unit of 9.0 days (SD = 17.68 days). At the time of the first dose of study drug, 17 patients (94.4%) were hospitalized due to aHUS.
Study 312, Cohort 2
For cohort 2, the main baseline demographics and disease characteristics of the 10 pediatric patients with stable TMA who were eculizumab-treated (for the FAS population) in the trial are summarized in Table 8 and Table 9. The median age (years) at time of first aHUS symptoms was 4.70 years (range, 0.4 years to 8.3 years). The median age at the time of first infusion was 12.5 years (range, 12 years to 15.5 years). Nine patients (90%) were male and 1 patient (10%) was female. A total of 50% of patients were white, and 40% were Asian. The median time from the first aHUS symptom to the first dose of ravulizumab was not reported.
The baseline median platelet level was 281.75 × 109/L (range = 207 × 109/L to 415.5 × 109/L). The baseline median serum LDH level was 206.50 U/L (range, 138.5 U/L to 356 U/L). The baseline median eGFR was 99.75 mL/min/1.73 m2 (range, 54 mL/min/1.73 m2 to 136.5 mL/min/1.73 m2 [normal range, ≥ 60 mL/min/1.73 m2]). No patients presented with CKD stage 5 or stage 4. No patient was on dialysis.
One patient had received a kidney transplant (related to aHUS) before entering the study. Prior to the study, no patients had received plasma exchange and/or plasma infusion related to the current TMA. All 10 patients received and responded to the eculizumab treatment.
One of the 10 patients had pretreatment extrarenal signs or symptoms of aHUS at baseline. None of the 10 patients had experienced a hospitalization and/or emergency room visit due to aHUS before start of screening.
Table 8: Baseline Demographic Characteristics (FAS)
Variable | Study 311 (N = 56) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
Age at time of first infusion (years) | |||
Median (minimum, maximum) | 40.1 (19.5, 76.6) | 5.2 (0.9, 17.3) | 12.5 (1.2, 15.5) |
Sex, n (%) | |||
Male | 19 (33.9) | 8 (44.4) | 9 (90) |
Female | 37 (66.1) | 10 (55.6) | 1 (10.0) |
Race, n (%)a | |||
Asian | 15 (26.8) | 5 (27.8) | 4 (40.0) |
White | 29 (51.8) | 9 (50.0) | 5 (50.0) |
Other | 4 (7.1) | 4 (22.3) | 1 (10.0) |
Unknown | 8 (14.3) | 1 (5.6) | 0 (0.0) |
Weight at time of first infusion (kg) | |||
n | 55 | 18 | 10 |
Median (minimum, maximum) | 67.7 (46.1, 111.6) | 16.7 (8.4, 69.3) | 47.8 (8.82, 69) |
Met TMA criteriab at day 1 (based on central laboratory results) | 30 (53.6) | NR | NR |
FAS = full analysis set; LDH = lactate dehydrogenase; LLN = lower limit of normal; NR = not reported; TMA = thrombotic microangiopathy; ULN = upper limit of normal.
aPatients can have multiple races selected.
bPlatelet count less than 150,000/μL; LDH greater than or equal to 1.5 times the ULN; hemoglobin less than or equal to the LLN; serum creatinine level greater than or equal to the ULN.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Table 9: Disease Characteristics (FAS)
Variable | Study 311 (N = 56) | Study 312 | Study 312 |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
Presence of gene mutations, n (%) | |||
Yes (ever positive) | 2 (3.6) | 0 (0.0) | NR |
No (always negative) | 52 (92.9) | 18 (100.0) | NR |
Unknown | 2 (3.6) | 0 (0.0) | NR |
Median age (years) at time of first aHUS symptoms (minimum, maximum) | 40.1 (9.3, 76.6) | 4.8 (0.8, 14.7) | 4.7 (0.4, 8.3) |
Median duration from first aHUS symptom to first dose of ravulizumab, months (minimum, maximum) | 0.28 (0, 215.0) | NR | NR |
Dialysis at baseline,a n (%) | 29 (51.8) | 6 (33.3) | 0 (0.0) |
Any kidney transplant before entering the study,b n (%) | 8 (14.3) | 1 (5.6) | 1 (10.0) |
Any kidney transplant before entering the studyb related to aHUS, n (%) | 0 (0.0) | 1 (100.0)c | 1 (100.0)c |
Number of patients with onset of TMA postpartum who entered the study with persistent evidence of TMA for > 3 days after childbirth | 8 (14.3) | NR | NR |
Baseline platelets (109/L) blood (normal range, 130 × 109/L to 400 × 109/L)d | |||
Mean (SD) | 118.52 (86.4) | 60.39 (32.6) | 287.90 (74.6) |
Median (minimum, maximum) | 95.25 (18, 4) | 51.25 (14, 125) | 281.75 (207, 415.5) |
Baseline LDH (U/L) serum (normal range, 120 U/L to 246 U/L)d | |||
Mean (SD) | 702.38 (557.9) | 2,223.47 (1,321.1) | 219.40 (56.9) |
Median (minimum, maximum) | 508.00 (229.5, 3,249) | 1963.00 (772, 4,985) | 206.5 (138.5, 356) |
Baseline hemoglobin (g/L) blood (overall normal range, 130 g/L to 175 g/L)d | |||
Mean (SD) | 86.26 (14.9) | 74.42 (17.4) | 131.50 (11.3) |
Median (minimum, maximum) | 85.00 (60.5, 140) | 74.25 (32, 106) | 132.00 (114.5, 148) |
Baseline eGFR (mL/min/1.73 m2) (normal range ≥ 60 mL/min/1.73 m2)d | |||
Mean (SD) | 15.86 (14.8) | 26.4 (21.2) | 104.90 (29.5) |
Median (minimum, maximum) | 10.00 (4, 80) | 22.0 (10, 84) | 99.75 (54, 136.5) |
Baseline serum creatine (μmol/L) | NR | NR | NR |
Baseline CKD stage, n (%)e | N = 54 | N = 18 | N = 10 |
1 | 0 (0.0) | 0 (0.0) | 8 (80.0) |
2 | 3 (5.6) | 2 (11.1) | 1 (10.0) |
3A | 1 (1.9) | 1 (5.6) | 1 (10.0) |
3B | 2 (3.7) | 1 (5.6) | 0 (0.0) |
4 | 9 (16.7) | 8 (44.4) | 0 (0.0) |
5 | 39 (72.2) | 6 (33.3) | 0 (0.0) |
Relevant prior medication and/or treatment | |||
Plasma exchange and/or plasma infusion related to the current TMA before the first dose of study drug | |||
Yes | 48 (82.8) | 0 (0.0) | 0 (0.0) |
No | 10 (17.2) | 10 (55.6) | 10 (100.0) |
Packed red blood cell transfusions | 17 (29.3) | 12 (57.1) | NR |
Platelet transfusions before the study | 6 (10.3) | 4 (19.0) | NR |
Selective immunosuppressants | 3 (5.2) | 1 (4.8) | 10 (100.0) |
Eculizumab | 0 (0.0) | 0 (0.0) | 10 (100.0) |
Glucocorticoids | 26 (44.8) | 4 (19.0) | NR (10.0) |
Baseline duration of dialysis | NR | NR | NR |
Duration of history of plasma therapy | NR | NR | NR |
Glucocorticoids for empiric treatment before the diagnosis of aHUS | 9 (15.5%) | NR | NR |
aHUS = atypical hemolytic uremic syndrome; CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; NR = not reported; SD = standard deviation; TMA = thrombotic microangiopathy.
aDialysis at baseline was recorded as “yes” for patients who had received dialysis within 5 days before study drug initiation.
bThe percentage was based on the total number of patients.
cThe percentage meant 1 of 1 patient (100%) who had a kidney transplant.
dThe normal value was provided in the Study 311 Clinical Study Report only for adult patients. The normal value was not provided in the Study 312 Clinical Study Report.
eBaseline CKD stage was available for 54 patients.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.12,22
Interventions
Study 311
A total 56 of 58 enrolled patients received the IV ravulizumab treatment. An interactive voice and/or web response system was used to assign vials containing ravulizumab to each patient. During the initial 26-week initial evaluation period, patients received a weight-based loading dose (refer to Figure 2) of ravulizumab IV on day 1, then every 8 weeks (all patients were ≥ 40 kg and received body weight-based maintenance doses on days 15, 71, and 127). After the 26-week initial evaluation period, all patients rolled into an extension period during which they received ravulizumab every 8 weeks (refer to Figure 2). A patient who discontinued and restarted ravulizumab on a scheduled study visit received a loading dose, a supplemental maintenance dose 2 weeks later, and a maintenance dose 6 weeks later, resuming an every-8-weeks regimen thereafter. If the decision to re-treat with ravulizumab occurred between scheduled study visits, the dosing regimen was to be determined by the Alexion medical monitor and the investigator. In the extension period, patients receive ravulizumab until product registration or approval (in accordance with country-specific regulations) or for up to 4.5 years, whichever occurs first (refer to Figure 2).
Study 312
In cohort 1, a total of 18 patients of 21 patients received the intended ravulizumab treatment. The dosing regimen was the same as that described for Study 311, except that the frequency of the body weight–based dosing regimen was every 8 weeks for patients weighing greater than or equal 20 kg and every 4 weeks for patients weighing less than 20 kg (refer to Figure 3).
In cohort 2, all 10 patients received the intended ravulizumab treatment. Day 1 of the study treatment occurred 14 days from the patient’s last dose of eculizumab. Changes in dosing regimen (i.e., dose amount [mg] or dose frequency [every 4 weeks or every 8 weeks]) were based on the same weight-based regimen (refer to Figure 3). Patients changing from every 4 weeks to every 8 weeks (i.e., those weighing 20 kg or more) or from every 8 weeks to every 4 weeks (i.e., those weighing less than 20 kg) were administered their first dose of the new scheduled dosing regimen (i.e., every 4 weeks or every 8 weeks) on the first ravulizumab administration day.
If the investigator and Alexion medical monitor mutually agreed that a patient would potentially benefit from a supplemental dose of ravulizumab, this supplemental dose may have been administered and the decision was documented. If the investigator and Alexion medical monitor mutually agreed that the infusion volume (120 mL) of the loading dose for patients weighing 5 kg to 9.9 kg (i.e., 600 mg) was too high for an individual patient, this dose may have been administered as 2 separate infusions no more than approximately 24 hours apart. This decision was also documented.
Concomitant Therapy
In both Study 311 and Study 312, concomitant medications were considered to be those the patient received from the first infusion of ravulizumab through 56 days after the patient’s last dose of the study drug. Any concomitant medication deemed necessary for the patient’s standard of care during the study, or for the treatment of any adverse event, was given at the discretion of the investigator.
All patients in the study (even those who had discontinued Study 311 and Study 312 in the extension period, but remained in the study) were prohibited from receiving any of the following medications and procedures from the first dose of the study drug until the completion of the study or early termination of the patient from the study:
eculizumab or other complement inhibitors
other investigational drugs or devices as part of a clinical trial
IV immunoglobulin (unless for an unrelated medical need, e.g., hypogammaglobinemia)
rituximab
plasma exchange and/or plasma infusion
new dialysis within the first 48-hour period following the first dose given in Study 311 and Study 312, unless there was a compelling medical need, such as in cases of hypervolemia unresponsive to diuretics, refractory electrolyte imbalance, or new-onset uremic encephalopathy (exceptions had to be approved on a case-by-case basis by Alexion before dialysis).
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 10. These end points are subsequently summarized. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 5.
The primary outcome for both Study 311 and Study 312, cohort 1 was complete TMA response (i.e., normalization of platelets and LDH and a 25% serum creatinine improvement). Patients must have met all the criteria for complete TMA response at 2 separate assessments obtained at least 4 weeks (28 days) apart, and at any measurement in between.
The secondary outcomes were hematological parameters (i.e., platelets, LDH, hemoglobin), time to complete TMA response, HRQoL, fatigue, renal function (e.g., eGFR, change in CKD stage, progression to end-stage renal disease), and dialysis-free status. The exploratory outcomes included symptom reduction and hospitalization.
The outcomes were measured at each visit.
Adverse events were assessed throughout both studies.21,22 A TEAE was defined as any adverse event that started during or after the first infusion of the study drug. Adverse events that started 56 days or more after the last dose of the study drug were not considered to be TEAEs. Adverse events were coded using MedDRA Version 21.0. The severity of adverse events was graded using version 4.03 of the Common Terminology Criteria for Adverse Events.21,22
Table 10: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 311 | Study 312 | |
|---|---|---|---|
Cohort 1 | Cohort 2 | ||
Mortality | Not reported as an efficacy outcome, but reported as an AE | ||
Complete TMA response at week 26 | Primary | Primary | Not applicable |
Hematologic normalization (platelet count and LDH) | Secondary | Secondary | Not applicable |
Hematologic TMA parameters (platelet count, LDH, and Hb) | Secondary | Secondary | Secondary |
Hemoglobin response (≥ 20 g/L from baseline) | Secondary | Secondary | NR |
Time to complete TMA response | Secondary | Secondary | None |
Presence of bleeding | NR | NR | NR |
Fatigue (FACIT-F) | Secondary | Secondary | Secondary |
HRQoL (3-Level EQ-5D) | Secondary | NR | NR |
Symptom reduction | Exploratory | Exploratory | Exploratory |
Renal function | |||
eGFR | Secondary | Secondary | Secondary |
CKD stage | Secondary | Secondary | Secondary |
Dialysis requirement status | Secondary | Secondary | Secondary |
Plasma therapy–free status | NR | NR | NR |
Packed red blood cell transfusion | NR | NR | NR |
Hospitalization | Exploratory | Exploratory | Exploratory |
Soluble membrane attack complex | NR | NR | NR |
AE = adverse event; CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; NR = not reported; TMA = thrombotic microangiopathy.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Statistical Analysis
The statistical analyses for Study 311 and Study 312 are described herein.
No formal hypothesis testing was planned in the 2 pivotal trials. An interim analysis was planned at the end of the initial 26-week evaluation period after all patients had completed or withdrawn from this phase of the study. This analysis allowed for the evaluation of the primary end point (i.e., complete TMA response). Additionally, a second analysis to summarize long-term efficacy and overall safety was performed at the data-cut-off dates of July 2, 2019, for Study 311 and December 3, 2019, for Study 312.
Power Calculation
Sample size and power calculations were not performed. However, the plan was to enrol approximately 55 patients in Study 311 and 23 to 28 patients in Study 312.
Primary Analysis
Statistical analysis methods for the efficacy outcomes are shown in Table 11. No multiplicity control was performed because there was no formal hypothesis testing. Efficacy analyses were performed using the FAS, the primary efficacy population.
The primary efficacy outcome (complete TMA response at week 26) was assessed by calculating the point estimate and a 95% CI for the proportion of complete TMA responders in patients treated with ravulizumab. The 95% CI was based on the asymptotic Gaussian approximation method with a continuity correction.
Secondary Outcomes
Hematologic Normalization
The number and proportion of patients who achieved hematologic normalization, defined as the normalization of both platelet count and LDH, was summarized over time with a 2-sided 95% CI for each postbaseline time point.
Hematologic TMA Parameters
Hematologic TMA parameters (platelets, LDH, and hemoglobin) were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. A mixed model for repeated measures (MMRM) with the fixed, categorical effect of visit and fixed, continuous effect of the specific test’s baseline value as covariates was performed to test whether changes differed from 0 at each time point.
Hemoglobin Response
The number and proportion of patients with an increase in hemoglobin of 20 g/L or more from baseline observed at 2 separate assessments at least 4 weeks apart (and at any measurement in between) was summarized over time by presenting the number and proportion of responders along with a 2-sided 95% CI for each postbaseline time point.
Time to Complete TMA Response
For the secondary efficacy end point of time to complete TMA response, Kaplan-Meier cumulative distribution curves were generated along with 2-sided 95% CIs. Patients who did not have a response were censored at the date of last visit or study discontinuation when the analysis was performed.
Health-Related Quality of Life
Quality of life was evaluated using the 3-Level EQ-5D. These data were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. An MMRM with the fixed, categorical effect of visit and fixed, continuous effect of the specific test’s baseline value as covariates was performed to test whether changes differed from 0 at each time point.
Symptoms Reduction
Fatigue was assessed using the FACIT-F version 4. The FACIT-F data were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. An MMRM with the fixed, categorical effect of visit and fixed, continuous effect of the specific test’s baseline value as covariates was performed to test whether changes differed from 0 at each time point.
Dialysis Requirement Status
For patients requiring dialysis within 5 days before ravulizumab treatment initiation, the proportion of patients no longer requiring dialysis was summarized over time using proportions. A 2-sided 95% CI for the proportion receiving dialysis was provided.
eGFR Value and Change From Baseline
Kidney function evaluated by eGFR was summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. A value of 10 mL/min/1.73 m2 for eGFR was imputed for patients requiring dialysis for acute kidney injury. This summary was repeated by kidney transplant status at enrolment. An MMRM with the fixed, categorical effect of visit and fixed, continuous effect of the baseline value as covariates was performed to test whether changes differed from 0 at each time point.
CKD Stage
CKD stage (refer to Table 38) was summarized over time by presenting the number and proportion of patients who improved (excluding those with stage 1 at baseline, given that they cannot improve), worsened (excluding those with stage 5 at baseline, given that they cannot worsen), and stayed the same compared to their CKD stage at baseline. Stage 5 was considered the worst category, while stage 1 was considered the best category. A 2-sided 95% CI for the proportion was provided for each category.
Other Outcomes
Mortality, presence of bleeding, packed RBC transfusions, and soluble MAC level were not assessed as efficacy outcomes in the 2 pivotal studies (Study 311 and Study 312). Symptoms (aside from fatigue) and hospitalization were reported on a by-patient basis in the 2 pivotal studies (CSRs) submitted by the sponsor; there were no summary data submitted. Therefore, symptom reduction and hospitalization have not been reported herein.
Subgroup and Sensitivity Analyses
Subgroup analyses were conducted only for the primary outcome, the complete TMA response. The primary efficacy analysis was repeated separately by the following main relevant subgroups: sex (male, female), age at enrolment (age 12 to 17 years, ≥ 18 years), kidney transplant history (yes, no), gene mutation status (ever positive, always negative), dialysis within 5 days before treatment initiation (yes, no), and whether or not they met all laboratory criteria for TMA, as determined by the central laboratory at day 1. Given that the number of patients in these subgroups may have been small, the CIs were based on exact confidence limits using the Clopper-Pearson method. No subgroup analyses were conducted for any secondary outcomes.
Sensitivity analyses were conducted only for the primary outcome (the complete TMA response). A sensitivity analysis was prespecified to evaluate a slightly modified version of complete TMA response. This modified complete TMA response applied only to the patients who were on dialysis at baseline (i.e., patients requiring dialysis within 5 days before the first dose of ravulizumab [N = 29 patients in Study 311 and N = 6 patients in Study 312, cohort 1]). For these patients, the criterion requiring an improvement from baseline of 25% or more in serum creatinine was replaced by a postbaseline change in dialysis status (from requiring dialysis at baseline to no longer requiring dialysis) that was maintained for at least 4 weeks. The definition of complete TMA response remained the same for all other patients (refer to Table 11). Primary and secondary end points were analyzed on the per protocol set as a sensitivity analysis to observe whether any substantial differences existed in the outcomes for this population compared to the FAS.
Handling of Dropouts and Missing Data
For the evaluation of complete TMA response during the 26-week initial evaluation period (i.e., the primary outcome), patients who missed an efficacy assessment that was part of the definition of complete TMA response while still in the study had their last observation carried forward. For patients who withdrew from the study before week 26, their data up to the time of withdrawal were used to assess complete TMA response. If a day 1 pretreatment assessment was missing, the screening assessment was used as the baseline assessment.
Safety Outcomes
Only descriptive statistics of safety were presented, with evidence summarized based on frequencies and proportion of total patients. Separate summaries were provided for all adverse events, SAEs, and adverse events leading to discontinuation and dose modification. Deaths and their primary causes were summarized.
Table 11: Statistical Analysis of Efficacy End Points (Study 311 and/or Study 312, Where Applicable)
End point | Statistical model | Missing data approach | Sensitivity analyses |
|---|---|---|---|
Complete TMA response | This analysis was performed by calculating the point estimate and a 95% CI for the proportion of complete TMA responders in ravulizumab-treated patients. The 95% CI was based on the asymptotic Gaussian approximation method with a continuity correction. | LOCF | 1. Assessed complete TMA response in patients who met all TMA criteria (active TMA) on day 1 (note: this is reported as a subgroup analysis in this report; refer to Table 14) 2. Evaluated a modified version of complete TMA response for patients who were on dialysis at baseline 3. Conducted a per-protocol analysis |
Time to complete TMA response | Kaplan-Meier cumulative distribution curves were generated along with 2-sided 95% CIs. | LOCF | None |
Hematologic normalization | The number and proportion of patients who achieved hematologic normalization was summarized over time with a 2-sided 95% CI for each postbaseline time point. | LOCF | Per-protocol analysis |
Hematologic TMA parameters | Hematologic TMA parameters were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. An MMRM with the fixed, categorical effect of visit and fixed, continuous effect of the specific test’s baseline value as covariates was performed to test whether changes differed from 0 at each time point. | LOCF | Per-protocol analysis |
Hemoglobin response | The number and proportion of patients with a hemoglobin response was summarized over time by presenting the number and proportion of responders along with a 2-sided 95% CI for each postbaseline time point. | LOCF | Per-protocol analysis |
Dialysis requirement status | For patients requiring dialysis within 5 days before ravulizumab treatment initiation, the proportion of patients no longer requiring dialysis after treatment was summarized over time. A 2-sided 95% CI for the proportion receiving dialysis was provided. | None | None |
eGFR value and change from baseline | Kidney function evaluated by eGFR was summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. A value of 10 mL/min/1.73 m2 for eGFR was imputed for patients requiring dialysis for acute kidney injury. An MMRM with the fixed, categorical effect of visit and fixed, continuous effect of the baseline value as covariates may have been performed to test whether changes differed from 0 at each time point. | None | None |
CKD stage | CKD stage was summarized over time by presenting the number and proportion of patients who improved, worsened, or stayed the same compared to their CKD stage at baseline. A 2-sided 95% CI for the proportion was provided for each category. | None | None |
HRQoL | Quality of life was evaluated using the 3-Level EQ-5D. Data were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. | Included only patients with available data | None |
Fatigue | Fatigue was evaluated using version 4 of the FACIT-F. FACIT-F data were summarized at baseline and at each postbaseline time point using descriptive statistics for continuous variables for the observed value as well as the change from baseline. | Included only patients with available data | None |
CI = confidence interval; CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; HRQoL = health-related quality of life; LOCF = last observation carried forward; MMRM = mixed model for repeated measures; TMA = thrombotic microangiopathy.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Analysis Populations
FAS: The FAS was based on a modified intention-to-treat approach in which confirmation of eligibility may have occurred after patients received the study drug. This applied specifically to the inclusion criterion of increased serum creatinine confirmed by a central laboratory and to the following 2 exclusion criteria: known familial or acquired ADAMTS13 confirmed by a central or local laboratory and known Shiga toxin–related HUS confirmed by a central or local laboratory. Accordingly, the FAS included all patients who were determined to have met the previously described criteria, had received at least 1 dose of ravulizumab, and had at least 1 postbaseline efficacy assessment.
PP set: The PP set included all patients in the FAS who received 100% of the planned number of infusions during the 26-week initial evaluation period; did not take any prohibited medications or undergo any prohibited procedures; met the inclusion criteria related to evidence of TMA (i.e., including thrombocytopenia, evidence of hemolysis, and kidney injury, based on laboratory findings, as detailed in Table 6 and Table 7); were willing and able to give written informed consent and comply with the study visit schedule; and did not meet the following exclusion criteria (refer to Table 6 and Table 7):
positive direct Coombs test
presence or suspicion of active and untreated systemic bacterial infection that, in the investigator’s opinion, could confound an accurate diagnosis of aHUS or impede the ability to manage the disease
acute kidney dysfunction within 4 weeks of transplant consistent with the diagnosis of acute AMR according to Banff 2013 criteria,39 among patients with a kidney transplant
among patients without a kidney transplant, history of kidney disease other than aHUS (such as known kidney biopsy finding suggestive of underlying disease other than aHUS), or known kidney ultrasound finding consistent with an alternative diagnosis to aHUS, or known family history and/or genetic diagnosis of noncomplement-mediated genetic renal disease
identified drug exposure–related HUS
plasma exchange and/or plasma infusion for 28 days or longer before the start of screening for the current TMA
bone marrow transplant and/or hematopoietic stem cell transplant within the past 6 months
hemolytic uremic syndrome related to known genetic defects of cobalamin C metabolism
known systemic sclerosis (scleroderma), systemic lupus erythematosus, or antiphospholipid antibody positivity or syndrome
chronic dialysis (defined as dialysis on a regular basis as renal replacement therapy for ESKD)
participation in another interventional treatment study or use of any experimental therapy within 30 days before initiation of the study drug on day 1 in this study or within 5 half-lives of that investigational product, whichever was greater
prior use of eculizumab or other complement inhibitors
use of tranexamic acid within 7 days before screening.
Safety set: The safety set included all patients who received at least 1 dose of the study drug.
Results
Patient Disposition
Data related to patient disposition in Study 311 and Study 312 are presented in Table 12.
In Study 311, a total of 74 patients were screened, and 58 patients were included. Two treated patients were withdrawn from the study after receiving the first dose of ravulizumab; both were deemed ineligible because they tested positive for Shiga toxin based on stool tests. Their results became known when the local laboratory results were made available following the first dose of the study drug. Among the 56 patients who received at least 1 dose of ravulizumab, 49 patients (84.5%) completed the initial 26-week period; 7 patients (12.5%) discontinued from the treatment; and 9 patients (15.5%) discontinued from the study during the initial 26-week period. At the time of the data cut-off date (July 2, 2019), no patient had completed the treatment, and 17 patients (29.3%) had discontinued the study. The main reasons for discontinuation were withdrawal by patient (n = 5, 8.6%), adverse event (n = 3, 5.2%), physician decision (n = 3, 5.2%), death (n = 2, 3.4%), being deemed ineligible posttreatment (n = 2, 3.4%), and protocol violation (n = 2, 3.4%). A total of 41 patients (70.1%) were continuing in the extension period; 38 of these patients (65.5%) were continuing to receive the study drug.
In Study 312, cohort 1, a total of 21 patients were screened and included. All 21 patients received at least 1 dose of ravulizumab. Seventeen patients (81.0%) completed the initial 26-week period and 4 patients (19.0%) discontinued the study during the initial 26-week period. At the time of the data cut-off date (December 3, 2019), no patient had completed the treatment, and 5 patients (23.8%) had discontinued from both the treatment and the study. The main reasons for discontinuation of treatment were being deemed ineligible posttreatment (n = 2, 9.5%), adverse event (n = 1, 4.8%), physician decision (n = 1, 4.8%), and protocol violation (n = 1, 4.8%). A total of 16 patients (76.2%) were continuing in the extension period.
In Study 312, cohort 2, at the time of the data cut-off date, all 10 included patients had completed the initial 26-week evaluation period and were continuing in the extension period.
Table 12: Disposition of Patients (All Enrolled Patients)
Category | Study 311 (N = 58) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 21) | Cohort 2 (N = 10) | ||
Screened, n | 74 | 21 | NR |
Enrolled, n | 58 | 21 | 10 |
Treated, n (%) | 58 (100.0) | 21 (100.0) | 10 (100.0) |
Completed the initial evaluation period, n (%) | 49 (84.5) | 17 (81.0) | 10 (100.0) |
Completed study, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Discontinued from study treatment, n (%) | 20 (34.5) | 5 (23.8) | 0 (0.0) |
Adverse event | 3 (5.2) | 1 (4.8) | 0 (0.0) |
Noncompliance with study drug | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Physician decision | 5 (8.6) | 1 (4.8) | 0 (0.0) |
Pregnancy | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Protocol violation | 2 (3.4) | 1 (4.8) | 0 |
Patient decision | 5 (8.6) | 0 (0.0) | 0 (0.0) |
Lack of efficacy | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Othera | 5 (8.6) | 2 (9.5) | 0 (0.0) |
Discontinued from study, n (%) | 17 (29.3) | 5 (23.8) | 0 (0.0) |
Adverse event | 3 (5.2) | 1 (4.8) | 0 (0.0) |
Death | 2 (3.4) | 0 (0.0) | 0 (0.0) |
Lost to follow-up | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Noncompliance with study drug | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Physician decision | 3 (5.2) | 1 (4.8) | 0 (0.0) |
Pregnancy | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Protocol violation | 2 (3.4) | 1 (4.8) | 0 (0.0) |
Withdrawal by patient | 5 (8.6) | 0 (0.0) | 0 (0.0) |
Lack of efficacy | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Deemed ineligible posttreatment | 2 (3.4) | 2 (9.5) | 0 (0.0) |
Initial evaluation period (week 26), n (%) | |||
Discontinued from the study in initial evaluation period | 9 (15.5) | 4 (19.0) | 0 (0.0) |
Adverse event | 3 (5.2) | 1 (4.8) | 0 (0.0) |
Death | 2 (3.4) | 0 (0.0) | 0 (0.0) |
Physician decision | 1 (1.7) | 0 (0.0) | 0 (0.0) |
Protocol violation | 1 (1.7) | 1 (4.8) | 0 (0.0) |
Deemed ineligible posttreatment | 2 (3.4) | 2 (9.5) | 0 (0.0) |
Extension period, n (%) | |||
Entered the extension period | 49 (84.5) | 17 (81.0) | 10 (100.0) |
Completed | 0 (0.0) | 0 (0.0) | 10 (100.0) |
Discontinued from the study during the extension period | 8 (13.8) | 1 (4.8) | 0 (0.0) |
Physician decision | 2 (3.4) | 1 (4.8) | 0 (0.0) |
Protocol violation | 1 (1.7) | 0 (0.0) | 0 (0.0) |
Withdrawal by patient | 5 (8.6) | 0 (0.0) | 0 (0.0) |
Number of patients ongoing in the extension period as of the data cut-off date (data as of July 2, 2019), n (%) | 41 (70.1) | 16 (76.2) | 10 (100.0) |
Number of patients ongoing and receiving ravulizumab in the extension period as of the data cut-off date (data as of July 2, 2019), n (%) | 38 (65.5) | NR | NR |
FAS, N (%) | 56 (96.7) | 18 (85.7) | 10 (100) |
PP, N (%) | 44 (75.9) | 18 (85.7) | 10 (100) |
Safety, N (%) | 58 (100) | 21 (100) | 10 (100) |
FAS = full analysis set; NR = not reported; PP = per protocol.
aTwo patients were discontinued from the study because they were deemed ineligible after receiving positive Shiga toxin test results (based on stool tests) after the first dose. Two patients discontinued the study due to death. One patient withdrew consent.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Exposure to Study Treatments
As of the data cut-off date, in Study 311, the median treatment duration in the safety set was 74.07 weeks (range, 0.57 weeks to 118.3 weeks) (refer to Table 40).
For Study 312, cohort 1, the median treatment duration in the safety set was 82.43 weeks (range, 1 week to 110.6 weeks) (refer to Table 40). For Study 312, cohort 2, the median treatment duration in the safety set was 52.29 weeks (range, 49.4 weeks to 58.7 weeks) (refer to Table 40).
Concomitant Treatments
In Study 311, a total of 17 patients (29.3% for the safety set) received packed RBC transfusions, and 3 patients (5.2%) received platelet transfusions; 3 patients (5.2%) received plasma exchange and/or plasma infusion during the study, which was prohibited per the protocol. These patients were discontinued from the study PP; 2 patients discontinued during the initial evaluation period and 1 patient discontinued during the extension period. Nine patients (15.5%) received concomitant blood substitutes and 9 patients (15.5%) received plasma protein fraction selective immunosuppressants. The main potential relevant concomitant medications used during the study are summarized in Table 37.
In Study 312, cohort 1, a total 4 patients (19%) received platelet transfusions. None of the patients received plasma exchange and/or plasma infusion during the study. A total of 2 patients (9.5%) received concomitant blood substitutes, and 6 patients (28.6%) received plasma protein fraction selective immunosuppressants (refer to Table 37).
In Study 312, cohort 2, 0 patients received plasma exchange and/or plasma infusion (refer to Table 37).
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 4 for detailed efficacy data.
Mortality
Mortality was not assessed as an efficacy outcome in the 2 pivotal studies (Study 311 and Study 312). The information on mortality was reported in the safety outcomes (refer to the Harms section and Table 27).21,22
Complete TMA Response
Study 311
At week 26, complete TMA response was observed in 30 patients of the 56 patients in the FAS (53.6%; 95% CI, 39.6% to 67.5%) (Table 13 and Figure 7). At the data cut-off date, complete TMA response was observed in 34 patients of the 56 patients in the FAS (60.7%; 95% CI, 47.0% to 74.4%).
Table 13: Complete TMA Response and Components of Complete TMA Response Analysis (Study 311 for Adults, FAS)
Outcomes | Week 26 | Extension period (median follow-up time: 75.6 weeks) | ||||
|---|---|---|---|---|---|---|
Total | Responder | Total | Responder | |||
N | n | Proportion, % (95% CI)a | N | n | Proportion, % (95% CI)a | |
Complete TMA response | 56 | 30 | 53.6 (39.6 to 67.5) | 56 | 34 | 60.7 (47.0 to 74.4) |
Components of complete TMA response | ||||||
Hematologic normalizationb | 56 | 41 | 73.2 (60.7 to 85.7) | 56 | 45 | 80.4 (69.1 to 91.7) |
Platelet count normalization | 56 | 47 | 83.9 (73.4 to 94.4) | 56 | 48 | 85.7 (75.7 to 95.8) |
LDH normalization | 56 | 43 | 76.8 (64.8 to 88.7) | 56 | 47 | 83.9 (73.4 to 94.4) |
≥ 25% improvement in serum creatinine from baseline | 56 | 33 | 58.9 (45.2 to 72.7) | 56 | 35 | 62.5 (48.9 to 76.1) |
CI = confidence interval; FAS = full analysis set; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
a95% CIs for the proportion were based on the asymptotic Gaussian approximation method with a continuity correction.
bHematologic normalization includes normalization of platelet count (≥ 150 × 109/L) and normalization of LDH (≤ 246 U/L).
Source: Study 311 Clinical Study Report.21
In the PP analysis set, the proportion of responders with complete TMA response was consistent with that in the primary analysis (FAS) at week 26 and at the data cut-off date (Table 42).
Study 311 Subgroup Analysis for Complete TMA Response
Prespecified subgroup analyses for complete TMA response at week 26 are presented in Table 14. At week 26, the complete TMA response rate was generally consistent across subgroups compared with the overall population (53.6%) (refer to Table 14 and Figure 9).
No subgroup analyses were conducted based on baseline platelet count, LDH level, serum creatinine, hemoglobin, duration of plasma therapy, or duration of dialysis before the study. Subgroup analysis was not conducted for the extension phase.
Table 14: Complete TMA Responder Analyses by Subgroups at Week 26 (FAS)
Subgroups | Study 311 (N = 56) | Study 312, cohort 1 | ||||
|---|---|---|---|---|---|---|
N | n | Responder, % (95%CI)a | N | n | Responder, % (95%CI)a | |
Overall primary analysis (FAS) | 56 | 30 | 53.6 (39.6 to 67.5) | 18 | 14 | 77.8 (52.4 to 93.6) |
Subgroups | ||||||
Presence of gene mutations | 54 | — | — | 18 | — | — |
Yes (ever positive) | 2 | 0 | 0.00 (0.00 to 84.2) | 0 | NA | NR |
No (always negative) | 52 | 30 | 57.7 (43.2 to 71.3) | 18 | 14 | 77.8 (52.4 to 93.6) |
Sex | 56 | — | — | 18 | — | — |
Female | 37 | 22 | 59.5 (42.1 to 75.2) | 10 | 7 | 70.0 (34.8 to 93.3) |
Male | 19 | 8 | 42.1 (20.3 to 66.5) | 8 | 7 | 87.5 (47.3 to 99.7) |
Active TMA on day 1 (baseline)b | 56 | — | NR | NR | NR | |
Yes | 30 | 14 | 46.7 (28.3 to 65.7) | NR | NR | NR |
No | 26 | 16 | 61.5 (40.6 to 79.8) | NR | NR | NR |
CKD stage | ||||||
Dialysis within 5 days before treatment initiation | 56 | — | — | 18 | — | — |
Yes | 29 | 14 | 48.3 (29.4 to 67.5) | 6 | 5 | 83.3 (35.9 to 99.6) |
No | 27 | 16 | 59.3 (38.8 to 77.6) | 12 | 9 | 75.0 (42.8 to 94.5) |
History of kidney transplant | 56 | — | — | 18 | — | — |
Yes | 8 | 2 | 25.0 (3.20 to 65.1) | 1 | 0 | 0.0 (0.0 to 97.5) |
No | 48 | 28 | 58.3 (43.2 to 72.4) | 17 | 14 | 82.4 (56.6 to 96.2) |
CI = confidence interval; CKD = chronic kidney disease; FAS = full analysis set; NA = not applicable; NR = not reported; TMA = thrombotic microangiopathy.
Notes: The proportion of complete TMA response is based on the responders among treated patients. The numerator is the number of patients achieving complete TMA response during the 26-week initial evaluation period, and the denominator is the number of patients in the FAS. Complete TMA response was not an outcome assessed for Study 312, cohort 2.
a95% CIs for the proportion are based on exact confidence limits using the Clopper-Pearson method.
bAlso reported as sensitivity analysis.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Study 311 Sensitivity Analyses for Complete TMA Response
A separate analysis was performed using a modified version of complete TMA response. This modified complete TMA response applied only to the patients who were on dialysis at baseline. For these patients, the criterion requiring an improvement from baseline of greater than or equal to 25% in serum creatinine was replaced by a postbaseline change in dialysis status (from requiring dialysis at baseline to no longer requiring dialysis after treatment) that was maintained for at least 4 weeks. The definition of complete TMA response remained the same for all other patients. At week 26, the modified complete TMA was observed in 32 patients of the 56 patients in the FAS (57.1%; 95% CI, 43.3% to 71.0%). As of the data cut-off date, modified complete TMA response was observed in 36 patients of the 56 patients in the FAS (64.3%; 95% CI, 50.8% to 77.7%).
Study 312, Cohort 1
In Study 312, cohort 1, at week 26, complete TMA response was observed in 14 patients of the 18 patients in the FAS (77.8%; 95% CI, 52.4% to 93.6%) (Table 15 and Figure 8). At the data cut-off date, complete TMA response was observed in 17 patients of the 18 patients in the FAS (94.4%; 95% CI, 72.7% to 99.9%).
Table 15: Complete TMA Response and Components of Complete TMA Response Analysis (Study 312, Cohort 1 FAS)
Outcomes | Week 26 | Extension period (median follow-up time = 84.2 weeks) | ||||
|---|---|---|---|---|---|---|
Responder | Responder | |||||
Total | n | Proportion, % (95% CI)a | Total | n | Proportion, % (95% CI)a | |
Complete TMA response | 18 | 14 | 77.8 (52.4 to 93.6) | 18 | 17 | 94.4 (72.7 to 99.9) |
Components of complete TMA response | ||||||
Hematologic normalization | 18 | 16 | 88.9 (65.3 to 98.6) | 18 | 17 | 94.4 (72.7 to 99.9) |
Platelet count normalization | 18 | 17 | 94.4 (72.7 to 99.9) | 18 | 17 | 94.4 (72.7 to 99.9) |
LDH normalization | 18 | 16 | 88.9 (65.3 to 98.6) | 18 | 17 | 94.4 (72.7 to 99.9) |
≥ 25% improvement in serum creatinine from baseline | 18 | 15 | 83.3 (58.6 to 96.4) | 18 | 17 | 94.4 (72.7 to 99.9) |
CI = confidence interval; FAS = full analysis set; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
a95% CIs for the proportion were based on exact confidence limits using the Clopper-Pearson method.
Source: Study 312 Clinical Study Report.22
The FAS number and PP set number were the same in Study 312. Therefore, no PP analysis was done.
Study 312, Cohort 2
TMA response was not relevant for this population.
Study 312, Cohort 1 Subgroup Analysis for Complete TMA Response
Prespecified subgroup analyses for complete TMA response at week 26 are presented in Table 14. At week 26, the complete TMA response rate was generally consistent across subgroups compared with the overall population (refer to Table 14 and Figure 10). No subgroup analyses were conducted based on baseline platelet count, LDH level, serum creatine, hemoglobin, duration of plasma therapy, or duration of dialysis before the study. Subgroup analysis was not conducted for the data cut-off date.
Study 312, Cohort 1 Sensitivity Analyses for Complete TMA Response
At week 26, the modified complete TMA was observed in 14 patients of the 18 patients in the FAS (77.8%; 95% CI, 52.4 to 93.6%). As of the data cut-off date, modified complete TMA response was observed in 17 patients of the 18 patients in the FAS (94.4%; 95% CI, 72.7% to 99.9%).
Complete TMA Response Status Over Time
Rates of complete TMA response status over time for Studies 311 and 312 are presented in Table 44 and Table 45, respectively. In Study 311, from the median time to complete TMA response (86 days), the proportion of responders was stable. After achieving complete TMA response, some patients had transient periods during which not all components of response continued to be met. In Study 312, for the 14 patients who achieved complete TMA response status during the initial evaluation period, these responses were sustained through the end of the 26-week initial evaluation period. After achieving complete TMA response, some patients had transient periods during which not all components of response continued to be met.
The complete TMA response components status over time for Study 311 and Study 312 are presented in Figure 11 to Figure 12 and Figure 13 to Figure 14, respectively (Appendix 4).
Hematologic Normalization
In Study 311, in the FAS, hematologic normalization was defined as the normalization of platelets and LDH. At week 26, hematologic normalization was observed in 41 patients of 56 patients in the FAS (73.2%; 95% CI, 60.7% to 85.7%) (Table 13 and Figure 11). As of the data cut-off date, hematologic normalization was observed in 45 patients of the 56 patients in the FAS (80.4%; 95% CI, 69.1% to 91.7%) (Table 13 and Figure 12). In the PP set, hematologic normalization was consistent with the primary analysis (FAS, Table 42).
In Study 312, cohort 1, at week 26, in the FAS, hematologic normalization was observed in 16 patients of the 18 patients (88.9%; 95% CI, 65.3% to 98.6%) (Table 15 and Figure 13). As of the data cut-off date, hematologic normalization was observed in 17 patients of the 18 patients in the FAS and PP sets (94.4%; 95% CI, 72.7% to 99.9%) (Table 15 and Figure 14).
Individual Hematologic Parameters
Study 311
Platelet Count: The mean platelet count improved after the initiation of ravulizumab treatment, increasing from 118.52 × 109/L (SD = 86.440 × 109/L) at baseline to 243.54 × 109/L (SD = 160.500 × 109/L) at day 8. The mean platelet count remained above 227 × 109/L at all subsequent visits in the 26-week period. The mean platelet count was 237.96 × 109/L (SD = 73.528 × 109/L) at day 183 (n = 48) and remained stable, at 205 × 109/L or higher, at all visits during the extension period. The mean platelet count was 241.56 × 109/L (67.523 × 109/L) at day 407 (n = 43).
Lactate Dehydrogenase: The mean LDH value decreased from baseline, with the majority of the decrease occurring during the first month of ravulizumab treatment; this mean reduction in LDH was sustained over a 26-week period. The mean LDH value decreased from 702.38 U/L (SD = 557.959) at baseline to 554.31 U/L (SD = 603.954 U/L) at day 8 and further to 293.27 U/L (SD = 156.999 U/L) at day 29. The mean LDH value remained below 250 U/L at all subsequent visits in the 26-week period. The mean LDH value was 194.46 U/L (SD = 58.099 U/L) at day 183 (n = 48) and remained below 215 U/L at all visits during the extension period. The mean LDH value was 192.86 U/L (SD = 67.536 U/L) at day 407 (n = 42).
Hemoglobin Change From Baseline: The mean hemoglobin value increased more gradually over time during the 26-week period. The mean hemoglobin value increased from 86.26 g/L (SD = 14.866 g/L) at baseline to 91.24 g/L (SD = 15.397 g/L) at day 15 and to 113.82 g/L (SD = 17.086 g/L) at day 57, with mean values remaining above this level at subsequent visits in the initial evaluation period. The mean hemoglobin value was 120.27 g/L (SD = 12.946 g/L) at day 183 (n = 48) and remained above 120 g/L at all visits during the extension period. The mean hemoglobin value was 125.21 g/L (SD = 15.557 g/L) at day 407 (n = 43).
Hemoglobin Response (> 20 g/L): During the initial evaluation period, 40 patients of the 56 patients in the FAS (71.4%; 95% CI, 58.7% to 84.2%) achieved a hemoglobin response (i.e., an increase in hemoglobin of ≥ 20 g/L compared to baseline with a confirmatory result) (Table 16). As of the data cut-off date, an additional 5 patients in the FAS achieved a hemoglobin response, bringing the total to 45 patients of 56 patients achieving a hemoglobin response (80.4%; 95% CI, 69.1% to 91.7%).
Table 16: Hemoglobin Response With a Confirmatory Result as of the Data Cut-Off Date (FAS, Study 311)
Parameter | Study 311 (N = 56) | Study 312, cohort 1(N = 18) | ||
|---|---|---|---|---|
n of N | Proportion (95% CI) | n of N | Proportion (95% CI) | |
Increase in hemoglobin of ≥ 20 g/L from baseline with a confirmatory result through week 26 | 40 of 56 | 0.714 (0.587 to 0.842)a | 16 of 18 | 0.889 (0.653 to 0.986) |
Increase in hemoglobin of ≥ 20 g/L from baseline with a confirmatory result through data cut-off date or EOS | 45 of 56 | 0.804 (0.691 to 0.917)a | 17 of 18 | 0.944 (0.727 to 0.999) |
Hemoglobin ≥ 20 g/L increase from baselineb | ||||
Day 8 | 0 of 53 | 0.000 (0.000 to 0.067) | 0 of 17 | 0.000 (0.000 to 0.195) |
Day 15 | 3/53 | 0.057 (0.012 to 0.157) | 3/17 | 0.176 (0.038 to 0.434) |
Day 183 | 37 of 49 | 0.755 (0.611 to 0.867) | 17 of 17 | 1.000 (0.805 to 1.000) |
Day 351 | 38 of 44 | 0.864 (0.726 to 0.948) | 16 of 17 | 0.941 (0.713 to 0.999) |
Day 743 | 7 of 8 | 0.875 (0.473 to 0.997) | 2 of 2 | 1.000 (0.158 to 1.000) |
Day 799 | 3 of 3 | 1.000 (0.292 to 1.000) | NR | NR |
CI = confidence interval; EOS = end of study; FAS = full analysis set; NR = not reported.
a95% CIs for the proportion are based on the asymptotic Gaussian approximation method with a continuity correction.
bFor hemoglobin increases of greater than or equal to 20 g/L at each visit, 95% CIs for the proportions were based on exact confidence limits using the Clopper-Pearson method.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Study 312, Cohort 1
Overall, patients in the FAS showed improvement in all hematologic TMA parameters (platelets, LDH, and hemoglobin) during the initial evaluation period.
Platelet Count: The mean platelet count increased from baseline early in treatment, and this mean improvement was sustained over the duration of the initial 26-week period. The mean platelet count improved after the initiation of ravulizumab treatment, increasing from 60.39 × 109/L (SD = 32.613 × 109/L) at baseline to 285.40 × 109/L (SD = 147.860 × 109/L) at day 8 and to 273.76 × 109/L (SD = 101.404 × 109/L) at day 22. The mean platelet count remained above 304 × 109/L at all subsequent visits in the initial 26 weeks. The mean platelet count was 304.94 × 109/L (SD = 75.711 × 109/L) at day 183 (n = 17) and remained greater than or equal to 218 × 109/L at all visits through the data cut-off date. The mean platelet count was 289.90 × 109/L (SD = 59.795 × 109/L) at day 575 (n = 10).
Lactate Dehydrogenase: The mean LDH value decreased from baseline, with the majority of the decrease occurring during the first month of ravulizumab treatment; this mean reduction in LDH was sustained over the duration of the initial 26 weeks. The mean LDH value decreased from 2,223.47 U/L (SD = 1,321.118 U/L) at baseline to 1,064.83 U/L (SD = 597.947 U/L) at day 8 and further to 326.94 U/L (SD = 121.606 U/L) at day 29. The mean LDH value remained at 311 U/L or lower at all subsequent visits in the initial 26 weeks. The mean LDH value was 262.41 U/L (SD = 59.995 U/L) at day 183 (n = 17) and remained at 262 U/L or lower at all visits through the data cut-off date. The mean LDH value was 248.18 U/L (SD = 53.822 U/L) at day 575 (n = 11).
Hemoglobin Change From Baseline: The mean hemoglobin value increased gradually over time during the initial 26 weeks. It increased from 74.42 g/L (SD = 17.387 g/L) at baseline to 86.93 g/L (SD = 16.589 g/L) at day 15 and 113.06 g/L (SD = 16.634 g/L) at day 57, with mean values remaining above this level at subsequent visits in the initial 26 weeks. The mean hemoglobin value was 120.06 g/L (SD = 8.011 g/L) at day 183 (n = 17) and remained above 114 g/L at all visits through the data cut-off date. The mean (SD) hemoglobin was 120.30 g/L (SD = 9.787 g/L) at day 575 (n = 10).
Hemoglobin Response (Increase ≥ 20 g/L): During the initial evaluation period, 16 patients of the 18 patients in the FAS (88.9%; 95% CI, 65.3% to 98.6%) (Table 16) had an increase in hemoglobin of greater than or equal to 20 g/L compared to baseline with a confirmatory result (i.e., a hemoglobin response). Of the 17 patients who completed the 26 weeks of ravulizumab treatment, 16 patients had a hemoglobin response as of day 99 (Table 16). As of the data cut-off date, 1 additional patient had achieved a hemoglobin response. At the day 575 visit, 10 patients of 11 patients (90.9%; 95% CI, 58.7% to 99.8%) had achieved a hemoglobin response. Of the 2 patients with data through day 743, each has maintained their hemoglobin response.
Study 312, Cohort 2
Hematologic TMA parameters (platelet count, LDH, and hemoglobin) remained stable for patients in cohort 2 during the initial 26 weeks as well as through the data cut-off date (refer to Figure 14, Figure 15, and Figure 16).
Time to Complete TMA Response
Study 311
As of the data cut-off date, complete TMA response was achieved at a median time of 86 days and occurred as early as 7 days following the first dose of ravulizumab (Figure 4). Four patients achieved a complete TMA response during the extension period. The latest response was observed at 401 days.
Figure 4: Time to Complete TMA Response — Kaplan-Meier Cumulative Distribution Curves as of Data Cut-Off Date (Study 311, FAS)
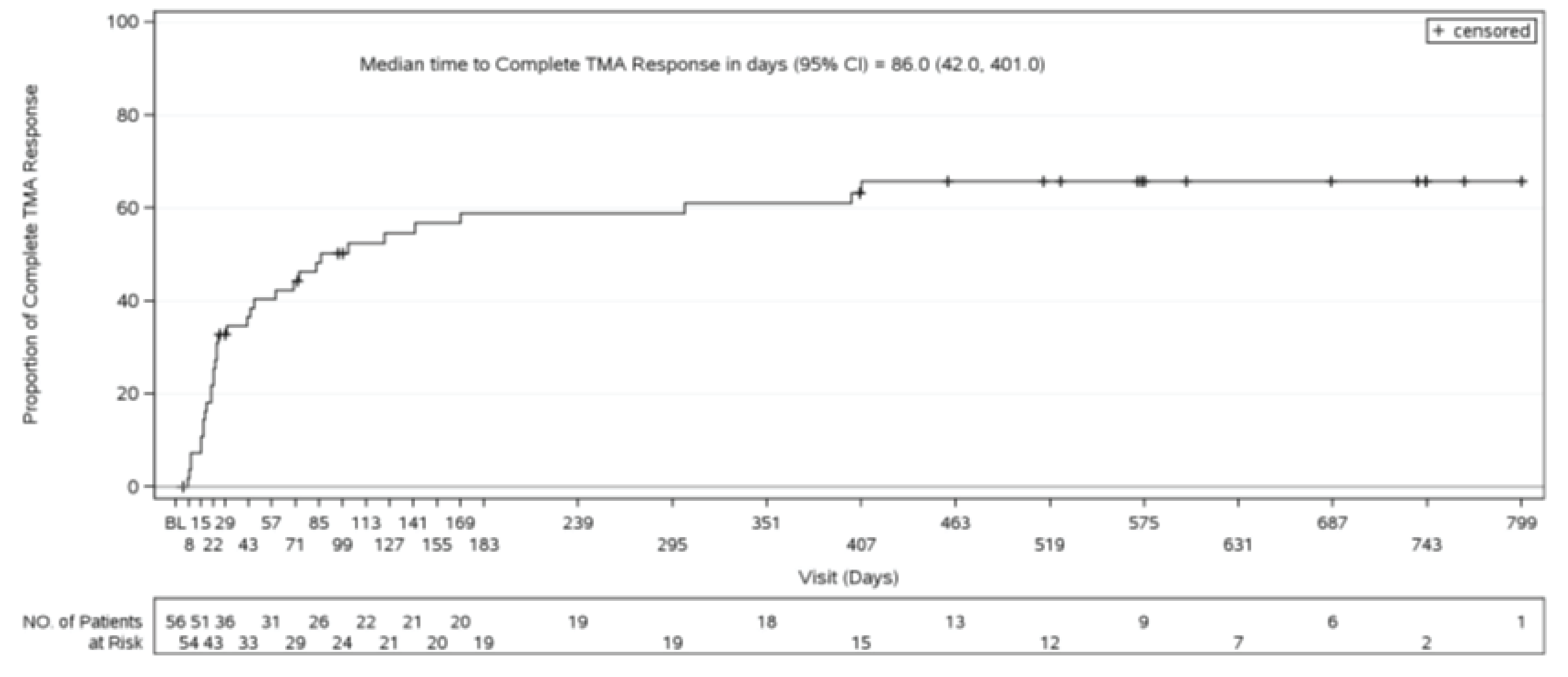
BL = baseline; CI = confidence interval; FAS = full analysis set; NO. = number; TMA = thrombotic microangiopathy.
Source: Study 311 Clinical Study Report.21
Study 312, Cohort 1
The median time to complete TMA response was 30 days and occurred as early as 15 days following the first dose of ravulizumab (Figure 5). Three patients achieved a complete TMA response during the extension period. The latest response was observed at 351 days.
Figure 5: Time to Complete TMA Response — Kaplan-Meier Cumulative Distribution Curves (Study 312, Cohort 1, FAS)
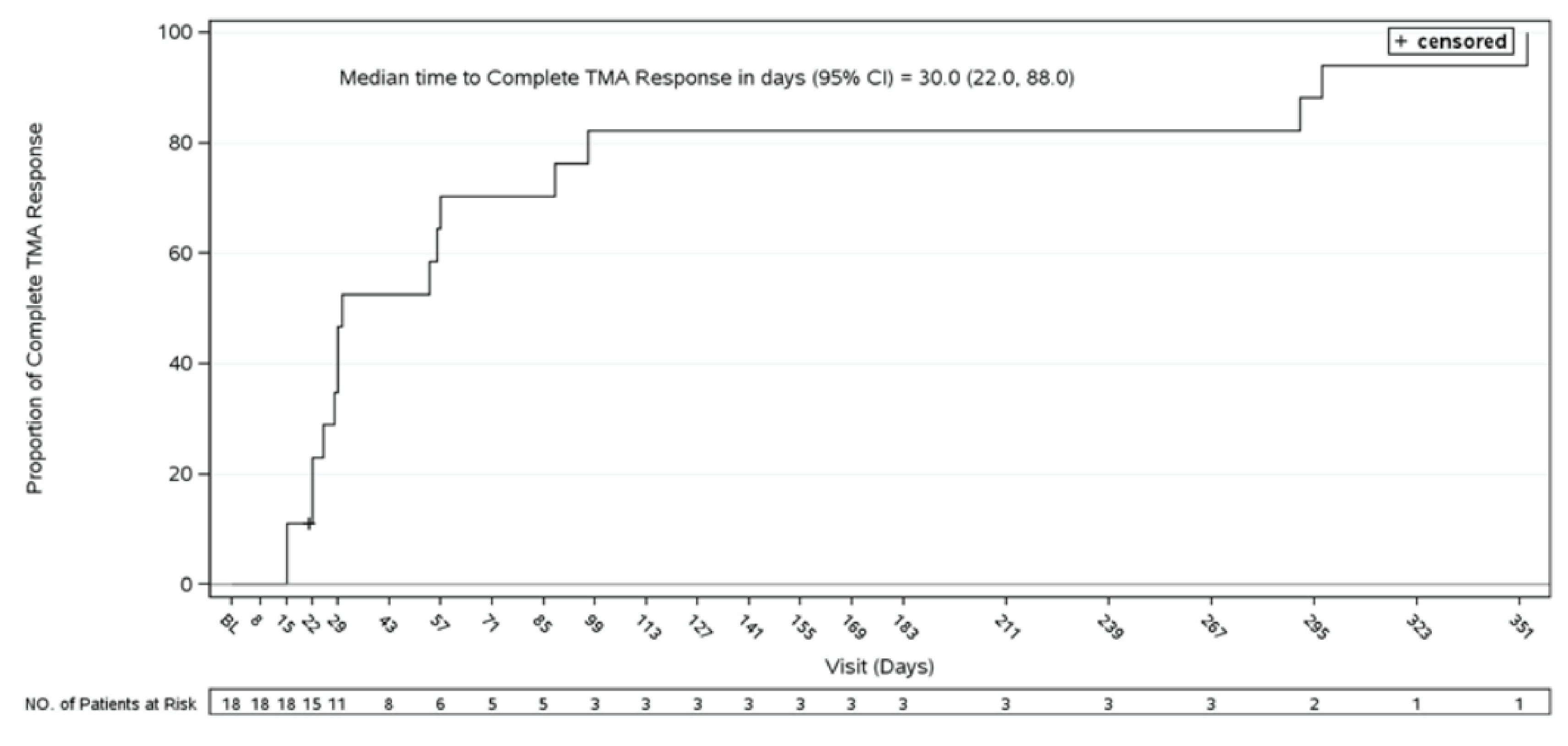
Note: Data as of December 3, 2019. Patients must have met all complete TMA response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart and at any measurement in between. The time of the event of a confirmed complete TMA response was considered as the first time point at which all the criteria for complete TMA response were met. Patients who did not have a response were censored at the date of the last visit or study discontinuation at the time when the analysis was performed.
Source: Study 312 Clinical Study Report.22
Presence of Severe Bleeding
Severe bleeding was not reported as an efficacy outcome in either Study 311 or Study 312.
HRQoL (Measured Using the 3-Level EQ-5D)
Study 311: At baseline, for the 53 patients in the FAS for whom data were available, the mean 3-Level EQ-5D score was 0.48 (SD = 0.271). Overall, patients in the FAS showed improvement in their 3-Level EQ-5D scores over time during the initial evaluation period, and this improvement continued into the extension period (Figure 24). At day 183, the 46 patients with available data had a mean change from baseline of 0.22 (SD = 0.247) (Table 17). At day 351, 42 patients with available data had a mean change from baseline of 0.25 (SD = 0.256). Observed and model-based values of changes in 3-Level EQ-5D scores over time (time trade-off value set for the US) for the initial evaluation period and during the extension period through the data cut-off date are presented in Figure 25. No reported MID was found for patients with aHUS.
Study 312: In Study 312, cohort 1 and cohort 2, HRQoL was not assessed.
Table 17: Outcomes in 3-Level EQ-5D (FAS)
Outcomes | Study 311 (N = 56) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
3-Level EQ-5D (US TTO) | |||
Baseline | |||
n | 53 | NA | NA |
Mean (SD) | 0.48 (0.27) | NA | NA |
Median (minimum, maximum) | 0.59 (–0.11, 0.75) | NA | NA |
Week 26 | 48 | NA | NA |
Mean (SD) | 0.71 (0.09) | NA | NA |
Median (minimum, maximum) | 0.74 (0.27, 0.75) | NA | NA |
Change from baseline at week 26 | 46 | NA | NA |
Mean (SD) | 0.22 (0.25) | NA | NA |
Median (minimum, maximum) | 0.15 (–0.14, 0.72) | NA | NA |
Week (day 351) | 44 | NA | NA |
Mean (SD) | 0.71 (0.06) | NA | NA |
Median (minimum, maximum) | 0.74 (0.47, 0.75) | NA | NA |
Change from baseline at day 351 | 42 | NA | NA |
Mean (SD) | 0.25 (0.26) | NA | NA |
Median (minimum, maximum) | 0.26 (–0.14, 0.71) | NA | NA |
Week (day 743), n | 9 | NA | NA |
Mean (SD) | 0.72 (0.03) | NA | NA |
Median (minimum, maximum) | 0.74 (0.66, 0.74) | NA | NA |
Change from baseline at day 743 | 8 | NA | NA |
Mean (SD) | 0.08 (0.17) | NA | NA |
Median (minimum, maximum) | –0.01 (–0.04, 0.40) | NA | NA |
NA = not assessed; FAS = full analysis set; SD = standard deviation; TTO = time trade-off.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Symptom Reduction
A by-patient listing of the patient-reported aHUS symptoms and extrarenal signs and symptoms of aHUS is provided in the CSRs for both Study 311 and Study 312. Atypical HUS symptoms were examined using a symptoms questionnaire, and results were reported for the FAS. However, the study level result was not summarized by the sponsor in the CSRs. Therefore, symptom reduction has not been reported herein. Fatigue was assessed using FACIT-F score as an HRQoL outcome (refer to the FACIT-F score assessment section).
Fatigue
Study 311
At baseline, the mean FACIT-F score for the 51 patients in the FAS for whom data were available was 24.03 (SD = 15.279). Overall, the patients in the FAS showed improvement in their FACIT-F scores over time during the initial 26 weeks, and this improvement continued into the extension period (Figure 18). At day 183, the 44 patients for whom data were available had a mean improvement from baseline in FACIT-F score of 19.15 (SD = 16.212) (Table 18). During the extension period, patients with available data had mean improvements from baseline in FACIT-F score of 19.29 at the day 351 visit (n = 40); 16.75 at the day 575 visit (n = 22); and 8.81 at the day 743 visit (n = 8) (Table 18).
An improvement of greater than or equal to 3 points in FACIT-F score, considered to be a clinically meaningful improvement,23 was observed in 37 patients of the 44 patients (84.1%) with available data at week 26. All had a 3-point improvement from baseline by day 29. During the extension period, 33 patients of the 40 patients (82.5%) with available data had a 3-point improvement from baseline at the day 351 visit (Table 18). Five patients of the 8 patients (62.5%; 95% CI, 24.5% to 91.5%) had an improvement from baseline of 3 points on day 743. The observed values of the changes in FACIT-F scores over time and of FACIT-F scores over time for the FAS are presented in Figure 19.
Study 312, Cohort 1
Among the 13 treated patients who were aged 5 years or older in this study, 9 patients had fatigue assessed using the pediatric FACIT-F scale. At the end of the initial 26 weeks (day 183), these 9 patients had a mean improvement in the pediatric FACIT-F score of 16.78 (SD = 14.704) compared to baseline (Table 18). During the extension period, patients with available data had a mean improvement from baseline in FACIT-F score of 16.67 (SD = 15.297) at the day 351 visit (n = 9) and of 17.40 (SD = 17.184) at the day 575 visit (n = 5). Observed values over time in pediatric FACIT-F scores are shown in Figure 20, with change from baseline presented in Figure 21.
Three patients of 9 patients (33.3%) had at least a 3-point improvement in FACIT-F total score from baseline at day 8; 7 patients (77.8%) had at least a 3-point improvement from baseline at day 29; and all 9 patients had at least a 3-point improvement from baseline by day 71 to day 351. At day 575, 5 of 5 patients had a 3-point improvement from baseline (Table 18).
Study 312, Cohort 2
For the 8 treated patients in cohort 2 who were aged 5 years or older, quality of life was assessed using the pediatric FACIT-F scale. There was no notable improvement or worsening compared to baseline in the pediatric FACIT-F scores for all 8 patients during the initial 26 weeks or through day 351 of the extension period (Table 18, Figure 22). During the extension period, the 8 patients had a mean improvement from baseline in FACIT-F score of –1.25 (SD = 2.712); this was observed at the day 351 visit. Observed values and change from baseline in the pediatric FACIT-F score over time for the FAS are presented in Figure 22 and Figure 23, respectively.
Table 18: Outcomes in Pediatric Quality of Life (FACIT-F, FAS)
Outcomes | Study 311 (N = 56) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
FACIT-F score | |||
Number of patients with ≥ 3 improvement | |||
Week 26 | |||
N of patients with data | 44 | 9 | NR |
Patients with ≥ 3-point improvement, n (%) | 37 (84.1) | 3 (33.3) | NR |
Extension period (day 351) | |||
N of patients with data | 40 | 9 | NR |
Patients with ≥ 3-point improvement, n (%) | 33 (82.5) | 9 (100) | NR |
FACIT-F score | |||
Baseline | |||
n | 51 | 9 | 8 |
FACIT-F score at baseline, mean (SD) | 24.03 (15.279) | 31.44 (13.648) | 48.88 (3.603) |
Median (minimum, maximum) | 24.00 (0, 51) | 35.00 (4, 44) | 50 (42, 52) |
Week 26 | |||
n | 48 | 9 | 8 |
FACIT-F score at week 26 (day 183), mean (SD) | 42.85 (8.796) | 48.22 (5.848) | 48.88 (5.410) |
Median (minimum, maximum) | 45.5 (12, 52) | 52.00 (36, 52) | 52 (37, 52) |
Change from baseline to week 26 | |||
n | 44 | 9 | 8 |
Mean (SD) | 19.15 (16.212) | 16.78 (14.704) | 0.00 (2.268) |
Median (minimum, maximum) | 20 (–16, 48) | 10.00 (4, 48) | 0.00 (–5, 3) |
Week (day 351), n | |||
n | 44 | 9 | 8 |
FACIT-F score at day 351, mean (SD) | 42.52 (9.802) | 48.11 (5.968) | 47.63 (4.470) |
Median (minimum, maximum) | 46.05 (12, 52) | 51.00 (35, 52) | 49 (40, 52) |
Change from baseline to day 351 | |||
n | 40 | 9 | 8 |
Mean (SD) | 19.29 (17.520) | 16.67 (15.297) | –1.25 (2.712) |
Median (minimum, maximum) | 16.5 (–17, 50) | 9.00 (3, 47) | –1 (–7, 2) |
Week (day 743 or day 575) | |||
n | Day 743 = 9 | Day 575 = 5 | NR |
FACIT-F score at day 743 and day 575, mean (SD) | 42.11 (10.635) | 45.00 (5.831) | NR |
Median (minimum, maximum) | 45.5 (17, 52) | 44.00 (39, 52) | NR |
Change from baseline to day 743 | 8 | 5 | NR |
Mean (SD) | 8.81 (15.140) | 17.40 (17.184) | NR |
Median (minimum, maximum) | 6.0 (–7, 34.455) | 8.00 (5, 46) | NR |
FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; NR = not reported; SD = standard deviation.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Renal Function
Estimated Glomerular Filtration Rate
Study 311: The mean eGFR gradually improved during the initial 26 weeks (Figure 26, Figure 27). Overall, the mean eGFR value at baseline was 15.86 mL/min/1.73 m2 (Table 19) and increased to 51.83 mL/min/1.73 m2 by the end of the initial week 26 (day 183, Table 19). During the extension period, the mean eGFR remained stable above 50.30 mL/min/1.73 m2 for the 43 patients who reached the day 407 visit.
Study 312, Cohort 1: The mean eGFR improved gradually during the initial 26 weeks (Figure 28, Figure 29). The mean change in eGFR over time for the FAS is presented Table 19 and in Figure 28 and Figure 29. Overall, the mean eGFR value at baseline was 26.4 mL/min/1.73m2 (SD = 21.17 mL/min/1.73m2) (Table 21). The eGFR was 108.5 mL/min/1.73m2 (SD = 56.87 mL/min/1.73m2) at the end of the initial 26 weeks (i.e., by day 183). During the extension period, the mean eGFR remained above 100 mL/min/1.73 m2 for the 14 patients who reached the day 407 visit.
In Study 312, cohort 2, the eGFR remained generally stable for all 10 patients during the initial 26-week period and through the data cut-off date (Figure 30 and Figure 31).
Table 19: Results for eGFR (FAS)
eGFR (mL/min/1.73m2) | Study 311 (N = 56) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
Baseline | — | — | — |
n | 55 | 18 | 10 |
Mean (SD) | 15.86 (14.815) | 26.4 (21.17) | 104.90 (29.545) |
Median (minimum, maximum) | 10 (4, 8) | 22 (10, 84) | 99.75 (54, 136.5) |
Week 26 (day 183) | |||
n | 48 | 17 | 10 |
Mean (SD) | 51.83 (39.162) | 108.5 (56.87) | 94.00 (35.131) |
Median (minimum, maximum) | 40 (2, 119) | 108 (10, 244) | 93.5 (40, 139) |
Change from baseline to week 26 | |||
n | 47 | 17 | 10 |
Mean (SD) | 34.80 (35.454) | 85.4 (54.33) | –10.90 (30.584) |
Median (minimum, maximum) | 29 (–13, 108) | 80 (0, 222) | –2 (–94, 18) |
Week 58 (day 407) | |||
n | 43 | 14 | 3 |
mean (SD) | 50.30 (36.722) | 109.9 (48.97) | 135.67 (5.859) |
Median (minimum, maximum) | 49 (3, 127) | 123.5 (31, 168) | 138.0 (129, 140) |
Change from baseline to week 58 | |||
n | 42 | 14 | 3 |
Mean (SD) | 33.30 (33.219) | 86.5 (45.89) | 1.50 (6.500) |
Median (minimum, maximum) | 21.5 (–13, 106) | 90.5 (9, 156) | –1.5 (–5, 8) |
Week 106 (day 743) | |||
n | 9 | 2 | NR |
mean (SD) | 41.56 (38.504) | 127.5 (26.16) | NR |
Median (minimum, maximum) | 10 (10, 91) | 127.5 (109, 146) | NR |
Change from baseline to week 106 (day 743) | 8 | 2 | NR |
Mean (SD) | 17.25 (31.404) | 109.0 (38.18) | NR |
Median (minimum, maximum) | 1.5 (–6, 78) | 109 (82, 136) | NR |
eGFR = estimated glomerular filtration rate; FAS = full analysis set; NR = not reported; SD = standard deviation.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
CKD Stage
Study 311
Most patients were in CKD stage 4 or stage 5 at baseline (Table 20). For the 47 patients with available baseline and week 26 (day 183) data, 32 patients of 47 patients in the FAS (68.1%) had improvement in their CKD stage compared to baseline: 6 patients improved by 5 stages (i.e., from ESKD to normal renal function); 7 patients improved by 4 stages; 5 patients improved by 3 stages; 4 patients improved by 2 stages; and 10 patients improved by 1 stage (Table 20). Two patients experienced worsening of their CKD stages. One of these patients worsened from stage 4 at baseline to stage 5 at day 8, received dialysis on day 16, and remained at stage 5 for the duration of the initial evaluation period. The other patient worsened from stage 4 at baseline to stage 5 at day 8 and remained at stage 5 for the duration of the initial evaluation period (except for 1 assessment of stage 4 at day 15). Nineteen of the 30 patients who had complete TMA responses continued to have improved renal function during the initial evaluation period after achieving complete TMA response, as assessed by an improvement in CKD stage from the time of complete TMA response to day 183 (Table 20). In the extension period, among the 42 patients with available baseline and day 407 data, 29 patients (69.0%) had improvement in CKD stage compared to baseline: 4 patients improved by 5 stages (i.e., from ESKD to normal renal function); 6 patients improved by 4 stages; 8 patients improved by 3 stages; 2 patients improved by 2 stages; and 9 patients improved by 1 stage (Table 21). The 2 patients who experienced worsening of their CKD stage during the initial evaluation period remained at stage 5 from day 183 through the last available visit during the extension period.
Table 20: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 311, at Week 26)
Baseline CKD stage (N = 47) | Postbaseline CKD stage at week 26 (N = 47) | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | n | 1 n (%) | 2 n (%) | 3A n (%) | 3B n (%) | 4 n (%) | 5 n (%) |
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2 | 3 | 2 (4.3) | 1 (2.1) | 0 | 0 | 0 | 0 |
3A | 1 | 1 (2.1) | 0 | 0 | 0 | 0 | 0 |
3B | 2 | 2 (4.3) | 0 | 0 | 0 | 0 | 0 |
4 | 7 | 1 (2.1) | 0 | 0 | 3 (6.4) | 1 (2.1) | 2 (4.3) |
5 | 34 | 6 (12.8) | 6 (12.8) | 3 (6.4) | 3 (6.4) | 5 (10.6) | 11 (23.4) |
Total | 47 (100) | 12 (25.5) | 7 (14.9) | 3 (6.4) | 6 (12.8) | 6 (12.8) | 13 (27.7) |
CKD = chronic kidney disease.
Note: Baseline data were derived based on the last available eGFR before starting treatment. Patients with both baseline data and at least 1 value at postbaseline visits were included in the summary.
Source: Study 311 Clinical Study Report.21
Table 21: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 311, on Day 407)
Baseline CKD stage (N = 42) | Postbaseline CKD stage at day 407 (N = 42) | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | n | 1 n (%) | 2 n (%) | 3A n (%) | 3B n (%) | 4 n (%) | 5 n (%) |
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2 | 2 | 1 (2.4) | 1 (2.4) | 0 | 0 | 0 | 0 |
3A | 1 | 1 (2.4) | 0 | 0 | 0 | 0 | 0 |
3B | 2 | 2 (4.8) | 0 | 0 | 0 | 0 | 0 |
4 | 7 | 0 | 1 (2.4) | 0 | 3 (7.1) | 1 (2.4) | 2 (4.8) |
5 | 30 | 4 (9.5) | 6 (14.3) | 5 (11.9) | 1 (2.4) | 5 (11.9) | 9 (21.4) |
Total | 42 (100) | 8 (19.0) | 8 (19.0) | 5 (11.9) | 4 (9.5) | 6 (14.3) | 11 (26.2) |
CKD = chronic kidney disease.
Note: Baseline data were derived based on the last available eGFR before starting treatment. Patients with both baseline data and at least 1 value at postbaseline visits were included in the summary.
Source: Study 311 Clinical Study Report.21
Study 312, Cohort 1
Of the patients with CKD stage data at both baseline and week 26, the majority (14 patients of 17 patients) evaluated at baseline were at CKD stage 4 or stage 5; 6 patients (35.3%) were at CKD stage 5 (Table 22). With the exception of 2 patients, all of these patients improved their CKD stage (i.e., shifted to a lower stage) from baseline through the end of the initial evaluation period (day 183); the shift was substantial, given that 14 patients improved by 2 or more stages. For the 17 patients with available data at the end of the initial evaluation period, 15 patients (88.2%) had improvement in their CKD stage compared to baseline (Table 22). Three of these patients improved by 5 stages; 7 patients improved by 4 stages; 2 patients improved by 3 stages; 2 patients improved by 2 stages; and 1 patient improved by 1 stage. Of the 2 patients who had no improvement in CKD stage during the initial evaluation period, 1 patient had a history of kidney transplant before the study. None of the patients worsened in CKD stage during the initial evaluation period. All 14 patients with available baseline and day 407 data had improvements in their CKD stage compared to baseline: 3 patients improved by 5 stages (i.e., from ESKD to normal renal function); 5 patients improved by 4 stages; 1 patient improved by 3 stages; 2 patients improved by 2 stages; and 3 patients improved by 1 stage (Table 23).
Table 22: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 312, Cohort 1, at Week 26)
Baseline CKD stage | Postbaseline CKD stage at week 26 (N = 18)a | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | N (%) | 1 n (%) | 2 n (%) | 3a n (%) | 3b n (%) | 4 n (%) | 5 n (%) |
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2 | 1 (5.9) | 1 (5.9) | 0 | 0 | 0 | 0 | 0 |
3a | 1 (5.9) | 1 (5.9) | 0 | 0 | 0 | 0 | 0 |
3b | 1 (5.9) | 1 (5.9) | 0 | 0 | 0 | 0 | 0 |
4 | 8 (47.1) | 5 (29.4) | 1 (5.9) | 1 (5.9) | 0 | 1 (5.9) | 0 |
5 | 6 (35.3) | 3 (17.6) | 2 (11.8) | 0 | 0 | 1 (5.9) | |
Total | 17 (100.0) | 11 (64.7) | 3 (17.6) | 1 (5.9) | 0 | 1 (5.9) | 1 (5.9) |
CKD = chronic kidney disease.
Note: Baseline data were derived based on the last available eGFR before starting treatment. Patients with both baseline data and at least 1 value at postbaseline visits were included in the summary.
aThe percentages for the postbaseline CKD stage at week 26 were based on the 17 patients with available data.
Source: Study 312 Clinical Study Report.22
Table 23: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 312, Cohort 1, on Day 407)
Baseline CKD stage | Postbaseline CKD stage at day 407 (N = 14) | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | n | 1 n (%) | 2 n (%) | 3a n (%) | 3b n (%) | 4 n (%) | 5 n (%) |
1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2 | 1 (7.1) | 1 (7.1) | 0 | 0 | 0 | 0 | 0 |
3a | 1 (7.1) | 1 (7.1) | 0 | 0 | 0 | 0 | 0 |
3b | 1 (7.1) | 1 (7.1) | 0 | 0 | 0 | 0 | 0 |
4 | 5 (35.7) | 3 (21.4) | 0 | 0 | 2 (14.2) | 0 | 0 |
5 | 6 (42.9) | 3 (21.4) | 2 (14.3) | 0 | 1 (7.1) | 0 | 0 |
Total | 14 (100.0) | 9 (64.3) | 2 (14.3) | 0 | 3 (21.4) | 0 | 0 |
CKD = chronic kidney disease.
Note: Baseline data were derived based on the last available eGFR before starting treatment. Patients with both baseline data and at least 1 value at postbaseline visits were included in the summary.
aThe percentages for the postbaseline CKD stage at week 26 were based on the 17 patients with available data.
Source: Study 312 Clinical Study Report.22
Study 312, Cohort 2
The majority of patients in cohort 2 (8 patients of 10 patients) were in CKD stage 1 at baseline; 1 patient was in stage 2, and 1 patient was in stage 3a (Table 24). By week 26, 7 of the patients had no change in their CKD stage; 2 patients had worsened by 1 stage; and 1 patient had worsened by 3 stages. During the extension period, all 10 patients had no change in CKD stage by day 351 compared to baseline (Table 25). The CKD stage remained unchanged for the 3 patients with data available at day 407.
Table 24: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 312, Cohort 2, at Week 26, FAS)
Baseline CKD stage | Postbaseline CKD stage at week 26 (N = 10) | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | n (%) | 1 n (%) | 2 n (%) | 3a n (%) | 3b n (%) | 4 n (%) | 5 n (%) |
1 | 8 (80.0) | 5 (50.0) | 2 (20.0) | 0 | 1 (10.0) | 0 | 0 |
2 | 1 (10.0) | 0 | 1 (10.0) | 0 | 0 | 0 | 0 |
3a | 1 (10.0) | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
3b | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Total | 10 (100.0) | 5 (50.0) | 3 (30.0) | 1 (10.0) | 1 (10.0) | 0 | 0 |
CKD = chronic kidney disease.
Source: Study 312 Clinical Study Report.22
Table 25: CKD Stage Shift From Baseline to End of Initial Evaluation Period (Study 312, Cohort 2, on Day 407, FAS)
Baseline CKD stage | Postbaseline CKD stage at day 407 (N = 10) | ||||||
|---|---|---|---|---|---|---|---|
CKD stage | n (%) | 1 n (%) | 2 n (%) | 3a n (%) | 3b n (%) | 4 n (%) | 5 n (%) |
1 | 8 (80.0) | 8 (80.0) | 0 | 0 | 0 | 0 | 0 |
2 | 1 (10.0) | 0 | 1 (10.0) | 0 | 0 | 0 | 0 |
3a | 1 (10.0) | 0 | 0 | 1 (10.0) | 0 | 0 | 0 |
3b | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Total | 10 (100.0) | 8 (80.0) | 1 (10.0) | 1 (10.0) | 0 | 0 | 0 |
CKD = chronic kidney disease.
Source: Study 312 Clinical Study Report.22
Dialysis Requirement Status
Study 311
At baseline, or within 5 days before the first dose of the study drug, 29 patients (51.8%) in the FAS had received renal dialysis (Table 26). During the initial 26 weeks, 17 patients of these 29 patients (58.6%) discontinued dialysis. As of the data cut-off date, 18 patients of these 29 patients (62.1%) had discontinued dialysis (Table 26). One of these patients discontinued within the first week of study treatment, then received dialysis at 3 time points (day 136, day 138, and day 141) during the initial evaluation period. Of the 27 patients who were not on dialysis at baseline, 7 patients (25.9%) initiated dialysis during the initial 26 weeks; 6 of these 7 patients remained on dialysis at day 183. As of the data cut-off date, 20 patients (74.0%) remained off dialysis.
Seven patients (25.9%) initiated dialysis after starting treatment: 3 patients started and stopped dialysis during the initial evaluation period; 3 patients started receiving dialysis during the initial evaluation period; and 1 patient started receiving dialysis during the extension period.
Study 312, Cohort 1
Of the 6 patients in the FAS who were receiving dialysis at baseline (within 5 days before the first dose of the study drug), 4 patients discontinued dialysis within the first 36 days of exposure to ravulizumab (Table 26). All 6 patients had discontinued dialysis by day 193. No patients initiated dialysis after starting treatment with study drug.
Study 312, Cohort 2
As of the data cut-off date, none of the 10 patients had initiated dialysis after starting treatment with the study drug (Table 23).
Table 26: Dialysis Requirement Status (Data Cut-Off Date)
Dialysis status | Study 311 (N = 56) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 18) | Cohort 2 (N = 10) | ||
Patients with dialysis at baseline, n (%) | 29 (51%) | 6 (33.3%) | 0 |
Discontinued dialysis during initial period | 17 (58.6) | 4 (22.2) | NA |
Extension period | 18 (62.1) | 6 (33.3) | NA |
Patients with no dialysis at baseline | 27 | 12 (66.8) | 10 |
Started dialysis during the initial period | 7 (25.9) | 0 | 0 |
Extension period | 6 (10.7) | 0 | 0 |
NA = not assessed.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Plasma Therapy–Free Status
Plasma therapy was prohibited during the trial and considered a protocol violation; thus, it was not an outcome assessed in the pivotal studies. However, plasma therapy was reported as a concomitant therapy. In Study 311, 3 patients (5.2%) received plasma therapy. No patient received plasma therapy in Study 312 (cohort 1 or cohort 2) (refer to Table 37).
Packed RBC Transfusions
RBC transfusions were reported as a concomitant treatment (refer to Table 37). In Study 311, 17 patients (29.3%) received packed RBC transfusions during the study. In Study 312 (cohort 1 and cohort 2), no information on packed RBC transfusions was reported (Table 37).
Hospitalizations
Hospitalizations were reported in the assessments of health resource utilization in both Study 311 and Study 312. However, there were no summaries or analyses of hospitalizations at the study level. Therefore, the data are not extractable and not reported in this review report.
Presence of Soluble MAC
Soluble MAC was not assessed in either Study 311 or Study 312 as an outcome.
Harms
Only those harms identified in the review protocol are reported. Refer to Table 27 for detailed harms data.
Adverse Events
In Study 311, as of the data cut-off date, all patients (N = 58, 100%) had experienced at least 1 TEAE. The most common adverse events (occurring in at least 30% of patients) were headache (n = 22; 37.9%), diarrhea (n = 19; 32.8%), and vomiting (n = 18; 31.0%) (refer to Table 27).
In Study 312, cohort 1, as of the data cut-off date, all patients (N = 21, 100%) had experienced at least 1TEAE. The most common adverse events (occurring in at least 30% of patients) were pyrexia (n = 10; 47.6%), headache (n = 7; 33.3%), diarrhea (n = 7; 33.3%), vomiting (n = 7; 33%), and nasopharyngitis (n = 7; 33.3%) (refer to Table 27).
In Study 312, cohort 2, as of the data cut-off date, all 10 patients (100.0%) had experienced at least 1 TEAE. The most common adverse event (occurring in at least 30% of patients) was oropharyngeal pain (n = 3; 30%) (Table 27).
Serious Adverse Events
In Study 311, as of the data cut-off date, a total of 33 patients (56.9%) had experienced an SAE (Table 27). Each SAE was reported in 1 patient, except for pneumonia and hypertension — both of which occurred in 3 patients (5.2%) — and septic shock, urinary tract infection, and malignant hypertension, each of which occurred in 2 patients (3.4%) (Table 27).
In Study 312, cohort 1, as of the data cut-off date, the SAEs occurring in 2 or more patients were gastroenteritis, viral infection and abdominal pain, each of which occurred in 2 patients (9.5%).
In Study 312, cohort 2, as of the data cut-off date, no SAE had been reported in more than 1 patient.
Withdrawals Due to Adverse Events
In Study 311, as of the data cut-off date, a total of 3 patients (5.2%) had experienced adverse events leading to study drug discontinuation (Table 27).
In Study 312, cohort 1, as of the data cut-off date, 1 patient (4.8%) had experienced adverse events leading to study drug discontinuation (Table 27).
In Study 312, cohort 2, as of the data cut-off date, no patient had experienced adverse events leading to study drug discontinuation (Table 27).
Mortality
In Study 311, 4 patients died during the initial 26-week evaluation period. One of these 4 patients died due a pretreatment SAE (cerebral arterial thrombosis); 3 patients (5.2%) died due to treatment-emergent SAEs that were considered not related to the study drug (2 due to septic shock and 1 due to intracranial hemorrhage)21 (refer to Table 27).
In cohort 1 and cohort 2 of Study 312, no patients had died due to adverse events as of the data cut-off date (Table 27).
Notable Harms
In Study 311, as of the data cut-off date, no meningococcal infection had been reported. Sepsis was reported in 1 patient (1.7%). Infusion-related reaction was not reported. Hypersensitivity was reported in 1 patient (1.7%) Antidrug antibodies were reported in 1 patient (1.7%) (Table 27).
In Study 312, cohort 1, as of the data cut-off date, 1 patient (4.8%) had reported hypersensitivity (Table 27). In cohort 2, as of the data cut-off date, no notable harms had been reported (Table 27).
Table 27: Summary of Harms (Safety Analysis, Data Cut-Off Date)
Adverse events | Study 311 N = 58 | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 21) | Cohort 2 (N = 10) | ||
Patients with ≥ 1 adverse event, n (%) | 58 (100.0) | 21 (100.0) | 10 (100) |
Most common events,a (≥ 30%), n (%) | |||
Diarrhea | 19 (32.8) | 7 (33.3) | NR |
Vomiting | 18 (31.0) | 7 (33.3) | 1 (10.0) |
Headache | 22 (37.9) | 7 (33.3) | |
Nasopharyngitis | 9 (15.5) | 7 (33.3) | 2 (20.0) |
Abdominal pain | 8 (13.8) | 6 (28.6) | NR |
Pyrexia | 12 (20.7) | 10 (47.6) | NR |
Oropharyngeal pain | NR | NR | 3 (30.0) |
Patients with ≥ 1 SAE, n (%) | 33 (56.9) | 14 (66.7) | 1 (10.0) |
Most common SAE eventsa (occurring in ≥ 2 patients), n (%) | |||
Pneumonia | 3 (5.2) | 1 (4.8) | 1 (4.8) |
Septic shock | 2 (3.4) | NR | NR |
Urinary tract infection | 2 (3.4) | NR | NR |
Atypical hemolytic uremic syndrome | 2 (3.4) | NR | NR |
Hypertension | 3 (5.2) | NR | NR |
Malignant hypertension | 2 (3.4) | NR | NR |
Gastroenteritis viral | NR | 2 (9.5) | NR |
Abdominal pain | 1 (1.7) | 2 (9.5) | NR |
AEs leading to infusion interruption, n, % | 0 | 2 (9.5) | 1(10) |
WDAE (patients who stopped treatment due to adverse events), n (%) | 3 (5.2) | 1 (4.8) | 0 |
Deaths, n (%) | 3 (5.2) | 0 | 0 |
Notable harms, n (%) | |||
Meningococcal disease | 0 | 0 | 0 |
Sepsis | 1 (1.7) | NR | NR |
Infusion-related reactions | NR | NR | NR |
Hypersensitivity to the drug | 1 (1.7) | 1 (4.8) | NR |
Antidrug antibodies | 1 (1.7) | 0 | 0 |
AE = adverse event; NR = not reported; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Note: The organisms involved in the 2 septic shock cases were Corynebacterium and Candida lusitaniae (n = 1) and Pseudomonas (n = 1). The organisms for pneumonia were not reported in the Clinical Study Report.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Critical Appraisal
Internal Validity
The main limitation of the 2 included pivotal studies (Study 311 and Study 312) is their single-arm design, which does not include a comparator arm. Due to the rare and severe nature of aHUS, a randomized control group was not likely to be feasible. Nonetheless, such a design, in addition to the lack of consideration for confounding variables, precludes causal inferences (i.e., the outcomes cannot be directly attributed to ravulizumab). Without an active comparator, standard of care, or statistical hypothesis testing, it is not possible to confirm the relative therapeutic benefit or safety of ravulizumab against other available treatments (such as eculizumab, in this population) or against standard care.
The clinical diagnosis of aHUS is challenging and relies on the exclusion of other conditions.11 Therefore, it is possible that some patients with other conditions that present similarly to aHUS were enrolled.12 If so, that would mean that not all of the included patients had a confirmed diagnose of aHUS. For example, in Study 311, 8 patients (14.3%) had a kidney transplant not related to aHUS before entering the study. The clinical experts consulted by CADTH for this review indicated that these patients (i.e., those who had a kidney transplant without a prior aHUS diagnosis) could potentially have aHUS, but that the diagnosis of aHUS in these patients could not be absolutely established. This is important because the clinical experts indicated that patients with a confirmed diagnosis of aHUS would be expected to respond better to ravulizumab than those patients without a confirmed diagnosis of aHUS. As a result, any bias associated with an uncertain diagnosis would be against ravulizumab. In other words, it would make ravulizumab appear less effective in terms of improving TMA parameters.
Furthermore, only 30 patients of 56 patients met all the TMA criteria (i.e., active criteria based on platelet, LDH, and serum creatinine levels) on day 1 of the trial. (All had met these criteria during the screening phase.) However, the subgroup analysis based on the TMA criteria on day 1 (yes or no) showed similar results for patients who met all of the TMA criteria on day 1 and those who did not, which minimizes any potential concern for bias being introduced.
In addition, both Study 311 and 312 were open-label trials, so the study investigators and patients were aware of their treatment status. This increases the risk of detection and performance biases that have the potential to influence outcome reporting. However, the primary end point (TMA response) and most of the secondary end points are considered to be objective response measurements for which the potential for bias due to the open-label design is low. The potential for bias is more of a concern for subjective end points, such as safety, symptoms (e.g., measured using the FACIT-F), and HRQoL (measured using the 3-Level EQ-5D). The direction of anticipated bias related to these outcomes is unclear. It is possible that known harms and anticipated benefits would be overreported.
The clinical experts consulted by CADTH for this review indicated that complete TMA response is usually used in clinical research, but not commonly used in clinical practice. An improvement in serum creatinine of greater than or equal to 25% is usually accepted as a component of the complete TMA response. However, for patients on dialysis, discontinuation of dialysis is more meaningful clinically. Within the pivotal trials, sensitivity analysis replacing the improvement in serum creatinine of greater than or equal to 25% with the discontinuation of dialysis showed a consistent and complete TMA response in the primary analysis.
For HRQoL (i.e., the 3-Level EQ-5D) and symptom scales (i.e., the FACIT-F), there is a potential risk of bias because a large number of patients did not have complete measures, especially during the extension period. There may have been differential recall bias, and/or those patients remaining in the study may have differed in some systematic way from those who remained in the study. Overall, the magnitude and direction of the impact of these missing data and recall bias on patient-reported outcomes, 3-Level EQ-5D, and FACIT-F is unknown. No MID was identified for the 3-Level EQ-5D in the aHUS population; therefore, the clinical importance of potential HRQoL improvements is unknown.
One additional potential limitation was that the efficacy assessment was not based on the intention-to-treat population (for Study 311 or Study 312, cohort 1). Instead, it included patients who received at least 1 dose of the study drug and at least 1 postbaseline efficacy assessment. A total of 2 patients (3.4%) in Study 311 and 3 patients (14.29%) in Study 312, cohort 1 were excluded from the primary FAS analysis. It is also noted that 43 patients (76.79%) in Study 311 and 14 patients (66.7%) in Study 312, cohort 1 experienced a major protocol violation; the majority (N = 25, 43.1% in Study 311 and N = 9, 42.9% in Study 312) were related to the eligibility criteria. A PP analysis (N = 44, 75.9% in Study 311 and N = 18, 85.7% for Study 312, cohort 1) was performed and showed results that were consistent with those of the FAS analysis. However, not all patients with a major protocol violation — especially those related to eligibility criteria — were excluded from the PP analysis.
It is worth mentioning that in Study 312, cohort 2, the main limitation was that the sample size (N = 10) was too small for the eculizumab-treated and TMA-stable pediatric patients with aHUS, which meant that the overall dataset was more sensitive to outliers and skewed distribution. However, this limitation is expected due to the rare nature of the disease.
External Validity
Overall, according to the clinical experts consulted by CADTH, the inclusion and exclusion criteria of 2 pivotal studies (Study 311 and 312) were reasonable, and the baseline patient characteristics, concomitant medications, and prohibited medications were reflective of patients they treat in clinical practice for the indication under review. No pediatric patients in Canada were included in Study 312. However, the clinical experts consulted by CADTH for this review indicated that they would not expect to find any important difference among different races or geographic regions in terms of the response to complement inhibitors, such as ravulizumab, for aHUS.
Patients who received plasma exchange and/or plasma infusion for 28 days or longer before the start of screening for the current TMA were excluded from the pivotal studies. Therefore, there is uncertainty as to whether the findings may be generalized to these populations. The clinical experts consulted by CADTH indicated that this would represent a small group of patients with catastrophic disease. This would be an uncommon scenario that is reasonable to exclude. There is no concern about generalizability because of this exclusion criteria.
No subgroup analysis was performed based on baseline platelet or LDH levels. No subgroup analysis based on the duration of prior plasma therapy was conducted, and patients with plasma exchange and/or plasma infusion for 28 days or longer before the start of trial were excluded from this study, as discussed.
Symptom reduction was identified as an important outcome for patients. However, symptom severity reduction at the study level was not assessed as a distinct outcome in the 2 pivotal studies. Instead, a list of patient-reported renal and extrarenal aHUS symptoms was included in the CSRs. It is understood that these symptoms can result in decreased HRQoL. Fatigue is an important symptom that patients with aHUS often experience, and it was assessed using the FACIT-F.
Furthermore, it is understood that aHUS is an extremely rare disease and that, as a result, it was not feasible to enrol large numbers of patients. However, it should be noted that the magnitude of the treatment effect estimates observed in a relatively small study sample may not be replicable in a larger study sample or generalizable to the target population in real-world clinical practice. Finally, given that all results are part of an interim analysis (i.e., at week 26 and during an extension period, with median follow-up times of 75.6 weeks for Study 311, 82.4 weeks for Study 312, cohort 1, and 52.3 weeks for Study 312, cohort 2). Based on the available data, it appears that the effects found at 26 weeks tended to be sustained through later time points.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Patients with aHUS in Canada are often treated with eculizumab or supportive care (e.g., plasma exchange or infusion, plasmapheresis). Evidence for ravulizumab is limited to single-arm trials in adult and pediatric populations; therefore, no direct comparisons are available to assess the efficacy and safety of ravulizumab relative to eculizumab. Direct comparisons between ravulizumab and eculizumab are likely to be infeasible due to the rare and severe nature of aHUS. Yet this comparison is important both clinically and economically. As such, an understanding the available ITCs may be useful to clinicians, patients, and pharmacoeconomic modelling groups.
For this submission, a systematic literature review was conducted to identify any sources of ITCs between ravulizumab and eculizumab or between ravulizumab and best supportive care. A single ITC, submitted by the sponsor and also published as a peer-reviewed publication,41 was identified.
Description of Indirect Treatment Comparison
A single sponsor-submitted study using stabilized inverse probability weighting to compare ravulizumab and eculizumab was reviewed for this submission. Although the submitted study has also been published as a peer-reviewed publication,41 the evidence for this submission was based on the sponsor-submitted report. Because the sponsor-submitted report contained greater details with respect to study rationale, sensitivity analyses, and methods, it was considered to be of greater use for this review. No discrepancies were noted with respect to the outcomes, methods, or overall interpretation between the sponsor-submitted study and the published article.
Methods of the Sponsor-Submitted Analysis
Objectives
The purpose of the sponsor-submitted analysis was to estimate the comparative efficacy and safety of ravulizumab and eculizumab for the treatment of aHUS.
Study Selection Methods
No systematic literature review was undertaken by the sponsor to identify eligible studies. No formal inclusion or exclusion criteria were applied with respect to the selection of studies for inclusion in the ITC. No details were provided on the data abstraction process, screening process, or quality assessment of included studies. No date was provided to indicate when the trials were assessed.
In total, 5 studies were identified by the sponsor as being appropriate for inclusion in the analysis: 3 studies of eculizumab (Studies aHUS-C08-002, aHUS-C10-003, and aHUS-C10-004) and 2 studies of ravulizumab (Studies 311 and 312). Two studies were excluded from analysis: Study aHUS-C08-003 was excluded on the basis of differences in disease history and plasma exchange status at baseline; no data were provided from Study aHUS-C08-003 to verify this assumption. Data from Study aHUS-C11-003 were excluded because it was a long-term extension study. Patient-level data were available for all patients within the analysis.
Patient populations were split into 4 groups: adults without kidney transplant (primary analysis), adults with kidney transplant, children without kidney transplant, and children with kidney transplant (not analyzed due to small sample size). Patients were considered for the primary analysis if baseline data were available for dialysis status, eGFR, platelet count, and LDH, and were required to have outcome data within 56 days of the 6-month study end point. No imputation was used for missing data; instead, patients with missing data at baseline or end point were excluded.
Analysis Methods
Because no direct evidence was available, the sponsor conducted a patient-level propensity score-adjusted analysis of outcome data, using several approaches (discussed in this section) to account for between-population differences. The sponsor’s primary analysis was performed using a stabilized weights approach, with 4 clinical characteristics that were reported to be chosen on the basis of clinical input: dialysis status at baseline, eGFR at baseline, platelet count at baseline, and LDH at baseline. The sponsor noted that systolic blood pressure was observed to be of importance at baseline, but that it was not included in the statistical model owing to similarities between the 2 eligible patient populations at baseline. The justification of stabilized weights was provided because the sponsor noted that the effective sample size calculation was subject to inflation owing to patients with unexpected propensity score values. Given that LDH levels were noted to remain imbalanced following the application of stabilized weights, the sponsor refactored baseline LDH values into both terciles and halves, identifying that balance was better for LDH refactored into halves.
Separate sensitivity analyses were performed using an inverse probability of treatment weighting approach and a propensity score matching approach. For propensity score matching approaches, 1:1 matching was used. A caliper width of 0.2 times the SD of the propensity score was used in random order, with sensitivity analyses performed using a more restrictive caliper of 0.01.
Stabilized weights were also applied to several sensitivity analyses, with restrictions on populations, outcome definitions, and missing data as follows (with the provided justifications, where available):
Outcome data to within 28 days (rather than 56 days) of the primary end point
Cases with complete final outcome data (i.e., for eGFR, LDH, creatinine, and platelet count)
Patients from South Korea, Taiwan, and Japan excluded
It was unclear whether between-country differences in overall health care might influence the comparative efficacy findings, given that these characteristics could not be accounted for in the primary model used.
Patients who died during the study excluded
Patients who died during the study did not have laboratory outcome data at the final end point.
Only patients aged under 65 years included
Patients who were older were considered to have worse prognoses overall.
Adult patients with kidney transplant
These patients were noted by the sponsor to be considered to have substantially different prognoses compared to adult patients without kidney transplant.
Pediatric patients without prior transplant
These patients were noted by the sponsor to be considered to have substantially different prognoses compared to pediatric patients with kidney transplant.
Data were provided on pediatric patients with transplant, but because of the small sample size (refer to the Results of the Sponsor-Submitted Analysis section), this analysis was not provided.
The study sponsor indicated that outcome definitions across the trials were the same except for dialysis at baseline and end point. For the primary analysis, end points were considered eligible if they were recorded within 56 days of 6 months’ follow-up from baseline; and a sensitivity analysis was performed restricting the outcome eligibility window to 28 days. To harmonize definitions within the constraints of patient-follow-up, the sponsor provided the following baseline and end point dialysis definitions for the following treatments and trials:
For ravulizumab in Study 311 and Study 312 and eculizumab in Studies C10-003 and C10-004, dialysis at baseline was recorded as “yes” for patients who received dialysis within 5 days before study drug initiation.
For eculizumab in Study C08-002, dialysis at baseline was recorded as “yes” for patients who received dialysis within 7 days before study drug initiation.
For ravulizumab in Studies 311 and 312, dialysis at the end points was recorded as “yes” for patients who received dialysis within 5 days before their end point measure.
For eculizumab in Studies C08-002, C10-003, and C10-004, dialysis at the end points was recorded as “yes” for patients who received dialysis within 7 days before their end point measure.
With respect to the statistical testing of differences between populations, Welch’s 2-way t-tests were performed for continuous variables, while chi-square tests were used to obtain P values for categorical variables. Statistical significance was determined to be at a P value of less than 0.1 for between-group differences at baseline and less than 0.05 for end point comparisons.
Details | Indirect treatment comparison 1 |
|---|---|
ITC methods | Stabilized inverse propensity score weights on patient-level data from pooled trials (primary) |
Outcomes | Dialysis at end point, death, CKD stage, change in CKD stage, creatinine in nondialysis patients, change in creatinine from baseline in nondialysis patients, improvement in creatinine in nondialysis patients, platelet count, change in platelet count from baseline, platelet count normalization, LDH, change in LDH from baseline, LDH normalization, eGFR, change in eGFR from baseline, improvement in eGFR, eGFR for nondialysis patients, change in eGFR from baseline for nondialysis patients, improvement in eGFR for nondialysis patients, systolic blood pressure, change in systolic blood pressure from baseline, fatigue (FACIT SS), change from baseline in FACIT SS, FACIT SS 3-point improvement from baseline, HRQoL (EQ-5D VAS), change from baseline in EQ-5D VAS, 10-point improvement in EQ-5D VAS from baseline, EQ-5D time trade-off, change from baseline in EQ-5D time trade-off, hematologic normalization, complete TMA response, time to complete TMA response |
Follow-up time points | Within 56 days of 6 months (primary); within 28 days of 6 months (sensitivity) |
Sensitivity analyses |
|
Subgroup analysis |
|
CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; FACIT = Functional Assessment of Chronic Illness Therapy; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; SS = subscale score; TMA = thrombotic microangiopathy; VAS = visual analogue scale.
Source: Sponsor-submitted analysis.42
Results of Sponsor-Submitted Analysis
Summary of Included Studies
The sponsor did not provide details on the characteristics of each individual trial included in the primary or sensitivity analyses. The application of restriction criteria for participants, as detailed in the analysis methods, resulted in a reduction in eligible sample size across all analysis populations studied, as demonstrated in Figure 6.
Figure 6: Patient Flow Chart Demonstrating Attrition Upon Application of ITC Criteria
FAS = full analysis set; ITC = indirect treatment comparison.
Note: Patient numbers for the final analysis represent patients with complete cases for propensity score variables, no more than 1 missing laboratory measure, and outcome data within 56 days of the 6-month end point. Children with kidney transplant were not included in the analysis due to low patient numbers.
Source: Sponsor-submitted analysis.42
As depicted in Figure 6, across the 3 major subpopulations of interest, there were variances with respect to the eligible and eventual analysis population sizes. For adult patients without kidney transplant (primary analysis), the ravulizumab primary analysis population represented 92% of the eligible patients, and the eculizumab population represented 95% of the eligible patients. For adult patients with kidney transplant, the ravulizumab population represented 87.5% of the eligible patients, and the eculizumab population represented 93.8% of the eligible patients. For pediatric patients without kidney transplant, the ravulizumab population represented 60% of the eligible patients, and the eculizumab population represented 95% of the eligible patient population.
For the primary analyses, Study 311 represents the entire analysis set for ravulizumab, and the unweighted patient demographics represent this trial alone among those eligible for analysis. The eculizumab data on adult populations without kidney transplant are informed by 2 trials, Study CO8-002 (21% of eculizumab patients in this cohort) and Study C10-004 (79% of eculizumab patients in this cohort). Data on the unadjusted demographic differences across these 2 populations are provided in Table 29. Data for eculizumab are merged across the 2 trials for which eculizumab data were available.
Overall, the 2 unweighted populations were comparable for most baseline covariates measured and assessed, with exceptions for: the proportion of patients from Japan, South Korea, and Taiwan (eculizumab = 0%, ravulizumab = 20%; 95% CI, 8 to 31); mean age (ravulizumab = 40 years, eculizumab = 35 years; 95% CI, 0 to 12); and LDH (eculizumab = 484 [SD = 518], ravulizumab = 714 [SD = 586]; 95% CI = –7 to 469).
Table 29: Patient Baseline Demographics, Unweighted Sample, Adults Without Kidney Transplant
Characteristic | Eculizumab | Ravulizumab | 95% CIa | |
|---|---|---|---|---|
Number of patients | — | 39 | 46 | — |
Trial name | ALXN-311 | 0 (0%) | 46 (100%) | |
ALXN-312 | 0 (0%) | 0 (0%) | ||
C08-002 | 8 (21%) | 0 (0%) | NR | |
C10-003 | 0 (0%) | 0 (0%) | ||
C10-004 | 31 (79%) | 0 (0%) | ||
Sex, n (%) | Female | 24 (62%) | 31 (67%) | –15% to 26% |
Male | 15 (38%) | 15 (33%) | ||
Region, n (%) | Japan, South Korea, Taiwan | 0 (0%) | 9 (20%) | (8% to 31%) |
All regions other than Japan, South Korea, Taiwan | 39 (100%) | 37 (80%) | ||
Baseline CKD stage, n (%) | 1 | 0 (0%) | 0 (0%) | |
2 | 0 (0%) | 3 (7%) | ||
3.1 | 3 (8%) | 1 (2%) | NR | |
3.2 | 4 (10%) | 2 (4%) | ||
4 | 6 (15%) | 5 (11%) | ||
5 | 26 (67%) | 35 (76%) | ||
Dialysis at baseline, n (%) | Yes | 19 (49%) | 26 (57%) | –13% to 29% |
No | 20 (51%) | 20 (43%) | ||
Age (years) | Mean | 35 | 40 | 0 to 12 |
Standard deviation | 13 | 15 | ||
Median | 31 | 37 | ||
Range | 50 | 58 | ||
N | 39 | 46 | ||
Aged 65+ years, n (%) | Yes | 2 (5%) | 5 (11%) | –6% to 17% |
No | 37 (95%) | 41 (89%) | ||
Height (cm) | Mean | 170 | 167 | –7 to 2 |
Standard deviation | 11 | 9 | ||
Median | 168 | 166 | ||
Range | 50 | 38 | ||
N | 39 | 45 | ||
Weight (kg) | Mean | 73 | 74 | –7 to 9 |
Standard deviation | 18 | 18 | ||
Median | 72 | 68 | ||
Range | 81 | 66 | ||
N | 38 | 45 | ||
BMI (kg/m2) | Mean | 25.2 | 26.4 | –2 to 4 |
Standard deviation | 6.6 | 5.8 | ||
Median | 23.4 | 25.1 | ||
Range | 28.7 | 22.7 | ||
N | 38 | 44 | ||
Creatinine in nondialysis patientsb | Mean | 348 | 405 | |
Standard deviation | 234 | 298 | –114 to 229 | |
Median | 243 | 295 | ||
Range | 875 | 956 | ||
N | 20 | 20 | ||
Platelet count at baseline (per μL) | Mean | 117 | 121 | –28 to 37 |
Standard deviation | 62 | 89 | ||
Median | 124 | 95 | ||
Range | 314 | 455 | ||
N | 39 | 46 | ||
LDH levels at baseline (per L) | Mean | 484 | 714 | –7 to 469 |
Standard deviation | 518 | 586 | ||
Median | 323 | 466 | ||
Range | 3,184 | 3,020 | ||
N | 39 | 46 | ||
eGFR at baseline (mL/min/1.73m2) | Mean | 17.4 | 16.2 | –7 to 5 |
Standard deviation | 13.2 | 16 | ||
Median | 10 | 10 | ||
Range | 53.2 | 76 | ||
N | 39 | 46 | ||
eGFR at baseline for nondialysis patients (mL/min/1.73m2) | Mean | 24.3 | 24.3 | –12 to 12 |
Standard Deviation | 15.5 | 22.1 | ||
Median | 23.8 | 17 | ||
Range | 53.2 | 76 | ||
N | 20 | 20 | ||
Systolic blood pressure at baseline (mm Hg) | Mean | 141 | 145 | –4 to 11 |
Standard Deviation | 18 | 17 | ||
Median | 142 | 141 | ||
Range | 85 | 80 | ||
N | 39 | 43 | ||
FACIT subscale score at baseline | Mean | 23 | 24 | |
Standard deviation | 14 | 14 | –7 to 7 | |
Median | 23 | 24 | ||
Range | 44 | 47 | ||
N | 28 | 38 | ||
EQ-5D VAS at baseline | Mean | 49 | 47 | –13 to 9 |
Standard deviation | 18 | 29 | ||
Median | 50 | 50 | ||
Range | 71 | 90 | ||
N | 35 | 39 | ||
EQ-5D TTO at baseline | Mean | 0.67 | 0.56 | –0.26 to 0.04 |
Standard deviation | 0.3 | 0.34 | ||
Median | 0.81 | 0.63 | ||
Range | 0.96 | 1.02 | ||
N | 35 | 40 | ||
BMI = body mass index; CI = confidence interval; CKD = chronic kidney disease, eGFR = estimated glomerular filtration rate; FACIT = Functional Assessment of Chronic Illness Therapy; LDH = lactate dehydrogenase; NR = not reported; TTO = time trade-off; VAS = visual analogue scale.
aRepresents the 95% CI of the mean difference between treatments for continuous variables, and the 95% CI of the mean difference in proportions for categorical variables. For categorical variables, 95% CI are presented for binary outcomes only and refer to the 95% CI around the difference between treatments for the first listed category (e.g., “yes” for dialysis at baseline).
bUnit not reported.
Source: Sponsor-submitted analysis.42
Following the application of stabilized weights, assessments were made of the balance of baseline characteristics among the study population, as demonstrated in Table 30. In this stabilized weights analysis of adult patients without kidney transplant, no statistically significant differences were noted between the treatment populations except for the proportion of patients from Japan, South Korea, and Taiwan (eculizumab = 0%, ravulizumab = 23%; 95% CI, 10 to 35; P = 0.002).
Table 30: Patient Baseline Demographics, Stabilized Weights Sample, Adults Without Kidney Transplant
Characteristic | Detail | Eculizumab | Ravulizumab |
|---|---|---|---|
Effective sample size, N | — | 39 | 46 |
Trial name | ALXN-311 | 0 (0%) | 46 (100%) |
ALXN-312 | 0 (0%) | 0 (0%) | |
C08-002 | 7.3 (19%) | 0 (0%) | |
C10-003 | 0 (0%) | 0 (0%) | |
C10-004 | 31.7 (81%) | 0 (0%) | |
Sex, n (%) | Female | 23.6 (61%) | 29.9 (65%) |
Male | 15.4 (39%) | 16.2 (35%) | |
Region, n (%) | Asia | 0 (0%) | 10.4 (23%) |
Ex-Asia | 39 (100%) | 35.6 (77%) | |
Baseline CKD stage, n (%) | 1 | 0 (0%) | 0 (0%) |
2 | 0 (0%) | 3.3 (7%) | |
3.1 | 2.4 (6%) | 1.3 (3%) | |
3.2 | 4.1 (11%) | 1.8 (4%) | |
4 | 5.2 (13%) | 5.5 (12%) | |
5 | 27.2 (70%) | 34.2 (74%) | |
Dialysis at baseline, n (%) | Yes | 20.5 (53%) | 24.2 (52%) |
No | 18.5 (47%) | 21.9 (48%) | |
Age (years) | Mean | 34 | 40 |
Standard deviation | 13 | 14 | |
Median | 31 | 37 | |
Range | 50 | 58 | |
N | 39 | 46 | |
Age 65+, n (%) | Yes | 1.5 (4%) | 4.8 (10%) |
No | 37.4 (96%) | 41.2 (90%) | |
Height, cm | Mean | 170 | 167 |
Standard deviation | 11 | 9 | |
Median | 168 | 167 | |
Range | 50 | 38 | |
N | 39 | 45 | |
Weight, kg | Mean | 72 | 73 |
Standard deviation | 16 | 18 | |
Median | 72 | 68 | |
Range | 81 | 66 | |
N | 38 | 45 | |
BMI, kg/m2 | Mean | 25 | 26.3 |
Standard deviation | 6.2 | 5.7 | |
Median | 23.4 | 25.4 | |
Range | 28.7 | 22.7 | |
N | 38 | 44 | |
Creatinine in nondialysis patientsa | Mean | 348 | 419 |
Standard deviation | 231 | 301 | |
Median | 278 | 326 | |
Range | 875 | 956 | |
N | 18 | 22 | |
Platelet count at baseline, per μL | Mean | 118 | 118 |
Standard deviation | 65 | 85 | |
Median | 125 | 95 | |
Range | 314 | 455 | |
N | 39 | 46 | |
LDH levels at baseline, per L | Mean | 534 | 664 |
Standard deviation | 549 | 568 | |
Median | 435 | 432 | |
Range | 3,184 | 3,020 | |
N | 39 | 46 | |
eGFR at baseline, mL/min/1.73m2 | Mean | 16.6 | 16.7 |
Standard deviation | 12.4 | 16.6 | |
Median | 10 | 10 | |
Range | 53.2 | 76 | |
N | 39 | 46 | |
eGFR at baseline for nondialysis patients, mL/min/1.73m2 | Mean | 24 | 24 |
Standard deviation | 15 | 22.1 | |
Median | 24.4 | 17 | |
Range | 53.2 | 76 | |
N | 18 | 22 | |
Systolic blood pressure at baseline, mm Hg | Mean | 143 | 145 |
Standard deviation | 17 | 16 | |
Median | 143 | 148 | |
Range | 85 | 80 | |
N | 39 | 43 | |
FACIT subscale score at baseline | Mean | 23 | 25 |
Standard deviation | 14 | 15 | |
Median | 23 | 24 | |
Range | 44 | 47 | |
N | 28 | 38 | |
EQ-5D VAS at baseline | Mean | 48 | 50 |
Standard deviation | 18 | 30 | |
Median | 50 | 60 | |
Range | 71 | 90 | |
N | 35 | 39 | |
EQ-5D TTO at baseline | Mean | 0.65 | 0.58 |
Standard deviation | 0.31 | 0.34 | |
Median | 0.79 | 0.71 | |
Range | 0.96 | 1.02 | |
N | 35 | 40 |
BMI = body mass index; CKD = chronic kidney disease; eGFR = estimated glomerular filtration rate; FACIT = Functional Assessment of Chronic Illness Therapy; LDH = lactate dehydrogenase; TTO = time trade-off; VAS = visual analogue scale.
aUnit not reported.
Source: Sponsor-submitted analysis.42
Efficacy Outcome Results
The sponsor-submitted study’s primary analysis was performed on the stabilized weights population of adult patients without kidney transplant, comparing the relative efficacy of ravulizumab to eculizumab, with outcomes assessed at 6 months plus or minus 56 days. A summary of the findings is presented in Table 31. The effective sample size for the eculizumab population was 39 and the effective sample size for the ravulizumab population was 46. Several outcomes of interest specified in the CADTH study protocol were unavailable: the presence of severe bleeding, hemoglobin concentration, plasma therapy–free status, packed RBC transfusions, hospitalizations, and presence of soluble MAC.
Briefly, the 95% CIs were generally too wide to conclude whether a difference existed between the treatments among the key outcomes of interest defined in the study review protocol. Additionally, the sponsor provided sensitivity analyses to cover subpopulations and scenarios, as described in Table 28. Broadly, these were consistent with the findings of the primary analysis.
Two other subpopulation analyses were presented by the sponsor: adults with kidney transplant at baseline, and pediatric patients without kidney transplant at baseline. For adults with kidney transplant, the effective sample sizes were 12.7 for eculizumab and 9.3 for ravulizumab, limiting the ability to draw conclusions. Similarly, the effective sample sizes for pediatric patients without transplant were limited to 21.3 for eculizumab and 10.7 for ravulizumab.
No safety outcomes were identified in the sponsor’s submitted ITC; therefore, no comparisons of relative safety between ravulizumab and eculizumab are possible.
Table 31: Efficacy Results of the Sponsor’s ITC, Stabilized Weights, Adult Patients With aHUS Without Renal Transplant, 26 Weeks
Results | Eculizumab (stabilized weights, Study C08-002 and Study C10-004) ESS = 39 | Ravulizumab (stabilized weights, Study ALXN-311) ESS = 46 |
|---|---|---|
Mortality | ||
Death in study period, % (95% CI) | 0% (0% to 9%) | 7% (2% to 18%) |
95% CI of mean difference in proportions | –1% to 14% | |
P value | 0.103 | |
Complete TMA response at 6 months | ||
Complete TMA response proportion | 70% (54% to 82%) | 61% (46% to 74%) |
95% CI of mean difference in proportions | –29% to 12% | |
P value | 0.398 | |
Time to complete TMA response | ||
N assessable | 39 | 43 |
Mean time to complete TMA response (SD) | 169 | 156 |
95% CI of mean complete TMA difference between treatments | –88 to 62 | |
P value | 0.728 | |
LDH at 6 months | ||
N assessable | 39 | 43 |
Mean LDH (SD) | 179 | 200 |
95% CI of mean LDH difference between treatments | –1 to 42 | |
P value | 0.059 | |
LDH change from baseline at 6 months | ||
N assessable | 39 | 43 |
Mean change in LDH from baseline (SD) | –355 | –475 |
95% CI of mean LDH change from baseline difference between treatments | –372 to 131 | |
P value | 0.344 | |
LDH normalization at 6 months | ||
LDH normalization, % (95% CI) | 95% | 89% |
95% CI of mean difference in proportions | –17%, 6% | |
P value | ||
Platelet count at 6 months | ||
N assessable | 39 | 43 |
Mean platelet count | 244 | 200 |
95% CI of mean platelet count difference between treatments | –33 to 31 | |
P value | 0.953 | |
Platelet count change from baseline at 6 months | ||
N assessable | 39 | 43 |
Mean change in platelet count from baseline (SD) | 126 | 122 |
95% CI of mean platelet count change from baseline difference between treatments | –50 to 41 | |
P value | 0.855 | |
Platelet count normalization at 6 months | ||
Platelet count normalization, % (95% CI) | 96% | 92% |
95% CI of mean difference in proportions | –14% to 6% | |
P value | 0.391 | |
EQ-5D VAS at 6 months | ||
N assessable | 37 | 41 |
Mean EQ-5D VAS (SD) | 74 | 79 |
95% CI of mean EQ-5D VAS difference between treatments | –4 to 13 | |
P value | 0.260 | |
EQ-5D VAS change from baseline at 6 months | ||
N assessable | 35 | 38 |
Mean change in EQ-5D VAS from baseline (SD) | 26 | 29 |
95% CI of mean EQ-5D VAS change from baseline difference between treatments | –9 to 15 | |
P value | 0.642 | |
10-point improvement in EQ-5D VAS from baseline at 6 months | ||
10-point improvement in EQ-5D VAS from baseline, % (95% CI) | 86% | 83% |
95% CI of mean difference in proportions | –20% to 13% | |
P value | 0.687 | |
FACIT subscale score at 6 months | ||
N assessable | 30 | 40 |
Mean FACIT subscale score (SD) | 40 | 43 |
95% CI of mean FACIT subscale score difference between treatments | –3 to 8 | |
P value | 0.382 | |
FACIT subscale score change from baseline at 6 months | ||
N assessable | 28 | 37 |
Mean change in FACIT subscale score from baseline,(SD) | 17 | 18 |
95% CI of mean FACIT subscale score change from baseline difference between treatments | –6 to 8 | |
P value | 0.803 | |
3-point improvement in FACIT subscale score from baseline at 6 months | ||
10-point improvement in FACIT subscale score from baseline, % (95% CI) | 88% | 84% |
95% CI of mean difference in proportions | –21% to 13% | |
P value | 0.623 | |
Dialysis at 6 months | ||
Dialysis, % (95% CI) | 8% | 22% |
95% CI of mean difference in proportions | –1% to 30% | |
P value | 0.07 | |
Improvement in CKD stage at 6 months | ||
Improvement in CKD stage, % (95% CI) | 81% | 69% |
95% CI of mean difference in proportions | NR | |
P value | 0.255 | |
Creatinine in nondialysis patients at 6 months | ||
N assessable | 36 | 33 |
Mean creatinine, (SD) | 152 | 179 |
95% CI of mean creatinine difference between treatments | –12 to 251 | |
P value | 0.595 | |
Change in creatinine from baseline in nondialysis patients at 6 months | ||
N assessable | 17 | 18 |
Mean creatinine change from baseline (SD) | –191 | –128 |
95% CI of mean creatinine change from baseline difference between treatments | –126 to 251 | |
P value | 0.531 | |
Improvement in eGFR at 6 months | ||
Improvement in eGFR, % (95% CI) | 64% | 59% |
95% CI of mean difference in proportions | –26% to 16% | |
P value | 0.662 | |
eGFR for nondialysis patients at 6 months | ||
N assessable | 36 | 33 |
Mean eGFR (SD) | 55 | 68.5 |
95% CI of mean eGFR difference between treatments | –3 to 30 | |
P value | 0.099 | |
eGFR change from baseline at 6 months | ||
N assessable | 17 | 18 |
Mean eGFR change from baseline (SD) | 34.4 | 39.7 |
95% CI of mean eGFR change from baseline difference between treatments | –15 to 26 | |
P value | 0.608 | |
CI = confidence interval; CKD = chronic kidney disease, eGFR = estimated glomerular filtration rate; ESS = effective sample size; FACIT = Functional Assessment of Chronic Illness Therapy; ITC = indirect treatment comparison; LDH = lactate dehydrogenase; SD = standard deviation; TMA = thrombotic microangiopathy; VAS = visual analogue scale.
Source: Sponsor-submitted ITC.42
Critical Appraisal of Sponsor-Submitted Analysis
A substantial limitation of the submitted analysis is the absence of safety data. Without these, it is not possible to compare the relative efficacy and safety; and given that the results of the analysis predominantly indicate uncertainty with respect to efficacy, treatment decisions may be heavily driven by safety data and patient preference. While naive unadjusted comparisons could be considered by observing published safety events, the sponsor-submitted analysis noted that population adjustment resulted in changes to comparative efficacy estimates. Accordingly, the influence of differences in patient populations with respect to safety events is unknown and remains an important gap in the available evidence when considering the safety of ravulizumab relative to eculizumab.
It is important to note that the provided propensity-adjusted analyses specifically incorporated only 4 covariates; as such, residual confounding from unmeasured characteristics may remain a concern with respect to the relative treatment effects observed. While the sponsor did indicate that covariate selection was based on clinical consultation, data are not provided on this process, and no quantification or exploration of the influence of these covariates on outcomes is provided. Some characteristics that may be important and quantifiable, such as the use of plasma therapy, were not reported to be available owing to inconsistent reporting across trials. Other subgroups of interest to this review were similarly unavailable, including the gene mutation status of patients and the severity of disease as defined by organ involvement. Separately, the sponsor noted a substantial time interval (approximately 10 years) between the eculizumab and ravulizumab trials. Temporal biases may include changes to standard of care, increased awareness of or capacity to diagnose disease, and changes in health care system capacity. These are all confounding factors that cannot be excluded from the current analysis. Indeed, the clinician input provided for this review indicated that improvements in access to genetic testing and other diagnostics have improved within Canada over the past 10 years to 15 years.
The sponsor also indicated that a fifth important characteristic, systolic blood pressure, was similar at baseline and, as a result, excluded from the propensity model. This is despite the fact that the systolic blood pressure difference demonstrated a similar difference (with respect to reported P value in the preweighting analysis) to other characteristics retained in the model, such as eGFR and platelet count. For example, among adult patients without transplant, baseline eGFRs were 17.4 in the eculizumab population and 16.2 in the ravulizumab population before weighting (P = 0.719). No further rationale is provided with respect to the exclusion of systolic blood pressure from subsequent propensity model-based analyses. While there is a balance to be met with respect to the number of covariates and the available sample size, it is also critical to incorporate all potentially clinically important covariates within a propensity model, regardless of between-group differences, to ensure appropriate inferences can be made.
No systematic review was undertaken, and the sponsor’s process for eliminating a trial of potential interest was unclear. Although the study population may be significantly different with respect to broad characteristics, data are not provided to back up this assertion; therefore, this cannot be assessed quantitatively or qualitatively. Because of the small available sample sizes for the primary analysis — and in particular, the subpopulation of interest — the influence of additional patient data may substantially influence the comparative effects observed in the sponsor-submitted ITC.
In terms of the applicability of the analysis to the population of patients in Canada, data were not presented with respect to the coverage of patients from Canada; therefore, the influence of systematic differences in health care provision between the included geographies of patients among the trial populations is unclear. The sponsor did provide a sensitivity analysis that excluded patients recruited in Japan, South Korea, and Taiwan, but this did not substantially alter the comparative efficacy estimates.
The submitted analysis provides no formal specification within the methods with respect to the estimand of interest used by the sponsor in its propensity-weighting models. As such, it is unclear whether the reported results correspond to the average treatment effect on the treated population or the average treatment effect within this analysis. Similarly, units of measurement are not provided for the outcomes and baseline characteristics of interest.
With respect to outcome data, it is important to note that all presented outcome analyses (except time-to-event outcomes) were limited to up to 6 months of follow-up time, plus or minus 56 days. As such, the present analysis does not permit the assessment of longer-term outcomes; uncertainty exists beyond the observed 6-month time window with respect to the efficacy of ravulizumab relative to eculizumab.
Summary
Overall, 1 study — a sponsor-submitted, stabilized, inverse propensity score–weighted analysis of pooled individual patient data — was available to assess the efficacy of ravulizumab relative to eculizumab. In a patient-level, propensity-based primary analysis with wide CIs, it was not possible to conclude whether differences exist between ravulizumab and eculizumab with respect to mortality, complete TMA response, LDH, platelets, HRQoL (EQ-5D VAS), fatigue (FACIT subscales), renal function, or dialysis status among adult patients with aHUS at 6 months. No safety data were available for review.
The 1 study submitted is subject to a number of limitations owing to the small available sample size, temporal biases between the comparator trial populations, and the absence of potentially significant clinical covariates in the model used. Further, because safety data were not presented for review, no conclusions can be drawn about the safety of ravulizumab relative to eculizumab. Accordingly, uncertainty remains with respect to the efficacy and safety of ravulizumab relative to eculizumab.
Other Relevant Evidence
No other relevant evidence was identified.
Discussion
Summary of Available Evidence
Two pivotal, sponsor-funded, prospective, multinational, phase III, single-arm trials (Study 311 for adults and Study 312 for children)21,22 were included in this review. An additional sponsor-submitted ITC using a patient-level, propensity score-adjusted analysis that compared the efficacy of ravulizumab with eculizumab in patients with aHUS was also included. No other relevant studies or ITCs were identified.
Study 311 is an ongoing, phase III, prospective, multicentre, single-arm, open-label trial that includes adult patients with aHUS treated with ravulizumab.21 The key objective is to evaluate the safety and efficacy of ravulizumab (administered through IV infusion) in adult patients (aged ≥ 18 years) with aHUS who are complement inhibitor treatment–naive. A total of 58 patients were included in this study, of whom 56 patients received at least 1 dose of ravulizumab. The primary outcome was complete TMA response during the 26-week initial evaluation period, which was defined as the normalization of hematologic parameters (platelet count and LDH) and an improvement of at least 25% in serum creatinine from baseline. The secondary outcomes were hematologic normalization (platelet count and LDH), hematologic TMA parameters (platelet count, LDH, hemoglobin), hemoglobin response (an increase of more than 2% increase), renal function (i.e., serum creatine, eGFR, dialysis status, and CKD stage change), fatigue (FACIT-F), and HRQoL (3-Level EQ-5D), as well as safety. Health care resource utilization, patient-reported aHUS symptoms, and extrarenal signs and symptoms of aHUS were reported as exploratory outcomes. At the data cut-off date (median follow-up periods = 75.5 weeks for Study 311, 82.4 weeks for Study 312, cohort 1, and 52.3 weeks for Study 312, cohort 2), the study was still ongoing and was expected to continue for up to 4.5 years.
Study 312 is an ongoing, phase III, prospective, multinational, single-arm, open-label trial that includes pediatric patients (aged < 18 years) with aHUS.22 Study 312 includes 2 cohorts (i.e., cohort 1 and cohort 2). Cohort 1 includes 21 children with aHUS who are complement inhibitor–naive. The key objective for cohort 1, Study 312, is to evaluate the safety and efficacy of ravulizumab (IV infusion) in pediatric patients with aHUS who are complement inhibitor treatment–naive. The outcomes assessed are the same as those in Study 311. Cohort 2 includes 10 children (aged < 18 years) with aHUS who previously responded to eculizumab with stable TMA parameters. The key objective for cohort 2, Study 312 is to evaluate the safety and efficacy of ravulizumab (administered through IV infusion) in children with aHUS following a switch from eculizumab to ravulizumab. The outcomes assessed in Study 312, cohort 2 were hematologic TMA parameters (platelet count, LDH, and hemoglobin), renal function, fatigue (FACIT-F), and safety.
Direct evidence comparing ravulizumab to eculizumab was unavailable; such comparisons are likely to be infeasible due to the rare and severe nature of aHUS. In the absence of direct comparative evidence, the sponsor submitted a propensity score–weighted comparison of the efficacy of ravulizumab versus eculizumab in the treatment of patients with aHUS. This ITC analysis estimated the mortality, complete TMA response, LDH, platelets, HRQoL (EQ-5D VAS), FACIT subscales, renal function, and dialysis status among adult patients with aHUS at 6 months’ follow-up.
Interpretation of Results
Efficacy
Among complement inhibitor treatment–naive adult and pediatric patients with aHUS who received a weight-based dosage of ravulizumab IV, at week 26, the majority were able to achieve a complete TMA response (54% in adult patients and 78% in pediatric patients), hematological normalization (73% in adult patients and 89% in pediatric patients), hemoglobin response (71% in adult patients and 89% in pediatric patients), and an improvement of at least 25% in serum creatinine from baseline (59% in adult patients and 83% in pediatric patients). In addition, the majority of patients experienced renal function improvement as measured by eGFR, CKD stage shifting, and dialysis status in both Study 311 and Study 312, cohort 1. Improvements in fatigue (FACIT-F) and HRQoL (3-Level EQ-5D) for adults in Study 311 and in fatigue (FACIT-F) for pediatrics in Study 312, cohort 1 were also observed in most patients. The apparent clinical benefits observed at week 26 were largely sustained and/or further improved through the extension period at the data cut-off date (median follow-up times = 75.57 weeks and 82.43 weeks for Study 311 and Study 312, cohort 1, respectively). It was also noted that in Study 311, 2 patients (3.5%) experienced worsening of CKD, and 7 patients of 27 patients (25.9%) who did not need dialysis at baseline started new dialysis during the trial.
For the majority of patients included in cohort 2, Study 312 (N = 10), at week 26 and through the data-cut-off date, hematological parameters, renal function, and fatigue findings appeared to be stable after patients switched from eculizumab to ravulizumab treatment. It should be noted that 2 patients of 10 patients (20%) experienced CKD stage worsening (by 1 stage); and 1 patient of 10 patients (10%) worsened by 3 stages during the initial 26 weeks, but returned to their original baseline normal CKD stage before the data-cut-off date.
Interpretation of the efficacy and safety findings is challenging in a single-arm trial, given that without a comparison group and no consideration of confounding variables, the observed efficacy results could potentially be confounded. Given that the trials were uncontrolled, and that it is unclear to what extent patients with poor prognoses were excluded from the trials, the impact of ravulizumab on TMA response is unclear. In addition, there was no formal hypothesis testing done in the 2 pivotal studies. Given that the trial was not designed to detect differences in treatment effects across subgroups, no conclusions should be drawn on the basis of prespecified subgroup results. Therefore, the findings should be interpreted with consideration for the limitations previously discussed.
However, it should be noted that, clinically, in patients with aHUS, acute, active onset of TMA is an extremely severe condition causing end organ damage (e.g., leading to CKD stage 4 or 5), often resulting in permanent disability and/or death. The majority of adult patients included in Study 311 and pediatric patients included in Study 312, cohort 1 were severely ill at study entry; most were hospitalized and had advanced kidney disease (i.e., CKD stage 4 or 5). In Study 311 and Study 312, cohort 1, 51.8% and 33% of patients, respectively, received dialysis in the 5 days before the study or when entering it. In addition, 82.8% of patients in Study 311 had received plasma exchange and/or plasma infusion related to their current TMA before receiving ravulizumab. Eight patients (14.3%) in Study 311 and 1 patient in Study 312, cohort 1 had already received a kidney transplant. All patients had aHUS symptoms (i.e., renal and extrarenal signs or symptoms). Furthermore, the clinical experts indicated that the complete TMA response to empirical plasma therapy for aHUS was reported in only 7% of patients. Therefore, the efficacy findings — complete TMA response, hematological normalization, kidney function improvement, discontinuation of dialysis, and change in CKD stage from severe (stage 5 or 4) to less severe (i.e., stage 3 or less) in both adult and pediatric patients with aHUS — appeared to be clinically meaningful, considering the nature of severe and life-threatening aHUS in this population. This view was echoed by the patient group input received from aHUS Canada, which noted that patients who had experience with ravulizumab reported more energy, less vein damage, fewer treatments, fewer symptom fluctuations, more freedom of choice, and less anxiety.
The diagnosis of aHUS is based on excluding other secondary TMA.10 Patients included in the 2 pivotal studies may potentially include some who did not have a confirmed diagnosis (such as 8 patients with kidney transplant not related to prior aHUS in Study 311). However, the clinical experts consulted by CADTH for this review indicated that ravulizumab is a C5 inhibitor, which is indicated for complement-mediated TMA (i.e., aHUS); it is expected that patients with a confirmed diagnosis of aHUS would respond better than those without a confirmed diagnosis of aHUS. Therefore, any bias associated with the uncertain diagnosis, if any, would be against ravulizumab, making it appear less effective at improving TMA parameters in this population. This might explain why the complete TMA response reported in Study 311 among adult patients appeared lower than would be expected by the clinical experts.
It is noted that during both studies, high proportions of patients experienced major protocol deviations. Many of these deviations were due to the complex eligibility criteria necessary to exclude, often in an acute setting, potential diagnoses other than complement-mediated TMA. For example, only 30 patients of the 56 patients in Study 311 met all of the TMA criteria at day 1 of the trial. Clinically, urgent treatment for a broad differential diagnosis needed to be initiated in this population. Also, the results for complete TMA response in the PP set and subgroup analysis for patients who met all of the laboratory criteria for TMA at day 1 were consistent with the results from the primary FAS analysis. This reduces concern about bias being introduced by the protocol deviations.
Both the patient advocacy groups and the clinical experts consulted by CADTH highlighted symptom reduction and HRQoL as important outcomes. Fatigue results (FACIT-F) in both Study 311 and Study 312, cohort 1 showed clinically meaningful improvements (≥ 3 points for both adult and children) in the majority of patients. HRQoL (3-Level EQ-5D) among adult patients in Study 311 also showed improvement overall. However, it remains uncertain whether the HRQoL improvement is clinically meaningful because the MID for the aHUS population is not known. Nevertheless, the patient groups and clinical experts consulted by CADTH expressed that they would expect to observe a substantial HRQoL improvement in patients with aHUS if ravulizumab were available, based on the substantially reduced frequency of IV injections required for ravulizumab compared with both eculizumab (the standard of care for aHUS in some jurisdictions) and supportive care (such as plasma therapy).
Among the 10 pediatric patients enrolled in Study 312, cohort 2, switching from eculizumab to ravulizumab appeared to result in sustained disease control (i.e., stable TMA parameters and renal function) as of the data cut-off date. However, because the sample size was small (N = 10), it is unclear whether the findings in this population can be generalized to all pediatric patients switching from eculizumab to ravulizumab. In a letter to editors, Ehren et al.43 reported real-world data from 6 definitively diagnosed pediatric patients with aHUS who switched from eculizumab to ravulizumab. The author indicated that a switch from eculizumab to ravulizumab in pediatric patients with aHUS was feasible in a real-life setting; however, no detailed efficacy or safety data were reported in the letter.43 In both adult and pediatric patients with aHUS, some limited real-world evidence for switching from eculizumab to ravulizumab is available as a conference abstract by Wang et al.44 However, at this time, due to several methodological limitations, it is not possible to draw strong conclusions from these early data. These limitations include the retrospective design, which may have affected data quality and completeness; the small sample size; the lack of a comparison group, with no adjustment for confounding; and the lack of formal statistical testing. It is unclear whether the results from these US patients would be generalizable to Canadian clinical practice.
The optimal duration of ravulizumab therapy, and the clinical conditions under which ravulizumab therapy may be discontinued, have not been well established. The long-term extension phase efficacy results — obtained at a median of 75.57 weeks for Study 311 among adults and 50.29 months to 82.43 months for Study 312 among children — were similar to those reported in the first 26 weeks of ravulizumab treatment. This may suggest that patients who respond in the first 6 months of therapy will likely maintain their response.
The sponsor-submitted propensity score–weighted analysis was inconclusive on the comparison between ravulizumab and eculizumab in terms of clinical efficacy outcomes due to wide CIs and a number of methodological limitations. The analysis did not report on safety outcomes. A noninferiority trial design would be valuable to compare the treatments; however, this is unlikely to be feasible due to the rare nature of the condition. Nevertheless, both ravulizumab and eculizumab are terminal complement inhibitors that specifically bind to the complement protein C5 with high affinity (i.e., the drugs have similar mechanisms of action). The clinical experts expected that ravulizumab would be considered as a first-line treatment for adults and pediatric patients with aHUS due to its substantially reduced frequency of IV administration compared with eculizumab. This would be expected to result in a decreased treatment burden and improved HRQoL for patients.
According to the clinical experts consulted by CADTH, patients recruited in the 2 pivotal trials were considered representative of patients in Canadian clinical practice. There were no major concerns about the generalizability of the findings to Canadian practice. The clinical experts anticipated that because of the mechanism of action and acceptable safety profile of ravulizumab, they would expect to find a benefit of treatment with ravulizumab for all patients with a confirmed diagnosis with aHUS.
Harms
The safety profile of ravulizumab has been well established in previous clinical trials for the treatment of paroxysmal nocturnal hemoglobinuria.18 Almost every patient in the 2 trials experienced at least 1 adverse event. The most common adverse events (reported in > 30% patients) were headache, diarrhea, vomiting, and oropharyngeal pain. These experiences were echoed in the patient group input received from aHUS Canada, which noted that patients who had experience with ravulizumab reported headache, nausea, and body aches right after their infusion or during the month after the infusion.
Treatment discontinuation due to TEAEs was relatively low (4.8% to 5.2% in the 2 trials). Three deaths due to TEAEs were reported, but these were considered unrelated to ravulizumab treatment.
The frequencies of TEAEs, SAEs, and notable adverse events reported in this trial appeared similar to the known safety profile of ravulizumab. No additional safety signals were identified with ravulizumab in the treatment of adult or pediatric patients with aHUS. With ravulizumab, there is the risk of developing meningitis; however, no meningitis was reported in either study. The reason may be that all patients received vaccination against meningitis before entering the studies. Because the vaccine takes 2 weeks to be effective, prophylactic antibiotic therapy was recommended when the 2-week window could not be met. Children (aged < 18 years) also needed to have received vaccination against Streptococcus pneumonia and Hemophilus influenza type B before the trial.
There was no direct evidence from a randomized controlled trial, nor any indirect evidence identified in this review, to inform conclusions about the safety of ravulizumab compared to eculizumab. The clinical experts consulted by CADTH agreed that the weight-based dosing regimen of ravulizumab safety profile observed in these 2 studies seemed generally manageable and consistent with the known safety profile of ravulizumab.
Conclusions
The evidence for the clinical benefits and harms of ravulizumab in the treatment of aHUS was based on the 2 sponsor-submitted, pivotal, multinational, single-arm, open-label, prospective phase III trials (Study 311 for adults with aHUS and Study 312 for pediatric patients with aHUS). The majority of pediatric and adult patients who were complement inhibitor treatment–naive experienced hematological normalization, improved renal function, and improved HRQoL with ravulizumab treatment. Despite uncertainty around the magnitude of the clinical benefit attributable to ravulizumab (given the limitations inherent in the single-arm trial design), the lack of formal hypothesis testing, and the relatively small sample size, the clinical experts indicated that the benefits observed in the 2 trials appeared clinically meaningful, considering that aHUS is an extremely rare and life-threatening disease. For adult patients who were complement inhibitor–experienced, no evidence was identified with the switching from eculizumab to ravulizumab. The expected benefit of switching lies in the reduced number of infusions required (because of the longer half-life of ravulizumab versus eculizumab). Although the 10 patients who switched from eculizumab to ravulizumab in Study 312 appeared to have a maintained TMA response, due to the small sample size, it remains unclear whether these findings are reflective of what would be observed in the larger population of patients with aHUS. The sponsor also submitted a propensity score–weighted analysis comparing ravulizumab with eculizumab; however, due to several methodological limitations, no robust conclusion could be drawn on the comparative efficacy and safety of ravulizumab versus eculizumab. The safety profile of ravulizumab observed in the 2 trials appeared consistent with the known safety profile of ravulizumab, and no additional safety signals were identified.
References
1.Fleming P, Cheung M, Sokol D. Complement-Mediated Thrombotic Microangiopathy: A Murky Presentation of a Rare Disease Entity. Blood. 2018;132(Supplement 1):5005.
2.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-1687. PubMed
3.Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-447. PubMed
4.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. PubMed
5.Schonermarck U, Ries W, Schroppel B, et al. Relative incidence of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome in clinically suspected cases of thrombotic microangiopathy. Clin Kidney J. 2020;13(2):208-216. PubMed
6.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859. PubMed
7.Fakhouri F, Vercel C, Fremeaux-Bacchi V. Obstetric nephrology: AKI and thrombotic microangiopathies in pregnancy. Clin J Am Soc Nephrol. 2012;7(12):2100-2106. PubMed
8.Raina R, Sethi SK, Dragon-Durey MA, et al. Systematic review of atypical hemolytic uremic syndrome biomarkers. Pediatr Nephrol. 2022;37(7):1479-1493. PubMed
9.George JN, Nester C. Thrombotic microangiopathies (TMAs) with acute kidney injury (AKI) in adults: CM-TMA and ST-HUS. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2022 Jun 27.
10.Fakhouri F, Hourmant M, Campistol JM, et al. Terminal Complement Inhibitor Eculizumab in Adult Patients With Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am J Kidney Dis. 2016;68(1):84-93. PubMed
11.Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551. PubMed
12.Rondeau E, Scully M, Ariceta G, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int. 2020;97(6):1287-1296. PubMed
13.Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of Atypical Hemolytic Uremic Syndrome: A Systematic Literature Review. Clin Epidemiol. 2020;12:295-305. PubMed
14.Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43(6):976-982. PubMed
15.Woodward L, Johnson S, Walle JV, et al. An innovative and collaborative partnership between patients with rare disease and industry-supported registries: the Global aHUS Registry. Orphanet J Rare Dis. 2016;11(1):154. PubMed
16.Lapeyraque AL, Bitzan M, Al-Dakkak I, et al. Clinical Characteristics and Outcome of Canadian Patients Diagnosed With Atypical Hemolytic Uremic Syndrome. Can J Kidney Health Dis. 2020;7:2054358119897229. PubMed
17.Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion [internal sponsor's package]. Vaughan (ON): Alexion Pharma Canada Corp.; 2022 Jun 23.
18.Ultomiris (ravulizumab): 10 mg/mL concentrate for solution for infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2021 Nov 29.
19.Ullman AJ, Marsh N, Mihala G, Cooke M, Rickard CM. Complications of Central Venous Access Devices: A Systematic Review. Pediatrics. 2015;136(5):e1331-1344. PubMed
20.Soliris (eculizumab for injection): 300 mg single-use vials each containing 30 mL of 10 mg/mL sterile solution for intravenous infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH 2021 Mar 25: https://pdf.hres.ca/dpd_pm/00060546.PDF. Accessed 2022 Jul 26.
21.Clinical Study Report: ALXN1210-aHUS-311. Single arm study of ALXN1210 in complement inhibitor treatment-naive adult and adolescent patients with atypical hemolytic uremic syndrome (aHUS) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals, Inc.; 2020 Feb 12.
22.Clinical Study Report: ALXNI210-aHUS-312. A phase 3, open-label, multicenter study of ALXN1210 in children and adolescents with atypical hemolytic uremic syndrome (aHUS) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals, Inc.; 2020 Mar 06.
23.Greenbaum LA, Licht C, Nikolaou V, et al. Functional Assessment of Fatigue and Other Patient-Reported Outcomes in Patients Enrolled in the Global aHUS Registry. Kidney Int Rep. 2020;5(8):1161-1171. PubMed
24.Schaefer F, Ardissino G, Ariceta G, et al. Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int. 2018;94(2):408-418. PubMed
25.Pugh D, O'Sullivan ED, Duthie FA, Masson P, Kavanagh D. Interventions for atypical haemolytic uraemic syndrome. Cochrane Database Syst Rev. 2021;3:CD012862. PubMed
26.Stahl AL, Vaziri-Sani F, Heinen S, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111(11):5307-5315. PubMed
27.Karpman D, Manea M, Vaziri-Sani F, Stahl AL, Kristoffersson AC. Platelet activation in hemolytic uremic syndrome. Semin Thromb Hemost. 2006;32(2):128-145. PubMed
28.Licht C, Pluthero FG, Li L, et al. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood. 2009;114(20):4538-4545. PubMed
29.Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-562. PubMed
30.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31(6):E1445-1460. PubMed
31.Fact Sheet. aHUS Canada; 2014: http://www.ahuscanada.org/wp-content/uploads/aHUS-Fact-Sheet-February-2014.pdf#:~:text=%EF%82%B7%20%20Atypical%20Hemolytic%20Uremic%20Syndrome%20%28aHUS%29%20is,500%2C000%20people%20per%20year%20in%20the%20United%20States1. Accessed 2022 Aug 20.
32.Atypical Hemolytic Uremic Syndrome: what you need to know. aHUS Canada; 2014: http://www.ahuscanada.org/wp-content/uploads/FINAL_aHUS-Canada-Brochure-ENG-May-9-2014.pdf. Accessed 2022 Aug 20.
33.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
34.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jun 24.
35.Ariceta G, Dixon BP, Kim SH, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int. 2021;100(1):225-237. PubMed
36.Barbour T, Scully M, Ariceta G, et al. Long-Term Efficacy and Safety of the Long-Acting Complement C5 Inhibitor Ravulizumab for the Treatment of Atypical Hemolytic Uremic Syndrome in Adults. Kidney Int Rep. 2021;6(6):1603-1613. PubMed
37.Gackler A, Schonermarck U, Dobronravov V, et al. Efficacy and safety of the long-acting C5 inhibitor ravulizumab in patients with atypical hemolytic uremic syndrome triggered by pregnancy: a subgroup analysis. BMC Nephrol. 2021;22(1):5. PubMed
38.Tanaka K, Adams B, Aris AM, et al. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr Nephrol. 2021;36(4):889-898. PubMed
39.Hassler J, Tanriover B, Ariyamutu V, Burguete D, Hendricks AR, Torrealba JR. 2013 Banff Criteria for Acute Antibody-Mediated Rejection Are Superior to 2007 Banff Criteria in the Diagnosis and Assessment of Renal Allograft Outcomes. Transplant Proc. 2019;51(6):1791-1795. PubMed
40.Schwartz GJ, Haycock GB, Edelmann CM, Jr., Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58(2):259-263. PubMed
41.Tomazos I, Hatswell AJ, Cataland S, et al. Comparative efficacy of ravulizumab and eculizumab in the treatment of atypical hemolytic uremic syndrome: An indirect comparison using clinical trial data. Clin Nephrol. 2022;97(5):261-272. PubMed
42.Indirect comparison of eculizumab and ravulizumab using propensity scoring [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughan (ON): Alexion Pharma Canada Corp.; 2022 Jun 23.
43.Ehren R, Habbig S. Real-world data of six patients with atypical hemolytic uremic syndrome switched to ravulizumab. Pediatr Nephrol. 2021;36(10):3281-3282. PubMed
44.Wang Y, Al-Dakkak I, Garlo K, Ong M-L, Tomazos I, Mahajerin A. Real-World Patient Characteristics, Treatment Patterns and Outcomes for Patients with Atypical Hemolytic Uremic Syndrome Who Have Switched from Eculizumab to Ravulizumab in the USA. Blood. 2021;138(Supplement 1):5003.
45.Legendre C, Rebecca Sberro S, Zuber J. Ravulizumab for the Treatment of aHUS in Adults: Improving Quality of Life. Kidney Int Rep. 2021;6(6):1489-1491. PubMed
46.Wu X, Szarzanowicz A, Garba A, Schaefer B, Waz WR. Blockade of the Terminal Complement Cascade Using Ravulizumab in a Pediatric Patient With Anti-complement Factor H Autoantibody-Associated aHUS: A Case Report and Literature Review. Cureus. 2021;13(11):e19476. PubMed
47.Chapter 1: Definition and classification of CKD. Kidney Int Suppl (2011). 2013;3(1):19-62.
48.Lai JS, Beaumont JL, Ogale S, Brunetta P, Cella D. Validation of the functional assessment of chronic illness therapy-fatigue scale in patients with moderately to severely active systemic lupus erythematosus, participating in a clinical trial. J Rheumatol. 2011;38(4):672-679. PubMed
49.Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74. PubMed
50.Chandran V, Bhella S, Schentag C, Gladman DD. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66(7):936-939. PubMed
51.Hewlett S, Dures E, Almeida C. Measures of fatigue: Bristol Rheumatoid Arthritis Fatigue Multi-Dimensional Questionnaire (BRAF MDQ), Bristol Rheumatoid Arthritis Fatigue Numerical Rating Scales (BRAF NRS) for severity, effect, and coping, Chalder Fatigue Questionnaire (CFQ), Checklist Individual Strength (CIS20R and CIS8R), Fatigue Severity Scale (FSS), Functional Assessment Chronic Illness Therapy (Fatigue) (FACIT-F), Multi-Dimensional Assessment of Fatigue (MAF), Multi-Dimensional Fatigue Inventory (MFI), Pediatric Quality Of Life (PedsQL) Multi-Dimensional Fatigue Scale, Profile of Fatigue (ProF), Short Form 36 Vitality Subscale (SF-36 VT), and Visual Analog Scales (VAS). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S263-286. PubMed
52.Mukherjee AA, Kandhare AD, Bodhankar SL. Evaluation of health-related quality of life in hemolytic uraemic syndrome patients treated with eculizumab: a systematic evaluation on basis of EMPRO. Ren Fail. 2018;40(1):107-118. PubMed
53.Nordin Å, Taft C, Lundgren-Nilsson Å, Dencker A. Minimal important differences for fatigue patient reported outcome measures - A systematic review. BMC Med Res Methodol. 2016;16(1). PubMed
54.Lai JS, Cella D, Kupst MJ, et al. Measuring fatigue for children with cancer: development and validation of the pediatric Functional Assessment of Chronic Illness Therapy-Fatigue (pedsFACIT-F). J Pediatr Hematol Oncol. 2007;29(7):471-479. PubMed
55.EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
56.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
57.Cella DF, Tulsky DS, Gray G, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. J Clin Oncol. 1993;11(3):570-579. PubMed
58.Bonomi AE, Cella DF, Hahn EA, et al. Multilingual Translation of the Functional Assessment of Cancer Therapy (FACT) Quality of Life Measurement System. Qual Life Res. 1996;5(3):309-320. PubMed
59.Eremenco S, Cella D, Arnold BJ. A Comprehensive Method for the Translation and Cross-Cultural Validation of Health Status Questionnaires. Eval Health Prof. 2005;28:212 - 232. PubMed
60.Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811-819. PubMed
61.Tinsley A, Macklin EA, Korzenik JR, Sands BE. Validation of the functional assessment of chronic illness therapy-fatigue (FACIT-F) in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2011;34(11-12):1328-1336. PubMed
62.Kosinski M, Gajria K, Fernandes AW, Cella D. Qualitative validation of the FACIT-fatigue scale in systemic lupus erythematosus. Lupus. 2013;22(5):422-430. PubMed
63.Signorovitch J, Brainsky A, Grotzinger KM. Validation of the FACIT-fatigue subscale, selected items from FACT-thrombocytopenia, and the SF-36v2 in patients with chronic immune thrombocytopenia. Qual Life Res. 2011;20(10):1737-1744. PubMed
64.Hagell P, Höglund A, Reimer J, et al. Measuring fatigue in Parkinson's disease: a psychometric study of two brief generic fatigue questionnaires. J Pain Symptom Manage. 2006;32(5):420-432. PubMed
65.Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89(3):701-711. PubMed
66.Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab Inhibits Thrombotic Microangiopathy and Improves Renal Function in Pediatric Patients with Atypical Hemolytic Uremic Syndrome: 1-Year Update. Blood. 2014;124(21):4986-4986.
67.Smets EMA, Garssen B, Bonke B, De Haes JCJM. The multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39(3):315-325. PubMed
68.Varni JW, Burwinkle TM, Katz ER, Meeske K, Dickinson P. The PedsQL in pediatric cancer: reliability and validity of the Pediatric Quality of Life Inventory Generic Core Scales, Multidimensional Fatigue Scale, and Cancer Module. Cancer. 2002;94(7):2090-2106. PubMed
69.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. New York (NY): Routledge; 1988.
70.Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution-based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. J Pain Symptom Manage. 2002;24(6):547-561. PubMed
Appendix 1: Ontario Eculizumab Reimbursement Criteria for aHUS
Note that this appendix has not been copy-edited.
Table 33: Reimbursement Status for Comparators for the Treatment of Adults and Pediatric Patients With aHUS to Inhibit Complement-Mediated TMA
Comparators | FPT public drug programs | CBS | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
BC1 | AB2 | SK | MB | ON3 | NB4 | NS5 | PE | NL6 | YT7 | NT8 | NIHB8 | CAF9 | VAC10 | CSC | ||
Soliris (eculizumab) | EX | EX | EX | EX | RES | NaB | NaB | NaB | NaB | NaB | NaB | NaB | NaB | NaB | NaB | NA |
AB = Alberta; BC = British Columbia; CAF = Canadian Armed Forces; CBS = Canadian Blood Services; CSC = Correctional Services Canada; EX = exception item for which coverage is determined on a case-by-case basis; FB = full benefit; FPT = federal, provincial, and territorial; MB = Manitoba; NaB = not a benefit; NIHB = Non-Insured Health Benefits Program; NL = Newfoundland and Labrador; NS = Nova Scotia; NT = Northwest Territories; ON = Ontario; PE = Prince Edward Island; RES = restricted benefit with specified criteria; SK = Saskatchewan; VAC = Veterans Affairs Canada; YT = Yukon.
Table 34: Reimbursement Criteria for Soliris (Eculizumab) for aHUS
Drug plan | Criteria for restricted benefit |
|---|---|
Ontario3 | A confirmed diagnosis of atypical hemolytic uremic syndrome is required for eculizumab funding. A patient must meet all 3 of the following criteria for initial treatment with eculizumab:
Continuation Criteria (6 months)
Continuation Criteria (12 months)
|
aHUS = atypical hemolytic uremic syndrome; eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; SrCr = serum creatine; STEC = Shiga toxin–producing E. coli; TMA = thrombotic microangiopathy.
Source: Sponsor’s submission.17
Appendix 2: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 7, 2022
Alerts: Biweekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Ultomiris* or ravulizumab* or ALXN-1810 or ALXN1810 or ALXN-1210 or ALXN1210 or C3VX249T6L).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*ravulizumab/
(Ultomiris* or ravulizumab* or ALXN-1810 or ALXN1810 or ALXN-1210 or ALXN1210).ti,ab,kf,dq.
or/3-4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Ultomiris OR ravulizumab OR ALXN-1810 OR ALXN1810 OR ALXN-1210 OR ALXN1210]
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
[Search terms -- (Ultomiris OR ravulizumab OR ALXN-1810 OR ALXN1810 OR ALXN-1210 OR ALXN1210) AND (hemolytic uremic syndrome OR aHUS)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- (Ultomiris OR ravulizumab OR ALXN-1810 OR ALXN1810 OR ALXN-1210 OR ALXN1210) AND (hemolytic uremic syndrome OR aHUS)]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- ravulizumab AND hemolytic uremic syndrome]
Grey Literature
Search dates: June 23, 2022 to June 30, 2022
Keywords: [Ultomiris OR ravulizumab OR ALXN-1810 OR ALXN1810 OR ALXN-1210 OR ALXN1210 | atypical hemolytic uremic syndrome OR aHUS OR familial hemolytic-uremic syndrome OR hereditary hemolytic-uremic syndrome OR Complement-Mediated Thrombotic Microangiopathy OR TMA]
Limits: Publication years: none
Updated: Search updated before the meeting of the CADTH Canadian Drug Expert Committee (CDEC)
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 3: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Ehren R, Habbig S. Real-world data of 6 patients with atypical hemolytic uremic syndrome switched to ravulizumab. Pediatr Nephrol. 2021;36(10):3281 to 3282.43 | Study design not of interest (case series) |
Legendre C, Rebecca Sberro S, Zuber J. Ravulizumab for the Treatment of aHUS in Adults: Improving Quality of Life. KI Rep. 2021;6(6):1489 to 1491.45 | Study design not of interest (review) |
Wu X, Szarzanowicz A, Garba A, Schaefer B, Waz WR. Blockade of the Terminal Complement Cascade Using Ravulizumab in a Pediatric Patient With Anti-complement Factor H Autoantibody-Associated aHUS: A Case Report and Literature Review. Cureus. 2021;13(11):e19476.46 | Study design not of interest (case report) |
Appendix 4: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 37: Pretreatment Extrarenal Signs or Symptoms of aHUS (FAS, ≥ 10%)
Sign or symptom | Study 311 (N = 56) | Study 312, cohort 1 (N = 18) |
|---|---|---|
Any pretreatment extrarenal signs or symptoms of aHUS, n (%) | 52 (92.9) | 13 (72.2) |
Hypertension | 34 (60.7) | 6 (33.3) |
Shortness of breath | 13 (23.2) | NR |
Pulmonary edema | 7 (12.5) | NR |
Pleural effusion | 9 (16.1) | 1 (5.6) |
Lethargy | 8 (14.3) | 3 (16.7) |
Irritability | 1 (1.8) | 2 (11.1) |
Headache | 17 (30.4) | 1 (5.6) |
Visual deficit | 9 (16.1) | NR |
Seizures | 1 (1.8) | 2 (11.1) |
Nausea | 21 (37.5) | 6 (33.3) |
Vomiting | 19 (33.9) | 7 (38.9) |
Diarrhea | 10 (17.9) | NR |
Abdominal pain | 8 (14.3) | 6 (33.3) |
Elevated transaminases (ALT and/or AST) | 7 (12.5) | 5 (27.8) |
Petechiae | 8 (14.3) | 5 (27.8) |
aHUS = atypical hemolytic uremic syndrome; ALT = alanine aminotransferase; AST = aspartate aminotransferase; FAS = full analysis set.
Note: In summarizing n (%), if a patient had multiple reports for a particular organ system/sign or symptom, they were counted only once for that organ system/sign or symptom. Patients may have been counted in more than 1 organ system/sign or symptom category.
Source: Clinical Study Reports.21,22
Table 38: Concomitant Medications and Treatments (Safety Set)
Medications or treatment | Study 311 (N = 58) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 21) | Cohort 2 (N = 10) | ||
Packed RBC transfusions, n (%) | 17 (29.3) | NR | NR |
Platelet transfusions, n (%) | 3 (5.2%) | 4 (19.0) | NR |
Plasma exchange or plasma infusion during the study, which was prohibited per the protocol, n (%) | 3 (5.2%) | 0 | 0 |
Blood substitutes and plasma protein fractions | 9 (15.5%) | 2 (9.5) | NR |
Selective immunosuppressants | 9 (15.5) | 6 (28.6%) | NR |
Mycophenolate mofetil | 6 (10.3) | 4 (19.0) | NR |
Sirolimus | 4 (6.9) | 0 | NR |
Mycophenolate sodium | 2 (3.4) | 0 | NR |
Antithymocyte immunoglobulin (rabbit) | 1 (1.7) | 0 | NR |
Belatacept | 1 (1.7) | 0 | NR |
Abatacept | 0 | 1 (4.8) | NR |
Eculizumab | 1 (1.7) | 1 (4.8) | NR |
Leflunomide | 1 (1.7) | 0 | NR |
Glucocorticoids | 30 (51) | 2 (9.5) | 3 (30.0) |
NR = not reported.Source: Clinical Study Reports.21,22
Table 39: Glomerular Filtration Rate Category and CKD Stage
CKD category or stage | GFR (mL/min/1.73 m2) | Terms |
|---|---|---|
1 | ≥ 90 | Normal or high |
2 | 60 to 89 | Mildly decreaseda |
3a | 45 to 59 | Mildly to moderately decreased |
3b | 30 to 44 | Moderately to severely decreased |
4 | 15 to 29 | Severely decreased |
5 | < 15 | Kidney failure |
CKD = chronic kidney disease; GFR = glomerular filtration rate.
aRelative to young adult level.
Note: In the absence of evidence of kidney damage, neither GFR category/stage G1 nor G2 fulfill the criteria for CKD.
Sources: KDIGO, 2012;47 Clinical Study Report.21
Table 40: Major Protocol Deviations (All Enrolled Patients)
Categories | Study 311 (N = 58) | Study 312, cohort 1 (N = 21) |
|---|---|---|
Major deviations, n (%) | 43 (74.1) | 14 (66.7) |
Eligibility and entry criteria | 25 (43.1) | 9 (42.9) |
Serious adverse event reporting criteria | 15 (25.9) | 7 (33.3) |
Study drug compliance | 13 (22.4) | 1 (4.8) |
Study procedures criteria | 7 (12.1) | NR |
Informed consent procedures | 7 (12.1) | 3 (14.3) |
Concomitant medication criteria | 4 (6.9) | NR |
Laboratory assessment criteria | 1 (1.7) | NR |
Source document criteria | 1 (1.7) | 1 (4.8) |
Deviations resulting in exclusion from the PP seta | 12 (20.7) | 3 |
Eligibility and entry criteria | 9 (15.5) | NR |
Concomitant medication criteria | 3 (5.2) | NR |
NR = not reported; PP = per protocol.
aPatients that did not meet this inclusion criteria or met these exclusion criteria were excluded from the PP set. In Study 312, the number of the PP set equals the number of the FAS.
Note: Percentages were based on the total number of patients. Patients could have been counted in more than 1 deviation category if the patient had more than 1 type of protocol deviation.
Note: In Study 312, cohort 2, 1 of the 10 patients in cohort 2 had a major protocol deviation. This patient had a major deviation related to informed consent procedures due to signature of an incorrect version of the informed consent form.
Source: Clinical Study Report.21,22
Table 41: Summary of Treatment Exposures and Follow-Up Durations as of Data Cut-Off (Safety Set)
Parameter | Study 311 (N = 58) | Study 312 | |
|---|---|---|---|
Cohort 1 (N = 21) | Cohort 2 (N = 10) | ||
Follow-up durationa (weeks) | |||
Mean (SD) | 70.05 (33.796) | 64.97 (34.755) | 52.69 (4.019) |
Median | 75.57 | 82.43 | 50.29 |
Min, max | 0.57, 118.29 | 1, 110.57 | 49.43, 58.71 |
Treatment durationb (weeks) | |||
Mean (SD) | 67.97 (34.374) | 64.96 (34.757) | 52.69 (4.019) |
Median | 74.07 | 82.40 | 50.29 |
Min, max | 0.57, 118.29 | 1, 110.6 | 49.43, 58.71 |
Number of infusions n, (%) | |||
1 | 3 (5.2) | 2 (9.5) | NR |
2 | 3 (5.2) | 2 (9.5) | NR |
3 | 2 (3.4) | NR | NR |
4 | 4 (6.9) | NR | NR |
5 | 1 (1.7) | NR | NR |
6 | 1 (1.7) | NR | NR |
7 | 1 (1.7) | NR | NR |
8 | 1 (1.7) | NR | 6 (60.0) |
9 | 5 (8.6) | 2 (9.5) | 2 (20.0) |
10 | 8 (13.8) | 1 (4.8) | 1 (10.0) |
11 | 6 (10.3) | 1 (4.8) | 1 (10.0) |
12 | 8 (13.8) | 2 (9.5) | NR |
13 | 4 (6.9) | 2 (9.5) | NR |
14 | 3 (5.2) | 1 (4.8) | NR |
15 | 5 (8.6) | NR | NR |
16 | 3 (5.2) | 1 (4.8) | NR |
21 | NR | 1 (4.8) | NR |
22 | NR | 1 (4.8) | NR |
23 | NR | 1 (4.8) | NR |
24 | NR | 2 (9.5) | NR |
28 | NR | 1 (4.8) | NR |
29 | NR | 1 (4.8) | NR |
Max = maximum; min = minimum; SD = standard deviation.
aFollow-up duration was defined as the number of weeks from date of first dose to completion of study or last available study visit or study discontinuation + 1 day.
bTreatment duration was defined as ([the date of last dose + 56 days] – [the date of first dose]) or ([study discontinuation date] – [the date of first dose]) if study discontinuation date is earlier than (the date of last dose + 56 days). The result was transferred to weeks.
Note: Percentages were based on the number of patients with nonmissing data.
Source: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Table 42: Treatment Exposures and Follow-Up Durations as of Data Cut-Off Date (Study 312, Cohort 1, Safety Set)
Parameter | Birth to < 6 years (N = 12) | 6 to < 18 years(N = 9) | Overall (N = 21) |
|---|---|---|---|
Follow-up durationa (weeks) | |||
Mean (SD) | 68.13 (35.511) | 60.75 (35.367) | 64.97 (34.755) |
Median | 82.50 | 74.14 | 82.43 |
Min, max | 3, 110.57 | 1, 90.14 | 1, 110.57 |
Treatment durationb (weeks) | |||
Mean (SD) | 68.13 (35.519) | 60.73 (35.360) | 64.96 (34.757) |
Median | 82.50 | 74.10 | 82.40 |
Min, max | 3, 110.6 | 1, 90.1 | 1, 110.6 |
Number of infusions | |||
1 | 0 | 2 (22.2) | 2 (9.5) |
2 | 2 (16.7) | 0 | 2 (9.5) |
9 | 1 (8.3) | 1 (11.1) | 2 (9.5) |
10 | 0 | 1 (11.1) | 1 (4.8) |
11 | 0 | 1 (11.1) | 1 (4.8) |
12 | 0 | 2 (22.2) | 2 (9.5) |
13 | 0 | 2 (22.2) | 2 (9.5) |
14 | 1 (8.3) | 0 | 1 (4.8) |
16 | 1 (8.3) | 0 | 1 (4.8) |
21 | 1 (8.3) | 0 | 1 (4.8) |
22 | 1 (8.3) | 0 | 1 (4.8) |
23 | 1 (8.3) | 0 | 1 (4.8) |
24 | 2 (16.7) | 0 | 2 (9.5) |
28 | 1 (8.3) | 0 | 1 (4.8) |
29 | 1 (8.3) | 0 | 1 (4.8) |
Compliance, n (%) | |||
≥ 100% | 12 (100.0) | 9 (100.0) | 21 (100.0) |
max = maximum; min = minimum; SD = standard deviation.
aFollow-up duration was defined as the number of weeks from date of first dose to completion of study or last available study visit or study discontinuation + 1 day.
bTreatment duration was defined as (the date of last dose- the date of first dose + 56) or (study discontinuation date – the date of first dose + 56 if discontinuation date is earlier than (the date of last dose + 56)). The result is presented in weeks.
Percentages were based on the number of patients with nonmissing data in each group. Patients received a weight-based loading dose of ravulizumab on day 1, followed by weight-based maintenance treatment on day 15 and every 8 weeks thereafter for patients weighing ≥ 20 kg, or every 4 weeks for patients weighing < 20 kg. Weight-based dosing was based on the patient’s body weight recorded on dosing regimen decision days.
Source: Study 312 Clinical Study Report.22
Table 43: Complete TMA Response and Components Analysis (Study 311, PP)
Outcomes | PP | |||||
|---|---|---|---|---|---|---|
Week 26 | Cut-off date (July 2, 2019) | |||||
Total | Responder | Total | Responder | |||
N | n | Proportion (95% CI)a | N | n | Proportion (95% CI)a | |
Complete TMA response | 44 | 22 | 0.500 (0.341, 0.659) | 44 | 26 | 0.591 (0.434, 0.748) |
Components of complete TMA response | ||||||
Platelet count normalization | 44 | 39 | 0.886 (0.781, 0.992) | 44 | 40 | 0.909 (0.813, 1.005) |
LDH normalization | 44 | 34 | 0.773 (0.638, 0.908) | 44 | 38 | 0.864 (0.751, 0.976) |
25% improvement in serum creatinine from baseline | 44 | 25 | 0.568 (0.410, 0.726) | 44 | 26 | 0.591 (0.434, 0.748) |
Hematologic normalization | 44 | 33 | 0.750 (0.611, 0.889) | 44 | 37 | 0.841 (0.721, 0.960) |
CI = confidence interval; LDH = lactate dehydrogenase; PP = per protocol; TMA = thrombotic microangiopathy.
a95% CIs for the proportion are based on the asymptotic Gaussian approximation method with a continuity correction.
Source: Study 311 Clinical Study Report.21
Figure 7: Number of Patients Who Achieved 1 or More Components of Complete TMA Response During the Initial Evaluation Period (Study 311, FAS)
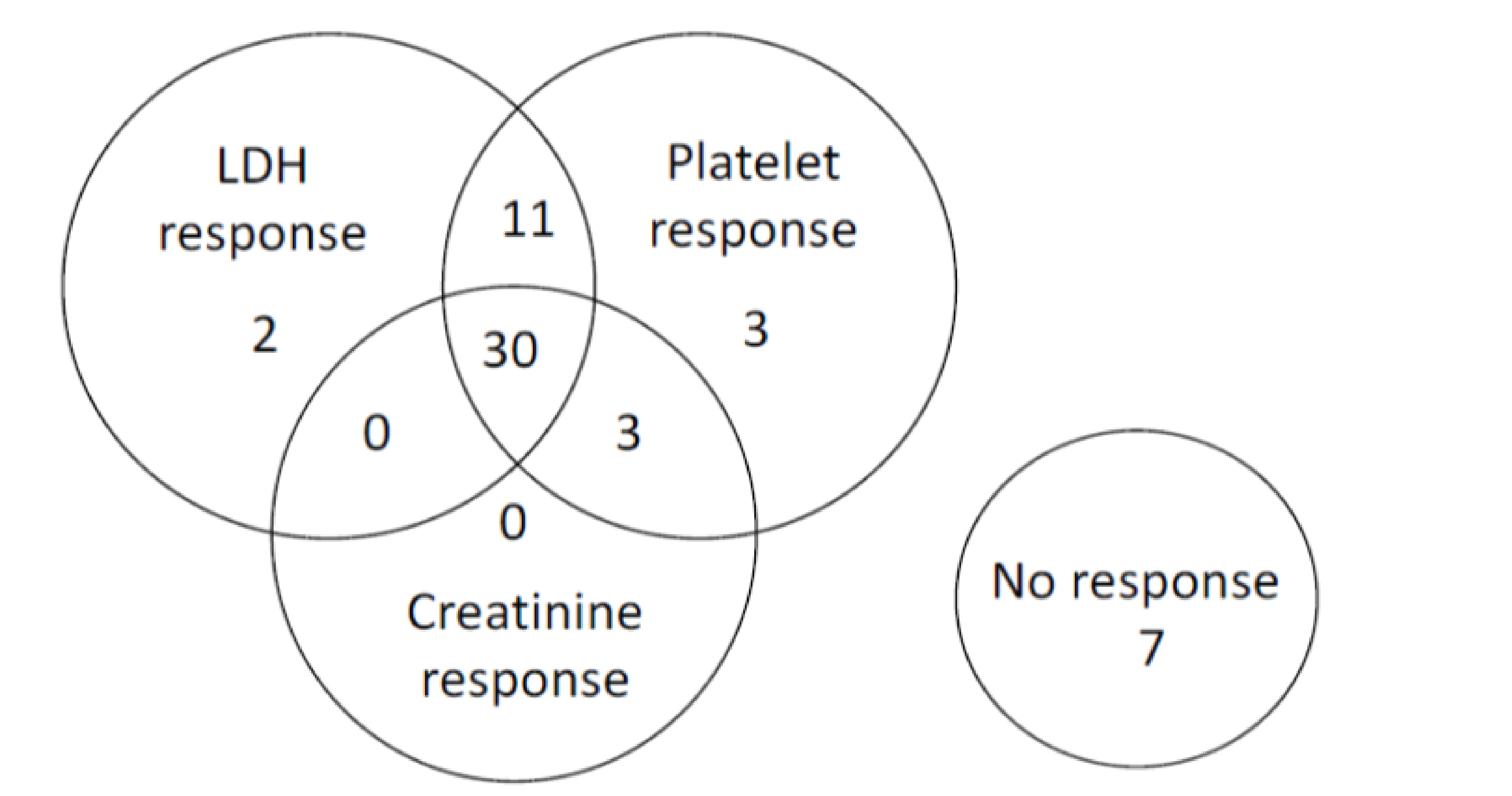
LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
Source: Study 311 Clinical Study Report.21
Figure 8: Number of Patients Who Achieved 1 or More Components of Complete TMA Response During Initial Evaluation Period (Study 312, Cohort 1 FAS)
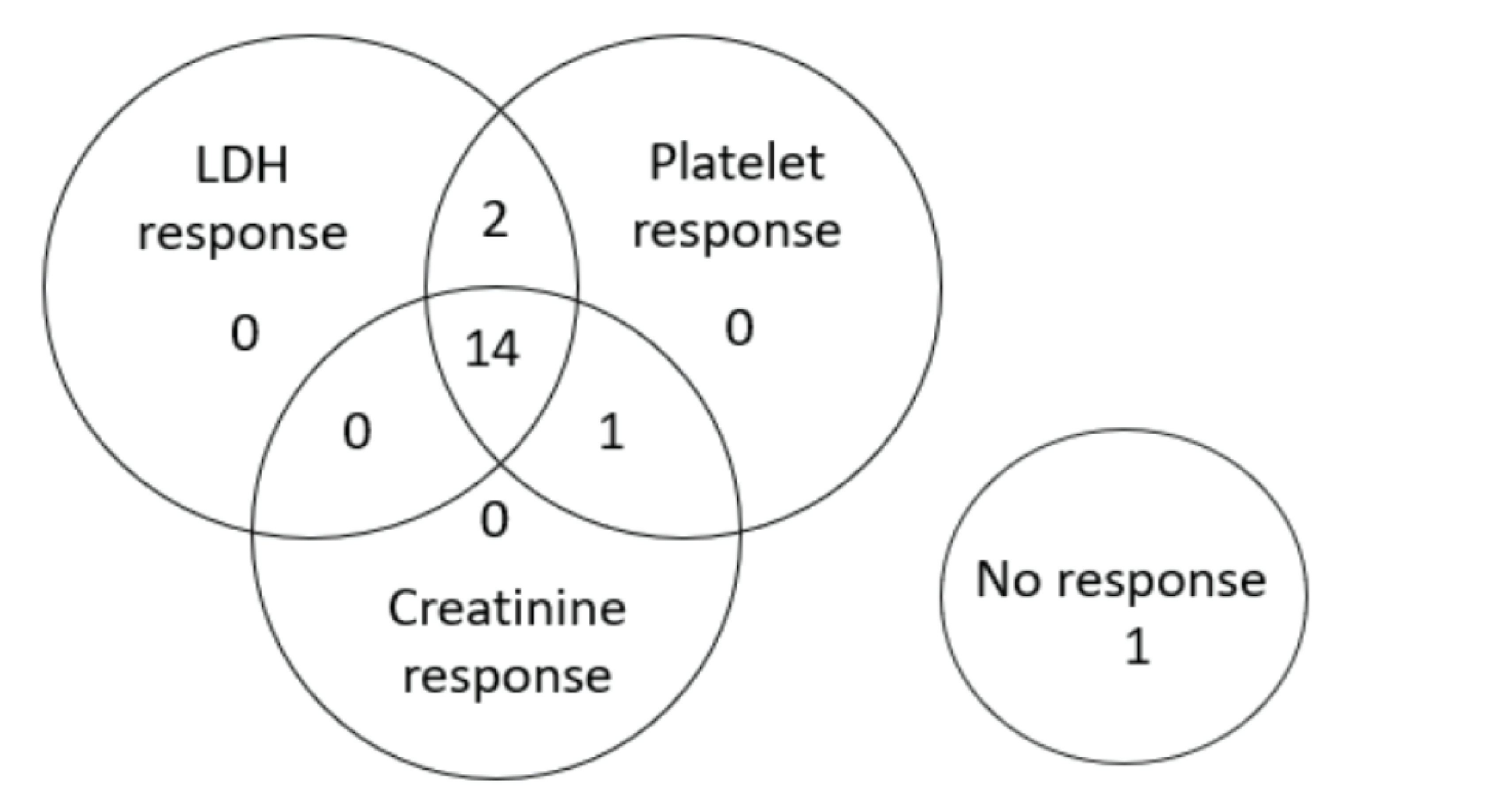
LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Figure 9: Forest Plot of Proportion and 95% CI of Complete TMA Response (Overall and by Subgroups) During the 26-Week Initial Evaluation Period (FAS, Study 311)
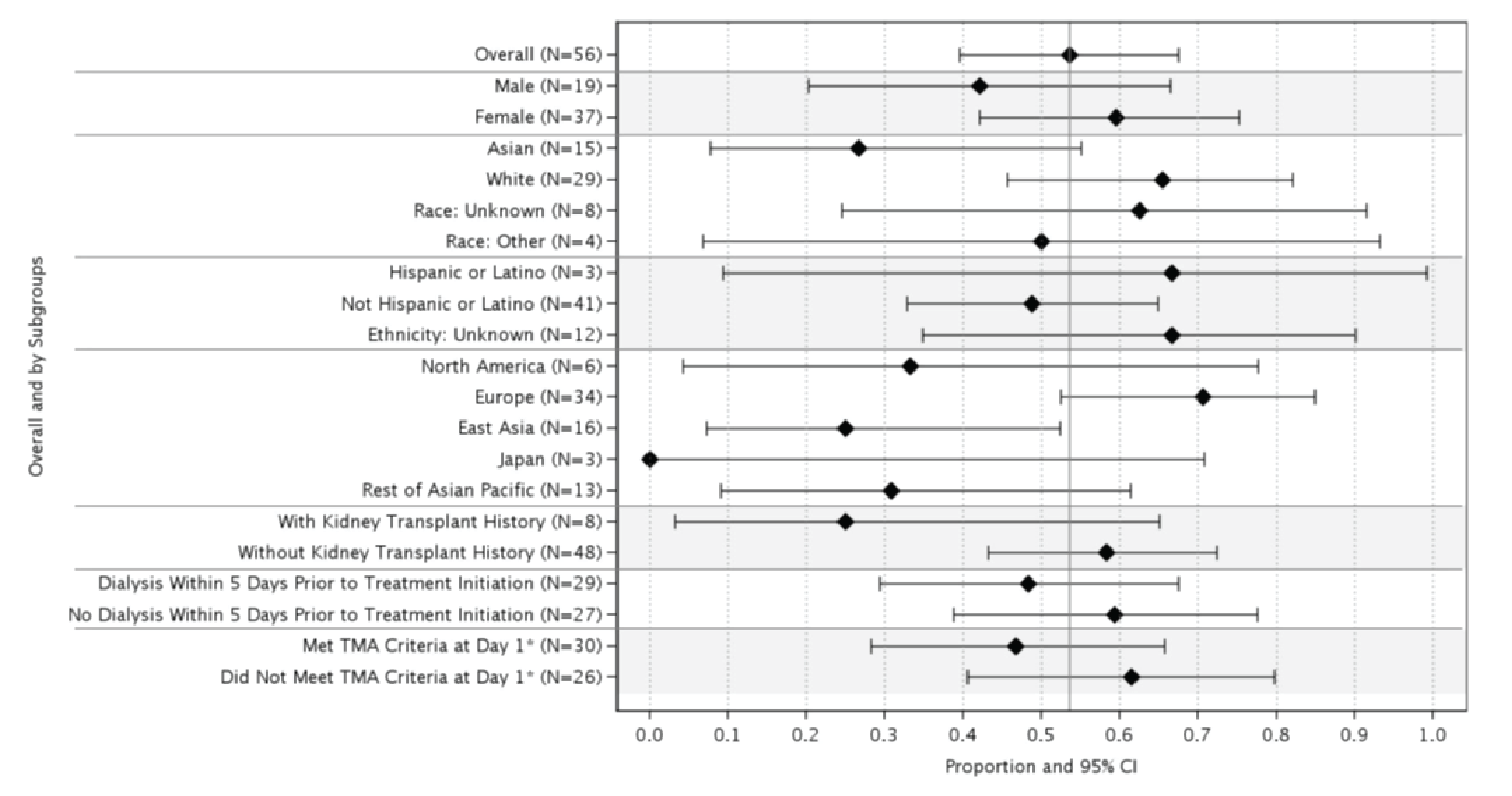
CI = confidence interval; LDH = lactate dehydrogenase; LLN = lower limit of normal; FAS = full analysis set; TMA = thrombotic microangiopathy; ULN = upper limit of normal.
* Based on central laboratory results. The TMA criteria at day 1 included platelet count < 150,000/μL, LDH ≥ 1.5 × ULN, hemoglobin ≤ LLN, and serum creatinine level ≥ ULN (or required dialysis for acute kidney injury).
Source: Study 311 Clinical Study Report.21
Table 44: Modified Complete TMA Response and Components Analysis (Study 311, Sensitivity Analysis, FAS)
Outcomes | Sensitivity analysis: Responder | |||||
|---|---|---|---|---|---|---|
Week 26 | Extension period | |||||
Total N | n | Proportion (95% CI)a | Total N | n | Proportion (95% CI)a | |
Modified complete TMA response | 56 | 32 | 0.571 (0.433, 0.710) | 44 | 24 | 0.545 (0.387, 0.704) |
Components of modified complete TMA response | ||||||
Platelet count normalization | 56 | 47 | 0.839 (0.734, 0.944) | 44 | 39 | 0.886 (0.781, 0.992) |
LDH normalization | 56 | 43 | 0.768 (0.648, 0.887) | 44 | 34 | 0.773 (0.638, 0.908) |
25% improvement in serum creatinine from baseline or postbaseline change in dialysis status with a confirmatory result | 56 | 35 | 0.625 (0.489, 0.761) | 44 | 27 | 0.614 (0.458, 0.769) |
Hematologic normalization | 56 | 41 | 0.732 (0.607, 0.857) | 44 | 33 | 0.750 (0.611, 0.889) |
CI = confidence interval; FAS = full analysis set; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
The modification to complete TMA response applies strictly to the patients on dialysis at baseline (i.e., patients requiring dialysis within 5 days before ravulizumab treatment initiation). For these patients, the criterion requiring an improvement from baseline of 25% or more in serum creatinine is replaced by a postbaseline change in dialysis status (from requiring dialysis at baseline to no longer requiring dialysis) that is maintained for at least 4 weeks. The definition of complete TMA response remains the same for all other patients. Patients must meet all modified complete TMA response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart, and any measurement in between. The proportion of modified complete TMA response is based on the responders among treated patients. The numerator is the number of patients achieving modified complete TMA response during the 26-week initial evaluation period and the denominator is the number of patients in the FAS.
a95%CI for the proportion are based on the asymptotic Gaussian approximation method with a continuity correction.
Hematologic normalization includes normalization of platelet count and normalization of LDH. Platelet values obtained from the day of a blood transfusion of platelets through 3 days after the transfusion are excluded from all analyses. All serum creatinine values obtained while a patient is on dialysis are excluded from all analyses. When a patient is on dialysis at baseline, then the first valid creatinine value to be used as the baseline value is the first assessment ≥ 6 days postdialysis. If a patient is on dialysis during the entire 26-week initial evaluation period, then the baseline creatinine is not calculated.
Source: Study 311 Clinical Study Report.21
Figure 10: Forest Plot of Proportion and 95% CI of Complete TMA Response (Overall and by Subgroups) During the 26-Week Initial Evaluation Period (Study 312, Cohort 1)
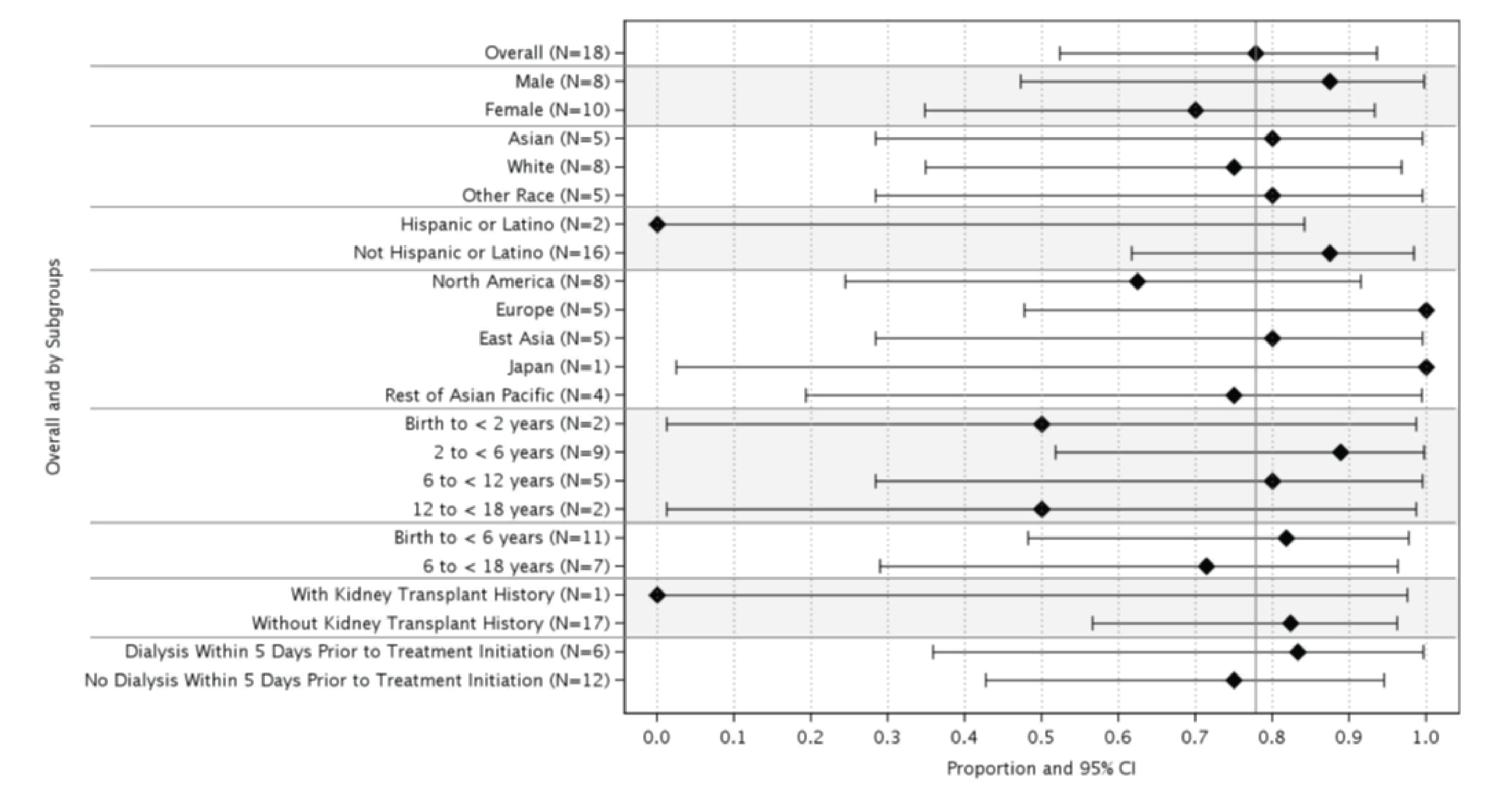
CI = confidence interval; LDH = lactate dehydrogenase; LLN = lower limit of normal; TMA = thrombotic microangiopathy; ULN = upper limit of normal.
* Based on central laboratory results. The TMA criteria at day 1 included platelet count < 150,000/μL, LDH ≥ 1.5 × ULN, hemoglobin ≤ LLN, and serum creatinine level ≥ ULN (or required dialysis for acute kidney injury).
Source: Study 312 Clinical Study Report.22
Table 45: Modified Complete TMA Response and Components Analysis (Study 312, Sensitivity Analysis FAS)
Outcomes | Sensitivity analysis: Responder | |||||
|---|---|---|---|---|---|---|
Week 26 (FAS) | Extension period (FAS) | |||||
Total | n | Proportion (95% CI) (a) | Total | n | Proportion (95% CI) (a) | |
Modified complete TMA response | 18 | 14 | 0.778 (0.524, 0.936) | 18 | 17 | 0.944 (0.727, 0.999) |
Components of modified complete TMA response | ||||||
Platelet count normalization | 18 | 17 | 0.944 (0.727, 0.999) | 18 | 17 | 0.944 (0.727, 0.999) |
LDH normalization | 18 | 16 | 0.889 (0.653, 0.986) | 18 | 17 | 0.944 (0.727, 0.999) |
25% improvement in serum creatinine from baseline or postbaseline change in dialysis status with a confirmatory result | 18 | 15 | 0.833 (0.586, 0.964) | 18 | 17 | 0.944 (0.727, 0.999) |
Hematologic normalization | 18 | 16 | 0.889 (0.653, 0.986) | 18 | 17 | 0.944 (0.727, 0.999) |
CI = confidence interval; FAS = full analysis set; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Table 46: Complete TMA Response Status Over Time With a Confirmatory Result as of Data Cut-Off Date (FAS, Study 311)
Visit | Study 311 (N = 56) | Study 312 | ||||
|---|---|---|---|---|---|---|
n | N | Proportion (95% CI)a | n | N | Proportion (95% CI)a | |
Day 8 | 4 | 53 | 0.075 (0.021, 0.182) | 0 | 17 | 0.000 (0.000, 0.195) |
Day 15 | 10 | 53 | 0.189 (0.094, 0.320) | 2 | 17 | 0.118 (0.015, 0.364) |
Day 22 | 18 | 53 | 0.340 (0.215, 0.483) | 5 | 17 | 0.294 (0.103, 0.560) |
Day 29 | 19 | 53 | 0.358 (0.231, 0.502) | 9 | 17 | 0.529 (0.278, 0.770) |
Day 43 | 22 | 53 | 0.415 (0.281, 0.559) | 9 | 17 | 0.529 (0.278, 0.770) |
Day 57 | 22 | 53 | 0.415 (0.281, 0.559) | 12 | 17 | 0.706 (0.440, 0.897) |
Day 71 | 23 | 53 | 0.434 (0.298, 0.577) | 10 | 17 | 0.588 (0.329, 0.816) |
Day 85 | 27 | 52 | 0.519 (0.376, 0.660) | 13 | 17 | 0.765 (0.501, 0.932) |
Day 99 | 28 | 51 | 0.549 (0.403, 0.689) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 113 | 27 | 50 | 0.540 (0.393, 0.682) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 127 | 24 | 50 | 0.480 (0.337, 0.626) | 13 | 17 | 0.765 (0.501, 0.932) |
Day 141 | 26 | 50 | 0.520 (0.374, 0.663) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 155 | 25 | 50 | 0.500 (0.355, 0.645) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 169 | 28 | 50 | 0.560 (0.413, 0.700) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 183 | 26 | 49 | 0.531 (0.383, 0.675) | 13 | 17 | 0.765 (0.501, 0.932) |
Day 239 | 23 | 46 | 0.500 (0.349, 0.651) | 13 | 17 | 0.765 (0.501, 0.932) |
Day 295 | 25 | 45 | 0.556 (0.400, 0.704) | 14 | 17 | 0.824 (0.566, 0.962) |
Day 351 | 22 | 44 | 0.500 (0.346, 0.654) | 15 | 17 | 0.882 (0.636, 0.985) |
Day 407 | 25 | 43 | 0.581 (0.421, 0.730) | 12 | 15 | 0.800 (0.519, 0.957) |
Day 463 | 22 | 37 | 0.595 (0.421, 0.752) | 10 | 12 | 0.833 (0.516, 0.979) |
Day 519 | 15 | 28 | 0.536 (0.339, 0.725) | 10 | 12 | 0.833 (0.516, 0.979) |
Day 575 | 11 | 24 | 0.458 (0.256, 0.672) | 9 | 11 | 0.818 (0.482, 0.977) |
Day 631 | 7 | 14 | 0.500 (0.230, 0.770) | 6 | 6 | 1.000 (0.541, 1.000) |
Day 687 | 4 | 11 | 0.364 (0.109, 0.692) | 2 | 2 | 1.000 (0.158, 1.000) |
Day 743 | 2 | 9 | 0.222 (0.028, 0.600) | 2 | 2 | 1.000 (0.158, 1.000) |
Day 799 | 1 | 3 | 0.333 (0.008, 0.906) | NR | NR | NR |
CI = confidence interval; NR = not reported; TMA = thrombotic microangiopathy.
a95% CIs for the proportion are based on exact confidence limits using the Clopper-Pearson method.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Figure 11: Complete TMA Response Components and Hematologic Normalization Status Over Time During the Initial Evaluation Period (FAS, Study 311)
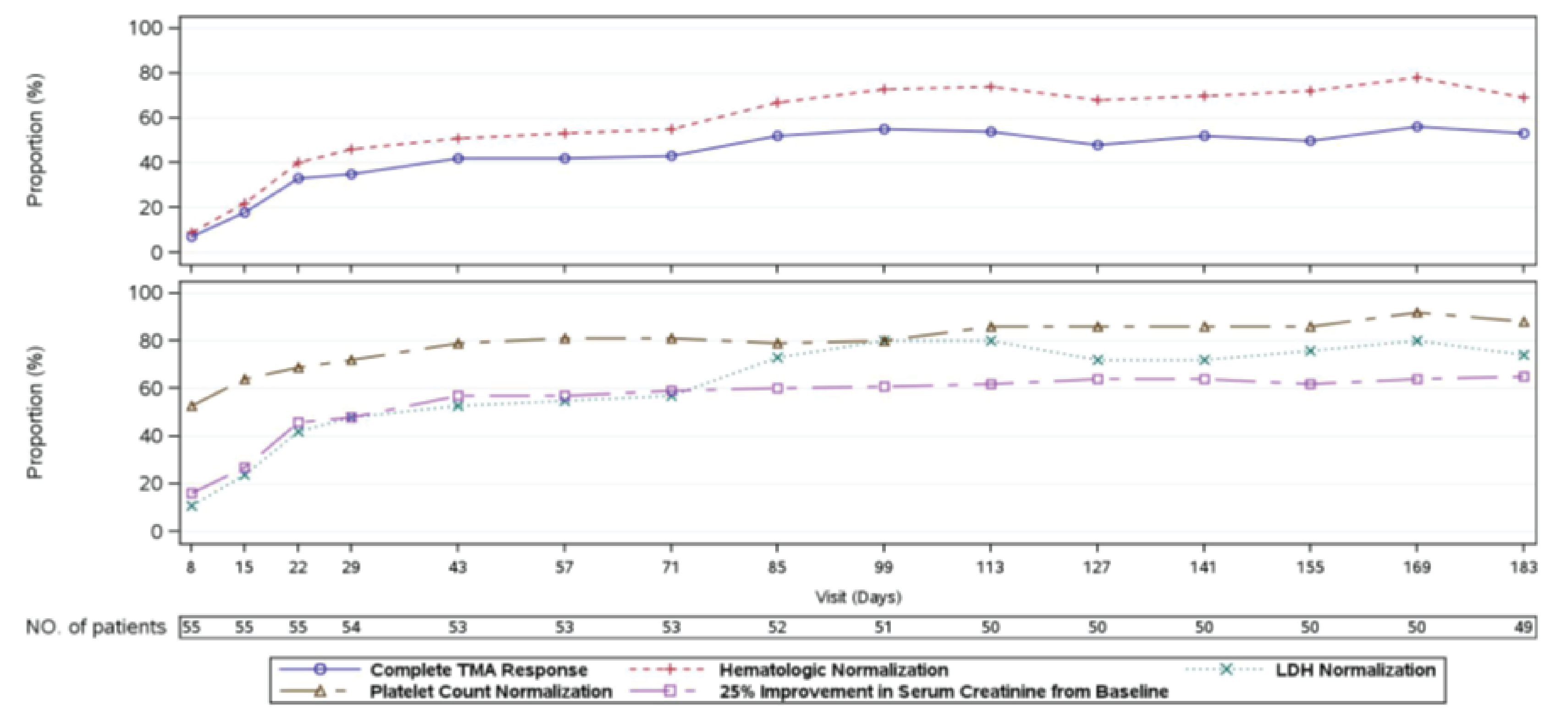
FAS = full analysis set; LDH = lactate dehydrogenase; NO. = number; TMA = thrombotic microangiopathy.
A patient was in the analysis for a specific postbaseline time point if it was possible for the result at that time point to be confirmed. Hematologic normalization includes normalization of platelets and LDH. Platelet values obtained from the day of a blood transfusion of platelets through 3 days after the transfusion were excluded from all analyses. All serum creatinine values obtained while a patient was on dialysis were excluded from all analyses. When a patient was on dialysis at baseline, then the first valid creatinine value to be used as the baseline value was the first assessment ≥ 6 days postdialysis. If a patient was on dialysis during the entire 26-week initial evaluation period, then the baseline creatinine was not calculated.
Source: Study 311 Clinical Study Report.21
Figure 12: Complete TMA Response Components and Hematologic Normalization Status Over Time During the Extension Period Through the Data Cut-Off Date (Study 311, FAS)
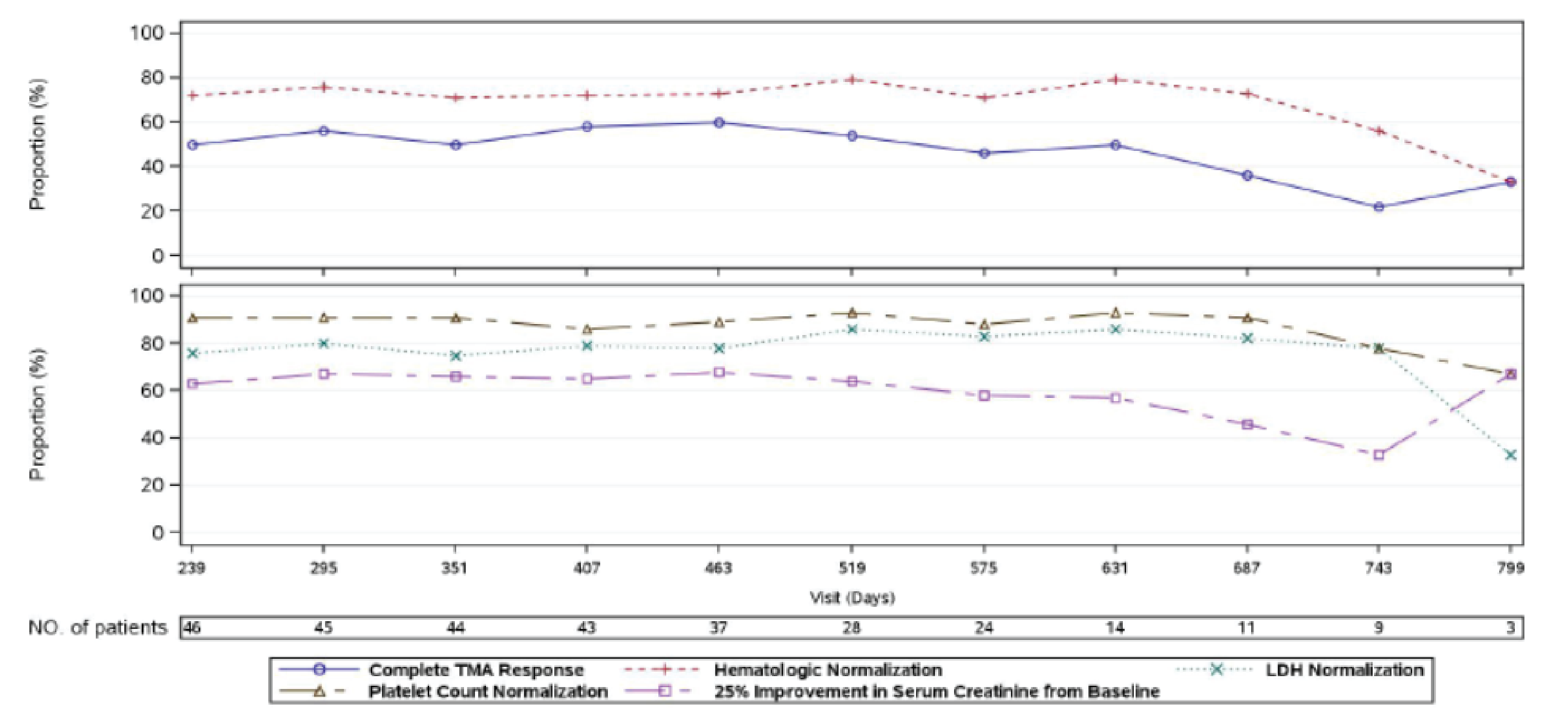
FAS = full analysis set; LDH = lactate dehydrogenase; NO. = number; TMA = thrombotic microangiopathy.
A patient was in the analysis for a specific postbaseline time point if it was possible for the result at that time point to be confirmed. Hematologic normalization includes normalization of platelets and LDH. Platelet values obtained from the day of a blood transfusion of platelets through 3 days after the transfusion were excluded from all analyses. All serum creatinine values obtained while a patient was on dialysis were excluded from all analyses. When a patient was on dialysis at baseline, then the first valid creatinine value to be used as the baseline value was the first assessment ≥ 6 days postdialysis. If a patient was on dialysis during the entire 26-week initial evaluation period, then the baseline creatinine was not calculated.
Source: Study 311 Clinical Study Report.21
Figure 13: Complete TMA Response, Hematologic Normalization, and Complete TMA Response Components Status Over Time During the Initial Evaluation Period (Study 312, Cohort 1, FAS)
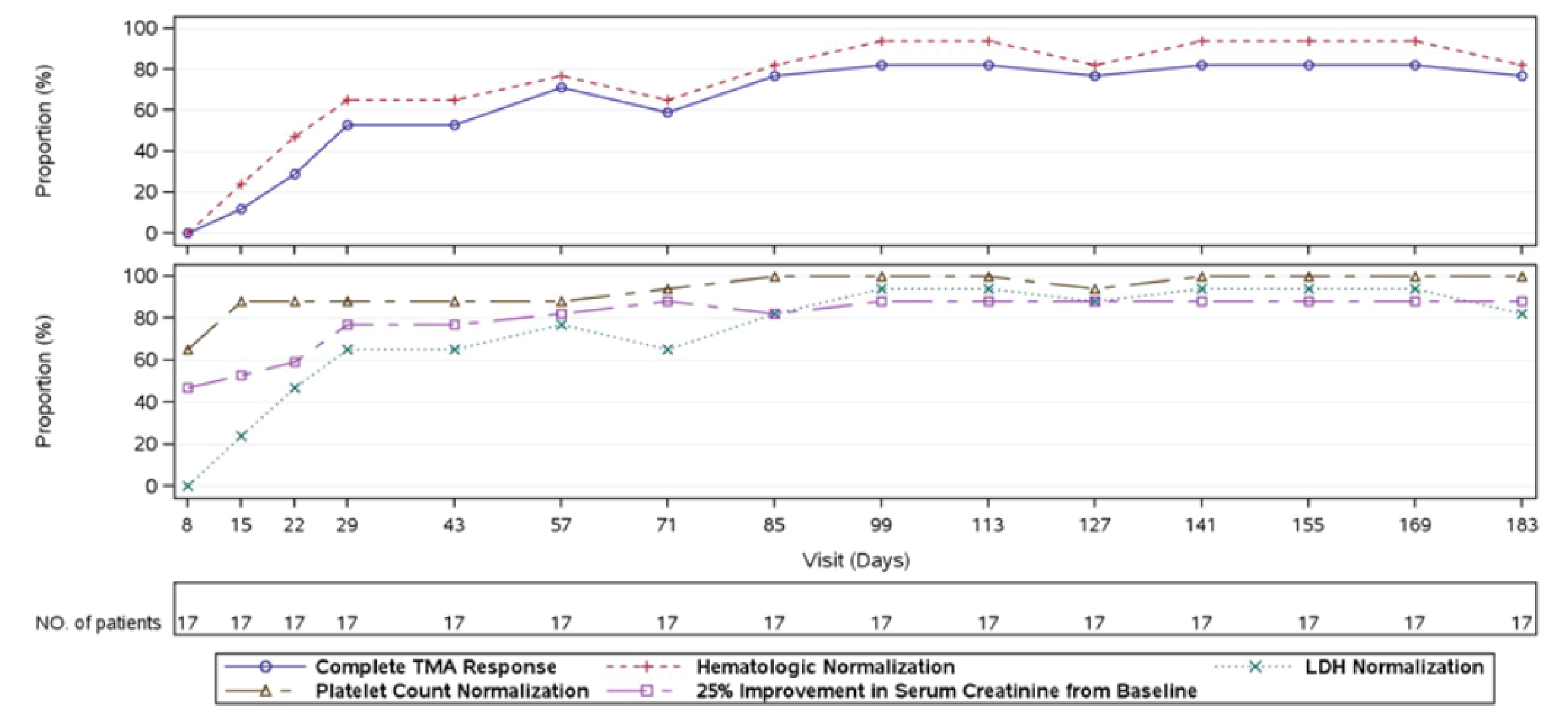
FAS = full analysis set; LDH = lactate dehydrogenase; NO. = number; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Figure 14: Complete TMA Response, Hematologic Normalization, and Complete TMA Response Components Status Over Time During the Extension Period Through the Data Cut-Off Date (Study 312, Cohort 1 FAS)
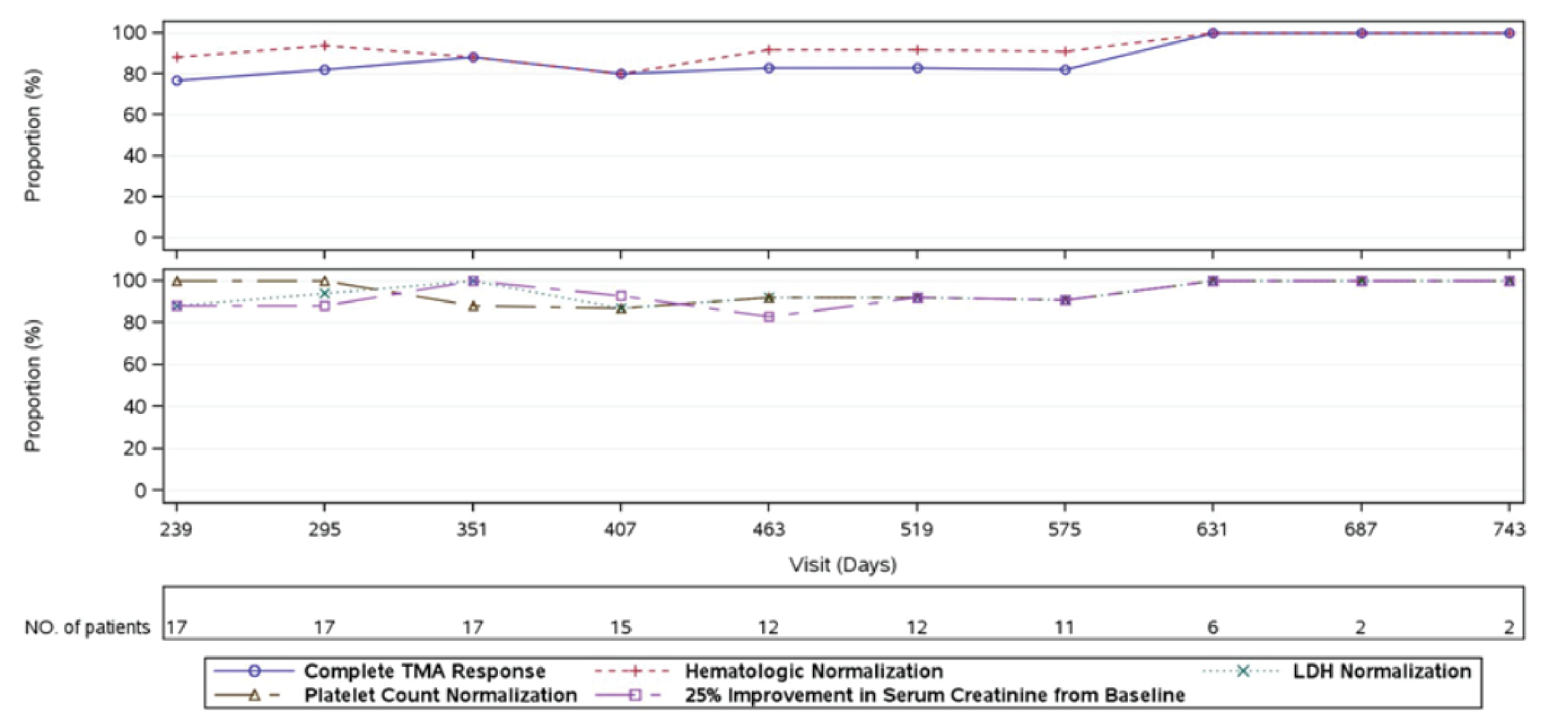
Source: Study 312 Clinical Study Report.22
Table 47: Concomitant Kidney Dialysis (Study 311, FAS and Safety Set)
Category | Study 311 | Study 312 | ||
|---|---|---|---|---|
Overall (FAS, N = 56) | Overall (Safety set, N = 58) | Overall (FAS, N = 18) | Overall (Safety set, N = 21) | |
Patients with concomitant kidney dialysis, n (%) | 32 (57.1) | 33 (56.9) | 6 (33.3) | 8 (38.1) |
Type | ||||
Hemodialysis | 32 (57.1) | 33 (56.9) | 5 (27.8) | 6 (28.6) |
Peritoneal dialysis | 2 (3.6) | 2 (3.4) | 1 (5.6) | 2 (9.5) |
Related to kidney failure caused by aHUS | ||||
Yes | 31 (55.4) | 31 (53.4) | 6 (33.3) | 8 (38.1) |
No | 3 (5.4) | 4 (6.9) | 0 (0.0) | 0 (0.0) |
aHUS = atypical hemolytic uremic syndrome; FAS = full analysis set.
Note: Concomitant kidney dialysis is defined as kidney dialysis that occurred while the patient also received study medication.
Source: Study 311 Clinical Study Report.21
Table 48: Concomitant Kidney Dialysis (Study 312, Cohort 1, FAS and Safety Set)
Category | FAS N = 18 | Safety set (N = 21) | ||||
|---|---|---|---|---|---|---|
Birth to < 6 years (N = 11) | 6 to < 18 years (N = 7) | Overall (N = 18) | Birth to < 6 years (N = 12) | 6 to < 18 years (N = 9) | Overall (N = 21) | |
Patients with Concomitant kidney dialysis, n, (%) | 2 (18.2) | 4 (57.1) | 6 (33.3) | 3 (25.0) | 5 (55.6) | 8 (38.1) |
Type | ||||||
Hemodialysis | 1 (9.1) | 4 (57.1) | 5 (27.8) | 1 (8.3) | 5 (55.6) | 6 (28.6) |
Peritoneal dialysis | 1 (9.1) | 0 (0.0) | 1 (5.6) | 2 (16.7) | 0 (0.0) | 2 (9.5) |
Related to kidney failure caused by aHUS | ||||||
Yes | 2 (18.2) | 4 (57.1) | 6 (33.3) | 3 (25.0) | 5 (55.6) | 8 (38.1) |
No | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
aHUS = atypical hemolytic uremic syndrome; FAS = full analysis set.
Note: Concomitant kidney dialysis is defined as kidney dialysis that occurred while the patient also received study medication.
Source: Study 312 Clinical Study Report.22
Table 49: Treatment-Emergent AEs Experienced by 20% or More Patients by System Organ Class and Preferred Term as of Data Cut-Off Date (Study 311, Safety Set)
AEs | Study 311 (N = 58) | Study 312, cohort 1 (N = 21) | Study 312, cohort 2 (N = 10) |
|---|---|---|---|
Any TEAE, n (%) | 58 (100.0) | 21 (100.0) | 10 (100.0) |
Diarrhea | 19 (32.8) | 7 (33.3) | 1 (10.0) |
Vomiting | 18 (31.0) | 7 (33.3) | 1 (10.0) |
Nausea | 15 (25.9) | 4 (19.0) | NR |
Abdominal pain | 8 (13.8) | 6 (28.6) | 1 (10.0) |
Nasopharyngitis | 9 (15.5) | 7 (33.3) | 2 (20.0) |
Otitis media | NR | NR | 2 (20.0) |
Oropharyngeal pain | NR | 3 (14.3) | 3 (30.0) |
Pharyngitis | NR | 2 (9.5) | 2 (20.0) |
Pyrexia | 12 (20.7) | 10 (47.6) | NR |
Cough | 10 (17.2) | 5 (23.8) | 1 (10.0) |
Headache | 22 (37.9) | 7 (33.3) | 1 (10.0) |
Arthralgia | 15 (25.9) | NR | NR |
Hypertension | 14 (24.1) | 6 (28.6) | 1 (10.0) |
AE = adverse event; NR = not reported; TEAE = treatment-emergent adverse event.
Note: Adverse events were coded using MedDRA Version 21.0. In summarizing n (%),if a patient had multiple events for a particular Preferred Terms, they were counted only once for that Preferred Terms. Patients may be counted in more than 1 Preferred Term category.
Sources: Study 311 Clinical Study Report21 and Study 312 Clinical Study Report.22
Figure 15: Observed Values of Platelets (109/L) Over Time (Study 312, Cohort 2, FAS)
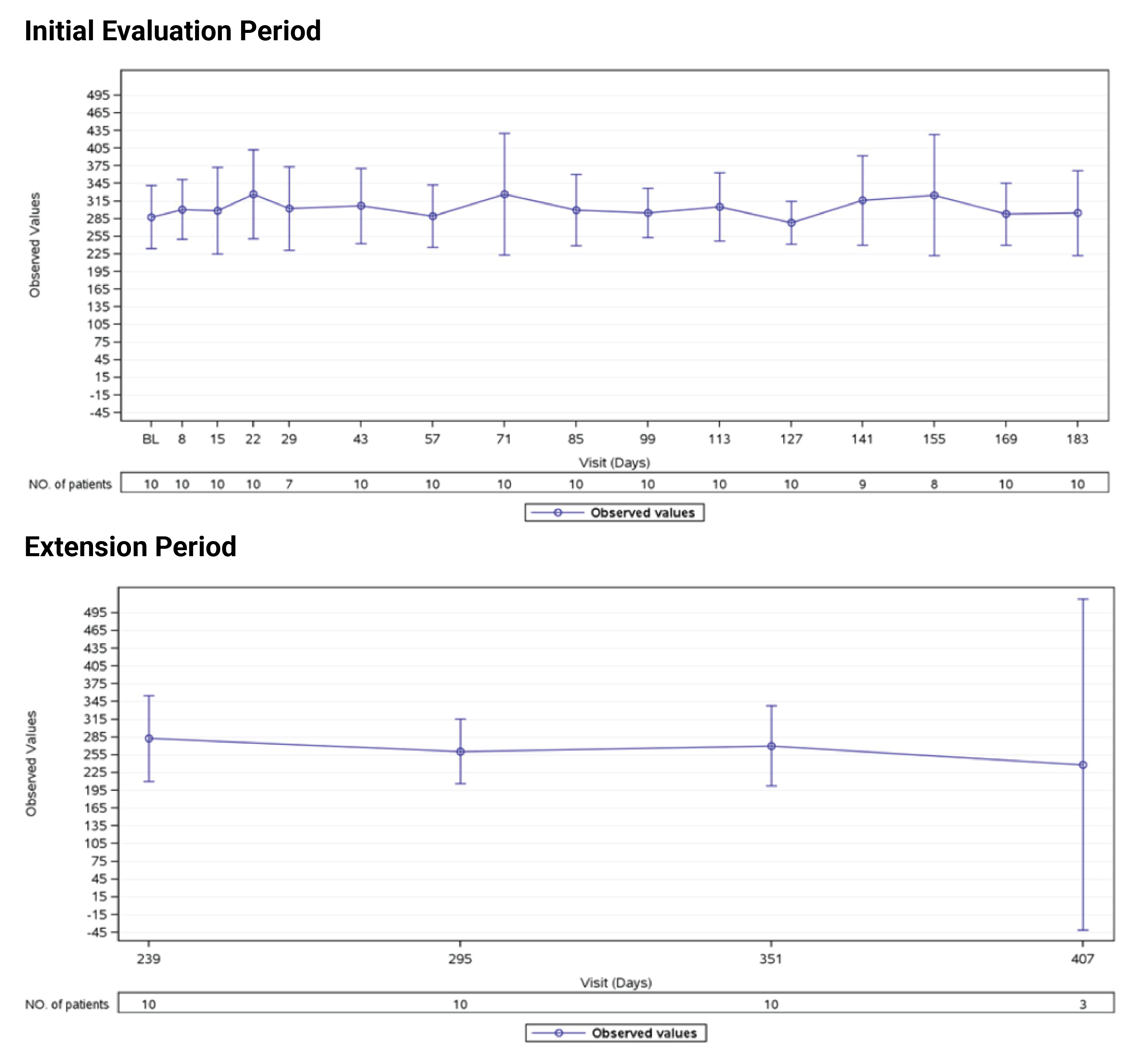
FAS = full analysis set; NO. = number; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Figure 16: Observed Values of LDH (U/L) Over Time (Study 312, Cohort 2, FAS)
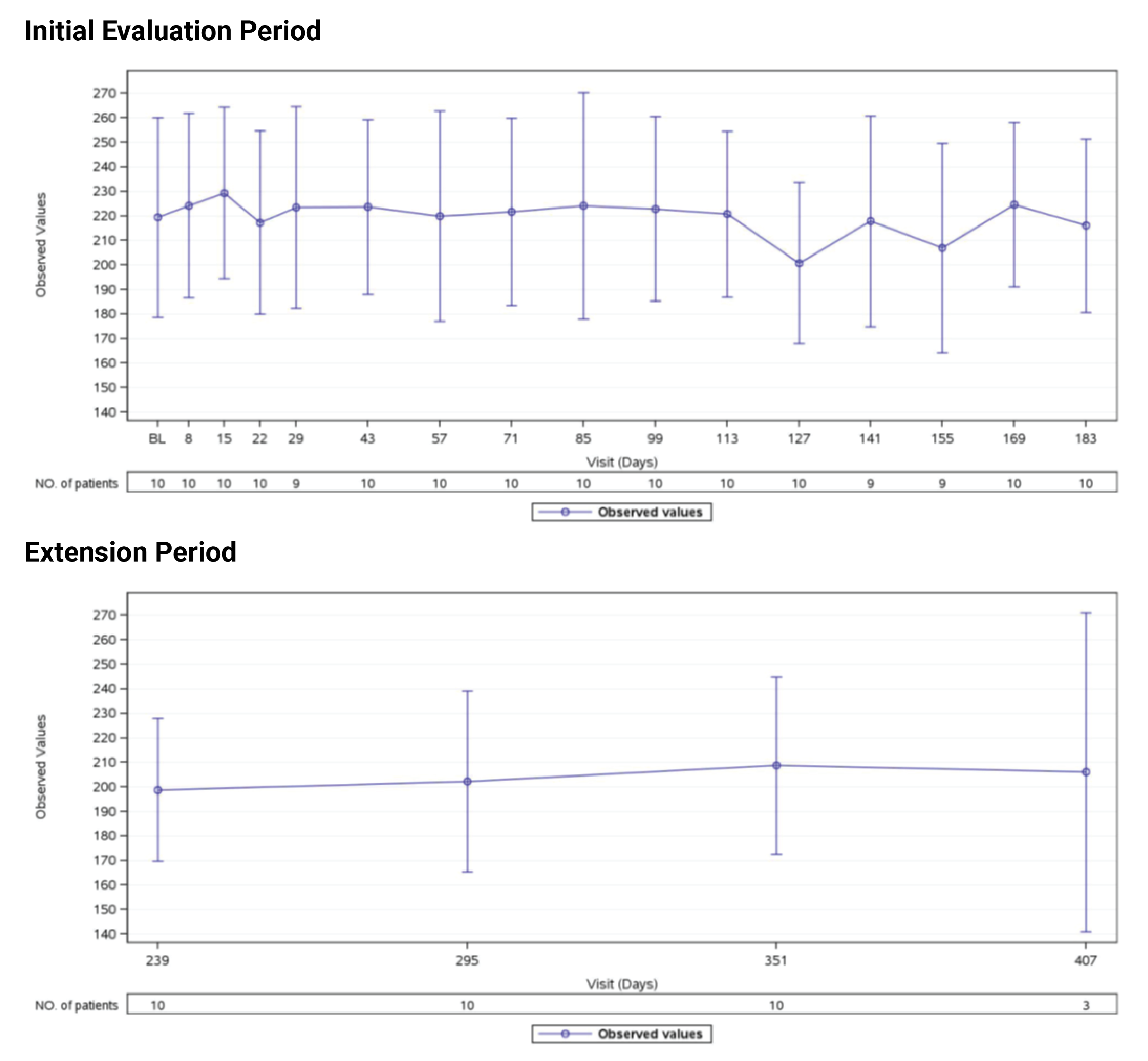
FAS = full analysis set; LDH = lactate dehydrogenase; NO. = number; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Figure 17: Observed Values of Hemoglobin (g/L) Over Time (Study 312, Cohort 2, FAS)
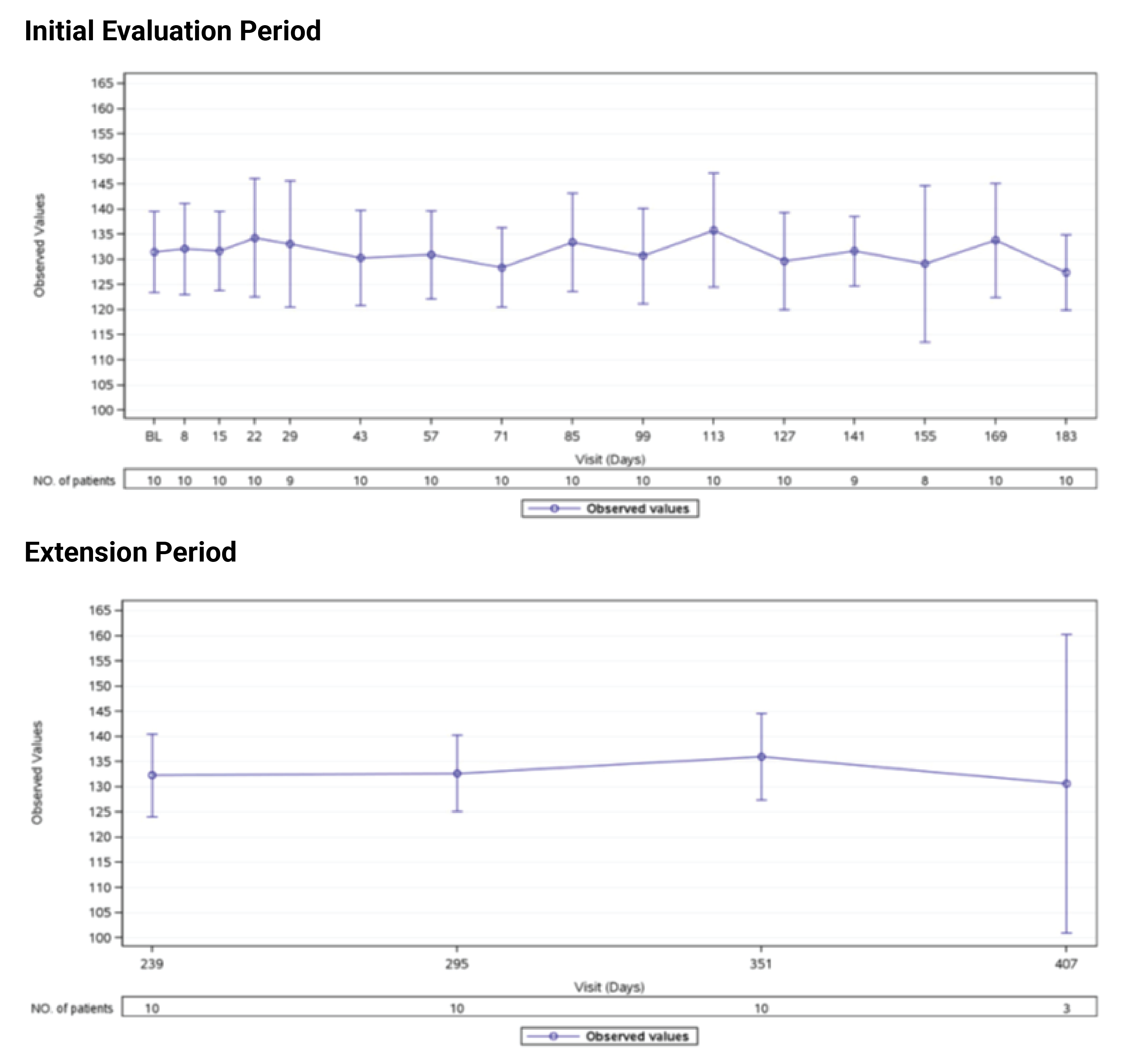
FAS = full analysis set; NO. = number; TMA = thrombotic microangiopathy.
Source: Study 312 Clinical Study Report.22
Figure 18: Observed Values of FACIT-F Scores Over Time as of Data Cut-Off Date (FAS, Study 311)
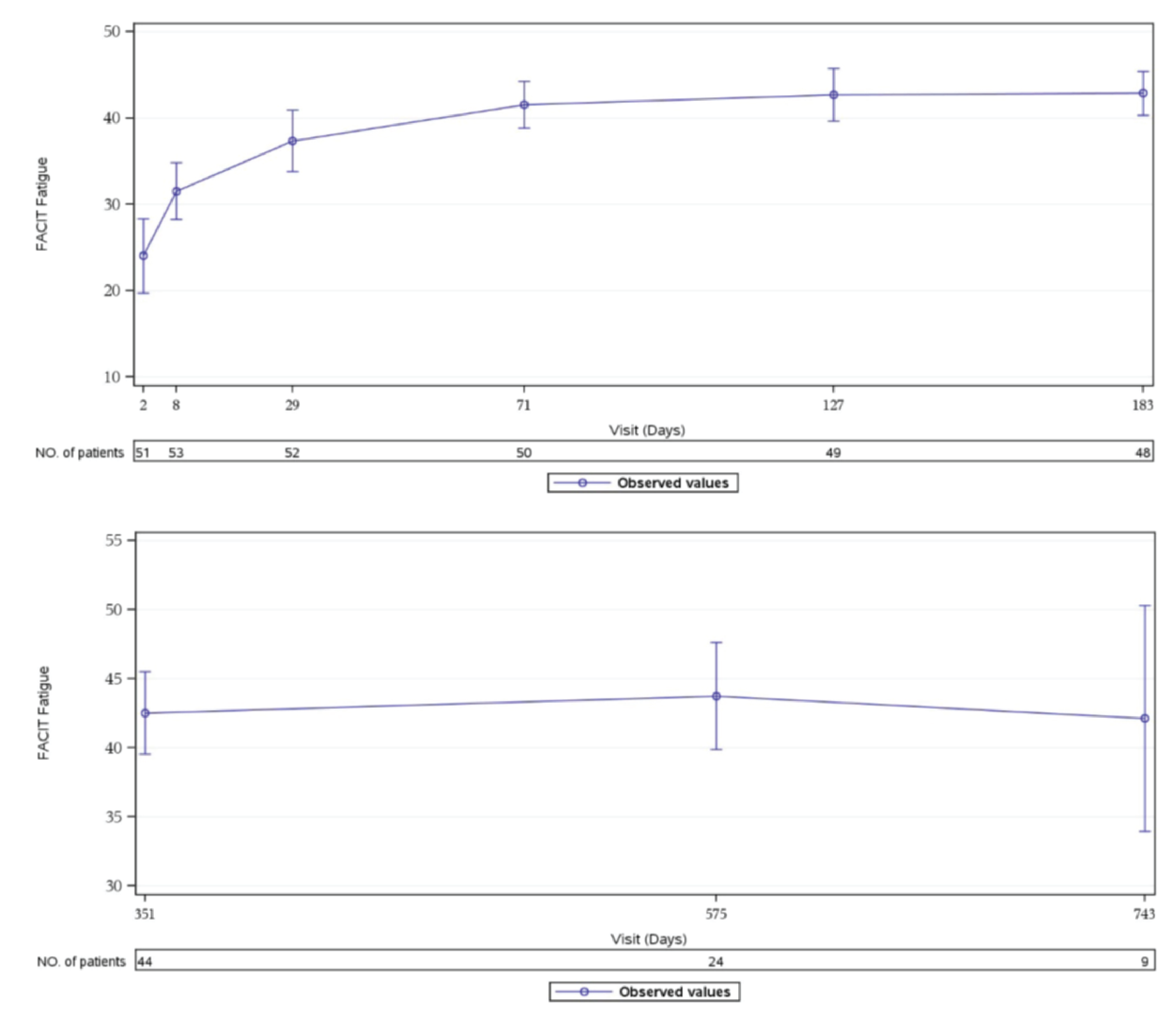
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue.; NO. = number.
Notes: Data as of July 2, 2019. Baseline was from the day 1 value. The FACIT-F questionnaire version 4 was used. The FACIT-F questionnaire at baseline and each postinfusion time point was scored using standard scoring algorithms. The FACIT-F score ranges from 0 to 52, with a higher score indicating less fatigue. Mean ± 95% CIs are displayed in the figure. Time points with fewer than 5 patients are not displayed on the figure. Source: Study 311 Clinical Study Report.21
Figure 19: Observed Values and Model-Based Values of Changes in FACIT-F Scores Over Time (Study 311, FAS)
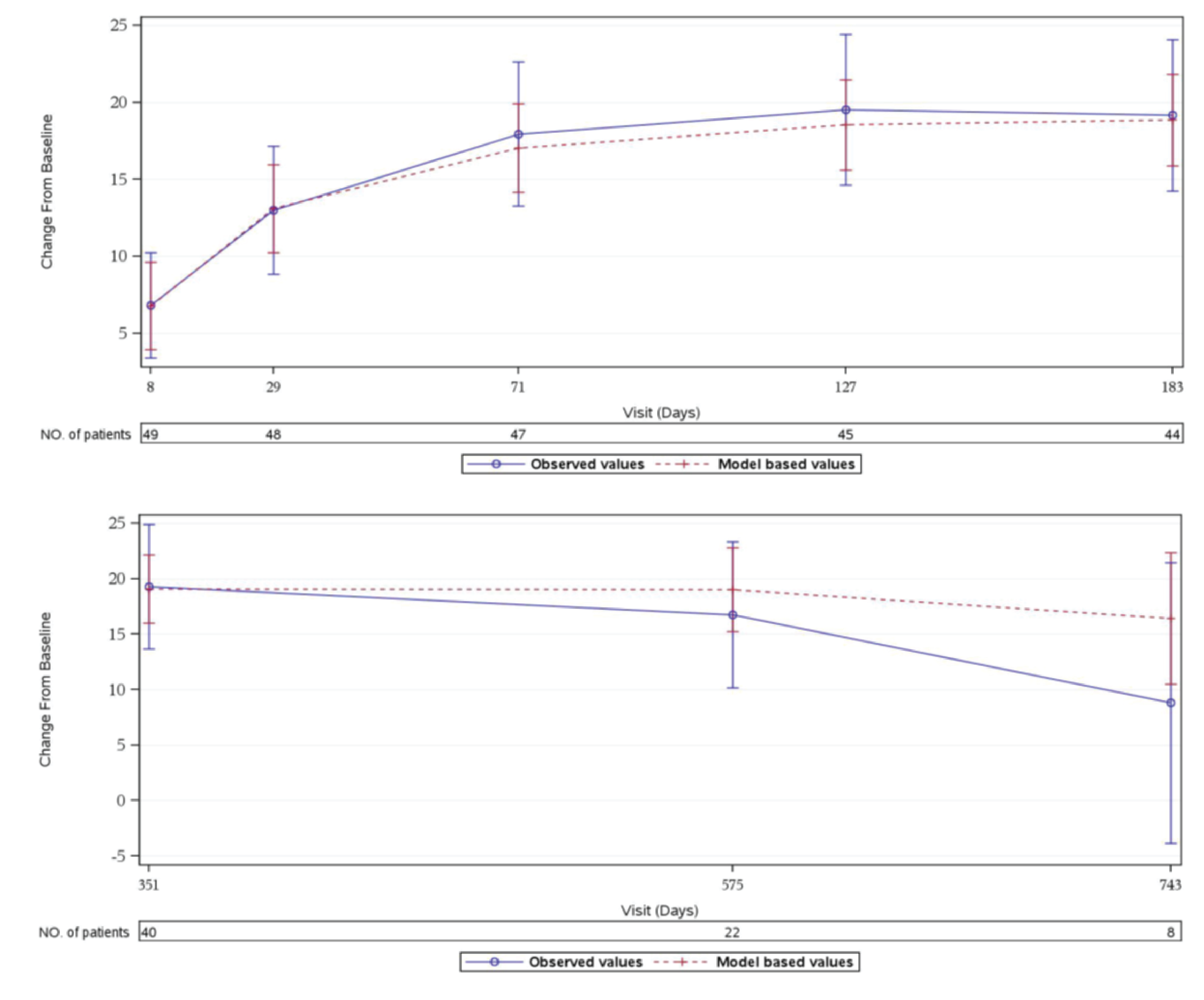
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; NO. = number.
Notes: Baseline is from the day 1 value. FACIT-F questionnaire version 4 is used. The FACIT-F questionnaire at baseline and each postinfusion time point is scored using standard scoring algorithms. For overall, a mixed model for repeated measures is used to improve the precision of estimation of changes over time, it includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A Toeplitz covariance structure is used to model the within patient errors. Time points with fewer than 5 patients are not displayed on the figure. FACIT score ranges from 0 to 52, with a higher score indicating less fatigue. Observed values: mean+/− 95% CI. Model-based values: mean +/− 95% CI.
Source: Study 311 Clinical Study Report.21
Figure 20: Observed Values of Pediatric FACIT-F Scores Over Time (Study 312, Cohort 1, FAS)
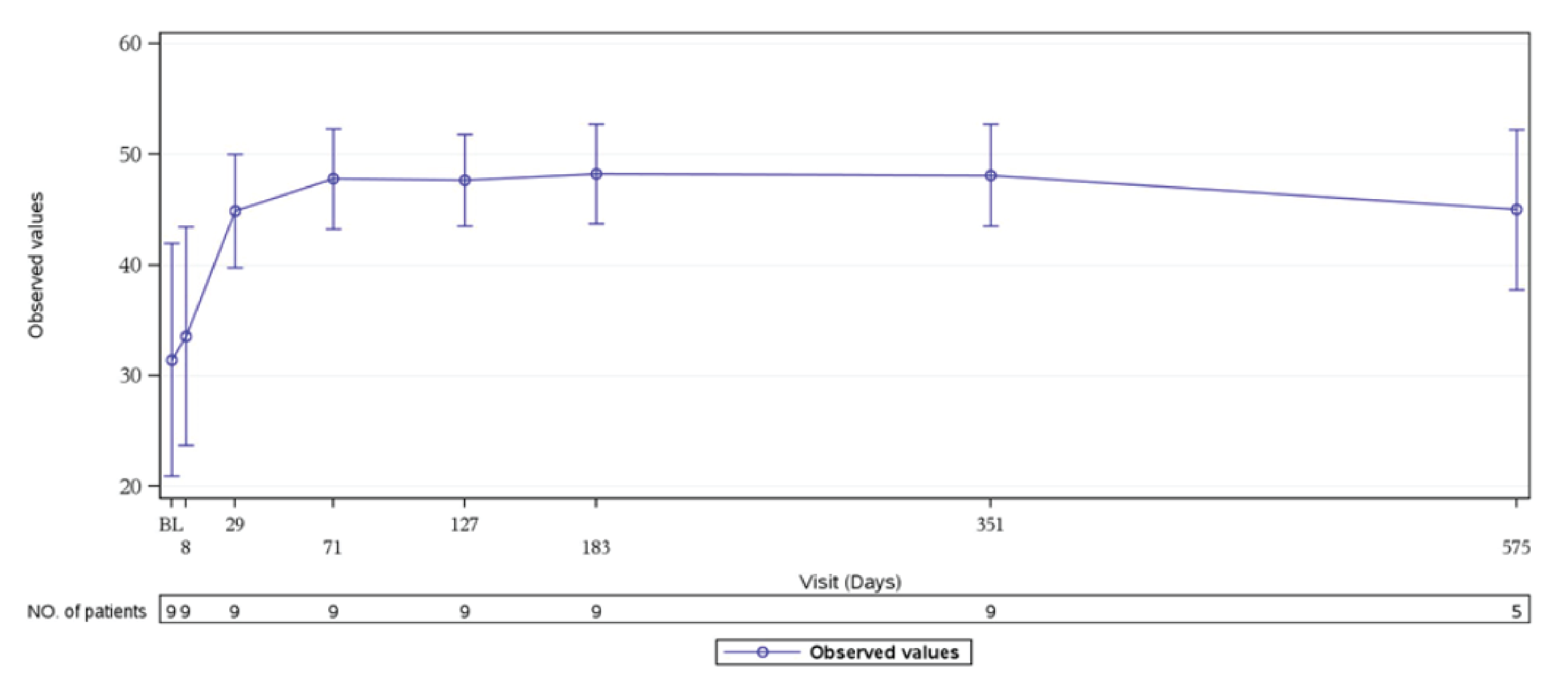
BL = baseline; CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; FAS = full analysis set; NO. = number.
Notes: Data as of December 3, 2019. Baseline was the day 1 value. The pediatric FACIT-F questionnaire was used. The pediatric FACIT-F questionnaire at baseline and each postinfusion time point was scored using standard scoring algorithms. The FACIT-F score ranged from 0 to 52, with a higher score indicating less fatigue. Values displayed are mean ± 95% CIs.
Source: Study 312 Clinical Study Report.22
Figure 21: Observed Values and Model-Based Values of Changes in Pediatric FACIT-F Scores Over Time (Study 312, Cohort 1, FAS)

CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; FAS = full analysis set; NO. = number.
Notes: Baseline is from the day 1 value. The pediatric FACIT-F questionnaire is used. The pediatric FACIT-F questionnaire at baseline and each postinfusion time point is scored using standard scoring algorithms. A mixed model for repeated measures is used, it includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A compound symmetry covariance structure is used to model the within patient errors. Time points with fewer than 5 patients are not displayed on the figure. FACIT score ranges from 0 to 52, with a higher score indicating less fatigue. Observed values: mean+/− 95% CI. Model-based values: mean +/− 95% CI.
Source: Study 312 Clinical Study Report.22
Figure 22: Observed Values of Pediatric FACIT-F Scores Over Time (Study 312, Cohort 2, FAS)
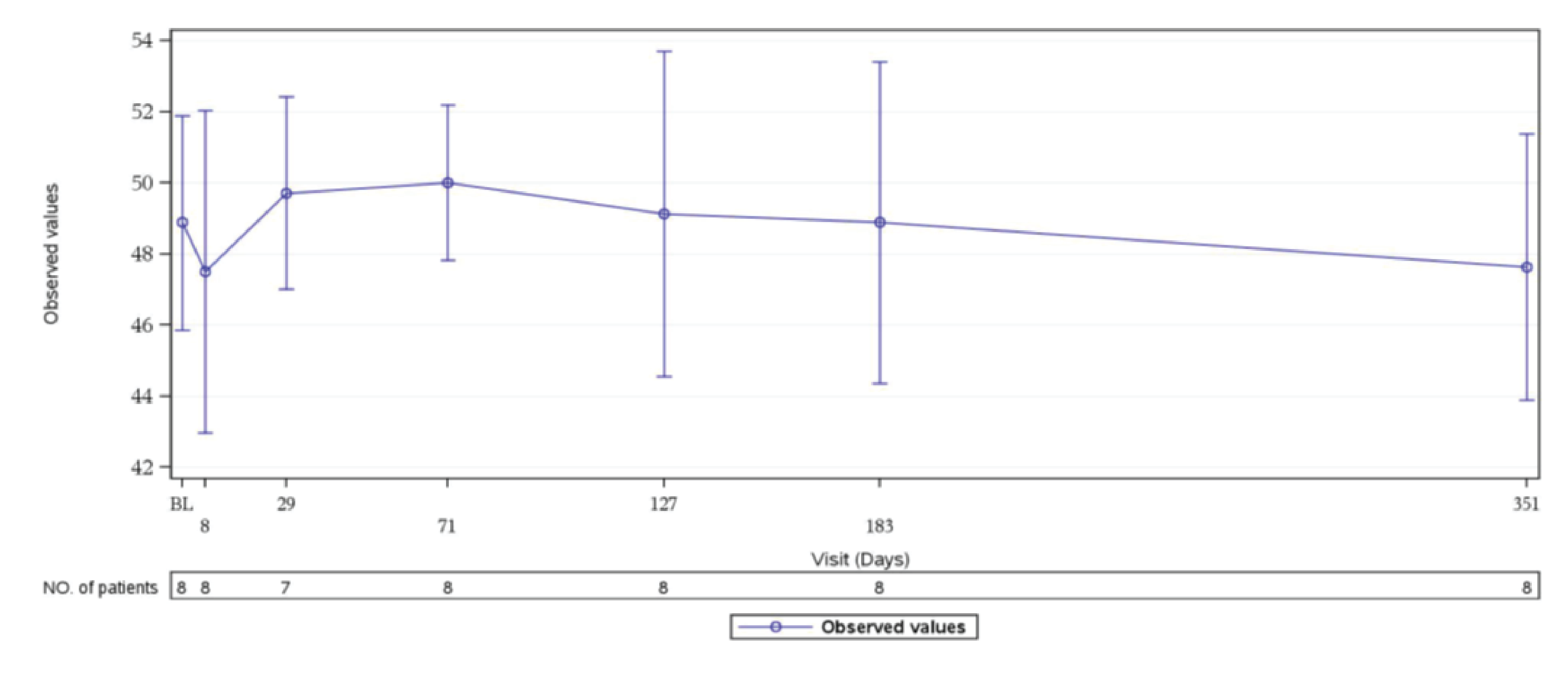
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; FAS = full analysis set; NO. = number.
Source: Study 312 Clinical Study Report.22
Figure 23: Observed Values and Model-Based Values of Changes in Pediatric FACIT-F Scores Over Time (Study 312, Cohort 2, FAS)
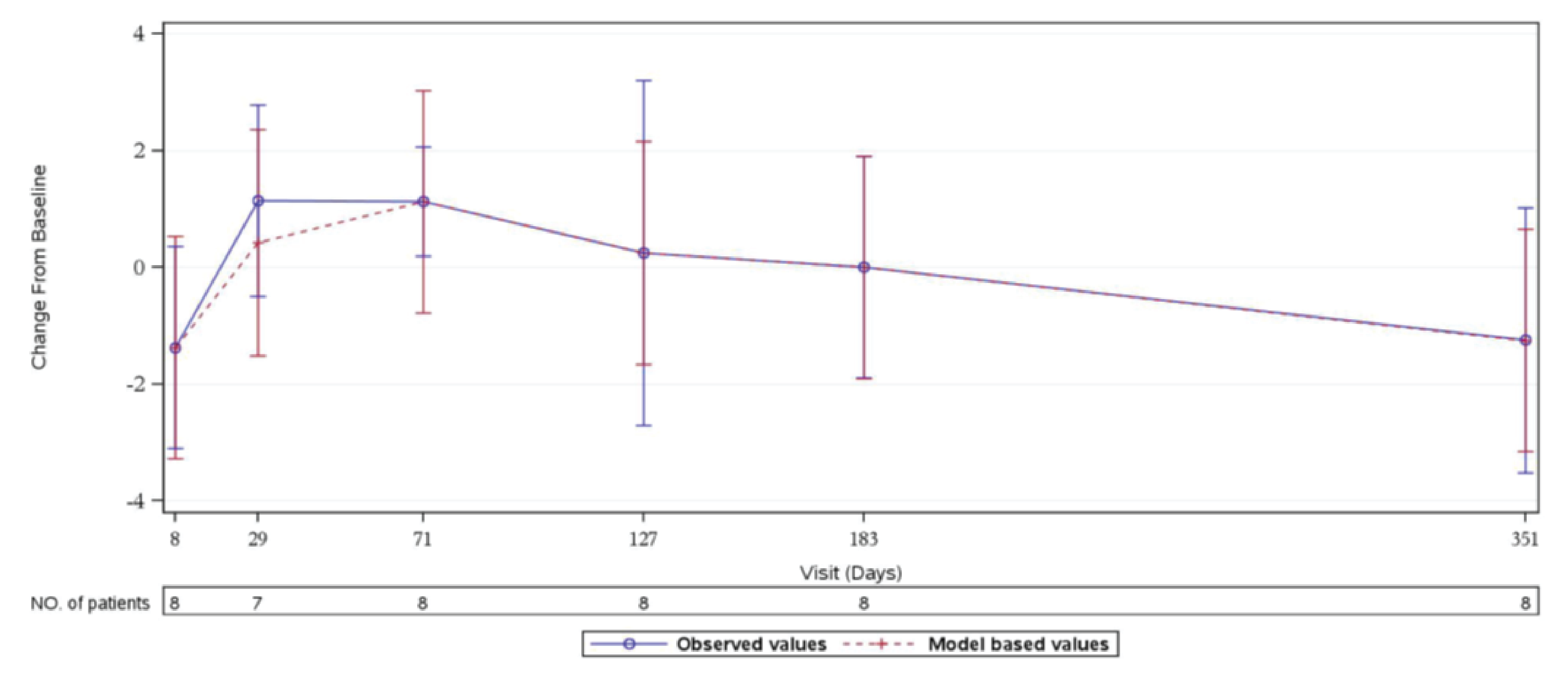
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy – Fatigue; FAS = full analysis set; NO. = number.
Source: Study 312 Clinical Study Report.22
Figure 24: Observed Values of 3-Level EQ-5D Scores Over Time as of Data Cut-Off Date (FAS, Study 311)
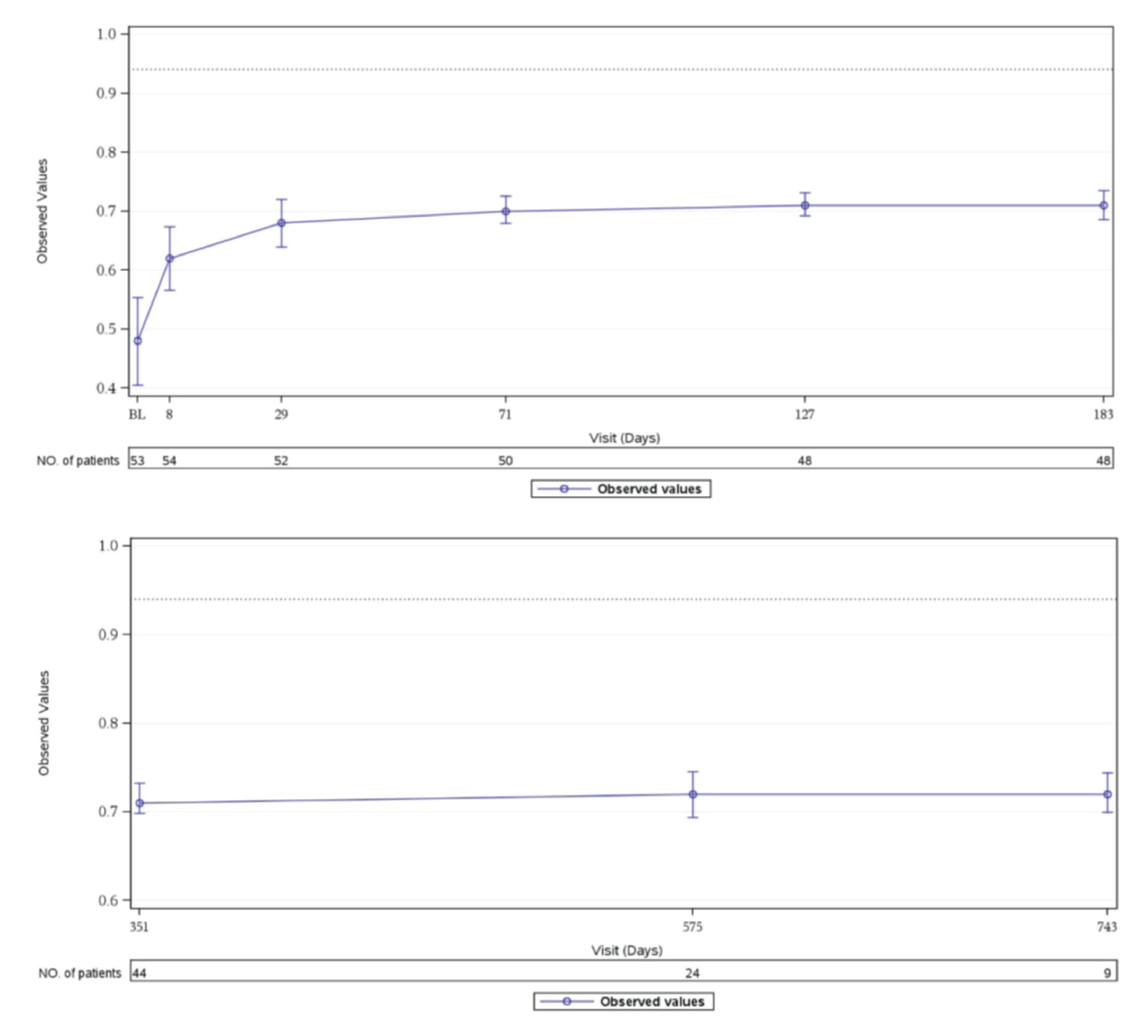
FAS = full analysis set; US TTO = time trade-off value set for the US; VAS = visual analogue scale.
Notes: Data as of July 2, 2019. Baseline was from the day 1 value. The 3-Level EQ-5D score was assessed using the index scored according to the US TTO as well as the response on the VAS question. The standard US TTO value set was used to assign a baseline index value as well as a value at each postinfusion time point, based on the health state indicated on the questionnaire. The US TTO > 0.94 indicates full health. Mean ± 95% CIs are displayed in the figure. Time points with fewer than 5 patients are not displayed in the figure.
Source: Study 311 Clinical Study Report.21
Figure 25: Observed Values and Model-Based Values of Changes in 3-Level EQ-5D (Time Trade-Off Value Set for the US) Over Time (Study 311, FAS)
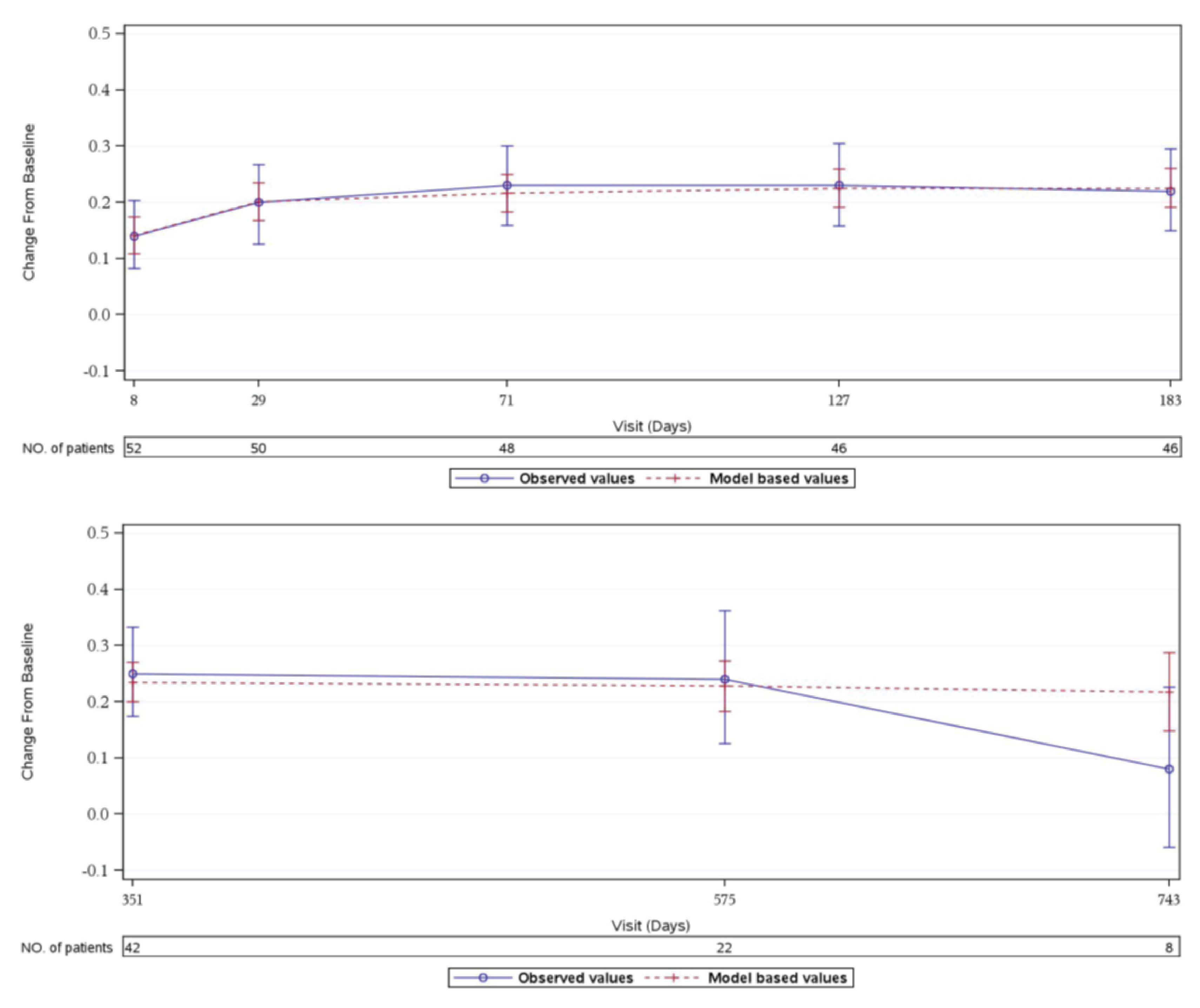
FAS = full analysis set; US TTO = time trade-off value set for the US; VAS = visual analogue scale.
Notes: Baseline is from the day 1 value. The 3-Level EQ-5D is assessed using the index scored according to the time trade-off value set for the US (US TTO) as well as the response on the visual analogue scale question. The standard US TTO value set is used to assign a baseline index value as well as a value at each postinfusion time point, based on the health state indicated on the questionnaire. US TTO > 0.94 indicates full health. For overall, a mixed model for repeated measures is used to improve the precision of estimation of changes over time, it includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A first-order autoregressive covariance structure is used to model the within patient errors. Time points with fewer than 5 patients are not displayed on the figure. Observed values: mean+/− 95% CI; Model-based values: mean +/− 95% CI.
Source: Study 311 Clinical Study Report.21
Figure 26: Observed Values of eGFR Over Time During the Initial Evaluation Period (FAS, Study 311)
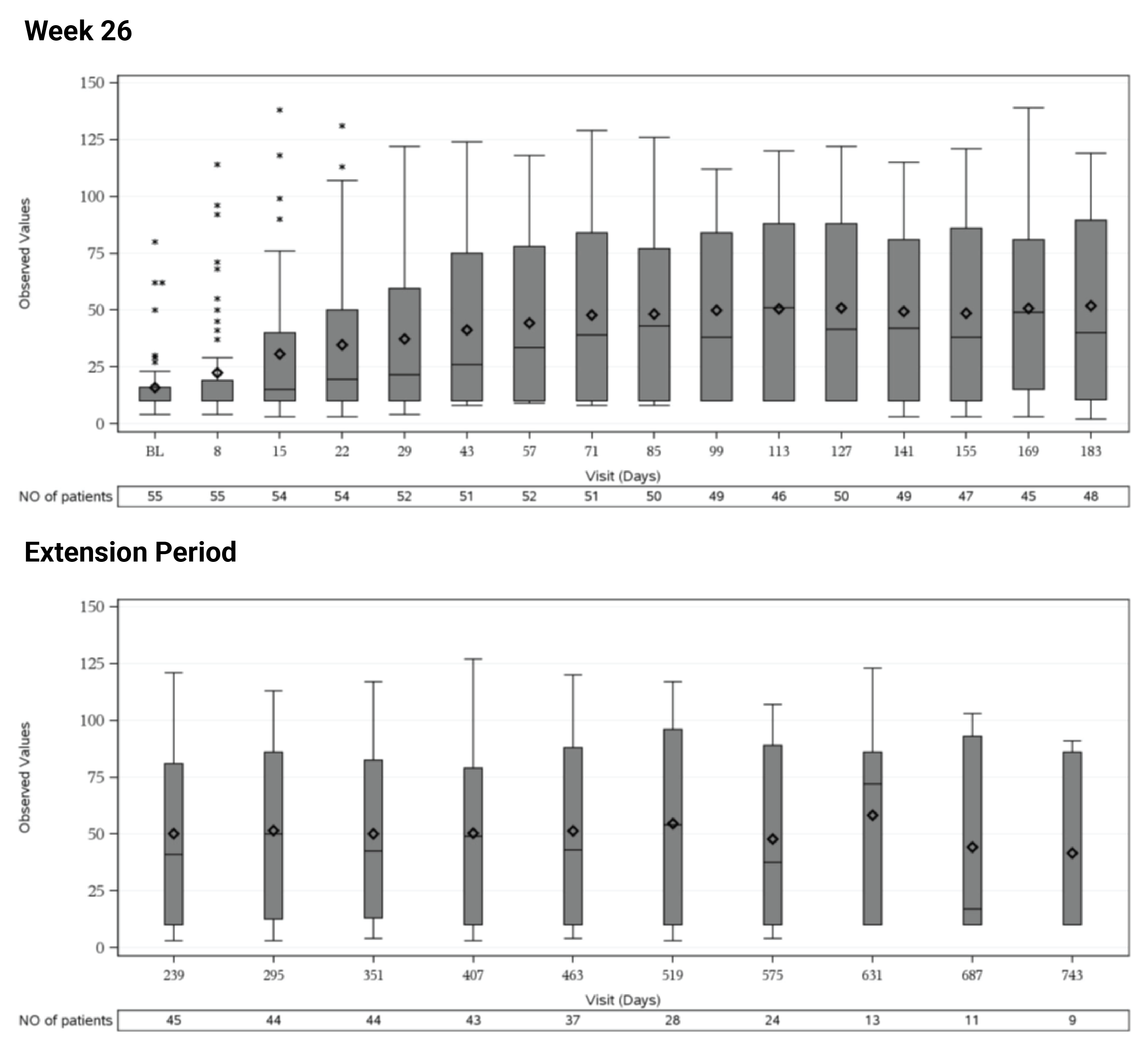
BL = baseline; eGFR = estimated glomerular filtration rate; FAS = full analysis set; NO. = number.
Note: Baseline value was defined as the average of the values from the assessments performed before the first study drug infusion (these could have included results from screening and the day 1 visit). For eGFR, 10 mL/min/1.73 m2 was imputed for patients requiring dialysis for acute kidney injury. The horizontal line in the middle of each box indicates the median, a diamond indicates the mean; the top and bottom borders of the box mark the 75th and 25th percentiles, respectively. The whiskers represent the highest and lowest values within 1.5 times the interquartile range from the lower quartile and upper quartile. Outliers are represented by asterisk beyond the whiskers.
Source: Study 311 Clinical Study Report.21
Figure 27: Observed Values and Model-Based Values of Changes in eGFR Over Time (Study, 311 FAS)
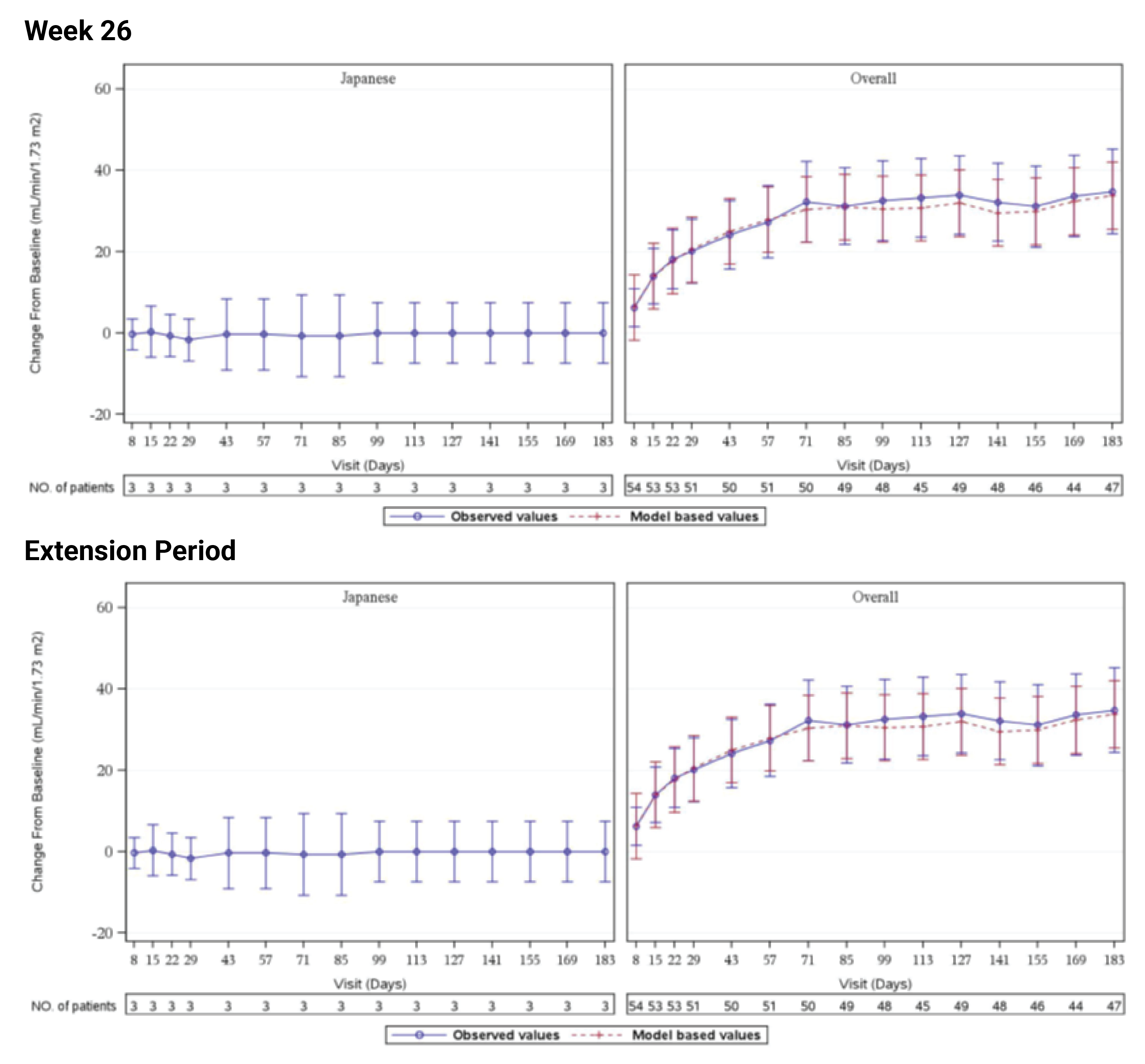
BL = baseline; eGFR = estimated glomerular filtration rate; FAS = full analysis set; NO. = number.
Note: Baseline value is defined as the average of the values from the assessments performed before the first study drug infusion (these can include results from screening and the day 1 visit); 10 mL/min/1.73 m2 for eGFR is imputed for patients requiring dialysis for acute kidney injury. For overall, a mixed model for repeated measures is used. It includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A compound symmetry covariance structure is used to model the within patient errors. Time points with fewer than 5 patients only present observed values (mean +/− 95% CI). Observed values: mean +/− 95% CI; Model-based values: mean +/− 95%.
Source: Study 311 Clinical Study Report.21
Figure 28: Observed Values of eGFR Over Time (Study 312, Cohort 1, FAS)
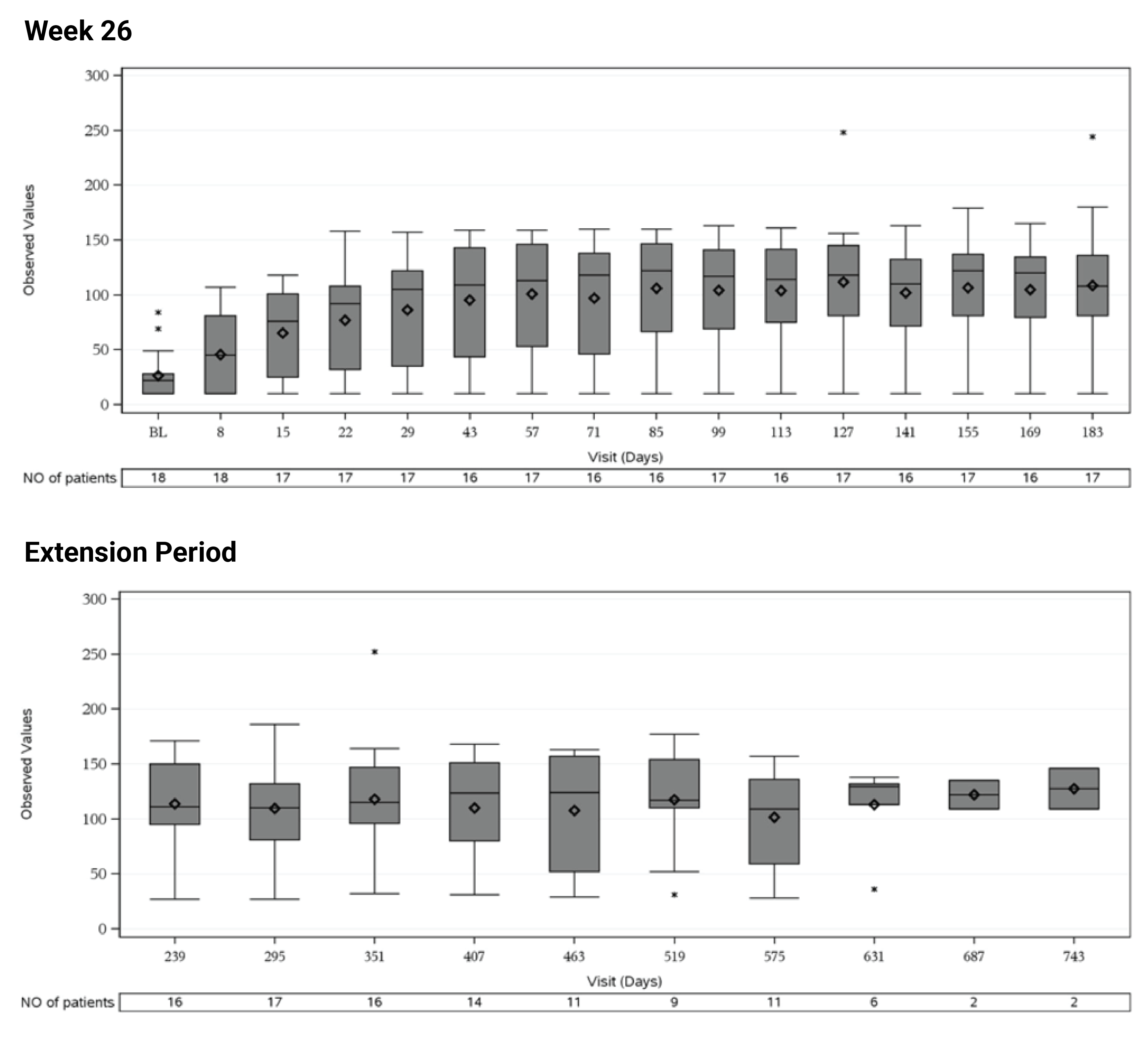
eGFR = estimated glomerular filtration rate; FAS = full analysis set; NO. = number.
Note: Data as of December 3, 2019. Baseline value was defined as the average of the values from the assessments performed before the first study drug infusion (these could include results from screening and the day 1 visit). For eGFR, 10 mL/min/1.73 m2 was imputed for patients requiring dialysis for acute kidney injury. The horizontal line in the middle of each box indicates the median, a diamond indicates the mean, and the top and bottom borders of the box mark the 75th and 25th percentiles, respectively. The whiskers represent the highest and lowest values within 1.5 times the interquartile range from the lower quartile and upper quartile. Outliers are represented by asterisk beyond the whiskers.
Source: Study 312 Clinical Study Report.22
Figure 29: Observed Values and Model-Based Values of Changes in eGFR Over Time (Study 312, Cohort 1, FAS)
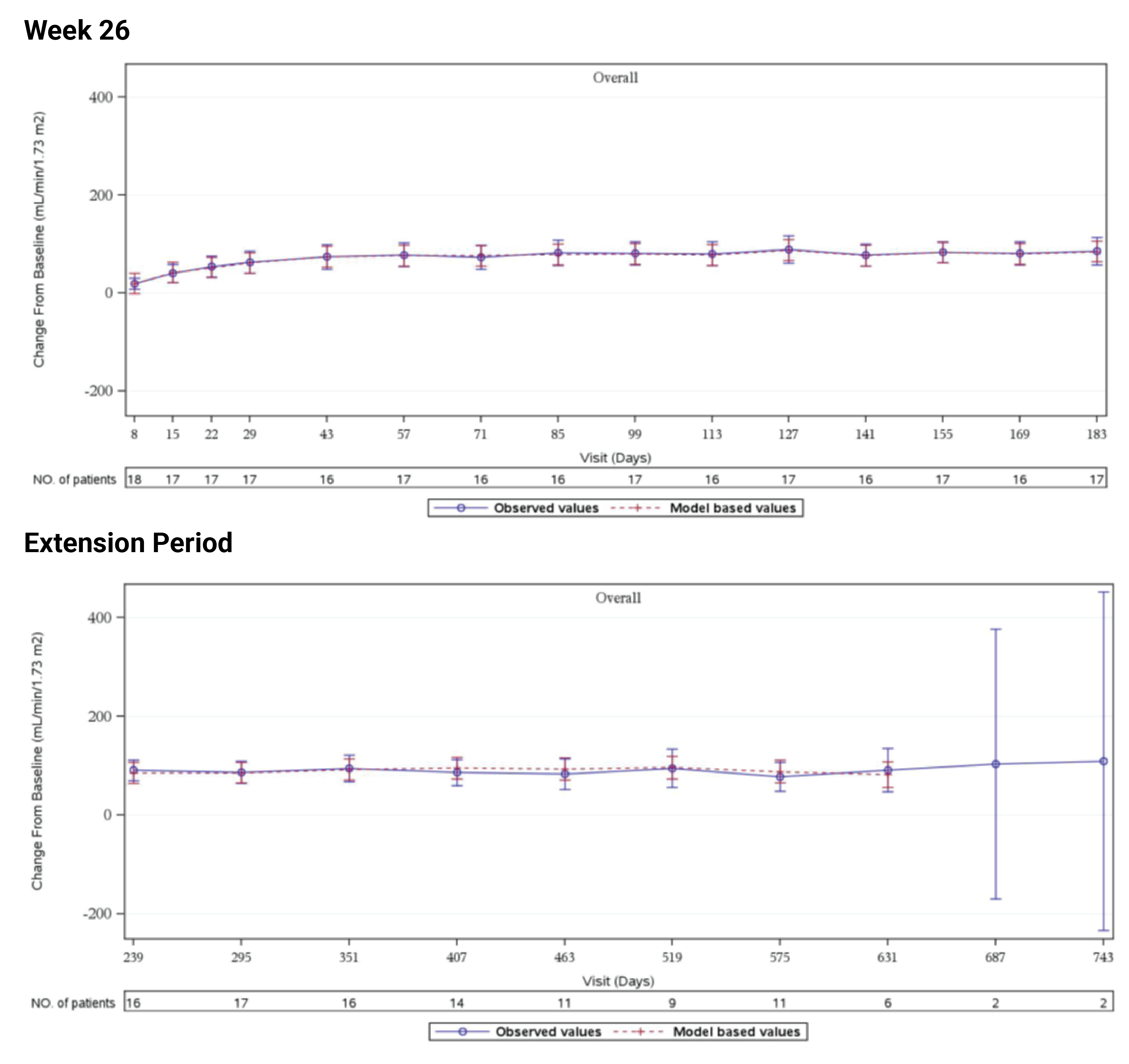
eGFR = estimated glomerular filtration rate; FAS = full analysis set; NO. = number.
Note: Baseline value is defined as the average of the values from the assessments performed before the first study drug infusion (these can include results from screening and the day 1 visit). 10 mL/min/1.73 m2 for eGFR is imputed for patients requiring dialysis for acute kidney injury. For overall, a mixed model for repeated measures is used. It includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A compound symmetry covariance structure is used to model the within patient errors. Time points with fewer than 5 patients only present observed values (mean+/− 95% CI). Observed values: mean+/− 95% CI; Model-based values: mean +/− 95% CI.
Source: Study 312 Clinical Study Report.22
Figure 30: Observed Values of eGFR Over Time (Study 312, Cohort 2, FAS)
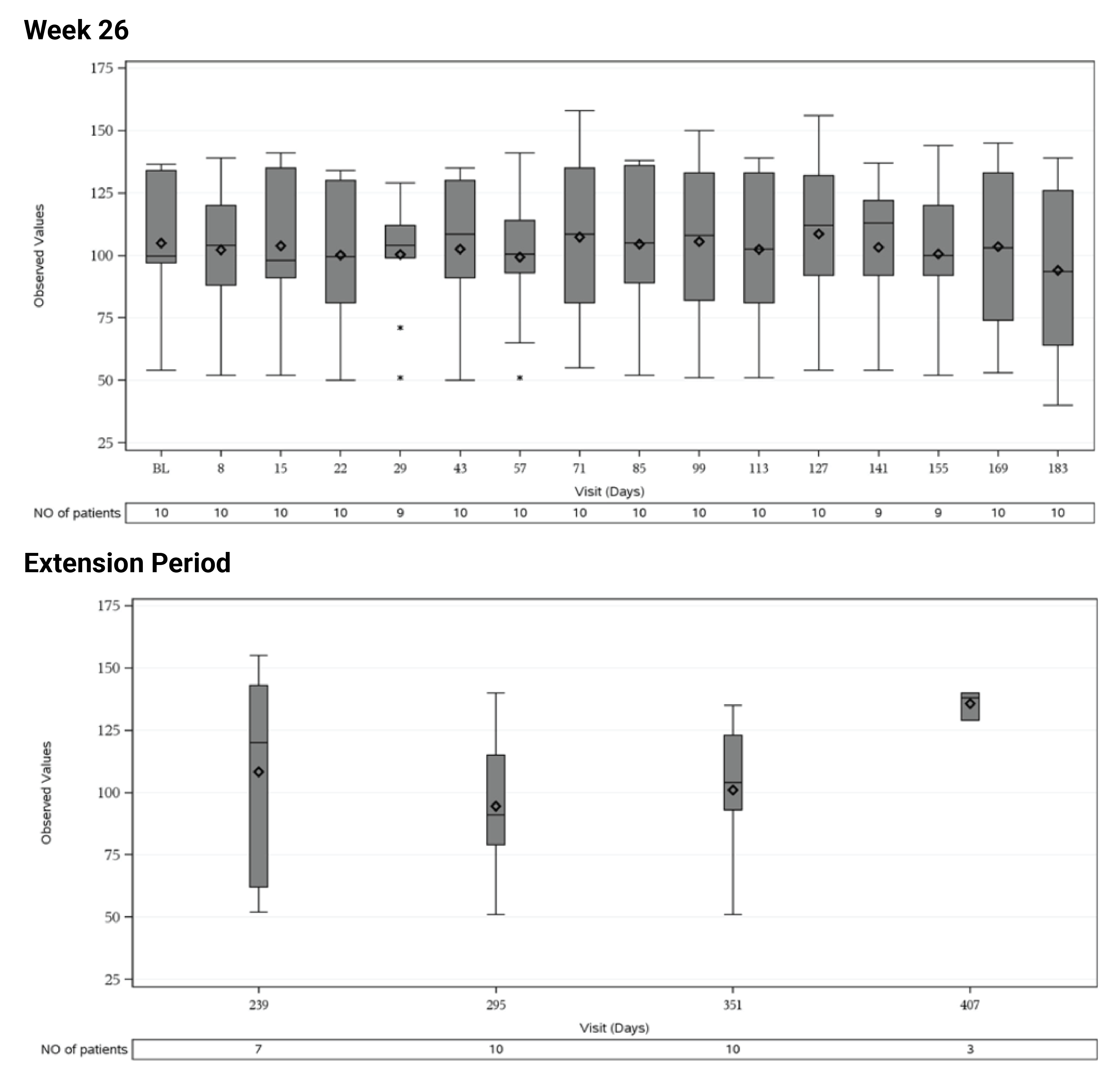
eGFR = estimated glomerular filtration rate; FAS = full analysis; NO. = number.
Note: Data as of December 3, 2019. Baseline value was defined as the average of the values from the assessments performed before the first study drug infusion (these could have included results from screening and the day 1 visit). For eGFR, 10 mL/min/1.73 m2 was imputed for patients requiring dialysis for acute kidney injury. The horizontal line in the middle of each box indicates the median, a diamond indicates the mean and the top and the bottom borders of the box mark the 75th and 25th percentiles, respectively. The whiskers represent the highest and lowest values within 1.5 times the interquartile range from the lower quartile and upper quartile. Outliers are represented by asterisk beyond the whiskers.
Source: Study 312 Clinical Study Report.22
Figure 31: Observed Values and Model-Based Values of Changes in eGFR Over Time (Study 312, Cohort 2 FAS)
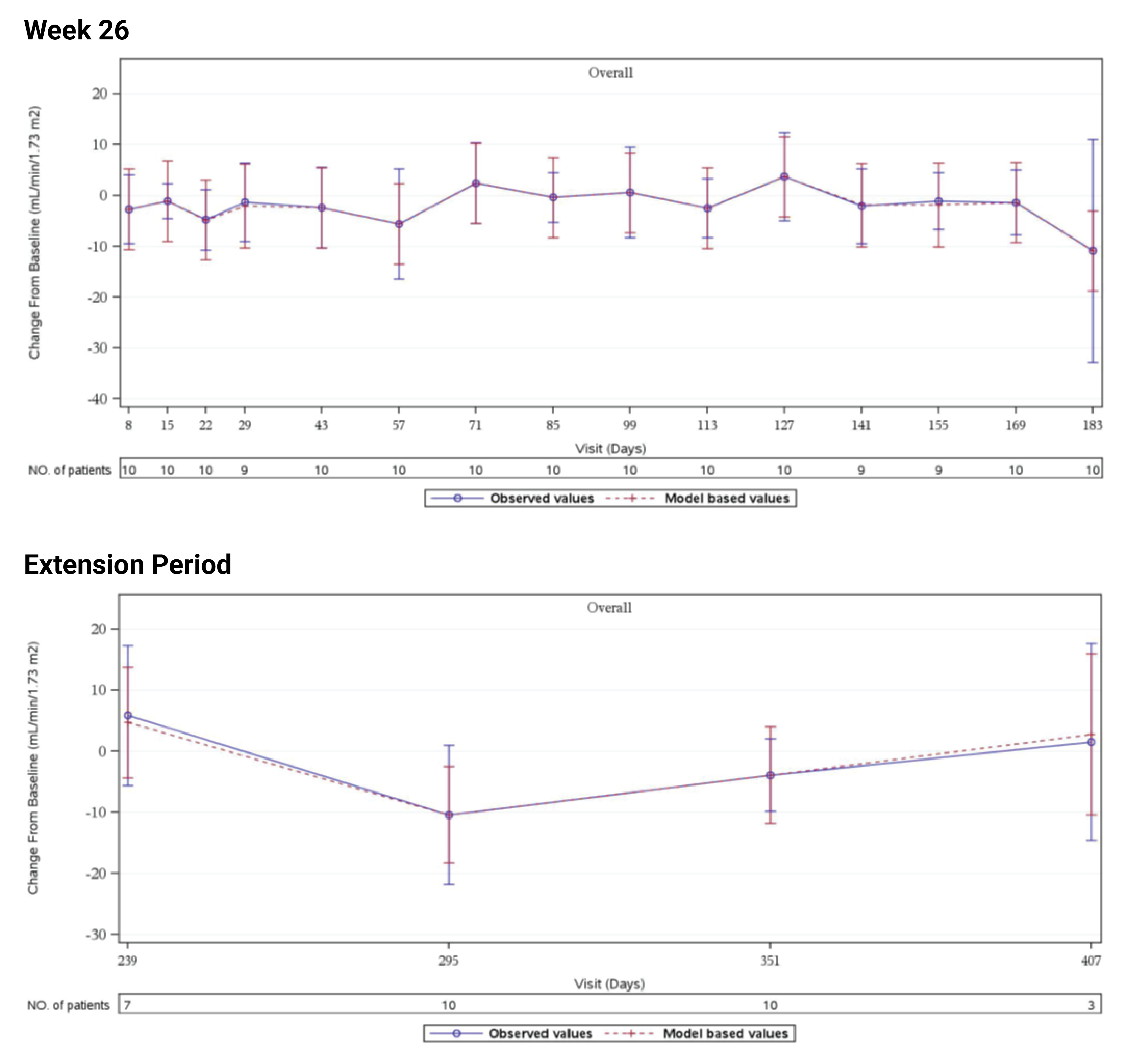
eGFR = estimated glomerular filtration rate; FAS = full analysis set; NO. = number.
Note: Baseline value is defined as the average of the values from the assessments performed before the first study drug infusion (these can include results from screening and the day 1 visit). 10 mL/min/1.73 m2 for eGFR is imputed for patients requiring dialysis for acute kidney injury. A mixed model for repeated measures is used. It includes the fixed, categorical effect of visit and the fixed, continuous effect of the baseline value as covariates. A compound symmetry covariance structure is used to model the within patient errors. Observed values: mean+/− 95% CI; Model-based values: mean +/− 95% CI.
Source: Study 312 Clinical Study Report.22
Appendix 5: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
The aim was to describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
FACIT-F scale and pediatric FACIT-F scale
3-Level EQ-5D.
Findings
Table 50: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
FACIT-F | A patient self-completed questionnaire to assess the intensity of fatigue (and its impact on daily life) during usual daily activities over the past week. It consists of a 13-item questionnaire that assesses self-reported tiredness, weakness, and difficulty conducting usual activities due to fatigue. The level of fatigue is measured on a 4-point Likert scale (4 = not at all fatigued to 0 = very much fatigued). The instrument scoring yields a range from 0 to 52, with higher scores representing better overall health status (i.e., less fatigue).48 | The instrument has good discriminant and convergent validity and good internal consistency reliability in other conditions (i.e., cancer, RA, psoriatic arthritis).49-51 Based on EMPRO scale, the reliability, validity, and responsiveness of FACIT-F instrument achieved scores of 25.00, 54.17, and 55.56, respectively, showing slightly above the overall threshold of acceptable validity and responsiveness.52 | A systematic review study conducted on MCIDs for patients with cancer, SLE, and RA showed MCIDs for FACIT-F score improvement ranged between 2.8 to 6.8.23,53 No reported MID was found for patients with aHUS. |
Pediatric FACIT-F | Developed with some unique areas relevant to children, the tool has 11 items evaluated on a 5-point Likert scale (from 0 = none of the time, to 4 = all of the time) for patients aged 8 years to 18 years with a recall period of 7 days. The maximum score is 44 and higher scores representing better overall health status (i.e., less fatigue).54 | The instrument has good concurrent validity and internal consistency reliability.54 However, no values were found from aHUS pediatric patients. | A difference > 4.7 points was considered of clinical importance during the tool’s development in patients with cancer.23,54 No reported MID was found for patients with aHUS. |
3-Level EQ-5D | A generic preference-based self-reported HRQoL instrument that has been applied to a wide range of health conditions and treatments. The 3-Level EQ-5D assesses 5 domains: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each domain has 3 levels: no problems, some problems, and severe problems. The 3-Level EQ-5D index score is generated by applying a multiattribute utility function to the descriptive system. The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores of less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively.55,56 | The validation 3-Level EQ-5D is available across countries around the world and for various conditions.55,56 No validity, reliability, and responsiveness information was found for patients with aHUS. | No reported MID was found for patients with aHUS. |
aHUS = atypical hemolytic uremic syndrome; CI = confidence interval; EMPRO = Evaluating Measures of Patient-Reported Outcomes; EORTC QLQ-C30 = European organization for research and treatment of cancer quality of life questionnaire core 30; EQ VAS = EQ visual analogue scale; FACT-B = Functional Assessment of Cancer Therapy-Breast Cancer; FACT-E = Functional Assessment of Cancer Therapy Scale-Esophageal Cancer; FACIT-F = Functional Assessment of Chronic Illness Therapy Fatigue Scale; FACT-G = : Functional Assessment of Cancer Therapy–General; HRQoL = health-related quality of life; HUS = health utility scores; MCID = minimal clinically important difference; MID = minimal important difference; PS = performance status; RA = rheumatoid arthritis; SLE = systemic lupus erythematosus.
FACIT-F Scale
The FACIT-F is a patient self-completed questionnaire to assess fatigue.49 It is a subscale of the general questionnaire, the FACIT-General.57 It was developed to assess fatigue associated with anemia with item content established by combined expert and patient input.49 The FACIT-F is completed by patients (or interviewer when applicable) to assess fatigue.50,51,58,59 The current version (v.4) was used in the submitted pivotal study.
The instrument includes questions about the intensity of fatigue (and its impact on daily life) during usual daily activities over the past week. Patients are presented with a list of 13 statements that assess self-reported tiredness, weakness, and difficulty conducting usual activities due to fatigue, and asked to rate each on a 5-point Likert scale (0 = not at all, 1 = a little bit, 2 = somewhat, 3 = quite a bit, 4 = very much) to indicate how true the statement was during the past 7 days. Examples of statements are “I feel fatigued” and “I feel weak all over.” In the scoring, the numbers are reversed so that higher scores denote better quality of life (i.e., 4 = not at all, 3 = a little bit, 2 = somewhat, 1 = quite a bit, and 0 = very much). For statements 7 (“I have energy”) and 8 (“I am able to do my usual activities”), the scores are not reversed. The total score is a sum of the individual items and ranges between 0 and 52 with a lower score representing a higher level of fatigue. FACIT-F questionnaire has been translated into 48 languages permitting cross-cultural comparisons of fatigue in patients of diverse backgrounds.50,51,58,59
Validity and Reliability
The FACIT-F scale was originally designed to assess the fatigue among cancer patients, showing good internal consistency reliability and discriminant and convergent validity.49 The FACIT-F instrument has been evaluated in rheumatoid arthritis and psoriatic arthritis, primary Sjogren’s syndrome, osteoarthritis, inflammatory bowel disease, chronic immune thrombocytopenia, Parkinson disease, and systemic lupus erythematosus, as well as many other long-term conditions (e.g., multiple sclerosis, cancer, neurologic disorders).48,50,51,60-64
In a systematic evaluation of quality of life in patients with aHUS treated with eculizumab based on the Evaluating Measures of Patient-Reported Outcomes tool, the psychometric determinants properties of FACIT-F instrument were assessed and rated. This rating was done on the basis on 3 studies where this instrument had been used among patients with aHUS to determine their HRQoL.10,65,66 Scores generated by the tool were considered reasonably acceptable when they exceeded ≥ 50 points, and the maximum theoretical points were 100. Based on this scale, the reliability, validity, and responsiveness of FACIT-F instrument achieved 25.00, 54.17, and 55.56, respectively, showing a low score for reliability and slightly above the overall threshold of acceptable validity and responsiveness.52
Minimal Clinically Important Difference
A systematic review study conducted on MCIDs for patients with cancer, systemic lupus erythematosus, and rheumatoid arthritis showed MCIDs for FACIT-F score improvement ranged between 2.8 to 6.8.53 No reported MID was found for patients with aHUS.
FACIT-F Scale in Pediatrics
The 11-item pediatric FACIT-F scale was developed to measure fatigue among children with cancer through literature review, feedback from patients, parents, and clinicians, face-to-face consensus, and the use of Rasch Analysis.54 Some of the pediatric FACIT-F scale items are unique to children, whereas others share the same concepts captured in the parallel adult version. The tool has 11 items evaluated on a 5-point Likert-type scale (from 0 = none of the time, to 4 = all of the time) for patients aged 8 years to 18 years with a recall period of 7 days. The maximum score is 44 and higher scores representing better overall health status (less fatigue).54
Concurrent validity of the pediatric FACIT-F scale has been examined in 1 study in children with cancer54 using Spearman r between scores on the pediatric FACIT-F scale and multidimensional fatigue scale.67,68 Moreover, analysis of variance was used to determine whether the pediatric FACIT-F scale differentiated between patients with different functional performance levels, anemic/nonanemic status, and risk levels (i.e., high, average, low). Analysis of variance results demonstrated significantly more severe fatigue among anemic patients compared to nonanemic patients, with a mean difference of 4.66 points in raw score units (effect size [ES] = 0.57; F [1,153] = 15.44; P < 0.001). The concurrent validity was confirmed with Spearman r = 0.86, 0.71, and 0.57 for general fatigue, sleep, and cognition, respectively. Acceptable internal consistency reliability was found when all patients were analyzed as a whole (Cronbach alpha = 0.89), and also when patients were analyzed separately by age group (Cronbach alpha = 0.85 and 0.91 for children and adolescents, respectively).
Minimal Important Difference
The MID of the pediatric FACIT-F scale was calculated by using anemia and functional performance status as clinical anchors among children with cancer. For the calculation of the MIDs for the pediatric FACIT-F scale, this study used ES, defined as mean difference divided by SD for each clinical anchor were calculated. An ES greater than 0.5 was considered moderate to large, based on previous literature.69,70 In addition to the previously stated analysis of variance results based on anemia level, lower fatigue scores had been reported for higher functioning patients (i.e., either Karnofsky or Lansky performance rating = 90 or 100) than patients with performance measures lower than 90, with a mean difference of 4.74 points (ES = 0.58; F [1,153] = 14.33; P < 0.001). A difference greater than 4.7 points was considered of clinical importance, based on these observed mean differences and an corresponding ES.54 No reported MID was found for patients with aHUS.
3-Level EQ-5D
Description
The 3-Level EQ-5D is a generic, preference-based HRQoL instrument that has been applied to a wide range of health conditions and treatments.55,56 The questionnaire consists of descriptive questions and a VAS.55 The first of 2 parts of the 3-Level EQ-5D is a descriptive system that classifies respondents (aged 12 years and older) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 3 possible levels (1, 2, or 3) representing “no problems,” “some problems,” and “extreme problems,” respectively. Respondents are asked to choose a level that reflects their own health state for each of the 5 dimensions. The 5 questions are scored and together contribute to the EQ-5D index (utility) score between 0 and 1, where 0 represents death, and 1 represents perfect health. A scoring function can be used to assign a value (3-Level EQ- 5D index score) to self-reported health states from a set of population-based preference weights.55,56 Hence, the 3-Level EQ-5D produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121, 33211
a population preference-weighted health index score based on the descriptive system
a self-reported current health status based on the EQ VAS that is used to assess the overall health of the respondent rather than selected dimensions of individuals’ health.
The 3-Level EQ-5D index score is generated by applying a multiattribute utility function to the descriptive system. Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The US algorithm was used in the pivotal studies.
Psychometric Properties
A literature search was conducted to identify validation information of the 3-Level EQ-5D in patients with aHUS and none were identified.
Minimal Important Difference
A literature search was conducted to identify the MID of the 3-Level EQ-5D in patients with aHUS and none was identified.
Pharmacoeconomic Review
Abbreviations
aHUS
atypical hemolytic uremic syndrome
BIA
budget impact analysis
BSC
best supportive care
CMA
cost-minimization analysis
ITC
indirect treatment comparison
PE
plasma exchange
PI
plasma infusion
TMA
thrombotic microangiopathy
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ravulizumab (Ultomiris) for IV infusion |
Submitted price | Ravulizumab 10 mg/mL solution for IV infusion: $7,296.67 per 30 mL single-use vial |
Indication | For the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard |
NOC date | November 1, 2022 |
Reimbursement request | As per indication |
Sponsor | Alexion Pharma GmBH |
Submission history | Previously reviewed: Yes Indication: For the treatment of adult patients with PNH Recommendation date: February 11, 2022 Recommendation: Recommended with clinical criteria and/or conditions |
aHUS = atypical hemolytic uremic syndrome; NOC = Notice of Compliance; PNH = paroxysmal nocturnal hemoglobinuria; TMA = thrombotic microangiopathy.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target populations | Adult and pediatric patients with aHUS |
Treatment | Ravulizumab |
Comparator | Eculizumab |
Perspective | Canadian publicly funded health care payer |
Time horizon | Undefined (year 1 and subsequent years) |
Key data source | A sponsor-commissioned ITC to establish the equivalent efficacy and safety of ravulizumab compared to eculizumab based on studies ALXN1210-aHUS-311 and ALXN1210-aHUS-312 (ravulizumab) and studies aHUS-C08-002, aHUS-C10-003, and aHUS-C10-004 (eculizumab)1,2 |
Costs considered | Drug acquisition costs |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
aHUS = atypical hemolytic uremic syndrome; █████ █████ ███████ █████████ ██████ ███████; BSC = best supportive care; CMA = cost-minimization analysis; ITC = indirect treatment comparison.
Conclusions
Assuming equal clinical efficacy and safety for ravulizumab and eculizumab, the sponsor submitted a cost-minimization analysis comparing drug costs alone. The CADTH clinical review determined that due to several methodological limitations, no robust conclusion could be drawn regarding the comparative efficacy and safety of ravulizumab versus eculizumab. Although evidence from the indirect treatment comparison (ITC) suggests no significant differences in efficacy between ravulizumab and eculizumab, no data were available with respect to several outcomes included in the CADTH clinical review protocol; as a result, conclusions regarding the equivalence of ravulizumab and eculizumab for these outcomes could not be drawn. Additionally, the submitted ITC did not include safety outcomes. Therefore, it is not possible to draw conclusions regarding the relative safety of ravulizumab versus eculizumab.
In the CADTH reanalysis that removed free doses of complement inhibitors given in the acute hospital setting, ravulizumab was associated with cost savings of $106,752 in year 1 of treatment and $184,436 in subsequent years of treatment in the adult population. In the pediatric population, ravulizumab was associated with cost savings of $53,977 in year 1 of treatment and $90,876 in subsequent years of treatment. The estimated incremental savings are based on publicly available list prices for eculizumab and may not reflect actual prices paid by Canadian public drug plans.
Limitations related to uncertainty surrounding comparative efficacy could not be addressed by CADTH. If ravulizumab confers differential safety or improved quality of life (due to less frequent treatment administrations compared to eculizumab), then a cost-minimization analysis (CMA) is insufficient to assess the cost-effectiveness of ravulizumab (refer to CADTH Appraisal of the Sponsor’s Economic Information for details); therefore, the true cost-effectiveness of ravulizumab compared to eculizumab would be unknown. Of note, eculizumab received a “do not list” recommendation by the CADTH Canadian Drug Expert Committee (CDEC) for atypical hemolytic uremic syndrome (aHUS).3 Therefore, best supportive care (BSC) may be the more suitable comparator in most jurisdictions. Given that the sponsor did not submit a cost-utility analysis comparing eculizumab to BSC, the cost-effectiveness of eculizumab compared with BSC is unknown.3
Economic Review
The current review is for ravulizumab (Ultomiris) for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated thrombotic microangiopathy (TMA).
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA for ravulizumab compared with eculizumab for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA from the perspective of a Canadian public health system.4 Separate analyses were conducted for the first year of treatment and subsequent years of treatment, with no discounting applied.
The sponsor assumed that ravulizumab would be equivalent to eculizumab in terms of efficacy and safety. This assumption was based on a sponsor-commissioned ITC that included the C08-002, C08-003, C10-003, and C10-004 single-arm trials for eculizumab and the ALXN-aHUS-311 and ALX-aHUS-312 single-arm trials for ravulizumab.2 Among the clinical outcomes considered in the sponsor’s ITC, no differences between treatment groups reached statistical significance.2 Therefore, the sponsor assumed equivalence between ravulizumab and eculizumab.4
Only drug acquisition costs were included in the model; all other costs were assumed to be equal across treatments in the sponsor’s base-case analysis.4 Given the weight-based dosing regimen for ravulizumab in both adults and pediatric patients, and for eculizumab in the pediatric population, treatment costs were calculated based on body weight for adult and pediatric patients. Weighted average annual costs for adult and pediatric patients were derived separately by multiplying the annual costs for treatment with ravulizumab for each weight group by the weight distribution for adult and pediatric patients. The weight distributions for adults at baseline for ravulizumab dosing were based on the ALXN-1210-aHUS-311 adult study (Table 9).4,5 The weight distributions for pediatric patients at baseline for ravulizumab and eculizumab doses were based on the ALXN-1210-aHUS-312 study of ravulizumab, and C08-003 and C10-003 studies of eculizumab (Table 10).6-8 The sponsor assumed that the estimated weight distributions at baseline from the aforementioned trials were generalizable to the population of patients in Canada.4 Because the dosage for eculizumab in adults is based on a fix-dose regimen, adults receiving eculizumab were assumed to weigh more than 40 kg in the calculation of treatment costs (Table 8).4
The sponsor assumed that all patients with aHUS require hospitalization upon presentation and that doses given at the hospital would be covered by the sponsor.4 Given that the sponsor assumed that patients presenting with aHUS would be hospitalized for 3 weeks, it was assumed that 3 loading doses of eculizumab would be administered at the hospital and covered by the sponsor.4 Similarly, for ravulizumab, it was assumed that 1 loading dose followed by 1 maintenance dose at the hospital would be covered by the sponsor.4 No treatment administration costs were included in the base-case analysis because it was assumed that all administration costs for both ravulizumab and eculizumab would be covered by the sponsor.4
At the submitted price of $7,296.67 per 300 mg vial, the sponsor estimated that, in the adult population, ravulizumab was associated with total drug costs of $477,320 and $516,732 per patient in the first year and subsequent years, respectively. In comparison with eculizumab, ravulizumab was associated with drug-cost savings of $190,475 and $184,436 per patient in the first year and subsequent years, respectively (Table 3).
In the pediatric population, at an estimated cost of $331,488 per patient in year 1 and $358,748 per patient in subsequent years, ravulizumab was associated with cost savings of $95,074 and $90,876 versus eculizumab in year 1 and subsequent years, respectively (Table 4).
Table 3: Summary of the Sponsor’s Economic Evaluation Results in Adult Patients
Drug | Total drug costs ($) | Incremental drug costs ($) |
|---|---|---|
Eculizumab | Year 1: 667,794 Year 1+: 701,168 | Reference |
Ravulizumab | Year 1: 477,320 Year 1+: 516,783 | Year 1: –190,475 Year 1+: –184,436 |
Note: The negative incremental costs represent cost savings. Results were presented deterministically because only treatment acquisition costs were included in the base case.
Source: Sponsor’s economic submission.4
Table 4: Summary of the Sponsor’s Economic Evaluation Results in Pediatric Patients
Drug | Total drug costs ($) | Incremental drug costs ($) |
|---|---|---|
Eculizumab | Year 1: 426,562 Year 1+: 449,624 | Reference |
Ravulizumab | Year 1: 331,488 Year 1+: 358,748 | Year 1: –95,074 Year 1+: –90,876 |
Note: The negative incremental costs represent cost savings. Results were presented deterministically because only treatment acquisition costs were included in the base case.
Source: Sponsor’s economic submission.4
The sponsor conducted a number of sensitivity analyses, including varying the number of free ravulizumab and eculizumab doses administered in hospital, varying the adult and pediatric weight distributions, including administration costs, and including productivity costs. Ravulizumab remained cost-saving compared to eculizumab in all scenarios.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
The cost-effectiveness of ravulizumab compared with BSC is unknown: The sponsor’s CMA compares ravulizumab to eculizumab only. As per the CADTH Guidelines for Economic Evaluations, all interventions currently used and potentially displaced should be considered in the analysis.9 CDEC issued a “do not list” recommendation for eculizumab for aHUS,3 and there is currently variability across jurisdictions in the funding status of eculizumab. In some jurisdictions, BSC would remain the only treatment option available to patients with aHUS. This finding was corroborated by the clinical experts consulted by CADTH. The experts noted that BSC (most frequently consisting of plasma exchange [PE] or plasma infusion [PI]) is often the only relevant treatment option in clinical practice. In the CADTH review of eculizumab, the sponsor submitted a cost analysis comparing total treatment costs for eculizumab versus BSC.3 Because the sponsor did not submit a cost-utility analysis comparing eculizumab to BSC, the cost-effectiveness of eculizumab compared with BSC is also unknown.3 Given that the clinical efficacy of ravulizumab versus BSC could not be assessed in the CADTH clinical review (due to the lack of available data because the submitted ravulizumab trials were single-arm, noncomparative studies), the cost-effectiveness of ravulizumab relative to BSC is unknown.
CADTH could not address this limitation in its reanalysis.
The sponsor’s assumption of clinical equivalence between ravulizumab and eculizumab is uncertain: The sponsor submitted a CMA on the basis of its conclusion that ravulizumab and eculizumab are clinically equivalent. In the absence of a head-to-head comparison between ravulizumab and eculizumab, the sponsor submitted an ITC of ravulizumab and eculizumab in adult and pediatric patients with aHUS using data from 5 trials2 to indirectly compare the relative effects of ravulizumab to eculizumab. The sponsor concluded that there are no differences between the 2 treatments. The CADTH clinical review determined that no robust conclusion could be drawn regarding comparative efficacy and safety based on the sponsor’s submitted ITC. The CADTH clinical review noted several limitations with the submitted ITC related to temporal biases between the comparator trial population, potential unmeasured confounding characteristics, and the absence of several reporting characteristics. No data were presented for some outcomes included in the CADTH clinical review protocol (e.g., the presence of severe bleeding, plasma therapy-free status, packed red blood cell transfusions, and hospitalizations); conclusions regarding equivalence between ravulizumab and eculizumab for these outcomes could not be drawn. Finally, the submitted ITC did not include safety outcomes. As such, it is not possible to draw conclusions regarding the relative safety of ravulizumab versus eculizumab.
Additionally, the clinical experts consulted by CADTH stated that a large proportion of pediatric patients receiving eculizumab require the use of venous access ports due to the frequency of treatment administrations and that less frequent infusions with ravulizumab may decrease the need for venous access ports. Because venous access ports can be associated with health complications and may influence patients’ quality of life, a CMA may be inappropriate.10 Additionally, both the clinical experts and patients noted in their input that less frequent infusions associated with ravulizumab would have important quality of life benefits for patients. Should there be differences in safety, disease management (e.g., treatment administration and venous port access) or quality of life between ravulizumab and eculizumab, the cost-effectiveness of ravulizumab compared to eculizumab would be unknown. Note that any additional quality of life benefits associated with not needing venous access ports or reduced frequency of treatment administration would favour ravulizumab.
CADTH could not address this limitation in its reanalysis due to the nature of the sponsor’s submission.
There is uncertainty surrounding the sponsor-covered doses of complement inhibitors administered in the hospital: The sponsor assumed that all patients with aHUS require hospitalization upon their initial disease presentation and that doses of complement inhibitors administered at the hospital would be covered by the sponsor, ██ ███ ███ ████ ███████ █████████ ██████ ███████ ████████4 █████ ██ ████ ███████, the sponsor assumes that all patients presenting with aHUS are hospitalized for 3 weeks and that, as a result, the costs of 3 doses of eculizumab and 1 loading dose plus 1 maintenance dose of ravulizumab would be covered by the sponsor. CADTH requested additional information ██ ███ █████████ ███████ ███ ███ ██████ █████████████ ████████ ██ █████ ███ ████████ ██ ███████████11 There was no information indicating that ███ █████ ███████ would be implemented for ravulizumab and ███ ████████ ████ ████ ███████ █████ ███████ ██ ████████ ████████████ ███████ ██████████ ████ ██ ██ ███ ███ ████████ ██ ██ ████████ ██ ███████ ████ █████ ███████ ███ █████11 ██████████ ████ ████ ██ ███████ ██ ███ ███████ ███ ███ █████████ ██ ████████ ████ ██████████ ████████ █████████ ██ █████ █████ ███ ████ ████████ ███ ██ ███ ███████ ██████████ ████ ██████████ ████ █████████11 If the hospital is liable for the costs of eculizumab or ravulizumab in the event of misdiagnosis, then this could represent notable costs to the Canadian health care system. ████████████ ███████ ███████ █████ ██ ██████████ ███ ███████████ ██ ███ ██████████ ██ ███████ ████ ██ ███ ████████11 According to clinical expert feedback received by CADTH, some patients may ███████ ██████████ ██ █████ ██████████ ███████, meaning free doses may not be realized for all aHUS patients. ███ ███████ ███ ██████ ████ ██ ███████ ██ ███ █████ ██ ██████████ ████ ███████ ██ ███ ███████ ███ ████████ ███████ ██████ ███ ██ ████████ ████████ ███ ██████ ████████ ██ ██ █████ █████ ███ ██ ██████████11 Because the base case assumed that each patient would receive 3 free doses of eculizumab and 2 free doses of ravulizumab, the number of free doses of complement inhibitor may be overestimated; consequently, so too would the estimated cost savings with ravulizumab.
Given the uncertainty associated with free doses of complement inhibitor, in its reanalysis, CADTH removed the assumption that the sponsor would cover the costs of all complement inhibitor doses given in hospitals.
In addition, CADTH conducted 2 scenario analyses to explore how sensitive the cost results would be to this assumption. These included: assuming a 2-week hospitalization period with free doses (1 dose of ravulizumab, 2 doses of eculizumab); and assuming that the sponsor provides free doses for eculizumab only, based on ██████████████████ ██████ ██████.
Analysis based on publicly available list prices: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for eculizumab. CADTH previously recommended that eculizumab not be reimbursed for aHUS because a clinical benefit could not be established.3 Despite this, eculizumab is listed for aHUS as a restricted benefit or exception on a case-by-case basis in some jurisdictions. In these jurisdictions, the confidentially negotiated price for eculizumab is unknown.
Because the confidentially negotiated price of eculizumab is unknown, CADTH conducted threshold analyses to investigate the price of eculizumab that would be required for ravulizumab to be cost-neutral (i.e., no longer cost-saving).
CADTH Reanalyses of the Economic Information
The CADTH reanalysis included 1 change to the sponsor’s base case: it removed the free complement inhibitor doses administered in hospital, rather than including the sponsor’s assumption that hospitals would cover the costs of 3 doses of eculizumab and 2 doses of ravulizumab per patient in year 1 of treatment. In the adult population, ravulizumab was associated with cost savings of $106,752 in year 1 of treatment and $184,436 in subsequent years (Table 5). In the pediatric population, ravulizumab was associated with cost savings of $53,977 in year 1 of treatment and $90,876 in subsequent years (Table 6).
Table 5: Summary of the CADTH Reanalysis Results in the Adult Population
Drug | Total drug costs ($) | Incremental drug costs ($) |
|---|---|---|
Eculizumab | Year 1: 728,472 Year 1+: 701,168 | Reference |
Ravulizumab | Year 1: 621,721 Year 1+: 516,732 | Year 1: –106,752 Year 1+: –184,436 |
Note: Reanalyses are based on publicly available prices of the comparator treatments. All analyses are deterministic. The negative incremental costs represent cost savings.
Table 6: Summary of the CADTH Reanalysis Results in the Pediatric Population
Drug | Total drug costs ($) | Incremental drug costs ($) |
|---|---|---|
Eculizumab | Year 1: 471,201 Year 1+: 449,624 | Reference |
Ravulizumab | Year 1: 417,224 Year 1+: 358,748 | Year 1: –53,977 Year 1+: –90,876 |
Note: Reanalyses are based on publicly available prices of the comparator treatments. All analyses are deterministic. The negative incremental costs represent cost savings.
CADTH conducted 3 additional scenario analyses, varying the number of free complement inhibitor doses administered in hospital and assessing the impact of including treatment administration costs. Additional details are available in Table 11 and Table 12 in Appendix 1.
When CADTH assumed a 2-week hospital stay, 1 dose of ravulizumab and 2 doses of eculizumab were assumed to be covered by the sponsor, resulting in cost savings of $171,655 and $74,568 for adult and pediatric patients, respectively, in the first year of treatment. When CADTH assumed that doses of eculizumab only (a total of 3) were covered by the sponsor during a 3-week hospital stay, the cost savings were $46,074 and $9,338 for adult and pediatric patients, respectively, in the first year of treatment. Cost savings in subsequent years were not affected when assessing the impact of initial free doses of complement inhibitors.
Lastly, the inclusion of administration costs resulted in increased cost savings compared to the CADTH base case because ravulizumab is administered less frequently than eculizumab. In the administration costs scenario, cost savings of $114,340 and $62,243 were observed for adult and pediatric patients, respectively, in the first year of treatment. Cost savings in subsequent years were estimated to be $191,956 and $98,997 for adult and pediatric patients, respectively. Note that both the sponsor’s and the CADTH treatment administration scenario analyses incorporate double counting due to the sponsor adding personnel time to the administration cost when this was already captured in the sponsor’s overhead cost estimates.12
Because confidential prices for eculizumab are unknown, CADTH conducted a threshold analysis using the CADTH base case to examine the price for eculizumab at which ravulizumab would no longer be considered cost-saving. In the adult population, a price reduction of 15% for eculizumab is required for ravulizumab to be cost-neutral in the first year of treatment (Table 7). The estimated price reduction required for eculizumab to achieve cost neutrality in subsequent years of treatment in adult patients is 26%. In the pediatric population, a price reduction of 11% for eculizumab is required for ravulizumab to be cost-neutral in the first year of treatment (Table 7). The estimated price reduction required for eculizumab to achieve cost neutrality in subsequent years of treatment in pediatric patients is 20%.
Table 7: CADTH Threshold Analyses of the Price of Eculizumab
Scenario | Eculizumab list price ($) | Eculizumab price reduction needed (%) | Reduced price of eculizumab ($) | Cost savings of ravulizumab relative to reduced price of eculizumaba ($) |
|---|---|---|---|---|
Price reduction required for eculizumab to result in no cost savings for ravulizumab in adult patients (year 1) | 6,742.00 | 15 | 5,753.56 | 0 |
Price reduction required for eculizumab to result in no cost savings for ravulizumab in adult patients (year 1+) | 6,742.00 | 26 | 4,968.58 | 0 |
Price reduction required for eculizumab to result in no cost savings for ravulizumab in pediatric patients (year 1) | 6,742.00 | 11 | 5,969.14 | 0 |
Price reduction required for eculizumab to result in no cost savings for ravulizumab in pediatric patients (year 1+) | 6,742.00 | 20 | 5,379.34 | 0 |
aSavings from the sponsor-submitted price per patient per year.
bRelative to publicly available list prices of eculizumab.
Issues for Consideration
Anticipated patent expiration of eculizumab: The patent for eculizumab is expected to expire on March 15, 2027.13 If eculizumab biosimilars become available and are considered clinically equivalent to eculizumab, ravulizumab is unlikely to remain less costly than these biosimilars. Consequently, the cost of ravulizumab at the submitted price would be less attractive to drug plans.
Comparator pricing based on publicly available prices: The modelled price of eculizumab is based on publicly accessible list prices and does not reflect any confidential pricing that may have been negotiated by public plans. The estimated cost savings associated with ravulizumab are likely less than estimated if confidential discounts have been negotiated for eculizumab.
Off-label use of complement inhibitors: Clinician input indicated that complement inhibitors may be used to treat patients with autoimmune disease where there is histological evidence of TMA due to secondary causes (e.g., malignant hypertension) in addition to evidence of complement dysregulation (e.g., select variants of lupus). The impact of off-label use on the cost-effectiveness of ravulizumab is unknown.
Uncertainty surrounding the diagnosis of aHUS patients: The CADTH clinical review noted that a serious challenge in treating aHUS patients is the difficulty of diagnosing them accurately. This is because diagnosis requires the presence of active TMA, which requires prompt treatment with PI, and also because there is no single diagnostic test that can confirm aHUS (instead, it is a diagnosis of exclusion for most patients). Therefore, overdiagnosis of the condition was indicated by clinicians to be a risk. The potential for unnecessary complement inhibitor treatment in patients with misdiagnoses of aHUS could result in potential negative impacts in the form of unnecessary costs.
Free doses in the acute hospital setting could potentially increase patient length of stay: Providing free doses of complement inhibitor only in hospitals could increase health care system costs by leading to longer hospital stays, if patients are hospitalized for longer than clinically required to receive these free doses. Hospitalization costs were not considered in the sponsor’s analysis.
Previous submission history of ravulizumab: Ravulizumab was reviewed by CADTH for paroxysmal nocturnal hemoglobinuria and received a recommendation to reimburse with clinical criteria and/or conditions.14 The recommendation concluded that ravulizumab is potentially less costly than eculizumab. However, there was notable uncertainty associated with this conclusion; due to loading-dose costs, any potential cost savings would be realized only after several decades of treatment. The submitted price for ravulizumab was the same across indications.
Supplemental dosing following PE or PI: The product monographs for eculizumab and ravulizumab indicate that supplemental dosing of complement inhibitor is required in the PE and/or PI setting to maintain the serum concentration of complement inhibitor.15,16 As per the product monograph, the supplemental dose of ravulizumab is approximately half of the “most recent ravulizumab dose,” whereas according to the product monograph for eculizumab, supplemental doses are equivalent to the most recent dose, with the exception of doses of 600 mg or more. Doses following PE and/or PI are capped at 600 mg.15,16
Conclusions
Assuming equivalent clinical efficacy and safety of ravulizumab and eculizumab, the sponsor submitted a CMA. The CADTH clinical review determined that, due to several methodological limitations, no robust conclusion could be drawn regarding the comparative efficacy and safety of ravulizumab versus eculizumab. Although evidence from the ITC suggests no significant differences in efficacy between ravulizumab and eculizumab, no data were available for several outcomes included in the CADTH clinical review protocol, and conclusions regarding the equivalence of ravulizumab and eculizumab for these outcomes could not be drawn. Furthermore, because adverse events were not included in the sponsor’s ITC, the impact of differences in safety events is unknown and remains a substantial gap in evidence when assessing the relative efficacy and safety of ravulizumab versus eculizumab.
In the CADTH reanalysis that removed free doses of complement inhibitors given in the acute hospital setting, ravulizumab was associated with cost savings of $106,752 in year 1 of treatment and $184,436 in subsequent years of treatment in the adult population. In the pediatric population, ravulizumab was associated with cost savings of $53,977 in year 1 of treatment and $90,876 in subsequent years of treatment. In scenario analyses assessing the implementation of free doses of complement inhibitors or the inclusion of administration costs, ravulizumab remained cost-saving. The estimated incremental savings are based on publicly available list prices for eculizumab and may not reflect actual prices paid by Canadian public drug plans. Because the confidentially negotiated price of eculizumab is unknown, CADTH conducted threshold analyses to determine the price of eculizumab at which ravulizumab would no longer be considered cost-saving. If the confidentially negotiated price of eculizumab is 15% less than its list price, ravulizumab would be cost-neutral in the first year of treatment in the adult population. In subsequent years of treatment, a price reduction of 26% for eculizumab would be required to achieve cost neutrality in adult patients. In the pediatric population, 11% and 20% reductions in the list price of eculizumab would result in ravulizumab being cost-neutral in the first and subsequent years of treatment, respectively. Notably, the patent for eculizumab is anticipated to expire on March 15, 2027.13 If eculizumab biosimilars become available, ravulizumab is unlikely to remain less costly than eculizumab biosimilars.
Numerous outstanding sources of uncertainty remain. Of note, eculizumab received a “do not list” recommendation by CDEC for aHUS.3 Therefore, BSC was noted to be a relevant comparator in this population, given that eculizumab may not be listed on all public formularies for the aHUS indication. Because the sponsor did not submit a cost-utility analysis comparing eculizumab to BSC, the cost-effectiveness of eculizumab compared with BSC is unknown.3 Limitations related to uncertainty surrounding comparative efficacy could not be addressed by CADTH. If ravulizumab confers differential safety or improved quality of life due to less frequent treatment administrations compared to eculizumab, then a CMA would be insufficient to assess the cost-effectiveness of ravulizumab; therefore, the true cost-effectiveness of ravulizumab compared to eculizumab would be unknown. A further source of uncertainty includes the duration for which the sponsor would cover in-hospital treatment administration costs. This duration would influence the magnitude of the cost savings likely to occur with the introduction of ravulizumab.
References
1.Tomazos I, Hatswell AJ, Cataland S, et al. Comparative efficacy of ravulizumab and eculizumab in the treatment of atypical hemolytic uremic syndrome: An indirect comparison using clinical trial data. Clinical nephrology. 2022;97(5):261-272. PubMed
2.Indirect comparison of eculizumab and ravulizumab using propensity scoring [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughan (ON): Alexion Pharma Canada Corp.; 2022 Jun 23.
3.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: eculizumab (Soliris). Ottawa (ON): CADTH; 2013: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_Soliris-aHUS_July-23-13.pdf. Accessed August 29, 2022.
4.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughan (ON): Alexion Pharma Canada Corp.; 2022 Jun 23.
5.Clinical Study Report: ALXN1210-aHUS-311. Single arm study of ALXN1210 in complement inhibitor treatment-naive adult and adolescent patients with atypical hemolytic uremic syndrome (aHUS) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals, Inc.; 2020 Feb 12.
6.Clinical Study Report: ALXNI210-aHUS-312. A phase 3, open-label, multicenter study of ALXN1210 in children and adolescents with atypical hemolytic uremic syndrome (aHUS) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals, Inc.; 2020 Mar 06.
7.Alexion Pharmaceuticals Inc. Data on file. Study C10-003 Clinical Study Report.
8.Alexion Pharmaceuticals Inc. Data on file. Study C08-003 Clinical Study Report.
9.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 1800 Jan 1.
10.Machat S, Eisenhuber E, Pfarl G, et al. Complications of central venous port systems: a pictorial review. Insights Imaging. 2019;10(1):86. PubMed
11.Alexion Pharma GmBH response to July 21, 2022 DRR request for additional information regarding ravulizmuab DRR review [internal additional sponsor's information]. Vaughan (ON): Alexion Pharma GmBH; 2022. Accessed July 26, 2022.
12.Shinder GA, Paradis PE, Posman M, et al. Patient and work flow and costs associated with staff time and facility usage at a comprehensive cancer centre in Quebec, Canada--a time and motion study. BMC Health Serv Res. 2012;12:370. PubMed
13.Health Canada Patent Register. Government of Canada; 2022. https://pr-rdb.hc-sc.gc.ca/pr-rdb/start-debuter.do?access=external&lang=en. Accessed August 23, 2022.
14.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: ravulizumab (Ultomiris). Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0700%20Ultomiris%20-%20Final%20CADTH%20Rec.pdf. Accessed August 23, 2022.
15.Ultomiris (ravulizumab): 10 mg/mL concentrate for solution for infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2021 Nov 29.
16.Soliris (eculizumab): 10 mg/mL concentrate for solution for infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH; August 20, 2018.
17.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020: https://open.alberta.ca/dataset/30add047-29c2-4fc7-83b5-a8ab78605cdd/resource/48d0dd0b-bb33-478d-a85c-85a86d4555ad/download/health-somb-medical-price-list-2020-03.pdf. Accessed 2021 Feb 23.
18.Lapeyraque AL, Bitzan M, Al-Dakkak I, et al. Clinical Characteristics and Outcome of Canadian Patients Diagnosed With Atypical Hemolytic Uremic Syndrome. Can J Kidney Health Dis. 2020;7:2054358119897229. PubMed
19.Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of Atypical Hemolytic Uremic Syndrome: A Systematic Literature Review. Clin Epidemiol. 2020;12:295-305. PubMed
20.Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20(1):125. PubMed
21.Bayer G, von Tokarski F, Thoreau B, et al. Etiology and Outcomes of Thrombotic Microangiopathies. Clin J Am Soc Nephrol. 2019;14(4):557-566. PubMed
22.Wuhl E, van Stralen KJ, Wanner C, et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA Registry. Nephrol Dial Transplant. 2014;29 Suppl 4:iv1-8. PubMed
Appendix 1: Additional Economic Information
Table 8: CADTH Cost Comparison Table for aHUS
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Ravulizumab (Ultomiris) | 10 mg/mL | 30 mL single dose vial of concentrate for solution for IV infusion | $7,296.6700 | Adult population: Loading dose, with maintenance doses given starting 2 weeks after, then administered every 8 weeks thereafter, based on weight as follows:a ≥ 40 kg to < 60 kg Loading: 2,400 mg Maintenance: 3,000 mg ≥ 60 kg to < 100 kg Loading: 2,700 mg Maintenance: 3,300 mg ≥ 100 kg Loading: 3,000 mg Maintenance: 3,600 mg | ≥ 40 kg to < 60 kg Year 1b: 1,559.29 Subsequent yearsc: 1,299.41 ≥ 60 kg to < 100 kg: Year 1b: 1,719.22 Subsequent yearsc: 1,429.35 ≥ 100 kg: Year 1b: 1,879.14 Subsequent yearsc: 1,559.29 | ≥ 40 kg to < 60 kg: Year 1b: 569,140 Subsequent yearsc: 474,284 ≥ 60 kg to < 100 kg: Year 1b: 627,514 Subsequent yearsc: 521,712 ≥ 100 kg: Year 1b: 685,887 Subsequent yearsc: 569,140 |
Pediatric populationa: ≥ 5 kg to < 10 kg Loading: 600 mg Maintenance: 300 mg 2 weeks after the loading dose, then administered every 4 weeks thereafter ≥ 10 kg to < 20 kg Loading: 600 mg Maintenance: 600 mg 2 weeks after the loading dose, then administered every 4 weeks thereafter ≥ 20 kg to < 30 kg Loading: 900 mg Maintenance: 2,100 mg 2 weeks after the loading dose, then administered every 8 weeks thereafter ≥ 30 kg to < 40 kg Loading: 1,200 mg Maintenance: 2,700 mg 2 weeks after the loading dose, then administered every 8 weeks thereafter | ≥ 5 kg to < 10 kg Year 1b: 319.85 Subsequent yearsc: 259.88 ≥ 10 kg to < 20 kg Year 1b: 599.73 Subsequent yearsc: 519.76 ≥ 20 kg to < 30 kg Year 1b: 1,039.53 Subsequent yearsc: 909.58 ≥ 30 kg to < 40 kg Year 1b: 1,339.39 Subsequent yearsc: 1,169.47 | ≥ 5 kg to < 10 kg Year 1b: 116,747 Subsequent yearsc: 94,857 ≥ 10 kg to < 20 kg Year 1b: 218,900 Subsequent yearsc: 189,713 ≥ 20 kg to < 30 kg Year 1b: 379,427 Subsequent yearsc: 331,998 ≥ 30 kg to < 40 kg Year 1b: 488,877 Subsequent yearsc: 426,855 | ||||
Complement inhibitor | ||||||
Eculizumab (Soliris) | 10 mg/mL | 300 mg single-use vial | 6,742.0000d | Adult population: Loading: 900 mg every 7 days for the first 4 weeks, then 1,200 mg for the fifth dose 1 week later Maintenance: 1,200 mg every 2 weeks thereafter | Year 1e: 1,994.89 Subsequent yearsf: 1,921.01 | Year 1e: 728,136 Subsequent yearsf: 701,168 |
Pediatric population: ≥ 5 kg to < 10 kg Loading: 300 mg for the first week, then 300 mg for the second dose 1 week later Maintenance: 300 mg every 3 weeks thereafter ≥ 10 kg to < 20 kg Loading: 600 mg for the first week, then 300 mg for the second dose 1 week later Maintenance: 300 mg every 2 weeks thereafter ≥ 20 kg to < 30 kg Loading: 600 mg every 7 days for the first 3 weeks Maintenance: 600 mg every 2 weeks thereafter ≥ 30 kg to < 40 kg Loading: 600 mg every 7 days for the first 2 weeks, then 900 mg for the third dose 1 week later Maintenance: 900 mg every 2 weeks thereafter | ≥ 5 kg to < 10 kg Year 1: 332.48g Subsequent years: 320.17h ≥ 10 kg to < 20 kg Year 1i: 517.19 Subsequent yearsf: 480.25 ≥ 20 kg to < 30 kg Year 1j: 997.45 Subsequent yearsf: 960.50 ≥ 30 kg to < 40 kg Year 1k: 1,459.23 Subsequent yearsf: 1,440.76 | ≥ 5 kg to < 10 kg Year 1: 121,356g Subsequent years: 116,861h ≥ 10 kg to < 20 kg Year 1i: 188,776 Subsequent yearsf: 175,292 ≥ 20 kg to < 30 kg Year 1j: 364,068 Subsequent yearsf: 350,584 ≥ 30 kg to < 40 kg Year 1k: 532,618 Subsequent yearsf: 525,876 | ||||
Note: All prices are obtained from the sponsor’s submission unless otherwise indicated, and do not include dispensing fees.4
aFor patients switching from eculizumab, the loading dose of ravulizumab is given 2 weeks after the last eculizumab infusion. Maintenance doses are then given every 4 or 8 weeks, starting 2 weeks after the loading dose, based on patient weight.
bYear 1 costs assume 1 loading dose and 7 maintenance doses. For the pediatric weight groups ≥ 5 kg to < 10 kg and ≥ 10 kg to < 20 kg, 14 maintenance doses are required.
cSubsequent year dosing are based on an average of 6.5 administrations (52/8) per year. For the pediatric weight groups ≥ 5 kg to < 10 kg and ≥ 10 kg to < 20 kg, 13 administrations are assumed.
dAlberta drug formulary (accessed July 2021).17
eYear 1 costs assume four 900 mg doses and 24 1,200 mg doses.
fSubsequent year costs assume 26 administrations per year.
gYear 1 costs assume 18 300 mg doses.
hSubsequent year costs assume 17.3 administrations per year.
iYear 1 costs assume one 600 mg dose and 26 300 mg doses.
jYear 1 costs assume 27 600 mg doses.
kYear 1 costs assume two 600 mg doses and 25 900 mg doses.
Note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Additional Details on the Sponsor’s Submission
Table 9: Weight Distributions at Baseline for Ravulizumab Dosing for the Adult Population
Weight range | Proportion of adults at baseline (%) |
|---|---|
≥ 10 to < 20 kg | 0.0 |
≥ 20 to < 30 kg | 0.0 |
≥ 30 to < 40 kg | 0.0 |
≥ 40 to < 60 kg | 19.3 |
≥ 60 to < 100 kg | 71.9 |
≥ 100 kg | 8.8 |
Source: Alexion Pharmaceuticals Inc. ALXN-1210-aHUS-311 Clinical Study Report.5
Table 10: Weight Distributions at Baseline for Ravulizumab and Eculizumab Dosing for the Pediatric Population
Weight range | Proportion of children at baseline (%) | |
|---|---|---|
Ravulizumab | Eculizumab | |
≥ 10 to < 20 kg | 32.1 | 32.1 |
≥ 20 to < 30 kg | 17.0 | 17.0 |
≥ 30 to < 40 kg | 13.2 | 13.2 |
≥ 40 to < 60 kg | 24.5 | 37.7a |
≥ 60 to < 100 kg | 13.2 | |
≥ 100 kg | 0.0 | |
aNote that for eculizumab, dosing for pediatric patients is the same for all patients ≥ 40 kg.
Source: Ravulizumab (ALXN210-aHUS-312) and eculizumab trials.6-8
Additional Details on the CADTH Reanalyses and Additional Analyses
Scenario Analyses
Table 11: Scenario Analyses in the Adult Population
Scenario analysis | Drug | Total costs ($) | Incremental costs ($) |
|---|---|---|---|
Free doses: 2-week hospitalization | Eculizumab | Year 1: 728,472 Year 1+: 701,168 | Reference |
Ravulizumab | Year 1: 556,817 Year 1+: 516,732 | Year 1: –171,655 Year 1+: –184,436 | |
Free doses: 3-week hospitalization eculizumab only | Eculizumab | Year 1: 667,794 Year 1+: 701,168 | Reference |
Ravulizumab | Year 1: 621,721 Year 1+: 516,732 | Year 1: –46,074 Year 1+: –184,436 | |
Administration costs | Eculizumab | Year 1: 740,274 Year 1+: 712,127 | Reference |
Ravulizumab | Year 1: 625,935 Year 1+: 520,171 | Year 1: –114,340 Year 1+: –191,956 |
Note: The negative incremental costs represent cost savings.
Table 12: Scenario Analyses in the Pediatric Population
Scenario analysis | Drug | Total costs ($) | Incremental costs ($) |
|---|---|---|---|
Free doses: 2-week hospitalization | Eculizumab | Year 1: 456,564 Year 1+: 449,624 | Reference |
Ravulizumab | Year 1: 381,996 Year 1+: 358,748 | Year 1: −74,568 Year 1+: −90,876 | |
Free doses: 3-week hospitalization eculizumab only | Eculizumab | Year 1: 426,562 Year 1+: 449,624 | Reference |
Ravulizumab | Year 1: 417,224 Year 1+: 358,748 | Year 1: −9,338 Year 1+: −90,876 | |
Administration costs | Eculizumab | Year 1: 484,596 Year 1+: 462,202 | Reference |
Ravulizumab | Year 1: 422,353 Year 1+: 363,205 | Year 1: −62,243 Year 1+: −98,997 |
Note: The negative incremental costs represent cost savings.
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing ravulizumab for the treatment of adult and pediatric patients with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy (TMA). The analysis took the perspective of CADTH-participating Canadian public drug plans using a top-down epidemiological approach and incorporated drug acquisition costs using a weighted annual cost based on pediatric and adult weight ranges. A time horizon of 3 years was taken. The target population size was estimated using prevalence of aHUS (2.7 patients per million),18 estimated incidence of aHUS (2.0 per million),19 proportion of those who are pediatric (40%) versus adult patients (60%),18 and public coverage (100%). Further specification related to discontinuation of complement inhibitor treatment,20 switching to ravulizumab from eculizumab, proportion of naive patients initiating with ravulizumab, and relapse of discontinued patients20 were included to determine the number of patients receiving eculizumab versus ravulizumab. The base-case analysis considers eculizumab in the reference scenario and the new drug scenario considered the reimbursement of ravulizumab. Key inputs to the BIA are documented in Table 14.
The sponsor’s submission included the following key assumptions:
The cost of a meningococcal vaccine was added to the cost of first year of treatment.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Prevalence rate for current aHUS patients18 | 2.7 per million |
Expected annual rate of incident aHUS patients19 | 2.0 per million |
Proportion of adult patients18 | 60% |
Proportion of pediatric patients18 | 40% |
Proportion of patients eligible for public coveragea | 100% |
Number of patients eligible for drug under review Pediatric18 Adult18 | 63 / 63 / 64 25 / 25 / 26 38 / 38 / 38 |
Expected annual rate of complement inhibitor discontinuation20,b | 45% |
Expected share of naive patients initiating with ravulizumaba,b | 80% |
Expected share of discontinued patients relapsing20,b | 50% |
Expected share of relapsing patients starting ravulizumaba,b | 100% |
Market uptake (3 years)a | |
Uptake (reference scenario) Eculizumab | 100% / 100% / 100% |
Uptake (new drug scenario) Ravulizumab Eculizumab | 80% / 85% / 90% 20% / 15% / 10% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year in adultsc Ravulizumab Eculizumab | $621,721d ($516,732) $728,136d ($701,504) |
Cost of treatment over 1 year in pediatricsc Ravulizumab Eculizumab | $417,224d ($358,748) $471,201d ($449,960) |
aHUS = atypical hemolytic uremic syndrome.
aBased on the sponsor’s assumption.
bThe following inputs were applied to the patient numbers calculated in the row titled “Number of patients eligible for drug under review.”
cCost of treatment for both adult and pediatric patients was presented as an annual cost weighted average based on the distribution of adult and pediatric weights observed in Table 9 and Table 10 in Appendix 1. Annual costs of maintenance treatment were presented in brackets.
dNote: Costs of treatment in year 1 include the $366 cost of a meningococcal vaccine, applied to patients receiving eculizumab or ravulizumab.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding ravulizumab for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA resulted in cost savings to the drug plans of $8,766,214 in year 1, $16,457,859 in year 2, and $19,595,551 in year 3, for a 3-year total of $44,819,623.
In the pediatric population, the total cost savings to drug plans across 3 years was estimated to be $11,044,426 and the cost savings across 3 years for the adult population was estimated to be $33,775,197.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the estimated target population: The sponsor estimated the incidence of aHUS from a systematic literature review that reported a range of incidence estimates from various countries.19 The sponsor used the upper-limit incidence estimate of 2.0 per million per year, which was derived from a study conducted in France encompassing patients of all ages.21 CADTH notes that incidence data specific to Canada was unavailable and it is uncertain whether the estimates sourced from literature are reflective of the patient population in Canada. Given the uncertainty in the true estimate of incidence, CADTH assessed the impact of decreasing the incidence rate to 0.39 per million per year, using a multinational study from the systematic review cited by the sponsor that pooled data from 815 patients of all ages from 11 countries.22 An overestimated target population would likely result in an overestimation of cost savings, given that drug costs of ravulizumab are estimated to be less costly than eculizumab at publicly available prices.
Expected share of treatment-naive patients initiating treatment with ravulizumab was likely underestimated: The sponsor estimated that 80% of treatment-naive patients would initiate treatment with ravulizumab. However, the clinical experts consulted by CADTH noted, should ravulizumab be reimbursed for aHUS, all treatment-naive patients would likely initiate with ravulizumab, unless differences in time to TMA response or remission were found in the CADTH clinical review. For example, if there are differences in induction time between ravulizumab and eculizumab, it could be feasible to assume that the remaining 20% of treatment-naive patients may opt to receive eculizumab to achieve treatment response more quickly. Given that the ITC did not find differences between ravulizumab and eculizumab with respect to time to TMA response, it is likely that all patients will initiate ravulizumab and therefore that this input was underestimated by the sponsor.
CADTH adjusted the share of treatment-naive patients initiating with ravulizumab to reach 100% in reanalysis.
Discontinuation rate of complement inhibitors and relapse rate of aHUS patients was likely overestimated: The sponsor derived annual rates of discontinuation and relapse from the eculizumab extension trial.20 Based on this trial, 45% of patients (42 of 93) had at least 1 off-treatment period during the 64.2 months of follow-up time.20 However, the sponsor assumes that this proportion can be used as an annual rate while failing to account for follow-up time. Additionally, reasons for discontinuation could not be discerned by CADTH, as this information was not available from the trial publication and was defined as at the discretion of physicians and patients. Similarly, the relapse rate of discontinued patients was estimated to be 50% (21 of 42 patients) based on the Soliris extension trial, across 65.4 months of follow-up time.20 This estimate is also applied as an annual rate of relapse without taking into account follow-up time.
Clinical experts consulted by CADTH noted that these estimates were uncertain. For example, patients without identifiable mutations may plan to discontinue treatment after 3 to 6 months, whereas patients with high-risk mutations may continue to be treated for a lifetime. Regarding relapse, the clinical experts consulted by CADTH also highlighted uncertainty surrounding the sponsor’s inputs. They noted that a 50% relapse rate would likely be applicable for high-risk patients, however the population being studied is highly heterogeneous. A proportion of patients may not have required complement inhibitor treatment and would discontinue; and conversely, if severe cases with identifiable causes or evidence of relapse were selected for, relapse rate would be higher. The expected relapse rate and discontinuation rates would be highly variable based on patient selection.
Overall, decreasing the annual discontinuation rate may overestimate cost savings, as a higher proportion of patients would remain treated with ravulizumab. An overestimation of relapse rate would likely result in overestimated cost savings, as the sponsor also assumed that all patients who relapse will receive treatment with ravulizumab. The true estimate of annual discontinuation and relapse rate remain associated with uncertainty.
Given the uncertainty surrounding these inputs, CADTH tested the impact of halving the sponsor’s assumed annual rate of discontinuation and relapse in scenario analyses.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown. Confidential negotiated prices for eculizumab may lead to budgetary savings being limited or eliminated.
This limitation could not be addressed by CADTH in reanalysis.
CADTH Reanalyses of the BIA
Table 15: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Proportion of treatment-naive patients initiating with ravulizumab | 80% | 100% |
CADTH base case | Reanalysis 1 | |
BIA = budget impact analysis.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses, and so on) that are not identified as limitations.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. The CADTH estimated budget impact of funding ravulizumab for the treatment of adult and pediatric patients with aHUS to inhibit complement-mediated TMA resulted in cost savings to the drug plans of $9,837,687 in year 1, $18,220,135 in year 2, and $21,453,528 in year 3, for a 3-year total of $49,511,350. In the pediatric population, the total cost savings to drug plans across 3 years was estimated to be $12,243,993 and the cost savings across 3 years for the adult population was estimated to be $37,267,357.
CADTH also conducted the following scenario analyses:
The annual incidence rate per million was reduced to 0.39 based on literature.18
The annual rate of treatment discontinuation of ravulizumab and eculizumab was halved (22.5%).
The annual rate of relapse following discontinuation was halved (25%).
The scenario analyses indicate that decreasing the annual incidence rate per million leads to considerable decreases in cost savings due to less patients being treated with ravulizumab. Decreasing the rate of complement inhibitor treatment discontinuation leads to increased cost savings due to less patients discontinuing treatment with ravulizumab. Decreasing the annual rate of relapse leads to a decrease in the anticipated cost savings with ravulizumab, as fewer relapsed patients are being treated with ravulizumab. All scenarios led to ravulizumab being cost-saving.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | –$44,819,623 |
CADTH reanalysis 1 | –$49,511,350 |
CADTH base case | –$49,511,350 |
BIA = budget impact analysis.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $66,216,790 | $86,444,481 | $101,188,038 | $114,310,663 | $301,943,182 |
New drug | $66,216,790 | $77,678,267 | $84,730,179 | $94,715,113 | $257,123,559 | |
Budget impact | $0 | –$8,766,214 | –$16,457,859 | –$19,595,551 | –$44,819,623 | |
CADTH base case | Reference | $66,216,790 | $86,444,481 | $101,188,038 | $114,310,663 | $301,943,182 |
New drug | $66,216,790 | $76,606,794 | $82,967,903 | $92,857,136 | $252,431,832 | |
Budget impact | $0 | –$9,837,687 | –18,220,135 | –$21,453,528 | –$49,511,350 | |
CADTH scenario analysis 1: Reduced incidence rate | Reference | $35,095,714 | $38,510,593 | $36,632,597 | $36,458,222 | $111,601,412 |
New drug | $35,095,714 | $34,312,708 | $29,723,497 | $29,368,828 | $93,405,033 | |
Budget impact | $0 | –$4,197,885 | –$6,909,100 | –$7,089,394 | –$18,196,379 | |
CADTH scenario analysis 2: Discontinuation rate reduced (22.5%) | Reference | $77,490,136 | $103,860,846 | $127,602,527 | $149,427,219 | $380,890,592 |
New drug | $77,490,136 | $92,946,172 | $103,095,757 | $118,694,604 | $314,736,534 | |
Budget impact | $0 | –$10,914,674 | –$24,506,769 | –$30,732,615 | –$66,154,059 | |
CADTH scenario analysis 3: Relapse rate reduced (25%) | Reference | $66,216,790 | $80,573,036 | $90,509,354 | $98,051,846 | $269,134,235 |
New drug | $66,216,790 | $71,539,167 | $74,085,649 | $79,439,900 | $225,064,716 | |
Budget impact | $0 | –$9,033,868 | –$16,423,705 | –$18,611,946 | –$44,069,519 |
BIA = budget impact analysis.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
Stakeholder Input
Patient Input
aHUS Canada
About aHUS Canada
aHUS Canada is a not-for-profit organization. Formed in November 2012, the mission of the organization is to support patients and families living with atypical Hemolytic Uremic Syndrome.
In addition to providing support to patients and caregivers, aHUS Canada strives to:
connect those affected by the condition to establish a Canadian aHUS community
build public awareness and understanding of this very rare and potentially fatal disease
advocate for the best possible care and treatment for patients
Website link: www.ahuscanada.org
Information Gathering -
Besides references to our submission on eculizumab in 2013, all data was collected in June of 2022 from inside and outside Canada through an online survey. We collected data from 19 caregivers and 41 patients, of which 19 had experience with Ravulizumab (further demographic breakdown available upon request).
Disease Experience
aHUS is an ultra-rare, life-threatening disease that progressively damages vital organs and affects both adults and children. aHUS is caused by chronic, uncontrolled activation of complement, a part of the body’s natural immune system, which leads to the formation of blood clots in small blood vessels throughout the body. These blood clots are known as thrombotic microangiopathy, or TMA and can lead to acute kidney failure, stroke, heart attack, and death. There is a very short timeline from the patient initially not feeling well and being admitted to ICU. At the onset of disease, acute kidney failure is most common but continual uncontrolled systemic thrombosis leads to the beginning of failure of other organs. During the acute phase, the patient is literally fighting for their life and therefore experiences little quality of life. The length of hospital stay is anywhere from 10 days to a couple of months depending upon the severity of the symptoms and the number of organs affected.
In the past, prior to Eculizumab, more than half of patients with aHUS died, required kidney dialysis, or had permanent renal damage within one year of diagnosis. Overall, the impact of aHUS on a patient's day to day life is overwhelming. Since Eculizumab, there has been renewed hope for patients. When given soon after initial attack, kidney failure may be avoided altogether and other patients have been able to discontinue dialysis or given the opportunity for a successful kidney transplant with the use of Eculizumab. It has also given patients and families less stress of relapses and the long term use of plasma treatments has greatly been reduced.
Impacts of the disease on patients (day to day life and quality of life)
Please be aware that the compiled information does not fully express the devastating, life threatening and ongoing experiences with aHUS.
In the acute phase, the primary clinical symptoms, which are the most threatening and difficult to control, are anemia, low platelet count and acute renal failure. The following impacts were common experiences of patients in the acute stage of the disease.
Initial flu-like symptoms, vomiting, general sickness accompanied by extreme fatigue.
Blood loss
Edema
Migraine
Kidney/organ failure
Internal bleeding
Loss of vision
Immediate plasmapheresis treatment
Immediate dialysis
Blood transfusion
In almost all cases, patients were admitted into ICU in life threatening condition
Uncontrolled blood pressure
Hospital stay ranging 10 days to months
The following information focuses on patients' experiences after they leave the hospital. For those who do not have access to Eculizumab or Ravulizumab, their experience is much different than those who do have access and feels more like a fight for survival. The same is true for patients on plasma treatments or dialysis.
Common Impacts of the Disease
While all patients shared different impacts based on the personal treatment or phase of the disease the following list is common experiences that people have while living with aHUS.
Lack of quality of life
Feeling helpless — no choice in treatment, fear of loss of funding
PTSD — many patients share they have PTSD from the experience and triggers can include getting a cold, nausea, lack of access to transportation, changes in doctors or medical professionals, accessing support in the emergency room.
Fatigue/exhaustion
Headache
High blood pressure
Inability to travel
Constant isolation (anxiety of getting sick)
Frequent hospital visits
Dealing with frequent blood tests, IV’s
Inability to work or attend school
Memory loss/brain fog
Daily oral medications
Dietary restrictions
Kidney issues/dialysis
aHUS dialysis patients needing a kidney transplant can have added complications as they are not eligible for transplant (in Canada) unless they receive Eculizumab infusions at transplant. In provinces where the government is not funding Eculizumab, the patients have feelings of hopelessness.
Caregivers’ experiences (day to day life and quality of life):
Two main challenges were described by caregivers: the emotional and the financial challenges, both creating a high amount of stress.
Not having access to Eculizumab or other alternatives can extend the patients time in hospital while caregivers find ways to advocate for access to the drug. This process is challenging and has looked different per province and in some cases has been unsuccessful.
The following are common experiences shared by caregivers:
Lack of knowledge of aHUS in the medical community makes it difficult to receive adequate care
This can cause delay in initial diagnosis
accessing care during cautious phase
fear of attending the emergency room as caregiver — can be the one who needs to educate medical team
lack of urgency during hospital visits due to lack of knowledge of disease
Dealing with fear and anxiety
not being able to support child due to the need to work
financial stress of the disease — this can include dietary needs, extensive childcare needs, cost of medications, travel for appointments, time off work, parking costs
fear of getting sick — common colds cause anxiety
difficulty in having teachers, school on board to understand the length of complications of those who are in cautious phase
unable to help
dealing with waiting on transplant lists
parents whose children are in hospital need to be there around the clock, this can mean not being able to parent other children
fear of losing work due to absenteeism — this is compounded when health insurance through work is covering the cost of treatments
Exhaustion
Feeling of isolation and lack of support
Aspects of the disease that are harder to control
Anemia, low platelet count and acute renal failure
Financial struggles
Anxiety about access to treatment.
Protecting organs
Exhaustion
Memory issues/brain fog
Experiences with Currently Available Treatments
Current therapy for aHUS is plasma therapy (FFP or plasmapheresis) or infusions every two weeks of Eculizumab (Soliris) or maintenance (no therapies or medications) or long-term dialysis in case the patient has experienced kidney failure. (Please refer to Ravulizumab Patients).
Plasmapheresis: Patients who do not have access to Eculizumab and Ravulizumab are often on plasmapheresis. This includes but is not limited to the following experiences:
Weekly/biweekly treatment in hospital
Treatment can take anywhere from 8 hours to an entire day
Common side effects experiences are nausea, headache, fatigue
Less common — anaphylactic reaction to plasma used (3 patients)
Less common — vein collapse 2 patients
Infection in port
Anxiety
Patients eventually became refractory to plasma therapy which results in their kidney failure.
Not being able to work due to frequency of treatment, length of travel for treatment, feeling of unwellness
Central line
Plasma therapy does not effectively control complement activity and thus control aHUS, all aHUS patients that are currently on dialysis are denied the opportunity of a kidney transplant. Eventually the few patients that are using plasma therapy to maintain their original kidney will fail as well.
Eculizumab (Soliris) Infusions: They receive their infusions every two weeks in a clinic or at home. The entire process takes about 1 hour and 20 minutes; the actual infusion takes approximately 40 minutes. Infusions are given intravenously or through a port/central line. The following are some of the common experiences but are not limited to what is listed:
Being able to stop plasmapheresis
Being able to stop dialysis
Eligible for a kidney transplant
Stability of the disease and return to home/work/school
Improved kidney function
Lessened overall symptoms
Being able to return to day-to-day life after first few treatments
Better controlled blood pressure
Patients still reported struggling with some issues as well as side effects. This included but was not limited to the following:
Issues in controlling blood pressure
Migraines
Exhaustion
Trouble with accessing healthy veins for bi-weekly infusion treatments
Nausea
Memory loss/brain fog
Anxiety (this can range from anxiety about the disease to anxiety about fear of losing access to funding for Eculizumab)
Less common — inability to take eculizumab due to an allergic reaction
Dialysis: Some patients who do not have access to Eculizumab or an alternative are still undergoing dialysis. The following common experiences were shared by patients:
Exhaustion
Central line issues
Infections
Dietary restrictions
Accessing treatment — amount of time spent at hospital
Inability to work/travel/attend school
Stress and anxiety, depression
Muscle cramps
Insomnia
Nausea
Abdominal pain
Fever & chills
Weight gain/weight loss
Improved Outcomes
Patients conclusively said that access to treatment and freedom for choice is a critical component of managing their disease. One patient quoted “freedom of schedule. Not everything is about the illness”. Quality of life was the most common outcome shared. This included aspects of choice in care, affordability of the drug, frequency of medical appointments. Many shared that the ability to travel, focus on family and to have more time between appointments was critical for their mental health. The following were the common examples shared:
Quality of life improvement due to infusions being once every two months
Makes the dream of traveling for longer than 2 weeks possible
Reduces time away from work and responsibilities for other family members, be it the patient or the caregiver
Less “stress” on veins.
Psychologically, the patient feels “safe”, that their organs are safe from another crisis
Improved mental health
Cost effective; cost savings to public and private health insurers
Less needle poking
Many patients shared the struggle with ongoing issues in frequent blood tests and IV therapies or ports. A caregiver shared that her daughter has significant problems with her vascular system due to the earlier fistulas and having a port-o-cath from the time she was 18 months until now as a young adult. Maintaining venous access is crucial for her continued access to Eculizumab. However, if she only had to receive the treatment every 8 weeks versus every 2 weeks, she likely could have her port removed. Furthermore, other patients have shared that they are not eligible for ports due to the damage to their veins from the disease and that less frequent treatments would probably be lifesaving long term.
Experience with Drug Under Review
Ravulizumab Patients
The responses from patients who use Ravulizumab as a treatment were predominantly positive. As Ravulizumab is not accessible in Canada where our patient base is located, we had a smaller number of responses from patients who have experience with Ravulizumab. Patients listed the following as some of the common benefits:
More energy
Less damage to veins
More time, less treatments
Returned to normal life
Fewer fluctuations in symptoms
Freedom of choice
Improved quality of life
More affordable treatments means better access
Less anxiety
Travel
Some patients did share that some side effects such as headache, nausea and body aches were worse right after their infusion or during the month yet shared that the overall benefits were worthwhile as these side effects were the same as or better than previous treatments. Furthermore, patients shared that being on Ravulizumab changed their life even when dealing with side effects. These side effects varied from patient to patient but were often just immediately after treatment.
One patient quoted:
“I get a pretty significant headache afterwards, but I did with Soliris as well & having it every 8 weeks rather than every 2 weeks is huge. When I was first sick, trying to find extra time or drivers was difficult as dialysis was 3 days/week also. Even after I was off dialysis, asking someone to drive me every 2 weeks was taxing for everyone. Now on Ultomiris, I am much better & can drive myself.”
When patients were asked if there was anything else they wanted us to know we received the following powerful statements:
“I have benefited from switching by spending less time going to & from infusions & less time at the infusion center each month. I almost forget that I even have aHUS because I don't have the constant reminder of the infusion’s multiple times during the month. It's allowed me more freedom to do what I want & need to do.”
“Ultomiris has allowed my family and myself with dealing with the side effects of this disease. I have felt better, more energy and less interruption of life plans. Would hate to ever have to go off. We also have a foundation that helps cover the cost left from insurance.”
“Every patient should have the option to choose, and the medicine should be made available across the world. It is necessary to save lives.”
“If I had to choose to stay on Ultomiris or switch back to Soliris, I would choose to stay on Ultomiris. The reason for this is because I enjoy the freedom associated with having my infusions less often.”
“It's hard for someone who isn't a patient or caregiver to appreciate the difference biweekly vs every 8 weeks would make in a person's life. I hope you can use all the responses to show the benefit. I also hope that this could be the start of reducing the price of treatment.”
“I have aHUS with factor H gene mutation. If I was not able to receive Ultomiris infusions, the chance of me relapsing and losing my kidneys is very high. I pray everyone someday is able to have access to this medication.”
Companion Diagnostic Test
We are unable to answer this question as we are unsure of what these tests are and no patients on Ravulizumab offered this information.
Anything Else?
We asked patients if there was anything else they wanted aHUS Canada to know. These are the responses:
“This medication saved my life.”
“This drug is so incredibly important to the aHUS community. It allows patients and caregivers to continue living life. Without it, the cost and toil for a patient and family would be far greater with dialysis, not able to work, complications and possible death.”
“Ultomiris has allowed my family and myself with dealing with the side effects of this disease. I have felt better, more energy and less interruption of life plans. Would hate to ever have to go off.”
“I have aHUS with factor H gene mutation. If I was not able to receive Ultomiris infusions, the chance of me relapsing and losing my kidneys is very high. I pray everyone someday is able to have access to this medication.”
“If I had to choose to stay on Ultomiris or switch back to Soliris, I would choose to stay on Ultomiris. The reason for this is because I enjoy the freedom associated with having my infusions less often.”
“Every patient should have the option to choose, and the medicine should be made available across the world. It is necessary to save lives.”
“These drugs should be available to everyone. Cost should not be the only determining factor. Also, governments need to work with the pharmaceutical companies to bring down the cost of these drugs.”
“It’s hard for someone who isn’t a patient or care giver to appreciate the difference biweekly vs every 8 weeks would make in a person’s life. I hope you can use all the responses to show the benefit. I also hope that this could be the start of reducing the price of treatment.”
“It’s a great overall medical treatment it will save you $.”
Conflict of Interest Declaration — aHUS Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No. We have no conflicts of interest nor financial disclosures.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No. We operate completely independently.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
We have not received any financial payment from any company or organization in the last two years.
Clinician Input
No clinician group input was received for this review.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.