Drugs, Health Technologies, Health Systems
Reimbursement Review
Osilodrostat (Isturisa)
Sponsor: Recordati Rare Diseases Canada Inc.
Therapeutic area: Cushing’s disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Summary
What Is Cushing Disease?
Cushing disease is a rare but serious condition caused by a pituitary tumour that leads to excessive production of adrenocorticotropic hormone (ACTH) and, consequently, cortisol. It accounts for the majority of endogenous Cushing syndrome cases. Cushing disease causes significant physical and psychological symptoms and increases the risk of long-term complications such as cardiovascular disease, diabetes, and osteoporosis. Even after treatment, many patients experience persistent health issues and reduced quality of life.
Globally, the estimated prevalence of Cushing disease is approximately 2.2 per 100,000 people, with an annual incidence of about 2.4 per million. No Canada-specific epidemiological data are available, but estimates from other regions are often extrapolated to Canada. Cushing disease occurs most frequently in adults aged between 30 and 50 years. It also occurs more commonly in women than men.
What Are the Treatment Goals and Current Treatment Options for Cushing Disease?
The primary treatment goal in Cushing disease is to normalize cortisol levels and eliminate the underlying pituitary tumour to prevent or reverse disease-related comorbidities.
Both the patient and clinician groups highlighted important outcomes, including normalization of cortisol levels (serum, salivary, or urinary), improvement in cortisol-related comorbidities (e.g., glycemia, blood pressure, weight), and enhanced quality of life and mental well-being. The clinical experts consulted for this review also emphasize the importance of reducing life-threatening complications such as myocardial infarction and sepsis.
Transsphenoidal surgery to remove the ACTH-secreting pituitary adenoma is the first-line treatment for most patients with Cushing disease. If surgery is not feasible or fails to achieve remission, second-line options include repeat surgery, radiotherapy, medical therapies (e.g., adrenal steroidogenesis inhibitors or pituitary-directed drugs) and, as a last resort, bilateral adrenalectomy. The clinical experts consulted for this review noted that ketoconazole is the most relevant medical comparator in Canada, while cabergoline is generally ineffective as monotherapy but may be used in combination with ketoconazole for ACTH-dependent Cushing disease.
What Is Isturisa and Why Did Canada’s Drug Agency Conduct This Review?
Isturisa is a drug that is available as an oral tablet. Isturisa has been approved by Health Canada for the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option.
Canada’s Drug Agency (CDA-AMC) reviewed Isturisa to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the Health Canada–approved indication.
How Did CDA-AMC Evaluate Isturisa?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Isturisa versus other treatments used in Canada for the treatment of adult patients with Cushing disease. Ketoconazole and the combination of ketoconazole plus cabergoline were considered as relevant treatments to compare with Isturisa when reviewing the clinical evidence.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence. The CDA-AMC review team also identified equity and ethical considerations relevant to Isturisa and Cushing disease.
The review was informed by 1 patient group submission and 1 clinician group submission in response to the CDA-AMC call for input, and by the input provided by the participating public drug programs around issues that may impact their ability to implement a recommendation.
Two clinical specialists with expertise in the diagnosis and management of Cushing disease were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
2 phase III trials compared Isturisa with placebo in adult patients with Cushing disease: LINC 3 (a randomized withdrawal study with 71 patients who had a complete response at week 24 and did not require uptitration between weeks 13 and 24), and LINC 4 (a randomized controlled trial with 73 patients).
1 indirect treatment comparison (ITC) using unadjusted naive comparisons that compared Isturisa and ketoconazole.
For the comparison of Isturisa versus placebo based on the LINC 3 and LINC 4 studies:
Isturisa likely increases both complete response rate (proportion of patients with a mean urinary free cortisol [mUFC] level equal to or below the upper limit of normal [ULN]) and overall response rate (proportion of patients with an mUFC equal to or below the ULN or with at least a 50% reduction from baseline) when compared with placebo. However, the validity of mUFC as a surrogate end point to reliably predict clinically important outcomes remains uncertain.
The effects of Isturisa on bone mineral density (L1 to L4 lumbar spine), hemoglobin A1C, low-density lipoprotein cholesterol, diastolic blood pressure, weight, and discontinuation due to adverse events are uncertain; no statistical comparisons were made.
Improvements in patient-reported outcome measures (the Cushing quality of life questionnaire, Beck Depression Inventory-II) were minimal or inconsistent, with no statistical testing, limiting interpretability.
No notable safety concerns were identified during the study period in the LINC 3 and LINC 4 studies.
Efficacy in the extension phases of the LINC 3 and LINC 4 studies was primarily assessed using mUFC response rates, suggesting Isturisa maintains biochemical control, although the uncontrolled design precludes causal interpretation.
No additional safety concerns with longer-term Isturisa treatment were identified during the extension phases of the LINC 3 and LINC 4 studies.
For the comparison of Isturisa versus ketoconazole based on the ITC:
No conclusions can be drawn from the naive comparison regarding the comparative efficacy and safety of Isturisa versus ketoconazole in the treatment of Cushing disease in adults due to a critical risk of bias. As such, the comparative efficacy and safety profile of Isturisa versus ketoconazole remains unknown.
There was no evidence to inform how Isturisa compares with ketoconazole plus cabergoline combination therapy.
Clinical Review
Abbreviations
ACTH
adrenocorticotropic hormone
AE
adverse event
AESI
adverse event of special interest
ALT
alanine aminotransferase
AST
aspartate aminotransferase
BDI-II
Beck Depression Inventory-II
BMD
bone mineral density
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CushingQoL
Cushing quality of life questionnaire
DBP
diastolic blood pressure
DXA
dual-energy x-ray absorptiometry
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
ITC
indirect treatment comparison
LC-MS/MS
liquid chromatography-tandem mass spectrometry
LDL
low-density lipoprotein
LTE
long-term extension
MID
minimal important difference
mUFC
mean urinary free cortisol
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
UFC
urinary free cortisol
ULN
upper limit of normal
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of osilodrostat 1 mg, 5 mg, and 10 mg oral film-coated tablets in the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option. The focus will be placed on comparing osilodrostat to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, and this focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the economic review is aligned with the scope of the clinical review, unless otherwise stated. For most reviews, CDA-AMC develops a base case that is informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Osilodrostat (Isturisa), 1 mg, 5 mg, 10 mg, film-coated tablets, oral |
Sponsor | Recordati Rare Diseases Canada Inc. |
Health Canada indication | For the treatment of adult patients with Cushing’s disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 3, 2025 |
Mechanism of action | Osilodrostat blocks cortisol synthesis via 11-beta-hydroxylase inhibition. |
Recommended dosage | Recommended starting dosage: 2 mg twice daily (can be gradually titrated by increments of 1 or 2 mg twice daily, no more frequently than once every 2 to 3 weeks), orally Maximum dosage: 30 mg twice daily |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $46.14 per 1 mg tablet $184.58 per 5 mg tablet $193.81 per 10 mg tablet |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: As defined in the Health Canada indication Subgroups: None Intervention: 1 mg to 30 mg, twice daily, orally Comparators:b Ketoconazole and ketoconazole plus cabergoline Outcomes: Complete response rate, overall response rate, bone mineral density at the lumbar spine, cardiovascular-related metabolic parameters associated with Cushing disease (hemoglobin A1C, LDL cholesterol, DBP, and weight), CushingQoL total score, BDI-II, and standard harms outcomes (AEs, SAEs, WDAEs, deaths, AESIs) |
AE = adverse event; AESI = adverse event of special interest; BDI-II = Beck Depression Inventory-II; CDA-AMC = Canada’s Drug Agency: CushingQoL = Cushing quality of life questionnaire; DBP = diastolic blood pressure; LDL = low-density lipoprotein; NOC = Notice of Compliance; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThe CDA-AMC economic review aligns with the scope of the clinical review, unless otherwise stated.
bCDA-AMC has not reviewed these drugs for reimbursement recommendations for any indications.
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed osilodrostat through the reimbursement review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from the clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each reimbursement review. One patient group submission from the Canadian Organization for Rare Disorders and 1 clinician group submission from the Canadian Society of Endocrinology and Metabolism (CSEM) were received. Patient group input was gathered through online questionnaires and interviews with 21 patients (12 from Canada and 9 from the US), including 18 individuals (86%) diagnosed with Cushing disease and 3 individuals (14%) with Cushing syndrome. Clinician group input was gathered through iterative review and discussion among all 12 CSEM clinician members, who were experts in the treatment of Cushing syndrome. The full submissions received are available on the CDA-AMC project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of the outcomes to include in the clinical review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two clinical specialists with expertise in the diagnosis and management of Cushing disease participated as part of the review team.
Disease Background
Cushing syndrome is a rare endocrine disorder resulting from excessive cortisol production, either due to elevated ACTH or autonomous adrenal activity.1-4 Cushing disease, the most common form of endogenous Cushing syndrome, arises from an ACTH-secreting pituitary adenoma and accounts for approximately 70% of cases.5-8 Unlike Cushing syndrome, which includes various causes of hypercortisolism, Cushing disease refers specifically to the pituitary-driven form. Key symptoms include progressive weight gain (especially in the face and trunk), purple striae, skin thinning, muscle weakness, fatigue, mood disturbances, hypertension, and glucose intolerance.6,9-12 These symptoms significantly impair quality of life and daily functioning. Chronic hypercortisolism affects patients physically, psychologically, and socially, leading to fatigue, emotional instability, cognitive impairment, and reduced work capacity.6,13-23 These burdens are compounded by increased mortality risk, primarily due to cardiovascular disease, infections, and psychiatric complications such as suicide.14,24-27 Longer exposure to elevated cortisol levels correlates with worse outcomes, and patients with persistent disease after treatment face the highest mortality rates.14,23-33
Cushing disease has an estimated global prevalence of 2.2 per 100,000 individuals and an annual incidence of approximately 2.4 per million.25,30,34-36 Regional data vary, with incidence rates ranging from 1.2 to 3.9 per million per year in Europe, and up to 7.6 per million per year in the US.25,30,35,36 Prevalence estimates include 39.1 per million in Spain and 55 per million in Belgium, which are often extrapolated to Canada due to the lack of national data.12,30,37 Cushing disease occurs most frequently in adults aged from 30 to 50 years.25,36-38 It also occurs more commonly in females than males, with a female-to-male ratio ranging from 3:1 to 4:1.39-42
Diagnosis of Cushing disease begins with biochemical tests to confirm hypercortisolism: 24-hour urinary free cortisol (UFC), late-night salivary cortisol, and low-dose dexamethasone suppression test.10,43 Once confirmed, plasma ACTH levels are measured to determine ACTH dependence.5,21 For ACTH-dependent cases, pituitary MRI and inferior petrosal sinus sampling are used to differentiate between pituitary and ectopic sources.4,43
Current Management
Treatment Goals
The patients with Cushing disease emphasized the challenges of delayed diagnosis and the severe physical and psychological toll of the disease, which also deeply affects family life. The clinician group, referencing the Endocrine Society’s guidelines,10,43 identifies the primary goals of treatment as eliminating the source of hypercortisolism, reducing cortisol levels, managing comorbidities, and achieving remission to improve quality of life for both patients and caregivers. The clinical experts consulted for this review support these goals and emphasize that tumour removal is essential to preventing or reversing disease-related complications. Both the clinician group and clinical experts agree that patients with severe disease require urgent and intensive intervention to rapidly normalize cortisol levels and reduce morbidity and mortality.
Current Treatment Options
Surgical resection remains the central therapeutic approach, with transsphenoidal surgery used for pituitary adenomas and unilateral adrenalectomy for adrenal lesions. The clinical experts emphasized the importance of surgery in achieving disease control. The clinician group noted that transsphenoidal surgery is not curative in all cases, with persistent hypercortisolism occurring in up to 30% of patients. When surgery is not feasible or fails to achieve achieve remission, both groups identify repeat surgery, medical therapy, radiotherapy, and bilateral adrenalectomy as appropriate alternatives.10,43
Medical therapy is used in several contexts: preoperatively in severe cases, postoperatively when surgery is unsuccessful, and as a bridging strategy during radiotherapy.6,12,38,43-45 The clinician group that provided input noted that ketoconazole and metyrapone are the primary adrenal steroidogenesis inhibitors, although neither is approved by Health Canada, and metyrapone is accessible only through the Special Access Program. If these drugs are ineffective, cabergoline and pasireotide may be considered. Cabergoline is not approved for Cushing syndrome and has limited efficacy, while pasireotide is not publicly funded and often inaccessible to patients without private insurance. The clinician group anticipated that osilodrostat would be used as a first-line medical therapy in patients for whom adrenal steroidogenesis inhibitors are appropriate, citing its efficacy, safety profile, and flexible dosing as advantages over existing options.
Key characteristics of osilodrostat are summarized with the other treatments available for Cushing disease in the Supplemental Material document available on the CDA-AMC project landing page, in the Key Characteristics table in Appendix 1.
Unmet Needs and Existing Challenges
The patient group input indicates that many found current treatments (surgery, radiation, and medications) only partially effective and often associated with severe side effects, highlighting the need for more tolerable and accessible therapies.
According to the clinician group providing input, treatment options for Cushing syndrome are constrained by suboptimal efficacy, serious side effects, limited accessibility, and a lack of long-term data. The clinician group noted that commonly used medical therapies such as ketoconazole, cabergoline, metyrapone, and pasireotide present various challenges, including off-label use, hepatotoxicity, modest efficacy, poor tolerability, and restricted availability or funding. Radiotherapy is slow to achieve remission and carries significant risks, while bilateral adrenalectomy is typically reserved as a last-resort option due to its high-risk profile and lifelong consequences. The clinician group indicated that these limitations prevent current treatments from achieving an optimal balance between efficacy and safety, underscoring the need for better-tolerated and effective therapies such as osilodrostat.
The clinical experts consulted for this review further noted that some patients may be unable to undergo surgery due to poor health or technical constraints that make surgery infeasible; for such patients, effective and well-tolerated nonsurgical treatments are needed.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from a clinician group. The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Supplemental Material document in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1. The following has been summarized by the review team.
Place in Therapy
The clinical experts consulted for this review noted that osilodrostat selectively inhibits cortisol synthesis, effectively lowering hormone levels without removing the underlying tumour. They emphasized that its selectivity offers an advantage over other therapies, which are often associated with more side effects due to broader mechanisms of action. Input from the clinician group further supported the use of osilodrostat as a first-line medical treatment, citing its consistent cortisol-lowering effects, improvements in metabolic and psychological parameters, and a manageable safety profile.
Patient Population
The clinical experts indicated that patients with active disease who are not candidates for surgery or who have not experienced a response with prior surgery — due to feasibility, ineffectiveness, or significant comorbidities — should be prioritized for medical treatment with osilodrostat. The clinical experts emphasized the importance of early and accurate diagnosis because Cushing disease is often underdiagnosed due to its clinical overlap with metabolic syndrome. The clinical experts also noted that diagnosis is frequently delayed by months or years, leading to complications that may limit the success or feasibility of surgery.
Assessing the Response to Treatment
The clinical experts consulted for this review indicated that key measures of treatment response include improvement in patient symptoms, control or reversal of comorbidities, and reduction in life-threatening complications such as myocardial infarction and serious infections or sepsis. Consistent with the input from the clinician group, the clinical experts noted that assessing treatment response can be challenging in patients with prolonged exposure to elevated cortisol levels, which results in a wide range of clinical manifestations.
Discontinuing Treatment
According to the clinical experts consulted for this review, discontinuation of osilodrostat may be considered when the drug is poorly tolerated due to the severity of clinical symptoms or documented adverse effects (e.g., liver dysfunction), or when the treatment fails to reduce cortisol levels and associated complications to a clinically satisfactory level.
The clinical experts emphasized that monitoring liver parameters during treatment with osilodrostat is important because liver dysfunction is common in patients with Cushing disease and may reflect disease–treatment interactions. They also emphasized the need to regularly assess drug effectiveness to ensure that continued use is clinically justified.
Prescribing Considerations
The clinical experts indicated that a specialist is required to diagnose and manage patients who may be treated with osilodrostat because Cushing disease is an uncommon condition that typically falls outside the scope of general practitioners or internists.
Clinical Review
Methods
The review team considered the studies included in the sponsor’s submitted systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extension studies (LTEs), ITCs, and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and IV RCTs. Relevant patient eligibility criteria and interventions were defined by the indication and the recommended dosage in the product monograph, respectively. The CDA-AMC review team did not identify any subgroups as potentially important for informing the reimbursement recommendation. Relevant comparators considered by the sponsor were drugs used in clinical practice in Canada to treat patients described in the indication under review. These included ketoconazole and the combination of ketoconazole plus cabergoline. LTEs of included pivotal studies and RCTs were included in the systematic review, regardless of whether there was a comparison group. ITCs and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time).
The review team selected outcomes (and follow-up times) for review, considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.46,47 The following outcomes were assessed using GRADE because they address important treatment goals for Cushing disease and are considered important to patients, per patient and clinician input:
Complete response rate: Proportion of randomized patients with mUFC equal to or below the ULN.
Overall response rate: Proportion of complete responders (mUFC equal to or below the ULN) plus partial responders (mUFC above the ULN with at least a 50% reduction from baseline); in other words, patients with an mUFC equal to or below the ULN or with at least a 50% reduction from baseline.
Cushing quality of life questionnaire (CushingQoL).
Beck Depression Inventory-II (BDI-II).
Treatment discontinuation due to adverse events (AEs).
Additionally, the clinical experts consulted for this review emphasized the importance of several surrogate outcomes in clinical practice, including bone mineral density (BMD) of the L1 to L4 lumbar spine, select cardiovascular-related metabolic parameters associated with Cushing disease (hemoglobin A1C, low-density lipoprotein [LDL] cholesterol, diastolic blood pressure [DBP], and weight), and liver function biochemical parameters. No between-group differences were available for these surrogate outcomes; therefore, the results for those outcomes were presented only descriptively. Given their clinical relevance, they are reported narratively in the main body of the report. However, they were not included in the GRADE assessment because they are considered less critical than other outcomes.
Methods for the data extraction, risk of bias appraisal, and certainty of evidence assessment are in the Supplemental Material document in Appendix 2.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
2 pivotal studies: LINC 3 (randomized withdrawal study) and LINC 4 (RCT)
2 LTEs: LINC 3 extension and LINC 4 extension
1 unadjusted (naive) indirect comparison of osilodrostat versus ketoconazole.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included studies are summarized in Table 2. Details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are in the Supplemental Material document in Appendix 3.
Two multicentre, phase III, double-blind, placebo-controlled studies — LINC 3 (randomized withdrawal trial) and LINC 4 (RCT) — provided evidence comparing osilodrostat with placebo in adult patients with Cushing disease. The LINC 3 study enrolled 137 patients. The core LINC 3 study included the single-arm, open-label, dose-titration administration of osilodrostat (2 mg to 5 mg to 10 mg to 20 mg to 30 mg) twice a day based on the mean of three 24-hour mUFC periods from week 0 to week 12, a maintenance period from week 12 to week 26, a double-blind randomized withdrawal period for either osilodrostat (1 mg to 30 mg, oral, twice daily) or placebo (at a 1:1 ratio) from week 26 to week 34, and an open-label, single-arm period from week 34 to week 48 in which all patients received osilodrostat. Osilodrostat dosing was individualized and titrated based on mUFC levels, with adjustments allowed to manage hypocortisolism; therefore, rescue medication was not applicable in the LINC 3 study. Additionally, dose increases were not permitted during the randomized withdrawal period.48,49 The LINC 4 study randomized 74 patients to receive either osilodrostat (1 mg to 30 mg, oral, twice daily) or placebo at a 2:1 ratio from week 0 to week 12 and, thereafter, all patients received open-label treatment with osilodrostat until either week 48 or the end of the optional extension phase (week 96). In the LINC 4 study, rescue medications were permitted throughout for the management of comorbid conditions, including hyperglycemia, hypertension, dyslipidemia, and hypokalemia.48,49 The maximum dosage of osilodrostat in the LINC 3 and LINC 4 studies was 30 mg twice daily.48,50
In both studies, the primary end point was the complete response rate (proportion of randomized patients with an mUFC equal to or below the ULN, i.e., 138 nmol per 24 hours) at the end of the placebo-controlled period. Secondary end points included overall response rate (proportion of patients with an mUFC equal to or below the ULN or with at least a 50% reduction from baseline), cardiovascular-related metabolic parameters, BMD, CushingQoL, BDI-II, as well as harms.48,50
Table 2: Characteristics of Studies Included in the Systematic Review
Study | LINC 3 | LINC 4 |
|---|---|---|
Study design, sample size | Multicentre, phase III, double-blind, placebo-controlled, randomized withdrawal study Total N (randomized withdrawal period, weeks 26 to 34) = 71 | Multicentre, phase III, double-blind RCT Total N = 74a |
Key inclusion criteria |
|
The rest of the key inclusion criteria matched those of the LINC 3 study. |
Key exclusion criteria |
| The key exclusion criteria in the LINC 4 study were consistent with those in the LINC 3 study. |
Intervention and comparatorb | All patients received dose titration of osilodrostat (2 mg to 5 mg to 10 mg to 20 mg to 30 mg twice daily from week 12), with a maintenance period to week 26 Intervention: Osilodrostat 1 mg to 30 mg tablet taken orally twice daily for 8 weeks (randomized withdrawal period) Comparator: Placebo taken orally twice daily for 8 weeks | Intervention: Osilodrostat 2 mg tablet taken orally twice daily for 12 weeks, with a dose-escalation sequence of 2 mg to 5 mg to 10 mg to 20 mg, administered twice daily Comparator: Placebo taken orally twice daily for 12 weeks |
Relevant end points | Primary:
Secondary:
Safety:
| Primary:
Secondary:
Safety:
|
ACTH = adrenocorticotropic hormone; AE = adverse event; AESI = adverse event of special interest; BDI-II = Beck Depression Inventory-II; BMD = bone mineral density; CushingQoL = Cushing quality of life questionnaire; mUFC = mean urinary free cortisol; QTc = QT corrected; RCT = randomized controlled trial; SAE = serious adverse event; UFC = urinary free cortisol; ULN = upper limit of normal.
Note: Normal range of mean 24-hour UFC is from 11 nmol per 24 hours to 138 nmol per 24 hours or from 4 mcg per 24 hours to 50 mcg per 24 hours. The ULN for mUFC is 138 nmol per 24 hours. Four additional reports were included: Pivonello et al. (2020),51 Fleseriu et al. (2022),51 Gadelha et al. (2022),52 and Gadelha et al. (2023).53 In the LINC 3 study, patients remain on open-label osilodrostat during the period between the end of week 24 and week 26 to ensure that sufficient time is allowed for central laboratory results (week 24 mUFC) to become available for all patients at all sites, and to standardize the time of randomization across sites. Entry into the extension period was optional in both the LINC 3 and LINC 4 studies.
aOne patient was randomized but not treated.
bThe maximum dosage of osilodrostat in the LINC 3 and LINC 4 studies was 30 mg twice daily. In the LINC 4 study, dosages below 2 mg twice daily (i.e., 1 mg twice daily, 1 mg once daily, or 1 mg every other day) were allowed, if necessary.
Sources: LINC 3 interim Clinical Study Report,48 LINC 3 final Clinical Study Report,49 LINC 4 primary analysis Clinical Study Report,50 and LINC 4 final Clinical Study Report.54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Statistical Testing and Analysis Populations
For the LINC 3 study, to detect a 40% difference in complete response rate between treatment arms (70% versus 30%), 33 patients per arm were required to achieve 87% power. Assuming that at least 50% of enrolled patients would be eligible for randomization, a total of 132 patients were enrolled. For the LINC 4 study, to detect a 45% difference in response rate (60% versus 15%) with 91% power, 63 patients were needed (42 in the osilodrostat arm and 21 in the placebo arm), with an additional 10% included to account for dropouts, resulting in approximately 69 patients planned for enrolment. The design provided greater than 98% probability that the lower bound of the 95% confidence interval (CI) would exceed 30%.
Key analysis populations of the LINC 3 and LINC 4 studies are presented in Appendix 3. The full analysis set included all enrolled patients who received at least 1 dose of osilodrostat in the LINC 3 study, or at least 1 dose of their assigned treatment (osilodrostat or placebo) in the LINC 4 study. In the LINC 3 study, efficacy analyses were based on the randomized analysis set, defined as all randomized patients who received at least 1 dose of their assigned treatment. In the LINC 4 study, efficacy analyses were conducted using the full analysis set. Safety analyses in both studies were based on the safety analysis set. Results from this set were analyzed according to the actual treatment received.
Patient Disposition
Patient disposition for each included study is summarized in the Supplemental Material, Appendix 4.
In the LINC 3 study, 19 of the 137 enrolled patients (13.9%) discontinued at or before week 26. Of the remaining 118 patients, 71 were randomized (36 to osilodrostat and 35 to placebo) and 47 nonrandomized patients continued on open-label osilodrostat treatment. Among the randomized patients, none in the osilodrostat group discontinued during the core phase (at or before week 48), whereas 2 patients (5.7%) in the placebo group discontinued due to AEs.
In the LINC 4 study, 1 patient who was randomized to the osilodrostat arm did not receive any study treatment (the patient discontinued prematurely from the study before receiving any study treatment). Of the remaining 73 patients in the LINC 4 study, 3 patients (6.3%) in the osilodrostat group and no patients in the placebo group discontinued the study by week 12. Discontinuation reasons included AEs (2.1% for osilodrostat versus 0 for placebo), and patient or guardian decision (4.2% versus 0).
In the LINC 3 study, protocol deviations were reported in 92.0% of patients (126/137), with the most common reasons being deviation from the protocol treatment plan (69.3%; 95/137) and use of prohibited concomitant medications (54.7%; 75/137), as reported in the primary Clinical Study Report. In the LINC 4 study, 68.5% of patients (50/73) in the all-patients population had at least 1 protocol deviation, including 66.7% (32/48) in the osilodrostat group and 72.0% (18/25) in the placebo group. The majority of deviations in the LINC 4 study fell under the category of “other deviation” (43.8%; 32/73), which, in most cases, were missed or delayed assessments, with similar rates in the osilodrostat group (43.8%; 21/48) and the placebo group (44.0%; 11/25).
Baseline Characteristics
A summary of key baseline patient characteristics in the LINC 3 and LINC 4 studies is presented in Table 3.
At the baseline of the LINC 3 study (randomized withdrawal period involving responders with no uptitration in the preceding weeks), the mean age was 44.3 years (standard deviation [SD] = 11.3) in the osilodrostat arm and 42.0 years (SD = 13.5 years) in the placebo withdrawal arm. The proportion of women was higher in the osilodrostat arm (83%) than in the placebo withdrawal arm (63%). Time since diagnosis of Cushing disease was shorter in the osilodrostat group (mean, 71.4 months) than in the placebo withdrawal group (mean, 88.3 months). Most patients had persistent or recurrent disease (89% in the osilodrostat arm and 94% in the placebo withdrawal arm). Adenoma size was not reported. At the baseline of the placebo-controlled period, the mUFC was higher in the osilodrostat arm (mean, 6.4 times the ULN, equivalent to 890 nmol per 24 hours; SD = 1,276 nmol per 24 hours) compared with the placebo withdrawal arm (4.1 times the ULN, equivalent to 560 nmol per 24 hours; SD = 549 nmol per 24 hours), respectively. Most patients had prior pituitary surgery (89% for osilodrostat versus 94% for placebo withdrawal) or irradiation (17% versus 14%) and medical therapy (97% versus 94%).
At the baseline of the LINC 4 study, the mean age was 42.3 years (SD = 13.8) in the osilodrostat arm and 38.9 years (SD = 12.3) in the placebo arm. The proportion of women was higher in the osilodrostat arm (90%) than in the placebo arm (72%). Adenoma classification showed a lower proportion of microadenomas in the osilodrostat arm (62.5%) compared to the placebo arm (80%), while macroadenomas were more frequent in the osilodrostat arm (35% versus 16%). Baseline mUFC levels were similar between groups, with a mean for osilodrostat of 3.1 times the ULN, equivalent to 421 nmol per 24 hours (SD = 291 nmol per 24 hours) versus 3.3 times the ULN for placebo, equivalent to 451.5 nmol per 24 hours (SD = 535 nmol per 24 hours). Most patients had prior pituitary surgery (85% for osilodrostat versus 92% for placebo) or irradiation (12.5% versus 12%), and prior medical therapy was more frequent in the placebo group (76%) than in the osilodrostat group (54%).
Table 3: Summary of Baseline Characteristics by Randomized Treatment From the LINC 3 and LINC 4 Studies (FAS)
Characteristic | LINC 3 trial | LINC 4 trial | ||
|---|---|---|---|---|
Osilodrostat (N = 36) | Placebo withdrawal (N = 35) | Osilodrostat (N = 48) | Placebo (N = 25) | |
Demographics | ||||
Age, years | ||||
Mean (SD) | 44.3 (11.27) | 42.0 (13.47) | 42.3 (13.82) | 38.9 (12.33) |
Median (range) | 41.0 (20.0 to 69.0) | 40.0 (19.0 to 68.0) | 41.0 (21.0 to 67.0) | 37.0 (19.0 to 63.0) |
Aged 18 to < 65 years, n (%) | 34 (94.4) | 34 (97.1) | 46 (95.8) | 25 (100) |
Aged ≥ 65 years, n (%)a | 2 (5.6) | 1 (2.9) | 2 (4.2) | 0 |
Sex, n (%) | ||||
Female | 30 (83.3) | 22 (62.9) | 43 (89.6) | 18 (72.0) |
Male | 6 (16.7) | 13 (37.1) | 5 (10.4) | 7 (28.0) |
Race or ethnic group, n (%) | ||||
Asian | 7 (19.4) | 7 (20.0) | 9 (18.8) | 8 (32.0) |
Black or African American | 0 | 3 (8.6) | 2 (4.2) | 0 |
White | 27 (75.0) | 23 (65.7) | 34 (70.8) | 15 (60.0) |
Other | 2 (5.6) | 2 (5.7) | 1 (2.1) | 1 (4.0) |
Unknown | 0 | 0 | 2 (4.2) | 1 (4.0) |
Weight, kg | ||||
Mean (SD) | 78.2 (19.02) | 83.4 (24.73) | 78.8 (17.46) | 77.3 (16.90) |
Median (range) | 73.6 (55.0 to 126.3) | 75.4 (50.8 to 141.0) | 80.1 (46.9 to 113.7) | 74.0 (53.5 to 114.5) |
Height, cm | ||||
Mean (SD) | 163.0 (9.01) | 163.9 (10.76) | 162.1 (6.61) | 160.6 (9.96) |
Median (range) | 160.2 (151.0 to 190.0) | 163.0 (142.0 to 185.3) | 162.0 (149.0 to 182.0) | 160.0 (136.0 to 177.0) |
BMI, kg/m2 | ||||
Mean (SD) | 29.6 (7.35) | 30.9 (8.37) | 29.9 (6.31) | 30.0 (6.25) |
Median (range) | 28.5 (18.8 to 47.7) | 29.0 (20.8 to 55.1) | 29.1 (18.4 to 50.0) | 29.0 (20.2 to 47.5) |
Baseline disease characteristics | ||||
Time since diagnosis, monthsb | ||||
Mean (SD) | 71.4 (63.54) | 88.3 (67.46) | 70.7 (55.94) | 73.9 (52.51) |
Median (range) | 53.6 (2.1 to 286.7) | 76.8 (2.9 to 277.7) | 69.9 (6.0 to 257.7) | 65.0 (11.2 to 215.9) |
CD status, n (%) | ||||
De novo | 4 (11.1) | 2 (5.7) | 3 (6.3) | 0 |
Persistent or recurrent | 32 (88.9) | 33 (94.3) | 45 (93.8) | 25 (100) |
Classification at time of CD diagnosis, n (%)c | ||||
Microadenoma | NR | NR | 30 (62.5) | 20 (80.0) |
Macroadenoma | NR | NR | 17 (35.4) | 4 (16.0) |
Missing | NR | NR | 1 (2.1) | 1 (4.0) |
mUFC at baseline of study, nmol/24 hours | ||||
Mean (SD; equivalent multiple value of ULN for mean) | 890.0 (1,275.66; 6.4 × ULN) | 560.0 (548.84; 4.1 × ULN) | 421.4 (291.25; 3.1 × ULN) | 451.5 (535.09; 3.3 × ULN) |
Median (range; equivalent multiple value of ULN for median) | 457.0 (35.6 to 5,719.5; 3.3 × ULN) | 357.9 (67.9 to 2,466.1; 2.6 × ULN) | 342.2 (90.1 to 1,720.0; 2.5 × ULN) | 297.6 (21.4 2 to 607.3; 2.2 × ULN) |
mUFC at randomization, nmol/24 hours | ||||
Mean (SD)d | 70.9 (43.53) | 79.1 (57.90) | Same as mUFC at baseline of study | |
Median (range)d | 57.6 (5.7 to 226.5) | 57.0 (8.9 to 245.0) | Same as mUFC at baseline of study | |
Previous treatment, n (%) | ||||
Previous pituitary surgery | 32 (88.9) | 33 (94.3) | 41 (85.4) | 23 (92.0) |
Previous medical therapy for CD | 35 (97.2) | 33 (94.3) | 26 (54.2) | 19 (76.0) |
Previous pituitary irradiation | 6 (16.7) | 5 (14.3) | 6 (12.5) | 3 (12.0) |
BMI = body mass index; CD = Cushing disease; FAS = full analysis set; mUFC = mean urinary free cortisol; NR = not reported; SD = standard deviation; ULN = upper limit of normal.
Note: Normal range of 24-hour mUFC: 11 nmol per 24 hours to 138 nmol per 24 hours or 4 mcg per 24 hours to 50 mcg per 24 hours. ULN for mUFC is 138 nmol per 24 hours.
aAged 65 to ≤ 75 years in the LINC 3 study or 65 to < 85 years in the LINC 4 study.
bTime (months) from diagnosis to first osilodrostat dose.
cPituitary adenomas < 10 mm in size are defined as microadenomas; those ≥ 10 mm in size are defined as macroadenomas.
dValues in the LINC 3 study were based on the randomized analysis set, i.e., 35 patients in the osilodrostat arm and 33 patients in the placebo withdrawal arm.
Sources: LINC 3 interim Clinical Study Report48 and LINC 4 primary analysis Clinical Study Report.50 Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure and use of concomitant medications in each included study are in the Supplemental Material document, Appendix 4.
In the LINC 3 study, during the 8-week randomized withdrawal period, the mean duration of exposure to osilodrostat was 8.5 weeks (SD = 1.3 weeks) and 7.0 weeks (SD = 2.5 weeks) for placebo withdrawal.48 In the LINC 4 study, during the 12-week placebo-controlled period, the mean duration of exposure was 11.6 weeks (SD = 1.8 weeks) in the osilodrostat arm and 12.2 weeks (SD = 0.4 weeks) in the placebo withdrawal arm.50
In the LINC 3 study, the majority of participants (96.4%) received concomitant medications and notable nonpharmacological interventions following study initiation, primarily to manage AEs. Among the 137 enrolled patients, 16.8% of patients received antidiabetic drugs, 9.5% received lipid-lowering therapies, and 52.6% were treated with medications for blood pressure control. During the initial 26-week treatment phase, 89.8% of patients received such therapies. During the randomized withdrawal phase, 61.1% of patients (22/36) in the osilodrostat group and 54.3% (19/35) in the placebo withdrawal group were administered concomitant medications.
During the LINC 4 study, 72 patients (98.6%) received at least 1 concomitant medication. The most frequently administered drugs (used by ≥ 10% of patients) included paracetamol (28.8%); spironolactone and cholecalciferol (21.9% each); calcium carbonate (20.5%); potassium chloride and metformin (17.8% each); amlodipine and levothyroxine (16.4% each); ibuprofen (15.1%); metformin hydrochloride and leucovorin calcium (13.7% each); and hydrocortisone, acetylsalicylic acid, atorvastatin, omeprazole, and levothyroxine sodium (12.3% each). Additionally, amoxicillin-clavulanate, hydrochlorothiazide, and unspecified vitamin D were each used by 11.0% of patients. According to the Anatomical Therapeutic Chemical classification, the most frequently used drug classes (≥ 30% of patients) were vitamin D and analogues (43.8%); ophthalmological drugs (39.7%); anilides and hydroxymethylglutaryl-coenzyme A reductase inhibitors (35.6% each); drugs for local oral treatment (34.2%); glucocorticoids (32.9%); and biguanides, corticosteroids, and homeopathic preparations (31.5% each), as well as topical anti-inflammatory drugs and plain corticosteroids (30.1% each).
Critical Appraisal
Internal Validity
Randomization in both the LINC 3 and LINC 4 trials was performed using an appropriate methodology with adequate allocation concealment, that is, an interactive response technology system. Randomization stratification was prespecified and was based on relevant prognostic factors, that is, osilodrostat dosage at week 24 (≤ 5 mg twice daily versus > 5 mg twice daily), and history of pituitary irradiation (yes versus no) in the LINC 3 study, and history of pituitary irradiation (yes versus no) in the LINC 4 study. For both the LINC 3 and LINC 4 trials, randomization and outcome data integrity during the placebo-controlled periods were adequately maintained, with no major concerns identified by the CDA-AMC review team. The LINC 3 study used a randomized withdrawal design, in which all patients were exposed to osilodrostat from week 1 to week 26 before randomized allocation. Only patients who achieved a response (defined as mUFC equal to or below the ULN) at week 24, without requiring uptitration in the preceding weeks (between weeks 13 and 24), were eligible for randomization withdrawal. A potential for carryover effects in the placebo group exists due to prior exposure to osilodrostat. The clinical experts consulted for this review noted that osilodrostat is short lived and rapidly reversible, suggesting that any carryover impact may be limited. This interpretation is based on clinical expert opinion and, in the absence of corroborating empirical evidence, should be considered a plausible but nonconclusive explanation. Overall, the baseline demographic and disease characteristics appeared to be reasonably balanced between the osilodrostat and placebo arms in the studies. Although a numerical between-group difference in baseline mUFC levels was noted in the LINC 3 study (6.4 times the ULN in the osilodrostat arm and 4.1 times the ULN in the placebo withdrawal arm), the clinical experts did not consider this imbalance, nor did they identify any other important imbalance in baseline characteristics to be of prognostic importance or likely to influence the interpretation of the study results.
In both trials, the double-blind approaches were adequate because they masked participants as well as investigators (including the outcome assessors) regarding treatment allocation from the time of random assignment until the time of unblinding, per the study protocols. During the placebo-controlled period (week 1 to week 12) in the LINC 4 study, a group of independent endocrinologists was responsible for managing dose titration (which was based on patient mUFC result from the previous visit) between visits; the independent endocrinologists communicated study drug dosing instructions for all patients in the osilodrostat and placebo arms to the site using the interactive response technology system in a blinded manner. The study drugs and placebo were identical in packaging, labelling, schedule of administration, appearance, and odour.
The clinical experts commented that the range of normal mUFC that was used in the LINC 3 and LINC 4 studies, that is, 11 nmol per 24 hours to 138 nmol per 24 hours (4 mcg per 24 hours to 50 mcg per 24 hours), with 138 nmol per 24 hours as the ULN for mUFC, was adequate. According to the sponsor, a reference for the specific range of normal UFC from 11 nmol per 24 hours to 138 nmol per 24 hours was not available. However, a threshold of 138 nmol per 24 hours was also used as the ULN in the phase III SONICS56 and LOGICS57 studies of levoketoconazole in treating patients with endogenous Cushing syndrome. The ULN used in the LINC studies was aligned with the normal range (30 nmol per 24 hours to 145 nmol per 24 hours) used in the study by Petersenn et al. (2014) to explore the variability in UFC values in patients with Cushing disease.58 The sponsor indicated that the regulatory authorities and local clinical experts who participated in the LINC studies considered the reference normal range of mUFC to be acceptable and appropriate.48,50,59
Patients had to have an mUFC of greater than 1.5 times the ULN and greater than 1.3 times the ULN at screening to be eligible for enrolment in the LINC 3 and LINC 4 studies, respectively. The clinical experts commented that the difference in the eligibility criteria between the 2 trials regarding the mUFC level (greater than 1.5 versus greater than 1.3 times the ULN) would not be a major concern that would impact the results in the 2 trials differently. The clinical experts pointed out that a healthy population could have an mUFC level of up to 1.2 times the ULN, and either patient inclusion criterion regarding mUFC level (greater than 1.5 or greater than 1.3 times the ULN) is adequate. The measurement of UFC in the LINC 3 and LINC 4 studies using liquid chromatography-tandem mass spectrometry (LC-MS/MS) was adequate.
The sponsor selected the proportion of patients who achieved cortisol normalization (mUFC equal to or below the ULN) as the primary efficacy end point for the LINC 3 and LINC 4 studies. The clinical experts suggested that the choice of this surrogate outcome was likely due to the infeasibility of measuring long-term clinical outcomes, which would require extended follow-up and larger sample sizes. Although international clinical practice guidelines and pituitary gland disorder experts consulted by the sponsor have emphasized cortisol reduction as a therapeutic goal,43,60 the clinical experts consulted by CDA-AMC noted that this outcome is not universally accepted as a validated surrogate for long-term morbidities of Cushing disease. It is indicated in the literature that hypercortisolism is associated with an increased risk of mortality compared to the general population,25,26,28 and that mortality risk in these patients is correlated with the duration of exposure to excess levels of cortisol.26,61,62 However, studies have shown that patients with Cushing disease who experience cortisol normalization may have mortality rates similar to the general population.24-28,31-33,63 While cortisol normalization is associated with improvements in other surrogate and clinical measures, such as cardiovascular and metabolic parameters, physical features, and health-related quality of life (HRQoL),61,62,64-77 these associations do not confirm its validity as a surrogate end point to reliably predict clinically meaningful treatment effects on the outcomes identified by the clinical experts consulted for this review. The clinical experts consulted for this review noted that a reduction in cortisol levels alone would not be sufficient, as a surrogate end point, to evaluate the effects of treatment on final outcomes that are important to patients. These outcomes include the risk of complications associated with hypercortisolism, such as cardiovascular diseases, diabetes, physical function (for example, mobility and performance status) and, ultimately, mortality.
The evidence from the LINC 3 and LINC 4 trials presented by the sponsor suggests there may be an association between cortisol normalization and reduced morbidity and mortality at a patient level. However, the preferred evidence for the validity of a surrogate end point is trial-based evidence that demonstrates the surrogate’s ability to predict a clinically relevant treatment effect (i.e., a between-group difference) on patient-important clinical outcomes (i.e., morbidity and mortality).78 This evidence was not available in this review for osilodrostat. It is worth noting that the FDA has considered normalization of mUFC to be an acceptable surrogate end point for disease control in patients with Cushing disease, based on its association with reductions in disease-related morbidity.79 Furthermore, cortisol normalization measured by mUFC has been used as an end point in studies for pasireotide, levoketoconazole, metyrapone, ketoconazole, mitotane, and cabergoline for the treatment of Cushing disease.43,60
During the core phase of the LINC 3 and LINC 4 trials, most patients received concomitant therapies, including antidiabetic, antihypertensive, and lipid-lowering medications. These were permitted per protocol, with stable dosing expected in the LINC 3 study, while the LINC 4 trial allowed rescue medications for comorbid conditions.48,50,51 At baseline, 12% to 17% of the patients across the different arms in the 2 trials had received previous pituitary irradiation. It is possible that these medications and/or the delayed effects of irradiation could have confounded the outcomes (including mUFC, BMD, and metabolic measures), thus complicating the attribution of observed effects solely to osilodrostat; it is uncertain how the findings in the pivotal studies may have been affected by this.
Overall, the statistical methods used in both trials are considered appropriate. The trials were powered on their primary end points. The full analysis set was used for analyses of most outcomes, including the patient-reported outcomes. Regarding outcomes of interest to this review, control for multiple comparisons accounted only for the complete response comparisons, as per study protocols. Formal statistical testing for other end points was not undertaken, although the risk for type I error (erroneously rejecting the null hypothesis) can be considered when the 95% CI excludes the null (i.e., BDI-II in the LINC 4 study). Most outcomes of interest were presented only descriptively per study group, without absolute between-group differences or CIs. Upon request, the sponsor was unable to supply these data, which were not a part of the statistical analysis plan. The lack of between-group differences and CIs challenged the meaningful interpretation of these outcomes, and judgments of imprecision relied solely on consideration of the sample size and numbers of events (which were small, as expected due to the rare nature of the condition), such that any definitive conclusions were not possible. Missing data were minimal for the outcomes assessed using the GRADE approach during the placebo-controlled periods of the LINC 3 and LINC 4 studies (Table 4, Table 5). However, substantial missing data were noted for BMD at the L1 to L4 lumbar spine at 48 weeks. In the LINC 3 study, 59% of all patients (81/137) and 72% of those with baseline data (81/113) had change-from-baseline values available. In the LINC 4 study, the corresponding figures were 66% (48/73) and 81% (48/59), respectively (Appendix 4). The handling of missing BMD data was not described, suggesting a risk of bias in the observed results.
External Validity
Patients in the LINC 3 and LINC 4 trials were recruited from multiple countries, including Canada, and the study populations were consistent with the Health Canada–approved indication. The clinical experts consulted for this review noted that the eligibility criteria of patients in both trials generally aligned with the diagnosis standards and treatment indication for Cushing disease in practice in Canada, and that the demographic characteristics were comparable to those typically seen in Canada. However, they emphasized that the severity of illness among the enrolled patients was lower than that of the patients observed in real-world clinical settings who would most need the treatment, particularly those at higher risk of Cushing disease–related complications. This introduces uncertainty regarding the generalizability of the findings to patients with more severe disease.
Importantly, the LINC 3 trial employed an enriched design for its randomized withdrawal phase, which included only patients who had experienced mUFC normalization without requiring uptitration between weeks 13 and 24. As noted by the FDA, this approach excluded patients whose disease was more difficult to control, potentially limiting the applicability of the results to the broader clinical population.80 This design estimates treatment effects in a subset of responders rather than evaluating efficacy in a treatment-naive population, and may overstate efficacy while understating harms. On the other hand, the LINC 4 trial featured an upfront, double-blind, placebo-controlled design that allows for a more conventional assessment of efficacy and safety. The FDA’s statistical review also suggested that a more moderate dose-titration strategy could reduce both efficacy and the incidence of withdrawal due to AEs, which may be relevant to clinical practice in Canada.81
The clinical experts further noted that the relatively low average dose of osilodrostat used in the studies, along with the low rate of serious adverse events (SAEs) and low dropout rates, suggests that the enrolled patients were likely at an earlier or milder disease stage. Whether this led to an over- or underestimation of the treatment effects remains unknown because the current review did not include any RCTs that enrolled patients with more severe disease. The investigators of the LINC 3 and LINC 4 studies selected a twice-daily dosing regimen for osilodrostat based on its half-life of 3 to 5 hours, with a maximum dosage of 30 mg twice daily (i.e., 60 mg daily). The clinical experts considered this dosing strategy reasonable for settings in Canada.
Both the LINC 3 and LINC 4 studies used placebo comparators rather than any of the active pharmacotherapies used in the Canadian context, such as ketoconazole or ketoconazole plus cabergoline. The comparative results submitted by the sponsor are summarized in the ITC section of this report.
The length of the placebo-controlled periods, that is, 8 weeks of randomized withdrawal in the LINC 3 study and 12 weeks of randomized withdrawal in the LINC 4 study, might not have been sufficient to observe effects on clinical symptoms, complications of hypercortisolism, or HRQoL outcomes. Although the core phase of each study was nearly 1 year long (48 weeks), much of this duration included single-arm treatment periods, which limit the ability to draw causal inferences between treatment and outcomes. Additionally, some AEs may take longer to emerge, underscoring the need for a longer-term randomization period to more fully assess the efficacy and safety of osilodrostat.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section (Table 4, Table 5). Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
Key results are presented subsequently.
mUFC Responders
Complete Response Rate
In the LINC 3 study, at the end of the 8-week randomized withdrawal period (week 34), a higher proportion of patients in the osilodrostat arm (86.1%) were complete responders compared with the placebo arm (29.4%), with an odds ratio of 13.71 (95% CI, 3.73 to 53.44; P < 0.001). Absolute between-group differences were not available.
In the LINC 4 study, at the end of the double-blind randomized period (week 12), a higher proportion of patients (77.1%) in the osilodrostat arm were complete responders compared with the placebo arm (8.0%), with an odds ratio of 43.40 (95% CI, 7.10 to 343.20; P < 0.0001). Absolute between-group differences were not available.
Overall Response Rate
The proportion of patients who were overall responders in the osilodrostat arm compared to the placebo arm in the LINC 3 study was █████ versus █████, and in the LINC 4 study was █████ versus █████ at the end of the double-blind period. Between-group differences were not available and were not tested statistically.
Bone Mineral Density
The BMD outcomes were measured at study baseline and week 48 in both the LINC 3 and LINC 4 studies, as specified in the study protocols. In the LINC 3 study, data were available for the all-patients population (N = 137), while in the LINC 4 study, BMD assessments at week 48 were available for 49 patients with at least 1 BMD assessment.
In the LINC 3 study, among patients with available data (n = 81), the mean percentage change from baseline in the BMD of the L1 to L4 lumbar spine at week 48 was 3.0% (SD = 6.45), with a mean change in the actual value of 0 g/cm2 (SD = 0.05).
In the LINC 4 study, among patients with available data (n = 48), the mean percentage change from baseline in the BMD of the L1 to L4 lumbar spine at week 48 was 1.5% (SD = 3.99), with a mean change in the actual value of 0 g/cm2 (SD = 0.04).
Cardiovascular-Related Metabolic Parameters Associated With Cushing Disease
In the LINC 3 study at week 34 (end of randomized withdrawal period), the mean changes from study baseline in the following parameters were: hemoglobin A1C, –0.5% (SD = 0.64%) in the osilodrostat arm (n = 36) and –0.4% (SD = 0.51%) in the placebo withdrawal arm (n = 33); LDL cholesterol, –0.2 mmol/L (SD = 0.67 mmol/L; n = 35) in the osilodrostat arm versus –0.2 mmol/L (SD = 0.85 mmol/L; n = 32) in the placebo withdrawal arm; DBP, –6.5 mm Hg (SD = 11.38 mm Hg; n = 36) in the osilodrostat arm versus –5.1 mm Hg (SD = 10.73 mm Hg; n = 34) in the placebo withdrawal arm; and weight, –2.3 kg (SD = 4.81 kg; n = 36) in the osilodrostat arm versus –2.6 kg (SD = 8.42 kg; n = 34) in the placebo withdrawal arm. Between-group differences were not reported for the randomized withdrawal period. The adjusted mean changes from baseline to week 48 in the all-patients population ranged from 0.71% to –0.60% for hemoglobin A1C, 0.36 mmol/L to –0.45 mmol/L for LDL cholesterol, –0.98 mm Hg to –6.56 mm Hg for DBP, and 0.50 kg to –4.09 kg for weight.
In the LINC 4 study, during the placebo-controlled period, there were greater improvements in all cardiovascular-related metabolic parameters in the osilodrostat arm compared with the placebo arm, except for triglycerides. However, there was an overall improvement in triglyceride values during the core phase. The mean changes from study baseline in the following parameters were: hemoglobin A1C, –0.2% (SD = 0.44%) in the osilodrostat arm (n = 46) and 0% (SD = 0.27%) in the placebo withdrawal arm (n = 24); LDL cholesterol, –0.5 mmol/L (SD = 0.80 mmol/L; n = 44) in the osilodrostat arm versus 0.1 mmol/L (SD = 0.47 mmol/L; n = 23) in the placebo arm; standing DBP, –4.8 mm Hg (SD = 11.14 mm Hg; n = 44) in the osilodrostat arm versus –1.4 mm Hg (SD = 9.84 mm Hg; n = 24) in the placebo arm; and weight, –0.8 kg (SD = 3.09 kg; n = 46) in the osilodrostat arm versus –0.1 kg (SD = 2.12 kg; n = 24) in the placebo arm. Between-group differences were not available and were not tested statistically.
Liver Function Biochemical Parameters
In the LINC 3 study, most liver abnormal parameter results occurred during the dose-titration period of the trial. During the placebo-controlled randomized withdrawal period, 5 patients (13.9%; 5/36) in the osilodrostat arm versus no patients in the placebo withdrawal arm had an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level that was greater than the ULN but did not exceed 3 times the ULN. During the 48-week core study period of the LINC 3 study, among the 137 patients in the all-patients population, 50 patients (36.5%) had an increase in the ALT or AST level that was greater than the ULN but did not exceed 3 times the ULN. Specifically, 5 patients (3.6%) had ALT or AST increases exceeding 3 times the ULN, 3 patients (2.2%) had results that exceeded 5 times the ULN, 2 patients (1.5%) had results that exceeded 8 times the ULN, and 1 patient (0.7%) had a result that exceeded 10 times the ULN. In 1 patient with liver metastases, ALT and AST did not normalize. No patients in the LINC 3 study had an ALT or AST level greater than 20 times the ULN.
In the LINC 4 study, abnormal liver enzyme results were infrequent, mild, and reversed spontaneously. During the placebo-controlled period (12 weeks), 2 patients (4.2%; 2/48) in the osilodrostat arm versus no patients in the placebo arm had an increase in ALT of more than 3 times the ULN. During the overall 48-week core study period, a total of 3 patients (4.1%; 3/73) had an increase in ALT of more than 3 times the ULN. In the LINC 4 study, no patients had an ALT of 5 times or the ULN or greater, and no patients had an increase in AST of more than 3 times the ULN during the overall study period. No patient had concurrent increases in AST, ALT, total bilirubin, and/or alkaline phosphatase. No patient met the criteria for Hy’s Law.
No patients in the LINC 3 and LINC 4 studies discontinued the study drug due to abnormal liver biochemical parameter results.
Patient-Reported Outcomes
CushingQoL Total Score (Range, 0 [Worst] to 100 [Best])
In the LINC 3 study at week 34 (end of the randomized withdrawal period), the mean change from study baseline in the CushingQoL total score was ████ points (SD = █████) in the osilodrostat arm (n = 36) and ███ points (SD = █████) in the placebo withdrawal arm (n = 34). A between-group difference was not reported.
In the LINC 4 study at week 12, the mean change from baseline in CushingQoL total score was ████ points (95% CI, ████ ██ ████) in the osilodrostat arm (n = 46) and ████ points (95% CI, ████ ██ █████) in the placebo arm (n = 24). The adjusted mean difference between the 2 treatment arms was █████ points (95% CI, █████ ██ ████).
BDI-II Total Score (Range, 0 [Best] to 63 [Worst])
In the LINC 3 study at week 34 (end of the randomized withdrawal period), the mean change from study baseline in BDI-II total score was ████ points (SD = ████) in the osilodrostat arm (n = 36) and ████ points (SD = ████) in the placebo withdrawal arm (n = 34). A between-group difference was not reported.
In the LINC 4 study at week 12, the mean change from baseline in BDI-II total score was █████ points (95% CI, █████ ██ ████) in the osilodrostat arm (n = 46) and █████ points (95% CI, █████ ██ █████) in the placebo arm (n = 24). The adjusted mean difference between the 2 treatment arms in the BDI-II total score was ████ points (95% CI, ████ ██ ████), favouring placebo.
Harms
Key results include the following:
During the double-blind randomized withdrawal period in the LINC 3 study, AEs were reported in 26 patients (72.2%) in the osilodrostat arm and 23 patients (65.7%) in the placebo withdrawal arm. The most commonly reported AEs were nausea (11.1% for osilodrostat versus 0 for placebo withdrawal), anemia (8.3% versus 8.6%), arthralgia (8.3% versus 0), and headache (8.3% versus 0).48 In the LINC 4 study, during the placebo-controlled period of the first 12 weeks, AEs were reported in 46 patients (95.8%) in the osilodrostat arm and 23 patients (92.0%) in the placebo arm. The most commonly reported AEs were arthralgia (35.4% versus 8.0%), decreased appetite (37.5% versus 16.0%), and fatigue (25.0% versus 16.0%).
During the double-blind randomized withdrawal period in the LINC 3 study, SAEs of cholelithiasis and neutropenia were each reported by 1 patient in the osilodrostat arm, while increased blood corticotrophin was reported as an SAE by 1 patient in the placebo withdrawal arm.48 During the placebo-controlled period in the LINC 4 study, SAEs were reported in 3 patients, 2 in the osilodrostat arm and 1 in the placebo arm.
During the double-blind randomized withdrawal period in the LINC 3 study, no patients in the osilodrostat arm and 2 patients in the placebo withdrawal arm discontinued the study due to AEs. During the placebo-controlled period in the LINC 4 study, 1 patient in the osilodrostat arm and no patients in the placebo arm discontinued the study drug due to AEs.
In the LINC 3 study, 1 patient (who was randomized to the placebo arm during the double-blind randomized withdrawal period) died by suicide during the extension period.48 No deaths were reported during the study period in the LINC 4 study.50
Adverse events of special interest (AESIs) included the following: hypocortisolism-related AEs, reported in 51.1% of patients in the LINC 3 study and 27.4% in the LINC 4 study; adrenal hormone precursor accumulation–related AEs (42.3% in the LINC 3 study and 61.6% in the LINC 4 study); pituitary tumour enlargement–related AEs (2.2% and 5.5%, respectively); and arrhythmogenic potential and QT prolongation–related AEs (4.4% and 4.1%, respectively).
During the double-blind randomized period of the LINC 4 study, a higher proportion of patients in the osilodrostat arm reported adrenal hormone precursor accumulation–related AEs compared to the placebo arm (43.8% for osilodrostat versus 36.0% for placebo), as well as hypocortisolism-related AEs (14.6% versus 0).
Summary of Findings and Certainty of the Evidence
Literature-based minimal important difference (MID) estimates were used as the thresholds for the CushingQoL total score (10.1 points based on within-group data); within-group estimates were used in the absence of available between-group estimates. Refer to the summary of outcome measures in Appendix 3 of the Supplemental Material document. In the absence of a known threshold, the certainty in the presence of a nonnull effect was rated for complete response rate. For all other outcomes, a specific threshold for a clinically important effect could not be established. However, with input from the clinical experts, the review team assessed whether the point estimates and corresponding CI bounds represented clinically important effects. According to the GRADE guidance, noncomparative evidence starts at very low.
Table 4: Summary of Findings for Osilodrostat Versus Placebo Withdrawal in Adult Patients With Cushing Disease With a Complete Response at Week 24 (mUFC Equal to or Below the ULN) and No Uptitration Between Weeks 13 and 24 (LINC 3 Trial)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo withdrawal | Osilodrostat | Difference | |||||
mUFC responders | |||||||
Complete response rate (proportion of patients with mUFC ≤ ULN) Follow-up: 8 weeks (RW period from week 26 to week 34) | 70 (1 randomized withdrawal study) | OR = 13.71 (3.73 to 53.44) | 294 per 1,000 | 861 per 1,000 (705 to 953 per 1,000) | NR | Moderatea (serious imprecision) | Osilodrostat likely results in an increase in the complete response rate when compared with placebo withdrawal; the clinical importance of the increase is uncertain. |
Overall response rate (proportion of patients with mUFC ≤ ULN or ≥ 50% reduction from baseline) Follow-up: 8 weeks (RW period from week 26 to week 34) | 71 (1 randomized withdrawal study) | NR | ██ | ██ | ██ | Lowb (very serious imprecision) | Osilodrostat may result in an increase in the overall response rate when compared with placebo withdrawal; the clinical importance of the increase is uncertain. |
Patient-reported outcomes (HRQoL and depression) | |||||||
CushingQoL total score (0 [worst] to 100 [best]), change from baseline, points Follow-up: 34 weeks | 70 (1 randomized withdrawal study) | NR | ██ | ██ | ██ | Lowb (very serious imprecision) | Osilodrostat may result in little to no difference in CushingQoL total score when compared with placebo withdrawal. |
BDI-II total score (0 [best] to 63 [worst]), change from baseline, points Follow-up: 34 weeks | 70 (1 randomized withdrawal study) | NR | ██ | ██ | ██ | Lowb (very serious imprecision) | Osilodrostat may result in little to no difference in BDI-II total score when compared with placebo withdrawal. |
Treatment discontinuation | |||||||
Treatment discontinuation due to AEsc Follow-up: 8 weeks (RW period from week 26 to week 34) | 71 (1 randomized withdrawal study) | NR | 57 per 1,000 | 0 | NR | Lowb (very serious imprecision) | Osilodrostat may result in a decrease in discontinuation due to AEs when compared with placebo withdrawal; the clinical importance of the decrease is uncertain. |
AE = adverse event; BDI-II = Beck Depression Inventory-II; CI = confidence interval; CushingQoL = Cushing quality of life questionnaire; HRQoL = health-related quality of life; MID = minimal important difference; mUFC = mean urinary free cortisol; NR = not reported; OR = odds ratio; RW = randomized withdrawal; ULN = upper limit of normal.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The clinical experts consulted for this review commented that the patients in this study had relatively mild disease and might have a relatively low risk of experiencing negative patient-important outcomes.
aNo literature-based MID was available and, in consultation with the clinical experts, an MID estimate could not be determined; therefore, the null was used as the threshold for “any” effect. No absolute between-group difference or CI was available; therefore, the relative effect was appraised. The level of evidence was rated down 1 level for serious imprecision; the sample size and number of events are small, raising concern for prognostic imbalance and that the estimated magnitude of effect and its CI may be unstable. The large size of the effect was considered only once when rating down.
bNo literature-based MID was available and in consultation with the clinical experts, an MID estimate could not be determined; therefore, the null was used as the threshold for “any” effect. No absolute between-group difference or CI was available; therefore, the rating of imprecision required consideration of the sample size and/or number of events. The level of evidence was rated down 2 levels for very serious imprecision; the sample size and number of events are small, raising concern for prognostic imbalance and that the estimated magnitude of effect and its CI may be unstable.
cAt 48 weeks, a total of 18 patients (13.1%) discontinued the study due to an AE.
Sources: LINC 3 interim Clinical Study Report,48 LINC 3 final Clinical Study Report,49 and sponsor’s submission.60
Table 5: Summary of Findings for Osilodrostat Versus Placebo for Adult Patients With Cushing Disease With mUFC Greater Than 1.3 Times the ULN (LINC 4 Trial)
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Osilodrostat | Difference | |||||
mUFC responders | |||||||
Complete response rate (proportion of patients with mUFC ≤ ULN) Follow-up: 12 weeks | 73 (1 RCT) | OR = 43.4 (7.06 to 343.19) | 80 per 1,000 | 771 per 1,000 (627 to 880 per 1,000) | NR | Moderatea (serious imprecision) | Osilodrostat likely results in an increase in complete response rate when compared with placebo. The clinical importance of the increase is uncertain. |
Overall response rate (proportion of patients with mUFC ≤ ULN or ≥ 50% reduction from baseline) Follow-up: 12 weeks | 73 (1 RCT) | NR | ██ | ██ | ██ | Moderateb (serious imprecision) | Osilodrostat likely results in an increase in overall response rate when compared with placebo. The clinical importance of the increase is uncertain. |
Patient-reported outcomes (HRQoL, and depression) | |||||||
CushingQoL total score (0 [worst] to 100 [best]), change from baseline, points Follow-up: 12 weeks | 70 (1 RCT) | NR | ██ | ██ | ██ | Lowc (very serious imprecision) | Osilodrostat may result in little to no difference in CushingQoL total score when compared with placebo. |
BDI-II total score (0 [best] to 63 [worst]), change from baseline, points Follow-up: 12 weeks | 70 (1 RCT) | NR | ██ | ██ | ██ | Moderated,e (serious imprecision) | Osilodrostat likely results in a smaller improvement in BDI-II total score compared with placebo. The clinical importance of the difference is uncertain. |
Treatment discontinuation | |||||||
Treatment discontinuation due to AEs Follow-up: 12 weeks | 74 (1 RCT) | NR | 0 | 21 per 1,000 (NR) | NR | Lowa,f (very serious imprecision) | Osilodrostat may result in little to no difference in treatment discontinuation due to AEs when compared with placebo. |
AE = adverse event; BDI-II = Beck Depression Inventory-II; CI = confidence interval; CushingQoL = Cushing quality of life questionnaire; HRQoL = health-related quality of life; MID = minimal important difference; mUFC = mean urinary free cortisol; NA = not applicable; NR = not reported; OR = odds ratio; RCT = randomized controlled trial; ULN = upper limit of normal.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. The clinical experts consulted for this review commented that the patients in this study had relatively mild disease and might have a relatively low risk of experiencing negative patient-important outcomes.
aNo literature-based MID was available and, in consultation with the clinical experts, an MID estimate could not be determined; therefore, the null was used as the threshold for “any” effect. No absolute between-group difference or CI was available; therefore, the relative effect was appraised. The level of evidence was rated down 1 level for serious imprecision; the sample size and number of events were small, raising concern for prognostic imbalance and that the estimated magnitude of effect and its CI may be unstable. The large size of the effect was considered only once when rating down.
bNo literature-based MID was available and, in consultation with the clinical experts, an MID estimate could not be determined; therefore, the null was used as the threshold for “any” effect. No absolute between-group difference or CI was available; therefore, rating of imprecision required consideration of the sample size and/or number of events. The level of evidence was rated down 1 level for serious imprecision; the sample size and number of events were small. The large size of the effect was considered only once when rating down.
cRated down 2 levels for very serious imprecision. Only a within-group literature-based MID was available; therefore, the null was used to assess certainty. The point estimate and the lower bound of the 95% CI suggested a decrease, while the upper bound of the 95% CI suggested an increase compared to placebo.
dNot controlled for multiple comparisons; there is an increased risk of type I error.
eRated down 1 level for serious imprecision. No MID was available; therefore, the null was used to assess certainty. The point estimate and the lower bound of the 95% CI suggested little to no differences, while the upper bound of the 95% CI suggested a smaller decrease from baseline compared to placebo.
fNo literature-based MID was available and, in consultation with the clinical experts, an MID estimate could not be determined; therefore, the null was used as the threshold for “any” effect. No absolute between-group difference or CI was available; therefore, rating of imprecision required consideration of the sample size and/or number of events. The level of evidence was rated down 2 levels for very serious imprecision; the sample size and number of events were small, raising concern for prognostic imbalance and that the estimated magnitude of effect and its CI may be unstable.
Sources: LINC 4 primary analysis Clinical Study Report50 and sponsor’s submission.60
Long-Term Extension Studies
Description of Studies
The LINC 3 trial extension was a single-arm open-label LTE of the LINC 3 study. Patients who continued to receive clinical benefit, as assessed by the study investigator, and who wished to enter the extension period had to reconsent at week 48 after study entry. Patients entered the extension period without any interruption of the study drug or assessments. The optional extension period ended 16 months after all patients completed week 72 unless the patient had discontinued the treatment earlier. At the end of the study, patients who continued to benefit from treatment were offered to participate in a separate long-term safety follow-up study. Details and results of this separate long-term safety follow-up study were not provided.
The LINC 4 trial extension was a single-arm open-label LTE of the LINC 4 study. At week 48 after randomization, patients had the option to enter an optional open-label extension phase. Patients who benefited from study treatment had the option to enter a separate long-term safety follow-up study or stop study treatment. Patients who did not enter the long-term safety follow-up study discontinued osilodrostat and concluded the study with a posttreatment (end of study) visit after 30 days off the study drug. During the optional extension phase, the dose of osilodrostat was maintained at the established effective dose unless a change was required based on the mUFC results collected at week 48 and, if applicable, at weeks 60, 72, and 84.
Patient Disposition
Patient disposition for each included study is summarized in the Supplemental Material document, Appendix 5.
The LINC 3 trial enrolled 137 patients. During the core period, 24 patients (17.5%) discontinued treatment and 7 patients (5.1%) did not enter the extension period. Of the 106 patients (77.4%) who entered the extension period, 72 (52.6%) completed it, while 34 (24.8%) discontinued. The most common reasons for discontinuation during the extension period were AEs, and patient or guardian decision (each 8.8%) followed by physician decision (3.6%). Other reasons included death (1.5%), withdrawal of consent (1.5%), and unsatisfactory therapeutic effect (0.7%).
In the LINC 4 extension phase, 65 (89.0%) of the 73 treated patients completed the core phase (week 48), and 60 (82.2%) entered the optional extension phase (week 48 and beyond). A total of 53 patients (72.6% of 73 patients) completed the extension phase, while 7 patients (9.6%) discontinued early, 6 (8.2%) due to AEs, and 1 (1.4%) due to physician decision.
Exposure to Study Treatments
In the LINC 3 trial, from study initiation to the data cut-off date, patients received a mean daily dose of 10.0 mg (SD = 8.5 mg). The mean dose administered for the longest duration was 10.6 mg/day (SD = 11.2 mg/day), and the mean highest dose was 18.4 mg/day (SD = 14.0 mg/day). The mean treatment duration was 80.3 weeks (SD = 44.0 weeks), with some patients receiving osilodrostat for more than 156 weeks. A total of 121 patients were treated for more than 24 weeks, and 105 patients were exposed to osilodrostat for at least 48 weeks.
In the LINC 4 trial, during the open-label treatment phase, the average osilodrostat dose was generally 4 mg/day or lower. From randomization to the data cut-off date, the mean daily dose was 8.7 mg (SD = 9.3 mg). Of the 73 patients, a starting dose of 4 mg/day resulted in an mUFC equal to or below the ULN (complete response) in 42.5% of patients, and represented the longest administered dose in 26.0% of patients. The most frequently recorded highest dose was 10 mg/day, observed in 37.0% of patients. Across the overall treatment period, the median exposure was 70.0 weeks (range, 2.0 to 112.7 weeks), with 80.8% of patients receiving osilodrostat for more than 46 weeks.
The median total daily dose of osilodrostat was 7.4 mg/day and 4.6 mg/day in the LINC 3 and LINC 4 studies, respectively.55
The use of concomitant medications during the open-label extension phases is not reported separately because this information is already presented in the randomized phase section.
Critical Appraisal
Internal Validity
Both the LINC 3 and LINC 4 extension studies were limited by their open-label and noncomparative design. Because there is no comparator, it is not possible to draw causal inferences about the longer-term effect of osilodrostat. The open-label design risks introducing bias in the collection of subjective outcomes (e.g., some harms). There were large reductions in sample size over time resulting in risk of bias due to missing outcome data.
External Validity
Both the LINC 3 and LINC 4 studies included rollover patients whose characteristics remained consistent with those at entry into the core study. It is reasonable to expect that similar limitations to generalizability of the study results are relevant to the open-label long-term safety extension phases of the LINC 3 and LINC 4 studies. The LTE of the LINC 3 study included all patients with continued clinical benefit, not only the enriched population of patients from the randomized withdrawal phase. The patient populations in both studies may have become more selective over time if the studies were preferentially including patients who were deriving benefit from the treatment without intolerable AEs. Information on patient-important outcomes such as HRQoL and depression was unavailable in the LTE studies.
Results
Efficacy
Detailed results for outcomes relevant to this review are in Appendix 5 in the Supplemental Material document. Key results include the following:
LINC 3 Extension Study
At the end of the core period (week 48), 75.9% of patients (104/137; 95% CI, 67.9% to 82.8%) met the predefined response criteria. Among these responders, 66.4% were complete responders and 9.5% were partial responders.
By week 72, of the 106 patients who entered the extension period, 88.7% (95% CI, 81.1% to 94.0%) met the response criteria, with 81.1% (86/106) classified as complete responders and 7.5% (8/106) as partial responders.
LINC 4 Extension Study
At the end of the core period (week 48), the overall response rate among all patients was 79.5% (58/73), with 68.5% (50/73) classified as complete responders and 11.0% (8/73) as partial responders.
At week 72, in the all-patients population, 65 patients had data available for the response outcome, including those who discontinued during the core phase. Among these patients, the overall response rate was 69.2% (45/65), with 61.5% (40/65) classified as complete responders and 7.7% (5/65) as partial responders.
At the end of the extension phase, 58 patients were evaluable for overall response, based on the longest available data for each individual. Among these patients, the overall response rate was 81.0% (47/58), with 72.4% (42/58) classified as complete responders and 8.6% (5/58) as partial responders.
Harms
Detailed results for harms are presented in Appendix 5 in the Supplemental Material document. Key results include the following:
All patients in the LINC 3 trial (100%) and almost all patients in the LINC 4 trial (98.6% of the 73 patients) reported at least 1 AE. The most frequently observed AEs (≥ 20%) included nausea (45.3% of patients in the LINC 3 study and 37.0% in the LINC 4 study), headache (36.5% and 34.2%), fatigue (32.8% and 39.7%), adrenal insufficiency (29.2% and 26.0%), and arthralgia (21.2% and 45.2%). Additional AEs reported among 20% or more patients in the LINC 3 study included vomiting (24.8%), nasopharyngitis (24.1%), back pain (21.2%), increased blood corticotrophin (20.4%), and glucocorticoid deficiency (20.4%). In the LINC 4 study, other frequently reported AEs were decreased appetite (46.6%), dizziness (30.1%), increased blood testosterone (24.7%), myalgia (24.7%), asthenia (23.3%), diarrhea (23.3%), hypertension (21.9%), and upper respiratory tract infection (21.9%).
Grade 3 or 4 AEs were reported in 60.6% of patients in the LINC 3 study and 38.4% of patients in the LINC 4 study. The most commonly reported grade 3 or 4 AE (≥ 5%) in both the LINC 3 and LINC 4 studies was hypertension, occurring in 11.7% and 12.3% of patients, respectively. In addition, myalgia was reported as a grade 3 or 4 event in 6.8% of patients in the LINC 4 study.
In the LINC 3 study, 55 patients (40.1%) experienced at least 1 SAE. The most commonly reported SAEs (≥ 2%) were adrenal insufficiency (5.8%); pituitary tumour (4.4%); adrenocortical insufficiency and gastroenteritis (2.9% each); and abdominal pain, headache, influenza, benign pituitary tumour, and sixth cranial nerve paralysis (2.2% each). All other SAEs occurred in no more than 2 patients. In the LINC 4 study, 10 patients (13.7%) experienced at least 1 SAE, with adrenal insufficiency being the only commonly reported SAE (≥ 2%), occurring in 4.1% of patients.
AEs leading to study drug discontinuation occurred in fewer than 20% of patients across both studies. In the LINC 3 study, 25 patients (18.2%) discontinued treatment due to an AE. The most commonly reported AEs leading to discontinuation were adrenal insufficiency, pituitary tumour, and benign pituitary tumour, each affecting 3.6% of patients. All other AEs leading to discontinuation occurred in no more than 2 patients. In the LINC 4 study, 9 patients (12.3%) discontinued treatment due to an AE. Adrenal insufficiency was the most frequent cause, reported in 4.1% of patients, while all other AEs leading to discontinuation were reported in a single patient each.
No additional deaths were reported during the open-label extension phases in the LINC 3 and LINC 4 studies.
AESIs included hypocortisolism-related AEs (54.0% of patients in the LINC 3 study and 28.8% in the LINC 4 study), adrenal hormone precursor accumulation–related AEs (58.4% and 61.6%, respectively), pituitary tumour enlargement–related AEs (16.1% and 5.5%, respectively), and arrhythmogenic potential and QT prolongation–related AEs (4.4% and 4.1%, respectively).
Indirect Evidence
Direct comparative evidence between osilodrostat and placebo is available from the LINC 3 and LINC 4 studies. However, there is a gap in evidence comparing osilodrostat with ketoconazole or with ketoconazole plus cabergoline. Indirect comparisons were required to address this gap.
Description of Indirect Comparison
The objective of this section is to summarize and critically appraise the available (sponsor-submitted) indirect evidence82,83 comparing osilodrostat with other relevant treatments (i.e., ketoconazole and ketoconazole plus cabergoline) in the treatment of patients with endogenous Cushing syndrome in settings in Canada. On July 3, 2025, Health Canada approved a slightly narrower indication for the treatment of adult patients with Cushing disease with persistent or recurrent hypercortisolism following primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option. This is more specific than the ITC evidence originally submitted in 2023, which was based on the broader population of patients with endogenous Cushing syndrome. The sponsor did not submit any new or updated ITC evidence reflecting the revised indication.
For the purpose of this review, the sponsor submitted an unadjusted (naive) indirect comparison to estimate the relative efficacy and safety of osilodrostat (as observed in the LINC 4 study) compared with ketoconazole (based on aggregate data from 4 published studies).84-87
Study Selection and Review Methods
The sponsor aimed to estimate the comparative efficacy and safety of osilodrostat versus the comparators the sponsor deemed relevant, including ketoconazole and ketoconazole plus cabergoline.
For the sponsor-submitted ITC, 2 primary sources of evidence were used: patient-level data from clinical trials of osilodrostat (LINC 4 study)54 and a systematic literature review82 (details provided in Appendix 6 in the Supplemental Material document). The systematic review included studies involving adults (aged 18 years or older) with endogenous Cushing syndrome, regardless of prior treatment. Eligible interventions included specific pharmacological treatments such as osilodrostat, pasireotide (subcutaneous and long-acting release types), ketoconazole, metyrapone, mitotane, etomidate, cabergoline, and mifepristone. Nonpharmacological treatments like surgery, radiotherapy, and adjuvants were excluded. Comparators could include placebo, best supportive care, or any pharmacological treatment. Outcomes of interest focused on clinical effectiveness and safety. Accepted study designs included randomized and nonrandomized trials, single-arm and cohort studies, long-term follow-ups, and systematic reviews. No restrictions were placed on language or publication date.
The sponsor indicated that a full ITC comparing osilodrostat with other existing therapies was considered not feasible due to limited data, such as small sample sizes in the comparator studies, missing patient characteristics information, and minimal overlap between available patient populations across comparator studies. However, a formal feasibility assessment report was not made available to CDA-AMC. The sponsor’s ITC report included an unadjusted (naive) indirect comparison conducted for the comparison of osilodrostat versus ketoconazole. Both the sponsor and the clinical experts consulted for this review noted that the addition of a tumour-targeting drug to a steroidogenesis inhibitor (i.e., ketoconazole plus) is commonly used in practice when patients experience an inadequate response to ketoconazole monotherapy.
ITC Analysis Methods
For additional information on the analysis methods of the sponsor-submitted ITC, refer to Appendix 6 in the Supplemental Material document.
Because time-to-event data (e.g., time to complete response and time to treatment failure post response, including discontinuation due to lack of efficacy, discontinuation due to AEs, and treatment escape) were not available, the outcomes assessed in the naive comparison were complete response (defined as mUFC ≤ 1.0 times the ULN), treatment escape, discontinuation due to any cause, discontinuation due to lack of efficacy, discontinuation due to AEs, and discontinuation due to lack of efficacy or AEs.
Odds ratios were derived to quantify the association between treatment and outcome.88 Hazard ratios (HRs) for osilodrostat versus ketoconazole were estimated using event proportions from comparator studies, following the method described by Woods et al.89 The number of events at a specified time was assumed to follow a binomial distribution. Log-hazards were estimated for each treatment, and the difference between them provided the log-HR, which was exponentiated to obtain the HR. The variance 𝑉 of the log-HR was calculated using the approach outlined by Tierney et al.90
Summary of Included Studies
Of the 46 studies identified in the sponsor’s systematic review, 5 studies54,84-87 were selected for the naive comparison comparing osilodrostat with ketoconazole. Patient-level data from the LINC 4 study54 were used to derive outcome results equivalent to those aggregate results available from ketoconazole publications.84-87 Characteristics for the LINC 4 study were described earlier in the pivotal studies section. The ketoconazole studies included 1 nonrandomized trial in Cushing disease (Correa-Silva et al. [2009]; N = 8) and 3 observational studies: Castinetti et al. (2014) (Cushing disease; N = 200), Young et al. (2018) (mixed Cushing syndrome; N = 191), and Castinetti et al. (2008) (Cushing disease; N = 38). The ketoconazole studies were not blinded. Study duration was not reported in most ketoconazole studies, with the exception of Correa-Silva et al. (2009), which had a 24-week treatment period.
Patient characteristics for the included studies are summarized in Appendix 6 of the Supplemental Material document. Characteristics for patients in the LINC 4 study were described earlier in the pivotal studies section. In the ketoconazole studies, patient profiles varied and, often, the characteristics of interest were unreported in the included studies; mean age ranged from 34 to 51 years, when reported, and the proportion of male patients ranged from 0% to 25%. Pituitary adenomas were present in 49% to 100% of patients, and prior surgery was reported in 15% to 72% of cases. Most patients had persistent or recurrent disease, although 1 study (Young et al. [2018]) reported that 85% of cases were de novo. In the only study reporting baseline mUFC (Correa-Silva et al. [2009]), the levels were higher than those observed in the osilodrostat group (614 nmol per 24 hours versus 421 nmol per 24 hours).
Critical Appraisal of ITC
According to the sponsor, eligible studies for the naive indirect comparison were identified using a systematic literature review. There was no evidence of a predefined protocol for the review nor a statistical analysis plan for the between-study comparisons. There is, therefore, an increased risk that the reported results were selected from multiple analyses of the data, based on a favourable direction, magnitude, or statistical significance of the effect estimates. Details of the literature search were not reported; therefore, it is not known whether it was comprehensive or up to date, introducing a risk that relevant studies were missed. The methods used to conduct the systematic literature review (screening, data extraction) were also unreported. Therefore, the possibility of bias and error in the review process cannot be excluded. The risk of bias of the included studies does not appear to have been assessed and was not reported; therefore, it is possible that bias within the studies influenced the indirect comparisons.
The sponsor provided no formal feasibility assessment to determine whether conducting a conventional ITC (e.g., network meta-analysis or matching-adjusted indirect comparison) was feasible for the comparison of osilodrostat versus ketoconazole. However, the sponsor rationalized conducting naive indirect comparisons due to the small sample sizes of the comparator studies, the limited availability of data on patient characteristics, and the limited overlap in patient populations. Naive indirect comparisons treat the results of treatment groups from different studies as though they came from a single trial, without any adjustment for differences in population (e.g., prognostic and treatment effect–modifying variables), design and methods (e.g., outcome definitions, length of follow-up), setting, and other characteristics that may impact the treatment effect. As a result, such comparisons are at critical risk of bias due to confounding and are prone to producing results that are misleading.
As noted in the sponsor’s ITC report, there were major differences across the 5 studies selected for the naive comparison in terms of the study design (e.g., RCT versus non-RCT versus observational study), baseline characteristics and outcome assessments, assessment time point, and definition of the outcomes (such as that for complete response). More specifically, the LINC 4 study was an RCT, whereas Correa-Silva et al. (2009) was a nonrandomized trial, and the 3 studies by Castinetti et al. (2014), Young et al. (2018), and Castinetti et al. (2008) were observational studies. The level of oversight in a clinical trial differs substantially from observational studies, resulting in differences in attributes such as the frequency of monitoring, timing of outcome collection, intervention adherence, the use of concomitant treatments, discontinuations from the study, and data quality. Only 8 patients were included in the study by Correa-Silva et al. (2009). The demographic (age, sex, race) and disease-related characteristics (e.g., baseline mUFC, time since diagnosis, previous treatments, Cushing disease status) varied from study to study and often were not reported. In addition, Young et al. (2018) enrolled patients with Cushing syndrome, which included both individuals with Cushing disease and those with non–Cushing disease etiologies. In contrast, the other 4 studies included only patients with Cushing disease, specifically those with pituitary adenomas. Therefore, the patient population in Young et al. (2018) was broader than the Health Canada–approved indication, which is restricted to adult patients with Cushing disease who have persistent or recurrent hypercortisolism following pituitary surgery and/or irradiation, or for whom surgery is not an option. Because the comparison study included patients outside this approved population, it is not directly comparable to the LINC 4 study. While approximately 65% of the 108 patients analyzed in Young et al. (2018)87 had Cushing disease, which reflects the typical prevalence of Cushing disease within the broader Cushing syndrome population, the inclusion of patients who do not have Cushing disease limits the generalizability of the findings to the approved indication. Finally, some results were presented for the entire duration of the contributing studies, the lengths of which may have varied.
The sponsor presented HRs that were derived using binary data reported in the individual studies using the Woods et al. method.91 This method assumes that the data from each study are complete (i.e., no censoring) and that the hazards are constant (i.e., exponential distribution) and proportional over time. This is a strong assumption that cannot be verified using the observed data. As a result, there is a risk that the presented HRs are biased, although the direction and extent cannot be predicted. No absolute between-group differences were reported. Owing to small population sizes and limited events across the included studies, many effect estimates are affected by important imprecision (i.e., wide CIs).
Finally, patient-important outcomes such as HRQoL and depression were not assessed in the naive comparison, limiting the ability to evaluate the broader patient-centred impact of the treatments. The time points presented were relatively short, and the long-term differences between treatments were not assessed. No ITC was provided by the sponsor to compare osilodrostat versus ketoconazole plus cabergoline; therefore, the relative efficacy and safety between these regimens could not be assessed, limiting the ability to draw conclusions regarding their comparative clinical value.
Efficacy and Harms Results of ITC
Key results of the ITCs are presented in Table 6, with additional details provided in Appendix 6 in the Supplemental Material document.
Between the comparison of osilodrostat and ketoconazole:
The results for complete response are inconclusive, with the treatment being ████████ varying by time point, and the odds ratio at ██████ being affected by serious imprecision. Osilodrostat was ████████ ███ █████████ ██████ ███ █████████ ███████████████ ██ █ ██████.
In analyses of various reasons for discontinuation, point estimates ██████ ████████████ but the CIs are often wide ███ ████████ █████ ██████████ ████ ██████ █████████ █████ ██ ████████.
No HRQoL was assessed in the naive comparison.
No ITC was provided by the sponsor to compare osilodrostat versus ketoconazole plus cabergoline combination therapy.
Table 6: Summary of Results of the Naive Comparison (Osilodrostat Versus Ketoconazole)
Outcome | Time point | Osilodrostat | Ketoconazole | Osilodrostat vs. ketoconazole | |||
|---|---|---|---|---|---|---|---|
Study | Patients with CR, n/N (%) | Study | Patients with CR, n/N (%) | OR (95% CI) | HR (95% CI) | ||
Complete responsea | 3 months | LINC 4 | 37/48 (77%) | Castinetti (2008) | 78/200 (39%) | ███ | ███ |
24.8 months | LINC 4 | 4/48 (8%) | Castinetti (2014) | 78/200 (39%) | ███ | ███ | |
6 months | LINC 4 | 37/48 (77%) | Correa-Silva (2009) | 6/8 (75%) | ███ | ███ | |
Treatment escape (trial defined)b | 6 months | LINC 4 | 0/45 (0%) | Correa-Silva (2009) | 1/5 (20%) | ███ | ███ |
Treatment discontinuationb | 6 months | LINC 4 | 4/48 (8%) | Young (2018) | 39/108 (36%) | ███ | ███ |
During studyc | LINC 4 | 11/48 (23%) | Castinetti (2014) | 118/160 (74%) | ███ | ███ | |
Discontinuation due to lack of efficacy (including physician’s decision)b | 6 months | LINC 4 | 0/48 (0%) | Young (2018) | 4/108 (4%) | ███ | ███ |
During studyc | LINC 4 | 1/48 (2%) | Castinetti (2014) | 43/160 (27%) | ███ | ███ | |
Discontinuation due to any AEb | 6 months | LINC 4 | 1/48 (2%) | Young (2018) | 7/108 (6%) | ███ | ███ |
During studyc | LINC 4 | 6/48 (13%) | Castinetti (2014) | 41/160 (26%) | ███ | ███ | |
Discontinuation due to lack of efficacy or any AE (including physician’s decision)b | 24 weeks | LINC 4 | 1/48 (2%) | Young (2018) | 11/108 (10%) | ███ | ███ |
During studyc | LINC 4 | 7/48 (15%) | Castinetti (2014) | 84/160 (53%) | ███ | ███ | |
AE = adverse event; CI = confidence interval; CR = complete response; HR = hazard ratio; NR = not reported; OR = odds ratio; vs. = versus.
aAn OR greater than 1 provides evidence of improved outcomes for osilodrostat compared with ketoconazole.
bAn OR less than 1 provides evidence of improved outcomes for osilodrostat compared with ketoconazole.
cThe specific time point was not reported. Discontinuation during whole study has been used.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were included in the review.
Discussion
This report summarizes the evidence for osilodrostat for the treatment of adults with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option, based on 2 phase III studies and their LTE phases and 1 ITC.
Efficacy
The LINC 3 and LINC 4 studies both showed that osilodrostat likely reduced mUFC compared to placebo at the end of the studies’ respective placebo-controlled periods. However, it is important to note that the 2 studies addressed this research question but involved distinct patient populations. The LINC 4 study evaluated the efficacy of osilodrostat versus placebo from baseline, whereas the LINC 3 study assessed the impact of continued treatment with osilodrostat following initial response compared to treatment withdrawal. Although the patient populations in both studies are comparable, they represent different stages of treatment and clinical decision-making, which should be considered when interpreting the results. Patients in the osilodrostat arm were more likely to achieve a complete response, defined as an mUFC equal to or below the ULN, as well as an overall response, which included either an mUFC equal to or below the ULN or a reduction of 50% or more from baseline. The sponsor selected mUFC normalization as the primary end point for the LINC 3 and LINC 4 studies, justifying this surrogate outcome based on its alignment with the therapeutic goals outlined in clinical guidelines and its association with improved metabolic parameters in studies of other therapies for Cushing disease.24-28,31-33,60-74 However, the clinical experts consulted for this review noted that mUFC response is a surrogate and not a validated predictor of the final outcomes that are important to patients, such as physical function, performance status, cardiovascular and metabolic morbidities, and mortality. With regard to the metabolic parameters associated with Cushing disease, such as hemoglobin A1C, LDL cholesterol, DBP, and weight, patients who received osilodrostat may have experienced slightly greater improvements compared to those who received placebo during the randomized or randomized withdrawal period. However, no between-group comparisons with CIs were reported, and no statistical testing was conducted, which precludes any definitive conclusions regarding these outcomes. Furthermore, these measures are surrogate end points, and it remains unclear how changes in these parameters would translate into patient-important clinical outcomes. The clinical experts commented that the extent to which osilodrostat can reduce the cortisol level and its related complications in different forms and severities of Cushing disease is unknown.
For patient-reported outcomes, evidence from the LINC 3 and LINC 4 studies suggests that osilodrostat may result in little to no difference in improvement from baseline in CushingQoL total score when compared with placebo or placebo withdrawal. While LINC 3 showed little to no different in depression as measured by the BDI-II total score, patients in the placebo arm of the LINC 4 study may have experienced a greater improvement (change from baseline) in BDI-II score compared to those in the osilodrostat arm at 12 weeks. However, it is unclear whether this observed difference is clinically meaningful, and the comparison was not controlled for within the multiple testing structure.
According to the clinical experts, the patient populations enrolled in the LINC 3 and LINC 4 studies generally reflected the demographic diversity of patients with Cushing disease seen in clinical settings in Canada. However, during the randomized withdrawal phase of the LINC 3 study, an enriched patient population (responders that did not require dose uptitration between week 13 and 24) were included. This design excluded patients with disease that was more difficult to control, which may limit the generalizability of the findings in the LINC 3 study to routine clinical practice.
Long-term efficacy data from the extension phase showed results that aligned with those observed in the core phase for both the LINC 3 and LINC 4 studies. However, these findings should be interpreted with caution due to limitations, including the single-arm design of the extension phase and the presence of missing outcome data, mainly due to patients being lost to follow-up.
The clinical experts noted key limitations in the evidence from the LINC 4 and LINC 3 studies, notably the use of mUFC as a surrogate outcome and a lack of patient-important final clinical outcomes. Furthermore, the enrolled patients in the LINC 3 and LINC 4 studies were at relatively low risk, raising uncertainty about the generalizability of the findings to patients with high risk who may have a greater need for treatment. The ability of mUFC to predict a clinically important treatment effect on long-term clinical outcomes that are important for patients and clinicians, such as morbidity and mortality, has not been established for osilodrostat. The lack of these clinical outcomes is a notable limitation.
The sponsor submitted naive indirect comparisons (i.e., unadjusted ITCs) comparing osilodrostat with ketoconazole based on osilodrostat data from the LINC 4 trial and ketoconazole data from 4 published studies. However, due to the significant methodological limitations of the naive comparison (such as the lack of adjustment for the prognostic factors or treatment effect modifiers such as baseline age, sex, baseline mUFC, prior surgery, prior medication, prior irradiation, Cushing disease status, time since diagnosis and the different follow-up times across the studies), its results are very uncertain. The lack of consideration of confounding and effect modification introduces a critical risk of bias; therefore, the results may be misleading (i.e., the reported effects are likely to systematically over- or underestimate the true effect). Therefore, a conclusion cannot be drawn from the naive comparison on the comparative efficacy and safety profile comparing osilodrostat versus ketoconazole in the treatment of endogenous Cushing syndrome in adults. No HRQoL was assessed in the naive comparison. No ITC was provided by the sponsor to compare osilodrostat versus ketoconazole plus cabergoline.
Harms
According to the clinical experts consulted for this review, there would be no major concerns about the safety profile of osilodrostat based on the harms outcomes reported in both the LINC 3 and LINC 4 studies. The clinical experts pointed out that liver dysfunction is common among patients with Cushing disease and can indicate patient tolerance to a treatment. Furthermore, liver function parameters can reflect disease–treatment interactions. Animal toxicity studies revealed the liver as 1 of the primary target organs of osilodrostat toxicity, and the observed liver dysfunction included hepatocellular hypertrophy (reversible), cytosolic vacuolation (partially reversible), and transient changes in ALT and/or AST.92 It is noted that patients with liver disease may need a lower initial dosage of osilodrostat (i.e., 1 mg twice daily).92 During the core phase of both the LINC 3 and LINC 4 studies (48 weeks), the elevation of ALT or AST in patients appeared to be infrequent. No patients discontinued the study drug due to abnormal liver biochemical parameter results. One possible explanation for this is that both clinical trials recruited patients with a lower risk of developing complications associated with Cushing disease, thereby representing a cohort with a comparatively milder manifestation of the condition. The clinical experts emphasized that, in practice, clinicians should closely monitor the liver function parameters during treatment of patients with Cushing disease.
No new safety signals were detected from the longer-term data.
Ethics and Equity Considerations
The LINC 3 and LINC 4 studies were conducted across Asia, Europe, North America, and South America. A total of 14 patients from Canada were enrolled in the 2 trials: 11 in the LINC 3 study (including 4 during the randomized withdrawal period) and 3 in the LINC 4 study. There was no available information indicating whether any of these individuals were identified as belonging to an Indigenous population in Canada. While study findings may be broadly generalizable, the absence of specific outcomes for diverse groups presents limitations for clinical and health system decision-making, raising important ethical considerations around equitable access and representation.
The clinician group providing input and the clinical experts consulted for this review noted that the overarching goal of treatment is to achieve disease remission, thereby alleviating the burden faced by patients and their caregivers. Qualitative insights that were provided through patient group input from the Canadian Organization for Rare Disorders highlight the lived experiences of 3 individuals with experience using osilodrostat. Two respondents, with 3 and 5 years of treatment experience with osilodrostat through clinical trials, reported meaningful and sustained improvements in blood pressure, weight, glucose control, and mood, along with manageable side effects such as occasional nausea, headache, and fatigue. One participant had recently initiated treatment. No patients from Canada were represented among those with experience using osilodrostat in the patient group input. This absence may be related to factors such as the limited availability of trial centres in Canada. This highlights the importance of inclusive and representative patient engagement strategies.
The clinical experts noted that diagnosing and managing Cushing disease requires specialist care because the condition is rare and typically outside the scope of general practice. While prevalence does not disproportionately affect marginalized populations, patients in remote or nonurban areas may face challenges accessing timely care due to limited specialist availability.
Patient input emphasized a strong preference for treatments that reduce cortisol with lasting benefits and minimal side effects. Respondents favoured oral therapies over surgery or radiation, citing convenience, affordability, and the ability to resume normal daily activities. The oral administration of osilodrostat may help reduce geographic barriers and lessen treatment burden. However, access challenges may persist due to the need for ongoing monitoring and broader systemic factors beyond geography.
Conclusion
Two phase III trials compared osilodrostat with placebo in adults with Cushing disease: LINC 3 (a randomized withdrawal study with patients who had a complete response and no uptitration in the preceding weeks), and LINC 4 (an RCT). Osilodrostat likely increases complete and overall response rates compared with placebo; however, the validity of mUFC as a surrogate end point to reliably predict clinically important outcomes remains uncertain, and the impact of mUFC on patient-important outcomes such as morbidity and mortality has yet to be demonstrated. When compared with placebo, the evidence is uncertain regarding the effect of osilodrostat on BMD (L1 to L4 lumbar spine), hemoglobin A1C, LDL cholesterol, DBP, weight, and treatment discontinuation due to AEs. These outcomes were not statistically compared between groups, and their relevance to long-term patient-important outcomes remains unclear. Improvements in patient-reported outcomes (CushingQoL and BDI-II scores) were minimal or inconsistent, and no statistical testing was conducted for between-group differences, limiting interpretability. Efficacy in the longer-term single-arm extension phases of the LINC 3 and LINC 4 studies was primarily assessed through overall mUFC response rates, which suggest that osilodrostat remains effective in maintaining biochemical control. However, these data are subject to a high risk of bias because of missing data and lack of ability to infer causality due to the uncontrolled design. No new safety signals were identified during the longer-term single-arm extension phases of the LINC 3 and LINC 4 studies. The clinical experts consulted for this review perceived the severity of illness in the trial populations to be lower than what is typically seen in patients most in need of treatment in real-world clinical settings. This introduces uncertainty regarding the generalizability of the study findings to patients at higher risk of complications from Cushing disease. Osilodrostat is an orally administered therapy that offers a convenient option for self-management, potentially reducing nonclinical treatment burden for patients and caregivers because many patients appear to prefer treatments that are easy to take and do not require hospital-based administration.
Due to critical risk of bias, a conclusion cannot be drawn from the naive indirect comparison of the efficacy and safety profile comparing osilodrostat versus ketoconazole in the treatment of Cushing disease in adults. No ITC was provided by the sponsor to compare osilodrostat versus ketoconazole plus cabergoline.
References
1.Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). 1932. Obes Res. 1994;2(5):486-508. doi:10.1002/j.1550-8528.1994.tb00097.x PubMed
2.Chabre O, Cristante J. Treatment of cushing’s syndrome: What place for medical treatment? Acta Endocrinol (Buchar). 2019;15(2):237-243. doi:10.4183/aeb.2019.237 PubMed
3.Aghi MK. Management of recurrent and refractory Cushing disease. Nat Clin Pract Endocrinol Metab. 2008;4(10):560-8. doi:10.1038/ncpendmet0947 PubMed
4.Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states. Endocr Rev. 1998;19(5):647-72. doi:10.1210/edrv.19.5.0346 PubMed
5.Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing's syndrome: diagnosis and therapy. Treat Endocrinol. 2002;1(2):79-94. doi:10.2165/00024677-200201020-00002 PubMed
6.Pivonello R, De Leo M, Cozzolino A, Colao A. The Treatment of Cushing's Disease. Endocr Rev. 2015;36(4):385-486. doi:10.1210/er.2013-1048 PubMed
7.Pivonello R, De Martino MC, De Leo M, Lombardi G, Colao A. Cushing's Syndrome. Endocrinol Metab Clin North Am. 2008;37(1):135-49, ix. doi:10.1016/j.ecl.2007.10.010
8.Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing's syndrome. Lancet. 2006;367(9522):1605-17. doi:10.1016/s0140-6736(06)68699-6 PubMed
9.Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing's syndrome. Lancet. 2015;386(9996):913-27. doi:10.1016/s0140-6736(14)61375-1 PubMed
10.Nieman LK. Recent Updates on the Diagnosis and Management of Cushing's Syndrome. Endocrinol Metab (Seoul). 2018;33(2):139-146. doi:10.3803/EnM.2018.33.2.139 PubMed
11.Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing's syndrome: state of the art. Lancet Diabetes Endocrinol. 2016;4(7):611-29. doi:10.1016/s2213-8587(16)00086-3 PubMed
12.Uum SV, Hurry M, Petrella R, Koch C, Dranitsaris G, Lacroix A. Management of patients with Cushing's disease: a Canadian cost of illness analysis. J Popul Ther Clin Pharmacol. 2014;21(3):e508-17. PubMed
13.Gotch PM. Cushing's syndrome from the patient's perspective. Endocrinol Metab Clin North Am. 1994;23(3):607-17. PubMed
14.Limumpornpetch P, Morgan AW, Tiganescu A, et al. The Effect of Endogenous Cushing Syndrome on All-cause and Cause-specific Mortality. The Journal of Clinical Endocrinology & Metabolism. 2022;107(8):2377-2388. doi:10.1210/clinem/dgac265 PubMed
15.Dekkers OM, Horvath-Puho E, Jorgensen JO, et al. Multisystem morbidity and mortality in Cushing's syndrome: a cohort study. J Clin Endocrinol Metab. 2013;98(6):2277-84. doi:10.1210/jc.2012-3582 PubMed
16.Ragnarsson O, Olsson DS, Papakokkinou E, et al. Overall and Disease-Specific Mortality in Patients With Cushing Disease: A Swedish Nationwide Study. J Clin Endocrinol Metab. 2019;104(6):2375-2384. doi:10.1210/jc.2018-02524 PubMed
17.van Aken MO, Pereira AM, Biermasz NR, et al. Quality of life in patients after long-term biochemical cure of Cushing's disease. J Clin Endocrinol Metab. 2005;90(6):3279-3286. PubMed
18.Webb SM, Badia X, Barahona MJ, et al. Evaluation of health-related quality of life in patients with Cushing's syndrome with a new questionnaire. Eur J Endocrinol. 2008;158(5):623-630. PubMed
19.Valassi E, Tabarin A, Brue T, et al. High mortality within 90 days of diagnosis in patients with Cushing's syndrome: results from the ERCUSYN registry. Eur J Endocrinol. 2019;181(5):461-472. doi:10.1530/eje-19-0464 PubMed
20.Sonino N, Fallo F, Fava GA. Psychosomatic aspects of Cushing's syndrome. Rev Endocr Metab Disord. 2010;11(2):95-104. doi:10.1007/s11154-009-9123-7 PubMed
21.Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2003;88(12):5593-602. doi:10.1210/jc.2003-030871 PubMed
22.Forget H, Lacroix A, Cohen H. Persistent cognitive impairment following surgical treatment of Cushing's syndrome. Psychoneuroendocrinology. 2002;27(3):367-83. doi:10.1016/s0306-4530(01)00059-2 PubMed
23.Pikkarainen L, Sane T, Reunanen A. The survival and well-being of patients treated for Cushing's syndrome. J Intern Med. 1999;245(5):463-468. PubMed
24.Dekkers OM, Biermasz NR, Pereira AM, et al. Mortality in patients treated for Cushing's disease is increased, compared with patients treated for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab. 2007;92(3):976-981. PubMed
25.Lindholm J, Juul S, Jorgensen JO, et al. Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86(1):117-23. doi:10.1210/jcem.86.1.7093 PubMed
26.Clayton RN, Raskauskiene D, Reulen RC, Jones PW. Mortality and morbidity in Cushing's disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J Clin Endocrinol Metab. 2011;96(3):632-42. doi:10.1210/jc.2010-1942 PubMed
27.Hammer GD, Tyrrell JB, Lamborn KR, et al. Transsphenoidal microsurgery for Cushing's disease: initial outcome and long-term results. J Clin Endocrinol Metab. 2004;89(12):6348-57. doi:10.1210/jc.2003-032180 PubMed
28.Graversen D, Vestergaard P, Stochholm K, Gravholt CH, Jorgensen JO. Mortality in Cushing's syndrome: a systematic review and meta-analysis. Eur J Intern Med. 2012;23(3):278-82. doi:10.1016/j.ejim.2011.10.013 PubMed
29.Ntali G, Asimakopoulou A, Siamatras T, et al. Mortality in Cushing's syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol. 2013;169(5):715-23. doi:10.1530/EJE-13-0569 PubMed
30.Etxabe J, Vazquez JA. Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin Endocrinol (Oxf). 1994;40(4):479-84. doi:10.1111/j.1365-2265.1994.tb02486.x PubMed
31.Bolland MJ, Holdaway IM, Berkeley JE, et al. Mortality and morbidity in Cushing's syndrome in New Zealand. Clin Endocrinol (Oxf). 2011;75(4):436-42. doi:10.1111/j.1365-2265.2011.04124.x PubMed
32.Hassan-Smith ZK, Sherlock M, Reulen RC, et al. Outcome of Cushing's Disease following Transsphenoidal Surgery in a Single Center over 20 years. J Clin Endocrinol Metab. 2012;97(4):1194-1201. PubMed
33.Yaneva M, Kalinov K, Zacharieva S. Mortality in Cushing's syndrome: data from 386 patients from a single tertiary referral center. Eur J Endocrinol. 2013;169(5):621-7. doi:10.1530/EJE-13-0320 PubMed
34.Giuffrida G, Crisafulli S, Ferraù F, et al. Global Cushing’s disease epidemiology: a systematic review and meta-analysis of observational studies. J Endocrinol Invest. 2022;45(6):1235-1246. doi:10.1007/s40618-022-01754-1 PubMed
35.Arnardottir S, Sigurjonsdottir HA. The incidence and prevalence of Cushing's disease may be higher than previously thought: results from a retrospective study in Iceland 1955 through 2009. Clin Endocrinol (Oxf). 2011;74(6):792-3. doi:10.1111/j.1365-2265.2010.03961.x PubMed
36.Broder MS, Neary MP, Chang E, Cherepanov D, Ludlam WH. Incidence of Cushing's syndrome and Cushing's disease in commercially-insured patients <65 years old in the United States. Pituitary. 2015;18(3):283-9. doi:10.1007/s11102-014-0569-6 PubMed
37.Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91(12):4769-75. doi:10.1210/jc.2006-1668 PubMed
38.Castinetti F, Morange I, Conte-Devolx B, Brue T. Cushing's disease. Orphanet J Rare Dis. 2012;7:41. doi:10.1186/1750-1172-7-41 PubMed
39.Esposito F, Dusick JR, Cohan P, et al. Clinical review: Early morning cortisol levels as a predictor of remission after transsphenoidal surgery for Cushing's disease. J Clin Endocrinol Metab. 2006;91(1):7-13. doi:10.1210/jc.2005-1204 PubMed
40.Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once-monthly pasireotide in Cushing's disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018;6(1):17-26. doi:10.1016/s2213-8587(17)30326-1 PubMed
41.Lase I, Strele I, Grönberg M, Kozlovacki G, Welin S, Janson ET. Multiple hormone secretion may indicate worse prognosis in patients with ectopic Cushing's syndrome. Hormones (Athens). 2020;19(3):351-360. doi:10.1007/s42000-019-00163-z PubMed
42.Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93(7):2454-62. doi:10.1210/jc.2007-2734 PubMed
43.Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847-875. doi:10.1016/s2213-8587(21)00235-7 PubMed
44.Capatina C, Hinojosa-Amaya JM, Poiana C, Fleseriu M. Management of patients with persistent or recurrent Cushing's disease after initial pituitary surgery. Expert Rev Endocrinol Metab. 2020;15(5):321-339. doi:10.1080/17446651.2020.1802243 PubMed
45.Gadelha MR, Vieira Neto L. Efficacy of medical treatment in Cushing's disease: a systematic review. Clin Endocrinol (Oxf). 2014;80(1):1-12. doi:10.1111/cen.12345 PubMed
46.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014. Epub 2019 Nov 9. PubMed
47.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015. Epub 2011 Jan 5. PubMed
48.Clinical Study Report: CLCI699C2301 (LINC 3), interim. A Phase III, multi-center, double-blind, randomized withdrawal study of LCI699 following a 24 week, single-arm, open-label dose titration and treatment period to evaluate the safety and efficacy of LCI699 for the treatment of patients with Cushing’s disease [internal sponsor's report]. Novartis; 2018 Sep 6.
49.Clinical Study Report: CLCI699C2301 (LINC 3), final. A Phase III, multi-center, double-blind, randomized withdrawal study of LCI699 following a 24 week, single-arm, open-label dose titration and treatment period to evaluate the safety and efficacy of LCI699 for the treatment of patients with Cushing’s disease [internal sponsor's report]. Novartis; 2020 May 27.
50.Clinical Study Report: LCI699C2302 (LINC 4), primary analysis. A Phase III, multi-center, randomized, double-blind, 48 week study with an initial 12 week placebo-controlled period to evaluate the safety and efficacy of osilodrostat in patients with Cushing’s disease [internal sponsor's report]. Novartis; 2020 Sep 24.
51.Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020;8(9):748-761. doi:10.1016/s2213-8587(20)30240-0 PubMed
52.Fleseriu M, Newell-Price J, Pivonello R, et al. Long-term outcomes of osilodrostat in Cushing's disease: LINC 3 study extension. Eur J Endocrinol. 2022;187(4):531-541. doi:10.1530/eje-22-0317 PubMed
53.Gadelha M, Bex M, Feelders RA, et al. Randomized Trial of Osilodrostat for the Treatment of Cushing Disease. J Clin Endocrinol Metab. 2022;107(7):e2882-e2895. doi:10.1210/clinem/dgac178 PubMed
54.Clinical Study Report: CLCI699C2302 (LINC 4), final. A Phase III, multi-center, randomized, double-blind, 48 week study with an initial 12 week placebo-controlled period to evaluate the safety and efficacy of osilodrostat in patients with Cushing’s disease [internal sponsor's report]. Novartis; 2021 Jul 28.
55.Sponsor Summary of Clinical Evidence: Isturisa (osilodrostat) for the treatment of endogenous Cushing’s syndrome in adults [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Isturisa (osilodrostat), 1 mg, 5 mg, and 10 mg oral film-coated tablets. Recordati Rare Diseases Canada Inc.; 2023 Oct 16, Revised 2025 Jul 18.
56.Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing's syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol. 2019;7(11):855-865. doi:10.1016/s2213-8587(19)30313-4 PubMed
57.Pivonello R, Zacharieva S, Elenkova A, et al. Levoketoconazole in the treatment of patients with endogenous Cushing's syndrome: a double-blind, placebo-controlled, randomized withdrawal study (LOGICS). Pituitary. 2022;25(6):911-926. doi:10.1007/s11102-022-01263-7 PubMed
58.Petersenn S, Newell-Price J, Findling JW, et al. High variability in baseline urinary free cortisol values in patients with Cushing's disease. Clin Endocrinol (Oxf). 2014;80(2):261-9. doi:10.1111/cen.12259 PubMed
59.Recordati Rare Diseases Canada Inc. response to December 8, 2023 request for additional information regarding Isturisa (osilodrostat) CADTH review [internal sponsor's report]. Recordati Rare Diseases Canada Inc.; 2023 Dec 15.
60.Recordati Rare Diseases Canada Inc. response to November 6, 2023 request for additional information regarding Isturisa (osilodrostat) CADTH review [internal sponsor's report]. Recordati Rare Diseases Canada Inc.; 2023 Nov 23.
61.Colao A, Pivonello R, Spiezia S, et al. Persistence of increased cardiovascular risk in patients with Cushing's disease after five years of successful cure. J Clin Endocrinol Metab. 1999;84(8):2664-72. doi:10.1210/jcem.84.8.5896 PubMed
62.Giordano R, Picu A, Marinazzo E, et al. Metabolic and cardiovascular outcomes in patients with Cushing's syndrome of different aetiologies during active disease and 1 year after remission. Clin Endocrinol (Oxf). 2011;75(3):354-60. doi:10.1111/j.1365-2265.2011.04055.x PubMed
63.Swearingen B, Biller BM, Barker FG, 2nd, et al. Long-term mortality after transsphenoidal surgery for Cushing disease. Ann Intern Med. 1999;130(10):821-4. doi:10.7326/0003-4819-130-10-199905180-00015 PubMed
64.Bakker RC, Gallas PR, Romijn JA, Wiersinga WM. Cushing's syndrome complicated by multiple opportunistic infections. J Endocrinol Invest. 1998;21(5):329-33. doi:10.1007/bf03350337 PubMed
65.Di Somma C, Pivonello R, Loche S, et al. Effect of 2 years of cortisol normalization on the impaired bone mass and turnover in adolescent and adult patients with Cushing's disease: a prospective study. Clin Endocrinol (Oxf). 2003;58(3):302-8. doi:10.1046/j.1365-2265.2003.01713.x PubMed
66.Faggiano A, Pivonello R, Spiezia S, et al. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing's disease during active disease and 1 year after disease remission. J Clin Endocrinol Metab. 2003;88(6):2527-33. doi:10.1210/jc.2002-021558 PubMed
67.Geer EB, Shen W, Strohmayer E, Post KD, Freda PU. Body composition and cardiovascular risk markers after remission of Cushing's disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab. 2012;97(5):1702-11. doi:10.1210/jc.2011-3123 PubMed
68.Graham BS, Tucker WS, Jr. Opportunistic infections in endogenous Cushing's syndrome. Ann Intern Med. 1984;101(3):334-8. doi:10.7326/0003-4819-101-3-334 PubMed
69.Kristo C, Jemtland R, Ueland T, Godang K, Bollerslev J. Restoration of the coupling process and normalization of bone mass following successful treatment of endogenous Cushing's syndrome: a prospective, long-term study. Eur J Endocrinol. 2006;154(1):109-18. doi:10.1530/eje.1.02067 PubMed
70.Manning PJ, Evans MC, Reid IR. Normal bone mineral density following cure of Cushing's syndrome. Clin Endocrinol (Oxf). 1992;36(3):229-34. doi:10.1111/j.1365-2265.1992.tb01437.x PubMed
71.Pereira AM, Delgado V, Romijn JA, Smit JW, Bax JJ, Feelders RA. Cardiac dysfunction is reversed upon successful treatment of Cushing's syndrome. Eur J Endocrinol. 2010;162(2):331-40. doi:10.1530/eje-09-0621 PubMed
72.Sonino N, Fava GA, Raffi AR, Boscaro M, Fallo F. Clinical correlates of major depression in Cushing's disease. Psychopathology. 1998;31(6):302-6. doi:10.1159/000029054 PubMed
73.Starkman MN, Giordani B, Berent S, Schork MA, Schteingart DE. Elevated cortisol levels in Cushing's disease are associated with cognitive decrements. Psychosom Med. 2001;63(6):985-93. doi:10.1097/00006842-200111000-00018 PubMed
74.Toja PM, Branzi G, Ciambellotti F, et al. Clinical relevance of cardiac structure and function abnormalities in patients with Cushing's syndrome before and after cure. Clin Endocrinol (Oxf). 2012;76(3):332-8. doi:10.1111/j.1365-2265.2011.04206.x PubMed
75.Newell-Price J, Pivonello R, Tabarin A, et al. Use of late-night salivary cortisol to monitor response to medical treatment in Cushing's disease. Eur J Endocrinol. 2020;182(2):207-217. doi:10.1530/eje-19-0695 PubMed
76.Biller BMK, Fleseriu M, Pivonello R, et al. THU-041 Clinical Improvements in Patients With Cushing's Disease Treated With Osilodrostat According to Urinary and Late-Night Salivary Cortisol Levels: Pooled Analysis from LINC 3 and LINC 4. Presented at the Endocrine Society Meeting (ENDO), June 15–18, 2023. 2023.
77.Biller BM, Fleseriu M, Pivonello R, et al. PMON160 Improvements in Clinical Signs of Hypercortisolism and Quality of Life According to Urinary and Late-Night Salivary Cortisol Levels in Patients with Cushing's Disease Treated with Osilodrostat. Journal of the Endocrine Society. 2022;6(Supplement_1):A545-A545.
78.Ciani O, Buyse M, Drummond M, Rasi G, Saad ED, Taylor RS. Time to Review the Role of Surrogate End Points in Health Policy: State of the Art and the Way Forward. Value Health. 2017;20(3):487-495. doi:10.1016/j.jval.2016.10.011 PubMed
79.Center for Drug Evaluation and Research. Medical review(s). Clinical Review of Osilodrostat, Isturisa. Application Number: 212801Orig1s000. U.S. Food and Drug Administration (FDA); 2020. Accessed 2023 Nov 23. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212801Orig1s000MedR.pdf
80.FDA Center for Drug Evaluation and Research. Clinical Review: Osilodrostat (Isturisa). NDA 212801. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212801Orig1s000MedR.pdf. Accessed 2025 September 18.
81.FDA Center for Drug Evaluation and Research. Statistical Review: Osilodrostat (Isturisa). NDA 212801. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212801Orig1s000StatR.pdf. Accessed 2025 September 18.
82.Results for Indirect Treatment Comparisons and Patient-Level Data Analyses for Osilodrostat in Cushing’s Syndrome. Version 1.0 — 18 November 2022. In: Drug Reimbursement Review sponsor submission: Isturisa (osilodrostat), film-coated tablets, 1 mg, 5 mg, 10 mg [internal sponsor's report]. Recordati Rare Diseases Canada Inc. Accessed 2023 Oct 16.
83.Recordati Rare Diseases Canada Inc. response to November 10, 2023 request for additional information regarding Isturisa (osilodrostat) CADTH review [internal sponsor's report]. Recordati Rare Diseases Canada Inc.; 2023 Nov 22.
84.Castinetti F, Morange I, Jaquet P, et al. Ketoconazole revisited: A preoperative or postoperative treatment in Cushing's disease. Eur J Endocrinol. 2008; 158(1):91-9. PubMed
85.Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing's disease: is it worth a try? The Journal of clinical endocrinology and metabolism. May 2014;99(5):1623-30. .1210/jc.2013- 3628. PubMed
86.Correa-Silva SR, Nascif SO, Molica P, Sá LBPC, Vieira JG, Lengyel AMJ. Partial restoration of GH responsiveness to ghrelin in Cushing's disease after 6 months of ketoconazole treatment: Comparison with GHRP-6 and GHRH. Eur J Endocrinol. 2009;161(5):681-686. PubMed
87.Young J, Bertherat J, Vantyghem MC, Chabre O. Hepatic safety of ketoconazole in Cushing’s syndrome: results of a Compassionate Use Programme in France. Eur J Endocrinol 2018;178(5):447-458. PubMed
88.Szumilas M. Explaining odds ratios. J Can Acad Child Adolesc Psychiatry. 2010; 19(3):227. PubMed
89.Woods BS, Hawkins N, Scott DA. Network meta-analysis on the log-hazard scale, combining count and hazard ratio statistics accounting for multi-arm trials: a tutorial. BMC Med Res Methodol. 2010;10(1):1-9. PubMed
90.Tierney JF, Stewart LA, Ghersi D, et al. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials. 2007; 8(1):16. PubMed
91.Woods BS, Hawkins N, Scott DA. Network meta-analysis on the log-hazard scale, combining count and hazard ratio statistics accounting for multi-arm trials: A tutorial. BMC Med Res Methodol. 2010;10(1):54. doi:10.1186/1471-2288-10-54 PubMed
92.ISTURISA (osilodrostat tablets): 1 mg, 5 mg, and 10 mg osilodrostat (as osilodrostat phosphate) [product monograph]. Recordati Rare Diseases Canada Inc.; 2025.
93.Apo-Ketoconazole (ketoconazole): 200 mg tablets [product monograph]. Apotex Inc.; 2021.
94.Dostinex (cabergoline): 0.5 mg tablets [product monograph]. Pfizer Canada Inc.; 2013.
95.Monaghan PJ, Keevil BG, Stewart PM, Trainer PJ. Case for the Wider Adoption of Mass Spectrometry-Based Adrenal Steroid Testing, and Beyond. The Journal of Clinical Endocrinology & Metabolism. 2014;99(12):4434-4437. doi:10.1210/jc.2014-2258 PubMed
96.Trainer PJ. Next generation medical therapy for Cushing's syndrome--can we measure a benefit? J Clin Endocrinol Metab. 2014;99(4):1157-60. doi:10.1210/jc.2014-1054 PubMed
97.Monaghan PJ, Owen LJ, Trainer PJ, Brabant G, Keevil BG, Darby D. Comparison of serum cortisol measurement by immunoassay and liquid chromatography-tandem mass spectrometry in patients receiving the 11β-hydroxylase inhibitor metyrapone. Ann Clin Biochem. 2011;48(Pt 5):441-6. doi:10.1258/acb.2011.011014 PubMed
98.Statistical Analysis Plan (SAP) for Final CSR: CLCI699C2301 (LINC 3). A Phase III, multi-center, double-blind, randomized withdrawal study of LCI699 following a 24 week, single-arm, open-label dose titration and treatment period to evaluate the safety and efficacy of LCI699 for the treatment of patients with Cushing’s disease [internal sponsor's report]. Novartis; 2019 Dec 2.
99.Statistical Analysis Plan (SAP): LCI699C2302 (LINC 4). A Phase III, multi-center, randomized, double-blind, 48 week study with an initial 12 week placebo-controlled period to evaluate the safety and efficacy of osilodrostat in patients with Cushing’s disease [internal sponsor's report]. Novartis; 2020 Dec 1.
100.Nelson LM, Forsythe A, McLeod L, et al. Psychometric evaluation of the Cushing's Quality-of-Life questionnaire. Patient. 2013;6(2):113-24. doi:10.1007/s40271-013-0012-5 PubMed
101.Center for Drug Evaluation and Research. Guidance for Industry. Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. U.S. Food and Drug Administration (FDA); 2009. Accessed 2017 Jul 27. https://www.fda.gov/downloads/drugs/guidances/ucm193282.pdf
102.Beck AT, Steer RA, Brown G. Manual for the Beck Depression Inventory-II. 2nd ed. Psychological Corporation; 1996.
103.Button KS, Kounali D, Thomas L, et al. Minimal clinically important difference on the Beck Depression Inventory--II according to the patient's perspective. Psychol Med. 2015;45(15):3269-79. doi:10.1017/s0033291715001270 PubMed
104.Wang YP, Gorenstein C. Psychometric properties of the Beck Depression Inventory-II: a comprehensive review. Braz J Psychiatry. 2013;35(4):416-31. doi:10.1590/1516-4446-2012-1048 PubMed
105.Meesters Y, Sijbrandij JJ, Visser E, de Beurs E. Sensitivity to change of the Beck Depression Inventory versus the Inventory of Depressive Symptoms. J Affect Disord. 2021;281:338-341. doi:10.1016/j.jad.2020.12.036 PubMed
Appendix 1: Characteristics of Treatment Options and Details of Considerations for Using the Drug Under Review
Please note that this appendix has not been copy-edited.
Table 7: Key Characteristics of Osilodrostat, Ketoconazole, and Cabergoline
Treatment | Mechanism of action | Indicationa | Recommended dosage and route of administration | Serious adverse effects or safety issues |
|---|---|---|---|---|
Osilodrostat | A cortisol synthesis inhibitor which inhibits 11-beta-hydroxylase (CYP11B1), the enzyme responsible for the final step of cortisol biosynthesis in the adrenal gland. | For the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option. | PO | QT interval prolongation, hypocortisolism, teratogenic risk, adrenal insufficiency, fatigue, edema, vomiting, nausea, decreased appetite, headache, dizziness, hypotension, arthralgia, myalgia, tachycardia, transaminases increased, and blood testosterone increased. |
Ketoconazole | Antifungal drug impairing ergosterol synthesis within fungal and yeast cell membranes. At high doses, ketoconazole also impairs the ability of the adrenal gland to produce cortisol by inhibiting several steroidogenic enzymes. | Not approved by Health Canada for Cushing syndrome. Ketoconazole tablets are indicated for the treatment of serious or life-threatening systemic fungal infections and should not be considered for mild to moderate infections. | PO | Liver toxicity, including liver failure and death; CYP3A4 inhibitor (high potential for drug interactions). |
Cabergoline | Cabergoline is a dopamine D2 receptor agonist that works by inhibiting prolactin secretion from the pituitary gland. | Not approved by Health Canada for Cushing syndrome. Cabergoline tablets are indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas. | PO | Cardiac valvulopathy, fibrotic disorders, and hypotension. |
PO = taken orally.
aHealth Canada–approved indication.
Sources: Isturisa,92 Apo-Ketoconazole (ketoconazole),93 and Dostinex (cabergoline)94 product monographs.
Table 8: Summary of Drug Program Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Were all relevant comparators considered? | The clinical experts consulted for this review indicated that ketoconazole is the primary comparator for this review. The clinical experts also indicated that ketoconazole plus cabergoline combination therapy may be used for patients who do not respond to the ketoconazole monotherapy. Therefore, combination therapy with ketoconazole plus cabergoline could be considered as a potential comparator in this subgroup of patients. No ITC was provided by the sponsor to compare osilodrostat vs. ketoconazole plus cabergoline combination therapy.59 |
Considerations for initiation of therapy | |
What would be the criteria for starting therapy? Where does osilodrostat fit in relation to other nonmedication therapies? (For example, the LINC 3 study included confirmed persistent or recurrent Cushing disease after pituitary surgery or irradiation or both, if they had not had previous surgery or radiotherapy and refused surgery or were not deemed to be surgical candidates. Patients were required to have evidence of a pituitary origin [based on specific criteria]. Patients could be receiving other medical therapies if the specified washout was achieved. The LINC 4 study included specific values for mUFC: greater than 1.3 × ULN and plasma ACTH above the lower limit of normal.) | According to the clinical experts consulted for this review, patients with active Cushing disease for whom surgery is not possible, feasible, or effective could be eligible for starting therapy with osilodrostat. The clinical experts pointed out that while osilodrostat can reduce the production of cortisol, it may not eliminate the structural presence of the tumour. For patients with inoperable Cushing disease, a medication that works through a selective enzyme inhibition to limit cortisol production (like osilodrostat) may be of significant advantage. |
Would there be a requirement to have any previous therapies before starting osilodrostat? | The clinical experts noted that surgical resection of the primary lesion(s) underlying the disease is considered the first-line treatment option for Cushing disease. For patients in whom surgery is not feasible, other medications that reduce cortisol production are available. According to the clinical experts, aside from surgery, most patients with persistently active Cushing disease would typically have received ketoconazole, with or without cabergoline, as part of their prior pharmacological management. Initiation of osilodrostat is generally considered appropriate only after documented failure or intolerance to these prior therapies. |
Considerations for continuation or renewal of therapy | |
What would the guidance be around continuing therapy? What is the marker for treatment success? The 2 studies (LINC 3 and LINC 4) used mUFC ≤ ULN. | The clinical experts noted that the criteria for continuing therapy would depend on patients’ response to osilodrostat. Some measures that are generally used by clinicians include patient symptoms, extent to which comorbidities are controlled or reversed, and reduced incidence of life-threatening complications such as myocardial infarcts and/or serious infections/sepsis. The clinical experts emphasized that the liver function parameters should be closely monitored among patients with Cushing disease because they are indicative of tolerance to the medication and reflective of disease–treatment interactions. |
Considerations for discontinuation of therapy | |
How would response to therapy be quantified? Is this a standard measure or would this depend on other individual patient factors? What parameters should be considered related to improvement of comorbid conditions? | The clinical experts noted that response to therapy in Cushing disease is typically quantified using normalization of mUFC, which is the primary end point in most clinical trials. However, they noted that mUFC alone is not considered sufficient to define treatment success in clinical practice. Instead, a broader evaluation that includes patient-important outcomes, such as the incidence of cardiovascular disease, incidence of diabetes, and physical function (for example, patient mobility and ECOG Performance Status and, ultimately, mortality, is more reflective of meaningful clinical benefit). The clinical experts noted that while mUFC is a standard measure in trials, its interpretation in clinical practice depends on individual patient factors. For example, changes in BMD at the lumbar spine can be a valuable indicator of treatment impact, particularly in relation to physical function. BMD can be measured precisely using validated techniques and normative values. However, the extent of BMD improvement may vary depending on the degree of cortisol reduction and the patient’s baseline condition, making it a context-dependent marker. In assessing improvement of comorbid conditions, clinical experts emphasized the importance of monitoring cardiovascular events, metabolic parameters, osteoporosis, and physical performance. |
System and economic issues | |
The primary comparison is vs. ketoconazole, which is not indicated in this population and is covered in most (but not all) formularies as a full benefit. Is this the most appropriate comparison or should other medications or surgery/radiation also be considered? | Clinical expert feedback suggested that osilodrostat was more likely to be used in patients for whom surgery was ineffective or not possible; and although it is unlikely to replace a currently available treatment, it may be preferred over currently available treatments. In addition to ketoconazole (as a potential first-line pharmacotherapy treatment comparator); the clinical experts expressed that the combination therapy of ketoconazole plus cabergoline and active surveillance as the most relevant comparators in patients in whom surgery was ineffective or not possible. Surgery may be an appropriate comparator in patients who had not had surgery. |
ACTH = adrenocorticotropic hormone; BMD = bone mineral density; ECOG = Eastern Cooperative Oncology Group; ITC = indirect treatment comparison; mUFC = mean urinary free cortisol; ULN = upper limit of normal; vs. = versus.
Appendix 2: Methods of the Clinical Review
Please note that this appendix has not been copy-edited.
Data Extraction and Critical Appraisal
One reviewer extracted study characteristics and results from the sponsor-submitted documents with verification by a second reviewer. One reviewer appraised the internal and external validity of the submitted evidence in consideration of inputs by the clinical experts, and patient and clinician groups, with input from a methodologist.
Certainty of Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, 1 reviewer assessed the certainty in the body of evidence for outcomes considered most important for informing expert committee recommendations using the GRADE instrument.46,47 A methodologist provided input on the certainty ratings. Standard wording was used to describe the results, including “may” alongside the direction of effect to describe results of low certainty and “likely” for those of moderate certainty. When the certainty in the evidence was very low, the evidence was described as “very uncertain.”
The certainty in the presence of a clinically important effect was rated when a threshold of clinical importance was available. In the absence of literature-based MID estimates, thresholds suggested by the clinical experts were used. In the absence of a known threshold, the certainty in the presence of a nonnull effect was rated. When possible, input from the clinical experts was leveraged to make judgments about the clinical importance of the effect estimate and its precision.
For the GRADE assessments, findings from the LINC 3 and LINC 4 studies were presented in 2 separate tables per outcome because the LINC 3 trial (a randomized withdrawal study randomizing only responders, i.e., patients who completed osilodrostat dose titration and had mUFC levels equal to or below to the ULN, resulting in an enriched population) differed from LINC 4 (an RCT involving patients with mUFC greater than 1.3 times the ULN) in design and population, although they were similar in terms of interventions, and outcome measures.
As the body of evidence relied on data from RCTs, the certainty of evidence started at high and could be rated down for study limitations, indirectness, imprecision, and publication bias.
Appendix 3: Methods of the Studies Included in the Systematic Review
Please note that this appendix has not been copy-edited.
Characteristics of the Included Study
Inclusion and Exclusion Criteria
Table 9: Details of the LINC 3 and LINC 4 Studies
Detail | LINC 3 trial | LINC 4 trial |
|---|---|---|
Designs and populations | ||
Locations | 66 sites in 19 countries Countries where the trial was conducted: Argentina, Austria, Bulgaria, Canada, China, Colombia, France, Germany, India, Italy, Japan, Netherlands, Republic of Korea, Russia, Spain, Thailand, Turkey, UK, US | 40 sites in 14 countries Countries where the trial was conducted: Belgium, Brazil, Canada, China, Costa Rica, Greece, Poland, Thailand, Turkey, Portugal, Russian Federation, Spain, Switzerland, US |
Patient enrolment dates | Start date:
End date:
| Start date:
End date:
|
Randomized (N) | Total N enrolled in the study = 137 Total randomized N = 71
| Total N = 74a
|
Inclusion criteria |
|
|
Exclusion criteria |
The following exclusion criteria were the same for the LINC 3 and LINC 4 trials
|
The rest of the exclusion criteria were the same as the common points for the LINC 3 and LINC 4 trials listed in the left column. |
Study duration | ||
Screening phase | Up to 5 weeks | Up to 8 weeks |
Run-in phase | 1 week to 6 months, depending on the current drug therapy for CD at screening, with details listed in the inclusion criteria | |
Open-label (single-arm) phase before randomization | 26 weeks
Patients remained on open-label osilodrostat from week 24 to week 26b | NA |
Randomized or randomized withdrawal phase (placebo-controlled period) | 8 weeks
| 12 weeks
|
Open-label (single-arm) period in core phase | 14 weeks
| 36 weeks
|
Optional extension phasec | This period ended 16 months after all patients completed week 72. | From end of week 48 to week 96, or until Protocol Amendment 02 approval or other treatment options became available. |
ACTH = adrenocorticotropic hormone; AE = adverse event; AIMAH = adrenocorticotropic hormone–independent macronodular adrenal hyperplasia; AIP = aryl hydrocarbon receptor interacting protein; ALT = alanine aminotransferase; AST = aspartate aminotransferase; BIPSS = bilateral inferior petrosal sinus sampling; BMD = bone mineral density; BMI = body mass index; BP = blood pressure; CD = Cushing disease; CRH = corticotropic-releasing hormone; CS = Cushing syndrome; CTCAE = Common Terminology Criteria for Adverse Events; C-SSRS = Colombia Suicide Severity Rating Scale; DBP = diastolic blood pressure; DDAVP = desmopressin; DXA = dual-energy x-ray absorptiometry; ECG = electrocardiogram; EOT = end of treatment; FPFV = first patient first visit; FPG = fasting plasma glucose; GFR = glomerular filtration rate; IUD = intrauterine device; IUS = intrauterine system; LPLV = last patient last visit; MDRD = Modification of Diet in Renal Disease trial; MEN-1 = multiple endocrine neoplasia type 1; MI = myocardial infarction; mUFC = mean urinary free cortisol; NA = not applicable; NYHA = New York Heart Association; PK = pharmacokinetic; PPAR = proliferator-activated receptor; PPNAD = Primary Pigmented Nodular Adrenal Dysplasia; QoL = quality of life questionnaire; QTc = QT corrected; RW = randomized withdrawal; SBP = systolic blood pressure; SC = subcutaneous; UFC = urinary free cortisol; ULN = upper limit of normal.
aOne patient was randomized but not treated.
bPatients remained on open-label osilodrostat during the period between the end of week 24 and week 26 to ensure that sufficient time is allowed for central laboratory results (week 24 mUFC) to become available for all patients at all sites, and to standardize the time of randomization across sites.
cEntry into the extension period was optional in both the LINC 3 and LINC 4 studies.
Sources: LINC 3 interim Clinical Study Report,48 LINC 3 final Clinical Study Report,49 LINC 4 primary analysis Clinical Study Report,50 and LINC 4 final Clinical Study Report.54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Description of Outcomes
Urinary Free Cortisol
UFC was assessed centrally and measured in three 24-hour urine samples averaged to obtain the mean UFC (mUFC) level.43,48-50,54,58 The 24-hour UFC measurements were chosen for the primary outcome measures for disease activity allowing for the circadian variability and pulsatile nature of ACTH secretion in patients with Cushing disease. Also, part of the variability in 24-hour UFC comes from the methodology and patient compliance.43,58
During the screening periods of the LINC 3 and LINC 4 studies, three 24-hour UFC samples were collected and sent to the central laboratory at least 14 days before day 1. These samples were used to assess eligibility of the patient. At the baseline of the LINC 3 and LINC 4 studies, another three 24-hour UFC samples were collected within 7 days before the first day of treatment to serve as the baseline value. In the LINC 3 study, during the treatment period patients collected three 24-hour UFC samples within 7 days before the next visit and with the last urine sample preferably collected the day before the visit at the site. During the study, patients were allowed to have unscheduled visits at any time if they report symptoms of hypercortisolism or hypocortisolism.48,49 In the LINC 4 study, during the treatment period, patients collected 2 24-hour UFC samples within 6 days before the next visit and with the last urine sample preferably collected the day before the visit at site. Patients collected three 24-hour UFC samples within 7 days before the next week 12, week 36, week 48 and if applicable, week 12 Posttreatment follow-up visits.
In both the LINC 3 and LINC 4 studies, LC-MS/MS was the method used at the central laboratory for the measurement of UFC. In contrast to immunoassays, LC-MS/MS has the advantage of measuring cortisol accurately and exclusively, without concern of interference by crossreactivity of accumulating cortisol precursors with osilodrostat therapy.95-97
In both the LINC 3 and LINC 4 studies, a range of 11 nmol per 24 hours to 138 nmol per 24 hours or 4 mcg per 24 hours to 50 mcg per 24 hours was considered as the reference of a normal range of mean 24-hour UFC.51 The reference ULN for mUFC is 138 nmol per 24 hours.51 The primary efficacy end point in the LINC 3 study was the proportion of randomized patients in each arm with an mUFC equal to or below ULN at the end of 8 weeks of randomized withdrawal (week 34), and who had neither discontinued nor had an osilodrostat dose increase above the level at week 26 during the randomized withdrawal period. The primary efficacy end point was the proportion of randomized patients with a complete response, that is, mUFC at or below the ULN at week 12. Partial response was defined as a 50% or greater reduction from baseline in mUFC, but an mUFC greater than the ULN at scheduled time points.55,98,99 Other secondary end points included partial response rate (a ≥ 50% reduction in mUFC from baseline and > ULN) in both studies, time to escape was defined as time from the first ULN equal to or below the first mUFC results that were greater than 1.5 times the ULN (in the LINC 3 study) or greater than 1.3 times the ULN (in the LINC 4 study), among others.48-50,54 The rationale for selecting 1.5 or 1.3 times the ULN as cut-off values for the eligibility criteria, and as an outcome in the LINC 3 and LINC 4 studies, respectively, was unclear.
In the LINC 3 study, patients who discontinued during the randomized withdrawal, placebo-controlled period were counted as nonresponders for the primary end point. In the LINC 4 study, patients who discontinued the study during the placebo-controlled period, or who had a missing mUFC assessment at week 12, were counted as nonresponders for the primary end point. In both the LINC 3 and LINC 4 studies, dose reductions and temporary dose interruptions for a safety reason during the placebo-controlled periods did not preclude patients from being complete responders for the primary end point.
BMD Assessments
BMD of the lumbar vertebrae (L1 to L4 or L2 to L4) was measured in both the LINC 3 and LINC 4 studies, using Lunar or Hologic dual-energy x-ray absorptiometry (DXA) instruments. A patient was scanned on the same DXA instrument throughout the study in each respective study. The BMD results were reported in actual density (g/cm2) and standardized against the peak bone mass in a healthy young adult population (BMD T-score). In the LINC 3 study, BMD results were reviewed centrally by the imaging vendor, while in the LINC 4 trial, the analysis and reporting of BMD results were conducted centrally by the imaging vendor. In the LINC 4 study, if the end of treatment occurred less than 6 months before the scheduled week 96 visit, an MRI (or CT) and DXA were not mandatory at the end of treatment. BMD assessments were not done in patients enrolled in Germany in the LINC 3 and LINC 4 studies.48-50,54
Patient-Reported Outcomes
CushingQoL: CushingQoL (version 1.0) was developed to evaluate quality of life in patients with Cushing syndrome.18 The CushingQoL comprises 12 items that capture patient responses on 7 concepts: daily activities, healing and pain, mood and self-confidence, social concerns, physical appearance, memory, and concern about the future. Content reliability, sensitivity to change and psychometric properties have been validated in patients with Cushing disease. Total score of CushingQoL ranges from 0 to 100, with higher scores indicating better HRQoL; a score of 0 reflects the poorest possible HRQoL, while a score of 100 represents the best.100
In the LINC 3 study and the core phase of the LINC 4 study, the CushingQoL was modified from the standard 4-week recall to a 1-week recall to measure shorter-term changes in patient quality of life, specifically during the randomized withdrawal period, where it was believed that changes in Cushing disease symptoms occurred rapidly once patients stopped treatment.48-50,54
An MID of 10.1 was initially estimated using a distribution-based method, specifically, a 0.5 SD unit for change-from-baseline data, based on data collected with the original 4-week recall period.100 However, anchor-based methods are generally considered more appropriate for interpreting changes in patient-reported outcome scores over time because they are based on individual-level change rather than group-level change.101 Given the importance of anchor-based MIDs and that the modification of the recall period to be 1 week, the sponsor proposed an anchor-based MID — estimated to be approximately 10 points — derived by Novartis from previous clinical trial data involving the CushingQoL, when feasible. This anchor-based MID reflects within-group differences.
BDI-II: The BDI-II is a patient-reported instrument that consists of 21 items designed to assess the intensity of depression in clinical and normal patients in the preceding 2 weeks. Each item is a list of 4 statements arranged in increasing severity about a particular symptom of depression. Items are rated on a 4-point severity scale of 0 (not at all) to 3 (extreme) for each symptom, with differing response options for each item. A global score ranging from 0 to 63 is calculated, with a higher score representing a greater level of depression. The following scoring guidelines for interpretation of the BDI-II have been suggested: a score of 0 to 13 suggests minimal depression, 14 to 19 suggests mild depression, 20 to 28 suggests moderate depression, and 29 to 63 suggests severe depression.102 A minimal clinically important difference for improvement in BDI-II scores was reported as a 17.5% reduction in scores from baseline among patients with depression; however, this was dependent on baseline severity.103
Harms Outcomes
In both the LINC 3 and LINC 4 studies, safety was monitored by assessing physical examination, vital signs, laboratory evaluations, radiological examinations, cardiac assessments, pregnancies (in the LINC 3 study only), as well as collecting the AEs according to the Common Terminology Criteria for Adverse Events version 4.03 at every visit. In the LINC 4 study only, safety was also monitored by assessments of imaging and the risk of suicide.48-50,54
In the LINC 3 and LINC 4 studies, AEs, laboratory result abnormalities that constitute an AE, SAE, or AESI were recorded together with their severity and relationship to the study drug. AESIs belonged to 1 of the following mechanistic groups:
adrenal hormone precursor accumulation–related AEs
hypocortisolism-related AEs
pituitary tumour enlargement–related AEs
QT prolongation–related AEs
arrhythmogenic potential AEs.
Table 10: Summary of Outcome Measures and Minimal Important Differences
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
mUFC | Twenty-four -hour collection of UFC concentrations are standard in patients with CD. ACTH secretion and cortisol are highly variable; therefore, 24-hour urinary collection was used to give an integrated measure of cortisol production during 24 hours. Part of the variability in 24-hour UFC comes from the methodological factors and patient adherence to collection procedures. | Validity, Reliability, and Responsiveness: Not applicable | An MID for patients with Cushing disease has not been estimated. |
CushingQoL | A 12-item questionnaire that captures patient responses on 7 concepts: daily activities, healing and pain, mood and self-confidence, social concerns, physical appearance, memory and concern about the future. In the LINC 3 study (randomized withdrawal period) and LINC 4 (periods 1 and 2), the CushingQoL was modified from the standard four-week recall to a 1-week recall to capture shorter-term changes in patient quality of life. | Reliability, validity, the ability to detect change were evaluated for the CushingQoL questionnaire, which has a 4-week recall period, using data from patients diagnosed with CD (n = 162) who participated in the phase III clinical trial designed to assess the safety and efficacy of different doses of pasireotide.100 This information is currently unavailable for the version of the instrument with a 1-week recall period. Validity: Construct validity hypotheses were in the anticipated direction. Changes in CushingQoL scores were moderately correlated with changes in mUFC levels, in BMI, and in weight.100 Reliability: Internal consistency reliability (Cronbach alpha = 0.87 to 0.88) and test-retest reliability (ICC = 0.87) were high.100 Responsiveness: Moderate Guyatt responsiveness effect sizes were observed for patients with reductions in weight, BMI, and waist circumference.100 | A within-group MID of 10.1 was estimated in the literature at the time the CSR was written based on the distribution method of an 0.5 SD unit change using baseline data from the pasireotide phase III trial (Nelson et al., 2013), with the original 4-week recall period.100 |
BDI-II | A patient-reported instrument consists of 21 items designed to assess the intensity of depression in clinical and normal patients in the preceding 2 weeks. Each item is a list of 4 statements arranged in increasing severity about a particular symptom of depression. | In a review of 118 studies that include nonclinical, psychiatric or institutionalized, and medical samples:104 Validity: The criterion-based validity of the BDI-II showed good sensitivity and specificity for detecting depression when compared against structured clinical interviews based on DSM-IV criteria, which served as the gold standard. Strong convergent validity was demonstrated between the BDI-I and BDI-II, with Pearson correlation coefficients ranging from 0.82 to 0.94. Regarding discriminant validity, research has shown weak associations (r < 0.4) between the BDI-II and measures of substance use and chronic pain. Interestingly, suicidal ideation — an essential symptom of depression and a specific item on the BDI-II — showed only limited to moderate correlation with the overall scale. The BDI-II exhibits acceptable content validity, although its scope is somewhat more limited compared to the original version.104 Reliability: The BDI-II showed strong internal consistency, with average alpha coefficients around 0.9. The retest reliability, assessed using Pearson’s correlation coefficients (r), ranged from 0.73 to 0.96.104 Responsiveness: The BDI-II has shown adequate responsiveness in detecting changes in depressive symptoms over time, particularly in response to treatment.103,105 | A minimal clinically important difference for improvement in BDI-II scores was reported as a 17.5% reduction in scores from baseline among patients with depression; however, this was dependent on baseline severity.103 |
BDI-II = Beck Depression Inventory-II; CD = Cushing disease; CushingQoL = Cushing quality of life questionnaire; DSM-IV = Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; ICC = intraclass correlation coefficient; MID = minimal important difference; mUFC = mean urinary free cortisol; SD = standard deviation.
Additional Details on Statistical Analysis
Subgroup of Interest and Data Cut-Off Date
For the LINC 3 study, subgroups of interest included the 2 stratification factors for randomization: history of pituitary irradiation (yes versus no), and osilodrostat dosage at week 24 (≤ 5 mg twice daily versus > 5 mg twice daily).98
For the LINC 4 study, the number of patients who completed or discontinued during the core phase, week 48 to 72, and after week 72 were tabulated by pandemic set. As per Novartis guidance, the start date in a given country or region was defined as the approximate time point at which, according to WHO situation reports and the Johns Hopkins database, the number of confirmed COVID-19 infections started to increase significantly (around 100 confirmed cases) and/or governments started to take measures (such as stay-at-home orders) to contain the epidemic, whichever occurred first (China: January 1, 2020; other countries with study sites: March 1, 2020). The pandemic impact was assessed based on 2 subsets: the before pandemic set (patients who completed or discontinued) the trial before the pandemic start date in their region/country), and the during pandemic set (patients with at least 1 on-treatment assessment or treatment-emergent event during the pandemic dates as defined for their region or country).99
Analysis Populations
Table 11: Analysis Populations of the LINC 3 and LINC 4 Studies
Study | Population | Definition | Application |
|---|---|---|---|
LINC 3 | RAS | All randomized patients who received at least 1 dose of their randomized treatment (osilodrostat or placebo). | Efficacy analyses. |
FAS | All enrolled patients who receive at least 1 dose of osilodrostat. | Efficacy analyses. | |
SAS | All patients who received at least 1 dose of osilodrostat and had at least 1 valid postbaseline safety assessment. | Safety analyses; analyzed according to actual treatment received. | |
SASR | Randomized patients who received at least 1 dose of randomized treatment (osilodrostat or placebo) and had at least 1 valid safety assessment during the randomized withdrawal period. | Safety analyses; analyzed according to actual treatment received. | |
PPRAS | A subset of the patients in the RAS who had no CSR-reportable protocol deviation. | Efficacy analyses. | |
PPFAS | A subset of the patients in the FAS who had no CSR-reportable protocol deviation. | Efficacy analyses. | |
LINC 4 | FAS | All randomized patients who received at least 1 dose of their randomized treatment (osilodrostat or placebo). | Efficacy analyses; analyzed according to the treatment and stratum assigned during randomization. |
SS | All patients who received at least 1 dose of their randomized treatment (osilodrostat or placebo). | Safety analyses; analyzed according to the study drug received. | |
PPS | A subset of the patients in the FAS who were compliant with the requirements of the clinical study protocol and had no CSR-reportable protocol deviation. | Efficacy analyses. |
CSR = Clinical Study Report; FAS = full analysis set; PAS = pharmacokinetic analysis set; PK = pharmacokinetic; PPFAS = per-protocol set for full analysis set; PPRAS = per-protocol set for randomized analysis set; PPS = per-protocol set; RAS = randomized analysis set; SAS = safety analysis set; SASR = safety analysis set for randomized withdrawal period; SS = safety set.
Sources: LINC 3 interim Clinical Study Report48 and LINC 4 primary analysis Clinical Study Report.50
Appendix 4: Results of the Studies Included in the Systematic Review
Please note that this appendix has not been copy-edited.
Patient Disposition in the Included Studies
Table 12: Summary of Patient Disposition by Randomized Treatment Group From the LINC 3 Trial (FAS)
Patient disposition, n (%) | Randomized to osilodrostat during RW (N = 36) | Randomized to placebo during RWa (N = 35) | Nonrandomized (N = 66) | All patients (N = 137) |
|---|---|---|---|---|
Patients enrolled and treated | 36 (100) | 35 (100) | 66 (100) | 137 (100) |
Discontinued at any timeb | 1 (2.8) | 5 (14.3) | 29 (43.9) | 35 (25.5) |
Primary reason for discontinuation at any time | ||||
Adverse event | 0 | 2 (5.7) | 18 (27.3) | 20 (14.6) |
Death | 0 | 1 (2.9) | 0 | 1 (0.7) |
Physician decision | 0 | 0 | 3 (4.5) | 3 (2.2) |
Patient withdrew consent | 1 (2.8) | 0 | 4 (6.1) | 5 (3.6) |
Patient/guardian decision | 0 | 2 (5.7) | 4 (6.1) | 6 (4.4) |
Discontinued at or before week 12 | 0 | 0 | 7 (10.6) | 7 (5.1) |
Primary reason for discontinuation at or before week 12 | ||||
Adverse event | 0 | 0 | 4 (6.1) | 4 (2.9) |
Patient withdrew consent | 0 | 0 | 2 (3.0) | 2 (1.5) |
Patient/guardian decision | 0 | 0 | 1 (1.5) | 1 (0.7) |
Discontinued at or before week 26 but after week 12 | 0 | 0 | 12 (18.2) | 12 (8.8) |
Primary reason for discontinuation at or before week 26, but after week 12 | ||||
Adverse event | 0 | 0 | 8 (12.1) | 8 (5.8) |
Physician decision | 0 | 0 | 2 (3.0) | 2 (1.5) |
Patient withdrew consent | 0 | 0 | 2 (3.0) | 2 (1.5) |
Discontinued before week 48 but after week 26 | 0 | 2 (5.7) | 3 (4.5) | 5 (3.6) |
Primary reason for discontinuation at or before week 48 but after WEEK 26 | ||||
Adverse event | 0 | 2 (5.7) | 1 (1.5) | 3 (2.2) |
Physician decision | 0 | 0 | 1 (1.5) | 1 (0.7) |
Patient/guardian decision | 0 | 0 | 1 (1.5) | 1 (0.7) |
Completed week 48 (core phase) | 36 (100) | 33 (94.3) | 44 (66.7) | 113 (82.5) |
Completed week 48 and did not enter the extension phaseb | 1 (2.8) | 3 (8.6) | 3 (4.5) | 7 (5.1) |
Randomized analysis set | 36 (100) | 34 (97.1)c | — | — |
Full analysis set | 36 (100) | 35 (100) | 66 (100) | 137 (100) |
Safety analysis set | 36 (100) | 35 (100) | 66 (100) | 137 (100) |
Safety analysis set for RW period | 36 (100) | 34 (97.1) | — | — |
Per-protocol set for RAS | 35 (97.2) | 33 (94.3) | — | — |
Per-protocol set for FAS | 36 (100) | 35 (100) | 64 (97.0) | 135 (98.5) |
FAS = full analysis set; RAS = randomized analysis set; RW = randomized withdrawal.
Note: % is based on N.
aFor patients randomized to placebo during the RW period and including all data while on either osilodrostat or placebo.
bPatients who completed week 48 and did not enter extension phase are not counted as discontinuations.
cOne patient was randomized to placebo but never received treatment and withdrew from study during the RW period on Day 220.
Source: LINC 3 interim Clinical Study Report.48
Table 13: Summary of Patient Disposition by Randomized Treatment Group From the LINC 4 Trial (FAS)
Patient disposition, n (%) | Osilodrostat (N = 49) | Placebo (N = 25) | All patients (N = 74) |
|---|---|---|---|
Patients randomized | 49 (100) | 25 (100) | 74 (100) |
Not treateda | 1 (2.0) | 0 | 1 (1.4) |
Treateda | 48 (98.0) | 25 (100) | 73 (98.6) |
Primary reason for not being treateda | |||
Adverse event | 1 (2.0) | 0 | 1 (1.4) |
Discontinued at or before week 12 | 3 (6.3) | 0 | 3 (4.1) |
Primary reason for discontinuation at or before week 12 | |||
Adverse event | 1 (2.1) | 0 | 1 (1.4) |
Patient or guardian decision | 2 (4.2) | 0 | 2 (2.7) |
Discontinued at or before week 48 but after week 12 | 3 (6.3) | 2 (8.0) | 5 (6.8) |
Primary reason for discontinuation at or before week 48 but after week 12 | |||
Adverse event | 0 | 2 (8.0) | 2 (2.7) |
Physician decision | 1 (2.1) | 0 | 1 (1.4) |
Patient or guardian decision | 2 (4.2) | 0 | 2 (2.7) |
Completed core phase | 42 (87.5) | 23 (92.0) | 65 (89.0) |
Completed core phase and did not enter extension phaseb | 4 (8.3) | 1 (4.0) | 5 (6.8) |
Completed core phase and entered extension phase | 38 (79.2) | 22 (88.0) | 60 (82.2) |
Full analysis set | 48 (100) | 25 (100) | 73 (100) |
Safety set | 48 (100) | 25 (100) | 73 (100) |
Per-protocol primary analysis set | 44 (91.7) | 23 (92.0) | 67 (91.8) |
Per-protocol secondary analysis set | 40 (83.3) | 20 (80.0) | 60 (82.2) |
FAS = full analysis set.
aThe percentages for these rows are based on N; the percentages for the remaining rows are based on randomized and treated patients.
bPatients who completed week 48 and did not enter the extension phase are not counted as discontinuations.
Source: LINC 4 primary analysis Clinical Study Report.50
Treatment Exposure and Concomitant Medications
Exposure to Study Treatments
In the LINC 3 study, during the first 26 weeks, the mean dose with the longest duration was 10.8 mg/day (SD = 10.01) and the mean highest dose was 17.8 mg/day (SD = 13.56). During the randomized withdrawal period, the mean dose with the longest duration was 10.1 mg/day (SD = 9.55) and the mean highest dose was 10.1 mg/day (SD = 9.55). Eighteen of 137 patients were exposed to osilodrostat for ≤ 24 weeks. The mean exposure during the first 26 weeks was 24.6 (SD = 5.40) weeks.48
In the LINC 4 study, during the placebo-controlled period, the mean average dose was 7.5 mg/day (SD = 4.22 mg/day) in the osilodrostat arm and 9.1 (SD = 3.52) mg/day in the placebo arm. In both arms, the dose with the longest duration was 4 mg/day in the majority of patients (osilodrostat arm: 26/48; 54.2%; placebo arm: 15/25; 60%). The highest dose in the majority of patients in osilodrostat arm was 10 mg/day (18/48; 37.5%), and 20 mg/day (13/25; 52.0%) in the placebo arm.50
Table 14: Dosing and Patient Exposure During the First 26 Weeks (Open Label) and the Randomized Withdrawal Period (Double Blind) From the LINC 3 Study (Safety Set)
Treatment exposure | First 26 weeks All patients (N = 137) | Randomized withdrawal period | |
|---|---|---|---|
Osilodrostat (N = 36) | Placebo withdrawal (N = 35) | ||
Highest dose, mg/day | |||
N | 137 | 36 | NA |
Mean (SD) | 17.8 (13.56) | 10.1 (9.55) | NA |
Median | 10.0 | 10.0 | NA |
Q1, Q3 | 10.0, 20.0 | 2.0, 13.0 | NA |
Range | 4.0 to 60.0 | 1.0 to 40.0 | NA |
Average dose, mg/day | |||
N | 137 | 36 | NA |
Mean (SD) | 10.0 (7.31) | 10.0 (9.56) | NA |
Median | 8.3 | 8.7 | NA |
Q1, Q3 | 4.5, 13.8 | 2.0, 13.0 | NA |
Range | 1.3 to 45.2 | 1.0 to 40.0 | NA |
Dose with longest duration, mg/day | |||
N | 137 | 36 | NA |
Mean (SD) | 10.8 (10.01) | 10.1 (9.55) | NA |
Median | 10.0 | 10.0 | NA |
Q1, Q3 | 4.0, 14.0 | 2.0, 13.0 | NA |
Range | 0.5 to 60.0 | 1.0 to 40.0 | NA |
Duration of exposure, weeks | |||
N | 137 | 36 | 34 |
Mean (SD) | 24.6 (5.40) | 8.5 (1.28) | 7.0 (2.48) |
Median | 26.0 | 8.1 | 8.0 |
Q1, Q3 | 25.7, 26.3 | 8.0, 9.9 | 5.1, 8.3 |
Range | 0.9 to 43.3 | 3.9 to 10.6 | 0.4 to 10.4 |
DB = double blind; NA = not applicable; Q1 = 25th percentile; Q3 = 75th percentile; SD = standard deviation.
Source: LINC 3 interim Clinical Study Report.48
Table 15: Dosing and Patient Exposure During Placebo-Controlled Period From the LINC 4 Trial (Safety Set)
Treatment exposure | Osilodrostat (N = 48) | Placebo (N = 25) |
|---|---|---|
Highest dose received, n (%) | ||
4 mg/day | 13 (27.1) | 3 (12.0) |
10 mg/day | 18 (37.5) | 6 (24.0) |
20 mg/day | 12 (25.0) | 13 (52.0) |
40 mg/day | 5 (10.4) | 3 (12.0) |
First dose leading to mUFC ≤ ULN | ||
Any dose | 45 (93.8) | 8 (32.0) |
2 mg/day | 1 (2.1) | 0 |
4 mg/day | 19 (39.6) | 6 (24.0) |
10 mg/day | 12 (25.0) | 1 (4.0) |
> 10 to < 20 mg/day | 1 (2.1) | 0 |
20 mg/day | 8 (16.7) | 1 (4.0) |
40 mg/day | 4 (8.3) | 0 |
Dose with longest duration | ||
2 mg/day | 3 (6.3) | 0 |
4 mg/day | 26 (54.2) | 15 (60.0) |
> 4 to < 10 mg/day | 1 (2.1) | 0 |
10 mg/day | 11 (22.9) | 4 (16.0) |
20 mg/day | 7 (14.6) | 6 (24.0) |
Average dose, mg/day | ||
Mean (SD) | 7.5 (4.22) | 9.1 (3.52) |
Median | 6.9 | 9.3 |
Q1, Q3 | 4.0, 10.7 | 6.2, 12.2 |
Duration of exposure, weeks | ||
N | 48 | 25 |
Mean (SD) | 11.6 (1.82) | 12.2 (0.41) |
Median | 12.0 | 12.0 |
Q1, Q3 | 11.9, 12.1 | 12.0, 12.4 |
Range | 2.0 to 13.0 | 11.7 to 13.7 |
mUFC = mean urinary free cortisol; Q1 = 25th percentile; Q3 = 75th percentile; SD = standard deviation; ULN = upper limit of normal (138 nmol per 24 hours).
Source: LINC 4 primary analysis Clinical Study Report.50
Concomitant Medications and Cointerventions
LINC 3 and LINC 4 studies summarized the data of concomitant medications and cointerventions using frequency counts and percentages.48,50
LINC 3 Study
In the LINC 3 study, 132 (96.4% of the enrolled patients) received concomitant medications and significant nondrug therapies after the start of the study. Concomitant medications were mainly prescribed to treat AEs. Of the 137 patients, the most common (in > 15% of patients) concomitant medications included paracetamol (40.1%), spironolactone (18.2%), hydrocortisone (17.5%), and potassium chloride (15.3%).48
During the first 26 weeks of treatment, 123 (89.8%) patients received concomitant medications and significant nondrug therapies. During the randomized withdrawal period, 22 patients (61.1%) on osilodrostat and 19 (54.3%) patients on placebo received concomitant medications.48
By Anatomical Therapeutic Chemical class and overall, 23 (16.8%) patients received antidiabetic concomitant medications. Thirteen (9.5%) patients received lipid-lowering concomitant medications. Seventy-two (52.6%) patients received blood pressure control concomitant medications.48
LINC 4 Trial
During the overall study period in the LINC 4 study, 72 patients (98.6%) received at least 1 concomitant medication. The most common concomitant medications other than steroids included drugs for the treatment of infections and inflammation, drugs for control of blood pressure, drugs for control of blood glucose, drugs for regulation of electrolytes, and vitamin and mineral supplements.50
Overall, the most common concomitant medications (≥ 10%) by preferred term were:
Paracetamol (28.8%)
Spironolactone, cholecalciferol (21.9%)
Calcium carbonate (20.5%)
Potassium chloride, metformin (17.8% each)
Amlodipine, levothyroxine (16.4% each)
Ibuprofen (15.1%)
Metformin hydrochloride, Lekovit CA (13.7% each)
Hydrocortisone, acetylsalicylic acid, atorvastatin, omeprazole, levothyroxine sodium (12.3% each)
Amoxi-clavulanico, hydrochlorothiazide, vitamin D NOS (11.0% each)
Overall, the most common Anatomical Therapeutic Chemical drug classes (≥ 30%) were:
Vitamin D and analogues (43.8%)
Other ophthalmologicals (39.7%)
Anilides, hydroxymethylglutaryl-coenzyme A reductase inhibitors (35.6% each)
Other drugs for local oral treatment (34.2%)
Glucocorticoids (32.9%)
Biguanides, corticosteroids, homeopathic preparations (31.5% each)
Anti-inflammatory preparations, including nonsteroids for topical use, plain corticosteroids (30.1% each)
Detailed Efficacy Results
Table 16: Summary of Primary End Point: Proportion of Complete Responders by Randomized Treatment From the LINC 3 (RAS) and LINC 4 (FAS) Trials
Variable | LINC 3 triala | LINC 4 trialb | ||
|---|---|---|---|---|
Osilodrostat (N = 36) | Placebo withdrawal (N = 35) | Osilodrostat (N = 48) | Placebo (N = 25) | |
Complete responder (mUFC ≤ ULN) at 8 weeks of RW period follow-up in the LINC 3 study and at 12 weeks of follow-up in the LINC 4 study | ||||
Number of patients included in the analysis | 36 | 34c | 48 | 25 |
Complete responder, n (%) | 31 (86.1) | 10 (29.4) | 37 (77.1) | 2 (8.0) |
95% CI of responder rate,d % | 70.50 to 95.33 | 15.10 to 47.48 | 62.7 to 88.0 | 1.0 to 26.0 |
RD (95% CI) | NR | NR | ||
OR (95% CI), stratified CMH exact test | 13.71 (3.73 to 53.44) | 43.40 (7.06 to 343.19) | ||
P value,e 2-sided | < 0.001 | < 0.0001 | ||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; FAS = full analysis set; ITT = intention to treat; mUFC = mean urinary free cortisol; NR = not reported; OR = odds ratio; RAS = randomized analysis set; RD = risk difference; RW = randomized withdrawal; ULN = upper limit of normal; vs. = versus.
Note: The normal range of 24-hour mUFC was 11 nmol per 24 hours to 138 nmol per 24 hours or 4 mcg per 24 hours to 50 mcg per 24 hours. The ULN for mUFC is 138 nmol per 24 hours.
aIn the LINC 3 study, a responder at the end of the RW period (week 34) is defined as a randomized patient who has mUFC ≤ ULN at week 34 and who was neither discontinued nor had an osilodrostat dose increase above the level at week 26 during the RW period of the study. Patients who discontinued during the RW period will be counted as nonresponders for the primary. In the LINC 3 study, the primary hypothesis was that complete response rates at week 34 were equal between osilodrostat and placebo. This was tested using a stratified Cochran-Mantel-Haenszel exact test on the RAS, following the ITT principle. The null hypothesis was rejected if the 2-sided P value was ≤ 0.05 and the OR favoured osilodrostat. Supportive analyses included unstratified Fisher exact tests and additional tests using the per-protocol set. Randomization was stratified by osilodrostat dose at week 24 (≤ 5 mg twice daily vs. > 5 mg twice daily) and history of pituitary irradiation (yes vs. no). These 2 factors were assumed to be independent. Based on these, 4 strata were defined: low dose with irradiation, low dose without irradiation, high dose with irradiation, and high dose without irradiation. The estimated distribution across these strata was 10%, 40%, 10%, and 40%, respectively.
bIn the LINC 4 study, a complete responder is defined as a patient who has mUFC ≤ ULN at the end of period 1 (week 12). Patients who discontinued before week 12, or who did not have a valid mUFC assessment at week 12, were counted as nonresponders. In the LINC 4 trial, the primary hypothesis was that the complete response rates at week 12 were equal between osilodrostat and placebo. This was also tested using a stratified Cochran-Mantel-Haenszel exact test. The null hypothesis was rejected if the 1-sided P value was ≤ 0.025 and the OR favoured osilodrostat. Supportive analyses included Fisher exact tests and per-protocol analyses. Although multiple imputation for missing data was planned, it was not performed due to the small number of missing cases. Patients without week 12 mUFC data were considered nonresponders. In the LINC 4 study, randomization was stratified by history of pituitary irradiation (yes versus no), with approximately 20% of patients expected to have received prior radiation based on the LINC 3 study data. This single stratification factor resulted in 2 strata: patients with a history of pituitary irradiation and those without.
cOne patient was randomized to placebo but never received treatment and withdrew from the study during the randomized withdrawal period on day 220.
dThe 2-sided 95% CIs were based on the exact (Clopper-Pearson) method.
eStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: LINC 3 interim Clinical Study Report,48 LINC 4 primary analysis Clinical Study Report,50 and sponsor’s submission.60
Bone Mineral Density
Table 17: Summary of Change in Bone Mineral Density From Baseline at Week 48 From the LINC 3 (FAS) and LINC 4 (FAS) Studies
Variable | Statistics | Actual (g/cm2) | Change from baseline, actual (g/cm2) | Change from baseline, percent (%) |
|---|---|---|---|---|
LINC 3, all patients, L1 to L4 lumbar spine | N | 137 | ||
Baseline | n | 113 | NA | NA |
Mean (SD) | 1.0 (0.18) | NA | NA | |
Median | 1.0 | NA | NA | |
Range | 0.6 to1.4 | NA | NA | |
Week 48 | n | 81 | 81 | 81 |
Mean (SD) | 1.0 (0.18) | 0 (0.05) | 3.0 (6.45) | |
Median | 1.0 | 0 | 2.1 | |
Range | 0.6 to 1.4 | −0.1 to 0.2 | −11.6 to 27.2 | |
95% CI | NA | (0.01 to 0.04) | (1.59 to 4.45) | |
LINC 4, all patients, L1 to L4 lumbar spine | N | 73 | ||
Baseline | n | 59 | NA | NA |
Mean (SD) | 1.0 (0.16) | NA | NA | |
Median | 1.0 | NA | NA | |
Range | 0.7 to 1.5 | NA | NA | |
Week 48 | n | 54 | 48 | 48 |
Mean (SD) | 1.0 (0.17) | 0 (0.04) | 1.5 (3.99) | |
Median | 1.0 | 0 | 1.8 | |
Range | 0.7 to 1.4 | −0.1 to 0.1 | −7.6 to 12.6 | |
95% CI | NA | (0 to 0) | (0.4 to 2.7) | |
BMD = bone mineral density; CI = confidence interval; DXA = dual-energy x-ray absorptiometry; FAS = full analysis set; NA = not applicable; SD = standard deviation.
Note: Assessment was performed using DXA scan. The 2-sided 95% CIs shown are on the mean change and mean percentage change from baseline. BMD assessments were not done in patients enrolled in Germany (number of study centres in Germany: 4; number of patients in Germany: not reported).
Source: LINC 3 interim Clinical Study Report.48
Cardiovascular-Related Metabolic Parameters
Table 18: Percentage Change From Baseline in Cardiovascular-Related Metabolic Parameters Associated With Cushing Disease at Selected Visits During the Core Period in the LINC 3 Study (FAS)
Parameter | All patients (N = 137) | |||||
|---|---|---|---|---|---|---|
Baseline | Week 12 | Week 24 | Week 34 (osilodrostat arm) | Week 34 (placebo withdrawal arm) | Week 48 | |
Mean (SD) | Mean (SD) % change from baseline (95% CI) | |||||
Fasting glucose (mg/dL) | n = 129 | n = 117 | n = 112 | n = 31 | n = 27 | n = 101 |
99.2 (29.83) | −7.0 (18.69) (−10.4 to −3.6) | −10.0 (15.74) (−13.0 to −7.1) | −10.7 (19.27) (−17.7 to −3.6) | −4.0 (12.29) (−8.9 to 0.9) | −7.1 (16.60) (−10.4 to −3.8) | |
Hemoglobin A1C (%) | n = 137 | n = 124 | n = 121 | n = 36 | n = 33 | n = 110 |
6.0 (0.96) | −5.0 (8.12) (−6.4 to −3.5) | −4.6 (8.80) (−6.2 to −3.0) | −6.8 (8.30) (−9.6 to −4.0) | −5.6 (6.99) (−8.1 to −3.1) | −5.4 (9.57) (−7.2 to −3.6) | |
Cholesterol (mmol/L) | n = 136 | n = 124 | n = 123 | n = 36 | n = 32 | n = 108 |
5.3 (1.16) | −8.9 (16.46) (−11.8 to −6.0) | −9.0 (17.13) (−12.1 to −6.0) | −7.3 (17.47) (−13.2 to −1.3) | −3.6 (16.88) (−9.7 to 2.5) | −8.8 (15.72) (−11.8 to −5.8) | |
LDL cholesterol (mmol/L) | n = 135 | n = 121 | n = 122 | n = 35 | n = 32 | n = 107 |
3.0 (0.95) | −5.0 (27.86) (−10.0 to 0) | −3.5 (30.69) (−9.0 to 2.0) | −3.0 (22.93) (−10.9 to 4.9) | −0.2 (0.85) (−0.5 to 0.2) | −5.4 (26.12) (−10.4 to −0.4) | |
HDL cholesterol (mmol/L) | n = 136 | n = 124 | n = 123 | n = 36 | n = 32 | n = 108 |
1.6 (0.45) | −19.9 (16.56) (−22.8 to −16.9) | −14.3 (15.05) (−17.0 to −11.6) | −15.9 (15.85) (−21.2 to −10.5) | −2.4 (12.26) (−6.8 to 2.0) | −14.4 (15.77) (−17.5 to −11.4) | |
Triglycerides (mmol/L) | n = 136 | n = 124 | n = 123 | n = 36 | n = 32 | n = 108 |
1.5 (1.31) | 15.2 (54.08) (5.6 to 24.8) | −1.8 (35.07) (−8.1 to 4.5) | 5.7 (38.29) (−7.2 to 18.7) | 2.8 (44.64) (−13.3 to 18.9) | 5.4 (102.02) (−14.0 to 24.9) | |
SBP (mm Hg) | n = 137 | n = 130 | n = 124 | n = 36 | n = 34 | n = 111 |
132.2 (15.14) | −4.8 (12.55) (−7.0 to −2.6) | −4.1 (11.85) (−6.2 to −2.0) | −5.2 (12.33) (−9.4 to −1.0) | −4.2 (12.0) (−8.4 to 0) | −6.8 (11.40) (−8.9 to −4.7) | |
DBP (mm Hg) | n = 137 | n = 130 | n = 124 | n = 36 | n = 34 | n = 111 |
85.3 (10.56) | −4.7 (12.99) (−7.0 to −2.5) | −3.8 (13.41) (−6.2 to −1.4) | −6.6 (13.03) (−11.0 to −2.2) | −5.4 (12.64) (−9.8 to −1.0) | −6.6 (12.72) (−9.0 to −4.2) | |
Weight (kg) | n = 137 | n = 130 | n = 124 | n = 36 | n = 34 | n = 112 |
80.8 (22.44) | −0.9 (4.11) (−1.6 to −0.2) | −3.0 (5.24) (−3.9 to −2.1) | −2.6 (6.08) (−4.7 to −0.6) | −3.4 (9.80) (−6.9 to 0) | −4.6 (6.72) (−5.8 to −3.3) | |
BMI (kg/m2) | n = 137 | n = 130 | n = 124 | n = 36 | n = 34 | n = 112 |
30.3 (7.77) | −0.9 (4.10) (−1.6 to −0.2) | −3.0 (5.24) (−3.9 to −2.1) | −2.6 (6.09) (−4.7 to −0.5) | −3.4 (9.81) (−6.8 to 0) | −4.6 (6.73) (−5.8 to −3.3) | |
Waist circumference (cm) | n = 133 | n = 125 | n = 116 | n = 35 | n = 32 | n = 109 |
103.4 (19.34) | −0.9 (6.54) (−2.1 to 0.2) | −2.6 (6.97) (−3.9 to −1.3) | −3.3 (5.01) (−5.1 to −1.6) | −3.7 (10.50) (−7.5 to 0.1) | −4.2 (7.63) (−5.7 to −2.8) | |
BMI = body mass index; DBP = diastolic blood pressure; FAS = full analysis set; HDL = high-density lipoprotein; LDL = low-density lipoprotein; SBP = systolic blood pressure; SD = standard deviation.
Source: LINC 3 interim Clinical Study Report.48
Table 19: Percentage Change From Baseline in Cardiovascular-Related Metabolic Parameters Associated With Cushing Disease at Selected Visits During the Core Period in the LINC 4 Study (FAS)
Parameter | Baseline | Week 12 (osilodrostat arm) | Week 12 (placebo arm) | Week 36 | Week 48 |
|---|---|---|---|---|---|
Mean (SD) | Mean (SD) % change from baseline (95% CI) | ||||
Fasting glucose (mg/dL) | n = 71 | n = 44 | n = 23 | n = 67 | n = 62 |
95.3 (17.30) | −3.1 (13.87) (−7.4 to 1.1) | −0.9 (11.30) (−5.8 to 4.0) | −3.3 (12.66) (−6.4 to −0.3) | −1.7 (14.60) (−5.4 to 2.0) | |
Hemoglobin A1C (%) | n = 73 | n = 46 | n = 24 | n = 69 | n = 62 |
5.9 (0.82) | −3.5 (6.36) (−5.4 to −1.6) | −0.7 (4.76) (−2.7 to 1.3) | −2.4 (7.78) (−4.3 to −0.6) | −1.1 (7.86) (−3.1 to 0.9) | |
Cholesterol (mmol/L) | n = 70 | n = 44 | n = 24 | n = 69 | n = 64 |
5.5 (1.26) | −12.8 (16.41) (−17.8 to −7.8) | 0.6 (13.28) (−5.0 to 6.2) | −11.7 (18.32) (−16.1 to −7.3) | −7.1 (23.72) (−13.0 to −1.1) | |
LDL cholesterol (mmol/L) | n = 69 | n = 44 | n = 23 | n = 68 | n = 62 |
3.3 (1.11) | −9.1 (35.64) (−19.9 to 1.7) | 4.5 (19.27) (−3.9 to 12.8) | −9.4 (27.90) (−16.1 to −2.6) | −6.8 (27.40) (−13.7 to 0.2) | |
HDL cholesterol (mmol/L) | n = 70 | n = 44 | n = 24 | n = 69 | n = 64 |
1.6 (0.37) | −19.9 (14.87) (−24.4 to −15.3) | 0.1 (17.45) (−7.2 to 7.5) | −15.2 (14.47) (−18.7 to −11.7) | −10.5 (16.23) (−14.5 to −6.4) | |
Triglycerides (mmol/L) | n = 70 | n = 44 | n = 24 | n = 69 | n = 64 |
1.6 (0.82) | 5.4 (43.70) (−7.8 to 18.7) | −7.1 (31.80) (−20.5 to 6.4) | −2.9 (34.40) (−11.1 to 5.4) | 2.9 (48.61) (−9.2 to 15.1) | |
Standing SBP (mm Hg) | n = 71 | n = 44 | n = 24 | n = 67 | n = 63 |
131.5 (18.57) | −4.1 (12.07) (−7.8 to −0.5) | −0.2 (9.53) (−4.2 to 3.9) | −5.4 (14.06) (−8.9 to −2.0) | −6.2 (14.51) (−9.9 to −2.6) | |
Supine SBP (mm Hg) | n = 73 | n = 46 | n = 24 | n = 69 | n = 64 |
130.4 (18.41) | −5.0 (12.21) (−8.6 to −1.3) | 2.4 (12.54) (−2.9 to 7.7) | −5.0 (13.73) (−8.3 to −1.7) | −4.6 (14.05) (−8.1 to −1.1) | |
Standing DBP (mm Hg) | n = 71 | n = 44 | n = 24 | n = 67 | n = 63 |
87.5 (12.03) | −4.6 (11.73) (−8.2 to −1.1) | −1.0 (11.13) (−5.7 to 3.7) | −5.2 (14.35) (−8.7 to −1.7) | −4.0 (14.07) (−7.5 to −0.4) | |
Supine DBP (mm Hg) | n = 73 | n = 46 | n = 24 | n = 69 | n = 64 |
83.0 (11.52) | −6.6 (12.02) (−10.1, −3.0) | 0.6 (10.03) (−3.6 to 4.9) | −6.4 (14.31) (−9.8 to −2.9) | −5.2 (13.07) (−8.5 to −2.0) | |
Weight (kg) | n = 73 | n = 46 | n = 24 | n = 69 | n = 64 |
78.3 (17.17) | −1.1 (3.96) (−2.3 to 0) | −0.2 (2.83) (−1.4 to 1.0) | −4.6 (6.88) (−6.3 to −2.9) | −5.4 (7.78) (−7.3 to −3.4) | |
Waist circumference (cm) | n = 73 | n = 46 | n = 24 | n = 69 | n = 64 |
102.8 (16.41) | −1.0 (4.35) (−2.3 to 0.3) | −0.4 (3.56) (−1.9 to 1.1) | −3.2 (6.44) (−4.7 to −1.6) | −4.4 (5.74) (−5.8 to −3.0) | |
CI = confidence interval; DBP = diastolic blood pressure; FAS = full analysis set; HDL = high-density lipoprotein; LDL = low-density lipoprotein; NR = not reported; SBP = systolic blood pressure; SD = standard deviation.
Note: 2-sided 95% CI shown are on the mean percentage change from baseline. n: Number of patients with data at baseline.
Source: LINC 4 primary analysis Clinical Study Report.50
Selected Liver Function Test Results
Table 20: Liver Biochemical Parameters From the LINC 3 Trial (Safety Set)
Characteristic | Randomized withdrawal period | Core study period | ||||
|---|---|---|---|---|---|---|
Osilodrostat (N = 36) | Placebo (N = 35) | Osilodrostat (osilodrostat arm) (N = 36) | Osilodrostat (placebo withdrawal arm) (N = 35) | Nonrandomized (N = 66) | All patients (N = 137) | |
Worst postbaseline values | ||||||
ALT category, n (%) | ||||||
ALT > 3 × ULN | 0 | 0 | 0 | 2 (5.7) | 2 (3.0) | 4 (2.9) |
ALT > 5 × ULN | NR | NR | 0 | 1 (2.9) | 2 (3.0) | 3 (2.2) |
ALT > 8 × ULN | NR | NR | 0 | 1 (2.9) | 1 (1.5) | 2 (1.5) |
ALT > 10 × ULN | NR | NR | 0 | 1 (2.9) | 0 | 1 (0.7) |
ALT > 20 × ULN | NR | NR | 0 | 0 | 0 | 0 |
AST category, n (%) | ||||||
AST > 3 × ULN | 0 | 0 | 0 | 1 (2.9) | 2 (3.0) | 3 (2.2) |
AST > 5 × ULN | NR | NR | 0 | 1 (2.9) | 0 | 1 (0.7) |
AST > 8 × ULN | NR | NR | 0 | 1 (2.9) | 0 | 1 (0.7) |
AST > 10 × ULN | NR | NR | 0 | 0 | 0 | 0 |
AST > 20 × ULN | NR | NR | 0 | 0 | 0 | 0 |
ALT or AST category, n (%) | ||||||
ALT or AST > ULN but ≤ 3 × ULN | 5 (13.9) | 0 | 11 (30.6) | 13 (37.1) | 26 (39.4) | 50 (36.5) |
ALT or AST > 3 × ULN | 0 | 0 | 0 | 2 (5.7) | 3 (4.5) | 5 (3.6) |
ALT or AST > 5 × ULN | 0 | 0 | 0 | 1 (2.9) | 2 (3.0) | 3 (2.2) |
ALT or AST > 8 × ULN | NR | NR | 0 | 1 (2.9) | 1 (1.5) | 2 (1.5) |
ALT or AST > 10 × ULN | 0 | 0 | 0 | 1 (2.9) | 0 | 1 (0.7) |
ALT or AST > 20 × ULN | 0 | 0 | 0 | 0 | 0 | 0 |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; NR = not reported; ULN = upper limit of normal.
Note: Categories are based on the worst postbaseline value for any specific parameter. Categories with multiple parameters are based on the worst postbaseline value for each parameter. The worst postbaseline value refers to the maximum postbaseline value. Patients with both an ALT > 3 × ULN and an AST ≤ 3 × ULN at the same or different time points were counted once in the ALT or AST > ULN but ≤ 3 × ULN category. Patients with both an AST > 3 × ULN and an ALT ≤ 3 × ULN at the same or different time points were counted once in the ALT or AST > ULN but ≤ 3 × ULN category. Unscheduled visits are considered for this analysis. Concurrent measurements are those occurring in the same assessment sample. Value for the ULN for ALT for patients aged 13 years and older = 0 U/L to 48 U/L (conventional). Value for the ULN for AST for patients aged 3 to 64 years = 0 U/L to 42 U/L (conventional). Value for the ULN for AST for patients aged 65 years and older = 0 U/L to 55 U/L (conventional).
Sources: LINC 3 interim Clinical Study Report,48 LINC 3 final Clinical Study Report,49 and sponsor’s submissions.59,60
Table 21: Liver Biochemical Parameters From the LINC 4 Trial (Safety Set)
Characteristic | Placebo-controlled period | Overall study period | |||
|---|---|---|---|---|---|
Osilodrostat (N = 48) | Placebo (N = 25) | Osilodrostat (Osilodrostat arm) (N = 48) | Osilodrostat (placebo arm) (N = 25) | All patients (N = 73) | |
Worst postbaseline values | |||||
ALT category, n (%) | |||||
ALT > 3 × ULN | 2 (4.2) | 0 | 2 (4.2) | 1 (4.0) | 3 (4.1) |
ALT > 5 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT > 8 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT > 10 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT > 20 × ULN | 0 | 0 | 0 | 0 | 0 |
AST category, n (%) | |||||
AST > 3 × ULN | 0 | 0 | 0 | 1 (4.0) | 1 (1.4) |
AST > 5 × ULN | 0 | 0 | 0 | 0 | 0 |
AST > 8 × ULN | 0 | 0 | 0 | 0 | 0 |
AST > 10 × ULN | 0 | 0 | 0 | 0 | 0 |
AST > 20 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT or AST category, n (%) | |||||
ALT or AST > 3 × ULN | 2 (4.2) | 0 | 2 (4.2) | 1 (4.0) | 3 (4.1) |
ALT or AST > 5 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT or AST > 8 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT or AST > 10 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT or AST > 20 × ULN | 0 | 0 | 0 | 0 | 0 |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal.
Note: The all-patients column excludes data in placebo arm collected during placebo-control period. The osilodrostat (placebo arm) column excludes data in the placebo arm that was collected during the placebo-controlled period. Concurrent measurements are those occurring in the same assessment sample. Value for ULN for ALT for patients aged 13 years and older = 0 U/L to 48 U/L (conventional). Value for ULN for AST for patients aged 3 to 64 years = 0 U/L to 42 U/L (conventional). Value for ULN for AST for patients aged 65 years and older = 0 U/L to 55 U/L (conventional).
Sources: LINC 4 primary analysis Clinical Study Report50 and sponsor’s submissions.59,60
Detailed Harms Results
Table 22: Summary of Harms Results From the LINC 3 and LINC 4 Trials (Safety Sets)
AEs | LINC 3 trial, all patients (N = 137) | LINC 4 trial, all patients (N = 73) |
|---|---|---|
Most common (> 10%) AEs, n (%) | ||
Patients with ≥ 1 AE | 137 (100) | 73 (100) |
Nausea | 57 (41.6) | 27 (37.0) |
Headache | 46 (33.6) | 24 (32.9) |
Adrenal insufficiency | 38 (27.7) | 18 (24.7) |
Nasopharyngitis | 31 (22.6) | 3 (4.1) |
Vomiting | 30 (21.9) | 9 (12.3) |
Glucocorticoid deficiency | 29 (21.2) | NR |
Arthralgia | 27 (19.7) | 33 (45.2) |
Back pain | 27 (19.7) | 10 (13.7) |
Diarrhea | 25 (18.2) | 17 (23.3) |
Influenza | 24 (17.5) | NR |
Asthenia | 23 (16.8) | 15 (20.5) |
Blood corticotrophin increased | 23 (16.8) | 2 (2.7) |
Edema peripheral | 21 (15.3) | 12 (16.4) |
Pyrexia | 20 (14.6) | 4 (5.5) |
Urinary tract infection | 20 (14.6) | 11 (15.1) |
Decreased appetite | 19 (13.9) | 33 (45.2) |
Dizziness | 19 (13.9) | 19 (26.0) |
Hormone level abnormal | 19 (13.9) | NR |
Myalgia | 19 (13.9) | 19 (26.0) |
Hypokalemia | 18 (13.1) | 7 (9.6) |
Rash | 18 (13.1) | 2 (2.7) |
Cough | 17 (12.4) | 4 (5.5) |
Hypertension | 17 (12.4) | 16 (21.9) |
Blood testosterone increased | 15 (10.9) | 18 (24.7) |
Dyspepsia | 14 (10.2) | 2 (2.7) |
Upper respiratory tract infection | 12 (8.8) | 15 (20.5) |
Fatigue | 39 (28.5) | 28 (38.4) |
Malaise | 9 (6.6) | 2 (2.7) |
Alanine aminotransferase increased | 4 (2.9) | 6 (8.2) |
Aspartate aminotransferase increased | 4 (2.9) | 5 (6.8) |
Constipation | 10 (7.3) | 2 (2.7) |
Pruritus | 7 (5.1) | 9 (12.3) |
SAEs (> 2%), n (%) | ||
Patients with ≥ 1 SAE | 50 (36.5) | 8 (11.0) |
Adrenal insufficiency | 8 (5.8) | 2 (2.7) |
Pituitary tumour | 5 (3.6) | NR |
Adrenocortical insufficiency acute | 3 (2.2) | NR |
Gastroenteritis | 3 (2.2) | NR |
Patients who stopped treatment due to AEs, n (%) | ||
Patients with ≥ 1 AE leading to study drug discontinuation | 18 (13.1) | 8 (11.0) |
Adrenal insufficiency | 4 (2.9) | 2 (2.7) |
Visual impairment | 1 (0.7) | NR |
Asthenia | 1 (0.7) | NR |
Fatigue | 1 (0.7) | NR |
Blood pressure diastolic increased | 1 (0.7) | NR |
Blood pressure systolic increased | 1 (0.7) | NR |
Electrocardiogram QT prolonged | 1 (0.7) | NR |
Hypokalemia | 1 (0.7) | 1 (1.4) |
Pain in extremity | 1 (0.7) | NR |
Pituitary tumour | 4 (2.9) | 1 (1.4) |
Pituitary tumour benign | 2 (1.5) | 1 (1.4) |
Malignant pituitary tumour | 1 (0.7) | NR |
Tumour invasion | 1 (0.7) | NR |
Headache | 1 (0.7) | 1 (1.4) |
Paresis cranial nerve | 1 (0.7) | NR |
VIth nerve paralysis | 1 (0.7) | NR |
Rash | 1 (0.7) | NR |
Hyperbilirubinemia | NR | 1 (1.4) |
Arthralgia | NR | 1 (1.4) |
Depression | NR | 1 (1.4) |
Death, n (%) | ||
Patients who died | 1 (0.7) | 0 |
Completed suicide | 1 (0.7) | 0 |
AEs of special interest, n (%) | ||
Hypocortisolism-related AEs | 70 (51.1) | 20 (27.4) |
Adrenal hormone precursor accumulation–related AEs | 58 (42.3) | 45 (61.6) |
Pituitary tumour enlargement–related AEsa | 3 (2.2) | 4 (5.5) |
Arrhythmogenic potential and QT prolongation AEs | 6 (4.4) | 3 (4.1) |
AE = adverse event; NR = not reported; SAE = serious adverse event.
Note: In both studies, a patient with multiple occurrences of an AE under one treatment was counted only once in the AE category for that treatment. In the LINC 3 study, a patient with multiple AEs was counted only once in the total row. In the LINC 4 trial, for AEs, the “all patients” column excludes data in placebo arm collected during placebo-control period.
aLINC 3 study: Includes only patients with diplopia, cranial nerve palsy, extraocular muscle paresis, pituitary infarction, and visual field defect.
Source: LINC 3 interim Clinical Study Report48 and LINC 4 primary analysis Clinical Study Report.50
Table 23: Summary of Harms Results From the LINC 4 Trial During the Placebo-Controlled Period by Treatment Group (Safety Set)
AEs | Osilodrostat (N = 48) | Placebo (N = 25) |
|---|---|---|
Most common (> 20%) AEs, n (%) | ||
Patients with ≥ 1 AE | 46 (95.8) | 23 (92.0) |
Decreased appetite | 18 (37.5) | 4 (16.0) |
Arthralgia | 17 (35.4) | 2 (8.0) |
Nausea | 15 (31.3) | 3 (12.0) |
Fatigue | 12 (25.0) | 4 (16.0) |
Myalgia | 11 (22.9) | 1 (4.0) |
Asthenia | 11 (22.9) | 0 |
Diarrhea | 10 (20.8) | 0 |
Dizziness | 9 (18.8) | 4 (16.0) |
Hypertension | 8 (16.7) | 7 (28.0) |
Headache | 7 (14.6) | 6 (24.0) |
Adrenal insufficiency | 7 (14.6) | 0 |
Blood testosterone increased | 5 (10.4) | 0 |
Upper respiratory tract infection | 5 (10.4) | 0 |
SAEs (> 2%), n (%) | ||
Patients with ≥ 1 SAEa | 2 (4.2) | 1 (4.0) |
Patients who stopped treatment due to AEs, n (%) | ||
Patients with ≥ 1 AE leading to study drug discontinuation | 1 (2.1) | 0 |
Death, n (%) | ||
Patients who died | 0 | 0 |
AEs of special interest, n (%) | ||
Adrenal hormone precursor accumulation–related AEs | 21 (43.8) | 9 (36.0) |
Hypocortisolism-related AEs | 7 (14.6) | 0 |
Pituitary tumour enlargement–related AEs | 0 | 0 |
Arrhythmogenic potential and QT prolongation AEs | 0 | 0 |
AE = adverse event; SAE = serious adverse event.
aTwo patients in the osilodrostat arm had 3 instances of SAEs (dengue fever, electrocardiogram T wave inversion, and erosive duodenitis). One patient in the placebo arm had 1 SAE (pneumonia).
Source: LINC 4 primary analysis Clinical Study Report.50
Appendix 5: Methods and Results of the Long-Term Extension Studies
Please note that this appendix has not been copy-edited.
Study Design and Objectives
Interventions
LINC 3 Study Extension
The study treatment consisted of osilodrostat in the form of film-coated tablets for oral administration, in the following strengths: 1 mg, 5 mg, 10 mg, and 20 mg.
Stable doses of concomitant medications (except those for hypercortisolism) were allowed during the study. Concomitant medications that are known for being strong inhibitors or inducers of CYP3A4/5 and CYP2D6 were to be used with caution with osilodrostat.
Eplerenone and glucocorticoids (e.g., prednisone, prednisolone, and dexamethasone), were prohibited except under certain conditions.
Preclinical and preliminary clinical data indicated there is a risk of corrected QT prolongation in humans. Therefore, the use of medications with a “known risk to cause Torsades de pointes (TdP)” and with “possible risk to cause TdP” concomitantly was prohibited. If a patient required a long-term medication from the 2 categories mentioned previously, and there was no appropriate alternative medication available, the patient was discontinued from the study.
Other drug treatments for Cushing disease were prohibited during the study.
LINC 4 Study Extension
Osilodrostat treatment continued until the end of the optional extension period, at the established effective dose in the core period unless a change was required based on mUFC results, or an AE occurred that required dose reduction or temporary withholding of osilodrostat.
All prescription medications and over-the-counter drugs taken before the start of the study and during the study, were recorded. Stable doses of concomitant medications (except those for hypercortisolism) were allowed during the study. Of note, eplerenone, spironolactone, cyproterone acetate, or finasteride, were allowed to be used for specific comorbidities.
Outcomes
The extensions of the LINC 3 and LINC 4 studies were similar to the core trials in terms of the efficacy and safety outcomes assessed.
Statistical Analysis
LINC 3 Extension
After the primary end point analysis, the interim Clinical Study Report was completed and additional protocol deviations were reported. These affected the per-protocol populations used to perform the per-protocol analyses of the primary and key secondary end points. As such, updated supportive analyses were conducted and reported in the final Clinical Study Report .
The study was powered based on the primary and key secondary analyses. All efficacy analyses presented in the final Clinical Study Report were considered supportive, and no formal statistical testing was performed.
LINC 4 Extension
Please refer to the LINC 4 trial information on statistical analyses for all end points other than those presented subsequently.
Overall Response Rate
The proportion of patients with a complete response (enrolled patients with mUFC ≤ ULN), partial response (enrolled patients with mUFC > ULN and at least a 50% reduction from baseline), and an overall response (enrolled patients with mUFC ≤ ULN or at least a 50% reduction in mUFC from baseline and > ULN) were summarized using point estimates for all scheduled visits in the core and extension phase when UFC was collected.
Due to the COVID-19 pandemic, planned visits could have been cancelled, potentially resulting in missing UFC evaluations. Patients at a visit with insufficient information due to any reason were considered to be a nonresponder for that visit. The numbers of patients who did not experience a response due to missing visits linked to the pandemic (recorded as a COVID-19–related protocol deviation) were summarized.
To explore the impact of the COVID-19 pandemic in the optional extension phase, individual patient doses and overall response status over time were graphically displayed together with the pandemic start date.
Patient-Reported Outcomes
The patient-reported outcome scales reported were CushingQoL score (primary patient-reported outcome variable of interest), EQ-5D utility index and visual analogue scale scores and BDI-II total score (secondary patient-reported outcome variables of interest). Descriptive statistics were used to summarize the actual and change from baseline for the scores at each scheduled assessment.
Safety Outcomes
The assessment of safety was based mainly on the frequency and severity of AEs and on the number of laboratory values that fell outside of predetermined ranges. Other safety data (e.g., ECGs, findings from Holter recordings, vital signs, and special tests) were also presented.
Safety analyses for the overall study period focused solely on the safety of osilodrostat treatment. These analyses tabulate data for the osilodrostat arm (excluding the placebo-control period), and overall patient data (excluding safety data from the placebo arm collected during the placebo-control period). As in the primary analysis, the overall observation period was divided into 3 mutually exclusive segments:
Pretreatment period: From the day of the patient’s informed consent to the day before the first dose of study medication.
On-treatment period: From the day of the first dose of study medication to 30 days after the last dose of study medication.
Posttreatment period: Starting at 31 days after the last dose of study medication.
Patient Disposition in the Included Studies
Table 24: Patient Disposition in the LINC 3 Trial Extension (FAS)
Disposition and reasons, n (%) | All patients (N = 137) |
|---|---|
Patients enrolled and treated | 137 (100) |
Discontinued at any timea | 58 (42.3) |
Primary reason for discontinuation at any time | |
Adverse event | 27 (19.7) |
Death | 2 (1.5) |
Physician decision | 8 (5.8) |
Patient withdrew consent | 6 (4.4) |
Patient or guardian decision | 14 (10.2) |
Unsatisfactory therapeutic effect | 1 (0.7) |
Completed week 48 (core phase) and entered extension phasea | 106 (77.4) |
Ongoing in extension phase | 0 |
Discontinued study in extension phase | 34 (24.8) |
Primary reason for discontinuation in the extension phase | |
Adverse event | 12 (8.8) |
Death | 2 (1.5) |
Physician decision | 5 (3.6) |
Patient withdrew consent | 2 (1.5) |
Patient/guardian decision | 12 (8.8) |
Unsatisfactory therapeutic effect | 1 (0.7) |
Discontinued at or before week 72 but after week 48 | 8 (5.8) |
Discontinued before week 96 but after week 72 | 8 (5.8) |
Discontinued after week 96 | 18 (13.1) |
Completed extension phase | 72 (52.6) |
FAS = full analysis set.
Note: The data presented is all data collected up to the data cut-off date of 4 December 2019. N is the total number of patients enrolled and treated. % based on N.
aPatients who completed week 48 and did not enter extension phase were not counted as discontinuations.
Source: LINC 3 final Clinical Study Report.49
Table 25: Patient Disposition in the LINC 4 Trial Extension (All Randomized Patients)
Disposition and reasons, n (%) | Osilodrostat (N = 49) | Placebo (N = 25) | All patients (N = 74) |
|---|---|---|---|
Patients randomized | 49 (100) | 25 (100) | 74 (100) |
Not treated | 1 (2.0) | 0 | 1 (1.4) |
Primary reason for not being treated | |||
Adverse event | 1 (2.0) | 0 | 1 (1.4) |
Treated | 48 (98.0) | 25 (100) | 73 (98.6) |
Completed the core phase | 42 (87.5) | 23 (92.0) | 65 (89.0) |
Completed the core phase and did not enter the extension phasea | 4 (8.3) | 1 (4.0) | 5 (6.8) |
Completed the core phase and entered the extension phase | 38 (79.2) | 22 (88.0) | 60 (82.2) |
Completed the extension phase | 33 (68.8) | 20 (80.0) | 53 (72.6) |
Discontinued at any time | 11 (22.9) | 4 (16.0) | 15 (20.5) |
Primary reason for discontinuation | |||
Adverse event | 6 (12.5) | 3 (12.0) | 9 (12.3) |
Physician decision | 1 (2.1) | 1 (4.0) | 2 (2.7) |
Patient or guardian decision | 4 (8.3) | 0 | 4 (5.5) |
Discontinued during the extension phase | 5 (10.4) | 2 (8.0) | 7 (9.6) |
Primary reason for discontinuation during the extension phase | |||
Adverse event | 5 (10.4) | 1 (4.0) | 6 (8.2) |
Physician decision | 0 | 1 (4.0) | 1 (1.4) |
Discontinued at or before week 72 but after week 48 | 4 (8.3) | 2 (8.0) | 6 (8.2) |
Discontinued after week 96 | 1 (2.1) | 0 | 1 (1.4) |
Note: N is the total number of randomized patients. The percentage for the first 3 rows is based on N; % for remaining rows is based on randomized and treated patients.
aPatients who completed week 48 and did not enter the extension phase were not counted as discontinuations.
Source: LINC 4 final Clinical Study Report.54
Detailed Efficacy Results
Table 26: Proportion of mUFC Responders at Selected Time Points During the Study in the LINC 3 Extension Trial (FAS)
Visit | All patients (N = 137) |
|---|---|
Week 48 (end of core period) | |
Complete responder: n/N’(%) 95% CI | 91/137 (66.4) (57.86, 74.26) |
Overall responder: n/N’ (%) 95% CI | 104/137 (75.9) (67.87, 82.80) |
Week 72 (End of extension period) | |
Complete responder: n/N’ (%) 95% CI | 86/106 (81.1) (72.38, 88.08) |
Overall responder: n/N’ (%) 95% CI | 94/106 (88.7) (81.06, 94.01) |
Week 120 | |
Complete responder: n/N’ (%) | 62/100 (62.0) |
Overall responder: n/N’ (%) | 73/100 (73.0) |
Last observed value | |
Complete responder: n/N’ (%) 95% CI | 86/137 (62.8) (54.11 to 70.87) |
Overall responder: n/N’ (%) 95% CI | 113/137 (82.5) (75.06 to 88.44) |
CI = confidence interval; FAS = full analysis set; mUFC = mean urinary free cortisol.
Note: The denominator (N’) is the FAS for all visits up to week 48, excluding assessments when patients were receiving placebo.
Beyond week 48, the denominator uses the following rules. First, patients who declined to enter the optional extension period after completion of the core phase were excluded. Second, patients who discontinued before the data cut-off date for the final database lock were included up to the furthest scheduled visit they could have completed if they had not discontinued early based on data cut-off and last completed based on the date of last completed schedule visit and analyses cut-off date.
If included in the analysis for calculating the proportion of responders at a given time point, patients who discontinued before this time point were counted as nonresponders. Patients with missing mUFC at a visit were counted as a nonresponder.
Two-sided 95% CIs for proportions are based on the exact (Clopper-Pearson) method.
Source: LINC 3 final Clinical Study Report.49
Table 27: Proportion of mUFC Responders at Week 48 and Beyond by Randomized Treatment in the LINC 4 Trial (FAS)
Visit | Osilodrostat (N = 48) | Placebo (N = 25) | All patients (N = 137) |
|---|---|---|---|
Week 48 | |||
Complete responders: n/N (%) | 34/48 (70.8) | 16/25 (64.0) | 50/73 (68.5) |
Partial responders: n/N (%) | 5/48 (10.4) | 3/25 (12.0) | 8/73 (11.0) |
Overall responders (complete or partial responders): n (%) | 39/48 (81.3) | 19/25 (76.0) | 58/73 (79.5) |
Nonresponders: n (%) | 9/48 (18.8) | 6/25 (24.0) | 15/73 (20.5) |
Week 72 | |||
Complete responders: n (%) | 25/41 (61.0) | 15/24 (62.5) | 40/65 (61.5) |
Partial responders: n (%) | 4/41 (9.8) | 1/24 (4.2) | 5/65 (7.7) |
Overall responders (complete or partial responders): n (%) | 29/41 (70.7) | 16/24 (66.7) | 45/65 (69.2) |
Nonresponders: n (%) | 12/41 (29.3) | 8/24 (33.3) | 20/65 (30.8) |
Insufficient information due to the pandemic: n (%) | 1/41 (2.4) | 3/24 (12.5) | 4/65 (6.2) |
Discontinued: n (%) | 8/41 (19.5) | 4/24 (16.7) | 12/65 (18.5) |
Insufficient information due to any other reason: n (%) | 1/41 (2.4) | 0 | 1/65 (1.5) |
mUFC not meeting overall response: n (%) | 2/41 (4.9) | 1/24 (4.2) | 3/65 (4.6) |
Week 96 | |||
Complete responders: n (%) | 4/19 (21.1) | 6/13 (46.2) | 10/32 (31.3) |
Partial responders: n (%) | 1/19 (5.3) | 0/13 | 1/32 (3.1) |
Overall responders (complete or partial responders): n (%) | 5/19 (26.3) | 6/13 (46.2) | 11/32 (34.4) |
Nonresponders: n (%) | 14/19 (73.7) | 7/13 (53.8) | 21/32 (65.6) |
Insufficient information due to the pandemic: n (%) | 4/19 (21.1) | 2/13 (15.4) | 6/32 (18.8) |
Discontinued: n (%) | 9/19 (47.4) | 4/13 (30.8) | 13/32 (40.6) |
Insufficient information due to any other reason: n (%) | 0 | 0 | 0 |
mUFC not meeting overall response: n (%) | 1/19 (5.3) | 1/13 (7.7) | 2/32 (6.3) |
End of extension phase | |||
Complete responders: n (%) | 28/38 (73.7) | 14/20 (70.0) | 42/58 (72.4) |
Partial responders: n (%) | 3/38 (7.9) | 2/20 (10.0) | 5/58 (8.6) |
Overall responders (complete or partial responders): n (%) | 31/38 (81.6) | 16/20 (80.0) | 47/58 (81.0) |
Nonresponders: n (%) | 7/38 (18.4) | 4/20 (20.0) | 11/58 (19.0) |
Insufficient information due to the pandemic: n (%) | 1/38 (2.6) | 1/20 (5.0) | 2/58 (3.4) |
Discontinued: n (%) | 0 | 0 | 0 |
Insufficient information due to any other reason: n (%) | 1/38 (2.6) | 0 | 1/58 (1.7) |
mUFC not meeting overall response: n (%) | 5/38 (13.2) | 3/20 (15.0) | 8/58 (13.8) |
CI = confidence interval; FAS = full analysis set; mUFC = mean urinary free cortisol.
Note: Up to and including week 48, the denominator is the FAS. Beyond week 48, patients who completed the extension phase were only included in the denominator until their individual end-of-study visit. Patients who discontinued before the data cut-off date for the final database lock were included for visits they could have completed if they had continued until week 96 or the data cut-off for the analysis (whatever occured earlier), but were excluded for further visits.
Complete responder:mUFC ≤ 1.0 × ULN; partial responder: mUFC > 1.0 × ULN but ≥ 50% reduction from baseline.
Overall responder: Either a complete or partial responder.
Nonresponder: Neither a complete nor partial responder.
If a patient has a missing mUFC at a visit, they were counted as nonresponders.
The 2-sided 95% CIs for proportions are based on the exact (Clopper-Pearson) method.
Source: LINC 4 final Clinical Study Report.54
Detailed Harms Results
Table 28: Summary of Harms Results From the Long-Term Extension Periods of the LINC 3 and LINC 4 Studies (Safety Set)
AEs | LINC 3 trial; (N = 137) | LINC 4 trial (N = 73) |
|---|---|---|
Most common (> 10%) AEs, n (%) | ||
≥ 1 AE | 137 (100) | 72 (98.6) |
Nausea | 62 (45.3) | 27 (37.0) |
Headache | 50 (36.5) | 25 (34.2) |
Fatigue | 45 (32.8) | 72 (98.6) |
Adrenal insufficiency | 40 (29.2) | 19 (26.0) |
Vomiting | 34 (24.8) | 9 (12.3) |
Nasopharyngitis | 33 (24.1) | 4 (5.5) |
Arthralgia | 29 (21.2) | 33 (45.2) |
Back pain | 29 (21.2) | 10 (13.7) |
Blood corticotrophin increased | 28 (20.4) | 2 (2.7) |
Glucocorticoid deficiency | 28 (20.4) | — |
Asthenia | 27 (19.7) | 17 (23.3) |
Diarrhea | 27 (19.7) | 17 (23.3) |
Dizziness | 26 (19.0) | 22 (30.1) |
Influenza | 26 (19.0) | 4 (5.5) |
Urinary tract infection | 25 (18.2) | 12 (16.4) |
Hypertension | 24 (17.5) | 16 (21.9) |
Decreased appetite | 22 (16.1) | 34 (46.6) |
Edema peripheral | 22 (16.1) | 12 (16.4) |
Pyrexia | 21 (15.3) | 5 (6.8) |
Rash | 21 (15.3) | 2 (2.7) |
Cough | 20 (14.6) | 4 (5.5) |
Myalgia | 20 (14.6) | 18 (24.7) |
Abdominal pain | 18 (13.1) | 12 (16.4) |
Hormone level abnormal | 18 (13.1) | — |
Hypokalemia | 18 (13.1) | 8 (11.0) |
Blood testosterone increased | 16 (11.7) | 18 (24.7) |
Anemia | 15 (10.9) | 3 (4.1) |
Dyspepsia | 15 (10.9) | 2 (2.7) |
Oropharyngeal pain | 14 (10.2) | 3 (4.1) |
Pain in extremity | 14 (10.2) | 5 (6.8) |
Upper respiratory tract infection | 14 (10.2) | 16 (21.9) |
Hypotension | 13 (9.5) | 12 (16.4) |
Urinary tract infection | 25 (18.2) | 12 (16.4) |
Acne | 13 (9.5) | 10 (13.7) |
Pruritis | — | 9 (12.3) |
Tachycardia | 8 (5.8) | 9 (12.3) |
Orthostatic hypotension | 4 (2.9) | 8 (11.0) |
Malaise | 10 (7.3) | 2 (2.7) |
Alanine aminotransferase increased | 4 (2.9) | 6 (8.2) |
Aspartate aminotransferase increased | 4 (2.9) | 5 (6.8) |
Constipation | 10 (7.3) | 2 (2.7) |
Pruritus | 13 (9.5) | 9 (12.3) |
Abdominal distension | 6 (4.4) | 4 (5.5) |
Abdominal pain upper | 9 (6.6) | 3 (4.1) |
Alopecia | 10 (7.3) | 5 (6.8) |
Anxiety | 13 (9.5) | 4 (5.5) |
Insomnia | 13 (9.5) | 2 (2.7) |
Iron deficiency anemia | 15 (10.9) | 3 (4.1) |
Edema peripheral | 22 (16.1) | 12 (16.4) |
Pyrexia | 21 (15.3) | 5 (6.8) |
Weight decreased | 9 (6.6) | 4 (5.5) |
Weight increased | 5 (3.6) | 3 (4.1) |
Most common (≥ 2%) SAEs, n (%) | ||
Patients with ≥ 1 SAE | 55 (40.1) | 10 (13.7) |
Adrenal insufficiency | 8 (5.8) | 3 (4.1) |
Pituitary tumour | 6 (4.4) | — |
Adrenocortical insufficiency | 4 (2.9) | — |
Gastroenteritis | 4 (2.9) | — |
Abdominal pain | 3 (2.2) | 1 (1.4) |
Headache | 3 (2.2) | — |
Influenza | 3 (2.2) | — |
Pituitary tumour benign | 3 (2.2) | — |
VI nerve paralysis | 3 (2.2) | — |
Patients who stopped treatment due to AEs, n (%) | ||
Patients with ≥ 1 AE leading to study drug discontinuation | 25 (18.2) | 9 (12.3) |
Adrenal insufficiency | 5 (3.6) | 3 (4.1) |
Hypokalemia | 1 (0.7) | 1 (1.4) |
Pituitary tumour | 5 (3.6) | 1 (1.4) |
Pituitary tumour benign | 5 (3.6) | 1 (1.4) |
Malignant pituitary tumour | 2 (1.5) | - |
Headache | 2 (1.5) | 1 (1.4) |
Sixth nerve paralysis | 2 (1.5) | — |
Deaths, n (%) | ||
Patients who died | 2 (1.4) | 0 |
Cardiorespiratory failure or cardiopulmonary failure | 1 (0.7) | — |
AEs of special interest, n (%) | ||
Hypocortisolism-related AEs | 74 (54.0) | 21 (28.8) |
Adrenal hormone precursor accumulation–related AEs | 80 (58.4) | 45 (61.6) |
Pituitary tumour enlargement–related AEs | 22 (16.1) | 4 (5.5) |
Arrhythmogenic potential and QT prolongation–related AEs | 6 (4.4) | 3 (4.1) |
AE = adverse event; SAE = serious adverse event.
Note: Numbers (n) represent counts of patients. For the LINC 3 study, the all-patients column excludes data in the placebo withdrawal arm collected during the placebo-control period.
Sources: LINC 3 final Clinical Study Report49 and LINC 4 final Clinical Study Report.54
Appendix 6: Methods and Results of the ITCs
Please note that this appendix has not been copy-edited.
Table 29: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Category | Inclusion criteria | Exclusion criteria |
|---|---|---|
Population | Adult patients (aged ≥ 18 years) with endogenous Cushing syndrome regardless of previous therapy |
|
Interventions |
|
|
Comparators |
|
|
Outcomes |
|
|
Study design |
|
|
Language | No limitb | None |
Time limit | No limit | None |
ITC = indirect treatment comparison; LAR = long-acting release; mUFC = mean urinary free cortisol.
aSystematic reviews were included and flagged for bibliography searches.
bNon-English citations that were potentially relevant to the SLR were flagged and discussed with Recordati to decide on their inclusion in the SLR. After discussion with Recordati, non-English language articles were excluded.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Table 30: Summary of Patient Baseline Characteristics From the Naive Comparison (Osilodrostat Versus Ketoconazole)
Characteristic | Osilodrostat | Ketoconazole | |||
|---|---|---|---|---|---|
LINC 4 trial (N = 48) | Correa-Silva (2009) (N = 8) | Castinetti (2014) (N = 200) | Young (2018)N = 108) | Castinetti (2008)a (N = 38) | |
Age, years | |||||
Mean (SD) | 42.3 (13.8) | 33.8 (8.9) | NR | 51.3 (NR) | NR |
Median (range) | 41 (21 to 67) | NR (19 to 41) | NR | NR (11 to 86) | NR |
Sex, n (%) | |||||
Female | 43 (89.6) | 8 (100.0) | 156 (78.0) | 81 (75.0) | NR |
Male | 5 (10.4) | 0 | 44 (22.0) | 27 (25.0) | NR |
Race or ethnic group, n (%) | |||||
Asian | 9 (18.8) | NR | NR | NR | NR |
White | 34 (70.8) | NR | NR | NR | NR |
Missing | 5 (10.4) | NR | NR | NR | NR |
Other demographic and disease characteristics | |||||
Pituitary adenoma, n (%) | 48 (100) | 8 (100) | 142 (71.0) | 23 (48.9) | NR |
BMI (kg/m2), mean (SD) | 29.9 (6.31) | 28.5 (2.2) | NR | NR | NR |
Baseline mUFC (nmol/24 hours), mean (SD) | 421.4 (291.3) | 613.8 (269.3)b | NR | NR | NR |
Time since diagnosis (months), mean (SD) | 70.7 (55.9) | NR | NR | NR | NR |
Previous treatment for CD, n (%) | |||||
Prior surgery | 41 (85.4) | NR | 144 (72.0) | 7 (14.9) | NR |
Prior medications | 26 (54.2) | NR | NR | NR | NR |
Prior pituitary irradiation | 6 (12.5) | NR | 47 (23.6) | NR | NR |
CD status, n (%) | |||||
N | 48 | NR | 200 | 47 | NR |
De novo | 3 (6.3) | NR | 56 (28.0) | 40 (85.1) | NR |
Persistent or recurrent | 45 (93.8) | NR | 144 (72.0) | 7 (14.9) | NR |
BMI = body mass index; CD = Cushing disease; ITC = indirect treatment comparison; mUFC = mean urinary free cortisol; NR = nor reported; SD = standard deviation.
aNR = not reported in the ITC summary.
bValues were converted from mcg per 24 hours to nmol per 24 hours by multiplying by 2.76.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.55
Appendix 7: Methods and Results of the Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were included in the review.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BLA
bilateral adrenalectomy
CDA-AMC
Canada’s Drug Agency
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
mUFC
mean urinary free cortisol
QALY
quality-adjusted life-year
TTCR
time to complete response
TTFPR
time to failure post response
ULN
upper limit of normal
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of osilodrostat compared to ketoconazole for adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option.1
Item | Description |
|---|---|
Drug product | Osilodrostat (Isturisa) 1 mg, 5 mg, and 10 mg, film-coated tablets, oral |
Indication | For the treatment of adult patients with Cushing’s disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option |
Submitted price | $46.14 per 1 mg tablet $184.58 per 5 mg tablet $193.81 per 10 mg tablet |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 3, 2025 |
Reimbursement request | Per indication |
Sponsor | Recordati Rare Diseases Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Summary
Isturisa is available as 1 mg, 5 mg, and 10 mg tablets.1 The median average dose was 4.6 mg and 7.4 mg per day in the LINC 4 and LINC 3 studies, respectively.2,3 At the submitted price of $46.14 per 1 mg tablet, $184.58 per 5 mg tablet, and $193.81 per 10 mg tablet,4 the annual cost of Isturisa is expected to range from $67,407 to $424,745 per patient, based on the Health Canada–recommended dosage (2 mg to 30 mg twice daily).4
The key clinical efficacy data (complete response rate, overall response rate) used to inform the economic model were derived from the LINC 4 trial,3 which compared Isturisa with placebo, and published literature for ketoconazole. Evidence submitted by the sponsor indicates that Isturisa is likely to improve complete response rate and overall response rate compared with placebo among adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option. Due to critical risk of bias, no conclusions can be drawn from the naive comparison regarding the comparative efficacy and safety of Isturisa versus ketoconazole in the treatment of Cushing disease in adults.
The results of the CDA-AMC base case suggest the following:
Isturisa is predicted to be associated with higher costs to the health care system than ketoconazole (incremental costs = $340,490), primarily driven by increased costs associated with drug acquisition.
Isturisa is predicted to be associated with a gain of 0.05 life-years compared to ketoconazole and may result in a gain of 0.44 quality-adjusted life-years (QALYs) compared to ketoconazole.
The incremental cost-effectiveness ratio (ICER) of Isturisa compared to ketoconazole was $742,689 per QALY gained in the CDA-AMC base case. The estimated ICER was highly sensitive to the long-term mortality benefits of Isturisa; approximately 95% of the benefit was predicted to be accrued after the treatment duration of the LINC 4 trial (study period of 11 months [48 weeks]).
CDA-AMC estimates that the budget impact of reimbursing Isturisa for the treatment of the indicated population will be approximately $30.5 million over the first 3 years of reimbursement compared to the amount currently spent on ketoconazole, with an estimated expenditure of $31 million on Isturisa over this period. The actual budget impact of reimbursing Isturisa will depend on the number of patients eligible.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of osilodrostat from the perspective of a public health care payer in Canada over a lifetime horizon (50 years).1 The modelled population comprised patients aged 18 to 75 years with confirmed Cushing disease that was persistent or recurrent, which is generally aligned with the Health Canada indication and was based on the participants in the LINC 4 trial.1,3 The sponsor’s base-case analysis included costs related to drug acquisition, management of adverse events (AEs), bilateral adrenalectomy (BLA) complications, and other comorbidities.1
In the sponsor’s base case, osilodrostat was associated with incremental costs of $339,028 and 0.69 incremental QALYs relative to ketoconazole. This resulted in an ICER of $491,746 per QALY gained.1 Of the incremental benefit compared to ketoconazole (0.69 incremental QALYs), approximately 92% of the benefit was predicted to be accrued after the treatment duration of the LINC 4 trial (study period of 11 months [48 weeks]).1 Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
Heterogeneous patient population. | The clinical experts consulted by CDA-AMC indicated Cushing disease is heterogeneous, and the presentation of Cushing disease or components of the condition upon diagnosis may also impact disease trajectory, related comorbidities, or treatment course. As a result, differences in disease presentation will likely result in differences in health outcomes and costs for patients. | CDA-AMC could not address this issue in the base case because the model was not programmed to evaluate different subgroups. | No scenario analysis was conducted. |
Appropriate comparator is uncertain. | The sponsor determined that ketoconazole is the only relevant comparator. However, the clinical experts consulted by CDA-AMC suggested that ketoconazole plus cabergoline may be the most relevant comparator. | CDA-AMC could not address this issue in the base case because the model considers ketoconazole only. | No scenario analysis was conducted. |
Comparative efficacy is unknown. | There is no direct evidence assessing the comparative efficacy and safety of osilodrostat and ketoconazole. Relative efficacy in the model was informed from a naive indirect comparison of the LINC 4 study with published literature.5,6 The modelled data suggested a relative benefit for osilodrostat; however, this finding could not be validated by CDA-AMC. | CDA-AMC could not address this issue in the base case due to a lack of comparative evidence. | In 3 scenario analyses, CDA-AMC tested the effects of setting the HR for TTCR and TTFPR to 1. |
Inappropriate use of surrogate outcome. | Patient outcomes were dichotomized into controlled and uncontrolled based on mUFC.1 Clinical experts consulted by CDA-AMC noted that response may not be dichotomized based on mUFC alone. | CDA-AMC could not address this issue in the base case due to the model structure. | No scenario analysis was conducted. |
BLA mortality and complications is overestimated. | A lifetime mortality increase was applied to patients who underwent a BLA.1 The clinical experts consulted by CDA-AMC noted there is an associated short-term mortality risk but not a lifetime risk. Additionally, the proportion of patients who develop Nelson syndrome was overestimated. | CDA-AMC set the long-term BLA mortality HR to 1 and reduced the prevalence of Nelson syndrome to 21% of patients. | No scenario analysis was conducted. |
BLA = bilateral adrenalectomy; CDA-AMC = Canada’s Drug Agency; HR = hazard ratio; mUFC = mean urinary free cortisol; TTCR = time to complete response; TTFPR = time to failure post response.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 7), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
Osilodrostat is predicted to be associated with additional health care costs compared to ketoconazole (incremental costs = $340,490). This increase in health care spending is primarily due to increased drug acquisition costs associated with osilodrostat (refer to Figure 1).
Figure 1: Impact of Osilodrostat vs. Ketoconazole on Health Care Costs
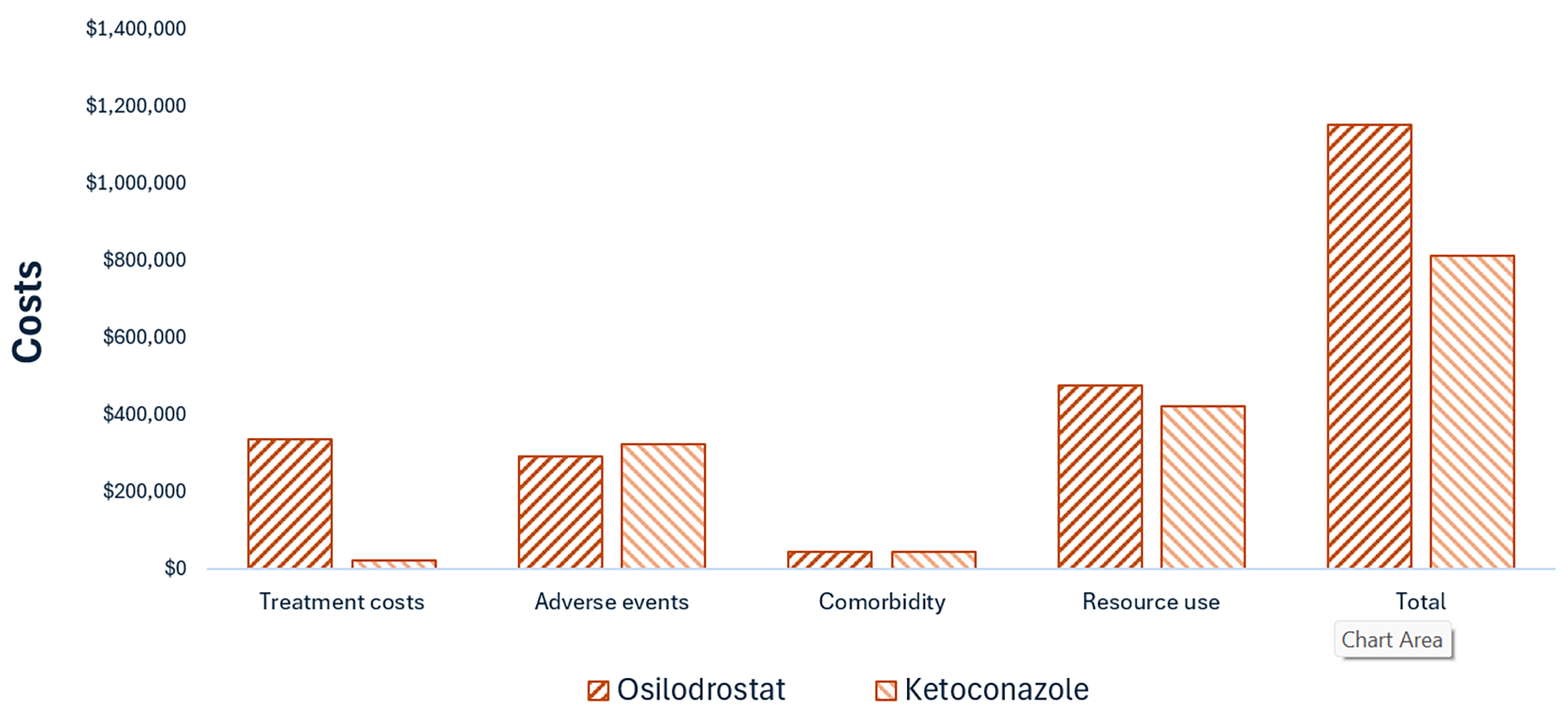
vs. = versus.
Note: Resource use includes costs associated with clinician visits, hospital visits, testing, and monitoring. Comorbidity includes costs associated with health care resource use and treatment for chronic comorbidities for patients with Cushing disease, as determined based on the input of the clinical experts consulted by the sponsor.
Impact on Health
Relative to ketoconazole, osilodrostat is predicted to increase the amount of time a patient remains in the controlled disease state by approximately 2.67 years (refer to Figure 2) and reduce the amount of time spent in the BLA health state by 2.48 years. Overall, taking into account time spent in each health state, osilodrostat is predicted to increase overall survival by 0.05 years. Considering the impact of treatment on both quality and length of life, osilodrostat is predicted to result in 0.44 additional QALYs per patient compared to ketoconazole. This is because quality of life is highest in the controlled disease state in which patients on osilodrostat spend more of their time relative to ketoconazole. Approximately 95% of the predicted incremental benefit was accrued based on extrapolation.
Figure 2: Impact of Osilodrostat vs. Ketoconazole on Patient Health
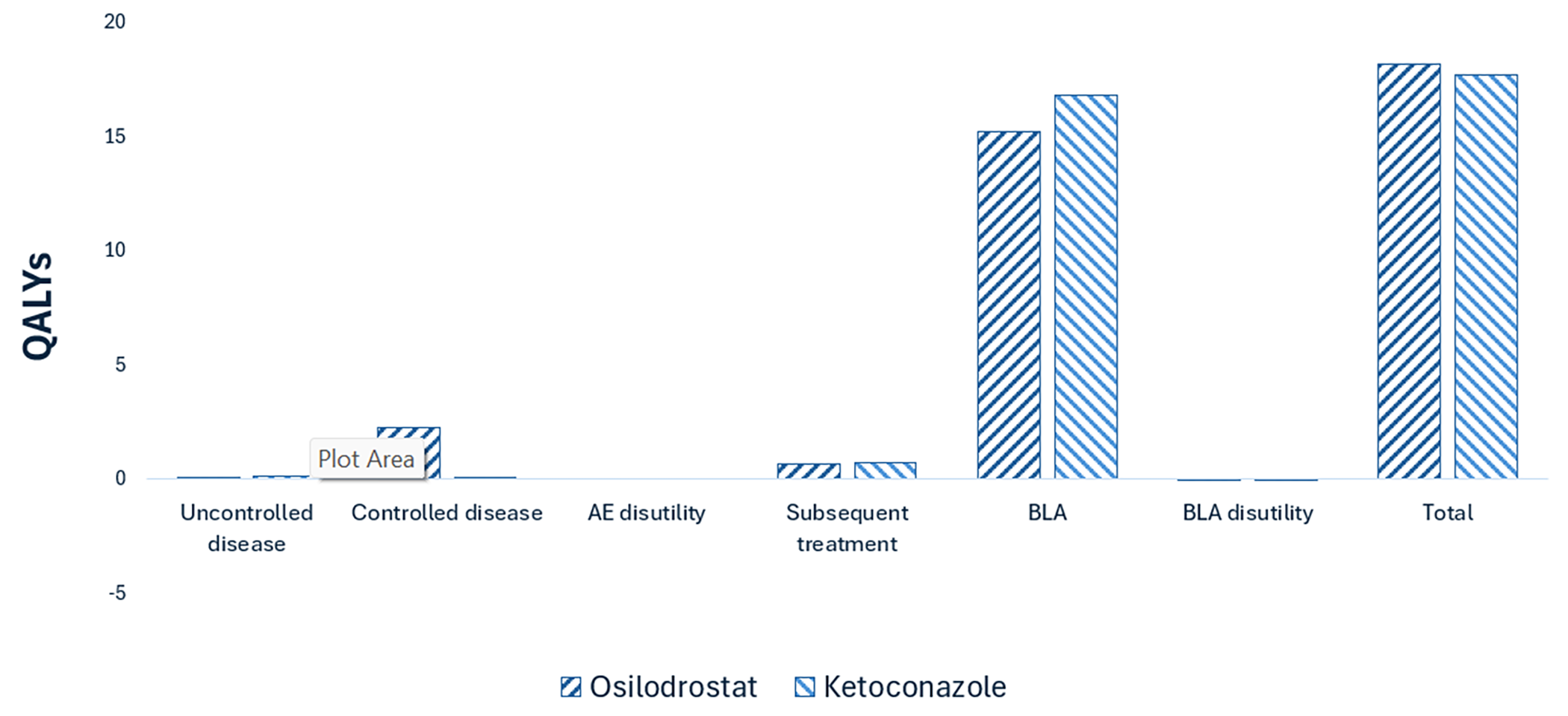
AE = adverse event; BLA = bilateral adrenalectomy; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $742,689 per QALY gained for osilodrostat compared to ketoconazole (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Ketoconazole | 811,911 | 17.73 | Reference |
Osilodrostat | 1,152,401 | 18.18 | 742,689 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty associated with comparative efficacy was explored by setting the hazard ratio (HR) for osilodrostat to 1 relative to ketoconazole for time to complete response (TTCR) and/or time to failure post response (TTFPR).
Scenario 1 (TTCR only) resulted in an ICER similar to the CDA-AMC base case; however, for scenario 2 (TTFPR) and scenario 3 (TTCR and TTFPR), the ICER ranged from ($4,945,020 per QALY to dominated).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (fourth quarter of 2024 to third quarter of 2027) budget impact of reimbursing osilodrostat for the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option.7 The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiological approach.7 The price of osilodrostat was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on publicly available list prices.7 Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with the clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that by year 3 of reimbursement, 129 patients would be eligible for osilodrostat; of these, 114 patients would be expected to receive osilodrostat. The estimated incremental budget impact of reimbursing osilodrostat is predicted to be approximately $30.5 million over the first 3 years, with an expected expenditure of $31 million on osilodrostat. The actual budget impact of reimbursing osilodrostat will depend on the number of patients eligible.
Conclusion
Based on the CDA-AMC base case, osilodrostat would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $742,689 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 10). The estimated cost-effectiveness of osilodrostat compared to ketoconazole is highly uncertain due to the lack of comparative evidence for osilodrostat against any comparator.
The budget impact of reimbursing osilodrostat to the public drug plans in the first 3 years is estimated to be approximately $30.5 million. The 3-year expenditure on osilodrostat (i.e., not accounting for any current expenditure on comparators) is estimated to be $31 million. The estimated budget impact is uncertain due to gaps in the evidence to determine an accurate patient population size and the uncertainty associated with the criteria for accessing osilodrostat, which may influence the eligible population.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
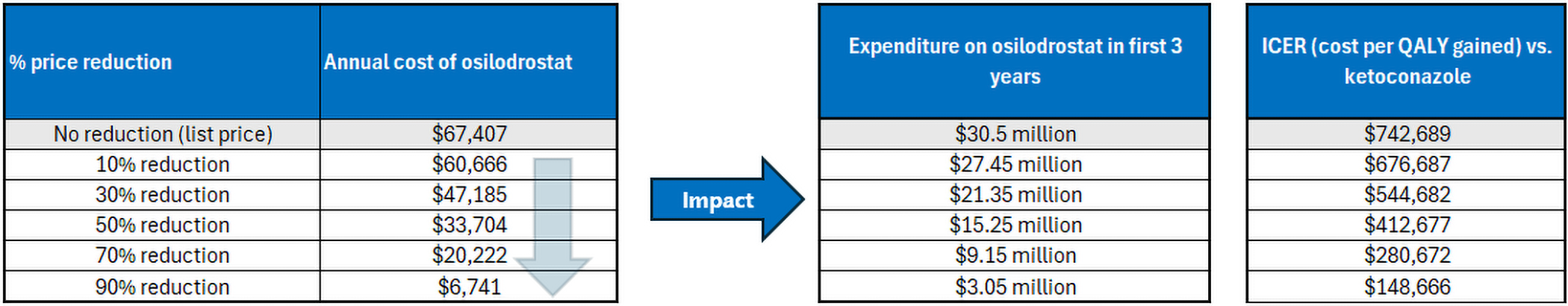
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Annual cost is based on the minimum recommended dosage for osilodrostat. Expenditure includes only the drug cost of osilodrostat.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Isturisa, 1 mg, 5 mg, and 10 mg Film-coated Tablets. Toronto (ON): Recordati Rare Disease Canada Inc; 2023 October 16. 2025.
2.Novartis. CLCI699C2301. Interim Clinical Study Report: A Phase III, multi-center, double-blind, randomized withdrawal study of LCI699 following a 24 week, single-arm, open-label dose titration and treatment period to evaluate the safety and efficacy of LCI699 for the treatment of patients with Cushing’s disease. Feb 21, 2018.
3.Novartis. CLCI699C2302. Primary Analysis Clinical Study Report: A Phase III, multi-center, randomized, double-blind, 48 week study with an initial 12 week placebo-controlled period to evaluate the safety and efficacy of osilodrostat in patients with Cushing’s disease. Sep 20, 2019.
4.Drug Reimbursement Review sponsor submission: osilodrostat (Isturisa) [internal sponsor's package]. Sponsor name; 2025.
5.Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing's disease: is it worth a try? J Clin Endocrinol Metab. 2014;99(5):1623-30. doi: 10.1210/jc.2013-3628 PubMed
6.Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing's disease. Eur J Endocrinol. 2008;158(1):91-9. doi: 10.1530/EJE-07-0514 PubMed
7.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Isturisa, 1 mg, 5 mg, and 10 mg Film-coated Tablets. Toronto (ON): Recordati Rare Disease Canada Inc; 2023 October 16.
8.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2025. Accessed 2025 September 1. https://www.formulary.health.gov.on.ca/formulary/
9.Fleseriu M, Auchus R, Bancos I, et al. Consensus on diagnosis and management of Cushing's disease: a guideline update. Lancet Diabetes Endocrinol. 2021;9(12):847-875. doi: 10.1016/s2213-8587(21)00235-7 PubMed
10.Health Canada's special access programs: Overview. Health Canada. 2023 Feb 15. Accessed 2023 Nov 18. https://www.canada.ca/en/health-canada/services/drugs-health-products/special-access.html
11.Saskatchewan Drug Plan: search formulary. 2025. Accessed 2025 September 1. http://formulary.drugplan.ehealthsask.ca/SearchFormulary
12.DeltaPA. IQVIA; 2025. Accessed 2025 September 1. https://www.iqvia.com/
13.Recordati Rare Diseases Canada Inc. Isturisa (Osilodrostat): 1 mg, 5 mg, and 10 mg film coated tablets [product monograph]. Recordati Rare Diseases Canada Inc.; 2025.
14.Novartis. Final Clinical Study Report: A Phase III, multi-center, randomized, double-blind, 48 week study with an initial 12 week placebo-controlled period to evaluate the safety and efficacy of osilodrostat in patients with Cushing’s disease. Jun 01, 2020.
15.Swearingen B, Wu N, Chen SY, Pulgar S, Biller BM. Health care resource use and costs among patients with cushing disease. Endocr Pract. 2011;17(5):681-90. doi: 10.4158/ep10368.or PubMed
16.Statistics C. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island. Table: 13-10-0114-01. 2019/2021.
17.Ragnarsson O, Olsson DS, Papakokkinou E, et al. Overall and Disease-Specific Mortality in Patients With Cushing Disease: A Swedish Nationwide Study. J Clin Endocrinol Metab. 2019;104(6):2375-2384. doi: 10.1210/jc.2018-02524 PubMed
18.Pivonello R, De Leo M, Cozzolino A, Colao A. The Treatment of Cushing's Disease. Endocr Rev. 2015;36(4):385-486. doi: 10.1210/er.2013-1048 PubMed
19.Almalki Z, Alatawi Y, Alharbi A, et al. Cost-Effectiveness of More Intensive Blood Pressure Treatment in Patients with High Risk of Cardiovascular Disease in Saudi Arabia: A Modelling Study of Meta-Analysis. Int J Hypertens. 2019;2019:6019401. doi: 10.1155/2019/6019401 PubMed
20.Doyle S, Lloyd A, Walker M. Health state utility scores in advanced non-small cell lung cancer. Lung Cancer. 2008;62(3):374-80. doi: 10.1016/j.lungcan.2008.03.019 PubMed
21.Hart HE, Redekop WK, Bilo HJ, Meyboom-de Jong B, Berg M. Health related quality of life in patients with type I diabetes mellitus: generic & disease-specific measurement. Indian J Med Res. 2007;125(3):203-16. PubMed
22.Huxley NC, L. Varley-Campbell, J., Tikhonova, I., Snowsill, T., Briscoe, S., Peters, J. Bond, M. Napier, M., Hoyle M. Cetuximab (review of TA176) and panitumumab (partial review of TA240) for the first line treatment of metastatic colorectal cancer. 2015. Accessed 23/06/2021. https://www.nice.org.uk/guidance/ta439/documents/colorectal-cancer-metastatic-cetuximab-review-ta176-and-panitumumab-part-review-ta240-1st-line-id794-assessment-report2
23.Lloyd A, Price D, Brown R. The impact of asthma exacerbations on health-related quality of life in moderate to severe asthma patients in the UK. Prim Care Respir J. 2007;16(1):22-7. doi: 10.3132/pcrj.2007.00002 PubMed
24.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health and quality of life outcomes. 2008;6:84. doi: 10.1186/1477-7525-6-84 PubMed
25.Sadatsafavi MM, C. Marra, F. Moran, O. Fitzgerald, J. M. Lynd, L. A quantitative benefit-risk analysis of isoniazid for treatment of latent tuberculosis infection using incremental benefit framework. Value Health. 2013;16(1)(66-75). doi: 10.1016/j.jval.2012.09.006 PubMed
26.Sullivan PW, Ghushchyan VH. EQ-5D Scores for Diabetes-Related Comorbidities. Value Health. 2016;19(8):1002-1008. doi: 10.1016/j.jval.2016.05.018 PubMed
27.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-4. doi: 10.1177/0272989x11401031 PubMed
28.Canadian Institute for Health Information. Data from: Patient Cost Estimator. 2020.
29.Government of Ontario MoH, Long-Term C, Government of Ontario MoH, Long-Term C. Ontario Schedule of Benefits. 2023. doi: http://www.health.gov.on.ca/en/
30.Hahner S, Spinnler C, Fassnacht M, et al. High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency: a prospective study. J Clin Endocrinol Metab. 2015;100(2):407-16. doi: 10.1210/jc.2014-3191 PubMed
31.Reincke M, Ritzel K, Osswald A, et al. A critical reappraisal of bilateral adrenalectomy for ACTH-dependent Cushing's syndrome. Eur J Endocrinol. 2015;173(4):M23-32. doi: 10.1530/EJE-15-0265 PubMed
32.Papakokkinou E, Piasecka M, Carlsen HK, et al. Prevalence of Nelson's syndrome after bilateral adrenalectomy in patients with cushing's disease: a systematic review and meta-analysis. Pituitary. 2021;24(5):797-809. doi: 10.1007/s11102-021-01158-z PubMed
33.Etxabe J, Vazquez JA. Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin Endocrinol (Oxf). 1994;40(4):479-84. doi: 10.1111/j.1365-2265.1994.tb02486.x PubMed
34.Sharma ST, Nieman LK, Feelders RA. Cushing's syndrome: epidemiology and developments in disease management. Clin Epidemiol. 2015;7:281-93. doi: 10.2147/clep.s44336 PubMed
35.Webb SM, Badia X, Barahona MJ, et al. Evaluation of health-related quality of life in patients with Cushing's syndrome with a new questionnaire. Eur J Endocrinol. 2008;158(5):623-630. PubMed
36.Petersenn S, Beckers A, Ferone D, et al. Therapy of endocrine disease: outcomes in patients with Cushing's disease undergoing transsphenoidal surgery: systematic review assessing criteria used to define remission and recurrence. Eur J Endocrinol. 2015;172(6):R227-39. doi: 10.1530/eje-14-0883 PubMed
37.Uum SV, Hurry M, Petrella R, Koch C, Dranitsaris G, Lacroix A. Management of patients with Cushing's disease: a Canadian cost of illness analysis. J Popul Ther Clin Pharmacol. 2014;21(3):e508-17. PubMed
38.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed 2025 September 1. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
39.Castinetti F, Morange I, Conte-Devolx B, Brue T. Cushing's disease. Orphanet J Rare Dis. 2012;7:41. doi: 10.1186/1750-1172-7-41 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and Canada’s Drug Agency–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans
Table 4: Cost Comparison for Cushing Disease
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Osilodrostat (Isturisa) | 1 mg 5 mg 10 mg | Tablet | 46.1378 184.5812 193.8147 | Initial dosage: 2 mg twice daily. The dosage can be gradually titrated (initially by increments of 1 or 2 mg twice daily) based on clinical response and patient tolerance. The maximum recommended dosage is 30 mg twice daily. | 2 mg to 4 mg twice daily: 184.55 to 369.10 Maximum 30 mg twice daily: 1,162.89 | 2 mg to 4 mg twice daily: 67,407 to 134,815 Maximum 30 mg twice daily: 424,745 |
Other adrenal steroidogenesis inhibitorsa | ||||||
Ketoconazole (generics) | 200 mg | Tablet | 0.93938 | 400 mg to 1,200 mg per day, in 2 doses.9 | 1.88 to 5.64 | 686 to 2,057 |
Other pharmaceutical therapies | ||||||
Cabergoline (generics) | 0.5 mg | Tablet | 7.2907b | 0.5 to 7 mg weekly9 | 1.04 to 14.58 | 380 to 5,326 |
Pasireotide (Signifor) | 0.3 mg/mL 0.6 mg/mL 0.9 mg/mL | 1 mL ampoule, solution for injection | 82.0952c 90.9181c 90.9181c | Initially 0.6 mg SC twice daily. Dose may be increased to 0.9 mg or decreased to 0.3 mg based on response and tolerance. | 178.19 to 181.84 | 65,039 to 66,370 |
Pasireotide (Signifor LAR) | 20 mg/2 mL 40 mg/2 mL 60 mg/2 mL | Injection kit with powder for suspension | 4,974.1500d 5,048.7623c 5,048.7623d | Initially 10 mg IM every 4 weeks; the dose may be titrated based on response and tolerability to a maximum of 40 mg every 4 weeks. | 177.65 to 180.31 | 64,886 to 65,859 |
IM = intramuscular; LAR = long-acting release; SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed September 2025), unless otherwise indicated, and do not include dispensing fees.
aAccording to clinical expert input, metyrapone is also used in Canada. Recommended dosing is 0.5 to 6 g per day in divided doses every 6 to 8 hours.9 Metyrapone is not approved in Canada and is only available through the Health Canada Special Access Program.10
bCabergoline is typically funded only for the treatment of hyperprolactinemic disorders.8,11
cAssociation québécoise des pharmaciens propriétaires list price as reported by IQVIA’s DeltaPA (accessed September 2025).12
dWholesale price as reported by IQVIA’s DeltaPA (accessed September 2025).12
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
The Canadian Organization for Rare Disorders (CORD) submitted input for this review. CORD collected information through a variety of sources including sending a survey to members, seeking input from Acromegaly Canada and the US-based Cushing’s Support and Research Foundation, and contacting 11 clinicians across 5 provinces (Nova Scotia, Quebec, Ontario, Alberta, and British Columbia). CORD gathered information from 21 patients (12 in Canada, 9 in US), 2 patients from Canada and 4 from the US were interviewed. Eighteen respondents identified as having Cushing disease due to pituitary tumour while the other 3 had Cushing syndrome. Responses highlighted 3 major concerns: 1) delayed diagnosis, taking up to 15 years and requiring numerous tests, hospitalizations, chance, and the patients’ perseverance; 2) debilitating physical, psychological, and cognitive symptoms impacting daily activities; and 3) “overwhelming” impact on their caregivers’ quality of life. Patients reported having hypertension, significant weight gain or obesity, osteoporosis, secondary diabetes, hypothyroidism, depression, fatigue, and low energy. All respondents had received surgery, 33% reported it as effective and 48% reported it as “not at all” or only “a little” effective. More than 66% reported surgery as having severe or very severe side effects. 33% of respondents received radiation therapy with 66% of those reporting is as effective, although a small percentage reported severe or very severe side effects. Two US-based patients had received pasireotide; 1 reported it as effective and the other reported severe side effects. Patients reported challenges in getting access to disability support and potentially helpful medications that are not approved and/or not reimbursed. All respondents wanted a treatment that reduced cortisol production with lasting benefits and few side effects, allowing them to normalcy, preferably an oral therapy not requiring hospital administration. All respondents stressed the importance that therapy be affordable to patients. Two US-based patients (none from Canada) had long-term osilodrostat experience via clinical trial, while 1 had started on it within the past 6 months. Side effects were reported as tolerable, including nausea, headache, and fatigue. Both long-term recipients reported significant improvement in blood pressure, weight loss, glucose levels, and mood, all maintained since the end of the trial.
The Canadian Society of Endocrinology and Metabolism (CSEM) submitted clinician group input for this review. CSEM stated that first-line treatment of patients with Cushing syndrome includes resection of the underlying lesion causing hypercortisolism, most commonly transsphenoidal surgery (TSS) for those with pituitary adenomas and unilateral adrenalectomy for those with adrenocortical adenomas, hyperplasia, and more rarely carcinomas. For patients in whom surgery is not feasible or not curative, the options include repeat TSS, radiotherapy, medical therapies, and BLA as a last resort. Current medical therapies include the adrenal steroidogenesis inhibitors ketoconazole, which is not indicated, of limited efficacy, and associated with hepatotoxicity, but widely funded and accessible, and metyrapone, which is difficult to use due to its 4 times daily administration and limited gastrointestinal tolerability, and requires special access. When adrenal steroidogenesis inhibitors fail, cabergoline or pasireotide may be used. Cabergoline is not indicated and often not funded for Cushing syndrome, but it may be used as monotherapy or as an adjunct to ketoconazole. Pasireotide is not funded by public plans. CSEM anticipates osilodrostat to be used as first-line medical treatment, preferred over adrenal steroidogenesis inhibitors, but does not consider it appropriate to use previous medical treatments to exclude reimbursing osilodrostat.
Drug plan input included questions regarding the most appropriate comparator for the economic model, and the definition of response to treatment.
Several of these concerns were addressed in the sponsor’s model:
Ketoconazole was included as the main comparator in the sponsor's model.
Cortisol levels, AEs, and BLA were included in the model.
CDA-AMC was unable to address the following concerns raised by the input from the patient and clinician groups and drug plans:
The sponsor only provided cost-effectiveness information for osilodrostat compared with ketoconazole. The sponsor's model did not allow comparison of osilodrostat with other relevant comparators, or the option to test different definitions of treatment response, other than the use of mean urinary free cortisol (mUFC) less than or equal to the upper limit of normal (ULN).
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and the best interpretation of the economic evidence based on the information provided.
For the review of osilodrostat, the sponsor provided a cost-utility analysis and a BIA.4 The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Osilodrostat (Isturisa), oral tablets (1 mg, 5 mg, 10 mg) |
Submitted price of drug under review | $46.14 per 1 mg tablet $184.58 per 5 mg tablet $193.81 per 10 mg tablet |
Regimen | Initially 2 mg twice daily; can be titrated in increments of 1 mg or 2 mg up to a maximum dose of 30 mg13 |
Annual cost of drug under review | $67,407 to $134,815 per patient based on a dosage of 2 to 4 mg twice daily4,a |
Model information | |
Type of economic evaluation | Cost-utility analysis1 Markov model1 |
Treatment | Osilodrostat1 |
Included comparator | Ketoconazole1 |
Perspective | Publicly funded health care payer perspective1 |
Time horizon | Lifetime (50 years)1 |
Cycle length | 7 days1 |
Modelled population | Patients aged 18 to 75 years with confirmed Cushing disease that was persistent or recurrent1 |
Characteristics of modelled population | Derived from the LINC 4 trial (mean age: 41.2 years, female: 83.6%; male: 16.4%)1,3 |
Model health states |
For additional information, refer to Model Structure. |
Data sources | |
Comparative efficacy | |
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities | |
Costs |
|
Summary of the submitted results | |
Base case results |
|
Scenario analysis results |
|
AE = adverse event; BLA = bilateral adrenalectomy; CDA-AMC = Canada’s Drug Agency; CIHI = Canadian Institute for Health Information; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; TTCR = time to complete response; TTFPR = time to failure post response; QALY = quality-adjusted life-years.
aThe maximum annual cost of osilodrostat is expected to be $374,096 based on a dosage of 30 mg twice daily.13
Model Structure
The sponsor submitted a Markov model with 5 distinct and mutually exclusive health states: uncontrolled disease on initial treatment, controlled on initial treatment, subsequent medical therapy, BLA, and dead. The cycle length was 7-day. Assignment to “controlled” and “uncontrolled” health states is dependent upon treatment response, with “controlled” defined by mUFC less than or equal to the ULN. All patients have uncontrolled disease (mUFC is greater than the ULN) when starting initial treatment. The LINC 4 study’s patient-level data were used to derive the TTCR Kaplan-Meier curve, and patients transition to the controlled health state (mUFC is less than or equal to ULN) according to the TTCR. Literature sources were used to inform transitions in the ketoconazole comparator arm. If no response occurs within 6 months, or if the treatment fails in patients with controlled disease, patients transition to subsequent medical treatment (ketoconazole + cabergoline) as indicated by sponsor’s clinical experts, regardless of their initial treatment. If patients’ disease is still uncontrolled after 12 months on subsequent treatment, patients will transition to the BLA state in which they will have a 100% response rate (controlled) and remain until death.1
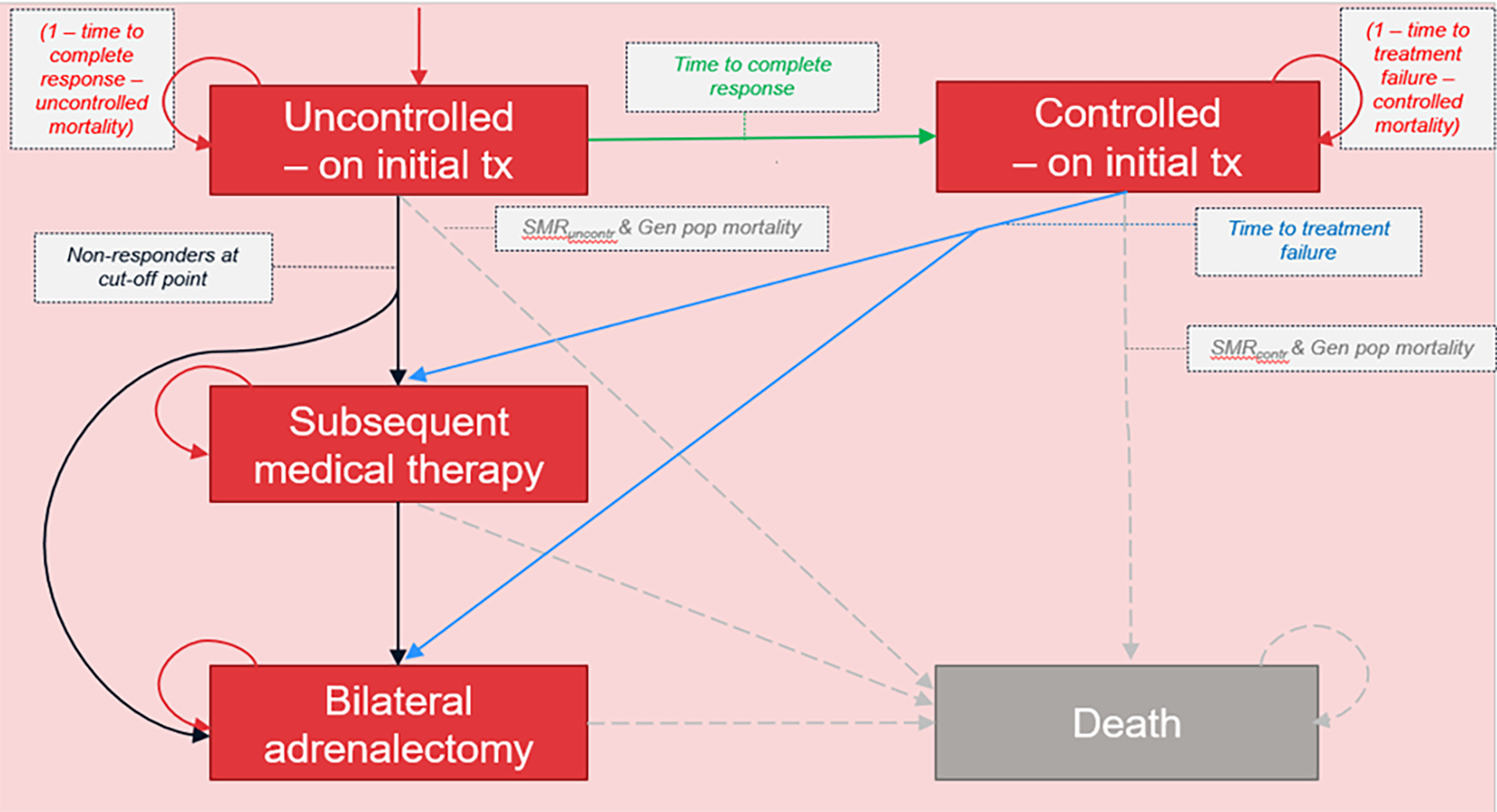
Contr = controlled; Gen pop = general population; SMR = standard mortality ratio; tx = treatment; uncontr = uncontrolled.
Source: Sponsor’s pharmacoeconomic submission.1
Table 6: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. ($/QALY) |
|---|---|---|---|---|---|
ketoconazole | 725,828 | Reference | 13.86 | Reference | Reference |
Osilodrostat | 1,064,856 | 339,028 | 14.55 | 0.69 | 491,746 |
ICER = incremental cost-utility ratio; QALY = quality-adjusted life-year.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC clinical review found that osilodrostat likely results in an increase in complete response rate and overall response rate compared to placebo. There have been no head-to-head trials of osilodrostat against any comparators, and only 1 unadjusted (naive) indirect comparison was submitted. No conclusions can be drawn from the naive comparison on the comparative efficacy and safety profile comparing osilodrostat versus ketoconazole in the treatment of Cushing syndrome in adults. Thus, the comparative efficacy and safety of osilodrostat versus ketoconazole or any comparator is unknown.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
Oversimplified model for heterogeneous patient population. Feedback from clinical experts consulted by CDA-AMC indicated that patients with Cushing disease are very heterogeneous by nature. The presentation of disease or components of the condition upon diagnosis may also impact disease trajectory, related comorbidities or treatment course (specifically, there appears to be differences in protocols between patients whose Cushing disease has pituitary involvement and those whose Cushing disease does not have pituitary involvement). Other disease or treatment modifiers were identified including disease severity (from mild to severe and life-threatening), or whether patients were newly diagnosed or had recurrent disease. Incorporating all patients into 1 single population in the model following the same sequence of treatments might oversimplify the complexity of the disease.
CDA-AMC noted that according to the clinical expert feedback obtained by CDA-AMC, the patients recruited in the LINC 4 trial were considered to have more mild disease and were at relatively low risk of incurring severe complications of Cushing disease or death compared to observed patient population in clinical practice in Canada. As such, because the clinical trajectory and outcomes may reflect patients with mild disease severity, it might not be generalizable to patients with severe Cushing disease.
The sponsor’s model was not programmed to adequately evaluate the cost-effectiveness of different subgroups, and therefore this limitation could not be addressed.
The most appropriate comparator for osilodrostat in practice in Canada is uncertain. The sponsor determined that ketoconazole was the only relevant comparator for osilodrostat for the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option Clinical expert feedback suggested that osilodrostat was more likely to be used in patients for whom surgery was ineffective or not possible; and although it is unlikely to replace a currently available treatment, it may be preferred over currently available treatments. In addition to ketoconazole (as a potential first-line pharmacotherapy treatment comparator), the CDA-AMC clinical experts suggested that the combination therapy of ketoconazole plus cabergoline and active surveillance may be among the most relevant comparators for patients diagnosed with the indication.
CDA-AMC could not address this limitation because the sponsor was unable to provide a comparison between osilodrostat and ketoconazole plus cabergoline.
The comparative efficacy of osilodrostat with relevant comparators is unknown. There is no direct evidence assessing the comparative efficacy and safety of osilodrostat and ketoconazole. Relative efficacy was derived by naive comparisons based on osilodrostat data from the LINC 4 trial and ketoconazole data from published literature; the sponsor's rationale was that due to the limited sample sizes and unavailability of patient characteristics for the comparator studies, an adjusted indirect comparison or network meta-analysis was not feasible. The sponsor’s base case derived HRs of ████ and ████ to model the TTCR and TTFPR for ketoconazole, respectively, based on retrospective studies of ██ patients (TTCR)6 and ███ patients (TTFPR).5 These HRs suggested a relative benefit for osilodrostat compared with ketoconazole. Clinical expert feedback obtained by CDA-AMC was unable to validate the assumption of greater clinical effectiveness with osilodrostat versus ketoconazole due to the lack of robust comparative effectiveness. This feedback also suggested that efficacy would be unlikely to attenuate over time if the drugs are effective in the patients (expectation of similar time to treatment failure between osilodrostat and ketoconazole). As such, using an all-cause discontinuation rate in the model, which includes discontinuation due to lack of efficacy, might overestimate the parameter of TTFPR.
In the CDA-AMC scenario analysis, the HR on TTCR and/or TTFPR were varied to include 1.0 to explore the impact on ICER, assuming the same efficacy between osilodrostat and ketoconazole.
Inappropriate use of surrogate outcome dichotomized into “controlled” and “uncontrolled.” The surrogate outcome of mUFC was used to define complete response in the LINC trials as well as in the economic model (mUFC ≤ ULN). The sponsor also assumed improved outcomes such as comorbidities, quality of life, mortality, and averted BLA based on the complete response status. The clinical experts consulted by CDA-AMC noted that response may be dichotomized based on a simple ULN threshold alone; the degree of mUFC reduction, or improvement of symptoms and comorbidities, is more clinically important than having the mUFC lower than a certain set threshold. Further, assigning clinical consequences based on dichotomized mUFC threshold may be misleading; for example, observational evidence describing clinical outcomes of mortality in subjects with uncontrolled disease (defined using multiple criteria) may differ from model-defined uncontrolled disease (for example, significant clinical improvement, with a large reduction in mUFC but not ≤ ULN).
The sponsor’s model was not programmed to adequately assess different response thresholds; therefore, this limitation could not be addressed.
BLA mortality and complications. The sponsor assumed an increased lifetime mortality HR of 2.7 for patients in remission after BLA versus patients in remission without BLA, additional to a 1-time 1.8% mortality risk during surgery. The clinical experts consulted by CDA-AMC stated that there is a temporary increase in mortality risk associated with undergoing BLA, but it would be unreasonable to assume that a patient who underwent a BLA would maintain an elevated mortality risk for their entire lifetime, in addition to the increased baseline mortality by Cushing disease. A review paper by Reincke et al31 also concluded that the long-term mortality after BLA is low. In addition, the sponsor assumed 43% of patients after BLA experienced Nelson syndrome, which the CDA-AMC clinical experts deemed to be too high.
In the CDA-AMC reanalysis, the long-term BLA mortality was set to 1 compared to control (no increased mortality risk after BLA versus patients without BLA). In addition, the prevalence of Nelson syndrome was reduced to 21% (95% CI, 18% to 26%) according to a recently published meta-analysis by Papakokkinou et al.32 Note that the CDA-AMC experts suggested the estimate in Canada is even lower.
Additional issues were identified but were not considered to be key issues:
Assessment of response and change in treatment was assessed at 6 months and 12 months for the initial treatment and subsequent medical therapy, respectively, based on clinician feedback. All patients received subsequent medical therapy before moving to BLA. Because patients with Cushing disease are highly heterogeneous, the clinical experts consulted by CDA-AMC stated that patients are unlikely to follow this treatment pathway. It is feasible that some patients might have BLA before medical therapies; or use medical therapy as a bridge to BLA.
A relative risk for uncontrolled versus controlled disease from published literature15 was applied to uncontrolled disease comorbidity probabilities; however, these patient groups are different from each other, and it was inappropriate to apply the published relative risks to the modelled populations.
All medical therapies have the same safety profile as osilodrostat.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 7). The impact of these changes, individually and collectively, is presented in Table 8.
Table 7: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Long-term BLA mortality HR | 2.7 | 1 |
2. Proportion of Nelson Syndrome for patient after BLA | 43% | 21% |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 |
BLA = bilateral adrenalectomy; CDA-AMC = Canada’s Drug Agency; HR = hazard ratio.
Note: CDA-AMC was unable to resolve the issues with the heterogeneous patient population, the most appropriate comparator, comparative efficacy, and usage of a surrogate outcome.
Table 8: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | Ketoconazole | 730,719 | 13.87 | Reference |
Osilodrostat | 1,055,995 | 14.53 | 495,853 | |
CDA-AMC reanalysis 1 | Ketoconazole | 887,828 | 17.68 | Reference |
Osilodrostat | 1,204,173 | 18.12 | 719,732 | |
CDA-AMC reanalysis 2 | Ketoconazole | 669,107 | 13.88 | Reference |
Osilodrostat | 1,000,590 | 14.54 | 505,968 | |
CDA-AMC base case: reanalysis 1 + 2 (deterministic) | Ketoconazole | 808,092 | 17.69 | Reference |
Osilodrostat | 1,131,673 | 18.13 | 737,858 | |
CDA-AMC base case: reanalysis 1 + 2 (probabilistic) | Ketoconazole | 811,911 | 17.73 | Reference |
Osilodrostat | 1,152,401 | 18.18 | 742,689 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 9: Disaggregated Results of the CDA-AMC Base Case
Parameter | Osilodrostat | Ketoconazole |
|---|---|---|
Discounted LYs | ||
Total | 27.25 | 27.20 |
Uncontrolled disease | 0.13 | 0.20 |
Controlled disease | 2.78 | 0.11 |
Subsequent treatment | 0.93 | 0.99 |
BLA | 23.42 | 25.90 |
Discounted QALYs | ||
Total | 18.18 | 17.73 |
Uncontrolled disease | 0.08 | 0.13 |
Controlled disease | 2.25 | 0.09 |
AE disutility | 0.00 | 0.00 |
Subsequent treatment | 0.67 | 0.72 |
BLA | 15.24 | 16.85 |
BLA disutility | −0.06 | −0.06 |
Discounted costs ($) | ||
Total | 1,152,401 | 811,911 |
Initial treatment health states (controlled or uncontrolled) | ||
Treatment costs | 317,964 | 432 |
AE costs | 38 | 4 |
Comorbidity costs | 4,751 | 670 |
Resource costs | 124,027 | 37,760 |
Subsequent treatment health state | ||
Treatment costs | 4,281 | 4,556 |
AE costs | 12 | 13 |
Comorbidity costs | 1,884 | 2,004 |
Resource costs | 96,655 | 102,680 |
BLA | ||
Treatment costs | 15,150 | 16,098 |
AE costs | 292,406 | 323,278 |
Comorbidity costs | 37,476 | 41,440 |
Resource costs | 257,756 | 282,975 |
AE = adverse event; BLA = bilateral adrenalectomy; CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
Note: The CDA-AMC base case is based on publicly available prices of comparators.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 10).
Table 10: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($)a | Annual cost ($)b | ICERs for osilodrostat vs. ketoconazole ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 1 mg tablet: $46.14 5 mg tablet: $184.58 10 mg tablet: $193.81 | 67,407 to 134,815 | 491,746 | 742,689 |
10% | 1 mg tablet: $36.60 5 mg tablet: $146.41 10 mg tablet: $153.74 | 60,666 to 121,334 | 447,856 | 676,687 |
20% | 1 mg tablet: $32.53 5 mg tablet: $130.15 10 mg tablet: $136.66 | 53,926 to 107,852 | 403,966 | 610,684 |
30% | 1 mg tablet: $28.46 5 mg tablet: $113.88 10 mg tablet: $119.57 | 47,185 to 94,371 | 360,077 | 544,682 |
40% | 1 mg tablet: $24.40 5 mg tablet: $97.61 10 mg tablet: $102.49 | 40,444 to 80,889 | 316,187 | 478,679 |
50% | 1 mg tablet: $20.33 5 mg tablet: $81.34 10 mg tablet: $85.41 | 33,704 to 67,408 | 272,297 | 412,677 |
60% | 1 mg tablet: $16.27 5 mg tablet: $65.07 10 mg tablet: $68.33 | 26,963 to 53,926 | 228,407 | 346,674 |
70% | 1 mg tablet: $12.20 5 mg tablet: $48.80 10 mg tablet: $51.25 | 20,222 to 40,445 | 184,518 | 280,672 |
80% | 1 mg tablet: $8.13 5 mg tablet: $32.54 10 mg tablet: $34.16 | 13,481 to 26,963 | 140,628 | 214,669 |
90% | 1 mg tablet: $4.07 5 mg tablet: $16.27 10 mg tablet: $17.08 | 6,741 to 13,482 | 96,738 | 148,666 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aSponsor’s submitted price for osilodrostat.4
bA range is provided for the annual cost due to the range in recommended dose (typically 4 mg to 8 mg per day, to a maximum of 60 mg per day). Using the sponsor’s dosing and average daily cost of osilodrostat ($264 per day) in the economic model, the annual cost was approximately $96,500 per patient (this does not account for discontinuation, dose changes, or duration of treatment).
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 11.
Uncertainty associated with comparative efficacy was explored by varying the HR for osilodrostat relative to ketoconazole for TTCR and/or TTFPR to 1.
Table 11: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | Ketoconazole | 808,092 | 17.69 | Reference |
Osilodrostat | 1,131,673 | 18.13 | 737,858 | |
CDA-AMC scenario 1a: same TTCR | Ketoconazole | 797,944 | 17.70 | Reference |
Osilodrostat | 1,131,673 | 18.13 | 777,037 | |
CDA-AMC scenario 1b: same TTFPR | Ketoconazole | 823,892 | 17.89 | Reference |
Osilodrostat | 980,546 | 17.92 | 4,945,020 | |
CDA-AMC scenario 1c: same TTCR and TTFPR | Ketoconazole | 815,585 | 17.92 | Reference |
Osilodrostat | 980,546 | 17.92 | More costly and no incremental QALYs |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TTCR = time to complete response; TTFPR = time to failure post response.
aDeterministic analyses.
Issues for Consideration
The sponsor identified key comparator, ketoconazole, is not indicated in this population although it is listed on most (but not all) CDA-AMC–participating public drug formularies as a full benefit. For jurisdictions in which ketoconazole is not publicly funded, there may be a greater additional cost to the public drug plan budget. Additionally, pasireotide was noted to be an off-label treatment for Cushing disease by clinical experts consulted by CDA-AMC. As previously noted, the cost-effectiveness of osilodrostat compared with other comparators is unknown.
Patient group feedback highlighted delayed diagnosis of Cushing disease, and the impact this has on patients and their caregivers’ well-being, as well as the amount of health care resources used during this time. Clinical expert feedback identified delay in diagnosis as leading to less success in subsequent treatments, including surgery.
According to the clinical experts consulted by CDA-AMC, a PET scan might also be a potential resource use by patients with Cushing disease, which was not accounted for in the economic model. Clinical expert feedback suggested the availability of osilodrostat is unlikely to impact the use of PET scans for patients with Cushing disease.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing osilodrostat for the treatment of adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (Q4 2024 to Q3 2027), with Q4 2023 to Q3 2024 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. The market uptake for osilodrostat was estimated using the sponsor’s internal forecasts. The key inputs to the BIA are documented in Table 12.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing osilodrostat would be $11,964,667 (year 1 = $3,133,242; year 2 = $3,999,143; year 3 = $4,832,282).
Table 12: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Adult population (18+) of participating jurisdictions (2023/2024) | 26,038,752 |
Prevalence of CS | 0.0039%33 |
Incidence of endogenous CS | 0.00016%34,a |
Proportion of patients with endogenous CS who have CD | 68%b |
Proportion with CD eligible for surgery | 84%35 |
CD surgery cure rate | 79%36,c |
CD annual relapse rate after first surgery | 7.79% within 2 years36,c |
Proportion of patients eligible for pharmacotherapy post CD surgery | 39%37 |
Proportion CD who are ineligible for surgery who are eligible for pharmacotherapy | 100%37 |
Public coverage rate | Varies by jurisdiction (25% to 100%)38 |
Annual population growth rate | Varies by jurisdiction (0.7% to 3.5%)16 |
Number of patients eligible for the drug under review | 57 / 58 / 59 |
Market shares (reference scenario) | |
Osilodrostat | 0% / 0% / 0% |
ketoconazole | 100% / 100% / 100% |
Market shares (new drug scenario) | |
Osilodrostat | 60% / 75% / 87.5% |
ketoconazole | 40% / 25% / 12.5% |
Cost of treatment (per patient per annum) | |
Osilodrostat | $93,012 |
ketoconazole | $1,371 |
ACTH = adrenocorticotropic hormone; CD = Cushing disease; CS = Cushing syndrome.
aThe midpoint of the range of CS incidences reported in Sharema et al. (2015) (0.7 to 2.4 per million).34
bAssumes that 85% of patients with Cushing syndrome have ACTH-dependent disease and, of these, 80% are due to ACTH production from a pituitary adenoma (i.e., Cushing disease). The remainder are assumed to have non-CD CS.34
cDerived from Peterson et al. (2015), who reported an overall recurrence of 15.2% with a mean time to recurrence of 50.8 months.36
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The deviation of the eligible patient population is uncertain. The sponsor’s deviation of the eligible patient population was informed by published literature. Clinical experts consulted by CDA-AMC indicated that the population was likely underestimated as parameters such as the prevalence of Cushing syndrome, the surgery cure rate, and relapse rate were lower than their expectations.
The sponsor estimated the prevalence of Cushing syndrome to be 39.1 per million population based on a 1994 Spanish study.33 However, this study estimated the prevalence of confirmed Cushing disease rather than Cushing syndrome. As the sponsor later estimated that of patients with Cushing syndrome, 68% have Cushing disease, the use of a Cushing syndrome prevalence rate reflecting only patients with Cushing disease is inappropriate. When the 39.1 per million estimate is inflated by assuming it represents only 68% of patients with Cushing syndrome, the resulting Cushing syndrome prevalence is 57.5 per million.
The sponsor estimated that the first surgery cure rate was 79%, and the relapse rate was 7.79%. Clinical experts consulted by CDA-AMC noted that these estimates were more optimistic compared to their clinical experience. The published literature used to inform the sponsor’s model may not be generalizable to clinical practice in Canada. The surgery cure rate was informed by a systematic review of clinical outcomes in patients with Cushing disease, which did not localize its search to outcomes in Canada, while the relapse rate was informed by a retrospective cohort study of 86 patients in southwestern Ontario. It is uncertain if the first surgery cure rate, informed by international outcomes, or the relapse rate, informed by care centres localized to southwestern Ontario, is generalizable to the indicated population in Canada. The sponsor used the proportion of the general population of each jurisdiction enrolled in public drug plans to estimate the proportion of the population with Cushing syndrome who would be eligible for public funding of osilodrostat.38 As uncontrolled Cushing syndrome has a high burden of illness, it is likely that patients eligible for osilodrostat will be less likely to have access to employer-sponsored private drug coverage than the general population. Additionally, at the submitted price and recommended dosing, osilodrostat is likely to qualify as a high-cost drug, further increasing the proportion of the population who would be eligible for public reimbursement if osilodrostat is funded.
CDA-AMC removed the percentage of patient with CS who have CD, as the published literature estimated the prevalence of CD.
CDA-AMC used the proportion of the 25 to 64 year old population who are eligible for public coverage38 as a proxy for the proportion of adults with Cushing syndrome who will be eligible for public reimbursement of osilodrostat, rather than the proportion already enrolled.
In a scenario analysis, CDA-AMC adjusted the first surgery cure rate and the relapse rate to 50%, and 16%, respectively, in line with clinical practice in Canada.
Use of different adherence rates to estimate drug costs is not appropriate: The sponsor assumed that patients receiving ketoconazole would be 100% adherent to treatment while those receiving osilodrostat would be 85% adherent, based on clinical expert feedback sought by the sponsor. This is inconsistent with the assumptions of the pharmacoeconomic model. The impact of reduced adherence on the clinical or cost-effectiveness of osilodrostat is unknown. Furthermore, prescriptions for osilodrostat may be filled and reimbursed regardless of treatment adherence.
CDA-AMC assumed 100% adherence for osilodrostat.
Uncertainty in the place in therapy for osilodrostat: The sponsor assumed that osilodrostat would be used for the same patient population as ketoconazole, i.e., for patients with Cushing disease who are not eligible for surgery or for whom surgery has failed or who have relapsed after surgery. However, clinical expert input obtained by CDA-AMC indicated that at least in the initial years of availability, clinicians may prefer continuing to use ketoconazole as the initial pharmacotherapy and reserve osilodrostat only for patients who do not respond to ketoconazole or for whom ketoconazole is intolerable or contraindicated. Clinician expert input to CDA-AMC indicated that approximately 50% of patients do not respond to ketoconazole or have intolerable side effects, which is consistent with reports in the literature on the proportion of patients with Cushing disease who achieve normalized cortisol levels with ketoconazole.39
CDA-AMC conducted a scenario exploring the impact of assuming that only patients for whom treatment had failed or who were unable to tolerate ketoconazole (50%) would receive osilodrostat.
The Non-Insured Health Benefits (NIHB) population was inappropriately calculated: For the purposes of modelling the budget impact of reimbursing osilodrostat, NIHB clients who live within Ontario who are aged younger than 25 years or 65 years or older are eligible for reimbursement by the Ontario Drug Benefit (ODB) program and thus should be counted as ODB clients and included in Ontario population estimates rather than as NIHB clients.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 13.
Table 13: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Proportion of patients with CD | 68% | 100% |
2. Adherence rates | 85% | 100% |
3. Public coverage | Proportion aged 25 to 64 enrolled in public drug plans | Proportion aged 25 to 64 eligible for public drug plans |
CDA-AMC base case | ― | (Reanalysis 1 + 2 + 3) |
BIA = budget impact analysis; CD = Cushing disease; CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 14, and a more detailed breakdown is presented in Table 15. In the CDA-AMC base case, the 3-year budget impact of reimbursing osilodrostat for adult patients with Cushing disease who have persistent or recurrent hypercortisolism after primary pituitary surgery and/or irradiation, or for whom pituitary surgery is not an option was $30,558,303 (year 1 = $8,021,653; year 2 = $10,243,514; year 3 = $12,293,135).
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 11,964,667 |
CDA-AMC reanalysis 1 | 17,551,328 |
CDA-AMC reanalysis 2 | 14,107,496 |
CDA-AMC reanalysis 3 | 17,653,013 |
CDA-AMC base case: reanalysis 1 + 2 + 3 | 30,558,303 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of comparators.
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing osilodrostat. The results are provided in Table 15.
Revised the first surgery cure rate and relapse rate to align with clinical practice.
Using osilodrostat only after a patient’s disease had failed to respond or they were unable to tolerate ketoconazole. In this scenario, costs for patients either remaining on ketoconazole or having an unsuccessful trial of ketoconazole are assumed equal between the reference and new drug scenario and are therefore not included.
Table 15: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 76,535 | 78,147 | 79,795 | 81,480 | 239,422 |
Osilodrostat | 0 | 0 | 0 | 0 | 0 | |
All comparators | 76,535 | 78,147 | 79,795 | 81,480 | 239,422 | |
New drug total | 76,535 | 3,211,389 | 4,078,938 | 4,913,762 | 12,204,089 | |
Osilodrostat | 0 | 3,180,131 | 4,058,989 | 4,903,577 | 12,142,696 | |
All comparators | 76,535 | 31,259 | 19,949 | 10,185 | 61,392 | |
Budget impact | 0 | 3,133,242 | 3,999,143 | 4,832,282 | 11,964,667 | |
CDA-AMC base case | Reference total | 166,098 | 169,679 | 173,341 | 177,088 | 520,108 |
Osilodrostat | 0 | 0 | 0 | 0 | 0 | |
All comparators | 166,098 | 169,679 | 173,341 | 177,088 | 520,108 | |
New drug total | 166,098 | 8,191,332 | 10,416,856 | 12,470,223 | 31,078,410 | |
Osilodrostat | 0 | 8,123,460 | 10,373,520 | 12,448,087 | 30,945,068 | |
All other comparators | 166,098 | 67,871 | 43,335 | 22,136 | 133,343 | |
Budget impact | 0 | 8,021,653 | 10,243,514 | 12,293,135 | 30,558,303 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: surgery and relapse rate | Reference total | 171,675 | 175,376 | 179,162 | 183,034 | 537,573 |
New drug total | 171,675 | 8,466,398 | 10,766,656 | 12,885,302 | 32,118,356 | |
Budget impact | 0 | 8,291,022 | 10,587,494 | 12,702,267 | 31,580,783 | |
Scenario 2: osilodrostat only after ketoconazole failure | Reference total | 0 | 0 | 0 | 0 | 0 |
New drug total | 0 | 4,061,730.05 | 5,186,760.22 | 6,224,043.53 | 15,472,534 | |
Budget impact | 0 | 4,061,730.05 | 5,186,760.22 | 6,224,043.53 | 15,472,534 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of comparators.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.