Drugs, Health Technologies, Health Systems
Reimbursement Review
Ruxolitinib (Opzelura)
Sponsor: Incyte Biosciences Canada Corporation
Therapeutic area: Atopic dermatitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic_Review
Clinical Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
BSA
body surface area
CDA-AMC
Canada’s Drug Agency
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CSPA
Canadian Skin Patient Alliance
DLQI
Dermatology Life Quality Index
EASI
Eczema Area and Severity Index
EASI-75
at least a 75% improvement in Eczema Area and Severity Index
ESC
Eczema Society of Canada
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IGA
Investigator’s Global Assessment
IGA-TS
Investigator’s Global Assessment treatment success
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
JAK
Janus kinase
LSM
least squares mean
MCID
minimal clinically important difference
MID
minimal important difference
mPP-NRS
modified Peak Pruritus Numeric Rating Scale
NMA
network meta-analysis
NRS
Numeric Rating Scale
NRS-4
at least a 4-point improvement in Numeric Rating Scale
OR
odds ratio
POEM
Patient Oriented Eczema Measure
PP-NRS
Peak Pruritus Numeric Rating Scale
PROMIS
Patient-Reported Outcomes Measurement Information System
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Ruxolitinib cream (Opzelura), 1.5% |
Sponsor | Incyte Biosciences Canada Corporation |
Indication | For the topical treatment of mild to moderate atopic dermatitis in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable.a |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 11, 2024 |
Recommended dose | Twice daily to affected skin areas up to a maximum of 20% of body surface area for each application |
NOC = Notice of Compliance; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
aThe product monograph states that the use of ruxolitinib cream in combination with other Janus kinase inhibitors, biological immunomodulators, or potent immunosuppressants has not been studied and is not recommended.
Introduction
Atopic dermatitis (AD) is a chronic relapsing-remitting skin condition characterized by itching, inflammation, dryness, recurrent eczematous lesions, erythematous papules, and lichenification.1,2 The intense itch associated with AD can lead to sleep disturbances, mental health burden, and reduced quality of life in patients and caregivers. The prevalence of AD in adults is estimated to vary from 1.8% to 3.5% in Canada,3,4 and the prevalence of AD in adolescents is estimated to vary from 9.4% to 15.8% in Canada.5,6
Patients who have uncontrolled disease despite nonpharmacological treatments (e.g., appropriate skin care practices and avoidance of symptom triggers and irritants) are recommended to initiate topical treatments, including topical corticosteroids (TCSs) and nonsteroidal topical treatments, such as topical calcineurin inhibitors (TCIs) (i.e., tacrolimus, pimecrolimus) and topical phosphodiesterase type 4 inhibitor (i.e., crisaborole; not widely used by clinicians).7 Patients who do not experience adequate disease control with topical treatments could receive phototherapy, off-label systemic immunosuppressant treatments (i.e., methotrexate, cyclosporine, mycophenolate mofetil, azathioprine), or advanced systemic therapies.8 Dupilumab, upadacitinib, and abrocitinib are advanced therapies currently reimbursed by the jurisdictions in Canada for the treatment of AD in adolescents and adults.
The objective of this report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ruxolitinib 1.5% cream in the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the Canada’s Drug Agency (CDA-AMC) call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
Input submissions were received from 2 patient groups. The Eczema Society of Canada (ESC) is a registered Canadian charity with a mission of support, education, awareness, and research for people living with eczema. The ESC gathered information from more than 3,000 patients living with AD in Canada, as well as their caregivers and/or family members, via survey questionnaires and one-on-one interviews. Another patient group input submission was jointly prepared by the Canadian Skin Patient Alliance (CSPA) and Eczema Quebec. CSPA is a national nonprofit organization that engages in collaboration, advocacy, and education for people affected by skin, hair, and nail conditions. Eczema Quebec is a nonprofit organization dedicated to providing support, resources, and education to individuals and families affected by eczema in Quebec. The joint input was based on information gathered between February and October 2023 from various sources, including a literature review, patients, and The Skin I’m In report, and through collaboration with an academic institution. Some patients (number not specified) surveyed by Eczema Quebec indicated that they had experience with ruxolitinib cream treatment.
Both submissions highlighted that the signs and symptoms of AD, such as dry, itchy, inflamed skin that can lead to cracks, oozing, bleeds, and thickening of skin, affect many aspects of patients’ lives, including physical, social, emotional, and professional aspects. Patients said itches can be extremely uncomfortable and painful and can require frequent medical visits, specialized treatments, and ongoing care. The joint input by CSPA and Eczema Quebec pointed out that AD is associated with other conditions, such as asthma, seasonal allergies, environmental allergies, food intolerances, sleep disorders, anxiety, and depression. The input submissions emphasized that caregivers and/or family members also share a significant burden of disease. The negative impact of AD on patients and their caregivers and/or families is amplified when AD is not well controlled despite optimization of the treatment regimen and when cycling through or switching to different therapies. According to the input by the ESC, uncontrolled AD or flares can lead to emergency department visits and hospitalizations. The input also highlighted that because AD can occur at a young age, it can cause significant impact on young people’s performance at school, their social life, and their mental health. In both input submissions, patients expressed a need for new treatments that are safe, improve the symptoms of AD, reduce flares, and improve quality of life (e.g., better sleep quality, less psychological burden, improved ability to carry out daily activities, and improved ability to establish and maintain intimate relationships), as well as reduce or eliminate potential complications and secondary infections associated with AD. Other key outcomes reported to be important to patients included fast and durable relief and reduced skin thickening; ease of medication use and affordability are also considered important. In addition, patients value treatments that do not require injections. Patients expressed a need for treatments suitable for application not only on the body but also on the face and sensitive areas of the body, for a simplified regimen. The patient groups acknowledged that AD is a heterogeneous disease and requires a variety of treatments to fill gaps in therapeutic needs.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts consulted for this review noted that currently available nonsteroidal topical treatments are not effective for all patients with AD and are inadequate for treating body areas with thick skin (e.g., palms, soles) or lichenification. These current nonsteroidal topical treatments are also associated with application site reactions (burning and stinging) and are costly. The clinical experts noted that the nonsteroidal topical treatment currently considered to be the most effective option (tacrolimus) is available in ointment formulation only, which is difficult to apply. One clinical expert anticipated that ruxolitinib would primarily serve as a second-line topical treatment following treatment failure with, or ineligibility for, TCS and/or TCI because of long-established treatment protocols favouring TCS and TCI, as well as anticipated access challenges with ruxolitinib cream due to its higher drug cost relative to currently reimbursed topical treatments. The other clinical expert anticipated that ruxolitinib cream could also be used as a first-line topical treatment. This clinical expert further explained that TCIs are associated with application site reactions and moderate efficacy. Provided that ruxolitinib cream is as or more effective and has fewer application site reactions than TCIs, the clinical expert thought it would be reasonable to use ruxolitinib cream ahead of TCIs, in particular for the face and groin, for which TCS treatment is inappropriate. The clinical experts noted that identifying the patients for whom use of TCS and TCI is advisable primarily depends on reaction to previous use (e.g., inadvisable in cases of intolerance). According to the clinical experts, there are very few contraindications to TCS and TCI (they would include the use of any TCS, including hydrocortisone, to the eyelids and the long-term use of corticosteroids more potent than hydrocortisone to the face and intertriginous skin). In the clinical experts’ experience, almost all patients who have not previously received topical treatment are eligible for TCS and TCI treatments. The clinical experts noted that depending on the severity of the symptoms and the treatment response in each anatomic location, ruxolitinib cream could be used as either monotherapy or in combination with other topical therapies (applied to different affected areas). The clinical experts noted that when used concurrently with other topical therapies, ruxolitinib cream could be applied to the same or different anatomic locations; most patients are expected to use 1 treatment at a time in a given location and apply different topicals to different parts of the body.
One clinical expert noted that patients with AD who have facial or intertriginous involvement, experience inadequate response to or intolerable adverse events (AEs) from TCS and/or TCI treatments, and have 10% or less body surface area (BSA) affected by AD are most suited for treatment with ruxolitinib cream as monotherapy. In the second clinical expert’s opinion, patients with AD could receive ruxolitinib cream treatment regardless of response to or eligibility for TCS and/or TCI treatments. Tthe second clinical expert felt that patients with more than 10% BSA affected might still be eligible for ruxolitinib cream provided that the cream is applied to no more than 10% to 20% BSA.
According to the clinical experts, there is no universal definition for adequate (or inadequate) response to TCS and TCI treatment. They noted that treatment response to TCS and TCI is typically determined by clinical judgment on a case-by-case basis and by patient satisfaction, although it might be reasonable to consider the initiation of ruxolitinib cream treatment for patients who do not experience skin improvements after 4 to 8 weeks of conventional topical therapy — including low-potency, mid-potency, or high-potency TCS, TCI, or crisaborole — as suggested in the sponsor’s submission. From a clinical perspective, the clinical experts noted that ruxolitinib cream could also be considered before the failure of existing topical treatments. The clinical experts noted that response to ruxolitinib cream treatment should similarly be assessed based on clinical judgment. They noted that at least a 75% improvement in Eczema Area and Severity Index (EASI-75), which is the benchmark currently applied to the renewal of systemic AD treatment reimbursement, may not be applicable to ruxolitinib cream. The clinical experts explained that, given that ruxolitinib cream may be used in combination with other topical treatments (applied to different affected areas), it is impossible to attribute changes in Eczema Area and Severity Index (EASI) score purely to ruxolitinib cream treatment in these scenarios. It would be reasonable to conduct a follow-up assessment at 8 weeks after treatment initiation, although a longer interval of 3 to 6 months may be more practical for patients with less severe disease, according to the clinical experts. Additionally, the clinical experts noted that given Canada’s medical resource constraints, particularly access to dermatology visits but also to family physician and other physician visits, shorter follow-up intervals may be impractical. The clinical experts noted that treatment discontinuation could be considered in patients who experience inadequate response or intolerable AEs on ruxolitinib cream treatment. The clinical experts noted that ruxolitinib cream could be prescribed by any health care provider with experience in diagnosing, treating, and monitoring patients with AD, principally general dermatologists, pediatricians, pediatric dermatologists, allergists, family practitioners, and nurse practitioners.
Clinician Group Input
The Canadian Dermatology Association, represented by 3 clinicians, and the Atlantic Dermatology Specialist Group, represented by 11 clinicians, prepared 2 separate input submissions. Consistent with the input from the clinical experts consulted by CDA-AMC, the clinician groups indicated that some patients receiving existing treatments experience uncontrolled disease, side effects, and poor tolerability with ointment formulation, or poor treatment adherence due to the need to apply different topical products to different body locations. The clinician groups agreed that there is an unmet need for a new topical therapy that is effective, better tolerated, and in a cream formulation. The clinician groups also noted that an effective topical therapy is needed to prevent the need to escalate to phototherapy or systemic treatments, which are associated with limitations (e.g., limited efficacy, accessibility, and drug coverage; side effects; monitoring requirements; and high treatment costs). Both clinician groups also agreed that the main treatment goals include reducing itch and inflammation (short-term and long-term), achieving skin clearance, minimizing tolerability and safety issues, and improving quality of life (e.g., sleep, anxiety, depression). The clinical groups and clinical experts agreed that an ideal topical treatment should be in a cosmetically appropriate base, convenient to use, and accessible.
In general, the 2 clinician groups and the clinical experts consulted by CDA-AMC agreed that ruxolitinib cream could be used in patients with AD that is not adequately controlled with topical prescription therapies (TCS or TCI) or when those therapies are not advisable. However, the Atlantic Dermatology Specialist Group and the clinical experts noted that there is potential for the ruxolitinib treatment to be used as a first-line treatment in some patients. The clinician groups noted that eligible patients include those with mild to moderate AD with up to 20% BSA affected, severe localized AD, or moderate to severe AD (EASI score > 16; ≥ 10% BSA affected), as well as those who cannot access or have contraindications to phototherapy or systemic therapies. Consistent with the input from the clinical experts consulted by CDA-AMC, the clinician groups noted that ruxolitinib cream could be used either as monotherapy or as an adjunct to systemic therapy (if eligible and tolerated) for continuous or as-needed use. While the clinician groups felt that ruxolitinib cream could be used on any body sites in patients with up to 20% BSA affected, 1 of the clinical experts consulted by CDA-AMC felt that use of ruxolitinib cream should be mainly limited to facial and intertriginous skin and applied to no more than 10% BSA due to potential systemic absorption and high treatment cost.
Both clinician groups and the clinical experts noted that the treatment response is typically assessed by signs and symptoms (e.g., itch and inflammation), BSA affected, extent of involvement of special sites (hands, feet, face, skin folds, or perineal area), and patient-reported outcomes (e.g., health-related quality of life [HRQoL], functional impact). The clinician groups and 1 of the clinical experts consulted by CDA-AMC agreed that after a trial period of 8 weeks, if there is an inadequate improvement in signs and symptoms of disease, recurrent flares, worsening of disease, or intolerance or side effects, then discontinuation of the treatment would be considered. The clinician groups’ input also indicated that a 3-month to 6-month follow-up for response assessment in patients receiving topical treatments could be more favourable in clinical practice, except for patients with more severe disease, for whom an 8-week assessment interval would be appropriate. The clinician groups agreed that generalist or primary care physicians as well as specialists (e.g., dermatologists, allergy and immunology specialists, and pediatricians) who are comfortable with the diagnosis and management of AD should prescribe, treat, and monitor patients who receive ruxolitinib cream.
Drug Program Input
The drug programs noted interest in the relationship between the Investigator’s Global Assessment (IGA) score (the primary end point in the submitted pivotal trials) and the EASI score, and the clinical experts consulted for this review provided context on the construct, scoring, and utility of these instruments in clinical trials and clinical practice. In response to the drug programs’ question about whether TCSs and TCIs should be considered comparators for ruxolitinib cream, the clinical experts noted that TCSs and TCIs are relevant comparators in their opinion. The clinical experts explained that even if a patient experiences inadequate response to a TCI treatment, that patient may remain on the same treatment or move to another topical treatment, such as ruxolitinib cream. Additionally, because patients should not need to experience the failure of all currently available topical therapies before they can move on to ruxolitinib cream, the clinical experts felt that any currently available topical therapy would be considered a relevant comparator. The clinical experts noted that ruxolitinib cream monotherapy would be appropriate in patients with mild to moderate AD, whereas systemic drugs would be appropriate in patients with moderate to severe AD. In the clinical experts’ opinion, systemic therapies and phototherapy are not relevant comparators for ruxolitinib cream. In response to the drug programs’ question about the anticipated average duration of treatment with ruxolitinib cream, the clinical experts expected patients to receive ruxolitinib cream at the lowest effective application frequency on an as-needed basis over months or years.
In response to the drug programs’ questions about the potential for combination use of ruxolitinib cream and other topical therapies, the clinical experts noted that such combination use is expected. The clinical experts noted that ruxolitinib cream could be applied to the same or different anatomic locations; most patients are expected to use 1 treatment at a time in a given location and apply different topical treatments to different parts of the body. Additionally, the drug programs inquired about the place in therapy and the appropriate prescribers for ruxolitinib cream; the clinical expert response is summarized in Table 4.
Clinical Evidence
Systematic Review
Description of Studies
The sponsor-conducted systematic literature review (SLR) identified 2 identically designed, pivotal, phase III, double-masked, randomized controlled trials (RCTs) (TRuE-AD1 trial [N = 631]; TRuE-AD2 trial [N = 618])9,10 aiming to assess the efficacy and safety of ruxolitinib cream relative to vehicle cream, as monotherapy, in adolescents and adults aged 12 years or older with AD of mild (IGA score of 2) or moderate (IGA score of 3) severity, and with 3% to 20% BSA affected by AD. Patients were randomized to receive ruxolitinib 1.5% cream, ruxolitinib 0.75% cream, or vehicle cream monotherapy in a 2:2:1 ratio for an 8-week vehicle-controlled period, followed by a 44-week long-term safety period. In the long-term safety period, patients who initially received vehicle cream in the vehicle-controlled period were rerandomized to 1 of the 2 ruxolitinib cream treatment groups to receive treatment on an as-needed basis, while patients who initially received ruxolitinib cream continued to receive the same intervention as needed. In both trials, the primary end point was the proportion of patients experiencing IGA treatment success (IGA-TS) (i.e., an IGA score of 0 or 1, with a ≥ 2-grade improvement from baseline) at week 8. The key secondary end points were the following at week 8: proportion of patients experiencing EASI-75 (i.e., at least a 75% improvement [i.e., reduction] from baseline in EASI score), at least a 4-point improvement (i.e., reduction) from baseline in Itch Numeric Rating Scale (NRS) score, at least a 6-point improvement (i.e., reduction) from baseline in Patient-Reported Outcomes Measurement Information System (PROMIS) Short Form–Sleep Disturbance (24-hour recall) score, and at least a 6-point improvement (i.e., reduction) from baseline in PROMIS Short Form–Sleep-Related Impairment (24-hour recall) score.
At baseline, the majority of patients in both trials were adults (TRuE-AD1 trial: 80.3%; TRuE-AD2 trial: 80.5%) and had an IGA score of 3 (TRuE-AD1 trial: 75.9%; TRuE-AD2 trial: 74.1%). The mean total percentage of BSA affected by AD was 9.5% for the patients in the TRuE-AD1 trial and 10.0% for the patients in the TRuE-AD2 trial. Prior TCI treatment was noted in 24.1% and 18.8% of patients in the TRuE-AD1 and TRuE-AD2 trials, respectively. Prior medium-potency, high-potency, and super-high-potency TCS treatment was noted in 43.7%, 34.9%, and 8.9% of patients, respectively, in the TRuE-AD1 trial and in 41.1%, 30.4%, 7.0% of patients, respectively, in the TRuE-AD2 trial. The proportion of patients who had experienced inadequate disease control with TCS and/or TCI, or for whom such treatments were not advisable, was not reported. A small proportion of patients had received prior systemic immunosuppressants, phototherapy, dupilumab, and/or systemic Janus kinase (JAK) inhibitor treatment.
Efficacy Results
The efficacy and safety results of the ruxolitinib 0.75% cream group are not presented in this report because this strength of ruxolitinib cream is not approved by Health Canada for the treatment of AD and is not of interest to this review. In addition, the study inclusion and exclusion criteria did not restrict entry based on prior experience with topical treatments, and the sponsor was unable to provide subgroup data in the patient population in accordance with the Health Canada indication (patients whose disease is not adequately controlled with conventional topical prescription therapies or when those therapies are not advisable) upon the review team’s request. However, the sponsor provided post hoc analyses using the pooled data from both trials by topical treatment history (for patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as for patients who had received any topical treatment in the 30 days before screening) as supportive evidence for select outcomes. The results for the full study population, along with the post hoc subgroup analyses, are presented in the following sections.
IGA Score
IGA Treatment Success
The proportion of patients experiencing IGA-TS at week 8 was the primary end point in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 38.7% (95% confidence interval [CI], 29.9% to 47.4%; P < 0.0001) in the TRuE-AD1 trial and 43.7% (95% CI, 35.6% to 51.8%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. The results of the prespecified exploratory subgroup and the sensitivity analyses were consistent in direction with the primary analysis in both trials. The subgroup analyses in both trials seem to suggest a higher IGA-TS response rate at week 8 in patients with a baseline IGA score of 3 (versus an IGA score of 2) and an EASI score greater than 7 (versus an EASI score ≤ 7), as well as patients in Europe (versus in North America).
A post hoc subgroup analysis by topical treatment history showed results consistent with the primary analysis across subgroups (patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received any topical treatment in the 30 days before screening).
IGA-TS was not assessed at week 52 in either trial.
IGA Score of 0 or 1
The proportion of patients who had an IGA score of 0 or 1 was a secondary end point at week 52 in both trials. At week 52, the proportions of patients who had an IGA score of 0 or 1 in the vehicle cream to ruxolitinib 1.5% cream group and the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group were 73.7% and 75.4%, respectively, in the TRuE-AD1 trial and 74.4% and 80.1%, respectively, in the TRuE-AD2 trial.
In a post hoc subgroup analysis by topical treatment history, the vehicle cream to ruxolitinib 1.5% cream group experienced a similar response rate in terms of IGA score of 0 or 1 at week 52 as the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group across patients who had received TCS only, TCI only, or TCS and TCI regardless of treatment time frame, as well as patients who had received any topical treatment in the 30 days before screening.
EASI Score
The proportion of patients who experienced EASI-75 at week 8 was a key secondary end point and was adjusted for multiplicity in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 37.5% (95% CI, 27.8% to 47.1%; P < 0.0001) in the TRuE-AD1 trial and 47.4% (95% CI, 38.5% to 56.4%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. In both trials, the results of the sensitivity analyses were consistent with the primary analysis. The results of the prespecified subgroup analyses showed a direction of effect consistent with the primary analysis. The subgroup analyses in both trials seem to suggest a higher EASI-75 response rate at week 8 in patients with a baseline EASI score greater than 7 (versus an EASI score ≤ 7).
A post hoc subgroup analysis by topical treatment history showed results consistent in direction with the primary analysis across all subgroups (patient who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received any topical treatment in the 30 days before screening).
EASI-75 was not assessed at week 52 in either trial.
Percentage of BSA Affected by AD
The change from baseline in the percentage of BSA affected by AD at week 8 and at week 52 were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group least squares mean (LSM) difference comparing ruxolitinib 1.5% cream with vehicle cream was –3.7% (95% CI, –4.7% to –2.8%) in the TRuE-AD1 trial and –4.5% (95% CI, –5.5% to –3.6%) in the TRuE-AD2 trial.
In both trials, a reduction in the percentage of BSA affected by AD was sustained at week 52 in patients who continued to receive ruxolitinib 1.5% cream in the long-term safety period (ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group). The vehicle cream to ruxolitinib 1.5% cream group experienced a percentage of BSA affected by AD similar to that of the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group at week 52 in both trials. In a post hoc subgroup analysis by topical treatment history, the vehicle cream to ruxolitinib 1.5% cream group experienced a similar total percentage of BSA affected by AD at week 52 as the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group across patients who had received TCS only, TCI only, or TCS and TCI regardless of treatment time frame, as well as patients who had received any topical treatment in the 30 days before screening.
Itch NRS Score
The proportion of patients with at least 4 points of improvement in Itch NRS from baseline at week 8 (among patients with a baseline score of at least 4 [vehicle cream: n = 78 in the TRuE-AD1 trial, n = 80 in the TRuE-AD2 trial; ruxolitinib: n = 161 in the TRuE-AD1 trial, n = 146 in the TRuE-AD2 trial]) was a key secondary end point and was adjusted for multiplicity in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 36.8% (95% CI, 25.7% to 47.9%; P < 0.0001) in the TRuE-AD1 trial and 34.4% (95% CI, 23.0% to 45.9%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. A post hoc subgroup analysis by topical treatment history showed results consistent in direction with the primary analysis across subgroups (patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening).
This end point was not assessed at week 52 in either trial.
Patient Oriented Eczema Measure
The change from baseline in Patient Oriented Eczema Measure (POEM) score at week 8 and at week 52 were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –6.3 (95% CI, –7.6 to –5.0) in the TRuE-AD1 trial and –5.9 (95% CI, –7.2 to –4.7) in the TRuE-AD2 trial.
In both trials, reduction in POEM score was sustained at week 52 in patients who continued to receive ruxolitinib 1.5% cream in the long-term safety period (ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group). The vehicle cream to ruxolitinib 1.5% cream group had a mean POEM score similar to that of the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group at week 52 in both trials.
PROMIS Short Form–Sleep Disturbance
The proportion of patients with at least a 6-point improvement in PROMIS Short Form–Sleep Disturbance score (24-hour recall) from baseline at week 8 (among patients with a baseline score of at least 6 [vehicle cream: n = 116 in the TRuE-AD1 trial, n = 110 in the TRuE-AD2 trial; ruxolitinib: n = 238 in the TRuE-AD1 trial, n = 211 in the TRuE-AD2 trial]) was a key secondary end point and was adjusted for multiplicity in both trials. The between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 12.8% (95% CI, 5.3% to 20.3%; P = 0.0039) in the TRuE-AD1 trial, in favour of ruxolitinib 1.5% cream. In the TRuE-AD2 trial, the between-group difference was 6.5% (95% CI, –2.9% to 15.9%; P = 0.2359), which did not favour either study intervention. As such, the end point of the proportion of patients with 6 or more points of improvement in the PROMIS Short Form–Sleep-Related Impairment score at week 8 was not tested for superiority as this end point is lower in the statistical testing hierarchy). This end point was not assessed at week 52 in either trial.
PROMIS Short Form–Sleep-Related Impairment
The proportion of patients with at least a 6-point improvement in PROMIS Short Form–Sleep-Related Impairment score (24-hour recall) from baseline at week 8 (among patients with a baseline score of at least 6 [vehicle cream: n = 114 in the TRuE-AD1 trial, n = 111 in the TRuE-AD2 trial; ruxolitinib: n = 245 in the TRuE-AD1 trial, n = 212 in the TRuE-AD2 trial]) was a key secondary end point in both trials. This end point was included in the statistical testing hierarchy, but no superiority testing was conducted due to prior failure in the hierarchy. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 8.4% (95% CI, 0.4% to 16.4%) in the TRuE-AD1 trial and 9.6% (95% CI, 1.4% to 18.4%) in the TRuE-AD2 trial. This end point was not assessed at week 52 in either trial.
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index Scores
Change From Baseline in Dermatology Life Quality Index
The change in the Dermatology Life Quality Index (DLQI) score from baseline at week 8 and at week 52 (among patients aged 16 years or older [vehicle cream: n = 82 in the TRuE-AD1 trial, n = 87 in the TRuE-AD2 trial; ruxolitinib: n = 201 in the TRuE-AD1 trial, n = 185 in the TRuE-AD2 trial]) were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –4.5 (95% CI, –5.6 to –3.4) in the TRuE-AD1 trial and –2.8 (95% CI, –3.7 to –1.8) in the TRuE-AD2 trial. The results of the so-called “responder analysis” (analysis of data from patients experiencing a ≥ 4-point improvement in DLQI score) at week 8 were similarly in favour of ruxolitinib cream in both trials ██████████ ████ █████ █ █████ ███ ███ ████ ██ █████ ██████████ █████████ ████ █████ █ █████ ███ ███ ████ ██ █████ █████████.
In both trials, improvement (i.e., reduction) from baseline in DLQI score was sustained at week 52 in patients who continued to receive ruxolitinib 1.5% cream in the long-term safety period (ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group). The vehicle cream to ruxolitinib 1.5% cream group experienced a mean DLQI score similar to that of the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group at week 52 in both trials.
Change From Baseline in Children’s Dermatology Life Quality Index
The change in Children’s Dermatology Life Quality Index (CDLQI) score from baseline at week 8 and at week 52 (among patients younger than 16 years [vehicle cream: n = 16 in the TRuE-AD1 trial, n = 11 in the TRuE-AD2 trial; ruxolitinib: n = 28 in the TRuE-AD1 trial, n = 25 in the TRuE-AD2 trial]) were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –2.3 (95% CI, –4.4 to –0.1) in the TRuE-AD1 trial and –3.1 (95% CI, –6.3 to –0.1) in the TRuE-AD2 trial.
In both trials, improvement (i.e., reduction) from baseline in CDLQI score was sustained at week 52 in patients who continued to receive ruxolitinib 1.5% cream in the long-term safety period (ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group). The vehicle cream to ruxolitinib 1.5% cream group experienced a mean CDLQI score similar to that of the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group at week 52 in both trials.
Harms Results
Adverse Events
In the vehicle-controlled period, the proportion of patients who reported at least 1 treatment-emergent AE (TEAE) was lower in the ruxolitinib 1.5% cream group than in the vehicle cream group in both trials (TRuE-AD1 trial: 29.2% versus 34.9%; TRuE-AD2 trial: 23.6% versus 31.5%). The difference appears to be partly attributable to including AD as a harm as no active treatment was given to patients in the vehicle group. In the long-term safety period, the proportion of patients who reported at least 1 TEAE in the ruxolitinib 1.5% cream group was higher than in the vehicle cream group in the TRuE-AD1 trial (ruxolitinib: 53.5%; vehicle cream: 48.9%) but lower than in the vehicle cream group in the TRuE-AD2 trial (ruxolitinib: 54.3%; vehicle cream: 65.4%).
The most common TEAEs in the ruxolitinib 1.5% cream group were upper respiratory tract infection, nasopharyngitis, and headache in the vehicle-controlled and long-term safety periods.
Serious AEs
In the vehicle-controlled period, serious TEAEs were reported in 2 patients (1.6%) in the vehicle cream group and 2 patients (0.8%) in the ruxolitinib 1.5% cream group in the TRuE-AD1 trial. In the TRuE-AD2 trial, no patient reported serious TEAEs in the vehicle cream group and 1 patient (0.4%) reported a serious TEAE in the ruxolitinib 1.5% cream group. A similarly low frequency of serious TEAEs was noted in both treatment arms of the trials in the long-term safety period.
Withdrawal Due to AEs
In the vehicle-controlled period, study treatment withdrawal due to TEAEs was reported in 5 patients (4.0%) in the vehicle cream group and 3 patients (1.2%) in the ruxolitinib 1.5% cream group in the TRuE-AD1 trial and in 3 patients (2.4%) in the vehicle cream group and 1 patient (0.4%) in the ruxolitinib 1.5% cream group in the TRuE-AD2 trial. TEAEs leading to patients discontinuing ruxolitinib cream treatment included papule, generalized pruritus, and urticaria (1 patient [0.4%] each in the TRuE-AD1 trial), as well as cerebrovascular accident (1 patient [0.4%] in the TRuE-AD2 trial). In the long-term safety period of both studies, no patient withdrew from study treatment due to TEAEs.
Mortality
No death was reported during the vehicle-controlled or long-term safety periods in either trial.
Critical Appraisal
The trials used adequate methods of randomization and allocation concealment. There were a few small baseline imbalances in patient characteristics, which may be compatible with chance and were not believed to substantially impact the study results. The trials were adequately masked to reduce bias; however, there is a small potential for bias in the measurement of patient-reported outcomes (i.e., Itch NRS, POEM, PROMIS Short Form–Sleep Disturbance, PROMIS Short Form–Sleep-Related Impairment, DLQI, and CDLQI scores) leading to inflated efficacy of ruxolitinib cream due to possible unmasking through patients becoming aware of their assignments based on treatment response. Responder analyses of IGA-TS, EASI-75, Itch NRS, and PROMIS Short Form–Sleep Disturbance and Sleep-Related Impairment scores at week 8 were controlled for multiplicity, while other outcomes (IGA score of 0 or 1 and change from baseline in percentage of BSA affected by AD and in POEM, DLQI, and CDLQI scores) were not and were at an increased risk of type I error (false-positive results). At least 30% of patients were excluded from each treatment group in the Itch NRS responder analysis (due to the baseline Itch NRS score being < 4 points), which could potentially impact randomization, although the extent and direction of the resulting bias is unclear. There is a risk of potential attrition bias in favour of ruxolitinib cream with respect to continuous secondary end points in the vehicle-controlled period given that study treatment discontinuation in the vehicle cream group was notably higher than in the ruxolitinib cream group. Implicit imputation using mixed models for repeated measures under the missing-at-random assumption was applied to account for missing data, although it is unclear if the missing-at-random assumption holds when the reasons for patient withdrawal (the most common reason for discontinuation) were not documented; no sensitivity analysis was conducted. The end point of change from baseline in CDLQI scores was based on a small sample size in both treatment groups, which could lead to instability of the treatment effect estimates. There is a lack of sample size consideration and control for multiplicity for subgroup analyses, which preclude definitive conclusions about subgroup effects. No firm conclusion can be drawn about the results of the long-term safety period due to the absence of a control group, potential selection bias, and sizable loss to follow-up (approximately 20%) in both trials.
The sponsor’s funding request (aligned with the Health Canada indication) was for the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable. However, the inclusion and exclusion criteria of the pivotal trials did not restrict entry based on prior experience with TCS and TCI treatment. Post hoc subgroup analyses in patients with a recent history of TCS and/or TCI treatment were submitted by the sponsor as supporting evidence. In consultation with the clinical experts, the review team considered that it is unclear if this subgroup population would adequately reflect most patients expected to receive ruxolitinib cream in clinical practice (i.e., patients with AD that is not adequately controlled with TCS and/or TCI treatment or for whom these treatments are not advisable). The clinical experts considered that the baseline patient characteristics in the pivotal studies were in general reflective of the patient population eligible for ruxolitinib cream in clinical practice, although the proportion of patients with mild disease (IGA score of 2), previous TCI treatment, and previous TCS treatment of medium, high, or super-high potency in the trials appear to be lower than would be expected in clinical practice. According to clinical expert input, the duration of the safety follow-up of 52 weeks was inadequate for capturing the long-term safety of ruxolitinib cream (including rare harms) given that AD is a lifelong condition requiring treatment over many years. The absence of head-to-head evidence comparing ruxolitinib cream with relevant comparators (systemic immunosuppressants, biologics, and JAK inhibitors) in patients with moderate AD and the absence of evidence for ruxolitinib cream in combination with other topical therapies represents gaps in evidence in the treatment of AD. The generalizability of the study results to the adolescent patient population in clinical practice could potentially be limited by the small proportion of adolescents enrolled in the trials (approximately 20%). However, a similarly small proportion of adolescent patients was observed in other clinical trials for AD treatments.11,12
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group. Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
severity and extent of AD (proportion of patients experiencing IGA-TS, IGA score of 0 or 1, or EASI-75; change from baseline in percentage of BSA affected by AD)
symptom control (proportion of patients experiencing ≥ 4-point improvement in Itch NRS score from baseline, ≥ 6-point improvement in PROMIS Short Form–Sleep Disturbance score from baseline, ≥ 6-point improvement in PROMIS Short Form–Sleep-Related Impairment score from baseline; change in POEM score from baseline)
HRQoL (change in DLQI and CDLQI scores from baseline)
harms (serious AEs [SAEs]).
The GRADE summary of findings for ruxolitinib 1.5% cream versus vehicle cream for the treatment of patients with AD is presented in Table 2.
Table 2: Summary of Findings for Ruxolitinib 1.5% Cream vs. Vehicle Cream for Patients With Mild to Moderate AD Not Adequately Controlled With Topical Therapies or for Whom Those Therapies Are Not Advisable
Outcome and follow-up | Patients (studies), N | Absolute effect | Certainty | What happens |
|---|---|---|---|---|
Extent and severity of disease | ||||
IGA-TS (i.e., IGA score 0 [clear] or 1 [almost clear] with ≥ 2-point reduction from baseline), proportion of patients experiencing IGA-TS (95% CI) Follow-up: 8 weeks | 725 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Moderatea,b | Ruxolitinib 1.5% cream likely results in a clinically important increase in IGA-TS response compared with placebo. |
IGA score (5-point scale, 0 [clear] to 4 [severe]), proportion of patients experiencing IGA score of 0 (clear) or 1 (almost clear) (95% CI) Follow-up: 52 weeks | 423 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on experiencing an IGA score of 0 or 1 compared with any comparator. |
EASI score (0 [clear] to 72 [very severe]), proportion of patients experiencing EASI-75 (i.e., at least a 75% reduction in EASI score from baseline) (95% CI) Follow-up: 8 weeks | 725 (2 RCTs) | TRuE-AD1 trial
TRuE-AD2 trial
| Moderatea,b | Ruxolitinib 1.5% cream likely results in a clinically important increase in EASI-75 response compared with placebo. |
LSM change from baseline in percentage of BSA affected by AD, % (95% CI) Follow-up: 8 weeks | 652 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowb,e,f | Ruxolitinib 1.5% cream may result in little to no clinically important difference in the percentage of BSA affected by AD compared with placebo. |
Change from baseline in percentage of BSA affected by AD, % (95% CI) Follow-up: 52 weeks | 424 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on the percentage of BSA affected by AD compared with any comparator. |
Symptom control | ||||
Itch NRS score (0 [no itch] to 10 [worst imaginable itch]), proportion of patients with ≥ 4-point improvement from baseline (95% CI) Follow-up: 8 weeks | 465 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowb,g | Ruxolitinib 1.5% cream may result in a clinically important increase in the proportion of patients with a ≥ 4-point improvement in Itch NRS score compared with placebo. |
POEM score (0 [clear] to 28 [very severe]), LSM change from baseline (95% CI) Follow-up: 8 weeks | 635 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowb,e,h | Ruxolitinib 1.5% cream may result in a clinically important improvement in POEM score compared with placebo. |
POEM score (0 [clear] to 28 [very severe]), change from baseline (95% CI) Follow-up: 52 weeks | 412 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on POEM score compared with any comparator. |
PROMIS Short Form–Sleep Disturbance score (8 [no disturbance] to 40 [severe disturbance]), proportion of patients with ≥ 6-point improvement (24-hour recall) from baseline (95% CI) Follow-up: 8 weeks | 675 (2 RCTs) | TRuE-AD1 trial
TRuE-AD2 trial
| Lowa,b,i | Ruxolitinib 1.5% cream may result in a clinically important increase in the proportion of patients with a ≥ 6-point improvement in PROMIS Short Form–Sleep Disturbance score compared with placebo. |
PROMIS Short Form–Sleep-Related Impairment score (8 [no impairment] to 40 [severe impairment]), proportion of patients with ≥ 6-point improvement (24-hour recall) from baseline (95% CI) Follow-up: 8 weeks | 682 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowa,b,k | Ruxolitinib 1.5% cream may result in a clinically important increase in the proportion of patients with a ≥ 6-point improvement in PROMIS Short Form–Sleep-Related Impairment score compared with placebo. |
Health-related quality of life | ||||
DLQI score (0 [best] to 30 [worst]), LSM change from baseline (95% CI) Follow-up: 8 weeks | 555 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowb,e,l | Ruxolitinib 1.5% cream may result in a clinically important improvement in DLQI score compared with placebo. |
DLQI score (0 [best] to 30 [worst]), change from baseline (95% CI) Follow-up: 52 weeks | 362 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on DLQI score compared with any comparator. |
CDLQI score (0 [best] to 30 [worst]), LSM change from baseline (95% CI) Follow-up: 8 weeks | 80 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Lowb,m | Ruxolitinib 1.5% cream may result in little to no clinically important improvement in CDLQI score compared with placebo. |
CDLQI score (0 [best] to 30 [worst]), change from baseline (95% CI) Follow-up: 52 weeks | 50 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on CDLQI score compared with any comparator. |
Harms | ||||
Serious adverse events Follow-up: 8 weeks | 749 (2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Moderaten | Ruxolitinib 1.5% cream likely results in little to no clinically important difference in serious adverse events compared with placebo. |
Serious adverse events Follow-up: 52 weeks | 545 (noncomparative from 2 RCTs) | TRuE-AD1 trial:
TRuE-AD2 trial:
| Very lowc | The evidence is very uncertain about the effect of ruxolitinib 1.5% cream on the frequency of serious adverse events compared with any comparator. |
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; LSM = least squares mean; MID = minimal important difference; NA = not applicable; NR = not reported; NRS = Numeric Rating Scale; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aDid not rate down for study limitations. Notable imbalance in mean number of flares in the past 12 months at baseline between treatment groups in the TRuE-AD1 trial, which could potentially result in bias in favour of ruxolitinib cream as per clinical expert input. Such imbalance was not observed in the identically designed TRuE-AD2 study, which showed results similar to the TRuE-AD1 trial. The imbalance noted in the TRuE-AD1 trial was compatible with chance.
bRated down 1 level for serious indirectness. The clinical experts consulted for this review anticipated that in most patients, ruxolitinib cream would be used when AD is inadequately controlled with topical corticosteroid and/or topical calcineurin inhibitor treatment or when these treatments are inadvisable. It is unclear if the trial population was reflective of the patient population in clinical practice because the inclusion and exclusion criteria of the trial did not restrict entry based on prior experience with topical corticosteroid or topical calcineurin inhibitor treatment. Other considerations included the smaller proportion of patients with mild disease (IGA score of 2) at baseline in trials vs. clinical practice. Baseline IGA score is a potential treatment effect modifier, as per clinical expert input.
cIn the absence of a comparator arm, conclusions about efficacy relative to any comparator cannot be drawn, and certainty of evidence started at “very low” without an opportunity to rate up.
dStatistical testing for this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
eRated down 1 level for serious study limitations. Study treatment discontinuation in the vehicle-controlled period was notably higher in the vehicle cream group than in the ruxolitinib cream group in both trials, which could potentially lead to attrition bias in favour of ruxolitinib cream. It is unclear if the imputation method used was appropriate to account for missing data. Did not rate down for imbalance in mean number of flares at baseline, which was considered by the review team to be compatible with chance.
fThe clinical experts consulted for this review indicated that a 5% to 10% between-group difference could be considered clinically important. Based on the lower limit of the MID estimates (i.e., 5% difference), did not rate down for imprecision; the 95% CI in the TRuE-AD2 trial included the possibility of benefit and no difference; however, this was not considered to be a source of serious imprecision due to its proximity to –5%. If the upper limit of the MID (i.e., 10% difference) was used instead, the review team would not rate down due to imprecision given that both 95% CIs excluded the possibility of benefit. The overall rating of certainty would remain as low.
gRated down 1 level for serious study limitations. A large proportion of patients with a baseline score of less than 4 (at least 30% in each treatment group) were excluded from the analysis, which could potentially impact randomization. The extent and direction of the resulting bias is, however, unclear.
hLiterature-identified MID estimates ranged between 3.4 and 5 points. Did not rate down due to imprecision regardless of whether the lower or upper limit of MID estimates was used. Based on the lower limit of MID estimates (i.e., 3.4-point difference), both 95% CIs included the possibility of benefit. Based on the upper limit of MID estimates (i.e., 5-point difference), although the upper boundary of the 95% CI in the TRuE-AD1 and TRuE-AD2 trials was –5.0 and –4.7, respectively, this was not considered to be a source of serious imprecision due to its proximity to –5.
iRated down 1 level for serious imprecision. Based on clinical expert input, 50 more patients per 1,000 experiencing at least a 6-point improvement (24-hour recall) from baseline in PROMIS Short Form–Sleep Disturbance score could be considered clinically important. The 95% CI in the TRuE-AD2 trial included the possibility of benefit and no difference.
jNo formal statistical testing was conducted due to prior failure in the statistical testing hierarchy (PROMIS Short Form–Sleep Disturbance score). The findings can be considered supportive.
kRated down 1 level for serious imprecision. Based on clinical expert input, 50 more patients per 1,000 experiencing at least a 6-point improvement (24-hour recall) from baseline in PROMIS Short Form–Sleep-Related Impairment score could be considered clinically important. Both 95% CIs included the possibility of benefit and no difference.
lLiterature-identified MID estimates ranged between 3 and 5 points. The review team considered the treatment effect to be clinically important, given that the point estimates in both trials were higher or in close proximity to the lower limit of the MID estimate (i.e., 3-point difference). The upper bound of the CI in the TRuE-AD1 trial indicates no clinically important difference, but the review team recognized that there is some uncertainty about whether the literature-identified MID could be reliably applied to the analysis of the between-group difference in change from baseline and thus did not rate down for imprecision.
mRated down 1 level for serious study limitations. There is a potential that the prognostic balance provided by the randomization is not fully preserved in this analysis because it was conducted in a small subset of patients, with no stratification involved. Literature-identified MID estimates ranged between 6 and 8 points. Based on the lower limit of MID estimates (i.e., 6-point difference), the review team did not rate down further for imprecision even though the lower boundary of the 95% CI was –6.3; this was not considered to be a source of serious imprecision due to its proximity to –6. The rating on imprecision remains the same if the upper limit of MID estimates (i.e., 8-point difference) was used instead.
nRated down 1 level for serious indirectness. The duration of follow-up of 8 weeks is inadequate for capturing potential rare serious adverse events associated with ruxolitinib cream as per clinical expert input.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Long-Term Extension Studies
Both the TRuE-AD1 and TRuE-AD2 trials had a 44-week extension phase assessing the efficacy and safety of ruxolitinib cream in patients who completed the 8-week vehicle-controlled period. Evidence from the extension phase was submitted as part of the pivotal trials and is summarized in the Systematic Review section.
Indirect Comparisons
Description of Study
In the absence of head-to-head evidence comparing ruxolitinib cream to other relevant therapies used in the treatment of mild to moderate AD, the sponsor submitted 1 indirect treatment comparison (ITC) indirectly comparing the treatment effect of ruxolitinib 1.5% cream to dupilumab, abrocitinib, and upadacitinib in patients with moderate AD — defined by the sponsor as having an IGA score of 3, an EASI score of 16 or higher, and a percentage of BSA affected of 10% or higher — via a frequentist network meta-analysis (NMA).16 Study outcomes included the proportion of patients experiencing IGA-TS, EASI-75, and an improvement in Itch NRS score of at least 4. No information on comparative harms was submitted. Eight studies were included in the NMA.
Efficacy Results
IGA Treatment Success
There was insufficient evidence to show a difference in terms of IGA-TS for ruxolitinib cream versus upadacitinib 30 mg and 15 mg and dupilumab 300 mg because the 95% CIs for the odds ratios (ORs) were wide (ruxolitinib 1.5% cream versus upadacitinib 30 mg: OR = 2.10 [95% CI, 0.10 to 44.41]; versus upadacitinib 15 mg: OR = 3.60 [95% CI, 0.17 to 76.04]; versus dupilumab 300 mg: OR = 6.69 [95% CI, 0.32 to 140.26]). Comparisons with abrocitinib 200 mg and 100 mg were not present in the evidence network for IGA-TS.
At Least a 75% Improvement in EASI Score
There was insufficient evidence to show a difference in terms of EASI-75 for ruxolitinib cream versus upadacitinib 30 mg and 15 mg, dupilumab 300 mg, and abrocitinib 200 mg and 100 mg because the 95% CIs for the ORs were wide (ruxolitinib 1.5% cream versus upadacitinib 30 mg: OR = 1.56 [95% CI, 0.22 to 11.03]; versus upadacitinib 15 mg: OR = 2.56 [95% CI, 0.36 to 18.12]; versus dupilumab 300 mg: OR = 3.36 [95% CI, 0.47 to 23.87]; versus abrocitinib 200 mg: OR = 1.52 [95% CI, 0.17 to 13.39]; versus abrocitinib 100 mg: OR = 3.10 [95% CI, 0.35 to 27.32]).
At Least 4-Point Improvement in Itch NRS
There was insufficient evidence to show a difference in terms of at least a 4-point improvement in Itch NRS (Itch NRS-4)for ruxolitinib cream versus upadacitinib 30 mg and 15 mg and dupilumab 300 mg because the 95% CIs for the ORs were wide (upadacitinib 30 mg versus ruxolitinib 1.5% cream: OR = 2.42 [95% CI, 0.46 to 12.79]; upadacitinib 15 mg versus ruxolitinib 1.5% cream: OR = 1.65 [95% CI, 0.31 to 8.74]; dupilumab 300 mg versus ruxolitinib 1.5% cream: OR = 1.19 [95% CI, 0.22 to 6.32]). Comparisons with abrocitinib 200 mg and 100 mg were not present in the evidence network for Itch NRS-4.
Harms Results
Harms outcomes were not assessed.
Critical Appraisal
The validity of the results of the NMA was uncertain because the key assumptions of the analysis, homogeneity and consistency, could not be determined due to insufficient reporting of baseline patient characteristics in the moderate AD–only subgroup and because of the sparse network without a closed loop connecting to ruxolitinib cream. For trials where the information was available, there was evidence of heterogeneity in patient populations (i.e., age group, history of disease control with or eligibility for topical AD treatment) between studies. Only 4 of the 12 included studies reported the baseline patient characteristics of the moderate AD–only subgroup; heterogeneity in disease severity and duration of AD diagnosis was noted between these studies and was not accounted for. These limitations result in uncertainty in the relative treatment effect estimates between ruxolitinib cream and the comparators. There is a risk of missing results in the synthesis given that close to half of the studies initially identified by the SLR were excluded due to the absence of available results for subgroups consisting solely of patients with moderate severity AD. In addition, the absence of comparative evidence between ruxolitinib cream monotherapy and systemic immunosuppressants in patients with moderate AD, and the absence of comparative evidence for ruxolitinib cream as a combination therapy (in combination with other topical treatments), represent gaps in the evidence in the treatment of AD.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
One phase II, open-label study (SCRATCH-AD) at a single Canadian site has been provided as supportive evidence of the short-term clinical benefits of ruxolitinib 1.5% cream in adults with AD to control itch and reduce severity.17 The maximum study duration per participant was approximately 80 days, including the run-in period in which participants had a baseline mean Peak Pruritus NRS (PP-NRS) score of at least 4.0 during days –7 to –1. Other key inclusion criteria of the study were 1% to 20% BSA affected and an IGA score of at least 2 on day 1. Key exclusion criteria were significant flares in the previous 4 weeks, known immune deficiency or immunocompromised condition, use of any systemic corticosteroids or phototherapy, use of JAK inhibitors in the previous 4 weeks, and use of dupilumab in the 26 weeks before the run-in period. Patients received ruxolitinib 1.5% cream, applied topically twice daily (morning and evening, approximately 12 hours between applications), from day 1 until the day before the day 29 visit. The primary end point was change from baseline in PP-NRS score at day 2 (24-hour recall period after the first application), and all data were analyzed descriptively. Concomitant use of emollient was permitted. No other concomitant AD treatments were permitted.
Of 84 individuals who were screened, 35 (41.7%) did not meet the screening criteria. Forty-nine participants applied ruxolitinib 1.5% cream at least once (safety population), and 46 patients completed the run-in period, met all entry criteria, and had a baseline and at least 1 postbaseline PP-NRS or modified PP-NRS (mPP-NRS) assessment (modified intention-to-treat [ITT] population). In the safety population (n = 49), the average age of the participants was 35.6 years (standard deviation [SD] = 14.77 years), and the majority of participants were female (71.4%) and white (85.7%). At baseline, the mean total percentage of BSA affected was 10.11% (SD = 5.34) and the mean baseline EASI score was 7.23 (SD = 3.21). The mean PP-NRS score at baseline was 6.83 (SD = 1.4), and the majority of participants (87.8%) had an IGA score of 3. During the study period, the median cumulative dose was 110.30 g (range, 2.4 g to 335.9 g; n = 48) and the majority of participants (73.5%) used emollients and protectives; participants also used other analgesics and antipyretics (36.7%), nonsteroidal anti-inflammatory drugs and antirheumatic products (28.6%), inhaled adrenergics (22.4%), and vitamins A and D, including combinations of the 2 vitamins (20.4%).
Efficacy Results
On day 2, a mean 3.37-point (SD = 1.85 points) or 50.57% (95% CI, 58.75% to 42.39%) reduction from baseline in PP-NRS (worst itch in the previous 24 hours) score was noted in the modified ITT population. The mean daily PP-NRS score decreased by 4.78 points (SD not reported) by day 7, with a continued decrease (i.e., 5.68-point reduction [SD not reported]) by day 29. In the modified ITT population, increasing proportions of participants experienced IGA-TS at days 8, 15, and 29: 45.5% (95% CI, 30.4% to 61.2%), 71.1% (95% CI, 55.7% to 83.6%), and 77.3% (95% CI, 62.2% to 88.5%), respectively. The mean change from baseline in IGA score at days 8, 15, and 29 was –1.4 (SD = 0.73), –2.0 (SD = 0.87), and –2.2 (SD = 0.90), respectively.
Harms Results
Approximately one-third of participants (n = 15; 30.6%) had at least 1 TEAE. The most frequently reported TEAEs were COVID-19 (6.1%) and back pain, nasopharyngitis, headache, and upper respiratory infection (4.1% each). One participant (2.0%) had an application site reaction (acne), which resolved with no change to study treatment. There were no deaths, SAEs, or TEAEs leading to study treatment interruption or discontinuation.
Critical Appraisal
The main limitation of the SCRATCH-AD study was the single-arm design. The lack of relevant comparator renders it impossible to draw causal conclusions about the comparative efficacy of ruxolitinib 1.5% cream with respect to other treatment options or to vehicle cream. Interpretation of the changes from baseline is complicated as they may be due to the intervention, concomitant treatments, a placebo effect, and/or natural history. Additionally, there is a potential risk of bias due to the open-label design of the study. Patients were aware of the treatment they were receiving and self-reported subjective outcomes, which may have resulted in overestimation of the change from baseline. The analyses were done in fewer than 50 patients (safety and modified ITT populations), which could add uncertainty to the efficacy results. As the SCRATCH-AD study was conducted in a single study site located in Quebec, Canada, its study findings generally have a good generalizability to clinical practice in Canada, except for less-than-ideal representation of Indigenous populations, in which AD is common.
Conclusions
Direct evidence from 2 pivotal double-masked RCTs demonstrated that 8 weeks of ruxolitinib 1.5% cream monotherapy likely results in a clinically important improvement in the severity and extent of AD (IGA-TS and EASI-75) in adults and adolescents with mild to moderate AD compared to placebo. Analyses of the percentage of BSA affected by AD and of symptoms (including itch and impact on sleep) in general favoured ruxolitinib cream. The results for these outcomes were associated with uncertainty due to methodological limitations but were considered supportive of a clinically important benefit with ruxolitinib cream treatment. The results were suggestive of little to no clinically important improvement in HRQoL with ruxolitinib cream treatment in adolescent patients and with a potentially clinically important improvement in HRQoL in adult patients. The benefits of ruxolitinib cream, in general, appeared to be sustained through week 52 in the trials, although the lack of a control group and sizable loss to follow-up beyond week 8 precluded firm conclusions versus any comparator, including vehicle cream. According to clinical expert input, in clinical practice ruxolitinib cream is anticipated to be primarily used in patients with mild to moderate AD that is not adequately controlled with TCS and/or TCI, which aligns with the Health Canada indication. Based on the submitted evidence (including the post hoc subgroup data), it is unclear if the study populations are generalizable to clinical practice given that study inclusion and exclusion criteria did not restrict entry based on prior experience with topical treatments. In addition, there was a smaller proportion of patients with mild AD (IGA score of 2) in the trials than would be expected in clinical practice, which could impact generalizability given that baseline IGA score is a potential treatment effect modifier according to clinical expert input. Indirect evidence from 1 ITC comparing the efficacy of ruxolitinib cream to dupilumab, upadacitinib, and abrocitinib in patients with moderate AD was inconclusive due to important limitations that prevented verification of whether the underlying assumptions of homogeneity and consistency were met, as well as due to imprecision. No definitive conclusion can be drawn from the supportive study (SCRATCH-AD) submitted by the sponsor with respect to the short-term clinical benefits of ruxolitinib cream due to limitations associated with the open-label, single-arm study design. The absence of direct or indirect comparisons of efficacy between ruxolitinib cream monotherapy and systemic immunosuppressants in patients with moderate AD and the absence of comparative evidence for ruxolitinib cream in combination with other topical therapies were gaps in the evidence for the treatment of AD. Ruxolitinib cream appeared to be well tolerated in adults and adolescents through week 52 in the pivotal trials, although a longer duration of follow-up is required to capture long-term safety data, particularly for potential rare harms. No comparative evidence for the harms of ruxolitinib cream versus relevant comparators was submitted.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ruxolitinib 1.5% topical cream in the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
AD causes itching, inflammation, dryness, recurrent eczematous lesions, erythematous papules (red bumps), and thickening and hardening of the skin known as lichenification.1,2 The severity of AD can vary from mild to severe. AD arises from an interplay of environmental factors, epidermal barrier dysfunction, and immune dysregulation.18-20 Patients with AD are also at a higher risk of developing other disorders, such as asthma and/or food allergies.21,22 AD is also associated with other comorbidities such as alopecia areata, cardiovascular diseases, diabetes, and gastrointestinal autoimmune diseases.23 The development of AD in infancy and the subsequent development of allergic rhinitis and asthma later in childhood is known as “atopic march.”22
Itching is the hallmark symptom of AD, which presents as itchy lesions on the skin. The lesions are symmetric with ill-defined margins and include patches or plaques, lichenification (a thickening and hardening of the skin that can also occur with repeated scratching),18 and erosion in the skin.24,25 AD rashes can occur on any part of the body and can weep fluid and bleed when scratched, which compromises the skin integrity as a barrier and can make the skin vulnerable to infection as external pathogens can more easily enter through the skin. Moisture loss and pathogen infiltration can inflame the skin further, perpetuating the need to scratch. Scratching then further damages the skin barrier, exacerbates dryness, and increases proinflammatory mediator release.18
AD is a chronic condition and has a waxing and waning clinical course. Some patients with AD may experience periods of remission (less disease or periods of clear skin) and may experience intermittent flares (periods of exacerbated disease), which are acute flares with clear skin in between flares. Some patients experience disease flares where the symptoms of AD (i.e., itch and pain) can be intolerable despite their best efforts to manage their disease and avoid triggers and irritants.26 Some patients may be in a constant state of flare, in which patients report some level of baseline AD at all times with additional varying periods and degrees of worsening.26 These patients never experience clear skin and always have a baseline level of disease activity. Some patients may experience a “flare crisis,” which is a significant flare that is not manageable and requires immediate medical intervention — this is more common in moderate or severe AD. A flare crisis not only causes a physical impact, including painful and extensive rashes, open wounds, and infection, but also takes a significant emotional toll on patients.26
The intense itch associated with AD has been shown to be related to poor sleep quality. AD is also associated with higher odds of fatigue, regular daytime sleepiness, and insomnia, which predict poorer overall health status and a greater number of sick days and doctor visits.27 Sleep disruption in children with AD can further result in several detrimental outcomes, including impaired neurocognitive function, higher rates of behavioural problems, changes in mood, attention-deficit/hyperactivity disorder, emotional and conduct problems, and short stature.28,29 The prevalence of anxiety and depression in patients with AD is higher than in the general population.30 The mental comorbidities associated with AD can significantly affect quality of life.5,19,23,30,31 Caretakers of children with AD similarly experience a mental health burden.32 An association between AD and suicidal ideation has also been identified.33 More than likely, the combination of itch, psychological stress, social isolation, depression, and anxiety results in a cycle for patients with AD that may result in suicidal ideation, suicide attempts, and even completed suicide.33
Indigenous Peoples are an underserved population in Canada who experience a high burden of AD.34 The atopic triad, starting with AD, is the most common chronic health condition in Indigenous children and youth. There is a high burden of poorly controlled, functionally debilitating AD, particularly in the Indigenous pediatric population, which is often exacerbated by skin infections. Factors such as compromised antimicrobial immune response, crowded housing, and limited access to care can amplify the infection risk in this population.34
The global prevalence of AD in 2022 was 223 million, and it has increased over the past 30 years.23,24,35 In Canada, the lifetime prevalence of AD is between 10% and 20%.7,21,36 According to the ESC, it is estimated that 11% of children and 7% of adults live with AD.26 However, estimates of prevalence vary for both the adult and pediatric populations. Studies have indicated that the prevalence of AD among adults living in Canada varies from 1.8% (period prevalence [2005 to 2015] in Ontario) to 3.5% (12-month point prevalence based on an international survey including Canada).3,4 Similarly, the prevalence of AD among children living in Canada varies from 9.9% (period prevalence [2005 to 2015] in Ontario) up to 25%.3,37 Silverberg et al. also report the prevalence for adolescents (aged 12 to < 18 years) living in Canada as being 15.8%.5 More specifically, according to a review conducted by the Institut national d’excellence en santé et en services sociaux (INESSS), the prevalence of AD among adolescents aged 12 to 18 years is estimated to be 9.4%.6 Overall, the majority of patients with AD have mild to moderate disease; however, the proportions vary by age group, with children younger than 18 years typically having milder disease. For example, a cross-sectional, international, web-based survey found that the proportion of adolescents aged 12 years to younger than 18 years (n = 106) in Canada with self-reported mild, moderate, and severe AD was 59.8%, 32.7%, and 7.5%, respectively.38 In comparison, using a cross-sectional web-based survey it was estimated that, based on the POEM score, the proportion of adults living in Canada with mild, moderate, and severe AD (n = 215) was 29%, 50%, and 21%, respectively.39 The true prevalence of AD among Indigenous Peoples in Canada may be underestimated for reasons such as poor health care and limited access to specialists and due to underrepresentation in research.34
Due to the heterogeneity of the disease, no specific laboratory biomarker or gold standard test exists to diagnose AD.40 AD is currently diagnosed by examining the skin for the distribution of lesions and associated clinical signs.25 In clinical practice, diagnosis of AD may be based on essential, important, and associated features (diagnostic standards of Hanifin and Rajika; 3 major features including itch and 3 minor features).25 Diagnosis also depends on excluding conditions such as seborrheic dermatitis, contact dermatitis (irritant or allergic), or psoriasis.25 The clinical experts consulted for this review agreed that dermatologists do not routinely apply Hanifin and Rajka criteria directly to diagnose AD in the Canadian clinical setting. They noted that dermatologists in Canada do consider the Hanifin and Rajka criteria when establishing the diagnosis of AD, which is made based on clinical history and physical examination. Occasionally, cases must be differentiated from other skin diseases that mimic AD, including ichthyoses, other dermatitis, photodermatoses, and cutaneous T-cell lymphoma. There are no specific diagnostic tests to define AD severity, so clinicians rely on clinical measures such as IGA, patient-reported symptoms, and quality of life outcomes.41
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
Patients with AD have indicated itch reduction as their top treatment goal.39,42 Other objectives of therapy for the treatment of AD are to reduce skin inflammation, restore skin barrier function, and improve quality of life.43,44 According to the clinical experts consulted for this review, treatment goals include achieving a rapid and significant reduction in itch and inflammation, improving sleep quality, and not only settling the current flare but also preventing or reducing the frequency and severity of future flares. The clinical experts also noted that treatments must be safe, convenient to use, well tolerated, accessible, and associated with improved HRQoL. Treatments for AD include general skin care such as moisturizing; topical prescription therapies such as TCSs, TCIs, and crisaborole; phototherapy; and systemic therapies.
Initial AD management involves patient education, appropriate skin care practices, and avoidance of symptom triggers and irritants. Appropriate daily skin care practices, including the use of moisturizers and emollients, and avoidance of symptom triggers and skin irritants are key principles in AD management.7,19,26,36,43 According to the clinical expert input, patients are advised to avoid overbathing and the use of strongly scented soaps and skin care products. Daily use of emollients is recommended as maintenance therapy for patients with AD to manage dry skin, prevent moisture loss, and restore the epidermal barrier to prevent disease exacerbations.7,19,26,36,43 The clinical experts consulted for this review agreed that daily or twice-daily application of lubricants is advised for patients with AD.
As per recent Canadian consensus documents, it is recommended that patients whose AD remains uncontrolled with appropriate skin care practices and through avoidance of symptom triggers and irritants be treated with a topical treatment.19,36,43,45 Patients will typically be prescribed an initial topical treatment such as a TCS.19,36 The clinical experts consulted for this review mentioned that crisaborole 2% ointment, a phosphodiesterase type 4 inhibitor, is an alternative initial therapy but that it is used much less commonly in Canada than other parts of the world. There are many forms of TCS available in Canada, including lotions, creams, ointments, or gels, which come in potencies ranging from low to ultra-high.36 When determining the appropriate potency and formulation of TCS, providers will consider patient age, the body area to be treated, and lesion characteristics such as lichenification.19,36,43 Providers aim to prescribe the least potent preparation, particularly to sensitive areas, to avoid the side effects of TCS including skin thinning, atrophy, acne, striae, and telangiectasia, which may be more common in sensitive areas.19,36 Systemic side effects may also occur, particularly with higher potency TCS.
TCIs are also recommended in the treatment of AD, specifically for patients who have experienced response to treatment with TCS, those with AD in sensitive areas, or those who require maintenance treatment.19,36,43,45 TCIs are a nonsteroidal topical option and may be prescribed after TCS if patients have not experienced disease control or have experienced frequent flares or for patients for whom steroid atrophy is a concern.19,36,43 TCIs may also be used as adjunctive or maintenance therapy alongside TCS.19,36,43 The clinical experts consulted for this review added that TCS and TCI are often used concomitantly. According to the clinical experts, TCIs and crisaborole have the advantage that they can be safely applied to eyelids and to periocular skin and creases whereas medium-potency or high-potency TCS cannot; however, the use of these drugs is limited by their cost and their tendency to induce a burning sensation. Two TCIs are currently approved in Canada: tacrolimus, which is indicated for patients with moderate to severe AD, and pimecrolimus, which is indicated for patients with mild to moderate AD. Both are indicated to be used as second-line therapy for short and intermittent long-term treatment of patients with AD who are not immunocompromised. The clinical experts also agreed that tacrolimus 0.1% ointment is on par with low to mid potency corticosteroids in terms of efficacy and that pimecrolimus is significantly less effective than tacrolimus. Tacrolimus is also indicated for maintenance therapy to prevent flares and prolong flare-free intervals in patients with moderate to severe AD who are experiencing a high frequency of flares (i.e., occurring 5 or more times per year) and who have had an initial response to tacrolimus twice daily within up to 6 weeks of treatment.46 Patient adherence to these treatments may be impacted by common AEs, such as skin burning and irritation over the applied area, and by the ointment formulation, which is not a preferred formulation compared to a cream, particularly on the face.36,43 If skin fails to improve with the current topical therapy, then an additional class of topical treatment may be added to the regimen to target a specific area.19,36,43,45 For example, topical antibiotics may be applied in combination with TCS when there are AD lesions with signs of infection, such as crusting, oozing, and pus.19,36,43 According to the clinical experts consulted for this review, topical antibiotics — usually fusidic acid and mupirocin — are used in instances of chronic impetiginization.
The 2018 Canadian practical guide for the treatment of AD includes TCS or TCI as initial treatment options.7 The practice guideline recommends that patients who do not experience disease control with multiple topical therapies be treated with phototherapy (if available) or with systemic treatments such as immunosuppressants (e.g.., methotrexate, cyclosporine), biologics (e.g., dupilumab, tralokinumab) and/or oral JAK inhibitors (e.g., upadacitinib, abrocitinib).8 The clinical experts consulted for this review stated that if a patient experiences treatment failure on topical therapy and phototherapy combined, discussions on treatment turn to systemic therapies.7
UV phototherapy may be recommended for patients who do not experience disease control with topical prescription therapies or for whom topical treatments are inadvisable.19,36,43 Typically, patients require 2 to 5 UV phototherapy treatments per week for 6 to 12 weeks, which can involve time away from work or school. While the efficacy of phototherapy has been demonstrated, it is limited by availability, patients’ ability to attend in-person appointments multiple times per week, and patient willingness and adherence.19,36,43 Phototherapy is not widely available in Canada and may be more easily accessible to patients located in urban centres than to those in rural areas.26 Home-based UV phototherapy may also be available and may be at least as effective as phototherapy provided in an outpatient clinic. However, home-based phototherapy is underused, in part due to issues associated with access and cost.47,48 The clinical experts consulted for this review agreed that if a patient’s response is judged to be inadequate with topical therapies, they may be offered UVB and/or UVA phototherapy, assuming they have access to a phototherapy clinic and no prior history with phototherapy failure.
In Canada, systemic immunosuppressant agents are used off-label in the treatment of moderate to severe AD and include, in the order of frequency of use, methotrexate, cyclosporine, mycophenolate mofetil, and azathioprine.36,43,49 These treatments are reserved for patients who do not experience control with topical prescription treatments or for whom these therapies are inadvisable.36 Due to potential side effects, it is essential to closely monitor patients taking these therapies for AEs, such as myelosuppression with azathioprine and decline in kidney and liver function with cyclosporine.8,36,43,50 Treatment with oral immunosuppressants therefore requires extensive baseline and follow-up monitoring, which may include hepatitis B and C testing, renal and liver function tests, and/or blood pressure and lipid monitoring, depending on the therapeutic option prescribed to the patient.43 Given these AEs and the risk of relapse following treatment discontinuation, oral immunosuppressants (e.g., cyclosporine) are typically reserved for short-term rescue or salvage therapy and are not typically used for more than 1 year.8,43 The evidence supporting the efficacy and safety of the use of immunosuppressants in AD is generally poor quality.8
Systemic corticosteroids may also be employed as a rescue therapy for patients experiencing acute severe flares and as a bridge to other therapies.8,36 Recent consensus statements highlight that evidence on the safety and efficacy of systemic corticosteroids is limited and that they should only be used for exceptional short-term cases.8,43 They are also associated with poor tolerance, a poor safety profile, and a risk of severe withdrawal rebound.8,43 Also, according to the clinical experts consulted, systemic antibiotics, chosen by the result of bacterial culture and sensitivity, are prescribed in cases of significant secondary infection.
Two biologics, dupilumab and tralokinumab, have received approval from Health Canada for the treatment of moderate to severe AD.51,52 Dupilumab is subcutaneously administered to adolescent or adult patients and may be used concomitantly with TCS.19,43 Patients taking dupilumab may be at increased risk of conjunctivitis.26,43 Tralokinumab is undergoing reimbursement review by CDA-AMC and it is not currently reimbursed by the jurisdictions in Canada for the treatment of AD.
Oral JAK inhibitors are a relatively new class of systemic therapy and include upadacitinib and abrocitinib. JAK inhibitors target the JAK-STAT pathway and are indicated in the treatment of moderate to severe AD.19,43 Treatment with oral JAK inhibitors requires additional treatment monitoring (i.e., full blood counts, differential liver function tests).53,54
Drug Under Review
The key characteristics of ruxolitinib 1.5% cream are summarized in Table 3, along with those of other treatments available for the treatment of AD in patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies or when those therapies are not advisable.
Ruxolitinib 1.5% cream is a topical treatment for AD, applied as a thin layer twice daily to affected skin areas up to a maximum of 20% BSA on each application.55 Patients are to discontinue treatment when signs and symptoms of AD (e.g., itch, rash, redness) resolve.55 If the signs and symptoms do not improve within 8 weeks, the health care provider should reevaluate treatment. The use of ruxolitinib cream in combination with other JAK inhibitors, biological immunomodulators, or potent immunosuppressants has not been studied and is not recommended, according to the Health Canada product monograph.
Ruxolitinib 1.5% cream acts by selectively inhibiting JAK1 and JAK2, providing both anti-inflammatory and antipruritic effects.37,40,56-58 Given its part in mediating inflammation and transmitting itch signals, the JAK-STAT pathway plays a key role in the pathogenesis of AD.37,40,56,59-62 JAK1 and JAK2 mediate the signalling of a number of cytokines and chemokines that promote inflammation and itch at the intracellular level (i.e., Th2 cytokines interleukin [IL]-4, IL-13, and IL-31; the Th22 cytokine IL-22; and thymic stromal lymphopoietin).63
The Health Canada indication for ruxolitinib 1.5% cream is for the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable. The Notice of Compliance was granted on October 11, 2024. The sponsor has requested the reimbursement of ruxolitinib cream per the indication. Ruxolitinib 1.5% cream has been approved by the FDA for topical short-term and noncontinuous chronic treatment of mild to moderate AD in patients 12 years of age and older who are not immunocompromised and whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Ruxolitinib is also approved by Health Canada for the topical treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older.
Table 3: Key Characteristics of Ruxolitinib Cream and Comparator Treatments
Characteristic | Mechanism of action | Indicationa | Route of administration and recommended dose | Serious AEs or safety issues |
|---|---|---|---|---|
Topical treatments | ||||
Ruxolitinib cream55 | Inhibits JAK1 and JAK2, blocking signalling of cytokines and chemokines. | For the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (topical corticosteroids, topical calcineurin inhibitors) or when those therapies are not advisable.b | Apply as a thin layer to the affected skin areas up to a maximum of 20% BSA on each application. Discontinue when signs and symptoms of AD (e.g., itch, rash, redness) resolve. If signs and symptoms do not improve within 8 weeks, consider reevaluation by the health care provider. | Serious infections, malignancies, MACE, and thrombosis. |
Exact mechanism of action in AD is unknown. Inhibits calcineurin, T-cell activation, and production and release of cytokines or lymphokines. | Tacrolimus: Second-line therapy for short- and long-term intermittent treatment of moderate to severe AD in patients who are not immunocompromised, and in whom the use of conventional therapies are deemed inadvisable because of potential risks, or who are not adequately responsive to or intolerant of conventional therapies. Pimecrolimus: Second-line therapy for short-term and intermittent long-term therapy of mild to moderate AD in patients who are not immunocompromised at 2 years of age and older, in whom the use of alternative, conventional therapies is deemed inadvisable because of potential risks, or in the treatment of patients who are not adequately responsive to or intolerant of alternative, conventional therapies. | Treatment Apply to affected area (including the face, neck, and eyelids for tacrolimus and the head, neck, and intertriginous areas for pimecrolimus) twice daily. Maintenance Tacrolimus — Patients who have a high frequency of flares (≥ 5 times per year) and are experiencing response within up to 6 weeks of acute treatment with tacrolimus ointment twice daily are suitable for maintenance treatment, applied once daily twice a week. There should be 2 to 3 days between applications. If flares recur, twice-daily treatment should be reinitiated. Pimecrolimus — Should be used for short or long intermittent periods of treatment. Therapy should be stopped upon clearance of the signs and symptoms of AD (e.g., pruritus, inflammation, erythema). If no improvement occurs after 3 weeks of treatment, or in cases of disease exacerbation, therapy should be discontinued. | Long-term safety of TCI has not been established. Although a causal relationship has not been established, rare cases of skin malignancy and lymphoma have been reported in patients treated with TCI. | |
Systemic treatments | ||||
Immunosuppressants — AZA, MMF, CYC, MTX, oral corticosteroids66,67 | AZA: antimetabolite, reduces proliferation of lymphocytes. MMF: inhibits purine synthesis, reduces lymphocyte proliferation, reduces antibody formation by B lymphocytes. CYC: inhibits IL-2 and T-cell activation. MTX: immune suppressive. Prednisone: exact mechanism not clearly understood; predominantly glucocorticoid and some mineralocorticoid properties. | AZA: off-label for AD MMF: off-label for AD CYC: off-label for AD MTX: off-label for AD Prednisone: AD | Approved or recommended dose for treatment of AD is not available for AZA, MMF, CYC, or MTX. AZA: oral MMF: oral and IV CYC: oral MTX: oral, SC Prednisone: Children aged > 6 years and adults: 5 mg to 60 mg orally per day as a single dose or in divided doses. | AZA: carcinogenic, leukopenia, thrombocytopenia, infection, hepatotoxicity. MMF: infection, lymphoma. CYC: infection, malignancy, nephrotoxicity, hypertension, hepatotoxicity, neurotoxicity. MTX: malignancy, serious rash, bone marrow suppression, vomiting, diarrhea, hepatotoxicity. Prednisone: infections, Cushingoid, cataract, glaucoma, hypertension, gastrointestinal and central nervous system AEs. |
Targeted biologics — dupilumab52 | A recombinant human IgG4 monoclonal antibody that inhibits both IL-4 and IL-13 signalling. | For the treatment of patients aged 6 months and older with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Dupilumab can be used with or without topical corticosteroids. | Adults: Initial dose of 600 mg,c followed by 300 mg every other week Children and adolescents (6 to 17 years):
Pediatric patients (6 months to 5 years):
| Conjunctivitis, eosinophilia, injection site reaction. |
Modulate cytokine signalling pathway at the point of JAKs (upadacitinib) or JAK1 (abrocitinib). | Upadacitinib: For the treatment of adults and adolescents 12 years of age and older with refractory moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., steroid or biologic) or when use of those therapies is inadvisable. Upadacitinib can be used with or without topical corticosteroids. Abrocitinib: For the treatment of patients 12 years and older with refractory moderate to severe AD, including the relief of pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advisable. Abrocitinib can be used with or without medicated topical therapies for AD. | Upadacitinib: 15 mg to 30 mg orally once daily Abrocitinib: 100 mg to 200 mg orally once daily | Serious warnings and precautions (“black box warning”): serious infections, malignancy, thrombosis, MACE. | |
AD = atopic dermatitis; AE = adverse event; AZA = azathioprine; BSA = body surface area; CYC = cyclosporine; IL = interleukin; JAK = Janus kinase; MACE = major adverse cardiovascular events; MMF = mycophenolate mofetil; MTX = methotrexate; SC = subcutaneous; TCI = topical calcineurin inhibitor.
aHealth Canada–approved indication.
bThe use of ruxolitinib cream in combination with other JAK inhibitors, biological immunomodulators, or potent immunosuppressants has not been studied and is not recommended.
cGiven as two 300 mg injections.
dGiven as two 200 mg injections.
Sources: Sponsor’s summary of clinical evidence;15 Opzelura FDA prescribing information;70 Health Canada product monographs for Apo-prednisone, azathioprine, Mar-mycophenolic acid, Sandoz cyclosporine,67 Apo-methotrexate,66 Protopic,65 Elidel,64 Dupixent,52 Rinvoq,68 Cibinqo.69
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) have been included in this section of this report. Input was received at the time of the CDA-AMC call for input based on the initial reimbursement request (i.e., for the topical treatment of AD in patients 12 years of age and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable), which predates the reimbursement request update provided by the sponsor (i.e., for the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies [TCS, TCI] or when those therapies are not advisable).
Two patient groups submitted input. The ESC is a registered Canadian charity with a mission of support, education, awareness, and research for people living with eczema. The ESC gathered information from more than 3,000 patients in Canada living with AD and from their caregivers and/or family members via survey questionnaires and one-on-one interviews. Another patient group input submission was jointly prepared by the CSPA and Ezema Quebec. The CSPA is a national nonprofit organization that engages in collaboration, advocacy, and education for people affected by skin, hair, and nail conditions. Eczema Quebec is a nonprofit organization dedicated to providing support, resources, and education to individuals and families affected by eczema in Quebec. The joint input was based on information gathered between February and October 2023 from various sources, including a literature review, patients, and The Skin I’m In report, and through collaboration with an academic institution. Some patients (number not specified) surveyed by Eczema Quebec indicated that they had experience with ruxolitinib cream treatment.
Both submissions highlighted that the signs and symptoms of AD, such as dry, itchy, inflamed skin that cracks, oozes, and bleeds, as well as the thickening of skin, affect many aspects of patients’ lives, including physical, social, emotional, and professional aspects. Patients said itches can be extremely uncomfortable and painful and can require frequent medical visits, specialized treatments, and ongoing care. The joint input by CSPA and Eczema Quebec pointed out that AD is associated with other conditions, such as asthma, seasonal allergies, environmental allergies, food intolerances, sleep disorders, anxiety, and depression. The input submissions emphasized that caregivers and/or family members also share a significant burden of disease. The negative impact of AD on patients and their caregivers and/or families is amplified when AD is not well controlled despite optimization of the treatment regimen and when cycling through or switching to different therapies. According to the input by the ESC, uncontrolled AD or flares can lead to emergency department visits and hospitalizations. The input also highlighted that because AD can occur at a young age, it can cause significant impact on young people’s performance at school, their social life, and their mental health. In both input submissions, patients expressed a need for new treatments that are safe, improve the symptoms of AD, reduce flares, and improve quality of life (e.g., better sleep quality, less psychological burden, improved ability to carry out daily activities, and improved ability to establish and maintain intimate relationships), as well as reduce or eliminate potential complications and secondary infections associated with AD. Other key outcomes reported to be important to patients include fast and durable relief and reduced skin thickening; ease of medication use and affordability are also considered important. In addition, patients value treatments that do not require injections. Patients expressed a need for treatments suitable for application not only on the body but also on the face and sensitive areas of the body, for a simplified regimen. The patient groups acknowledged that AD is a heterogeneous disease and requires a variety of treatments to fill gaps in therapeutic needs.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of AD.
Unmet Needs
The clinical experts noted several needs that are not being met by currently available nonsteroidal topical treatments for AD. First, not all patients respond to available nonsteroidal topical treatments, according to the clinical experts. Second, the clinical experts noted that tacrolimus 0.1% ointment, the most effective TCI to date, is about as effective as moderate-potency steroids and is generally inadequate for areas of the body with thicker skin, such as the palms and soles, and for areas with thick, lichenified AD; a more potent nonsteroidal topical treatment is needed. Third, the clinical experts noted that some patients develop an application site reaction (e.g., burning and stinging) that results in reduced adherence to therapy. Fourth, the clinical experts noted that the most effective currently available nonsteroidal topical therapy (tacrolimus) is available only in ointment formulation, which could influence patient adherence. According to the clinical experts, cream formulation is preferred by many patients, particularly for application to face and intertriginous areas, given that cream in general has less visible residue after application and is easier to apply than ointment. Fifth, the high costs associated with available nonsteroidal topical therapies could be a barrier to access to treatments for some patients, according to the clinical experts.
Place in Therapy
One clinical expert anticipated that, in the majority of patients, ruxolitinib cream will likely be initiated following inadequate disease response to TCS and/or TCI and will continue to be used as an adjunctive treatment in patients who have moved on to phototherapy and/or systemic therapy. This clinical expert noted that ruxolitinib would primarily serve as a second-line topical treatment following the failure of TCS and/or TCI because of long-established treatment protocols favouring TCS and TCI as well as the anticipated access challenges with ruxolitinib cream due to the higher drug cost relative to currently reimbursed topical treatments. The other clinical expert noted that while TCS is expected to remain as the first-line treatment for most patients, ruxolitinib cream could sometimes be used as a first-line topical treatment (before trialling TCS and/or TCI) as well. This clinical expert noted that TCIs are associated with application site reactions and moderate efficacy; provided that ruxolitinib cream is similarly or more effective and has fewer application site reactions than TCIs, the clinical expert thought it would be reasonable to use ruxolitinib cream ahead of TCIs, in particular for the face and groin, for which TCS treatment is inappropriate. The clinical experts agreed that it may be reasonable to consider ruxolitinib cream treatment in patients who are reluctant to use TCS and/or TCI due to the perceived risk of AEs.
The clinical experts expected ruxolitinib cream to be used as either monotherapy, primarily for facial and intertriginous skin, or in combination with other topical therapies (applied to different affected areas). The clinical experts noted that when used concurrently with other topical therapies, ruxolitinib cream could be applied to the same or different anatomic locations; most patients are expected to use 1 treatment at a time in a given location and apply different topicals to different parts of the body. The clinical experts did not think that ruxolitinib cream represents a new line of therapy as proposed by the sponsor. The clinical experts noted that not all patients who experienced inadequate response to, or were ineligible for TCS and/or TCI, are expected to receive ruxolitinib cream before trying phototherapy and systemic treatments — only some would. Neither clinical expert considered ruxolitinib as a step-up therapy; rather, in their opinion, ruxolitinib cream would add 1 more nonsteroidal option to the topical treatment arsenal. The clinical experts agreed that ruxolitinib cream is not expected to cause a shift in the current treatment paradigm.
Patient Population
One clinical expert noted that patients with AD who have facial or intertriginous involvement, experience inadequate response to or intolerable AEs from TCS and/or TCI treatments, and have 10% or less BSA affected by AD are the most suited for treatment with ruxolitinib cream as monotherapy. The clinical expert remarked that in the clinical trials of ruxolitinib cream, the study populations had a mean percentage BSA affected of approximately 10%. Due to potential significant systemic absorption leading to harms and high anticipated treatment cost, the clinical expert felt that it would be reasonable to use ruxolitinib cream as monotherapy in patients with 10% or less BSA affected. In the second clinical expert’s opinion, patients could receive ruxolitinib treatment regardless of response to or eligibility for TCS and/or TCI treatments. The second clinical expert also felt that patients with more than 10% BSA affected by AD would be candidates for ruxolitinib cream. In these patients, the clinical expert noted, ruxolitinib cream would be used in combination with other treatments and should not be applied to more than 10% to 20% BSA. Both clinical experts noted that patients who are averse to steroids are potential candidates for ruxolitinib cream treatment.
The clinical experts noted that there is no universal definition for adequate (or inadequate) response to TCS and TCI treatment and that treatment response is typically determined by clinical judgment on a case-by-case basis and by patient satisfaction, although they felt it might be reasonable to consider the initiation of ruxolitinib cream treatment in patients whose skin fails to improve after 4 to 8 weeks of conventional topical therapy, including low-potency, midpotency, or high-potency TCS, TCI, or crisaborole (which has very limited use in Canada), as suggested in the sponsor’s submission. From a clinical perspective, the clinical experts felt that ruxolitinib cream treatment might also be considered before the failure of existing topical treatments (i.e., in the first-line setting). One clinical expert noted that because AD is often a chronic disease with periods of flares, even in patients who experience otherwise good response to topical treatment, ruxolitinib cream could be considered as a treatment option for patients who generally do well with TCS or TCI but who are interested in trying a different treatment. The other clinical expert acknowledged patients’ need for additional treatment options but was concerned about the additional financial burden for public and private payers from the use of ruxolitinib cream in such patients, given the cost of this treatment.
The clinical experts noted that determining for which patients the use of TCS and TCI is advisable primarily depends on reaction to previous use (i.e., their use is inadvisable in cases of intolerance). According to the clinical experts, there are very few contraindications to TCS and TCI (e.g., the use of all TCS, including hydrocortisone, on the eyelids, the long-term use of corticosteroids more potent than hydrocortisone to the face and intertriginous skin). In their experience, almost all patients who have not previously received topical treatment are eligible for TCS and TCI treatments.
Patients with a baseline IGA score of 4 (severe AD) were not included in the pivotal trials of ruxolitinib cream. The clinical experts commented that these patients could potentially benefit from ruxolitinib cream either as monotherapy, in patients with localized severe areas of AD, or in combination with other topical treatments (applied to different affected areas).
Assessing Response to Treatment
The clinical experts noted that most clinicians would assess response to treatment based on improvement in lesional inflammation (reduced area, reduced thickness, reduced redness, reduced lichenification, reduced oozing), patient-reported improvement in pruritus (supported by the observation of reduced excoriation and reduced lichenification), and patient’s global assessment of treatment success. The clinical experts noted that BSA affected by AD and Peak Pruritus NRS are measured by some clinicians in clinical practice. IGA-TS is not assessed in clinical practice, and DLQI and EASI scores are, in general, only generated for reimbursement purposes, according to the clinical experts.
The definition of a clinically meaningful response to treatment is expected to vary widely among physicians and also across patient populations, according to the clinical experts. In the clinical experts’ opinion, the EASI-75 benchmark currently applied to the renewal of a systemic drug for AD is not applicable to the assessment of a treatment response to ruxolitinib cream, and treatment response to ruxolitinib cream should be based on clinical judgment. The clinical experts explained that ruxolitinib cream may be used in combination with other topical treatments; therefore, the patient’s total EASI score may not be attributable to ruxolitinib cream treatment. One clinical expert noted that there are concerns about the suitability of EASI in assessing patients with mild AD.
The clinical experts noted that the follow-up intervals tend to be dictated by the severity of AD; many patients with mild AD would be returned to the referring physician for follow-up after the consultation visit, while patients with more severe disease would continue to be seen by a specialist at monthly intervals or even more frequently. One clinical expert commented that the initial assessment time point in the pivotal trials of ruxolitinib cream at 8 weeks would correspond quite well to the first follow-up visit for many patients. The other clinical expert commented that in their clinical practice, the 8-week follow-up interval is applicable to patients with relatively severe disease treated with topical therapy and that for most patients treated with topical therapy there may be no specialist follow-up or a lower frequency of follow-up (3 to 6 months).
Discontinuing Treatment
The clinical experts noted that treatment discontinuation could be considered in patients who experience an inadequate response (as judged by the clinician or patient) or intolerable AEs (e.g., application site reaction) to ruxolitinib cream treatment.
Prescribing Considerations
The clinical experts noted that ruxolitinib cream could be prescribed by any health care provider with experience in diagnosing, treating, and monitoring patients with AD. This would include, principally, general dermatologists, pediatricians, pediatric dermatologists, allergists, family practitioners, and nurse practitioners.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group input(s) have been included in the Perspectives of Patients, Clinicians, Drug Programs section of this report. Input was received at the time of the CDA-AMC call for input based on the initial reimbursement request (i.e., for the topical treatment of AD in patients 12 years of age and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable), which predates the reimbursement request update provided by the sponsor (i.e., for the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies [TCS, TCI] or when those therapies are not advisable).
The Canadian Dermatology Association, represented by 3 clinicians, and the Atlantic Dermatology Specialist Group, represented by 11 clinicians, made 2 separate input submissions. Consistent with the input from the clinical experts consulted by CDA-AMC, the clinician groups indicated that some patients receiving existing treatments experience uncontrolled disease, side effects, or poor tolerability of ointment formulation or have poor treatment adherence due to the need to apply different topical products to different body locations. The clinician groups agreed that there is an unmet need for a new topical therapy that is effective, better tolerated, and in cream formulation. They also noted that an effective topical therapy is needed to prevent the need to escalate to phototherapy or systemic treatments, which are associated with limitations (e.g., limited efficacy, accessibility, and drug coverage; side effects; monitoring requirements; high treatment cost). Both clinician groups agreed that the main treatment goals include reducing itch and inflammation (short term and long term), achieving skin clearance, minimizing tolerability and safety issues, and improving quality of life (e.g., sleep, anxiety, depression). The clinical groups and clinical experts agreed that an ideal topical treatment should be in a cosmetically appropriate base, convenient to use, and accessible.
In general, the 2 clinician groups and the clinical experts consulted by CDA-AMC agreed that ruxolitinib cream could be used for patients with AD whose disease is not adequately controlled with topical prescription therapies (TCS or TCI) or for whom those therapies are not advisable. The Atlantic Dermatology Specialist Group and the clinical experts noted that there is potential for ruxolitinib treatment to be used as a first-line treatment in some patients. The clinician groups noted that this includes patients with mild to moderate AD, patients with up to 20% BSA affected, patients with severe localized AD, and patients with moderate to severe AD (EASI score > 16 and ≥ 10% BSA affected), as well as patients who cannot access or have contraindications to phototherapy or systemic therapies. Consistent with the input from the clinical experts consulted by CDA-AMC, the clinician groups noted that ruxolitinib cream could be used as either monotherapy or adjunct therapy (to systemic therapy if eligible and tolerated) for continual or as-needed use. While the clinician groups felt that ruxolitinib cream could be used on any body sites in patients with up to 20% BSA affected, 1 clinical expert felt that use of ruxolitinib cream should be mainly limited to facial and intertriginous skin only and applied to no more than 10% BSA due to potential systemic absorption and high treatment cost.
Both clinician groups and the clinical experts noted that responses to treatment are assessed by signs and symptoms (e.g., itch and inflammation), BSA affected, extent of involvement of special sites (hands, feet, face, skin folds, or perineal area), and patient-reported outcomes (e.g., HRQoL, functional impact). The clinician groups and 1 of the clinical experts agreed that after a trial period of 8 weeks, if there is an inadequate improvement in the signs and symptoms of disease, recurrent flares, worsening of disease, or intolerance or side effects, then discontinuation would be considered. The second clinical expert noted that in their clinical practice, a lower frequency of follow-up of 3 to 6 months would apply to most patients receiving topical treatments, except patients with more severe disease for whom an 8-week assessment interval would be appropriate. All clinician groups and the clinical experts agreed that generalist or primary care physicians as well as specialists (e.g., dermatologists, allergy and immunology specialists, and pediatricians) who are comfortable with the diagnosis and management of AD should prescribe, treat, and monitor patients who receive ruxolitinib cream.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Double-masked, randomized, vehicle-controlled studies to evaluate efficacy and safety. Vehicle was the comparator. | This was a comment from the public drug plans to inform CDEC deliberation. |
Considerations for initiation of therapy | |
Primary outcome in clinical trials was improvement in IGA score. Some newer therapies for AD included EASI-75. How does the IGA score relate to the EASI score? | The clinical experts noted that the IGA is a 5-point scale that provides a global clinical assessment of AD severity ranging from 0 to 4, where 0 indicates clear, 1 indicates almost clear, 2 indicates mild, 3 indicates moderate, and 4 indicates severe. The IGA was designed for, and is commonly used for, clinical trials but is not used in clinical practice, according to the clinical experts. The clinical experts noted that the EASI score is primarily used in clinical trials but is also used in practice, if required, or for reimbursement purposes. It assesses the severity of 4 disease characteristics of AD (erythema, infiltration, excoriation, and lichenification) along with an estimation of the area of involvement. According to the clinical experts, it is generally felt that the severity of AD based on EASI is as follows:
The clinical experts noted that a key difference between EASI and IGA is that many IGAs do not include an assessment of the extent of involvement (i.e., amount of the body affected) or, if they include it, it is not a major part of the score. In contrast, EASI combines lesional severity with extent. The clinical experts noted that EASI-75 is the typical threshold used for reimbursement renewal for systemic medications. |
Steroids and calcineurin inhibitors are topical options that can be used before systemic therapies. These were not included in the list of comparators, likely due to the indication (i.e., for use after topical steroids and calcineurin inhibitors or if those treatments are inappropriate). Should topical corticosteroids and topical calcineurin inhibitors be considered comparators? Phototherapy is not readily available in all situations. Systemic immunosuppressants (methotrexate, cyclosporine) are not ideal for all patients and are used off-label. Cyclosporine is not a benefit in all jurisdictions. New biologics are available for varying age groups. (dupilumab, tralokinumab). JAK inhibitors upadacitinib and abrocitinib are oral options for the treatment of AD. For other recent recommendations for drugs for AD, specific drugs, dosages, and durations were specified. Depending on the place in therapy for ruxolitinib cream, it may also be helpful to specify those for the recommendation of ruxolitinib cream. For example, history of treatment failure and contraindication or intolerance to prior therapies (i.e., TCS or TCI) would help with implementation of criteria, if that is the determined place in therapy. | The clinical experts consulted for this review noted that from a clinical perspective, currently available topical therapies, including TCS and TCI, are potential comparators for ruxolitinib cream. The clinical experts noted that even if a patient experiences an inadequate response to a TCI treatment, they may remain on the same treatment or move to another topical treatment, such as ruxolitinib cream. Additionally, in the clinical experts’ opinion, patients should not need to experience treatment failure on all currently available topical therapies to move on to ruxolitinib cream, and as such, any topical therapy may be considered a relevant comparator for ruxolitinib cream. The review team acknowledged the clinical experts’ input while noting that the indication positions ruxolitinib cream as a treatment subsequent to the use of TCS and TCI (or when these treatments are inadvisable); this renders comparisons of ruxolitinib cream with TCS and TCI to be irrelevant for the purpose of this review. Based on the clinical expert input, phototherapy, systemic immunosuppressants, biologics, and JAK inhibitors are currently used in clinical practice following inadequate disease response to, or patient ineligibility for, TCS and TCI. |
Clinical trials of ruxolitinib cream were conducted for up to 52 weeks. What would the average duration of use be? Would consistent use be the norm for these patients, or would the drug be used only as needed? | Because AD is a chronic condition in most patients, the clinical experts noted that long-term use of ruxolitinib cream over months and years is expected. The clinical experts noted that patients will be treated on an as-needed basis and are expected to reduce application frequency to the lowest effective frequency. The quantity of the product used is expected to fluctuate over time based on the frequency and extent of flares, according to the clinical experts. |
Considerations for prescribing of therapy | |
Supplied as a 100 g tube. To be applied as a thin layer twice daily to affected areas up to a maximum of 20% BSA. (It could be difficult to accurately determine 20% BSA.) | This was a comment from the public drug plans for CDEC consideration. |
Who would be considered appropriate prescribers? Specialists or family practitioners? | The clinical experts noted that ruxolitinib cream could be prescribed by any health care provider with experience in diagnosing, treating, and monitoring patients with AD. This would principally include general dermatologists, pediatricians, pediatric dermatologists, allergists, family practitioners, and nurse practitioners, according to the clinical experts. |
There is potential for combination use of ruxolitinib cream with systemic therapies (immunosuppressants and biologics). Is there potential for combination use of ruxolitinib cream and other topical therapies (i.e., TCS, TCI)? | The clinical experts noted that there is potential for concurrent use of ruxolitinib cream with TCS and/or TCI. This may occur in the same anatomic location, but most patients would only use 1 treatment at a time in a given location and would be applying different topicals to different parts of the body. |
Ruxolitinib cream is a first-in-class therapy for AD. What is the appropriate place in therapy for this product? | One clinical expert anticipated that, in the majority of patients, ruxolitinib cream will likely be initiated following inadequate disease response to TCS and/or TCI and will continue to be used as an adjunctive treatment in patients who move on to phototherapy and/or systemic therapy. This clinical expert noted that ruxolitinib will most likely serve as a second-line topical treatment following failure of TCS and/or TCI because of long-established treatment protocols favouring TCS and TCI and anticipated access challenges for ruxolitinib cream due to the higher drug cost relative to currently reimbursed topical treatments. The other clinical expert noted that while TCS is expected to remain the first-line treatment for most patients, ruxolitinib cream might be considered for some patients as a first-line topical treatment (before trialling TCS and/or TCI). This clinical expert noted that TCIs are associated with application site reactions and moderate efficacy; provided that ruxolitinib cream is similarly or more effective and has fewer application site reactions than TCIs, the clinical expert thought it would be reasonable to use ruxolitinib cream ahead of TCIs, in particular for the face and groin, for which TCS treatment is inappropriate. The clinical experts expected ruxolitinib cream to be used as either monotherapy, primarily for facial and intertriginous skin, or in combination with other topical therapies (applied to different affected areas). |
Generalizability | |
Dupilumab is approved for patients aged 6 months and older. Abrocitinib, upadacitinib, and tralokinumab are approved for patients aged 12 years and older. This may be a consideration for the place in therapy for ruxolitinib cream. | This was a comment from the public drug plans to inform CDEC deliberation. |
Care provision issues | |
Considerations related to history of failure of prior therapies may be helpful for implementation of criteria. | This was a comment from the public drug plans to inform CDEC deliberation. |
System and economic issues | |
Patients reporting a reduction in itch within the first 8 weeks of therapy. If ruxolitinib cream is prescribed in combination with JAK inhibitors or biologics while they are taking effect, it may have significant budget impact. | This was a comment from the public drug plans to inform CDEC deliberation. |
The product is only available in a 100 g tube, which costs $1,075.9708 per tube. This is a significant cost if the patient does not tolerate this therapy. | This was a comment from the public drug plans to inform CDEC deliberation. |
Dupilumab, upadacitinib, and abrocitinib have proceeded through pCPA. Tralokinumab and lebrikizumab received a do not reimburse recommendation from CDEC in 2024. | This was a comment from the public drug plans to inform CDEC deliberation. |
This review was submitted pre-NOC. The NOC for ruxolitinib cream was granted on October 11, 2024. | This was a comment from the public drug plans to inform CDEC deliberation. |
AD = atopic dermatitis; BSA = body surface area; CDEC = Canadian Drug Expert Committee; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; JAK = Janus kinase; NOC = Notice of Compliance; pCPA = pan-Canadian Pharmaceutical Alliance; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
Clinical Evidence
The objective of the Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ruxolitinib 1.5% cream in the topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable. The focus will be placed on comparing ruxolitinib cream to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ruxolitinib cream is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes the pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section typically includes sponsor-submitted long-term extension studies; however, such evidence was not submitted for this review. The third section includes indirect evidence from the sponsor. The fourth section includes an additional study that was considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
2 pivotal RCTs identified in the sponsor-conducted systematic review (TRuE-AD1 and TRuE-AD2 trials)9,10
1 sponsor-submitted ITC16
1 additional study addressing gaps in evidence (SCRATCH-AD study).17
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The characteristics of the studies included in the systematic review are summarized in Table 5.
Two identically designed, pivotal, phase III, double-masked RCTs (TRuE-AD1 trial: N = 631; TRuE-AD2 trial: N = 618) aiming to assess the efficacy and safety of ruxolitinib cream relative to vehicle cream in adolescents and adults aged 12 years or older with AD were identified by the systematic review conducted by the sponsor. The trials were conducted in study sites in Europe and North America (TRuE-AD1 trial: 78 sites in total, including 7 in Canada; TRuE-AD2 trial: 65 sites in total, including 3 in Canada). Patients were enrolled between December 2018 and December 2020 in the TRuE-AD1 trial, and between December 2018 and November 2020 in the TRuE-AD2 trial. The studies are now complete. The study design is shown in Figure 1. The studies consisted of the following study periods:
Screening period (28 days): Patients were assessed for study eligibility.
Vehicle-controlled period (8 weeks): Enrolled patients were randomized in a 2:2:1 ratio to receive either ruxolitinib 0.75% cream twice a day, ruxolitinib 1.5% cream twice a day, or vehicle cream twice a day for 8 weeks. Randomization was conducted using a central interactive response technology system and was stratified by baseline IGA score (2 or 3) and region (North America or other).
Long-term safety period (44 weeks): Patients who completed the vehicle-controlled period with an IGA score of 0 to 4, 0% to 20% BSA affected, and no safety concerns could continue into the double-masked long-term safety period. Patients who initially received the vehicle cream in the vehicle-controlled period were rerandomized in a masked manner in a 1:1 ratio to 1 of the 2 active treatment groups (ruxolitinib 0.75% or 1.5% cream twice a day), receiving treatment as needed. Patients who initially received the active treatments during the vehicle-controlled period remained on the same regimen on an as-needed basis during the long-term safety period.
Safety follow-up period (30 days): Safety follow-up assessments were conducted.
The results of the ruxolitinib 0.75% cream treatment group are not of interest to this review and will not be presented in this report because this strength of ruxolitinib cream was not approved by Health Canada for the indication under review.
Table 5: Details of Studies Included in the Systematic Review
Characteristic | TRuE-AD1 | TRuE-AD2 |
|---|---|---|
Designs and populations | ||
Study design | Phase III, randomized, double-masked, VC trial | |
Locations | 78 sites in Europe and North America (including 7 in Canada) | 65 sites in Europe and North America (including 3 in Canada) |
Patient enrolment dates | Start date: December 20, 2018 End date: December 1, 2020 | Start date: December 20, 2018 End date: November 9, 2020 |
Randomized (N) | N = 631
| N = 618
|
Key inclusion criteria |
| |
Key exclusion criteria |
| |
Drugs | ||
Intervention |
| |
Comparator(s) | Vehicle cream, topically b.i.d.h | |
Study duration | ||
Screening phase | 28 days | |
Treatment phase | 8 weeks in VC period; 44 weeks in LTS period | |
Follow-up phase | 30 ( + 7) days safety follow-up | |
Outcomes | ||
Primary end point | The proportion of patients experiencing IGA-TS, defined as an IGA score of 0 or 1 (clear or almost clear) with a ≥ 2-grade improvement from baseline, at week 8 | |
Secondary and exploratory end points | Key secondary:
Other secondary:
Exploratory:
| |
Publication status | ||
Publications | ||
Clinical trial record number | NCT03745638 | NCT03745651 |
AD = atopic dermatitis; AE = adverse event; b.i.d. = twice a day; BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = at least a 50% improvement in Eczema Area and Severity Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; EASI-90 = at least a 90% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; LTS = long-term safety; NRS = Numeric Rating Scale; PGI-C = Patient Global Impressions–Change; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System; QoL = quality of life; SCORAD = SCORing Atopic Dermatitis; TEWL = transepidermal water loss; VC = vehicle controlled; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
aThe results of the ruxolitinib 0.75% group are not of interest to this review and are not presented in the report because this strength is not approved by Health Canada.
bThe sponsor noted that the patients in Canada were aged 18 years or older, given Health Canada feedback regarding preclinical juvenile toxicity data and related safety margins. However, a phase III trial of ruxolitinib 1.5% cream and ruxolitinib 0.75% cream in children aged 2 to 11 years with AD is currently ongoing and includes patients from Canada.
cAD was diagnosed using the Hanifin and Rajka criteria.
dPatients enrolled in the VC period were required to have an IGA score of 2 or 3 at screening and baseline. Patients with an IGA score of 0 to 4 at week 8 could enter the LTS period.
ePatients enrolled in the VC period were required to have 3% to 20% BSA affected at screening and baseline. Patients with 0% to 20% BSA affected at week 8 could enter the LTS period.
fThe lesion must be representative of the patient’s disease state and not be located on the hands, feet, or genitalia.
gAn “unstable course of AD” is defined as spontaneously improving or rapidly deteriorating AD, as determined by the investigator. The baseline was defined as the last nonmissing measurement obtained before or on the day of the first application of ruxolitinib cream or vehicle cream.
hA thin film was applied to the affected area in the morning and evening (at least 1 hour before bedtime), at least 8 hours apart.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2;13,14 Papp et al. (2021);9 Papp et al. (2023).10 Details included in the table are from the sponsor’s summary of clinical evidence.15
Figure 1: TRuE-AD1 and TRuE-AD2 Study Design
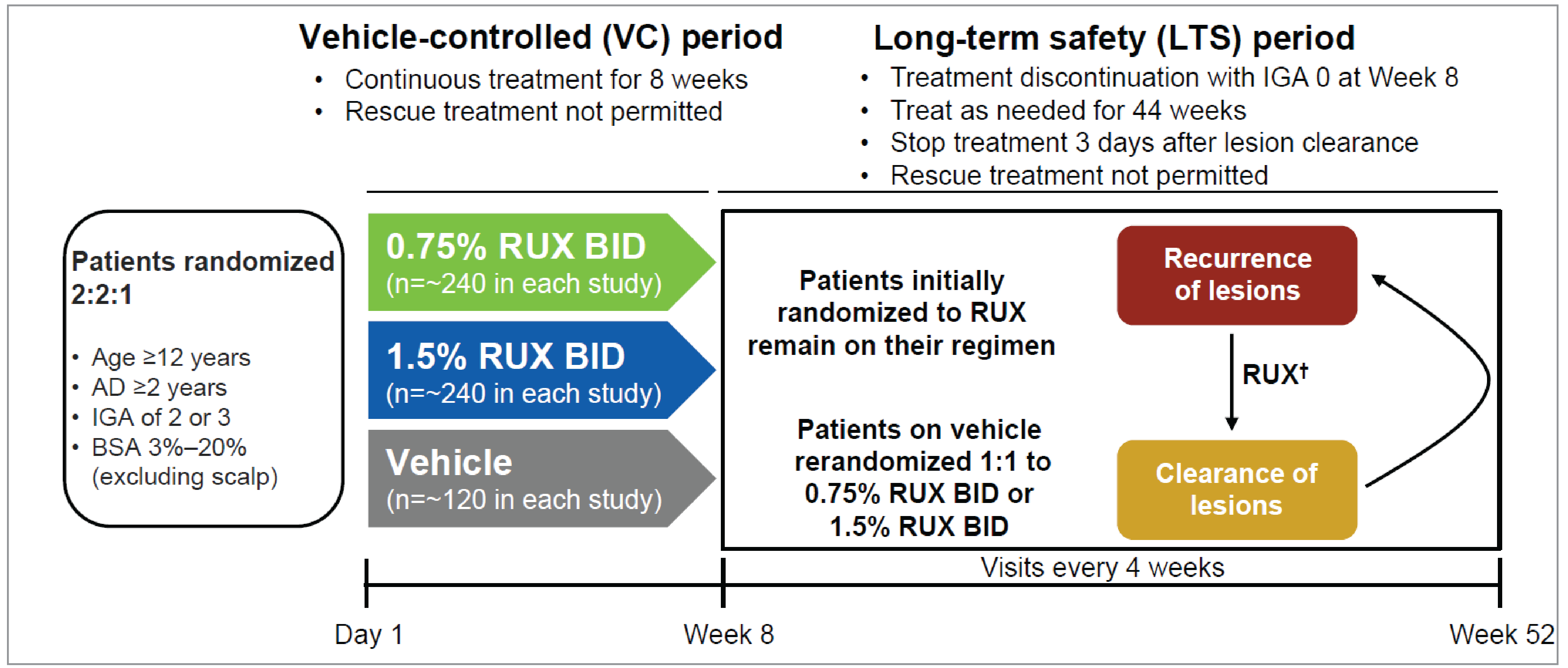
AD = atopic dermatitis; BID = twice a day; BSA = body surface area; IGA = Investigator’s Global Assessment; LTS = long-term safety; RUX = ruxolitinib; VC = vehicle controlled.
†Patients self-evaluated the recurrence of lesions between study visits and treated lesions with active AD (not to exceed 20% BSA). If lesions cleared between study visits, patients stopped treatment 3 days after lesion disappearance. If new lesions were extensive or appeared in new areas, patients contacted the investigator to determine if an additional visit was needed.
Source: Papp K, Szepietowski JC, Kircik L, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: Results from 2 phase III, randomized, double-blind studies. Journal of the American Academy of Dermatology. 2021;85(4):863 to 872. Copyright 2021 by the authors. Available from: https://www.jaad.org/article/S0190-9622(21)00931-2/fulltext. Reprinted in accordance with Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International Licence (CC BY-NC-ND 4.0): https://creativecommons.org/licenses/by-nc-nd/4.0/.9
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria of the TRuE-AD1 and TRuE-AD2 trials are presented in Table 5. The studies included patients aged 12 years or older who had been diagnosed with AD for at least 2 years, had an IGA score of 2 or 3, and had 3% to 20% BSA affected. Patients who had experienced an unstable course of AD (spontaneously improving or rapidly deteriorating) in the 4 weeks before baseline were excluded. Patients who had previously received systemic or topical JAK inhibitors, as well as those who had received biologic treatment (e.g., dupilumab) in the 12 weeks (or 5 half-lives, whichever was longer) before baseline, systemic immunosuppressants or immunomodulators in the 2 weeks before baseline, or topical treatment for AD (e.g., TCS, TCI) in the week before baseline were excluded.
Interventions
In the 8-week vehicle-controlled period, enrolled patients were randomized 2:2:1 to receive ruxolitinib 0.75% cream, ruxolitinib 1.5% cream, or a matching vehicle cream. The interventions were applied topically as a thin film to the affected areas twice a day, in the morning and in the evening (at least 1 hour before bedtime), at least 8 hours apart. Patients applied the masked study drug twice a day to all areas identified for treatment at baseline, even if those areas began to improve throughout the vehicle-controlled period. Patients who developed additional areas of AD could treat these additional areas with approval from the investigator.
In the 44-week long-term safety period, patients who initially received the active treatments during the vehicle-controlled period remained on the same regimen. Patients who initially received the vehicle cream in the vehicle-controlled period were rerandomized (1:1) to receive either the ruxolitinib 1.5% or 0.75% cream. Treatments were administered on an as-needed basis. At each visit (every 4 weeks), AD lesions were evaluated by the investigator to confirm whether the patient required continuation of therapy (IGA score > 1) or could otherwise (re)enter the observation or no-treatment cycle. Between study visits, patients self-evaluated the recurrence of AD and applied the study drug twice a day to areas of the skin with active lesions (not to exceed 20% BSA). If the lesions cleared between study visits, patients would stop treatment application 3 days after the lesions had disappeared.
TCS, TCI (i.e., tacrolimus, pimecrolimus), topical phosphodiesterase 4 inhibitor (i.e., crisaborole), other topical treatments for AD (except for bland emollients), systemic corticosteroid, immunosuppressants (e.g., methotrexate, cyclosporine, azathioprine), biological therapies, allergen immunotherapy, and phototherapy were prohibited during the study.
The study interventions were administered in a double-masked manner. The vehicle cream had all the same excipients as ruxolitinib cream but did not include the ruxolitinib. The vehicle cream looked and felt the same as ruxolitinib cream and absorbed into the skin just as ruxolitinib cream would.
Outcomes
A list of the efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by the review team and input from patient groups, clinician groups, and public drug programs. Using the same considerations, the review team selected the end points that were considered to be most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE.
The considerations that informed the selection of the efficacy outcomes to be summarized and assessed using GRADE include the following:
The IGA and EASI scales are used to measure the extent and severity of AD, which are considered important outcomes according to input from patient groups, clinician groups, and the clinical experts. IGA-TS, an IGA score of 0 or 1, and EASI-75 are the most commonly used thresholds in the assessment of response to treatment in clinical trials of AD.
The percentage of BSA affected by AD and Itch NRS are commonly assessed in clinical practice according to clinical expert input. Itch was a main symptom of AD noted by the patient groups.
POEM is an AD-specific measure used to assess multiple symptoms of AD noted in the patient group input (e.g., dryness, itching, cracking, flaking, and bleeding of skin). It is commonly used in clinical trials of AD. While Skin Pain NRS was assessed in the trials, it was not considered to be most relevant for clinical decision-making according to the clinical expert input.
The impact of AD on sleep was noted to be an important outcome by patient groups and was assessed using PROMIS Short Form–Sleep-Related Impairment and PROMIS Short Form–Sleep Disturbance scales in the trials. Responder analysis was considered by the review team to be the most relevant analysis for decision-making, based on clinical expert input.
HRQoL was identified as an important outcome in patients with AD, based on input from patient groups, clinician groups, and the clinical experts. Of the HRQoL measures included in the trials, AD-specific measures, including DLQI and CDLQI, were included in the GRADE assessment. EQ-5D-5L is a generic HRQoL measure. It is not included in GRADE assessment but is summarized in Appendix 1 given that it informs the sponsor’s pharmacoeconomic model.
EASI, POEM, Itch NRS, DLQI, and CDLQI are outcome measures recommended to be reported in AD clinical trials.71
Harms were considered important outcomes according to input from patient groups, clinician groups, and the clinical experts. Occurrence of SAEs was selected for inclusion in the GRADE assessment.
For all efficacy end points, the baseline was defined as the last nonmissing measurement obtained before or on the day of the first application of ruxolitinib cream or vehicle cream. For patients who crossed over from vehicle cream to ruxolitinib treatment in the long-term safety period, the baseline was defined as the last nonmissing measurement obtained before or on the day of the first application of study treatment in the long-term safety period.
Table 6: Outcomes Summarized From TRuE-AD1 and TRuE-AD2 Trials
Outcome measure | Time point | TRuE-AD1 and TRuE-AD2 |
|---|---|---|
Extent and severity of AD and AD lesions | ||
IGA-TS (i.e., IGA score of 0 or 1 with ≥ 2-grade improvement from baseline) | Week 8 | Primarya |
IGA score of 0 or 1 | Week 52 | Secondary |
EASI-75 | Week 8 | Key secondarya |
Change from baseline in percentage of BSA affected by AD | Weeks 8 and 52 | Secondary |
Symptom reduction | ||
Proportion of patients with ≥ 4-point improvement in Itch NRS from baseline | Week 8 | Key secondarya |
Change from baseline in POEM score | Weeks 8 and 52 | Secondary |
Proportion of patients with ≥ 6-point improvement (24-hour recall) from baseline in PROMIS Short Form–Sleep Disturbance score | Week 8 | Key secondarya |
Proportion of patients with ≥ 6-point improvement (24-hour recall) from baseline in PROMIS Short Form–Sleep-Related Impairment score | Week 8 | Key secondarya |
Health-related quality of life | ||
Change from baseline in DLQI | Weeks 8 and 52 | Secondary |
Change from baseline in CDLQI | Weeks 8 and 52 | Secondary |
Safety outcomes | ||
Serious adverse events | Weeks 8 and 52 | Secondary |
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; NRS = Numeric Rating Scale; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System.
aStatistical testing for these end points was adjusted for multiple comparisons.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Investigator’s Global Assessment
The IGA score is an investigator-reported assessment of overall AD severity.72 The score is based on an overall assessment of the degree of erythema, induration, or papulation and of oozing or crusting. It is a 5-point scale that ranges from 0 (clear) to 4 (severe). Evidence for the validity and reliability of this instrument is summarized in Table 7. No minimal important difference (MID) in adult or adolescent patients with AD was identified from the literature. Based on input from the clinical experts consulted for this review, a difference of at least 10% between the ruxolitinib cream group and the vehicle cream group could be considered clinically important with respect to the proportion of patients experiencing IGA-TS and the proportion of patients experiencing an IGA score of 0 or 1.
IGA-TS was defined as an IGA score of 0 or 1 with at least a 2-grade improvement from baseline.
Eczema Area and Severity Index
The EASI score is an investigator-reported assessment of the severity and extent of AD.73 Four disease characteristics of AD (erythema, infiltration or papulation, excoriations, and lichenification) are assessed for severity on a 4-point scale ranging from 0 (none or absent) to 3 (severe). The total EASI score ranges from 0 to 72 points, with higher values indicating more severe and/or more extensive condition. Evidence for the validity, reliability, and responsiveness of this instrument in patients with AD is summarized in Table 7. Based on the input from the clinical experts consulted for this review, a difference of at least 10% between the ruxolitinib cream group and the vehicle cream group with respect to the proportion of patients experiencing EASI-75 could be considered clinically important.
EASI-75 was defined as at least a 75% improvement (i.e., reduction) from baseline in EASI score.
Percentage of BSA Affected by AD
BSA assessment was administered by the investigator and was approximated to the nearest 0.1% using the Palmar method as a guide: the palm plus 5 digits, with fingers tucked together and thumb tucked to the side (handprint) as 1% BSA, and the thumb as 0.1% BSA. No evidence for the validity, reliability, responsiveness, and MID of this instrument was identified. According to the clinical expert input, a difference of at least 5% to 10% between the ruxolitinib cream group and the vehicle cream group with respect to the change from baseline in the percentage of BSA affected by AD could be considered clinically important.
Itch NRS
Itch NRS is a daily patient-reported measure of the worst level of itch intensity. Patients were asked to rate the itching severity of their AD by selecting a number from 0 (no itch) to 10 (worst imaginable itch) to best describe their worst level of itching in the past 24 hours. Evidence for the validity, reliability, and responsiveness of this instrument in patients with AD is summarized in Table 7. Patients with a decrease in Itch NRS score of at least 4 points from baseline were considered as experiencing a response in the TRuE-AD1 and TRuE-AD2 trials. Findings from Simpson et al. (2017)74 and Yosipovitch et al.75 provided support for the use of 4-point change in assessing clinically important difference. Based on input from the clinical experts consulted by CDA-AMC, a difference of at least 10% between the ruxolitinib cream group and the vehicle cream group with respect to the proportion of patients experiencing at least a 4-point improvement in Itch NRS score could be considered clinically important.
The proportion of patients with at least a 4-point improvement (i.e., reduction) in Itch NRS score from baseline at week 8 was assessed in patients with a baseline Itch NRS of at least 4.
Patient Oriented Eczema Measure
POEM is a 7-item, AD-specific symptom questionnaire.76 Based on patient-reported frequency of occurrence during the past week, the 7 items (itching, sleep, bleeding, weeping, cracking, flaking, and dryness) are assessed using a 5-point scale (0 = no days; 1 = 1 to 2 days; 2 = 3 to 4 days; 3 = 5 to 6 days; 4 = every day). The score of this assessment ranges from 0 to 28, with a higher score being indicative of worse disease severity. Evidence for the validity, reliability, and responsiveness of POEM is summarized in Table 7. The MID has been estimated in AD as 3.4 points in adults77 and from 3.0 to 3.9 points in children.78 Another study estimated 5 points as the MID for adults using the global severity of AD as the anchor.76
PROMIS Short Form
The PROMIS Short Form–Sleep Disturbance questionnaire is used to assess patient-reported perceptions of sleep quality, sleep depth, and restoration associated with sleep and was collected in the morning during the vehicle-controlled period. The PROMIS Short Form–Sleep-Related Impairment questionnaire is used to assess patient-reported perceptions of alertness, sleepiness, and tiredness during usual waking hours and the perceived functional impairments during wakefulness associated with sleep problems and impaired alertness. This questionnaire was collected in the evening during the vehicle-controlled period. Both questionnaires had a range of scores between 8 and 40, with a higher score indicating a greater level of disturbance or impairment. Evidence for the reliability, validity, and responsiveness of these instruments in patients with AD is summarized in Table 7. No MID in patients with AD was identified from a targeted review of the literature. The sponsor submitted additional evidence, including post hoc analyses using data from the TRuE-AD1 and TRuE-AD2 trials, which estimated the MID for both instruments to be 1 to 3 using a distribution-based method.79 Using anchor-based methods, the analyses estimated the MID to be 2 with Patient Global Impressions–Change as the anchor and to be 6 with Itch NRS as the anchor.79 Based on clinical expert input, a difference of at least 5% between the ruxolitinib cream group and the vehicle cream group could be considered clinically important with respect to the proportion of patients experiencing at least a 6-point improvement in PROMIS Short Form–Sleep Disturbance score and the proportion of patients experiencing at least a 6-point improvement in PROMIS Short Form–Sleep-Related Impairment score.
The proportion of patients with at least a 6-point improvement (i.e., reduction) in PROMIS Short Form–Sleep Disturbance and Sleep-Related Impairment scores from baseline at week 8 was assessed in patients with a baseline score of at least 6.
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
The DLQI is a widely used dermatology-specific HRQoL instrument for use in adults. It is a 10-item questionnaire on which patients self-report 6 aspects that may affect quality of life (symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and the effect of treatment).80-82 The DLQI score ranges between 0 and 30. The higher the score, the more quality of life is impaired. Evidence for the validity, reliability, and responsiveness of the DLQI instrument in patients with AD is summarized in Table 7. Estimates of the MID for a variety of skin conditions have ranged from 2.2 to 6.9 points. For inflammatory skin diseases specifically, MID estimates of between 3 and 5 were reported, but no information about an AD-specific MID was found.80-82
The CDLQI questionnaire is based on the adult version (the DLQI) and is designed and validated for patients aged 3 to 16 years with dermatological conditions.83-85 The CDLQI is available in text and cartoon versions. The questionnaire consists of 10 items addressing the patient’s perception of the impact of their skin disease on various aspects of their quality of life over the last week, including dermatology-related symptoms and feelings, leisure, school, friendships, and sleep, as well as the impact of treatment. The total score ranges from 0 to 30, with a higher score being indicative of a poor quality of life.83-85 Evidence for the validity, reliability, and responsiveness of this instrument is summarized in Table 7. In adolescents with moderate to severe AD, a reduction in CDLQI score of 6 to 8 points has been suggested as the clinically relevant threshold for within-person change in CDLQI.83
Change from baseline in DLQI was assessed in patients aged 16 years or older. Change from baseline in CDLQI was assessed in patients younger than 16 years.
Harms Outcomes
Harms outcomes including AEs, SAEs, withdrawal due to AEs, and death were assessed. Based on clinical expert input, a difference of at least 10% between the ruxolitinib cream group and the vehicle cream group in the frequency of SAEs was considered clinically important.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
IGA/PGA | Investigator-reported assessment instrument used in clinical trials to rate AD severity. It is a 5-point scale, ranging from 0 (clear) to 4 (severe) with distinct morphological descriptors for each category.72 IGA-TS is defined as an IGA score of 0 or 1 with ≥ 2-grade improvement from baseline. | Validity: Moderate to strong correlation with EASI (r = 0.66 to 0.72) in adult patients with AD.72 Reliability: Moderate intrarater (ICC = 0.54; SD = 0.28) and inter-rater reliability (CV = 33.0; SD = 12.3) in adult patients with AD.72 Responsiveness: No evidence identified. | No MID was identified in patients with AD. |
EASI | A physician-administered, composite index that assesses the severity and extent of AD in patients aged 8 years or older.73 The severity of 4 AD disease characteristics (erythema, induration/papulation, excoriation, and lichenification) on the 4 body regions (head/neck, trunk, upper extremities, and lower extremities) is assessed by the investigator on a 4-point scale ranging from 0 (none/absent) to 3 (severe). The EASI score equals the sum of the weighted scores obtained for each body region. Scores range from 0 to 72, with higher values indicating more severe and/or more extensive condition.73 EASI-75 represent a ≥ 75% improvement from baseline in EASI score. | Validity: In adult patients with AD, moderate to strong correlation with SCORAD (r = 0.84 to 0.93) was found.77,86 In pediatric patients with AD, including those older than 12 years, EASI was correlated strongly with IGA (r > 0.8 at day 43 and 6 months).73 Reliability: In adult patients with AD, the internal consistency of EASI is adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94, respectively.87 Test-retest reliability was also adequate (intrarater and inter-rater reliability kappa = 0.76),87 whereas the reliability of each component of EASI ranged from 0.38 (ICC, lichenification) to 0.75 (ICC, area), indicating poor to good intrarater reliability.72 No evidence of reliability in adolescent patients with AD was identified. Responsiveness: In a study with adult patients with AD (MAcAD trial), responsiveness to improvement and decline in global severity based on IGA over 24 weeks was also demonstrated (AUC = 0.67; 95% CI, 0.60 to 0.76).77 In pediatric patients with AD, sensitivity to change in disease status from baseline to day 8 was judged as adequate (P < 0.001; n = 1,068).73 | In a study of mainly adult and an unknown number of adolescent patients with AD, the overall MID was reported to be 6.6.77 |
Itch NRS | A patient-reported measure of intensity of itch assessed using an 11-point scale (0 = no itch; 10 = worst itch imaginable).75 Patients were asked in the evening to rate the maximum intensity of itch (“worst” or “highest level” of itch) over the 24-hour recall period.88 | Psychometric assessment was performed in adult patients with moderate to severe AD from clinical trial populations (SOLO 1 and SOLO 2 trials).75 Validity: Content validity has been ensured through concept elicitation during development and in-depth, one-to-one patient interviews (n = 14). The construct validity with instruments with similar constructs (PCS, DLQI itch item, SCORAD itch VAS) was strong (Pearson r = 0.61 to 0.77), whereas with instruments with dissimilar constructs (EASI, IGA) it was weak to moderate (r = 0.09 to 0.24). Known-group validity has been established: patients with “absent” or “mild” itch based on PCS, “no impact” based on DLQI, or a rating of “excellent” based on PGADS had a significantly lower score on NRS (P < 0.0001).75 Reliability: Test-retest reliability over 1 week was adequate (ICC = 0.95 to 0.96).75 Responsiveness: Change from baseline at week 16 in NRS was correlated well with PCS (Pearson r = 0.71), DLQI itch item (0.66), and SCORAD itch VAS (0.77) and less well with EASI (0.50) and IGA (0.50).75 No psychometric assessment in the adolescent population with AD has been identified. | Improvement on the Peak Pruritus NRS of at least 3 to 4 points from baseline is regarded as a clinically meaningful change, which was calculated using anchor-based and distribution-based methods in adult patients with moderate to severe AD.74 In adult patients with moderate to severe AD, the MID was estimated to be between 2 and 4 based on anchor-based methods (EASI, IGA, PCS) and 1.0 based on distribution-based methods (based on 0.5 SD).75 |
POEM | A patient-reported, AD-specific, symptom questionnaire with a recall period of the past 7 days.76 Consists of 7 items (itching, sleep, bleeding, weeping, cracking, flaking, and dryness), each assessed on a 5-point categorical response scale (0 = no days; 1 = 1 to 2 days; 2 = 3 to 4 days; 3 = 5 to 6 days; 4 = every day). The total score is the sum of the 7 items ranging from 0 to 28. A higher score indicates worse symptoms, reflecting a greater degree of disease-related morbidity (0 to 2 = clear or almost clear; 3 to 7 = mild, 8 to 16 = moderate; 17 to 24 = severe; 25 to 28 = very severe eczema).76 | Validity: In adult patients, concurrent validity was reported in those with moderate to severe self-reported AD severity (Spearman r = 0.53); however, weak correlation (r = 0.39) with clear to mild AD was found. Convergent validity with DLQI (r = 0.59), correlation with EASI (r = 0.52), and weaker correlation with worst Itch NRS (r = 0.45) were found in adult patients with AD.76 Reliability: Internal consistency was acceptable (Cronbach alpha = 0.88), and test-retest reliability was acceptable, with 95% of scores falling within 2.6 points on repeat testing (mean score difference = 0.04; SD = 1.32) in adult patients with AD.76 Responsiveness: In the Prove trial conducted in adult patients with AD, POEM was responsive to change in global severity as measured by the PGA (AUC = 0.67; 95% CI, 0.59 to 0.75).77 | The MID was estimated in AD as 3.4 points (SD = 4.8) in adults and from 3.0 to 3.9 points in children.77,78 Other studies have established 5 points as the MID for adults using the global severity of AD as the anchor.76 |
PROMIS Short Form | PROMIS is a set of self-administered questionnaires measuring key health-related outcome domains in a variety of chronic diseases. PROMIS Short Form is a modified form with a diary to be completed by the patient on a daily basis. Sleep-Related Impairment (8a): Assessed in the evening via an 8-item questionnaire where each item is rated on a 5-point scale (1 = not at all; 5 = very much). Raw scores on the 8 items are summed to obtain a total raw score, which ranges from 8 to 40, with higher scores indicating greater severity of sleep-related impairment. The recall period was 24 hours for the VC period and the past 7 days for the LTS period.88 Sleep Disturbance (8b): Assessed in the morning. Generic, self-reported perceptions of sleep quality, depth, and restoration on a 5-point scale, with a range of scores from 8 to 40, with higher scores indicating greater severity of sleep disturbance. Each item asks the patient to rate the severity of sleep disturbance. The recall period was the past 24 hours for the VC period and the past 7 days for the LTS period.88 | Psychometric properties for the PROMIS Short Form have been assessed in patients from the TRuE-AD1 and TRuE-AD2 studies.79 Validity: Content validity has been ensured during developmental stage. Both scales demonstrated moderate convergent validity with Itch NRS, POEM, DLQI, and CDLQI total scores (Pearson r = 0.33 to 0.46). Known-groups validity has been also demonstrated (P < 0.05) as both scales could differentiate distinct health states defined by IGA, DLQI/CDLQI, POEM, EASI, and Itch NRS. Reliability: Internal consistency was excellent for both scales: alpha of 0.95 to 0.98 for the 8a scale and alpha of 0.94 to 0.95 for the 8b scale. Test-retest reliability was found to be high (n = 802): ICC of 0.83 for the 8a scale and ICC of 0.82 for the 8b scale. Responsiveness to change: Both scales were able to detect changes from baseline to week 8 in POEM, DLQI, and Itch NRS (r > 0.3). | The MID (within-patient meaningful change) was estimated to be between 1 (SEM) and 3 (based on 0.5 SD) using the distribution-based method, and between 2 (based on PGI-C) and 6 (based on Itch NRS) using the anchor-based method. |
DLQI (≥ 16 years) | A patient-reported, dermatology-specific HRQoL instrument for use in adults. Consists of 10 items addressing the patient’s perception of the impact of their skin disease on the following 5 aspects of quality of life, each scored on a 4-point Likert scale (0 = not at all or not relevant; 1 = only a little; 2 = quite a lot; 3 = very much or prevented work or studying):
The total score is the sum of the 10 items (0 to 30), with a higher score being indicative of a poorer quality of life (0 to 1 = no effect; 2 to 5 = small effect; 6 to 10 = moderate effect; 11 to 20 = very large effect; 21 to 30 = extremely large effect). Recall period is the past 1 week.80-82 | Validity: Content validity has been ensured with input from adult patients with AD (n = 9; other eczema, n = 10) during the development phase.89 Construct validity has been demonstrated by a strong correlation with POEM (r = 0.78) and a moderate correlation with SCORAD (r = 0.42). Reliability: In patients with stable AD, test-retest reliability was adequate (ICC > 0.7). Among adult patients with mixed skin diseases including AD, internal consistency was acceptable (Cronbach alpha = 0.75 to 0.92).90-92 Responsiveness: In patients aged 16 years or older with a variety of skin conditions, including AD (n = 192; patients with eczema = 12.5%), an improved DLQI score was observed in those whose disease severity decreased over a 1 to 3 month period (P < 0.0001).82 | Estimates of the MID ranged from 2.2 to 6.9 for a variety of skin conditions. For inflammatory skin diseases specifically, MID estimates of between 3 and 5 were reported, but no information about AD-specific MID was identified.80-82 |
CDLQI (≥ 3 years to < 16 years) | A patient-reported, dermatology-specific questionnaire. Based on the adult version (DLQI) and designed for patients aged 3 years to 16 years with dermatological conditions. Available in text and cartoon versions. Consists of 10 items addressing the patient’s perception of the impact of their skin disease on various aspects of their HRQoL over the last week, including dermatology-related symptoms and feelings, leisure, school, friendships, sleep, and the impact of treatment.83-85 Each question is scored on a 4-point Likert scale (0 = not at all; 1 = only a little; 2 = quite a lot; 3 = very much or prevented school). The total score is the sum of the 10 items (0 to 30), with a higher score being indicative of a worse HRQoL (0 to 1 = no effect; 2 to 6 = small; 7 to 12 = moderate; 13 to 18 = very large; 19 to 30 = extremely large effect on child’s life).83-85 | Validity: A review including 3 studies demonstrated concurrent validity, 2 between CDLQI and the Cardiff Acne Disability Index and 1 between CDLQI and the Childhood Atopic Dermatitis Impact Scale.85 Convergent construct validity and divergent construct validity of CDLQI were demonstrated in 45 and 6 studies, respectively.85 Reliability: Good internal consistency of the CDLQI has been shown (examined in 6 studies), with Cronbach alpha values ranging from 0.82 to 0.92.84,85 Test-retest reliability is adequate, with the Spearman rank order correlation coefficient calculated in 4 studies (range, 0.74 to 0.97).84,85 One study also confirmed the test-retest reliability (ICC = 0.80).84,85 Responsiveness: A review of 26 studies demonstrated responsiveness to a change of CDLQI.85 | In adolescent patients with moderate to severe AD, a reduction of 6.0 to 8.0 points has been suggested as the clinically relevant threshold for within-patient change corresponding to improvement in anchors.83 |
AD = atopic dermatitis; AUC = area under curve; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence internal; CV = coefficient of variation; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; HRQoL = health-related quality of life; ICC = intraclass correlation; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; LTS = long-term safety; MID = minimal important difference; NRS = Numeric Rating Scale; PCS = Pruritus Categorical Scale; PGA = Physician’s Global Assessment; PGADS = Patient Global Assessment of Disease Status; PGI-C = Patient Global Impressions–Change; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System; SCORAD = SCORing Atopic Dermatitis; SD = standard deviation; SEM = standard error of the mean; VAS = visual analogue scale; VC = vehicle controlled.
Sources: True-AD1 study protocol;88 Bozek and Reich;72 Barbier et al.;73 Schmitt et al.;87 Schram et al.;77 Hanifin et al.;86 Yosipovitch et al.;75 Simpson et al. (2017);74 Silverberg et al.;76 Howells et al.;78 Shikiar et al.;80 Basra et al. (2015);82 Basra et al. (2008);90 Heinl et al.;81 Simpson et al. (2019);83 Simpson et al. (2021);93 Lewis-Jones and Finlay;84 Salek et al.;85 Badia et al.92
Statistical Analysis
The statistical analysis methods used in the TRuE-AD1 and TRuE-AD2 trials are summarized in Table 8.
Sample Size and Power Calculation
A sample size calculation determined that approximately 600 randomized patients were required in each study to demonstrate a statistically significant difference between each of the 2 treatments arms (ruxolitinib 1.5% and 0.75% cream) and the vehicle cream arm with respect to the primary end point of IGA-TS at week 8, at a 2-sided significance level of 0.025 with more than 95% power. This estimate assumed that IGA-TS response rates in the ruxolitinib 1.5% cream, ruxolitinib 0.75% cream, and vehicle cream groups were 45%, 30%, and 10%, respectively, based on the results from a phase II, randomized, dose-ranging study.58 The sample size was determined to also provide an adequate database for safety evaluation, according to the sponsor, although the assumptions were not outlined.
Statistical Model
All efficacy analyses in the TRuE-AD1 trial were based on the ITT population. Efficacy analyses in the TRuE-AD2 trial were based on the ITT population █████████ ██ ████████ ██ ████ ███ █████ ██ ███████ █████████████ ████ ███ ████████ ███ ████████ ████ ████████ ████████ ██████ ████████ █████████ ███ █ ████████████ ██ ███ ████. The difference between ruxolitinib cream and vehicle cream with respect to the primary and key secondary end points was analyzed at an overall 2-sided significance level of 0.05.
Binary end points were tested using logistic regression, including treatment group, baseline IGA, and region as adjustment factors. The unadjusted P values between each treatment group versus vehicle cream were calculated based on the Wald test.
Continuous end points including change from baseline in percentage of BSA affected by AD, POEM score, and DLQI or CDLQI score at week 8 were analyzed using the analysis of covariance model, including treatment group, baseline IGA, region, and baseline scores as covariates (where applicable). All continuous outcomes at week 52 were presented using descriptive statistics.
Harms Outcomes
Harms outcomes were presented descriptively.
Multiple Testing Procedure
A graphical procedure with a gatekeeping testing strategy for the primary and key secondary analyses was implemented to control for type I error at a 2-sided significance level of 0.05, as outlined in Figure 2.
Figure 2: Statistical Testing Hierarchy in TRuE-AD1 and TRuE-AD2 Trials
EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA-TS = Investigator’s Global Assessment treatment success; NRS = Numeric Rating Scale; PROMIS = Patient-Reported Outcomes Measurement Information System
Notes: H11 = proportion of participants who experience IGA-TS for ruxolitinib 1.5% versus vehicle cream; H12 = proportion of participants who experience EASI-75 for ruxolitinib 1.5% versus vehicle cream; H13 = proportion of participants with at least a 4-point improvement in Itch NRS over baseline for ruxolitinib 1.5% versus vehicle cream; H14 = proportion of participants with at least a 6-point improvement in the PROMIS Short Form–Sleep Disturbance score for ruxolitinib 1.5% versus vehicle cream; H21 = proportion of participants who experience IGA-TS for ruxolitinib 0.75% versus vehicle cream; H22 = proportion of participants who experience EASI-75 for ruxolitinib 0.75% versus vehicle cream; H23 = proportion of participants with at least a 4-point improvement in Itch NRS over baseline for ruxolitinib 0.75% versus vehicle cream; H24 = proportion of participants with at least a 6-point improvement in the PROMIS Short Form–Sleep Disturbance score for ruxolitinib 0.75% versus vehicle cream; H31 = proportion of participants with at least a 6-point change from baseline in the PROMIS Short Form–Sleep-Related Impairment score for ruxolitinib 1.5% cream twice a day versus vehicle cream; H32 = proportion of patients with at least a 6-point change from baseline in the PROMIS Short Form–Sleep-Related Impairment score for ruxolitinib 0.75% cream twice a day versus vehicle cream.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Handling of Missing Data and Imputation
For binary end points, patients with missing postbaseline values were imputed as not experiencing a response in the primary analysis. For continuous end points, any patients with missing postbaseline values were adjusted for implicitly using the mixed models for repeated measures approach.
Subgroup Analyses
Prespecified subgroup analyses (by age, sex, race, baseline IGA score, baseline EASI score, and region) were conducted with respect to IGA-TS and EASI-75 during the vehicle-controlled period. Multiplicity was not accounted for in these analyses. No statistical testing was performed for treatment-by-subgroup interaction. In addition, the following post hoc subgroup analyses were conducted using the pooled data from both trials:
previous medication history (TCS, TCI, systemic treatment, no previous treatment, no previous TCS)
subset of patients with moderate AD at baseline (IGA = 3, EASI ≥ 16, BSA affected ≥ 10%).
Sensitivity Analyses
Four sensitivity analyses with respect to IGA-TS and EASI-75 were conducted to adjust for dependence underlying the hierarchical multilevel data structure (longitudinal logistic regression with repeated measures analysis) and to assess the impact of missing data assumptions (multiple imputation analysis, last observation carry forward analysis, and tipping point analysis).
Table 8: Statistical Analysis of Efficacy End Points in TRuE-AD1 and TRuE-AD2 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Proportion of patients who experienced:
| Logistic regression |
| All patients who were missing postbaseline values were imputed as not experiencing a response |
|
Proportion of patients who experienced:
| Logistic regression |
| All patients who were missing postbaseline values were imputed as not experiencing a response | None |
Proportion of patients who experienced IGA score of 0 or 1 at week 52 | Descriptive statistics | NA | NA | NA |
Change from baseline in:
| Week 8: ANCOVA with MMRM Week 52: descriptive statistics |
| Implicit via MMRM | None |
AD = atopic dermatitis; ANCOVA = analysis of covariance; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; MMRM = mixed models for repeated measures; NA = not applicable; NRS = Numeric Rating Scale; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Analysis Populations
The analysis populations of the TRuE-AD1 and TRuE-AD2 trials are summarized in Table 9.
Table 9: Analysis Populations of Interest in TRuE-AD1 and TRuE-AD2 Trials
Population | Definition | Application |
|---|---|---|
ITT | Includes all randomized patients. | Demographics, baseline characteristics, patient disposition, and all efficacy analyses |
Safety | Includes all patients who applied at least 1 dose of study drug or vehicle cream. Treatment groups for this population were determined according to the actual treatment the patient applied on day 1. | Safety analyses |
LTS evaluable population | Includes all patients who applied at least 1 dose of ruxolitinib 0.75% or 1.5% cream during the LTS period. The treatment groups for this population were determined according to the actual treatment the patient applied for the first time during the LTS period. | All analyses for the LTS period |
ITT = intention to treat; LTS = long-term safety.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Results
The results of the ruxolitinib 1.5% cream and vehicle cream groups are presented in this report. The results of the ruxolitinib 0.75% cream group are not of interest to this review and are not presented because this strength has not been approved by Health Canada for the treatment of AD.
Patient Disposition
Patient disposition in the TRuE-AD1 and TRuE-AD2 trials is summarized in Table 10 (vehicle-controlled period) and Table 11 (long-term safety period).
Of the 631 patients enrolled in the TRuE-AD1 trial, 126 (20.0%) and 253 (40.1%) were randomized to receive vehicle cream and ruxolitinib 1.5% cream, respectively, in the vehicle-controlled period. Of the 618 patients enrolled in the TRuE-AD2 trial, 124 (20.1%) and 246 (39.8%) were randomized to receive vehicle cream and ruxolitinib 1.5% cream, respectively, in the vehicle-controlled period. Study treatment discontinuation in the vehicle-controlled period was notably higher in the vehicle cream group than in the ruxolitinib 1.5% cream group in both studies (TRuE-AD1 trial: 19.8% versus 8.3%; TRuE-AD2 trial: 14.5% versus 4.5%), with the primary reason being withdrawal by patient in both groups.
Table 10: Patient Disposition in TRuE-AD1 and TRuE-AD2 Trials — VC Period
Patient disposition | TRuE-AD1a | TRuE-AD2a | ||
|---|---|---|---|---|
Vehicle cream | Ruxolitinib 1.5% cream | Vehicle cream | Ruxolitinib 1.5% cream | |
Screened, N | NR | NR | ||
Enrolled, N | 631 | 618 | ||
Randomized, N | 126 | 253 | 124 | 246 |
Discontinued from study drug during the VC period, n (%) | 25 (19.8) | 21 (8.3) | 18 (14.5) | 11 (4.5) |
Adverse events | 5 (4.0) | 2 (0.8) | 3 (2.4) | 1 (0.4) |
Lack of efficacy | 1 (0.8) | 0 (0) | 1 (0.8) | 0 (0) |
Lost to follow-up | 5 (4.0) | 7 (2.8) | 3 (2.4) | 4 (1.6) |
Physician decision | 0 (0) | 0 (0) | 1 (0.8) | 1 (0.4) |
Pregnancy | 1 (0.8) | 0 (0) | 0 (0) | 0 (0) |
Patient withdrawal | 12 (9.5) | 12 (4.7) | 9 (7.3) | 5 (2.0) |
Other | 1 (0.8) | 0 (0) | 1 (0.8) | 0 (0) |
Completed treatment during the VC period, n (%) | 101 (80.2) | 232 (91.7) | 106 (85.5) | 235 (95.5) |
ITT set, n (%) | 126 (100) | 253 (100) | 124 (100) | 246 (100) |
ITT excluding site 461,b n (%) | NA | NA | 118 (95.2) | 228 (92.7) |
Safety set, n (%) | 126 (100) | 253 (100) | 124 (100) | 246 (100) |
LTS evaluable set, n (%) | 95 (75.4) | 225 (88.9) | 105 (84.7) | 221 (89.8) |
ITT = intention to treat; LTS = long-term safety; NA = not applicable; NR = not reported; VC = vehicle controlled.
aThe results of the ruxolitinib 0.75% cream group are not presented in this table.
b████ ████ ████████ ██ ████ ███ ██ █ ███ ████ ███████ ████ ███ ████████ ████████ ███ ████████ █████ ██ ███████ █████████████ ████ ███ ████████ ███ ████████ ████ ████████ ████████ ██████ ████████ █████████ ███ █ ████████ ██ ███ ████.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
In both trials, the ITT and the safety analysis populations consisted of all randomized patients. The efficacy analyses in the TRuE-AD2 trial were conducted in the ITT population █████████ ████████ ██ ████ ███ █████ ██ ███████ █████████████ ████ ███ ████████ ███ ████████ ████ ████████ ████████; thus, 118 of the 124 randomized patients (95.2%) and 228 of the 246 randomized patients (92.7%) were included in the efficacy analyses of the vehicle cream group and the ruxolitinib 1.5% cream group, respectively.
In both trials, the majority of patients (88.9% in the TRuE-AD1 trial; 89.8% in the TRuE-AD2 trial) randomized to ruxolitinib 1.5% cream in the vehicle-controlled period continued to receive ruxolitinib 1.5% cream in the long-term safety period. The majority of patients randomized to vehicle cream in the vehicle-controlled period entered the long-term safety period (75.4% in the TRuE-AD1 trial; 84.7% in the TRuE-AD2 trial), and approximately half of these patients were rerandomized to receive ruxolitinib 1.5% cream in the long-term safety period.
Table 11: Patient Disposition in TRuE-AD1 and TRuE-AD2 Trials — LTS Period
Patient disposition | TRuE-AD1a | TRuE-AD2a | ||
|---|---|---|---|---|
Vehicle cream to ruxolitinib 1.5% cream | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream | Vehicle cream to ruxolitinib 1.5% cream | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream | |
Entered the LTS period, N | 47 | 225 | 52 | 221 |
Treated during the LTS period, n (%) | 47 (100) | 225 (100) | 52 (100) | 221 (100) |
Discontinued from study drug during the LTS period, n (%) | 8 (17.0) | 49 (21.8) | 10 (19.2) | 48 (21.7) |
Adverse events | 0 (0) | 1 (0.4) | 0 (0) | 0 (0) |
Lack of efficacy | 0 (0) | 2 (0.9) | 1 (1.9) | 4 (1.8) |
Lost to follow-up | 5 (10.6) | 22 (9.8) | 2 (3.8) | 7 (3.2) |
Physician decision | 1 (2.1) | 0 (0) | 0 (0) | 1 (0.5) |
Pregnancy | 0 (0) | 0 (0) | 0 (0) | 1 (0.5) |
Noncompliance with study drug | 0 (0) | 2 (0.9) | 0 (0) | 1 (0.5) |
Withdrawal by participant | 2 (4.3) | 21 (9.3) | 7 (13.5) | 34 (15.4) |
Other | 0 (0) | 1 (0.4) | 0 (0) | 0 (0) |
Completed treatment in the LTS period, n (%) | 39 (83.0) | 176 (78.2) | 42 (80.8) | 173 (78.3) |
LTS = long-term safety period.
aThe results of the vehicle cream to ruxolitinib 0.75% cream group and the ruxolitinib 0.75% cream to ruxolitinib 0.75% cream group are not presented in this table.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Baseline Characteristics
The baseline demographics, disease characteristics, and treatment history of the ITT population in the TRuE-AD1 and TRuE-AD2 trials are summarized in Table 12. The characteristics outlined are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The baseline patient characteristics were similar in both the trials. At baseline, the mean age of the study population was 35.2 years (SD = 18.2 years) in the TRuE-AD1 trial and 36.4 years (SD = 18.4 years) in the TRuE-AD2 trial. The majority of patients were adults (TRuE-AD1 trial: 80.3%; TRuE-AD2 trial: 80.5%), female (TRuE-AD1 trial: 62.0%; TRuE-AD2 trial: 61.5%), and white (TRuE-AD1 trial: 68.9%; TRuE-AD2 trial: 70.7%) and had an IGA score of 3 (TRuE-AD1 trial: 75.9%; TRuE-AD2 trial: 74.1%). The mean total percentage of BSA affected by AD was 9.5% in the TRuE-AD1 trial and 10.0% in the TRuE-AD2 trial. Approximately half the patients had an EASI score of 7 or less in both trials. Prior TCI use was noted in 24.1% and 18.8% of patients in the TRuE-AD1 trial and the TRuE-AD2 trial, respectively. Prior medium-potency, high-potency, and super-high-potency TCS was noted in 43.7%, 34.9%, and 8.9% of patients, respectively, in the TRuE-AD1 trial and in 41.1%, 30.4%, and 7.0% of patients, respectively, in the TRuE-AD2 trial. Most patients had not received prior systemic immunosuppressants, phototherapy, or dupilumab.
There is, in general, no notable imbalance in the baseline patient characteristics of the ITT population in either trial, except that in the TRuE-AD1 trial, the ruxolitinib 1.5% cream group, compared with the vehicle cream group, had a lower mean number of AD flare episodes (vehicle cream: 9.4 [SD = 35.2]; ruxolitinib: 6.0 [SD = 23.3]) and a lower proportion of patients with an EASI score of 7 or less (vehicle cream: 56.3%; ruxolitinib: 47.8%) and who had previously received a low-potency TCS treatment (vehicle cream: 67.5%; ruxolitinib: 54.9%). In addition, in the TRuE-AD2 trial, the ruxolitinib 1.5% cream group, compared with the vehicle cream group, had a higher proportion of patients with an EASI score of 7 or less (vehicle cream: 45.2%; ruxolitinib: 50.0%) and a lower proportion of patients who had received prior TCI (vehicle cream: 22.6%; ruxolitinib: 16.7%) and high-potency TCS treatments (vehicle cream: 33.9%; ruxolitinib: 24.8%).
In the TRuE-AD2 trial, the baseline characteristics of the ITT population excluding patients at site 461 was in general consistent with the ITT population, and no notable between-group imbalance was observed in this analysis set, except for the proportion of patients who had an EASI score of 7 or less, prior TCI use, and prior high-potency TCS use, as similarly noted in the ITT population.
Exposure to Study Treatments
Study drug exposure in the TRuE-AD1 and TRuE-AD2 trials is summarized in Table 13 (vehicle-controlled period) and Table 14 (long-term safety period). There is no notable between-group difference in study drug exposure in the vehicle-controlled and long-term safety periods in either study.
Efficacy
Key efficacy results from the TRuE-AD1 and TRuE-AD2 trials are summarized in Table 15 (vehicle-controlled period) and Table 16 (long-term safety period).
Table 12: Baseline Patient Characteristics From TRuE-AD1 and TRuE-AD2 Trials — ITT Population
Characteristic | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream (N = 126) | Ruxolitinib 1.5% cream (N = 253) | Vehicle cream (N = 124) | Ruxolitinib 1.5% cream (N = 246) | |
Demographics | ||||
Age (years), mean (SD) | 35.2 (18.1) | 33.7 (17.2) | 38.9 (18.9) | 35.9 (18.0) |
Age group, n (%) | ||||
12 to 17 years | 23 (18.3) | 47 (18.6) | 22 (17.7) | 45 (18.3) |
≥ 18 years | 103 (81.7) | 206 (81.4) | 193 (77.8) | 201 (81.7) |
Female, n (%) | 79 (62.7) | 158 (62.5) | 80 (64.5) | 150 (61.0) |
Race, n (%) | ||||
White/Caucasian | 85 (67.5) | 177 (70.0) | 85 (68.5) | 178 (72.4) |
Black/African American | 29 (23.0) | 56 (22.1) | 32 (25.8) | 57 (23.2) |
Asian | 8 (6.3) | 14 (5.5) | 2 (1.6) | 6 (2.4) |
Other | 4 (3.2) | 6 (2.4) | 5 (4.0) | 5 (2.0) |
Region, n (%) | ||||
North America | 88 (69.8) | 176 (69.6) | 84 (67.7) | 165 (67.1) |
Europe | 38 (30.2) | 77 (30.4) | 40 (32.3) | 81 (32.9) |
Disease characteristics | ||||
Years since onset of initial diagnosis of atopic dermatitis, mean (SD) | 20.4 (16.1) | 18.8 (13.9) | 21.4 (16.9) | 20.2 (14.7) |
Number of atopic dermatitis episodes/flares over the last 12 months, mean (SD) | 9.4 (35.2) | 6.0 (23.3) | 5.1 (8.1) | 5.9 (8.5) |
Facial involvement of atopic dermatitis, n (%) | 52 (41.3) | 118 (46.6) | 41 (33.1) | 79 (32.1) |
Total percentage of BSA affected in current atopic dermatitis episode, mean (SD) | 9.2 (5.1) | 9.3 (5.2) | 10.1 (5.8) | 9.9 (5.4) |
IGA score, n (%) | ||||
2 | 31 (24.6) | 60 (23.7) | 33 (26.6) | 63 (25.6) |
3 | 95 (75.4) | 193 (76.3) | 91 (73.4) | 183 (74.4) |
EASI score, mean (SD) | 7.4 (4.3) | 7.9 (4.6) | 8.2 (5.2) | 7.8 (4.9) |
≤ 7, n (%) | 71 (56.3) | 121 (47.8) | 56 (45.2) | 123 (50.0) |
> 7, n (%) | 55 (43.7) | 132 (52.2) | 68 (54.8) | 123 (50.0) |
Itch NRS score,a mean (SD) | 5.1 (2.5) | 5.2 (2.5) | 5.1 (2.4) | 4.9 (2.5) |
PROMIS Short Form–Sleep-Related Impairment score,b mean (SD) | 16.9 (6.1) | 17.6 (6.4) | 16.6 (6.1) | 17.4 (6.0) |
PROMIS Short Form–Sleep Disturbance score,c mean (SD) | 18.1 (5.3) | 19.0 (5.8) | 19.2 (6.2) | 19.0 (6.4) |
Prior atopic dermatitis therapy | ||||
Topical treatment, n (%) | ||||
TCI | 32 (25.4) | 60 (23.7) | 28 (22.6) | 41 (16.7) |
TCS | ||||
Low potency | 85 (67.5) | 139 (54.9) | 55 (44.4) | 98 (39.8) |
Medium potency | 49 (38.9) | 107 (42.3) | 49 (39.5) | 100 (40.7) |
High potency | 43 (34.1) | 85 (33.6) | 42 (33.9) | 61 (24.8) |
Super-high potency | 9 (7.1) | 25 (9.9) | 9 (7.3) | 17 (6.9) |
Systemic treatment, n (%) | ||||
Corticosteroid | 23 (18.3) | 45 (17.8) | 18 (14.5) | 44 (17.9) |
Immunosuppressants | ||||
Methotrexate | 1 (0.8) | 3 (1.2) | 2 (1.6) | 0 (0) |
Azathioprine | 0 (0) | 1 (0.4) | 0 (0) | 0 (0) |
Mycophenolate | 1 (0.8) | 1 (0.4) | 0 (0) | 0 (0) |
Cyclosporine | 5 (4.0) | 7 (2.8) | 2 (1.6) | 5 (2.0) |
Phototherapy, n (%) | 9 (7.1) | 23 (9.1) | 11 (8.9) | 18 (7.3) |
Dupilumab, n (%) | 1 (0.8) | 1 (0.4) | 0 (0) | 0 (0) |
BSA = body surface area; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; ITT = intention to treat; NRS = Numeric Rating Scale; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
aIn The TRuE-AD1 trial, baseline Itch NRS score was reported in 118 and 245 of patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively. In the TRuE-AD2 trial, baseline Itch NRS score was reported in 117 and 233 patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively.
bIn The TRuE-AD1 trial, baseline PROMIS Short Form–Sleep-Related Impairment score was reported in 114 and 245 of patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively. In the TRuE-AD2 trial, baseline Itch NRS score was reported in 117 and 230 patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively.
cIn The TRuE-AD1 trial, baseline PROMIS Short Form–Sleep Disturbance score was reported in 116 and 245 of patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively. In the TRuE-AD2 trial, baseline PROMIS Short Form–Sleep Disturbance score was reported in 116 and 229 patients in the vehicle cream and ruxolitinib 1.5% cream groups, respectively.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Table 13: Study Drug Exposure From TRuE-AD1 and TRuE-AD2 Trials — Vehicle-Controlled Period (Safety Population)
Exposure | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream (N = 126) | Ruxolitinib 1.5% cream (N = 253) | Vehicle cream (N = 124) | Ruxolitinib 1.5% cream (N = 246) | |
Duration of treatment (days) | ||||
Mean (SD) | ████ █ | ████ █ | ████ █ | ████ █ |
Median (range) | ████ █ | ████ █ | ████ █ | ████ █ |
Average daily amount of study drug applied (g) | ||||
Mean (SD) | ████ █ | ████ █ | ████ █ | ████ █ |
Median (range) | ████ █ | ████ █ | ████ █ | ████ █ |
Application adherence (mean %) | 96.1 | 97.9 | 97.8 | 96.7 |
SD = standard deviation.
Note: This table presents a cumulative summary of exposure during the vehicle-controlled period for patients who applied ruxolitinib cream during that period. If study drug tubes were not returned at a specific study visit, it was assumed that all the cream had been used on the skin during the relevant period. This may have occasionally led to an overestimation of study drug use in some patients.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Table 14: Study Drug Exposure From TRuE-AD1 and TRuE-AD2 Trials — Long-Term Safety Period (Long-Term Safety Evaluable Population)
Exposure | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream to ruxolitinib 1.5% cream (N = 47) | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream (N = 225) | Vehicle cream to ruxolitinib 1.5% cream (N = 52) | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream (N = 221) | |
Duration of treatment (days) | ||||
Mean (SD) | ████ █ | ████ █ | ████ █ | ████ █ |
Median (range) | ████ █ | ████ █ | ████ █ | ████ █ |
Average daily amount of study drug applied (g) | ||||
Mean (SD) | ████ █ | ████ █ | ████ █ | ████ █ |
Median (range) | ████ █ | ████ █ | ████ █ | ████ █ |
SD = standard deviation.
Note: This table presents a summary of exposure during the long-term safety period for patients in the long term safety evaluable population. If study drug tubes were not returned at a specific study visit, it was assumed that all the cream had been used on the skin during the relevant period. This may have occasionally led to an overestimation of study drug use in some participants.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
IGA Score
IGA Treatment Success
The proportion of patients experiencing IGA-TS at week 8 was the primary end point in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 38.7% (95% CI, 29.9% to 47.4%; P < 0.0001) in the TRuE-AD1 trial and 43.7% (95% CI, 35.6% to 51.8%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. The results of the prespecified subgroup analyses (Figure 9 and Figure 10 in Appendix 1) and the sensitivity analyses were consistent with the primary analysis in both trials. In both trials, the prespecified exploratory subgroup analyses showed a direction of effect consistent with the primary analysis. However, in both trials the subgroup analyses seem to suggest a higher IGA-TS response rate at week 8 in patients with a baseline IGA score of 3 (versus an IGA score of 2), patients with an EASI score greater than 7 (versus an EASI score of 7 or less), and patients in Europe (versus in North America).
Table 15: Key Efficacy Results From TruE-AD1 and TruE-AD2 Trials — Vehicle-Controlled Period (ITT Population)
Efficacy outcome | TruE-AD1 | TruE-AD2 | ||
|---|---|---|---|---|
Vehicle cream N = 126 | Ruxolitinib 1.5% cream N = 253 | Vehicle cream N = 118 | Ruxolitinib 1.5% cream N = 228 | |
IGA-TS at week 8 | ||||
Patients who experienced IGA-TS, n (%) | 19 (15.1) | 136 (53.8) | 9 (7.6) | 117 (51.3) |
OR vs. vehicle cream (95% CI) | 7.5 (4.2 to 14.0) | 15.8 (7.4 to 38.1) | ||
Difference in response rate vs. vehicle cream, % (95% CI)a | 38.7 (29.9 to 47.4) | 43.7 (35.6 to 51.8) | ||
P value | < 0.0001 | < 0.0001 | ||
EASI-75 at week 8 | ||||
Patients who experienced EASI-75, n (%) | 31 (24.6) | 157 (62.1) | 17 (14.4) | 141 (61.8) |
OR vs. vehicle cream (95% CI) | 5.2 (3.1 to 8.8) | 10.7 (5.8 to 20.7) | ||
Difference in response rate vs. vehicle cream, % (95% CI)a | 37.5 (27.8 to 47.1) | 47.4 (38.5 to 56.4) | ||
P value | < 0.0001 | < 0.0001 | ||
Change from baseline in percentage of BSA affected by AD at week 8 | ||||
Patients contributing to the analysis, n (%) | 101 (80.2) | 232 (91.7) | 101 (85.6) | 218 (95.6) |
Baseline BSA affected (%), mean (SD) | 9.2 (5.1) | 9.3 (5.2) | 9.9 (5.7) | 9.5 (5.1) |
Change from baseline (%), LSM (SE) | –3.0 (0.42) | –6.7 (0.3) | –2.2 (0.4) | –6.7 (0.3) |
Difference vs. vehicle cream (%), LSM (95% CI)b | –3.7 (–4.7 to –2.8) | –4.5 (–5.5 to –3.6) | ||
P valuec | < 0.0001 | < 0.0001 | ||
≥ 4-point improvement in Itch NRS score at week 8 | ||||
Patients contributing to analysis, Nd | 78 | 161 | 80 | 146 |
Patients with ≥ 4-point improvement in Itch NRS score from baseline, n (%) | 12 (15.4) | 84 (52.2) | 13 (16.3) | 74 (50.7) |
OR vs. vehicle cream (95% CI) | 6.0 (2.9 to 13.2) | 5.8 (2.8 to 12.7) | ||
Difference in response rate vs. vehicle cream, % (95% CI)a | 36.8 (25.7 to 47.9) | 34.4 (23.0 to 45.9) | ||
P valuec | < 0.0001 | < 0.0001 | ||
Change from baseline in POEM score at week 8 | ||||
Patients contributing to the analysis, n (%) | 99 (78.6) | 228 (90.1) | 98 (83.1) | 210 (92.1) |
Baseline score, mean (SD) | 14.9 (6.9) | 16.2 (6.3) | 15.0 (6.2) | 14.8 (6.2) |
Change from baseline, LSM (SE) | –5.2 (0.6) | –11.5 (0.4) | –4.0 (0.6) | –10.0 (0.4) |
Difference vs. vehicle cream, LSM (95% CI)b | –6.3 (–7.6 to –5.0) | –5.9 (–7.2 to –4.7) | ||
P valuec | < 0.0001 | < 0.0001 | ||
PROMIS Short Form–Sleep Disturbance score at week 8 | ||||
Patients contributing to analysis, Ne | 116 | 238 | 110 | 211 |
Patients with ≥ 6-point improvement in PROMIS Short Form–Sleep Disturbance (24-hour recall), n (%) | 11 (9.5) | 53 (22.3) | 21 (19.1) | 54 (25.6) |
OR vs. vehicle cream (95% CI) | 2.7 (1.3 to 6.1) | 1.5 (0.8 to 2.7) | ||
Difference in response rate vs. vehicle cream, % (95% CI)a | 12.8 (5.3 to 20.3) | 6.5 (–2.9 to 15.9) | ||
P value | 0.0039 | 0.2359 | ||
PROMIS Short Form–Sleep-Related Impairment score at week 8 | ||||
Patients contributing to analysis, Ne | 114 | 245 | 111 | 212 |
Patients with ≥ 6-point improvement in PROMIS Short Form–Sleep-Related Impairment (24-hour recall), n (%) | 15 (13.2) | 53 (21.6) | 15 (13.5) | 49 (23.1) |
OR vs. vehicle cream (95% CI) | 1.8 (0.9 to 3.7) | 2.0 (1.0 to 4.0) | ||
Difference in response rate vs. vehicle cream, % (95% CI)a | 8.4 (0.4 to 16.4)a | 9.6 (1.4 to 18.4)a | ||
P value | 0.0746f | 0.0472f | ||
Change from baseline in DLQI score at week 8 | ||||
Patients contributing to the analysis, n of N (%)g | 82 of 107 (76.6) | 201 of 223 (90.1) | 87 of 105 (82.9) | 185 of 202 (91.6) |
Baseline score, mean (SD) | 9.7 (5.8) | 10.0 (6.6) | 9.2 (7.0) | 8.9 (6.3) |
Change from baseline, LSM (SD) | –3.1 (0.5) | –7.5 (0.3) | –3.5 (0.4) | –6.3 (0.3) |
Difference vs. vehicle cream, LSM (95% CI)b | –4.5 (–5.6 to –3.4) | –2.8 (–3.7 to –1.8) | ||
P valuec | < 0.0001 | < 0.0001 | ||
Change from baseline in CDLQI score at week 8 | ||||
Patients contributing to the analysis, n of N (%)h | 16 of 19 (84.2) | 28 of 30 (93.3) | 11 of 13 (84.6) | 25 of 26 (96.2) |
Baseline score, mean (SD) | 7.4 (5.8) | 10.3 (6.2) | 8.9 (5.9) | 7.4 (6.3) |
Change from baseline, LSM (SD) | –3.8 (0.9) | –6.1 (0.7) | –1.3 (1.5) | –4.4 (1.0) |
Difference vs. vehicle cream, LSM (95% CI)b | –2.3 (–4.4 to –0.1) | –3.1 (–6.3 to 0.1) | ||
P valuec | 0.0378 | 0.0542 | ||
AD = atopic dermatitis; b.i.d. = twice a day; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; ITT = intention to treat; LSM = least squares mean; NRS = Numeric Rating Scale; OR = odds ratio; POEM = Patient Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; SE = standard error; vs. = versus.
Note: All analyses of the TRuE-AD2 trial were conducted in the ITT population █████████ ██ ████████ ████████ ██ ████ ███ █████ ██ ███████ █████████████ ████ ███ ████████ ███ ████████ ████ ████████ ████████ ██████ ████████ █████████ ███ █ ████████ ██ ███ ████. The baseline was defined as the last nonmissing measurement obtained before or on the day of the first application of ruxolitinib cream or vehicle cream.
aThis end point was analyzed using logistic regression, with treatment groups, baseline IGA, and region as stratification factors. Patients who had missing data were imputed as not experiencing a response.
bThis end point was analyzed using analysis of covariance, with treatment groups, region, baseline IGA, and baseline score as adjustment factors.
cThis end point was not adjusted for multiplicity.
dConducted in patients with a baseline Itch NRS score of at least 4.
eConducted in patients with a baseline score of at least 6.
fNo formal statistical testing was conducted due to prior failure in the statistical testing hierarchy (PROMIS Short Form–Sleep Disturbance score).
gConducted in patients aged 16 years or older at baseline.
hConducted in patients younger than 16 years at baseline.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Table 16: Key Efficacy Results From TRuE-AD1 and TRuE-AD2 Trials — Long-Term Safety Period (Long-Term Safety Evaluable Population)
Efficacy outcomes | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 47 | Ruxolitinib 1.5% cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 225 | Vehicle cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 49 | Ruxolitinib 1.5% cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 203 | |
Proportion of patients with an IGA score of 0 or 1 at week 52 | ||||
Patients contributing to the analysis, n (%) | 38 (80.9) | 171 (76.0) | 43 (87.8) | 171 (84.2) |
Patients with an IGA score of 0 or 1, n (%) | 28 (73.7) | 129 (75.4) | 32 (74.4) | 137 (80.1) |
Change from baseline in percentage of BSA affected at week 52 | ||||
Patients contributing to the analysis, n (%) | 38 (80.9) | 171 (76.0) | 43 (87.8) | 172 (84.7) |
Baseline BSA affected (%), mean (SD) | 5.9 (5.5) | 9.5 (5.2) | 8.9 (6.1) | 9.7 (5.1) |
Week 52 BSA affected (%), mean (SD) | 1.0 (2.0) | 1.5 (2.4) | 2.2 (3.3) | 1.4 (2.4) |
Change from baseline, mean (SD) | –4.9 (5.8) | –8.1 (4.9) | –6.8 (5.8) | –8.4 (5.0) |
Change from baseline in POEM score at week 52 | ||||
Patients contributing to the analysis, n (%) | 38 (80.9) | 170 (75.6) | 43 (87.8) | 161 (79.3) |
Baseline score, mean (SD) | 10.7 (8.3) | 16.4 (6.0) | 11.5 (6.7) | 15.0 (6.1) |
Week 52 score, mean (SD) | 3.3 (4.3) | 5.9 (6.2) | 4.7 (5.6) | 4.4 (5.5) |
Change from baseline, mean (SD) | –7.0 (8.8) | –10.6 (7.1) | –6.3 (7.3) | –10.7 (6.7) |
Change from baseline in DLQI score at week 52 | ||||
Patients contributing to the analysis, n of N (%) | 31 of 38 (81.6) | 149 of 200 (74.5) | 35 of 41 (85.4) | 147 of 180 (81.7) |
Baseline score, mean (SD) | 7.2 (6.6) | 10.0 (6.4) | 5.5 (5.8) | 9.1 (6.4) |
Week 52 score, mean (SD) | 2.3 (4.1) | 2.5 (3.7) | 2.0 (2.9) | 2.3 (4.1) |
Change from baseline, mean (SD) | –4.8 (6.7) | –7.7 (6.4) | –3.2 (4.2) | –7.3 (6.2) |
Change in CDLQI score at week 52 | ||||
Patients contributing to the analysis, n of N (%) | 7 of 9 (77.8) | 21 of 25 (84.0) | 8 of 8 (100) | 14 of 23 (60.9) |
Baseline score, mean (SD) | 3.9 (4.4) | 10.8 (6.5) | 7.3 (8.7) | 7.8 (6.3) |
Week 52 score, mean (SD) | 2.3 (4.3) | 1.9 (1.8) | 0.9 (1.4) | 1.9 (2.6) |
Change from baseline, mean (SD) | –0.4 (5.0) | –9.7 (6.3) | –6.4 (9.0) | –6.6 (6.0) |
b.i.d. = twice a day; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; POEM = Patient Oriented Eczema Measure; SD = standard deviation.
Notes: Descriptive statistics were used to present the data in this table. The baseline was defined as the last nonmissing measurement obtained before or on the day of the first application of ruxolitinib 1.5% cream twice a day in the study.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
A post hoc subgroup analysis by topical treatment history (Table 28 in Appendix 1) showed results consistent in direction with the primary analysis across subgroups (patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening).
IGA-TS was not assessed at week 52 in either trial.
IGA Score of 0 or 1
The proportion of patients who had an IGA score of 0 or 1 was a secondary end point at week 52 in both trials. At week 52, the proportions of patients experiencing an IGA score of 0 or 1 in the vehicle cream to ruxolitinib 1.5% cream group and the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group were 73.7% and 75.4%, respectively, in the TRuE-AD1 trial and 74.4% and 80.1%, respectively, in the TRuE-AD2 trial.
In a post hoc subgroup analysis by topical treatment history (Table 29 in Appendix 1), the vehicle cream to ruxolitinib 1.5% cream group experienced a similar IGA score of 0 or 1 response rate at week 52 as the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group across patients who had received TCS only, TCI only, and TCS and TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening.
EASI Score
The proportion of patients experiencing EASI-75 at week 8 was a key secondary end point and was adjusted for multiplicity in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 37.5% (95% CI, 27.8% to 47.1%; P < 0.0001) in the TRuE-AD1 trial and 47.4% (95% CI, 38.5% to 56.4%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. In both trials, the results of the sensitivity analyses were consistent with those of the primary analysis. The results of the prespecified subgroup analyses were consistent in direction with the primary analysis (Figure 11 and Figure 12 in Appendix 1). In both trials, the subgroup analyses seem to suggest a higher EASI-75 response rate at week 8 in patients with a baseline EASI score greater than 7 (versus an EASI score of 7 or less).
A post hoc subgroup analysis by topical treatment history (Table 28 in Appendix 1) showed results consistent in direction with the primary analysis across subgroups (patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening).
EASI-75 was not assessed at week 52 in either trial.
Percentage of BSA Affected by AD
The change from baseline in the percentage of BSA affected by AD at week 8 and at week 52 were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –3.7% (95% CI, –4.7% to –2.8%) in the TRuE-AD1 trial and –4.5% (95% CI, –5.5% to –3.6%) in the TRuE-AD2 trial.
At week 52, the change from baseline in the percentage of BSA affected by AD in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group was –8.1% (SD = 4.9%) and –8.4% (SD = 5.0%) in the TruE-AD1 and TruE-AD2 trials, respectively. The vehicle cream to ruxolitinib 1.5% cream group experienced a percentage of BSA affected by AD (TRuE-AD1 trial: 1.0% [SD = 2.0%]; TRuE-AD2 trial: 2.2% [SD = 3.3%]) similar to that in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group (TRuE-AD1 trial: 1.5% [SD = 2.4%]; TRuE-AD2 trial: 1.4% [SD = 2.4%]) at week 52 in both trials. In a post hoc subgroup analysis by topical treatment history (Table 30 in Appendix 1), the vehicle cream to ruxolitinib 1.5% cream group experienced a similar total percentage of BSA affected by AD at week 52 as the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group across patients who had received TCS only, TCI only, and TCS and TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening.
Itch NRS Score
The proportion of patients with at least 4 points of improvement in Itch NRS from baseline at week 8 (among patients with a baseline score of at least 4 [vehicle cream: n = 78 in the TRuE-AD1 trial, n = 80 in the TRuE-AD2 trial; ruxolitinib: n = 161 in the TRuE-AD1 trial, n = 146 in the TRuE-AD2 trial]) was a key secondary end point and was adjusted for multiplicity in both trials. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 36.8% (95% CI, 25.7% to 47.9%; P < 0.0001) in the TRuE-AD1 trial and 34.4% (95% CI, 23.0% to 45.9%; P < 0.0001) in the TRuE-AD2 trial, both of which were in favour of ruxolitinib 1.5% cream. A post hoc subgroup analysis by topical treatment history (Table 28 in Appendix 1) showed results consistent in direction with the primary analysis across subgroups (patients who had received TCS only, TCI only, or TCS plus TCI regardless of treatment time frame, as well as patients who had received topical treatment in the 30 days before screening).
This end point was not assessed at week 52 in either trial.
Patient Oriented Eczema Measure
The change from baseline in POEM score at week 8 and at week 52 were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –6.3 (95% CI, –7.6 to –5.0) in the TRuE-AD1 trial and –5.9 (95% CI, –7.2 to –4.7) in the TRuE-AD2 trial.
At week 52, the change from baseline in POEM score in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group was –10.6 (SD = 7.1) and –10.7 (SD = 6.7) in the TruE-AD1 trial and TruE-AD2 trial, respectively. The vehicle cream to ruxolitinib 1.5% cream group experienced a mean POEM score (TRuE-AD1 trial: 3.3 [SD = 4.3]; TRuE-AD2 trial: 4.7 [SD = 5.6]) similar to that in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group (TRuE-AD1 trial: 5.9 [SD = 6.2]; TRuE-AD2 trial: 4.4 [SD = 5.5]) at week 52 in both trials.
PROMIS Short Form–Sleep Disturbance
The proportion of patients with at least 6 points of improvement in the PROMIS Short Form–Sleep Disturbance score (24-hour recall) from baseline at week 8 (among patients with a baseline score of at least 6 [vehicle cream: n = 116 in the TRuE-AD1 trial, n = 110 in the TRuE-AD2 trial; ruxolitinib: n = 238 in the TRuE-AD1 trial, n = 211 in the TRuE-AD2 trial]) was a key secondary end point and was adjusted for multiplicity in both trials. The between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 12.8% (95% CI, 5.3% to 20.3%; P = 0.0039) in the TRuE-AD1 trial, in favour of ruxolitinib 1.5% cream. In the TRuE-AD2 trial, the between-group difference was 6.5% (95% CI, –2.9% to 15.9%; P = 0.2359), which did not favour either study intervention. No superiority testing was conducted for the efficacy end point lower in the statistical testing hierarchy (i.e., proportion of patients with ≥ 6 points of improvement in the PROMIS Short Form–Sleep-Related Impairment score at week 8).
This end point was not assessed at week 52 in either trial.
PROMIS Short Form–Sleep-Related Impairment
The proportion of patients with at least 6 points of improvement in the PROMIS Short Form–Sleep-Related Impairment score (24-hour recall) from baseline at week 8 (among patients with a baseline score of at least 6 [vehicle cream: n = 114 in the TRuE-AD1 trial, n = 111 in the TRuE-AD2 trial; ruxolitinib: n = 245 in the TRuE-AD1 trial, n = 212 in the TRuE-AD2 trial]) was a key secondary end point in both trials. This end point was included in the statistical testing hierarchy, but no superiority testing was conducted due to prior failure in the hierarchy. At week 8, the between-group difference comparing ruxolitinib 1.5% cream with vehicle cream was 8.4% (95% CI, 0.4% to 16.4%) in the TRuE-AD1 trial and 9.6% (95% CI, 1.4% to 18.4%) in the TRuE-AD2 trial.
This end point was not assessed at week 52 in either trial.
DLQI and CDLQI Scores
Change From Baseline in DLQI
The change in DLQI score from baseline at week 8 and at week 52 (among patients aged 16 years or older [vehicle cream: n = 82 in the TRuE-AD1 trial, n = 87 in the TRuE-AD2 trial; ruxolitinib: n = 201 in the TRuE-AD1 trial, n = 185 in the TRuE-AD2 trial]) were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –4.5 (95% CI, –5.6 to –3.4) in the TRuE-AD1 trial and –2.8 (95% CI, –3.7 to –1.8) in the TRuE-AD2 trial. The results of the responder analysis (proportion of patients experiencing at least a 4-point improvement in DLQI score) at week 8 were similarly in favour of ruxolitinib cream in both trials ██████████ ████ █████ █ █████ ███ ███ ████ ██ █████ ██████████ █████████ ████ █████ █ █████ ███ ███ ████ ██ █████ ██████████.
At week 52, the mean change from baseline in DLQI score in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group was –7.7 (SD = 6.4) and –7.3 (SD = 6.2) in the TruE-AD1 trial and TruE-AD2 trial, respectively. The vehicle cream to ruxolitinib 1.5% cream group experienced a mean DLQI score (TRuE-AD1 trial: 2.3 [SD = 4.1]; TRuE-AD2 trial: 2.0 [SD = 2.9]) similar to that in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group (TRuE-AD1 trial: 2.5 [SD = 3.7]; TRuE-AD2 trial: 2.3 [SD = 4.1]) at week 52 in both trials.
Change From Baseline in CDLQI
The change in CDLQI score from baseline at week 8 and at week 52 (among patients younger than 16 years [vehicle cream: n = 16 in the TRuE-AD1 trial, n = 11 in the TRuE-AD2 trial; ruxolitinib: n = 28 in the TRuE-AD1 trial, n = 25 in the TRuE-AD2 trial]) were secondary end points in both trials and were not adjusted for multiplicity in either trial. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –2.3 (95% CI, –4.4 to –0.1) in the TRuE-AD1 trial and –3.1 (95% CI, –6.3 to –0.1) in the TRuE-AD2 trial.
At week 52, the mean change from baseline in CDLQI score in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group was –9.7 points (SD = 6.3) and –6.6 points (SD = 6.0) in the TruE-AD1 trial and TruE-AD2 trial, respectively. The vehicle cream to ruxolitinib 1.5% cream group experienced a mean CDLQI score (TRuE-AD1 trial: 2.3 [SD = 4.3]; TRuE-AD2 trial: 0.9 [SD = 1.4]) similar to that in the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group (TRuE-AD1 trial: 1.9 [SD = 1.8]; TRuE-AD2 trial: 1.9 [SD = 2.6]) at week 52 in both trials.
Harms
Harms outcomes from the TRuE-AD1 and TRuE-AD2 studies are summarized in Table 17 (vehicle-controlled period) and Table 18 (long-term safety period).
Vehicle-Controlled Period
Adverse Events
The proportion of patients who reported at least 1 TEAE was lower in the ruxolitinib 1.5% cream group than in the vehicle cream group in both trials (TRuE-AD1 trial: 29.2% versus 34.9%; TRuE-AD2: 23.6% versus 31.5%). The difference appears to be partly attributable to including AD as a harm. The most common TEAEs in the ruxolitinib 1.5% cream group were nasopharyngitis (3.6% in the TRuE-AD1 trial; 1.6% in the TRuE-AD2 trial), upper respiratory tract infection (3.6% in the TRuE-AD1 trial; 1.2% in the TRuE-AD2 trial), and headache (2.4% in the TRuE-AD1 trial; 2.0% in the TRuE-AD2 trial).
Serious AEs
Serious TEAEs were reported in 2 patients (1.6%) in the vehicle cream group and 2 patients (0.8%) in the ruxolitinib 1.5% cream group in the TRuE-AD1 trial. In the TRuE-AD2 trial, no patient reported a serious TEAE in the vehicle cream group and 1 patient (0.4%) reported a serious TEAE in the ruxolitinib 1.5% cream group.
Withdrawals Due to AEs
Study treatment withdrawal due to TEAEs was reported in 5 patients (4.0%) in the vehicle cream group and 3 patients (1.2%) in the ruxolitinib 1.5% cream group in the TRuE-AD1 trial and in 3 patients (2.4%) in the vehicle cream group and 1 patient (0.4%) in the ruxolitinib 1.5% cream group in the TRuE-AD2 trial. TEAEs leading to discontinuation of ruxolitinib cream treatment included papule, generalized pruritus, urticaria (1 patient [0.4%] each in the TRuE-AD1 trial), as well as cerebrovascular accident (1 patient [0.4%] in the TRuE-AD2 trial).
Mortality
No deaths were reported during the vehicle-controlled period in either trial.
Notable Harms
No notable harms were identified.
Long-Term Safety Period
Adverse Events
The proportion of patients who reported at least 1 TEAE in the ruxolitinib 1.5% cream group was higher than in the vehicle cream group in the TRuE-AD1 trial (ruxolitinib: 53.5%; vehicle cream: 48.9%) but was lower than in the vehicle cream group in the TRuE-AD2 trial (ruxolitinib: 54.3%; vehicle cream: 65.4%). Upper respiratory tract infection, nasopharyngitis, and headache were the most common TEAEs in the ruxolitinib 1.5% cream group, consistent with the results from the vehicle-controlled period.
Serious AEs
Serious TEAEs were reported in 1 patient (2.1%) in the vehicle cream group and 3 patients (1.3%) in the ruxolitinib 1.5% cream group in the TRuE-AD1 trial. In the TRuE-AD2 trial, no patient reported a serious TEAE in the vehicle cream group and 3 patients (1.4%) reported a serious TEAE in the ruxolitinib 1.5% cream group.
Withdrawals Due to AEs
No patient withdrew from the study treatment due to TEAEs in either study.
Mortality
No deaths were reported during the long-term safety period in either trial.
Notable Harms
No notable harms were identified.
Table 17: Harms Results From TRuE-AD1 and TRuE-AD2 Trials — Vehicle-Controlled Period (Safety Population)
Adverse event | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream b.i.d. N = 126 | Ruxolitinib 1.5% cream b.i.d. N = 253 | Vehicle cream b.i.d. N = 124 | Ruxolitinib 1.5% cream b.i.d. N = 246 | |
TEAEs | ||||
Patients with ≥ 1 TEAE, n (%) | 44 (34.9) | 74 (29.2) | 39 (31.5) | 58 (23.6) |
Most common TEAEs,a n (%) | ||||
Application site pain | 5 (4.0) | 2 (0.8) | 8 (6.5) | 2 (0.8) |
Atopic dermatitisb | 7 (5.6) | 2 (0.8) | 3 (2.4) | 0 (0) |
Headache | 5 (4.0) | 6 (2.4) | 0 (0) | 5 (2.0) |
Application site pruritus | 2 (1.6) | 0 (0) | 5 (4.0) | 1 (0.4) |
Nasopharyngitis | 2 (1.6) | 9 (3.6) | 0 (0) | 4 (1.6) |
Upper respiratory tract infection | 4 (3.2) | 9 (3.6) | 1 (0.8) | 3 (1.2) |
Pruritus | 4 (3.2) | 3 (1.2) | 0 (0) | 0 (0) |
Serious TEAEs | ||||
Patients with ≥ 1 serious TEAE, n (%) | 2 (1.6) | 2 (0.8) | 0 (0) | 1 (0.4) |
Study treatment discontinuation due to a TEAE | ||||
Patients who discontinued study treatment due to a TEAE, n (%) | 5 (4.0) | 3 (1.2) | 3 (2.4) | 1 (0.4) |
Most common reason for discontinuation,c n (%) | ||||
Atopic dermatitisb | 4 (3.2) | 1 (0.4) | 0 (0) | 0 (0) |
Application site pain | 0 (0) | 0 (0) | 2 (1.6) | 0 (0) |
Deaths | ||||
Deaths, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
b.i.d. = twice a day; TEAE = treatment-emergent adverse event.
aPresent in ≥ 3% of patients.
bReported term: worsening of atopic dermatitis.
cPresent in ≥ 2 patients.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Table 18: Harms Results From TRuE-AD1 and TRuE-AD2 Trials — Long-Term Safety Period (Long-Term Safety Evaluable Population)
Adverse event | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 47 | Ruxolitinib 1.5% cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 225 | Vehicle cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 52 | Ruxolitinib 1.5% cream b.i.d. to ruxolitinib 1.5% cream b.i.d. N = 221 | |
TEAEs | ||||
Patients with ≥ 1 TEAE, n (%) | 23 (48.9) | 120 (53.5) | 34 (65.4) | 120 (54.3) |
Most common TEAEs,a n (%) | ||||
Upper respiratory tract infection | 2 (4.3) | 27 (12.0) | 5 (9.6) | 16 (7.2) |
Nasopharyngitis | 6 (12.8) | 19 (8.4) | 8 (15.4) | 17 (7.7) |
Headache | 3 (6.4) | 10 (4.4) | 1 (1.9) | 3 (1.4) |
Neutropenia | 3 (6.4) | 5 (2.2) | 1 (1.9) | 1 (0.5) |
Bronchitis | 2 (4.3) | 5 (2.2) | 3 (5.8) | 6 (2.7) |
Influenza | 2 (4.3) | 8 (3.6) | 5 (9.6) | 3 (1.4) |
Serious TEAEs | ||||
Patients with ≥ 1 serious TEAE,b n (%) | 1 (2.1) | 3 (1.3) | 0 (0) | 3 (1.4) |
Study treatment discontinuation due to a TEAE | ||||
Patients who discontinued study treatment due to a TEAE, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Deaths | ||||
Deaths, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
b.i.d. = twice a day; TEAE = treatment-emergent adverse event.
aPresent in ≥ 4% of patients.
bNone of the reported serious TEAEs was present in more than 1 patient.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Critical Appraisal
Internal Validity
The TRuE-AD1 and TRuE-AD2 trials were randomized, double-masked, vehicle-controlled trials. The method used for the initial randomization and re-randomization across trials consisted of a central interactive response technology system, which enabled concealment of the allocation sequence. The baseline demographics and disease characteristics were balanced in general, with the exception of a few characteristics — including AD flares; the proportion of patients with an EASI score of 7 or less; and the proportion of patients with prior TCI, low-potency TCS, and high-potency TCS use — which were imbalanced between treatment groups in at least 1 trial. Of these imbalances, the clinical experts consulted by CDA-AMC noted that the higher mean number of AD flares in the vehicle cream group in the TRuE-AD1 trial could potentially have introduced bias in favour of ruxolitinib cream because a higher number of AD flares is indicative of more severe disease, which tends to be less responsive to treatment. However, such imbalance was not observed in the TRuE-AD2 trial, which showed similar results to the TRuE-AD1 trial. The clinical experts noted that the impact of other imbalances on the study results is likely to be insignificant.
The masking of patients and study personnel was appropriately maintained. However, given that a vehicle cream was used in the trials, there is a small potential that patients may have become unmasked or aware of their assignments through improvement in their symptoms over the study period, which could have introduced bias to the results in favour of ruxolitinib cream for patient-reported outcomes (Itch NRS, POEM, PROMIS Short Form–Sleep Disturbance, PROMIS Short Form–Sleep-Related Impairment, DLQI, and CDLQI scores). IGA and EASI were assessed by the investigators; these instruments involved some level of subjectivity, resulting in some potential for bias, but the risk is probably low. The risk of bias in the measurement of outcome is also low for the percentage of BSA affected by AD, which is an objective measure.
Efficacy analyses in the TRuE-AD1 trial were conducted in the ITT population, which is the ideal approach to assess the effect of assignment to the intervention. Efficacy analyses in the TRuE-AD2 trial were conducted in the ITT population █████████ ██████████ ████████ ██ ████ ███ █████ ██ ███████ █████████████ ████ ███ ████████ ██ ███ ████; 4.8% and 7.3% of randomized patients were excluded from the efficacy analysis of the vehicle cream and ruxolitinib 1.5% cream groups, respectively. The exclusion of randomized patients from the analysis set could potentially impact randomization and hence study results; however, given that the proportion of excluded patients was small and was not notably different between the treatment groups, and given that the baseline patient characteristics in this analysis set were also, in general, balanced between the treatment groups, the impact on the study results is likely small. Responder analysis of Itch NRS, PROMIS Short Form–Sleep Disturbance, and PROMIS Short Form–Sleep-Related Impairment scales were conducted in patients with specific baseline scores, which could affect randomization. The proportion of patients excluded from the PROMIS Short Form responder analyses was small (< 10%) and was balanced between treatment groups, and thus the impact on the study results is likely small. However, a large proportion of patients (≥ 30% in each treatment group) were excluded from the Itch NRS responder analysis, which could potentially have had a more substantial impact on randomization. The extent and direction of the resulting bias is, however, unclear. Analyses of change from baseline in DLQI and CDLQI scores were both conducted in a subset of patients (adults only and adolescents only, respectively), with no stratification involved; therefore, there is a potential that the prognostic balance provided by the randomization was not fully preserved, particularly for the CDLQI analysis, which was informed by a small proportion of the initially randomized population.
Study treatment discontinuation in the vehicle-controlled period was notably higher in the vehicle cream group than in the ruxolitinib 1.5% cream group in both trials (TRuE-AD1 trial: 19.8% versus 8.3%; TRuE-AD2 trial: 14.5% versus 4.5%), primarily due to withdrawal by patient, which could potentially lead to attrition bias in favour of ruxolitinib cream. In the primary analysis of binary outcomes, patients with missing data were imputed as not experiencing a response, which is considered a conservative approach. Sensitivity analyses were conducted for IGA-TS and EASI-75 outcomes to assess the impact of different imputation methods; these analyses showed results consistent with the primary analysis. Missing data for continuous secondary end points (change from baseline in percentage of BSA affected, and POEM, DLQI, and CDLQI scores) were implicitly imputed using mixed models for repeated measures under the missing-at-random assumption. It is unclear if this assumption holds because most treatment discontinuations were primarily a result of withdrawal of consent by the patient and the reasons for withdrawal were not documented. No sensitivity analyses were conducted to assess the impact of imputation on these outcomes. In addition, the end point of change from baseline in CDLQI score was based on a small sample size in both treatment groups, which could lead to instability of the treatment effect estimates.
A hierarchal testing procedure was appropriately used to account for multiplicity in the primary and key secondary end points. Analyses of other secondary end points were not part of the statistical hierarchy and are at an increased risk of type I error (false-positive results). Subgroup analyses by age, sex, race, baseline IGA and EASI scores, and region were specified a priori, and the subgroup analysis by previous treatment history was conducted on a post hoc basis. There was a lack of sample size consideration and control for multiplicity for these subgroup analyses, which preclude the formation of definitive conclusions on subgroup effects.
All patients who entered the long-term safety period received ruxolitinib cream treatment. In the absence of a control arm, it could not be determined whether the observed treatment effects could be attributed to ruxolitinib cream treatment alone. In addition, because patients could only enter the long-term safety period after completing the vehicle-controlled period, there is a risk of selection bias given that patients who better tolerated the treatment or perceived the treatment as benefiting them were more likely to enter the long-term safety period. Approximately 20% of patients were lost to follow-up in both trials, which adds to the uncertainty of the results.
The primary and key secondary end points were assessed using instruments that have evidence for validity in patients with AD (IGA, EASI, Itch NRS, PROMIS Short Form–Sleep Disturbance and PROMIS Short Form–Sleep-Related Impairment scales), though evidence for these PROMIS Short Form scales was established based on the TRuE-AD1 and TRuE-AD2 trial populations. EASI-75 responder analysis was based on a threshold of a 75% reduction in EASI score from baseline, which is a commonly accepted threshold in the treatment of AD among clinicians, according to the clinical experts. Responder analyses of Itch NRS and PROMIS Short Form–Sleep Disturbance and PROMIS Short Form–Sleep-Related Impairment scores were based on thresholds of 4-point and 6-point improvements in score, respectively, which are consistent with literature-identified MID estimates for these instruments. Evidence for the validity of other instruments, including POEM, DLQI, and CDLQI, were also identified in the literature. MID estimates for these instruments were available; the estimates for POEM and CDLQI were AD specific, but the estimates for DLQI were not.
External Validity
The clinical experts consulted for the purpose of this review anticipated that, in most cases, ruxolitinib cream would be used for the treatment of patients with mild to moderate AD that is inadequately controlled with TCS and/or TCI treatment or when these treatments are inadvisable, which aligns with the Health Canada indication. However, the inclusion and exclusion criteria for the TRuE-AD1 and TRuE-AD2 trials did not restrict entry based on prior experience with TCS or TCI treatment. The sponsor submitted post hoc subgroup analyses in patients with a recent history of TCS and/or TCI treatment as supporting evidence. It is unclear if this subgroup population would adequately reflect the patient population expected to receive ruxolitinib cream in clinical practice according to clinical expert input. One clinical expert noted that adequate (or inadequate) disease response to, and patient eligibility for, TCS and TCI are determined largely based on clinical judgment in the absence of universal definitions. This clinical expert felt that this subgroup population (patients with a recent history of TCS and/or TCI treatment) is reasonably reflective of the patients expected to receive ruxolitinib cream in practice. The other clinical expert noted that having received TCS and/or TCI treatment within the 30 days before screening does not establish inadequate control with TCS and/or TCI and that the lack of evidence for the use of ruxolitinib cream in patients for whom TCS and TCI treatments failed is a concern. The clinical experts also commented that the inclusion criteria of the TRuE-AD1 and TRuE-AD2 trials with respect to age (12 years and older), IGA score (2 and 3, corresponding to mild and moderate severity, respectively), and percentage of BSA affected (3% to 20%) were in general reflective of the patient population eligible for ruxolitinib cream in clinical practice, although they anticipated that patients with an IGA score of 4 (severe disease) and patients with 0% to 3% BSA affected, who were not included in the trials, would be candidates for ruxolitinib cream treatment and that patients would likely be eligible for ruxolitinib cream regardless of the duration of their AD diagnosis. As well, a recent history of unstable AD or a history of AD treatment, which were exclusion criteria of the trials, would not preclude eligibility for ruxolitinib cream in clinical practice, according to the clinical experts.
The clinical experts noted that the baseline characteristics of the trial population were in general reflective of the patient population eligible for ruxolitinib cream in Canada, with the exception that the trial population had a higher proportion of patients who are Black or African American and a lower proportion of patients who are Asian. The clinical experts additionally noted that the proportions of patients with mild disease (IGA score of 2), previous TCI treatment, and previous TCS treatment of medium, high, or super-high potency in the trials appear to be lower than expected in clinical practice.
Patients in the clinical trials were instructed to apply ruxolitinib cream on affected skin areas, up to 20% BSA for each application, twice daily for 8 weeks and on an as-needed basis in the long-term safety period. This is consistent with the product monograph, which recommends twice-daily application and for patients to be re-evaluated by a health care provider if the signs and symptoms do not improve within 8 weeks. The clinical experts noted that, in clinical practice, some patients who respond to ruxolitinib cream are expected to taper to as-needed use before 8 weeks, while others are expected to require twice-daily application long term. The product monograph recommends that ruxolitinib cream not be used in combination with other JAK inhibitors, biological immunomodulators, or potent immunosuppressants but does not restrict the use of ruxolitinib cream in combination with other topical treatments for AD. Although the pivotal trials assessed the use of ruxolitinib cream monotherapy only, the clinical experts expected that ruxolitinib cream would also be prescribed in combination with TCS (applied to different affected areas) in clinical practice.
The efficacy outcomes assessed in the study were of clinical importance to patients and clinicians and included the severity and extent of AD, itch symptoms, sleep quality, and HRQoL. The clinical experts noted that the percentage of BSA affected, EASI, DLQI, and Itch NRS are assessed in clinical practice, although DLQI and EASI scores are in general only determine for reimbursement purposes. IGA-TS and POEM are commonly used in clinical trials but not in clinical practice. According to the clinical experts, the duration of follow-up of 8 weeks in the vehicle-controlled period was adequate for the assessment of the efficacy of ruxolitinib; however, the clinical experts felt that a longer follow-up, beyond 52 weeks, would be required to capture the long-term safety of ruxolitinib cream, including potential rare AEs, given that AD is a lifelong condition requiring treatment over many years.
The TRuE-AD1 and TRuE-AD2 trials — the phase III RCT evidence submitted for this review — are both vehicle-controlled studies. The absence of head-to-head evidence comparing ruxolitinib cream with relevant comparators (systemic immunosuppressants, biologics, and JAK inhibitors) in patients with moderate AD, is an evidence gap in the treatment of AD. Another gap is the absence of evidence for ruxolitinib cream in combination with other topical therapies. In addition, the generalizability of the study results to the adolescent patient population in clinical practice could potentially be limited by the small proportion of adolescents enrolled in the trials (approximately 20%). A similarly small proportion of adolescent patients has been observed in other clinical trials for AD treatments.11,12
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:94
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was based on the presence or absence of an important effect as informed by thresholds identified in the literature (for change in POEM, DLQI, and CDLQI scores from baseline) and through clinical expert input (for proportion of patients experiencing IGA-TS, IGA score of 0 or 1, EASI-75, at least a 4-point improvement in Itch NRS score from baseline, at least a 6-point improvement in PROMIS Short Form–Sleep Disturbance score from baseline, at least a 6-point improvement in PROMIS Short Form–Sleep-Related Impairment score from baseline, SAEs, and change from baseline in percentage of BSA affected by AD).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for ruxolitinib 1.5% cream versus vehicle cream in patients with AD.
Long-Term Extension Studies
Both the TRuE-AD1 and TRuE-AD2 trials had a 44-week long-term safety period assessing the efficacy and safety of ruxolitinib cream in patients who completed the 8-week vehicle-controlled period. Evidence from the long-term safety period was submitted as part of the pivotal trials and was summarized in the Systematic Review section.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the indirect evidence comparing ruxolitinib 1.5% cream with alternative treatments for AD. An ITC was required to address a gap in pivotal and RCT evidence in the absence of studies directly comparing ruxolitinib 1.5% cream with treatments for AD.
Description of the Sponsor-Submitted ITC
One ITC conducted by the sponsor was included in this report.16 The efficacy of ruxolitinib 1.5% cream was compared against dupilumab, upadacitinib 15 mg and 30 mg, and abrocitinib 100 mg and 200mg in patients with moderate AD, defined as having an IGA score of 3, an EASI score of 16 or higher, and a percentage of BSA affected of 10% or higher, via a frequentist NMA. The outcomes assessed included the proportion of patients experiencing EASI-75, an IGA score of 0 or 1, and an improvement in Itch NRS score of 4 or more.
Objectives
The primary objective of the ITC was to determine the comparative efficacy of ruxolitinib 1.5% cream (as assessed in the TRuE-AD1 and TRuE-AD2 trials) versus 1 or more comparators in the treatment of AD in patients aged 12 years and older.
Study Selection Methods
An SLR was conducted to identify the studies for inclusion in the ITC. The selection criteria for the SLR are summarized in Table 19. Relevant studies were identified by searching databases up to February 2023. The databases searched included Excerpta Medica dataBASE (Embase), Medical Literature Analysis and Retrieval System Online (MEDLINE), and Cochrane Central Register of Controlled Trials (CENTRAL). Other materials were identified via handsearch of the US National Institutes of Health Clinical Trial Registry, National Institute for Health and Care Excellence committee papers, CADTH Recommendation reports, and European Medicines Agency European Public Assessment Reports.
Article screening was performed independently by 2 reviewers, with a third reviewer resolving disagreements. Data extraction was performed by 1 reviewer, followed by quality control completed by a second reviewer. Quality assessment of the selected studies was conducted by 1 reviewer, followed by quality control by a second reviewer, to assess the risk of bias using the National Institute for Health and Care Excellence risk of bias tool.
Table 19: Study Selection Criteria and Methods for ITC Submitted by the Sponsor
Characteristics | Selection criteria |
|---|---|
Population | Adult and adolescent patients (≥ 12 years) with AD Potential subgroups of interest:
|
Intervention | Regimens approved (or anticipating approval in near future) by Health Canada or recommended by CDA-AMC, as monotherapy, in combination with one another, or with concomitant background topical treatment:
|
Comparator |
|
Outcome | Efficacy outcomes:
Patient-reported outcomes:
Safety outcomes:
Time points included the primary end point of each trial, the time point closest to week 8, and the end of study to capture the longest follow-up outcome data |
Study designs | Phase III or IV RCTs |
Publication characteristics | Only studies published in English |
Exclusion criteria |
|
Databases searched | Excerpta Medica dataBASE (Embase), Medical Literature Analysis and Retrieval System Online (MEDLINE), and Cochrane Central Register of Controlled Trials (CENTRAL) |
Selection process | Abstract and full-text selection of studies was performed by 2 independent investigators, and data extraction of the included studies was performed by a single reviewer (quality checked by a senior reviewer). Any unresolved discrepancies that occurred between the 2 investigators were resolved by involving a third investigator and reaching a consensus. |
Data extraction process | One reviewer extracted data on study characteristics, interventions, patient characteristics, and outcomes for the final list of included studies, followed by quality control by a senior independent reviewer. |
Quality assessment | One reviewer assessed the study quality, followed by quality control by a senior independent reviewer. The NICE risk of bias tool was used to assess the risk of bias in the included clinical trials. |
AD = atopic dermatitis; AE = adverse event; BSA = body surface area; CDA-AMC = Canada’s Drug Agency; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; ISGA = Investigator’s Static Global Assessment; JAK = Janus kinase; NICE = National Institute for Health and Care Excellence; NRS = Numeric Rating Scale; PDE4 = phosphodiesterase type 4; PROMIS = Patient-Reported Outcomes Measurement Information System; RCT = randomized controlled trial; SC = subcutaneous; SCORAD = SCORing Atopic Dermatitis; vIGA-AD = validated Investigator’s Global Assessment for Atopic Dermatitis.
Sources: Network meta-analysis technical report.16 Details included in the table are from the sponsor’s summary of clinical evidence.15
ITC Analysis Methods
Analysis Model
The sponsor selected the frequentist approach as the main analysis due to the anticipated challenges in model fitting and convergence associated with a small number of comparators and the potential for sparsity of the resulting evidence network. The frequentist NMA was conducted using a penalized likelihood NMA. A penalized likelihood NMA was used to attempt to reduce the bias of the maximum likelihood estimate known to occur in the presence of rare events such as an IGA score of 0 or 1 in certain treatment arms. Model fit was assessed based on the Akaike information criterion and residual deviance.
A fixed-effect Bayesian NMA was also conducted where feasible. The feasibility of a Bayesian NMA was evaluated using conventional diagnostics including model convergence (r-hat < 1.01) and an effective sample size per chain (> 400 per parameter). Where these diagnostic measures were inadequate, models were refitted using more iterations and burn-ins. In cases where a Bayesian NMA was deemed infeasible, the results are not presented.
Assessment of Consistency
Consistency was not assessed in the absence of a closed loop connecting ruxolitinib 1.5% cream and comparators in the evidence network.
Feasibility Assessment
A feasibility assessment of the full trial population was conducted and included a review of the study characteristics, the patient eligibility criteria, the baseline patient characteristics, the outcome definitions and time points, and the baseline risk associated with the studies identified by the SLR. Based on the results of the feasibility assessment, it was deemed that an analysis with the full trial populations would not be feasible as the trial eligibility criteria (age, severity, and prior treatment) and population baseline characteristics (AD duration and severity) differed substantially between the identified trials. Therefore, a feasibility assessment was repeated for the moderate-only subgroup (as defined by an IGA score of 3, an EASI score of ≥ 16, and ≥ 10% BSA affected). The results suggested that an NMA using the moderate-only subgroup data was feasible for outcomes including IGA score of 0 or 1, EASI-75, and Itch NRS-4. Therefore, studies without results for subgroups consisting of solely of patients with moderate severity disease were excluded. Further, the identified studies used placebo or placebo in combination with TCS as the comparators. Because a larger placebo effect is typically seen across all considered efficacy outcomes in comparator trials where placebo is combined with TCS or other prescription topicals, the authors of the sponsor-conducted NMA determined that it was not appropriate to combine placebo and placebo plus TCS comparators or to compare against placebo plus TCS. Therefore, trials without a link to the ruxolitinib 1.5% cream through the shared placebo arm in the network were also excluded from the analysis. An adolescent-only trial was excluded from the main analysis to improve the comparability of study populations.
Outcomes
The choice of outcomes for the NMA was informed by data availability and relevance for payers and reimbursement agencies. The outcomes included in the NMA included IGA score of 0 or 1, EASI-75, and Itch NRS-4. Insufficient data were available to conduct analyses of safety outcomes as the active comparator trials did not report AE rates specific to the subgroup of interest. Given the lack of reporting for the specific subgroup, an NMA of safety outcomes was not conducted.
Construction of Nodes
Each drug and respective dose was handled as a separate node, except for sensitivity analysis 2 in which pooled dosing was tested for abrocitinib and upadacitinib.
Sensitivity Analyses
Two sensitivity analyses were conducted to assess the impact of the excluded adolescent-only trial in the main analysis and of pooling doses, where relevant:
moderate-only subgroup (IGA = 3, EASI ≥ 16, BSA affected ≥ 10%), including adolescent-only trials (LIBERTY AD ADOL trial)
moderate-only subgroup (IGA = 3, EASI ≥ 16, BSA affected ≥ 10%), including pooled doses for abrocitinib (100 mg and 200 mg) and upadacitinib (15 mg and 30 mg).
Table 20: ITC Analysis Methods
Methods | Ruxolitinib 1.5% cream vs. dupilumab 300 mg | Ruxolitinib 1.5% cream vs. abrocitinib 100 mg and 200 mg | Ruxolitinib 1.5% cream vs. upadacitinib 15 mg and 30 mg |
|---|---|---|---|
Main analysis method | Frequentist PL-NMAa | ||
Assessment of model fit | Akaike information criterion and residual deviance | ||
Assessment of consistency | Not conducted because there is no closed loop connecting ruxolitinib 1.5% cream and relevant comparators | ||
Outcomes |
| EASI-75 |
|
Follow-up time points | Week 8 for the TRuE-AD trial population | ||
Week 16 for dupilumab studies | Week 12 for abrocitinib studies | Week 16 for upadacitinib studies | |
Construction of nodes | Each drug and respective dose was a separate node | ||
Sensitivity analyses |
| ||
BSA = body surface area; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; ITC = indirect treatment comparison; NRS = Numeric Rating Scale; NRS-4 = at least a 4-point improvement in Numeric Rating Scale; PL-NMA = penalized likelihood network meta-analysis; vs. = versus.
aAn additional analysis with a fixed-effect Bayesian network meta-analysis was determined to be feasible for EASI-75 and Itch NRS-4 outcomes. Model fit was assessed based on the deviance information criterion and the total residual deviance.
Sources: NMA technical report.16 Details included in the table are from the sponsor’s summary of clinical evidence.15
Results of ITC
Summary of Included Studies
Twenty-three unique studies were identified by the SLR, 12 of which reported outcomes for moderate-only population subgroups (IGA = 3, EASI ≥ 16, and ≥ 10% BSA affected) and were considered for inclusion in the NMA, including 2 studies of ruxolitinib 1.5% cream (TRuE-AD1 and TRuE-AD2 trials), 6 studies of dupilumab (LIBERTY AD ADOL, LIBERTY AD CAFÉ, LIBERTY AD CHRONOS, SOLO1, SOLO2, and Zhao [2021]96 trials), 3 studies of upadacitinib (Measure Up 1, Measure Up 2, and AD Up trials), and 1 study of abrocitinib (JADE MONO-1 trial). Subsequently, 3 studies (LIBERTY AD CAFÉ, LIBERTY AD CHRONOS, and AD Up trials) were excluded because they were not connected with ruxolitinib 1.5% cream through the common placebo arm and 1 adolescent-only study (LIBERTY AD ADOL trial) was excluded to improve comparability between the trials. In total, 8 studies representing 4 unique interventions (ruxolitinib 1.5% cream, upadacitinib, abrocitinib, and dupilumab) were included in the main analysis. The characteristics of the 8 included studies are presented in Table 21. A risk of bias assessment conducted at the full study population level by the sponsor suggested that the included studies were associated with a low risk of bias.
Table 21: Overview of Trials Included in the NMA
Study name | Phase | Age (years) | Moderate subgroup | Intervention | Comparator | Outcomes of interest | Assessment time point | ||
|---|---|---|---|---|---|---|---|---|---|
IGA 0 or 1 | EASI-75 | Itch NRS-4 | |||||||
TRuE-AD19 | III | ≥ 12 | IGA = 3; BSA affected ≥ 10%; EASI ≥ 16 at baseline | Ruxolitinib cream 0.75%a Ruxolitinib cream 1.5% | Vehicle | Yes | Yes | Yes | Week 8 |
TRuE-AD29 | III | ≥ 12 | IGA = 3; BSA affected ≥ 10%; EASI ≥ 16 at baseline | Ruxolitinib cream 0.75%a Ruxolitinib cream 1.5% | Vehicle | Yes | Yes | Yes | Week 8 |
SOLO195 | III | ≥ 18 | IGA = 3 (IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Dupilumab 300 mg q.w. Dupilumab 300 mg q.2.w. | Placebo | Yes | Yes | Yes | Week 16 |
SOLO295 | III | ≥ 18 | IGA = 3 (IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Dupilumab 300 mg q.w. Dupilumab 300 mg q.2.w. | Placebo | Yes | Yes | Yes | Week 16 |
Zhao (2021)96 | III | ≥ 18 | IGA = 3 (IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Dupilumab 300 mg q.2.w. | Placebo | Yes | NR | NR | Week 16 |
JADE MONO-197 | III | ≥ 12 | EASI 16 to 25 (IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Abrocitinib 100 mg Abrocitinib 200 mg | Placebo | NR | Yes | NR | Week 12 |
Measure Up 111 | III | ≥ 12 | v-IGA = 3 (v-IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Upadacitinib 15 mg Upadacitinib 30 mg | Placebo | Yes | Yes | Yes | Week 16 |
Measure Up 211 | III | ≥ 12 | v-IGA = 3 (v-IGA ≥ 3; EASI ≥ 16; BSA affected ≥ 10%) | Upadacitinib 15 mg Upadacitinib 30 mg | Placebo | Yes | Yes | Yes | Week 16 |
BSA = body surface area; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; NR = not reported; NRS-4 = at least a 4-point improvement in Numeric Rating Scale; q.2.w. = every 2 weeks; q.w. = every week; v-IGA = validated Investigator’s Global Assessment.
aThis refers to the definition of moderate subgroup as per eligibility criteria of each trial.
Sources: NMA technical report.16 Details included in the table are from the sponsor’s summary of clinical evidence.15
All included studies were phase III, double-masked, placebo-controlled studies. They were conducted globally, except for the Zhao (2021) study, which was conducted in 1 country only. Three studies (SOLO1, SOLO2, and Zhao [2021] studies) included adult patients only (aged 18 years or older), and 5 included both adolescent (aged 12 to younger than 18 years) and adult patients. All studies evaluated the comparative efficacy and safety of the study intervention relative to placebo as monotherapy. The assessment time point varied between the studies (week 8 for the ruxolitinib studies; week 16 for the dupilumab and upadacitinib studies; week 12 for the abrocitinib study).
All patients in the moderate-only subgroup had a baseline IGA of 3; the lone exception was the subgroup from the JADE MONO-1 trial, where patients had an EASI score of 16 to 25 at baseline. The baseline patient characteristics of the moderate-only subgroup were available from 4 studies (TRuE-AD1, TRuE-AD2, SOLO1, and SOLO2 trials). Notable differences in EASI score and the duration of AD were observed between these studies. The baseline EASI score was higher in the SOLO1 and SOLO2 studies (pooled analysis; dupilumab: 25.1 [SD = 8.09]; placebo: 25.7 [SD = 8.46]) than in the TRuE-AD1 and TRuE-AD2 studies (pooled analysis; ruxolitinib: 19.3 [SD = 2.9]; placebo: 20.2 [SD = 2.9]). The duration of AD also varied between the treatment groups and between the studies (pooled analysis of TRuE-AD1 and TRuE-AD2 studies: median 18.2 years versus 30.1 years for ruxolitinib versus placebo; pooled analysis of SOLO1 and SOLO2 studies: mean 25.1 years versus 28.6 years for ruxolitinib versus placebo).
Table 22: Assessment of Homogeneity for the 8 Studies Included in the NMA — Moderate-Only Subgroup
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Age | Most studies included adults and adolescents aged 12 years or older, except for the dupilumab studies (SOLO1, SOLO2, and Zhao [2021]), which included adult patients (18 years and older) only. For studies conducted in patients aged 12 years and older, the distribution of adults and adolescents in the moderate-only subgroup was not reported. |
Disease severity |
|
Treatment history | All studies enrolled patients who had a history of inadequate control of AD with topical treatment or for whom topical treatment was not advisable, except for the TRuE-AD1 and TRuE-AD2 studies, which did not have such an inclusion criterion. The proportion of patients in the moderate-only subgroup who had received prior phototherapy and/or systemic immunosuppressant therapy in each of these studies was not reported. |
Comparator | Placebo or vehicle cream only in all studies. |
Definitions of end points | The outcome definitions were generally aligned across trials. There were minor differences in the Itch outcome (peak pruritus vs. worst pruritus); however, these outcomes were considered comparable.
|
Timing of end point evaluation | Week 8 for ruxolitinib cream studies; week 16 for dupilumab and upadacitinib studies; week 12 for abrocitinib study. |
Study design | Phase III, double-blind RCTs; conducted across multiple countries for all studies except for Zhao 2021 (1 country). |
AD = atopic dermatitis; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; NMA = network meta-analysis; NRS = Numeric Rating Scale; RCT = randomized controlled trial; SD = standard deviation; vs. = versus.
Sources: NMA technical report.16 Details included in the table are from the sponsor’s summary of clinical evidence.15
In terms of treatment history, all studies enrolled patients who had a history of inadequate control of AD with topical treatment or for whom topical treatment was not advisable, except for the TRuE-AD1 and TRuE-AD2 studies, which did not have such an inclusion criterion. The proportion of patients in the moderate-only subgroup who had received prior phototherapy and/or systemic immunosuppressant therapy in each of these studies was not reported.
There is no notable difference in the definition of the end points assessed.
Results
IGA Treatment Success
The evidence network for IGA-TS in the moderate-only subgroup is illustrated in Figure 3. The network consisted of 7 studies evaluating 4 interventions — ruxolitinib 1.5% cream, upadacitinib 30 mg, upadacitinib 15 mg, and dupilumab 300 mg — connected by a common placebo comparator. One closed loop was formed by a single trial connecting upadacitinib 15 mg, upadacitinib 30 mg, and placebo.
The ORs and 95% CIs across various comparisons in relation to experiencing IGA-TS in the moderate-only subgroup are summarized in Figure 4. The IGA-TS response rate of ruxolitinib 1.5% cream was favoured over placebo (OR = 39.00; 95% CI, 1.92 to 793.43). There was insufficient evidence to show a difference when comparing ruxolitinib cream with upadacitinib 30 mg, upadacitinib 15 mg, and dupilumab 300 mg, because the 95% CIs for the ORs were wide.
A fixed-effect Bayesian NMA was not feasible for this outcome. The results of the sensitivity analyses (including LIBERTY AD ADOL; pooled doses of upadacitinib and abrocitinib) were consistent with the main analysis.
Figure 3: Network Diagram for the IGA-TS Analysis in the Moderate-Only Subgroup
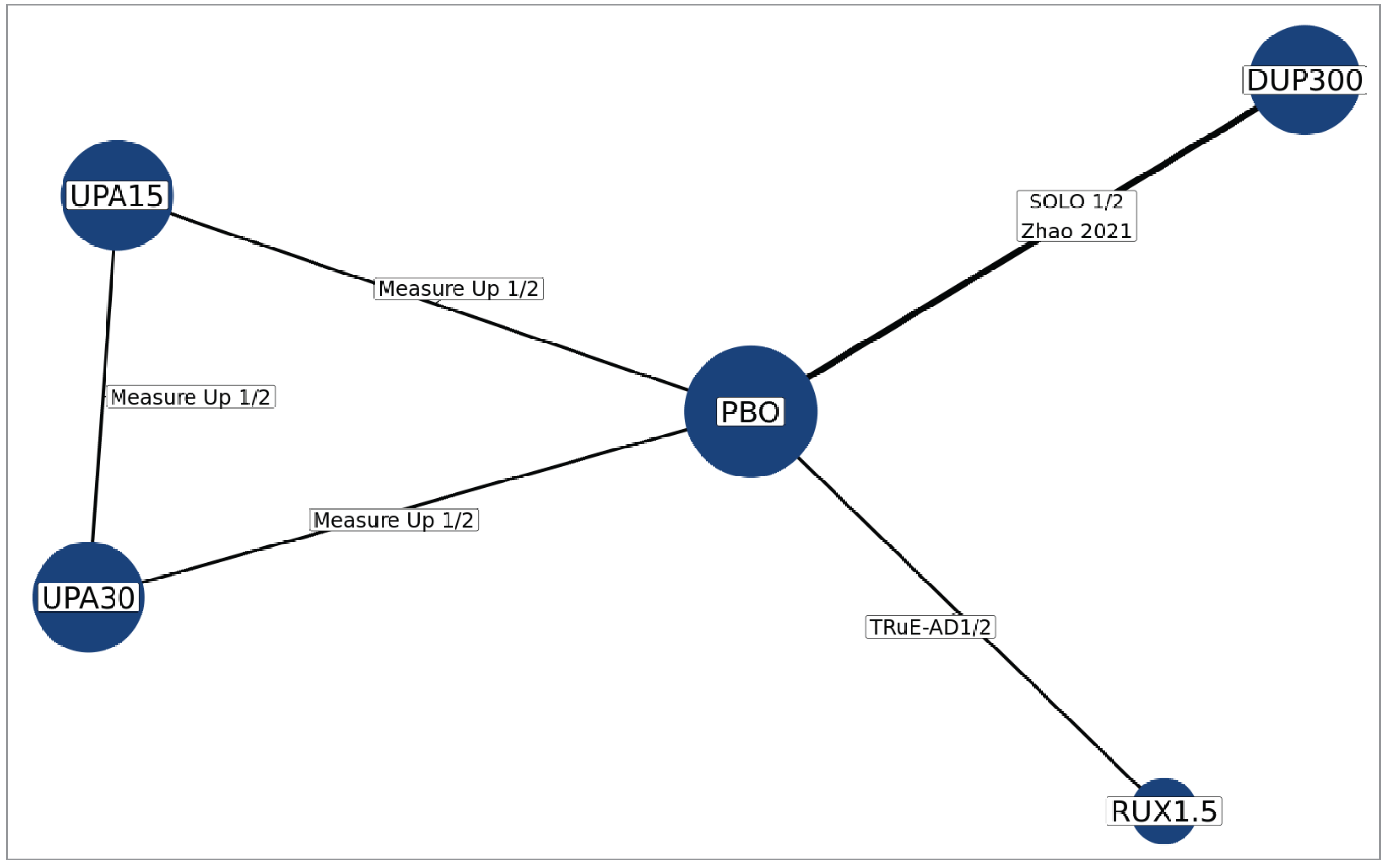
DUP = dupilumab; IGA-TS = Investigator’s Global Assessment treatment success; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Source: Network meta-analysis technical report.16
Figure 4: Odds Ratio (95% CI) Across Comparisons for IGA-TS in the Moderate-Only Subgroup
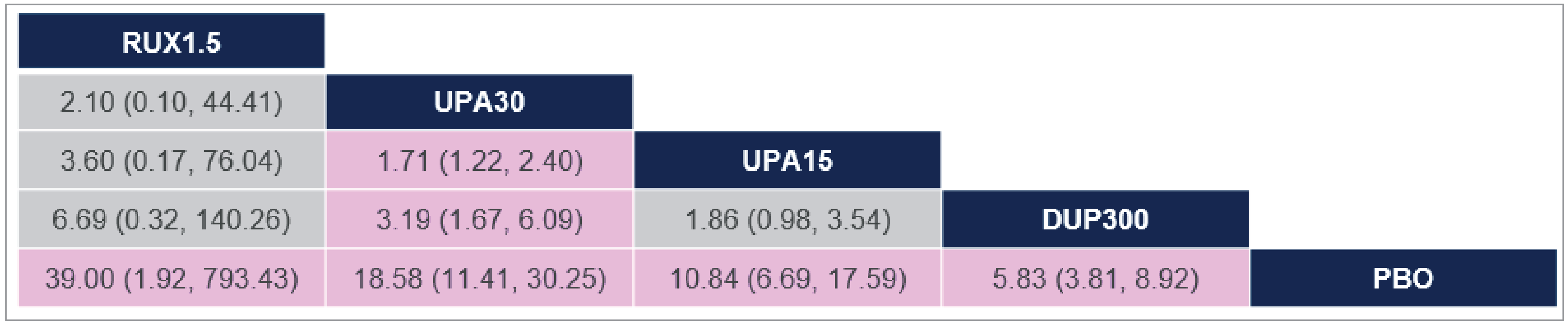
CI = confidence interval; DUP = dupilumab; IGA-TS = Investigator’s Global Assessment treatment success; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Notes: The results are presented as odds ratio and associated 95% CIs. Pink cells indicate results where 1 treatment is statistically significantly favoured over the other.
Source: Network meta-analysis technical report.16
At Least a 75% Improvement in EASI
The evidence network for EASI-75 in the moderate-only subgroup is illustrated in Figure 5. The network consisted of 7 studies evaluating 6 interventions — ruxolitinib 1.5% cream, upadacitinib 30 mg, upadacitinib 15 mg, dupilumab 300 mg, abrocitinib 100 mg, and abrocitinib 200 mg — connected by a common placebo comparator. Two closed loops were formed between the connections of a single trial of upadacitinib 15 mg, upadacitinib 30 mg, and placebo and a single trial of abrocitinib 200 mg, abrocitinib 100 mg, and placebo.
The ORs and 95% CIs across various comparisons in relation to experiencing EASI-75 in the moderate-only subgroup are summarized in Figure 6. The EASI-75 response rate of ruxolitinib 1.5% cream was favoured over placebo (OR = 20.61; 95% CI, 3.03 to 140.17). There was insufficient evidence to show a difference when comparing ruxolitinib cream with upadacitinib 30 mg, upadacitinib 15 mg, dupilumab 300 mg, abrocitinib 100 mg, and abrocitinib 200 mg, because the 95% CIs for the ORs were wide.
A fixed-effect Bayesian NMA was feasible for this outcome, and the results were consistent with the main analysis. The results of the sensitivity analyses (including the LIBERTY AD ADOL trial; pooled doses of upadacitinib and abrocitinib) were consistent with the main analysis.
Figure 5: Network Diagram for the EASI-75 Analysis in the Moderate-Only Subgroup
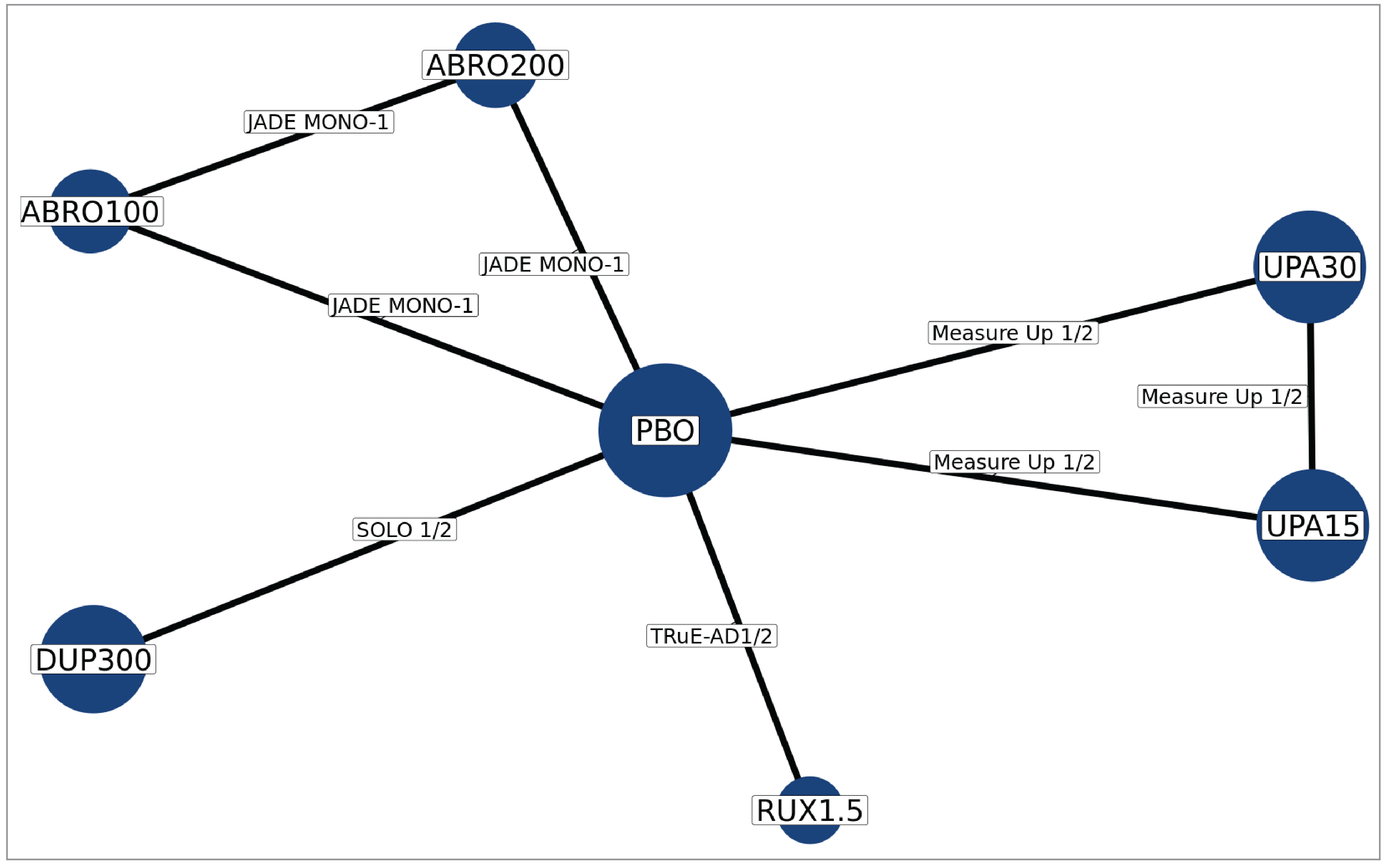
ABRO = abrocitinib; DUP = dupilumab; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Source: Network meta-analysis technical report.16
Figure 6: Odds Ratio (95% CI) Across Comparisons for EASI-75 in the Moderate-Only Subgroup
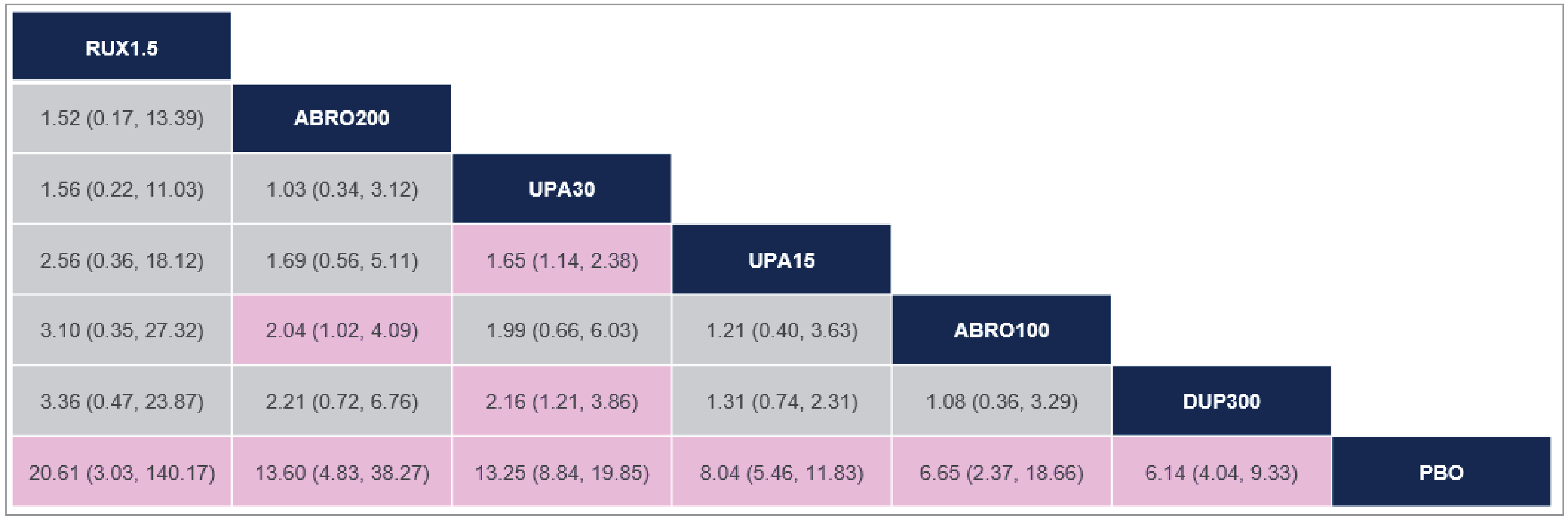
ABRO = abrocitinib; CI = confidence interval; DUP = dupilumab; EASI-75 = at least a 75% improvement in Eczema Area and Severity Index; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Notes: The results are presented as odds ratios and associated 95% CIs. Pink cells indicate results where 1 treatment is statistically significantly favoured over the other.
Source: Network meta-analysis technical report.16
Itch NRS-4
The evidence network for Itch NRS-4 in the moderate-only subgroup is illustrated in Figure 7. The network consisted of 6 studies evaluating 4 interventions — ruxolitinib 1.5% cream, upadacitinib 30 mg, upadacitinib 15 mg, and dupilumab 300 mg — connected by a common placebo comparator. One closed loop was formed by a single trial connecting upadacitinib 15 mg, upadacitinib 30 mg, and placebo.
The ORs and 95% CIs across various comparisons for Itch NRS-4 in the moderate-only subgroup are summarized in Figure 8. There was insufficient evidence to show a difference when comparing ruxolitinib cream with any of the interventions (including placebo) because the 95% CIs for the ORs were wide.
A fixed-effect Bayesian NMA was feasible for this outcome, and the results were consistent with the main analysis. The results of the sensitivity analyses (including the LIBERTY AD ADOL trial; pooled doses of upadacitinib and abrocitinib) were consistent with the main analysis.
Figure 7: Network Diagram for the Itch NRS-4 Analysis in the Moderate-Only Subgroup
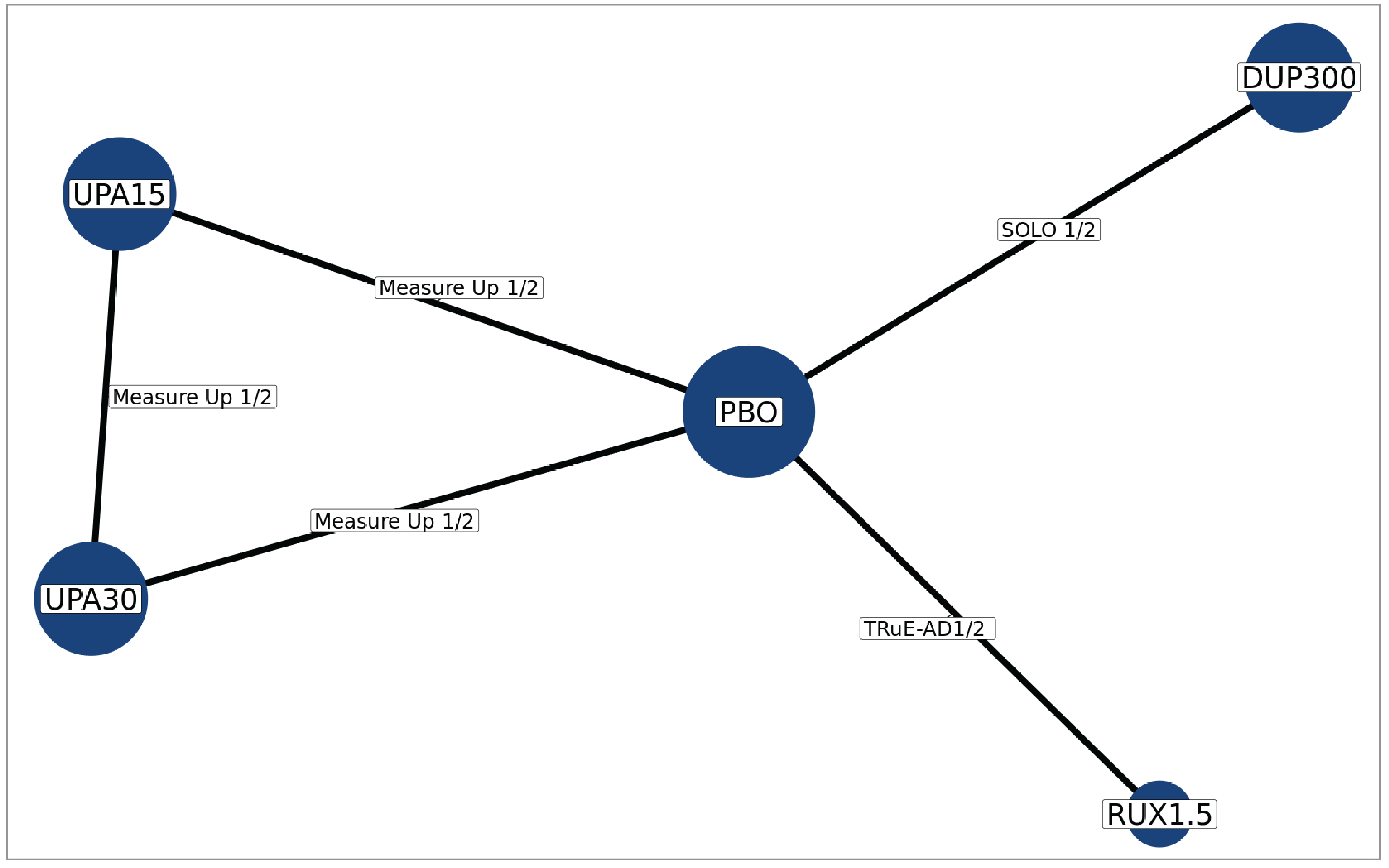
DUP = dupilumab; NRS-4 = 4-point improvement in Numeric Rating Scale; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Source: Network meta-analysis technical report.16
Figure 8: Odds Ratio (95% CI) Across Comparisons for Itch NRS-4 in the Moderate-Only Subgroup
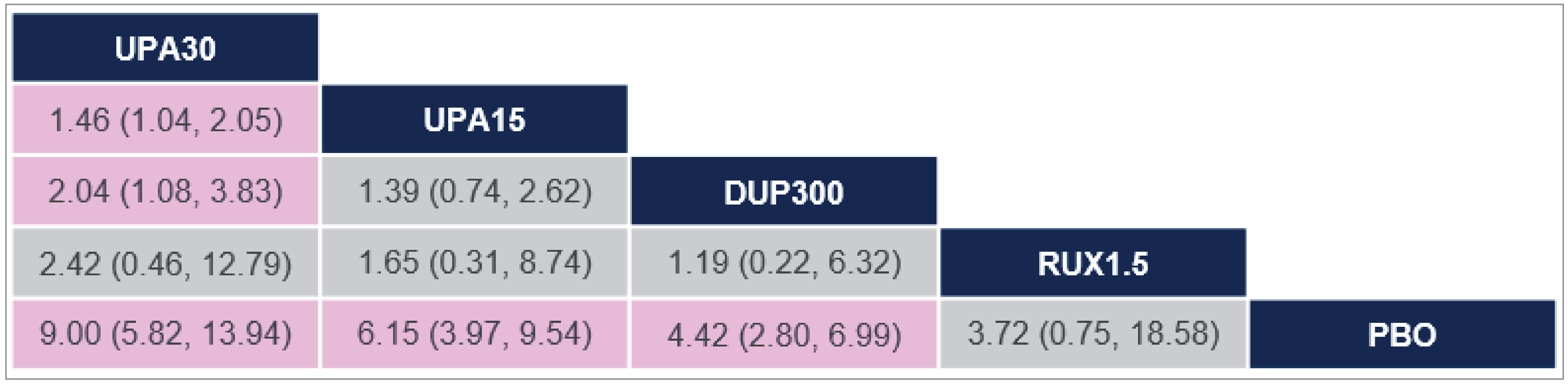
CI = confidence interval; DUP = dupilumab; NRS-4 = 4-point improvement in Numeric Rating Scale; PBO = placebo; RUX = ruxolitinib cream; UPA = upadacitinib.
Notes: The results are presented as odds ratio and associated 95% CIs. Pink cells indicate results where 1 treatment is statistically significantly favoured over the other.
Source: Network meta-analysis technical report.16
Critical Appraisal of ITC
Study inclusion was informed by an SLR, with appropriate methods to reduce the risk of bias and error in study selection and data extraction (e.g., a priori PICOS [population, intervention, comparison, results and study design] selection criteria, duplicate independent reviewers for article screening, data extraction by a single reviewer with quality check by another reviewer). Although a risk of bias assessment of included studies was performed, it was conducted in the full study population, rather than in the subgroup of interest; it is unclear if randomization was preserved in the moderate-only subgroup population of the included studies. Additionally, risk of bias appraisals were performed at the study level, which does not account for potential differences in risk of bias across the 3 included outcomes. A study protocol was available for the SLR but not the NMA. There is a risk of bias in the selection of the reported NMA results in the absence of a protocol because there is a possibility that the most favourable analysis was chosen from multiple alternative analyses of the data. The exclusion of additional studies to enable data to be analyzed in the moderate-only subgroup was informed by a feasibility assessment to promote the homogeneity of the included studies, and the reasons for exclusion were noted. However, with close to half the 23 initially identified studies being excluded due to the absence of results for subgroups consisting solely of patients with moderate severity disease; there is a risk of bias in the NMA findings due to missing results in the synthesis.
All included studies were phase III, double-masked RCTs in patients with AD assessing outcomes including the proportion of patients experiencing IGA-TS, EASI-75, and an improvement of at least 4 points in peak pruritus (or worst pruritus) scores. Based on the review team’s assessment, multiple sources of heterogeneity in the study designs were identified between the studies included in the NMA. While most studies included adult and adolescent patients aged 12 years or older, 3 studies of dupilumab (SOLO1, SOLO2, and Zhao [2021] trials) were conducted in adult patients only. All studies enrolled patients who had a history of inadequate control of AD with topical treatment or for whom topical treatment was not advisable, except for the TRuE-AD1 and TRuE-AD2 studies, which did not have such an inclusion criterion. Further, the timing of end point evaluation differed between the included studies (week 8 for the ruxolitinib cream studies; week 12 for the abrocitinib study; and week 16 for the dupilumab and upadacitinib studies), although the clinical experts consulted by CDA-AMC noted that the differences in timing are unlikely to be a concern for bias because they correspond to the time course of clinical improvement expected for each drug. Although these differences in study design could not be adjusted for in the analysis, these inherent differences suggest that the transitivity assumption of the NMA likely has not been met.
Between-study differences in baseline patient characteristics were identified. All patients were included in the moderate-only subgroup based on a baseline IGA score of 3, with the lone exception of the patients from the JADE MONO-1 trial, who were included based on an EASI score of 16 to 25 at baseline. The clinical experts consulted by CDA-AMC noted that the definition of moderate AD is commonly accepted as an EASI score of 7.1 to 21. The severity of AD in patients with an EASI score of 16 to 25 could be greater than for those with an IGA score of 3 (moderate AD), according to the clinical expert. Such differences in disease severity between the trial populations were not adjusted for in the NMA modelling. In addition, select baseline patient characteristics for the moderate-only subgroup were reported from 4 studies (TRuE-AD1, TRuE-AD2, SOLO1, and SOLO2 studies), and notable differences in baseline EASI scores and duration of AD diagnosis were noted between these studies. The clinical experts noted that baseline EASI score and duration of AD diagnosis are possible prognostic factors or treatment effect modifiers and could potentially introduce bias if not accounted for. The percentage of BSA affected and previous medication history are also possible treatment effect modifiers, according to the clinical experts; however, it is unclear if heterogeneity exists between the studies for these baseline characteristics because they were not reported. The lack of reporting of baseline patient characteristics for the other 8 studies also means that a robust appraisal of the homogeneity of the groups being compared was not possible. No information was given on the details of the assessment of statistical heterogeneity.
The NMA was informed by 12 studies, although not all studies were included in the evidence network for each outcome due to the absence of readily available subgroup data from some studies; 7 studies were included in the analyses of IGA-TS and EASI-75, and 6 studies were included in the analysis of Itch NRS-4. The resulting networks were sparse, with no closed loop connecting ruxolitinib 1.5% cream and comparators, rendering an assessment of the consistency of the results between direct and indirect comparisons not possible. Comparisons with ruxolitinib 1.5% cream were informed solely by indirect evidence and thus are associated with increased uncertainty. The 95% CIs for all outcomes were wide, which precluded conclusions about the comparative efficacy of ruxolitinib 1.5% cream versus dupilumab, abrocitinib, and upadacitinib.
The main analysis was conducted using a frequentist framework. Model fit was assessed based on the Akaike information criterion and residual deviance. Additional analyses with a fixed-effect Bayesian NMA were determined by the sponsor to be feasible for EASI-75 and Itch NRS-4 outcomes; the results were consistent with the frequentist NMA. Sensitivity analyses were conducted assessing the impact of including an adolescent-only study (LIBERTY AD ADOL trial) and of pooled doses of abrocitinib and upadacitinib, with results consistent with the main analysis. Sensitivity analyses assessing the potential impact of other identified trial heterogeneity were not conducted, which adds to the uncertainty in the relative treatment estimates.
The study assessed efficacy outcomes on the extent and severity of AD (IGA-TS, EASI-75) and itch symptoms (Itch NRS-4), which are important to patients and clinicians. However, only a limited comparison to abrocitinib was possible due to a lack of available data for IGA-TS and Itch NRS-4. Other outcomes of importance, including HRQoL and harms, were not measured. The study population of the NMA was patients with moderate disease only; patients with mild AD were not assessed. Additionally, the proportion of patients aged 12 to 18 years in each trial was unclear, and trials of dupilumab included data for adults only. As well, comparisons in the moderate-only subgroup were measured relative to dupilumab, abrocitinib, and upadacitinib monotherapy; comparisons with systemic immunosuppressants, which also are relevant comparators of ruxolitinib cream, were not assessed. The review team acknowledged that the sponsor’s feasibility assessment deemed comparisons with methotrexate and cyclosporin not feasible, although the absence of comparative evidence (both direct and indirect) between ruxolitinib cream monotherapy and systemic immunosuppressants in patients with moderate AD represents a gap in the evidence for the treatment of AD. Additionally, ruxolitinib cream is expected to be used in combination with other topical treatments (applied to different affected areas) according to clinical expert input; no direct or indirect comparative evidence for ruxolitinib cream as a combination therapy was submitted.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Table 23: Summary of Gaps in the Systematic Review Evidence
Evidence gap | SCRATCH-AD | Summary of key results |
|---|---|---|
Clinician and patient groups indicated that rapid itch relief is 1 of the desirable outcomes important to patients. The pivotal trials assessed the proportion of patients with ≥ 4-point reduction in PP-NRS at 8 weeks, but not earlier. | Phase II, open-label, single-arm study at a single Canadian site. | The mean change from baseline in PP-NRS on day 2, the primary end point of the study, was –3.37 (SD = 1.85) and continued to decrease through day 29. The mean change from baseline in mPP-NRS at 15 minutes posttreatment was –2.3 (SD = 2.34), peaking at –4.2 (SD = 2.12) at 4 hours. The efficacy for reducing itch at these time points vs. a relevant comparator is unknown. |
mPP-NRS = modified Peak Pruritus Numeric Rating Scale; PP-NRS = Peak Pruritus Numeric Rating Scale; SD = standard deviation.
Source: SCRATCH-AD Clinical Study Report.98
Description of Study
One phase II, open-label study (SCRATCH-AD trial) at a single Canadian site has been summarized to provide evidence regarding the short-term clinical benefits of ruxolitinib 1.5% cream in adults with AD to control itch and reduce disease severity.17 The primary and secondary objectives of the SCRATCH-AD study were to evaluate the short-term efficacy (pruritis and IGA) and safety of ruxolitinib 1.5% cream in participants with AD. The maximum study duration per participant was approximately 80 days, including a 37-day screening period, a 28-day treatment period (± 2 days), and a 15-day safety follow-up period (± 2 days). The screening period includes the run-in period, where participants had a baseline mean PP-NRS score of at least 4.0 during days –7 to –1 and continued to meet all inclusion criteria and none of the exclusion criteria.
Table 24: Details of Study Addressing Gap in the Systematic Review Evidence
Detail | SCRATCH-AD |
|---|---|
Design and population | |
Study design | Open-label, single-arm, interventional |
Enrolled, N | 49 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Ruxolitinib 1.5% cream applied topically twice daily (morning and evening, approximately 12 hours between applications) from day 1 until the day before the day 29 visit |
Comparator(s) | None |
Outcomes | |
Primary end point | Change from baseline in PP-NRS at day 2 (24-hour recall period after the first application) |
Secondary end points |
|
Notes | |
Publications | Bissonnette, et al.17 |
AD = topic dermatitis; BSA = body surface area; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; JAK = Janus kinase; MCID = minimal clinically important difference; mPP-NRS = modified Peak Pruritus Numeric Rating Scale; PP-NRS = Peak Pruritus Numeric Rating Scale; PUVA = psoralen plus UVA.
aModified PP-NRS measures current itch intensity as opposed to the 24 hours after the first application.
bIGA-TS is defined as an IGA score of 0 or 1 with at least a 2-grade reduction from baseline.
Sources: Clinicaltrials.gov, NCT04839380;99 SCRATCH-AD Clinical Study Report;98 Bissonette et al.17
Populations
The key inclusion and exclusion criteria are described in Table 24. The inclusion criteria were adults aged 18 to 65 years with at least a 6-month history of AD, 1% to 20% BSA affected, chronic itch for at least the past 3 months, a baseline PP-NRS score of 4 or greater, an IGA score of at least 2, and at least 1 target lesion. To be eligible for treatment, patients were required to have a mean PP-NRS score of 4 or greater during the 7-day run-in period.
Interventions
Ruxolitinib 1.5% cream was applied in an open-label fashion on all AD lesions, with a maximum treated area of 20% BSA as a thin film, twice daily, for 28 (± 2) days. There was no comparator.
Outcomes
The primary end point was change from baseline in PP-NRS at day 2 (24-hour recall period after the first application). The secondary end points were change from baseline in PP-NRS from day 3 through day 29 and proportion of participants experiencing at least a 1-grade or 2-grade decrease from baseline in PP-NRS as measured from day 3 through day 29. Time to minimal clinically important difference (MCID) (≥ 2-grade reduction in PP-NRS from baseline); change from baseline in IGA score at days 8, 15, and 29; and proportion of participants experiencing IGA-TS (IGA score of 0 or 1 with ≥ 2-grade reduction from baseline) at days 8, 15, and 29 were also measured as secondary end points. The safety and tolerability of ruxolitinib 1.5% cream was measured by the incidence and severity of local and systemic AEs.
Statistical Analysis
No formal statistical comparisons were performed. All data were analyzed descriptively. The 95% CIs are provided for the mean change, the percent change, and the proportions. In the analyses of time to MCID (e.g., ≥ 2-grade reduction in PP-NRS and mPP-NRS from baseline), a participant was considered censored (at their last available mPP-NRS or PP-NRS score) if they did not have the event of interest at the end of the study or if they were lost to follow-up at the last observation.
Results
Patient Disposition
Of the 84 participants who were screened, 35 (41.7%) did not meet the screening criteria. Forty-nine patients applied ruxolitinib 1.5% cream at least once (safety population). Forty-six participants completed the run-in period and met all entry criteria (ITT population) and had baseline and at least 1 postbaseline PP-NRS or mPP-NRS assessment (modified ITT population). In the ITT population, 44 participants (95.7%) completed the treatment or study. Two participants (4.3%) withdrew consent and discontinued the study treatment.
Baseline Characteristics
In the safety population (N = 49), the average age of the participants was 35.6 years (SD = 14.77 years), and the majority of participants were female (71.4%) and white (85.7%). At baseline, the mean total percentage of BSA affected was 10.11% (SD = 5.34%) and the mean EASI score was 7.23 (SD = 3.21). The mean PP-NRS score was 6.83 (SD = 1.4) and the mean mPP-NRS score was 6.6 (SD = 1.77) during the run-in period. The majority of participants (87.8%) had an IGA score of 3, and 6 participants (12.2%) had an IGA score of 2. The mean duration of AD was 27.53 years (SD = 15.56 years), and all participants had chronic itch. Forty-seven participants in the safety population had at least 1 comorbidity, and 49% of participants had taken at least 1 prior medication for an indication other than AD — emollients and protectives (10.2%), other analgesics and antipyretics and viral vaccines (8.2% each), and antihistamines for systemic use (6.1%) — in the 28 days before the screening period.
Exposure to Study Treatments
In the safety population, participants (N = 49) applied ruxolitinib 1.5% cream twice daily for a median of 28 days (range, 7 to 31 days). During the same period, the median cumulative dose was ██████ █ ██████ █ ███ ██ ███████). During the study period, ███ ████████ ██ ████████████ ███████ ████ ██████████ ███ ████████████ █████ ██████████ ███ ████████████ ████████ ██████ ███ ██████████████ ████████ ████████ ███████ ███████████ ████████ ███ ████████ █ ███ ██ █████████ ████████████ ██ ███ █ ███████.
Efficacy
At day 2, a mean 3.37-point reduction (SD = 1.85 points) or 50.57% reduction (95% CI, 58.75% to 42.39%) from baseline in PP-NRS score (worst itch in the previous 24 hours) was noted. The mean daily PP-NRS score decreased by 4.78 points (SD not reported) by day 7, and there was a 5.68-point (SD not reported) reduction by day 29. The proportion of participants experiencing at least a 1-grade decrease from baseline in PP-NRS score was 91.3% on day 2 and 100.0% on day 11. Thereafter, 91.3% of participants maintained at least a 1-grade decrease from baseline in PP-NRS score up until day 29. The proportion of participants who experienced at least a 2-grade decrease from baseline in PP-NRS score was 69.6% on day 2, 97.8% on day 16, and 89.1% on day 29. The Kaplan-Meier estimate of the median time to MCID (≥ 2-grade reduction) for PP-NRS was 1.0 day (95% CI not evaluable). A mean decrease from baseline in mPP-NRS score was observed at all time points on day 1, with the greatest decrease occurring 4 hours postapplication on day 1 (a 4.2-point reduction [SD = 2.12 points] or a 65.9% reduction [SD = 26.17%]). The proportion of participants who experienced at least a 1-grade and 2-grade decrease from baseline in mPP-NRS score increased from 82.6% and 58.7%, respectively, at 15 minutes postapplication to 100.0% and 95.7%, respectively, at 4 hours postapplication on day 1. The Kaplan-Meier estimate of the median time to MCID (≥ 2-grade reduction from baseline) for mPP-NRS was 0.250 hours (95% CI, 0.250 to 0.500).
Table 25: Key Efficacy Results — Modified ITT Population
Efficacy results | SCRATCH-AD N = 46 |
|---|---|
PP-NRSa | |
Number of patients at baselineb | 46 |
Mean score (SD) | 6.72 (1.36) |
Median score (range) | 6.86 (4.0 to 9.3) |
Day 2, n | 45 |
Mean change from baseline, % (95% CI) | –50.57 (–58.75 to –42.39) |
Median change from baseline, % (range) | –50.0 (–100.0 to 13.5) |
Day 2 through day 29 | |
≥ 1-grade decrease from baseline, n (%) | |
Day 2 | 42 (91.3) |
Day 16 | 45 (97.8) |
Day 29 | 42 (91.3) |
≥ 2-grade decrease from baseline, n (%) | |
Day 2 | 32 (69.6) |
Day 16 | 45 (97.8) |
Day 29 | 41 (89.1) |
mPP-NRSa | |
Day 1 time point | |
15 minutes postapplication | |
Mean change from baseline (SD) | –2.3 (2.34) |
Percent change from baseline (SD) | –33.6 (39.87) |
30 minutes postapplication | |
Mean change from baseline (SD) | –3.0 (2.38) |
Percent change from baseline (SD) | –45.8 (36.62) |
1 hour postapplication | |
Mean change from baseline (SD) | –3.3 (2.45) |
Percent change from baseline (SD) | –48.9 (35.65) |
2 hours postapplication | |
Mean change from baseline (SD) | –3.8 (2.16) |
Percent change from baseline (SD) | –59.7 (27.02) |
4 hours postapplication | |
Mean change from baseline (SD) | –4.2 (2.12) |
Percent change from baseline (SD) | –65.9 (26.17) |
6 hours postapplication | |
Mean change from baseline (SD) | –4.2 (2.04) |
Percent change from baseline (SD) | –65.4 (25.31) |
12 hours postapplication | |
Mean change from baseline (SD) | –3.1 (2.0) |
Percent change from baseline (SD)d | –47.9 (27.74) |
Day 1 time point, proportion of participants | |
≥ 1-grade decrease from baseline, n (%) | |
15 minutes postapplication | 38 (82.6) |
30 minutes postapplication | 41 (89.1) |
1 hour postapplication | 41 (89.1) |
2 hours postapplication | 45 (97.8) |
4 hours postapplication | 46 (100.0) |
6 hours postapplication | 45 (97.8) |
12 hours postapplication | 39 (90.7) |
≥ 2-grade decrease from baseline, n (%) | |
15 minutes postapplication | 27 (58.7) |
30 minutes postapplication | 31 (67.4) |
1 hour postapplication | 33 (71.7) |
2 hours postapplication | 42 (91.3) |
4 hours postapplication | 44 (95.7) |
6 hours postapplication | 43 (93.5) |
12 hours postapplication | 33 (76.7) |
IGA | |
Number of patients at baseline, nb | 46 |
Mean score (SD) | 2.9 (0.31) |
Median score (range) | 3.0 (2 to 3) |
Day 8, n | 44 |
Mean change from baseline, % (95% CI) | –48.1 (–55.37 to –40.84) |
Day 15, n | 45 |
Mean change from baseline, % (95% CI) | –67.0 (–75.41 to –58.67) |
Day 29, n | 44 |
Mean change from baseline, % (95% CI) | –75.0 (–82.98 to –66.02) |
IGA-TSc | |
Day 8, n (%; 95% CI) | 20 (45.5; 30.4 to 61.2) |
Day 15, n (%; 95% CI) | 32 (71.1; 55.7 to 83.6) |
Day 29, n (%; 95% CI) | 34 (77.3; 62.2 to 88.5) |
CI = confidence interval; IGA = Investigator’s Global Assessment; IGA-TS = Investigator’s Global Assessment treatment success; ITT = intention to treat; mPP-NRS = modified Peak Pruritus Numeric Rating Scale; PP-NRS = Peak Pruritus Numeric Rating Scale; SD = standard deviation.
aPP-NRS has a 24-hour recall period after the first application. The baseline is defined as the average of all nonmissing PP-NRS scores. The mPP-NRS measures current itch intensity.
bThe baseline is defined as the last nonmissing assessment before or on day 1 and before the first application of the study drug (including unscheduled assessments). Percentages are based on the number of participants with a response at the specified visit and time point.
cIGA-TS is defined as an IGA score of 0 or 1, with at least a 2-grade reduction from baseline.
Source: SCRATCH-AD Clinical Study Report.98
The mean changes from baseline in IGA score at days 8, 15, and 29 were –1.4 (–48.1%; 95% CI, –55.37% to –40.84%), –2.0 (–67.0%; 95% CI, –75.41% to –58.67%), and –2.2 (–75.0%; 95% CI, –82.98% to –66.02%), respectively. Increasing proportions of participants experienced IGA-TS at days 8, 15, and 29: 45.5% (95% CI, 30.4% to 61.2%), 71.1% (95% CI, 55.7% to 83.6%), and 77.3% (95% CI, 62.2% to 88.5%), respectively. The mean EASI scores decreased over time during the treatment period, from 6.92 at baseline; on days 15 and 29, the mean changes from baseline in EASI score were −6.22 (SD = 2.82) and −6.58 (SD = 3.06), respectively. The total percentage of BSA affected by AD decreased over time from 9.51% at baseline to 0.74% (SD = 1.05%) by day 15 and to 0.52% (SD = 1.37%) by day 29 (91.01% [SD = 14.02%] and 92.18% [SD = 20.60%] reductions, respectively).
Harms
Approximately one-third of participants (n = 15; 30.6%) had at least 1 TEAE. The most frequently reported TEAEs were COVID-19 (6.1%) and back pain, nasopharyngitis, headache, and upper respiratory infection (4.1% each). One participant (2.0%) had an application site reaction (acne), which resolved with no change to study treatment. There were no deaths, SAEs, or TEAEs leading to study treatment interruption or discontinuation.
Critical Appraisal
Internal Validity
The main limitation of the SCRATCH-AD study was the single-arm design. The lack of a relevant comparator renders it impossible to draw causal conclusions about the comparative efficacy of ruxolitinib 1.5% cream with respect to other treatment options or to vehicle cream. Interpretation of the changes from baseline is complicated, as they may be due to the intervention, concomitant treatments, a placebo effect, and/or natural history. Additionally, there is a potential risk of bias due to the open-label design. Patients were aware of the treatment they were receiving and self-reported subjective outcomes, which may have resulted in overestimation of the change from baseline. The analyses were conducted for fewer than 50 patients (safety and modified ITT populations), which could add uncertainty to the efficacy results.
External Validity
As the SCRATCH-AD study was conducted in a single study site located in Quebec, Canada, its findings in general have good generalizability to the Canadian clinical setting, except for less-than-ideal representation of Indigenous populations, in which AD is common.
Discussion
This report summarizes the evidence for ruxolitinib 1.5% cream in the treatment of AD based on 2 phase III RCTs, 1 ITC, and 1 phase II, open-label, single-arm study.
Summary of Available Evidence
Two studies, the TRuE-AD1 trial (N = 631) and the TRuE-AD2 trial (N = 618),9,10 met the inclusion criteria for the systematic review conducted by the sponsor. The TRuE-AD1 and TRuE-AD2 trials were identically designed, pivotal, phase III, double-masked, randomized, placebo-controlled trials that aimed to assess the efficacy and safety of ruxolitinib cream relative to vehicle cream, as monotherapy, in adolescents and adults aged 12 years or older with mild or moderate AD (IGA score of 2 or 3) and 3% to 20% BSA affected. In the 8-week vehicle-controlled period, the following were assessed: the proportion of patients experiencing IGA-TS (primary end point), EASI-75, at least a 4-point reduction from baseline in Itch NRS score, and at least a 6-point reduction from baseline in PROMIS Short Form–Sleep Disturbance and PROMIS Short Form–Sleep-Related Impairment scores (key secondary end points). Efficacy and safety outcomes from the 44-week long-term safety period were also presented. At baseline, the majority of patients were adults and had an IGA score of 3 (approximately 80% and 75%, respectively). The mean total percentage of BSA affected by AD was approximately 10%. Prior TCI treatment and/or medium-potency, high-potency, or super-high-potency TCS treatment were each reported in no more than 50% of patients. A small proportion of patients had received prior systemic immunosuppressants, phototherapy, dupilumab, or systemic JAK inhibitor treatment.
In the absence of head-to-head evidence comparing ruxolitinib cream to other relevant therapies used in the treatment of mild to moderate AD, the sponsor submitted 1 NMA indirectly comparing the treatment effects of ruxolitinib 1.5% cream to dupilumab, abrocitinib, and upadacitinib in patients with moderate AD with respect to the proportion of patients experiencing IGA-TS, EASI-75, and at least a 4-point reduction from baseline in Itch NRS score, based on 8 included studies.16 No comparative information on harms was submitted.
One phase II, open-label, single-arm study (SCRATCH-AD trial;17 N = 49) at a single site in Canada was submitted by the sponsor as supporting evidence for the short-term clinical benefits of ruxolitinib 1.5% cream in controlling itch (based on PP-NRS assessment) and reducing the severity of AD (based on IGA assessment) in a 29-day treatment period in adults with AD of at least mild severity (IGA ≥ 2) and no more than 20% BSA affected at baseline.
Interpretation of Results
Efficacy
Direct comparative evidence from the TRuE-AD1 and TRuE-AD2 trials supported the superiority of ruxolitinib 1.5% cream over placebo with respect to IGA-TS and EASI-75 at week 8 in adults and adolescents with mild to moderate AD, addressing a key treatment outcome noted by patients and clinicians: improvement in the extent and severity of AD. The magnitude of the benefits of ruxolitinib cream on IGA-TS and EASI-75 was considered to be clinically important by the clinical experts consulted by CDA-AMC. Prespecified subgroup analysis of IGA-TS and EASI-75 seem to suggest a higher response rate in patients with a baseline EASI score greater than 7 (moderate AD) than in patients with a score of 7 or less (mild AD); however, due to the lack of sample size consideration, control for multiplicity, and statistical testing for treatment-by-subgroup interaction, no definitive conclusions can be drawn on subgroup effects. Change in the percentage of BSA affected, a standalone secondary outcome measuring the extent of AD, which was not adjusted for multiplicity, was assessed at week 8 as supportive evidence and showed results in favour of ruxolitinib cream, although there is some uncertainty in the findings due to potential attrition bias associated with a notable imbalance in study treatment discontinuation between groups, which could result in inflated benefits with ruxolitinib cream. Nonetheless, the magnitude of benefit did not meet the threshold of clinical importance of between 5% and 10% noted in the clinical expert input. According to clinical expert input, both the extent and severity of AD (as reflected by EASI and IGA scores) should be considered when assessing response to treatment. According to the clinical experts, patients who report an overall improvement in AD may experience a reduction in the severity of AD but not the extent of the disease, or vice versa; therefore, the clinical experts did not find the lack of a clinically important reduction in the percentage of BSA affected alone to be particularly concerning given the favourable results for IGA-TS and EASI-75.
Reduction of AD symptoms, an important goal in the treatment of AD, was assessed using patient-reported outcomes — including Itch NRS, POEM, and PROMIS Short Form–Sleep Disturbance and PROMIS Short Form–Sleep-Related Impairment scales — at week 8 in the TRuE-AD1 and TRuE-AD2 trials. Evidence for the validity of these instruments in patients with AD was available, though evidence for the 2 PROMIS Short Form scales was established based on the TRuE-AD1 and TRuE-AD2 trial populations. There was evidence of low certainty that ruxolitinib cream may result in a clinically important improvement in these outcomes relative to placebo. A large proportion of patients (at least 30%) were excluded from both treatment groups in the Itch NRS responder analysis, which could potentially have impacted randomization. The end point of change in POEM score was associated with a risk of attrition bias due to an imbalance in study treatment withdrawal and with an increased risk of type I error due to lack of control for multiplicity. No formal statistical testing was performed on the PROMIS Short Form–Sleep-Related Impairment responder analysis due to prior failure in the statistical testing hierarchy. There is also evidence of imprecision in the PROMIS Short Form–Sleep-Related Impairment and PROMIS Short Form–Sleep Disturbance responder analyses (the 95% CI included the possibility of benefit and no clinically important difference based on the MID estimate of 5% in at least 1 trial). There is also a small potential for bias in the measurement of these patient-reported outcomes, potentially resulting in inflated efficacy of ruxolitinib cream, though the impact on the study results is unlikely to be significant.
DLQI and CDLQI, dermatology-specific HRQoL instruments, were used to assess the HRQoL of patients in the studies as supportive evidence (not controlled for multiplicity). The questionnaires captured the impact of AD on the physical, social, and emotional aspects of quality of life, as well as on the ability to carry out daily activities and to establish and maintain intimate relationships, which were noted to be important by patients. There was evidence of low certainty that ruxolitinib cream may result in a clinically important improvement in DLQI score at week 8 in adult patients when compared with placebo. The study results were associated with a risk of attrition bias due to a notable imbalance in study treatment withdrawal, potentially in favour of ruxolitinib. There was evidence of low certainty that ruxolitinib cream may result in little to no clinically important improvement in CDLQI score at 8 weeks relative to placebo in adolescent patients. There is potential that the prognostic balance provided by the randomization is not fully preserved in this analysis because it was conducted in a small subset of patients, with no stratification involved. As well, there is a small risk of inflated efficacy of ruxolitinib cream due to potential bias in the measurement of the outcomes, as with other patient-reported outcomes, although it is not expected to have a substantial impact on the results.
The longer-term efficacy of ruxolitinib cream, beyond 8 weeks, with respect to IGA scores of 0 or 1, the percentage of BSA affected, and POEM, DLQI, and CDLQI scores was assessed in the long-term safety period of the pivotal trials. The results suggested that the efficacy of ruxolitinib cream was sustained; however, the results were uncertain due to the absence of a control group (which makes it impossible to definitively attribute the treatment effects to ruxolitinib cream) and due to a sizable loss to follow-up (approximately 20%) in both trials.
The clinical experts consulted for this review anticipated that ruxolitinib cream would primarily be used in patients with AD whose disease is inadequately controlled with TCS and/or TCI treatment. However, the inclusion and exclusion criteria of the submitted trials did not restrict entry based on prior experience with TCS and TCI treatment. Therefore, there is no evidence to inform how the results of the TRuE-AD1 and TRuE-AD2 trials may be generalizable to patients whose disease is inadequately controlled with TCS and/or TCI treatment. In consultation with the clinical experts, the review team considered that it is unclear if the post hoc subgroup analyses submitted by the sponsor (comparing the treatment effect in patients with and patients without a recent history of TCS and/or TCI treatment) could adequately reflect the patient population expected to receive ruxolitinib cream in clinical practice based on the Health Canada indication. Additionally, there was a lower proportion of patients with mild AD (IGA score of 2) in the trials than in clinical practice, which could potentially impact generalizability given that baseline IGA score is a potential treatment effect modifier, according to the clinical expert input.
The TRuE-AD1 and TRuE-AD2 trials have been the only RCTs of ruxolitinib cream to date. In an effort to address the evidence gap in the absence of head-to-head evidence comparing ruxolitinib cream with relevant comparators, the sponsor submitted an NMA indirectly comparing ruxolitinib cream with dupilumab, upadacitinib, and abrocitinib in patients with moderate AD. The validity of the NMA results was overall uncertain because the key assumptions of the analysis (i.e., homogeneity and consistency) could not be determined because of insufficient reporting of baseline patient characteristics in the moderate-only subgroup and because of the sparse network without a closed loop connecting ruxolitinib cream. It is also impossible to make conclusions with any certainty on the efficacy of ruxolitinib cream versus the comparators because the results of the submitted NMA were imprecise. In addition, there are concerns about missing results in the synthesis: close to half the studies initially identified by the SLR were excluded due to the absence of results for subgroups consisting solely of patients with moderate severity disease.
Although evidence from the SCRATCH-AD study suggested that ruxolitinib cream could provide rapid itch relief (an outcome noted to be important by the clinician and patient groups) and lead to IGA-TS in the first month of treatment, the results were uncertain due to the open-label study design, which could potentially lead to overestimation of treatment effects, and due to the single-arm study design, which precluded conclusions on efficacy versus any relevant comparator or vehicle cream.
Overall, despite the evidence submitted by the sponsor, there remain evidence gaps in the treatment of AD. These gaps include the absence of comparative evidence for ruxolitinib cream monotherapy versus systemic immunosuppressants in patients with moderate AD and the absence of comparative evidence for ruxolitinib cream in combination with other topical therapies.
Harms
Evidence from the TRuE-AD1 and TRuE-AD2 trials showed that the safety profile of ruxolitinib 1.5% cream was similar to that of vehicle cream in the 8-week vehicle-controlled period. The most frequently reported TEAEs of ruxolitinib cream were nasopharyngitis, upper respiratory tract infection, and headache; all of which were reported at a low frequency, were not serious, and did not lead to treatment withdrawal or death. No new safety signals were identified in the 44-week long-term safety period. The study results at week 8 suggested that ruxolitinib cream likely resulted in little to no clinically important difference in SAEs compared with placebo. There is some uncertainty in the findings because, according to the clinical expert input, a longer duration of follow-up for safety (beyond 52 weeks) is required to adequately capture the long-term safety of ruxolitinib cream, particularly with respect to rare harms. A review of postmarketing safety data from the sponsor’s global safety database and the FDA Adverse Event Reporting System in the first year of FDA approval of ruxolitinib cream for the treatment of AD and nonsegmental vitiligo (2021 to 2022) showed 4 cases of SAEs, including skin cancer, pericarditis, and thrombocytopenia, and there were no reports of SAEs associated with the class warning for JAK inhibitors (serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis).100 Comparative evidence for the harms of ruxolitinib cream versus relevant comparators was not identified.
Conclusion
Direct evidence from 2 pivotal double-masked RCTs demonstrated that 8 weeks of ruxolitinib 1.5% cream monotherapy likely results in a clinically important improvement in the severity and extent of AD (IGA-TS and EASI-75) in adults and adolescents with mild to moderate AD compared to placebo. Analyses of the percentage of BSA affected by AD and of the symptoms experienced (including itch and impact on sleep) in general favoured ruxolitinib cream; however, the results for these outcomes were associated with uncertainty due to methodological limitations and were considered supportive of a clinically important benefit with ruxolitinib cream treatment. The results were suggestive of little to no clinically important improvement in HRQoL with ruxolitinib cream treatment in adolescent patients and of a potentially clinically important improvement in HRQoL in adult patients. The benefits of ruxolitinib cream, in general, appeared to be sustained through week 52 in the trials, although the lack of a control group and sizable loss to follow-up beyond week 8 precluded firm conclusions versus any comparator, including vehicle cream. According to clinical expert input, in clinical practice ruxolitinib cream is anticipated to be primarily used in patients with mild to moderate AD that is not adequately controlled with TCS and/or TCI, which aligns with the Health Canada indication. Based on the submitted evidence (including the post hoc subgroup data), it is unclear if the study populations are generalizable to clinical practice given that study inclusion and exclusion criteria did not restrict entry based on prior experience with topical treatments. In addition, there was a lower proportion of patients with mild AD (IGA score of 2) in the trials than in clinical practice, which could impact generalizability given that baseline IGA score is a potential treatment effect modifier according to clinical expert input. Indirect evidence from 1 ITC comparing the efficacy of ruxolitinib cream versus dupilumab, upadacitinib, and abrocitinib in patients with moderate AD was inconclusive due to important limitations that prevented verification of whether the underlying assumptions of homogeneity and consistency were met, as well as due to imprecision. No definitive conclusion can be drawn from the supportive study submitted by the sponsor (SCRATCH-AD study) with respect to the short-term clinical benefits of ruxolitinib cream due to limitations associated with the open-label, single-arm study design. The absence of direct or indirect comparisons of the efficacy of ruxolitinib cream monotherapy versus systemic immunosuppressants in patients with moderate AD and the absence of comparative evidence for ruxolitinib cream in combination with other topical treatments were gaps in the evidence for the treatment of AD. Ruxolitinib cream appeared to be well tolerated in adults and adolescents through week 52 in the pivotal trials, although a longer duration of follow-up is required to capture the long-term safety, particularly for potential rare harms. No comparative evidence for the harms of ruxolitinib cream versus relevant comparators was submitted.
References
1.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109-1122. doi:10.1016/S0140-6736(15)00149-X PubMed
2.Howe W. Post TW, ed. Atopic dermatitis (eczema): Pathogenesis, clinical manifestations, and diagnosis [sponsor-supplied reference]. UpToDate; 2022. Accessed January 12, 2024.
3.Drucker AM, Bai L, Eder L, et al. Sociodemographic characteristics and emergency department visits and inpatient hospitalizations for atopic dermatitis in Ontario: a cross-sectional study. CMAJ Open. 2022;10(2):E491-E499. doi:10.9778/cmajo.20210194 PubMed
4.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy. 2018;73(6):1284-1293. doi:10.1111/all.13401 PubMed
5.Silverberg JI, Gelfand JM, Margolis DJ, et al. Patient burden and quality of life in atopic dermatitis in US adults: A population-based cross-sectional study. Ann Allergy Asthma Immunol. 2018;121(3):340-347. doi:10.1016/j.anai.2018.07.006 PubMed
6.Institut national d’excellence en santé et en services sociaux (INESSS). Extrait de l'Avis au ministre sur Dupixent Adolescent [sponsor-supplied reference]. 2020. Accessed January 12, 2024. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Juillet_2020/Dupixent_Ado_2020_06.pdf
7.Lynde C, Barber K, Claveau J, et al. Canadian practical guide for the treatment and management of atopic dermatitis. J Cutan Med Surg. 2005;8 Suppl 5:1-9. doi:10.1007/s10227-005-8080-3 PubMed
8.Lynde CW, Bourcier M, Gooderham M, et al. A Treatment Algorithm for Moderate to Severe Atopic Dermatitis in Adults. J Cutan Med Surg. 2018;22(1):78-83. doi:10.1177/1203475417733460 PubMed
9.Papp K, Szepietowski JC, Kircik L, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: Results from 2 phase 3, randomized, double-blind studies. J Am Acad Dermatol. 2021;85(4):863-872. doi:10.1016/j.jaad.2021.04.085 PubMed
10.Papp K, Szepietowski JC, Kircik L, et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: Results from two phase 3 studies. J Am Acad Dermatol. 2023;88(5):1008-1016. doi:10.1016/j.jaad.2022.09.060 PubMed
11.Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151-2168. doi:10.1016/S0140-6736(21)00588-2 PubMed
12.Reich K, Teixeira HD, de Bruin-Weller M, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397(10290):2169-2181. doi:10.1016/S0140-6736(21)00589-4 PubMed
13.Incyte Corporation. Clinical Study Report: INCB 18424-304 (TRuE-AD2). A Phase 3, Double-Blind, Randomized, 8-Week, Vehicle-Controlled Efficacy and Safety Study of Ruxolitinib Cream Followed by a Long-Term Safety Extension Period in Adolescents and Adults With Atopic Dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2021.
14.Incyte Corporation. Clinical Study Report: INCB 18424-303 (TRuE-AD1). A Phase 3, Double-Blind, Randomized, 8-Week, Vehicle-Controlled Efficacy and Safety Study of Ruxolitinib Cream Followed by a Long-Term Safety Extension Period in Adolescents and Adults With Atopic Dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2021.
15.Incyte Biosciences Canada. Clinical Evidence Template: Ruxolitinib [as ruxolitinib phosphate] 1.5% cream for the Treatment of Atopic Dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2023.
16.Incyte Biosciences Canada Corporation. Ruxolitinib [as Ruxolitinib Phosphate] Cream 1.5% Indirect Treatment Comparisons versus Systemic Therapies in Atopic Dermatitis (≥12 Years) - Network Meta-analyses Technical Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. October 27, 2023.
17.Bissonnette R, Haobo R, Nawaz H, Halden P, Saint-Cyr Proulx E. Rapid, Substantial, and Sustained Reduction of Itch in Adults With Atopic Dermatitis Applying Ruxolitinib Cream 1.5% (SCRATCH-AD). Br J Dermatol. 2023;188(3)doi:10.1093/bjd/ljad162.021
18.National Eczema Association. Atopic Dermatitis [sponsor-supplied reference]. 2023. Accessed January 12, 2024. https://nationaleczema.org/eczema/types-of-eczema/atopic-dermatitis/
19.Fleming P, Yang YB, Lynde C, O'Neill B, Lee KO. Diagnosis and Management of Atopic Dermatitis for Primary Care Providers. J Am Board Fam Med. 2020;33(4):626-635. doi:10.3122/jabfm.2020.04.190449 PubMed
20.National Eczema Association. Types of Eczema [sponsor-supplied reference]. 2023. Accessed January 12, 2024. https://nationaleczema.org/eczema/types-of-eczema/
21.Spergel JM. Epidemiology of atopic dermatitis and atopic march in children. Immunol Allergy Clin North Am. 2010;30(3):269-80. doi:10.1016/j.iac.2010.06.003 PubMed
22.Bantz SK, Zhu Z, Zheng T. The Atopic March: Progression from Atopic Dermatitis to Allergic Rhinitis and Asthma. J Clin Cell Immunol. 2014;5(2)doi:10.4172/2155-9899.1000202 PubMed
23.Paller A, Jaworski JC, Simpson EL, et al. Major Comorbidities of Atopic Dermatitis: Beyond Allergic Disorders. Am J Clin Dermatol. 2018;19(6):821-838. doi:10.1007/s40257-018-0383-4 PubMed
24.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. doi:10.1038/s41572-018-0001-z PubMed
25.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-32. doi:10.1016/j.jaad.2014.03.023 PubMed
26.Eczema Society of Canada. The Atopic Dermatitis Patient Journey [sponsor-supplied reference]. 2020. Accessed January 12, 2024. https://eczemahelp.ca/wp-content/uploads/2023/06/ESC-Atopic-Dermatitis-Patient-Journey-Report-2020.pdf
27.Silverberg JI, Garg NK, Paller AS, Fishbein AB, Zee PC. Sleep disturbances in adults with eczema are associated with impaired overall health: a US population-based study. J Invest Dermatol. 2015;135(1):56-66. doi:10.1038/jid.2014.325 PubMed
28.Lee DG, Gui XY, Mukovozov I, Fleming P, Lynde C. Sleep Disturbances in Children With Atopic Dermatitis: A Scoping Review. J Cutan Med Surg. 2023;27(2):157-164. doi:10.1177/12034754231159337 PubMed
29.Chang YS, Chiang BL. Mechanism of Sleep Disturbance in Children with Atopic Dermatitis and the Role of the Circadian Rhythm and Melatonin. Int J Mol Sci. 2016;17(4):462. doi:10.3390/ijms17040462 PubMed
30.Hong CH, Sussman G, Turchin I, Wiseman M, Gooderham MJ. Approach to the Assessment and Management of Adult Patients With Atopic Dermatitis: A Consensus Document. Section III: Evaluation of Atopic Dermatitis Patients for Comorbidities. J Cutan Med Surg. 2018;22(1_suppl):17s-20s. doi:10.1177/1203475418805709 PubMed
31.Davis DMR, Drucker AM, Alikhan A, et al. American Academy of Dermatology Guidelines: Awareness of comorbidities associated with atopic dermatitis in adults. J Am Acad Dermatol. 2022;86(6):1335-1336.e18. doi:10.1016/j.jaad.2022.01.009 PubMed
32.Eczema Society of Canada. Atopic Dermatitis Quality of Life Report. Moderate-to-Severe Disease. 2016/2017 Survey Results [sponsor-supplied reference]. 2017. Accessed January 12, 2024. http://altogethereczema.org/images/files/ESC_QUALITY_OF_LIFE_REPORT_2017.pdf
33.Rønnstad ATM, Halling-Overgaard AS, Hamann CR, Skov L, Egeberg A, Thyssen JP. Association of atopic dermatitis with depression, anxiety, and suicidal ideation in children and adults: A systematic review and meta-analysis. J Am Acad Dermatol. 2018;79(3):448-456.e30. doi:10.1016/j.jaad.2018.03.017 PubMed
34.Asiniwasis RN, Chu DK. Atopic Dermatitis and Canadian Indigenous Peoples: Burdens, Barriers, and Potential for Solutions. Canadian Allergy & Immunology Today. 2022;2(3):26-31. doi:10.58931/cait.2022.2337
35.Global Atopic Dermatitis Atlas. Global Report on Atopic Dermatitis 2022. 2022. Accessed January 12, 2024. https://www.eczemacouncil.org/assets/docs/global-report-on-atopic-dermatitis-2022.pdf
36.Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):52. doi:10.1186/s13223-018-0281-6 PubMed
37.Howell MD, Parker ML, Mustelin T, Ranade K. Past, present, and future for biologic intervention in atopic dermatitis. Allergy. 2015;70(8):887-96. doi:10.1111/all.12632 PubMed
38.Silverberg JI, Barbarot S, Gadkari A, et al. Atopic dermatitis in the pediatric population: A cross-sectional, international epidemiologic study. Ann Allergy Asthma Immunol. 2021;126(4):417-428 e2. doi:10.1016/j.anai.2020.12.020 PubMed
39.Silverberg JI, Mohawk JA, Cirulli J, et al. Burden of Disease and Unmet Needs in Atopic Dermatitis: Results From a Patient Survey. Dermatitis. 2023;34(2):135-144. doi:10.1089/derm.2022.29015.jsi PubMed
40.Lee H, Ryu WI, Kim HJ, et al. TSLP Down-Regulates S100A7 and ss-Defensin 2 Via the JAK2/STAT3-Dependent Mechanism. J Invest Dermatol. 2016;136(12):2427-2435. doi:10.1016/j.jid.2016.07.027 PubMed
41.Gooderham MJ, Bissonnette R, Grewal P, Lansang P, Papp KA, Hong CH. Approach to the Assessment and Management of Adult Patients With Atopic Dermatitis: A Consensus Document. Section II: Tools for Assessing the Severity of Atopic Dermatitis. J Cutan Med Surg. 2018;22(1_suppl):10S-16S. doi:10.1177/1203475418803628 PubMed
42.Canadian Skin Patient Alliance, Eczéma Québec. The Skin I'm In: 2022 update. A national report of the patient and caregiver experience with atopic dermatitis [sponsor-supplied reference]. 2022. Accessed January 12, 2024. https://eczemaquebec.com/wp-content/uploads/2022/12/2022-11-12_AD-Report-2022_CB-3.pdf
43.Dhadwal G, Albrecht L, Gniadecki R, et al. Approach to the Assessment and Management of Adult Patients With Atopic Dermatitis: A Consensus Document. Section IV: Treatment Options for the Management of Atopic Dermatitis. J Cutan Med Surg. 2018;22(1_suppl):21S-29S. doi:10.1177/1203475418805721 PubMed
44.Gooderham MJ, Hong CH, Albrecht L, et al. Approach to the Assessment and Management of Adult Patients With Atopic Dermatitis: A Consensus Document. J Cutan Med Surg. 2018;22(1_suppl):3S-5S. doi:10.1177/1203475418803627 PubMed
45.Lynde C, Bergman JN, Fiorillo L, et al. Optimal Use of Crisaborole in Atopic Dermatitis – An Expert Guidance Document [sponsor-supplied reference]. Skin Therapy Letter. 2021;16(1)
46.Fleischer AB, Jr., Abramovits W, Breneman D, Jaracz E. Tacrolimus ointment is more effective than pimecrolimus cream in adult patients with moderate to very severe atopic dermatitis. J Dermatolog Treat. 2007;18(3):151-7. doi:10.1080/09546630701287332 PubMed
47.Ontario Health. Home Narrowband Ultraviolet B Phototherapy for Photoresponsive Skin Conditions: Recommendation [sponsor-supplied reference]. 2020. Accessed January 12, 2024. https://www.hqontario.ca/Portals/0/Documents/evidence/reports/recommendation-home-narrowband-ultraviolet-en.pdf
48.Hum M, Kalia S, Gniadecki R. Prescribing Home Narrowband UVB Phototherapy: A Review of Current Approaches. J Cutan Med Surg. 2019;23(1):91-96. doi:10.1177/1203475418800947 PubMed
49.CADTH. CADTH Upadacitinib Clinical and Pharmacoeconomic Combined Report. (Rinvoq - AbbVie Corporation) [sponsor-supplied reference]. 2022.
50.Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J Am Acad Dermatol. 2017;77(4):623-633. doi:10.1016/j.jaad.2017.06.042 PubMed
51.Leo Pharma, Inc. Adtralza (tralokinumab): 150 mg/1 mL and 300 mg/2 mL single-use, pre-filled syringe, solution for subcutaneous injection [product monograph; sponsor-supplied reference]. 2023.
52.Sanofi-aventis Canada, Inc. Dupixent (dupilumab): 200 mg/1.14 mL and 300 mg/2 mL single-use syringe, injection [product monograph; sponsor-supplied reference]. 2022.
53.CADTH. CADTH Canadian Drug Expert Committee Recommendation (Final). Abrocitinib (Cibinqo - Pfizer Canada ULC.) [sponsor-supplied reference]. 2022.
54.CADTH. CADTH Canadian Drug Expert Committee Recommendation (Final). Upadacitinib (Rinvoq -AbbVie Corporation) [sponsor-supplied reference]. 2022.
55.Incyte Corporation. Opzelura (ruxolitinib phosphate): 1.5% w/w cream, topical [product monograph]. October 11, 2024.
56.Howell MD, Kuo FI, Smith PA. Targeting the Janus Kinase Family in Autoimmune Skin Diseases. Front Immunol. 2019;10:2342. doi:10.3389/fimmu.2019.02342 PubMed
57.Amano W, Nakajima S, Kunugi H, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015;136(3):667-677 e7. doi:10.1016/j.jaci.2015.03.051 PubMed
58.Kim BS, Howell MD, Sun K, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol. 2020;145(2):572-582. doi:10.1016/j.jaci.2019.08.042 PubMed
59.Solimani F, Meier K, Ghoreschi K. Emerging Topical and Systemic JAK Inhibitors in Dermatology. Front Immunol. 2019;10:2847. doi:10.3389/fimmu.2019.02847 PubMed
60.Howell MD, Fitzsimons C, Smith PA. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann Allergy Asthma Immunol. 2018;120(4):367-375. doi:10.1016/j.anai.2018.02.012 PubMed
61.Bao L, Shi VY, Chan LS. IL-4 regulates chemokine CCL26 in keratinocytes through the Jak1, 2/Stat6 signal transduction pathway: Implication for atopic dermatitis. Mol Immunol. 2012;50(1-2):91-7. doi:10.1016/j.molimm.2011.12.008 PubMed
62.Borriello F, Longo M, Spinelli R, et al. IL-3 synergises with basophil-derived IL-4 and IL-13 to promote the alternative activation of human monocytes. Eur J Immunol. 2015;45(7):2042-51. doi:10.1002/eji.201445303 PubMed
63.Hoy SM. Ruxolitinib Cream 1.5%: A Review in Mild to Moderate Atopic Dermatitis. Am J Clin Dermatol. 2023;24(1):143-151. doi:10.1007/s40257-022-00748-2 PubMed
64.Bausch Health Canada Inc. Elidel (pimecrolimus): 1% cream, topical [product monograph; sponsor-supplied reference]. 2020.
65.LEO Pharma Inc. Protopic (tacrolimus): 0.03% and 0.1% (w/w) ointment, topical [product monograph]. 2020. Accessed January 12, 2024. https://pdf.hres.ca/dpd_pm/00057788.PDF
66.Pfizer Canada, Inc. Apo-methotrexate (methotrexate): 2.5 mg tablet, oral [product monograph; sponsor-supplied reference]. 2017.
67.Sandoz Canada Inc. Sandoz Cyclosporine (cyclosporine): 25 mg, 50 mg, 100 mg soft gelatin capsule, oral [product monograph]. 2015. Accessed January 12, 2024. https://pdf.hres.ca/dpd_pm/00029804.PDF
68.AbbVie Corporation. Rinvoq (upadacitinib): 15 mg, 30 mg, and 45 mg extended-release tablets, oral [product monograph; sponsor-supplied reference]. 2023.
69.Pfizer Canada, U. L. C. Cibinqo (abrocitinib): 50 mg, 100 mg, and 200 mg tablets, oral [product monograph; sponsor-supplied reference]. 2023.
70.Incyte Corporation. Prescribing information: Opzelura (ruxolitinib) 1.5% cream. U.S. Food and Drug Administration; 2022. Accessed November 24, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215309s001lbl.pdf
71.Williams HC, Schmitt J, Thomas KS, et al. The HOME Core outcome set for clinical trials of atopic dermatitis. J Allergy Clin Immunol. 2022;149(6):1899-1911. doi:10.1016/j.jaci.2022.03.017 PubMed
72.Bozek A, Reich A. Assessment of Intra- and Inter-Rater Reliability of Three Methods for Measuring Atopic Dermatitis Severity: EASI, Objective SCORAD, and IGA. Dermatology. 2017;233(1):16-22. doi:10.1159/000472711 PubMed
73.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. doi:10.1111/j.1365-2133.2004.05696.x PubMed
74.Simpson E, Beck LA, Gadkari A, et al. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate-to-severe atopic dermatitis: Detailed analysis from randomized trials of dupilumab. J Am Acad Dermatol. 2017;76(6):AB93. doi:10.1016/j.jaad.2017.04.376
75.Yosipovitch G, Reaney M, Mastey V, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. doi:10.1111/bjd.17744 PubMed
76.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs Eczema Area and Severity Index and other measures of atopic dermatitis: A validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. doi:10.1016/j.anai.2020.03.006 PubMed
77.Schram ME, Spuls PI, Leeflang MM, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. doi:10.1111/j.1398-9995.2011.02719.x PubMed
78.Howells L, Ratib S, Chalmers JR, Bradshaw L, Thomas KS. How should minimally important change scores for the Patient-Oriented Eczema Measure be interpreted? A validation using varied methods. Br J Dermatol. 2018;178(5):1135-1142. doi:10.1111/bjd.16367 PubMed
79.Incyte Corporation. Evidence Dossier. Ruxolitinib cream - Sleep Disturbance and Sleep-related Impairment Measures [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2020.
80.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. doi:10.1186/1477-7525-3-36 PubMed
81.Heinl D, Prinsen CA, Deckert S, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: a systematic review. Allergy. 2016;71(3):358-70. doi:10.1111/all.12806 PubMed
82.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. doi:10.1159/000365390 PubMed
83.Simpson EL, de Bruin-Weller M, Eckert L, et al. Responder Threshold for Patient-Oriented Eczema Measure (POEM) and Children's Dermatology Life Quality Index (CDLQI) in Adolescents with Atopic Dermatitis. Dermatol Ther (Heidelb). 2019;9(4):799-805. doi:10.1007/s13555-019-00333-2 PubMed
84.Lewis-Jones MS, Finlay AY. The Children's Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-9. doi:10.1111/j.1365-2133.1995.tb16953.x PubMed
85.Salek MS, Jung S, Brincat-Ruffini LA, et al. Clinical experience and psychometric properties of the Children's Dermatology Life Quality Index (CDLQI), 1995-2012. Br J Dermatol. 2013;169(4):734-59. doi:10.1111/bjd.12437 PubMed
86.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10(1):11-8. doi:10.1034/j.1600-0625.2001.100102.x PubMed
87.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-47. doi:10.1016/j.jaci.2013.07.008 PubMed
88.Incyte Corporation. Clinical Study Protocol: Topical Ruxolitinib Evaluation in Atopic Dermatitis Study 1 (TRuE-AD1). A Phase 3, Double-Blind, Randomized, 8-Week, Vehicle-Controlled Efficacy and Safety Study of Ruxolitinib Cream Followed by a Long-Term Safety Extension Period in Adolescents and Adults With Atopic Dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2019.
89.Barrett A, Hahn-Pedersen J, Kragh N, Evans E, Gnanasakthy A. Patient-Reported Outcome Measures in Atopic Dermatitis and Chronic Hand Eczema in Adults. Patient. 2019;12(5):445-459. doi:10.1007/s40271-019-00373-y PubMed
90.Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994-2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159(5):997-1035. doi:10.1111/j.1365-2133.2008.08832.x PubMed
91.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-80. doi:10.1111/j.1087-0024.2004.09113.x PubMed
92.Badia X, Mascaro JM, Lozano R. Measuring health-related quality of life in patients with mild to moderate eczema and psoriasis: clinical validity, reliability and sensitivity to change of the DLQI. The Cavide Research Group. Br J Dermatol. 1999;141(4):698-702. doi:10.1046/j.1365-2133.1999.03112.x PubMed
93.Simpson EL, de Bruin-Weller M, Bansal A, et al. Definition of Clinically Meaningful Within-Patient Changes in POEM and CDLQI in Children 6 to 11 Years of Age with Severe Atopic Dermatitis. Dermatol Ther (Heidelb). 2021;11(4):1415-1422. doi:10.1007/s13555-021-00543-7 PubMed
94.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
95.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med. 2016;375(24):2335-2348. doi:10.1056/NEJMoa1610020 PubMed
96.Zhao Y, Wu L, Lu Q, et al. The efficacy and safety of dupilumab in Chinese patients with moderate-to-severe atopic dermatitis: a randomized, double-blind, placebo-controlled study. Br J Dermatol. 2022;186(4):633-641. doi:10.1111/bjd.20690 PubMed
97.Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255-266. doi:10.1016/S0140-6736(20)30732-7 PubMed
98.Incyte Corporation. Clinical Study Report INCB 1824-901. An Open-Label, Single-Arm Study to Evaluate the Effect of Ruxolitinib 1.5% Cream on Itch in Adult Participants With Atopic Dermatitis (SCRATCH-AD) [sponsor-supplied reference]. 2023.
99.Incyte Corporation. NCT04839380: The Purpose of the Study is to Evaluate the Effect of Ruxolitinib Cream on Itch in Participants With Atopic Dermatitis (SCRATCH-AD). ClinicalTrials.gov. October 31, 2023. Accessed January 12, 2024. https://clinicaltrials.gov/study/NCT04839380
100.Hu W, Thornton M, Livingston RA. Real-World Use of Ruxolitinib Cream: Safety Analysis at 1 Year. Am J Clin Dermatol. 2024;25(2):327-332. doi:10.1007/s40257-023-00840-1 PubMed
101.Incyte Biosciences Canada. Incyte Biosciences Canada response to December 15, 2023 CADTH request for additional information regarding Ruxolitinib Cream CADTH review: Subgroup Data from TRuE-AD1 and TRuE-AD2 [internal additional sponsor's information]. January 5, 2024.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Supportive outcome data from the TRuE-AD1 and TRuE-AD2 trials are summarized in Table 26 (vehicle-controlled period) and Table 27 (long-term safety period).
Change From Baseline in EASI Score
Change from baseline in EASI score at week 8 was a secondary end point and was not adjusted for multiplicity in both trials. The between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –3.4 (95% CI, –4.4 to −2.4) in the TRuE-AD1 trial and –3.9 (95% CI, –4.6 to –3.3) in the TRuE-AD2 trial.
Change From Baseline in Itch NRS Score
Change from baseline in Itch NRS score at week 8 was a secondary end point and was not adjusted for multiplicity in both trials. The between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was –2.0 (95% CI, –2.6 to –1.4) in the TRuE-AD1 trial and –1.7 (95% CI, –2.3 to –1.1) in the TRuE-AD2 trial.
Change From Baseline in EQ-VAS Score
Change from baseline in EQ-VAS score at week 8 was a secondary end point and was not adjusted for multiplicity in both trials. At week 8, the between-group LSM difference comparing ruxolitinib 1.5% cream with vehicle cream was 5.7 (95% CI, 2.5 to 9.0) in the TRuE-AD1 trial and 5.1 (95% CI, 1.6 to 8.5) in the TRuE-AD2 trial. At week 52, change from baseline in EQ-VAS score in the vehicle cream to ruxolitinib 1.5% cream group and the ruxolitinib 1.5% cream to ruxolitinib 1.5% cream group was 10.1 (SD = 19.1) and 10.5 (SD = 17.5), respectively, in the TRuE-AD1 trial; and 7.3 (SD = 15.0) and 12.2 (SD = 18.1), respectively, in the TRuE-AD2 trial. No statistical analysis was conducted to assess the between-group difference at week 52.
Table 26: Supportive Data From TRuE-AD1 and TRuE-AD2 Trials — Vehicle-Controlled Period (ITT)
Efficacy outcome | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream N = 126 | Ruxolitinib 1.5% cream N = 253 | Vehicle cream N = 118 | Ruxolitinib 1.5% cream N = 228 | |
Change from baseline in EASI score at week 8 | ||||
Number of patients contributing to the analysis, n (%) | 101 (80.2) | 232 (91.7) | 101 (85.6) | 218 (95.6) |
Baseline, mean (SD) | 7.4 (4.3) | 7.9 (4.6) | 8.2 (5.3) | 7.6 (5.0) |
Change from baseline, LSM (SE) | –2.8 (0.4) | –6.2 (0.3) | 5.5 (0.3) | 1.6 (0.2) |
Difference vs. vehicle (%), LSM (95% CI)a | –3.4 (–4.4 to –2.4) | –3.9 (–4.6 to –3.3) | ||
P valueb | < 0.0001 | < 0.0001 | ||
Change from baseline Itch NRS at week 8 | ||||
Number of patients contributing to the analysis, n (%) | 80 (63.5) | 203 (80.2) | 83 (70.3) | 187 (82.0) |
Baseline, mean (SD) | 5.1 (2.5) | 5.2 (2.5) | 5.3 (2.4) | 5.0 (2.5) |
Change from baseline, LSM (SE) | –1.5 (0.3) | –3.5 (0.2) | –1.4 (0.3) | –3.1 (0.2) |
Difference vs. vehicle, LSM (95% CI)a | –2.0 (–2.6 to –1.4) | –1.7 (–2.3 to –1.1) | ||
P valueb | < 0.0001 | < 0.0001 | ||
Change from baseline in EQ-VAS score at week 8 | ||||
Number of patients contributing to the analysis, n (%) | ███ █████ | ███ ███ | ███ ████ | ███ ████ |
Baseline, mean (SD) | ████ ████ | ████ ██ | ████ ██ | ████ ██ |
Week 8, mean (SD) | ████ ██ | ████ ██ | ████ ██ | ████ ██ |
Change from baseline, LSM (SE) | ███ ████ | ███ ███ | ███ ███ | ███ ███ |
Difference vs. vehicle (%), LSM (95% CI)c | ███ ████ ██ ████ | ███ ████ ██ ████ | ||
P valueb | ██████ | ██████ | ||
b.i.d. = twice a day; CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention-to-treat; LSM = least squares mean; NRS = Numeric Rating Scale; SD = standard deviation; SE = standard error; VAS = visual analogue scale. VC = vehicle-controlled.
Note: All analyses of the TRuE-AD2 trial were conducted in the ITT population █████████ ██ ████████ ████████ ██ ████ ███ █████ ██ ███████ █████████████ ████ ███ ████████ ███ ████████ ████ ████████ ████████ ██████ ████████ █████████ ███ █ ████████ ██ ███ ████.
aThis end point was analyzed using mixed models for repeated measures including treatment groups, region, baseline IGA, visit, and treatment-by-visit interaction as adjustment factors.
bThis end point was not adjusted for multiplicity.
cThis end point was analyzed using analysis of covariance including treatment groups, region, baseline IGA, and baseline score as adjustment factors.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Table 27: Supportive Data on EQ-VAS From TRuE-AD1 and TRuE-AD2 Trials — Long-Term Safety Period (Long-Term Safety Evaluable Population)
Efficacy outcome | TRuE-AD1 | TRuE-AD2 | ||
|---|---|---|---|---|
Vehicle cream to ruxolitinib 1.5% cream N = 47 | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream N = 225 | Vehicle cream to ruxolitinib 1.5% cream N = 49 | Ruxolitinib 1.5% cream to ruxolitinib 1.5% cream N = 203 | |
Change from baseline in EQ-VAS score at week 52 | ||||
Number of patients contributing to the analysis, n (%) | ██ ██████ | ███ ██████ | ██ ██████ | ███ ████ |
Baseline, mean (SD) | ████ ███ | ████ ███ | ████ ████ | ████ ████ |
Week 52, mean (SD) | ████ | ████ | ████ ████ | ████ ████ |
Change from baseline, mean (SD) | ████ ██ | ████ ███ | ███ █████ | ████ ████ |
b.i.d. = twice daily; SD = standard deviation; VAS = visual analogue scale.
Note: No formal statistical analysis was conducted to test the between-group difference. Descriptive statistics by treatment group was summarized.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Details included in the table are from the sponsor’s summary of clinical evidence.15
Figure 9: Prespecified Subgroup Analyses of Proportion of Patients Experiencing IGA-TS at Week 8 (Ruxolitinib 1.5% vs. Vehicle Cream) in TRuE-AD1 Trial (ITT)
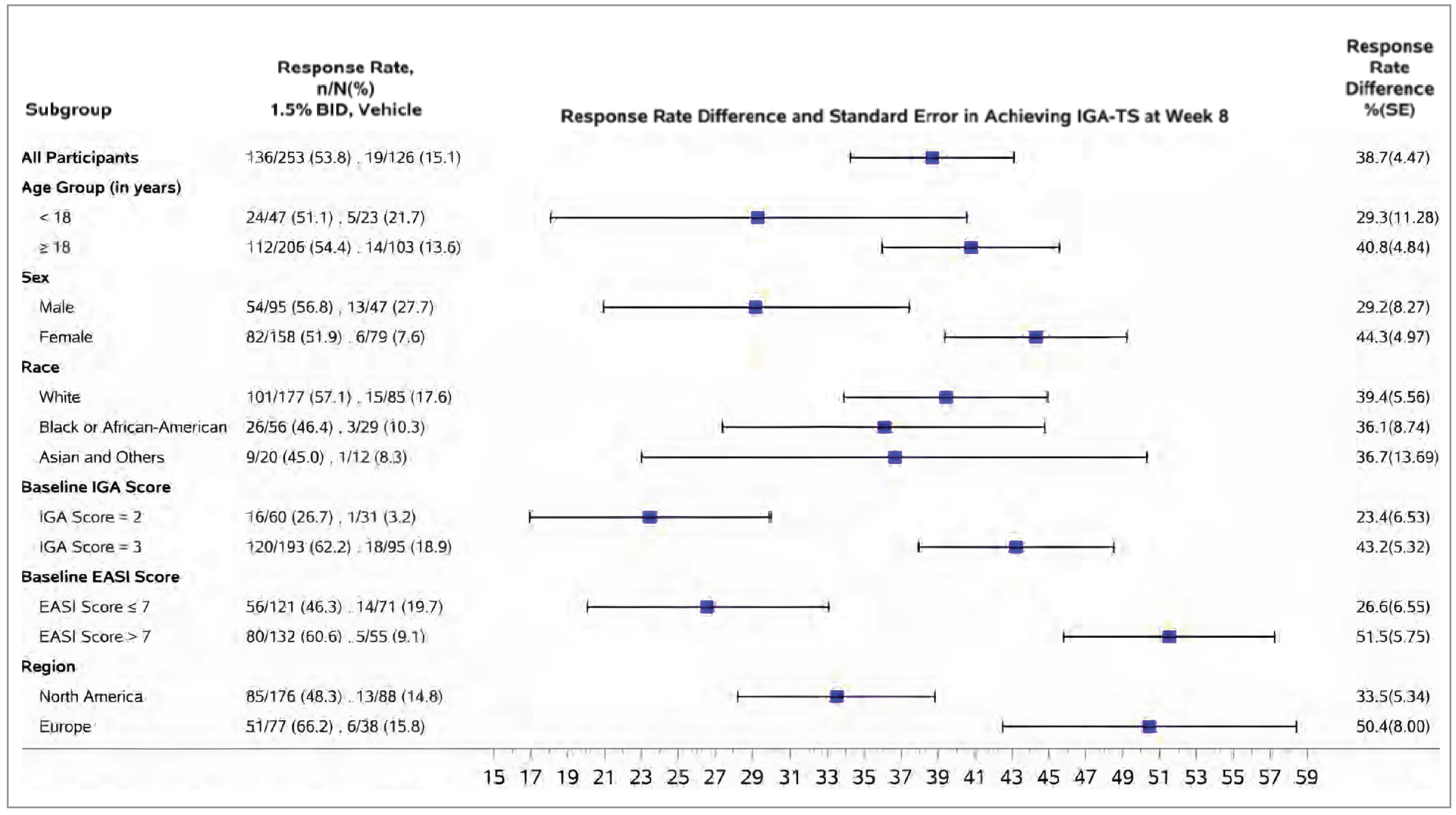
BID = twice a day; IGA-TS = Investigator’s Global Assessment–Treatment Success; ITT = intention-to-treat; IGA = Investigator’s Global Assessment; EASI = Eczema Area Severity Index; SE = standard error; vs. = versus.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14
Figure 10: Prespecified Subgroup Analyses of Proportion of Patients Experiencing IGA-TS at Week 8 (Ruxolitinib 1.5% vs. Vehicle Cream) in TRuE-AD2 Trial (ITT Excluding Site 461)
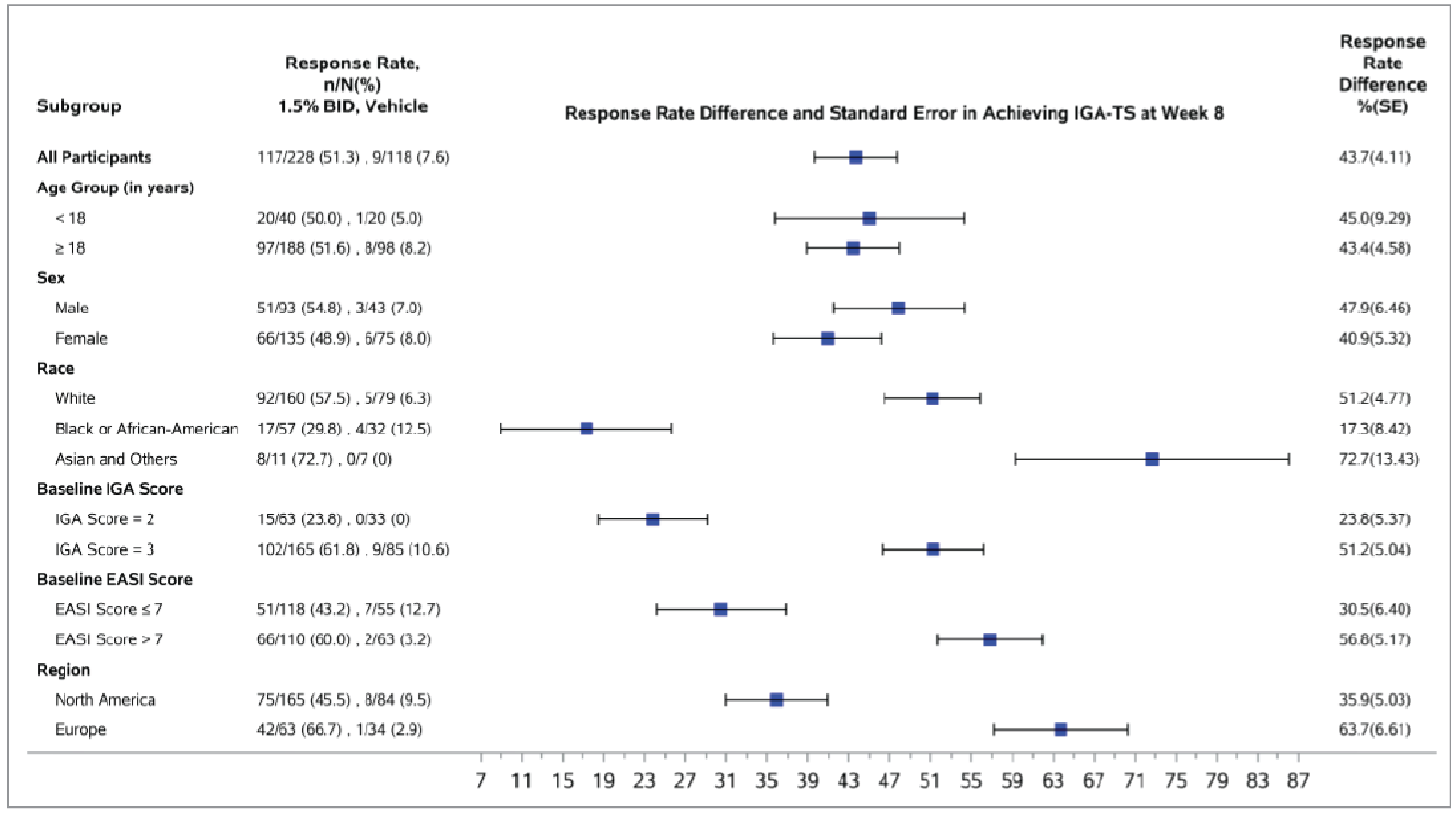
BID = twice a day; IGA-TS = Investigator’s Global Assessment–Treatment Success; ITT = intention-to-treat; IGA = Investigator’s Global Assessment; EASI = Eczema Area Severity Index; SE = standard error; vs. = versus.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14
Figure 11: Prespecified Subgroup Analyses of Proportion of Patients Experiencing EASI-75 at Week 8 (Ruxolitinib 1.5% vs. Vehicle Cream) in TRuE-AD1 Trial (ITT)
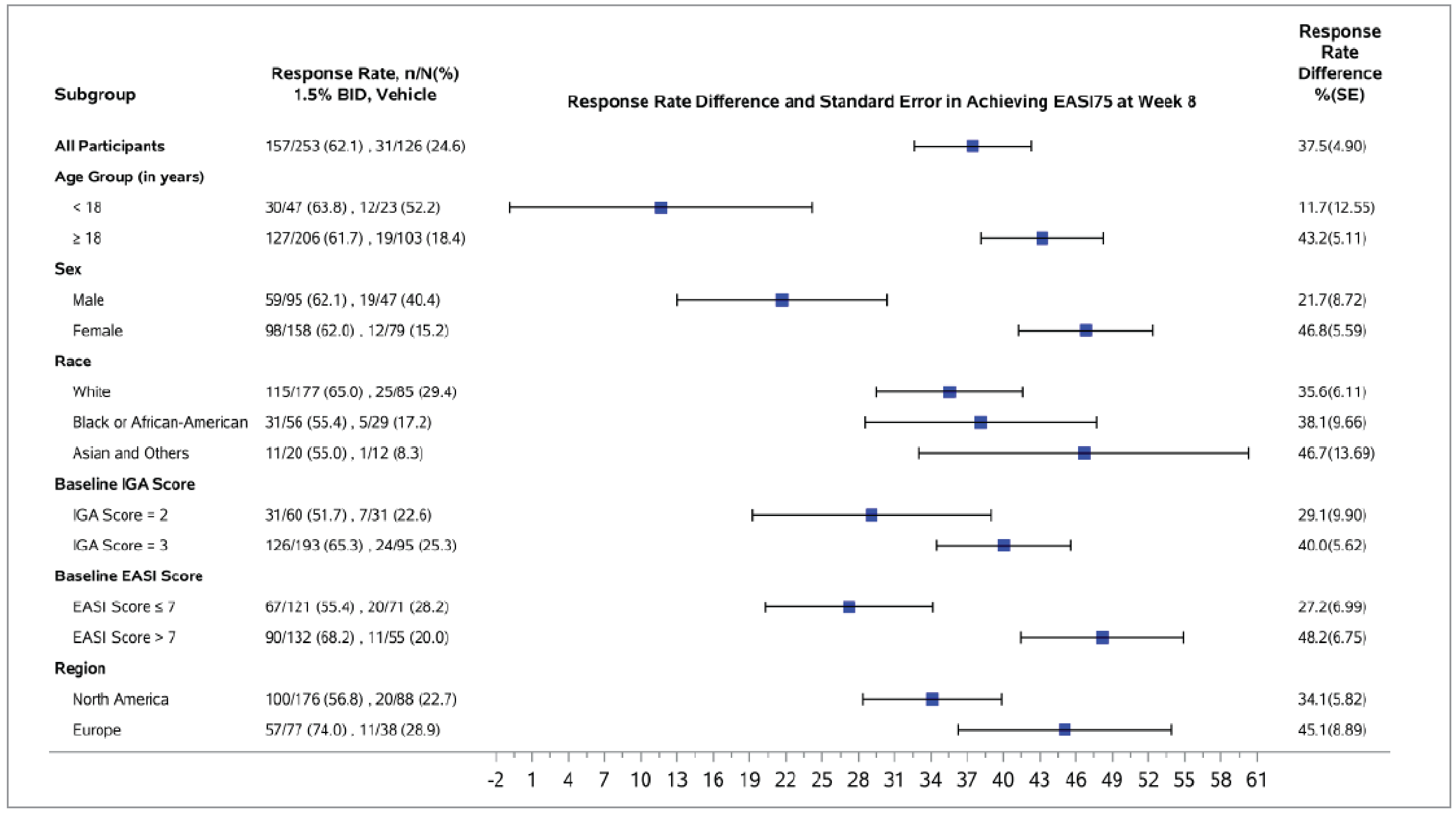
BID = twice a day; ITT = intention-to-treat; IGA = Investigator’s Global Assessment; EASI = Eczema Area Severity Index; SE = standard error; vs. = versus.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14
Figure 12: Prespecified Subgroup Analyses of Proportion of Patients Experiencing EASI-75 at Week 8 (Ruxolitinib 1.5% vs. Vehicle Cream) in TRuE-AD2 Trial (ITT Excluding Site 461)
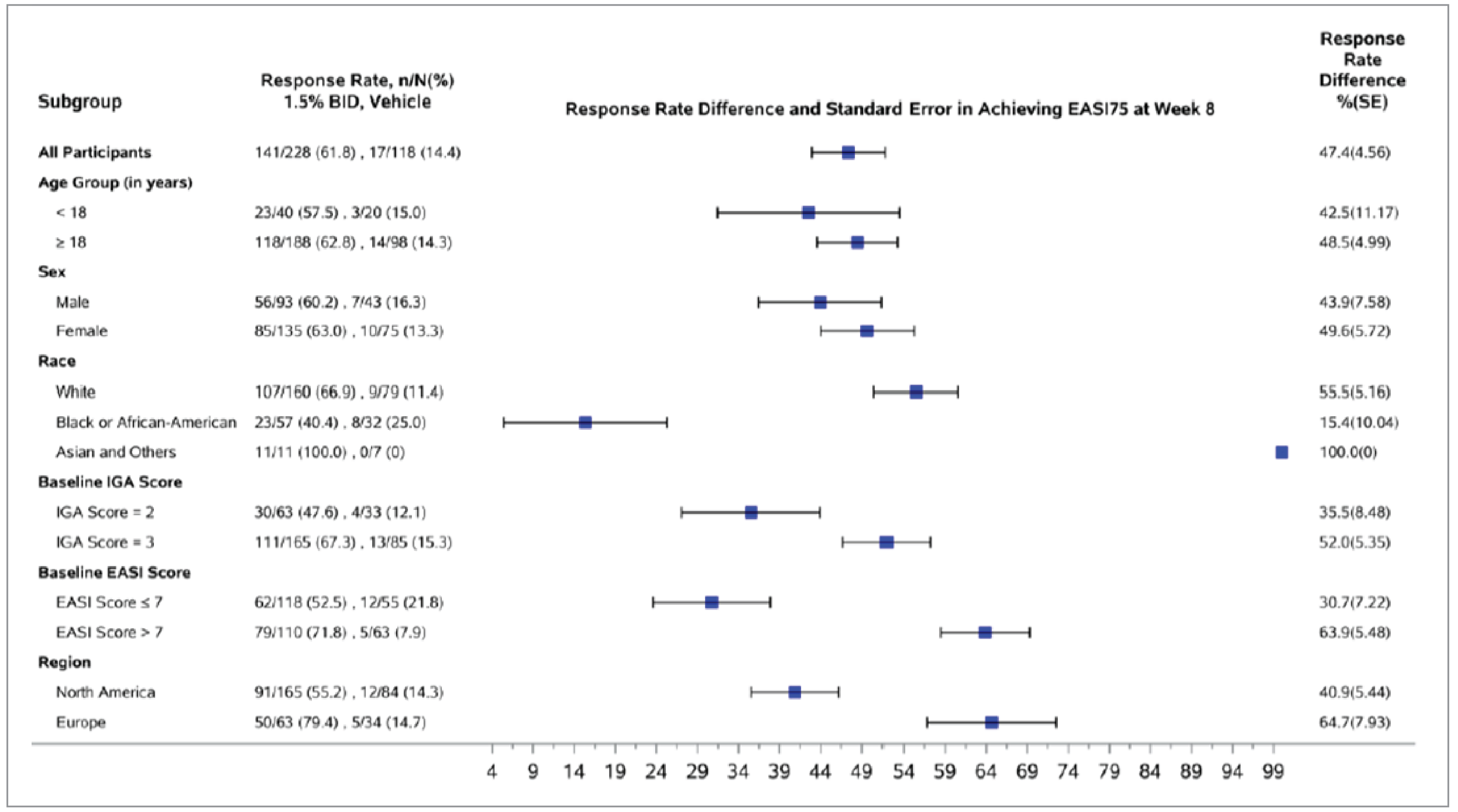
BID = twice a day; ITT = intention-to-treat; IGA = Investigator’s Global Assessment; EASI = Eczema Area Severity Index; SE = standard error; vs. = versus.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14
Post Hoc Subgroup Efficacy Results by History of Topical Treatment (Pooled Analysis of TRuE-AD1 and TRuE-AD2 Trials)
Table 28: Subgroup Analysis by History of Topical Treatment — Week 8 (ITT Population, Pooled Analysis of TRuE-AD1 and TRuE-AD2 Trials)
Subgroup | History of topical treatment use regardless of time frame | History of topical treatment use in the last 30 days before screening | ||||||
|---|---|---|---|---|---|---|---|---|
Total N | Response rate, n (%) | OR (95% CI) | P value | Total N | Response rate, n (%) | OR (95% CI) | P value | |
IGA-TS at week 8 | ||||||||
Overall | ||||||||
VEH | ███ | ██ ███ | █████ | ████ | ██ | ██ | ██ | ██ |
RUX | ███ | ███ ██ | ████ █ | ████ | ██ | ██ | ██ | ██ |
Previously received TCS | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ███ ██ | ████ █ | ████ | ███ | ███ ██ | ████ ██ | ███ |
Previously received TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCS and TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
No previous AD treatment | ||||||||
VEH | ██ | ████ | ██ | ██ | ██ | ██ | ██ | ██ |
RUX | ██ | ██ ██ | ██ | ██ | ██ | ██ | ██ | ██ |
EASI75 at week 8 | ||||||||
███████ | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCS | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCS and TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
No previous AD treatment | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Itch NRS-4 at week 8 | ||||||||
Overall | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCS | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
Previously received TCS and TCI | ||||||||
VEH | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
RUX | ███ | ██ ███ | █████ | ███ | ███ | ██ ██ | █████ | ████ |
AD = atopic dermatitis; CI = confidence interval; EASI = Eczema Area and Severity Index; IGA-TS = Investigator’s Global Assessment–Treatment Success; ITT = intention-to-treat; OR = odds ratio; NR = not reported; NRS = Numeric Rating Scale; RUX = ruxolitinib 1.5% cream; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; VEH = vehicle cream.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Additional Information Submitted by the Sponsor.101
Table 29: Subgroup Analysis of IGA 0/1 at Week 52 by History of Topical Treatment (Long-Term Safety Population, Pooled Analysis of TRuE-AD1 and TRuE-AD2 Trials)
Subgroup | History of topical treatment use regardless of time frame | History of topical treatment use in the last 30 days before screening | ||
|---|---|---|---|---|
Total N | Response rate, n (%) | Total N | Response rate, n (%) | |
Overall | ||||
VEH to RUX | ██ | ██ ██████ | ██ | ██ |
RUX to RUX | ███ | ███ ██████ | ██ | ██ |
Previously received TCS | ||||
VEH to RUX | ██ | ██ ██████ | ██ | ██ ██████ |
RUX to RUX | ███ | ███ ██████ | ███ | ███ ██████ |
Previously received TCI | ||||
VEH to RUX | ██ | ██ ██████ | ██ | ██ ██████ |
RUX to RUX | ██ | ██ ██████ | ██ | ██ ██████ |
Previously received TCS and TCI | ||||
VEH to RUX | ██ | ██ ██████ | ██ | ██ ██████ |
RUX to RUX | ██ | ██ ██████ | ██ | ██ ██████ |
IGA = investigator global assessment; NR = not reported; NRS = Numeric Rating Scale; RUX = ruxolitinib 1.5% cream; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; VEH = vehicle cream.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Additional information submitted by the sponsor.101
Table 30: Subgroup Analysis of Total Percent BSA Affected by Atopic Dermatitis at Week 52 by History of Topical Treatment (Long-Term Safety, Pooled Analysis of TRuE-AD1 and TRuE-AD2 Trials)
Subgroup | History of topical treatment use regardless of time frame | History of topical treatment use in the last 30 days before screening | ||||||
|---|---|---|---|---|---|---|---|---|
Total N | Baseline value | Week 52 value | Change from baseline | Total N | Baseline value | Week 52 value | Change from baseline | |
Overall | ||||||||
VEH to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
RUX to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
Previously received TCS | ||||||||
VEH to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
RUX to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
Previously received TCI | ||||||||
VEH to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
RUX to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
Previously received TCS and TCI | ||||||||
VEH to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
RUX to RUX | ██ | ███ | ████ | ████ | ██ | ██ | ██ | ██ |
BSA = body surface area; NR = not reported; RUX = ruxolitinib 1.5% cream; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; VEH = vehicle cream.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-AD2.13,14 Additional information submitted by the sponsor.101
Table 31: Subgroup Analysis of Harms by History of Topical Treatment (Pooled Analysis of TRuE-AD1 and TRuE-AD2 Trials)
Subgroup | History of topical treatment use regardless of time frame | History of topical treatment use in the last 30 days before screening | ||
|---|---|---|---|---|
VEH (N = 250)a or VEH to RUX (N = 96)b | RUX (N = 499)a or VEH to RUX (N = 428)b | VEH (N = 250)a or VEH to RUX (N = 96)b | RUX (N = 499)a or VEH to RUX (N = 428)b | |
VC period — week 8 (safety population) | ||||
Overall | ||||
n | ███ | ███ | ██ | ██ |
TEAE | ██ ██████ | ███ ██████ | ██ | ██ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCS | ||||
n | ███ | ███ | ███ | ███ |
TEAE | ██ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCI | ||||
n | ██ | ███ | ██ | ██ |
TEAE | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCS and TCI | ||||
n | ██ | ██ | ██ | ██ |
TEAE | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
LTS period — week 52 (LTS population) | ||||
Overall | ||||
n | ██ | ███ | ██ | ██ |
TEAE | ██ ██████ | ███ ██████ | ██ | ██ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCS | ||||
n | ██ | ███ | ██ | ███ |
TEAE | ██ ██████ | ███ ██████ | ██ ██████ | ███ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCI | ||||
n | ██ | ██ | ██ | ██ |
TEAE | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
Previously received TCS and TCI | ||||
n | ██ | ██ | ██ | ██ |
TEAE | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Serious TEAE | █████ | █████ | ██ | ██ |
WDAE | █████ | █████ | ██ | ██ |
b.i.d. = twice daily; LTS = long-term safety; NR = not reported; NRS = Numeric Rating Scale; RUX = ruxolitinib 1.5% cream; TEAE = treatment-emergent adverse event; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; VC = vehicle-controlled; VEH = vehicle cream; WDAE = withdrawal due to adverse events.
aApplicable to the vehicle-controlled period.
bApplicable to the long-term safety period.
Sources: Clinical Study Reports for TRuE-AD1 and TRuE-fAD2.13,14 Additional information submitted by the sponsor.101
Pharmacoeconomic Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
BIA
budget impact analysis
BSA
body surface area
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IGA
Investigator’s Global Assessment
ITC
indirect treatment comparison
NMA
network meta-analysis
QALY
quality-adjusted life-year
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ruxolitinib cream (Opzelura), 1.5% topical cream |
Indication | Topical treatment of mild to moderate AD in adult and pediatric patients 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (topical corticosteroids, topical calcineurin inhibitors) or when those therapies are not advisable |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 11, 2024 |
Reimbursement request | As per indication |
Sponsor | Incyte Biosciences Canada Corporation |
Submission history | Previously reviewed: Noa |
AD = atopic dermatitis; NOC = Notice of Compliance.
aThe oral form of ruxolitinib has been evaluated for other indications.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population |
|
Treatment | Ruxolitinib cream |
Dose regimen | Applied twice daily to affected skin areas (maximum of 20% BSA for each application) for 8 weeks and as needed thereafter |
Submitted price | $1,075.97 per 100 g tube |
Submitted treatment costs | First year: ██████ per person (based on ████ tubes per year)a Subsequent years: ██████ per person (based on ████ tubes per year)a |
Comparators | Base case: No active treatmentb Moderate AD scenario:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (50 years) |
Key data source | Effectiveness of ruxolitinib cream was informed by the TRuE-AD 1 and TRuE-AD2 trials in the sponsor’s base case. In the moderate AD scenario analysis, the effectiveness of abrocitinib, dupilumab, and upadacitinib was informed by the sponsor-conducted NMA. |
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
AD = atopic dermatitis; AE = adverse event; BSA = body surface area; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vs. = versus.
aTreatment cost was estimated by the sponsor based on observations from the TRuE-AD trials (BSA affected by AD, cream used per BSA affected, reduction in BSA affected by AD, and percentage of days on treatment) (refer to Table 12).
bIn the base case, the sponsor compared ruxolitinib cream to vehicle cream (from the TRuE-AD trials), which contained no active ingredient (i.e., ruxolitinib).
cTreatment response was assessed by use of EASI-75 (75% improvement in the Eczema Area and Severity Index). The sponsor additionally submitted a scenario analysis using the Investigator’s Global Assessment instrument; however, abrocitinib was excluded from that analysis.
Conclusions
The Canada’s Drug Agency (CDA-AMC) Clinical Review of the TRuE-AD1 and TRuE-AD2 trials found that, compared to vehicle cream (i.e., placebo), ruxolitinib cream likely results in clinically important improvements in the severity and extent of atopic dermatitis (AD) in patients aged 12 years and older with mild to moderate AD. Changes in the percentage of body surface area (BSA) affected by AD and changes in symptoms (including itch and impact on sleep) favoured ruxolitinib cream; however, these results were considered supportive owing to methodological limitations. Results from the pivotal trials suggest little to no clinically important improvement in health-related quality of life (HRQoL) with ruxolitinib cream in adolescent patients, and the evidence is very uncertain about the effect of ruxolitinib cream on HRQoL in adult patients. The results of an indirect treatment comparison (ITC) submitted by the sponsor comparing the efficacy of ruxolitinib cream versus dupilumab, upadacitinib, and abrocitinib in patients with moderate AD were considered inconclusive due to methodologic limitations and imprecision.
Clinical expert input received by CDA-AMC for this review indicated that patients who do not experience adequate disease control with topical treatments would generally try additional treatments, including systemic immunosuppressants (e.g., methotrexate, cyclosporine) and advanced systemic therapies (i.e., abrocitinib, upadacitinib, dupilumab), depending in part on disease severity. As such, the sponsor’s base case, which compared ruxolitinib to vehicle cream (i.e., no active treatment) is not informative for decision-making. No evidence was submitted by the sponsor comparing ruxolitinib cream to systemic immunosuppressants, and the sponsor’s ITC comparing ruxolitinib cream to the advanced systemic treatments indicated for moderate AD were inconclusive.
CDA-AMC identified additional important limitations with the economic submission, including uncertainty as to whether the efficacy data from the TRuE-AD trials adequately reflect the indicated population (i.e., patients who experience inadequate response or intolerance to topical corticosteroid [TCS] or topical calcineurin inhibitor [TCI]), uncertainty in the long term effectiveness and acquisition costs of ruxolitinib cream, logical fallacies in the modelling of discontinuation and subsequent therapy, limited efficacy data pertaining to adolescents, and poor modelling practices.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis, which included aligning the time horizon with the TRuE-AD trials (1 year), given the clinical uncertainty, and aligning the number of ruxolitinib tubes in year 1 with data from the TRuE-AD trials. Results of the CDA-AMC reanalyses suggest that, at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained, ruxolitinib cream is not a cost-effective option for the treatment of mild to moderate AD compared with no active treatment. A price reduction of at least 68% would be required for ruxolitinib cream to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. Owing to the remaining limitations, which CDA-AMC was unable to address, additional price reductions may be required.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process, specifically the information that pertains to the economic submission.
Patient input was received from the Eczema Society of Canada and the Canadian Skin Patient Alliance, which collected the perspectives of caregivers and patients with AD through surveys, interviews, and informal conversations. Patients with AD reported dry and itchy skin, painful lesions, and sleep disruption. Patients reported using TCSs, TCIs, topical phosphodiesterase type 4 inhibitors (crisaborole), systemic immunosuppressants (methotrexate, cyclosporine, azathioprine, oral steroids), and advanced systemic treatments (e.g., dupilumab) to treat AD. Treatment goals included improvement of itch, dryness, inflammation, blistering, and cracked skin; sleep improvement; fewer AD flares; decreased short-term and long-term adverse events (AEs); and improved quality of life. Patients who had experience with ruxolitinib cream noted that stinging or burning after the application of ruxolitinib cream was rare.
Clinician input was received from the Canadian Dermatology Association and the Atlantic Dermatology Specialist Group. The clinician input noted that current treatments for patients with AD included TCSs, TCIs, and topical phosphodiesterase type 4 inhibitors (crisaborole). For moderate to severe AD, additional options were noted to include phototherapy, systemic immunosuppressants, and advanced systemic treatments (dupilumab, tralokinumab, upadacitinib, abrocitinib). The clinicians noted that the goals of treatment included the short-term and long-term management of AD, the resolution of itch, skin clearance, reduced AEs, and improved quality of life and that response to treatment would be assessed by signs and symptoms after 8 weeks of induction therapy. The input noted that physician-reported clinical scoring systems such as Investigator’s Global Assessment (IGA), IGA treatment success, or a 75% improvement in the Eczema Area and Severity Index may also be used to evaluate treatment response. The clinicians noted that ruxolitinib cream may be used as monotherapy or as an adjunct to systemic therapy and that treatment should be continued until inadequate response after induction, loss of response, recurrent AD flares, or AEs.
The drug plans highlighted concerns with the relevant comparators and the outcomes used to measure treatment response. The plans raised concerns with the long-term dosing of ruxolitinib cream and its use in combination with other systemic therapies.
Several of these concerns were addressed in the sponsor’s model:
Costs and disutilities associated with some AEs, including AD flares, were included.
Quality of life was incorporated in the sponsor’s model by use of EQ-5D data collected in the TRuE-AD trials.
In scenario analyses, the cost-effectiveness of ruxolitinib cream was compared to advanced systemic treatments; however, no comparison to systemic immunosuppressants was provided.
CDA-AMC was unable to address the following concerns raised from the relevant input:
The cost-effectiveness of ruxolitinib cream compared to systemic immunosuppressants (e.g., methotrexate, cyclosporine) owing to the lack of submitted clinical data.
The cost-effectiveness of ruxolitinib cream when used in combination with other treatments for AD; however, CDA-AMC notes that the draft Health Canada monograph recommends against concomitant use with “other JAK [Janus kinase] inhibitors, biological immunomodulators or potent immunosuppressants.”
Economic Review
The current review is for ruxolitinib cream (Opzelura) for the topical treatment of mild to moderate AD in patients aged 12 years and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or for whom those therapies are not advisable.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of ruxolitinib cream 1.5% (from now on referred to as ruxolitinib cream) compared with no active treatment (represented by vehicle cream in the pivotal trials) in the indicated population (mild to moderate AD), aligned with the TRuE-AD1 and TRuE-AD2 trials, as well as a scenario analysis in which ruxolitinib cream was compared with dupilumab, upadacitinib, and abrocitinib among patients with moderate AD. The Health Canada indication specifies that ruxolitinib cream should be used by patients with mild to moderate AD that is inadequately controlled with TCS or TCI (or for patients for whom those treatments are not advisable); however, the TRuE-AD trial populations were not restricted based on prior treatment use.
Ruxolitinib cream is supplied in 100 g tubes, with a recommended application of a thin layer twice daily to affected areas (up to 20% of BSA for each application) until the signs or symptoms (e.g., itch, rash, redness) of AD resolve.1 The monograph specifies that if the signs or symptoms do not resolve within 8 weeks, re-evaluation by a health care provider is warranted. In the model, the sponsor assumed that ruxolitinib cream would be used “as needed” beyond 8 weeks. At the submitted price of $1,076 per 100 g tube, the sponsor estimated the annual per-patient cost of ruxolitinib cream among patients with mild or moderate AD to be ██████ in the first year (based on ████ tubes) and ██████ in subsequent years (████ tubes), while “no active treatment” had no cost. Among patients with moderate AD (scenario analysis), the sponsor estimated the annual per-patient cost of ruxolitinib cream to be ███████ in the first year (based on █████ tubes) and ██████ based on ████ tubes in subsequent years, while the cost of dupilumab, upadacitinib, and abrocitinib ranged from $17,776 to $27,029.
The analysis was undertaken from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years, QALYs) were estimated over a lifetime time horizon (50 years), discounted at an annual rate of 1.5%. The cycle length was 1 month.
Model Structure
The sponsor submitted a Markov model that included a short-term induction phase (8 weeks) and a long-term maintenance phase (Figure 1). In the sponsor’s base case, patients with mild to moderate AD receive ruxolitinib cream or no active treatment for 8 weeks, after which all patients enter the maintenance phase, receiving either ruxolitinib cream or no active treatment. During model cycle 3 (representing weeks 8 to 12), patients who experience a response to treatment (defined as an IGA score of 0 or 1 with a ≥ 2-grade improvement from baseline [IGA treatment success]) remain in their initial treatment group (ruxolitinib cream or no active treatment), while those who experience no or an inadequate response move to subsequent treatment, assumed to be dupilumab for all patients. In each cycle, after week 12 the sponsor assumed that 0.3% of ruxolitinib patients would discontinue treatment and move to subsequent treatment and that 2% of patients who received no active treatment would initiate subsequent treatment.
Model Inputs
The baseline characteristics in the model were derived from the TRUE-AD1 and TRuE-AD2 trials (mean patient age: 35.8 years; 61.7% female and 38.3% male; mean percentage of BSA affected: 9.4%).2,3 In the moderate AD scenario analysis, patients had a higher starting percentage of BSA affected by AD (18%).
The clinical efficacy inputs (the proportion of patients who experienced response to treatment; the proportion of patients who discontinued treatment) were derived from the TRuE-AD1 and TRuE-AD2 trials.2,3 In the base case, the proportion of patients who experienced response to subsequent treatment (i.e., dupilumab) was obtained from the sponsor-submitted network meta-analysis (NMA). Similarly, the proportion of patients who experienced response to treatment (dupilumab, upadacitinib, and abrocitinib) in the moderate AD scenario analysis was obtained from the sponsor’s NMA.4 The sponsor assumed that treatment did not affect mortality risk. Age-specific and sex-specific mortality rates were based on general population life tables from Statistics Canada.5
Health state utility values were derived from patient-level EQ-5D-5L data from the ruxolitinib cream (1.5%) arm of the TRuE-AD1 and TRuE-AD2 trials, valued using UK tariffs.2,3 The sponsor included AEs that occurred in more than 1% of patients in either the TRuE-AD1 or TRuE-AD2 trial in the first 8 weeks of treatment with ruxolitinib cream or no active treatment (nasopharyngitis, upper respiratory tract infection, headache, application site reactions, and AD flares). Additional AEs included in the model were herpes simplex, conjunctivitis, neutropenia, and injection site reactions. Disutility values were obtained from the literature.6-10 The sponsor assumed that AD flares were AEs associated with a temporary decrease in HRQoL. In the moderate AD subgroup analysis, the rate of AEs was based on a naive comparison of clinical trial data.2,3,11-14
Costs in the sponsor’s base case included those associated with drug acquisition, AEs, and disease management and monitoring. Drug acquisition costs for ruxolitinib were based on the sponsor’s submitted price, with ruxolitinib cream usage based on observations from the TRuE-AD1 and TRuE-AD2 trials.2,3 During the 8-week induction phase, the sponsor assumed that ████ tubes would be used (█████ g of ruxolitinib cream, to treat ██████ ████ ███ m2 of BSA). During the maintenance phase, the sponsor assumed that the proportion of mean total BSA treated would decrease from 2.93% in the first year to 1.41% in subsequent years. The sponsor estimated that ████ tubes of ruxolitinib cream would be used during the 8 week induction period (based on █████ of mean total BSA [█████]), with ████ tubes used in the maintenance period in the first year (████ tubes total in the first year) and ████ tubes per year in subsequent years. The sponsor assumed no administration cost for ruxolitinib cream. Costs related to AEs included family physician visits, and AD flares were assumed to be managed by a visit to a dermatologist and a 100 g tube of high-potency TCS (fluocinonide 0.05% cream). Disease management costs included dermatology consultation during induction, and complex assessment, repeat consultation, and phototherapy during maintenance. Resource use was estimated based on clinical expert opinion, published literature, and product monographs.15-18 Monitoring costs included physician visits and laboratory tests, which were obtained from the Ontario Schedule of Benefits for Physician Services and the Ontario Schedule of Benefits for Laboratory Services.19,20 The sponsor assumed that best supportive care (BSC) would include phototherapy as part of disease management.
In the scenario analysis involving patients with moderate AD, the cost of dupilumab was obtained from the IQVIA DeltaPA Database and the cost of upadacitinib and abrocitinib was obtained from previous CADTH reviews.21-23 The dosing for each drug was obtained from their monographs.17,18,24 The sponsor included an administration cost for dupilumab. Patients who received upadacitinib or abrocitinib were assumed to incur monthly monitoring costs during induction and every 3 months during maintenance.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (2,000 iterations). The results of the deterministic and probabilistic analyses were similar; the results of the probabilistic analyses are presented in the following section. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base case, which considered both mild and moderate AD, ruxolitinib cream was more effective (incremental QALYs: 0.93) and less costly (incremental savings: $5,295) than no active treatment (Table 3). At the end of the 50-year time horizon, approximately 63% of patients remained alive in each treatment group.
Results were driven by the lower cost of subsequent therapy among patients who received ruxolitinib cream (incremental savings: $23,618), which offset the acquisition cost of ruxolitinib cream ($19,801) (Table 13).
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Mild to Moderate AD
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no active treatment ($/QALY gained) |
|---|---|---|---|---|---|
No active treatment | 110,106 | Reference | 21.72 | Reference | Reference |
Ruxolitinib cream | 104,811 | –5,295 | 22.65 | 0.93 | Dominant |
AD = atopic dermatitis; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.15
Sensitivity and Scenario Analysis Results
The sponsor conducted several additional scenario analyses, including restricting usage to patients with moderate AD only, assuming that patients would use 2 tubes of ruxolitinib cream per year, removing subsequent treatment costs, adopting alternative utility values,25 adopting a shorter time horizon, and adopting different discount rates. The incremental cost-effectiveness ratio (ICER) was sensitive to assumptions about patient population and subsequent therapy. When subsequent therapy costs were removed (i.e., all patients were assumed to receive BSC after initial therapy, which the sponsor assumed had zero cost), ruxolitinib cream was no longer cost saving and was associated with an ICER of $17,602 per QALY gained.
In the scenario that included only patients with moderate AD, ruxolitinib cream was more effective (higher QALYs) than dupilumab, upadacitinib, and abrocitinib and was less costly than dupilumab and upadacitinib (Table 14). In sequential analyses, ruxolitinib cream was associated with an ICER of $11,807 per QALY gained compared with abrocitinib.
The sponsor additionally conducted a scenario analysis from a societal perspective that included additional costs associated with productivity loss. The results of this scenario analysis were similar to the sponsor’s base case using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
It is unclear whether the modelled population adequately reflects the Health Canada indication. Ruxolitinib cream is indicated for use by patients with mild or moderate AD that is not adequately controlled with topical prescription therapies (TCI, TCS) or when those therapies are not advisable. In the sponsor’s economic model, the clinical efficacy of ruxolitinib cream (i.e., the proportion of patients who experience a treatment response) was informed by observations from the TRuE-AD1 and TRuE-AD2 trials. Although the sponsor submitted post hoc subgroup analyses in patients with a recent history of TCS and/or TCI treatment as supporting clinical evidence, it is unclear whether these patients experienced an inadequate response to prior exposure, and these data were not used in the sponsor’s economic evaluation.
CDA-AMC was unable to address this limitation owing to uncertainty in the clinical trial population.
The sponsor’s base case compared ruxolitinib cream to no active treatment. The sponsor’s submitted base case compared ruxolitinib cream to vehicle cream from the TRuE-AD clinical trials. As described in the CDA-AMC Clinical Review, vehicle cream looked and felt the same as ruxolitinib cream but contained no active ingredient (i.e., ruxolitinib). Clinical expert input received by CDA-AMC for this review noted that patients who do not experience adequate disease control with topical treatments generally try additional treatments, including systemic immunosuppressants (e.g., methotrexate, cyclosporine), advanced systemic therapies (i.e., abrocitinib, upadacitinib, dupilumab), and phototherapy. As such, the comparison of ruxolitinib cream to no active treatment is not relevant to the decision problem.
CDA-AMC notes that the sponsor requested a deviation from the CDA-AMC pharmacoeconomic requirements to exclude all comparators from the economic model, citing that an ITC was not feasible for the indicated population. Although this request was approved by CDA-AMC, the sponsor was advised that this would be considered a major limitation of its submission. Instead of modelling the patient population as a whole (i.e., mild to moderate AD), it may have been more appropriate to consider the cost-effectiveness of ruxolitinib cream in each subgroup separately. Although the sponsor submitted a scenario analysis for patients with moderate AD, in which ruxolitinib cream was compared to dupilumab, abrocitinib, and upadacitinib, no corresponding analysis was provided for mild AD. As such, the cost-effectiveness of ruxolitinib cream among patients with mild AD is unknown.
The sponsor’s base case comparing ruxolitinib cream to no active treatment is not informative for decision-making. Although the sponsor-provided scenario analysis suggests that ruxolitinib cream may be cost-effective in patients with moderate AD, the cost-effectiveness of ruxolitinib cream among patients with mild AD, and hence the full indication, is unknown. CDA-AMC was unable to address this limitation.
The comparative effectiveness and safety of ruxolitinib cream is uncertain. Given that the TRuE-AD1 and TRuE-AD2 trials compared ruxolitinib cream to vehicle cream, there has been no head-to-head comparison of ruxolitinib cream to a relevant comparator (e.g., systemic immunosuppressants, advanced systemic treatments), and the sponsor deemed an ITC for the full indicated population unfeasible. To inform efficacy for the scenario analysis that included the use of dupilumab, upadacitinib, and abrocitinib among patients with moderate AD, the sponsor conducted an NMA to estimate the relative proportion of patients who experience a treatment response. As noted in the CDA-AMC Clinical Review, the results of this NMA were inconclusive due to important limitations that prevented CDA-AMC from verifying whether the underlying assumptions of homogeneity and consistency were met, as well as imprecision in the estimates. Notably, the credible intervals from the sponsor’s NMA suggest that there may be no meaningful difference in treatment response between ruxolitinib cream and advanced systemic treatments.
The sponsor incorporated AEs in the economic model via naive comparison, directly using data from the TRuE-AD trials for ruxolitinib cream and from the respective clinical trials for dupilumab, abrocitinib, and upadacitinib.2,3,11-14 Owing to the direct use of trial data, it is not possible to determine if any observed differences between therapies are due to the treatment or due to bias or confounding factors (e.g., differences in study populations, definitions of outcomes, or study designs). Further, although the scenario analysis involving these treatments was for moderate AD only, the sponsor used AE data from the entire trial populations, rather than data specific to patients with moderate AD. Finally, the long-term comparative safety of these treatments is unknown owing to a lack of data. Although the CDA-AMC Clinical Review notes that ruxolitinib cream appeared to be well tolerated through week 52 in the pivotal trials, a longer duration of follow-up is required to capture long-term safety, particularly for potential rare harms.
Given the lack of direct evidence for ruxolitinib cream compared to any comparator and limitations with the sponsor’s ITC, it remains uncertain whether ruxolitinib cream provides a net clinical benefit over currently used treatments for either mild or moderate AD.
The long-term effectiveness of ruxolitinib cream is highly uncertain. In the pharmacoeconomic submission, the sponsor assumed that patients would use ████ tubes of ruxolitinib cream per year starting in year 2 (Table 12), based on the assumption that observations from the TRuE-AD trials (up to 52 weeks) will be maintained over the lifetime analysis horizon (50 years). Because of an absence of long-term data, it is highly uncertain whether these benefits (e.g., reduction in BSA affected by AD) will be realized in clinical practice. Further, the sponsor’s model did not consider effectiveness waning. If long-term effectiveness is lower than anticipated by the sponsor (i.e., if effectiveness waning occurs), patients who continue to use ruxolitinib cream may require more tubes per year, and the drug acquisition costs will be higher than assumed by the sponsor.
In the CDA-AMC reanalysis, the analysis horizon was assumed to be 1 year (aligned with the duration of the pivotal trial plus the long-term safety phase) owing to the lack of long-term data about the effectiveness (and hence treatment costs) of ruxolitinib cream beyond 1 year.
The acquisition cost of ruxolitinib cream is highly uncertain. In the economic model, the sponsor assumed that patients with mild to moderate AD would use ████ tubes of ruxolitinib cream in year 1 and ████ in subsequent years. Although these estimates were based on observations from the TRuE-AD trials (pivotal trials plus the long-term extension study), the number of tubes that will be used in clinical practice is highly uncertain. First, observations from the TRuE-AD trials (mean patient BSA, daily cream per BSA treated, and mean percentage of BSA affected by AD) were used to derive the number of tubes used in the induction and maintenance health states. However, observations from the TRuE-AD trials suggest that ████ tubes would be required in the first year (mean weight of ruxolitinib cream over 52 weeks: TRuE-AD1 ███ g, TRuE-AD2 ███ g), which is greater than the amount calculated based on the individual trial inputs. As noted in the previously discussed limitation (about long-term effectiveness), in the second year (and thereafter) the usage of ruxolitinib cream will depend on its long-term efficacy, and the clinical expert input received by CDA-AMC noted that the assumptions incorporated by the sponsor for mean percentage of BSA affected by AD and the number of days treated in the maintenance period may be underestimated. CDA-AMC additionally notes that at least 2 tubes will be required per patient per year, as the sponsor has indicated that each tube should be used within 6 months of opening, based on an in-house stability study.
Second, the sponsor included AD flares in the model as an AE with an assigned utility and treatment cost (dermatologist visits and use of a high-potency TCS); however, the clinical expert input indicated that the amount of topical creams used, including ruxolitinib cream, is expected to vary based on the frequency and extent of AD flares. The rate of flares in the sponsor’s model was based on data from the 8-week TRuE-AD trials and was assumed to stay constant over the remainder of the 50-year analysis horizon.
Third, the amount of cream used in the trial took patient adherence into account (approximately 97% in the TRuE-AD trials), which may be lower in clinical practice than in a trial setting. Clinical expert input received by CDA-AMC indicates that adherence to topical treatments, especially by adolescents, may not be optimal. If adherence is lower in practice, the drug cost would remain the same, but the BSA affected may not decrease to the extent observed in the trial. Finally, the sponsor did not consider drug wastage in its calculation of drug costs. Expert feedback obtained for this review noted that drug wastage should be anticipated with ruxolitinib cream and some comparators as a result of not all product being squeezed from the tube or too much product being squeezed from the tube during each application.
In the CDA-AMC reanalysis, patients were assumed to require 8 tubes of ruxolitinib in year 1.
The modelling of subsequent therapy does not adequately reflect clinical practice. In the sponsor’s base case, all patients who do not experience an adequate response at the end of the induction period or who discontinue initial treatment (ruxolitinib cream or no active treatment) receive dupilumab as subsequent therapy. Dupilumab is indicated for the treatment of moderate to severe AD, thus the sponsor has implicitly assumed that patients with mild AD will have progressed to moderate AD after 8 weeks. The sponsor’s disaggregated base-case results (Table 13) suggest that the predicted cost savings associated with ruxolitinib cream compared to no active treatment are due to the lower drug acquisition costs of subsequent treatment; however, whether there will be cost savings, and the magnitude of any such savings, in clinical practice is highly uncertain, as several treatments that are less costly than dupilumab could be used as subsequent therapy (Appendix 1).
In the sponsor’s subgroup analysis involving only patients with moderate AD, patients were assumed to receive BSC (emollients, at zero cost) after discontinuing initial treatment with ruxolitinib cream, dupilumab, upadacitinib, or abrocitinib. Expert feedback indicates that, in clinical practice, patients would likely try additional treatments, including alternative advanced therapies or ruxolitinib cream in combination with other systemic treatments. The direction of bias resulting from this assumption is uncertain and will depend, at least in part, on confidentially negotiated drug prices.
In the CDA-AMC reanalysis, costs related to subsequent therapy were removed.
The modelling of treatment discontinuation lacks face validity. In its base case, the sponsor assumed that in each model cycle after cycle 3 (week 12 onward, for the remainder of the 50-year horizon), 0.3% of patients who received ruxolitinib cream and 2% of patients who received no active treatment would discontinue treatment. This assumption lacks face validity for several reasons. First, it is not possible to discontinue no active treatment. Second, the sponsor noted that patients could discontinue treatment due to either AEs or loss of response. While discontinuing because of AEs may be reasonable for ruxolitinib cream, AEs are unlikely to occur for patients who do not receive active treatment, and discontinuation because of a lack of response is accounted for in the model at the end of the induction period (week 8). Third, the sponsor applied the 8-week pivotal trial discontinuation rates to the 50-year model horizon. It would have been more appropriate to apply the discontinuation rates from the long-term safety study for ruxolitinib; however, there are no data to inform the discontinuation of ruxolitinib cream versus any active treatment.
In the CDA-AMC reanalysis, the discontinuation rate for no active treatment from 12 weeks onward was removed for the no active treatment group.
The cost-effectiveness of ruxolitinib cream among adolescents is uncertain. Ruxolitinib cream is indicated for patients aged 12 years and older. However, the cost-effectiveness among adolescents is uncertain for several reasons. First, efficacy in the pharmacoeconomic model was informed by observations from the TRuE-AD1 and TRuE-AD2 trials, in which the majority of the patients were adults (approximately 80% in each). As noted in the CDA-AMC Clinical Review, the generalizability of the study results to adolescents in clinical practice could potentially be limited by the small proportion of adolescents enrolled in the trials. Second, clinical expert input received by CDA-AMC indicates that adherence to topical treatments may be lower among adolescents, which may affect drug acquisition costs and hence cost-effectiveness estimates. Third, the results from the TRuE-AD1 pivotal trials suggest that there may be little to no clinically important improvement in HRQoL with ruxolitinib cream among adolescent patients, whereas a benefit was observed in adults (although considered very uncertain by CDA-AMC). Finally, the sponsor used recommended dosing for adults in calculating the costs of some comparators (i.e., dupilumab, in the moderate AD scenario analysis).
CDA-AMC was unable to address this limitation. The cohort starting age of the model is 36 years, and no subgroup analyses were provided for adolescents. Whether the cost-effectiveness findings apply equally to adolescents and to adults is unknown.
Poor modelling practices were employed. The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors. CDA-AMC additionally identified several programming issues in the model, including a discount rate being applied in year 1 and coding errors in the formulas used to sum life-years, QALYs, and costs over the model horizon.
CDA-AMC was unable to address the use of IFERROR statements in the model and notes that a thorough validation of the sponsor’s model was not possible. In its reanalysis, CDA-AMC removed the discount rate in year 1 and corrected errors in the Excel syntax used to sum life-years, QALYs, and costs over the model horizon.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Ruxolitinib cream will be used by patients with mild to moderate AD that is not adequately controlled with topical prescription therapies or when those therapies are not advisable. | Uncertain. Clinical expert input received for this review noted that there is a lack of standardized definition of inadequate control in clinical practice. In practice, response to treatment is generally assessed by clinician judgment and patient reports as to whether symptoms have improved. |
Treatment response is assessed by use of IGA and EASI scores. | Uncertain. The clinical experts consulted for this review indicated that these instruments are not commonly used in clinical practice. IGA was designed for and is commonly used in clinical trials but is not used in practice, whereas the EASI score may be used in practice. Most clinicians, however, assess response based on improvement in lesional inflammation (reduced area, reduced thickness, reduced redness, reduced lichenification, reduced oozing), patient-reported improvement in pruritus (supported by observation of reduced excoriation and reduced lichenification), and patient’s global assessment of treatment response. |
Patients receive 2 dermatology assessments each year. | Uncertain. The clinical expert input noted that the sponsor’s estimated number of dermatology assessments during disease management may have been overestimated because in clinical practice patients are not seen by clinics as frequently as modelled. |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency; EASI = Eczema Area and Severity Index; IGA = Investigator's Global Assessment.
CDA-AMC Reanalyses of the Economic Evaluation
Reanalysis Results
CDA-AMC was unable to derive a base-case analysis because of the highly uncertain nature of the clinical data and the limitations of the model structure. Instead, reanalyses were conducted to address some of the identified limitations in the sponsor’s analysis. CDA-AMC notes that the magnitude of clinical benefit estimated for ruxolitinib cream in this reanalysis may still be overestimated. The reanalyses are summarized in Table 5; these were derived using input from the clinical experts.
CDA-AMC was unable to address the other identified limitations, including uncertainty as to whether the modelled population adequately reflects the Health Canada–indicated population, the comparison to no active treatment and the exclusion of relevant comparators, the uncertainty in the comparative effectiveness data, and the lack of long-term efficacy data.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1. Discounting of costs and benefits | Starting at year 0 | Starting at year 1 |
2. Error in Excel syntax used to sum the LYs, QALYs, and costs over the model horizona | =ROUNDUP(SUM(AB13:OFFSET(AB13, horizon*(365.25/(cyc_length*7)),0))/num_ pts,10) | = sum(AB13:AB664)/num_pts |
Changes to derive the CDA-AMC base case | ||
1. Time horizon | Lifetime | 1 year, aligned with TRuE-AD trial duration (pivotal trials plus long-term extension study) |
2. Subsequent treatment | Included | Excludedb |
3. Tubes of ruxolitinib cream in year 1 | ████ tubes | 8 tubes |
4. Discontinuation rate from no active treatment, week 12 onward | Included | Excluded |
CDA-AMC reanalysis | ― | 1 + 2 + 3 + 4 |
CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
aAn example syntax is presented in this table. Changes were applied to model engines for ruxolitinib cream and all comparators. The number of rows shown in the example syntax (CDA-AMC correction) pertains to a horizon of 50 years.
bThe costs and efficacy of subsequent treatment were excluded.
CDA-AMC undertook a stepped analysis, incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 6; disaggregated results are presented in Appendix 4).
Results from the CDA-AMC reanalysis suggest that ruxolitinib cream is associated with higher costs (incremental costs: $6,747) and higher QALYs (incremental QALYs: 0.04) than no active treatment, resulting in an ICER of $151,282 per QALY gained over a 1-year horizon. Results were driven by the drug acquisition cost of ruxolitinib cream (incremental costs: $6,710; Table 16).
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | No active treatment | 110,106 | 21.72 | Reference |
Ruxolitinib cream | 104,811 | 22.65 | Dominant | |
Sponsor’s base case (deterministic) | No active treatment | 108,194 | 21.71 | Reference |
Ruxolitinib cream | 103,448 | 22.65 | Dominant | |
Sponsor’s base case (deterministic; corrected) | No active treatment | 108,090 | 21.42 | Reference |
Ruxolitinib cream | 103,052 | 22.35 | Dominant | |
CDA-AMC reanalysis 1 | No active treatment | 12,632 | 0.79 | Reference |
Ruxolitinib cream | 11,917 | 0.83 | Dominant | |
CDA-AMC reanalysis 2 | No active treatment | 655 | 21.00 | Reference |
Ruxolitinib cream | 21,366 | 22.03 | 3,012 | |
CDA-AMC reanalysis 3 | No active treatment | 108,090 | 21.42 | Reference |
Ruxolitinib cream | 104,668 | 22.35 | Dominant | |
CDA-AMC reanalysis 4 | No active treatment | 96,941 | 21.74 | Reference |
Ruxolitinib cream | 103,052 | 22.35 | 10,019 | |
CDA-AMC base case (1 + 2 + 3 + 4) | No active treatment | 168 | 0.78 | Reference |
Ruxolitinib cream | 6,915 | 0.82 | 151,361 | |
CDA-AMC reanalysis (1 + 2 + 3 + 4) (probabilistic) | No active treatment | 167 | 0.78 | Reference |
Ruxolitinib cream | 6,915 | 0.82 | 151,282 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The results of all steps are presented deterministically unless otherwise indicated.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s results and the CDA-AMC base case (Table 7). The CDA-AMC base case suggests that a 68% price reduction would be required for ruxolitinib cream to be considered cost-effective at a WTP threshold of $50,000 per QALY relative to no active treatment.
Table 7: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for ruxolitinib cream vs. no active treatment ($/QALY) | |
|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis | ||
No price reduction | 1,076 | Dominant | 151,361 |
10% | 968 | NA | 136,308 |
20% | 861 | NA | 121,254 |
30% | 753 | NA | 106,201 |
40% | 646 | NA | 91,148 |
50% | 538 | NA | 76,095 |
60% | 430 | NA | 61,042 |
70% | 323 | NA | 45,988 |
80% | 215 | NA | 30,935 |
90% | 108 | NA | 15,882 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
CDA-AMC conducted a scenario analysis to evaluate the impact of greater ruxolitinib cream usage in year 1 (11 tubes) on the cost-effectiveness analysis. The results from this analysis, which included revisions aligned with the CDA-AMC reanalysis, are presented in Table 17. In this analysis, ruxolitinib cream was associated with an incremental cost of $9,975 and an incremental QALY gain of 0.04 compared with no active treatment, resulting in an ICER of $223,776 per QALY gained for the first year of treatment.
CDA-AMC conducted an additional scenario analysis exploring the cost-effectiveness of ruxolitinib cream in moderate AD (Table 18). Results of the sequential analysis suggest that ruxolitinib cream is more effective than no active treatment (incremental QALYs: 0.07) with an ICER of $159,238 per QALY gained for the first year; the other treatments were dominated by ruxolitinib cream (i.e., it was less costly and more effective). In this analysis, which assumed equal efficacy and safety between ruxolitinib cream and dupilumab, abrocitinib, and upadacitinib (based on the findings of the sponsor’s NMA and the CDA-AMC critical appraisal), ruxolitinib cream was associated with higher QALYs (incremental QALYs: 0.01) than these advanced treatments. This finding was entirely driven by the timing of the initial response assessment — that is, if response is assessed at the same time point for all treatments, incremental QALYs would be zero. The results of this analysis suggest that ruxolitinib cream is less costly than abrocitinib, upadacitinib, and dupilumab (cost savings: $3,977 to $11,677); however, because the costs of the comparators are based on public list prices (i.e., confidential negotiated discounts are not accounted for), it is highly uncertain whether these savings will be realized in practice. Thus, on the basis of this analysis, it is uncertain whether the use of ruxolitinib cream will result in improved QALYs or reduced costs relative to other advanced treatments for moderate AD.
Issues for Consideration
The Health Canada monograph specifies that ruxolitinib cream should be used on no more than 20% of a patient’s BSA;1 however, it is unclear whether patients will adhere to this guidance in practice.
Ruxolitinib cream is available only as a 100 g tube (submitted price: $1,076 per tube). If a patient discontinues treatment (e.g., because of AEs or inadequate response), the full acquisition cost would still be incurred by the drug plans.
The sponsor has indicated that each tube of ruxolitinib cream, once opened, should be used within 6 months based on the results of an in-house stability study. As such, each patient would require at least 2 tubes of ruxolitinib cream per year, regardless of the amount of drug used.
Clinical expert input received by CDA-AMC indicated that in clinical practice ruxolitinib cream may used in combination with other treatments (e.g., with systemic treatments). The cost-effectiveness of ruxolitinib cream when used in combination with other treatments is unknown owing to the lack of clinical data. CDA-AMC notes that the draft Health Canada monograph recommends against concomitant use of ruxolitinib cream with “other JAK [Janus kinase] inhibitors, biological immunomodulators or potent immunosuppressants.”
Abrocitinib and upadacitinib have successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance for the treatment of AD. Dupilumab has successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance for the treatment of adults with AD and is under consideration for negotiations for the treatment of pediatric patients with AD. It is therefore likely that these treatments are reimbursed, or will be reimbursed, by jurisdictional drug plans at confidential prices that are less than the publicly available list prices.
Overall Conclusions
The CDA-AMC Clinical Review of the TRuE-AD1 and TRuE-AD2 trials found that, compared to vehicle cream (i.e., placebo), ruxolitinib cream likely results in clinically important improvements in the severity and extent of AD in patients aged 12 years and older with mild to moderate AD. Changes in the percentage of BSA affected by AD and changes in symptoms (including itch and impact on sleep) favoured ruxolitinib cream; however, these results were considered supportive owing to methodological limitations. The results from the pivotal trials suggest little to no clinically important improvement in HRQoL with ruxolitinib cream in adolescent patients, and the evidence is very uncertain about the effect of ruxolitinib cream on HRQoL in adult patients. The results of an ITC submitted by the sponsor comparing the efficacy of ruxolitinib cream versus dupilumab, upadacitinib, and abrocitinib in patients with moderate AD were considered inconclusive due to methodologic limitations and imprecision.
Clinical expert input received by CDA-AMC for this review indicated that patients who do not experience adequate disease control with topical treatments would generally try additional treatments, including systemic immunosuppressants (e.g., methotrexate, cyclosporine, mycophenolate mofetil, azathioprine) and advanced systemic therapies (i.e., abrocitinib, upadacitinib, dupilumab), depending in part on disease severity. As such, the sponsor’s base case, which compared ruxolitinib to vehicle cream (i.e., no active treatment) is not informative for decision-making. No evidence was submitted by the sponsor comparing ruxolitinib cream to treatments used for patients with mild AD (e.g., systemic immunosuppressants), and the sponsor’s ITC comparing ruxolitinib cream to the advanced systemic treatments indicated for moderate AD were inconclusive.
CDA-AMC identified additional important limitations with the sponsor’s base case, including uncertainty as to whether the efficacy data from the TRuE-AD trials adequately reflect the indicated population (i.e., patients who experience inadequate response or intolerance to TCS or TCI), uncertainty in the long term effectiveness and acquisition costs of ruxolitinib cream, logical fallacies in the modelling of discontinuation and subsequent therapy, limited efficacy data pertaining to adolescents, and poor modelling practices.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis, which included aligning the time horizon with the TRuE-AD trials (1 year), given the clinical uncertainty, and aligning the number of ruxolitinib tubes in year 1 with data from the TRuE-AD trials. Results of the CDA-AMC reanalyses suggest that, at a WTP threshold of $50,000 per QALY gained, ruxolitinib cream is not a cost-effective option for the treatment of mild to moderate AD compared with no active treatment. A price reduction of at least 68% would be required for ruxolitinib to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. Owing to the remaining limitations, which CDA-AMC was unable to address, additional price reductions may be required.
CDA-AMC conducted an additional scenario analysis exploring the cost-effectiveness of ruxolitinib cream in moderate AD. Results of the sequential analysis suggest that ruxolitinib cream is more effective than no active treatment (incremental QALYs: 0.07) with an ICER of $159,238 per QALY gained over 1 year. In this analysis, which assumed equal efficacy and safety between ruxolitinib cream and dupilumab, abrocitinib, and upadacitinib (based on the findings of the sponsor’s NMA and the CDA-AMC critical appraisal), ruxolitinib cream was associated with higher QALYs (incremental QALYs: 0.01) than advanced systemic treatments. This finding was entirely driven by the timing of the initial response assessment — that is, if response is assessed at the same time point for all treatments, incremental QALYs would be zero. The results of this analysis suggest that ruxolitinib cream is less costly than abrocitinib, upadacitinib, and dupilumab (cost savings: $3,977 to $11,677); however, because the cost of the comparators are based on public list prices (i.e., confidential negotiated discounts are not accounted for), it is highly uncertain whether these savings will be realized in practice. Thus, on the basis of this analysis, it is highly uncertain whether the use of ruxolitinib cream will result in improved QALYs or reduced costs relative to other advanced treatments for moderate AD.
References
1.Incyte Corporation. Opzelura ruxolitinib (as ruxolitinib phosphate) [draft product monograph; sponsor-supplied reference]. 2023.
2.Incyte Corporation. Clinical Study Report - Topical Ruxolitinib Evaluation in Atopic Dermatitis Study 1 (TRuE-AD1) [sponsor-supplied reference]. 2021.
3.Incyte Corporation. Clinical Study Report -Topical Ruxolitinib Evaluation in Atopic Dermatitis Study 2 (TRuE-AD2) [sponsor-supplied reference]. 2021.
4.Incyte Biosciences Canada. Ruxolitinib [as Ruxolitinib Phosphate] Cream 1.5% Indirect Treatment Comparisons versus Systemic Therapies in Atopic Dermatitis (≥12 Years) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. October 27, 2023.
5.Statistics Canada. Life Tables, Canada, Provinces and Territories [sponsor-supplied reference]. 2016. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310071001
6.Zimmermann M, Rind D, Chapman R, Kumar V, Kahn S, Carlson J. Economic Evaluation of Dupilumab for Moderate-to-Severe Atopic Dermatitis: A Cost-Utility Analysis [sponsor-supplied reference]. J Drugs Dermatol. 2018;17(7):750-756. https://jddonline.com/articles/economic-evaluation-of-dupilumab-for-moderate-to-severe-atopic-dermatitis-a-cost-utility-analysis-S1545961618P0750X/ PubMed
7.You S, Yaesoubi R, Lee K, et al. Lifetime quality-adjusted life years lost due to genital herpes acquired in the United States in 2018: a mathematical modeling study. Lancet Reg Health Am. 2023;19:100427. doi:10.1016/j.lana.2023.100427 PubMed
8.Geale K, Saridogan E, Lehmann M, Arriagada P, Hultberg M, Henriksson M. Repeated intermittent ulipristal acetate in the treatment of uterine fibroids: a cost-effectiveness analysis. Clinicoecon Outcomes Res. 2017;9:669-676. doi:10.2147/CEOR.S143557 PubMed
9.Chirikov V, Ma I, Joshi N, et al. Cost-Effectiveness of Alemtuzumab in the Treatment of Relapsing Forms of Multiple Sclerosis in the United States. Value Health. 2019;22(2):168-176. doi:10.1016/j.jval.2018.08.011 PubMed
10.Institute for Clinical Economic Review. Novel Agents to Prevent Chemotherapy-Induced Neutropenia and Other Myelosuppressive Effects Revised Evidence Report March 17, 2022 [sponsor-supplied reference]. 2022.
11.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med. 2016;375(24):2335-2348. doi:10.1056/NEJMoa1610020 PubMed
12.Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151-2168. doi:10.1016/S0140-6736(21)00588-2 PubMed
13.Eichenfield LF, Flohr C, Sidbury R, et al. Efficacy and Safety of Abrocitinib in Combination With Topical Therapy in Adolescents With Moderate-to-Severe Atopic Dermatitis: The JADE TEEN Randomized Clinical Trial. JAMA Dermatol. 2021;157(10):1165-1173. doi:10.1001/jamadermatol.2021.2830 PubMed
14.Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255-266. doi:10.1016/S0140-6736(20)30732-7 PubMed
15.Incyte Biosciences Canada. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2024.
16.Health Quality Ontario. Ontario Health: Home Narrowband Ultraviolet B Phototherapy for Photoresponsive Skin Conditions [sponsor-supplied reference]. November 2020. Accessed June 20, 2023. https://www.hqontario.ca/evidence-to-improve-care/health-technology-assessment/reviews-and-recommendations/home-narrowband-ultraviolet-b-phototherapy-for-photoresponsive-skin-conditions
17.AbbVie Corporation. Rinvoq (upadactinib) [product monograph; sponsor-supplied reference]. July 19, 2023.
18.Pfizer Canada, U. L. C. Cibinqo (abrocitinib) [product monograph; sponsor-supplied reference]. June 30, 2023.
19.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Schedule of Benefits, Physician Services Under the Health Insurance Act [sponsor-supplied reference]. 2023. Accessed April 2023. http://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20160401.pdf
20.Ontario Ministry of Health. Schedule of benefits for laboratory services: effective July 1, 2020 [sponsor-supplied reference]. 2020. Accessed April 2023. http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf
21.IQVIA. Delta PA Ontario Costs - January 2023 [sponsor-supplied reference]. 2022. https://www.iqvia.com/
22.CADTH. CADTH Upadacitinib Clinical and Pharmacoeconomic Combined Report. (Rinvoq - AbbVie Corporation) [sponsor-supplied reference]. 2022.
23.CADTH. CADTH Abrocitinib Clinical and Pharmacoeconomic Combined Report. (Cibinqo - Pfizer Canada ULC) [sponsor-supplied reference]. 2022.
24.Sanofi-aventis Canada, Inc. Dupixent (dupilumab) [product monograph; sponsor-supplied reference]. July 12, 2023.
25.Silverberg JI, Gelfand JM, Margolis DJ, et al. Health Utility Scores of Atopic Dermatitis in US Adults. J Allergy Clin Immunol Pract. 2019;7(4):1246-1252 e1. doi:10.1016/j.jaip.2018.11.043 PubMed
26.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022. Accessed October 1, 2024. https://www.formulary.health.gov.on.ca/formulary/
27.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Exceptional Access Program (EAP). 2022. Accessed October 15, 2024. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
28.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis. J Am Acad Dermatol. 2014;71(1):116-32. doi:10.1016/j.jaad.2014.03.023 PubMed
29.CADTH. CADTH Dupilumab Pharmacoeconomic Report. (Dupixent - Sanofi-Aventis Canada Inc.) [sponsor-supplied reference]. 2018.
30.Nonino A, Kim H, Flora APADS. PCN87 Budget Impact Analysis (BIA) of Ruxolitinib for Treatment of Intermediate-2 or High Risk IPSS Myelofibrosis in the Brazilian Public Healthcare System (SUS). Value in Health. 2021;24(Supplement 1):S35-S36. doi:10.1016/j.jval.2021.04.179
31.Bausch Health Canada Inc. Elidel (pimecrolimus): 1% cream [product monograph]. 2020. Accessed October 1, 2024. https://bauschhealth.ca/wp-content/uploads/pdf/Elidel%20PM-E-2020-01-10.pdf
32.Teva Canada Limited. Ratio-amcinonide (amcinonide): amcinonide cream, USP 0.1% w/w; amcinonide ointment, USP 0.1% w/w; amcinonide lotion 0.1% w/w [product monograph]. 2017. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00039966.PDF
33.Merck Canada Inc. Diprosone (betamethasone): betamethasone dipropionate cream, Merck standard, 0.05% w/w betamethasone (as dipropionate); betamethasone dipropionate ointment, Merck standard, 0.05% w/w betamethasone (as dipropionate); betamethasone dipropionate lotion, USP, 0.05% w/w betamethasone (as dipropionate) [product monograph]. 2017. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00042539.PDF
34.American Society of Health-System Pharmacists. Betamethasone, Betamethasone Benzoate, Betamethasone Dipropionate, Betamethasone Valerate (Topical) [product monograph]. Drugs.com; 2023. Accessed October 1, 2024. https://www.drugs.com/monograph/betamethasone-topical.html
35.Teva Canada Limited. Teva-clobetasol (clobetasol 17-propionate): 0.05% cream, ointment and scalp lotion [product monograph]. 2017. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00042697.PDF
36.PENDOPHARM, Division of Pharmascience Inc. Desonide (desonide cream and ointment): 0.05% (w/w) [product monograph]. 2014. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00026056.PDF
37.Valeant Canada LP. Topicort (desoximetasone): Topicort (desoximetasone cream, USP, 0.25%); Topicort Mild (desoximetasone cream, USP, 0.05%); Topicort Gel (desoximetasone gel, USP, 0.05%); Topicort Ointment (desoximetasone ointment, USP, 0.25%) [product monograph]. 2012. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00015890.PDF
38.TaroPharma, A Division of Taro Pharmaceuticals Inc. Tiamol (fluocinonide): emollient base cream [product monograph]. 2003. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00001696.PDF
39.Bausch Health, Canada Inc. Ultravate (halobetasol propionate): cream 0.05% w/w; ointment 0.05% w/w; lotion 0.01% w/w [product monograph]. 2020. Accessed October 1, 2024. https://bauschhealth.ca/wp-content/uploads/pdf/Ultravate%20PM-E-2020-10-07.pdf
40.American Society of Health-System Pharmacists. Hydrocortisone (topical) [product monograph]. Drugs.com; 2023. Accessed October 1, 2024. https://www.drugs.com/monograph/hydrocortisone-topical.html
41.Paladin Labs Inc. Topiderm HC 2% (hydrocortisone acetate): cream 2% w/w [product monograph]. 2014. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00026112.PDF
42.TaroPharma, A Division of Taro Pharmaceuticals Inc. HydroVal (hydrocortisone valerate): cream & ointment 0.2% [product monograph]. 2008. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00006434.PDF
43.Organon Canada Inc. Elocom (mometasone furoate): Cream, BP; ointment, Organon standard; lotion [product monograph]. 2021. Accessed October 1, 2024. https://www.organon.com/canada-en/wp-content/uploads/sites/5/2021/05/ELOCOM-CI_E.pdf
44.Taro Pharmaceuticals Inc. Viaderm K.C. cream, Viaderm K.C. ointment (triamcinolone acetonide): triamcinolone acetonide 1 mg/g, nystatin 100,000 units/g, neomycin base (as sulphate) 25 mg/g, and gramicidin 0.25 mg/g [product monograph]. 2014. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00025807.PDF
45.LEO Pharma Inc. Protopic (tacrolimus): ointment 0.03% and 0.1% (w/w) [product monograph]. 2020. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00057788.PDF
46.Pfizer Canada ULC. Eucrisa (crisaborole): ointment, 2% for topical use [product monograph]. 2021. Accessed October 1, 2024. https://pdf.hres.ca/dpd_pm/00060960.PDF
47.Saskatchewan Drug Plan: search formulary. 2022. Accessed October 1, 2024. http://formulary.drugplan.ehealthsask.ca/SearchFormulary
48.Government of Alberta. Interactive drug benefit list. 2022. Accessed October 1, 2024. https://idbl.ab.bluecross.ca/idbl/load.do
49.British Columbia. British Columbia Formulary 2022. Accessed October 1, 2024. https://pharmacareformularysearch.gov.bc.ca/
50.CADTH. CADTH Pharmacoeconomic Report. Crisaborole (Eucrisa - Pfizer Canada Inc.) [sponsor-supplied reference]. 2019.
51.CADTH. CADTH Canadian Drug Expert Committee Recommendation (Final). Crisaborole (Eucrisa - Pfizer Canada Inc.) [sponsor-supplied reference]. 2019.
52.IQVIA. DeltaPA. 2022. Accessed October 1, 2024. https://www.iqvia.com/
53.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). 2022. Accessed January 18, 2024. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
54.Incyte Biosciences Canada. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib phosphate), 1.5% w/w cream, topical. 2023.
55.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy. 2018;73(6):1284-1293. doi:10.1111/all.13401 PubMed
56.eHealth Saskatchewan. Saskatchewan Covered Population Reports [sponsor-supplied reference]. 2015. https://opendata.ehealthsask.ca/MicroStrategyPublic/asp/Main.aspx
57.B. C. Government. BC PharmaCare formulary search [sponsor-supplied reference]. 2022. Accessed October 20, 2024. https://pharmacareformularysearch.gov.bc.ca
58.Government of Alberta. Alberta Health Care Insurance Plan statistical supplement [sponsor-supplied reference]. 2021. https://open.alberta.ca/publications/0845-4775
59.Government of Canada. Non-Insured Health Benefits Program - First Nations and Inuit Health Branch: Annual Reports [sponsor-supplied reference]. 2021. https://www.sac-isc.gc.ca/eng/1576790320164/1576790364553
60.Government of Manitoba. Manitoba Health Population Reports [sponsor-supplied reference]. 2020. https://www.gov.mb.ca/health/population/index.html
61.Incyte Corporation. Data on File [sponsor-supplied reference]. 2023.
62.Nova Scotia Ministry of Health. Direct data request of eligible beneficiaries by age group (2008-2011) [sponsor-supplied reference]. 2011.
63.Ontario Public Drug Programs. Direct data request of elgible beneficiaries by age group (2007-2014) [sponsor-supplied reference]. 2014.
64.Silverberg JI, Barbarot S, Gadkari A, et al. Atopic dermatitis in the pediatric population: A cross-sectional, international epidemiologic study. Ann Allergy Asthma Immunol. 2021;126(4):417-428 e2. doi:10.1016/j.anai.2020.12.020 PubMed
65.CADTH. Drug Reimbursement Expert Review Committee final recommendation: dupilumab (Dupixent - Sanofi-Aventis Canada Inc.). April 24, 2020. Accessed October 16, 2024. https://www.cda-amc.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
According to clinical expert input received for this review, the following treatments may also be used to treat mild to moderate AD (Table 9).
Table 8: CDA-AMC Cost Comparison Table for AD
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Indicated for mild to moderate AD | ||||||
Ruxolitinib (Opzelura) | 1.5% | Cream, 100 g tubea | 1,075.9708b | Thin layer applied twice daily, up 20% of BSA | Year 1: 23.57 to 35.35 Subsequent years: 5.89 to 23.57 | Year 1: 8,608 to 12,912 (8 to 12 tubes)c Subsequent years: 2,152 to 8,608 (2 to 8 tubes)d |
Indicated for moderate to severe AD | ||||||
Abrocitinib (Cibinqo) | 50 mg 100 mg 200 mg | Tablet | 48.6667e 48.6667e 54.4667e | 100 mg or 200 mg orally once daily | 48.67 to 54.47 | 17,776 to 19,894 |
Dupilumab (Dupixent) | 200 mg/ 1.14 mL 300 mg / 2 mL | Prefilled syringe Single-use pen or single-use syringe | 978.7000e 978.7000e | Adolescents < 60 kg: 400 mg as an initial dose, followed by 200 mg every 2 weeks Adolescents ≥ 60 kg and adults: 600 mg as an initial dose, followed by 300 mg every 2 weeks | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,425 Year 2+: 25,446 |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Extended release tablets | 51.6810e 76.9600e 101.8100e | Adolescents (12 to 17 years) ≥ 40 kg: 15 mg once daily Adults: 15 mg or 30 mg once daily | 51.68 to 76.96 | 18,876 to 28,110 |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency.
Note: The recommended dosages for comparators were obtained from respective product monographs.17,18,24 BSA was assumed to be 1.8 m2. Prices from the Ontario Drug Benefit Formulary list price (October 1, 2024)26 unless stated otherwise.
aAlthough ruxolitinib cream is available in 60 g and 100 g tubes, the sponsor has indicated that only the 100 g size will be marketed in Canada.
bPrice submitted by sponsor.15
cLower end of the estimated range assumes 10% BSA affected at baseline, with a reduction to 3% at the end of the induction period. Upper end assumes 20% BSA affected at baseline, with a reduction to 5% at the end of induction. Actual number of tubes needed annually will depend on, for example, patient BSA, % of BSA with AD at baseline, amount of cream per application, duration of use, and wastage.
dLower end of the estimated range based on expiration of tubes within 6 months of opening. Upper end assumes 5% BSA at the start of year 2 and that ruxolitinib cream will be used for 274 days per year.
eOntario Exceptional Access Program formulary list price (accessed October 15, 2024).27
Table 9: CDA-AMC Cost Comparison Table for Systemic Therapy of AD (Not Indicated for AD)
Treatment | Strength / concentration | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | 24-week course cost ($) |
|---|---|---|---|---|---|---|
Immunosuppressantsa | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.5185 | Pediatric: 1.0 to 4.0 mg/kg per day Adult: 1.0 to 3.0 mg/kg per day | Pediatric: 0.52 to 2.07 Adult: 1.04 to 3.11 | Pediatric: 87 to 348 Adult: 174 to 523 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7526 0.7870 1.5350 3.0720 | Pediatric: 3.0 to 6.0 mg/kg per day Adult: 150 to 300 mg per day | Pediatric: 3.86 to 7.68 Adult: 4.61 to 9.22 | Pediatric: 648 to 1,290 Adult: 774 to 1,548 |
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | Pediatric: 0.2 to 0.7 mg/kg per week Adult: 7.5 to 25 mg per week | Pediatric: 0.14 to 0.43 Adult: 0.11 to 0.36 | Pediatric: 24 to 72 Adult: 18 to 60 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | Pediatric: 30.0 to 50.0 mg/kg per day Adult: 2,000 to 13,000 mg daily | Pediatric: 1.86 to 2.97 Adult: 2.97 to 19.30 | Pediatric: 312 to 499 Adult: 499 to 3,242 |
Retinoidsb | ||||||
Acitretin (Soriatane) | 10 mg 25 mg | Capsule | 1.2965 2.2770 | 10 to 50 mg once daily, maximum of 75 mg daily | 1.30 to 6.83 | 218 to 1,148 |
Alitretinoin (Toctino) | 10 mg | Capsule | 16.9868 | 30 mg once daily, dose may be reduced to 10 mg if unacceptable side effects | 50.96 | 8,561 |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency.
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary (accessed October 1, 2024),26 unless otherwise indicated, and do not include dispensing fees. Annual period assumes 365.25 days, average weight of 85 kg for adults and 40 kg for adolescents.
aRecommended dosage based on the American Atopic Dermatology Guidelines.28
bRecommended dosage aligned with the previous CADTH Pharmacoeconomic Review of dupilumab.29 According the clinical expert consulted by CADTH for a previous review, retinoids are primarily used to treat dermatitis on the hands of adults, not adolescents.
Table 10: CDA-AMC Cost Comparison Table of Topical Treatments for AD
Treatment | Strength | Dosage form | Price per tube ($) | Price per gram or mL ($) | Recommended dose |
|---|---|---|---|---|---|
Topical corticosteroids | |||||
Amcinonide (generics) | 0.1% | Cream 60 g | 27.1320 | 0.4522 | Thin amount to affected area twice daily, max 5 days on face, axillae, scrotum or scalp, 2 to 3 weeks elsewhere. |
Ointment 60 g | 21.6540 | 0.3609a | |||
Lotion 60 mL | 17.9820 | 0.2997a | |||
Betamethasone dipropionate (generic) | 0.05% | Cream 50 g | 10.2400 | 0.2048 | Thin film to affected area twice daily, duration of therapy varies; need should be reassessed at least every 4 weeks. |
Ointment 50 g | 10.7600 | 0.2152 | |||
Lotion 75 mL | 14.8500 | 0.1980 | |||
Betamethasone valerate (generic) | 0.05% | Cream Not available | Not available | 0.0596 | Apply to affected area 1 to 3 times daily. |
Ointment Not available | Not available | ||||
0.1% | Cream Not available | Not available | 0.0889 | ||
Ointment Not available | |||||
Lotion Not available | Not available | 0.3125 | |||
Clobetasol propionate (generic) | 0.05% | Cream 15 g 50 g 450 g | 3.4185 11.3950 102.5550 | 0.2279 | Thin amount to affected area twice daily. Weekly application should not exceed 50 g, and limited to 2 consecutive weeks. |
Ointment 15 g 50 g 450 g | |||||
Lotion 20 mL 60 mL | 3.9800 11.9400 | 0.1990 | |||
Desonide (generic) | 0.05% | Cream 15 g 60 g 454 g | 4.2855 17.1420 129.7078 | 0.2857 | Thin amount to affected area twice daily, may be increased in refractory cases. |
Ointment 15 g 60 g | 4.2795 17.1180 | 0.2853 | |||
Desoximetasone (Topicort) | 0.05% | Cream 20 g 60 g | 11.5200 34.5600 | 0.5760a | Thin amount to affected area twice daily. |
0.25% | Cream 20 g 60 g | 16.2020 48.6060 | 0.8101a | ||
0.25% | Ointment 60 g | 48.7440 | 0.8124a | ||
0.05% | Gel 60 g | 37.8120 | 0.6302a | ||
Fluocinonide (Lidemol, Lyderm, Lidex) | 0.05% | Cream 15 g 60 g 400 g | 3.8250 15.3000 102.000 | 0.2550 | Thin amount to affected area twice daily. Weekly application should not exceed 45 g, and limited to 2 weeks.30 |
Ointment 60 g | 20.2200 | 0.3370 | |||
Gel 60 g | 21.1020 | 0.3517 | |||
Fluocinonide (Tiamol) | 0.05% | Emollient Cream 25 g 100 g | 4.9500 19.8000 | 0.1980 | Thin amount 2 to 4 times daily. |
Halobetasol propionate (Ultravate, Bryhali) | 0.01% | Lotion 45 g 60 g 100 g | 44.1720 58.8960 98.1600 | 0.9816 | Thin amount to affected area twice daily, limited to 50 g weekly and 2 weeks without re-evaluation. |
0.05% | Ointment Cream 50 g | 66.9300 | 1.3386c | ||
Hydrocortisone (generics) | 1.0% | Cream Not available | Not available | 0.2056 | Apply appropriate cream, lotion, ointment, or solution sparingly 1 to 4 times daily. |
Lotion Not available | Not available | 0.1587 | |||
Ointment Not available | Not available | 0.0390 | |||
0.5% | Ointment Not available | Not available | 0.1400 | ||
Hydrocortisone acetate | 1% | Cream 30 g | 6.1680 | 0.2056 | Twice-daily application is generally recommended initially; intermittent use 1 to 2 times per week on areas that commonly flare for maintenance therapy. |
Hydrocortisone valerate (Hydroval) | 0.2% | Ointment Cream 15 g 45 g 60 g | 2.5005 7.5015 10.0020 | 0.1667 | Small amount to affected area twice daily. Discontinue as soon as lesions heal or if no response. |
Mometasone furoate (generic) | 0.1% | Cream 15 g 50 g | 8.3130 27.7100 | 0.5542 | Thin film to affected areas twice daily. |
Ointment 50 g | 11.2600 | 0.2252 | |||
Lotion 75 mL | 27.2025 | 0.3627 | |||
Triamcinolone acetonide (various) | 0.1% | Cream 15 g 30 g 60 g | 0.7995 1.5990 3.1980 | 0.0533 | Small amount to affected area twice or thrice daily. |
Ointment 15 g 30 g | 2.6700 5.3400 | 0.1780 | |||
Topical calcineurin inhibitors | |||||
Pimecrolimus (Elidel)d | 1% | Cream 30 g 60 g | 87.1560 174.3120 | 2.9052 | Thin layer to affected area twice daily, discontinue when resolved or after 3 weeks if no improvement or exacerbation. |
Tacrolimus | 0.03% | Ointment 30 g 60 g 100 g | 94.8060 189.6120 316.0200 | 3.1602 | Thin layer to affected area twice daily. Discontinue after 6 weeks if no improvement or exacerbation. |
0.10% | Ointment 30 g 60 g 100 g | 101.4120 202.8240 338.0400 | 3.3804 | ||
Phosphodiesterase type 4 inhibitor | |||||
Crisaborole (Eucrisa)e | 2% | Ointment 30 g 60 g 100 g | 72.4500 144.9000 241.5000 | 2.4150f | Thin layer to affected area twice daily. |
Phototherapy | |||||
UV light therapy | NA | NA | NA | 7.85 per treatmentg | Administered 3 to 5 times per weekh |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency.
Note: Prices are obtained from the Ontario Drug Benefit Formulary (accessed October 1, 2024),26 and recommended doses from respective product monographs, unless otherwise indicated.31-46
aSaskatchewan Formulary list price (accessed October 1, 2024).47
bAlberta Formulary list price (accessed October 1, 2024).48
cBritish Columbia Formulary list price (accessed October 1, 2024).49
dPimecrolimus is indicated for treatment of mild to moderate atopic dermatitis in patients 2 years of age and older.
eCrisaborole received a do not reimburse recommendation from CDEC in March 2019 for treatment of mild to moderate atopic dermatitis in patients 2 years of age and older who have failed or are intolerant to a topical corticosteroid treatment.50,51
fCost obtained from IQVIA DELTA PA database (accessed October 1, 2024).52
gOntario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy.”53
hMinimum frequency of phototherapy sessions required per week for successful maintenance. Length of maintenance period varies between individuals.28
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CDA-AMC appraisal section “Relevant comparators were excluded.” No active treatment (informed by vehicle from the TRuE-AD trials) is not informative for decision-making. |
Model has been adequately programmed and has sufficient face validity | No | The model includes numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The uncertainty around ruxolitinib dosing and treatment cost lacks face validity. The sponsor’s probabilistic results show that there is low uncertainty that treatment with ruxolitinib costs less than treatment with placebo (as most of the scatter lies below the ICER scatter plot), however, this is not corroborated with the trial data. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Refer to CDA-AMC appraisal section “The treatment dosing and cost of ruxolitinib is uncertain.” |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
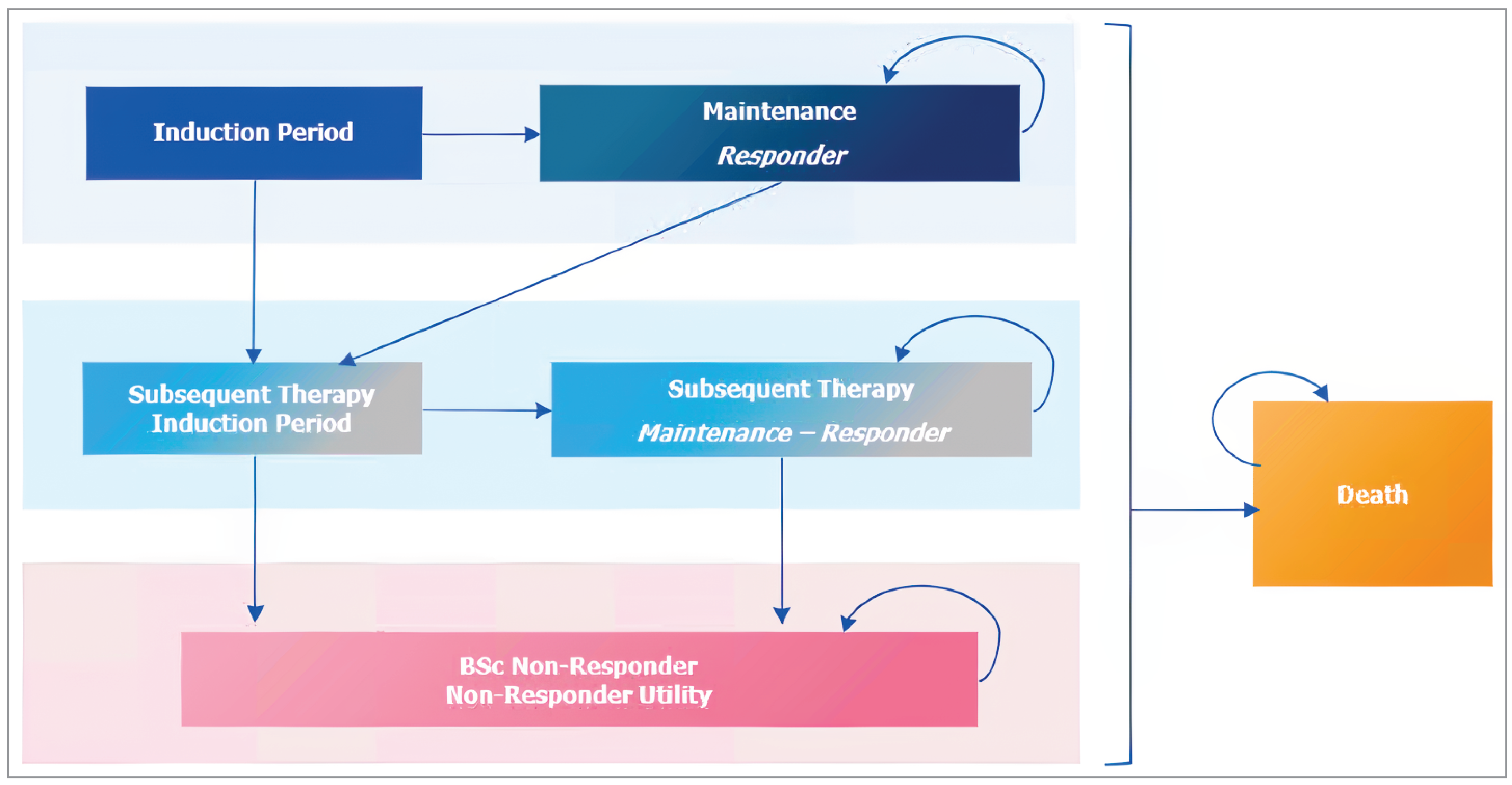
Table 12: Sponsor’s Calculation of Number of Ruxolitinib Cream Tubes Required per Year Results — Mild to Moderate AD
Parameter | Year 1 | Year 2+ | |
|---|---|---|---|
Induction period | Maintenance period | ||
BSA with AD, mean, % | ████ | ████ | ████ |
BSA treated, mean, m2 | ████ | ████ | ████ |
Daily cream per BSA treated (g/m2) | █████ | █████ | █████ |
Ruxolitinib cream, per day | ████ | ████ | ███ |
Duration of use | █████ ███ | █████ ███ | █████ ██ |
Ruxolitinib cream, grams per period | ██████ █ █ | █████ █ ██ | █████ █ █ |
Ruxolitinib cream, tubes per period | ████ ███ █ | ████ ████ | ████ ███ |
Total number of tubes, annually per patient | ████ | ████ | |
Note: The number of tubes was estimated per 4-week interval to algin with the cycle length of the sponsor’s model. Inputs are rounded for presentation. A starting BSA of ███ was assumed by the sponsor. Inputs based on observations from the TRuE-AD trials.
Source: Sponsor’s pharmacoeconomic submission.15
Detailed Results of the Sponsor’s Base Case
Table 13: Disaggregated Summary of Sponsor’s Economic Evaluation Results — Mild to Moderate AD
Parameter | No active treatment | Ruxolitinib | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 28.04 | 28.04 | 0.00 |
Discounted QALYs | |||
Total | 21.72 | 22.65 | 0.93 |
Initial treatment | 0.46 | 7.34 | 6.88 |
Subsequent treatment | 21.50 | 15.53 | –5.97 |
Adverse events | –0.24 | –0.21 | 0.03 |
Discounted costs ($) | |||
Total | 110,106 | 104,811 | −5,295 |
Drug acquisition and administration | 100,683 | 96,865 | –3,817 |
Initial treatment | 0 | 19,801 | 19,801 |
Subsequent treatment | 100,683 | 77,064 | –23,618 |
Resource useb | 8,711 | 7,227 | –1,484 |
Initial treatment | 173 | 1,095 | 922 |
Subsequent treatment | 8,539 | 6,133 | –2,406 |
Adverse events | 712 | 718 | 6 |
AD = atopic dermatitis; BSC = best supportive care; LY = life-year; QALY = quality-adjusted life-year.
aIncludes up to 2 lines of subsequent treatment
bIncludes costs associated with disease management and monitoring.
Source: Sponsor’s pharmacoeconomic submission.15
Detailed Results of the Sponsor’s Scenario Analyses
Table 14: Summary of the Sponsor’s Economic Evaluation Results — Moderate AD
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
No active treatment | 9,273 | 19.73 | Reference |
Ruxolitinib 1.5% cream | 68,873 | 21.41 | 35,476 |
Dominated treatments | |||
Abrocitinib | 50,814 | 19.88 | Extendedly dominated by no active treatment and ruxolitinib 1.5% cream |
Upadacitinib | 105,903 | 20.37 | Dominated by ruxolitinib 1.5% cream |
Dupilumab | 154,283 | 20.34 | Dominated by ruxolitinib 1.5% cream |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Treatment response was based on EASI-75 in this analysis. The sponsor provided an additional scenario analysis in which treatment response was based on IGA score; however, abrocitinib could not be included in that analysis owing to a lack of reported data for patients with moderate AD.
Source: Sponsor’s pharmacoeconomic submission.15
Table 15: Disaggregated Summary of Sponsor’s Economic Evaluation Results — Moderate AD
Parameter | No active treatment | Ruxolitinib | Dupilumab | Upadacitinib | Abrocitinib |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 26.46 | 26.46 | 26.46 | 26.46 | 26.46 |
Discounted QALYsc | |||||
Total | 19.73 | 21.41 | 20.34 | 20.37 | 19.88 |
Initial treatment | 0.66 | 10.98 | 4.99 | 4.80 | 2.01 |
Subsequent treatmenta | 19.26 | 10.59 | 15.61 | 15.77 | 18.11 |
Adverse events | –0.19 | –0.16 | –0.25 | –0.19 | –0.24 |
Discounted costs ($) | |||||
Total | 9,273 | 68,873 | 154,283 | 105,903 | 50,814 |
Drug acquisition and | 0 | 61,998 | 145,722 | 97,041 | 41,460 |
Initial treatment | 0 | 61,998 | 145,722 | 97,041 | 41,460 |
Subsequent treatmenta | 0 | 0 | 0 | 0 | 0 |
Resource useb | 8,802 | 6,317 | 7,797 | 8,317 | 8,722 |
Initial treatment | 201 | 1,591 | 824 | 1,276 | 634 |
Subsequent treatment | 8,602 | 4,726 | 6,973 | 7,040 | 8,088 |
Adverse events | 470 | 558 | 765 | 545 | 632 |
AD = atopic dermatitis; BSC = best supportive care; LY = life-year; QALY = quality-adjusted life-year.
Note: Treatment response was based on EASI-75 in this analysis. The sponsor provided an additional scenario analysis in which treatment response was based on IGA score; however, abrocitinib could not be included in that analysis owing to a lack of reported data for patients with moderate AD.
aSubsequent treatment was assumed by the sponsor to be BSC.
bIncludes costs associated with disease management and monitoring.
cUtility values for this analysis were derived from patients with IGA = 3 from the TRuE-AD trial.
Source: Sponsor’s pharmacoeconomic submission.15
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the CDA-AMC Reanalysis
Table 16: Disaggregated Summary of CDA-AMC Reanalysis Results — Mild to Moderate AD
Parameter | No active treatment | Ruxolitinib cream | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.00 | 1.00 | 0 |
Discounted QALYs | |||
Total | 0.78 | 0.82 | 0.04 |
Initial treatment | 0.24 | 0.53 | 0.29 |
Subsequent treatment | 0.54 | 0.30 | −0.25 |
Adverse events | –0.01 | –0.01 | 0 |
Discounted costs ($) | |||
Total | 167 | 6,915 | 6,747 |
Drug acquisition and administration | 0 | 6,710 | 6,710 |
Initial treatment | 0 | 6,710 | 6,710 |
Subsequent treatment | 0 | 0 | 0 |
Resource usea | 144 | 182 | 39 |
Initial treatment | 144 | 182 | 39 |
Subsequent treatment | 0 | 0 | 0 |
Adverse events | 22 | 24 | –2 |
AD = atopic dermatitis; BSC = best supportive care; LY = life-year; QALY = quality-adjusted life-year.
aIncludes costs associated with disease management and monitoring.
Results of CDA-AMC Scenario Analyses
Scenario Analyses
Table 17: Summary of the CDA-AMC Scenario Analyses — Mild to Moderate AD
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY)a |
|---|---|---|---|---|---|
No active treatment | 168 | Reference | 0.78 | Reference | Reference |
Ruxolitinib creama | 10,142 | 9,975 | 0.82 | 0.04 | 223,766 |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Deterministic analysis.
aAnalysis assumes 7 tubes in the induction period (based 20% BSA affected by AD at baseline, the maximum recommended BSA in the Health Canada monograph) and 4 tubes in the maintenance period (based on 3% BSA at the end of the induction period).
Table 18: Summary of the CDA-AMC Scenario Results — Moderate AD
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
No active treatment | 179 | 0.77 | Reference |
Ruxolitinib cream | 12,001 | 0.84a | 159,238 |
Dominated treatments | |||
Abrocitinib | 15,977 | 0.83 | Dominated by ruxolitinib cream |
Upadacitinib | 16,944b | 0.83 | Dominated by ruxolitinib cream |
Dupilumab | 23,678 | 0.83 | Dominated by ruxolitinib cream |
AD = atopic dermatitis; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Notes: Treatment response was based on EASI-75 in this analysis. The sponsor provided an additional scenario analysis in which treatment response was based on IGA score; however, abrocitinib could not be included in that analysis owing to a lack of reported data for patients with moderate AD.
In this scenario, CDA-AMC made changes in alignment with the CDA-AMC reanalysis for mild to moderate AD (1 year horizon, costs associated with subsequent treatment excluded, removal of discontinuation rate for no active treatment). CDA-AMC assumed that 11 tubes of ruxolitinib cream would be used in year 1 (20% BSA affected at baseline), and assumed equal efficacy and safety between ruxolitinib cream, abrocitinib, upadacitinib, and dupilumab.
Costs of comparators are based on publicly available list prices and do not take confidential negotiated prices into account.
aDifferences in QALYs between ruxolitinib cream and abrocitinib, upadacitinib, and dupilumab in this analysis (0.01 QALYs) are entirely due to the timing of outcome assessment (8 weeks for ruxolitinib cream; 16 weeks for abrocitinib, upadacitinib, and dupilumab). If initial response to treatment is assessed at either 8 or 16 weeks for all treatments, incremental QALYs are zero, and cost-effectiveness is determined by drug acquisition costs.
bCost of upadacitinib was corrected to $51.6810 per 15-mg tablet.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 19: Summary of Key Take-Aways
Key Take-aways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) assessed the expected budgetary impact of reimbursing ruxolitinib for the topical treatment of mild to moderate AD in patients aged 12 years of age and older whose disease is not adequately controlled with conventional topical prescription therapies (TCS, TCI) or when those therapies are not advisable.54 The BIA was undertaken from the perspective of the Canadian public drug plans at base year (2024) and over a three-year time horizon (2025 to 2027). The sponsor’s pan-Canada estimates reflected the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 20.
The sponsor estimated the number of eligible patients for ruxolitinib using an epidemiologic approach with data obtained from provincial public drug programs, published literature and sponsor’s clinical experts.55-64 The sponsor estimated the number of beneficiaries using data from the public drug programs for adolescents (aged 12 to 17 years) and adults (18 years and above) and narrowed the population to those treated with AD and not adequately controlled with conventional topical prescription therapies such as TCS and TCI or in whom these therapies were not advisable. Drug acquisition costs of immunosuppressants (methotrexate and cyclosporin), dupilumab and oral Janus kinase inhibitors (upadacitinib and abrocitinib) were included; BSC was assumed to comprise nonprescription products (e.g., emollients) and have zero cost to the drug plans. The sponsor assumed ruxolitinib would capture market share from immunosuppressants (methotrexate and cyclosporin), biologic (dupilumab) and Janus kinase inhibitor (abrocitinib), and BSC.
Table 20: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) |
|---|---|
Target population | |
Number of beneficiaries (excluding Quebec and territories) | 10,154,772 / 10,534,212 / 10,935,794 |
Proportion of patients | |
Adolescents | 6.9% |
Adults | 93.1% |
Proportion diagnosed with AD | |
Adolescents | 15.8% |
Adults | 3.5% |
Proportion with disease severity | |
Adolescents | 85.0% |
Adults | 79.0% |
Proportion receiving treatment | 74.7% |
Proportion not adequately controlled with conventional topical prescription therapies such as TCS and TCI or in whom these therapies are inadvisable | 39.6% |
Number of patients eligible for drug under review | 102,870 / 106,560 / 110,474 |
Market uptake (3 years) | |
Uptake (reference scenario)a Methotrexate Cyclosporine Dupilumab Abrocitinib Upadacitinib BSC | 21% / 21% / 21% 2% / 2% / 2% 30% / 30% / 30% 3% / 3% / 3% 6% / 6% / 6% 38% / 38% / 38% |
Uptake (new drug scenario)a Ruxolitinib Methotrexate Cyclosporine Dupilumab Abrocitinib Upadacitinib BSC | 3% / 8% / 10% 21% / 19% / 19% 1% / 1% / 1% 29% / 28% / 27% 3% / 2% / 2% 6% / 6% / 6% 37% / 35% / 34% |
Cost of treatment, annual per patient | |
Ruxolitinib Methotrexate (adolescents / adults) Cyclosporine (adolescents / adults) Dupilumab (year 1 / year 2+) Abrocitinib Upadacitinib BSCb | $2,152 $22 / $36 $929 / $1,275 $26,425 / $25,446 $17,446 $17,780 $0 |
BSC = best supportive care.
Note: Costs for Ontario are presented. The sponsor estimated treatment cost for each jurisdiction based on respective listed prices.
aDifferent market share estimates were incorporated for British Columbia.
bAssumed to comprise nonprescription products (e.g., emollients) and have zero cost to the drug plans.
Summary of the Sponsor’s BIA Results
The sponsor estimated that reimbursing ruxolitinib for the topical treatment of mild to moderate AD in patients aged 12 years of age and older whose disease is not adequately controlled with TCS or TCI or when those therapies are not advisable will be cost saving, with a total savings of $111,047,612 over the first 3 years (year 1: $15,356,603; year 2: $41,916,566; year 3: $53,774,443).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The treatment cost of ruxolitinib is underestimated. In the BIA, the sponsor assumed that all patients would use 2 tubes of ruxolitinib cream annually, regardless of AD severity or year of treatment. This is not aligned with the number of tubes that the sponsor assumed in their economic evaluation or supported by data from the TRuE-AD trials. As noted in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation, it is more likely that patients will use, on average, approximately 8 tubes in the first year of treatment and at least 2 tubes in each subsequent year, while patients with moderate AD will require additional tubes. The actual amount of ruxolitinib cream used (and hence the number of tubes needed) will depend on, for example, patient BSA, percentage of BSA with AD, amount of cream per application, duration of use, and wastage.
In reanalysis, CDA-AMC assumed that 8 tubes ruxolitinib cream would be used in year 1.
The structure of the sponsor’s model did not permit higher costs to be incurred for new ruxolitinib users. As noted in the previous limitation, the amount of ruxolitinib cream used by patients in their first year of treatment is anticipated to be higher than in subsequent years. In the sponsor’s base case, all patients were assumed to use 2 tubes of ruxolitinib cream per year, regardless of how long they had been on treatment.
CDA-AMC was unable to address this limitation in reanalysis because of the structure of the sponsor’s model. The cost of ruxolitinib cream is underestimated in the CDA-AMC reanalysis, as patients newly entering the model in year 2 and 3 (e.g., because of population growth or new diagnosis) would be expected to use 8 tubes in their first year of treatment.
Uncertainty about the market share of comparator treatments. In the sponsor’s base case, the market share of comparators was estimated based on a survey of clinicians in Canada. Clinical experts consulted by CDA-AMC indicated that the market share in the reference case of comparators is uncertain and will depend, at least in part, on age group. Further, as noted in the CDA-AMC Appraisal of the Sponsor’s Economic model, treatment approaches differ depending on disease severity. For example, dupilumab, abrocitinib, and upadacitinib are indicated for treatment of moderate and severe AD, but not mild. Finally, clinical expert input received by CDA-AMC indicated that it is unlikely that the market share of abrocitinib and upadacitinib will remain static over the 3-year analysis horizon.
CDA-AMC was unable to address this limitation owing to the structure of the sponsor’s model (e.g., an inability to separately consider mild or moderate AD) and the absence of alternative estimates for each subgroup.
Uncertainty about ruxolitinib market capture from comparators. The sponsor assumed that the 10% of market share anticipated for ruxolitinib cream by year 3 will taken from BSC (i.e., no active treatment; 3%), methotrexate (2%), cyclosporine (1%), abrocitinib (1%) or dupilumab (22%). The sponsor did not justify the assumption that no market share would be taken from upadacitinib and no data were submitted supporting the sponsor’s assumptions about displacement.
CDA-AMC explored the impact of alternative displacement assumptions in scenario analyses.
The prices paid by public drug plans is uncertain: The prices for comparators were based on publicly available list prices and may not reflect the actual prices paid by public drug plans, as any potential confidential rebates are not reflected in this analysis.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by aligning the eligible population with the Health Canada indication, adopting a higher number of ruxolitinib tubes in year 1, and correcting the price of dupilumab.
Table 21: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Number of ruxolitinib tubes in Year 1 | 2 | 8 |
CDA-AMC base case | Reanalysis 1 | |
AD = atopic dermatitis; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 22 and a more detailed breakdown is presented in Table 23.
In the CDA-AMC base case, reimbursing ruxolitinib cream for mild to moderate AD is predicted to be cost saving, with 3-year savings of $39,727,424 (year 1: -$4,566,758; year 2: -$6,805,318; year 3: -$37,488,864).
Table 22: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | –111,047,612 |
CDA-AMC reanalysis 1: 8 tubes ruxolitinib cream in year 1 | –39,727,424 |
CDA-AMC base case (1) | –39,727,424 |
AD = atopic dermatitis; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC also conducted additional scenario analyses to address remaining uncertainty, using the CDA-AMC base case. Results are provided in Table 23.
Assuming a confidential price of dupilumab that is 54% lower than the public list price, aligned with the CDEC pricing recommendation.65
Assuming ruxolitinib cream will displace only systemic immunosuppressants (i.e., cyclosporine, methotrexate). Treatments for moderate AD (i.e., dupilumab, abrocitinib, upadacitinib) were excluded from this analysis, to approximate the impact of reimbursing ruxolitinib for the treatment of mild AD.
Table 23: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 898,357,892 | 904,246,473 | 937,960,969 | 973,725,879 | 3,714,291,214 |
New drug | 898,357,892 | 888,889,871 | 896,044,403 | 919,951,436 | 3,603,243,602 | |
Budget impact | 0 | –15,356,603 | –41,916,566 | –53,774,443 | –111,047,612 | |
CDA-AMC base case | Reference | 898,357,892 | 904,246,473 | 937,960,969 | 973,725,879 | 3,714,291,214 |
New drug | 898,357,892 | 908,813,231 | 931,155,651 | 936,237,015 | 3,674,563,789 | |
Budget impact | 0 | 4,566,758 | –6,805,318 | –37,488,864 | –39,727,424 | |
CDA-AMC scenario 1: Dupilumab price reductiona | Reference | 506,981,781 | 512,807,761 | 531,624,227 | 551,585,359 | 2,102,999,128 |
New drug | 506,981,781 | 526,380,038 | 549,428,082 | 545,699,204 | 2,128,489,105 | |
Budget impact | 0 | 13,572,277 | 17,803,855 | –5,886,155 | 25,489,977 | |
CDA-AMC scenario 1: Mild AD onlyb | Reference | 47,095,817 | 48,203,909 | 49,384,553 | 50,642,797 | 195,327,076 |
New drug | 47,095,817 | 73,047,358 | 96,360,655 | 82,326,912 | 298,830,742 | |
Budget impact | 0 | 24,843,450 | 46,976,102 | 31,684,115 | 103,503,667 |
AD = atopic dermatitis; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
aAnalysis assumes a 54% price reduction for dupilumab, in line with the CDEC pricing recommendation.65
bAnalysis assumes ruxolitinib cream will be used for the treatment of mild AD. Treatments indicated for moderate AD were removed from the analysis (i.e., dupilumab, abrocitinib, upadacitinib), with their market shares distributed proportionally to methotrexate and cyclosporine.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.