Drugs, Health Technologies, Health Systems
Reimbursement Review
Cabotegravir (Apretude)
Sponsor: ViiV Healthcare ULC
Therapeutic area: HIV-1 infection, pre-exposure prophylaxis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
APAA
Africans in Partnership Against AIDS
CBRC
Community-Based Research Centre
CI
confidence interval
CrI
credible interval
DBS
dried blood spot
gbMSM
gay, bisexual, and other men who have sex with men
HIV Edmonton
HIV Network of Edmonton Society
HR
hazard ratio
INSTI
integrase strand transfer inhibitor
ISR
injection site reaction
ITC
indirect treatment comparison
ITT
intent to treat
LA
long-acting
mITT
modified intent to treat
MSM
men who have sex with men
NMA
network meta-analysis
NNRTI
non-nucleoside reverse transcriptase inhibitor
NRTI
nucleoside reverse transcriptase inhibitor
OBSP
on blinded study product
PI
protease inhibitor
PK
pharmacokinetic
PLHIV
people living with HIV
POSSE
Peer Outreach Support Services and Education
PP
per protocol
PrEP
pre-exposure prophylaxis
PWID
people who inject drugs
PY
person-years
RCT
randomized controlled trial
RR
relative risk
SAE
serious adverse event
SLR
systematic literature review
STI
sexually transmitted infection
TAF-FTC
tenofovir alafenamide fumarate-emtricitabine
TDF
tenofovir disoproxil fumarate
TDF-FTC
tenofovir disoproxil fumarate-emtricitabine
TFV
tenofovir
TFV-DP
tenofovir diphosphate
TGW
transgender women
VOICE
Vaginal and Oral Interventions to Control the Epidemic
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Cabotegravir (Apretude), 30 mg oral tablets and 200 mg/mL (600 mg/3 mL), extended-release injectable suspension |
Sponsor | ViiV Health care ULC |
Indication | For at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg for PrEP to reduce the risk of sexually acquired HIV-1 infection |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | May 10, 2024 |
Recommended dose | For individuals weighing at least 35 kg. When an oral lead-in is used, the recommended dose is as follows:
When cabotegravir LA injection is initiated directly, the recommended dose is as follows:
|
LA = long-acting; NOC = Notice of Compliance; PrEP = pre-exposure prophylaxis.
aIndividuals may be given cabotegravir LA injection up to 7 days before or after the date the individual is scheduled to receive the injections.
Introduction
HIV is a retrovirus that impairs the human immune system, and is transmitted through bodily fluids via sex or vertically (i.e., from mother to child during pregnancy, childbirth, and/or breastfeeding).1 Without treatment, HIV infections can progress from acute through clinical latency to AIDS, making people living with HIV (PLHIV) more vulnerable to opportunistic infections and diseases.2-6 At the end of 2020, the prevalence of PLHIV in Canada was estimated to be 62,790 (with an associated range of uncertainty of 55,200 to 70,300 reported) PLHIV in Canada, a prevalence rate of approximately 170 per 100,000 persons, and representing a 3.6% increase from the estimated 60,600 PLHIV reported at the end of 2018.7 In 2022, 1,833 new HIV infections were diagnosed in Canada, representing a 4.7 incidence per 100,000, and a 24.9% increase from estimates reported in 2021 according to the Public Health Agency of Canada. The estimated rate of new HIV infections in males was 6.3 per 100,000 and 3.1 per 100,000 females (excluding cases for trans individuals or for whom sex was not reported).8
Canada has adopted an integrated approach toward HIV management and prevention.1 Pre-exposure prophylaxis (PrEP), which involves the use of antiretroviral drugs to prevent HIV infection, is an effective tool when used in combination with other strategies in the prevention of HIV for at-risk persons.1 However, the effectiveness of any PrEP option depends on key behavioural factors that impact efficacy such as medication adherence and participation in clinical follow-up.1 There are currently 2 PrEP options in Canada, tenofovir disoproxil fumarate-emtricitabine (TDF-FTC) (Truvada) and tenofovir alafenamide fumarate-emtricitabine (TAF-FTC) (Descovy). TDF-FTC is an oral therapy, reimbursed by most jurisdictions in Canada while TAF-FTC is not indicated for individuals at risk from receptive vaginal sex and is only reimbursed through the Canadian Armed Forces Drug Benefit List.1 Although PrEP usage in Canada has increased over the past years, it is most commonly used by gay, bisexual, and other men who have sex with men (gbMSM), driven by education and awareness initiatives for the use of PrEP.9 Almost all (98%) PrEP users in Canada are male.10 There is a need for options that are convenient for individuals and which promote adherence, according to the clinical expert consulted during the CDA-AMC review.
Cabotegravir is an antiretroviral medication which inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral DNA integration which is essential for the HIV replication cycle. Cabotegravir long-acting (LA) is indicated for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg for PrEP to reduce the risk of sexually acquired HIV-1 infection. The sponsor’s reimbursement request is consistent with the Health Canada–approved indication. The recommended dosing depends on whether an oral lead-in is used or the cabotegravir LA injection is administered initially (Table 1). Cabotegravir LA monotherapy has not been previously reviewed by CDA-AMC for PrEP for HIV-1 prevention. However, cabotegravir (tablets and injectable forms) in combination with rilpivirine has been previously reviewed for the treatment of HIV-1 in infected patients.
The objective of CDA-AMC’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of cabotegravir (Apretude), 30 mg oral tablets, and 200 mg/mL (600 mg/3 mL), extended-release injectable solution as PrEP for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg to reduce the risk of sexually acquired HIV-1 infection.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CDA-AMC’s call for input and from clinical expert(s) consulted by CDA-AMC for the purpose of this review.
Patient Input
Five patient groups submitted inputs on the indication being reviewed: Africans in Partnership Against AIDS (Toronto) (APAA), HIV Network of Edmonton Society (HIV Edmonton), CATIE, Community-Based Research Centre (CBRC), and Peer Outreach Support Services and Education (POSSE).
All patient groups highlighted the stigma related to HIV and discrimination at the system level, in both laws and institutions, within the medical profession and communities at large, and between individuals or by oneself in the form of shame and guilt. The stigma often leads to isolation and fear of disclosure and affects treatment maintenance or medication drop-off and the quality of life of those affected by HIV. According to the Sex Now 2021 online survey, the awareness of PrEP for HIV as a medication to prevent HIV varies among at-risk populations. Patients from 2 groups (APAA and CATIE) identified racism and cultural and linguistic barriers deter African Caribbean Black PLHIV from accessing treatment. Other identified barriers included homophobia, limited information and access to health care facilities, and financial constraints. Other groups highlighted challenges such as side effects of oral medications on the digestive and intestinal systems, pill burden, and the impact of daily medication on lifestyle, which can affect treatment adherence. One input noted that youth struggle with adherence to medications and require solutions reducing adherence requirements.
Patient groups highlighted that there are stigma and adherence issues associated with current oral PrEP options (i.e., TDF-FTC and TAF-FTC). The patient groups noted that remembering to take oral pills can be challenging for people who use substances or for people dealing with competing priorities. The groups highlighted concerns relating to the safe storage of medications, especially for persons in need of shelter, and the renewal of prescriptions.
Patients expressed their preference for injectable PrEP, according to 1 survey (Sex Now 2022). The advantages of injectable PrEP identified by patients include the reduced stigma experienced in multiple settings due to reduced exposure to health services or systems where stigmatizing experiences occur, increased privacy and discretion, decreased risk of treatment interruptions during travel, increased adherence to treatment, reduced impact on digestive-related issues from consuming pill treatment, and improved quality of life (e.g., improved autonomy and self-determination by having a choice in treatment decisions and not being encumbered by medication regimen schedules).
Patients expect new PrEP therapies to demonstrate improved access to treatment, improved treatment adherence, decreased breakthrough infection and risk of resistance, sustained viral suppression, and increased level of comfort with the treatment.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical expert consulted during the CDA-AMC review highlighted that an important goal in the management of HIV in Canada is to prevent persons from acquiring HIV infections sexually using different strategies. The expert noted that PrEP is an important tool available to at-risk persons for the prevention of HIV-1 infections; however, current options (these include the oral therapies TDF-FTC and TAF-FTC) do not cater to all populations. Therefore, there is an unmet need for newer treatments that are convenient and promote adherence in all at-risk populations. The clinical expert anticipates that cabotegravir LA injectable would provide an alternative to daily oral treatment for individuals hoping to access PrEP options. According to the clinical expert, all persons considered at risk of acquiring HIV sexually will benefit from cabotegravir LA as a PrEP option. The expert noted that cabotegravir LA will be less suitable for individuals who cannot tolerate injections. The expert indicated that response to treatment will be assessed based on whether persons remain HIV-negative during routine follow-up tests, which are typically performed every 3 months to 6 months. The expert highlighted that factors such as individual intolerance to treatment and the acquisition of HIV will lead to treatment discontinuation. Although injection reactions are frequently observed, the expert noted that patients usually tolerate these adverse events (AEs); however, a severe injection site reaction (ISR) may precipitate changes in treatment modality. The clinical expert highlighted cabotegravir LA can be prescribed by any clinician who provides PrEP care and follow-up (these include clinicians at sexual health clinics, physicians, primary care providers, or infectious diseases specialists).
Clinician Group Input
Two inputs were submitted on the indication being reviewed: 1 clinician group of 6 clinicians from the Vancouver Coastal Health Regional HIV Program, which is a public health program that aims to reduce the rate of HIV infection among the 1.25 million people living in the region, and 1 clinician, Dr. Philippe El-Helou.
Inputs from clinician groups are in line with the clinical expert consulted by CDA-AMC. The inputs discussed that oral PrEPs are currently available for individuals who are at higher risk of acquiring HIV, the treatment goal is to decrease the incidence of newly acquired HIV infections, and there remains an unmet need to improve treatment compliance and convenience. Both the clinical expert consulted by CDA-AMC and the clinician groups agreed that cabotegravir LA would be an alternative to daily oral PrEP and the patients best suited for cabotegravir LA would be individuals who are at risk of sexually acquired HIV. Clinicians from the Vancouver Coastal Health Regional HIV Program specified that individuals in whom adherence to oral daily HIV PrEP is difficult are best suited to LA injectable HIV PrEP. The clinical expert consulted by CDA-AMC was aligned with the clinician groups in using incident HIV infections as an outcome to determine patients’ response to treatment in clinical practice. Inputs from clinician groups highlighted that oral PrEP along with a robust monitoring and follow-up strategy are crucial. Clinician groups stated that cabotegravir LA should be prescribed and monitored by various health care providers (e.g., family doctors, nurse practitioners, and specialists in HIV care) in community, hospital, and specialty clinics where individuals can access PrEP prescriptions, HIV testing, and ongoing care.
Drug Program Input
Input was obtained from drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for cabotegravir LA: relevant comparators, considerations for initiation of therapy, consideration for prescribing of therapy, generalizability, and system and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
Two pivotal trials (HPTN 083 and HPTN 084) provided evidence on the safety and efficacy of cabotegravir LA compared to daily oral TDF-FTC for PrEP in key at-risk populations.
The HPTN 083 trial is an ongoing phase IIb/III, multicentre, double-blind, randomized, noninferiority trial designed to evaluate the efficacy and safety of injectable cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years and older) cisgender men who have sex with men (MSM) and transgender women (TGW) who have sex with men. In total, 4,570 participants enrolled at 43 study centres (no study sites were in Canada) were randomized in a 1:1 ratio to receive either daily oral cabotegravir (30 mg tablets) and oral placebo TDF-FTC for up to 5 weeks (step 1), followed by cabotegravir LA injection (600 mg, intramuscular injection at weeks 5, 9, and every 8 weeks thereafter) plus daily oral placebo as step 2 (n = 2,283); or daily oral TDF-FTC (300 mg-200 mg tablets) and oral placebo cabotegravir for up to 5 weeks (step 1), followed by daily oral TDF-FTC plus placebo intramuscular injection at weeks 5, 9, and every 8 weeks thereafter as step 2 (n = 2,287). Of the participants randomized in each group, 4,566 were treated (2,281 participants in the cabotegravir LA group and 2,285 in the TDF-FTC group).11 The majority of participants enrolled were aged 30 years or younger. The findings presented in this submission are from the first preplanned interim analysis at the May 14, 2020, data cut-off date.
The HPTN 084 study is an ongoing phase III, multicentre, double-blind, randomized, superiority trial designed to evaluate the efficacy and safety of injectable cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years to 45 years) cisgender women. In total, 3,224 participants from 20 study centres were randomized in a 1:1 ratio to receive either daily oral cabotegravir (30 mg tablets) and oral placebo TDF-FTC for up to 5 weeks (step 1), followed by cabotegravir LA injection (600 mg intramuscular injection at weeks 5, 9, and every 8 weeks thereafter) plus daily oral placebo as step 2 (n = 1,614); or daily oral TDF-FTC (300 mg-200 mg tablets) and oral placebo cabotegravir LA for up to 5 weeks (step 1), followed by daily oral TDF-FTC plus placebo intramuscular injection at weeks 5, 9, and every 8 weeks thereafter as step 2 (n = 1,610).11 All participants enrolled were cisgender females and more than 99% were Black and aged younger than 35 years. The findings presented in this submission are from the second preplanned interim analysis conducted at the November 5, 2020, data cut-off date.
Both trial designs included an oral lead-in phase (step 1), an injection phase (step 2), and an open-label extension phase (step 3). Key primary and secondary outcomes investigated were similar for both trials and included documented incident HIV infections in steps 1 and 2 and number of participants experiencing grade 2 or higher clinical and laboratory AEs. Other important outcomes assessed across trials included documented incident HIV infections in step 2, resistance mutations to study products, adherence to study product during step 2, and the incidence of sexually transmitted infections (STIs). Patient-reported outcomes were assessed using an acceptability scale questionnaire and survey of attitudes and willingness to use cabotegravir and TDF-FTC using the Study Medication Satisfaction Questionnaire (the HPTN study 083 only). The blinded phase in both trials was amended to an open-label design following results from planned interim analyses.11 All participants included in the data analyses for this submission were blinded to study treatments.
Efficacy Results
Incident HIV Infections in Steps 1 and 2
Based on the primary analysis in the HPTN 083 and HPTN 084 studies that evaluated incident HIV-1 infections at steps 1 and 2 of the trials, the risk of HIV-1 infection was lower in the cabotegravir LA group than in the TDF-FTC group. More specifically, in the HPTN 083 study, 13 HIV-1 infections were reported in the cabotegravir LA group (incidence rate per 100 person-years [PY] = 0.40; 95% confidence interval [CI], 0.22 to 0.69) versus 39 in the TDF-FTC group (incidence rate per 100 PY = 1.22; 95% CI, 0.87 to 1.67) after 6,404 PY of accumulated follow-up by the May 14, 2020, interim cut-off date. The between-group difference in incidence rates was in favour of cabotegravir LA relative to TDF-FTC (–0.82 per 100 PY; 95% CI, –1.26 to –0.38). Noninferiority of cabotegravir LA to TDF-FTC was demonstrated and the estimated hazard ratio (HR) was 0.34 (95% CI, 0.18 to 0.62; P = 0.0005), suggesting a 66% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. A revised data analysis from additional testing confirmed 12 HIV-1 infections in the cabotegravir LA group and 40 in the TDF-FTC group (new bias-adjusted HR = 0.31; 95% CI, 0.16 to 0.58). Supportive analyses conducted on blinded study product (OBSP) were consistent with the primary analysis (estimated HR = 0.164; 95% CI, 0.06 to 0.47), suggesting an 83.6% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group (P = 0.0008).
In the HPTN 084 study, superiority of cabotegravir LA was demonstrated by the November 5, 2020, interim cut-off date. In total, 40 incident HIV-1 infections were identified: 4 infections occurred in the cabotegravir LA group (incidence rate per 100 PY = 0.20; 95% CI, 0.06 to 0.52) and 36 occurred in the TDF-FTC group (incidence rate per 100 PY = 1.85; 95% CI, 1.3 to 2.56) after 3,907 PY of accumulated follow-up. The between-group difference also favoured cabotegravir LA relative to TDF-FTC (–1.65 per 100 PY; 95% CI, –2.28 to –1.01). The estimated HR was 0.12 (95% CI, 0.05 to 0.31; P < 0.0001), suggesting an 88% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. A revised data analysis from additional testing confirmed 39 incident HIV-1 infections, 3 occurring in the cabotegravir LA group (bias-adjusted HR = 0.1; 95% CI, 0.04 to 0.27), indicating a 90% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. Findings from 2 supportive analyses were consistent with the primary analysis, suggesting a 95% and 89% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group in the blinded study product analysis and per-protocol (PP) analysis, respectively.
Incident HIV-1 Infections in Step 2 Only
Both the HPTN 083 and HPTN 084 studies met the secondary end point, incident HIV-1 infections in step 2 only. In the HPTN 083 study, 8 HIV-1 infections were identified in the cabotegravir LA group and 37 in the TDF-FTC group in step 2 only by the May 14, 2020, interim cut-off date. The incidence rate per 100 PY in the cabotegravir LA group was 0.27 (95% CI, 0.12 to 0.54) and 1.29 (95% CI, 0.91 to 1.77) in the TDF-FTC group (HR = 0.210; 95% CI, 0.10 to 0.45), suggesting a 79% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. The between-group difference in incidence rates favoured cabotegravir LA over TDF-FTC (–1.01 per 100 PY; 95% CI, –1.47 to –0.56).
In the HPTN 084 study, 2 HIV-1 infections were identified in the cabotegravir LA group and 34 in the TDF-FTC group in step 2 only by the November 5, 2020, interim cut-off date. The incidence rate per 100 PY in the cabotegravir LA group was 0.11 (95% CI, 0.01 to 0.41) compared to 1.94 (95% CI, 1.35 to 2.72) in the TDF-FTC group (HR = 0.06; 95% CI, 0.01 to 0.24), suggesting a 94% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. The between-group difference in incidence rates also favoured cabotegravir LA over TDF-FTC (–1.83 per 100 PY; 95% CI, –2.5 to –1.16).
Viral Genotyping for Drug Resistance
Viral genotyping of participants who were seroconverters was assessed as a secondary end point in the HPTN 083 study, and a tertiary end point in the HPTN 084 study. No new resistance mutations were reported among seroconverters for the 2 drugs in both trials. In the cabotegravir LA group of the HPTN 083 study, HIV genotyping results were obtained for 12 of the 15 incidents of HIV-1 infections in people receiving cabotegravir LA (1 failed analysis and 2 had no viremic visits). Integrase resistance mutations were identified in 3 participants and non-nucleoside reverse transcriptase inhibitor (NNRTI) resistance was identified in 3 other participants at the first viremic visit, including 1 case of resistance to a nucleoside reverse transcriptase inhibitor (NRTI).
In the TDF-FTC group of the HPTN 083 study, HIV genotyping results were presented for 40 of the 42 HIV cases reported in the TDF-FTC group (2 cases had no viremic visit). There were no resistance patterns identified in participants who had HIV-1 at baseline. Twelve participants showed resistance at the first viremic visit (7 had NNRTI resistance only, 1 had NRTI resistance only, 1 had single protease inhibitor [PI] resistance mutation only, and 3 had NNRTI and NRTI resistance). Ten participants identified with NNRTI resistance had 1 or more of the following mutations: K103N/S, Y181C, G190A/S, H221Y, and P225H. In 4 participants with NRTI resistance (including 3 who had multiclass resistance), 3 had M184V/I and K65R mutations.
In the cabotegravir LA group of the HPTN 084 study, HIV genotyping results were available for 3 of 4 cabotegravir LA participants with HIV-1 infections (1 case with no viremic sample). One of 3 had an integrase mutation at the first viremic visit (L74l), which is considered a polymorphism, and also detected in participants in the TDF-FTC group.11
In the TDF-FTC group of the HPTN 084 study, HIV genotyping results were obtained for 33 of the 36 incident infections in the TDF-FTC group (2 failed testing; 1 had no viremic sample). A major NRTI mutation (M184V) was identified in 1 participant in addition to an NNRTI resistance with the K103N mutation. Eight other participants had NNRTI resistance only (6 had K103N alone, or with E138A or P225H; 1 had K101E alone; and 1 had E138A alone). Integrase strand transfer inhibitor (INSTI) mutations/polymorphisms were detected in 10 samples (L74I, L74M, T97A, V151I, E157Q, and G193E). For 1 participant with a dual-class resistance (NRTI and NNRTI), resistance observed in the first viremic visit was the same as the first site-positive visit (at step 2, week 17; 33 days after the first HIV-positive visit).
Adherence (Measured Through Pharmacokinetics)
Adherence was assessed as a tertiary end point assessed within a subset of participants for each study medication in both trials. In the HPTN 083 study, adherence to cabotegravir LA injections was assessed in a random subset of 170 participants. Injection coverage was 91.5% of all PY contributions for the subsample. Adherence to TDF-FTC was assessed in a random subset of 390 participants using plasma tenofovir (TFV) concentrations and intraerythrocytic TFV diphosphate (TFV-DP) concentrations collected as dried blood spot (DBS) in the HPTN 083 study. In total, 74.2% of participants had TFV concentrations consistent with daily dosing (i.e., ≥ 40 ng/mL) and more than 86% had detectable TFV (≥ 0.31 ng/mL). Findings based on DBS showed that 73% of samples yielded TFV-DP concentrations consistent with 4 or more doses per week.
In a random subset of 150 participants in the HPTN 084 study, injection coverage in the cabotegravir LA group was 93% of all PY contributions for the subsample. TDF-FTC assessments were conducted in a random subset of 409 participants, of which 41.9% had TFV concentrations consistent with daily dosing (≥ 40 ng/mL, corresponding to expected daily use concentration of TDF-FTC) and 55.9% had detectable TFV (≥ 0.31 ng/mL).
Harms
The proportion of participants reporting at least 1 AE in the safety population set (OBSP steps 1 and 2) was generally similar in both groups across trials. In the HPTN 083 study, 95% versus 94% in the cabotegravir LA group and TDF-FTC group, respectively, reported at least 1 AE, and in the HPTN 084 study, 96% in both the cabotegravir LA group and TDF-FTC groups reported at least 1 AE. Commonly reported AEs included injection site pain, creatinine clearance decreased, blood creatine phosphokinase increased, blood creatinine increased, and nasopharyngitis. Serious AEs (SAEs) were reported by 5% of patients in each group in the HPTN 083 trial, and 2% each in each group in the HPTN 084 trial.
In total, 10 deaths were reported in the HPTN 083 study, in the combined steps 1 and 2 (4 in the cabotegravir LA group and 6 in the TDF-FTC group), and 1 additional death was reported in step 3. In the HPTN 084 study, 3 participants in the cabotegravir LA group died due to AEs. No deaths were reported in the TDF-FTC group. Withdrawals due to AEs were generally low in the 2 groups in the 2 studies (6% versus 4% in the cabotegravir LA and TDF-FTC groups, respectively, in the HPTN 083 study and 1% in each group in the HPTN 084 study).
Notable harms commonly reported in both trials included ISRs, hepatotoxicity, hypersensitivity reactions, rash, and neuropsychiatric events. ISRs were higher in the cabotegravir LA group in both trials (76% versus 32% in the cabotegravir LA and TDF-FTC groups, respectively, in the HPTN 083 study and 38% versus 11% in the cabotegravir LA and TDF-FTC groups, respectively, in step 2 of the HPTN 084 study).
Critical Appraisal
The HPTN 083 and HPTN 084 trials were multicentre trials with centres in the US, South America, Asia, and Sub-Saharan Africa. There were no sites in Canada. The methods for randomization, allocation concealment, and double-blinding maintenance were appropriate. Randomization was stratified by study site, and permuted blocks were used to ensure balance in treatment assignments within study sites. The use of placebo and the blinding of patients and outcome assessors mitigated concerns related to the risk of bias due to deviations from the intended interventions. The inclusion and exclusion criteria and patient characteristics at baseline were considered generalizable to Canada. Overall, the primary and key secondary outcomes assessed in both trials were considered appropriate and relevant to decision-making; they also adequately reflected measures of both efficacy and harms assessed in clinical practice. There were no notable imbalances in baseline demographics between treatment groups indicating that randomization was effective.
The use of the Poisson model to estimate the rate of HIV infection in both trials was deemed appropriate by the CDA-AMC review team but subject to 2 critical assumptions for the rate of infection with HIV. First, that the rate of infection within the population is at a constant rate and second, that the withdrawal and censoring were noninformative of an individual’s potential future infection. The HPTN 083 study was a noninferiority trial and the HR margin (M2) was selected based on evidence from prior placebo-controlled trials (the iPrex,12 iPERGAY,13 and PROUD studies14). In the HPTN 084 study, the superiority of cabotegravir LA was demonstrated by an improvement in the incidence of HIV infection. The analyses conducted were preplanned interim analyses, which can lead to an increased risk of overestimating the treatment effects (only 30% and 35% of the total targeted preplanned infections for powering the HPTN 083 and HPTN 084 studies, respectively, were achieved at both interim data cut-offs). Missing data for the primary outcome across trials were addressed using noninformative censoring, supported by prespecified sensitivity analyses. Adjustments for type I error were accounted for in key primary and secondary outcomes assessed in both trials. Neither study was powered for subgroup evaluations and no adjustments were made for multiple testing subgroup analyses. Treatment adherence was assessed using pharmacokinetic (PK) blood concentrations of study drugs in a random subset of participants for each treatment. There were differences between the 2 treatment groups in both trials which may have impacted the efficacy of the primary outcome. There were notable differences in treatment adherence between the 2 groups within each trial, and between the 2 trials. However, PK assessments of plasma for drug concentrations may not be a comprehensive evaluation of adherence in participants due to known variabilities in drug metabolism across individuals. Both trials provided direct evidence of the comparative efficacy of cabotegravir LA compared to an available PrEP option in Canadian practice; however, there is a lack of evidence on the long-term therapeutic benefit and safety of cabotegravir LA beyond the duration of both trials, which is a source of uncertainty. The dosing regimen of TDF-FTC in both trials aligned with Canadian practice. The duration of follow-up was considered appropriate and adequate to identify HIV-1 events and a difference between the 2 groups. Although follow-up frequencies and adherence measurements assessed during the trials were considered appropriate, they may not be reflective of current Canadian guideline recommendations. There were no concerns with the concomitant medications administered that may have impacted on cabotegravir LA’s efficacy.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the CDA-AMC’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.15,16
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of a clinically important effect based on a threshold informed by the clinical expert consulted by the CDA-AMC for documented incident HIV infections. There is no established minimally important difference and the clinical expert consulted by the CDA-AMC could not provide a threshold of important difference so the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect. Other targets for the certainty of evidence assessment were the presence or absence of any effect for the proportion of patients reporting SAEs and ISRs.
Results of GRADE Assessments
The GRADE assessments included an evaluation of the main outcomes considered important by clinicians, patient groups, and stakeholders. The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following outcomes were finalized in consultation with expert committee members: documented incident HIV infections and harms (SAEs and ISRs). Two outcomes included in the report (resistance mutations to study products among seroconverters and adherence to study product) were not included on the GRADE table.
Table 2 and Table 3 present the GRADE summary of findings for cabotegravir LA versus TDF-FTC for cisgender MSM and TGW who have sex with men, and cisgender women, respectively, at risk of acquiring HIV-1 infection.
Table 2: Summary of Findings for Cabotegravir LA Versus TDF-FTC for Cisgender MSM and TGW Who Have Sex With Men at Risk of Acquiring HIV-1 Infection (Study HPTN 083)
Outcome and follow-up | Patients (studies), N | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|
Cabotegravir LA | TDF-FTC | Difference | ||||
Documented HIV-1 infections | ||||||
Incidence rate of HIV-1 infections in steps 1 and 2 Follow-up: 6,404 total PY | 4,561 (1 RCT) | 0.40 per 100 PY (0.22 to 0.69) | 1.22 per 100 PY (0.87 to 1.67) | 0.82 fewer incident HIV-1 infections per 100 PY (0.38 to 1.26 fewer) | Higha | Cabotegravir LA results in a reduction in the incidence of HIV-1 when compared to TDF-FTC in cisgender MSM and TGW. The clinical importance of the reduction is unclear. |
Harms | ||||||
Proportion of participants with SAEs Follow-up: approximately 160 weeks cumulative follow-up (before data cut-off) | 4,566 (1 RCT) | 5 per 100 PY (NR) | 5 per 100 PY (NR) | 0.36 more SAEs per 100 PY (0.9 fewer to 1.6 more) | Moderateb | Cabotegravir LA likely results in fewer to more SAEs when compared to TDF-FTC in cisgender MSM and TGW. The clinical importance of the reduction is unclear. |
Proportion of participants with ISRs Follow-up: approximately 160 weeks cumulative follow-up (before data cut-off) | 4,198 (1 RCT) | 82 per 100 PY (NR) | 35 per 100 PY (NR) | 47.4 more ISRs per 100 PY (44.8 to 50 more ISRs) | Highc | Cabotegravir LA likely results in more ISRs when compared to TDF-FTC in cisgender MSM and TGW. |
CI = confidence interval; ISR = injection site reaction; LA = long-acting; MID = minimally important difference; MSM = men who have sex with men; NR = not reported; PY = person-years; RCT = randomized controlled trial; SAE = serious adverse event; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TGW = transgender women.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The HPTN 083 study is a noninferiority phase IIb/II study which enrolled HIV-1 uninfected cisgender men and TGW who have sex with men, at risk of acquiring HIV-1 infection.
aThere is no established MID and the clinical expert consulted by CDA-AMC could not provide a threshold of important difference, therefore the null was used. Not rated down for imprecision as CI of the difference between the 2 groups did not overlap with the null (0). The CDA-AMC review team judged that the point estimate and the 95% CI for the between-group difference suggested a benefit. Although results were from an interim analysis, certainty of evidence was not rated down by the CDA-AMC team because appropriate methods (i.e., Lan DeMets modification of the O’Brien-Fleming stopping bounds method) were used to account for alpha spending before interim analysis.
bThere is no established MID and the clinical expert consulted by CDA-AMC could not provide a threshold of important difference, therefore the null was used. Rated down 1 level for serious imprecision. The lower bound of the 95% CI for the between-group difference was < 0 while the upper bound was > 0 and suggested no clinically important difference between the 2 groups.
cThere is no established MID and the clinical expert consulted by CDA-AMC did not provide a threshold of important difference. The CDA-AMC review team judged that the MID of harm for ISR was null given that both treatments consist of 2 formulations: an oral medication and intramuscular injections. Not rated down for imprecision as CI of the difference between the 2 groups did not overlap with the null (0) and fell beyond the clinically meaningful benefit threshold, indicating harm.
Source: HPTN 083 Clinical Study Report.17,18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.11
Table 3: Summary of Findings for Cabotegravir LA Versus TDF-FTC for Cisgender Women at Risk of Acquiring HIV-1 Infection (Study HPTN 084)
Outcome and follow-up | Patients (studies), N | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|
Cabotegravir LA | TDF-FTC | Difference | ||||
Documented HIV-1 infections | ||||||
Incidence rate of HIV-1 infections in steps 1 and 2 Follow-up: 3,907 total PY | 3,224 (1 RCT) | 0.20 per 100 PY (0.06 to 0.52) | 1.85 per 100 PY (1.30 to 2.56) | 1.65 fewer incident HIV-1 infections per 100 PY (1.01 to 2.28 fewer) | Higha | Cabotegravir LA results in a reduction in the incidence of HIV-1 when compared to TDF-FTC in cisgender women. The clinical importance of the reduction is unclear. |
Harms | ||||||
Proportion of participants with SAEs Follow-up: approximately 158 weeks cumulative follow-up up (before data cut-off) | 3,224 (1 RCT) | 2 per 100 PY(NR) | 2 per 100 PY (NR) | 0.005 fewer SAEs per 100 (0.98 fewer to 0.97 more) | Moderateb | Cabotegravir LA likely results in fewer to more SAEs when compared to TDF-FTC in cisgender women. The clinical importance of the reduction is unclear. |
Proportion of participants with ISRs Follow-up: approximately 158 weeks cumulative follow-up (before data cut-off) | 3,035 (1 RCT) | 38 per 100 PY (NR) | 11 per 100 PY (NR) | 27.1 more ISRs per 100 PY (24.2 to 30 more ISRs) | Highc | Cabotegravir LA likely results in more ISRs when compared to TDF-FTC in cisgender women. |
CI = confidence interval; ISR = injection site reaction; LA = long-acting; MID = minimally important difference; NR = not reported; PY = person-years; RCT = randomized controlled trial; SAE = serious adverse event; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The HPTN 084 study is a phase III superiority trial which enrolled HIV-uninfected cisgender women at risk of acquiring HIV-1 infections.
aThere is no established MID and the clinical expert consulted by CDA-AMC could not provide a threshold of important difference, therefore the null was used. Not rated down for imprecision as the CI of the difference between the 2 groups did not overlap with the null (0). The CDA-AMC review team judged that the point estimate and the 95% CI for the between-group difference suggested a benefit. Although results were from an interim analysis, certainty of evidence was not rated down by the CDA-AMC team because appropriate methods (i.e., Lan DeMets modification of the O’Brien-Fleming stopping bounds method) were used to account for alpha spending before interim analysis.
bThere is no established MID and the clinical expert consulted by CDA-AMC could not provide a threshold of important difference, therefore the null was used. Rated down 1 level for serious imprecision. The lower bound of the 95% CI for the between-group difference was < 0 while the upper bound was > 0 and suggested no clinically important difference between the 2 groups.
cThere is no established MID and the clinical expert consulted by CDA-AMC did not provide a threshold of important difference. The CDA-AMC review team judged that the MID of harm for ISR was null given that both treatments consist of 2 formulations: an oral medication and intramuscular injections. Not rated down for imprecision as the CI of the difference between the 2 groups did not overlap with the null (0) and fell beyond the clinically meaningful benefit threshold, indicating harm.
Source: HPTN 084 Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.11
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparisons
Description of Studies
One sponsor-conducted indirect treatment comparison (ITC) compared cabotegravir LA to placebo or no PrEP with respect to the effectiveness for reducing HIV transmission using a Bayesian network meta-analysis (NMA).
Efficacy Results
In the Bayesian fixed-effect NMA, based on 10 trials, cabotegravir LA demonstrated improved effectiveness in reducing HIV transmission compared to placebo or no PrEP (drug effectiveness = 91.10%; 95% credible interval [CrI], 82.87% to 95.95% in the HPTN 083 study population [cisgender MSM and TGW] and drug effectiveness = 92.52%; 95% Crl, 83.02% to 97.38% in the HPTN 084 study population [cisgender women]).
Harms Results
No harm results were reported in the sponsor-submitted NMA.
Critical Appraisal
The validity of the NMA results is dependent on key assumptions (e.g., homogeneity and consistency). Network homogeneity, and consistency could not be determined based on insufficient reporting of study characteristics and a sparse linear network without a closed loop. Based on the available information, there was evidence of heterogeneity between the included studies based on study designs (e.g., blinding), patient populations (e.g., mixing people who inject drugs [PWID] and non-PWID), and trial characteristics that were unaccounted for in the analysis. These limitations result in uncertainty in the magnitude of the relative treatment effect estimates between cabotegravir LA versus placebo or no PrEP.
Studies Addressing Gaps in the Evidence from the Systematic Review
Two studies conducted in adolescent populations were submitted for this review. The HPTN 083 to 01 and HPTN 084 to 01 studies were both open-label, single-arm, phase IIb substudies of the main pivotal trials (HPTN 083 and HPTN 084) assessing the safety, tolerability, and acceptability of cabotegravir LA in HIV-1-negative adolescent participants (aged < 18 years), cisgender females, and males (identifying as MSM or TGW) at risk of acquiring HIV-1.
Efficacy Results
No efficacy outcomes were assessed in both trials.
Harms
No new safety concerns were identified. ISRs reported in both studies were of grade 1 and 2 and did not result in study drug discontinuations. Cabotegravir LA injections were also well-tolerated with no participant discontinuing treatment prematurely due to intolerability of injection or burden of study procedures.
Critical Appraisal
There is uncertainty about whether the sample size and power calculations for both studies were sufficient to assess the efficacy of cabotegravir LA in the 2 studies (N = 9 in the HPTN 083 to 01 study and N = 55 in the HPTN 084 study). The lack of a comparative and the absence of any assessments related to primary efficacy outcomes limited the interpretability of the magnitude of the benefit of cabotegravir LA reducing HIV-1 infections in adolescent populations. Thus, no definitive conclusions could be drawn; however, no safety signals were identified.
Conclusions
Evidence from 2 pivotal, multicentre, double-blind, RCTs (HPTN 083 and HPTN 084) demonstrated the efficacy and safety of cabotegravir LA administered by intramuscular injections compared to oral TDF-FTC for PrEP in adult (aged18 years and older) HIV-negative cisgender MSM, TGW who sex with men, and cisgender women. Noninferiority and superiority of cabotegravir LA to TDF-FTC was demonstrated in the HPTN 083 trial and superiority of cabotegravir LA to TDF-FTC was demonstrated in the HPTN 084 study.
The totality of evidence from the interim analyses of both trials suggests that cabotegravir LA reduces the incidence of HIV-1 infections in participants at risk of sexually acquired HIV-1 infection compared to oral TDF-FTC. The certainty of evidence was considered high in both trials, with a reported risk difference in favour of cabotegravir LA for participants receiving drug during the combined oral lead-in and injection phases, and injection phase only. However, it is unclear whether the observed between-group differences are of clinical importance. Adherence assessments reported in a subset of participants in each treatment group in both studies showed higher coverage for cabotegravir LA injections compared to drug concentrations of TDF-FTC consistent with daily dosing and could be the driving factor for the observed reduction in HIV acquisition risk. Key integrase resistance mutations to cabotegravir LA were identified in some participants receiving cabotegravir LA who tested positive for HIV-1 during treatment. Other mutations detected included NNRTIs and NRTIs, which do not contribute to cabotegravir LA resistance. According to the expert, mutations may impact the subsequent choice of treatment for HIV. The safety profile of cabotegravir LA observed in both trials was considered manageable with no new safety signals identified.
Evidence from the sponsor-conducted NMA on the comparative effectiveness suggests benefits of cabotegravir LA over placebo and no PrEP in reducing HIV-1 infections. Data were lacking for TAF-FTC, another treatment available to eligible populations in Canada. The sponsor’s NMA did not assess harms that may impact on the safety profile of cabotegravir LA. Overall, there is uncertainty in the NMA findings due to several limitations preventing the assessment of key assumptions of the analyses. Evidence of long-term safety and efficacy beyond the pivotal trials was not available for this review; however, long-term extension studies for both trials are currently ongoing with no expected completion date at the time of this review. Data in adolescent populations were lacking; however, according to the clinical expert Canada’s Drug Agency (CDA-AMC) consulted, the findings observed in the adults will be generalizable in adolescents.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of cabotegravir 30 mg oral tablets, and 200 mg/mL (600 mg/3 mL) extended-release injectable solution, as PrEP to reduce the risk of sexually acquired HIV-1 for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
HIV is a retrovirus that impairs the human immune system, and is transmitted through bodily fluids via sex or vertically (i.e., from mother to child during pregnancy, childbirth, and/or breastfeeding).1 Without treatment, HIV infections progress from acute through clinical latency to AIDS, making PLHIV more vulnerable to opportunistic infections and diseases.2-6 PLHIV face discrimination and stigma, leading to increased psychosocial and emotional burden and an overall decreased quality of life.19-22
The number of PLHIV in Canada remains high and the incidence of new HIV infections continues to rise in Canada. At the end of 2020, it was estimated that there were 62,790 PLHIV cases in Canada, with a prevalence rate of approximately 170 per 100,000 persons.23 Of the 62,790 prevalent HIV infections in Canada at the end of 2020, half (50.3%) were among gbMSM, 13.3% were PWID, 32.8% were heterosexual people, 24.6% were female, and 10.3% were Indigenous peoples.23 The provinces with the highest estimated number of PLHIV at the end of 2020 were Ontario, Quebec, and British Columbia, and gbMSM accounted for more than half of the prevalent HIV infections in these provinces.23 Heterosexual people made up more than half of all PLHIV in Manitoba and Alberta while more than two-thirds of all PLHIV in Saskatchewan were PWID. In Atlantic Canada, gbMSM made up more than half of all PLHIV while 1 in 4 PLHIV were heterosexual people. In 2022, there were 1,833 new HIV infections in Canada resulting in an incidence of 4.7 per 100,000 persons.8 This represents a 24.9% increase from the estimates for 2021. The rate of new HIV infections was 6.3 per 100,000 males and 3.1 per 100,000 females (excludes cases for whom sex was reported as transgender or not reported).8
There are no companion diagnostic tests to identify patients who would be eligible for PrEP; however, as stated in the cabotegravir product monograph, individuals considering treatment with PrEP must have had a documented negative HIV-1 test, in accordance with applicable guidelines, before initiating cabotegravir LA. Testing for HIV-1 is performed by blood test in an accredited laboratory.24-27
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
Canada has adopted an integrated approach toward HIV management and prevention.1 PrEP, which refers to the use of antiretroviral medication to prevent HIV infection, is considered an effective treatment strategy, and is the standard of care for HIV prevention.1 However, the effectiveness of any PrEP option depends on the modification of key behavioural factors that impact efficacy such as medication adherence, participation in a clinical follow-up in eligible individuals, and the use of other HIV prevention strategies (such as condom use, safer injection equipment, risk reduction counselling, and regular HIV and STI testing).1 The Canadian guideline on HIV management (2017)1 recommends PrEP in combination with behavioural interventions (e.g., condoms, counselling on risk reduction, partner reduction), biomedical interventions (e.g., treatment of HIV-positive partners, testing and treatment of STIs), and attention to syndemic conditions that may predispose people to increased risk-taking behaviour (e.g., depression, substance use) for gbMSM and TGW individuals who report condomless anal sex, HIV-negative partners in heterosexual serodiscordant relationships reporting condomless vaginal or anal sex where the HIV-positive partner has a substantial risk of having transmissible HIV, and for PWID if they are sharing injection drug use paraphernalia with a person with a non-negligible risk of HIV infection.1
There are currently 2 PrEP options in Canada: TDF-FTC (Truvada) and TAF-FTC (Descovy). TDF-FTC is an oral therapy, reimbursed by most jurisdictions in Canada.1 TAF-FTC (Descovy) is not indicated for individuals at risk from receptive vaginal sex and is only reimbursed through the Canadian Armed Forces Drug Benefit List.1 There is a need for options that are convenient for individuals, and which promote adherence in all at-risk populations according to the clinical expert consulted during the CDA-AMC review.
Drug Under Review
Key characteristics of cabotegravir LA are summarized in Table 4 with other PrEP treatments available for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg to reduce the risk of sexually acquired HIV-1 infection.
Cabotegravir is an antiretroviral medication, which inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral DNA integration which is essential for the HIV replication cycle. Cabotegravir tablets may be used as an oral lead-in to assess tolerability of cabotegravir before administration of cabotegravir LA injections or as short-term oral PrEP in individuals who will miss planned dosing with cabotegravir LA injections. Cabotegravir LA is indicated for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg for PrEP to reduce the risk of sexually acquired HIV-1 infection. The sponsor’s reimbursement request aligns with the Health Canada–approved indication. Cabotegravir LA monotherapy has not been previously reviewed by CDA-AMC for PrEP for HIV-1 prevention. However, cabotegravir LA in combination with rilpivirine has been previously reviewed for the treatment of HIV-1 in infected patients.28 Cabotegravir LA has been approved in the US and European Union for the same proposed indication in Canada.29,30
Table 4: Key Characteristics of Cabotegravir LA, TDF-FTC, and TAF-FTC
Characteristic | Cabotegravir LA | TDF-FTC | TAF-FTC |
|---|---|---|---|
Mechanism of action | Cabotegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral DNA integration which is essential for the HIV replication cycle. | Emtricitabine is a nucleoside HIV‐1 reverse transcriptase inhibitor, and tenofovir disoproxil fumarate is a nucleotide analogue reverse transcriptase inhibitor and is the prodrug of tenofovir. | Emtricitabine is a nucleoside HIV‐1 reverse transcriptase inhibitor. Tenofovir alafenamide is a nucleotide reverse transcriptase inhibitor and is a prodrug of tenofovir. |
Indicationa | For at-risk adults and adolescents aged ≥ 12 years and weighing at least 35 kg for PrEP to reduce the risk of sexually acquired HIV-1 infection. | In combination with safer sex practices for PrEP to reduce the risk of sexually acquired HIV-1 infection in adults at high risk. | TAF-FTC (Descovy) is indicated for PrEP to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing ≥ 35 kg, excluding individuals at risk from receptive vaginal sex. |
Route of administration | Intramuscular injection Option for oral lead-in | Oral | Oral |
Recommended dose | When an oral lead-in is used, the recommended dose is as follows:
When cabotegravir LA injection is initiated directly, the recommended dose is as follows:
| One tablet (containing 200 mg of emtricitabine and 300 mg of tenofovir disoproxil fumarate) once daily taken orally with or without food. | 200 mg-25 mg once daily with or without food |
Serious adverse events or safety issues |
|
|
|
Other | Individuals should be counselled periodically to strictly adhere to the recommended cabotegravir LA dosing schedule to reduce the risk of HIV-1 acquisition and the potential development of resistance. It is essential to clinically reassess individuals for risk of HIV-1 acquisition and to frequently test to confirm HIV-1 negative status to minimize the risk of developing resistance to cabotegravir LA. | TDF-FTC used for a PrEP indication must only be prescribed to individuals confirmed to be HIV-negative immediately before initial use and periodically (at least every 3 months) during use. Do not initiate TDF-FTC for a PrEP indication if signs or symptoms of acute HIV infection are present unless negative infection status is confirmed. | TAF-FTC used for HIV-1 PrEP must only be prescribed to individuals confirmed to be HIV-negative immediately before initiating and at least every 3 months during use. Do not initiate TAF-FTC for HIV-1 PrEP if signs or symptoms of acute HIV-1 infection are present unless negative infection status is confirmed. |
LA = long-acting; PrEP = pre-exposure prophylaxis; TAF-FTC = tenofovir alafenamide fumarate-emtricitabine; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
aHealth Canada–approved indication.
bindividuals may be given cabotegravir LA injection up to 7 days before or after the date the individual is scheduled to receive the injections.
Sources: Cabotegravir product monograph,11 Truvada product monograph,31 and Descovy product monograph.32
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) received by CDA-AMC have been included in the Stakeholder section of this report.
Five patient groups submitted inputs on the indication being reviewed: APAA, which is a nonprofit AIDS service organization; HIV Edmonton, which provides programs, services and engagement activities with and for people living with, or impacted by, HIV and AIDS and other sexually transmitted blood borne infections; CATIE, which is Canada’s HIV, hepatitis C, and sexually transmitted blood borne infection knowledge exchange broker and a trusted national source for up-to-date, unbiased information about HIV, hepatitis C, and related sexually transmitted blood borne infections; CBRC, which promotes the health of people of diverse sexualities and genders through research and intervention development; and POSSE, which is a youth-driven, harm reduction and human rights training and peer outreach project, for youth between the ages of 15 to 35 years. APAA collected verbal and written inputs from their clients through surveys, evaluation questionnaires, focus group discussions, and regular client meetings, and did not provide the number of patients that participated in this submission. The HIV Edmonton research team collected data through 27 interviews with 18 PLHIV. HIV Edmonton also submitted a testimony from an older adult and long-term HIV survivor who shared the barriers and challenges they had experienced. CATIE gathered information through education evaluation activities, consultations with community-based organizations, people affected by HIV, and patient groups through partnership and collaboration and multiple projects and research projects that focus on HIV prevention; CATIE did not provide the number of patients that participated in this submission. CBRC gathered information through Sex Now (2021 and 2022 [N = 144]), which is CBRC’s principal community-based research initiative and Canada’s largest and longest-running survey of gay, bisexual, queer men (cisgender and transgender), nonbinary, and Two-Spirit people’s health; CBRC did not provided the number of patients that participated in the 2021 survey. POSSE obtained inputs from staff and service users; POSSE did not provide the number of patients that participated in this submission.
HIV-related stigma and discrimination at the system level in our laws and institutions, within the medical profession and communities at large, and between individuals or by oneself in the form of shame and guilt were highlighted by all patient groups. The stigma often leads to isolation and fear of disclosure and affects treatment maintenance or medication drop-off and the quality of life of those affected by HIV. According to the Sex Now 2021 online survey, the awareness of HIV PrEP as a medication to prevent HIV varied among at-risk populations. For example, approximately 90% of 2SLGBTQ+ men were aware of HIV PrEP while only 50% of Indigenous people were aware that the medication is covered by the Non-Insured Health Benefits Program. Inputs from CATIE highlighted that oral PrEP uptake remains low in Canada. According to the Sex Now 2021 online survey, 17% of HIV-negative 2SLGBTQ+ men were using HIV PrEP and only an additional 9% had ever used HIV PrEP before. Patients from APAA and CATIE identified racism and cultural and linguistic barriers deter African Caribbean Black PLHIV from accessing treatment; other barriers included homophobia, limited information and access to health care facilities, and financial constraints. Patients from HIV Edmonton, APAA, and CBRC highlighted challenges such as side effects of oral medications on the digestive and intestinal systems, pill burden, and the impact of daily medication on lifestyle, which can affect treatment adherence. Inputs from POSSE mentioned that youth struggle with adherence to medications and require solutions reducing adherence requirements.
Patient groups highlighted that there are stigma and adherence issues associated with current oral PrEP options (i.e., TDF-FTC and TAF-FTC). The patient groups noted that remembering to take oral pills can be challenging for people who use substances or for people dealing with competing priorities. The groups highlighted concerns relating to the safe storage of medications for especially persons in need of shelter and the renewal of prescriptions.
Patients have been using cabotegravir LA recently according to inputs from CBRC and CATIE. Patients expressed their preference for injectable PrEPs, according to the Sex Now 2022 survey. Among PLHIV (n = 144), only 19% preferred taking daily oral pills versus an injectable medication taken every 2 months, with 47% preferring the injectable. The advantages of injectable PrEPs identified by patients include the reduced stigma experienced in multiple settings due to reduced exposure to health services or systems where stigmatizing experiences occur, increased privacy and discretion, decreased risk of treatment interruptions when they travel, increased adherence to treatment, reduced impact on digestive-related issues from consuming pill treatment, and improved quality of life (e.g., improved autonomy and self-determination by having a choice in treatment decisions and not being encumbered by medication regimen schedules).
Outcomes that patients would like to improve include improved access to treatment, improved treatment adherence, decreased breakthrough infection and risk of resistance, sustained viral suppression, and increased level of comfort in the treatment.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of HIV.
Unmet Needs
The clinical expert consulted during the review noted that a current treatment goal for HIV is to prevent persons from acquiring HIV through sexual transmission. The expert noted that although there are PrEP options currently available in Canada (TDF-FTC and TAF-FTC), these regimens, mostly administered orally, may not benefit all at-risk populations. According to the expert, there is an unmet need for options that are convenient to individuals, and which promote adherence.
Place in Therapy
The clinical expert noted that cabotegravir LA, which is administered intramuscularly every 2 months, would provide an alternative to daily oral treatment for individuals.
Patient Population
According to the clinical expert, anyone at risk of contracting sexually acquired HIV-1 will benefit from cabotegravir LA. Individuals who will be less suitable will include those who cannot tolerate injections.
Assessing the Response Treatment
According to the clinical expert, the most reliable way to assess treatment response is to assess whether individuals remain HIV-negative during routine follow-up tests, typically performed every 3 months to 6 months.
Discontinuing Treatment
The clinical expert highlighted that factors that may lead to the discontinuation of cabotegravir LA will include individual tolerance to treatment and acquisition of HIV-1 (the need for transition to HIV treatment). Although injection reactions are frequently observed, the expert noted that the injections are usually well-tolerated. However, a severe ISR (pain) may precipitate changes in treatment modality.
Prescribing Considerations
The clinical expert noted that any clinician who provides PrEP care can prescribe and monitor the use of cabotegravir LA for PrEP. These would include clinicians at sexual health clinics, physicians, primary care providers, or infectious diseases specialists. The expert noted that training and medical expertise is required to administer intramuscular injections, necessitating a nurse practitioner or physician or an appropriately licensed professional to provide the treatment. This requirement limits the setting for injection administration but does not restrict the prescription or monitoring of patients on the treatment.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group input(s) received by CDA-AMC have been included in the Stakeholder section of this report.
Two inputs were submitted on the indication being reviewed: 1 clinician group of 6 clinicians from the Vancouver Coastal Health Regional HIV Program, which is a public health program that aims to reduce the rate of HIV infection among the 1.25 million people living in the region, and 1 clinician, who is an associate professor in the Division of Infectious Diseases in the Faculty of Health Sciences at McMaster University.
Inputs from clinician groups are in line with the clinical expert consulted by CDA-AMC. The inputs discussed that oral PrEPs are currently available for individuals who are at higher risk of acquiring HIV, the treatment goal is to decrease the incidence of newly acquired HIV infections, and there remains an unmet need to improve treatment compliance and convenience. Both the clinical expert consulted by CDA-AMC and the clinician groups agreed that cabotegravir LA would be an alternative to daily oral PrEP and the best-suited patients for cabotegravir LA would be individuals who are at risk of sexually acquired HIV. Clinicians from the Vancouver Coastal Health Regional HIV Program specified that individuals in whom adherence to oral daily HIV PrEP is difficult are best suited to LA injectable HIV PrEP. Incident HIV infections is the outcome used to determine whether a patient is responding to treatment in clinical practice by the clinical expert consulted by CDA-AMC and the clinician groups. Inputs from clinician groups highlighted oral PrEP along with a robust monitoring and follow-up strategy are crucial after patients have discontinued cabotegravir LA. Clinician groups stated that cabotegravir LA should be prescribed and monitored by various health care providers (e.g., family doctors, nurse practitioners, and specialists in HIV care) in community, hospital, and specialty clinics where individuals can access PrEP prescriptions, HIV testing, and ongoing care.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 5.
Table 5: Summary of Drug Program Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There are 2 PrEP modalities available in Canada, TDF-FTC (Truvada) and TAF-FTC (Descovy). Both clinical trials (HPTN 083 and HPTN 084) used TDF-FTC (Truvada) as the comparator which is the therapy reimbursed by the federal, provincial, and territory jurisdictions. TAF-FTC (Descovy) is only funded in 1 Canadian jurisdiction. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Clinical trials included participants who were at high risk for HIV acquisition. How should “high risk” be defined (i.e., use Canadian PrEP guidelines definition)? | The clinical expert considered the definition of high-risk individuals in the Canadian guidelines on PrEP as appropriate to be used in clinical practice. The definition of “high risk” according to the guidelines depends on type of exposure (anal, vaginal, or oral sex, or percutaneous). The guidelines define exposures at higher risk for HIV transmission to include condomless receptive anal sex and needle sharing. Exposures conferring moderate risk include condomless insertive anal sex and vaginal sex. |
The HPTN 083 study was conducted in MSM and TGW aged ≥ 18 years who are HIV-uninfected and at high risk for HIV acquisition and the HPTN 084 study was conducted in cisgender women aged 18 years to 45 years who are at high risk for HIV acquisition. Could this be considered for use in patients aged < 18 years? | The clinical expert did not consider age as a determining factor for the use of cabotegravir LA since the studies were driven by weight (weight > 35 kg). According to the expert, adolescent persons weighing ≥ 35 kg will be eligible to receive cabotegravir LA if they are considered at risk of acquiring HIV-1 sexually. |
Cabotegravir tablets may be used as an oral lead-in to assess tolerability of cabotegravir before administration of cabotegravir LA injections or as short-term oral PrEP in individuals who will miss planned dosing with cabotegravir LA injections. Should lead-in with cabotegravir oral tablets be required to assess tolerability before administration of cabotegravir LA injections? | The clinical expert consulted anticipates a variation in the use of the oral lead-in option for cabotegravir LA in practice. The expert noted that given the familiarity of clinicians with cabotegravir LA’s safety profile (observed from its use in combination therapies for HIV-1 infections), an oral lead-in during initiation may not be required for all candidates. The expert noted that cabotegravir LA has a tolerable safety profile; therefore, health care professionals prescribing the drug for PrEP may recommend the oral lead-in tablets to individuals with concerns related to safety of the IM injection at initiation. |
For consistency with initiation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space, consider aligning with criteria for Truvada for PrEP. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
Should this be restricted to prescribers in the context of a sexual health program or by a specialist experienced in the diagnosis and management of HIV? | The clinical expert indicated that any health care professional who provides PrEP care and monitoring is eligible to prescribe and monitor the use of cabotegravir LA in practice. These would include sexual health clinics, physicians, primary care providers, or infectious diseases specialists. The expert noted that access to treatment is clinic specific and although any prescriber could prescribe cabotegravir LA, intramuscular drug administration will typically be performed in clinics with trained personnels for intramuscular injections. |
For consistency with prescribing criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space, consider aligning with criteria for TDF-FTC (Truvada). | This is a comment from the drug plans to inform CDEC deliberations. |
Generalizability | |
Could pediatric patients and/or patients weighing < 35 kg be considered eligible? | The clinical expert noted that pediatric patients weighing > 35 kg considered at risk of acquiring HIV-1 infections sexually will be eligible to receive cabotegravir LA given that the efficacy of the drug is weight-dependent and not age-dependent. |
System and economic issues | |
Cabotegravir LA may have a significant budget impact. For participating drug plans, it was estimated that there will be █████ ████ and ████ patients treated with cabotegravir LA in years 1 to 3, respectively. In the scenario where cabotegravir LA is funded, the total drug cost of cabotegravir LA is anticipated to be $16,954,205, $35,006,553, and $40,205,665 in years 1 to 3, respectively. The resulting incremental budget impact from a drug program perspective was calculated to be $14,269,064, $28,293,702, and $30,136,388 in years 1 to 3, respectively. | This is a comment from the drug plans to inform CDEC deliberations. |
CDEC = Canadian Drug Expert Committee; IM = intramuscular; LA = long-acting; MSM = men who have sex with men; PrEP = pre-exposure prophylaxis; TAF-FTC = tenofovir alafenamide fumarate-emtricitabine; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TGW = transgender women.
Clinical Evidence
The objective of CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of cabotegravir, 30 mg oral tablets, and 200 mg/mL (600 mg/3 mL), extended-release injectable solution for PrEP to reduce the risk of sexually acquired HIV-1 infections in at-risk individuals. The focus will be placed on comparing cabotegravir LA to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of cabotegravir LA is presented in 4 sections with CDA-AMC’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following were included in the CDA-AMC review and appraised in this document:
2 pivotal RCTs identified in systematic review
1 long-term extension study
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Category | HPTN 083 | HPTN 084 |
|---|---|---|
Designs and populations | ||
Study design | Phase IIb/III, randomized, multicentre, double-blind, double-dummy, noninferiority study conducted in gbMSM and TGW | Phase III, randomized, multicentre, double-blind, double-dummy, open-label, superiority study conducted in cisgender women |
Locations | US (27 centres), Peru (5 centres), Brazil (4 centres), Argentina (2 centres), Thailand (3 centres), Vietnam (1 centre), and South Africa (1 centre) | 20 centres in Sub-Saharan Africa: Botswana, Kenya, Malawi, South Africa, Eswatini, Uganda, and Zimbabwe |
Patient enrolment dates | Start date: December 19, 2016 End date: March 16, 2020 | Start date: November 27, 2017 End date: November 4, 2020 |
Randomized | Randomized, N = 4,570 Cabotegravir LA and placebo TDF-FTC, n = 2,283 TDF-FTC and placebo cabotegravir LA, n = 2,287 | Randomized, N = 3,224 Cabotegravir LA and placebo TDF-FTC, n = 1,614 TDF-FTC and placebo cabotegravir LA, n = 1,610 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention arm | Step 1: Oral cabotegravir, 30 mg tablets, q.d. for up to 5 weeksc Step 2: Cabotegravir LA, 600 mg as a single IM injection at 2 time points 4 weeks apart and every 8 weeks thereafter to week 153 Step 3: Open-label extension phase oral TDF-FTC, 300 mg-200 mg fixed-dose combination tablets, q.d. for 48 weeks | Step 1: Oral cabotegravir, 30 mg tablets, q.d. for up to 5 weeksd Step 2: Cabotegravir LA, 600 mg in 1 3 mL IM injection at 2 time points 4 weeks apart and every 8 weeks thereafter until the required number of end points (114 events) was reached Step 3: Open-label extension phase oral TDF-FTC, 300 mg-200 mg tablets, q.d. for up to 48 weeks |
Comparator arm | Step 1: Oral TDF-FTC, 300 mg-200 mg fixed-dose combination tablets, q.d. for 5 weeks Step 2: Oral TDF-FTC, 300 mg-200 mg fixed-dose combination tablets, q.d. to week 153 Step 3: Open-label extension phase oral TDF-FTC, 300 mg-200 mg fixed-dose combination tablets, q.d. for 48 weeks | Step 1: Oral TDF-FTC, 300 mg-200 mg tablets, q.d. for up to 5 weeks Step 2: Oral TDF-FTC, 300 mg-200 mg tablets, q.d. until the required number of end points (114 events) was reached Step 3: Open-label TDF-FTC, 300 mg-200 mg tablets, q.d. for up to 48 weeks |
Study duration | ||
Screening phase | Up to 45 days | Up to 45 days |
Run-in phase | Step 1: Up to 5 weeks | Step 1: Up to 5 weeks |
Treatment phase | Step 2: 153 weeks | Step 2: Up to 185 weeks |
Follow-up phasec | Step 3: 48 weekse | Step 3: 48 weekse |
Outcomes | ||
Primary end point | Efficacy: Number of documented incident HIV infections in steps 1 and 2 Safety: Number of participants experiencing ≥ grade 2 clinical and laboratory AEs | Efficacy: Number of documented incident HIV infections in steps 1 and 2 Safety: Number of participants experiencing ≥ grade 2 clinical and laboratory AEs |
Secondary and tertiary end points | Secondary:
Tertiary:
| Secondary:
Tertiary:
|
Publication status | ||
Publications | ||
Ab = antibody; AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; CABG = coronary artery bypass grafting; CMC = clinical management committee; CPK = creatine phosphokinase; CT = chlamydia; DBS = dried blood spot; DXA = dual-energy X-ray absorptiometry; gbMSM = gay, bisexual, and other men who have sex with men; GC = Neisseria gonorrhea; HBsAg = hepatitis B virus surface antigen; HCG = human chorionic gonadotropin; HCV = hepatitis C virus; HSV = herpes simplex virus; IM = intramuscular; IoR = investigator of record; IUD = intrauterine device; IUS = intrauterine system; LA = long-acting; PTCA = percutaneous transluminal coronary angioplasty; q.d. = every day; QTc = QT corrected for heart rate; SSP = study-specific procedure; STI = sexually transmitted infection; Tbili = total bilirubin; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TFV = tenofovir; TFV-DP = intraerythrocytic tenofovir diphosphate; TGW = transgender women; ULN = upper limit of normal; VOICE = Vaginal and Oral Interventions to Control the Epidemic; vs. = versus.
aProtocol version 1.0 (March 2, 2017) in the HPTN 084 trial permitted enrolment of women who scored > 2 using a modified VOICE risk score. Protocol version 1.0 was updated on November 6, 2019, to permit enrolment of women who scored ≥ 5 using a modified VOICE risk score to target women at higher risk of HIV acquisition.
bIn the HPTN 084 study, HIV-uninfected was defined based on an HIV test result obtained at screening and just before randomization at the enrolment visit. All HIV test results from the screening visit were required to be obtained and be negative or nonreactive. This included testing for acute HIV infection, which was performed within 14 days of enrolment. In addition, at least 1 HIV test result using blood drawn at the enrolment visit was required before randomization into the study and had to be negative or nonreactive. Individuals who had ≥ 1 reactive or positive HIV test result(s) were not enrolled, even if subsequent confirmatory testing indicated that they were not HIV-infected (as described in the SSP manual). Those with any enrolment-positive HIV test result proceeded through the HIV algorithm per the SSP but were not able to receive study product regardless of subsequent test results.
cTo allow for any delays in return of week 4 results.
dFive weeks of oral cabotegravir was supplied to monitor tolerability and safety over 4 weeks and to allow for any delays in testing results.
eThe blinded phases of the HPTN 083 and HPTN 084 studies were ended prematurely following a recommendation from the data safety monitoring board based on superior efficacy data. Following this, the protocol of each trial was amended (HPTN 083 Protocol V5 [April 2022]; HPTN 084 Protocol V4.0 [November 2022]) to offer open-label cabotegravir LA or TDF-FTC in step 3.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Two pivotal trials (HPTN 083 and HPTN 084) met the inclusion criteria for the sponsor’s systematic review, providing evidence on the safety and efficacy of cabotegravir LA compared to daily oral TDF-FTC for PrEP in key at-risk populations.
The HPTN 083 study is an ongoing phase IIb/III, multicentre, double-blind, randomized, noninferiority trial designed to evaluate the efficacy and safety of injectable cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years and older) cisgender MSM and TGW who have sex with men and who were at increased risk of sexual acquisition of HIV infections. In total, 4,570 participants enrolled at 43 study centres (no study sites were in Canada) were randomized in a 1:1 ratio to receive either daily oral cabotegravir (30 mg tablets) and oral placebo TDF-FTC for up to 5 weeks (step 1), followed by cabotegravir LA injection (600 mg, intramuscular at weeks 5, 9, and every 8 weeks thereafter) plus daily oral placebo as step 2 (n = 2,283); or daily oral TDF-FTC (300 mg-200 mg tablets) and oral placebo cabotegravir for up to 5 weeks (step 1), followed by daily oral TDF-FTC plus placebo intramuscular injection at weeks 5, 9, and every 8 weeks thereafter as step 2 (n = 2,287). Of the total randomized, 4,566 participants were treated (2,281 participants in the cabotegravir LA group and 2,285 in the TDF-FTC group). Randomization was stratified by study site, and a permuted block design was used to ensure balanced treatment assignments within each study site. Participant enrolment occurred over approximately 3.25 years with the first patient enrolled on December 19, 2016, and the last patient enrolled on March 16, 2020. Data presented in this submission are from the first preplanned interim analysis, with a data cut-off date of May 14, 2020.11
The HPTN 084 study is an ongoing phase III, multicentre, double-blind, randomized, superiority trial, designed to evaluate the efficacy and safety of injectable cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years to 45 years) cisgender women who were at increased risk of sexual acquisition of HIV infection. In total, 3,224 participants from 20 study centres were randomized in a 1:1 ratio to receive either daily oral active cabotegravir (30 mg tablets) and oral placebo TDF-FTC for up to 5 weeks (step 1), followed by cabotegravir LA injection (600 mg intramuscular injection at weeks 5, 9, and every 8 weeks thereafter) plus daily oral placebo in step 2 (n = 1,614); or daily oral active TDF-FTC (300 mg-200 mg tablets) and oral placebo cabotegravir for up to 5 weeks (step 1), followed by daily oral TDF-FTC plus placebo intramuscular injection at weeks 5, 9, and every 8 weeks thereafter in step 2 (n = 1,610). Randomization was conducted using a permuted block design to ensure balanced treatment assignments within study site. Participants were enrolled over a period of approximately 3 years, with the first patient enrolled on November 27, 2017, and the last patient enrolled on November 4, 2020. Findings presented in this review are from the second interim analysis data cut-off date of November 5, 2020.11
Both trial designs included an oral lead-in phase (step 1), an injection phase (step 2), and an open-label extension phase (step 3). Key primary and secondary outcomes investigated were similar in both trials and included the number of documented incident HIV infections in steps 1 and 2 and the number of participants experiencing grade 2 or higher clinical and laboratory AEs. Other important outcomes assessed included the number of documented incident HIV infections in step 2, resistance mutations to study products, adherence to study product during step 2, and the incidence of STIs. Patient-reported outcomes assessed included acceptability scale assessments using the Study Medication Satisfaction Questionnaire and survey of attitudes and willingness to use cabotegravir LA and TDF-FTC. The blinded phases in both trials were ended prematurely based on efficacy results obtained at planned interim analyses, and study protocols in both studies were amended to offer open-label cabotegravir LA or TDF-FTC at step 2 (HPTN 083 Protocol V5. April 2022 and HPTN 084 Protocol V4.0. November 2022).11 All participants included in both study analyses for this submission were blinded to the study treatments. Figure 1 presents the original study designs of the 2 trials.
Figure 1: Schematic Presentation of the Original Study Design of the HPTN 083 and HPTN 084 Trials

CAB = cabotegravir; IM = intramuscular; LA = long-acting; MSM = men who have sex with men; Q8W = every 8 weeks; QD = every day; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine; TGW = transgender women.
Note: The blinded phases of the HPTN 083 and HPTN 084 studies were ended prematurely following a recommendation from the data safety monitoring board based on superior efficacy data. Following this, the protocol of each trial was amended (HPTN 083 Protocol V5 [April 2022]; HPTN 084 Protocol V4.0 [November 2022]) to offer open-label cabotegravir LA or TDF/FTC in step 3.
Special recruitment emphasis on participants aged < 30 years, TGW, and Black MSM (US sites only).
† Oral tablets received QD for 5 weeks to verify safety of the study project before injections.
‡ Active and placebo tablets and injections look alike to ensure blinding of staff and participants.
§ First 2 injections are 4 weeks apart, then every 8 weeks thereafter.
¶ Participants were offered open-label daily oral TDF/FTC or cabotegravir LA to participants after the early stopping of the trial, depending on whether the participant wished to initiate or continue on cabotegravir LA, which group the participant was originally randomized to, and whether they had completed the oral lead-in of cabotegravir.
Source: Sponsor’s drug submission package.40
Populations
Inclusion and Exclusion Criteria
The HPTN 083 trial enrolled adult (aged 18 years and older) cisgender MSM and TGW who have sex with men, who were at high risk for acquiring an HIV infection, had a negative HIV serologic test at enrolment, had undetectable blood HIV RNA viral load within 14 days before trial entry, and had a creatinine clearance of 60 mL or more per minute. Patients were excluded if they had used illicit IV drugs within 90 days before enrolment, had previously participated in an active treatment group of an HIV vaccine trial, had coagulopathy, buttock implants, or fillers, had a seizure disorder, or had a corrected QT interval of greater than 500 msec. Participants who had a positive hepatitis B virus surface antigen test or hepatitis C virus antibody test were also excluded.11
The HPTN 084 trial enrolled adult (aged 18 years to 45 years) cisgender women who had reported at least 2 episodes of vaginal intercourse in the previous 30 days, were at risk of HIV acquisition (based on a modified Vaginal and Oral Interventions to Control the Epidemic [VOICE] risk score to target women at higher risk of HIV acquisition), and had agreed to use a LA reversible contraceptive method with a failure rate of less than 1%. Patients were excluded if they were pregnant or breastfeeding; had substantial renal, hepatic, or cardiovascular disease; had a history of seizures, coagulopathy, or allergy to any of the study products; or if they were previously enrolled in an HIV vaccine or monoclonal antibody trial.11
Interventions
HPTN 083 and HPTN 084 Studies
Arm A (cabotegravir):
In step 1 (oral lead-in phase), participants received daily oral cabotegravir (30 mg tablets) and oral placebo TDF-FTC for 4 weeks (up to 5 weeks was allowed for any delays in testing results).
In step 2 (injection phase), participants received cabotegravir LA (600 mg as a single intramuscular injection at 2 time points, 4 weeks apart and every 8 weeks thereafter) and daily oral placebo TDF-FTC to week 153.
Arm B (TDF-FTC):
In step 1 (oral lead-in phase), participants received daily oral TDF-FTC (300 mg-200 mg tablets) and oral placebo cabotegravir for 4 weeks (up to 5 weeks was allowed for any delays in testing results).
In step 2 (injection phase), participants received daily oral TDF-FTC (300 mg-200 mg tablets) and intramuscular placebo (at 2 time points 4 weeks apart and every 8 weeks thereafter) to week 153.
Participants who became HIV-infected during step 1 were to permanently discontinue the study drug, terminate from the study, and be referred for HIV-related care. Participants who became HIV-infected during step 2 were to permanently discontinue study drug, be placed on immediate suppressive antiretroviral therapy, and be followed at quarterly intervals for 52 weeks after their last injection before diagnosis of HIV to test for safety parameters, as well as CD4+ cell count and HIV viral load. After 52 weeks, the participants were to be terminated from the study and transitioned to continued HIV-related care.11
Participants who did not become HIV-infected during step 1 or step 2, could enter the protocol-planned transition to step 3 in which open-label daily oral TDF-FTC was offered at week 153 (last day of step 2 and first day of step 3) and continued for 48 weeks. This was considered as entering “early step 3” for scenarios where participants discontinued study drug during step 2 (i.e., before week 153) and began open-label TDF-FTC. Following a protocol amendment, participants were subsequently offered open-label cabotegravir LA or TDF-FTC in step 3.11
Prior and Concomitant Medications
HPTN 083 and HPTN 084 Studies
All concomitant medications and preparations (prescription and nonprescription) including alternative or complementary medications or preparations (e.g., herbs, vitamins, and so forth) taken within 30 days before enrolment and any time after during study participation were collected on study case report forms. Alcohol and recreational or street drug use reported by participants during the study were included in the study database.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted by CDA-AMC and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform the CDA-AMC’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select efficacy end points and notable harms outcomes considered important for informing the CDA-AMC’s expert committee deliberations were assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time interval | HPTN 083 | HPTN 084 |
|---|---|---|---|
Number of documented incident HIV infections in steps 1 and 2 | Step 1 (5 weeks): time between randomization and the first injection date or study product discontinuation and/or study termination date, whichever occurred first. Step 2: time from the first injection through the termination of the study | Primary | Primary |
≥ Grade 2 clinical and laboratory adverse events | Throughout the study | Primary | Primary |
Number of documented incident HIV infections in step 2 | Step 2: time from the first injection through the termination of the study | Secondary | NA |
Resistance mutations to study products among seroconverters | Step 1 (5 weeks): time between randomization and the first injection date or study product discontinuation and/or study termination date, whichever occurred first. Step 2: time from the first injection through the termination of the study | Secondary | Tertiary |
Adherence to study product during step 2 for cabotegravir LA or placebo cabotegravir LA scheduled injections received and for TDF-FTC or placebo and TDF-FTC pill dispensing | Step 2: Time from the first injection through the termination of the study | Tertiary | NA |
LA = long-acting; NA = not applicable; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
HIV End Points
The number of documented incident HIV infections was the primary end point of both trials. A blinded special end point adjudication committee determined when an HIV infection occurred (i.e., HIV infections occurring after enrolment and during primary analysis follow-up). Prior to each data safety monitoring board interim monitoring efficacy review, and before unblinding of the final analysis, the special end point adjudication committee engaged in a blinded review of all cases with positive or reactive HIV test results to adjudicate study end points.11
Patients with a reactive HIV test were first identified at the trial site via a HPTN 083 study seroconverter alias, which assists managing the care of potential seroconverters, including additional testing and study drug hold. HIV status confirmation for patients with a reactive test was performed by subsequent testing at the laboratory centre incorporating results from before, at, and after visits. An end point review was performed to finalize decisions of all cases of HIV acquisition and data related to the timing of HIV infection.11 The following parameters were considered during the review of HIV end points in both trials:
total number of cases designated as infected at enrolment
number of cases reported at a visit before detection of infection by the site, including the review of infections first detected in step 3 that were subsequently determined to have occurred in step 2
all cases where site testing did not follow the HIV testing algorithm for determining infection for any reason
any other unusual case determined by the laboratory centre, statistical centre, or seroconverter alias.
Adherence (Measured Through PKs)
Plasma and DBS samples needed to determine drug concentrations were collected throughout the study from all participants, although PK testing was limited to a subset of the samples. Plasma and DBS samples were processed and frozen locally for subsequent shipment to the HPTN study laboratory centre following procedures outlined in the study-specific procedure manual. Pharmacology testing was performed at the HPTN study laboratory centre. The primary pharmacologic assessments were performed using assays that have been validated and approved by the Clinical Pharmacology Quality Assurance program. Plasma was stored for possible future testing, for example, to test for the presence of other antiretroviral drugs or other substances.11
Resistance Mutations
The HPTN study LC performed additional testing for all participants with any site reactive or positive HIV test result, including testing to determine HIV infection status and the timing of HIV infection. This testing included the following tests for participants who acquired HIV infection: HIV viral load, HIV drug resistance testing, HIV subtyping. Other tests may have been performed in some cases to characterize HIV viruses and the host response to HIV infection. Results were not returned to the sites or study participants, except for HIV testing (if results obtained at the HPTN study LC did not agree with site results). Resistance testing and HIV subtyping were performed retrospectively at Monogram Biosciences. Because real-time resistance testing may have been needed for clinical management in the event of HIV infection, each site had an standard operating procedure as to how they would accomplish real-time local or regional resistance testing to assist with clinical decision-making; separate specimens were collected for that testing.11
Harms End Points
Safety assessments conducted included the monitoring of AEs, clinical laboratory tests (including HIV testing, hepatitis testing, Neisseria gonorrhea and chlamydia testing, blood chemistry, hematology, urinalysis, and fasting glucose and lipids), vital signs, electrocardiograms, physical examinations, ISRs, and bone, cardiovascular, and renal biomarkers. Grade 2 or higher clinical and laboratory AEs were assessed as primary outcomes in both trials.11 Safety assessments were summarized using the Medical Dictionary for Regulatory Activities (version 23.1) System Organ Class and Preferred Terms.
Statistical Analysis
The statistical analyses for trial end points are summarized in Table 8.
Sample Size and Power Calculation
HPTN 083 Study
The sample size calculation aimed to achieve 90% power to reject the noninferiority margin set at a type I error of 0.025. The alternative hypothesis was that cabotegravir LA was 25% more effective than TDF-FTC in the treatment population. The number of enrolled participants needed to observe the target number of HIV infections was dependent on the following: background incidence of HIV transmission in the study populations, the efficacy of cabotegravir LA and TDF-FTC in these populations, the dropout rate, and the study duration. Assuming an annual HIV incidence of 2.0% for the TDF-FTC group, cabotegravir LA was said to be 25% more protective than TDF-FTC, resulting in approximately 172 observed HIV infections. At an annual dropout rate of 7.5%, approximately 5,000 individuals were needed for the study to achieve events at a planned follow-up duration of 2.5 years.11
A noninferiority HR margin of 1.23 — referred to as M2 — was calculated based on an inverse-variance weighting of a meta-analysis of 3 RCTs of TDF-FTC compared to placebo in MSM (the iPrex,12 iPERGAY,13 and PROUD14 studies). The calculation built upon a first HR margin of 1.39 — M1 — derived based on the lower limit of the 95% CI around the placebo versus active control HR estimate from a meta-analysis of prior studies conducted in this setting. M2 was defined as the reduced bound designed to preserve a clinically acceptable amount of benefit provided by the active control (TDF-FTC). M2 was set at a conservative value of 50% of M1 (on the log scale in the case of HRs). A decision to establish noninferiority would be made if the estimated HR of cabotegravir LA versus TDF-FTC was approximately 0.90 or less (indicating a 10% or better advantage of cabotegravir LA over TDF-FTC) after a predefined number of HIV events was observed. A decision to establish superiority under the same assumptions would be made if the estimated HR was 0.74 or less (indicating a 26% or better advantage of cabotegravir LA over TDF-FTC) at a 47% power.
A Lan DeMets modification of the O’Brien-Fleming stopping bounds was used to control alpha spending for a total of 4 planned interim analyses. Trial termination would occur at the first occurrence of any of the following: the accrual of 172 incident HIV end points, completion of 11,800 PY of follow-up, or crossing a stopping boundary.11
HPTN 084 Study
A sample size of 3,200 participants at 90% power was planned for the HPTN 084 trial, using the following assumptions: a background HIV incidence of 3.5% per year in the absence of any PrEP; an 85% effectiveness of both cabotegravir LA and TDF-FTC when used at 100% adherence; a 2.5% 1-sided type I error rate and 90% power at the indicated alternative; an average follow-up duration of 2.6 years (range, 1.6 years to 3.6 years); and a maximum 5% lost to follow-up per year. The sponsor reported 5 scenarios associated with total sample sizes. The 2 scenarios based on the most conservative assumptions were used. The superiority design assumed an 80% participant adherence rate to cabotegravir LA for both scenarios and a 45% and 48% adherence to the oral TDF-FTC treatment in each of the 2 scenarios, respectively. Adherence rates proposed for TDF-FTC were based on prior trials which showed lower adherence rates to TDF-FTC (the iPrEX,12 TDF2,41 Partners-PrEP,42 VOICE,43 FEM-PrEP, Bangkok,44 and IPERGAY13 studies). A sample size calculation of at least 3,128 participants was considered robust in assessing the uncertainties in adherence rates to cabotegravir LA and TDF-FTC in the trial.
A total of 114 events or a crossed stopping boundary was required to detect a HR of 0.54 for the incidence of HIV infections in the cabotegravir LA group compared to TDF-FTC group, at a 1-sided significance level of 0.025. An O’Brien-Fleming boundary was considered for early stopping of the trial for efficacy and for the 5 planned interim analyses (4 interim and 1 final). Trial termination would be triggered at the first occurrence of either of the following: the accrual of 114 incident HIV end points or a stopping boundary was crossed.11
Statistical Testing
Primary End Points
HPTN 083 Study
The primary end point of the HPTN 083 study was time to HIV-1 infection occurring after study enrolment. The time to HIV infection was calculated as the time from randomization to the midpoint between the first visit where an HIV infection was present, and the most recent prior visit where HIV infection was not detectable. Follow up was censored at the completion of the blinded injection phase of study (i.e., week 153 or the study-wide transition to step 3 or end of the blinded phase of the study, whichever occurred first). The number of participants, number of infections, and cumulative PY were presented by arm and overall. Incidence rates in each group were calculated as the number of HIV infections divided by the total observed person-time. CIs were computed under an assumed Poisson distribution of HIV infections. A Kaplan-Meier estimator was used to estimate the cumulative probability of acquiring an HIV infection over time. The cumulative incidence of HIV detection at 12, 24, and 36 months was reported by study arm and overall, with a Wald-type 95% CI computed using the estimated pointwise asymptotic standard errors. Participants who were randomized and received the study product but later determined to have been infected at enrolment were not included in this analysis. A bias-adjusted HR, 95% CI, and P value were determined for the primary end point to account for the group sequential trial design and for any potential bias due to early termination of the study. The adjusted HR (the median unbiased estimate), and the CI and P value were calculated based on the maximum likelihood estimate ordering of the sample space. The interim estimated HR was compared to prespecified group sequential stopping boundaries instead of to the noninferiority margin.11
Eligible participants not infected at enrolment were included and were classified according to randomized study group based on the intent-to-treat (ITT) approach. The primary modified intent-to-treat (mITT) population included participants who stopped injections early and could have subsequently initiated open-label TDF-FTC. Person-time was defined as the time from enrolment to the first of either the midpoint of the interval where HIV infection was detected or the last HIV test included in the primary analysis period. Study time was censored at the last HIV test in the primary analysis follow-up for participants who did not acquire HIV infection.11 A Cox regression model stratified by region, with the study arm as the only covariate, was used to estimate the HR of HIV infection in treatment groups and the 95% Wald-based CI was derived.
Handling of Missing Data
Analyses for the primary end points were conducted assuming noninformative censoring if participants dropped out of the study or missed visits. Additional sensitivity analyses were planned in the case of a loss to follow-up of more than 20% between the 2 groups (≥ 5% of the data points). An inverse probability censoring weighting analysis45 was also planned to adjust for loss to follow-up and to compare the adjusted treatment effect to the unadjusted treatment effect. In addition, a tipping-point analysis was planned to examine how variations in the patterns of the missing data could meaningfully change the interpretation of the results. Treatment differences were estimated for participants lost to follow-up assuming they had stopped taking PrEP.
Supportive Analysis of the Primary Efficacy
An efficacy analysis restricted to time while OBSP was conducted as a supportive analysis to verify the consistency of findings with the ITT analysis and to understand the potential mechanisms for lack of consistency. Study time was censored at the first time during the blinded injection phase of study follow-up when study injections were not received on the protocol schedule for any reason. The HRs of HIV infection in the treatment groups were estimated using a Cox regression model, stratified by region, with study arm as the only covariate and the 95% CIs were Wald-based.
HPTN 084 Study
The primary end point of the HPTN 084 study, HIV-1 infection occurring after study enrolment during step 1 and 2, was conducted on the mITT population. HIV-1 status was assumed negative for any missing visits before the first positive HIV-1 test (unless defined differently by the end points committee before analysis). Person-time and HIV events were included in the analysis based on scheduled participation in steps 1 and 2, as determined at randomization. Specifically, individuals who refused or discontinued injections, pills, or both, or who received open-label study drug (e.g., due to pregnancy) were included in their original randomization arm for the duration of their originally scheduled participation in steps 1 and 2. Participants who dropped out of the study and refused further testing prior and those who died before follow-up completion were censored at the last valid HIV test date.11
The HIV incidence rate was calculated as the total number of participants with confirmed incident HIV-1 infection during steps 1 and 2 of study follow-up, divided by the PY accumulated in each arm. Corresponding 95% CIs were computed under an assumed Poisson distribution of HIV infections. Cumulative incidence over follow-up for each arm was computed using product limit estimates, plotted with the 95% CIs.11 A Cox model for time-to-HIV stratified by site was used to estimate an intervention HR. Consistent with an ITT analysis, participants were included in the analysis in their original randomization group whenever the end point information was available within the prespecified follow-up time frame. Participants who remained HIV-1 uninfected were censored at their last negative HIV test. In the scenario of early trial termination (before the final analysis), the bias-adjusted mean HR (calculated using symmetric group sequential tests as outlined by the Emerson and Fleming study46) and adjusted 95% CI were reported for the primary analysis.11
A supportive analysis for the primary outcome was conducted using the OBSP population, censoring in the injection (step 2) efficacy population, where study follow-up was censored when a participant did not receive blinded injection on time.
Missing Data Imputations
The analyses for the primary outcomes (the HPTN 084 study) were conducted assuming noninformative censoring if participants dropped out of study or missed visits. Sensitivity analyses to the assumptions of missing data were planned if loss to follow-up of more than 20% was observed between the 2 groups; otherwise, no further analyses were planned if loss to follow-up was low or similar between the 2 groups. An inverse probability censoring weighting analysis45 was planned to adjust for loss to follow-up and to compare the adjusted treatment effect to the unadjusted treatment effect.
Secondary End Points
HPTN 083 Study
HIV Incidence in Step 2 Only (Step 2 Efficacy Population)
This analysis included only participants who received an injection (first active injection for the cabotegravir LA group or first placebo injection for the TDF-FTC arm). The statistical hypothesis tested for step 2 paralleled the primary analysis.11 The analyses included only participants who received an injection, with time 0 starting at the time of the first injection (first active injection for the cabotegravir LA arm or first placebo injection for the TDF-FTC arm). A formal statistical comparison between study arms was performed using the Wald hypothesis test, at a 2-sided alpha of 0.05. The HIV incidence rate was calculated as the total number of participants with confirmed incident HIV-1 infection during step 2 of study follow-up, divided by the PY accumulated in each arm. Corresponding 95% CIs were computed under an assumed Poisson distribution of HIV infections. Cumulative incidence over follow-up for each arm was computed using product limit estimates and plotted with 95% CIs. A Cox model for time-to-HIV stratified by site was used to estimate an intervention HR. A sensitivity analysis using OBSP censoring time followed the same plan as the primary analyses.
Resistance Mutations to Study Products Among Seroconverters (Seroconverter Population)
The number of participants with HIV drug resistance at their first HIV-positive visit was identified by the presence of mutations known to be associated with cabotegravir (oral or LA and including T66I, E92Q/M, F121Y, Y143R/H/C, S147G, Q148H/K/R, N155H, T97I/A, G140S, and a 5AA duplication at 232), TDF-FTC (including K65R, K70E, and M184V/I), and nonstudy drugs (all others). Resistance was reported separately for those infected before enrolment and during primary analysis follow-up, compared to step 3. The proportion of seroconverter participants who had drug resistance mutations associated with cabotegravir (oral or LA) or TDF-FTC at the first HIV-positive visit was compared using chi-square tests (or exact binomial tests when sample size is small).11
HPTN 084 Study
Rates of HIV Drug Resistance Among Participants Who Acquired HIV Infection
The primary seroconverter population was used for the analysis and data from steps 1, 2, and 3 were included. The number of cases of drug resistance was summarized by arm and step.
Subgroup Analyses
Prespecified subgroup analyses were performed in both the HPTN 083 and HPTN 084 studies. Neither study was powered for any of the individual subgroup evaluations and no adjustments were made for multiple testing.
HPTN 083 Study
Documented HIV-1 infections in step 2 were assessed in the primary ITT population. Estimated HRs were reported for subgroups in both study groups. The presence of effect modification was tested using a subgroup-by-arm interaction.11
Important participant subgroups were prespecified in the HPTN 083 trial:
region: US versus Latin America versus Asia versus Africa
age: younger than 30 years versus aged 30 years or older
race, for US only: Black versus non-Black
ethnicity: Hispanic versus non-Hispanic
baseline risk:
greater than the median number of sexual partners
greater than the median number of occurrences of condomless receptive anal sex.
gender identity: cisgender MSM or TGW who have sex with men
baseline risk for the number of male and or transgender female partners with whom the participant reported having anal or vaginal sex within the past month; subgroups were those with the median number of sexual partners or fewer and those with more than the median number of sexual partners
baseline risk for the number of times the participant reported having receptive anal sex in the past month without using a condom; subgroups were those with the median number or less of occurrences of condomless anal sex and those with more than the median number of occurrences of condomless anal sex.
HPTN 084 Study
Important participant subgroups were prespecified:
age: younger than 25 years versus aged 25 years or older
body mass index less than 30 kg/m2 versus 30 kg/m2 or more.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
HPTN 083 study | ||||
Primary efficacy analysis: number of documented incident HIV infections in steps 1 and 2 | Cox proportional hazards regression with treatment arm as the only covariate | Stratified by region and adjusted for early stopping | HIV infection status was assumed to be negative at all missing visits, if any, before the first reactive or positive HIV test | Supportive analysis: OBSP efficacy analysis |
Number of documented incident HIV infections in step 2 | Cox proportional hazards regression with treatment arm as the only covariate | Stratified by region and adjusted for early stopping | HIV infection status was assumed to be negative at all missing visits, if any, before the first reactive or positive HIV test | OBSP censoring in the injection (step 2) efficacy population |
Resistance mutations to study products among seroconverters | Chi-square distribution tests (or exact binomial tests) | NA | NA | NA |
Safety | Summarized using MedDRA (version 23.1) System Organ Class and Preferred Terms | NA | For a participant who never receives an injection, AEs were censored when the onset date falls after the earliest of 120 days after randomization or at the termination or permanent oral product discontinuation date + 1 | NA |
HPTN 084 study | ||||
Primary efficacy analysis: number of documented incident HIV infections in steps 1 and 2 | Cox proportional hazards model with treatment arm as the only covariate | Stratified by site using data from steps 1 and 2, cutting the data at the time the blinded portion of the trial was stopped | HIV-1 status was assumed to be negative for any missing visits before the first positive HIV-1 test | Supportive analysis: OBSP efficacy analysis |
Number of documented incident HIV infections in step 2 | Cox proportional hazards model with treatment arm as the only covariate | Stratified by site using data from steps 1 and 2, cutting the data at the time the blinded portion of the trial was stopped | HIV-1 status was assumed to be negative for any missing visits before the first positive HIV-1 test | Not performed |
Resistance mutations to study products among seroconverters | Number of cases of drug resistance was summarized by arm and step | NA | NA | NA |
Safety | Summarized using MedDRA (version 23.1) System Organ Class and Preferred Terms | NA | For a participant who never receives an injection, AEs were censored when the onset date falls after the earliest of 120 days after randomization, date of unblinding, or at the termination or permanent oral product discontinuation date | NA |
AE = adverse event; MedDRA = Medical Dictionary for Regulatory Activities; NA = not applicable; OBSP = on blinded study product.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Analysis Populations
The analysis populations of the HPTN 083 and HPTN 084 studies are presented in Table 9.
Table 9: Analysis Populations of the HPTN 083 and HPTN 084 Studies
Study | Population | Definition | Application |
|---|---|---|---|
HPTN 083 | ITT | All participants who were randomized, excluding those who were inappropriately enrolled. | Efficacy analyses |
mITT | The ITT population, excluding those who were found to be HIV-infected at randomization. Analysis period: primary analysis follow-up data included study time through the completion of the blinded injection phase of study follow-up (i.e., week 153 or the study-wide transition to step 3, or end of the blinded phase of the study, whichever occurred first). Person-time and end point events were included in the primary analysis regardless of whether participants remained on their blinded study product, including when participants moved to open-label TDF-FTC (step 3) early. | Efficacy analyses; mITT was the primary assessment for efficacy comparison | |
PP | The mITT population excluding all participants with protocol violations that were judged to be exclusionary from the PP population. | Efficacy analyses | |
Injection (step 2) efficacy population | The mITT population who received at least 1 injection and were uninfected at the time of the first injection. Analysis period: follow-up time included primary analysis study time from the time of the first injection through the completion of the blinded injection phase of study follow-up. | Efficacy analyses | |
Step 3 population | All mITT participants who were uninfected at the start of step 3 follow-up (i.e., the week 153 and study-wide transition to step 3). | Efficacy analyses | |
Safety population (primary analysis) | All ITT participants who received any oral or injectable product. All safety events occurring on study were reported. Step 1 AEs included all AEs occurring until the first injection date, or 120 days postrandomization, whichever occurred first. | Safety analyses | |
Injection (step 2) safety population | All safety population participants who received at least 1 injection. Step 2 safety included all AEs occurring from the first injection date through 48 weeks after the last injection. | Safety analyses | |
HPTN 084 | Randomized population | All participants who were randomized. | Efficacy analyses |
ITT | All participants who were randomized, excluding those who were inappropriately enrolled. | Efficacy analyses | |
mITT | The ITT population, excluding those who were found to be HIV-infected at randomization. | Efficacy analyses; mITT was the primary assessment for efficacy comparison | |
PP | The mITT population excluding all participants with protocol violations that were judged to be exclusionary from the PP population. | Efficacy analyses | |
Injection (step 2) efficacy population | The mITT population who received at least 1 injection and were uninfected at the time of the first injection. Analysis period: follow-up time will include primary analysis study time from the time of the first injection through the completion of the blinded injection phase of study follow-up. | Efficacy analyses | |
Safety | All ITT participants who received at least 1 dose of oral or injectable product. | Safety analyses | |
Injection (step 2) safety population | All safety population participants who received at least 1 injection during step 2. | Safety analyses |
AE = adverse event; ITT = intent to treat; mITT = modified intent to treat; PP = per protocol; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Results
Patient Disposition
The disposition of participants in the HPTN 083 and HPTN 084 trials are summarized in Table 10.
A total of 6,449 participants underwent screening in the HPTN 083 trial, of which 1,879 were not enrolled. The most common reason for screening failure reported was HIV-1 infection or participants did not meet inclusion or exclusion criteria. Following confirmation of eligibility, a total of 4,570 participants were randomized to receive either treatment regimen. In total, 67 (3%) versus 78 (3%) patients in the cabotegravir LA and TDF-FTC groups, respectively, discontinued from the study.11
In the HPTN 084 study, a total of 4,775 participants underwent screening, of which 1,551 were not enrolled. The most common reason for screening failure reported was did not meet inclusion or exclusion criteria. In total, 3,224 participants were randomized (1,614 and 1,610 in the cabotegravir LA and TDF-FTC groups, respectively). Overall, 25 (2%) and 17 (1%) participants in the cabotegravir LA and TDF-FTC groups, respectively, discontinued the HPTN 084 study.11
Table 10: Patient Disposition in the HPTN 083 and HPTN 084 Trials
Patient disposition | HPTN 083 | HPTN 084 | ||
|---|---|---|---|---|
Cabotegravir LA n = 2,283 | TDF-FTC n = 2,287 | Cabotegravir LA n = 1,614 | TDF-FTC n = 1,610 | |
Screened, n | 6,649 | 4,475 | ||
Reason for screening failure, n | 1,879 | 1,551 | ||
HIV-1 infected | 231 | — | ||
Did not meet inclusion or exclusion criteria | 1,648 | 1,093 | ||
Unable to be contacted or no show | — | 250 | ||
Other | — | 155 | ||
Changed their mind | — | 51 | ||
Declined to receive treatment | — | 2 | ||
Randomized, n | 2,283 | 2,287 | 1,614 | 1,610 |
Randomized and treated, n (%) | 2,281 (99.9) | 2,285 (99.9) | 1,614 (100) | 1,610 (100) |
Ongoinga,,n (%) | 2,210 (97) | 2,203 (96) | 1,586 (98) | 1,586 (99) |
Completed, n (%) | 6 (< 1)b | 6 (< 1)b | 3 (< 1)c | 7 (< 1)c |
Discontinued from study, n (%)d | 67 (3) | 78 (3) | 25 (2) | 17 (1) |
Reason for discontinuation, n (%) | ||||
Scheduled exit visit or end of study | 2 (< 1) | 1 (< 1) | 0 | 0 |
Death | 4 (< 1) | 7 (< 1) | 3 (< 1) | 0 |
Participant refused further participation | 54 (2) | 54 (2) | 20 (1) | 15 (< 1) |
Participant relocated; no follow-up planned | 2 (< 1) | 4 (< 1) | 2 (< 1) | 0 |
Investigator decision | 1 (< 1) | 3 (< 1) | 0 | 1 (< 1) |
Inappropriate enrolment | 1 (< 1) | 3 (< 1) | 0 | 0 |
Other | 3 (< 1) | 6 (< 1) | 0 | 0 |
HIV infection, step 1d | 0 | 0 | 0 | 1 (< 1) |
Randomized population, n | 2,283 | 2,287 | 1,614 | 1,610 |
ITT population, n | 2,282 | 2,284 | 1,614 | 1,610 |
mITT population, n | 2,280 | 2,281 | 1,614 | 1,610 |
Injection step 2 efficacy population, n (%) | 2,109 (92) | 2,069 (90) | 1,495 (93) | 1,494 (93) |
PP population, n | 2,268 | 2,276 | 1,598 | 1,600 |
Safety population, n | 2,281 | 2,285 | 1,614 | 1,610 |
Injection step 2 safety population, n | 2,117 | 2,081 | 1,519 | 1,516 |
Step 3 population, n (%) | 281 (12) | 225 (10) | — | — |
ITT = intent to treat; LA = long-acting; mITT = modified intent to treat; PP = per protocol; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
aOngoing in the study includes participants who are not actively attending scheduled visits but have not formally discontinued study participation.
bCompleted the study indicates a participant has seroconverted or has entered step 3 after reaching week 145 visit and was subsequently followed for 48 weeks in step 3 without prior investigational product discontinuation or study termination or has entered step 3 after discontinuation of IP and was subsequently followed for 48 weeks and was followed for a total of 3 years from enrolment.
cAny seroconverters (per site results or end point adjudication committee) in step 1 and seroconverters in step 2 who have completed 48 weeks of follow-up.
dExcludes participants who completed the study.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Baseline Characteristics
At baseline, in the HPTN 083 trial, the mean age of participants was 28 years. In the cabotegravir LA group, 27% of participants were white, 27% were American Indian or Alaska Native, and 25% were Black or African American. In the TDF-FTC group, 28% of participants were white, 26% were American Indian or Alaska Native, and 25% were Black or African American. Twelve percent of the participants in the cabotegravir LA group vs 13% in the TDF-FTC group were TGW. Only participants assigned male at birth were enrolled.
Most participants in the HPTN 084 trial were Black (> 99%), aged younger than 35 years, and had a screening modified VOICE risk score of 5 or more (80% versus 79% in the cabotegravir LA and TDF-FTC groups, respectively). Only participants who were female (assigned at birth) were enrolled. Baseline characteristics were generally balanced between treatment groups in both trials (Table 11).
Table 11: Summary of Baseline Characteristics of the HPTN 083 and HPTN 084 Trials
Characteristic | HPTN 083 | HPTN 084 | ||
|---|---|---|---|---|
Cabotegravir LA n = 2,283 | TDF-FTC n = 2,287 | Cabotegravir LA n = 1,614 | TDF-FTC n = 1,610 | |
Age | ||||
Mean (SD) | 28.0 (8.17) | 28.2 (8.14) | 26.0 (5.7) | 26.0 (5.8) |
Median | 26.0 | 26.0 | 25.0 | 25.0 |
Cohort, n (%) | ||||
MSM | 2014 (88) | 1982 (87) | — | — |
TGWa | 266 (12) | 304 (13) | — | — |
Prefer not to answer | 3 (< 1) | 1 (< 1) | — | — |
Race, n (%) | ||||
American Indian or Alaska Native b | 616 (27) | 600 (26) | 0 | 0 |
Asian | 417 (18) | 406 (18) | 2 (< 1) | 3 (< 1) |
Black or African American | 565 (25) | 569 (25) | 1,612 (> 99) | 1,606 (> 99) |
Mixed race | 49 (2) | 54 (2) | 0 | 0 |
Native Hawaiian or Other Pacific Islander | 5 (< 1) | 2 (< 1) | 0 | 0 |
Unknown | 13 (< 1) | 7 (< 1) | 0 | 0 |
White | 618 (27) | 649 (28) | 0 | 1 (< 1) |
Ethnicity, n (%) | ||||
Hispanic or Latino | 1,043 (46) | 1,067 (47) | 0 | 0 |
Not Hispanic or Latino | 1,240 (54) | 1,219 (53) | 1,614 (100) | 1,610 (100) |
Not Reported | 0 | 1 (< 1) | 0 | 0 |
Sex assigned at birth, n (%) | ||||
Male | — | — | 0 | 0 |
Female | — | — | 1,614 (100) | 1,610 (100) |
Self-identified gender, n (%) | ||||
Man | — | — | 0 | 3 (< 1) |
Woman | — | — | 1,612 (> 99) | 1,607 (> 99) |
Transgender male (female to male) | — | — | 2 (< 1) | 0 |
SexPro score, n (%)c | ||||
≤ 16 | 1,555 (68) | 1,571 (69) | — | — |
VOICE risk score at screening, n (%) | ||||
< 5 | — | — | 327 (20) | 345 (21) |
≥ 5 | — | — | 1,287 (80) | 1,265 (79) |
BMI, kg/m2 | ||||
Mean (SD) | 25.5 (5.6) | 25.4 (5.4) | 27 (6.2) | 27 (5.9) |
Median (min, max) | 24.40 (14.7, 91.0) | 24.50 (14.3, 67.4) | 25.7 (16.4, 54.3) | 25.6 (15.0, 51.3) |
< 30, n (%) | 1,904 (83) | 1,939 (85) | 1,149 (71) | 1,180 (73) |
≥ 30, n (%) | 373 (16) | 344 (15) | 465 (29) | 430 (27) |
BMI = body mass index; LA = long-acting; max = maximum; min = minimum; MSM = men who have sex with men; SD = standard deviation; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TGW = transgender women; VOICE = Vaginal and Oral Interventions to Control the Epidemic.
aThe TGW cohort included participants who self-identified at baseline as female, transgender female, genderqueer, gender variant, gender non-conforming, or gender fluid.
bAmerican Indian or Alaska Native was defined as a person having origins in any of the original peoples of North and South America (including Central America).
cSexPro Score was only collected in North and South America. SexPro is a web-based tool for estimating personalized HIV risk score. Scores can range from 1 (highest risk) to 20 (lowest risk), with a score of 16 or less indicating high risk of HIV infection.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Exposure to Study Treatments
A summary of patient exposure in the pivotal trials is presented in Table 12.
By the May 14, 2020, data cut-off date, 2,281 participants in the HPTN 083 study received at least 1 dose of oral cabotegravir or cabotegravir LA injection (2,117 participants for a total of 20,286 cabotegravir LA injections). The median exposure was 457 days (range, 1 days to 1,093 days) for the cabotegravir LA group and 457 days (range, 1 days to 1,131 days) for the TDF-FTC group. The median time (maximum, minimum) of exposure of the oral lead-in was similar in both treatment groups (cabotegravir LA group = 29 days; range, 1 days to 115 days; TDF-FTC group = 29 days; range, 1 days to 485 days).
By the November 5, 2020, data cut-off date, 1,614 participants in the HPTN 084 study received at least 1 dose of oral cabotegravir or cabotegravir LA injection (1,519 participants, for a total of 13,068 cabotegravir LA injections). The median exposure was 452.5 days for both groups (cabotegravir LA [oral or LA] range, 1 day to 1,072 days; TDF-FTC range, 1 days to 1,018 days). The median time of exposure of the oral lead-in was similar in both treatment groups (cabotegravir LA group = 29 days; range, 1 days to 334 days and TDF-FTC group = 29 days; range, 1 days to 224 days).
Table 12: Patient Exposure to Study Treatments in the HPTN 083 and HPTN 084 Trials
Exposure | HPTN 083 | HPTN 084 | ||
|---|---|---|---|---|
Cabotegravir LA n = 2,283 | TDF-FTC n = 2,287 | Cabotegravir LA n = 1,614 | TDF-FTC n = 1,610 | |
Oral phase, exposure (days) | ||||
Median (minimum, maximum) | 29.0 (1.0, 115.0) | 29.0 (1.0, 485.0) | 29.0 (1, 334)a | 29.0 (1, 224)a |
Injection phase, exposure (number of injection visits) | ||||
Median (minimum, maximum) | 9.0 (1.0, 20.0) | 9.0 (1.0, 15.0) | 9.0 (1, 20) | 9.0 (1, 19) |
Overall, exposure (days) | ||||
Median (minimum, maximum) | 457.0 (1.0, 1093.0) | 457.0 (1.0, 1131.0) | 452.5 (1, 1,072)b | 452.5 (1, 1,018)b |
LA = long-acting; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
aParticipants were not provided enough oral product to cover daily oral dosing for more than 60 days.
bOverall exposure includes confirmed pregnancy days.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Adherence
Majority of participants in the cabotegravir LA and TDF-FTC groups across the HPTN 083 and HPTN 084 studies showed greater than 90% adherence to the oral study product at week 4 based on pill counts. Most injection visits were within the allowable window of 7 days before or after the due date (Table 13). Less than 1% of injection visits were missed in either treatment group before discontinuing randomized treatment.
Table 13: Summary of Adherence to Cabotegravir LA Injection Dosing Schedule in the HPTN 083 and HPTN 084 Studies (Injection Step 2 Safety Population)
Timelines of injections relative to date of projected dosing visits | HPTN 083 | HPTN 084 | ||
|---|---|---|---|---|
Cabotegravir LA n = 2,117 | TDF-FTC n = 2,081 | Cabotegravir LA n = 1,519 | TDF-FTC n = 1,516 | |
Number of injection visits (% of injections of the total injection visits)a | 20,294 | 20,191 | 14,249 | 14,105 |
< –14 days | 107 (< 1) | 107 (< 1) | 502 (4) | 399 (3) |
–14 days to –8 days | 305 (2) | 334 (2) | 620 (4) | 576 (4) |
–7 days to –4 days | 3,100 (15) | 3,049 (15) | 922 (6) | 859 (6) |
–3 days to –2 days | 1,581 (8) | 1,488 (7) | 828 (6) | 795 (6) |
–1 day | 1,463 (7) | 1,477 (7) | 1,022 (7) | 1,034 (7) |
0 day | 4,957 (24) | 4,951 (25) | 5,798 (41) | 5,942 (42) |
1 day | 1,587 (8) | 1,746 (9) | 874 (6) | 905 (6) |
2 days to 3 days | 1,657 (8) | 1,629 (8) | 542 (4) | 552 (4) |
4 days to 7 days | 2,348 (12) | 2,374 (12) | 863 (6) | 877 (6) |
8 days to 14 days | 1,296 (6) | 1,243 (6) | 523 (4) | 516 (4) |
> 14 days | 1,885 (9) | 1,788 (9) | 574 (4) | 543 (4) |
Missed injectionb | 8 (< 1) | 5 (< 1) | 1,091 (8) | 1,037 (7) |
Missed injection due to confirmed pregnancy | NA | NA | 90 (< 1) | 70 (< 1) |
Early out-of-window injections (> 7 days early relative to projected visit date), number of days relative to the projected visit date | ||||
n | 412 | 441 | 1,122 | 975 |
Mean (SD) | 13.0 (5.32) | 13.5 (8.11) | 15.3 (5.95) | 14.8 (5.97) |
Median (minimum, maximum) | 12.0 (8.0, 42.0) | 11.0 (8.0, 54.0) | 14.0 (8.0, 41.0) | 14.0 (8.0, 27.0) |
Q1 | 9.0 | 8.0 | 10.0 | 9.0 |
Q3 | 15.0 | 14.0 | 21.0 | 20.0 |
Late out-of-window injections (> 7 days late relative to projected visit date), number of days relative to the projected visit date | ||||
n | 3,181 | 3,031 | 1,097 | 1,059 |
Mean (SD) | 22.7 (17.74) | 22.9 (18.68) | 16.7 (7.09) | 16.8 (7.03) |
Median (minimum, maximum) | 17.0 (8.0, 406.0) | 17.0 (8.0, 376.0) | 15.0 (8.0, 56.0) | 15.0 (8.0, 54.0) |
Q1 | 12.0 | 12.0 | 11.0 | 11.0 |
Q3 | 29.0 | 29.0 | 22.0 | 22.0 |
LA = long-acting; NA = not applicable; Q = quarter; SD = standard deviation; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Notes: The percentages are based on number of injection visits.
Missed injection was defined as when there is a gap between injection number or any projected injection missed on or before study discontinuation (e.g., lost to follow-up).
Projected visit date is calculated from randomization date for the HPTN 083 study. Projected visit date is calculated from date of week 5.
Days are calculated by using “actual injection visit date – projected visit date.”
Each injection visit is only counted once. If multiple injections are given in the same injection visit or injections split into > 1 visit (within the same visit window), it was counted only once.
Missed injection due to confirmed pregnancy was calculated as the number of expected scheduled injection visits missed.
aThe sum of each participant’s total number of expected injection visits following the first injection visit up to and including the last available planned (i.e., scheduled) injection visit during step 2.
bMissed injection excludes injections missed after discontinuing randomized treatment.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Concomitant Medications and Co-Interventions
Concomitant medication use was generally similar in the 2 groups in the HPTN 083 and HPTN 084 trials. In the HPTN 083 study, a greater proportion of participants in the cabotegravir LA group compared to the TDF-FTC group reported the use of paracetamol (34% versus 25%, respectively), ibuprofen (36% versus 23%, respectively), diclofenac (11% versus 6%, respectively), naproxen (7% versus 4%, respectively), and ketoprofen (4% versus 1%, respectively). In the HPTN 084 study, the most commonly reported concomitant medications were medroxyprogesterone acetate (cabotegravir LA = 59% versus TDF-FTC = 58%) and paracetamol (cabotegravir LA = 47% versus TDF-FTC = 44%). Hepatitis B vaccine was provided to participants who did not have evidence of immunity to hepatitis B virus (negative hepatitis B virus surface antibody) at screening and recorded as a prior or concomitant medication for the majority of study participants (78%).11
Concomitant medication use reported for 5% or more of participants is presented on Table 14 for the HPTN 083 study and Table 15 for the HPTN 084 study.
Table 14: Concomitant Medications in the HPTN 083 Study (Occurring in ≥ 5% of Participants)
Category | HPTN 083 | |
|---|---|---|
Cabotegravir LA n = 2,283 | TDF-FTC n = 2,287 | |
Number of participants with ≥ 1 concomitant medication | 2,083 (91) | 2,033 (89) |
Anti-infectives for systemic use | 1,651 (72) | 1,659 (73) |
Azithromycin | 869 (38) | 864 (38) |
Ceftriaxone | 454 (20) | 454 (20) |
Benzathine benzylpenicillin | 408 (18) | 415 (15) |
Hepatitis B vaccine | 285 (12) | 275 (12) |
Doxycycline | 205 (9) | 260 (11) |
Amoxicillin | 241 (11) | 217 (9) |
Influenza vaccine | 175 (8) | 185 (8) |
Hepatitis A vaccine | 162 (7) | 172 (8) |
Nervous system | 1,387 (61) | 1,196 (52) |
Paracetamol | 780 (34) | 562 (25) |
Metamizole sodium | 134 (6) | 100 (4) |
Alimentary tract and metabolism | 1,032 (45) | 1,025 (45) |
Multivitamins, other combinations | 199 (9) | 244 (11) |
Ascorbic acid | 175 (8) | 157 (7) |
Dietary | 124 (5) | 141 (6) |
Omeprazole | 122 (5) | 133 (6) |
Musculoskeletal system | 187 (52) | 855 (37) |
Ibuprofen | 26 (36) | 536 (23) |
Diclofenac | 62 (11) | 126 (6) |
Naproxen | 49 (7) | 93 (4) |
Respiratory system | 873 (38) | 859 (38) |
Loratadine | 177 (8) | 162 (7) |
Cetirizine | 110 (5) | 110 (5) |
Dermatologicals | 515 (23) | 486 (21) |
Various | 481 (21) | 463 (20) |
Genitourinary system and sex hormones | 316 (14) | 338 (15) |
Cardiovascular system | 292 (13) | 336 (15) |
Systemic hormonal preparations, excluding sex hormones and insulins | 232 (10) | 208 (9) |
Blood and blood-forming organs | 141 (6) | 148 (6) |
Antiparasitic products, insecticides, and repellents | 132 (6) | 148 (6) |
Sensory organs | 98 (4) | 113 (5) |
LA = long-acting; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: Values are presented as n (%).
Source: HPTN 08317 Clinical Study Report.
Table 15: Concomitant Medications in the HPTN 084 Study (Occurring in ≥ 5% of Participants)
Category | HPTN 084 | |
|---|---|---|
Cabotegravir LA n = 1,614 | TDF-FTC n = 1,610 | |
Number of participants with ≥ 1 concomitant medication, n (%) | 1,591 (99) | 1,595 (> 99) |
Genitourinary system and sex hormones | 1,550 (96) | 1,555 (97) |
Medroxyprogesterone | 949 (59) | 935 (58) |
Levonorgestrel | 295 (18) | 315 (20) |
Etonogestrel | 289 (18) | 282 (18) |
Contraceptives | 210 (13) | 201 (12) |
Norethisterone | 198 (12) | 197 (12) |
Clotrimazole | 176 (11) | 197 (12) |
Norethisterone | 171 (11) | 155 (10) |
Intrauterine | 100 (6) | 96 (6) |
Ethinylestradiol and norgestrel | 83 (5) | 90 (6) |
Ethinylestradiol and levonorgestrel | 59 (4) | 74 (5) |
Anti-infectives for systemic use | 1,446 (90) | 1,435 (89) |
Hepatitis B vaccine | 1,021 (63) | 948 (59) |
Azithromycin | 621 (38) | 576 (36) |
Ceftriaxone | 265 (16) | 240 (15) |
Ciprofloxacin | 236 (15) | 216 (13) |
Amoxicillin | 187 (12) | 203 (13) |
Hepatitis B vaccine rHBsAg (yeast) | 180 (11) | 170 (11) |
Doxycycline | 127 (8) | 156 (10) |
Cefixime | 129 (8) | 119 (7) |
Fluconazole | 112 (7) | 114 (7) |
Hepatitis B vaccine rHBsAg | 92 (6) | 92 (6) |
Nervous system | 941 (58) | 900 (56) |
Paracetamol | 762 (47) | 707 (44) |
Codeine phosphate and paracetamol | 198 (12) | 177 (11) |
Lidocaine hydrochloride | 82 (5) | 71 (4) |
Musculoskeletal system | 647 (40) | 581 (36) |
Ibuprofen | 437 (27) | 389 (24) |
Diclofenac | 230 (14) | 197 (12) |
Respiratory system | 616 (38) | 599 (37) |
Chlorphenamine | 288 (18) | 282 (18) |
Chlorphenamine maleate | 119 (7) | 124 (8) |
Chlorphenamine maleate, paracetamol, and phenylephrine hydrochloride | 80 (5) | 68 (4) |
Antiparasitic products, insecticides, and repellents | 592 (37) | 563 (35) |
Metronidazole | 489 (30) | 458 (28) |
Alimentary tract and metabolism | 515 (32) | 545 (34) |
Omeprazole | 105 (7) | 113 (7) |
Magnesium | 82 (5) | 95 (6) |
Vitamin | 78 (5) | 76 (5) |
Dermatologicals | 348 (22) | 327 (20) |
Clotrimazole | 82 (5) | 79 (5) |
Hydrocortisone | 89 (6) | 65 (4) |
Blood and blood-forming organs | 193 (12) | 220 (14) |
Sensory organs | 104 (6) | 102 (6) |
Cardiovascular system | 71 (4) | 69 (4) |
LA = long-acting; rHBsAg = recombinant hepatitis B surface antigen; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: Values are presented as n (%).
Source: HPTN 08418 Clinical Study Report.
Efficacy
Incident HIV Infections in Steps 1 and 2
HPTN 083 Study
At the May 14, 2020, interim cut-off date, cabotegravir LA demonstrated a benefit in preventing the acquisition of HIV-1 compared to TDF-FTC in steps 1 and 2 combined (Table 16). The upper bound of the 95% CI excluded the noninferiority margin of 1.23 and the superiority margin of 1, with a P value of 0.0005.
There were 52 HIV-1 infections reported in total (representing 30% of the total infections targeted to power the study [n = 172 events expected for the final analysis]), 13 infections were reported in the cabotegravir LA group (incidence rate per 100 PY = 0.40; 95% CI, 0.22 to 0.69) versus 39 (incidence rate per 100 PY = 1.22; 95% CI, 0.87 to 1.67) in the TDF-FTC group after 6,404 PY of accumulated follow-up. A bias-adjusted HR, 95% CI, and P value were determined for the primary end point to account for the group sequential trial design and for any potential bias due to early termination of the study. The bias-adjusted HR (corrected for early stopping) was 0.34 (95% CI, 0.18 to 0.62; P = 0.0005), indicating a 66.0% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group. A revised data analysis from additional testing confirmed 12 HIV-1 infections in the cabotegravir LA group and 40 in the TDF-FTC group (new bias-adjusted HR = 0.31; 95% CI, 0.16 to 0.58).11
Figure 7 (Appendix 1) presents the cumulative rates of acquired HIV-1 infections in the mITT population (steps 1 and 2 combined) by randomized group.
Table 16: Time to HIV-1 Infection in Steps 1 and 2 in the HPTN 083 Study — Cox Proportional Hazards Regression Model (mITT Population)
Category | Cabotegravir LA n = 2,280 | TDF-FTC n = 2,281 |
|---|---|---|
Number of HIV infections | 13 | 39 |
Hazard ratio (95% CI), Cox regression | 0.328a (0.18 to 0.616) | |
Noninferiority P value, Cox regression | < 0.0001 | |
Superiority P value, Cox regressionb | 0.0005 | |
Hazard ratio (95% CI), bias-adjustedc | 0.34 (0.18 to 0.62) | |
Superiority P value, bias-adjustedc | 0.0005 | |
Noninferiority P value, bias-adjustedc | < 0.0001 | |
PY | 3,211 | 3,193 |
Incidence rate (95% CId), per 100 PY | 0.40 (0.22 to 0.69) | 1.22 (0.87 to 1.67) |
Between-group treatment difference (95% CId), per 100 PY | –0.82 (–1.26 to –0.38) | |
CI = confidence interval; LA = long-acting; mITT = modified intent to treat; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: A hazard ratio < 1.0 indicates a lower risk on cabotegravir LA compared to TDF-FTC; noninferiority margin of 1.23. The P values are 2-sided. The trial was stopped based on a breach of the first interim stopping bound (z = –4.00; P = 0.000063), which was derived from an O’Brien-Fleming design with 3 planned interim analyses plus 1 final analysis. Data cut-off date: May 14, 2020.
aThe unadjusted hazard ratio is based on a Cox proportional hazards model stratified by region.
bSuperiority was established if the hazard ratio point estimate was at least 0.74 (indicating a 26% or better advantage of cabotegravir LA over TDF-FTC).
cThe bias-adjusted hazard ratio, CI, and P value account for the group sequential trial design and the early stopping time. The adjusted point estimate is the median unbiased estimate, and the CI and P value are based on the maximum likelihood estimate ordering of the sample space.
dThe 95% CI for incidence rate is calculated using the exact Poisson method.
Source: HPTN 083 Clinical Study Report.17
Supportive Analysis: Incident HIV-1 Infections While OBSP
The supportive analysis conducted using the OBSP censoring of the injection step 2 efficacy population was consistent with the primary analysis (estimated HR = 0.164; 95% CI, 0.06 to 0.47; P value = 0.0008) indicating an 83.6% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to the TDF-FTC group. Table 25 and Figure 8 (Appendix 1) present a summary of the supportive analysis in the OBSP and the cumulative rates of acquired HIV-1 infections.11
HPTN 084 Study
At the November 5, 2020, interim cut-off date, cabotegravir LA was shown to be superior to TDF-FTC in preventing the acquisition of HIV-1 in steps 1 and 2 combined (P < 0.0001). There were 40 incident HIV-1 infections identified in total (representing 35% of the total infections targeted for powering the study [n = 114 events expected for the final analysis]), 4 occurred in the cabotegravir LA group (incidence rate per 100 PY = 0.20; 95% CI, 0.06 to 0.52) and 36 occurred in the TDF-FTC group with approximately 3,907 PY of accumulated follow-up. A revised data analysis from additional testing confirmed 39 incident HIV-1 infections, 3 occurring in the cabotegravir LA group (bias-adjusted HR = 0.1; 95% CI, 0.04 to 0.27), indicating a 90% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC group.11 The cumulative rates are provided in Figure 6 (Appendix 1).
Table 17: Time to HIV-1 Infection in Steps 1 and 2 in the HPTN 084 Trial — Cox Proportional Hazards Regression Model (mITT Population)
Category | Cabotegravir LA n = 1,614 | TDF-FTC n = 1,610 |
|---|---|---|
Number of HIV infections | 4 | 36 |
HR (95% CI), Cox regression | 0.11 (0.04 to 0.31) | |
Superiority P valuea, Cox regression | < 0.0001 | |
HR (95% CI), bias-adjustedb | 0.12 (0.05 to 0.31) | |
Superiority P valuea, bias-adjustedb | < 0.0001 | |
PY | 1,961 | 1,946 |
Incidence rate, (95% CIc), per 100 PY | 0.20 (0.06 to 0.52) | 1.85 (1.30 to 2.56) |
Between-group treatment difference (95% CIc), per 100 PY | –1.65 (–2.28 to –1.01) | |
CI = confidence interval; HR = hazard ratio; LA = long-acting; mITT = modified intent to treat; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: The P values are 2-sided. Efficacy analyses using the mITT population included data from steps 1 and 2 as well as from participants who discontinued study product altogether and moved to annual follow-up in step 1 or 2. Data cut-off date: November 5, 2020.
aHR < 1.0 indicates a lower risk on cabotegravir LA compared to TDF-FTC. The HR is based on a Cox proportional hazards model stratified by site.
bBias-adjusted, corrected for early stopping. The bias-adjusted HR, CI, and P value account for the group sequential trial design and the decision to stop the trial at the second interim analysis.
cThe 95% CI for the incidence rate was calculated using the exact Poisson method.
Source: HPTN 084 Clinical Study Report.18
Supportive Analysis: Incident HIV-1 Infections in the While OBSP Population
The supportive analysis using the OBSP censoring of the injection step 2 efficacy population showed that cabotegravir LA treatment was superior (P = 0.0034) to oral TDF-FTC for the prevention of HIV-1 acquisition (Table 26, Appendix 1). The estimated HR was 0.05 (95% CI, 0.01 to 0.37), indicating a 95% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to the TDF-FTC group.11 The cumulative rates of acquired HIV-1 infections for each group are presented in Figure 7 (Appendix 1).
Supportive Analysis: Incident HIV Infections in Steps 1 and 2 (PP Set)
The planned supportive analysis on the PP population supported the primary analysis, demonstrating that cabotegravir LA was superior to oral TDF-FTC (P < 0.0001) for the prevention of HIV-1 acquisition during steps 1 and 2 (estimated HR = 0.11; 95% CI, 0.04 to 0.31), indicating an 89% reduction in the incidence of HIV-1 infections in the cabotegravir LA group relative to TDF-FTC. The incidence rate in the PP population for the cabotegravir LA group and TDF-FTC groups was 0.21 per 100 PY versus 1.91 per 100 PY, respectively.11
Secondary Analyses
Incident HIV-1 Infections in Step 2 Only
HPTN 083 Study
By the May 14, 2020, interim cut-off date, 8 HIV-1 infections were identified in the cabotegravir LA group and 37 in the TDF-FTC group during step 2 only (Table 18). The incidence rate per 100 PY in the cabotegravir LA group was 0.27 (95% CI, 0.12 to 0.54) and 1.29 (95% CI, 0.91 to 1.77) for the TDF-FTC group (HR = 0.210; 95% CI, 0.10 to 0.45).
Table 18: Time to HIV-1 Infection in Step 2 in the HPTN 083 Trial — Cox Proportional Hazards Regression Model (Injection Step 2 Efficacy Population)
Category | Cabotegravir LA n = 2,114 | TDF-FTC n = 2,079 |
|---|---|---|
Number of HIV infections | 8 | 37 |
Hazard ratio (95% CI)a | 0.21 (0.10 to 0.45) | |
Superiority P value | < 0.0001 | |
PY | 2,923 | 2,877 |
Incidence rate, (95% CIb), per 100 PY | 0.27 (0.12 to 0.54) | 1.29 (0.91 to 1.77) |
Between-group treatment difference, (95% CIb), per 100 PY | –1.01 (–1.47 to –0.56) | |
CI = confidence interval; LA = long-acting; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: The P values are 2-sided. Data cut-off date: May 14, 2020.
aHazard ratio of < 1.0 indicates a lower risk on cabotegravir LA compared to TDF-FTC. The hazard ratio is based on a Cox proportional hazards model stratified by region.
bThe 95% CI for incidence rate was calculated using the exact Poisson method.
Source: HPTN 083 Clinical Study Report.17
HPTN 084 Study
By the November 5, 2020, interim cut-off date, 2 HIV-1 infections were identified in the cabotegravir LA group and 34 in the TDF-FTC group in step 2 only. The incidence rate per 100 PY in the cabotegravir LA group was 0.11 (95% CI, 0.01 to 0.41) compared to 1.94 (95% CI, 1.35 to 2.72) in the TDF-FTC group (HR = 0.06; 95% CI, 0.01 to 0.24) (Table 19).11
Table 19: Time to HIV-1 Infection in Step 2 Only in the HPTN 084 Trial — Cox Proportional Hazards Regression Model (Injection Step 2 Efficacy Population)
Category | Cabotegravir LA n = 1,495 | TDF-FTC n = 1,494 |
|---|---|---|
Number of HIV infections | 2 | 34 |
Hazard ratio (95% CI)a | 0.06 (0.01 to 0.24) | |
Superiority P value | < 0.0001 | |
PY | 1,766 | 1,750 |
Incidence rate (95% CIb), per 100 PY | 0.11 (0.01 to 0.41) | 1.94 (1.35 to 2.72) |
Between-group treatment difference, (95% CIb), per 100 PY | –1.83 (–2.50 to –1.16) | |
CI = confidence interval; LA = long-acting; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: The P values are 2-sided. Data cut-off date: November 5, 2020.
aHazard ratio < 1.0 indicates a lower risk on cabotegravir LA compared to TDF-FTC. The hazard ratio is based on a Cox proportional hazards model stratified by site.
bThe 95% CI for incidence rate was calculated using the exact Poisson method.
Source: HPTN 084 Clinical Study Report.18
Viral Genotyping for Drug Resistance
HPTN 083 Study
At the May 14, 2020, interim cut-off date, 52 HIV-1 new infections were identified overall. Of the 15 infections reported in the cabotegravir LA group, 2 occurred at baseline, 5 infections occurred with no recent cabotegravir LA exposure, 3 infections occurred during the oral lead-in phase, and 5 infections occurred despite on-time cabotegravir LA injections. Of the 42 infections reported in the TDF-FTC group, 3 occurred at baseline and 39 were incident infections during treatment. Four of the incident infections occurred during a prolonged period where TDF-FTC was not dispensed, or study visits were overdue. HIV genotyping (WT, HIV-1 strain NL4 to 3) was performed at the first visit where the HIV viral load was greater than 500 copies/mL (first viremic visit, based on data on the timing of HIV infection that were available at the time of the primary analysis).
Cabotegravir LA Group
HIV genotyping results were obtained for 12 of the 15 cabotegravir LA-reported HIV-1 cases (1 failed analysis and 2 had no viremic visits). Integrase resistance mutations were identified in 3 participants; NNRTI resistance was identified in 3 other participants at the first viremic visit, including 1 case of resistance to an NRTI.11
TDF-FTC Group
HIV genotyping results were presented for 40 of the 42 HIV cases reported in the TDF-FTC group (2 cases had no viremic visit). There were no resistance patterns identified in participants who had contracted HIV-1 at baseline. Twelve participants with incident infections showed resistance at the first viremic visit (7 had NNRTI resistance only, 1 had NRTI resistance only, 1 had single PI resistance mutation only, and 3 had NNRTI and NRTI resistance). Ten participants identified with NNRTI resistance had 1 or more of the following mutations: K103N/S, Y181C, G190A/S, H221Y, and P225H. In 4 participants with NRTI resistance (including 3 who had multiclass resistance), 3 had M184V/I and K65R mutations.11
HPTN 084 Study
At the November 5, 2020, interim analysis, 40 HIV-1 new infections were identified in the 2 groups. Two infections occurred in women without any recent oral cabotegravir LA exposure and no injections, and 2 occurred during the injection phase of the study. Thirty-six infections occurred in the TDF-FTC group.11 HIV genotyping was performed at the first visit where HIV viral load was greater than 500 copies/mL (first viremic visit, based on viral load data available at the time of the primary analysis).
Cabotegravir LA Group
HIV genotyping results were available for 3 of 4 cabotegravir LA participants with HIV-1 (1 participant had no viremic sample). One of the 3 participants had an integrase mutation at the first viremic visit (L74l), which was considered a polymorphism, also detected in several participants in the TDF-FTC group.11
TDF-FTC Group
HIV genotyping results were obtained for 33 of the 36 incident infections in the TDF-FTC group (2 failed testing, 1 had no viremic sample). A major NRTI mutation (M184V) was identified in 1 participant in addition to NNRTI resistance with the K103N mutation. Eight other participants had NNRTI resistance only (6 had K103N alone or with E138A or P225H, 1 had K101E alone, 1 had E138A mutations alone). INSTI mutations or polymorphisms were detected in 10 samples (L74I, L74M, T97A, V151I, E157Q, G193E). For 1 participant with reported dual-class resistance (NRTI and NNRTI), resistance observed in the first viremic visit was identical to the first site-positive visit (at step 2, week 17, 33 days after the first HIV-positive visit).11
Adherence
Drug concentrations were measured in plasma and DBS and PK testing was limited to a subset of the samples.
HPTN 083 Study
A random subset of 170 participants was used to assess adherence to cabotegravir LA injections (using the cabotegravir LA longitudinal PK population). Injection coverage (defined as receiving an injection within less than 2 weeks delay of scheduled administration) in the cabotegravir LA group was 91.5% of all PY contributions for the subsample (Figure 2).
Adherence to TDF-FTC was assessed in a random subset of 390 participants based on plasma TFV concentrations and intraerythrocytic TFV-DP concentrations collected as DBS. Overall, 84% (1,700 of 2,025) of the evaluated samples yielded plasma TFV concentrations consistent with 4 or more doses per week; this adherence benchmark decreased from 92% (361 of 390) at week 4 to 76% (127 of 169) at week 81. In total, 74.2% of participants had TFV concentrations consistent with daily dosing (≥ 40 ng/mL, corresponding to expected TDF-FTC daily use concentration) and more than 86% had detectable TFV (≥ 0.31 ng/mL). When assessed using TFV-DP concentrations, 73% of evaluated DBS samples yielded concentrations consistent with 4 or more doses per week.
Figure 2: Adherence Data in the HPTN 083 Study
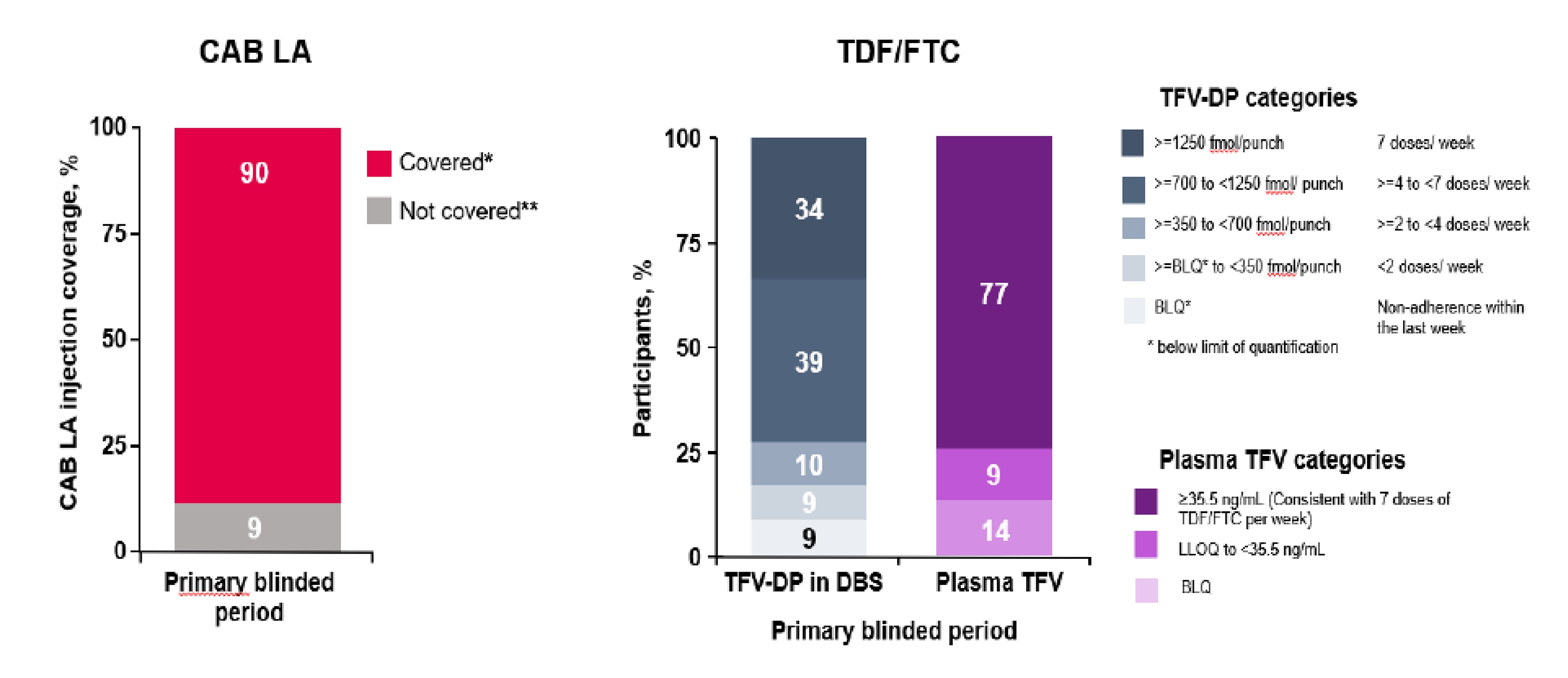
BLQ = below the limit of quantification; CAB = cabotegravir; DBS = dried blood spot; DP = diphosphate; LA = long-acting; LLOQ = lower limit of quantification; PK = pharmacokinetic; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine; TFV = tenofovir.
Note: Coverage is defined as receiving an injection within less than 2 weeks delay of scheduled administration.
*Covered: injections received with a delay up to 14 days
**Not covered: injections received with a delay of more than 14 days.
Source: Sponsor’s drug submission package.40
HPTN 084 Study
A random subset of 150 participants was used to assess adherence in the cabotegravir LA group (using the cabotegravir LA longitudinal PK population). Injection coverage in the cabotegravir LA group was 93% of all PY contributions in the subsample (Figure 3). In a random subset of those randomized to TDF-FTC (n = 409), 41.9% had TFV concentrations consistent with daily dosing (≥ 40 ng/mL, corresponding to expected daily use concentration of TDF-FTC) and 55.9% had detectable TFV (≥ 0.31 ng/mL).
Figure 3: Adherence Data in the HPTN 084 Study
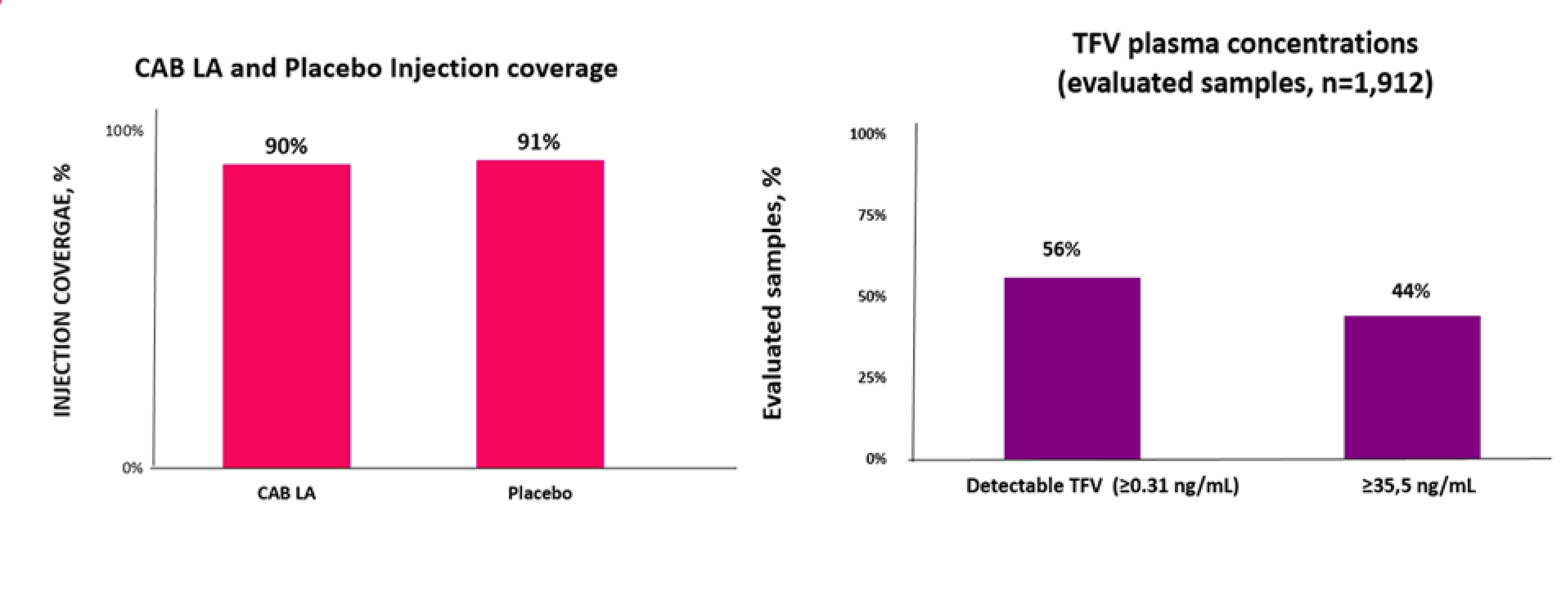
CAB = cabotegravir; LA = long-acting; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TFV = tenofovir.
Source: Sponsor’s drug submission package.40
Harms
A summary of AEs reported in the HPTN 083 and HPTN 084 trials is presented in Table 20. Safety analyses were conducted using the safety population set (OBSP). AEs reported in step 1 included all AEs occurring until the first injection date, or 120 days postrandomization, whichever occurred first. The injection step 2 safety population was also used in the safety analysis which included all AEs occurring from the first injection date through 48 weeks after the last injection.11
Adverse Event
There were similarities in the proportion of participants reporting at least 1 AE in both groups in the 2 trials. In the HPTN 083 study, 95% versus 94% in the cabotegravir LA group and TDF-FTC groups, respectively, reported at least 1 AE and 96% in both the cabotegravir LA group and TDF-FTC groups reported at least 1 AE in the HPTN 084 trial. Commonly reported AEs included injection site pain (in the HPTN 083 study, 75% versus 30% in the cabotegravir LA and TDF-FTC groups, respectively; in the HPTN 084 study, 32% versus 9% in the cabotegravir LA and TDF-FTC groups, respectively), creatinine renal clearance decreased (in the HPTN 083 study, 69% versus 73% in the cabotegravir LA and TDF-FTC groups, respectively; in the HPTN 084 study, 72% versus 74% in the cabotegravir LA and TDF-FTC groups, respectively), blood creatine phosphokinase increased (in the HPTN 083 study, 22% in both the cabotegravir LA and TDF-FTC groups; in the HPTN 084 study, 15% versus 16% in the cabotegravir LA and TDF-FTC groups, respectively), blood creatinine increased (in the HPTN 083 study, 17% versus 19% in the cabotegravir LA and TDF-FTC groups, respectively; in the HPTN 084 study, 22% in both the cabotegravir LA and TDF-FTC groups), and nasopharyngitis (in the HPTN 083 study, 17% in the cabotegravir LA and TDF-FTC groups, respectively; in the HPTN 084 study, 5% versus 6% in the cabotegravir LA and TDF-FTC groups, respectively).11
Grade 3 or higher AEs reported in more than 1% of participants in either treatment group in the 2 trials included blood creatine phosphokinase increased, creatinine renal clearance decreased, blood creatinine increased, lipase increased, aspartate aminotransferase increased, injection site pain, and alanine aminotransferase increased.11
Serious AEs
SAEs were reported by 5% of participants in the cabotegravir LA and TDF-FTC groups in the HPTN 083 trial, and 2% in the cabotegravir LA and TDF-FTC groups in the HPTN 084 trial. The most common SAEs reported in the HPTN 083 trial were suicide attempt (< 1% in both groups), suicidal ideation (< 1% in both groups), and dengue fever (< 1% in both groups). The most reported SAEs in the HPTN 084 trial were suicide attempt (< 1% in the TDF-FTC group, 0% in the cabotegravir LA group), depression (< 1% in both treatment groups), pelvic inflammatory disease (< 1% in both treatment groups), and malaria (< 1% in both treatment groups).11
Withdrawals Due to AEs
In the HPTN 083 study, 135 (6%) participants in the cabotegravir LA group and 91 (4%) participants in the TDF-FTC group had AEs that led to drug discontinuation. Four participants (1%) discontinued study drug during the step 1 and had never received intramuscular cabotegravir LA injection. In the HPTN 084 study, 17 (1%) participants in the cabotegravir LA group and 22 (1%) in the TDF-FTC group had AEs leading to discontinuation. Four participants (< 1%) in the cabotegravir LA group and 6 (< 1%) in the TDF-FTC group discontinued study drug at step 1.11
Deaths
In the HPTN 083 study, 10 deaths were reported during steps 1 and 2 (4 in the cabotegravir LA group, 6 in the TDF-FTC group, and 1 additional death reported in step 3). In the HPTN 084 study, 3 participants in the cabotegravir LA group died due to AEs, 2 during step 2 and 1 during step 2 of the non-OBSP group. No deaths were reported in the TDF-FTC group.11
AEs of Special Interest
Injection Site Reactions
In the HPTN 083 trial, a total of 1,740 (82%) participants reported at least 1 drug-related ISR in the cabotegravir LA group. Injection site pain was the most reported ISR (81% of cabotegravir LA group participants), followed by injection site nodule, induration, and swelling (> 10% of cabotegravir LA participants).11
In the HPTN 084 study, a total of 575 (38%) participants reported at least 1 drug-related ISR in the cabotegravir LA group. Injection site pain was the most common (34% of cabotegravir LA participants) followed by injection site swelling, injection site nodule, and injection site induration (≥ 5% of cabotegravir LA participants). All other drug-related ISRs were reported in 2% or less of cabotegravir LA participants.11
Hepatotoxicity
In the HPTN 083 trial, hepatic steatosis was the most frequently reported hepatotoxicity in both groups, occurring in less than 1% of participants. In the HPTN 084 study, acute hepatitis was reported in 1 participant in the cabotegravir LA group, and hepatotoxicity and liver disorder were each reported in 1 participant in the TDF-FTC group.11
Hypersensitivity Reactions
In the HPTN 083 study, hypersensitivity was the most reported AE of special interest in both groups (1% of participants in the cabotegravir LA group and < 1% of participants in the TDF-FTC group). In the HPTN 084 study, hypersensitivity was the most reported AE of special interest in the cabotegravir LA (n = 8; < 1% of participants) and TDF-FTC (n = 10; < 1% of participants) groups.11
Rash
In the HPTN 083 study, rash was the most reported AE of special interest in both groups (3% in both the cabotegravir LA and TDF-FTC groups). All other rash occurred in less than 1% of participants in either group. In the HPTN 084 study, rash was the most reported AE of special interest in both groups (2% in both the cabotegravir LA and TDF-FTC groups).11
Neuropsychiatric Events
In the HPTN 083 study, neuropsychiatric AEs occurring in more than 1% participants included sleep disorder (10% in the cabotegravir LA group and 11% in the TDF-FTC group), followed by depression (5% in both groups), and anxiety (4% in both groups). In the HPTN 084 study, sleep disorder was reported in 5% of participants in both groups.11
Table 20: Harms Reported During Steps 1 and 2 of the HPTN 083 and HPTN 084 Trials (Safety Population)
AEs, n (%) | HPTN 083 | HPTN 084 | ||
|---|---|---|---|---|
Cabotegravir LA | TDF-FTC | Cabotegravir LA | TDF-FTC | |
Most common AEs (occurring in ≥ 5% in either treatment group) | ||||
≥ 1 AE | 2,174 (95) | 2,157 (94) | 1,556 (96) | 1,540 (96) |
Injection site pain | 1,713 (75) | 688 (30) | 522 (32) | 147 (9) |
Creatinine renal clearance decreased | 1,576 (69) | 1,661 (73) | 1,160 (72) | 1,192 (74) |
Blood creatine phosphokinase increased | 506 (22) | 497 (22) | 237 (15) | 263 (16) |
Blood creatinine increased | 379 (17) | 426 (19) | 363 (22) | 347 (22) |
Nasopharyngitis | 383 (17) | 379 (17) | 82 (5) | 96 (6) |
Headache | 377 (17) | 356 (16) | 377 (23) | 373 (23) |
Diarrhea | 328 (14) | 336 (15) | 101 (6) | 119 (7) |
Anal chlamydia infection | 264 (12) | 297 (13) | — | — |
Upper respiratory tract infection | 264 (12) | 271 (12) | 268 (17) | 293 (18) |
SAEs | ||||
Participants with ≥ 1 SAE | 109 (5) | 104 (5) | 25 (2) | 33 (2) |
Suicide attempt | 7 (< 1) | 9 (< 1) | 0 | 2 (< 1) |
Suicidal ideation | 6 (< 1) | 6 (< 1) | — | — |
Dengue fever | 5 (< 1) | 3 (< 1) | — | — |
AEs leading to treatment discontinuation | ||||
Number of participants with at least ≥ 1 AE leading to discontinuation | 135 (6) | 91 (4) | 17 (1) | 22 (1) |
Alanine aminotransferase increased | 29 (1) | 31 (1) | 12 (< 1) | 15 (< 1) |
Aspartate aminotransferase increased | 7 (< 1) | 8 (< 1) | — | — |
Lipase increased | 5 (< 1) | 4 (< 1) | 0 | 1 (< 1) |
Deaths | ||||
Participants who died | 4 (< 1) | 6 (< 1) | 3 (< 1) | 0 (0) |
AEs of special interest | ||||
ISRs (occurring in > 1% in either treatment group) | ||||
Number of participants with at least 1 ISR AE | 1,740 (76) | 726 (32) | 578 (38) | 166 (11) |
Hepatotoxicity | 8 (< 1) | 8 (< 1) | 1 (< 1) | 2 (< 1) |
Hypersensitivity reactions | 52 (2) | 38 (2) | 14 (< 1) | 16 (< 1) |
Rash | 94 (4) | 98 (4) | 71 (4) | 66 (4) |
Neuropsychiatric events | ||||
Sleep disorders | 217 (10) | 248 (11) | 81 (5) | 76 (5) |
Depression | 115 (5) | 108 (5) | 7 (< 1) | 12 (< 1) |
Anxiety | 99 (4) | 97 (4) | 16 (< 1) | 11 (< 1) |
Mood disorders | 30 (1) | 19 (< 1) | 9 (< 1) | 6 (< 1) |
Suicidal ideation and behaviour | 25 (1) | 23 (1) | 3 (< 1) | 6 (< 1) |
AE = adverse events; ISR = injection site reaction; LA = long-acting; SAE = serious adverse event; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Sources: HPTN 083 Clinical Study Report17 and HPTN 084 Clinical Study Report.18
Critical Appraisal
Internal Validity
The HPTN 083 and HPTN 084 studies were multicentre, randomized trials. Randomization was stratified by study site, and permuted blocks were used to ensure balance in treatment assignments within study sites. Placebo tablets were film-coated to visually match TDF-FTC tablets and placebo injections were visually similar to the active injection to ensure treatment allocation concealment. Study staff and participants were masked to study group allocation, except site pharmacists. There were no notable imbalances in baseline demographics in the 2 treatment groups across trials, indicating randomization was successful. Body mass index, which was considered an important baseline characteristic by the clinical expert, showed no differences between the 2 groups in both trials. Both study designs were double blinded to allow for a direct comparison of the 2 drugs in the absence of the additional differential effect of adherence to daily pills and differences in risk behaviour. Adjudication of HIV end points in the 2 trials was performed by blinded personnel, mitigating the risk of assessment bias for the primary outcome.
Treatment adherence was assessed using PK blood concentrations of study drugs in a random subset of participants for each treatment. There were differences between the 2 treatment groups in both trials which may have impacted the efficacy of the primary outcome. Injection coverage in PY reported for participants receiving cabotegravir LA was high in both trials (91.5% versus 93% PY coverage in the subsample of participants in the HPTN 083 and HPTN 084 trials, respectively) compared to TDF-FTC (concentrations consistent with daily dosing [≥ 40 ng/mL)] were 74.3% versus 41.9% in the HPTN 083 and HPTN 084 trials, respectively). The expert highlighted that drug concentration measurements using PK parameters may not be truly representative of treatment adherence due to known variabilities in drug metabolism across individuals, resulting in variable plasma concentrations at the time of sampling. The expert also noted that low drug concentrations at a given time does not necessarily translate to drug inefficacy and the risk of acquiring HIV infection. In addition, the sample size of both subsets for the analysis was considered small relative to the total number of participants in each group for each study. As a result, no definitive conclusions could be drawn from the differences observed in the 2 trials.
The use of the Poisson model to estimate the rate of HIV infection in both trials was deemed appropriate by CDA-AMC’s review team but subject to 2 important limitations: the assumption of a constant rate (i.e., the rate of infection within the population is constant) and the use of noninformative censoring (that the withdrawal and censoring were noninformative of an individual’s potential future infection). These limitations may impact the interpretation of the efficacy results due to a lack of generalizability of the results outside of the trial setting. The HPTN 083 study was a noninferiority trial and the HR margin (M2) was selected based on evidence from prior placebo-controlled trials (the iPrex,12 iPERGAY,13 and PROUD14 studies). The clinical expert noted that the relative risk (RR) and relative efficacy obtained from the individual trials were different based on the enrolled population; however, the composite estimate derived from the evidence was likely appropriate to use in the context of the HPTN 083 and HPTN 084 trials. The mITT population (consisting of the ITT population excluding those who were found to be HIV-infected at randomization) was used to assess the primary efficacy end point in the HPTN 083 study. A supportive analysis conducted on the OBSP population to verify the consistency of the ITT analysis was consistent with the primary analysis. The HR for the relative incidence of HIV infection was estimated using the Cox regression stratified according to predefined factors. Interim analyses were prespecified and occurred using O’Brien-Fleming method; the derived HR was compared to a prespecified group sequential stopping boundary. Missing data were handled by noninformative censoring and event rates were compared between treatment groups using the same model at a prespecified type I error rate of 5%, which was considered appropriate. Secondary analyses to assess the incidence HIV infections in step 2 only were conducted using the same models as the primary outcome and adjusted for type I error and early stopping; however, no sensitivity analyses were performed. Overall, the methods used were appropriate to test for a difference in incidence of HIV infection across treatment groups.
The HPTN 084 study was a superiority trial and the HR and 95% CI estimates for the primary outcome were derived using a Cox proportional hazards model, stratified by site. The primary analyses were conducted on the ITT dataset and supplemented with 2 sensitivity analyses (the PP analyses and supportive analysis using the OBSP population) conducted on the mITT population which was consistent with the primary analyses. Four interim analyses and a final analysis were prespecified, and the O’Brien-Fleming boundary was considered for early stopping. Missing data were handled by noninformative censoring and subject to bias (it assumed that the withdrawal and censoring were noninformative of an individual’s potential future infection). Event rates were compared between treatment groups using the same model at a prespecified type I error rate of 5%, which was considered appropriate. Secondary analyses to assess the incidence HIV infections in step 2 only were conducted using the same models as the primary outcome and adjusted for type I error and early stopping; however, no sensitivity analyses were performed.
Evidence presented for this submission for both trials were from preplanned interim analyses, both of which met the criteria for stopping based on efficacy. There is a potential source of uncertainty in the efficacy of cabotegravir LA for the primary outcome because interim analyses have the potential of increasing the risk, overestimating treatment effects47 (in the HPTN 083 study, only approximately 30% [52 of 172 events] and 35% [40 of 114 events] in the HPTN 084 study of the total targeted preplanned infections for powering the studies was achieved at both data cut-offs of the preplanned interim analyses). Subgroup analyses were preplanned in both studies; however, both trials were not powered to detect a statistically significant difference between groups. No adjustments were made for multiple testing in the subgroup analyses.
Discontinuation from the studies was infrequent in both trials, with no more than 3% of participants in any treatment group discontinuing from study. However, the clinical expert noted that real-world evidence has shown that adherence rates for daily pills is typically lower in the real world than in clinical trials, where patients are motivated and subject to frequent follow-ups.
The proportion of participants that reported 1 or more protocol deviations was as follows in both studies. In the HPTN 083 study, a total of 18% of participants in the TDF-FTC group reported protocol deviations compared to 16% in the cabotegravir LA group. There were more protocol deviations (> 15%) in both groups in the HPTN 084 study (33% in the cabotegravir LA group and 31% in the TDF-FTC group). The impact of protocol deviations on the findings were considered minimal.
External Validity
The HPTN 083 and HPTN 084 studies were multicentre RCTs conducted in the US, South America, Asia, and Sub-Saharan Africa. There were no sites in Canada. The inclusion and exclusion criteria of both trials were considered very inclusive according to the clinical expert consulted, and the at-risk population defined aligned with the Canadian HIV guideline recommendations. The baseline characteristics reported in both trials were considered generalizable to the population of people living in Canada.
There were no concerns identified for the HIV testing methods implemented in both trials that may have impacted HIV-1 identification in the trial populations. The expert indicated that the serological methods and assays utilized in both trials for HIV testing aligned with test generations available to Canada and internationally at the time of trial coordination. The expert noted that current tests methods available in Canada are fifth-generation assays which have a higher sensitivity of early detection of HIV (as early as 2 weeks postexposure48).
Overall, the outcomes assessed in both trials were considered important. Key outcomes highlighted by the expert as clinically relevant for decision-making included documented HIV-1 infections, adherence, and resistance mutations among seroconverters. Patient-reported outcomes were also highlighted as important by the clinical expert and patient advocacy and clinician groups participating in the review. The clinical expert highlighted the need for more evidence on Indigenous populations, a key population impacted in Canada.
The duration of follow-up in both trials was considered appropriate and long enough to identify HIV events for the population enrolled. The expert indicated that there is no fixed duration of exposure to drug to assess PrEP benefits, since cabotegravir LA, including other PrEP options are designed to prevent infections and not for HIV treatment. The expert also noted that individuals receiving PrEP in practice will be followed every 3 months after treatment is initiated. However, adherence assessments using PK drug concentrations may not be applicable to current practice for logistical reasons. Some trial procedures implemented in both designs may not be applicable to real-world practice, for example, the frequency of HIV screening and the methods for measuring adherence. The expert noted that monitoring adherence to cabotegravir LA is not anticipated to be an issue in clinical practice because the intramuscular injections will be administered under supervision at a health clinic.
The dosing of TDF-FTC in both trials was reflective of Canadian practice according to the expert. The expert did not anticipate additional dose adjustments for cabotegravir LA in practice. However, the expert anticipates a variation in the use of an oral lead-in with cabotegravir LA tablets in practice. Given the familiarity of the use of cabotegravir LA (in combination therapies for HIV-1 treatment) and the tolerable safety profile observed in the trials, the expert indicated that the oral lead-in may only be suggested for persons having concerns related to cabotegravir LA intramuscular safety at initiation.
Both trials provided direct evidence of the comparative efficacy of cabotegravir LA compared to other PrEP options in Canada. However, there is a lack of evidence on the long-term therapeutic benefit and safety of cabotegravir LA beyond the duration of both trials. There are also limited efficacy data for the key primary end point in adolescent populations and definitive conclusions could not be made in the absence of efficacy data for the primary outcome. The expert noted that although both trials were conducted in adults aged 18 years and older, cabotegravir LA will be effective in adolescents weighing at least 35 kg if they are perceived to be at risk of HIV-1 acquisition. No dose adjustment is also anticipated for these populations weighing at least 35 kg.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDA-AMC’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.15,49
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of a clinically important effect based on threshold informed by the clinical expert consulted by CDA-AMC for documented incident HIV infections. There is no established minimally important difference and the clinical expert consulted by CDA-AMC could not provide a threshold of important difference so the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect. Other targets for the certainty of evidence assessment were the presence or absence of any effect for the proportion of patients reporting SAEs and ISRs.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for cabotegravir LA versus TDF-FTC.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
One ITC submitted by the sponsor comparing cabotegravir LA with alternative treatments for PrEP was included in this report. An ITC was included to address a gap in pivotal and RCT evidence in the absence of studies directly comparing cabotegravir LA with no PrEP and inform the pharmacoeconomic model.
Description of Indirect Comparison
A systematic literature review (SLR) was first conducted by the sponsor to identify evidence for inclusion in the ITC. The relative efficacy of cabotegravir LA from the HPTN 083 and HPTN 084 trials was converted to effectiveness and indirectly compared with patients with no PrEP who are at risk of sexually acquired HIV-1 infection via Bayesian NMA using meta-regression. Comparators of interest for the sponsor-submitted NMA included TDF-FTC and no PrEP (i.e., placebo or no treatment). Effectiveness in reducing the risk of HIV acquisition was the outcome of interest.
ITC Design
Objectives
The objective of the analysis presented in this report is to estimate the comparative effectiveness of oral cabotegravir LA, TDF-FTC, and no PrEP (i.e., placebo or no treatment), using an NMA.
Study Selection Methods
As of June 2023, the most recently published SLR was conducted by Huic and Reinsperger with the Austrian Institute for Health Technology Assessment (searches conducted November 2022), which identified populations of “HIV-negative people at risk for HIV.”50 The Huic and Reinsperger SLR used the Murchu et al. SLR of oral PrEP (in “populations at substantial risk of HIV”)51 and the Fonner 2022 SLR (included in the WHO guidelines) of cabotegravir LA (in “populations at substantial risk of HIV”)52 as their base, which they updated to capture any newly published relevant data. The Huic and Reinsperger SLR is also the only SLR to include both oral and LA PrEP.
The sponsor conducted an SLR in November 2023. Search dates for identified published SLRs ranged from July 2020 to November 2022. To bridge the gap between search dates of the published SLRs and now, a search was run from the beginning of 2022 to November 2023 to identify any RCTs that had been published since these SLRs were conducted. An SLR was conducted using MEDLINE (In-Process), Embase, and the Cochrane Library to identify RCTs published for all time until the date of search (i.e., November 1, 2022). Abstracts from the following 3 conferences from the past 3 years (2020 to 2023) were searched to identify trials which may have not yet published their data in peer-reviewed articles: International AIDS Society; International AIDS conference; International AIDS Society Conference on HIV Science; IDWeek; Conference on Retroviruses and Opportunistic Infections; European AIDS Clinical Society European AIDS Conference; and HIV Glasgow Congress. Clinical trial registries (i.e., Clinicaltrials.gov and International Clinical Trials Registry Platform) were searched to identify ongoing trials. Of the 19 unique RCTs included in the SLR, 10 met the additional NMA eligibility criteria of reporting TDF-FTC PrEP adherence based on plasma sampling (or pill count data for the relevant sensitivity analysis).
Article screening was performed independently by 2 reviewers at 2 stages (title and abstracts, and then full text), with a third reviewer to resolve disagreements. The study selection criteria and methods are shown in Table 21. Reasons for exclusion were documented. Data extraction was performed by 1 reviewer, with quality check on all data by a second reviewer. Quality assessment of the selected studies was conducted to assess the risk of bias using the Centre for Reviews and Dissemination risk of bias tool;53 the sponsor did not specify the number of reviewers participating in the quality assessment process. The measure of effectiveness was the reduction in risk of HIV acquisition. The results of the indirect comparison were reported on the percentage effectiveness scale.
Table 21: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population |
|
Intervention |
|
Comparator |
|
Outcome | Incidence of HIV infection Cases of HIV infection averted Adherence to PrEP Adverse events Incidence of other STIs Behavioural changes (including condom use) Drug resistance |
Study designs | Randomized controlled trials |
Publication characteristics | English language only |
Exclusion criteria | Reasons for exclusion were coded according to the primary reason for exclusion (i.e., population, interventions, outcomes) |
Databases searched | MEDLINE and MEDLINE In-Process (via Embase.com) Embase (via Embase.com) Cochrane (via cochranelibrary.com) Manual search of abstracts from: International AIDS Society conference International AIDS Society, International AIDS conference, International AIDS Society Conference on HIV Science, IDWeek, Conference on Retroviruses and Opportunistic Infections, European AIDS Clinical Society European AIDS Conference, and HIV Glasgow Congress. Manual searches of the following clinical trial registries: Clinicaltrials.gov International clinical trials registry platform. |
Selection process | The final list of abstracts was reviewed independently by 2 systematic reviewers against the eligibility criteria. The results of the reviews were then compared and where discrepancies arose, consensus between reviewers was reached by mutual consent. Where consensus could not be reached between reviewers, a third member of the team reviewed the reference and cast a deciding vote. For those selected for inclusion, the double review process was repeated using the full-text articles. Reasons for exclusion at the full-text review stage were recorded. |
Data extraction process | Data from the included studies were extracted into a data extraction sheet. Data were extracted by a single reviewer and fully validated by a second reviewer. |
Quality assessment | A risk of bias assessment was conducted on the included studies using the tool suggested by the Centre for Reviews and Dissemination. The following were assessed.
|
ITC = indirect treatment comparison; LA = long-acting; PrEP = pre-exposure prophylaxis; STI = sexually transmitted infection; TAF-FTC = tenofovir alafenamide fumarate-emtricitabine; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: Systematic Literature Review Technical Report.54 Details included in the table are from the sponsor’s Summary of Clinical Evidence.11
ITC Analysis Methods
A summary of the analysis methods for the NMA is presented in Table 22. All analyses were conducted using Bayesian models. Fixed-effects and random-effects models were evaluated for the logarithmic model, which was the base-case analysis. Vague prior distributions on model parameters were used. These were selected using the recommended priors in the NICE technical support document 2.55 Posterior outcome distributions were based on at least 50,000 simulations after a burn-in of at least 50,000 using Markov chain Monte Carlo sampling. Three chains were run starting from different initial values. Adequate convergence was assessed byR squared statistic and Brooks Gelman Rubin plots.56
The measure of effectiveness was the reduction in risk of HIV acquisition. The indirect comparison presented the drug effectiveness in reducing HIV transmission compared to a patient population that received a placebo in place of PrEP yet was in some way comparable to the trial populations included in the HPTN 083 and HPTN 084 studies. The rate of HIV transmission in the placebo group was estimated using the comparator trial results, and effectiveness defined as equal to (1 – RR of HIV acquisition) × 100.
The indirect comparison of the effectiveness of cabotegravir LA versus no PrEP in preventing HIV acquisition was estimated for each trial population representing cisgender MSM and TGW who have sex with men (i.e., the HPTN 083 study) and cisgender women (i.e., the HPTN 084 study). Fixed-effects and random-effects models were fitted and compared on deviance information criteria to determine the better fitting model (lower deviance information criteria values indicate better fit to the data). For each trial population representing cisgender MSM and TGW who have sex with men (i.e., the HPTN 083 study) and cisgender women (i.e., the HPTN 084 study), the fixed-effects model was favoured as the random treatment effects models have a higher deviance information criteria than the corresponding fixed-effects model indicating that the inclusion of the random treatment effects does not improve the model fit.
Each treatment was considered a separate node in the evidence network. According to the sponsor, results of the included studies reflect the difference in cumulative HIV incident infections between arms over the duration of the follow-up period.
Sensitivity analyses were conducted based on multiple scenarios. Refer to Table 22 for details of the sensitivity analyses. No subgroup analysis was conducted. Assessment for consistency was not possible due to the lack of closed loops.
Table 22: Sponsor-Submitted NMA Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian meta-regression with a trial adherence variable |
Priors | Vague prior distributions, N (0,104), were used for the model parameters |
Assessment of model fit | Model selection was assessed using deviance information criteria |
Assessment of consistency | Not applicable |
Assessment of convergence | Convergence was judged sufficient if the R2 statistic is < 1.05 and by applying the “blue finger test” to trace plots;56 Brooks Gelman Rubin plots were also examined56 |
Accuracy of estimation | The accuracy of estimation was judged sufficient if the Monte Carlo standard error divided by posterior standard deviation for all parameters was < 5% |
Outcomes | The measure of effectiveness was the reduction in risk of HIV acquisition The results of the ITC were reported on the percentage effectiveness scale, where: % effectiveness = (1 – relative risk of HIV acquisition) × 100 |
Follow-up time points | Ranged from 9.3 months to 36.0 months |
Construction of nodes | Each treatment was a separate node |
Sensitivity analyses |
|
Subgroup analysis | Not conducted |
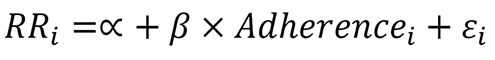
ITC = indirect treatment comparison; LA = long-acting; NMA = network meta-analysis; PrEP = pre-exposure prophylaxis; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: ITC Technical Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.11
Results of Sponsor-Submitted NMA
Summary of Included Studies
A total of 10 trials were eligible for this analysis to form a network, including 2 studies (HPTN 083 and HPTN 084) contributing to the direct estimates of effectiveness of cabotegravir LA versus TDF-FTC, and 8 studies assessed the comparative efficacy between TDF-FTC and placebo (the iPrEx, IPERGAY, FEM-PrEP, TENOFOVIR2, the Partners PrEP, Bangkok Tenofovir Study, and VOICE studies) or delayed PrEP (PROUD).
An overview of the assessment of homogeneity of the 10 studies included in the ITC is presented in Table 23. There was 1 phase IIb trial (VOICE) and 1 phase IV trial (PROUD); the remaining trials were phase III trials. Among the included studies, PROUD was open label and VOICE was partially blinded; the remaining trials were double blinded. The HPTN 083, HPTN 084, and iPrEx trials were conducted globally, PROUD and IPERGAY were conducted in Europe, the Bangkok Tenofovir Study was conducted in Asia, and the remaining studies were conducted in Africa. All trials included patients who were at risk of HIV-1 infection. Median age at baseline in a study treatment arm ranged from 23 years (range, 18 years to 35 years) to 35 years (range not reported). The included studies can be split into 3 populations: the gbMSM and TGW populations were analyzed by the HPTN 083, iPrEx, IPERGAY, and PROUD studies; the cisgender women population was analyzed by the HPTN 084, Partners PrEP, FEM-PrEP, VOICE, and TENOFOVIR2 studies, (of note, the TENOFOVIR2 study also included cisgender men); and the PWID population was analyzed by the Bangkok Tenofovir Study. The treatment group was TDF in the Bangkok Tenofovir Study, event-driven TDF-FTC in the IPERGay study, cabotegravir LA in the HPTN 083 and HPTN 084 studies, and the remaining trials used TDF-FTC. Most of the included studies used placebo as the comparator group while the PROUD study used deferred PrEP and the HPTN 083 and HPTN 084 studies used TDF-FTC. The median follow-up of the included studies ranged from 9.3 months to 36.0 months. The RR of HIV acquisition in a study treatment arm ranged from 0.14 (95% CI, 0.02 to 0.6) to 1.04 (95% CI, 1.49 to 0.29). The adherence rate to TDF-FTC (detectable in plasma) in a study treatment arm ranged from 0.29 to 0.88.
The sponsor considered the between-trial differences in TDF-FTC adherence to be clinically meaningful and could affect the comparability of the studies’ treatment effect within the NMA; thus, adjustments for heterogeneity in TDF-FTC adherence were made.
The quality assessment of included studies concluded that the trials were generally well-conducted, with only minor concerns raised across 4 studies: iPrEx, PROUD, FEM-PrEP, and VOICE. Refer to the Appendix for details of the quality assessment of included studies.
Table 23: Assessment of Homogeneity for Sponsor-Submitted NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Not applicable |
Participant characteristics | The mean age of participants in the included studies ranged from 23 to 35 years. Participants in the studies included men who have sex with men, male cisgender, female cisgender, male and female drug users, and transgender women (HPTN 083 only). |
Treatment history | Not applicable |
Trial eligibility criteria | Not reported |
Dosing of comparators | Not reported |
Treatment effect estimates | Among the studies comparing TDF-FTC to placebo, the relative risk of HIV acquisition ranged from 0.14 to 1.04. Among the studies comparing cabotegravir LA to placebo, the relative risk of HIV acquisition was 0.12 in the HPTN 084 study and 0.34 in the HPTN 083 study. |
Definitions of end points | Not reported |
Timing of end point evaluation | HIV acquisition could occur throughout the duration of the studies |
Withdrawal frequency | Not reported |
Adherence to TDF-FTC | TDF-FTC adherence ranged in the included studies from 0.29 to 0.88 |
Clinical trial setting | Not reported |
Study design | Double-blind and open-label studies |
ITC = indirect treatment comparison; LA = long-acting; NMA = network meta-analysis; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: ITC Technical Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.11
Results
Ten studies were included in the sponsor-submitted NMA (Figure 4). The fixed-effects model was selected as the base case for the estimated of effectiveness, based on the lower reported deviance information criteria. Results of the fixed-effects model and random-effects model are summarized in Table 24.
Figure 4: Network Diagram of Studies Included in Sponsor-Submitted NMA
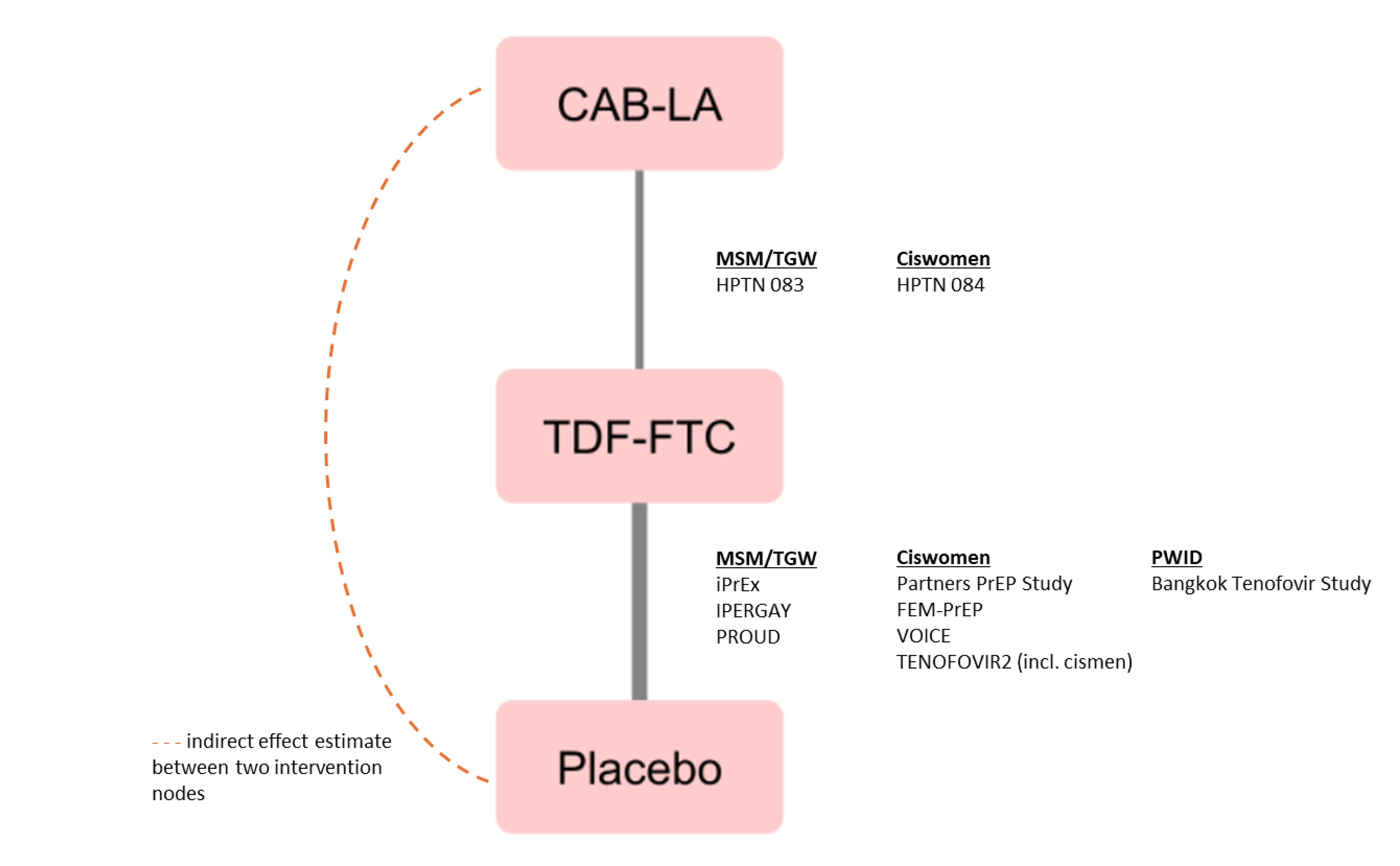
CAB = cabotegravir; LA = long-acting; MSM = men who have sex with men; NMA = network meta-analysis; PWID = people who inject drugs; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine; TGW = transgender women.
HPTN 083 Population (Cisgender MSM and TGW Who Have Sex With Men)
Cabotegravir LA was favoured over placebo or no PrEP (drug effectiveness = 91.10%; 95% Crl, 82.87% to 95.95%) in the base-case analysis (fixed-effects model). Similar results were observed in the random-effects model. Generally, the results of all sensitivity analysis were consistent with the base case.
HPTN 084 Population (Cisgender Women)
Cabotegravir LA was favoured over placebo or no PrEP (drug effectiveness = 92.52%; 95% Crl, 83.02% to 97.38%) in the base-case analysis (fixed-effects model). Similar results were observed in the random-effects model. Generally, the results of all sensitivity analysis were consistent with the base case.
Table 24: Results of Sponsor-Submitted NMA
Comparisons | Mean effectiveness, % (95% CrI) | |
|---|---|---|
Fixed-effect model (base case) | Random effect model | |
HPTN 083 study population (gbMSM and TGW) | ||
Cabotegravir LA vs. placebo or no PrEP | 91.10 (82.87 to 95.95) | 91.14 (81.02 to 96.79) |
HPTN 084 study population (cisgender women) | ||
Cabotegravir LA vs. placebo or no PrEP | 92.52 (83.02 to 97.38) | 92.36 (81.14 to 97.69) |
CrI = credible interval; gbMSM = gay, bisexual, and other men who have sex with men; LA = long acting; NMA = network meta-analysis; PrEP = pre-exposure prophylaxis; TGW = transgender women; vs. = versus.
Source: ITC Technical Report.23
Critical Appraisal of ITC 1
The sponsor conducted a systematic search of published SLRs to inform the indirect comparisons and an additional search of individual RCTs to bridge the gap between search dates of the published SLRs and the time of this submission. Although the results of the literature review were reported, there was no information available on the timing of end point evaluation and censoring rules used when estimating the HR across the studies included in the sponsor-submitted NMA. According to the sponsor, results of the included studies reflect the difference in cumulative HIV incident infections between arms over the duration of the follow-up period and patients were essentially censored at the end of follow-up. The outcome included in the NMA, drug effectiveness in reducing HIV transmission, was relevant to provide the source for key inputs in the pharmacoeconomic model submitted to CDA-AMC. Other outcomes that are of interest to this review, including incidence of HIV infection and AEs, were not assessed.
One study (the DISCOVER trial) that assessed TDF-FTC compared to TAF-FTC, a relevant comparator of cabotegravir LA as per feedback from the clinical expert, was not eligible for inclusion into the NMA. As such, the comparative efficacy of cabotegravir LA and TAF-FTC remains unknown.
An assessment of the degree of heterogeneity between the included studies based on the sponsor-provided technical report was difficult due to limited reporting of study design and patient characteristics. Aside from description of interventions, sample size, country, and duration of follow-up, there is no reporting of other trial characteristics (e.g., the inclusion and exclusion criteria, treatment withdrawal frequency, handling of missing data, and the various censoring rules used when estimating the HR, and so forth) within the report. The sponsor-submitted NMA included 1 study (the Bangkok Tenofovir Study) that focused on PWID; the clinical expert consulted by CDA-AMC confirmed that PWID are relevant to this review as there will be patients in that population who are best served by cabotegravir LA. Of note, the CDA-AMC review team noted that, as per feedback from the clinical expert, the risk profiles of PWID and non-PWID populations are different in terms of socioeconomic risk factors and behaviour, which may result in different risk of HIV acquisition. The CDA-AMC review team considered that these factors increased the heterogeneity of the studies included in the NMA. Additionally, there were differences in study design with respect to blinding, where 1 study was open label, 1 was partially blinded, and 8 were double blind. Although these difference in study design could not be adjusted for in the analysis, these inherent differences suggest that the transitivity assumption of the NMA likely have not been met.
The sponsor considered the between-trial differences in TDF-FTC adherence to be clinically meaningful and could affect the comparability of the studies’ treatment effect within the NMA. The clinical expert consulted by CDA-AMC confirmed that adherence is an effect modifier but pointed out that the real-world adherence level of cabotegravir LA and oral TDF-FTC may not be as optimistic as in trial settings given studies in PrEP follow-up in other countries showed significant loss to follow-up and low adherence after the initial phase of treatment (i.e., after the clinical study period is over). The CDA-AMC review team noted that the meta-regression prediction of TDF-FTC versus placebo or no PrEP relies solely on adherence as a prediction variable. No adjustment was made in the modelling of the NMA to account for the potential interaction between the patient characteristics and treatment efficacy of cabotegravir LA. Additionally, the proposed estimates of efficacy of cabotegravir LA relies on the HPTN 083 and HPTN 084 trials and their reported adherence to cabotegravir LA instead of their adherence to oral TDF-FTC. The efficacy and adherence may be greatly different in practice, which increased uncertainty in the findings.
The NMA was informed by 10 studies, including 2 studies that contributed to the direct estimates of effectiveness of cabotegravir LA versus TDF-FTC, and 8 studies that assessed the comparative efficacy between TDF-FTC and placebo. The resulting networks were sparse with no closed loop; as such, it was not possible to assess consistency of results between direct and indirect comparisons. Results of the NMA were informed solely by indirect evidence, and thus are associated with increased uncertainty.
The NMA was conducted using a Bayesian framework. Fixed-effect models were chosen for the base-case analyses for all assessed outcomes given that random treatment effects models have higher deviance information criteria than the corresponding fixed-effects model indicating that the inclusion of the random treatment effects does not improve the model fit. The fixed-effect model relies on the assumption that there was no between-trial heterogeneity, which unlikely holds true as previously discussed. No definitive conclusions could be reached regarding the effects of cabotegravir LA versus placebo or no PrEP due to considerable uncertainty in the relative treatment effect estimates.
Studies Addressing Gaps in the Systematic Review Evidence
Two studies conducted in adolescent populations were identified for this review.
HPTN 083 to 01 Trial
The HPTN 083 to 01 study is an ongoing open-label, unblinded, single-arm, phase IIb trial designed to assess the safety, tolerability, and acceptability of cabotegravir LA for the prevention of HIV-1 among adolescent males aged < 18 years, with body weight of 35 kg or more. The HPTN 083 to 01 study is a substudy of the HPTN 083 study, which enrolled 9 participants to receive cabotegravir LA. One participant discontinued from study treatment and terminated from the study after 1 injection. The mean age of those enrolled was 16.4 years, most were white (n = 5 [56%]) and Black (n = 3 [33%]).
The oral run-in phase consisted of the administration of 30 mg oral cabotegravir daily as a lead-in for up to 5 weeks and an injection phase consisting of 5 injections of 3 mL (600 mg) of cabotegravir LA administered at 8-week intervals after a loading dose at week 4. All 9 participants reported at least 1 AE in steps 1 and 2 that were grade 2 to 5 (100%), of whom one-third (33%) had AEs that were considered by the investigator to be drug-related; 22% of participants reported AEs in step 1 and 2 that were grade 3 to 5. No participants reported an SAE or AE leading to study drug discontinuation during step 1 and 2. There were no fatal SAEs reported. No new safety findings were identified and ISRs reported were of grade 1 and 2, which did not result in drug discontinuation. Cabotegravir LA injections were well-tolerated in participants. Cabotegravir LA was considered acceptable in 7 of 9 (78%) of participants who entered the injection phase receiving all scheduled injections and 67% of participants who would consider using injectable (with or without condoms) for HIV prevention in the future.
HPTN 084 to 01 Trial
The HPTN 084 to 01 study is an ongoing open-label, unblinded, single-arm, phase IIb trial designed to assess the safety, tolerability, and acceptability of cabotegravir LA for the prevention of HIV-1. The HPTN 084 to 01 study is a substudy of the HPTN 084 study, which enrolled HIV-uninfected at-risk female adolescents aged < 18 years and with a body weight of 35 kg or more. There was no treatment randomization.
The oral run-in phase consisted of the administration of oral cabotegravir 30 mg daily as a lead-in for up to 5 weeks and the injection phase consisted of 5 injections of 3 mL (600 mg) of cabotegravir LA administered at 8-week intervals after a loading dose at week 4. A total of 69 participants were screened, of which 14 were not enrolled. Of the 14 who were not enrolled, 10 failed to meet 1 or more of the inclusion criteria and 4 met 1 or more of the exclusion criteria. A total of 55 participants were enrolled and all 55 received study drug. All participants were Black heterosexual female adolescents (aged 12 years to 17 years). Most participants weighed at least 50 kg. The proportion of participants who discontinued study treatment or terminated study was overall low. The most common reason for treatment discontinuation was AEs. At the time of the data cut-off for this report (July 21, 2022), 15 (27%) participants are ongoing in the study. Approximately half of all participants (58%) had at least 1 protocol deviation during the study, the majority of which had lab assessment deviations. In the HPTN 084 to 01 study, cabotegravir LA resulted in a safety profile consistent with that observed in the HPTN 084 study. No new safety concerns were identified. ISRs reported were grade 1 and 2 and did not result in study drug discontinuations. Cabotegravir LA injections were also well-tolerated with no participant discontinuing treatment prematurely due to intolerability of injection or burden of study procedures. Cabotegravir LA injections were also considered acceptable with 100% of participants who entered the injection phase receiving all scheduled injections and 62% of participants highlighting they would consider using injectable cabotegravir LA (with or without condoms) for HIV prevention in the future. The reported PK concentrations of cabotegravir LA in participants were consistent with the adult population in the HPTN 084 study.
Critical Appraisal
There is uncertainty about whether the sample size and power calculations for both studies were sufficient to assess the efficacy of cabotegravir LA in the 2 studies (total sample size of participants enrolled for the HPTN 083 to 01 study was n = 9 and n = 55 for the HPTN 084 study). The lack of a comparative and the absence of any assessments related to primary efficacy outcomes limited the interpretability of the magnitude of the benefit of cabotegravir LA in reducing HIV-1 infections in adolescent populations. Thus, no definitive conclusions could be drawn; however, no safety signals were identified.
Discussion
Summary of Available Evidence
Two pivotal trials (HPTN 083 and HPTN 084) included in the sponsor’s systematic review provided evidence for this submission.
The HPTN 083 study is an ongoing phase IIb/III, multicentre, double-blind, randomized, noninferiority trial designed to assess the efficacy and safety of cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years and older) cisgender MSM and TGW who have sex with men who were at increased risk of sexual acquisition of HIV infection. A total of 4,570 participants enrolled at 43 study centres in 7 countries (no study sites identified in Canada) were randomized in a 1:1 ratio to receive either treatment. The HPTN 084 study is an ongoing phase III, multicentre, double-blind, randomized, superiority trial designed to evaluate the efficacy and safety of cabotegravir LA compared to oral TDF-FTC for PrEP in HIV-negative adult (aged 18 years to 45 years) cisgender women who were at increased risk of sexual acquisition of HIV infection. A total of 3,224 participants at 20 study sites were randomized in a 1:1 ratio to receive either treatment. Both trials consisted of an oral lead-in phase (step 1) during which oral cabotegravir (30 mg tablets) plus daily oral TDF-FTC placebo or TDF-FTC (300 mg-200 mg tablets) plus daily oral cabotegravir placebo were administered to participants for up to 5 weeks. This was followed by an injection phase (step 2) where cabotegravir LA injection plus daily oral placebo or daily oral TDF-FTC plus placebo intramuscular injection were administered at weeks 5, 9, and every 8 weeks thereafter, and an open-label extension phase (step 3) where open-label cabotegravir LA injection or TDF-FTC was administered. Key primary outcomes investigated were similar in both trials and included documented incidence of HIV infections in steps 1 and 2 and number of participants experiencing grade 2 or higher clinical and laboratory AEs. Other important outcomes assessed included documented incidence of HIV infections in step 2 (the HPTN 083 study only), resistance mutations to study products, adherence to study product during step 2, and patient-reported outcomes such as acceptability scale assessments, and a survey of attitudes and willingness to use cabotegravir LA and TDF-FTC (the HPTN 084 study only).
The treatment groups in both trials were generally well-balanced in baseline characteristics and demographics. The HPTN 083 study enrolled most participants aged 30 years and younger, at least 10% of whom were TGW, and at least 50% of whom were Black gbMSM (in US sites) who were assigned males at birth. The HPTN 084 study enrolled participants who were assigned female at birth. More than 99% were Black and aged younger than 35 years. Overall, both studies were well-conducted. Potential sources of uncertainty for the primary outcome were related to the data presented. Evidence assessed from both trials in this submission was from preplanned interim analyses, which could potentially increase the risk of overestimating treatment effects and introduces a source of uncertainty of the magnitude of efficacy of cabotegravir LA. However, appropriate methods (O’Brien-Fleming boundary) were established to account for alpha spending during each analysis.
Two studies (the HPTN 083 to 01 and HPTN 084 to 01 studies) conducted in adolescent populations were included in the submission. However, due to limited data on efficacy outcomes (incidence of HIV-1 infections), no definitive conclusions could be drawn. Safety data presented from these substudies were consistent with the HPTN 083 and HPTN 084 studies.
One NMA, which evaluated the comparative efficacy of cabotegravir LA versus placebo or no PrEP on effectiveness in reducing HIV transmission in at-risk individuals based on 10 included studies, was conducted by the sponsor in the absence of direct comparative evidence of cabotegravir LA and no use of PrEP.
Interpretation of Results
Efficacy
Input from patient advocacy and clinician groups highlighted the importance of PrEP options that can reduce new HIV infections, reduce drug resistance, sustain viral suppression, and increase comfort level among individuals taking the treatment. In consultation with the clinical expert involved in the CDA-AMC review, the following outcomes assessed in the trials were considered important for decision-making: documented HIV-1 infection, adherence, and resistance mutations among seroconverters. Therefore, results from the analyses of these 3 outcomes were summarized and assessed using GRADE.
The HPTN 083 study provided evidence to support the noninferiority of cabotegravir LA to TDF-FTC in reducing the incidence of HIV-1 infections in HIV-1 negative adult (aged ≥ 18 years) cisgender MSM and TGW who have sex with men who were at increased risk of sexually acquired HIV-1. Findings from the HPTN 084 trial supported the superiority of cabotegravir LA compared to TDF-FTC in reducing the incidence of HIV-1 infections in adult (aged 18 years to 45 years) cisgender women who are negative for HIV-1 and at increased risk of sexually acquired HIV-1. Both trials measured outcomes that were considered important for at-risk populations in Canada.
Evidence from both trials demonstrated that cabotegravir LA was beneficial in reducing the risk of incident HIV-1 infections relative to TDF-FTC in the combined oral and injection phases (steps 1 and 2) of both trials. The primary analyses in the HPTN 083 trial showed a 66.0% reduction in the risk of incident HIV-1 infections after 6,404 PY of accumulated follow-up, and results of the HPTN 084 study revealed an 89% reduction in the risk of incident HIV-1 infections, after 3,907 PY of accumulated follow-up. Findings from the supplementary and sensitivity analyses were also consistent with the primary analyses. The clinical expert consulted during the CDA-AMC review discussed that any treatment in the HIV setting that can substantially reduce the incidence of HIV-1 infections by at least 80% (risk reduction of ≥ 80%) compared to no treatment in persons exposed to HIV-1, is clinically meaningful. Similarly, cabotegravir LA was shown to reduce the risk of incident HIV-1 infections in the injection phase only (step 2) in both trials (the estimated HR in the HPTN 083 and HPTN 084 studies suggested 79% and 94% reduction in the risk of incident HIV-1 infections, respectively, for the cabotegravir LA group relative to the TDF-FTC group). However, the evidence presented from both trials should be interpreted with caution given that both trials were concluded early after stopping rules were met and continued as open label following results from the preplanned interim analyses. Interim analyses could potentially increase the risk of overestimating treatment effects (as only a fraction of the targeted number of events for the final analyses were achieved during the interim analyses for both trials). Thus, it introduces a source of uncertainty of the magnitude of efficacy observed for cabotegravir LA.
Following the GRADE approach, the certainty of evidence from the HPTN 083 and HPTN 084 studies was considered high for the primary outcome despite the potential increased risk of treatment overestimation in the interim analyses as previously highlighted. There were fewer incident HIV-1 infections per 100 PY among participants in the cabotegravir LA group compared to the TDF-FTC group in the combined steps 1 and 2 in both trials (in the HPTN 083 trial, the risk difference observed was 0.82 fewer HIV-1 infections [range, 1.26 to 0.38] and in the HPTN 084 trial, the risk difference observed was 1.65 fewer HIV-1 infections [range, 2.28 to 1]). Similar findings were reported in both trials in the injection phase only, and the certainty of evidence was considered high. The incidence rate of new HIV-1 infections per 100 PY was lower in the cabotegravir LA group compared to the TDF-FTC group. In the HPTN 083 study, the incidence rate was 1 fewer (range, 0.5 to 1.4) and in the HPTN 084 study, the incidence rate was 1.8 fewer (range, 2.5 to 1.16). However, in the absence of a clinically meaningful threshold of important difference, it is unclear whether the observed between-group difference is of clinical importance.
The clinical expert consulted during the CDA-AMC review highlighted the importance of treatment adherence on the efficacy of PrEP options, which aligns with the concerns underscored by current guidelines on HIV management in Canada with regards to current PrEP options. Differences in treatment benefit in the 2 trials may have been influenced by participant adherence in each group. There were notable differences in adherence between the 2 groups reported for both trials. Injection coverage in PY, although similar in the cabotegravir LA group in both trials (91.5% versus 93% PY coverage in the HPTN 083 and HPTN 084 studies, respectively) the plasma concentrations of TDF-FTC differed (concentrations consistent with daily dosing [≥ 40 ng/mL] was 74.3% versus 41.9% in the HPTN 083 and HPTN 084 trials, respectively). Regardless, definitive conclusions could not be drawn from the evidence to support a correlation between adherence and cabotegravir LA benefit for the primary efficacy outcome due to several limitations. Adherence analyses were conducted in a subset sample for each group in both trials and results may not be representative of the entire population enrolled; the small sample sizes in the subset may have resulted in an overestimation of the effect. Worth noting, the clinical expert highlighted that PK measurements of drug levels in participants, although informative in the trial setting, may not be a true representation of treatment adherence (especially for oral TDF-FTC) due to existing biological variability in drug metabolism between individuals across populations. The expert also noted that low levels of drug concentrations at any given moment may not necessarily translate to drug inefficacy in preventing HIV acquisition.
Participants who were seroconverters to HIV-1 within the 2 treatment groups in both trials were tested retrospectively for potential resistance mutations. There were no new signals on resistance mutations identified that may impact subsequent treatment selection for seroconverters for both drugs, according to the expert consulted during the review. Key integrase resistance mutations to cabotegravir LA were identified in some participants receiving cabotegravir LA who tested positive for HIV-1 during treatment. Other mutations detected included NNRTIs and NRTIs, which do not contribute to cabotegravir LA resistance. In the HPTN 083 trial, integrase resistance and NNRTI and NRTI mutations were identified in participants receiving cabotegravir LA, and in the HPTN 084 study, integrase mutations (e.g., L74l, which is considered a polymorphism, and also detected in participants in the TDF-FTC group), were observed. For participants in the TDF-FTC group, NNRTI resistance mutations (K103N/S, Y181C, G190A/S, H221Y, and P225H), PI resistance mutations, and NRTI resistance mutations were observed in the HPTN 083 study. In the HPTN 084 study, mutations reported in participants in the TDF-FTC group who tested positive for HIV-1 included major NRTI mutation (M184V) and NNRTI resistance (6 had K103N, alone or with E138A or P225H; 1 had K101E alone; 1 had E138A alone), INSTI mutations or polymorphisms, and dual-class resistance (i.e., NRTI and NNRTI). The clinical expert consulted during the CDA-AMC review considered the evidence related to resistance patterns among seroconverters important as it will guide decision-making processes in practice regarding the selection of subsequent therapy for individuals who contract HIV-1 while receiving cabotegravir LA as PrEP.
Evidence gaps identified included data on the long-term benefits and harmful effects of cabotegravir LA in the enrolled population beyond each trial’s duration. The HPTN 08317 and HPTN 08418 trial designs included an open-extension phase, which will allow participants to choose between cabotegravir LA or TDF-FTC. The open-label phase is currently ongoing, with no additional information on key primary outcomes or completion date available at the time of this review. The sponsor has provided evidence on the safety of cabotegravir LA in persons aged younger than 18 years who were cisgender women or cisgender MSM and TGW who have sex with men. Given that both studies had a small sample size, lacked a comparative arm, and that efficacy outcomes were not assessed, no definitive conclusions could be drawn on the benefit of cabotegravir LA in reducing HIV-1 infections in adolescent populations; however, no safety signals were identified. Further, the clinical expert consulted by CDA-AMC did not have any concerns about safety or efficacy when generalizing the results of PrEP in adults to adolescents who are at risk of sexually acquired HIV-1 infection. Subgroup analyses conducted in both trials showed benefit of cabotegravir LA compared to TDF-FTC in the subgroups of interest, including age, gender, race, body mass index, ethnicity, and region in both trials. However, these analyses were considered exploratory by the CDA-AMC review team as neither study was powered for any of the individual subgroup evaluations and no adjustments were made for multiple testing subgroup analyses, and as such, no conclusions could be drawn.
The results of the sponsor-submitted NMA agreed with the trial comparison that cabotegravir LA was associated with improved effectiveness in reducing HIV transmission compared with TDF-FTC and placebo or no PrEP. However, the validity of the results of the NMA are uncertain because of important limitations preventing assessment of the key assumptions for the analyses. Because of limited available studies, the NMA included only 3 interventions, with cabotegravir LA connected with placebo or no PrEP through TDF-FTC as the central connection in the linear network. It is not possible to evaluate consistency between direct and indirect comparisons in networks with this geometry (a sparse network with no closed loop). As well, the limited number of studies and limited reporting of study characteristics (including baseline patient characteristics) in the technical report hampered the assessment homogeneity. Of the available information, there appeared to be important sources of heterogeneity in study designs and patient populations between studies that were unaccounted for and differences between adherence in real-world and trial settings, which result in increased uncertainty in the comparative treatment effect estimates. Therefore, no concrete conclusions could be drawn on the comparative effectiveness and safety of cabotegravir LA and placebo or no PrEP.
Further, direct or indirect evidence between cabotegravir LA and TAF-FTC is absent in the submission. The sponsor suggested that TAF-FTC is not an appropriate comparator on the basis that TAF-FTC is only listed on the Canadian Armed Forces Drug Benefit List under a special authorization criterion where there is a contraindication to the use of TDF-FTC and its generic versions and TAF-FTC is not listed for PrEP on any provincial drug plan. Additionally, the sponsor stated that there was only 1 trial, the DISCOVER study, that evaluated the used of TAF-FTC as PrEP; however, the trial did not report adherence data based on plasma samples, and as such it is not possible to incorporate these data into the analysis as these measures are not comparable to other studies included in the NMA. Further, the composition of the DISCOVER study participants in some key populations of interest including MSM of colour and TGW differed when compared to other PrEP trials included in the NMA. Therefore, the sponsor concluded that the comparison of cabotegravir LA and TAF-FTC was inadvisable. The clinical expert noted that TAF-FTC was a relevant comparator to cabotegravir LA. The CDA-AMC review team acknowledges that there are practical considerations that guide treatment choice; however, the lack of comparative evidence between cabotegravir LA and TAF-FTC represents a gap in evidence given the shared place in therapy for PrEP.
Harms
The proportion of participants reporting at least 1 AE was similar across groups in both trials (95% versus 94% in the cabotegravir LA and TDF-FTC groups, respectively, in the HPTN 083 study and 96% in each group in the HPTN 084 study). The most common AEs reported in both trials were injection site pain, creatinine clearance decreased, blood creatine phosphokinase increased, blood creatinine increased, and nasopharyngitis.
SAEs were reported in 5% of participants in each group in the HPTN 083 trial, and 2% in each group in the HPTN 084 trial. There were 4 deaths in the cabotegravir LA group and 6 in the TDF-FTC group in the HPTN 083 study compared to 3 in the cabotegravir LA group versus 0 in the TDF-FTC group in the HPTN 084 study. ISRs are very common for drugs with intramuscular routes of administration, which may impact treatment discontinuation in a clinical setting, according to the expert CDA-AMC consulted. In the HPTN 083 trial, at least 76% and 32% of participants in the cabotegravir LA and TDF-FTC groups, respectively, reportedly experienced ISRs compared to at least 38% and 11% of participants the cabotegravir LA and TDF-FTC groups, respectively, in step 2 of the HPTN 084 trial. However, the clinical expert noted that ISR events are manageable with no meaningful consequences to individuals, and they tend to tolerate injections over the course of treatment. The expert noted that patients may value intramuscular injections over taking oral pills daily differently based on perceived convenience.
The clinical expert considered the safety profile of cabotegravir LA manageable with no new safety signals identified. The NMA did not assess any harms for cabotegravir LA relative to other treatments that may impact cabotegravir LA’s safety profile.
Conclusion
Evidence from 2 pivotal, multicentre, double-blind RCTs (the HPTN 083 and HPTN 084 studies) demonstrated the efficacy and safety of cabotegravir LA administered by intramuscular injection compared to oral TDF-FTC for PrEP in adult (aged 18 years and older) cisgender MSM and TGW who have sex with men (the HPTN 083 study) and cisgender women negative for HIV-1 (the HPTN 084 study). Noninferiority and superiority of cabotegravir LA to TDF-FTC was demonstrated in the HPTN 083 trial and superiority of cabotegravir LA to TDF-FTC was demonstrated in the HPTN 084 trial.
The totality of evidence from the interim analyses of both trials suggests that cabotegravir LA reduces the incidence of HIV-1 infections in participants at risk of sexually acquired HIV-1 infection compared to oral TDF-FTC. The certainty of evidence was considered high in both trials, with a reported risk difference in favour of cabotegravir LA for participants receiving drug during the combined oral lead-in and injection phases and injection phase only. However, it is unclear whether the observed between-group differences are of clinical importance. Adherence assessments reported in a subset of participants in each treatment group in both studies showed higher coverage for cabotegravir LA injections compared to drug concentrations of TDF-FTC consistent with daily dosing and could be the driving factor for the observed reduction in HIV acquisition risk. Key integrase resistance mutations to cabotegravir LA were identified in some participants receiving cabotegravir LA who tested positive for HIV-1 during treatment. Other mutations detected included those to NNRTIs and NRTIs, which do not contribute to cabotegravir LA resistance. According to the expert, mutations may impact the subsequent choice of treatment for HIV. The safety profile of cabotegravir LA observed in both trials was considered manageable with no new safety signals identified.
Evidence from the sponsor-conducted NMA on the comparative effectiveness suggests benefits of cabotegravir LA over placebo and no PrEP in reducing HIV-1 infections. Data were lacking for TAF-FTC, another treatment available to eligible populations in Canada. The sponsor’s NMA did not assess harms that may impact the safety profile of cabotegravir LA. Overall, there is uncertainty in the NMA findings due to several limitations preventing the assessment of key assumptions of the analyses. Evidence of long-term safety and efficacy beyond the pivotal trials was not available for this review; however, long-term extension studies for both trials are currently ongoing with no expected completion date at the time of this review. Data on adolescent populations was lacking; however, according to the clinical expert CDA-AMC consulted, the findings observed in the adults will be generalizable to adolescents.
References
1.Tan DHS, Hull MW, Yoong D, et al. Canadian guideline on HIV pre-exposure prophylaxis and nonoccupational postexposure prophylaxis. CMAJ. 2017;189(47):E1448-E1458. PubMed
2.Health Canada clarimail: (cabotegravir tablets and extended-release injectable solution) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare ULC; 2023 Dec 5.
3.ViiV Healthcare ULC response to 2023 Nov 21 Health Canada clarimail regarding cabotegravir tablets and extended-release injectable solution CADTH review [internal additional sponsor's information]. Montreal (QC): ViiV Healthcare; 2023.
4.Pantaleo G, Graziosi C, Fauci AS. The immunopathogenesis of human immunodeficiency virus infection. N Engl J Med. 1993;328(5):327-335. PubMed
5.Wingfield T, Wilkins E. Opportunistic infections in HIV disease. Br J Nurs. 2010;19(10):621-627. PubMed
6.Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041. PubMed
7.Public Health Agency of Canada. Estimates of HIV incidence, prevalence and Canada's progress on meeting the 90-90-90 HIV targets, 2020. 2022; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/estimates-hiv-incidence-prevalence-canada-meeting-90-90-90-targets-2020.html. Accessed 2024 Apr 1.
8.Public Health Agency of Canada. HIV in Canada: 2022 surveillance highlights 2023; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/hiv-2022-surveillance-highlights.html. Accessed 2024 Apr 1.
9.Ontario HIV Treatment Network. HIV pre-exposure prophylaxis (PrEP) in Ontario, 2020. 2022; https://www.ohtn.on.ca/hiv-pre-exposure-prophylaxis-prep-in-ontario-2020, 2024 Apr 1.
10.Public Health Agency of Canada. Trends in Pre-Exposure Prophylaxis (PrEP) use in 9 Canadian provinces-2019–2022 (infographic). 2023; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/trends-pre-exposure-prophylaxis-use-in-9-canadian-provinces-2019-2022-infographic.html. Accessed 2024 Apr 1.
11.Cabotegravir: 30 mg cabotegravir (as cabotegravir sodium) oral tablets and cabotegravir extended-release injectable suspension: 200 mg cabotegravir/mL (600 mg/3mL) [product monograph]. Montreal (QC): ViiV Healthcare ULC; 2024 May 10.
12.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587-2599. PubMed
13.Molina J-M, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373(23):2237-2246. PubMed
14.McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387(10013):53-60. PubMed
15.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
16.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
17.Clinical Study Report: HPTN 083. A phase IIb/III double blind safety and efficacy study of injectable cabotegravir compared to daily oral tenofovir disoproxil fumarate/emtricitabine (TDF/FTC), for pre-exposure prophylaxis in HIV uninfected cisgender men and transgender women who have sex with men [internal sponsor's report]. Montreal (QC): ViiV Healthcare ULC; 2021.
18.Clinical Study Report: HPTN 084. A phase 3 double blind safety and efficacy study of long-acting injectable cabotegravir compared to daily oral TDF/FTC for pre-exposure prophylaxis in HIV-uninfected Women (HPTN 084)[internal sponsor's report]. Montreal (QC): ViiV Healthcare ULC; 2021.
19.Briongos Figuero LS, Bachiller Luque P, Palacios Martin T, Gonzalez Sagrado M, Eiros Bouza JM. Assessment of factors influencing health-related quality of life in HIV-infected patients. HIV Med. 2011;12(1):22-30. PubMed
20.Miners A, Phillips A, Kreif N, et al. Health-related quality-of-life of people with HIV in the era of combination antiretroviral treatment: a cross-sectional comparison with the general population. Lancet HIV. 2014;1(1):e32-40. PubMed
21.CADTH reimbursement review: fostemsavir (Rukobia) for human immunodeficiency virus type 1. Can J Health Technol. 2023;3(7).
22.CADTH common drug review. Clinical review report: cabotegravir tablets, cabotegravir extended-release injectable suspension, and rilpivirine extended-release injectable suspension: vocabria (Cabenuva - ViiV Healthcare ULC). Ottawa (ON): CADTH; 2020.
23.Indirect Treatment Comparison of the effectiveness of cabotegravir long acting (CAB LA) for PrEP with no pre-exposure prophylaxis option (no PrEP) for HIV prevention technical report Version 4 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare ULC; 2024 Jan 19.
24.CATIE. HIV testing and diagnosis. 2022; https://www.catie.ca/hiv-testing-technologies. Accessed 2024 Apr 1.
25.Bureau de normalisation du Quebec. Evaluation de laboratoires. https://bnq.qc.ca/fr/laboratoires.html. Accessed 2024 Apr 1.
26.Accredidation Canada Diagnostics. Accredited facilities. 2024; https://diagnostics.accreditation.ca/Diagnostic-Assessment-Programs/Medical-Laboratory-and-Diagnostic-Imaging/Accredited-Facilities. Accessed 2024 Apr 1.
27.Standards Council of Canada. SSC accreditation program for medical testing laboratories. 2024; https://www.scc.ca/en/accreditation/programs/medical-labs. Accessed 2024 Apr 1.
28.Drug Reimbursement Review clinical guidance report: Cabotegravir tablets (Vocabria) in combination with rilpivirine tablets (Edurant) for the treatment of HIV-1 infection in adults who are virologically suppressed. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0628-vocabria%2Bcabenuva-clinical-review-report.pdf. Accessed 2024 April 1.
29.Center for Drug Evaluation Research. Approval letter. Cabotegravir 600 mg/3 ml. Company: ViiV Healthcare ULC. Application No.:215499. Approval date: 12/20/2021 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2021: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/215499Orig1s000Approv.pdf. Accessed 2024 April 1.
30.Committee for Medicinal Products for Human Use. Assessment report: Apretude (cabotegravir). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2023: https://www.ema.europa.eu/en/documents/overview/apretude-epar-medicine-overview_en.pdf. Accessed 2024 April 1.
31.Truvada (emtricitabine/tenofovir disoproxil fumarate): 200 mg/300 mg oral tablets [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc; 2018.
32.Descovy (emtricitabine/tenofovir alafenamide tablets): 200 mg emtricitabine/ 10 mg and 25 mg tenofovir alafenamide, oral [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc; 2016.
33.Landovitz RJ, Donnell D, Clement ME, et al. Cabotegravir for HIV prevention in cisgender men and transgender women. N Engl J Med. 2021;385(7):595-608. PubMed
34.Marzinke MA, Fogel JM, Wang Z, et al. Extended analysis of HIV infection in cisgender men and transgender women who have sex with men receiving injectable cabotegravir for HIV prevention: HPTN 083. Antimicrob Agents Chemother. 2023;67(4):e0005323. PubMed
35.Marzinke MA, Grinsztejn B, Fogel JM, et al. Characterization of Human Immunodeficiency Virus (HIV) infection in cisgender men and transgender women who have sex with men receiving injectable cabotegravir for HIV prevention: HPTN 083. J Infect Dis. 2021;224(9):1581-1592. PubMed
36.Marzinke MA, Hanscom B, Wang Z, et al. Efficacy, safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir for HIV pre-exposure prophylaxis in transgender women: a secondary analysis of the HPTN 083 trial. Lancet HIV. 2023;10(11):e703-e712. PubMed
37.Landovitz RJ, Hanscom BS, Clement ME, et al. Efficacy and safety of long-acting cabotegravir compared with daily oral tenofovir disoproxil fumarate plus emtricitabine to prevent HIV infection in cisgender men and transgender women who have sex with men 1 year after study unblinding: a secondary analysis of the phase 2b and 3 HPTN 083 randomised controlled trial. Lancet HIV. 2023;10(12):e767-e778. PubMed
38.Delany-Moretlwe S, Hughes JP, Bock P, et al. Cabotegravir for the prevention of HIV-1 in women: results from HPTN 084, a phase 3, randomised clinical trial. Lancet. 2022;399(10337):1779-1789. PubMed
39.Eshleman SH, Fogel JM, Piwowar-Manning E, et al. The Levi syndrome: characteristics of early HIV Infection with cabotegravir for PrEP. Abstract #160. Presented at CROI, February 19-22, 2023, Seattle, Washington.
40.Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension [internal sponsor's package]. Montreal, Quebec: ViiV Healthcare ULC; 2024 Feb 1.
41.Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367(5):423-434. PubMed
42.Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367(5):399-410. PubMed
43.Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372(6):509-518. PubMed
44.Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2013;381(9883):2083-2090. PubMed
45.Cain LE, Cole SR. Inverse probability-of-censoring weights for the correction of time-varying noncompliance in the effect of randomized highly active antiretroviral therapy on incident AIDS or death. Stat Med. 2009;28(12):1725-1738. PubMed
46.Emerson SS, Fleming TR. Symmetric group sequential test designs. Biometrics. 1989;45(3):905-923. PubMed
47.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-1187. PubMed
48.Alexander TS. Human immunodeficiency virus diagnostic testing: 30 years of evolution. Clin Vaccine Immunol. 2016;23(4):249-253. PubMed
49.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
50.Huic M, Reinsperger I. Oral and parenteral preexposure prophylaxis (PrEP) to prevent HIV in people at risk. HTA Austria – Austrian Institute for Health Technology Assessment GmbH; 2023. Report No.: 152.
51.Eamon O Murchu, Marshall L, Teljeur C, et al. Oral pre-exposure prophylaxis (PrEP) to prevent HIV: a systematic review and meta-analysis of clinical effectiveness, safety, adherence and risk compensation in all populations. BMJ Open. 2022;12(5):e048478. PubMed
52.World Health Organization. Guidelines on long-acting injectable cabotegravir for HIV prevention. 2022; https://www.who.int/publications/i/item/9789240054097. Accessed 2024 Apr 1.
53.Systematic reviews York (GB): Centre for Reviews and Dissemination, University of York; 2009.
54.The efficacy of and adherence to HIV pre-exposure prophylaxis systematic literature review report version 3 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare ULC; 2023.
55.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. London (GB): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/pubmed/27466657. Accessed 2023 Feb 8.
56.Brooks SP, Gelman A. General methods for monitoring convergence of iterative simulations. J Comput Graph Stat. 1998;7(4):434-455.
57.Hanscom B, Hughes JP, Williamson BD, Donnell D. Adaptive non-inferiority margins under observable non-constancy. Stat Methods Med Res. 2019;28(10-11):3318-3332. PubMed
58.Parienti JJ. On-demand PrEP efficacy: forgiveness or timely dosing. Lancet HIV. 2020;7(2):e79-e80. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 5: Cumulative Rates of Acquired HIV-1 Infections by Group in HPTN 083 (mITT Population)
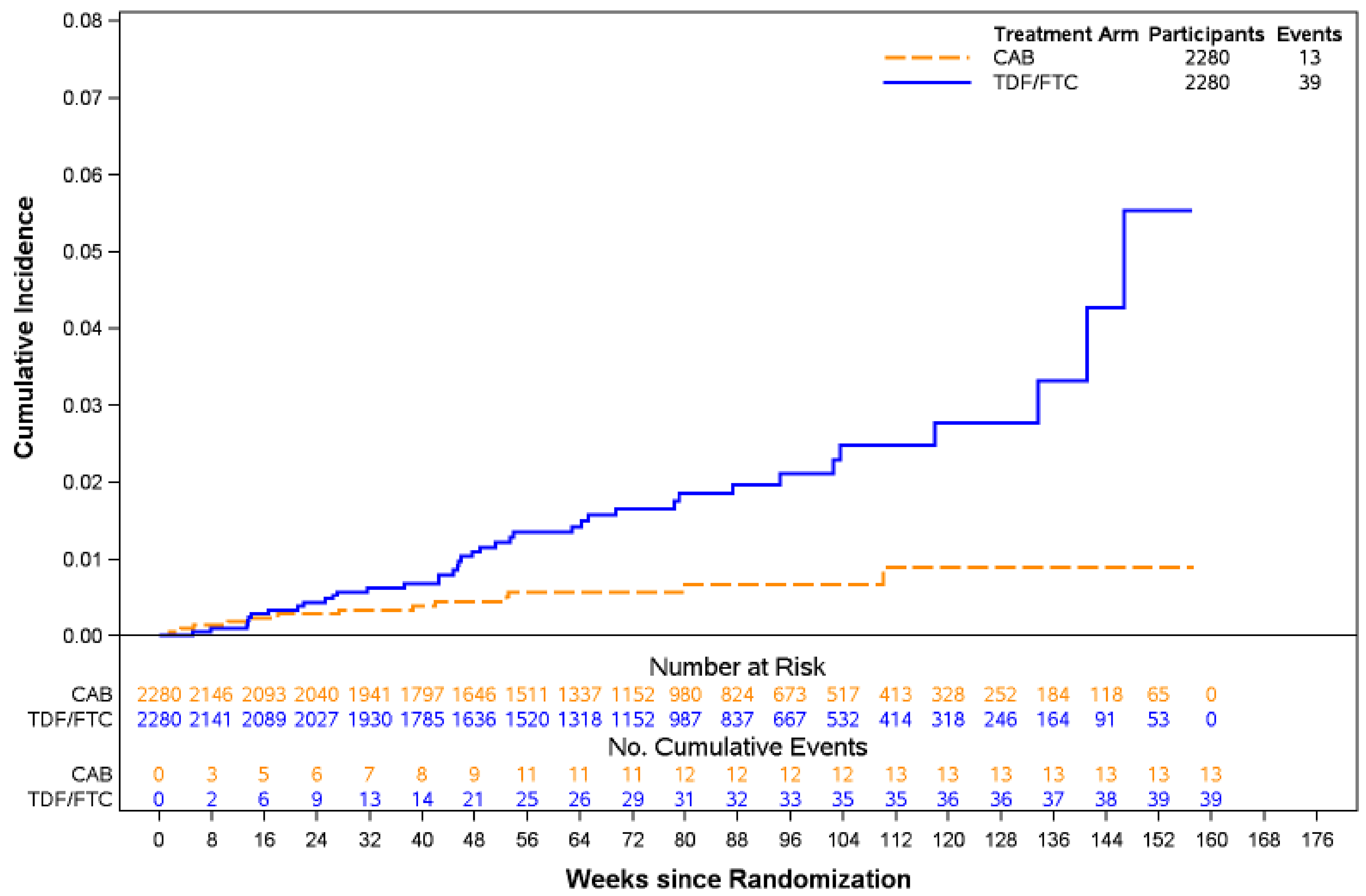
CAB = cabotegravir; mITT = modified intent to treat; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine.
Data cut-off date: May 14, 2020.
Source: HPTN 083 Clinical Study Report.17
Table 25: Time to HIV-1 Infection in Step 2 for the HPTN 083 Study (Efficacy Population OBSP) — Cox Proportional Hazards Regression Model
Category | Cabotegravir LA n = 2,109 | TDF-FTC n = 2,069 |
|---|---|---|
Number of HIV infections | 4 | 24 |
Hazard ratio (95% CI)a | 0.164 (0.06, 0.47) | |
Superiority P value | 0.0008 | |
PY | 2,459 | 2,445 |
Incidence rate (95% CIb) (per 100 PY) | 0.16 (0.04 to 0.42) | 0.98 (0.63 to 1.46) |
CI = confidence interval; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: The P values are two-sided.
aHazard ratio < 1.0 indicates a lower risk on cabotegravir LA as compared to TDF-FTC. The hazard ratio is based on a Cox proportional hazards model stratified by region.
bThe 95% CI for incidence rate was calculated using the exact Poisson method.
Source: HPTN 083 Clinical Study Report.17
Figure 6: Cumulative Rates of Acquired HIV-1 Infections by Group in HPTN 084 (mITT Population)
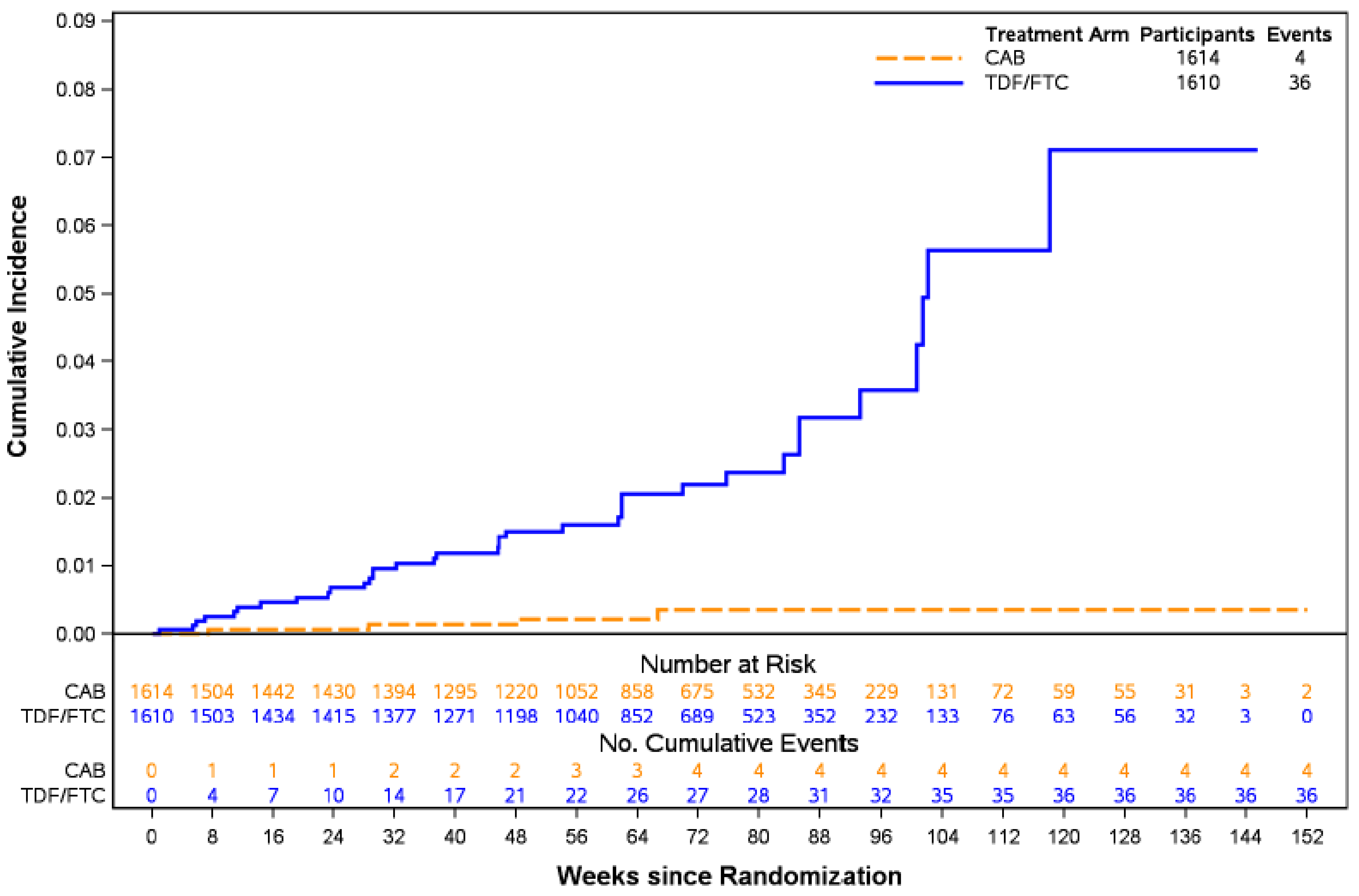
CAB = cabotegravir; mITT = modified intent to treat; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine.
Data cut-off: November 5, 2020.
Source: HPTN 084 Clinical Study Report.18
Table 26: Time to HIV-1 Infection in the HPTN 084 Study (Injection Step 2 Efficacy Population OBSP) — Cox Proportional Hazards Regression Model
Category | Cabotegravir LA n = 1,495 | TDF-FTC n = 1,494 |
|---|---|---|
Number of HIV infections | 1b | 20 |
Hazard ratio (95% CI)a | 0.05 (0.01, 0.37) | |
Superiority P value | 0.0034 | |
PY | 1,413 | 1,431 |
Incidence rate (95% CIc) (per 100 PY) | 0.07 (0.00 to 0.39) | 1.40 (0.85 to 2.16) |
CI = confidence interval; PY = person-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Note: The P values are two-sided.
aHazard ratio < 1.0 indicates a lower risk on cabotegravir LA as compared to TDF-FTC. The hazard ratio is based on a Cox proportional hazards model stratified by site.
bThe analysis with OBSP censoring did not count 1 of the 2 total participants who seroconverted during step 2; 1 participant had several delayed injections outside of the protocol allowance windows, and the OBSP analysis follow-up time was censored at the last nondelayed injection, resulting in the seroconversion event for this patient not being counted in the OBSP analysis.
cThe 95% CI for incidence rate was calculated using the exact Poisson method.
Source: HPTN 084 Clinical Study Report18
Figure 7: Cumulative Rates of Acquired HIV-1 Infections With OBSP Censoring by Group in the HPTN 084 Study (Injection Step 2 Efficacy Population)
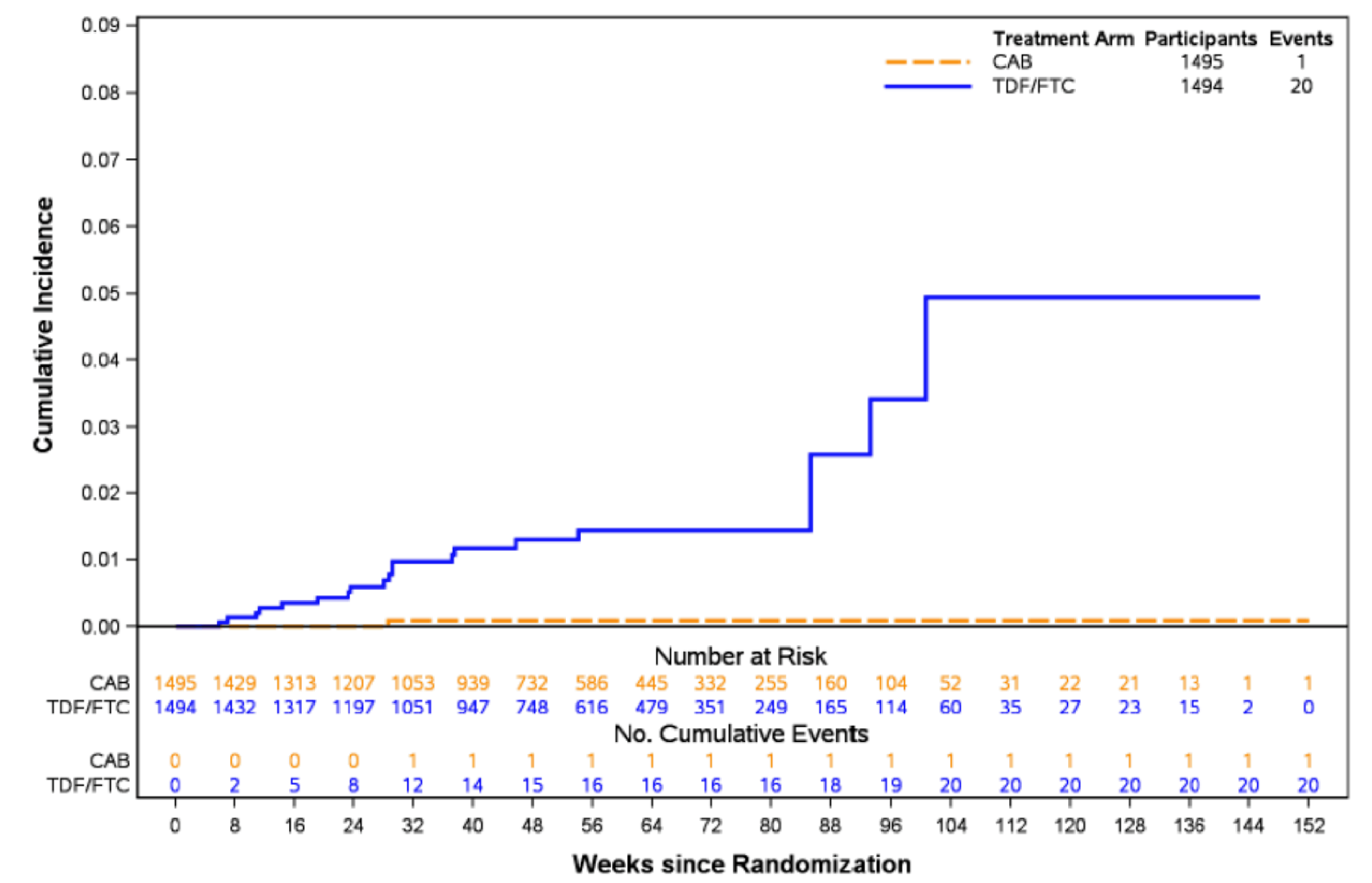
CAB = cabotegravir LA; mITT = modified intent to treat; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: HPTN 084 Clinical Study Report.18
Figure 8: Cumulative Rates of Acquired HIV-1 Infections With OBSP Censoring by Group in HPTN 083 (Injection Step 2 Efficacy Population)
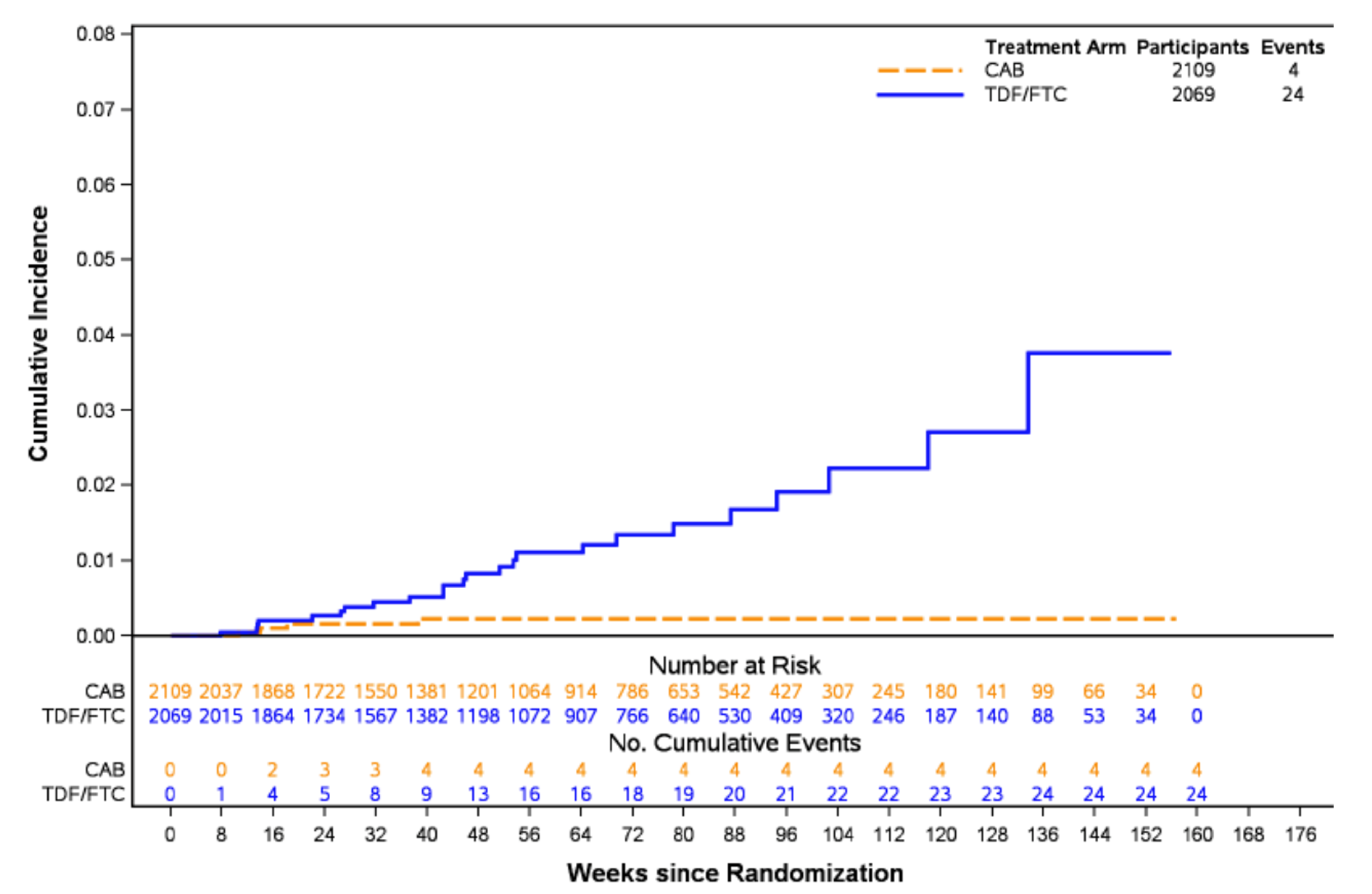
CAB = cabotegravir; mITT = modified intent to treat; TDF/FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: HPTN 083 Clinical Study Report.17
Table 27: Risk of Bias Summary of Studies Included in the NMA
Study | Randomization | Allocation concealment | Similar baseline | Dropouts | Outcome reporting | Outcome reporting | Intention to treat | Declarations |
|---|---|---|---|---|---|---|---|---|
HPTN 083 | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
iPrEx | Yes | Unsure | Yes | Unsure | No | No | Yes | Yes |
PROUD | Yes | No | Yes | No | No | No | Yes | Yes |
IPERGAY | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
HPTN 084 | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
FEM-PrEP | Unsure | Yes | Yes | Yes | No | No | Unsure | Yes |
TENOFOVIR2 | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
VOICE | Unsure | Unsure | Yes | Unsure | No | No | Yes | Yes |
Partners PrEP Study | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
Partners PrEP Study Continuation | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
Bangkok Tenofovir Study | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
Source: Systematic Literature Review Technical Report.54
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CrI
credible interval
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LA
long-acting
LY
life-year
MSM
men who have sex with men
NMA
network meta-analysis
PrEP
pre-exposure prophylaxis
QALY
quality-adjusted life-year
TAF-FTC
tenofovir alafenamide fumarate-emtricitabine
TDF-FTC
tenofovir disoproxil fumarate-emtricitabine
TGW
transgender women
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Cabotegravir (Apretude), 30 mg tablet or 200 mg/mL extended-release injectable suspension |
Indication | Cabotegravir is indicated for at-risk adults and adolescents aged 12 years and older and weighing at least 35 kg for PrEP to reduce the risk of sexually acquired HIV-1 infection. |
Health Canada approval status | Approved |
Health Canada review pathway | Priority review |
NOC date | May 10, 2024 |
Reimbursement request | As per indication |
Sponsor | ViiV Health care ULC |
Submission history | Previously reviewed: State: Yes Indication: Cabotegravir in combination with rilpivirine as a complete regimen for short-term treatment of HIV-1 infection in adults who are virologically stable and suppressed (HIV-1 ribonucleic acid < 50 copies/mL) Recommendation date: July 22, 2020 Recommendation: Reimburse with clinical criteria and/or conditions |
NOC = Notice of Compliance; PrEP = pre-exposure prophylaxis.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults at increased risk (men who have sex with men, transgender women, and cisgender women) of acquiring HIV-1 infection and eligible to receive PrEP |
Treatment | Cabotegravir |
Dose regimen |
|
Submitted price | Cabotegravir 30 mg tablet: $30.08 per tablet Cabotegravir 600 mg/3 mL extended-release injectable solution: $1,710 per vial |
Submitted treatment cost | Year 1: $11,252 (with oral lead-in), year 2 and thereafter: $10,260; $10,260 per year without oral lead-in |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (100 years) |
Key data sources | Clinical trials: HPTN 083 and HPTN 084 |
Submitted results | Cabotegravir was less costly and more effective than TDF-FTC (incremental cost savings = $17,481; incremental QALYs = 0.19) and no PrEP (incremental cost savings = $86,835; incremental QALYs = 0.57). |
Key limitations |
|
CDA-AMC reanalysis results |
|
ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; PrEP = pre-exposure prophylaxis; QALY = quality-adjusted life-year; TAF-FTC = tenofovir alafenamide fumarate-emtricitabine; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Conclusions
The CDA-AMC clinical review of the sponsor-submitted network meta-analysis (NMA) found that cabotegravir long acting (LA) provided a benefit compared to tenofovir disoproxil fumarate-emtricitabine (TDF-FTC) or no pre-exposure prophylaxis (PrEP) in reducing the risk of sexually acquired HIV infection in adults who are considered high risk and weigh at least 35 kg. Findings from the direct comparison with TDF-FTC were consistent with those obtained in the sponsor-submitted NMA. However, considerable uncertainty was observed in the NMA from wide credible intervals (CrIs). Meanwhile, the CDA-AMC clinical review could not reach a definitive conclusion on the relative effectiveness of cabotegravir LA compared with no PrEP. The uncertainty around relative efficacy contributes uncertainty to the economic evaluation.
CDA-AMC identified several additional limitations with the sponsor’s submitted economic evaluation. These involved the source of evidence used for the baseline rate of HIV infection, the exclusion of treatment administration costs, the consideration of spillover effects (which lacked face validity), and an approach to the characterization of parameter uncertainty that was inconsistent with recommended practice. In addition, the sponsor assumed that adherence would act as an effect modifier for oral PrEP but not injectable forms. Clinical experts consulted by CDA-AMC confirmed that the effectiveness of an injectable, as with an oral tablet, would depend on the level of adherence to the prescribed regimen. These limitations resulted in an economic evaluation that may have overestimated the incremental quality-adjusted life-year (QALY) gains and cost savings of cabotegravir relative to oral PrEP, introducing a bias that favoured cabotegravir. CDA-AMC attempted to address some of these limitations through reanalysis. Modifications were made to the submitted model to incorporate treatment administration costs, remove spillover effects, change the source of the baseline HIV incidence rate, and ensure the effectiveness of cabotegravir LA and TDF-FTC take a consistent approach to the effect modification from treatment adherence.
At the sponsor’s submitted price, results from the CDA-AMC base case indicated that cabotegravir LA and TDF-FTC were the only comparators on the efficiency frontier. Cabotegravir LA was more costly and more effective than TDF-FTC, with an estimated incremental cost-effectiveness ratio (ICER) of $29,283 per QALY gained. At a threshold of $50,000 per QALY gained, a price reduction was not necessary to achieve cost-effectiveness. The cost-effectiveness results were highly sensitive to the adherence assumption. If adherence to cabotegravir LA is higher than adherence to oral PrEP, the cost-effectiveness of cabotegravir is likely to be more favourable. Conversely, if patients prefer oral PrEP to cabotegravir LA, there may be insufficient evidence to justify the increased drug cost compared to oral PrEP.
The drug acquisition cost and total treatment cost of cabotegravir LA is greater than the drug acquisition cost and total treatment cost of TDF-FTC. The CDA-AMC reanalysis suggests that cabotegravir LA is associated with increased QALYs compared to TDF-FTC. The changes made to derive the CDA-AMC base case revealed greater uncertainty regarding cabotegravir’s status as the optimal cost-effective alternative. However, the results from the CDA-AMC base case do not fully represent the uncertainty in the evidence base used to estimate the costs and QALYs of each treatment. There remains unmeasured uncertainty, particularly for the relative effectiveness estimates from the submitted indirect treatment comparison (ITC), which may further influence the results. CDA-AMC expects that a properly specified analysis would still lead to the conclusion that treatment with cabotegravir LA is more costly and more effective than treatment with TDF-FTC.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from 5 groups. This included the HIV Network of Edmonton Society, Africans in Partnership Against AIDS (Toronto), CATIE, Community-Based Research Centre (British Columbia), and Peer Outreach Support Services and Education. Information from these submissions was obtained from the director of the organization or individuals living with HIV. One submission (HIV Network of Edmonton Society) conducted interviews and focus groups with people living with HIV. Meanwhile, data from the Community-Based Research Centre were collected using their Sex Now survey, which is Canada’s largest and longest-running survey of gay, bisexual, queer men (cisgender and transgender), nonbinary, and Two-Spirit people’s health. In the 2021 Sex Now survey, only 17% of HIV-negative 2SLGBTQ+ people were using HIV PrEP and only 26% had ever used PrEP. Some responses indicated that an injectable PrEP option may be preferable because its administration can be more discrete than taking a once-daily pill. This may be preferrable to avoid a stigma associated with the alternatives used as treatment for HIV. One submission reported knowledge of patients who had travelled to another country to get access to cabotegravir LA. These individuals appreciated the infrequent basis of treatment administration. Apart from temporary pain and injection site reactions, side effects were not reported.
Clinician input was received from the Vancouver Coastal Health Regional HIV Program, 2 infectious disease specialists practising in Ontario and Alberta, and an HIV pharmacist. A consensus was that the current pathway of care was the administration of PrEP for individuals considered to be high risk for HIV-1 acquisition. This can include individuals who are in partnership with someone with HIV, men who have sex with men (MSM), and people who inject drugs, among others. Oral PrEP is viewed as an important component of HIV prevention for individuals who can adhere to a regimen of daily oral medication. There was agreement among all submissions that an injectable, LA PrEP using cabotegravir would add another effective and appropriate tool to prevent HIV acquisition. In addition, clinical input suggested it may be preferable to oral PrEP among individuals who have trouble adhering to daily oral PrEP. The input suggested that the LA formulation can improve adherence, which is a key component for the efficacy of any PrEP intervention.
Drug plan input raised concerns regarding the anticipated budget impact. This was attributed to the sponsor’s submitted budget impact analysis (BIA), which suggested a 3-year net budget impact of $72,699,154. In addition, the drug plans noted that existing oral PrEP options were subject to confidential negotiated prices.
The sponsor’s model directly linked the effectiveness of oral PrEP to the degree of adherence to the daily medication. However, the submitted model applied this assumption to oral PrEP but not an injectable such as cabotegravir.
Economic Review
The current review is for cabotegravir (Apretude) LA for PrEP treatment to reduce the risk of sexually acquired HIV-1 infection among at-risk adults weighing at least 35 kg.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing cabotegravir LA to TDF-FTC or no PrEP. The model population comprised adults weighing at least 35 kg considered to be at high risk of being infected with HIV-1 by sexual transmission. This population comprised MSM, transgender women (TGW), and cisgender women.1 The target population was aligned with the Health Canada–approved indication and reimbursement request.
Cabotegravir LA is available as a 30 mg tablet or 200 mg/mL solution for injection. The submitted price was $30.08 per tablet and $1,710 per 3 mL vial. For the indicated population, the recommended dosage is an initiation injection of 1 3 mL vial, repeated 1 month later, and then followed by continuation injections of 1 3 mL vial every 2 months thereafter. Tablets may be used as an oral lead-in to assess tolerability before the initiation of injections or as a short-term option for patients who will miss planned dosing. The recommended dose for the oral lead-in is a single 30 mg tablet daily for 28 days, with the initiation of injections within 3 days of completing the oral lead-in. The sponsor calculated that cabotegravir LA would cost $11,252 ($938 per month) in the first year of treatment and $10,260 ($855 per month) in every subsequent year.1
Two comparators were considered in the submitted base case: TDF-FTC or no PrEP. TDF-FTC is available as a 300 mg-200 mg combined dose tablet, at a publicly listed price of $7.30 per unit.1,2 At a recommended dose of 1 tablet daily, the sponsor calculated that TDF-FTC would cost $2,629 per year ($219 per month). The no PrEP comparator was defined as the absence of prophylactic treatment to prevent HIV infection. In addition, the sponsor considered tenofovir alafenamide fumarate-emtricitabine (TAF-FTC) as a comparator in a scenario analysis for the MSM and TGW subpopulation. TAF-FTC is available as a 200 mg-20 mg combined dose tablet and has a recommended dose of 1 tablet daily. At a cost of $26 per tablet, the sponsor calculated that TAF-FTC would cost $9,397 per year ($783 per month).1
Modelled outcomes included life-years (LYs) and QALYs. Costs were estimated from the perspective of the Canadian public health care payer. Model outputs were generated over a lifetime horizon of 100 years, with a cycle length of 1 month (30.44 days). Costs and outcomes were discounted at 1.5% per annum.1
Model Structure
The sponsor submitted a Markov model which tracked a hypothetical cohort of patients across 5 health states. A summary of the model structure is presented in Figure 1. At model entry, patients initiated 1 of the eligible comparators (cabotegravir, TDF-FTC, or no PrEP). Patients could remain in their initial state, discontinue PrEP, or transition to the HIV or death states. Following discontinuation of cabotegravir, some patients were eligible to initiate second-line TDF-FTC. This was meant to reflect a situation known as the pharmacokinetic tail in which residual concentrations of cabotegravir LA remain in systemic circulation for an extended period. Treatment switching was not permitted for patients who initiated TDF-FTC at model entry. It was assumed that patients would not be eligible for any other PrEP alternatives following entry to the no PrEP state, where patients remained at risk for HIV infection and death.
Transitions between the health states were informed by a series of time-dependent transition probabilities. However, it was assumed that patients were only eligible for PrEP during a period where they would be at an elevated risk of HIV acquisition. This was assumed to be 5 years in the base case, after which there would be no more incident cases of HIV and all patients on PrEP were switched to no PrEP to reflect the change in treatment eligibility.
In addition to the primary cohort, the model also tracked a secondary cohort which represented the partners of the cohort who were eligible for PrEP. The purpose of this secondary cohort was to capture the benefits of PrEP in terms of HIV infections that were avoided because of primary prevention. Partners were tracked in a 3-state structure comprised of no PrEP (no HIV), HIV, and death. Transitions were informed by the all-cause mortality risk and the risk of secondary HIV infection.
Model Inputs
In response to heterogeneity within the at-risk HIV-1 population, costs and effects were estimated using 2 homogeneous subgroups: MSM and TGW and cisgender women. The results were subsequently combined using a weighted average, using weights described (MSM and TGW = 98%, cisgender women = 2%) in a report published by the Public Health Agency of Canada.3 Subgroup-specific data were obtained from the HPTN 083 and HPTN 084 clinical trials.4,5 These studies were randomized phase III trials which involved the direct comparison of cabotegravir and TDF-FTC for PrEP to reduce the risk of sexually acquired HIV-1 infection.4,5 Data of interest included: mean age (MSM and TGW = 26 years; cisgender women = 25 years), the proportion with detectable tenofovir (MSM and TGW = 86.0%; cisgender women = 55.9%), injection site reactions, and PrEP-related antiretroviral breakthrough resistance.4,5
Estimates of relative efficacy for the economic evaluation were obtained from the sponsor-submitted systematic review and NMA.1,6 This ITC was necessary as the HPTN 084 and HPTN 084 trials did not include a direct comparison with no PrEP. Studies of interest included PrEP interventions in cisgender women, MSM, and TGW aged 18 years and older who are at an increased risk of acquiring HIV-1 infection.1,6 The 10 identified trials formed a connected network that used TDF-FTC as a common comparator between cabotegravir and no PrEP.1,4-14 The outcome of interest for the NMA, as it relates to the economic evaluation, was the reduction in the risk of HIV acquisition.1,6 To reflect the fact that oral PrEP effectiveness is dependent on the level of adherence, the sponsor leveraged a meta-regression to adjust the estimated treatment effectiveness value to an adherence parameter which reflected the proportion with detectable tenofovir. Based on assessment of model fit statistics, the meta-regression equation was fitted on the log-scale.1,6
For the primary cohort, transition probabilities to inform the estimates of state membership were calculated using 5 distinct parameters: HIV acquisition risk, mortality risk, probability of first- and second-line treatment discontinuation, and proportion of patients with residual cabotegravir concentrations (and therefore eligible for second-line TDF-FTC).
PrEP-specific estimates of the HIV acquisition risk were calculated using the background incidence of HIV infection, the sponsor-submitted NMA, and the degree of adherence to oral PrEP. Consistent with the original TDF-FTC CDA-AMC submission, the background incidence (no PrEP) of HIV infection was assumed to be 4.3 and 3.1 cases per 100 person-years for the MSM and TGW and cisgender women subgroups respectively.7,9,15 These values, along with the relative efficacy data from the ITC, were used to calculate treatment-specific annual incidence rates which were subsequently converted to per cycle (1 month) probabilities.
In each cycle of the model, patients faced an all-cause mortality risk that increased with age. It was assumed that HIV-negative patients would have an all-cause mortality risk equivalent to that of the general population. Age- and gender-specific general population mortality risks were obtained from Canadian Life Tables published by Statistics Canada.16 Meanwhile, HIV-positive individuals experienced losses in life expectancy, as reflected from the application of standardized mortality ratios.1 The latter were calculated on the assumption that MSM and TGW and cisgender women would have remaining life expectancies of 39.0 years and 37.0 years, respectively.9
The probability of first-line treatment discontinuation differed for oral PrEP and cabotegravir. Persistence with oral PrEP (TAF-FTC or TDF-FTC) was obtained from an observational study of patients living in the US over a 12-month period.1,17 In this study, 70.2% and 57.4% of patients remained on treatment after 6 months and 12 months, respectively.1,17 The complement of these values, converted to monthly probabilities, were subsequently used to reflect the first-line discontinuation risk.1 For cabotegravir, it was assumed that adherence to treatment would be 20% higher than oral PrEP. Following first-line cabotegravir discontinuation, the sponsor assumed 50% of patients would initiate TDF-FTC to cover the pharmacokinetic tail. Among those patients, a 20% per cycle second-line discontinuation risk was assumed. In the submitted base case, it was assumed patients would not initiate second-line TAF-FTC following discontinuation of TDF-FTC or cabotegravir.1
For the secondary cohort of partners, the transition probabilities were calculated under the assumption that a secondary infection would occur in the same cycle as the primary infection. A secondary infection rate of 0.80 was assumed, based on the findings of a study of patients living in the US by Farnham et al.1,18 Consistent with the primary cohort, the secondary cohort of partners faced an all-cause mortality risk stratified by HIV status.1
Health-related quality of life was captured in the model by combining health state utilities with disutilities associated with HIV and injection site reactions. For all non-HIV health states, it was assumed that the cohorts in the model had utilities that were consistent with the general population of the UK.1,19 For patients receiving cabotegravir, it was assumed that the disutility associated with an injection site reaction (–0.011) would occur in the first cycle on treatment.1,20 Utilities for the HIV state were calculated as the difference between the general population utility and the disutility associated with HIV (–0.11).1,21
The submission considered costs associated with the acquisition, administration, and monitoring of PrEP therapy. Treatment acquisition costs were calculated from the price per unit consumed following the recommended dosage for each alternative considered in the model. Unit prices reflected the sponsor’s submitted price, values from the Ontario Drug Benefit Formulary/Comparative Drug Index, or the IQVIA DeltaPA wholesale price.1,2,22 Trial data were used to adjust the acquisition costs for oral PrEP but not cabotegravir. In the submitted base case, the administration cost for injectable cabotegravir was set to $0. It was assumed those costs would be covered by the sponsor’s patient support program.1 PrEP monitoring costs were intended to capture routine physician visits and recommended laboratory testing for individuals receiving PrEP. Costs associated with laboratory testing and physician visits were obtained from the Ontario Schedule of Benefits for Physician or Laboratory Services.1,23,24
In addition, the model considered costs associated with the management of injection site reactions, HIV infection, and PrEP-related breakthrough resistance. The costs associated with injection site reactions were restricted to severe cases which would require an extra general practitioner visit. Meanwhile, HIV management costs included the cost of antiretroviral treatment, physician follow-up, and monitoring. Costs of physician follow-up for patients living with HIV were obtained from a published source on quarterly physician billing and hospitalization costs.1,25 It was assumed 81% of patients living with HIV without PrEP-related breakthrough resistance would receive bictegravir-emtricitabine-tenofovir alafenamide ($29.23 per tablet) and the remaining 19% would receive dolutegravir-lamivudine ($32.33 per tablet).1,2 Patients with PrEP-related breakthrough resistance were assumed to require alternate first-line regimens. Those with integrase strand transfer inhibitor resistance were assumed to receive TDF-FTC plus darunavir-cobicistat (75%) or TDF-FTC plus darunavir and ritonavir (25%). Those with nucleoside reverse transcriptase inhibitor resistance were assumed to receive bictegravir-emtricitabine-tenofovir alafenamide (40%), TDF-FTC plus dolutegravir (20%), or dolutegravir plus darunavir-cobicistat (40%). The proportion of patients receiving each antiretroviral regimen were based on expert opinion solicited by the sponsor.1
Summary of Sponsor’s Economic Evaluation Results
All analysis were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following.
Base-Case Results
The submitted analysis was based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3. Cabotegravir LA was less costly and more effective than TDF-FTC and no PrEP (i.e., cabotegravir LA dominates both comparators). At a willingness-to-pay (WTP) threshold of $50,000 per QALY, cabotegravir had a 100% probability of cost-effectiveness.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Cabotegravir LA | 174,847 | 36.86 | — |
Dominated treatments | |||
TDF-FTC | 192,328 | 36.67 | Dominated by cabotegravir |
No PrEP | 261,682 | 36.29 | Dominated by cabotegravir |
ICER = incremental cost-effectiveness ratio; LA = long-acting; PrEP = pre-exposure prophylaxis; QALY = quality-adjusted life-year; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
In addition to the submitted base case, the sponsor considered 19 distinct scenario analyses. These scenarios considered alternate inputs or assumptions relating to the subgroup of interest, time horizon, the duration at which patients are at elevated risk for HIV-1 acquisition, the mortality risk relative to the general population, oral PrEP adherence, treatment switching following cabotegravir discontinuation, inclusion of administration costs, sources of HIV disutility, and discount rates. While each scenario had a slight impact on the expected costs and benefits, none had a meaningful effect on the conclusion for the cost-effectiveness of cabotegravir.
The sponsor conducted a scenario analysis from a societal perspective; this analysis included additional costs associated with the productivity loss from clinic visits and HIV infection. Consistent with the sponsor’s base-case analysis using a health care payer perspective, cabotegravir LA dominated both TDF-FTC and no PrEP.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The magnitude of clinical effectiveness of cabotegravir LA compared to no PrEP is highly uncertain: The submitted economic evaluation compared cabotegravir LA with relevant alternatives that could be used as PrEP to reduce the risk of sexually acquired HIV infection. In the base case, these alternatives included TDF-FTC and no PrEP; TAF-FTC was included as an additional comparator in a scenario analysis. In the absence of direct comparative information, relative treatment efficacy in the economic evaluation was estimated from the sponsor-submitted systematic review and NMA, using separate networks for the MSM and TGW and cisgender women subgroups. The results of the sponsor-submitted NMA indicated that cabotegravir was associated with improvements in HIV prevention compared with TDF-FTC and no PrEP. Relative to TDF-FTC, improvements in HIV prevention of 65% (95% CrI, 38% to 82%) and 86% (95% CrI, 69% to 95%) were estimated for the MSM and TGW and cisgender women subgroups, respectively. However, it was noted that these estimates were subject to considerable uncertainty as reflected by the wide CrIs. Relative to no PrEP, improvements in HIV prevention of 91% (95% CrI, 83% to 96%) and 93% (95% CrI, 83% to 97%) were estimated for the MSM and TGW and cisgender women subgroups, respectively. The CDA-AMC clinical review noted that most of the studies included in the NMA compared TDF-FTC with placebo, rather than no PrEP. Additional uncertainty in the validity of the results was attributed to limitations in the submitted NMA which prevented the assessment of several key assumptions. Most prominently, the limited number of studies and sparse reporting of study and baseline patient characteristics by the sponsor prevented the clinical review from appraising patient heterogeneity. Based on the available information, important differences were identified with respect to study design and patient populations which were not accounted for in the analysis. This prevented the CDA-AMC clinical review from drawing definitive conclusions on the relative effectiveness of cabotegravir LA and no PrEP.
These limitations in the underlying clinical evidence could not be addressed through reanalysis.
Treatment adherence assumptions introduce bias: The sponsor used meta-regression to adjust for heterogeneity in treatment adherence for TDF-FTC. In the context of the economic model, this meant that the assumed level of adherence for TDF-FTC had a direct impact on the effectiveness of this PrEP option. Clinical experts consulted by CDA-AMC confirmed that adherence is an effect modifier and raised concerns that the relative difference in effectiveness may be overestimated. This was attributed to the fact that real-world adherence levels of PrEP may not be as optimistic as those reported in more controlled trial settings. An additional concern was that adherence was only considered to be an effect modifier for oral PrEP but not cabotegravir LA. Clinical experts consulted by CDA-AMC confirmed that the same adherence concerns would apply to an injectable like cabotegravir, as there is no guarantee that patients will attend every appointment to receive an injection. The inconsistent consideration of an adherence-based effect modification (i.e., adjustment for 1 treatment, not for another) has a meaningful impact on the economic evaluation, as the results from the NMA were used to calculate the treatment-specific risk of HIV infection. This inconsistency made the results of the economic evaluation more favourable to cabotegravir LA.
The CDA-AMC reanalysis used a consistent approach to adherence for TDF-FTC and cabotegravir. In the absence of a mechanism to link adherence to cabotegravir LA efficacy, adherence to oral PrEP was assumed to be 100%. While cabotegravir LA remained more effective than TDF-FTC, this modification served to decrease the magnitude of the relative difference in treatment effectiveness.
Uncertainty in the baseline rate of HIV infection: In the MSM and TGW subgroup, the baseline rate of HIV infection was 4.3 per 100 person-years. This value was obtained from the TDF-FTC submission to CDA-AMC which the sponsor claimed was representative of the placebo arm of the iPrEx study.1,7,15 For the cisgender women subgroup, the baseline rate of HIV infection was 3.1 per 100 person-years, as reported in the TDF2 trial.1,9,15 The assumption that the incidence of infection would follow either input is subject to considerable uncertainty. The baseline infection rates from both trials reflected the placebo group which was provided with HIV prevention services such as testing, counselling, treatment of sexually transmitted infections, and free condoms. In other words, the incidence estimates do not reflect the proportion of the population seen in regular clinical practice who have never been offered HIV prevention services. Consequently, the reported baseline infection values may underestimate what might be expected in clinical practice. In addition, the assumed baseline rate of HIV infection for each subgroup only reflected 2 of the 10 trials included in the ITC, where the baseline rate of HIV was also estimated. The mean baseline risk across all included trials was estimated to be 5.01 cases per 100 person-years (95% confidence interval, 2.96 to 7.86) for the MSM and TGW subgroup, and 3.47 cases per 100 person-years (95% confidence interval, 2.31 to 4.93) for the cisgender women subgroup. Therefore, reliance on inputs from a prior CDA-AMC submission may not capture a more updated estimate and the associated uncertainty.
In reanalysis, CDA-AMC used estimates obtained from the submitted ITC.
Exclusion of cabotegravir administration costs: The total costs in the submitted base case do not include the costs associated with the administration of cabotegravir. These costs were excluded by the sponsor on the basis that this would be covered as part of the sponsor’s patient support program. CDA-AMC guidelines require the consideration of all costs that are relevant to the payer. This is particularly relevant given that the comparators to cabotegravir LA are orally administered and have administration costs of $0. Therefore, the total cabotegravir costs in the sponsor’s base case were underestimated.
CDA-AMC modified the submission to include administration costs for all treatments.
Consideration of spillover effects in the submitted base case: In addition to the primary cohort of PrEP-eligible individuals, the model considered a secondary cohort of sexual partners. The purpose of this secondary cohort was to capture any spillover effects from PrEP use, in terms of additional cases of HIV that were prevented. This was implemented as an assumption that there was an 80% chance of secondary infection in the same cycle as the primary HIV infection, based on results from a modelling study. CDA-AMC accepts the sponsor’s assertion that the benefits of HIV prophylaxis will likely extend beyond those benefits experienced by people receiving the prophylaxis themselves. However, CDA-AMC’s review of the sponsor’s model identified several concerns with the approach the sponsor employed to quantify these benefits. First, the inclusion of spillover effects in the submitted base case was inconsistent with CDA-AMC guidelines for the economic evaluation of health technologies.26 The consideration of spillover effects, like the prevention of secondary infections, is encouraged as part of a scenario analysis. In other words, the base-case analysis should always reflect the target population for the alternatives considered in a decision problem. Second, the sponsor’s approach for the consideration of spillover effects should have no impact on the assessment of relative cost-effectiveness. The assumption that 80% of the secondary cohort would acquire HIV will translate to an increase in the total costs, LYs, and QALYs gained for each comparator. However, this linear relationship means that the relative differences in each outcome will continue to be driven by the HIV infection risk in the primary cohort. Therefore, the only way the inclusion of the secondary cohort can influence the assessment of relative cost-effectiveness is in the presence of an error in the sponsor’s programming. Third, the implementation of the secondary cohort underestimated the total costs and benefits. This was attributable to the sponsor’s omission of individuals in the secondary cohort who never acquired HIV. Fourth, the sponsor’s approach to modelling the secondary prevention necessitated the inclusion of several questionable assumptions. For example, it was assumed that the individuals in the primary cohort were in monogamous relationships. The sponsor also assumed that individuals in the secondary cohort did not have any other sexual partners. Clinical experts consulted by CDA-AMC confirmed that having multiple sexual partners is a key factor which is considered in PrEP eligibility assessments. Finally, the sponsor assumed that individuals in the secondary cohort were not receiving or eligible for PrEP. Clinical experts consulted by CDA-AMC expressed doubt this would be the case as members of the secondary cohort would come from the same underlying populations as the primary cohort.
In reanalysis, CDA-AMC excluded the effect of secondary prevention from the base case. Given the identified concerns regarding the face validity of the spillover effects, any estimate produced using this approach would not be sufficiently rigorous to reflect the secondary effects of PrEP on costs and outcomes. Consequently, a scenario analysis which included secondary prevention was not performed.
Improper characterization of uncertainty: To address the fact that the true value of a parameter may not be known, CDA-AMC guidelines require the probabilistic evaluation of economic models.26 This involves the repeated estimation of costs and QALYs for each alternative using values selected at random from an assumed distribution for each parameter.26-28 It is therefore critical to ensure that this is done in a way that considers the imprecision in all relevant model input parameters. Approaches which do not capture the uncertainty in the underlying evidence base may affect the full range of costs and benefits estimated by the model.26-28 While the impact from the improper characterization of uncertainty will depend on the modelling context, in some circumstances it may be severe enough to have a meaningful influence on the conclusion regarding a specific intervention’s relative cost-effectiveness.
The sponsor’s approach to characterizing parameter uncertainty did not follow recommended practice.26-28 This was reflected by the assumption that several model inputs were not subject to any uncertainty. Examples of such parameters include: general population utility weights, mean life expectancy stratified by HIV status, baseline HIV incidence rate, and relative treatment effectiveness estimated from the NMA.7,9,19,29 For several of these inputs, this omission can be attributed to a choice to characterize the uncertainty associated with an intermediate value rather than the evidence used as its inputs. For example, the treatment-specific estimates of HIV incidence were assumed to follow a beta distribution. These values were calculated from 2 other sources of evidence: the baseline incidence and the relative effectiveness value estimated from the submitted NMA. The treatment-specific estimates of HIV incidence should have been calculated after sampling a random value for the baseline incidence and treatment effectiveness parameters. Given that the HIV incidence parameter affected the calculation of HIV risk in the model, it is possible that the conclusions from the economic evaluation will be sensitive to the approach used to characterize the uncertainty in the evidence base. The model therefore likely fails to properly reflect the parameter uncertainty surrounding the adoption decision.
CDA-AMC was unable to address this limitation. The necessary modifications to facilitate the proper characterization of uncertainty would have involved a complete redevelopment of the submitted spreadsheet. Such activity is beyond the scope of CDA-AMC reviews.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC conducted a reanalysis of the economic evaluation which addressed some of the key limitations identified in the sponsor’s submission. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. A summary of each independent modification to the submitted economic evaluation is presented in Table 4. The costs and effects for the CDA-AMC base case were generated using a Monte Carlo simulation of 1,000 iterations.
Table 4: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Exclusion of treatment administration costs | Excluded on the basis that it will be covered as part of a patient access program | Included for consistency with CDA-AMC guidelines |
2. Spillover effects in the base case | 80% chance of secondary infection in the same cycle as the primary HIV infection | Removal of secondary cohort (0% chance of secondary infection) |
3. Baseline HIV incidence rate | MSM and TGW: 4.3 cases per 100 person-years7 Cisgender women: 3.1 cases per 100 person-years9 | Estimated values from the submitted ITC: MSM and TGW (mean) = 5.01 cases per 100 person-years; cisgender women (mean) = 3.47 cases per 100 person-years |
4. Oral PrEP adherence | Heterogeneity in adherence is only a matter of concern for the efficacy of oral PrEP; all other forms are unaffected | Heterogeneity in adherence is a matter of concern for all forms of PrEP, including cabotegravir; in the absence of a mechanism to establish this link in the ITC, oral PrEP adherence is assumed to be 100% |
CDA-AMC base case | ― | 1 + 2 + 3 + 4 |
ITC = indirect treatment comparison; MSM = men who have sex with men; PrEP = pre-exposure prophylaxis; TGW = transgender women.
Results from the CDA-AMC base case are presented in Table 5. Cabotegravir LA and TDF-FTC were both less costly and more effective than no PrEP. However, cabotegravir LA was more costly and more effective than TDF-FTC as reflected in the estimated ICER of $29,283. At a threshold of $50,000 per QALY gained, cabotegravir LA had a 55% probability of being cost-effective.
The results from the CDA-AMC base case differ from those in the sponsor’s base case. While the sponsor suggested that cabotegravir LA was less costly and more effective than TDF-FTC, CDA-AMC’s reanalysis suggests that cabotegravir LA is associated with higher costs as well as greater QALYs. Both the sponsor’s and CDA-AMC’s base case found that cabotegravir LA is less costly and more effective than no PrEP. Due to the structural limitations identified and discussed previously, both the sponsor’s base case and CDA-AMC’s reanalysis likely do not properly reflect the uncertainty surrounding the decision. The 2 changes which most likely influenced this shift in expected values were the use of the submitted ITC values for the baseline HIV incidence rate and setting oral PrEP adherence to 100%. Both modifications were directly linked to the model’s ability to incorporate uncertain input parameter values in the calculation of costs and QALYs for each treatment. As illustrated in Figure 2, the CDA-AMC base case reflected a much different characterization of uncertainty returning lower probabilities of cost-effectiveness for both PrEP options. Furthermore, the results from the CDA-AMC base case may not fully represent the uncertainty in the evidence base used to reach conclusions regarding the cost-effectiveness of cabotegravir. As discussed previously, the sponsor’s approach to the characterization of parameter uncertainty did not always follow recommended practice. This unmeasured uncertainty, particularly for the relative effectiveness estimates from the submitted ITC, may affect the magnitude of the reported ICER but is unlikely to change the conclusion from the economic evaluation (i.e., cabotegravir LA would likely remain more costly and more effective than TDF-FTC).
Additional details summarizing the CDA-AMC base case are presented in Appendix 4. Even though the ICER changed in the second reanalysis, the removal of the secondary cohort was not influential to the conclusion regarding cost-effectiveness. As discussed in the limitation regarding spillovers, this may be indicative of an error in the spreadsheet’s programming.
Disaggregated results from the CDA-AMC base case are reported in Table 10. Compared with the sponsor base case, the changes for the CDA-AMC base case resulted in a reduction in the expected LYs gained for each treatment. This contributed a portion of the reduction in QALYs, which were also affected by the uncertainty in the probability of HIV risk calculated from the baseline HIV incidence, oral PrEP adherence, and other parameters. Changing oral PrEP adherence to 100% resulted in the smallest possible difference in HIV risk between cabotegravir LA and TDF-FTC. This led to meaningful reductions in HIV management costs compared to the sponsor’s base case. While HIV management costs remained higher for TDF-FTC than cabotegravir, the incremental difference decreased from $38,671 in the sponsor’s base case to $17,978 in the CDA-AMC base case.
Table 5: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor base case (probabilistic) | |||
Cabotegravir LA | 174,847 | 36.86 | Reference |
Dominated treatments | |||
TDF-FTC | 192,328 | 36.67 | Dominated by cabotegravir |
No PrEP | 261,682 | 36.29 | Dominated by cabotegravir |
CDA-AMC base case (probabilistic) | |||
TDF-FTC | 108,357 | 31.62 | Reference |
Cabotegravir LA | 111,135 | 31.71 | 29,283 |
Dominated treatments | |||
No PrEP | 150,244 | 31.37 | Dominated by cabotegravir and TDF-FTC |
ICER = incremental cost-effectiveness ratio; LA = long-acting; PrEP = pre-exposure prophylaxis; QALY = quality-adjusted life-years; TDF-FTC = tenofovir disoproxil fumarate-emtricitabine.
Source: Sponsor’s pharmacoeconomic submission.1
Scenario Analysis Results
Given the results of the sponsor’s base case and the CDA-AMC base case, no price reduction is required to obtain an ICER below a WTP threshold of $50,000 per QALY gained.
Issues for Consideration
TAF-FTC was excluded from the submitted economic evaluation. This was justified by the fact that it is not covered by most public drug plans in Canada and would therefore not apply to the public payer perspective. The only exception is the Canadian Armed Forces Drug Benefit List, where TAF-FTC is available under special authorization where there is a contraindication to the use of TDF-FTC.
Overall Conclusions
The CDA-AMC clinical review of the sponsor-submitted NMA found that cabotegravir LA provided a benefit compared to TDF-FTC and no PrEP in reducing the risk of sexually acquired HIV infection in adults who are considered high risk and weigh at least 35 kg. Findings from the direct comparison with TDF-FTC were consistent with those obtained in the sponsor-submitted NMA. However, considerable uncertainty was observed in the NMA from wide CrIs. Meanwhile, the CDA-AMC clinical review could not reach a definitive conclusion on the magnitude of benefit of cabotegravir LA compared with no PrEP. The uncertainty around relative efficacy contributes uncertainty to the economic evaluation.
CDA-AMC identified several additional limitations with the sponsor’s submitted economic evaluation. These involved the source of evidence used for the baseline rate of HIV infection, the exclusion of treatment administration costs, the consideration of spillover effects (which lacked face validity), and an approach to the characterization of parameter uncertainty which was inconsistent with recommended practice. In addition, the sponsor assumed that adherence would act as an effect modifier for oral PrEP but not injectable forms. Clinical experts consulted by CDA-AMC confirmed that the effectiveness of an injectable, as with an oral tablet, would depend on the level of adherence to the prescribed regimen. These limitations resulted in an economic evaluation which may have overestimated the incremental QALY gains and cost savings of cabotegravir relative to oral PrEP, introducing a bias that favoured cabotegravir. CDA-AMC attempted to address some of these limitations through reanalysis. Modifications were made to the submitted model to incorporate treatment administration costs, remove spillover effects, change the source of the baseline HIV incidence rate, and ensure the effectiveness of cabotegravir LA and TDF-FTC take a consistent approach to the effect modification from treatment adherence.
At the sponsor’s submitted price, results from the CDA-AMC base case indicated that cabotegravir LA and TDF-FTC were the only comparators on the efficiency frontier. Cabotegravir LA was more costly and more effective than TDF-FTC, with an estimated ICER of $29,283 per QALY gained. At a threshold of $50,000 per QALY gained, a price reduction was not necessary to achieve cost-effectiveness. The cost-effectiveness results were highly sensitive to the adherence assumption. If adherence to cabotegravir LA is higher than adherence to oral PrEP, the cost-effectiveness of cabotegravir is likely to be more favourable. Conversely, if patients prefer oral PrEP to cabotegravir LA, there may be insufficient evidence to justify the increased drug cost compared to oral PrEP.
The drug acquisition cost and total treatment cost of cabotegravir LA is greater than the drug acquisition cost and total treatment cost of TDF-FTC. The CDA-AMC reanalysis suggests that cabotegravir LA is associated with increased QALYs compared to TDF-FTC. The changes made to derive the CDA-AMC base case revealed greater uncertainty regarding cabotegravir’s status as the optimally cost-effective alternative. However, the results from the CDA-AMC base case do not fully represent the uncertainty in the evidence base used to estimate the costs and QALYs of each treatment. There remains unmeasured uncertainty, particularly for the relative effectiveness estimates from the submitted ITC, which may further influence the results. CDA-AMC expects that a properly specified analysis would still lead to the conclusion that treatment with cabotegravir LA is more costly and more effective than treatment with TDF-FTC.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare; 2024 Feb 1.
2.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 Feb 1.
3.Public Health Agency of Canada. Trends in HIV Pre-Exposure Prophylaxis [HIV-PrEP] use in 9 Canadian provinces, 2019 – 2022 [Infographic]. Ottawa (ON): Government of Canada; 2023: https://www.canada.ca/en/public-health/services/publications/diseases-conditions/trends-pre-exposure-prophylaxis-use-in-9-canadian-provinces-2019-2022-infographic.html. Accessed 2023.
4.Landovitz R, Donnell D, Clement ME, Hanscom B, Cottle L, Coelho R. Cabotegravir for HIV Prevention in Cisgender Men and Transgender Women. N Engl J Med. 2021;385(7):595-608. PubMed
5.Delany-Moretlwe S, Hughes JP, Bock P, et al. Cabotegravir for the prevention of HIV-1 in women: results from HPTN 084, a phase 3, randomised clinical trial. The Lancet. 2022;399(10337):1779-1789. PubMed
6.Indirect Treatment Comparison of the effectiveness of cabotegravir long acting (CAB-LA) for PrEP with no pre-exposure prophylaxis option (no PrEP) for HIV prevention [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir, 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare; 2024 Feb 1.
7.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587-2599. PubMed
8.Molina J-M, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373(23):2237-2246. PubMed
9.Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367(5):423-434. PubMed
10.Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367(5):411-422. PubMed
11.McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. The Lancet. 2016;387(10013):53-60. PubMed
12.Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367(5):399-410. PubMed
13.Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet. 2013;381(9883):2083-2090. PubMed
14.Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372(6):509-518. PubMed
15.Canadian Agency for Drugs and Technologies in Health. Common Drug Review Pharmacoeconomic Review Report: emtricitabine/tenofovir disoproxil fumarate (Truvada). Ottawa (ON): CADTH; 2016.
16.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2023: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2024 Feb 22.
17.Oglesby A, Germain G, Laliberte F, et al. Real-World Persistency of Patients Receiving Tenofovir-Based Pre-Exposure Prophylaxis for the Prevention of HIV Infection in the US. Paper presented at: Open Forum Infectious Diseases; 2021, 2021.
18.Farnham PG, Gopalappa C, Sansom SL, et al. Updates of lifetime costs of care and quality-of-life estimates for HIV-infected persons in the United States: late versus early diagnosis and entry into care. JAIDS. 2013;64(2):183-189. PubMed
19.Alava MH, Pudney S, Wailoo A. Estimating EQ-5D by age and sex for the UK [sponsor supplied reference]. 2022.
20.Boye KS, Matza LS, Walter KN, Van Brunt K, Palsgrove AC, Tynan A. Utilities and disutilities for attributes of injectable treatments for type 2 diabetes. Eur J Health Econ. 2011;12:219-230. PubMed
21.Miners A, Phillips A, Kreif N, et al. Health-related quality-of-life of people with HIV in the era of combination antiretroviral treatment: a cross-sectional comparison with the general population. The Lancet HIV. 2014;1(1):e32-e40. PubMed
22.DeltaPA. Ottawa (ON): IQVIA; 2023.
23.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2024 Feb 22.
24.Schedule of benefits for laboratory services (Effective July 24, 2023). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/sob_lab_2023.pdf. Accessed 2024 Feb 22.
25.Nosyk B, Min JE, Lima VD, Hogg RS, Montaner JSG. Cost-effectiveness of population-level expansion of highly active antiretroviral treatment for HIV in British Columbia, Canada: a modelling study. The Lancet HIV. 2015;2(9):e393-e400. PubMed
26.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2024 Feb 22.
27.Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation [sponsor supplied reference]. Oup Oxford; 2006.
28.Claxton K. Exploring uncertainty in cost-effectiveness analysis. Pharmacoeconomics. 2008;26:781-798. PubMed
29.Trickey A, Sabin CA, Burkholder G, et al. Life expectancy after 2015 of adults with HIV on long-term antiretroviral therapy in Europe and North America: a collaborative analysis of cohort studies. The Lancet HIV. 2023;10(5):e295-e307. PubMed
30.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cabotegravir 30 mg tablet and 200 mg/mL (600 mg/3 mL) extended-release injectable suspension. Montreal (QC): ViiV Healthcare; 2024 Feb 1.
31.Statistics Canada. Population estimates on July 1st, by age and sex Ottawa (ON): Government of Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2024 Jan 18.
32.Cox J, Apelian H, Moodie EEM, et al. Use of HIV pre-exposure prophylaxis among urban Canadian gay, bisexual and other men who have sex with men: a cross-sectional analysis of the Engage cohort study. CMAJ Open. 2021;9(2):E529-E538. PubMed
33.Colyer S, Lachowsky NJ, Schmidt AJ, et al. Measures of HIV Pre-exposure Prophylaxis Uptake Among Gay, Bisexual, and Other Men Who Have Sex with Men in Canada and Demographic Disparities Among Those at Elevated Likelihood for HIV Acquisition. AIDS Behav. 2021;25(11):3638-3650. PubMed
34.Patented Medicine Prices Review Board. Budget Impact Analysis Guidelines: Guidelines for Conducting Pharmaceutical Budget Impact Analyses for Submission to Public Drug Plans in Canada. Ottawa (ON): Government of Canada; 2020: https://www.canada.ca/en/patented-medicine-prices-review/services/reports-studies/budget-impact-analysis-guidelines.html. Accessed 2024 Apr 1.
Appendix 1: Cost Comparison Table
Please note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CDA-AMC Cost Comparison Table for PrEP of Sexually Acquired HIV-1 Infection
Treatment | Strength/concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
cabotegravir LA | 30 mg 200 mg/mL | Tablet Single-dose vial | 30.0750 1,710.000 | Injection only: One 3mL (600 mg) injection on days 1 and 28. Followed by 1 3 mL (600 mg) injection every 2 months thereafter. Oral lead-in: One 30 mg tablet per day for 28 days. Injections initiated within 3 days of completing oral lead-in. | With oral lead-in: 30.81 (Year 1) 28.09 (Year 2 and thereafter) Injection only: 28.09 | With oral lead-in: 11,252 (Year 1) 10,260 (Year 2 and thereafter) Injection only: 10,260 |
Alternative Treatments for PrEP | ||||||
tenofovir disoproxil fumarate-emtricitabine (Generic) | 200 mg/300 mg | Tablet | 7.3035 | One tablet daily | 7.30 | 2,668 |
tenofovir disoproxil fumarate-emtricitabine (Truvada) | 200 mg/300 mg | Tablet | 29.2140 | One tablet daily | 29.21 | 10,670 |
tenofovir alafenamide fumarate-emtricitabine (Descovy) | 25 mg/200 mg | Tablet | 26.1020 | One tablet daily | 26.10 | 9,534 |
LA = long acting.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed February 2024), unless otherwise indicated, and do not include dispensing fees.
Appendix 2: Submission Quality
Please note this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to limitation: Uncertainty in estimates of relative effectiveness, Improper characterization of decision uncertainty |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to limitation: Improper characterization of decision uncertainty |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note this appendix has not been copy-edited.
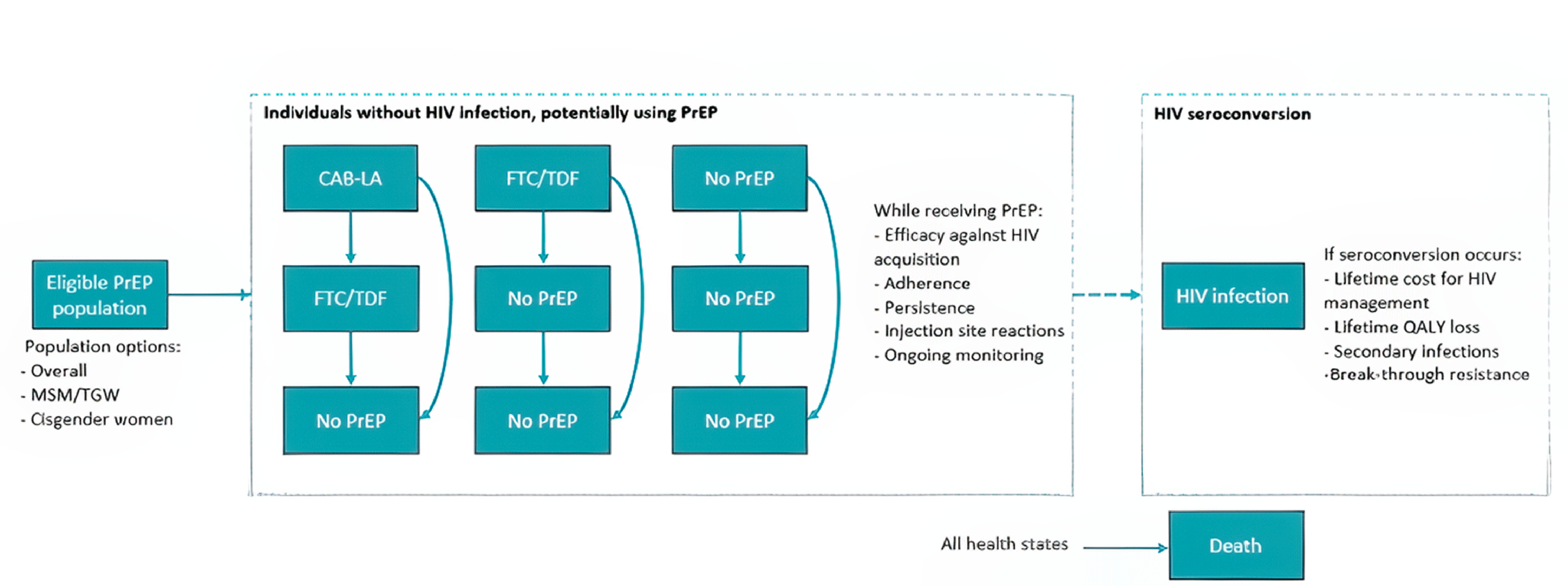
CAB-LA = cabotegravir; FTC = emtricitabine; MSM = men who have sex with men; PrEP = pre-exposure prophylaxis; QALY = quality-adjusted life-year; TDF = tenofovir disoproxil fumarate; TGW = transgender women.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Cabotegravir LA | TDF-FTC | No PrEP |
|---|---|---|---|
Discounted LYs | |||
Total | 43.24 | 43.23 | 43.22 |
Discounted QALYs | |||
Total | 36.86 | 36.67 | 36.29 |
By cohort | |||
Primary Cohort | 31.773 | 31.671 | 31.476 |
Secondary Cohort | 5.090 | 4.996 | 4.816 |
Discounted costs ($) | |||
Total | 174,847 | 192,328 | 261,682 |
Acquisition | 24,027 | 3,369 | 0 |
Administration and Visits | 1,033 | 635 | 0 |
PrEP Monitoring | 1,206 | 796 | 0 |
Injection Site Reaction Costs | 2 | 0 | 0 |
Breakthrough Resistance | −195 | 42 | 0 |
HIV Management | 148,775 | 187,486 | 261,682 |
LY = life-year; QALY = quality-adjusted life-year; LA = long-acting; PrEP = pre-exposure prophylaxis; TDF = tenofovir disoproxil fumarate; FTC = emtricitabine.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
The CDA-AMC base case reflects a series of modifications to the sponsor’s submission to address some of the identified limitations with the economic evaluation. The specific changes that were applied are detailed in Table 4, and the results obtained from each revision are presented in Table 9. Disaggregated results from the CDA-AMC base case are included in Table 10.
Table 9: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor base case | Cabotegravir LA | 175,622 | 36.83 | Reference |
TDF-FTC | 192,411 | 36.61 | Dominated by cabotegravir | |
No PrEP | 263,054 | 36.19 | Dominated by cabotegravir | |
CDA-AMC reanalysis 1 | Cabotegravir LA | 175,716 | 36.83 | Reference |
TDF-FTC | 192,411 | 36.61 | Dominated by cabotegravir | |
No PrEP | 263,054 | 36.19 | Dominated by cabotegravir | |
CDA-AMC reanalysis 2 | TDF-FTC | 98,630 | 31.63 | Reference |
Cabotegravir LA | 100,788 | 31.74 | 19,582 | |
No PrEP | 131,637 | 31.41 | Dominated by cabotegravir and TDF-FTC | |
CDA-AMC reanalysis 3 | Cabotegravir LA | 198,086 | 37.50 | Reference |
TDF-FTC | 220,366 | 37.25 | Dominated by cabotegravir | |
No PrEP | 301,299 | 36.77 | Dominated by cabotegravir | |
CDA-AMC reanalysis 4 | Cabotegravir LA | 174,944 | 36.83 | Reference |
TDF-FTC | 184,542 | 36.66 | Dominated by cabotegravir | |
No PrEP | 263,054 | 36.19 | Dominated by cabotegravir | |
CDA-AMC base case (deterministic) 1 + 2 + 3 + 4 | TDF-FTC | 108,345 | 31.57 | Reference |
Cabotegravir LA | 111,715 | 31.68 | 33,595 | |
No PrEP | 150,780 | 31.30 | Dominated by cabotegravir and TDF-FTC | |
CDA-AMC base case (probabilistic) 1 + 2 + 3 + 4 | TDF-FTC | 108,357 | 31.62 | Reference |
Cabotegravir LA | 111,135 | 31.71 | 29,283 | |
No PrEP | 150,244 | 31.37 | Dominated by cabotegravir and TDF-FTC |
FTC = emtricitabine; ICER = incremental cost-effectiveness ratio; LA = long-acting; PrEP = pre-exposure prophylaxis; QALY = quality-adjusted life-years; TDF = tenofovir disoproxil fumarate; Ref. = reference; vs. = versus.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Table 10: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results
Parameter | Cabotegravir LA | TDF-FTC | No PrEP |
|---|---|---|---|
Discounted LYs | |||
Total | 36.81 | 36.81 | 36.78 |
Discounted QALYs | |||
Total | 31.71 | 31.62 | 31.37 |
By cohort | |||
Primary Cohort | 31.71 | 31.62 | 31.37 |
Secondary Cohort | 0.00 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 111,135 | 108,357 | 150,244 |
Acquisition | 24,077 | 3,967 | 0 |
Administration and Visits | 1,128 | 635 | 0 |
PrEP Monitoring | 1,206 | 799 | 0 |
Injection Site Reactions | 2 | 0 | 0 |
Breakthrough Resistance | −222 | 34 | 0 |
HIV Management | 84,943 | 102,921 | 150,244 |
LY = life-year; QALY = quality-adjusted life-year; LA = long-acting; PrEP = pre-exposure prophylaxis; TDF = tenofovir disoproxil fumarate; FTC = emtricitabine.
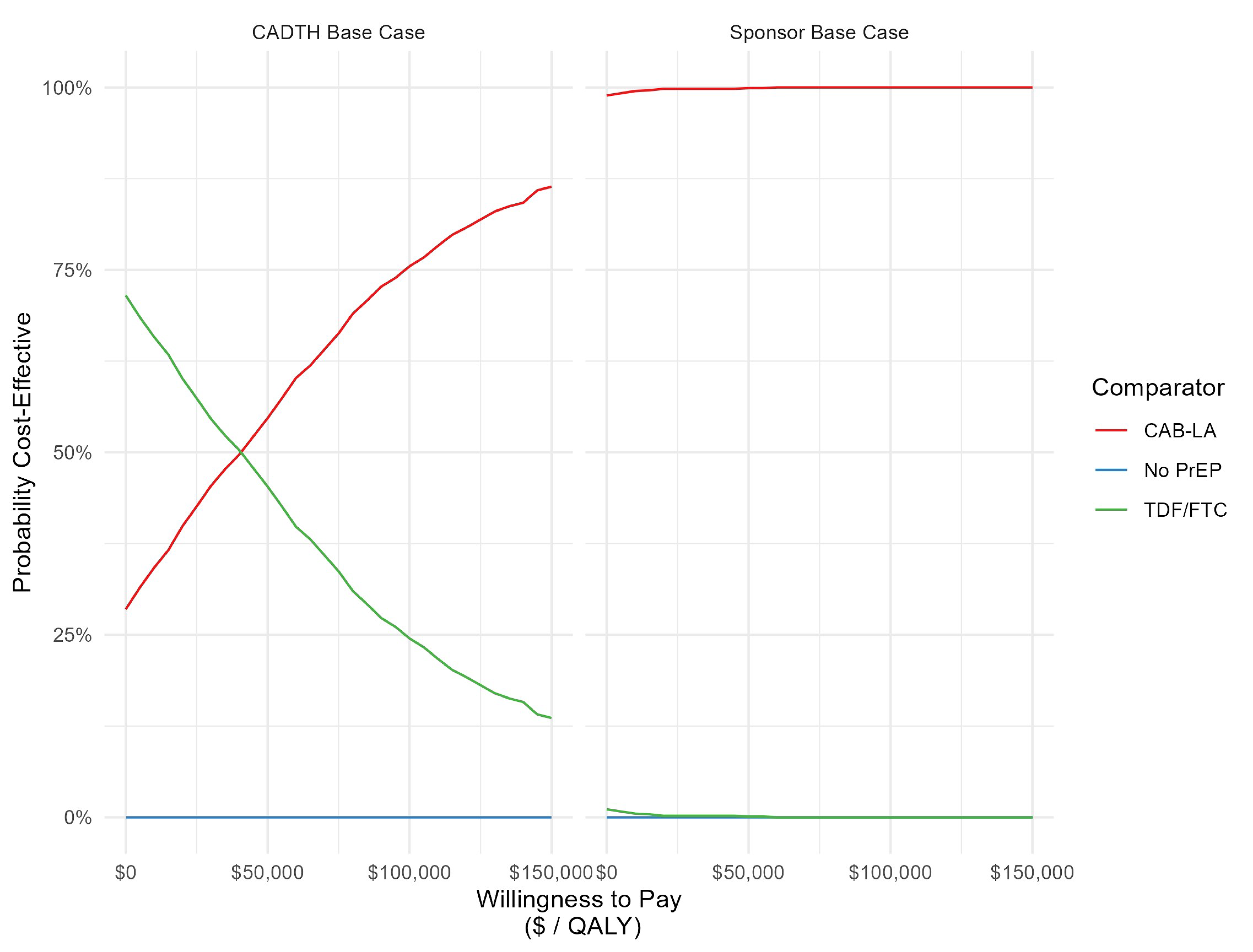
Figure 2 describes the difference in the characterization of decision uncertainty within (left) the CDA-AMC base case and (right) the sponsor’s base case. While the sponsor’s results suggest that CAB-LA is the optimal decision at all levels of WTP for an additional QALY, the CDA-AMC base case suggests that there is considerable uncertainty surrounding the cost-effectiveness of CAB-LA compared to TDF-FTC. It is important to note that neither the sponsor’s base case nor the CDA-AMC base case can accurately reflect the amount of uncertainty surrounding the adoption decision due to structural limitations within the sponsor-submitted model.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note this appendix has not been copy-edited.
Table 11: Summary of Key Take Aways
Key take aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the expected budget impact of reimbursing cabotegravir LA for PrEP to reduce the risk of sexually acquired HIV-1 infection among at-risk adults weighing at least 35 kg. The BIA was undertaken from the perspective of Canadian public drug plans (excluding Quebec) over a three-year time horizon. An epidemiological approach was used to estimate the eligible number of patients in each year of the analysis (Figure 3). Key inputs to the BIA are documented in Table 12.
In the reference scenario, it was assumed that all patients eligible for treatment would receive current PrEP options. These included: tenofovir disoproxil fumarate-emtricitabine (TDF-FTC), tenofovir alafenamide fumarate-emtricitabine (TAF-FTC), and no PrEP. In the new drug scenario, it was assumed that cabotegravir would displace market share from the existing alternatives.
State the key assumptions:
It was assumed that 0.45% of adults would be eligible for PrEP each year. This value was calculated by multiplying the total population of people living in Canada (38,929,902) by the incidence of PrEP use (89 per 100,000) divided by the proportion assumed to be on PrEP (Baseline combined market share of 30%).3,30,31 The estimate obtained was subsequently restricted to the adult population by dividing by the total population of people living in Canada aged older than 18 years (25,634,001).3,30,31
It was assumed that 43% of PrEP prescriptions will be covered by the public payer. This was based on an analysis published by the Public Health Agency of Canada in November, 2023.3
It was assumed that there is no growth in the size of the population eligible for PrEP in each year of the BIA.30
TAF-FTC was assumed to have 0% market share.30 This was attributable to the fact that it is not covered by most public health insurance plans in Canada and would therefore not apply to the public payer perspective. The only exception is the Canadian Armed Forces Drug Benefit List, where TAF-FTC is available under special authorization where there is a contraindication to the use of TDF-FTC.
Estimates of market share in the reference scenario were obtained from Canadian-specific studies. The first was a cross-sectional analysis of the Engage study cohort on sexually active MSM. The second source was a published analysis of a survey among MSM in Canada.30,32,33
In the new drug scenario, it was assumed that cabotegravir will take market share from TDF-FTC and no PrEP. This is based on an expectation that cabotegravir update will come from PrEP-eligible individuals who are not indicated for oral PrEP or are willing to consider an injectable over an orally administered tablet. Over the 3 years captured in the BIA, it is assumed that cabotegravir market share will increase from ███% to ███%.30
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population

PrEP = pre-exposure prophylaxis.
Source: Sponsor-submitted BIA.30
Table 12: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Total Canadian population, excluding Quebec (2022) | 38,929,902 |
Total adult population (18+) | 25,634,001 |
% adults eligible for PrEP | 0.45% |
Total adults eligible to receive PrEP | 115,492 |
Eligible for public coverage | 43.0% |
Number of patients eligible for drug under review | 49,662/49,662/49,662 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
No PrEP | 68%/66%/64% |
TAF-FTC | 0%/0%/0% |
TDF-FTC | 32%/34%/36% |
Uptake (new drug scenario) | |
Cabotegravir LA | ████ █ ████ █ ████ |
No PrEP | █████ █ █████ █ █████ |
TAF-FTC | ██ █ ██ █ ██ |
TDF-FTC | ███ █ ███ █ █████ |
Cost of treatment (per patient, per year)a | |
Cabotegravir LA | $11,692.55 (First Year); $10,312.98 (Subsequent Years) |
No PrEP | $0 |
TAF-FTC | $9,569.59 |
TDF-FTC | $2,703.44 |
LA = long acting; PrEP = pre-exposure prophylaxis; TAF = tenofovir alafenamide fumarate; TDF = tenofovir disoproxil fumarate; FTC = emtricitabine.
aCosts include dispensing fees of 61.81 (Cabotegravir, Year 1); 52.98 (Cabotegravir, Year 2+); 35.84 (oral tablets).
Summary of the Sponsor’s BIA Results
The net budget impact of cabotegravir was $14,269,064 in Year 1, $28,293,702 in Year 2, and $30,136,388 in Year 3. The three-year net budget impact of cabotegravir was estimated to be $72,699,154.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Underestimation of the market size: In estimating the eligible population for treatment, the sponsor assumed that 0.45% of adults in Canada would be eligible for PrEP each year. This value was obtained by multiplying the total population size by the incidence of PrEP use (89 per 100,000) divided by the proportion assumed to be on PrEP (30%). This value was subsequently divided by the total number of adults in Canada to obtain the proportion of adults eligible for PrEP. Clinical experts consulted by CDA-AMC raised concerns that this value may underestimate the proportion of adults who may be eligible for PrEP each year by as much as half. This would increase the total expenditures for all PrEP options considered in the BIA. Therefore, the underestimation of the market size may have some impact on the anticipated budget impact from the introduction of cabotegravir.
CDA-AMC conducted a scenario analysis to explore the effect of a larger market size on the budget impact of cabotegravir. For this analysis, the proportion of adults eligible for PrEP was assumed to be double that from the sponsor’s base case.
The analysis did not consider an open population: The sponsor assumed that there would be no growth in the size of the population eligible for PrEP in each year of the BIA. This is reflected by the absence of a mechanism to incorporate population growth over time. Guidelines for developing BIAs published by the Patented Medicine Prices Review Board recommend that an open population should be used to capture the forecasted growth of the target population in the estimates of the budget impact.34 Therefore, the estimates of the budget impact may have been underestimated.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the parameters of the BIA, the sponsor’s submitted base case was maintained. CDA-AMC expects that the budget impact of cabotegravir LA will be sensitive to more reliable inputs which may affect the market size calculation. This is reflected in a scenario analysis conducted by CDA-AMC which explored how an increase in PrEP eligibility would affect the budget impact. In this scenario, it was assumed that 0.90% of adults living in Canada would be eligible for PrEP each year.
Table 13: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Underestimation of market size | 0.45% | 0.90% |
The results of the submitted BIA and CDA-AMC scenario analysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15. All CDA-AMC reanalyses were based on publicly available prices of the comparator treatments. In the CDA-AMC scenario analysis, the three-near net budget impact of cabotegravir LA was estimated to be $145,398,309. This increase illustrates how the budget impact is sensitive to the proportion of patients who may be eligible for PrEP.
Table 14: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 72,699,154 |
Table 15: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 47,277,106 | 42,962,247 | 45,647,387 | 48,332,527 | 136,942,161 |
New drug | 47,277,106 | 57,231,311 | 73,941,089 | 78,468,916 | 209,641,316 | |
Budget impact | 0 | 14,269,064 | 28,293,702 | 30,136,388 | 72,699,154 | |
CDA-AMC scenario analysis: 0.90% of Adults Eligible for PrEP | Reference | 80,554,212 | 85,924,493 | 91,294,774 | 96,665,055 | 354,438,535 |
New drug | 80,554,212 | 114,462,222 | 147,882,178 | 156,937,831 | 499,836,843 | |
Budget impact | 0 | 28,538,129 | 56,587,404 | 60,272,776 | 145,398,309 |
BIA = budget impact analysis; PrEP = pre-exposure prophylaxis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.