Drugs, Health Technologies, Health Systems
Reimbursement Review
Faricimab (Vabysmo)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Retinal vein occlusion
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
APTC
Antiplatelet Trialists’ Collaboration
BCVA
best-corrected visual acuity
BRVO
branch retinal vein occlusion
CI
confidence interval
CrI
credible interval
CRT
central retinal thickness
CRVO
central retinal vein occlusion
CST
central subfield thickness
DIC
deviance information criterion
DMC
data monitoring committee
DME
diabetic macular edema
ETDRS
Early Treatment Diabetic Retinopathy Study
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
HRVO
hemiretinal vein occlusion
ITC
indirect treatment comparison
ITT
intention to treat
LOCF
last observation carried forward
MMRM
mixed model for repeated measures
nAMD
neovascular age-related macular degeneration
NMA
network meta-analysis
NEI-VFQ-25
National Eye Institute Visual Functioning Questionnaire–25
OCT
optical coherence tomography
PP
per protocol
PRN
pro re nata (as needed)
RCT
randomized controlled trial
RVO
retinal vein occlusion
SAE
serious adverse event
SD
standard deviation
SD-OCT
spectral-domain optical coherence tomography
SLR
systematic literature review
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Faricimab injection (Vabysmo) 6 mg/0.05 mL solution for intravitreal injection |
Sponsor | Hoffmann-La Roche Limited |
Indication | For the treatment of macular edema secondary to retinal vein occlusion |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 24, 2024 |
Recommended dose | The recommended dose is 6 mg (0.05 mL) administered by intravitreal injection every 4 weeks (approximately every 28 days ± 7 days) for 6 months. Patients should be assessed regularly. Monitoring between the dosing visits should be scheduled based on the patient’s status and at the physician’s discretion. |
NOC = Notice of Compliance.
Sources: Product monograph for faricimab injection (Vabysmo).1 Details included in the table are from the sponsor’s summary of clinical evidence.2
Introduction
Retinal vein occlusion (RVO) develops when a thrombus blocks the venous outflow of the retina, resulting in macular edema (fluid accumulation at the back of the eye).3 Macular edema can lead to significant retinal thickening, hemorrhage, and leakage.4,5 Patients with RVO usually experience acute, painless visual symptoms, including vision loss or varying degrees of visual alteration due to the edema.4-6 There are 3 subtypes of RVO that are classified according to the location of the occlusion: branch (involving a complete or partial obstruction at a branch or tributary of the central retinal vein), central (involving obstruction of the retinal vein at or posterior to the optic nerve head), and hemicentral (involving occlusion occurring at the disc that commonly involves half of the neurosensory retinal venous drainage, either the superior or inferior hemifield).4,6
In Canada, the annual estimated incidence rate of visual impairment due to macular edema secondary to branch retinal vein occlusion (BRVO) and central retinal vein occlusion (CRVO) was 0.056% and 0.021%, respectively (56 and 21 per 100,000, respectively).7 Based on pooled data from 11 population-based studies from the US, Europe, Asia, and Australia, the estimated prevalence rates of RVO, BRVO, and CRVO were 0.52%, 0.44%, and 0.08%, respectively (520, 440, and 80 people per 100,000, respectively).8
The Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus guidelines (published in 2015)7 advise the following:
BRVO with OCT evidence of macular edema: If visual acuity is greater than 20/40, then observation and close follow-up are suggested. Alternatively, anti-VEGF therapy can be considered in patients with relatively good functional vision and optical coherence tomography (OCT) evidence of minimal subclinical macular edema (i.e., 1 to 2 small intraretinal cysts). If there is no foveal involvement, then focal laser therapy is also an option. If visual acuity is less than 20/40 with subfoveal involvement, then treatment with anti-VEGF monotherapy is advised. According to the Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus, most clinicians manage macular edema secondary to hemiretinal vein occlusion (HRVO) similarly to BRVO.7
CRVO with OCT evidence of macular edema: If visual acuity is greater than 20/40, then observation and close follow-up are suggested. Otherwise, treatment with anti-VEGF monotherapy is advised.7
Patients with RVO have expressed the need for new treatments that prevent or slow down further vision loss and restore vision. The clinical expert identified the need for therapies to treat patients whose condition does not respond to available treatments or becomes refractory to current treatment options. They also noted the need for treatments that can reverse the course of disease (none currently exist) and address key outcomes, and for treatments that are better tolerated and improve adherence and convenience. Additionally, the clinical expert indicated that treatments that reduce treatment frequency and associated socioeconomic burden are needed. The clinician groups identified a need for efficacious, durable, and long-lasting treatments that can minimize treatment burden compared with existing options.
Faricimab was previously reviewed in 2022 for the following indications: the treatment of neovascular (wet) age-related macular degeneration (nAMD) and the treatment of diabetic macular edema (DME).
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of faricimab 6 mg/0.05 mL solution for intravitreal injection for the treatment of RVO.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call for input and from the clinical expert consulted for this review.
Patient Input
Input from 1 joint patient advocacy group (comprising Fighting Blindness Canada, the Canadian Council of the Blind, and Vision Loss Rehabilitation Canada) has been summarized for this report. The patient input submission included a summary of the results of an online survey made available from March to April 2024 to people in Canada living with RVO, supplemented with qualitative interviews with 3 patients diagnosed with RVO conducted in March and April 2024. The joint survey gathered information about patient-lived experiences associated vision loss and current treatments for RVO. In total, 32 patients living in Canada (62.5% of respondents had CRVO and 21.9% had BRVO; 15.6% did not know what type of RVO they had been diagnosed with) responded to the survey. Most respondents worried about vision worsening (53%). Respondents to the survey revealed that RVO significantly impacted their day-to-day lives and psychological well-being. Some respondents expressed being anxious about their RVO diagnosis, while others expressed feelings of fear, isolation, anger, and/or a loss of confidence or self-worth. Due to the sudden nature of RVO and its severity, respondents expressed ongoing fear of progression or that the unaffected eye could one day be affected. Overall, 60.0% of respondents indicated they had received anti-VEGF injections for their condition.
When patients were asked how they felt about their current ongoing treatments, the main reasons for stress were anxiety about injections (83%), symptoms from the injections (50%), and travel to appointments (33%). Respondents primarily expressed that treatment improved their vision or made their vision stable. Survey respondents highlighted the need for new treatments that prevent or slow down further vision loss and restore vision. Patients also expressed that they would like treatments that lower out-of-pocket costs, lower side effects, require fewer injections, and are effective.
The joint patient group noted that RVO leads to visual complications that render certain daily activities — such as reading or driving — either problematic or impossible. The joint input noted that while the current anti-VEGF treatments on the market have shown high levels of effectiveness in slowing or halting vision loss, they also come with highly burdensome regular intravitreal injections, creating challenges for many patients, such as the painfulness of the injection, both during and after the procedure, and their difficulties managing their bidirectional commute for their appointments. Patients expressed that they preferred treatment options that could be administered less frequently and supported treatments that could be made available to patients regardless of their province.
Clinician Input
Input From the Clinical Expert Consulted
The clinical expert identified the need for therapies to treat patients whose condition does not respond to available treatments or becomes refractory to current treatment options. They also noted the need for treatments that can reverse the course of disease (none currently exist) and address key outcomes, and for treatments that are better tolerated and improve adherence and convenience. Additionally, the clinical expert indicated that treatments that reduce treatment frequency and associated socioeconomic burden (i.e., treatment burden for clinicians and associated costs for patients and their families, such as transportation costs and lost income due to missed workdays) are needed. The clinical expert indicated that the anticipated place in therapy for faricimab is as an alternative to other currently available anti-VEGF therapies (i.e., the clinical expert did not expect a shift in the current treatment paradigm for macular edema secondary to RVO). The clinical expert noted that clinical practice, treatment history (suboptimal response with other treatments), access, and drug costs can influence treatment decisions. The clinical expert indicated that the anticipated target population for faricimab includes all patients with RVO, regardless of subtype, severity, symptoms, and so forth. Additionally, the clinical expert indicated that patients currently being treated with an anti-VEGF therapy may also be considered candidates for treatment with faricimab.
Based on clinical expert input, the diagnosis of RVO and treatment with faricimab should ideally be performed by a retina specialist. In situations where a retina specialist is not available, such as in remote areas, the clinical expert advised that the diagnosis of RVO and treatment with faricimab should ideally be performed by a well-trained general ophthalmologist. The clinical expert further advised that an outpatient setting that is well equipped for ophthalmic examination and OCT imaging is an appropriate setting for treatment with faricimab.
The clinical expert acknowledged the goal of treatment is to improve visual acuity; however, in practice, the clinical expert indicated that an OCT quantitative measurement of macular edema (central subfield thickness [CST] measurement) is the most important outcome used to assess response to treatment. The clinical expert noted that some clinicians also use qualitative parameters to assess treatment response, such as the presence and size of cystoid spaces. For visual acuity, the clinical expert indicated that a difference of more than 1 line of acuity in a Snellen chart is typically considered clinically meaningful (in the context of a comparison with a similar treatment). However, depending on variability in the light conditions, technician factors, and patient concentration, the clinical expert indicated that a difference of at least 2 Snellen lines can be considered clinically meaningful. For macular edema measured by OCT, the clinical expert indicated that a difference of 10% is typically considered clinically meaningful. The clinical expert indicated that assessment of treatment response usually coincides with treatment schedule (i.e., initially, once per month). When the condition is stabilized and the patient enters into the treat-and-extend phase, the clinical expert indicated that both the treatment and the response assessment are extended accordingly.
The clinical expert suggested the following scenarios in which the discontinuation of faricimab could be considered: when the underlying pathology has resolved, when the presence or absence of macular edema shows no difference in acuity, and when the treat-and-extend protocol allows an extension to more than 4 to 6 months between injections.
Clinician Group Input
Inputs from 4 clinician groups were summarized for this review: Toronto Retina Institute, Southeastern Ontario Community Ophthalmologists, Southwestern Ontario Community Ophthalmologists, and Northeastern Ontario Community Ophthalmologists. In total, 19 ophthalmologists contributed to the clinician input submission. Treatment goals highlighted by the groups included extending the treatment intervals, reducing macular edema, preserving visual acuity, improving visual acuity, reducing VEGF levels, and preventing neovascularization and neovascular glaucoma in patients with RVO. According to the groups, an ideal treatment is one that has demonstrated efficacy in sustaining improvements in visual acuity over the long-term and is durable in reducing the treatment burden associated with repetitive intravitreal therapy (i.e., requiring fewer injections, reducing the frequency of patient visits, and lowering costs and the burden on the health care system). There is an unmet need for patients whose condition does not achieve durable responses to existing treatment options. Therefore, there is a need for efficacious, durable, and long-lasting treatments that can minimize treatment burden compared with existing ones and extend treatment intervals while maintaining efficacy. The clinician groups anticipate that faricimab will be used as a first-line option for patients newly diagnosed with RVO based on its bispecific action mechanism and likelihood of generating a greater response. According to the clinician groups, faricimab would be suited for any patient with RVO, particularly those whose condition has failed to respond to other treatment options, although caution would be exercised for patients who have inflammation from other pre-existing conditions. The clinician groups did not anticipate any misdiagnoses. Any improvement in swelling, determined with an OCT scan, was considered a clinically meaningful improvement. The groups noted the following outcomes to assess whether a patient’s condition has responded to treatment: stable or improved visual acuity (improved vision); reduced presence of fluid assessed by an OCT scan; improved clinical exam measures of retinal hemorrhages, ischemia, and neovascularization; and fewer injections required and/or an increased interval between injections. The clinician groups highlighted that the factors considered for treatment discontinuation would be similar to those for currently approved therapies. These included no response or the presence of irreversible macular damage. One group highlighted that if a patient responds well and the treatment extension has increased to 4 months or more, clinicians would assess whether to stop treatment and would ensure the patient undergoes reasonably close observation. Faricimab can be administered in any outpatient setting and should preferably be administered by trained retina specialists.
Drug Program Input
Input was obtained from the drug programs that participate in the Reimbursement Review process. The following items were identified as key factors that could potentially impact the implementation of a recommendation of faricimab:
relevant comparators
initiation of therapy
continuation or renewal of therapy
discontinuation of therapy
prescribing of therapy
system and economic issues.
The clinical expert consulted for this review provided advice on the potential implementation issues raised by the drug programs, which is presented in Table 5.
Clinical Evidence
Systematic Review
Description of Studies
BALATON and COMINO were phase III, multicentre, randomized, double-masked, active comparator–controlled, parallel-group, 2-part studies. Part 1 evaluated the efficacy, safety, and pharmacokinetics of intravitreal faricimab 6 mg every 4 weeks compared with intravitreal aflibercept 2 mg every 4 weeks in patients with macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial) from day 1 through week 24 (24 weeks of treatment). Part 2 evaluated the efficacy, durability, safety, and pharmacokinetics of faricimab administered at masked treatment intervals of every 4, 8, 12, or 16 weeks based on personalized treatment interval dosing criteria, without an active control from week 24 through week 72 (48 weeks of treatment). Of note, there was no comparator in part 2 of the studies because all patients from part 1 received faricimab intravitreal injections according to a personalized treatment interval dosing regimen plus sham procedures to maintain masking of the treatment intervals.
The studies included patients with foveal centre–involved macular edema in the study eye due to BRVO (BALATON study), or CRVO or HRVO (COMINO study), diagnosed no longer than 4 months before screening and confirmed by the central reading centre based on spectral-domain optical coherence tomography (SD-OCT) (or swept-source OCT) images. The studies excluded patients with a history of previous episodes of macular edema due to RVO or persistent macular edema due to RVO diagnosed greater than 4 months before screening, as well as any prior or current treatment for macular edema due to RVO, including anti-VEGF intravitreal injections, in the study eye.
In BALATON, the mean age of patients was 64.3 years (standard deviation [SD] = 10.7 years; range, 35 to 93 years) in the faricimab group and 63.8 years (SD = 10.6 years; range, 28 to 88 years) in the aflibercept group. The mean best-corrected visual acuity (BCVA) in the study eye was 57.50 letters (SD = 13.04 letters; range, 19.0 to 76.0 letters) in the faricimab group and 57.64 letters (SD = 12.15 letters; range, 21.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 558.32 µm (SD = 177.03 µm; range, 281.0 µm to 1,154.0 µm) in the faricimab group and 558.12 µm (SD = 180.26 µm; range, 290.0 µm to 1,208.0 µm) in the aflibercept group. A total of 2.9% of patients (8 of 276 patients) in the faricimab group and 5.8% of patients (16 of 277 patients) in the aflibercept group had experience with at least 1 prior targeted ocular therapy or treatment in the study eye. A total of 17.4% of patients (48 of 276 patients) in the faricimab group and 16.6% of patients (46 of 277 patients) in the aflibercept group had at least 1 prior ocular surgery or procedure in the study eye, with the most common being cataract surgery.
In the COMINO trial, the mean age of patients was 65.6 years (SD = 13.1 years; range, 22 to 100 years) in the faricimab group and 64.7 years (SD = 13.3 years; range, 27 to 95 years) in the aflibercept group. A total of 83.0% of patients (303 of 366 patients) randomized to receive faricimab and 81.9% of patients (294 of 363 patients) randomized to receive aflibercept were reported with CRVO. A total of 17.0% of patients (62 patients) randomized to receive faricimab and 18.1% of patients (65 patients) randomized to receive aflibercept were reported with HRVO. The mean BCVA in the study eye was 50.25 letters (SD = 16.25 letters; range, 19.0 to 87.0 letters) in the faricimab group and 50.71 letters (SD = 16.34 letters; range, 19.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 702.21 µm (SD = 244.00 µm; range, 266.0 µm to 1,500.0 µm) in the faricimab group and 721.07 µm (SD = 242.86 µm; range, 281.0 µm to 1,419.0 µm) in the aflibercept group. A total of 5.7% of patients (21 patients) in the faricimab group and 5.0% of patients (18 patients) in the aflibercept group had experience with at least 1 prior targeted ocular therapy or treatment in the study eye. A total of 17.5% of patients (64 patients) in the faricimab group and 15.2% of patients (55 patients) in the aflibercept group had at least 1 prior ocular surgery or procedure in the study eye, with the most common being cataract surgery.
The BALATON and COMINO studies were ongoing at the time of the primary analysis; that analysis report reflected a data cut-off date of July 6, 2022, and August 9, 2022, respectively, when all patients from the global enrolment phase had either completed the study through week 24 or had discontinued from the study before week 24. This report also presented data from the final analysis through week 72, corresponding to the last patient last visit date of June 12, 2023, in the BALATON trial and July 12, 2023, in the COMINO trial (global enrolment phase only).
Note that efficacy and safety data from the BALATON and COMINO studies were available up to week 68; however, these data were not summarized in this report because the results at week 24 (with the exception of the treatment interval) were considered to be the most relevant for the purpose of this review to inform expert committee deliberations.
Efficacy Results
Visual Acuity Outcomes
The assessments of visual acuity that were determined to be the most relevant for this review were change in BCVA and the proportion of patients with an improvement in BCVA. These outcomes provide information on the degree of improvement in visual acuity and the proportion of patients with an improvement in visual acuity, respectively. Scores are based on the number of letters read correctly on an Early Treatment Diabetic Retinopathy Study (ETDRS) chart, with higher letter scores indicating better visual acuity (the maximum score is 100).
The primary end point in both studies was change from baseline in BCVA at week 24. If statistical significance was achieved on the noninferiority test, then the test for superiority could proceed; the noninferiority margin was 4 letters.
BALATON trial: The treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.6 letters (95% confidence interval [CI], −2.2 to 1.1 letters; P value for superiority test = 0.4978). A sensitivity analysis was performed for this outcome using multiple imputation to handle missing data differently. Two supplementary analyses were also performed: an analysis of the per-protocol (PP) population and an analysis using a hypothetical strategy for all intercurrent events. The results of the sensitivity and supplementary analyses were generally supportive of the primary analysis results.
The treatment difference in the estimated proportion of patients with a gain in BCVA of 15 letters or greater in the study eye from baseline at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −4.3% (95% CI, −12.3% to 3.8%; P = 0.3023). The supplementary analysis result was generally supportive of the primary analysis result.
COMINO trial: The treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.4 letters (95% CI, −2.5 to 1.6 letters; P value for superiority test = 0.6715). The sensitivity and supplementary analysis results were generally supportive of the primary analysis results.
A subgroup analysis based on baseline RVO status (CRVO and HRVO) was performed for the change from baseline in BCVA in the study eye at week 24. In the subgroup of patients with CRVO, the treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 0.2 letters (95% CI, −2.1 to 2.6 letters). In the subgroup of patients with HRVO, the treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −3.8 letters (95% CI, −7.3 to −0.4 letters).
The treatment difference in the estimated proportion of patients with a gain in BCVA of 15 letters or greater in the study eye from baseline at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −1.5% (95% CI, −8.4% to 5.3%; P = 0.6661). The supplementary analysis result was generally supportive of the primary analysis result.
Anatomic Outcomes
The assessments of the anatomy of the study eye that were determined to be the most relevant to this review were change in CST and the proportion of patients with an absence of macular edema and fluid. According to the clinical expert, these outcomes provide information on the extent of improvement in tissue swelling or edema, the physiological environment (i.e., re-establishment of the blood–retina barrier), and the presence or absence of cystoid spaces, respectively.
In both studies, CST was defined as the distance measured between the internal limiting membrane and Bruch membrane, standardized to Spectralis SD-OCT; the absence of macular edema was defined as a CST of less than 325 µm, and the absence of both intraretinal fluid and subretinal fluid was measured in the central subfield (within the 1 mm diameter centre of the macula).
BALATON trial: The treatment difference in the mean change from baseline in CST in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −7.0 µm (95% CI, −14.1 to 0.0 µm; P value for superiority test = 0.0495).
The treatment difference in the estimated proportion of patients with an absence of macular edema at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 1.4% (95% CI, −2.3% to 5.0%; P value for superiority test = 0.4742).
The treatment difference in the estimated proportion of patients with an absence of both intraretinal fluid and subretinal fluid at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 5.3% (95% CI, −2.7% to 13.3%; P value for superiority test = 0.1967).
COMINO trial: The treatment difference in the mean change from baseline in CST in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −12.8 µm (95% CI, −26.7 to 1.0 µm; P value for superiority test = 0.0684).
The treatment difference in the estimated proportion of patients with an absence of macular edema at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 1.7% (95% CI, −2.0% to 5.4%; P value for superiority test = 0.3589).
The treatment difference in the estimated proportion of patients with an absence of both intraretinal fluid and subretinal fluid at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 6.5% (95% CI, 0.1% to 13.0%; P value for superiority test = 0.0489).
Vision-Related Functioning and Health-Related Quality of Life Outcome
The assessment of vision-related functioning and health-related quality of life (HRQoL) that was determined to be the most relevant to this review was change in the National Eye Institute Visual Functioning Questionnaire–25 (NEI-VFQ-25) composite score. This outcome provides information on the degree of improvement in vision-related functioning and HRQoL from the patient’s perspective. Specifically, subscales include general vision, ocular pain, near activities, distance activities, social functioning, mental health, role difficulties, dependency, driving, colour vision, and peripheral vision. The composite score ranges from 0 to 100, with higher scores indicating better vision-related functioning and HRQoL.
BALATON trial: The treatment difference in the mean change from baseline in the NEI-VFQ-25 composite score at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.4 (95% CI, −1.9 to 1.1; P value for superiority test = 0.6370).
COMINO trial: The treatment difference in the mean change from baseline in the NEI-VFQ-25 composite score at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −1.2 (95% CI, −2.7 to 0.3; P value for superiority test = 0.1088).
Treatment Interval Outcomes
Treatment interval was identified as a relevant outcome for this review because the clinician groups indicated that an ideal treatment demonstrates a durable effect (i.e., demonstrates efficacy in sustaining improvement in visual acuity over the long-term) measured by a reduction in the treatment burden associated with repetitive intravitreal injections.
The treatment interval for a patient at week 68 was defined as the treatment interval decision made at week 68. The algorithm for the interactive web-based response system that determined the personalized treatment interval dosing regimen for faricimab in part 2 of the BALATON and COMINO studies is presented in Table 7.
BALATON trial: The proportions of patients on an extended treatment interval at week 68 were as follows (patients randomized to receive faricimab every 4 weeks in part 1 versus patients randomized to receive aflibercept every 4 weeks in part 1):
Faricimab every 8 weeks: 13.3% (95% CI, 9.1% to 17.5%) versus 18.0% (95% CI, 13.2% to 22.9%), respectively.
Faricimab every 12 weeks: 11.7% (95% CI, 7.7% to 15.7%) versus 9.4% (95% CI, 5.8% to 13.1%), respectively.
Faricimab every 16 weeks: 52.4% (95% CI, 46.2% to 58.6%) versus 47.5% (95% CI, 41.3% to 53.8%), respectively.
COMINO trial: The proportions of patients on an extended treatment interval at week 68 were as follows (patients randomized to receive faricimab every 4 weeks in part 1 versus patients randomized to receive aflibercept every 4 weeks in part 1):
Faricimab every 8 weeks: 20.0% (95% CI, 15.7% to 24.3%) versus 17.5% (95% CI, 13.3% to 21.7%), respectively.
Faricimab every 12 weeks: 8.5% (95% CI, 5.5% to 11.5%) versus 11.1% (95% CI, 7.6% to 14.6%), respectively.
Faricimab every 16 weeks: 37.0% (95% CI, 31.8% to 42.2%) versus 39.0% (95% CI, 33.7% to 44.4%), respectively.
Harms Results
At the primary analysis, safety was assessed through a descriptive summary based on data through week 24. At the final analysis, safety was also assessed through a descriptive summary based on data through week 72 for to the various predefined groups (due to the crossover) (refer to Table 26).
Adverse Events Through Week 24
BALATON trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 16.3% of those (45 of 276 patients) randomized to receive faricimab and 20.4% of those (56 of 274 patients) randomized to receive aflibercept were reported with at least 1 ocular adverse event (AE) in the study eye. Each type of ocular AE in the study eye was reported in less than 4.0% of patients in each group.
An independent clinical events coding committee adjudicated the thromboembolic events reported during the study. Antiplatelet Trialists’ Collaboration (APTC) events were defined as nonfatal strokes or nonfatal myocardial infarctions, or vascular deaths (including deaths of unknown cause). Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% of those (3 patients) randomized to receive faricimab and 1.5% of those (4 patients) randomized to receive aflibercept were reported with at least 1 adjudicated APTC-defined AE. Each type of adjudicated APTC-defined AE was reported in less than 1.0% of patients in each group.
COMINO trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 23.0% of those (84 of 365 patients) randomized to receive faricimab and 27.7% of those (100 of 361 patients) randomized to receive aflibercept were reported with at least 1 ocular AE in the study eye. Each type of ocular AE in the study eye was reported in less than 4.0% of patients in each group.
Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% of those (4 patients) randomized to receive faricimab and 1.4% of those (5 patients) randomized to receive aflibercept were reported with at least 1 adjudicated APTC-defined AE. Each type of adjudicated APTC-defined AE was reported in less than 1.0% of patients in each group.
Serious Adverse Events Through Week 24
BALATON trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% of those (3 patients) randomized to receive faricimab and 0.7% of those (2 patients) randomized to receive aflibercept were reported with at least 1 serious ocular AE in the study eye. Each type of serious ocular AE in the study eye was reported in less than 1.0% of patients in each group.
COMINO trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 2.5% of those (9 patients) randomized to receive faricimab and 3.3% of those (12 patients) randomized to receive aflibercept were reported with at least 1 serious ocular AE in the study eye. Each type of serious ocular AE in the study eye was reported in less than 1.0% of patients in each group.
Withdrawals Due to Adverse Events Through Week 24
BALATON trial: Of the patients who received at least 1 injection of the active study drug in the study eye, no patients stopped study treatment due to ocular AEs.
COMINO trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 0.8% of patients (3 patients) randomized to receive faricimab and 0.6% of patients (2 patients) randomized to receive aflibercept stopped study treatment due to ocular AEs. Each ocular AE that led to a patient stopping their study treatment was reported in less than 1.0% of patients in each group.
Mortality Through Week 24
BALATON trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 0.4% of patients (1 patient due to cerebrovascular accident) randomized to receive faricimab and no patients randomized to receive aflibercept died during the study through week 24.
COMINO trial: Of the patients who received at least 1 injection of the active study drug in the study eye, 0.3% (1 patient due to pneumonia) of patients randomized to receive faricimab and 0.6% of patients (2 patients due to myocardial infarction) randomized to receive aflibercept died during the study through week 24.
Notable Harms Through Week 24
BALATON trial: Of the patients who received at least 1 injection of the active study drug in the study eye, no patients were reported with endophthalmitis in the study eye.
COMINO trial: Of the patients who received at least 1 injection of the active study drug in the study eye, no patients randomized to receive faricimab and 0.3% of patients (1 patient) randomized to receive aflibercept were reported with endophthalmitis in the study eye.
Critical Appraisal
Internal Validity
Part 1 of the BALATON and COMINO studies was appropriately designed and powered to evaluate the efficacy of faricimab relative to aflibercept. The methods for randomization and allocation concealment were appropriate, and the review team judged that the risk of bias arising from the randomization process is unlikely. Part 2 of the studies did not have a relevant comparison group; therefore, conclusions about the number of injections relative to aflibercept or any other active comparator could not be drawn.
There is a lack of evidence in the literature to inform the measurement properties of BCVA as measured by ETDRS charts, CST as measured by OCT, and vision-related functioning and HRQoL as measured by NEI-VFQ-25 in patients with RVO. However, there was also no evidence in the literature to suggest there are concerns with these tools. Because the studies were masked, the risk of bias in the measurement of the outcomes is likely low.
As noted in guidance by the FDA,9 there was no impact on the type I error rate for the superiority test following the noninferiority test.10 The review team judged that the methods for deciding the 4-letter noninferiority margin were appropriate. Further, the clinical expert agreed that a difference of 5 letters or 1 Snellen line can be considered clinically meaningful in the context of comparisons with a similar treatment. Because no formal superiority tests were performed for the secondary end points and the subgroup analysis of the primary end point, these results are considered supportive evidence only. For statistically significant results, there is an increased risk that the null hypothesis was rejected erroneously.
The number of patients with HRVO available for the subgroup analysis was relatively low (< 20%) and, as such, the small sample size introduced uncertainty in the results (i.e., whether they would be replicated in a larger sample). There was no formal statistical approach for testing subgroup differences by RVO type. Although the estimated effect was statistically significant in the HRVO subgroup (and not in the CRVO subgroup), this contrast is not sufficient for inferring effect modification.11
Although major protocol deviation rates through week 24 were approximately 30% for each group from each study, the rates were generally balanced between groups. The most frequent type of major protocol deviation in both studies was procedural-related; however, each procedural-related protocol deviation was reported in less than 10% of patients in each group from both studies. As such, it was concluded that the risk of bias due to deviations from the intended intervention in part 1 of the studies is low. However, more than 50% of patients in each group from both studies were reported with at least 1 major protocol deviation through week 72, with more than 40% related to procedures. As such, it was concluded that the risk of bias (of unknown direction and magnitude) due to deviations from the intended interventions in part 2 of the studies is high.
Missing data were implicitly imputed by the mixed model for repeated measures (MMRM) model, assuming missing at random for both the primary end point of change from baseline in BCVA and the secondary end point of change from baseline in CST at week 24. A sensitivity analysis (in which missing data were assumed to be missing not at random and were assumed to have worse outcomes compared with the rest of the study population) was performed for the primary end point only. Because the results were consistent with the main analysis, the review team judged that the risk of bias due to missing outcomes data for this end point was low.
The assumptions for missing outcomes data (missing at random for change from baseline in CST at week 24 and last observation carried forward [LOCF] for categorical secondary outcomes at week 24) are likely not plausible and, for change from baseline in the NEI-VFQ-25 composite score at week 24 and treatment intervals at week 68, missing data were not imputed. Nonetheless, the amounts of missing outcomes data were generally low and balanced between groups in both studies, so the risk of bias due to missing outcomes data was considered low. The exception was for the proportion of patients with an absence of both intraretinal fluid and subretinal fluid in the BALATON trial, where the amount of missing outcome data was relatively high (13% to 16%). The clinical expert stated that the assumption that fluid status would stay constant over time is likely not plausible, so there is a potential for risk of bias due to missing outcomes data for this end point.
External Validity
The inclusion criteria in the BALATON and COMINO studies included the population of interest identified in the indication for faricimab, which is for the treatment of macular edema secondary to RVO. Notably, less than 20% of patients in the COMINO trial had HRVO; therefore, the generalizability of the study results to patients with HRVO is less certain.
The clinical expert indicated that the inclusion criteria adequately captured all patients who would be considered candidates for faricimab in practice. Further, the clinical expert indicated that the study population was generally representative of the patients typically seen in practice who would be candidates for treatment with faricimab. Of note, the clinical expert noted that patients with RVO generally present with uncontrolled blood pressure; cardiovascular disease, including stroke and myocardial infarction; diabetic retinopathy; and complications of cataract surgery. The clinical expert advised that these patients would be considered candidates for treatment with faricimab in practice.
In general, the Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus (published in 2015)7 advises on the use of anti-VEGF therapy in patients with RVO with OCT evidence of macular edema. This is aligned with input from the clinician groups and the clinical expert consulted for this review regarding the current treatment options that are available in practice. Therefore, the comparator in the studies (aflibercept) is relevant for the purpose of the present review; however, direct evidence for the effect of faricimab versus other anti-VEGF treatments (e.g., ranibizumab and bevacizumab) in the treatment of patients with macular edema secondary to RVO is lacking.
In consultation with the clinical expert, it was concluded that the outcome measures are generally reflective of assessments of treatment response in practice. Because the goal of treatment is to improve visual acuity, the clinical expert advised that if treatment response is demonstrated on imaging (CST measurement) but there is no change in visual acuity, then the clinician will consider discontinuing treatment. However, if treatment response is demonstrated in visual acuity with change in macular edema status, then the clinician will likely use the imaging results (CST as assessed by OCT) as an objective approach to determine whether to extend, maintain, or reduce the treatment interval in practice.
The clinical expert indicated that the common practice in Canada is to treat and extend, but it can also be a fixed treatment interval if extending is not possible. However, the clinical expert noted that the criteria used to determine a personalized treatment interval dosing regimen are not used uniformly by clinicians in practice.
In consultation with the clinical expert, it was concluded that the assessment time point at week 24 is considered appropriate for evaluating treatment effect in the therapeutic area of macular edema secondary to RVO. The exception is the outcome measure of treatment interval (proportion of patients on extended treatment intervals), for which the clinical expert suggested an assessment time point at month 24 (versus at week 68 in the studies).
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:12,13
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
For single-arm trials: Although GRADE guidance is not available for noncomparative studies, the review team assessed pivotal single-arm trials for study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up. In the current review, 68-week data from both trials were appraised as single-arm data, given the lack of a relevant comparator at this time point.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review. For the primary outcome of change from baseline in BCVA at week 24, the noninferiority margin used in the trials was the threshold.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with the clinical expert, and the input received from the patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
visual acuity (BCVA)
anatomic outcomes (CST, absence of macular edema, absence of both intraretinal and subretinal fluid)
vision-related functioning and HRQoL (NEI-VFQ-25)
extended treatment interval (every 8 to 16 weeks)
notable harms (endophthalmitis).
For the GRADE assessments, the BALATON and COMINO studies were assessed individually because the BALATON study had a patient population with macular edema secondary to BRVO and the COMINO study had a patient population with macular edema secondary to CRVO or HRVO.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for faricimab versus aflibercept in patients with macular edema secondary to BRVO.
Table 3 presents the GRADE summary of findings for faricimab versus aflibercept in patients with macular edema secondary to CRVO or HRVO.
Table 2: Summary of Findings for Faricimab vs. Aflibercept for Patients With Macular Edema Secondary to Branch Retinal Vein Occlusion
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Aflibercept | Faricimab (95% CI) | Difference (95% CI) | |||||
Visual acuity | |||||||
Change from baseline in BCVA in the study eye (ETDRS letter score), adjusted mean Follow-up: Week 24 | 553 (1 RCT) | NA | 17.5 | 16.9 (15.7 to 18.1) | −0.6 (−2.2 to 1.1) | Higha | Faricimab results in little to no difference in BCVA when compared with aflibercept. |
Proportion of patients gaining ≥ 15 letters in BCVA in the study eye from baseline, weighted estimate Follow-up: Week 24 | 553 (1 RCT) | NR | 60 per 100 | 56 per 100 (50 to 62 per 100) | 4 fewer per 100 | Moderateb | Faricimab likely results in little to no difference in the proportion of patients gaining ≥ 15 letters in BCVA when compared with aflibercept. |
Anatomic | |||||||
Change from baseline in CST in the study eye (µm), adjusted mean Follow-up: Week 24 | 553 (1 RCT) | NA | −304.4 | −311.4 (−316.4 to −306.4) | −7.0 (−14.1 to 0) | Highc | Faricimab results in little to no difference in CST when compared with aflibercept. |
Proportion of patients with an absence of macular edema defined as CST < 325 µm, weighted estimate Follow-up: Week 24 | 553 (1 RCT) | NR | 94 per 100 | 95 per 100 (93 to 98 per 100) | 1 more per 100 (2 fewer to 5 more per 100) | Highc | Faricimab results in little to no difference in the proportion of patients with an absence of macular edema when compared with aflibercept. |
Proportion of patients with an absence of both intraretinal fluid and subretinal fluid, weighted estimate Follow-up: Week 24 | 553 (1 RCT) | NR | 61 per 100 | 66 per 100 (61 to 72 per 100) | 5 more per 100 (3 fewer to 13 more per 100) | Lowd | Faricimab may result in little to no difference in the proportion of patients with an absence of both intraretinal fluid and subretinal fluid when compared with aflibercept. |
Vision-related functioning and HRQoL | |||||||
Change from baseline in the NEI-VFQ-25 composite score, adjusted mean Follow-up: Week 24 | 497 (1 RCT) | NA | 5.9 | 5.6 (4.5 to 6.7) | −0.4 (−1.9 to 1.1) | Highc | Faricimab results in little to no difference in vision-related functioning and HRQoL as assessed by NEI-VFQ-25 when compared with aflibercept. |
Treatment interval | |||||||
Proportion of patients on an extended treatment interval with faricimab Follow-up: Week 68 | 492 (1 RCT) | q.8.w. treatment interval:
q.12.w. treatment interval:
q.16.w. treatment interval:
| Very lowe | The evidence is very uncertain about the effect of faricimab on an extended treatment interval when compared with aflibercept. | |||
Harms | |||||||
Patients with an AE of endophthalmitis in the study eye Follow-up: Week 24 | 550 (1 RCT) | NR | 0 per 100 | 0 per 100 (NR) | NR | Lowf | Faricimab may result in little to no difference in endophthalmitis when compared with aflibercept. |
AE = adverse event; BCVA = best-corrected visual acuity; CI = confidence interval; CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; HRQoL = health-related quality of life; NA = not applicable; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; NR = not reported; PTI = personalized treatment interval; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The primary end point was change from baseline in BCVA at week 24; if statistical significance was achieved on the noninferiority test, then the test for superiority could proceed. No formal superiority tests were performed for the secondary end points; therefore, these results were considered to be supportive evidence only.
aThe noninferiority margin of 4 letters was used as the threshold of importance for assessing imprecision.
bRated down 1 level for serious imprecision. There was no known threshold for a clinically important effect, and the clinical expert consulted for this review could not estimate the threshold of a clinically important difference. The review team considered the 95% CI to include the potential for both little to no difference and clinically relevant comparative harm.
cThere was no known threshold for a clinically important effect, and the clinical expert consulted for this review could not estimate the threshold of a clinically important difference. The review team considered the 95% CI to include the potential for little to no difference only.
dRated down 1 level for serious study limitations. Missing outcome data were relatively high (13% to 16%), and it is unclear whether the reasons for missingness are balanced between groups. In consultation with the clinical expert, it was concluded that the assumption that fluid status would stay constant over time is likely not plausible. Therefore, it was concluded that there are some concerns for risk of bias due to missing outcome data. Rated down 1 level for serious imprecision. There was no known threshold for a clinically important effect, and the clinical expert consulted for this review could not estimate the threshold of a clinically important difference. The review team considered the 95% CI to include the potential for both little to no difference and clinically relevant comparative benefit.
eIn the absence of a relevant comparison group, conclusions about the number of injections relative to aflibercept or any other active comparator could not be drawn and the certainty of evidence started at very low. Rated down 1 level for serious study limitations. A relatively large proportion of patients (> 50%) were reported with at least 1 major protocol deviation through week 72, with the majority of the major protocol deviations (> 40%) related to procedures. Therefore, it was concluded that the risk of bias (of unknown direction and magnitude) due to deviations from the intended intervention in part 2 of the studies is high. Rated down 1 level for serious indirectness. Per the clinical expert consulted for this review, the criteria for extending the treatment interval in the trials were not reflective of clinical practice in Canada.
fRated down 2 levels for very serious imprecision. No events were observed; therefore, there was an inadequate number of events to inform a higher-certainty judgment.
Sources: Primary14 and final15 Clinical Study Reports for GR41984 (BALATON). Details included in the table are from the sponsor’s summary of clinical evidence.
Table 3: Summary of Findings for Faricimab vs. Aflibercept for Patients With Macular Edema Secondary to Hemiretinal and Central Retinal Vein Occlusion
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Aflibercept | Faricimab (95% CI) | Difference (95% CI) | |||||
Visual acuity | |||||||
Change from baseline in BCVA in the study eye (ETDRS letter score), adjusted mean Follow-up: Week 24 | 729 (1 RCT) | NA | 17.3 | 16.9 (15.4 to 18.3) | −0.4 (−2.5 to 1.6) | Higha | Faricimab results in little to no difference in BCVA when compared with aflibercept. |
Proportion of patients gaining ≥ 15 letters in BCVA in the study eye from baseline, weighted estimate Follow-up: Week 24 | 729 (1 RCT) | NR | 58 per 100 | 57 per 100 | 1 less per 100 (8 less to 5 more per 100) | Highb | Faricimab results in little to no difference in the proportion of patients gaining ≥ 15 letters in BCVA when compared with aflibercept. |
Anatomic | |||||||
Change from baseline in CST in the study eye (µm), adjusted mean Follow-up: Week 24 | 729 (1 RCT) | NA | −448.8 | −461.6 (−471.4 to −451.9) | −12.8 | Highb | Faricimab results in little to no difference in CST when compared with aflibercept. |
Proportion of patients with an absence of macular edema defined as CST < 325 µm, weighted estimate Follow-up: Week 24 | 729 (1 RCT) | NR | 92 per 100 | 94 per 100 (91 to 96 per 100) | 2 more per 100 (2 less to 5 more per 100) | Highb | Faricimab results in little to no difference in the proportion of patients with an absence of macular edema when compared with aflibercept. |
Proportion of patients with an absence of both intraretinal fluid and subretinal fluid, weighted estimate Follow-up: Week 24 | 729 (1 RCT) | NR | 69 per 100 | 75 per 100 (71 to 79 per 100) | 6 more per 100 (0 to 13 more per 100) | Moderatec | Faricimab likely results in little to no difference in the proportion of patients with an absence of both intraretinal fluid and subretinal fluid when compared with aflibercept. |
Vision-related functioning and HRQoL | |||||||
Change from baseline in the NEI-VFQ-25 composite score, adjusted mean Follow-up: Week 24 | 669 (1 RCT) | NA | 8.1 | 6.9 (5.8 to 8.0) | −1.2 (−2.7 to 0.3) | Highb | Faricimab results in little to no difference in vision-related functioning and HRQoL as assessed by NEI-VFQ-25 when compared with aflibercept. |
Treatment interval | |||||||
Proportion of patients on an extended treatment interval with faricimab Follow-up: Week 68 | 645 (1 RCT) | q.8.w. treatment interval:
q.12.w. treatment interval:
q.16.w. treatment interval:
| Very lowd | The evidence is very uncertain about the effect of faricimab on an extended treatment interval when compared with aflibercept. | |||
Harms | |||||||
Patients with an AE of endophthalmitis in the study eye Follow-up: Week 24 | 726 (1 RCT) | NR | 3 per 1,000 | 0 per 1,000 (NR) | NR | Lowe | Faricimab may result in little to no difference in endophthalmitis when compared with aflibercept. |
AE = adverse event; BCVA = best-corrected visual acuity; CI = confidence interval; CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; HRQoL = health-related quality of life; NA = not applicable; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; NR = not reported; PTI = personalized treatment interval; q.4.w. = every 4 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
The primary end point was change from baseline in BCVA at week 24; if statistical significance was achieved on the noninferiority test, then the test for superiority could proceed. No formal superiority tests were performed for the secondary end points; therefore, these results were considered to be supportive evidence only.
aThe noninferiority margin of 4 letters was used as the threshold of importance for assessing imprecision.
bThere was no known threshold for a clinically important effect, and the clinical expert consulted for this review could not estimate the threshold of a clinically important difference. The review team considered the 95% CI to include the potential for little to no difference only.
cRated down 1 level for serious imprecision. There was no known threshold for a clinically important effect, and the clinical expert consulted for this review could not estimate the threshold of a clinically important difference. The review team considered the 95% CI to include the potential for both little to no difference and clinically relevant comparative benefit.
dIn the absence of a relevant comparison group, conclusions about the number of injections relative to aflibercept or any other active comparator could not be drawn and the certainty of evidence started at very low. Rated down 1 level for serious study limitations. A relatively large proportion of patients (> 50%) were reported with at least 1 major protocol deviation through week 72, with the majority of the major protocol deviations (> 40%) related to procedures. Therefore, it was concluded that the risk of bias (of unknown direction and magnitude) due to deviations from the intended intervention in part 2 of the studies is high. Rated down 1 level for serious indirectness. Per the clinical expert consulted for this review, the criteria for extending the treatment interval in the trials were not reflective of clinical practice in Canada.
eRated down 2 levels for very serious imprecision. Few to no events were observed; therefore, there was an inadequate number of events to inform a higher-certainty judgment.
Sources: Primary16 and updated17 Clinical Study Reports for GR41986 (COMINO). Details included in the table are from the sponsor’s summary of clinical evidence.
Long-Term Extension Studies
The sponsor did not submit any long-term extension studies.
Indirect Comparisons
Description of Studies
One sponsor-conducted indirect treatment comparison (ITC) compared faricimab (6 mg every 4 weeks) with other anti-VEGF treatments, dexamethasone, and laser therapy for the treatment of RVO. The main comparators of interest identified in the systematic literature review (SLR) were anti-VEGF treatments (aflibercept 2 mg, ranibizumab, and bevacizumab), specifically those given in flexible regimens such as pro re nata (PRN) (as needed), which are typically used in clinical practice. A Bayesian approach under the random-effects model as the principal analysis and fixed-effects model for sensitivity was implemented. The outcomes assessed included the mean change from baseline in BCVA, proportion of patients gaining 15 or more letters, CST, and mean number of injections. The difference in the proportion of patients with any serious adverse events (SAEs) as well as all-cause discontinuations were also assessed. Treat-and-extend regimens could not be investigated due to a lack of connected studies.
Efficacy Results
Mean Change From Baseline in BCVA
Compared with other anti-VEGFs, the point estimates for the difference in mean change from baseline BCVA at 6 months mostly suggested little to no difference compared with faricimab 6 mg every 4 weeks. The point estimate for the comparison with bevacizumab 1.25 mg PRN favoured faricimab 6 mg every 4 weeks. In most comparisons, the 95% credible intervals (CrIs) were wide and included the possibility of clinically important effects favouring either treatment being compared. The mean changes and 95% CrIs for faricimab against anti-VEGFs administered PRN were as follows: aflibercept 2 mg PRN, 1.87 (95% CrI, −7.43 to 11.16); ranibizumab PRN, 3.59 (95% CrI, −2.94 to 10.17); and bevacizumab PRN, 5.22 (95% CrI, −3.35 to 13.80). The median between-study heterogeneity estimate (tau) was 2.85 (95% Crl, 1.397 to 3.911), indicating a low level of heterogeneity.
Mean Change From Baseline in CST
For change from baseline in CST at 6 months, faricimab 6 mg every 4 weeks was favoured over bevacizumab 1.25 mg PRN; however, the 95% CrI for the between-group difference included the possibility of little to no difference between the 2 treatments. In the comparisons with all other anti-VEGFs, the point estimates for between-group differences favoured faricimab 6 mg every 4 weeks; however, the 95% CrI included the possibility that either treatment could be favoured. The mean changes and 95% CrIs for faricimab against anti-VEGFs administered PRN were as follows: aflibercept 2 mg PRN, −37.3 (95% CrI, −107.99 to 35.72); ranibizumab PRN, −20.08 (95% CrI, −70.53 to 32.35); and bevacizumab PRN, −68.95 (95% CrI, −133.02 to −1.48). The between-study heterogeneity estimate (tau) had a median of 9.518 (95% Crl, 0.334 to 23.977), indicating a low level of heterogeneity.
Proportion of Patients Gaining at Least 15 ETDRS Letters
Results specific to the proportion of patients gaining at least 15 ETDRS were not reported in the sponsor-submitted ITC.
Mean Number of Injections
The network of studies for faricimab and anti-VEGF treatments allowing for a flexible (PRN) treatment regimen was connected with sham injections only; therefore, no treatment effect with regard to flexible regimens could be estimated.
Harms Results
Ocular Adverse Events
For ocular AEs, for comparisons with all anti-VEGFs, the 95% CrIs for the odds ratios were too wide to inform on which treatment might be favoured. The between-study heterogeneity estimate (tau) reported a median of 0.351 (95% Crl, 0.025 to 1.350). The odds ratio for faricimab against the anti-VEGF ranibizumab administered PRN was 0.64 (95% CrI, 0.14 to 2.80).
Serious Ocular AEs
For serious ocular AEs, for comparisons with all anti-VEGFs, the 95% CrIs for the odds ratios were too wide to inform on which treatment might be favoured. The odds ratio for faricimab against the anti-VEGF ranibizumab administered PRN was 0.53 (95% CrI, 0.03 to 10.5).
All-Cause Discontinuation
For comparisons with all other anti-VEGFs, the 95% CrIs for the odds ratios were too wide to inform about which treatment may be favoured. The between-study heterogeneity estimate (tau) had a median of 0.632 (95% Crl, 0.160 to 1.377). The odds ratios and 95% CrIs for faricimab against anti-VEGFs were as follows: aflibercept 2 mg every 4 weeks, 1.28 (95% CrI, 0.39 to 5.14); ranibizumab PRN, 1.00 (95% CrI, 0.16 to 6.53); and bevacizumab every 4 weeks, 0.79 (95% CrI, 0.11 to 6.20).
Critical Appraisal
There was variability in baseline characteristics (age, baseline BCVA, retinal thickness measurements, treatment patterns, number of injections administered, prior therapy, concomitant or background medications, intraocular pressure) across the studies included in the network meta-analysis (NMA) feasibility assessment. There was also a lack of reporting on several key study characteristics of interest for RVO (e.g., blood pressure, diabetes, concurrent diabetic retinopathy, coagulability, blood viscosity, and anemia) that could be potential effect modifiers. As such, there is uncertainty as to whether the assumptions related to homogeneity were met for the NMA. There was also a lack of clarity on the number of studies included in the network that enrolled treatment-experienced or treatment-naive patients with RVO. Prior treatment for macula edema with anti-VEGFs potentially negatively impacts treatment response. This adds uncertainty to the results and limits conclusions on the relative effect of faricimab against the anti-VEGFs commonly used as PRN regimens in practice. In addition, the CrIs for between-group comparisons were wide, often including the potential that either treatment being compared could be favoured.
Studies Addressing Gaps in the Evidence From the Systematic Review
Sixteen studies were submitted by the sponsor to address gaps in the RCTs submitted for faricimab for the treatment of RVO. These studies were excluded from the report because patients enrolled across studies included nAMD and DME populations, which differ from the sponsor-submitted reimbursement population. One matched cohort study, which matched patients with RVO from 2 registries (Vestrum and Medisoft) with the baseline characteristics of patients enrolled in the 2 pivotal trials submitted for this review (BALATON and COMINO trials), was also submitted by the sponsor. However, given that the therapies evaluated, in association with the outcomes of interest, did not include faricimab, the study was excluded from the report.
Conclusion
Two phase III, randomized, multicentre, double-masked, active comparator–controlled, parallel-group, 2-part trials were submitted for this review. Part 1 evaluated the efficacy, safety, and pharmacokinetics of intravitreal faricimab 6 mg every 4 weeks compared with intravitreal aflibercept 2 mg every 4 weeks in patients with macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial) from day 1 through week 24 (24 weeks of treatment). Part 2 evaluated the efficacy, durability, safety, and pharmacokinetics of faricimab administered at masked treatment intervals of every 4, 8, 12, or 16 weeks based on personalized treatment interval dosing criteria, without an active control from week 24 through week 72 (48 weeks of treatment). Based on input from patients and clinicians, visual acuity, anatomic outcomes, vision-related functioning and HRQoL, and treatment interval are important outcomes. The studies demonstrated that 24 weeks of treatment with intravitreal faricimab results in little to no difference in visual acuity when compared with intravitreal aflibercept, based on change from baseline in BCVA and the proportion of patients gaining 15 letters or greater in BCVA. Faricimab was statistically noninferior, but not superior, to aflibercept for change from baseline in BCVA. The studies also suggested that 24 weeks of treatment with faricimab likely results in little to no difference in anatomic outcomes when compared with aflibercept, based on the change from baseline in CST, the proportion of patients with an absence of macular edema, and the proportion of patients with an absence of both intraretinal and subretinal fluid. Overall, uncertainty in these efficacy end point results showing little to no difference in treatment effect was primarily due to concerns of risk of bias due to missing outcome data and imprecision of the 95% CIs. The studies also suggested that 24 weeks of treatment with faricimab results in little to no difference in vision-related functioning and HRQoL when compared with aflibercept, based on change from baseline in the NEI-VFQ-25 composite score. The evidence informed by the trials is very uncertain about the effect of faricimab on an extended treatment interval (i.e., every 8 to 16 weeks) at week 68 when compared with any active comparator, primarily due to the absence of a relevant comparison group. Further, there are concerns with the generalizability of the results because the criteria used to determine a personalized treatment interval dosing regimen are not used uniformly by clinicians in Canada.
Indirect evidence from the sponsor-submitted NMA provided evidence for faricimab relative to anti-VEGFs other than aflibercept. The evidence from the NMA suggested that faricimab may have little to no difference in treatment effect (in BCVA and CST) compared to other anti-VEGF therapies administered as needed in patients with macular edema secondary to RVO. However, there is uncertainty in the NMA results due to wide 95% CrIs, uncertain plausibility of the homogeneity assumption (i.e., variability in potential effect modifiers), a sparse network of evidence, and a potential for risk of bias in the included studies (e.g., uncertain handling of missing data).
With regard to safety, the frequency of AEs through week 24 was generally similar between the faricimab and aflibercept groups from each trial and was considered relatively low. The studies suggested that 24 weeks of treatment with faricimab may result in little to no difference in endophthalmitis when compared with aflibercept. Although endophthalmitis can be assessed within 3 to 4 days of injection, uncertainty in this safety end point remained primarily due to concerns of imprecision because there were few to no events observed to inform a higher-certainty judgment. Evidence from the NMA was insufficient to inform on the relative harms of faricimab compared with other anti-VEGFs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of faricimab 6 mg/0.05 mL solution for intravitreal injection for the treatment of RVO.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
RVO is a multifactorial disease that develops when a thrombus blocks the venous outflow of the retina, affecting the branch retinal veins, central vein, or hemicentral vein resulting in macula edema (fluid accumulation at the back of the eye).3 Macula edema can lead to significant retinal thickening, hemorrhage, and leakage.4,5 Patients with RVO typically experience acute, painless visual symptoms (vision loss or varying degrees of visual alteration) due to the edema.4-6 If left untreated, macula edema may cause blurred vision, eventually leading to blindness. RVO represents a significant cause of visual disability, frequently associated with systemic cardiovascular pathologies.5,18
There are 3 forms of RVO, classified according to the location of the occlusion: branch (involving a complete or partial obstruction at a branch or tributary of the central retinal vein), central (involving obstruction of the retinal vein at or posterior to the optic nerve head), and hemicentral RVO (involving occlusion occurring at the disc that commonly involves half of the neurosensory retinal venous drainage, either the superior or inferior hemifield).4,6 RVO can be further subclassified as ischemic RVO or nonischemic RVO, depending on the degree of blockage and blood supply restriction. Ischemic RVO is a serious form of RVO involving inadequate blood supply (ischemia) to the retina, with substantial and often irreversible damage. Nonischemic RVO is less serious, occurring when there is still adequate blood supply to the retina, which is associated with better outcomes.5,19
In Canada, the yearly incidence rate of visual impairment caused by macular edema secondary to BRVO and CRVO was 0.056% and 0.021%, respectively (56 and 21 per 100,000).7 Globally, there were about 28 million persons with RVO in 2015, with BRVO and CRVO accounting for 83.3% and 16.7% of cases, respectively.20 While Canadian prevalence data for RVO is difficult to source, pooled data from 11 population-based studies from the US, Europe, Asia, and Australia showed prevalence rates of approximately 0.52% for any RVO, 0.44% for BRVO, and 0.08% for CRVO corresponding to 520, 440, and 80 people per 100,000 respectively.8
RVO is diagnosed based on clinical assessments and imaging techniques, including eye exams, blood pressure and glucose tests, a complete blood count, and an erythrocyte sedimentation rate analysis.6 Common retinal imaging techniques used in practice include OCT, which is crucial for evaluating neovascularization and the extent of macula edema in the disease; OCT angiography; and fluorescein angiography. The Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus recommends using OCT angiography alongside OCT, and fluorescein angiography is recommended at initial assessment.7 Fluorescein angiography is also valuable for an in-depth diagnosis and classification of RVO because it offers detailed views of the retinal circulation, pinpointing capillary nonperfusion areas and assisting in distinguishing between ischemic and nonischemic RVO types.2
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus (published in 2015) advises on the following:7
BRVO with OCT evidence of macular edema: If visual acuity is greater than 20/40, then observation and close follow-up are suggested. Alternatively, anti-VEGF therapy can be considered in patients with relatively good functional vision and OCT evidence of minimal subclinical macular edema (i.e., 1 to 2 small intraretinal cysts). If there is no foveal involvement, then focal laser therapy is also an option. If visual acuity is less than 20/40 with subfoveal involvement, then treatment with anti-VEGF monotherapy is advised.7
If vision is stable and/or there is no OCT evidence of fluid after 3 monthly intravitreal injections of anti-VEGF, then as-needed treatment with frequent monitoring is advised. If there is some improvement in vision and/or OCT evidence of fluid after 3 monthly intravitreal injections of an anti-VEGF, continuation with anti-VEGF therapy for 3 monthly doses is advised. If there is no improvement or a worsening in vision after 3 monthly intravitreal injections of an anti-VEGF, then fluorescein angiography is advised to assess for ischemia and other complications and/or causes of a suboptimal treatment response to guide subsequent therapeutic approaches (i.e., sector panretinal photocoagulation, focal macular laser, switching the anti-VEGF, steroids).7 The clinical expert consulted for this review noted that with the introduction of anti-VEGF therapy, laser treatment is used less frequently for BRVO in practice but may still be used as an adjunct to anti-VEGF therapy to reduce the number of injections.
According to the Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus, most clinicians manage macular edema secondary to HRVO similarly to BRVO.7
CRVO with OCT evidence of macular edema: If visual acuity is greater than 20/40, then observation and close follow-up are suggested. Otherwise, treatment with anti-VEGF monotherapy is advised.7
If vision is stable and/or there is no OCT evidence of fluid after 3 monthly intravitreal injections of an anti-VEGF, then as-needed treatment with frequent monitoring is advised. If there is some improvement in vision and/or OCT evidence of fluid after 3 monthly intravitreal injections of an anti-VEGF, continuation with anti-VEGF therapy is advised, with consideration for assessment using fluorescein angiography to guide subsequent therapeutic approaches (i.e., panretinal photocoagulation, focal macular laser, switching the anti-VEGF). If there is no improvement or worsening in vision after 3 monthly intravitreal injections of an anti-VEGF, then intravitreal steroids or switching the anti-VEGF can be considered.7 The clinical expert indicated that laser therapy is not usually used for CRVO in practice.
The clinical expert advised that the mainstay of treatment for macular edema associated with RVO is anti-VEGF therapy. The clinical expert indicated that all anti-VEGF drugs (aflibercept, ranibizumab and its biosimilars, bevacizumab) are generally effective (i.e., they reduce macular edema and improve visual acuity in a majority of patients), with a relatively low and acceptable risk of complications. The clinical expert further indicated that continued injections at a reduced frequency are needed in most patients with RVO.
The clinical expert noted that triamcinolone suspension or dexamethasone implant, which are commonly associated with complications such as cataract and elevated intraocular pressure, are used infrequently (i.e., primarily for a suboptimal response to anti-VEGF therapy).
Based on the clinical expert input, the most important goal of therapy is to improve vision, thereby improving the HRQoL of patients with macular edema secondary to RVO. In addition to improving visual acuity, based on the clinician group input, the goals of therapy include extending the treatment intervals, reducing macular edema, preserving visual acuity, reducing VEGF levels, and preventing neovascularization and neovascular glaucoma.
Drug Under Review
Key characteristics of faricimab are summarized in Table 4 with other treatments available for RVO.
Faricimab is a humanized bispecific immunoglobulin G1 antibody that acts through the inhibition of both Ang-2 and VEGF-A. Faricimab suppresses endothelial cell proliferation, neovascularization, and vascular permeability by inhibiting VEGF-A. By inhibiting Ang-2, faricimab is thought to increase vascular stability and desensitize blood vessels to the effects of VEGF-A. Ang-2 levels are increased in some patients with wet age-related macular degeneration, DME, and RVO.1
The recommended dose of faricimab for RVO is 6 mg (0.05 mL), administered intravitreally every 4 weeks (approximately every 28 days plus or minus 7 days) for 6 months. Patients are required to be assessed regularly. Monitoring between the dosing visits should be scheduled based on the patient’s status and at the physician’s discretion.1
Faricimab was reviewed by Health Canada for the treatment of macular edema secondary to RVO and granted a Notice of Compliance on July 24, 2024. Faricimab was previously reviewed for nAMD and DME. Faricimab has been approved by the FDA for the treatment of macular edema following RVO and is currently under review by the European Medicines Agency.
Table 4: Key Characteristics of Faricimab, Aflibercept 2 mg, Ranibizumab, and Bevacizumab
Characteristic | Faricimab | Aflibercept 2 mg | Ranibizumab | Bevacizumab |
|---|---|---|---|---|
Mechanism of action | A bispecific antibody that simultaneously targets and neutralizes Ang-2 and VEGF-A. | VEGF inhibitor (soluble decoy receptor; targets VEGF-A and PlGF). | VEGF inhibitor (mAb; targets VEGF-A isoforms). | VEGF inhibitor (mAb; targets VEGF). |
Indicationa | For the treatment of macular edema secondary to RVO. | For the treatment of visual impairment due to macular edema secondary to:
| For the treatment of visual impairment due to macular edema secondary to RVO. | Noneb |
Route of administration | IVT | IVT | IVT | IVT |
Recommended dose | 6 mg (0.05 mL) administered by IVT injection every 4 weeks (approximately every 28 days ± 7 days) for 6 months. Patients should be assessed regularly. Monitoring between the dosing visits should be scheduled based on the patient’s status and at the physician’s discretion. | 2 mg (0.05 mL or 50 µl) administered by IVT injection once every month (every 4 weeks). The interval between 2 doses should not be shorter than 1 month. The treatment interval may be extended up to 3 months (12 weeks) based on visual and anatomic outcomes. Prescribers are advised to periodically (every 1 to 2 months) assess the need for continued therapy. Clinical trial experience with a monthly dosing regimen of 2 mg aflibercept beyond 6 months for CRVO and BRVO indications is limited. The dosing regimen of once every 4 weeks was changed at 24 weeks to a regimen that allowed for the extension of the treatment interval based on visual and anatomic outcomes in the CRVO clinical trials and to once every 8 weeks in the BRVO clinical trial. | Monthly and continued until maximum VA is achieved, confirmed by stable VA for 3 consecutive monthly assessments performed while on ranibizumab treatment. Thereafter, patients should be monitored monthly for VA. Treatment is resumed with monthly injections when monitoring indicates a loss of VA due to ME secondary to RVO and continued until stable VA is reached again for 3 consecutive monthly assessments. | None |
Serious adverse effects or safety issues |
|
|
|
|
ATE = arterial thromboembolic event; BRVO = branch retinal vein occlusion; CRVO = central retinal vein occlusion; IOP = intraocular pressure; IVT = intravitreal; mAb = monoclonal antibody; ME = macular edema; PlGF = placental growth factor; RVO = retinal vein occlusion; VA = visual acuity.
aHealth Canada–approved indication.
bBevacizumab is used off label in the treatment of RVO.
Sources: Product monograph for Vabysmo,1 Eylea,21 Lucentis,22 and Avastin.23
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Input from a joint patient group comprising 3 not-for-profit organizations (Fighting Blindness Canada, Canadian Council of the Blind, and Vision Loss Rehabilitation Canada) was summarized for this report. The 3 organizations cater to research and activities promoting vision health and the well-being of patients with RVO and other vision-related conditions. Information from this input was summarized from the results of an online survey made available from March to April 2024 to people in Canada living with RVO, supplemented with qualitative interviews with 3 patients diagnosed with RVO that were conducted in March and April 2024.
The joint survey gathered information about patient-lived experiences, associated vision loss, and current treatments for RVO. In total, 32 patients living in Canada (62.5% of respondents with central RVO, 21.9% with branch RVO, and 15.6% who did not know what type of RVO they were diagnosed with) responded to the survey. The mean age of respondents was 69 years and most respondents (71.0%) had RVO in 1 eye only. A total of 51.9% of respondents were from Ontario followed by British Columbia (22.2%) and Alberta (11.1%). The remaining respondents were from Newfoundland, Nova Scotia, Quebec, and Saskatchewan (3.7% each). Overall, 70% of respondents lived in urban areas. Most respondents worried about vision worsening (53%). Respondents to the survey revealed that RVO significantly impacted their day-to-day lives and psychological well-being. Some respondents expressed being anxious about their RVO diagnosis, while others expressed feelings of fear, isolation, anger, and/or loss of confidence or self-worth. Due to the sudden nature of RVO and its severity, respondents expressed ongoing fear of progression or that the unaffected eye may 1 day be affected. Patients noted that RVO diagnosis is lengthy and complicated because an ophthalmologist may not be the first point of contact. Respondents of working age expressed concerns about the lack of treatment coverage and difficulties attending multiple appointments, coupled with the negative impact that vision loss may have on their careers.
Overall, 60.0% of respondents indicated they had received anti-VEGF injections for their condition, 20.0% indicated they had received eye injections but were unable to identify the drug, 26.7% indicated they had also received laser treatment, and 13.3% stated they had never received treatment for RVO. When patients were asked how they felt about their current ongoing treatments, 78% of those who received injections reported feeling pain or discomfort 24 hours following treatment, and 52.1% of respondents found it stressful or difficult to attend appointments. The main reasons for the stress were anxiety about injections (83%), symptoms from the injections (50%), and travel to appointments (33%). Overall, 68% of respondents with experience with eye injections were satisfied with the injections. Respondents primarily expressed that treatment improved their vision or that treatment made their vision stable. Respondents who indicated dissatisfaction with their treatment listed pain or side effects after injections, lack of vision improvement, and the lack of coverage by drug plans as reasons.
Respondents to the survey highlighted the need for new treatments that prevent or slow down further vision loss and restore vision. Patients also expressed that they would like treatments that lower out-of-pocket costs, lower side effects, require fewer injections, and are effective. Four of the survey respondents and 2 of the individuals interviewed had experience with faricimab. One patient who had switched from another anti-VEGF did not notice a difference after 1 injection but noted that their doctor saw a difference on an OCT scan. Another interviewee expressed hope after treatment because they had experienced fewer blind spots following treatment.
The joint patient group input noted that RVO leads to visual complications that render certain daily activities — such as reading or driving — either problematic or impossible. In addition to causing diminishing visual acuity, RVO can present acute emotional and psychological burdens. When asked about their experience of the disease and its treatment, patients noted that treatments with less demanding injection regimes would help ease some of the burden associated with RVO. The joint input noted that while the current anti-VEGF treatments on the market have shown high levels of effectiveness in slowing or halting vision loss, they also come with highly burdensome regular intravitreal injections, creating challenges for many patients, such as painfulness of the injection, both during and after the procedure, and making it difficult to manage their bidirectional commute for their appointments. The group expressed challenges, especially for patients living in rural areas and remote parts of the country, such as travelling significantly long distances to their appointments and the frequent inaccessibility of transportation for low-vision individuals, both of which may lead to reliance and dependency on caregivers for travelling to and from appointments. They also noted challenges in managing daily living and tasks that are rendered very difficult by RVO, and short-term visual complications due to intravitreal injections. Patients expressed that they preferred treatment options that could be administered less frequently and supported treatments that could be made available to patients regardless of their province.
Clinician Input
Input From the Clinical Expert Consulted
All review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of macular edema secondary to RVO.
Unmet Needs
The clinical expert identified the need for therapies to treat patients whose condition does not respond to available treatments or becomes refractory to current treatment options. They also noted the need for treatments that can reverse the course of disease (none currently exist) and address key outcomes, and for treatments that are better tolerated and improve adherence and convenience. Additionally, the clinical expert indicated that treatments that reduce treatment frequency and associated socioeconomic burden (i.e., treatment burden for clinicians and associated costs for patients and their families, such as transportation costs and lost income due to missed workdays) are needed.
Place in Therapy
The clinical expert indicated that the anticipated place of faricimab in therapy is as an alternative to other currently available anti-VEGF therapies (i.e., the clinical expert did not expect a shift in the current treatment paradigm for macular edema secondary to RVO). The clinical expert noted that clinical practice, treatment history (suboptimal response with other treatments), access, and drug costs can influence treatment decisions.
Patient Population
The clinical expert indicated that the anticipated target population for faricimab includes all patients with RVO, regardless of subtype, severity, symptoms, and so forth. Additionally, the clinical expert indicated that patients currently being treated with another anti-VEGF therapy may also be considered candidates for treatment with faricimab.
Per the clinical expert input, the diagnosis of RVO involves a detailed retinal examination. To determine the subtype of RVO, supplementary information is required, including an OCT, OCT angiography, and fluorescein angiography. OCT is used to diagnose and quantify macular edema, while OCT angiography and fluorescein angiography are used to assess the degree of ischemia and identify whether there is a need for panretinal photocoagulation.
Assessing Treatment Response
The clinical expert acknowledged the goal of treatment is to improve visual acuity; however, in practice, the clinical expert indicated that the OCT quantitative measurement of macular edema (measurement of CST) is the most important outcome used to assess response to treatment. The clinical expert noted that some clinicians also use qualitative parameters to assess treatment response, such as the presence and size of cystoid spaces.
For visual acuity, the clinical expert indicated that a difference of more than 1 Snellen line of acuity is typically considered clinically meaningful (in the context of a comparison with a similar treatment). However, depending on variability in the light conditions, technician factors, and patient concentration, the clinical expert indicated that a difference of at least 2 Snellen lines can be considered clinically meaningful.
For macular edema measured by OCT, the clinical expert indicated that a difference of 10% is typically considered clinically meaningful.
The clinical expert indicated that assessment of treatment response usually coincides with treatment schedule (i.e., initially, once per month). When the condition is stabilized and the patient enters into the treat-and-extend phase, the clinical expert indicated that both the treatment and response assessment are extended accordingly.
Discontinuing Treatment
The clinical expert suggested the following scenarios in which the discontinuation of faricimab could be considered:
When the underlying pathology has resolved (in some cases, there may be disease resolution due to recanalization of the thrombotic process).
When the presence or absence of macular edema shows no difference in acuity due to retinal atrophy, chronic cystoid degeneration, or macular ischemia.
When the treat-and-extend protocol allows an extension to more than 4 to 6 months between injections.
Prescribing Considerations
Based on clinical expert input, the diagnosis of RVO and treatment with faricimab should ideally be performed by a retina specialist. In situations where a retina specialist is not available, such as in remote areas, the clinical expert advised that the diagnosis of RVO and treatment with faricimab should ideally be performed by a well-trained general ophthalmologist.
The clinical expert further advised that an outpatient setting that is well equipped for ophthalmic examination and OCT imaging is an appropriate setting for treatment with faricimab.
Clinician Group Input
This section was prepared by the review team based on the input provided by the clinician groups.
Inputs from 4 clinician groups were summarized for this review: Toronto Retina Institute (comprising a group of 10 retina specialists in multiple locations across the Greater Toronto Area), Southeastern Ontario Community Ophthalmologists (comprising 2 practising ophthalmologists with a community practice in Southeastern Ontario), Southwestern Ontario Community Ophthalmologists (comprising community-based ophthalmologists practising in Southwestern Ontario), and Northeastern Ontario Community Ophthalmologists (comprising ophthalmologists practising in Northeastern Ontario). In total, 19 ophthalmologists contributed to the clinician input submission. Input across groups was generally sourced from virtual meetings and discussions, emails, and phone calls.
The treatment goals highlighted for RVO were similar across the participating clinician groups. These goals included extending the treatment intervals, reducing macular edema, preserving visual acuity, improving visual acuity, reducing VEGF levels, and preventing neovascularization and neovascular glaucoma in patients with RVO. According to the groups, an ideal treatment is 1 with demonstrated efficacy in sustaining improvements in visual acuity over the long-term and is durable in reducing the treatment burden associated with repetitive intravitreal therapy (i.e., requiring fewer injections, reducing the frequency of patient visits, and lowering costs and the burden on the health care system). According to the clinician groups, the current treatments options for RVO in practice include anti-VEGF treatments (aflibercept 2 mg, ranibizumab, and bevacizumab) and steroids (including dexamethasone and subtenon Kenalog). The clinician groups noted that RVO has a large inflammatory component, and some studies have demonstrated that the Ang-2 pathway is more active in patients with RVO.
The unmet needs identified in the clinician group input submissions were also consistent across groups. The groups highlighted that RVO treatment requires repeated visits (every 4, 5, 6, 7, or 8 weeks) regardless of the type of treatment, which poses a significant burden to patients. There is an unmet need for patients whose condition does not achieve durable responses to existing treatment options. Therefore, there is a need for efficacious, durable, and long-lasting treatments that can minimize treatment burden compared with existing therapies and extend treatment intervals while maintaining efficacy. The clinician groups anticipate that faricimab will offer a valuable additional anti-VEGF and Ang-2 option to potentially extend the treatment interval.
The clinician groups anticipate that faricimab will be used as a first-line option for patients newly diagnosed with RVO based on its bispecific action mechanism. The clinician groups anticipate that faricimab will likely expand treatment options for patients with RVO that fails other anti-VEGF treatments. According to the clinician groups, faricimab would be suited for any patient with RVO, particularly those whose condition has failed to respond to other treatment options, although caution would be exercised for patients who have inflammation from other pre-existing conditions. The clinician groups did not anticipate any misdiagnoses because fluid in the retina is identifiable through OCT; thus, the introduction of faricimab will not impact diagnostic paradigms.
According to the clinician groups, any improvement in swelling, determined with an OCT scan, was considered a clinically meaningful improvement. The groups listed the following outcomes to assess whether a patient’s condition has responded to treatment: stable or improved visual acuity (improved vision); reduced presence of fluid assessed by an OCT scan; improved clinical exam measures of retinal hemorrhages, ischemia, and neovascularization; and fewer injections required and/or an increased interval between injections.
One clinician group highlighted that clinicians use a treat-and-extend protocol to determine whether the interval between treatments can be adjusted and whether treatment response approaches are fairly standardized across the retinal medical community. The clinician groups highlighted that the factors considered for treatment discontinuation would be similar to those considered for currently approved therapies. These include no response or the presence of irreversible macular damage. One group highlighted that if a patient responds well and the treatment extension has increased to 4 months or more, clinicians would assess whether to stop treatment and would ensure the patient undergoes reasonably close observation. However, a new treatment option would be considered if the patient’s treatment interval declines, or they experience progressive vision loss. Another group noted that the discontinuing of faricimab may occur due to patient-related factors such as better perceived efficacy on another therapy or difficulty travelling to the clinic for treatment. According to the groups, faricimab can be administered in any outpatient setting and should preferably be administered by a trained retina specialist.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
In the BALATON and COMINO studies, faricimab 6.0 mg every 4 weeks was compared with aflibercept 2.0 mg every 4 weeks through week 20. Active control is considered appropriate because the dose for aflibercept is specified in the product monograph. | This is a comment from the drug plans to inform CDEC deliberations. |
Different programs providing access to anti-VEGFs exist across drug plans:
| This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Is RVO similar to macular edema secondary to BRVO (i.e., same indication)? Note that aflibercept was initially reviewed for BRVO. | The clinical expert indicated that the subtypes of RVO are similar but also have important clinical differences. However, when considering treatment response, there may be less need to differentiate between the subtypes. The clinical expert advised that the mainstay of treatment for macular edema associated with RVO is anti-VEGF therapy. The clinical expert noted some differences in the treatment approach between the subtypes of RVO. For example, in certain cases of BRVO, laser treatment may be started to avoid anti-VEGF treatment. In other cases of BRVO, intravitreal injections may be supplemented with laser treatment to reduce the number of injections. In contrast to BRVO, CRVO generally does not respond to laser treatment. |
Switching between anti-VEGF treatments is a consideration. | This is a comment from the drug plans to inform CDEC deliberations. |
Given differences in the reimbursement landscape across drug plans, consider implementation guidance to note that faricimab, if determined to be therapeutically equivalent, could be initiated, renewed, discontinued, and prescribed in a manner similar to the practices followed for other anti-VEGF drugs for RVO, as per the reimbursement criteria for each public drug plan. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Treat and extend is an important consideration for drug plans and may be beneficial from a health system perspective. By week 72 of the trials, a portion of patients transitioned to extended dosing intervals with faricimab (i.e., q.12.w. and q.16.w.). | This is a comment from the drug plans to inform CDEC deliberations. |
1. What is the importance of the extended intervals? | 1. The clinical expert suggested that the advantage of extended treatment intervals is a reduction in the burden of treatment for patients and clinicians as well as a reduced socioeconomic burden. |
2. Are the extended intervals maintained over time? | 2. The clinical expert indicated that, in most cases, the extended interval is maintained over time, but also highlighted the fact that treatment intervals may be reduced, maintained, or extended in practice (i.e., there is greater flexibility in practice compared with the criteria used in the pivotal trials to determine the personalized treatment interval dosing regimen). |
3. In practice, how many patients could be treated at an extended interval? | 3. The clinical expert estimated that 40% to 50% of patients (as the upper limit) could be treated at an extended interval (i.e., every 4 months). |
Considerations for discontinuation of therapy | |
Is there a role for faricimab in patients whose condition has failed to respond to previous anti-VEGF therapies? | The clinical expert indicated there is presently no evidence to support this approach. |
Considerations for prescribing of therapy | |
Is there a potential to extend intervals for other anti-VEGF therapies for RVO?
| The clinical expert indicated that extending the treatment intervals to every 4 months or beyond and stopping therapy for other anti-VEGF therapies are both possible in patients with RVO. As noted previously, the clinical expert indicated that, in most cases, the extended interval is maintained over time, but also highlighted the fact that treatment intervals can be reduced, maintained, or extended in practice. The clinical expert further advised that such changes also depend on systemic factors (e.g., whether diabetes, hypertension, or other cardiovascular disease is controlled). |
Intraocular injection requires administration by a retina specialist. Is access to specialists appropriate to support patient populations and ongoing treatment regimens? | The clinical expert agreed that, ideally, treatment should be performed by a retina specialist. The expert also noted that in situations where a retina specialist is not available, such as in remote areas, the diagnosis of RVO and treatment with faricimab could be performed by a well-trained general ophthalmologist; however, they noted that the definition of a well-trained individual may differ between jurisdictions. |
Is the treat-and-extend approach an important consideration to help support ongoing access to treatment and support the growing patient population? | Please refer to the response to the question under “Considerations for continuation or renewal of therapy.” |
Is there potential for combination use of different anti-VEGF therapies? | The clinical expert did not anticipate combination use of different anti-VEGF therapies. |
System and economic issues | |
| This is a comment from the drug plans to inform CDEC deliberations. |
Certain programs in place within jurisdictions may affect the economic impact of faricimab. Anti-VEGFs are administered via intravitreal injection either at a hospital, ophthalmology clinic, or in a private ophthalmology clinic, and this varies across provinces and jurisdictions in Canada. | This is a comment from the drug plans to inform CDEC deliberations. |
AMD = age-related macular degeneration; BRVO = branch retinal vein occlusion; CDEC = Canadian Drug Expert Committee; DME = diabetic macular edema; pCPA = pan-Canadian Pharmaceutical Alliance; PM = pathologic myopia; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RVO = retinal vein occlusion.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of faricimab 6 mg/0.05 mL solution for intravitreal injection in the treatment of macular edema secondary to RVO. The focus will be placed on comparing faricimab injection with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of faricimab injection is presented in 2 sections, with the review team’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The review team’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 pivotal studies identified in the systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies, BALATON and COMINO, are summarized in Table 6.
BALATON and COMINO were phase III, multicentre, randomized, double-masked, active comparator–controlled, parallel-group, 2-part studies. The first part of the studies (day 1 through week 24) evaluated the efficacy, safety, and pharmacokinetics of faricimab every 4 weeks compared with aflibercept every 4 weeks in patients with macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial). The second part of the studies (week 24 through week 72) evaluated the efficacy, durability, safety, and pharmacokinetics of faricimab administered at masked treatment intervals of every 4, 8, 12, or 16 weeks based on personalized treatment interval dosing criteria, without an active control. Of note, there was no comparator in part 2 of the studies because all patients from part 1 received faricimab intravitreal injections according to a personalized treatment interval dosing regimen, plus sham procedures to maintain masking of the treatment intervals. The study design is depicted in Figure 1.
Table 6: Details of Studies Included in the Systematic Review
Detail | BALATON trial | COMINO trial |
|---|---|---|
Design and populations | ||
Study design | Phase III, randomized, double-masked, active comparator–controlled, parallel-group, 2-part study | |
Locations | Multicentre study conducted at 149 sites across 22 countries (0 sites in Canada) | Multicentre study conducted at a total of 192 sites across 22 countries (0 sites in Canada) |
Patient enrolment dates | First patient enrolled: March 2, 2021 Last patient last visit: June 12, 2023 | First patient enrolled: March 2, 2021 Last patient last visit (global enrolment phase only): July 12, 2023 |
Randomized (N) | N = 553:
| N = 729:
|
Inclusion criteria | General criteria:
Ocular criteria for the study eye:
| |
Exclusion criteria | General criteria:
Ocular criteria for both eyes:
Ocular criteria for study eye:
Ocular criteria for fellow nonstudy eye:
| |
Drugs | ||
Intervention | Part 1: Day 1 through week 24 Faricimab 6 mg intravitreal injections every 4 weeks for a total of 6 injections Part 2: Week 24 through week 72 Faricimab 6 mg intravitreal injections according to a personalized treatment interval dosing regimen (i.e., every 4, 8, 12, or 16 weeks) for a total of 3 to 12 injections A sham procedure that mimicked an intravitreal injection was conducted at visits when no faricimab injection was administered, as per the personalized treatment interval regimen | |
Comparator | Part 1: Day 1 through week 24 Aflibercept 2 mg intravitreal injections every 4 weeks for a total of 6 injections Part 2: Week 24 through week 72 No comparator; all patients received faricimab 6 mg intravitreal injections according to a personalized treatment interval dosing regimen plus sham procedures to maintain masking of the treatment intervals | |
Study duration | ||
Screening phase | Up to 28 days for baseline assessment | |
Treatment phase | Approximately 68 weeks followed by the final study visit at week 72 | |
Outcomes | ||
Primary end point | Change from baseline in BCVA at week 24 | |
Secondary, exploratory, and safety end points | Secondary End points assessed at both primary analysis (week 24) and final analysis (week 72):
End point assessed at the primary analysis:
End points assessed at the final analysis:
End points assessed in reference to week 24 at the final analysis:
Exploratory End points assessed at both the primary analysis and final analysis:
Safety
| |
Publication status | ||
Publication | Tadayoni, R., Paris, L.P., Danzig, C.J., et al., Efficacy and Safety of Faricimab for Macular Edema Due to Retinal Vein Occlusion: 24-Week Results From the BALATON and COMINO Trials. Ophthalmology. 2024;131(8):950-960. | |
ClincalTrials.gov identifier | NCT04740905 | NCT04740931 |
AE = adverse event; BCVA = best-corrected visual acuity; BRVO = branch retinal vein occlusion; C3F8 = octafluoropropane (perfluoropropane); CRC = central reading centre; CRVO = central retinal vein occlusion; CST = central subfield thickness; DME = diabetic macular edema; DR = diabetic retinopathy; ETDRS = Early Treatment Diabetic Retinopathy Study; FFA = fundus fluorescein angiography; HRVO = hemiretinal vein occlusion; nAMD = neovascular age-related macular degeneration; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RVO = retinal vein occlusion; SD-OCT = spectral-domain optical coherence tomography; SS-OCT = swept-source optical coherence tomography.
aBRVO was defined by retinal hemorrhages, telangiectatic capillary bed, dilated venous system, or other biomicroscopic evidence of RVO (neovascularization, vitreous hemorrhages) in no more than 1 quadrant of the retina drained by the affected vein.
bCRVO and HRVO were defined by retinal hemorrhages, telangiectatic capillary bed, dilated venous system or other biomicroscopic evidence of RVO (neovascularization, vitreous hemorrhages) in the entire retina (CRVO) or 2 quadrants of the retina (HRVO).
cCST was defined as the distance (measured in µm) between the internal limiting membrane and Bruch membrane, as assessed by CRC.
dCST for Cirrus SD-OCT and Topcon SD-OCT were standardized to Spectralis SD-OCT by the CRC.
eTreatment intervals at week 68 were defined as the treatment interval decision followed at week 68.
fAbsence was defined as an area of ischemic nonperfusion within the macula of 0 mm2 to 0.1 mm2.
gAbsence was defined as an area of leakage within the macular of 0 mm2.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO),16 final Clinical Study Report for GR41984 (BALATON),15 updated Clinical Study Report for GR41986 (COMINO),17 and version 1 of the statistical analysis plan for GR41984 and GR41986.24 Details included in the table are from the sponsor’s summary of clinical evidence.2
Figure 1: Design of the BALATON and COMINO Studies
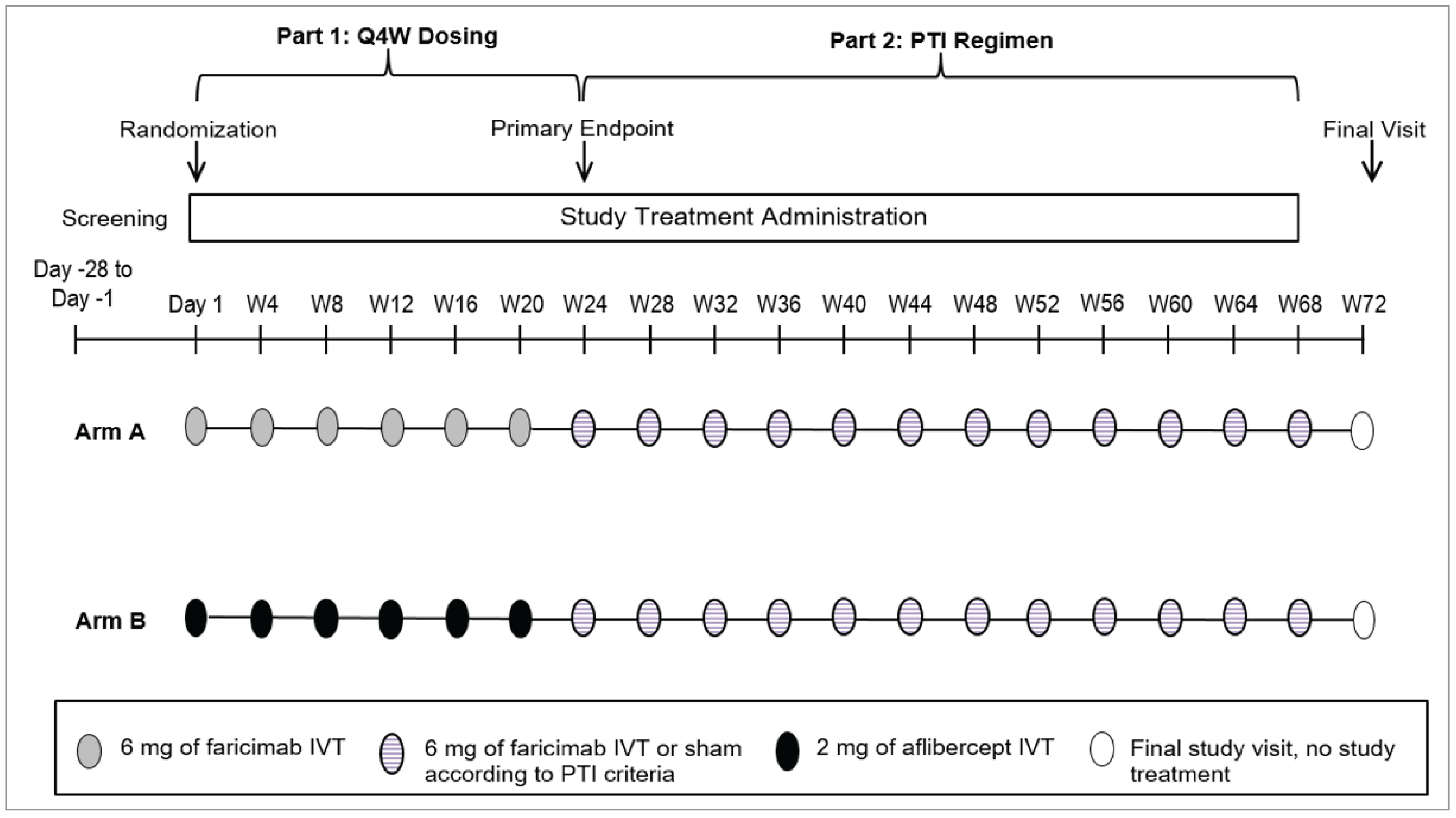
IVT = intravitreal; PTI = personalized treatment interval; Q4W = every 4 weeks; W = week.
Sources: Version 2 of the study protocols for GR41984 (BALATON)25 and GR41986 (COMINO).26
Patients were assessed for eligibility during screening, including through the review of colour fundus photographs and SD-OCT (or swept-source OCT) scans by the central reading centre. The diagnosis of macular edema due to RVO was confirmed by the central reading centre. Only 1 eye was assigned as the study eye. If both eyes were eligible, then the eye with the worse BCVA at screening was selected as the study eye.
In part 1 of each study, eligible patients were randomized in a 1:1 ratio through an interactive web-based response system to receive either faricimab 6 mg intravitreal injections every 4 weeks or aflibercept 2 mg intravitreal injections every 4 weeks. Randomization was stratified by the baseline BCVA ETDRS letter score (≥ 55 letters versus ≤ 54 letters in the BALATON trial and (≤ 34 letters, 35 to 54 letters, and ≥ 55 letters in the COMINO trial) and region (US and Canada, Asia, and the rest of the world). Notably, there were no study sites in Canada. In part 2 of each study, all patients received faricimab 6 mg intravitreal injections according to a personalized treatment interval dosing regimen (i.e., no active control was administered during this period). Study visits were scheduled at every 4 weeks for the entire study duration of 72 weeks.
An independent data monitoring committee (DMC) monitored safety and study conduct until the primary analysis was completed. A DMC evaluation of unmasked ocular and systemic safety events was prepared by an independent data coordinating centre every 6 months to provide recommendations to the sponsor regarding ongoing study conduct (e.g., stopping the study early for safety reasons).
Ophthalmology imaging and grading charters and a reading centre charter were prepared to outline the methods and procedures for independently grading medical images and supportive clinical data; the design, conduct, and reporting of grading imaging data; and the roles and responsibilities of the centre. Three central reading centres were used to conduct masked ocular imaging analyses and for storage.
An extension of the global enrolment phase specifically for China could be established (prespecified in the protocol) because patient recruitment was expected to take longer in China. After the global enrolment phase of the COMINO study was completed for the planned sample size for the primary analysis, additional patients continued to be enrolled at sites in China to reach sufficient enrolment to support registration in that country. The COMINO study’s China extension was ongoing at the time of the sponsor’s submission; therefore, data from patients enrolled during the China extension were not included in the primary analyses. However, all patients who were enrolled at sites in China during the global enrolment phase of COMINO were included in the primary analysis. A China extension was not needed for the BALATON trial because sufficient enrolment to support registration in China was reached before the global enrolment phase was completed.
The BALATON and COMINO studies were ongoing at the time of the primary analysis; the present report reflects a data cut-off date of July 6, 2022, and August 9, 2022, respectively, when all patients from the global enrolment phase had either completed the study through week 24 or had discontinued from the study before week 24.
The present report also presents data from the final analysis through week 72, corresponding to the last patient last visit date of June 12, 2023, in the BALATON trial and July 12, 2023, in the COMINO trial (global enrolment phase only).
Populations
Inclusion and Exclusion Criteria
Both the BALATON and COMINO studies included patients aged 18 years and older with foveal centre–involved macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial) diagnosed no longer than 4 months before screening and confirmed by the central reading centre based on SD-OCT (or swept-source OCT) images. The BCVA of the study eye had to be 73 to 19 letters, inclusive, as assessed on the ETDRS visual acuity chart at a starting test distance of 4 m on day 1. Further, the CST of the study eye at screening had to be 325 μm or greater, as measured on Spectralis SD-OCT, or 315 μm or greater, as measured on Cirrus SD-OCT or Topcon SD-OCT.
Both the BALATON and COMINO studies excluded patients with a history of retinal detachment or a macular hole (stage 3 or 4) in the study eye. Further, patients were excluded if they had received any prior treatment, or were receiving any current treatment, in the study eye for macular edema due to RVO or for other causes, macular neovascularization, or vitreomacular-interface abnormalities, or intravitreal treatment for any other retinal diseases that can lead to macular edema complications, or prior macular laser. Additionally, patients with a nonfunctioning fellow eye, defined as either BCVA of 20/320 or worse, or no physical presence of a fellow eye were excluded from the trials.
Interventions
Study Treatments
All intravitreal injections of the study drug were performed onsite. Investigators and patients were masked to treatment assignment in part 1 and to both the original treatment assignment and faricimab treatment interval in part 2.
At least 2 investigators were required per site to fulfill the masking requirements. One investigator was masked to treatment assignment and performed ocular assessments, including predose assessments and assessments at screening, at day 7, and at the final or early termination visit. One other investigator was unmasked and performed study treatments as well as post-dose administration safety assessments (i.e., finger counting, hand-motion and/or light-perception tests, intraocular pressure measurements) and treatment of AEs that occurred during or shortly after study treatment administration. Depending on the masked investigator’s decision, patients could self-administer predose and postdose antimicrobials.
Part 1 of the BALATON and COMINO Trials: Day 1 Through Week 24
Patients received intravitreal injections of either faricimab 6 mg or aflibercept 2 mg every 4 weeks for a total of 6 injections.
Dose modifications for faricimab and aflibercept were not permitted in the study. Study treatment interruption and patient discontinuation from study treatment due to AEs were permitted according to the prespecified criteria listed in the protocol.
Part 2 of the BALATON and COMINO Trials: Week 24 Through Week 72
All patients received faricimab 6 mg intravitreal injections every 4, 8, 12, or 16 weeks according to a personalized treatment interval dosing regimen for a total of 3 to 12 injections.
Starting at week 24, all patients received faricimab every 4 weeks until their CST value met the predefined threshold of less than 325 µm for Spectralis SD-OCT or less than 315 µm for Cirrcus SD-OCT and Topcon SD-OCT (initial reference CST), as determined by the central reading centre. If the established reference CST value was stable (i.e., did not increase or decrease by > 10%) with no associated loss of vision of 10 letters or greater relative to their reference BCVA (defined as the mean of the 3 best BCVA scores obtained at any prior dosing visit), then the patient was eligible to have their faricimab dosing interval extended. Thereafter, the treatment intervals were maintained or adjusted based on changes from the reference CST and BCVA values at the faricimab dosing visits. Decisions on the personalized treatment interval were automatically determined by the interactive web-based response system according to the criteria presented in Table 7.
If a patient’s dosing interval, previously extended, needed shortening due to disease progression, the patient was generally not permitted to return to a longer interval later in the study. For example, if the interval was reduced from every 12 weeks to every 8 weeks, then the interval was not extended beyond every 8 weeks for the remainder of the treatment period. However, if a patient’s interval was reduced to every 4 weeks, it could be extended again, but by no more than 4 weeks shorter than the patient’s maximum prior extension. For example, if the interval was reduced from every 16 weeks to every 4 weeks, then the interval could be extended up to every 12 weeks but could not be extended back to every 16 weeks.
Throughout the 72-week study duration, all patients were scheduled for study visits every 4 weeks. To maintain masking of the faricimab treatment intervals, a sham procedure that mimicked an intravitreal injection was conducted during visits when no faricimab injection was administered, per their personalized treatment interval regimen. The sham procedure involved the blunt end of an empty syringe, without a needle, being pressed against an anesthetized eye.
Permitted Therapies
The therapies permitted during the study included maintenance therapies, including, but not limited to, the following:
Treatment for onset of ocular hypertension or glaucoma in the study eye, as indicated.
Vitrectomy could be performed at the discretion of the masked investigator in the event that the study eye developed a sight-threatening vitreous hemorrhage or retinal detachment. These conditions were recorded as SAEs and the study treatment would be interrupted and restarted based on patient status, after discussion with the medical monitor.
Treatment for the onset of cataract or posterior capsular opacification in either eye, as indicated, was permitted. Dose interruption criteria could apply with cataract surgery.
The short-term use of topical ocular corticosteroids after cataract surgery, yttrium-aluminum-garnet capsulotomy, peripheral iridotomy, argon or selective laser trabeculoplasty, or the presence of ocular allergic conditions in either eye was allowed.
Complete, sector, or local panretinal photocoagulation in either eye was permitted if needed for the treatment of ischemic RVO or new peripheral neovascularization, after discussion with the medical monitor. Both conditions were recorded as SAEs.
At the discretion of the masked investigator, the fellow eye could be treated with an anti-VEGF therapy licensed for ocular use if the patient were diagnosed with an ocular condition for which the selected anti-VEGF therapy had been approved by the country’s regulatory agency. Masking was maintained by having the unmasked physician administer treatment with an anti-VEGF if it was to be provided to the fellow eye at the same visit as the study eye treatment.
Table 7: Algorithm for the IxRS-Determined Faricimab Personalized Treatment Interval Dosing Regimen in Part 2 of the BALATON and COMINO Studies
Personalized treatment interval | Criteria |
|---|---|
Interval extended by 4 weeks | If the CST value increased or decreased by ≤ 10% without an associated ≥ 10-letter decrease in BCVA |
Interval maintained | If any of the following criteria were met:
|
Interval reduced by 4 weeks | If any of the following criteria were met:
|
Interval reduced to every 4 weeks | If the CST value increased > 10% with an associated ≥ 10-letter BCVA decrease |
BCVA = best-corrected visual acuity; CST = central subfield thickness; IxRS = interactive web-based response system; SD-OCT = spectral-domain optical coherence tomography.
Notes: The algorithm used by the IxRS for interval decision-making is presented in the preceding table. The criteria were based on the relative change of the CST and BCVA values at the faricimab dosing visits compared with the reference CST and BCVA values.
The initial reference CST value was defined as the CST value when the CST threshold was initially met (< 325 µm for Spectralis SD-OCT or < 315 µm for Cirrcus SD-OCT and Topcon SD-OCT, as determined by the central reading centre), but no earlier than week 20. Reference CST was adjusted if CST decreased by > 10% from the previous reference CST for 2 consecutive faricimab dosing visits and the values obtained were within 30 µm. The CST value obtained at the latter visit served as the new reference CST, starting immediately at that visit.
The reference BCVA was defined as the mean of the 3 best BCVA scores obtained at any prior dosing visit.
Sources: Version 2 protocols for the GR41984 (BALATON)25 and GR41986 (COMINO)26 studies.
Prohibited Therapies
The therapies prohibited during the study included the following:
systemic anti-VEGF therapy
bevacizumab (highlighted by the investigator because it is not licensed for ophthalmic use in any participating country)
systemic drugs known to cause macular edema (fingolimod and tamoxifen)
intravitreal anti-VEGF drugs other than study-assigned aflibercept and faricimab in the study eye
intravitreal or periocular steroid implants, or chronic topical ocular corticosteroids in the study eye
treatment with verteporfin in the study eye
administration of micropulse and focal or grid laser in the study eye
other experimental therapies, with the exception of vitamins and minerals.
Outcomes
A list of the efficacy end points assessed in this Clinical Review Report is provided in Table 8 followed by descriptions of the outcome measures. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical expert consulted, and input from the patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All of the efficacy end points that were summarized were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
The following outcome measures and their measurement properties are presented in Table 9.
Table 8: Outcomes Summarized From the BALATON and COMINO Studies
Outcome measure | Time point | Type of end point |
|---|---|---|
Visual acuity | ||
Change from baseline in BCVA | At week 24 | Primary |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline | At week 24 | Secondary |
Anatomic | ||
Change from baseline in CSTa | At week 24 | Secondary |
Proportion of patients with an absence of macular edema, defined as CST of < 325 µmb | ||
Proportion of patients with an absence of both intraretinal fluid and subretinal fluidc | ||
Vision-related functioning and health-related quality of life | ||
Change from baseline in the NEI-VFQ-25 composite score | At week 24 | Secondary |
Treatment interval | ||
Proportion of patients on a q.4.w., q.8.w., q.12.w., or q.16.w. treatment interval | At week 68d | Secondary |
Notable harms | ||
Endophthalmitis | At week 24 | Safety |
BCVA = best-corrected visual acuity; CST = central subfield thickness; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SD-OCT = spectral-domain optical coherence tomography.
aCST was defined as the distance between the internal limiting membrane and Bruch membrane as assessed by the central reading centre.
bCST for Cirrus SD-OCT or Topcon SD-OCT were standardized to Spectralis SD-OCT by the central reading centre.
cIntraretinal fluid and subretinal fluid were as measured in the central subfield (centre 1 mm).
dTreatment intervals at week 68 were defined as the treatment interval decision followed at week 68.
Sources: Version 1 of the statistical analysis plan for the GR41984 (BALATON) and GR41986 (COMINO) studies.24 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ETDRS charts (letters) | A chart that measures visual acuity. ETDRS charts present a series of 5 letters of equal reading difficulty in each row, with standardized spacing between letters and rows comprising a total of 14 lines (70 letters). Letters range from 58.18 mm to 2.92 mm in height, corresponding to Snellen visual acuity fractions of 20/200 to 20/10, respectively. Charts are used in a standard light box. The standard testing distance is 4 m. Visual acuity is documented as the smallest line read by each eye in the absence of any errors.27 Scores are based on the number of letters correctly read by the patient. An ETDRS letter score can be calculated when 20 or more letters are read correctly at 4.0 m, i.e., the visual acuity letter score is equal to the total number of letters read correctly at 4.0 m plus 30. A maximum score is 100.28 Higher scores indicate better visual acuity. | Validity: No relevant evidence of validity found in patients with RVO. Reliability: No relevant evidence of reliability found in patients with RVO. Responsiveness: No relevant evidence of responsiveness found in patients with RVO. | No MID has been estimated in the ETDRS in patients with RVO. |
CST as assessed by OCT | OCT is a noninvasive instrument commonly used in clinical practice and trial settings for the diagnosis, management, and follow-up of optic nerve and retinal disorders. OCT is commonly used to measure retinal thickness, an important outcome in retinal diseases. OCT creates cross-sectional maps of the retinal structures, identifying differences in the reflectance of retinal layers and borders.29 Of note, OCT technology has shifted from TD-OCT to SD-OCT because the latter can acquire data at a higher speed with better image resolution and reduced motion artifacts.30 | Validity: No relevant evidence of validity found in patients with RVO. Reliability: No relevant evidence found in patients with RVO. Responsiveness: No relevant evidence found in patients with RVO. | No MID has been estimated in CST in patients with RVO. |
NEI-VFQ-25 | The NEI-VFQ-25 assesses the impact of visual impairment on vision-targeted health-related quality of life across a broad range of eye conditions.31 The NEI-VFQ-25 (a shortened version of the original 51-item questionnaire) is administered as an interview and consists of 25 items relevant to 11 subscales, in addition to a single-item general health component.32 Each subscale score is the average score of all items in the subscale transformed to a 0 to 100 scale, with 0 indicating the worst possible score and 100 indicating the best possible score. The composite score is the unweighted average score of all items except for the general health rating, which is considered a standalone item representing overall health status.32 | Validity: No relevant evidence found in patients with RVO. Reliability: No relevant evidence found in patients with RVO. Responsiveness: No relevant evidence found in patients with RVO. | No MID has been estimated in the NEI-VFQ-25 in patients with RVO. |
CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; MID = minimal important difference; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; OCT = optical coherence tomography; RVO = retinal vein occlusion; SD-OCT = spectral-domain optical coherence tomography; TD-OCT = time-domain optical coherence tomography.
Visual Acuity
The assessments of visual acuity that were determined to be the most relevant for this review were change in BCVA and the proportion of patients with an improvement in BCVA. These outcomes provide information on the degree of improvement in visual acuity and the proportion of patients with an improvement in visual acuity, respectively.
Change from baseline in BCVA at week 24 was the primary end point, and the proportion of patients gaining 15 letters or greater in BCVA at week 24 was a secondary end point in both studies. At each study visit, BCVA was assessed by masked examiners using the ETDRS visual acuity chart at a starting test distance of 4 m for both eyes. Scores are based on the number of letters read correctly on the ETDRS chart, with higher scores indicating better visual acuity (the maximum score is 100 letters).
Anatomic
The assessments of the anatomy of the study eye that were determined to be the most relevant for this review were change in CST as measured by OCT and the proportion of patients with an absence of macular edema and fluid. Based on the clinical expert input, these outcomes provide information on the extent of improvement in tissue swelling or edema, the physiological environment (i.e., re-establishment of the blood–retina barrier), and the presence or absence of cystoid spaces, respectively.
Change from baseline in the CST at week 24, the proportion of patients with an absence of macular edema (defined as CST of < 325 µm) at week 24, and the proportion of patients with an absence of both intraretinal fluid and subretinal fluid (as measured in the central subfield; centre 1 mm) at week 24 were secondary end points in both studies. CST was defined as the distance measured between the internal limiting membrane and Bruch membrane, standardized to Spectralis SD-OCT. SD-OCT (or swept-source OCT, if applicable) imaging was performed for both eyes at each study visit by masked OCT technicians. Images were forwarded to the central reading centres for independent analysis; the analysis was based on the same type of scan and evaluated over time within a patient. The data resulting from this masked review of ocular images were forwarded to the sponsor and the SD-OCT CST values were forwarded to the interactive web-based response system for the determination of the treatment interval.
Vision-Related Functioning and HRQoL
The assessment of vision-related functioning and HRQoL that was determined to be the most relevant for this review was the change in the NEI-VFQ-25 composite score. The NEI-VFQ-25 is used to assess a patient’s perception of vision-related functioning and HRQoL. The questionnaire comprises 11 vision-related subscales (25 items) and a question on general health (1 item). Subscales include general vision, ocular pain, near activities, distance activities, social functioning, mental health, role difficulties, dependency, driving, colour vision, and peripheral vision. Specific to the BALATON and COMINO studies, an additional 6 appendix items were included in the near activities and distance activities subscales.
Change from baseline in the NEI-VFQ-25 composite score at week 24 was a secondary end point in both studies. The NEI-VFQ-25 was administered by the masked site staff at the clinic or over the telephone. Specifically, the NEI-VFQ-25 was administered before the patient received their disease status, the performance of other assessments, and the administration of study treatment, unless otherwise specified. The composite and subscale scores range from 0 to 100, with higher scores indicating better vision-related functioning and HRQoL.
Treatment Intervals
Treatment interval was identified as a relevant outcome for this review because the clinician groups indicated that an ideal treatment demonstrates a durable effect (i.e., demonstrates efficacy in sustaining improvement in visual acuity over the long-term) as measured by a reduction in the treatment burden associated with repetitive intravitreal injections.
The proportion of patients on each treatment interval of every 4, 8, 12, or 16 weeks at week 68 was a secondary end point in both studies. The treatment intervals at week 68 were defined as the treatment interval decision followed at week 68. The algorithm for the interactive web-based response system that determined the personalized treatment interval dosing regimen for faricimab in part 2 of the BALATON and COMINO studies is presented in Table 7 and discussed in detail in the preceding interventions section.
Safety
Safety assessments consisted of monitoring and recording AEs, including SAEs and AEs of special interest; performing protocol-specified safety laboratory assessments; measuring protocol-specified vital signs; and conducting other protocol-specified tests. AEs were assessed at each study visit.
An independent clinical events coding committee adjudicated the thromboembolic events reported during the study. APTC events were defined as nonfatal strokes or nonfatal myocardial infarctions or vascular deaths (including deaths of unknown cause).
Statistical Analysis
Statistical Hypotheses and Multiple Testing Procedure
Two hypotheses on the primary end point of change from baseline in BCVA at week 24 were tested in the following order:
The noninferiority of faricimab every 4 weeks compared with aflibercept every 4 weeks at week 24 in the intention-to-treat (ITT) population at a 1-sided significance level of 0.02485.
The superiority of faricimab every 4 weeks compared with aflibercept every 4 weeks at week 24 in the ITT population at a 2-sided significance level of 0.0497.
If statistical significance was achieved on the noninferiority test, then the test for superiority could proceed.
If the lower bound of a 2-sided 95.03% CI for the difference between treatments was greater than –4 letters (noninferiority margin), intravitreal faricimab 6 mg every 4 weeks was considered noninferior to intravitreal aflibercept 2 mg every 4 weeks.
If the lower bound of a 2-sided 95.03% CI for the difference between treatments was greater than 0 letters, then the test was considered positive, and intravitreal faricimab 6 mg every 4 weeks was considered superior to intravitreal aflibercept 2 mg every 4 weeks.
Statistical testing for the secondary end points was undertaken without adjustment for multiple testing.
A nominal type I error penalty of 0.0001 (2-sided) was applied for each time the DMC reviewed unmasked data before the formal analysis of the primary efficacy end point. The investigators did not anticipate that this type I error adjustment would impact sample size and power. At the time of the primary analysis, it was estimated the DMC would conduct 3 reviews of interim safety data; therefore, the efficacy analyses would be performed with a familywise significance level of 0.0497. The actual adjustment was dependent on the actual number of interim safety reviews conducted by the DMC.
Noninferiority Margin
The 4-letter noninferiority margin was selected based on the statistical rationale for preserving approximately 50% of the benefit compared with sham control, as estimated by the lower limit of a 95% CI, based on the aflibercept pivotal trials in BRVO and CRVO.33-35 More specifically, based on the results of the aflibercept pivotal trials, the 4-letter noninferiority margin preserved between 44% and 77% of the least estimated benefit of aflibercept relative to a control (sham or grid laser).
Sample Size and Power Calculation
The sample size was determined based on the number of patients enrolled in the global enrolment phase; approximately 570 patients for the BALATON trial and 750 patients for the COMINO trial were planned and then randomized in a 1:1 ratio to receive treatment with either faricimab or aflibercept.
The investigators indicated that a sample size of 285 patients per group for the BALATON trial and 375 patients per group for the COMINO trial would provide greater than 90% power to demonstrate the noninferiority of faricimab to aflibercept based on change in BCVA at week 24 in the ITT population, using the 4-letter noninferiority margin and under the following assumptions:
no difference in the mean change from baseline in BCVA between the treatment groups
an SD of 13 letters for the BALATON trial and 15 letters for the COMINO trial for the change from baseline in BCVA at week 24
2-sample t tests
2.5% 1-sided type I error rate
10% dropout rate.
Further, the investigators indicated that the planned sample sizes would provide greater than 80% power to demonstrate a superiority of 3.5 letters for faricimab over aflibercept, under the same SD, test, and dropout assumptions as shown in the preceding list, and a 2-sided type I error rate of 5%.
The investigators also indicated that the planned sample size would provide adequate power for the PP analysis accounting for the potential impact of COVID-19; however, the sponsor decided to stop recruitment at an under-recruitment level of 3% because the impact of COVID-19 was less than anticipated. Enrolment ended after 553 patients were enrolled in the BALATON trial and 730 patients were enrolled in the COMINO trial. The final sample size provided greater than 90% power for the noninferiority assessment and greater than 80% power for the superiority test.
For the China extension, the investigators anticipated that 62 patients would be enrolled in the BALATON trial and 82 patients would be enrolled in the COMINO trial; this included patients who had already been enrolled during the global enrolment phase. The data from patients enrolled at study sites in China during the global enrolment phase of the study were included in the primary analysis; although the data from the patients enrolled at study sites in China during the China extension were not included in the primary analyses, these data will be included in the China subpopulation analysis.
Statistical Model
General Considerations
The analyses described in this section were based on the patients enrolled during the global enrolment phase, excluding the China extension, unless otherwise specified. The efficacy analysis at the primary analysis was based on data through week 24; the efficacy end points at week 24 were measured before the week 24 injection and were included in the comparative analysis.
The analyses of the efficacy outcome measures were stratified by baseline BCVA ETDRS letter score as assessed on day 1 (≥ 55 letters versus ≤ 54 letters for the BALATON trial; ≤ 34 letters versus 35 to 54 letters versus ≥ 55 letters for the COMINO trial) and region (US and Canada, Asia, and the rest of the world).
Continuous outcomes were analyzed using an MMRM. The binary secondary end points were analyzed using a stratified estimation for binomial proportions. The estimates and associated CIs were provided for the mean for continuous variables and the proportion for binary variables for each treatment group, and for the respective differences between aflibercept and faricimab, where applicable. All CIs were 2-sided and at the 95.03% level.
Invalid BCVA values that were confirmed to be inaccurate because the BCVA test was performed incorrectly were excluded from the analyses. Nonstandard BCVA data were included in the analyses, including as assessed by ETDRS BCVA testing with prior visit refraction, and a BCVA test performed by an unmasked certified ETDRS BCVA assessor or by an uncertified experienced ETDRS BCVA assessor. A supplementary PP analysis was performed with the nonstandard BCVA data excluded.
Intercurrent events were defined as the discontinuation of study treatment due to AEs or a lack of efficacy, and the use of any prohibited systemic treatment or prohibited therapy in the study eye, as per protocol. For the primary and secondary end points, intercurrent events were handled by applying a treatment policy strategy.
Primary End Point Analysis
The main analysis for the primary end point of change from baseline in BCVA at week 24 was performed using an MMRM. The model included the following categorical covariates as fixed effects: treatment group, visit, visit by treatment group interactions, baseline BCVA (continuous), and randomization stratification factors. The MMRM model assumed an unstructured covariance structure. It there were convergence problems in the model, then a heterogenous compound symmetry or an autoregressive covariance structure could be fitted.
No interim efficacy or futility analyses were planned or conducted.
Data Imputation Methods
Data for patients with missing baseline assessments were not imputed.
Specific to the primary end point, missing data were implicitly imputed by the MMRM model assuming missing at random.
Sensitivity Analysis
To evaluate the robustness of the main analysis finding, a sensitivity analysis using multiple imputation was performed for the primary efficacy end point (a different approach to handling missing data). Missing BCVA data were assumed to be missing not at random. Each group was imputed separately for patients with missing BCVA data at week 24. An approximate Bayesian bootstrap method was used for imputation. These patients were assumed to have worse outcomes compared with the rest of the population and were imputed from patients with nonmissing BCVA data with the worst outcomes. The number of imputations was set to 100. The analysis was performed using an analysis of covariance model with adjustment for the following covariates: treatment group, baseline BCVA (continuous), and randomization stratification factors.
Supplementary Analyses
To further the understanding of treatment effect, the following supplementary analyses were performed for the primary efficacy end point: a PP analysis, an analysis distinguishing COVID-19–related and non-COVID-19–related intercurrent events, and an analysis using a hypothetical strategy for all intercurrent events.
Of interest to this review were the PP analysis and the analysis using a hypothetical strategy for all intercurrent events.
The PP analysis was based on the PP population; patients with major protocol deviations that impacted the efficacy evaluation were excluded from the analysis.
In the analysis using a hypothetical strategy for all intercurrent events, all values were censored after the occurrence of the intercurrent event to estimate a treatment effect in the absence of intercurrent events. Data censored after intercurrent events as per the hypothetical strategy, and missing data due to reasons other than intercurrent events were both implicitly imputed by the MMRM model assuming missing at random.
Subgroup Analyses
The following subgroups were analyzed with respect to the primary end point using the same method described previously:
baseline BCVA (note that the baseline covariate consisting of the BCVA stratification factor was removed from the model)
BALATON trial (BRVO): 54 letters or fewer and 55 letters or greater
COMINO trial (CRVO and HRVO): 34 letters or fewer, 35 to 54 letters, and 55 letters or greater
low vision of 23 letters or fewer and 24 letters or greater (analysis was performed if a sufficient sample size was available to draw meaningful conclusions)
region (note that baseline covariate consisting of region stratification factor was removed from the model)
US and Canada, Asia, and the rest of the world
baseline age: younger than 65 years and 65 years and older
sex: female and male
race: white, Asian, and other
baseline RVO status (CRVO and HRVO; applicable only to the COMINO trial).
A forest plot was presented to summarize the results. The subgroup categories could be combined if there was insufficient representation of a specific subpopulation.
Of the subgroups listed previously, baseline RVO status was identified as the most relevant for the purpose of this review to inform expert committee deliberations.
Secondary Outcomes
No formal superiority test was performed for secondary end points. No sensitivity analysis was performed for any secondary end points.
Secondary End Points at the Primary Analysis
At the primary analysis, nominal superiority P values were provided for secondary end points, where applicable, for reference purposes only.
The continuous secondary end points at the primary analysis were analyzed using the same analysis methods used for the main analysis of the primary end point, except for change from baseline in the NEI-VFQ-25 composite score, which was analyzed using an analysis of covariance. The missing data for the change from baseline in the NEI-VFQ-25 composite score at week 24 were not imputed; all observed available data were included in the analysis regardless of intercurrent events.
Binary secondary end points at the primary analysis were analyzed using Cochran-Mantel-Haenszel. The proportion of patients in each treatment group and the difference in proportions between treatment groups were estimated using the weighted average of the observed proportions and the differences in observed proportions over the strata defined by the randomization stratification factor for the baseline BCVA score and region using the Cochran-Mantel-Haenszel weights.36,37 The CIs were calculated using the normal approximation to the weighted proportions.38 If the response rate was low, an unstratified analysis could be performed. Missing data were imputed using LOCF.
For the proportion of patients gaining 15 letters or greater in BCVA from baseline at week 24, which was a secondary end point, a supplementary analysis was also performed at the primary analysis. The supplementary analysis was performed using the same estimand and analysis method that was used for the main analysis of the binary secondary end points but with the following differences:
Handling of intercurrent events: A composite variable strategy was applied in which it was assumed that the disease of those patients with an intercurrent event had failed treatment after the intercurrent event.
Handling of missing data (without intercurrent events): If BCVA at weeks 16, 20, and 24 were all missing, then the disease of those patients was assumed to have failed treatment at week 24; otherwise, missing data were imputed using LOCF.
Secondary End Points at the Final Analysis
At the final analysis, no comparisons between treatment groups were made because all patients received faricimab during part 2 of each study. The proportions of patients on a treatment interval of every 4 weeks, every 8 weeks, every 12 weeks, and every 16 weeks at week 68 were analyzed using descriptive statistics, with the percentage based on the patients who had not discontinued the study at week 68.
Safety Analysis
At the primary analysis, safety was assessed through a descriptive summary based on data obtained before week 24 and the assessments at week 24 that were measured before the administration of the week 24 injection (i.e., data through week 24).
At the final analysis, safety was assessed through a descriptive summary based on data through week 72 according to the following groups: faricimab every 4 weeks before week 24, aflibercept every 4 weeks before week 24, faricimab personalized treatment interval (faricimab every 4 weeks in part 1) on and after week 24, faricimab personalized treatment interval (aflibercept every 4 weeks in part 1) on and after the first dose of faricimab, and all patients who had received at least 1 dose of faricimab with safety data on and after the first dose of faricimab.
In consultation with the clinical expert, no notable harm (in the context of RVO and faricimab) monitored at week 72 was identified. As such, the safety summary through week 72 was not included in this report.
Analysis Populations
Efficacy analyses were based on the ITT population, defined as all patients who were randomized in the study. Supplementary analysis of the primary end point was based on the PP population, defined as all patients randomized in the study who received at least 1 dose of the study treatment and who did not have a major protocol deviation that impacted the efficacy evaluation. Before study unmasking, protocol deviations were reviewed and a determination of the definition of the population for PP analysis was made.
Results
Patient Disposition
A summary of patient disposition through week 24 and week 72 from the BALATON and COMINO studies is presented in Table 12 and Table 13, respectively.
BALATON Trial
A total of 768 patients were screened, of which 215 patients were not randomized in the study due to screening failure; the reasons for screening failure were not reported.
Table 10: Statistical Analysis of Efficacy End Points From the BALATON and COMINO Studies
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Change from baseline in BCVA at week 24 | MMRM | Categorical covariates as fixed variables:
| Missing baseline assessments were not imputed Missing data were implicitly imputed by the MMRM model assuming missing at random | A sensitivity analysis using multiple imputation was performed as a different approach to handling missing data:
The analysis was performed using ANCOVA adjusted for the following covariates:
|
Change from baseline in CST at week 24 | MMRM | Categorical covariates as fixed variables:
| Missing baseline assessments were not imputed Missing data were implicitly imputed by the MMRM model assuming missing at random | Not performed |
Change from baseline in the NEI-VFQ-25 composite score at week 24 | ANCOVA | Categorical covariates as fixed variables:
| Missing data were not imputed | Not performed |
Proportion of patients with an absence of macular edema, defined as CST of < 325 µm at week 24 | CMH | Randomization stratification factors:
| Missing data were imputed using LOCF | Not performed |
Proportion of patients with an absence of both intraretinal fluid and subretinal fluid at week 24 | CMH | Randomization stratification factors:
| Missing data were imputed using LOCF | Not performed |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline at week 24 | CMH | Randomization stratification factors:
| Missing data were imputed using LOCF | Not performed |
Proportion of patients on a q.4.w., q.8.w., q.12.w., or q.16.w. treatment interval at week 68 | Descriptive statistics | Not applicable | Not applicable | Not applicable |
ANCOVA = analysis of covariance; BCVA = best-corrected visual acuity; CMH = Cochran-Mantel-Haenszel; CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; LOCF = last observation carried forward; MMRM = mixed model for repeated measures; NEI-VFQ-25 = National Eye Institute Visual Functioning Questionnaire–25; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; vs. = versus.
Sources: Version 1 of the statistical analysis plan for GR41984 (BALATON) and GR41986 (COMINO).24 Details included in the table are from the sponsor’s summary of clinical evidence.2
A total of 276 patients were randomized to receive faricimab every 4 weeks and 277 patients were randomized to receive aflibercept every 4 weeks. Of those who were randomized, 1.4% (4 patients) in the faricimab group and 1.1% (3 patients) in the aflibercept group discontinued the study before week 24. Each reason for early discontinuation from the study, including lost to follow-up, AEs, withdrawal by patient, and protocol deviation, was reported in less than 2.0% of patients who were randomized in each group. Of those who were randomized, 11.2% (31 patients) in the group of patients who were randomized to receive faricimab in part 1 and 11.9% (33 patients) in the group of patients who were randomized to receive aflibercept in part 1 discontinued the study before week 72. Each reason for early discontinuation from the study was reported in approximately 5.0% of patients or less who were randomized in each group for part 1 of the study.
Table 11: Analysis Populations of the BALATON and COMINO Studies
Population | Definition | Application |
|---|---|---|
ITT | All patients who were randomized in the studya | Efficacy analyses |
PP | All patients randomized in the study who received at least 1 dose of the study treatment and who did not have a major protocol violation that impacted the efficacy evaluationb,c | Supplementary analysis of the primary end point |
Safety-evaluable | All patients who received at least 1 injection of the active study drug (faricimab or aflibercept) in the study eyeb | Safety analyses |
PP = per protocol; ITT = intention to treat.
aPatients were grouped according to the treatment assigned at randomization. Patients who were incorrectly randomized to 1 study, discontinued without treatment, and were then randomized to the other study (COMINO or BALATON), were included in the latter study only.
bPatients were grouped according to the actual treatment received through week 20. If, by error, a patient received a combination of different active study drugs (faricimab and aflibercept) in the study eye, the patient’s treatment group was as randomized.
cBefore study unmasking, protocol deviations were reviewed and a determination of the definition of the population for PP analysis was made.
Sources: Version 1 of the statistical analysis plan for the GR41984 (BALATON) and GR41986 (COMINO) trials.24 Details included in the table are from the sponsor’s summary of clinical evidence.2
Of those who received treatment, 3.3% (9 of 276 patients) in the faricimab group and 1.1% (3 of 274 patients) in the aflibercept group discontinued study treatment in their study eye before week 24. Each reason for early discontinuation of the study treatment, including lost to follow-up, withdrawal by patient, AEs, and death, was reported in less than 2.0% of patients who received treatment in each group. Of those who received treatment, 11.6% (32 patients) in the group of patients who were randomized to receive faricimab in part 1 and 10.2% (28 patients) in the group of patients who were randomized to receive aflibercept in part 1 discontinued study treatment in their study eye before week 72. Each reason for early discontinuation of the study treatment was reported in approximately 5.0% of patients or less who received treatment in each group.
COMINO Trial
A total of 1,004 patients were screened, of which 274 patients were not randomized in the study due to screening failure; the reasons for screening failure were not reported.
A total of 366 patients were randomized to receive faricimab every 4 weeks and 363 patients were randomized to receive aflibercept every 4 weeks. Of those who were randomized, 1.6% (6 patients) in the faricimab group and 2.8% (10 patients) in the aflibercept group discontinued from the study before week 24. Each reason for early discontinuation, including lost to follow-up, AEs, withdrawal by patient, protocol deviation, death, noncompliance with study drug, physician decision, and other reasons, was reported in less than 2.0% of patients who were randomized in each group. Of those who were randomized, 9.0% (33 patients) in the group of patients who were randomized to receive faricimab in part 1 and 11.0% (40 patients) in the group of patients who were randomized to receive aflibercept in part 1 discontinued the study before week 72. Each reason for early discontinuation from the study was reported in less than 5.0% of patients who were randomized in each group for part 1 of the study.
Of those who received treatment, 3.0% (11 of 365 patients) in the faricimab group and 4.2% (15 of 361 patients) in the aflibercept group discontinued the study treatment in their study eye before week 24. Each reason for early discontinuation of the study treatment, including lost to follow-up, withdrawal by patient, AEs, death, physician decision, noncompliance with study drug, and other reasons, was reported in less than 2.0% of patients who received treatment in each group. Of those who received treatment, 9.9% (36 patients) in the group of patients who were randomized to receive faricimab in part 1 and 11.6% (42 patients) in the group of patients who were randomized to receive aflibercept in part 1 discontinued study treatment in their study eye before week 72. Each reason for early discontinuation of the study treatment was reported in less than 5.0% of patients who received treatment in each group.
Table 12: Summary of Patient Disposition Through Week 24 From the BALATON and COMINO Studies
Patient disposition | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab | Aflibercept | Faricimab | Aflibercept | |
Screened, N | 768 | 1,004 | ||
Screening failure, N | 215 | 274 | ||
Reason for screening failure, n (%) | NR | NR | ||
Randomized, N | 276 | 277 | 366 | 363 |
Treated, n (%) | 276 (100) | 274 (98.9) | 365 (99.7) | 361 (99.4) |
Discontinued study treatment in the study eye before week 24, n (%)a | 9 (3.3) | 3 (1.1) | 11 (3.0) | 15 (4.2) |
Reason for discontinuation, n (%)a | ||||
Lost to follow-up | 5 (1.8) | 2 (0.7) | 0 | 4 (1.1) |
Withdrawal by patient | 2 (0.7) | 1 (0.4) | 3 (0.8) | 6 (1.7) |
Adverse events | 1 (0.4) | 0 | 2 (0.5) | 2 (0.6) |
Death | 1 (0.4) | 0 | 1 (0.3) | 2 (0.6) |
Physician decision | 0 | 0 | 2 (0.5) | 1 (0.3) |
Noncompliance with study drug | 0 | 0 | 2 (0.5) | 0 |
Other | 0 | 0 | 1 (0.3) | 0 |
Discontinued study before week 24, n (%)b | 4 (1.4) | 3 (1.1) | 6 (1.6) | 10 (2.8) |
Reason for discontinuation, n (%)b | ||||
Lost to follow-up | 2 (0.7) | 0 | 0 | 1 (0.3) |
Adverse events | 1 (0.4) | 0 | 1 (0.3) | 1 (0.3) |
Withdrawal by patient | 1 (0.4) | 0 | 0 | 4 (1.1) |
Protocol deviation | 0 | 3 (1.1) | 0 | 1 (0.3) |
Death | 0 | 0 | 1 (0.3) | 2 (0.6) |
Noncompliance with study drug | 0 | 0 | 2 (0.5) | 0 |
Physician decision | 0 | 0 | 0 | 1 (0.3) |
Other | 0 | 0 | 2 (0.5) | 0 |
ITT, N | 276 | 277 | 366 | 363 |
PP, N | 241 | 243 | 328 | 311 |
Safety, N | 276 | 274 | 365 | 361 |
ITT = intention to treat; NR = not reported; PP = per protocol.
Note: The data presented in the preceding table include discontinuations occurring before day 154 (first day of week 24 analysis visit window).
aPercentages are based on number of patients treated.
bPercentages are based on number of patients randomized.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 13: Summary of Patient Disposition Through Week 72 From the BALATON and COMINO Studies
Patient disposition | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI | Aflibercept q.4.w. to faricimab PTI | Faricimab q.4.w. to faricimab PTI | Aflibercept q.4.w. to faricimab PTI | |
Randomized, N | 276 | 277 | 366 | 363 |
Discontinued study treatment in the study eye before week 72, n (%)a | 32 (11.6) | 28 (10.2) | 36 (9.9) | 42 (11.6) |
Reason for discontinuation, n (%)a | ||||
Withdrawal by patient | 14 (5.1) | 11 (4.0) | 11 (3.0) | 15 (4.2) |
Lost to follow-up | 10 (3.6) | 6 (2.2) | 3 (0.8) | 10 (2.8) |
Death | 2 (0.7) | 2 (0.7) | 4 (1.1) | 3 (0.8) |
Noncompliance with study drug | 2 (0.7) | 1 (0.4) | 4 (1.1) | 0 |
Lack of efficacy | 2 (0.7) | 0 | 0 | 0 |
Adverse events | 1 (0.4) | 4 (1.5) | 8 (2.2) | 10 (2.8) |
Physician decision | 0 | 4 (1.5) | 4 (1.1) | 3 (0.8) |
Other | 1 (0.4) | 0 | 2 (0.5) | 1 (0.3) |
Discontinued study before week 72, n (%)b | 31 (11.2) | 33 (11.9) | 33 (9.0) | 40 (11.0) |
Reason for discontinuation, n (%)b | ||||
Withdrawal by patient | 14 (5.1) | 12 (4.3) | 11 (3.0) | 15 (4.1) |
Lost to follow-up | 9 (3.3) | 6 (2.2) | 3 (0.8) | 10 (2.8) |
Death | 2 (0.7) | 2 (0.7) | 5 (1.4) | 3 (0.8) |
Noncompliance with study drug | 2 (0.7) | 1 (0.4) | 3 (0.8) | 0 |
Adverse events | 1 (0.4) | 4 (1.4) | 6 (1.6) | 6 (1.7) |
Protocol deviation | 0 | 3 (1.1) | 0 | 1 (0.3) |
Physician decision | 0 | 4 (1.4) | 2 (0.5) | 2 (0.6) |
Other | 3 (1.1) | 1 (0.4) | 3 (0.8) | 3 (0.8) |
PTI = personalized treatment interval; q.4.w. = every 4 weeks.
Note: The data presented in the preceding table include the discontinuations that occurred through the end of each study.
aPercentages are based on number of patients treated.
bPercentages are based on number of patients randomized.
Sources: Final Clinical Study Report for GR41984 (BALATON)15 and updated Clinical Study Report for GR41986 (COMINO).17
Protocol Deviations
A summary of protocol deviations through week 24 and week 72 from the BALATON and COMINO studies is presented in Table 14 and Table 15, respectively.
BALATON Trial
A total of 27.2% of patients (75 of 276 patients) randomized to receive faricimab every 4 weeks and 26.4% of patients (73 of 277 patients) randomized to receive aflibercept every 4 weeks had at least 1 major protocol deviation through week 24. The most frequently reported category of protocol deviation was procedural-related, reported for 25.7% of patients (71 patients) in the faricimab group and 24.5% of patients (68 patients) in the aflibercept group. The most common procedural-related protocol deviations (frequency > 5.0% of patients in either group), including missing weeks 20 and 24 and major issues with images of the study eye, were each reported in less than 8.0% of patients in each group. Other categories of major protocol deviations, relating to medication and inclusion and exclusion criteria, were each reported in less than 2.0% of patients in each group.
A total of 52.9% of patients (146 of 276 patients) randomized to receive faricimab in part 1 and 50.9% of patients (141 of 277 patients) randomized to receive aflibercept in part 1 had at least 1 major protocol deviation through week 72. The most frequently reported category of protocol deviation continued to be procedural-related, which was reported for 131 patients (47.5%) randomized to receive faricimab in part 1 and 123 patients (44.4%) randomized to receive aflibercept in part 1. The most common procedural-related protocol deviations were each reported in less than 15.0% of patients in each group. Other categories of major protocol deviations, relating to medication and inclusion and exclusion criteria, were each reported in less than 9.0% of patients in each group.
COMINO Trial
A total of 27.3% of patients (100 of 366 patients) randomized to receive faricimab every 4 weeks and 32.2% of patients (117 of 363 patients) randomized to receive aflibercept every 4 weeks had at least 1 major protocol deviation through week 24. The most frequently reported category of protocol deviation was procedural-related, reported for 25.4% of patients (93 patients) in the faricimab group and 30.6% of patients (111 patients) in the aflibercept group. The most common procedural-related protocol deviations (frequency > 5.0% of patients in either group), including missing weeks 20 and 24 and major issues with images of the study eye, were each reported in less than 6.0% of patients in each group. Other categories of major protocol deviations, relating to medication and inclusion and exclusion criteria, were each reported in approximately 2.0% of patients or less in each group.
Table 14: Summary of Protocol Deviations Through Week 24 From the BALATON and COMINO Studies (ITT Population)
Major protocol deviation | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 277) | Faricimab (N = 366) | Aflibercept (N = 363) | |
At least 1 major protocol deviation, n (%) | 75 (27.2) | 73 (26.4) | 100 (27.3) | 117 (32.2) |
At least 1 major procedural-related protocol deviation, n (%) | 71 (25.7) | 68 (24.5) | 93 (25.4) | 111 (30.6) |
Most common protocol deviations,a n (%) | ||||
Week 24 missed | 12 (4.3) | 22 (7.9) | 17 (4.6) | 20 (5.5) |
Week 20 missed | 15 (5.4) | 17 (6.1) | 17 (4.6) | 20 (5.5) |
Major issues with images of study eyeb | 17 (6.2) | 6 (2.2) | 10 (2.7) | 16 (4.4) |
At least 1 major medication-related protocol deviation,c n (%) | 4 (1.4) | 5 (1.8) | 5 (1.4) | 8 (2.2) |
At least 1 major exclusion criteria–related protocol deviation,c n (%) | 2 (0.7) | 5 (1.8) | 5 (1.4) | 7 (1.9) |
At least 1 major inclusion criteria–related protocol deviation,c n (%) | 4 (1.4) | 0 | 1 (0.3) | 0 |
ITT = intention to treat.
Note: Multiple occurrences of the same deviation in an individual patient were counted only once.
aFrequency greater than 5.0% of patients in either group.
bMajor issues with images included images not acquired according to study protocol, images not acquired at an attended study visit, and images not submitted to or rejected by reading centre, imaging performed by non–study-certified personnel.
cNo major protocol deviation was reported in greater than 5.0% of patients in either group.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16
Table 15: Summary of Protocol Deviations Through Week 72 From the BALATON and COMINO Studies (ITT Population)
Major protocol deviation | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI (N = 276) | Aflibercept q.4.w. to faricimab PTI (N = 277) | Faricimab q.4.w. to faricimab PTI (N = 366) | Aflibercept q.4.w. to faricimab PTI (N = 363) | |
At least 1 major protocol deviation, n (%) | 146 (52.9) | 141 (50.9) | 186 (50.8) | 199 (54.8) |
At least 1 major procedural-related protocol deviation, n (%) | 131 (47.5) | 123 (44.4) | 172 (47.0) | 179 (49.3) |
Most common protocol deviations,a n (%) | ||||
Major issues with images of study eyeb | 40 (14.5) | 30 (10.8) | 50 (13.7) | 49 (13.5) |
Week 60 missed | 17 (6.2) | 22 (7.9) | 18 (4.9) | 27 (7.4) |
Week 68 missed | 17 (6.2) | 10 (3.6) | 18 (4.9) | 20 (5.5) |
Week 20 missed | 15 (5.4) | 17 (6.1) | 18 (4.9) | 23 (6.3) |
Week 24 missed | 15 (5.4) | 27 (9.7) | 19 (5.2) | 22 (6.1) |
Week 72 missed | 14 (5.1) | 9 (3.2) | 13 (3.6) | 12 (3.3) |
Week 64 missed | 12 (4.3) | 20 (7.2) | 13 (3.6) | 20 (5.5) |
Other significant procedural deviation issues | 19 (6.9) | 15 (5.4) | 27 (7.4) | 30 (8.3) |
At least 1 major medication-related protocol deviation,c n (%) | 21 (7.6) | 26 (9.4) | 36 (9.8) | 41 (11.3) |
Most common protocol deviations,a n (%) | ||||
Time window between 2 consecutive doses is < 21 days | 12 (4.3) | 12 (4.3) | 18 (4.9) | 22 (6.1) |
At least 1 major exclusion criteria–related protocol deviation,c n (%) | 3 (1.1) | 8 (2.9) | 5 (1.4) | 8 (2.2) |
At least 1 major inclusion criteria–related protocol deviation,c n (%) | 4 (1.4) | 0 | 1 (0.3) | 2 (0.6) |
ITT = intention to treat; PTI = personalized treatment interval; q.4.w. = every 4 weeks.
Note: Multiple occurrences of the same deviation in an individual patient were counted only once.
aFrequency greater than 5.0% of patients in either group.
bMajor issues with images included images not acquired according to study protocol, images not acquired at an attended study visit, images not submitted to or rejected by reading centre, and imaging performed by non–study-certified personnel.
cNo major protocol deviation was reported in greater than 5.0% of patients in either group.
Sources: Final Clinical Study Report for GR41984 (BALATON)15 and updated Clinical Study Report for GR41986 (COMINO).17
A total of 50.8% of patients (186 of 366 patients) randomized to receive faricimab in part 1 and 54.8% of patients (199 of 363 patients) randomized to receive aflibercept in part 1 had at least 1 major protocol deviation through week 72. The most frequently reported category of protocol deviation continued to be procedural-related, which was reported for 172 patients (47.0%) randomized to receive faricimab in part 1 and 179 patients (49.3%) randomized to receive aflibercept in part 1. The most common procedural-related protocol deviations were each reported in less than 14.0% of patients in each group. Other categories of major protocol deviations, relating to medication and inclusion and exclusion criteria, were each reported in approximately 11.0% of patients or less in each group.
Baseline Characteristics
The baseline characteristics outlined in Table 16 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
BALATON Trial
The mean age of patients was 64.3 years (SD = 10.7 years; range, 35 to 93 years) in the faricimab group and 63.8 years (SD = 10.6 years; range, 28 to 88 years) in the aflibercept group. The distribution of patients by age based on a cut-off of 65 years was well balanced between groups; approximately 50.0% of patients were in each age group (< 65 years and ≥ 65 years). The mean time since RVO diagnosis in the study eye was 1.26 months (SD = 1.88 months; range, 0.2 to 27.7 months) in the faricimab group and 1.65 months (SD = 7.27 months; range, 0.1 to 120.6 months) in the aflibercept group.
The mean BCVA in the study eye was 57.50 letters (SD = 13.04 letters; range, 19.0 to 76.0 letters) in the faricimab group and 57.64 letters (SD = 12.15 letters; range, 21.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 558.32 µm (SD = 177.03 µm; range, 281.0 µm to 1,154.0 µm) in the faricimab group and 558.12 µm (SD = 180.26 µm; range, 290.0 µm to 1,208.0 µm) in the aflibercept group.
The mean intraocular pressure in the study eye was 14.57 mm Hg (SD = 2.88 mm Hg; range, 7.0 mm Hg to 21.0 mm Hg) in the faricimab group and 14.48 mm Hg (SD = 2.94 mm Hg; range, 7.0 mm Hg to 22.0 mm Hg) in the aflibercept group. A total of 1.8% of patients (5 of 276 patients) randomized to receive faricimab and 1.4% of patients (4 of 277 patients) randomized to receive aflibercept were reported with nonproliferative diabetic retinopathy in the study eye. A total of 0.4% of patients (1 patient) randomized to receive faricimab and no patients randomized to receive aflibercept were reported with proliferative diabetic retinopathy in the study eye. A total of 5.8% of patients (16 patients) randomized to receive faricimab and 5.4% of patients (15 patients) randomized to receive aflibercept were reported with glaucoma in the study eye.
The distribution of patients by blood pressure categories (> 160 systolic or > 90 diastolic, ≤ 140 systolic and ≤ 85 diastolic, ≤ 120 systolic and ≤ 80 diastolic) was well balanced between groups, with approximately 20.0%, 50.0%, and 9.0% of patients in each category, respectively. A total of 17.8% of patients (49 of 276 patients) randomized to receive faricimab and 16.6% of patients (46 of 277 patients) randomized to receive aflibercept were reported with type 2 diabetes.
COMINO Trial
The mean age of patients was 65.6 years (SD = 13.1 years; range, 22 to 100 years) in the faricimab group and 64.7 years (SD = 13.3 years; range, 27 to 95 years) in the aflibercept group. The distribution of patients by age based on a cut-off of 65 years was well balanced between groups; approximately 55.0% of patients were aged 65 years and older. The mean time since RVO diagnosis in the study eye was 1.57 months (SD = 6.39 months; range, 0.1 to 120.3 months) in the faricimab group and 1.10 months (SD = 0.95 months; range, 0.1 to 8.9 months) in the aflibercept group. A total of 83.0% of patients (303 of 366 patients) randomized to receive faricimab and 81.9% of patients (294 of 363 patients) randomized to receive aflibercept were reported with CRVO. A total of 17.0% of patients (62 patients) randomized to receive faricimab and 18.1% of patients (65 patients) randomized to receive aflibercept were reported with HRVO.
The mean BCVA in the study eye was 50.25 letters (SD = 16.25 letters; range, 19.0 to 87.0 letters) in the faricimab group and 50.71 letters (SD = 16.34 letters; range, 19.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 702.21 µm (SD = 244.00 µm; range, 266.0 µm to 1,500.0 µm) in the faricimab group and 721.07 µm (SD = 242.86 µm; range, 281.0 µm to 1,419.0 µm) in the aflibercept group.
The mean intraocular pressure in the study eye was 14.89 mm Hg (SD = 3.06 mm Hg; range, 7.0 mm Hg to 25.0 mm Hg) in the faricimab group and 14.97 mm Hg (SD = 3.17 mm Hg; range, 7.0 mm Hg to 26.0 mm Hg) in the aflibercept group. A total of 2.5% of patients (9 of 366 patients) randomized to receive faricimab and 2.5% of patients (9 of 363 patients) randomized to receive aflibercept were reported with nonproliferative diabetic retinopathy in the study eye. A total of 0.3% of patients (1 patient) randomized to receive faricimab and no patients randomized to receive aflibercept were reported with proliferative diabetic retinopathy in the study eye. A total of 10.7% of patients (39 patients) randomized to receive faricimab and 9.4% of patients (34 patients) randomized to receive aflibercept were reported with glaucoma in the study eye.
The distribution of patients by blood pressure categories (> 160 systolic or > 90 diastolic, ≤ 140 systolic and ≤ 85 diastolic, ≤ 120 systolic and ≤ 80 diastolic) was well balanced between groups, with approximately 16.0%, 54.0%, and 14.0% of patients in each category, respectively. A total of 20.2% of patients (74 of 366 patients) randomized to receive faricimab and 21.8% of patients (79 of 363 patients) randomized to receive aflibercept were reported with type 2 diabetes.
Table 16: Summary of Baseline Characteristics From the BALATON and COMINO Studies (ITT Population)
Characteristic | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 277) | Faricimab (N = 366) | Aflibercept (N = 363) | |
Baseline demographics | ||||
Age (years), mean (SD) | 64.3 (10.7) | 63.8 (10.6) | 65.6 (13.1) | 64.7 (13.3) |
Range, years | 35 to 93 | 28 to 88 | 22 to 100 | 27 to 95 |
Age group, n (%) | ||||
< 65 years | 133 (48.2) | 144 (52.0) | 162 (44.3) | 158 (43.5) |
≥ 65 years | 143 (51.8) | 133 (48.0) | 204 (55.7) | 205 (56.5) |
Sex, n (%) | ||||
Male | 143 (51.8) | 130 (46.9) | 193 (52.7) | 200 (55.1) |
Female | 133 (48.2) | 147 (53.1) | 173 (47.3) | 163 (44.9) |
Race, n (%) | ||||
White | 172 (62.3) | 172 (62.1) | 243 (66.4) | 253 (69.7) |
Asian | 90 (32.6) | 94 (33.9) | 89 (24.3) | 88 (24.2) |
Black or African American | 6 (2.2) | 7 (2.5) | 10 (2.7) | 13 (3.6) |
American Indian or Alaska Native | 3 (1.1) | 0 | 2 (0.5) | 3 (0.8) |
Native Hawaiian or other Pacific Islander | 1 (0.4) | 0 | 0 | 1 (0.3) |
Multiple | NR | NR | 1 (0.3) | 0 |
Unknown | 4 (1.4) | 4 (1.4) | 21 (5.7) | 5 (1.4) |
Ocular baseline characteristics in the study eye | ||||
Study eye, n (%) | ||||
Left eye | 140 (50.7) | 127 (45.8) | 180 (49.2) | 181 (49.9) |
Right eye | 136 (49.3) | 150 (54.2) | 186 (50.8) | 182 (50.1) |
Time since RVO diagnosis in the study eye, months, mean (SD) | 1.26 (1.88) | 1.65 (7.27) | 1.57 (6.39) | 1.10 (0.95) |
Range, months | 0.2 to 27.7 | 0.1 to 120.6 | 0.1 to 120.3 | 0.1 to 8.9 |
Unknown, n (%) | 5 (1.8) | 2 (0.7) | 6 (1.6) | 11 (3.0) |
Number of BCVA letters, mean (SD) | 57.50 (13.04) | 57.64 (12.15) | 50.25 (16.25) | 50.71 (16.34) |
Range, letters | 19.0 to 76.0 | 21.0 to 73.0 | 19.0 to 87.0 | 19.0 to 73.0 |
Missing or invalid, n (%) | 0 | 0 | 0 | 0 |
Nonstandard BCVA data,a n (%) | 0 | 2 (0.7) | 1 (0.3) | 1 (0.3) |
BCVA categories, n (%) | ||||
≤ 34 letters (20/200 or worse) | NR | NR | 79 (21.6) | 80 (22.0) |
> 34 letters to < 55 letters (better than 20/200 to worse than 20/80) | NR | NR | 106 (29.0) | 105 (28.9) |
≤ 54 letters (20/80 or worse) | 89 (32.2) | 90 (32.5) | NR | NR |
≥ 55 letters (20/80 or better) | 187 (67.8) | 187 (67.5) | 181 (49.5) | 178 (49.0) |
CST, µm, mean (SD) | 558.32 (177.03) | 558.12 (180.26) | 702.21 (244.00) | 721.07 (242.86) |
Range, µm | 281.0 to 1,154.0 | 290.0 to 1,208.0 | 266.0 to 1,500.0 | 281.0 to 1,419.0 |
Missing or ungradable, n (%) | 1 (0.4) | 2 (0.7) | 7 (1.9) | 4 (1.1) |
Macular ischemic nonperfusion, n (%) | 49 (17.8) | 48 (17.3) | 38 (10.4) | 37 (10.2) |
Missing or ungradable | 157 (56.9) | 163 (58.8) | 134 (36.6) | 149 (41.0) |
Intraocular pressure, mm Hg, mean (SD) | 14.57 (2.88) | 14.48 (2.94) | 14.89 (3.06) | 14.97 (3.17) |
Range, mm Hg | 7.0 to 21.0 | 7.0 to 22.0 | 7.0 to 25.0 | 7.0 to 26.0 |
Lens status, n (%) | ||||
Phakic | 236 (85.5) | 239 (86.3) | 308 (84.2) | 314 (86.5) |
Pseudophakic | 40 (14.5) | 36 (13.0) | 57 (15.6) | 48 (13.2) |
Aphakic | 0 | 1 (0.4) | 1 (0.3) | 1 (0.3) |
RVO subtype, n (%) | ||||
CRVO | NR | NR | 303 (83.0) | 294 (81.9) |
HRVO | NR | NR | 62 (17.0) | 65 (18.1) |
Nonocular baseline characteristics | ||||
Blood pressure by category, mm Hg | ||||
N | 276 | 273 | 365 | 360 |
> 160 systolic or > 90 diastolic, n (%) | 55 (19.9) | 51 (18.7) | 60 (16.4) | 61 (16.9) |
≤ 140 systolic and ≤ 85 diastolic, n (%) | 138 (50.0) | 132 (48.4) | 202 (55.3) | 188 (52.2) |
≤ 120 systolic and ≤ 80 diastolic, n (%) | 24 (8.7) | 23 (8.4) | 49 (13.4) | 51 (14.2) |
Missing or invalid, n | 0 | 4 | 1 | 3 |
History of ocular medical conditions in the study eye | ||||
At least 1 prior targeted ocular condition in the study eye, n (%) | 85 (30.8) | 82 (29.6) | 156 (42.6) | 145 (39.9) |
Cystoid macular edema | 55 (19.9) | 43 (15.5) | 102 (27.9) | 93 (25.6) |
Dry AMD | 5 (1.8) | 5 (1.8) | 5 (1.4) | 9 (2.5) |
Nonproliferative diabetic retinopathy | 5 (1.8) | 4 (1.4) | 9 (2.5) | 9 (2.5) |
Diabetic macular edema | 1 (0.4) | 3 (1.1) | 2 (0.5) | 2 (0.6) |
Wet AMD | 1 (0.4) | 1 (0.4) | 0 | 2 (0.6) |
Active neovascularization | 1 (0.4) | 0 | 5 (1.4) | 4 (1.1) |
Neovascular glaucoma | 1 (0.4) | 0 | 0 | 4 (1.1) |
Proliferative diabetic retinopathy | 1 (0.4) | 0 | 1 (0.3) | 0 |
BRVO without macular edema | 37 (13.4) | 38 (13.7) | NR | NR |
CRVO or HRVO without macular edema | NR | NR | 55 (15.0) | 48 (13.2) |
CRVO or HRVO with macular edema | NR | NR | 2 (0.5) | 1 (0.3) |
At least 1 ocular condition in the study eye, n (%) | 151 (54.7) | 156 (56.3) | 175 (47.8) | 190 (52.3) |
Most common condition,a n (%) | ||||
Cataract | 115 (41.7) | 112 (40.4) | 132 (36.1) | 139 (38.3) |
Retinal vein occlusion | 34 (12.3) | 33 (11.9) | 16 (4.4) | 19 (5.2) |
Retinopathy — hypertensive | 15 (5.4) | 17 (6.1) | 21 (5.7) | 24 (6.6) |
Glaucoma | 16 (5.8) | 15 (5.4) | 39 (10.7) | 34 (9.4) |
Dry eye disease | 13 (4.7) | 14 (5.1) | 16 (4.4) | 17 (4.7) |
History of nonocular general medical conditions | ||||
At least 1 prior targeted nonocular general medical history, n (%) | 181 (65.6) | 190 (68.6) | 236 (64.5) | 238 (65.6) |
Most common condition,b n (%) | ||||
Systemic hypertension | 150 (54.3) | 167 (60.3) | 200 (54.6) | 204 (56.2) |
Type 2 diabetes | 49 (17.8) | 46 (16.6) | 74 (20.2) | 79 (21.8) |
Uncomplicated diabetes mellitus | 28 (10.1) | 20 (7.2) | 34 (9.3) | 41 (11.3) |
Atrial fibrillation | 11 (4.0) | 17 (6.1) | 26 (7.1) | 20 (5.5) |
Coronary artery disease | 15 (5.4) | 10 (3.6) | 26 (7.1) | 22 (6.1) |
Solid tumour | 7 (2.5) | 11 (4.0) | 19 (5.2) | 12 (3.3) |
AMD = age-related macular degeneration; BCVA = best-corrected visual acuity; BRVO = branch retinal vein occlusion; CRVO = central retinal vein occlusion; CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; HRVO = hemiretinal vein occlusion; ITT = intention to treat; NR = not reported; RVO = retinal vein occlusion; SD = standard deviation.
aNonstandard BCVA data, such as assessed by ETDRS BCVA testing with prior visit refraction and BCVA test performed by an unmasked certified ETDRS BCVA assessor or by an uncertified experienced ETDRS BCVA assessor, were included in the analyses.
bFrequency greater than 5.0% of patients in either group.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16 Details included in the table are from the sponsor’s summary of clinical evidence.2
Prior Ocular Medications
A summary of prior ocular therapies and surgeries from the BALATON and COMINO studies is presented in Table 17.
BALATON Trial
A total of 2.9% of patients (8 of 276 patients) randomized to receive faricimab and 5.8% of patients (16 of 277 patients) randomized to receive aflibercept used at least 1 targeted ocular therapy or treatment in the study eye before the start of the trial. Each applicable therapy or treatment (topical steroids, panretinal photocoagulation laser, dietary supplement, other laser photocoagulation, and all other therapies and treatments) was reported in less than 5.0% of patients in each group.
A total of 17.4% of patients (48 patients) randomized to receive faricimab and 16.6% of patients (46 patients) randomized to receive aflibercept underwent at least 1 ocular surgery or procedure in the study eye before the start of the trial. The most common surgery or procedure (frequency > 5.0% of patients in either group) was cataract surgery, reported for 14.5% of patients (40 patients) in the faricimab group and 11.9% of patients (33 patients) in the aflibercept group.
COMINO Trial
A total of 5.7% of patients (21 of 366 patients) randomized to receive faricimab and 5.0% of patients (18 of 363 patients) randomized to receive aflibercept used at least 1 targeted ocular therapy or treatment in the study eye before the start of the trial. Each applicable therapy or treatment (topical steroids, panretinal photocoagulation laser, dietary supplement, other laser photocoagulation, and all other therapies and treatments) was reported in less than 5.0% of patients of patients in each group.
A total of 17.5% of patients (64 patients) randomized to receive faricimab and 15.2% of patients (55 patients) randomized to receive aflibercept underwent at least 1 ocular surgery or procedure in the study eye before the start of the trial. The most common surgery or procedure (frequency > 5.0% of patients of patients in either group) was cataract surgery, reported for 15.0% of patients (55 patients) in the faricimab group and 11.6% of patients (42 patients) in the aflibercept group.
Table 17: Summary of Prior Ocular Therapies and Surgeries From the BALATON and COMINO Studies (ITT Population)
Category of therapy or treatment | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 277) | Faricimab (N = 366) | Aflibercept (N = 363) | |
Prior targeted ocular therapies and treatments in the study eye | ||||
At least 1 applicable therapy or treatment, n (%) | 8 (2.9) | 16 (5.8) | 21 (5.7) | 18 (5.0) |
Topical steroids | 0 | 1 (0.4) | 0 | 1 (0.3) |
Panretinal photocoagulation laser | 0 | 1 (0.4) | 0 | 1 (0.3) |
Dietary supplement | 0 | 1 (0.4) | 1 (0.3) | 1 (0.3) |
Other laser photocoagulation | 2 (0.7) | 1 (0.4) | 2 (0.5) | 0 |
Other | 6 (2.2) | 13 (4.7) | 18 (4.9) | 15 (4.1) |
Prior ocular surgeries and procedures in the study eye | ||||
At least 1 surgery or procedure, n (%) | 48 (17.4) | 46 (16.6) | 64 (17.5) | 55 (15.2) |
Most common condition,a n (%) | ||||
Cataract surgery | 40 (14.5) | 33 (11.9) | 55 (15.0) | 42 (11.6) |
ITT = intention to treat.
Note: Multiple occurrences of the same therapy, treatment, surgery, or procedure in an individual patient were counted only once.
aFrequency greater than 5.0% of patients in either group.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16
Exposure to Study Treatments
A summary of study treatment exposure in the study eye through week 24 and week 72 from the BALATON and COMINO studies is presented in Table 18 and Table 19, respectively.
BALATON Trial
The mean number of study drug administrations through week 24 was 5.8 administrations (SD = 0.72 administrations; range, 1 to 6 administrations) in the faricimab every 4 weeks group and 5.8 administrations (SD = 0.46 administrations; range, 3 to 6 administrations) in the aflibercept every 4 weeks group. Of the patients who received at least 1 injection of the study drug in the study eye, 1.1% (3 of 276 patients) in the faricimab group and 1.8% (5 of 274 patients) in the aflibercept group had at least 1 interrupted dose through week 24.
The mean number of study drug administrations through week 72 was 10.3 administrations (SD = 3.01 administrations; range, 1 to 18 administrations) in the group of patients who were randomized to receive faricimab in part 1 and 10.6 administrations (SD = 2.96 administrations; range, 4 to 18 administrations) in the group of patients who were randomized to receive aflibercept in part 1. Of the patients who received at least 1 injection of the study drug in the study eye, 6.5% (18 patients) in the group of patients who were randomized to receive faricimab in part 1 and 6.9% (19 patients) in the group of patients who were randomized to receive aflibercept in part 1 had at least 1 interrupted dose through week 72. Each specific reason for an interrupted dose listed in Table 19 was reported in less than 1.0% of patients in each group, while “other” reasons were reported in 4.3% (12 patients) of the patients in the faricimab group and 6.6% (18 patients) of the patients in the aflibercept group.
COMINO Trial
The mean number of study drug administrations through week 24 was 5.7 administrations (SD = 0.68 administrations; range, 1 to 6 administrations) in the faricimab every 4 weeks group and 5.7 administrations (SD = 0.72 administrations; range, 1 to 6 administrations) in the aflibercept every 4 weeks group. Of the patients who received at least 1 injection of the study drug in the study eye, a total of 4.1% (15 of 365 patients) of patients in the faricimab group and 3.0% (11 of 361 patients) of patients in the aflibercept group had at least 1 interrupted dose through week 24.
The mean number of study drug administrations through week 72 was 11.2 administrations (SD = 3.29 administrations; range, 1 to 18 administrations) in the group of patients who were randomized to receive faricimab in part 1 and 10.9 administrations (SD = 3.31 administrations; range, 1 to 18 administrations) in the group of patients who were randomized to receive aflibercept in part 1. Of the patients who received at least 1 injection of the study drug in the study eye, 15.3% (56 patients) in the group of patients who were randomized to receive faricimab in part 1 and 10.8% (39 patients) in the group of patients who were randomized to receive aflibercept in part 1 had at least 1 interrupted dose through week 72. Each specific reason for an interrupted dose listed in Table 19 was reported in less than approximately 2.0% of patients in each group, while “other” reasons were reported in 11.2% (41 patients) and 8.0% (29 patients) of patients in the faricimab and aflibercept groups, respectively.
Table 18: Summary of Study Treatment Exposure in the Study Eye Through Week 24 From the BALATON and COMINO Studies (Safety-Evaluable Population)
Exposure | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 274) | Faricimab (N = 365) | Aflibercept (N = 361) | |
Treatment duration (weeks), mean (SD) | 19.7 (2.63) | 20.0 (1.46) | 19.9 (2.06) | 19.7 (2.25) |
Range, weeks | 0 to 21 | 8 to 21 | 0 to 22 | 0 to 22 |
Number of study drug administrations, mean (SD) | 5.8 (0.72) | 5.8 (0.46) | 5.7 (0.68) | 5.7 (0.72) |
Range, n | 1 to 6 | 3 to 6 | 1 to 6 | 1 to 6 |
At least 1 interrupted dose, n (%) | 3 (1.1) | 5 (1.8) | 15 (4.1) | 11 (3.0) |
Cataract surgery in the study eye | 1 (0.4) | 0 | 0 | 0 |
Intraocular surgery in the study eye | 1 (0.4) | 0 | 0 | 0 |
Intraocular inflammation | 0 | 1 (0.4) | 2 (0.5) | 1 (0.3) |
BCVA decrease | 0 | 0 | 3 (0.8) | 0 |
Elevated intraocular pressure | 0 | 0 | 1 (0.3) | 1 (0.3) |
Active or suspected infection | 0 | 0 | 1 (0.3) | 1 (0.3) |
Rhegmatogenous retinal break | 0 | 0 | 1 (0.3) | 0 |
Vitrectomy | 0 | 0 | 1 (0.3) | 0 |
Other | 2 (0.7) | 5 (1.8) | 11 (3.0) | 8 (2.2) |
BCVA = best-corrected visual acuity; SD = standard deviation.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 19: Summary of Study Treatment Exposure in the Study Eye Through Week 72 From the BALATON and COMINO Studies (Safety-Evaluable Population)
Exposure | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI (N = 276) | Aflibercept q.4.w. to faricimab PTI (N = 274) | Faricimab q.4.w. to faricimab PTI (N = 365) | Aflibercept q.4.w. to faricimab PTI (N = 361) | |
Treatment duration (weeks), mean (SD) | 64.2 (13.05) | 64.8 (10.85) | 64.9 (11.38) | 63.9 (13.12) |
Range, weeks | 0 to 70 | 13 to 70 | 0 to 70 | 0 to 70 |
Number of study drug administrations, mean (SD) | 10.3 (3.01) | 10.6 (2.96) | 11.2 (3.29) | 10.9 (3.31) |
Range, n | 1 to 18 | 4 to 18 | 1 to 18 | 1 to 18 |
At least 1 interrupted dose, n (%) | 18 (6.5) | 19 (6.9) | 56 (15.3) | 39 (10.8) |
Cataract surgery in the study eye | 2 (0.7) | 2 (0.7) | 3 (0.8) | 3 (0.8) |
Active or suspected infection | 2 (0.7) | 1 (0.4) | 3 (0.8) | 4 (1.1) |
Intraocular surgery in the study eye | 2 (0.7) | 0 | 1 (0.3) | 1 (0.3) |
BCVA decrease | 2 (0.7) | 0 | 5 (1.4) | 1 (0.3) |
Rhegmatogenous retinal break | 1 (0.4) | 0 | 1 (0.3) | 0 |
Vitrectomy | 1 (0.4) | 0 | 2 (0.5) | 0 |
Elevated intraocular pressure | 0 | 1 (0.4) | 2 (0.5) | 4 (1.1) |
Intraocular inflammation | NR | NR | 9 (2.5) | 4 (1.1) |
Rhegmatogenous retinal detachment or macular hole | NR | NR | 1 (0.3) | 1 (0.3) |
On-study prohibited medications | NR | NR | 0 | 1 (0.3) |
Other | 12 (4.3) | 18 (6.6) | 41 (11.2) | 29 (8.0) |
BCVA = best-corrected visual acuity; NR = not reported; PTI = personalized treatment interval; q.4.w. = every 4 weeks; SD = standard deviation.
Sources: Final Clinical Study Report for GR41984 (BALATON)15 and updated Clinical Study Report for GR41986 (COMINO).17
Concomitant Ocular Medications
A summary of concomitant therapies and surgeries through week 24 and week 72 from the BALATON and COMINO studies is presented in Table 20 and Table 21, respectively.
BALATON Trial
Of the patients who received at least 1 injection of the study drug in the study eye, 13.8% (38 of 276 patients) randomized to receive faricimab and 20.1% (55 of 274 patients) randomized to receive aflibercept also received at least 1 concomitant ocular medication in the study eye through week 24. Through week 72, 24.3% of patients (67 patients) randomized to receive faricimab in part 1 and 26.3% of patients (72 patients) randomized to receive aflibercept in part 1 received at least 1 concomitant ocular medication in the study eye. No single type of concomitant ocular medication was reported in greater than 5.0% of patients in either group.
A total of 1.1% of patients (3 of 276 patients) randomized to receive faricimab and no patients randomized to receive aflibercept had at least 1 concurrent ocular surgery or procedure in the study eye through week 24. No single type of concurrent ocular surgery or procedure was reported in greater than 5.0% of patients in either group; no patients underwent a vitrectomy. In part 2 of the studies, 3.7% of patients (10 of 270 patients) randomized to receive faricimab in part 1 and 2.6% of patients (7 of 274 patients) randomized to receive aflibercept in part 1 had at least 1 concurrent ocular surgery or procedure in the study eye through week 72. No single type of concurrent ocular surgery or procedure was reported in greater than 5.0% of patients in either group; only 0.4% of patients (1 patient) randomized to receive faricimab in part 1 underwent a vitrectomy.
Of the patients who received at least 1 injection of the study drug in the study eye, 85.9% (237 patients) randomized to receive faricimab and 86.9% (238 patients) randomized to receive aflibercept also received at least 1 concomitant nonocular medication through week 24. Of note, 13.8% (38 patients) and 17.2% (47 patients) of patients, respectively, used at least 1 drug for diabetes. The most common drugs used for diabetes (frequency > 5.0% of patients in either group) were metformin, used by 3.6% of patients (10 patients) in the faricimab group and 8.4% of patients (23 patients) in the aflibercept group, and metformin hydrochloride, used by 5.1% (14 patients) and 4.7% (13 patients) of patients, respectively. The most common drugs used for diabetes (frequency > 5.0% of patients in either group) continued to be metformin and metformin hydrochloride through week 72.
COMINO Trial
Of the patients who received at least 1 injection of the study drug in the study eye, 24.7% (90 of 365 patients) randomized to receive faricimab and 25.2% (91 of 361 patients) randomized to receive aflibercept also received at least 1 concomitant ocular medication in the study eye through week 24. No single type of concomitant ocular medication was reported in greater than 5.0% of patients in either group. Through week 72, 38.4% of patients (140 patients) randomized to receive faricimab in part 1 and 37.7% of patients (136 patients) randomized to receive aflibercept in part 1 received at least 1 concomitant ocular medication in the study eye. The most common concomitant ocular medications used in the study eye (frequency > 5.0% of patients in either group) were dorzolamide hydrochloride plus timolol maleate, and latanoprost.
A total of 1.4% of patients (5 of 366 patients) randomized to receive faricimab and 3.3% of patients (12 of 361 patients) randomized to receive aflibercept had at least 1 concurrent ocular surgery or procedure in the study eye through week 24. No single type of concurrent ocular surgery or procedure was reported in greater than 5.0% of patients in either group; vitrectomy was reported in 0.3% of patients (1 patient) in each group. In part 2 of the studies, 4.2% of patients (15 of 359 patients) randomized to receive faricimab in part 1 and 4.5% of patients (16 of 352 patients) randomized to receive aflibercept in part 1 had at least 1 concurrent ocular surgery or procedure in the study eye through week 72. No single type of concurrent ocular surgery or procedure was reported in greater than 5.0% of patients in either group; 0.3% of patients (1 patient) in each group underwent vitrectomy.
Table 20: Summary of Concomitant Therapies and Surgeries Through Week 24 From the BALATON and COMINO Studies (Safety-Evaluable Population)
Category of therapy or treatment | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 274) | Faricimab (N = 365) | Aflibercept (N = 361) | |
Concomitant ocular medications in the study eye through week 24 | ||||
At least 1 treatment received,a n (%) | 38 (13.8) | 55 (20.1) | 90 (24.7) | 91 (25.2) |
Concurrent ocular surgeries and procedures in the study eye through week 24 (ITT population) | ||||
N | 276 | 277 | 366 | 363 |
At least 1 applicable concurrent procedure,a n (%) | 3 (1.1) | 0 | 5 (1.4) | 12 (3.3) |
Vitrectomy,b n (%) | 0 | 0 | 1 (0.3) | 1 (0.3) |
Anti-VEGF administration in the fellow eye through week 24 | ||||
At least 1 dose of anti-VEGF in the fellow eye,c n (%) | 5 (1.8) | 4 (1.5) | 8 (2.2) | 7 (1.9) |
Aflibercept | 3 (1.1) | 4 (1.5) | 7 (1.9) | 5 (1.4) |
Ranibizumab | 2 (0.7) | 0 | 1 (0.3) | 2 (0.6) |
Bevacizumab | 1 (0.4) | 0 | 0 | 0 |
Concomitant nonocular medications through week 24 | ||||
At least 1 treatment received, n (%) | 237 (85.9) | 238 (86.9) | 313 (85.8) | 315 (87.3) |
At least 1 drug used for diabetes,b n (%) | 38 (13.8) | 47 (17.2) | 75 (20.5) | 78 (21.6) |
Most common drug used for diabetes,d n (%) | ||||
Metformin | 10 (3.6) | 23 (8.4) | 36 (9.9) | 33 (9.1) |
Metformin hydrochloride | 14 (5.1) | 13 (4.7) | 22 (6.0) | 21 (5.8) |
ITT = intention to treat.
aNo single type of treatment or procedure was reported in greater than 5.0% of patients in either group.
bKey ocular surgery and procedure or key nonocular medication identified by the clinical expert consulted for this review. The clinical expert advised that anti-VEGF therapies are less effective after vitrectomy and consequently can impact treatment decision-making (i.e., extending the treatment interval would be difficult). The clinical expert also advised that drugs used for the treatment of diabetes can cause macular edema.
cAnti-VEGF treatments before baseline were excluded.
dFrequency greater than 5.0% of patients in either group.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO),16 and final Clinical Study Report for GR41984 (BALATON).15
Of the patients who received at least 1 injection of the study drug in the study eye, 85.8% (313 patients) randomized to receive faricimab and 87.3% (315 patients) randomized to receive aflibercept also received at least 1 concomitant nonocular medication through week 24. Of note, a total of 20.5% (75 patients) and 21.6% (78 patients) of patients, respectively, used at least 1 drug for diabetes. The most common drugs used for diabetes (frequency > 5.0% of patients in either group) were metformin, used by 9.9% (36 patients) and 9.1% (33 patients) of patients in the faricimab and aflibercept groups, respectively, and metformin hydrochloride, used by 6.0% (22 patients) and 5.8% (21 patients) of patients in the faricimab and aflibercept groups, respectively. The most common drugs used for diabetes (frequency > 5.0% of patients in either group) continued to be metformin and metformin hydrochloride through week 72.
Efficacy
A summary of key efficacy results from the BALATON (in patients with BRVO) and COMINO (in patients with CRVO and HRVO) studies is presented in Table 22 and Table 23.
Visual Acuity Outcomes
BALATON Trial
The treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.6 letters (95% CI, −2.2 to 1.1 letters; P value for superiority test = 0.4978). Based on the analysis for noninferiority followed by the test for superiority, faricimab was statistically noninferior, but not superior, to aflibercept for this end point. A sensitivity analysis was performed for this outcome using multiple imputation to handle missing data differently. Two supplementary analyses were also performed: a PP analysis of the PP population and an analysis using a hypothetical strategy for all intercurrent events. The sensitivity and supplementary analysis results were generally supportive of the primary analysis results.
The treatment difference in the estimated proportion of patients with a gain in BCVA of 15 letters or greater in the study eye from baseline at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −4.3% (95% CI, −12.3% to 3.8%; P = 0.3023). The supplementary analysis result was generally supportive of the primary analysis result.
COMINO Trial
The treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.4 letters (95% CI, −2.5 to 1.6 letters; P value for superiority test = 0.6715). Three analyses were performed for this outcome: a sensitivity analysis using multiple imputation to handle missing data differently, a supplementary analysis of the PP analysis population, and a supplementary analysis using a hypothetical strategy for all intercurrent events. The sensitivity and supplementary analysis results were generally supportive of the primary analysis results.
Table 21: Summary of Concomitant Ocular Therapies and Surgeries Through Week 72 From the BALATON and COMINO Studies (Safety-Evaluable Population)
Category of therapy or treatment | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI (N = 276) | Aflibercept q.4.w. to faricimab PTI (N = 274) | Faricimab q.4.w. to faricimab PTI (N = 365) | Aflibercept q.4.w. to faricimab PTI (N = 361) | |
Concomitant ocular medications in the study eye through week 72 | ||||
At least 1 treatment received, n (%) | 67 (24.3) | 72 (26.3) | 140 (38.4) | 136 (37.7) |
Most common treatment,a n (%) | ||||
Dorzolamide hydrochloride; timolol maleate | 11 (4.0) | 7 (2.6) | 24 (6.6) | 17 (4.7) |
Latanoprost | 7 (2.5) | 5 (1.8) | 25 (6.8) | 30 (8.3) |
Concurrent ocular surgeries and procedures in the study eye through week 72 (ITT population) | ||||
N | 276 | 277 | NR | NR |
At least 1 applicable concurrent procedure,b n (%) | 13 (4.7) | 9 (3.2) | NR | NR |
Vitrectomy,c n (%) | 1 (0.4) | 0 | NR | NR |
N in part 2 | 270 | 274 | 359 | 352 |
At least 1 applicable concurrent procedure,b n (%) | 10 (3.7) | 7 (2.6) | 15 (4.2) | 16 (4.5) |
Vitrectomy,c n (%) | 1 (0.4) | 0 | 1 (0.3) | 1 (0.3) |
Anti-VEGF administration in the fellow eye through week 72 | ||||
At least 1 dose of anti-VEGF in the fellow eye,d n (%) | 9 (3.3) | 5 (1.8) | 12 (3.3) | 9 (2.5) |
Aflibercept | 7 (2.5) | 5 (1.8) | 9 (2.5) | 7 (1.9) |
Ranibizumab | 2 (0.7) | 0 | 3 (0.8) | 2 (0.6) |
Bevacizumab | 1 (0.4) | 0 | 0 | 1 (0.3) |
Concomitant nonocular medications through week 24 | ||||
At least 1 treatment received, n (%) | 257 (93.1) | 259 (94.5) | 344 (94.2) | 340 (94.2) |
At least 1 drug used for diabetes,c n (%) | 46 (16.7) | 59 (21.5) | 91 (24.9) | 96 (26.6) |
Most common drug used for diabetes,a n (%) | ||||
Metformin | 13 (4.7) | 25 (9.1) | 40 (11.0) | 39 (10.8) |
Metformin hydrochloride | 17 (6.2) | 18 (6.6) | 24 (6.6) | 26 (7.2) |
ITT = intention to treat; PTI = personalized treatment interval; q.4.w. = every 4 weeks.
Notes: Part 1 included procedures performed before week 24 treatment or dose hold or, if no dose hold, performed before day 168 (target day of week 24 visit).
Part 2, for the faricimab q.4.w. to faricimab PTI arm, included procedures performed on or after the week 24 treatment or dose hold or, if no dose hold, performed on or after day 168 through week 72. Part 2 for the aflibercept q.4.w. to faricimab PTI arm included procedures performed on or after the date of the first faricimab dose through week 72.
aFrequency greater than 5.0% of patients in either group.
bNo single type of treatment or procedure was reported in greater than 5.0% of patients in either group.
cKey ocular surgery and procedure, or key nonocular medication, identified by the clinical expert consulted for this review. The clinical expert advised that anti-VEGF therapies are less effective after vitrectomy and consequently can impact treatment decision-making (i.e., extending the treatment interval would be difficult). The clinical expert also advised that drugs used for the treatment of diabetes can cause macular edema.
dAnti-VEGF treatments before baseline were excluded.
Sources: Final Clinical Study Report for GR41984 (BALATON)15 and updated Clinical Study Report for GR41986 (COMINO).17
A summary of the relevant subgroup (based on baseline RVO status) analysis results for the primary end point from the COMINO study is presented in Table 24. In the subgroup of patients with CRVO, the treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 0.2 letters (95% CI, −2.1 to 2.6 letters). In the subgroup of patients with HRVO, the treatment difference in the mean change from baseline in BCVA in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −3.8 letters (95% CI, −7.3 to −0.4 letters).
The treatment difference in the estimated proportion of patients with a gain in BCVA of 15 letters or greater in the study eye from baseline at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −1.5% (95% CI, −8.4% to 5.3%; P value = 0.6661). The supplementary analysis result was generally supportive of the primary analysis result.
Anatomic Outcomes
BALATON Trial
The treatment difference in the mean change from baseline in CST in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −7.0 µm (95% CI, −14.1 µm to 0.0 µm; P value for superiority test = 0.0495).
The treatment difference in the estimated proportion of patients with an absence of macular edema (defined as CST < 325 µm) at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 1.4% (95% CI, −2.3% to 5.0%; P value for superiority test = 0.4742).
Table 22: Summary of Key Efficacy Results From the BALATON and COMINO Studies (ITT Population)
Variable | BALATON trial | COMINO trial | |||
|---|---|---|---|---|---|
Faricimab q.4.w. (N = 276) | Aflibercept q.4.w. (N = 277) | Faricimab q.4.w. (N = 366) | Aflibercept q.4.w. (N = 363) | ||
Visual acuity | |||||
Change from baseline in BCVA in the study eye at week 24 (main analysis; ITT population)a | |||||
Baseline | |||||
n | 276 | 277 | 366 | 363 | |
BCVA (ETDRS letter score), mean (SE) | 57.5 (0.78) | 57.6 (0.73) | 50.2 (0.85) | 50.7 (0.86) | |
At week 24 | |||||
n | 254 | 247 | 340 | 331 | |
Change from baseline (ETDRS letter score), adjusted mean (95% CI) | 16.9 (15.7 to 18.1) | 17.5 (16.3 to 18.6) | 16.9 (15.4 to 18.3) | 17.3 (15.9 to 18.8) | |
Treatment group difference vs. aflibercept (letters), adjusted mean (95% CI)b | −0.6 (−2.2 to 1.1) | −0.4 (−2.5 to 1.6) | |||
P value for superiority test | 0.4978 | 0.6715 | |||
Change from baseline in BCVA in the study eye at week 24 (supplementary analysis; PP population)a | |||||
N | 241 | 243 | 328 | 311 | |
Baseline | |||||
n | 241 | 243 | 328 | 311 | |
BCVA (ETDRS letter score), mean (SE) | 57.6 (0.83) | 57.1 (0.80) | 49.8 (0.90) | 50.2 (0.93) | |
At week 24 | |||||
n | 228 | 224 | 312 | 296 | |
Change from baseline (ETDRS letter score), adjusted mean (95% CI) | 17.1 (15.8 to 18.3) | 17.4 (16.2 to 18.6) | 17.3 (15.8 to 18.8) | 18.4 (16.9 to 19.9) | |
Treatment group difference vs. aflibercept (ETDRS letter score), adjusted mean (95% CI) | −0.3 (−2.1 to 1.4) | −1.1 (−3.2 to 0.9) | |||
P value for superiority test | 0.7006 | 0.2812 | |||
Proportion of patients gaining ≥ 15 letters in BCVA in the study eye from baseline at week 24c | |||||
Patients with outcome, n | 155 | 167 | 207 | 211 | |
CMH weighted percent estimate, (95% CI) | 56.1 (50.4 to 61.9) | 60.4 (54.7 to 66.0) | 56.6 (51.7 to 61.5) | 58.1 (53.3 to 62.9) | |
Treatment group difference in CMH weighted estimate vs. aflibercept, % (95% CI) | −4.3 (−12.3 to 3.8) | −1.5 (−8.4 to 5.3) | |||
P value for superiority testd | 0.3023 | 0.6661 | |||
Missing, n (%) | 18 (6.5) | 27 (9.7) | 20 (5.5) | 22 (6.1) | |
Discontinued study before week 24, n (%) | 4 (1.4) | 3 (1.1) | 6 (1.6) | 10 (2.8) | |
Anatomic | |||||
Change from baseline in CST in the study eye at week 24e | |||||
Baseline | |||||
n | 275 | 275 | 359 | 359 | |
CST (µm), mean (SE) | 558.3 (10.68) | 558.1 (10.87) | 702.2 (12.88) | 721.1 (12.82) | |
At week 24 | |||||
n | 252 | 243 | 329 | 326 | |
Change from baseline (µm), adjusted mean (95% CI) | −311.4 (−316.4 to −306.4) | −304.4 (−309.3 to −299.4) | −461.6 (−471.4 to −451.9) | −448.8 (−458.6 to −439.0) | |
Treatment group difference vs. aflibercept (µm), adjusted mean (95% CI) | −7.0 (−14.1 to 0.0) | −12.8 (−26.7 to 1.0) | |||
P value for superiority testd | 0.0495 | 0.0684 | |||
Missing, n (%) | 20 (7.2) | 31 (11.2) | 31 (8.5) | 27 (7.4) | |
Discontinued study before week 24, n (%) | 4 (1.4) | 3 (1.1) | 6 (1.6) | 10 (2.8) | |
Proportion of patients with an absence of macular edema, defined as CST < 325 µm at week 24c | |||||
Patients with outcome, n | 263 | 260 | 343 | 334 | |
CMH weighted percent estimate, (95% CI) | 95.3 (92.8 to 97.7) | 93.9 (91.2 to 96.6) | 93.7 (91.2 to 96.2) | 92.0 (89.2 to 94.7) | |
Treatment group difference in CMH weighted estimate vs. aflibercept, % (95% CI) | 1.4 (−2.3 to 5.0) | 1.7 (−2.0 to 5.4) | |||
P value for superiority testd | 0.4742 | 0.3589 | |||
Missing, n (%) | 19 (6.9) | 31 (11.2) | 25 (6.8) | 24 (6.6) | |
Discontinued study before week 24, n (%) | 4 (1.4) | 3 (1.1) | 6 (1.6) | 10 (2.8) | |
Proportion of patients with an absence of both intraretinal fluid and subretinal fluid at week 24c | |||||
Patients with outcome, n | 183 | 169 | 275 | 249 | |
CMH weighted percent estimate, (95% CI) | 66.3 (60.8 to 71.9) | 61.0 (55.3 to 66.7) | 75.1 (70.8 to 79.5) | 68.6 (63.8 to 73.4) | |
Treatment group difference in CMH weighted estimate vs. aflibercept, % (95% CI) | 5.3 (−2.7 to 13.3) | 6.5 (0.1 to 13.0) | |||
P value for superiority testd | 0.1967 | 0.0489 | |||
Missing, n (%) | 37 (13.4) | 45 (16.2) | 33 (9.0) | 27 (7.4) | |
Discontinued study before week 24, n (%) | 4 (1.4) | 3 (1.1) | 6 (1.6) | 10 (2.8) | |
Vision-related functioning and health-related quality of life | |||||
Change from baseline in the NEI-VFQ-25 composite score at week 24f | |||||
Baseline | |||||
n | 276 | 274 | 364 | 362 | |
Score, mean (SE) | 80.7 (0.81) | 80.2 (0.88) | 76.6 (0.86) | 76.9 (0.86) | |
At week 24 | |||||
n | 253 | 244 | 339 | 330 | |
Change from baseline, adjusted mean (95% CI) | 5.6 (4.5 to 6.7) | 5.9 (4.8 to 7.1) | 6.9 (5.8 to 8.0) | 8.1 (7.0 to 9.2) | |
Treatment group difference vs. aflibercept, adjusted mean (95% CI) | −0.4 (−1.9 to 1.1) | −1.2 (−2.7 to 0.3) | |||
P value for superiority testd | 0.6370 | 0.1088 | |||
Missing, n (%) | 19 (6.9) | 30 (10.8) | 22 (6.0) | 24 (6.6) | |
Discontinued study before week 24, n (%) | 4 (1.4) | 3 (1.1) | 5 (1.4) | 9 (2.5) | |
AE = adverse event; ANCOVA = analysis of covariance; BCVA = best-corrected visual acuity; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CST = central subfield thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; ITT = intention to treat; LOCF = last observation carried forward; MMRM = mixed model for repeated measures; NEI-VFQ-25 = National Eye Institute Visual Function Questionnaire-25; PP = per protocol; PTI = personalized treatment interval; q.4.w. = every 4 weeks; SE = standard error; vs. = versus.
Note: For missing outcomes that were not due to early study discontinuation, reasons for missing data include study visits missed, and outcomes not measured although patients attended the visit.
aFor the primary analysis, an MMRM analysis was performed; the model was adjusted for treatment group, visit, visit by treatment group interaction, baseline BCVA (continuous), baseline BCVA (≤ 54 letters vs. ≥ 55 letters in the BALATON trial and ≤ 34 letters, 35 to 54 letters, and ≥ 55 letters in the COMINO trial), and region (US and Canada, Asia, and the rest of the world). An unstructured covariance structure was used. Observed BCVA assessments were used regardless of the occurrence of intercurrent events (i.e., discontinued study treatment due to AEs or lack of efficacy or received any prohibited systemic treatment or prohibited therapy in the study eye). Missing data were implicitly imputed by MMRM. Invalid BCVA values were excluded from analysis. The 95% CI was a rounding of 95.03% CI.
bFor the primary analysis, if the lower bound of the 2-sided 95% CI for the difference in the adjusted means of the 2 treatments was greater than −4 letters (the noninferiority margin), then faricimab was considered noninferior to aflibercept.
cThe weighted estimate was based on CMH weights stratified by baseline BCVA score (≤ 54 letters vs. ≥ 55 letters in the BALATON trial and ≤ 34 letters, 35 to 54 letters, and ≥ 55 letters in the COMINO trial) and region (US and Canada, Asia, and the rest of the world). All observed values were used regardless of the occurrence of the intercurrent event. Missing assessments were imputed by LOCF. Proportion was calculated after LOCF imputation. CI was a rounding of 95.03% CI and estimates below 0% or above 100% were imputed as 0% or 100%, respectively. Invalid BCVA values were excluded from the analysis. Absence of macular edema was defined as CST < 325 µm. Intraretinal fluid and subretinal fluid were as measured in the central subfield (centre 1 mm).
dNo formal superiority test was performed for secondary end points. At the primary analysis, nominal superiority P values were provided for secondary end points, where applicable, for reference purposes only.
eFor the MMRM analysis, the model was adjusted for treatment group, visit, visit by treatment group interaction, baseline CST (continuous), baseline BCVA score (≤ 54 letters vs. ≥ 55 letters in the BALATON trial and ≤ 34 letters, 35 to 54 letters, and ≥ 55 letters in the COMINO trial) and region (US and Canada, Asia, and the rest of the world). An unstructured covariance structure was used. Observed assessments were used regardless of the occurrence of intercurrent events. Missing data were implicitly imputed by MMRM. The 95% CI was a rounding of 95.03% CI. CST was defined as the distance between the internal limiting membrane and Bruch membrane as assessed by the central reading centre.
fFor the ANCOVA analysis, the model used the nonmissing change from baseline in the NEI-VFQ-25 composite score at week 24 as the response variables adjusted for the treatment group, baseline NEI-VFQ-25 composite score (continuous), baseline BCVA score (≤ 54 letters vs. ≥ 55 letters in the BALATON trial and ≤ 34 letters, 35 to 54 letters, and ≥ 55 letters in the COMINO trial), and region (US and Canada, Asia, and the rest of the world). Observed NEI-VFQ-25 assessments were used regardless of the occurrence of intercurrent events. Missing data were not imputed. Two patients discontinued the COMINO study before week 24; however, they completed the NEI-VFQ-25 assessment at their end-of-study visit; therefore, they were counted as patients with available data. The 95% CI was a rounding of the 95.03% CI.
Sources: Primary Clinical Study Reports for GR41984 (BALATON trial)14 and GR41986 (COMINO trial),16 final Clinical Study Report for GR41984 (BALATON trial),15 updated Clinical Study Report for GR41986 (COMINO trial),17 and the sponsor’s October 21, 2024, response to the request by CDA-AMC on October 16, 2024, for additional information.39 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 23: Summary of Personalized Treatment Interval Outcome at Week 68 From the BALATON and COMINO Studies (ITT Population)
Variable | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI (N = 276) | Aflibercept q.4.w. to faricimab PTI (N = 277) | Faricimab q.4.w. to faricimab PTI (N = 366) | Aflibercept q.4.w. to faricimab PTI (N = 363) | |
Treatment interval | ||||
Proportion of patients on a q.4.w., q.8.w., q.12.w., or q.16.w. treatment interval at week 68a | ||||
n | 248 | 244 | 330 | 315 |
Patients on a q.4.w. faricimab treatment interval, n (%) | 56 (22.6) | 61 (25.0) | 114 (34.5) | 102 (32.4) |
95% CI | 17.4 to 27.8 | 19.6 to 30.4 | 29.4 to 39.7 | 27.2 to 37.6 |
Patients on a q.8.w. faricimab treatment interval, n (%) | 33 (13.3) | 44 (18.0) | 66 (20.0) | 55 (17.5) |
95% CI | 9.1 to 17.5 | 13.2 to 22.9 | 15.7 to 24.3 | 13.3 to 21.7 |
Patients on a q.12.w. faricimab treatment interval, n (%) | 29 (11.7) | 23 (9.4) | 28 (8.5) | 35 (11.1) |
95% CI | 7.7 to 15.7 | 5.8 to 13.1 | 5.5 to 11.5 | 7.6 to 14.6 |
Patients on a q.16.w. faricimab treatment interval, n (%) | 130 (52.4) | 116 (47.5) | 122 (37.0) | 123 (39.0) |
95% CI | 46.2 to 58.6 | 41.3 to 53.8 | 31.8 to 42.2 | 33.7 to 44.4 |
CI = confidence interval; ITT = intention to treat; PTI = personalized treatment interval; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
aPercentages were based on patients who were randomized and who had not discontinued the study at week 68. Proportions are descriptive and were unadjusted. The treatment interval at week 68 was defined as the treatment interval decision followed at that visit. Patients received either intravitreal faricimab 6 mg q.4.w. or intravitreal aflibercept 2 mg q.4.w. to week 20. From week 24 (when all patients were scheduled to receive faricimab) onward, patients were treated according to the PTI dosing regimen up to week 68. The 95% CI was a rounding of the 95.03% CI.
Sources: Primary Clinical Study Reports for GR41984 (BALATON trial)14 and GR41986 (COMINO trial),16 final Clinical Study Report for GR41984 (BALATON trial),15 updated Clinical Study Report for GR41986 (COMINO trial),17 and the sponsor’s October 21, 2024, response to the request by CDA-AMC on October 16, 2024, for additional information.39 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 24: Summary of Subgroup Analysis Results of the Primary End Point From the COMINO Study (ITT Population)
Variable | Faricimab q.4.w. | Aflibercept q.4.w. |
|---|---|---|
Change from baseline in BCVA in the study eye at week 24 in subgroup of patients with CRVO | ||
Baseline | ||
n | 303 | 294 |
BCVA (ETDRS letter score), mean (SE) | 50.0 (0.94) | 50.5 (0.97) |
At week 24 | ||
n | 279 | 270 |
Change from baseline (ETDRS letter score), adjusted mean (95% CI) | 16.4 (14.7 to 18.0) | 16.1 (14.5 to 17.8) |
Treatment group difference vs. aflibercept (letters), adjusted mean (95% CI) | 0.2 (−2.1 to 2.6) | |
P value for superiority testa | 0.8545 | |
Change from baseline in BCVA in the study eye at week 24 in the subgroup of patients with HRVO | ||
Baseline | ||
n | 62 | 65 |
BCVA (ETDRS letter score), mean (SE) | 52.4 (1.88) | 52.4 (1.85) |
At week 24 | ||
n | 60 | 59 |
Change from baseline (ETDRS letter score), adjusted mean (95% CI) | 18.5 (16.0 to 20.9) | 22.3 (19.9 to 24.7) |
Treatment group difference vs. aflibercept (letters), adjusted mean (95% CI) | −3.8 (−7.3 to −0.4) | |
P value for superiority testa | 0.0310 | |
BCVA = best-corrected visual acuity; CI = confidence interval; CRVO = central retinal vein occlusion; ETDRS = Early Treatment Diabetic Retinopathy Study; HRVO = hemiretinal vein occlusion; MMRM = mixed model for repeated measures; ITT = intention to treat; q.4.w. = every 4 weeks; SE = standard error; vs. = versus.
Note: For the MMRM analysis, the model was adjusted for treatment group, visit, visit by treatment group interaction, baseline BCVA (continuous), baseline BCVA (≤ 34 letters, 35 to 54 letters, and ≥ 55 letters), and region (US and Canada, Asia, and the rest of the world). An unstructured covariance structure was used. Observed BCVA assessments were used regardless of the occurrence of intercurrent events. Missing data were implicitly imputed by MMRM. Invalid BCVA values were excluded from the analysis. The 95% CI was a rounding of the 95.03% CI.
aThe P value for the superiority test was not adjusted for multiple comparisons.
Source: Primary Clinical Study Report for GR41986 (COMINO).16
The treatment difference in the estimated proportion of patients with an absence of both intraretinal fluid and subretinal fluid (as measured in the central subfield; centre 1 mm) at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 5.3% (95% CI, −2.7% to 13.3%; P value for superiority test = 0.1967).
COMINO Trial
The treatment difference in the mean change from baseline in CST in the study eye at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −12.8 µm (95% CI, −26.7 µm to 1.0 µm; P value for superiority test = 0.0684).
The treatment difference in the estimated proportion of patients with an absence of macular edema (defined as CST < 325 µm) at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 1.7% (95% CI, −2.0% to 5.4%; P value for superiority test = 0.3589).
The treatment difference in the estimated proportion of patients with an absence of both intraretinal fluid and subretinal fluid (as measured in the central subfield; centre 1 mm) at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was 6.5% (95% CI, 0.1% to 13.0%; P value for superiority test = 0.0489).
Vision-Related Functioning and Health-Related Quality of Life Outcome
BALATON Trial
The treatment difference in the mean change from baseline in the NEI-VFQ-25 composite score at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −0.4 (95% CI, −1.9 to 1.1; P value for superiority test = 0.6370).
COMINO Trial
The treatment difference in the mean change from baseline in the NEI-VFQ-25 composite score at week 24 between faricimab every 4 weeks versus aflibercept every 4 weeks was −1.2 (95% CI, −2.7 to 0.3; P value for superiority test = 0.1088).
Treatment Interval Outcomes
BALATON Trial
The proportion of patients on an extended treatment interval at week 68 were as follows (patients randomized to receive faricimab every 4 weeks in part 1 and patients randomized to receive aflibercept every 4 weeks in part 1):
Faricimab every 8 weeks: 13.3% (95% CI, 9.1% to 17.5%) and 18.0% (95% CI, 13.2% to 22.9%), respectively.
Faricimab every 12 weeks: 11.7% (95% CI, 7.7% to 15.7%) and 9.4% (95% CI, 5.8% to 13.1%), respectively.
Faricimab every 16 weeks: 52.4% (95% CI, 46.2% to 58.6%) and 47.5% (95% CI, 41.3% to 53.8%), respectively.
COMINO Trial
The proportion of patients on an extended treatment interval at week 68 were as follows (patients randomized to receive faricimab every 4 weeks in part 1 and patients randomized to receive aflibercept every 4 weeks in part 1):
Faricimab every 8 weeks: 20.0% (95% CI, 15.7% to 24.3%) and 17.5% (95% CI, 13.3% to 21.7%), respectively.
Faricimab every 12 weeks: 8.5% (95% CI, 5.5% to 11.5%) and 11.1% (95% CI, 7.6% to 14.6%), respectively.
Faricimab every 16 weeks: 37.0% (95% CI, 31.8% to 42.2%) and 39.0% (95% CI, 33.7% to 44.4%), respectively.
Harms
A summary of safety results through week 24 from the BALATON and COMINO studies is presented in Table 25.
Additionally, a summary of safety results from baseline through week 72 from the BALATON and COMINO studies is presented in Table 26. Note that there was no comparison group because all patients received faricimab 6 mg intravitreal injections (and sham procedures to maintain masking of the treatment intervals) according to a personalized treatment interval dosing regimen in part 2 of the studies.
AEs Through Week 24
BALATON Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 16.3% (45 of 276 patients) randomized to receive faricimab and 20.4% (56 of 274 patients) randomized to receive aflibercept were reported with at least 1 ocular AE in the study eye. Each ocular AE in the study eye was reported in less than 4.0% of patients in each group.
Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% (3 patients) randomized to receive faricimab and 1.5% (4 patients) randomized to receive aflibercept were reported with at least 1 adjudicated APTC-defined AE. Each adjudicated APTC-defined AE was reported in less than 1.0% of patients in each group.
COMINO Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 23.0% (84 of 365 patients) randomized to receive faricimab and 27.7% (100 of 361 patients) randomized to receive aflibercept were reported with at least 1 ocular AE in the study eye. Each ocular AE in the study eye was reported in less than 4.0% of patients in each group.
Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% (4 patients) randomized to receive faricimab and 1.4% (5 patients) randomized to receive aflibercept were reported with at least 1 adjudicated APTC-defined AE. Each adjudicated APTC-defined AE was reported in less than 1.0% of patients in each group.
Serious Ocular AEs Through Week 24
BALATON Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 1.1% (3 patients) randomized to receive faricimab and 0.7% (2 patients) randomized to receive aflibercept were reported with at least 1 serious ocular AE in the study eye. Each serious ocular AE in the study eye was reported in less than 1.0% of patients in each group.
COMINO Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 2.5% (9 patients) randomized to receive faricimab and 3.3% (12 patients) randomized to receive aflibercept were reported with at least 1 serious ocular AE in the study eye. Each serious ocular AE in the study eye was reported in less than 1.0% of patients in each group.
Withdrawals Due to AEs Through Week 24
BALATON Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, no patients stopped study treatment due to ocular AEs.
COMINO Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 0.8% (3 patients) randomized to receive faricimab and 0.6% (2 patients) randomized to receive aflibercept stopped study treatment due to ocular AEs. Each ocular AE that led to a patient stopping their study treatment was reported in less than 1.0% of patients in each group.
Mortality Through Week 24
BALATON Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 0.4% (1 patient due to cerebrovascular accident) randomized to receive faricimab and no patients randomized to receive aflibercept died during the study through week 24.
COMINO Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, 0.3% (1 patient due to pneumonia) randomized to receive faricimab and 0.6% (2 patients due to myocardial infarction) randomized to receive aflibercept died during the study through week 24.
Notable Harms Through Week 24
BALATON Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, no patients were reported with endophthalmitis in the study eye.
COMINO Trial
Of the patients who received at least 1 injection of the active study drug in the study eye, no patients randomized to receive faricimab and 0.3% (1 patient) randomized to receive aflibercept were reported with endophthalmitis in the study eye.
Table 25: Summary of Harms Results Through Week 24 From the BALATON and COMINO Studies (Safety-Evaluable Population)
AEs | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab (N = 276) | Aflibercept (N = 274) | Faricimab (N = 365) | Aflibercept (N = 361) | |
AEs | ||||
Most common ocular AEs,a n (%) | ||||
≥ 1 ocular AE in the study eye | 45 (16.3) | 56 (20.4) | 84 (23.0) | 100 (27.7) |
Conjunctival hemorrhage | 8 (2.9) | 10 (3.6) | 10 (2.7) | 14 (3.9) |
Vitreous floaters | 6 (2.2) | 6 (2.2) | 6 (1.6) | 6 (1.7) |
Dry eye | 5 (1.8) | 9 (3.3) | 5 (1.4) | 6 (1.7) |
Pain | 5 (1.8) | 0 | 1 (0.3) | 1 (0.3) |
Vitreous detachment | 4 (1.4) | 2 (0.7) | 11 (3.0) | 9 (2.5) |
Cataract | 3 (1.1) | 1 (0.4) | 2 (0.5) | 7 (1.9) |
Epiretinal membrane | 2 (0.7) | 3 (1.1) | 3 (0.8) | 2 (0.6) |
Retinal exudates | 2 (0.7) | 3 (1.1) | 1 (0.3) | 4 (1.1) |
Intraocular pressure increasedb | 1 (0.4) | 7 (2.6) | 8 (2.2) | 13 (3.6) |
Retinal vein occlusion | 1 (0.4) | 3 (1.1) | 1 (0.3) | 5 (1.4) |
Eye pain | 0 | 4 (1.5) | 6 (1.6) | 2 (0.6) |
Ocular hypertension | 0 | 1 (0.4) | 7 (1.9) | 2 (0.6) |
Cystoid macular edema | 0 | 1 (0.4) | 3 (0.8) | 5 (1.4) |
Retinal tear | NR | NR | 2 (0.5) | 4 (1.1) |
≥ 1 ocular AE in the fellow eye | 26 (9.4) | 20 (7.3) | 28 (7.7) | 34 (9.4) |
Dry eye | 3 (1.1) | 7 (2.6) | 2 (0.5) | 4 (1.1) |
Retinal hemorrhage | 3 (1.1) | 0 | 1 (0.3) | 1 (0.3) |
Retinal vein occlusion | 3 (1.1) | 0 | 2 (0.5) | 1 (0.3) |
Visual acuity reduced | 3 (1.1) | 0 | 1 (0.3) | 2 (0.6) |
Cataract | 2 (0.7) | 3 (1.1) | 1 (0.3) | 5 (1.4) |
Most common nonocular AEs,c n (%) | ||||
≥ 1 nonocular AE | 90 (32.6) | 97 (35.4) | 121 (33.2) | 134 (37.1) |
Hypertension | 17 (6.2) | 7 (2.6) | 13 (3.6) | 10 (2.8) |
COVID-19 | 10 (3.6) | 16 (5.8) | 16 (4.4) | 12 (3.3) |
Nasopharyngitis | 6 (2.2) | 6 (2.2) | 5 (1.4) | 7 (1.9) |
Back pain | 2 (0.7) | 10 (3.6) | 5 (1.4) | 8 (2.2) |
Upper respiratory tract infection | NR | NR | 3 (0.8) | 8 (2.2) |
SAEs | ||||
Most common serious ocular AEs,d n (%) | ||||
≥ 1 serious ocular AE in the study eye | 3 (1.1) | 2 (0.7) | 9 (2.5) | 12 (3.3) |
Retinal ischemia | 2 (0.7) | 0 | 1 (0.3) | 2 (0.6) |
Cystoid macular edema | NR | NR | 2 (0.5) | 2 (0.6) |
Retinal artery occlusion | NR | NR | 2 (0.5) | 1 (0.3) |
Uveitis | NR | NR | 2 (0.5) | 0 |
≥ 1 serious ocular AE in the fellow eye | 0 | 0 | 1 (0.3) | 1 (0.3) |
Cataract | NR | NR | 1 (0.3) | 0 |
Retinal vein occlusion | NR | NR | 0 | 1 (0.3) |
Most common serious nonocular AEs,a n (%) | ||||
≥ 1 serious nonocular AE | 9 (3.3) | 16 (5.8) | 22 (6.0) | 23 (6.4) |
Cerebral infarction | 3 (1.1) | 0 | NR | NR |
Adjudicated APTC-defined AEse | ||||
≥ 1 adjudicated APTC-defined AE, n (%) | 3 (1.1) | 4 (1.5) | 4 (1.1) | 5 (1.4) |
Nonfatal stroke, n (%) | 2 (0.7) | 2 (0.7) | 3 (0.8) | 2 (0.6) |
Cerebral infarction | 2 (0.7) | 1 (0.4) | NR | NR |
Cerebrovascular accident | 0 | 1 (0.4) | 1 (0.3) | 1 (0.3) |
Retinal artery occlusion | NR | NR | 2 (0.5) | 1 (0.3) |
Nonfatal MI, n (%) | 1 (0.4) | 2 (0.7) | 1 (0.3) | 2 (0.6) |
Acute myocardial infarction | 0 | 2 (0.7) | 1 (0.3) | 1 (0.3) |
Myocardial infarction | 1 (0.4) | 0 | 0 | 1 (0.3) |
Death, n (%) | NR | NR | 0 | 1 (0.3) |
Myocardial infarction | NR | NR | 0 | 1 (0.3) |
Patients who stopped study treatment due to AEs | ||||
Patients who stopped study treatment due to ocular AEs, n (%) | ||||
Patients who stopped study treatment due to ocular AEs | 0 | 0 | 3 (0.8) | 2 (0.6) |
Uveitis | NR | NR | 2 (0.5) | 0 |
Endophthalmitis | NR | NR | 0 | 1 (0.3) |
Retinal vein occlusion | NR | NR | 0 | 1 (0.3) |
Rhegmatogenous retinal detachment | NR | NR | 1 (0.3) | 0 |
Patients who stopped study treatment due to nonocular AEs, n (%) | ||||
Patients who stopped study treatment due to nonocular AEs | 1 (0.4) | 1 (0.4) | 0 | 1 (0.3) |
Cerebral infarction | 1 (0.4) | 0 | NR | NR |
Multiple sclerosis | 0 | 1 (0.4) | NR | NR |
Squamous cell carcinoma of the oral cavity | NR | NR | 0 | 1 (0.3) |
Deaths | ||||
Patients who died, n (%) | 1 (0.4) | 0 | 1 (0.3) | 2 (0.6) |
Cerebrovascular accident | 1 (0.4) | 0 | NR | NR |
Myocardial infarction | NR | NR | 0 | 2 (0.6) |
Pneumonia | NR | NR | 1 (0.3) | 0 |
AEs of special interest | ||||
Endophthalmitis in study eye | 0 | 0 | 0 | 1 (0.3) |
AE = adverse event; APTC = Antiplatelet Trialists’ Collaboration; MI = myocardial infarctions; NR = not reported; SAE = serious adverse event.
Notes: Multiple occurrences of the same AE in 1 individual patient were counted only once.
Includes AEs with onset before week 24 treatment or dose hold or, if no dose hold, before day 168.
aFrequency ≥ 1% in any study drug group.
bTiming of the event was not specified (i.e., unknown if this was chronic increase in intraocular pressure). Intraocular pressure was measured in the study eye at 30 minutes by the unmasked investigator after the study treatment was performed. If the intraocular pressure value was of concern to the unmasked investigator, the patient remained in the clinic and was managed according to the clinical judgment of the unmasked investigator.
cFrequency ≥ 2% in any study drug group.
dFrequency ≥ 0.5% in any study drug group.
eAPTC events were defined as nonfatal strokes or nonfatal myocardial infarctions or vascular deaths (including deaths of unknown cause). If no events occurred for a certain category, the category was not presented.
Sources: Primary Clinical Study Reports for GR41984 (BALATON)14 and GR41986 (COMINO).16 Details included in the table are from the sponsor’s summary of clinical evidence.2
Critical Appraisal
Internal Validity
In general, part 1 of the BALATON and COMINO studies were appropriately designed and powered to evaluate the efficacy of faricimab relative to aflibercept. Methods for randomization and allocation concealment were appropriate and there were no notable imbalances in baseline patient disease and demographic characteristics and prior and concomitant ocular therapies and surgeries through week 24. The exception was that patients in the BALATON study had relatively greater baseline BCVA and lower CST values compared with patients in the COMINO study. The investigator indicated these observed baseline ocular differences between patients with BRVO and CRVO or HRVO were expected based on historical pivotal trials in RVO; the clinical expert agreed with this statement. As such, it was concluded that the risk of bias arising from the randomization process is unlikely. In contrast to part 1 of the studies, part 2 of the studies did not have a relevant comparison group; therefore, conclusions about the number of injections relative to aflibercept or any other active comparator could not be drawn (i.e., challenges the ability to draw causal inferences).
Table 26: Summary of Harms Results From Baseline Through Week 72 From the BALATON and COMINO Studies (Safety-Evaluable Population)
AEs | BALATON trial | COMINO trial | ||
|---|---|---|---|---|
Faricimab q.4.w. to faricimab PTI (N = 276) | Aflibercept q.4.w. to faricimab PTI (N = 274) | Faricimab q.4.w. to faricimab PTI (N = 365) | Aflibercept q.4.w. to faricimab PTI (N = 361) | |
AEs | ||||
Most common ocular AEs,a n (%) | ||||
≥ 1 ocular AE in the study eye | 100 (36.2) | 112 (40.9) | 177 (48.5) | 172 (47.6) |
Conjunctival hemorrhage | 17 (6.2) | 19 (6.9) | 20 (5.5) | 20 (5.5) |
Intraocular pressure increasedb | 13 (4.7) | 13 (4.7) | 21 (5.8) | 25 (6.9) |
Vitreous detachment | 11 (4.0) | 12 (4.4) | 17 (4.7) | 27 (7.5) |
Cataract | 11 (4.0) | 11 (4.0) | 17 (4.7) | 24 (6.6) |
Retinal vein occlusion | 10 (3.6) | 11 (4.0) | 13 (3.6) | 14 (3.9) |
Macular edema | 10 (3.6) | 6 (2.2) | 11 (3.0) | 14 (3.9) |
Dry eye | 9 (3.3) | 14 (5.1) | 14 (3.8) | 10 (2.8) |
Vitreous floaters | 7 (2.5) | 13 (4.7) | 7 (1.9) | 12 (3.3) |
Eye pain | 7 (2.5) | 4 (1.5) | 11 (3.0) | 5 (1.4) |
Epiretinal membrane | 4 (1.4) | 10 (3.6) | 7 (1.9) | 15 (4.2) |
Cystoid macular edema | 4 (1.4) | 3 (1.1) | 19 (5.2) | 12 (3.3) |
Glaucoma | 3 (1.1) | 1 (0.4) | 5 (1.4) | 12 (3.3) |
≥ 1 ocular AE in the fellow eye | 53 (19.2) | 49 (17.9) | 90 (24.7) | 79 (21.9) |
Cataract | 8 (2.9) | 9 (3.3) | 11 (3.0) | 10 (2.8) |
Dry eye | 6 (2.2) | 11 (4.0) | 13 (3.6) | 7 (1.9) |
Most common nonocular AEs,c n (%) | ||||
≥ 1 nonocular AE | 168 (60.9) | 167 (60.9) | 237 (64.9) | 242 (67.0) |
COVID-19 | 43 (15.6) | 42 (15.3) | 60 (16.4) | 57 (15.8) |
Hypertension | 32 (11.6) | 16 (5.8) | 26 (7.1) | 20 (5.5) |
Nasopharyngitis | 12 (4.3) | 15 (5.5) | 14 (3.8) | 22 (6.1) |
Upper respiratory tract infection | 8 (2.9) | 15 (5.5) | 10 (2.7) | 13 (3.6) |
Back pain | 7 (2.5) | 15 (5.5) | 14 (3.8) | 12 (3.3) |
Serious AEs | ||||
Most common serious ocular AEs,d n (%) | ||||
≥ 1 serious ocular AE in the study eye | 7 (2.5) | 5 (1.8) | 34 (9.3) | 27 (7.5) |
Retinal ischemia | 2 (0.7) | 2 (0.7) | 2 (0.5) | 3 (0.8) |
Retinal vein occlusion | 1 (0.4) | 1 (0.4) | 7 (1.9) | 4 (1.1) |
Macular edema | 1 (0.4) | 0 | 3 (0.8) | 1 (0.3) |
Macular ischemia | 1 (0.4) | 0 | 2 (0.5) | 0 |
Vitreous hemorrhage | 0 | 1 (0.4) | 2 (0.5) | 0 |
Cystoid macular edema | NR | NR | 7 (1.9) | 3 (0.8) |
Uveitis | NR | NR | 3 (0.8) | 0 |
Retinal artery occlusion | NR | NR | 2 (0.5) | 3 (0.8) |
Retinal tear | NR | NR | 1 (0.3) | 2 (0.6) |
Endophthalmitis | NR | NR | 0 | 2 (0.6) |
≥ 1 serious ocular AE in the fellow eye | 0 | 1 (0.4) | 2 (0.5) | 1 (0.3) |
Ocular vascular disorder | 0 | 1 (0.4) | NR | NR |
Cataract | NR | NR | 1 (0.3) | 0 |
Macular edema | NR | NR | 1 (0.3) | 0 |
Retinal vein occlusion | NR | NR | 0 | 1 (0.3) |
Most common serious nonocular AEs,a n (%) | ||||
≥ 1 serious nonocular AE | 32 (11.6) | 38 (13.9) | 49 (13.4) | 59 (16.3) |
Adjudicated APTC-defined AEse | ||||
≥ 1 adjudicated APTC-defined AE, n (%) | 6 (2.2) | 12 (4.4) | 12 (3.3) | 11 (3.0) |
Nonfatal stroke, n (%) | 2 (0.7) | 7 (2.6) | 7 (1.9) | 5 (1.4) |
Cerebral infarction | 2 (0.7) | 2 (0.7) | NR | NR |
Cerebrovascular accident | 0 | 3 (1.1) | 1 (0.3) | 1 (0.3) |
Cerebral hematoma | 0 | 1 (0.4) | NR | NR |
Cerebral thrombosis | 0 | 1 (0.4) | NR | NR |
Retinal artery occlusion | NR | NR | 5 (1.4) | 3 (0.8) |
Ischemic stroke | NR | NR | 1 (0.3) | 1 (0.3) |
Nonfatal MI, n (%) | 2 (0.7) | 4 (1.5) | 4 (1.1) | 4 (1.1) |
Myocardial infarction | 2 (0.7) | 1 (0.4) | 2 (0.5) | 1 (0.3) |
Acute myocardial infarction | 0 | 3 (1.1) | 1 (0.3) | 3 (0.8) |
Stress cardiomyopathy | NR | NR | 1 (0.3) | 0 |
Death, n (%) | 2 (0.7) | 2 (0.7) | 2 (0.5) | 2 (0.6) |
Myocardial infarction | 1 (0.4) | 0 | 0 | 1 (0.3) |
Cerebrovascular accident | 1 (0.4) | 0 | NR | NR |
Coronary artery disease | 0 | 1 (0.4) | NR | NR |
Death | 0 | 1 (0.4) | 1 (0.3) | 0 |
Aortic dissection | NR | NR | 1 (0.3) | 0 |
Cardiac failure | NR | NR | 0 | 1 (0.3) |
Patients who stopped study treatment due to AEs | ||||
Patients who stopped study treatment due to ocular AEs, n (%) | ||||
Patients who stopped study treatment due to ocular AEs | 0 | 1 (0.4) | 8 (2.2) | 6 (1.7) |
Epiretinal membrane | 0 | 1 (0.4) | NR | NR |
Uveitis | NR | NR | 3 (0.8) | 0 |
Vitritis | NR | NR | 2 (0.5) | 0 |
Cystoid macular edema | NR | NR | 1 (0.3) | 1 (0.3) |
Retinal vein occlusion | NR | NR | 1 (0.3) | 1 (0.3) |
Rhegmatogenous retinal detachment | NR | NR | 1 (0.3) | 0 |
Endophthalmitis | NR | NR | 0 | 1 (0.3) |
Choroidal neovascularization | NR | NR | 0 | 1 (0.3) |
Glaucoma | NR | NR | 0 | 1 (0.3) |
Macular hole | NR | NR | 0 | 1 (0.3) |
Patients who stopped study treatment due to nonocular AEs, n (%) | ||||
Patients who stopped study treatment due to nonocular AEs | 1 (0.4) | 3 (1.1) | 3 (0.8) | 4 (1.1) |
Cerebral infarction | 1 (0.4) | 0 | 0 | 1 (0.3) |
Ischemic stroke | 0 | 1 (0.4) | NR | NR |
Multiple sclerosis | 0 | 1 (0.4) | NR | NR |
Death | NR | NR | 2 (0.5) | 0 |
Gastrointestinal inflammation | NR | NR | 1 (0.3) | 0 |
Pneumonia | NR | NR | 1 (0.3) | 0 |
Cerebrovascular accident | NR | NR | 0 | 1 (0.3) |
Squamous cell carcinoma of the oral cavity | NR | NR | 0 | 1 (0.3) |
Asthenia | NR | NR | 0 | 1 (0.3) |
Deaths | ||||
Patients who died, n (%) | 2 (0.7) | 2 (0.7) | 5 (1.4) | 3 (0.8) |
Cerebrovascular accident | 1 (0.4) | 0 | NR | NR |
Myocardial infarction | 1 (0.4) | 0 | 0 | 2 (0.6) |
Coronary artery disease | 0 | 1 (0.4) | NR | NR |
Death | 0 | 1 (0.4) | 2 (0.5) | 0 |
Pneumonia | NR | NR | 1 (0.3) | 0 |
Aortic dissection | NR | NR | 1 (0.3) | 0 |
COVID-19 | NR | NR | 1 (0.3) | 0 |
Cardiac failure | NR | NR | 0 | 1 (0.3) |
AE of special interest | ||||
Endophthalmitis in study eyef | NR | NR | NR | NR |
AE = adverse event; APTC = Antiplatelet Trialists’ Collaboration; MI = myocardial infarction; NR = not reported; PTI = personalized treatment interval; q.4.w. = every 4 weeks.
Notes: Multiple occurrences of the same AE in 1 individual patient were counted only once.
Includes AEs with an onset on or after the date of the first dose through week 72.
aFrequency ≥ 3% in any study drug group.
bTiming of the event was not specified (i.e., unknown if this was a chronic increase in intraocular pressure). Intraocular pressure was measured in the study eye at 30 minutes by the unmasked investigator after the study treatment was performed. If the intraocular pressure value was of concern to the unmasked investigator, the patient remained in the clinic and was managed according to the clinical judgment of the unmasked investigator.
cFrequency ≥ 5% in any study drug group.
dFrequency ≥ 0.5% in any study drug group.
eAPTC events were defined as nonfatal strokes or nonfatal myocardial infarctions or vascular deaths (including deaths of unknown cause). If no events occurred for a certain category, the category was not presented.
fAEs of endophthalmitis in the study eye were not reported from baseline through week 72. In part 2 of the BALATON study, no cases of endophthalmitis in the study eye were reported in any study drug group. In part 2 of the COMINO study, 1 case of endophthalmitis in the study eye was reported in the aflibercept q.4.w. to faricimab PTI group.
Sources: Final Clinical Study Report for GR41984 (BALATON)15 and updated Clinical Study Report for GR41986 (COMINO).17 Details included in the table are from the sponsor’s summary of clinical evidence.2
Investigators and patients were masked to treatment assignment in part 1 and to both the original treatment assignment and faricimab treatment interval in part 2 of the studies (with the use of sham procedures). All intravitreal injections of the study drug were performed onsite by the unmasked investigator, while the other masked investigator evaluated ocular assessments and administered the NEI-VFQ-25. BCVA was assessed on the ETDRS visual acuity chart by masked BCVA examiners. Imaging was performed by masked OCT technicians, and central reading centres were used to conduct masked ocular imaging analyses and for storage. As such, it was concluded that any possible impact on the interpretation of the efficacy results due to unmasking is unlikely. Further, no notable difference was identified in efficacy or harms that could have contributed to unmasking (i.e., inferring treatment assignment).
There is a lack of evidence in the literature to inform the measurement properties of BCVA as measured by ETDRS charts, CST as measured by OCT, and vision-related functioning and HRQoL as measured by the NEI-VFQ-25 (i.e., studies that investigate the measurement properties and estimate the minimal important difference) as outcome measures in patients with RVO. However, there was also no evidence in the literature to suggest there are concerns with these tools.
The investigators indicated there was no impact on the type I error rate for the superiority test following the noninferiority test (i.e., a claim of superiority after noninferiority can be made without multiplicity adjustment).10 This assertion aligns with FDA guidance9 indicating there is no concern for an increased risk of type I error if superiority is tested after noninferiority in a noninferiority trial.
Based on the results of the aflibercept pivotal trials in BRVO and CRVO,33-35 the investigator indicated that the 4-letter noninferiority margin preserved between 44% and 77% of the least estimated benefit of aflibercept relative to a control. The investigator further stated that a loss of 5 letters (1 ETDRS line) between treatments was considered clinically relevant; therefore, a 4-letter noninferiority margin ensured no important loss of efficacy with the new treatment over the reference treatment. The clinical expert agreed with the rationale provided by the investigator on the use of a 4-letter noninferiority margin (i.e., a difference of 5 letters or 1 Snellen line can be considered clinically meaningful in the context of comparisons with a similar treatment).
Because no formal superiority tests were performed for the secondary end points and the subgroup analysis of the primary end point, these results are considered supportive evidence only. For statistically significant results (i.e., change from baseline in CST in the study eye at week 24 in the BALATON trial, and proportion of patients with an absence of both intraretinal fluid and subretinal fluid at week 24 and the change from baseline BCVA among the subgroup of patients with HRVO in the COMINO trial), there is an increased risk that the null hypothesis was erroneously rejected. Further, the number of patients with HRVO available for the subgroup analysis was relatively low (< 20%) and, as such, the small sample size likely introduced uncertainty in the results. There was no formal statistical approach for testing subgroup differences (e.g., test for treatment by subgroup interaction). Although the estimated effect was statistically significant in the HRVO subgroup (and not in the CRVO subgroup), comparing statistical significance across subgroups is not a valid method for inferring effect modification.11
Study and study treatment discontinuation rates before week 24, including due to lost to follow-up and AEs, were less than 5% for each group from each study and, as such, the risk of attrition bias is unlikely.
Although major protocol deviation rates through week 24 were approximately 30% for each group from each study, the rates were generally balanced between groups. The most frequent type of major protocol deviation in both studies was procedural-related; however, each procedural-related protocol deviation was reported in less than 10% of patients in each group from both studies. As such, it was concluded that the risk of bias due to deviations from the intended intervention in part 1 of the studies is low. Moreover, 98.9% of patients or greater in each group from each study received treatment through week 24; the mean number of study drug administrations in the study eye through week 24 was 5.8 in each group in the BALATON trial and 5.7 in each group in the COMINO trial (note that a total of 6 injections were planned for each group in part 1 of the studies). However, it should be noted that a relatively large proportion of patients (> 50%) in each group from both studies were reported with at least 1 major protocol deviation through week 72, with the majority of the major protocol deviations (> 40%) related to procedures. As such, it was concluded that the risk of bias (of unknown direction and magnitude) due to deviations from the intended intervention in part 2 of the studies is high.
Missing data were implicitly imputed by the MMRM model assuming missing at random for both the primary end point of change from baseline in BCVA, and the secondary end point of change from baseline in CST at week 24. A sensitivity analysis (in which a multiple imputation was performed where missing BCVA data were assumed to be missing not at random and were assumed to have worse outcomes compared with the rest of the study population) was performed for the primary end point only. Overall, the sensitivity analysis results were generally consistent with the primary analysis results of the primary end point in both studies and, as such, no major concern was identified with this approach to handling missing outcomes data.
The assumptions for missing outcomes data (missing at random for change from baseline in CST at week 24 and LOCF for the categorical secondary outcomes at week 24) are likely not plausible. For change from baseline in the NEI-VFQ-25 composite score at week 24, the missing data were not imputed. For the treatment intervals at week 68, the percentages were based on patients who had not discontinued the study at week 68. Nonetheless, missing outcomes data and data on the patients who discontinued from the study before week 24 were generally low and balanced between groups in both studies. Therefore, the risk of bias due to missing outcomes data was considered low. The exception was for the proportion of patients with an absence of both intraretinal fluid and subretinal fluid in the BALATON trial, where the amount of missing outcome data was relatively high (13% to 16%). Reasons for missingness, aside from early study discontinuation, included study visits missed and outcome not measured at a study visit; it is unclear whether the reasons for missingness are balanced between groups. Further, in consultation with the clinical expert, it was concluded that the assumption that fluid status would stay constant over time is likely not plausible. Therefore, it was concluded that there are some concerns for risk of bias due to missing outcome data for the proportion of patients with an absence of both intraretinal fluid and subretinal fluid.
Intercurrent events were defined as a discontinuation of study treatment due to AEs or lack of efficacy, and use of any prohibited systemic treatment or prohibited therapy in the study eye, as per protocol. For the primary and secondary end points, intercurrent events were handled by applying a treatment policy strategy. Of the therapies prohibited in the studies, the clinical expert advised that the administration of micropulse and focal or grid laser, as well as steroid implant and triamcinolone injections may be used in practice as adjunct therapies (to anti-VEGF therapies and faricimab), particularly in cases of suboptimal treatment effect; therefore, no major concern with the application of a treatment policy strategy was identified. Overall, the results of the supplementary analyses (a PP analysis and an analysis using a hypothetical strategy for all intercurrent events) were generally consistent with the primary analysis results of the primary end point in both studies.
External Validity
The inclusion criteria in the BALATON and COMINO studies included the population of interest identified in the indication for faricimab, which is for the treatment of macular edema secondary to RVO. Notably, less than 20% of patients in the COMINO trial had HRVO; therefore, the generalizability of the study results to patients with HRVO is less certain.
The clinical expert indicated that the inclusion criteria adequately captured all patients who would be considered candidates for faricimab in practice. Further, the clinical expert indicated that the study population was generally representative of the patients typically seen in practice who would be candidates for treatment with faricimab.
From the perspective of conducting a clinical trial in RVO, it was concluded that no patient who could be considered a candidate for treatment with faricimab was excluded due to any exclusion criteria. However, the clinical expert noted that patients with RVO generally present with uncontrolled blood pressure; cardiovascular disease, including stroke and myocardial infarction; diabetic retinopathy; and complications of cataract surgery. The clinical expert advised that these patients would be considered candidates for treatment with faricimab in practice.
In general, the Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus (published in 2015)7 advises on the use of anti-VEGF therapy in patients with RVO with OCT evidence of macular edema. This is aligned with input from the clinician groups and the clinical expert consulted for this review regarding the current treatment options that are available in practice. Therefore, the comparator in the studies (aflibercept) is relevant for the purpose of the present review; however, direct comparative evidence for the effect of faricimab versus other anti-VEGF treatments (e.g., ranibizumab and bevacizumab) in the treatment of patients with macular edema secondary to RVO is lacking.
In consultation with the clinical expert, it was concluded that the outcome measures are generally reflective of assessments of treatment response in practice (i.e., the outcomes are clinically meaningful based on the clinical expert input). Because the goal of treatment is to improve visual acuity, the clinical expert advised that if treatment response is demonstrated on imaging (CST measurement) but there is no change in visual acuity, then the clinician will consider discontinuing treatment (because this could indicate significant macular ischemia, and this state of edema does not affect visual acuity). However, if treatment response is demonstrated in visual acuity with change in macular edema status, then the clinician will likely use the imaging results (CST as assessed by OCT) as an objective approach to determine whether to extend, maintain, or reduce the treatment interval in practice.
The clinical expert indicated that the common practice in Canada is to treat and extend, but it can also be a fixed treatment interval if extending is not possible. However, the clinical expert noted that the criteria used to determine a personalized treatment interval dosing regimen are not used uniformly by clinicians in practice.
In consultation with the clinical expert, it was concluded that the assessment time point at week 24 is considered appropriate for evaluating treatment effect in the therapeutic area of macular edema secondary to RVO. The exception is the outcome measure of treatment interval (proportion of patients on extended treatment intervals), for which the clinical expert suggested an assessment time point at month 24 (versus at week 68 in the studies).
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:12,13
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
For single-arm trials: Although GRADE guidance is not available for noncomparative studies, the review team assessed pivotal single-arm trials for study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up. In the current review, 68-week data from both trials were appraised as single-arm given the lack of relevant comparator at this time point.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on the thresholds informed by the clinical expert consulted for this review. For the primary outcome of change from baseline in BCVA at week 24, the noninferiority margin used in the trials was the threshold.
For the GRADE assessments, the BALATON and COMINO studies were assessed individually because the BALATON study had a patient population with macular edema secondary to BRVO and the COMINO study had a patient population with macular edema secondary to CRVO or HRVO.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for faricimab versus aflibercept in patients with macular edema secondary to BRVO.
Table 3 presents the GRADE summary of findings for faricimab versus aflibercept in patients with macular edema secondary to CRVO or HRVO.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The included studies did not provide evidence on the efficacy or safety of faricimab against other interventions beyond aflibercept 2 mg administered every 4 weeks. The ITC was conducted to investigate the noninferior efficacy and safety of faricimab versus other anti-VEGF treatments, dexamethasone, and laser therapy for the treatment of RVO. This section aims to provide an overview of the conduct, the results, and the critical appraisal of the sponsor-submitted ITC.
Description of Indirect Comparison
Methods for the Search, Selection, Data Extraction, and Risk-of-Bias Appraisal
A targeted literature search by the sponsor was conducted on April 3, 2023, and updated in December 2023. An SLR was conducted to identify RCTs with relevant treatments, and a feasibility assessment was conducted to compare faricimab with relevant comparator treatments in patients with RVO, with CRVO and BRVO split as a sensitivity analysis. Methods for the systematic review are in Table 25. The population of interest was limited to adult patients (18 years and older) with macula edema secondary to RVO; no specific geography restrictions were applied.
As part of the feasibility assessment, the investigators compared patient baseline characteristics and study designs for candidate RCTs qualitatively to determine whether they were sufficiently homogeneous to combine in an NMA and whether they formed a connected network for each outcome of interest.
The sponsor-submitted ITC was conducted using a Bayesian approach under a random-effects model as the principal analysis and a fixed-effects model for sensitivity to compare faricimab 6 mg every 4 weeks with aflibercept 2.0 mg every 4 weeks (reference treatment) in patients with RVO. Of the outcomes investigated in the sponsor’s ITC, the following are relevant to the current review: BCVA, proportion of patients gaining at least 15 letters, mean change in CST, safety (analyzed as the proportion of patients with SAEs as well as all-cause discontinuations), and mean number of injections. The outcome of mean number of injections was not considered feasible to investigate; thus, no findings were presented. The main comparators of interest to the current review were anti-VEGF treatments, specifically those given in flexible regimens such as PRN, which are typically used in clinical practice.
Objectives
The objective of the NMA was to assess the noninferior comparative efficacy and safety of faricimab 6 mg every 4 weeks compared with other anti-VEGF treatments, and to assess efficacy compared with dexamethasone and laser therapy using the populations and end points defined in Table 27.
Table 27: Study Selection Criteria and Methods for the Systematic Review Portion of the ITC Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients (18 years and older) with ME secondary to RVO:
|
Intervention |
|
Comparator |
|
Outcome | Vision outcomes:
Anatomic outcomes:
Safety outcomes:
Injection frequency:
|
Study designs | Randomized controlled trials (phases II to IV):
|
Publication characteristics | Full publications with no restrictions. Conference abstracts from January 2019 to December 2023. Only English-language sources were eligible. |
Exclusion criteria |
|
Databases searched | The electronic databases, including Embase, MEDLINE, and the Cochrane Library, were queried in April and December 2023. Additional searches comprised conference proceedings, HTA agency websites, clinical trial registries, regulatory reports, reference lists of included publications, and related SLRs. |
Selection process | Duplicate citations were removed, and titles and abstracts of the remaining citations were screened independently by 2 reviewers (first pass) using the predefined eligibility criteria. Two reviewers independently examined full papers (second pass), and the project lead verified the final inclusion and exclusion of citations. Disputes as to eligibility were referred to a strategic advisor. |
Data extraction process | Data extraction occurred based on country, study design, baseline characteristics, interventions, outcomes reported, results, and limitations. A single reviewer extracted the data, and quality was checked by a senior reviewer. |
Quality assessment | Two reviewers performed the study-level quality assessment of the final studies included in the SLR. Any disagreements were resolved by discussion and/or additional referees. The quality (risk of bias) assessment of the studies was conducted using the 7-criteria checklist provided in section 2.5 of the NICE single technology appraisal user guide. This approach is based on guidance provided by the Centre for Reviews and Dissemination for assessing the quality of studies included in SLRs and assesses the likelihood of selection, performance, attrition, and detection bias. |
AE = adverse event; BCVA = best-corrected visual acuity; BRVO = branch retinal vein occlusion; CRVO = central retinal vein occlusion; CST = central subfield thickness; HRVO = hemiretinal vein occlusion; HTA = health technology assessment; ITC = indirect treatment comparison; MA = meta-analysis; ME = macular edema; NICE = National institute for Health and Care Excellence; PRN = pro re nata (as needed); q.4.w. = every 4 weeks; RCT = randomized controlled trial; RVO = retinal vein occlusion; SAE = serious adverse event; SLR = systematic literature review.
Sources: Sponsor-submitted ITC.40 Details included in the table are from the sponsor’s summary of clinical evidence.2
NMA Design
NMA Analysis Methods
A feasibility assessment compared faricimab with relevant comparator treatments in patients with RVO (split into CRVO and BRVO as a sensitivity analysis). The structure of the networks of evidence was assessed, and network plots were drawn to visualize the evidence base. Analyses of AEs were conducted using the difference in the proportion of patients with SAEs and all causes of discontinuation.
The mean change from baseline in BCVA and CST were analyzed using a Bayesian NMA with normal likelihood. AEs, including the proportion of patients discontinuing studies, were analyzed using a binomial likelihood and logit link function. Changes in visual acuity were also analyzed using the outcome of categorical gains or losses (e.g., gaining at least 15 letters).
In some instances, the fixed-effects model had a lower deviance information criterion (DIC) than the random-effects model; however, no DIC was deemed meaningful (> 5-point difference), so the random-effects model with an informative prior was selected as the best fit for the base case throughout in the RVO population. To avoid the truncation of tau in the CRVO and BRVO subgroups, the second sensitivity analysis listed in Table 28 was conducted using a widened prior. The structure of the networks of evidence was assessed, and network plots were drawn to visualize the current evidence base. The total residual deviance was calculated and compared against the total number of independent data points to determine absolute fit. For the random-effects model, the estimate and 95% CI for the between-study SD were reported. As applicable, median odds ratios, treatment differences, and the corresponding 95% CrIs were reported. There was no evidence of inconsistency in the results of the node-splitting random-effects model (Table 28).
Methodological heterogeneity was assessed qualitatively, comparing study design, baseline characteristics, and end points with a focus on 6 key outcomes: mean change from baseline in BCVA and CST, categorical vision changes from baseline (i.e., at least a 15-letter gain or loss), (serious) ocular AEs, number of injections, and all-cause discontinuations.
Change from baseline in BCVA score and CST were modelled as continuous data, using arm-level mean change from baseline as the outcome. The treatment effects for BCVA and CST were calculated using appropriate methods. Variances were derived from the CI using standard methods based on the normal distribution if they were not reported explicitly. If neither the CI nor the variance in the change from baseline were reported but estimates of baseline and follow-up values along with variances were available, computations of the values were made, where appropriate. The median change was used and reported in scenarios where the mean change was not reported or could not be derived using the methods defined in the NMA. If the variance of the change was not reported or could not be derived, it was estimated using a pooled variance (pooled SD squared) of the change across all studies and arms with values in the relevant network. In the case of nonconvergence due to rare events, a continuity correction was applied by adding 0.5 to all study cells with 0 events, and analysis was performed on the log odds ratio scale using a normal likelihood. Studies that did not report on the outcome of interest were excluded from the NMA for that outcome. Outcome definitions in the feasibility assessment were harmonized as follows: the NMA was conducted at the 6-month time point, and time points of 6 months, 24 weeks, and 180 days were interpreted interchangeably.
Table 28: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian network meta-analysis (random- and fixed-effects models based on the deviance information criterion) through a Markov chain Monte Carlo simulation |
Priors | Vague (noninformative) |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | Node-splitting method as described by National Institute for Health and Care Excellence Decision Support Unit Technical Support Document 4 |
Computation | Trace plots and the Brooks-Gelman-Rubin statistics The simulation models used 40,000 iterations with a burn-in of 10,000 and a thinning parameter of 10, except for CST, where the iterations were multiplied by 10 to improve convergence diagnostics41 |
Assessment of convergence | Trace plots and the Brooks-Gelman-Rubin statistics |
Outcomes | Mean change from baseline in BCVA and CST, proportion of patients gaining or losing BCVA letters, proportion of patients with any adverse events and serious adverse events, number of injections, and all-cause discontinuations |
Follow-up time points | 24 (± 4) weeks |
Construction of nodes | Each treatment and schedule were included in the network multiple connections were informed by more than 1 study |
Sensitivity analyses | Sensitivity analysis around model assumptions:
|
BCVA = baseline change in visual acuity; CST = central subfield thickness; FE = fixed effects; ITC = indirect treatment comparison; RE = random effects.
Sources: Sponsor-submitted ITC.40 Details included in the table are from the sponsor’s summary of clinical evidence.2
Results of the Sponsor-Submitted ITC
Summary of Included Studies
In total, 39 published RCTs met the inclusion criteria for the systematic review, of which 19 were excluded following the feasibility assessment. Of the 20 studies, 10 were conducted in patients with BRVO only, 9 in patients with CVRO, and 1 in patients with CRVO or BRVO. An overview of the assessment of homogeneity for the ITC is presented in Table 29. The 20 studies provided data on the 6 interventions: aflibercept, bevacizumab, dexamethasone, faricimab, ranibizumab, and laser therapy. Eleven of the included studies assessed ranibizumab,42-52 5 studies48,49,51,53,54 assessed bevacizumab, 7 studies33-35,46,54-57 assessed aflibercept with other treatments, and 2 studies assessed faricimab versus aflibercept.55,56
There was variability observed in the baseline demographic characteristics across the studies included in the feasibility assessment representing a source of heterogeneity. The number of patients enrolled ranged from 29 to 1,267; the mean age per treatment group ranged from 52.9 to 72.4 years; there were more male patients enrolled, with the proportion of male patients ranging from 30% to 68.42%. There was variability in the regions where the trials were conducted. Ten studies enrolled patients from a single country. Trial start dates also varied across studies, ranging from 2004 to 2021, and the end dates ranged from 2008 to 2022. Two studies were open-label, 17 studies were double-masked, and 1 study did not report on masking. Prior therapy at baseline was reported in 2 studies and included anti-VEGF therapy and laser therapy. Concomitant medications and background treatments were reported in 5 studies.
The treatment regimens evaluated across the studies included a fixed-interval treatment (every 4, 6, or 12 weeks), PRN, and a standard dose schedule. Data on number of injections from baseline to 6 months were available for 16 studies included in the NMA. The mean number of injections reported ranged from 1.7 to 5.94. There was limited information included in the feasibility assessment on the number of treatment-naive patients enrolled across studies, presenting a source of uncertainty. The treatments reported included anti-VEGFs, laser, and other ocular therapies. The outcome of mean number of injections was not considered feasible to investigate.
The mean time of RVO diagnosis was reported in 14 studies and ranged from 1.1 (SD = 0.95) to 10.5 (SD = 26.6) months across studies. Eleven studies included the BRVO population or reported a subgroup for this population and 12 studies included the CRVO population or reported a subgroup for this population. Heterogeneity was also observed in the baseline BCVA measurements and there was variability observed in the study definitions of visual acuity and units of measurement. Twenty studies provided data on mean change from baseline in BCVA, which was measured in ETDRS letters or using the Snellen LogMAR (logarithm of the minimum angle of resolution) scale; 10 and 12 studies reported results for patients with BRVO and CRVO, respectively. Baseline mean BCVA scores (terminology reported across studies included letters, ETDRS letters, ETDRS visual acuity testing charts, and ETDRS visual acuity) ranged from 41 to 64.6, with 2 studies having no unit of outcome reported for BCVA. BCVA measured in Snellen LogMAR ranged from 0.62 to 0.87. There was also variability in the time point for assessing efficacy and safety end points (range of 1 to 15 months). There was also variability in how retinal thickness was measured across the 20 studies included in the NMA. Retinal thickness was measured using CST, centre-point thickness, and central retinal thickness (CRT). Out of these, 14 reported the SD or CIs. Mean retinal thickness was reported in 1 study and ranged from 268.59 μm to 289.87 μm. Mean change from baseline in CRT varied across studies and ranged from −457.2 μm to −119 μm. The mean CST at baseline was presented in 4 studies and ranged from 222.1 μm to 451.6 μm (unit of measurement not reported in 1 study).
Heterogeneity was also observed in the NEI-VFQ-25 scale measurements assessed across studies included in the feasibility assessment. The proportion of patients who withdrew or discontinued the study was reported in 17 studies included in the feasibility assessment. Intraocular pressure at baseline was reported in only 5 studies and the mean ranged from 14.8 mm Hg to 18 mm Hg.
Table 29: Assessment of Homogeneity for the Sponsor-Submitted ITC
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history |
|
Trial eligibility criteria | Similar inclusion and exclusion criteria across studies. |
Dosing of comparators |
|
Placebo response | 8 studies included in the network included sham (placebo) treatment arms. |
Definitions of end points |
|
Timing of end point evaluation or trial duration | Time points were reported as 24 (± 4) weeks; 24 weeks and 180 days were interpreted interchangeably. Time periods of 6 to 7 months were grouped as 6 months. |
Withdrawal frequency | Reported in 17 studies. Treatment withdrawal was unclear overall. |
Clinical trial setting | Similar due to the nature of the injection. |
Study design |
|
BCVA = best-corrected visual acuity; BRVO = branch retinal vein occlusion; CRT = central retinal thickness; CRVO = central retinal vein occlusion; ETDRS = Early Treatment Diabetic Retinopathy Study; ITC = indirect treatment comparison; vs. = versus.
Sources: Sponsor-submitted ITC.40 Details included in the table are from the sponsor’s summary of clinical evidence.2
Risk of Bias
Overall, the risk of bias for the studies included in the feasibility assessment ranged from low to moderate or unclear. Details on randomization and allocation concealment were lacking for some studies (there was unclear judgment in 9 studies). The risk of bias was considered low for 11 of the studies included in the NMA. Most studies provided details on the patient population and prognostic factors at baseline; however, key methodological details pertaining to the treatment regimens and how missing data (e.g., using LOCF) was addressed were lacking. Participants and assessors were masked in the majority of the studies. Selective outcome reporting is not suspected across the included studies.
Results
Mean Change From Baseline in BCVA
In total, 20 trials were included in the base-case analysis for BCVA. There were several connections consisting of aflibercept 2 mg, ranibizumab, bevacizumab, dexamethasone 0.7 mg, faricimab, and sham or placebo given PRN, given according to standard care practices, or given every 4 weeks. The networks were sparse, with many nodes connected by just 1 or 2 studies. The analysis was conducted under a random-effects model, which was considered the best-fit model by DIC (DIC random effects = 174.713). Results from the fixed-effects model (sensitivity analysis 1) and the random-effects model using a vague prior for the RVO population were consistent with the base case.
Compared with anti-VEGFs administered PRN, the point estimates for the difference in mean change from baseline in BCVA at 6 months mostly suggested little to no difference (i.e., the point estimate for the difference was less than 4 letters) compared with faricimab 6 mg every 4 weeks. One exception was the comparison with bevacizumab 1.25 mg PRN, where the point estimate for the difference favoured faricimab 6 mg every 4 weeks. The 95% CrIs for comparison between faricimab and ant-VEGFs administered PRN were wide, including the possibility of clinically important effects for either treatment being compared. The between-study heterogeneity estimate (tau) assessed reported a median of 2.85 (95% Crl, 1.397 to 3.911), indicating a low level of heterogeneity. The estimates for mean change from baseline in BCVA for faricimab 6 mg every 4 weeks and other comparators are presented in Table 30. The sensitivity analyses conducted using the fixed-effects model (sensitivity analysis 1) and a vague prior (sensitivity analysis 2) were consistent with findings from the base-case analysis (Table 31). Inconsistency was investigated using node-splitting fixed- and random-effects models. There was no evidence of inconsistency from the node-splitting random-effects model results.
Mean Change From Baseline in CST
The network for mean change from baseline in CST was informed by 17 studies. There were several connections consisting of aflibercept 2 mg, ranibizumab, bevacizumab, dexamethasone 0.7 mg, faricimab, or sham or placebo given PRN, given according to standard care practices, or given every 4 weeks. However, the networks were sparse, with many nodes connected by just 1 or 2 studies. The base-case analysis under a random-effects model was considered the best fit by DIC (DIC random effects = 339.279). Results from the sensitivity analyses fixed-effects model (sensitivity analysis 1) and the random-effects model using a vague prior for the RVO population were consistent with the base-case analysis.
Faricimab 6 mg every 4 weeks was favoured over bevacizumab 1.25 mg PRN in the NMA findings; however, the 95% CrI for the between-group difference included the possibility of little to no difference between the 2 treatments (Table 30). In the comparisons with all other anti-VEGFs, the point estimates for between-group differences favoured faricimab 6 mg every 4 weeks; however, the 95% CrI included the possibility that either treatment could be favoured. There is uncertainty as to the clinical relevance of the point estimates and values within the 95% CrIs. The between-study heterogeneity estimate (tau) had a median of 9.518 (95% Crl, 0.334 to 23.977), indicating some heterogeneity. Additionally, networks for studies investigating CRVO and BRVO populations separately were analyzed, and the results were consistent with the base-case findings.
Patients Gaining at Least 15 ETDRS Letters
Results specific to the proportion of patients gaining at least 15 ETDRS were not reported in the sponsor-submitted ITC.
Mean Number of Injections
The network of studies for faricimab and anti-VEGF treatments allowing for a flexible (PRN) treatment regimen was connected with sham injections only; therefore, no treatment effect with regard to flexible regimens could be estimated.
Harms
Ocular Adverse Events
For ocular AEs, 10 studies were included in the network. There were several connections consisting of aflibercept 2 mg, ranibizumab, bevacizumab, dexamethasone 0.7 mg, faricimab, and placebo that were given PRN, given according to standard care practices, or given every 4 weeks. However, the networks were sparse, with many nodes connected by just 1 or 2 studies. The base case under the random-effects model was considered the best fit by DIC (DIC random effects = 140.99). Results from the fixed-effects model were consistent with the base-case analysis. The network contained 1 closed loop, and some connections were informed by more than 1 study.
For comparisons with anti-VEGFs administered PRN, the 95% CrIs for the odds ratios were too wide to inform a conclusion about which treatment might be favoured (Table 30). The between-study heterogeneity estimate (tau) reported a median of 0.351 (95% Crl, 0.025 to 1.350).
Serious Ocular Adverse Events
For serious ocular AEs, 10 studies were included in the network. The base case under the random-effects model was considered the best fit by DIC (DIC random effects = 58.635). There were several connections consisting of aflibercept 2 mg, ranibizumab, bevacizumab, dexamethasone 0.7 mg, faricimab, and placebo that were given PRN, given according to standard care practices, or given every 4 weeks. However, the networks were sparse, with many nodes connected by just 1 or 2 studies.
For comparisons with anti-VEGFs administered PRN, the 95% CrIs for the odds ratios were too wide to inform a conclusion about which treatment might be favoured (Table 30).
All-Cause Discontinuation
In total, 16 studies were included in the network for all-cause discontinuation. There were several connections in the network consisting of aflibercept 2 mg, ranibizumab, bevacizumab, dexamethasone 0.7 mg, faricimab, and placebo that were given PRN, given according to standard care practices, or given every 4 weeks. However, the networks were sparse, with many nodes connected by just 1 or 2 studies. The base-case analysis under the random-effects model was considered the best fit based on the DIC (DIC random effects = 188.744). Results from the fixed-effects model were consistent with the base-case model. For comparisons with all other anti-VEGFs, the 95% CrIs for the odds ratios were too wide to inform a conclusion about which treatment might be favoured (Table 30). The between-study heterogeneity estimate (tau) reported a median of 0.632 (95% Crl, 0.160 to 1.377).
Table 30: ITC Results for Faricimab Versus Anti-VEGF Comparators Administered PRN
Characteristic | Faricimab vs. comparator |
|---|---|
BCVA | |
BCVA change from baseline at 6 months, mean difference (95% CrI) | |
Bevacizumab 1.25 mg, PRN | 5.22 (−3.35 to 13.80) |
Ranibizumab 0.5 mg, PRN | 3.59 (−2.94 to 10.17) |
Aflibercept 2 mg, PRN | 1.87 (−7.43 to 11.16) |
Patients gaining at least 15 ETDRS letters | Not assessed |
Central subfield thickness | |
Change from baseline to 6 months, mean difference (95% CrI) | |
Bevacizumab 1.25 mg, PRN | −68.95 (−133.02 to −1.48) |
Ranibizumab 0.5 mg, PRN | −20.08 (−70.53 to 32.35) |
Aflibercept 2 mg, PRN | −37.30 (−107.99 to 35.72) |
Treatment interval | |
Mean number of injections | Not assessed |
Ocular adverse events | |
Odds ratio, change from baseline to 6 months (95% CrI) | |
Bevacizumab 1.25 mg, PRN | NR |
Ranibizumab 0.5 mg, PRN | 0.64 (0.14 to 2.80) |
Aflibercept 2 mg, PRN | NR |
Serious ocular adverse events | |
Odds ratio, change from baseline to 6 months (95% CrI) | |
Bevacizumab 1.25 mg, PRN | NR |
Ranibizumab 0.5 mg, PRN | 0.53 (0.03 to 10.50) |
Aflibercept 2 mg, PRN | NR |
All-cause discontinuation | |
Odds ratio, change from baseline to 6 months (95% CrI) | |
Bevacizumab 1.25 mg, PRN | NR |
Ranibizumab 0.5 mg, PRN | 1.00 (0.16 to 6.53) |
Aflibercept 2 mg, PRN | NR |
BCVA = best-corrected visual acuity; CrI = credible interval; ETDRS = Early Treatment Diabetic Retinopathy Study; ITC = indirect treatment comparison; NR = not reported; PRN = pro re nata (as needed); vs. = versus.
Sources: Sponsor-submitted ITC.40 Details included in the table are from the sponsor’s summary of clinical evidence.2
Critical Appraisal of ITC
Studies included in the sponsor-submitted ITC were identified through an SLR approach based on prespecified criteria. The research question and inclusion criteria for the systematic review and feasibility assessment aligned with the protocol. The search was adequately comprehensive and the methods for the SLR were generally appropriate to reduce the risk of error and bias in study selection, data extraction, and risk-of-bias appraisal. Risk of bias was assessed at the study level rather than the outcome level. The risk of bias for individual effect estimates reported in each study could differ; therefore, the study-level risk-of-bias assessments may not apply equally to all study results.
The inclusion criteria for the SLR allowed a population relevant to settings in Canada to be included in the NMA. All comparators of interest were included in the NMA. The outcomes included in the feasibility assessment were considered clinically relevant and appropriate; however, outcomes such as vision-related functioning and HRQoL and injection frequency (for mean number of injections) were not investigated in the NMA despite being identified as important to patients and having been reported in some studies identified in the SLR.
The treatment regimens evaluated across studies included in the ITC were fixed-interval treatments (every 4, 6, or 12 weeks), treatment as needed (PRN), and standard dose schedule. Although highlighted in the SLR, it was not considered feasible to assess injection frequency (i.e., the mean number of injections) in the sponsor-submitted ITC. The network for studies reporting on mean number of injections showed that faricimab and other anti-VEGF treatments administered PRN were connected based on comparisons with sham treatment only. Therefore, treatment effects with respect to flexible regimens could not be estimated and definitive conclusions could not be drawn. Preplanned fixed schedules may not be reflective of current Canadian clinical practice, where it is common to use a treat-and-extend approach. According to the clinical expert consulted for this review, PRN dosing is beneficial in real-world practice because it reduces clinical and patient burden and produces acceptable clinical outcomes compared with fixed monthly schedules. Therefore, the PRN regimens align with clinical practice procedures.
The use of a Bayesian NMA and the random-effects model was appropriate. The random-effects model provided the best model fit and accounts for within-study and between-study variability. There was variability observed in the baseline characteristics across the trials included in the feasibility assessment, and the degree of heterogeneity was difficult to assess due to the lack of reporting on several key study characteristics of interest for RVO that could be potential effect modifiers (for example, blood pressure, diabetes, concurrent diabetic retinopathy, coagulability, blood viscosity, and anemia). There was considerable variability in study design, year of conduct and sample size, age, and dosing in the included trials. The studies contributing to the NMA were considered at low or unclear risk of bias by the investigators, and there was information lacking about methods for allocation concealment and handling missing data for some studies included. According to the sponsor, studies from the relevant network that did not report the outcome of interest were excluded. This approach may potentially increase the risk of bias due to missing evidence in the synthesis, particularly for studies where the outcome would be expected to have been measured but was not reported. Of the available information, variability existed in of the several baseline characteristics reported, including age, baseline BCVA, proportion of treatment-experienced patients, retinal thickness and intraocular pressure measurements, treatment patterns, number of injections administered, and use of prior therapies and concomitant or background medications. As such, there is uncertainty as to whether the assumptions related to homogeneity underlying the NMA methodology were met. There was a lack of clarity on the number of studies included in the network that enrolled treatment-experienced or treatment-naive patients. Prior treatment for macula edema with anti-VEGFs potentially impacts treatment response negatively in patients with RVO. This adds uncertainty to the results and limits conclusions on the relative effect of faricimab versus the anti-VEGFs commonly used in practice.
The CrIs for the comparative effect estimates for mean change from baseline in BCVA, mean change from baseline in CRT, ocular AEs, serious ocular AEs, and all-cause discontinuation were wide, often precluding conclusions as to which treatment being compared could be favoured. Although there were some closed loops for some networks, overall, the networks were sparse with many comparisons connected by few trials. The geometry of the networks likely contributed to uncertainty in the comparative effect estimates and to the level of imprecision in many comparisons. The connection between faricimab and anti-VEGF treatments given in a PRN regimen for CST and BCVA was based on comparisons without an active comparator (sham comparison only) or with therapies of suboptimal efficacy (laser), which could increase bias because unobserved imbalances in the study populations may affect the active treatments arms differently compared with patient responses in the placebo (sham) study arms. However, the sham comparison for CST was informed by 2 trials and an additional loop via single-dose dexamethasone, and the sham comparison for BCVA was informed by 3 trials, which reduced the uncertainty for both outcomes.
Some studies were excluded from the analysis of CST (BLOSSOM and BRAVO studies comparing ranibizumab and sham in BRVO) due to smaller baseline mean retinal thickness. According to the authors, the COMRADE-B trial, which investigated dexamethasone versus sham in patients with BRVO, was excluded because it did not report baseline CST values. This increases the uncertainty of the results due to fewer studies included in the network. However, these 3 trials were included in the BRVO subgroup network.
There was considerable variability in how retinal thickness was defined, measured, and reported across the studies included in the NMA, which may contribute considerable heterogeneity to the ITC. RVO subtype was identified as a treatment modifier by the clinical expert consulted. A possible limitation is the pooling of patients with CRVO and BRVO in 1 analysis. Anti-VEGF treatments are very effective in drying the retina and there is a limit to how dry the retina can get. Therefore, in studies with lower baseline mean retinal thickness and a sham comparator, the active treatment arm may hit the maximum drying threshold, making the relative effect versus the sham comparator look smaller than in a study with patients who have a greater retinal thickness at baseline. Studies with a BRVO population tended to have a lower CRT at baseline compared with studies that included only patients with CRVO. While the sponsor did not consider this to be an effect modifier, comparisons between active treatments could lead to bias in comparisons versus sham treatment. While this, in general, is just a reflection of 2 types of RVO (BRVO versus CRVO), it can lead to bias if not all treatments are compared equally against sham in BRVO and CRVO. The sponsor further conducted a scenario analysis for the 2 populations for key clinical end points. Although the splitting of the RVO populations into BRVO and CRVO populations was done to supplement the base-case analysis in the overall population, these results are subject to increased uncertainty due to the smaller number of studies, leading to less robust networks of evidence.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Sixteen studies were submitted by the sponsor to address gaps in the RCTs that were submitted for faricimab for the treatment of RVO. These studies were excluded from the report because the patients enrolled across the studies included nAMD and DME populations, which differ from the sponsor-submitted reimbursement population. One matched cohort study, which matched patients with RVO from 2 registries (Vestrum and Medisoft) with the baseline characteristics of patients enrolled in the 2 pivotal trials submitted for this review (BALATON and COMINO studies), was also submitted by the sponsor. However, because the therapies evaluated, in association with the outcomes of interest, did not include faricimab, the study was excluded from the report.
Discussion
Summary of Available Evidence
BALATON and COMINO were phase III, multicentre, randomized, double-masked, active comparator–controlled, parallel-group, 2-part studies. Part 1 evaluated the efficacy, safety, and pharmacokinetics of intravitreal faricimab 6 mg every 4 weeks compared with intravitreal aflibercept 2 mg every 4 weeks in patients with macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial) from day 1 through week 24 (24 weeks of treatment). The primary end point in both studies was the change from baseline in BCVA at week 24. Part 2 evaluated the efficacy, durability, safety, and pharmacokinetics of faricimab administered at masked treatment intervals every 4, 8, 12, or 16 weeks based on personalized treatment interval dosing criteria, without an active control from week 24 through week 72 (48 weeks of treatment). Of note, there was no comparator in part 2 of the studies because all patients from part 1 received faricimab intravitreal injections according to a personalized treatment interval dosing regimen plus sham procedures to maintain masking of the treatment intervals.
In the BALATON trial, the mean age of patients was 64.3 years (SD = 10.7 years; range, 35 to 93 years) in the faricimab group and 63.8 years (SD = 10.6 years; range, 28 to 88 years) in the aflibercept group. The mean BCVA in the study eye was 57.50 letters (SD = 13.04 letters; range, 19.0 to 76.0 letters) in the faricimab group and 57.64 letters (SD = 12.15 letters; range, 21.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 558.32 µm (SD = 177.03 µm; range, 281.0 µm to 1,154.0 µm) in the faricimab group and 558.12 µm (SD = 180.26 µm; range, 290.0 µm to 1,208.0 µm) in the aflibercept group.
In the COMINO trial, the mean age of patients was 65.6 years (SD = 13.1 years; range, 22 to 100 years) in the faricimab group and 64.7 years (SD = 13.3 years; range, 27 to 95 years) in the aflibercept group. A total of 83.0% of patients (303 of 366 patients) randomized to receive faricimab and 81.9% of patients (294 of 363 patients) randomized to receive aflibercept were reported with CRVO. A total of 17.0% of patients (62 patients) randomized to receive faricimab and 18.1% of patients (65 patients) randomized to receive aflibercept were reported with HRVO. The mean BCVA in the study eye was 50.25 letters (SD = 16.25 letters; range, 19.0 to 87.0 letters) in the faricimab group and 50.71 letters (SD = 16.34 letters; range, 19.0 to 73.0 letters) in the aflibercept group. The mean CST in the study eye was 702.21 µm (SD = 244.00 µm; range, 266.0 µm to 1,500.0 µm) in the faricimab group and 721.07 µm (SD = 242.86 µm; range, 281.0 µm to 1,419.0 µm) in the aflibercept group.
Note that efficacy and safety data from the BALATON and COMINO studies were available up to week 68; however, these data were not summarized in this report because the results at week 24 (with the exception of the treatment interval) were considered to be the most relevant for the purpose of this review to inform expert committee deliberations.
The sponsor submitted 1 NMA that evaluated the comparative effectiveness and safety of faricimab compared with anti-VEGF treatments (specifically those administered in flexible regimens such as PRN) and other therapies commonly used in practice to treat patients with RVO. The NMA was based on a systematic review of the literature and data from 39 published articles (20 individual studies) were used to inform the Bayesian model with random effects as the base-case model, and fixed effects for sensitivity analyses.
Interpretation of Results
Efficacy
Patients have expressed the need for new treatments that prevent or slow down further vision loss and restore vision. The clinician groups and the clinical expert have identified improvement in and preservation of visual acuity as an important goal of therapy and a measure of treatment response. The BALATON and COMINO studies demonstrated that 24 weeks of treatment with intravitreal faricimab 6 mg every 4 weeks results in little to no difference in visual acuity (measured by BCVA) when compared with intravitreal aflibercept 2 mg every 4 weeks in patients with macular edema secondary to BRVO and CRVO or HRVO. In both studies, statistically, intravitreal faricimab 6 mg every 4 weeks was noninferior (based on a noninferiority margin of –4 letters), but not superior, to intravitreal aflibercept 2 mg every 4 weeks for change from baseline in BCVA at 24 weeks. The PP analysis results of the change from baseline in BCVA at week 24 from the BALATON and COMINO studies provide supportive evidence for the primary analysis results. Further, the studies suggested that 24 weeks of treatment with faricimab likely results in little to no difference in patients with a gain in BCVA of 15 letters or greater when compared with aflibercept. Uncertainty in the evidence (from the BALATON study only) for little to no difference in treatment effect was primarily due to concerns of imprecision of the 95% CI, per the review team’s assessment of the end point using GRADE. When interpreting the results, the following limitations discussed by Beck et al.28 related to creating a categorical outcome from a continuous measure, could be considered: loss of information (i.e., by giving the same weight to all scores on the same side of a threshold), misclassification of the outcome due to variability in measurements or due to bias, and increased susceptibility to a ceiling or floor effect (relative to the threshold).
The subgroup analysis results of the change from baseline in BCVA at week 24 from the COMINO study provide supportive evidence for the treatment effect of faricimab versus aflibercept in patients with macular edema secondary to CRVO. The small sample size of the subset of patients with HRVO limits the ability to interpret the results for this subgroup. Differences in the statistical significance for the estimated effects in the subgroups by RVO subtype are insufficient to inform effect modification.11 Because less than 20% of patients in the COMINO trial had HRVO, there is uncertainty as to whether the results could be generalized to this subgroup of patients. However, the clinical expert advised that the difference between subtypes of RVO is not a major factor for consideration in the discussion of a treatment plan for macular edema because RVO is treated with anti-VEGFs in practice (i.e., the clinical expert advised that patients with HRVO are expected to respond to treatment for macular edema similarly to patients with CRVO).
The clinician groups identified a reduction in macular edema as a goal of therapy and indicated that any improvement in swelling measured by OCT is considered clinically meaningful. The clinical expert advised that OCT (for measuring CST) is an objective, clinically important quantitative measure of macular edema and degree of treatment response. The clinical expert further advised that the absence of macular edema indicates a re-establishment of the physiological environment (blood–retina barrier), while the absence of both intraretinal fluid and subretinal fluid indicates the absence of degenerative cysts or fluid. Overall, the studies suggested that 24 weeks of treatment with faricimab likely results in little to no difference in anatomic outcomes (CST measurement), patients with an absence of macular edema, patients with an absence of both intraretinal fluid and subretinal fluid) when compared with aflibercept. Uncertainty in the evidence for little to no difference in treatment effect was primarily due to some concerns for risk of bias due to missing outcome data, as discussed in the critical appraisal section, and imprecision of the 95% CI, per the review team’s assessment of the end point using GRADE (for absence of both intraretinal fluid and subretinal fluid, low certainty for the BRVO population and moderate certainty for the CRVO and HRVO populations).
Patients have indicated that RVO significantly impacts their day-to-day lives and psychological well-being. More specifically, patients have indicated that RVO leads to visual complications that make daily activities, such as reading and driving, challenging or impossible. Patients have also expressed feeling anxious about their diagnosis of RVO, while others have expressed feelings of fear, isolation, anger, and loss of confidence. The studies suggested that 24 weeks of treatment with faricimab results in little to no difference in vision-related functioning and HRQoL (measured by NEI-VFQ-25) when compared with aflibercept.
Patients with RVO have expressed that they would like treatments that require fewer injections. Specifically, patients have indicated that although currently available anti-VEGF treatment options are efficacious in slowing vision loss, they are associated with burdens, including commuting to and from their appointments for regular intravitreal injections and associated side effects. Similarly, the clinical expert indicated that treatments that reduce treatment frequency and associated socioeconomic burden (i.e., treatment burden for clinicians and associated costs for patients and their families, such as transportation costs and lost income due to missed workdays) are needed. The clinician groups indicated that an ideal treatment demonstrates a durable effect (i.e., demonstrates efficacy in sustaining improvement in visual acuity over the long-term) measured by a reduction in the treatment burden associated with repetitive intravitreal injections; this was also identified as an unmet need by the clinician groups. The evidence informed by the BALATON and COMINO studies is very uncertain about the effect of faricimab administered at an extended treatment interval (i.e., every 8 to 16 weeks) at week 68 when compared with aflibercept or any other active comparator, primarily due to an absence of a relevant comparison group. The clinical expert acknowledged that a relatively high proportion of patients (approximately 50%) were on the extended treatment interval (faricimab every 16 weeks) in the BALATON study. However, the clinical expert noted that the criteria used to determine a personalized treatment interval dosing regimen are not used uniformly by clinicians in Canada and, as such, the results may not be generalizable to practice.
The clinician groups suggested that faricimab would be suitable for any patient with RVO, particularly patients with RVO that has failed to respond to other treatment options, including anti-VEGF therapies. Similarly, the clinical expert indicated that the anticipated target population for faricimab will include all patients with RVO, regardless of subtype, severity, symptoms, and so forth. Additionally, the clinical expert indicated that patients currently being treated with another anti-VEGF therapy may also be considered candidates for treatment with faricimab. Of note, the drug programs commented that switching between anti-VEGF treatments is a consideration for initiation of therapy. However, the BALATON and COMINO studies included patients with macular edema due to RVO that had been diagnosed no longer than 4 months before screening. The studies excluded patients with a history of previous episodes of macular edema due to RVO or persistent macular edema due to RVO diagnosed greater than 4 months before screening, as well as patients with any prior or current treatment for macular edema due to RVO, including anti-VEGF intravitreal injections, in the study eye. From the perspective of providing care to patients in practice, the clinical expert suggested there was no concern with generalizing the efficacy results to patients with chronic macular edema due to RVO and patients with experience with treatment for macular edema due to RVO. Moreover, the clinical expert highlighted that patients who received aflibercept in part 1 were switched to faricimab in part 2 of the studies; however, the studies were not designed to inform on treatment switching (i.e., there was no relevant comparator group for this part of the trials).
The NMA results suggest that faricimab may provide efficacy comparable to other anti-VEGFs administered PRN with respect to change from baseline in BCVA and CST. However, the 95% CrIs for the comparative effect estimates were often too wide to inform a strong conclusion of similarity because they included the potential that faricimab may be more effective or less effective compared with other anti-VEGFs for these outcomes. Several sources of heterogeneity were noted across trials, including age, baseline BCVA, proportion of treatment-experienced patients, retinal thickness and intraocular pressure measurements, treatment patterns, number of injections administered, and use of prior therapies and concomitant or background medications. There was also a lack of reporting on several key study characteristics of interest for RVO that could be potential effect modifiers. Due to this heterogeneity, there is uncertainty as to whether the assumptions related to homogeneity were met. Despite the heterogeneity observed in key prognostic factors in RVO such as baseline BCVA (higher BCVA was reported to be associated with better visual outcomes), age, sex, and retinal thickness measurements (although not prognostic, retinal thickness could influence visual outcomes because greater macular thickness variations are often associated with worse visual outcomes),58 the findings from the NMA suggest that faricimab may provide benefits comparable to anti-VEGFs administered to patients diagnosed with RVO in practice. The ITC did not assess other efficacy outcomes of interest to this review, including vision-related function and HRQoL. Further, the sponsor noted it was not feasible to investigate the mean number of injections in the NMA. Thus, data are lacking on safety, impacts on vision-related functioning and HRQoL, and number of injections compared with other anti-VEGFs.
Harms
Patients have expressed that they would like treatments that are associated with lower rates of side effects, highlighting injection pain. In both the BALATON and COMINO studies, the proportions of patients who were reported to have at least 1 ocular AE in the study eye, at least 1 serious ocular AE in the study eye or at least 1 adjudicated APTC-defined AE, or were reported to have stopped study treatment due to ocular AEs, and the proportions who died during the studies through week 24, were similar between the study drug groups. Notably, the frequencies of ocular AEs reported as pain and eye pain in the study eye at week 24 were similar between groups. The clinical expert suggested that, in general, the frequency of AEs reported appears within expectations for treatment with faricimab and is considered to be relatively low. The clinical expert noted that retinal hemorrhage, retinal ischemia, and cystoid macular edema are part of RVO; therefore, it would be difficult to assess whether these AEs are associated with treatment or the natural trajectory of RVO.
According to the product monograph for faricimab, transient increases in intraocular pressure (observed within 60 minutes of intravitreal injections) and sustained increases in intraocular pressure (present at 2 or more consecutive visits; > 21 mm Hg) associated with treatment have been reported.1 The clinical expert advised that an increase in intraocular pressure typically follows an intravitreal injection but is a temporary event (as opposed to a harmful effect). Instead, the clinical expert advised that a chronic increase in intraocular pressure as a result of medication (usually from a steroid), or repeated injections (usually due to silicone droplets from silicone-coated syringes) is an important AE to consider in treatment decision-making; this can be assessed within a few weeks to a few months following the initiation of treatment. At week 24, increased intraocular pressure was reported in few patients in each study drug group from each trial.
According to the product monograph for faricimab, endophthalmitis is associated with intravitreal injections.1 The clinical expert advised that endophthalmitis is an important AE to consider for all intravitreal injection treatments, including faricimab, and is important in treatment decision-making; endophthalmitis can be assessed within 3 to 4 days of injection. The studies suggested that 24 weeks of treatment with faricimab may result in little to no difference in endophthalmitis when compared with aflibercept. Uncertainty in the evidence was primarily due to concerns of imprecision because there were few to no events observed; therefore, there was an inadequate number of events to inform a higher-certainty judgment.
The evidence informed by the BALATON and COMINO studies is very uncertain about the effect of faricimab on AEs through week 72 when compared with aflibercept or any other active comparator, primarily due to an absence of a relevant comparison group. In the context of RVO and faricimab, no notable harms that would be assessed at week 72 were identified by the clinical expert.
Evidence from the sponsor-submitted NMA was insufficient to determine whether faricimab would be favoured over other anti-VEGFs for relevant harms. The 95% CrIs for the comparative effect estimates (odds ratios) for ocular AEs and SAEs, and treatment discontinuations were too wide to inform any conclusion as to whether faricimab or the comparator anti-VEGFs would be favoured for these outcomes.
Conclusion
Two phase III, randomized, multicentre, double-masked, active comparator–controlled, parallel-group, 2-part trials were submitted for this review. Part 1 evaluated the efficacy, safety, and pharmacokinetics of intravitreal faricimab 6 mg every 4 weeks compared with intravitreal aflibercept 2 mg every 4 weeks in patients with macular edema secondary to BRVO (in the BALATON trial) or CRVO or HRVO (in the COMINO trial) from day 1 through week 24 (24 weeks of treatment). Part 2 evaluated the efficacy, durability, safety, and pharmacokinetics of faricimab administered at masked treatment intervals every 4, 8, 12, or 16 weeks based on personalized treatment interval dosing criteria, without an active control from week 24 through week 72 (48 weeks of treatment). Based on input from patients and clinicians, visual acuity, anatomic outcomes, vision-related functioning and HRQoL, and treatment interval are important outcomes. The studies demonstrated that 24 weeks of treatment with intravitreal faricimab results in little to no difference in visual acuity when compared with intravitreal aflibercept, based on change from baseline in BCVA and proportion of patients gaining 15 letters or greater in BCVA. Faricimab was statistically noninferior, but not superior, to aflibercept for change from baseline in BCVA. The studies also suggested that 24 weeks of treatment with faricimab likely results in little to no difference in anatomic outcomes when compared with aflibercept, based on the change from baseline in CST, the proportion of patients with an absence of macular edema, and the proportion of patients with an absence of both intraretinal and subretinal fluid. Overall, uncertainty in these efficacy end point results showing little to no difference in treatment effect was primarily due to concerns of risk of bias due to missing outcome data and imprecision of the 95% CIs. The studies also suggested that 24 weeks of treatment with faricimab results in little to no difference in vision-related functioning and HRQoL when compared with aflibercept, based on change from baseline in the NEI-VFQ-25 composite score. The evidence informed by the trials is very uncertain about the effect of faricimab administered at an extended treatment interval (i.e., every 8, 12, or 16 weeks) at week 68 when compared with any active comparator, primarily due to the absence of a relevant comparison group. Further, there are concerns with the generalizability of the results because the criteria used to determine a personalized treatment interval dosing regimen are not used uniformly by clinicians in Canada.
Indirect evidence from the sponsor-submitted NMA provided evidence for faricimab relative to anti-VEGFs other than aflibercept. The evidence from the NMA suggested that faricimab may have little to no difference in treatment effect (in BCVA and CST) compared to other anti-VEGF therapies administered as needed in patients with macular edema secondary to RVO. However, there is uncertainty in the NMA results due to wide 95% CrIs, uncertain plausibility of the homogeneity assumption (i.e., variability in potential effect modifiers), a sparse network of evidence, and the potential for risk of bias in the included studies (e.g., uncertain handling of missing data).
With regard to safety, the frequency of AEs through week 24 was generally similar between the faricimab and aflibercept groups from each trial and was considered relatively low. The studies suggested that 24 weeks of treatment with faricimab may result in little to no difference in endophthalmitis when compared with aflibercept. Although endophthalmitis can be assessed within 3 to 4 days of injection, uncertainty in this safety end point remained primarily due to concerns of imprecision because there were few to no events observed to inform a higher-certainty judgment. Evidence from the NMA was insufficient to inform on the relative harms of faricimab compared with other anti-VEGFs.
References
1.Vabysmo (faricimab injection): Single-use vials 6 mg/0.05 mL solution for intravitreal injection [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2024.
2.Clinical Evidence for Vabysmo (faricimab) for the Treatment of Patients with Macular Edema (ME) Secondary to Retinal Vein Occlusion (RVO) [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Faricimab injection (Vabysmo) 6 mg/0.05 mL solution for intravitreal injection. Mississauga (ON): Hoffmann-La Roche Limited; 2024.
3.Royal College of Ophthalmologists. Clinical Guidelines: Retinal Vein Occlusion. 2022; https://www.rcophth.ac.uk/wp-content/uploads/2015/07/Retinal-Vein-Occlusion-Guidelines-2022.pdf. Accessed 2023 Feb 27.
4.Karia N. Retinal vein occlusion: pathophysiology and treatment options. Clin Ophthalmol. 2010;4:809-816. PubMed
5.Schmidt-Erfurth U, Garcia-Arumi J, Gerendas BS, et al. Guidelines for the Management of Retinal Vein Occlusion by the European Society of Retina Specialists (EURETINA). Ophthalmologica. 2019;242(3):123-162. PubMed
6.American Academy of Ophthalmology. Diagnosis and Management of Central Retinal Vein Occlusion. 2018; https://www.aao.org/eyenet/article/diagnosis-of-central-retinal-vein-occlusion. Accessed 2024 Feb 27.
7.Berger AR, Cruess AF, Altomare F, et al. Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus. Ophthalmologica. 2015;234(1):6-25. PubMed
8.Rogers S, McIntosh RL, Cheung N, et al. The prevalence of retinal vein occlusion: pooled data from population studies from the United States, Europe, Asia, and Australia. Ophthalmology. 2010;117(2):313-319.e311. PubMed
9.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Noninferiority clinical trials to establish effectiveness guidance for industry. 2016; https://www.fda.gov/media/78504/download. Accessed 2024 Oct 1.
10.Ke C, Ding B, Jiang Q, Snapinn SM. The issue of multiplicity in noninferiority studies. Clin Trials. 2012;9(6):730-735. PubMed
11.Cochrane Handbook for Systematic Reviews of Interventions. Chapter 10: Analysing data and undertaking meta-analyses. Cochrane: https://training.cochrane.org/handbook/current/chapter-10#section-10-11-3-1. Accessed 17 Oct 2024.
12.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
13.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
14.Clinical Study Report: GR41984. Primary CSR Study GR41984, (BALATON) A Phase III, Multicenter, Randomized, Double-Masked, Active Comparator-Controlled Study to Evaluate the Efficacy and Safety of Faricimab in Patients with Macular Edema Secondary to Branch Retinal Vein Occlusion. Report No. 1115182. February, 2023 [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2023.
15.Clinical Study Report: GR41984 (BALATON). A Phase III, Multicenter, Randomized, Double-Masked, Active Comparator-Controlled Study to Evaluate the Efficacy and Safety of Faricimab in Patients with Macular Edema Secondary to Branch Retinal Vein Occlusion. Report No. 1126074. January 2024. [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2024.
16.Clinical Study Report: GR41986. Primary CSR Study GR41986, (COMINO) A Phase III, Multicenter, Randomized, Double-Masked, Active Comparator-Controlled Study to Evaluate the Efficacy and Safety of Faricimab in Patients with Macular Edema Secondary to Central Retinal or Hemiretinal Vein Occlusion. Report No. 1115183. February, 2023 [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2023.
17.Clinical Study Report: GR41986 (COMINO). A Phase III, Multicenter, Randomized, Double-Masked, Active Comparator-Controlled Study to Evaluate the Efficacy and Safety of Faricimab in Patients with Macular Edema Secondary to Central Retinal or Hemiretinal Vein Occlusion. Report No. 1126075. January 2024. [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2024.
18.O’Mahoney PR, Wong DT, Ray JG. Retinal vein occlusion and traditional risk factors for atherosclerosis. Arch Ophthalmol. 2008;126(5):692-699. PubMed
19.Lattanzio R, Torres Gimeno A, Battaglia Parodi M, Bandello F. Retinal vein occlusion: current treatment. Ophthalmologica. 2011;225(3):135-143. PubMed
20.Song P, Xu Y, Zha M, Zhang Y, Rudan I. Global epidemiology of retinal vein occlusion: a systematic review and meta-analysis of prevalence, incidence, and risk factors. J Glob Health. 2019;9(1):010427. PubMed
21.Eylea (aflibercept injection): single use pre-filled syringes and single use vials for the treatment of a single eye. 2 mg / 0.05 mL solution for intravitreal injection [product monograph]. Mississauga (ON): Bayer Inc; 2022 Feb 10: https://pdf.hres.ca/dpd_pm/00064757.PDF. Accessed 2022 Apr 26.
22.Lucentis (ranibizumab injection): single use vials. Single use pre-filled syringes. 10 mg/mL solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Dec 21: https://pdf.hres.ca/dpd_pm/00064117.PDF. Accessed 2022 Apr 26.
23.Avastin (bevacizumab for injection): 100 mg and 400 mg vials (25 mg/mL solution for injection) [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Jan 14: https://pdf.hres.ca/dpd_pm/00059621.PDF. Accessed 2024 Sep 13.
24.Statistical Analysis Plan: GR41984 (BALATON) and GR41986 (COMINO). A phase III, multicenter, randomized, double-masked, active comparator-controlled studies to evaluate the efficacy and safety of faricimab in patients with macular edema secondary to branch retinal vein occlusion, and macular edema secondary to central retinal or hemiretinal vein occlusion (BALATON and COMINO) [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2020.
25.Protocol: GR41984 (BALATON). A phase III, multicenter, randomized, double-masked, active comparator-controlled study to evaluate the efficacy and safety of faricimab in patients with macular edema secondary to branch retinal vein occlusion [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2020.
26.Protocol: GR41986 (COMINO). A phase III, multicenter, randomized, double-masked, active comparator-controlled study to evaluate the efficacy and safety of faricimab in patients with macular edema secondary to central retinal or hemiretinal vein occlusion [internal sponsor’s report]. Basel (CH): F. Hoffmann-La Roche Ltd; 2020.
27.Kniestedt C, Stamper RL. Visual acuity and its measurement. Ophthalmol Clin North Am. 2003;16(2):155-170, v. PubMed
28.Beck RW, Maguire MG, Bressler NM, Glassman AR, Lindblad AS, Ferris FL. Visual acuity as an outcome measure in clinical trials of retinal diseases. Ophthalmology. 2007;114(10):1804-1809. PubMed
29.Bhende M, Shetty S, Parthasarathy MK, Ramya S. Optical coherence tomography: A guide to interpretation of common macular diseases. Indian J Ophthalmol. 2018;66(1):20-35. PubMed
30.Optical coherence tomography for age-related macular degeneration and diabetic macular edema: an evidence-based analysis. Ont Health Technol Assess Ser. 2009;9(13):1-22. PubMed
31.Mangione CM, Berry S, Spritzer K, et al. Identifying the content area for the 51-item National Eye Institute Visual Function Questionnaire: results from focus groups with visually impaired persons. Arch Ophthalmol. 1998;116(2):227-233. PubMed
32.Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol. 2001;119(7):1050-1058. PubMed
33.Campochiaro PA, Clark WL, Boyer DS, et al. Intravitreal aflibercept for macular edema following branch retinal vein occlusion: the 24-week results of the VIBRANT study. Ophthalmology. 2015;122(3):538-544. PubMed
34.Boyer D, Heier J, Brown DM, et al. Vascular endothelial growth factor Trap-Eye for macular edema secondary to central retinal vein occlusion: six-month results of the phase 3 COPERNICUS study. Ophthalmology. 2012;119(5):1024-1032. PubMed
35.Holz FG, Roider J, Ogura Y, et al. VEGF Trap-Eye for macular oedema secondary to central retinal vein occlusion: 6-month results of the phase III GALILEO study. Br J Ophthalmol. 2013;97(3):278-284. PubMed
36.Cochran WG. Some Methods for Strengthening the Common χ 2 Tests. Biometrics. 1954;10(4).
37.Mantel N, Haenszel W. Statistical Aspects of the Analysis of Data From Retrospective Studies of Disease. JNCI: Journal of the National Cancer Institute. 1959;22(4). PubMed
38.Mehrotra DV, Railkar R. Minimum risk weights for comparing treatments in stratified binomial trials. Stat Med. 2000;19(6):811-825. PubMed
39.Roche response to 2024 October 16 CDA-AMC request for additional information regarding Vabysmo CDA-AMC review [internal additional sponsor’s information]. Hoffmann-La Roche Limited; 2024 October 21.
40.Network meta-analysis of treatments in retinal vein occlusion (RVO) Roche compound of interest: faricimab/Vabysmo [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Faricimab injection (Vabysmo) 6 mg/0.05 mL solution for intravitreal injection.: Hoffmann-La Roche Limited; 2024 Feb. 2024. Accessed August 30, 2024.
41.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE Decision Support Unit Technical Support Documents. NICE DSU Technical Support Document 4: Inconsistency in Networks of Evidence Based on Randomised Controlled Trials. London: National Institute for Health and Care Excellence (NICE); 2014 [sponsor submitted reference].
42.Brown DM, Campochiaro PA, Singh RP, et al. Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology. 2010;117(6):1124-1133.e1121. PubMed
43.Campochiaro PA, Heier JS, Feiner L, et al. Ranibizumab for macular edema following branch retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology. 2010;117(6):1102-1112.e1101. PubMed
44.Hattenbach LO, Feltgen N, Bertelmann T, et al. Head-to-head comparison of ranibizumab PRN versus single-dose dexamethasone for branch retinal vein occlusion (COMRADE-B). Acta Ophthalmol. 2018;96(1):e10-e18. PubMed
45.Hoerauf H, Feltgen N, Weiss C, et al. Clinical Efficacy and Safety of Ranibizumab Versus Dexamethasone for Central Retinal Vein Occlusion (COMRADE C): A European Label Study. Am J Ophthalmol. 2016;169:258-267. PubMed
46.Hykin P, Prevost AT, Vasconcelos JC, et al. Clinical Effectiveness of Intravitreal Therapy With Ranibizumab vs Aflibercept vs Bevacizumab for Macular Edema Secondary to Central Retinal Vein Occlusion: A Randomized Clinical Trial. JAMA Ophthalmol. 2019;137(11):1256-1264. PubMed
47.Kinge B, Stordahl PB, Forsaa V, et al. Efficacy of ranibizumab in patients with macular edema secondary to central retinal vein occlusion: results from the sham-controlled ROCC study. Am J Ophthalmol. 2010;150(3):310-314. PubMed
48.Narayanan R, Panchal B, Das T, Chhablani J, Jalali S, Ali MH. A randomised, double-masked, controlled study of the efficacy and safety of intravitreal bevacizumab versus ranibizumab in the treatment of macular oedema due to branch retinal vein occlusion: MARVEL Report No. 1. Br J Ophthalmol. 2015;99(7):954-959. PubMed
49.Rajagopal R, Shah GK, Blinder KJ, et al. Bevacizumab Versus Ranibizumab in the Treatment of Macular Edema Due to Retinal Vein Occlusion: 6-Month Results of the CRAVE Study. Ophthalmic Surg Lasers Imaging Retina. 2015;46(8):844-850. PubMed
50.Tadayoni R, Waldstein SM, Boscia F, et al. Individualized Stabilization Criteria-Driven Ranibizumab versus Laser in Branch Retinal Vein Occlusion: Six-Month Results of BRIGHTER. Ophthalmology. 2016;123(6):1332-1344. PubMed
51.Vader MJC, Schauwvlieghe AME, Verbraak FD, et al. Comparing the Efficacy of Bevacizumab and Ranibizumab in Patients with Retinal Vein Occlusion: The Bevacizumab to Ranibizumab in Retinal Vein Occlusions (BRVO) study, a Randomized Trial. Ophthalmol Retina. 2020;4(6):576-587. PubMed
52.Wei W, Weisberger A, Zhu L, Cheng Y, Liu C. Efficacy and Safety of Ranibizumab in Asian Patients with Branch Retinal Vein Occlusion: Results from the Randomized BLOSSOM Study. Ophthalmol Retina. 2020;4(1):57-66. PubMed
53.Gado AS, Macky TA. Dexamethasone intravitreous implant versus bevacizumab for central retinal vein occlusion-related macular oedema: a prospective randomized comparison. Clin Exp Ophthalmol. 2014;42(7):650-655. PubMed
54.Scott IU, VanVeldhuisen PC, Ip MS, et al. Effect of Bevacizumab vs Aflibercept on Visual Acuity Among Patients With Macular Edema Due to Central Retinal Vein Occlusion: The SCORE2 Randomized Clinical Trial. JAMA. 2017;317(20):2072-2087. PubMed
55.Study GR41984 (BALATON) Primary CSR. Roche data on file [sponsor submitted reference].
56.Study GR41986 (COMINO) Primary CSR. Roche data on file. [sponsor submitted reference].
57.Brown DM, Heier JS, Clark WL, et al. Intravitreal aflibercept injection for macular edema secondary to central retinal vein occlusion: 1-year results from the phase 3 COPERNICUS study. Am J Ophthalmol. 2013;155(3):429-437.e427. PubMed
58.Yin S, Cui Y, Jiao W, Zhao B. Potential Prognostic Indicators for Patients With Retinal Vein Occlusion. Front Med. 2022;9. PubMed
Appendix 1: Detailed Outcome Data for Indirect Evidence
Please note that this appendix has not been copy-edited.
Table 31: Sensitivity Analyses for Faricimab 6 mg q.4.w. vs. Anti-VEGFs Administered PRN for BCVA Mean Change From Baseline at 6 Months
Characteristic | Faricimab vs. comparator |
|---|---|
Sensitivity 1, FE model | |
BCVA change from baseline at 6 months, mean difference (95% CrI) | — |
Bevacizumab 1.25 mg, PRN | 4.84 (0.43 to 9.36) |
Ranibizumab 0.5 mg, PRN | 2.77 (−0.45 to 5.99) |
Aflibercept 2 mg, PRN | 1.64 (−3.23 to 6.45) |
Sensitivity 2, RE model, alternative prior for tau | |
Change from baseline to 6 months, mean difference (95% CrI) | — |
Bevacizumab 1.25 mg, PRN | 5.14 (−4.70 to 15.11) |
Ranibizumab 0.5 mg, PRN | 3.52 (−4.40 to 11.14) |
Aflibercept 2 mg, PRN | 1.93 (−9.15 to 12.91) |
BCVA = best-corrected visual acuity; FE = fixed effects; NR = not reported; PRN = pro re nata (as needed); q.4.w. = every 4 weeks; RE = random effects; vs. = versus.
Sources: Sponsor-submitted indirect treatment comparison.40 Details included in the table are from the sponsor’s summary of clinical evidence.2
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BCVA
best-corrected visual acuity
BIA
budget impact analysis
BRVO
branch retinal vein occlusion
CDA-AMC
Canada’s Drug Agency
CRVO
central retinal vein occlusion
DME
diabetic macular edema
ICER
incremental cost-effectiveness ratio
nAMD
neovascular age-related macular degeneration
NMA
network meta-analysis
RVO
retinal vein occlusion
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Faricimab (Vabysmo), solution for intravitreal injection |
Submitted price | Faricimab, 6 mg per 0.05 mL, single-use vial: $1,350.00 |
Indication | For the treatment of macular edema secondary to retinal vein occlusion |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 24, 2024 |
Reimbursement request | As per indication |
Sponsor | Hoffmann-La Roche Limited |
Submission history | Previously reviewed: Yes Indication: Diabetic macular edema Recommendation date: October 14, 2022 Recommendation: Recommended with clinical criteria and/or conditions Indication: Neovascular (wet) age-related macular degeneration Recommendation date: August 12, 2022 Recommendation: Recommended with clinical criteria and/or conditions |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients with retinal vein occlusion |
Treatment | Faricimab (Vabysmo) administered by intravitreal injection every 4 weeks for 6 monthsa |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (25 years) |
Key data source |
|
Submitted results | The ICER for faricimab vs. bevacizumab was $242,712 per QALY gained (incremental costs: $67,293; incremental QALYs: 0.28). Faricimab was more effective and less costly (dominant) compared with ranibizumab and aflibercept 2 mg. |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year.
aInjection schedule beyond 6 months not specified in the product monograph.
Conclusions
Based on the clinical review by Canada’s Drug Agency (CDA-AMC), data from the BALATON and COMINO trials suggest that faricimab is noninferior, but not superior, to aflibercept 2 mg in terms of best-corrected visual acuity (BCVA) among patients with macular edema secondary to retinal vein occlusion (RVO). Indirect evidence submitted by the sponsor suggests there may be little to no difference in BCVA with faricimab compared with other anti-VEGF treatments administered using flexible injection schedules. Limitations with the submitted indirect evidence include variability in the baseline patient characteristics across trials, a lack of reporting of important patient characteristics, and imprecision in the effect estimates.
Injection frequencies for faricimab and comparators were incorporated directly from clinical trials involving faricimab, aflibercept 2 mg, or ranibizumab, without adjustment or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine whether any observed differences are due to the treatment rather than to confounding factors.
Given that the indirect evidence submitted by the sponsor suggests there may be little to no difference in BCVA between faricimab and other anti-VEGFs, there is insufficient evidence to suggest that faricimab should be priced higher than the other currently available anti-VEGFs used in the treatment of macular edema secondary to RVO.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process (specifically, information that pertains to the economic submission).
CDA-AMC received patient input via a joint submission from Fighting Blindness Canada, the Canadian Council of the Blind, and Vision Loss Rehabilitation Canada, with input derived from a survey completed by people in Canada with RVO and from interviews conducted in March and April 2024. Of the 32 respondents, most (63%) had central retinal vein occlusion (CRVO) and 22% had branch retinal vein occlusion (BRVO), while 16% indicated they did not know the type. Most respondents indicated they were worried about vision worsening, noting that RVO significantly affects their daily lives and psychological well-being. Most respondents reported receiving anti-VEGF injections, which they found effective in stabilizing their vision but stressful due to anxiety about the injections, side effects, and the burden of frequent travel to appointments. Patients expressed a need for treatments that not only prevent or slow further vision loss but also lower side effects, require fewer injections, and are accessible across provinces. The joint patient advocacy group emphasized the challenges posed by regular intravitreal injections, highlighting the importance of developing new therapies that are both effective and more convenient.
Clinical input was received from 4 clinician groups: Toronto Retina Institute, Southeastern Ontario Community Ophthalmologists, Southwestern Ontario Community Ophthalmologists, and Northeastern Ontario Community Ophthalmologists. Their input noted that the treatment goals for RVO include extending the intervals between injections, reducing macular edema, preserving and improving visual acuity, lowering VEGF levels, and preventing complications such as neovascularization and glaucoma. Ideal treatments would sustain visual improvements long-term, reduce the burden of repetitive intravitreal injections, and be more durable for patients whose disease has been unresponsive to current options. The clinician input noted that faricimab may be used as a first-line treatment for patients with newly diagnosed RVO and as later-line treatment for patients with RVO that has had an inadequate response to other treatments. Treatment response is generally assessed by changes in visual acuity, decrease in retinal thickness or fluid accumulation, and injection frequency.
The participating drug plans noted that funding of anti-VEGFs and biosimilar-switching policies vary across jurisdictions. The drug plans inquired about the proportion of patients who would be expected to receive injections at extended intervals. The plans questioned whether patients would switch to faricimab from other anti-VEGFs, and whether faricimab would result in longer injection intervals compared with currently available treatments for RVO.
The potential for extended dosing intervals with faricimab was noted as being of particular importance in the patient, clinician, and drug plan input. Owing to uncertainty in the comparative clinical efficacy for faricimab and the lack of comparative data for injection frequency, CDA-AMC was unable to address these concerns.
Economic Review
The current review is for faricimab (Vabysmo) for patients with macular edema secondary to RVO.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of faricimab compared with other anti-VEGFs (aflibercept 2 mg, bevacizumab, ranibizumab) in patients with macular edema secondary to RVO.1 The modelled population is aligned with the Health Canada indication and the patients enrolled in the BALATON and COMINO trials.2,3 As noted in the CDA-AMC Clinical Review, the BALATON trial enrolled patients with BRVO, while the COMINO trial enrolled patients with CRVO or hemiretinal RVO.
Faricimab is supplied in single-use vials containing 28.8 mg of faricimab in a 0.24 mL solution (6 mg per 0.05 mL). The recommended dose of faricimab is 6 mg administrated by intravitreal injection every 4 weeks (28 days plus or minus 7 days) for 6 months, with monitoring between dosing visits, which are scheduled based on the patient’s status and at the physician’s discretion.4 Note that the product monograph does not indicate a recommended dosing schedule beyond the first 6 months. Using the submitted price for faricimab ($1,350.00 per vial), the sponsor estimated the per-patient cost to be $13,190 in the first year (█████████████ ██ ██████████ ██ ████ |), and $10,179 in subsequent years (███ ██████████ ███ ████). The annual per-patient costs for comparators estimated by the sponsor ranged from $199 (bevacizumab, based on █████ ██████████) to $14,733 (aflibercept 2 mg, █████ ██████████) in the first year, while the annual per-patient costs in subsequent years ranged from $191 (bevacizumab, █████ ██████████) to $12,450 (aflibercept 2 mg | ████ ██████████).
The clinical outcomes were life-years and quality-adjusted life-years (QALYs), estimated over a lifetime time horizon (25 years; 4-week cycle length) from the perspective of Canada’s publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum, and no half-cycle correction was applied.
Model Structure
The sponsor submitted a Markov model that consisted of 36 health states that were defined based on BCVA. The model considered both eyes and assumed that disease progression in each eye was independent of the other eye. Patients entered the model at the start of treatment in the first eye with RVO. In each 4-week cycle, a patient’s first-eye BCVA could change (based on the results of the sponsor’s network meta-analysis [NMA]), and patients could move either up or down a maximum of 2 BCVA-based health states per cycle or remain in the same health state. Death was an absorbing health state in the sponsor’s model, and patients remained in that health state until the end of the model’s time horizon.
Model Inputs
The baseline characteristics and initial distribution of patients across the BCVA-based health states were based on those of patients enrolled in the BALATON and COMINO trials.2,3 In both studies, patients were randomized to receive either faricimab every 4 weeks or aflibercept 2 mg every 4 weeks for 24 weeks, after which time all patients received faricimab at a personalized treatment interval.
Clinical inputs to inform the economic model (changes in BCVA, severe adverse events [AEs], and discontinuations) were obtained from a sponsor-submitted NMA. The probability of death was based on age-specific Canadian background morality rates,5 to which the sponsor applied a mortality multiplier of 1.54 to account for higher mortality among patients who are blind in both eyes and a multiplier of 1.23 to account for impaired vision in at least 1 eye.6
In the model, the sponsor assumed that all patients would receive injections every 4 weeks for the 6 months of treatment (induction phase) for faricimab and comparators. From 6 months onward (maintenance phase), the sponsor adopted a treat-and-extend approach for faricimab, informed by observations from the BALATON and COMINO trials. The injection frequencies for comparators were incorporated directly from clinical trials. Based on these inputs, the sponsor calculated that ████ faricimab injections would be required annually (after the first 6 months), with ████ injections for aflibercept 2 mg, █████ injections for bevacizumab, and █████ injections for ranibizumab.
Utility values for each BCVA state were estimated by using regression coefficients from a published study that simulated visual impairment in healthy volunteers from the UK.7 Disutilities associated with AEs and the duration of AEs were obtained from the literature.5,7 The sponsor assumed that half of patients would experience zero utility on an injection day.7
The economic model included costs related to drugs (acquisition, administration), monitoring, AEs, and vision loss. The sponsor estimated treatment costs using the drug and administration costs per injection and the estimated number of injections per year. Public list prices were used for faricimab and comparators.8,9 The acquisition cost of bevacizumab and aflibercept 2 mg was based on the branded forms, while the price of ranibizumab was based on its biosimilar. The sponsor assumed that all vials were single-use and that any unused product would be wasted, except for bevacizumab, for which vial sharing was assumed. The administration costs per injection included costs associated with intravitreal injection, optical coherence tomography (OCT), and ophthalmology consultation, with the price of each obtained from the Ontario Schedule of Benefits.10 Costs of treating AEs were obtained from either the Ontario Schedule of Benefits or the Canadian Institute for Health Information Patient Cost Estimator.10,11 Costs of vision loss were obtained from the Cost of Vision Loss and Blindness in Canada report.12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s base case, faricimab was associated with an estimated cost of $94,877 and 11.30 QALYs over a 25-year horizon. In sequential analysis, faricimab was associated with an incremental cost-effectiveness ratio (ICER) of $242,712 per QALY gained compared with bevacizumab (incremental costs: $67,293; incremental QALYs: 0.28), with a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Results were driven by drug acquisition costs.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Bevacizumab | 27,584 | 11.03 | Reference |
Faricimab | 94,877 | 11.30 | 242,712 vs. bevacizumab |
Dominated treatments | |||
Ranibizumab | 97,588 | 11.12 | Dominated by faricimab |
Aflibercept 2 mg | 112,256 | 11.21 | Dominated by faricimab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including adopting alternative discount rates and alternative time horizons, and considering either BRVO or CRVO subgroups, alternative assumptions about ranibizumab biosimilar use, and alternative assumptions about injection frequency. The most impactful changes were adopting a 15-year analysis horizon (ICER = $340,327 per QALY gained versus bevacizumab) and considering BRVO (ICER = $430,435 versus bevacizumab) and CRVO ($317,036 per QALY versus bevacizumab) separately.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy and safety of faricimab versus other anti-VEGFs are uncertain. There is a lack of head-to-head evidence comparing faricimab with anti-VEGF drugs other than aflibercept 2 mg. The results of the BALATON and COMINO trials2,3 suggest that faricimab is noninferior, but not superior, compared with aflibercept 2 mg for improvement in visual acuity (i.e., change from baseline in BCVA). In the absence of head-to-head evidence for most comparators, the sponsor conducted NMAs to provide comparative evidence for BCVA and severe ocular AEs in the economic model. As noted in the CDA-AMC Clinical Review, the indirect evidence submitted by the sponsor suggests there may be little to no difference in BCVA with faricimab compared with other anti-VEGFs administered using a flexible injection schedule. Limitations with the submitted indirect evidence include variability in baseline patient characteristics across trials, a lack of reporting of important patient characteristics, and imprecision. Evidence from the NMA was insufficient to inform on the relative harms of faricimab compared with other anti-VEGFs.
Given the lack of direct evidence for faricimab relative to anti-VEGF drugs other than aflibercept 2 mg and limitations with the sponsor’s NMA, whether faricimab improves visual acuity compared with other anti-VEGFs is uncertain.
The relative frequency of anti-VEGF injections is uncertain. In the pharmacoeconomic model, the number of annual injections (and hence drug acquisition and administration costs) for faricimab and comparators were incorporated directly from clinical trials involving faricimab, aflibercept 2 mg, or ranibizumab, without adjustment or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine whether any observed differences in injection frequency between the therapies are solely due to the treatment or, rather, due to bias or confounding factors (e.g., differences in study populations, definitions of outcomes, or study designs). Further, the clinical expert input received for this review indicated that, in clinical practice, all anti-VEGF therapies are administered using a treat-and-extend approach that is personalized for each patient.
CDA-AMC could not address this limitation owing to the lack of comparative data. As such, it is highly uncertain whether faricimab will result in fewer injections compared with other anti-VEGFs used in clinical practice.
Drug acquisition costs may be overestimated. In its economic model, the sponsor used the price of branded bevacizumab (Avastin). A biosimilar bevacizumab product (Mvasi) is available in Canada at a reduced price compared with the originator brand. In some jurisdictions, Avastin is reimbursed by the public drug plans only up to the list price for Mvasi; this is the case, for example, in the Yukon, the jurisdiction from which the sponsor obtained bevacizumab pricing.
Faricimab has successfully undergone price negotiations for the treatment of neovascular age-related macular degeneration (nAMD) and diabetic macular edema (DME), and it is likely that the unit cost paid by public drug plans is lower than the price incorporated in the sponsor’s model. Similarly, the prices of other anti-VEGFs in the sponsor’s model do not reflect any existing confidential prices negotiated by public plans.
CDA-AMC explored the impact of biosimilar bevacizumab pricing in sensitivity analyses. If all other inputs in the sponsor’s base case are kept constant, the ICER for faricimab versus biosimilar bevacizumab is $286,757 per QALY gained. CDA-AMC could not address the impact of confidential negotiated prices.
Issues for Consideration
Biosimilars for aflibercept are currently under review by Health Canada. The introduction of such biosimilars may affect the cost-effectiveness of faricimab versus aflibercept 2 mg, depending on the list price.
Faricimab has successfully undergone price negotiations for the treatment of nAMD and DME, and it is likely that the unit cost paid by public drug plans is lower than the price submitted in the current review. Similarly, the prices of other anti-VEGFs in the sponsor’s model do not reflect any existing confidential prices negotiated by public plans.
Aflibercept 8 mg is currently being actively negotiated with the pan-Canadian Pharmaceutical Alliance for the treatment of DME. The clinical expert input received by CDA-AMC for this review indicated that aflibercept 8 mg may be used off-label in clinical practice to treat RVO. The cost-effectiveness of faricimab relative to aflibercept 8 mg is unknown.
Overall Conclusions
Based on the CDA-AMC clinical review, data from the BALATON and COMINO trials suggest that, over a 24-week period, faricimab administered every 4 weeks is noninferior, but not superior, to aflibercept 2 mg every 4 weeks, in terms of visual acuity (measured by BCVA) in patients with macular edema secondary to BRVO (in the BALATON trial) and CRVO or hemiretinal RVO (in the COMINO trial). Observations from these trials additionally suggest that compared with aflibercept 2 mg, faricimab likely results in little to no difference in anatomic outcomes and health-related quality of life. Indirect evidence submitted by the sponsor suggests there may be little to no difference in BCVA with faricimab compared with other anti-VEGFs administered using a flexible injection schedule. However, there is uncertainty in these findings owing to identified limitations with the sponsor-submitted indirect evidence (e.g., variability in baseline patient characteristics across trials, imprecision).
Evidence from the BALATON and COMINO trials is insufficient to ascertain the relative injection frequency for faricimab versus other anti-VEGFs due to the absence of a comparison group. In the economic model, the sponsor incorporated the injection frequency for each anti-VEGF directly from clinical trials, without adjustment or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine whether any observed differences are due to the treatment. In clinical practice, the injection frequency is guided by treatment response and is not determined by the treatment received.
Given that the indirect evidence submitted by the sponsor suggests there may be little to no difference in BCVA between faricimab and other anti-VEGFs, and the lack of comparative evidence to support longer injection intervals with faricimab, there is insufficient evidence to suggest that faricimab should be priced higher than other currently available anti-VEGFs used in the treatment of RVO.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission:Vabysmo (faricimab): 6 mg/0.05 mL solution for intravitreal injection. Mississauga (ON): Hoffmann-La Roche Limited; 2024 Jul 31.
2.Study GR41984 BALATON Primary CSR, Report 1115182. Roche data on file. [sponsor submitted reference].
3.Study GR41986 COMINO Primary CSR, Report 1115183. Roche data on file. [sponsor submitted reference].
4.Hoffmann-La Roche Limited. Vabysmo (faricimab) product monograph. 2022 [sponsor submitted reference].
5.Brown GC, Sharma S, Brown MM, Kistler J. Utility values and age-related macular degeneration. Arch Ophthalmol. 2000;118(1):47-51. PubMed
6.Christ SL, Lee DJ, Lam BL, Zheng DD, Arheart KL. Assessment of the effect of visual impairment on mortality through multiple health pathways: structural equation modeling. Invest Ophthalmol Vis Sci. 2008;49(8):3318-3323. PubMed
7.Czoski-Murray C, Carlton J, Brazier J, Young T, Papo NL, Kang HK. Valuing condition-specific health states using simulation contact lenses. Value Health. 2009;12(5):793-799. PubMed
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 Oct 1.
9.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2024 Oct 1.
10.Ontario Ministry of Health and Long-Term Care. Schedule of Benefits and Fees for Physician Services. 2023 [sponsor submitted reference].
11.Patient cost estimator. 2024; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2024 Oct 1.
12.Deloitte. The cost of vision loss and blindness in Canada. 2021; https://www.fightingblindness.ca/wp-content/uploads/2021/12/Deloitte-Cost-of-vision-loss-and-blindness-in-Canada-report-May-2021.pdf. [sponsor submitted reference].
13.CADTH Reimbursement Review Faricimab (Vabysmo). Diabetic macular edema. 2023; https://www.cadth.ca/sites/default/files/DRR/2022/SR0729-Vabysmo_combined.pdf. [sponsor submitted reference].
14.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug Reimbursement Review sponsor submission:Vabysmo (faricimab): 6 mg/0.05 mL solution for intravitreal injection. Mississauga (ON): Hoffmann-La Roche Limited; 2024 Jul 31.
15.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex. 2024; https://doi.org/10.25318/1710000501-eng. . Accessed 2024 Jan 15.
16.Berger AR, Cruess AF, Altomare F, et al. Optimal Treatment of Retinal Vein Occlusion: Canadian Expert Consensus. Ophthalmologica. 2015;234(1):6-25. PubMed
17.Roche. Data on File. 2024 [sponsor submitted reference].
18.Wong TY, Scott IU. Clinical practice. Retinal-vein occlusion. N Engl J Med. 2010;363(22):2135-2144. PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CDA-AMC Cost Comparison Table for Retinal Vein Occlusion
Treatment | Strength or concentration | Form | Price | Recommended dosageb | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Faricimab (Vabysmo) | 120 mg/mL | 0.05 mL Solution for intravitreal injection | 1,350.0000a | 6 mg every 4 weeks for first 6 doses followed by 6 mg at a dosing interval of 8 to 16 weeks | Year 1: 25.87 to 36.96 Subsequent: 14.78 to 25.87 | Year 1: 9,450 to 13,500 (7 to 10 inj.) Subsequent: 5,400 to 9,450 (4 to 7 inj.) |
Anti-VEGF inhibitors | ||||||
Aflibercept 2 mg (Eylea) | 40 mg/mL | 0.05 mL Solution for intravitreal injection | 1,418.0000a | 2 mg every 4 weeks for first 6 doses followed by 2 mg every 8 to 16 weeks | Year 1: 27.18 to 38.82 Subsequent: 15.53 to 27.18 | Year 1: 9,926 14,180 (7 to 10 inj.) Subsequent: 5,672 to 9,926 (4 to 7 inj.) |
Bevacizumab (Avastin) | 25 mg/mL | 4 mL 16 mL Solution for intravitreal injection | 519.1800c 2,076.7104c | 1.25 mg every 4 weeks for first 6 doses followed by 1.25 mg every 6 to 8 weeksd | Year 1: 0.38 to 0.47 Subsequent: 0.33 to 0.43 | Year 1: 138 to 173 (8 to 10 inj.) Subsequent: 121 to 156 (7 to 9 inj.) |
Bevacizumab (Mvasi) | 25 mg/mL | 4 mL 16 mL Solution for intravitreal injection | 347.0000c 1,388.0000c | 1.25 mg every 4 weeks for first 6 doses followed by 1.25 mg every 6 to 8 weeksd | Year 1: 0.25 to 0.32 Subsequent: 0.22 to 0.29 | Year 1: 93 to 116 (8 to 10 inj.) Subsequent: 81 to 104 (7 to 9 inj.) |
Ranibizumab (Lucentis) | 10 mg/mL | 0.23 mL Solution for intravitreal injection | 1,710.3000a | 0.5 mg every month for first 6 doses followed by 0.5 mg every 1 or 3 months | Year 1: 28.10 to 56.19 Subsequent: 23.41 to 56.19 | Year 1: 10,262 to 20,524 (6 to 12 inj.) Subsequent: 8,552 to 20,524 (5 to 12 inj.) |
Ranibizumab (biosimilar) | 10 mg/mL | 0.23 mL Solution for intravitreal injection | 990.0000a | 0.5 mg every month for first 6 doses followed by 0.5 mg every 1 or 3 months | Year 1: 16.26 to 32.53 Subsequent: 13.55 to 32.53 | Year 1: 5,490 to 11,880 (6 to 12 inj.) Subsequent: 4,950 to 11,880 (5 to 12 inj.) |
CDA-AMC = Canada’s Drug Agency; RVO = retinal vein occlusion.
Note: All prices do not include dispensing fees. Annual costs are based on 52 weeks per year.
aPrice obtained from the Ontario Drug Benefit Formulary (accessed October 2024).8
bRecommended doses are from the respective product monographs, unless otherwise indicated.
cPrice obtained from the IQVIA Delta PA database (accessed October 2024).9
dBevacizumab is used off-label for the treatment of RVO. Dosage and number of administrations per vial (30 per 4 mL vial) were obtained from a previous CADTH review13 and validated by clinical expert input.
Appendix 2: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 5: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; RVO = retinal vein occlusion.
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) to estimate the incremental budget impact of reimbursing faricimab for the treatment of macular edema secondary to RVO.14 The BIA was undertaken from the perspective of the pan-Canadian drug plans over a 3-year time horizon (2025 to 2027). The sponsor estimated the size of the eligible population using an epidemiologic approach, with data obtained from the literature. The sponsor estimated the market shares of comparators and uptake of faricimab using internal market estimates and clinical expert opinion. The sponsor’s base case included drug acquisition costs, with pharmacy fees and administration costs included in a scenario analysis. Faricimab and comparators were assumed to be administered every 4 weeks for 6 months (6 injections). After the first 6 months of treatment, the annual injection frequency was assumed by the sponsor to be ████ injections of faricimab, ████ injections of aflibercept, █████ injections of ranibizumab, and █████ injections of bevacizumab. Key inputs to the BIA are documented in Table 6.
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Adult population in Canada Percentage of adults with RVO Percentage of patients diagnosed Percentage of patients receiving anti-VEGF treatment Percentage of patients eligible for public coverage | 16,403,05115 90%19 90%17 95%19 |
Number of patients eligible for the drug under review | 66,787 / 67,962 / 69,124 |
Market uptake (3 years) | |
Uptake (reference scenario) Aflibercept 2 mg Bevacizumab Ranibizumab (Lucentis) Ranibizumab (biosimilars) | 52% / 52% / 52% 31% / 31% / 31% 15% / 14% / 13% 2% / 3% / 4% |
Uptake (new-drug scenario) Faricimab Aflibercept 2 mg Bevacizumab Ranibizumab (Lucentis) Ranibizumab (biosimilars) | 1% / 7% / 12% 50% / 45% / 40% 31% / 31% / 31% 15% / 14% / 13% 2% / 3% / 4% |
Annual cost of treatment per patient per eyea (induction year / subsequent years) | |
Faricimab Aflibercept 2 mg Bevacizumab Ranibizumab (Lucentis) Ranibizumab (biosimilars) | $13,190 / $10,179 $14,733 / $12,450 $199 / $191 $18,526 / $17,653 $10,314 / $10,865 |
RVO = retinal vein occlusion.
aTreatment cost differs for patients with bilateral disease.
Summary of the Sponsor’s BIA Results
The sponsor estimated the incremental 3-year budget impact of reimbursing faricimab for the treatment of adult patients with macular edema secondary to RVO to result in cost savings of $2,252,064 in year 1, $10,660,462 in year 2, $19,250,763 in year 3. The 3-year estimated budgetary savings was $32,163,288.
Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The frequency of anti-VEGF injections is uncertain. In the BIA, injection frequencies were incorporated directly from clinical trials. As noted in the Appraisal of the Sponsor’s Economic Evaluation, owing to the direct use of clinical trial data, it is not possible to determine whether any observed differences are due to the treatment or, rather, due to bias or confounding factors (e.g., differences in study populations, definitions of outcomes, or study designs). Clinical expert input received by CDA-AMC indicated that in clinical practice all anti-VEGF therapies are administered using a treat-and-extend approach that is personalized for each patient.
In the absence of comparative data to support reduced injection frequency with faricimab compared with other anti-VEGFs, CDA-AMC conducted a scenario analysis that assumed an equal injection frequency for all treatments.
The price of drugs paid by public drug plans is uncertain: The analyses conducted by the sponsor and CDA-AMC are based on publicly available list prices for all treatments, including faricimab. Faricimab has successfully undergone price negotiations for the treatment of nAMD and DME, and it is likely that the unit cost paid by public drug plans is lower than the price submitted in the current review. Similarly, the prices of other anti-VEGFs in the sponsor’s model do not reflect existing confidential prices negotiated by public plans.
The sponsor incorporated both branded and biosimilar ranibizumab in the BIA, assuming that branded captures 13% of total anti-VEGF market share by year 3, while the biosimilar form captures 4%. In jurisdictions with a biosimilar switch policy, the market share of biosimilar ranibizumab may be higher than assumed by the sponsor (versus branded ranibizumab), which may increase the incremental impact of reimbursing faricimab for RVO if faricimab displaces ranibizumab.
CDA-AMC was unable to incorporate the presence of confidential negotiated prices in reanalysis. Depending on the negotiated prices of faricimab and comparators, reimbursing faricimab for the treatment of macular edema secondary to RVO may lead to lower or no cost savings compared with currently available anti-VEGFs.
The displacement of other anti-VEGFs by faricimab is uncertain. The sponsor assumed that faricimab will displace only aflibercept 2 mg (i.e., faricimab will not take market share from other anti-VEGFs used to treat macular edema secondary to RVO). Clinical expert feedback received by CDA-AMC agreed that faricimab is likely to capture market share from aflibercept 2 mg; however, it is also reasonable to expect that faricimab will also capture market share from bevacizumab and ranibizumab. As such, displacement assumptions in the sponsor’s analysis are associated with uncertainty.
CDA-AMC was unable to address this limitation. If faricimab displaces less costly comparators, the cost savings associated with reimbursing faricimab for RVO will be less than predicted by either the sponsor’s base case or CDA-AMC scenario 1.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CDA-AMC conducted a scenario analysis to explore uncertainty in injection frequency, by assuming an equal injection frequency for all treatments. Results of this analysis suggest that the budgetary impact of reimbursing faricimab for the treatment of macular edema secondary to RVO is highly sensitive to injection frequency (Table 7).
Table 7: Detailed Breakdown of the Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 643,574,106 | 654,872,301 | 662,543,727 | 668,639,267 | 2,629,629,401 |
New drug | 643,574,106 | 652,620,237 | 651,883,265 | 649,388,504 | 2,597,466,112 | |
Budget impact | 0 | −2,252,064 | −10,660,462 | −19,250,763 | −32,163,288 | |
CDA-AMC scenario analysis 1: equal injection frequency | Reference | 527,072,385 | 536,325,347 | 543,037,394 | 548,620,684 | 2,155,055,810 |
New drug | 527,072,385 | 535,784,550 | 540,477,453 | 543,997,918 | 2,147,332,306 | |
Budget impact | 0 | −540,797 | −2,559,941 | −4,622,765 | −7,723,504 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.