Drugs, Health Technologies, Health Systems
Reimbursement Review
Ruxolitinib (Opzelura)
Sponsor: Incyte Biosciences Canada Corporation
Therapeutic area: Nonsegmental vitiligo
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BSA
body surface area
CDA-AMC
Canada’s Drug Agency
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
DLQI
Dermatology Life Quality Index
F-VASI
Facial Vitiligo Area Scoring Index
F-VASI25
25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index
F-VASI50
50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index
F-VASI75
75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index
F-VASI90
90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IFN
interferon
ITT
intention to treat
JAK
Janus kinase
LTE
long-term extension
MCID
minimal clinically important difference
NE
not evaluable
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
TEAE
treatment-emergent adverse event
TSQM
Treatment Satisfaction Questionnaire for Medication
T-VAS
Total Vitiligo Area Scoring Index
T-VASI50
50% or more improvement from baseline on the Total Vitiligo Area Scoring Index
T-VASI75
75% or more improvement from baseline on the Total Vitiligo Area Scoring Index
VASI
Vitiligo Area Scoring Index
VitiQoL
Vitiligo-Specific Quality of Life Instrument
VNS
Vitiligo Noticeability Scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Ruxolitinib (Opzelura) 1.5% topical cream |
Sponsor | Incyte Biosciences Canada Corporation |
Indication | Topical treatment of nonsegmental vitiligo in adult and pediatric patients 12 years of age and older |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 11, 2024 |
Recommended dosage | A thin layer of ruxolitinib applied twice daily to affected skin areas up to a maximum of 10% of body surface area for each application. |
NOC = Notice of Compliance.
Introduction
Vitiligo is a chronic autoimmune disorder that causes progressive depigmentation of the skin due to the loss of melanocytes,1-4 affecting up to 2% of the global population,4,5 with an estimated prevalence in Canada of between 0.5% and 1%.6 It is categorized into nonsegmental vitiligo (presenting as symmetric patches), segmental vitiligo (affecting 1 side of the body), and mixed forms (displaying characteristics of both forms),4 with nonsegmental vitiligo being the most common form, accounting for approximately 80% of vitiligo cases.5 It can also have an unpredictable progression, often starting before age 12 and peaking around age 30.4 The pathogenesis of vitiligo involves autoimmune mechanisms targeting melanocytes, driven by increased oxidative stress and inflammatory pathways, leading to immune-mediated destruction.1,7
Lesions often appear on the face, hands, and genital areas,8 and are often triggered by stress. Flares are also common, especially in individuals with more extensive skin involvement or darker skin tones.7,9,10 Additionally, about 25% of patients have autoimmune comorbidities,11 with thyroid disease being the most frequent.12 The psychosocial impact of vitiligo is profound, often leading to depression, anxiety, and social stigma,13-15 particularly when lesions are visible.16,17 Children13,18 and those with darker skin tones9,14 or from cultures with stronger stigma about skin tone are particularly vulnerable.19 Vitiligo also carries both direct and indirect economic costs, including treatment expenses and lost productivity.20,21
Diagnosis is based on physical exams, clinical history, and laboratory tests, with biopsies used in rare cases.4,12,22-24 Because of the association with autoimmune conditions, thyroid function and the potential for other autoimmune disorders is often assessed.23
Ruxolitinib in the form of a 1.5% cream was approved by Health Canada for the topical treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older (Notice of Compliance received on October 11, 2024). The sponsor’s reimbursement request aligns with this indication. This is the first review of ruxolitinib for this indication.
The recommended dosage is a thin layer of 1.5% ruxolitinib cream applied twice daily to affected skin areas, covering up to a maximum of 10% of the patient’s body surface area (BSA) per application.25 Satisfactory repigmentation may require more than 24 weeks of treatment. If meaningful repigmentation is not observed by 24 weeks, re-evaluation by a health care provider is recommended.25
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by Canada’s Drug Agency (CDA-AMC) for this review.
Patient Input
Input was submitted by the Canadian Skin Patient Alliance in collaboration with Vitiligo Voices Canada regarding the current review of ruxolitinib for nonsegmental vitiligo, based on a survey conducted in Canada between September 26 and October 15, 2024, with 19 respondents.
Participants were primarily from Ontario and Alberta, with others from Newfoundland, Nova Scotia, New Brunswick, Quebec, British Columbia, and Yukon. Survey respondents were predominantly white, and about one-half were aged 55 years or older.
According to the input, vitiligo’s impact extends beyond physical symptoms, affecting identity, emotional well-being, sense of belonging, and social interactions. Young individuals face bullying and social stigma, while many reported struggles with anxiety due to the condition’s unpredictable nature. Severe mental health consequences, such as depression and suicidal thoughts, were also reported, with challenges exacerbated for individuals with darker skin tones, some of whom faced heightened judgment due to cultural beauty standards.
Current treatment options were seen as inadequate. Patients who responded to the survey reported frustration with inconsistent results, side effects, high costs, and access barriers, particularly for patients living in rural areas, due to the need for frequent clinic visits. The patients who provided input through the patient groups noted that topical corticosteroids, the first-line therapy, were largely ineffective, as were other treatments, such as vitamin D derivatives and immunomodulators. Some patients reported limited effectiveness with depigmentation therapy (monobenzone) and narrowband UV-B phototherapy, although responses varied, and these treatments posed challenges due to the need for frequent clinic appointments and additional costs. None of those who responded had experience with ruxolitinib.
Those who responded to the survey emphasized the need for a treatment that is effective, easy to use, and capable of delivering reliable repigmentation with lasting results. They also prioritized fewer side effects, increased affordability, and greater accessibility, ideally through at-home or simpler solutions.
Clinician Input
Input From Clinical Experts Consulted for This Review
Vitiligo can range from being barely perceptible to cosmetically distressing, with patients having different perceptions of their condition. The disease site and degree of repigmentation matter, as areas of the skin affected by vitiligo in visible sites will be more difficult to hide and therefore have a greater effect on the patient. Partial repigmentation is not necessarily associated with improvement in health-related quality of life (HRQoL), as the visibility of vitiligo can still negatively affect patients’ HRQoL.
The clinical experts consulted for this review emphasized that the choice of treatment will be based on the impact that the disease has on a patient’s life. Options include conservative camouflage, which is usually not readily accepted by patients and does not modify disease mechanisms or provide any degree of disease improvement. The mainstays of treatment are topical corticosteroids and calcineurin inhibitors, which address the underlying inflammatory attack on the melanocytes. Other alternative treatments include phototherapy, which may be combined with topical treatments. However, according to the clinical experts, approximately one-half of the patients routinely seen in clinical practice become refractory due to disease resistance, while some other patients discontinue treatments due to unacceptable toxicity.
As none of the other currently available therapies are approved by Health Canada for the treatment of nonsegmental vitiligo, the clinical experts indicated that topical ruxolitinib has the potential to be a first-line therapy for nonsegmental vitiligo, or it could be reserved as a second-line option for patients refractory or intolerant to current mainstay treatments. They noted that the absence of comparative evidence against the drugs currently used in clinical practice was considered a substantial limitation. The experts also emphasized that the place in therapy for ruxolitinib would depend on whether it can provide clinically meaningful improvements for patients who have the highest unmet need. These patients were identified as those with darker skin tones; those likely to experience a greater decline in HRQoL due to the increased visibility of the condition; and those with affected lip-tip, periocular, and/or perioral regions, which are highly visible and often resistant to repigmentation.
The clinical experts expected treatment with ruxolitinib to be discontinued if there was a lack of efficacy or disease progression after 24 weeks or once the skin was fully repigmented. The clinical experts noted that referring patients to a specialist, such as a dermatologist, was preferable. They also indicated that it would be appropriate to restrict the amount of medication used by patients, limiting administration to 10% of the BSA.
Clinician Group Input
Two clinician groups, comprising 12 clinicians, provided input for this review: the Canadian Dermatology Association in collaboration with the Dermatologist Association of Ontario and Dermatology Association of Saskatchewan (with 8 clinicians contributing to the input), and the Southwestern Ontario Dermatologists Group (with 4 clinicians contributing).
Both groups agreed that, aside from ruxolitinib, no effective treatments for vitiligo are available in Canada. Current options include off-label use of corticosteroids, calcineurin inhibitors, and narrowband UV-B phototherapy, with systemic drugs and surgical grafting considered for more severe cases. Despite these treatments, vitiligo remains difficult to manage, with no cure available and a high recurrence rate. The core treatment goals, as outlined by both groups, are to achieve visible repigmentation and halt disease progression, although attaining high levels of repigmentation remains challenging. The Canadian Dermatology Association emphasized the need for maintenance therapy to prevent relapse, while the Southwestern Ontario group focused on reducing stress on melanocytes to improve outcomes.
Both groups described 1.5% ruxolitinib cream as a transformative first-line therapy for vitiligo in patients aged 12 years and older. It targets the underlying disease mechanisms and has a favourable safety profile, allowing for prolonged maintenance use.
Treatment response for vitiligo is typically assessed based on repigmentation and disease progression. The primary outcome measures include the Vitiligo Area Scoring Index (VASI), Investigator’s Global Assessment, Vitiligo Noticeability Scale (VNS), and global impression scales.
Regarding treatment discontinuation, input from the Canadian Dermatology Association suggested stopping treatment after 6 months without repigmentation, while others recommended extending it to 18 months. The Southwestern Ontario group stressed that, after 1 year, an inadequate response based on patient-reported outcomes, physician assessments, disease progression, and adverse events (AEs) should lead to discontinuation. Both groups agreed that vitiligo can be diagnosed by any physician, but dermatologists are ideally suited for diagnosis, treatment selection, and monitoring to ensure the best long-term outcomes.
Drug Program Input
Drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review process by identifying issues that may affect their ability to implement a recommendation. For this review, the drug plans provided questions pertaining primarily to initiation and prescribing of therapy. These questions were addressed by the clinical experts consulted for this review. The clinical experts’ responses are included in the Drug Program Input section (Table 4).
Clinical Evidence
Systematic Review
Description of Studies
Two studies were reviewed: TRuE-V1 (n = 330) and TRuE-V2 (n = 344) were phase III, multicentre, double-blind, vehicle-controlled randomized controlled trials (RCTs) identically designed to evaluate the efficacy and safety of 1.5% ruxolitinib topical cream, applied twice daily to depigmented areas, for the treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older.
The primary outcome was the proportion of patients achieving an improvement in the Facial Vitiligo Area Scoring Index (F-VASI) score of at least 75% from baseline at week 24. Additional levels of VASI thresholds were assessed as secondary end points in the trials. The VASI score evaluates the objective response to treatment, capturing the overall surface area of vitiligo involvement and the degree of repigmentation. However, while the VASI score is a validated instrument, it is not routinely used in clinical practice. Evidence in the literature suggests that a 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index (F-VASI75), and 50% or more improvement from baseline on the Total Vitiligo Area Scoring Index (T-VASI50), would likely result in a clinically meaningful change in repigmentation for patients with nonsegmental vitiligo.26 Patient-reported noticeability was assessed as a key secondary outcome using the VNS, a validated instrument for which scores of 4 (a lot less noticeable) or 5 (no longer noticeable) have been used as the minimal clinically important difference (MCID).
HRQoL was an exploratory outcome in both studies and was assessed using the Dermatology Life Quality Index (DLQI) and the Children’s Dermatology Life Quality Index (CDLQI), as well as the Vitiligo-Specific Quality of Life Instrument (VitiQoL). The DLQI is a 10-item questionnaire designed to assess the impact of skin conditions on an adult’s life, while the CDLQI is a similar questionnaire for children. The instrument covers domains such as symptoms, daily activities, relationships, work or school, and emotional well-being. The maximum total score on either index is 30, with higher scores denoting a greater negative impact on quality of life. The VitiQoL is a specialized, patient-reported HRQoL assessment tool designed to measure the impact of vitiligo on patients’ lives. The total score can range from 0 to 90, with higher scores denoting a greater reduction in quality of life.
Efficacy Results
Vitiligo Area Scoring Index
For the primary outcome, which was the proportion of patients achieving an F-VASI75, treatment with ruxolitinib was associated with between-group differences in the response rate of 22.3% (95% confidence interval [CI], 14.2 to 30.5; P < 0.0001) in the TRuE-V1 trial and 19.5% (95% CI, 10.5 to 28.4; P = 0.0004) in the TRuE-V2 trial over 24 weeks versus vehicle treatment. In absolute effects in the TRuE-V1 trial, 60 patients (30.8%) who applied ruxolitinib achieved the outcome compared to 7 patients (7.8%) who applied the vehicle treatment. In the TRuE-V2 trial, 62 patients (31.2%) who applied ruxolitinib achieved the outcome compared to 11 patients (11.2%) who applied the vehicle cream.
An F-VASI75 has been reported in the literature as a threshold for treatment success based on perceptions of patients with vitiligo and dermatologists; however, no MCID for between-group differences were reported. The presence of an important effect was determined by the clinical experts consulted for this review. The difference between treatments in terms of repigmentation was considered clinically meaningful, but the clinical experts noted that the overall impact was difficult to assess. In clinical practice, partial repigmentation as measured by the F-VASI may not necessarily be associated with a meaningful change for patients, as long as the disease remains visible. The minimal clinically important objective response can be highly variable across patients depending on how the disease affects their daily lives. Evidence of moderate certainty suggests that treatment with ruxolitinib likely results in a clinically important increase in the proportions of patients achieving an F-VASI75 compared to vehicle treatment.
Treatment with ruxolitinib was also likely associated with a clinically important increase in the proportions of patients achieving other VASI thresholds, such as a 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index (F-VASI90), over 24 weeks compared to vehicle treatment. A total of 31 patients (15.9%) who applied ruxolitinib in the TRuE-V1 trial achieved an F-VASI90 compared with 2 patients (2.2%) who applied the vehicle cream; in the TRuE-V2 trial, 33 patients (16.6%) who applied ruxolitinib achieved the outcome compared with 1 patient (1.0%) who applied the vehicle cream. The between-group differences in the response rate were 13.2% (95% CI, 7.5% to 18.8%; P = 0.0038) in the TRuE-V1 trial and 15.0% (95% CI, 9.3% to 20.7%; P = 0.0065) in the TRuE-V2 trial. Results for T-VASI50 responses were consistent with those for F-VASI90 responses. However, results for the more conservative threshold of a 75% or more improvement from baseline on the Total Vitiligo Area Scoring Index (T-VASI75) were deemed by the clinical experts to be unlikely to constitute a meaningful change for the patients.
Vitiligo Noticeability Scale
Patient-reported noticeability, assessed using the VNS, suggests that treatment with ruxolitinib likely results in a clinically important increase in the proportions of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) over 24 weeks compared to vehicle treatment. Among patients who applied ruxolitinib, the proportions of patients who responded to treatment were 25% in the TRuE-V1 trial and 21% in the TRuE-V2 trial, resulting in between-group differences in the response rate versus vehicle treatment of 21.2% (95% CI, 14.3% to 28.1%; P = 0.0002) in the TRuE-V1 trial and 15.5% (95% CI, 8.5% to 22.6%; P = 0.0013) in the TRuE-V2 trial. Uncertainty was introduced by the absence of a reported MCID for differences between treatments, and by the fact that, as was the case for the VASI, a less noticeable condition may not necessarily be associated with a meaningful change for patients, as long as the disease remains visible.
Health-Related Quality of Life
The clinical experts consulted for this review indicated that treatment of vitiligo is targeted at improving current and future HRQoL rather than focusing on surface area of involvement and degree of repigmentation. This was also consistent with patient and clinician input, which emphasized the importance of improving the psychosocial impact of the disease on quality of life. In the trials, HRQoL was assessed as an exploratory outcome. Statistical testing was not adjusted for multiplicity, and the results should be considered supportive evidence.
Results suggest that treatment with ruxolitinib may not result in a clinically important improvement in HRQoL, as measured with the VitiQoL over 24 weeks compared to vehicle treatment. The mean between-group differences in change from baseline through 24 weeks in VitiQoL were −0.28 (95% CI, −4.51 to 3.95; P = 0.8976) in the TRuE-V1 trial and −3.52 (95% CI, −7.60 to 0.57; P = 0.0915) in the TRuE-V2 trial. Results for HRQoL assessed using the DLQI and CDLQI were deemed not clinically meaningful. There is currently no MCID established for these instruments in patients with vitiligo, and the absence of an important effect was determined by the clinical experts consulted for this review. This indicates that, despite observing an objective response to ruxolitinib in terms of overall surface area of involvement and degree of repigmentation, which can make the condition less noticeable in some patients, ruxolitinib did not improve the impact of the disease on HRQoL in the overall study population. Findings from post hoc analyses that compared the change in VitiQoL and DLQI from baseline to week 24 among patients who received ruxolitinib and achieved various levels of F-VASI responses and those who did not achieve such responses suggest that patients may observe improvement in their HRQoL with at least an F-VASI75; however, whether the improvement in HRQoL is clinical meaningful is uncertain. In addition, interpretation of these findings is limited by the post hoc nature of the analyses.
Harms Results
A relatively large proportion of patients receiving ruxolitinib in the TRuE-V1 trial (46%) and the TRuE-V2 trial (50%) experienced at least 1 AE. The most common treatment-emergent adverse events (TEAEs) were related to application site reactions (acne, pruritus, rash, and exfoliation) and infections. Serious adverse events (SAEs) were uncommon. Treatment with ruxolitinib appeared to be well tolerated, as few discontinuations were due to AEs. No deaths were reported throughout the trials’ duration. Findings for the treatment extensions in the TRuE-V1 and TRuE-V2 trials, as well as from the TRuE-V long-term extension (LTE) study, were consistent with those from the pivotal trials. Overall, the clinical experts indicated that the harms profile of ruxolitinib did not raise any new safety signals or any particular safety concerns. However, as with most clinical trials, the studies were not powered to detect infrequent AEs, or those with a lag time.
Critical Appraisal
Interpretation of the findings is limited by the fact that the key efficacy evidence for ruxolitinib is focused on objective response to treatment. Because vitiligo can range from being barely perceptible to cosmetically distressing, and because different individuals are likely to have different priorities and objectives when assessing the magnitude of response to treatment, the clinical meaningfulness of an objective response is uncertain. As the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence comparing ruxolitinib to other therapies currently used for vitiligo. The comparative effectiveness and safety of ruxolitinib relative to other available treatment options, which were considered well accepted overall and routinely prescribed according to the clinical experts, are therefore unknown.
The TRuE-V1 and TRuE-V2 trials may be generalizable to a selected sample of individuals living in Canada with vitiligo. The majority of patients included in the studies were white and had a lighter skin colour. However, vitiligo is particularly visible in patients with darker skin tones and, as such, is likely to present with an increased impact on quality of life in these patients. Because few patients with darker skin tones were included in the studies, the effect of ruxolitinib in these patients is uncertain. In addition, there is a possibility that the trial population was not representative of patients whose condition interferes substantially with their daily life, considering the lower than expected use of prior therapies despite a long-lasting disease duration, as well as the relatively low level of HRQoL impairment at baseline. Few adolescents were enrolled in the trials, and there are limited data to interpret in this younger age group. The follow-up duration of 24 weeks was considered relatively short, as the disease generally improves over a longer period of time. Although the clinical experts considered the follow-up period sufficient to capture improvements in objective response, treatment with ruxolitinib is likely to continue over the long term, and evidence beyond the studies’ follow-up duration is limited.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined by a process outlined by the GRADE Working Group.27,28
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
For the GRADE assessments, findings from the TRuE-V1 and TRuE-V2 trials were considered together and summarized narratively per outcome because the populations, interventions, designs, and outcome measures of these studies were similar.
The selection of outcomes for the GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
improvements in F-VASI
patient-reported decrease in noticeability (VNS)
HRQoL (VitiQoL)
harms.
Table 2 presents the GRADE summary of findings for ruxolitinib versus vehicle cream.
Table 2: Summary of Findings for Ruxolitinib Versus Vehicle Cream for Patients With Vitiligo
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Vehicle cream | Ruxolitinib | Difference (95% CI) | |||||
F-VASI | |||||||
Proportions of patients achieving F-VASI75 Follow-up: 24 weeks | N = 394, ruxolitinib N = 188, vehicle cream (2 RCTs) | TRuE-V1: OR = 5.28 (2.341 to 11.903) TRuE-V2: OR = 3.45 (1.737 to 6.835) | TRuE-V1: 74 per 1,000 patients TRuE-V2: 114 per 1,000 patients | TRuE-V1: 298 per 1,000 patients TRuE-V2: 309 per 1,000 patients | TRuE-V1: 223 more per 1,000 patients (142 to 305) TRuE-V2: 195 more per 1,000 patients (105 to 284) | Moderatea | Ruxolitinib likely results in a clinically important increase in the proportions of patients achieving F-VASI75 over 24 weeks compared to vehicle cream. |
Proportions of patients achieving F-VASI90 Follow-up: 24 weeks | N = 394, ruxolitinib N = 188, vehicle cream (2 RCTs) | TRuE-V1: OR = 8.49 (1.997 to 36.048) TRuE-V2: OR = 15.29 (2.150 to 108.739) | TRuE-V1: 22 per 1,000 patients TRuE-V2: 13 per 1,000 patients | TRuE-V1: 153 per 1,000 patients TRuE-V2: 163 per 1,000 patients | TRuE-V1: 132 more per 1,000 patients (75 to 188) TRuE-V2: 150 more per 1,000 patients (93 to 207) | Moderatea | Ruxolitinib likely results in a clinically important increase in the proportions of patients achieving F-VASI90 over 24 weeks compared to vehicle cream. |
VNS | |||||||
Proportion of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) Follow-up: 24 weeks | N = 394, ruxolitinib N = 188, vehicle cream (2 RCTs) | TRuE-V1: OR = 9.53 (2.900 to 31.290) TRuE-V2: OR = 4.86 (1.851 to 12.755) | TRuE-V1: 33 per 1,000 patients TRuE-V2: 49 per 1,000 patients | TRuE-V1: 245 per 1,000 patients TRuE-V2: 205 per 1,000 patients | TRuE-V1: 212 more per 1,000 patients (143 to 281) TRuE-V2: 155 more per 1,000 patients (85 to 226) | Moderateb | Ruxolitinib likely results in a clinically important increase in the proportions of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) over 24 weeks compared to vehicle cream. |
HRQoL | |||||||
Change from baseline in VitiQoL Follow-up: 24 weeks | N = 394, ruxolitinib N = 188, vehicle cream (2 RCTs) | NA | TRuE-V1: LSM = −6.18 (SE = 1.77) TRuE-V2: LSM = −2.66 (SE = 1.70) | TRuE-V1: LSM = −6.45 (SE = 1.21) TRuE-V2: LSM = −6.18 (SE = 1.20) | TRuE-V1: LSM difference = −0.28 (−4.51 to 3.95) TRuE-V2: LSM difference = −3.52 (−7.60 to 0.57) | Lowc | Ruxolitinib may not result in a clinically important improvement in HRQoL as measured with the VitiQoL over 24 weeks compared to vehicle cream. |
Harms | |||||||
Patients with SAEs Follow-up: 24 weeks | N = 449, ruxolitinib N = 224, vehicle (2 RCTs) | NR | TRuE-V1: 9 per 1,000 patients TRuE-V2: 0 per 1,000 patients | TRuE-V1: 27 per 1,000 patients TRuE-V2: 9 per 1,000 patients | TRuE-V1: 18 more per 1,000 patients TRuE-V2: 9 more per 1,000 patients | Moderated | Ruxolitinib likely did not result in a clinically important increase in SAEs over 24 weeks compared to vehicle cream. |
CI = confidence interval; F-VASI = Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; HRQoL = health-related quality of life; LSM = least squares means; MCID = minimal clinically important difference; NA = not applicable; NR = not reported; OR = odds ratio; RCT = randomized controlled trial; SAEs = serious adverse event; SE = standard error; VASI = Vitiligo Area Severity Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aVASI: Rated down 1 level for imprecision, due to uncertainty surrounding the outcome measure and MCID. The F-VASI score is not used in clinical practice; the instrument was developed specifically for clinical trial assessment. Validation studies were identified from the literature. While a 75% improvement in F-VASI has been suggested as a threshold for treatment success based on perceptions of patients with vitiligo and dermatologists, no MCID for between-group differences were reported. The presence of an important effect was informed by the clinical experts consulted for this review, but was deemed difficult to assess, as partial repigmentation as measured by the F-VASI score may not necessarily be associated with a meaningful change for patients as long as the disease remains visible.
bVNS: Rated down 1 level for imprecision, due to uncertainty surrounding the outcome measure and MCID. While the VNS is a validated instrument, for which scores of 4 (a lot less noticeable) or 5 (no longer noticeable) have been used as the MCID, no MCID for between-group differences has been reported. The presence of an important effect was determined by the clinical experts consulted for this review, but was deemed difficult to assess, as a less noticeable condition may not necessarily be associated with a meaningful change for patients as long as the disease remains visible.
cHRQoL: Rated down 2 levels for imprecision. The VitiQoL was assessed as an exploratory outcome. Statistical testing for the VitiQoL was not adjusted for multiplicity in the trial and should be considered as supportive evidence. In addition, there is currently no MCID established for this instrument in the literature; the absence of an important effect was informed by the clinical experts consulted for this review. The uncertainty surrounding the MCID precluded a definitive judgment on whether the bounds of the CI suggest a meaningful effect on either side of the null.
dHarms: Rated down 1 level for imprecision, because of the low number of events in the study.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
LTE Studies
TRuE-V1 and TRuE-V2 Trials Open-Label Treatment Extension
Description of Studies
In the pivotal RCTs (TRuE-V1 and TRuE-V2), the double-blind controlled period was followed by a 28-week open-label treatment extension. Patients initially randomized to vehicle cream crossed over to ruxolitinib, while patients initially randomized to ruxolitinib received an additional 28 weeks of treatment with the active drug, as long as they completed the week 24 assessments with no safety concerns. During the treatment-extension period, patients continued to treat depigmented areas identified for treatment at baseline even if the area achieved full repigmentation.
Efficacy Results
The proportions of patients who achieved all the predefined thresholds in the VASI score (i.e., a 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index [F-VASI25], 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index [F-VASI50], F-VASI75, F-VASI90, and T-VASI75) were numerically higher in all treatment arms when compared to the corresponding proportions from the double-blind period. However, in the treatment-extension period, no statistical analysis was reported to assess whether the change from week 24 to week 52 was statistically significant, or to assess the magnitude of the between-group difference. Similar results were obtained in the proportions of patients who achieved a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable). According to the clinical experts, results for HRQoL, assessed by the DLQI or CDLQI and the VitiQoL, suggest that observed within-group changes from baseline to week 52 were small and not clinically meaningful.
Harms Results
The proportions of patients who experienced at least 1 AE during the treatment extension ranged from 33.7% to 41.2% across treatment arms in the 2 trials. Few patients experienced SAEs. One patient discontinued due to application site eczema. No deaths were reported during the treatment extension.
Critical Appraisal
Conclusions regarding the efficacy and safety of ruxolitinib in the longer term are noncomparative due to the single-arm nature of the TRuE-V1 and TRuE-V2 trial open-label treatment extensions. The same limitations pertaining to the uncertain clinical impact of the F-VASI score and selected patient population, which were highlighted for the double-blind controlled period of the studies, also apply to the extension period.
TRuE-V LTE Study
Description of Studies
The TRuE-V LTE31 is a phase III, double-blind, vehicle-controlled, randomized withdrawal trial designed to assess the long-term efficacy and safety of ruxolitinib cream in patients with vitiligo. It follows the TRuE-V1 and TRuE-V2 studies and includes 2 cohorts: cohort A, which evaluates the duration of response after withdrawing ruxolitinib cream, and cohort B, which assesses the maintenance of response with continued treatment. The LTE study had a duration of 52 weeks, followed by a 30-day safety follow-up.
Cohort A involved a randomized withdrawal design, providing data on the duration of response after discontinuation and the maintenance of response with continued treatment. Participants who achieved complete or near-complete facial repigmentation (greater than or equal to an F-VASI90) at week 52 in either the TRuE-V1 or TRuE-V2 trial were assigned to this cohort. They were randomized in a 1:1 ratio to either continue 1.5% ruxolitinib cream or switch to vehicle cream during the LTE. Participants in cohort A who experienced a disease relapse (defined as less than an F-VASI75) received open-label ruxolitinib cream as rescue treatment until week 104 or the end of the trial. Cohort B included participants who did not achieve a response greater than or equal to an F-VASI90 at week 52 in the parent studies. These participants continued treatment with 1.5% ruxolitinib cream for the entire LTE period. Both clinician groups remained blinded in cohort A until after the primary analysis (week 104), while the treatment in cohort B was open-label.31
The primary outcome of the TRuE-V LTE study was the time to relapse in cohort A, defined as a loss of an F-VASI75 response. The key secondary outcome was the time to maintain an F-VASI90. Additional secondary outcomes included the proportion of patients achieving an F-VASI50, F-VASI75, F-VASI90, a VNS score of 4 (a lot less noticeable) or 5 (no longer noticeable), or a T-VASI75, and the time to regain F-VASI90 and F-VASI75 in patients who experienced disease relapse. Exploratory outcomes included changes in DLQI, CDLQI, and VitiQoL from week 52, and time to regain an F-VASI75 and F-VASI90 following a relapse. Safety outcomes were consistent with the TRuE-V1 and TRuE-V2 parent studies.
Statistical analyses were exploratory with no alpha control, and 95% CIs were used. Data from participants with noncompliance or incorrect randomization were excluded. Primary and secondary analyses used the intention to treat (ITT) extension population, and time to event data were analyzed using Kaplan-Meier and Cox models. Relapse incidence, subgroup analyses, and safety outcomes were summarized descriptively.31
Efficacy Results
Primary end point: Time to relapse (less than an F-VASI75) — In cohort A, a smaller proportion of patients on ruxolitinib experienced a disease relapse (14.5%) compared to the vehicle cream group (28.6%). The risk of relapse was lower in the ruxolitinib cream group (hazard ratio [HR] = 0.422; 95% CI, 0.180 to 0.990; P = 0.0414).31
Key secondary end point: Time to maintain an F-VASI90 — The majority of patients who achieved complete or near-complete repigmentation of the face in cohort A in the parent studies maintained this level of repigmentation with continued application of ruxolitinib cream beyond week 52. Of the cohort of patients who received vehicle cream, 55.4% lost their F-VASI90 response. The median time to loss of an F-VASI90 in the group of patients who received vehicle cream was 195.0 days (95% CI, 113.0 days to 372.0 days). Of the cohort of patients who applied ruxolitinib cream in the double-blind period then continued treatment with ruxolitinib and achieved an F-VASI90 response, 23.6% lost their F-VASI90 response. The median time to loss of an F-VASI90 response in this cohort was not evaluable. The risk of losing an F-VASI90 response was lower for patients who continued to use ruxolitinib cream compared with patients who applied vehicle cream (HR = 0.316; 95% CI, 0.165 to 0.606; P = 0.0003).
Additional secondary and exploratory end points — In cohort B, 86.4%, 66.1%, and 33.9% achieved an F-VASI50, F-VASI75, and F-VASI90, respectively, with ruxolitinib cream at week 104, compared to 69.9%, 47.3%, and 28.0% of those who switched to ruxolitinib, respectively.
In cohort A, the proportions of participants continuing on 1.5% ruxolitinib cream who achieved a T-VASI75 were 42.1% at week 52 and 55.3% at week 104. Among participants using vehicle cream, 38.6% achieved a T-VASI75 at week 52 and 39.1% achieved it at week 104.
In cohort B, for those who continued using 1.5% ruxolitinib cream, at week 52, 12.2% achieved a T-VASI75 and by week 104, 30.5% achieved this threshold. Among participants initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the TRuE-V LTE study, 3.4% achieved a T-VASI75 at week 52 and 18.3% reached the threshold at week 104.
In cohort A, the proportions of participants receiving blinded treatment who reported a VNS score of 4 (a lot less noticeable) or 5 (no longer noticeable) had their response remain generally stable compared to that of week 52. Among participants receiving 1.5% ruxolitinib cream, 50.0% reported a score of 4 or 5 at week 104, compared to 42.1% at week 52. Among participants receiving vehicle cream, 56.5% reported a score of 4 or 5 at week 104, compared to 49.1% at week 52.
In cohort B, the proportions of participants achieving a VNS score of 4 or 5 for those who continued using 1.5% ruxolitinib cream had their response remain generally stable (43.3% at week 104 versus 35.3% at week 52). The VNS score of 4 or 5 increased from week 52 to week 104 for those initially randomized to the vehicle cream who switched to 1.5% ruxolitinib cream during the TRuE-V LTE study (30.1% at week 104 versus 11.9% at week 52).
██ ███ ███████ ██ █████ █████ ████ ██ ███████████ ███████ ██ ███ ███████████ ████ ███████ █████ ██████ ██████ ███ ██████ ██ █████ ██████ █ ██████ ██████ ████████████ ██ ██████████ ███████████ ████ ███████ █████ ███████ ███████ ██████████ ██ ███████ ███ ████████ ██ ██████.
In exploratory end points, the median time to regain an F-VASI75 was 85.0 days for patients who experienced disease relapse and switched to open-label rescue treatment, whereas 62.5% of patients on ruxolitinib regained an F-VASI75 after a median of 205.0 days.31
Harms Results
The overall incidences of TEAEs and application site reactions for cohort A were higher among patients who applied ruxolitinib cream (55.2% and 6.9%, respectively) compared with the vehicle cream treatment group (36.2% and 3.4%, respectively). One participant treated with 1.5% ruxolitinib cream in cohort A (1.7%) had a serious TEAE, and no participant had a TEAE with a fatal outcome or a TEAE leading to study drug discontinuation.
The overall incidences of TEAEs and application site reactions in cohort B were 50.9% and 8.5%, respectively, among patients who continued receiving 1.5% ruxolitinib cream compared to 50.0% and 5.1% for participants initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the LTE study. The incidences of serious TEAEs were 3.1% in patients treated with ruxolitinib cream and 3.4% in those initially treated with vehicle cream. The only TEAE leading to study drug discontinuation in cohort B was ████ ███████ accident (involving 1 participant) that the investigator assessed was unlikely to be related to the study drug.
Critical Appraisal
Internal validity: Cohort A of the TRuE-V LTE trial employed appropriate random allocation using an interactive response technology system. Allocation concealment was ensured, and both patients and investigators remained blinded to treatment assignment until the study’s conclusion. Baseline characteristics between groups were well balanced, supporting the validity of comparisons. The single-arm design of cohort B introduced potential bias in assessing efficacy outcomes, as both participants and investigators were aware of the treatment being administered. This lack of blinding could lead to detection bias. Additionally, the single-arm nature of the study inherently carries a high risk of bias, which may influence the assessment of subjective treatment outcomes. No conclusions can be made on the comparative efficacy and safety.
Participants selected for the TRuE-V LTE trial represented a subsample of the parent trials, consisting of those who completed the parent trials without safety concerns following ruxolitinib use. This selection process may have introduced a risk of selection bias, as it could limit the representativeness of the wider patient population.
The TRuE-V LTE trial had a high dropout rate and, unlike the pivotal trials, imputation methods to address missing data were not employed, increasing the risk of attrition bias. The substantial number of missing participants may have skewed the results and affected interpretation of the findings.
External validity: The TRuE-V LTE trial consisted of patients who took part in the pivotal studies (TRuE-V1 and TRuE-V2), and it is reasonable to expect that the same strengths and limitations regarding generalizability apply to the extension studies. While the studies were conducted in centres in Europe and North America, the patient population of those studies may be reflective of the patient population in Canada and the clinical evidence is generalizable to Canada.
Indirect Comparisons
As the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence comparing ruxolitinib to the currently used off-label therapies for vitiligo to inform the reimbursement question. In addition, the sponsor submitted no indirect evidence. Although potential studies were identified to perform an indirect treatment comparison, the sponsor rated the feasibility of conducting robust evidence synthesis as low, limiting the feasibility of such a comparison. As a result, the comparative efficacy and safety of ruxolitinib compared with any off-label therapies for the treatment of vitiligo is unknown.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps were submitted by the sponsor.
Conclusions
In patients with nonsegmental vitiligo, findings of moderate certainty from the TRuE-V1 and TRuE-V2 trials suggest that treatment with ruxolitinib likely results in a clinically important improvement in the F-VASI score compared with vehicle treatment, based on thresholds identified in the literature for a clinically meaningful change in repigmentation. The clinical experts consulted for this review indicated that the VASI score can be relevant to inform on the objective response to treatment by capturing the overall surface area of vitiligo involvement and degree of repigmentation. However, given that vitiligo can range from being barely perceptible to cosmetically distressing, different individuals are likely to have different priorities and objectives when assessing the magnitude of response to treatment. The clinical impact of objective response on patients’ daily lives is therefore uncertain and, as noted by clinician and patient input, should be interpreted together with findings on HRQoL. Based on results from exploratory analyses, patients who applied ruxolitinib in the studies did not experience a clinically meaningful improvement in their HRQoL. This suggests that, despite making the condition less noticeable, treatment with ruxolitinib did not improve the negative impact of the disease on patients’ lives in the overall study population as measured by HRQoL metrics. In patients who received ruxolitinib, analyses of change in HRQoL among patients achieving an F-VASI threshold and those who did not suggest that patients may observe an improvement in their HRQoL with a response of at least an F-VASI75. However, whether the improvement in HRQoL is clinical meaningful is uncertain, and interpretation of these findings is limited by the post hoc nature of the analyses. A relatively large proportion of patients in the TRuE-V1 and TRuE-V2 trials experienced AEs, most notably involving application site reactions and infections, although ruxolitinib appeared to be well tolerated, with few reports of SAEs and withdrawals due to AEs. The overall harms profile did not raise any particular safety signal.
Special consideration should be given to the fact that vitiligo can have a profound effect on patients. Historically, this condition has been perceived negatively, carrying significant cultural implications, with patients and families facing stigma and social isolation. The condition is particularly visible in patients with darker skin tones, for whom it can be associated with a loss of identity and lowered self-esteem, resulting in a marked reduction in quality of life. The patient input noted dissatisfaction with current treatment options, creating a need for more effective, accessible, and tolerable therapies. However, external validity issues in the TRuE-V1 and TRuE-V2 trials preclude making definitive conclusions about the effect of ruxolitinib in individuals with the greatest unmet need. The majority of patients included in the studies were white and had a lighter skin colour, based on the Fitzpatrick Scale skin type classification. In addition, it is likely that the trials included patients whose condition did not interfere substantially with their daily life, given the lower than expected use of prior therapies despite long-lasting disease and relatively low level of HRQoL impairment at baseline.
Evidence is limited beyond the studies’ follow-up duration, even though treatment with ruxolitinib is likely to continue over the long term. However, results from extension studies suggest that the findings are consistent with those from the pivotal evidence over the 2-year follow-up. Because the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence comparing ruxolitinib to other currently used therapies for vitiligo to inform the reimbursement question. The comparative effectiveness and safety of ruxolitinib relative to other treatment options are therefore unknown.
Introduction
This report reviews and critically appraises the evidence submitted by the sponsor on the beneficial and harmful effects of 1.5% ruxolitinib topical cream for the treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
Vitiligo is a chronic autoimmune disorder characterized by the progressive loss of the pigment-producing cells in the skin known as melanocytes, leading to depigmented patches on various parts of the body.1-4 These lesions are typically white and nonscaly.4 The global prevalence of vitiligo is estimated to range from 0.5% to 2%,4,5 with the prevalence in Canada estimated at between 0.5% and 1%.6
Vitiligo is classified into 3 primary types: nonsegmental vitiligo, which is the most common form, presenting as symmetric patches; segmental vitiligo, which affects 1 side of the body and usually develops earlier; and mixed vitiligo, which displays characteristics of both forms.4 Nonsegmental vitiligo can be further categorized into subtypes, including generalized (symmetric patches), acrofacial (affecting the face, hands, and feet), mucosal (affecting the genital and oral mucosa), focal (isolated lesions), and universal (affecting 80% to 90% of the BSA).4,32 Nonsegmental vitiligo accounts for approximately 80% of vitiligo cases5 and can have an unpredictable progression, often beginning before the age of 12 years and peaking around age 30.4
The pathogenesis of nonsegmental vitiligo is driven by autoimmune mechanisms that target melanocytes. These cells experience stress due to protein misfolding and increased production of reactive oxygen species,1 which triggers inflammatory pathways, particularly the Janus kinase (JAK) pathway activated by interferon (IFN)-gamma.7 This leads to the release of chemokines CXCL9 and CXCL10, which recruit autoreactive CD8+ T cells to melanocytes, initiating immune-mediated destruction. The accumulation of these T cells correlate with disease severity and is central to vitiligo’s pathogenesis.7
Vitiligo lesions, which may progress to complete depigmentation, can have smooth or irregular borders, and may appear red, inflamed, or brown due to hyperpigmentation. Itching is common in some cases.33,34 Flares are often triggered by stress, with two-thirds of patients reporting flare-ups during such periods, particularly those with more extensive skin involvement, darker skin tones, or facial lesions.7,9,10 Common lesion sites include the face, hands, and genital areas,8 with facial vitiligo significantly affecting patient self-esteem.35 In nonsegmental vitiligo, children often develop facial lesions, while adults tend to have lesions on the arms. Early-onset vitiligo (before the age of 12 years) typically affects the eyelids and lower extremities, while later-onset vitiligo (after the age of 12 years) is more common on the upper extremities.36
Approximately 25% of patients with vitiligo have at least 1 autoimmune comorbidity,11 with thyroid disease being the most common.12 Other associated autoimmune conditions include atopic dermatitis, psoriasis, alopecia areata,37,38 and, less frequently, rheumatoid arthritis, inflammatory bowel disease, and lymphoma.39
The psychosocial impact of vitiligo is profound, often leading to depression, anxiety, and social stigma,13-15 particularly when depigmentation occurs in visible areas.16,17 Some studies found that individuals with vitiligo experience higher rates of suicidal ideation and attempts compared with the general population.40-42 Children are particularly vulnerable to bullying and stigmatization,13,18 while those with darker skin tones9,14 or from cultures in which stronger stigma are more common face heightened challenges.19 These issues can lead to social isolation, low self-esteem, and discrimination, particularly in public-facing jobs.13,18,43
Economically, vitiligo incurs direct costs for treatments such as phototherapy, medication, and psychological support, as well as for over-the-counter medications, clothing, and camouflage.20,21 Indirect costs, such as absenteeism and decreased productivity, further exacerbate the financial burden on both patients and society.21
The diagnosis of vitiligo relies on physical examination, clinical history, laboratory tests, and, in rare cases, biopsies.4,12,22-24 A Wood’s lamp may be used to detect depigmented areas, and biopsies are performed in atypical cases to rule out other conditions.4 Laboratory tests primarily assess thyroid function and test for autoimmune disorders, given the high prevalence of such conditions in patients with vitiligo.23
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
The clinical experts consulted for this review indicated that the choice of treatment for vitiligo is based on the impact that the disease has on a patients’ life. There are no Canadian guidelines for the treatment of vitiligo, and no approved therapeutics in Canada indicated for repigmentation in patients with vitiligo. The current treatment modalities are therefore used off-label.
Nonpharmacological options include conservative camouflage products, including cover-up makeup, skin tanners, and clothing, but the experts consulted for this review noted that these are usually not well accepted by patients. Camouflage only reduces the visibility of the vitiligo and, while targeting the social and quality of life aspects, does not modify disease mechanisms or provide disease improvement.
The mainstays of treatment are topical corticosteroids and calcineurin inhibitors. These address the underlying inflammatory attack on the melanocytes. However, the use of topical corticosteroids may be limited by skin atrophy, especially with chronic use. Other alternative treatments include phototherapy, which may be also combined with topical treatments. Oral therapies, such as corticosteroids, may be used in rare, acute circumstances.
According to the clinical experts, although the off-label treatments currently used are typically well accepted, some patients experience a lack of efficacy, which can include incomplete and/or uneven repigmentation. In addition, approximately one-half of the patients routinely seen in clinical practice become refractory due to disease resistance, while some other patients discontinue treatments due to unacceptable toxicity. The patient input emphasized the lack of satisfaction with current treatment options and the need for more effective, accessible, and tolerable therapies for vitiligo.
Drug Under Review
Key characteristics of 1.5% ruxolitinib cream are summarized in Table 3, along with other treatments available for nonsegmental vitiligo.
Ruxolitinib in the form of 1.5% cream was approved by Health Canada for the topical treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older (Notice of Compliance received on October 11, 2024). The sponsor’s reimbursement request aligns with this indication. This is the first review of ruxolitinib for this indication.
Table 3: Key Characteristics of Ruxolitinib
Characteristic | Ruxolitinib |
|---|---|
Mechanism of action | Ruxolitinib is a JAK inhibitor that selectively targets the JAK1 and JAK2 isoforms, blocking cytokine and chemokine signalling. |
Indicationa | Topical treatment of nonsegmental vitiligo in patients 12 years of age and older |
Route of administration | Topical |
Recommended dose | The recommended dose is a thin layer of cream applied twice daily to affected skin areas up to a maximum of 10% of BSA for each application. Ten percent of BSA is about 10 times the size of the hand (including the palm and fingers). Satisfactory patient response may require treatment with ruxolitinib for more than 24 weeks. If the patient does not find the repigmentation meaningful by 24 weeks, consider re-evaluation by the health care provider. |
Serious adverse effects or safety issues | Serious infections, malignancies, major adverse cardiovascular events, and thrombosis |
Other | Not applicable |
BSA = body surface area; JAK = Janus kinase.
aHealth Canada–approved indication.
Source: Ruxolitinib (Opzelura) product monograph.25
The recommended dosage is a thin layer of 1.5% ruxolitinib cream applied twice daily to affected skin areas, covering up to a maximum of 10% of the BSA per application.25 Ten percent of the BSA is roughly 10 times the size of the patient’s hand, including the palm and fingers.44,45 Satisfactory repigmentation may require more than 24 weeks of treatment. If meaningful repigmentation is not observed by 24 weeks, re-evaluation by a health care provider is recommended.25
Ruxolitinib is a JAK inhibitor that selectively targets the JAK1 and JAK2 isoforms. By inhibiting JAK signalling, it modulates gene expression through the recruitment of signal transducers and activators of transcription to cytokine receptors. In vitiligo, autoimmune cytotoxic T lymphocytes that produce IFN-gamma are thought to destroy melanocytes.46 These lymphocytes are recruited to lesional skin via IFN-gamma–dependent chemokines such as CXCL10. Ruxolitinib reduces CXCL10 levels by inhibiting JAK1-dependent and JAK2-dependent signalling.46
Although topical corticosteroids and 0.1% tacrolimus are prescribed in Canada for the treatment of vitiligo, these therapies are prescribed off-label, as there are no approved therapeutics in Canada indicated for repigmentation in patients with vitiligo.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on input provided by patient groups.
Input was submitted by the Canadian Skin Patient Alliance in collaboration with Vitiligo Voices Canada regarding the current review of ruxolitinib for nonsegmental vitiligo. This submission was based on data collected from a patient and caregiver survey conducted between September 26 and October 15, 2024.
The survey received responses from 19 participants across Canada, with the majority from Ontario (36.84%), followed by Alberta (21.05%) and smaller groups from Newfoundland and Labrador and British Columbia (10.53% each), and from Nova Scotia, New Brunswick, Quebec, and Yukon (5.26% each). No survey respondents were from Prince Edward Island, Manitoba, Saskatchewan, Northwest Territories, or Nunavut.
Among the survey respondents who provided demographic details (n = 13), 53% were aged 55 years and older, 23% were aged 25 to 34 years, 15% were aged 35 to 44 years, and 8% were aged 45 to 54 years. In terms of ethnicity, 61.5% identified as white, 15.4% as South Asian, and 1 respondent identified as Southern European, Southeast Asian, and West Asian or Middle Eastern. The group included 10 females and 3 males, with 2 survey respondents serving as caregivers. Among those who reported the duration of their vitiligo (n = 12), 33% had the condition for less than 5 years, 25% for 5 to 10 years, and another 25% for more than 20 years. Disease severity was most commonly rated as moderate (33%) or severe (33%). Common comorbidities included autoimmune diseases (50%) and mental health conditions such as depression or anxiety (30%).
According to the input, vitiligo has significant impacts on individuals and their families. Seventy-five percent of survey respondents reported that it affected their sense of identity, while 42% noted its impact on daily activities and social life. Additionally, 25% experienced a diminished sense of belonging, 33% faced challenges in intimate relationships, and 25% reported issues in family relationships. Young individuals in particular reportedly struggled with bullying and social stigma. The input stated that the unpredictable progression of vitiligo heightens anxiety and helplessness and, in severe cases, suicidal ideation and attempts can occur. Sociocultural beauty standards intensify the emotional strain of the disease, particularly for those with darker skin tones, leading to feelings of alienation and judgment.
Moreover, the input indicated that patients with vitiligo and their caregivers face significant challenges with current treatment options. Emotional exhaustion stemming from inconsistent treatment results was reported by 40% of respondents, adversely affecting family dynamics and heightening the psychological burden on caregivers, often leaving them feeling helpless. While 3 patients who responded had never used any treatment and none had experience with the drug under review, 9 participants reported trying various options, including topical corticosteroids (commonly considered the first-line therapy), vitamin D derivatives, oral steroids, topical immunomodulators, and calcineurin inhibitors. None of these 9 participants found topical corticosteroids effective, with 44.4% stating they “did not work at all” and 22.2% indicating they “did not work very well.” Vitamin D derivatives, oral steroids, immunomodulators, calcineurin inhibitors, tattooing, and various transplant surgeries were similarly deemed ineffective by those who responded. Although some patients reported limited effectiveness from narrowband UV-B phototherapy and depigmentation therapy with monobenzone, responses varied significantly.
Patients also reported various treatment-related side effects, such as skin thinning (n = 1), irritation (n = 2), and burning (n = 1), leading some to discontinue treatment due to adverse reactions, inconvenience, high costs, or access challenges — particularly for rural patients requiring frequent clinic visits. Treatments such as phototherapy posed specific barriers due to the necessity for regular clinic appointments and additional costs, which were particularly burdensome for those in remote areas. This cycle of ineffective treatments has left many patients feeling exhausted and disillusioned; 10 out of 12 patients expressed interest in new treatments, while only 1 out of 11 patients reported satisfaction with current options, underscoring the need for more effective, accessible, and tolerable therapies.
Survey respondents articulated distinct goals for the new treatments. The majority (83%) prioritized efficacy, seeking reliable repigmentation that reduces patchiness and delivers lasting, comprehensive results. They also hoped for treatments with fewer side effects (75%), increased affordability (67%), and greater accessibility, ideally through at-home or simpler solutions. Preferences included ease of use (50%), flexibility for daily life (33%), and consistent results across diverse skin tones to minimize psychological burdens.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of vitiligo.
Unmet Needs
Vitiligo can range from being barely perceptible to cosmetically distressing. Experience from clinical practice suggests that patients will have different perceptions of their condition. The clinical management of vitiligo is therefore based on assessing its impact on the daily life of each individual patient.
There are currently no Health Canada–approved therapies for nonsegmental vitiligo. According to the clinical experts consulted for this review, although the off-label treatments currently used are typically well accepted, approximately one-half of the patients routinely seen in clinical practice become refractory due to disease resistance while some other patients discontinue treatments due to unacceptable toxicity. In addition, response varies by anatomic site. For example, while the cheeks and neck are typically more responsive to therapies, lip-tip, periocular, and perioral regions tend to be resistant, making repigmentation more difficult to achieve when these areas are affected. The disease site and degree of repigmentation matter, as areas affected by vitiligo in visible sites will be more difficult to hide and therefore have a greater impact on the patient. Partial repigmentation is not necessarily associated with improvement in HRQoL, which can be affected as long as the disease is visible.
The clinical experts emphasized that there is a need for effective and well-tolerated therapies for the treatment of vitiligo.
Place in Therapy
The choice of treatment for vitiligo is based on the impact of the disease on a patient’s life. Conservative camouflage options, including cover-up makeup, skin tanners, and clothing, can be a first step, but the clinical experts noted that such measures are usually not well accepted by patients. Camouflage only reduces the visibility of the vitiligo and, while targeting the social and quality of life aspects, does not modify disease mechanisms or provide any sort of disease improvement.
The mainstays of treatment are topical corticosteroids and calcineurin inhibitors, which address the underlying inflammatory attack on melanocytes. However, the use of topical corticosteroids may be limited by skin atrophy, particularly with chronic use. Other alternative treatments include phototherapy, which may be also combined with topical treatments. The use of oral therapies such as corticosteroids is considered rare. Assessing the presence or absence of other concomitant autoimmune diseases, and ensuring knowledge of sun protection, are also important parts of the clinical evaluation.
Topical ruxolitinib would constitute a new class of medication for the treatment of nonsegmental vitiligo. Its overarching anti-inflammatory mechanism of action would be similar to that of topical corticosteroids and calcineurin inhibitors; however, its individual immunosuppressant action through the JAK-inhibition pathway would be unique. As none of the other current therapies are approved by Health Canada, the clinical experts indicated that topical ruxolitinib may be a first-line therapy for nonsegmental vitiligo or reserved as a second-line option for patients refractory or intolerant to current mainstay treatments. The experts did not expect that ruxolitinib would cause a shift in the current treatment paradigm. They noted that the absence of comparative evidence against the drugs currently used in clinical practice was considered a substantial limitation. The experts also highlighted that the place in therapy for ruxolitinib would depend on its potential to provide clinically meaningful improvements for patients, as certain highly visible disease sites pose greater treatment challenges, to the point that HRQoL may not be deemed superior if repigmentation is only partial. Finally, it is unknown whether ruxolitinib can be safely combined with phototherapy without increasing the risk of skin cancer.
Patient Population
Patients whose characteristics are similar to those of patients included in the pivotal clinical studies would be best suited for treatment with ruxolitinib.
Although the impacts of the disease vary from patient to patient, the clinical experts indicated that those with darker skin tones are likely to experience a greater impact on quality of life because of the increased visibility of vitiligo. However, such patients constituted only a small proportion of the studies’ populations.
Assessing the Response Treatment
Treatment of vitiligo is targeted at improving the current and future impact on quality of life, as the effect of visible dermatoses can be profound. The goals of therapy include repigmentation and stopping disease progression to ultimately reduce the psychosocial impacts on quality of life.
The key outcomes in the clinical trials assessed repigmentation and disease progression; however, HRQoL was assessed both as a secondary and as an exploratory outcome. The instruments used were scores developed specifically for clinical trials of vitiligo treatments, and according to the clinical experts, are not used in clinical practice. In their experience, the most important parameter to assess efficacy would be HRQoL, as partial repigmentation may not be meaningful for all patients and the disease has a greater impact in certain sites, which is not captured by the VASI score.
The experts indicated that response to treatment should be assessed after 24 weeks, although some level of improvement may continue to be observed over time. An F-VASI75 was identified as a reasonable threshold for objective response to treatment, although the F-VASI does not inform on the level of improvement in HRQoL.
Discontinuing Treatment
In the absence of unacceptable toxicity, the clinical experts expected that patients and providers would discontinue ruxolitinib if there were a lack of efficacy or disease progression, which would be assessed after a minimum of 24 weeks, or once the skin is fully repigmented. However, the experts noted that the clinical trials showed some evidence of improvement in repigmentation up to 52 weeks. They advised that ruxolitinib should continue to be reimbursed if disease recurs after treatment is discontinued because of full skin repigmentation.
Prescribing Considerations
The clinical experts noted that referring patients to a specialist, such as a dermatologist, would be preferable. Although the diagnosis is generally straightforward, experience in managing vitiligo is considered important. The experts noted that patients referred to them sometimes have other hypopigmented conditions, such as postinflammatory hypopigmentation, pityriasis alba, pityriasis versicolour, nevoid hypopigmented or depigmented conditions, idiopathic hypomelanosis, or trauma-induced depigmentation. The experts indicated that it would be appropriate to consider restrictions on the amount of medication used by patients so that the treatment area does not exceed 10% of the BSA.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Two clinician groups, comprising 12 clinicians, provided input for this review: the Canadian Dermatology Association in collaboration with the Dermatologist Association of Ontario and Dermatology Association of Saskatchewan (with 8 clinicians contributing to the input), and the Southwestern Ontario Dermatologists Group (with 4 contributing clinicians). Input from the Canadian Dermatology Association drew on clinical experience, research expertise, medical literature, and insights from national and international meetings. In contrast, input from the Southwestern Ontario Dermatologists Group was based on a review of articles focused on vitiligo and its treatments.
The clinician groups stated that, aside from the recently approved ruxolitinib, no effective treatments for vitiligo are available in Canada. The dermatologists from Southwestern Ontario reported that most current treatment modalities are used off-label. These include topical corticosteroids, calcineurin inhibitors, and prostaglandin analogues that modulate immunity and promote melanocyte proliferation, while narrowband UV-B phototherapy is the primary option for extensive cases. Laser treatments are effective but limited in accessibility. Systemic drugs such as corticosteroids and immunosuppressants are used to stabilize progression when topical treatments fail, and surgical grafting is reserved for those whose condition remains stable.
While the Canadian Dermatology Association noted the absence of formal guidelines for vitiligo, both clinician groups agreed that core treatment goals include visible repigmentation and halting disease progression, although achieving high levels of repigmentation can be challenging. The Canadian Dermatology Association focused on the need for maintenance therapy to prevent recurrence after successful treatment, while the dermatologists from Southwestern Ontario emphasized the importance of reducing intrinsic stress on melanocytes.
Both clinician groups acknowledged that vitiligo is among the most challenging conditions to treat, with off-label therapies showing limited effectiveness and tolerability. Current options, such as topical corticosteroids, cannot be used over the long term due to side effects, and some treatments are not covered by public health plans, reducing both adherence and outcomes. The Canadian Dermatology Association also noted that no existing treatments reverse the disease, with around 40% of patients experiencing recurrence. One Southern Ontario dermatologist added that patients with vitiligo often face delayed diagnoses and misdiagnoses and tend to believe that health care providers underestimate the severity of their condition, leading to frustration and low compliance. Both groups emphasized the urgent need for new, safe, accessible, and effective long-term therapies to address these unmet needs.
Both clinician groups described 1.5% ruxolitinib cream, which is a targeted monotherapy option for patients aged 12 years and older, as a transformative first-line therapy for vitiligo. It directly addresses the disease’s underlying mechanisms by inhibiting CD8+ T-cell activity and the IFN-gamma pathway, while also improving repigmentation by inhibiting the JAK-STAT protein pathway. With a favourable safety profile, it allows for prolonged maintenance use. The Canadian Dermatology Association noted the potential of ruxolitinib to be combined with narrowband UV-B phototherapy or antioxidants for enhanced effects against IFN-gamma and reactive oxygen species. The Southwestern Ontario Dermatologists Group stated that ruxolitinib cream is best suited for patients with mild to moderate vitiligo, particularly those with visible lesions on areas that significantly affect daily life, such as the face, hands, and genitals. However, patients with rapidly spreading vitiligo affecting more than 50% of their BSA may initially require systemic immunosuppressants and are not ideal candidates for this treatment.
Both clinician groups agreed that treatment response for vitiligo is assessed based on how well it results in repigmentation and halts disease progression. The Southwestern Ontario Dermatologists Group noted that the VASI is a suitable end point in clinical trials, with a clinically meaningful response defined as a 50% improvement in both F-VASI and T-VASI scores after 1 year. The Canadian Dermatology Association panel, while agreeing that a F-VASI75 at 24 weeks is an appropriate end point for a clinical trial, emphasized that it should not be used as a strict cut-off in practice, as it may overlook meaningful improvements, particularly for patients with visible lesions. They suggested reassessing treatment response at 52 weeks or longer. Additionally, the Southwestern Ontario Dermatologists Group considered the Investigator’s Global Assessment and patient-reported outcomes, such as the VNS and global impression scales, to be the most effective measures of treatment outcomes for vitiligo.
Regarding treatment discontinuation, input from the Canadian Dermatology Association indicated that some experts suggest discontinuing treatment after 6 months with no repigmentation, while others recommend extending this period to 18 months. The Southwestern Ontario Dermatologists Group stressed that, after 1 year, an inadequate response based on patient-reported outcomes and physician assessments, along with factors such as disease progression and AEs should be considered for discontinuation. Both clinician groups agreed that, while vitiligo can be diagnosed by any physician and treated in various settings, dermatologists are ideally suited for diagnosis, treatment selection, and monitoring to ensure optimal long-term outcomes.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Patients in trials are required to have the following:
Do you anticipate any challenges with implementing these scales in clinical practice? | The clinical experts indicated that the F-VASI and T-VASI scores are not routinely used in clinical practice. It is therefore likely that there would be a learning curve for clinicians in implementing these scales for reimbursement purposes. However, this would be feasible. |
What if the total BSA of vitiligo is greater than 10%? Should these patients avoid treatment with ruxolitinib given the black box warnings in the ruxolitinib product monograph including serious infections, malignancies, thrombosis, and MACE? | According to the clinical experts, ruxolitinib should be made available for patients who have a total BSA of vitiligo of greater than 10%. Based on the pivotal trials and on the product monograph, treatment should focus on a maximum of 10% of BSA. Whether this will be respected in clinical practice is uncertain. |
What is the role of immunomodulators in the treatment of autoimmune disease that causes skin depigmentation? Should these therapies be considered for patients with an affected BSA of greater than 10%? | Although some patients may use systemic therapies for acute treatments of aggressive vitiligo, the clinical experts noted that this rarely occurs in clinical practice. |
Considerations for prescribing of therapy | |
The product monograph indicates dosing should not exceed one 60 g tube per week or one 100 g tube per 2 weeks. The recommended dose is a thin layer of ruxolitinib applied twice daily to affected skin areas up to a maximum of 10% of BSA for each application. Can dosing be increased to 3 to 4 times daily in the event of a lack of response? | The clinical experts noted that there is no evidence regarding the effect of increasing the dosage of ruxolitinib. Ruxolitinib would therefore be unlikely to be used more than twice a day, according to the product monograph. |
Should prescribing be limited to dermatologists? | One clinical expert indicated that referral to a dermatologist may be preferable. Both experts agreed that experience in the diagnosis and management of vitiligo is important for prescribing ruxolitinib treatment, as there are differential diagnoses that can mimic vitiligo presentation. |
Care provision issues | |
Phase III studies indicate that ruxolitinib is associated with acne and pruritus at the application site. Larger and longer trials are required to determine the effect and safety of ruxolitinib cream in patients with vitiligo. Would adverse effects be treated with a course of topical steroids and/or antiacne medications (e.g., topical benzyl peroxide)? | According to the clinical experts, adverse events such as acne would be treated in the same way as standard acne. Depending on the impact on the patient, first-line options could include topical antiacne treatments. |
BSA = body surface area; F-VASI = Facial Vitiligo Area Scoring Index; MACE = major adverse cardiovascular event; T-VASI = Total Vitiligo Area Scoring Index.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of 1.5% ruxolitinib topical cream for the treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older. The focus will be placed on comparing ruxolitinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ruxolitinib is presented in 2 sections, with CDA-AMC’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted LTE studies. No indirect evidence or additional studies addressing important gaps in the systematic review evidence were submitted.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
2 pivotal RCTs identified in the systematic review
1 open label treatment-extension period from the 2 pivotal RCTs
1 long-term extension study.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
Description of Studies
Two studies were identified and included in the systematic review. The TRuE-V1 study (n = 330) and the TRuE-V2 study (n = 344) were phase III, multicentre, double-blind, vehicle-controlled RCTs designed to evaluate the efficacy and safety of 1.5% ruxolitinib topical cream, applied twice daily to depigmented areas to treat nonsegmental vitiligo in adult and pediatric patients aged 12 years and older. The 2 studies had identical designs. The primary outcome was the proportion of patients achieving an F-VASI75 at week 24.
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | TRuE-V1 | TRuE-V2 |
|---|---|---|
Designs and populations | ||
Study design | Phase III, double-blind, vehicle-controlled RCTs | |
Locations | 45 study centres in North America and Europe, with 3 study sites (n = 18 patients) specific to Canada | 49 study centres in North America and Europe, with 5 study sites (n = 49 patients) specific to Canada |
Patient enrolment dates | Start date: September 20, 2019 End date: October 21, 2021 | Start date: October 3, 2019 End date: October 1, 2021 |
Randomized (N) | 330 patients randomized:
| 344 patients randomized:
|
Key inclusion criteria |
Patients who agree to discontinue all drugs used to treat vitiligo were screened through the final safety follow-up visit; over-the-counter preparations deemed acceptable by investigator and camouflage makeups were permitted. | |
Key exclusion criteria |
| |
Drugs | ||
Intervention | 1.5% ruxolitinib cream applied as a thin film twice daily to depigmented areas for 52 weeks | |
Comparator(s) | Vehicle cream applied as a thin film twice daily to depigmented areas for 24 weeks followed by 1.5% ruxolitinib cream applied as a thin film twice daily to depigmented areas for 28 weeks | |
Study duration | ||
Screening phase | Up to 32 days | |
Treatment phase | 24 weeks in the double-blind vehicle-controlled period 28 weeks in the open-label treatment-extension period | |
Follow-up phase | 30 (+ 7) days of safety follow-up | |
Outcomes | ||
Primary end point | Proportion of patients achieving F-VASI75 at week 24 | |
Secondary and exploratory end points | Key secondary
Additional secondary
Exploratory
| |
Publication status | ||
Publications | Ezzedine et al. (2022),47 Pandya et al. (2022),48 and Rosmarin et al. (2022)49 | |
Clinical trial record number | NCT04052425 | NCT04057573 |
AE = adverse event; DB = double-blind; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; F-PaGIC-V = Facial-Patient’s Global Impression of Change-Vitiligo; F-PhGVA = Facial-Physician Global Vitiligo Assessment; F-VASI = Facial Vitiligo Area Scoring Index; F-VASI25 = 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI50 = 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; HADS = Hospital Anxiety and Depression Scale; RCT = randomized controlled trial; TE = treatment-extension; T-PaGIC-V = Total-Patient’s Global Impression of Change-Vitiligo; T-PhGVA = Total-Physician Global Vitiligo Assessment; TSQM = Treatment Satisfaction Questionnaire for Medication; T-VASI = Total Vitiligo Area Scoring Index; T-VASI25 = 25% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI50 = 50% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI75 = 75% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI90 = 90% or more improvement from baseline on the Total Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life Instrument; VNS = Vitiligo Noticeability Scale; WHO-5 = World Health Organization–Five Well-Being Index.
Source: Incyte Corporation (2021),29,30 ClinicalTrials.gov (2022).50,51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The study design for the TRuE-V1 and TRuE-V2 trials is illustrated in Figure 1. Eligible patients were randomized in a 2:1 ratio to receive either ruxolitinib or a vehicle cream over the 24-week controlled treatment period. Randomization was performed using interactive response technology and stratified by baseline skin type (Fitzpatrick Scale skin type I and type II versus type III to type VI) and by region (North America or Europe). The double-blind controlled period was followed by a 28-week open-label treatment extension, in which patients who had been randomized to the vehicle cream crossed over to ruxolitinib. Results from the treatment-extension period (i.e., at 52 weeks) are presented in the LTE Studies section of this report.
Figure 1: TRuE-V1 and TRuE-V2 Study Design
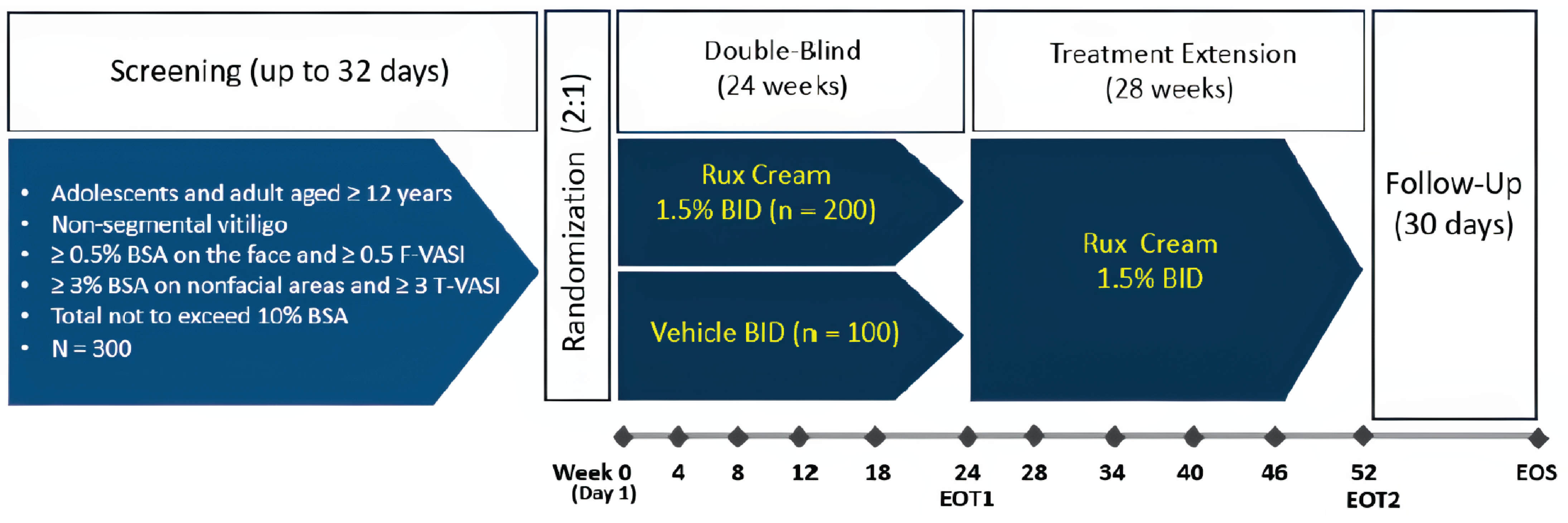
BID = twice a day; BSA = body surface area; EOT1 = first end of treatment; EOT2 = second end of treatment; EOS = end of safety; F-VASI = Facial Vitiligo Area Scoring Index; Rux = ruxolitinib; T-VASI = Total Vitiligo Area Scoring Index.
Source: TRuE-V1 and TRuE-V2 Clinical Study ReportsCSRs.29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Patients aged 12 years and older were eligible for the trial if they had nonsegmental vitiligo with depigmented areas covering at least 0.5% of the BSA on the face (F-VASI score of ≥ 0.5) and at least 3% of the BSA on nonfacial areas (T-VASI score of ≥ 3). The maximum total body vitiligo area allowed was 10% of the BSA, including both facial and nonfacial areas.
Patients were excluded from the trial if they had segmental vitiligo, complete leukotrichia within any lesions on the face, or a dermatologic disease confounding vitiligo assessment. Other key exclusion criteria included previous use of JAK inhibitors, as well as recent use of any biological or experimental therapy, phototherapy, immunomodulating treatments, or topical treatments.
Interventions
Patients received either 1.5% ruxolitinib topical cream or a matching administration-vehicle cream that did not contain the active drug. Both were applied as a thin film twice daily to the depigmented vitiligo areas identified for treatment at baseline and were continued even if the areas began to improve. Further vitiligo areas could be treated if they were created through an expansion of existing areas and as long as the total BSA affected did not exceed 10%.
No rescue therapy was allowed. Patients needed to discontinue all drugs used for the treatment of vitiligo from screening through the final safety follow-up visit. Over-the-counter preparations deemed acceptable by the investigator, as well as camouflage makeups, were permitted.
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important by the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, end points were selected that were considered to be most relevant to inform expert committee deliberations and end points were finalized in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From Studies Included in the Systematic Review
Outcome measure | Time point | TRuE-V1 andTRuE-V2 |
|---|---|---|
Primary outcome in the studies | ||
Proportion of patients achieving F-VASI75 | At week 24 | Primarya,b |
Key secondary outcomes in the studies | ||
Proportion of patients achieving F-VASI50 | At week 24 | Secondaryb |
Proportion of patients achieving F-VASI90 | Secondarya,b | |
Proportion of patients achieving T-VASI50 | Secondaryb | |
Proportion of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) at week 24 | Secondarya,b | |
Change from baseline in facial BSA | Secondaryb | |
Additional secondary outcomes in the studies | ||
Proportion of patients achieving F-VASI25 | At week 24 | Secondary |
Proportion of patients achieving T-VASI75 | ||
Change from baseline in DLQI | ||
Change from baseline in CDLQI | ||
Exploratory patient-reported outcomes in the studies | ||
TSQM | At week 24 | Exploratory |
Change from baseline in VitiQoL | Exploratorya | |
Safety outcomes in the studies | ||
Number of patients with AEs and SAEs | At week 24 | Secondarya |
AE = adverse event; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; F-VASI25 = 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI50 = 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; SAE = serious adverse event; TSQM = Treatment Satisfaction Questionnaire for Medication; T-VASI50 = 50% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI75 = 75% or more improvement from baseline on the Total Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale.
aOutcomes included in the Grading of Recommendations Assessment, Development and Evaluation assessment.
bStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Vitiligo Area Scoring Index
A VASI score may be determined for facial vitiligo (F-VASI) or total body vitiligo (T-VASI).
Facial Vitiligo Area Scoring
The primary outcome in the TRuE-V1 and TRuE-V2 studies was the proportion of patients achieving an F-VASI75 at week 24. The F-VASI is measured by the percentage of vitiligo involvement on the face, as a proportion of the BSA, and the degree of depigmentation.
The percentage of the BSA involved is assessed using the palmar method, in which the hand unit is based on the patient’s hand size. As such, a handprint represents 1% of BSA, while a thumbprint corresponds to 0.1% of BSA. For each site on the face with vitiligo involvement, the degree of depigmentation is then estimated; complete depigmentation results in a score of 100%, while the absence of any depigmentation yields a score of 0%. The percentage of BSA is multiplied by the degree of depigmentation and the products are summed together for each affected site on the face to produce a score from 0 to 3, with higher scores indicating higher involvement.
Total Vitiligo Area Scoring Index
The T-VASI includes contributions from all body regions, divided into 6 separate sites: head and neck, hands, upper extremities, trunk, lower extremities, and feet. For each body region, the percentage of BSA affected is determined and multiplied by the degree of depigmentation in the same way that was outlined for the F-VASI. The scores obtained for each of the 6 body regions are summed to produce a score between 0 and 100, with higher scores indicating higher involvement.
The VASI score was developed for clinical trial assessments. The clinical experts indicated that the VASI score is not used in clinical practice. The sponsor reported that the VASI score is a validated, sensitive, and quantitative approach for assessing the extent of depigmentation. Part of the validation analyses were performed using studies from the ruxolitinib drug development program, and additional validation studies were identified from the literature. A 75% improvement in F-VASI has been suggested as a threshold for treatment success based on perceptions of patients with vitiligo and dermatologists.52 In addition, evidence in the literature from a mixed-method study suggests that an F-VASI75 and T-VASI50 would likely result in a clinically meaningful change in repigmentation for patients with nonsegmental vitiligo.26 However, there are no known MCID values for between-group differences.
Vitiligo Noticeability Scale
The VNS, which measures patient-rated vitiligo noticeability on a 5-point scale, is a validated outcome designed to assess changes in the visibility of vitiligo lesions over time from a patient’s perspective.53 Patients compare their current skin appearance to its state before treatment and rate it on a scale where 1 indicates “more noticeable,” 2 “as noticeable,” 3 “slightly less noticeable,” 4 “a lot less noticeable,” and 5 “no longer noticeable.” A successful treatment outcome is typically reflected by a score of 4 or 5, signalling significant improvement in noticeability.53
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
The DLQI is a nonspecific, 10-item questionnaire that measures how much the skin condition has affected the patient over the previous 7 days. The instrument covers domains such as symptoms, daily activities, relationships, work or school, and emotional well-being. Each item is scored on a 4-point Likert scale (0 = not at all, 1 = a lot, 2 = a little, 3 = very much). The maximum total DLQI score is 30. Higher scores denote a greater negative impact on quality of life.
The DLQI has evidence to support its validity and reliability, but not responsiveness, in patients with vitiligo. Based on a review of the literature, no MCID has been established for this instrument in patients with vitiligo. However, in patients with various inflammatory skin conditions, the use of an anchor-based approach suggests that a within-group change of at least 4 points from baseline in the DLQI may be consistent with clinically meaningful improvements for patients.54
The CDLQI is similar to the DLQI but designed for children. It was used in the TRuE-V1 and TRuE-V2 trials to assess HRQoL in patients aged at least 12 years but younger than 16 years.
Vitiligo-Specific Quality of Life
The VitiQoL is a 15-item, specialized, patient-reported HRQoL assessment tool requiring patients to rate various aspects of their condition using a 7-point scale, ranging from 0 (“Not at all”) to 6 (“All of the time”). Questions relate to participation limitation, stigma, and behaviour. The total score can range from 0 to 90, with higher scores indicative of a greater negative impact on quality of life.55 The VitiQoL has evidence to support the validity and reliability, but not responsiveness, in patients with vitiligo. According to the literature, no MCID has been established for this instrument in patients with vitiligo.
Harms
The safety analysis included AEs, SAEs, and AEs leading to study discontinuation.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MCID |
|---|---|---|---|
VASI | The VASI is a clinical tool used to assess the extent of vitiligo by quantifying the depigmentation across various body regions. The body is divided into 6 regions: head and neck, upper limbs, trunk, lower limbs, hands, and feet; each contributing a specific percentage to the BSA. The extent of depigmentation within each region is estimated visually, and the score is based on the percentage of affected skin, ranging from 0% (no depigmentation) to 100% (complete depigmentation). The scoring also includes fractional values, such 0.5 for 50%. The VASI score is calculated by multiplying the percentage of affected area in each region by the fractional depigmentation score, with the total score being the sum of these regional scores. The scale helps to track the severity of vitiligo over time, providing a standardized method for monitoring treatment progress. VASI scores typically range from 0 (no depigmentation) to 100 (complete depigmentation). A version called F-VASI focuses on localized areas such as the face for a more targeted evaluation.56,57 | From the sponsor (studies on TRUE-V): The validity and reliability of F-VASI and T-VASI was assessed by measuring correlation to the facial and total PaGIC-V (7-point scale) and PhGVA (5-point scale) instruments.58,59 Validity: F-VASI and T-VASI differentiated well among most PhGVA categories (i.e., mild, moderate, severe) at baseline, demonstrating known-group validity.58,59 Reliability: Among patients whose condition remained stable (i.e., no change from baseline to week 24) according to the PaGIC-V, reliability was moderate to good for F-VASI (ICC = 0.891) and T-VASI (ICC = 0.768). Among patients whose condition remained stable as measured by the PhGVA, reliability was also moderate to good for F-VASI (ICC = 0.739) and T-VASI (ICC = 0.686).58,59 | In 1 study assessing the perceptions of patients with vitiligo and those of dermatologists, the vast majority of participants indicated that a 75% improvement in an F-VASI score over time would represent treatment success.52 One mixed-method study suggested that an F-VASI75 and T-VASI50 would be the MCID repigmentation threshold in patients with nonsegmental vitiligo.26 Lower thresholds have been reported in other studies. A post hoc analysis using data from the phase II study of ruxolitinib cream reported a respective change from baseline of 57% on the F-VASI as indicating a clinically meaningful change.45,60 |
VNS | The VNS is a 5-point patient-reported measure designed to assess changes in the visibility of vitiligo lesions over time, particularly during treatment. Patients compare their current skin appearance to its state before treatment and rate it on a scale where 1 indicates “more noticeable,” 2 “noticeable,” 3 “slightly less noticeable,” 4 “a lot less noticeable,” and 5 “no longer noticeable.” A successful treatment outcome is typically reflected by a score of 4 or 5, signalling significant improvement in noticeability.61 | A study53 using data from the HI-Light Vitiligo trial (N = 287) evaluated the validity, reliability, and acceptability of the VNS among participants with vitiligo. Validity: Construct validity was supported by a positive association between VNS scores and participant-reported global treatment success (kappa = 0.41 for success defined as ≥ 4; kappa = 0.71 for success defined as ≥ 3). The association was stronger for VNS (kappa = 0.41) compared to clinician-reported percentage repigmentation (kappa = 0.17), although blinded patient and public involvement panel assessments had weaker associations (kappa = 0.28).53 Reliability: Test-retest reliability was strong, with kappa = 0.69 (95% CI, 0.63 to 0.74). Age and skin phototype did not influence the interpretation of VNS scores.53 Responsiveness: Not estimated in patients with vitiligo. | In the study assessing correlation between global treatment success and VNS score, VNS scores of 4 and 5 were highly associated with treatment success among both patients and clinicians.53 |
DLQI | The DLQI is a 10-item questionnaire designed to assess the impact of skin conditions on an adult’s life over the past week. It covers a range of domains, including symptoms, daily activities, relationships, work and school, and emotional well-being. The CDLQI is a similar questionnaire designed to measure the impact of any skin disease on the lives of children. Each item is scored on a 4-point Likert scale (0 = not at all, 1 = a lot, 2 = a little, 3 = very much), with the total score ranging from 0 to 30. A higher score indicates a greater impact on quality of life. | A study of 70 vitiligo patients in Iran tested the reliability and validity of the 10-item DLQI questionnaire. Validity: Convergent validity was assessed, and the study found statistically significant differences in the mean scores of the PR scale based on the involvement of covered vs. uncovered areas (P = 0.016)62 Reliability: Reliability analysis showed satisfactory results, with a Cronbach alpha coefficient of 0.77.62 Reliability was also assessed in the original validation study of the DLQI by Finlay and Khan63 in a population of various skin diseases. The test-retest reliability correlation coefficients were high for both the overall score (Spearman rank correlation of 0.99) and for individual questions (0.95 to 0.98). Slightly lower correlation coefficients (ranging from 0.56 to 0.99) were reported in a later systematic review by Basra et al.54 Responsiveness: Not estimated in patients with vitiligo. | The literature includes no established MCID for the DLQI instrument in patients with NSV. However, a study by Basra et al. (2015)54 aimed to determine the MCID of DLQI in patients with inflammatory skin diseases such as psoriasis, eczema, and acne (N = 192) using an anchor-based approach. The study found that the mean change in DLQI was 3.3, indicating an MCID. These findings align with previous literature,64-66 which has established an MCID range for the DLQI of 2.2 to 5.7. |
VitiQoL | VitiQoL is a specialized, patient-reported HRQoL assessment tool designed to measure the impact of vitiligo on patients’ lives. It consists of 15 items, with each question rated on a 7-point scale, ranging from 0 (“Not at all”) to 6 (“All of the time”), to assess various aspects of the disease’s effect. The total score, which can range from 0 to 90, reflects the level of HRQoL, with higher scores indicating worse quality of life.55 | A study55 involving 90 patients with vitiligo aimed to develop and validate a vitiligo-specific HRQoL instrument, VitiQoL. Validity: Convergent validity was demonstrated by strong correlations between VitiQoL and external dermatology scales measuring similar constructs (Skindex-16, r = 0.82; DLQI, r = 0.83). Known-groups validity was evidenced by significant differences in the VitiQoL behaviour subscale between individuals with exposed vs. unexposed patches (P = 0.01). Concurrent validity was supported by a moderate correlation between self-reported severity and VitiQoL scores (r = 0.51).55 Reliability: The instrument showed high internal consistency, with a Cronbach alpha coefficient of 0.935.55 Responsiveness: Not estimated in patients with vitiligo. | There is currently no established MCID for the VitiQoL instrument in patients with NSV in the literature. |
BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; F-VASI = Facial Vitiligo Area Scoring Index; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MCID = minimal clinically important difference; NSV = nonsegmental vitiligo; PaGIC-V = Patient’s Global Impression of Change-Vitiligo; PhGVA = Physician Global Vitiligo Assessment; T-VASI = Total Vitiligo Area Scoring Index; VASI = Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale; vs. = versus.
Statistical Analysis
A summary of the statistical analyses is presented in Table 8.
End Point Analyses in the Studies and Handling of Missing Data
The primary analysis was based on the ITT population. Exact logistic regression was used to test the primary hypothesis for superiority of ruxolitinib compared with vehicle treatment on the percentage of patients achieving an F-VASI75 at 24 weeks. This model included the treatment group and stratification factors (geographic region and skin type). The unadjusted P value between ruxolitinib versus vehicle cream was compared at a 2-sided alpha of 0.05. The odds ratio and 95% CI in response rates (ruxolitinib versus vehicle cream) at week 24 were also computed. Rescue therapy was not allowed in the studies; as such, patients who received rescue therapy were considered to have a protocol violation and were imputed as a patient who did not respond to treatment. All patients who were missing the F-VASI assessment at a given visit in the double-blind period were handled using multiple imputation under a missing at random assumption. Missing scores were imputed by fully conditional specification. The multiple imputation method used treatment and observed stratification factors as predicators.
For the primary end point, 3 sensitivity and supportive analyses were laid out in the statistical analysis plan: the nonresponder imputation, which was used as an alternative method to handle missing data; the last observation carried forward, which was used for patients who were missing postbaseline values and where the last observed nonmissing postbaseline value was used to fill in missing values at week 24; and a tipping point analysis, which was conducted to examine the potential effects of missing data. In the latter analysis, the missing F-VASI75 response at week 24 in each treatment group was replaced by a range of values from the most conservative to the most aggressive case. The most conservative case was that all missing patients who applied ruxolitinib were patients who did not respond to treatment and all missing patients who applied the vehicle cream were patients who did respond to treatment, while the opposite was true for the most aggressive case. For each scenario, between-treatment comparisons were performed using a Fisher exact test.
Key secondary efficacy end points (e.g., F-VASI50, F-VASI90, and VNS response) were analyzed using methods similar to those outlined for primary efficacy end points. For other secondary efficacy analyses, summary statistics were presented. Continuous outcomes were planned to be analyzed using a mixed-effect model with repeated measurements, if applicable. Categorical outcomes were planned to be analyzed using exact logistic regression models similar to those specified in the primary and key secondary analyses, if applicable.29,30
Sample Size and Power Calculation
For the TRuE-V1 and TRuE-V2 trials, a sample size of approximately 300 patients randomized in a 2:1 ratio to ruxolitinib or vehicle cream and stratified by baseline skin type and region was determined to provide sufficient statistical power (> 85%) to detect a difference between ruxolitinib and vehicle cream in primary and key secondary end points. A Fisher exact test with a 2-sided alpha of 0.05 was used to provide a conservative evaluation of statistical power and was accurate when the expected number of patients who did respond to treatment in the vehicle group was small.
Statistical Testing and Multiplicity
Data from the 2 treatment groups were compared using exact logistic regression for binary end points, presented with relative risk, and were compared with the use of an analysis of covariance model for facial BSA affected by vitiligo.
A gatekeeping testing strategy for the primary and key secondary analyses was implemented to control the overall type I error rate. These end points were tested in a fixed sequence at a 2-sided alpha of 0.05 in the following order:
Proportion of patients achieving F-VASI75 at week 24.
Proportion of patients achieving F-VASI50 at week 24.
Proportion of patients achieving F-VASI90 at week 24.
Proportion of patients achieving T-VASI50 at week 24.
VNS response at week 24.
Percent change from baseline in facial BSA at week 24.
Each end point was tested only if the null hypothesis for the primary end point and the secondary end points in previous steps was rejected. All additional secondary and exploratory end points were reported as observed without adjustments for multiplicity and analyses were summarized with the use of descriptive statistics.
Subgroup Analyses
Subgroup analyses were performed for the primary end point based on the following patient characteristics and baseline variables:
skin type (Fitzpatrick Scale skin types I and II versus types III, IV, V, and VI)
age category I (12 to 17 years, ≥ 18 to 64 years, and ≥ 65 years)
age category II (≤ 40 years and > 40 years)
region (North America and Europe)
sex
race (Asian, Black or African American, white, not reported, and other)
facial BSA (< 1.5% and ≥ 1.5%).
Subgroup efficacy analyses were prespecified; however, the statistical tests that were performed to evaluate differences in effects between subgroups were not reported. Whether the comparability of the treatment arms was checked, and whether multiplicity was taken into account, was not reported for either trial.
Changes in Statistical Methods and Planned Analyses After Unblinding or Database Lock
The data from patients at TRuE-V2 study site 710 (N = 13) were removed from all efficacy analyses performed on the ITT population because of noncompliance with the study protocol resulting in serious concerns with the data quality. However, data from these patients were included in all safety analyses because all patients applied the study drug at least once.
Table 8: Statistical Analysis of Efficacy End Points in the TRuE-V1 and TRuE-V2 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
TRuE-V1 and TRuE-V2 trials | ||||
F-VASI75 | Exact logistic regression | Treatment group, geographic region, skin type, baseline, and postbaseline F-VASI | Patients who were missing an assessment at the given time point were considered to have missing data. Imputation was performed as multiple imputation under the missing at random assumption | Nonresponder imputation, last observation carried forward, tipping point |
F-VASI25, F-VASI50, FVASI90 T-VASI25, T-VASI50 T-VASI90 VNS response | ||||
Percentage change from baseline in facial BSA | ANCOVA | Treatment group, stratification factors, and baseline value | NA | |
Change from baseline in F-VASI and T-VASI, facial BSA and total BSA, DLQI and CDLQI | Summary statistics (sample size, mean, standard deviation, median, minimum and maximum, quartiles, 95% CI) | NA | NR | NA |
Change from baseline in VitiQoL | Descriptive statistics | NA | NR | NA |
TEAEs | Descriptive statistics | NA | NR | NA |
ANCOVA = analysis of covariance; BSA = body surface area; CI = confidence interval; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; F-VASI = Facial Vitiligo Area Scoring Index; F-VASI25 = 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI50 = 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; NA = not applicable; NR = not reported; TEAE = treatment-emergent adverse event; T-VASI = Total Body Vitiligo Area Scoring Index; T-VASI25 = 25% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI50 = 50% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI90 = 90% or more improvement from baseline on the Total Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
Table 9: Analysis Populations of the TRuE-V1 and TRuE-V2 Trials
Population | Definition | Application |
|---|---|---|
Intention to treat | All patients who were randomized; treatment groups were defined according to the treatment assignment at the time of randomization | Study population and characteristics, and efficacy data |
Full analysis set | All patients who were randomized and had baseline and any postbaseline assessments; treatment groups were defined according to the treatment assignment at the time of randomization | Summary of analyses of demographics, baseline characteristics, participant disposition and all efficacy dataa |
Per protocol | Patients in the full analysis set population who were sufficiently compliant with the protocol | Supportive sensitivity analyses for primary efficacy end point in the double-blind period |
Safety | All patients who applied ruxolitinib cream or vehicle cream at least once; treatment groups were defined according to the treatment applied on day 1 | Safety data |
aThe primary analysis population for demographics, baseline characteristics, and efficacy analyses was changed from the full analysis set to the intention-to-treat population after unblinding and the database lock based on feedback from the FDA.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Of the 452 patients screened for the TRuE-V1 trial, 122 (27.0%) did not meet the eligibility criteria while 330 (73.0%) were randomized in the study. In the TRuE-V2 trial, 138 (28.6%) of the 482 patients screened did not meet the eligibility criteria and 344 (71.4%) were initially randomized. However, 13 patients from 1 study site, including 7 patients (3.2%) who applied ruxolitinib and 6 patients (5.5%) who applied vehicle cream, were excluded from the efficacy analyses due to serious protocol violations. No further details were reported. As a result, the randomized efficacy population consisted of 331 patients.
Numerically more patients who did not receive active treatment discontinued the TRuE-V1 study (12.7% with ruxolitinib and 17.4% with vehicle cream), and these proportions were similar in the TRuE-V2 trial (13.1% with ruxolitinib and 13.8% with vehicle cream). The most frequent reasons for discontinuation were withdrawal by patient, which was numerically higher in patients who applied vehicle cream in both studies, as well as those lost to follow-up. Details are supplied in Table 10.
Table 10: Summary of Patient Disposition From Studies Included in the Systematic Review
Patient disposition | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib (N = 221) | Vehicle cream (N = 109) | Ruxolitinib (N = 222) | Vehicle cream (N = 109) | |
Screened, N | 452 | 482 | ||
Did not meet eligibility criteria, n | 122 | 138 | ||
Initially randomized, n | 221 | 109 | 229 | 115 |
Study site excluded post hoc due to serious protocol violation, n | 0 | 0 | 7 | 6 |
Randomized and included in the efficacy analyses, N | 221 | 109 | 222 | 109 |
Not treated, n | 0 | 0 | 1 | 0 |
Discontinued the study during the double-blind period, n (%) | 28 (12.7) | 19 (17.4) | 29 (13.1) | 15 (13.8) |
Reason for discontinuation, n | ||||
Adverse events | 0 | 1 (0.9) | 2 (0.9) | 0 |
Lack of efficacy | 0 | 1 (0.9) | 0 | 1 (0.9) |
Protocol violation | 1 (0.5) | 0 | 0 | 0 |
Lost to follow-up | 14 (6.3) | 7 (6.4) | 11 (5.0) | 4 (3.7) |
Withdrawal by patient | 9 (4.1) | 10 (9.2) | 10 (4.5) | 8 (7.3) |
Physician decision | 1 (0.5) | 0 | 1 (0.5) | 0 |
COVID-19 pandemic | 3 (1.4) | 0 | 3 (1.4) | 1 (0.9) |
Pregnancy | 0 | 0 | 1 (0.5) | 0 |
Other | 0 | 0 | 1 (0.5) | 1 (0.9) |
Intention to treat (without excluded study site), N | 221 | 109 | 222 | 109 |
Per protocol, N | 191 | 89 | 203 | 99 |
Safety, N | 221 | 109 | 228 | 115 |
Source: Incyte Corporation (2021).29,30
Baseline Characteristics
The baseline characteristics that are most relevant to this review or that may affect the interpretation of the study results are outlined in Table 11.
The mean age of patients was 40.2 years (standard deviation [SD] = 15.85 years) in the TRuE-V1 trial and 38.7 years (SD = 14.46 years) in the TRuE-V2 trial, with patients ranging in age from 12 to 79 years. A total of 36 patients were aged younger than 18 years in the TRuE-V1 trial (25 patients who applied ruxolitinib and 11 patients who applied vehicle cream) as well as in the TRuE-V2 trial (30 patients who applied ruxolitinib and 6 patients who applied vehicle cream). The majority of patients included in the studies were white, had a Fitzpatrick Scale skin type of II, III, or IV, and had a facial BSA involvement of less than 1.5%. The median times since diagnosis were 11.1 years in the TRuE-V1 trial and 13.0 years in the TRuE-V2 trial, with 58% of patients in the TRuE-V1 trial and 65% of patients in the TRuE-V2 trial having received prior therapy for vitiligo.
Overall, baseline characteristics appeared to be balanced between treatment groups in the TRuE-V1 and TRuE-V2 trials. Although there was a slight numerical difference in the proportions of male and female patients in the TRuE-V1 trial, the clinical experts did not expect this to affect the course of the disease and/or response to treatment. However, according to experts, the impact of a visible skin condition such as vitiligo on quality of life may vary according to several different factors, including sex. The clinical experts indicated that patients included in the trials likely constituted a selected sample of the patient population seen in clinical practice, particularly as the proportions of patients with darker skin tones were small. Although vitiligo affects all races and types of skin tones, the experts noted that patients with darker skin tones are likely to present with an increased impact on quality of life, which can be attributed to the increased visibility of the condition.
Table 11: Summary of Baseline Characteristics From Studies Included in the Systematic Review (ITT Population)
Characteristic | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib (N = 221) | Vehicle cream (N = 109) | Ruxolitinib (N = 222) | Vehicle cream (N = 109) | |
Age (years) | ||||
Mean (SD) | 40.5 (15.44) | 39.7 (16.71) | 38.4 (15.44) | 39.4 (12.29) |
Median (minimum to maximum) | 40.0 (12 to 79) | 38.0 (12 to 79) | 38.0 (12 to 77) | 39.0 (13 to 68) |
Age category (years) | ||||
< 18, n (%) | 25 (11.3) | 11 (10.1) | 30 (13.5) | 6 (5.5) |
Sex, n (%) | ||||
Female | 136 (61.5) | 50 (45.9) | 110 (49.5) | 58 (53.2) |
Male | 85 (38.5) | 59 (54.1) | 112 (50.5) | 51 (46.8) |
Fitzpatrick Scale skin type,a n (%) | ||||
I | 10 (4.5) | 3 (2.8) | 2 (0.9) | 0 (0.0) |
II | 74 (33.5) | 40 (36.7) | 56 (25.2) | 32 (29.4) |
III | 89 (40.3) | 43 (39.4) | 86 (38.7) | 43 (39.4) |
IV | 34 (15.4) | 15 (13.8) | 55 (24.8) | 23 (21.1) |
V | 11 (5.0) | 7 (6.4) | 16 (7.2) | 9 (8.3) |
VI | 3 (1.4) | 1 (0.9) | 7 (3.2) | 2 (1.8) |
Race,b n (%) | ||||
Asian | 5 (2.3) | 4 (3.7) | 11 (5.0) | 6 (5.5) |
American Indian or Alaska Native | 1 (0.5) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
Black or African American | 11 (5.0) | 4 (3.7) | 11 (5.0) | 5 (4.6) |
Native Hawaiian or Pacific Islander | NR | NR | 2 (0.9) | 0 (0.0) |
White | 180 (81.4) | 96 (88.1) | 178 (80.2) | 88 (80.7) |
Not reported | 16 (7.2) | 3 (2.8) | 3 (1.4) | 3 (2.8) |
Other | 8 (3.6) | 2 (1.8) | 16 (7.2) | 7 (6.4) |
Years since diagnosis | ||||
Median (minimum to maximum) | 10.60 (0.0 to 60.5) | 11.96 (0.1 to 47.5) | 12.28 (0.0 to 50.3) | 14.87 (0.0 to 59.5) |
Disease status, n (%) | ||||
Stable | 165 (74.7) | 80 (73.4) | 159 (71.6) | 82 (75.2) |
Progressive | 56 (25.3) | 29 (26.6) | 63 (28.4) | 27 (24.8) |
F-VASI score | ||||
Mean (SD) | 0.932 (0.5813) | 0.999 (0.5942) | 0.898 (0.5256) | 0.834 (0.5342) |
Median (minimum to maximum) | 0.690 (0.40 to 3.00) | 0.740 (0.50 to 2.70) | 0.700 (0.45 to 3.00) | 0.600 (0.50 to 3.00) |
Facial BSA involvement, proportion of the total body | ||||
Mean (SD) | 1.05 (0.692) | 1.15 (0.710) | 0.98 (0.571) | 0.92 (0.582) |
Median (minimum to maximum) | 0.80 (0.5 to 3.0) | 0.90 (0.5 to 3.0) | 0.80 (0.5 to 3.0) | 0.70 (0.5 to 3.0) |
Facial BSA involvement, n (%) | ||||
< 1.5% | 172 (77.8) | 86 (78.9) | 183 (82.4) | 93 (85.3) |
1.5% | 49 (22.2) | 23 (21.1) | 39 (17.6) | 16 (14.7) |
T-VASI score | ||||
Mean (SD) | 6.489 (2.0228) | 6.424 (1.9241) | 6.790 (2.0435) | 6.979 (2.1953) |
Median (minimum to maximum) | 6.380 (3.01 to 10.00) | 6.250 (3.06 to 10.00) | 7.125 (3.00 to 10.00) | 7.290 (3.10 to 10.00) |
Total BSA involvement, proportion of the total body | ||||
Mean (SD) | 7.28 (2.033) | 7.22 (2.008) | 7.38 (2.025) | 7.66 (2.040) |
Median (minimum to maximum) | 7.70 (3.2 to 10.0) | 7.30 (3.7 to 10.0) | 7.70 (3.5 to 10.0) | 8.30 (3.6 to 10.1) |
Prior therapy for vitiligo, n (%) | ||||
No | 90 (40.7) | 48 (44.0) | 81 (36.5) | 34 (31.2) |
Yes | 131 (59.3) | 61 (56.0) | 141 (63.5) | 75 (68.8) |
BSA = body surface area; F-VASI = Facial Vitiligo Area Scoring Index; ITT = intention to treat; SD = standard deviation; T-VASI = Total Vitiligo Area Scoring Index.
aFitzpatrick Scale: Type I: always burns, never tans (pale peach; blond or red hair; blue eyes; freckles); type II: usually burns, tans minimally (peach; fair; blond or red hair; blue, green, or hazel eyes); type III: sometimes mild burn, tans uniformly (light brown; fair with any hair or eye colour); type IV: burns minimally, always tans well (moderate brown); type V: very rarely burns, tans very easily (dark brown); type VI: never burns, always tans (deeply pigmented dark brown to darkest brown).
bCategories are as reported in study.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Exposure to study treatment is outlined in Table 12. The median exposure duration during the double-blind treatment period was 168 days in both studies, which was consistent with the planned duration of 24 weeks (168 days). Patient exposure suggests notable variation among patients in the amount of the study drug applied, which appeared to be inconsistent with the usage of topical treatments in clinical practice according to the clinical experts. The experts consulted for this review indicated that it is uncertain if drug usage observed in the trials would be mirrored in real-world practice, but that it is likely that drug usage will be higher in the real-world setting. No additional detail was provided by the sponsor to explain the high level of variability observed.
Table 12: Summary of Patient Exposure From Studies Included in the Systematic Review (ITT Population) [Redacted]
████████ | ███████ | ███████ | ||
|---|---|---|---|---|
██████ | █████ | ████████ | ███████ | |
████████ ██ █████████ ██████ ███ ██ ██████ ██████ | ||||
██████ | ██████ | ██████ | ██████ | ██████ |
██████ | ██████ | ██████ | ██████ | ██████ |
██████ ██ █████ ████ ███████ ██████ ███ ██ ██████ ███ | ||||
██████ | ||||
██████ | ██████ | ██████ | ██████ | ██████ |
██████ | ██████ | ██████ | ██████ | ██████ |
█████ | ||||
██████ | ██████ | ██████ | ██████ | ██████ |
██████ | ██████ | ██████ | ██████ | ██████ |
███████████ ██████████ ███ | ||||
██████ | ██████ | ██████ | ██████ | ██████ |
██ █ █████████████ ███ █ ████████ ███ █ ████████ ██ █ ████████ ███████████
ITT = intention to treat.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Prior Medications
The most frequently used prior medications for vitiligo in the TRuE-V1 trial were tacrolimus (█████), pimecrolimus (████), triamcinolone (████), clobetasol (████), hydrocortisone (████), and mometasone furoate (████). Similarly, in the TRuE-V2 trial, these were tacrolimus (█████), pimecrolimus (████), hydrocortisone (████), and clobetasol (████). In both trials, the most common reason for discontinuing prior vitiligo treatment was lack of efficacy (█████ ███ █████, respectively).
Concomitant Medications
Most patients received concomitant medications in the studies; the most commonly used are presented in Table 13. Slight numerical differences were evident between treatment arms. However, overall the clinical experts did not expect these differences to have a clinically meaningful impact on the disease course or the treatment response.
Table 13: Summary of Most Commonly Used Concomitant Medications From Studies Included in the Systematic Review (ITT Population) [Redacted]
████████ | ███████ | ███████ | ||
|---|---|---|---|---|
████████ | ██████ | █████████ | █████ | |
███████████ ███████████ ██████ ███ ██ ███████ █ ███ | ||||
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
████ ████████ █████ █ ███ | ||||
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
███ █ ██████████████████████████████ ██ █ ██████ ██████ ███████ ████████████████████████ ███
ITT = intention to treat.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Detailed efficacy results for the double-blind treatment period, at 24 weeks, are presented in Table 14. Results for the open-label treatment-extension period, at 52 weeks, are presented in the LTE Studies section of this report.
Vitiligo Area Scoring Index
Proportions of Patients Achieving F-VASI75
Throughout the TRuE-V1 24-week double-blind treatment period, 60 patients (30.8%) who applied ruxolitinib achieved an F-VASI75, compared with 7 patients (7.8%) who applied vehicle cream. The TRuE-V2 trial yielded consistent results, with 62 patients (31.2%) who applied ruxolitinib achieving an F-VASI75, compared with 11 patients (11.2%) who applied vehicle cream. The between-group differences in the response rate were 22.3% (standard error [SE] = 4.15; 95% CI, 14.2 to 30.5; P < 0.0001) in the TRuE-V1 trial and 19.5% (SE = 4.56; 95% CI, 10.5 to 28.4; P = 0.0004) in the TRuE-V2 trial. The use of an F-VASI75, which was identified in the literature as the threshold for clinically meaningful change in repigmentation,26 was considered relevant by the clinical experts to assess the objective response to treatment.
Sensitivity Analyses
The various sensitivity analyses aligned with the main analysis. In the TRuE-V1 trial, the between-group differences in response rate were 20.7% (SE = 3.80%) for the nonresponder imputation analysis and 21.5% (SE = 4.00%) for the last observation carried forward analysis. In the TRuE-V2 trial, the between-group differences in the response rate were 17.8% (SE = 4.17%) for the nonresponder imputation analysis and 18.0% (SE = 4.41%) for the last observation carried forward analysis.
The tipping point at which significance would have been lost required extreme assumptions in both studies. In the TRuE-V1 trial, 63.2% of patients (12 of 19) with missing data in the vehicle group would need to be patients who did respond to treatment, while the corresponding proportion would need to be no more than 3.8% of patients (1 of 26) in the ruxolitinib group. In the TRuE-V2 trial, 81.8% of patients (9 of 11) with missing data in the vehicle group would need to be patients who did respond to treatment, while the corresponding proportion would need to be no more than 4.3% (1 of 23) in the ruxolitinib group.
Subgroup Analyses
Subgroup analyses identified by the clinical experts relevant in the context of vitiligo included race (Asian; Black or African American) and skin type (Fitzpatrick Scale skin type III, IV, V, and VI). In the TRuE-V1 trial, 5 of 11 patients (45.5%) of Black or African American ethnicity who applied ruxolitinib achieved an F-VASI75, compared with none of the 3 patients (0.0%) who applied the vehicle cream (response rate difference of 45.5%; SE = 15.01%). In patients of Asian ethnicity, 2 of 4 patients (50.0%) who applied ruxolitinib and none of the 2 patients (0.0%) who applied the vehicle cream achieved the outcome (response rate difference of 50.0%; SE = 25.00%). Finally, in patients with Fitzpatrick Scale skin type III, IV, V, and VI skin, the corresponding proportions were 41 of 120 patients (34.2%) who applied ruxolitinib, compared with 3 of 55 patients (5.5%) who applied the vehicle cream (response rate difference of 28.7%; SE = 5.30%).
In the TRuE-V2 trial, 4 of 10 patients (40.0%) of Black or African American ethnicity who applied ruxolitinib achieved an F-VASI75, compared with 1 of 3 patients (33.3%) who applied the vehicle cream (response rate difference of 6.7%; SE = 31.32%). In patients of Asian ethnicity, 3 of 11 patients (27.3%) who applied ruxolitinib and none of the 5 patients (0.0%) who applied the vehicle cream achieved the outcome (response rate difference of 27.3%; SE = 13.43%). Finally, in patients with Fitzpatrick Scale skin type III, IV, V, and VI skin, the corresponding proportion was 45 of 147 patients (30.6%) who applied ruxolitinib, compared with 10 of 70 patients (14.3%) who applied the vehicle cream (response rate difference of 16.3%; SE = 5.65%).
Proportions of Patients Achieving F-VASI90
In the TRuE-V1 trial, 31 patients (15.9%) who applied ruxolitinib achieved an F-VASI90, compared with 2 patients (2.2%) who applied the vehicle cream. The TRuE-V2 trial produced consistent results with 33 patients (16.6%) who applied ruxolitinib achieving an F-VASI90, compared with 1 patient (1.0%) who applied the vehicle cream. The between-group differences in the response rate were 13.2% (SE = 2.89; 95% CI, 7.5 to 18.8; P = 0.0038) in the TRuE-V1 trial and 15.0% (SE = 2.92; 95% CI, 9.3 to 20.7; P = 0.0065) in the TRuE-V2 trial.
Proportions of Patients Achieving F-VASI50
In the TRuE-V1 trial, 102 patients (52.3%) who applied ruxolitinib achieved an F-VASI50, compared with 15 patients (16.7%) who applied the vehicle cream. Results in the TRuE-V2 trial were consistent, with 102 patients (51.3%) who applied ruxolitinib achieving an F-VASI50, compared with 20 patients (20.4%) who applied the vehicle cream. The between-group differences in the response rate were 34.2% (SE = 5.18; 95% CI, 24.1 to 44.4; P < 0.0001) in the TRuE-V1 trial and 30.6% (SE = 5.36; 95% CI, 20.0 to 41.1; P < 0.0001) in the TRuE-V2 trial.
Proportions of Patients Achieving F-VASI25
In the TRuE-V1 trial, 138 patients (70.8%) who applied ruxolitinib achieved an F-VASI25, compared with 25 (27.8%) who applied the vehicle cream. In the TRuE-V2 trial, 125 patients (62.8%) who applied ruxolitinib achieved an F-VASI25, compared with 31 (31.6%) who applied the vehicle cream. The between-group differences in the response rate were 39.8% (SE = 5.74; 95% CI, 28.6 to 51.1; P < 0.0001) in the TRuE-V1 trial and 31.9% (SE = 5.74; 95% CI, 20.7 to 43.2; P < 0.0001) in the TRuE-V2 trial.
Proportions of Patients Achieving T-VASI75
In the TRuE-V1 trial, 9 patients (4.6%) who applied ruxolitinib achieved a T-VASI75, compared with 2 patients (2.2%) who applied the vehicle cream. In the TRuE-V2 trial, 17 patients (8.5%) who applied ruxolitinib achieved a T-VASI75, compared with 2 patients (2.0%) who applied the vehicle cream. The between-group differences in response rate were 2.3% (SE = 1.86; 95% CI, −1.4 to 5.9; P = 0.2921) in the TRuE-V1 trial and 6.1% (SE = 2.25; 95% CI, 1.7 to 10.5; P = 0.0436) in the TRuE-V2 trial.
Proportions of Patients Achieving T-VASI50
In the TRuE-V1 trial, 43 patients (22.1%) who applied ruxolitinib achieved a T-VASI50, compared with 4 patients (4.4%) who applied the vehicle cream. In the TRuE-V2 trial, 49 patients (24.6%) who applied ruxolitinib achieved a T-VASI50, compared with 7 patients (7.1%) who applied the vehicle cream. The between-group differences in the response rate were 15.5% (SE = 3.63; 95% CI, 8.3 to 22.6; P = 0.0020) in the TRuE-V1 trial and 17.1% (SE = 3.87; 95% CI, 9.5 to 24.7; P = 0.0006) in the TRuE-V2 trial.
Facial Body Surface Area
Percent Change From Baseline in Facial BSA
The least squares mean percent changes from baseline through 24 weeks in facial BSA were −28.9% (SE = 2.22%) in patients who applied ruxolitinib and −9.5% (SE = 3.25%) in patients who applied the vehicle cream in the TRuE-V1 trial, for a between-group difference of −19.3% (95% CI, −27.05% to −11.64%; P < 0.0001). Similarly, in the TRuE-V2 trial, the corresponding changes from baseline were −26.4% (SE = 2.57%) in patients who applied ruxolitinib and −7.0% (SE = 3.82%) in patients who applied the vehicle cream, for a between-group difference of −19.5% (95% CI, −28.46% to −10.45%; P < 0.0001).
Vitiligo Noticeability Scale
Proportions of Patients Achieving a VNS of “A Lot Less Noticeable” or “No Longer Noticeable”
In the TRuE-V1 trial, 49 patients (25.1%) who applied ruxolitinib achieved a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) compared with 3 patients (3.3%) who applied the vehicle cream. The TRuE-V2 trial yielded consistent results, with 41 patients (20.6%) who applied ruxolitinib achieving the outcome, compared with 5 patients (5.1%) who applied the vehicle cream. The between-group differences in response rate were 21.2% (SE = 3.54; 95% CI, 14.3 to 28.1; P = 0.0002) in the TRuE-V1 trial and 15.5% (SE = 3.58; 95% CI, 8.5 to 22.6; P = 0.0013) in the TRuE-V2 trial.
Treatment Satisfaction Questionnaire for Medication
Treatment Satisfaction Questionnaire for Medication Score at Week 24
The least squares mean Treatment Satisfaction Questionnaire for Medication (TSQM) scores at week 24 were 65.68 (SE = 1.62) in patients who applied ruxolitinib and 51.47 (SE = 2.37) in patients who applied the vehicle cream in the TRuE-V1 trial, for a between-group difference of 14.21 (95% CI, 8.6 to 19.9; P < 0.0001). However, in the TRuE-V2 trial, the corresponding TSQM scores were 61.09 (SE = 1.66) in patients who applied ruxolitinib and 48.51 (SE = 2.36) in patients who applied the vehicle cream, for a between-group difference of 12.59 (95% CI, 6.9 to 18.3; P < 0.0001).
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
Change From Baseline in DLQI (Aged 16 Years)
The mean changes from baseline through 24 weeks in DLQI were −1.15 (SD = 3.796) in patients who applied ruxolitinib and −0.79 (SD = 3.355) in patients who applied the vehicle cream in the TRuE-V1 trial, for a between-group difference of −0.32 (95% CI, −1.26 to 0.62; P = 0.4970). Similarly, in the TRuE-V2 trial, the corresponding changes from baseline were −1.19 (SD = 4.075) in patients who applied ruxolitinib and −0.71 (SD = 3.988) in patients who applied the vehicle cream, for a between-group difference of −0.60 (95% CI, −1.60 to 0.39; P = 0.2350). The maximum total DLQI score was 30, with higher scores denoting a greater negative impact on quality of life. The clinical experts consulted for this review did not consider the magnitude of the between-group difference to be clinically meaningful.
Change From Baseline in CDLQI (Aged Younger Than 16 Years)
The mean changes from baseline through 24 weeks in CDLQI were −0.25 (SD = 2.113) in the 16 patients aged younger than 16 years who applied ruxolitinib and 0.00 (0.00) in the 3 patients in this age group who applied the vehicle cream in the TRuE-V1 trial. In the TRuE-V2 trial, the corresponding changes from baseline were 0.00 (SD = 1.837) in the 17 patients who applied ruxolitinib and −2.33 (SD = 8.505) in the 3 patients who applied the vehicle cream. No between-group difference was reported for this outcome in either study.
Vitiligo-Specific Quality of Life
Change From Baseline in VitiQoL
The mean changes from baseline through 24 weeks in VitiQoL were −6.34 (SD = 17.433) in patients who applied ruxolitinib and −6.37 (SD = 15.988) in patients who applied the vehicle cream in the TRuE-V1 trial, for a between-group difference of −0.28 (95% CI, −4.51 to 3.95; P = 0.8976). In the TRuE-V2 trial, the corresponding changes from baseline were −6.07 (SD = 18.148) in patients who applied ruxolitinib and −2.64 (SD = 14.462) in patients who applied the vehicle cream, for a between-group difference of −3.52 (95% CI, −7.60 to 0.57; P = 0.0915). VitiQoL scores range from 0 to a maximum of score of 90, where higher scores denote a greater negative impact on quality of life. The clinical experts consulted for this review did not consider the magnitude of the between-group difference to be clinically meaningful.
Post Hoc HRQoL Analyses
Findings from post hoc analyses in patients who received ruxolitinib compared the change in VitiQoL and DLQI from baseline to week 24 between patients who achieved various levels of F-VASI and those who did not. Results suggest that patients who achieved at least an F-VASI75 may observe an improvement in their HRQoL; however, whether this improvement is clinically meaningful is uncertain. In addition, interpretation of these findings is limited by the post hoc nature of the analyses. Detailed results are presented in Appendix 1.
Table 14: Summary of Key Efficacy Results From Studies Included in the Systematic Review (ITT Population)
Variable | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 221 | Vehicle cream N = 109 | Ruxolitinib N = 222 | Vehicle cream N = 109 | |
Number of patients contributing to the analyses, N | 195 | 90 | 199 | 98 |
Vitiligo Area Scoring Index | ||||
Proportions of patients achieving F-VASI75 at week 24 (primary outcome in the studies) | ||||
Achieving response, n (%) | 60 (30.8) | 7 (7.8) | 62 (31.2) | 11 (11.2) |
Achieving response, % (SE)a | 29.8 (3.21) | 7.4 (2.65) | 30.9 (3.27) | 11.4 (3.20) |
Difference in response rate (SE) | 22.3 (4.15) | 19.5 (4.56) | ||
95% CI | 14.214 to 30.471 | 10.537 to 28.420 | ||
Difference odds ratio (95% CI)b | 5.28 (2.341 to 11.903) | 3.45 (1.737 to 6.835) | ||
P valuec | < 0.0001 | 0.0004 | ||
Proportions of patients achieving F-VASI90 at week 24 | ||||
Achieving response, n (%) | 31 (15.9) | 2 (2.2) | 33 (16.6) | 1 (1.0) |
Achieving response, % (SE) | 15.3 (2.50) | 2.2 (1.51) | 16.3 (2.62) | 1.3 (1.25) |
Difference in response rate (SE) | 13.2 (2.89) | 15.0 (2.92) | ||
95% CI | 7.497 to 18.839 | 9.250 to 20.702 | ||
Difference odds ratio (95% CI)b | 8.49 (1.997 to 36.048) | 15.29 (2.150 to 108.739) | ||
P valuec | 0.0038 | 0.0065 | ||
Proportions of patients achieving F-VASI50 at week 24 | ||||
Achieving response, n (%) | 102 (52.3) | 15 (16.7) | 102 (51.3) | 20 (20.4) |
Achieving response, % (SE) | 51.2 (3.46) | 16.9 (3.89) | 51.4 (3.50) | 20.9 (4.06) |
Difference in response rate (SE) | 34.2 (5.18) | 30.6 (5.36) | ||
95% CI | 24.092 to 44.408 | 20.048 to 41.061 | ||
Difference odds ratio (95% CI)b | 5.18 (2.831 to 9.482) | 3.99 (2.296 to 6.949) | ||
P valuec | < 0.0001 | < 0.0001 | ||
Proportions of patients achieving F-VASI25 at week 24 | ||||
Achieving response, n (%) | 138 (70.8) | 25 (27.8) | 125 (62.8) | 31 (31.6) |
Achieving response, % (SE) | 69.8 (3.23) | 30.0 (4.68) | 63.9 (3.41) | 32.0 (4.58) |
Difference in response rate (SE) | 39.8 (5.74) | 31.9 (5.74) | ||
95% CI | 28.590 to 51.108 | 20.699 to 43.193 | ||
Difference odds ratio (95% CI)b | 5.56 (3.226 to 9.578) | 3.76 (2.267 to 6.246) | ||
P valuee | < 0.0001 | < 0.0001 | ||
Proportions of patients achieving T-VASI75 at week 24 | ||||
Achieving response, n (%) | 9 (4.6) | 2 (2.2) | 17 (8.5) | 2 (2.0) |
Achieving response, % (SE) | 4.1 (1.34) | 1.8 (1.29) | 8.0 (1.85) | 1.8 (1.29) |
Difference in response rate (SE) | 2.3 (1.86) | 6.1 (2.25) | ||
95% CI | −1.371 to 5.906 | 1.729 to 10.547 | ||
Difference odds ratio (95% CI)b | 2.30 (0.489 to 10.823) | 4.59 (1.045 to 20.192) | ||
P valuee | 0.2921 | 0.0436 | ||
Proportions of patients achieving T-VASI50 at week 24 | ||||
Achieving response, n (%) | 43 (22.1) | 4 (4.4) | 49 (24.6) | 7 (7.1) |
Achieving response, % (SE) | 20.6 (2.76) | 5.1 (2.34) | 23.9 (2.97) | 6.8 (2.50) |
Difference in response rate (SE) | 15.5 (3.63) | 17.1 (3.87) | ||
95% CI | 8.339 to 22.592 | 9.538 to 24.721 | ||
Difference odds ratio (95% CI)b | 4.93 (1.795 to 13.566) | 4.29 (1.865 to 9.853) | ||
P valuec | 0.0020 | 0.0006 | ||
Facial body surface area | ||||
Percent change from baseline in facial BSA at week 24 | ||||
Baseline facial BSA, mean (SD) | 1.05 (0.692) | 1.15 (0.710) | 0.98 (0.571) | 0.92 (0.582) |
Facial BSA at week 24, mean (SD) | 0.72 (0.629) | 1.00 (0.699) | 0.67 (0.474) | 0.87 (0.613) |
Change from baseline, mean (SD) | −0.30 (0.439) | −0.10 (0.270) | −0.30 (0.493) | −0.09 (0.33) |
Change from baseline, %, mean (SD) | −30.22 (34.044) | −9.66 (27.586) | −26.82 (37.002) | −8.81 (26.886) |
Change from baseline, %, LSM (SE) | −28.9 (2.22) | −9.5 (3.25) | −26.4 (2.57) | −7.0 (3.82) |
LSM difference (SE)d | −19.3 (3.93) | −19.5 (4.59) | ||
95% CI | −27.05 to −11.64 | −28.46 to −10.45 | ||
Between-group P valuec | < 0.0001 | < 0.0001 | ||
Vitiligo Noticeability Scale | ||||
Proportions of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) at week 24 | ||||
Achieving response, n (%) | 49 (25.1) | 3 (3.3) | 41 (20.6) | 5 (5.1) |
Achieving response, % (SE) | 24.5 (3.03) | 3.3 (1.85) | 20.5 (2.85) | 4.9 (2.17) |
Difference in response rate (SE) | 21.2 (3.54) | 15.5 (3.58) | ||
95% CI | 14.271 to 28.143 | 8.515 to 22.561 | ||
Difference odds ratio (95% CI)b | 9.53 (2.900 to 31.290) | 4.86 (1.851 to 12.755) | ||
P valuec | 0.0002 | 0.0013 | ||
Proportions of patients achieving each VNS response category, n (%) | ||||
4 – A lot less noticeable | 47 (24.1) | 3 (3.3) | 41 (20.6) | 4 (4.1) |
5 – No longer noticeable | 2 (1.0) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
Treatment Satisfaction Questionnaire For Medication | ||||
Score at week 24 | ||||
LSM (SE) | 65.68 (1.62) | 51.47 (2.37) | 61.09 (1.66) | 48.51 (2.36) |
95% CI | 62.50 to 68.86 | 46.81 to 56.14 | 57.83 to 64.36 | 43.87 to 53.14 |
LSM difference (SE) | 14.21 (2.87) | 12.59 (2.88) | ||
95% CI | 8.56 to 19.85 | 6.91 to 18.26 | ||
P valuee | < 0.0001 | < 0.0001 | ||
Dermatology Life Quality Index | ||||
Change from baseline (aged 16 years) | ||||
Number of patients contributing to the analysis, N | 178 | 87 | 182 | 94 |
Baseline DLQI, mean (SD) | 4.63 (4.446) | 4.59 (4.871) | 4.37 (4.485) | 5.38 (4.876) |
DLQI at week 24, mean (SD) | 3.56 (3.850) | 3.77 (3.624) | 3.34 (3.468) | 4.62 (4.774) |
Change from baseline, mean (SD) | −1.15 (3.796) | −0.79 (3.355) | −1.19 (4.075) | −0.71 (3.988) |
Change from baseline, LSM (SE) | −1.17 (0.27) | −0.85 (0.39) | −1.19 (0.30) | −0.59 (0.41) |
LSM difference (SE) | −0.32 (0.48) | −0.60 (0.51) | ||
95% CI | −1.26 to 0.62 | −1.60 to 0.39 | ||
Between-group P valuee | 0.4970 | 0.2350 | ||
Change from baseline in CDLQI (age < 16 years) | ||||
Number of patients contributing to the analysis, N | 16 | 3 | 17 | 3 |
Baseline CDLQI, mean (SD) | 2.50 (2.805) | 1.25 (1.893) | 1.29 (2.201) | 6.33 (10.116) |
CDLQI at week 24, mean (SD) | 2.18 (2.325) | 1.33 (2.309) | 1.29 (1.404) | 4.00 (2.646) |
Change from baseline, mean (SD) | −0.25 (2.113) | 0.00 (0.000) | 0.00 (1.837) | −2.33 (8.505) |
Change from baseline, LSM (SE) | NR | NR | NR | NR |
LSM difference (SE) | NR | NR | ||
95% CI | ||||
Between-group P valuee | ||||
Vitiligo-Specific Quality of Life | ||||
Change from baseline | ||||
Baseline VitiQoL, mean (SD) | 36.32 (22.254) | 36.63 (23.229) | 36.48 (24.300) | 41.61 (24.634) |
VitiQoL at week 24, mean (SD) | 30.17 (21.780) | 32.34 (22.818) | 31.18 (22.660) | 38.93 (24.192) |
Change from baseline, mean (SD) | −6.34 (17.433) | −6.37 (15.988) | −6.07 (18.148) | −2.64 (14.462) |
Change from baseline, LSM (SE) | −6.45 (1.21) | −6.18 (1.77) | −6.18 (1.20) | −2.66 (1.70) |
LSM difference (SE) | −0.28 (2.15) | −3.52 (2.08) | ||
95% CI | −4.51 to 3.95 | −7.60 to 0.57 | ||
Between-group P valuee | 0.8976 | 0.0915 | ||
ANCOVA = analysis of covariance; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; F-VASI = Facial Vitiligo Area Scoring Index; F-VASI25 = 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI50 = 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; ITT = intention to treat; LSM = least squares mean; NR = not reported; SD = standard deviation; SE = standard error; T-VASI50 = 50% or more improvement from baseline on the Total Vitiligo Area Scoring Index; T-VASI75 = 75% or more improvement from baseline on the Total Vitiligo Area Scoring Index; VASI = Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale.
aMultiple imputation: missing F-VASI score was imputed by fully conditional specification. Multiple imputation method used treatment and observed stratification factors as predicators.
bAn exact logistic regression statistical model was used to estimate the difference between groups for all VASI and VNS outcomes reported in the table.
cStatistical testing at week 24 for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
dAn ANCOVA statistical model was used to estimate the difference between groups in change from baseline in facial BSA.
eStatistical testing for these end points was not controlled for multiple comparisons.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Data for harms outcomes included in the review are presented in Table 15.
Adverse Events
In the TRuE-V1 trial, 45.7% of patients who applied ruxolitinib and 38.5% of patients who applied vehicle cream experienced at least 1 AE; in the TRuE-V2 trial, the corresponding proportions were 50.0% with ruxolitinib and 33.9% with vehicle cream, respectively. The most common TEAEs across the 2 studies included application site acne and pruritus, headache, and AEs related to infections.
Serious Adverse Events
In the TRuE-V1 trial, 6 patients (2.7%) who applied ruxolitinib and 1 patient (0.9%) who applied vehicle cream experienced SAEs. Serious TEAEs reported with active treatment were anal fistula, appendicitis, concussion, hepatic infectious mononucleosis, kidney contusion, and myocarditis.
In the TRuE-V2 trial, 2 patients (0.9%) who applied ruxolitinib experienced SAEs, that included coronary artery stenosis and ureterolithiasis. No patient who applied vehicle cream experienced an SAE.
Withdrawals Due to AEs
In the TRuE-V1 trial, 1 patient (0.5%) who applied ruxolitinib and 1 patient (0.9%) who applied vehicle cream discontinued due to AEs; the reasons for withdrawal were fatigue, nausea, and headache. In the TRuE-V2 trial, 1 patient (0.4%) who applied ruxolitinib discontinued due to an AE of application site rash. No patient who applied vehicle cream discontinued treatment.
Mortality
No deaths were reported in the studies.
Notable Harms
The sponsor identified application site reactions as a harm of special interest.
In the TRuE-V1 trial, 38 patients (17.2%) who applied ruxolitinib and 7 patients (6.4%) who applied vehicle cream reported an application site reaction. The most frequently reported were acne (5.9% and 0%, respectively), pruritus (5.0% and 3.7%), and rash (2.3% and 0.9%).
In the TRuE-V2 trial, 30 patients (13.2%) who applied ruxolitinib and 7 patients (6.1%) who applied vehicle cream reported an application site reaction. The most frequently reported were acne (5.7% and 2.6%, respectively) and pruritus (5.3% and 1.7%).
Table 15: Summary of Harms Results From Studies Included in the Systematic Review (Safety Population)
Adverse events | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 221 | Vehicle cream N = 109 | Ruxolitinib N = 228 | Vehicle cream N = 115 | |
Most common TEAEs, n (%) | ||||
Patients with ≥ 1 AE | 101 (45.7) | 42 (38.5) | 115 (50.0) | 39 (33.9) |
Application site acne | 13 (5.9) | 0 (0.0) | 13 (5.7) | 3 (2.6) |
Application site pruritus | 11 (5.0) | 4 (3.7) | 12 (5.3) | 2 (1.7) |
Nasopharyngitis | 9 (4.1) | 4 (3.7) | 10 (4.4) | 1 (0.9) |
Headache | 6 (2.7) | 2 (1.8) | 11 (4.8) | 4 (3.5) |
Upper respiratory tract infection | 6 (2.7) | 5 (4.6) | 7 (3.1) | 0 |
Application site rash | 5 (2.3) | 1 (0.9) | 1 (0.4) | 0 |
Urinary tract infection | 5 (2.3) | 1 (0.9) | NR | NR |
Sinusitis | 4 (1.8) | 3 (2.8) | 6 (2.6) | 2 (1.7) |
COVID-19 | 3 (1.4) | 5 (4.6) | 10 (4.4) | 2 (1.7) |
Application site exfoliation | NR | NR | 5 (2.2) | 1 (0.9) |
Pyrexia | 1 (0.5) | 0 (0.0) | 5 (2.2) | 0 |
Most common treatment-emergent SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 6 (2.7) | 1 (0.9) | 2 (0.9) | 0 (0.0) |
Anal fistula | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Appendicitis | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Concussion | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Coronary artery stenosis | 0 (0.0) | 0 (0.0) | 1 (0.4) | 0 (0.0) |
Hepatic infectious mononucleosis | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Kidney contusion | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Myocarditits | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Tibia fracture | 0 (0.0) | 1 (0.9) | 0 (0.0) | 0 (0.0) |
Ureterolithiasis | 0 (0.0) | 0 (0.0) | 1 (0.4) | 0 (0.0) |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients who stopped treatment | 1 (0.5) | 1 (0.9) | 1 (0.4) | 0 (0.0) |
Application site rash | 0 (0.0) | 0 (0.0) | 1 (0.4) | 0 (0.0) |
Fatigue | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Nausea | 0 (0.0) | 1 (0.9) | 0 (0.0) | 0 (0.0) |
Headache | 0 (0.0) | 1 (0.9) | 0 (0.0) | 0 (0.0) |
Deaths, n (%) | ||||
Patients who died | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
AE = adverse event; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Study Design
The TRuE-V1 and TRuE-V2 RCTs were designed to evaluate the efficacy and safety of ruxolitinib compared to a vehicle cream over a 24-week controlled treatment period. Patients were allocated to treatment groups using an appropriate methodology (interactive response technology), with randomization stratified by relevant and clinically important potential prognostic factors, particularly baseline skin type. The double-blind controlled period was followed by a 28-week open-label treatment extension, in which all patients received ruxolitinib. This provided uncontrolled and complementary findings regarding the long-term efficacy and safety of the drug.
Patient Population
Most baseline characteristics appeared to be balanced between treatment groups in both studies, except in the proportions of male and female patients in the TRuE-V1 trial, in which some imbalance between treatment groups was observed. The clinical experts consulted for this review indicated that sex is not expected to affect the course of the disease and/or response to treatment. Although the impact of a visible skin condition such as vitiligo on quality of life may vary according to different factors, including sex, the experts agreed that this baseline difference was unlikely to be clinically important enough to bias the results.
There were numerically more discontinuations in patients who applied the vehicle cream in the TRuE-V1 trial, which could introduce bias against ruxolitinib if patients did not respond to treatment; however, most of the discontinuations were due to withdrawal by patient or loss to follow-up. This concern was also mitigated by the use of multiple imputations to account for missing data. Patient disposition was balanced between treatment groups in the TRuE-V2 trial. In this latter study, patients from 1 study site were excluded from the efficacy analyses due to a serious protocol violation, representing 3.2% of patients who applied ruxolitinib and 5.5% of patients who applied the vehicle cream. No further details were reported. Nevertheless, as the discontinuation rate was low and the difference between treatment arms was small, it is not expected that this would affect the findings.
Interventions
As both the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence comparing ruxolitinib to other currently used therapies for vitiligo. This absence restricts the trials’ ability to assess comparative effectiveness and safety relative to other available treatment options.
No rescue therapy was allowed and patients needed to discontinue all drugs used for the treatment of vitiligo, minimizing the impact of this potential confounding factor. Both ruxolitinib and vehicle cream were to be continued, even if the affected area improved. The clinical experts consulted for this review did not expect the use of the reported concomitant medications to have a clinically meaningful impact on the disease course or on the treatment response.
Outcome Measures
The primary outcome in the TRuE-V1 and TRuE-V2 trials was an F-VASI75. Additional levels of F-VASI and T-VASI thresholds were assessed as secondary end points in the trials. The clinical experts indicated that, while the VASI score was not used in clinical practice, the instrument has been developed specifically for clinical trial assessment. The sponsor reported that the VASI score was validated. Part of the validation analyses was performed using studies from the ruxolitinib drug development program, and validation studies were identified from the literature. As for the MCID, a 75% improvement in the F-VASI has been suggested as a threshold for treatment success based on a mixed-method study and on perceptions of patients with vitiligo and dermatologists; however, no MCID for between-group differences was reported. Patient-reported noticeability was also assessed as a key secondary outcome using the VNS, a validated instrument for which scores of 4 (a lot less noticeable) or 5 (no longer noticeable) have been used as the MCID.
The use of the VASI score was considered relevant by the clinical experts to assess the objective response to treatment, as the VASI score captures the overall surface area of vitiligo involvement and the degree of repigmentation. However, a substantial limitation to the interpretation of the findings is that this instrument does not inform on the level of improvement in HRQoL, which was identified by the clinical experts as the main treatment goal of vitiligo. Experience from clinical practice suggests that partial repigmentation may not necessarily be associated with a meaningful change for patients, and HRQoL may be affected for as long as the disease is visible. For example, the clinical experts noted that improvements seen in responsive areas, such as the cheeks and neck, may be of little importance if other known resistant sites, such as the periocular and perioral areas, cannot be repigmented. Areas affected by vitiligo in these highly visible sites will be more difficult to hide and therefore be more consequential for the patient, particularly when compared to other areas on the body.
However, the studies were not designed to test primarily for differences in HRQoL. This outcome was measured using the DLQI and CDLQI as well as the VitiQoL and was assessed as an exploratory outcome. Evidence is available supporting the validity and reliability but not the responsiveness, of the DLQI and CDLQI as well as the VitiQoL in patients with vitiligo. As the DLQI and CDLQI are not disease-specific instruments, not all of their items are relevant to the clinical assessment of vitiligo, and they may not be sufficiently sensitive to capture nuanced details of how vitiligo patients handle their disease burden, a shortcoming that was confirmed by the clinical experts. Based on the literature, there is currently no MCID established for these instruments in patients with vitiligo. However, the use of an anchor-based approach for patients with various inflammatory skin conditions suggests that a within-group change from baseline of at least 4 points in the DLQI or CDLQI may be consistent with clinically meaningful improvements for patients.
Statistical Analysis
All patients with missing F-VASI assessments were handled using multiple imputations under a missing at random assumption. The key assumptions regarding missing data were tested in prespecified sensitivity analyses (i.e., nonresponder imputation, last observation carried forward, and tipping point analysis) and results of these sensitivity analyses demonstrated the robustness of the findings from the primary analysis to violations in the assumed missing data mechanism.
The TRuE-V1 and TRuE-V2 studies used a gatekeeping testing strategy for the primary and key secondary analyses to control the overall type I error rate. Other outcomes of interest to this review (e.g., proportions of patients achieving an F-VASI25 and T-VASI 75, as well as HRQoL as measured by the DLQI, CDLQI, and VitiQoL) were not controlled for multiplicity of testing.
Prespecified subgroup analyses included those for skin type and race, which were considered by the clinical experts to be particularly relevant to the treatment of vitiligo. However, because so few patients with darker skin tones were included, these subgroups were underpowered. The statistical tests that were performed to evaluate the differences in effects between subgroups, as well as whether the comparability of the treatment arms was checked and whether multiplicity was taken into account, were not reported for either trial. As such, the findings should be viewed as supplementary.
As with most clinical trials, the studies were not powered to detect infrequent AEs or those with a lag time.
External Validity
Population
Because patients with leukotrichia and segmental vitiligo were excluded from the trial, its findings are not generalizable to these patients.
The majority of patients included in both studies were white and had a Fitzpatrick Scale skin type of II, III, or IV (i.e., ranging from fair peach to moderate brown). Although vitiligo affects all races and types of skin colour, the clinical experts indicated that patients with darker skin tones are likely to present with an increased impact on quality of life, which can be attributed to the increased visibility of the condition. Of the 661 patients in the TRuE-V1 and TRuE-V2 trials combined, 31 patients (4.7%) were Black or African American, 26 (3.9%) were Asian, and 2 (0.3%) were Native Hawaiian or Pacific Islander. As for skin colour according to the Fitzpatrick Scale skin type, 43 patients (6.5%) had skin type V (i.e., dark brown) and 13 patients (2.0%) had skin type VI (i.e., deeply pigmented dark brown to darkest brown). The fact that the patients with potentially the greatest unmet need for an effective treatment were not adequately represented in the studies is a significant concern for external validity of the findings.
Given the natural trajectory of the disease, the clinical experts indicated that a longer disease duration is likely to be associated with a more severe, refractory condition. In both the TRuE-V1 and TRuE-V2 trials, the median time since diagnosis exceeded 10 years. However, despite a long-lasting disease, an important proportion of patients in both trials never received treatment aiming to improve their condition. Overall, 62% of patients had received prior therapy, a proportion that the clinical experts described as low. The experts expected that, in clinical practice, virtually all patients who sustain a negative impact from vitiligo would seek and receive treatment, particularly when the long-lasting diagnosis of patients in the studies is considered. According to the experts, this suggests that the trial population may not be representative of patients whose condition interferes substantially with their daily life. This is consistent with the relatively low level of HRQoL impairment observed at baseline, as measured by the DLQI, CDLQI, and VitiQoL. Of the patients who received treatment in the trials, 40% reported discontinuing therapy due to lack of efficacy.
Overall, only 11% of patients enrolled in the TRuE-V1 and TRuE-V2 trials combined were adolescents; there are therefore limited data to interpret in this younger age group.
Intervention and Comparator
Patient exposure to ruxolitinib and vehicle cream in the trials indicates notable variation among patients in the amount of the study drug applied, which appeared to be inconsistent with the usage patterns of topical treatments in clinical practice, according to the clinical experts. No additional details were provided by the sponsor to explain the high level of variability observed. The clinical experts consulted for this review indicated that it is uncertain if the amounts of the drug used in the trials would be mirrored in real-world practice, but they noted that it is likely that drug usage rates will be higher in the real-world setting.
Overall, the treatment options currently available in clinical practice for vitiligo are well accepted and routinely prescribed, even if they are being used off-label, according to the clinical experts, who noted that patients who first seek treatment will typically be offered a few options. Over time, approximately one-half of patients may become refractory due to disease resistance, and others will discontinue treatments due to unacceptable toxicity. The absence of comparative evidence between ruxolitinib and these drugs was therefore considered a substantial limitation.
Outcomes
The clinical experts noted that the primary clinical assessment of vitiligo in the trials using the VASI score was not representative of routine clinical practice in Canada, where treatment is targeted at improving current and future quality of life rather than only focusing on reducing the surface area of involvement and degree of repigmentation. This was also consistent with patient and clinician input, all of which emphasized the importance of improving the psychosocial impact of the disease on quality of life. The experts pointed out that several additional key issues of vitiligo have an important impact on HRQoL, including the disease site and associated visibility, the level of heterogeneity in repigmentation, and differences between the colour of the affected, repigmented area and background skin tone, as well as family and cultural influences.
Because the treatment of vitiligo in clinical practice is highly individualized, the clinical experts indicated that the clinical meaningfulness of the difference between the 2 treatments, when expressed solely in terms of VASI score improvements, can be highly variable among patients, depending on how the disease affects their daily lives. As such, the overall impact of achieving the VASI score MCID on daily life was difficult to assess according to the clinical experts, as different individuals are likely to have different priorities and objectives when assessing the response to treatment, and as the instrument is not used in clinical practice.
Follow-up
The follow-up duration of 24 weeks was considered relatively short, as the disease generally improves over a longer period of time. However, it was considered sufficient to assess changes in the VASI outcomes and capture improvements in objective response to treatment. Nevertheless, treatment is likely to continue over the long-term and evidence from beyond the studies follow-up duration is limited.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:27,28
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
For the GRADE assessments, findings from the TRuE-V1 and TRuE-V2 trials were considered together and summarized narratively per outcome because populations, interventions, designs, and outcome measures of the 2 studies were similar.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for ruxolitinib versus vehicle cream.
LTE Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
TRuE-V1 and TRuE-V2 Trials Open-Label Treatment Extension
Description of TRuE-V1 and TRuE-V2 Trials Open-Label Treatment Extension
In the pivotal RCTs (TRuE-V1 and TRuE-V2), the double-blind controlled period was followed by a 28-week open-label treatment extension. The study design for TRuE-V1 and TRuE-V2 is presented in Figure 1. Patients initially randomized to the vehicle cream crossed over to ruxolitinib, while patients initially randomized to ruxolitinib received an additional 28 weeks of treatment with the active drug, as long as they completed the week 24 assessments with no safety concerns. During the treatment-extension period, patients continued to treat depigmented areas identified for treatment at baseline even if the area had fully repigmented. The evaluable population of the treatment extension consisted of all patients who applied ruxolitinib cream at least once in the treatment-extension period; this analysis set was used for all efficacy and safety assessments.
Results
Details of patient disposition are provided in Table 16. The proportions of patients who discontinued the study during the treatment extension ranged from 9.8% to 19.4% across treatment arms in the 2 trials.
Exposure to study treatment is outlined in Table 17. The median exposure duration during the treatment extension was ███ days in both studies, which was consistent with the planned duration of 28 weeks (196 days). Patient exposure suggests notable variation among patients in the amount of study drug applied, particularly in the TRuE-V2 trial.
The proportions of patients who received concomitant medications in the studies ranged from █████ ██ █████ across treatment arms in the 2 trials. The most commonly used medications are presented in Table 18.
Table 16: Summary of Patient Disposition (Treatment-Extension Period)
Patient disposition | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 193 | Vehicle cream to ruxolitinib N = 90 | Ruxolitinib N = 199 | Vehicle cream to ruxolitinib N = 98 | |
Discontinued the study during the treatment-extension period, n (%) | 19 (9.8) | 10 (11.1) | 21 (10.6) | 19 (19.4) |
Reason for discontinuation, n | ||||
Adverse events | 1 (0.5) | 0 | 1 (0.5) | 0 |
Lack of efficacy | 1 (0.5) | 0 | 1 (0.5) | 0 |
Lost to follow-up | 5 (2.6) | 1 (1.1) | 9 (4.5) | 10 (10.2) |
Withdrawal by patient | 10 (5.2) | 7 (7.8) | 9 (4.5) | 9 (9.2) |
Physician decision | 0 | 1 (1.1) | 0 | 0 |
Other | 2 (1.0) | 1 (1.1) | 1 (0.5) | 0 |
Source: Incyte Corporation (2021).29,30
Table 17: Summary of Patient Exposure (Treatment-Extension Period) [Redacted]
Exposure | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 193 | Vehicle cream to ruxolitinib N = 90 | Ruxolitinib N = 199 | Vehicle cream to ruxolitinib N = 98 | |
████████ ██ █████████ ████ | ||||
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
██████ ██ █████ ████ ███████ | ||||
████ ████ | ||||
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
█████ | ||||
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
███████████ ██████████ ███ | ||||
████ ████ | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Max = maximum; Min = minimum; SD = standard deviation.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 18: Summary of Most Commonly Used Concomitant Medications (Treatment-Extension Period) [Redacted]
Exposure | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 193 | Vehicle cream to ruxolitinib N = 90 | Ruxolitinib N = 199 | Vehicle cream to ruxolitinib N = 98 | |
███████████ ███████████ ████ | ||||
██████████████ █████ | █████ | █████ | █████ | █████ |
████ ████████ █████ █ ███ | ||||
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
██████████████ █████ | █████ | █████ | █████ | █████ |
ACE = angiotensin-converting-enzyme; DB = double-blind; HMG-CoA = 3-hydroxyl-3-methyl-glutaryl-coenzyme A.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Detailed efficacy results for the treatment-extension period, at 52 weeks, are presented in Table 19.
Throughout the TRuE-V1 and TRuE-V2 trials’ treatment extensions, which ranged from week 24 to week 52, the proportions of patients who achieved all predefined thresholds of reduction in the VASI score (i.e., F-VASI25, F-VASI50, F-VASI75, F-VASI90, and T-VASI75) were numerically higher in all treatment arms when compared to the corresponding proportions during the double-blind period (i.e., from start to week 24). Similar results were obtained in the proportions of patients who achieved a VNS score of 4 (a lot less noticeable) or 5 (no longer noticeable). However, in the treatment-extension period, no statistical analysis was reported to assess whether the change from week 24 to week 52 was significant, or to assess the magnitude of the between-group difference. Results for HRQoL, assessed using the DLQI, CDLQI, and VitiQoL, suggest that the observed within-group changes from baseline to week 52 were small and not clinically meaningful, according to the clinical experts. However, no between-group statistical analysis was reported.
Table 19: Summary of Key Efficacy Results (Treatment-Extension Period)
Variable | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 221 | Vehicle cream to ruxolitinib N = 109 | Ruxolitinib N = 222 | Vehicle cream to ruxolitinib N = 109 | |
Number of patients contributing to the analyses, N | 173 | 82 | 177 | 81 |
Vitiligo Area Scoring Index | ||||
Proportions of patients achieving F-VASI75 at week 52 | ||||
Achieving response, n (%) | 91 (52.6) | 22 (26.8) | 85 (48.0) | 24 (29.6) |
SE | 3.80 | 4.89 | 3.76 | 5.07 |
95% CI | 44.9 to 60.2 | 17.6 to 37.8 | 40.5 to 55.6 | 20.0 to 40.8 |
Proportions of patients achieving F-VASI90 at week 52 | ||||
Achieving response, n (%) | 57 (32.9) | 10 (12.2) | 49 (27.7) | 13 (16.0) |
SE | 3.57 | 3.61 | 3.36 | 4.08 |
95% CI | 26.0 to 40.5 | 6.0 to 21.3 | 21.2 to 34.9 | 8.8 to 25.9 |
Proportions of patients achieving F-VASI50 at week 52 | ||||
Achieving response, n (%) | 130 (75.1) | 46 (56.1) | 131 (74.0) | 40 (49.4) |
SE | 3.29 | 5.48 | 3.30 | 5.56 |
95% CI | 68.0 to 81.4 | 44.7 to 67.0 | 66.9 to 80.3 | 38.1 to 60.7 |
Proportions of patients achieving F-VASI25 at week 52 | ||||
Achieving response, n (%) | 155 (89.6) | 61 (74.4) | 146 (82.5) | 58 (71.6) |
SE | 2.32 | 4.82 | 2.86 | 5.01 |
95% CI | 84.1 to 93.7 | 63.6 to 83.4 | 76.1 to 87.8 | 60.5 to 81.1 |
Proportions of patients achieving T-VASI75 at week 52 | ||||
Achieving response, n (%) | 35 (20.2) | 8 (9.8) | 37 (20.9) | 7 (8.6) |
SE | 3.05 | 3.28 | 3.06 | 3.12 |
95% CI | 14.5 to 27.0 | 4.3 to 18.3 | 15.2 to 27.6 | 3.5 to 17.0 |
Vitiligo Noticeability Scale | ||||
Proportions of patients achieving a VNS of 4 (a lot less noticeable) at week 52 | ||||
Achieving response, n (%) | 68 (39.3) | 16 (19.5) | 57 (32.2) | 11 (13.6) |
Proportions of patients achieving a VNS of 5 (no longer noticeable) at week 52 | ||||
Achieving response, n (%) | 1 (0.6) | 0 | 1 (0.6) | 0 |
Dermatology Life Quality Index | ||||
Change from baseline (aged 16 years) | ||||
Number of patients contributing to the analysis, N | 157 | 79 | 161 | 78 |
Baseline DLQI, mean (SD) | 4.63 (4.446) | 4.59 (4.871) | 4.37 (4.485) | 5.38 (4.876) |
DLQI at week 52, mean (SD) | 3.31 (3.743) | 3.29 (3.259) | 3.55 (3.991) | 4.31 (4.210) |
Change from baseline, mean (SD) | −1.40 (4.087) | −1.37 (3.617) | −0.84 (3.976) | −1.15 (4.264) |
Change from baseline in CDLQI (age < 16 years) | ||||
Number of patients contributing to the analysis, N | 15 | 3 | 16 | 3 |
Baseline DLQI, mean (SD) | 2.50 (2.805) | 1.25 (1.893) | 1.29 (2.201) | 6.33 (10.116) |
DLQI at week 52, mean (SD) | 1.63 (1.544) | 1.33 (1.528) | 2.50 (4.502) | 5.33 (6.110) |
Change from baseline, mean (SD) | −1.00 (2.507) | 0.00 (1.000) | 1.19 (4.665) | −1.00 (4.583) |
Vitiligo-Specific Quality of Life | ||||
Change from baseline | ||||
Baseline VitiQoL, mean (SD) | 36.32 (22.254) | 36.63 (23.229) | 36.48 (24.300) | 41.61 (24.634) |
VitiQoL at week 52, mean (SD) | 27.35 (22.015) | 31.59 (21.094) | 29.98 (21.792) | 35.93 (24.481) |
Change from baseline, mean (SD) | −9.39 (19.034) | −7.78 (17.141) | −7.13 (18.606) | −6.89 (17.076) |
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; F-VASI25 = 25% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI50 = 50% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; SD = standard deviation; SE = standard error; T-VASI75 = 75% or more improvement from baseline on the Total Vitiligo Area Scoring Index; VitiQoL = Vitiligo-Specific Quality of Life; VNS = Vitiligo Noticeability Scale.
Source: Incyte Corporation (2021)29,30 and the sponsor’s Summary of Clinical Evidence.
Harms data are presented in Table 20. The proportions of patients who experienced at least 1 AE during the treatment extension ranged from 33.7% to 41.2% across treatment arms in the 2 trials. Few patients experienced SAEs. One patient discontinued due to application site eczema. No deaths were reported during the treatment extension.
Table 20: Summary of Harms (Treatment-Extension Period)
Adverse events | TRuE-V1 | TRuE-V2 | ||
|---|---|---|---|---|
Ruxolitinib N = 193 | Vehicle cream to ruxolitinib N = 90 | Ruxolitinib N = 199 | Vehicle cream to ruxolitinib N = 98 | |
Most common TEAEs, n (%) | ||||
Patients with ≥ 1 AE | 65 (33.7) | 31 (34.4) | 82 (41.2) | 38 (38.8) |
COVID-19 | 11 (5.7) | 1 (1.1) | 9 (4.5) | 5 (5.1) |
Application site acne | 1 (0.5) | 0 (0.0) | 3 (1.5) | 5 (5.1) |
Headache | 3 (1.6) | 2 (2.2) | 6 (3.0) | 1 (1.0) |
Nasopharyngitis | 3 (1.6) | 2 (2.2) | 4 (2.0) | 3 (3.1) |
Application site dermatitis | 1 (0.5) | 0 (0.0) | 5 (2.5) | 0 (0.0) |
Hypertension | NR | NR | 3 (1.5) | 2 (2.0) |
Upper respiratory tract infection | 2 (1.0) | 3 (3.3) | 0 (0.0) | 2 (2.0) |
Oral herpes | 2 (1.0) | 2 (2.2) | 0 (0.0) | 1 (1.0) |
Acne | 0 (0.0) | 3 (3.3) | 2 (1.0) | 1 (1.0) |
Neutropenia | NR | NR | 1 (0.5) | 2 (2.2) |
Oral candidiasis | NR | NR | 1 (0.5) | 2 (2.0) |
Procedural pain | NR | NR | 1 (0.5) | 2 (2.0) |
Hypothyroidism | 1 (0.5) | 2 (2.2) | NR | NR |
Most common treatment-emergent SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 1 (0.5) | 1 (1.1) | 2 (1.0) | 2 (2.0) |
Hypersensitivity | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Prostate cancer | 0 (0.0) | 1 (1.1) | 0 (0.0) | 0 (0.0) |
Subacute combined cord degeneration | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Appendiceal abscess | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
Joint dislocation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
Papillary thyroid cancer | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
Rhabdomyolysis | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients who stopped | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
Application site eczema | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
Deaths, n (%) | ||||
Patients who died | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
AE = adverse event; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Source: Incyte Corporation (2021).29,30 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Due to the single-arm nature of the TRuE-V1 and TRuE-V2 trials open-label treatment extensions, the conclusions that can be drawn regarding the efficacy and safety of ruxolitinib in the longer term are noncomparative. The same limitations pertaining to the uncertain clinical impact of the F-VASI score that were highlighted for the double-blind controlled period of the studies also apply to the extension period. In addition, findings from both the TRuE-V1 and TRuE-V2 studies may be considered generalizable to a selected sample of the individuals in Canada with vitiligo. The majority of patients included in the studies were white and had lighter skin tones; however, vitiligo is particularly visible in patients with darker skin tones and is therefore likely to have a greater impact on the quality of life in these patients. Considering disease duration, prior therapies, and baseline HRQoL, the trial population may not be representative of patients whose condition interferes substantially with their daily life.
TRuE-V LTE Study
Description of TRuE-V LTE Study
The TRuE-V LTE31 is a phase III, double-blind, vehicle-controlled, randomized withdrawal LTE trial of the TRuE-V1 and TRuE-V2 studies, designed to assess the long-term efficacy and safety of ruxolitinib cream in patients with vitiligo. The study aimed to evaluate the duration of response following withdrawal of 1.5% ruxolitinib cream (cohort A) and the safety and maintenance of response with continued use of the cream (cohort B). The LTE study had a duration of 52 weeks, followed by a 30-day safety follow-up.
Eligible participants were assigned to either cohort A or cohort B based on their F-VASI responses at the time of enrolment in the LTE study (week 52).
Cohort A followed a randomized withdrawal design, providing data on the duration of response after discontinuation and the maintenance of response with continued treatment. Participants who achieved complete or near-complete facial repigmentation (greater than or equal to an F-VASI90) at week 52 in either the TRuE-V1 or TRuE-V2 trial were assigned to this cohort. They were randomized in a 1:1 ratio to either continue 1.5% ruxolitinib cream or switch to the vehicle cream during the LTE. Members of cohort A who experienced a disease relapse (defined as less than an F-VASI75) received open-label ruxolitinib cream as rescue treatment until week 104 or the end of the trial.31
Cohort B included participants who did not achieve a response of greater than or equal to an F-VASI90 at week 52 in the parent studies. These participants continued treatment with 1.5% ruxolitinib cream for the entire LTE period (Figure 2).
In cohort A, the participant, investigator, and sponsor remained blinded to treatment assignment until after database lock for the primary analysis (week 104); the treatment for cohort B was open-label.31
Figure 2: TRuE-V LTE Study Design
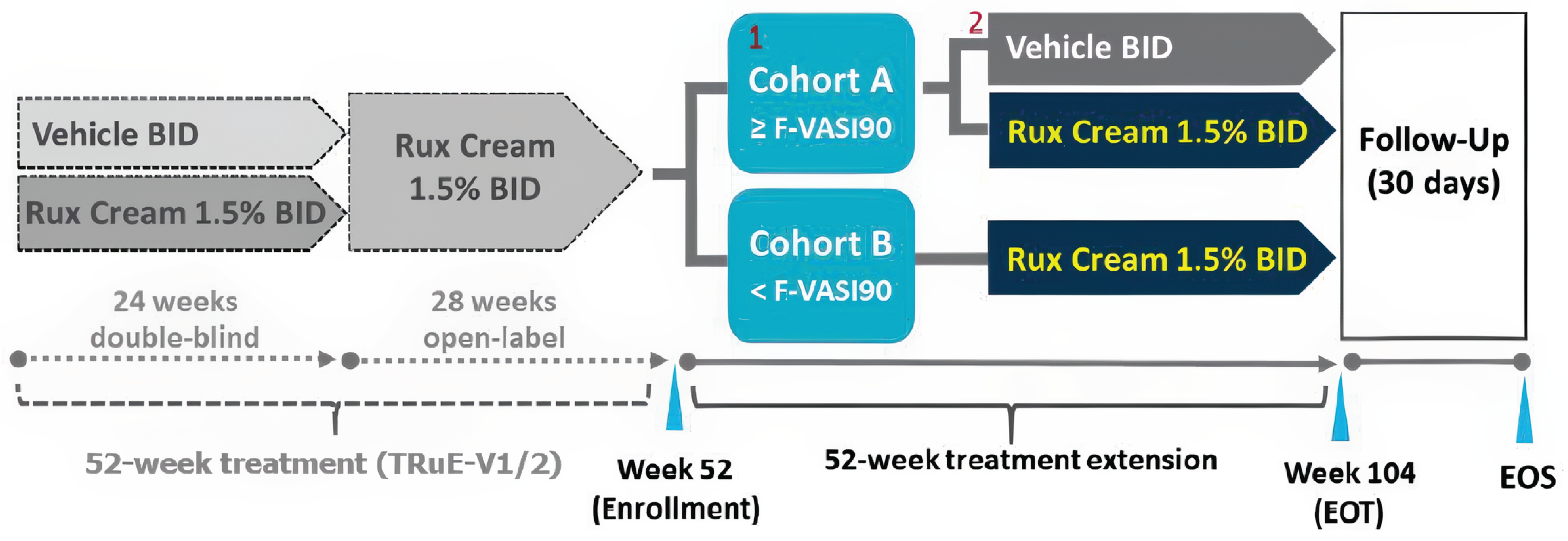
BID = twice a day; EOS = end of study; EOT = end of treatment; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; LTE = long-term extension; Rux = ruxolitinib.
1All participants in cohort A used their randomly assigned treatment (either vehicle cream or 1.5% ruxolitinib cream) on both the face and total body.
2Rescue treatment: If, at any time, a participant in cohort A lost a clinically meaningful response on the face (< F-VASI75), the participant received open-label 1.5% ruxolitinib cream twice a day until week 104 or EOT.
Source: TRuE-V LTE Clinical Study Report.31
Outcomes
The primary outcome of the study was the time to relapse in cohort A. Relapse was defined as loss of an F-VASI75 response assessed as percentage improvement from baseline (day 1 of the parent study) to an F-VASI of less than 75%. The time to relapse was defined as the number of days from the week 52 randomization date to the first evaluation date at which the participant met the definition of relapse. The key secondary outcome was the time to maintain a response greater than or equal to an F-VASI90, defined as the number of days from week 52 randomization to the first evaluation date at which the participant achieved an F-VASI90.
Additional secondary outcomes that are included in this report:
proportion of patients who achieve an F-VASI50, F-VASI75, or F-VASI90 during the LTE period
proportion of patients achieving a VNS of 4 (a lot less noticeable) or 5 (no longer noticeable) during the LTE
proportion of patients who achieve T-VASI75 during the LTE period
time to regain at least an F-VASI90 and at least an F-VASI75 in patients whose disease relapsed after entering the LTE.
Exploratory outcomes included changes from week 52 in DLQI, CDLQI, and VitiQoL and time to regain a response greater than or equal to an F-VASI75 and greater than or equal to an F-VASI90 following disease relapse. Safety outcomes were consistent with those defined in the TRuE-V1 and TRuE-V2 parent studies.
Statistical Analysis
All statistical analyses were exploratory, with no alpha control implemented. Unless otherwise specified, all CIs were at the 95% level. The sample size was not determined by statistical power calculations. Three participants from cohort A were excluded from the ITT extension population due to incorrect randomization. Data from 2 participants at site 710 were excluded from both the ITT extension and cohort A efficacy analyses due to protocol noncompliance. Similarly, in cohort B, data from 2 participants at site 710 were excluded from efficacy analyses due to noncompliance.
The primary and key secondary analyses were conducted on the ITT extension population (participants achieving a response greater than or equal to an F-VASI90 at week 52). For participants who discontinued early, completed the study without meeting the event criteria, or received open-label rescue treatment without meeting event criteria, the time to event was censored at the last assessment date or the date of rescue treatment. Time to event data were analyzed using the Kaplan-Meier method, with treatment comparisons between 1.5% ruxolitinib cream twice a day and vehicle cream twice a day performed via the log-rank test. HRs and 95% CIs were estimated using a Cox proportional hazards model, accounting for treatment groups and stratification factors (skin type and region). Relapse incidence was summarized by treatment, and swimmer plots, including subgroup analyses by baseline characteristics, were provided. Secondary efficacy analyses for cohorts A and B used specific criteria for data inclusion. Safety outcomes, including TEAEs, lab values, and vital signs, were summarized descriptively.
Results
Baseline Characteristics
Baseline characteristics for cohort A and cohort B of the TRuE-V LTE study are summarized in Table 21. In cohort A, participants applying vehicle cream twice a day were primarily adults, with ████ aged 12 to younger than 18 years; their mean age was 39.3 years (SD = 12.49 years). Most were white (72.4%), had Fitzpatrick Scale skin type II or III (█████), and were female (53.4%). This group had a long history of vitiligo, with a mean time since diagnosis of █████ █████ ███ █ █████, and █████ had stable disease at baseline. Key disease metrics included F-VASI scores ranging from ████ ██ ████ ███████ █ █████, facial BSA involvement ranging from ████ ██ ████ ███████ █ ██████, T-VASI scores ranging from ████ ██ █████ ███████ █ █████, and total BSA involvement ranging from ████ ██ █████ ███████ █ ███████.31
Similarly, participants in the 1.5% ruxolitinib cream twice a day group were predominantly adults, with █████ aged 12 to younger than 18 years and a mean age of 42.9 years (SD = 15.95 years). The majority were white (82.8%), had Fitzpatrick Scale skin type II or III skin (█████), and were female (56.9%). They also had long-standing vitiligo, with a mean time since diagnosis of █████ █████ ███ █ ██████, and ███ had stable disease at baseline. Their baseline disease metrics were comparable: F-VASI scores ranged from ████ ██ ████ ███████ █ █████, facial BSA involvement ranged from ████ ██ ████ ███████ █ ██████, T-VASI scores ranged from ████ ██ █████ ███████ █ █████, and total BSA involvement ranged from ████ ██ █████ ███████ █ ██████.31
Members of cohort B (participants who did not achieve a response greater than or equal to an F-VASI90) were initially treated with the vehicle cream and then switched to ruxolitinib cream. It included █████ adults, with ████ aged 12 years to younger than 18 years; the mean age was 39.7 years (SD = 14.62 years). The majority were white (90.7%), had Fitzpatrick Scale skin type II or III skin ███████, and were female (51.7%). Most participants had long-standing vitiligo, with a mean time since diagnosis of █████ █████ ███ █ ██████, and █████ had stable disease at baseline. Disease metrics at baseline included F-VASI scores ranging from ████ ██ ████ ███████ █ █████, facial BSA involvement from ████ ██ ████ ███████ █ ██████, T-VASI scores from ████ ██ ████ ███████ █ █████, and total BSA involvement ████ ████ ██ █████ ███████ █ ██████.31
For participants in cohort B who continued with ruxolitinib cream, █████ were adults, and █████ were aged 12 to younger than 18 years; the mean age was 39.3 years (SD = 16.45 years). The majority were white (80.4%), had Fitzpatrick Scale type II or III skin ███████, and were female (57.6%). Like the previous group, this cohort comprised primarily individuals with long-standing vitiligo, with a mean time since diagnosis of █████ █████ ███ █ ██████, and █████ had stable disease at baseline. Baseline F-VASI scores ranged from ████ ██ ████ ███████ █ █████, facial BSA involvement ranged from ████ ██ ████ ███████ █ █████), T-VASI scores ranged from ████ ██ ████ ███████ █ █████, and total BSA involvement ranged from ████ ██ █████ ███████ █ ██████.31
Table 21: Summary of Baseline Demographic and Disease Characteristics of the TRuE-V LTE Study (FAS)
Characteristic | Cohort A vehicle cream b.i.d. (N = 58) | Cohort A 1.5% ruxolitinib cream b.i.d. (N = 58) | Cohort B vehicle cream to 1.5% ruxolitinib cream b.i.d (N = 118)a | Cohort B 1.5% ruxolitinib cream b.i.d. to 1.5% ruxolitinib b.i.d.b (N = 224) |
|---|---|---|---|---|
Age (years) | ||||
Mean (SD) | 39.3 (12.49) | 42.9 (15.95) | 39.7 (14.62) | 39.3 (16.45) |
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Age category (years) | ||||
< 18, n (%) | █████ | █████ | █████ | █████ |
Sex, n (%) | ||||
Female | 31 (53.4) | 33 (56.9) | 61 (51.7) | 129 (57.6) |
Male | 27 (46.6) | 25 (43.1) | 57 (48.3) | 95 (42.4) |
Fitzpatrick Scale skin type,a n (%) | ||||
|---|---|---|---|---|
I | █████ | █████ | █████ | █████ |
II | █████ | █████ | █████ | █████ |
III | █████ | █████ | █████ | █████ |
IV | █████ | █████ | █████ | █████ |
V | █████ | █████ | █████ | █████ |
VI | █████ | █████ | █████ | █████ |
Race,c n (%) | ||||
Asian | 4 (6.9) | 3 (5.2) | 3 (2.5) | 8 (3.6) |
American Indian or Alaska Native | NR | NR | 0 (0.0) | 1 (1.04) |
Black or African American | 5 (8.6) | 4 (6.9) | 3 (2.5) | 11 (4.9) |
Native Hawaiian or Pacific Islander | 1 (1.7) | 0 (0.0) | 0 (0.0) | 1 (1.04) |
White | 42 (72.4) | 48 (82.8) | 107 (90.7) | 180 (80.4) |
Not reported | 2 (3.4) | 1 (1.7) | 3 (2.5) | 13 (5.8) |
Other | 4 (6.9) | 2 (3.4) | 2 (1.7) | 10 (4.5) |
Years since diagnosis | ||||
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Disease status, n (%) | ||||
Stable | █████ | █████ | █████ | █████ |
Progressive | █████ | █████ | █████ | █████ |
F-VASI score | ||||
Mean (SD) | █████ | █████ | █████ | █████ |
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Facial BSA involvement, proportion of the total body | ||||
Mean (SD) | █████ | █████ | █████ | █████ |
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Facial BSA involvement category, n (%) | ||||
< 1.5% | █████ | █████ | █████ | █████ |
1.5% | █████ | █████ | █████ | █████ |
T-VASI score | ||||
Mean (SD) | █████ | █████ | █████ | █████ |
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Total BSA involvement, proportion of the total body | ||||
Mean (SD) | █████ | █████ | █████ | █████ |
Median (minimum to maximum) | █████ | █████ | █████ | █████ |
Prior therapy for vitiligo, n (%) | ||||
No | █████ | █████ | █████ | █████ |
Yes | █████ | █████ | █████ | █████ |
History of acne vulgaris, n (%) | ||||
No | █████ | █████ | █████ | █████ |
Yes | █████ | █████ | █████ | █████ |
Concomitant acne vulgaris on the face, n (%) | ||||
No | █████ | █████ | █████ | █████ |
Yes | █████ | █████ | █████ | █████ |
b.i.d. = twice a day; BSA = body surface area; FAS = full analysis set; F-VASI = Face Vitiligo Area Scoring Index; LTE = long-term extension; SD = standard deviation; T-VASI = Total Vitiligo Area Scoring Index
aPatients randomized to vehicle cream in the double-blind period of the TRuE-V1 or TRuE-V2 trials who crossed over to receive 1.5% ruxolitinib cream after 24 weeks.
bPatients who received 1.5% ruxolitinib cream since study start.
cCategories are as reported in study.
Source: TRuE-V LTE Clinical Study Report.31
Patient Disposition
Patient disposition of the TRuE-V LTE study is summarized in Table 22. A total of 116 participants in cohort A and 342 in cohort B were enrolled across North America and Europe. In cohort A, 77.6% completed treatment, while 22.4% discontinued the study drug, primarily due to participant withdrawal or being lost to follow-up. Cohort B had a 78.9% treatment-completion rate, with 21.1% discontinuing the study drug, mostly for similar reasons, although 2 participants cited a lack of efficacy and 1 discontinued due to an AE.31
Table 22: Patient Disposition of the TRuE-V LTE Study
Patient disposition | Cohort A vehicle cream b.i.d. | Cohort A 1.5% ruxolitinib cream b.i.d. | Cohort B vehicle cream to 1.5% ruxolitinib cream b.i.d.a | Cohort B 1.5% ruxolitinib cream b.i.d. to 1.5% ruxolitinib b.i.d.b |
|---|---|---|---|---|
Randomized, N | 116 | 116 | 342 | 342 |
Received treatment, N (%) | 58 (100.0) | 58 (100.0) | 118 (100.0) | 224 (100.0) |
Completed treatment, N (%) | 41 (70.74) | 49 (84.5) | 93 (78.8) | 177 (79.0) |
Discontinued treatment, N (%) | 17 (29.3%) | 9 (15.5%) | 25 (21.2%) | 47 (21.0%) |
Adverse event | 0 (0.00) | 0 (0.00) | 1 (0.8) | 1 (0.4) |
Death | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Lack of efficacy | 0 (0.00) | 0 (0.00) | 0 (0.0) | 2 (0.9) |
Lost to follow-up | 3 (5.2) | 3 (5.2) | 2 (1.7) | 8 (3.6) |
Physician decision | 0 (0.00) | 0 (0.00) | 1 (0.8) | 1 (0.4) |
Pregnancy | 0 (0.00) | 0 (0.00) | 2 (1.7) | 2 (0.9) |
Protocol violation | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Noncompliance with study drug | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Study terminated by sponsor | 1 (1.7) | 1 (1.7) | 0 | 2 (0.9) |
Withdrawal by patient | 13 (22.4) | 5 (8.6) | 17 (14.4) | 31 (13.8) |
Discontinued treatment due to COVID-19 pandemic | 0 (0.00) | 0 (0.00) | 1 (0.8) | 0 (0.00) |
Other | 0 (0.00) | 0 (0.00) | 1 (0.8) | 0 (0.00) |
FAS, N (%) | 57 (98.3) | 57 (98.3) | 118 (100.0) | 222 (99.1) |
ITT extension, N (%) | 56 (96.6) | 55 (94.8) | NA | NA |
b.i.d. = twice daily; FAS = full analysis set; ITT = intention to treat; LTE = long-term extension; NA = not applicable.
aPatients randomized to vehicle cream in the double-blind period of the TRuE-V1 or TRuE-V2 trials who crossed over to receive 1.5% ruxolitinib cream after 24 weeks.
bPatients who received 1.5% ruxolitinib cream since the study start.
Source: TRuE-V LTE Clinical Study Report.31
Exposure to Study Treatments
Participants in cohort A (those who achieved a response of greater than or equal to an F-VASI90 at week 52) applied the study cream for durations that varied by their treatment group. Participants who were initially randomized to vehicle cream twice a day applied the study cream for a median of ██████ days. Those who experienced a disease relapse (less than an F-VASI75), applied 1.5% ruxolitinib cream twice daily for a median of ██████ days. Participants randomized to 1.5% ruxolitinib cream twice daily applied the study cream for a median of 364.00 days31 (Table 23).
The median daily amount of study drug applied was comparable across groups: ████ g for those randomized to 1.5% ruxolitinib cream twice daily, ████ g for those initially on vehicle cream twice a day who switched to ruxolitinib following relapse, and similar values for those who remained on vehicle cream twice a day. Study drug application compliance was high, with a median of ██████ ██████ █ █████ ██████ for participants applying 1.5% ruxolitinib cream twice a day and ██████ ██████ █ █████ ██████ for those on vehicle cream twice daily31 (Table 23).
In cohort B (participants who had not achieved a response greater than or equal to an F-VASI90 at week 52), the median duration of study drug exposure was ██████ days for both treatment groups. Among participants initially treated with vehicle cream and later switched to 1.5% ruxolitinib cream, the median daily amount of ruxolitinib cream applied was ████ g, with a median study drug application compliance rate of ███████ In the group that continued ruxolitinib cream throughout the study, the median daily amount applied was ████ g, with a median study drug compliance rate of ███████31 (Table 24).
Table 23: Summary of Patient Exposure in the Cohort A (FAS)
Exposure | TRuE-V LTE | |||
|---|---|---|---|---|
Vehicle cream b.i.d. (N = 58)a | Vehicle cream b.i.d. to 1.5% ruxolitinib cream b.i.d. (N = 23)b | 1.5% ruxolitinib cream b.i.d. (N = 58)c | 1.5% ruxolitinib cream b.i.d. total (N = 81)d | |
Duration of treatment (days) | ||||
Mean (SD) | ██████ | ██████ | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ | ██████ | ██████ |
Average weight of study drug (g) applied daily during the LTE period | ||||
Mean (SD) | ██████ | ██████ | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ | ██████ | ██████ |
Total weight of study drug (g) applied daily during the LTE period | ||||
Mean (SD) | ██████ | ██████ | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ | ██████ | ██████ |
b.i.d. = twice a day; FAS = full analysis set; LTE = long-term extension; SD = standard deviation.
aThis represents the total group of patients randomized to vehicle cream, which includes those who remained on the vehicle cream for the entire study, and those whose disease relapsed and switched to 1.5% ruxolitinib cream.
bThese patients were initially randomized to vehicle cream and then were switched onto 1.5% ruxolitinib cream after their disease relapsed.
cThese patients received 1.5% ruxolitinib cream for the entire study period.
dThis is the combined group of patients who received 1.5% ruxolitinib cream at any point during the study period, which includes those initially randomized to the vehicle cream and then were switched onto 1.5% ruxolitinib cream after their disease relapsed.
Source: TRuE-V LTE Clinical Study Report.31
Table 24: Summary of Patient Exposure in the Cohort B (FAS)
Exposure | Vehicle cream b.i.d. to (N = 118)a | Ruxolitinib 1.5% cream b.i.d. to 1.5% ruxolitinib cream b.i.d. (N = 224)b |
|---|---|---|
Duration of treatment (days) | ||
Mean (SD) | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ |
Average weight of study drug (g) applied daily during the LTE period | ||
Mean (SD) | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ |
Total weight of study drug (g) applied daily during the LTE period | ||
Mean (SD) | ██████ | ██████ |
Median (minimum to maximum) | ██████ | ██████ |
b.i.d. = twice a day; FAS = full analysis set; SD = standard deviation; LTE = long-term extension.
aPatients randomized to vehicle cream in the double-blind period of the TRuE-V1 or TRuE-V2 trial who crossed over to receive 1.5% ruxolitinib cream after 24 weeks.
bPatients who received 1.5% ruxolitinib cream since study start.
Source: TRuE-V LTE Clinical Study Report.31
Concomitant Medications
Concomitant medications used in the TRuE-V LTE study are listed in Table 25 by WHO drug class. In cohort A, medications used by 10% or more of participants included other viral vaccines, thyroid hormones and anilides, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, propionic acid derivatives, and progestogens and estrogens in fixed combinations. The most frequently reported concomitant medications by WHO drug term were tozinameran (█████) and paracetamol (█████). Other medications used by greater than or equal to ██ of participants included elasomeran, ethinyl estradiol or levonorgestrel, ibuprofen, levothyroxine, salbutamol, vitamin B12, and other unspecified vitamins.31
Table 25: Most Commonly Used Concomitant Medications During the TRUE-V LTE Study
Medication, n (%) | Cohort A vehicle cream b.i.d. | Cohort A 1.5% ruxolitinib cream b.i.d. | Cohort B vehicle cream to 1.5% ruxolitinib cream b.i.d.a | Cohort B 1.5% ruxolitinib cream b.i.d. to ruxolitinib 1.5% b.i.d.b |
|---|---|---|---|---|
Any concomitant medication, n (%) | █████ | █████ | █████ | █████ |
Other viral vaccines | █████ | █████ | █████ | █████ |
Thyroid hormones | █████ | █████ | █████ | █████ |
Anilides | █████ | █████ | █████ | █████ |
HMG-CoA reductase inhibitors | █████ | █████ | █████ | █████ |
Propionic acid derivatives | █████ | █████ | █████ | █████ |
Progestogens and estrogens, fixed combinations | █████ | █████ | █████ | █████ |
Vitamin D and analogues | █████ | █████ | █████ | █████ |
b.i.d. = twice a day; HMG-CoA = 3-hydroxy-3-methyl glutaryl coenzyme A; LTE = long-term extension.
aPatients randomized to vehicle cream in the double-blind period of the TRuE-V1 or TRuE-V2 trial who crossed over to receive 1.5% ruxolitinib cream after 24 weeks.
bPatients who received 1.5% ruxolitinib cream since study start.
Source: TRuE-V LTE Clinical Study Report.31
In cohort B, medications used by 10% or more of participants included other viral vaccines, thyroid hormones and anilides, propionic acid derivatives, and vitamin D and analogues. Additional medications used by at least 5% of participants included ascorbic acid, colecalciferol, other unspecified vitamins, elasomeran, levothyroxine, metformin, and vitamin D. The most frequently used medications were similar between participants initially randomized to vehicle cream and those randomized to 1.5% ruxolitinib cream in the parent study and were generally consistent with prestudy medication use.31
Efficacy
Primary End Point — Time to Relapse (Less Than an F-VASI75)
The majority of patients who achieved complete or near-complete repigmentation of the face (cohort A, patients who had achieved greater than or equal to F-VASI90 at week 52) in the parent studies did not relapse (less than an F-VASI75) while on study. The median time to relapse (less than an F-VASI75) was not evaluable (NE) for either the ruxolitinib cream group (95% CI, NE to NE) or the vehicle cream treatment group (95% CI, 238.0 to NE) due to the small number of patients who experienced disease relapse (Table 26 and Figure 3).31 Fewer patients experienced disease relapse in the ruxolitinib cream group (14.5%) compared with the vehicle cream group (28.6%), and the risk of relapse was lower for patients who continued to apply ruxolitinib cream beyond week 52 compared with patients who applied vehicle cream after week 52 (HR = 0.422; 95% CI, 0.180 to 0.990; P = 0.0414).31
Table 26: Summary and Analysis of Time to Relapse (Less Than an F-VASI75) (ITT Extension Population)
Variable | Vehicle cream b.i.d. (N = 56) | Ruxolitinib 1.5% cream b.i.d. (N = 55) |
|---|---|---|
Number (%) of participants with event | 16 (28.6) | 8 (14.5) |
Number (%) of participants censored | 40 (71.4) | 47 (85.5) |
End of treatment | 22 (39.3) | 38 (69.1) |
Treatment discontinuation | 13 (23.2) | 7 (12.7) |
Rescue treatment without relapse | 5 (8.9) | 2 (3.6) |
Median time to relapse | ||
95% CI | NE (238.0 to NE) | NE (NE to NE) |
Treatment versus vehiclea | ||
HR | 0.422 | |
95% CI | 0.180 to 0.990 | |
P value | 0.0414 | |
b.i.d. = twice a day; CI = confidence interval; F-VASI = Facial Vitiligo Area Scoring Index; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; HR = hazard ratio; ITT = intention to treat; NE = not evaluable.
Note: Relapse was defined as a loss of response, assessed as improvement in the F-VASI score at baseline (day 1 of the parent study) to less than 75%.
aCox regression model stratified by stratification factor (treatment assignment in the parent studies) was conducted to compare the difference in hazard rate between the treatment and vehicle cream. The P value was based on the log-rank test stratified by randomization stratification factor between treatment and vehicle cream.
Source: TRuE-V LTE Clinical Study Report.31
Figure 3: Kaplan-Meier Curve of the Time to Relapse (ITT Extension Population)
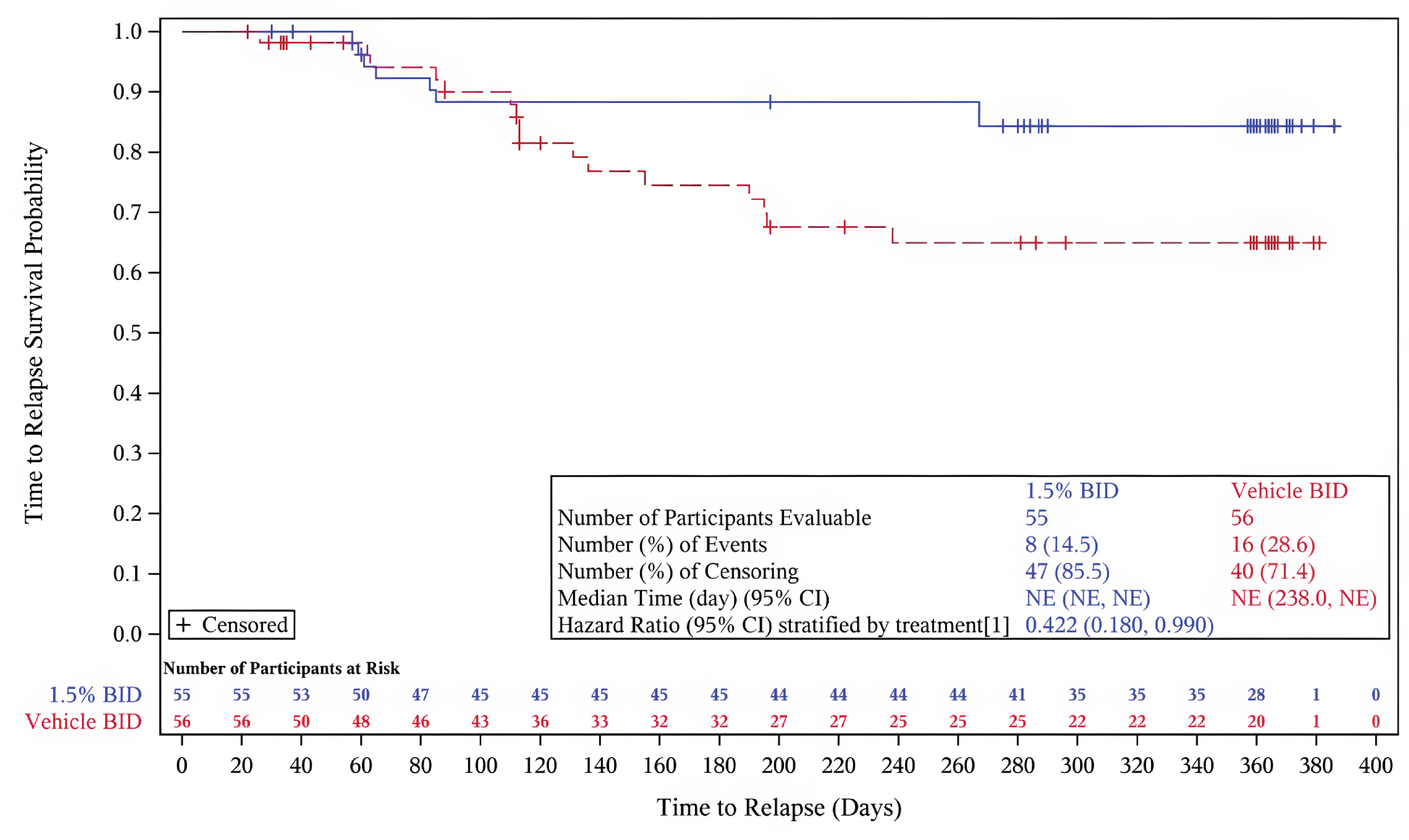
BID = twice a day; CI = confidence interval; ITT = intention to treat; NE = not evaluable.
Note: Relapse is defined as a loss of an F-VASI75 response, assessed as percentage improvement in the F-VASI score at baseline (day 1 of the parent study) to less than 75%.
Source: TRuE-V LTE Clinical Study Report.31
Key Secondary End Points
Time to Maintain F-VASI90 Response
The majority of patients who achieved a complete or near-complete repigmentation of the face (cohort A, patients who had achieved a response of greater than or equal to an F-VASI90 at week 52) in the parent studies maintained this level of repigmentation with continued application of ruxolitinib cream beyond week 52. Of the cohort of patients who received vehicle cream, 55.4% lost their F-VASI90 response.31 The median time to loss of an F-VASI90 in the group of patients who received the vehicle cream was 195.0 days (95% CI, 113.0 days to 372.0 days). Of the cohort of patients who applied ruxolitinib cream in the double-blind period then continued treatment with ruxolitinib and achieved an F-VASI90 response, 23.6% lost their F-VASI90 response31 (Table 27). The median time to loss of an F-VASI90 response in this cohort was not evaluable.31 The risk of losing an F-VASI90 response was lower for patients who continued to use ruxolitinib cream compared with patients who applied the vehicle cream (HR = 0.316; 95% CI, 0.165 to 0.606; P = 0.0003).31
Figure 4: Kaplan-Meier Curve for Maintenance of an F-VASI90 Response (ITT Extension Population)
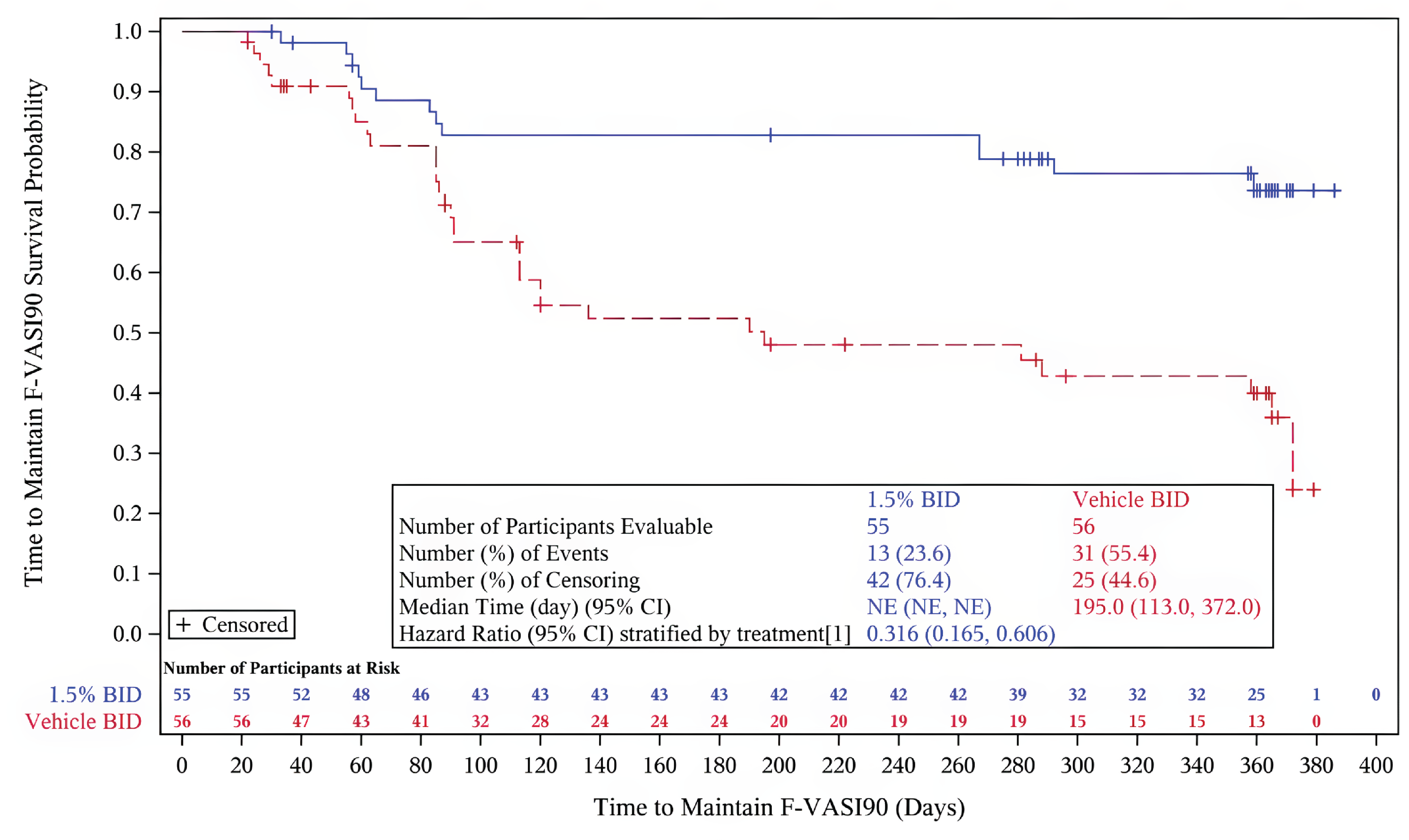
BID = twice a day; CI = confidence interval; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; ITT = intention to treat; NE = not evaluable.
Note: Loss of adequate response is defined as a loss of F-VASI90 response, assessed as percentage improvement in the Facial Vitiligo Area Scoring Index score at baseline (day 1 of the parent study) to less than 90%. The hazard ratio stratification factor was the initial treatment assigned in the parent studies.
Source: TRuE-V LTE Clinical Study Report.31
Table 27: Summary and Analysis of Time to Maintain an F-VASI90 Response (ITT Extension Population)
Variable | Vehicle cream b.i.d. (N = 56) | 1.5% ruxolitinib cream b.i.d. (N = 55) |
|---|---|---|
Number (%) of participants with event | 31 (55.4) | 13 (23.6) |
Number (%) of participants censored | 25 (44.6) | 42 (76.4) |
End of treatment | 12 (21.4) | 34 (61.8) |
Treatment discontinuation | 10 (17.9) | 6 (10.9) |
Rescue treatment without lost response | 3 (5.4) | 2 (3.6) |
Median time to loss of adequate response (days) | ||
95% CI | 195 (113.0 to 372.0) | NE (NE to NE) |
Treatment versus vehiclea | ||
HR | 0.316 | |
95% CI | 0.165 to 0.606 | |
P value | 0.0003 | |
b.i.d. = twice a day; CI = confidence interval; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; HR = hazard ratio; ITT = intention to treat; NE = not evaluable.
Note: Loss of adequate response is defined as a loss of F-VASI90 response, assessed as percentage improvement in the Facial Vitiligo Area Scoring Index score at baseline (day 1 of the parent study) to less than 90%.
aA Cox regression model stratified by stratification factor (treatment assignment in the parent studies) was conducted to compare the difference in hazard rate between treatment and vehicle cream. The P value was based on the log-rank test stratified by randomization stratification factor between treatment and vehicle cream.
Source: TRuE-V LTE Clinical Study Report.31
Additional Secondary End Points
Vitiligo Area Scoring Index
Proportions of patients achieving an F-VASI 50, F-VASI75, or F-VASI90: In cohort B, at week 104, for those who continued using 1.5% ruxolitinib cream, 86.4%, 66.1%, and 33.9% achieved an F-VASI50, F-VASI75, or F-VASI90, respectively. Among those initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the TRuE-V LTE study, the respective rates were 69.9%, 47.3%, and 28.0%.31
Proportions of patients achieving T-VASI75: In cohort A, the proportions of participants continuing on 1.5% ruxolitinib cream who achieved a T-VASI75 were 42.1% at week 52 and 55.3% at week 104. Among participants using vehicle cream, 38.6% achieved a T-VASI75 at week 52 and 39.1% at week 104.
In cohort B at week 52, for those who continued using 1.5% ruxolitinib cream, 12.2% achieved a T-VASI75, and by week 104, 30.5% achieved this threshold. Among participants initially randomized to the vehicle cream who switched to 1.5% ruxolitinib cream during the TRuE-V LTE study, 3.4% achieved a T-VASI75 at week 52 and 18.3% at week 104.31
Vitiligo Noticeability Scale
Proportions of patients achieving a VNS of “a lot less noticeable” or “no longer noticeable”: In cohort A, the proportions of participants receiving blinded treatment who reported a VNS score of 4 (a lot less noticeable) or 5 (no longer noticeable) remained generally stable compared to week 52. Among participants receiving 1.5% ruxolitinib cream, 50.0% reported a score of 4 or 5 at week 104, compared to 42.1% at week 52. Among participants receiving the vehicle cream, 56.5% reported a score of 4 or 5 at week 104, compared to 49.1% at week 52.31
In cohort B, the proportion of participants achieving a VNS score of 4 or 5 for those who continued using 1.5% ruxolitinib cream remained generally stable (43.3% at week 104 versus 35.3% at week 52). The VNS score of 4 or 5 increased from week 52 to week 104 for those initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the TRuE-V LTE study (30.1% at week 104 versus 11.9% at week 52).31
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
█████ ███ ██ █████ ███████ ██ ██████ ██ ████ █████ ██████ ███ ████ ████████ ██████ ████████ ██ ████ ██ ██████ ███ ███████ █████████ ██████ ███ ████████████ ██ ██████ █ ███ ████ ██ ███████ █████████ ███ ████████████ ██ ██████.
For cohort A, irrespective of treatment group, participants had mean total scores between ████ and ████ throughout the 52-week treatment period. At week 104 in cohort A, the mean changes from week 52 in total score were █████ in the 1.5% ruxolitinib cream treatment group and ████ in the vehicle cream twice a day treatment group.31
For cohort B, irrespective of initial randomization, participants had mean total scores between ████ and ████ throughout the 52-week treatment period. At week 104 in cohort B, the mean changes from week 52 in the total score were █████ for participants initially randomized to 1.5% ruxolitinib cream in the parent study and █████ for participants initially randomized to vehicle cream and then switched to 1.5% ruxolitinib cream.31
Change From Baseline in CDLQI (Aged Younger Than 16 Years)
CDLQI scores were reported for a small number of participants: 9 participants in cohort A (6 applying 1.5% ruxolitinib cream and 3 applying vehicle cream) and 26 participants in cohort B (all applying 1.5% ruxolitinib cream twice a day). Due to the small sample size, no definitive conclusions could be drawn from the data.
At week 104 in cohort A, the mean change from week 52 in CDLQI score was ████ in participants who applied 1.5% ruxolitinib cream and ████ in participants who applied vehicle cream.31
At week 104 in cohort B (participants who had not achieved a response greater than or equal to an F-VASI90 at week 52), the mean changes from week 52 in CDLQI score were █████ in participants initially randomized to 1.5% ruxolitinib cream in the parent study and █████ in participants initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during TRuE-V LTE study.31
Vitiligo-Specific Quality of Life
There was no clear pattern of change in VitiQoL total or subscale scores for participants in cohort A who were receiving blinded treatment during the 52-week treatment period. At week 104 in cohort A, the mean changes from week 52 in total score were █████ in the 1.5% ruxolitinib cream twice a day treatment group and ████ in the vehicle cream twice a day treatment group.31
Participants in cohort B (participants who had not achieved a response greater than or equal to an F-VASI90 at week 52) who continued to apply 1.5% ruxolitinib cream showed improvement in VitiQoL total scores relative to week 52; at week 104, the mean changes from week 52 were █████ for participants initially randomized to 1.5% ruxolitinib cream in the parent study and █████ for participants initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the LTE study.31
Exploratory End Points
Time to Regain F-VASI Response
The median time to regain an F-VASI75 for patients randomized to vehicle cream in cohort A (patients who had achieved a response greater than or equal to an F-VASI90 at week 52) who experienced disease relapse (less than an F-VASI75) and received open-label rescue treatment was 85.0 days (95% CI, 43.0 days to 106.0 days) (Figure 5).31 Although some patients randomized to 1.5% ruxolitinib cream in cohort A experienced disease relapse, the majority (5 of 8 patients [62.5%]) regained an F-VASI75 with continued application of ruxolitinib cream; the median time to regain an F-VASI75 was 205.0 days (95% CI, 27.0 days to NE).31
The median time to regain an F-VASI90 for patients randomized to the vehicle cream in cohort A who experienced disease relapse and received open-label rescue treatment was 106.0 days (95% CI, 50.0 days to NE)31 (Figure 6). For patients randomized to ruxolitinib cream in cohort A who experienced disease relapse and continued to apply ruxolitinib cream, the median time to regain an F-VASI90 was 205.0 days (95% CI, 36.0 days to NE).31
Figure 5: Kaplan-Meier Curve of the Time to Regain an F-VASI75 Response (RES)
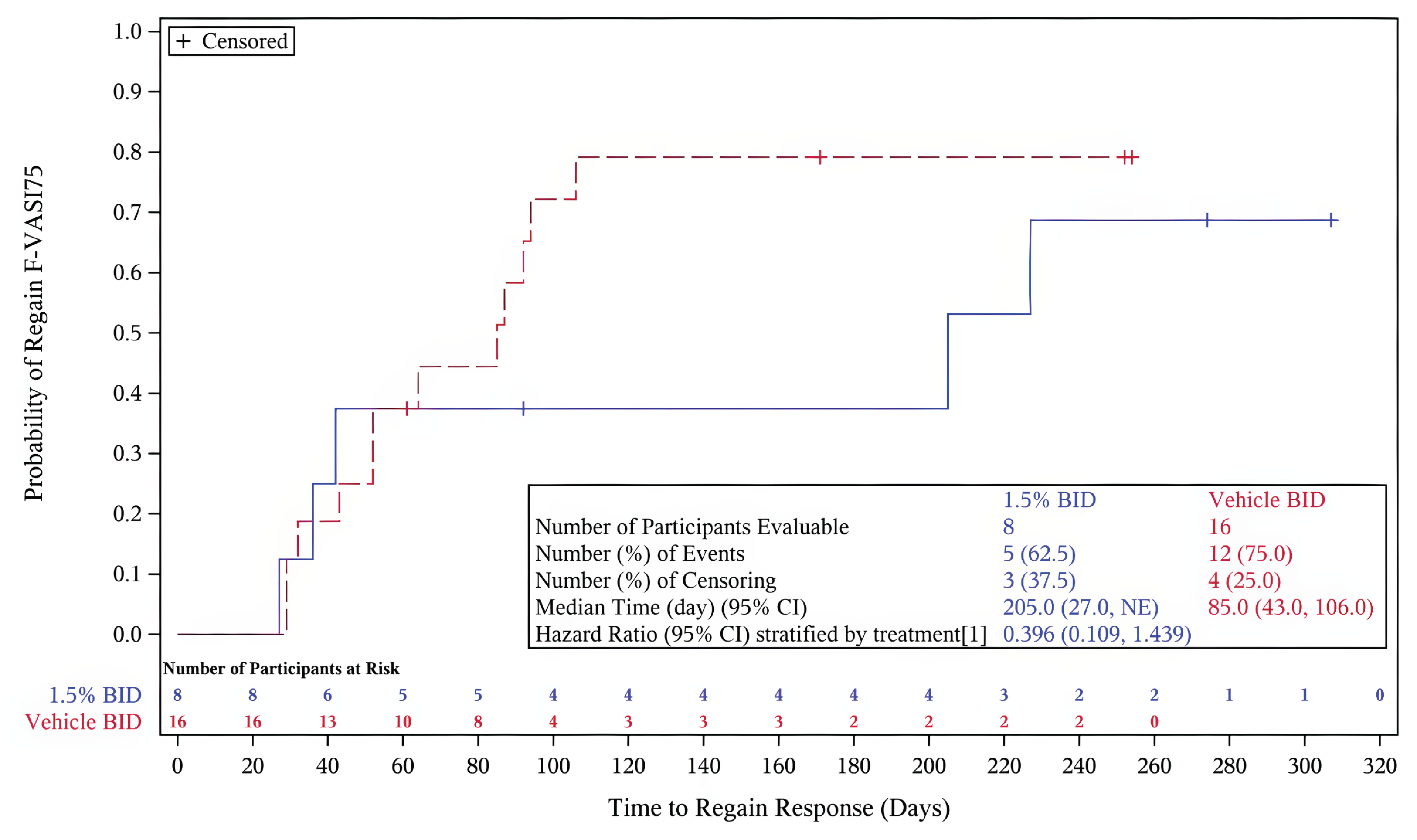
BID = twice a day; CI = confidence interval; F-VASI75 = 75% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; NE = not evaluable.
Note: The hazard ratio stratification factor was the initial treatment assigned in the parent studies.
Source: TRuE-V LTE Clinical Study Report.31
Figure 6: Kaplan-Meier Curve of the Time to Regain F-VASI90 Response (RES)
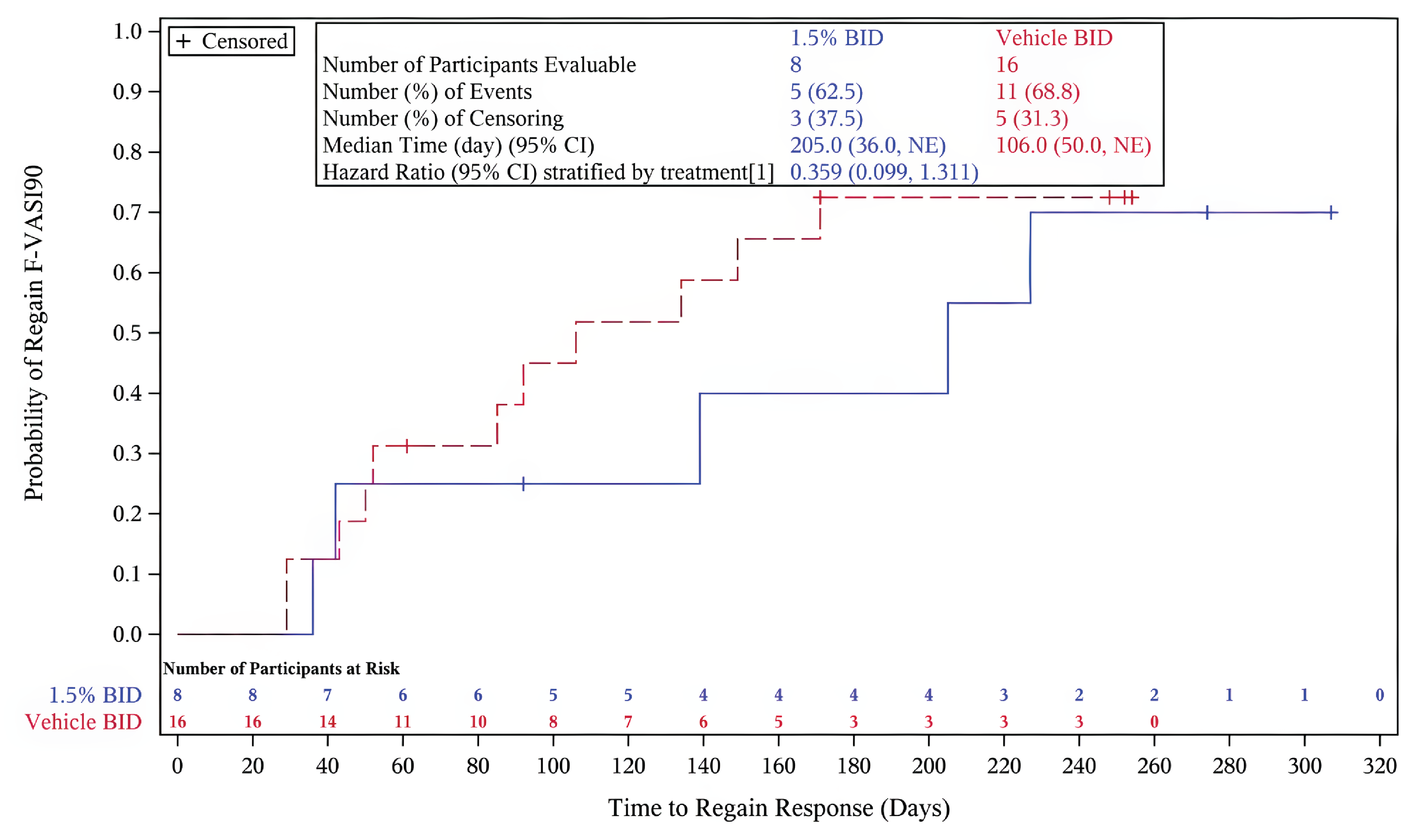
BID = twice a day; CI = confidence interval; F-VASI90 = 90% or more improvement from baseline on the Facial Vitiligo Area Scoring Index; NE = not evaluable.
Note: The hazard ratio stratification factor was the initial treatment assigned in the parent studies.
Source: TRuE-V LTE Clinical Study Report.31
Harms
An overall summary of TEAEs for participants in TRuE-V LTE (cohorts A and B) is presented in Table 28.
The overall incidences of TEAEs (all causality) and application site reactions for cohort A were higher among patients who applied ruxolitinib cream (55.2% and █████ respectively) compared with the vehicle cream treatment group (36.2% and ████, respectively).
One participant treated with 1.5% ruxolitinib cream in cohort A (1.7%) had a serious TEAE, and no participant had a TEAE with a fatal outcome or a TEAE leading to study drug discontinuation.31
The overall incidences of TEAEs and application site reactions in cohort B were 50.9% and ████, respectively, among patients who continued receiving 1.5% ruxolitinib cream, compared to 50.0% and ████ for participants initially randomized to vehicle cream who switched to 1.5% ruxolitinib cream during the LTE study.31 The incidence of serious TEAEs was 3.1% in patients treated with 1.5% ruxolitinib cream and 3.4% in those initially treated with vehicle cream. The only TEAE leading to discontinuation of the study drug in cohort B was ████ ███████ accident (in 1 participant) that the investigator assessed as unlikely to be related to the study drug.31
Table 28: Summary of Harms From TRuE-V LTE Study (FAS)
Harms | Cohort A vehicle cream (N = 58) | Cohort A 1.5% ruxolitinib cream (N = 58) | Cohort B vehicle cream to 1.5% ruxolitinib cream (N = 118)a | Cohort B 1.5% ruxolitinib cream to 1.5% ruxolitinib cream (N = 224)b |
|---|---|---|---|---|
Most common TEAEs, n (%) | ||||
≥ 1 adverse event | 21 (36.2) | 32 (55.2) | 59 (50.0) | 114 (50.9) |
COVID-19 | 6 (10.3) | 9 (15.5) | 11 (9.3) | 34 (15.2) |
Anemia | 0 | 2 (3.4) | NR | NR |
Upper respiratory tract infection | 0 | 4 (6.9) | 6 (5.1) | 4 (1.8) |
Urinary tract infection | NR | NR | 1 (0.8) | 7 (3.1) |
Viral infection | NR | NR | 4 (3.4) | 1 (0.4) |
Application site dermatitis | 0 | 2 (3.4) | 1 (0.8) | 4 (1.8) |
Application site pruritis | NR | NR | 2 (1.7) | 2 (0.9) |
Application site rash | 0 | 2 (3.4) | NR | NR |
Application site acne | NR | NR | 2 (1.7) | 4 (1.8) |
Gastroenteritis | NR | NR | 1 (0.8) | 5 (2.2) |
Hemorrhoids | 1 (1.7) | 2 (3.4) | NR | NR |
Diarrhea | NR | NR | 1 (0.8) | 4 (1.8) |
Arthralgia | NR | NR | 1 (0.8) | 3 (1.3) |
Headache | 2 (3.4) | 2 (3.4) | 1 (0.8) | 5 (2.2) |
Sinusitis | NR | NR | 1 (0.8) | 5 (2.2) |
Anxiety | NR | NR | 0 | 5 (2.2) |
Nasopharyngitis | 2 (3.4) | 2 (3.4) | 5 (4.2) | 11 (4.9) |
Pharyngitis | NR | NR | 1 (0.8) | 4 (1.8) |
Toothache | 2 (3.4) | 1 (1.7) | NR | NR |
Bronchitis | 2 (3.4) | 0 | 3 (2.5) | 1 (0.4) |
Cough | 2 (3.4) | 0 | NR | NR |
Increased blood creatine phosphokinase | NR | NR | 1 (0.8) | 3 (1.3) |
Muscle strain | 2 (3.4) | 0 | NR | NR |
Skin papilloma | 2 (3.4) | 0 | NR | NR |
Pyrexia | NR | NR | 2 (1.7) | 2 (0.9) |
Tachycardia | NR | NR | 1 (0.8) | 3 (1.3) |
Patients who had application site reaction | 2 (3.4) | 4 (6.9) | 6 (5.1) | 19 (8.5) |
Serious TEAEs, n (%) | ||||
Patients with ≥ 1 SAE | 0 | 1 (1.7) | 4 (3.4) | 7 (3.1) |
Uterine leiomyoma | 0 | 1 (1.7) | NR | NR |
Acute respiratory failure | NR | NR | 0 | 1 (0.4) |
Angina pectoris | NR | NR | 0 | 1 (0.4) |
Bipolar I disorder | NR | NR | 1 (0.08) | 0 |
COVID-19 pneumonia | NR | NR | 1 (0.08) | 0 |
Cholelithiasis | NR | NR | 0 | 1 (0.4) |
Cystocele | NR | NR | 0 | 1 (0.4) |
Hip fracture | NR | NR | 0 | 1 (0.4) |
Intervertebral disc disorder | NR | NR | 0 | 1 (0.4) |
Mental status changes | NR | NR | 0 | 1 (0.4) |
Otosclerosis | NR | NR | 0 | 1 (0.4) |
Pelvic prolapse | NR | NR | 1 (0.8) | 0 |
Rectocele | NR | NR | 0 | 1 (0.4) |
Spinal fracture | NR | NR | 1 (0.8) | 0 |
Spinal osteoarthritis | NR | NR | 0 | 1 (0.4) |
Uterine prolapse | NR | NR | 0 | 1 (0.4) |
TEAEs of interest | ||||
Any erythropenia TEAE | 0 | 2 (3.4) | 2 (1.7) | 2 (0.9) |
Any neutropenia TEAE | 0 | 1 (1.7) | 0 | 2 (0.9) |
Any thrombocytopenia TEAE | 0 | 0 | 0 | 1 (0.43) |
TEAEs leading to discontinuation of study drug, n (%) | ||||
Patients who stopped | 0 | 0 | 1 (0.8) | 0 |
████ ███████ ██ | 0 | 0 | 1 (0.8) | 0 |
Deaths, n (%) | ||||
Patients who died | 0 | 0 | 0 | 0 |
FAS = full analysis set; LTE = long-term extension; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aPatients randomized to vehicle cream in the double-blind period of the TRuE-V1 or TRuE-V2 trial who crossed over to receive 1.5% ruxolitinib cream after 24 weeks.
bPatients who received 1.5% ruxolitinib cream since study start.
Source: TRuE-V LTE Clinical Study Report.31
Critical Appraisal
Internal Validity
Cohort A of the TRuE-V LTE study used appropriate random allocation in the form of an interactive response technology system. Allocation concealment was ensured, and both patients and investigators remained blinded to treatment assignment until the study’s conclusion. Baseline characteristics between groups were well balanced, supporting the validity of comparisons. The single-arm design of cohort B introduced a potential bias in assessing efficacy outcomes, as both participants and investigators were aware of the treatment being administered. This lack of blinding could lead to detection bias. Additionally, the single-arm nature of the study inherently carries a high risk of bias, which may influence the assessment of subjective treatment outcomes, and no conclusion can be made on the comparative efficacy and safety.
Participants selected for the TRuE-V LTE study represented a subsample of the parent trials’ populations, consisting of those who completed the parent trials without safety concerns following ruxolitinib use. This selection process may have introduced a risk of selection bias: it could limit the representativeness of the wider population as patients enrolled in this study are likely overrepresented by patients who will respond well to treatment because they had to make it through the trial first.
The TRuE-V LTE study had a high dropout rate and, unlike the pivotal trials, imputation methods to address missing data were not employed, increasing the risk of attrition bias. The substantial number of missing participants may have skewed the results and affected the interpretation of the findings.
External Validity
The TRuE-V LTE study consisted of patients who took part in the pivotal studies (the TRuE-V1 and TRuE-V2 trials); it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies. While the studies were conducted in centres in Europe and North America, the patient population of those studies may be reflective of the population in Canada, and the clinical evidence is generalizable to the setting in Canada.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
No indirect evidence was submitted by the sponsor and reviewed by the review team.
The sponsor indicated that it conducted a feasibility assessment to determine whether it was possible to establish the comparative efficacy and safety of ruxolitinib compared with the following identified off-label therapies for the treatment of vitiligo: topical or oral JAK inhibitors (e.g., ruxolitinib); topical or oral corticosteroids; topical or oral calcineurin inhibitors (e.g., tacrolimus); phototherapy; laser therapy; topical vitamin D analogues; and combinations of phototherapy with topical therapies (corticosteroid or calcineurin inhibitor). The sponsor rated the feasibility of conducting robust evidence synthesis as low due to the low quality and small sample sizes of the identified studies, considerable between-study heterogeneity, and lack of information or incomplete reporting on outcomes of interest to the ruxolitinib review, all of which resulting in disconnected and sparse evidence networks. The sponsor reported conducting 3 exploratory analyses, each of which yielded highly uncertain results. The efficacy and safety of ruxolitinib compared with any off-label therapies for the treatment of vitiligo are therefore unknown.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the review team.
No studies addressing gaps in the systematic review evidence were reviewed.
Discussion
Summary of Available Evidence
The 2 reviewed studies, TRuE-V1 (n = 330) and TRuE-V2 (n = 344), were phase III, multicentre, double-blind, vehicle-controlled RCTs identically designed to evaluate the efficacy and safety of 1.5% ruxolitinib topical cream, applied twice a day to depigmented areas, for the treatment of nonsegmental vitiligo in adult and pediatric patients aged 12 years and older. The primary outcome was the proportion of patients achieving a response of at least an F-VASI75 at week 24.
Input from clinical experts suggested that findings from the TRuE-V1 and TRuE-V2 trials may be generalizable to a selected sample of individuals in Canada with vitiligo. The majority of patients included in the studies were white and had a lighter skin tone and, considering disease duration, prior therapies, and baseline HRQoL, there is a possibility that the trial population may not be representative of patients whose condition interferes substantially with their daily life.
Additional evidence assessed in the ruxolitinib review included the open-label treatment-extension period from the TRuE-V1 and TRuE-V2 trials, as well as the TRuE-V LTE study.
Interpretation of Results
Efficacy
The primary outcome in the TRuE-V1 and TRuE-V2 trials was improvement in the F-VASI score, which captures the overall surface area of vitiligo involvement and the degree of repigmentation in the face. Results from the 2 studies were consistent in suggesting that a larger proportion of patients who applied ruxolitinib achieved a response of an F-VASI75 compared with patients who applied vehicle cream over 24 weeks, which was identified in the literature as the threshold for clinically meaningful change in repigmentation.26 Among patients who applied ruxolitinib, the proportions of those who responded and achieved an F-VASI75 was 31% in both trials, resulting in between-group differences in the response rate versus vehicle treatment of 22.3% (SE = 4.15; 95% CI, 14.2 to 30.5; P < 0.0001) in the TRuE-V1 trial and 19.5% (SE = 4.56; 95% CI, 10.5 to 28.4; P = 0.0004) in the TRuE-V2 trial. According to the clinical experts consulted for this review, the F-VASI75 response was relevant to inform on the objective response to treatment, as measured by the level of disease regression and skin repigmentation. Results for other levels of F-VASI improvements were consistent with those for the primary outcome and suggested that a larger proportion of patients who applied ruxolitinib achieved each level of improvement in VASI score compared with patients who applied vehicle cream over 24 weeks. Results for T-VASI responses were of a smaller magnitude; however, the clinical experts noted that vitiligo on the body is more difficult to treat than on facial areas.
The difference between treatments was considered clinically meaningful in terms of repigmentation, but the clinical experts noted that the overall impact of the objective response was difficult to assess. The experts noted that different individuals are likely to have different priorities and objectives when assessing response to treatment and, as such, the minimal objective response required to have a clinically meaningful impact can be highly variable among patients, depending on how the disease affects their daily lives. Vitiligo can range from being barely perceptible to cosmetically distressing. In clinical practice, treatment is targeted at improving the current and future quality of life, which is not captured by the VASI score, therefore going beyond the surface area of involvement and degree of repigmentation. This was also consistent with patient and clinician input, all of which highlighted the importance of improving the psychosocial impact of the disease on quality of life by seeking reliable repigmentation that reduces patchiness and delivers lasting, comprehensive results. Partial repigmentation, such as improvements in responsive areas such as the cheeks and neck, may not necessarily be associated with a meaningful change if other known resistant sites, such as periocular or perioral areas, cannot be repigmented. In the clinical experts’ experience, HRQoL may be affected for as long as the disease is visible.
Results for patient-reported noticeability, assessed as a key secondary outcome using the VNS, suggest that a larger proportion of patients who applied ruxolitinib achieved MCID scores of 4 (a lot less noticeable) or 5 (no longer noticeable) versus patients who applied vehicle cream over 24 weeks. Among patients who applied ruxolitinib, the proportions of those who responded were 25% in the TRuE-V1 trial and 21% in the TRuE-V2 trial, resulting in between-group differences in response rate versus vehicle treatment of 21.2% (SE = 3.54; 95% CI, 14.3 to 28.1; P = 0.0002) in the TRuE-V1 trial and 15.5% (SE = 3.58; 95% CI, 8.5 to 22.6; P = 0.0013) in the TRuE-V2 trial. Results for the TSQM suggest that patients who applied ruxolitinib showed increased treatment satisfaction versus patients who received vehicle cream in both studies.
HRQoL was measured using the DLQI or CDLQI as well as the VitiQoL and assessed as an exploratory outcome. The studies were not designed to test for differences in HRQoL and any interpretation of these findings should take this into account. Results from both trials did not suggest benefits for HRQoL from ruxolitinib. The differences between ruxolitinib and vehicle cream, as well as the within-group changes from baseline observed with the use of ruxolitinib, were not considered clinically meaningful by the clinical experts consulted for this review. This indicates that, despite observing an objective response to ruxolitinib in terms of overall surface area of involvement and degree of repigmentation, which made the condition less noticeable in some patients, ruxolitinib did not reduce the impact of the disease on HRQoL in the overall study population. Findings from post hoc analyses in patients who received ruxolitinib comparing the change in VitiQoL and DLQI (from baseline to week 24 among patients who achieved various levels of F-VASI responses and those who did not) suggest that patients may observe an improvement in their HRQoL with at least an F-VASI75; however, whether the improvement in HRQoL is clinical meaningful is uncertain. In addition, interpretation of these findings is limited by the post hoc nature of the analyses. The experts emphasized that several additional key issues of vitiligo that were not captured in the VASI score may have an important impact on patients’ daily life, such as the disease site and associated visibility, the level of heterogeneity in repigmentation, differences between the colour of the repigmented area and background skin tone, as well as family and cultural influences. Of note, the DLQI, CDLQI, and VitiQoL baseline values suggest that there may have been a relatively low level of HRQoL impairment at baseline in the trial populations. Given the long-lasting condition (the median time since diagnosis exceeded 10 years in both studies) and the fact that 38% of patients never received prior therapy for vitiligo, the clinical experts cautioned that the trial population may not be representative of patients whose condition interferes substantially with their daily life, as it would be expected in clinical practice that virtually all patients who sustain a negative impact from vitiligo would have sought and received treatment.
Prespecified subgroup analyses included skin type and race, which were considered by the clinical experts to be particularly relevant to the treatment of vitiligo. Although vitiligo affects all races and types of skin colour, the condition is particularly visible in patients with darker skin tones, who therefore are likely to experience a greater impact on quality of life. However, few patients with darker skin tones were included in the studies. Of the 661 patients in the TRuE-V1 and TRuE-V2 trials combined, 31 (4.7%) were Black or African American, 26 (3.9%) were Asian, and 2 (0.3%) were Native Hawaiian or Pacific Islander. As for skin colour according to the Fitzpatrick Scale, 43 patients (6.5%) had skin type V (dark brown) and 13 (2.0%) had skin type VI (deeply pigmented dark brown to darkest brown). Subgroups were therefore underpowered, and these findings should be viewed as supplemental to the primary results.
Evidence gaps also include the fact that patients were followed for 24 weeks, while treatment is more likely to continue over the long-term. To inform this gap, the double-blind controlled period in the TRuE-V1 and TRuE-V2 trials was followed by a 28-week open-label treatment extension, for a total follow-up of 52 weeks. In addition, the TRuE-V LTE, a vehicle-controlled, randomized withdrawal extension trial of the TRuE-V1 and TRuE-V2 studies, was conducted to assess the long-term efficacy and safety of ruxolitinib over a total follow-up of 104 weeks. The median duration of exposure to ruxolitinib was 364 days. Overall, findings were consistent with those from the pivotal evidence. The objective response, as measured by F-VASI and T-VASI scores, appeared to continue to improve over time. In addition, ruxolitinib was shown to offer maintained response of repigmentation even after discontinuation among patients who achieved an F-VASI90 before treatment discontinuation. In patients who experienced disease relapse, responses were regained in a median of 12 weeks upon re-treatment with ruxolitinib. In patients who did not achieve an F-VASI90 at week 52, treatment response continued to improve with longer term use of ruxolitinib. However, these improvements in F-VASI did not correlate with HRQoL, for which no clinically meaningful improvement was observed. Uncertainty remains due to the uncontrolled, open-label nature of extension studies, and evidence is limited beyond the studies follow-up duration.
As both the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence to inform the comparative effectiveness and safety of ruxolitinib relative to other currently therapies currently in use for the treatment of vitiligo. No indirect evidence was submitted. The sponsor identified potential studies in an attempt to perform an indirect treatment comparison but rated the feasibility of conducting robust evidence synthesis as low. The absence of comparative evidence was considered by the clinical experts to be a substantial limitation, as the treatment options currently available in clinical practice for vitiligo are considered well accepted overall and routinely prescribed, even if they are being used off-label. The efficacy and safety of ruxolitinib relative to any of these options are unknown.
Harms
A relatively high proportion of patients receiving ruxolitinib in the TRuE-V1 and TRuE-V2 trials experienced at least 1 AE during the studies. The most common TEAEs were related to application site reactions (acne, pruritus, rash, and exfoliation) and infections. SAEs were uncommon. Ruxolitinib appeared to be well tolerated, as there were few discontinuations due to AEs. No deaths were reported throughout the trials’ duration. Findings for the treatment extensions in the TRuE-V1 and TRuE-V2 trials, as well as from the TRuE-V LTE study, were consistent with those from the pivotal trials.
The drug product monograph for ruxolitinib includes several serious warnings and precautions regarding toxicities reported in patients treated with oral JAK inhibitors, including serious infections, malignancies, thrombosis, and major adverse cardiovascular events. Ruxolitinib holds a Health Canada indication for the treatment of vitiligo as a topical cream. At the time of this review, there is no evidence regarding these toxicities associated with the use of a topical JAK inhibitor.
Overall, the clinical experts indicated that the harms profile of ruxolitinib did not raise new safety signals or any particular safety concern. However, as with most clinical trials, the studies were not powered to detect infrequent AEs, or those with a lag time.
Other Considerations
Unmet needs and equity considerations were raised by the sponsor and agreed upon by the clinical experts consulted for this review. Visible dermatoses such as vitiligo can affect patients profoundly. Vitiligo specifically is regarded as a historically negative disease with significant cultural impacts. In certain cultural groups, there is a degree of fear and loathing of the condition. Patients as well as their entire families can become stigmatized and socially isolated. The condition is particularly visible in patients with darker skin tones and is therefore likely to present with an increased impact on quality of life in these patients. The physical effect of depigmentation leads to the key emotional factors of loss of identity, decreased self-esteem, and decrease self-confidence.13,67 As a result, patients have a tendency to experience depression and anxiety,13-15 as well as an increased risk of suicide compared to the general population.40-42 Vitiligo also comes with an economic burden for patients through over-the-counter medications, clothing, and camouflage makeup, as well as time away from work and loss of productivity.20,21
Although the off-label treatments currently in use for vitiligo are typically well accepted, there are limitations to these therapies. Some patients face a lack of efficacy, which can include incomplete and/or uneven regimentation, as well as loss of response over time. According to the clinical experts, approximately one-half of patients routinely seen in clinical practice become refractory due to disease resistance, while some others discontinue treatments due to unacceptable toxicity. The patient input emphasized lack of satisfaction with current treatment options and the need for more effective, accessible, and tolerable therapies for vitiligo.
There is therefore an unmet need for additional, effective treatment options, particularly in patients with darker skin tones who could not achieve satisfactory improvements with the currently available therapies, and who experience negative effects on their quality of life. According to the clinical experts, whether ruxolitinib can address these important unmet needs is uncertain due to the lack of improvement in HRQoL and limitations in the external validity of the available evidence.
Conclusion
In patients with nonsegmental vitiligo, findings of moderate certainty from the TRuE-V1 and TRuE-V2 trials suggest that treatment with ruxolitinib likely results in a clinically important improvement in the F-VASI score compared with vehicle treatment, based on thresholds identified in the literature for a clinically meaningful change in repigmentation. The clinical experts consulted for this review indicated that the VASI score can be relevant to inform on the objective response to treatment by capturing the overall surface area of vitiligo involvement and degree of repigmentation. However, given that vitiligo can range from being barely perceptible to cosmetically distressing, different individuals are likely to have different priorities and objectives when assessing the magnitude of response to treatment. The clinical impact of objective response on patients’ daily lives is therefore uncertain and, as noted by clinician and patient input, should be interpreted together with findings on HRQoL. Based on results from exploratory analyses, patients who applied ruxolitinib in the studies did not experience a clinically meaningful improvement in their HRQoL. This suggests that, despite making the condition less noticeable, treatment with ruxolitinib did not improve the negative impact of the disease on patients’ lives in the overall study population as measured by HRQoL metrics. In patients who received ruxolitinib, analyses of change in HRQoL among patients achieving an F-VASI threshold and those who did not suggest that patients may observe an improvement in their HRQoL with a response of at least an F-VASI75. However, whether the improvement in HRQoL is clinical meaningful is uncertain, and interpretation of these findings is limited by the post hoc nature of the analyses. A relatively large proportion of patients in the TRuE-V1 and TRuE-V2 trials experienced AEs, most notably involving application site reactions and infections, although ruxolitinib appeared to be well tolerated, with few reports of SAEs and withdrawals due to AEs. The overall harms profile did not raise any particular safety signal.
Special consideration should be given to the fact that vitiligo can have a profound effect on patients. Historically, this condition has been perceived negatively, carrying significant cultural implications, with patients and families facing stigma and social isolation. The condition is particularly visible in patients with darker skin tones, for whom it can be associated with a loss of identity and lowered self-esteem, resulting in a marked reduction in quality of life. The patient input noted dissatisfaction with current treatment options, creating a need for more effective, accessible, and tolerable therapies. However, external validity issues in the TRuE-V1 and TRuE-V2 trials preclude making definitive conclusions about the effect of ruxolitinib in individuals with the greatest unmet needs. The majority of patients included in the studies were white and had a lighter skin colour, based on the Fitzpatrick Scale skin type classification. In addition, it is likely that the trials included patients whose condition did not interfere substantially with their daily life, given the lower than expected use of prior therapies despite long-lasting disease and relatively low level of HRQoL impairment at baseline.
Evidence is limited beyond the studies’ follow-up duration, even though treatment with ruxolitinib is likely to continue over the long term. However, results from extension studies suggest that the findings are consistent with those from the pivotal evidence over the 2-year follow-up. Because the TRuE-V1 and TRuE-V2 trials included a vehicle control group, there is no direct evidence comparing ruxolitinib to other currently used therapies for vitiligo to inform the reimbursement question. The comparative effectiveness and safety of ruxolitinib relative to other treatment options are therefore unknown.
References
1.Rodrigues M, Ezzedine K, Hamzavi I, Pandya AG, Harris JE, Vitiligo Working G. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77(1):1-13. doi:10.1016/j.jaad.2016.10.048 PubMed
2.Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. 2015;386(9988):74-84. doi:10.1016/S0140-6736(14)60763-7 PubMed
3.Picardo M, Bastonini E. A New View of Vitiligo: Looking at Normal-Appearing Skin. J Invest Dermatol. 2015;135(7):1713-1714. doi:10.1038/jid.2015.92 PubMed
4.Bergqvist C, Ezzedine K. Vitiligo: A Review. Dermatology. 2020;236(6):571-592. doi:10.1159/000506103 PubMed
5.Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of Vitiligo Among Adults in the United States. JAMA Dermatol. 2022;158(1):43-50. doi:10.1001/jamadermatol.2021.4724 PubMed
6.Canadian Skin Patient Alliance. Vitiligo Overview [sponsor supplied reference]. 2023. https://www.canadianskin.ca/vitiligo
7.Bergqvist C, Ezzedine K. Vitiligo: A focus on pathogenesis and its therapeutic implications. J Dermatol. 2021;48(3):252-270. doi:10.1111/1346-8138.15743 PubMed
8.Linthorst Homan MW, Spuls PI, de Korte J, Bos JD, Sprangers MA, van der Veen JP. The burden of vitiligo: patient characteristics associated with quality of life. J Am Acad Dermatol. 2009;61(3):411-20. doi:10.1016/j.jaad.2009.03.022 PubMed
9.Hamzavi IH, Bibeau K, Grimes P, et al. Exploring the natural and treatment history of vitiligo: perceptions of patients and healthcare professionals from the global VALIANT study. Br J Dermatol. 2023;189(5):569-577. doi:10.1093/bjd/ljad245 PubMed
10.Rashighi M, Harris JE. Vitiligo Pathogenesis and Emerging Treatments. Dermatol Clin. 2017;35(2):257-265. doi:10.1016/j.det.2016.11.014 PubMed
11.Sheth VM, Guo Y, Qureshi AA. Comorbidities associated with vitiligo: a ten-year retrospective study. Dermatology. 2013;227(4):311-5. doi:10.1159/000354607 PubMed
12.Eleftheriadou V, Atkar R, Batchelor J, et al. British Association of Dermatologists guidelines for the management of people with vitiligo 2021. Br J Dermatol. 2022;186(1):18-29. doi:10.1111/bjd.20596 PubMed
13.Ezzedine K, Eleftheriadou V, Jones H, et al. Psychosocial Effects of Vitiligo: A Systematic Literature Review. Am J Clin Dermatol. 2021;22(6):757-774. doi:10.1007/s40257-021-00631-6 PubMed
14.Bibeau K, Ezzedine K, Harris JE, et al. Mental Health and Psychosocial Quality-of-Life Burden Among Patients With Vitiligo: Findings From the Global VALIANT Study. JAMA Dermatol. 2023;159(10):1124-1128. doi:10.1001/jamadermatol.2023.2787 PubMed
15.Wang G, Qiu D, Yang H, Liu W. The prevalence and odds of depression in patients with vitiligo: a meta-analysis. J Eur Acad Dermatol Venereol. 2018;32(8):1343-1351. doi:10.1111/jdv.14739 PubMed
16.Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173(2):607-9. doi:10.1111/bjd.13839 PubMed
17.Ezzedine K, Sheth V, Rodrigues M, et al. Vitiligo is not a cosmetic disease. J Am Acad Dermatol. 2015;73(5):883-5. doi:10.1016/j.jaad.2015.07.039 PubMed
18.Grimes PE, Miller MM. Vitiligo: Patient stories, self-esteem, and the psychological burden of disease. Int J Womens Dermatol. 2018;4(1):32-37. doi:10.1016/j.ijwd.2017.11.005 PubMed
19.Thompson AR, Clarke SA, Newell RJ, Gawkrodger DJ, Appearance Research C. Vitiligo linked to stigmatization in British South Asian women: a qualitative study of the experiences of living with vitiligo. Br J Dermatol. 2010;163(3):481-6. doi:10.1111/j.1365-2133.2010.09828.x PubMed
20.Bickers DR, Lim HW, Margolis D, et al. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55(3):490-500. doi:10.1016/j.jaad.2006.05.048 PubMed
21.Ezzedine K, Soliman AM, Li C, Camp HS, Pandya AG. Economic Burden among Patients with Vitiligo in the United States: A Retrospective Database Claims Study. J Invest Dermatol. 2024;144(3):540-546 e1. doi:10.1016/j.jid.2023.08.025 PubMed
22.Ludmann P. Vitiligo: Diagnosis and treatment. American Academy of Dermatology Association. 2023. Accessed 04 December, 2024. https://www.aad.org/public/diseases/a-z/vitiligo-treatment
23.Taieb A, Alomar A, Bohm M, et al. Guidelines for the management of vitiligo: the European Dermatology Forum consensus. Br J Dermatol. 2013;168(1):5-19. doi:10.1111/j.1365-2133.2012.11197.x PubMed
24.van Geel N, Speeckaert R, Taieb A, et al. Worldwide expert recommendations for the diagnosis and management of vitiligo: Position statement from the International Vitiligo Task Force Part 1: towards a new management algorithm. J Eur Acad Dermatol Venereol. 2023;37(11):2173-2184. doi:10.1111/jdv.19451 PubMed
25.Incyte. Ruxolitinib Cream 1.5% [sponsor supplied reference]. 2024.
26.Ezzedine K, Soliman AM, Camp HS, et al. Psychometric Properties and Meaningful Change Thresholds of the Vitiligo Area Scoring Index. JAMA Dermatol. 2025;161(1):39-46. doi:10.1001/jamadermatol.2024.4534 PubMed
27.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
28.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
29.Incyte Corporation. Clinical Study Report: INCB 18424-306. Topical Ruxolitinib Evaluation in Vitiligo Study 1 (TRuE-V1): A Phase 3, Double-Blind, Randomized, Vehicle-Controlled, Efficacy and Safety Study of Ruxolitinib Cream Followed by an Extension Period in Participants With Vitiligo [internal sponsor's report]. March 24, 2022.
30.Incyte Corporation. Clinical Study Report: INCB 18424-307. Topical Ruxolitinib Evaluation in Vitiligo Study 2 (TRuE-V2): A Phase 3, Double-Blind, Randomized, Vehicle-Controlled, Efficacy and Safety Study of Ruxolitinib Cream Followed by an Extension Period in Participants With Vitiligo [internal sponsor's report]. March 21, 2022.
31.A Double-Blind, Vehicle-Controlled, Randomized Withdrawal and Treatment-Extension Study to Assess the Long-Term Efficacy and Safety of Ruxolitinib Cream in Participants With Vitiligo (TRuE-V LTE) [sponsor supplied reference]. 2023.
32.Ezzedine K, Lim HW, Suzuki T, et al. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25(3):E1-13. doi:10.1111/j.1755-148X.2012.00997.x PubMed
33.Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2015(2):CD003263. doi:10.1002/14651858.CD003263.pub5 PubMed
34.Talsania N, Lamb B, Bewley A. Vitiligo is more than skin deep: a survey of members of the Vitiligo Society. Clin Exp Dermatol. 2010;35(7):736-9. doi:10.1111/j.1365-2230.2009.03765.x PubMed
35.Merhi S, Salameh P, Abboud M, et al. Facial involvement is reflective of patients' global perception of vitiligo extent. Br J Dermatol. 2023;189(2):188-194. doi:10.1093/bjd/ljad109 PubMed
36.Nicolaidou E, Antoniou C, Miniati A, et al. Childhood- and later-onset vitiligo have diverse epidemiologic and clinical characteristics. J Am Acad Dermatol. 2012;66(6):954-8. doi:10.1016/j.jaad.2011.07.010 PubMed
37.Lee JH, Ju HJ, Seo JM, et al. Comorbidities in Patients with Vitiligo: A Systematic Review and Meta-Analysis. J Invest Dermatol. 2023;143(5):777-789 e6. doi:10.1016/j.jid.2022.10.021 PubMed
38.Dahir AM, Thomsen SF. Comorbidities in vitiligo: comprehensive review. Int J Dermatol. 2018;57(10):1157-1164. doi:10.1111/ijd.14055 PubMed
39.Hadi A, Wang JF, Uppal P, Penn LA, Elbuluk N. Comorbid diseases of vitiligo: A 10-year cross-sectional retrospective study of an urban US population. J Am Acad Dermatol. 2020;82(3):628-633. doi:10.1016/j.jaad.2019.07.036 PubMed
40.Ramakrishna P, Rajni T. Psychiatric morbidity and quality of life in vitiligo patients. Indian J Psychol Med. 2014;36(3):302-3. doi:10.4103/0253-7176.135385 PubMed
41.Baidya S, Dey P, Mohanty R. Assessment of quality of life in vitiligo patients attending a tertiary care hospital - A cross sectional study. Ind Psychiatry J. 2021;30(1):62-66. doi:10.4103/ipj.ipj_16_20 PubMed
42.Phan K, Shumack S, Gupta M. Association between vitiligo and risk of suicide and suicidal ideation. Pigment International. 2022;9(2):127-130. doi:10.4103/pigmentinternational.pigmentinternational_69_20
43.Salama AH, Alnemr L, Khan AR, Alfakeer H, Aleem Z, Ali-Alkhateeb M. Unveiling the Unseen Struggles: A Comprehensive Review of Vitiligo's Psychological, Social, and Quality of Life Impacts. Cureus. 2023;15(9):e45030. doi:10.7759/cureus.45030 PubMed
44.Rhodes J, Clay C, Phillips M. The surface area of the hand and the palm for estimating percentage of total body surface area: results of a meta-analysis. Br J Dermatol. 2013;169(1):76-84. doi:10.1111/bjd.12290 PubMed
45.Rosmarin D, Pandya AG, Lebwohl M, et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet. 2020;396(10244):110-120. doi:10.1016/S0140-6736(20)30609-7 PubMed
46.Haq M, Adnan G. Ruxolitinib. StatPearls. StatPearls Publishing; 2023. Accessed December 04, 2024. https://www.ncbi.nlm.nih.gov/books/NBK570600/
47.Ezzedine K, van Geel N, Butler K, Bibeau K, Gao J, Pandya AG. CO108 Subgroup Analysis of Quality of Life and Treatment Satisfaction By Disease Duration and Use of PRIOR Treatment: Pooled Results from Two Randomized Phase 3 Studies of Ruxolitinib Cream for the Treatment of Vitiligo. Value in Health. 2022;25(7):S324. doi:10.1016/j.jval.2022.04.204
48.Pandya AG, van Geel N, Butler K, Bibeau K, Gao J, Ezzedine K. CO130 Subgroup Analysis of Quality-of-Life Outcomes in Patients Achieving F-VASI75: Pooled Results from Two Randomized Phase 3 Studies of Ruxolitinib Cream for the Treatment of Vitiligo. Value in Health. 2022;25(7):S328. doi:10.1016/j.jval.2022.04.225
49.Rosmarin D, Passeron T, Pandya AG, et al. Two Phase 3, Randomized, Controlled Trials of Ruxolitinib Cream for Vitiligo. N Engl J Med. 2022;387(16):1445-1455. doi:10.1056/NEJMoa2118828 PubMed
50.Incyte Corporation. NCT04052425: Topical Ruxolitinib Evaluation in Vitiligo Study 1 (TRuE-V1). ClinicalTrials.gov; 2022. Accessed 04 December, 2024. https://www.clinicaltrials.gov/study/NCT04052425
51.Incyte Corporation. NCT04057573: Topical Ruxolitinib Evaluation in Vitiligo Study 2 (TRuE-V2). ClinicalTrials.gov; 2022. Accessed 04 December, 2024. https://www.clinicaltrials.gov/study/NCT04057573
52.Kitchen H, Wyrwich KW, Carmichael C, et al. Meaningful Changes in What Matters to Individuals with Vitiligo: Content Validity and Meaningful Change Thresholds of the Vitiligo Area Scoring Index (VASI). Dermatol Ther (Heidelb). 2022;12(7):1623-1637. doi:10.1007/s13555-022-00752-8 PubMed
53.Batchelor JM, Gran S, Leighton P, et al. Using the Vitiligo Noticeability Scale in clinical trials: construct validity, interpretability, reliability and acceptability. Br J Dermatol. 2022;187(4):548-556. doi:10.1111/bjd.21671 PubMed
54.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. doi:10.1159/000365390 PubMed
55.Lilly E, Lu PD, Borovicka JH, et al. Development and validation of a vitiligo-specific quality-of-life instrument (VitiQoL). J Am Acad Dermatol. 2013;69(1):e11-8. doi:10.1016/j.jaad.2012.01.038 PubMed
56.Hamzavi I, Jain H, McLean D, Shapiro J, Zeng H, Lui H. Parametric modeling of narrowband UV-B phototherapy for vitiligo using a novel quantitative tool: the Vitiligo Area Scoring Index. Arch Dermatol. 2004;140(6):677-83. doi:10.1001/archderm.140.6.677 PubMed
57.Komen L, da Graca V, Wolkerstorfer A, de Rie MA, Terwee CB, van der Veen JP. Vitiligo Area Scoring Index and Vitiligo European Task Force assessment: reliable and responsive instruments to measure the degree of depigmentation in vitiligo. Br J Dermatol. 2015;172(2):437-43. doi:10.1111/bjd.13432 PubMed
58.Bibeau K, Butler K, Sun K, Skaltsa K, Hamzavi IH. Assessment of Measurement Properties of the Facial and Total Vitiligo Area Scoring Index Instruments in the Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V) Phase 3 Studies [sponsor supplied reference]. 2022. Accessed March 24.
59.Bibeau K, Butler K, Wang M, Skaltsa K, Hamzavi IH. Psychometric Evaluation of the Facial and Total Vitiligo Area Scoring Index Instruments in the TRuE-V Phase 3 Studies. Dermatol Ther (Heidelb). 2024;14(8):2223-2234. doi:10.1007/s13555-024-01223-y PubMed
60.Rosmarin D, Pandya AG, Lebwohl M, et al. Supplementary Appendix: Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet. 2020;396(10244):110-120. doi:10.1016/S0140-6736(20)30609-7 PubMed
61.Batchelor JM, Tan W, Tour S, Yong A, Montgomery AA, Thomas KS. Validation of the Vitiligo Noticeability Scale: a patient-reported outcome measure of vitiligo treatment success. Br J Dermatol. 2016;174(2):386-94. doi:10.1111/bjd.14208 PubMed
62.Aghaei S, Sodaifi M, Jafari P, Mazharinia N, Finlay AY. DLQI scores in vitiligo: reliability and validity of the Persian version. BMC Dermatol. 2004;4:8. doi:10.1186/1471-5945-4-8 PubMed
63.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-6. doi:10.1111/j.1365-2230.1994.tb01167.x PubMed
64.Khilji FA, Gonzalez M, Finlay A. Clinical meaning of change in Dermatology Life Quality Index scores. Br J Dermatol. 2002;147(Suppl 62):50. doi:10.1046/j.1365-2133.147.s62.16.x
65.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. doi:10.1186/1477-7525-3-36 PubMed
66.Shikiar R, Willian MK, Okun MM, Thompson CS, Revicki DA. The validity and responsiveness of three quality of life measures in the assessment of psoriasis patients: results of a phase II study. Health Qual Life Outcomes. 2006;4:71. doi:10.1186/1477-7525-4-71 PubMed
67.U.S. Food and Drug Administration (FDA). The Voice of the Patient: Vitiligo. 2021. Accessed 04 December, 2024. https://www.fda.gov/media/155068/download
68.Incyte. Data on file (Post hoc QoL responder analyses) [sponsor supplied reference]. 2023.
Appendix 1: Post Hoc Analysis
Please note that this appendix has not been copy-edited.
The role of repigmentation in HRQoL improvement was investigated by the sponsor in post hoc analyses in patients who received ruxolitinib. These were conducted to compare the change in VitiQoL and DLQI from baseline to week 24 among patients who achieved an F-VASI50, F-VASI75, or F-VASI90 in those who responded to treatment and those who did not. Detailed results are presented in Table 29.
███ ████ ██████ ████ ████████ ██ ███████ ██ ████████ ███ ████████ █████████ █████████ ███ ████████ ██ ████ ███ ████████ ██ █████ ████████ ███ ███ ████ ███ █████ ████ ██ ██████ ██████ ██████████ █████ ████ ██ ██████ ██████ ██████████ ███ █████ ████ ██ ███████ ██████ ██████████ █████████████████ ████ ██████ ████ ████████ ██ ████ ██ ████████ ███ ████████ █████████ █████████ ███ ████████ ██ ████ ███ ████████ ██ █████ ████████ ███ ███ ████ ███ █████ ████ ██ ██████ █████ ██████████ █████ ████ ██ ██████ ██████ ██████████ ███ █████ ████ ██ ██████ ██████ ██████████ █████████████.
The sponsor reported that these results were sustained when stratified by skin type, for both the DLQI and VitiQoL.
████████ ███████ | ██████ ███████ █ ███████ | |
|---|---|---|
█████████████ | █████████ | |
█████████████████████████████████████████████████ | ||
██████ ████ ████████ ██ ███████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██████ ████ ████████ ██ ███████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██████ ████ ████████ ██ ███████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██████████████████████████████████████ | ||
██████ ████ ████████ ██ ████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██████ ████ ████████ ██ ████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██████ ████ ████████ ██ ████ ██ ████ ██ █████████ █ █████████ | ||
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | █████ ██ |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
███ ██████████ ████ | █████ ██ | |
██ █ ██████████ █████████ ████ █ ███████████ ████ ███████ ██████ ██████ █ ██████ ████ ███████ █████ ████████ █ ████ ███████████ ██ ███████ ██ █ ███ █████████ ██ █ ████████ ██████████ ██ █ ████████ ██████ ███████ █ ████████ ███████ ██ ████ ███████ ████████ ████████ ████ ████ █████████ ██ ██████ ███████ ███ ███████ ████ ████ ████████ ███ ████ ██████████ ██ █████████████.
Source: Incyte Corporation (2023).68 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
F-VASI
Facial Vitiligo Area Scoring Index
ICER
incremental cost-effectiveness ratio
LTE
long-term extension
NSV
nonsegmental vitiligo
QALY
quality-adjusted life-year
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ruxolitinib cream (Opzelura), 1.5% |
Indication | Topical treatment of nonsegmental vitiligo in adult and pediatric patients 12 years of age and older |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 11, 2024 |
Reimbursement request | As per indication |
Sponsor | Incyte Biosciences Canada Corporation |
Submission history | Previously reviewed: Yesa Indication: For topical treatment of mild to moderate atopic dermatitis in adult and pediatric patients aged 12 years and older whose disease is not adequately controlled with conventional topical prescription therapies (topical corticosteroids, topical calcineurin inhibitors) or when those therapies are not advisable. Recommendation date: TBD Recommendation: TBD |
NOC = Notice of Compliance; TBD = to be determined.
aThe tablet form of ruxolitinib has also been evaluated for other indications by Canada’s Drug Agency.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients aged 12 years and older with nonsegmental vitiligo |
Treatment | Ruxolitinib cream |
Dose regimen | Applied twice daily to affected skin areas (maximum of 10% of BSA for each application) for 24 weeks and as needed thereafter |
Submitted price | $1,075.97 per 100 g tube |
Submitted treatment cost | $1,156.88 per 28-day cycle or $15,091 per year |
Comparator | No active treatment (i.e., vehicle cream)a |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (61 years) |
Key data sources |
|
Submitted results | ICER = $85,787 per QALY gained versus no active treatment (incremental costs: $20.958; incremental QALYs: 0.24) |
Key limitations |
|
CDA-AMC reanalysis results |
|
BSA = body surface area; CDA-AMC = Canada’s Drug Agency; F-VASI = Facial Vitiligo Area Scoring Index; ICER = incremental cost-effectiveness ratio; LTE = long-term extension; LY = life-year; NSV = nonsegmental vitiligo; QALY = quality-adjusted life-year; WTP = willingness to pay.
aIn the base case, using data from the TRuE-V1 and TRuE-V2 trials, the sponsor compared ruxolitinib cream to vehicle cream that contained no active ingredient.
Conclusions
The Clinical Review by Canada’s Drug Agency (CDA-AMC) of the TRuE-V1 and TRuE-V2 trials found that, based on findings of moderate certainty, ruxolitinib cream likely results in clinically important improvements in the Facial Vitiligo Area Scoring Index (F-VASI) compared with vehicle cream in patients aged 12 years and older with nonsegmental vitiligo (NSV). However, the clinical impact of the objective response as measured by the Vitiligo Area Scoring Index on patients’ daily lives is uncertain. According to results from the pivotal trials’ exploratory analysis, patients who applied ruxolitinib in the studies did not experience a clinically important improvement in health-related quality of life. This suggests that, despite making the condition less noticeable, treatment with ruxolitinib did not reduce the negative impact of the disease on patients’ lives as measured by these metrics. The Clinical Review also noted concerns with the generalizability of the trial findings to individuals with the greatest unmet need, such as patients with a pronounced contrast between depigmented and unaffected skin, patients who do not achieve satisfactory improvements with the currently available therapies, and those whose condition interferes substantially with their daily lives.
Clinical expert input received by CDA-AMC for this review indicated that, in clinical practice, multiple alternative treatments, including topical corticosteroids (TCSs), topical calcineurin inhibitors (TCIs), phototherapy, and combination therapy, are used to manage patients with NSV. The sponsor’s base case, which compared ruxolitinib to vehicle cream (i.e., no active treatment) is therefore not informative for decision-making. The comparative effectiveness and safety of ruxolitinib relative to these therapies for vitiligo is unknown. Notably, these treatments are less costly than ruxolitinib at publicly available list prices. While the cost per gram of ruxolitinib is $10.76, the costs for TCSs and TCIs range from $0.06 to $0.55 and from $2.47 to $2.75 per gram, respectively.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis, which included assuming the utility for patients who did not respond to treatment was the same as for the baseline and aligning the daily dose of ruxolitinib with the product monograph. Results of the CDA-AMC reanalyses align with those of the sponsor’s submitted analysis, indicating that, at a willingness to pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained, ruxolitinib cream is not a cost-effective option for the treatment of NSV compared with no active treatment. In the CDA-AMC base case, ruxolitinib cream was associated with an incremental cost-effectiveness ratio (ICER) of $535,419 per QALY gained compared with no active treatment in the patient population aligned with the TRuE-V trials (incremental costs = $39,038; incremental QALYs = 0.07). A price reduction of approximately 90% would be required for ruxolitinib to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. This would reduce the price of ruxolitinib cream from $1,076 to $108 per 100 g tube. With this price reduction, the annual drug acquisition costs for ruxolitinib would be approximately $2,905 per patient.
CDA-AMC was unable to address concerns regarding the appropriateness of the F-VASI scores, which informed the model structure and assumptions on changes in clinical practice and disease management. Furthermore, concerns remain about whether the TRuE-V trial results can be generalized to patient groups with the greatest unmet needs, given the concerns about external validity raised in the Clinical Review. CDA-AMC notes that the utility values of patients who did respond to treatment are greater than that of the general population in both the sponsor’s analysis and the CDA-AMC base-case reanalysis. The incremental QALYs may have been overestimated, resulting in a lower ICER that favours ruxolitinib. These factors introduced uncertainty regarding the cost-effectiveness of ruxolitinib cream in clinical practice and limits the scope of the interpretation of economic results. Several key limitations remained unresolved and, because the reanalysis performed by CDA-AMC was associated with uncertainty, higher price premiums may be required.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from a joint submission by the Canadian Skin Patient Alliance and Vitiligo Voices Canada, which collected perspectives of caregivers and patients aged 25 years and older with vitiligo through an online survey across Canada. Patients with vitiligo reported a negative impact on psychological health, emotional health, social interactions, and the ability to carry out daily activities. Patients also reported experiencing fear stemming from the unpredictable progression of the disease. Patients who responded to the survey described experience with a variety of treatments, including TCIs, TCSs, topical immunomodulators, topical vitamin D derivatives, oral steroids, depigmentation therapy with monobenzone for advanced vitiligo, transplant surgeries, and phototherapy, although some patients reported not seeking any treatment for vitiligo. Patients reported that these treatments had limited efficacies. Treatment goals described by patients included establishing sustained repigmentation, reducing the appearance of patches, and fewer side effects. No patients who responded to the survey had experience with ruxolitinib.
Clinician input was received from multiple clinician groups. This included a joint submission by the Canadian Dermatology Association, the Dermatologist Association of Ontario, and the Dermatology Association of Saskatchewan and a submission from the Southwestern Ontario Dermatologists Group. Both inputs noted that no treatments have been approved in Canada for NSV. First-line therapy for patients often includes TCIs (such as tacrolimus and pimecrolimus) and TCSs (such as betamethasone valerate, clobetasol propionate, dexamethasone, mometasone furoate, prednisone, and triamcinolone acetonide). Phototherapy may be a first-line option for patients with extensive vitiligo, although it may not be widely available. Other treatment options, such as laser surgery, systemic therapy with immunomodulators, and JAK inhibitors, may also be considered. One clinician group noted latanoprost may also be used off-label. The clinician input noted that long-acting corticosteroids may be administered as an oral mini-pulse or in low, intermittent doses over a short period. Clinician input reiterated that currently available treatments have limited efficacy, and long-term use is often associated with adverse events (AEs). Clinically meaningful treatment goals included attaining repigmentation and preventing relapse following successful repigmentation. There was limited consensus regarding the degree of response needed to qualify as a clinically meaningful response to treatment, but clinician input consistently noted that any visible sign of repigmentation would justify continuation of therapy and would be assessed through patient-reported experiences and clinician judgment. With respect to ruxolitinib, the clinician input noted it would serve as first-line treatment for vitiligo and identified transient acneiform eruption, exacerbation of acne, nasopharyngitis, pruritus at application site, mild erythema, and myelosuppression as potential side effects. The submissions agreed that treatment response takes a minimum of 6 months or longer. Treatment may be discontinued if no visible repigmentation is observed.
The drug plans highlighted concerns with the long-term efficacy and safety of ruxolitinib and its anticipated budget impact. There is further uncertainty about how dosing would affect the cost of treatment.
CDA-AMC addressed concerns about the uncertainty with ruxolitinib dosing and evaluated its impact on treatment costs.
CDA-AMC was unable to address the concern regarding the unknown cost-effectiveness of ruxolitinib cream compared with topical therapies (TCIs and TCSs) and phototherapy because of a lack of direct and indirect comparative clinical evidence.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of ruxolitinib cream compared with no active treatment (represented by vehicle cream in the TRuE-V1 and TruE-V2 trials).1,2 The modelled population was aligned with the TRuE-V1 and TRuE-V2 trials, which included patients aged 12 years or older with NSV. The modelled population was also aligned with the Health Canada indication and reimbursement request.
The sponsor submitted a new pharmacoeconomic model and report on November 2024. The inputs and results that follow are reflective of these new submissions.
Ruxolitinib cream is supplied in 100 g tubes, with a recommended application of a thin layer twice daily to affected areas (up to 10% of the body surface area for each application).3 The monograph specifies that satisfactory patient response may require treatment with ruxolitinib cream for more than 24 weeks, but re-evaluation by a health care provider is recommended if the patient does not achieve meaningful repigmentation by 24 weeks.3 Using data from in the TRuE-V1 and TRuE-V2 trials,1,2 the sponsor estimated that an average of 3.84 g of ruxolitinib cream would be used daily. At the submitted price of $1,076 per 100 g tube, the sponsor estimated the annual drug acquisition cost of ruxolitinib to be $15,091 per year, assuming no drug wastage. Vehicle cream was assumed to incur no treatment costs by the sponsor.
The analysis was undertaken from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and QALYs) were estimated over a lifetime time horizon of 61 years and discounted at an annual rate of 1.5%. The cycle length was 4 weeks.
Model Structure
The sponsor submitted a Markov model with 7 health states: “Initial period,” “Maintenance period,” “Stable,” “Re-treated,” “Stable re-treated,” “Non-response” and “Death” (Figure 1). All patients entered the model in the “Initial period” health state, in which they received either ruxolitinib or vehicle cream for 24 weeks. Treatment response, defined as an improvement of 25 points or more from baseline on the Facial Vitiligo Area Scoring Index, was assessed at week 24. Patients who responded to treatment moved to the maintenance period and continued on treatment while patients who achieved a change in F-VASI score by 90 points or more were moved to the “Stable” health state, in which it was assumed all treatment (e.g., active and concomitant medication) would be discontinued. Patients who discontinued treatment moved to the “Non-response” health state, in which they received best supportive care (BSC) consisting of TCSs and phototherapy. Patients could transition to the “Death” state from all other health states at any time.
The maintenance period stratified health states by the change in F-VASI scores from baseline (i.e., F-VASI25 to 49, F-VASI50 to 74, and F-VASI75 to 89). Response to treatment was assessed at 2 further time points: week 52 and week 104. Similar to the first assessment time point at week 24, patients who had a change in F-VASI score between 25 to 89 points remained in the maintenance phase and continued treatment. Patients who achieved a change in F-VASI score by 90 points or greater transitioned to the “Stable” health state, and those with an F-VASI score of less than 25 points transitioned to the “Non-response” health state. After the second response assessment at week 104, patients remaining on maintenance treatment were assumed to discontinue treatment at a constant rate and were moved to the “Non-response” health state.
Patients whose condition has not changed could experience a disease relapse, which was defined as the loss of 75% of the repigmentated area and enter the “Re-treated” health state in which they resumed their initial treatment. If patients experienced a disease relapse and regained response (i.e., F-VASI ≥ 90), they moved to the “Stable re-treated” health state, and those who did not regain a response (i.e., F-VASI < 90) transitioned to the “Non-response” health state where they received BSC. Patients whose condition has been stable, who were re-treated, and who lost their regained response (i.e., F-VASI < 75) the second time were assumed to transition to the “Non-response” health state and would remain on lifelong BSC.
Model Inputs
The baseline characteristics in the model were derived by pooling the phase III, double-blinded, randomized, vehicle-controlled TRuE-V1 and TRuE-V2 trials (mean age: 39.6% years; 53.1% female).1,2
Comparative clinical efficacy in terms of response, defined as an improvement of 25 points or more from baseline on the F-VASI, was derived using data pooled from the double-blinded phase of the TRuE-V1 and TRuE-V2 trials at week 24.1,2 Response rates for ruxolitinib (i.e., proportions of patients who had achieved F-VASI scores of < 25, 25 to 49, 50 to 74, 75 to 89, and 90) at week 52 were derived using pooled data from the open-label phase of the TRuE-V1 and TRuE-V2 trials. Response rates at week 104 were based on data from cohort B of the TRuE-V long-term extension (LTE) study, in which all patients received ruxolitinib cream twice daily for an additional 52 weeks.1,2,4 Because no comparative clinical efficacy data were gathered beyond week 24, the risk ratio in the vehicle group compared to ruxolitinib derived at week 24 informed the response rates for patients receiving no active treatment at week 52 and week 104. The sponsor estimated the rates of relapse and regaining of response using time to relapse (i.e., time to achieve F-VASI < 75) and time to regain response following disease relapse (i.e., time to achieve F-VASI ≥ 90) data from the vehicle arm of cohort A in the TRuE-V LTE study.4 The proportion of patients who did not regain a response following disease relapse was based on data collected at week 104 from cohort B of the TRuE-V LTE study and was assumed to be the same for ruxolitinib and no active treatment.4 The sponsor assumed that the odds a patient receiving ruxolitinib would not respond to treatment when re-treated, having experienced a disease relapse, and regained a response following disease relapse were the same as for patients receiving no active treatment.
Discontinuation was applied distinctly at 3 periods within the model: from the start of the treatment to week 24 (the initial period), between weeks 24 and 52, and at week 52 onwards. The discontinuation rates during the initial and maintenance periods were based on pooled data from the TRuE-V1 and TRuE-V2 trials.1,2 Discontinuation during the initial period captured discontinuation due to all causes, excluding lack of efficacy, while discontinuation during the maintenance period captured discontinuation from all causes, including lack of efficacy. Because patients receiving vehicle cream switched to ruxolitinib cream after the double-blind phase of the TRuE-V trials, the discontinuation rate for weeks 24 to 52 for patients receiving no active treatment were derived from discontinuation data collected from week 0 to 24. All-cause discontinuation rates at week 52 onward were based on TRuE-V LTE study cohort B.4
The sponsor included any treatment-emergent AEs that occurred in 4% or greater of patients in any group of the safety population from the 24-week double-blind period pooled from the TRuE-V1 and TRuE-V2 trials. Age-specific and sex-specific mortality rates were based on general population life tables from Statistics Canada.5
Health state and baseline utility values were derived using data collected using the F-VASI and Vitiligo-Specific Quality of Life Instruments, respectively. These were pooled from the TruE-V1 and TruE-V2 trials and then transformed to EQ-5D values using mapping algorithms developed by Begum et al. (2023).1,2,6 The “Non-response” health state was associated with a decrement to baseline utility while health states corresponding to an improvement in F-VASI scores (i.e., 25 to 49, 50 to 74, 75 to 89, and 90) were associated with increases to baseline utility values. Utility values were not adjusted for age, and disutilities from AEs were not included.
Costs in the model included drug acquisition, disease management, and AEs. Drug acquisition costs for ruxolitinib were based on the sponsor’s submitted price, with the average daily dose of ruxolitinib cream based on the mean of a log-normal distribution applied to the pooled data from the TRuE-V1 and TRuE-V2 trials.1,2 The sponsor assumed that all patients receiving treatment (whether ruxolitinib or vehicle cream) accrued the cost of concomitant therapy consisting of TCSs and hospital-based narrowband UV-B phototherapy. The cost of concomitant therapy was estimated as an average of TCSs and phototherapy costs, weighted by the proportions of patients assumed to use each therapy. The price of TCSs was an average of the prices of hydrocortisone, hydrocortisone acetate, betamethasone dipropionate, and betamethasone valerate cream obtained from the Ontario Drug Benefit Formulary.7 Phototherapy costs were based on the price per session of light therapy listed in the Ontario Schedule of Benefits for Physician Services.8 For patients in the “Non-response” health state, the cost of BSC (i.e., TCSs and narrowband UV-B) was the same as that of concomitant therapy. The sponsor assumed that patients in the “Stable” and “Stable re-treated” health states did not accrue the cost of any treatment and health care resources. For patients receiving treatment and those not responding to treatment, resource use pertaining to disease management was informed by expert opinion gathered by the sponsor and a cost-effectiveness analysis by Sach et al. (2021).9 This included the cost of dermatologist visits, which was obtained from the Ontario Schedule of Benefits for Physician Services.8 For AEs, the sponsor included only the cost of treating upper respiratory tract infections, which was assumed to be managed by a general physician visit, with costs obtained from the Ontario Schedule of Benefits for Physician Services.8
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically for 2,000 iterations. The results of the deterministic and probabilistic results were similar; the results of the probabilistic analyses are presented in the following section. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base case, ruxolitinib cream was more effective (incremental QALYs = 0.24) and more costly (incremental costs = $20,958) compared with no active treatment, resulting in an ICER of $85,787 per QALY gained (Table 3). Ruxolitinib treatment was not associated with any additional life-years gained compared with no active treatment. At a WTP of $50,000 per QALY gained, the probability of ruxolitinib cream being cost-effective compared with no active treatment was 0.2%.
Nearly all (99.7%) of the incremental QALYs gained for ruxolitinib cream were derived after the randomized, double-blind period of 24 weeks. Results were driven primarily by the increased duration of the time patients receiving ruxolitinib cream spent in the “Stable” health state (i.e., the incremental time with an F-VASI score of 90 was 1.05 years) and the higher drug acquisition cost of ruxolitinib cream (incremental drug costs = $21,336) (Table 12). Drug acquisition costs accounted for nearly all (99.9%) of the incremental costs for ruxolitinib.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no active treatment ($/QALY) |
|---|---|---|---|---|---|
No active treatment | 9,462 | Reference | 24.15 | Reference | Reference |
Ruxolitinib cream | 30,420 | 20,958 | 24.40 | 0.24 | 85,787 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.10
Sensitivity and Scenario Analysis Results
The sponsor conducted several additional scenario analyses, including assuming alternative definitions of treatment response, including the cost of 0.1% tacrolimus as part of BSC, adopting age-related decline in utility, applying an alternative utility value for the “Non-response” health state and evaluating different comparative efficacy parameters for sustained response, disease relapse, no response, and regaining response. The ICER was sensitive to the definition of treatment response and health utility value for the “Non response” health state. When a higher F-VASI threshold was adopted to define the treatment response, the ICERs increased to $118,896 and $124,271 per QALY gained when the initial response was defined as an F-VASI score of 75 or greater at week 24 and 50 or greater at week 52, respectively. When a higher utility estimate was selected for the “Non- response” health state, the ICER increased to $119,591 per QALY gained.
The sponsor also conducted a scenario analysis from a societal perspective that included additional costs associated with over-the-counter medications for concomitant therapy or BSC (such as the cost of 0.1% tacrolimus, vitamin D supplements, camouflage cream, fixing powder, sunscreen, and home-based UV-B therapy), and the management of AEs, productivity loss due to days of missed work, and out-of-pocket costs for psychological support. The results of this scenario analysis were similar to the sponsor’s base case using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The sponsor’s base case compared ruxolitinib cream to no active treatment, which is not a relevant comparator for decision-making: The sponsor’s submitted base case compared ruxolitinib cream to vehicle cream, based on the results of the TRuE-V1 and TRuE-V2 clinical trials. As described in the CDA-AMC Clinical Review, the vehicle cream looked and felt the same as ruxolitinib cream, but contained no active ingredient (i.e., ruxolitinib). However, according to clinical expert input received by CDA-AMC for this review and published literature,11 multiple alternative treatments are used to manage patients with NSV. These treatments include TCSs such as clobetasol propionate, TCIs such as tacrolimus and pimecrolimus, and phototherapy (narrowband UV-B).11 These comparators were not included in the sponsor’s analysis, which is problematic because it does not assess the relative cost-effectiveness of ruxolitinib among all relevant treatment options. The sponsor’s choice of no active treatment as the only relevant comparator has limited relevance to the decision problem.
CDA-AMC notes that the sponsor requested a deviation from the CDA-AMC pharmacoeconomic requirements to exclude all relevant comparators from the economic model, proposing that an indirect treatment comparison was not feasible for the indicated population. Although this request was approved by CDA-AMC, the sponsor was advised that the exclusion of relevant comparators would be considered a major limitation of its submission. The sponsor’s decision to exclude all relevant comparators is problematic because it does not fully capture the comparative cost-effectiveness of ruxolitinib cream. The cost-effectiveness of ruxolitinib cream against existing therapies used for patients with NSV remains unknown.
The sponsor’s base case comparing ruxolitinib to no active treatment is not informative for decision-making. Several other treatments are used to manage patients with NSV in clinical practice that, at publicly available list prices, are notably less costly than ruxolitinib cream (Appendix 1).
The model structure does not appropriately capture all aspects of the condition: An appropriate economic evaluation model structure should capture relevant and meaningful underlying clinical and/or biological processes.12 Health states within the submitted economic model were defined based on the F-VASI score, and changes in F-VASI scores were assumed to affect clinical practice decisions on when to continue, discontinue, and resume treatment. As noted in the CDA-AMC Clinical Review, there are concerns with the appropriateness and meaningfulness of the F-VASI measurement. In clinical practice, the focus of treatment is not restricted to an overall numerical reduction in the depigmentation area but includes improving patient quality of life. Clinical expert input received by CDA-AMC emphasized several key issues associated with vitiligo that affect patients’ quality of life, such as the site of depigmentation and associated visibility, the level of heterogeneity in repigmentation, differences between the colours of the affected area, repigmented area, and background skin tone. However, these are not adequately captured by the F-VASI instrument. The expert feedback also indicated that F-VASI scores were designed specifically to support the development of ruxolitinib and are not used in current clinical practice to guide treatment decisions.
CDA-AMC was unable to address the limitations regarding the use of changes in F-VASI scores to inform the model structure.
The limited external validity of the TruE-V1 and TruE-V2 trials affects the scope of the economic model: Clinical efficacy within the submitted model was informed by the TruE-V1 and TruE-V2 trials. However, as noted in the CDA-AMC Clinical Review, the results have limited generalizability. According to expert opinion, the patient populations with potentially the greatest unmet needs include those with a pronounced contrast between depigmented and unaffected skin, patients who do not achieve a satisfactory improvement with currently available therapies, and patients whose condition interferes substantially with their daily lives. The fact that these patient populations were not adequately represented in the TRuE-V1 and TRuE-V2 trials was a significant problem when assessing the external validity of the trial findings. Because the modelled population was aligned with the TRuE-V1 and TRuE-V2 trials, which may not fully represent patients with the greatest unmet need, the cost-effectiveness of ruxolitinib may not be applicable to the population in whom ruxolitinib is most likely to be used in clinical practice.
CDA-AMC was unable to address this limitation. The generalizability of the effects of ruxolitinib in these patients is uncertain and, given that this informs the cost-effectiveness model, the cost-effectiveness of ruxolitinib may not be applicable to its use in patient populations in whom ruxolitinib is most likely to be used in clinical practice.
The health utilities lack face validity: The sponsor’s submitted pharmacoeconomic model incorporated health state utilities by adapting published mapping algorithms and data from the TRuE-V1 and TRuE-V2 trials.1,2,6 The sponsor estimated health utilities for health states (i.e., defined by F-VASI scores of < 25, 25 to 49, 50 to 74, 75 to 89, and 90), and at baseline by mapping F-VASI scores and Vitiligo-Specific Quality of Life scores, respectively, to EQ-5D utilities.6,10 The considerable uncertainty associated with adopting mapped utilities is further compounded by the fact that the mapping algorithm proposed by Begum et al. (2023) is based on a different scoring tool, and interchangeability was assumed between vitiligo repigmentation scores and F-VASI scores.6 No published evidence supports this assumption. The sponsor’s derived health utilities also lack face validity. The utilities at baseline and for the response health states are higher than that reported for the general population in Canada.13 According to input from the clinical experts, this is implausible given patients with NSV seeking treatment may have a psychological burden higher than that of the general population in Canada. In fact, the TRuE-V1 and TRuE-V2 trial evidence showed no improvement in patient-reported quality of life, calling into question whether the mapped utilities have been overestimated for these health states.
In addition, the healthy utility derived for patients who did not respond to treatment lacks face validity. In the sponsor’s pharmacoeconomic model, those who did not respond to treatment experienced a lower utility compared with their baseline values. However, according to input from the clinical experts received by CDA-AMC for this review, patients who do not respond to treatment are likely to have the same health utility as their baseline values.
In a reanalysis, the utilities for patients who did not respond to treatment were set to be identical to the baseline value. As the reanalysis did not change the utility value for the response health states, these health utilities remained greater than that of the general population and the reanalysis likely overestimated the expected incremental QALYs, resulting in a lower ICER that favours ruxolitinib.
The acquisition cost of ruxolitinib cream is highly uncertain. The sponsor applied a log-normal distribution to the drug-exposure data from the TruE-V1 and TruE-V2 trials to estimate that patients with NSV would use 3.84 g of ruxolitinib cream per day.1,2 However, as noted in the CDA-AMC Clinical Review, there is considerable variability in the amount of drug used by patients in the TruE-V1 and TruE-V2 trials. The mean daily dose of ruxolitinib ranged from 5.82 g to 8.86 g per day during the double-blind controlled period of the trials (24-week duration), and from ████ grams to █████ grams per day in the extension period of the trials (28-week duration).1,2,4 Expert input received by CDA-AMC indicated that the drug use in the trials may be an underestimate of what is used by real-world patients. The product monograph for ruxolitinib cautions against using more than one 100 g tube of ruxolitinib every 2 weeks, which translates into 7.14 g per day.3
The sponsor also did not consider drug wastage in its calculation of drug costs. However, expert feedback obtained for this review noted that wastage of ruxolitinib cream should be anticipated because not all of the product is always squeezed from the tube and too much product may be squeezed from the tube during an application.
In a reanalysis, CDA-AMC assumed that 7.14 g of ruxolitinib cream would be used daily. CDA-AMC could not incorporate different amounts of drug used for patients who continued treatment into subsequent years due to limitations of the parameterization of the sponsor’s model. The sponsor’s assumption that the daily dose of TCSs is the same as the ruxolitinib dose was retained.
Additionally, the following key assumptions made by the sponsor were appraised by CDA-AMC (Table 5).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Individuals do not experience a disutility associated with AEs. | Inappropriate but unlikely to affect the results. Although the cost of treating upper respiratory tract infections was included, the impact of this AE on health utility was not modelled. The sponsor’s approach overlooked the impact of AEs on quality of life. However, given the short duration of upper respiratory tract infections, it is not expected to have a large impact on the cost-effectiveness results. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC undertook a stepped analysis, sequentially incorporating each adjustment outlined in Table 5 into the sponsor’s model, to demonstrate the impact of each change. These included setting utilities for patients who did not respond to treatment to be identical to baseline utilities and adopting a higher daily dose for ruxolitinib. A summary of the results of the reanalyses is presented in Table 6.
CDA-AMC was unable to address other key limitations, including the exclusion of relevant comparators, concerns with the appropriateness of F-VASI scores, and the overall external validity of the pivotal trials, which limited the relevancy of the base case to decision-making.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Change in health utility for no response | −0.123 | 0 |
2. Dose per day | 3.84 g | 7.14 g |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency.
In the CDA-AMC base case, ruxolitinib cream was associated with an ICER of $535,419 per QALY gained compared with no active treatment (incremental costs = $39,038; incremental QALYs = 0.07) (Table 6). Results were driven by the drug acquisition cost of ruxolitinib cream (incremental costs = $39,061; Table 14).
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | No active treatment | 9,462 | 24.15 | Reference |
Ruxolitinib cream | 30,420 | 24.40 | 85,787 | |
CDA-AMC reanalysis 1 | No active treatment | 9,452 | 27.92 | Reference |
Ruxolitinib cream | 30,365 | 27.99 | 286,150 | |
CDA-AMC reanalysis 2 | No active treatment | 10,974 | 24.16 | Reference |
Ruxolitinib cream | 50,006 | 24.40 | 162,385 | |
CDA-AMC base case (1 + 2) (probabilistic) | No active treatment | 10,926 | 27.93 | Reference |
Ruxolitinib cream | 49,964 | 28.00 | 535,419 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated.
Scenario Analysis Results
CDA-AMC undertook price-reduction analyses based on the sponsor’s results and the CDA-AMC base case (Table 7). The CDA-AMC base case suggests that a price reduction of approximately 90% (from $1,076 to $108 per 100 g tube) would be required for ruxolitinib cream to be considered cost-effective at a WTP threshold of $50,000 per QALY relative to no active treatment.
Table 7: CDA-AMC Price-Reduction Analyses
Analysis price reduction | Unit drug cost ($) | ICER for ruxolitinib cream vs. no active treatment ($/QALY) | |
|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis | ||
No price reduction | 1,076 | 85,787 | 535,419 |
10% | 968 | 77,140 | 481,545 |
20% | 861 | 68,493 | 427,670 |
30% | 753 | 59,845 | 373,795 |
40% | 646 | 51,198 | 319,920 |
50% | 538 | 42,551 | 266,045 |
60% | 430 | 33,904 | 212,170 |
70% | 323 | 25,257 | 158,295 |
80% | 215 | 16,610 | 104,420 |
90% | 108 | 7,962 | 50,545 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
Ruxolitinib cream is available to be dispensed only as a 100 g tube (at a submitted price of $1,076 per tube). If a patient discontinues treatment (e.g., because of AEs or an inadequate response), the cost of the full 100 g tube would be incurred by the drug plans. The sponsor further reported that an in-house stability study found that each tube of ruxolitinib cream, once opened, should be used within 6 months. As such, each patient would require at least 2 tubes of ruxolitinib cream per year, regardless of the amount of drug used.
Clinical expert input received by CDA-AMC indicated that ruxolitinib may be used in clinical practice in combination with phototherapy, TCSs, or TCIs. The cost-effectiveness of ruxolitinib cream when used in combination with these treatments is unknown because of a lack of comparative clinical data.
Ruxolitinib cream is currently under review at CDA-AMC for topical treatment of mild to moderate atopic dermatitis in adult and pediatric patients aged 12 years and older whose disease is not adequately controlled with conventional topical prescription therapies (TCSs and TCIs) or when those therapies are not advisable.14
Overall Conclusions
The CDA-AMC Clinical Review of the TRuE-V1 and TRuE-V2 trials found that, based on findings of moderate certainty, ruxolitinib cream likely results in clinically important improvements in the F-VASI score compared with vehicle cream in patients aged 12 years and older with NSV. However, the clinical impact of the objective response on patients’ daily lives as measured by the Vitiligo Area Scoring Index is uncertain. According to results from the pivotal trials’ exploratory analysis, patients who applied ruxolitinib in the studies did not experience a clinically important improvement in health-related quality of life. This suggests that, despite making the condition less noticeable, treatment with ruxolitinib did not reduce the negative impact of the disease on patients’ lives as measured by these metrics. The CDA-AMC Clinical Review also noted concerns with the generalizability of the effect of ruxolitinib in individuals with the greatest unmet needs, such as patients with a pronounced contrast between depigmented and unaffected skin, patients who do not achieve satisfactory improvements with the currently available therapies, and patients whose condition interferes substantially with their daily lives.
Clinical expert input received by CDA-AMC for this review indicated that multiple alternative treatments are used to manage patients with NSV in clinical practice, including TCSs, TCIs, phototherapy, and combination therapy. As such, the sponsor’s base case, which compared ruxolitinib to vehicle cream (i.e., no active treatment) is not informative for decision-making. The comparative effectiveness and safety of ruxolitinib relative to these therapies for vitiligo is unknown. These treatments are less costly than ruxolitinib at publicly available list prices. While the cost per gram of ruxolitinib is $10.76, the costs of TCSs and TCIs range from $0.06 to $0.55 and from $2.47 to $2.75 per gram, respectively.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis. This included assuming the utility for patients who did not respond to treatment was the same as for the baseline and aligning the daily dose of ruxolitinib with that in the product monograph. The results of the CDA-AMC reanalyses align with those of the sponsor’s submitted analysis, indicating that, at a WTP threshold of $50,000 per QALY gained, ruxolitinib cream is not a cost-effective treatment option for the treatment of NSV compared with no active treatment. In the CDA-AMC base case, ruxolitinib cream was associated with an ICER of $535,419 per QALY gained compared with no active treatment in the patient population in the TRuE-V trials (incremental costs = $39,038; incremental QALYs = 0.07). A price reduction of approximately 90% would be required for ruxolitinib to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. This would reduce the price of ruxolitinib cream from $1,076 to $108 per 100 g tube. With this price reduction, the annual drug acquisition costs for ruxolitinib would be approximately $2,905 per patient.
CDA-AMC was unable to address concerns regarding the appropriateness of using F-VASI scores to inform the model structure and the assumptions about changes in clinical practice and disease management. Concerns also remain as to whether the TRuE-V trial results can be generalized to patient groups with the greatest unmet needs, given the external validity concerns raised in the CDA-AMC Clinical Review. CDA-AMC notes that the utility values of patients who did respond to treatment remain greater than those of the general population in both the sponsor’s analysis and the CDA-AMC base-case reanalysis. As a result, the incremental QALYs gained may have been overestimated, producing a lower ICER that favours ruxolitinib. These factors introduce uncertainty regarding the cost-effectiveness of ruxolitinib cream in clinical practice and limit the scope of the interpretation of economic results. As several key limitations remained unresolved, the reanalysis performed by CDA-AMC is associated with uncertainty and, as such, higher price premiums may be required.
References
1.TRuE-V2 - A Phase 3, Double-Blind, Randomized, Vehicle-Controlled, Efficacy and Safety Study of Ruxolitinib Cream Followed by an Extension Period in Participants With Vitiligo, Pub. L. No. INCB 18424-307, (2022).
2.TRuE-V1 - A Phase 3, Double-Blind, Randomized, Vehicle-Controlled, Efficacy and Safety Study of Ruxolitinib Cream Followed by an Extension Period in Participants With Vitiligo, Pub. L. No. INCB 18424-306, (2022).
3.Pr Opzelura®(ruxolitinib cream): Cream, 1.5% w/w ruxolitinib (as ruxolitinib phosphate), Topical Janus Kinase (JAK) Inhibitor [product monograph]. Innomar Strategies Inc.; 2024.
4.A Double-Blind, Vehicle-Controlled, Randomized Withdrawal and Treatment-Extension Study to Assess the Long-Term Efficacy and Safety of Ruxolitinib Cream in Participants With Vitiligo (TRuE-V LTE), Pub. L. No. INCB 18424-308, (2023).
5.Table 13-10-0114-01 Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island (2024).
6.Begum R, Crott R, Martina R, Loizidou EM, Khan I. Estimating health related quality of life effects in vitiligo. Mapping EQ-5D-5 L utilities from vitiligo specific scales: VNS, VitiQoL and re-pigmentation measures using data from the HI-Light trial. Health Qual Life Outcomes. Aug 10 2023;21(1):85. doi:10.1186/s12955-023-02172-4 PubMed
7.Government of O. Ontario Drug Benefit Formulary. May 17, 2024 2024;(May 29, 2024)
8.Schedule of Benefits, Physician Services Under the Health Insurance Act (2024).
9.Sach TH, Thomas KS, Batchelor JM, et al. An economic evaluation of the randomized controlled trial of topical corticosteroid and home-based narrowband ultraviolet B for active and limited vitiligo (the HI-Light Vitiligo Trial). Br J Dermatol. May 2021;184(5):840-848. doi:10.1111/bjd.19554 PubMed
10.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib), 1.5% cream [internal sponsor's package]. Saint-Laurent (QC): Incyte Biosciences Canada Corporation; 2024.
11.Eleftheriadou V, Atkar R, Batchelor J, et al. British Association of Dermatologists guidelines for the management of people with vitiligo 2021. British Journal of Dermatology. 2022;186(1):18-29. doi:https://doi.org/10.1111/bjd.20596 PubMed
12.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. CDA-AMC; 2017. Accessed 1800 Jan 1. https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition
13.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. Feb 2024;25(1):147-155. doi:10.1007/s10198-023-01570-1 PubMed
14.Drug Reimbursement Review: Ruxolitinib (Opzelura) for Atopic dermatitis. CDA-AMC; 2024. Accessed 2025 Jan 14. https://www.cda-amc.ca/ruxolitinib-1
15.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. Accessed 2025 Jan 03, https://www.formulary.health.gov.on.ca/formulary/
16.ELOCOM® (Mometasone Furoate) [product monograph]. Organon Canada Inc; 2021. https://www.organon.com/canada-en/wp-content/uploads/sites/5/2021/05/ELOCOM-CI_E.pdf
17.PrELIDEL (Pimecrolimus) [product monograph]. Valeant Canada LP; 2019. https://pdf.hres.ca/dpd_pm/00053301.PDF
(Mometasone Furoate 0.1% Ointment) [product monograph]. Teva Canada Limited; 2018. https://pdf.hres.ca/dpd_pm/00043401.PDF
19.Protopic (tacrolimus ointment) [product monograph]. LEO Pharma Inc; 2016. https://pdf.hres.ca/dpd_pm/00035397.PDF
(Clobetasol 17-propionate) [product monograph]. Teva Canada Limited; 2017. https://pdf.hres.ca/dpd_pm/00042697.PDF
21.DIPROSONE®(Betamethasone Dipropionate) [product monograph]. Merck Canada Inc; 2017. https://pdf.hres.ca/dpd_pm/00042539.PDF
22.Seirafi H, Farnaghi F, Firooz A, Vasheghani-Farahani A, Alirezaie NS, Dowlati Y. Pimecrolimus cream in repigmentation of vitiligo. Dermatology. 2007;214(3):253-9. doi:10.1159/000099592 PubMed
23.Iraji F, Banihashemi SH, Faghihi G, Shahmoradi Z, Tajmirriahi N, Jazi SB. A Comparison of Betamethasone Valerate 0.1% Cream Twice Daily Plus Oral Simvastatin Versus Betamethasone Valerate 0.1% Cream Alone in the Treatment of Vitiligo Patients. Adv Biomed Res. 2017;6:34. doi:10.4103/2277-9175.203159 PubMed
24.Kumaran MS, Kaur I, Kumar B. Effect of topical calcipotriol, betamethasone dipropionate and their combination in the treatment of localized vitiligo. J Eur Acad Dermatol Venereol. Mar 2006;20(3):269-73. doi:10.1111/j.1468-3083.2006.01420.x PubMed
25.Köse O, Arca E, Kurumlu Z. Mometasone cream versus pimecrolimus cream for the treatment of childhood localized vitiligo. J Dermatolog Treat. May 2010;21(3):133-9. doi:10.3109/09546630903266761 PubMed
26.Silpa-Archa N, Nitayavardhana S, Thanomkitti K, Chularojanamontri L, Varothai S, Wongpraparut C. Comparison of the efficacy and safety of 0.1% tacrolimus ointment and 0.1% mometasone furoate cream for adult vitiligo: A single-blinded pilot study. Dermatologica Sinica. 2016/12/01/ 2016;34(4):177-179. doi:https://doi.org/10.1016/j.dsi.2016.05.005
27.Lepe V, Moncada B, Castanedo-Cazares JP, Torres-Alvarez MB, Ortiz CA, Torres-Rubalcava AB. A Double-blind Randomized Trial of 0.1% Tacrolimus vs 0.05% Clobetasol for the Treatment of Childhood Vitiligo. Archives of Dermatology. 2003;139(5):581-585. doi:10.1001/archderm.139.5.581 PubMed
28.Saskatchewan Drug Plan: search formulary. Accessed 2025 Jan 20, https://formulary.drugplan.ehealthsask.ca/SearchFormulary
29.Government of A. Interactive drug benefit list. Accessed 1800 Jan 1, https://idbl.ab.bluecross.ca/idbl/load.do
30.Table 23: Measured weight, by age and sex, household population, Canada, 2009 to 2011 (2015).
31.Lei T-C, Xu A-E, Gao T-W, et al. Consensus on the Diagnosis and Treatment of Vitiligo in China (2021 Revision)#. International Journal of Dermatology and Venereology. 2021;4(1):10-15. doi:10.1097/jd9.0000000000000151
32.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Ontario Ministry of Health; 2023. Accessed 2025 Jan 3. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
33.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Opzelura (ruxolitinib), 1.5% cream [internal sponsor's package]. Saint-Laurent (QC): Incyte Biosciences Canada Corporation; 2024.
34.Bergqvist C, Ezzedine K. Vitiligo: A Review. Dermatology. 2020;236(6):571-592. doi:10.1159/000506103 PubMed
35.BC PharmaCare Annual Performance Reports and BC PharmaCare Trends (2021).
36.Ontario Public Drug P. Direct data request of eligible beneficiaries by age group (2007-2014). 2014.
37.Nova Scotia Ministry of H. Direct data request of eligible beneficiaries by age group (2008-2011). 2011.
38.Manitoba Health Population Reports (2020).
39.Non-Insured Health Benefits Program - First Nations and Inuit Health Branch: Annual Reports (2021).
40.Alberta Health Care Insurance Plan statistical supplement (2021).
41.Saskatchewan Covered Population Reports (2015).
42.Government of O. Ontario Drug Benefit Formulary. Updated May 17, 2024. Accessed May 29,2024, https://www.formulary.health.gov.on.ca/formulary/
43.Quebec Clinical E. Clinical Expert Opinion. Quebec2022.
44.Ontario Clinical E. Clinical Expert Opinion. Ontario2022.
45.British Columbia Clinical E. Clinical Expert Opinion. British Columbia2022.
46.Incyte. Vitiligo Patient Journey Online Community. In: Incyte, editor. Qualitative Market Research Report2023.
47.Incyte. Canadian Results - The VALIANT Study. 2021.
48.Incyte response to 14-Nov-2024 request for additional information regarding Ruxolitinib (Opzelura) CDA-AMC review: Source for market share assumptions CDA-AMC; 1800. Accessed 1800 Jan 1. URL.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Nonsegmental Vitiligo
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Ruxolitinib (Opzelura) | 1.5% | Cream, 100 g tubea | 1,075.9708b | Thin layer applied twice daily, up 10% of BSA | 79.54c | 29,051c |
BSA = body surface area; CDA-AMC = Canada’s Drug Agency.
Note: A year was assumed to have 365.25 days, or 52.12 weeks.
aAlthough ruxolitinib cream is available in 60 g and 100 g tubes, the sponsor has indicated that only the 100 g size will be marketed in Canada.
bPrice submitted by sponsor.10
cA daily application of approximately 7.14 g of ruxolitinib cream was assumed to estimate treatment cost. The daily dose was based on the maximum use of one 100 g tube per 2 weeks (i.e., 14 days) as noted in the product monograph.3 Based on this assumption, 27 tubes would be required per year. CDA-AMC notes that at minimum, the cost of 2 tubes over 12 months would be incurred based on expiration date of 6 months after opening a tube, which comes out to an annual cost of $2,152 or daily cost of $5.89 per patient.
According to clinical expert input received for this review, the following treatments may also be used to treat patients with nonsegmental vitiligo (Table 9 and Table 10).
Table 9: CDA-AMC Cost Comparison Table of Topical Treatments for Nonsegmental Vitiligo (Not Indicated for Nonsegmental Vitiligo)
Treatment | Strength | Dosage form | Price per tube ($) | Price per gram or mL ($) | Recommended dose |
|---|---|---|---|---|---|
Topical corticosteroids | |||||
Betamethasone dipropionate (generic) | 0.05% | Cream (50 g) | 10.24 | 0.20 | Apply a thin layer to affected area twice daily. |
Ointment (50 g) | 10.76 | 0.22 | |||
Lotion (75 mL) | 14.85 | 0.20 | |||
Betamethasone valerate (generic) | 0.05% | Cream (Not available) | Not available | 0.06 | Apply a thin layer to affected area twice daily. |
Ointment (Not available) | Not available | ||||
0.1% | Cream (Not available) | Not available | 0.09 | ||
Ointment Not available | |||||
Scalp lotion Not available | Not available | 0.09 | |||
Clobetasol propionate (generic) | 0.05% | Cream 15 g 50 g 450 g | 3.42 11.40 102.56 | 0.23 | Apply a thin layer to affected area twice daily for 2 months on and 2 months off. |
Ointment 15 g 50 g 450 g | |||||
Scalp lotion 20 mL 60 mL | 3.98 11.94 | 0.20 | |||
Mometasone furoate (generic) | 0.1% | Cream 15 g 50 g | 8.31 27.71 | 0.55 | Apply thin layer to affected area once or twice daily for 2 months on and 2 months off. |
Ointment 15 g 50 g | 3.38 11.26 | 0.23 | |||
Lotion 75 mL | 27.20 | 0.36 | |||
Topical calcineurin inhibitorsa | |||||
Pimecrolimus (Elidel) | 1% | Cream 20 g 60 g | 55.00 165.00 | 2.75b | Apply a thin layer to affected area twice daily. |
Tacrolimus (protopic) | 0.03% | Ointment 30 g 60 g 100 g | 73.97 147.94 246.57 | 2.47c | Apply a thin layer to affected area twice daily continuously. |
0.10% | Ointment 30 g 60 g 100 g | 78.53 157.06 261.77 | 2.62c | ||
CDA-AMC = Canada’s Drug Agency.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed January 3, 2025),15 and drug specifications (i.e., strength and form) from respective product monographs.16-21 Recommended doses were obtained from published literature and validated with clinical expert input.22-27
aClinical expert feedback indicated that topical calcineurin inhibitors are used in clinical practice, although they may not be covered by public drug plans for vitiligo.
bSaskatchewan formulary list price, accessed January 20, 2025.28
cAlberta formulary list price, accessed January 14, 2025.29
Table 10: CDA-AMC Cost Comparison Table for Systemic Therapy of NSV (Not Indicated for NSV)
Treatment | Strength | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Systemic corticosteroidsa | ||||||
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | 10 mg weeklyb | 0.14 | 52 |
Prednisone (Winpred, generic) | 1 mg 5 mg 50 mg | Tablet | 0.1276 0.0220 0.1735 | 0.3 mg/kg once dailyc | 0.09 | 32 |
Phototherapy | ||||||
UV light therapy | NA | NA | 7.85 per treatmentd | Administered 2 to 3 times per weekc | ||
CDA-AMC = Canada’s Drug Agency; NA = not applicable; NSV = nonsegmental vitiligo.
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary (accessed January 3, 2025),15 unless otherwise indicated, and do not include dispensing fees. A weight of 60 kg was assumed,30 and a year was assumed to have 365.25 days.
aAccording to clinical expert feedback received for this review, systemic corticosteroids are not commonly used for long-term treatment of patients with NSV in clinical practice.
bDosage was informed by clinician input received for this review.
cRecommended dosage was obtained from published literature.31
dOntario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy.”32
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CDA-AMC appraisal section. There are several relevant treatments that were not included. |
Model has been adequately programmed and has sufficient face validity | No | The model includes numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Changes to the user-defined input cells were not propagated into the probabilistic sensitivity analysis as the submitted model reverted to sponsor default values. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | There were several instances of hard coding in the pharmacoeconomic model that made it challenging to validate model inputs. Additionally, there was no guidance provided on changing inputs for the probabilistic sensitivity analysis. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
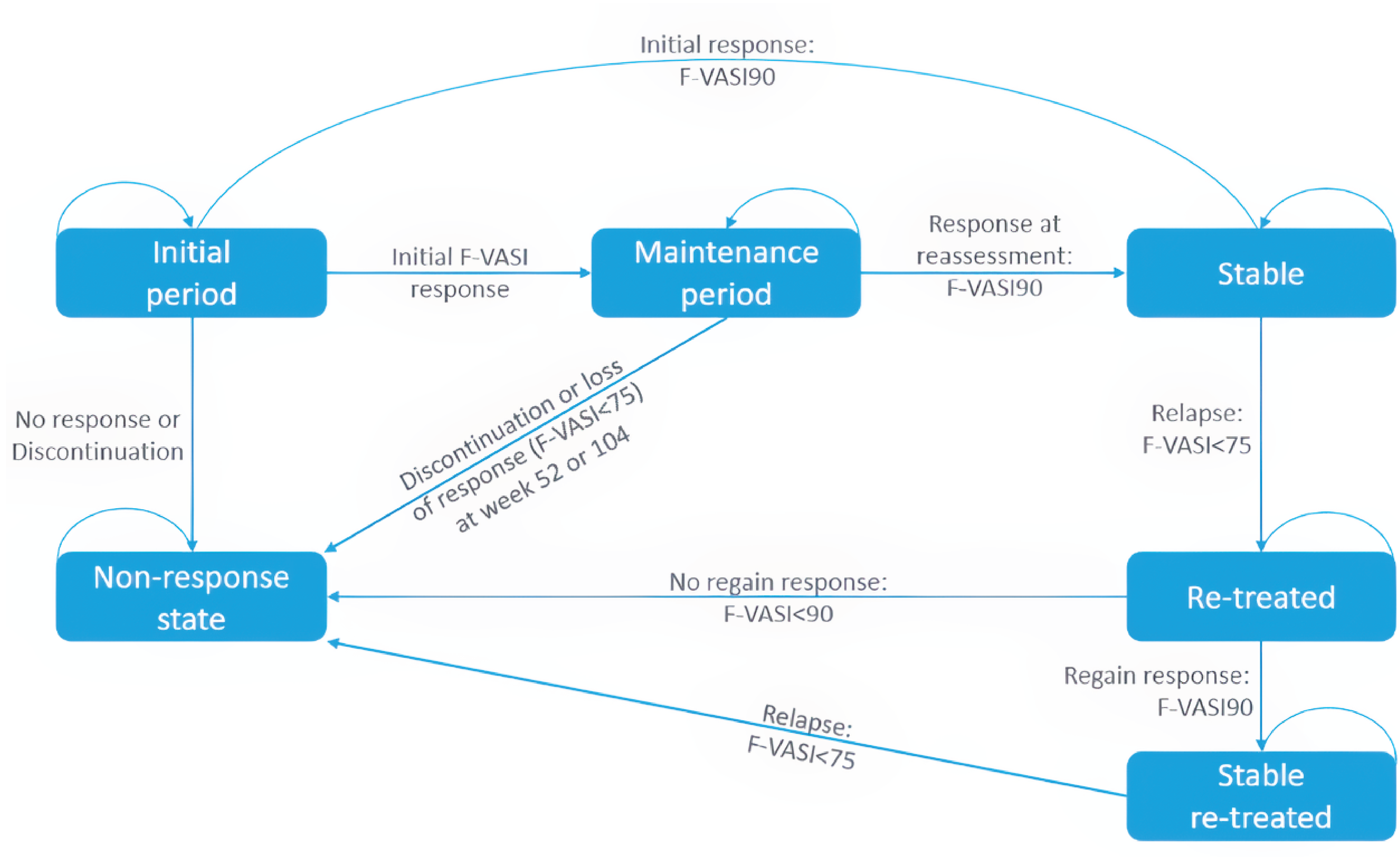
F-VASI = Facial Vitiligo Area Scoring Index; F-VASI<75 = less than 75% improvement in the Facial Vitiligo Area Scoring Index; F-VASI90 = 90% or greater improvement from baseline in the Facial Vitiligo Area Scoring Index; F-VASI<90 = less than 90% improvement in the Facial Vitiligo Area Scoring Index.
Source: Sponsor’s pharmacoeconomic submission.10
Detailed Results of the Sponsor’s Base Case
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Ruxolitinib | No active treatment |
|---|---|---|
Discounted LYs | ||
Total | 31.67 | 31.67 |
Total time in F-VASI90 (years) | 1.34 | 0.29 |
Discounted QALYs | ||
Total | 24.40 | 24.15 |
By health state | ||
Initial period – F-VASI (< 25) | 0.45 | 0.44 |
Maintenance period – F-VASI25 to 49 | 0.11 | 0.06 |
Maintenance period – F-VASI50 to 74 | 0.28 | 0.08 |
Maintenance period – F-VASI75 to 89 | 0.36 | 0.10 |
Stable | 0.62 | 0.12 |
Re-treatment | 0.07 | 0.01 |
Stable re-treated | 0.29 | 0.06 |
AE disutility | 0.00 | 0.00 |
No response | 22.22 | 23.28 |
Discounted costs ($) | ||
Total | 30,420 | 9,462 |
Treatment costs | 28,993 | 8,011 |
Disease management costs | 1,419 | 1,448 |
AE costs | 8 | 3 |
AE = adverse event; F-VASI = Facial Vitiligo Area Scoring Index; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.10
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 14: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Ruxolitinib | No active treatment |
|---|---|---|
Discounted LYs | ||
Total | 31.69 | 31.69 |
Total time in F-VASI90 (years) | 1.34 | 0.29 |
Discounted QALYs | ||
Total | 28.00 | 27.93 |
By health state | ||
Initial period – F-VASI(< 25) | 0.45 | 0.44 |
Maintenance period – F-VASI25 to 49 | 0.11 | 0.06 |
Maintenance period – F-VASI50 to 74 | 0.28 | 0.08 |
Maintenance period – F-VASI75 to 89 | 0.36 | 0.10 |
Stable | 0.62 | 0.12 |
Re-treatment | 0.07 | 0.01 |
Stable re-treated | 0.29 | 0.06 |
AE disutility | 0.00 | 0.00 |
No response | 25.83 | 27.05 |
Discounted costs ($) | ||
Total | 49,964 | 10,926 |
Treatment costs | 48,547 | 9,486 |
Disease management costs | 1,409 | 1,437 |
AE costs | 8 | 3 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; F-VASI = Facial Vitiligo Area Scoring Index; LY = life-year; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency; NSV = nonsegmental vitiligo.
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) assessed the expected budgetary impact of reimbursing ruxolitinib for the topical treatment of patients aged 12 years and older with NSV.33 The BIA was undertaken from the perspective of the Canadian public drug plans at base year (2025) and over a 3-year time horizon (2026 to 2028). The sponsor’s pan-Canadian estimates reflected the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 16.
The sponsor estimated the number of patients eligible for ruxolitinib using an epidemiologic approach with data obtained from published literature and sponsor’s clinical experts input.33,34 The sponsor estimated the number of beneficiaries for each jurisdiction using data from the public drug programs for individuals aged 12 years and older and narrowed the population to those seeking care for NSV.35-41 Comparators included TCSs and BSC. BSC was assumed to include nonprescription products (e.g., sunscreen and camouflage makeup) and have zero cost to the drug plans. Prices for TCSs were obtained from the Ontario Drug Benefit Formulary.42 Dispensing fees and mark-ups were not included. The total treatment cost of all drugs was adjusted by a compliance rate of 50%. Market share assumptions were informed by sponsor-sought clinical experts.43-45The sponsor assumed that market share of ruxolitinib would be captured equally from TCSs and BSC.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) |
|---|---|
Target population | |
Number of beneficiaries (excluding Quebec and territories) | 10,512,679 / 10,914,534 / 11,339,930 |
Prevalence of vitiligo, % | 0.75% |
Patients with NSV, % | 86.0% |
Patients with NSV seeking care, % | 44.1% |
Number of patients eligible for drug under review | 29,903 / 31,046 / 32,256 |
Market uptake (3 years) | |
Uptake (reference scenario) TCS BSC | 50% / 50% / 50% 50% / 50% / 50% |
Uptake (new drug scenario) Ruxolitinib TCS BSC | 3% / 6% / 10% 49% / 47% / 45% 49% / 47% / 45% |
Cost of treatment (per patient, per year) | |
Ruxolitinib TCSa BSC | $8,607.77 $120.34 $0 |
BSC = best supportive care; NSV = nonsegmental vitiligo; TSC = topical corticosteroid.
aThe cost attributed to topical corticosteroids was the average price of the lowest cost of hydrocortisone, hydrocortisone acetate, betamethasone dipropionate, and betamethasone valerate sourced from Ontario Drug Benefit Formulary and assuming a 50 g tube.33
Summary of the Sponsor’s BIA Results
The sponsor estimated that reimbursing ruxolitinib for the topical treatment of patients aged 12 years and older with NSV will be $51,160,968 over the first 3 years (year 1: $7,667,911; year 2: $15,922,044; year 3: $27,571,014).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The proportion of patients seeking care is underestimated: The sponsor estimated that approximately 44.1% of all patients with NSV sought care based on internal market research.46,47 However, this estimate lacks face validity according to the sponsor-sought clinician input in which 2 out of 3 clinicians estimated that 75% to 80% of patients with NSV would seek treatment.44,45 According to sponsor-sought clinician input and the clinical expert feedback obtained by CDA-AMC, the sponsor’s estimated proportion of patients with NSV seeking care is an underestimate.
Market share of ruxolitinib cream may be underestimated: Based on internal assumptions from the sponsor,48 ruxolitinib market share was 3% in year 1, 6% in year 2, and 10% in year 3. According to clinical expert feedback received by CDA-AMC for this review, the market share of ruxolitinib is uncertain. Clinical expert feedback noted that, while its uptake would depend on its efficacy demonstrated in real-world conditions, the uptake of ruxolitinib would be higher than sponsor’s estimate because it is the first on-label treatment for NSV.
In reanalysis, ruxolitinib market share was increased to 30% in year 1, 40% in year 2, and 50% in year 3 based on clinical expert feedback obtained by CDA-AMC for this review. The impact of assuming a constant 30% market share was explored in a scenario analysis.
Uncertainty about how ruxolitinib will capture market shares from its comparators. In addition to the issue previously noted regarding not all comparators adequately captured in the model, the sponsor assumed that market displacement from the introduction of ruxolitinib would be equal between BSC (i.e., consisting of sunscreen and camouflage makeup) and TCSs. The sponsor’s estimate lacks face validity. According to the clinical expert input received for this review, the majority of market share for ruxolitinib is expected to be captured from TCSs rather than BSC because patients receiving BSC are less likely to initiate treatment. As such, if ruxolitinib is reimbursed, its uptake among patients receiving BSC would likely be lower compared to those receiving TCSs.
In reanalysis, 75% of market shares of ruxolitinib was captured from TCSs and 25% from BSC.
The treatment cost of ruxolitinib is underestimated: In the BIA, the sponsor estimated that all patients would use 3.84 g of ruxolitinib cream per day over the 3-year time horizon. As noted in the appraisal of the economic evaluation, there is considerable variability in the amount of drug used in the TruE-V1 and TruE-V2 trials. The mean daily dose of ruxolitinib ranged from 5.82 g to 8.86 g per day during the double-blind controlled period of the trials (24-week duration), and from ████ grams to █████ grams per day in the extension period of the trials (28-week duration).1,2,4 CDA-AMC notes that the product monograph for ruxolitinib cream recommends using a maximum of one 100 g tube per 2 weeks, which translates into 7.14 g per day.3
In reanalysis, CDA-AMC assumed that 7.14 g of ruxolitinib cream would be used daily. CDA-AMC could not incorporate different amounts of drug used for patients who continued treatment into subsequent years given the model structure. If the amount of ruxolitinib cream required becomes lower over time, the budget impact would be lower than estimated. The sponsor’s assumption that the same amount of TCSs as ruxolitinib would be used daily was retained.
Use of compliance rate to estimate actual drug costs is not appropriate: The sponsor adjusted the total treatment cost of ruxolitinib and TCSs by assuming a compliance rate of 50%. The sponsor’s approach effectively assumes that only 50% of the cream products would be used and decreases the treatment cost incurred by public drug plans. This consideration of compliance rate is problematic because the cost of full tube of cream products is incurred by public drug plans once the tube is dispensed, regardless of actual cream usage. The sponsor’s assumption also introduced misalignment between the BIA and cost-utility analysis because the sponsor’s pharmacoeconomic analysis assumed 100% compliance for all drugs.
In reanalysis, a compliance rate of 100% was assumed.
Relevant comparators are excluded: The sponsor-submitted BIA included BSC (consisting of sunscreen and camouflage makeup) and TCSs as treatments used currently by patients with NSV. Other commonly used treatments, such as TCIs, phototherapy, and combination therapy, were identified as relevant by both sponsor-sought and CDA-AMC-sought clinical expert input. However, these treatments were not included in the BIA. As such, the sponsor’s analysis does not adequately capture the budget impact of ruxolitinib in terms of displacing all relevant treatments.
CDA-AMC was unable to address this limitation. If these comparators were included, the budget impact would be lower than estimated. Because these comparators are much less costly than ruxolitinib cream at publicly available prices, displacing them is unlikely to be a key driver of the results.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by increasing the proportion of patients with NSV seeking care, increasing the market share of ruxolitinib, adjusting the market capture of ruxolitinib to reflect expert feedback, adopting a higher daily dose and assuming a 100% compliance rate for all drugs.
Table 17: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Proportion of patients with NSV seeking care | 44.1% | 75.0% |
2. Market share of ruxolitinib (year 1 / year 2 / year 3) | 3% / 6% / 10% | 30% / 40% / 50% |
3. Market capture of ruxolitinib (year 1 / year 2 / year 3) | TCS: 50% BSC: 50% | TCS: 75% BSC: 25% |
4. Daily dose (gram per day) | Ruxolitinib: 3.84 TCS: 3.84 | Ruxolitinib: 7.14 TCS: 7.14 |
5. Compliance rate | 50% | 100% |
CDA-AMC base-case | Reanalysis 1 + 2 + 3 + 4 + 5 | |
BIA = budget impact analysis; BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; NSV = nonsegmental vitiligo; TCS = topical corticosteroid.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19.
In the CDA-AMC reanalysis, the 3-year budget impact of reimbursing ruxolitinib cream for topical treatment of patients with NSV aged 12 years and older increased to $1,833,254,114 (year 1: $438,355,337; year 2: $606,815,718; year 3: $788,083,059). The estimated budget impact is primarily driven by market share of ruxolitinib and key parameters informing drug usage such as daily dose and compliance rates.
Table 18: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 51,160,968 |
CDA-AMC reanalysis 1 | 87,008,450 |
CDA-AMC reanalysis 2 | 320,681,135 |
CDA-AMC reanalysis 3 | 50,980,901 |
CDA-AMC reanalysis 4 | 89,513,688 |
CDA-AMC reanalysis 5 | 95,905,808 |
CDA-AMC base case | 1,833,254,114 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted a scenario analysis of assuming ruxolitinib has a constant market share of 30% across all 3 years to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 19).
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 1,734,224 | 1,799,214 | 1,867,990 | 1,940,795 | 5,607,998 |
New drug | 1,734,224 | 9,467,124 | 17,790,034 | 29,511,809 | 56,768,967 | |
Budget impact | 0 | 7,667,911 | 15,922,044 | 27,571,014 | 51,160,968 | |
CDA-AMC base case | Reference | 10,421,077 | 10,811,601 | 11,224,882 | 11,662,373 | 33,698,856 |
New drug | 10,421,077 | 449,166,937 | 618,040,600 | 799,745,432 | 1,866,952,970 | |
Budget impact | 0 | 438,355,337 | 606,815,718 | 788,083,059 | 1,833,254,114 | |
CDA-AMC scenario analysis 1: 30% market share of ruxolitinib | Reference | 10,421,077 | 10,811,601 | 11,224,882 | 11,662,373 | 33,698,856 |
New drug | 10,421,077 | 449,166,937 | 466,336,670 | 484,512,209 | 1,400,015,816 | |
Budget impact | 0 | 438,355,337 | 455,111,789 | 472,849,836 | 1,366,316,961 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.