Drugs, Health Technologies, Health Systems
Reimbursement Review
Olopatadine Hydrochloride and Mometasone Furoate Nasal Spray (Ryaltris)
Sponsor: Bausch Health, Canada Inc.
Therapeutic area: Seasonal allergic rhinitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AR
allergic rhinitis
CI
confidence interval
CrI
credible interval
DIC
deviance information criterion
FAS
full analysis set
FE
fixed effects
GRADE
Grading of Recommendations Assessment, Development and Evaluation
IgE
immunoglobulin E
ITC
indirect treatment comparison
iTNSS
instantaneous Total Nasal Symptom Score
LS
least squares
MID
minimal important difference
mometasone NS
mometasone furoate nasal spray
NMA
network meta-analysis
NR
not reported
NS
nasal spray
olopatadine-mometasone
olopatadine hydrochloride and mometasone furoate nasal spray
PAR
perennial allergic rhinitis
PRQLQ
Paediatric Rhinoconjunctivitis Quality of Life Questionnaire
RCT
randomized controlled trial
RE
random effects
RQLQ(S)
Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities
rTNSS
reflective Total Nasal Symptom Score
rTOSS
reflective Total Ocular Symptom Score
SAR
seasonal allergic rhinitis
SD
standard deviation
SE
standard error
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
TNSS
Total Nasal Symptom Score
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product, strength, formulation | Olopatadine hydrochloride and mometasone furoate nasal spray suspension (Ryaltris): 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate (as monohydrate) per delivered dose |
Sponsor | Bausch Health, Canada Inc. |
Indication | For the symptomatic treatment of moderate to severe seasonal allergic rhinitis (SAR) and associated ocular symptoms in adults, adolescents, and children aged 6 years and older |
Reimbursement request | As per indication, for the symptomatic treatment of moderate to severe seasonal allergic rhinitis (SAR) and associated ocular symptoms in adults, adolescents, and children aged 6 years and older |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | September 21, 2022 |
NOC = Notice of Compliance.
Sources: Sponsor’s summary of clinical evidence1 and product monograph.2
Introduction
Allergic rhinitis (AR) is an immunoglobulin E (IgE)–mediated inflammation of the nasal mucosa triggered by exposure to allergens.3,4 AR has been categorized as seasonal allergic rhinitis (SAR) or perennial allergic rhinitis (PAR). AR is estimated to affect approximately 500 million people globally, irrespective of age or ethnic group.5 Studies suggest that the prevalence of AR varies by country but generally affects more than 25% of children and 40% of adults around the world.6 In Canada, AR is highly prevalent, affecting up to 20% to 25% of the population.7 SAR accounts for approximately 76.7% of AR cases. Based on these data, estimates of the prevalence of SAR in Canada range from 12.9% to 19.2%,8,9 affecting approximately 3.5 million people. Patients often describe 1 or more of the following symptoms of AR: nasal congestion (stuffiness), nasal itching, rhinorrhea, sneezing, and cough.4,10 AR is often accompanied by allergic conjunctivitis and is collectively known as allergic rhinoconjunctivitis,11 which also includes ocular symptoms, such as itchiness, redness, and irritation of the eye.11 Approximately 80% of SAR symptoms develop before the age of 20 years and peak at age 20 to 40 years before progressively declining.12
According to the clinical experts consulted by the review team, the management of moderate to severe SAR involves a comprehensive approach, with the goals of alleviating symptoms, improving quality of life, and minimizing symptom exacerbations. The clinical experts consulted by the review team noted that the goals of treatment are generally consistent across age groups (i.e., adults, adolescents, and children aged 6 years and older), but the approach to treatment and consideration of medication choices may vary across these age groups. Intranasal corticosteroids can be used alone or combined with oral or intranasal antihistamines.13-15 According to the clinical experts consulted by the review team, intranasal corticosteroids alone or in combination with intranasal antihistamines are considered a first-line treatment option for moderate to severe SAR and generally preferred over oral antihistamines alone. The clinical experts consulted by the review team noted that fluticasone propionate, mometasone furoate, budesonide, ciclesonide, beclomethasone dipropionate, and fluticasone furoate are widely used or prescribed to reduce nasal inflammation and associated symptoms. Oral antihistamines are also used to manage itching, sneezing, and ocular symptoms, and would be considered as adjunctive therapy. According to the clinical experts consulted by the review team, second-generation nonsedating antihistamines, such as cetirizine, loratadine, desloratadine, fexofenadine, bilastine, and rupatadine, are used for the treatment of SAR instead of first-generation antihistamines. The clinical experts consulted by the review team noted that leukotriene receptor antagonists can be considered for the treatment of AR, particularly in patients who have concomitant asthma or those whose condition does not respond adequately to other therapies. The clinical experts consulted for this review noted that other pharmaceutical therapies that can be used in patients with AR include ocular antihistamines, mast cell stabilizers, and allergen immunotherapy or desensitization. Nonpharmacological management includes educating patients regarding allergen avoidance measures and environmental control measures, as well as saline nasal irrigation to help alleviate nasal symptoms and reduce the need for pharmacological treatments, according to the clinical experts consulted by the review team.
Olopatadine hydrochloride and mometasone furoate nasal spray (NS) (Ryaltris) contains 2 active substances: olopatadine hydrochloride and mometasone furoate. Olopatadine hydrochloride acts by selectively inhibiting the histamine-1 receptor and stabilizing mast cells to attenuate the inflammatory and allergic response experienced by patients with SAR.16 Mometasone furoate possesses anti-inflammatory, vasoconstrictive, and antipruritic properties.17 Mometasone furoate crosses cellular membranes to repress inflammatory gene transcription either directly or by activating the transcription of anti-inflammatory factors. Used together, olopatadine may trigger fast relief from the symptoms of SAR, while mometasone furoate may act to suppress the underlying allergic inflammatory reaction.18,19 (In this Clinical Review Report, olopatadine hydrochloride and mometasone furoate NS is referred to as olopatadine-mometasone, and mometasone furoate NS is referred to as mometasone NS.)
The objective of this Clinical Review Report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of olopatadine-mometasone for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the review team’s call for input and from the clinical experts consulted by the review team for the purpose of this review.
Patient Input
This review received patient group submissions from Asthma Canada and Allergy Quebec. Asthma Canada is a national charity focusing on improving the quality of life and health of people with asthma and respiratory allergies. Allergy Quebec is the main reference centre in Quebec for patients with food allergies and brings together allergists, nutritionists, pharmacists, institutions, and companies in the food sector. Asthma Canada collected patient input using its 2024 Annual Asthma Survey from a total of 1,407 patients and caregivers. Asthma Canada also conducted 2 one-on-one interviews with patients with AR who were selected at random from the participants who completed the AR section of the survey and provided their contact information. Allergy Quebec did not collect any input data from patients.
Both patient groups noted that AR can cause uncomfortable symptoms, including runny and/or itchy nose, nasal congestion, swollen and/or itchy eyes, headaches, sinus pain and/or pressure, and tiredness that negatively impact patients’ daily activities and quality of life. In total, 82% of the respondents to the Asthma Canada survey indicated that the physical symptoms are the most difficult and/or frustrating aspect of living with AR. Patients stated that finding a solution and/or treatment to eliminate or significantly lessen the symptoms of AR would be important for them, in particular, the elimination of rhinorrhea and relief of other symptoms, and more effective medications that do not trigger asthma flare-ups. Based on the survey data from Asthma Canada, just 43% of the participants reported that their current treatments can, or most of the time can, control their allergic symptoms, while 57% reported that current treatments do not control their symptoms. Based on the interview results from Asthma Canada, patient concerns included the lack of efficacy or lack of sustained efficacy, the undesired side effects (e.g., drowsiness, stuffy or dry nose), and the cost and problems with accessing some antihistamines (e.g., due to lack of insurance coverage or availability at local pharmacies).
Clinician Input
Input From the Clinical Experts Consulted by the Review Team
According to the clinical experts consulted by the review team, the main goals of the management of moderate to severe SAR include alleviating symptoms, improving quality of life, and minimizing symptom exacerbations. According to the clinical experts consulted by the review team, there were several unmet needs. For instance, not all patients respond adequately to the currently available treatments, particularly intranasal corticosteroids and oral antihistamines. A patient’s condition can also become refractory to current treatment options over time (e.g., due to escalation of eosinophilic inflammation that does not respond to first-line treatment with antihistamines). The clinical experts also noted the need for treatment options that offer better tolerability and can improve adherence.
According to the clinical experts consulted by the review team, olopatadine-mometasone can be used as a first-line treatment option, based on individual patient needs and treatment responses, by providing a dual-action therapy that combines an intranasal corticosteroid with an antihistamine. The clinical experts consulted by the review team noted that in clinical practice, intranasal corticosteroids alone are usually given to patients first because they can be given once daily and may be sufficient to treat symptoms. Intranasal corticosteroids combined with antihistamines are usually reserved for when intranasal corticosteroids alone are insufficient because the combination therapy is generally more costly, requires twice-daily administration, and may not be tolerated due to taste. Of note, the clinical experts consulted by the review team also noted that it is not necessary to trial monotherapy with an antihistamine or nasal corticosteroid before using olopatadine-mometasone.
According to the clinical experts consulted by the review team, the patients most suitable or most likely to respond to olopatadine-mometasone include the following:
patients who are experiencing moderate to severe symptoms of SAR, have had an inadequate response to monotherapy with intranasal corticosteroids or antihistamines, and require both anti-inflammatory (intranasal corticosteroids) and antihistaminic or mast cell–stabilizing effects to effectively manage their symptoms
patients whose quality of life is significantly impacted by SAR symptoms, affecting daily activities, sleep, and overall well-being.
According to the clinical experts consulted by the review team, the patients most suitable for treatment with olopatadine-mometasone would be identified through a clinical evaluation and symptom assessment, and noted that the assessment of symptom severity would occur through a physical examination and by obtaining a patient history. Conversely, the patients least suitable for olopatadine-mometasone include those with mild symptoms of SAR that are well controlled with monotherapy (either intranasal corticosteroids or antihistamines alone). The clinical experts consulted by the review team noted that allergy testing, such as skin prick tests or specific IgE testing, can identify the allergens triggering symptoms but is not required specifically for the initiation of olopatadine-mometasone.
According to the clinical experts consulted by the review team, in clinical practice, determining treatment response involves assessing various outcomes that reflect improvements in symptom control and overall quality of life. The clinical experts consulted by the review team noted that the typical outcomes used to assess response include reductions in the frequency and severity of nasal and ocular symptoms, such as congestion, sneezing, itching, rhinorrhea, and eye redness or watering. The clinical experts consulted by the review team noted that the extent to which these symptoms interfere with daily activities, sleep patterns, and productivity is evaluated, and assessments are conducted regularly, especially at the beginning of treatment and during peak allergy seasons, to ensure efficacy and to adjust therapy as needed. According to the clinical experts consulted by the review team, the outcomes used in clinical practice are generally aligned with those in clinical trials and include measurement of symptom scores, medication usage, and quality of life assessments. According to the clinical experts consulted by the review team, a clinically meaningful response to treatment varies according to many factors, including the patient population, the severity of initial symptoms, and the patient’s expectations, and may even vary among physicians based on their clinical experience.
The clinical experts consulted by the review team noted several situations when the discontinuation of olopatadine-mometasone should be considered, including lack of effectiveness, intolerable or persistent adverse events (AEs), or patient preference or adherence.
According to the clinical experts consulted by the review team, olopatadine-mometasone is suitable for treatment in various clinical settings, including in community settings, outpatient clinics in hospitals, and specialty allergy clinics. The clinical experts consulted by the review team noted that primary care physicians can diagnose and initiate treatment for patients with SAR and monitor treatment response through regular follow-up visits and adjust therapy as needed. According to the clinical experts consulted by the review team, although specialists such as allergists and immunologists or otolaryngologists may offer additional expertise in managing severe or refractory cases of AR, their involvement is not always required for routine diagnosis and management with olopatadine-mometasone.
Clinician Group Input
No clinician group input was received by the review team for this review.
Drug Program Input
Input was obtained from the drug programs that participate in the Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a recommendation for olopatadine-mometasone:
relevant comparators
consideration for initiation of therapy
consideration for prescribing of therapy
generalizability
system and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
Three sponsor-conducted pivotal studies, GSP301-301, GSP301-304, and GSP301-305, were included in the sponsor-submitted systematic literature review (SLR). Both the GSP301-301 (N = 1,176) and GSP301-304 (N = 1,180) trials were phase III, double-blind randomized controlled trials (RCTs) that enrolled adolescent and adult patients (aged 12 years and older) with SAR. The primary objective of the GSP301-301 and GSP301-304 trials was to compare the efficacy of olopatadine-mometasone with placebo and the individual constituent monotherapies (i.e., olopatadine hydrochloride NS and mometasone NS) at the same dose in the same vehicle, and to assess the efficacy of olopatadine hydrochloride NS and mometasone NS versus placebo over 14 days of study treatment. Of note, olopatadine hydrochloride NS is not relevant to this Reimbursement Review because it is currently unavailable in Canada; therefore, results for olopatadine hydrochloride NS are not presented in this Clinical Review Report. The GSP301-305 trial (N = 446) was a phase III, double-blind, RCT investigating children (aged ≥ 6 to < 12 years) with SAR. The primary objective of the GSP301-305 trial was to assess the efficacy of olopatadine-mometasone relative to placebo over 14 days of study treatment. The primary end point of all 3 pivotal trials was patient-reported 12-hour reflective Total Nasal Symptom Score (rTNSS). The secondary efficacy and safety outcomes reported in the 3 pivotal trials included patient-reported 12-hour instantaneous Total Nasal Symptom Score (iTNSS), patient-reported 12-hour reflective Total Ocular Symptom Score (rTOSS), and harms, i.e., treatment-emergent adverse events (TEAEs), treatment-emergent serious adverse events (TESAEs), withdrawals, and deaths. The health-related quality of life (HRQoL) outcomes evaluated in the trials included the Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities (RQLQ[S]) in the GSP301-301 and GSP301-304 trials, and the Paediatric Rhinoconjunctivitis Quality of Life Questionnaire (PRQLQ) in the GSP301-305 trial.
In the GSP301-301 and GSP301-304 trials, the mean age of patients was 39.3 years (standard deviation [SD] = 15.3 years) and 39.6 years (SD = 14.81 years), respectively. Across trials, most patients were female (64.6% and 62.9%, respectively). In the GSP301-301 trial, the baseline rTNSS score was the same across the olopatadine-mometasone group, the mometasone NS group, and the placebo group (mean = 10.1; SD = 1.2). In the GSP301-304 trial, the baseline mean rTNSS score was 10.1 (SD = 1.2) for the olopatadine-mometasone group, 10.3 (SD = 1.3) for the mometasone NS group, and 10.3 (SD = 1.2) for the placebo group. In the GSP301-305 trial, the mean age of the study population was 8.7 years (SD = 1.7 years), and there were slightly more males (56.0%) in the olopatadine-mometasone group, while in the placebo group, the proportion of male and female patients was similar (50.7% versus 49.3%). In the GSP301-305 trial, the baseline mean rTNSS score was 8.83 (SD = 1.41) for the olopatadine-mometasone group and 8.84 (SD = 1.66) for the placebo group.
Efficacy Results
Twelve-Hour rTNSS Over the 14-Day Treatment Period
In the full analysis set (FAS) for the GSP301-301 trial, the within-group least squares (LS) mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (standard error [SE] = not reported [NR]) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −0.98 points (95% confidence interval [CI], −1.38 to −0.57) between the olopatadine-mometasone group and the placebo group and −0.39 points (95% CI, −0.79 to 0.01) between the olopatadine-mometasone group and the mometasone NS group, with both point estimates of the LS mean difference favouring the olopatadine-mometasone group.
In the FAS of the GSP301-304 trial, the within-group LS mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −1.09 points (95% CI, −1.49 to −0.69) between the olopatadine-mometasone group and the placebo group and −0.47 points (95% CI, −0.86 to −0.08) between the olopatadine-mometasone group and the mometasone NS group, with both point estimates of the LS mean difference in favour of the olopatadine-mometasone group.
In the FAS of the GSP301-305 trial, the within-group LS mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in both treatment groups: ████ ██████ (SE = 0.18) in the olopatadine-mometasone group and ███ ████ ██████ (SE = 0.17) in the placebo group. The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −0.6 points (95% CI, −0.9 to −0.2) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Twelve-Hour iTNSS Over the 14-Day Treatment Period
In the FAS of the GSP301-301 trial, the within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and ███ █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.93 points (95% CI, −1.28 to −0.58) between the olopatadine-mometasone group and the placebo group and −0.36 points (95% CI, −0.71 to −0.01) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
In the FAS of the GSP301-304 trial, the within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.94 points (95% CI, −1.32 to −0.56) between the olopatadine-mometasone group and the placebo group and −0.51 points (95% CI, −0.88 to −0.13) between the olopatadine-mometasone group and the mometasone NS group, with both point estimates of the LS mean difference favouring the olopatadine-mometasone group.
In the FAS of the GSP301-305 trial, the within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in both treatment groups: ████ ██████ (SE = 0.17) in the olopatadine-mometasone group and ████ ██████ (SE = 0.17) in the placebo group. The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.6 points (95% CI, −1.0 to −0.3) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Twelve-Hour rTOSS Over the 14-Day Treatment Period
In the FAS of the GSP301-301 trial, the within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and ███ █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.49 points (95% CI, −0.79 to −0.19) between the olopatadine-mometasone group and the placebo group and −0.19 points (95% CI, −0.49 to 0.11) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
In the FAS of the GSP301-304 trial, the within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.52 points (95% CI, −0.84 to −0.20) between the olopatadine-mometasone group and the placebo group and −0.35 points (−0.66 to −0.03) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
In the FAS of the GSP301-305 trial, the within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in both treatment groups: | ████ ██████ ███ █ █████ in the olopatadine-mometasone group and ████ ██████ ███ █ █████ in the placebo group. The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.2 points (95% CI, −0.6 to 0.1) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
RQLQ(S) Overall Score on Day 15
In the FAS of the GSP301-301 trial, the within-group LS mean change from baseline in the RQLQ(S) overall score at day 15 showed an improvement in all 3 treatment groups: █████ ██████ (SE = NR) in the olopatadine-mometasone group, █████ ██████ (SE = NR) in the mometasone NS group, and ███ █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the RQLQ(S) overall score at day 15 was −0.43 points (95% CI, −0.64 to −0.21) between the olopatadine-mometasone group and the placebo group and −0.20 points (95% CI, −0.41 to 0.02) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
In the FAS of the GSP301-304 trial, the within-group LS mean change from baseline in the RQLQ(S) overall score at day 15 showed an improvement in all 3 treatment groups: | █████ █████ (SE = NR) in the olopatadine-mometasone group, | █████ ██████ (SE = NR) in the mometasone NS group, and ███ █████ ██████ (SE = NR) in the placebo group. The between-group LS mean difference in the RQLQ(S) overall score at day 15 was −0.45 points (95% CI, −0.68 to −0.22) between the olopatadine-mometasone group and the placebo group and −0.09 points (95% CI, −0.32 to 0.14) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
PRQLQ Overall Score on Day 15
In the FAS of the GSP301-305 trial, the within-group LS mean change from baseline in the PRQLQ overall score at day 15 showed an improvement in both treatment groups: | ████ ██████ ███ █ █████ in the olopatadine-mometasone group and ████ ██████ ███ █ █████ in the placebo group. The between-group LS mean difference in the PRQLQ overall score at day 15 was −0.3 points (95% CI, −0.5 to −0.1) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Harms Results
Treatment-Emergent Adverse Events
In the safety analysis set of the GSP301-301 trial, the proportion of patients experiencing TEAEs was 12.9% (39 out of 302) in the olopatadine-mometasone group, which was higher than in the mometasone NS group (7.1%; 21 out of 294) or in the placebo group (9.4%; 27 out of 287). The proportion of patients who had dysgeusia was 3.3% (10 out of 302) in the olopatadine-mometasone group, 0.7% (2 out of 287) in the placebo group, and 0% in the mometasone NS group. Headache occurred in 2.8% (8 out of 287) of the patients in the placebo group, a higher proportion than in the olopatadine-mometasone group (0.7%; 2 out of 302) or in the mometasone NS group (0.7%; 2 out of 294). █████████ ███ ████████ ██ ████ ███████ of the patients in the olopatadine-mometasone group, in 0.7% (2 out of 294) of the patients in the mometasone NS group, and ██ ████ ███████ of the patients in the placebo group.
In the safety analysis set of the GSP301-304 trial, the proportion of patients experiencing TEAEs was 15.6% (46 out of 294) in the olopatadine-mometasone group, higher than in the mometasone NS group (9.6%; 28 out of 293) or in the placebo group (9.5%; 28 out of 294). Dysgeusia was reported in 3.7% (11 out of 294) of the patients in the olopatadine-mometasone group and in 0% of patients in the mometasone NS and placebo groups. ████████ ████████ ██ ████ ███████ of the patients in the mometasone NS group, in ████ ███████ of the patients in the placebo group, and in 0 patients in the olopatadine-mometasone group. The proportion of patients who had epistaxis was 0.7% (2 out of 294) in the olopatadine-mometasone group, 1.0% (3 out of 293) in the mometasone NS group, and 1.0% (3 out of 294) in the placebo group.
In the safety analysis set of the GSP301-305 trial, the proportion of patients experiencing TEAEs was 12.0% (27 out of 225) in the olopatadine-mometasone group and 10.4% (23 out of 221) in the placebo group. The most common TEAE in the olopatadine-mometasone group was epistaxis (2.3%; 5 out of 225), while 0.9% (2 out of 221) of the patients in the placebo group had epistaxis. Dysgeusia was reported in 1.3% (3 out of 225) of the patients in the olopatadine-mometasone group and in 0% of patients in the placebo group. Headache occurred in 1.3% (3 out of 225) of the patients in the olopatadine-mometasone group and 0.5% (1 out of 221) of the patients in the placebo group.
Treatment-Emergent Serious Adverse Events
In the safety analysis set of the GSP301-301 trial, only 1 patient had a TESAE (0.3%), which was 1 spontaneous abortion in the olopatadine-mometasone group.
In the safety analysis set of the GSP301-304 trial, no patients in the olopatadine-mometasone group had a TESAE. One patient in the mometasone NS group (0.3%) had 1 TESAE (i.e., peritonsillar abscess), and 1 patient in the placebo group (0.3%) had 3 TESAEs (including 1 instance each of osteomyelitis, syncope, and foot fracture).
In the safety analysis set of the GSP301-305 trial, there was only 1 TESAE (i.e., meningitis), which was reported in 1 patient (0.5%) in the placebo group.
Withdrawals Due to TEAEs
In the safety analysis set of the GSP301-301 trial, no patients withdrew due to TEAEs in the olopatadine-mometasone group, while 4 patients in the mometasone NS group and 1 patient in the placebo group withdrew. The specific reasons for withdrawal were NR.
In the safety analysis set of the GSP301-304 trial, no patients withdrew due to TEAEs in the olopatadine-mometasone group or in the mometasone NS group. One patient in the placebo group (0.3%) discontinued due to a foot fracture.
In the safety analysis set of the GSP301-305 trial, 4 patients in the olopatadine-mometasone group (1.8%) withdrew due to TEAEs (1 instance each of conjunctivitis, acute otitis media, and sinusitis, and 1 upper respiratory tract infection) and 1 patient in the placebo group (0.5%) due to otitis media.
Mortality
No deaths were reported in the GSP301-301, GSP301-304, or GSP301-305 trials.
Critical Appraisal
The risk of bias arising from the randomization process was determined to be low for all 3 pivotal trials — that is, the GSP301-301 and GSP301-304 studies in adolescents and adults (aged 12 years and older) and the GSP301-305 study in children (aged ≥ 6 to < 12 years). The randomization processes were based on a computer-generated randomization scheme. Both the review team and the clinical experts consulted by the review team determined that the baseline characteristics were generally balanced across treatment groups within each of the 3 pivotal trials. The risk of performance bias due to the knowledge of treatment assignment was considered to be low by the review team because all 3 pivotal trials adopted a double-blind design, which masked the trial participants and trial personnel. An adherence rate of between 75% and 125% (i.e., twice a day for 14 days = 100%; twice a day for 17 days = 125%) was achieved by more than 90% of patients in each treatment group. The risk of bias due to missing outcome data was determined to be low for all 3 pivotal trials. Based on patient disposition information, a small proportion of patients in each treatment group of the 3 pivotal trials discontinued from the study for various reasons (e.g., loss to follow-up, withdrawal by patients, nonadherence). In all 3 pivotal trials, the analyses in the per-protocol analysis set, which excluded patients who had not adhered to the study protocol (defined as a major protocol violation), and the sensitivity analyses for rTNSS, which assumed the data missing were missing not at random, showed results consistent with those from the FAS (results NR) according to study investigators. Definitions for patient-reported symptom scores, including rTNSS (primary efficacy end point), iTNSS, and rTOSS were consistent across the 3 pivotal trials and considered accurate by the clinical experts consulted by the review team. However, because reflective and instantaneous symptom scales were designed primarily for assessment in adults, young children might need the assistance of a proxy to assess and report the severity of their symptoms. In the GSP301-305 trial, children assessed their symptoms with the assistance of their parents, guardians, or caregivers as needed. The possibility of underestimating the treatment difference between olopatadine-mometasone and placebo due to the assistance of a proxy remains unclear for the GSP301-305 trial. A gatekeeping strategy was used for rTNSS, iTNSS, and rTOSS in the GSP301-301 and GSP301-304 trials to adjust for multiplicity; however, multiplicity was not adjusted for RQLQ(S) in these 2 trials. In the GSP301-305 trial, adjustment for multiplicity was not carried out for any outcome.
Overall, the clinical experts consulted by the review team noted that the results from the 3 sponsor-submitted pivotal trials were generalizable to the Canadian context despite some potential issues. First, the Health Canada–approved indication is for patients with moderate to severe SAR. None of the 3 pivotal trials explicitly used the term “moderate to severe” in the trial eligibility criteria; rather, disease severity in the GSP301-301 and GSP301-304 trials was defined as patients with an rTNSS of 8 or greater (out of a possible 12 points) and a congestion score of 2 or more at the a.m. assessment at the screening visit, and patients with an rTNSS of 6 or greater (out of a possible 12 points) and a congestion score of 2 or more at the a.m. assessment at the screening visit in the GSP301-305 trial. According to the clinical experts consulted by the review team, these symptom score thresholds correctly reflect “moderate to severe” disease and were appropriate in the clinical trial setting to define patients with moderate to severe SAR. However, the clinical experts consulted by the review team also noted that in the clinical setting, these symptom score thresholds are typically not required to determine a patient’s disease severity. Instead, a determination of disease severity relies on a clinician’s judgment based on the extent to which patients are impacted by their symptoms. Second, the clinical experts consulted by the review team noted that, from the perspective of real-world clinical practice, the exclusion criteria of the 3 pivotal trials were restrictive. For instance, according to the clinical experts, patients with nasal structural abnormalities and patients with a history of significant rhinitis medicamentosa were excluded from the 3 pivotal trials while, in clinical practice, these patients might still be eligible for and benefit from olopatadine-mometasone. Despite these potential concerns, the experts consulted by the review team noted that the trial eligibility criteria were still reflective of the patients they would see in the real world and may be generalized to a broader population. The clinical experts also noted that the 14-day treatment duration used in the pivotal trials might not be reflective of the duration of treatment in the real-world clinical setting, where patients are often given treatment for a longer period. Furthermore, the clinical experts highlighted that adherence to treatment in all 3 pivotal trials was higher than they would expect to see in the real world, which may overestimate the treatment effect that would be observed in a real-world setting.
GRADE Summary of Findings and Certainty of the Evidence
Following the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty-of-evidence assessment for the rTNSS, iTNSS, rTOSS, RQLQ(S), and PRQLQ were set according to the presence of an important effect based on thresholds agreed upon by the clinical experts consulted by the review team for this review. For harm events, the certainty of evidence was summarized narratively.
For the GRADE assessments, the findings from the GSP301-301 and GSP301-304 trials were considered together and summarized narratively per outcome and per comparison because these studies were similar in population, interventions, design, and outcome measures. The findings from the GSP301-305 trial were assessed individually because the GSP301-305 trial had a child population (aged ≥ 6 to < 12 years), while the GSP301-301 and GSP301-304 trials had an adolescent and adult population (aged 12 years and older).
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Nasal symptoms: 12-hour rTNSS, 12-hour iTNSS
Ocular symptoms: 12-hour rTOSS
HRQoL outcomes: RQLQ(S), PRQLQ
Harms: TESAEs.
Results of GRADE Assessments
Olopatadine-Mometasone Versus Placebo
Table 2 presents the GRADE summary of findings for olopatadine-mometasone versus placebo for adolescent and adult patients (aged 12 years and older) with SAR.
Table 4 presents the GRADE summary of findings for olopatadine-mometasone versus placebo for children (aged ≥ 6 to < 12 years) with SAR.
Olopatadine-Mometasone Versus Mometasone Monotherapy
Table 3 presents the GRADE summary of findings for olopatadine-mometasone versus mometasone NS for adolescent and adult patients (aged 12 years and older) with SAR.
Table 2: Summary of Findings for Olopatadine-Mometasone Versus Placebo for Adolescent and Adult Patients (Aged 12 Years and Older) With SAR
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Nasal symptoms | ||||
12-hour rTNSS:
Follow-up: 14 days | N = 1,163 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Higha | Olopatadine-mometasone results in a clinically important improvement in the 12-hour rTNSS over 14 days compared with placebo. |
12-hour iTNSS:
Follow-up: 14 days | N = 1,163 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Higha | Olopatadine-mometasone results in a clinically important improvement in 12-hour iTNSS over 14 days compared with placebo. |
Ocular symptoms | ||||
12-hour rTOSS:
Follow-up: 14 days | N = 1,163 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderateb | Olopatadine-mometasone likely results in an improvement in the 12-hour rTOSS over 14 days compared with placebo. |
HRQoL | ||||
RQLQ(S) overall score:
Follow-up: Day 15 | N = 1,140 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderatec | Olopatadine-mometasone likely results in little to no difference in the RQLQ(S) overall score at day 15 compared with placebo. |
Harms | ||||
TESAEs | N = 1,177 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderated | Olopatadine-mometasone likely results in little to no difference in TESAEs compared with placebo. |
CI = confidence interval; HRQoL = health-related quality of life; iTNSS = instantaneous Total Nasal Symptom Score; LS = least squares; MID = minimal important difference; mometasone NS = mometasone furoate nasal spray; NR = not reported; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RCT = randomized controlled trial; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SAR = seasonal allergic rhinitis; TESAE = treatment-emergent serious adverse event.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aCertainty of evidence was not rated down because there were no serious concerns in risk of bias, indirectness, inconsistency, and imprecision.
bRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour rTOSS in both the GSP301-301 and GSP301-304 trials crossed the MID, with point estimates favouring olopatadine-mometasone, despite that the point estimates were very close to the MID.
cRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of LS mean change from baseline in the RQLQ(S) overall score in both the GSP301-301 and GSP301-304 trials crossed the MID, with point estimates favouring olopatadine-mometasone.
dRated down 1 level for imprecision due to small number of events.
Sources: Clinical Study Reports for the GSP301-30120 and GSP301-30421 trials and sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Table 3: Summary of Findings for Olopatadine-Mometasone Versus Mometasone NS for Adolescent and Adult Patients (Aged 12 Years and Older) With SAR
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Nasal symptoms | ||||
12-hour rTNSS:
Follow-up: 14 days | N = 1,177 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderatea | Olopatadine-mometasone likely result in little to no difference in the 12-hour rTNSS over 14 days compared with mometasone NS. |
12-hour iTNSS:
Follow-up: 14 days | N = 1,177 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderateb | Olopatadine-mometasone likely results little to no difference in 12-hour iTNSS over 14 days compared with mometasone NS. |
Ocular symptoms | ||||
12-hour rTOSS:
Follow-up: 14 days | N = 1,177 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Lowc | Olopatadine-mometasone may result in little to no difference in the 12-hour rTOSS over 14 days compared with mometasone NS. |
HRQoL | ||||
RQLQ(S) overall score:
Follow-up: Day 15 | N = 1,154 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Highd | Olopatadine-mometasone results in little to no difference in the RQLQ(S) overall score at day 15 compared with mometasone NS. |
Harms | ||||
TESAEs | N = 1,177 (2 RCTs) | GSP301-301 trial:
GSP301-304 trial:
| Moderatee | Olopatadine-mometasone likely results in little to no difference in TESAEs compared with mometasone NS. |
CI = confidence interval; HRQoL = health-related quality of life; iTNSS = instantaneous Total Nasal Symptom Score; LS = least squares; MID = minimal important difference; mometasone NS = mometasone furoate nasal spray; NR = not reported; NS = nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RCT = randomized controlled trial; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; TESAE = treatment-emergent serious adverse event.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour rTNSS in the GSP301-301 and GSP301-304 trials included the MID, with point estimates favouring olopatadine-mometasone.
bRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour iTNSS in both the GSP301-301 and GSP301-304 trials included the MID, with point estimates favouring olopatadine-mometasone.
cRated down 1 level for inconsistency: The point estimate of the LS mean change from baseline in average a.m. and p.m. 12-hour rTOSS was near the no-effect line (i.e., 0) for the GSP301-301 trial and near the MID (i.e., 0.5) specified by the clinical experts consulted by the review team for the GSP301-304 trial. A fair proportion of the 95% CI crossed the no-effect line for the GSP301-301 trial, while the 95% CI excluded the no-effect line for the GSP301-304 trial. Rated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour rTOSS in the GSP301-301 and GSP301-304 trials included the MID, with point estimates favouring olopatadine-mometasone.
dCertainty of evidence was not rated down as there were no serious concerns in risk of bias, indirectness, inconsistency, and imprecision.
eRated down 1 level for imprecision due to small number of events.
Sources: Clinical Study Reports for the GSP301-30120 and GSP301-30421 trials and sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Table 4: Summary of Findings for Olopatadine-Mometasone Versus Placebo for Children (Aged From 6 Years to Younger Than 12 Years) With SAR
Outcome and follow-up | Patients, N (studies) | Effect (GSP301-305 trial) | Certainty | What happens |
|---|---|---|---|---|
Nasal symptoms | ||||
12-hour rTNSS:
Follow-up: 14 days | N = 441 (1 RCT) |
| Moderatea | Olopatadine-mometasone likely results in an improvement in the 12-hour rTNSS over 14 days compared with placebo. |
12-hour iTNSS:
Follow-up: 14 days | N = 441 (1 RCT) |
| Moderateb | Olopatadine-mometasone likely results in an improvement in 12-hour iTNSS over 14 days compared with placebo. |
Ocular symptoms | ||||
12-hour rTOSS:
Follow-up: 14 days | N = 441 (1 RCT) |
| Moderatec | Olopatadine-mometasone likely result in little to no difference in the 12-hour rTOSS over 14 days compared with placebo. |
HRQoL | ||||
PRQLQ overall score:
Follow-up: Day 15 | N = 441 (1 RCT) |
| Moderated | Olopatadine-mometasone likely results in little to no difference in the PRQLQ overall score at day 15 compared with placebo. |
Harms | ||||
TESAEs | N = 446 (1 RCT) |
| Moderatee | Olopatadine-mometasone likely results in little or no difference in TESAEs compared with placebo. |
CI = confidence interval; HRQoL = health-related quality of life; iTNSS = instantaneous Total Nasal Symptom Score; LS = least squares; MID = minimal important difference; NR = not reported; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RCT = randomized controlled trial; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SAR = seasonal allergic rhinitis; TESAE = treatment-emergent serious adverse event.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour rTNSS in the GSP301-305 trial included the MID, with the point estimate favouring olopatadine-mometasone and excluding MID.
bRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour iTNSS in the GSP301-305 trial included the MID, with the point estimate favouring olopatadine-mometasone and excluding MID.
cRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in average a.m. and p.m. 12-hour rTOSS in the GSP301-305 trial included the MID, with the point estimate favouring olopatadine-mometasone.
dRated down 1 level for imprecision: According to the clinical experts consulted by the review team, a between-group difference of > 0.5 points was considered clinically important (i.e., MID). The upper bound of the 95% CI of the LS mean change from baseline in the PRQLQ overall score in the GSP301-305 trial included the MID, with the point estimate favouring olopatadine-mometasone.
eRated down 1 level for imprecision due to small number of events.
Sources: Clinical Study Report for the GSP301-30523 trial and sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Long-Term Extension Studies
A long-term extension study that evaluated the long-term (52 weeks) safety, tolerability, and efficacy of olopatadine-mometasone in adults and adolescents (aged 12 years and older) with PAR was submitted by the sponsor. However, given that the Health Canada–approved indication is for the treatment of SAR, not PAR, the long-term study submitted by the sponsor was not considered relevant to this review and was therefore not appraised.
Indirect Comparisons
Description of Studies
The indirect treatment comparisons (ITCs) submitted by the sponsor included 2 network meta-analyses (NMAs). One NMA evaluated the efficacy of olopatadine-mometasone compared with placebo, intranasal corticosteroids, and oral antihistamines in adolescent and adult patients (aged 12 years and older) with SAR. The other NMA assessed the efficacy of olopatadine-mometasone relative to placebo and intranasal corticosteroids in children (aged ≥ 6 to < 12 years) with SAR. The NMA for adolescent and adult patients was based on 13 RCTs identified from a sponsor-conducted SLR, while the NMA for children was based on 4 RCTs. Efficacy was measured using 12-hour rTNSS in both NMAs.
Efficacy Results
The NMA in Adolescent and Adult Patients Aged 12 Years and Older
In the base-case analysis, the mean or LS mean difference in the 12-hour rTNSS was −1.26 points (95% credible interval [CrI], −1.86 to −0.67) between the olopatadine-mometasone and placebo arms, −0.27 points (95% CrI, −0.87 to 0.33) between the olopatadine-mometasone and intranasal corticosteroids arms, and −0.91 points (95% CrI, −1.91 to 0.06) between the olopatadine-mometasone and oral antihistamines arms. Results from the sensitivity analyses were generally consistent with the results in the base-case analysis.
The NMA in Adolescent and Child Patients Aged From 6 Years to Younger Than 12 Years
In the base-case analysis, the mean or LS mean difference in the 12-hour rTNSS was −1.21 points (95% CrI, −1.86 to −0.56) between the olopatadine-mometasone and placebo arms and −0.94 points (95% CrI, −1.63 to −0.26) between the olopatadine-mometasone and intranasal corticosteroids arms. No sensitivity analyses were conducted.
Harms Results
Harms data were not examined in either NMA submitted by the sponsor.
Critical Appraisal
The 2 NMAs submitted by the sponsor defined the review questions (i.e., population, intervention, comparator, outcomes, and study design) a priori. With respect to comparators in the SLR protocol, the sponsor listed several active comparators under 2 drug classes: intranasal corticosteroids and oral antihistamines. The clinical experts consulted by the review team noted that some relevant comparators that were approved by Health Canada for the treatment of the symptoms of SAR were missing from the 2 classes in the protocol, including fluticasone furoate, bilastine, and rupatadine fumarate. No rationale was provided for why these comparators were not included. Consequently, these missing relevant comparators from the SLR protocol might have resulted in missing evidence in the subsequent NMAs, although the impact of this potential bias remains unknown. In addition, there is a possibility that missing comparators may jeopardize the generalizability of the NMA results to these missing comparator therapies.
To form a network, the individual treatments identified from the included studies were categorized into corresponding nodes: olopatadine-mometasone, intranasal corticosteroids, oral antihistamines, and placebo. The sponsor assumed that individual drugs in the same drug class were equivalent in terms of clinical efficacy (intraclass clinical equivalency), which was considered reasonable by the clinical experts consulted for this review. However, it was noted that within some nodes, there were only 1 or 2 individual drugs included due to a lack of eligible studies, which was beyond the sponsor’s control. For instance, only loratadine was available and included in the oral antihistamine node in the adolescents and adults NMA. In the children NMA, the intranasal corticosteroid node consisted only of mometasone and ciclesonide. The review team determined that there was a concern and associated uncertainty regarding whether only 1 or 2 individual therapies would properly represent the corresponding drug class in terms of efficacy. Thus, the interpretation of the efficacy of olopatadine-mometasone relative to the intranasal corticosteroid class and to the oral antihistamine class should be made with caution.
The clinical experts consulted by the review team generally agreed with the sponsor’s evaluation and identified no serious heterogeneity arising from the patient and disease characteristics examined in the NMAs (i.e., age, sex, disease duration, baseline symptom scores, comorbidity). However, the clinical experts consulted by the review team also noted that some patient or disease characteristics that might be potential sources of heterogeneity were missing from the sponsor-conducted NMAs, including urban versus rural living conditions, genetic predisposition, family history of atopic diseases, and smoking or vaping status. Thus, some uncertainty concerning the results of the NMA is warranted due to these potential sources of heterogeneity; however, the inclusion of these variables was beyond the sponsor’s control, given the limited data available in the included studies.
Studies Addressing Gaps in the Evidence from the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were submitted by the sponsor.
Conclusions
The sponsor submitted 3 phase III, double-blind, RCTs: GSP301-301, GSP301-304, and GSP301-305. Both the GSP301-301 (N = 1,176) and GSP301-304 (N = 1,180) trials compared the efficacy of olopatadine-mometasone with placebo and the individual constituent monotherapies (i.e., olopatadine hydrochloride NS and mometasone NS) in adolescent and adult patients (aged 12 years and older) with SAR, while the GSP301-305 trial (N = 446) assessed the efficacy of olopatadine-mometasone relative to placebo in children (aged ≥ 6 to < 12 years) with SAR. Compared with placebo, the evidence from the 3 pivotal trials showed added clinical benefits of olopatadine-mometasone in reducing nasal and ocular symptoms. Results from the GSP301-301 and GSP301-304 trials suggested with moderate to high certainty that olopatadine-mometasone, compared with placebo, results in an improvement in the 12-hour rTNSS, 12-hour iTNSS, and 12-hour rTOSS in adolescent and adult patients with SAR. Evidence from the GSP301-305 trial also suggested improvements in rTNSS and iTNSS with olopatadine-mometasone compared with placebo with moderate certainty, but not for ocular symptoms. Compared with mometasone NS in adolescent and adult patients, although the results for nasal and ocular symptoms favoured olopatadine-mometasone, they were not clinically meaningful. Olopatadine-mometasone was considered safe in adults, adolescents, and children with SAR because the harms observed in patients treated with olopatadine-mometasone were expected and manageable, given the known safety profiles of the individual excipients.
The indirect evidence submitted by the sponsor included 1 NMA in adolescent and adult patients with SAR and 1 NMA in children with SAR. The results of the NMAs were consistent with the pivotal trial, favouring olopatadine-mometasone over placebo in both analysis populations. In the NMA in children, olopatadine-mometasone was favoured over intranasal corticosteroids, but there was no difference between these treatments in the adolescents and adults NMA. There was also no difference between olopatadine-mometasone and oral antihistamines in the adolescents and adults NMA. Overall, the findings from the 2 NMAs were uncertain due to limitations surrounding the comparators considered and whether there was appropriate representation in the comparator drug classes in terms of efficacy.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of olopatadine-mometasone (Ryaltris), 25 mcg mometasone NS, and placebo for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
AR is an IgE-mediated inflammation of the nasal mucosa triggered by exposure to allergens.3,4 AR has been categorized as SAR or PAR. Generally, SAR is induced by pollen whereas PAR is induced by allergens, such as those from animals and dust mites.3,4,11 It is estimated to affect approximately 500 million people globally, irrespective of age or ethnic group.5 Studies suggest that the prevalence of AR varies by country, but generally affects more than 25% of children and 40% of adults around the world.6 It is also widely acknowledged that the prevalence of SAR is increasing globally, as it has been shown to have risen significantly in the past 2 decades.9,10,13 This trend has been attributed to several factors, such as global urbanization, changing global climate conditions, improvements in hygiene, changes in diet, and increased obesity.9,10
Patients often describe 1 or more of the following classic symptoms of AR: nasal congestion (stuffiness), nasal itching, rhinorrhea, sneezing, and cough.4,10 AR is often accompanied by allergic conjunctivitis and is collectively known as allergic rhinoconjunctivitis,11 which also includes ocular symptoms, such as itchiness, redness, and irritation of the eye.11 The incidence rate of AR in children over the first 5 years of life was reported to be 17.2%, with a peak age at diagnosis of between 24 and 29 months (2.5%).24 Meta-analysis studies demonstrate that there are sex-specific differences in the prevalence of AR, namely a male predominance in childhood but a female predominance in adolescents.25,26 Approximately 80% of SAR symptoms develop before the age of 20 years and peak at age 20 to 40 years before progressively declining.12 AR impairs patients’ quality of life, social life, and attendance and productivity at school and work, and is associated with substantial economic costs.7
In Canada, AR is highly prevalent, adversely affecting up to 20% to 25% of the population.7 SAR accounts for approximately 76.7% of AR cases. Based on these data, estimates of the prevalence of SAR in Canada range from 12.9% to 19.2%.8,9 Considering this, it is estimated that approximately 3.5 million people in Canada may be affected by moderate to severe SAR.27 However, this number may be larger because it has been suggested that there is substantial underdiagnosis of AR in Canada.7
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
According to the clinical experts consulted for this review, the management of moderate to severe SAR involves a comprehensive approach, with the goals of alleviating symptoms, improving quality of life, and minimizing symptom exacerbations. The clinical experts consulted by the review team noted that the goals of treatment are generally consistent across age groups (i.e., adults, adolescents, and children aged 6 years and older), but the approach to treatment and consideration of medication choices may vary across these age groups. According to the clinical experts consulted by the review team, the choice of medications in children and adolescents should consider factors such as age-appropriate formulations (e.g., nasal sprays versus pills versus syrup), appropriate dosing, safety profiles, potential impact on growth and development, and caregiver involvement in adherence to treatment plans. For instance, the clinical experts consulted by the review team noted that monitoring for side effects, especially growth suppression with long-term intranasal corticosteroids use, is important in younger patients. Consideration of how AR affects sleep, school attendance, social activities, and overall well-being is also important.
Several options for pharmaceutical management are available for moderate to severe SAR, including intranasal corticosteroids, oral antihistamines, leukotriene receptor antagonists, and allergen immunotherapy or desensitization.15 Intranasal corticosteroids can be used alone or combined with oral or intranasal antihistamines.13-15 According to the clinical experts consulted by the review team, intranasal corticosteroids alone or combined with intranasal antihistamines are considered as first-line treatment options for moderate to severe SAR and are generally preferred over oral antihistamines alone. The clinical experts consulted by the review team noted that fluticasone propionate, mometasone furoate, budesonide, ciclesonide, beclomethasone dipropionate, and fluticasone furoate are widely used or prescribed to reduce nasal inflammation and associated symptoms.
Oral antihistamines are also used to manage itching, sneezing, and ocular symptoms. According to the clinical experts consulted by the review team, oral antihistamines would be considered as adjunctive therapy due to being less effective than intranasal corticosteroids for nasal congestion. First-generation antihistamines (e.g., diphenhydramine and hydroxyzine) are no longer suggested due to various adverse side effects impacting the central nervous system, anticholinergic side effects, and cardiac toxicity.15 According to the clinical experts consulted by the review team, second-generation nonsedating antihistamines, such as cetirizine, loratadine, desloratadine, fexofenadine, bilastine, and rupatadine, are used for the treatment of SAR.
Leukotriene receptor antagonists, such as montelukast, are also approved in Canada for the treatment of AR.16 The clinical experts consulted by the review team noted that leukotriene receptor antagonists are generally considered for patients with AR who have concomitant asthma or those whose condition does not respond adequately to other therapies. However, the clinical experts consulted for this review noted that leukotriene receptor antagonists have become less preferred in clinical practice due to their side effect profile, including sleep and mood disturbances.
According to the clinical experts consulted by the review team, other pharmaceutical therapies that may be used in patients with AR include ocular antihistamines, mast cell stabilizers, and allergen immunotherapy or desensitization. The clinical experts consulted by the review team noted that ocular antihistamines and mast cell stabilizers are used for managing allergic conjunctivitis symptoms as an adjunct therapy, and olopatadine and ketotifen have been approved by Health Canada for allergic conjunctivitis and are commonly used in clinical practice. According to the clinical experts, allergen immunotherapy or desensitization may be used to treat patients with AR when specific criteria are met, such as when the aforementioned medications are not enough to alleviate symptoms, the patient wishes to decrease medication use, or the patient is intolerant of the aforementioned medications.
According to the clinical experts consulted by the review team, nonpharmacological management includes educating patients about allergen avoidance measures and environmental control measures, as well as the use of saline nasal irrigation to help alleviate nasal symptoms and reduce the need for pharmacological treatments.
Drug Under Review
Olopatadine-mometasone (Ryaltris) contains 2 active substances: olopatadine hydrochloride and mometasone furoate. Olopatadine acts by selectively inhibiting the histamine-1 receptor and stabilizing mast cells to attenuate the inflammatory and allergic response experienced by patients with SAR.16 As an anti-inflammatory drug, when applied intranasally, olopatadine reduces itchy and/or runny nose and sneezing.28 Mometasone furoate, like other corticosteroids, possesses anti-inflammatory, vasoconstrictive, and antipruritic properties.17 Mometasone furoate crosses cellular membranes to repress inflammatory gene transcription either directly or by activating the transcription of anti-inflammatory factors. In vitro, mometasone furoate blocks the synthesis and release of interleukin-1, interleukin-6, and tumour necrosis factor alpha from CD4+ T cells,29 and inhibits the production of T helper 2 cytokines, interleukin-4, and interleukin-5, from CD4+ T cells.30 By blocking these pathways, the intranasal corticosteroid mometasone reduces SAR symptoms of nasal itching and congestion, sneezing, and rhinorrhea by inhibiting the release of inflammatory mediators.30 As a monotherapy, mometasone has a well-documented safety profile with minimal systemic effects.30 Mechanistically, olopatadine may trigger fast relief from the symptoms of SAR, while mometasone furoate may act to suppress the underlying allergic inflammatory reaction.18,19
On September 21, 2022, olopatadine hydrochloride and mometasone furoate monohydrate received a Notice of Compliance from Health Canada for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older.2 The sponsor’s reimbursement request is in line with the Health Canada indication. Olopatadine-mometasone has not been previously reviewed in Canada. Olopatadine-mometasone received approval from the FDA in January 2022 for the treatment of symptoms of SAR in adults and pediatric patients aged 12 years and older.31,32
The recommended dosage is 2 sprays (665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate per delivered dose) for patients who are aged 12 years and older, and 1 spray for patients aged 6 to 11 years in each nostril twice daily.2
Key characteristics of olopatadine-monohydrate and other treatments available for SAR are summarized in Table 5.
Table 5: Key Characteristics of Olopatadine-Mometasone and Main Comparators
Drug and comparators | Mechanism of action | Indicationa | Route of administration and recommended dosage | Serious adverse effects and/or safety issues |
|---|---|---|---|---|
Drug under review | ||||
Olopatadine-mometasone (Ryaltris)2 | Olopatadine is an H1 receptor antagonist. Mometasone furoate is a glucocorticosteroid with local anti-inflammatory properties at doses that are minimally systemically active. | For the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older. | Each delivered dose (1 spray) contains 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate. Dosage:
|
|
Comparators | ||||
H1 receptor antagonist | ||||
Oral cetirizine hydrochloride33 | Cetirizine hydrochloride, an active human metabolite of hydroxyzine, is an H1 receptor antagonist antiallergic compound. It also has a carboxylic acid function. |
| Tablets (taken with or without food):
|
|
Oral desloratadine34 | Desloratadine is a nonsedating long-acting antihistamine with selective peripheral H1 receptor antagonist activity that has demonstrated antiallergic, antihistaminic, and anti-inflammatory activity. |
| One desloratadine 5 mg tablet daily, regardless of mealtime. For oral use. | Very rare cases of hypersensitivity reactions, including anaphylaxis and rash have been reported during the marketing of desloratadine. Cases of tachycardia, palpitations, psychomotor hyperactivity, seizures, elevations of liver enzymes, hepatitis, increased bilirubin, and increased appetite have been reported very rarely. |
Oral fexofenadine hydrochloride35 | Fexofenadine, the predominant human and animal active metabolite of terfenadine, is a selective H1 receptor antagonist. Both enantiomers of fexofenadine display approximately equipotent antihistaminic effects. | For the relief of symptoms associated with SAR and PAR in adults and children aged 12 years and older. | SAR:
| The most common adverse reactions reported in clinical trials were headache, nausea, drowsiness, fatigue, epistaxis, and abdominal pain. |
Oral loratadine36 | Loratadine is a long-acting tricyclic antihistamine with selective peripheral H1 receptor antagonistic activity. |
| Adults and children aged 12 years and older: One 10 mg tablet taken orally once daily. |
|
Corticosteroid for nasal use | ||||
Beclomethasone dipropionate37 | Beclomethasone dipropionate is a potent anti-inflammatory steroid with strong topical and weak systemic activity. When inhaled intranasally at therapeutic doses, it has a direct anti-inflammatory action within the nasal mucosa, the mechanism of which is not yet completely defined. | For treatment of PAR and SAR that is poorly responsive to conventional treatment. | The usual dosage of beclomethasone dipropionate for patients who have received no previous systemic steroid is 2 sprays (100 mcg beclomethasone dipropionate) into each nostril twice daily. A dosage regimen of 1 spray into each nostril 3 or 4 times daily may be preferred. | AEs associated with the nasal mucous membranes (e.g., sensations of irritation and burning in the nose following the use of beclomethasone dipropionate, and occasional sneezing attacks). |
Budesonide38 | Budesonide is a potent synthetic glucocorticosteroid with strong topical and weak systemic effects. | For the treatment of SAR and allergic and nonallergic perennial and vasomotor rhinitis unresponsive to conventional therapy in patients aged 6 years and older. |
|
|
Ciclesonide39 | Ciclesonide is a prodrug with a low glucocorticoid receptor affinity and is pharmacologically inactive. Following intranasal application, ciclesonide is enzymatically converted by esterases in the nasal mucosa to the pharmacologically active metabolite, 21 des-methylpropionyl-ciclesonide (des-ciclesonide, des-CIC, M1). | For the treatment of SAR, including hay fever, and PAR in adults and adolescents aged 12 years and older. | Adults and adolescents (aged 12 years and older): 200 mcg per day administered as 2 sprays (50 mcg per spray) in each nostril once daily. | The most common AEs reported in clinical trials were epistaxis, nasal passage irritation, and headache. |
Mometasone furoate40 | Mometasone furoate is a topical glucocorticosteroid with local anti-inflammatory properties at doses that are minimally systemically active. |
| Mometasone furoate (50 mcg), administered as 2 sprays per nostril or mometasone furoate (25 mcg), administered as 2 sprays per nostril. |
|
Fluticasone propionate41 | Fluticasone propionate is a synthetic trifluorinated corticosteroid with anti-inflammatory activity. |
| Adults (aged 18 years and older): 2 sprays (50 mcg each) in each nostril once a day (total daily dose of 200 mcg). If the patient’s symptoms are under control after 1 week of use, the dose should be lowered to 1 spray in each nostril once a day. | The most frequently reported AEs in clinical trials were nasal burning, pharyngitis, runny nose, blood in nasal mucus, epistaxis, sneezing, nasal ulcer, and headache. |
Triamcinolone acetonide42 | Triamcinolone acetonide is a potent anti-inflammatory steroid with strong topical and weak systemic activity. Triamcinolone acetonide is a more potent derivative of triamcinolone. When administered intranasally in therapeutic doses, it has a direct anti-inflammatory action on the nasal mucosa, the mechanism of which is not yet completely defined. | Children aged 4 to 12 years: For the topical treatment of the symptoms of PAR and SAR unresponsive to conventional treatment. | Children aged 4 to 12 years: Starting from 110 mcg per day given as 1 spray in each nostril once daily. Patients who do not achieve maximum symptom control may benefit from a dose of 220 mcg given as 2 sprays in each nostril once daily. Once symptoms are controlled, patients should be maintained on 110 mcg (1 spray in each nostril) once daily. |
|
Fluticasone furoate43 | Fluticasone furoate is a synthetic trifluorinated corticosteroid with potent anti-inflammatory activity. The precise mechanism through which fluticasone furoate affects rhinitis symptoms is not known. Corticosteroids have been shown to have a wide range of actions on multiple cell types (e.g., mast cells, eosinophils, neutrophils, macrophages, and lymphocytes) and mediators (e.g., histamine, eicosanoids, leukotrienes, and cytokines) involved in inflammation. These anti-inflammatory actions of corticosteroids may contribute to their efficacy in rhinitis. | For the treatment of the symptoms of SAR and PAR in patients aged 2 years and older. |
| Nose bleeds; nasal ulcers; pain, burning, irritation, soreness, or dryness in the inside of the nose; sore throat; upper respiratory tract infection; fever; bronchitis; cough; stuffy nose. |
Bilastine44 | Bilastine is an antihistamine; its principal effects are mediated via selective inhibition of peripheral H1 receptors. The antihistaminic activity of bilastine has been documented in a variety of animal and human models. It shows moderate to high affinity for H1 receptors and no affinity for muscarinic, serotonergic, dopaminergic, and noradrenergic receptors. Bilastine has been demonstrated to have limited distribution to the brain following oral administration. | For the symptomatic relief of nasal and non-nasal symptoms of SAR in patients aged 12 years and older. | Aged ≥ 12 years: 20 mg (1 tablet) once daily. Tablet should be swallowed with water on an empty stomach to achieve optimal exposure to bilastine. The bilastine tablet should be taken without food or grapefruit juice or other fruit juices, as these dietary compounds may decrease the effect of bilastine. Patients should be instructed to take the tablet and wait for 1 hour before taking food or fruit juice, or if food or fruit juice has been taken, to wait for 2 hours before taking the tablet. | Headache, sleepiness, dizziness, stomach pain. |
Rupatadine45 | Rupatadine is a second-generation antihistamine, a long-acting histamine antagonist with selective peripheral H1 receptor and platelet activating factor antagonistic activities. Some of the metabolites (desloratadine and its hydroxylated metabolites) retain antihistaminic activity and may partially contribute to the overall efficacy of the drug, maintaining activity for up to 24 hours. | For the symptomatic relief of nasal and non-nasal symptoms of SAR and PAR in patients aged 2 years and older. | Tablet (adults and adolescents 12 years of age and older): One 10 mg tablet once daily with or without food. Maximum 10 mg per day. Oral solution (children aged 2 to 11 years): Body weight of 10 to 25 kg: 2.5 mL (2.5 mg) once daily with or without food. Maximum 2.5 mL (2.5 mg) per day. Body weight > 25 kg: 5 mL (5 mg) once daily with or without food. Maximum 5 mL (5 mg) per day. | Sleepiness; headache; dizziness; dry mouth; sensation of weakness; fatigue; nausea; pain in the upper or lower abdomen (belly); abnormal blood test results (increase in an enzyme called creatine phosphokinase); symptoms of the common cold and/or flu; red eyes; vomiting and diarrhea; mucus in the back of the nose, throat, or sinuses; throat pain; tonsil pain; pain when swallowing; cough; pain during menstruation; muscle pain; and/or muscle weakness. |
AE = adverse event; AGEP = acute generalized exanthematous pustulosis; AR = allergic rhinitis; CNS = central nervous system; H1 = histamine-1; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PAR = perennial allergic rhinitis; SAR = seasonal allergic rhinitis.
aHealth Canada–approved indication.
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups. The full original patient input(s) received by the review team have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
This review received patient group submissions from Asthma Canada and Allergy Quebec. Asthma Canada is a national charity focusing on improving the quality of life and health for people with asthma and respiratory allergies. Allergy Quebec is the main reference centre in Quebec for patients with food allergies and brings together allergists, nutritionists, pharmacists, institutions, and companies in the food sector.
Asthma Canada collected patient input using its 2024 Annual Asthma Survey from a total of 1,407 patients and caregivers. To gain more in-depth knowledge of the impact of AR on quality of life, Asthma Canada also conducted 2 one-on-one interviews with patients with AR who were selected at random from the participants who completed the AR section of the survey and provided their contact information. Allergy Quebec did not collect any input data from patients.
Both patient groups noted that AR can cause uncomfortable symptoms, including runny and/or itchy nose, nasal congestion, swollen and/or itchy eyes, headaches, sinus pain and/or pressure, and tiredness that negatively impact daily activities and quality of life. In total, 82% of the survey respondents indicated that the most difficult and/or frustrating aspect of living with AR is the physical symptoms. Patients stated that finding a solution and/or treatment to eliminate or significantly lessen the symptoms of AR would be important for them, in particular, elimination of rhinorrhea, relief of other symptoms, and more effective medications that do not trigger asthma flare-ups.
The patient groups noted that treatments for AR consist of prescription and nonprescription options, including corticosteroids, anticholinergics, oral and intranasal antihistamines, immunotherapy, and over-the-counter decongestants. Based on the survey data from Asthma Canada, just 43% of the participants reported that their current treatments can, or most of the time can, control their allergic symptoms, while 57% reported that current treatments do not control their symptoms. Based on the interview results from Asthma Canada, patient concerns included the lack of efficacy or lack of sustained efficacy, the undesired side effects (e.g., drowsiness, stuffy, or dry nose), and the cost and problems with accessing some antihistamines (e.g., due to lack of insurance coverage or availability at local pharmacies). No patients with experience using olopatadine-mometasone were reported by either patient group.
Clinician Input
Input From the Clinical Experts Consulted by the Review Team
All Canada’s Drug Agency review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of SAR in adult patients or pediatric patients.
Unmet Needs
According to the clinical experts consulted by the review team, the main goals of the management of moderate to severe SAR include alleviating symptoms, improving quality of life, and minimizing symptom exacerbations. The clinical experts consulted by the review team noted several unmet needs in the treatment of SAR:
Not all patients respond adequately to currently available treatments, particularly intranasal corticosteroids and oral antihistamines, while some patients may experience only partial symptom relief.
A patient’s condition can become refractory to current treatment options over time, e.g., due to an escalation of eosinophilic inflammation that does not respond to first-line treatment with antihistamines.
There is a lack of treatment options that can modify the underlying disease mechanism of SAR. Allergen immunotherapy (e.g., sublingual immunotherapy) is currently the only treatment option that modifies the disease mechanism. Most treatments focus on symptom management rather than altering the disease progression.
While current treatments aim to alleviate symptoms like nasal congestion and pruritis, there is a need for therapies that address broader outcomes, such as reducing the frequency and severity of exacerbations, improving sleep quality, and enhancing overall quality of life. The specific outcomes of importance may vary slightly between adults and younger patients, such as school attendance and social activities in children and adolescents.
There is a need for treatment options that can offer better treatment tolerance. Some patients experience AEs with currently available treatments, such as nasal irritation or epistaxis with intranasal corticosteroids, or drowsiness with oral antihistamines. Younger patients may be more sensitive to side effects or have difficulty with medication administration, especially children, who may struggle with the proper administration of NSs or dislike the taste or smell of some oral and intranasal medications.
The clinical experts consulted by the review team noted that most intranasal corticosteroids do not provide instant relief of symptoms, which may affect patients’ adherence. There is a need for treatment options that can improve treatment adherence.
There is a need for more convenient formulations that enhance ease of use and patient acceptance, such as once-daily dosing options, user-friendly devices for NSs, or novel delivery systems.
Place in Therapy
According to the clinical experts consulted by the review team, olopatadine-mometasone can be used as a first-line or combination treatment option based on individual patient needs and treatment responses, by providing a dual-action therapy that combines an intranasal corticosteroid with an antihistamine. The clinical experts consulted by the review team noted that in clinical practice, intranasal corticosteroids alone are usually given to patients first because they can be given once daily and may be sufficient to treat symptoms. Intranasal corticosteroids with antihistamines are usually reserved for when intranasal corticosteroids alone are insufficient because the combination therapy is generally more costly, requires twice-daily administration, and may not be tolerated due to taste. However, the clinical experts consulted by the review team also noted that it is not necessary to trial monotherapy with an antihistamine or nasal corticosteroid before using olopatadine-mometasone. According to the clinical experts consulted by the review team, generally, 50% of the dose of intranasal corticosteroid is preferred for children aged between 6 and 12 years, and olopatadine-mometasone containing intranasal mometasone 25 mcg will allow clinicians to prescribe a more appropriate dose for children.
According to the clinical experts consulted by the review team, Dymista (fluticasone propionate 50 mcg plus azelastine 137 mcg NS) is already available in Canada, although it received a Do Not List recommendation for reimbursement in 2015. Dymista, like olopatadine-mometasone, requires twice-daily administration due to the duration of effect of the antihistamine.
Patient Population
According to the clinical experts consulted by the review team, the patients most suitable or most likely to respond to olopatadine-mometasone include the following:
Patients who are experiencing moderate to severe symptoms of SAR, have had an inadequate response to monotherapy with intranasal corticosteroids or antihistamines, and require both anti-inflammatory (intranasal corticosteroids) and antihistaminic or mast cell–stabilizing effects to effectively manage their symptoms.
Patients whose quality of life is significantly impacted by SAR symptoms, affecting daily activities, sleep, and overall well-being. Additionally, the clinical experts consulted by the review team noted that patients who want a quicker onset of action and struggle with treatment adherence or patients who are most troubled by eye symptoms may be suitable for olopatadine-mometasone.
According to the clinical experts consulted by the review team, the patients best suited for olopatadine-mometasone would be identified through a clinical evaluation and symptom assessment. The clinical experts consulted by the review team noted that assessment of symptom severity would occur by obtaining a patient history and conducting a physical examination. The clinical experts consulted by the review team also noted that a nasal endoscopy or rhinoscopy may help evaluate the degree of nasal inflammation or identify structural causes (e.g., septal deviation) that may be contributing to nasal inflammation, but is not essential to identify AR or stratify its severity. Furthermore, allergy testing, such as skin prick tests or specific IgE testing, can identify allergens triggering symptoms, but is not required specifically for initiation of olopatadine-mometasone. The clinical experts consulted by the review team noted some issues related to a diagnosis of SAR, particularly that underdiagnosis of AR may occur when symptoms are not fully explored or when seasonal triggers are not adequately considered, and symptoms of SAR can overlap with other conditions, such as nonallergic rhinitis or chronic sinusitis. A companion diagnostic test is not required for the use of olopatadine-mometasone.
According to the clinical experts consulted by the review team, patients least suitable for olopatadine-mometasone include those with mild symptoms of AR that are well controlled with monotherapy (either intranasal corticosteroids or antihistamines alone).
Assessing the Response Treatment
According to the clinical experts consulted by the review team, in clinical practice, determining treatment response involves assessing various outcomes that reflect improvements in symptom control and overall quality of life. The clinical experts consulted by the review team noted that the typical outcomes used to assess response include reductions in the frequency and severity of nasal and ocular symptoms, such as congestion, sneezing, itching, rhinorrhea, and eye redness or watering. The clinical experts consulted by the review team noted that the extent to which these symptoms interfere with daily activities, sleep patterns, and productivity is evaluated, with assessments conducted regularly, especially at the beginning of treatment and during peak allergy seasons, to ensure efficacy and to adjust therapy as needed. Per the clinical experts consulted for this review, the outcomes used in clinical practice are generally aligned with those used in clinical trials, which measure symptom scores, medication usage, and quality of life. However, in clinical practice, emphasis may be placed on patient-reported outcomes, such as subjective improvement in symptoms and the impact on daily living, which may not always be captured in trials.
According to the clinical experts consulted by the review team, a clinically meaningful response to treatment varies among individuals and may depend on the severity of initial symptoms and the patient’s expectations, while the threshold for a clinically meaningful response can vary among physicians based on their clinical experience, the patient population, and treatment goals. The 2 clinical experts consulted by the review team noted that at least a 50% improvement in baseline scores in terms of a reduction in symptom frequency or severity would be considered a clinically meaningful response, while a 20% improvement would be considered the threshold for clinical significance.
Discontinuing Treatment
The clinical experts consulted by the review team noted several situations when the discontinuation of olopatadine-mometasone should be considered:
Lack of effectiveness: When there is persistence in or worsening of symptoms despite adherence to treatment. If the patient’s symptoms are not adequately controlled or continue to interfere significantly with daily activities despite optimized therapy, a re-evaluation of treatment effectiveness is warranted.
AEs: When AEs (e.g., nasal irritation, epistaxis) are intolerable or persist despite management strategies. The frequency, severity, and impact of AEs on the patient’s quality of life should be assessed. If patients develop chronic rhinosinusitis with nasal polyposis, clinicians may want to increase the intranasal corticosteroid to the maximum dose (generally mometasone 100 mcg in each nostril twice daily). Although rare, patients may develop a septal perforation, which may be irritated by the intranasal corticosteroid.
Patient preference or adherence: When a patient has difficulty with administration (e.g., improper technique when using an NS) or expresses dissatisfaction with treatment despite adequate symptom control, alternative therapeutic options should be discussed.
Prescribing Considerations
According to the clinical experts consulted by the review team, olopatadine-mometasone is suitable for treatment in various clinical settings, including community settings, outpatient clinics in hospitals, and specialty allergy clinics. In the community setting, primary care physicians can diagnose and initiate treatment for patients with SAR based on presenting symptoms and allergy history, as well as monitor treatment response through regular follow-up visits and adjust therapy as needed. Similarly, the clinical experts consulted by the review team noted that hospital outpatient clinics provide an appropriate environment for managing more complex cases or patients requiring specialist consultation. Although specialists such as allergists, immunologists, or otolaryngologists may offer additional expertise in managing severe or refractory cases of SAR, their involvement is not always required for routine diagnosis and management with olopatadine-mometasone.
Clinician Group Input
No clinician group input was received by the review team for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by the review team are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Three trials (GSP301-301, GSP301-304, and GSP301-305) were submitted to evaluate the efficacy, safety, and tolerability of olopatadine-mometasone (Ryaltris). The GSP301-301 and GSP301-304 trials compared olopatadine-mometasone with placebo NS and with individual monotherapies vs. placebo NS in patients aged 12 years and older. The GSP301-305 trial compared olopatadine-mometasone with placebo NS in children aged 6 years to younger than 12 years. The studies used olopatadine NS and mometasone NS as their individual monotherapy comparators. Olopatadine nasal spray is not available in Canada. | This is a comment from the drug plans to inform CDEC deliberations. |
The only other antihistamine nasal spray marketed in Canada is the combination antihistamine plus corticosteroid product azelastine hydrochloride and fluticasone propionate (Dymista), which received a Do No List recommendation in 2015 and, according to the pCPA website, negotiations were not pursued. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
According to multiple guidelines, AR is confirmed with positive diagnostic testing (skin prick test or serum-specific IgE). Question: Is this routinely done in clinical practice? | The clinical experts highlighted that AR should be confirmed with objective evidence of sensitivity to an allergen by skin prick test and/or serum-specific IgE. However, they also noted that there are individuals with “local AR” who will not demonstrate IgE sensitivity on skin or serum testing but will react to nasal provocation testing, which is not an accessible test. In addition, the clinical experts noted that due to allergist supply–demand mismatch, particularly in rural areas, not everyone has access to different testing methods. The clinical experts noted that in clinical practice, it is common that a history of symptoms (e.g., stuffy nose) would be the only consideration and patients would get prescribed drugs without confirming AR. Per the clinical experts, there are particular features in clinical history (e.g., pruritus of the nose or eyes, seasonality of symptoms, relief of symptoms with antihistamines, family history of atopy), any of which can help establish a diagnosis of AR without objective testing. |
The indication under review is specifically for SAR. Traditionally, AR has been categorized as seasonal (occurs during a specific season) or perennial (occurs throughout the year). However, not all patients fit into this classification scheme. For example, some allergic triggers, such as pollen, may be seasonal in cooler climates but perennial in warmer climates, and patients with multiple “seasonal” allergies may have symptoms throughout most of the year. Therefore, AR is now classified according to symptom duration (intermittent or persistent) and severity (mild, moderate, or severe). Question: Will the indication specific to SAR be an issue for requests that are for “AR” or “PAR”? Can this drug also be used for patients with PAR or other types of allergic rhinitis? | The clinical experts noted that the pivotal trials included only patients with SAR; however, they considered the results potentially generalizable to other types of AR, including PAR. As such, the clinical experts highlighted that the indication would not likely be restricted to only SAR. |
Current guidelines state that oral second-generation antihistamines and nasal corticosteroids are first-line treatment options for moderate to severe SAR, and there are improved outcomes in patients treated with a combination of these drugs. Question: Currently, does a patient have to trial an antihistamine or nasal corticosteroid as monotherapy before using combination therapy? Incoming Canadian guidelines suggest that patients with a confirmed positive diagnostic test of AR can be prescribed a fixed combination of intranasal corticosteroids and antihistamine spray as first-line before the trial of monotherapies and/or oral antihistamines. | The clinical experts noted that olopatadine-mometasone can be given to patients as first-line treatment, and it is not necessary to trial monotherapy with an antihistamine or nasal corticosteroid before using olopatadine-mometasone. |
Considerations for prescribing of therapy | |
The recommended dose of olopatadine-mometasone is 2 sprays in each nostril b.i.d. for adults and adolescents aged 12 years and older, and 1 spray in each nostril b.i.d. for children aged 6 to 11 years. | This is a comment from the drug plans to inform CDEC deliberations. |
System and economic issues | |
At 8 actuations per day (adult dosing), the cost is $1.8704 per day, resulting in a monthly cost of $56.11. Ryaltris sales: Year 1, $779,361; year 2, $1,094,432; year 3, $1,431,101. Net impact of listing Ryaltris: Year 1, $356,728; year 2, $500,072; year 3, $653,463. | This is a comment from the drug plans to inform CDEC deliberations. |
Negotiations were not pursued for the similar combination product Dymista. | This is a comment from the drug plans to inform CDEC deliberations. |
AR = allergic rhinitis; b.i.d. = twice a day; CDEC = Canadian Drug Expert Committee; IgE = immunoglobulin E; NS = nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; pCPA = pan-Canadian Pharmaceutical Alliance; PAR = perennial allergic rhinitis; SAR = seasonal allergic rhinitis; vs. = versus.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of olopatadine-mometasone (665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate per dose, delivered by NS) for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older. The focus will be placed on comparing olopatadine-mometasone with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of olopatadine-mometasone is presented in 2 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Of note, a long-term extension study that evaluated the long-term (52 weeks) safety, tolerability, and efficacy of olopatadine-mometasone in adults and adolescents (aged 12 years and older) with PAR was submitted by the sponsor. However, given that the Health Canada–approved indication and reimbursement request are for the treatment of SAR, the long-term study submitted by the sponsor was not considered relevant to this Clinical Review Report and has therefore not been summarized and appraised.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
3 pivotal, phase III, double-blind, RCTs (GSP301-301, GSP301-304, GSP301-305)
2 sponsor-submitted NMAs.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Three pivotal studies, GSP301-301, GSP301-304, and GSP301-305, all of which were conducted by the sponsor, met the inclusion criteria for the sponsor-submitted SLR. Characteristics of the included studies are summarized in Table 7.
Both the GSP301-301 and GSP301-304 trials were phase III randomized, double-blind, parallel-group studies evaluating the efficacy and safety of olopatadine-mometasone in adult and adolescent patients (aged 12 years and older) with SAR. Both studies consisted of 2 phases, a placebo run-in period (spanning 7 to 10 days from the screening visit to the randomization visit) and a treatment period (lasting 15 to 17 days from the randomization visit to the final treatment visit). The primary objective of both studies was to compare the efficacy of olopatadine-mometasone with placebo and the individual constituent monotherapies at the same dose in the same vehicle, and to assess the efficacy of these individual constituent monotherapies versus placebo over 14 days of study treatment.
The GSP301-301 study enrolled patients across 43 sites in the US. After completion of the placebo run-in period, 1,176 patients were randomized equally into 4 groups based on a computer-generated randomization scheme: 294 patients to olopatadine-mometasone, 294 patients to olopatadine hydrochloride NS, 294 patients to mometasone NS, and 294 patients to placebo NS.
The GSP301-304 study enrolled patients across 37 study sites in the US. After completion of the placebo run-in period, 1,180 patients were randomized to 4 treatment groups, with 302 patients assigned to olopatadine-mometasone, 297 patients to olopatadine hydrochloride NS, 294 patients to mometasone NS, and 287 patients to placebo NS.
GSP301-305 was a phase III double-blind, randomized, parallel-group, placebo-controlled study evaluating the use of olopatadine-mometasone in pediatric patients (aged ≥ 6 to < 12 years) with SAR. The GSP301-305 study comprised a single-blind placebo run-in period lasting 7 to 10 days from the screening visit to the randomization visit followed by a 14-day double-blind treatment period, with the final or discontinuation visit scheduled for day 15. Upon completion of the placebo run-in period, 446 patients were randomized to olopatadine-mometasone (N = 225) or placebo (N = 221). The primary objective of the GSP301-305 trial was to assess the efficacy of olopatadine-mometasone relative to placebo over 14 days of study treatment.
Table 7: Details of Studies Included in the Systematic Review
Detail | GSP301-301 trial (NCT02631551) | GSP301-304 trial (NCT02870205) | GSP301-305 trial (NCT03463031) |
|---|---|---|---|
Designs and populations | |||
Study design | A phase III, double-blind, randomized, parallel-group study. | A phase III randomized, double-blind, parallel-group study. | A phase III, double-blind, randomized, parallel-group study. |
Locations | 37 sites in the US | 43 sites in the US | 32 sites in the US |
Patient enrolment dates | Start date: March 2016 End date: NR | Start date: August 2016 End date: NR | Start date: March 16, 2018 End date: NR |
Randomized (N) | N = 1,180:
| N = 1,176:
| N = 446:
|
Inclusion criteria | Males and nonpregnant females aged ≥ 12 years. Signed informed consent or assent form (signed by the patient and/or parent, caregiver, or legal guardian) that met all criteria of the current FDA or local regulations. Documented clinical history of SAR for at least 2 years preceding the screening visit (visit 1) with exacerbations (clinical evidence of active symptoms) for the relevant seasonal allergen during the spring allergy season (tree or grass pollen) and exhibiting a documented skin prick test result positive for spring allergens (i.e., wheal diameter at least 5 mm greater than the negative diluent control wheal). Documentation of a positive result within 12 months before the screening visit (visit 1) was acceptable. A 12-hour rTNSS ≥ 8 out of a possible 12 and a congestion score ≥ 2 for the a.m. assessment at the screening visit (visit 1). General good health and free of any disease or concomitant treatment that could interfere with the interpretation of study results, as determined by the investigator. Patients must have been able to demonstrate the correct NS application technique at the screening visit (visit 1). Patients must have been willing and able to comply with all aspects of the protocol. | Males and nonpregnant females aged ≥ 12 years. Signed informed consent or assent form (signed by the patient and/or parent, caregiver, or legal guardian) that met all criteria of the current FDA or local regulations. Documented clinical history of SAR for at least 2 years preceding the screening visit (visit 1) with exacerbations (clinical evidence of active symptoms) for the relevant seasonal allergen during the fall or mountain cedar allergy seasons (e.g., ragweed or mountain cedar pollen). A skin prick test positive for the allergen (i.e., wheal diameter at least 5 mm greater than negative diluent control wheal) relevant to the fall or mountain cedar season must have been exhibited and documented. Documentation of a positive result within 12 months before the screening visit (visit 1) was acceptable. A 12-hour rTNSS ≥ 8 out of a possible 12 and a congestion score ≥ 2 for the a.m. assessment at the screening visit (visit 1). General good health and free of any disease or concomitant treatment that could interfere with the interpretation of study results, as determined by the investigator. Patients must have been able to demonstrate the correct NS application technique at the screening visit (visit 1). Patients must have been willing and able to comply with all aspects of the protocol. | Male or nonpregnant female patients aged ≥ 6 to < 12 years at the screening visit (visit 1). Signed informed consent or assent form (signed by the patient and/or parent, caregiver, or legal guardian) that met all criteria of the current FDA or local regulations. Documented clinical history of SAR for at least 2 years preceding the screening visit (visit 1) with exacerbations (clinical evidence of active symptoms) during the spring or fall allergy seasons for the relevant seasonal allergen (e.g., tree or grass pollen, ragweed pollen). The SAR must have been of sufficient severity to have required treatment (either continuous or intermittent) in the past and, in the investigator’s judgment, was expected to require treatment throughout the study period. Demonstrated sensitivity to at least 1 seasonal allergen (e.g., tree or grass pollen, ragweed pollen) known to induce SAR through a documented positive skin prick test (wheal diameter at least 5 mm greater than the negative control) for a relevant seasonal allergen. Documentation of a positive result within 12 months before the screening visit (visit 1) was acceptable. The patient’s positive allergen must have been consistent with their medical history of SAR. Additionally, the patient was expected to be adequately exposed for the entire duration of the study to the SAR allergen that had generated a positive test result. A 12-hour rTNSS ≥ 6 (out of a possible 12) for the a.m. assessment at the screening visit (visit 1). General good health and free of any disease or concomitant treatment that could interfere with the interpretation of study results, as determined by the investigator. Able to demonstrate the correct NS application technique (with the help of parents, guardians, or caregivers, if needed) at the screening visit (visit 1). Willing and able to comply with all aspects of the protocol (with the help of parents, guardians, or caregivers, if needed). |
Exclusion criteria | Had a positive pregnancy test or an established pregnancy, was breastfeeding, or planned to become pregnant during the study. Female patients of childbearing potential (as judged by the investigator) who did not agree to remain abstinent or use medically acceptable methods of contraception (e.g., implants, injectables, combined oral contraceptives, intrauterine devices, double-barrier protection) during the study. Male participants who did not agree to use a condom with spermicide during intercourse (if not surgically sterilized) during the study. Plans to travel outside the known pollen area for the investigational site for 24 hours or longer during the last 7 days of the run-in period. Plans to travel outside the known pollen area for the investigational site for 2 or more consecutive days OR 3 or more days in total between the randomization visit (visit 2) and the final visit (visit 4). History of significant atopic dermatitis or rhinitis medicamentosa within 60 days before the screening visit (visit 1). Treatment with any known potent CYP3A4 inducers (e.g., carbamazepine, dexamethasone, phenytoin, rifabutin, rifampin, pioglitazone) or potent inhibitors (e.g., azole antifungals, macrolide antibiotics) within 30 days before or during the study. Nonvaccinated exposure to or active infection with chickenpox or measles within the 21 days preceding the screening visit (visit 1). Known hypersensitivity to any corticosteroids or antihistamines or to the study drug or its excipients. History of anaphylaxis and/or other severe local reaction(s) to skin testing. History of alcohol or drug dependence within 2 years preceding the screening visit (visit 1). History of a positive test for HIV, hepatitis B, or hepatitis C infection. Evidence of acute or significant chronic sinusitis or chronic purulent postnasal drip. Any of the following conditions (including but not limited to the following) that were judged by the investigator to be clinically significant and/or to affect the patient’s ability to participate in this study:
Any major surgery (as assessed by investigator) within 4 weeks of the screening visit (visit 1). Requirement for the chronic use of tricyclic antidepressants. Dependence (in the opinion of the investigator) on nasal, oral, or ocular decongestants, nasal topical antihistamines, or nasal steroids. Active pulmonary disorder or infection (including but not limited to bronchitis, pneumonia, or influenza) or upper respiratory tract or sinus infection within the 14 days before the screening visit (visit 1) or the development of respiratory infections during the run-in period. Patients with mild asthma were allowable on the condition that treatment was limited to inhaled short-acting beta-agonists only (up to 8 puffs per day). Use of antibiotic therapy for acute conditions within 14 days before the screening visit (visit 1). Low doses of antibiotics taken for prophylaxis were allowed if the therapy was started before the screening visit (visit 1) and was expected to continue at the same stable dose throughout the clinical study duration. Had posterior subcapsular cataracts or glaucoma, or any other ocular disturbances, or other listed related conditions, including:
Known history of hypothalamic-pituitary-adrenal axis impairment. Existence of any significant surgical or medical condition or clinically significant physical finding (e.g., significant nasal polyps or other clinically significant respiratory tract malformations or nasal structural abnormalities, significant nasal trauma such as a nasal piercing or significant nasal septal deviation) that, in the opinion of the investigator or sponsor’s medical monitor, significantly interfered with the absorption, distribution, metabolism, or excretion of the study medication or significantly interfered with nasal air flow or interfered with the patient’s ability to complete or reliably complete the AR assessment diary. Participation in any investigational nonbiological drug clinical study in the 30 days, or investigational biological drug in the 120 days, preceding the screening visit (visit 1) or planned participation in another investigational clinical study at any time during the current study. Initiation of immunotherapy injections or immunosuppressive or immune-modulator medications (except topical pimecrolimus cream or tacrolimus ointment if initiated at least 30 days before screening and maintained at a stable dose) within 60 days preceding the screening visit (visit 1). A 180-day washout period was required following the last dose of sublingual immunotherapy (investigational or other) before the screening visit (visit 1). Use of topical corticosteroids in concentrations in excess of 1% hydrocortisone or equivalent within 30 days before the screening visit (visit 1), use of a topical hydrocortisone or equivalent in any concentration covering greater than 20% of the body surface, or the presence of an underlying condition (as judged by the investigator) that could reasonably be expected to require treatment with such preparations during the clinical study duration. Study participation by clinical investigational site employees and/or their immediate relatives. Study participation by more than 1 patient from the same household at the same time. However, after the completion or discontinuation by 1 patient in the household, another patient from the same household could have been screened. Known to have failed to experience symptom improvement with any approved or marketed monotherapy component of olopatadine-mometasone (i.e., NASONEX NS or PATANASE NS or both), as judged by the investigator. Previous participation in an olopatadine-mometasone NS study as a randomized patient. | Eligible females of childbearing potential who were known to be sexually active or pregnant were excluded and referred for appropriate evaluation. If a young female had reached puberty and achieved menarche (as determined by the investigator), their parents, guardians, or caregivers were consulted to obtain consent for pregnancy testing and permission to counsel the patient, followed by the counselling of the patient by the investigator regarding the possible unknown risks associated with the study medication during pregnancy. A urine pregnancy test had to be negative at the screening visit (visit 1). Male patients who were known to be sexually active were excluded and referred appropriately. Planned to travel outside the known pollen area for the investigational site for 24 hours or longer during the last 7 days of the screening or run-in period. Planned to travel outside the known pollen area for the investigational site for 2 or more consecutive days OR 3 or more days in total between the randomization visit (visit 2) and the final treatment visit (visit 4). History of significant (based on investigator’s judgment) atopic dermatitis or rhinitis medicamentosa within 60 days before the screening visit (visit 1). Treatment with any known strong CYP3A4 inducers (e.g., carbamazepine, phenytoin, rifabutin, rifampin, pioglitazone) or strong inhibitors (e.g., azole antifungals, macrolide antibiotics) within 30 days before the screening visit (visit 1) or during the study. Nonvaccinated exposure to or active infection with chickenpox or measles within the 21 days preceding screening visit (visit 1). A known hypersensitivity to any corticosteroids or antihistamines or to either of the drug components of the investigational product or its excipients. History of anaphylaxis and/or other severe local reaction(s) to skin testing. Any history or current use of alcohol or drug dependence at the screening visit (visit 1), as determined by the investigator. History of positive test for HIV, hepatitis B, or hepatitis C infection (parents, guardians, or caregivers were consulted to obtain consent). History and evidence of acute or significant chronic sinusitis or chronic purulent postnasal drip at the screening visit (visit 1). Any of the following conditions (including but not limited to the following) that were judged by the investigator to be clinically significant and/or to affect the patient’s ability to participate in this study:
Any major surgery (as assessed by the investigator) within 4 weeks before the screening visit (visit 1). A requirement for the chronic use of tricyclic antidepressants. Dependence (in the opinion of the investigator) on nasal, oral, or ocular decongestants; nasal topical antihistamines; or nasal steroids. Active pulmonary disorder or infection (including but not limited to bronchitis, pneumonia, or influenza), upper respiratory tract or sinus infection within the 14 days before the screening visit (visit 1), or the development of respiratory infections during the placebo run-in period. Patients with mild asthma (as judged by the investigator) were allowed on the condition that treatment was limited to inhaled short-acting beta-agonists only (up to 8 puffs per day). Use of antibiotic therapy for acute conditions within 14 days before screening visit (visit 1). Low doses of antibiotics taken for prophylaxis were allowed if the therapy was started before the screening visit (visit 1) and was expected to continue at the same stable dose throughout the clinical study duration. Posterior subcapsular cataracts or glaucoma, or any other ocular disturbances or other listed related conditions (as applicable), including:
Known history of hypothalamic-pituitary-adrenal axis impairment. Existence of any significant surgical or medical condition, or clinically significant physical finding (e.g., significant nasal polyps or other clinically significant respiratory tract malformations or nasal structural abnormalities, significant nasal trauma [such as nasal piercing] or significant nasal septal deviation) which, in the opinion of the investigator (or in consultation with the sponsor’s medical monitor or designee), significantly interfered with the absorption, distribution, metabolism, or excretion of the study medication or significantly interfered with nasal air flow or interfered with the patient’s ability to reliably complete the AR assessment diary. Participation in any investigational nonbiological drug clinical study in the 30 days, or investigational biological drug in the 120 days, preceding the screening visit (visit 1) or planned participation in another investigational clinical study at any time during the current study. Initiation of immunotherapy injections or immunosuppressive or immune-modulator medications within 60 days preceding the screening visit (visit 1) and/or currently undergoing treatment with immunotherapy or immunosuppressive or immune-modulator medications. Topical pimecrolimus cream or tacrolimus ointment treatment if initiated at least 30 days before screening and maintained on a stable dose was acceptable. A 180-day washout period was required following the last dose of sublingual immunotherapy (investigational or other) before the screening visit (visit 1). Use of topical corticosteroids in concentrations in excess of 1% hydrocortisone, or equivalent, within 30 days before the screening visit (visit 1), use of a topical hydrocortisone or equivalent in any concentration covering greater than 20% of the body surface, or the presence of an underlying condition (as judged by the investigator) that was reasonably expected to require treatment with such preparations over the clinical study duration. Previous participation in another GSP-301 NS study as a randomized patient. Clinical study participation by employees of the clinical investigator site and/or their immediate relatives. Study participation by more than 1 patient from the same household at the same time. However, after the completion or discontinuation by 1 patient in the household, another patient from the same household could have been screened. Known to have not experienced symptom improvement with any approved or marketed monotherapy component of olopatadine-mometasone (i.e., NASONEX NS, PATANASE NS, or both), as judged by the investigator. | |
Drugs | |||
Intervention | 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate intranasal treatments: 2 sprays per nostril twice daily for 14 days. | 665 mcg olopatadine hydrochloride and 25 mcg of mometasone furoate intranasal treatments: 2 sprays per nostril twice daily for 14 days. | 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate: 1 spray in each nostril twice daily. |
Comparators |
|
| Placebo (GSP301 vehicle): 1 spray per nostril twice daily. |
Study duration | |||
Screening phase | The screening visit (visit 1) occurred between days −10 and −7 before the randomization visit on day 1 (visit 2). | The screening visit (visit 1) occurred between days −10 and −7 before the randomization visit on day 1 (visit 2). | The screening visit (visit 1) was performed between days −10 and −7 before the randomization visit on day 1 (visit 2). |
Run-in phase | The placebo run-in period (7 to 10 days from the screening visit to the randomization visit). | The placebo-run-in period (7 to 10 days from the screening visit to the randomization visit). | Single-blind placebo run-in period (7 to 10 days from the screening visit to the randomization visit). |
Treatment phase | The treatment period (15 to 17 days from the randomization visit to the final treatment visit). | The treatment period (15 to 17 days from the randomization visit to the final treatment visit). | The treatment visit (visit 3) was performed on day 8 (plus 2 days). |
Follow-up phase | The final or discontinuation visit (visit 4) occurred on approximately day 15. No scheduled post-treatment follow-up visit was planned for this study. | The final or discontinuation visit (visit 4) occurred on approximately day 15. No scheduled post-treatment follow-up visit was planned for this study. | The final or discontinuation visit (visit 4) was performed on day 15 (plus 2 days). No scheduled post-treatment follow-up visit was planned for this study. |
Outcomes | |||
Primary end point | The mean change from baseline to the end of the 14-day treatment period in average a.m. and p.m. 12-hour rTNSS. | The mean change from baseline to the end of the 14-day treatment period in average a.m. and p.m. 12-hour rTNSS. | Change from baseline in average a.m. and p.m. patient-reported 12-hour rTNSS over the 14-day treatment period. |
Secondary and exploratory end points | Secondary
Tertiary
| Secondary
Tertiary
| Secondary
Tertiary
|
Publication status | |||
Publications | Hampel (2019),46 NCT02631551 | Gross (2019),47 NCT02870205 | Prenner (2022),48 NCT03463031 |
ADHD = attention-deficit/hyperactivity disorder; AR = allergic rhinitis; CYP3A4 = cytochrome P450 3A4; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total Ocular Symptom Score; mometasone NS = mometasone furoate nasal spray; NR = not reported; NS = nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PNSS = Physician-Assessed Nasal Symptom Score; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RCAT = Rhinitis Control Assessment Test; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SAR = seasonal allergic rhinitis.
aOlopatadine hydrochloride NS is not available in Canada.
Sources: Sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22 Details included in the table are from the sponsor’s summary of clinical evidence.1
Populations
Inclusion and Exclusion Criteria
The GSP301-301 trial enrolled male and female patients aged 12 years and older with a documented clinical history of SAR (for at least 2 years preceding the screening visit) with exacerbations (clinical evidence of active symptoms) during the spring allergy season (tree or grass pollen) or with a positive skin prick test result for relevant allergens (wheal diameter at least 5 mm greater than the negative control) during the fall or mountain cedar allergy seasons (e.g., ragweed pollen, mountain cedar pollen). At the screening visit, patients were required to demonstrate a minimum average morning (a.m.) and evening (p.m.) 12-hour rTNSS of 8 or higher out of 12, along with an a.m. congestion score of 2 or higher.
The GSP301-305 trial enrolled patients who were aged between 6 and 12 years with a clinical history of SAR for at least 2 years before screening, with exacerbations during the spring or fall allergy seasons for the relevant seasonal allergen (e.g., tree or grass pollen or ragweed pollen) and a positive skin prick test result (wheal diameter at least 5 mm greater than the negative control) consistent with their medical history of SAR. Patients were required to have a 12-hour rTNSS of 6 points or greater for the a.m. assessment at the screening visit.
Interventions
For both the GSP301-301 and GSP301-304 trials, the intervention group received intranasal olopatadine-mometasone (665 mcg of olopatadine hydrochloride and 25 mcg of mometasone furoate) for 14 days (2 sprays per nostril twice daily). For comparison,1 group was administered intranasal olopatadine hydrochloride (at a dose of 665 mcg), another group received mometasone NS (at a dose of 25 mcg), and a third group received placebo (vehicle comprising inactive ingredients identical to those used in the active treatments) for a period of 14 days (2 sprays per nostril twice daily). Of note, olopatadine hydrochloride NS is not relevant to this Reimbursement Review because it is currently unavailable in Canada; therefore, results for olopatadine hydrochloride are not presented in this Clinical Review Report.
For the GSP301-305 trial, the intervention group was administered intranasal olopatadine-mometasone (consisting of 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate), with 1 spray administered in each nostril twice daily for a duration of 14 days. Placebo (vehicle) was used as the comparator, with 1 spray delivered in each nostril twice daily for the same 14-day period.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 8, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by the review team and the input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 8: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | GSP301-301 trial | GSP301-304 trial | GSP301-305 trial |
|---|---|---|---|---|
Change from baseline in average a.m. and p.m. patient-reported 12-hour rTNSS | Over a 14-day treatment period | Primary | Primary | Primary |
Change from baseline in average a.m. and p.m. patient-reported 12-hour iTNSS | Over a 14-day treatment period | Secondary | Secondary | Secondary |
Change from baseline in average a.m. and p.m. patient-reported 12-hour rTOSS | Over a 14-day treatment period | Secondary | Secondary | Secondary |
Change from baseline in the RQLQ(S) overall score | Day 15 | Secondary | Secondary | NA |
Change from baseline in the PRQLQ overall score | Day 15 | NA | NA | Secondary |
iTNSS = instantaneous Total Nasal Symptom Score; NA = not applicable; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22 Details included in the table are from the sponsor’s summary of clinical evidence.1
The following are descriptions of the efficacy and safety outcomes presented in studies GSP301-301,20 GSP301-304,21 and GSP301-30523 and appraised in this Clinical Review.
Efficacy Outcomes
Patient-Reported 12-Hour rTNSS and iTNSS
The rTNSS and iTNSS were assessed by scoring the severity of 4 nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching) over the past 12 hours before the recording of the score (i.e., reflective) and just before each dosing (i.e., instantaneous), respectively.
Scores for each symptom ranged from 0 (no signs or symptoms evident) to 3 (severe signs or symptoms that are hard to tolerate). The overall rTNSS score was calculated as the sum of the self-reported severity scores for each of the 4 symptoms and ranged from 0 (no signs or symptoms evident) to 12 (severe signs or symptoms that are hard to tolerate). The a.m. assessment was performed before bathing, consumption of food or beverages, or strenuous activities. The p.m. assessment was performed approximately 12 hours after the a.m. assessment.
Patient-Reported 12-Hour rTOSS
The rTOSS was defined as the sum of 3 eye-related symptom scores: itching or burning eyes, tearing or watering eyes, and redness of eyes. Each symptom was scored on a scale of 0 to 3 with a maximum rTOSS score of 9. A higher score indicated worse symptoms.
Health-Related Quality of Life
Patient-reported HRQoL was measured by RQLQ(S) in the GSP301-301 and GSP301-304 trials and by PRQLQ in the GSP301-305 study. The measurement properties of the instruments are shown in Table 9.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
RQLQ(S) | RQLQ or RQLQ(S) consists of 28 questions related to impairment of QoL in 7 domains: activity limitations, sleep disturbance, non-nose or eye symptoms, practical problems, nasal symptoms, eye problems, and emotional problems.49-51 Patients recall how they have been during the previous week. Overall QoL score is expressed as the mean of the 28 questions. Each question as well as the overall RQLQ score is scored on a 7-point scale from 0 to 6, where 0 represents no impairment and 6 represents maximum impairment.49,51,52 In RQLQ(S), the 3 patient-specific activity questions were replaced by generic activities.50,51 | For both RQLQ and RQLQ(S):49-51,53 Validity Validity and responsiveness were tested upon development of the test. In terms of validity, the study showed that in both treatment arms, QoL scores improved with treatment, and the Pearson correlation coefficients between QoL domains and diary symptom scores were very close to those predicted.49-51,53 Reliability A study that included 100 adults with rhinoconjunctivitis showed that reliability was high and almost identical, with an intraclass correlation coefficient of 0.97.51 Responsiveness Responsiveness was assessed and showed that the test is responsive to within-patient change in QoL during a clinical trial in ragweed pollen season and that it is able to measure change in impairment over time. The test was also able to detect differences in effect between 2 active medications.49-51,53 | For adult patients with severe ragweed pollen–induced rhinoconjunctivitis, the reported estimate of MID (for within-group change) is approximately 0.5 points for the RQLQ score on a 7-point scale (0 = no impairment, 6 = maximum impairment).52 Juniper and colleagues also indicated that an MID of 0.5 points could be applicable to adolescent patients (aged 12 to 17 years) with seasonal allergic rhinoconjunctivitis.52,53 |
PRQLQ | The PRQLQ is a validated, disease-specific, QoL questionnaire developed to measure the physical, emotional, and social impairments that are experienced by children (aged ≥ 6 to < 12 years) with rhinoconjunctivitis.54 The PRQLQ has 23 questions in 5 domains (nose symptoms, eye symptoms, practical problems, activity limitations, and other symptoms). Patients recall how they have been during the previous week and respond to each question on a 7-point scale, with scores ranging from 6 (extremely bothered), 5 (very bothered), 4 (quite bothered), 3 (somewhat bothered), 2 (bothered a bit), 1 (hardly bothered at all), to 0 (not bothered).54 The overall PRQLQ score is the mean of all 23 patient-reported responses. The overall PRQLQ score ranges from 0 to 6, where 0 represents no impairment and 6 represents maximum impairment.54 | Juniper and colleagues conducted a study that included 75 children with SAR, with the findings of validation for PRQLQ shown as follows.54 Validity Correlations between the PRQLQ and diary scores were close to predicted and supported both the cross-sectional and longitudinal validity of the PRQLQ.54 Reliability The PRQLQ showed good reliability (intraclass correlation coefficient = 0.93).54 Responsiveness PRQLQ is very responsive to change (P < 0.001). The PRQLQ is able to capture changes in overall QoL and in each domain in children whose SAR changed between weeks 1 and 3. PRQLQ is able to detect a difference in overall QoL between those children whose SAR remained stable and those whose SAR changed (P < 0.005). All of the specific domains showed P values of less than 0.09.54 | Not identified. |
MID = minimal important difference; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; QoL = quality of life; RQLQ = Rhinoconjunctivitis Quality-of-Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; SAR = seasonal allergic rhinitis.
Harms Outcomes
The harms outcomes assessed in the GSP301-301, GSP301-304, and GSP301-305 trials mainly included TEAEs, TESAEs, withdrawal due to TEAEs, and deaths.
Statistical Analysis
Details on the statistical analysis of the efficacy end points in the GSP301-301, GSP301-304, and GSP301-305 trials are presented in Table 10.
GSP301-301 and GSP301-304 Trials
To evaluate the superiority of olopatadine-mometasone in comparison with placebo, a sample size of 279 patients per treatment group was determined to provide 94% power to detect a between–treatment group difference of 0.9 units (assuming an SD of 3.0) in the absolute change from baseline in average a.m. and p.m. 12-hour rTNSS over the 14-day treatment period (assuming a 2-sided alpha of 5%). Additionally, this sample size could afford a 94% power to detect a between–treatment group difference of 0.6 units (assuming an SD of 2.0) in the absolute change from baseline in average a.m. and p.m. 12-hour rTNSS over the same treatment period when comparing olopatadine-mometasone with each monotherapy (i.e., olopatadine hydrochloride NS or mometasone NS). Finally, 279 patients per treatment group could provide 90% power to detect a treatment difference between each monotherapy and placebo of 0.55 units (assuming an SD of 2.0) in the absolute change from baseline in average a.m. and p.m. 12-hour rTNSS over the 14-day treatment period.
Efficacy end points were analyzed utilizing a mixed model for repeated measures, which was adjusted for covariates, including treatment, site, baseline clinical measures, and study day as the within-patient effect, with the assumption that any missing data were missing at random. To adjust for multiple comparisons, a gatekeeping strategy was adopted for rTNSS, iTNSS, and rTOSS. Statistical significance was set at a P value of less than 0.05 for all analyses. The gatekeeping strategy for rTNSS and rTOSS is shown in Figure 1.
Figure 1: Gatekeeping Strategy in the GSP301-301 and GSP301-304 Trials
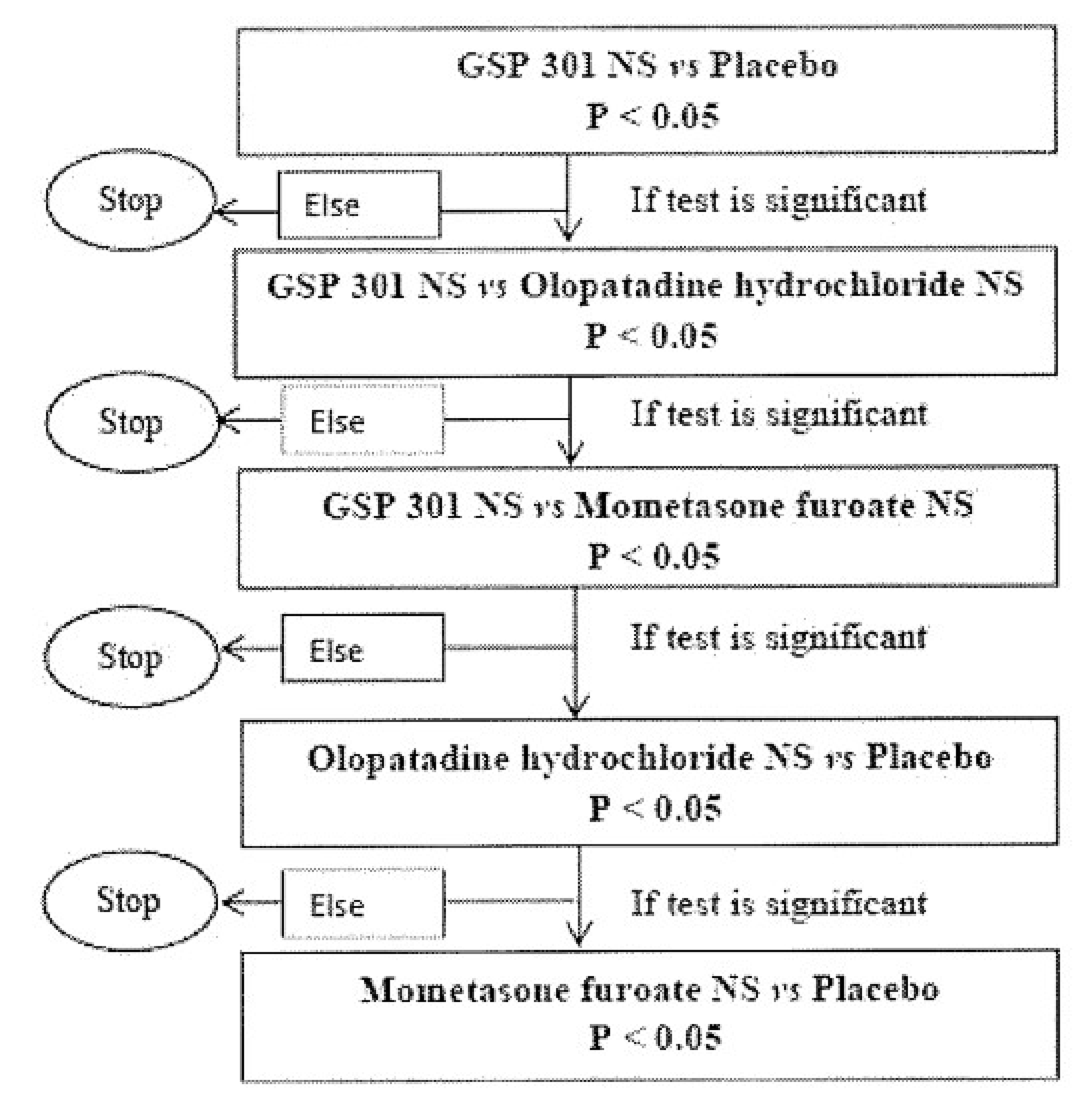
NS = nasal spray; vs = versus.
Note: GSP 301 in the figure refers to olopatadine hydrochloride and mometasone furoate nasal spray.
Source: Sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
GSP301-305 Trial
To assess the superiority of olopatadine-mometasone over placebo, a sample size of 382 patients was determined to provide 90% power to detect a between-group mean difference of 1.0 unit (assuming an SD of 3.0 and a 2-sided alpha of 5%) in the absolute change from baseline in average a.m. and p.m. 12-hour rTNSS over the 14-day treatment period.
Efficacy end points were analyzed utilizing a mixed model for repeated measures that was adjusted for covariates, including treatment, site, baseline clinical measures, and study day as the within-patient effect, with the assumption that any missing data were missing at random. No multiplicity adjustment was made.
Table 10: Statistical Analysis of Efficacy End Points for Included Studies
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity and subgroup analyses |
|---|---|---|---|---|
GSP301-301 trial | ||||
12-hour rTNSS | 12-hour rTNSS was analyzed using an MMRM. | Treatment, site, baseline 12-hour rTNSS, and study day as the within-patient effect. | Missing data were assumed as MAR under MMRM. | Sensitivity analyses:
Subgroup analyses:
|
12-hour iTNSS | 12-hour iTNSS was analyzed using an MMRM. | Treatment, site, baseline 12-hour rTNSS, and study day as the within-patient effect. | Missing data were assumed as MAR under MMRM. | Sensitivity analysis:
Subgroup analyses:
|
12-hour rTOSS | 12-hour rTOSS was analyzed using an MMRM. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. | Missing data were assumed as MAR under MMRM. | Sensitivity analysis:
Subgroup analyses:
|
RQLQ | RQLQ was analyzed using an ANCOVA model. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. | NR | Sensitivity analysis:
Subgroup analysis:
|
GSP301-304 trial | ||||
12-hour rTNSS | 12-hour rTNSS was analyzed using an MMRM. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. |
| Sensitivity analyses:
Subgroup analyses:
|
12-hour iTNSS | 12-hour iTNSS was analyzed using an MMRM. | Treatment, site, baseline 12-hour rTNSS, and study day as the within-patient effect. |
| Sensitivity analysis:
Subgroup analyses:
|
12- hour rTOSS | 12-hour rTOSS was analyzed using an MMRM. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. |
| Sensitivity analysis:
Subgroup analysis:
|
RQLQ | RQLQ was analyzed using an ANCOVA model. | Treatment, site, baseline RQLQ(S). |
| Not performed. |
GSP301-305 trial | ||||
12-hour rTNSS | 12-hour rTNSS was analyzed using an MMRM. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. |
| Sensitivity analysis:
Subgroup analysis:
|
12-hour iTNSS | 12-hour iTNSS was analyzed using an MMRM. | Treatment, site, baseline 12-hour rTNSS, and study day as the within-patient effect. |
| Sensitivity analysis:
Subgroup analysis:
|
12- hour rTOSS | 12-hour rTOSS was analyzed using an MMRM. | Treatment, site, baseline clinical measures, and study day as the within-patient effect. |
| Sensitivity analysis:
Subgroup analysis:
|
PRQLQ | PRQLQ was analyzed using an ANCOVA model. | Treatment, site, baseline PRQLQ. |
| Sensitivity analysis:
Subgroup analysis:
|
ANCOVA = analysis of covariance; iTNSS = instantaneous Total Nasal Symptom Score; MAR = missing at random; MMRM = mixed model for repeated measures; MNAR = missing not at random; NR = not reported; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score.
Sources: Clinical Study Reports for GSP301-301,20 GSP301-304,21 and GSP301-30523 and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Analysis Populations
The analysis populations for the GSP301-301, GSP301-304, and GSP301-305 trials are summarized in Table 11.
Table 11: Analysis Populations for the GSP301-301, GSP301-304, and GSP301-305 Trials
Study | Population | Definition | Application |
|---|---|---|---|
GSP301-301 and GSP301-304 | FAS | All randomized patients who received 1 dose or more of the study drug and completed 1 postbaseline primary efficacy assessment | 12-hour rTNSS, iTNSS, 12-hour rTOSS, RQLQ(S) |
Safety analysis set | Included all patients who received 1 dose or more of study medication | Safety assessment | |
GSP301-305 | FAS | All randomized patients who received at least 1 dose of the study drug or placebo and had at least 1 postbaseline primary efficacy assessment | 12-hour rTNSS, iTNSS, 12-hour rTOSS, PRQLQ |
Safety analysis set | All randomized patients who received at least 1 dose of the study drug | Safety assessment |
FAS = full analysis set; iTNSS = instantaneous Total Nasal Symptom Score; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Protocol Amendments and Deviations
GSP301-301 Trial
In total, there were 3 versions of the clinical study protocol for the GSP301-301 trial. The original study protocol (version 1.0) was issued on October 26, 2015. Protocol version 2.0 and version 3.0 were issued on December 10, 2015, and January 25, 2016, respectively; all issue dates were before the start date for patient recruitment (i.e., March 2016). In summary, the main changes to the protocol included sample size increases and changes to the eligibility criteria, including the addition of various medical conditions (e.g., clinically significant respiratory tract malformations, nasal structural abnormalities).
During the randomized treatment period in the safety analysis set, the proportions of patients who had protocol deviations were similar across treatment groups: 15.9% (48 out of 302) in the olopatadine-mometasone group, 17.3% (51 out of 294) in the mometasone NS group, and 15.3% (44 out of 287) in the placebo group. The most commonly reported deviation was “outside visit window” (67 deviations in total: 17 in the olopatadine-mometasone group, 19 each in the olopatadine group and mometasone NS group, and 12 in the placebo group).
GSP301-304 Trial
There were 3 versions of the clinical study protocol for the GSP301-304 trial. The original study protocol (version 1.0) was issued on June 22, 2015, while protocol version 2.0 was issued on September 6, 2016, and protocol version 3.0 was issued on November 15, 2016. After the start of patient recruitment (i.e., August 2016), changes were made to the inclusion criteria to extend the recruitment period to include the season for mountain cedar pollen to meet the target randomization number.
During the randomized treatment period in the safety analysis set, the proportions of patients who had protocol deviations were generally similar across treatment groups: 21.8% (64 out of 294) in the olopatadine-mometasone group, 22.2% (65 out of 293) in the mometasone NS group, and 19.7% (58 out of 294) in the placebo group. The most commonly reported deviation was “other protocol deviation” (total of 148 deviations).
GSP301-305 Trial
There were 3 versions of the clinical study protocol for the GSP301-305 trial. The original study protocol (version 1.0) was issued on January 8, 2018. Protocol version 2.0 was issued on April 5, 2018, and protocol version 3.0 was issued on July 18, 2018. After the start of patient recruitment (i.e., March 2018), changes were made to clarify season-related information (i.e., spring and fall allergy seasons) for the study.
During the randomized treatment period in the safety analysis set, the proportion of patients who had protocol deviations was slightly higher in the olopatadine-mometasone group (25.8%; 58 out of 225), compared with the placebo group (22.2%; 49 out of 221). The most commonly reported protocol deviation was “assessment not performed per protocol” (44 deviations in the olopatadine-mometasone group versus 35 deviations in the placebo group).
Results
Patient Disposition
A summary of patient disposition in the GSP301-301, GSP301-304, and GSP301-305 trials is shown in Table 12.
In the GSP301-301 trial, 1,696 participants were screened and 1,180 were randomized. In the GSP301-304 trial, 1,808 participants were screened and 1,176 were randomized. In the GSP301-305 trial, 616 participants were screened and 446 were randomized. The reasons for screening failure were NR for all 3 trials. In the GSP301-301 trial, 4.3% of patients in the olopatadine-mometasone group, 3.83% of patients in the placebo group, and 3.74% of patients in the mometasone NS group discontinued from the study. In the GSP301-304 trial, 1.7% of patients in the olopatadine-mometasone group, 2.38% of patients in the mometasone NS group, and 3.4% of patients in the placebo group discontinued from the study. The proportion of patients who discontinued from the GSP301-305 trial was 4.44% for the olopatadine-mometasone group and 2.26% for the placebo group.
Baseline Characteristics
The baseline characteristics outlined in Table 13 are limited to those that were considered most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Overall, the baseline demographic and disease characteristics were generally similar across treatment groups within each of the 3 trials (GSP301-301, GSP301-304, and GSP301-305).
The baseline demographic and disease characteristics were similar overall between the GSP301-301 and GSP301-304 trials. In the GSP301-301 trial, the mean age of the study population was 39.3 years (SD = 15.3 years). The majority of patients were female (64.6%) and white (77.5%). In the GSP301-304 trial, the mean age of the patients was 39.6 years (SD = 14.81 years); the majority of the trial population was female (62.9%) and white (81.6%).
In the GSP301-305 trial, the mean age of the study population was 8.7 years (SD = 1.7 years). There were slightly more males (56.0%) in the olopatadine-mometasone group, while in the placebo group, the percentages of males and females were similar (50.7% versus 49.3%). The majority of the trial population was white (> 76.5%).
Exposure to Study Treatments
For the GSP301-301 and GSP301-304 trials, treatment duration was 14 plus 2 days. Twenty-eight doses (twice a day for 14 days) was considered 100% adherence, 35 or more doses (i.e., beyond 17 days of treatment due to out-of-window visits) was considered an adherence rate of 125% or more, and 21 or fewer doses was considered less than 75% adherence. Thus, it was possible for patients to have a treatment adherence of greater than 100% and still be compliant with the protocol. In the safety analysis set of the GSP301-301 trial, an adherence rate of between 75% and 125% was achieved by 94.4% of patients in the olopatadine-mometasone group, 94.2% in the mometasone NS group, and 95.1% in the placebo group. In the GSP301-304 trial, an adherence rate of between 75% and 125% was achieved by 99.6% of patients in the olopatadine-mometasone group, 99.0% in the mometasone NS group, and 98.6% in the placebo group.
For the GSP301-305 trial, treatment duration was 14 days but could have been up to 17 days based on permitted windows for study visits. Therefore, 28 doses (i.e., twice a day for 14 days) was considered 100% adherence. It was possible for patients to have a treatment adherence of greater than 100% and still be compliant with the protocol. In the safety analysis set of the GSP301-305 trial, the proportions of patients achieving an adherence rate of between 75% and 100% or between 100% and 125% were 81.3% and 15.6% in the olopatadine-mometasone group, respectively, and 79.6% and 17.6% in the placebo group, respectively.
Table 12: Summary of Patient Disposition From the Studies Included in the Systematic Review
Patient disposition | GSP301-301 trial | GSP301-304 trial | GSP301-305 trial | |||||
|---|---|---|---|---|---|---|---|---|
Olopatadine-mometasone | Mometasone NS | Placebo | Olopatadine-mometasone | Mometasone NS | Placebo | Olopatadine-mometasone | Placebo | |
Screened, N | 1,696 | 1,808 | 616 | |||||
Randomized, N | 302 | 294 | 287 | 294 | 294 | 294 | 225 | 221 |
Discontinued from study, N (%) | 13 (4.30) | 11 (3.74) | 11 (3.83) | 5 (1.70) | 7 (2.38) | 10 (3.40) | 10 (4.44) | 5 (2.26) |
Reason for discontinuation, N (%) | ||||||||
Adverse events | 1 (0.33) | 4 (1.36) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (0.34) | 4 (1.78) | 1 (0.45) |
Lost to follow-up | 1 (0.33) | 0 (0.00) | 3 (1.05) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (0.44) | 1 (0.45) |
Nonadherence | 1 (0.33) | 1 (0.34) | 1 (0.35) | 3 (1.02) | 1 (0.34) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Withdrawal by patient | 2 (0.66) | 0 (0.00) | 5 (1.74) | 1 (0.34) | 1 (0.34) | 4 (1.36) | 4 (1.78) | 2 (0.90) |
Removed by investigator | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 4 (1.36) | 2 (0.68) | 0 (0.00) | 1 (0.45) |
Protocol deviation | 3 (0.99) | 3 (1.02) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (0.34) | 0 (0.00) | 0 (0.00) |
Randomization failure | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (0.34) | 1 (0.34) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
Other | 5 (1.66) | 2 (0.68) | 2 (0.70) | 0 (0.00) | 0 (0.00) | 2 (0.68) | 1 (0.44) | 0 (0.00) |
Lack of efficacy | 0 (0.00) | 1 (0.34) | 0 (0.00) | 0 (0.00) | 0(0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
FAS, N | 299 | 294 | 283 | 291 | 293 | 290 | 222 | 219 |
Safety analysis set, N | 302 | 294 | 287 | 294 | 293 | 294 | 225 | 221 |
FAS = full analysis set; mometasone NS = mometasone furoate nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Table 13: Summary of Baseline Characteristics From Studies Included in the Systematic Review (Safety Analysis Set)
Characteristic | GSP301-301 trial | GSP301-304 trial | GSP301-305 trial | |||||
|---|---|---|---|---|---|---|---|---|
Olopatadine-mometasone (n = 302) | Mometasone NS (n = 294) | Placebo (n = 287) | Olopatadine-mometasone (n = 294) | Mometasone NS (n = 293) | Placebo (n = 294) | Olopatadine-mometasone (n = 225) | Placebo (n = 221) | |
Age, years: Mean (SD) | 39.5 (15.4) | 38.7 (16.3) | 39.4 (14.8) | 39.9 (14.9) | 39.2 (14.9) | 39.6 (14.9) | 8.7 (1.6) | 8.6 (1.7) |
Age, years: Median (range) | 39.5 (12.0 to 81.0) | 38.0 (12.0 to 84.0) | 41.0 (12.0 to 83.0) | 40.0 (12.0 to 76.0) | 38.0 (12.0 to 77.0) | 40.0 (12.0 to 82.0) | 9.0 (6.0 to 11.0) | 9.0 (6.0 to 11.0) |
Age group, years: n (%) | ||||||||
≥ 6 and < 9 | NA | NA | NA | NA | NA | NA | 100 (44.4) | 99 (44.8) |
≥ 9 and < 12 | NA | NA | NA | NA | NA | NA | 125 (55.6) | 122 (55.2) |
≥ 12 and < 18 | There were 115 patients aged 12 to 17 years, 1,016 patients aged 18 to 65 years, and 39 patients aged 65 years included in the FAS in this study. | 25 (8.5) | 27 (9.2) | 22 (7.5) | NA | NA | ||
≥ 18 and < 65 | 258 (88.1) | 252 (86.0) | 255 (87.0) | NA | NA | |||
≥ 65 | 10 (3.4) | 14 (4.8) | 16 (5.5) | NA | NA | |||
Female, n (%) | 201 (66.6) | 198 (67.3) | 182 (63.4) | 202 (68.9) | 170 (58.0) | 176 (60.1) | 99 (44.0) | 109 (49.3) |
Male, n (%) | 101 (33.4) | 96 (32.7) | 105 (36.6) | 91 (31.1) | 123 (42.0) | 117 (39.9) | 126 (56.0) | 112 (50.7) |
Race, n (%) | ||||||||
White | 241 (79.8) | 224 (76.2) | 231 (80.5) | 251 (85.7) | 232 (79.2) | 229 (78.2) | 170 (75.6) | 171 (77.4) |
Black | 56 (18.5) | 59 (20.1) | 48 (16.7) | 30 (10.2) | 50 (17.1) | 60 (20.5) | NR (NR) | NR (NR) |
Asian | 2 (0.7) | 7 (2.4) | 7 (2.4) | 7 (2.4) | 8 (2.7) | 3 (1.0) | 8 (3.6) | 5 (2.3) |
Other | 3 (1.0) | 4 (1.4) | 1 (0.3) | 5 (1.7) | 3 (1.0) | 1 (0.3) | 1 (0.4) | 1 (0.5) |
Average a.m. and p.m. rTNSS mean (SD) | 10.1 (1.2) | 10.2 (1.2) | 10.2 (1.2) | 10.1 (1.2) | 10.2 (1.3) | 10.3 (1.2) | 8.83 (1.41) | 8.84 (1.66) |
Average a.m. and p.m. iTNSS mean (SD) | 9.2 (1.7) | 9.3 (1.7) | 9.3 (1.7) | 9.2 (1.8) | 9.4 (1.8) | 9.6 (1.8) | 7.89 (1.93) | 7.82 (2.04) |
Average a.m. and p.m. rTOSS mean (SD) | 7.1 (1.4) | 7.0 (1.5) | 7.2 (1.3) | 7.0 (1.5) | 7.0 (1.5) | 7.2 (1.4) | 3.84 (2.45) | 3.59 (2.52) |
Average a.m. and p.m. iTOSS mean (SD) | 6.6 (1.7) | 6.6 (1.8) | 6.8 (1.6) | 6.5 (1.8) | 6.7 (1.7) | 6.8 (1.6) | 3.54 (2.40) | 3.26 (2.46) |
PNSS | 9.6 (2.1) | 9.7 (1.8) | 9.6 (1.9) | 9.2 (2.1) | 9.5 (2.0) | 9.6 (1.8) | 7.8 (2.3) | 7.8 (2.2) |
RQLQ(S) score | 4.0 (1.1) | 3.9 (1.2) | 3.9 (1.2) | 4.0 (1.2) | 4.0 (1.3) | 4.0 (1.2) | NR (NR) | NR (NR) |
PRQLQ (overall score) | NR (NR) | NR (NR) | NR (NR) | NR (NR) | NR (NR) | NR (NR) | 2.51 (1.08) | 2.42 (1.00) |
FAS = full analysis set; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total Ocular Symptom Score; mometasone NS = mometasone furoate nasal spray; NA = not applicable; NR = not reported; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PNSS = Physician-Assessed Nasal Symptom Score; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SD = standard deviation.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22
Concomitant Medications and Subsequent Treatment
At the discretion of the investigator and in consultation with the sponsor, patients were allowed to use any concomitant medications that were not prohibited by the study protocol.
In the GSP301-301 trial, the number of patients who used concomitant medications was 63 in the olopatadine-mometasone group, 65 in the mometasone NS group, and 65 in the placebo group. Some of the frequently used concomitant medications included ibuprofen (12 in the olopatadine-mometasone group, 8 in the mometasone NS group, and 13 in the placebo group), albuterol (6 in the olopatadine-mometasone group, 7 in the mometasone NS group, and 3 in the placebo group), naproxen (3 in the olopatadine-mometasone group, 0 in the mometasone NS group, and 5 in the placebo group), multivitamin (3 in the olopatadine-mometasone group, 0 in the mometasone NS group, and 3 in the placebo group), levothyroxine (1 in the olopatadine-mometasone group, 3 in the mometasone NS group, and 1 in the placebo group), Tylenol (1 in the olopatadine-mometasone group, 2 in the mometasone NS group, and 2 in the placebo group), and metformin (0 in the olopatadine-mometasone group, 0 in the mometasone NS group, and 2 in the placebo group).
No concomitant medications–related information summarized at the treatment-arm level was available for the GSP301-304 and GSP301-305 trials. None of the 3 pivotal trials reported any data on any subsequent treatment used.
Efficacy
Key efficacy results in the FAS during the randomized period are presented in Table 14. Given that olopatadine hydrochloride NS is currently unavailable in Canada, it was not considered relevant to this Reimbursement Review; thus, results for olopatadine hydrochloride are not presented.
Table 14: Summary of Key Efficacy Results From Studies Included in the Systematic Review (FAS)
Efficacy outcomes | GSP301-301 trial | GSP301-304 trial | GSP301-305 trial | |||||
|---|---|---|---|---|---|---|---|---|
Olopatadine-mometasone (n = 299) | Mometasone NS (n = 294) | Placebo (n = 283) | Olopatadine-mometasone (n = 291) | Mometasone NS (n = 293) | Placebo (n = 290) | Olopatadine-mometasone (n = 222) | Placebo (n = 219) | |
12-hour rTNSS over 14-day treatment period | ||||||||
Baseline, mean (SD) | 10.1 (1.2) | 10.2 (1.2) | 10.2 (1.2) | 10.09 (1.219) | 10.20 (1.309) | 10.32 (1.215) | 8.83 (1.41) | 8.84 (1.66) |
Number of patients contributing to LS mean change from baseline, n | 299 | 294 | 283 | 291 | 293 | 290 | 222 | 219 |
LS mean change from baseline (SE) | −3.48 (NR) | −3.09 (NR) | −2.50 (NR) | −3.52 (NR) | −3.05 (NR) | −2.44 (NR) | −1.6 (0.18) | −2.2 (0.17) |
LS mean difference between treatment groups (95% CI) | — | −0.39 (−0.79 to 0.01) | −0.98 (−1.38 to −0.57) | — | −0.47 (−0.86 to −0.08) | −1.09 (−1.49 to −0.69) | — | −0.6 (−0.9 to −0.2) |
P value | — | 0.0587 | < 0.0001 | — | 0.019 | < 0.001 | — | 0.001 |
12-hour iTNSS over 14-day treatment period | ||||||||
Baseline, mean (SD) | 9.2 (1.7) | 9.3 (1.7) | 9.3 (1.7) | 9.17 (1.800) | 9.42 (1.821) | 9.58 (1.767) | 7.89 (1.93) | 7.82 (2.04) |
Number of patients contributing to LS mean change from baseline, n | 299 | 294 | 283 | GSP301-301 291 | 293 | 290 | 222 | 219 |
LS mean change from baseline (SE) | −3.03 (NR) | −2.67 (NR) | −2.10 (NR) | −3.11 (NR) | −2.60 (NR) | −2.16 (NR) | −1.1 (0.17) | −1.8 (0.17) |
LS mean difference between treatment groups (95% CI) | — | −0.36 (−0.71 to −0.01) | −0.93 (−1.28 to −0.58) | — | −0.51 (−0.88 to −0.13) | −0.94 (−1.32 to −0.56) | — | −0.6 (−1.0 to −0.3) |
P value | — | 0.0413 | < 0.0001 | — | 0.008 | < 0.001 | — | < 0.001 |
12-hour rTOSS over 14-day treatment period | ||||||||
Baseline, mean (SD) | 7.1 (1.4) | 7.0 (1.5) | 7.2 (1.3) | 6.95 (1.476) | 7.04 (1.491) | 7.21 (1.406) | 3.84 (2.45) | 3.59 (2.52) |
Number of patients contributing to LS mean change from baseline, n | 299 | 294 | 283 | 291 | 293 | 290 | 222 | 219 |
LS mean change from baseline (SE) | −2.23 (NR) | −2.04 (NR) | −1.74 (NR) | −2.36 (NR) | −2.01 (NR) | −1.84 (NR) | −0.6 (0.13) | −0.8 (0.14) |
LS mean difference between treatment groups (95% CI) | — | −0.19 (−0.49 to 0.11) | −0.49 (−0.79 to −0.19) | — | −0.35 (−0.66 to −0.03) | −0.52 (−0.84 to −0.20) | — | −0.2 (−0.6 to 0.1) |
P value | — | 0.2113 | 0.0014 | — | 0.030 | 0.001 | — | 0.233 |
RQLQ(S) overall score | ||||||||
Baseline, mean (SD) | 4.0 (1.1) | 3.9 (1.2) | 3.9 (1.2) | 3.96 (1.235) | 4.04 (1.260) | 4.00 (1.241) | NA | NA |
Number of patients contributing to LS mean change from baseline, n | 298 | 293 | 279 | 283 | 280 | 280 | NA | NA |
LS mean change from baseline, (95% CI) | −1.54 (NR) | −1.34 (NR) | −1.11 (NR) | −1.67 (NR) | −1.22 (NR) | −1.58 (NR) | NA | NA |
LS mean difference between treatment groups (95% CI) | — | −0.20 (−0.41 to 0.02) | −0.43 (−0.64 to −0.21) | — | −0.09 (−0.32 to 0.14) | −0.45 (−0.68 to −0.22) | — | NA |
P value | — | 0.0692 | < 0.0001 | — | 0.4236 | 0.0001 | — | NA |
PRQLQ overall score | ||||||||
Baseline, mean (SD) | NA | NA | NA | NA | NA | NA | 2.51 (1.078) | 2.42 (1.001) |
Number of patients contributing to LS mean from baseline, n | NA | NA | NA | NA | NA | NA | 222 | 219 |
LS mean change from baseline, (95% CI) | NA | NA | NA | NA | NA | NA | −0.8 (0.08) | −0.5 (0.08) |
LS mean difference between treatment groups (95% CI) | NA | NA | NA | NA | NA | NA | — | −0.3 (−0.5 to −0.1) |
P value | NA | NA | NA | NA | NA | NA | — | < 0.001 |
CI = confidence interval; FAS = full analysis set; iTNSS = instantaneous Total Nasal Symptom Score; LS = least squares; mometasone NS = mometasone furoate nasal spray; NA = not applicable; NR = not reported; NS = nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PRQLQ = Paediatric Rhinoconjunctivitis Quality of Life Questionnaire; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SD = standard deviation; SE = standard error.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22 Details included in the table are from the sponsor’s summary of clinical evidence.1
Twelve-Hour rTNSS Over the 14-Day Treatment Period
GSP301-301 Trial
The mean baseline rTNSS score was 10.1 (SD = 1.2) in the olopatadine-mometasone group and 10.2 (SD = 1.2) in both the mometasone NS and placebo groups. The within-group LS mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −3.48 points (SE = NR) in the olopatadine-mometasone group, −3.09 points (SE = NR) in the mometasone NS group, and −2.50 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −0.98 points (95% CI, −1.38 to −0.57) between the olopatadine-mometasone group and the placebo group and −0.39 points (95% CI, −0.79 to 0.01) between the olopatadine-mometasone group and the mometasone NS group, with both point estimates of the LS mean difference favouring the olopatadine-mometasone group.
GSP301-304 Trial
The mean baseline rTNSS score was 10.09 (SD = 1.2) in the olopatadine-mometasone group, 10.2 (SD = 1.2) in the mometasone NS group, and 10.3 (SD = 1.2) in the placebo group. The within-group LS mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −3.52 points (SE = NR) in the olopatadine-mometasone group, −3.05 points (SE = NR) in the mometasone NS group, and −2.44 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −1.09 points (95% CI, −1.49 to −0.69) between the olopatadine-mometasone group and the placebo group and −0.47 points (95% CI, −0.86 to −0.08) between the olopatadine-mometasone group and the mometasone NS group; both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
GSP301-305 Trial
The mean baseline rTNSS score was 8.83 (SD = 1.4) in the olopatadine-mometasone group and 8.84 (SD = 1.7) in the placebo group. The within-group LS mean change from baseline in the 12-hour rTNSS over the 14-day treatment period showed an improvement in both treatment groups: −1.6 points (SE = 0.18) in the olopatadine-mometasone group and −2.2 points (SE = 0.17) in the placebo group.
The between-group LS mean difference in the 12-hour rTNSS over the 14-day treatment period was −0.6 points (95% CI, −0.9 to −0.2) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Twelve-Hour iTNSS Over the 14-Day Treatment Period
GSP301-301 Trial
The mean baseline iTNSS score was 9.2 (SD = 1.7) in the olopatadine-mometasone group and 9.3 (SD = 1.7) in both the mometasone NS and placebo groups. The within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −3.03 points (SE = NR) in the olopatadine-mometasone group, −2.67 points (SE = NR) in the mometasone NS group, and −2.10 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.93 points (95% CI, −1.28 to −0.58) between the olopatadine-mometasone group and the placebo group and −0.36 points (95% CI, −0.71 to −0.01) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
GSP301-304 Trial
The mean baseline iTNSS score was 9.17 (SD = 1.8) in the olopatadine-mometasone group, 9.42 (SD = 1.8) in the mometasone NS group, and 9.58 (SD = 1.8) in the placebo group. The within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −3.11 points (SE = NR) in the olopatadine-mometasone group, −2.60 points (SE = NR) in the mometasone NS group, and −2.16 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.94 points (95% CI, −1.32 to −0.56) between the olopatadine-mometasone group and the placebo group and −0.51 points (95% CI, −0.88 to −0.13) between the olopatadine-mometasone group and the mometasone NS group, with both point estimates of the LS mean difference favouring the olopatadine-mometasone group.
GSP301-305 Trial
The mean baseline iTNSS score was 7.89 (SD = 1.9) in the olopatadine-mometasone group and 7.82 (SD = 2.0) in the placebo group. The within-group LS mean change from baseline in the 12-hour iTNSS over the 14-day treatment period showed an improvement in both treatment groups: −1.1 points (SE = 0.17) in the olopatadine-mometasone group and −1.8 points (SE = 0.17) in the placebo group.
The between-group LS mean difference in the 12-hour iTNSS over the 14-day treatment period was −0.6 points (95% CI, −1.0 to −0.3) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Twelve-Hour rTOSS Over the 14-Day Treatment Period
GSP301-301 Trial
The mean baseline rTOSS score was 7.1 (SD = 1.4) in the olopatadine-mometasone group, 7.0 (SD = 1.5) in the mometasone NS group, and 7.2 (SD = 1.3) in the placebo group. The within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −2.23 points (SE = NR) in the olopatadine-mometasone group, −2.04 points (SE = NR) in the mometasone NS group, and −1.74 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.49 points (95% CI, −0.79 to −0.19) between the olopatadine-mometasone group and the placebo group and −0.19 points (95% CI, −0.49 to 0.11) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
GSP301-304 Trial
The mean baseline rTOSS score was 6.95 (SD = 1.48) in the olopatadine-mometasone group, 7.04 (SD = 1.49) in the mometasone NS group, and 7.21 (SD = 1.41) in the placebo group. The within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in all 3 treatment groups: −2.36 points (SE = NR) in the olopatadine-mometasone group, −2.01 points (SE = NR) in the mometasone NS group, and −1.84 points (SE = NR) in the placebo group.
The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.52 points (95% CI, −0.84 to −0.20) between the olopatadine-mometasone group and the placebo group and −0.35 points (−0.66 to −0.03) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
GSP301-305 Trial
The mean baseline rTOSS score was 3.84 (SD = 2.45) in the olopatadine-mometasone group and 3.59 (SD = 2.52) in the placebo group. The within-group LS mean change from baseline in the 12-hour rTOSS over the 14-day treatment period showed an improvement in both treatment groups: −0.6 points (SE = 0.13) in the olopatadine-mometasone group and −0.8 points (SE = 0.14) in the placebo group.
The between-group LS mean difference in the 12-hour rTOSS over the 14-day treatment period was −0.2 points (95% CI, −0.6 to 0.1) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
RQLQ(S) Overall Score on Day 15
GSP301-301 Trial
The mean baseline RQLQ(S) score was 4.0 (SD = 1.1) in the olopatadine-mometasone group and 3.9 (SD = 1.2) in both the mometasone NS and placebo groups. The within-group LS mean change from baseline in the RQLQ(S) overall score at day 15 showed an improvement in all 3 treatment groups: −1.54 points (SE = NR) in the olopatadine-mometasone group, −1.34 points (SE = NR) in the mometasone NS group, and −1.11 points (SE = NR) in the placebo group.
The between-group LS mean difference in the RQLQ(S) overall score at day 15 was −0.43 points (95% CI, −0.64 to −0.21) between the olopatadine-mometasone group and the placebo group and −0.20 points (95% CI, −0.41 to 0.02) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
GSP301-304 Trial
The mean baseline RQLQ(S) score was 3.96 (SD = 1.24) in the olopatadine-mometasone group, 4.04 (SD = 1.26) in the mometasone NS group, and 4.0 (SD = 1.24) in the placebo group. The within-group LS mean change from baseline in the RQLQ(S) overall score at day 15 showed an improvement in all 3 treatment groups: −1.67 points (SE = NR) in the olopatadine-mometasone group, −1.22 points (SE = NR) in the mometasone NS group, and −1.58 points (SE = NR) in the placebo group.
The between-group LS mean difference in the RQLQ(S) overall score at day 15 was −0.45 points (95% CI, −0.68 to −0.22) between the olopatadine-mometasone group and the placebo group and −0.09 points (95% CI, −0.32 to 0.14) between the olopatadine-mometasone group and the mometasone NS group, and both point estimates of the LS mean difference were in favour of the olopatadine-mometasone group.
PRQLQ Overall Score on Day 15
GSP301-305 Trial
The mean baseline PRQLQ score was 2.51 (SD = 1.08) in the olopatadine-mometasone group and 2.42 (SD = 1.0) in the placebo group. The within-group LS mean change from baseline in the PRQLQ overall score at day 15 showed an improvement in all 3 treatment groups: −0.8 points (SE = 0.08) in the olopatadine-mometasone group and −0.5 points (SE = 0.08) in the placebo group.
The between-group LS mean difference in the PRQLQ overall score at day 15 was −0.3 points (95% CI, −0.5 to −0.1) between the olopatadine-mometasone group and the placebo group, which favoured the olopatadine-mometasone group.
Harms
Harms data from the GSP301-301, GSP301-304, and GSP301-305 trials are shown in Table 15.
Table 15: Summary of Harms Results From the Studies Included in the Systematic Review (Safety Analysis Set)
Detail | GSP301-301 trial | GSP301-304 trial | GSP301-305 trial | |||||
|---|---|---|---|---|---|---|---|---|
Olopatadine-mometasone (n = 302) | Mometasone NS (n = 294) | Placebo (n = 287) | Olopatadine-mometasone | Mometasone NS | Placebo | Olopatadine-mometasone (n = 225) | Placebo (n = 221) | |
TEAE, n (%) | ||||||||
Patients with any TEAEa | 39 (12.9) | 21 (7.1) | 27 (9.4) | 46 (15.6) | 28 (9.6) | 28 (9.5) | 27 (12.0) | 23 (10.4) |
Dysgeusia | 10 (3.3) | 0 (0.0) | 2 (0.7) | 11 (3.7) | 0 (0.0) | 0 (0.0) | 3 (1.3) | 0 (0.0) |
Headache | 2 (0.7) | 2 (0.7) | 8 (2.8) | 0 (0.0) | 4 (1.4) | 2 (0.7) | 3 (1.3) | 1 (0.5) |
Epistaxis | 4 (1.3) | 2 (0.7) | 1 (0.3) | 2 (0.7) | 3 (1.0) | 3 (1.0) | 5 (2.3) | 2 (0.9) |
TESAE, n (%) | ||||||||
Patients with any TESAE | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.3) | 1 (0.3) | 0 (0.0) | 1 (0.5) |
Discontinuation of treatment due to TEAEs, n (%) | ||||||||
Patients who discontinued treatment | 0 (0.0) | 4 (1.4) | 1 (0.3) | 0 (0.0) | 0 (0.0) | 1 (0.3) | 4 (1.8) | 1 (0.5) |
Death, n (%) | ||||||||
Patients who died | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
mometasone NS = mometasone furoate nasal spray; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event.
aReported in ≥ 2% of patients in any treatment group.
Sources: Clinical Study Reports for the GSP301-301,20 GSP301-304,21 and GSP301-30523 trials and the sponsor’s response to the June 26, 2024, request by Canada’s Drug Agency for additional information.22 Details included in the table are from the sponsor’s summary of clinical evidence.1
Treatment-Emergent Adverse Events
GSP301-301 Trial
The proportion of patients experiencing TEAEs was 12.9% (39 out of 302) in the olopatadine-mometasone group, which was higher than in the mometasone NS group (7.1%; 21 out of 294) and in the placebo group (9.4%; 27 out of 287). The proportion of patients who had dysgeusia was 3.3% (10 out of 302) in the olopatadine-mometasone group, 0.7% (2 out of 287) in the placebo group, and 0% in the mometasone NS group. Headache occurred in 2.8% (8 out of 287) of the patients in the placebo group, higher than in the olopatadine-mometasone group (0.7%; 2 out of 302) and in the mometasone NS group (0.7%; 2 out of 294). Epistaxis was reported in 1.3% (4 out of 302) of the patients in the olopatadine-mometasone group, 0.7% (2 out of 294) of the patients in the mometasone NS group, and 0.3% (1 out of 287) of the patients in the placebo group.
GSP301-304 Trial
The proportion of patients experiencing TEAEs was 15.6% (46 out of 294) in the olopatadine-mometasone group, higher than in the mometasone NS group (9.6%; 28 out of 293) and in the placebo group (9.5%; 28 out of 294). Dysgeusia was reported in 3.7% (11 out of 294) of the patients in the olopatadine-mometasone group and in 0% of patients in both the mometasone NS and placebo groups. Headache occurred in 1.4% (4 out of 293) of the patients in the mometasone NS group, 0.7% (2 out of 294) of the patients in the placebo group, and 0% of patients in the olopatadine-mometasone group. The proportion of patients who had epistaxis was 0.7% (2 out of 294) in the olopatadine-mometasone group, 1.0% (3 out of 293) in the mometasone NS group, and 1.0% (3 out of 294) in the placebo group.
GSP301-305 Trial
The proportion of patients experiencing TEAEs was 12.0% (27 out of 225) in the olopatadine-mometasone group and 10.4% (23 out of 221) in the placebo group. The most common TEAE in the olopatadine-mometasone group was epistaxis (2.3%; 5 out of 225), while 0.9% (2 out of 221) of the patients in the placebo group had epistaxis. Dysgeusia were reported in 1.3% (3 out of 225) of the patients in the olopatadine-mometasone group and in 0% of patients in the placebo group. Headache occurred in 1.3% (3 out of 225) of the patients in the olopatadine-mometasone group and 0.5% (1 out of 221) of the patients in the placebo group.
Treatment-Emergent Serious Adverse Events
GSP301-301 Trial
Only 1 patient had a TESAE (0.3%) in the GSP301-301 trial, which was 1 spontaneous abortion in the olopatadine-mometasone group.
GSP301-304 Trial
No patients had a TESAE in the olopatadine-mometasone group. One patient in the mometasone NS group (0.3%) had 1 TESAE (i.e., peritonsillar abscess), and 1 patient in the placebo group (0.3%) had 3 TESAEs (1 instance each of osteomyelitis, syncope, and foot fracture).
GSP301-305 Trial
There was only 1 TESAE (i.e., meningitis), which was reported in 1 patient (0.5%) in the placebo group.
Withdrawals Due to TEAEs
GSP301-301 Trial
No patients withdrew due to TEAEs in the olopatadine-mometasone group compared with 4 in the mometasone NS group and 1 in the placebo group. The specific reasons for withdrawal were NR.
GSP301-304 Trial
No patients withdrew due to TEAEs in the olopatadine-mometasone group or in the mometasone NS group. One patient (0.3%) in the placebo group discontinued due to a foot fracture.
GSP301-305 Trial
There were 4 patients (1.8%) who withdrew due to TEAEs (1 instance each of conjunctivitis, acute otitis media, and sinusitis, and 1 upper respiratory tract infection) in the olopatadine-mometasone group and 1 patient (0.5%) in the placebo group who withdrew due to otitis media.
Mortality
No deaths were reported in the GSP301-301, GSP301-304, or GSP301-305 trials.
Critical Appraisal
Internal Validity
This Clinical Review Report assesses 3 pivotal studies submitted by the sponsor, including 2 phase III RCTs (i.e., GSP301-301 and GSP301-304) in adolescents and adults (aged 12 years and older) with SAR, and 1 phase III RCT (i.e., GSP301-305) in children (aged ≥ 6 to < 12 years) with SAR. In the GSP301-301 trial, 1,696 participants were screened and 1,180 were randomized. In the GSP301-304 trial, 1,808 participants were screened and 1,176 were randomized. In the GSP301-305 trial, 616 participants were screened and 446 were randomized. None of the 3 trials reported the reasons for screening failure. All 3 pivotal trials used a computer-generated randomization scheme that was reviewed and approved by a statistician. Information on allocation concealment was unclear for the GSP301-301 and GSP301-304 trials, whereas central randomization was performed for the GSP301-305 study, which was considered adequate to conceal allocation. The baseline characteristics were generally balanced across treatment groups within each of the 3 pivotal trials. Consequently, it was determined that the overall risk of bias arising from the randomization process was low.
All 3 pivotal trials adopted a double-blind design, which masked the trial participants and trial personnel. Blinding was achieved by packaging the active products and placebo in identical bottles and outer cartons for all 3 pivotal trials. The risk of performance bias due to the awareness of treatment assignment was considered low by the review team. An adherence rate of between 75% and 125% (i.e., from twice a day for 14 days to twice a day for up to 17 days) was achieved by more than 90% of the patients in each treatment group, which the clinical experts considered to be higher than clinical practice. The clinical experts consulted by the review team considered the use of concomitant medications generally appropriate. There were no major concerns with respect to protocol amendments in any of the 3 pivotal trials.
The risk of bias due to missing outcome data was determined to be low for all 3 pivotal trials. Based on the patient disposition information, a small proportion of patients in each treatment group of the 3 pivotal trials discontinued from the study for various reasons (e.g., loss to follow-up, withdrawal by patients, nonadherence). In all 3 pivotal trials, analyses in the per-protocol analysis set, which excluded patients who had not adhered to the study protocol (defined as a major protocol violation), showed an LS mean difference in the rTNSS similar to that in the FAS. Moreover, the mixed model for repeated measures used in the primary analysis assumed the data missing were missing at random. The GSP301-301 and GSP301-304 studies conducted sensitivity analyses for the rTNSS using the jump-to-reference approach and/or tipping-point approach, which assumed the data missing were missing not at random. According to the study investigators, the results of these sensitivity analyses were in agreement with the primary analysis; however, the results were NR and could not be validated, although the small amount of missing data did not raise concerns for this assumption.
The patient-reported symptom scores evaluated in the pivotal studies included rTNSS (primary efficacy end point), iTNSS, and rTOSS. The definitions of these symptom scores were consistent across the 3 pivotal trials and considered accurate by the clinical experts consulted by the review team. However, because reflective and instantaneous symptom scales were designed primarily for assessment in adults, young children might need the assistance of a proxy to assess and report the severity of their symptoms. In the GSP301-305 trial, children assessed their symptoms with the assistance of their parents, guardians, or caregivers as needed. Inconsistency between children-reported symptom severity and proxy-reported symptom severity has been reported, where children with asthma, rhinitis, or eczema tended to report more symptoms than their parents55 and, with an increase in the extent of child self-rating, there is a greater treatment difference between intervention and placebo, favouring the intervention.56 However, the GSP301-305 trial did not conduct a subgroup analysis of rater difference for any efficacy outcomes. As a result, the possibility of underestimating the treatment difference between olopatadine-mometasone and placebo due to the assistance of a proxy remains unclear for the GSP301-305 trial.
Lastly, a gatekeeping strategy to adjust for multiplicity was used for the primary (rTNSS), and secondary outcomes (iTNSS and rTOSS) in the GSP301-301 and GSP301-304 trials; however, multiplicity was not adjusted for the secondary outcomes of RQLQ(S) in these 2 trials. In the GSP301-305 trial, adjustment for multiplicity was not carried out for any outcome, so results should be considered with respect to the potential for type I error.
External Validity
The Health Canada–approved indication of olopatadine-mometasone is for patients with moderate to severe SAR; however, none of the 3 pivotal trials explicitly used the term “moderate to severe” to describe disease severity in the trial eligibility criteria. Instead, in the GSP301-301 and GSP301-304 trials, severity was defined as patients with an rTNSS of 8 or greater (out of a possible 12 points) and a congestion score of 2 or greater for the a.m. assessment at the screening visit. In the GSP301-305 trial, severity was defined as patients with an rTNSS of 6 or greater (out of a possible 12 points) and a congestion score of 2 or greater for the a.m. assessment at the screening visit. According to the clinical experts consulted by the review team, the aforementioned symptom scores thresholds correctly reflect “moderate to severe” disease severity and were appropriate in the clinical trial setting to define patients with moderate to severe SAR. However, the clinical experts also noted that in the clinical setting, these symptom score thresholds are typically not required to determine a patient’s disease severity. Instead, determination of disease severity relies on a clinician’s judgment based on the extent to which patients are impacted by their symptoms. The clinical experts noted there are patients with SAR who do not meet these minimum threshold scores for moderate to severe disease but could still benefit from olopatadine-mometasone if they are seriously impacted by their symptoms.
According to the clinical experts consulted by the review team, the eligibility criteria of all 3 pivotal trials were, in general, appropriate from the perspective of selecting patients for clinical trials. However, the clinical experts noted that from the perspective of real-world clinical practice, the exclusion criteria of the 3 pivotal trials were restrictive. For instance, per the clinical experts, patients with nasal structural abnormalities and/or patients with a history of significant rhinitis medicamentosa were excluded while, in clinical practice, these patients might benefit from olopatadine-mometasone. Despite these potential concerns, the experts consulted by the review team noted that the trial eligibility criteria were still reflective of the patients they would see in the real world and may be generalized to a broader population.
The clinical experts consulted by the review team noted that the dose and duration of treatments used in the 3 pivotal trials were, in general, appropriate and adequate in the clinical trial setting. However, according to the clinical experts, the treatment duration used in the pivotal trials might not be reflective of the duration of treatment in the real-world setting, where patients are often given treatment for a longer period. The clinical experts consulted by the review team noted that patient adherence to treatment in all 3 pivotal trials was higher than they would expect to see in the real world, which may overestimate the treatment effect that would be observed in a real-world setting.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the expert committee deliberations by Canada’s Drug Agency, and a final certainty rating was determined as outlined by the GRADE Working Group:57,58
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty-of-evidence assessment for the rTNSS, iTNSS, rTOSS, RQLQ(S), and PRQLQ were set according to the presence of an important effect based on thresholds agreed upon by the clinical experts consulted by the review team for this review. For harms outcomes, the certainty of evidence was summarized narratively.
For the GRADE assessments, the findings from the GSP301-301 and GSP301-304 trials were considered together and summarized narratively per outcome and per comparison because these studies were similar in population, interventions, design, and outcome measures. The findings from the GSP301-305 trial were assessed individually because that trial had a child population (aged ≥ 6 years and < 12 years), while the GSP301-301 and GSP301-304 trials had an adolescent and adult population (aged 12 years and older).
Results of GRADE Assessments
Olopatadine-Mometasone Versus Placebo
Table 2 presents the GRADE summary of findings for olopatadine-mometasone versus placebo for adolescent and adult patients (aged 12 years and older) with SAR.
Table 4 presents the GRADE summary of findings for olopatadine-mometasone versus placebo for children (aged ≥ 6 to < 12 years) with SAR.
Olopatadine-Mometasone Versus Mometasone NS
Table 3 presents the GRADE summary of findings for olopatadine-mometasone versus mometasone NS for adolescent and adult patients (aged 12 years and older) with SAR.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Long-term extension studies of olopatadine-mometasone in moderate to severe SAR were not available.
Instead, the sponsor submitted a double-blind, randomized, parallel-group study (NCT02709538)59,60 to evaluate the long-term (52 weeks) safety, tolerability, and efficacy outcomes of olopatadine-mometasone in adults and adolescents (aged 12 years and older) with PAR, a condition that was not included in either the Health Canada–approved indication for olopatadine-mometasone or in the reimbursement request.1 Therefore, the long-term extension study was considered not relevant to this review and was not appraised. A brief description of the sponsor-submitted long-term extension study follows.
In NCT02709538, a total of 593 patients were randomized 4:1:1 to olopatadine-mometasone (N = 393), which has a pH value of 3.7, or to a placebo with a pH value of either 3.7 (N = 99) or 7.0 (N = 101). The overall TEAE rates at week 52 were numerically greater in the placebo pH 7.0 group than in the other 2 treatment groups.1 A total of 326 patients were treated with olopatadine-mometasone for 6 months, and 250 patients were treated with olopatadine-mometasone for 1 year.2 The incidence of TEAEs reported in 2% or more of patients was greater among the patients in the placebo pH 3.7 group (i.e., matches the pH of olopatadine-mometasone) compared with the patients treated with olopatadine-mometasone. These TEAEs were upper respiratory tract infection (6.4% in the olopatadine-mometasone group versus 6.1% in the placebo pH 3.7 group), epistaxis (4.6% versus 2.0%), headache (4.1% versus 3.0%), nasal discomfort (2.8% versus 2.0%), viral upper respiratory tract infection (2.3% versus 2.0%), urinary tract infection (2.3% versus 2.0%), cough (2.3% versus 2.0%), and dysgeusia (2.0% versus 0).2,61 At week 52, a higher proportion of patients in the placebo pH 3.7 group reported nasopharyngitis (5.1%) compared with the olopatadine-mometasone group (3.1%).1,61 Over 52 weeks, serious AEs were experienced by 11 patients, 7 in the olopatadine-mometasone group (1.8%), 2 in the placebo pH 3.7 group (2.0%), and 2 in the placebo pH 7.0 group (2.0%).1,61 No deaths occurred during the study.1,61
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The pivotal trials submitted by the sponsor (i.e., GSP301-301, GSP301-304, and GSP301-305) provided head-to-head comparisons between olopatadine-mometasone versus placebo, olopatadine hydrochloride NS, and mometasone NS. However, the relative efficacy between olopatadine-mometasone and the general class of corticosteroids and antihistamines is unclear. Therefore, an ITC is warranted to address this evidence gap.
Description of the Indirect Comparisons
Two ITCs,62 both of which were NMAs, were submitted by the sponsor. According to the clinical experts consulted by the sponsor, it was not appropriate to combine studies in adolescent and adult patients (i.e., aged 12 years and older) with studies in children (i.e., aged ≥ 6 to < 12 years). Therefore, the sponsor conducted NMAs for 2 age groups separately.
The NMA for patients with SAR aged 12 years and older assessed the efficacy of olopatadine-mometasone relative to placebo, intranasal corticosteroids, and oral antihistamines, while the NMA for patients with SAR aged from 6 years to younger than 12 years compared olopatadine-mometasone relative to placebo and intranasal corticosteroids.
Indirect Treatment Comparison Design
Objectives
The objectives of the 2 sponsor-submitted NMAs were to assess the relative efficacy for olopatadine-mometasone versus the existing relevant comparators in patients with SAR aged 12 years and older and in patients with SAR aged from 6 years to younger than 12 years, respectively. Of note, descriptions in this section apply to both NMAs, unless specified otherwise.
Study Selection Methods
The criteria for study selection and the methods for the 2 ITCs submitted by the sponsor are shown in Table 16. An SLR conducted by the sponsor served as the evidence base. Studies eligible for the ITCs were identified according to the eligibility criteria, as outlined in Table 16. The literature searches, last updated on March 24, 2024, were conducted in multiple electronic databases, conference proceedings, and trial registries. Searches of relevant systematic reviews and meta-analyses, including NMAs, were performed to ensure the initial searches captured all relevant clinical studies. The study screening and selection process was conducted by 2 independent reviewers, with disagreement resolved by consultation with a third reviewer. Data were extracted by 1 reviewer and verified by a second reviewer. A risk-of-bias assessment for each included study was carried out using the revised Cochrane risk-of-bias tool.63
ITC Analysis Methods
The details with respect to the analysis methods for the sponsor-submitted ITCs are shown in Table 17.
Table 16: Study Selection Criteria and Methods for the ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | The target population for the NMA included children (aged from 6 years to younger than 12 years), adolescents (aged 12 to 17 years), and adults (aged 18 years and older) with moderate to severe SAR. |
Intervention | Olopatadine-mometasone |
Comparators | The following treatments were considered relevant comparators. Oral antihistamines:
Nasal corticosteroids:
|
Outcomes | The outcomes of interest for the NMA at 2 weeks (14 or 15 days) were as follows:
|
Study designs | RCT |
Publication characteristics | Published in the English language |
Exclusion criteria |
|
Databases searched |
Bibliographic searches of key systematic reviews and meta-analyses, including NMAs, were performed to ensure the initial searches captured all of the relevant clinical studies. Searches were also performed to identify potentially relevant conference posters or podium presentations. The following conferences were included: International Conference on Allergy, Asthma and Clinical Immunology; International Conference on Otolaryngology: ENT Surgery; and Asia Pacific Association of Allergy, Asthma, and Clinical Immunology. To ensure all relevant trials were captured, searches of the following registry websites for both completed and incomplete studies were also undertaken using a 2-year look back: clinicaltrials.gov, the International Clinical Trials Registry Platform, and the European Union’s Clinical Trials Register. The completed studies identified were cross-checked against the available publications to ensure all relevant information was captured. Date of search (last update): March 24, 2024. |
Selection process | All records identified through the searches were exported to EndNote bibliographic management software. After removing duplicates, citations were exported to a Microsoft Excel spreadsheet. All records were reviewed based on their abstracts and titles against the set of predefined eligibility criteria. The abstracts were screened independently for inclusion and exclusion by 2 reviewers. In case of ambiguity, a third (senior) reviewer was consulted, and a decision was reached by consensus. All papers included at the end of this stage were retained for full-text review. Full-text publications were independently assessed by 2 reviewers, and discrepancies were resolved by consulting a third reviewer. |
Data extraction process | Data extraction was performed by 1 researcher and checked by another independent researcher. A standard data template in the format of a Microsoft Excel workbook was developed to facilitate data collection. For the studies identified that met the inclusion criteria, the following variables were extracted:
No calculations were made except for very straightforward calculations of percentages (e.g., male and female proportions). Where data were reported in graphs, the GetData Graph Digitizer64 was used to extract relevant data. Data extraction was performed by 1 researcher and checked by another independent researcher. Where relevant, the units (e.g., years or months), type of statistic (e.g., mean or median), indication of data spread (e.g., SD, SE, or IQR), and definition of outcome was reported. |
Quality assessment | The quality of each study identified in the SLR was assessed to ensure that the conclusions and findings of this review were based on the best available evidence and that any potential sources of bias in the data were identified. The quality of RCTs retained for data extraction was assessed using the revised Cochrane risk-of-bias tool.63 |
IQR = interquartile range; ITC = indirect treatment comparison; NMA = network meta-analysis; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RCT = randomized controlled trial; rTNSS = reflective Total Nasal Symptom Score; SAR = seasonal allergic rhinitis; SD = standard deviation; SE = standard error; SLR = systematic literature review; TNSS = Total Nasal Symptom Score.
Sources: Sponsor-submitted ITCs.62 Details included in the table are from the sponsor’s summary of clinical evidence.1
Table 17: Analysis Methods for the Sponsor-Submitted ITCs
Methods | Description |
|---|---|
Feasibility assessment | During the feasibility assessment of an ITC, the studies identified in the SLR evaluating interventions of interest were assessed for between-study heterogeneity. The following were assessed for the presence and extent of between-study heterogeneity:
|
Analysis methods | The planned analyses consisted of continuous data (mean change from baseline). Therefore, a normal model with an identity link function was employed. Analyses were performed under a Bayesian framework and were conducted in Stan through R (version 4.3.3) using the RStudio interface and the “multinma” package. Analyses were repeated in OpenBUGS version 3.2.3 (OpenBUGS Foundation) to calculate the RM between interventions. |
Priors | Vague priors were specified for all model parameters,65 except in cases where model convergence could not be achieved. Weakly informative priors were used in the event of divergent transitions occurring in the post warm-up phase of the Hamiltonian Monte Carlo sampling algorithm or if the Markov chain Monte Carlo algorithm in OpenBUGS did not converge.66 |
Assessment of model fit | The selection of the best-fitting model was performed using the DIC. DIC provides a measure of model fit that penalizes model complexity — lower values of the DIC suggest a more parsimonious model — and is calculated as the sum of total residual deviance, which represents goodness of fit, and leverage, which represents the effective number of parameters. |
Assessment of consistency | The consistency assumption could not be assessed due to no closed loops or multiarm design of the included studies. |
Assessment of convergence | Convergence was assessed by visual inspection of the trace and the autocorrelation plots. All the results presented were based on a further sample of at least 5,000 simulations. A convergence diagnosis and output analysis for Markov chain Monte Carlo values were generated for RM by the NMA runs. |
Assessment of heterogeneity | Heterogeneity was assessed qualitatively for the main baseline characteristics, including age, sex, duration of disease, rTNSS, iTNSS, rTOSS, iTOSS, PNSS, RQLQ, and comorbidity in children and in adolescents and adults. Heterogeneity was also assessed quantitatively using 𝜏2 with an RE model. |
Outcomes | Average 12-hour rTNSS at 2 weeks (14 or 15 days). For the purposes of economic analysis and to align with the cost-effectiveness model requirements, the RM was defined as an additional scale of analysis. The model specified for FE or RE in the results section was extended to calculate the RM and to compare the effects between interventions. The RM between any pair of interventions 𝑐, 𝑘 was defined as: 𝑅𝑀𝑐𝑘 = 𝑇𝑐/ 𝑇𝑘 where 𝑇𝑘 = 𝐴 + 𝑑1𝑘 is the absolute effect of the treatment 𝑘, defined as the linear combination of the absolute effect of the control 𝐴 and the basic parameter (relative effect) 𝑑1𝑘. The absolute effect of the control 𝐴 ~ 𝑁 (𝜇𝛢, 𝜎𝛢2) is the normally distributed effect of the Ryaltris study arm used as an anchor to calculate the absolute mean effects. Based on the baseline scores of TNSS and rTNSS reported in the articles and comparing these with the 12-hour rTNSS values, an assumption was made that TNSS and rTNSS could be considered equivalent to the average 12-hour rTNSS. Consequently, these studies were included in the NMA. Additionally, the included trials varied in their data reporting formats; some reported data as mean (SD), while others presented findings as LS mean. To ensure consistency and facilitate comparison, all results were converted to mean data. |
Follow-up time points | 2 weeks (14 or 15 days) |
Construction of nodes | Treatment nodes represent different treatment classes. For the NMA in patients aged 12 years and older, the pooled treatment nodes were placebo, oral antihistamines, intranasal corticosteroids, and olopatadine-mometasone. For the NMA in children aged ≥ 6 to < 12 years, the pooled treatment nodes were placebo, intranasal corticosteroids, and olopatadine-mometasone because there were no studies with oral antihistamines that met the inclusion criteria. |
Base-case analysis | The NMA in patients aged 12 years and older included 13 RCTs, meeting the predefined eligibility criteria and adopted mean difference as the effect measure for the 12-hour rTNSS at 2 weeks. The NMA in children aged ≥ 6 to < 12 years included 4 RCTs and adopted mean difference as the effect measure for the 12-hour rTNSS at 2 weeks. |
Sensitivity analyses | For the NMA in patients aged 12 years and older:
For the NMA in children aged ≥ 6 to < 12 years, no sensitivity analysis was conducted. |
DIC = deviance information criterion; FE = fixed effects; ITC = indirect treatment comparison; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total ocular symptom scores; LS = least squares; NMA = network meta-analysis; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; PNSS = Physician-Assessed Nasal Symptom Score; RCT = randomized controlled trial; RE = random effects; RM = ratio of means; RQLQ = Rhinoconjunctivitis Quality-of-Life Questionnaire; rTNSS = reflective Total Nasal Symptom Score; rTOSS = Reflective Total Ocular Symptom Score; SD = standard deviation; SLR = systematic literature review; TNSS = Total Nasal Symptom Score.
Sources: Sponsor-submitted ITCs.62 Details included in the table are from the sponsor’s summary of clinical evidence.1
Results of ITC in Patients Aged 12 Years and Older
Summary of Included Studies
In total, 13 studies were included in the ITC for patients aged 12 years and older.46,47,67-77 The characteristics of the 13 included studies are presented in Table 18, and the assessment of potential heterogeneity is shown in Table 19.
The risk of bias of the 13 RCTs included in the ITC for patients aged 12 years and older was assessed using the revised Cochrane risk-of-bias tool (i.e., RoB 2). The overall risk of bias was determined to be low for 3 included studies,67,75,76 and there were some concerns of bias for 10 included studies.46,47,68-74,77 The concerns for the 10 included RCTs arose mainly from the risk of bias in the randomization process.
Table 18: Characteristics of the Included Studies in the Sponsor-Submitted ITC for Patients Aged 12 Years and Older
Study | Design | Total, N | Intervention | Comparators | Primary end point(s) | Outcomes analyzed in ITC |
|---|---|---|---|---|---|---|
NCT0388204770 | Phase III, double-blind RCT | 313 | Mometasone furoate nasal spray: 200 mcg once daily for 15 days |
| TNSS (participant-rated) | TNSS mean change from baseline The mean change from baseline at study day 15 was calculated for TNSS as assessed by the investigator. TNSS was a composite of the individual nasal symptom scores of discharges (rhinorrhea), stuffiness, sneezing, and itching. The 4 individual nasal symptom scores were rated by the investigator at the visit as follows: 0 = none, 1 = mild, 2 = moderate, 3 = severe. TNSS was the sum of the 4 individual nasal symptom scores (range = 0 to12, with a higher score indicating more frequent and/or severe nasal symptoms). For each participant, individual scores were totalled and used to calculate the change from baseline in TNSS at the visit. Participant changes were then used to calculate the mean change for each treatment group at that visit. Change from baseline = visit score minus baseline score (at day 1 visit). Negative changes indicate a decrease in symptom severity. |
NCT0385518971 | Phase III, triple-blind RCT | 345 | Mometasone furoate nasal spray: 200 mcg once daily for 29 days |
|
| TNSS mean change from baseline TNSS evaluated participant nasal symptoms of discharge (rhinorrhea), stuffiness, sneezing, and itching. The 4 individual symptom scores were rated by the physician at the visit as follows: 0 = none, 1 = mild, 2 = moderate, 3 = severe. TNSS was the sum of the 4 individual symptom scores (range, 0 to 12, with higher score indicating more frequent or severe nasal symptoms). For each participant, individual scores were totalled and used to calculate the change from baseline in TNSS at the visit. Participant changes were then used to calculate the mean change for each treatment group at that visit. Change from baseline = visit score minus baseline score (day 1 visit). Negative changes from baseline indicate a decrease in symptom severity. |
Anolik (2008)67 | Phase III, double-blind RCT | 702 | Mometasone furoate nasal spray: 200 mcg once daily for 15 days |
|
| TNSS mean change from baseline and percent reduction Patient-rated average change in TNSS (nasal discharge plus stuffiness plus sneezing plus itching) using data from patients’ a.m. and p.m. diary records of TNSSs during the 15 days of therapy. |
Gawchik (2003)74 | Double-blind RCT (phase not reported) | 254 | Mometasone furoate nasal spray: 200 mcg once daily for 14 days | Placebo | Cough severity | TNSS mean change from baseline The sum of both a.m. and p.m. TNSS recorded over the 3 consecutive days before visit 3 (including the morning of the visit) had to be ≥ 36 points (maximum possible score of 72 points). |
Meltzer (2011)75 | Phase III, double-blind RCT | 684 | Mometasone furoate nasal spray: 200 mcg once daily for 15 days | Placebo |
| TNSS LS mean change from baseline The change from baseline in average a.m. or p.m. nasal congestion score recorded 12 hours prior and 1 of the 4 symptoms that comprise the TNSS, averaged over days 1 to 15. |
NCT0224692072 | Phase III, double-blind RCT | 1,474 | Fluticasone propionate nasal spray: 200 mcg once daily for 14 days |
| rTNSS (average a.m. and p.m.) | rTNSS LS mean change from baseline TNSS is defined as the sum of the patient-rated nasal symptom severity scores for the following 4 allergy symptoms (categories): runny nose, nasal congestion, itchy nose, and sneezing. The severity score for each symptom was based on a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe). The higher the score, the worse the symptoms in the 4 allergy categories. The primary analysis for determining the therapeutic equivalence of the test and reference treatments was based on each treatment’s mean change from baseline for average rTNSS over the 2-week randomization treatment period. |
Gross (2019),47 i.e., GSP301-304 | Phase III, double-blind RCT | 1,176 | Olopatadine-mometasone: 665 mcg plus 25 mcg per delivered dose, twice daily for 14 days |
| 12-hour rTNSS (average a.m. and p.m.) | 12-hour rTNSS LS mean change from baseline The mean change from baseline to the end of the 14-day treatment period in average a.m. and p.m. 12-hour rTNSS. The rTNSS was assessed by scoring the severity of 4 nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching) over the past 12 hours before the recording of the score (i.e., reflective) and just before each dosing (i.e., instantaneous). |
Hampel (2019),46 i.e., GSP301-301 | Phase III, double-blind RCT | 1,180 | Olopatadine-mometasone: 665 mcg plus 25 mcg per delivered dose, twice daily for 14 days |
| 12-hour rTNSS (average a.m. and p.m.) | 12-hour rTNSS LS mean change from baseline The mean change from baseline to the end of the 14-day treatment period in average a.m. and p.m. 12-hour rTNSS. The rTNSS was assessed by scoring the severity of nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching) over the past 12 hours before the recording of the score (i.e., reflective) and just before each dosing (i.e., instantaneous). |
Prenner (2010)76 | Phase III, double-blind RCT | 429 | Mometasone furoate nasal spray: 200 mcg once daily for 15 days | Placebo |
| TNSS mean change from baseline Daily scores for the rTNSS were obtained from averaging the reflective a.m. and p.m. individual symptom scores. Patients rated the severity of their individual symptoms, including rhinorrhea, nasal congestion, nasal itching, sneezing, redness of eyes, itching or burning eyes, and tearing or watering eyes, on a 4-point scale (i.e., 0 = none, 1 = mild, 2 = moderate, and 3 = severe) twice daily in patient diaries (in the a.m. before dosing and approximately 12 hours later, i.e., in the p.m.). Instantaneous and reflective (over the previous 12 hours) scores were captured. |
Ratner (2006)77 | Double-blind RCT (phase not reported) | 327 | Ciclesonide: 200 mcg once daily for 28 days | Placebo | TNSS (average a.m. and p.m.) | 12-hour rTNSS LS mean change from baseline rTNSS measures nasal symptom severity over the past 12 hours and is calculated as the sum of 4 nasal symptoms (runny nose, itchy nose, sneezing, and nasal congestion), each rated on a scale of 0 (no signs or symptoms evident) to 3 (signs or symptoms that interfere with daily activities). |
NCT0102460869 | Phase III, double-blind RCT | 340 | Beclomethasone dipropionate hydrofluoroalkane: 320 mcg once daily for 14 days | Placebo | 12-hour rTNSS (average a.m. and p.m.) | 12-hour rTNSS LS mean change from baseline Participants recorded the severity of their nasal symptoms (sneezing, runny nose, nasal itching and nasal congestion) over the past 12 hours (before the assessment) twice daily (a.m. and p.m.) using the following scale: 0 = absent (no sign or symptom present); 1 = mild (sign or symptom present, easily tolerated); 2 = moderate (definite awareness of sign or symptom, bothersome but tolerable); 3 = severe (hard to tolerate, interferes with daily activities and/or sleeping). The rTNSS (sum of 4 symptom scores) ranges from 0 to 12 (worst symptoms). A negative change from baseline score indicates symptom improvement. |
NCT0185082368 | Phase III, double-blind RCT | 880 | Mometasone furoate nasal spray (Watson Laboratories, Inc.): 200 mcg twice daily for 14 days |
| 12-hour rTNSS (average a.m. and p.m.) | 12-hour rTNSS mean change from baseline Superiority analysis of change from baseline in rTNSS for both active arms compared with placebo arm. The TNSS scale was used. Patients rated each of the following 4 symptoms on a scale from 0 to 3: nasal congestion, runny nose, sneezing, and itchy nose twice daily, approximately 12 hours apart. |
NCT0073238173 | Phase III, quadruple-blind RCT | 351 | Mometasone furoate nasal spray: 200 mcg once daily for 15 days | Placebo | Nasal congestion score recorded 12 hours prior (average a.m. and p.m.) | The rTNSS mean change from baseline TNSS is a composite of 4 symptoms; each is scored on a scale of 0 = none, 1 = mild, 2 = moderate, 3 = severe. The total can range from 0 to 12. |
AE = adverse event; ITC = indirect treatment comparison; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total Ocular Symptom Score; LS = least squares; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RCT = randomized controlled trial; rTNSS = reflective Total Nasal Symptom Score; TNSS = Total Nasal Symptom Score; TOSS = Total Ocular Symptom Score.
Sources: Sponsor-submitted ITCs.62
Table 19: Assessment of Homogeneity of the Sponsor-Submitted ITC for Patients Aged 12 Years and Older
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Baseline characteristics | Age: The average age of participants across the 13 included studies ranged from 25 to 41 years. No information was provided in the sponsor-submitted ITC regarding the proportion of patients in different age groups (e.g., ≥ 12 and < 17 years, ≥ 18 years). Sex: Ten studies included more female participants (i.e., > 50%) than male participants except for 3 studies that had fewer female patients (NCT03882047, 47.1%; NCT03855189, 44.8%; Anolik [2008], 48.29%). Race: Most of the included studies involved predominantly white participants, with the proportion varying between 68.4% and 85.7%. This was followed by Black participants, whose proportion ranged from 10.2% to 27.4%. Duration of SAR: Most of the included studies did not report on duration of disease except 2 studies: 13 to 14 years in Anolik (2008) and 16.3 to 16.8 years in Gawchik (2003). Baseline symptom scores: Out of the 13 included studies, 6 reported data for rTNSS. The baseline rTNSS mean scores ranged from 7.3 (SD = 2.2) to 10.3 (SD = 1.2). Only 3 studies reported scores for the following:
Only 2 studies reported scores for PNSS. Comorbidity: Comorbidity could not be assessed due to poor reporting in the individual included studies, according to the sponsor. |
Treatment history | No relevant information was provided in the sponsor-submitted ITC report. |
Trial eligibility criteria | No relevant information was provided in the sponsor-submitted ITC report. |
Comparators | In general, the dosing and treatment duration for the same comparator treatment were similar across the included studies. The sponsor-submitted ITC used 3 classes (i.e., intranasal corticosteroids, oral histamines, placebo) to group comparator treatments in the network geometry. According to the clinical experts consulted by the review team, the grouping was appropriate from a clinical perspective. |
Placebo response | No relevant information was provided in the sponsor-submitted ITC report. |
End point | According to the ITC report submitted by the sponsor, all 13 included studies reported changes in TNSS and rTNSS over a 14-day period, including 12-hour intervals. Seven studies measured TNSS, while 6 measured rTNSS. Out of the 13 included studies, the TNSS in 11 studies was assessed by scoring the severity of 4 nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching). For the remaining 2 included studies, i.e., Gawchik (2003) and Meltzer (2011), no relevant information was found in the sponsor-submitted ITC report. Eight studies measured outcomes using mean change from baseline, while 5 used LS mean change from baseline. |
Withdrawal frequency | No relevant information was provided in the sponsor-submitted ITC report. |
Clinical trial setting | No relevant information was provided in the sponsor-submitted ITC report. |
Study design | All 13 included studies were RCTs with 11 being phase III trials. Two studies did not report on study phase. Eleven included studies used double-blind masking, 1 study used triple-blind masking, and 1 study used quadruple-blind masking. Ten included studies were conducted in the US while 2 studies did not provide relevant location information. The sample sizes in the included studies vary significantly, ranging from 245 to 1,176 participants. |
ITC = indirect treatment comparison; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total Ocular Symptom Score; LS = least squares; PNSS = Physician-Assessed Nasal Symptom Score; RCT = randomized controlled trial; RQLQ(S) = Rhinoconjunctivitis Quality-of-Life Questionnaire – Standardized Activities; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SAR = seasonal allergic rhinitis; SD = standard deviation.
Source: Sponsor-submitted ITC.62
Results
Twelve-Hour rTNSS
The base-case network diagram for the ITC for patients aged 12 years and older is presented in Figure 2.
Figure 2: Network Diagram for the Sponsor-Submitted ITC for Patients Aged 12 Years and Older (Base-Case Analysis)
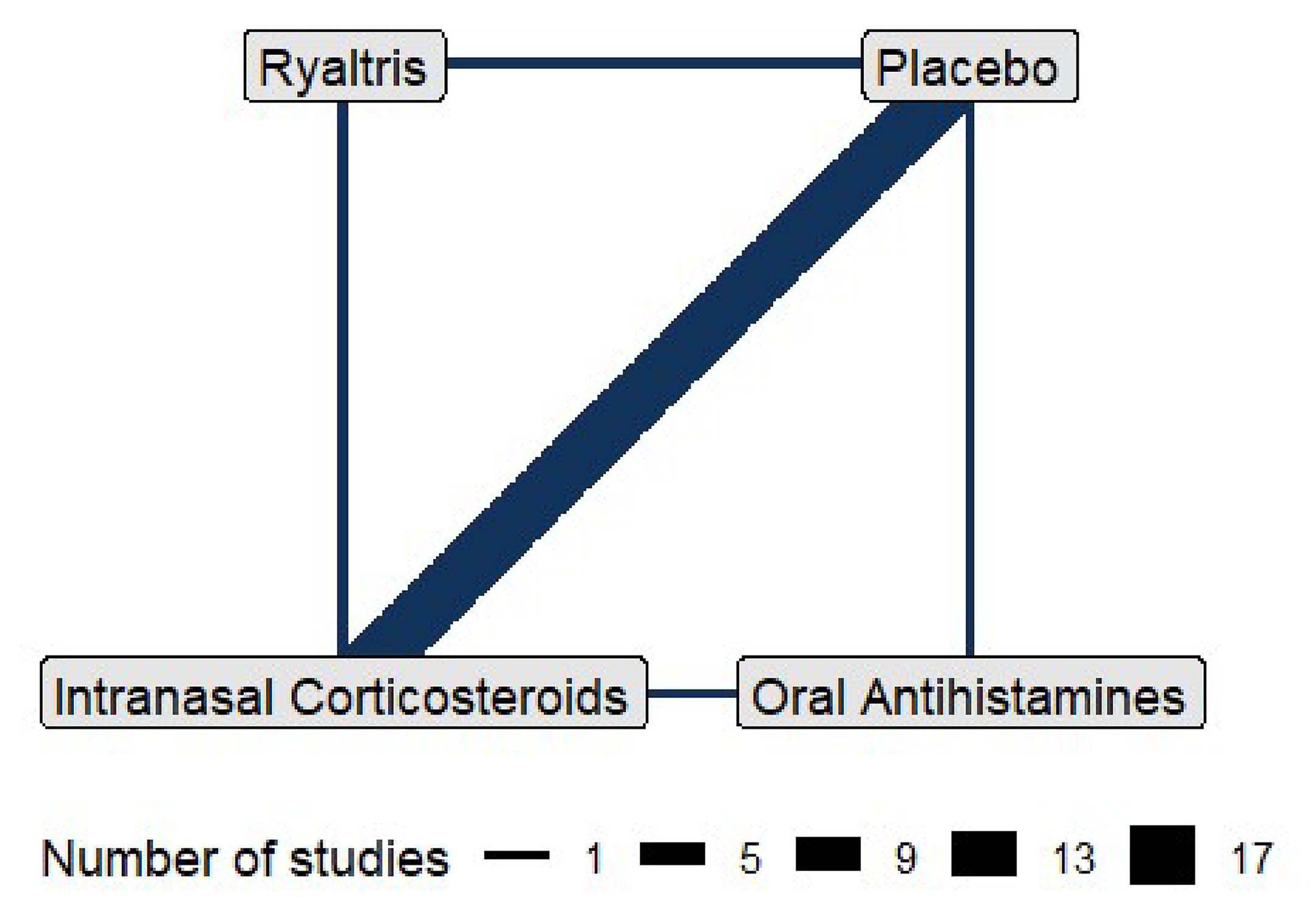
ITC = indirect treatment comparison.
Source: Sponsor-submitted ITCs.62
Both the fixed-effects (FE) and random-effects (RE) models were examined. The deviance information criterion (DIC) for the FE model was 82.60, while the DIC for the RE model was 63.15. The RE model was considered by the investigators to be a better fit than the FE model in the sponsor-submitted ITC; therefore, results from this model were used as the primary results. The results generated from the RE model, presented in Table 20, illustrate the estimated relative effects (mean or LS mean difference in symptom scores between the treatments) together with the 95% CrI. Negative values indicated a reduction in the Total Nasal Symptom Score (TNSS), which suggests that a treatment is more effective than its comparator.
In the base-case analysis, the mean or LS mean difference in the 12-hour rTNSS was −1.26 points (95% CrI, −1.86 to −0.67) between the olopatadine-mometasone and placebo arms, −0.27 points (95% CrI, −0.87, 0.33) between the olopatadine-mometasone and intranasal corticosteroids arms, and −0.91 points (95% CrI, −1.91 to 0.06) between the olopatadine-mometasone and oral antihistamine arms. Results from the sensitivity analyses were generally consistent with the results in the base-case analysis.
Table 20: Network Estimates for 12-Hour rTNSS (Patients Aged 12 Years and Older, RE Model)
Comparison | Mean or LS mean difference in symptom scores between treatments (95% CrI) | ||
|---|---|---|---|
Base-case analysis | Sensitivity analysis: Removing 2 trials with missing values for SEs | Sensitivity analysis: Including only 5 studies with LS mean change from baseline as an end point | |
Olopatadine-mometasone vs. placebo | −1.26 (−1.86 to −0.67) | −1.26 (−1.92 to −0.61) | −1.19 (−1.53 to −0.88) |
Olopatadine-mometasone vs. oral antihistamines | −0.91 (−1.91 to 0.06) | −0.91 (−1.99 to 0.17) | NA |
Olopatadine-mometasone vs. intranasal corticosteroids | −0.27 (−0.87 to 0.33) | −0.27 (−0.91 to 0.38) | −0.34 (−0.67 to −0.02) |
CrI = credible interval; LS = least squares; NA = not applicable; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RE = random effects; rTNSS = reflective Total Nasal Symptom Score; SE = standard error; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.62
Results of the ITC in Patients Aged From 6 Years to Younger Than 12 Years
Summary of Included Studies
In total, 4 studies were included in the ITC for children (aged ≥ 6 years and < 12 years).48,78-80 The characteristics of the 4 included studies are presented in Table 21.
The risk of bias of the 4 RCTs included in the ITC for patients aged from 6 years to younger than 12 years was assessed using the revised Cochrane risk-of-bias tool (i.e., RoB 2). All 4 included RCTs48,78-80 were rated as having some concerns, mainly deriving from the randomization process.
Table 21: Characteristics of Included Studies in the Sponsor-Submitted ITC for Patients Aged 6 Years to Younger Than 12 Years
Study | Design | Total N | Intervention | Comparator | Primary end point(s) | Outcomes analyzed in ITC |
|---|---|---|---|---|---|---|
NCT0387977280 | Phase III, triple-blind RCT | 679 | Mometasone furoate nasal spray: 25 mcg once daily for 28 days |
|
| TNSS mean CFB The mean CFB at study day 15 was calculated for TNSS as assessed by the investigator. TNSS was a composite of the individual nasal symptom scores for discharges (rhinorrhea), stuffiness, sneezing, and itching. The 4 individual nasal symptom scores were rated by the investigator at the visit as follows: 0 = none, 1 = mild, 2 = moderate, 3 = severe. TNSS was the sum of the 4 individual nasal symptom scores (range = 0 to 12, with higher scores indicating more frequent or severe nasal symptoms). For each participant, individual scores were totalled and used to calculate the CFB in TNSS at the visit. Participant changes were then used to calculate the mean change for each treatment group at that visit. CFB = visit score minus baseline score (at day 1 visit). Negative changes indicate a decrease in symptom severity. |
NCT0145827579 | Phase III, double-blind RCT | 847 | Ciclesonide nasal aerosol: 37 mcg once daily for 14 days |
| rTNSS (average a.m. and p.m.) | rTNSS LS mean CFB TNSS is the sum of individual symptoms of runny nose, sneezing, itchy nose, and nasal congestion. Patients assess each individual symptom on a scale of 0 to 3 where 0 = absent, 1 = mild, 2 = moderate, and 3 = severe. Therefore, rTNSS values range from 0 to 12 (with 0 representing an absence of symptoms and higher scores reflecting more severe symptoms). The rTNSS measures these symptoms over the previous 12-hour time interval. Difference was calculated as the 2-week treatment average minus baseline. Greater reductions in the CFB score indicate greater improvement. |
Prenner (2022),48 i.e., GSP301-305 | Phase III, double-blind RCT | 446 | Olopatadine-mometasone: 665 mcg plus 25 mcg per delivered dose twice daily for 14 days | Placebo | 12-hour rTNSS (average a.m. and p.m.) | 12-hour rTNSS mean CFB, LS mean difference This is the CFB in average a.m. and p.m. patient-reported 12-hour rTNSS over the 14-day treatment period. The rTNSS was assessed by scoring the severity of 4 nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching) over the past 12 hours before the recording of the score (i.e., reflective) and just before each dosing (i.e., instantaneous). |
Berger (2008)78 | Double-blind RCT (phase not reported) | 254 | Ciclesonide nasal aerosol: 200 mcg once daily for 14 days |
| Cough severity | rTNSS LS mean CFB, mean difference rTNSS measures nasal symptom severity over the previous 12 hours and is calculated as the sum of the 4 individual nasal symptoms, each rated on a scale of 0 (no signs or symptoms evident) to 3 (signs or symptoms that interfere with daily activities). |
CFB = change from baseline; ITC = indirect treatment comparison; LS = least squares; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; RCT = randomized controlled trial; rTNSS = reflective Total Nasal Symptom Score; TNSS = Total Nasal Symptom Score.
Source: Sponsor-submitted ITC.62
Table 22: Assessment of Homogeneity of the Sponsor-Submitted ITC for Patients Aged 6 Years to Younger Than 12 Years
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Baseline characteristics | Age: Three out of the 4 included studies reported the mean age of patients. The mean age of participants across the 3 studies ranged from 8.5 to 8.8 years. Sex: More than 50% of the participants in all 4 included studies were male (range, 50.7% to 63%). Race: Three out of the 4 included studies reported data on race. All 3 of these studies included predominantly white participants, with the proportion varying between 75.6% and 86.2%. Duration of SAR: No included studies reported data on the duration of SAR. Baseline symptom scores: All 4 of the included studies reported data for rTNSS. The baseline rTNSS mean scores ranged from 7.8 (SD = 1.7) to 8.84 (SD = 1.66). Only 1 study reported scores for iTNSS, rTOSS, iTOSS, and PNSS. Comorbidity: Comorbidity could not be assessed due to poor reporting in individual included studies, according to the sponsor. |
Treatment history | No relevant information was provided in the sponsor-submitted ITC report. |
Trial eligibility criteria | No relevant information was provided in the sponsor-submitted ITC report. |
Comparators | The sponsor-submitted ITC used 2 classes (i.e., intranasal corticosteroids, placebo) to group comparator treatments in the network geometry because no studies included oral antihistamines. It was noted that the dose of ciclesonide used in 2 included studies was different: 100 mcg and 200 mcg once daily for 14 days in Berger (2008), vs. 37 mcg and 74 mcg once daily for 14 days in NCT01458275. |
Placebo response | No relevant information was provided in the sponsor-submitted ITC report. |
End points | The study NCT03879772 reported the mean change from baseline in TNSS. NCT01458275 assessed the LS mean change from baseline in rTNSS. Prenner (2022) evaluated the 12-hour rTNSS with LS mean change from baseline and LS mean difference. Berger (2008) reported the LS mean change from baseline and the mean difference in rTNSS. TNSS or rTNSS was assessed by scoring the severity of 4 nasal symptoms (i.e., rhinorrhea, sneezing, nasal congestion, and nasal itching) in 3 included studies. The information on end points in 1 study (Berger 2008) was not clear. |
Withdrawal frequency | No relevant information was provided in the sponsor-submitted ITC report. |
Clinical trial setting | No relevant information was provided in the sponsor-submitted ITC report. |
Study design | All 4 included studies were phase III RCTs. All included studies used double-blind masking except 1 study that adopted triple-blind masking. Three included studies were conducted in the US, while 1 study did not provide relevant information on study design. |
ITC = indirect treatment comparison; iTNSS = instantaneous Total Nasal Symptom Score; iTOSS = instantaneous Total Ocular Symptom Score; LS = least squares; PNSS = Physician-Assessed Nasal Symptom Score; RCT = randomized controlled trial; rTNSS = reflective Total Nasal Symptom Score; rTOSS = reflective Total Ocular Symptom Score; SAR = seasonal allergic rhinitis; SD = standard deviation; vs. = versus.
Source: Sponsor-submitted ITC.62
Results
Twelve-Hour rTNSS
The base-case network diagram for the ITC for children (aged ≥ 6 to < 12 years) is presented in Figure 3.
Figure 3: Network Diagram for the Sponsor-Submitted ITC for Patients Aged 6 Years to Younger Than 12 Years (Base-Case Analysis)
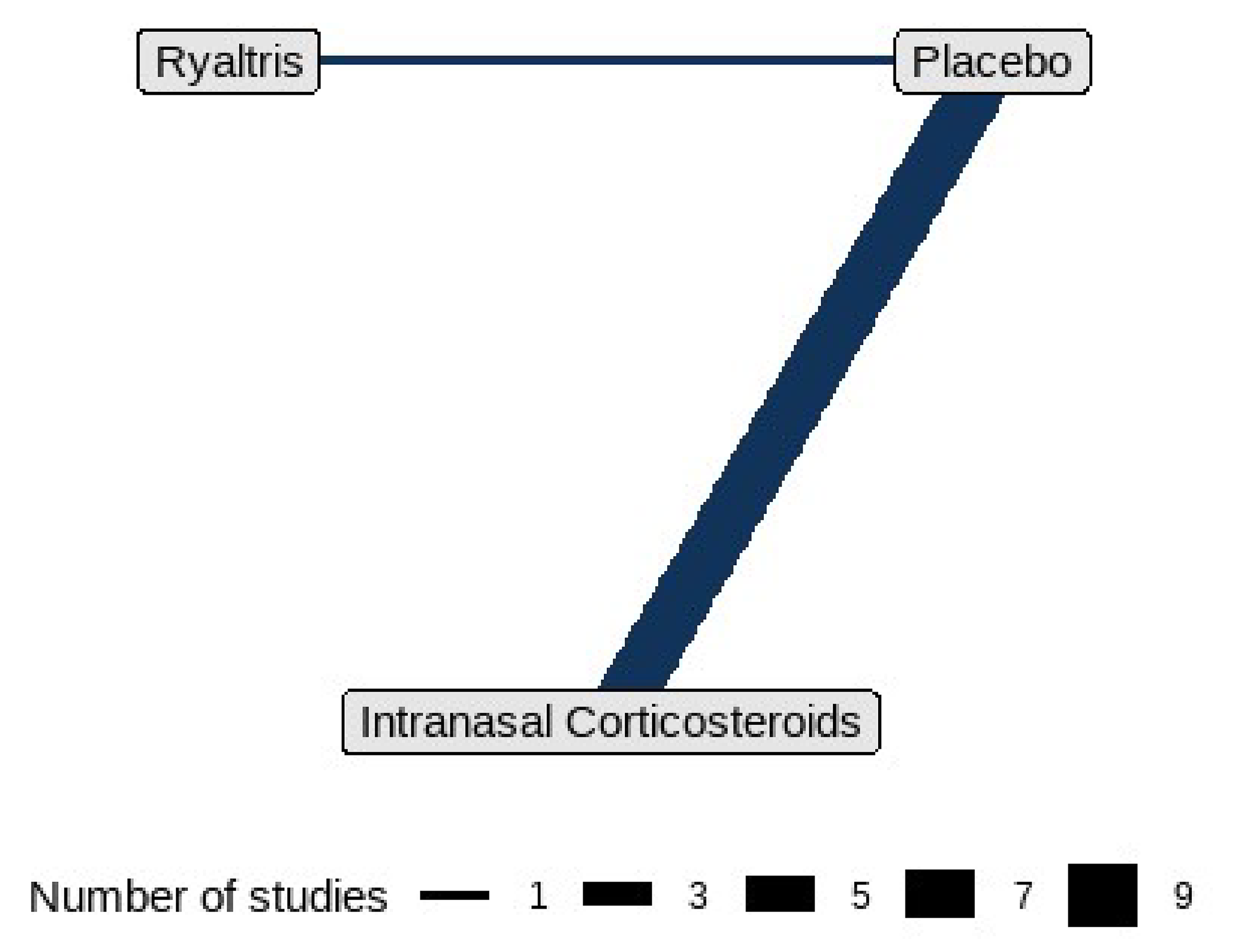
ITC = indirect treatment comparison.
Sources: Sponsor-submitted ITCs.62
The DIC for the FE model was 21.47, while the DIC for the RE model was 22.51. The FE model was considered by the investigators to be a better fit than the RE model in the sponsor-submitted ITC; therefore, results from this model were presented as the primary results for this comparison. The results generated from the FE model are presented in Table 23 and illustrate the estimated relative effects (mean or LS mean difference in symptom scores between the treatments) together with the 95% CrI. Negative values indicate a reduction in the TNSS, which suggests that a treatment is more effective than its comparator.
In the base-case analysis, the mean or LS mean difference in the 12-hour rTNSS was −1.21 points (95% CrI, −1.86 to −0.56) between the olopatadine-mometasone and placebo arms and −0.94 points (95% CrI, −1.63 to −0.26) between the olopatadine-mometasone and intranasal corticosteroids arms. The mean or LS mean difference in the 12-hour rTNSS yielded by the RE model was −1.21 points (95% CrI, −2.03 to −0.39) between the olopatadine-mometasone and placebo arms and −0.91 points (95% CrI, −1.76 to −0.01) between the olopatadine-mometasone and intranasal corticosteroids arms.
Table 23: Network Estimates for the 12-Hour rTNSS (Patients Aged 6 Years to Younger Than 12 Years, FE Model)
Comparison | Base case: Mean or LS mean difference in symptom scores between treatments (95% CrI) |
|---|---|
Olopatadine-mometasone vs. placebo | −1.21 (−1.86 to −0.56) |
Olopatadine-mometasone vs. intranasal corticosteroids | −0.94 (−1.63 to −0.26) |
CrI = credible interval; FE = fixed effects; LS = least squares; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; rTNSS = reflective Total Nasal Symptom Score; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.62
Critical Appraisal of the 2 NMAs Submitted by the Sponsor
Two NMAs were submitted by the sponsor: 1 targeting patients with SAR aged 12 years and older (referred to as the adolescents and adults NMA) that assessed the efficacy of olopatadine-mometasone compared with placebo, intranasal corticosteroids, and oral antihistamines, and 1 targeting patients with SAR aged 6 (inclusive) to younger than 12 years (referred to as the children NMA) that compared olopatadine-mometasone relative to placebo and intranasal corticosteroids.
The adolescents and adults NMA and the children NMA were based on 13 and 4 RCTs, respectively, and were identified from a sponsor-conducted SLR. The literature search for the SLR, which involved multiple electronic databases, clinical trial registries, conference proceedings, and reference lists of key systematic reviews and meta-analyses existing in the literature, was considered appropriate. Errors in the study selection and data extraction processes were minimized by carrying out the processes in duplicate. The risk of bias in the individual RCTs included in the NMAs was assessed using the revised Cochrane risk-of-bias tool (i.e., RoB 2).63 There were some concerns regarding the randomization process for 10 out of 13 studies in the adolescents and adults NMA and for all 4 studies included in the children NMA, which might have introduced some uncertainty in the NMA estimates.
Both NMAs defined the review questions (i.e., population, intervention, comparator, outcomes, and study design) a priori. With respect to the comparators in the SLR protocol, the sponsor listed several active comparators under 2 drug classes: intranasal corticosteroids and oral antihistamines. The clinical experts consulted by the review team noted that some relevant comparators were missing from the 2 classes in the protocol. For instance, fluticasone furoate (approved by Health Canada for the treatment of the symptoms of SAR in patients aged 2 years and older), which is widely used or prescribed to reduce nasal inflammation and associated symptoms, was missing from intranasal corticosteroids. Bilastine (approved by Health Canada for the symptomatic relief of nasal and non-nasal symptoms of SAR in patients aged 4 years and older with a body weight of at least 16 kg) and rupatadine fumarate (approved by Health Canada for the symptomatic relief of nasal and non-nasal symptoms of SAR in patients aged 2 years and older), both of which can be used in patients with moderate to severe SAR, were missing from the oral antihistamines in the SLR protocol. The ITC report did not provide rationales for why these comparators were not included. Yet, missing relevant comparators from the SLR protocol might have resulted in missing evidence in the subsequent NMAs, although the impact of this potential bias remains unknown. In addition, there is a possibility that missing comparators may jeopardize the generalizability of the NMA results to these missing comparator therapies.
The network of the adolescents and adults NMA comprised 4 nodes, representing olopatadine-mometasone, intranasal corticosteroids, oral antihistamines, and placebo. The network of the children NMA comprised 3 nodes, which represented olopatadine-mometasone, intranasal corticosteroids, and placebo. The individual treatments identified from the included studies were categorized into corresponding nodes. The sponsor assumed that individual drugs in the same drug class were equivalent in terms of clinical efficacy (intraclass clinical equivalency), which was considered reasonable overall by the clinical experts consulted by the review team. However, it was noted that within some nodes, there were only 1 or 2 individual drugs included due to a lack of eligible studies, which was beyond the sponsor’s control. For instance, only loratadine was available and included in the oral antihistamine node in the adolescents and adults NMA, while other drugs specified in the protocol, such as cetirizine, desloratadine, and fexofenadine, were not. In the children’s NMA, the intranasal corticosteroid node included only mometasone and ciclesonide. Despite the assumption of intraclass clinical equivalency being reasonable, there was concern and associated uncertainty regarding whether only 1 or 2 individual therapies would properly represent the corresponding drug class in terms of efficacy. Thus, the interpretation of the efficacy of olopatadine-mometasone relative to the intranasal corticosteroid class and to the oral antihistamine class should be made with caution.
Out of 4 included studies in the NMA for children, 3 studies had multiple treatment arms that involved different doses of the same treatment. For instance, in Berger (2008),78 patients with SAR were randomized to once-daily ciclesonide 100 mcg or 200 mcg or placebo for 2 weeks, while in NCT01458275,79 patients received once-daily ciclesonide nasal aerosol 37 mcg or 74 mcg or placebo for 2 weeks. In the sponsor-submitted NMA report, no information was provided on which dosage groups of ciclesonide were selected to generate the NMA treatment estimates. In addition, the clinical experts consulted by the review team noted the large variance between the dose of ciclesonide (i.e., 100 mcg or 200 mcg once daily for 2 weeks) used in Berger (2008)78 and the dose (i.e., 37 mcg or 74 mcg once daily for 2 weeks) used in NCT01458275.79 As a result, this limitation represents an additional source of uncertainty in the treatment effects estimates for the children NMA.
The clinical experts consulted by the review team identified several patient and disease characteristics as potential sources of heterogeneity that might bias the NMA estimates. Some of these characteristics (i.e., age, sex, disease duration, baseline symptom scores, comorbidity) were examined by the sponsor in both the adolescents and adults NMA and the children NMA by the sponsor. In general, the clinical experts consulted by the review team agreed with the sponsor’s evaluation and identified no serious heterogeneity arising from the aforementioned patient and disease characteristics. However, the clinical experts consulted by the review team also noted some patient and disease characteristics that might be potential sources of heterogeneity were missing from the sponsor-conducted NMAs, including urban versus rural living conditions, genetic predisposition, family history of atopic diseases, and smoking or vaping status. Thus, some uncertainty concerning the results of the NMA is warranted due to these potential sources of heterogeneity; however, the inclusion of these variables was beyond the sponsor’s control, given the limited data available in the included studies.
The individual studies included in the adolescents and adults NMA and the children NMA reported TNSS, rTNSS, and 12-hour TNSS. Both NMAs used 12-hour rTNSS as the efficacy end point and assumed that TNSS and rTNSS could be considered equivalent to the average 12-hour rTNSS. The clinical experts consulted by the review team determined that this assumption was acceptable.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Discussion
Summary of Available Evidence
Three sponsor-conducted pivotal studies, GSP301-301, GSP301-304, and GSP301-305, were included in this review. Both GSP301-301 (N = 1,176) and GSP301-304 (N = 1,180) were phase III, double-blind RCTs that enrolled adolescent and adult patients (aged 12 years and older) with SAR. The primary objective of the GSP301-301 and GSP301-304 trials was to compare the efficacy of olopatadine-mometasone with placebo and the individual constituent monotherapies (i.e., olopatadine hydrochloride NS and mometasone NS) at the same dose in the same vehicle, and to assess the efficacy of olopatadine hydrochloride NS and mometasone NS versus placebo over 14 days of study treatment. Of note, olopatadine hydrochloride NS is currently unavailable in Canada and therefore not relevant to this Reimbursement Review. As such, results comparing olopatadine-mometasone with olopatadine hydrochloride NS are not presented in this report. The GSP301-305 trial (N = 446) was a phase III, double-blind, RCT investigating the efficacy and safety of olopatadine-mometasone compared with placebo over 14 days in children (aged ≥ 6 to < 12 years) with SAR. The primary end point of all 3 pivotal trials was change from baseline in the patient-reported 12-hour rTNSS. The secondary efficacy and safety outcomes reported in the 3 pivotal trials included change from baseline in the patient-reported 12-hour iTNSS, patient-reported 12-hour rTOSS, and harms (i.e., TEAEs, TESAEs, withdrawals, and deaths). HRQoL was evaluated in the GSP301-301 and GSP301-304 trials via the RQLQ(S), while the GSP301-305 trial used the PRQLQ. All 3 pivotal trials were completed.
The mean age of patients in the GSP301-301 and GSP301-304 trials was 39.3 years (SD = 15.3 years) and 39.6 years (SD = 14.81 years), while the mean age in the GSP301-305 trial was 8.7 years (SD = 1.7 years). In the GSP301-301 and GSP301-304 trials, most patients were female (64.6% and 62.9%, respectively). In the GSP301-305 trial, there were slightly more males (56.0%) in the olopatadine-mometasone group while, in the placebo group, the proportions of male and female patients were similar (50.7% versus 49.3%). In the GSP301-301 trial, the baseline rTNSS score was the same across the olopatadine-mometasone group, the mometasone NS group, and the placebo group (mean = 10.1; SD = 1.2). In the GSP301-304 trial, the baseline mean rTNSS score was 10.1 (SD = 1.2) for the olopatadine-mometasone group, 10.3 (SD = 1.3) for the mometasone NS group, and 10.3 (SD = 1.2) for the placebo group. In the GSP301-305 trial, the baseline mean rTNSS score was 8.83 (SD = 1.41) for the olopatadine-mometasone group and 8.84 (SD = 1.66) for the placebo group.
Indirect evidence from 2 NMAs was submitted by the sponsor. One NMA evaluated the efficacy of olopatadine-mometasone compared with placebo, intranasal corticosteroids, and oral antihistamines among adolescent and adult patients (aged 12 years and older) with SAR. The other NMA assessed the efficacy of olopatadine-mometasone relative to placebo and intranasal corticosteroids in children (aged ≥ 6 to < 12 years) with SAR. The NMA for adolescent and adult patients was based on 13 RCTs identified from a sponsor-conducted SLR, while the NMA for children was based on 4 RCTs. Efficacy was measured using the 12-hour rTNSS in both NMAs.
The sponsor also submitted a long-term extension study that evaluated the long-term (52 weeks) safety, tolerability, and efficacy of olopatadine-mometasone in adults and adolescents (aged 12 years and older) with PAR. However, given that the Health Canada–approved indication is for SAR, not PAR, the long-term study submitted by the sponsor was not considered relevant to this review and was therefore not appraised.
Interpretation of Results
Efficacy
The patient group input highlighted that the key treatment goal for patients with SAR was to eliminate or significantly lessen AR symptoms, in particular, nasal symptoms. Relief of other symptoms and improvement of quality of life were also considered important goals of treatment by patients. Outcomes of the pivotal trials, GSP301-301, GSP301-304, and GSP301-305, were in line with the needs identified by patients, assessing nasal symptoms (12-hour rTNSS, 12-hour iTNSS), ocular symptoms (12-hour rTOSS), and HRQoL (RQLQ[S] as well as PRQLQ). In the included trials, baseline nasal and ocular symptom scores were lower for children (i.e., in the GSP301-305 trial) than those of adolescents and adults. For instance, the baseline 12-hour rTNSS score was 8.83 and 8.84 for the olopatadine-mometasone and placebo groups in the GSP301-305 trial, whereas the rTNSS score was greater than 10 across treatment groups in the adolescents and adults enrolled in the GSP301-301 and GSP301-304 trials. This is likely due to the difference in inclusion criteria, as the GSP301-305 trial required children to have a minimum rTNSS score of 6, while the GSP301-301 and GSP301-304 trials required a minimum score of 8; however, the actual reason for and overall impact of this difference in criteria remain unclear. According to the clinical experts consulted by the review team, lower baseline scores might also have contributed to the smaller treatment effect observed in the GSP301-305 trial and a possible underestimation of the treatment effect, because lower scores leave a smaller margin for effect sizes in children.
The GSP301-301 and GSP301-304 studies had similar study populations, interventions, comparators, outcomes, and study designs, and focused on the adolescent and adult patient populations (aged 12 years and older). Evidence from both trials demonstrated with high certainty that, compared with placebo, olopatadine-mometasone results in a statistically significant improvement in nasal symptoms as measured by the 12-hour rTNSS and 12-hour iTNSS. The LS mean difference in the 12-hour rTNSS was consistent between the GSP301-301 and GSP301-304 trials in terms of the direction and magnitude of the treatment effect, with LS mean differences of −0.98 points (95% CI, −1.38 to −0.57) in the GSP301-301 trial and −1.09 points (95% CI, −1.49 to −0.69) in the GSP301-304 trial. Using the threshold of a minimal important difference (MID) of 0.5 points, described by the clinical experts, the results also indicated a clinically meaningful improvement favouring olopatadine-mometasone over placebo. Evidence from the GSP301-301 and GSP301-304 trials also indicated that, compared with placebo, treatment with olopatadine-mometasone in adolescent and adult patients likely results in an improvement in ocular symptoms; however, there was uncertainty in the clinical meaningfulness of the results because the 95% CIs included the MID threshold, suggesting that the results were consistent with both a clinically meaningful benefit and no meaningful benefit for this outcome.
The GSP301-305 trial compared olopatadine-mometasone with placebo in children (aged ≥ 6 years and < 12 years) with SAR. Evidence from the GSP301-305 trial was generally in line with the evidence from the GSP301-301 and GSP301-304 trials, demonstrating that olopatadine-mometasone likely results in an improvement in nasal symptoms as measured by the 12-hour rTNSS (LS mean difference = −0.6 points; 95% CI, −0.9 to −0.2) and 12-hour iTNSS (LS mean difference = −0.6 points; 95% CI, −1.0 to −0.3), but likely results in little or no difference in improving ocular symptoms as measured by the rTOSS (LS mean difference = −0.2 points; 95% CI, −0.6 to 0.1). None of these outcomes were considered clinically meaningful because the 95% CIs included the MID of 0.5 points. The clinical experts consulted by the review team noted there was a possibility of underestimating the relative efficacy between olopatadine-mometasone and placebo in children because the caregivers assisting children may report symptom scores that inaccurately assess symptom severity.
When comparing olopatadine-mometasone with mometasone NS in the GSP301-301 and GSP301-304 trials, there was inconsistency in the statistical significance and clinical meaningfulness of the efficacy end points across trials. The results for nasal or ocular symptom outcomes (i.e., rTNSS, iTNSS, rTOSS) were statistically significantly in favour of olopatadine-mometasone in the GSP301-304 trial, but not in the GSP301-301 trial. The evidence from these trials indicated with low to moderate certainty that the difference in improving nasal or ocular symptoms, although favouring olopatadine-mometasone, was not clinically meaningful. Across trials, there was inconsistency in the results for rTNSS and rTOSS between the GSP301-301 and GSP301-304 trials; however, only the inconsistency in rTOSS was determined to be serious, based on the relative position of the point estimates and the extent to which the 95% CIs overlapped, which might have introduced further uncertainty in the certainty of evidence. The LS mean difference in the 12-hour rTOSS between olopatadine-mometasone and mometasone NS was −0.19 points (95% CI, −0.49 to 0.11) in the GSP301-301 trial, with the point estimate lying near the no-effect line (i.e., between-group difference = 0) and with the 95% CI crossing the no-effect line. The LS mean difference in the 12-hour rTOSS between olopatadine-mometasone and mometasone NS was −0.35 points (95% CI, −0.66 to −0.03) in the GSP301-304 trial, with the point estimate lying near the MID (i.e., a between-group difference of > 0.5 points) and with the 95% CI excluding the no-effect line.
HRQoL was investigated in all 3 pivotal trials using the RQLQ(S) in the adolescent and adult population, and the PRQLQ in the child population, both of which are validated measurement instruments in these populations. The MID for within-group change in the Rhinoconjunctivitis Quality-of-Life Questionnaire has been reported for both the adolescent and adult population.52,53 However, there is no validated MID for between-group changes identified from the literature. A between-group difference of 0.5 was provided by the clinical experts as the MID for both the RQLQ(S) and PRQLQ. In general, the results of the RQLQ(S) in the adolescent and adult population between olopatadine-mometasone versus placebo and mometasone NS, as well as the results of the PRQLQ in the child population between olopatadine-mometasone versus placebo, favoured olopatadine-mometasone, based on the point estimate and 95% CI with moderate to high certainty. However, these results were not clinically meaningful (i.e., MID was crossed by the 95% CIs).
In the 3 pivotal trials, treatment with olopatadine-mometasone was administered for up to 17 days. According to the clinical experts consulted by the review team, patients in the real world are often given therapy for longer than the duration used in the trials. As such, the long-term efficacy of olopatadine-mometasone beyond 17 days remains unknown.
The sponsor-submitted ITCs included 1 NMA in adolescents and adults (aged 12 years and older) and 1 NMA in children (aged ≥ 6 to < 12 years) that evaluated the relative efficacy of olopatadine-mometasone compared with placebo and 2 drug classes, including intranasal corticosteroids and oral antihistamines. Echoing the evidence from the pivotal trials, olopatadine-mometasone was favoured over placebo in terms of improving the 12-hour rTNSS in the adolescent and adult population (mean or LS mean difference of −1.26 points; 95% CrI, −1.86 to −0.67) and in the child population (mean or LS mean difference of −1.21 points; 95% CrI, −1.86 to −0.56). In addition, the robustness of the results from the base-case analysis in the adolescent and adult population was supported by 2 sensitivity analyses that removed 2 trials with missing values for SEs or that included only 5 trials with LS mean change from baseline as an end point. In the base-case analyses of adolescents and adults, although the point estimates of mean or LS mean difference in the 12-hour rTNSS favoured olopatadine-mometasone over intranasal corticosteroids or oral antihistamines, the 95% CrIs of the mean or LS mean difference crossed the no-effect line (i.e., difference = 0), suggesting there was no difference between olopatadine-mometasone and these treatments. In the NMA for children with SAR, olopatadine-mometasone was favoured over the intranasal corticosteroids category (mean or LS mean difference of −0.94 points; 95% CrI, −1.63 to −0.26) in the 12-hour rTNSS. However, uncertainty existed in these NMA findings due to limitations of the NMAs, which is discussed in the critical appraisal section. For instance, relevant comparators were missing from the SLR protocol that might have resulted in bias due to missing evidence. Additionally, the fact that only loratadine was included in the oral antihistamine node in the adolescents and adults NMA, and only mometasone and ciclesonide were included in the intranasal corticosteroid node in the children NMA, raised concerns about whether only 1 or 2 individual therapies would properly represent the corresponding drug class in terms of efficacy.
The clinical experts also noted that Dymista antihistamine NS (i.e., azelastine hydrochloride) and corticosteroid NS (i.e., fluticasone propionate) were not included in the NMA conducted by the sponsor, yet were considered relevant comparators to olopatadine-mometasone. Dymista is approved by Health Canada for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in adults, adolescents, and children aged 6 years and older; however, it is not reimbursed by the public drug plans in Canada; thus, this remains a gap in the evidence.
Harms
In the GSP301-301, GSP301-304, and GSP301-305 trials, dysgeusia, which is a known side effect of olopatadine, was the most common TEAE across the olopatadine-mometasone groups, although frequencies in the 3 trials were low (3.3%, 3.7%, and 1.3%, respectively). Across all groups in the GSP301-301 trial, only 1 patient (0.3%) in the olopatadine-mometasone group had a TESAE, which was a spontaneous abortion. In the GSP301-304 trial, no patients in the olopatadine-mometasone group had TEAEs, while 1 patient in the mometasone NS group (0.3%) had 1 TESAE (i.e., peritonsillar abscess), and 1 patient in the placebo group (0.3%) had 3 TESAEs (including 1 instance each of osteomyelitis, syncope, and foot fracture). In the GSP301-305 trial, no patients in the olopatadine-mometasone group had a TEAE, and 1 patient (0.5%) in the placebo group had a TESAE. No patients in the olopatadine-mometasone group discontinued from the study due to TEAEs in either the GSP301-301 or GSP301-304 trials, while 4 patients (1.8%) in the olopatadine-mometasone group in the GSP301-305 trial withdrew due to TEAEs, including due to 1 instance each of conjunctivitis, acute otitis media, and sinusitis, and 1 instance of upper respiratory tract infection. No deaths were reported in any of the pivotal trials. Overall, the clinical experts consulted by the review team noted that olopatadine-mometasone was safe in adults, adolescents, and children with SAR, and the harms observed in patients treated with olopatadine-mometasone were expected and manageable.
Harms were not examined in the sponsor-submitted NMA; thus, there is a gap in the evidence with respect to the safety of olopatadine-mometasone relative to intranasal corticosteroids and oral antihistamines. However, this was not considered by the clinical experts consulted by the review team to be a critical gap, who stated there was no reason to suspect additional or new safety concerns with olopatadine-mometasone based on the safety data available for individual components and other intranasal corticosteroids with similar excipients.
Conclusion
The sponsor submitted 3 phase III, double-blind, RCTs: GSP301-301, GSP301-304, and GSP301-305. Both the GSP301-301 (N = 1,176) and GSP301-304 (N = 1,180) trials compared the efficacy of olopatadine-mometasone with placebo and the individual constituent monotherapies (i.e., olopatadine hydrochloride NS and mometasone NS) in adolescent and adult patients (aged 12 years and older) with SAR, while the GSP301-305 trial (N = 446) assessed the efficacy of olopatadine-mometasone relative to placebo in children (aged ≥ 6 to < 12 years) with SAR. Compared with placebo, the evidence from the 3 pivotal trials showed added clinical benefits of olopatadine-mometasone in reducing nasal and ocular symptoms. Results from the GSP301-301 and GSP301-304 trials suggested with moderate to high certainty that olopatadine-mometasone, compared with placebo, results in an improvement in the 12-hour rTNSS, 12-hour iTNSS, and 12-hour rTOSS in adolescent and adult patients with SAR. Evidence from the GSP301-305 trial also suggested improvements in rTNSS and iTNSS with olopatadine-mometasone compared with placebo with moderate certainty, but not for ocular symptoms. Compared with mometasone NS in adolescent and adult patients, although the results for nasal and ocular symptoms favoured olopatadine-mometasone, they were not clinically meaningful. Olopatadine-mometasone was considered safe in adolescents, adults, and children with SAR because the harms observed in patients treated with olopatadine-mometasone were expected and manageable, given the known safety profiles of the individual excipients.
The indirect evidence submitted by the sponsor included 1 NMA in adolescent and adult patients with SAR and 1 NMA in children with SAR. The results of the NMAs were consistent with the pivotal trial, favouring olopatadine-mometasone over placebo in both analysis populations. In the NMA in children, olopatadine-mometasone was favoured over intranasal corticosteroids, but there was no difference between these treatments in the adolescents and adults NMA. There was also no difference between olopatadine-mometasone and oral antihistamines in the adolescents and adults NMA. Overall, the findings from the 2 NMAs were uncertain due to limitations surrounding the comparators considered and whether there was appropriate representation in the comparator drug classes in terms of efficacy.
References
1.Bausch Health, Canada Inc. Sponsor’s clinical summary [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Olopatadine hydrochloride and mometasone furoate (Ryaltris), 665 µg olopatadine hydrochloride and 25 µg mometasone furoate per delivered dose, nasal spray. June 21, 2024.
2.Bausch Health, Canada Inc. Ryaltris (olopatadine hydrochloride and mometasone furoate nasal spray suspension): 665 µg olopatadine hydrochloride and 25 µg mometasone furoate (as monohydrate) per delivered dose, nasal spray [product monograph]. September 21, 2022.
3.Meadows A, Kaambwa B, Novielli N, et al. A systematic review and economic evaluation of subcutaneous and sublingual allergen immunotherapy in adults and children with seasonal allergic rhinitis. Health Technol Assess. 2013;17(27):vi, xi-xiv, 1-322. doi:10.3310/hta17270
4.Peden D. Post TW, ed. An overview of rhinitis. UpToDate; 2023. Accessed July 19, 2024. https://www.uptodate.com/
5.Al-Digheari A, Mahboub B, Tarraf H, et al. The clinical burden of allergic rhinitis in five Middle Eastern countries: results of the SNAPSHOT program. Allergy Asthma Clin Immunol. 2018;14:63. doi:10.1186/s13223-018-0298-x PubMed
6.Nur Husna SM, Tan HT, Md Shukri N, Mohd Ashari NS, Wong KK. Allergic Rhinitis: A Clinical and Pathophysiological Overview. Front Med (Lausanne). 2022;9:874114. doi:10.3389/fmed.2022.874114 PubMed
7.Keith PK, Desrosiers M, Waserman S, Schellenberg RR. Burden of Illness of Allergic Rhinitis in Canada. J Allergy Clin Immunol. 2007;119(1):S227. doi:10.1016/j.jaci.2006.12.261
8.Canuel M, Lebel G. Epidemiology of allergic rhinitis in Quebec: from a 2008 population-based survey. Chronic Dis Inj Can. 2014;34(2-3):163-8. doi:10.24095/hpcdp.34.2/3.11 PubMed
9.Keith PK, Desrosiers M, Laister T, Schellenberg RR, Waserman S. The burden of allergic rhinitis (AR) in Canada: perspectives of physicians and patients. Allergy Asthma Clin Immunol. 2012;8(1):7. doi:10.1186/1710-1492-8-7 PubMed
10.Li J, Wang H, Chen Y, Zheng J, Wong GW, Zhong N. House dust mite sensitization is the main risk factor for the increase in prevalence of wheeze in 13- to 14-year-old schoolchildren in Guangzhou city, China. Clin Exp Allergy. 2013;43(10):1171-9. doi:10.1111/cea.12157 PubMed
11.Rak S, Yang WH, Pedersen MR, Durham SR. Once-daily sublingual allergen-specific immunotherapy improves quality of life in patients with grass pollen-induced allergic rhinoconjunctivitis: a double-blind, randomised study. Qual Life Res. 2007;16(2):191-201. doi:10.1007/s11136-006-9110-3 PubMed
12.Skoner DP. Allergic rhinitis: definition, epidemiology, pathophysiology, detection, and diagnosis. J Allergy Clin Immunol. 2001;108(1 Suppl):S2-8. doi:10.1067/mai.2001.115569 PubMed
13.Dykewicz MS, Wallace DV, Amrol DJ, et al. Rhinitis 2020: A practice parameter update. J Allergy Clin Immunol. 2020;146(4):721-767. doi:10.1016/j.jaci.2020.07.007 PubMed
14.Nathan RA. The burden of allergic rhinitis. Allergy Asthma Proc. 2007;28(1):3-9. doi:10.2500/aap.2007.28.2934 PubMed
15.Small P, Keith PK, Kim H. Allergic rhinitis. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):51. doi:10.1186/s13223-018-0280-7 PubMed
16.Hashimoto T, Ishii N, Hamada T, et al. Effects of olopatadine hydrochloride, a histamine h(1) receptor antagonist, on histamine-induced skin responses. Dermatol Res Pract. 2010;2010:638051. doi:10.1155/2010/638051 PubMed
17.Prakash A, Benfield P. Topical mometasone. A review of its pharmacological properties and therapeutic use in the treatment of dermatological disorders. Drugs. 1998;55(1):145-163. doi:10.2165/00003495-199855010-00009 PubMed
18.Derendorf H, Meltzer EO. Molecular and clinical pharmacology of intranasal corticosteroids: clinical and therapeutic implications. Allergy. 2008;63(10):1292-1300. doi:10.1111/j.1398-9995.2008.01750.x PubMed
19.Linton S, Hossenbaccus L, Ellis AK. Evidence-based use of antihistamines for treatment of allergic conditions. Ann Allergy Asthma Immunol. 2023;131(4):412-420. doi:10.1016/j.anai.2023.07.019 PubMed
20.Bausch Health, Canada Inc. Clinical Study Report: GSP 301-301. A double-blind, randomized, parallel-group, comparative study to evaluate the efficacy, safety and tolerability of a fixed dose combination GSP 301 nasal spray compared with placebo nasal spray and individual monotherapy formulations of olopatadine hydrochloride nasal spray and mometasone furoate nasal spray in subjects (aged 12 years and older) with seasonal allergic rhinitis (SAR) [internal sponsor’s report]. October 26, 2017.
21.Bausch Health, Canada Inc. Clinical Study Report: GSP 301-304. A double-blind, randomized, parallelgroup seasonal allergic rhinitis (sar) study to evaluate the efficacy, safety and tolerability of GSP 301 nasal spray compared with placebo nasal spray and individual monotherapy formulations (olopatadine hydrochloride nasal spray and mometasone furoate nasal spray) in adult and adolescent subjects (12 years of age and older) [internal sponsor’s report]. January 5, 2018.
22.Bausch Health, Canada Inc. Bausch Health, Canada Inc. response to June 26, 2024 CDA-AMC request for additional information regarding Olopatadine hydrochloride and mometasone furoate CDA-AMC review: Requests for study protocols and statistical analysis plans [internal additional sponsor’s information]. August 2, 2024.
23.Bausch Health, Canada Inc. Clinical Study Report: GSP 301-305. A double-blind, randomized, parallel group study to evaluate the efficacy, safety and tolerability of fixed dose combination GSP 301 nasal spray compared with placebo nasal spray in pediatric subjects (aged 6 to under 12 years) with seasonal allergic rhinitis (SAR) [internal sponsor’s report]. May 6, 2019.
24.Hill DA, Grundmeier RW, Ram G, Spergel JM. The epidemiologic characteristics of healthcare provider-diagnosed eczema, asthma, allergic rhinitis, and food allergy in children: a retrospective cohort study. BMC Pediatr. 2016;16:133. doi:10.1186/s12887-016-0673-z PubMed
25.Frohlich M, Pinart M, Keller T, et al. Is there a sex-shift in prevalence of allergic rhinitis and comorbid asthma from childhood to adulthood? A meta-analysis. Clin Transl Allergy. 2017;7(44):44. doi:10.1186/s13601-017-0176-5 PubMed
26.Pinart M, Keller T, Reich A. Sex-Related Allergic Rhinitis Prevalence Switch from Childhood to Adulthood: A Systematic Review and Meta-Analysis. Int Arch Allergy Immunol. 2017;172(4):224-235. doi:10.1159/000464324 PubMed
27.Statistics Canada. Census of Population [sponsor-supplied reference]. Accessed February 9, 2023. https://www12.statcan.gc.ca/census-recensement/index-eng.cfm
28.Fairchild CJ, Meltzer EO, Roland PS, Wells D, Drake M, Wall GM. Comprehensive report of the efficacy, safety, quality of life, and work impact of Olopatadine 0.6% and Olopatadine 0.4% treatment in patients with seasonal allergic rhinitis. Allergy Asthma Proc. 2007;28(6):716-23. doi:10.2500/aap.2007.28.3062 PubMed
29.Barton BE, Jakway JP, Smith SR, Siegel MI. Cytokine inhibition by a novel steroid, mometasone furoate. Immunopharmacol Immunotoxicol. 1991;13(3):251-61. doi:10.3109/08923979109019704 PubMed
30.Brannan M, Seiberling M, Cutler D, Cuss F, Affrime M. Lack of systemic activity with intranasal mometasone furoate. J Allergy Clin Immunol. 1996;97(1):198-198. doi:10.1016/S0091-6749(96)80280-6
31.Glenmark Specialty SA. Prescribing information: Ryaltris (olopatadine hydrochloride and mometasone furoate) nasal spray. U.S. Food and Drug Administration (FDA); 2022. Accessed July 19, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/211746s000lbl.pdf
32.Center for Drug Evaluation Research. NDA Approval Letter: Ryaltris (olopatadine hydrochloride and mometasone furoate) nasal spray. Company: Glenmark Specialty SA. Application No.: 211746. Approval date: 01/13/2022 (FDA approval package). U.S. Food and Drug Administration (FDA); 2022. Accessed July 19, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/211746Orig1s000ltr.pdf
33.McNeil Consumer Healthcare, division of Johnson & Johnson Inc. Reactine (cetirizine hydrochloride): tablets, 5 mg, 10 mg, oral; Reactine® Fast Melt® Junior (cetirizine hydrochloride): orally disintegrating tablets, 10 mg, oral; Reactine® Rapid Dissolve (cetirizine hydrochloride): orally disintegrating tablets, 10 mg, oral; Reactine® Allergy Liquid Gels (cetirizine hydrochloride): capsules, 10 mg, oral; Reactine® (cetirizine hydrochloride): syrup 5 mg/5 ml, oral; Reactine® Allergy (cetirizine hydrochloride): syrup 5 mg/5 ml, oral [product monograph]. December 15, 2023. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00073921.PDF
34.Pharmascience Inc. Desloratadine Allergy Control (desloratadine): tablet, 5mg, oral [product monograph]. September 12, 2023. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00072647.PDF
35.Sanofi Consumer Health Inc. Allegra® 12 Hour (fexofenadine hydrochloride): tablets, 60 mg, oral; Allegra® 24 Hour (fexofenadine hydrochloride): tablets, 120 mg, oral; Allegra® Hives (fexofenadine hydrochloride): tablets, 60 mg, oral [product monograph]. October 5, 2021. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00063260.PDF
36.Apotex Inc. Loratadine soft gel capsules (loratadine): capsules, 10 mg [product monograph]. May 31, 2019. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00051857.PDF
37.Apotex Inc. Apo-beclomethasone nasal spray (beclomethasone dipropionate aqueous suspension): 50 mcg / ACT (0.05% w / w) [product monograph]. March 11, 2021. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00060436.PDF
38.Mylan Pharmaceuticals ULC. Mylan-budesonide AQ (budesonide aqueous nasal spray): 100 mcg per metered dose [product monograph]. May 5, 2020. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00065699.PDF
39.Innomar Strategies Inc. Omnaris (ciclesonide nasal spray): 50 mcg/metered spray [product monograph]. February 11, 2021. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00059998.PDF
40.Apotex Inc. Apo-mometasone (mometasone furoate aqueous nasal spray): 50 mcg/metered spray [product monograph]. November 2, 2022. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00068391.PDF
41.Apotex Inc. 24hr nasal allergy relief (fluticasone propionate aqueous nasal spray): 50 mcg / metered spray [product monograph]. January 8, 2020. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00054939.PDF
42.Apotex Inc. Apo-triamcinolone AQ (triamcinolone acetonide aqueous nasal spray): aqueous nasal spray, 55 mcg/metered spray, nasal [product monograph]. May 8, 2023. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00070872.PDF
43.GlaxoSmithKline Inc. Avamys (fluticasone furoate nasal spray): 27.5 ug/metered spray [product monograph]. December 2, 2021. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00063816.PDF
44.Aralez Pharmaceuticals Canada Inc. Blexten (bilastine): orodispersible tablets 10 mg; tablets 20 mg; oral solution 2.5 mg/mL [product monograph]. August 10, 2021. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00062419.PDF
45.Medexus Inc. Rupall (Rupatadine): tablet, 10 mg rupatadine (as rupatadine fumarate), oral; oral solution, 1 mg/mL rupatadine (as rupatadine fumarate), oral [product monograph]. January 26, 2023. Accessed March 28, 2025. https://pdf.hres.ca/dpd_pm/00069453.PDF
46.Hampel FC, Pedinoff AJ, Jacobs RL, Caracta CF, Tantry SK. Olopatadine-mometasone combination nasal spray: Evaluation of efficacy and safety in patients with seasonal allergic rhinitis. Allergy Asthma Proc. 2019;40(4):261-272. doi:10.2500/aap.2019.40.4223 PubMed
47.Gross GN, Berman G, Amar NJ, Caracta CF, Tantry SK. Efficacy and safety of olopatadine-mometasone combination nasal spray for the treatment of seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 2019;122(6):630-638 e3. doi:10.1016/j.anai.2019.03.017 PubMed
48.Prenner BM, Amar NJ, Hampel FC, Jr., Caracta CF, Wu W. Efficacy and safety of GSP301 nasal spray in children aged 6 to 11 years with seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 2022;129(5):618-626 e2. doi:10.1016/j.anai.2022.07.029 PubMed
49.Juniper EF, Guyatt GH. Development and testing of a new measure of health status for clinical trials in rhinoconjunctivitis. Clin Exp Allergy. 1991;21(1):77-83. doi:10.1111/j.1365-2222.1991.tb00807.x PubMed
50.Juniper EF, Riis B, Juniper BA. Development and validation of an electronic version of the Rhinoconjunctivitis Quality of Life Questionnaire. Allergy. 2007;62(9):1091-3. doi:10.1111/j.1398-9995.2007.01370.x PubMed
51.Juniper EF, Thompson AK, Ferrie PJ, Roberts JN. Validation of the standardized version of the Rhinoconjunctivitis Quality of Life Questionnaire. J Allergy Clin Immunol. 1999;104(2 Pt 1):364-9. doi:10.1016/s0091-6749(99)70380-5 PubMed
52.Juniper EF, Guyatt GH, Griffith LE, Ferrie PJ. Interpretation of rhinoconjunctivitis quality of life questionnaire data. J Allergy Clin Immunol. 1996;98(4):843-5. doi:10.1016/s0091-6749(96)70135-5 PubMed
53.Juniper EF, Guyatt GH, Dolovich J. Assessment of quality of life in adolescents with allergic rhinoconjunctivitis: development and testing of a questionnaire for clinical trials. J Allergy Clin Immunol. 1994;93(2):413-23. doi:10.1016/0091-6749(94)90349-2 PubMed
54.Juniper EF, Howland WC, Roberts NB, Thompson AK, King DR. Measuring quality of life in children with rhinoconjunctivitis. J Allergy Clin Immunol. 1998;101(2 Pt 1):163-70. doi:10.1016/s0091-6749(98)70380-x PubMed
55.Danell CS, Bergström A, Wahlgren CF, Hallner E, Böhme M, Kull I. Parents and school children reported symptoms and treatment of allergic disease differently. J Clin Epidemiol. 2013;66(7):783-9. doi:10.1016/j.jclinepi.2013.02.006 PubMed
56.Berger W, Meltzer EO, Amar N, et al. Efficacy of MP-AzeFlu in children with seasonal allergic rhinitis: Importance of paediatric symptom assessment. Pediatr Allergy Immunol. 2016;27(2):126-33. doi:10.1111/pai.12540 PubMed
57.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
58.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
59.Segall N, Prenner B, Lumry W, Caracta CF, Tantry SK. Long-term safety and efficacy of olopatadine-mometasone combination nasal spray in patients with perennial allergic rhinitis. Allergy Asthma Proc. 2019;40(5):301-310. doi:10.2500/aap.2019.40.4233 PubMed
60.Glenmark Specialty S.A. NCT02709538: Long Term Safety and Efficacy of Fixed Dose Combination GSP 301 Nasal Spray (NS) in the Treatment of Perennial Allergic Rhinitis (PAR) (GSP 301-303). ClinicalTrials.gov; 2020. Accessed July 19, 2024. https://clinicaltrials.gov/study/NCT02709538
61.Bausch Health, Canada Inc. Clinical Study Report: GSP 301-303. Double-blind, randomized, parallelgroup study to evaluate long-term safety, tolerability, and efficacy of a fixed dose combination GSP 301 nasal spray compared with two placebo nasal spray formulations in subjects (aged 12 years and older) with perennial allergic rhinitis (PAR) [internal sponsor’s report]. March 23, 2018.
62.Bausch Health, Canada Inc. Indirect treatment comparison for Ryaltris for the treatment of moderate to severe seasonal allergic rhinitis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Olopatadine hydrochloride and mometasone furoate nasal spray suspension (Ryaltris): 665 µg olopatadine hydrochloride and 25 µg mometasone furoate (as monohydrate) per delivered dose; nasal spray. June 21, 2024.
63.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. doi:10.1136/bmj.l4898 PubMed
64.Fedorov S. GetData graph digitizer [sponsor-supplied reference]. 2020. https://getdata-graph-digitizer.com/index.php
65.Dias S, Welton NJ, Sutton AJ, Ades AE. A generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials (NICE Decision Support Unit Technical Support Documents) [sponsor-supplied reference]. National Institute for Health and Care Excellence (NICE); 2014. Accessed 2014. https://www.ncbi.nlm.nih.gov/books/NBK310366/
66.Gabry J, Simpson D, Vehtari A, Betancourt M, Gelman A. Visualization in bayesian workflow. Journal of the Royal Statistical Society Series A: Statistics in Society. 2019;182(2):389-402. doi:10.1111/rssa.12378
67.Anolik R, Mometasone Furoate Nasal Spray With Loratadine Study Group. Clinical benefits of combination treatment with mometasone furoate nasal spray and loratadine vs monotherapy with mometasone furoate in the treatment of seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 2008;100(3):264-71. doi:10.1016/S1081-1206(10)60452-8 PubMed
68.Actavis Inc. NCT01850823: Clinical Equivalence Study of Mometasone Nasal Spray [sponsor-supplied reference]. ClinicalTrials.gov; 2020. Accessed 2020. https://clinicaltrials.gov/ct2/show/NCT01850823
69.Teva Branded Pharmaceutical Products R&D, Inc. NCT01024608: A Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Phase 3 Study to Assess the Efficacy and Safety of Beclomethasone Dipropionate Hydrofluoroalkane (BDP HFA) Nasal Aerosol in Subjects 12 Years of Age and Older With SAR [sponsor-supplied reference]. ClinicalTrials.gov; 2021. Accessed December 1, 2021. https://clinicaltrials.gov/study/NCT01024608
70.Organon and Co. NCT03882047: Efficacy and Safety of Mometasone Furoate Aqueous Nasal Spray vs Placebo and Flonase® (Fluticasone Propionate) in Seasonal Allergic Rhinitis Patients (I94-001) [sponsor-supplied reference]. ClinicalTrials.gov; 2022. Accessed February 7, 2022. https://clinicaltrials.gov/study/NCT03882047
71.Organon and Co. NCT03855189: Safety and Efficacy of SCH 32088 vs Beclomethasone Dipropionate (Vancenase AQ) and Placebo in Seasonal Allergic Rhinitis [sponsor-supplied reference]. ClinicalTrials.gov; 2022. Accessed February 7, 2022. https://clinicaltrials.gov/study/NCT03855189
72.Teva Pharmaceuticals USA. NCT02246920: A Double-Blind, Randomized, Placebo-Controlled, Parallel Group, Multi-Site Study to Compare the Therapeutic Equivalence of Fluticasone Propionate Nasal Spray, 50 mcg With Flonase® Nasal Spray in the Relief of the Signs and Symptoms of Seasonal Allergic Rhinitis [sponsor-supplied reference]. ClinicalTrials.gov; 2023. Accessed May 11, 2023. https://clinicaltrials.gov/study/NCT02246920
73.Organon and Co. NCT00732381: Placebo-Controlled Study of Mometasone Furoate Nasal Spray (MFNS) 200 mcg QD in the Relief of Nasal Congestion Associated With Seasonal Allergic Rhinitis (SAR) [sponsor-supplied reference]. ClinicalTrials.gov; 2024. Accessed May 8, 2024. https://clinicaltrials.gov/study/NCT00732381
74.Gawchik S, Goldstein S, Prenner B, John A. Relief of cough and nasal symptoms associated with allergic rhinitis by mometasone furoate nasal spray. Ann Allergy Asthma Immunol. 2003;90(4):416-21. doi:10.1016/S1081-1206(10)61826-1 PubMed
75.Meltzer EO, Shekar T, Teper AA. Mometasone furoate nasal spray for moderate-to-severe nasal congestion in subjects with seasonal allergic rhinitis. Allergy Asthma Proc. 2011;32(2):159-67. doi:10.2500/aap.2011.32.3424 PubMed
76.Prenner BM, Lanier BQ, Bernstein DI, Shekar T, Teper A. Mometasone furoate nasal spray reduces the ocular symptoms of seasonal allergic rhinitis. J Allergy Clin Immunol. 2010;125(6):1247-1253 e5. doi:10.1016/j.jaci.2010.03.004 PubMed
77.Ratner PH, Wingertzahn MA, van Bavel JH, Hampel F, Darken PF, Shah T. Efficacy and safety of ciclesonide nasal spray for the treatment of seasonal allergic rhinitis. J Allergy Clin Immunol. 2006;118(5):1142-8. doi:10.1016/j.jaci.2006.07.050 PubMed
78.Berger WE, Nayak A, Lanier BQ, et al. Efficacy and safety of once-daily ciclesonide nasal spray in children with allergic rhinitis. Pediatr Asthma Allergy Immunol. 2008;21(2):73-82. doi:10.1089/pai.2007.0022.73
79.Sumitomo Pharma America, Inc. NCT01458275: A 2-Week Randomized, Double-Blind, Placebo-Controlled, Parallel Group, Safety and Efficacy Study of Ciclesonide Nasal Aerosol in Subjects 6 to 11 Years With Seasonal Allergic Rhinitis [sponsor-supplied reference]. ClinicalTrials.gov; 2014. Accessed April 16, 2014. https://clinicaltrials.gov/study/NCT01458275
80.Organon and Co. NCT03879772: Dose-Ranging Study of Mometasone Furoate Nasal Spray (SCH 32088) in the Treatment of Children (Ages 6-11) With Seasonal Allergic Rhinitis [sponsor-supplied reference]. ClinicalTrials.gov; 2022. Accessed February 7, 2022. https://clinicaltrials.gov/study/NCT03879772
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AH
antihistamine
AR
allergic rhinitis
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
HRQoL
health-related quality of life
INCS
intranasal corticosteroids
NMA
network meta-analysis
olopatadine-mometasone
olopatadine hydrochloride and mometasone furoate nasal spray
QALY
quality-adjusted life-year
rTNSS
reflective Total Nasal Symptom Score
rTOSS
reflective Total Ocular Symptom Score
SAR
seasonal allergic rhinitis
SLR
systematic literature review
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Olopatadine hydrochloride and mometasone furoate nasal spray suspension (Ryaltris): 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate per delivered dose |
Indication | For the symptomatic treatment of moderate to severe seasonal allergic rhinitis (SAR) and associated ocular symptoms in adults, adolescents, and children aged 6 years or older |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 21, 2022 |
Reimbursement request | As per indication |
Sponsor | Bausch Health, Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Patients aged 6 years and older experiencing an episode of moderate to severe SAR |
Treatment | Olopatadine-mometasone, daily use during an episode of SAR |
Dose regimen |
|
Submitted price | Olopatadine-mometasone: $56.11 per bottle (240 metered sprays) |
Submitted treatment cost |
|
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs |
Time horizon | 28 days |
Key data sources | Efficacy of olopatadine-mometasone informed by the GSP301-301 and GSP301-304 trials for adolescents and adults (compared with placebo and mometasone), and by the GSP301-305 trial for children (compared with placebo). The efficacy of oral AHs and INCSs were informed by sponsor-submitted NMAs. |
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
AH = antihistamine; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; INCS = intranasal corticosteroid; NMA = network meta-analysis; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; QALY = quality-adjusted life-year; SAR = seasonal allergic rhinitis.
aIn the economic model, the sponsor considered INCSs to be represented by mometasone furoate, beclomethasone dipropionate, budesonide, ciclesonide, and fluticasone propionate. Costing for this group was based on the least costly generic drug (mometasone furoate). Efficacy for oral AHs from the sponsor’s NMA for children was represented by mometasone and ciclesonide, with the assumption that efficacy would be the same for all drugs in the class.
bIn the economic model, the sponsor considered oral AHs to be represented by cetirizine, desloratadine, fexofenadine, and loratadine. Costing for this group was based on the least costly generic drug (cetirizine). Efficacy for oral AHs from the sponsor’s NMA was represented by loratadine, with the assumption that efficacy would be the same for all drugs in the class.
Conclusions
Based on the Canada’s Drug Agency (CDA-AMC) Clinical Review of the GSP301-301, GSP301-304, and GSP301-305 trials, the use of olopatadine hydrochloride and mometasone furoate nasal spray (olopatadine-mometasone) by adolescents and adults (aged 12 years and older) with moderate or severe seasonal allergic rhinitis (SAR) over a 14-day treatment course improves nasal and ocular symptoms, based on the 12-hour reflective Total Nasal Symptom Score (rTNSS) and reflective Total Ocular Symptom Score (rTOSS) outcomes compared with placebo. However, compared with mometasone furoate nasal spray, olopatadine-mometasone did not result in a clinically meaningful improvement in symptoms in this age group. Among children (aged 6 to 11 years), the trial evidence suggests that olopatadine-mometasone improves nasal symptoms compared with placebo over 14 days of treatment. In both age groups, olopatadine-mometasone had a nonstatistically significant impact on health-related quality of life (HRQoL) after 14 days of treatment.
There have been no head-to-head trials of olopatadine-mometasone versus oral antihistamines (AHs) or intranasal corticosteroids (INCSs) other than mometasone furoate nasal spray in the adolescent and adult subgroup. The indirect evidence submitted by the sponsor suggests there may be no meaningful difference in nasal symptoms between olopatadine-mometasone and oral AHs or INCSs in adults. Among children, the sponsor’s indirect evidence suggests that olopatadine-mometasone may improve nasal symptoms compared with INCSs; however, no evidence comparing olopatadine-mometasone with oral AHs was provided. The CDA-AMC Clinical Review noted that the findings from the sponsor’s network meta-analysis (NMA) should be viewed as uncertain, owing to the identified limitations. Ocular symptoms, HRQoL, and safety were not assessed in the sponsor’s NMA.
CDA-AMC was unable to address several key limitations with the sponsor’s economic submission, including uncertainty in the comparative clinical data, as well as methodological and conceptual limitations related to the model structure. These limitations prevented CDA-AMC from deriving a base-case estimate of the cost-effectiveness of olopatadine-mometasone for the treatment of moderate to severe SAR and associated ocular symptoms. The sponsor’s analysis suggests that the drug acquisition cost of olopatadine-mometasone will be offset by savings in health care costs; however, whether these predicted savings will be realized in clinical practice is highly uncertain, owing to the lack of clinical data to support reduced health care resource use with olopatadine-mometasone. Overall, there is insufficient clinical and economic evidence to suggest that olopatadine-mometasone should be priced higher than currently reimbursed treatments for moderate to severe SAR and associated ocular symptoms.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
Patient group input was provided by Asthma Canada and Allergy Quebec. Asthma Canada gathered feedback through its 2024 Annual Asthma Survey, which involved 1,407 patients and caregivers. Among these, 37% reported having allergic rhinitis (AR) as a comorbidity with asthma, and 63% had experienced SAR. They also conducted 2 one-on-one interviews with AR patients selected from survey respondents. Allergy Quebec did not perform any data collection from patients. Both organizations indicated that AR is associated with symptoms, including runny or itchy nose, nasal congestion, itchy or swollen eyes, headaches, sinus pressure, and fatigue, which can adversely affect daily life and quality of life. Respondents noted that the physical symptoms of AR are the most challenging aspect of their condition and emphasized the need for better treatments to reduce symptoms, including treatments that address rhinorrhea and do not trigger asthma flare-ups. Many respondents noted that their current treatments did not effectively control their allergic symptoms. Interviewed patients expressed concerns about treatment efficacy and side effects, including drowsiness and nasal dryness, and issues related to cost and accessibility, including insurance coverage and local pharmacy availability.
No clinician group input was received for this review.
The CDA-AMC participating drug plans sought clarification on the definition of the seasonal aspect of SAR, as some allergic triggers may be seasonal in some climates but perennial in others, with symptoms throughout most of the year. Additionally, the drug plans questioned whether diagnostic testing (e.g., skin prick or serum-specific immunoglobulin E test) is routinely done in clinical practice for SAR. The drug plans noted that the only other AH nasal spray in Canada (combination AH and corticosteroid product azelastine hydrochloride and fluticasone propionate [Dymista]) received a Do Not List recommendation from the Canadian Drug Expert Committee in 2015 and did not undergo negotiations with the pan-Canadian Pharmaceutical Alliance. Plans additionally noted the presence of confidential negotiated prices for comparators.
Several of these concerns were addressed in the sponsor’s model:
Nasal symptoms associated with SAR were included in the sponsor’s model. Other symptoms noted to be important by patients (e.g., itchy and swollen eyes, headaches, fatigue) were not included in the model.
In addition, CDA-AMC addressed some of these concerns, as follows:
CDA-AMC compared the acquisition costs of olopatadine-mometasone and relevant comparators.
CDA-AMC was unable to address the following concerns raised from the input:
CDA-AMC was unable to assess the cost-effectiveness of olopatadine-mometasone for the treatment of SAR-associated ocular symptoms and, hence, for the full Health Canada indication.
The CDA-AMC cost comparison is based on publicly available prices and does not consider the presence of confidential negotiated prices.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing costs and outcomes for olopatadine-mometasone compared with oral AHs, INCSs, and placebo.1 The sponsor assumed intraclass clinical equivalency between products from the same class. The modelled population was patients aged 6 years and older experiencing an episode of moderate to severe SAR, with separate analyses submitted for patients aged 6 to 11 years (hereafter referred to as children), and aged 12 years and older (hereafter referred to as adolescents and adults). The modelled population was aligned with the olopatadine-mometasone pivotal trials, which enrolled patients with a morning congestion score of at least 2 and an rTNSS score of at least 6 or 8 (≥ 6 in the GSP301-301 and GSP301-304 trials and ≥ 8 in the GSP301-305 trial). The modelled population is aligned with the Health Canada indication for olopatadine-mometasone.
Olopatadine-mometasone is available as a nasal spray containing a fixed dose of 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate in each spray (bottle of 240 metered sprays). The recommended dosage of olopatadine-mometasone is 2 sprays in each nostril twice daily for adolescents and adults and 1 spray in each nostril twice daily for children.2 At the submitted price of $56.11 per bottle, the daily per-patient cost of olopatadine-mometasone was estimated by the sponsor to be $0.94 for children and $1.87 for adolescents and adults.
The sponsor’s model predicts clinical outcomes in terms of quality-adjusted life-days, which the sponsor converted to quality-adjusted life-years (QALYs). The economic evaluation was conducted over a 28-day time horizon from the perspective of a Canadian public health care payer. No discounting was applied.
Model Structure
The sponsor’s submitted model was a decision tree with 3 health states: SAR, recovered from SAR, and extended SAR (Figure 1). All patients entered the model in the SAR health state and received olopatadine-mometasone, INCSs, or oral AHs (in adolescents and adults only). Patients who had a response to treatment within the first 2 weeks were assumed to have moved to the recovered from SAR state and to maintain that response for the remainder of the 28-day horizon. Patients who did not have a treatment response by day 14 were assumed to have moved to the extended SAR health state and to receive an additional 14 days of treatment. By day 28, all patients were expected to have responded. The sponsor assumed that no patients would be at risk of death due to SAR.
Model Inputs
Model populations were obtained from the GSP301-301 and GSP301-304 trials for the adolescent and adult subgroup (patients aged 12 years and older with a documented clinical history of SAR) and the GSP301-305 trial for the children subgroup (patients aged 6 to 11 years with a clinical history of SAR, diagnosed with a positive skin prick test within 12 months before screening). In the GSP301-301 and GSP301-304 trials, the mean age of patients was 39.3 and 39.6 years, respectively, and approximately 63% to 65% were female. In GSP301-305, the mean age of the study population was 8.7 years, and 56% of the patients in the olopatadine-mometasone group were male. Clinical efficacy inputs estimated treatment by the sponsor as follows: first, the sponsor used the pivotal trial data to determine the proportion of patients who had a 50% or greater reduction in rTNSS for olopatadine-mometasone and INCS comparators.3 Second, the sponsor used the results from the submitted NMA to estimate the relative risk (using the change from baseline for the 12-hour rTNSS) of responding to the therapies included in the model for both subgroups. Because the sponsor identified no relevant data for oral AHs in the children subgroup, the model did not include oral AHs for this subgroup.4
Adverse events (AEs) were reported by at least 1% of patients, based on pooled estimates from the GSP301-301 and GSP301-304 trials for olopatadine-mometasone and INCSs. The model considered only AEs that were reported in more than 1% of patients and AEs with a high impact on costs and outcomes. AEs for oral AHs were obtained from Kuna et al. (2009), based on a threshold of at least 1%.5
Health state utility values were obtained from the literature. The utility for the SAR health state was obtained from Retzler (2018).6 The health state utility value for the recovered from SAR state was obtained from Pollock (2023) and Ara (2011).7,8 The sponsor estimated the utility value for the extended SAR state by taking the average of the utility values for the SAR and the recovered from SAR states.
The costs in the model included those related to drug acquisition, resource use, diagnostic testing, and AEs. Drug acquisition costs were calculated by the sponsor using the submitted price for olopatadine-mometasone and IQVIA DeltaPA database prices for comparators.9 The sponsor assumed that adolescents and adults would receive 2 sprays of olopatadine-mometasone in each nostril twice daily and that children would receive 1 spray in each nostril twice daily, as per the olopatadine-mometasone product monograph. Resource use costs included those associated with visits to general physicians, specialists, and the emergency department for SAR, as well as costs related to inpatient stays for SAR. Diagnostic testing costs included skin prick tests, allergen testing, chest radiographs, and electrocardiograms. Costs for resource use and diagnostic testing were obtained from the Ontario Schedule of Benefits and the literature.10,11 Costs for the management of AEs were obtained from the Ontario Case Costing Initiative (2018).12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations), and the probabilistic results were similar to the deterministic results. The probabilistic findings are presented subsequently. The submitted analyses were based on the publicly available pricing for olopatadine hydrochloride and mometasone furoate monohydrate and other comparators. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
Among adolescents and adults, olopatadine-mometasone was associated with a total treatment cost of $274 over the 28-day horizon. Of this, 11% ($29) was associated with drug acquisition and 89% ($245) was associated with health care resource use. Olopatadine-mometasone was predicted by the sponsor to be less costly (incremental savings = $20 to $27) compared with INCSs and AHs, owing to reduced health care resource use (reduced health care costs = $45 to $46), despite increased drug acquisition costs (increased acquisition costs = $20 to $25). The sponsor’s model additionally predicted that olopatadine-mometasone would be more effective (incremental gain in QALYs = 0.00031 to 0.00032) compared with INCSs and oral AHs. At a willingness-to-pay threshold of $50,000 per QALY gained, the probability of olopatadine-mometasone being cost-effective was 62%.
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Adolescents and Adults)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Olopatadine-mometasone | 274 | 0.06812 | Reference |
Dominated treatments | |||
Oral antihistamines | 294 | 0.06781 | Dominated by olopatadine-mometasone |
Intranasal corticosteroids | 301 | 0.06780 | Dominated by oral antihistamines |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Placebo was included as a comparator in the sponsor’s submission to align with trial data. Results that align with trial data are presented in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.1
Among children, olopatadine-mometasone was associated with a total treatment cost of $244 over the 28-day time horizon. Of this, 6% ($14) was associated with drug acquisition and 93% ($228) was associated with health care resource use. Olopatadine-mometasone was predicted by the sponsor to be less costly (incremental savings = $47) compared with INCSs, owing to reduced health care resource use (reduced health care costs = $59), despite increased drug acquisition costs (increased acquisition costs = $12). The sponsor’s model additionally predicted that olopatadine-mometasone will be more effective (incremental gain in QALYs: 0.0004) compared with INCSs.
Table 4: Summary of the Sponsor’s Economic Evaluation Results (Children)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Olopatadine-mometasone | 244 | 0.0681 | Reference |
Dominated treatments | |||
Intranasal corticosteroids | 291 | 0.0677 | Dominated by olopatadine-mometasone |
ICER = incremental cost-effectiveness ratio; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; QALY = quality-adjusted life-year.
Note: Oral antihistamines were not included as comparators in the subgroup of children aged 6 to 11 years.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor provided deterministic scenario analyses exploring the impact of modelling response rates using NMA results and adopting alternative sources to assign utility weights to health states. The scenario analyses with the greatest impact on the incremental cost-effectiveness ratio (ICER) were changes to the source of efficacy. In the adolescents and adults subgroup, when the source of efficacy was derived from the NMA results, olopatadine-mometasone was more costly than INCSs, resulting in an ICER of $80,807 per QALY gained. Alternatively, in the children subgroup, this scenario analysis resulted in outputs similar to the sponsor’s base-case analysis (i.e., olopatadine-mometasone dominated all other comparators).
The sponsor additionally conducted a scenario analysis from a societal perspective that included costs related to absenteeism and presenteeism for adults and costs of informal caregiving for children. The results of this analysis were similar to the sponsor’s base case for both subgroups.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative efficacy and safety of olopatadine-mometasone is highly uncertain. In the economic evaluation, olopatadine-mometasone was compared with INCSs and oral AHs, with efficacy informed by the sponsor-submitted NMAs. There have been no head-to-head trials of olopatadine-mometasone comparing it with any INCSs or oral AHs, and the CDA-AMC Clinical Review identified notable limitations with the sponsor’s NMAs. Notably, some nodes in the NMA were represented by 1 or 2 drugs. For example, in the adolescents and adults NMA, the efficacy of oral AHs was represented entirely by loratadine (i.e., it was the only treatment included within the oral AH node). However, in the sponsor-submitted model, the cost and recommended dosage of cetirizine were used to determine the treatment costs of oral AHs. Although the sponsor assumed that all drugs within a class are equally effective, the clinical experts consulted by CDA-AMC noted that some relevant comparators were not included in the sponsor’s systematic literature review (SLR) protocol. For example, fluticasone furoate, bilastine, and rupatadine fumarate are indicated and prescribed for the treatment of moderate to severe SAR. As noted in the CDA-AMC Clinical Review, the sponsor’s SLR report did not provide any rationale for why these treatments were not included. The omission of relevant comparators from the sponsor’s SLR protocol may have led to missing evidence in the subsequent NMAs. The impact of this potential bias on the results of the NMA and, hence, the economic evaluation, remains unknown. Overall, the NMA was considered too uncertain by the CDA-AMC Clinical Review team to support decision-making.
In the economic model, the sponsor included AEs that occurred in at least 1% of the patients in their respective pivotal trials. These included dysgeusia, epistaxis, nasal discomfort, headaches, and drowsiness. Data to inform the rate of these AEs were incorporated in the model through a naive comparison, without adjustment or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine whether any observed differences between the therapies are solely due to the treatment or, rather, due to bias or confounding factors.
The sponsor’s model predicts that olopatadine-mometasone will be associated with an incremental gain of approximately 0.0003 QALYs over the 28-day model horizon compared with oral AHs and INCSs in the adolescents and adults subgroup, and with an incremental gain of approximately 0.0004 QALYs compared with INCSs in the children subgroup. Given the lack of direct evidence for olopatadine-mometasone versus relevant comparators, limitations with the sponsor’s indirect treatment comparison, and the use of a naive comparison for AEs, it is highly uncertain whether olopatadine-mometasone provides a net clinical benefit relative to currently available treatments.
Health care resource use is highly uncertain. The sponsor’s model included health care resource use for patients with moderate to severe SAR (i.e., emergency department visits, physician visits, specialist visits, inpatients costs), both in the first 14 days of treatment as well as in the second 14 days among patients without an initial treatment response (i.e., extended SAR period). CDA-AMC notes that the cost savings predicted by the sponsor’s model with olopatadine-mometasone are driven by savings in health care resource use, particularly in the extended SAR period. Health care resource use was not an outcome in any of the included trials (GSP301-301, GSP301-304, GSP301-305), and resource use in the sponsor’s model (proportion of patients who require each resource, number of resources used by each patient) was based on clinical expert input obtained by the sponsor. The clinical expert input received by CDA-AMC indicated that some resource use estimates in the model may be overestimated, in particular, the proportion of patients who visit an emergency department (15%) or who are admitted to hospital (5%) in the extended SAR period.
CDA-AMC was unable to address this limitation. In the absence of robust studies of health care resource use for patients with SAR in Canada, health care resource use and, hence, the savings predicted by the sponsor’s model should be considered highly uncertain.
SAR-associated ocular symptoms were not considered in the sponsor’s model. The indication for olopatadine-mometasone is for symptomatic treatment of moderate to severe SAR and associated ocular symptoms; however, the sponsor’s model included health states related only to treatment response in terms of nasal symptoms. As such, the sponsor’s model does not consider efficacy or costs related to ocular symptoms. The clinical expert input received by CDA-AMC for this review indicated that ocular symptoms are common among patients with SAR and can result in additional resource use. Data from the olopatadine-mometasone pivotal trials suggest that olopatadine-mometasone likely improves ocular symptoms (assessed via rTOSS) compared with placebo in adolescents and adults, with little to no difference among children. Ocular symptoms were not included in either the sponsor’s NMA or the comparative efficacy; hence, the cost-effectiveness of olopatadine-mometasone for the treatment of SAR-associated ocular symptoms is unknown.
CDA-AMC was unable to address this limitation. The omission of SAR-associated ocular symptoms from the economic model increases uncertainty as to the incremental benefits and costs associated with the use of olopatadine-mometasone for the full Health Canada indication.
Oral AHs were not included as comparators in the child subgroup. The sponsor justified the exclusion of oral AHs in this subgroup by stating that no relevant data were identified for oral AHs in the child population. However, as noted in an earlier appraisal point, the sponsor’s SLR protocol omitted some relevant oral AHs (i.e., bilastine and rupatadine). As such, CDA-AMC is unable to comment on the validity of this statement. The clinical expert input received by CDA-AMC indicated that oral AHs are prescribed to children with moderate to severe SAR; as such, the omission of oral AHs does not represent Canadian clinical practice.
CDA-AMC was unable to address this limitation. The cost-effectiveness of olopatadine-mometasone relative to oral AHs in children is unknown.
The impact of olopatadine-mometasone on HRQoL is uncertain. The impact of olopatadine-mometasone on HRQoL and, hence QALYs, predicted by the sponsor’s model, is uncertain for several reasons. First, as noted in the CDA-AMC Clinical Review, olopatadine-mometasone likely results in little to no difference in either HRQoL after 15 days of treatment compared with either placebo or mometasone alone in adolescents and adults or compared with placebo in children. No data were provided beyond 15 days; as such, the HRQoL impact of olopatadine-mometasone in the extended SAR phase (days 14 to 28) is unknown.
Second, there is considerable uncertainty associated with the health state utility values adopted by the sponsor, as a result of using utility values from multiple sources and averaging utility values. For example, for the SAR health state, which reflects patients with either moderate or severe SAR, the sponsor averaged the reported utilities for moderate SAR (0.86 for the adult subgroup and 0.68 for the child subgroup) and severe SAR (0.83 for the adult subgroup and 0.65 for the child subgroup) to inform the model health states. Although the exact utility for this health state is uncertain, the utility value lacks face validity (i.e., does not reflect the utility of either moderate or severe SAR), as the clinical experts consulted by CDA-AMC indicated that patients with moderate SAR generally have different HRQoL compared with patients with severe SAR. The use of an average weight additionally makes the implicit assumption that there is an equal distribution of patients with moderate and severe SAR, which is uncertain. Additionally, to inform the utility weights for the extended SAR health state (patients whose condition does not respond to treatment in the first 14 days and receive an additional 14 days of treatment), the sponsor took the average of utilities for the SAR health state (0.87) and the recovered SAR health state (0.92), which results in a utility of 0.88 for patients whose condition does not respond to initial treatment. Although the exact utility for this health state is uncertain, the clinical experts consulted by CDA-AMC indicated that a higher utility for those whose condition does not respond to initial treatment lacks face validity and indicated that patient HRQoL in the second 14 days of treatment (extended SAR health state) would likely be the same or worse compared with the first 14 days of treatment (SAR health state).
The sponsor’s model predicts that olopatadine-mometasone will be associated with an incremental gain of approximately 0.0003 QALYs over a 28-day period. Whether this marginal gain will be realized in practice is highly uncertain owing to uncertainty in the underlying clinical evidence.
Adherence to treatment was not considered. The sponsor’s model does not consider adherence to olopatadine-mometasone or comparators, thus implicitly making the assumption that patients will be fully adherent to treatment. The clinical expert input received by CDA-AMC indicated that patients may not fully adhere to treatment in practice, for example, if they perceive no or insufficient improvement after using the treatment a few times. In the olopatadine-mometasone pivotal trials, an adherence rate of between 75% and 125% (i.e., twice a day for 14 days = 100%; twice a day for up to 17 days = 125%) was achieved by more than 90% of patients in each treatment group. If adherence is lower in clinical practice, efficacy may be lower than estimated with no impact on costs.
CDA-AMC was unable to explore uncertainty in the impact of adherence on the cost-effectiveness of olopatadine-mometasone due to the structure of the sponsor’s model. The directionality of impact on the cost-effectiveness of olopatadine-mometasone is unknown because of a lack of adherence data for comparators.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patients whose condition does not respond to treatment in the first 14 days will continue to receive the same therapy for an additional 14 days. | Uncertain. The clinical expert feedback received by CDA-AMC indicated that patients whose symptoms do not respond to treatment within 14 days may change therapies, either within or between classes. |
Diagnostic testing costs (skin prick and serum allergen–specific IgE tests) were included in the model. | Reasonable, although the frequency of testing may be lower than estimated by the sponsor (100% of patients receive a skin prick test, 3% receive serum allergen testing). The clinical experts consulted by CDA-AMC indicated that, in clinical practice, a diagnosis is generally based on symptoms (e.g., stuffy nose, pruritus of nose or eyes, seasonality of symptoms) and that diagnostic testing is not always performed. |
CDA-AMC = Canada’s Drug Agency; IgE = immunoglobulin E.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address several key limitations with the sponsor’s submission, including uncertainty in the comparative clinical data as well as methodological and conceptual limitations related to the model structure. These limitations prevented CDA-AMC from deriving a base-case estimate of the cost-effectiveness of olopatadine-mometasone for the treatment of moderate to severe SAR and associated ocular symptoms.
As noted in the CDA-AMC Clinical Review, the findings of the NMA should be viewed with uncertainty; that is, it is unclear whether olopatadine-mometasone will result in improved outcomes compared with INCSs or AHs in either subgroup. Thus, CDA-AMC undertook a comparison of drug acquisition costs for olopatadine-mometasone and relevant comparators (Table 6 and Table 7). Readers should note that this analysis assumes equal efficacy between olopatadine-mometasone, which is uncertain, owing to the limitations identified with the submitted indirect evidence (refer to CDA-AMC Clinical Review).
In the CDA-AMC cost comparison, the drug acquisition cost of olopatadine-mometasone for a 14-day treatment course was $26 for adolescents and adults and $13 for children, based on the Health Canada–recommended dosage. It should be noted, however, that olopatadine-mometasone is available in Canada only as a 240-spray bottle at a submitted price of $56.11 per bottle. Thus, the acquisition cost for olopatadine-mometasone for a first course of treatment would be $56.11.
The cost of oral AHs for a 14-day treatment course ranged from $3 to $17 for adolescents and adults and from $3 to $11 for children, while a 14-day treatment course of INCSs ranged from $4 to $15 in adolescents and adults and from $2 to $7 for children (note that costs may be higher cost owing to per-bottle costs for INCSs; refer to Appendix 1). The price reductions required for the treatment cost of olopatadine-mometasone for a 14-day treatment course to be equivalent to that of the lowest-cost comparators in both the adult and children subgroups are shown in Table 6 and Table 7.
Table 6: Summary of the CDA-AMC Cost Comparison Analysis (Adolescents and Adults)
Comparator | Cost per 14-day course of comparator ($) | Olopatadine-mometasone price reduction needed (%) | Savings in treatment costs ($) |
|---|---|---|---|
Oral antihistamines | |||
Bilastine | 17 | 36 | 9 |
Cetirizine | 3 to 12 | 53 to 88 | 14 to 23 |
Desloratadine | 14 | 47 | 12 |
Fexofenadine | 10 | 62 to 63 | 16 to 17 |
Loratadine | 9 | 66 | 17 |
Rupatadine fumarate | 15 | 42 | 11 |
Intranasal corticosteroids | |||
Mometasone furoate | 17 | 34 | 9 |
Beclomethasone dipropionate | 7 to 10 | 61 to 74 | 16 to 19 |
Budesonide 65 mcg | 3 to 5 | 81 to 90 | 21 to 23 |
Budesonide 100 mcg | 4 | 83 | 22 |
Ciclesonide | 13 | 50 | 13 |
Fluticasone furoate | 15 | 45 | 11 |
Fluticasone propionate | 10 | 61 | 16 |
CDA-AMC = Canada’s Drug Agency; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray.
Note: The cost of olopatadine-mometasone per 14-day treatment course is estimated to be $26 for adolescents and adults (≥ 12 years) based on the sponsor’s submitted price ($56.11 per bottle of 240 metered sprays). At the monograph-recommended dosages, each 240-spray bottle contains sufficient product for 30 days for adolescents and adults (must be used within 2 months of opening). The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
Table 7: Summary of the CDA-AMC Cost Comparison Analysis (Children)
Comparator | Cost per 14-day course of comparator ($) | Olopatadine-mometasone price reduction needed (%) | Savings in treatment costs ($) |
|---|---|---|---|
Oral antihistamines | |||
Bilastine | 10 or 11 | 18 or 26 | 2 or 3 |
Cetirizine | NA | NA | NA |
Desloratadine | 3 to 5 | 61 to 81 | 8 to 10 |
Fexofenadine | NA | NA | NA |
Loratadine | 4 | 68 | 9 |
Rupatadine fumarate | 6 to 11 | 13 to 57 | 2 to 7 |
Intranasal corticosteroids | |||
Mometasone furoate | 4 | 67 | 9 |
Beclomethasone dipropionate | 7 | 48 | 6 |
Budesonide 65 mcg | 3 to 5 | 61 to 81 | 8 to 10 |
Budesonide 100 mcg | 2 to 4 | 67 to 83 | 9 to 11 |
Ciclesonide | NA | NA | NA |
Fluticasone furoate | 7 | 45 | 6 |
Fluticasone propionate | NA | NA | NA |
CDA-AMC = Canada’s Drug Agency; NA = not applicable; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray.
Note: Cost of olopatadine-mometasone per 14-day treatment course is estimated to be $13 for children (6 to 11 years) based on the sponsor’s submitted price ($56.11 per 240 metered spray bottle). At the monograph-recommended dosages, each 240-spray bottle contains sufficient product for 60 days for children (must be used within 2 months of opening). The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. NA indicates that the treatment was excluded as a comparator in the subgroup of children aged 6 to 11 years.
Issues for Consideration
Each bottle of olopatadine-mometasone contains 240 metered sprays, which reflects 1 month of dosing for adults or 2 months of dosing for children. Although the duration of treatment for 1 episode of SAR may be approximately 14 days (based on the duration of the olopatadine-mometasone pivotal trials and the clinical expert input received by CDA-AMC), the full cost of 1 bottle would be incurred by payers (submitted price = $56.11). The olopatadine-mometasone monograph indicates that a bottle must be used within 2 months of first opening.
The sponsor’s cost-utility analysis and the CDA-AMC cost comparison rely on publicly accessible list prices and do not reflect the existing confidential prices negotiated by public plans. It is likely that the prices paid by public drug plans are lower than the values used in each analysis.
The olopatadine-mometasone pivotal trials included only patients with SAR. The clinical experts consulted by CDA-AMC noted that the results of these trials may be potentially generalizable to other types of AR, including perennial AR, and that off-label use may occur in practice.
Overall Conclusions
Based on the CDA-AMC Clinical Review of the GSP301-301, GSP301-304, and GSP301-305 trials, use of olopatadine-mometasone by adolescents and adults (aged 12 years and older) with moderate or severe SAR over a 14-day treatment course improves nasal and ocular symptoms (based on 12-hour rTNSS and rTOSS outcomes) compared with placebo. However, compared with mometasone furoate nasal spray, olopatadine-mometasone did not result in a clinically meaningful improvement in symptoms in this age group. Among children (aged 6 to 11 years), trial evidence suggests that olopatadine-mometasone improves nasal symptoms compared with placebo over 14 days of treatment. In both age groups, olopatadine-mometasone had a nonstatistically significant impact on HRQoL after 14 days of treatment.
There have been no head-to-head trials comparing olopatadine-mometasone with oral AHs or INCSs (other than mometasone furoate nasal spray), and the submitted indirect evidence was deemed by the CDA-AMC review team to be uncertain due to limitations surrounding the comparators considered and whether there was appropriate representation in the comparator drug classes in terms of efficacy, and a possibility of overestimation of the treatment effect estimates in some analyses. CDA-AMC identified additional important limitations with the sponsor’s economic evaluation that precluded reanalysis, including (but not limited to) uncertainty related to SAR-related health care resource use, the lack of consideration of SAR-related ocular symptoms, and the exclusion of oral AHs from the child subgroup. Due to limitations with the available comparative evidence and the sponsor’s economic evaluation, CDA-AMC was unable to provide a more robust estimate of the cost-effectiveness of olopatadine-mometasone compared with relevant comparators.
The sponsor’s model predicts that olopatadine-mometasone will be associated with a marginal incremental gain of QALYs (approximately 0.0003 to 0.0004), and that the increased drug acquisition costs of olopatadine-mometasone will be offset by cost savings from a reduction in health care resource use. Whether olopatadine-mometasone will reduce health care costs among patients with moderate to severe SAR is highly uncertain, given that health care resource use was not an outcome in the olopatadine-mometasone pivotal trials. Further, this reduction in health care usage is predicated on differences in treatment response between olopatadine-mometasone and comparators; however, as noted previously, the findings of the sponsor’s NMA should be considered highly uncertain, and it is unclear whether there will be differences in treatment outcomes in clinical practice. If the efficacy of olopatadine-mometasone is not different from that of comparators, the predicted savings in health care costs will not be realized, and the reimbursement of olopatadine-mometasone will result in additional costs to the health care system.
Overall, the clinical and economic evidence submitted by the sponsor is insufficient to suggest that olopatadine-mometasone should be priced higher than currently reimbursed treatments for moderate to severe SAR and associated ocular symptoms.
References
1.Bausch Health, Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ryaltris (olopatadine hydrochloride and mometasone furoate), suspension, 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate (as monohydrate) per delivered dose; nasal spray. June 21, 2024.
2.Glenmark Pharmaceuticals Canada Inc. Ryaltris (olopatadine hydrochloride and mometasone furoate): suspension, 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate (as monohydrate) per delivered dose; nasal spray [product monograph]. September 21, 2022.
3.Carr W, Bernstein J, Lieberman P, et al. A novel intranasal therapy of azelastine with fluticasone for the treatment of allergic rhinitis. J Allergy Clin Immunol. 2012;129(5):1282-1289 e10. doi:10.1016/j.jaci.2012.01.077 PubMed
4.Hampel FC, Pedinoff AJ, Jacobs RL, Caracta CF, Tantry SK. Olopatadine-mometasone combination nasal spray: Evaluation of efficacy and safety in patients with seasonal allergic rhinitis. Allergy Asthma Proc. 2019;40(4):261-272. doi:10.2500/aap.2019.40.4223 PubMed
5.Kuna P, Bachert C, Nowacki Z, et al. Efficacy and safety of bilastine 20 mg compared with cetirizine 10 mg and placebo for the symptomatic treatment of seasonal allergic rhinitis: a randomized, double-blind, parallel-group study. Clin Exp Allergy. 2009;39(9):1338-47. doi:10.1111/j.1365-2222.2009.03257.x PubMed
6.Retzler J, Grand TS, Domdey A, Smith A, Romano Rodriguez M. Utility elicitation in adults and children for allergic rhinoconjunctivitis and associated health states. Qual Life Res. 2018;27(9):2383-2391. doi:10.1007/s11136-018-1910-8 PubMed
7.Pollock RF, Slaettanes AK, Brandi H, Grand TS. A Cost-Utility Analysis of SQ((R)) Tree SLIT-Tablet versus Placebo in the Treatment of Birch Pollen Allergic Rhinitis from a Swedish Societal Perspective. Clinicoecon Outcomes Res. 2023;15:69-86. doi:10.2147/CEOR.S377399 PubMed
8.Ara R, Brazier JE. Using health state utility values from the general population to approximate baselines in decision analytic models when condition-specific data are not available. Value Health. 2011;14(4):539-45. doi:10.1016/j.jval.2010.10.029 PubMed
9.IQVIA. DeltaPA database [sponsor-supplied reference]. 2024.
10.Ontario Ministry of Health. Schedule of benefits for laboratory services [sponsor-supplied reference]. 2020. https://files.ontario.ca/moh-schedule-of-benefits-laboratory-services-2020.pdf
11.Mtibaa M, Gupta S, Muthukumar M, et al. Cost-Effectiveness of Once-Daily, Single-Inhaler Indacaterol Acetate/ Glycopyrronium Bromide/ Mometasone Furoate in Patients with Uncontrolled Moderate-to-Severe Asthma in Canada. Clinicoecon Outcomes Res. 2021;13:957-967. doi:10.2147/CEOR.S336915 PubMed
12.Ontario Health and Long-Term Care. Ontario Case Costing Initiative (OCCI) [sponsor-supplied reference]. 2017. https://hsim.health.gov.on.ca/hdbportal/user/21878
13.Government of Ontario. Ontario Drug Benefit Formulary [sponsor-supplied reference]. 2024. https://www.formulary.health.gov.on.ca/formulary/
14.Bausch Health, Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ryaltris (olopatadine hydrochloride and mometasone furoate), suspension, 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate (as monohydrate) per delivered dose; nasal spray. June 21, 2024.
15.IQVIA. IQVIA Pharmastat [sponsor-supplied reference]. 2024.
16.CADTH. Drug Reimbursement Review pharmacoeconomic report: azelastine hydrochloride and fluticasone propionate nasal spray (Dymista) for moderate-to-severe seasonal allergic rhinitis and associated ocular symptoms [sponsor-supplied reference]. 2016. https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0408_Dymista_PE_Report.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Moderate to Severe SAR
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost ($) | Cost per 14-day treatment course ($) |
|---|---|---|---|---|---|---|
Olopatadine hydrochloride and mometasone furoate monohydrate (Ryaltris) | 665 mcg olopatadine hydrochloride and 25 mcg mometasone furoate | Nasal spray (240 sprays per bottle)a | 56.1120 |
|
|
|
Oral antihistamines | ||||||
Bilastine (Blexen) |
| Tablet 120 mL oral solution |
|
|
|
|
Cetirizine (generic) |
| Tablet |
| ≥ 12 years: 5 to 20 mg daily. Maximum duration of 14 days for ages 12 to 18 years | ≥ 12 years: 0.22 to 0.89 | ≥ 12 years: 3 to 12 |
Desloratadine (generic) |
| Tablet 100 mL syrup |
|
|
|
|
Fexofenadine (Allegra 12 hour) | 60 mg | Tablet | 0.3549 | ≥ 12 years: one 60 mg tablet every 12 hours | ≥ 12 years: 0.71 | ≥ 12 years: 10 |
Fexofenadine (Allegra 24 hour) | 120 mg | Tablet | 0.6833 | ≥ 12 years: one 120 mg tablet daily | ≥ 12 years: 0.68 | ≥ 12 years: 10 |
Loratadine (generic) | 10 mg | Tablet | 0.6267 | ≥ 2 years: one 10 mg tablet daily | 0.63 | 9 |
Rupatadine fumarate (Rupall) |
|
|
|
|
|
|
Intranasal corticosteroids | ||||||
Mometasone furoate (generic) | 50 mcg per metered spray | Nasal spray (140 sprays) | 21.6860a |
|
|
|
Beclomethasone dipropionate (generic) | 50 mcg per metered spray | Nasal spray (200 sprays) | 12.2600b |
|
|
|
Budesonide (generic) | 65 mcg per metered spray | Nasal spray (120 sprays) | 11.0400b |
|
|
|
Budesonide (generic) | 100 mcg per metered spray | Nasal spray (165 sprays) | 12.7400b |
|
|
|
Ciclesonide (Omnaris) | 50 mcg per metered spray | Nasal spray (60 sprays) | 14.0820 | ≥ 12 years: 2 sprays in each nostril once daily (200 mcg) | ≥ 12 years: 0.94 | ≥ 12 years: 13 |
Fluticasone furoate (Avamys) | 27.5 mcg per metered spray | Nasal spray (30 sprays) | 7.7760 |
|
|
|
Fluticasone propionate (generic) | 50 mcg per metered spray | Nasal spray (60 sprays) | 10.8180 | ≥ 18 years: 2 sprays in each nostril once daily (200 mcg) | ≥ 18 years: 0.72 | ≥ 18 years: 10 |
CDA-AMC = Canada’s Drug Agency; olopatadine-mometasone = olopatadine hydrochloride and mometasone furoate nasal spray; SAR = seasonal allergic rhinitis.
Note: All prices are from IQVIA DeltaPA (accessed September 2024),9 unless otherwise indicated, and do not include dispensing fees. Cost estimates were based on 14 days of treatment, i.e., the duration of an average course of treatment for SAR. All treatments are indicated for SAR and do not specify severity.
aOne bottle of olopatadine-mometasone (240 metered sprays) contains up to 1 month of dosing for adults or 2 months of dosing for children. The daily cost and cost for 1 course of olopatadine-mometasone presented here reflect the cost over a 14-day course of treatment, based on the duration of the olopatadine-mometasone pivotal trials and on clinical expert input. However, the full cost of 1 bottle would be incurred by payers (submitted price = $56.11). As per the olopatadine-mometasone monograph, a bottle must be used within 2 months of first opening.
bCosting from Ontario Drug Benefit Formulary (accessed September 2024).13
cMometasone furoate is indicated for SAR for children between the ages of 3 and 11 years. Use of mometasone furoate in adolescents and adults > 12 years is off-label.
dAge range not specified in product monograph.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to the CDA-AMC critical appraisal |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | No | Refer to the CDA-AMC critical appraisal |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 10: Utility Values Used in the Sponsor's Economic Evaluation
Health state | Adults and adolescents | Children | Source |
|---|---|---|---|
SAR | 0.85 | 0.67 | Retzler (2018)6 |
Extended SAR | 0.88 | 0.80 | Assumption derived as the average of utilities for the SAR and recovered from SAR states |
Recovered from SAR | 0.92 | 0.92 |
SAR = seasonal allergic rhinitis.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results (Adolescents and Adults)
Parameter | Olopatadine- mometasone | INCSs | Oral AHs | Placebo |
|---|---|---|---|---|
QALYs | ||||
Adolescents and adults | ||||
Total | 0.06812 | 0.06780 | 0.06781 | 0.06753 |
SAR | 0.02072 | 0.02298 | 0.02276 | 0.02503 |
Recovered from SAR | 0.03196 | 0.02628 | 0.02642 | 0.02180 |
Extended SAR | 0.01543 | 0.01855 | 0.01863 | 0.02070 |
Costs ($) | ||||
Adolescents and adults | ||||
Total | 274 | 301 | 294 | 322 |
Drug acquisition | 29 | 9 | 4 | 0 |
Resource use | 245 | 291 | 290 | 322 |
Adverse events | 1 | 1 | 0 | 0 |
AH = antihistamine; INCS = intranasal corticosteroid; QALY = quality-adjusted life-year; SAR = severe allergic rhinitis.
Note: SAR refers to days 1 to 14 of treatment. Extended SAR refers to days 14 to 28 of treatment. Recovered from SAR assumes that the patient is no longer on treatment. Resource use includes visits to an emergency department for SAR, visits to a general physician for SAR, visits to a specialist for SAR, and hospital admission for SAR.
Source: Sponsor’s pharmacoeconomic submission (probabilistic analysis).1
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results (Children)
Parameter | Olopatadine- mometasone | INCSs | Oral AHs | Placebo |
|---|---|---|---|---|
QALYs | ||||
Children | ||||
Total | 0.0681 | 0.6775 | NA | 0.0676 |
SAR | 0.0216 | 0.0238 | NA | 0.0244 |
Recovered from SAR | 0.0325 | 0.0259 | NA | 0.0239 |
Extended SAR | 0.0140 | 0.0180 | NA | 0.0193 |
Costs ($) | ||||
Children | ||||
Total | 244 | 291 | NA | 303 |
Drug acquisition | 14 | 2 | NA | 0 |
Resource use | 228 | 287 | NA | 303 |
Adverse events | 1 | 1 | NA | 0 |
AH = antihistamine; INCS = intranasal corticosteroid; NA = not applicable; QALY = quality-adjusted life-year; SAR = severe allergic rhinitis.
Note: SAR refers to days 1 to 14 of treatment. Extended SAR refers to days 14 to 28 of treatment. Recovered from SAR assumes that the patient is no longer on treatment. Resource use includes visits to an emergency department for SAR, visits to a general physician for SAR, visits to a specialist for SAR, and hospital admission for SAR.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Given the identified limitations within the clinical evidence as well as the submitted pharmacoeconomic model, CDA-AMC was unable to conduct any additional analyses to assess the relative cost-effectiveness of olopatadine-mometasone for the treatment of moderate to severe SAR and associated ocular symptoms.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) assessing the expected budgetary impact of reimbursing olopatadine-mometasone for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in patients aged 6 years and older.14 The BIA was undertaken from the perspective of the pan-Canadian drug plans over a 3-year time horizon (2025 to 2027). The sponsor described their approach as claims-based using cost data.
In their analysis, the sponsor based the total cost for each comparator on forecasted pan-Canadian public claims data from IQVIA Pharmastat.15 IQVIA cost was translated into days procured based on the cost per day for each comparator. The sponsor estimated future costs for comparators which were then translated into an estimated number of days procured based on the cost per day, which was calculated using the unit cost and average daily dosage.
The dosage regimen per day were informed by Health Canada product monographs The reference case forecasted the number of days procured for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms in people aged 6 years and older in the olopatadine-mometasone, and the new-drug scenario forecasted the number of days procured for the symptomatic treatment of moderate to severe SAR and associated ocular symptoms should olopatadine-mometasone be reimbursed.
Key inputs to the BIA are documented in Table 14.
The following key assumptions were made by the sponsor:
The proportion of costs attributable to SAR ranged from 31% to 48% across individual AH and INCS treatments, based on expert opinion obtained by the sponsor.
The sponsor assumed the reimbursement of olopatadine-mometasone would not result increase the number of patients receiving treatment for moderate to severe SAR.
The estimated market uptake olopatadine-mometasone was based on internal forecasting by the sponsor.
Olopatadine-mometasone was assumed to proportionately displace market shares from comparators.
Figure 2: Schematic of the Sponsor’s BIA
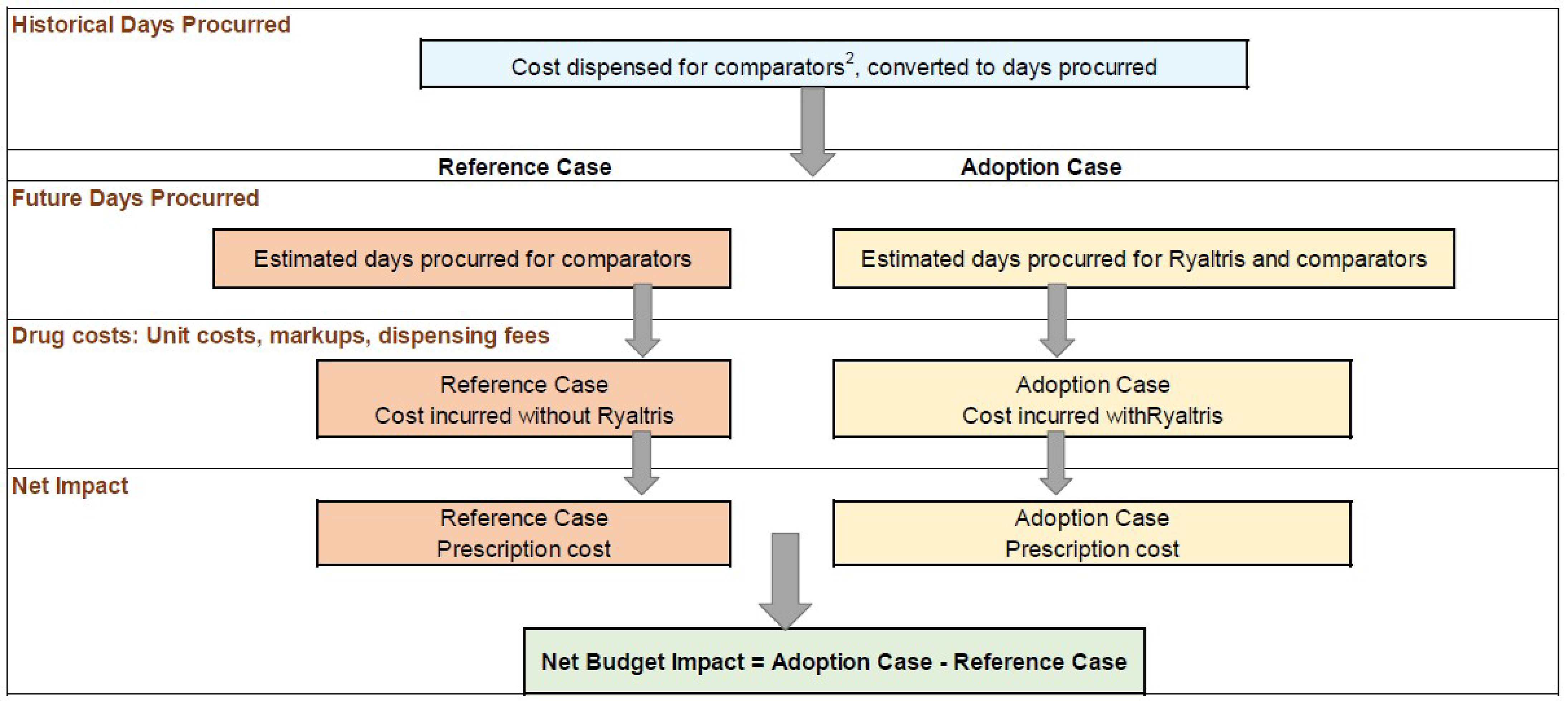
BIA = budget impact analysis.
Source: Sponsor’s submitted BIA.14
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3) |
|---|---|
Target population | |
Number of claims (people aged 6 years and older) | Not reporteda |
Market uptake (3 years) | |
Uptake (reference scenario):
| 0% / 0% / 0% Not reported Not reported |
Uptake (new-drug scenario):
| 7.70% / 10.04% / 12.22% Not reported Not reported |
Daily cost of treatment, per patient | |
Olopatadine-mometasone Oral AHs INCSs | $1.87 $0.18 to 0.98 $0.05 to 0.94 |
AH = antihistamine; INCS = intranasal corticosteroid.
aSize of the eligible population or number of claims not provided by the sponsor. The sponsor estimates that the number of days procured will be 4,129,471 in year 1, 4,447,53 in year 2, and 4,777,994 in year 3.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing olopatadine-mometasone for the treatment of moderate to severe SAR and associated ocular symptoms among patients aged 6 years and older to be $8,222,757 over the first 3 years (year 1 = $1,958,164; year 2 = $2,723,295; year 3 = $3,541,293).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The approach used by the sponsor to estimate the budgetary impact of reimbursing olopatadine-mometasone is associated with considerable uncertainty. In their calculation of estimated budgetary impact, the sponsor utilized historical cost data adjusted by the estimated proportion of claims attributable to SAR. These historical costs were then used to project future costs using linear regression, which were converted to days procured based on cost per day for each comparator. In the new-drug scenario, the sponsor estimated days procured for olopatadine-mometasone based on their predicted market uptake multiplied by the total estimated days procured for each comparator, and, after estimating the number of days procured for olopatadine-mometasone, the sponsor redistributed the remaining days procured proportionally among model comparators refer to (Figure 2). This approach is associated with considerable uncertainty.
First, the sponsor’s approach to estimating days procured for olopatadine-mometasone and comparators did not provide an estimate of the eligible population size, which is a key aspect of determining the budgetary impact. Instead, the sponsor estimated the total annual expenditure to the CDA-AMC–participating drug plans by multiplying the estimated total days procured by the cost per day. This approach does not provide sufficient transparency or information for decision-making.
Second, the sponsor assumed that between 31% and 48% of historical costs were attributable to SAR, based on expert opinion. Whether this accurately reflects usage is highly uncertain, owing to the multiple indications and uses for the included drugs. Clinical experts consulted by CDA-AMC indicated that the drugs included in the model are used for multiple non-SAR indications, including eosinophilic rhinitis, chronic obstructions, urticaria, and nasal polyposis. Thus, it is challenging to properly estimating the proportion of claims owing to SAR. Additionally, this approach would be insufficient to separate out claims for moderate or severe SAR from all claims for SAR.
CDA-AMC was unable to address this limitation owing to the structure of the sponsor’s model and the lack of alternative information. The eligible population size is thus unknown.
Additional limitations were identified but could not be addressed by CDA-AMC owing to the modelling approach adopted by the sponsor and a lack of alternative inputs:
The market uptake of olopatadine-mometasone is uncertain: The sponsor derived market shares for olopatadine-mometasone based on internal market research and product forecasting. The sponsor used Dymista as an analogue, which it justified as it being the only other combination therapy on the market and used Dymista market shares at 18 months post launch. The sponsor adjusted the Dymista market shares by a factor of 2 “given the improved attributes of RYALTRIS, as seen through patient preference studies and clinician feedback.” Clinical expert feedback received by CDA-AMC for this review suggests that the sponsor’s estimated market share for olopatadine-mometasone (12% by year 3) may be underestimated and indicated that olopatadine-mometasone may capture up to 50% of the market. Additionally, because Dymista received a do not reimburse recommendation from the Canadian Drug Expert Committee in 201516 and is not reimbursed by the public formularies, the use of its uptake to forecast uptake of olopatadine-mometasone in the context of public drug plan reimbursement introduces additional uncertainty in the budget impact.
Adult dosages applied to all patients regardless of age: The sponsor calculated days procured by use of the average daily doses for adult patients for each treatment. This does not take the lower dosages for children into account in the BIA.
The price of drugs paid by public drug plans is uncertain: The sponsor’s analysis was based on publicly available list prices for comparators. The prices paid by public drug plans are not known.
CDA-AMC Reanalyses of the BIA
CDA-AMC was unable to provide a reliable estimate of the true budgetary impact of reimbursing olopatadine-mometasone, owing to the modelling approach used by the sponsor and a lack of alternative inputs. The impact of reimbursing olopatadine-mometasone is thus highly uncertain.
Table 15: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 24,768,253 | 26,493,678 | 28,457,869 | 30,514,632 | 110,234,433 |
New drug | 24,768,253 | 28,451,842 | 31,181,165 | 34,055,925 | 118,457,185 | |
Budget impact | 0 | 1,958,164 | 2,723,295 | 3,541,293 | 8,222,752 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.