Drugs, Health Technologies, Health Systems
Reimbursement Review
Crovalimab (Piasky)
Sponsor: Hoffmann-La Roche Limited
Therapeutic area: Paroxysmal nocturnal hemoglobinuria
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAMAC
Aplastic Anemia & Myelodysplasia Association of Canada
AE
adverse event
AESI
adverse event of special interest
BTH
breakthrough hemolysis
CCOD
clinical cut-off date
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
DTDC
drug-target-drug complex
ECG
electrocardiogram
EORTC
European Organisation for Research and Treatment of Cancer
EORTC IL-40
European Organisation for Research and Treatment of Cancer – Item Library 40
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FACIT-F
Functional Assessment of Chronic Illness Therapy–Fatigue
GEE
generalized estimating equation
GHS
global health status
GPI
glycosylphosphatidylinositol
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ITC
indirect treatment comparison
LDH
lactate dehydrogenase
MAVE
major adverse vascular event
MCMC
Markov Chain Monte Carlo
MDS
myelodysplastic syndrome
MFS
multidimensional fatigue scale
MID
minimal important difference
MMRM
mixed model of repeated measures
NIM
noninferiority margin
NMA
network meta-analysis
OLE
open-label extension
OR
odds ratio
PD
pharmacodynamic
PedsQL
Pediatric Quality of Life Inventory
PK
pharmacokinetic
PNH
paroxysmal nocturnal hemoglobinuria
PPQ
Patient Preference Questionnaire
pRBC
packed red blood cell
PRO
patient-reported outcome
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SLR
systematic literature review
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Crovalimab (Piasky), solution, 340 mg/2 mL (170 mg/mL) for SC injection and IV infusion |
Sponsor | Hoffmann-La Roche Limited |
Indication | For the treatment of PNH in adults and adolescents 13 years of age and older with a body weight of at least 40 kg |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | June 4, 2025 |
Recommended dosage | Recommended maintenance dosage: 680 mg (body weight ≥ 40 kg to < 100 kg) or 1,020 mg (≥ 100 kg) by SC injection once every 4 weeks, after 1 loading dose of 1,000 mg (≥ 40 kg to < 100 kg) or 1,500 mg (≥ 100 kg) by IV infusion and 4 additional weekly doses of 340 mg by SC injection |
NOC = Notice of Compliance; PNH = paroxysmal nocturnal hemoglobinuria; SC = subcutaneous.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, life-threatening, complement-mediated hematological disorder. PNH is the primary manifestation of chronic intravascular hemolysis, associated with bone marrow failure and thrombophilia.1 It develops when hematopoietic cells acquire somatic mutations in the X-linked gene of PIGA.2 Key clinical signs and symptoms of PNH include anemia, fatigue, thrombosis, esophageal spasms, male erectile dysfunction, hemoglobinuria, abdominal pain, and dyspnea.3 Patients with PNH may experience impaired health-related quality of life (HRQoL), and may struggle to complete normal everyday activities.1,4 The mortality rate of patients with PNH who receive supportive care alone is approximately 20% to 35% within 5 years to 10 years of diagnosis.4 Chronic kidney disease, pulmonary hypertension, and venous or arterial thromboembolic events are life-threatening complications of PNH.4 Thromboembolic events were the leading cause of death in patients with PNH (40% to 67% of deaths with known cause).5 An allogeneic bone marrow transplant is considered a curative treatment for PNH.6,7 However, a bone marrow transplant is not usually offered as a first-line treatment for classic PNH, owing to the risks of transplant-related morbidity and mortality, and is usually reserved for younger patients with PNH with concomitant severe aplastic anemia or other causes of bone marrow failure.4,8,9 In Canada, patients with PNH receive C5 inhibitors as standard first-line therapy to reduce uncontrolled complement activation in the terminal complement cascade, and the primary C5 inhibitor therapies are ravulizumab and eculizumab, both administered through IV infusion.4
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of crovalimab 340 mg/2 mL for subcutaneous (SC) injection following 1 loading dose by IV infusion in the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call by Canada’s Drug Agency (CDA-AMC) for input and from clinical experts consulted for the purpose of this review.
Patient Input
CDA-AMC received 1 joint input from the Canadian Association of PNH Patients and the Aplastic Anemia & Myelodysplasia Association of Canada (AAMAC). The patient groups gathered personal experiences from 1 patient living in Canada who had received crovalimab and provided information from the crovalimab clinical trial to highlight the trial’s findings and demonstrate the potential efficacy of crovalimab. The patient group input stated that PNH is a complex and multifaceted disorder that requires comprehensive management to address the various aspects of the disease and improve the quality of life (QoL) for those affected. The chronic nature of PNH means that patients must manage their condition over a lifetime, dealing with the physical, emotional, and financial burdens associated with the disease. The impact on QoL is profound, as patients must cope with the unpredictability of symptoms, the side effects of treatments, and the constant threat of serious complications. Regular monitoring and supportive care are essential to managing the disease and improving patient outcomes. According to the patient group input, 1 patient with PNH in Canada reported that the disease started with the patient feeling constantly exhausted, followed by dark urine in the morning and chest pain. The patient explained feeling terrified of the risk of blood clots and not knowing the effect of the disease on life, job, and family, along with accepting that this is a lifelong condition. Based on the patient group input, 1 significant challenge with current treatments is that they require regular clinic visits every 2 weeks to 8 weeks for IV infusions, which can be time-consuming and disruptive. The patient group stated that crovalimab has the potential to greatly improve patients’ QoL. It reduces the physical, emotional, and logistical burdens associated with traditional IV therapies, allowing patients to regain a sense of freedom and control over their lives. Fewer clinic visits and less travel for IV injections mean lower health care costs and less financial burden for patients. This more convenient and less invasive approach can significantly improve not only patients’ QoL but also their overall well-being and adherence to treatment, leading to better health outcomes.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review noted that crovalimab may address an unmet need for some patients with a preference for an SC route of drug administration, particularly for those with poor venous access and who are opposed to the insertion of a port-a-cath (a tube that is inserted into a vein and connected to a port that is placed under the skin). The clinical experts noted that crovalimab may be a safer alternative for patients living in isolated geographic locations that preclude the guaranteed delivery of eculizumab, and some patients may be more comfortable with self-administration than having to travel to a clinic for IV therapy (which is associated with transportation costs and exposure to potential infections at a clinic). The clinical experts noted that where ravulizumab is funded, most patients with PNH would choose to start ravulizumab as the first-line therapy over eculizumab or crovalimab (of note, ravulizumab is not funded in some jurisdictions such as British Columbia). However, per clinical guidelines, eculizumab would be preferred for patients who are pregnant. Furthermore, the clinical experts pointed out that patients who prefer to be more independent in receiving therapy, have significant needle phobia, or find it difficult to visit a clinic due to geographic location may choose crovalimab as first-line therapy. The clinical experts noted that switching from ravulizumab or eculizumab to crovalimab could be uncommon as there is a risk of transient immune complex formation that only occurs in patients switching to or from other C5 inhibitors. The clinical experts further pointed out that in children, due to the high association with aplastic or hypoplastic anemia, management would be driven by treating aplastic or hypoplastic anemia. The clinical experts noted that the rare cases of isolated PNH would be treated along the same lines as for adults, except that PNH clone size is smaller in children and only clones greater than 10% would be eligible for C5 inhibition therapy. The clinical experts noted that patients who are fully motivated to self-administer the SC injections and those known to have genetic polymorphism that obviates efficacy with eculizumab or ravulizumab would be best suited for treatment with crovalimab. The clinical experts pointed out that the outcomes used to assess the treatment response would be the same as for eculizumab or ravulizumab, including improvements in hemoglobin levels, transfusion avoidance, and renal function, along with reductions in smooth muscle spasm, less fatigue, the normalization of lactate dehydrogenase (LDH), and the avoidance of breakthrough hemolysis (BTH). The clinical experts noted that frequent laboratory and patient assessments are required during the early stages of crovalimab, followed by standard assessments once the laboratory results stabilize. The clinical experts noted that if patients treated with crovalimab are not showing any clinical responses by 3 weeks to 4 weeks, a transition back to ravulizumab or eculizumab and then monitoring for the immune responses should be considered. The clinical experts pointed out that crovalimab should be discontinued when thrombosis occurs, or when there is ongoing LDH elevation or ongoing symptoms of PNH.
Clinician Group Input
CDA-AMC received 1 clinician group input from the Canadian PNH Network. Information for this submission was obtained via publicly available documents, congress abstracts, the published literature, and members of the Canadian PNH Network who were invited to contribute to the various sections. Three clinicians contributed to this submission. According to the clinician group input, the current standard of care for patients with hemolytic PNH is terminal complement inhibition with a C5 blockade; eculizumab and ravulizumab remain the only first-line therapies across Canada. The only curative treatment for PNH is an allogeneic hematopoietic stem cell transplant, which is reserved for patients with predominant or progressive bone marrow failure (e.g., aplastic anemia), which can coincide with, precede, or follow a diagnosis of PNH.
The Canadian PNH Network highlighted that crovalimab is expected to address important treatment goals unmet by current therapies, including offering a C5 inhibitory strategy that does not require IV access, enabling self-administration by patients or administration by caregivers, and providing an option for rare cases of resistance to eculizumab or ravulizumab due to a C5 polymorphism. Additionally, the clinician group expected that crovalimab would be another first-line option for patients with PNH, either switching from an IV C5 inhibitor or starting from being treatment-naive. Patients who favour the freedom and reduced treatment burden of SC administration would very likely select this therapy. On the other hand, patients with PNH least suitable would be those who are not accepting SC drug delivery, or those who develop clinically significant extravascular hemolysis, which necessitates proximal complement inhibition strategies.
The Canadian PNH Network stated that response to treatment focuses on LDH reduction, which reduces hemolysis and the risk of thrombosis, and may also improve hemoglobin levels and reduce transfusion dependence in patients with PNH. Clinical outcomes related to the response included decreased fatigue and transfusion requirements as well as improved QoL and overall survival. Efficacy outcomes would typically be assessed every 2 weeks to 4 weeks initially after starting a new therapy or switching, but follow-up would be required less often (e.g., every 3 months to 6 months) as a patient becomes established on the drug and does not show evidence of side effects or other concerns.
Based on the input, some of the factors that should be considered to discontinue the treatment include adverse events (AEs), type III immune complex reactions, poor compliance, and pregnancy. The clinician group added that treatment with crovalimab is most likely going to be done either entirely at the patient’s home or, in the case of loading doses, at a local infusion clinic.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a Canadian Drug Expert Committee recommendation for crovalimab:
considerations for the initiation of therapy
considerations for the prescribing of therapy.
The clinical experts consulted for this review provided advice on the potential implementation issues raised by the drug programs. Refer to Table 5 for further details.
Clinical Evidence
Systematic Review
Description of Studies
Two multicentre, phase III, randomized, open-label, active-controlled trials submitted by the sponsor were included, comparing crovalimab (maintenance dose is 680 mg [body weight ≥ 40 kg to < 100 kg] or 1,020 mg [≥ 100 kg] by SC injection once every 4 weeks) with eculizumab (maintenance dose is 900 mg by IV infusion once every 2 weeks) in adult patients with PNH who had not been previously treated with a complement inhibitor (the COMMODORE 2 study, randomized N = 204) and patients with documented treatment with complement inhibitors (the COMMODORE 1 study, randomized N = 89). Patients were randomized to receive either crovalimab (arm A) or eculizumab (arm B) in a 2:1 ratio in the COMMODORE 2 study, and in a 1:1 ratio in the COMMODORE 1 study. In addition, both trials enrolled a nonrandomized descriptive arm (arm C) where patients only received crovalimab (with 6 patients with PNH aged younger than 18 years in the COMMODORE 2 study and 38 patients on a complement inhibitor treatment, including 1 patient with PNH aged under 18 years, in the COMMODORE 1 study, at the clinical cut-off date of November 16, 2022). In both trials, the primary treatment period was 24 weeks for all arms. After 24 weeks, all patients had the opportunity to continue or switch to crovalimab in a 46-week safety follow-up period. In the COMMODORE 2 trial, transfusion avoidance and hemolysis control (measured by an LDH level ≤ 1.5 × upper limit of normal [ULN] at a central laboratory) were coprimary outcomes. Secondary outcomes were BTH, stabilized hemoglobin, and the Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) version 4 score. The mean percentage change in LDH levels, maintenance of a minimum hemoglobin level, and the number of pRBC units transfused, among others, were also reported as exploratory outcomes. HRQoL assessed with the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) version 3 tool and selected symptoms of the European Organisation for Research and Treatment of Cancer – Item Library 40 (EORTC IL-40) were evaluated. Patient preference for crovalimab or eculizumab, and harms were also reported. In the COMMODORE 1 trial, due to the introduction of ravulizumab to the treatment landscape and a reduced pool of patients treated with eculizumab over time, randomization was stopped in November 2022 per the amended protocol, which was earlier than initially planned in the study. As a result, the evaluation of safety became the primary objective and all efficacy objectives became exploratory end points. The specific safety and efficacy outcomes in the COMMODORE 1 trial were similar to those in the COMMODORE 2 trial.
At baseline, the median age of the randomized population in the crovalimab and eculizumab arms was 36 years (range, 18 years to 76 years) and 38 years (range, 17 years to 78 years), respectively, in the COMMODORE 2 study, and 42 years (range, 21 years to 81 years) and 49 years (range, 22 years to 85 years), respectively, in the COMMODORE 1 study. The proportions of females and males were similar between the 2 arms in both studies (43% to 49% were females and 51% to 57% were males in the COMMODORE 2 study, and 50% to 53% were females and 47% to 50% were males in the COMMODORE 1 study). Most patients were Asian (64% in the crovalimab arm and 74% in the eculizumab arm) or white (33% in the crovalimab arm versus 23% in the eculizumab arm), followed by other or unknown (3%) in the COMMODORE 2 trial. In the COMMODORE 1 trial, most patients were white (76% in the crovalimab arm versus 73% in the eculizumab arm), followed by Asian (20% in the crovalimab arm versus 16% in the eculizumab arm), and other or unknown (4% in the crovalimab arm versus 11% in the eculizumab arm). In the COMMODORE 2 trial, the baseline mean LDH level was 7.6 multiplied by ULN (standard deviation [SD] = 3.4 × ULN) and 7.8 multiplied by ULN (SD = 3.5 × ULN) in the crovalimab and eculizumab arms, respectively. In the COMMODORE 2 study, a total of 77% of patients in the crovalimab arm had received a pRBC transfusion within the 12 months before screening, with a mean of 6.5 (SD = 8.3) transfused pRBC units; 74% of patients in the eculizumab arm had received a pRBC transfusion with a mean of 6.6 (SD = 8.7) transfused pRBC units (half of the patients received > 0 to ≤ 6 transfused pRBC units in the prior 6 months in both arms). In the COMMODORE 1 trial, 23% of the patients in the crovalimab arm had received a mean of 1.6 (SD = 3.7) transfused pRBC units, and 25% of the patients in the eculizumab arm had received a mean of 2.3 (SD = 5.4) transfused pRBC units. In both studies, the median of the PNH clone size was smaller in the crovalimab arm than in the eculizumab arm for erythrocytes (25% in the crovalimab arm versus 45% in the eculizumab arm in the COMMODORE 2 study and 45% in the crovalimab arm versus 47% in the eculizumab arm in the COMMODORE 1 trial), for granulocytes (60% in the crovalimab arm versus 75% in the eculizumab arm in the COMMODORE 2 trial and 67% in the crovalimab arm versus 68% in the eculizumab arm in the COMMODORE 1 trial), and for monocytes (91% in the crovalimab arm versus 95% in the eculizumab arm in the COMMODORE 2 trial and 89% in the crovalimab arm versus 96% in the eculizumab arm in the COMMODORE 1 trial). The main PNH-relevant conditions in patient history were aplastic anemia (39% in the crovalimab arm versus 38% in the eculizumab arm in the COMMODORE 2 study and 33% in the crovalimab arm versus 36% in the eculizumab arm in the COMMODORE 1 study) and major vascular events (16% in the crovalimab arm versus 15% in the eculizumab arm in the COMMODORE 2 study and 22% in the crovalimab arm versus 23% in the eculizumab arm in the COMMODORE 1 study).
Efficacy Results
The key efficacy results from the COMMODORE 2 and COMMODORE 1 studies are summarized in Table 2 and Table 3, respectively, ordered by importance and with categories suggested by the clinical experts consulted for this review. In the COMMODORE 1 study, all efficacy outcomes are exploratory as randomization was stopped early, and there was not sufficient power for formal statistical noninferiority or superiority testing.
Hemolysis Outcomes
Hemolysis control: In the COMMODORE 2 study, crovalimab demonstrated noninferiority compared with eculizumab for hemolysis control as measured by a central LDH level of 1.5 or less multiplied by ULN (odds ratio [OR] = 1.02; 95% confidence interval [CI], 0.57 to 1.82; P value = ████) based on a predefined noninferiority margin (NIM) of 0.2 for the lower limit of the 95% CI. The mean proportion of patients with hemolysis control from week 5 through week 25 was 79.3% (95% CI, 72.9% to 84.5%) in the crovalimab arm and 79.0% (95% CI, 69.7% to 86.0%) in the eculizumab arm.
In the COMMODORE 1 study, the proportion of patients with a central LDH level of 1.5 or less multiplied by ULN at baseline was 93.2% for crovalimab and 95.2% for eculizumab. The mean proportion of patients who achieved hemolysis control (a central LDH level ≤ 1.5 × ULN) during the primary treatment period (i.e., from baseline through week 25) was 92.9% (95% CI, 86.6% to 96.4%) in the crovalimab arm and 93.7% (95% CI, 87.3% to 97.0%) in the eculizumab arm.
BTH: In the COMMODORE 2 study, crovalimab showed noninferiority efficacy compared with eculizumab in the proportion of patients with a BTH event from baseline through week 25 with the crovalimab arm at 10.4% (95% CI, 6.0% to 17.2%) versus the eculizumab arm at 14.5% (95% CI, 7.5% to 25.5%) with a weighted difference of −3.9% (95% CI, −14.8% to 5.3%; P value = ████) based on a predefined NIM of 20% for the upper limit of the 95% CI.
In the COMMODORE 1 study, the proportion of patients with BTH from baseline through week 25 was 10.3% (95% CI, 3.3% to 25.2%) in the crovalimab arm and 13.5% (95% CI, 5.1% to 29.6%) in the eculizumab arm.
Mean percentage change in LDH levels: In the COMMODORE 2 study, the mean percentage reduction in LDH levels from baseline to week 25 was −73.6% (95% CI, −78.95% to −68.2%) in the crovalimab arm and −64.1% (95% CI, −71.4% to −56.8%) in the eculizumab arm.
In the COMMODORE 1 study, the mean percentage change in the central LDH level from baseline to the average over week 21, week 23, and week 25 was 16.6% (95% CI, ███ ██ ████) in the crovalimab arm and 4.5% (95% CI, ████ ██ ████) in the eculizumab arm.
Hemoglobin Outcomes
Stabilized hemoglobin: In the COMMODORE 2 study, crovalimab showed noninferiority efficacy compared with eculizumab in the proportion of patients reaching hemoglobin stabilization (avoidance of a ≥ 2 g/dL decrease in the hemoglobin level from baseline, in the absence of transfusion) from baseline through week 25 with the crovalimab arm at 63.4% (95% CI, 54.6% to 71.5%) versus the eculizumab arm at 60.9% (95% CI, 48.4% to 72.2%) with a weighted difference of 2.2% (95% CI, −11.4% to 16.3%; P value = ████) based on a predefined NIM of −20% for the lower limit of the 95% CI.
In the COMMODORE 1 study, the proportion of patients with stabilized hemoglobin from baseline through week 25 was 59.0% (95% CI, 42.2% to 74.0%) in the crovalimab arm versus 70.3% (95% CI, 52.8% to 83.6%) in the eculizumab arm.
Patients Who Reached and Maintained a Minimum Hemoglobin Level (Hemoglobin ≥ 10 g/dL Without a Subsequent Decrease < 9 g/dL)
The proportion of patients who reached and maintained a minimum hemoglobin level from baseline through week 25 was █████ ████ ███ ████ ██ █████ in the crovalimab arm and █████ ████ ███ ████ ██ █████ in the eculizumab arm in the COMMODORE 2 study; in the COMMODORE 1 study, the proportion was █████ ████ ███ ████ ██ █████ in the crovalimab arm and █████ ████ ███ ████ ██ █████ in the eculizumab arm.
Transfusion Outcomes
Transfusion avoidance: In the COMMODORE 2 study, crovalimab demonstrated noninferiority compared with eculizumab for transfusion avoidance from baseline through week 25 with a weighted difference in the proportion of patients with transfusion avoidance of −2.8% (95% CI, −15.7% to 11.1%, P = ████), based on the predefined NIM of −20% for the lower limit of the 95% CI. In the crovalimab arm, 65.7% (95% CI, 56.9% to 73.5%) of patients were transfusion-free from baseline through week 25 compared with 68.1% (95% CI, 55.7% to 78.5%) of patients in the eculizumab arm.
In the COMMODORE 1 study, the proportion of patients who achieved transfusion avoidance from baseline through week 25 was 79.5% (95% CI, 63.1% to 90.1%) for the crovalimab arm and 78.4% (95% CI, 61.3% to 89.6%) for the eculizumab arm.
Red blood cell transfusions (packed red blood cell [pRBC] units transfused): In the COMMODORE 2 study, during the 24-week randomized primary treatment period, 45 (33.6%) patients in the crovalimab arm and 22 (31.9%) patients in the eculizumab arm had at least 1 transfusion. The mean number of units of pRBCs transfused was 2.3 units (95% CI, 1.3 units to 3.4 units) and 2.2 units (95% CI, 1.0 unit to 3.4 units) in the crovalimab and eculizumab arms, respectively.
In the COMMODORE 1 study, 8 (20.5%) patients in the crovalimab arm and 7 (18.9%) patients in the eculizumab arm had at least 1 transfusion, and the mean number of pRBC units transfused from baseline to week 25 in all randomized patients was 0.97 unit (95% CI, 0.2 unit to 1.7 units) in the crovalimab arm and 1.9 units (95% CI, 0.5 unit to 3.3 units) in the eculizumab arm.
Patient-Reported Outcomes
FACIT-F: In both the COMMODORE 2 and COMMODORE 1 studies, FACIT-F (range, 0 to 52, with a higher score indicative of less fatigue) was assessed in adult patients only (≥ 18 years).
In the COMMODORE 2 study, FACIT-F data were evaluable in 95.5% of adult patients in the crovalimab arm and 95.7% of adult patients in the eculizumab arm at each visit from baseline through week 25. The adjusted mean change (improvement) from baseline at week 25 in FACIT-F was 7.8 points (95% CI, 6.5 points to 9.1 points) in the crovalimab arm compared to 5.2 points (95% CI, 3.4 points to 6.9 points) in the eculizumab arm, with a between-group difference of 2.6 points (95% CI, 0.7 point to 4.6 points). By week 25, 58.6% of patients in the crovalimab arm and 54.5% of patients in the eculizumab arm had an improvement in fatigue severity of at least 5 points.
In the COMMODORE 1 study, FACIT-F data were evaluable in 86.4% of adult patients in the crovalimab arm and 76.2% of adult patients in the eculizumab arm at each visit from baseline through week 25. The adjusted mean change in FACIT-F scores from baseline to week 25 was 1.1 points (95% CI, −1.5 points to 3.7 points) in the crovalimab arm and −2.6 points (95% CI, −5.4 points to 0.1 point) in the eculizumab arm, with a between-group difference of 3.7 points (95% CI, 0.05 point to 7.4 points). The proportion of patients with a 5-point or greater improvement from baseline in the FACIT-F score was not reported for the COMMODORE 1 study.
EORTC QLQ-C30 global health status (GHS) and QoL: In the COMMODORE 2 study, the mean change in the EORTC QLQ-C30 GHS and QoL score from baseline to week 25 was 13.4 points (95% CI, 10.1 points to 16.7 points) for the crovalimab arm and 9.9 points (95% CI, 4.8 points to 14.9 points) for the eculizumab arm. Mean values at week 25 were similar to normative population values.
In the COMMODORE 1 study, the mean change in the EORTC QLQ-C30 GHS and QoL score from baseline to week 25 was 5.7 points (95% CI, −2.4 points to 13.8 points) in the crovalimab arm and −1.0 point (95% CI, −6.9 points to 4.9 points) in the eculizumab arm.
Harms Results
During the 24-week primary safety period, in the COMMODORE 2 study, there were similar proportions of patients in the 2 treatment arms who reported at least 1 AE, with 78% of patients in the crovalimab arm and 80% of patients in the eculizumab arm. A total of 10% of patients in the crovalimab arm and 13% of patients in the eculizumab arm reported at least 1 serious adverse event (SAE). The most common AEs were any infections (24% in the crovalimab arm and 36% in the eculizumab arm), infusion-related reactions (16% in the crovalimab arm versus 13% in the eculizumab arm), hypokalemia (11% in the crovalimab arm versus 13% in the eculizumab arm), hypersensitivity other than type III immune complex reactions (type III hypersensitivity reactions, ██ ███ ████), and injection-related reactions (5% in the crovalimab arm versus not applicable in the eculizumab arm). The most common SAEs were infections and infestations (3% in the crovalimab arm versus 7% in the eculizumab arm).
In the COMMODORE 1 study, a higher proportion of patients in the crovalimab arm compared with the eculizumab arm reported at least 1 AE (77% versus 67%, respectively) or at least 1 SAE (14% versus 2%, respectively). The most common AEs in the crovalimab arm compared with the eculizumab arm were any infections (41% versus 36%, respectively), infusion-related reactions (14% versus 0%, respectively), hypersensitivity other than type III immune complex reactions (██ ███), and injection-related reactions (7% versus not applicable, respectively). The most common SAEs were infections and infestations (7% in the crovalimab arm versus 2% in the eculizumab arm).
In both trials, no cases of infection with Neisseria meningitidis, including meningococcal meningitis, were reported in either arm.
One patient each from the crovalimab arm (0.7%) and the eculizumab arm (1.4%) in the COMMODORE 2 study and no patients in the COMMODORE 1 study experienced AEs leading to withdrawal from treatment with crovalimab or eculizumab.
In the COMMODORE 2 study, no adverse events of special interest (AESIs) of type III immune complex reactions related to drug-target-drug complexes (DTDCs) were reported in either arm during the primary treatment period as the patients were treatment-naive. In the COMMODORE 1 study, 7 (15.9%) patients in the crovalimab arm versus no patients in the eculizumab arm experienced type III immune complex reactions, because these AEs were expected only in patients who switched from eculizumab to crovalimab who are at risk of developing DTDC-associated type III immune complex reactions. In both trials, there were no AESIs reported for abnormal liver function tests or suspected transmission of an infectious drug by the study drug in either arm.
In the COMMODORE 2 study, death was reported in 2 (1.5%) patients in the crovalimab arm (1 patient died of respiratory tract hemorrhage and the other patient died of myocardial infarction) and 1 (1.4%) patient in the eculizumab arm (this patient died of ischemic stroke). In the COMMODORE 1 study, no deaths were reported during the primary safety period.
Critical Appraisal
Methods for randomization and allocation concealment were appropriate in both the COMMODORE 2 and COMMODORE 1 trials. In both trials, most baseline characteristics were similar between the randomized crovalimab and eculizumab arms except for numeric differences in a few of the characteristics. In the COMMODORE 2 study, they included race (64% Asian and 33% white in the crovalimab arm, and 74% Asian and 23% white in the eculizumab arm), the median PNH clone size for erythrocytes (25% in the crovalimab arm and 45% in the eculizumab arm) and for granulocytes (60% in the crovalimab arm versus 75% in the eculizumab arm), as well as the history of myelodysplastic syndrome (MDS) before enrolment (4% in the crovalimab arm versus 9% in the eculizumab arm); in the COMMODORE 1 trial, numeric differences in certain characteristics included the median PNH clone size for monocytes (89% in the crovalimab arm versus 96% in the eculizumab arm). Nevertheless, the clinical experts did not consider that the between-group imbalance in these characteristics would impact the efficacy and safety results in the studies. In the COMMODORE 2 study, the predefined NIMs for the efficacy outcomes were considered appropriate. In the COMMODORE 1 study, randomization was stopped early (in November 2022 per protocol amendment version 6) due to the introduction of ravulizumab to the treatment landscape, and a reduced pool of patients treated with eculizumab over time.10 The initially targeted sample size for the randomized arms of approximately 200 patients could not be reached, providing insufficient statistical power for efficacy analyses. The results of the exploratory efficacy analyses were reported descriptively, with no formal statistical noninferiority or superiority testing, limiting the interpretation and certainty of efficacy results of the COMMODORE 1 study.
The clinical experts consulted for this review noted that the maintenance dose of eculizumab (900 mg maintenance every 2 weeks for IV infusion) in both pivotal studies aligned with its product monograph.11 In clinical practice, when BTH occurs repetitively, increasing the eculizumab dose may be considered, according to the clinical experts. For the COMMODORE 2 study, the lack of permitted dosage adjustments may have biased the efficacy results in favour of crovalimab relative to how eculizumab is dosed in clinical practice; however, the magnitude of this potential bias is unclear. In the pivotal trials, there was no randomized comparative evidence of crovalimab versus eculizumab at a higher dose than that recommended by Health Canada in patients who are C5 inhibitor–naive and in patients who switched from eculizumab.
Fatigue and HRQoL results were evaluable in most patients in the COMMODORE 2 study (96% of patients) and in 76% to 86% of patients in the COMMODORE 1 study. For these outcomes, the impact of missing outcome data in the COMMODORE 2 study is minimal; in the COMMODORE 1 study, the impact of missing outcome data (14% in the crovalimab arm and 24% in the eculizumab arm) is unclear. The open-label design of both trials may have impacted the reporting of subjective patient-reported outcomes (PROs), including the FACIT-F score, the EORTC QLQ-C30 GHS and QoL score, EORTC IL-40 symptoms, and patient treatment preference.
The interpretation of results of arm C in both trials is limited due to the lack of a comparator, the limited number of patients and events, and the descriptive summary of data. Similarly, the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of crovalimab versus any comparator during the extension periods beyond week 25 in both the COMMODORE 2 and COMMODORE 1 trials.
The clinical experts noted that overall, the eligibility criteria of patients in both trials aligned with the diagnosis standard and treatment indication for PNH in clinical settings, and the demographic and disease characteristics (including LDH levels and history of transfusion) of the patients were mostly aligned with the patients seen in clinical practice in Canada. The clinical experts consulted for this review noted that the patients excluded from the COMMODORE 2 and COMMODORE 1 trials due to not meeting 1 of the patient inclusion criteria (granulocyte or monocyte clone size ≥ 10%) are typically asymptomatic and would less likely be considered for treatment. If these patients were showing other disease manifestations indicating a therapy, crovalimab could also be used with anticipated similar efficacy and safety results as those seen in the 2 trials as per the clinical experts.
The sample size of pediatric patients was small in the COMMODORE 2 study (n = 6) and in the COMMODORE 1 study (n = 1), and only descriptive results were available. The clinical experts expected that pediatric patients would have efficacy results similar to those in the main trial arms; however, there is a need for enhanced safety consideration regarding the risk of infections, including meningitis in pediatric patients. The recommended body weight–based doses of crovalimab once every 4 weeks via SC injection were considered as adequate and reasonable in clinical practice in Canada by the clinical experts. The clinical experts pointed out the importance of monitoring patients for any occurrence of kidney dysfunction or infections (particularly meningitis) during the treatment of crovalimab. According to the clinical experts and the clinician group, given that compliance may impact treatment effects, longer-term comparative evidence on the durability of the effectiveness of crovalimab would be informative. Likewise, the occurrence of some AEs, especially rare ones (e.g., meningitis), may take longer than 24 weeks to be identified. Comparative longer-term follow-up to assess safety between crovalimab and other complement inhibitors would be preferred.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) framework was used to assess the certainty of the evidence for outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.12,13 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
clinical outcomes — hemolysis control, BTH, mean percentage change in LDH levels, stabilized hemoglobin, having reached and maintained a minimum hemoglobin level, transfusion avoidance, and pRBC units transfused
HRQoL outcomes — FACIT-F score and EORTC QLQ-C30 GHS and QoL score
harms outcomes — infections and deaths.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on the threshold (minimal important difference [MID]) in the literature for the FACIT-F score in both trials.
For the COMMODORE 2 study, the target of the certainty of evidence assessment was the presence or absence of a predefined NIM per study protocol for hemolysis control, BTH, stabilized hemoglobin, having reached and maintained a minimum hemoglobin level, and transfusion avoidance. Due to the lack of a formal MID estimate or NIM value, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for the mean percentage change in LDH levels, pRBC units transfused, the EORTC QLQ-C30 GHS and QoL score, infections, and deaths.
For the COMMODORE 1 study, due to the lack of a formal MID estimate or NIM (after amendment of the COMMODORE 1 study, all efficacy end points became exploratory and descriptive, and thus NIM values were not applicable), the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for hemolysis control, BTH, mean percentage change in LDH levels, stabilized hemoglobin, having reached and maintained a minimum hemoglobin level, transfusion avoidance, pRBC units transfused, the EORTC QLQ-C30 GHS and QoL score, infections, and deaths.
For the GRADE assessments, the COMMODORE 2 and COMMODORE 1 studies were assessed individually because the COMMODORE 2 study included patients naive to complement inhibition and the COMMODORE 1 study included complement inhibition–experienced patients.
Results of GRADE Assessments
Table 2 and Table 3 present the GRADE summary of findings for crovalimab versus eculizumab for patients with PNH who were naive to and had experience with complement inhibitors, respectively.
Long-Term Extension Studies
Description of Studies
The phase I and phase II COMPOSER trial (ClinicalTrials.gov identification number NCT03157635) consisted of 4 sequential parts and an open-label extension (OLE). This section includes a summary of the OLE period that evaluated the safety, tolerability, PKs, and PDs of crovalimab in patients with PNH, aged 18 years to 75 years, who were treatment-naive or who switched from eculizumab. Participants were enrolled at 14 sites in 6 countries: Germany (3 sites), Japan (5 sites), France (1 site), Hungary (2 sites), Korea (2 sites), and Italy (1 site). The study did not include Canadian sites.
Efficacy Results
Hemolysis Control
The proportion of patients reaching an LDH level of 1.5 or less multiplied by ULN per visit suggested there was little to no difference during the OLE period, with 80% to 100% of the evaluable patients at each visit having an LDH level of 1.5 or less multiplied by ULN.
The mean normalized LDH level was generally maintained below 1.5 multiplied by ULN during the OLE phase. The point estimate of the mean normalized LDH level by visit ranged from 1.09 to 1.24 multiplied by ULN.
Table 2: Summary of Findings for Crovalimab Versus Eculizumab for Complement Inhibitor–Naive Patients With PNH
Outcome and follow-up | Patients (study), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Eculizumab | Crovalimab | Difference | |||||
Hemolysis outcomes | |||||||
Mean proportion of patients with hemolysis control (measured by LDH ≤ 1.5 × ULN) from week 5 through week 25 Follow-up: 24 weeks | 203 (1 RCT) | OR = 1.02 (0.57 to 1.82) | 790 per 1,000 | 793 per 1,000 (729 to 845 per 1,000) | NR | Higha,b | Crovalimab results in little to no difference (i.e., a noninferior effect) in the proportion of patients with hemolysis control compared with eculizumab. |
Proportion of patients with breakthrough hemolysis from baseline to week 25 Follow-up: 24 weeks | 203 (1 RCT) | NR | 145 per 1,000 | 104 per 1,000 (60 to 172 per 1,000) | 39 fewer per 1,000 (from 148 fewer to 53 more per 1,000) | Higha,c | Crovalimab results in little to no difference (i.e., a noninferior effect) in the proportion of patients with breakthrough hemolysis when compared with eculizumab. |
Mean percentage change in LDH levels from baseline through week 25 Follow-up: 24 weeks | 203 (1 RCT) | NA | −64.1% | −73.6% (−79.0% to −68.2%) | −9.5% (−18.4% to −0.6%) | Moderated | Crovalimab likely results in a reduction in the mean percentage in LDH levels when compared with eculizumab; the clinical importance of the reduction is uncertain. |
Hemoglobin outcomes | |||||||
Proportion of patients with stabilized hemoglobin from baseline through week 25 Follow-up: 24 weeks | 203 (1 RCT) | NR | 609 per 1,000 | 634 per 1,000 (546 to 715 per 1,000) | 22 more per 1,000 (114 fewer to 163 more per 1,000) | Higha,e | Crovalimab results in little to no difference (i.e., a noninferior effect) in the proportion of patients with stabilized hemoglobin when compared with eculizumab. |
Proportion of patients who reached and maintained a minimum hemoglobin level from baseline through week 25 Follow-up: 24 weeks | ███ █ | ███ ███ | ███ ██ | ███ ███ ██ | ██ ████ | ████ | ██████████ ███ |
Transfusion outcomes | |||||||
Proportion of patients who achieved transfusion avoidance from baseline through week 25 Follow-up: 24 weeks | 203 (1 RCT) | NR | 681 per 1,000 | 657 per 1,000 (569 to 735 per 1,000) | 28 fewer per 1,000 (157 fewer to 111 more per 1,000) | Higha,g | Crovalimab results in little to no difference (i.e., a noninferior effect) in the proportion of patients who achieved transfusion avoidance when compared with eculizumab. |
Mean units of pRBC transfused from baseline through week 25 Follow-up: 24 weeks | 203 (1 RCT) | NA | 2.2 | 2.3 (1.3 to 3.4) | NR | Moderateh,i | Crovalimab likely results in little to no difference in mean units of pRBC transfused when compared with eculizumab. |
Patient-reported outcomes | |||||||
Adjusted mean change from baseline in FACIT-F (0 [worst] to 52 [best]) Follow-up: 24 weeks | 194 (1 RCT) | NA | 5.2 | 7.8 (6.5 to 9.1) | 2.6 (0.7 to 4.6) | Moderatej,k | Crovalimab likely results in little to no clinically important difference in FACIT-F score when compared with eculizumab. |
Absolute change in EORTC QLQ-C30 GHS and QoL score from baseline (0 [worst] to 100 [best]) Follow-up: 24 weeks | 194 (1 RCT) | NA | 9.9 | 13.4 (10.1 to 16.7) | NR | Lowh,j | Crovalimab may result in little to no difference in the EORTC QLQ-C30 GHS and QoL score compared with eculizumab. |
Harms | |||||||
Proportion of patients with infections (including Neisseria meningitidis) Follow-up: 24 weeks | 204 (1 RCT) | NR | 362 per 1,000 | 237 per 1,000 (NR) | 125 fewer per 1,000 (from 259 fewer to 9 more per 1,000)l | Lowm | Crovalimab may result in a reduction in the proportion of patients with infections compared with eculizumab. |
Deaths | |||||||
Proportion of patients who died Follow-up: 24 weeks | 204 (1 RCT) | NR | 14 per 1,000 | 15 per 1,000 (NR) | NR | Lown | Crovalimab may result in little to no difference in the proportion of patients who died when compared with eculizumab. |
CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GHS = global health status; LDH = lactate dehydrogenase; MID = minimal important difference; NA = not applicable; NIM = noninferiority margin; NR = not reported; OR = odds ratio; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; QoL = quality of life; RCT = randomized controlled trial; ULN = upper limit of normal.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the Table 2 footnotes. Data presented in this table were based on analyses at the clinical cut-off date of November 16, 2022. The outcomes — patients who reached and maintained a minimum hemoglobin level, mean percentage change in LDH levels, mean units of pRBC transfused, and EORTC QLQ-C30 GHS and QoL score — were not adjusted for multiplicity and should be considered as supportive evidence.
aImprecision did not result in the level of certainty being rated down. The point estimate and both the lower and upper boundaries of the 95% CI of the between-group comparison indicate trivial or no clinically meaningful difference according to the clinical experts consulted for this review. The 95% CI of the effect excludes the predefined NIM.
bFor hemolysis control, the NIM for the lower 95% CI limit of the OR was 0.2. In the absence of available data for the between-group difference in the mean proportion of patients with hemolysis control, the judgment of imprecision is based on the 95% CI for the OR of crovalimab versus eculizumab using the predefined NIM of 0.2 as the threshold (the lower limit of the 95% CI was above 0.2, declaring noninferiority of eculizumab).
cBreakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin < 10 g/dL], a major adverse vascular event [including thrombosis], dysphagia, or erectile dysfunction) in the presence of elevated LDH of 2 or more multiplied by ULN after a prior reduction of LDH to 1.5 or less multiplied by ULN on treatment. For breakthrough hemolysis, the NIM for the upper 95% CI limit for the weighted difference in proportions was 20%.
dThe level of evidence was rated down 1 level for serious imprecision. There was no known MID and clinical experts consulted by CDA-AMC could not provide a threshold of important difference. The CDA-AMC review team judged that the upper bound of the 95% CI was close to the null and unlikely to include an important effect.
eStabilized hemoglobin was defined as the avoidance of a 2 g/dL or more decrease in hemoglobin level from baseline, in the absence of transfusion. For stabilized hemoglobin, the NIM for the lower 95% CI limit of the weighted difference in proportions was −20%.
fPatients who reached and maintained a minimum hemoglobin level were defined as patients who reached a hemoglobin level of at least 10 g/dL, without a subsequent decrease below 9 g/dL, in the absence of transfusion. For this outcome, the NIM was −20%.
gTransfusion avoidance was defined as patients who were pRBC transfusion–free and who did not require a transfusion per protocol-specified guidelines. For transfusion avoidance, the NIM for the lower 95% CI limit of the weighted difference in proportions was −20%.
hThe level of evidence was rated down 1 level for serious imprecision. Serious imprecision was defined as being unable to conclusively determine whether the between-group difference included the null threshold of 0, which would have been used to assess certainty given that the review team was unable to identify the MID to assess a between-group difference from the literature or the clinical experts consulted for this review. The within-group CIs overlapped and the overall sample size was small.
iThis outcome presents the total number of pRBC units transfused among the full population.
jThe level of evidence was rated down 1 level for serious risk of bias in favour of crovalimab (maintenance dose based on body weight every 4 weeks, SC injections) compared with eculizumab (maintenance dose of 900 mg every 2 weeks, IV infusion) arising from the open-label nature of the study and the subjective nature of the outcome.
kFACIT-F was assessed in adult patients only (≥ 18 years). Imprecision did not result in the level of certainty being rated down. Based on the MID identified in the literature (5 points),14 which the clinical experts consulted for this review regarded as reasonable, the point estimate and both the lower and upper boundaries of the 95% CI of the between-group comparison suggested little to no difference.
lThis analysis was not part of the sponsor's statistical analysis plan and was requested by CDA-AMC to facilitate a certainty of evidence appraisal.
mThis outcome refers to the proportion of patients with at least 1 infection. No patients in either group had N. meningitidis. The level of evidence was rated down 2 levels for very serious imprecision. The review team was unable to identify the MID to assess a between-group difference from literature or the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CI of the absolute effect included the null threshold of 0.
nThe level of evidence was rated down 2 levels for very serious imprecision. There was a very small number of events captured.
Sources: COMMODORE 2 Primary Clinical Study Report15 and sponsor’s submission.16
Table 3: Summary of Findings for Crovalimab Versus Eculizumab for Complement Inhibitor–Experienced Patients With PNH
Outcome and follow-up | Patients (study), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Eculizumab | Crovalimab | Difference | |||||
Hemolysis outcomes | |||||||
Mean proportion of patients with hemolysis control (measured by LDH ≤ 1.5 × ULN) from baseline through week 25 Follow-up: 24 weeks | 76 (1 RCT) | OR = 0.88 (0.28 to 2.77) | 937 per 1,000 | 929 per 1,000 (866 to 964 per 1,000) | NR | Lowa | Crovalimab may result in little to no difference in the proportion of patients with hemolysis control when compared with eculizumab. |
Proportion of patients with breakthrough hemolysis from baseline to week 25 Follow-up: 24 weeks | 76 (1 RCT) | NR | 135 per 1,000 | 103 per 1,000 (33 to 252 per 1,000) | 35 fewer per 1,000 (from 192 fewer to 117 more per 1,000) | Lowb,c | Crovalimab may result in little to no difference in the proportion of patients with breakthrough hemolysis when compared with eculizumab. |
Mean percentage change in LDH levels from baseline to average of week 21, week 23, and week 25 Follow-up: 24 weeks | 76 (1 RCT) | NA | 4.5% | 16.6% (████ █) | 12.1% (█ ██ █) | Lowc | Crovalimab may result in an increase in the mean percentage change in LDH levels when compared with eculizumab. |
Hemoglobin outcomes | |||||||
Proportion of patients with stabilized hemoglobin from baseline through week 25 Follow-up: 24 weeks | 76 (1 RCT) | NR | 703 per 1,000 | 590 per 1,000 (422 to 740 per 1,000) | 108 fewer per 1,000 (308 fewer to 104 more per 1,000) | Lowc,d | Crovalimab may result in a decrease in the proportion of patients with stabilized hemoglobin when compared with eculizumab. |
Proportion of patients who reached or maintained a minimum hemoglobin level from baseline through week 25 Follow-up: 24 weeks | ██ ██ ███ | ██ | ███ ███ | ███ ███ ██ | ███ █████ | ███ | ████████ |
Transfusion outcomes | |||||||
Proportion of patients with transfusion avoidance from baseline through week 25 Follow-up: 24 weeks | 76 (1 RCT) | NR | 784 per 1,000 | 795 per 1,000 (631 to 901 per 1,000) | 18 more per 1,000 (167 fewer to 199 more per 1,000) | Lowc,f | Crovalimab may result in little to no difference in the proportion of patients with transfusion avoidance when compared with eculizumab. |
Mean units of pRBC transfused from baseline through week 25 Follow-up: 24 weeks | 76 (1 RCT) | NA | 1.9 | 1.0 (0.2 to 1.7) | NR | Lowg,h | Crovalimab may result in little to no difference in the mean units of pRBC transfused when compared with eculizumab. |
Patient-reported outcomes | |||||||
Adjusted mean change from baseline in FACIT-F (0 [worst] to 52 [best]) Follow-up: 24 weeks | 70 (1 RCT) | NA | −2.6 | 1.1 (−1.5 to 3.7) | 3.7 (0.05 to 7.4) | Lowi,j,k | Crovalimab may result in little to no clinically important difference in FACIT-F scores when compared with eculizumab. |
Absolute change in EORTC QLQ-C30 GHS and QoL score from baseline (0 [worst] to 100 [best]) Follow-up: 24 weeks | 70 (1 RCT) | NA | −1.0 | 5.7 (−2.4 to 13.8) | NR | Very lowh,i,k | The evidence is very uncertain about the effect of crovalimab on the EORTC QLQ-C30 GHS and QoL score compared with eculizumab. |
Harms | |||||||
Proportion of patients with infections (including Neisseria meningitidis) Follow-up: 24 weeks | 86 (1 RCT) | NR | 357 per 1,000 | 409 per 1,000 (NR) | 52 more per 1,000 (from 153 fewer to 257 more per 1,000)l | Lowm | Crovalimab may result in little to no difference in the proportion of patients with infections compared with eculizumab. |
Deaths | |||||||
Number of deaths Follow-up: 24 weeks | 86 (1 RCT) | NR | 0 | 0 | NA | Lown | Crovalimab may result in little to no difference in the proportion of patients who died when compared with eculizumab. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GHS = global health status; LDH = lactate dehydrogenase; MID = minimal important difference; NA = not applicable; NR = not reported; OR = odds ratio; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; QoL = quality of life; RCT = randomized controlled trial; SC = subcutaneous; ULN = upper limit of normal.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the Table 3 footnotes. Data presented in this table were based on analyses at the clinical cut-off date of November 16, 2022. The COMMODORE 1 trial was originally planned to recruit 200 patients to the randomized arms to be sufficiently powered to assess the efficacy of crovalimab. However, due to the introduction of ravulizumab to the treatment landscape and a reduced pool of patients treated with eculizumab over time, randomization to the crovalimab and eculizumab arms was stopped in November 2022 per protocol amendment version 6. The evaluation of safety became the primary objective and all efficacy objectives became exploratory. The results of the exploratory efficacy analyses were only reported descriptively because there was not sufficient power for formal statistical noninferiority or superiority testing. All outcomes in this table were not adjusted for multiplicity and should be considered as supportive evidence.
aThe level of evidence was rated down 2 levels for very serious imprecision. The review team was unable to identify the MID to assess a between-group difference from literature or the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CI of the OR included the null threshold of 1.
bBreakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin < 10 g/dL], a major adverse vascular event [including thrombosis], dysphagia, or erectile dysfunction) in the presence of an elevated LDH level of 2 or more multiplied by ULN after prior reduction of the LDH level to 1.5 or less multiplied by ULN on treatment.
cThe level of evidence was rated down 2 levels for very serious imprecision. The review team was unable to identify the MID to assess a between-group difference from literature or the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CI of the absolute effect included the null threshold of 0.
dStabilized hemoglobin was defined as the avoidance of a 2 g/dL or greater decrease in the hemoglobin level from baseline, in the absence of transfusion.
ePatients who reached or maintained a minimum hemoglobin level were defined as patients who reached or maintained a hemoglobin level of at least 10 g/dL, without a subsequent decrease below 9 g/dL, in the absence of transfusion.
fTransfusion avoidance was defined as patients who were pRBC transfusion–free and did not require a transfusion per protocol-specified guidelines.
gThis outcome presents the total number of pRBC units transfused among the full population.
hThe level of evidence was rated down 2 levels for very serious imprecision because the within-group CIs overlapped, the overall sample size was small, and the review team was unable to conclusively determine whether the between-group difference included the null threshold of 0, which would have been used to assess certainty given that the review team was unable to identify the MID to assess a between-group difference from the literature or the clinical experts consulted for this review.
iThe level of evidence was rated down 1 level for serious risk of bias in favour of crovalimab (maintenance dose based on body weight every 4 weeks, SC injections) compared with eculizumab (900 mg every 2 weeks, IV infusion) arising from the open-label nature of the study and the subjective nature of the outcome.
jFACIT-F was assessed in adult patients only (≥ 18 years). The level of evidence was rated down 1 level for serious imprecision. Based on the MID identified in the literature (5 points)14 that the clinical experts consulted for this review regarded as reasonable, the point estimate and the lower boundary of the 95% CI suggested little to no difference, and the 95% CI for the between-group difference crossed the MID threshold.
kThe level of evidence was further rated down 1 level for a potential risk of bias due to missing data (14% in the crovalimab arm and 24% in the eculizumab arm). The impact of these missing outcome data is unclear.
lThis analysis was not part of the sponsor's statistical analysis plan and was requested by CDA-AMC to facilitate a certainty of evidence appraisal.
mThis outcome refers to the proportion of patients with at least 1 infection. No patients in either group had N. meningitidis. This analysis was not part of the sponsor's statistical analysis plan and was requested by CDA-AMC to facilitate a certainty of evidence appraisal. The level of evidence was rated down 2 levels for very serious imprecision. There was a very small number of events captured.
nThe level of evidence was rated down 2 levels for very serious imprecision. There was a very small number of events captured.
Sources: COMMODORE 1 Primary Clinical Study Report17 and sponsor’s submission.16
Transfusion Avoidance
The proportion of patients with transfusion avoidance remained relatively stable over time during the OLE phase, with 82.9% to 91.7% of all patients avoiding transfusion across the 24-week intervals. Seven of the 11 patients who received a blood transfusion during the OLE phase reported a history of aplastic anemia and/or anemia.
Stabilized Hemoglobin
The proportion of patients reaching hemoglobin stabilization also remained relatively stable over time during the OLE phase, with 79.5% to 87.5% of all patients reaching hemoglobin stabilization across the 24-week intervals.
Breakthrough Hemolysis
The proportion of patients with BTH events was low during the OLE phase, with 0% to 4.9% of all patients reporting a BTH event across the 24-week intervals. One patient who had a BTH event discontinued the study before completing the full interval from week 20 to week 44, during which the BTH event occurred. Consequently, this BTH event was reported and included in the computation of the rate of BTH events adjusted for patient-years at risk but is not included in the calculation of the proportion of patients with BTH events per 24-week interval analyses.
Harms Results
███████████ of the 44 patients who were enrolled in part 2, part 3, and part 4 of the study entered the OLE phase, and █████ patients (██████ naive patients and ██████ patients who switched from eculizumab) were ongoing on crovalimab treatment at the time of the most recent clinical cut-off date (Table 23) (August 28, 2023). Overall, a total of ██████ patients discontinued from the OLE phase (██████ patient originally enrolled in part 2 [naive], █████ patients originally enrolled in part 3 [switch], ██████ patients originally enrolled in part 4, arm A [naive], and ██████ patient originally enrolled in part 4, arm B [switch]). No patients discontinued from the study due to an AE. The median treatment duration from baseline to the CCOD was ████ years.
A total of ██ ███████ patients experienced at least 1 AE from baseline to the CCOD. There was 1 death (in a patient who switched from eculizumab) that occurred in the OLE phase, in the period since the publication of the previous Update Clinical Study Report, and the death was not considered by the investigators to be related to study treatment. No AEs resulted in the withdrawal of a patient from the study. A total of ██ ███████ patients experienced at least 1 SAE. SAEs were reported in █████ of patients in the treatment-naive group and in █████ of patients in the switched-from-eculizumab group. The most frequently reported AEs by System Organ Class (with at least a 50% incidence in the total population) were infections and infestations (█████), gastrointestinal disorders (█████), and musculoskeletal and connective tissue disorders (█████).
The most frequently reported AEs with at least a 15% incidence in the total population were nasopharyngitis (█████), COVID-19 (█████), upper respiratory tract infection (█████), headache (█████), back pain (█████), pyrexia (█████), arthralgia (█████), cough (█████), bronchitis (█████), and dizziness (█████).
A total of ██ ███████ patients experienced at least 1 infection up to the CCOD (this was an increase from 36 [81.8%] patients as reported in the Update Clinical Study Report).
Critical Appraisal
The OLE period of the COMPOSER study was conducted to evaluate the longer-term safety and exploratory efficacy outcomes in 44 patients with PNH, beyond the 20-week primary evaluation period. Patients were enrolled in the extension period if study investigators determined that the patient derived benefit from treatment with crovalimab in the 20-week primary study period. The open-label design may have increased the risk of bias in patient selection and outcome ascertainment for end points that included more subjective assessments because the lack of blinding may have impacted investigators and patients’ expectations of treatment. The direction and magnitude of potential bias remains unclear. Given the design, the long-term extension was noncomparative because all patients in part 2, part 3, and part 4 received crovalimab. As a result, the comparative safety and efficacy of crovalimab to relevant comparators were not addressed and precluded conclusions about the comparative safety and efficacy of crovalimab. The small number of patients included in each study part, the single-arm setting, and the open-label design introduced significant uncertainty to study results and conclusions. There was variability in visit schedules across study parts, which may have introduced confounding to the study and limited the number of patients available at each follow-up time point.
The efficacy-evaluable population included all efficacy-evaluable patients who completed the primary treatment period in part 2, part 3, or part 4 of the study and were enrolled in the OLE period (18 treatment-naive patients and 25 switched patients). The study analysis included patients who were both treatment-naive and treatment-experienced — populations that differed in disease characteristics, such as transfusion history and LDH level. The impact of including both naive and experienced patients in the same analysis on the results is unclear.
During the OLE period, patients who were not receiving dosing every 4 weeks were required to switch to receive either 680 mg SC every 4 weeks (body weight ≥ 40 kg to < 100 kg) or 1,020 mg SC every 4 weeks (body weight ≥ 100 kg), which is aligned with the product monograph recommendation.
No method for the imputation of missing values was reported, and the attrition rate was 11.6%.
The study included diverse geographical representation and populations with varying demographics and health care systems, which may have enhanced the generalizability of the results. However, the included patients were mostly men (60% to 86%), with ethnicity of “not Hispanic or Latino,” and included patients with median ages ranging from 44 years to 56 years; therefore, results may not be generalizable to a broader population. Patients with conditions such as a psychiatric disorder, hereditary complement deficiency, a history of meningococcal meningitis, active primary or secondary immunodeficiency, a known history of HIV infection, chronic active hepatitis C, malignant disease, and alcohol, drug, or chemical abuse within 1 year before screening were excluded, and the results may not be generalizable to these groups of patients.
The dose-escalation approach, which allowed for the inclusion of different dosing frequencies, may have enhanced the generalizability across patient subgroups; however, frequent modifications and variations between groups increases intergroup variability and impacts the certainty and interpretation of study results.
Indirect Comparisons
Description of Studies
Due to the scarcity of direct evidence comparing crovalimab with other existing relevant therapies, including ravulizumab, eculizumab biosimilar, and the standard of care without a C5 inhibitor for PNH, the sponsor conducted a network meta-analysis (NMA) comparing crovalimab with ravulizumab, eculizumab biosimilar, and the standard of care without a C5 inhibitor for PNH.
Efficacy Results
The NMA suggested little to no difference between crovalimab and ravulizumab for the mean difference in the proportion of patients with transfusion avoidance, hemoglobin stabilization, and number of pRBC units transfused; however, the 95% credible intervals (CrIs) were wide and included the possibility that either treatment may be favoured. The NMA suggested little to no difference between crovalimab and ravulizumab for the change in the FACIT-F score; however, the CIs were wide and included the possibility of a clinically important effect that favoured crovalimab.
Harms Results
███ ███ ██████████ ██ ██████████ ████ ███████████ ███ ███ ████ ██ ████████████ ███ ███████ ███████ ███ █████ ████████ ████████ ████████████ ████████ ███ ███ ███ ███ ████ ███ ████████ ███ █████████ ████ ██████ █████████ ███ ██ █████████.
Critical Appraisal
Overall, the NMA was conducted according to accepted methodological guidance. A key limitation of the NMA was the heterogeneity in effect modifiers and prognostic factors across the included studies. The base-case analysis included patients who were both treatment-naive and treatment-experienced — populations that differed in disease characteristics such as transfusion history and LDH level. This heterogeneity suggests that the assumption of similarity across the included studies may not hold true for the NMA, increasing the uncertainty about the validity of the results for determining the effectiveness of crovalimab compared to ravulizumab. In a subgroup analysis of treatment-naive and treatment-experienced patients, the results were overall consistent with base-case results. Other limitations included the fact that the evidence networks were sparse, the connections between treatment nodes were informed by 1 or 2 trials, and the total number of patients in the included studies was relatively small. The aforementioned limitations may have increased the potential for biased treatment-effect estimates, and the results of the NMA should be interpreted with consideration of the limitations.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Conclusions
Two phase III, multicentre, open-label RCTs evaluated the efficacy and safety of SC injection of crovalimab at a weight-based dose once every 4 weeks compared with IV infusion of eculizumab 900 mg once every 2 weeks in adult patients with PNH after 24 weeks of treatment. Evidence from the COMMODORE 2 trial in patients with PNH who were naive to C5 inhibitors demonstrated that crovalimab was noninferior to eculizumab across the coprimary end points (hemolysis control [measured by LDH ≤ 1.5 × ULN] and transfusion avoidance) and secondary outcomes (BTH and stabilized hemoglobin). Other secondary (FACIT-F score) and exploratory end points were supportive of the noninferiority results, suggesting overall little to no difference between the treatment groups. Efficacy outcomes were rated as being of high to moderate certainty (except the EORTC QLQ-C30 GHS and QoL score, which was rated as low certainty), using the GRADE approach.
Results from the COMMODORE 1 trial in patients with PNH who were exposed to eculizumab were overall supportive of the results observed in the COMMODORE 2 trial, suggesting that crovalimab results in little to no difference in hemolysis control, BTH, transfusion avoidance, and the FACIT-F score compared with eculizumab. Efficacy analyses in the COMMODORE 1 trial were descriptive without formal statistical testing and should be considered as supportive evidence. The evidence was rated as being of low certainty, using the GRADE approach.
Descriptive analyses on long-term efficacy and safety based on the OLE phases of the COMMODORE 1 and COMMODORE 2 trials (an additional 24 weeks of treatment with crovalimab) and an OLE study (the COMPOSER study, with crovalimab treatment for a median duration of 3.40 years) appeared consistent with the randomized 24-week treatment periods of the pivotal trials for the crovalimab group, suggesting an ongoing benefit of crovalimab.
Based on the results of the COMMODORE 2 and COMMODORE 1 studies, the overall safety profile of crovalimab was consistent with that expected for a C5 inhibitor, and no additional safety signals were identified from the longer-term, single-arm COMPOSER study; however, long-term comparative data were not available. Approximately 1 in 5 patients who switched to receive crovalimab from eculizumab reported type III immune complex–mediated reactions.
The sample size of pediatric patients was small in the COMMODORE 2 study (n = 6) and in the COMMODORE 1 study (n = 1), and only descriptive results were available. The clinical experts anticipated that pediatric patients would have efficacy results similar to those of the main trial arms; however, there is a need for enhanced safety consideration regarding the risk of infections, including meningitis in pediatric patients.
Indirect evidence from the sponsor-submitted NMA suggested that crovalimab may provide efficacy and safety comparable to that of ravulizumab. Despite the analyses suggesting overall little to no difference between crovalimab compared to ravulizumab, the evidence was insufficient (i.e., a limited number of included studies, heterogeneity in patient characteristics across trials, and CrIs that crossed the null) to draw definitive conclusions on the relative efficacy of crovalimab compared to ravulizumab. The clinical experts consulted by the review team indicated that results suggesting little to no difference are plausible given that the 2 comparators belong to the same group of drugs and ravulizumab and eculizumab have demonstrated similar benefits in adult patients with PNH in RCTs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of crovalimab (Piasky) 340 mg/2 mL for SC injection following the 1 loading dose by IV infusion in the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg.
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
PNH is a rare, life-threatening, complement-mediated hematological disorder. PNH is the primary manifestation of chronic intravascular hemolysis, associated with bone marrow failure and thrombophilia.1 It develops when hematopoietic cells acquire somatic mutations in the X-linked gene of PIGA.2
PNH can develop at any age, but it is most commonly diagnosed in the third decade of life, with no ethnic, geographic, or sex preferences.4 The mortality rate of patients with PNH who receive supportive care alone is approximately 20% to 35% within 5 years to 10 years of diagnosis.4 Chronic kidney disease, pulmonary hypertension, and venous or arterial thromboembolic events are life-threatening complications of PNH.4 Thromboembolic events were the leading cause of death in patients with PNH (40% to 67% of deaths with known cause) and their risk is high even among patients with no clinical evidence of thromboembolism or those under prophylactic anticoagulation therapy.5 In 1 study, the disease-specific cumulative incidence of mortality at 10 years was reported as 5.2%, with a transplant considered as a competing risk.5
Key clinical signs and symptoms of PNH include anemia, fatigue, thrombosis, esophageal spasms, male erectile dysfunction, hemoglobinuria, abdominal pain, and dyspnea.1,4 Patients with PNH and their caregivers may experience impaired HRQoL and may struggle to complete normal everyday activities.18
Although epidemiological data are sparse, the previously estimated 15-year prevalence of PNH is 15.9 patients per 1 million population and the incidence is estimated at approximately 1.3 cases per million individuals per year.19 As of January 2018, the Canadian PNH Network followed 67 patients on therapy as well as 97 patients with small clones in the context of bone marrow failure; up to 52% of these patients were female and the median age of disease onset was 43 years.3
A diagnosis of PNH may be suspected if a patient presents with hemoglobinuria, cytopenia, thrombosis, aplastic anemia and/or myelodysplasia, or Coombs-negative hemolysis, with no known cause.3 A diagnosis of PNH is made by a combination of clinical examination, patient history, a physical exam, and specialized tests.3
A diagnosis of PNH will be confirmed by peripheral blood flow cytometry to detect the loss of glycosylphosphatidylinositol (GPI)-linked structures.3
The International PNH Interest Group has classified disease with GPI-deficient PNH clones into 3 subtypes: classic PNH, PNH in the context of other primary bone marrow disorders, and subclinical PNH.20 In the classic PNH subcategory, there is clinical evidence of intravascular hemolysis; however, there is no evidence of another defined bone marrow abnormality. Patients with PNH in the context of another specified bone marrow disorder have clinical and laboratory evidence of hemolysis plus a history of marrow abnormality. In the last category, patients with subclinical PNH have no clinical or laboratory evidence of hemolysis, but there are small populations of hematopoietic cells deficient in GPI-anchored protein.20
A Belgian expert panel recommended that patients with a granulocyte clone size of greater than 0.01% need to be monitored regularly, with frequency determined by clinical manifestations and clone size. In the absence of clinical manifestations, recommendations suggest that those with a clone size of less than 1% should be tested every year, whereas those with a clone size of greater than 1% should be tested every 6 months.21
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The goals of PNH treatment are to reduce mortality, the number of thromboembolic events, and the risk of intravascular hemolysis; slow the progression of pulmonary pressures and renal impairment; alleviate PNH symptoms; improve patients’ HRQoL; and help patients regain better functional status, enabling a return to prediagnosis activities and employment.22 The clinical experts consulted for this review also pointed out that for pediatric patients with PNH, an additional treatment goal would be to control the enhanced complications risks of intravascular hemolysis and thromboembolic events because of aplastic or hypoplastic anemia, the condition that is often associated with PNH in children.
An allogeneic bone marrow transplant is considered a curative treatment for PNH.6,7 However, a bone marrow transplant is not usually offered as a first-line treatment for classic PNH, owing to the risks of transplant-related morbidity and mortality, and is usually reserved for pediatric or younger adult patients with PNH with concomitant severe aplastic anemia or other causes of bone marrow failure.4,8,9 The clinical experts consulted for this review noted that immunosuppressive therapy can be a treatment option when there is no bone marrow donor.
In Canada, patients with PNH receive C5 inhibitors as standard first-line therapy to reduce uncontrolled complement activation in the terminal complement cascade, and the primary C5 inhibitor therapies are ravulizumab (IV infusion administration every 8 weeks as a maintenance weight-based dose after a single loading weight-based dose) and eculizumab (IV infusion administration every 14 days as a maintenance dose after weekly doses over the first 5 weeks after initiation).4 The clinical experts consulted for this review noted the treatment algorithm for patients with PNH in Canada as follows: once a patient has developed the criteria for high disease activity (an elevated LDH level > 1.5 × ULN and 1 of the symptoms of fatigue, thrombosis, renal insufficiency, transfusions, and anemia), an application for eculizumab or ravulizumab would be submitted to the provincial or territorial special authorization drug plan. The current funding criteria for eculizumab vary across provinces and territories, but are generally aligned to initiate complement inhibition in patients with a leukocyte PNH clone of greater than 10%, laboratory evidence of significant intravascular hemolysis, and at least 1 of the following: symptomatic anemia (regardless of transfusion dependence), thrombosis, renal insufficiency, pulmonary insufficiency or hypertension, abdominal pain requiring admission, or opioid analgesia.3 The clinical experts consulted for this review noted that the majority of patients would start on C5 inhibition in the upfront setting, with ravulizumab being preferred due to the convenience to the patient and a lower risk of BTH, but eculizumab is likely preferred in patients who are pregnant. Eculizumab and immunosuppressive therapy can be used concurrently in situations where both are felt to be indicated.23 Of note, ravulizumab is not reimbursed in all Canadian jurisdictions (e.g., British Columbia), leaving eculizumab as the only C5 inhibitor option for some patients.23
In Canada, patients receiving C5 inhibitors who are diagnosed with extravascular hemolysis may be switched to pegcetacoplan.24,25 Pegcetacoplan, a proximal C3 inhibitor, was approved by Health Canada in December 2022 and is indicated for the treatment of adults with PNH who have an inadequate response to, or are intolerant of, a C5 inhibitor.26 It is administered subcutaneously twice a week via a syringe system infusion pump.25 Pegcetacoplan has been previously reviewed by CDA-AMC with a recommendation of reimburse with conditions (April 2023).24
Supportive care may be necessary for some patients with PNH, comprising additional transfusions, iron and folic acid supplementation, and analgesia to manage abdominal pain and smooth muscle dystonia.4
Drug Under Review
Key characteristics of crovalimab are summarized in Table 4 with other treatments available for the treatment of patients with PNH.
Crovalimab solution, 340 mg/2 mL (170 mg/mL) for injection and infusion, is indicated for the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg. It has not been previously reviewed by CDA-AMC.
The recommended dosing regimen consists of 1 loading dose administered by IV infusion (on day 1), followed by 4 additional weekly loading doses administered by SC injection (on day 2, day 8, day 15, and day 22). The maintenance dose starts on day 29 and is then administered every 4 weeks by SC injection.
Loading dose:
On day 1, the dose is 1,000 mg (IV) for a body weight of 40 kg to less than 100 kg, and 1,500 mg (IV) for a body weight of 100 kg or more.
On day 2, day 8, day 15, and day 22, the dose is 340 mg (SC) for a body weight of 40 kg or more.
Maintenance dose: On day 29 and every 4 weeks thereafter, the dose is 680 mg (SC) for a body weight of 40 kg to less than 100 kg, and 1,020 mg (SC) for a body weight of 100 kg or more.
Crovalimab is a recombinant humanized immunoglobulin G1–based monoclonal antibody that specifically binds with high affinity to component 5 of the complement system (C5), inhibiting its cleavage into C5a and C5b and thus preventing the formation of the membrane attack complex. In patients with PNH, crovalimab inhibits terminal complement-mediated intravascular hemolysis. Crovalimab binds to a different C5 epitope compared to other C5 inhibitors ravulizumab and eculizumab.
The sponsor’s reimbursement request is for the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg; this is aligned with the Health Canada indication.
Crovalimab is approved by FDA for the treatment of adult and pediatric patients 13 years and older with PNH and a body weight of at least 40 kg. In the European Union, crovalimab as monotherapy is indicated for the treatment of adult and pediatric patients aged 12 years or older with a weight of 40 kg or more with PNH:
in patients with hemolysis with clinical symptom(s) indicative of high disease activity
in patients who are clinically stable after having been treated with a C5 inhibitor for at least the past 6 months.
In the European Union, crovalimab is under additional monitoring, meaning that it is monitored even more intensively than other medicines.27
Table 4: Key Characteristics of Crovalimab, Eculizumab, and Ravulizumab
Characteristic | Crovalimab | Eculizumab | Ravulizumab |
|---|---|---|---|
Mechanism of action | C5 inhibitor that inhibits terminal complement-mediated intravascular hemolysis and prevents formation of the membrane attack complex | C5 inhibitor that inhibits terminal complement-mediated intravascular hemolysis | C5 inhibitor that inhibits terminal complement-mediated intravascular hemolysis and prevents formation of the membrane attack complex |
Indicationa | For the treatment of PNH in adults and adolescents 13 years of age and older with a body weight of at least 40 kg | PNH, atypical hemolytic uremic syndrome, generalized myasthenia gravis, neuromyelitis optica spectrum disorder | PNH, atypical hemolytic uremic syndrome, generalized myasthenia gravis, neuromyelitis optica spectrum disorder |
Route of administration | Undiluted for SC injection or diluted for IV infusion | IV infusion | IV infusion |
Recommended dosage | One loading dose IV on day 1 of 1,000 mg (for body weight of ≥ 40 kg to < 100 kg) or 1,500 mg (for body weight of ≥ 100 kg), 4 weekly loading doses SC of 340 mg (for body weight of ≥ 40 kg) on day 2, day 8, day 15, and day 22 Maintenance dose of 680 mg (for body weight of ≥ 40 kg to < 100 kg) or 1,020 mg (for body weight of ≥ 100 kg), starts on day 29 and is then every 4 weeks SC | 600 mg every 7 days for the first 4 weeks, followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter | Depending on body weight, a loading dose of 600 mg to 3,000 mg, and a maintenance dose of 300 mg to 3,600 mg Dosing interval of every 4 weeks for body weight ≥ 5 kg to < 20 kg, and every 8 weeks for body weight ≥ 20 kg |
Serious adverse effects or safety issues | Serious or fatal meningococcal infections and/or sepsis. Serious infections other than meningococcal infection. Serious hemolysis after drug discontinuation | Serious or fatal meningococcal infections, and allergy and/or infusion reactions. Patients who discontinue should be carefully monitored. | Life-threatening meningococcal infections and/or sepsis, infusion reactions, and systemic infections |
Other | Patients must be vaccinated against meningococcal infections, Streptococcus pneumoniae, and Haemophilus influenzae type b. | Patients must be vaccinated against meningococcal infections, S. pneumoniae, and H. influenzae type b. | Patients must be vaccinated against meningococcal infections. Patients younger than 18 years must be vaccinated against H. influenzae and pneumococcal infections. |
PNH = paroxysmal nocturnal hemoglobinuria; SC = subcutaneous.
aHealth Canada–approved indication.
Sources: Product monograph for crovalimab (Piasky),28 product monograph for eculizumab (Soliris),11 and product monograph for ravulizumab (Ultomiris).29
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received by CDA-AMC are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 1 joint input from the Canadian Association of PNH Patients and AAMAC. The Canadian Association of PNH Patients is a nonprofit Canadian organization dedicated to advocating for optimal patient care, ensuring access to the latest tools and information for managing the condition effectively, raising awareness, and offering support to caregivers. AAMAC’s mission is to offer comprehensive support to people in Canada affected by aplastic anemia, myelodysplasia, or PNH, including patients, family members, friends, and health care providers.
Due to the limited size of the clinical trial for crovalimab, the patient groups were able to receive input from only 1 patient living in Canada. This patient had experience with crovalimab treatment. In this submission, the patient groups also provided information from the crovalimab clinical trial to highlight the trial’s findings and demonstrate the potential efficacy of crovalimab.
The patient groups stated that PNH is a complex and multifaceted disorder that requires comprehensive management to address the various aspects of the disease and improve the QoL of those affected. The chronic nature of PNH means that patients must manage their condition over a lifetime, dealing with the physical, emotional, and financial burdens associated with the disease. The impact on QoL is profound, as patients must cope with the unpredictability of symptoms, the side effects of treatments, and the constant threat of serious complications. Regular monitoring and supportive care are essential to managing the disease and improving patient outcomes.
According to the patient group input, 1 patient with PNH in Canada reported that the disease started with feeling constantly exhausted, followed by dark urine in the morning and chest pain. The patient explained feeling terrified of the risk of blood clots, and not knowing the effect of the disease on life, job, and family, along with having to accept that this is a lifelong condition.
Based on the input, 1 significant challenge with current treatments is that they require regular clinic visits every 2 weeks to 8 weeks for IV infusions, which can be time-consuming and disruptive. Because crovalimab can be given as a small SC injection, it is more convenient for patients than IV treatments. Based on the patient group input, the patient with PNH reported that the SC crovalimab treatment that only requires administration once every 4 weeks was much more convenient than a treatment that requires regular IV infusions. Furthermore, the patient reported that the SC injections are quick and easy, and can be administered at home. This patient reported that crovalimab significantly reduced hemolysis, improved energy levels within the first few months, reduced fatigue, increased independence, and alleviated the fear of blood clots. The patient also noted that the side effects of crovalimab were minimal.
The patient groups stated that crovalimab has the potential to greatly improve QoL. According to the patient groups, crovalimab reduces the physical, emotional, and logistical burdens associated with traditional IV therapies, allowing patients to regain a sense of freedom and control over their lives. Fewer clinic visits and less travel for IV injections mean lower health care costs and less financial burden for patients. The more convenient and less invasive approach can significantly improve not only patients’ QoL but also patients’ overall well-being and adherence to treatment, leading to better health outcomes.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PNH.
Unmet Needs
The clinical experts consulted for this review noted that crovalimab may fulfill an unmet need for some patients with a preference for an SC route of drug administration, particularly for those with poor venous access and who are opposed to the insertion of a port-a-cath (a tube that is inserted into a vein and connected to a port that is placed under the skin). The clinical experts noted that crovalimab may be a safer alternative for patients living in isolated geographic locations that preclude the guaranteed delivery of eculizumab, and some patients may be more comfortable with self-administration than having to travel to a clinic for IV therapy (which is associated with transportation costs and exposure to potential infections at a clinic).
Place in Therapy
According to the clinical experts consulted for this review, most patients with PNH would choose to start ravulizumab as the first-line therapy over eculizumab or crovalimab where ravulizumab is funded (of note, ravulizumab is not funded in some jurisdictions such as British Columbia). However, eculizumab would be preferred for patients who are pregnant. Furthermore, the clinical experts noted that patients who prefer to be more independent in receiving therapy, have significant needle phobia, or find it difficult to visit a clinic due to geographic locations may choose crovalimab as first-line therapy. The clinical experts noted that switching from ravulizumab or eculizumab to crovalimab could be uncommon because there is a risk of transient immune complex formation that only occurs in patients switching to or from other C5 inhibitors.
The clinical experts further pointed out that in children with PNH, due to the high association with aplastic or hypoplastic anemia, the management would be driven by treating aplastic or hypoplastic anemia and allogeneic hematopoietic stem cell transplant is commonly considered a treatment option in children with PNH. The clinical experts noted that the rare cases of isolated PNH would be treated along the same lines as those of adults, except that PNH clone size is smaller in children and only clones of greater than 10% would be eligible for C5 inhibition therapy. The clinical experts noted that crovalimab would be indicated in the rare setting of PNH without aplastic anemia.
Patient Population
The clinical experts noted that patients who are fully motivated to self-administer the SC injections and those known to have genetic polymorphism that obviates efficacy with eculizumab or ravulizumab would be best suited for treatment with crovalimab.
Assessing the Response to Treatment
The clinical experts pointed out that crovalimab is another C5 inhibitor. Thus, the outcomes used to assess the treatment response would be the same as for eculizumab or ravulizumab, including improvements in hemoglobin levels, transfusion avoidance, and renal function, along with reductions in smooth muscle spasm, less fatigue, the normalization of LDH levels, and avoidance of BTH. The clinical experts noted that frequent laboratory and patient assessments are required during the early stages of crovalimab initiation, followed by standard assessments once the laboratory results stabilize.
Discontinuing Treatment
The clinical experts noted that if patients treated with crovalimab are not showing any clinical responses by 3 weeks to 4 weeks, a transition back to ravulizumab or eculizumab monitoring for the immune responses should be considered. The clinical experts pointed out that crovalimab should be discounted when any thrombosis occurs, or when there is an ongoing LDH elevation or ongoing symptoms of PNH.
Prescribing Considerations
The clinical experts commented that ideally, crovalimab should be prescribed by hematologists treating patients with PNH and the management of patients receiving crovalimab should take place at a centre of excellence.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One clinician group input was received from the Canadian PNH Network. The Canadian PNH Network is a group of hematologists in Canada with a special interest and expertise in the care of patients with PNH; the members represent centres of expertise from Newfoundland and Labrador, Nova Scotia, Quebec, Ontario, Alberta, and British Columbia. Information for this submission was obtained via publicly available documents, congress abstracts, and the published literature, and members of the Canadian PNH Network were invited to contribute to the various sections. A total of 3 clinicians contributed to this submission.
According to the clinician group input, the current standard of care for patients with hemolytic PNH is terminal complement inhibition with a C5 blockade; as such, eculizumab and ravulizumab remain the only first-line therapies across Canada. The only curative treatment for PNH at this time is an allogeneic hematopoietic stem cell transplant. It should be noted, however, that this is reserved for patients with predominant or progressive bone marrow failure (e.g., aplastic anemia), which can coincide with, precede, or follow a diagnosis of PNH. More recently, danicopan received a positive conditional CDA-AMC recommendation (November 2024); it is used in combination with a continued C5 inhibitor, either eculizumab or ravulizumab. The CDA-AMC review team notes that at the time of receiving the crovalimab submission, danicopan was still under review with CDA-AMC.
The Canadian PNH Network highlighted that crovalimab is expected to address important treatment goals unmet by current therapies, including offering a C5 inhibitory strategy that does not require IV access, enabling self-administration by patients or administration by caregivers, and providing an option for rare cases of resistance to eculizumab or ravulizumab due to a C5 polymorphism. Additionally, the clinician group expected that crovalimab would be another first-line option for patients with PNH.
In terms of patients who are best suited for treatment with crovalimab, the Canadian PNH Network noted that crovalimab would be offered as first-line treatment to patients with PNH, either switching from an IV C5 inhibitor or starting from being treatment- naive. Patients who favour the freedom and reduced treatment burden of SC administration would very likely select crovalimab therapy. On the other hand, patients with PNH least suitable would be those who are not accepting SC drug delivery, or those who develop clinically significant extravascular hemolysis, which necessitates proximal complement inhibition strategies.
The Canadian PNH Network stated that response to treatment focuses on LDH reduction, which reduces hemolysis and the risk of thrombosis and may improve hemoglobin as well as transfusion dependence in patients with PNH. Clinical outcomes related to the response included decreased fatigue and transfusion requirements as well as improved QoL and overall survival. Efficacy outcomes would typically be assessed every 2 weeks to 4 weeks initially after starting a new therapy or switching, but follow-up would be required less often (e.g., every 3 months to 6 months) as a patient becomes established on the drug and does not show evidence of side effects or other concerns.
Based on the input, some of the factors that should be considered to discontinue the treatment include AEs, type III immune complex reactions, poor compliance, and pregnancy. The clinician group added that treatment with crovalimab is most likely going to be done either entirely at the patient’s home or, in the case of loading doses, at a local infusion clinic.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation question | Clinical expert response |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trials
| This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Prior therapies required for eligibility: A first-line option like Soliris (eculizumab) and Ultomiris (ravulizumab) Question for the clinical experts: Should patients having experienced a drug of the same class (C5 inhibitors), such as first-line Soliris (eculizumab) or Ultomiris (ravulizumab), be eligible for crovalimab? Consider alignment with prescribing criteria for Soliris (eculizumab) and Ultomiris (ravulizumab). | The clinical experts consulted for this review noted that patients who have experienced a drug of the same class, (i.e., a C5 inhibitor such as eculizumab or ravulizumab) are eligible for crovalimab. The clinical experts noted that patients who switch between different C5 inhibitors are at risk of developing DTDC-associated type III immune complex reactions (e.g., in the COMMODORE and COMPOSER studies, approximately 18% of the patients who switched from eculizumab to crovalimab over an observation period of 44 weeks developed this), which are generally self-limiting, or manageable with appropriate treatment. |
Soliris (eculizumab) is covered in New Brunswick, Ontario, Manitoba, Alberta, and British Columbia, as well as by the NIHB program. For consistency, consider alignment with initiation criteria for Soliris and Ultomiris (initial approval of 6 months). Question for the clinical experts: To reduce the risk for meningococcal disease (McNamara et el. [2017]),30 should there be a consideration for an antimicrobial prophylaxis with oral antibiotics for the duration of therapy? | The clinical experts consulted for this review recommended that patients must be fully vaccinated against meningococcal infections (per the guidelines, the vaccinations for meningitis are given every 3 years). With regard to an antimicrobial prophylaxis with oral antibiotics for the duration of therapy with crovalimab, the clinical experts were concerned about the adverse effects associated with a long-term antimicrobial prophylaxis and recommended against the universal use of oral antibiotics as a routine prophylaxis among the patient target population. However, the clinical experts noted that the treating physician should assess the risk of infections, especially meningitis, based on the patients’ characteristics, comorbidities, and testing results, and decide whether an antimicrobial prophylaxis is required during the therapy. |
Considerations for continuation or renewal of therapy | |
For consistency, consider alignment with prescribing criteria for Soliris and Ultomiris. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for discontinuation of therapy | |
For consistency, consider alignment with prescribing criteria for Soliris and Ultomiris. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
The weight-based loading dose is administered intravenously on day 1, followed by additional weight-based loading doses administered subcutaneously on days 2, 8, 15, and 22. Question for the clinical experts: Are there any recommendations if post–day 1 loading doses are missed? | The clinical experts consulted for this review suggested that patients should strictly follow the schedule of crovalimab administration and if any post–day 1 loading doses are missed, the patients should take the missed dose as soon as possible before the day of the next scheduled dose. The clinical experts noted that if it is almost time for the next dose, the patient should skip the missed dose and continue their regular dosing schedule without taking a double dose on the same day to make up for a missed one. The clinical experts’ recommended criteria for missed doses are in line with the criteria about missing a dose of crovalimab in the COMMODORE 2 and COMMODORE 1 studies. |
System and economic issues | |
The BIA seems to be underestimating the number of clients starting or switched to crovalimab. Given the subcutaneous administration, believe the higher usage of crovalimab due to patient and prescriber preference. Patent expiry for Soliris is 2027 and for Ultomiris is 2035. If patients transition to the new, more convenient C5 inhibitor, then savings that could be obtained by the entry of biosimilars will be lost. | This is a comment from the drug plans to inform CDEC deliberations. |
Confidential price agreements exist for Ultomiris and Soliris in PNH. | This is a comment from the drug plans to inform CDEC deliberations. |
BIA = budget impact analysis; CDEC = Canadian Drug Expert Committee; DTDC = drug-target-drug complex; NIHB = Non-Insured Health Benefits; PNH = paroxysmal nocturnal hemoglobinuria; PT = province and territory; VAC = Veterans Affairs Canada.
Note: In the Canadian Expert Drug Advisory Committee final recommendation for eculizumab (Soliris),31 the recommendation was do not list at the submitted price. In CADTH Reimbursement Recommendation: Ravulizumab (Ultomiris),32 the Canadian Drug Expert Committee recommends that ravulizumab be reimbursed for the treatment of adult patients with PNH with clinical criteria and/or conditions, including for the treatment’s initiation, renewal, discontinuation, and prescribing. The clinical criteria and/or conditions are as follows: 1) list in a similar manner to eculizumab for initiation, renewal, discontinuation, and prescribing, with the addition of condition 2 for initiation and condition 3 for prescribing; 2) patients with insufficient initial response or who did not experience improvement with treatment with eculizumab at the Health Canada–recommended dosage are not eligible for reimbursement of ravulizumab; 3) ravulizumab should only be prescribed at the Health Canada–recommended dosage; 4) ravulizumab should be negotiated so that it does not exceed the drug program cost of treatment with eculizumab reimbursed for the treatment of adult patients with PNH; and 5) the feasibility of the adoption of ravulizumab must be addressed.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of crovalimab 340 mg/2 mL for SC injection following 1 loading dose by IV infusion in the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg. The focus will be placed on comparing crovalimab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of crovalimab is presented in 3 sections with CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Assessment by CDA-AMC of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. No additional studies were submitted to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 pivotal studies (RCTs) identified in systematic review
1 long-term extension study
1 indirect treatment comparison (ITC).
Systematic Review
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
The COMMODORE 2 and COMMODORE 1 studies are pivotal phase III RCTs evaluating the safety and efficacy of crovalimab compared to eculizumab in patients with PNH who had not been previously treated with a complement inhibitor (the COMMODORE 2 study) and patients with documented treatment with complement inhibitors (the COMMODORE 1 study).
COMMODORE 2 Study
The COMMODORE 2 trial was a multicentre, randomized, open-label, active-controlled study comparing the efficacy and safety of crovalimab compared with eculizumab in patients diagnosed with PNH who had not been previously treated with a complement inhibitor therapy, and who had a body weight of 40 kg or more. Adult patients were randomized 2:1 to receive crovalimab (arm A, n = 135) or eculizumab (arm B, n = 69). A central randomization procedure was used, where a block-based randomization was stratified by the most recent locally measured LDH level (2 to 4 × ULN, and > 4 × ULN) before randomization and number of pRBC units (0, > 0 to ≤ 6, and > 6) administered within the 6 months before randomization. Six additional patients were enrolled in arm C (descriptive arm that received crovalimab), which enrolled pediatric patients with PNH who were younger than 18 years. The primary treatment period was 24 weeks for all arms. After 24 weeks, all patients had the opportunity to continue or switch to crovalimab in the extension period (Figure 1). Patients were enrolled across 67 sites in 25 countries. The study did not include sites in Canada. The CCOD as submitted in the aligned review to Health Canada was November 16, 2022.15,35
Table 6: Details of Studies Included in the Systematic Review
Detail | COMMODORE 2 study | COMMODORE 1 study |
|---|---|---|
Designs and populations | ||
Study design | Phase III, open-label, active-controlled, multicentre RCT | Phase III, open-label, active-controlled, multicentre RCT |
Locations | 67 centres in 25 countries from continents of Asia, Europe, and South America | 70 sites in 25 countries from continents of Asia, Europe, North America (Canada = 1 site with 1 patient), and South America |
Patient enrolment dates | Start date: October 8, 2020 End date: May 31, 2022 | Start date: September 29, 2020 End date: May 2, 2024 |
Study population (N) | Total N = 210 | Total N = 127 |
Randomized (N) | Randomized N = 204
| Randomized N = 89
|
Nonrandomized (N) | Nonrandomized N = 6
| Nonrandomized N = 38
|
Key inclusion criteria | All arms (arm A, arm B, and arm C):
Additional criterion for patients in randomized arm A and arm B:a
Additional criterion for patients in descriptive arm C:
| All arms (arm A, arm B, and arm C):
Arm A and arm B:
Arm C:
|
Key exclusion criteria |
|
|
Drugs | ||
Intervention | Arm A and arm C (crovalimab):
| Arm A (crovalimab):
Arm C (crovalimab, nonrandomized arm):
|
Comparator | Arm B (eculizumab):
| Arm B (eculizumab):
|
Study duration | ||
Screening phase | 4 weeks | Up to 4 weeks |
Treatment phase | 24 weeks | 24 weeks |
Extension or follow-up phase |
| Same as the COMMODORE 2 study |
Outcomes | ||
Primary end point | Arm A and arm B (crovalimab vs. eculizumab):
| All arms (arm A, arm B, and arm C):c
|
Secondary and exploratory end points | Secondary end points Arm A and arm B (crovalimab vs. eculizumab):
Exploratory end points Arm A and arm B (crovalimab vs. eculizumab):
Arm C (crovalimab, descriptive):
Safety:
| Secondary end points NA Exploratory end pointsd All arms (arm A, arm B, and arm C):
Arm C only (crovalimab, descriptive):
|
Publication status | ||
Publications | Roth et al. (2024)33 ClinicalTrials.gov identifier: NCT04434092 | Scheinberg et al. (2024)34 ClinicalTrials.gov identifier: NCT04432584 |
AE = adverse event; ANC = absolute neutrophil count; CTCAE v5 = Common Terminology Criteria for Adverse Events, Version 5; DTDC = drug-target-drug complex; EORTC = European Organisation for Research and Treatment of Cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; ICF = informed consent form; IPSS-R = Revised International Prognostic Scoring System; LDH = lactate dehydrogenase; MAVE = major adverse vascular event; MFS = multidimensional fatigue scale; NA = not applicable; NCI = National Cancer Institute; PedsQL = Pediatric Quality of Life Inventory; PGI-S = Patient Global Impressions–Severity; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; QLQ-AA/PNH = Quality of Life Questionnaire – Aplastic Anemia and/or Paroxysmal Nocturnal Hemoglobinuria; RCT = randomized controlled trial; SC = subcutaneous; TSQM-9 = Treatment Satisfaction Questionnaire for Medication 9; ULN = upper limit of normal; WBC = white blood cell; vs. = versus.
Note: LDH level was assessed by the central laboratory in the COMMODORE 2 and COMMODORE 1 trials. Transfusion avoidance was defined as patients who are pRBC transfusion–free and who do not require a transfusion per protocol-specified guidelines. Breakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, dyspnea, anemia [Hb < 10 g/dL], a MAVE, dysphagia, or erectile dysfunction) in the presence of an elevated LDH level of 2 or more multiplied by ULN after the prior reduction of the LDH level to 1.5 or less multiplied by ULN on treatment. Stabilized hemoglobin was defined as the avoidance of a 2 g/dL or greater decrease in hemoglobin level from baseline, in the absence of transfusion. Two additional reports were included (Roth et al. [2024]33 and Scheinberg et al. [2024]34).
aTwo pediatric patients were randomized to eculizumab in arm B, aged 17 years.
bThis exclusion criterion was added as an exclusion criterion to the COMMODORE 2 trial following the protocol amendment on January 24, 2022.
cThe primary objective of the study was evaluating the safety and tolerability of crovalimab compared to eculizumab.
dClinical outcomes were listed. Pharmacokinetic and pharmacodynamic outcomes were not listed in Table 6.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
Figure 1: COMMODORE 2 Study Design
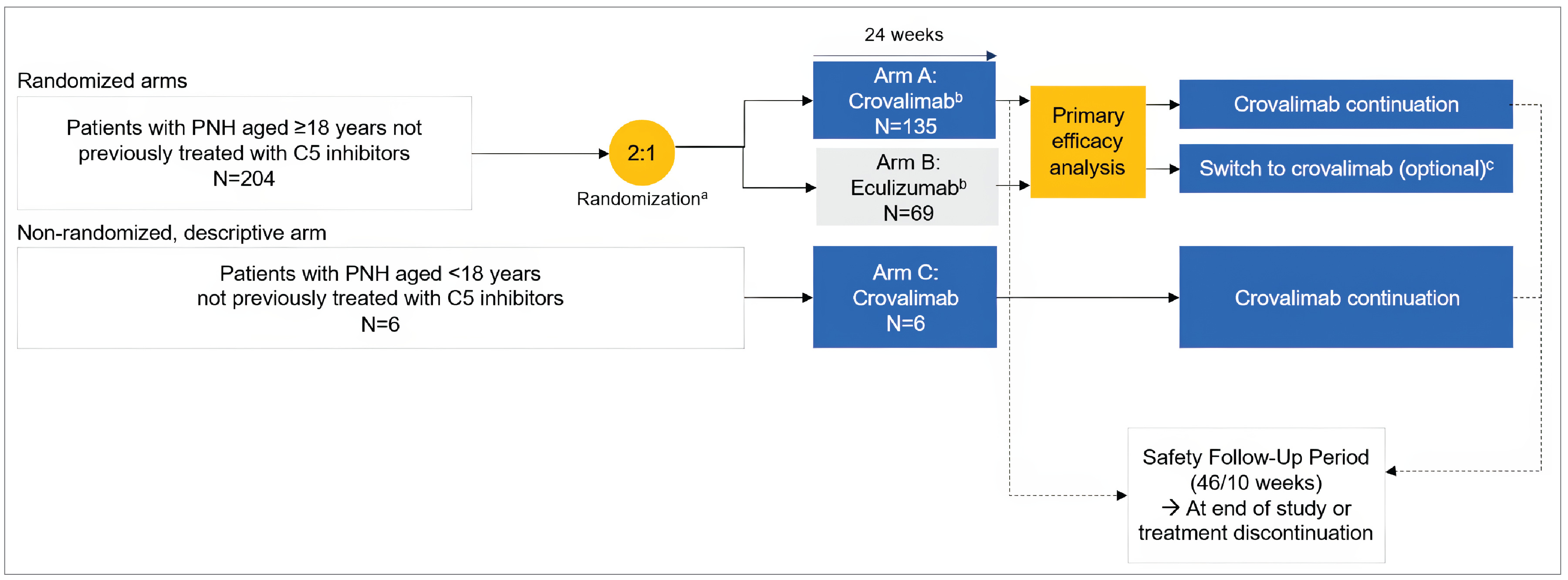
LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; RBC = red blood cell; ULN = upper limit of normal.
aRandomization was stratified based on the most recent LDH value (2 to 4 × ULN, and > 4 × ULN) and packed RBC transfusion history (0 unit, > 0 to 6 units, and > 6 units) within 6 months before randomization. Patients were randomized 2:1 to crovalimab or eculizumab.
bPatients who discontinued crovalimab entered the safety follow-up period of 46 weeks. Patients who discontinued eculizumab entered the safety follow-up period of 10 weeks.
cAfter completing 24 weeks of treatment, patients randomized to eculizumab could continue treatment or switch to crovalimab. If they discontinued treatment, the safety follow-up period for eculizumab patients was 10 weeks.
Source: Sponsor’s submission.16
COMMODORE 1 Study
The COMMODORE 1 study is a multicentre, randomized, open-label, active-controlled study to evaluate the safety, pharmacokinetics (PKs), pharmacodynamics (PDs), and efficacy of crovalimab compared with eculizumab in patients diagnosed with PNH who were treated with a complement inhibitor therapy, and who had a body weight of 40 kg or more. Patients were enrolled across 70 sites in 25 countries, including 1 site in Canada. The COMMODORE 1 study was originally planned to recruit 200 patients to the randomized arms to be sufficiently powered to assess the efficacy of crovalimab. However, due to the introduction of ravulizumab to the treatment landscape and a reduced pool of patients treated with eculizumab over time, randomization to arm A and arm B was stopped in November 2022 per protocol amendment version 6.10 The evaluation of safety became the primary objective and all efficacy objectives became exploratory. As a result, 89 adult participants were randomized 1:1 to receive crovalimab (arm A, n = 45) or eculizumab (arm B, n = 44) in the modified design of the COMMODORE 1 study. A central randomization procedure was used. Randomization was stratified according to a patient’s transfusion history (received a transfusion of pRBCs within 12 months before randomization [yes or no]) using a block-based method. After completing 24 weeks of crovalimab or eculizumab treatment, all patients had the opportunity to continue or switch to crovalimab in the extension period (Figure 2).10,17
The nonrandomized arm C originally intended to recruit about 50 patients in different clinically relevant cohorts. Following the protocol amendment to stop randomization due to a reduced pool of patients treated with eculizumab, adult patients (≥ 18 years) who had been receiving eculizumab at the approved dose for at least 24 weeks before study entry (i.e., the same population as those who would have been enrolled in the randomized arms) were able to enter arm C to continue access to crovalimab in a nonrandomized setting. At the CCOD of primary analysis, 38 patients were enrolled in arm C: 1 patient in the pediatric cohort, 21 patients in the prior ravulizumab cohort, 10 patients in the prior eculizumab at a higher-than-approved dose cohort, and 6 patients in the C5 polymorphism cohort.10,17 Enrolment in arm C is ongoing.
The CCOD of the COMMODORE 1 study as submitted in the aligned review to Health Canada was November 16, 2022, which was the same date as for the COMMODORE 2 study.10,17
Populations
Inclusion and Exclusion Criteria
COMMODORE 2 Study
Eligible patients had clinically significant disease activity at screening, demonstrated by an LDH level of 2 or more multiplied by ULN and the presence of 1 or more PNH-related signs or symptoms in the past 3 months. Patients with current or previous treatment with a complement inhibitor were excluded. Vaccination against N. meningitidis serotypes A, C, W, and Y less than 3 years before the initiation of study treatment was required. Randomized arm A and arm B enrolled adult patients, and descriptive arm C enrolled patients younger than 18 years at the time of signing the informed consent form.
Figure 2: COMMODORE 1 Study Design
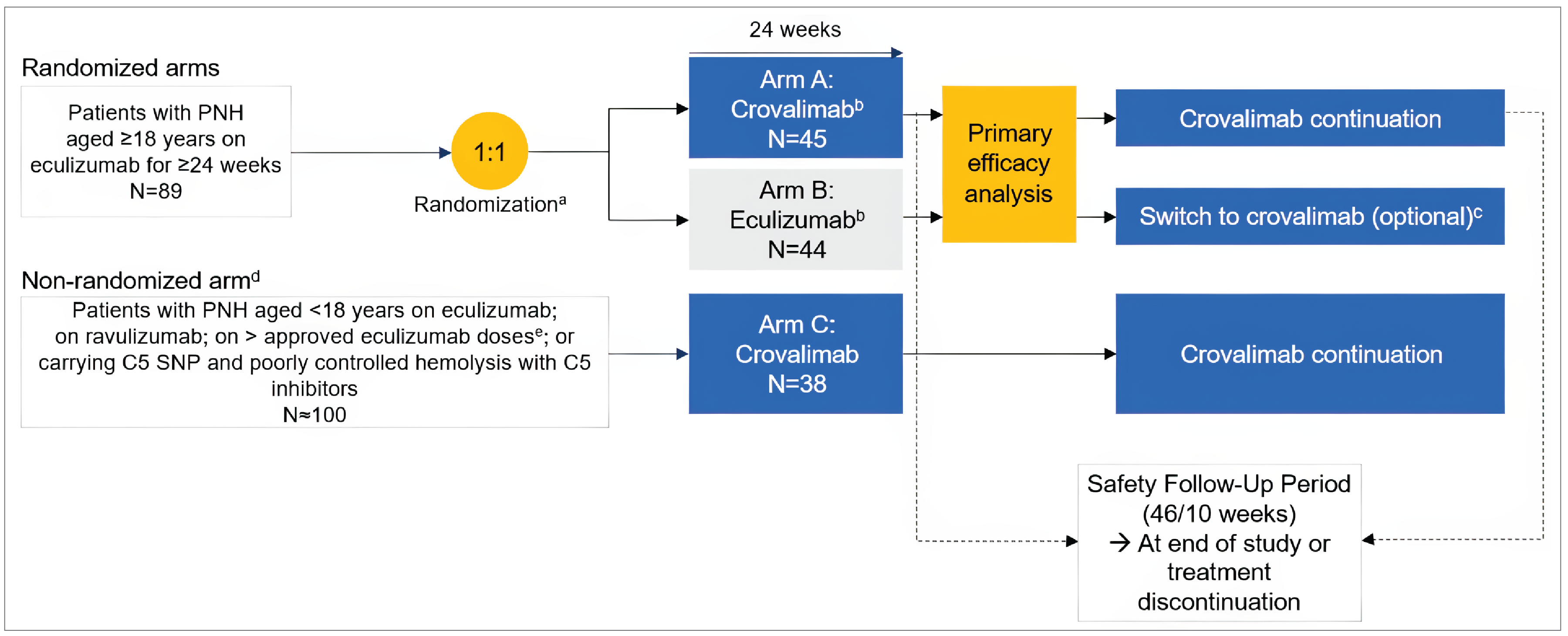
PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; q.2.w. = every 2 weeks; SNP = single nucleotide polymorphism.
aStratification: History of pRBC transfusion within 1 year before randomization (yes or no).
bPatients who discontinued eculizumab or crovalimab entered a safety follow-up period of 10 weeks for eculizumab and 46 weeks for crovalimab.
cAfter completing 24 weeks of treatment, patients randomized to eculizumab could continue treatment or switch to crovalimab.
dFollowing closure of the randomized arms to recruitment, the nonrandomized arm was opened to adults receiving eculizumab at the approved dose for 24 weeks or more before study entry.
eHigher-than-approved doses of eculizumab: More than 900 mg per dose and/or more frequently than q.2.w.
Source: Sponsor’s submission.16
COMMODORE 1 Study
Eligibility criteria in the COMMODORE 1 study (randomized arm A and arm B) are identical to those of the COMMODORE 2 study with the following exceptions:
patients must have documented treatment with eculizumab according to the approved dosing recommended for PNH (900 mg every 2 weeks) and completion of a minimum of 24 weeks of treatment before day 1
patients must have an LDH level of 1.5 or less multiplied by ULN at screening (indicating hemolysis control on current therapy, enabling a stable switch in the randomized period)
patients are excluded if they have a pre-enrolment hemoglobin value of 7 g/dL or less, or a pre-enrolment hemoglobin value of more than 7 g/dL and up to 9 g/dL with concurrent signs and symptoms of anemia (of note, this exclusion criterion was also added as an exclusion criterion to the COMMODORE 2 trial following the protocol amendment on January 24, 2022).
Eligibility criteria in arm C of the COMMODORE 1 trial differs from those of arm C of the COMMODORE 2 trial, as outlined in Table 6.
Interventions
COMMODORE 2 and COMMODORE 1 Studies
Crovalimab was administered with the same dosing regimen in both the COMMODORE 2 and COMMODORE 1 studies. This is also the same dosing regimen as in the reimbursement request and the submission to Health Canada (Table 1 and Table 6).23,28
In both studies, the crovalimab group received a weight-based tiered dosing regimen of crovalimab consisting of a loading series (an IV dose on day 1 [1,000 mg for a body weight of 40 kg to < 100 kg or 1,500 mg for a weight of ≥ 100 kg] followed by SC injection doses on day 2, day 8, day 15, and day 22 [340 mg]) and maintenance dosing (SC injection every 4 weeks starting on day 29 [680 mg for a body weight of 40 kg to < 100 kg or 1,020 mg for a weight of ≥ 100 kg]). For patients 40 kg to less than 100 kg receiving an initial IV loading dose of 1,000 mg, the infusion was delivered over 60 (± 10) minutes. For patients receiving an initial IV loading dose of 1,500 mg, the infusion was delivered over 90 (± 10) minutes. Patients were observed by a health care professional during the IV infusion and for 60 minutes following the completion of the IV infusion. Crovalimab self-administration (in patients ≥ 12 years only) or administration by a caregiver was permitted starting at week 9, after training and confirmation of proficiency by the health care professional. Patients who did not wish to self-inject or did not have assistance could continue to have crovalimab administered by the study site staff. If a patient experienced signs and symptoms of their underlying PNH, such as BTH (which may be due to an acute event such as acute illness, trauma or surgery), 1 or more additional IV doses of crovalimab could be administered as a rescue dose (340 mg IV regardless of body weight infused over 30 minutes).15,17
The eculizumab group received eculizumab per local guidelines or per pharmacy manual: induction doses of 600 mg on day 1, day 8, day 15, and day 22, followed by maintenance doses of 900 mg on day 29 and every 2 weeks thereafter. No eculizumab dose modifications were permitted in the COMMODORE 2 and COMMODORE 1 studies.15,17 Patients who discontinued treatment entered a safety follow-up period.
Concomitant Medications
In both the COMMODORE 2 and COMMODORE 1 studies, patients were permitted to use the following therapies: oral contraceptives with a failure rate of less than 1% per year, immunosuppressant therapy, corticosteroids, iron supplements, and folic acid.10,35
Patients could receive other concomitant medication, which was recorded. Investigators could provide supportive therapies as clinically indicated, per local standard practice. Infusion-associated symptoms could be treated symptomatically with acetaminophen, ibuprofen, diphenhydramine, and/or histamine H2 receptor antagonists (e.g., famotidine, cimetidine), or equivalent medications per local standard practice. Serious infusion-associated events manifested by dyspnea, hypotension, wheezing, bronchospasm, tachycardia, reduced oxygen saturation, or respiratory distress were managed with supportive therapies as clinically indicated (e.g., supplemental oxygen and beta2-adrenergic agonists).15,17
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, CDA-AMC selected end points that were considered to be most relevant to informing expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using the GRADE tool. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | COMMODORE 2 study | COMMODORE 1 study |
|---|---|---|---|
Proportion of patients with hemolysis control (measured by LDH level ≤ 1.5 × ULN at the central laboratory) | From week 5 through week 25 | Coprimary for arm A and arm B Exploratory for arm C | Exploratory |
Proportion of patients with breakthrough hemolysis | From baseline through week 25 | Secondary for arm A and arm B Exploratory for arm C | Exploratory |
Mean percentage change in LDH levels | From baseline through week 25 | Exploratory | Exploratory (change from baseline averaged over week 21, week 23, and week 25) |
Proportion of patients with stabilized hemoglobin | From baseline through week 25 | Secondary for arm A and arm B Exploratory for arm C | Exploratory |
Proportion of patients who reached and maintained a minimum hemoglobin level | From baseline through week 25 | Exploratory | Exploratory |
Proportion of patients who achieved transfusion avoidance | From baseline through week 25 | Coprimary for arm A and arm B Exploratory for arm C | Exploratory |
Total number of pRBC units transfused | 24 weeks | Exploratory | Exploratory |
Mean change in FACIT-F scores | From baseline through week 25 | Secondary for arm A and arm B | Exploratory |
Proportion of patients who achieved a ≥ 5-point improvement from baseline in FACIT-F scores | At week 25 | Exploratory for arm A and arm B | NR |
Mean change in EORTC QLQ-C30 (physical functioning score, role functioning score, GHS and QoL score) | From baseline through week 25 | Exploratory for arm A and arm B | Exploratory |
Mean change in EORTC IL-40 symptoms for subscales (dyspnea, dysphagia, chest pain, headaches, abdominal pain, erectile dysfunction) | From baseline through week 25 | Exploratory | Exploratory |
Patient preference for crovalimab or eculizumab, measured with PPQ | Week 17 (the COMMODORE 1 study), switch week 17 (the COMMODORE 1 and COMMODORE 2 studies) | Exploratory (arm B: for patients ≥ 18 years randomized to eculizumab who switch to crovalimab) | Exploratory (arm A and arm C: for patients ≥ 12 years) |
Safety (AEs, SAEs, AEs leading to study drug discontinuation, AESIs) | 24 weeks | Secondary | Primary |
AE = adverse event; AESI = adverse event of special interest; EORTC IL-40 = European Organisation for Research and Treatment of Cancer – Item Library 40; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GHS = global health status; LDH = lactate dehydrogenase; NR = not reported; PPQ = Patient Preference Questionnaire; pRBC = packed red blood cell; QoL = quality of life; SAE = serious adverse event; ULN = upper limit of normal.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
Efficacy Outcomes
The COMMODORE 2 and COMMODORE 1 trials investigated the same outcomes at week 25 (i.e., after a 24-week treatment period). A description of the efficacy outcome measures and the measurement properties that were used in both the COMMODORE 2 and COMMODORE 1 trials are presented in this section (Table 8).
Hemolysis Control
Serum LDH is a biochemical marker of intravascular hemolysis. The cut-off for a clinically meaningful threshold of improvement in both the COMMODORE 2 and COMMODORE 1 studies was an LDH level of 1.5 or less multiplied by ULN (as measured at the central laboratory), based on the available literature supporting its clinical relevance as the most sensitive and specific predictor of mortality and thromboembolic events.36-39
Breakthrough Hemolysis
BTH was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin < 10 g/dL], a major adverse vascular event (MAVE) [including thrombosis, dysphagia, or erectile dysfunction]) in the presence of an elevated LDH level of 2 or more multiplied by ULN after a prior reduction of the LDH level to 1.5 or less multiplied by ULN on treatment. PRO instruments were self-administered at the clinic at specified time points during the studies.
Stabilized Hemoglobin
Stabilized hemoglobin was defined as avoidance of a 2 g/dL or greater decrease in hemoglobin level from baseline, in the absence of transfusion. Baseline hemoglobin was defined as the latest available hemoglobin measurement before the first on-study drug administration of the study drug.
Transfusion Avoidance
Transfusion avoidance is a disease-related event for patients with PNH, defined as patients who are pRBC transfusion–free and do not require transfusions per protocol-specified guidelines. In both trials, transfusions were administered per the investigators' clinical judgment. In general, pRBC transfusions were recommended when a patient had hemoglobin of 9 g/dL or less with clinical signs and symptoms of sufficient severity to warrant transfusion or hemoglobin of 7g/dL or less regardless of clinical signs or symptoms.
FACIT-F (Version 4)
FACIT-F is a validated, reliable, self-report measure for use in a variety of conditions, including anemia.40,41 The content validation research supports its use in patients with PNH, as it comprehensively covers PNH-related fatigue.42 The FACIT-F consists of 13 items that assess fatigue using a 7-day recall period. Items are scored on a response scale that ranges from 0 (“not at all”) to 4 (“very much so”). Relevant items are reverse-scored and all items are summed to create a total score ranging from 0 to 52, with higher scores indicating lower fatigue.43,44 The threshold for a clinically meaningful change is 5 points or more.44 FACIT-F was administered to adult patients only in both the COMMODORE 2 and COMMODORE 1 studies.
EORTC QLQ-C30 (Version 3)
EORTC QLQ-C30 is a validated, reliable, self-report measure.45 Although the measure was originally developed for use in patients undergoing cancer treatment, recent content validation research supports its use in patients with PNH.42 It consists of 30 questions that assess 5 aspects of patient functioning (physical, emotional, role, cognitive, and social), 3 symptom scales (fatigue, nausea and vomiting, and pain), GHS and QoL, and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), with a recall period of 1 week. Scale scores can be obtained for the multi-item scales. The functioning and symptom items are scored on a 4-point scale that ranges from “not at all” to “very much,” and the GHS and QoL items are scored on a 7-point scale that ranges from “very poor” to “excellent.” This PRO tool was administered in a truncated version of the measure that included only the physical functioning, role functioning, and GHS and QoL scales. Each scale ranges from 0 to 100, with higher scores indicating better physical functioning, role functioning, or GHS and QoL, respectively.42 EORTC QLQ-C30 was administered to adult patients only in both the COMMODORE 2 and COMMODORE 1 studies.
EORTC Item Library: PNH Symptoms
The EORTC Item Library is a database of items used in fully and partially validated EORTC QoL questionnaires. Eight items from the library were used to assess disease-related symptoms that are relevant to patients with PNH that are not sufficiently covered in the other PRO measures (EORTC IL-40). Selected symptoms included headache, dyspnea at rest and upon exertion, dysphagia, abdominal pain, chest pain, and erectile dysfunction.39,42,46 All items are scored on a 4-point scale that ranges from “not at all” to “very much,” with higher scores indicative of higher symptom severity. All symptom items will be scored individually unless they comprise a scale (i.e., dyspnea). This measure was administered to adult patients only in both trials.
Patient Preference Questionnaire
The Patient Preference Questionnaire (PPQ) is a 2-item measure of treatment preference developed by the sponsor. Patients were asked to indicate their preference for IV eculizumab or ravulizumab (the COMMODORE 1 study’s arm C only) or SC crovalimab and indicate the top 3 reasons for their preference from a list of 13 potential reasons.33,34 In the COMMODORE 2 study, this measure was administered to patients aged 18 years or older in arm B. In the COMMODORE 1 study, this measure was administered to patients aged 12 years or older at week 17 in arm A and arm C, and administered at week 41 to all 3 arms.
Pediatric Quality of Life Inventory Multidimensional Fatigue Scale
The Pediatric Quality of Life Inventory (PedsQL) Multidimensional Fatigue Scale (MFS) is a valid, reliable measure for assessing fatigue in healthy children and adolescents, and those with a range of acute and chronic health conditions.47-50 Self-report versions are available for children and adolescents that contain developmentally appropriate language for defined age groups (i.e., 5 years to 7 years, 8 years to 12 years, and 13 years to 17 years). This version contains 18 items that are scored in 3 respective domains: general fatigue (6 items), sleep/rest fatigue (6 items), and cognitive fatigue (6 items). Domains can be further combined into an overall total fatigue score. Domain and total scores are converted to a 0 to 100 scale, with higher scores indicative of lower fatigue. For the COMMODORE 2 and COMMODORE 1 studies, the self-report version with a 1-week recall period (i.e., acute version) was used for patients aged 8 years to 17 years.
PedsQL Generic Core Scales (Version 4)
The PedsQL Generic Core Scales are valid, reliable measures for assessing HRQoL in healthy children and adolescents, and those with acute or chronic health conditions.51,52 Self-report versions are available for children and adolescents that contain developmentally appropriate language for defined age groups (i.e., 5 years to 7 years, 8 years to 12 years, and 13 years to 17 years). This version contains 23 items that are scored in 4 respective domains: physical functioning (8 items), emotional functioning (5 items), social functioning (5 items), and school functioning (5 items). Physical and psychosocial health summary scores, as well as an overall total score, can be created by further combining domains. Domain and total scores are converted to a 0 to 100 scale, with higher scores indicative of better functioning. Only the physical functioning scale of the self-report version with a 1-week recall period (i.e., acute version) was used for patients aged 8 years to 17 years.
Harms Outcomes
In the COMMODORE 2 and COMMODORE 1 studies, safety was assessed through descriptive summaries of exposure to each study treatment, AEs, changes in laboratory test results, and changes in vital signs and an electrocardiogram (ECG). Safety analyses were performed on the safety-evaluable population, defined as all enrolled participants who received at least 1 dose of study drug (crovalimab, eculizumab, or placebo), with patients grouped according to treatment received.
Considerations that informed the selection of efficacy and safety outcomes to be summarized and assessed using GRADE included the following:
the proportion of patients with hemolysis control was identified as important by the patient and clinician group input, and by the clinical experts consulted for this review
the proportion of patients with BTH was identified as important by the clinical experts
the mean percentage change in LDH levels was identified as important by the clinical experts
the proportion of patients with stabilized hemoglobin was identified as important by the clinical experts
the proportion of patients who reached and maintained a minimum hemoglobin level was identified as important by the clinical experts
the proportion of patients who achieved transfusion avoidance was identified as important by the clinical experts
the mean number of total pRBC units transfused was identified as important by the clinical experts
HRQoL outcomes were identified by the patient input, and the clinical experts and clinician group input specified to include the FACIT-F and the EORTC QLQ-C30 GHS and QoL measures
the proportion of patients with infections (including meningococcal meningitis) and the proportion of deaths were identified as important safety outcomes by referring to other relevant reviews.
The following outcomes were considered relevant in providing supplementary data in assessing the efficacy and safety of crovalimab for PNH by the clinical experts, but only needed to be summarized in the report without using the GRADE tool for assessment:
the proportion of patients who achieved a 5-point or greater improvement from baseline in the FACIT-F score at week 5
the mean change in EORTC IL-40 symptoms for 6 subscales (dyspnea, dysphagia, chest pain, headaches, abdominal pain, and erectile dysfunction)
the proportion of patients with a preference for crovalimab or eculizumab
the proportion of patients with a MAVE
the proportion of patients with an immune complex–mediated reaction (only applicable among the patients who switched from other C5 inhibitors to crovalimab).
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusion about measurement properties | MID |
|---|---|---|---|
FACIT-F | A 13-item, patient-reported, fatigue-specific QoL questionnaire using a 5-point Likert scale. It assesses tiredness, weakness, and difficulty conducting usual activities as a result of fatigue over the past week.53 The 13-item scale ranges from 0 (extreme fatigue) to 52 (no fatigue). Higher scores indicate less fatigue.53 | Patients with PNH:
Responsiveness:
| Per the submission, a change in score of 5 points is considered clinically important in the PNH population.14 |
EORTC QLQ-C30 | A 30-item, patient-reported, cancer-specific QoL questionnaire using 4-point and 7-point Likert scales. It consists of 5 multi-item functional scales (physical, role, emotional, cognitive, and social), 3 multi-item symptom scales (fatigue, nausea and vomiting, and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item GHS and QoL scale. A 1-week recall period is used to assess the items.56 Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A higher score on the functional scales represents better functioning, a higher score on the symptom scales represents a higher level of symptomatology, and a higher score on the GHS and QoL scale represents a higher HRQoL.56 | Patients with PNH:
Convergent validity between EORTC QLQ-C30 scales and Hb, ARC, and indirect bilirubin were as follows.
Responsiveness:
| No MID was identified in patients with PNH. Patients with cancer:57
|
EORTC Item Library: PNH symptoms | The EORTC Item Library is a database of items used in fully or partially validated EORTC QoL questionnaires. PNH-relevant symptoms include headache, dyspnea at rest and upon exertion, dysphagia, abdominal pain, chest pain, and erectile dysfunction on a 4-point Likert scale (where 1 = not at all and 4 = very much). For each subscale, the score is then converted to a scale that ranges from 0 to 100, with a higher score indicating higher symptom severity. | Content validity of individual library items has been established in the items’ instrument(s) of origin. The validity of individual items abstracted from their instrument of origin may not be retained.59 | Not established |
ARC = absolute reticulocyte count; EORTC = European Organisation for Research and Treatment of Cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GHS = global health status; Hb = hemoglobin; HRQoL = health-related quality of life; MID = minimal important difference; PNH = paroxysmal nocturnal hemoglobinuria; QoL = quality of life.
Statistical Analysis
The statistical analysis of efficacy end points in the COMMODORE 2 and COMMODORE 1 trials is summarized in Table 9.
Sample Size and Power Calculation
COMMODORE 2 Study
The sample size estimation for the randomized portion of the study (arm A and arm B) was based on the noninferiority assessment of the coprimary end points of hemolysis control, as assessed by centrally measured LDH levels (the assumption of the rate of hemolysis control was 86% in both the crovalimab and the eculizumab arms), and the proportion of patients who achieve transfusion avoidance (the assumption of the rate of transfusion avoidance was 67% in the crovalimab arm and 66% in the eculizumab arm) during the efficacy period. The final target sample size corresponds to the end point that requires the larger number of patients (i.e., transfusion avoidance from baseline to week 25). A total of 204 adult patients were randomly assigned in a 2:1 ratio to receive either crovalimab (n = 135) or eculizumab (n = 69) to ensure approximately 180 evaluable patients, assuming a 10% dropout rate. This sample size provided 80% power to demonstrate the noninferiority of crovalimab to eculizumab with respect to transfusion avoidance, using a NIM of –20% and 1-sided type I error rate of 2.5%.15
The NIM for transfusion avoidance was determined based on the data (protocol of ALXN1210-PNH-301 [the 301 study])60 comparing eculizumab-treated patients with untreated patients from the Global PNH Patient Registry for eculizumab-treated patients (i.e., patients treated with eculizumab showed a benefit over untreated patients), with a difference of approximately 40% (with a transfusion avoidance proportion of 57.1% and 18.6%, respectively), after adjustment for the history of transfusions 12 months before enrolment. It was assumed that a difference in proportions of –20%, the NIM, would preserve at least 50% of the control treatment effect. This NIM was also defined considering the rarity of PNH. A more conservative NIM would have resulted in the estimated sample size being too large and infeasible. Lee et al. reported a proportion of patients with transfusion avoidance of 66.1% (95% CI, 65.9% to 74.6%) in treatment-naive patients treated with eculizumab.60
With regard to hemolysis control, 116 patients were required in a 2:1 ratio to test the noninferiority of crovalimab versus eculizumab, with a NIM of 0.2 in the OR scale, 80% power, and 1-sided type I error rate of 2.5%. Incidentally, a similar sample size was required to test for noninferiority in the probability scale when the NIM was –0.2. The 301 study also reported a proportion of LDH normalization below 1.0 multiplied by ULN of 49.4%.60 Under the assumption of LDH being distributed via a log-normal method, the expected proportion below 1.5 multiplied by ULN was 86%. The same proportion was assumed for crovalimab. The NIM in the OR scale was obtained as 1 divided by OR,0.5 where OR equals 24.6 assuming 86% of patients receiving eculizumab will reach an LDH level of 1.5 or less multiplied by ULN compared to an upper bound of the 95% CI of the proportion among placebo-treated patients of 20%. Note that both proportions are approximately twice the ones used in the 301 study60 for an LDH level of 1 or less multiplied by ULN, and a fraction of 0.5 of that effect is retained.61 Assuming a 10% dropout, the total needed sample size would be 128 patients (85 patients randomized to crovalimab and 43 patients to eculizumab). With 180 evaluable patients expected in the study, the power for this end point would be 94%. Hence, the joint power for both transfusion avoidance and LDH would be 75% if they were uncorrelated. It is likely that coprimary end points would be correlated and hence, joint power would be higher. If there were no dropouts and all the approximately 200 patients (the calculated sample size) contributed at least 1 LDH sample, then the power for transfusion avoidance would be 84% and for LDH 96%.
Pediatric patients were enrolled in the descriptive arm (arm C) throughout the duration of the study. No target sample size was specified.
The primary analysis was performed once the last randomized patient on trial completed 24 weeks of study treatment or discontinued early, whichever happened first.15
COMMODORE 1 Study
The COMMODORE 1 study was originally planned to recruit 200 patients to the randomized arms to be sufficiently powered to assess the efficacy of crovalimab. Considering the evolving treatment landscape and a reduced pool of patients treated with eculizumab over time, key design features of the study had to be modified in the protocol amendment version 6. As per protocol amendment version 6, the safety objective was the only primary objective of the COMMODORE 1 study and all efficacy analyses were considered descriptive without any formal statistical testing. In the modified design, the randomized arms were expected to randomize approximately 90 adult patients (aged ≥ 18 years) in a 1:1 ratio to crovalimab (arm A) or eculizumab (arm B). The nonrandomized arm C originally intended to recruit about 50 patients in different clinically relevant cohorts. With protocol amendment version 6, arm C was also further expanded to include an additional cohort of adult patients (≥ 18 years) who had been receiving eculizumab at the approved dose for at least 24 weeks before study entry (i.e., the same population as those who would have been enrolled in the randomized arms, to continue study access for such patients in a nonrandomized setting). With this, the total sample size of arm C was increased to approximately 100 patients.17
In line with protocol amendment version 6, the primary analysis for this study was conducted at the same time as the primary analysis of the COMMODORE 2 study, which was the pivotal trial assessing crovalimab in naive patients with PNH. Patients had to have received at least 1 dose of crovalimab or eculizumab to be included in the primary analysis of the COMMODORE 1 study.17
Statistical Testing
COMMODORE 2 Study
Coprimary end point: Transfusion avoidance — The percentage of patients with transfusion avoidance from baseline through week 25 (after 24 weeks on treatment) was computed. As a conservative approach, patients who prematurely withdrew from the study before week 25 were assumed to have undergone a transfusion. The difference in the percentage of patients with transfusion avoidance in the 2 treatment arms was calculated, along with a 95% CI for the difference using the stratified Newcombe CI method.62 The difference between the 2 treatment arms was computed as a weighted combination of the differences between the crovalimab and eculizumab arms within the stratification indicators of transfusion history and the LDH level before randomization using Mantel-Haenszel weights.63 Noninferiority with respect to transfusion avoidance was to be concluded if the lower limit of the 95% CI for the difference between crovalimab and eculizumab was greater than the predefined NIM of −20% (alternative hypothesis).
Coprimary end point: Hemolysis control — A generalized estimating equation (GEE) model was used to estimate the adjusted log OR of hemolysis control (LDH level ≤ 1.5 × ULN) due to treatment, accounting for the intraindividual correlation between LDH control statuses across visits. Covariates included the categorical effects of treatment and visit, visit by treatment interaction, continuous baseline LDH level, and number of pRBC units administered within the 6 months before randomization. The baseline LDH level was defined as the mean of all central LDH values taken during screening, and the LDH value at week 1, day 1, collected before the first dose administration of either crovalimab or eculizumab. The primary analysis applied an unstructured covariance matrix, which imposed minimal assumptions; however, it required estimating a large number of parameters, which may prevent model convergence. The sensitivity analysis accounted for this (described as follows in the sensitivity analyses section).
Measurements taken every 2 weeks from week 5 through week 25 corresponded to the period of stability in treatment-naive patients and were used in the statistical analysis, given that treatment-naive patients were expected to reach a stable LDH plateau by the end of the first month of treatment. For each patient at each visit, a binary variable was created if the central LDH level was less than or equal to 1.5 times the ULN or if the central LDH level was more than 1.5 times the ULN, and it was considered missing if LDH was not measured. Unscheduled central LDH measurements were mapped to the nearest scheduled assessment visit using time windowing. If more than 1 measurement was attributed to a visit window, the average was taken. Central LDH samples affected by tabletop hemolysis were excluded from analyses. The noninferiority of crovalimab relative to eculizumab was concluded if the lower limit of the 2-sided 95% CI for the OR of crovalimab compared to eculizumab was greater than the predefined NIM of 0.2 (alterative hypothesis).
Secondary end points: If noninferiority was established for the coprimary end points, superiority testing of primary and secondary end points was conducted using the hierarchical order as described in Table 9 to ensure that the familywise 1-sided type I error rate was controlled at the 2.5% level. A closed testing procedure was used and the lack of significance of an end point in the hierarchy precluded the analysis of subsequent end points.
Table 9: Hierarchical Statistical Testing of Primary and Secondary Efficacy End Points in the COMMODORE 2 Study
End point in COMMODORE 2 study | Test |
|---|---|
Proportion of patients with TA from baseline through week 25a | Noninferiority |
Hemolysis control from week 5 through week 25a | Noninferiority |
Proportion of patients with BTH from baseline through week 25 | Noninferiority |
Proportion of patients with stabilization of hemoglobin from baseline through week 25 | Noninferiority |
Proportion of patients with TA from baseline through week 25 | Superiority |
Hemolysis control from week 5 through week 25 | Superiority |
Proportion of patients with BTH from baseline through week 25 | Superiority |
Proportion of patients with stabilization of hemoglobin from baseline through week 25 | Superiority |
Mean change from baseline to week 25 in FACIT-F scale (for adults aged ≥ 18 years) | Noninferiority |
BTH = breakthrough hemolysis; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; TA = transfusion avoidance.
aDenotes coprimary efficacy end points that were tested together.
Source: Sponsor’s submission.23
BTH: The proportion of patients with BTH from baseline through week 25 was analyzed using the same methodology as for transfusion avoidance. As a conservative approach, patients withdrawing from study treatment before week 25 were assumed to have experienced a BTH event in the unobserved period. If the upper limit of the 95% CI for the difference between crovalimab and eculizumab in the proportion of patients with BTH was less than the predefined NIM of 20%, then crovalimab was considered noninferior to eculizumab.
Hemoglobin stabilization: The stabilization of hemoglobin was analyzed using the same approach as for transfusion avoidance. As a conservative approach, patients who withdrew from study treatment before week 25 were assumed to not have met hemoglobin stabilization criteria. If the lower limit of the 95% CI for the difference between crovalimab and eculizumab in the proportion of patients with stabilized hemoglobin was greater than the predefined NIM of −20%, then crovalimab was to be considered noninferior to eculizumab.
FACIT-F: The change from baseline to week 25 was analyzed using a mixed model for repeated measures (MMRM) assuming normally distributed scores, with adjustment for stratification factors, and the baseline FACIT-F score. An unstructured covariance matrix was used to model the within-patient errors. If the model did not converge with the unstructured covariance matrix, a more parsimonious structure was considered in the following order until model convergence was achieved: Toeplitz, first-order autoregressive, and compound symmetry. If the lower limit of the 95% CI for the difference between crovalimab and eculizumab in the mean change from baseline to week 25 in fatigue was greater than the predefined NIM of −5 points, then crovalimab was considered noninferior to eculizumab and the end point was to be tested for superiority (Table 9).
Exploratory efficacy analysis: All exploratory efficacy end points were analyzed descriptively, without any formal statistical testing. No adjustment for multiplicity was conducted. Patient preference was assessed in arm B patients at switch week 17 (Week 41). The intermediate time point was chosen to allow patients sufficient time on treatment with crovalimab to be able to compare to eculizumab while preventing recall bias.23
COMMODORE 1 Study
As the anticipated enrolment was not expected to provide sufficient power for the statistical testing of efficacy data from the randomized arms, all efficacy objectives and related end points became exploratory and descriptive. There was no change to the safety end points and the safety analysis was conducted as per the COMMODORE 2 study.23
For all exploratory efficacy end points, summary statistics (including means, median, range, SDs, and proportions where appropriate) were presented by treatment arm using tables, listings, and graphs as appropriate. Proportions were reported with 95% CIs. In graphs, CIs were displayed for a treatment arm when the sample size was at least 10.
In nonrandomized arm C, the same analyses as for the randomized arms are repeated, except for the LDH level percentage change. Change in LDH levels from baseline to LDH levels averaged over week 21, week 23, and week 25 were computed. In contrast to patients in arm A and arm B, not all patients in arm C were required to have hemolysis control (defined as an LDH level ≤ 1.5 × ULN) at study entry, and therefore the mean change in the LDH level from baseline is not a meaningful analysis for arm C.
Exploratory Efficacy End Points for Randomized Arm A and Arm B
Percentage change from baseline in LDH levels:
Baseline LDH was defined as the mean of all central LDH values taken during screening (i.e., within 4 weeks before the first on-study drug administration of either crovalimab or eculizumab), and the LDH value at week 1, day 1, collected before the first dose administration of either crovalimab or eculizumab. All postbaseline LDH values included in this analysis were based on the central laboratory LDH measurements. All LDH measurements (up to week 25) were used in the statistical model. Unscheduled central LDH measurements were mapped to the nearest scheduled assessment visit using time windowing. If more than 1 measurement was attributed to a visit window, the average was taken. Central LDH samples affected by tabletop hemolysis were excluded from analyses. One additional LDH sample was taken at week 1, day 2, only for patients on the crovalimab arm following the week 1, day 2, crovalimab loading dose. This assessment was not included in the statistical model because there was no corresponding LDH sample (week 1, day 2) for patients in the eculizumab arm.
The percentage change in LDH levels from baseline averaged over week 21, week 23, and week 25 between the crovalimab arm (arm A) and the eculizumab arm (arm B) were analyzed using the MMRM with fixed categorical effects of treatment, visit, and visit by treatment arm interaction, as well as the continuous fixed covariate of baseline LDH level, baseline LDH level by visit interaction, and the stratification factor of transfusion history (yes or no).64 An unstructured covariance matrix was used to model the within-patient errors. If the model did not converge with the unstructured covariance matrix, a more parsimonious structure was considered in the following order until model convergence was achieved: Toeplitz, first-order autoregressive, and compound symmetry. The point estimate for the difference between crovalimab and eculizumab, along with a 2-sided 95% CI, was presented. The percentage change in LDH levels from baseline averaged over week 21, week 23, and week 25 was estimated using contrasts. The model included data from all patients in the efficacy-evaluable population. Missing data were assumed as data missing at random as part of the MMRM model.17,23
Hemolysis control:
A GEE model was used to estimate the adjusted log OR of an LDH level of 1.5 or less multiplied by ULN due to treatment, taking account of the intraindividual correlation between LDH control statuses across visits. The GEE model was adjusted for baseline covariates and correlation matrix structure in a manner similar to the MMRM. Similarly, unscheduled LDH assessments were included in the analysis via windowing, and LDH samples suspected of being affected by tabletop hemolysis were excluded from analyses.
Transfusion avoidance:
The percentage of patients with transfusion avoidance from baseline through week 25 (after 24 weeks on treatment) was computed. Note that, as a conservative approach, patients who prematurely withdraw from study treatment were assumed to have undergone a transfusion. For the descriptive comparison of transfusion avoidance between the randomized arms, a difference in the percentage of patients with transfusion avoidance in the 2 treatment arms was calculated, along with a 95% CI for the difference using the stratified Newcombe CI method. 62 The difference between the 2 treatment arms was computed as a weighted combination of the differences between the crovalimab and eculizumab arms within the stratification indicator of transfusion history using Mantel-Haenszel weights. 63 The total number of units (based on a local equivalent) of pRBCs transfused per patient by week 25 was presented with 95% CI per treatment arm. Transfusion avoidance analysis included only patients who had completed the 24 weeks of treatment, or who had discontinued from study but had their first dose of the relevant study treatment at least 24 weeks before CCOD.
Breakthrough hemolysis:
The proportion of patients with BTH from baseline through week 25 was analyzed using the same approach as for transfusion avoidance. As a conservative approach, patients withdrawing from study treatment before week 25 were assumed to have experienced BTH in the unobserved period.
Hemoglobin stabilization and other analyses of proportion:
The proportion of patients with stabilization of hemoglobin from baseline through week 25 was analyzed using the same approach as for transfusion avoidance. As a conservative approach, patients withdrawing from study treatment before week 25 were assumed to not have met hemoglobin stabilization criteria. Similar analyses of proportions were performed for other end points (e.g., the proportion of patients who had experienced a MAVE).
Analysis of PRO end points:
Changes from baseline in HRQoL end points were calculated at each visit. Summary statistics describe the raw scores and mean change (95% CI) from baseline at each visit. Plots of average scores with dispersion bars were presented over time and by treatment arm.
The FACIT-F score was further analyzed as change from baseline to week 25 using the MMRM approach.
For patient preference for crovalimab, or eculizumab or ravulizumab, the percentage of patients preferring each treatment and the reasons for preference were summarized per treatment arm.
Sensitivity Analyses
COMMODORE 2 Study
Various sensitivity analyses were performed to assess the robustness of the coprimary end point results against the following.
Efficacy population definition: Hemolysis control and transfusion avoidance were assessed in the following alternative efficacy populations —
randomized (ITT) population
per-protocol population.
Impact of missing data: The following multiple imputation approaches were performed to assess the robustness of the GEE model against various missing data assumptions in the analysis of hemolysis control —
A Markov Chain Monte Carlo (MCMC) model was used assuming missing data are missing at random.
Pattern mixture models and tipping point analyses were used assuming data are missing not at random. Given that the pattern mixture model imputes missing observations to follow the distribution of the control arm, the approach is considered anticonservative. The tipping point analysis was performed by first assuming a worst case scenario (LDH level ≤ 1.5 × ULN not met for all missing central LDH values) in the crovalimab arm and the reverse for the eculizumab arm (LDH level ≤ 1.5 × ULN met for all missing central LDH values). Only if this worst case scenario was not consistent with the primary analysis would intermediary imputations be performed.
It was expected that all transfusions were recorded.
COMMODORE 1 Study
The sensitivity analyses in the COMMODORE 1 study were not reported.
Subgroup Analyses
COMMODORE 2 Study
The robustness of the treatment effect of crovalimab compared to eculizumab — assessed via the coprimary efficacy end points of hemolysis control and transfusion avoidance — was investigated in predefined subgroups based on key baseline demographic and disease characteristics (age, sex, region [Europe/Central, South, and North America; or Japan and the rest of Asia-Pacific], eculizumab available in region [yes or no], race, pRBC units transfused in the 6 months before baseline, local LDH level at randomization, body weight, and prior diagnosis of aplastic anemia).15,35
Table 10: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factor | Handling of missing data | Sensitivity analysis |
|---|---|---|---|---|
COMMODORE 2 study | ||||
Proportion of patients achieving TA | The difference between proportions was computed using Mantel-Haenszel weights and its 95% CIs using the stratified Newcombe method. | Adjustment for stratification factors of transfusion history and baseline LDH | NA; analysis did not have missing data | By different populations in analysis: randomized (ITT) population, per-protocol population, 24-week efficacy-evaluable population (i.e., primary analysis population) |
Proportion of patients with hemolysis control | A GEE model was used to estimate adjusted log odds ratios of LDH level ≤ 1.5 × ULN. | Independent covariates are categorical effects of treatment and visit, visit by treatment interaction, continuous baseline LDH level, and number of pRBC units administered within the 6 months before randomization. | Implicitly imputed assuming missing at random. Patients who withdrew from the study were assumed to have a BTH event. | Different methods assessing the impact of missing central LDH values: MCMC algorithm, PMM algorithm, tipping point analysis |
Proportion of patients with BTH | Same as TA | Adjustment for stratification factors of transfusion history and baseline LDH level | NA; analysis did not have missing data | NA |
Proportion of patients with stabilized hemoglobin | Same as TA | Adjustment for stratification factors of transfusion history and baseline LDH level | NA; analysis did not have missing data | NA |
Change from baseline in FACIT-F scores | MMRM | Adjustment for stratification factors of transfusion history and baseline LDH level and baseline FACIT-F score | Implicitly imputed by the MMRM model, assuming missing at random | NA |
Safety | Safety was assessed through descriptive summaries of exposure to each study treatment, AEs, changes in laboratory test results, and changes in vital signs and ECG. | NA | NA | NA |
COMMODORE 1 study | ||||
Percentage change in LDH levels from baseline | MMRM | Fixed categorical effects of treatment, visit, and visit by treatment arm interaction, as well as a continuous fixed covariate of baseline LDH level, baseline LDH level by visit interaction, and the stratification of transfusion history (yes or no) | Implicitly imputed assuming missing at random | NA |
Proportion of patients achieving TA | The difference between proportions was computed using Mantel-Haenszel weights and its 95% CIs using the stratified Newcombe method. | The model included the stratification factor of transfusion history. | NA; analysis did not have missing data | NA |
Proportion of patients with hemolysis control | A GEE model was used to estimate adjusted log odds ratios of LDH level ≤ 1.5 × ULN. | Baseline covariates similar as LDH change | Implicitly imputed assuming missing at random | NA |
Proportion of patients with BTH | Same as TA | Same as TA | NA; analysis does not have missing data | NA |
Proportion of patients with stabilized hemoglobin | Same as TA | Same as TA | NA; analysis does not have missing data | NA |
Change from baseline in FACIT-F scores | MMRM | Adjustment for stratification factor of transfusion history | Implicitly imputed by the MMRM model, assuming missing at random | NA |
Safety | Descriptive summaries of exposure to each study treatment, AEs, changes in laboratory test results, and changes in vital signs and ECG | NA | NA | NA |
AE = adverse event; BTH = breakthrough hemolysis; CI = confidence interval; ECG = electrocardiogram; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GEE = generalized estimating equation; ITT = intention to treat; LDH = lactate dehydrogenase; MCMC = Markov Chain Monte Carlo; MMRM = mixed model of repeated measures; NA = not applicable; PMM = predictive mean matching; pRBC = packed red blood cell; TA = transfusion avoidance; ULN = upper limit of normal.
Sources: COMMODORE 2 Primary Clinical Study Report,15 COMMODORE 1 Primary Clinical Study Report,17 and sponsor’s submission.16
COMMODORE 1 Study
As the study evolved into a safety study, subgroup analyses for efficacy were not considered or summarized in this review.
Analysis Populations
The definitions for the different analysis populations in the COMMODORE 2 and COMMODORE 1 trials are summarized in Table 11.
Table 11: Analysis Populations of the COMMODORE 2 and COMMODORE 1 Studies
Study | Population | Definition | Application |
|---|---|---|---|
COMMODORE 2 study | ITT (randomized) population | The randomized population (i.e., ITT) was defined as all randomized patients from arm A and arm B (crovalimab and eculizumab, respectively) with analyses performed as randomized. | For the primary and key secondary efficacy analyses to evaluate the noninferiority of crovalimab compared with eculizumab |
PAP | The PAP included all randomized patients in arm A and arm B who received at least 1 dose of treatment with crovalimab or eculizumab and had at least 1 centrally processed LDH level assessment after the first IV infusion. | ||
PP population | The PP population was composed of all randomized patients who fulfill the following criteria:
| The primary efficacy end point analyses and the secondary end point analyses were repeated on the PP population as sensitivity analyses. | |
24-week crovalimab efficacy population (arm B switch) | Patients had to have received at least 1 dose of crovalimab from week 25, and had to have at least 1 valid postswitch LDH level assessment by the central laboratory after the first IV infusion. | Exploratory efficacy data from arm B patients switching from eculizumab to crovalimab after 24 weeks of treatment (arm B switch) were summarized separately for the time period under crovalimab treatment.a | |
Efficacy-evaluable population (arm C) | All patients who received at least 1 dose of treatment with crovalimab and had at least 1 central LDH level assessment after the first IV infusion | Descriptive efficacy analyses for patients enrolled in arm C | |
Safety-evaluable population | All randomized patients who received at least 1 dose of study drug, with patients grouped according to treatment received | Safety analyses | |
COMMODORE 1 study | ITT (randomized) population | Same as COMMODORE 2 study | Efficacy end points analyses |
24-week efficacy population | All randomized patients in arm A and arm B who received at least 1 dose of treatment with crovalimab or eculizumab and had at least 1 centrally processed LDH level assessment after the first IV infusion; this was restricted to patients recruited at least 24 weeks before CCOD | Exploratory | |
24-week crovalimab efficacy population (arm B switch) | Patients had to have received at least 1 dose of crovalimab from week 25, and had to have at least 1 valid postswitch LDH level assessment by the central laboratory after the first IV infusion. | Exploratory efficacy data from arm B patients switching from eculizumab to crovalimab after 24 weeks of treatment (arm B switch) were summarized separately for the time period under the crovalimab treatment.a | |
Efficacy-evaluable population (arm C) | All patients who received at least 1 dose of treatment with crovalimab and had at least 1 central LDH level assessment after the first IV infusion | Exploratory efficacy results were presented by cohorts (prior ravulizumab, prior high-dose eculizumab, and C5 SNP) for those patients who had the opportunity to complete 24 weeks of treatment before CCOD. | |
Safety-evaluable population | All randomized patients who received at least 1 dose of study drug, with patients grouped according to treatment received | Safety analyses |
CCOD = clinical cut-off date; ITT = intention to treat; LDH = lactate dehydrogenase; PAP = primary analysis population; PNH = paroxysmal nocturnal hemoglobinuria; PP = per protocol; SNP = single nucleotide polymorphism; ULN = upper limit of normal; WBC = white blood cell.
aFor analysis of end points defined over 24-week treatment intervals, only patients with at least 24 weeks of crovalimab treatment were included. However, listings and by visit summary analysis of efficacy end points included all available efficacy data.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
Results
Patient Disposition
Patient disposition for the COMMODORE 2 and COMMODORE 1 trials is summarized in Table 12.
COMMODORE 2 Study
Of the 239 patients in the randomized population screened in the COMMODORE 2 study, 29 (12%) patients were those who did not advance past screening, leading to an enrolment of 210 patients in total. The main reasons for not advancing past screening were poor willingness to comply (62%) and absolute neutrophil count of less than 500 cells/µL (14%). A total of 204 patients were randomized. Of the 135 patients randomized to crovalimab (arm A), 129 (95.6%) patients completed 24 weeks of treatment in the primary treatment period and continued to receive crovalimab in the crovalimab extension period. Of these, 127 patients continued to receive crovalimab treatment up to the CCOD. Of the 69 patients randomized to eculizumab (arm B), 68 (98.6%) patients completed 24 weeks of treatment in the primary treatment period and switched to crovalimab treatment in the crovalimab extension period. Of these 68 patients, 65 continued to receive crovalimab treatment up to the CCOD; the other 3 patients discontinued study treatment and did not enter the safety follow-up. All patients in the randomized arms were 18 years or older, with the exception of 2 adolescent patients (both aged 17 years at the time of randomization), who were randomized to the eculizumab arm before the opening of the separate pediatric-specific descriptive arm C in protocol version 3, and switched to crovalimab in the crovalimab extension period after completing the primary treatment period. One of the 2 adolescent patients had already turned 18 years at the time of switching to crovalimab in the extension period while the other patient was still aged 17 years at the time of switching.15
Table 12: Summary of Patient Disposition From the COMMODORE 2 and COMMODORE 1 Studies
Patient disposition | COMMODORE 2 study | COMMODORE 1 study | ||||
|---|---|---|---|---|---|---|
Arm A: Crovalimab (N = 135) | Arm B: Eculizumab (N = 69) | Arm C: Descriptive pediatric (N = 6) | Arm A: Crovalimab (N = 45) | Arm B: Eculizumab (N = 44) | Arm C: Nonrandomized (N = 38) | |
Screened, N | 239 | NR | 146 | |||
Did not advance past screening, N (%) | 29 (12.1) | 19 (13.0) | ||||
Reason for not advancing past screening, N (%) | ||||||
Poor willingness to comply | 18 (27.6) | 6 (31.6) | ||||
ANC < 500 cells/µL | 3 (10.3) | 0 | ||||
Documented eculizumab treatment as per label | NA | 4 (21.1) | ||||
Lack of signed informed consent form | 2 (6.9) | 4 (21.1) | ||||
Other | 6 (20.7) | 5 (26.3) | ||||
Randomized, N | 135 | 69a | 6 | 45 | 44 | 38 |
No treatment received, N (%) | 0 | 0 | 0 | 1 (2.2) | 2 (4.5) | 0 |
Discontinued from study before 24 weeks, N (%) | 6 (4.4)b | 1 (1.4)c | 0 | 0 | 2 (4.5) | 4 (10.5) |
Reason for discontinuation before 24 weeks, N (%) | ||||||
Physician decision | 2 (1.5) | 0 | 0 | 0 | 0 | 0 |
Withdrawal by patient | 2 (1.5) | 0 | 0 | 0 | 0 | 3 (7.9) |
Death | 1 (0.7)d | 1 (0.7)e | 0 | 0 | 0 | 0 |
Lost to follow-up | 1 (0.7) | 0 | 0 | 0 | 0 | 0 |
Protocol deviation | 0 | 0 | 0 | 0 | 1 (2.3) | 0 |
Adverse event | 0 | 0 | 0 | 0 | 0 | 1 (2.6) |
Other | 0 | 0 | 0 | 0 | 1 (2.3) | 0 |
Completed primary treatment period, N (%) | 129 (95.6) | 68 (98.6) | 6 (100) | 39 (86.7)f | 35 (79.5)g | 29 (76.3)h |
Discontinued from study at or after 24 weeks, N (%) | 2 (1.5) | 3 (4.3) | 0 | 2 (4.4) | 3 (6.8) | 1 (2.6) |
Reason for discontinuation at or after 24 weeks, N (%) | ||||||
Withdrawal by patient | 2 (1.5) | 2 (2.9) | 0 | 1 (2.2) | 1 (2.3) | 0 |
Adverse event | 0 | 1 (1.4) | 0 | 0 | 1 (2.3) | 0 |
Death | 0 | 0 | 0 | 1 (2.2)i | 0 | 0 |
Physician decision | 0 | 0 | 0 | 0 | 1 (2.3) | 0 |
Lack of efficacy | 0 | 0 | 0 | 0 | 0 | 1 (2.6) |
Completed 24 weeks of treatment in extension, N | 79 | 40 | 6 | 24 | 26 | 11 |
Ongoing extension treatment period (as of CCOD), N | 127 | 65 | 6 | 37 | 32 | 28 |
ITT, N | 134j | 69 | 6 | 39 | 37 | 34 |
PP, N | 112 | 59 | 0 | 0 | 0 | 0 |
Safety, N | 135 | 69 | 6 | 44 | 42 | 38 |
Arm B switch patientsk, N | NA | 68 | NA | NA | 35 | NA |
Arm B switched to crovalimab ≥ 24 weeks before CCOD, N | NA | 43 | NA | NA | 28 | NA |
ANC = absolute neutrophil count; CCOD = clinical cut-off date; ITT = intention to treat; LDH = lactate dehydrogenase; NA = not applicable; NR = not reported; PP = per protocol.
Note: Data presented in Table 12 were based on analyses at the CCOD of November 16, 2022.
aTwo pediatric patients were enrolled in randomized arm B before separate descriptive arm C was opened in protocol version 3.
bThree patients did not enter the safety follow-up.
cPatient did not enter the safety follow-up.
dDeath due to myocardial infarction at day 2, which was unrelated to the study drug.
eDeath due to fatal ischemic stroke at day 71, which was unrelated to the study drug.
fSix patients were ongoing in the arm A primary treatment period at CCOD.
gFive patients were ongoing in the arm B primary treatment period at CCOD.
hFive patients were ongoing in the arm C primary treatment period at CCOD.
iDeath due to colorectal cancer, which was unrelated to the study drug.
jThe patient who died in arm A (crovalimab) was excluded from the primary analysis population for not fulfilling the criteria of at least 1 postbaseline LDH level assessment.
kArm B switch patients were defined as all patients in arm B who received at least 1 dose of crovalimab from week 25, and had at least 1 valid postswitch LDH level assessment by the central laboratory after the first IV infusion.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
COMMODORE 1 Study
Of the 146 patients in the randomized population screened in the COMMODORE 1 study, 19 (13%) patients did not advance past screening due to not meeting the inclusion criteria, leading to an enrolment of 127 patients in total. The main reasons for not advancing past screening were poor willingness to comply (32%) and documented eculizumab treatment as per label (21%). In the nonrandomized arm C, 38 patients were enrolled at the time of the CCOD. Of the 45 patients randomized to the crovalimab arm, 39 (86.7%) patients completed 24 weeks of treatment and then continued to receive crovalimab treatment in the crovalimab extension period. Of these, 37 patients continued to receive crovalimab treatment up to the CCOD. Of the 45 patients in the crovalimab arm, 5 patients were still ongoing in the primary treatment period as of the CCOD. Of the 44 patients randomized to the eculizumab arm, 35 (79.5%) patients completed 24 weeks of eculizumab treatment and switched to crovalimab treatment upon entering the crovalimab extension period. Of these, 32 patients continued to receive crovalimab treatment up to the CCOD. Of the 44 patients in the eculizumab arm, 5 patients were still ongoing in the primary treatment period as of the CCOD.17
Baseline Characteristics
The baseline characteristics outlined in Table 13 are limited to those that are most relevant to this review or were relevant for the interpretation of the study results.
COMMODORE 2 Study
In the COMMODORE 2 study, the randomized population had a median age of 36 years (crovalimab arm) or 38 years (eculizumab arm), and most patients were within the age range of 18 years to 64 years (90% in the crovalimab arm and 84% in the eculizumab arm).15 Approximately 43% to 49% were females and 51% to 57% of the patients were males. The majority of patients were Asian (64% to 74%) or white (23% to 33%), followed by other or unknown (3%). Most patients weighed between 40 kg and less than 100 kg at baseline (97% in the crovalimab arm versus 96% in the eculizumab). The baseline mean LDH level was 7.6 multiplied by ULN and 7.8 multiplied by ULN in the crovalimab and eculizumab arms, respectively. Most patients had an LDH level of more than 4 multiplied by ULN (83% in the crovalimab arm versus 84% in the eculizumab arm). At baseline, 74% to 77% of the patients had received a pRBC transfusion with mean transfused pRBCs within the 12 months before screening of 6.5 (SD = 8.3) units in the crovalimab arm and 6.6 (SD = 8.7) units in the eculizumab arm, with half of the patients receiving up to 6 units of pRBC transfusion in the prior 6 months in both arms. The median of the PNH clone size was smaller in the crovalimab arm than in the eculizumab arm for erythrocytes (25% and 45%, respectively) and granulocytes (60% versus 75%, respectively). The main PNH-relevant conditions in patient history were aplastic anemia (39% in the crovalimab arm and 38% in the eculizumab arm), major vascular events (16% versus 15%, respectively), and MDS (4% versus 9%, respectively).
Table 13: Summary of Baseline Characteristics From the COMMODORE 2 and COMMODORE 1 Studies
Characteristic | COMMODORE 2 study | COMMODORE 1 study | |||||||
|---|---|---|---|---|---|---|---|---|---|
Arm A: Crovalimab (N = 135) | Arm B: ECU (N = 69) | Arm C: Descriptive pediatric (N = 6) | Arm A: Crovalimab (N = 45) | Arm B: ECU (N = 44) | Arm C: < 18 years (N = 1) | Arm C: Prior RAVU (N = 21) | Arm C: Prior high- dose ECU (N = 10) | Arm C: C5 SNP (N = 6) | |
Age, years | |||||||||
Mean (SD) | 40.5 (15.2) | 41.9 (16.0) | 16.0 (1.5) | 44.4 (15.6) | 49.5 (14.8) | 16.0 (NE) | 45.5 (11.6) | 35.5 (13.6) | 58.2 (17.3) |
Median (range) | 36.0 (18 to 76) | 38.0 (17 to 78) | 16.5 (13 to 17) | 42.0 (21 to 81) | 49.0 (22 to 85) | 16.0 (NA) | 45.0 (27 to 70) | 32.0 (20 to 58) | 58.0 (38 to 80) |
< 18 years, n (%) | 0 | 2 (2.9)a | 6 (100) | 0 | 0 | 1 (100) | 0 | 0 | 0 |
≥ 18 years, n (%) | 135 (100) | 67 (97.1) | 0 | 45 (100) | 44 (100) | 0 | 21 (100) | 10 (100) | 6 (100) |
Sex, n (%) | |||||||||
Female | 58 (43.0) | 34 (49.3) | 2 (33.3) | 24 (53.3) | 22 (50.0) | 0 | 9 (42.9) | 6 (60.0) | 4 (66.7) |
Male | 77 (57.0) | 35 (50.7) | 4 (66.7) | 21 (46.7) | 22 (50.0) | 1 (100) | 12 (57.1) | 4 (40.0) | 2 (33.3) |
Race, n (%) | |||||||||
Asian | 86 (63.7) | 51 (73.9) | 5 (83.3) | 9 (20.0) | 7 (15.9) | 0 | 11 (52.4) | 0 | 6 (100) |
White | 45 (33.3) | 16 (23.2) | 0 | 34 (75.6) | 32 (72.7) | 1 (100) | 9 (42.9) | 6 (60.0) | 0 |
Other or unknown | 4 (2.9) | 2 (2.8) | 1 (16.7) | 2 (4.4) | 5 (11.4) | 0 | 1 (4.8) | 4 (40.0) | 0 |
Weight, kg | |||||||||
Mean (SD) | 68.3 (15.8) | 67.1 (15.3) | 69.7 (17.8) | 77.0 (17.5) | 76.5 (18.0) | 53.0 (NE) | 69.2 (13.9) | 67.95 (11.2) | 65.9 (19.6) |
Median (range) | 66.1 (42.0 to 140.3) | 62.2 (47.0 to 122.0) | 67.8 (50.0 to 98.5) | 80.0 (45.2 to 120.0) | 75.1 (47.2 to 126.4) | 53.0 (NA) | 69.5 (46.0 to 91.0) | 65.95 (48.1 to 82.0) | 66.2 (44.0 to 89.2) |
PNH history, years | |||||||||
n | 135 | 69 | NR | 43 | 42 | 1 | 20 | 10 | 6 |
Mean age at PNH diagnosis (SD) | 35.8 (15.5) | 37.4 (16.4) | NR | 36.2 (15.4) | 39.1 (14.8) | 13.0 (NE) | 34.3 (12.1) | 24.7 (9.1) | 53.3 (19.5) |
Mean time from PNH diagnosis to enrolment (SD) | 5.2 (7.4) | 4.97 (5.9) | NR | 8.0 (6.6) | 11.2 (7.1) | 3.8 (NE) | 12.3 (11.6) | 11.4 (10.6) | 5.3 (4.8) |
Baseline central LDH levelb | |||||||||
n | 134 | 69 | NR | 44 | 42 | 1 | 21 | 10 | 6 |
LDH value, mean (SD), U/L | 1,770.61 (790.02) | 1,817.50 (928.09) | NR | 249.16 (65.54) | 234.20 (55.27) | 358.50 (NE) | 234.07 (46.51) | 229.10 (52.49) | 1,826.00 (1,585.41) |
LDH level, mean (SD), × ULN | 7.57 (3.38) | 7.77 (3.54) | NR | 1.06 (0.28) | 1.00 (0.24) | 1.53 (NE) | 1.00 (0.20) | 0.98 (0.22) | 7.80 (6.78) |
Stratification factor (COMMODORE 2 study): Categorical local LDH level at baseline, n (%) | |||||||||
2 to 4 × ULN | 24 (17.8) | 11 (15.9) | NR | NR | NR | NR | NR | NR | NR |
> 4 × ULN | 111 (82.8) | 58 (84.1) | NR | NR | NR | NR | NR | NR | NR |
Baseline hemoglobin level | |||||||||
n | 135 | 69 | NR | 44 | 42 | 1 | 21 | 10 | 6 |
Hemoglobin, mean (SD), g/L | 87.18 (14.06) | 99.68 (87.86)c | NR | 109.74 (19.96) | 107.27 (17.66) | 149.00 (NE) | 109.76 (20.02) | 97.36 (19.47) | 89.83 (19.50) |
Baseline pRBC transfusion history ≤ 12 months before screening | |||||||||
n | 133 | 68 | NR | 44 | 44 | 1 | 21 | 10 | 6 |
Number of patients with pRBC transfusion, n (%) | 103 (77.4) | 50 (73.5) | NR | 10 (22.7) | 11 (25.0) | 0 | 3 (14.3) | 4 (40.0) | 3 (50.0) |
n | 132 | 67 | NR | 44 | 44 | 1 | 21 | 10 | 6 |
Number of pRBC units transfused, mean (SD) | 6.47 (8.27) | 6.63 (8.70) | NR | 1.55 (3.72) | 2.32 (5.43) | 0 (NE) | 0.57 (1.80) | 1.80 (3.08) | 14.00 (21.39) |
Stratification factor (COMMODORE 2 study): Categorical pRBC units received ≤ 6 months before randomization, n (%) | |||||||||
0 units | 33 (24.4) | 17 (24.6) | NR | NA | NA | NA | NA | NA | NA |
> 0 to 6 units | 68 (50.4) | 34 (49.3) | NR | NA | NA | NA | NA | NA | NA |
> 6 units | 34 (25.2) | 18 (26.1) | NR | NA | NA | NA | NA | NA | NA |
PNH clone size, median (range), % | |||||||||
n | 77 | 41 | NR | 41 | 35 | 1 | 21 | 9 | 6 |
Erythrocytes | 25.1 (3.5 to 96.0) | 44.6 (0.1 to 88.9) | NR | 44.6 (2.6 to 100) | 46.5 (1.3 to 100) | NE (99.99 to 99.99) | 72.65 (1.90 to 99.94) | 47.07 (16.92 to 94.37) | 36.59 (8.47 to 48.05) |
n | 75 | 41 | NR | 40 | 36 | NAd | 21 | 9 | 6 |
Granulocytes | 60.3 (0.8 to 96.1) | 74.6 (1.3 to 95.2) | NR | 66.5 (1.7 to 92.4) | 67.9 (2.2 to 97.8) | NAd | 57.94 (1.17 to 95.60) | 74.02 (7.72 to 94.13) | 57.37 (19.26 to 86.53) |
n | 76 | 41 | NR | 40 | 36 | NAd | 21 | 9 | 6 |
Monocytes | 90.8 (42.5 to 100) | 95.1 (41.5 to 99.9) | NR | 88.6 (13.8 to 100) | 96.3 (7.6 to 99.9) | NAd | 87.76 (9.61 to 99.97) | 91.83 (69.47 to 99.04) | 87.18 (37.61 to 99.37) |
History of PNH-relevant conditions before enrolment or first dose administration, n (%) | |||||||||
Aplastic anemia | 53 (39.3) | 26 (37.7) | NR | 15 (33.3) | 16 (36.4) | 0 | 9 (42.9) | 2 (20.0) | 1 (16.7) |
Major vascular events | 21 (15.6) | 10 (14.5) | NR | 10 (22.2) | 10 (22.7) | 0 | 2 (9.5) | 0 | 3 (50.0) |
MDS | 6 (4.4) | 6 (8.7) | NR | 0 | 0 | 0 | 0 | 0 | 0 |
ECU = eculizumab; LDH = lactate dehydrogenase; MDS = myelodysplastic syndrome; NA = not applicable; NE = not estimable; NR = not reported; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; RAVU = ravulizumab; SD = standard deviation; SNP = single nucleotide polymorphism; ULN = upper limit of normal.
Note: Data presented in Table 13 were based on analyses at the clinical cut-off date of November 16, 2022. Only patients with available data were included in the calculations of percentages.
aTwo patients aged 17 years were randomized to the ECU arm before the opening of the separate pediatric-specific nonrandomized arm.
bBaseline LDH level was defined as the mean of all central LDH values, collected within 28 days before the first on-study drug administration including the predose value from day 1. The switch baseline LDH value was defined as the mean of all central LDH values, collected within 28 days of the first dose of crovalimab including the predose value from switch day 1.
cThe maximum baseline hemoglobin value in the ECU arm of 810 g/L was a result of erroneous data entry.
dNo patients in this cohort had data available for the PNH clone size (%) for granulocytes or monocytes.
Sources: COMMODORE 2 Primary Clinical Study Report,15 COMMODORE 1 Primary Clinical Study Report,17 and sponsor’s submission.23
COMMODORE 1 Study
In the COMMODORE 1 study, the randomized population had a median age of 42 years (crovalimab arm) or 49 years (eculizumab arm), and as in the COMMODORE 2 study, most patients were within the age range of 18 years to 64 years (89% in the crovalimab arm and 84% in the eculizumab arm). Approximately half of the patients were female (50% to 53%) and half were male (47% to 50%). Unlike the COMMODORE 2 trial, the majority of patients in the COMMODORE 1 trial were white (76% in the crovalimab arm and 73% in the eculizumab arm), followed by Asian (20% versus 16%, respectively), and other or unknown (4% versus 11%, respectively). Most patients weighed between 40 kg and less than 100 kg (93% in the crovalimab arm versus 91% in the eculizumab arm). The mean time from PNH diagnosis to enrolment was shorter in the crovalimab arm (8.0 [SD = 6.6] years) than in the eculizumab arm (11.2 [SD = 7.1] years). At baseline, 23% to 25% of the patients had received a pRBC transfusion with mean pRBC units transfused within the 12 months before screening of 1.6 (SD = 3.7) units in the crovalimab arm and 2.3 (5.4) units in the eculizumab arm. The median of PNH clone size for monocytes was larger in the eculizumab arm (96%) than in the crovalimab arm (89%). The main PNH-relevant conditions in patient history were aplastic anemia (33% in the crovalimab arm and 36% in the eculizumab arm) and major vascular events (22% versus 23%, respectively).
Exposure to Study Treatments
The extent of patient exposure to the study drugs in the COMMODORE 2 and COMMODORE 1 trials is summarized in Table 14.
COMMODORE 2 Study
The median treatment duration during the primary treatment period was similar in the crovalimab and eculizumab arms (20.1 weeks versus 22.1 weeks, respectively). The median number of doses administered in the crovalimab arm at 10 (range, 1 to 12) was lower than in the eculizumab arm at 14 (range, 6 to 16). The median total cumulative dose in the crovalimab arm at 5,760 mg (range, 1,000 mg to 9,390 mg) was lower than in the eculizumab arm at 11,400 mg (range, 4,200 mg to 13,200 mg).
In the crovalimab arm up to the CCOD (November 16, 2022), the median treatment duration was 48.3 weeks, with 57% of patients having had a treatment duration of at least 48 weeks. Analysis of serum concentrations showed that 5 patients had partial loss of crovalimab exposure and 2 patients had complete loss of exposure.15
At the CCOD, the median treatment duration with crovalimab for eculizumab (arm B) switch patients was 24.1 weeks, with a median of 11 doses administered. The median total cumulative dose was 6,440 mg (range, 1,340 mg to 15,280 mg). Overall, 40 (59%) patients had a treatment duration of at least 24 weeks and 41% of patients had a treatment duration of less than 24 weeks. One patient reached more than 72 weeks of treatment duration at the CCOD.15
In arm C, treatment duration in the 6 pediatric patients ranged between 21 weeks and 57 weeks. All patients completed the 24-week primary treatment period per the dose recommendation in the study protocol and were ongoing in the crovalimab extension period as of the CCOD. For 1 pediatric patient, the SC dose of crovalimab was increased from 680 mg to 1,020 mg from week 13 onward in the context of weight increase to more than 100 kg (from 98.5 kg at baseline); however, this weight increase did not meet the more than 10% change criterion and a major protocol deviation in relation to this modification was recorded; no new safety signal was detected in relation to this increased crovalimab dose. The patient remained on the 1,020 mg dose at the time of the CCOD.15
COMMODORE 1 Study
The median treatment duration during the primary safety period was 20.1 weeks in the crovalimab arm and 22.1 weeks in the eculizumab arm. The median number of doses administered was 10 (range, 4 to 11) and 12 (range, 1 to 14) in the crovalimab and eculizumab arms, respectively. The median total cumulative dose in the crovalimab arm at 5,760 mg (range, 2,020 mg to 7,960 mg) was lower than in the eculizumab arm at 10,800 mg (range, 900 mg to 12,600 mg).
In the crovalimab arm up to the CCOD, the median treatment duration was 52 weeks. The majority of patients (54.5%) had a treatment duration of at least 48 weeks. At the CCOD, the median treatment duration with crovalimab for eculizumab (arm B) switch patients was 32 weeks, with a median of 13 doses administered. The median total cumulative dose was 9,160 mg (range, 2,360 mg to 17,140 mg). The majority of patients (74%) had a treatment duration of at least 24 weeks and 26% of patients had a treatment duration of less than 24 weeks.17
In arm C, the median treatment durations in the crovalimab safety population were 2.1 weeks (range, 2.1 weeks to 2.1 weeks), 36.0 weeks (range, 2.3 weeks to 80.1 weeks), 38.1 weeks (range, 0.3 weeks to 64.1 weeks), and 50.2 weeks (range, 28.1 weeks to 100.1 weeks) in the pediatric, prior ravulizumab, prior high-dose eculizumab, and C5 polymorphism cohorts, respectively. One patient in the pediatric cohort, 6 patients in the prior ravulizumab cohort, and 2 patients in the prior high-dose eculizumab cohort, respectively, had a treatment duration of less than 24 weeks at the time of primary analysis. The median number of doses administered was 4.0 (range, 4.0 to 4.0), 14.0 (range, 4.0 to 25.0), 14.5 (range, 2.0 to 20.0), and 17.5 (range, 12.0 to 30.0) in the pediatric, prior ravulizumab, prior high-dose eculizumab, and C5 polymorphism cohorts, respectively. The median total cumulative doses were 2,020 mg (range, 2,020 mg to 2,020 mg), 8,480 mg (range, 2,020 mg to 15,960 mg), 8,820 mg (range, 1,340 mg to 12,255 mg), and 10,860 mg (range, 7,120 mg to 19,360 mg) in the pediatric, prior ravulizumab, prior high-dose eculizumab, and C5 polymorphism cohorts, respectively. The median IV and SC dose intensity across these cohorts was 100.0% (IV range, 100.0% to 137.5%; SC range, 85.7% to 102.4%).17
Table 14: Summary of Patient Exposure From the COMMODORE 2 and COMMODORE 1 Studies (Primary Safety Period, Randomized Safety Population)
Patient exposure | COMMODORE 2 study | COMMODORE 1 study | ||
|---|---|---|---|---|
Arm A: Crovalimab (N = 135) | Arm B: Eculizumab (N = 69) | Arm A: Crovalimab (N = 44) | Arm B: Eculizumab (N = 42) | |
Treatment duration, weeks | ||||
Duration, mean (SD) | 19.72 (2.77) | 22.02 (2.01) | 19.1 (3.7) | 20.4 (5.7) |
Duration, median (IQR) | 20.14 (20.00 to 20.29) | 22.14 (22.14 to 22.29) | 20.1 (20.1 to 22.3) | 22.1 (22.1 to 22.1) |
> 0 to < 12, n (%) | 3 (2.2) | 1 (1.4) | 2 (4.5) | 4 (9.5) |
12 to < 24, n (%) | 132 (97.8) | 67 (97.1) | 42 (95.5) | 35 (83.3) |
24 to < 36, n (%) | 0 | 1 (1.4) | 0 | 3 (7.1) |
Total cumulative dose, mg | ||||
Mean (SD) | 5,714.4 (776.3) | 11,226.1 (933.8) | 5,695.1 (829.6) | 9,985.7 (2,582.6) |
Median (range) | 5,760.0 (1,000 to 9,390) | 11,400.0 (4,200 to 13,200) | 5,760.0 (2,020 to 7,960) | 10,800.0 (900 to 12,600) |
Total IV dose intensity, % | ||||
Mean (SD) | 101.5 (8.9) | 100.0 (0) | 101.7 (7.9) | 100.0 (0) |
Median (range) | 100.0 (95.0 to 175.0) | 100.0 (100.0 to 100.0) | 100.0 (99.7 to 137.5) | 100.0 (100.0 to 100.0) |
Total SC dose intensity, % | ||||
n | 134 | 0 | 44 | 0 |
Mean (SD) | 99.99 (2.12) | NE (NE) | 100.0 (0) | NE (NE) |
Median (range) | 100.0 (82.1 to 116.7) | NE (NE to NE) | 100.0 (100.0 to 100.0) | NE (NE to NE) |
Number of doses administered, n | ||||
Mean (SD) | 9.8 (1.1) | 13.8 (1.1) | 9.7 (1.1) | 11.1 (2.9) |
Median (range) | 10.0 (1.0 to 12.0) | 14.0 (6.0 to 16.0) | 10.0 (4.0 to 11.0) | 12.0 (1.0 to 14.0) |
Total | 1,328.0 | 952.0 | 428.0 | 466.0 |
Number of IV scheduled doses, n | ||||
Mean (SD) | 1.0 (0.1) | 13.8 (1.1) | 1.0 (0) | 11.1 (2.9) |
Median (range) | 1.0 (1.0 to 2.0) | 14.0 (6.0 to 16.0) | 1.0 (1.0 to 1.0) | 12.0 (1.0 to 14.0) |
Total | 136.0 | 952.0 | 44.0 | 466.0 |
Number of IV unscheduled doses, n (%) | ||||
0 | 131 (97.0) | 69 (100) | 42 (95.5) | 42 (100) |
1 | 2 (1.5) | 0 | 2 (4.5) | 0 |
2 | 2 (1.5) | 0 | NA | NA |
Number of SC doses, n | ||||
n | 134 | 0 | 44 | 0 |
Mean (SD) | 8.9 (0.7) | NE (NE) | 8.7 (1.1) | NE (NE) |
Median (range) | 9.0 (3.0 to 10.0) | NE (NE to NE) | 9.0 (3.0 to 10.0) | NE (NE to NE) |
Total | 1,186.0 | NE | 382.0 | NE |
IQR = interquartile range; NA = not applicable; NE = not estimable; SC = subcutaneous; SD = standard deviation.
Note: Data presented in Table 14 were based on analyses at the clinical cut-off date of November 16, 2022. IV dose intensity is a percentage based on actual IV dose per total planned IV dose. SC dose intensity is a percentage based on actual SC dose per total planned SC dose.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
Adherence to Self-Administration in COMMODORE 2 and COMMODORE 1 Studies
Crovalimab self-administration or drug administration by a caregiver was permitted in both the COMMODORE 2 and COMMODORE 1 studies starting at week 9, after training and confirmation of proficiency by the health care provider. Between week 9 and week 25, the majority of SC injections were administered by health care professionals with an increasing proportion of patients with SC administration of crovalimab done by the patient or their caregiver up to week 25 (range, 17.1% to 31.0%, in arm A of the COMMODORE 2 study; range, 11.6% to 15.8%, in arm A of the COMMODORE 1 study; and range, 21.6% to 27.6%, in arm C of the COMMODORE 1 study). After week 25, the rate of administration by the patient or caregiver was generally rising, with an increase in offsite administrations. No medication errors specifically due to self-administration were reported up to the CCOD in either study.15
Concomitant Medications and Cointerventions in COMMODORE 2 and COMMODORE 1 Studies
The most frequently used (≥ 50% in either arm) previous or concomitant medications in the 2 trials are listed in Table 15.
In the COMMODORE 2 study, the majority of patients in the crovalimab and eculizumab arms had at least 1 previous medical condition (74.1% versus 81.2%, respectively).15 The most frequent previous medical conditions were hypertension (22.2% in the crovalimab arm versus 14.5% in the eculizumab arm), cholelithiasis (6.7% versus 8.7%, respectively), and MDS (4.4% versus 8.7%, respectively).15 Most patients in each arm received at least 1 previous or concomitant treatment (99.3% in the crovalimab arm versus 98.6% in the eculizumab arm). The most common concomitant medications were ophthalmologicals (78.5% in the crovalimab arm versus 87.5% in the eculizumab arm), antibacterials for systemic use (76.3% versus 75.4%, respectively), and otologicals (62.2% versus 68.1%, respectively) (Table 15).
In the COMMODORE 1 study, the majority of patients in the crovalimab and eculizumab arms had at least 1 previous medical condition (75.6% versus 72.7%, respectively).17 The most frequent previous medical conditions were hypertension (15.6% in the crovalimab arm versus 27.3% in the eculizumab arm), cholelithiasis (8.9% versus 11.4%, respectively), and hypothyroidism (8.9% versus 6.8%, respectively).17 The proportion of patients who received at least 1 concomitant medication in the crovalimab arm and eculizumab arm was 93.3% and 86.4%, respectively. The most common concomitant medications were topical products for joint and muscular pain (73.3% in the crovalimab arm versus 72.7% in the eculizumab arm), ophthalmologicals (64.4% versus 61.4%, respectively) and antibacterials for systemic use (71.1% versus 50.0%, respectively) (Table 15).
At baseline, medications of special interest included immunosuppressive therapies, systemic corticosteroids, and antithrombotic drugs. Overall, the use of these selected therapeutics was comparable between the randomized arm A (crovalimab) and arm B (eculizumab) in the COMMODORE 2 study while in the COMMODORE 1 study, the rate of some medications in arm A was higher than in arm B: systemic corticosteroid therapy (34.1% versus 36.2%, respectively, in the COMMODORE 2 trial and 11.1% versus 6.8%, respectively, in the COMMODORE 1 trial), immunosuppressants (17.0% versus 18.8%, respectively, in the COMMODORE 2 trial and 42.2% versus 27.3%, respectively, the COMMODORE 1 trial), ██████████ ████ █████████ █ █████ █████ ███ ██████, and anticoagulants (25.9% versus 24.6%, respectively, in the COMMODORE 2 trial and 26.7% versus 22.7%, respectively, in the COMMODORE 1 trial).15,17
In the COMMODORE 2 study, arm C (pediatric), systemic antibiotic therapeutics, immunosuppressants, and erythrocyte-stimulating drugs were administered to 1 pediatric patient each before enrolment. One patient received systemic antibacterial therapeutics on day 1.15
In the COMMODORE 1 study, arm C, at baseline, the proportion of patients receiving immunosuppressants or antithrombotic drugs was 63.2% and 15.8%, respectively. No patients received corticosteroids for systemic use at baseline.17
The majority of patients was immunized with Neisseria meningitidis serotypes A, C, W, and Y, with rates in the COMMODORE 2 study for arm A (crovalimab, 96.3%), arm B (eculizumab, 98.6%), and arm C (pediatric, 100%), and in the COMMODORE 1 study for arm A (crovalimab, 91.1%), arm B (eculizumab, 95.5%), and arm C (nonrandomized, 92.1%).15,17
Table 15: Summary of Previous or Concomitant Treatment From the COMMODORE 2 Study (Arm A and Arm B) and the COMMODORE 1 Study (All Arms), Full Reporting Period
Medication, % | COMMODORE 2 study | COMMODORE 1 study | |||
|---|---|---|---|---|---|
Arm A: Crovalimab (N = 135) | Arm B: Eculizumab (N = 69) | Arm A: Crovalimab (N = 45) | Arm B: Eculizumab (N = 44) | Arm C: Nonrandomized (N = 38) | |
At least 1 previous or concomitant treatment, n (%) | 134 (99.3) | 68 (98.6) | 42 (93.3) | 38 (86.4) | 36 (94.7) |
Treatment, % | |||||
Ophthalmologicals | 78.5 | 87.0 | 64.4 | 61.4 | 78.9 |
Antibacterials for systemic use | 76.3 | 75.4 | 71.1 | 50.0 | 63.2 |
Otologicals | 62.2 | 68.1 | 55.6 | 27.3 | 65.8 |
Antianemic preparations | 58.5 | 59.4 | 55.6 | 63.6 | 47.4 |
Topical products for joint and muscular pain | 54.8 | 60.9 | 73.3 | 72.7 | 65.8 |
Stomatological preparations | 51.9 | 60.9 | 42.2 | 45.5 | 47.4 |
Diagnostic drugs | 50.4 | 52.2 | 51.1 | 56.8 | 36.8 |
Drugs for acid-related disorders | 46.7 | 59.4 | 31.1 | 36.4 | 47.4 |
Corticosteroids, dermatological preparations | 39.3 | 66.7 | 33.3 | 34.1 | 47.4 |
Corticosteroids for systemic use | 39.3 | 65.2 | 26.7 | 27.3 | 44.7 |
Nasal preparations | 42.2 | 50.7 | 24.4 | 22.7 | 44.7 |
Analgesics | 46.7 | 33.3 | 55.6 | 56.8 | 63.2 |
Vaccines | 41.5 | 31.9 | 44.4 | 56.8 | 50.0 |
Ophthalmological and otological preparations | 41.5 | 49.3 | 48.9 | 13.6 | 55.3 |
Note: Data presented in Table 15 were based on analyses at the clinical cut-off date of November 16, 2022.
Sources: COMMODORE 2 Primary Clinical Study Report15 and COMMODORE 1 Primary Clinical Study Report.17
Efficacy
A summary of the key efficacy results for the randomized comparison of crovalimab (arm A) versus eculizumab (arm B) in the primary treatment periods of the COMMODORE 2 and COMMODORE 1 studies is shown in Table 16 and Figure 12 (Appendix 1). The efficacy analyses included all randomized patients in arm A and arm B who were recruited at least 24 weeks before the CCOD of November 16, 2022.15,17 Exploratory efficacy data from arm B patients switching from eculizumab to crovalimab beyond week 25 of the trials are summarized under each efficacy outcome for the time period under crovalimab treatment, following the switch. Depending on the availability and relevance of data, the results of the extension period are reported for arm B switch patients (68 patients in the COMMODORE 2 study and 35 patients in the COMMODORE 1 study) and/or arm B switch patients who switched to crovalimab at least 24 weeks before the data cut-off (43 patients in the COMMODORE 2 study and 28 patients in the COMMODORE 1 study).15,17
Hemolysis Outcomes
Hemolysis Control
COMMODORE 2 study: Crovalimab demonstrated noninferiority compared with eculizumab treatment for hemolysis control as measured by a central LDH value of 1.5 or less multiplied by ULN from week 5 through week 25. The OR for hemolysis control (crovalimab versus eculizumab) was 1.02 (95% CI, 0.57 to 1.82). The lower limit of the 95% CI for the OR of 0.57 was greater than the predefined NIM of 0.2. The mean proportion of patients with hemolysis control as measured by a central LDH value of 1.5 or less multiplied by ULN from week 5 through week 25 was similar between the crovalimab arm at 79.3% (95% CI, 72.9% to 84.5%) and the eculizumab arm at 79.0% (95% CI, 69.7% to 86.0%; P = 0.95).
The proportion of patients who achieved hemolysis control increased from 0% in both treatment arms at baseline to 81.0% of patients (95% CI, 73.0% to 87.4%) in the crovalimab arm and 83.8% of patients (95% CI, 72.9% to 91.6%) in the eculizumab arm at week 5. These proportions remained between 75.2% and 83.8% in the crovalimab arm and between 73.8% and 85.1% in the eculizumab arm through week 25 (Figure 3). Overall, the results of the different sensitivity analyses using intention-to-treat and per-protocol populations and different statistical models were consistent with the primary analysis of hemolysis control. Specifically, a total of 7.5% missing central LDH values was reported from week 5 through week 25 (9.5% in the crovalimab arm versus 3.7% in the eculizumab arm). The results from the respective imputation-based sensitivity analyses (MCMC algorithm, predictive mean matching algorithm, and tipping point analysis) were consistent with those from the primary analysis.15,23
Point estimates in the crovalimab arm were generally consistent across subgroups for hemolysis control. █████████ █████████ ██████ ██ ██████████ ██████ ██████████ ███ ████ ██ ███ █████ █████████ ███ █████████ ███████ ██ ███████ █████ █████ █████████ ███████████ █████ ███ ███ ████ ██ █████ ███ ██ █████ █████ ██████ █████ ███ ███ ████ ██ ██████.15 A different treatment effect by sex was not observed for the coprimary end point of transfusion avoidance. The sponsor reported that differences based on sex were not expected.
In the COMMODORE 2 trial, following the 24-week randomized, primary treatment period, of the 68 eculizumab-treated patients who switched to crovalimab (arm B switch patients), 50 patients (proportion of patients = 73.5%; 95% CI, 61.4% to 83.5%) had hemolysis control at the time of the switch (switch baseline). From switch baseline to 25 weeks later (switch week 25), the proportion of patients with hemolysis control ranged from 77.3% to 89.7% at each visit (Appendix 1, Figure 13).15,23
Figure 3: COMMODORE 2 Study — Proportion of Patients With Hemolysis Control (Central LDH Value ≤ 1.5 × ULN) Through Week 25 by Visit, With 95% CI (Primary Analysis Population)
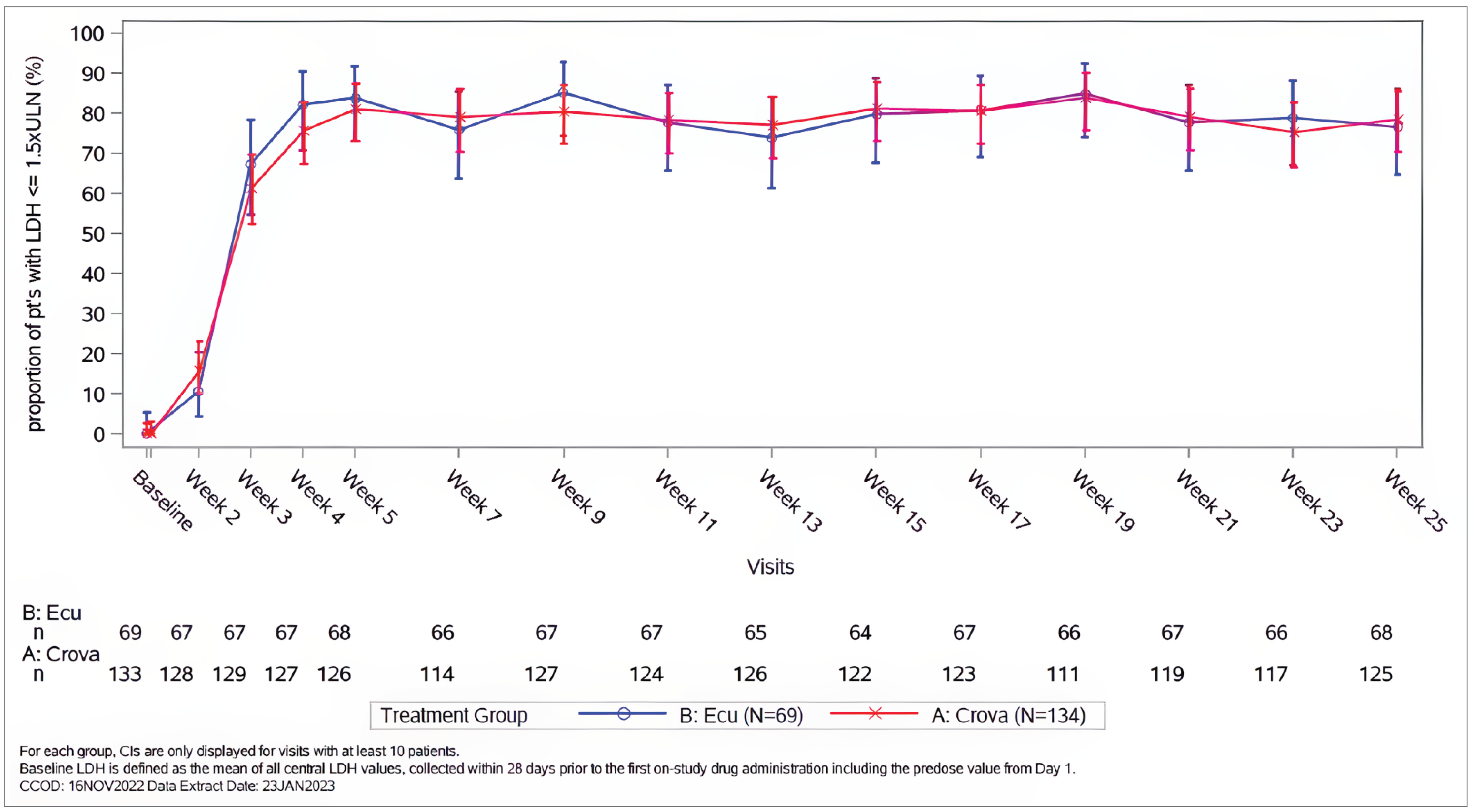
CI = confidence interval; Crova = crovalimab; Ecu = eculizumab; LDH = lactate dehydrogenase; pt = patient; ULN = upper limit of normal.
Note: Data shown in Figure 3 were based on analyses at the clinical cut-off date of November 16, 2022. For each group, CIs are displayed only for visits with at least 10 patients. The baseline LDH value was defined as the mean of all central LDH values, collected within 28 days before the first on-study drug administration including the predose value from day 1.
Source: COMMODORE 2 Primary Clinical Study Report.15
COMMODORE 1 study: The mean proportion of patients who achieved hemolysis control (a central LDH value ≤ 1.5 × ULN) from baseline through week 25 was 92.9% (95% CI, 86.6% to 96.4%) for the crovalimab arm versus 93.7% (95% CI, 87.3% to 97.0%) for the eculizumab arm (Table 16). The mean proportion of patients with a central LDH value of 1.5 or less multiplied by ULN at baseline was 93.2% in the crovalimab arm versus 95.2% in the eculizumab arm. These proportions ranged from 76.9% to 100.0% of patients in the crovalimab arm and 86.1% to 97.2% of patients in the eculizumab arm up to week 25.17
Of the 35 arm B patients who switched to crovalimab (arm B switch patients), 31 patients (proportion of patients = 88.6%; 95% CI, 73.3% to 96.8%) had hemolysis control at switch baseline. The proportion of patients with hemolysis control ranged between 89.7% and 100.0% across all following visits up to switch week 25.17 In the arm B switch patients who switched to crovalimab for at least 24 weeks (n = 28), the mean proportion of patients who achieved hemolysis control by the CCOD was 95.6% (95% CI, 87.3% to 98.6%).17
Breakthrough Hemolysis
COMMODORE 2 study: The proportion of patients with a BTH event from baseline through week 25 was 10.4% (95% CI, 6.0% to 17.2%) in the crovalimab arm compared with 14.5% (95% CI, 7.5% to 25.5%) in the eculizumab arm. The weighted difference in the proportions of patients with BTH (crovalimab versus eculizumab) was −3.9% (95% CI, −14.8% to 5.3%). The upper limit of the 95% CI for the difference in the proportions was 5.3%, which is lower than the predefined NIM of 20% (Table 16).
Of the 43 arm B switch patients, a BTH event was attributed to 7 patients (proportion of patients = 16.3%; 95% CI, 7.3% to 31.3%) between switch baseline and switch week 25. Of these, 4 patients had recorded a BTH event while the other 3 patients were considered to have had a BTH event as a conservative analysis approach.15
COMMODORE 1 study: The proportion of patients with BTH from baseline through week 25 was 10.3% (95% CI, 3.3% to 25.2%) for the crovalimab arm versus 13.5% (95% CI, 5.1% to 29.6%) for the eculizumab arm (Table 16). Two patients in the eculizumab arm without a BTH event discontinued treatment before week 25 and were therefore conservatively assumed as having had a BTH event.
Of the 28 arm B switch patients who switched to crovalimab for at least 24 weeks, a BTH event was attributed to 5 (17.9%) patients between switch baseline and switch week 25. Of these, 3 patients were considered to have had a BTH event as a conservative analysis approach.17
Mean Percentage Change in LDH Levels
COMMODORE 2 study: The mean percentage reduction in LDH levels (central assessment) from baseline to week 25 was −73.6% (95% CI, −79.0% to −68.2%) in the crovalimab arm compared with −64.1% (95% CI, −71.4% to −56.8%) in the eculizumab arm (Table 16).
Results of this outcome were not reported for the arm B switch patients in the extension period.
COMMODORE 1 study: The mean percentage change in the central LDH value from baseline to the average over week 21, week 23, and week 25 was 16.6% (95% CI, ███ ██ ████) for the crovalimab arm versus 4.5% (95% CI, ████ ██ ████) for the eculizumab arm (Table 16). While the percentage change (increase) in the central LDH value from baseline was higher in the crovalimab arm compared with the eculizumab arm, the mean central LDH value remained at 1.5 or less multiplied by ULN for both the crovalimab and eculizumab arms throughout the primary treatment period (Appendix 1, Figure 14) and up to the CCOD. The mean proportion of patients with a central LDH value of 1.5 or less multiplied by ULN by visit through week 25 was also similar between the 2 arms (refer to Table 16).17,23
Results of this outcome were not reported for the arm B switch patients in the extension period.
Hemoglobin Outcomes
Stabilized Hemoglobin
COMMODORE 2 study: The proportion of patients reaching hemoglobin stabilization (the avoidance of a ≥ 2 g/dL decrease in hemoglobin level from baseline, in the absence of transfusion) from baseline through week 25 was 63.4% (95% CI, 54.6% to 71.5%) in the crovalimab arm compared with 60.9% (95% CI, 48.4% to 72.2%) in the eculizumab arm. The weighted difference in the proportion of patients with hemoglobin stabilization (crovalimab versus eculizumab) was 2.2% (95% CI, −11.4% to 16.3%) and the lower limit of the 95% CI of −11.4% was higher than the predefined NIM of −20% (Table 16).
In arm B switch patients, 27 of 43 patients (proportion of patients = 62.8%; 95% CI, 46.7% to 76.6%) achieved hemoglobin stabilization from switch baseline to switch week 25. Of these, 2 patients who were hemoglobin-stabilized while on study discontinued treatment before week 25 and were therefore conservatively assumed as not having stabilized hemoglobin.15
COMMODORE 1 study: The proportion of patients with stabilized hemoglobin from baseline through week 25 was 59.0% (95% CI, 42.2% to 74.0%) in the crovalimab arm versus 70.3% (95% CI, 52.8% to 83.6%) in the eculizumab arm (Table 16).
A total of 18 of the 28 (64.3%) arm B switch patients in the 24-week crovalimab efficacy population achieved hemoglobin stabilization from switch baseline to switch week 25.
Patients Who Reached and Maintained a Minimum Hemoglobin Level (Hemoglobin ≥ 10 g/dL Without Subsequent Decrease < 9 g/dL)
In the COMMODORE 2 study, the proportion of patients who reached and maintained a hemoglobin level of at least 10 g/dL (without a subsequent decrease below 9 g/dL, in the absence of transfusion) from baseline through week 25 was █████ ████ ███ ████ ██ █████ in the crovalimab arm and █████ ████ ███ ████ ██ █████ in the eculizumab arm.
In the COMMODORE 1 study, the proportion of patients who reached and maintained a hemoglobin level of at least 10 g/dL (without a subsequent decrease below 9 g/dL, in the absence of transfusion) from baseline through week 25 was █████ ████ ███ ████ ██ █████ in the crovalimab arm versus █████ ████ ███ ████ ██ █████ in the eculizumab arm.
In both the COMMODORE 2 and COMMODORE 1 trials, results of this outcome were not reported for the arm B switch patients in the extension period.
Transfusion Outcomes
Transfusion Avoidance
COMMODORE 2 study: Crovalimab demonstrated noninferiority compared with eculizumab for transfusion avoidance from baseline through week 25. The weighted difference in the proportion of patients with transfusion avoidance (crovalimab versus eculizumab) was −2.8% (95% CI, −15.7% to 11.1%; P = ███) with a lower limit of the 95% CI of −15.7%, which was higher than the predefined NIM of −20% (Table 16). In the crovalimab arm, 65.7% (95% CI, 56.9% to 73.5%) of patients were transfusion-free from baseline through week 25 compared with 68.1% (95% CI, 55.7% to 78.5%) of patients in the eculizumab arm. One patient in the crovalimab arm discontinued treatment before week 25 without a transfusion and was conservatively assumed to have had a transfusion. Overall, the results of the sensitivity analyses using different analysis population definitions were consistent with the results from the primary analysis.15,23
Of the 43 arm B switch patients, 33 patients (proportion of patients = 76.7%; 95% CI, 61.0% to 87.7%) achieved transfusion avoidance in the period after switching to crovalimab through switch week 25. Two arm B switch patients discontinued treatment before switch week 25 without a transfusion and were conservatively assumed to have had a transfusion.15,23
Point estimates in the crovalimab arm were generally consistent across subgroups for transfusion avoidance.15
COMMODORE 1 study: The proportion of patients who achieved transfusion avoidance from baseline through week 25 was 79.5% (95% CI, 63.1% to 90.1%) for the crovalimab arm versus 78.4% (95% CI, 61.3% to 89.6%) for the eculizumab arm (Table 16). One patient in the eculizumab arm discontinued treatment before week 25 without a transfusion and was conservatively assumed as having had a transfusion.
Of the 28 arm B switch patients who switched to crovalimab for at least 24 weeks, 23 (82.1%) patients achieved transfusion avoidance after switching to crovalimab through switch week 25.17
Red Blood Cell Transfusions
COMMODORE 2 study: Among the primary analysis population, the mean number of units of pRBC transfused from baseline through week 25 in the primary analysis population was similar between the crovalimab arm at 2.3 units (95% CI, 1.3 units to 3.4 units) and the eculizumab arm at 2.2 units (95% CI, 1.0 unit to 3.4 units) (Table 16). During the primary treatment period, 45 (33.6%) patients in the crovalimab arm and 22 (31.9%) patients in the eculizumab arm had at least 1 transfusion. Among patients who received at least 1 transfusion, the mean number of units of pRBC transfused was similar with 6.9 units (95% CI, 4.3 units to 9.6 units) in the crovalimab arm and 6.9 units (95% CI, 4.1 units to 9.8 units) in eculizumab arm.
In the 43 arm B switch patients who switched to crovalimab for at least 24 weeks, 8 (18.6%) patients had at least 1 transfusion from switch baseline up to switch week 25. The mean number of units of pRBC transfused among these 43 patients was 1.9 units (95% CI, 0.4 unit to 3.4 units) from switch baseline through switch week 25.15,23 Among the arm B switch patients with at least 1 transfusion (n = 8), the mean number transfused was 10.3 units (95% CI, 4.9 units to 15.6 units).15
COMMODORE 1 study: Among the efficacy-evaluable population, 8 (20.5%) patients in the crovalimab arm and 7 (18.9%) patients in the eculizumab arm had at least 1 transfusion, and the mean number of pRBC units transfused from baseline to week 25 in all randomized patients was 0.97 unit (95% CI, 0.24 unit to 1.70 units) in the crovalimab arm and 1.89 units (95% CI, 0.53 unit to 3.25 units) in the eculizumab arm (Table 16). The mean units transfused were not reported among patients who had at least 1 transfusion.
The mean number of units of pRBC transfused among the 28 arm B switch patients who switched to crovalimab for at least 24 weeks was 1.0 (SD = 2.96) unit(s).17
Patient-Reported Outcomes
Functional Assessment of Chronic Illness Therapy–Fatigue
In both the COMMODORE 2 and COMMODORE 1 studies, FACIT-F was assessed in adult patients only (≥ 18 years).
Table 16: Summary of Key Efficacy Results From the COMMODORE 2 Study (Primary Analysis Population) and COMMODORE 1 Study (24-Week Efficacy Population)
Outcome | COMMODORE 2 study | COMMODORE 1 study | ||
|---|---|---|---|---|
Arm A: Crovalimab (N = 134) | Arm B: Eculizumab (N = 69) | Arm A: Crovalimab (N = 39) | Arm B: Eculizumab (N = 37) | |
Hemolysis outcomes (LDH) | ||||
Proportion of patients who achieved hemolysis control (central LDH level ≤ 1.5 × ULN), %, mean (95% CI)a | From week 5 through week 25 | From baseline through week 25 | ||
79.27 (72.86 to 84.48) | 78.96 (69.66 to 85.99) | 92.93 (86.62 to 96.39) | 93.74 (87.26 to 97.04) | |
Odds ratio (95% CI) | 1.02 (0.57 to 1.82)b NIM for lower 95% CI limit = 0.2 | 0.88 (0.28 to 2.77) | ||
Difference in proportions, % (95% CI) | NR (NR) | NR (NR) | ||
P valuec | ██████d | NR | ||
Proportion of patients with BTH from baseline through week 25, n (%) | 14 (10.4)e | 10 (14.5)e | 4 (10.3) | 5 (13.5)e |
95% CI for proportion, % | 6.04 to 17.21 | 7.54 to 25.50 | 3.34 to 25.16 | 5.08 to 29.57 |
Weighted difference in proportions, % (95% CI) | –3.9 (–14.82 to 5.26) NIM for upper 95% CI limit = 20% | –3.5 (–19.20 to 11.68) | ||
P valuec | 0.4358d | NR | ||
Mean percentage change in LDH levels, % (95% CI)a | From baseline to week 25 | From baseline to average of week 21, week 23, and week 25 | ||
–73.58 (–78.95 to –68.22) | –64.06 (–71.36 to –56.75) | 16.60 (█████ █) | 4.53 (███████) | |
Difference in proportions, % (95% CI) | –9.53 (–18.42 to –0.63) | 12.07 (–7.44 to 31.58) | ||
P value | NR | NR | ||
Hb outcomes | ||||
Proportion of patients with stabilized Hb from baseline through week 25, n (%) | 85 (63.4)f | 42 (60.9) | 23 (59.0) | 26 (70.3) |
95% CI for proportion, % | 54.63 to 71.45 | 48.35 to 72.17 | 42.19 to 74.02 | 52.83 to 83.56 |
Weighted difference in proportions, % (95% CI) | 2.2 (–11.37 to 16.31) NIM for lower 95% CI limit = –20% | –10.8 (–30.84 to 10.39) | ||
P valuec | 0.7496d | NR | ||
Proportion of patients who reached and maintained a minimum Hb level from baseline through week 25, n (%) | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
95% CI for proportion, % | ██████ | ███████ █ | ███████ | ███████ |
Weighted difference in proportions, % (95% CI) | ███ ████████ ███ | █████ ████████ ███ | ||
P value | ██ ██████ | ██ ██████ | ||
Transfusion outcomes | ||||
Transfusion avoidance from baseline through week 25, n (%) | 88 (65.7)g | 47 (68.1) | 31 (79.5) | 29 (78.4)g |
95% CI for proportion | 56.91 to 73.52 | 55.67 to 78.53 | 63.06 to 90.13 | 61.34 to 89.58 |
Weighted difference in proportions, % (95% CI) | –2.8 (–15.67 to 11.14) NIM for lower 95% CI limit = –20% | 1.8 (–16.67 to 19.94) | ||
P valuec | 0.6655d | NR | ||
Patients with at least 1 transfusion, n (%) | 45 (33.6) | 22 (31.9) | 8 (20.5) | 7 (18.9) |
Total number of pRBC units transfused among the full population, mean (SD) | 2.33 (6.02) | 2.20 (4.83) | 0.97 (2.25) | 1.89 (4.09) |
95% CI for mean | 1.30 to 3.36 | 1.04 to 3.36 | 0.24 to 1.70 | 0.53 to 3.25 |
Median (range) | 0 (0 to 55.0) | 0 (0 to 25.0) | 0 (0 to 10.0) | 0 (0 to 13.0) |
Difference in means (95% CI) | NR (NR) | NR (NR) | ||
P value | NR | NR | ||
FACIT-F scores | ||||
Baseline, N | 134 | 67 | 39 | 37 |
FACIT-F score at baseline, mean (SE) | 36.02 (0.877) | 35.09 (1.408) | 39.10 (1.623) | 40.05 (1.444) |
Week 25, N | 128 | 66 | 38 | 32 |
Adjusted mean change in FACIT-F score from baseline to week 25, mean (SE)h,i | 7.79 (0.661) | 5.15 (0.880) | 1.09 (1.285) | –2.61 (1.373) |
95% CI for adjusted mean change | 6.49 to 9.09 | 3.42 to 6.89 | –1.47 to 3.65 | –5.35 to 0.12 |
Difference in adjusted means change from baseline (95% CI) | 2.64 (0.68 to 4.60) | 3.71 (0.05 to 7.36) | ||
P value | 0.0087d | NR | ||
EORTC QLQ-C30 GHS and QoL scores | ||||
Baseline, N | 134 | 67 | 44 | 42 |
EORTC QLQ-C30 GHS and QoL score at baseline, mean (SD) | 62.69 (19.60) | 62.44 (19.86) | 63.45 (23.59) | 71.43 (20.42) |
Week 25, N | 128 | 66 | 38 | 32 |
EORTC QLQ-C30 GHS and QoL score at week 25, mean (SD) | 76.11 (15.91) | 72.22 (18.10) | 69.30 (23.82) | 67.71 (18.90) |
Absolute change in EORTC QLQ-C30 GHS and QoL score from baseline to week 25, mean (SD) | 13.41 (18.89) | 9.85 (20.46) | 5.70 (24.52) | –1.04 (16.36) |
Difference in mean change from baseline (95% CI) | NR (NR) | NR (NR) | ||
P value | NR | NR | ||
BTH = breakthrough hemolysis; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GEE = generalized estimating equation; GHS = global health status; Hb = hemoglobin; LDH = lactate dehydrogenase; MMRM = mixed model of repeated measures; NIM = noninferiority margin; NR = not reported; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; QoL = quality of life; RCT = randomized controlled trial; SD = standard deviation; SE = standard error; ULN = upper limit of normal.
Note: Data presented in Table 16 were based on analyses at the clinical cut-off date of November 16, 2022. In the COMMODORE 2 study, the outcomes of patients who reached and maintained a minimum Hb level, a mean percentage change in LDH levels, mean units of pRBC transfused, and an EORTC QLQ-C30 GHS and QoL score were not adjusted for multiplicity. In the COMMODORE 1 study, no adjustment for multiplicity was conducted for all outcomes. Definitions of the key efficacy outcomes are as follows. BTH was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [Hb < 10 g/dL], a major adverse vascular event [including thrombosis], dysphagia, or erectile dysfunction) in the presence of elevated LDH of 2 or more multiplied by ULN after the prior reduction of LDH to 1.5 or less multiplied by ULN on treatment. Stabilized Hb was defined as avoidance of a 2 g/dL or greater decrease in Hb level from baseline, in the absence of transfusion. Patients who reached and maintained a minimum Hb level were defined as patients who reached an Hb level of at least 10 g/dL, without a subsequent decrease below 9 g/dL, in the absence of transfusion. Transfusion avoidance was defined as patients who were pRBC transfusion–free and did not require a transfusion per protocol-specified guidelines. The FACIT-F tool consists of 13 items that assess fatigue using a 7-day recall period. Items are scored on a response scale that ranges from 0 (“not at all”) to 4 (“very much so”); relevant items are reverse-scored and all items are summed to create a total score, with a higher score indicative of better functioning (i.e., less fatigue). The total score ranges from 0 to 52. MMRMs were used in analysis. The EORTC QLQ-C30 GHS and QoL scale was assessed in adult patients only; the scale ranges from 0 to 100, with higher scores indicating better GHS and QoL.
aEstimates were based on a GEE model.
bAn odds ratio of greater than 1 favours crovalimab.
cThe P value was based on the Mantel-Haenszel test.
dThe predefined statistical testing hierarchy was broken before superiority testing could be conducted. Therefore, the P value reported is descriptive only.
eFour patients in the crovalimab arm and 1 patient in the eculizumab arm of the COMMODORE 2 study, as well as 2 patients in the eculizumab arm of the COMMODORE 1 study, discontinued their respective studies before week 25 and were considered to have experienced a BTH event as a conservative analysis approach.
fOne patient in the crovalimab arm of the COMMODORE 2 study discontinued the study before week 25 with Hb stabilization and was conservatively assumed to have not had Hb stabilization.
gOne patient in the crovalimab arm of the COMMODORE 2 study and 1 patient in the eculizumab arm of the COMMODORE 1 study discontinued their respective studies before week 25 without a transfusion and were conservatively assumed to have had a transfusion.
hFACIT-F was assessed in adult patients only (crovalimab arm = 134 adult patients; eculizumab arm = 67 adult patients). The total FACIT-F score ranges from 0 to 52 with higher scores indicating lower fatigue severity. The threshold for a clinically meaningful change is 5 points or more.
iNoninferiority testing of FACIT-F was planned to occur only after successful superiority testing of all the other coprimary and secondary efficacy end points. Given the outcome of this superiority testing, the comparative results of FACIT-F are descriptive only. The summary of MMRM FACIT-F scores are presented in Table 16.
Sources: COMMODORE 2 Primary Clinical Study Report,15 COMMODORE 1 Primary Clinical Study Report,17 and sponsor’s submission.16
COMMODORE 2 study: Due to the break in the statistical testing hierarchy, noninferiority testing for FACIT-F was not performed and the results were considered descriptive only. FACIT-F data were evaluable in 95.5% of adult patients in the crovalimab arm and 95.7% of adult patients in the eculizumab arm at each visit from baseline through week 25. The mean FACIT-F scores at baseline were below normative values and similar in the crovalimab arm at 36.0 points (95% CI, 34.29 points to 37.76 points) and in the eculizumab arm at 35.1 points (95% CI, 32.28 points to 37.90 points) (Table 16 as well as Appendix 1, Figure 15). Improvement in fatigue was observed by week 2, with the mean score increasing to 40.9 points (95% CI, 39.57 points to 42.28 points) in the crovalimab arm compared with 38.9 points (95% CI, 36.35 points to 41.47 points) in the eculizumab arm. Further improvement in levels of fatigue were reported up to week 25, with the mean score increasing to 44.3 points (95% CI, 43.15 points to 45.52 points) in the crovalimab arm compared with 41.4 points (95% CI, 39.29 points to 43.47 points) in the eculizumab arm. The adjusted mean change from baseline at week 25 in the FACIT-F score was 7.8 points (95% CI, 6.49 points to 9.09 points) in the crovalimab arm compared with 5.2 points (95% CI, 3.42 points to 6.89 points) in the eculizumab arm.
For the proportion of patients with a 5-point or greater improvement from baseline in the FACIT-F score, improvement of fatigue was observed by week 2, with 41.7% of patients (95% CI, 33.25% to 50.57%) in the crovalimab arm and 40.3% of patients (95% CI, 28.72% to 53.00%) in the eculizumab arm exceeding the threshold for clinically meaningful change (≥ 5 points) (Appendix 1, Table 34). By week 25, 58.6% of patients in the crovalimab arm and 54.5% of patients in the eculizumab arm had an improvement in fatigue severity of at least 5 points.15
In 36 of the 43 arm B switch patients who switched to crovalimab for at least 24 weeks and with available data for this outcome, the adjusted mean change from switch baseline to switch week 25 in the FACIT-F score was 0.18 points (95% CI, −2.72 points to 3.09 points).15
COMMODORE 1 study: FACIT-F data were evaluable in 86.4% of adult patients in the crovalimab arm (n = 38) and 76.2% of adult patients in the eculizumab arm (n = 32) at each visit from baseline through week 25. The adjusted mean change in FACIT-F scores from baseline to week 25 was 1.1 points (95% CI, −1.47 points to 3.65 points) for the crovalimab arm and −2.6 points (95% CI, −5.35 points to 0.12 point) for the eculizumab arm (Table 16 as well as Appendix 1, Figure 16).
The proportion of patients with a 5-point or greater improvement from baseline in the FACIT-F score was not reported for the COMMODORE 1 study.17,23
In 26 of the 35 arm B switch patients who had completed the assessment at both visits, the adjusted mean change from switch baseline to switch week 25 in the FACIT-F score was 2.50 (95% CI, 0.03 to 4.98).17
EORTC QLQ-C30 and EORTC IL-40 Populations
EORTC QLQ-C30 and EORTC IL-40 results were evaluable in:
the COMMODORE 2 study — 95.5% of adult patients in arm A (crovalimab) (n = 128) and 95.7% of adult patients in arm B (eculizumab) (n = 66) at each visit from baseline through week 25
the COMMODORE 1 study — 86.4% of adult patients in arm A (crovalimab) (n = 38) and 76.2% of adult patients in arm B (eculizumab) (n = 32) at each visit from baseline through week 25.
EORTC QLQ-C30 GHS and QoL
COMMODORE 2 study: The mean score at baseline was comparable between the crovalimab arm at 62.7 points (95% CI, 59.34 points to 66.03 points) and the eculizumab arm at 62.4 points (95% CI, 57.59 points to 67.28 points), indicating moderate levels of GHS and QoL. The mean change from baseline to week 25 was 13.4 points (95% CI, 10.11 points to 16.72 points) for the crovalimab arm and 9.9 points (95% CI, 4.82 points to 14.88 points) for the eculizumab arm. Mean values at week 25 were similar to normative population values.15,65,66
From switch baseline to switch week 25, the GHS and QoL scores remained relatively stable in arm B switch patients of the crovalimab efficacy population.15
COMMODORE 1 study: The mean change in EORTC GHS and QoL score from baseline to week 25 was 5.7 (95% CI, −2.36 to 13.76) in the crovalimab arm and −1.0 (95% CI, −6.94 to 4.86) in the eculizumab arm.17
Up to switch week 25, the GHS and QoL scores remained relatively stable in the 26 arm B switch patients of the crovalimab efficacy population.17
EORTC Item Library 40
COMMODORE 2 study: Overall, there was an improvement in dyspnea, dysphagia, chest pain, abdominal pain, and erectile dysfunction scores after starting treatment with either crovalimab or eculizumab, up to week 25 (Appendix 1, Table 35). In both treatment arms, the EORTC IL-40 headache score was numerically larger at week 2 compared to baseline, with a change from baseline of 4.3 (SD = 29.2; 95% CI, −0.74 to 9.33) in the crovalimab arm and 9.5 (SD = 25.8; 95% CI, 3.15 to 15.75) in the eculizumab arm. However, the scores decreased from week 5 onward, and the change from baseline at week 25 in the crovalimab arm was −6.8 (SD = 25.2; 95% CI, −11.18 to −2.36) and in the eculizumab arm was −4.6 (SD = 18.4; 95% CI, −9.06 to −0.03).15
From switch baseline to switch week 25, all symptoms assessed using the EORTC IL-40 tool (dyspnea, dysphagia, chest pain, headaches, abdominal pain, and erectile dysfunction) remained relatively stable in the arm B switch patients.15
COMMODORE 1 study: Overall, the point estimates suggested there was little to no difference in dyspnea, dysphagia, chest pain, headaches and abdominal pain scores from baseline to week 25 in both the crovalimab and eculizumab arms (Appendix 1, Table 36). Erectile dysfunction scores were numerically larger (worsening) from baseline to week 25 in the crovalimab arm (change in mean [SD] = 6.7 [38.2]) and the eculizumab arm (change in mean [SD] = 7.1 [32.5]).17
In arm C, up to week 25, the outcome data suggested little to no difference in point estimates of dysphagia, chest pain and abdominal pain assessed by EORTC IL-40 in the patients from the prior ravulizumab, prior high-dose eculizumab, and C5 polymorphism arm C cohorts from baseline to week 25. The study results showed a small numeric increase (worsening) in dyspnea scores and a small numeric decrease (improvement) in headache scores compared with baseline in the cohort of patients who switched from ravulizumab. The sample size of male patients (n = 8 in the prior ravulizumab cohort, n = 3 in the prior high-dose eculizumab cohort, and n = 2 in the C5 polymorphism cohort) was too low to draw firm conclusions from the summary data for erectile dysfunction.17,23
From switch baseline to switch week 25, all symptoms assessed using the EORTC IL-40 tool (dyspnea, dysphagia, chest pain, headaches, abdominal pain, and erectile dysfunction) remained relatively stable in arm B switch patients.17
Proportion of Patients With a Preference for Crovalimab per PPQ
COMMODORE 2 study: At switch week 17, 84% of the arm B switch patients (32 of 38 patients) in the crovalimab efficacy population who switched from eculizumab treatment to crovalimab treatment preferred treatment with crovalimab. The remaining patients preferred eculizumab (10.5%) or had no preference (5.3%) (Table 17). The top 3 reasons for crovalimab preference were “(t)he way treatment was given was easier,” “(f)ewer hospital visits associated with treatment,” and “(t)ime to administer treatment was shorter.”15
COMMODORE 1 study: At week 17 of treatment, 85% of patients in the crovalimab arm (33 of 39 patients) preferred treatment with crovalimab, 2.6% of patients preferred eculizumab, and 12.8% of patients had no preference (Table 17). The top 3 reasons given for patient preference were “(t)he way treatment was given was easier,” “(t)ime to administer treatment was shorter,” and “(f)ewer hospital visits associated with treatment.”17
At switch week 17, most of the arm B switch patients (27 of 28 [96.4%] patients) in the crovalimab efficacy population who switched from eculizumab treatment to crovalimab treatment preferred treatment with crovalimab (Table 17). The remaining patient had no preference between eculizumab and crovalimab treatments. The top 3 reasons for preference for crovalimab were “(f)ewer hospital visits associated with treatment,” “(b)etter quality of life,” and “(t)he way treatment was given was easier.”17
Table 17: Proportion of Patients With a Preference for Crovalimab or Eculizumab (Arm B, Period After Switch to Crovalimab in the COMMODORE 2 Study, and Arm A and Arm B, Primary Efficacy Period in the COMMODORE 1 Study)
Week 17 treatment preference, n (%) | COMMODORE 2 study | COMMODORE 1 study | |
|---|---|---|---|
Arm B: After switch from eculizumab to crovalimaba (N = 38)b | Arm A: Crovalimabc (N = 39)b | Arm B: After switch from eculizumab to crovalimaba (N = 28)b | |
Preferred crovalimab | 32 (84.2) | 33 (84.6) | 27 (96.4) |
Preferred eculizumab | 4 (10.5) | 1 (2.6) | 0 |
No preference | 2 (5.3) | 5 (12.8) | 1 (3.6) |
Note: Data presented in Table 17 were based on analyses at the clinical cut-off date of November 16, 2022.
aPatients who were initially randomized to eculizumab had the opportunity to switch to crovalimab in the extension period after completing 24 weeks of eculizumab treatment.
bOnly participants with available data (having completed the questionnaire) were included in the calculations of percentages.
cPatients were under treatment with eculizumab before enrolment and were randomized to arm A, crovalimab.
Sources: COMMODORE 2 Primary Clinical Study Report,15 COMMODORE 1 Primary Clinical Study Report,17 and sponsor’s submission.23
At week 17, 57% of the patients in arm C who were pretreated with eculizumab (the higher-than-approved dose eculizumab or C5 polymorphism patients) preferred treatment with crovalimab (8 of 14 patients). Smaller proportions of patients had no preference (4 of 14 [28.6%] patients) or preferred eculizumab (2 of 14 [14.3%] patients). At week 17, 60% of the patients in arm C pretreated with ravulizumab preferred treatment with crovalimab (9 of 15 patients). Smaller proportions had no preference (2 of 15 [3.3%] patients) or preferred ravulizumab (4 of 15 [26.7%] patients).17
Summary of Efficacy in Arm C of COMMODORE 2 and COMMODORE 1 Studies
COMMODORE 2 Study, Arm C (Nonrandomized, Descriptive, and Pediatric [Patients < 18 Years])
Hemolysis control: In the 6 pediatric patients, the normalized LDH value (central assessment) ranged between 3.8 and 23.4 multiplied by ULN at baseline. All 6 patients reached a central LDH value of 1.5 or less multiplied by ULN between week 2 and week 4 of treatment with crovalimab, and this was sustained across the first 24 weeks of treatment for all except 1 patient (Appendix 1, Figure 17). In this 1 patient, a central LDH value of 1.5 or less multiplied by ULN was reached by week 4; however, starting at week 7, the LDH value increased to 1.6 multiplied by ULN and thereafter ranged between 1.41 and 1.81 multiplied by ULN through to week 25.15
Transfusion avoidance: Four of the 6 pediatric patients achieved transfusion avoidance from baseline through to week 25. During the primary treatment period, 2 patients had at least 1 transfusion postbaseline. One patient with a documented history of aplastic anemia had a pRBC transfusion at a single time point on day 2. A second patient with a history of aplastic anemia and a history of transfusion in the 12 months before study start (with 40.5 pRBC units transfused in the prior 12 months) had multiple transfusions on study starting at day 24.15
BTH: No BTH events were observed in the 6 pediatric patients from baseline to week 25.15
Stabilized hemoglobin: Four of 6 pediatric patients achieved hemoglobin stabilization from baseline to week 25. The 2 patients who did not achieve hemoglobin stabilization were the same 2 patients who received a transfusion during the primary treatment period.15
Mean change in PedsQL MFS and physical functioning scale of the PedsQL Generic Core Scales: PedsQL Generic Core Scales and PedsQL MFS were evaluable in all 6 pediatric patients at each visit from baseline through week 25.
Overall fatigue scores from the PedsQL MFS tool were variable across time points for all 6 pediatric patients. Most patients experienced an overall improvement in fatigue over the course of treatment. One patient had a similar score at their last time point (week 49) compared to baseline.15
Physical functioning scores were variable across time points for all 6 pediatric patients. Most patients experienced an overall improvement in physical functioning. One patient had a similar score at their last time point (week 29) compared to baseline, and 1 patient experienced a decline in physical functioning.15
COMMODORE 1 Study, Arm C (Nonrandomized Cohorts)
Analysis of exploratory efficacy end points included only patients who had had the opportunity to complete 24 weeks of treatment (i.e., were enrolled more than 24 weeks before CCOD). This corresponded to 19 patients in the prior ravulizumab cohort, 9 patients in the prior high-dose eculizumab cohort, and 6 patients in the C5 polymorphism cohort.17
The summary of exploratory efficacy outcomes from the COMMODORE 1 study are presented in Table 18.
Table 18: Summary of Exploratory Efficacy Outcomes From the COMMODORE 1 Study, Arm C (Nonrandomized Arm)
Exploratory efficacy outcome | Arm C: Prior ravulizumab (N = 19) | Arm C: Prior high-dose eculizumab (N = 9) | Arm C: C5 SNP (N = 6) |
|---|---|---|---|
Mean proportion of patients with hemolysis control (central LDH level ≤ 1.5 × ULN) from baseline through week 25, % (95% CI) | 95.8 (89.11 to 98.43) | 91.0 (71.49 to 97.60) | 58.3 (29.68 to 82.22) |
Proportion of patients who achieved transfusion avoidance from baseline through week 25, % (95% CI)a | 57.9 (33.97 to 78.88) | 33.3 (9.04 to 69.08) | 50.0 (13.95 to 86.05) |
Proportion of patients with BTH from baseline through week 25, % (95% CI)b | 36.8 (17.23 to 61.37) | 33.3 (9.04 to 69.08) | 16.7 (0.88 to 63.52) |
Proportion of patients with stabilized hemoglobin from baseline through week 25, % (95% CI)c | 52.6 (29.50 to 74.79) | 22.2 (3.95 to 59.81) | 50.0 (13.95 to 86.05) |
Proportion of patients with at least 1 transfusion from baseline to week 25, n (%) | 6 (31.6) | 5 (55.6) | 3 (50.0) |
Total number of pRBC units transfused, mean (95% CI) | 1.5 (0.13 to 2.82) | 2.0 (–0.61 to 4.61) | 4.7 (–2.44 to 11.77) |
BTH = breakthrough hemolysis; C5 = complement component 5; CI = confidence interval; LDH = lactate dehydrogenase; pRBC = packed red blood cell; SNP = single nucleotide polymorphism; ULN = upper limit of normal.
aTwo patients in the prior ravulizumab cohort and 1 patient in the high-dose eculizumab cohort did not have a transfusion on study but as a conservative analysis approach, they were regarded as having had a transfusion given that they discontinued the study treatment before week 25.
bThree patients in the prior ravulizumab cohort and 1 patient in the prior high-dose eculizumab cohort without a BTH discontinued treatment before week 25 and were therefore conservatively considered to have had a BTH event.
cTwo patients in the prior ravulizumab cohort and 1 patient in the prior high-dose eculizumab cohort discontinued crovalimab treatment before week 25 and as a conservative analysis approach were therefore regarded as not having had stabilized hemoglobin.
Source: COMMODORE 1 Primary Clinical Study Report.17
FACIT-F: Scores in patients from arm C cohorts suggested that there was little to no difference postbaseline up to week 25.17
EORTC QLQ-C30: Physical functioning, role functioning, and GHS and QoL scores suggested that there was little to no difference in patients from all arm C cohorts postbaseline up to week 25.17
EORTC IL-40: From baseline to week 25, dysphagia, chest pain, and abdominal pain scores suggested that there was little to no difference in in the patients from all arm C cohorts. The study results showed a small numeric increase (worsening) in dyspnea scores and a small numeric decrease (improvement) in headache scores as compared to baseline in the cohort of patients who switched from ravulizumab. The sample size of male patients for the assessment of erectile dysfunction was very small (n = 8 in the prior ravulizumab cohort, n = 3 in the prior high-dose eculizumab cohort, and n = 2 in the C5 polymorphism cohort).17
PPQ: At week 17, most patients in arm C who had been pretreated with eculizumab (high-dose eculizumab or C5 polymorphism patients) preferred treatment with crovalimab (8 of 14 [57.1%] patients). Smaller proportions of patients had no preference (4 of 14 [28.6%] patients) or preferred eculizumab (2 of 14 [14.3%] patients). At week 17, a similar proportion of patients in arm C pretreated with ravulizumab preferred treatment with crovalimab (9 of 15 [60.0%] patients). Smaller proportions of patients had no preference (2 of 15 [13.3%] patients) or preferred ravulizumab (4 of 15 [26.7%] patients).17
PedsQL for pediatric patients: One pediatric patient was enrolled approximately 2 weeks before the CCOD and therefore data for this patient were limited.17
Harms
A summary of harms reported in the randomized safety populations during the 24-week primary safety period in the COMMODORE 2 and COMMODORE 1 trials is provided in Table 19.
Adverse Events
In the COMMODORE 2 study, the proportion of patients with at least 1 AE was comparable between the crovalimab arm (77.8%) and the eculizumab arm (79.7%). Infusion-related reaction was the most commonly reported AE by preferred term in the crovalimab arm (15.6%). The most frequent AEs in the eculizumab arm were also infusion-related reaction, together with urinary tract infection, and hypokalemia (all 13.0%). Injection-related reactions occurred in the crovalimab arm (5.2%) but not in the eculizumab arm due to SC administration being unique to the crovalimab arm. The proportion of patients who experienced infusion-related reactions was comparable between the crovalimab arm (15.6%) and the eculizumab arm (13.0%). The proportion of patients who experienced at least 1 infection was lower in the crovalimab arm (23.7%) compared with the eculizumab arm (36.2%). No cases of infection with N. meningitidis, including meningococcal meningitis, were reported in either arm. Eight (5.9%) patients in the crovalimab arm versus no patients in the eculizumab arm reported hypersensitivity other than type III immune complex reactions.
In the COMMODORE 1 study, the proportion of patients with at least 1 AE in the crovalimab arm (77.3%) was higher than in the eculizumab arm (66.7%). This difference was driven by type III immune complex events, injection-related reactions, and infusion-related reactions. The most frequent AE in the randomized safety population was COVID-19, with 13.6% of patients in the crovalimab arm and 16.7% of patients in the eculizumab arm experiencing it. Infusion-related reactions occurred only in the crovalimab arm (13.6%) but not in the eculizumab arm as these reactions were less likely to occur in the eculizumab arm due to patients starting the study stabilized on eculizumab treatment. A similar proportion of patients experienced infections in the crovalimab arm (40.9%) and the eculizumab arm (35.7%), and there were no cases of meningococcal meningitis in either arm. Four (9.1%) patients in the crovalimab arm versus no patients in the eculizumab arm reported hypersensitivity other than type III immune complex reactions.
Serious Adverse Events
In the COMMODORE 2 study, a similar proportion of patients reported at least 1 SAE in the crovalimab arm (10.4%) and the eculizumab arm (13.0%). In total, 3.0% of patients in the crovalimab arm and 1.4% of patients in the eculizumab arm experienced SAEs considered by the investigators to be related to study treatment.15
In the COMMODORE 1 study, a higher proportion of patients experienced SAEs in the crovalimab arm (13.6%) compared with the eculizumab arm (2.4%). In the crovalimab arm, the most frequent SAEs by System Organ Class were infections and infestations (6.8%). There was no obvious pattern in the SAEs observed and none of the SAEs were considered by the investigators to be related to crovalimab or eculizumab.17
Withdrawals Due to Adverse Events
In the COMMODORE 2 study, 1 patient each from the crovalimab arm (0.7%) and the eculizumab arm (1.4%) experienced AEs leading to the discontinuation of treatment.
In the COMMODORE 1 study, no patients experienced AEs leading to the withdrawal from the crovalimab or eculizumab arms.
Mortality
In the COMMODORE 2 study, death was reported in 2 (1.5%) patients in the crovalimab arm and 1 (1.4%) patient in the eculizumab arm. No deaths were related to treatment per the study investigators’ assessment.15
In the COMMODORE 1 study, no deaths were reported during the primary safety period.
Notable Harms
In the COMMODORE 2 study, no AESIs of type III immune complex reactions related to DTDCs were reported in either arm during the primary treatment period because the patients were treatment-naive. No AESIs of abnormal liver function tests or suspected transmission of an infectious drug by the study drug were reported in either arm. No clinically significant changes from baseline in vital signs and ECGs were observed during treatment with crovalimab or eculizumab.15
In the COMMODORE 1 study, 7 (15.9%) patients in the crovalimab arm versus no patients in the eculizumab arm experienced an AESI of type III immune complex reactions, because type III immune complex reactions were expected only in patients who switched from eculizumab to crovalimab who were at risk of developing DTDC-associated type III immune complex reactions. There were no AESIs reported for abnormal liver function tests or suspected transmission of an infectious drug by the study drug. Overall, in both treatment arms, no clinically significant changes from baseline for vital signs and ECGs were observed.17
Percentage of Patients Experiencing MAVE From Baseline to Week 25
COMMODORE 2 Study
One patient on crovalimab and 1 patient on eculizumab had a MAVE during the study; both patients had a prior history of MAVEs. One patient in the crovalimab arm had a MAVE of a myocardial infarction on study day 1 and subsequently died on study day 2 as a consequence of this AE. Retrospective analysis of laboratory data indicated that this patient had a myocardial infarction before crovalimab dosing. One patient in the eculizumab arm had a MAVE of a transient ischemic attack on study day 49. This AE occurred 6 days after the last dose of study drug administration (on day 43). The patient received treatment for this AE but subsequently died on day 71. In the investigators’ opinion, neither of the AEs in the 2 arms was related to treatment with the study drug.15
No MAVEs were reported in arm B switch patients from switch baseline through switch week 25.15
COMMODORE 1 Study
No patients in the crovalimab arm and 3 patients in the eculizumab arm were regarded as having a MAVE during the primary treatment period from baseline to week 25. In the eculizumab arm, 1 of the 3 patients experienced a MAVE (a transient ischemic attack). This patient experienced this MAVE on study day 20. The patient received treatment and the event resolved on study day 21. This patient did not have a prior history of a MAVE before entering the study. In the investigators’ opinion, this AE was not related to treatment with eculizumab. The other 2 patients in the eculizumab arm were conservatively considered as having a MAVE due to treatment discontinuation before 24 weeks without an actual MAVE reported.17
Table 19: Summary of Harms Results From the COMMODORE 2 and COMMODORE 1 Studies (24-Week Primary Safety Period, Randomized Safety Population)
AE | COMMODORE 2 study | COMMODORE 1 study | ||
|---|---|---|---|---|
Crovalimab (N = 135) | Eculizumab (N = 69) | Crovalimab (N = 44) | Eculizumab (N = 42) | |
Most common AEs, n (%) | ||||
≥ 1 AE | 105 (77.8) | 55 (79.7) | 34 (77.3) | 28 (66.7) |
Infections | 32 (23.7) | 25 (36.2) | 18 (40.9) | 15 (35.7) |
RD, % (95% CI) | −12.5 (−25.9 to 0.9) | 5.2 (−15.3 to 25.7) | ||
Upper respiratory tract infection | 11 (8.1) | 9 (13.0) | 3 (6.8) | 1 (2.4) |
COVID-19 | 11 (8.1) | 4 (5.8) | 6 (13.6) | 7 (16.7) |
Urinary tract infection | 2 (1.5) | 4 (5.8) | 2 (4.5) | 3 (7.1) |
Influenza | 4 (3.0) | 0 | 2 (4.5) | 3 (7.1) |
Neisseria meningitidis, including meningococcal meningitis | 0 | 0 | 0 | 0 |
Infusion-related reaction | 21 (15.6) | 9 (13.0) | 6 (13.6) | 0 |
Hypokalemia | 15 (11.1) | 9 (13.0) | 1 (2.3) | 0 |
Hypersensitivity other than type III hypersensitivity (type III immune complex reactions) | 8 (5.9) | 0 | 4 (9.1) | 0 |
Injection-related reactionsa | 7 (5.2) | NA | 3 (6.8) | NA |
Injection site reactions | 1 (0.7) | NA | 1 (2.3) | NA |
SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 14 (10.4) | 9 (13.0) | 6 (13.6) | 1 (2.4) |
Infections and infestations | 4 (3.0) | 5 (7.2) | 3 (6.8) | 1 (2.4) |
Aplastic anemia | 2 (1.5) | 1 (1.4) | NR | NR |
Thrombocytopenia | 1 (0.7) | 1 (1.4) | NR | NR |
Febrile neutropenia | 0 | 1 (1.4) | NR | NR |
Cardiac disorders | 1 (0.7) | 1 (1.4) | NR | NR |
Infusion-related reaction | 1 (0.7) | 0 | NR | NR |
AEs leading to treatment discontinuation, n (%) | ||||
Patients who stopped | 1 (0.7) | 1 (1.4) | 0 | 0 |
Thrombocytopenia | 1 (0.7) | 0 | 0 | 0 |
Ischemic stroke | 0 | 1 (1.4) | 0 | 0 |
Deaths, n (%) | ||||
Patients who died | 2 (1.5) | 1 (1.4) | 0 | 0 |
RD, % (95% CI) | NR (NR) | NR (NR) | ||
Respiratory tract hemorrhage | 1 | 0 | 0 | 0 |
Myocardial infarction | 1 | 0 | 0 | 0 |
Ischemic stroke | 0 | 1 | 0 | 0 |
AESIs, n (%) | ||||
Type III hypersensitivity reactions (type III immune complex reactions) | 0b | 0b | 7 (15.9) | 0 |
Abnormal liver function tests (Hy’s Law) | 0 | 0 | 0 | 0 |
Suspected transmission of an infectious drug by the study drug | 0 | 0 | 0 | 0 |
MAVE | ||||
n | 134c | 69 | 39 | 37 |
MAVE, n (%) | 1 (0.75) | 1 (1.45) | 0 | 3 (8.11) |
95% CI for proportion | NR | NR | (0 to 11.17) | (2.12 to 23.02) |
RD, % (95% CI) | NR (NR) | NR (NR) | ||
AE = adverse event; AESI = adverse event of special interest; CCOD = clinical cut-off date; CI = confidence interval; DTDC = drug-target-drug complex; MAVE = major adverse vascular event; NA = not applicable; NR = not reported; RD = relative difference; SAE = serious adverse event.
Note: Data presented in Table 19 were based on analyses at the CCOD of November 16, 2022. Only treatment-emergent AEs are displayed.
aInjection-related reactions were part of the “hypersensitivity other than type III immune complex reactions” group preferred term classification approach.
bDTDCs only occur in patients switching to or from other C5 inhibitors, which bind to different epitopes than crovalimab does, and therefore were not relevant or expected in this part of the COMMODORE 2 study. Accordingly, no AESIs of type III immune complex reactions related to DTDCs were reported during the primary safety period in the randomized safety population that was treatment-naive or in patients who continued in the crovalimab arm after the primary safety period up to the CCOD.
cMAVE was an exploratory end point being assessed in the primary analysis population in the COMMODORE 2 study. One of the 135 patients in arm A (crovalimab) was excluded from the primary analysis population for not fulfilling the criteria of at least 1 postbaseline LDH level assessment.
Sources: COMMODORE 2 Primary Clinical Study Report,15 COMMODORE 1 Primary Clinical Study Report,17 and sponsor’s submission.16
Summary of Harms in Arm C of COMMODORE 2 and COMMODORE 1 Studies
In the COMMODORE 2 study, overall, 83.3% (5 of 6) of pediatric patients experienced at least 1 AE (all were grade 1 to grade 2 in intensity). No deaths were reported. No pediatric patients experienced SAEs. One (16.7%) pediatric patient experienced a treatment-related AE of fatigue (grade 1), which resolved without treatment and dose modification. No pediatric patients had an AE leading to the withdrawal of treatment or dose modification. No pediatric patients experienced selected AEs (injection site reactions, infusion-related reactions, infections, and hypersensitivity reactions other than type III immune complex reactions) or AESIs. No clinically significant changes from baseline for vital signs and ECGs were observed. No MAVEs were reported in the 6 pediatric patients from baseline through week 25.15
In the COMMODORE 1 study, overall, 18 (85.7%) patients in the prior ravulizumab cohort, 10 (100%) patients in the prior high-dose eculizumab cohort, and 5 (83.3%) patients in the C5 polymorphism cohort experienced at least 1 AE. The single patient enrolled in the pediatric cohort (enrolled approximately 2 weeks before CCOD) did not experience any AEs. No deaths were reported in arm C. There was 33.3% of patients in the prior ravulizumab cohort and 20.0% of patients in the prior high-dose eculizumab cohort who experienced at least 1 SAE. No patients in the C5 polymorphism cohort experienced SAEs. One (4.8%) patient in the prior ravulizumab cohort had a grade 3 AE of sepsis leading to the withdrawal of treatment. Selected AEs of injection site reactions, infusion-related reactions, and hypersensitivity other than type III immune complex reactions were experienced in a small number of patients across cohorts. Infections were experienced in 42.9% (9) of patients, 50.0% (5) of patients, and 50.0% (3) of patients in the prior ravulizumab, prior high-dose eculizumab, and C5 polymorphism cohorts, respectively. There were no cases of meningococcal meningitis across all arm C cohorts. Overall, 23.8% of patients in the prior ravulizumab cohort and 20.0% of patients in the prior high-dose eculizumab cohort experienced at least 1 AESI of type III immune complex event. There were no AESIs reported for abnormal liver function tests or suspected transmission of an infectious drug by the study drug. Overall, no clinically significant changes from baseline for vital signs and ECGs were observed across all arm C cohorts. The number of patients with at least 1 MAVE was 3 of 19 patients (proportion of patients = 15.8%; 95% CI, 4.17% to 40.49%) in the prior ravulizumab cohort and 1 of 9 patients (proportion of patients = 11.1%; 95% CI, 0.58% to 49.33%) in the prior high-dose eculizumab cohort. These 4 patients discontinued crovalimab treatment before 24 weeks and, as a conservative analysis approach, were regarded as having had a MAVE. There were no patients with MAVE in the C5 polymorphism cohort.17
Summary of the Pooled Safety Data From COMMODORE 1, COMMODORE 2, and COMMODORE 3 Studies
██████ ██████ ████████ █████ ██ ████ ████ █████████ ██ █████████ ██ ███ █ ██████████ █████ ██████████ ██ ███ ████ █████████ ██ ███ ████████ ███ ███ █████ ██████████ ██████████ ██ █ ████ ███ ███ █████ ██████████ ██████████ ██ █ █████ ███ ██████████ ██████████ ███ ███████ ███████ ██ █████████ ███████ ██████████ █████████ █████████ ███████ ██████████ █████ ██ █ ███ ███ ██████████ ██████ ██ █ ████ ██ ███ ██████ ██████ █████████ ███ ██████ █████████████ ███████ ███████ ██████████ █████ ███ ██████████ ██████ █████ ██████████ ██████████████ █ ███████ ██████████ ██ ████████ ███████████ ██ █████ ███ ██ ██████ ███ ███████ █████ █ ██ ██ ██████ ███ ███████ ████ ██████ ███ ███████ █████████████████ ████ █████ ███ █████ ██ █████ █████ ███ ██ ███ ██████████ ██████ ████████████ █████████████ █ ██████ ██████████ ██ ████ ███████████ █████████████████ ███ ██████ ███ ██████ ██ ███ ██████████ █████ ████████████ ██ ███ ██████████ ██████ ███████████ ████████████ ███████████.
A pooled safety analysis from COMMODORE 1, COMMODORE 2, and a single-arm study (COMMODORE 3) was submitted by the sponsor. Across the 3 trials, a total of 11 pediatric patients (9 treatment-naive and 2 switch patients with PNH) weighing 40 kg or more were treated with crovalimab up to the CCOD. Of these, 9 (81.8%) pediatric patients reported at least 1 AE. No pediatric patients reported a fatal AE, an SAE, or an AE that led to drug interruption and/or modification or withdrawal.10,35
Critical Appraisal
Internal Validity
Methods for randomization and allocation concealment were appropriate in both the COMMODORE 2 and COMMODORE 1 trials (i.e., a central, block-based randomization procedure was used), where randomization was stratified based on prespecified relevant prognostic factors (i.e., the LDH level and number of pRBC units administered before randomization in the COMMODORE 2 trial and transfusion history in the COMMODORE 1 trial). The clinical experts consulted for this review noted that the selection of these stratification factors was appropriate given their prognostic value for the efficacy end points, including hemolysis control and transfusion avoidance. In both trials, most baseline characteristics were similar between the randomized crovalimab and eculizumab arms. However, there were numeric differences in a few of the characteristics. In the COMMODORE 2 study, they included race (64% Asian and 33% white in the crovalimab arm; 74% Asian and 23% white in the eculizumab arm), the median PNH clone size for erythrocytes (25% in the crovalimab arm and 45% in the eculizumab arm) and for granulocytes (60% in the crovalimab arm versus 75% in the eculizumab arm), as well as the history of MDS before enrolment (4% in the crovalimab arm versus 9% in the eculizumab arm); in the COMMODORE 1 study, numeric differences in certain characteristics included the median PNH clone size for monocytes (89% in the crovalimab arm versus 96% in the eculizumab arm). Nevertheless, the clinical experts did not consider that the between-group imbalances in these characteristics would impact the efficacy and safety results in the studies. For the COMMODORE 2 study, the inclusion criteria focused on patients with clinically significant disease activity. By excluding those previously treated with complement inhibitors, the COMMODORE 2 study minimized confounding from prior therapies, supporting the study’s aim of isolating crovalimab effects.
In the COMMODORE 2 trial, the predefined NIMs for the coprimary end points in the comparison of crovalimab to eculizumab (i.e., the proportion of patients with transfusion avoidance [the NIM for 95% CI lower limit was −20%] and hemolysis control [the NIM for 95% CI lower limit of the OR was 0.2]) were determined based on data reported in protocol of the 301 study,60 comparing eculizumab-treated patients with untreated patients from the Global PNH Patient Registry database. Based on the CDA-AMC review team’s assessment, considering the rarity of PNH and that no prior studies compared crovalimab to eculizumab, the determination of these NIMs for the efficacy end points of transfusion avoidance and hemolysis control in the COMMODORE 2 trial were considered appropriate. For the change in FACIT-F score, the NIM of −5 points was considered appropriate for a population with PNH based on the literature.14 Detailed methods were not reported regarding the selection of the NIMs for the proportion of patients with BTH (the NIM for 95% CI upper limit was 20%) and the proportion of patients with stabilized hemoglobin (the NIM for 95% CI lower limit was −20%). Concerns about the level set for the NIMs were alleviated by the fact that for all 3 outcomes, a more conservative margin would have been met. In addition, all of the NIMs in the COMMODORE 2 trial were predefined, which is appropriate.
No major protocol deviations were noted in either group in the COMMODORE 2 study. The sensitivity analyses and per-protocol analyses (these data are not included in this report) were completed and supported the conclusion of noninferiority for the coprimary end points (transfusion avoidance and hemolysis control).
The COMMODORE 1 study originally planned to recruit 200 patients to the crovalimab and eculizumab arms with a primary efficacy objective of a noninferiority assessment of mean percentage change in LDH levels from baseline to week 25, and with secondary objectives of a noninferiority assessment for end points including transfusion avoidance, BTH, hemoglobin stabilization, and fatigue. Also, it was planned that the superiority of crovalimab versus eculizumab would be evaluated provided that noninferiority had first been demonstrated for all primary and secondary end points in the COMMODORE 1 trial protocol version 1.10 However, the randomization was stopped early (in November 2022 per protocol amendment version 6) due to the introduction of ravulizumab to the treatment landscape and a reduced pool of patients treated with eculizumab over time.10 The review team and the clinical experts consulted for this review assessed that the early termination of randomization in the COMMODORE 1 trial likely did not bias the study results because the reason for termination was unrelated to the observed intervention effect (e.g., when early results show a noninferiority or statistically significant superiority effect),67 and the baseline characteristics were generally comparable between the randomization arms. Nonetheless, with this change (randomization was terminated in November 2022), the initially targeted sample size for the randomized arms of approximately 200 patients could not be reached, providing insufficient statistical power for efficacy analyses. The results of the exploratory efficacy analyses were reported descriptively, with no formal statistical noninferiority or superiority testing, limiting the interpretation and certainty of efficacy results of the COMMODORE 1 study.
Although a between-group difference was noted in the proportions of patients who discontinued from the study before 24 weeks (4.4% in the crovalimab arm versus 1.4% in the eculizumab arm in the COMMODORE 2 study, and 0% in the crovalimab arm versus 4.5% in the eculizumab arm in the COMMODORE 1 trial), the CDA-AMC review team and the clinical experts consulted for this review did not consider that the difference would impact the study results because the numbers of events were small, and the top reasons for discontinuation were physician decision or withdrawal by study participants in the crovalimab arm (Table 12). The impact of missing data was minimal in the COMMODORE 2 study because most patients completed the randomized, 24-week, primary treatment period (95.6% in the crovalimab arm and 98.6% in the eculizumab arm). In the COMMODORE 1 study, the data of 86.7% of patients in the crovalimab arm and 79.5% of patients in the eculizumab arm who completed the randomized treatment period were available for analysis. Of note, in the COMMODORE 1 study, 5 patients in each treatment arm were ongoing in the study at the CCOD; thus, the proportions of patients with missing data due to discontinuation during the randomized period were 4.4% in the crovalimab arm and 6.8% in the eculizumab arm. The impact of missing data in the COMMODORE 1 study was considered as likely being small.
There were imbalances for some previous or concomitant treatments (Table 15). In the COMMODORE 2 study, between-group differences were observed in the proportions of patients who had corticosteroids for dermatological preparations (39% in the crovalimab arm and 67% in the eculizumab arm) and systemic use (39% in the crovalimab arm versus 65% in the eculizumab arm), which was considered unexpected by the clinical experts; in the COMMODORE 1 study, the differences were noted for antibacterials for systematic use (71% in the crovalimab arm versus 50% in the eculizumab arm), otological treatments (56% in the crovalimab arm versus 27% in the eculizumab arm), and ophthalmological and otological preparations (49% in the crovalimab arm versus 14% in the eculizumab arm). Nonetheless, the clinical experts noted that the between-group imbalance for these previous or concomitant treatments in the 2 trials likely did not impact the efficacy and safety results.
The clinical experts consulted for this review noted that the maintenance dose of eculizumab (900 mg maintenance every 2 weeks for IV infusion) in both pivotal studies aligned with its product monograph11 and clinical guidelines. The clinical experts further noted that approximately one-third of patients require higher doses of eculizumab than recommended by Health Canada to properly control symptoms and signs associated with PK breakthrough. Dose modifications for eculizumab were not permitted in the COMMODORE 1 and COMMODORE 2 trials (similar to the noninferiority 301 study60 comparing ravulizumab versus eculizumab). For the COMMODORE 2 trial, the lack of permitted dosage adjustments may have biased the efficacy results in favour of crovalimab relative to how eculizumab is dosed in clinical practice; however, the magnitude of this potential bias is unclear. The clinical experts noted that in clinical practice, patients are observed on eculizumab treatment for at least 3 months after initiation before considering a dose increase for patients in whom the 900 mg dose is considered to be inadequate. The clinical experts noted that the randomized assessment period (24 weeks) in the COMMODORE 2 trial was likely too short to explore the impact of increasing the eculizumab dose. For the COMMODORE 2 trial, there was no randomized comparative evidence of crovalimab versus eculizumab at a higher dose than that recommended by Health Canada in patients who are C5 inhibitor–naive. For the COMMODORE 1 trial, the inclusion criteria specified treatment with eculizumab according to the approved dosing recommended for PNH (900 mg maintenance every 2 weeks) and completion of a minimum of 24 weeks of treatment before day 1. It is likely that patients requiring a higher or more frequent dose of eculizumab beyond the product monograph–recommended dosage would have been excluded from the COMMODORE 1 trial. For the COMMODORE 1 study, there is no randomized evidence to inform the efficacy or safety of crovalimab in patients who switched from eculizumab given at higher doses than recommended by Health Canada.
The clinical experts consulted for this review noted that LDH is a sensitive marker for intravascular hemolysis that was assessed at multiple time points, and hemolysis control defined as an LDH value of 1.5 or less multiplied by ULN from week 5 (in the COMMODORE 2 trial) and from baseline (in the COMMODORE 1 study) through the end of the primary treatment period was appropriate. FACIT-F and EORTC QLQ-C30 GHS and QoL are validated measurements, but the CDA-AMC review team was unable to find studies about validation of EORTC IL-40 symptoms or the PPQ developed by the sponsor. Fatigue and HRQoL results were evaluable in most patients in the COMMODORE 2 trial (96%) and in 76% to 86% of the patients in the COMMODORE 1 study. For these outcomes, the impact of missing outcome data in the COMMODORE 2 study is minimal; in the COMMODORE 1 study, the impact of missing outcome data (14% in the crovalimab arm and 24% in the eculizumab arm) is unclear.
Both the COMMODORE 2 and COMMODORE 1 trials used open-label study designs, which could have potentially increased the risk of bias during outcome ascertainment. While laboratory outcomes such as LDH and hemoglobin are likely at a low risk of bias due to central measurement, the open-label design of the COMMODORE 1 and COMMODORE 2 trials may have impacted the reporting of subjective PROs, including FACIT-F, EORTC QLQ-C30 GHS and QoL, EORTC IL-40 symptoms, and patient treatment preference. The magnitude and direction of potential bias for the FACIT-F, EORTC QLQ-C30 GHS and QoL, and EORTC IL-40 outcomes were uncertain. Data about patient preference were collected among the patients who first received eculizumab and then switched to crovalimab. Most patients (84% to 96%) preferred crovalimab, with the top reasons including that crovalimab was easier and less time-consuming to administer, and was associated with fewer hospital visits compared to eculizumab. Due to the open-label design, the patient treatment preference outcome was particularly prone to bias in favour of crovalimab. Furthermore, the sponsor developed a PPQ that included a list of 13 predefined reasons for the preference, which may have introduced bias if predefined reasons did not fully capture the range of opinions or preferences across patients included in the studies.
Overall, the statistical methods used in both trials were appropriate. The COMMODORE 2 study was powered on its coprimary outcomes (hemolysis control and transfusion avoidance); however, the subgroup analyses based on key baseline demographic and disease characteristics for these 2 coprimary outcomes were likely underpowered to identify or confirm subgroup differences. For hemolysis control in the COMMODORE 2 study, a total of 7.5% of missing central LDH values were reported from week 5 through week 25 (9.5% in the crovalimab arm versus 3.7% in the eculizumab arm). The results from the respective imputation-based sensitivity analyses (MCMC algorithm, predictive mean matching algorithm, and tipping point analysis) were consistent with those from the primary analysis, supporting the robustness of the primary analysis results. For the analysis of transfusion avoidance, there were no missing data and imputation methods were not applicable.
The interpretation of results of arm C in both the COMMODORE 2 and COMMODORE 1 trials was limited due to the lack of a comparator, the limited number of patients and events, and the descriptive summary of data.
Results in the extension periods beyond week 25 (from switch baseline up to switch week 25) in the COMMODORE 2 and COMMODORE 1 trials were limited by the open-label and descriptive nature of the extension study, the lack of comparative analyses, and missing data (of patients in the arm B groups of the COMMODORE 2 and COMMODORE 1 trials — approximately 63% of patients in each trial were in the arm B switch to crovalimab cohort for at least 24 weeks before the data cut-off for analysis sets).
External Validity
Patients in the 2 pivotal trials were recruited from multiple countries. No patients were enrolled from Canada in the COMMODORE 2 study. One patient (assigned in the eculizumab arm) was from 1 site in Canada in the COMMODORE 1 study. The majority of patients enrolled in the COMMODORE 2 study were Asian and in the COMMODORE 1 study were white. There were relatively low rates of those who did not advance past screening in the COMMODORE 2 study (12%) and the COMMODORE 1 study (13%), mainly due to a lack of willingness to comply. The clinical experts did not regard them as factors that would influence the generalizability of the studies. The clinical experts noted that overall, the eligibility criteria of patients in both trials aligned with the diagnosis standard and treatment indication for PNH in clinical settings, and the demographic and disease characteristics of the patients (including LDH levels and history of transfusion) were mostly aligned with the patients seen in clinical practice in Canada. Of note, in both trials, 1 patient inclusion criterion was having a granulocyte or monocyte clone size of 10% or more. Fewer than 10 patients had a baseline PNH clone size for granulocytes of less than 10%, and no patients had a baseline PNH clone size for monocytes of less than 10%.15 In the COMMODORE 1 trial, the number of patients with a PNH clone size of less than 10% for granulocytes or monocytes was not reported, but based on the median and range values, the CDA-AMC review team estimates that the numbers of such patients are likely to be small. There may be some uncertainties in the generalizability of results from the 2 pivotal studies in patients with a PNH clone size of less than 10% for these 2 biomarkers. The clinical experts consulted for this review noted that the patients excluded from the COMMODORE 2 and COMMODORE 1 trials due to not fulfilling the inclusion criteria of having a granulocyte or monocyte clone size of 10% or more are typically asymptomatic and would less likely be considered for treatment. If these patients were showing other disease manifestations indicating a therapy, crovalimab could also be used with anticipated similar efficacy and safety results as seen in the 2 trials as per the clinical experts.
The sample size of pediatric patients was small in the COMMODORE 2 trial (n = 6) and in the COMMODORE 1 trial (n = 1), and only descriptive results were available. The clinical experts anticipated that pediatric patients would have efficacy results similar to those in the main trial arms; however, there is a need for enhanced safety consideration regarding the risk of infections, including meningitis in pediatric patients.
The recommended body weight–based doses of crovalimab once every 4 weeks via SC injection were considered as adequate and reasonable in clinical practice in Canada by the clinical experts. The clinical experts pointed out the importance of monitoring patients for any occurrence of kidney dysfunction or infections (particularly meningitis) during the treatment of crovalimab.
The trials included outcomes that were important to patients, including hemolysis control assessed by LDH relative to the ULN, stabilized hemoglobin and maintenance, transfusion outcomes, fatigue measurement, and HRQoL. These outcomes, together with other outcomes reported in this review (i.e., EORTC IL-40 selected symptoms scales) and patient preference for crovalimab or eculizumab, were considered appropriate by the clinical experts and the clinician group. The randomized period of the 2 trials was 24 weeks. According to the clinical experts and the clinician group, given that compliance may impact treatment effects, longer-term comparative evidence on the durability of the effectiveness of crovalimab would be informative. Likewise, the occurrence of some AEs, especially rare ones (e.g., meningitis), may take longer than 24 weeks to be identified. Comparative longer-term follow-up to assess safety between crovalimab and other complement inhibitors would be preferred.
There was a lack of direct comparison between crovalimab and ravulizumab in C5-naive patients and in patients who switched from ravulizumab to crovalimab. Ravulizumab and eculizumab have the same mechanism of action and ravulizumab has demonstrated similar benefits in adult patients with PNH compared with eculizumab in 2 RCTs (the 301 study and the 302 study). The clinical experts anticipated similar efficacy between crovalimab and ravulizumab and highlighted the novel route of administration and schedule for crovalimab. Based on the results from the COMMODORE 1 and COMMODORE 2 trials, the clinical experts supported initiating crovalimab or switching a patient from a currently available C5 inhibitor (eculizumab or ravulizumab) to crovalimab in patients believed to benefit from crovalimab’s SC administration and schedule (for patients who plan for pregnancy or are pregnant, eculizumab would be used).
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:12,13
“High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word ‘likely’ for evidence of moderate certainty (e.g., ‘X intervention likely results in Y outcome’).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word ‘may’ for evidence of low certainty (e.g., ‘X intervention may result in Y outcome’).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as ‘very uncertain.’”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on threshold (MID) in the literature for the FACIT-F score in both trials. For the COMMODORE 2 study, the target of the certainty of evidence assessment was the presence or absence of a predefined NIM per study protocol for hemolysis control, BTH, stabilized hemoglobin, having reached and maintained a minimum hemoglobin level, and transfusion avoidance. Due to the lack of a formal MID estimate or NIM value, the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for mean percentage change in LDH levels, pRBC units transfused, EORTC QLQ-C30 GHS and QoL score, infections, and deaths. For the COMMODORE 1 study, due to the lack of a formal MID estimate or NIM (after amendment of the COMMODORE 1 study, all the efficacy end points became exploratory and descriptive; thus, NIM values were not applicable), the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for hemolysis control, BTH, mean percentage change in LDH levels, stabilized hemoglobin, having reached and maintained a minimum hemoglobin level, transfusion avoidance, pRBC units transfused, EORTC QLQ-C30 GHS and QoL score, infections, and deaths.
For the GRADE assessments, the COMMODORE 2 and COMMODORE 1 studies were assessed individually because the COMMODORE 2 study included patients naive to complement inhibition and the COMMODORE 1 study included complement inhibition–experienced patients.
Results of GRADE Assessments
Table 2 and Table 3 present the GRADE summary of findings for crovalimab versus eculizumab for patients with PNH who were naive to and had experienced complement inhibitors, respectively.
Long-Term Extension Studies
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The phase I and phase II COMPOSER trial (NCT03157635) consisted of 4 sequential parts and an OLE. This section includes a summary of the OLE period that evaluated the safety, tolerability, PKs, and PDs of crovalimab in patients with PNH aged 18 years to 75 years who were treatment-naive or who switched from eculizumab. Participants were enrolled at 14 sites in 6 countries: Germany (3 sites), Japan (5 sites), France (1 site), Hungary (2 sites), Korea (2 sites), and Italy (1 site). The study did not include sites in Canada.
Part 1 of the COMPOSER trial was designed to evaluate the safety, tolerability, PKs, and PDs of crovalimab in healthy volunteers and part 2, part 3, part 4, and the OLE period evaluated safety, tolerability, PKs, and PDs of crovalimab in patients with PNH. Part 1 evaluated the safety and tolerability of single doses of crovalimab in healthy volunteers and assessed PD and PK outcomes. Methods and results for part 1 are not further summarized in this CDA-AMC Clinical Review Report. Part 2, part 3, and part 4 comprised a dose-escalation study (planned to enrol 6 patients), a multidose, single-arm study (planned to enrol 18 patients), and a multidose, 2-arm study (planned to enrol 10 patients) in patients with PNH, respectively. Results for part 2, part 3, and part 4 are not further described in this CDA-AMC Clinical Review Report.
The OLE was initiated once the first patient in part 2 was eligible for the OLE. Patients transitioning from part 2, part 3, or part 4 initially remained on the same SC dose regimen as they were receiving in part 2, part 3, and part 4, respectively.
Populations
This is an OLE for patients who participated in part 2, part 3, and part 4 and who, in the opinion of the investigators, derived benefit from treatment with crovalimab. Part 2, part 3, and part 4 of the COMPOSER study enrolled patients with PNH of a population similar to that of the pivotal trials.
Interventions
The OLE phase was available for patients after completing 20 weeks of part 2, part 3, or part 4. Patients enrolling in the OLE period from part 2, part 3, and part 4 initially stayed on the previously assigned treatment schedule. Following analysis of the primary treatment period for part 2, part 3, and part 4, weight-based tiered dosing was introduced in the OLE phase to ensure that all patients received a comparable drug exposure across the body weight continuum. With the implementation of the tiered weight dosing, all patients in the OLE phase who were not currently receiving dosing every 4 weeks were required to switch to the dosing every 4 weeks regimen. Patients either received 680 mg SC every 4 weeks (body weight ≥ 40 kg to < 100 kg) or 1,020 mg SC every 4 weeks (body weight ≥ 100 kg).
Dose modification was required in patients whose body weight changed by 10% or more (compared with screening or the visit when the latest dose modification occurred, whichever was later) to exceed or become equal to 100 kg or to fall below 100 kg during the course of therapy.
Patients with 2 or more qualifying intravascular hemolysis events in 24 weeks were considered for an increased crovalimab maintenance dose with approval from the medical monitor. Qualifying intravascular hemolysis events were defined as symptoms of intravascular hemolysis that occurred within a week before the next maintenance dose without an identifiable trigger. If approved by the medical monitor, patients with a body weight of 40 kg to less than 100 kg would increase their maintenance dose from 680 mg every 4 weeks to 1,020 mg every 4 weeks; patients with a body weight of 100 kg or more would increase their maintenance dose from 1,020 mg every 4 weeks to 1,360 mg every 4 weeks.
If, based on investigators’ assessment, the patient or caregiver was able to self-administer or administer crovalimab-vial SC at home, after successful SC self-administration or the administration of crovalimab-vial training, the patient or caregiver received permission by the investigators to self-administer or administer a crovalimab vial at home.
Although 1 or more additional IV doses of crovalimab could be administered as a rescue dose (340 mg IV regardless of body weight infused over 30 minutes), none were administered during the COMPOSER study.
Outcomes
The efficacy results in the OLE period (i.e., from week 20 to the CCOD of November 1, 2021) are presented in text as a pooled total study population from part 2, part 3, and part 4. At the time of enrolment in the OLE period, all patients had been on stable crovalimab maintenance during the 20-week primary treatment period and were therefore considered comparable from a safety and efficacy standpoint, irrespective of naive or switch status at the time of enrolment in the study.
The exploratory long-term efficacy end points that were analyzed in the OLE period included the following:
hemolysis control (change in LDH) reported from week 20 to the CCOD using the mean normalized LDH level by visit and the proportion of patients by visit achieving an LDH value of 1.5 or less multiplied by ULN
the proportion of patients achieving transfusion avoidance by 24-week intervals from week 20 to the CCOD
the proportion of patients with stabilized hemoglobin levels by 24-week intervals from week 20 to the CCOD
the proportion of patients with BTH events by 24-week intervals from week 20 to the CCOD
the rate of BTH events from week 20 to the CCOD adjusted for patient-years at risk.
Safety data included AEs, reasons for withdrawal from the study, laboratory data, ECGs, concomitant medications, vital signs, and physical examination results. AESIs for this study included abnormal liver function tests, type III immune complex reactions, and suspected transmission of an infectious drug by the study drug.
Statistical Analysis
For part 2, part 3, and part 4 of the study, the efficacy analysis population was the same as the safety analysis population.
For the OLE period, the efficacy-evaluable population included all efficacy-evaluable patients who completed the primary treatment period in part 2, part 3, or part 4 of the study and were enrolled in the OLE period (18 treatment-naive patients and 25 switched patients).
The safety-evaluable population was defined as all healthy volunteers or patients who received at least 1 administration of the study drug (crovalimab or placebo) and included 18 treatment-naive patients and 26 switched patients.
At the time of the CCOD for the Update Clinical Study Report, patients had a maximum of approximately 4 years of follow-up in the OLE period. Hemolysis control (change in LDH levels) was analyzed by visit. To support pooled analysis of LDH by visit, time windows were defined to identify corresponding visits across the study parts, which have nonidentical visit schedules.
The proportion of patients with transfusion avoidance, hemoglobin stabilization, and BTH events was analyzed over sequential 24-week intervals, starting at week 20, to evaluate whether the efficacy of crovalimab, as measured by these end points, remained stable over time.
The analysis of a specific 24-week interval only included patients who had completed the entire 24-week interval at the time of the CCOD (November 1, 2021). Additionally, as the occurrence of BTH events is expected to be rare, the rate of BTH was also calculated over the entire OLE period and adjusted for patient-years at risk to take into account the individual patients’ exposure.
Due to different long-term follow-up times per patient at the CCOD, the data for some visits or intervals were based on small sample sizes (i.e., n < 10), which results in wide 95% CIs and significant uncertainty in the evidence. Therefore, for the purpose of the interpretation of the results obtained from the exploratory efficacy end points, only visits or intervals where the sample size was n equals 10 or more were considered in the following discussion.
Results
Patient Disposition
Overall, 56 patients with PNH were screened. In total, 44 patients with PNH were enrolled in part 2 (N = 10), part 3 (N = 19), and part 4 (N = 15). Of the 44 patients,18 individuals were treatment-naive and 26 patients switched from eculizumab. One patient in part 3 of the study elected not to proceed to the OLE phase. As of the CCOD (November 1, 2021), 38 patients were ongoing in the OLE period (17 treatment-naive patients and 21 patients who switched from eculizumab). Overall, a total of 5 patients were discontinued from the OLE phase (4 patients originally enrolled in part 3 and 1 patient originally enrolled in part 4, arm A) (Table 20).
The efficacy-evaluable population for the OLE period consisted of 42 patients (treatment-naive, n = 18; switched, n = 25); 1 patient in PART 3 completed the primary treatment period but elected not to enrol in the OLE phase (Table 21).
Table 20: Patient Disposition From the COMPOSER Study
Patient disposition | COMPOSER study | ||
|---|---|---|---|
Part 2 (N = 10) | Part 3 (N = 19) | Part 4 (N = 15) | |
Screened, all cohorts, N | 56 | ||
Reason for not advancing past screening during primary treatment period, N | |||
Halted recruitment | 6 | ||
Did not meet eligibility criteria | 3 | ||
Withdrew consent | 2 | ||
Experienced intensive hemolysis despite taking eculizumab | 1 | ||
Enrolled, N | 44 | ||
Safety, N | 44 | ||
Completed primary treatment period, N | 44 | ||
Enrolled in OLE (FAS), N | 43 | ||
Discontinued from OLE, N | |||
Withdrawal by patient | 3 | ||
Lack of efficacy | 2 | ||
FAS = full analysis set; OLE = open-label extension.
Note: One patient in part 3 completed the primary treatment period but did not enrol in the OLE period; this is not captured as a study discontinuation.
Sources: Sponsor’s submission.23
Table 21: Summary of Baseline Characteristics From the COMPOSER Study
Characteristic | Part 2 (N = 10) | Part 3 (N = 19) | Part 4 (N = 15) | |
|---|---|---|---|---|
Eculizumab-naive | Previous treatment with eculizumab (switched) | Eculizumab-naive (N = 8) | Previous treatment with eculizumab (switched) (N = 7) | |
Age | ||||
Mean age (SD), years | 53.9 (11.8) | 49.5 (11.0) | 56.6 (10.6) | 44.0 (11.0) |
Median age (range), years | 52.5 (35 to 74) | 46.0 (33 to 69) | 55.5 (42 to 73) | 44.0 (29 to 57) |
Sex | ||||
Male, n (%) | 6 (60.0) | 13 (68.0) | 6 (75.0) | 6 (85.7) |
Race, n (%) | ||||
Asian | 3 (30.0) | 7 (36.8) | 4 (50.0) | 2 (28.6) |
White | 7 (70.0) | 9 (47.4) | 3 (37.5) | 3 (42.9) |
Unknown | 0 | 3 (15.8) | 1 (12.5) | 2 (28.6) |
Ethnicity, n (%) | ||||
Not Hispanic or Latino | 10 (100) | 15 (80) | 7 (87.5) | 5 (71.4) |
Weight | ||||
Mean weight (SD), kg | 75.11 (15.76) | 80.11 (26.03) | 76.58 (15.98) | 81.56 (19.19) |
Median weight (range), kg | 66.9 (58.9 to 98.0) | 78.2 (40.6 to 131.5) | 80.1 (56.7 to 100.0) | 79.8 (60.4 to 114.0) |
RBC transfusion, n (%) | ||||
History of RBC transfusiona | 3 (30.0) | 8 (42.1) | 4 (50) | 2 (28.6) |
Median RBC units transfuseda | 2 (2 to 6) | 5 (1 to 21) | 7 (2 to 198) | 3 (1 to 5) |
Other characteristics | ||||
Median baseline PNH granulocyte clone size (range), % | 82.18 (14.8 to 94.4) | 92.40 (71.6 to 99.9) | 86.00 (37.0 to 97.0) | 92.32 (17.9 to 99.6) |
Median baseline PNH erythrocyte clone size (range), % | 49.00 (14.3 to 71.4) | 64.80 (12.4 to 99.1) | 17.00 (5.0 to 58.4) | 23.42 (8.7 to 87.4) |
History of aplastic anemia, n (%) | 1 (10.0) | 4 (21.1) | 3 (37.5) | 2 (28.6) |
Mean baseline normalized LDH level × ULN (SD) | 5.59 (3.06) | 1.56 (1.11) | 6.99 (5.86) | 1.09 (0.19) |
Mean baseline hemoglobin (SD), g/L | 95.2 (14.70) | 104.68 (17.66) | 96.38 (14.91) | 106.29 (20.81) |
LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; RBC = red blood cell; SD = standard deviation; ULN = upper limit of normal.
aTransfusion events that occurred within a year before the first dose.
Sources: Sponsor’s submission.23
Exposure to Study Treatments
Treatment-Naive Patients With PNH
From baseline to the November 1, 2021, CCOD, treatment-naive patients with PNH received crovalimab treatment for a median duration of 3.40 years (range, 0.9 year to 4.4 years). The median number of doses was 83.5 (range, 19 to 163).
Patients With PNH Switching From Eculizumab
From baseline to the November 1, 2021, CCOD, patients with PNH switching from eculizumab received crovalimab treatment for a median duration of 2.88 years (range, 0.4 year to 3.9 years). The median number of doses was 48.5 (range, 11 to 129) (Table 22).
Table 22: Summary of Treatment Exposure (Safety-Evaluable Population, Part 2, Part 3, and Part 4 of the COMPOSER Study, as of CCOD of November 1, 2021)
Measure of exposure | Treatment-naive patients (n = 18) | Switch patientsa (n = 26) | Total (n = 44) |
|---|---|---|---|
Treatment duration, years | |||
Mean (SD) | 3.17 (1.07) | 2.64 (1.10) | 2.86 (1.11) |
Median (range) | 3.40 (0.9 to 4.4) | 2.88 (0.4 to 3.9) | 3.03 (0.4 to 4.4) |
Total cumulative dose, mg | |||
Mean (SD) | 30,786.61 (9,043.33) | 26,223.35 (10,309.62) | 28,090.14 (9,963.87) |
Median (range) | 31,880.0 (11,949.0 to 41,485.0) | 28,030.0 (4,400.0 to 42,480.0) | 28,880.0 (4,400.0 to 42,480.0) |
Number of doses administered, total | |||
Mean (SD) | 88.3 (55.7) | 52.4 (28.1) | 67.1 (44.8) |
Median (range) | 83.5 (19.0 to 163.0) | 48.5 (11.0 to 129.0) | 51.0 (11.0 to 163.0) |
Number of doses administered, IV | |||
Mean (SD) | 2.22 (1.00) | 1.38 (1.77) | 1.73 (1.55) |
Median (range) | 3.0 (1.0 to 3.0) | 1.0 (1.0 to 10.0) | 1.0 (1.0 to 10.0) |
Number of doses administered, SC | |||
Mean (SD) | 86.11 (54.88) | 51.00 (28.54) | 65.36 (44.37) |
Median (range) | 80.50 (16.0 to 160.0) | 47.50 (10.0 to 128.0) | 50.00 (10.0 to 160.0) |
CCOD = clinical cut-off date; SC = subcutaneous; SD = standard deviation.
aPatients who switched from eculizumab.
Sources: Sponsor’s submission.23
Concomitant Medications and Cointerventions
In addition to the study treatment, participants in part 2, part 3, and part 4 were permitted to continue receiving immunosuppressant therapy (with the exception of azathioprine and IV immunoglobulin), corticosteroids, iron supplements, and folic acid (with the exception of erythrocyte-stimulating drugs such as erythropoietin).
During the OLE phase, in addition to the study treatment, patients were permitted to concomitantly continue to receive immunosuppressant therapy (with the exception of IV immunoglobulin and azathioprine), corticosteroids, iron supplements, and folic acid (with the exception of erythrocyte-stimulating drugs such as erythropoietin). For patients in the OLE phase, revaccination considered as necessary was based on standard of care or local guidance.
Efficacy
Hemolysis Control
The proportion of patients reaching an LDH level of 1.5 or less multiplied by ULN per visit suggested there was little to no difference across visits during the OLE period, with 80% to 100% of the evaluable patients at each visit having an LDH level of 1.5 or less multiplied by ULN.
The mean normalized LDH level was generally maintained below 1.5 multiplied by ULN during the OLE phase. The point estimate of the mean normalized LDH level by visit ranged from 1.09 to 1.24 multiplied by ULN.
Transfusion Avoidance
The proportion of patients with transfusion avoidance remained relatively stable over time during the OLE period, with 82.9% to 91.7% of all patients avoiding transfusion across the 24-week intervals. Seven of the 11 patients who received a blood transfusion during the OLE period reported a history of aplastic anemia and/or anemia.
Stabilized Hemoglobin
The proportion of patients reaching hemoglobin stabilization also remained relatively stable over time during the OLE phase, with 79.5% to 87.5% of all patients reaching hemoglobin stabilization across the 24-week intervals.
Breakthrough Hemolysis
The proportion of patients with BTH events was low during the OLE period, with 0% to 4.9% of all patients reporting a BTH event across the 24-week intervals. One patient who had a BTH event discontinued the study before completing the full interval from week 20 to week 44 in which the BTH event occurred. Consequently, this BTH event was reported and included in the computation of the rate of BTH events adjusted for patient-years at risk but is not included in the calculation of the proportion of patients with BTH events per 24-week interval analyses.
Harms
From baseline to the CCOD (November 1, 2021), a total of 42 (95.5%) patients experienced at least 1 AE. There were no deaths and no AEs that resulted in withdrawal from the study. A total of 14 (31.8%) patients experienced at least 1 SAE over a median treatment duration of 3.03 years.
AEs of any grade were reported in 17 (94.4%) treatment-naive patients and in 25 (96.2%) patients who switched from eculizumab. The most frequently reported AEs were nasopharyngitis (29.5%), upper respiratory tract infection (22.7%), and pyrexia (22.7%).
Fourteen (31.8%) patients experienced SAEs — 6 (33.3%) treatment-naive patients and 8 (30.8%) patients who switched from eculizumab. In the treatment-naive patients, all SAEs occurred in single patients. In the patients who switched from eculizumab, the SAE that occurred in 2 or more patients was BTH (7.7%). All other SAEs occurred in single patients. In the treatment-naive group, 1 (5.6%) patient experienced an SAE of BTH considered by the investigators to be related to study treatment. In the switch group, 1 (3.8%) patient experienced an SAE of upper respiratory tract infection assessed as related to study treatment by the investigators. No patients experienced SAEs that led to dose modification. One treatment-naive patient experienced an SAE of cardiac failure that led to dose interruption; it was deemed not related to study treatment as assessed by the investigators.
From baseline to the November 1, 2021, CCOD, 2 AESIs of abnormal liver function tests were reported in 2 (4.5%) patients with PNH (1 treatment-naive patient [increased alanine aminotransferase] and 1 patient who switched from eculizumab [a drug-induced liver injury]). In both cases, the events were of moderate severity and not considered related to study treatment as assessed by the investigators.
Of the 26 patients who switched from eculizumab, 2 (7.7%) patients experienced AESIs of type III immune complex reactions. Both of these AESIs were mild to moderate in severity and occurred during the primary treatment period. No additional type III immune complex reactions occurred during the OLE period. One patient in the treatment-naive group experienced an AESI of type III immune complex reactions at the time of switching from crovalimab to eculizumab, after discontinuing from the study due to a lack of efficacy. This AESI was mild in severity.
Other than type III immune complex reactions, no other hypersensitivity events were reported in patients with PNH during this study up to the November 1, 2021, CCOD. In addition, no AESIs of suspected transmission of an infectious drug by the study drug or cases of meningococcal meningitis were reported.
A total of 2 (4.5%) patients experienced at least 1 injection site reaction up to the CCOD (1 treatment-naive patient and 1 patient who switched from eculizumab). Three (11.5%) patients who switched from eculizumab reported 5 events of infusion-related reaction during the primary treatment period. The infusion-related reaction that occurred in 2 or more patients was paresthesia. No treatment-naive patients reported infusion-related reactions.
A total of 36 (81.8%) patients (77.8% of treatment-naive patients and 84.6% of switched patients) experienced at least 1 infection up to the CCOD.
There were 3 pregnancies in partners of patients reported in the study, with no reported detectable detrimental consequences to the infants (Table 23).
Table 23: Summary of Harms Results From the Long-Term Extension Study of the COMPOSER Study
AE | Crovalimab (naive) (N = 18) | Crovalimab (switch) (N = 26) | Crovalimab total (N = 44) |
|---|---|---|---|
Most common AEs in more than 20% of patients in each group, n (%) | |||
≥ 1 AE | 17 (94.4) | 25 (96.2) | 42 (95.5) |
Nasopharyngitis | 4 (22.2) | 9 (34.6) | 13 (29.5) |
Upper respiratory tract infection | 5 (27.8) | 5 (19.2) | 10 (22.7) |
Pyrexia | 2 (11.1) | 8 (30.8) | 10 (22.7) |
Headache | 3 (16.7) | 6 (23.1) | 9 (20.5) |
Cough | 1 (5.6) | 7 (26.9) | 8 (18.2) |
Diarrhea | 0 (0) | 6 (23.1) | 6 (13.6) |
SAEs, n (%) | |||
Patients with ≥ 1 SAE | 6 (33.3) | 8 (30.8) | 14 (31.8) |
Breakthrough hemolysis | 1 (5.6) | 2 (7.7) | 3 (6.8) |
Patients who stopped treatment due to AEs, n (%) | |||
Patients who stopped | 0 | 0 | 0 |
Deaths, n (%) | |||
Patients who died | 0 | 0 | 0 |
AEs of special interest, n (%) | |||
Alanine aminotransferase, increased | 1(5.6) | 0 | 1 (2.3) |
Drug-induced liver injury | 0 | 1 (3.8) | 1 (2.3) |
Type III hypersensitivity reactions (type III immune complex reactions) | 1 (5.6) | 2 (7.7) | 3 (6.8) |
Meningococcal meningitis | 0 | 0 | 0 |
Nasopharyngitis | 4 (22.2) | 9 (34.6) | 13 (29.5) |
Upper respiratory tract infection | 5 (27.8) | 5 (19.2) | 10 (22.7) |
Bronchitis | 1 (5.6) | 5 (19.2) | 6 (13.6) |
Gastroenteritis | 2 (11.1) | 3 (11.5) | 5 (11.4) |
Influenza | 2 (11.1) | 3 (11.5) | 5 (11.4) |
Injection site reactions | 1 (5.6) | 1 (3.8) | 2 (4.5) |
Infusion-related reactions | 0 | 3 (11.5) | 3 (6.8) |
AE = adverse event; SAE = serious adverse event.
Sources: Sponsor’s submission.23
The Supplemental Results Report provides additional long-term safety data from the treatment-naive and switch patients participating in the OLE phase of the COMPOSER study up to a CCOD of August 28, 2023 (i.e., approximately an additional 2 years of PNH safety data compared to the analysis presented in the Update Clinical Study Report that was submitted in the initial Marketing Authorisation Application).16 Long-term safety analyses were conducted covering both the primary treatment period (20 weeks) as well as the OLE period of the COMPOSER study, providing a median treatment duration of approximately 5 years. Safety analyses report cumulative results from baseline to the CCOD of August 28, 2023.
Forty-three of the 44 patients who were enrolled in part 2 to part 4 of the study entered the OLE phase, and 34 patients (15 naive patients and 19 patients who switched from eculizumab) were ongoing on crovalimab treatment at the time of the most recent CCOD (August 28, 2023). Overall, a total of 9 patients discontinued from the OLE phase (1 patient originally enrolled in part 2 [naive], 5 patients originally enrolled in part 3 [switch], 2 patients originally enrolled in part 4, arm A [naive], and 1 patient originally enrolled in part 4, arm B [switch]). No patients discontinued the study due to an AE. The median treatment duration from baseline to the CCOD was 4.69 years.
Overall, crovalimab’s safety profile remained unchanged as compared with the safety data presented for the November 1, 2021, CCOD. No new safety signals or new adverse drug reactions were identified in the updated safety analyses. A total of 43 (97.7%) patients experienced at least 1 AE from baseline to the CCOD. There was 1 death (in a patient who switched from eculizumab), which occurred in the OLE period since the publication of the previous Update Clinical Study Report and it was not considered by the investigators to be related to study treatment. No AEs resulted in the withdrawal of a patient from the study. A total of 19 (43.2%) patients experienced at least 1 SAE. SAEs were reported in 44.4% of patients in the treatment-naive group and in 42.3% of patients in the switched-from-eculizumab group. The most frequently reported AEs by System Organ Class (at least 50% incidence in the total population) were infections and infestations (93.2%), gastrointestinal disorders (52.3%), and musculoskeletal and connective tissue disorders (50.0%).
The most frequently reported AEs by preferred term with at least 15% incidence in the total population were nasopharyngitis (38.6%), COVID-19 (34.1%), upper respiratory tract infection (29.5%), headache (25.0%), back pain (22.7%), pyrexia (22.7%), arthralgia (20.5%), cough (20.5%), bronchitis (15.9%), and dizziness (15.9%).
A total of 41 (93.2%) patients experienced at least 1 infection up to the CCOD; this was an increase from 36 (81.8%) patients as reported in the Update Clinical Study Report.
Critical Appraisal
Internal Validity
The OLE period of the COMPOSER study was conducted to evaluate the longer-term safety and exploratory efficacy outcomes in 44 patients with PNH, beyond the 20-week primary evaluation period. Patients were enrolled in the extension if study investigators determined that the patient derived benefit from treatment with crovalimab in the 20-week primary study period. The open-label design may have increased the risk of bias in patient selection and outcome ascertainment for end points that include more subjective assessments because the lack of blinding may have impacted investigators and patients’ expectations of treatment. The direction and magnitude of potential bias remain unclear. Given the design, the long-term extension is noncomparative as all patients in part 2, part 3, and part 4 received crovalimab. As a result, the comparative safety and efficacy of crovalimab to relevant comparators were not addressed and this precludes conclusions about the comparative safety and efficacy of crovalimab. The small number of patients included in each study part, the single-arm setting, and the open-label design introduced significant uncertainty into study results and conclusions. There was variability in visit schedules across study parts, which may have introduced confounding to the analyses and limited the number of patients available at each follow-up time point.
The efficacy-evaluable population included all efficacy-evaluable patients who completed the primary treatment period in part 2, part 3, or part 4 of the study and were enrolled in the OLE period (18 treatment-naive patients and 25 switched patients). The study analysis included patients who were both treatment-naive and treatment-experienced, populations that differed in disease characteristics, such as transfusion history and LDH level. The impact of including both naive and experienced patients in the same analysis on the results is unclear.
During the OLE period, patients who were not receiving dosing every 4 weeks were required to switch to receive either 680 mg SC every 4 weeks (body weight ≥ 40 kg to < 100 kg) or 1,020 mg SC every 4 weeks (body weight ≥ 100 kg), which is aligned with the product monograph recommendation.
No method for the imputation of missing values was reported, and the attrition rate was 11.6%.
External Validity
The study included diverse geographical representation and populations with varying demographics and health care systems, which may enhance the generalizability of the results. However, the included patients were mostly men (60% to 86%), with ethnicity of “not Hispanic or Latino,” and included patients with median ages ranging from 44 years to 56 years; therefore, results may not be generalizable to a broader population. Patients with conditions such as psychiatric disorder, hereditary complement deficiency, a history of meningococcal meningitis, active primary or secondary immunodeficiency, a known history of HIV infection, chronic active hepatitis C, malignant disease, and alcohol, drug, or chemical abuse within 1 year before screening were excluded, and the results may not be generalizable to these groups of patients.
The dose-escalation approach that allowed for the inclusion of different dosing frequencies may enhance the generalizability across patient subgroups; however, frequent modifications and variations between groups increases intergroup variability and impacts the certainty and interpretation of study results.
Indirect Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
In the absence of head-to-head trial data of crovalimab compared with other existing therapies for the treatment of PNH in adults and adolescents aged 13 years and older with a body weight of at least 40 kg, the sponsor conducted an ITC using an NMA. The objective of this section is to summarize and critically appraise the sponsor-submitted indirect ITC evidence comparing crovalimab to other relevant comparators in settings in Canada.
Description of Indirect Comparison
Objectives
The objective of the NMA was to estimate the comparative efficacy and safety of crovalimab compared with ravulizumab, with eculizumab biosimilar, and with standard of care without C5 treatment.
Study Selection Methods
The NMA included trials identified through a systematic literature review (SLR) of published literature up to December 6, 2022, and then updated on March 1, 2024. The SLR included phase III trials of adult patients with PNH who were C5-naive or treated with a C5 inhibitor. The SLR was supplemented by searches of conference abstracts from 2020 to 2022, trial registries, and bibliographic searches of published systematic reviews. The study selection criteria and methods are summarized in Table 24.
With data from the SLR and the COMMODORE studies, 2 feasibility assessments were conducted to compare crovalimab with relevant comparator treatments in patients with previously untreated PNH and in patients with PNH previously treated with C5 inhibitors. A qualitative heterogeneity assessment was then performed to compare study design, study definitions of end points, and baseline characteristics (or inclusion and exclusion criteria where baseline characteristics were unavailable). Potential prognostic factors or treatment-effect modifiers were identified from previous studies and compared across studies included in the SLR. The assessment focused on 6 key end points reported in the COMMODORE trials: transfusion avoidance, BTH, hemoglobin stabilization, pRBC transfusion, and FACIT-F score and harms (i.e., any AEs). The feasibility assessment identified that the TRIUMPH study enrolled patients who were transfusion-dependent (at least 4 transfusions in the last 12 months before study entry) and acknowledged transfusion history as a key effect modifier. The TRIUMPH study was included in the ITC. The submission did not include a sensitivity analysis excluding the trial from the NMA.
Table 24: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients with PNH who are C5-experienced or C5-naive |
Intervention |
|
Comparator |
|
Outcome | Efficacy
Safety
|
Study designs | Published and unpublished (but data available) phase III trials |
Publication characteristics | Published literature, conference proceedings, trial registries |
Exclusion criteria |
|
Databases searched |
|
Selection process | Implementation and reporting followed the approach recommended by the PRISMA statement. Articles were screened independently by 2 reviewers and any discrepancies were resolved by a third reviewer. |
Data extraction process | Data from the final set of included studies were extracted in a Microsoft® Excel data extraction sheet. Each study was extracted by 2 independent reviewers, and discrepancies were resolved by a third reviewer. |
Quality assessment | Critical appraisal and quality assessment of the included studies were carried out using the assessment criteria for RCTs (NICE manufacturer’s template for RCTs) or for single-arm trials (AHRQ single-arm quality assessment tool). |
AE = adverse event; AHRQ = Agency for Healthcare Research and Quality; BTH = breakthrough hemolysis; C5 = complement component 5; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; NICE = National Institute for Health and Care Excellence; pRBC = packed red blood cell; PRISMA = Preferred Reporting Items for Systematic Reviews and Meta-Analyses; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; TA = transfusion avoidance.
Sources: Sponsor’s submission for indirect comparison evidence.68
ITC Analysis Methods
Table 25 provides an overview of the analysis methods used in the NMA. The NMA was conducted using a Bayesian framework and both fixed-effects and random-effects models. The random-effects model was selected as the base case because the absence of heterogeneity between studies was considered unlikely. In some instances, the fixed-effects model had a lower deviance information criterion than the random-effects model; however, the difference was not deemed meaningful (> 5-point difference). Meaningful priors were used to estimate study heterogeneity. No closed loops exist in the network, so no inconsistency analysis was performed. The base case presents results pooling C5-naive and C5-experienced studies, and subgroup analyses were provided for both populations.
The proportion of patients with transfusion avoidance, BTH, hemoglobin stabilization, the number of pRBC units transfused, and the change in FACIT-F score from baseline was analyzed using a normal likelihood and identity link. The submission outlined that efficacy end points could have also been modelled using a binomial likelihood; however, a normal likelihood was selected to allow calculating the probability that crovalimab is comparable to ravulizumab. The submission did not include a sensitivity analysis comparing results modelled using a binomial likelihood. The proportion of patients with any AE was modelled using a binomial likelihood and logit link function. If the percentage of patients with an event was missing but the number of responders and the sample size were available, this was imputed. If the CI was missing but the number of responders and the sample size were available, the Wilson method with continuity correction was applied to impute the CI. If the standard error was missing but the CI was available, the range rule was applied to calculate the standard error.
Table 25: Summary of ITC Methods
Method | Description |
|---|---|
Analysis methods | The NMA was conducted in a Bayesian framework using fixed-effects and random-effects models, with random-effects models selected as the base case. The starting point was using 50,000 iterations with a burn-in of 5,000 and a thinning factor of 10, and values were to be increased until model convergence was achieved. Continuous and dichotomous outcomes were assessed using normal and binomial models. |
Priors | As the NMA was based on a small set of studies with a small sample size, meaningfully informative priors were used to estimate between-study heterogeneity in all analysis (Table 26). Empirical priors established in the literature were used where applicable. Otherwise, the scale of the end point as well as the studies investigating ravulizumab vs. eculizumab were used to inform the prior, given that all 3 active treatments use the same mechanism of action. A log-normal distribution was assumed to ensure tau > 0. No sensitivity analysis was performed for choice of priors. |
Assessment of model fit | DIC, residual deviance, number of parameters |
Assessment of convergence | Model convergence was assessed using the following:
n.eff: A sufficient number of iterations was used to achieve n.eff, allowing for posterior inference (typically n.eff > 1,000 if credible intervals were sought). |
Outcomes | Efficacy outcomes: The mean difference in the proportion of patients with transfusion avoidance, breakthrough hemolysis, hemoglobin stabilization, the number of packed red blood cell transfusions, and the mean difference in change in FACIT-F score from baseline. Safety was analyzed using the difference in the proportion of patients with any adverse events. |
Follow-up time points | Up to week 25 or week 26, depending on the trial |
Construction of nodes | The network of evidence is star-shaped and does not provide closed loops. It allows for comparisons of crovalimab with ravulizumab, eculizumab biosimilar, and standard of care without C5 inhibitors. |
Sensitivity analyses | Fixed-effects model provided as sensitivity analysis |
Subgroup analysis | The base case presents C5-naive and C5-experienced studies pooled. Subgroup analysis is presented for both populations (C5-naive and C5-experienced). |
C5 = complement component 5; DIC = deviance information criterion; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; ITC = indirect treatment comparison; n.eff = effective sample size; NMA = network meta-analysis; PSRF = Potential Scale Reduction Factor; vs. = versus.
Sources: Sponsor’s submission for indirect comparison evidence.68
Table 26: Meaningfully Informative Priors for Between-Study Heterogeneity by End Point
End point | Reference | Steps |
|---|---|---|
Proportion of patients with TA | Scale of end point, the 301 study, and the 302 study | 1. A proportion is naturally limited [0, 1]. 2. Assuming a normal distribution, the SD is approximately (1 to 0) ÷ 4 = 0.25. 3. The SD of the effect between ravulizumab and eculizumab for naive and experienced patients is very small (< 0.02). 4. Considering this, using 0.1 as the upper limit is interpreted as a reasonably meaningful informative prior. With this, tau will have a mean of 0.05 and an SD of (0.1 to 0) ÷ 4 = 0.025. |
Proportion of patients with BTH | Scale of end point, the 301 study, the 302 study, and the 303 study | 1. A proportion is naturally limited [0, 1]. 2. Assuming a normal distribution, the SD is approximately (1 to 0) ÷ 4 = 0.25. 3. The SD of the effect between ravulizumab and eculizumab for naive and experienced patients is very small (< 0.01). 4. Considering this, using 0.1 as the upper limit is interpreted as a reasonably meaningful informative prior. With this, tau will have a mean of 0.05 and an SD of (0.1 to 0) ÷ 4 = 0.025. |
Proportion of patients with HS | Scale of end point, the 301 study, the 302 study, and the 303 study | 1. A proportion is naturally limited [0, 1]. 2. Assuming a normal distribution, the SD is approximately (1 to 0) ÷ 4 = 0.25. 3. The SD of the effect between ravulizumab and eculizumab for naive and experienced patients is very small (< 0.02). However, the 303 study comparing ravulizumab IV vs. ravulizumab SC in C5-experienced patients shows a relevant difference (approximately 10 percentage points) between the same drug. 4. Considering this, using 0.15 as the upper limit is interpreted as a reasonably meaningful informative prior. With this, tau will have a mean of 0.075 and an SD of (0.15 to 0) ÷ 4 = 0.0375. |
Number of pRBC units transfused | Scale of end point, the TRIUMPH study, the 301 study, the 302 study, and the 303 study | 1. The number of transfusions in the symptomatic treatment arm of the TRIUMPH study was 11, so we assume it is limited to between 0 and 20. 2. Assuming a normal distribution, the SD is approximately (20 to 0) ÷ 4 = 5. 3. The SD of the effect between ravulizumab and eculizumab for naive and experienced patients is very small (< 0.01). 4. Considering this, using 1 as the upper limit seems like a reasonable meaningful informative prior. With this, tau will have a mean of 0.5 and an SD of (1 to 0) ÷ 4 = 0.25. |
Change in FACIT-F score from baseline | Scale of end point, the 301 study, the 302 study, and the 303 study | 1. The FACIT-F scale is limited between 0 and 52. 2. Assuming a normal distribution, the SD is approximately (52 to 0) ÷ 4 = 13. 3. The SD of the effect between ravulizumab and eculizumab for naive and experienced patients is very small (< 1). 4. Considering this, using 5 as the upper limit seems like a reasonable meaningful informative prior. With this, tau will have a mean of 2.5 and an SD of (5 to 0) ÷ 4 = 1.25. |
Proportion of patients with any adverse event | Empirical prior for the log odds ratio taken from Turner et al. (2012)69 | NA |
BTH = breakthrough hemolysis; C5 = complement component 5; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; NA = not applicable; pRBC = packed red blood cell; SC = subcutaneous; SD = standard deviation; TA = transfusion avoidance; vs. = versus.
Sources: Sponsor's submission for indirect comparison evidence.68
Results of ITC
Summary of Included Studies
The SLR identified 13 published and unpublished (but data available) trials. Of the 13 published and data available trials, 7 were excluded in the feasibility assessment. The CLNP023X2204, PRINCE, and R3918-PNH-2092 trials were excluded because the studies were not connected to the network. The ALXN1210-PNH-303 study was excluded because the primary end point was evaluated at 10 weeks instead of 24 weeks. The PEGASUS and APPLY-PNH studies were excluded because patient characteristics were not comparable to the COMMODORE 1 trial. The ECU-PNH-III study was excluded because the trial included both treatment-naive and previously treated populations without reporting results separately for the 2 subgroups and randomization may have been impaired by the small sample size of the trial. Overall, this resulted in 6 phase III trials being included in the indirect comparison, including 4 trials (the 301 study, the TRIUMPH study, the SB12-3003 study, and the COMMODORE 2 study) among patients who were treatment-naive and 2 trials (the 302 study and the COMMODORE 1 trial) among patients previously treated with a C5 inhibitor (Table 27).
Table 27: List of Studies Included in the NMA
Study | Population | Intervention vs. comparator |
|---|---|---|
301 study60 | Treatment-naive | Ravulizumab vs. eculizumab |
TRIUMPH study70 | Treatment-naive | Eculizumab vs. standard of care |
SB12-3003 study71 | Treatment-naive | Eculizumab biosimilar vs. eculizumab |
COMMODORE 2 study15 | Treatment-naive | Crovalimab vs. eculizumab |
302 study72 | Pretreated | Ravulizumab vs. eculizumab |
COMMODORE 1 study17 | Pretreated | Crovalimab vs. eculizumab |
NMA = network meta-analysis; vs. = versus.
Variability was observed in baseline characteristics, including potential prognostic factors and effect modifiers, across studies included in the NMA (Table 28). Across studies, the history of transfusions ranged from 12.2% to 82.6%, LDH levels ranged from 228.0 U/L to 2,258.0 U/L, the presence of aplastic anemia ranged from 14.0% to 39.8%, and the history of MAVEs ranged from 14.0% to 29.0% (Table 28).
Table 28: Baseline Characteristics of Studies Included in the NMA
Study | Intervention | Prior transfusions | Hb level (g/dL) | Age (years) | LDH levels (U/L) | Aplastic anemia | History of MAVE |
|---|---|---|---|---|---|---|---|
COMMODORE 2 study | Crovalimab | 77% | Mean (SD) = 8.7 (1.4) | Mean (SD) = 40.5 (15.2) | Mean (SD) = 1,770.6 (790.02) | 39% | 16% |
Eculizumab | 74% | Mean (SD) = 10.0 (8.8) | Mean (SD) = 41.9 (16.0) | Mean (SD) = 1,817.5 (829.09) | 38% | 15% | |
COMMODORE 1 study | Crovalimab | 23% | Mean (SD) = 11.0 (2.0) | Mean (SD) = 44.4 (15.6) | Mean = 249.2 | 33% | 22% |
Eculizumab | 25% | Mean (SD) = 10.7 (1.8) | Mean (SD) = 49.5 (14.8) | Mean = 234.2 | 36% | 23% | |
301 study | Ravulizumab | 82.4% | Mean (SD) = 9.4 (1.46) | Mean (SD) = 44.8 (15.2) | Mean (SD) = 1,633.5 (778.8) | 32.8% | 14% |
Eculizumab | 82.6% | Mean (SD) = 9.6 (1.41) | Mean (SD) = 46.2 (16.2) | Mean (SD) = 1,578.3 (727.1) | 31.4% | 21% | |
302 study | Ravulizumab | 13.4% | Mean = 11.1 | Mean = 46.6 | Mean = 228.0 | 35.1% | 22% |
Eculizumab | 12.2% | Mean = 10.9 | Mean = 48.8 | Mean = 235.2 | 39.8% | 29% | |
TRIUMPH study | Eculizumab | NR | Mean = 10.0 | Median (range) = 41 (20 to 85) | Mean = 2,199.7 | 14% | 21% |
Placebo | NR | Mean = 9.7 | Median (range) = 35 (18 to 78) | Mean = 2,258.0 | 27% | 18% | |
SB12-3003 study | Eculizumab (SB12; biosimilar) | 64% | NR | Mean (SD) = 40.0 (13.4) | Mean (SD) = 2,220.2 (2,001.6) | NR | NR |
Eculizumab (Soliris) | 56% | NR | Mean (SD) = 36.3 (13.7) | Mean (SD) = 2,156.0 (1,750.6) | NR | NR |
Hb = hemoglobin; LDH = lactate dehydrogenase; MAVE = major adverse vascular event; NMA = network meta-analysis; NR = not reported; SD = standard deviation.
Efficacy
Overall Network Structure
The evidence network is provided for C5-naive studies (Figure 4) and C5-experienced studies (Figure 5).
Figure 4: Evidence Network in Treatment-Naive Patients With PNH Based on Published Evidence and Data From the COMMODORE 2 Study
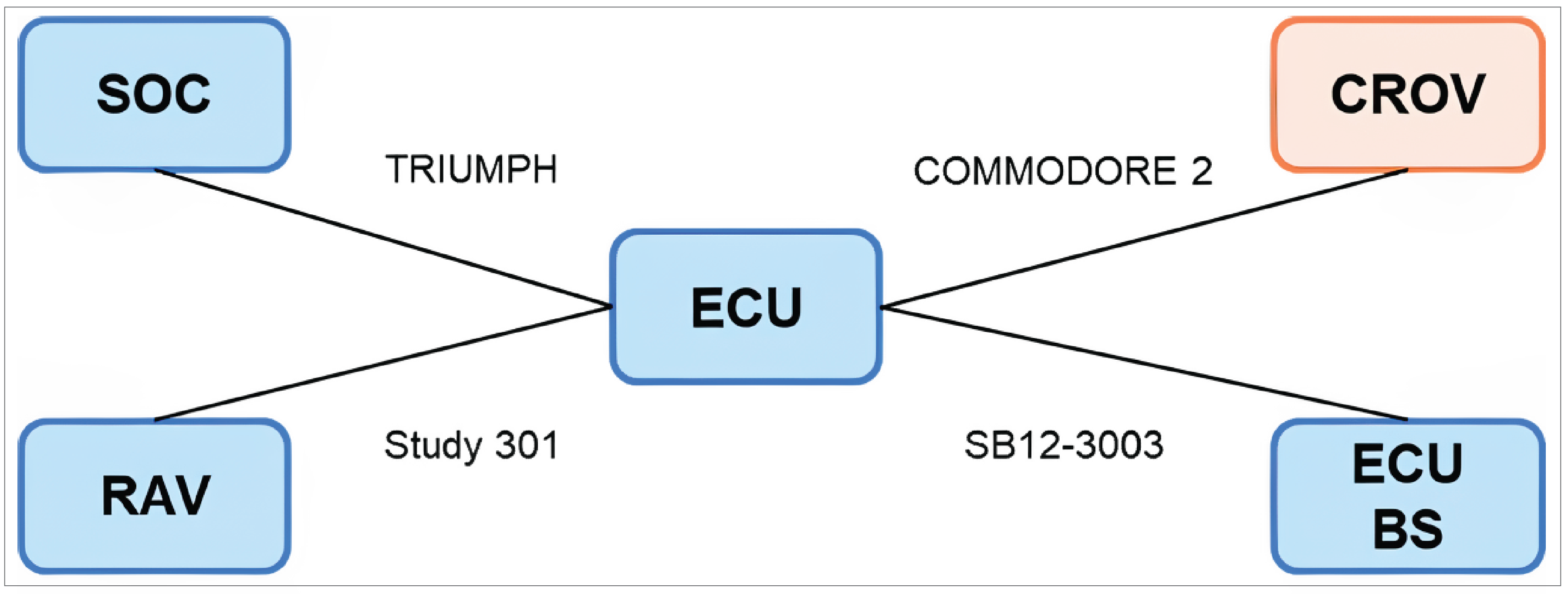
BS = biosimilar; CROV = crovalimab; ECU = eculizumab; PNH = paroxysmal nocturnal hemoglobinuria; RAV = ravulizumab; SOC = standard of care without complement component 5 inhibitor.
Figure 5: Evidence Network in Previously Treated Patients With PNH Based on Published Evidence and Data From the COMMODORE 1 Study

CROV = crovalimab; ECU = eculizumab; PNH = paroxysmal nocturnal hemoglobinuria; RAV = ravulizumab.
Network Structure for Each Outcome
Evidence networks are presented for each efficacy outcome, including transfusion avoidance (Figure 6), BTH (Figure 7), hemoglobin stabilization (Figure 8), the number of pRBC units transfused (Figure 9), and the FACIT-F score (Figure 10).
Transfusion Avoidance: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the TRIUMPH study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 6: Network for Transfusion Avoidance Based on the Percentage of Patients With an Event — C5-Naive and C5-Experienced Populations, Pooled
C5 = complement component 5; SoC = standard of care.
BTH: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 7: Network for BTH Based on the Percentage of Patients With an Event — C5-Naive and C5-Experienced Populations, Pooled
C5 = complement component 5.
Hemoglobin Stabilization: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the TRIUMPH study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 8: Network for Hemoglobin Stabilization Based on the Percentage of Patients With an Event — C5-Naive and C5-Experienced Populations, Pooled
C5 = complement component 5; SoC = standard of care without complement component 5 inhibitor.
Number of pRBC Units Transfused: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the TRIUMPH study, the SB12-3003 study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 9: Network for pRBC Transfusion — C5-Naive and C5-Experienced Populations, Pooled
BS = biosimilar; C5 = complement component 5; pRBC = packed red blood cell; SoC = standard of care without complement component 5 inhibitor.
FACIT-F Score: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the TRIUMPH study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 10: Network for FACIT-F Score Change From Baseline — C5-Naive and C5- Experienced Populations, Pooled
C5 = complement component 5; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; SoC = standard of care without complement component 5 inhibitor.
NMA Results
The following sections present ITC results for the pooled C5-naive and C5-experienced population for each outcome using the base-case analysis employing random-effects models because the absence of between-study heterogeneity was considered unlikely. Results for additional analyses including a fixed-effects model for the pooled population, and C5-naive and C5-experienced subgroup populations using random-effects and fixed-effects models, will be discussed briefly.
Base-case analysis: The base case presented here is pooling C5-naive and C5-experienced studies using random-effects models (Table 29). Sensitivity analysis of the pooled C5-naive and C5-experienced studies was conducted using fixed-effects models (Table 30). The deviance information criteria (model fit statistics) for random-effects and fixed-effects models are presented in Table 31. The results of the subgroup analysis splitting the 2 populations (C5-naive and C5-experienced) are presented in Table 32 and Table 33.
Base-Case Analysis (i.e., Random-Effects Model)
Transfusion Avoidance: C5-Naive and C5-Experienced Populations, Pooled
The point estimate for the percentage of patients who achieved transfusion avoidance suggested little to no difference (i.e., the difference was less than 20%) between crovalimab and ravulizumab (mean difference = 7.7%; 95% CI, −9.0% to 24.0%); however, the CrI was wide and included the possibility that ravulizumab was favoured (Table 29). The NMA suggested crovalimab was favoured over best supportive care in the absence of C5 treatment (mean difference = −49.0%; 95% CI, −73.0% to −27.0%) (Table 29).
Table 29: Summary of ITC Results for Crovalimab Versus Relevant Comparators (Random-Effects Model), C5-Naive and C5-Experienced Populations, Pooled
Outcome | Random-effects model | |||
|---|---|---|---|---|
Ravulizumab vs. crovalimab | Eculizumab vs. crovalimab | Best supportive care in the absence of C5 treatment vs. crovalimab | Eculizumab biosimilar vs. crovalimab | |
TA, %, MD (95% CrI) | 7.7 (−9.0 to 24.0) | 1.7 (−11.0 to 15.0) | −49.0 (−73.0 to −27.0) | NA |
BTH, %, MD (95% CrI) | −2.9 (−17.0 to 10.0) | 2.9 (−8.0 to 13.0) | NA | NA |
HS, %, MD (95% CrI) | 5.1 (−15.0 to 27.0) | 3.0 (−13.0 to 20.0) | −46.0 (−73.0 to −18.0) | NA |
Number of pRBC units transfused, MD (95% CrI) | 0.44 (−1.2 to 2.0) | 0.39 (−0.77 to 1.5) | 8.4 (5.5 to 11.0) | 0.59 (−1.4 to 2.6) |
Change in FACIT-F score from baseline, MD (95% CrI) | −2.0 (−6.9 to 3.0) | −3.1 (−6.8 to 0.52) | −13.0 (−20.0 to −6.7) | NA |
Proportion of patients with any AE (all AEs), odds ratio (95% CrI) | 0.94 (0.35 to 2.40) | 0.88 (0.43 to 1.70) | NA | NA |
AE = adverse event; BTH = breakthrough hemolysis; C5 = complement component 5; CrI = credible interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; ITC = indirect treatment comparison; MD = mean difference; NA = not applicable; pRBC = packed red blood cell; TA = transfusion avoidance; vs. = versus.
BTH: C5-Naive and C5-Experienced Populations, Pooled
The point estimate for the percentage of patients with a BTH event suggested little to no difference (i.e., the difference was less than 20%) between crovalimab and ravulizumab (mean difference = 2.9%; 95% CrI, −17.0% to 10.0%) (Table 29).
Hemoglobin Stabilization: C5-Naive and C5-Experienced Populations, Pooled
The point estimate for the percentage of patients who achieved hemoglobin stabilization suggested little to no difference (i.e., the difference was less than 20%) between crovalimab and ravulizumab (mean difference = 5.1%; 95% CI, −15.0% to 27.0%); however, the CrI was wide and included the possibility that ravulizumab was favoured (Table 29). The NMA suggested crovalimab was favoured over best supportive care in the absence of C5 treatment (mean difference = −46.0%; 95% CrI, −73.0% to −18.0%) (Table 29).
Number of pRBC Units Transfused: C5-Naive and C5-Experienced Populations, Pooled
The point estimate for the number of pRBC units transfused favoured ravulizumab compared to crovalimab (mean difference = 0.44 units; 95% CrI, −1.2 units to 2.0 units); however, the 95% CrI included the possibility that either treatment may be favoured (Table 29). The point estimate for the number of pRBC units transfused was larger in the best supportive care group compared to crovalimab (mean difference = 8.4 units; 95% CrI, 5.5 units to 11.0 units) (Table 29).
FACIT-F Score: C5-Naive and C5-Experienced Populations, Pooled
The point estimate for the change in FACIT-F score from baseline suggested little to no difference (i.e., the difference was less than 5) between crovalimab and ravulizumab (mean difference = −2.0 points; 95% CrI, −6.9 points to 3.0 points); however, the CrI was wide and included the possibility of a clinically important effect that favoured crovalimab (Table 29). The NMA suggested crovalimab was favoured over best supportive care in the absence of C5 treatment (mean difference = −13.0 points; 95% CrI, −20.0 points to −6.7 points) (Table 29).
Sensitivity Analysis (i.e., Fixed-Effects Model)
The results of the ITC based on the fixed-effects model were overall consistent with findings from the analysis using random-effects models (Table 30).
Table 30: Summary of ITC Results for Crovalimab Versus Relevant Comparators (Fixed-Effects Model), C5-Naive and C5-Experienced Populations, Pooled
Outcome | Fixed-effects model | |||
|---|---|---|---|---|
Ravulizumab vs. crovalimab | Eculizumab vs. crovalimab | Best supportive care in the absence of C5 treatment vs. crovalimab | Eculizumab biosimilar vs. crovalimab | |
TA, %, MD (95% CrI) | 7.7 (−6.2 to 21.0) | 1.6 (−9.7 to 13.0) | −49.0 (−69.0 to −30.0) | NA |
BTH, %, MD (95% CrI) | −2.4 (−12.0 to 6.7) | 3.3 (−5.0 to 11.0) | NA | NA |
HS, %, MD (95% CrI) | 4.3 (−11.0 to 19.0) | 2.0 (−10.0 to 14.0) | −47.0 (−67.0 to −27.0) | NA |
Number of pRBC units transfused, MD (95% CrI) | 0.32 (0.16 to 0.48) | 0.19 (0.058 to 0.33) | 8.2 (6.1 to 10.0) | 0.39 (0.019 to 0.75) |
Change in FACIT-F score from baseline, MD (95% CrI) | −1.8 (−4.4 to 0.69) | −3.0 (−5.0 to −0.92) | −13.0 (NR) | NA |
Proportion of patients with any AE (all AEs), odds ratio (95% CrI) | 0.94 (0.42 to 2.10) | 0.89 (0.51 to 1.60) | NA | NA |
AE = adverse event; BTH = breakthrough hemolysis; C5 = complement component 5; CrI = credible interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; ITC = indirect treatment comparison; MD = mean difference; NA = not applicable; NR = not reported; pRBC = packed red blood cell; TA = transfusion avoidance; vs. = versus.
Model Fit
The model fit was similar when using random-effects models and fixed-effects models, with a deviance information criterion difference of less than 3 points for all outcomes, except for the number of pRBC units transfused where the random-effects model provided a better fit (Table 31).
Table 31: Summary of DICs (Model Fit Statistics)
Outcome | DIC | |
|---|---|---|
Fixed-effects model | Random-effects model | |
TA, %, MD | 16.11 | 17.17 |
BTH, %, MD | 12.85 | 14.37 |
HS, %, MD | 17.76 | 18.53 |
Number of pRBC units transfused, %, MD | 460.2 | 24 |
Change in FACIT-F score from baseline, MD | 16.55 | 18.62 |
Proportion of patients with any AE (all AEs), odds ratio | 13.23 | 13.72 |
AE = adverse event; BTH = breakthrough hemolysis; DIC = deviance information criterion; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; MD = mean difference; pRBC = packed red blood cell; TA = transfusion avoidance.
Subgroup Analysis (C5-Naive and C5-Experienced)
The findings in the subgroup analysis examining the results in both the C5-naive and C5-experienced populations were largely consistent with results reported in the base-case analysis pooling C5-naive and C5-experienced populations using random-effects models (Table 32 and Table 33). The subgroup results using fixed-effects models were similar to those using random-effects models.
Table 32: Summary of ITC Results for Crovalimab Versus Relevant Comparators in the C5-Naive Subgroup Population (Random-Effects Model)
Outcome | C5-naive population (random-effects model) | |||
|---|---|---|---|---|
Ravulizumab vs. crovalimab | Eculizumab vs. crovalimab | Best supportive care in the absence of C5 treatment vs. crovalimab | Eculizumab biosimilar vs. crovalimab | |
TA, %, MD (95% CrI) | 9.3 (−15.0 to 33.00) | 1.7 (−16.0 to 20.0) | −49.0 (−76.0 to −23.0) | NA |
BTH, %, MD (95% CrI) | −1.2 (−22.0 to 19.0) | 5.6 (−11.0 to 21.0) | NA | NA |
HS, %, MD (95% CrI) | 0.23 (−30.0 to 30.0) | −3.3 (−26.0 to 19.0) | −52.0 (−84.0 to −20.0) | NA |
Number of pRBC units transfused, MD (95% CrI) | −0.93 (−2.6 to 0.66) | −0.13 (−1.3 to 1.0) | 7.9 (5.3 to 10.0) | 0.059 (−1.6 to 1.7) |
Change in FACIT-F score from baseline, MD (95% CrI) | −1.9 (−11.0 to 6.7) | −2.6 (−8.8 to 3.6) | −13 (−22 to −3.7) | NA |
Proportion of patients with any AE (all AEs), odds ratio (95% CrI) | 1.3 (0.35 to 4.90) | 1.2 (0.46 to 2.90) | NA | NA |
AE = adverse event; BTH = breakthrough hemolysis; C5 = complement component 5; CrI = credible interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; ITC = indirect treatment comparison; MD = mean difference; NA = not applicable; pRBC = packed red blood cell; TA = transfusion avoidance; vs. = versus.
Table 33: Summary of ITC Results for Crovalimab Versus Relevant Comparators in the C5-Experienced Subgroup Population (Random-Effects Model)
Outcome | C5-experienced population (random-effects model) | |||
|---|---|---|---|---|
Ravulizumab vs. crovalimab | Eculizumab vs. crovalimab | Best supportive care in the absence of C5 treatment vs. crovalimab | Eculizumab biosimilar vs. crovalimab | |
TA, %, MD (95% CrI) | 7.2 (−19.0 to 34.0) | 2.2 (−20.0 to 24.0) | NA | NA |
BTH, %, MD (95% CrI) | −7.3 (−31.0 to 17.0) | −2.3 (−22.0 to 18.0) | NA | NA |
HS, %, MD (95% CrI) | 14.0 (−20.0 to 49.0) | 14.0 (−13.0 to 41.0) | NA | NA |
Number of pRBC units transfused, MD (95% CrI) | 1.8 (0.16 to 3.4) | 0.91 (−0.27 to 2.1) | NA | NA |
Change in FACIT-F score from baseline, MD (95% CrI) | −2.3 (−11 to 6.6) | −3.7 (−10 to 3.0) | NA | NA |
Proportion of patients with any AE (all AEs), odds ratio (95% CrI) | 0.58 (0.11 to 3.0) | 0.59 (0.17 to 2.0) | NA | NA |
AE = adverse event; BTH = breakthrough hemolysis; C5 = complement component 5; CrI = credible interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HS = hemoglobin stabilization; ITC = indirect treatment comparison; MD = mean difference; NA = not applicable; pRBC = packed red blood cell; TA = transfusion avoidance; vs. = versus.
Harms
Any AEs: C5-Naive and C5-Experienced Populations, Pooled
Network structure: The 301 study, the COMMODORE 2 study, the 302 study, and the COMMODORE 1 study were included in the network.
Figure 11: Network for Any Adverse Events — C5-Naive and C5-Experienced Populations, Pooled
███ ███ ██████████ ██ ██████████ ████ ███████████ ███ ███ ████ ██ ████████.
Critical Appraisal of ITC
Overall, the NMAs were conducted according to accepted methodological guidance. An SLR was used as the basis for selecting relevant studies. The SLR searched multiple databases and grey literature sources, conducted study selection and data extraction using accepted methods, and provided a Preferred Reporting Items for Systematic Reviews and Meta-Analyses diagram of the study selection. The risk of bias of each included study was assessed using the criteria in the National Institute for Health and Care Excellence manufacturer’s template for RCTs. The inclusion criteria for the SLR included studies with populations that are relevant to the context in Canada. A feasibility assessment was performed to identify RCTs with the aim of minimizing intertrial heterogeneity and increasing the validity of the results. A prespecified analysis plan for conducting NMAs was used to guide the analyses. Geometry of the evidence networks was provided for each outcome analysis. Sensitivity analyses using fixed-effects models were conducted to further validate findings from the base analysis, which applied random-effects modelling. Subgroup analyses among C5-naive and C5-experienced populations were conducted to further examine if the efficacy and safety between the subpopulations and the pooled population (i.e., base case) were consistent. The potential limitations of the NMA are discussed as follows.
One of the limitations of the ITC was the heterogeneity in study-level patient demographic and disease characteristics across the included studies. Age, transfusion history, LDH levels, hemoglobin levels, the history of MAVE, and PNH subtype (aplastic anemia) were considered potential effect modifiers and these characteristics differed across included studies. For instance, enrolment in the TRIUMPH study was limited to patients who were transfusion-dependent. No additional sensitivity analysis excluding the TRIUMPH study was conducted. The base-case analysis included studies of patients who were both treatment-naive and treatment-experienced, populations that differed in disease characteristics, such as transfusion history and LDH level. The differences in patient characteristics across included studies may threaten the transitivity assumption for the NMA analysis. The assumption of similarity across the included studies may not hold true for the NMA, increasing the likelihood of bias and uncertainty about the validity of the results estimating the effectiveness of crovalimab compared to other existing therapies. The submission included subgroup analysis examining the results in both C5-naive and C5-experienced populations, and results were overall consistent with the results of the base case. No meta-regression analyses to adjust for factors that may bias comparisons were conducted, and the limited number of trials included in the NMA preclude the effective use of meta-regression to adjust for sources of between-trial heterogeneity.
The small number of trials with a relatively small sample size and the structure of constructed evidence networks were other key limitations. All evidence networks were sparsely connected, and the connections between treatment nodes were informed by 1 or 2 trials. There were no closed loops between nodes, inherently limiting that ability to examine inconsistency. Given the small number of trials, meaningfully informative priors were used to estimate between-study heterogeneity in all analysis. No additional sensitivity analyses exploring the impact of varying priors or using an informative prior compared to a noninformative prior were conducted.
The NMA was conducted using random-effects models as the base case, regardless of the model fit statistic (i.e., deviance information criterion). The sponsor justified the use of random-effects models due to the heterogeneity across the included studies. Given the sparseness of the network, the random-effects models may overfit the data while the fixed-effects models may underestimate the between-study heterogeneity leading to overly precise estimates. Nevertheless, in a sensitivity analysis comparing the results of fixed-effects models to random-effects models, the results are largely consistent using both methods. The deviance information criteria between the random-effects and fixed-effects models differed by less than 3 points for all outcomes, except for the number of pRBC units transfused where the random-effects model provided a better fit.
The threshold of 20% — which was used to assess the results of the NMA for the BTH, stabilized hemoglobin, and transfusion avoidance outcomes — is based on the predefined NIMs from the COMMODORE 2 trial; it included patients naive to C5 inhibitors. It is unclear if it is appropriate to apply the same threshold to a patient population with different characteristics (i.e., a pooled population of patients with PNH who are C5-naive and C5-experienced) or to the subgroup of patients who are C5 inhibitor–experienced. However, assessing the results in the absence of a threshold (i.e., using the null threshold) leads to a similar overall assessment in the pooled and C5-experienced populations. Using the null as the threshold, the point estimates for transfusion avoidance and stabilized hemoglobin favoured ravulizumab compared to crovalimab; however, the Crls were wide and included the possibility that either treatment may be favoured (in the pooled and C5-experienced populations). Using the null as the threshold, the point estimate for BTH favoured crovalimab compared to ravulizumab; however, the CrI was wide and included the possibility that either treatment may be favoured (in the C5-experienced population).
Despite the analyses suggesting overall little to no difference between crovalimab compared to ravulizumab across the majority of outcomes in the pooled and subgroup analyses, the effect estimates were imprecise (i.e., had wide CrIs that spanned the null, including the possibility of harm, benefit, or both) and the evidence was insufficient to draw definitive conclusions on the relative efficacy of crovalimab compared to ravulizumab. However, the clinical experts consulted by the review team indicated that results suggesting little to no difference is plausible given that crovalimab and ravulizumab belong to the same group of drugs and ravulizumab has demonstrated similar benefits in adult patients with PNH compared with eculizumab in 2 RCTs (the 301 study and the 302 study).
Finally, the proportion of patients with hemolysis control (i.e., an LDH level ≤ 1.5 × ULN at the central laboratory), which was the coprimary outcome in the COMMODORE 2 trial, was not reported in the ravulizumab trials and therefore was not able to be assessed in the NMA. As such, comparative evidence for this outcome for crovalimab versus ravulizumab is not available. The NMA provided no information on HRQoL, an outcome of importance to patients.
Studies Addressing Gaps in the Systematic Review Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Discussion
Summary of Available Evidence
Two multicentre, phase III, randomized, open-label, active-controlled trials submitted by the sponsor were included comparing crovalimab (maintenance dose of 680 mg [body weight ≥ 40 kg to < 100 kg] or 1,020 mg [≥ 100 kg] by SC injection once every 4 weeks) with eculizumab (maintenance dose of 900 mg by IV infusion once every 2 weeks) in adult patients with PNH who had not been previously treated with a complement inhibitor (the COMMODORE 2 study, randomized N = 204) and patients with documented treatment with complement inhibitors (the COMMODORE 1 study, randomized N = 89). Patients were randomized to receive either crovalimab (arm A) or eculizumab (arm B) in a 2:1 ratio in the COMMODORE 2 study, and in a 1:1 ratio in the COMMODORE 1 study. In addition, both trials enrolled a nonrandomized descriptive arm (arm C) with patients who only received crovalimab (6 patients with PNH aged younger than 18 years in the COMMODORE 2 trial, and 38 patients on a complement inhibitor treatment including 1 patient with PNH aged younger than 18 years in the COMMODORE 1 trial, at the CCOD of November 16, 2022). In both trials, the primary treatment period was 24 weeks for all arms. After that, all patients had the opportunity to continue or switch to crovalimab in a 46-week safety follow-up period. In the COMMODORE 2 trial, transfusion avoidance and hemolysis control (measured by an LDH level ≤ 1.5 × ULN at a central laboratory) were coprimary outcomes. Secondary outcomes were BTH, stabilized hemoglobin, and the FACIT-F score. The mean percentage change in LDH levels, the maintenance of a minimum hemoglobin level, and the number of pRBC units transfused, among others, were also reported as exploratory outcomes. HRQoL assessed with EORTC QLQ-C30, and EORTC IL-40 selected symptoms, were compared. Patient preference for crovalimab or eculizumab, and harms were also reported. In the COMMODORE 1 trial, due to the introduction of ravulizumab to the treatment landscape and a reduced pool of patients treated with eculizumab over time, randomization was stopped in November 2022 per the amended protocol, which was earlier than the initial plan of study. As a result, the evaluation of safety became the primary objective and all efficacy objectives became exploratory. The specific safety and efficacy outcomes in the COMMODORE 1 trial were similar to those in the COMMODORE 2 trial.
At baseline, the median age of the randomized population in the crovalimab and eculizumab arms was 36 years and 38 years, respectively, in the COMMODORE 2 study, and 42 years and 49 years, respectively, in the COMMODORE 1 study. The difference between the proportions of females and males was small in both studies. Most patients were Asian (64% in the crovalimab arm versus 74% in the eculizumab arm) or white (33% in the crovalimab arm versus 23% in the eculizumab arm), followed by other or unknown (3%) in the COMMODORE 2 study, while in the COMMODORE 1 study, most patients were white (76% in the crovalimab arm versus 73% in the eculizumab arm), followed by Asian (20% in the crovalimab arm versus 16% in the eculizumab arm), and other or unknown (4% in the crovalimab arm versus 11% in the eculizumab arm). In the COMMODORE 2 study, the baseline mean LDH level was 7.6 multiplied by ULN and 7.8 multiplied by ULN in the crovalimab and eculizumab arms, respectively; 77% of the patients in the crovalimab arm had received a pRBC transfusion with a mean transfused pRBC within the 12 months before screening of 6.5 units and 74% of the patients in the eculizumab arm had received a pRBC transfusion with a mean of 6.6 units, with half of the patients receiving up to 6 units of pRBC transfusion in the prior 6 months in both arms. In the COMMODORE 1 study, 23% of the patients in the crovalimab arm (mean pRBC transfused = 1.6 units) and 25% of the patients in the eculizumab arm (mean pRBCs transfused = 2.3 units) had received pRBC transfusion. In both studies, the median of the PNH clone size was smaller in the crovalimab arm than in the eculizumab arm for erythrocytes, granulocytes, and monocytes. The main PNH-relevant conditions in patient history were aplastic anemia (33% to 39%) and major vascular events (15% to 23%).
There is no direct evidence comparing crovalimab with ravulizumab. The sponsor submitted an NMA to compare crovalimab with available relevant therapies including eculizumab, ravulizumab, eculizumab biosimilar, and the standard of care without a C5 inhibitor. According to the clinical experts consulted by CDA-AMC for this review, the currently available first-line treatment for PNH is C5 inhibitor therapy either with eculizumab or ravulizumab, where funded. Treatment without a C5 inhibitor is no longer the standard of care for PNH in clinical practice in Canada. Furthermore, eculizumab biosimilar is not marketed in Canada. Therefore, the most relevant comparator for indirect comparison in this review is ravulizumab. The ITC summary for this review has focused on comparing crovalimab with ravulizumab.
Interpretation of Results
Efficacy
PNH is a chronic lifelong disease. Eculizumab and ravulizumab are first-line therapies used to treat PNH in patients, based on their ability to meet the treatment goals of reducing intravascular hemolysis, thrombosis risk, and transfusion needs, and improving survival. Like eculizumab and ravulizumab, crovalimab is also a terminal complement inhibitor that targets C5.23,33,34 In addition, crovalimab is engineered with Sequential Monoclonal Antibody Recycling Technology and has properties of high solubility, extended half-life, and high bioavailability. As such, for crovalimab, the delivery of a low-volume maintenance dose by SC injection every 4 weeks is possible, allowing for caregiver-assisted administration or self-administration at off-clinic settings.23,34 The clinician group highlighted that crovalimab is expected to address important treatment goals unmet by current therapies, including offering a C5 inhibitory strategy that does not require IV access, enabling self-administration by patients or administration by caregivers, and providing an option for rare cases of resistance to eculizumab or ravulizumab due to a C5 polymorphism. The patient group provided input that crovalimab helps reduce treatment burden (in particular, clinic visits associated with the treatment if frequent IV administrations are required) and improves patient convenience. According to the patient group, an easy and quick administration of treatment may be of particular importance in the context of this lifelong disease.
The COMMODORE 2 study showed that crovalimab administered subcutaneously every 4 weeks was noninferior to eculizumab administered by IV every 2 weeks in C5 inhibitor–naive patients across coprimary and secondary outcomes. Based on the testing hierarchy specified in Table 9, noninferiority of crovalimab was demonstrated for the coprimary end points of hemolysis control and transfusion avoidance and the secondary end points of BTH and hemolysis stabilization. The testing hierarchy broke given that the superiority of crovalimab to eculizumab was not met for transfusion avoidance. The significance level was not met for superiority (the superiority of crovalimab to eculizumab was not met for transfusion avoidance) and no further testing was performed.
Regarding the disease characteristics (potential effect modifiers) at baseline, all patients in the COMMODORE 2 trial had an LDH level of 2 or more multiplied by ULN, and more than 80% of patients had an LDH level of 4 or more multiplied by ULN. Approximately three-quarters of the patients had received a blood transfusion within the 12 months before screening, including approximately a quarter of patients receiving more than 6 pRBC units in the prior 6 months, indicating uncontrolled hemolysis and a high level of transfusion dependence before treatment in the study patients. Nearly 40% of patients had a history of aplastic anemia. The COMMODORE 2 trial showed that treatment with crovalimab results in little to no difference in efficacy outcomes when compared to eculizumab. The evidence was rated as being of high or moderate certainty across outcomes, except for EORTC QLQ-C30, infections, and deaths, which were rated as being of low certainty, using the GRADE approach. Confidence in the between-group differences for efficacy outcomes in the COMMODORE 2 trial were limited due to imprecision (indicated by the associated CIs that included small effects close to the null or that crossed the null and a small sample size) and the open-label design of the analyses.
The clinician group and clinical experts regarded fatigue and HRQoL outcomes to be of importance. In the COMMODORE 2 study, based on the FACIT-F tool — a validated measurement that assesses fatigue in patients with PNH — both crovalimab and eculizumab demonstrated a clinically meaningful (at least 5 points) improvement in fatigue from baseline to week 25 (Table 16 and Table 34) (the proportion of patients who achieved at least a 5-point improvement from baseline). The noninferiority of the change in fatigue with crovalimab versus eculizumab was not formally tested due to the break in the predefined statistical testing hierarchy. In the COMMODORE 2 study, patients in the crovalimab and eculizumab arms showed similar improvements in the EORTC QLQ-C30 GHS and QoL score and EORTC IL-40 symptoms (dyspnea, dysphagia, chest pain, headaches, abdominal pain, erectile dysfunction) from baseline to week 25 (between-group effects estimates were not reported) (Table 16 and Table 35). However, this outcome was rated as low certainty evidence using the GRADE assessment.
In the COMMODORE 1 trial, exploratory efficacy results during the 24-week, randomized primary treatment period were overall supportive of the results observed in the COMMODORE 2 trial, suggesting that crovalimab overall maintains disease control in patients with PNH switching from eculizumab in hemolysis control (defined as a central LDH value ≤ 1.5 × ULN), transfusion avoidance, BTH, and self-reported fatigue (Table 16). Results suggested that crovalimab may result in little to no difference in hemolysis control, a reduction in BTH, transfusion avoidance, number of pRBC units transfused, FACIT-F score, infections, and deaths. ███ ███ ██████████ ██ ████████ ███ ███████████████ █████████████ ███ ███ ██████████ ██ ████████ ███ ███████ ███ ██████████ █ ████████████████ ██████ ███ ███████ ████ ███████████ ██████ ██ ███ ██████████ ████ ████████ ████ ███████████ ██ █ ██████████ ██ █ ████████ ██████ █████ ██████ ███████ ███ █████ █████ ███ ████ ██ ███████ ████████ ██████ ███. While the percentage change in the central LDH value from baseline was numerically higher in the crovalimab arm compared with the eculizumab arm, the mean proportion of patients with a central LDH value of 1.5 or less multiplied by ULN by visit through week 25 was similar between the 2 arms at 92.93% for the crovalimab arm (95% CI, 86.62% to 96.39%) versus 93.74% for the eculizumab arm (95% CI, 87.26% to 97.04%). The clinical experts consulted by the review team agreed that the difference between groups in change in central LDH levels did not raise clinical concern, noting that results were in line with clinician expectations; clinicians would not anticipate seeing large changes in LDH levels in patients who switch to crovalimab from a stable dose of another C5 inhibitor. The rating of certainty was low for all outcomes in the COMMODORE 1 trial, except for the EORTC QLQ-C30 GHS and QoL score, which was rated as very low certainty using the GRADE assessment. Confidence in the between-group differences for outcomes was limited due to imprecision (indicated by the associated CIs that crossed the null) and relatively small sample sizes. There is some uncertainty about the clinical importance of the estimates, in the absence of identified thresholds. All efficacy analyses were descriptive without formal statistical testing and should be considered as supportive evidence.
For patients from all arm C cohorts in the COMMODORE 1 study, there was little to no difference in the EORTC QLQ-C30 physical functioning, role function, and GHS and QoL scores, as well as EORTC IL-40 dysphagia, chest pain, and abdominal pain scores postbaseline to week 25. Overall, the COMMODORE 2 study arm C (n = 6) suggested favourable efficacy and HRQoL results of crovalimab in pediatric patients (median age = 16.5 years [range, 13 years to 17 years]). However, the interpretation and application of study results were limited due to small sample size and the single-arm descriptive analysis of this cohort. In the COMMODORE 1 study, 1 pediatric patient (aged ██ years) was enrolled about 2 weeks before the CCOD and therefore data for this patient were limited. In addition, no pediatric patients with a body weight of under 40 kg were enrolled in either pivotal study. There is a lack of evidence in patients who are aged younger than 13 years and those with a body weight of less than 40 kg. The Health Canada indication covers adults and adolescents aged 13 years and older with a body weight of at least 40 kg. The clinical experts consulted for this review pointed out that being a rare disease, the incidence of PNH in children is even lower than in adults. The clinical experts expected that pediatric patients would have similar efficacy results with crovalimab treatment as adults in a comparative study.
The clinical experts consulted for this review noted that using eculizumab as a comparator in the 2 pivotal RCTs was appropriate. Although there have been recent additions to the treatments available for patients with PNH, eculizumab is the standard of care in most of the countries where patients were enrolled at the time of study initiation. The clinical experts noted that ravulizumab is more commonly used in North America compared to the rest of the world; however, for patients who plan for pregnancy or who are pregnant, eculizumab would be used per clinical guidelines.
Despite the sponsor-submitted NMA analyses suggesting overall little to no difference between crovalimab compared to ravulizumab, the evidence was insufficient (i.e., a limited number of included studies, heterogeneity in patient characteristics across trials, and CrIs that crossed the null) to draw definitive conclusions on the relative efficacy of crovalimab compared to ravulizumab. The effect estimates were imprecise (i.e., had wide CrIs that spanned the null including the possibility of harm, benefit, or both). However, the clinical experts consulted by the review team indicated that results suggesting little to no difference are plausible given that crovalimab and ravulizumab belong to the same group of drugs and ravulizumab has demonstrated similar benefits in adult patients with PNH compared with eculizumab in 2 RCTs (the 301 study and the 302 study).
In the COMMODORE 2 and COMMODORE 1 trial extension, during safety follow-up periods (i.e., from switch baseline up to switch week 25), similar efficacy outcomes appeared consistent with the randomized 24-week treatment periods of the pivotal trials for the crovalimab group suggesting an ongoing benefit of crovalimab. However, further interpretation of these data was limited by the open-label and descriptive nature of the extension study, the lack of comparative analyses, and some missing data.
Efficacy data from the OLE study (the COMPOSER trial) suggested that patients treated with crovalimab maintained disease control during approximately 4 years of follow-up time, based on end points related to hemolysis control (change in LDH levels), transfusion avoidance, hemoglobin stabilization, and BTH. However, further interpretation of these data was limited by the noncomparative, open-label, and descriptive nature of the extension study as well as the small sample size (N = 42).
Harms
Based on the results of the COMMODORE 2 and COMMODORE 1 studies, the overall safety profile of crovalimab was consistent with that expected for a C5 inhibitor, and no additional safety signals were identified from the longer-term, single-arm study (the COMPOSER trial). The most common AEs in the crovalimab and eculizumab arms were infections and infestations, infusion-related or injection-related reactions, hypokalemia, and hypersensitivity other than type III immune complex reactions. Of note, injection-related reactions were reported and were expected to occur only in the crovalimab arm due to SC administration being unique to crovalimab. These injection-related reactions usually occurred during or within 24 hours of infusion in the COMMODORE studies15,17 and patients would recover without a need for treatment or with simple symptomatic treatment.
PNH is associated with complications such as thromboembolic events, renal function impairment, pulmonary arterial hypertension, and abdominal pain. In patients who do not receive an adequate treatment for PNH, thromboembolic events can be the primary cause of death. Two patients in the crovalimab arm and 1 patient in the eculizumab arm in the COMMODORE 2 study died during the 24-week primary treatment period but the reasons for death were likely unrelated to the study drug, according to the clinical experts consulted for this review. No deaths were reported during the primary treatment period in the COMMODORE 1 study. The clinical experts also emphasized the observation of risk of meningitis due to its significant impact on life. In both trials, no cases of meningitis were reported, likely because all patients received vaccinations before being enrolled in the trials. And the observation of deaths and some rare AEs like meningitis would require a longer length of follow-up. MAVE occurred infrequently in the crovalimab arm in both trials.
During the primary safety period, the safety profile of crovalimab was comparable to that of eculizumab, with key safety parameters being similar between the 2 treatment arms in patients with PNH who were naive to a C5 inhibitor in the COMMODORE 2 trial.
In the COMMODORE 1 study that enrolled patients who had completed at least 24 weeks of treatment with eculizumab for PNH, beside the safety profile known for C5 inhibitors, there was a newly identified risk of type III immune complex reactions, which only occur in patients who switch between crovalimab and another C5 inhibitor. During the primary safety period of the COMMODORE 1 study, the proportion of patients in the crovalimab arm was higher than in the eculizumab arm for some AEs (e.g., any AEs in the crovalimab arm = 77% versus any AEs in the eculizumab arm = 67%; any infections in the crovalimab arm = 41% versus any infections in the eculizumab arm = 36%; SAEs in the crovalimab arm = 14% versus SAEs in the eculizumab arm = 2%; and serious infections and infestations in the crovalimab arm = 7% versus serious infections and infestations in the eculizumab arm = 2%) (Table 19). Nonetheless, the events that underlie these between-group imbalances were mostly reflective of risks unique to the crovalimab arm (e.g., type III immune complex reactions, injection site reactions); thus, patients who were allocated to the eculizumab arm (i.e., who continued to take and were stabilized on eculizumab during the 24-week randomized period in the COMMODORE 1 study) reported no events of such AEs. In addition, infusion-related reactions, which were probably related to a single IV loading dose, occurred only in the crovalimab arm (14% versus 0% with eculizumab). This was likely due to the steady receipt of eculizumab for at least 24 weeks before study enrolment was interrupted by the loading doses of a different treatment (crovalimab) and at different intervals. Overall, the results in the nonrandomized arm C showed a consistent safety profile of crovalimab.
A total of 16% of the patients in the crovalimab arm (i.e., those who switched from eculizumab, n = 44) (Table 19) during the 24-week primary treatment period in the COMMODORE 1 trial and 18% of the patients who were switched to crovalimab from a C5 inhibitor in a pooled safety analysis (n = 185) (Table 38) reported type III immune complex reactions. Type III immune complex reactions are expected only in crovalimab-switch patients because crovalimab binds to different epitopes on C5 compared to eculizumab and ravulizumab, and when both drugs are present in the circulation, DTDCs — which is a transient immune complex formation (eculizumab or ravulizumab-C5-crovalimab motifs) — may form.33,34 Hence, patients who switched from eculizumab to crovalimab in the COMMODORE studies and the COMPOSER study (or patients switching to or from other C5 inhibitors in clinical practice, such as from eculizumab or ravulizumab to crovalimab or vice versa) are at risk of developing DTDC-associated type III immune complex reactions.15,17,33,34 In the COMMODORE 1 trial, type III immune complex reactions manifested clinically as arthralgia and/or rashes of mild or moderate severity in most patients, and symptomatic treatment and/or corticosteroid therapy were only required for more severe cases.17,34 The clinical experts consulted for this review noted that the type III immune complex reactions appeared manageable such that patient education about these types of AEs would be adequate, but recommended that such AEs be evaluated and monitored by health care providers and more data with a longer period of observation on type III immune complex reactions is preferred.
Based on the results of the COMMODORE 2 trial, overall, the safety results of the 6 pediatric patients in arm C during the crovalimab safety period indicated that crovalimab was well tolerated in pediatric patients with PNH because most only experienced grade 1 to grade 2 AEs, and no SAEs or AEs leading to the withdrawal of treatment or dose modification were reported. In the COMMODORE 1 trial, the single pediatric patient enrolled approximately 2 weeks before CCOD did not experience any AEs. The clinical experts noted a need for enhanced safety consideration regarding the risk of infections, including meningitis in pediatric patients because of the significant adverse impact of meningitis and because children often have associated cytopenias.
██ ███ ████ ███ ███ ██████████ ██ ██████████ ████ ███████████ ███ ███ ████ ██ ████████████ ███ ███████ ███████ ███ █████ ████████ ████████ ████████████ ████████ ███ ███ ███ ███ ████ ███ ████████ ███ █████████ ████ ██████ █████████ ███ ██ █████████.
No new signals in safety outcomes were identified from the OLE study (the COMPOSER trial) or the 46-week safety follow-up periods of the COMMODORE 2 and COMMODORE 1 trials. However, further interpretation of their results was limited by the lack of a comparator arm, by being open-label, and due to descriptive reporting.
Other Considerations
Data about patient preference were collected using a sponsor-developed questionnaire among the patients who first received eculizumab and then switched to crovalimab in the 2 pivotal trials. Most patients (84% to 96%) preferred crovalimab, with the top reasons including easier and less time-consuming drug administration, fewer hospital visits associated with treatment, and better QoL compared to eculizumab. The patient preference outcome has a risk of bias in favour of crovalimab because of the open-label design of the trials.
In the COMMODORE 2 and COMMODORE 1 studies, patients were able to self-administer crovalimab, although the overall proportions of SC administration of crovalimab done by the patient or their caregivers (range, 12% to 31%) during the 24-week randomized period were not high. In both trials, no medication errors specifically due to self-administration were reported up to the CCOD.15,17,33,34
Based on input from the patient and clinician groups, 1 significant challenge with current treatments is that patients with PNH require regular clinic visits every 2 weeks to 8 weeks for IV infusions, which can be time-consuming and disruptive. The groups noted that crovalimab can be given as a small SC injection, thus helping to reduce the physical, emotional, and logistical burdens associated with traditional IV therapies, allowing patients to regain a sense of freedom and control over their lives. The clinical experts consulted for this review noted that crovalimab may fulfill an unmet need for some patients with a preference for a SC route of drug administration, particularly for those with poor venous access, and who are opposed to a port-a-cath insertion. In addition, the clinical experts noted that crovalimab may be a safer alternative for patients living in isolated geographic locations that preclude the guaranteed delivery of eculizumab.
Conclusion
Two phase III, multicentre, open-label RCTs evaluated the efficacy and safety of SC injection of crovalimab at a weight-based dose once every 4 weeks compared with IV infusion of eculizumab 900 mg once every 2 weeks in adult patients with PNH after 24 weeks of treatment. Evidence from the COMMODORE 2 trial in patients with PNH who were naive to C5 inhibitors demonstrated that crovalimab was noninferior to eculizumab across the coprimary end points (hemolysis control [measured by an LDH level ≤ 1.5 × ULN] and transfusion avoidance) and secondary outcomes (BTH and stabilized hemoglobin). Other secondary (FACIT-F score) and exploratory end points were supportive of the noninferiority results, suggesting overall little to no difference between the treatment groups. Efficacy outcomes were rated as being of high to moderate certainty (except for the EORTC QLQ-C30 GHS and QoL score, which was rated as low certainty), using the GRADE approach.
Results from the COMMODORE 1 trial in patients with PNH who were exposed to eculizumab were overall supportive of the results observed in the COMMODORE 2 trial, suggesting that crovalimab results in little to no difference in hemolysis control, BTH, transfusion avoidance, and the FACIT-F score compared with eculizumab. Efficacy analyses in the COMMODORE 1 trial were descriptive without formal statistical testing and should be considered as supportive evidence. The evidence was rated as being of low certainty, using the GRADE approach.
Descriptive analyses on long-term efficacy and safety based on the OLE phases of the COMMODORE 1 and COMMODORE 2 studies (an additional 24 weeks of treatment with crovalimab) and an OLE study (the COMPOSER trial; crovalimab treatment for a median duration of 3.40 years) appeared consistent with the randomized 24-week treatment periods of the pivotal trials for the crovalimab group, suggesting an ongoing benefit of crovalimab.
Based on the results of the COMMODORE 2 and COMMODORE 1 studies, the overall safety profile of crovalimab was consistent with that expected for a C5 inhibitor, and no additional safety signals were identified from the longer-term, single-arm study (the COMPOSER trial); however, long-term comparative data were not available. Approximately 1 in 5 patients who switched to receive crovalimab from eculizumab reported type III immune complex–mediated reactions.
The sample size of pediatric patients was small in the COMMODORE 2 study (n = 6) and in the COMMODORE 1 study (n = 1), and only descriptive results were available. The clinical experts anticipated that pediatric patients would have efficacy results similar to those in the main trial arms; however, there is a need for enhanced safety consideration regarding the risk of infections, including meningitis in pediatric patients.
Indirect evidence from the sponsor-submitted NMA suggested that crovalimab may provide efficacy and safety comparable to ravulizumab. Despite the analyses suggesting overall little to no difference between crovalimab compared to ravulizumab, the evidence was insufficient (i.e., a limited number of included studies, heterogeneity in patient characteristics across trials, and CrIs that crossed the null) to draw definitive conclusions on the relative efficacy of crovalimab compared to ravulizumab. The clinical experts consulted by the CDA-AMC review team indicated that results suggesting little to no difference are plausible given that the 2 comparators belong to the same group of drugs and that ravulizumab and eculizumab have demonstrated similar benefits in adult patients with PNH in RCTs.
References
1.Schrezenmeier H, Roth A, Araten DJ, et al. Baseline clinical characteristics and disease burden in patients with paroxysmal nocturnal hemoglobinuria (PNH): updated analysis from the International PNH Registry. Ann Hematol. 2020;99(7):1505-1514. doi: 10.1007/s00277-020-04052-z PubMed
2.Shah N, Bhatt H. Paroxysmal nocturnal hemoglobinuria [sponsor supplied reference]. StatPearls. Treasure Island (FL)2022.
3.Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: A consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36-52. doi: 10.1111/ejh.13176 PubMed
4.Oliver M, Patriquin CJ. Paroxysmal Nocturnal Hemoglobinuria: Current Management, Unmet Needs, and Recommendations. J Blood Med. 2023;14:613-628. doi: 10.2147/JBM.S431493 PubMed
5.Socie G, Schrezenmeier H, Muus P, et al. Changing prognosis in paroxysmal nocturnal haemoglobinuria disease subcategories: an analysis of the International PNH Registry. Intern Med J. 2016;46(9):1044-53. doi: 10.1111/imj.13160 PubMed
6.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804-11. doi: 10.1182/blood-2014-02-522128 PubMed
7.Risitano AM, Marotta S, Ricci P, et al. Anti-complement Treatment for Paroxysmal Nocturnal Hemoglobinuria: Time for Proximal Complement Inhibition? A Position Paper From the SAAWP of the EBMT. Front Immunol. 2019;10:1157. doi: 10.3389/fimmu.2019.01157 PubMed
8.Brodsky RA. Stem cell transplantation for paroxysmal nocturnal hemoglobinuria. Haematologica. 2010;95(6):855-6. doi: 10.3324/haematol.2010.023176 PubMed
9.Peffault de Latour R, Schrezenmeier H, Bacigalupo A, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11):1666-73. doi: 10.3324/haematol.2012.062828 PubMed
10.Hoffmann-La Roche Limited. Clinical Study Protocol: Study BO42161, A Phase III, Randomized, Open-Label, Active- Controlled, Multicenter Study Evaluating the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of Crovalimab versus Eculizumab in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) Currently Treated with Complement Inhibitors. Report No. 1109894. Version 6 [internal sponsor's report]. May 9, 2023.
11.Alexion Pharma GmbH. Eculizumab SOLIRIS® [product monograph] [sponsor supplied reference]. 2021.
12.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
13.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
14.Cella D, Johansson P, Ueda Y, et al. Clinically important change for the FACIT-Fatigue scale in paroxysmal nocturnal hemoglobinuria: a derivation from international PNH registry patient data. J Patient Rep Outcomes. 2023;7(1):63. doi: 10.1186/s41687-023-00609-4 PubMed
15.Hoffmann-La Roche Limited. Clinical Study Report: Primary CSR - Study BO42162 (COMMODORE 2), A Phase III, Randomized, Open-Label, Active-Controlled, Multicenter Study Evaluating The Efficacy And Safety Of Crovalimab Versus Eculizumab In Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH) Not Previously Treated With Complement Inhibitors. Report No. 1109893 [internal sponsor's report]. April 2023.
16.Hoffmann-La Roche Limited. Hoffmann-La Roche Limited response to November 1, 2024 CADTH request for additional information regarding crovalimab CADTH review [internal additional sponsor's information]. November 8, 2024.
17.Hoffmann-La Roche Limited. Clinical Study Report: Primary CSR - Study BO42161, A Phase III, Randomized, Open-Label, Active- Controlled, Multicenter Study Evaluating the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of Crovalimab versus Eculizumab in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) Currently Treated with Complement Inhibitors. Report No. 1109894 [internal sponsor's report]. April 2023.
18.Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3:17028. doi: 10.1038/nrdp.2017.28 PubMed
19.Hill A, Platts PJ, Smith A, et al. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood. 2006;108:985. doi: 10.1182/blood.V108.11.985.985
20.Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-709. doi: 10.1182/blood-2005-04-1717 PubMed
21.Devos T, Meers S, Boeckx N, et al. Diagnosis and management of PNH: Review and recommendations from a Belgian expert panel. Eur J Haematol. 2018;101(6):737-749. doi: 10.1111/ejh.13166 PubMed
22.Kulasekararaj AG, Brodsky RA, Nishimura JI, Patriquin CJ, Schrezenmeier H. The importance of terminal complement inhibition in paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol. 2022;13:20406207221091046. doi: 10.1177/20406207221091046 PubMed
23.Hoffmann-La Roche Limited. PIASKY (crovalimab) for the treatment of patients with paroxysmal nocturnal hemoglobinuria (PNH): clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: crovalimab, 340 mg/2 mL (170 mg/mL) for injection/infusion. Oct 1, 2024.
24.CADTH Reimbursement Recommendation: Pegcetacoplan (Empaveli). Can J Health Technol. 2023;3(4). doi:10.51731/cjht.2023.614
25.Swedish Orphan Biovitrium A. B. EMPAVELI™ [product monograph] [sponsor supplied reference]. 2022.
26.Health Canada. Summary Basis of Decision for Empaveli. 2024. Accessed July 17, 2024. https://dhpp.hpfb-dgpsa.ca/review-documents/resource/SBD1682951781384
27.European Medicines Agency. Committee for Medicinal Products for Human Use. Assessment report: Piasky (crovalimab). 2025. Accessed February 4, 2025. https://www.ema.europa.eu/en/medicines/human/EPAR/piasky
28.Hoffmann-La Roche Limited. PIASKY (crovalimab): solution, 340 mg/2 mL (170 mg/mL) for subcutaneous injection/intravenous infusion [product monograph] [sponsor supplied reference]. 2025.
29.Alexion Pharma GmbH. Ravulizumab ULTOMIRIS® [product monograph] [sponsor supplied reference]. 2023.
30.McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734-737. doi: 10.15585/mmwr.mm6627e1 PubMed
31.CEDAC Drug Reimbursement Expert Review Committee final recommendation: aculizumab (Soliris - Alexion Pharmaceuticals, Inc.). CADTH; 2010. Accessed November 22, 2024. https://www.cda-amc.ca/sites/default/files/cdr/complete/cdr_complete_Soliris_February_18_2010.pdf
32.CADTH Reimbursement Recommendation: Ravulizumab (Ultomiris). Can J Health Technol. 2022;2(3). doi:10.51731/cjht.2022.278
33.Roth A, He G, Tong H, et al. Phase 3 randomized COMMODORE 2 trial: Crovalimab versus eculizumab in patients with paroxysmal nocturnal hemoglobinuria naive to complement inhibition. Am J Hematol. 2024;99(9):1768-1777. doi: 10.1002/ajh.27412 PubMed
34.Scheinberg P, Cle DV, Kim JS, et al. Phase 3 randomized COMMODORE 1 trial: Crovalimab versus eculizumab in complement inhibitor-experienced patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2024;99(9):1757-1767. doi: 10.1002/ajh.27413 PubMed
35.Hoffmann-La Roche Limited. Clinical Study Protocol: Study BO42162 (COMMODORE 2), A Phase III, Randomized, Open-Label, Active-Controlled, Multicenter Study Evaluating The Efficacy And Safety Of Crovalimab Versus Eculizumab In Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH) Not Previously Treated With Complement Inhibitors. Report Number: 1109893. Version 6 [internal sponsor's report]. May 9, 2023.
36.Jang JH, Kim JS, Yoon SS, et al. Predictive Factors of Mortality in Population of Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH): Results from a Korean PNH Registry. J Korean Med Sci. 2016;31(2):214-21. doi: 10.3346/jkms.2016.31.2.214 PubMed
37.Lee JW, Jang JH, Kim JS, et al. Uncontrolled Complement Activation and the Resulting Chronic Hemolysis As Measured by LDH Serum Level At Diagnosis As Predictor of Thrombotic Complications and Mortality in a Large Cohort of Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH). Blood. 2011;118(21):3166-3166. doi: 10.1182/blood.V118.21.3166.3166
38.Lee JW, Jang JH, Kim JS, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749-57. doi: 10.1007/s12185-013-1346-4 PubMed
39.Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99(5):922-9. doi: 10.3324/haematol.2013.093161 PubMed
40.Lai JS, Beaumont JL, Ogale S, Brunetta P, Cella D. Validation of the functional assessment of chronic illness therapy-fatigue scale in patients with moderately to severely active systemic lupus erythematosus, participating in a clinical trial. J Rheumatol. 2011;38(4):672-9. doi: 10.3899/jrheum.100799 PubMed
41.Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74. doi: 10.1016/s0885-3924(96)00274-6 PubMed
42.Weitz I, Meyers G, Lamy T, et al. Cross-sectional validation study of patient-reported outcomes in patients with paroxysmal nocturnal haemoglobinuria. Intern Med J. 2013;43(3):298-307. doi: 10.1111/j.1445-5994.2012.02924.x PubMed
43.Cella D, Lai JS, Chang CH, Peterman A, Slavin M. Fatigue in cancer patients compared with fatigue in the general United States population. Cancer. 2002;94(2):528-38. doi: 10.1002/cncr.10245 PubMed
44.Cella D, Johansson P, Ueda Y, et al. Clinically Important Difference for the FACIT-Fatigue Scale in Paroxysmal Nocturnal Hemoglobinuria: A Derivation from International PNH Registry Patient Data. Blood. 2021;138(Supplement 1):1952-1952. doi: 10.1182/blood-2021-153127
45.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLQ-C30 Scoring Manual (3rd Edition) [sponsor supplied reference]. European Organisation for Research and Treatment of Cancer. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf
46.Yenerel MN, Muus P, Wilson A, Szer J. Clinical course and disease burden in patients with paroxysmal nocturnal hemoglobinuria by hemolytic status. Blood Cells Mol Dis. 2017;65:29-34. doi: 10.1016/j.bcmd.2017.03.013 PubMed
47.Panepinto JA, Torres S, Bendo CB, et al. PedsQL Multidimensional Fatigue Scale in sickle cell disease: feasibility, reliability, and validity. Pediatr Blood Cancer. 2014;61(1):171-7. doi: 10.1002/pbc.24776 PubMed
48.Varni JW, Burwinkle TM, Katz ER, Meeske K, Dickinson P. The PedsQL in pediatric cancer: reliability and validity of the Pediatric Quality of Life Inventory Generic Core Scales, Multidimensional Fatigue Scale, and Cancer Module. Cancer. 2002;94(7):2090-106. doi: 10.1002/cncr.10428 PubMed
49.Varni JW, Burwinkle TM, Szer IS. The PedsQL Multidimensional Fatigue Scale in pediatric rheumatology: reliability and validity. J Rheumatol. 2004;31(12):2494-500. PubMed
50.Varni JW, Limbers CA, Bryant WP, Wilson DP. The PedsQL Multidimensional Fatigue Scale in type 1 diabetes: feasibility, reliability, and validity. Pediatr Diabetes. 2009;10(5):321-8. doi: 10.1111/j.1399-5448.2008.00482.x PubMed
51.Varni JW, Limbers CA, Burwinkle TM. How young can children reliably and validly self-report their health-related quality of life?: an analysis of 8,591 children across age subgroups with the PedsQL 4.0 Generic Core Scales. Health Qual Life Outcomes. 2007;5:1. doi: 10.1186/1477-7525-5-1 PubMed
52.Varni JW, Seid M, Kurtin PS. PedsQL 4.0: reliability and validity of the Pediatric Quality of Life Inventory version 4.0 generic core scales in healthy and patient populations. Med Care. 2001;39(8):800-12. doi: 10.1097/00005650-200108000-00006 PubMed
53.Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) Scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-9. PubMed
54.Cella D, Nowinski CJ. Measuring quality of life in chronic illness: the functional assessment of chronic illness therapy measurement system. Arch Phys Med Rehabil. 2002;83(12 Suppl 2):S10-7. doi: 10.1053/apmr.2002.36959 PubMed
55.Cella D, Sarda SP, Hsieh R, et al. Changes in hemoglobin and clinical outcomes drive improvements in fatigue, quality of life, and physical function in patients with paroxysmal nocturnal hemoglobinuria: post hoc analyses from the phase III PEGASUS study. Ann Hematol. 2022;101(9):1905-1914. doi: 10.1007/s00277-022-04887-8 PubMed
56.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-76. doi: 10.1093/jnci/85.5.365 PubMed
57.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the Significance of Changes in Health-Related Quality-of-Life Scores. J Clin Oncol. 2023;41(35):5345-5350. doi: 10.1200/JCO.22.02776 PubMed
58.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-21. doi: 10.1016/j.ejca.2012.02.059 PubMed
59.Piccinin C, Kuliś D, Bottomley A, et al. EORTC Quality Of Life Group Item Library User Guidelines, First Edition. EORTC; 2022. Accessed November 22, 2024. https://qol.eortc.org/manual/eortc-item-library-user-guidelines
60.Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530-539. doi: 10.1182/blood-2018-09-876136 PubMed
61.Ng TH. Noninferiority hypotheses and choice of noninferiority margin. Stat Med. 2008;27(26):5392-406. doi: 10.1002/sim.3367 PubMed
62.Yan X, Su XG. Stratified Wilson and Newcombe Confidence Intervals for Multiple Binomial Proportions. Stat Biopharm Res. 2010;2(3):329-335. doi: 10.1198/sbr.2009.0049
63.Agresti A. Categorical Data Analysis, 3rd edition [accessed by sponsor]. John Wiley & Sons; 2013.
64.Ratitch, O’Kelly. Clinical Trials with Missing Data: A Guide for Practitioners [sponsor supplied reference]. 2014.
65.Nolte S, Liegl G, Petersen MA, et al. General population normative data for the EORTC QLQ-C30 health-related quality of life questionnaire based on 15,386 persons across 13 European countries, Canada and the United States. Eur J Cancer. 2019;107:153-163. doi: 10.1016/j.ejca.2018.11.024 PubMed
66.Scott N, Fayers P, Aaronson N, et al. EORTC QLQ-C30 Reference Values Manual. 2nd ed. Brussels, Belgium: EORTC Quality of Life Group, 2008; p 427 [sponsor supplied reference].
67.Chapter 8: Assessing risk of bias in included studies. In: Julian PT Higgins J, Altman D, eds. Cochrane Handbook for Systematic Reviews of Interventions. Version 5.0.2 [updated September 2009]. Accessed November 22, 2024. https://handbook-5-1.cochrane.org/v5.0.2/index.htm#chapter_8/8_14_1_2_early_stopping.htm
68.Bührer C. Network meta-analysis of treatments in paroxysmal nocturnal hemoglobinuria (PNH) [sponsor supplied reference]. 2023.
69.Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta-analysis, using empirical data from the Cochrane Database of Systematic Reviews. Int J Epidemiol. 2012;41(3):818-27. doi: 10.1093/ije/dys041 PubMed
70.Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-43. doi: 10.1056/NEJMoa061648 PubMed
71.Jang JH, Gomez RD, Bumbea H, et al. A phase III, randomised, double-blind, multi-national clinical trial comparing SB12 (proposed eculizumab biosimilar) and reference eculizumab in patients with paroxysmal nocturnal haemoglobinuria. EJHaem. 2023;4(1):26-36. doi: 10.1002/jha2.632 PubMed
72.Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540-549. doi: 10.1182/blood-2018-09-876805 PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Additional Efficacy Results From COMMODORE 2 and COMMODORE 1 Studies
Figure 12: COMMODORE 2 Study — Graphical Representation of Coprimary and Secondary Efficacy End Point Results (Primary Analysis Population)
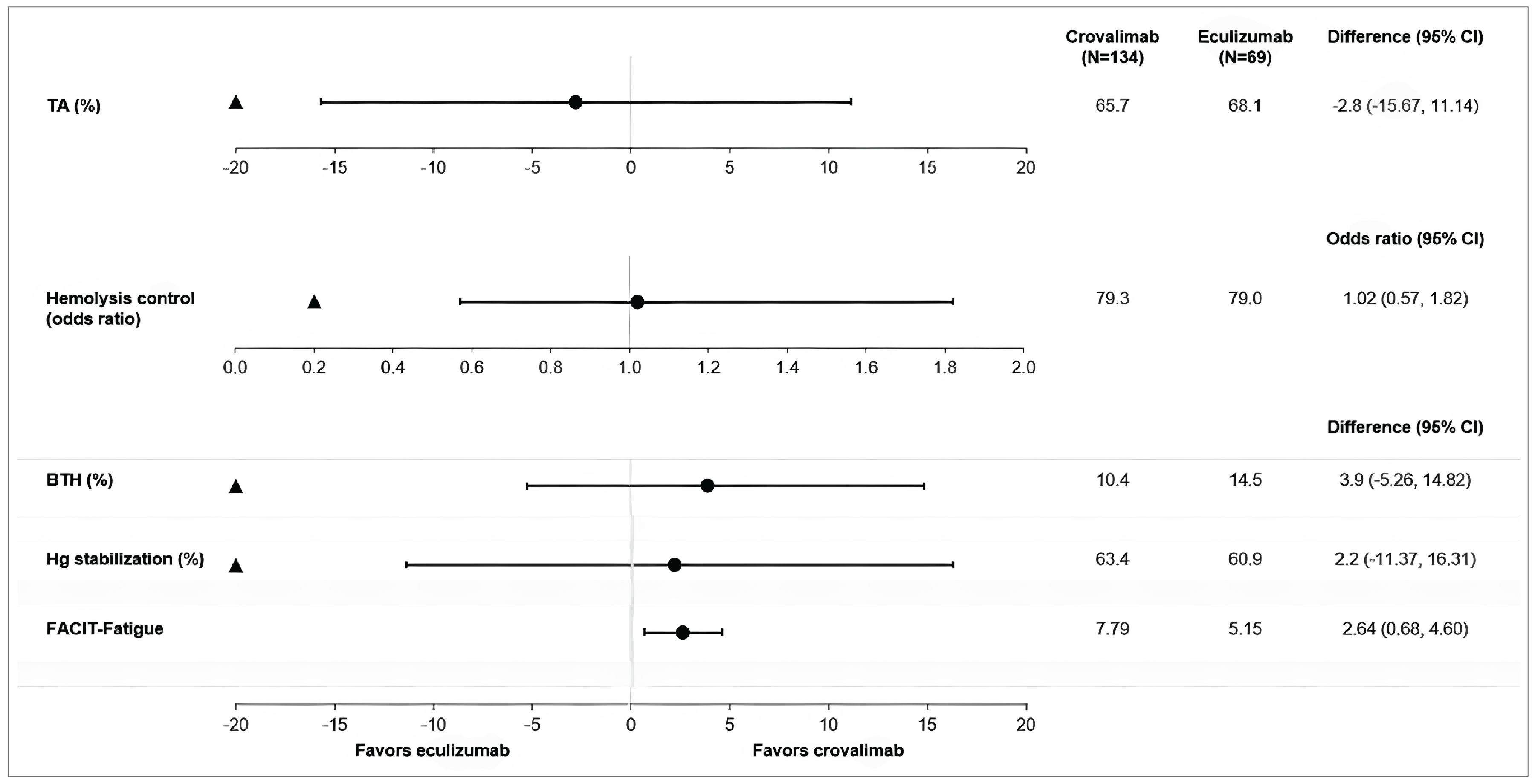
BTH = breakthrough hemolysis; CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hg = hemoglobin; TA = transfusion avoidance.
Note: Data shown in this figure were based on analyses at clinical cut-off date of November 16, 2022. The black triangles indicate the predefined NIMs. Black dots indicate point estimates, and the lines indicate the respective 95% CIs.
Source: Sponsor’s submission.23
Table 34: COMMODORE 2 Study — Proportion of Patients Who Achieved a 5-Point or Greater Improvement From Baseline in FACIT-F Scores by Visit (Primary Analysis Population)
Outcome | Arm A: Crovalimab (N = 134) | Arm B: Eculizumab (N = 69) |
|---|---|---|
Baseline | ||
Patients at visit, n | 134 | 67 |
Week 2 | ||
Patients at visit, n | 132 | 67 |
Patients who achieved a ≥ 5-point improvement from baseline, n (%) | 55 (41.7) | 27 (40.3) |
95% CI | 33.3, 50.6 | 28.7, 53.0 |
Week 5 | ||
Patients at visit, n | 130 | 66 |
Patients who achieved a ≥ 5-point improvement from baseline, n (%) | 62 (47.7) | 33 (50.0) |
95% CI | 38.9, 56.6 | 37.6, 62.4 |
Week 9 | ||
Patients at visit, n | 132 | 66 |
Patients who achieved a ≥ 5-point improvement from baseline, n (%) | 68 (51.5) | 37 (56.1) |
95% CI | 42.7, 60.2 | 43.4, 68.1 |
Week 17 | ||
Patients at visit, n | 129 | 66 |
Patients who achieved a ≥ 5-point improvement from baseline, n (%) | 74 (57.4) | 31 (47.0) |
95% CI | 48.4, 65.9 | 34.7, 59.6 |
Week 25 | ||
Patients at visit, n | 128 | 66 |
Patients who achieved a ≥ 5-point improvement from baseline, n (%) | 75 (58.6) | 36 (54.5) |
95% CI | 49.5, 67.1 | 41.9, 66.7 |
CI = confidence interval; FACIT–Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue.
Note: Data presented in this table were based on analyses at clinical cut-off date of November 16, 2022. The FACIT-F consists of 13 items that assess fatigue using a 7-day recall period. Items are scored on a response scale that ranges from 0 (“not at all”) to 4 (“very much so”). Relevant items are reverse-scored, and all items are summed to create a total score, with a higher score indicative of better functioning (i.e., less fatigue). The total score ranges from 0 to 52.
Source: COMMODORE 2 Primary Clinical Study Report.15
Figure 13: COMMODORE 2 Study — Proportion of Patients (95% CI) Achieving Hemolysis Control (Central LDH ≤ 1.5 × ULN) by Visit (Crovalimab Efficacy Population, Arm B Switch)
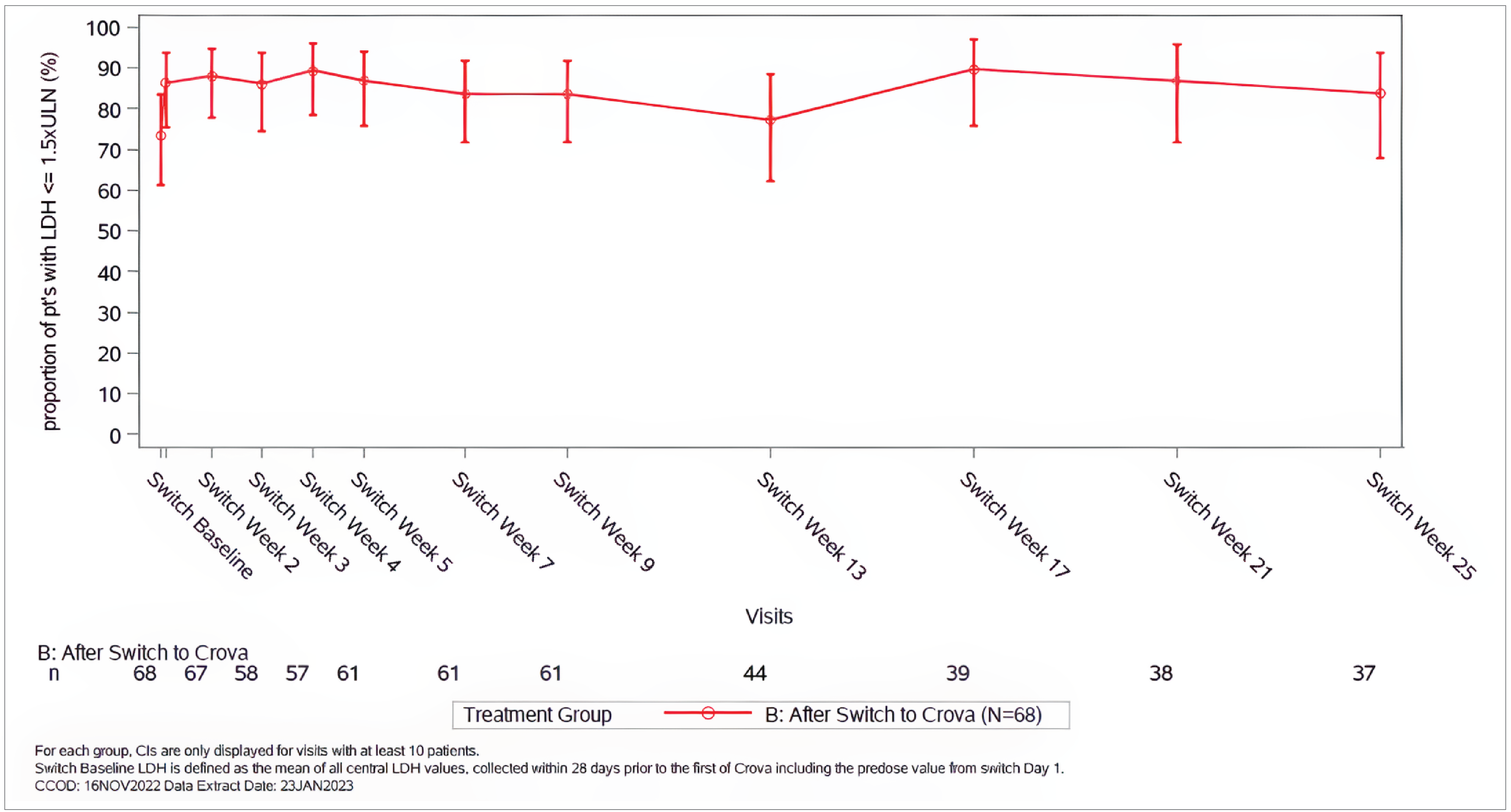
CI = confidence interval; Crova = crovalimab; LDH = lactate dehydrogenase; pt = patient; ULN = upper limit of normal.
Note: Data shown in this figure were based on analyses at clinical cut-off date of November 16, 2022. For each group, CIs are displayed only for visits with at least 10 patients. Switch baseline LDH was defined as the mean of all central LDH values, collected within 28 days before the first dose of crovalimab including the predose value from switch day 1.
Source: COMMODORE 2 Primary Clinical Study Report.15
Table 35: COMMODORE 2 Study — Summary of Absolute Scores and Change From Baseline to Week 25, EORTC IL-40 Symptoms (Primary Analysis Population)
Disease-related symptom | Arm A: Crovalimab (n = 134) | Arm B: Eculizumab (n = 69) | ||
|---|---|---|---|---|
Value at visit | Absolute change from baseline | Value at visit | Absolute change from baseline | |
Dyspnea | ||||
Baseline, n | 134 | NA | 67 | NA |
Mean (SD) | 30.60 (20.59) | NA | 30.51 (23.16) | NA |
95% CI | 27.08, 34.12 | NA | 24.87, 36.16 | NA |
Week 25, n | 128 | 128 | 66 | 66 |
Mean (SD) | 17.97 (15.53) | −13.37 (19.88) | 16.16 (15.12) | −14.81 (20.66) |
95% CI | 15.25, 20.69 | −16.85, −9.89 | 12.44, 19.88 | −19.89, −9.74 |
Dysphagia | ||||
Baseline, n | 134 | NA | 67 | NA |
Mean (SD) | 7.21 (17.03) | NA | 10.94 (23.49) | NA |
95% CI | 4.30, 10.12 | NA | 5.22, 16.67 | NA |
Week 25, n | 128 | 128 | 66 | 66 |
Mean (SD) | 2.86 (11.85) | −4.43 (18.88) | 5.05 (12.04) | −6.06 (24.74) |
95% CI | 0.79, 4.94 | −7.73, −1.12 | 2.09, 8.01 | −12.14, 0.02 |
Chest pain | ||||
Baseline, n | 134 | NA | 67 | NA |
Mean (SD) | 8.71 (17.31) | NA | 13.43 (23.25) | NA |
95% CI | 5.75, 11.66 | NA | 7.76, 19.10 | NA |
Week 25, n | 128 | 128 | 66 | 66 |
Mean (SD) | 4.43 (12.80) | −4.69 (17.11) | 5.56 (13.82) | −8.08 (21.13) |
95% CI | 2.19, 6.67 | −7.68, −1.69 | 2.16, 8.95 | −13.28, −2.89 |
Headaches | ||||
Baseline, n | 134 | NA | 67 | NA |
Mean (SD) | 17.66 (24.07) | NA | 15.92 (20.40) | NA |
95% CI | 13.55, 21.77 | NA | 10.94, 20.89 | NA |
Week 25, n | 128 | 128 | 66 | 66 |
Mean (SD) | 11.46 (19.37) | −6.77 (25.22) | 11.11 (15.83) | −4.55 (18.38) |
95% CI | 8.07, 14.84 | −11.18, −2.36 | 7.22, 15.00 | −9.06, −0.03 |
Abdominal pain | ||||
Baseline, n | 134 | NA | 67 | NA |
Mean (SD) | 12.93 (21.98) | NA | 14.93 (26.13) | NA |
95% CI | 9.18, 16.69 | NA | 8.55, 21.30 | NA |
Week 25, n | 128 | 128 | 66 | 66 |
Mean (SD) | 4.17 (11.07) | −8.85, (22.71) | 8.08 (15.54) | −7.07 (27.12) |
95% CI | 2.23, 6.10 | −12.83, −4.88 | 4.26, 11.90 | −13.74, −0.40 |
Erectile dysfunction | ||||
Baseline, n | 68 | NA | 31 | NA |
Mean (SD) | 30.88 (33.74) | NA | 30.11 (30.25) | NA |
95% CI | 22.72, 39.05 | NA | 19.01, 41.20 | NA |
Week 25, n | 73 | 65 | 34 | 30 |
Mean (SD) | 14.16 (27.17) | −17.95 (26.40) | 18.63 (24.88) | −10.00 (24.99) |
95% CI | 7.82, 20.49 | −24.49, −11.41 | 9.95, 27.31 | −19.33, −0.67 |
CI = confidence interval; EORTC IL-40 = European Organisation for Research and Treatment of Cancer – Item Library 40; NA = not applicable; SD = standard deviation.
Note: Data presented in this table were based on analyses at clinical cut-off date of November 16, 2022. EORTC IL-40 items were assessed in adult patients only. Eight items from the EORTC Item Library were used to assess 6 disease-related symptoms that are relevant to patients with PNH, including dyspnea, dysphagia, chest pain, headaches, abdominal pain, and erectile dysfunction. All items were scored on a 4-point scale that ranges from “not at all” to “very much.” Scores were transformed to a 0 to 100 scale in accordance with EORTC scoring guidelines, with higher scores indicative of higher symptom severity.
Source: COMMODORE 2 Primary Clinical Study Report.15
Figure 14: COMMODORE 1 Study — Mean Percentage Change in Central LDH Levels From Baseline to Average of Week 21, Week 23, and Week 25 (24-Week Efficacy Population)
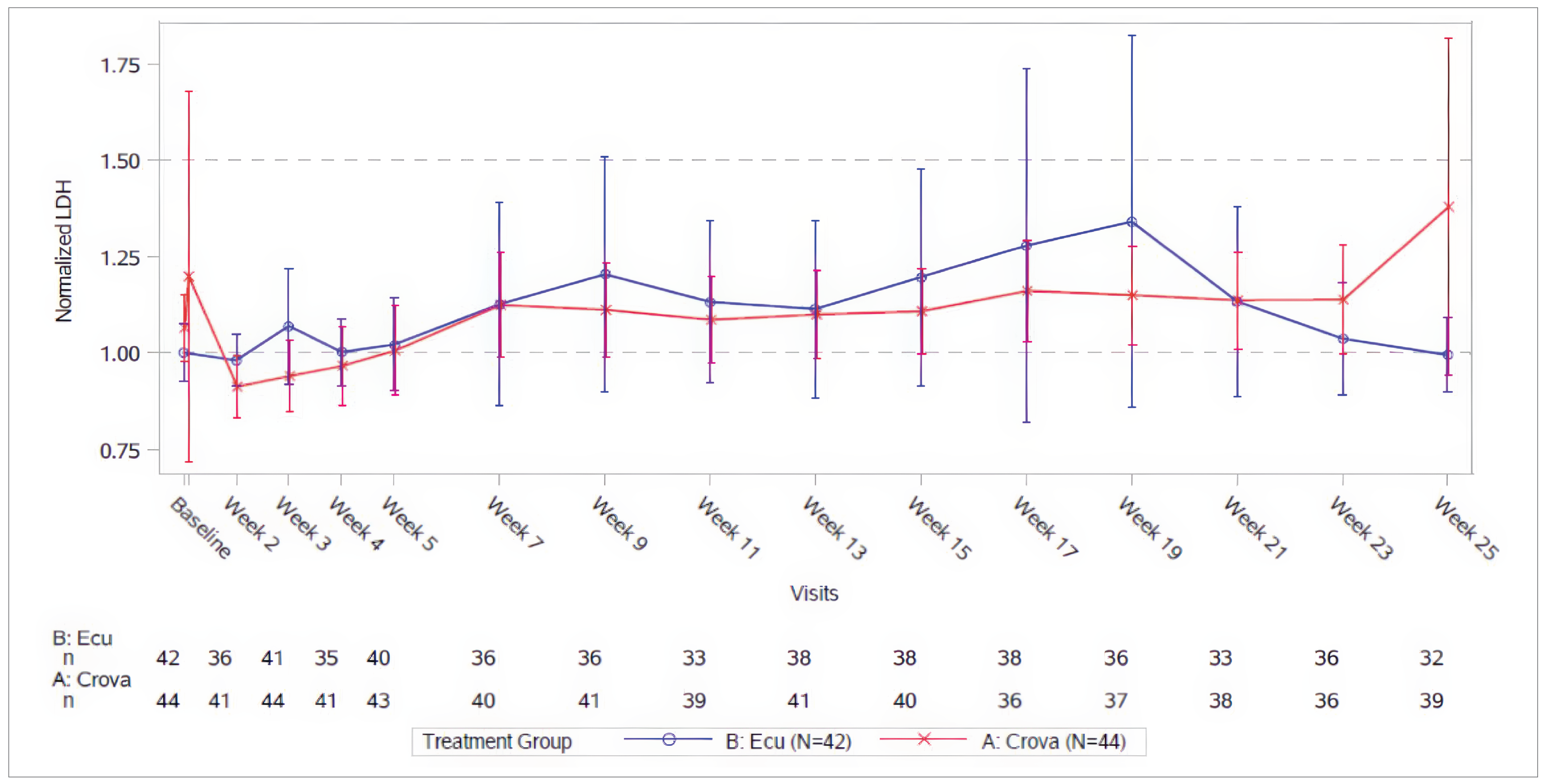
CCOD = clinical cut-off date; CI = confidence interval; Crova = crovalimab; Ecu = eculizumab; LDH = lactate dehydrogenase; ULN = upper limit of normal.
Note: Data shown in this figure were based on analyses at CCOD of November 16, 2022. For each group, CIs are only displayed for visits with at least 10 patients.
Source: COMMODORE 1 Primary Clinical Study Report.17
Figure 15: COMMODORE 2 Study — FACIT-F Scores Through Week 25 by Visit (Primary Analysis Population)
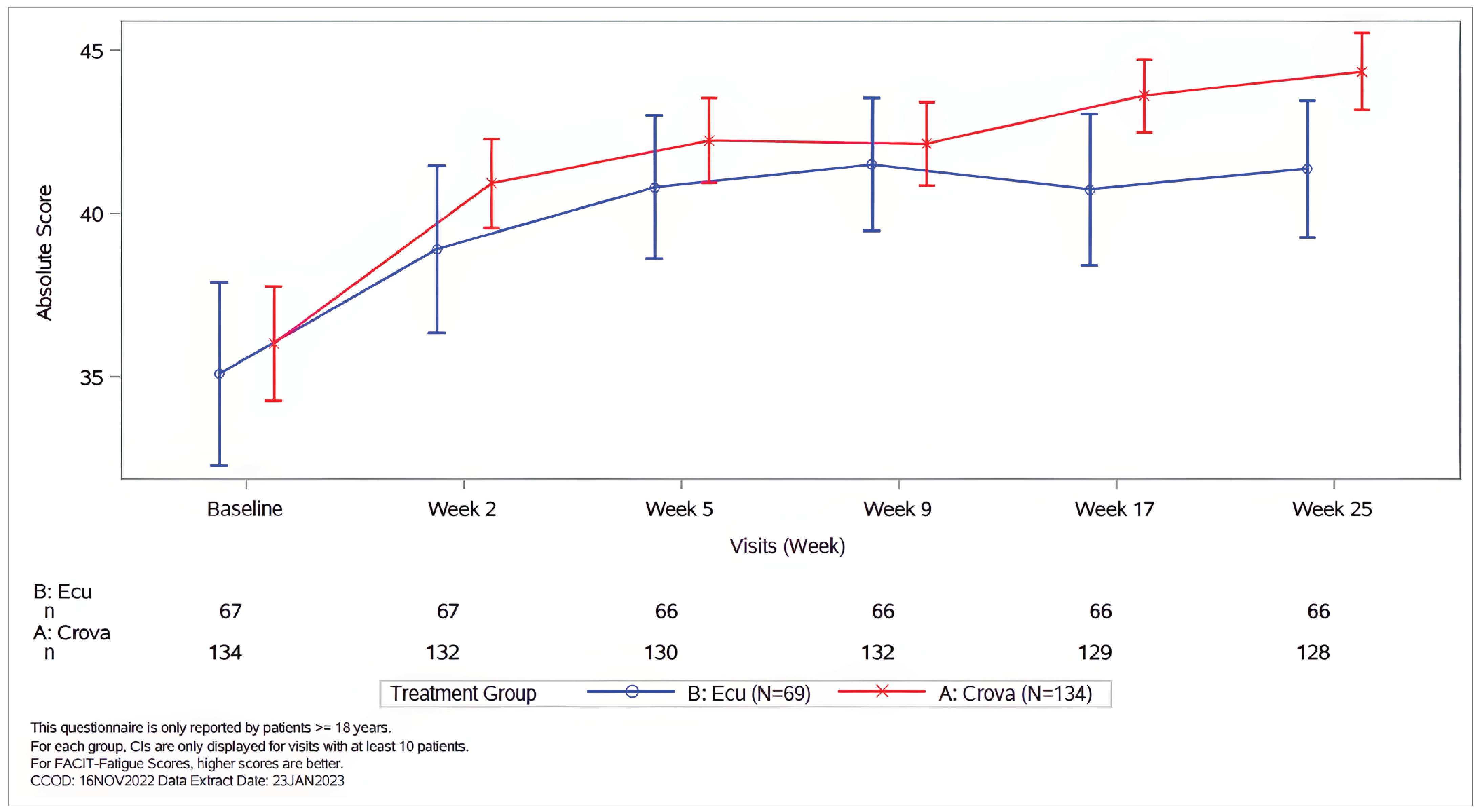
CI = confidence interval; Crova = crovalimab; Ecu = eculizumab; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue.
Note: Data shown in this figure were based on analyses at clinical cut-off date of November 16, 2022. For each group, CIs are only displayed for visits with at least 10 patients.
Source: COMMODORE 2 Primary Clinical Study Report.15
Table 36: COMMODORE 1 Study — Summary of Absolute Scores and Change From Baseline to Week 25, EORTC IL-40 Symptoms (24-Week Efficacy Population)
Disease-related symptom | Arm A: Crovalimab (N = 44) | Arm B: Eculizumab (N = 42) | ||
|---|---|---|---|---|
Value at visit | Absolute change from baseline | Value at visit | Absolute change from baseline | |
Dyspnea | ||||
Baseline, n | 44 | NA | 42 | NA |
Mean (SD) | 16.9 (18.3) | NA | 20.9 (21.0) | NA |
95% CI | 11.35, 22.49 | NA | 14.36, 27.44 | NA |
Week 25, n | 38 | 38 | 32 | 32 |
Mean (SD) | 19.3 (22.0) | 3.22 (19.66) | 24.0 (22.2) | −0.4 (14.0) |
95% CI | 12.06, 26.54 | −3.25, 9.68 | 15.94, 31.98 | −5.38, 4.69 |
Dysphagia | ||||
Baseline, n | 44 | NA | 42 | NA |
Mean (SD) | 5.3 (14.3) | NA | 8.7 (22.2) | NA |
95% CI | 0.96, 9.64 | NA | 1.82, 15.64 | NA |
Week 25, n | 38 | 38 | 32 | 32 |
Mean (SD) | 4.4 (11.4) | 0.9 (12.2) | 6.3 (15.7) | −4.2 (14.0) |
95% CI | 0.63, 8.14 | −3.14, 4.89 | 0.59, 11.91 | −9.23, 0.90 |
Chest pain | ||||
Baseline, n | 44 | NA | 42 | NA |
Mean (SD) | 7.6 (22.6) | NA | 4.8 (11.8) | NA |
95% CI | 0.71, 14.44 | NA | 1.08, 8.44 | NA |
Week 25, n | 38 | 38 | 32 | 32 |
Mean (SD) | 3.5 (10.4) | 0 (7.8) | 6.3 (15.7) | 0 (14.7) |
95% CI | 0.10, 6.92 | −2.55, 2.55 | 0.59, 11.91 | −5.29, 5.29 |
Headaches | ||||
Baseline, n | 44 | NA | 42 | NA |
Mean (SD) | 20.5 (24.1) | NA | 18.3 (24.6) | NA |
95% CI | 13.13, 27.77 | NA | 10.57, 25.93 | NA |
Week 25, n | 38 | 38 | 32 | 32 |
Mean (SD) | 17.5 (20.1) | −1.8 (23.2) | 21.9 (21.8) | −1.0 (28.7) |
95% CI | 10.93, 24.15 | −9.37, 5.86 | 14.03, 29.72 | −11.39, 9.30 |
Abdominal pain | ||||
Baseline, n | 44 | NA | 42 | NA |
Mean (SD) | 11.4 (20.3) | NA | 7.9 (14.4) | NA |
95% CI | 5.20, 17.52 | NA | 3.46, 12.41 | NA |
Week 25, n | 38 | 38 | 32 | 32 |
Mean (SD) | 9.7 (20.4) | −0.9 (21.2) | 6.3 (12.2) | −2.1 (18.8) |
95% CI | 2.95, 16.34 | −7.85, 6.09 | 1.48, 11.01 | −8.87, 4.70 |
Erectile dysfunction | ||||
Baseline, n | 16 | NA | 19 | NA |
Mean (SD) | 16.7 (21.1) | NA | 24.6 (29.1) | NA |
95% CI | 5.43, 27.90 | NA | 10.55, 38.57 | NA |
Week 25, n | 19 | 15 | 15 | 14 |
Mean (SD) | 22.8 (31.5) | 6.7 (38.2) | 35.6 (38.8) | 7.1 (32.5) |
95% CI | 7.61, 38.00 | −14.50, 27.83 | 14.09, 57.02 | −11.62, 25.91 |
CI = confidence interval; EORTC IL-40 = European Organisation for Research and Treatment of Cancer – Item Library 40; NA = not applicable; SD = standard deviation.
Note: Data presented in this table were based on analyses at clinical cut-off date of November 16, 2022. EORTC IL-40 items were assessed in adult patients only. Eight items from the EORTC Item Library were used to assess 6 disease-related symptoms that are relevant to patients with PNH, including dyspnea, dysphagia, chest pain, headaches, abdominal pain, and erectile dysfunction. All items were scored on a 4-point scale that ranges from “not at all” to “very much.” Scores were transformed to a 0 to 100 scale in accordance with EORTC scoring guidelines, with higher scores indicative of higher symptom severity.
Source: COMMODORE 1 Primary Clinical Study.17
Figure 16: COMMODORE 1 Study — FACIT-F Scores Through Week 25 by Visit (24-Week Efficacy Population)
CI = confidence interval; Crova = crovalimab; Ecu = eculizumab; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue.
Note: Data shown in this figure were based on analyses at clinical cut-off date of November 16, 2022. For each group, CIs are only displayed for visits with at least 10 patients.
Source: COMMODORE 1 Primary Clinical Study Report.17
Figure 17: COMMODORE 2 Study — Plot of Individual Patient Profiles, Normalized LDH (Efficacy-Evaluable Population, Arm C [the Pediatric Patients])
LDH = lactate dehydrogenase; ULN = upper limit of normal.
Note: Data shown in this figure were based on analyses at clinical cut-off date of November 16, 2022.
Source: COMMODORE 2 Primary Clinical Study Report.15
Pooled Safety Data From COMMODORE 1, COMMODORE 2, and COMMODORE 3 Studies
Safety Evaluation Plan
██████ ██████████ ███████████ ██████ ████ ████ ████ █████████ ███ █ ████████ █████ ██████████ ████ █████████ ██ █████████ ██ ███ █████████ █ ██ ██ █████ ███████ ████ ████████ ███ ███ █████ ██████████ ██████████ ██ █ ████ ███ ███ █████ ██████████ ██████████ ██ █ █████ ██ ████ ██ ███████ ███████ ██ █████████ ███████ ██████████ █████████ █████████ ███████ ██████████ █████ ██ █ ████ ███ ██████████ ██████ ██ █ ███████ ███████████ ██ █ ███████████████ █████████ █████████ ██ █████████ █ ████ █ ███ █ ████ ██████████ ███ █████████ █ ███ █████████████████████ ██████████ █████████ ██ █████████ █ ███ ████████ ███ ██████████ ███ █████████ █ ████ █ ███ █ ███ ██████████ ███ █████████ █ ███ ████████ ███ ████████████████ ███████████ ██ █ ███████████████ ███████ ██████ █████████ ████████████ ██████████████ ██████ █████████ ███ ████████ ████████ ██ ███ █ ██ ███████ █████████ █ ███ █████████ █ ███ ████ ███████████ ██ ████ ███ ██████████ ███ ██████████ ██████ ████████████ ██ ████ ███████ ██████████ ██████ ███ ███████ █████████ ██████ ██ ██ ██████ ████ ███ ██ ██████ ██ ██████ ██ ██████████ █████████ ██ ███ █████████ ██████████ █████████ ██ ████████ █████ ██ █ ███ █████████ ████ ██ ██████ ███ ███ ████ ██ ████████ ███████ ███ ███ ████████ ████████ ██ ████ ███████ ██ ███ ████████ ██ ████ ██ ███ ██████ ████████ ███ ██ ████ ██ ███ ██████ ██████ ██████████ ██ █ ██████████ █████ ██ ███ ██ ███ ████████ ██ ████████ ██ ███ ██████ ██ ███████████ ██████ ██████ ██████████ ███████████ ███ ████ █████████ ████████ ██ █████ ██████████ ██████████ ██████ ██████ ███ ██████ ███ █████ ██ ████████ ██ ███ █████ ██████████ ██████████ ██████ ██████ ███ ██████ ███ ██████ ██████ ██ █████ ████████████ ██ ██ ████ ██ ███ █████ ██████████ ██████████ ███ ██ ███████ █ ██ ██ ██████ ███ ██ █████ ██████████ ██████████ ███ ██ ███████ █ ██ ██ ███████ ███ ████████ ██████████ ██ ███ ██████ █████████ █████████ ███ ██ █████████ ██ ███ ████ ████ ███ █████ ██████████ ██████████ ████████ ███ ██████████ █████████ ████ ██████ ███ ███████ █████████ ██████ ██ ██ █████ ██ ███████ █████████ █ ███ █████████ █ ████████ ██ ███ ████████ ██ ███ █████ ██████████ ██████████ ███ ████████ ██████████ █████████ ██████ ███ ███████ █████████ ██████ ██ ████ ██ ██████ ███ █████████ ██████ ██ ██ ████ ██ ███ █ ███████ ███████ ███ ███ █ ██ █████████ █ ███ █████████ ███ ████ ██████████ ██ █████████ ████████ █████ ██ ██ █████ ████ █████████████ ████ ████████ █████████ ██████████ ██ █████████ █████ ███████ ███ █████ ██████████ ███ █████.
Table 37: Pooled Safety Analysis of COMMODORE 2, COMMODORE 1, and COMMODORE 3 Studies — Overview of AEs (Safety-Evaluable Population, Eculizumab and Crovalimab Total) [Redacted]
███ █████ █████ | ███ █████ █████ | ███ █████ █████ |
|---|---|---|
███ █████ ██████ | ███ █████ ██████ | ███ █████ █████ |
███ █████ █████ | ███ █████ ██████ | ███ █████ ██████ |
Note: This table has been redacted.
Source: Sponsor’s submission.23
Adverse Events
██████ ██████████ ███████████ ██████ ████ ████ ████ █████████ ███ ██████████████████ ████ █████████ ██ █████████ ██ ███ █████████ █ ██ ██ █████ ███████ ████ ████████ ███ ███ █████ ██████████ ██████████ ██ █ ████ ███ ███ █████ ██████████ ██████████ ██ █ █████ ██ ████ ██ ███████ ███████ ██ █████████ ███████ ██████████ █████████ █████████ ███████ ██████████ █████ ██ █ ████ ███ ██████████ ██████ ██ █ █████████ ███████████ ███████████ ██████████ █████████ ██ █████████ █ ████ █ ███ █ ████ ██████████ ███ █████████ ████████ ██████████ █████████ ██ █████████ █ ███ ████████ ███ ██████████ ███ █████████ █ ████ █ ███ █ ███ ██████████ ███ █████████ █ ███ ████████ ██████████████ ███████████ ██ ████████ ████████ ██████ █████████ ███████████ ███████████████ ██████ █████████ ███ ████████ ████████ ██ ███ █ ██ ███████ █████████ █ ███ █████████ █ ███ ████ ███████████ ██ ████ ███ ██████████ ███ ██████████ ██████ ████████████ ██ ████ ███████ ██████████ ██████ ███ ███████ █████████ ██████ ██ ██ ██████ ████ ███ ██ ██████ ██ ████████████████ █████ ██ ███ ██ ███ ████████ ██ ████████ ██ ███ ██████ ████████████████ ███████████████ ███ ████ █████████ ████████ ██ █████ ██████████ ██████████ ██████ ██████ ███ ██████ ███ █████ ██ ████████ ██ ███ █████ ██████████ ██████████ ██████ ██████ ███ ██████ ███ ██████.
Table 38: Pooled Safety Analysis of COMMODORE 2, COMMODORE 1, and COMMODORE 3 Studies — Summary of AEs With an Incidence Rate of 5% or More by System Organ Class (Safety-Evaluable Population, Eculizumab and Crovalimab Total) [Redacted]
███ █████ █████ | ███ █████ █████ | ███ █████ █████ |
|---|---|---|
███ █████ ██████ | ███ █████ ██████ | ███ █████ █████ |
███ █████ █████ | ███ █████ ██████ | ███ █████ ██████ |
Note: This table has been redacted.
Source: Sponsor’s submission.23
██████ ██████████ ███████████ ██████ ████ ████ ████ █████████ ███ ██████████████████ ████ █████████ ██ █████████ ██ ███ █████████ █ ██ ██ █████ ███████ ████ ████████ ███ ███ █████ ██████████ ██████████ ██ █ ████ ███ ███ █████ ██████████ ██████████ ██ █ █████ ██ ████ ██ ███████ ███████ ██ █████████ ███████ ██████████ █████████ █████████ ███████ ██████████ █████ ██ █ ████ ███ ██████████ ██████ ██ █ ██████████ ███████████ ██ ███████ █████████ █████████ ██ █████████ █ ████ █ ███ █ ████ ██████████ ███████████ █ ████████ ███████████ █████████ ██ █████████ █ ██████████ ███ ██████████ ██████████ █ ████ █ ███ █ ███ ██████████ ███ █████████ █ ███ ████████ ██████████████ ███████████ ██ █ ███████████████ █████████ ██████ █████████ ███ █████████████ ███████████████ ██████ █████████ ███ ████████ ████████ ██ ███ █ ██ ███████ █████████ █ ███ █████████ █ ███ ████ ███████████ ██ ████ ███ ██████████ ███ ██████████ ██████ ████████████ ██████ ███████ ██████████ ██████ ███ ███████ █████████ ██████ ██ ██ ██████ ████ ███ ██ ██████ ██ ██████ ██ ██████████ █████████ ██ ████████████ █████████ ██ █ ██████████ █████ ██ █ ██████████ ████ ██ ██████ ███ ███ ████ ██████████ ███████ ███ ███ ████████ ████████ ██ ████ ███████ ██ ███ ████████ ██ ████ ██ ███ ██████ ████████ ███ ██ ████ ██ ███ █████████ ██ █ ██████████ █████ ██ ███ ██ ███ ████████ ██ ████████ ██ ███ ██████ ██ █████████████ ██████ ██████ ████████████████████ ███ ████ █████████ ████████ ██ █████ ██████████ ██████████ ██████ ██████ ███ ██████ ███ █████ ██ ████████ ██ ███ ███████████████████ ██████ ██████ ███ ██████ █████████ ██████ ██ █████ ████████████ ██ ██ ████ ██ ███ █████ ██████████ ██████████ ███ ████████ █ ██ ██ ██████ ███ ██ █████ ██████████ ██████████ ███ ██ ███████ █ ██ ██ ███████ ████████ ██████████ ██ ███ ██████ █████████ █████████ ███ ██ █████████ ██ ███ ████ ████ ███ █████ ██████████ ██████████ ████████ ██████████ █████████ ████ ██████ ███ █████████████ ██████ ██ ██ █████ ██ ███████ █████████ █ ███ █████████ █ ████████ ██ ███ ████████ ██ ███ █████ ██████████ ██████████ ███ ████████ ██████████ █████████ ██████ ███████████████ ██████ ██ ████ ██ ██████ ███ █████████ ██████ ██ ██ ████ ██ ███ █ ███████ ███████ ███ ███ █ ██ █████████ █ ███ █████████ ███ ████ ██████████ ██ █████████ ████████ █████ ██ ██████ ████ █████████████ ███████ █████████ ██████████ ██ █████████ █████ ███████ ███ █████ ██████████ ███ █████.
AESI: Type III Immune Complex Reactions
Crovalimab and other C5 inhibitors bind different epitopes on C5 such that transient immune complexes consisted of the antibodies bridged by C5 may form when both are present in the circulation. These transient immune complexes, also referred to as DTDCs, can comprise 1 or more units of C5 bound to both crovalimab and to another C5 inhibitor. These transient immune complexes may be cleared after a longer duration in the case of switch from a C5 inhibitor with an extended half-life such as ravulizumab. In some patients, the formation of these complexes can result in type III immune complex–mediated reactions, also referred to as transient immune complex reactions. In general, type III immune complex reactions may manifest in skin, joints, and kidneys. In patients switching from another C5 inhibitor therapy, a transient increase in clearance is observed due to the formation of transient immune complexes, leading to a faster elimination of crovalimab. However, this transient increase in clearance is not clinically relevant and did not require dose adjustment in patients switching from another C5 inhibitor.
Up to the CCOD, 17.8% of patients in the crovalimab-switch population experienced type III immune complex reactions (Table 38). The majority of events were grade 1 to grade 2, and 7.0% of patients had grade 3 events. One (0.5%) patient also reported an AE of grade 3 axonal neuropathy. Two patients each experienced 2 events of type III immune complex reaction. Both patients experienced the first event when switching from eculizumab/ravulizumab to crovalimab in the study, and the second event after they discontinued treatment with crovalimab and switched to ravulizumab.
In the crovalimab-switch population, the most frequently reported (≥ 5% of switch patients) symptoms of type III immune complex reactions were arthralgia (9.2%) and rash (5.9%). None of the type III immune complex reactions had renal manifestations. The majority of symptoms were grade 1 to grade 2. Grade 3 symptoms were experienced in 6.5% of patients.23
In the crovalimab-switch population, the median time to onset of the first type III immune complex reaction was 1.6 weeks (range, 0.7 to 4.4 weeks) and the median duration of type III immune complex reactions was 1.86 weeks (range, 0.4 to 34.1 weeks).23
Of the 2 patients who had relatively longer type III immune complex events (≥ 12 weeks): 1 patient had signs and symptoms of rash, neck pain and pain in extremity (grade 2), which started on day 15. The rash subsided after 15 days, but neck pain and pain in extremity persisted for 239 days. The event resolved with no change in crovalimab treatment, and treatments received were paracetamol and loxoprofen.
One patient had signs and symptoms of pyrexia (grade 1) and arthralgia (grade 2) starting on day 10, and rash on legs and feet (grade 2) starting on day 13. Pyrexia and rash subsided after 6 and 10 days, respectively; however, arthralgia persisted for 89 days. The event resolved with no change in crovalimab treatment, and treatments received were levofloxacin and ketoprofen.
Median time to onset and median duration were also comparable between patients who switched from eculizumab and ravulizumab. Median time to onset for eculizumab switch patients was 1.57 weeks (range, 0.7 to 4.4 weeks) and median duration was 1.86 weeks (range, 0.4 to 34.1 weeks), whereas in ravulizumab switch patients median time to onset was 2.0 weeks (range, 1.3 to 4.3 weeks) and median duration was 1.71 weeks (range, 1.0 to 11.9 weeks).23
Other AESIs
No AESIs of abnormal liver function tests were reported. No AESIs of suspected transmission of an infectious drug by the study drug were reported.
Safety in Patients Switching from Ravulizumab Treatment
As of the CCOD, 21 ravulizumab-treated patients were enrolled in arm C of the COMMODORE 1 study. Key safety results for these patients included:
Overall, 85.7% of prior ravulizumab-treated patients experienced at least 1 AE.
The most frequently reported (≥ 10%) AEs were: pyrexia (28.6%), type III immune complex–mediated reaction (23.8%), headache (19.0%), injection-related reaction (19.0%), myalgia (19.0%), nausea (19.0%), arthralgia (14.3%), COVID-19 (14.3%), hemolysis (14.3%) and infusion-related reaction (14.3%).
Treatment-related AEs were reported in 47.6% of prior ravulizumab-treated patients. The only AEs reported as treatment-related in 10% or more of patients were type III immune complex–mediated reaction (23.8%) and injection-related reaction (19.0%).
No fatal AEs (grade 5 AEs) occurred in prior ravulizumab-treated patients. One patient discontinued from study treatment due to AE (sepsis) and 2 patients experienced an AE leading to dose modification/interruption (nasopharyngitis and infusion-related reaction). Grade 3 to grade 5 AEs were reported in 42.9% of prior ravulizumab-treated patients, with the only AE reported as grade 3 to grade 5 in more than 1 patient being type III immune complex–mediated reaction (14.3%).
SAEs were reported in 33.3% of prior ravulizumab-treated patients, with the only AE reported as an SAE in more than 1 patient being type III immune complex–mediated reaction (14.3%), all of which were considered related to treatment. The only other serious treatment-related AEs were sepsis and axonal neuropathy (which was indicated as being a type III immune complex reaction), which occurred in 1 patient each.
The 21 ravulizumab-treated patients were also included in the crovalimab-switch population of the pooled safety analysis, as it is not anticipated that the safety profile will vary depending on whether patients are switching from eculizumab or ravulizumab. Underlying AEs by preferred terms observed in these patients were consistent with the safety profile observed for the total crovalimab population, with the exception of type III immune complex reactions which was consistent with the general crovalimab-switch population.
Safety in Pediatric Patients
Of the total crovalimab population, 2.9% of patients were younger than 18 years at study enrolment.
Across the phase III studies, a total of 11 pediatric patients (9 treatment-naive and 2 switch patients with PNH) weighing 40 kg or more were treated with crovalimab up to the CCOD:
One patient in arm B (eculizumab) of the COMMODORE 2 study who received crovalimab in the extension period (switch patient)
Six treatment-naive patients in arm C of the COMMODORE 2 study
Three treatment-naive patients in the COMMODORE 3 study
One switch patient in arm C of the COMMODORE 1 study
Enrolment in arm C of the COMMODORE 1 study is still ongoing.
Of the total 11 pediatric patients, 9 (81.8%) pediatric patients reported at least 1 AE, and the majority of AEs were of grade 1 to grade 2. Two (18.2%) patients experienced AEs of grade 3 to grade 5. Five (45.5%) patients experienced AEs that were assessed as related to study treatment by the investigators. No pediatric patients reported a fatal AE, an SAE or an AE that led to drug interruption/modification or withdrawal.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BTH
breakthrough hemolysis
CDA-AMC
Canada’s Drug Agency
CMA
cost-minimization analysis
NMA
network meta-analysis
PNH
paroxysmal nocturnal hemoglobinuria
SC
subcutaneous
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Crovalimab (Piasky), solution for infusion or injection, 340 mg/2 mL vial (170 mg/mL) |
Submitted price | Crovalimab, 170 mg/mL, solution for infusion, $15,980 per 340 mg vial |
Indication | For the treatment of PNH in adults and adolescents 13 years of age and older with a body weight of at least 40 kg |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | June 4, 2025 |
Reimbursement request | As per indication |
Sponsor | Hoffmann-La Roche Limited |
Submission history | Previously reviewed: No |
CDA-AMC = Canada’s Drug Agency; NOC = Notice of Compliance; PNH = paroxysmal nocturnal hemoglobinuria.
Note: The sponsor’s application was filed on a pre-NOC basis and the pharmacoeconomic submission and budget impact analysis is reflective of the indication that was initially submitted to Health Canada and CDA-AMC. The sponsor’s submission included a broader age range and weight range than in the final indication.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Patients with PNH |
Treatment | Crovalimab |
Regimen | For patients weighing between 40 kg and less than 100 kg: One loading dose of 1,000 mg administered intravenously, followed by 4 weekly loading doses of 340 mg subcutaneously starting on day 2 of week 1. Maintenance dosing starts on day 29 and is 680 mg administered subcutaneously every 28 days. For patients weighing 100 kg or more: One loading dose of 1,500 mg administered intravenously, followed by 4 weekly doses of 340 mg subcutaneously starting on day 2 of week 1. Maintenance dosing starts on day 29 and is 1,020 mg administered subcutaneously every 28 days. |
Submitted price | Crovalimab: $15,980.00 per 340 mg vial |
Submitted treatment costs | Year 1: $544,247 per patient annually Year 2 and beyond: $424,060 per patient annually |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Time horizon | Lifetime (60 years) |
Key data source | COMMODORE 1 and COMMODORE 2 studies were phase III, randomized controlled trials assessing crovalimab in comparison to eculizumab in treatment-experienced and treatment-naive patients, supplemented by an NMA to inform efficacy of crovalimab in comparison to ravulizumab |
Costs considered | Drug acquisition, administration, blood transfusion, and medical resource usage costs (hospitalizations, dialysis treatments, and physician visits) |
Submitted results | Crovalimab was associated with cost-savings compared to eculizumab ($3,987,169 per patient) and ravulizumab ($2,468,868 per patient) over a lifetime time horizon |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; NMA = network meta-analysis; PNH = paroxysmal nocturnal hemoglobinuria.
Conclusions
According to the clinical review, in patients with paroxysmal nocturnal hemoglobinuria (PNH) who are naive to C5 inhibitors, crovalimab was noninferior to eculizumab in hemolysis control, transfusion avoidance, breakthrough hemolysis (BTH), and stabilized hemoglobin. In the C5 inhibitor–experienced population, the clinical review concluded that there may be little to no difference between crovalimab and eculizumab in terms of hemolysis control, BTH, and transfusion avoidance. The sponsor’s submitted network meta-analysis (NMA) results suggest that crovalimab may provide comparable efficacy and safety to ravulizumab in a C5 inhibitor–experienced, C5 inhibitor–naive, and mixed population. However, definitive conclusions on the relative efficacy of crovalimab and ravulizumab cannot be drawn due to limitations in the NMA (i.e., a limited number of included studies, heterogeneity in patient characteristics across trials, and credible intervals that crossed the null).
The sponsor’s cost-minimization analysis (CMA) is based on the assumption of similar clinical efficacy and safety between crovalimab and eculizumab and ravulizumab. The Canada’s Drug Agency (CDA-AMC) clinical review concluded that crovalimab demonstrated similarity versus eculizumab in C5 inhibitor–naive patients based on the COMMODORE 2 trial and that these results were supported by exploratory analyses of crovalimab versus eculizumab in the C5 inhibitor–experienced patients based on the COMMODORE 1 trial. As such, the claim of similarity between crovalimab and eculizumab may be plausible but is associated with uncertainty in the C5 inhibitor–experienced population. While the clinical review determined that the NMA evidence was insufficient to conclude whether crovalimab is similar to ravulizumab, based on clinical expert input received by CDA-AMC for this review, the claim may be plausible given ravulizumab is in the same drug class. No revisions were undertaken to the sponsor’s base case, which suggested that, at the sponsor’s submitted price for crovalimab and list prices for comparators, crovalimab was cost-saving compared with eculizumab (a cost-saving of $3,987,169 per patient) and ravulizumab (a cost-saving of $2,468,868 per patient) over a lifetime time horizon. All cost-savings associated with crovalimab are attributable to drug acquisition costs (i.e., there was no difference in blood transfusion or medical resource use, and administration costs were similar among treatments).
CDA-AMC notes that the cost-savings estimated for crovalimab are dependent on participating drug plan prices for comparators. If actual prices paid by plans for eculizumab and ravulizumab are 23% and 16% lower than list prices, respectively, crovalimab will no longer be cost-saving. Additionally, the magnitude of cost-savings are dependent on the time period over which they are used. Annual cost-savings associated with crovalimab versus ravulizumab are estimated to be $55,556 and $83,268 per year in year 1 and year 2, respectively ($41,987 and $98,402 per year in year 1 and year 2, respectively, when crovalimab is compared with eculizumab).
Economic Review
The current review is for crovalimab (Piasky) for patients with PNH.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA comparing crovalimab with eculizumab and ravulizumab.1 Crovalimab’s proposed Health Canada indication is for the treatment of patients with PNH.2 The modelled population is consistent with the COMMODORE 1 and COMMODORE 2 trials but narrower than that of the Health Canada indication, which is not restricted by granulocyte threshold.
Crovalimab is available in a 170 mg/mL 2 mL single-use vial.2 The recommended dosage of crovalimab for those weighing between 40 kg to less than 100 kg is a 1,000 mg IV loading dose on day 1, followed by 4 weekly doses of 340 mg via subcutaneous (SC) injection from day 2 of week 1, and 680 mg SC on day 29 every 4 weeks thereafter for maintenance.2 Patients weighing 100 kg or more receive a 1,500 mg loading dose on day 1, followed by 4 weekly doses of 340 mg SC on day 2 of week 1, and then 1,020 mg SC on day 29 every 4 weeks thereafter for maintenance.2 At the submitted price of $15,980 per 340 mg vial, the annual cost of crovalimab for PNH was estimated to be $544,247 and $424,060 in year 1 and in year 2 and onward, respectively, in the sponsor’s base case.1 The annual costs of eculizumab and ravulizumab in year 1 were $586,234 and $599,803, respectively. In subsequent years, the annual costs of eculizumab and ravulizumab were $522,461 and $507,328, respectively. In the sponsor’s CMA, a weight distribution of 96.6% of patients weighing between 40 kg and less than 100 kg and 3.4% of patients weighing 100 kg or more, informed based on pooled data from the COMMODORE 1 and COMMODORE 2 studies, was used to estimate drug costs by weight category to derive cost comparisons for the overall PNH population.3-5
The sponsor assumed that crovalimab has similar clinical efficacy and safety to eculizumab and ravulizumab. This was supported by data from 2 pivotal phase III, randomized controlled trials evaluating the safety and efficacy of crovalimab versus eculizumab in patients with PNH who had not been previously treated with a complement inhibitor (the COMMODORE 2 study) and patients with documented treatment with complement inhibitors (the COMMODORE 1 study).3,4 In the absence of head-to-head evidence comparing crovalimab with ravulizumab, an NMA was conducted to support the sponsor's assumption that crovalimab has similar clinical efficacy and safety to ravulizumab.6 The sponsor’s CMA model included BTH events, which were assumed to be equal across all treatments, with the probability of occurrence being sourced from a Dutch cost-effectiveness study.5 Because safety was assumed to be similar across treatments, adverse events were not included. Background mortality, informed by general population life tables, was included in the analysis and was equal across all treatments. Baseline patient characteristics in the model were based on pooled data from the COMMODORE 1 and COMMODORE 2 studies and were used to inform background mortality (average patient age = 40.97 years; female = 50%, male = 50%).3-5
The sponsor’s analysis adopted a publicly funded health care system perspective and considered a lifetime horizon (60 years), assuming a maximum age of 100 years, with costs discounted at 1.5% annually.
The model included drug acquisition, administration, blood transfusion, and health care resource costs (including hospitalizations, dialysis, and physician visits). Unit drug costs for crovalimab were provided by the sponsor, and unit costs for comparators were sourced from IQVIA DeltaPA.7 Single up-doses for both crovalimab and eculizumab were included in the model based on the occurrence of BTH events. In addition, based on the input of the sponsor’s clinical expert, it was assumed that 20% of patients receiving eculizumab required continuous up-dosing to manage BTH.1 Administration costs were calculated based on the recommended infusion duration specified in each drug’s product monograph. For ravulizumab and eculizumab, the sponsor assumed that the public payer is responsible for administration costs associated with the loading dose and first maintenance dose; thereafter, administration costs were assumed to be covered by a manufacturer patient support program.1 For crovalimab, the sponsor assumed the public payer would cover the IV administration costs associated with the first dose and the cost of the first 3 SC administrations; thereafter, crovalimab was assumed to be administered by patients, which was not associated with health care administration costs.1 Blood transfusion costs were included in the model, with rates differing for whether a BTH event occurred or not (transfusion probability per 2-week cycle of 30% during BTH events and 9% in their absence), based on data from a Dutch cost-effectiveness study.5 Costs associated with blood transfusions were sourced from a ravulizumab reimbursement review.8 The sponsor estimated medical resource use costs based on patient presentations to health care facilities during BTH events. These costs encompassed general hospital and intensive care unit stays, dialysis, and 2 annual hematologist visits, as informed by input from the sponsor’s clinical experts. The frequencies of general hospitalizations, intensive care unit visits, dialysis, and consultant visits were derived from a Dutch study, based on a patient cohort from the Netherlands.5 The costs for BTH events were informed by the Canadian Institute for Health Information Patient Cost Estimator.9 The sponsor assumed that blood transfusion and medical resource use costs were equal across all treatments and, therefore, these did not contribute to differences in costs between treatments.
A summary of the sponsor’s base case is presented in Table 3. The sponsor’s submitted base case estimated that, in patients with PNH, crovalimab would be associated with total costs of $13,290,078. Total costs for eculizumab and ravulizumab were $17,277,247 and $15,758,945, respectively. Over a lifetime time horizon, this resulted in incremental cost-savings of $3,987,169 and $2,468,868 per patient compared to eculizumab and ravulizumab, respectively. Disaggregated results of the sponsor’s analysis can be found in Table 5. Cost-savings associated with crovalimab were driven entirely by differences in drug acquisition costs.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|
Crovalimab | 13,128,441 | Reference | 13,290,078 | Reference |
Eculizumab | 17,121,148 | –3,992,707 | 17,277,247 | −3,987,169 |
Ravulizumab | 15,603,797 | −2,475,356 | 15,758,945 | −2,468,868 |
Source: Sponsor’s economic submission.1
The sponsor conducted a number of scenario analyses. Though the model was sensitive to assumptions regarding time horizon, discount rate, model starting age, continuous up-dosing parameterization, and the distribution of patient weights, in all scenarios crovalimab was associated with cost-savings compared with both eculizumab and ravulizumab.
CDA-AMC Appraisal of the Sponsor’s Economic Information
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The sponsor’s assumption of clinical equivalence between crovalimab and other C5 inhibitors is uncertain: The sponsor submitted a CMA assuming clinical similarity between crovalimab, eculizumab, and ravulizumab.1 For the comparison with eculizumab, this assumption was based on 2 phase III trials examining the efficacy and safety of crovalimab versus eculizumab.3,4 In the C5 inhibitor–naive patient population, the CDA-AMC clinical review concluded that crovalimab was noninferior to eculizumab across coprimary end points (hemolysis control and transfusion avoidance) and secondary outcomes (BTH and stabilized hemoglobin). The evidence was rated as being of high to moderate certainty, using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. In the C5 inhibitor–experienced population, the clinical review concluded that crovalimab may result in little to no difference in hemolysis control, BTH, and transfusion avoidance compared with eculizumab. The evidence in the C5 inhibitor–experienced population was rated as being of low certainty, using the GRADE approach. As such, there is high to moderate certainty that crovalimab is similar to eculizumab in the C5 inhibitor–naive population; however, conclusions in the C5 inhibitor–experienced population are uncertain.
For the comparison with ravulizumab, the assumption of clinical equivalence was based on a sponsor-submitted NMA.6 According to the CDA-AMC clinical review, the NMA results suggested that crovalimab may provide comparable efficacy and safety compared with ravulizumab. However, definitive conclusions on the comparative efficacy of ravulizumab and crovalimab cannot be drawn due to limitations in the NMA, including heterogeneity, a small number of trials in the network, and credible intervals that crossed the null. Finally, hemolysis control (a coprimary outcome of the COMMODORE 2 trial) was not reported in the ravulizumab trials and therefore was not able to be assessed in the NMA; as such, comparative evidence for this outcome for crovalimab versus ravulizumab is not available. As such, CDA-AMC was unable to conclude whether crovalimab is similar to ravulizumab.
Taken together, while there is some uncertainty regarding whether crovalimab is clinically similar to other C5 inhibitors, the conclusion that there is little to no difference between crovalimab and the 2 comparators was deemed to be plausible by clinical experts consulted by CDA-AMC given that they belong to the same group of drugs and eculizumab and ravulizumab have demonstrated similar benefits in adult patients with PNH in randomized controlled trials. If differences in crovalimab and comparators exist, a CMA would be insufficient to assess cost-effectiveness.
CDA-AMC did not address this limitation through reanalyses.
Continuous up-dosing among patients receiving eculizumab is uncertain: The sponsor assumed that 20% of patients receiving eculizumab would require continuous up-dosing to 1,200 mg to maintain disease control, which was informed by Quist et al. and validated by the sponsor’s experts.1,5 According to the CDA-AMC clinical review, the dose for patients receiving eculizumab in the COMMODORE 1 and COMMODORE 2 trials was fixed and dose changes were not allowed. Therefore, the proportion of patients requiring up-dosing could not be informed by clinical trial evidence. Clinical experts consulted by CDA-AMC noted that while a proportion of patients may require continuous up-dosing, the proportion that will receive it may vary based on differences in reimbursement for eculizumab among jurisdictions. This assumption has implications for the economic analysis because continuous up-dosing increases eculizumab treatment costs, which increases the cost-savings associated with crovalimab treatment in the sponsor’s analysis.
CDA-AMC conducted a scenario analysis to explore the impact of assuming all patients receiving eculizumab received the 900 mg dose.
Comparator pricing is based on publicly available prices: The prices used in the CMA for eculizumab and ravulizumab are based on publicly available list prices and do not account for potential confidential pricing agreements negotiated by public drug plans.7,10,11 Given that the projected cost-savings for crovalimab are based on public list prices for comparators, price reductions may be necessary for crovalimab to achieve cost-parity if the actual prices of the comparators are lower than their listed prices.
CDA-AMC conducted threshold analyses to investigate the price of eculizumab and ravulizumab that would be required for crovalimab to be cost-neutral (i.e., no longer cost-saving).
The administration costs included for treatments are uncertain: The sponsor assumed that the health system would bear 1-time administration costs for loading doses in both the crovalimab and comparator arms. Clinical experts consulted by CDA-AMC indicated that administration costs for ravulizumab and eculizumab are typically covered through manufacturer patient support programs, which apply comprehensively to both loading and maintenance doses.
Administrative costs constitute a small proportion of the total treatment costs for ravulizumab and eculizumab (approximately $25,000 or 0.26% and 0.14% of the total cost-savings, respectively) and have a negligible impact on cost differences between therapies (Table 5). As such, this limitation was not addressed by CDA-AMC in reanalyses.
CDA-AMC Reanalyses of the Economic Information
CDA-AMC did not undertake a base-case reanalysis and accepted the sponsor’s submitted base case. CDA-AMC conducted a scenario analysis to evaluate the impact of including pediatric patients on the overall results, with parameters informed by clinical expert input. In this scenario, incremental savings estimated for crovalimab were less than anticipated in the sponsor’s base case (incremental savings of $3,369,570 and $2,432,628 compared with eculizumab and ravulizumab, respectively) (Table 6). CDA-AMC also conducted a scenario assuming no up-dosing for patients treated with eculizumab. In this scenario, cost-savings estimated for crovalimab were also lower than estimated in the sponsor’s base case (incremental savings of $2,921,559 versus eculizumab) (Table 6).
Issues for Consideration
Anticipated patent expiration of eculizumab: The patent for eculizumab is expected to expire on March 15, 2027.12 If eculizumab biosimilars become available and are considered clinically equivalent to eculizumab, it is uncertain whether crovalimab will be less costly than eculizumab biosimilars. Consequently, the cost of crovalimab at the submitted price may be less attractive to drug plans.
Proposed Health Canada indication for crovalimab is broader than model population: CDA-AMC accepted a deviation request from the sponsor to focus the budget impact analysis (BIA) on patients with PNH who have a granulocyte clone size of 10% or more, which is consistent with the inclusion criteria for the COMMODORE 1 and COMMODORE 2 trials. CDA-AMC notes that the proposed Health Canada indication for crovalimab is not restricted by granulocyte count. Clinical expert input received by CDA-AMC for this review indicated that crovalimab is unlikely to be used in those with a granulocyte clone size of less than 10%.
Conclusions
According to the clinical review, in patients with PNH who are naive to C5 inhibitors, crovalimab was noninferior to eculizumab in hemolysis control, transfusion avoidance, BTH, and stabilized hemoglobin. In the C5 inhibitor–experienced population, the clinical review concluded that there may be little to no difference between crovalimab and eculizumab in terms of hemolysis control, BTH, and transfusion avoidance. The sponsor’s submitted NMA results suggest that crovalimab may provide comparable efficacy and safety to ravulizumab in a C5 inhibitor–experienced, C5 inhibitor–naive, and mixed population. However, definitive conclusions on the relative efficacy of crovalimab and ravulizumab cannot be drawn due to limitations in the NMA (i.e., a limited number of included studies, heterogeneity in patient characteristics across trials, and credible intervals that crossed the null).
The sponsor’s CMA is based on the assumption of similar clinical efficacy and safety between crovalimab and eculizumab and ravulizumab. The CDA-AMC clinical review concluded that crovalimab demonstrated similarity versus eculizumab in C5 inhibitor–naive patients based on the COMMODORE 2 trial and these results were supported by exploratory analyses of crovalimab versus eculizumab in C5 inhibitor–experienced patients based on the COMMODORE 1 trial. As such, the claim of similarity between crovalimab and eculizumab may be plausible but is associated with uncertainty in the C5 inhibitor–experienced population. While the clinical review determined that the NMA evidence was insufficient to conclude whether crovalimab is similar to ravulizumab, based on the clinical expert input received by CDA-AMC for this review, the claim may be plausible given ravulizumab is in the same drug class. No revisions were undertaken to the sponsor’s base case, which suggested that, at the sponsor’s submitted price for crovalimab and list prices for comparators, crovalimab was cost-saving compared with eculizumab (a cost-saving of $3,987,169 per patient) and ravulizumab (a cost-saving of $2,468,868 per patient) over a lifetime time horizon. All cost-savings associated with crovalimab are attributable to drug acquisition costs (i.e., there was no difference in blood transfusion or medical resource use, and administration costs were similar among treatments).
CDA-AMC notes that the cost-savings estimated for crovalimab are dependent on participating drug plan prices for comparators. If actual prices paid by plans for eculizumab and ravulizumab are 23% and 16% lower than list prices, respectively, crovalimab will no longer be cost-saving. Additionally, the magnitude of cost-savings is dependent on the time period over which they are used. Annual cost-savings associated with crovalimab versus ravulizumab are estimated to be $55,556 and $83,268 per year in year 1 and year 2, respectively ($41,987 and $98,402 per year in year 1 and year 2, respectively, compared with eculizumab).
References
1.Hoffmann-La Roche Limited. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Piasky (crovalimab injection), solution, 340 mg/2 mL (170 mg/mL) for injection/infusion. October 1, 2024.
2.Hoffmann-La Roche Limited. Piasky (crovalimab injection): solution, 340 mg/2 mL (170 mg/mL) for injection/infusion [product monograph]. March 28, 2024.
3.Hoffmann-La Roche Limited. Clinical Study Report: BO42161. Crovalimab CSR PNH Switch COMMODORE 1 [internal sponsor's report]. 2023.
4.Hoffmann-La Roche Limited. Clinical Study Report: BO42162. Crovalimab CSR PNH Naive COMMODORE 2 [internal sponsor's report]. 2023.
5.Quist SW, Postma AJ, Myren KJ, de Jong LA, Postma MJ. Cost-effectiveness of ravulizumab compared with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria in the Netherlands. Eur J Health Econ. 2023;24(9):1455-1472. doi: 10.1007/s10198-022-01556-5PubMed
6.Hoffmann-La Roche Limited. Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Piasky (crovalimab injection), solution, 340 mg/2 mL (170 mg/mL) for injection/infusion. October 1, 2024.
7.IQVIA. DeltaPA. 2023. Accessed February 24, 2025. https://www.iqvia.com/
8.CADTH Reimbursement Review: Ravulizumab (Ultomiris). Can J Health Technol. 2022;2(4). doi:10.51731/cjht.2022.319
9.Canadian Institute for Health Information. Patient cost estimator. 2024. Accessed November 21, 2024. https://www.cihi.ca/en/patient-cost-estimator
10.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: Ultomiris (ravulizumab). CADTH. Accessed January 3, 2025. https://www.pcpacanada.ca/
11.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: Soliris (eculizumab). CADTH. Accessed January 3, 2025. https://www.pcpacanada.ca/
12.Health Canada. Patent register (Ecuzlizumab). Accessed January 8, 2025. https://pr-rdb.hc-sc.gc.ca/pr-rdb/patent_result-resultat_brevet.do?action=search_recherche&formId=10092&din=02322285&drugId=2548&lang=fr&patentNumber_numeroBrevet=2645810
13.IQVIA. DeltaPA. 2024. Accessed August 16, 2024. https://www.iqvia.com/
14.Alexion Pharma GmbH. ULTOMIRIS [product monograph] [sponsor supplied reference]. 2023.
15.Hoffmann-La Roche Limited. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Piasky (crovalimab injection), solution, 340 mg/2 mL (170 mg/mL) for injection/infusion. October 1, 2024.
16.Hill A, Platts PJ, Smith A, et al. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood. 2006;108:985. doi: 10.1182/blood.V108.11.985.985
17.Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: A consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36-52. doi: 10.1111/ejh.13176 PubMed
18.IQVIA. PharmaStat. 2024. Accessed September 6, 2024. https://www.iqvia.com/
19.IQVIA. PharmaStat. 2023. Accessed January 6, 2025. https://www.iqvia.com/
20.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed January 8, 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
21.Alexion Pharma GmbH. SOLIRIS [product monograph] [sponsor supplied reference]. 2021.
22.Canadian Institute for Health Information. Pan-Canadian Prescription Drug Data Landscape. 2024. Accessed January 6, 2025. https://www.cihi.ca/sites/default/files/document/pan-canadian-prescription-drug-data-landscape-report-en.pdf
23.Liu H, Xia L, Weng J, et al. COMMODORE 3: Supporting information [sponsor supplied reference]. Am J Hematol. 2023.
Appendix 1: Additional Economic Information
Please note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CDA-AMC Cost Comparison Table for Paroxysmal Nocturnal Hemoglobinuria
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost (S) |
|---|---|---|---|---|---|---|
Crovalimab (Piasky) | 170 mg / mL | 340 mg / 2 mL Single-use vial for IV infusion or subcutaneous use | 15,980.0000 | ≥ 40 kg to < 100 kg Loading: 1,000 mg on day 1, followed by 340 mg on days 2, 8, 15, and 22 Maintenance: 680 mg on day 29 and every 4 weeks thereafter | Year 1: 1,360.18 Subsequent years: 1,141.43 | Year 1: 496,807 Subsequent years: 416,907 |
≥ 100 kg Loading: 1,500 mg on day 1, followed by 340 mg on days 2, 8, 15, and 22 Maintenance: 1,020mg on day 29 and every 4 weeks thereafter | Year 1: 1,974.65 Subsequent years: 1,712.14 | Year 1: 721,240 Subsequent years: 625,360 | ||||
Eculizumab | ||||||
Eculizumab | 10 mg / mL | 300 mg / 30 mL Single-use vial for IV infusion | 6,675.3000 | ≥ 40 kg Loading:
Maintenance: 900 in week 5 every 2 weeks thereafter | Year 1: 1,494.39 Subsequent years: 1,430.42 | Year 1: 545,825 Subsequent years: 522,461 |
Ravulizumab | ||||||
Ravulizumab | 10 mg / mL 100 mg / mL | 300 mg / 30 mL 300 mg / 3 mL 1,100 mg / 11 mL Single-use vial for IV infusion | 7,282.1500 7,282.1500 26,701.1998 | Loading dose, with maintenance doses given starting 2 weeks after, then administered every 8 weeks thereafter, based on weight as follows: ≥ 40 kg to < 60 kg Loading: 2,400 mg Maintenance: 3,000 mg | Year 1: 1,555.12a Subsequent years: 1,295.93b | Year 1: 568,008a Subsequent years: 473,340b |
≥ 60 kg to < 100 kg Loading: 2,700 mg Maintenance: 3,300 mg | Year 1: 1,714.62a Subsequent years: 1,425.53b | Year 1: 626,265a Subsequent years: 520,674b | ||||
≥ 100 kg Loading: 3,000 mg Maintenance: 3,600 mg | Year 1: 1,874.12a Subsequent years: 1,555.12b | Year 1: 684,522a Subsequent years: 568,008b | ||||
CDA-AMC = Canada’s Drug Agency.
Note: All prices are from IQVIA DeltaPA (accessed November 2024), unless otherwise indicated, and do not include dispensing fees.13
aYear 1 ravulizumab costs assume 1 loading dose and 7 maintenance doses.
bSubsequent year ravulizumab dosing are based on an average of 6.5 administrations (52/8) per year.
Additional Details on the Sponsor’s Submission
Table 5: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Cost category | Crovalimab ($) | Eculizumab ($) | Ravulizumab ($) | Incremental vs. crovalimab ($) | |
|---|---|---|---|---|---|
Eculizumab | Ravulizumab | ||||
Drug acquisition costs (total) | 13,128,441 | 17,121,148 | 15,603,797 | −3,992,707 | –2,475,356 |
Drug acquisition costs: Regular treatment | 13,085,330 | 12,830,023 | 15,603,797 | 255,307 | –2,518,467 |
Drug acquisition costs: Single up‑dosing | 43,111 | 18,009 | 0 | 25,102 | 43,111 |
Drug acquisition costs: Continuous up‑dosing | 0 | 4,273,116 | 0 | –4,273,116 | 0 |
Drug administration | 31,110 | 25,573 | 24,622 | 5,537 | 6,488 |
Blood transfusions | 121,803 | 121,803 | 121,803 | 0 | 0 |
Medical resource use | 8,723 | 8,723 | 8,723 | 0 | 0 |
Total costs | 13,290,078 | 17,277,247 | 15,758,945 | –3,987,169 | –2,468,868 |
vs. = versus.
Source: Sponsor’s economic submission.14
Additional Details on the Additional CDA-AMC Analyses
Table 6: Scenario Analyses Conducted by CDA-AMC on the Sponsor’s Base Case
Scenario analysis | Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|---|
Sponsor’s base case | Crovalimab | 13,128,441 | Reference | 13,290,078 | Reference |
Eculizumab | 17,121,148 | –3,992,707 | 17,277,247 | −3,987,169 | |
Ravulizumab | 15,603,797 | −2,475,356 | 15,758,945 | −2,468,868 | |
CDA-AMC scenario 1: No continuous up-dosing for eculizumab | Crovalimab | 13,128,441 | Reference | 13,290,078 | Reference |
Eculizumab | 16,055,537 | −2,927,096 | 16,211,636 | −2,921,559 | |
Ravulizumab | 15,603,797 | −2,475,356 | 15,758,945 | −2,468,868 | |
CDA-AMC scenario 2: Including pediatric patient weights in the model | Crovalimab | 13,105,096 | Reference | 13,266,710 | Reference |
Eculizumab | 16,480,180 | −3,375,084 | 16,636,279 | −3,369,570 | |
Ravulizumab | 15,544,276 | −2,439,180 | 15,699,338 | −2,432,628 |
CDA-AMC = Canada’s Drug Agency.
Appendix 2: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 7: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the 3-year budgetary impact of reimbursing crovalimab for patients with PNH with granulocyte of 10% or more. CDA-AMC approved a deviation request from the sponsor to focus the BIA on this population. The analysis was conducted from the perspective of the pan-Canadian public drug plans, over a 3-year time horizon (2025 to 2027).
An epidemiologic approach was used to determine the eligible population, with the analysis including both treatment-naive and experienced patients (Table 8). Reference scenario market shares for the incident population were based on a previous CADTH review and validated by the sponsor’s clinical experts.8,15 Reference scenario market shares for the prevalent population were also informed by a previous CADTH review and validated by the sponsor’s clinical experts, alongside IQVIA Pharmastat data.7,8,15 Market shares in the new drug scenario for both incident and prevalent patients were informed by the opinions of the sponsor’s clinical experts.15 Drug costs were calculated as the sum of incident and prevalent patients, multiplied by their respective loading and maintenance doses based on weight distributions from the COMMODORE 1 and COMMODORE 2 studies.3,4 Wastage was not considered for crovalimab patients because loading doses would become negligible over the extended treatment period. Key inputs to the BIA are documented in Table 8.
Table 8: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Target population | ||
Incidence of PNH | 0.13 per 100,00016 | |
Prevalence of PNH | 1.59 per 100,00016 | |
Proportion with granulocyte ≥ 10% | 53.60%17 | |
Proportion treated for PNH | 59.62%17 | |
Rate of public coverage | 65.18%18 | |
Number of patients eligible for drug under review | 125 / 126 / 127 | |
Market uptake, pan-Canadian excluding British Columbia (3 years, incident patients) | ||
Uptake (reference scenario) Eculizumab Ravulizumab | 5% / 5% / 5% 95% / 95% / 95% | |
Uptake (new drug scenario) Crovalimab Eculizumab Ravulizumab | 10% / 15% / 20% 5% / 5% / 5% 85% / 80% / 75% | |
Market uptake, pan-Canadian excluding British Columbia (3 years, prevalent patients) | ||
Uptake (reference scenario) Eculizumab Ravulizumab | 25% / 5% / 5% 75% / 95% / 95% | |
Uptake (new drug scenario) Crovalimab Eculizumab Ravulizumab | 5% / 10% / 15% 25% / 5% / 5% 70% / 85% / 80% | |
British Columbia market uptake (3 years, incident patients) | ||
Uptake (reference scenario) Eculizumab Ravulizumab | 100% / 100% / 100% 0% / 0% / 0% | |
Uptake (new drug scenario) Crovalimab Eculizumab Ravulizumab | 10% / 25% / 50% 90% / 75% / 50% 0% / 0% / 0% | |
British Columbia market uptake (3 years, prevalent patients) | ||
Uptake (reference scenario) Eculizumab Ravulizumab | 100% / 100% / 100% 0% / 0% / 0% | |
Uptake (new drug scenario) Crovalimab Eculizumab Ravulizumab | 10% / 25% / 50% 90% / 75% / 50% 0% / 0% / 0% | |
Cost of treatment (per patient) | ||
Cost of treatment, annually Crovalimab Eculizumab Ravulizumab | Loading year: $509,670 $570,643 $551,101 | Maintenance years: $430,452 $559,962 $507,328 |
PNH = paroxysmal nocturnal hemoglobinuria.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing crovalimab for patients with PNH to be –$2,738,865 (year 1: –$129,340; year 2: –$844,604; year 3: –$1,764,921).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Continuous up-dosing among patients receiving eculizumab is uncertain: As in the submitted CMA, for the BIA, the sponsor assumed that 20% of patients receiving eculizumab require continuous up-dosing to 1,200 mg to maintain disease control.1 As outlined in the critical appraisal of the CMA (refer to limitation “Continuous up-dosing among patients receiving eculizumab is uncertain”), this assumption is uncertain and increases treatment acquisition costs for eculizumab compared with assuming no up-dosing.
CDA-AMC conducted a scenario analysis to explore the impact of assuming that all patients receiving eculizumab received the 900 mg dose.
The rate of public drug coverage is uncertain: The sponsor assumed a public coverage rate of 64.11% across all provinces, with 100% coverage for the Non-Insured Health Benefits program population, resulting in a pan-Canadian average coverage rate of 65.18%.15 The 64.11% of patients being covered under public funding was based on the split of public versus private claims data for eculizumab and ravulizumab from IQVIA Pharmastat data.15,19 This approach is uncertain for several reasons. One, while the output of the approach is being applied to the patient population to refine it to those who are eligible for crovalimab through public drug plans, the value being used as a proxy for this is, of the total claims for ravulizumab and eculizumab, what proportion are public claims versus private claims. Whether this is an adequate proxy for the proportion who are eligible for public drug plan coverage is uncertain. Two, CDA-AMC was unable to validate this approach because the proportion of claims that were public from 2021 to 2024 ranged from 73% to 82% rather than 64%.18 Three, the approach does not account for differences in coverage rates by age group which is a known determinant of public coverage.20 Four, while eculizumab and ravulizumab have several other indications, the approach assumes that all claims would be specific to PNH; whether this assumption is reasonable is unknown given that claims data are not indication specific.14,21 Taking 3 and 4 together, if the age distribution for the other indications that eculizumab and ravulizumab are reimbursed for differ from that of PNH, the proportion of claims that are public may not be representative of the proportion of patients with PNH eligible for public coverage. Five, the approach does not account for differences in public coverage eligibility by jurisdiction, which is also a known determinant of public coverage;20 therefore, the proportion eligible may not be representative of individual jurisdictional reimbursement contexts. Finally, the approach does not account for the public drug program supports that some jurisdictions may have to assist with coverage for expensive drugs and/or for drugs for rare diseases.22
CDA-AMC conducted a scenario analysis to explore the impact of assuming 100% of patients have public drug coverage.
The price of drugs paid by public drug plans is uncertain. Analyses from both the sponsor and CDA-AMC are based on publicly available list prices for all comparators. Actual prices paid by drug plans are unknown. Confidential negotiated prices for eculizumab and ravulizumab may lead to the budgetary savings associated with crovalimab reimbursement to be limited or eliminated.10,11
CDA-AMC was unable to incorporate confidentially negotiated prices in reanalyses. Confidential negotiated prices for comparators may reduce the potential cost-savings for crovalimab.
Other limitations were identified by were not considered to be key limitations. This includes that the modelled population is reflective of those with greater than 10% granulocyte clone size threshold, which is narrower than the Health Canada indication.3,4,17,23 CDA-AMC conducted a scenario analysis to explore the budget impact of reimbursing crovalimab in the entire PNH population (i.e., not restricting by granulocyte count).
CDA-AMC Reanalyses of the BIA
CDA-AMC did not undertake base-case reanalyses on the sponsor’s base case. CDA-AMC conducted several scenario analyses to address remaining uncertainty, using the sponsor’s base case. The results are provided in Table 9. These included:
Assuming all patients received the 900 mg dose of eculizumab.
Assuming 100% of patients are eligible for public coverage.
Not restricting reimbursement by granulocyte count (i.e., reimbursement of crovalimab for the entire Health Canada indication).
Table 9: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $61,998,390 | $65,945,655 | $65,503,450 | $66,056,504 | $197,505,609 |
New drug | $61,998,390 | $65,816,315 | $64,658,846 | $64,291,582 | $194,766,744 | |
Budget impact | $0 | –$129,340 | –$844,604 | –$1,764,921 | –$2,738,865 | |
CDA-AMC scenario analysis 1: no continuous eculizumab updosing | Reference | $59,755,044 | $64,350,423 | $64,567,239 | $65,112,923 | $194,030,586 |
New drug | $59,755,044 | $64,296,119 | $63,911,721 | $63,729,084 | $191,936,925 | |
Budget impact | $0 | –$54,304 | –$655,518 | –$1,383,839 | –$2,093,661 | |
CDA-AMC scenario analysis 2: 100% public coverage | Reference | $95,135,798 | $101,184,138 | $100,494,513 | $101,325,276 | $303,003,927 |
New drug | $95,135,798 | $100,982,976 | $99,189,871 | $98,597,791 | $298,770,637 | |
Budget impact | $0 | –$201,162 | –$1,304,642 | –$2,727,485 | –$4,233,290 | |
CDA-AMC scenario analysis 3: No restriction based on granulocyte count | Reference | $115,668,639 | $123,032,939 | $122,207,930 | $123,239,746 | $368,480,615 |
New drug | $115,668,639 | $122,791,633 | $120,632,176 | $119,946,983 | $363,370,795 | |
Budget impact | $0 | –$241,306 | –$1,575,753 | –$3,292,764 | –$5,109,823 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.