Drugs, Health Technologies, Health Systems
Reimbursement Review
Nemolizumab (Nemluvio)
Sponsor: Galderma Canada Inc.
Therapeutic area: Prurigo nodularis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
AESI
adverse event of special interest
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
COPD
chronic obstructive pulmonary disease
CrI
credible interval
CSPA
Canadian Skin Patient Alliance
DAO
Dermatology Association of Ontario
DCS
dual-chamber syringe
DLQI
Dermatology Life Quality Index
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IAC
independent adjudication committee
IGA
Investigator’s Global Assessment
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
LS
least square
LTE
long-term extension
NMA
network meta-analysis
NRS
numerical rating scale
PN
prurigo nodularis
PP-NRS
Peak Pruritus Numerical Rating Scale
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SD-NRS
Sleep Disturbance Numerical Rating Scale
SE
standard error
SLR
systematic literature review
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Nemolizumab (Nemluvio) lyophilized powder and solvent solution for SC injection available as:
|
Sponsor | Galderma Canada Inc. |
Indication | For the treatment of adults with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
Reimbursement request | For the treatment of moderate to severe PN when disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | December 18, 2025 |
Recommended dose | Patients weighing < 90 kg: Initial dose of 60 mg (two 30 mg injections), followed by 30 mg given every 4 weeks Patients weighing ≥ 90 kg: Initial dose of 60 mg (two 30 mg injections), followed by 60 mg given every 4 weeks |
NOC = Notice of Compliance; PN = prurigo nodularis; SC = subcutaneous.
Sources: Nemluvio Product Monograph;1 sponsor’s Summary of Clinical Evidence.2
Introduction
Prurigo nodularis (РΝ) is a rare, neuroimmune, inflammatory skin disease characterized by the presence of chronic (≥ 6 weeks) itch; history and signs of repeated scratching, such as excoriation and scars; and multiple, localized or generalized, hyperkeratotic, symmetrically distributed pruritic nodules on the extensor surfaces of the extremities and trunk.3-7 Exact incidence and prevalence estimates for Canada are unknown; however, based on the expected prevalence of PN published in a study by Bahloul et al. (2024),8 there are an estimated 6,000 patients with moderate to severe PN in Canada. ΡN can occur in all age groups, without sex preferences, but it primarily affects older adults,4 with approximately 69% of patients aged 51 years or older.5,9-11 PN leads to significantly reduced quality of life (QoL) because symptoms like itch and skin lesions affect patients’ personal and social lives.12,13 The negative impact on QoL from PN is primarily driven by the increased incidence and intensity of pruritus.14 Furthermore, patients with PN may be affected by insomnia and sleep disturbances, a higher level of absenteeism at work (full or partial days), decreased productivity or presenteeism, and lower work performance.15 PN symptoms also affect patients’ ability to get dressed, engage in self-care or personal hygiene, plan activities, and complete chores.13 PN is associated with the presence of chronic obstructive pulmonary disease (COPD), chronic hepatitis C, HIV, and atopic dermatitis (AD),14 resulting in an increased burden for these patients. It is estimated that approximately 43% of patients with PN are initially misdiagnosed.16 Therefore, additional bloodwork may be necessary to ascertain the underlying causes of symptoms (e.g., chronic kidney disease, liver disease, thyroid disease, HIV infection, parasitic infection, or malignancy).4,17,18 However, clinical experts consulted by Canada’s Drug Agency (CDA-AMC) for this review noted that if PN is diagnosed by a dermatologist, a biopsy is not required to confirm the diagnosis. Given the burden of PN and its associated comorbidities, patients with PN have high rates of health care and specialty care utilization.19
According to clinical experts consulted by CDA-AMC, PN is often initially managed with high-potency topical corticosteroids (TCSs). Other initial treatment options include topical calcineurin inhibitors (TCIs) or anesthetics, oral antihistamines, intralesional corticosteroid injections, topical capsaicin, and UV light treatment (i.e., phototherapy). The clinical experts commented that systemic immunosuppressants, such as cyclosporine and methotrexate, may be prescribed for severe or treatment-resistant disease. Other medications that may be considered to treat PN include gabapentin, pregabalin, carbamazepine, doxepin, mirtazapine, mycophenolate mofetil, and low-dose naltrexone. Clinical experts highlighted that PN is difficult to treat and that the aforementioned treatment options may provide only partial or short-term symptomatic relief; in addition, the use of these treatments is often limited by side effects and feasibility issues.
International guidelines detail several medication options for the treatment of PN, most of which have limited evidence supporting efficacy and are used off-label.20-22 In Canada, dupilumab is currently the only treatment indicated for PN.2,23 Clinical experts agreed with patient and clinician groups that there is a need for highly effective, disease-modifying, systemic PN therapies that provide sustained relief, are safe for long-term use, and are convenient and accessible.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of nemolizumab 30 mg per 0.49 mL for subcutaneous (SC) injection in the treatment of moderate to severe PN. The approved Health Canada indication is for nemolizumab to be used for the treatment of adults with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. This review is based on the sponsor’s initial reimbursement request for nemolizumab to be used for the treatment of moderate to severe PN. The sponsor has revised the reimbursement request to align with the approved Health Canada indication; however, the revision did not warrant a substantial update to the report.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to our call for input and from the clinical expert(s) consulted for the purpose of this review.
Patient Input
A single patient group, the Canadian Skin Patient Alliance (CSPA), provided input for this submission. CSPA is a national charity that supports the health and well-being of people across Canada affected by skin, hair, and nail conditions through collaboration, advocacy, and education. Information regarding the experiences of people living with PN was compiled from a patient and caregiver survey conducted from September 12, 2024, to November 29, 2024. A total of 9 survey responses were received from participants in Canada. None of the patients had experience with nemolizumab.
Five survey respondents indicated that they had had PN for less than 5 years; most reported severe (n = 3) or moderate (n = 1) PN. Respondents indicated that their PN diagnosis affected their family relationships, intimate relationships, work life, mental health, social life, daily activities, sleep, self-esteem, finance, and sex life. Respondents highlighted the following symptoms: itchy skin, itchy bumps (nodules), burning or stinging skin, scratching, pain, and hyperpigmentation (dark spots).
The caregiver respondent reported that the disease affected their loved one with regards to family balance and relationships, mental health, and intimacy. Additionally, they shared that it was difficult having to encourage their loved one to continue taking treatments for PN.
The respondents pointed to effectiveness, lack of side effects, and affordability as the top 3 unmet needs. They also said there is a need for treatments that are easy to take or apply and conducive to patients’ schedules. The caregiver respondent noted the cost of medication as an important unmet need.
Clinician Input
Input From Clinical Experts Consulted for This Review
Clinical experts consulted by CDA-AMC expressed an unmet need for disease-modifying, systemic therapies for PN that are highly effective in achieving treatment goals (e.g., itch relief, treatment of lesions), provide sustained disease control and symptom relief, are safe for short- and long-term use across diverse patient populations (e.g., all ages, those with comorbidities), and are convenient and accessible. Clinical experts commented that, along with dupilumab, nemolizumab is expected to cause a shift in the current treatment paradigm for PN in Canada. Clinical experts expected that traditional therapies (e.g., TCSs) would still be used for the initial management of PN; they stated that nemolizumab would be best suited for patients with moderate to severe or refractory PN, particularly those with persistent pruritus and many chronic lesions for whom conventional treatments have failed. According to clinical experts, the patients least suited for nemolizumab would be those with mild PN or mild symptoms that can be managed with conventional treatments. Clinical experts stated that nemolizumab or dupilumab would likely be the preferred systemic treatment options over systemic immunosuppressants. They also noted that nemolizumab would likely be combined with some existing PN treatments, such as topical or intralesional corticosteroids. According to clinical experts, treatment response in patients with PN is assessed in clinical practice based on improvement in the number of lesions, reduction in pruritus, and improvement in QoL; physicians may prioritize these outcomes differently. They noted that assessments of treatment response in clinical practice do not usually use the grading tools used in clinical trials as stringently. Clinical experts stated that response would typically be assessed 3 to 6 months after initiating PN treatment and at the same interval while the disease is active, with annual follow-up if the disease is controlled. When deciding to discontinue treatment with nemolizumab, the clinical experts would evaluate whether the patient had achieved the desired clinical outcomes within a 6-month time frame. Clinical experts identified that a clinically important response would be a decrease in lesion count from 20 or more at the start of therapy to 5 or fewer, or a decrease in peak itch severity of at least 4 points out of 10 on the numerical rating scale (NRS). Clinical experts also acknowledged that that the decision to stop treatment in a patient with a partial response would consider whether the patient wants to continue therapy and if any other effective treatment options are available. Additional factors for discontinuation may include serious adverse events (SAEs) or the emergence of comorbidities requiring other treatments. Clinical experts agreed that due to the complexity of diagnosing PN, it is essential that treatment of PN with nemolizumab is prescribed only by dermatologists.
Clinician Group Input
Two clinician groups, the Atlantic Dermatology Group and the Dermatology Association of Ontario (DAO), provided input for this submission. Input was provided by 5 clinicians, and information was gathered from sessions related to PN at the September 2024 European Academy of Dermatology and Venerology in Amsterdam as well as from the literature. Input from the DAO was provided by 7 clinicians, and information was gathered from clinical trial data, available literature retrieved through PubMed, and experience with the use of nemolizumab from clinical trialists in Canada.
Both clinician groups highlighted that currently, there are no specific clinical practice guidelines for PN in Canada. They indicated that current therapies include topical treatments, such as corticosteroids and emollients, dupilumab, and off-label regimens (i.e., systemic immunosuppressants, thalidomide, lenalidomide, tricyclic antidepressants, Janus kinase inhibitors, neurokinin-1 receptor antagonists, and monoclonal antibodies targeting interleukin [IL]-4 pathways). The DAO also highlighted that nondrug treatments, such as UVB phototherapy and psychosocial approaches, had been attempted with limited availability and success in addressing the psychological burden of PN and improving overall QoL. The Atlantic Dermatology Group further highlighted evidence from a systematic review, which concluded that while phototherapy, corticosteroids, cyclosporin, and methotrexate are viable options, the potential benefits are limited because of the risks of relapse and potential side effects.
The DAO highlighted the following unmet needs in PN treatment: therapies that provide rapid and sustained relief from severe itch; improve skin lesions; address the psychosocial impacts of the disease; improve tolerance and/or convenience compared with current treatments; and target the underlying neuroimmune mechanisms driving PN. Atlantic Dermatology Group clinicians noted the need for therapy that provides clinically significant reductions in itch, improved physician global assessment of skin lesions, and improved QoL.
The Atlantic Dermatology Group indicated that nemolizumab will cause a shift in the current treatment paradigm because it can be used as a first-line therapy in patients with PN, in patients who are intolerant to dupilumab, TCSs, or phototherapy, and in patients for whom these treatments have failed. They also noted that nemolizumab would be used as a stand-alone treatment. However, the DAO indicated that nemolizumab would also fit as a targeted second- or third-line option in patients for whom topical therapies and phototherapy produced an insufficient disease response or for whom these treatments have failed.
The DAO clinician group noted that the patients best suited for treatment with nemolizumab would be those with moderate to severe PN who did not achieve adequate relief with topical therapies or phototherapy or those unable to access or tolerate these treatments. This would include those who have persistent and intense pruritus, widespread nodular lesions, and/or significant impairment in QoL. The Atlantic Dermatology Group highlighted that patients with moderate to severe PN with an itch score of 7 or higher with at least 20 lesions would be in most need and be best suited for treatment. The DAO further noted that patients with contraindications (such as known hypersensitivity to nemolizumab), patients with milder disease that responds to topical steroids, and patients with conditions that significantly compromise immune function (unless the benefits outweigh the risks) would not be suitable for treatment with nemolizumab.
The DAO clinician group noted that to identify the patients most likely respond to nemolizumab, a clinician assessment of the intensity and persistence of itch, extent of nodular lesions, and impact on daily functioning and QoL was sufficient. However, the Atlantic Dermatology Group noted that no clinical, pathological, or chemical markers exist to identify the patients most likely to exhibit a response to treatment.
The Atlantic Dermatology Group noted that misdiagnoses are unlikely if patients are managed by dermatologists in clinical practice. The group emphasized the consideration of other diagnoses, including pemphigoid nodularis, mastocytosis, lichen planus, nodular scabies, arthropod bites, lymphocytoma cutis, lymphomatoid papulosis, amyloidosis, reticulohistiocytosis, and cutaneous T-cell lymphoma. However, the DAO indicated that PN may be underdiagnosed or misdiagnosed as other pruritic dermatoses, and that clinician education and awareness are key to improving diagnostic accuracy.
Both clinician groups noted that a 4-point improvement in a patient’s Peak Pruritus Numerical Rating Scale (PP-NRS) score should be used to determine if a patient is responding to treatment. The Atlantic Dermatology Group and the DAO noted that a physician global assessment or Investigator’s Global Assessment (IGA) of clear or almost clear and a reduction of at least 2 points from baseline, respectively, are the outcomes used to determine treatment response. In addition, the Atlantic Dermatology Group noted that the Dermatology Life Quality Index (DLQI) should be used, and the DAO noted that subjective feedback on daily comfort and sleep quality should be assessed.
Both clinician groups agreed that treatment discontinuation should be considered if there is a lack of meaningful reduction in PP-NRS or if there are persistent nodular lesions as assessed by the IGA. The Atlantic Dermatology Group noted that treatment should be discontinued after 6 months of therapy if there is a lack of response. Additional criteria for discontinuation were noted by the DAO, including significant adverse events (AEs) (i.e., hypersensitivity, injection-site reaction, or long-term safety issues, including recurrent infections). Disease progression (e.g., worsening lesions), complications such as skin infections, and patient-reported dissatisfaction were also noted as important factors requiring treatment reassessment. In cases where systemic immunosuppressants or other biologics are required to manage refractory disease, nemolizumab may no longer be an optimal choice.
The Atlantic Dermatology Group indicated that PN should be managed by dermatologists experienced in both the diagnosis and management of PN and the use of biologics. The group also noted that dermatologists should be experienced in measuring DLQI, IGA, and PP-NRS, as well as counting nodules. The DAO noted that, in addition to dermatologists, allergists or immunologists may also be involved in managing patients with PN where PN overlaps with other atopic or immune-mediated conditions. The group highlighted that while the diagnosis, treatment initiation, and monitoring of patients requires the expertise and oversight of specialists, nemolizumab may be prescribed in various clinical settings (such as community clinics, hospital outpatient departments, and specialty dermatology or allergist clinics) and administered by patients at home.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The following items were identified as key factors that could affect the implementation of a recommendation for nemolizumab: relevant comparators; considerations for the prescription, initiation, continuation, renewal, and discontinuation of therapy; generalizability; care provision issues; and system and economic issues. The details of the drug program input along with advice from the clinical experts consulted for this review are available in Table 4.
Clinical Evidence
Systematic Review
Description of Studies
The OLYMPIA 1 (N = 286) and OLYMPIA 2 (N = 274) trials were both phase III, randomized, double-blind, placebo-controlled studies investigating the efficacy and safety of nemolizumab versus placebo for the treatment of adult patients with PN. The studies were conducted in a total of 16 countries (77 sites for the OLYMPIA 1 trial and 55 sites for the OLYMPIA 2 trial), including 8 sites in Canada that randomized a total of 44 patients. In both trials, patients were randomized 2 to 1 to receive nemolizumab or matched placebo by SC injection every 4 weeks. The treatment periods were 24 weeks and 16 weeks in the OLYMPIA 1 and OLYMPIA 2 trials, respectively; follow-up was 8 weeks in each study. The primary end points in the OLYMPIA 1 and OLYMPIA 2 trials were the proportion of patients with at least a 4-point improvement from baseline in PP-NRS at week 16 and the proportion of patients reporting IGA success, defined as an IGA response of 0 (clear) or 1 (almost clear) and at least a 2-point reduction from baseline, at week 16. Key secondary and other secondary end points included measures of improvement in Sleep Disturbance NRS (SD-NRS), itch reduction (as measured using the PP-NRS), lesion clearance (IGA success), health-related QoL (HRQoL) (as measured using the DLQI). Harms outcomes were also assessed.
Patients eligible for participation in both of the OLYMPIA pivotal trials were adults aged 18 years or older with a clinical diagnosis of PN for at least 6 months with pruriginous nodular lesions on the upper limbs, lower limbs, and/or trunk; at least 20 nodules on the entire body, with bilateral distribution; and an IGA score of at least 3 (indicating moderate or severe global severity). Eligible patients were also required to have severe pruritus, defined as a PP-NRS score of at least 7. In the OLYMPIA 1 and OLYMPIA 2 trials, most patients were female, white, and aged 18 to 65 years (details are provided in Table 13). Patients were not eligible for participation in the trials if they had uncontrolled or exacerbated asthma, COPD, chronic bronchitis, certain infections, or active AD within the previous 3 months.
Efficacy Results
Improvement of 4 or More Points From Baseline in PP-NRS Score at Weeks 16 and 24
In the OLYMPIA 1 trial, the proportions of patients with improvements of 4 or more points from baseline in weekly average PP-NRS score at week 16 were 58.4% in the nemolizumab group and 16.7% in the placebo group (strata-adjusted proportion difference = 40.1%; 95% confidence interval [CI], 29.4 to 50.8; P < 0.0001). For the same end point in the OLYMPIA 2 trial, the proportions of patients were 56.3% in the nemolizumab group and 20.9% in the placebo group (strata-adjusted proportion difference = 37.4%; 95% CI, 26.3 to 48.5; P < 0.0001). The proportion difference between the groups was statistically significant in favour of nemolizumab in both trials, and clinical experts consulted by CDA-AMC considered the differences to be clinically meaningful. The results at week 24 in the OLYMPIA 1 trial roughly maintained those reported at week 16.
IGA Success at Weeks 16 and 24
In the OLYMPIA 1 trial, the proportions of patients with IGA success at week 16 were 26.3% in the nemolizumab group and 7.3% in the placebo group (strata-adjusted proportion difference = 14.6%; 95% CI, 6.7 to 22.6; P = 0.0025). For the same end point in the OLYMPIA 2 trial, the proportions of patients with IGA success were 37.7% in the nemolizumab group and 11.0% in the placebo group (strata-adjusted proportion difference = 28.5%; 95% CI, 18.8 to 38.2; P < 0.0001). The proportion difference between groups was statistically significant in favour of nemolizumab in both trials, and clinical experts consulted by CDA-AMC considered the differences to be clinically meaningful. The week 24 results in the OLYMPIA 1 trial were consistent with those reported at week 16.
Improvement of 4 or More Points From Baseline in SD-NRS Score at Week 16
In the OLYMPIA 1 trial, the proportions of patients with improvement of 4 or more points from baseline in weekly average SD-NRS score at week 16 were 50.0% in the nemolizumab group and 11.5% in the placebo group (strata-adjusted proportion difference = 38.0%; 95% CI, 27.8 to 48.2; P < 0.0001). For the same end point in the OLYMPIA 2 trial, the proportions of patients with improvement of 4 or more points from baseline in weekly average SD-NRS score were 51.9% in the nemolizumab group and 20.9% in the placebo group (strata-adjusted proportion difference = 31.9%; 95% CI, 20.7 to 43.2; P < 0.0001). The proportion difference between groups was statistically significant in favour of nemolizumab in both trials. (Input regarding the clinical meaningfulness of these differences was not received from clinical experts consulted by CDA-AMC.)
Improvement of 4 or More Points From Baseline in DLQI Score
In the OLYMPIA 1 trial, at week 16, the proportions of patients with an improvement of 4 or more points from baseline in DLQI total score were 70.5% in the nemolizumab group and 42.7% in the placebo group (strata-adjusted proportion difference = 27.5%; 95% CI, 15.8 to 39.2); at week 24, the proportions were 71.1% and 35.4%, respectively (strata-adjusted proportion difference = 35.5%; 95% CI, 23.9 to 47.2). In the OLYMPIA 2 trial, at week 16, the proportions of patients with an improvement of 4 or more points from baseline in DLQI total score were 74.9% in the nemolizumab group and 39.6% in the placebo group (strata-adjusted proportion difference = 37.4%; 95% CI, 25.7 to 49.0). The clinical experts consulted by CDA-AMC considered these differences to be clinically meaningful.
There were no published between-group minimum important difference values provided by the sponsor for the PP-NRS, IGA success, and DLQI end points assessed using Grading of Recommendations Assessment, Development and Evaluation (GRADE); as such, the thresholds used to judge the target of certainty in the GRADE assessment (Table 2) are based on input from the clinical experts consulted by CDA-AMC.
Harms Results
In the OLYMPIA 1 trial, at least 1 AE was reported in 71.7% of patients in the nemolizumab group and 65.3% of patients in the placebo group. The most common AEs (nemolizumab versus placebo) were COVID-19 (8.0% versus 14.7%), nasopharyngitis (6.4% versus 8.4%), headache (7.0% versus 2.1%), cough (4.8% versus 5.3%), dyspnea (3.2% versus 5.3%), neurodermatitis (9.6% versus 20.0%), and eczema (5.3% versus 1.1%). At least 1 SAE was reported in 8.6% of patients in the nemolizumab group and 10.5% of patients in the placebo group; AEs leading to study drug withdrawal occurred in 4.8% and 3.2% of patients, respectively. One patient in the placebo group died during the OLYMPIA 1 trial. At least 1 adverse event of special interest (AESI) was reported in 17.1% of patients in the nemolizumab group and 18.9% of patients in the placebo group. The most commonly reported AESI in the nemolizumab and placebo groups was infection (10.7% versus 16.8%). The AESI of newly diagnosed asthma or worsening of asthma was reported in 3.7% of patients in the nemolizumab group and in 2.1% of patients in the placebo group.
In the OLYMPIA 2 trial, at least 1 AE was reported in 61.2% and 53.8% of patients in the nemolizumab and placebo groups, respectively. In the OLYMPIA 2 trial, the most common AEs were headache (6.6% versus 4.4%), neurodermatitis (3.8% versus 11.0%), and AD (5.5% versus 0%). At least 1 SAE was reported in 2.2% of patients in the nemolizumab group and 5.5% of patients in the placebo group; AEs leading to study drug withdrawal occurred in 2.7% and 2.2% of patients, respectively. No patient deaths occurred during the OLYMPIA 2 trial. At least 1 AESI was reported in 11.5% of patients in the nemolizumab group and in 9.9% of patients in the placebo group. The most commonly reported AESI in the nemolizumab and placebo groups was infection (5.5% versus 6.6%). The AESI of newly diagnosed asthma or worsening of asthma was reported in 2.7% of patients in the nemolizumab group and in 1.1% of patients in the placebo group.
Critical Appraisal
The OLYMPIA 1 and OLYMPIA 2 trials were both phase III, randomized, double-blind, placebo-controlled, multicentre studies. Methods of randomization and treatment allocation in the trials were adequate. Reported baseline characteristics were generally balanced across the nemolizumab and placebo groups in both trials. Some differences between arms were noted; however, clinical experts consulted for this review did not anticipate that these differences would affect the interpretation of the results. For both pivotal trials, the prespecified sample size was achieved, and the screening failure rates approached or slightly exceeded the expected rates. Both trials were powered for the primary end points of the proportion of patients with at least a 4-point improvement in PP-NRS and the proportion of patients with IGA success (both at week 16). The multiple testing procedure included all primary and key secondary end points. Analyses of other secondary end points were not adjusted for multiplicity; therefore, conclusions cannot be drawn regarding statistical significance for these end points. Loss to follow-up was low, and rates of discontinuation were similar between treatment arms in both studies. Due to differences in treatment response between arms (e.g., reduction in pruritus), patients and investigators could have become aware of treatment allocation, which may have introduced bias with respect to the subjective end points, such as PP-NRS and DLQI. In both trials, there was a disproportionate use of rescue treatment between study arms, with a higher percentage of patients in the placebo arms requiring rescue therapy than in the nemolizumab arms. This imbalance in rescue treatment could have exaggerated the itch reduction response with placebo, thereby minimizing the comparative benefit of nemolizumab. However, the clinical experts consulted by CDA-AMC did not expect the differences in rescue therapy use between groups to affect the interpretation of the efficacy results.
The clinical experts stated that the characteristics of the patients randomized in the OLYMPIA pivotal trials were a reasonable reflection of patients who would receive nemolizumab for moderate to severe PN in Canada. In both OLYMPIA trials, most patients were white, and patients with certain medical conditions (e.g., active AD, uncontrolled asthma, chronic infections) were not eligible. These factors present potential generalizability limitations. The trial populations do not adequately represent the racial diversity of patients with PN. In addition, the clinical experts noted that PN generally affects older people with comorbidities, such as eczema, severe asthma, or hepatitis, who may qualify for treatment with nemolizumab in clinical practice. Topical and systemic medications and procedures were prohibited during the trials, and nemolizumab was given as monotherapy (unless rescue therapy was used). Clinical experts expressed that, although nemolizumab may be used on its own in clinical practice, it is likely that concomitant treatments, such as topical or intralesional corticosteroids, would also be prescribed as needed. An important limitation of the OLYMPIA 1 and OLYMPIA 2 trials is that nemolizumab was compared to placebo, which does not represent the standard of care for treatment of PN. The clinical experts noted that dupilumab would be the most relevant comparator to nemolizumab for the indication under review. However, dupilumab was not indicated for treatment of PN at the time of the OLYMPIA 1 and OLYMPIA 2 trials. The main efficacy and harms outcomes assessed in the OLYMPIA 1 and OLYMPIA 2 trials align with the outcomes of importance identified by patients and clinicians.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
pruritus response (i.e., the proportion of patients with an improvement of ≥ 4 from baseline in PP-NRS)
global severity of PN (i.e., the proportion of patients with IGA success)
HRQoL (i.e., the proportion of patients with an improvement of ≥ 4 in DLQI)
harms (i.e., AESI of newly diagnosed asthma or worsening of asthma).
Table 2: Summary of Findings for Nemolizumab vs. Placebo for Patients With Moderate to Severe PN
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
Pruritus response | ||||
Proportion of patients with an improvement of ≥ 4 from baseline in PP-NRS scorea Follow-up: 16 weeks | 560 (2 RCTs) | OLYMPIA 1 trial:
OLYMPIA 2 trial:
| Highb | Nemolizumab results in a clinically important increase in the proportion of patients with at least a 4‑point improvement in PP‑NRS score at 16 weeks compared with placebo. |
Global severity of PN | ||||
Proportion of patients with IGA successc Follow-up: 16 weeks | 560 (2 RCTs) | OLYMPIA 1 trial:
OLYMPIA 2 trial:
| Highb | Nemolizumab results in a clinically important increase in the proportion of patients with IGA success at 16 weeks compared with placebo. |
HRQoL | ||||
Proportion of patients with an improvement of ≥ 4 in DLQI scored Follow-up: 24 weeks | 286 (1 RCT) | OLYMPIA 1 trial:
| Moderateb,e | Nemolizumab likely results in a clinically important increase in the proportion of patients with at least a 4‑point improvement in DLQI score at 24 weeks compared with placebo. |
Proportion of patients with an improvement of ≥ 4 in DLQI scored Follow-up: 16 weeks | 274 (1 RCT) | OLYMPIA 2 trial:
| Moderateb,e | Nemolizumab likely results in a clinically important increase in the proportion of patients with at least a 4‑point improvement in DLQI score at 16 weeks compared with placebo. |
Harms | ||||
Proportion of patients with newly diagnosed asthma or worsening of asthma Follow-ups:
| 556 (2 RCTs) | OLYMPIA 1 trialf:
OLYMPIA 2f:
| Lowg | Nemolizumab may result in an increase in newly diagnosed asthma or worsening of asthma when compared with placebo. The clinical importance of the increase is unclear. |
CI = confidence interval; DLQI = Dermatology Life Quality Index; HRQoL = health-related quality of life; IGA = Investigator’s Global Assessment; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; RCT = randomized controlled trial; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe PP-NRS is a patient-reported, daily scale that asks patients for a unit score on an 11-point scale (0 to 10) where 0 is “no itch” and 10 is “worst itch imaginable.” The PP-NRS was used to assess the maximum intensity of pruritus during the previous 24 hours. It asked patients to rate their itch at the worst moment during that time period. Based on clinical expert input, the threshold for a clinically important between-group difference was 200 per 1,000 for the proportion of patients with at least a 4-point reduction from baseline at 16 weeks.
bThe thresholds used to judge the target of certainty in the GRADE assessments were based on input from clinical experts.
cThe IGA is a 5-point scale used to evaluate the global severity of PN. Based on a review of the patient’s skin, the investigator assigned a score of 0 (clear), 1 (almost clear), 2 (mild), 3 (moderate), or 4 (severe). Treatment response (i.e., IGA success) was defined as 0 (clear) or 1 (almost clear) and at least a 2-point improvement from baseline. Based on clinical expert input, the threshold for a clinically important between-group difference was 150 per 1,000 for the proportion of patients with IGA success at 16 weeks.
dThe DLQI is a patient-reported, 10-item questionnaire covering the domains of symptoms and feelings, daily activities, leisure, work and school, personal relationships, and treatment to measure how much a patient’s skin problem has affected their life over the last week. Patients rate each question from 0 (not at all) to 3 (very much). The DLQI total score is calculated as the sum of the score of each question, resulting in a minimum score of 0 and a maximum score of 30; a higher total score indicates greater impairment in quality of life. Based on clinical expert input, the thresholds for a clinically important between-group difference for the proportion of patients with at least a 4-point reduction from baseline were 200 per 1,000 at 24 weeks and 150 to 200 per 1,000 at 16 weeks.
eNo statistical tests were performed. Rated down 1 level due to serious study limitations (i.e., the potential for unblinding with a self-reported measure).
fBetween-group difference and corresponding 95% CI results were not available.
gNo statistical tests were performed. There was no known minimum important difference; therefore, the target of certainty appraisal was any effect. Rated down 2 levels due to very serious imprecision; between-group differences and CIs were not available. Therefore, it is unknown if the CIs include the possibility of no difference, fewer harms, or increased harms. Clinical experts consulted by Canada’s Drug Agency commented that a greater incidence of worsening of asthma may occur in clinical practice versus what was reported in the OLYMPIA pivotal trials.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report;25 sponsor’s Response to Request for Additional Information.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
Description of Studies
The OLYMPIA long-term extension (LTE) study (NCT04204616) is an ongoing, phase III, prospective, single-arm, multicentre, open-label study to evaluate the long-term safety and efficacy of nemolizumab in adult patients with PN. The efficacy results summarized in this review are from Interim Analysis 2 (data available as of July 21, 2024) supplemented with data from Interim Analysis 1 (data cut-off date: March 10, 2023). Patients from a previous phase IIa study who had completed all screening assessments 28 days before the baseline visit or first dose of study drug were eligible to enrol. In addition, patients from the phase III trials (i.e., the OLYMPIA 1 and OLYMPIA 2 trials) were eligible to enrol within 56 days following their last visit.
Each patient’s participation in the study will be up to 196 weeks. The study consists of a screening period (up to 4 weeks), a treatment period (up to 184 weeks with final dose administered), and an 8-week follow-up period.
The dose received in the LTE study (1 or 2 injections) is based on the patient’s body weight and previous treatment assignment in the nemolizumab PN studies.
For patients enrolling from the phase IIa study:
Patients who weighed less than 90 kg at baseline receive 30 mg nemolizumab (with a 60 mg loading dose at baseline) every 4 weeks.
Patients who weighed 90 kg or more at baseline receive 60 mg nemolizumab (as 2 injections of 30 mg, with no loading dose) every 4 weeks.
For patients enrolling from the phase III studies:
The day 1 or baseline dose in the LTE study was based on the blinded study treatment assigned during the pivotal trial to maintain the blinding of the study. Therefore, patients received either 2 blinded 30 mg injections of nemolizumab or 1 blinded 30 mg injection of nemolizumab and 1 blinded injection of placebo.
After day 1, the same dosing regimen is used as in the phase III study (i.e., 1 or 2 SC injections of 30 mg nemolizumab, based on patient weight at baseline in the phase III study).
Key efficacy outcomes include IGA success (defined as an IGA score of 0 [clear] or 1 [almost clear]) and improvements in PP‑NRS, SD-NRS, and DLQI scores.
No sample size calculation was performed. However, the plan was to enrol approximately 450 patients in this study, depending on the rollover rate from the lead-in studies. The safety population consisted of all patients who received at least 1 dose of nemolizumab (blinded or open label). All efficacy analyses were performed on the safety population and were descriptive in nature. The efficacy analyses were carried out using observed cases without imputing missing data. For binary secondary end points, efficacy data collected on or after the use of rescue therapy were treated as failure of treatment, with the exception of observed cases analysis (in which observed data are used regardless of the use of rescue therapy).
All efficacy assessments were summarized by LTE study treatment and previous treatment at each analysis visit. Data were summarized under the following headings for efficacy:
continuous nemolizumab (i.e., patients with a < 12-week interval between the last nemolizumab dose in the lead-in study and the first dose in the LTE study [N = 307]; used to evaluate the persistence of nemolizumab effect)
placebo to nemolizumab (includes patients who had never received nemolizumab before the LTE study)
re-treatment (i.e., patients with a ≥ 12-week interval between the last nemolizumab dose in the lead-in study and the first dose in the LTE study [N = 27]; used to assess any loss of response).
Overall, 508 patients were enrolled in the LTE study. The mean age was 55.40 years, and the mean body weight was 82.44 kg. Most of the patients were white (87.20%) and female (60.40%). Overall, 32.10% of patients had a moderate IGA score and 15.70% of patients had a severe IGA score. A higher proportion of patients were in the placebo-to-nemolizumab group (moderate: 43.10%; severe: 31.60%) than in the continuous nemolizumab group (moderate: 26.10%; severe: 6.20%). At the lead-in baseline, patients’ weekly average PP-NRS score was 8.25. Their weekly average SD-NRS score was 7.09, and the mean DLQI total score was 16.90. The majority of patients had no atopy background (continuous nemolizumab group: 64.20%; placebo-to-nemolizumab group: 64.90%). The average time since PN diagnosis was 101.63 months.
Efficacy Results
IGA Success From LTE Study Baseline
The proportions of patients with IGA success (defined as an IGA of 0 [clear] or 1 [almost clear]) at the LTE study baseline were 40.1% and 12.6% for patients who were continuing treatment with nemolizumab (i.e., the continuous nemolizumab group) and patients who transitioned from placebo to treatment with nemolizumab (i.e., the placebo-to-nemolizumab group), respectively. At week 28, the proportions of patients with IGA success were similar across the 2 groups (continuous nemolizumab group: 53.3%; placebo-to-nemolizumab group: 56.3%). Continuous improvements in skin clearance at week 100 were observed among the 2 groups (continuous nemolizumab group: 73.4%; placebo-to-nemolizumab group: 75.2%).
Improvement of 4 or More Points in PP-NRS Score From Baseline Lead-In
More than 80% of patients experienced an improvement of 4 or more points in PP-NRS score at week 28 in the continuous nemolizumab group (88.8%) and the placebo-to-nemolizumab group (82.4%). At week 100, results indicated consistent improvement in itch relief in the continuous nemolizumab group (92.1%) and the placebo-to-nemolizumab group (94.1%); however, the sample sizes steadily decreased over time.
Improvement of 4 or More Points in SD-NRS Score From Baseline Lead-In
Following nemolizumab treatment in the LTE study, the proportion of patients with an improvement of 4 or more points in SD-NRS score from the lead-in baseline generally increased over time in all patients and in each group. A consistent improvement of greater than or equal to 4 points from baseline lead-in was observed across the groups at week 100 (continuous nemolizumab group: 86.4%; placebo-to-nemolizumab group: 87.3%). However, the sample sizes steadily decreased over time.
Improvement of 4 or More Points in DLQI Score From Baseline Lead-In
At week 28, the proportions of patients achieving an improvement of 4 or more points from baseline lead-in were 87.8% versus 90.5% in the continuous nemolizumab group and the placebo-to-nemolizumab group, respectively. Continuous improvements were observed in patients in both groups at week 52 (continuous nemolizumab group: 90.1%; placebo-to-nemolizumab group: 91.0%) and week 100 (continuous nemolizumab group: 89.9%; placebo-to-nemolizumab group: 93.1%).
Harms Results
For Interim Analysis 1, a total of 407 patients (80.1%) experienced at least 1 AE during the overall study period. The most frequently reported AEs included infections and infestations (54.3%), skin and SC tissue disorders (36.8%), musculoskeletal and connective tissue disorders (26.0%), respiratory, thoracic, and mediastinal disorders (17.0%), and nervous system disorders (15.4%). SAEs were reported in 54 patients (10.6%). SAEs experienced by more than 1 patient were neurodermatitis (4 patients [0.8%]), myocardial infarction (3 patients [0.6%]), and angina pectoris, cardiac failure congestive, cholelithiasis, pneumonia, osteoarthritis, and carotid artery stenosis (2 patients [0.4%] each). A total of 35 patients (6.9%) had an AE leading to withdrawal from the study. AESIs occurred among 319 patients (62.8%). The most commonly reported AESIs were infections (54.5%) and injection-related reactions (31.7%). Newly diagnosed asthma or worsening of asthma was reported in 5.7% of patients. Two patients (0.4%) experienced AEs leading to death. The causes of death were myocardial infarction and end-stage renal disease; however, both patients had medical histories of comorbidities, and neither death was considered related to the study drug.
Critical Appraisal
The OLYMPIA LTE study is a phase III, prospective, single-arm, multicentre, open-label study. The single-arm design of the study limits the ability to draw conclusions about the long-term efficacy of nemolizumab. The open-label nature of the study may increase the risk of bias in the evaluation of subjective outcomes. The cumulative exposure to study treatment and treatment adherence were not summarized by group (i.e., continuous nemolizumab, re-treatment, or placebo to nemolizumab); this further compromised our ability to judge the potential patterns of correlation between treatment exposure and outcome over the long term. Patients receive treatment in the LTE study based on their previous enrolment in nemolizumab studies. The impact of these differing dosing regimens on efficacy results is unknown.
Data from patients contributing to the analyses steadily decreased over time as patients discontinued treatment (around 30% at Interim Analysis 2). Those patients who discontinued the LTE study (17.9% versus 14.9% in the continuous nemolizumab and placebo-to-nemolizumab groups, respectively) could be systematically different from those who remained in the study. Therefore, the trend of relative stable treatment effect over time could be biased as a result of survival or attrition bias. The sample size in the re-treatment group was small, limiting the ability to draw conclusions regarding efficacy results. Most patients received at least 1 concomitant therapy during the treatment period; the effect of these on efficacy outcomes cannot be determined. The harms data aligned with the evidence from the pivotal studies.
Indirect Comparisons
Description of Studies
Due to the lack of direct evidence comparing nemolizumab with other existing, relevant therapies for the treatment of patients with PN, the sponsor submitted 1 indirect treatment comparison (ITC), a network meta-analysis (NMA) comparing nemolizumab with dupilumab, vixarelimab, nalbuphine, and placebo. The outcomes assessed in the NMA included PP-NRS change from baseline, PP‑NRS response, IGA success, composite PP-NRS and IGA response, DLQI, and AEs at time points ranging from week 4 to week 24. This ITC summary focuses on the comparison of nemolizumab with dupilumab because vixarelimab and nalbuphine are not relevant in clinical practice settings in Canada.
Efficacy Results
The NMA demonstrated a favourable benefit of nemolizumab when compared with dupilumab. However, the favourable effect was not consistent. It was influenced by the outcomes measured (such as PP-NRS change from baseline, PP-NRS response, IGA success, composite PP-NRS and IGA response, and DLQI change from baseline), the time points assessed, and the choice of the model fit (i.e., fixed-effects model versus random-effects model). Overall, although the findings of the NMA are subject to considerable limitations due to the significant heterogeneity between the trials included in the analyses, the clinical experts consulted for this review indicated that the results overall aligned with their expectations that the effects of nemolizumab would be comparable to those of dupilumab in clinical practice.
Sensitivity analyses were conducted based on the non-TCS and/or non-TCI populations. Based on the fixed-effects model, for PP‑NRS response at week 12, a favourable effect of nemolizumab was reported. No favourable effect of nemolizumab was reported for other outcomes. Based on the random-effects model, a favourable effect for nemolizumab was observed for PP-NRS response at week 12, while no favourable effect for nemolizumab was observed for other outcomes (e.g., composite PP-NRS and IGA response at week 24).
Harms Result
Based on both the fixed- and random-effects models, the NMA did not show a favourable safety profile for nemolizumab compared with dupilumab.
Critical Appraisal
Overall, the NMA was conducted according to accepted methodological guidance. The potential key limitations of the NMA were the considerable heterogeneity across the included studies in terms of study and patient characteristics (e.g., eligibility criteria, population, and trial duration). For example, the dupilumab trials required patients to have a history of a 2-week course of a medium-potent to superpotent TCS having failed, or for it to be medically inadvisable for them to use a TCS; however, this was not required in the nemolizumab trials. Therefore, the NMA results may be biased in favour of nemolizumab because patients with a history of treatment refractoriness (in the dupilumab trial) may have a relatively poorer response during the trial than patients without such a history. The significant differences in patient and trial characteristics across the included studies may threaten the transitivity assumption for the NMA analysis.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
Evidence from the OLYMPIA 1 and OLYMPIA 2 trials demonstrated that, compared to placebo, treatment with nemolizumab resulted in greater improvements in itch response, lesion clearance, and HRQoL in adult patients with moderate to severe PN. The GRADE assessment suggested that, at 16 weeks, compared to placebo, nemolizumab results in a clinically meaningful increase in the proportion of patients with at least a 4-point improvement in PP-NRS score and in the proportion of patients with IGA success. The GRADE assessment also suggested that nemolizumab likely results in a clinically meaningful increase in the proportion of patients with at least a 4-point improvement in DLQI score compared to placebo at 16 weeks (evidence from the OLYMPIA 2 trial) and 24 weeks (evidence from the OLYMPIA 1 trial).
In the OLYMPIA 1 and OLYMPIA 2 trials, most patients in the nemolizumab and placebo groups experienced at least 1 AE. In both trials, the proportions of patients who experienced the AESI of newly diagnosed asthma or worsening of asthma were low, and the GRADE assessment suggested low certainty regarding the effect of nemolizumab on this AESI compared to placebo.
The ability to draw conclusions regarding the long-term efficacy of nemolizumab is limited by the single-arm, open-label design of the OLYMPIA LTE study and by the patient attrition that occurred throughout the study. The harms results in the LTE study aligned with those of the pivotal trials, with no new AEs identified.
The NMA demonstrated a favourable benefit of nemolizumab compared with dupilumab. However, the favourable effect was not consistent, and it was influenced by the outcomes measured and time points assessed. The NMA may suggest a comparable clinical efficacy and overall safety profile between nemolizumab and dupilumab in the treatment of patients with PN; however, the findings of the NMA are subject to considerable limitations.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of nemolizumab 30 mg per 0.49 mL for SC injection in the treatment of moderate to severe PN. The approved Health Canada indication is for nemolizumab to be used for the treatment of adults with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. This review is based on the sponsor’s initial reimbursement request for nemolizumab to be used for the treatment of moderate to severe PN. The sponsor has revised the reimbursement request to align with the approved Health Canada indication; however, the revision did not warrant a significant update to the report.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
PN is a rare, neuroimmune, inflammatory skin disease characterized by the presence of chronic (≥ 6 weeks) itch; history and signs of repeated scratching, such as excoriation and scars; and multiple, localized or generalized, hyperkeratotic, and symmetrically distributed pruritic nodules on the extensor surfaces of the extremities and trunk.3-7 РΝ is frequently associated with a history of AD.4 The etiology and pathophysiology of PN are still unclear; however, it is thought to be associated with IL-31–driven neuroimmune dysfunction, immune and neuronal dysregulation, and skin tissue remodelling.27 Itch is the most burdensome symptom for patients with PN.28 Itching and scratching cause severe skin barrier damage, thickening and crusting of nodules, and scarring once the nodules heal.29 Nodules resulting from PN are disfiguring and affect large body surface areas.30 Different etiologies may trigger pruritus (induction phase), which leads to the scratching that leads to neuronal sensitization (chronicity phase) and the development of the pruriginous lesions (disease stage).20,31
The exact incidence and prevalence of PN in Canada are unknown. In an epidemiological study of adults in the US aged 18 to 64 years, the estimated prevalence was 72 per 100,000 persons.4 ΡN can occur in all age groups and without sex preferences. However, it primarily affects older adults,4 with approximately 69% of patients aged 51 years or older.5,9-11 In addition, in the US, PN seems to be more common among African Americans than among people from other ethnic origins.4
PN is associated with an increased prevalence of comorbidities of dermatologic, systemic, neurologic, and/or psychiatric origin.14,32,33 PN leads to significantly reduced QoL because the symptoms, such as itch and skin lesions, affect patients’ personal and social lives.12,13 PN interrupts sleep quality due to the nocturnal itch-scratch cycle; it is strongly associated with fatigue.15 It also disrupts day-to-day activities, such as getting dressed, self-care or personal hygiene, planning activities, and performing chores.13
Diagnosis of PN requires a physical examination and assessment of clinical symptoms; the symptoms may be similar to those associated with other chronic itch or excoriating disorders. It is estimated that 43% of patients with PN are initially misdiagnosed;16 additional bloodwork may be required to differentiate patients with an unknown underlying cause (e.g., chronic kidney disease, liver disease, thyroid disease, HIV infection, parasitic infection, or malignancy). 4,17,18 However, the clinical experts consulted by CDA‑AMC noted that if PN is diagnosed by a dermatologist, a biopsy is not required to confirm the diagnosis. Given the burden of PN and its associated comorbidities, patients with PN have high rates of health care and specialty care utilization.19
Standards of Therapy
The contents of this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
According to clinical experts consulted by CDA-AMC, the primary goals of therapy for patients with PN are to reduce the severity of pruritus, treat existing lesions (i.e., reduce both the number and size of nodules), prevent the formation of new lesions, and improve HRQoL while minimizing any adverse effects of treatment. In Canada, no formal practice guidelines for the management of PN exist. International guidelines and published literature describe the use of various medications and procedures for the treatment of PN using a stepwise and multimodal approach, considering factors such as the patient’s age, comorbidities, the severity of PN, QoL, concomitant medications, and the treatment side effect profile.10,20-22 The guideline recommendations include emollients for PN of any severity and topical medications (e.g., corticosteroids, calcineurin inhibitors, capsaicin), intralesional corticosteroids, and H1 antihistamines as initial-tier treatment options. Guidelines state that various systemic medications (e.g., methotrexate, cyclosporine, gabapentinoids, thalidomide, dupilumab) and UV phototherapy are used as later-tier options, but note that that patients may be considered for treatment at any tier, based on clinical presentation. Most medications for PN are used off-label and have limitations with respect to evidence of efficacy; certain treatment options (e.g., methotrexate and cyclosporine) are also associated with safety concerns.20-22 In Canada, dupilumab is currently the only treatment with an approved indication for the treatment of PN.2,23
Clinical experts consulted by CDA-AMC stated that PN is often managed initially with high-potency TCSs to reduce inflammation and alleviate itching. Other initial treatment options include TCIs, topical anesthetics, and oral antihistamines. Clinical experts noted that intralesional corticosteroid injections are often effective for flattening nodules and reducing itch, but that these treatments may not be suitable for multiple lesions. Other therapies that may be considered for PN include topical capsaicin, gabapentin, pregabalin, carbamazepine, doxepin, mirtazapine, mycophenolate mofetil, low-dose naltrexone, and UV light treatment (i.e., phototherapy). Clinical experts commented that systemic immunosuppressants (i.e., methotrexate or cyclosporine) may be considered for severe or treatment-resistant disease, but noted that these may cause significant side effects and may not be appropriate for long-term use, older patients, or those with polypharmacy. Clinical experts highlighted that PN is difficult to treat; in addition, the currently available treatment options may provide only partial and/or short-term symptomatic relief, and the use of these treatments is often limited by side effects, safety risks, and access and feasibility issues. Clinical experts expressed the need for highly effective, disease-modifying, systemic PN therapies that provide sustained relief, are safe for long-term use, and are convenient and accessible.
Drug Under Review
The key characteristics of nemolizumab are summarized in Table 3 along with other treatments available for PN.
According to the product monograph,1 nemolizumab is available as an SC injection, and the dosing is weight-based. For patients who weigh less than 90 kg, nemolizumab is recommended to be administered at an initial dose of 60 mg through 2 injections of 30 mg each, followed by 30 mg administered every 4 weeks. For patients who weigh 90 kg or more, nemolizumab is recommended to be administered at an initial dose of 60 mg through 2 injections of 30 mg, followed by 60 mg every 4 weeks. The patient may self-inject or the caregiver may administer nemolizumab. Before the first injection, patients and/or caregivers should be given proper instructions for preparing and administering nemolizumab.
Nemolizumab is a humanized immunoglobulin G2 monoclonal antibody that inhibits IL-31 signalling by binding selectively to the IL‑31 receptor alpha. IL-31 is a cytokine that is involved in pruritus, inflammation, epidermal dysregulation, and fibrosis. Although the mechanism of action of nemolizumab has not been fully established, the drug inhibits IL-31–induced responses, including the release of proinflammatory cytokines, such as IL-6.1
Nemolizumab has not been previously reviewed by CDA-AMC.
Nemolizumab has been approved for the treatment of adults with PN by the FDA in the US.34 It has also been approved by the European Medicines Agency (in the European Union) for the treatment of adults with moderate to severe PN who are candidates for systemic therapy.35 Nemolizumab is currently under review for the treatment of PN in adults by the National Institute for Health and Care Excellence in England.36
At the time of this CDA-AMC review, nemolizumab was also under review by Health Canada and CDA-AMC for AD, with the following requested reimbursement criteria submitted to CDA-AMC: treatment of moderate to severe AD in patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and/or who are refractory to or ineligible for systemic immunosuppressant therapies.37
Table 3: Key Characteristics of Nemolizumab and Dupilumab
Characteristic | Nemolizumab | Dupilumab |
|---|---|---|
Mechanism of action | Inhibition of interleukin-31 signalling by binding selectively to interleukin-31 receptor A | Inhibition of interleukin-4 and interleukin‑13 |
Indicationa | For the treatment of adults with moderate to severe prurigo nodularis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable | Treatment of adult patients with moderate to severe prurigo nodularis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
Route of administration | SC | SC |
Recommended dose | For patients who weigh < 90 kg: Initial dose of 60 mg (2 injections of 30 mg each) followed by 30 mg every 4 weeks For patients who weigh ≥ 90 kg: Initial dose of 60 mg (2 injections of 30 mg each) followed by 60 mg every 4 weeks | 600 mg initial dose followed by 300 mg every 2 weeks |
Serious adverse effects or safety issues | Nemolizumab is contraindicated in patients who are hypersensitive to the drug or to any ingredient in the formulation, including any nonmedicinal ingredient or any component of the container. Warnings and precautions: Hypersensitivity reactions (immediate or delayed), vaccinations (avoid use of live vaccines and consider completion of all age-appropriate vaccinations), respiratory reactions (i.e., cases of worsening asthma in patients with pre-existing asthma were observed in clinical studies) | Hypersensitivity reactions, serious systemic eosinophilia, conjunctivitis, and keratitis |
SC = subcutaneous.
aHealth Canada–approved indication.
Sources: Product monographs for nemolizumab1 and dupilumab.23
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
A single patient group, the CSPA, provided input for this submission. The CSPA is a national charity that supports the health and well-being of people across Canada affected by skin, hair, and nail conditions through collaboration, advocacy, and education. The CSPA gathered information regarding the experience of people living with PN through a patient and caregiver survey conducted from September 12, 2024, to November 29, 2024. Nine survey responses (from 8 patients and 1 caregiver) were received from people in Canada (New Brunswick [n = 1], Quebec [n = 1], Ontario [n = 5], Alberta [n = 1], and Northwest Territories [n = 1]). None of the survey respondents had experience with nemolizumab.
Five respondents provided their ages. All were older than 35 years, with more than half older than 55 years. Of those who answered the question, 5 respondents had had PN for fewer than 5 years; most respondents reported having severe (n = 3) or moderate (n = 1) PN, and none reported mild PN. All survey respondents reported their arms and backs being affected by the condition, while 75% reported impacts on their legs and 50% reported impacts on their buttocks. All the patients reported itchy skin, itchy bumps (nodules), burning or stinging skin, scratching, pain, and hyperpigmentation (dark spots). Three patients reported skin scarring because of PN. One patient reported experiencing side effects of flares and hypopigmentation (light spots).
The survey respondents mentioned that PN affects the following aspects of their lives: family relationships, intimate relationships, work life, mental health, social life, daily activities, sleep, self-esteem, finance, and sex. Twenty-five percent of respondents shared that they miss work 5 to 10 times monthly. The CSPA highlighted that caregivers were also affected, given that witnessing their loved ones endure emotional pain, insecurity, and social withdrawal caused a psychological burden and left them feeling helpless. Additionally, the caregiver in the survey shared that it can be difficult to encourage their loved one to continue with treatments for PN.
All respondents had used TCSs in the past and rated their experience as either “did not work very well” (60%) or “no change” (40%). In addition, 80% of respondents indicated that they had used topical capsaicin, oral antihistamines, and methotrexate, and 60% had tried TCIs, narrow-band UVB phototherapy, and medical cannabis. However, they noted that these did not work very well or that there was no change. Two respondents reported the use of dupilumab in the past, but they had “no change” in their condition.
Patients reported experiencing side effects with currently available treatments, including a racing heart, skin irritation, nausea, vomiting, hypopigmentation, and hyperpigmentation. One respondent stopped treatment for PN because of side effects, and 3 respondents stopped their treatments because of a lack of efficacy. The CSPA highlighted that 4 of 5 respondents strongly agreed that they would be interested in a new treatment for PN and that they wished there was a better PN treatment option for them. Of the 5 respondents, 3 respondents disagreed — and 2 respondents strongly disagreed — with the statement that they were satisfied with their current treatment for PN. Respondents also shared that financial challenges affected their access to PN treatments.
The respondents identified effectiveness, affordability, and lack of side effects as the top priority characteristics of a new PN treatment. Others included the treatment being easy to take or apply and conducive to the patient’s schedule. According to the patient group input, in addition to managing itch, psychological and social relief are the motivations underlying these desired outcomes. The caregiver respondent disclosed that the cost of medication is the most important aspect of a new treatment for them.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the drug’s potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PN.
Unmet Needs
Clinical experts consulted by CDA-AMC expressed that there is an unmet need for disease-modifying, systemic therapies that address the underlying pathophysiological mechanisms of PN; are highly effective in achieving treatment goals (e.g., itch relief, treatment of lesions); provide sustained disease control and symptom relief; are safe for short- and long-term use; and are convenient and accessible. Clinical experts noted a need for treatments that are safe across diverse patient populations (e.g., all ages, particularly older patients; those with comorbidities, such as chronic kidney disease, chronic liver disease, HIV, and hepatitis; and patients who are pregnant). Clinical experts highlighted the limitations of traditional treatment options. For example, topical therapies (e.g., high-potency topical steroids) may provide limited benefit, may be impractical or unsafe for extensive disease, and may be inconvenient. Intralesional corticosteroid injections may be painful and cannot address multiple lesions simultaneously. Systemic immunosuppressants (e.g., methotrexate and cyclosporine) lack disease specificity, have limited efficacy, require regular monitoring, and are associated with significant adverse effects and contraindications that preclude their use in the population of patients typically affected by PN. Phototherapy has significant limitations in terms of access and patients’ ability to adhere to treatment. Clinical experts highlighted that resistance to treatments can occur, with patients experiencing recurrent symptoms or worsening of disease.
Place in Therapy
Clinical experts commented that, along with dupilumab, nemolizumab is expected to cause a shift in the current treatment paradigm for PN in Canada. Clinical experts expect that, as novel drugs targeting pathways that play a major role in PN pathogenesis, biologic therapies will redefine the treatment approach, providing benefit beyond the symptomatic management offered by conventional treatments. Clinical experts expected that traditional, less expensive therapies (e.g., TCSs) would still be used for the initial management of PN because these are generally well-tolerated and can effectively manage mild to moderate cases; nemolizumab would be an appropriate choice for patients with moderate to severe PN who do not respond to these treatments. According to clinical experts, nemolizumab would also be expected to be used in patients with more severe, chronic, or refractory disease when standard therapies have failed to provide adequate relief. Clinical experts noted that it would be reasonable for patients to try TCSs or other first-line therapies initially, but that for patients with moderate to severe PN who do not respond, initiating nemolizumab earlier would be appropriate to avoid unnecessary delays in relief. Clinical experts stated that nemolizumab or dupilumab would likely be the preferred systemic treatment options over systemic immunosuppressants because biologic therapies offer safer, more targeted options. Clinical experts stated that, based on their experience with previously approved biologic medications across different indications, there are instances where the biologic on its own is sufficient, and this may be the case with nemolizumab. However, they noted that nemolizumab would likely be combined with TCSs (especially when starting nemolizumab) and/or intralesional corticosteroids (as needed). Clinical experts also noted that, although less likely, nemolizumab could be combined with phototherapy if patients are not experiencing an adequate response and that nemolizumab may be combined with methotrexate in very severe cases. (However, combining nemolizumab and cyclosporine therapy would be very unlikely.)
Patient Population
Clinical experts stated that nemolizumab would best suit patients with moderate to severe or refractory PN, particularly those with persistent pruritus and large numbers of chronic lesions for whom conventional treatments have failed. Clinical experts also commented that the patients most in need of treatment are those who have chronic disease that significantly affects their QoL (e.g., severe itch, sleep disruption, social isolation) as well as those who have contraindications to conventional systemic therapies. According to clinical experts, the patients who are least suited for nemolizumab would be those with mild PN or mild symptoms that can be managed with conventional treatments; nemolizumab may not be recommended for individuals with compromised immune function or those with active infections, although clinical experts noted that this can be debated. In clinical practice, suitable patients would be identified using clinical judgment, based on an assessment of the number of lesions, symptom severity, and QoL. Clinical experts expressed that PN is a clinical diagnosis; they noted that skin biopsy may be performed, but PN on biopsy can be similar to other chronic itch or excoriating disorders, and biopsy results are not entirely specific to PN. Clinical experts also commented that underdiagnosis of PN is possible due to its similarities with other pruritic skin conditions (e.g., eczema, psoriasis).
Assessing the Response to Treatment
According to clinical experts, treatment response in patients with PN is assessed in clinical practice based on improvement in the number of lesions, reduction in pruritus, and improvement in QoL; physicians may prioritize these outcomes differently. The clinical experts noted that assessment of treatment response in clinical practice does not usually rely on the grading tools used in clinical trials as stringently. Disease severity may be assessed using lesion count (including subjective improvement) or scoring systems like physician global assessment, with a clinically meaningful response being a score of 0 or 1 (i.e., 5 or fewer nodules). Clinical experts noted that itch intensity may be measured using an NRS, with a reduction of 4 or more points considered clinically meaningful; however, it may also be measured subjectively, with patient-reported “good to excellent” improvement being considered clinically meaningful. QoL may be assessed using the DLQI or other indicators, such as improvement in daily life, sleep, and social well-being. Clinical experts indicated that response would typically be assessed 3 to 6 months after the initiation of PN treatment and at the same interval while the disease is active, with annual follow-up if the disease is controlled (with or without therapy). The experts indicated that the initial treatment response for a biologic would usually be assessed after 6 months.
Discontinuing Treatment
When deciding to discontinue treatment with nemolizumab, the clinical experts would evaluate whether the patient had achieved the desired clinical outcomes within a 6-month time frame. Specifically, a clinically important response would be a decrease in the lesion count from 20 or more at the start of therapy to 5 or fewer, or a decrease in peak itch severity by at least 4 points out of 10 on the NRS. The clinical experts emphasized that some patients may not meet these specific parameters, but for patients experiencing some improvement who wish to continue, therapy would not be stopped. In patients with a partial response to therapy, the decision to stop treatment would consider whether any other effective treatment options were available. Moreover, clinical experts expressed that they take a patient-centred approach and consider patient-reported happiness when deciding whether to continue therapy. The experts also noted that pruritus may improve before lesions or vice versa, and that both may not always show simultaneous improvement. If neither outcome is achieved, discontinuation due to lack of improvement would be considered. Additional factors supporting discontinuation may include SAEs or the emergence of comorbidities requiring other treatments; however, the primary focus is on achieving clinical milestones related to disease severity and symptom control.
Regarding the possibility of withdrawing nemolizumab in patients who achieve treatment goals, clinical experts noted that PN is a chronic, immune-mediated condition; as such, there may be risk of relapse if nemolizumab treatment is stopped. This has been observed in patients with PN who have stopped other therapies. As such, clinical experts expected that, based on current clinical knowledge, patients would continue to take nemolizumab over the long term to treat PN. No dose adjustment (i.e., lowering of the dose) would be expected to occur in patients who respond well to maintenance therapy with nemolizumab.
Prescribing Considerations
Both clinical experts agreed that, due to the complexity of diagnosing PN, nemolizumab treatment for PN should be prescribed only by dermatologists. PN can be underdiagnosed, overdiagnosed, or misdiagnosed by other providers. PN has a broad differential that requires the clinician to rule out other potential causes. Patients with PN can also have other underlying diagnoses. Clinical experts noted that dermatologists are best equipped to maximize nemolizumab treatment outcomes by optimizing the use of concurrent conventional therapies and that academic (hospital) and community dermatologists are well-equipped to diagnose PN and prescribe nemolizumab.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Two clinician groups, the Atlantic Dermatology Group and the DAO, provided their input for this submission. The Atlantic Dermatology Group is a group of dermatologists practising in the Atlantic provinces (in hospitals, in academic positions in medical schools, and as investigators with extensive experience in clinical trials and research). Input was provided by 5 clinicians, and information was gathered from sessions related to PN at the September 2024 European Academy of Dermatology and Venerology in Amsterdam and from the literature. The DAO consists of community dermatologists as well as nationally and internally recognized experts in the treatment of acne. It provides a unified voice for Ontario dermatologists in promoting better patient care, promoting dermatology in Ontario, and supporting research and education within the community. Input was provided by 7 clinicians, and information was gathered from clinical trial data, available literature retrieved through PubMed, and experience with the use of nemolizumab from clinical trialists in Canada.
Both clinician groups highlighted that, currently, there are no specific clinical practice guidelines for PN in Canada. They indicated that current therapies include topical treatments, such as corticosteroids and emollients, dupilumab (approved by Health Canada for PN, but not publicly reimbursed), and off-label regimens (i.e., systemic immunosuppressants, such as methotrexate, cyclosporine, thalidomide, lenalidomide, tricyclic antidepressants, Janus kinase inhibitors, neurokinin-1 receptor antagonists, and monoclonal antibodies targeting IL-4 pathways). The DAO also highlighted that nondrug treatments, such as UVB phototherapy and psychosocial approaches, have been attempted, but that these have been associated with limited availability and success in addressing the psychological burden of PN and improving overall QoL. The Atlantic Dermatology Group further highlighted evidence from a systematic review that concluded that while phototherapy, corticosteroids, cyclosporin, and methotrexate represent viable options, the potential benefits are limited because of the risk of relapse and potential side effects.
The DAO clinician group highlighted the following unmet needs in PN treatment: therapies that provide rapid and sustained relief from severe itch, improve skin lesions, and address the psychosocial impacts of the disease (e.g., sleep disturbances and mental health issues). The group highlighted the poor tolerance and/or inconvenience associated with other current treatments and the need to target the underlying neuroimmune mechanisms that drive PN. The Atlantic Dermatology Group clinicians noted the need for a therapy that provides clinically significant reductions in itch and improves physician global assessment of skin lesions along with QoL.
The Atlantic Dermatology Group indicated that nemolizumab will shift the current treatment paradigm because it can be used as first-line therapy in patients with PN, including those who are intolerant to dupilumab, TCSs, or phototherapy, or for whom these treatments have failed. The group also noted that nemolizumab would be used as a stand-alone treatment. However, the DAO clinician group indicated that nemolizumab would fit as a targeted, second-or third-line option in patients for whom topical therapies and phototherapy produced an insufficient disease response or for whom these treatments have failed.
The DAO clinician group noted that the patients best suited for treatment with nemolizumab would be those with moderate to severe PN who did not achieve adequate relief from topical therapies or phototherapy, or those unable to access or tolerate these treatments. This would include those who have persistent and intense pruritus, widespread nodular lesions, or significant impairment in QoL. The Atlantic Dermatology Group highlighted that those patients with moderate to severe PN and an itch score of 7 or higher with at least 20 lesions would be in most need and best suited for treatment. The DAO clinician group further noted that patients with contraindications (such as a known hypersensitivity to nemolizumab), those with milder disease that responds to topical steroids, and those with conditions that significantly compromise their immune function (unless the benefits outweigh the risks) would not be suitable for treatment with nemolizumab.
The DAO clinician group noted that to identify the patients who would most likely respond to nemolizumab, a clinician assessment of the intensity and persistence of itch, the extent of nodular lesions, and the impact on daily functioning and QoL was sufficient. The Atlantic Dermatology Group noted that no clinical, pathological, or chemical markers exist to help clinicians identify the patients most likely to exhibit a response to treatment.
The Atlantic Dermatology Group noted that misdiagnoses were unlikely for patients managed by dermatologists in clinical practice. It emphasized the consideration of other diagnoses (i.e., pemphigoid nodularis, mastocytosis, lichen planus, nodular scabies, arthropod bites, lymphocytoma cutis, lymphomatoid papulosis, amyloidosis, reticulohistiocytosis, and cutaneous T-cell lymphoma). However, the DAO clinician group indicated that PN may be underdiagnosed or misdiagnosed as other pruritic dermatoses and that clinician education and awareness are key to improving diagnostic accuracy.
Both clinician groups noted that a 4-point improvement in PP-NRS score should be used to determine if a patient is responding to treatment. The Atlantic Dermatology Group and the DAO noted that a physician’s IGA finding of clear or almost clear and a reduction of at least 2 points from baseline are the outcomes used to determine treatment response. In addition, the Atlantic Dermatology Group highlighted that DLQI should be measured, while the DAO highlighted the importance of assessing subjective feedback on daily comfort and sleep quality.
Both clinician groups agreed that treatment discontinuation should be considered if there is a lack of meaningful reduction in PP-NRS or persistent nodular lesions as assessed by IGA. The Atlantic Dermatology Group noted that treatment should be discontinued after 6 months of therapy if there is a lack of response. Additional criteria for discontinuation were noted by the DAO, including significant AEs (i.e., hypersensitivity, injection-site reaction, and long-term safety issues, including recurrent infections). Disease progression (such as worsening lesions), complications such as skin infections, and patient-reported dissatisfaction were also noted as important factors requiring treatment reassessment. In cases in which systemic immunosuppressants or other biologics are required to manage refractory disease, nemolizumab may no longer be an optimal choice.
The Atlantic Dermatology Group indicated that PN should be managed by dermatologists experienced in both the diagnosis and management of PN and the use of biologics. The group also noted that dermatologists should be experienced in counting nodules and assessing patients through the DLQI, IGA, and PP-NRS. The DAO clinician group noted that, in addition to dermatologists, allergists or immunologists may be involved in managing patients with PN where PN overlaps with other atopic or immune-mediated conditions. The group highlighted that while the diagnosis, treatment initiation, and monitoring of patients requires the expertise and oversight of specialists, nemolizumab may be prescribed in various clinical settings (such as community clinics, hospital outpatient departments, and specialty dermatology or allergist clinics) and administered by patients at home.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Other implementation issues regarding relevant comparators (e.g., access and funding, covered populations) Comments for awareness:
Question for clinical experts:
| Clinical experts commented that dupilumab would be the most relevant comparator to nemolizumab; there are no other drugs with similar clinical effects that should be considered relevant comparators. |
Considerations for initiation of therapy | |
Disease diagnosis and scoring or staging for eligibility Key inclusion criteria for the pivotal trials:
The sponsor notes: “Diagnosis of PN requires a physical examination and assessment of clinical presentation of symptoms. In some cases, a biopsy may be required to examine tissue for confirmation of diagnosis, and to distinguish from other skin conditions.” Questions for clinical experts and/or CDEC:
| Clinical experts commented that a having a PN diagnosis for at least 6 months would be reasonable, given that most patients would have had this condition for months to years. Clinical experts commented that, when determining patient eligibility, a specific itch score requirement may not be necessary; however, the criterion of severe itching would be suitable because patients with moderate to severe PN would be experiencing severe itch. However, clinical experts acknowledged that using a PP-NRS score at baseline may be useful to allow for objective follow-up in determining clinical improvement in itch. Clinical experts expressed that the fulfillment of both disease severity criteria may not be necessary because patients with 20 or more lesions would also have an IGA score of 3 or more. Clinical experts suggested that the eligibility criteria could state, “≥ 20 pruriginous nodular lesions on the body with a bilateral distribution and/or IGA ≥ 3,” or state only 1 of these criteria. Clinical experts noted that PN is a clinical diagnosis. Clinical experts commented that if PN is diagnosed by a dermatologist, a biopsy is not required to confirm the diagnosis. PN on biopsy can be similar to other chronic itch or excoriating disorders, and biopsy results are not entirely specific to PN. |
Prior therapies required for eligibility Relatively small proportions of patients in the pivotal trials received prior therapies for PN, aside from topical corticosteroids. Question for clinical experts and/or CDEC:
| Clinical experts stated that it is reasonable that all patients should have tried moderate- to high-potency topical corticosteroids before starting nemolizumab, especially considering that these are likely to have been prescribed by family physicians before patients were able to consult a dermatologist. Clinical experts commented that patients should not be required to have received methotrexate or cyclosporine due to the high failure rate, side effects, and monitoring requirements associated with these medications. (In addition, cyclosporine is not a safe long-term treatment option in PN.) Clinical experts would prefer to offer patients nemolizumab before prescribing methotrexate or cyclosporine. Clinical experts commented that it is not reasonable to require patients to have tried phototherapy before receiving treatment with nemolizumab. There are barriers to access, and not all patients are able to adhere to treatment. |
Consistency with initiation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space It is expected that a CDA-AMC recommendation will be issued for dupilumab before nemolizumab has been reviewed by CDEC. Comment for CDEC:
| Comment from the drug programs to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Challenges related to assessing and monitoring therapeutic response The primary end points in the pivotal trials were:
The draft product monograph notes: “Some patients with initial partial response may subsequently improve with continued treatment beyond 16 weeks.” Questions for clinical experts and/or CDEC:
| Clinical experts commented that itch response is the key consideration for continuation of therapy. Criteria for continuation of therapy could include itch response and/or improvement in the number of lesions; however, patients should not be required to fulfill both criteria to continue treatment. Clinical experts considered itch response to be at least a 4-point improvement in PP-NRS score from baseline. One clinical expert commented that improvement in the number of lesions could be measured as an IGA score of 0 or 1. The other clinical expert suggested that an IGA of 0 or 1 or a 2-point improvement in IGA score from baseline could be used as a measure of improvement in the number of lesions. Clinical experts commented that assessment for treatment renewal (i.e., response to treatment) should occur 4 to 6 months following the initiation of nemolizumab. Clinical experts noted that assessment at 6 months is preferable; 1 clinical expert specified that assessment of lesions should occur at this time point, not at 4 months. For patients with good response to treatment at the initial assessment, the clinical experts commented that the second follow-up assessment would occur at 6 months or 1 year (experts suggested different time points), and annually thereafter. Clinical experts agreed that for patients who do not maintain good response to therapy (as previously defined), treatment with nemolizumab should be discontinued. Regarding continued treatment beyond 16 weeks in patients with partial response, 1 clinical expert commented that patients should instead be assessed at 24 weeks; if treatment response (as previously defined) has not been achieved at this point, improvement thereafter is unlikely. The other clinical expert commented that all patients should be allowed up to 24 weeks to become full responders; patients with partial response at 16 weeks should be seen again at 24 weeks, at which point treatment response (as previously defined) should be achieved to be able to continue treatment. |
Consistency with renewal criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space It is expected that a CDA-AMC recommendation will be issued for dupilumab before nemolizumab is reviewed by CDEC. Comment for CDEC:
| Comment from the drug programs to inform CDEC deliberations. |
Considerations for discontinuation of therapy | |
Consistency with discontinuation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space It is expected that a CDA-AMC recommendation will be issued for dupilumab before nemolizumab is reviewed by CDEC. Comment for CDEC:
| Comment from the drug programs to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
Concerns related to combination usage Patients in the pivotal trials received nemolizumab monotherapy. Question for clinical experts and/or CDEC:
| Clinical experts commented that it is likely that nemolizumab would be combined with topical corticosteroids and intralesional corticosteroids. Although less likely, nemolizumab could be combined with phototherapy if patients are not experiencing an adequate response. Nemolizumab may be combined with methotrexate in very severe cases. Combining nemolizumab and cyclosporine therapy would be very unlikely. It is also very unlikely that multiple biologics would be combined. |
Consistency with prescribing criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space It is expected that a CDA-AMC recommendation will be issued for dupilumab before nemolizumab is reviewed by CDEC. Comment for CDEC:
| Comment from the drug programs to inform CDEC deliberations. |
Generalizability | |
Populations of interest that match the indication, but for whom there are insufficient data Patients with any of the following were excluded from the pivotal trials:
Question for clinical experts and/or CDEC:
| One clinical expert noted that approximately 30% of patients with PN also have AD, and that the presence of AD should not preclude eligibility for treatment with nemolizumab in patients with moderate to severe PN; however, patients with AD alone (i.e., without moderate to severe PN) would not be eligible. The other clinical expert commented that all patients with AD and PN should be eligible to start nemolizumab. One clinical expert commented that patients with chronic pruritus resulting from another active condition in the absence of moderate to severe PN should be ineligible to receive nemolizumab, noting that PN can occur alongside other conditions that cause itching (e.g., uremic pruritus, AD). Regarding the use of nemolizumab in patients with chronic pruritus resulting from an active condition other than PN, the other clinical expert believed that this decision should be left to the discretion of the dermatologist. The clinical expert highlighted this as a reason why nemolizumab should be prescribed only by dermatologists who are able to manage these other conditions, adding that dermatologists would not unsafely prescribe nemolizumab if a patient were diagnosed with another itch-related dermatosis. Both clinical experts stated that their aforementioned comments would also be applicable in the context of neuropathic and psychogenic pruritus. |
Populations outside the indication or reimbursement request, but of interest to jurisdictions The pivotal trials included only adult patients. The draft product monograph notes: “The safety and efficacy of Nemluvio in pediatric patients with PN have not been established.” Question for clinical experts and/or CDEC:
| One clinical expert stated that for the time being, pediatric patients should be ineligible to receive nemolizumab, noting a lack of phase III studies conducted in the pediatric population. The clinical expert highlighted that PN is extremely rare in children. The other clinical expert stated that they cannot comment on the use of nemolizumab in pediatric patients. |
Care provision issues | |
Management of adverse effects In the pivotal trials, AD was reported as an AE with nemolizumab more frequently than with placebo (which seems odd, given that the sponsor is also seeking an NOC for AD). Question or comment for clinical experts and/or CDEC:
| One clinical expert commented that patients who develop AD while receiving nemolizumab for PN should have their treatment switched to another drug; for example, because dupilumab can be used for both AD and PN, it would not make sense to add dupilumab to nemolizumab when dupilumab monotherapy would be sufficient. The other clinical expert also noted that combining multiple biologics would be unlikely; the likely approach would be to find 1 biologic that is effective for both conditions. |
System and economic issues | |
Concerns regarding the anticipated budget impact and sustainability Comment for awareness:
| Comment from the drug programs to inform CDEC deliberations. |
AD = atopic dermatitis; AE = adverse event; CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; IGA = Investigator’s Global Assessment; NOC = Notice of Compliance; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of nemolizumab 30 mg per 0.49 mL powder and solvent solution for SC injection in the treatment of moderate to severe PN. The focus will be on comparing nemolizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of nemolizumab is presented in 4 sections, with our critical appraisal of the evidence included at the end of each. The first section, the Systematic Review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted LTE studies. The third section includes indirect evidence from the sponsor. The sponsor did not include additional studies addressing important gaps in the systematic review evidence (fourth section).
Included Studies
Clinical evidence from the following studies is included in the review and appraised in this document:
2 pivotal RCTs identified in the systematic review
1 LTE study
1 ITC.
Systematic Review
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | OLYMPIA 1 trial | OLYMPIA 2 trial |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, double-blind, placebo-controlled, randomized, parallel-group study | Phase III, multicentre, double-blind, placebo-controlled, randomized, parallel-group study |
Locations | 77 study sites in 10 countries (Austria, Canada, Denmark, Germany, Hungary, Italy, Poland, Sweden, the UK, and the US) | 55 study sites in 9 countries (Belgium, Canada, France, the Netherlands, Poland, South Korea, Spain, Switzerland, and the US) |
Patient enrolment dates | First patient randomized: October 21, 2020 Last patient’s last visit: February 21, 2023 | First patient randomized: October 29, 2020 Last patient’s last visit: March 30, 2022 |
Randomized (N) | N = 286 Nemolizumab group: n = 190 Placebo group: n = 96 | N = 274 Nemolizumab group: n = 183 Placebo group: n = 91 |
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention |
| |
Comparator | Matched placebo | |
Study duration | ||
Screening phase | 4 weeks | 4 weeks |
Treatment phase | 24 weeks | 16 weeks |
Follow-up phase | 8 weeks | 8 weeks |
Outcomes | ||
Primary end points |
| |
Secondary end points | Key secondary:
Secondary:b
| |
Publication status | ||
Publications | Stander et al. (2024)38 Clinical Trial IDs: NCT04501666 RD.06.SPR.202685 2019-004293-25 | Kwatra et al. (2023)39 Clinical Trial IDs: NCT04501679 RD.06.SPR.203065 2019-004789-17 |
ACT = asthma control test; AD = atopic dermatitis; AP-NRS = Average Pruritus Numerical Rating Scale; DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; HBcAb = hepatitis B core antibody; HBsAg = hepatitis B surface antigen; HCV = hepatitis C virus; HRQoL = health-related quality of life; IGA = Investigator’s Global Assessment; PAS = Prurigo Activity Score; PCR = polymerase chain reaction; PEF = peak expiratory flow; PGAD = Patient Global Assessment of Disease; PGAT = Patient Global Assessment of Treatment; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; PRO = patient-reported outcome; SD-NRS = Sleep Disturbance Numerical Rating Scale; TB = tuberculosis; ULN = upper limit of normal; WASO = wakefulness after sleep onset.
aIGA success was defined as an IGA of 0 (clear) or 1 (almost clear) and an improvement of 2 or more points from baseline.
bSecondary end points were assessed up to 24 weeks for the OLYMPIA 1 trial and up to 16 weeks for the OLYMPIA 2 trial.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 1 Clinical Study Protocol;40 OLYMPIA 2 Clinical Study Report;25 OLYMPIA 2 Clinical Study Protocol.41 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The OLYMPIA 1 and OLYMPIA 2 trials were both phase III, randomized, double-blind, placebo-controlled, parallel-group, multicentre studies investigating the efficacy and safety of nemolizumab versus placebo for the treatment of adult patients with PN. In the OLYMPIA 1 trial (N = 286) and OLYMPIA 2 trial (N = 274), patients were randomized 2 to 1 to receive nemolizumab or placebo across 77 sites (10 countries) and 55 sites (9 countries), respectively, including a total of 8 sites in Canada that randomized a total of 44 patients.2,24,25 Randomization was done by interactive response technology and stratified by study site and baseline body weight (< 90 kg and ≥ 90 kg) using an interactive response technology system. Each study comprised 3 phases: screening for enrolment (4 weeks in each study), treatment (24 weeks in the OLYMPIA 1 trial and 16 weeks in the OLYMPIA 2 trial), and follow-up (8 weeks in each study).2,40,41 The trial designs for the OLYMPIA 1 and OLYMPIA 2 studies are presented in Figure 1 and Figure 2, respectively. At the end of the 24-week OLYMPIA 1 or 16-week OLYMPIA 2 treatment periods, patients were eligible to enter an active treatment LTE study; participants in the LTE study were not required to complete the follow-up visit.42,43
The primary end points in the OLYMPIA 1 and OLYMPIA 2 trials were the proportions of patients with improvements of at least 4 points from baseline in PP-NRS score at week 16 and the proportions of patients reporting IGA success (defined as an IGA response of 0 [clear] or 1 [almost clear] and at least a 2-point reduction from baseline) at week 16. The key secondary end points were the proportions of patients with an improvement of at least 4 points from baseline in PP-NRS score at week 4; a PP-NRS score of less than 2 at week 16; an improvement of at least 4 points from baseline in SD-NRS score at week 16; an improvement of at least 4 points from baseline in SD-NRS score at week 4; and a PP-NRS score of less than 2 at week 4.2,40,41 Efficacy and safety assessments were performed at visits throughout the screening and treatment periods.42,43
Throughout both studies, patient safety was reviewed and monitored by an independent data monitoring committee. In addition, an independent adjudication committee (IAC) reviewed all asthma-related events and assessed whether these represented new diagnoses or worsening of asthma; it also assessed severity as mild, moderate, or severe. The IAC did not assess relationship to study drug. For the OLYMPIA 1 trial, the results presented in this review are based on a database lock of April 11, 2023; for the OLYMPIA 2 trial, the final database lock was on June 16, 2022.2,24,25
Protocol Amendments
Original protocols for both the OLYMPIA 1 and OLYMPIA 2 studies were finalized on February 21, 2020. Both trial protocols were amended a total of 4 times, and amendments occurred at the same time for both. The first 2 amendments (May 4, 2020, and June 25, 2020) were conducted before patient enrolment. The second 2 amendments (December 22, 2022, and November 19, 2021) occurred following the start of the studies and included updates to the eligibility criteria, clarifications of the protocol and statistical analysis plan, and the addition of the proportion of patients with PP-NRS score improvement of at least 4 points from baseline and IGA success at week 16, week 20, and week 24 as a secondary efficacy end point (week 20 and week 24 time points for the OLYMPIA 1 trial only). Amendments occurred before study unblinding and were not considered by the sponsor to affect the interpretation of the study results.2,24,25
Figure 1: OLYMPIA 1 Trial Design
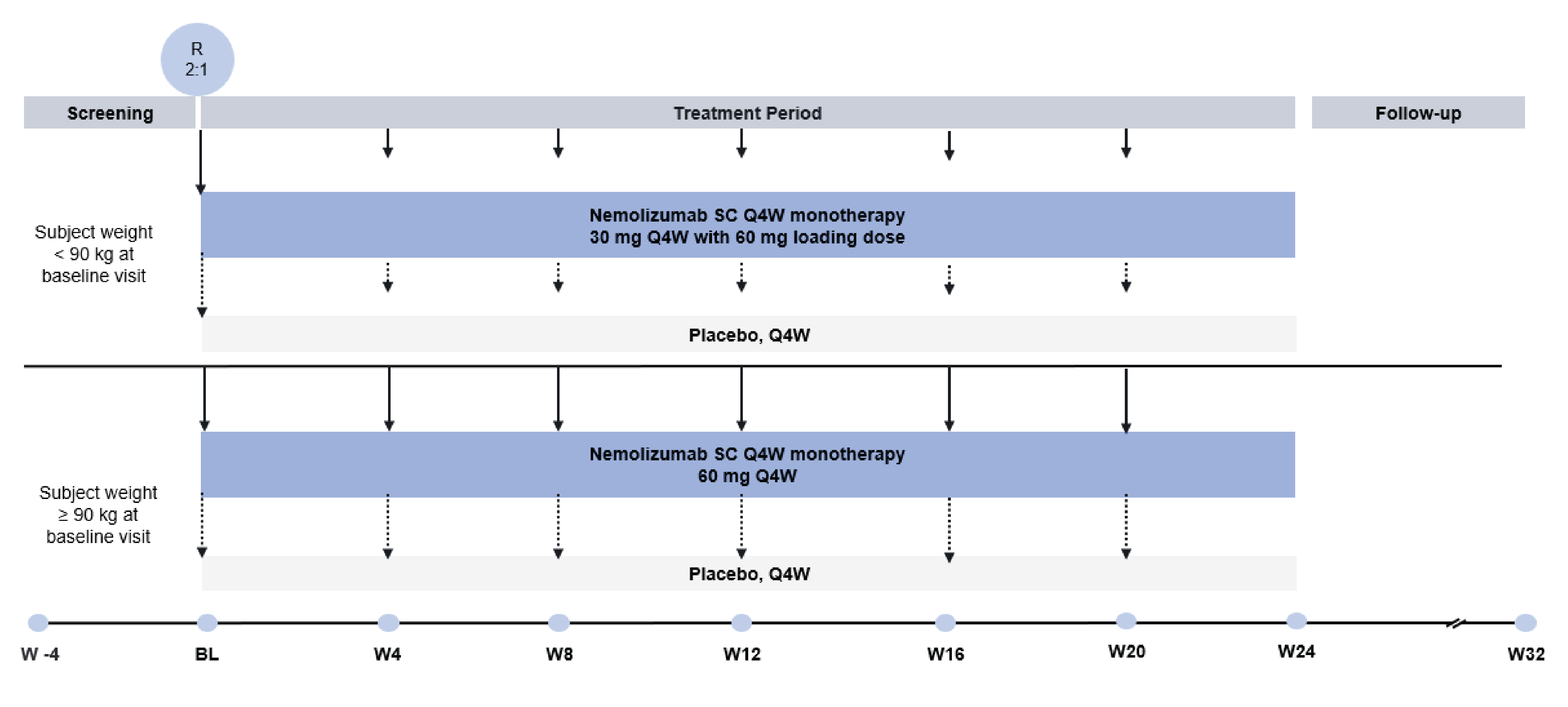
BL = baseline; Q4W = every 4 weeks; R = randomized; SC = subcutaneous; W = week.
Note: This schematic representation of the OLYMPIA 1 study reports the initial screening phase followed by the treatment period and follow-up period. The schematic was organized into 2 treatments groups of patients, those who weighed less than 90 kg and those who weighed 90 kg or more. The loading dose was 60 mg for patients who weighed less than 90 kg, while no loading dose was administered to patients who weighed 90 kg or more.44
Sources: Sponsor’s Summary of Clinical Evidence;2 sponsor’s data on file.45
Populations
Inclusion and Exclusion Criteria
The inclusion and exclusion criteria were the same for the OLYMPIA 1 and OLYMPIA 2 trials; the key eligibility criteria are provided in Table 5. For both studies, those eligible for participation were female or male patients aged 18 years or older with a clinical diagnosis of PN for at least 6 months with pruriginous nodular lesions on the upper limbs, trunk, and/or lower limbs, at least 20 nodules on the entire body (with bilateral distribution), and an IGA score of at least 3. Eligible patients were also required to have had severe pruritus, defined as a PP-NRS score of at least 7 at the screening and baseline visits. Exclusion criteria included body weight less than 30 kg, chronic pruritus from an active condition other than PN, unilateral lesions of prurigo, presence or history of certain infections or comorbidities, and the use of restricted therapies. The exclusion criteria also included the presence of uncontrolled or exacerbated asthma, the presence of COPD and/or chronic bronchitis, and active AD within the previous 3 months.2,40,41
Figure 2: OLYMPIA 2 Trial Design
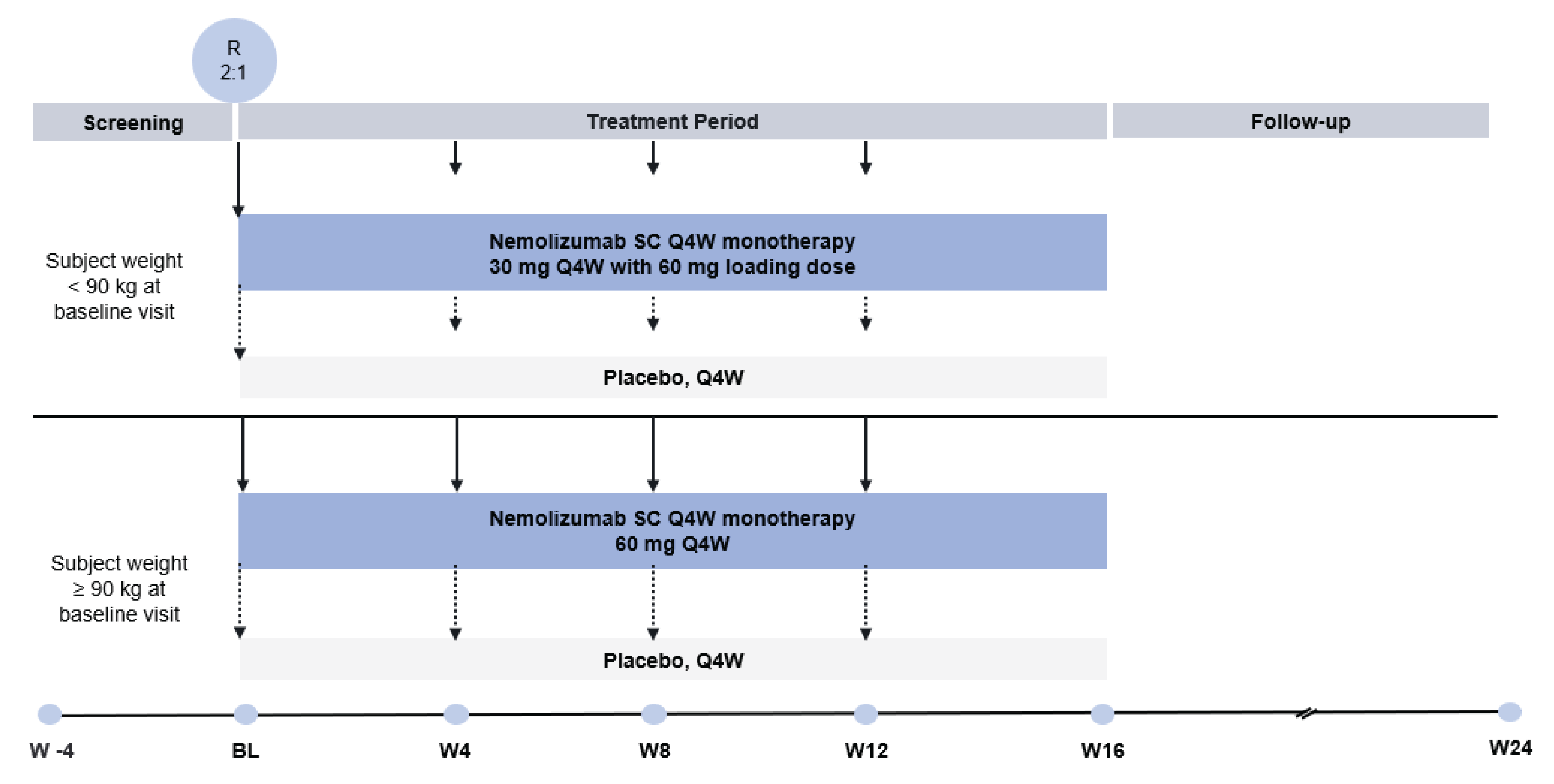
BL = baseline; Q4W = every 4 weeks; R = randomized; SC = subcutaneous; W = week.
Note: This schematic representation of the OLYMPIA 2 study reports the initial screening phase followed by the treatment period and follow-up period. The schematic was organized into 2 treatments groups, 1 with patients who weighed less than 90 kg and 1 with those who weighed 90 kg or more. The loading dose was 60 mg for patients who weighed less than 90 kg, while no loading dose was administered to patients who weighed 90 kg or more.44
Sources: Sponsor’s Summary of Clinical Evidence;2 sponsor’s data on file.45
Interventions
In both the OLYMPIA 1 and OLYMPIA 2 trials, the investigational treatment was nemolizumab. Patients in this treatment group were administered nemolizumab as an SC injection, with an initial dose of 60 mg, followed by either 30 mg every 4 weeks (for patients who weighed less than 90 kg at baseline) or 60 mg every 4 weeks (for patients who weighed 90 kg or more at baseline). Two 30 mg injections were used to administer a 60 mg dose.2,40,41 Dose modifications were not permitted.40,41 Nemolizumab was provided as a lyophilized powder in a dual-chamber syringe (DCS) for solution for injection. Patients randomized to the placebo arm of the OLYMPIA 1 and OLYMPIA 2 trials received matched SC placebo injections (also as lyophilized powder in a DCS for solution for injection) every 4 weeks, with the number of injections determined according to patient body weight at baseline, as done in the nemolizumab arm.2,40,41 The treatment duration in the OLYMPIA 1 trial was 24 weeks (with the last injection at week 20); in the OLYMPIA 2 trial, the duration was 16 weeks (with the last injection at week 12).40,41 Patients were trained on self-injection of the study drug and were permitted to do so at all subsequent visits while at the study centre and under staff supervision; for patients who did not wish to self-inject, study staff could administer the injection at each visit.2,40,41 The pharmacist or other qualified personnel performed the DCS preparation, including reconstitution. All study drug injections occurred at the study centre.40,41
Prohibited therapies for both the OLYMPIA 1 and OLYMPIA 2 trials are listed in Table 6. Except for these prohibited therapies, all therapies were authorized, including basic skin care (cleansing and bathing), moisturizers, bleach baths, and topical anesthetics. Patients could use daily moisturizers (if these did not contain any ingredients with anti-itch effect) starting from the screening visit; however, they should not have changed their moisturizers or emollients, nor should they have applied products for itch relief during the course of the trial. Antidepressants and sedatives (Table 6) were permitted, provided that these had been administered at a stable dose for at least 3 months before screening baseline and that dose changes were not planned during the trial.2,40,41
Rescue therapies were permitted to be prescribed to patients during the studies if the investigator deemed that these were medically necessary (e.g., to control intolerable PN signs or symptoms). Per the protocol general guideline and per the judgment of the individual investigator, rescue therapy should not have been prescribed within the first 4 weeks of the study to allow a minimum time for study drug exposure. Rescue therapies included TCSs, TCIs, oral antihistamines, systemic or intralesional corticosteroids, biologics (including their biosimilars), systemic nonsteroidal immunosuppressants and/or immunomodulators, phototherapy, and gabapentinoids.2,40,41 Investigators should first have prescribed topical medications or oral antihistamines before escalating to other systemic therapies, whenever possible. Unless the investigator judged there to be a concern, patients who received topical treatments, oral antihistamines, or UVB phototherapy as rescue therapy could continue to receive the study drug. Permanent discontinuation of study drug administration was required for patients who received systemic rescue therapy (other than oral antihistamines), intralesional corticosteroids, or oral psoralen plus UVA treatment; these patients were encouraged to complete the remaining scheduled study visits.24,25,40,41 In the efficacy analysis, treatment was deemed a failure for patients who received any rescue therapy. Investigator efficacy assessments (including as unscheduled visits for patients needing therapy between scheduled visits) should have been performed before initiating rescue treatment.2,40,41
Table 6: Prohibited Therapies in the OLYMPIA 1 and OLYMPIA 2 Trials
Treatments | Time frame | |
|---|---|---|
Before baseline and/or day 1 | Day 1 to week 32 (OLYMPIA 1 trial), day 1 to week 24 (OLYMPIA 2 trial) | |
Topical calcineurin inhibitors (tacrolimus, pimecrolimus) and topical corticosteroids | 2 weeks | Prohibiteda |
Vitamin D analogues and PDE-4 inhibitors | 2 weeks | Prohibited |
Any other topical treatment other than moisturizer (e.g., capsaicin, cryotherapy for treatment of PN) | 2 weeks | Prohibited |
Emollients or moisturizers with menthol, polidocanol, or others claiming to be anti-itch | 1 week | Prohibited |
Systemic or intralesional corticosteroids (corticosteroid inhalers were permitted) | 4 weeks | Prohibiteda |
Oral antihistamines (unless taken at a stable dose for 3 months before screening or for a seasonal allergy) | 1 week | Prohibiteda |
Drugs with sedative effects, such as benzodiazepines, imidazopyridines, barbiturates, sedative antidepressants (e.g., amitriptyline), SSRIs (e.g., paroxetine), or SNRIs (unless taken at a stable dose for at least 3 months before screening) | 1 week | Prohibited |
Phototherapy | 4 weeks | Prohibiteda |
Tanning beds | 4 weeks | Prohibited |
Immunosuppressive or immunomodulatory drugs (e.g., cyclosporine A, methotrexate, thalidomide, oral tacrolimus, cyclophosphamide, azathioprine, mycophenolate mofetil, JAK inhibitors) | 8 weeks or 5 half-lives (whichever is longer) | Prohibiteda |
Biologics and their biosimilars (e.g., dupilumab, etanercept, adalimumab, infliximab, omalizumab) | 8 weeks or 5 half-lives (whichever is longer) | Prohibiteda |
Systemic retinoids | 8 weeks or 5 half-lives (whichever is longer) | Prohibited |
Systemic roxithromycin, erythromycin | 1 week | Prohibited |
Opioid antagonists (e.g., naltrexone, naloxone), opioid partial and/or mixed agonists (e.g., nalbuphine, butorphanol), or opioid agonists (except when used for short-term and/or acute pain); NK1 receptor antagonists (e.g., aprepitant, serlopitant) | 4 weeks or 5 half-lives (whichever is longer) | Prohibited |
Gabapentinoids, unless used at a stable dose for at least 6 months or for nonprurigo conditions | 4 weeks | Prohibiteda |
Cannabinoids (e.g., dronabinol) | 2 weeks | Prohibited |
Investigational topical or systemic medication | 12 weeks or 5 half-lives (whichever is longer) | Prohibited |
Alternative medicine for PN (e.g., traditional Chinese medicine) | 2 weeks | Prohibited |
Live vaccines | 12 weeks | Prohibited |
Nonlive vaccines | 4 weeks | Prohibited (exceptions apply) |
JAK = Janus kinase; NK1 = neurokinin-1; PDE-4 = phosphodiesterase type 4; PN = prurigo nodularis; SNRI = serotonin-norepinephrine reuptake inhibitor; SSRI = selective serotonin reuptake inhibitor.
aUnless used as rescue therapy during the study.
Sources: OLYMPIA 1 Clinical Study Protocol;40 OLYMPIA 2 Clinical Study Protocol.41
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as on any outcomes identified as important to this review, according to the clinical expert(s) consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected the end points that were considered most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select efficacy end points and notable harms outcomes considered important for informing expert committee deliberations were assessed using GRADE.
Clinical experts consulted by CDA-AMC identified intensity of pruritus as the symptom of PN that has the greatest impact on patients. The proportion of patients with at least a 4-point improvement in PP-NRS at 16 weeks was considered a clinically relevant end point by the clinical experts and was a primary end point in both OLYMPIA pivotal trials. Disease severity, focusing on the clearance of lesions (i.e., IGA success), was also identified by clinical experts as an outcome of importance; the proportion of patients with IGA success at 16 weeks was a primary end point in both OLYMPIA trials. Similarly, clinician and patient groups identified reduction in itch and clearance of lesions as important outcomes for patients with PN. Clinical experts, clinician groups, and patient groups highlighted that PN may have a substantial impact on patients’ HRQoL; as such, the proportion of patients with at least a 4-point improvement from baseline in DLQI at 16 weeks (in the OLYMPIA 2 trial) or 24 weeks (in the OLYMPIA 1 trial) was selected as a key end point. Clinical experts consulted by CDA-AMC identified newly diagnosed asthma or worsening of asthma as the AESI of greatest clinical relevance. The aforementioned end points were assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time pointa | OLYMPIA 1 trial | OLYMPIA 2 trial |
|---|---|---|---|
Proportion of patients with an improvement of ≥ 4 from baseline in PP-NRS score | Week 16 | Primaryb | Primaryb |
Proportion of patients with IGA success (defined as an IGA of 0 [clear] or 1 [almost clear] and a ≥ 2-point improvement from baseline) | Week 16 | Primaryb | Primaryb |
Proportion of patients with an improvement of ≥ 4 from baseline in SD-NRS score | Week 16 | Key secondaryb | Key secondaryb |
Proportion of patients with an improvement of ≥ 4 from baseline in PP-NRS score | Week 12, week 24 | Secondary | Secondary |
IGA success rate at each visit | Week 24 | Secondary | Secondary |
Proportion of patients with PP-NRS score improvement of ≥ 4 from baseline and IGA success | Week 16, week 24 | Secondary | Secondary |
Proportion of patients with an improvement of ≥ 4 in DLQI score | Week 16, week 24 | Secondary | Secondary |
Change from baseline in DLQI score | Week 16, week 24 | Secondary | Secondary |
DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; PP-NRS = Peak Pruritus Numerical Rating Scale; SD-NRS = Sleep Disturbance Numerical Rating Scale.
aSecondary end points were assessed at up to 24 weeks in the OLYMPIA 1 trial. Given that the OLYMPIA 2 trial was 16 weeks in length, end points were assessed up to 16 weeks only.
bStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 1 Clinical Study Protocol;40 OLYMPIA 2 Clinical Study Report;25 OLYMPIA 2 Clinical Study Protocol.41 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The composite end point of the proportion of patients with at least a 4-point improvement in PP-NRS score and IGA success has been included in this review report because it informs the economic model. The results for the proportion of patients with at least a 4-point improvement in SD-NRS are also presented in this review report because improvement in sleep was identified as an important outcome by clinician and patient groups.
Peak Pruritus Numerical Rating Scale
The PP-NRS was completed by patients once daily in the evenings. The scale asked patients for a unit score on an 11-point scale (0 to 10) where 0 was no itch and 10 was the worst itch imaginable. The PP-NRS was used as an assessment of the maximum intensity of pruritus during the previous 24 hours, asking patients to rate their itch at the worst moment during that time period. Patients who did not complete the scale in the evening before a scheduled visit were permitted to complete the assessment at the clinic visit the following day.2,40,41 The PP-NRS baseline score was determined based on the average of daily PP-NRS scores during the 7 days immediately preceding baseline; a minimum of 4 daily scores out of the 7 days immediately preceding baseline were required for this calculation. If there were fewer than 4 nonmissing assessments in the baseline diary window, the interval lower bound could be extended up to 7 additional days, 1 day at a time, to obtain the most recent 4 nonmissing values. The PP-NRS score was determined by an average at every week from baseline to week 24 (OLYMPIA 1 trial) or week 16 (OLYMPIA 2 trial).42,43
Investigator’s Global Assessment
The IGA is a 5-point scale used by an investigator to evaluate the global severity of PN, as shown in Table 8. Based on review of the patient’s skin, the investigator assigned a score of 0 (clear), 1 (almost clear), 2 (mild), 3 (moderate), or 4 (severe). Treatment response (IGA success) was defined as 0 (clear) or 1 (almost clear) and at least a 2-point improvement from baseline.2,24,25,40,41
Table 8: Investigator’s Global Assessment
Score | Category | Description |
|---|---|---|
0 | Clear | No nodules |
1 | Almost clear | Rare palpable pruriginous nodules |
2 | Mild | Few palpable pruriginous nodules |
3 | Moderate | Many palpable pruriginous nodules |
4 | Severe | Abundant palpable pruriginous nodules |
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Protocol;40 OLYMPIA 2 Clinical Study Protocol.40
Sleep Disturbance Numerical Rating Scale
The SD-NRS was completed by patients once daily in the morning (within 1 hour of getting out of bed, if possible) to report the degree of PN-related sleep loss. The scale asked patients for a unit score on an 11-point scale (0 to 10) in answer to the following question: “On a scale of 0 to 10, with 0 being ‘no sleep loss related to the symptoms of my skin disease (PN)’ and 10 being ‘I did not sleep at all due to the symptoms of my skin disease (PN),’ how would you rate your sleep last night?” Patients who did not complete the scale in the morning before a scheduled visit were permitted to complete the assessment at the clinic visit.2,40,41 The baseline SD-NRS score was determined based on the average of the daily SD-NRS score during the 7 days up to the treatment start; a minimum of 4 daily scores out of the 7 days up to baseline study day were required for this calculation. If there were fewer than 4 nonmissing assessments in the baseline diary window, the interval lower bound could be extended up to 7 additional days, 1 day at a time, to obtain the most recent 4 nonmissing values.42,43
QoL Questionnaire
DLQI: The DLQI is a 10-item questionnaire covering domains including symptoms and/or feelings, daily activities, leisure, work and/or school, personal relationships, and treatment. The aim is to measure how much a patient’s skin problem has affected their life over the preceding week.2,40,41,46 Patients rated each question from 0 (not at all) to 3 (very much). The DLQI total score was calculated as the sum of the score of each question, resulting in a minimum score of 0 and a maximum score of 30; a higher total score indicated greater impairment in QoL.2,40,41
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
PP-NRS score | The PP-NRS is an 11-point scale assessing the maximum itch intensity in that period. It asks patients: “On a scale of 0 to 10, with 0 being ‘no itch’ and 10 being the ‘worst itch imaginable,’ how would you rate your itch at the worst moment during the previous 24 hours?” | Validity: The validity of the PP‑NRS was evaluated in a sample of 67 patients with moderate to severe PN.47 The authors found that the PP-NRS average weekly score correlated strongly with the PP‑VRS average weekly score (Spearman rank order correlation, r = 0.75), the AP‑VRS average weekly score (r = 0.73), the DLQI item 1 score (r = 0.54), and the AP‑NRS average weekly score (r = 0.85); it correlated moderately with the DLQI total score (r = 0.36) and the IGA score (r = 0.33).47 Reliability: In a sample of patients with stable PN (defined as those with no change in IGA score or in the number of lesions in a representative area from baseline to week 4 or score change of ≤ 1 point on the AP-VRS or PP-VRS), the authors noted that the test-retest reliability of the PP-NRS was substantial based on patient-reported outcomes (i.e., the ICC values were 0.76 for the AP-VRS [n = 16] and 0.73 for the PP-VRS [n = 15]) and weak to fair based on clinician-reported outcomes (i.e., ICC values were 0.20 [n = 26] for the PAS number of lesions and 0.25 [n = 37] for IGA score).47 Responsiveness: The authors noted that the PP-NRS demonstrated responsiveness based on changes in other outcome measures from baseline to week 4.47 Decreases in mean PP-NRS weekly average scores were significantly higher in patients classified as “improved” (−3.8 to −4.8) than in those classified as “worsened or no change” (−0.2 to −2.1; P = 0.0010 for PAS lesions with excoriations and/or crusts; P < 0.0001 for all other outcome measures).47 | A 2- to 5-point decrease in PP-NRS score was considered a meaningful within-patient change threshold.47 |
IGA | The IGA is a 5-point scale used by an investigator to evaluate the global severity of PN. The investigator reviewed the patient’s skin and assigned a score of 0 (clear), 1 (almost clear), 2 (mild), 3 (moderate), or 4 (severe). Treatment response and/or success was defined as 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline. | Psychometric properties of the IGA-CPG and IGA-CNPG stages (which are identical to the IGA PNS) were evaluated in a sample of 187 patients with chronic prurigo.48 Validity: The authors noted statistically significant correlations (P < 0.001) between IGA-CPG stage, IGA-CNPG stage, and IGA-CPG activity. The IGA stage scales (i.e., the IGA-CPG and IGA-CNPG) correlated with each other, and the correlation was considered strong (i.e., Kendall’s tau-b = 0.62). The IGA activity scale was considered moderately correlated with the IGA stage scales (i.e., Kendall’s tau-b = 0.47).48 It was also noted that IGA scores increased with greater pruritus intensity. Strong correlations were noted between IGA-CPG stage and PAS item 2 (the estimation of the total number of pruriginous lesions; t = 0.794; P < 0.01) and the total counted number of all pruriginous lesions (t = 0.729; P < 0.01); a moderate correlation was noted with NRS worst 24 hours (t = 0.343; P < 0.01); and a weak correlation was noted with DLQI (t = 0.265; P < 0.01).48 The authors also noted that IGA-CPG activity was strongly correlated with PAS item 5a (pruriginous lesions with excoriations and/or crusts on top; t = 0.759; P < 0.01).48 Reliability: Intrarater test-retest reliability was evaluated by 5 experienced dermatologist raters who completed the 3 IGA instruments for 8 randomly chosen patients twice within 1 hour. The authors considered the results to be excellent for all items (ICC for IGA-CPG stage = 0.915; ICC-CPNG = 0.874; and ICC for IGA-CPG activity = 0.972).48 The authors considered the congruence among raters as very good in IGA-CPG activity (i.e., Kendall’s W = 0.837) and good in the IGA-CPG stage (i.e., Kendall’s W = 0.747) and IGA-CNPG stage (i.e., Kendall’s W = 0.747). Responsiveness: No information was submitted by the sponsor. | No relevant information was submitted on MID by the sponsor. |
SD-NRS | An NRS relating to sleep disturbance was completed by the patient once daily in the morning and, if possible, within 1 hour of getting out of bed to report the degree of sleep loss related to PN. The SD-NRS asked for a unit score on an 11-point scale (0 to 10). The question asked was: “On a scale of 0 to 10, with 0 being ‘no sleep loss related to the symptoms of my skin disease (PN)’ and 10 being ‘I did not sleep at all due to the symptoms of my skin disease (PN),’ how would you rate your sleep last night?” | Psychometric properties of SD‑NRS were evaluated in a sample of 67 patients with moderate to severe PN.49 Validity: The authors noted Spearman’s rank order correlation coefficients of 0.32 to 0.76 between the SD-NRS and the AP-NRS, AP-VRS, PP-NRS, PP-VRS, and DLQI; these were considered moderate to strong.49 Known-group validity was demonstrated by higher (worse) SD-NRS scores in participants with worse scores on the AP-NRS (P < 0.0001), AP-VRS (0 < 0.0001), PP‑VRS (P = 0.0004), and DLQI (P = 0.0233).49 Reliability: In the sample of patients described, the authors noted the test-retest reliability as substantial (ICC = 0.76) based on the PP-VRS average weekly score and near-perfect (ICC = 0.87) based on the AP-VRS average weekly score.49 Responsiveness: In the sample of patients described, the authors noted that decreases in mean SD-NRS weekly average scores were significantly higher in participants classified as improved) (−2.9 to −4.2) than in those classified as worsened or unchanged (−0.1 to −1.7; P = 0.0029 for AP-NRS average weekly score; P = 0.0005 for dynamic pruritus score; P = 0.0002 for PAS lesions with excoriations and/or crusts and healed lesions; and P < 0.0001 for all other outcomes).49 | A 2- to 4-point decrease on the 11-point SD-NRS scale was identified as a meaningful within-patient change.49 |
DLQI | The DLQI is a 10-item questionnaire covering the domains of symptoms and/or feelings, daily activities, leisure, work and/or school, personal relationships, and treatment.46 Patients rated each ranging from 0 (not at all) to 3 (very much). The DLQI total score was calculated by summing each questionnaire. The total score could have a maximum of 30 and a minimum of 0; a higher total score indicated poorer QoL. | The psychometric properties of the DLQI were evaluated in patients with PN (N = 311) based on the pooled ITT patient population from the PRIME and PRIME2 trials.38,50 Validity: For DLQI total scores, moderate to strong correlations (absolute r = 0.33 to 0.50) between scores were expected to be closely related (e.g., WI‑NRS score, Skin Pain NRS score, sleep NRS score, PGIS, EQ-5D pain and discomfort items, EQ VAS, HADS-A, and HADS-D).38 Reliability: The authors observed adequate test-retest reliability when stable patients were defined using PGIS and PGIC for DLQI total scores (ICC range, 0.64 to 0.87); however, internal consistency was considered good to excellent (Cronbach alpha = 0.88).38 Responsiveness: Moderate to strong correlations were observed for the change in DLQI scores, with changes in PGIS, PGIC, WI-NRS score, sleep NRS score, and Skin Pain NRS score from baseline to week 24 (absolute r = 0.38 to 0.60).50 | Based on the anchor-based analysis, 9.0 (range, 8.00 to 10.00) was considered a clinically meaningful within-patient improvement threshold.50 A 4.0-point change (range, 3.5 to 6.5) between groups was considered a meaningful improvement.38 |
AP-NRS = Average Pruritus Numerical Rating Scale; AP-VRS = Average Pruritus Verbal Rating Scale; DLQI = Dermatology Life Quality Index; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; ICC = intraclass correlation coefficient; IGA = Investigator’s Global Assessment; IGA-CNPG = Investigator’s Global Assessment of Chronic Nodular Prurigo; IGA-CPG = Investigator’s Global Assessment of Chronic Prurigo; ITT = intention to treat; MID = minimal important difference; NRS = numerical rating scale; PAS = Prurigo Activity Score; PGIC = Patient Global Impression of Change; PGIS = Patient Global Impression of Severity; PNS = Prurigo Nodularis Stage; PP-NRS = Peak Pruritus Numerical Rating Scale; PP-VRS = Peak Pruritus Verbal Rating Scale; QoL = quality of life; SD-NRS = Sleep Disturbance Numerical Rating Scale; t = Kendall’s correlation coefficient; WI-NRS = Worst Itch Numerical Rating Scale.
Adverse Events
AE assessments were performed beginning at the screening visit and throughout the treatment and follow-up periods at protocol-specified visits.2,40,41 Treatment-emergent adverse events (TEAEs) were defined as AEs occurring after the first administration of study drug until the last study visit.2,42,43 AEs were coded using the Medical Dictionary for Regulatory Activities Version 25.0.42,43 Patient safety was reviewed and monitored by an independent data monitoring committee. In addition, the IAC reviewed all asthma-related events and assessed whether these represented new diagnoses or worsening of asthma; it also assessed severity (mild, moderate, or severe). However, the IAC did not assess relationship to study drug. For the purpose of the adjudication, asthma-related AEs were retrieved using the standardized Medical Dictionary for Regulatory Activities query, “asthma/bronchospasm.”2,24,25 AESIs were considered noteworthy TEAEs for the study drug that should be monitored closely and reported promptly. These could be considered serious or nonserious; the following were considered AESIs in both the OLYMPIA 1 and OLYMPIA 2 trials:24,25,42,43
injection-related reactions
newly diagnosed asthma or worsening of asthma
infections
peripheral edema (limbs, bilateral)
facial edema
elevated alanine aminotransferase (ALT) or aspartate aminotransferase (AST) (i.e., > 3 × upper limit of normal in combination with elevated bilirubin [> 2 × upper limit of normal]).
An SAE was defined as any untoward medical occurrence that was life-threatening; resulted in death, inpatient hospitalization, prolongation of existing hospitalization, or persistent or significant disability or incapacity; or was a congenital anomaly or birth defect.24,25
Statistical Analysis
Statistical analyses for the OLYMPIA 1 and OLYMPIA 2 trial end points presented in this review report are summarized in Table 10.
Sample Size and Power Calculation
To achieve at least 90% power for both primary end points at a 5% significance level, 270 patients (180 in the nemolizumab group and 90 in the placebo group) were to be randomized to detect the following differences between treatment groups in the primary end points with a 2:1 randomization ratio, assuming a 15% dropout rate during the treatment period:
NRS responders (≥ 4-point reduction from baseline): Based on phase IIa data, it was expected that the NRS response at week 16 would be 50% in the nemolizumab group and 20% in the placebo group.
IGA response (0 or 1): It was expected that the IGA response at week 16 would be 30% in the nemolizumab group and 10% in the placebo group.2,42,43
No power analyses were planned for the key secondary end points. No interim analyses were planned or performed for the OLYMPIA 1 and OLYMPIA 2 trials.2,24,25
Multiple Testing Procedure
A fixed sequential testing approach was used to control the type I error at 5%. The primary end points were first tested in order (the PP-NRS end point followed by the IGA success end point) at a 5% significance level; testing of the key secondary end points would start only if both primary end points were successful at a 5% level of significance. Key secondary end points were tested in the following order, and testing was planned to be stopped when an end point did not test statistically significant (P > 0.05):
proportion of patients with an improvement of 4 or more points from baseline in PP-NRS at week 4
proportion of patients with a PP-NRS score of less than 2 at week 16
proportion of patients with an improvement of 4 or more points from baseline in SD-NRS at week 16
proportion of patients with an improvement of 4 or more points from baseline in SD-NRS at week 4
proportion of patients with a PP-NRS score of less than 2 at week 4.2,42,43
Other secondary end points were not adjusted for multiplicity.2
Subgroup Analyses
Subgroup analyses were prespecified and conducted for the following groups for the primary and key secondary end points:
region (Europe and North America in the OLYMPIA 1 trial; Asia Pacific, Europe, and North America in the OLYMPIA 2 trial)
age group (18 to 65 years and > 65 years)
sex (female, male)
race (African American or Black, Alaska Native or American Indian, Asian, Native Hawaiian or Pacific Islander, white, or other [including multiple]—wording of original sources)42,43
weight at randomization (< 90 kg, ≥ 90 kg)
The subgroup analyses were performed for the intention-to-treat (ITT) population and conducted in the same manner as for the primary end points. A forest plot was presented along with the subgroup analysis results.2,42,43 Subgroup analyses were not adjusted for multiplicity.2
Table 10: Statistical Analysis of Efficacy End Points in the OLYMPIA 1 and OLYMPIA 2 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end points | ||||
Proportion of patients with an improvement of ≥ 4 from baseline in PP-NRS score at week 16 | CMH test: The estimate of the treatment difference and corresponding 2-sided 95% CI and P values were presented. The CIs were based on the Wald statistic controlling for stratification variables. Strata-adjusted proportion differences were obtained using a weighted average of stratum-specific proportion using CMH. In addition, an unadjusted CMH test was performed. Bar charts for the proportions of patients with improvement or success from baseline to week 16 were presented. | Randomization strata, analysis centre, and body weight at randomization (< 90 kg, ≥ 90 kg) | Patients with missing data at week 16 were considered nonresponders.a If a patient was in receipt of rescue medication at any point on or before week 16, data on or after the receipt of rescue medication were regarded as failure of treatment. |
|
Proportion of patients with IGA successb at week 16 | As previously noted | As previously noted | As previously noted | As previously noted |
Key secondary end points | ||||
Proportion of patients with an improvement of ≥ 4 from baseline in SD-NRS score at week 16 | As previously noted | As previously noted | As previously noted | As previously noted |
Other secondary end points | ||||
IGA successb rate at each visit | CMH test: The estimate of treatment-unadjusted and strata-adjusted differences with the corresponding 2-sided 95% CIs and P values from the CMH test were presented. Line plots for the proportions of patients with IGA success using nonrespondera imputation were generated for the ITT and per-protocol populations. | As previously noted | OC, missing as nonresponder,a LOCF | None |
Proportion of patients with improvement in PP-NRS score of ≥ 4 from baseline at each visit | CMH test: The estimate of treatment-unadjusted and strata-adjusted differences with the corresponding 2-sided 95% CIs and P values from the CMH test were presented. Line plots for the proportion of patients with PP-NRS score improvement of ≥ 4 using nonrespondera imputation were generated for the ITT and per-protocol populations. | Randomization strata, analysis centre, and body weight at randomization (< 90 kg, ≥ 90 kg) | OC, missing as nonresponder,a LOCF | None |
Proportion of patients with PP‑NRS score improvement of ≥ 4 from baseline and IGA success | CMH | Analyses were conducted without adjustment and strata-adjusted | OC, missing as nonresponder,a LOCF | None |
Proportion of patients with an improvement of ≥ 4 in DLQI score at each visit | CMH test: A line plot for the proportion of patients with a DLQI total score improvement of ≥ 4 using nonrespondera imputation was presented for the ITT population. | Analyses were conducted without adjustment and strata-adjusted | OC, missing as nonrespondera | None |
Change from baseline in DLQI at each visit | ANCOVA | Randomization strata, analysis centre, and body weight at randomization (< 90 kg, ≥ 90 kg) | MI assuming MAR, MMRM | None |
ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITT = intention to treat; LOCF = last observation carried forward; MAR = missing at random; MI = multiple imputation; MMRM = mixed-effect models for repeated measures; OC = observed case; PP-NRS = Peak Pruritus Numerical Rating Scale; SD-NRS = Sleep Disturbance Numerical Rating Scale.
aWording of the original source.
bIGA success was defined as an IGA of 0 (clear) or 1 (almost clear) and an improvement of 2 or more grades from baseline.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Statistical Analysis Plan;42 OLYMPIA 2 Statistical Analysis Plan.43 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
A summary of the analysis populations in the OLYMPIA 1 and OLYMPIA 2 trials is presented in Table 11.
Table 11: Analysis Populations in the OLYMPIA 1 and OLYMPIA 2 Trials
Study | Population | Definition | Application |
|---|---|---|---|
OLYMPIA 1 and OLYMPIA 2 trials | ITT population:
| The ITT population consisted of all randomized patients. Patients were included in the treatment group to which they were randomized. | The ITT population was the primary population for the efficacy analyses. |
Safety population:
| The safety population consisted of all randomized patients who received at least 1 administration of study drug. The treatment group assignment in this population was defined by the treatment actually received. | The safety population was used for the analyses of safety. | |
Per-protocol population:
| The per-protocol population consisted of all patients in the ITT population who had no major protocol deviations that would have had a significant effect on the efficacy of the study treatment. Patients were analyzed under the treatment group as randomized. | Only the primary and key secondary end points were analyzed using the per-protocol population. |
ITT = intention to treat.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Protocol;40 OLYMPIA 2 Clinical Study Protocol.41 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition in the OLYMPIA 1 and OLYMPIA 2 trials is presented in Table 12.
In the OLYMPIA 1 trial, a total of 482 patients were screened, of whom 286 were randomized to nemolizumab (N = 190) and placebo (N = 96) arms. Three patients (1.6%) in the nemolizumab arm and 1 patient (1.0%) in the placebo arm were randomized, but did not receive treatment.2,24 In the OLYMPIA 2 trial, a total of 424 patients were screened, of whom 274 were randomized and received treatment with nemolizumab (N = 183) or placebo (N = 91).2,25 In both the OLYMPIA 1 and OLYMPIA 2 trials, most patients who were screened out did not meet the eligibility criteria (94.4% and 92.7%, respectively). A similar percentage of patients completed treatment in the nemolizumab and placebo arms in the OLYMPIA 1 trial (88.4% and 88.5%, respectively); this was also seen in the OLYMPIA 2 trial (i.e., 95.6% in both arms). The most common reasons for discontinuation of treatment in the OLYMPIA 1 trial were AEs (nemolizumab: 5.8%; placebo: 4.2%) and patient request (4.2% in each arm). In the OLYMPIA 2 trial, the most common reasons for discontinuation of treatment were AEs (2.2% in each arm). The most common reasons for discontinuation from the study in the OLYMPIA 1 trial were AEs (nemolizumab: 6.3%; placebo: 4.2%) and patient request (nemolizumab: 5.3%; placebo: 6.3%); in the OLYMPIA 2 trial, the most common reasons for discontinuation from the trial were AEs (2.2% in each arm). In the OLYMPIA 1 trial, 81.6% of patients in the nemolizumab arm and 81.3% of patients in the placebo arm rolled over to the LTE study. In the OLYMPIA 2 trial, 93.4% and 94.5% of patients, respectively, rolled over to the LTE study.2
Table 12: Patient Disposition in the OLYMPIA 1 and OLYMPIA 2 Trials (ITT Population)
Patient disposition | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 190 | Placebo N = 96 | Nemolizumab N = 183 | Placebo N = 91 | |
Screened, N | 482 | 424 | ||
Screened out, N | 196 | 150 | ||
Reason for being screened out, N (%) | ||||
Inclusion or exclusion criteria not met | 185 (94.4) | 139 (92.7) | ||
Patient request | 7 (3.6) | 11 (7.3) | ||
Adverse event | 1 (0.5) | 0 | ||
Enrolment closed | 1 (0.5) | 0 | ||
Sample not taken by DHL (logistics company) | 1 (0.5) | 0 | ||
Patient to get approval from GI specialist to participate in trial appointment out of screening window | 1 (0.5) | 0 | ||
Randomized, N (%) | 190 (100) | 96 (100) | 183 (100) | 91 (100) |
Completed treatment | 168 (88.4) | 85 (88.5) | 175 (95.6) | 87 (95.6) |
Discontinued treatment | 19 (10.0) | 10 (10.4) | 8 (4.4) | 4 (4.4) |
Primary reason for discontinuation of treatment | ||||
Pregnancy | 0 | 0 | 0 | 1 (1.1) |
Lack of efficacy | 0 | 0 | 1 (0.5) | 1 (1.1) |
Adverse event | 11 (5.8) | 4 (4.2) | 4 (2.2) | 2 (2.2) |
Patient request | 8 (4.2) | 4 (4.2) | 1 (0.5) | 0 |
Lost to follow-up | 0 | 0 | 0 | 0 |
Protocol deviation | 0 | 0 | 0 | 0 |
Physician or primary investigator decisiona | 0 | 1 (1.0) | 1 (0.5) | 0 |
Sponsor decision | 0 | 0 | 0 | 0 |
Other: COVID-19 | 0 | 0 | 0 | 0 |
Other: Site permanently closing | 0 | 1 (1.0) | 0 | 0 |
Other: Inability to attend appointments | 0 | 0 | 1 (0.5) | 0 |
Completed the study | 166 (87.4) | 83 (86.5) | 174 (95.1) | 88 (96.7) |
Discontinued from the study | 24 (12.6) | 13 (13.5) | 9 (4.9) | 3 (3.3) |
Primary reason for discontinuation from the study | ||||
Pregnancy | 0 | 0 | 0 | 1 (1.1) |
Lack of efficacy | 0 | 0 | 0 | 0 |
Adverse event | 12 (6.3) | 4 (4.2) | 4 (2.2) | 2 (2.2) |
Patient request | 10 (5.3) | 6 (6.3) | 2 (1.1) | 0 |
Lost to follow-up | 0 | 0 | 2 (1.1) | 0 |
Protocol deviation | 2 (1.1) | 1 (1.0) | 0 | 0 |
Physician or primary investigator decisiona | 0 | 1 (1.0) | 1 (0.5) | 0 |
Sponsor decision | 0 | 0 | 0 | 0 |
Other: COVID-19 | 0 | 0 | 0 | 0 |
Other: Site permanently closing | 0 | 1 (1.0) | 0 | 0 |
Rolled over to long-term extension | 155 (81.6) | 78 (81.3) | 171 (93.4) | 86 (94.5) |
Completed follow-up | 19 (10.0) | 10 (10.4) | 6 (3.3) | 3 (3.3) |
ITT, N | 190 (100) | 96 (100) | 183 (100) | 91 (100) |
Safety, N | 187 (98.4) | 95 (99.0) | 183 (100) | 91 (100) |
Per protocol, N | 166 (87.4) | 84 (87.5) | 159 (86.9) | 83 (91.2) |
Pharmacokinetic analysis population, N (%) | 183 (96.3) | 0 | 180 (98.4) | 0 |
DHL = Dalsey, Hillblom, and Lynn; GI = gastrointestinal; ITT = intention to treat.
aPatient 5489-001 was terminated from the study drug and the study at the investigator’s discretion.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report;25 sponsor’s Response to Request for Additional Information.51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
A summary of baseline characteristics for the OLYMPIA 1 and OLYMPIA 2 trials is presented in Table 13.
In the OLYMPIA 1 trial, baseline characteristics were generally balanced across the nemolizumab and placebo groups, with exceptions subsequently described. There was a lower proportion of patients who were of Hispanic or Latino ethnicity in the nemolizumab arm than in the placebo arm (2.1% versus 5.2%), and the mean weight and proportion of patients who weighed 90 kg or more at baseline were higher in the nemolizumab group (87.07 kg and 38.4%, respectively) compared to the placebo group (80.81 kg and 30.2%, respectively). Imbalances between the nemolizumab and placebo groups existed for smoking status for people who had never smoked (57.4% versus 45.8%) and people who currently smoked (16.8% versus 25.0%). In addition, the proportion of patients with an IGA category of severe (i.e., 4) was higher in the nemolizumab arm (43.7%) than in the placebo arm (35.4%), and the mean time since PN diagnosis was shorter in the nemolizumab arm compared to the placebo arm (86.92 months versus 100.61 months).2
In the OLYMPIA 1 trial, most patients were female (58.0%), white (84.3%), not Hispanic or Latino (95.1%), and aged 18 to 65 years (71.3%), with a mean age of 57.5 years (details are provided in Table 13). Most participants were in Europe (74.1%) while the rest were in North America (25.9%). Most patients (64.13%) weighed less than 90 kg at baseline and had either never smoked (53.5%) or had previously smoked (26.9%). Overall, 59.1% of patients had moderate disease (i.e., IGA category 3), and 40.9% of patients had severe disease (i.e., IGA category 4). The mean DLQI score at baseline was 17.0. Most patients did not have a background of atopy (67.5%). The mean time since PN diagnosis was 91.51 months.24,25
In the OLYMPIA 2 trial, baseline characteristics were generally balanced across the nemolizumab and placebo groups (exceptions are subsequently described). The proportion of patients older than 65 years was higher in the nemolizumab arm (25.1%) than in the placebo arm (15.4%). There was a lower proportion of patients of Hispanic or Latino ethnicity in the nemolizumab arm than in the placebo arm (2.7% versus 7.7%) and a lower proportion of Black of African American patients in the nemolizumab arm than in the placebo arm (2.7% versus 7.7%). Imbalances between the nemolizumab and placebo groups existed for smoking status for participants who had never smoked (59.6% versus 67.0%) and those who currently smoked (15.8% versus 11.0%). The proportion of patients with an IGA category of severe (i.e., 4) was lower in the nemolizumab arm (41.0%) than in the placebo arm (47.3%). In addition, the mean time since PN diagnosis was shorter in the nemolizumab arm compared to the placebo arm (104.16 months versus 108.60 months).2
In the OLYMPIA 2 trial, most patients were female (61.3%), white (78.5%), not Hispanic or Latino (92.0%), and aged 18 to 65 years (78.1%), with a mean age of 52.7 years (details are provided in Table 13). There were higher proportions of participants in Europe (66.8%) and North America (25.2%) than in the Asia Pacific region (8.0%). Most patients (72.6%) weighed less than 90 kg at baseline and had either never smoked (62.0%) or had previously smoked (23.7%). Overall, 56.9% and 43.1% of patients had moderate and severe disease, respectively. The mean DLQI score at baseline was 16.7. Most patients did not have a background of atopy (67.9%). The mean time since PN diagnosis was 105.64 months.24,25
In both trials, all patients (100%) were reported to have at least 1 previous and ongoing medical condition, most commonly neurodermatitis and hypertension.24,25
Table 13: Summary of Baseline Characteristics From the OLYMPIA 1 and OLYMPIA 2 Trials (ITT Population)
Characteristic | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 190 | Placebo N = 96 | Nemolizumab N = 183 | Placebo N = 91 | |
Age (years) | ||||
Mean (SD) | 57.5 (12.77) | 57.6 (13.36) | 53.7 (14.41) | 50.8 (15.00) |
Age group, n (%) | ||||
18 to 65 years | 136 (71.6) | 68 (70.8) | 137 (74.9) | 77 (84.6) |
> 65 years | 54 (28.4) | 28 (29.2) | 46 (25.1) | 14 (15.4) |
Sex, n (%) | ||||
Female | 110 (57.9) | 56 (58.3) | 113 (61.7) | 55 (60.4) |
Male | 80 (42.1) | 40 (41.7) | 70 (38.3) | 36 (39.6) |
Region, n (%) | ||||
Asia Pacific | 0 | 0 | 14 (7.7) | 8 (8.8) |
Europe | 141 (74.2) | 71 (74.0) | 122 (66.7) | 61 (67.0) |
North America | 49 (25.8) | 25 (26.0) | 47 (25.7) | 22 (24.2) |
Ethnicity, n (%) | ||||
Hispanic or Latino | 4 (2.1) | 5 (5.2) | 5 (2.7) | 7 (7.7) |
Not Hispanic or Latino | 184 (96.8) | 88 (91.7) | 173 (94.5) | 79 (86.8) |
Unknown | 1 (0.5) | 0 | 0 | 0 |
Not reported | 1 (0.5) | 3 (3.1) | 5 (2.7) | 5 (5.5) |
Race, n (%) | ||||
Alaska Native or American Indian | 1 (0.5) | 0 | 0 | 0 |
Asian | 10 (5.3) | 2 (2.1) | 23 (12.6) | 14 (15.4) |
Black or African American | 18 (9.5) | 10 (10.4) | 5 (2.7) | 7 (7.7) |
Multiple | 0 | 0 | 0 | 0 |
Native Hawaiian or other Pacific Islander | 0 | 0 | 2 (1.1) | 0 |
White | 160 (84.2) | 81 (84.4) | 147 (80.3) | 68 (74.7) |
Other | 2 (1.0) | 2 (2.1) | 5 (2.7) | 2 (2.2) |
Not reported | 0 | 1 (1.0) | 1 (0.5) | 0 |
Height at baseline (cm) | ||||
Mean (SD) | 169.97 (9.48) | 168.89 (9.95) | 167.89 (8.47) | 167.67 (10.81) |
Weight at baseline (kg) | ||||
Mean (SD) | 87.07 (21.78) | 80.81 (17.76) | 79.65 (17.76) | 80.83 (22.31) |
Weight at baseline < 90 kg | ||||
n (%) | 117 (61.6) | 67 (69.8) | 132 (72.1) | 67 (73.6) |
Weight at baseline ≥ 90 kg | ||||
n (%) | 73 (38.4) | 29 (30.2) | 51 (27.9) | 24 (26.4) |
Body mass index (kg/m2) | ||||
Mean (SD) | 30.00 (6.54) | 28.20 (5.21) | 28.16 (5.35) | 28.49 (5.90) |
Smoking status, n (%) | ||||
Never | 109 (57.4) | 44 (45.8) | 109 (59.6) | 61 (67.0) |
Former | 49 (25.8) | 28 (29.2) | 45 (24.6) | 20 (22.0) |
Current | 32 (16.8) | 24 (25.0) | 29 (15.8) | 10 (11.0) |
IGA category, n (%) | ||||
Clear (0) | 0 | 0 | 0 | 0 |
Almost clear (1) | 0 | 0 | 0 | 0 |
Mild (2) | 0 | 0 | 0 | 0 |
Moderate (3) | 107 (56.3) | 62 (64.6) | 108 (59.0) | 48 (52.7) |
Severe (4) | 83 (43.7) | 34 (35.4) | 75 (41.0) | 43 (47.3) |
Weekly average PP-NRS | ||||
n | 184 | 96 | 183 | 91 |
Mean (SD) | 8.50 (0.94) | 8.44 (0.99) | 8.47 (0.90) | 8.37 (0.99) |
Weekly average AP-NRS | ||||
n | 184 | 94 | 178 | 90 |
Mean (SD) | 8.21 (1.10) | 8.23 (1.11) | 8.29 (0.95) | 8.21 (1.09) |
Weekly average SD-NRS | ||||
n | 190 | 96 | 182 | 91 |
Mean (SD) | 7.01 (2.37) | 6.89 (2.33) | 7.19 (2.21) | 7.31 (2.23) |
Pain frequency, n (%) | ||||
Never | 15 (7.9) | 3 (3.1) | 7 (3.8) | 2 (2.2) |
Less than once a week | 11 (5.8) | 3 (3.1) | 9 (4.9) | 9 (9.9) |
1 to 2 days a week | 6 (3.2) | 11 (11.5) | 11 (6.0) | 3 (3.3) |
3 to 4 days a week | 25 (13.2) | 7 (7.3) | 24 (13.1) | 8 (8.8) |
5 to 6 days a week | 19 (10.0) | 4 (4.2) | 16 (8.7) | 5 (5.5) |
Every day | 114 (60.0) | 68 (70.8) | 116 (63.4) | 64 (70.3) |
Pain intensity | ||||
Mean (SD) | 7.1 (2.90) | 7.6 (2.45) | 7.7 (2.37) | 7.8 (2.32) |
PAS item 4 (number of lesions in representative area) | ||||
Mean (SD) | 23.0 (18.36) | 17.7 (13.30) | 21.7 (18.52) | 25.5 (22.00) |
PAS item 5a (excoriation and/or crusts), n (%) | ||||
0% | 0 | 0 | 1 (0.5) | 0 |
1% to 25% | 9 (4.7) | 7 (7.3) | 11 (6.0) | 11 (12.1) |
26% to 50% | 39 (20.5) | 17 (17.7) | 34 (18.6) | 20 (22.0) |
51% to 75% | 54 (28.4) | 23 (24.0) | 64 (35.0) | 29 (31.9) |
76% to 100% | 88 (46.3) | 49 (51.0) | 73 (39.9) | 31 (34.1) |
PAS item 5b (healed lesion stages), n (%) | ||||
100% | 0 | 0 | 0 | 0 |
76% to 99% | 1 (0.5) | 2 (2.1) | 3 (1.6) | 0 |
51% to 75% | 22 (11.6) | 12 (12.5) | 23 (12.6) | 13 (14.3) |
26% to 50% | 41 (21.6) | 23 (24.0) | 47 (25.7) | 26 (28.6) |
0% to 25% | 126 (66.3) | 59 (61.5) | 110 (60.1) | 52 (57.1) |
DLQI total score at baseline | ||||
Mean (SD) | 17.1 (6.97) | 16.9 (6.74) | 16.5 (6.79) | 17.1 (6.60) |
Atopy background, n (%) | ||||
Yes | 60 (31.6) | 33 (34.4) | 57 (31.1) | 31 (34.1) |
No | 130 (68.4) | 63 (65.6) | 126 (68.9) | 60 (65.9) |
Time since PN diagnosis (months) | ||||
Mean (SD) | 86.92 (85.28) | 100.61 (98.57) | 104.16 (100.72) | 108.60 (114.93) |
AP-NRS = Average Pruritus Numerical Rating Scale; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITT = intention to treat; PAS = Prurigo Activity Score; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; SD = standard deviation; SD-NRS = Sleep Disturbance Numerical Rating Scale.
Notes: Percentages were based on the number of patients in each treatment group.
Weekly average PP-NRS scores and weekly average AP-NRS scores were calculated as the average of 7 consecutive days of data up to the target study day (excluding) and set to missing, if fewer than 4 days of data were available.
Weekly average SD-NRS scores were calculated as the average of 7 consecutive days of data up to the target study day (inclusive) and set to missing, if fewer than 4 days of data were available.
Baseline was defined as the last nonmissing value or weekly value before the first dose of study drug.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Prior PN Therapies
Selected commonly used prior PN medications and procedures are presented in Table 14. Treatment with at least 1 medication for PN before study entry had been received by 63.7% of patients in the nemolizumab arm and 62.5% of patients in the placebo arm in the OLYMPIA 1 trial and by 83.6% and 86.8% of patients, respectively, in the OLYMPIA 2 trial. In both trials, the most commonly used prior PN therapies included TCSs (51.6% in the nemolizumab arm and 55.2% in the placebo arm of the OLYMPIA 1 trial; 78.1% and 79.1%, respectively, in the OLYMPIA 2 trial); systemic oral antihistamines (24.2% and 13.5% in the OLYMPIA 1 trial; 38.8% and 36.3% in the OLYMPIA 2 trial); systemic corticosteroids (14.7% and 11.5% in the OLYMPIA 1 trial; 19.7% and 23.1% in the OLYMPIA 2 trial); systemic nonsteroidal immunosuppressants (12.1% and 12.5% in the OLYMPIA 1 trial; 19.1% and 18.7% in the OLYMPIA 2 trial); and TCIs (10.0% and 8.3% in the OLYMPIA 1 trial; 15.3% and 12.1% in the OLYMPIA 2 trial). At least 1 prior PN procedure had been received by 22.6% of patients in the nemolizumab arm and 16.7% of patients in the placebo arm of the OLYMPIA 1 trial and by 28.4% and 33.0% of patients, respectively, in the OLYMPIA 2 trial. The most commonly received prior PN procedures were phototherapy (22.6% and 15.6% in the OLYMPIA 1 trial; 27.3% and 28.6% in the OLYMPIA 2 trial) and UV light therapy (20.0% and 13.5% in the OLYMPIA 1 trial; 21.3% and 20.9% in the OLYMPIA 2 trial).2
Table 14: Selected Commonly Used Prior PN Medications and Procedures (ITT Population)
Medicationa or procedure | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 190 n (%) | Placebo N = 96 n (%) | Nemolizumab N = 183 n (%) | Placebo N = 91 n (%) | |
Patients with at least 1 selected prior PN medication | 121 (63.7) | 60 (62.5) | 153 (83.6) | 79 (86.8) |
Topical corticosteroids | 98 (51.6) | 53 (55.2) | 143 (78.1) | 72 (79.1) |
Clobetasol propionate | 33 (17.4) | 21 (21.9) | 67 (36.6) | 39 (42.9) |
Mometasone furoate | 17 (8.9) | 13 (13.5) | 34 (18.6) | 17 (18.7) |
Betamethasone dipropionate; gentamicin sulphate | 5 (2.6) | 4 (4.2) | 30 (16.4) | 11 (12.1) |
Clobetasol | 15 (7.9) | 7 (7.3) | 19 (10.4) | 7 (7.7) |
Betamethasone dipropionate | 7 (3.7) | 4 (4.2) | 18 (9.8) | 6 (6.6) |
Betamethasone valerate | 12 (6.3) | 3 (3.1) | 15 (8.2) | 8 (8.8) |
Triamcinolone | 12 (6.3) | 4 (4.2) | < 4% | < 4% |
Hydrocortisone | < 4% | < 4% | 14 (7.7) | 7 (7.7) |
Betamethasone | 10 (5.3) | 4 (4.2) | 10 (5.5) | 4 (4.4) |
Diflucortolone valerate | < 4% | < 4% | 10 (5.5) | 8 (8.8) |
Methylprednisolone aceponate | 9 (4.7) | 6 (6.3) | 10 (5.5) | 10 (11.0) |
Prednicarbate | 8 (4.2) | 1 (1.0) | < 4% | < 4% |
Dexamethasone | < 4% | < 4% | 9 (4.9) | 7 (7.7) |
Betamethasone valerate; fusidic acid | < 4% | < 4% | 5 (2.7) | 4 (4.4) |
Topical calcineurin inhibitors | 19 (10.0) | 8 (8.3) | 28 (15.3) | 11 (12.1) |
Tacrolimus monohydrate | < 4% | < 4% | 24 (13.1) | 8 (8.8) |
Pimecrolimus | 3 (1.6) | 4 (4.2) | < 4% | < 4% |
Topical, other | 18 (9.5) | 7 (7.3) | 18 (9.8) | 11 (12.1) |
Systemic corticosteroids | 28 (14.7) | 11 (11.5) | 36 (19.7) | 21 (23.1) |
Prednisolone | 12 (6.3) | 9 (9.4) | < 4% | < 4% |
Methylprednisolone | < 4% | < 4% | 12 (6.6) | 7 (7.7) |
Prednisone | < 4% | < 4% | 10 (5.5) | 7 (7.7) |
Systemic biologics | 8 (4.2) | 2 (2.1) | 4 (2.2) | 3 (3.3) |
Systemic nonsteroidal immunosuppressants | 23 (12.1) | 12 (12.5) | 35 (19.1) | 17 (18.7) |
Methotrexate | 15 (7.9) | 7 (7.3) | 24 (13.1) | 13 (14.3) |
Ciclosporin | 13 (6.8) | 4 (4.2) | 7 (3.8) | 4 (4.4) |
Systemic gabapentinoids | 14 (7.4) | 3 (3.1) | 6 (3.3) | 8 (8.8) |
Pregabalin | < 4% | < 4% | 1 (0.5) | 5 (5.5) |
Systemic oral antihistamines | 46 (24.2) | 13 (13.5) | 71 (38.8) | 33 (36.3) |
Bilastine | < 4% | < 4% | 15 (8.2) | 6 (6.6) |
Fexofenadine hydrochloride | < 4% | < 4% | 13 (7.1) | 8 (8.8) |
Hydroxyzine | < 4% | < 4% | 12 (6.6) | 5 (5.5) |
Hydroxyzine hydrochloride | 15 (7.9) | 7 (7.3) | 10 (5.5) | 8 (8.8) |
Desloratadine | 13 (6.8) | 3 (3.1) | 9 (4.9) | 5 (5.5) |
Epinastine hydrochloride | < 4% | < 4% | 8 (4.4) | 6 (6.6) |
Bepotastine besilate | < 4% | < 4% | 7 (3.8) | 5 (5.5) |
Loratadine | 9 (4.7) | 3 (3.1) | 7 (3.8) | 4 (4.4) |
Rupatadine fumarate | < 4% | < 4% | 7 (3.8) | 4 (4.4) |
Systemic, other | 22 (11.6) | 5 (5.2) | 9 (4.9) | 7 (7.7) |
Naloxone hydrochloride | 12 (6.3) | 2 (2.1) | < 4% | < 4% |
Intralesional corticosteroids (group) | 5 (2.6) | 2 (2.1) | 8 (4.4) | 5 (5.5) |
Patients with at least 1 selected prior PN procedure | 43 (22.6) | 16 (16.7) | 52 (28.4) | 30 (33.0) |
Phototherapyb | 43 (22.6) | 15 (15.6) | 50 (27.3) | 26 (28.6) |
UV light therapy | 38 (20.0) | 13 (13.5) | 39 (21.3) | 19 (20.9) |
PUVA | 4 (2.1) | 2 (2.1) | 13 (7.1) | 6 (6.6) |
Phototherapyc | 2 (1.1) | 0 | 2 (1.1) | 2 (2.2) |
Other | 6 (3.2) | 2 (2.1) | 4 (2.2) | 4 (4.4) |
Balneotherapy | 5 (2.6) | 2 (2.1) | 0 | 0 |
Laser therapy | 1 (0.5) | 0 | 0 | 1 (1.1) |
Skin lesion removal | 0 | 0 | 1 (0.5) | 0 |
ITT = intention to treat; PN = prurigo nodularis; PUVA = psoralen plus UVA.
aMost commonly reported medications were reported based on an incidence of 4% or higher in either treatment group.
bRefers to the group term that covers all types of phototherapies (i.e., group of procedures).
cRefers to the preferred term that was used for coding when further information on the specific type of phototherapy was not available.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report;25 sponsor’s Response to Request for Additional Information.51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Treatment exposure during the OLYMPIA 1 and OLYMPIA 2 trials is summarized in Table 15.
In the OLYMPIA 1 trial, the treatment period was 24 weeks. The mean treatment durations were 131.1 days in the nemolizumab arm and 134.2 days in the placebo arm. The mean total nemolizumab dose administered was 248.5 mg. The proportions of patients who missed at least 1 dose were 9.1% in the nemolizumab arm and 8.4% in the placebo arm. There were more missed doses of the assigned study drug in the nemolizumab arm than in the placebo arm (29 doses and 19 doses, respectively).
In the OLYMPIA 2 trial, the treatment period was 16 weeks. The mean treatment durations were 82.4 days in the nemolizumab arm and 81.0 days in the placebo arm. The mean total nemolizumab dose administered was 170.5 mg. The proportions of patients who missed at least 1 dose were 2.2% in the nemolizumab arm and 8.8% in the placebo arm. There were fewer missed doses in the nemolizumab arm than in the placebo arm (5 doses and 12 doses, respectively).
Table 15: Summary of Patient Exposure in the OLYMPIA 1 and OLYMPIA 2 Trials (Safety Population)
Characteristic | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 187 | Placebo N = 95 | Nemolizumab N = 183 | Placebo N = 91 | |
Total dose administered (mg),a mean (SD) | 248.5 (85.87) | 0.0 (0.00) | 170.5 (44.56) | 0.0 (0.00) |
Total dose planned (mg),b mean (SD) | 253.2 (82.83) | 0.0 (0.00) | 171.3 (44.23) | 0.0 (0.00) |
Patients who missed at least 1 dose, n (%) | 17 (9.1) | 8 (8.4) | 4 (2.2) | 8 (8.8) |
Patients who missed at least 1 dose due to COVID‑19, n (%) | 8 (4.3) | 7 (7.4) | 2 (1.1) | 2 (2.2) |
Number of doses missed, n | 29 | 19 | 5 | 12 |
Number of doses missed due to COVID-19, n | 10 | 17 | 3 | 2 |
Treatment duration (days), mean (SD) | 131.1 (32.79) | 134.2 (27.78) | 82.4 (15.49) | 81.0 (14.96) |
SD = standard deviation.
Note: Treatment duration was calculated as: (date of last treatment minus date of first treatment) + 1.
aThe total dose administered was calculated as the sum of all doses of study drug administered.
bThe total dose planned was calculated as the sum of all doses of study drug planned (dispensed) according to the treatment schedule of the treatment group.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report;25 sponsor’s Response to Request for Additional Information.51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
In the OLYMPIA 1 trial, at least 1 concomitant medication was received by 88.9% of patients in the nemolizumab group and 94.8% of patients in the placebo group, with the most commonly received medications being tozinameran (14.7% and 11.5%, respectively), atorvastatin (9.5% and 15.6%, respectively), and ibuprofen (11.6% and 9.4%, respectively). In the OLYMPIA 2 trial, at least 1 concomitant medication was received by 89.6% of patients in the nemolizumab group and 87.9% of patients in the placebo group, with the most commonly received medications being tozinameran (23.0% and 23.1%, respectively), paracetamol (13.7% and 8.8%, respectively), and acetylsalicylic acid (7.7% and 8.8%, respectively).24,25
Rescue Treatment
Rescue medications and procedures received by patients in the OLYMPIA 1 and OLYMPIA 2 trials are summarized in Table 16. In both trials, the incidence of rescue therapy consisting of any topical or systemic medication was higher in the placebo group than in the nemolizumab group (OLYMPIA 1 trial: nemolizumab 6.3%, placebo 19.8%; OLYMPIA 2 trial: 4.9% and 15.4%, respectively), with the most commonly received rescue medications being TCSs.24,25 In the OLYMPIA 1 trial, the incidence of any procedure as rescue therapy was 2.1% in the placebo group; no patients in the nemolizumab group received this rescue therapy.24 No patients met the criteria for concomitant procedures as rescue therapy in the OLYMPIA 2 trial.25
Table 16: Incidence of Rescue Therapy in the OLYMPIA 1 and OLYMPIA 2 Trials (ITT Population)
Rescue therapy | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 190 | Placebo N = 96 | Nemolizumab N = 183 | Placebo N = 91 | |
Any medication, n (%) | 12 (6.3) | 19 (19.8) | 9 (4.9) | 14 (15.4) |
Topical | 11 (5.8) | 16 (16.7) | 9 (4.9) | 11 (12.1) |
Corticosteroids | 10 (5.3) | 15 (15.6) | 8 (4.4) | 11 (12.1) |
Calcineurin inhibitors | 1 (0.5) | 2 (2.1) | — | — |
Other | 1 (0.5) | 0 | 1 (0.5) | 2 (2.2) |
Systemic | 2 (1.1) | 7 (7.3) | 3 (1.6) | 7 (7.7) |
Corticosteroids | 0 | 1 (1.0) | — | — |
Nonsteroidal immunosuppressants | 0 | 1 (1.0) | 0 | 1 (1.1) |
Oral antihistamines | 2 (1.1) | 5 (5.2) | 3 (1.6) | 6 (6.6) |
Other | 0 | 1 (1.0) | — | — |
Any procedure, n (%) | 0 | 2 (2.1) | — | — |
Phototherapy | 0 | 2 (2.1) | — | — |
ITT = intention to treat.
Note: Patients are counted once where more than 1 rescue therapy was reported.
Sources: OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report.25
Treatment Adherence
In the OLYMPIA 1 and OLYMPIA 2 trials, mean treatment adherence was greater than 97% in both treatment arms, as presented in Table 17.
Table 17: Treatment Adherence in the OLYMPIA 1 and OLYMPIA 2 Trials (Safety Population)
Treatment adherence | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 187 | Placebo N = 95 | Nemolizumab N = 183 | Placebo N = 91 | |
Mean (SD) | 97.75 (8.22) | 97.90 (8.67) | 99.46 (3.69) | 97.55 (8.31) |
Median | 100.00 | 100.00 | 100.00 | 100.00 |
Q1 to Q3 | 100.00 to 100.00 | 100.00 to 100.00 | 100.00 to 100.00 | 100.00 to 100.00 |
Minimum to maximum | 50.0 to 100.0 | 33.3 to 100.0 | 66.7 to 100.0 | 60.0 to 100.0 |
Q1 = first quartile; Q3 = third quartile; SD = standard deviation.
Notes: Treatment adherence was calculated as the ratio (percentage) between the total number of actual injections and the total number of expected injections multiplied by 100.
The total number of actual injections was counted based on collected study drug administration data.
The total number of expected injections was counted based on the dosage schedule and dispensed as per protocol.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Results for the primary, key secondary, and secondary end points of interest for this review are summarized in Table 18.
Table 18: Summary of Primary, Key Secondary, and Secondary Efficacy End Points in the OLYMPIA 1 and OLYMPIA 2 Trials (ITT Population)
End point | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 190 | Placebo N = 96 | Nemolizumab N = 183 | Placebo N = 91 | |
Primary end pointsa | ||||
Improvement of ≥ 4 from baseline in PP-NRS score at week 16 | ||||
n (%) | 111 (58.4) | 16 (16.7) | 103 (56.3) | 19 (20.9) |
Unadjusted proportion difference, (%) | 41.8 | — | 35.4 | — |
Unadjusted 95% CI | 31.5 to 52.0 | — | 24.4 to 46.4 | — |
Unadjusted P value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 40.1 | — | 37.4 | — |
Strata-adjusted 95% CI | 29.4 to 50.8 | — | 26.3 to 48.5 | — |
Strata-adjusted P value | < 0.0001 | — | < 0.0001 | — |
IGA successb at week 16 | ||||
n (%) | 50 (26.3) | 7 (7.3) | 69 (37.7) | 10 (11.0) |
Unadjusted proportion difference, (%) | 19.0 | — | 26.7 | — |
Unadjusted 95% CI | 10.9 to 27.2 | — | 17.2 to 36.2 | — |
Unadjusted P value | 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 14.6 | — | 28.5 | — |
Strata-adjusted 95% CI | 6.7 to 22.6 | — | 18.8 to 38.2 | — |
Strata-adjusted P value | 0.0025 | — | < 0.0001 | — |
Key secondary end pointsa | ||||
Improvement of ≥ 4 from baseline in SD-NRS score at week 16 | ||||
n (%) | 95 (50.0) | 11 (11.5) | 95 (51.9) | 19 (20.9) |
Unadjusted proportion difference, (%) | 38.5 | — | 31.0 | — |
Unadjusted 95% CI | 29.0 to 48.1 | — | 20.0 to 42.1 | — |
Unadjusted P value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 38.0 | — | 31.9 | — |
Strata-adjusted 95% CI | 27.8 to 48.2 | — | 20.7 to 43.2 | — |
Strata-adjusted P value | < 0.0001 | — | < 0.0001 | — |
Secondary end pointsc | ||||
Improvement of ≥ 4 from baseline in PP-NRS score at week 24 | ||||
n (%) | 90 (47.4) | 14 (14.6) | — | — |
Unadjusted proportion difference, (%) | 32.8 | — | — | — |
Unadjusted 95% CI | 22.8 to 42.8 | — | — | — |
Unadjusted P value | < 0.0001 | — | — | — |
Strata-adjusted proportion difference, (%) | 30.7 | — | — | — |
Strata-adjusted 95% CI | 20.4 to 41.1 | — | — | — |
Strata-adjusted P value | < 0.0001 | — | — | — |
IGA successb at week 24 | ||||
n (%) | 58 (30.5) | 9 (9.4) | — | — |
Unadjusted proportion difference, (%) | 21.2 | — | — | — |
Unadjusted 95% CI | 12.4 to 29.9 | — | — | — |
Unadjusted P value | < 0.0001 | — | — | — |
Strata-adjusted proportion difference, (%) | 19.2 | — | — | — |
Strata-adjusted 95% CI | 10.3 to 28.1 | — | — | — |
Strata-adjusted P value | 0.0002 | — | — | — |
Improvement of ≥ 4 from baseline in PP-NRS score and IGA successb at week 16 | ||||
n (%) | 43 (22.6) | 2 (2.1) | 54 (29.5) | 5 (5.5) |
Unadjusted proportion difference, (%) | 20.5 | — | 24.0 | — |
Unadjusted 95% CI | 13.9 to 27.1 | — | 15.9 to 32.1 | — |
Unadjusted P value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 16.0 | — | 26.1 | — |
Strata-adjusted 95% CI | 9.5 to 22.5 | — | 17.4 to 34.8 | — |
Strata-adjusted P value | 0.0002 | — | < 0.0001 | — |
Improvement of ≥ 4 from baseline in PP-NRS score and IGA successb at week 24 | ||||
n (%) | 43 (22.6) | 4 (4.2) | — | — |
Unadjusted proportion difference, (%) | 18.5 | — | — | — |
Unadjusted 95% CI | 11.3 to 25.6 | — | — | — |
Unadjusted P value | < 0.0001 | — | — | — |
Strata-adjusted proportion difference, (%) | 16.6 | — | — | — |
Strata-adjusted 95% CI | 9.1 to 24.1 | — | — | — |
Strata-adjusted P value | 0.0002 | — | — | — |
Improvement of ≥ 4 from baseline in DLQI score at week 16 | ||||
n (%) | 134 (70.5) | 41 (42.7) | 137 (74.9) | 36 (39.6) |
Unadjusted proportion difference, (%) | 27.8 | — | 35.3 | — |
Unadjusted 95% CI | 16.0 to 39.6 | — | 23.5 to 47.2 | — |
Unadjusted P value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 27.5 | — | 37.4 | — |
Strata-adjusted 95% CI | 15.8 to 39.2 | — | 25.7 to 49.0 | — |
Strata-adjusted P value | < 0.0001 | — | < 0.0001 | — |
Improvement of ≥ 4 from baseline in DLQI score at week 24 | ||||
n (%) | 135 (71.1) | 34 (35.4) | — | — |
Unadjusted proportion difference, (%) | 35.6 | — | — | — |
Unadjusted 95% CI | 24.1 to 47.2 | — | — | — |
Unadjusted P value | < 0.0001 | — | — | — |
Strata-adjusted proportion difference, (%) | 35.5 | — | — | — |
Strata-adjusted 95% CI | 23.9 to 47.2 | — | — | — |
Strata-adjusted P value | < 0.0001 | — | — | — |
Change from baseline in DLQI score at week 16, week 24 | ||||
Baseline (observed cases) | — | — | — | — |
n | 190 | 96 | 183 | 91 |
LS mean (SD) | 17.1 (6.97) | 16.9 (6.74) | 16.5 (6.79) | 17.1 (6.60) |
Week 16 | — | — | — | — |
n | 190 | 96 | 183 | 91 |
LS mean (SE) (MI estimate) | −8.57 (0.62) | −2.18 (0.87) | −8.91 (0.69) | −0.75 (0.91) |
95% CI (MI estimate) | −9.79 to −7.35 | −3.90 to −0.47 | −10.25 to −7.56 | −2.54 to 1.03 |
LS mean difference (95% CI) (MI estimate) | −6.39 (−8.43 to −4.35) | — | –8.15 (−10.16 to −6.14) | — |
P value | < 0.0001 | — | < 0.0001 | — |
Week 24 | — | — | — | — |
n | 190 | 96 | — | — |
LS mean (SE) (MI estimate) | −8.99 (0.64) | −0.56 (0.91) | — | — |
95% CI (MI estimate) | −10.25 to −7.73 | −2.34 to 1.22 | — | — |
LS mean difference (95% CI) (MI estimate) | −8.43 (−10.54 to −6.31) | — | — | — |
P value | < 0.0001 | — | — | — |
CI = confidence interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITT = intention to treat; LS = least square; MI = multiple imputation; PP-NRS = Peak Pruritus Numerical Rating Scale; SD = standard deviation; SD-NRS = Sleep Disturbance Numerical Rating Scale; SE = standard error.
aThe P value has been adjusted for multiple testing.
bIGA success was defined as an IGA of 0 (clear) or 1 (almost clear) and an improvement of 2 or more points from baseline.
cThe P values for secondary end points are nominal because the analyses have not been controlled for multiplicity.
Sources: OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report.25 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Primary End Points
Improvement of 4 or More Points From Baseline in PP-NRS Score at Week 16
OLYMPIA 1 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in weekly average PP-NRS scores were 58.4% in the nemolizumab group and 16.7% in the placebo group (strata-adjusted proportion difference = 40.1%; 95% CI, 29.4 to 50.8; P < 0.0001).2 The results of the sensitivity analyses were consistent with those of the primary analysis.24
OLYMPIA 2 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in weekly average PP-NRS scores were 56.3% in the nemolizumab group and 20.9% in the placebo group (strata-adjusted proportion difference = 37.4%; 95% CI, 26.3 to 48.5; P < 0.0001). The results of the sensitivity analyses were consistent with those of the primary analysis.25
IGA Success at Week 16
OLYMPIA 1 trial: At week 16, the proportions of patients with IGA success were 26.3% in the nemolizumab group and 7.3% in the placebo group (strata-adjusted proportion difference = 14.6%; 95% CI, 6.7 to 22.6; P = 0.0025). The results of the sensitivity analyses were consistent with those of the primary analysis.24
OLYMPIA 2 trial: At week 16, the proportions of patients with IGA success were 37.7% in the nemolizumab group and 11.0% in the placebo group (strata-adjusted proportion difference = 28.5%; 95% CI, 18.8 to 38.2; P < 0.0001). The results of the sensitivity analyses were consistent with those of the primary analysis.25
Key Secondary End Points
Improvement of 4 or More Points From Baseline in SD-NRS Score at Week 16
OLYMPIA 1 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in weekly average SD-NRS scores were 50.0% in the nemolizumab group and 11.5% in the placebo group (strata-adjusted proportion difference = 38.0%; 95% CI, 27.8 to 48.2; P < 0.0001). The results of the sensitivity analyses were consistent with those of the primary analysis.24
OLYMPIA 2 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in weekly average SD-NRS scores were 51.9% in the nemolizumab group and 20.9% in the placebo group (strata-adjusted proportion difference = 31.9%; 95% CI, 20.7 to 43.2; P < 0.0001). The results of the sensitivity analyses were consistent with those of the primary analysis.25
Other Secondary End Points
Improvement of 4 or More Points From Baseline in PP-NRS Score at Week 24
OLYMPIA 1 trial: At week 24, the proportions of patients with improvements of 4 or more points from baseline in weekly average PP-NRS scores were 47.4% in the nemolizumab group and 14.6% in the placebo group (strata-adjusted proportion difference = 30.7%; 95% CI, 20.4 to 41.1).24
IGA Success at Week 24
OLYMPIA 1 trial: At week 24, the proportions of patients with IGA success were 30.5% in the nemolizumab group and 9.4% in the placebo group (strata-adjusted proportion difference = 19.2%; 95% CI, 10.3 to 28.1).24
Improvement of 4 or More Points From Baseline in PP-NRS Score and IGA Success
OLYMPIA 1 trial: At week 16, the proportions of patients with both improvements of 4 or more points from baseline in weekly average PP-NRS scores and IGA success were 22.6% in the nemolizumab group and 2.1% in the placebo group (strata-adjusted proportion difference = 16.0%; 95% CI, 9.5 to 22.5). At week 24, the proportions were 22.6% and 4.2%, respectively (strata-adjusted proportion difference = 16.6%; 95% CI, 9.1 to 24.1).24
OLYMPIA 2 trial: At week 16, the proportions of patients with both improvements of 4 or more points from baseline in weekly average PP-NRS scores and IGA success were 29.5% in the nemolizumab group and 5.5% in the placebo group (strata-adjusted proportion difference = 26.1%; 95% CI, 17.4 to 34.8).25
Dermatology Life Quality Index
OLYMPIA 1 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in DLQI total score were 70.5% in the nemolizumab group and 42.7% in the placebo group (strata-adjusted proportion difference = 27.5%; 95% CI, 15.8 to 39.2). At week 24, the proportions were 71.1% and 35.4%, respectively (strata-adjusted proportion difference = 35.5%; 95% CI, 23.9 to 47.2).2,24
At baseline, the least squares (LS) mean DLQI total scores were 17.1 (standard deviation [SD] = 6.97) in the nemolizumab group and 16.9 (SD = 6.74) in the placebo group. At week 16, the LS mean changes from baseline were −8.57 (standard error [SE] = 0.62) in the nemolizumab group and −2.18 (SE = 0.87) in the placebo group (LS mean difference = −6.39; 95% CI, −8.43 to −4.35). At week 24, the LS mean changes from baseline were −8.99 (SE = 0.64) in the nemolizumab group and −0.56 (SE = 0.91) in the placebo group (LS mean difference = −8.43; 95% CI, −10.54 to −6.31).2,24
OLYMPIA 2 trial: At week 16, the proportions of patients with improvements of 4 or more points from baseline in DLQI total score were 74.9% in the nemolizumab group and 39.6% in the placebo group (strata-adjusted proportion difference = 37.4%; 95% CI, 25.7 to 49.0).2,25
At baseline, the LS mean DLQI total scores were 16.5 (SD = 6.79) in the nemolizumab group and 17.1 (SD = 6.60) in the placebo group. At week 16, the LS mean changes from baseline were −8.91 (SE = 0.69) in the nemolizumab group and −0.75 (SE = 0.91) in the placebo group (LS mean difference = −8.15; 95% CI, −10.16 to −6.14).2,25
Harms
A summary of key harms data from the OLYMPIA 1 and OLYMPIA 2 trials is presented in Table 19.
Adverse Events
In the OLYMPIA 1 trial, at least 1 AE was reported in 71.7% of patients in the nemolizumab group and 65.3% of patients in the placebo group; in the OLYMPIA 2 trial, at least 1 AE was reported in 61.2% and 53.8% of patients, respectively. In both trials, for the nemolizumab arm compared to the placebo arm, the most commonly reported AEs by system organ class were skin and SC tissue disorders (OLYMPIA 1 trial: 31.6% versus 29.5%; OLYMPIA 2 trial: 24.0% versus 23.1%) and infections and infestations (OLYMPIA 1 trial: 31.0% versus 29.5%; OLYMPIA 2 trial: 21.9% versus 20.9%). In the OLYMPIA 1 trial, the most common AEs (nemolizumab versus placebo) were COVID-19 (8.0% versus 14.7%), nasopharyngitis (6.4% versus 8.4%), headache (7.0% versus 2.1%), cough (4.8% versus 5.3%), dyspnea (3.2% versus 5.3%), neurodermatitis (9.6% versus 20.0%), and eczema (5.3% versus 1.1%). In the OLYMPIA 2 trial, the most common AEs were headache (6.6% versus 4.4%), neurodermatitis (3.8% versus 11.0%), and AD (5.5% versus 0%).2
Serious AEs
In the OLYMPIA 1 trial, at least 1 SAE was reported in 8.6% of patients in the nemolizumab group and 10.5% of patients in the placebo group; in the OLYMPIA 2 trial, at least 1 SAE was reported in 2.2% and 5.5% of patients in these groups, respectively. In the OLYMPIA 1 trial, the most common SAE was neurodermatitis (nemolizumab group: 1.6%; placebo group: 2.1%).2 In the OLYMPIA 2 trial, none of the SAEs reported occurred in more than 1 patient in either treatment group.2,25
Withdrawals Due to AEs
In the OLYMPIA 1 trial, AEs leading to study drug withdrawal occurred in 4.8% of patients in the nemolizumab group and in 3.2% of patients in the placebo group; in the OLYMPIA 2 trial, AEs leading to study drug withdrawal occurred in 2.7% and 2.2% of patients, respectively.2
In the OLYMPIA 1 trial, AEs leading to study discontinuation occurred in 4.8% of patients in the nemolizumab group and 4.2% of patients in the placebo group; in the OLYMPIA 2 trial, AEs leading to study discontinuation occurred in 2.2% of patients in each arm.2,51
Mortality
In the OLYMPIA 1 trial, 1 patient in the placebo group died during the study. No patient deaths occurred during the OLYMPIA 2 trial.2,24,25
Notable Harms
In the OLYMPIA 1 trial, at least 1 AESI was reported in 17.1% of patients in the nemolizumab group and 18.9% of patients in the placebo group; in the OLYMPIA 2 trial, at least 1 AESI was reported in 11.5% and 9.9% of patients, respectively. In both trials, the most commonly reported AESI in the nemolizumab and placebo groups was infections (OLYMPIA 1 trial: 10.7% versus 16.8%; OLYMPIA 2 trial: 5.5% versus 6.6%). In the OLYMPIA 1 trial, the AESI of newly diagnosed asthma or worsening of asthma was reported in 3.7% of patients in the nemolizumab group and in 2.1% of patients in the placebo group; in the OLYMPIA 2 trial, this AESI was reported in 2.7% and 1.1% of patients, respectively.2
Table 19: Summary of Harms Results in the OLYMPIA 1 and OLYMPIA 2 Trials (Treatment Period; Safety Population)
Adverse events | OLYMPIA 1 trial | OLYMPIA 2 trial | ||
|---|---|---|---|---|
Nemolizumab N = 187 | Placebo N = 95 | Nemolizumab N = 183 | Placebo N = 91 | |
Most common AEs, n (%) | ||||
≥ 1 AE | 134 (71.7) | 62 (65.3) | 112 (61.2) | 49 (53.8) |
Gastrointestinal disorders | 12 (6.4) | 11 (11.6) | 11 (6.0) | 5 (5.5) |
Gastritis | 1 (0.5) | 2 (2.1) | 0 | 1 (1.1) |
Diarrhea | 3 (1.6) | 1 (1.1) | 3 (1.6) | 2 (2.2) |
General disorders and administration-site conditions | 18 (9.6) | 7 (7.4) | 15 (8.2) | 7 (7.7) |
Fatigue | 8 (4.3) | 3 (3.2) | 6 (3.3) | 2 (2.2) |
Injection-site erythema | 1 (0.5) | 2 (2.1) | 0 | 1 (1.1) |
Edema, peripheral | 3 (1.6) | 1 (1.1) | 4 (2.2) | 1 (1.1) |
Immune system disorders | 2 (1.1) | 3 (3.2) | 1 (0.5) | 0 |
Seasonal allergy | 2 (1.1) | 2 (2.1) | 1 (0.5) | 0 |
Infections and infestations | 58 (31.0) | 28 (29.5) | 40 (21.9) | 19 (20.9) |
COVID-19 | 15 (8.0) | 14 (14.7) | 9 (4.9) | 3 (3.3) |
Nasopharyngitis | 12 (6.4) | 8 (8.4) | 5 (2.7) | 4 (4.4) |
Investigations | 12 (6.4) | 8 (8.4) | 8 (4.4) | 1 (1.1) |
Aspartate aminotransferase increased | 2 (1.1) | 2 (2.1) | 0 | 0 |
Alanine aminotransferase increased | 1 (0.5) | 2 (2.1) | 0 | 0 |
Blood creatine phosphokinase increased | 1 (0.5) | 4 (4.2) | 1 (0.5) | 0 |
Musculoskeletal and connective tissue disorders | 22 (11.8) | 7 (7.4) | 21 (11.5) | 7 (7.7) |
Back pain | 5 (2.7) | 0 | 3 (1.6) | 2 (2.2) |
Myalgia | 2 (1.1) | 2 (2.1) | 3 (1.6) | 2 (2.2) |
Pain in extremity | 3 (1.6) | 1 (1.1) | 4 (2.2) | 0 |
Nervous system disorders | 21 (11.2) | 9 (9.5) | 17 (9.3) | 9 (9.9) |
Headache | 13 (7.0) | 2 (2.1) | 12 (6.6) | 4 (4.4) |
Dizziness | 2 (1.1) | 1 (1.1) | 2 (1.1) | 2 (2.2) |
Psychiatric disorders | 1 (0.5) | 3 (3.2) | 1 (0.5) | 1 (1.1) |
Depression | 0 | 2 (2.1) | 1 (0.5) | 0 |
Renal and urinary disorders | 2 (1.1) | 3 (3.2) | 2 (1.1) | 0 |
Proteinuria | 0 | 2 (2.1) | 1 (0.5) | 0 |
Respiratory, thoracic, and mediastinal disorders | 21 (11.2) | 12 (12.6) | 13 (7.1) | 5 (5.5) |
Cough | 9 (4.8) | 5 (5.3) | 5 (2.7) | 2 (2.2) |
Dyspnea | 6 (3.2) | 5 (5.3) | 3 (1.6) | 1 (1.1) |
Asthma | 4 (2.1) | 4 (4.2) | 2 (1.1) | 1 (1.1) |
Skin and subcutaneous tissue disorders | 59 (31.6) | 28 (29.5) | 44 (24.0) | 21 (23.1) |
Neurodermatitis | 18 (9.6) | 19 (20.0) | 7 (3.8) | 10 (11.0) |
Eczema | 10 (5.3) | 1 (1.1) | 4 (2.2) | 3 (3.3) |
Eczema nummular | 7 (3.7) | 0 | 6 (3.3) | 0 |
Dermatitis atopic | 7 (3.7) | 1 (1.1) | 10 (5.5) | 0 |
Vascular disorders | 7 (3.7) | 3 (3.2) | 6 (3.3) | 2 (2.2) |
Hypertension | 4 (2.1) | 2 (2.1) | 5 (2.7) | 2 (2.2) |
SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 16 (8.6) | 10 (10.5) | 4 (2.2) | 5 (5.5) |
Cardiac disorders | 2 (1.1) | 2 (2.1) | 1 (0.5) | 2 (2.2) |
Infections and infestations | 4 (2.1) | 2 (2.1) | 1 (0.5) | 1 (1.1) |
Nervous system disorders | 2 (1.1) | 0 | 0 | 0 |
Skin and subcutaneous tissue disorders | 4 (2.1) | 2 (2.1) | 2 (1.1) | 1 (1.1) |
Neurodermatitis | 3 (1.6) | 2 (2.1) | 0 | 0 |
AEs leading to study drug withdrawal | ||||
Patients with ≥ 1 AE leading to study drug withdrawal | 9 (4.8) | 3 (3.2) | 5 (2.7) | 2 (2.2) |
Cardiac disorders | 0 | 1 (1.1) | 0 | 1 (1.1) |
Cardiac sarcoidosis | 0 | 1 (1.1) | 0 | 0 |
Atrial flutter | 0 | 0 | 0 | 1 (1.1) |
Myocardial infarction | 0 | 0 | 0 | 1 (1.1) |
Ear and labyrinth disorders | 1 (0.5) | 0 | 0 | 0 |
Vertigo, positional | 1 (0.5) | 0 | 0 | 0 |
Infections and infestations | 2 (1.1) | 0 | 1 (0.5) | 0 |
Nasopharyngitis | 1 (0.5) | 0 | 0 | 0 |
Staphylococcal skin infection | 1 (0.5) | 0 | 0 | 0 |
COVID-19 | 0 | 0 | 1 (0.5) | 0 |
Musculoskeletal and connective tissue disorders | 1 (0.5) | 0 | 0 | 0 |
Myalgia | 1 (0.5) | 0 | 0 | 0 |
Neoplasms (benign, malignant, and unspecified, including cysts and polyps) | 1 (0.5) | 0 | 0 | 0 |
Squamous cell carcinoma | 1 (0.5) | 0 | 0 | 0 |
Skin and subcutaneous tissue disorders | 4 (2.1) | 2 (2.1) | 4 (2.2) | 1 (1.1) |
Neurodermatitis | 1 (0.5) | 1 (1.1) | 0 | 0 |
Atopic dermatitis | 0 | 1 (1.1) | 2 (1.1) | 0 |
General disorders and administration-site conditions | 0 | 0 | 1 (0.5) | 0 |
Face edema | 0 | 0 | 1 (0.5) | 0 |
Edema, peripheral | 0 | 0 | 1 (0.5) | 0 |
AEs leading to study discontinuation | ||||
Patients with ≥ 1 AE leading to study discontinuation | 9 (4.8) | 4 (4.2) | 4 (2.2) | 2 (2.2) |
Cardiac disorders | 0 | 2 (2.1) | 0 | 1 (1.1) |
Cardiac sarcoidosis | 0 | 1 (1.1) | 0 | 0 |
Cardiogenic shock | 0 | 1 (1.1) | 0 | 0 |
Myocardial infarction | 0 | 0 | 0 | 1 (1.1) |
Ear and labyrinth disorders | 1 (0.5) | 0 | 0 | 0 |
Vertigo, positional | 1 (0.5) | 0 | 0 | 0 |
Infections and infestations | 2 (1.1) | 0 | 0 | 0 |
Nasopharyngitis | 1 (0.5) | 0 | 0 | 0 |
Staphylococcal skin infection | 1 (0.5) | 0 | 0 | 0 |
Musculoskeletal and connective tissue disorders | 1 (0.5) | 0 | 0 | 0 |
Myalgia | 1 (0.5) | 0 | 0 | 0 |
Neoplasms (benign, malignant, and unspecified, including cysts and polyps) | 1 (0.5) | 0 | 0 | 0 |
Squamous cell carcinoma | 1 (0.5) | 0 | 0 | 0 |
Skin and subcutaneous tissue disorders | 4 (2.1) | 2 (2.1) | 4 (2.2) | 1 (1.1) |
Neurodermatitis | 1 (0.5) | 1 (1.1) | 0 | 0 |
Dermatitis atopic | 0 | 1 (1.1) | 2 (1.1) | 0 |
Generalized disorders and administration-site conditions | 0 | 0 | 1 (0.5) | 0 |
Face edema | 0 | 0 | 1 (0.5) | 0 |
Edema peripheral | 0 | 0 | 1 (0.5) | 0 |
Deaths, n (%) | ||||
Patients who died | 0 | 1 (1.1) | 0 | 0 |
Cardiogenic shock | 0 | 1 (1.1) | 0 | 0 |
AESIs, n (%) | ||||
Patients with ≥ 1 AESI | 32 (17.1) | 18 (18.9) | 21 (11.5) | 9 (9.9) |
Injection-related reactions | 2 (1.1) | 0 | 0 | 0 |
Newly diagnosed asthma or worsening of asthma | 7 (3.7) | 2 (2.1) | 5 (2.7) | 1 (1.1) |
Infections | 20 (10.7) | 16 (16.8) | 10 (5.5) | 6 (6.6) |
Peripheral edema: limbs, bilateral; facial edema | 5 (2.7) | 1 (1.1) | 6 (3.3) | 2 (2.2) |
AE = adverse event; AESI = adverse event of special interest; SAE = serious adverse event.
Sources: Sponsor’s Summary of Clinical Evidence;2 OLYMPIA 1 Clinical Study Report;24 OLYMPIA 2 Clinical Study Report;25 sponsor’s Response to Request for Additional Information.51 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The OLYMPIA 1 and OLYMPIA 2 trials were both phase III, randomized, double-blind, placebo-controlled, multicentre studies. The trials’ methods of randomization and treatment allocation (by interactive response technology) were adequate. Reported baseline characteristics were generally balanced across the groups in both trials; however, differences were noted (e.g., age group, IGA category, and time since PN diagnosis). Nevertheless, clinical experts consulted on this review did not anticipate that those differences would affect the interpretation of the efficacy or safety results. Because the incidence of AEs was similar between groups in both studies, it was unlikely that unblinding would have occurred due to AEs. However, due to differences in treatment response between arms (e.g., reduction in pruritus), patients and investigators could have become aware of treatment allocation, which may have introduced bias for subjective end points, such as PP-NRS and DLQI.
For both OLYMPIA pivotal trials, the prespecified sample size was achieved, and the screening failure rates approached or slightly exceeded the expected rates. The OLYMPIA 1 and OLYMPIA 2 trials were powered for the primary end points of the proportion of patients with at least a 4-point improvement in PP-NRS score and the proportion of patients with IGA success (both at week 16). The multiple testing procedure included all primary and key secondary end points and used a fixed sequential testing approach with a plan to stop testing if an end point did not test statistically significant. Analyses of other secondary end points were not adjusted for multiplicity; therefore, conclusions cannot be drawn regarding statistical significance for these end points.
In both trials, loss to follow-up was low and did not occur in most treatment arms; the exception was the nemolizumab arm of the OLYMPIA 2 trial, in which 1.1% of patients were lost to follow-up in the discontinuation from study disposition data. In both trials, the percentages of patients who discontinued treatment (including the percentage discontinuing due to AEs) were similar between the nemolizumab and placebo arms. However, a higher proportion of patients discontinued treatment in the OLYMPIA 1 trial (nemolizumab: 10.0%; placebo: 10.4%) than in the OLYMPIA 2 trial (4.4% in each arm). Clinical experts consulted by CDA-AMC noted that the rates of discontinuation were generally aligned with what would be anticipated for treatment with nemolizumab, but that the rates in the OLYMPIA 1 trial may be slightly higher than expected.
In both OLYMPIA pivotal trials, the mean treatment durations were similar for the nemolizumab and placebo arms. The overall treatment exposure was higher in the OLYMPIA 1 trial than in the OLYMPIA 2 trial due to the trials’ different treatment period lengths (24 weeks and 16 weeks, respectively). In both trials, the mean total dose administered closely approached the mean total dose planned. Mean treatment adherence was similar between treatment arms for OLYMPIA 1 and OLYMPIA 2 and was greater than 97% across all arms in both trials.
During the OLYMPIA pivotal trials, a range of topical and systemic medications and procedures were prohibited (some were allowed as rescue therapy), with washout periods required before study baseline. Basic skin care products (for cleansing and bathing), moisturizers, bleach baths, and topical anesthetics were permitted. In both trials, the proportions of patients requiring the use of concomitant rescue therapy (particularly TCSs and oral antihistamines) were higher in the placebo arms than in the nemolizumab arms. This imbalance in rescue treatment could have exaggerated the itch reduction response with placebo, thereby minimizing the comparative benefit of nemolizumab. In contrast, clinical experts consulted by CDA-AMC did not expect the differences between rescue therapy use between groups to affect the interpretation of the efficacy results.
For the OLYMPIA 1 and OLYMPIA 2 trials’ primary and key secondary end points reported in this review, several sensitivity analyses were performed, and the results were consistent with those of the primary analysis. In both trials, tipping point and multiple imputation analyses for these end points revealed that the primary results were robust to missing data. Sensitivity analyses were not performed for other secondary end points. The approach to handling missing data for other secondary end points was conservative. The results of subgroup analyses were not presented by the sponsor, and clinical experts consulted by CDA-AMC did not identify any subgroups of patients with moderate to severe PN that would be more or less likely to respond to treatment with nemolizumab.
Overall, the outcome measures in the OLYMPIA pivotal trials reported in this review have been validated in patients with PN or chronic prurigo, and the thresholds used to indicate clinically meaningful change in these measures are supported by the literature and/or by the clinical experts consulted by CDA-AMC.
External Validity
Clinical experts consulted by CDA-AMC commented that the inclusion criteria used in the OLYMPIA 1 and OLYMPIA 2 trials appropriately captured patients who would be considered candidates for nemolizumab in practice and reflected patients with a diagnosis of moderate to severe PN. Patients with certain medical conditions, such as uncontrolled asthma, active AD, and certain chronic infections, were not eligible for the OLYMPIA pivotal trials. Clinical experts noted that PN generally affects older people with comorbidities and expected that such patients may require treatment with nemolizumab in clinical practice. For instance, clinical experts stated that it would not be reasonable to exclude all patients with eczema from receiving treatment with nemolizumab. They also noted that it may be difficult to screen patients for severe asthma and exclude these patients in clinical practice. Clinical experts also expressed that the exclusion of patients with chronic infections, such as hepatitis or HIV, is standard in a clinical trial setting, but expected that patients with these conditions may require the use of nemolizumab in future practice, given that these patients may be more prone to having PN. Regarding the characteristics of the patients randomized in the OLYMPIA pivotal trials, clinical experts stated that these were a reasonable reflection of patients who would receive nemolizumab for moderate to severe PN in Canada and patients with PN whom clinical experts treat in practice. Clinical experts noted that the time since PN diagnosis was quite long and reflected the current lack of effective treatments for PN; this duration would be expected to decrease over time with the availability of medications such as dupilumab and nemolizumab. In both trials, most patients were white, and patients with certain comorbidities were excluded; these factors present potential generalizability limitations because the trial populations did not adequately represent the racial diversity or concomitant conditions of patients with PN. Of note, clinical experts identified that approximately one-third of patients with PN also have atopic diatheses (e.g., AD, asthma, allergic rhinitis). Both the OLYMPIA 1 and OLYMPIA 2 trials included study sites in Canada; clinical experts expressed that they did not have concerns regarding the generalizability of the trial findings based on study locations.
Most patients in both OLYMPIA pivotal trials had received prior medications and procedures for PN treatment, with washout required before study baseline. Clinical experts commented that these prior PN therapies were generally a reasonable reflection of what patients with moderate to severe PN would receive in practice, noting that a higher percentage of patients (80% to almost 100%) would be expected to have received a TCS. As previously mentioned, topical and systemic medications and procedures were prohibited during the trials, and nemolizumab was given as monotherapy (unless rescue therapy was used). Clinical experts stated that, based on experience with previously approved biologic medications across different indications, there are instances where a biologic medication on its own is sufficient to treat the condition, and that this may be true with nemolizumab. However, they noted that it is likely that patients prescribed nemolizumab would also require TCSs, especially when initiating nemolizumab; use of TCSs would be expected to decrease over time, but these drugs could still be used to spot-treat new lesions. Clinical experts also commented that intralesional corticosteroids may also be used in combination with nemolizumab as necessary, and that while it is less likely, concomitant phototherapy or methotrexate (only for very severe cases) may be used. Regarding the prohibition of concomitant therapies, clinical experts did not have concerns about this having an impact on the generalizability of the trial evidence. Regarding concomitant rescue therapies used in the OLYMPIA 1 and OLYMPIA 2 trials, clinical experts acknowledged that these would be used in patients who experience flares in PN or inadequate treatment response while receiving nemolizumab. An important limitation of the OLYMPIA 1 and OLYMPIA 2 trials is that nemolizumab was compared to placebo, which does not represent the standard of care for treatment of PN. Currently, dupilumab is approved by Health Canada for the treatment of patients with moderate to severe PN.23 Clinical experts stated that dupilumab would be the most relevant comparator to nemolizumab for the indication under review. However, dupilumab was not indicated for treatment of PN at the time of the OLYMPIA 1 and OLYMPIA 2 trials.
According to clinical experts consulted by CDA-AMC, the primary goals of therapy for patients with PN are to reduce the severity of pruritus, treat existing lesions, prevent the formation of new lesions, and improve HRQoL while minimizing adverse effects of treatment. Similarly, outcomes of importance identified by clinician and patient groups included reduction in itch, clearance of lesions, improvement in sleep and HRQoL, and minimization of adverse effects. As such, the main efficacy and harms outcomes assessed in the OLYMPIA pivotal trials align with the key outcomes of importance to patients and clinicians.
According to clinical experts, outcomes used in clinical practice to assess response to treatment in patients with PN are improvement in the number of lesions, reduction in pruritus, and improvement in QoL; physicians may prioritize these outcomes differently. Clinical experts stated that assessments of treatment response in clinical practice do not usually rely on the grading tools used in clinical trials as stringently. They noted that disease severity may be assessed using lesion count (including subjective improvement) or scoring systems and that itch intensity may be measured subjectively or using an NRS. Clinical experts stated that QoL may be assessed using the DLQI or other indicators, such as improvement in daily life, sleep, and social well-being. According to clinical experts, 16 to 24 weeks is an appropriate amount of time to assess the trials’ primary end point outcomes (i.e., itch reduction and clearing of lesions); they noted that while 24 weeks may be ideal, 16 weeks is sufficient.
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:52,53
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The thresholds used for the presence or absence of an important effect were based on thresholds informed by the clinical experts consulted for this review for the PP-NRS, IGA success, and DLQI end points. For newly diagnosed asthma or worsening of asthma, the presence or absence of any non-null effect was used.
For the GRADE assessments, findings from the OLYMPIA 1 and OLYMPIA 2 trials were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for nemolizumab versus placebo.
LTE Studies
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Description of Studies
The OLYMPIA LTE study (NCT04204616) is an ongoing, phase III, prospective, single-arm, multicentre, open-label study to evaluate the long-term safety and efficacy of nemolizumab in adult patients with PN. The efficacy results summarized in this review are from Interim Analysis 2 (data available as of July 21, 2024) supplemented with data from Interim Analysis 1 (data cut-off date of March 10, 2023). Patients were eligible to enrol in the LTE study from the following previous studies of nemolizumab for PN:
Phase IIa study (NCT03181503): Patients previously randomized to this study were considered for eligibility in the LTE study. Patients who enrolled into the open-label extension study from this phase IIa study must have completed all screening assessments within 28 days before the baseline visit or first dose of study drug.
Phase III OLYMPIA 1 (NCT04501666) and OLYMPIA 2 (NCT04501679) trials: Patients completing the treatment periods of these studies may have been eligible to enrol in the open-label extension study immediately. Patients were not eligible to enrol in the LTE study more than 56 days following their last visit in the lead-in OLYMPIA studies.
At week 52, clinical responders, defined as patients with an IGA score of 0 or 1 and an improvement in PP-NRS score of 4 or more points from baseline of the lead-in study, may have been eligible to exit the LTE study and enter a 24-week durability study (NCT05052983). Patients who completed week 24 of the durability study or met the criteria for disease relapse at any point during the treatment period of the durability study were eligible to re-enter the LTE study. For re-entered patients, data were summarized by nemolizumab every 4 weeks overall (N = 17) from the durability study as well as by patients who experienced disease relapse (N = 9) and those who did not experience relapse (N = 8).
Each patient’s participation in the LTE study will be up to 196 weeks. The study consists of a screening period (up to 4 weeks), a treatment period (up to 184 weeks, with the final dose administered at week 180), and an 8-week follow-up period (12 weeks after the last injection of study drug). The primary end point is long-term safety (i.e., incidence and severity of AEs, including AESIs, TEAEs, and SAEs).
Populations
In addition to the inclusion criteria outlined in Table 5 of this report, to be enrolled in the LTE, patients must have been those who may benefit from study participation, in the opinion of the investigator, and participated in a prior nemolizumab study for PN by doing any of the following:
completing the treatment period of a phase III pivotal study (i.e., the OLYMPIA 1 trial or OLYMPIA 2 trial) and enrolled within 56 days
undergoing randomization in the phase IIa study of nemolizumab for PN (i.e., NCT03181503)
completing week 24 of the phase IIIb durability study (NCT05052983) or exited the study due to relapse (these patients may have been eligible to re-enter the LTE study within 28 days at select countries and sites).
Patients were excluded from the LTE study if they experienced any AEs during any of the lead-in nemolizumab studies that may have resulted in unreasonable risk for patients, if they had a body weight of less than 30 kg, or if they received any restricted treatments within the prespecified time frame before baseline.
Interventions
The dose received in the LTE study (i.e., 1 or 2 injections) was based on the body weight and the previous treatment assignment in the nemolizumab PN studies (refer to Appendix 1, Table 36 and Table 37).
For patients who enrolled from the phase IIa study:
Patients who weighed less than 90 kg at baseline received open-label 30 mg nemolizumab (with a 60 mg loading dose at baseline) every 4 weeks.
Patients who weighed 90 kg or more at baseline received 60 mg nemolizumab (2 injections of 30 mg, with no loading dose) every 4 weeks.
For patients who enrolled from the phase III studies:
The day 1 or baseline dose in the LTE study was based on the blinded study treatment assigned during the pivotal trial; this was done to maintain the blinding of the study. Therefore, patients received either 2 blinded 30 mg injections of nemolizumab or 1 blinded 30 mg injection of nemolizumab and 1 blinded injection of placebo.
After day 1, the same dosing regimen was used as in the pivotal study (i.e., 1 or 2 SC injections of 30 mg nemolizumab, based on patient weight at baseline of the lead-in study).
For patients re-entering from the durability study:
Patients received the same dose of nemolizumab that was assigned to them in the durability study
Outcomes
The efficacy end points assessed included IGA success (defined as an IGA of 0 [clear] or 1 [almost clear]) and improvements in PP‑NRS, SD-NRS, and DLQI scores. A detailed description of these end points is provided in the Systematic Review section.
Statistical Analysis
No formal sample size calculations were performed for this LTE study. The study planned to enrol approximately 450 patients, depending on the rollover rate from the lead-in studies, from approximately 160 study sites in Asia Pacific, Europe, and North America.
The safety population consisted of all patients who received at least 1 dose of nemolizumab (blinded or open label). All efficacy analyses were performed on the safety population and were descriptive in nature. The efficacy analyses were carried out using observed cases without imputing missing data. All efficacy assessments were summarized by LTE treatment and previous treatment at each analysis visit. Scheduled, unscheduled, and early termination visits were windowed based on the analysis visit window. No hypothesis testing was performed.
Data are summarized for all patients in the safety population (N = 508) under the following headings:
Continuous nemolizumab: includes patients with an interval of less than 12 weeks between the last nemolizumab dose in the lead-in study and the first dose in the LTE study (N = 307); continuous nemolizumab was used to evaluate the persistence of the nemolizumab effect.
Re-treatment: includes patients with an interval of 12 or more weeks between the last nemolizumab dose in the lead-in study and the first dose in the LTE study (N = 27); re-treatment was used to assess any loss of response. This group included patients from the phase III studies (i.e., the OLYMPIA 1 or OLYMPIA 2 studies) or phase IIa studies with an interval of 12 weeks or more after the last nemolizumab dose.
Placebo to nemolizumab: includes patients who had never received nemolizumab before the LTE (e.g., patients who were on placebo during the lead-in or who enrolled directly in the LTE study [N = 174]).
Sensitivity Analyses
For binary secondary end points, efficacy data collected on or after the use of rescue therapy were classified as indicative of treatment failure, except in the observed cases analysis, in which observed data were used regardless of rescue therapy use. Continuous variables were set to the worst-case value, and the patient’s binary response was based on the underlying continuous value before the imputation of missing data. Continuous secondary end points were analyzed using multiple imputation, assuming that missing data were missing at random.
Results
Patient Disposition
Patients were enrolled from January 11, 2021, to March 10, 2023. A total of 510 patients were screened at 120 study sites, and 508 of those patients were enrolled and received the study drug. Of patients enrolled in the LTE, the majority were from the OLYMPIA 1 trial (N = 232) and the OLYMPIA 2 trial (N = 255). Given that this assessment is from an interim analysis, no patients had completed the LTE study; however, less than 20% had discontinued primarily due to AEs, patient request, or lack of efficacy (Table 20). Further, the proportion of patients who had discontinued treatment was similar among patients in the continuous nemolizumab and placebo-to-nemolizumab groups (18.9% and 17.2%, respectively), and slightly lower in the re-treated group (14.8%; Table 20). At Interim Analysis 2, ███% and ███% of patients in the continuous nemolizumab and placebo-to-nemolizumab groups, respectively, discontinued from treatment (Appendix 1, Table 38).
Table 20: Patient Disposition by Previous Treatment (Safety Population)
Number of patients | Nemolizumab q.4.w. N = 508 n (%) | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 n (%) | ||||
All N = 334 n (%) | Continuous nemolizumab N = 307 n (%) | Re-treatment N = 27 n (%) | |||
Treated | 508 (100.0) | 334 (100.0) | 307 (100.0) | 27 (100.0) | 174 (100.0) |
Completed treatment | 0 | 0 | 0 | 0 | 0 |
Discontinued treatment | 92 (18.1) | 62 (18.6) | 58 (18.9) | 4 (14.8) | 30 (17.2) |
Primary reason for discontinuation of treatment | |||||
Pregnancy | 0 | 0 | 0 | 0 | 0 |
Lack of efficacy | 13 (2.6) | 9 (2.7) | 9 (2.9) | 0 | 4 (2.3) |
Adverse event | 35 (6.9) | 25 (7.5) | 25 (8.1) | 0 | 10 (5.7) |
Patient request | 27 (5.3) | 19 (5.7) | 16 (5.2) | 3 (11.1) | 8 (4.6) |
Lost to follow-up | 8 (1.6) | 5 (1.5) | 5 (1.6) | 0 | 3 (1.7) |
Protocol deviation | 1 (0.2) | 1 (0.3) | 1 (0.3) | 0 | 0 |
Physician or primary investigator decision | 4 (0.8) | 1 (0.3) | 1 (0.3) | 0 | 3 (1.7) |
Sponsor decision | 0 | 0 | 0 | 0 | 0 |
Other | |||||
COVID-19 | 0 | 0 | 0 | 0 | 0 |
PEF criteria not met | 1 (0.2) | 1 (0.3) | 0 | 1 (3.7) | 0 |
Site closure | 2 (0.4) | 1 (0.3) | 1 (0.3) | 0 | 1 (0.6) |
Visit schedulea | 1 (0.2) | 0 | 0 | 0 | 1 (0.6) |
Completed the study | 0 | 0 | 0 | 0 | 0 |
Discontinued from the study | 84 (16.5) | 58 (17.4) | 55 (17.9) | 3 (11.1) | 26 (14.9) |
Primary reason for discontinuation from the study | |||||
Pregnancy | 0 | 0 | 0 | 0 | 0 |
Lack of efficacy | 9 (1.8) | 6 (1.8) | 6 (2.0) | 0 | 3 (1.7) |
Adverse event | 34 (6.7) | 24 (7.2) | 24 (7.8) | 0 | 10 (5.7) |
Patient request | 26 (5.1) | 19 (5.7) | 17 (5.5) | 2 (7.4) | 7 (4.0) |
Lost to follow-up | 8 (1.6) | 5 (1.5) | 5 (1.6) | 0 | 3 (1.7) |
Protocol deviation | 1 (0.2) | 1 (0.3) | 1 (0.3) | 0 | 0 |
Physician decision | 2 (0.4) | 1 (0.3) | 1 (0.3) | 0 | 1 (0.6) |
Sponsor decision | 0 | 0 | 0 | 0 | 0 |
Other | |||||
COVID-19 | 0 | 0 | 0 | 0 | 0 |
PEF criteria not meta | 1 (0.2) | 1 (0.3) | 0 | 1 (3.7) | 0 |
Site closure | 2 (0.4) | 1 (0.3) | 1 (0.3) | 0 | 1 (0.6) |
Visit scheduleb | 1 (0.2) | 0 | 0 | 0 | 1 (0.6) |
Rolled over to study NCT05052983 | 33 (6.5) | 23 (6.9) | 18 (5.9) | 5 (18.5) | 10 (5.7) |
Re-entered from study NCT05052983 | 17 (3.3) | 12 (3.6) | 8 (2.6) | 4 (14.8) | 5 (2.9) |
Completed follow-up | 37 (7.3) | 27 (8.1) | 26 (8.5) | 1 (3.7) | 10 (5.7) |
LTE = long-term extension; PEF = peak expiratory flow; q.4.w. = every 4 weeks.
Note: Percentages were based on the number of patients.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12-weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with at least a 12-week interval between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE study.
aPatient 828-5433-005 did not meet the PEF criterion at baseline; therefore, they should not have been enrolled. The reason for discontinuation should have been noted as a protocol deviation.
bDue to a misunderstanding regarding the visit schedule/early termination, the patient did not complete the treatment per protocol.
Source: LTE Clinical Study Report.54
Baseline Characteristics
Overall, the patients had a mean age and body weight of 55.4 years and 82.59 kg, respectively (Table 21). The majority were female (60.40%) and white (76.0%). The duration between the last lead-in study dose and the first LTE study dose was 72.0 days. Details of baseline and demographic characteristics by specific groups can be found in Table 21.
Disease Characteristics
Baseline disease characteristics, grouped by previous treatment, are summarized in Table 22. At the LTE study baseline, the overall proportions of patients with moderate and severe IGA were 32.10% and 15.70%, respectively. There was a higher proportion of patients in the placebo-to-nemolizumab group (moderate: 43.10%; severe: 31.60%) than in the continuous nemolizumab group (moderate: 26.10%; severe: 6.20%). At the lead-in baseline, patients’ weekly average PP-NRS score had a mean of 8.49; their weekly average SD‑NRS score had a mean of 7.09; and their mean DLQI total score was 16.90. Most had no atopy background (65.20%). The average time since PN diagnosis was 106.63 months.
Table 21: Demographic and Baseline Characteristics by Previous Treatment (Safety Population) — Interim Analysis 1
Characteristic | Nemolizumab q.4.w. N = 508 | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 | ||||
All N = 334 | Continuous nemolizumab N = 307 | Re-treatment N = 27 | |||
Age (years) | |||||
Mean (SD) | 55.4 (14.04) | 56.0 (13.63) | 56.0 (13.42) | 56.0 (16.06) | 54.5 (14.78) |
Age group, n (%) | |||||
18 to 65 years | 375 (73.80) | 244 (73.10) | 223 (72.60) | 21 (77.80) | 131 (75.30) |
> 65 years | 133 (26.20) | 90 (26.90) | 84 (27.40) | 6 (22.20) | 43 (24.70) |
Sex, n (%) | |||||
Female | 307 (60.40) | 202 (60.50) | 185 (60.30) | 17 (63.00) | 105 (60.30) |
Male | 201 (39.60) | 132 (39.50) | 122 (39.70) | 10 (37.00) | 69 (39.70) |
Region, n (%) | |||||
Asia Pacific | 19 (3.70) | 11 (3.30) | 9 (2.90) | 2 (7.40) | 8 (4.60) |
Europe | 372 (73.20) | 241 (72.20) | 216 (70.40) | 25 (92.60) | 131 (75.30) |
North America | 117 (23.00) | 82 (24.60) | 82 (26.70) | 0 | 35 (20.10) |
Ethnicity, n (%) | |||||
Hispanic or Latino | 17 (3.30) | 8 (2.40) | 8 (2.60) | 0 | 9 (5.20) |
Not Hispanic or Latino | 443 (87.20) | 297 (88.90) | 273 (88.90) | 24 (88.90) | 146 (83.90) |
Unknown | 0 | 0 | 0 | 0 | 0 |
Not reported | 48 (9.40) | 29 (8.70) | 26 (8.50) | 3 (11.10) | 19 (10.90) |
Race, n (%) | |||||
Alaska Native or American Indian | 1 (0.20) | 1 (0.30) | 1 (0.30) | 0 | 0 |
Asian | 41 (8.10) | 27 (8.10) | 25 (8.10) | 2 (7.40) | 14 (8.00) |
Black or African American | 26 (5.10) | 14 (4.20) | 14 (4.60) | 0 | 12 (6.90) |
Native Hawaiian or other Pacific Islander | 1 (0.20) | 1 (0.30) | 1 (0.30) | 0 | 0 |
White | 386 (76.00) | 258 (77.20) | 236 (76.90) | 22 (81.50) | 128 (73.60) |
Multiple | 0 | 0 | 0 | 0 | 0 |
Not reported | 46 (9.10) | 29 (8.70) | 26 (8.50) | 3 (11.10) | 17 (9.80) |
Other | 7 (1.40) | 4 (1.20) | 4 (1.30) | 0 | 3 (1.70) |
Height (cm) at baseline | |||||
n | 507 | 333 | 306 | 27 | 174 |
Mean (SD) | 168.61 (9.46) | 168.96 (8.91) | 169.08 (8.96) | 167.53 (8.30) | 167.96 (10.42) |
Weight (kg) | |||||
Mean (SD) | 82.59 (19.80) | 83.32 (19.65) | 83.74 (19.55) | 78.56 (20.58) | 81.18 (20.08) |
Weight subgroup, n (%) | |||||
< 90 kg without change | 346 (68.10) | 223 (66.80) | 202 (65.80) | 21 (77.80) | 123 (70.70) |
≥ 90 kg without change | 153 (30.10) | 105 (31.40) | 99 (32.20) | 6 (22.20) | 48 (27.60) |
< 90 kg with change | 4 (0.80) | 1 (0.30) | 1 (0.30) | 0 | 3 (1.70) |
≥ 90 kg with change | 5 (1.00) | 5 (1.50) | 5 (1.60) | 0 | 0 |
Weight (kg) at LTE study baseline | |||||
Mean (SD) | 82.44 (19.66) | 83.24 (19.53) | 83.66 (19.43) | 78.35 (20.41) | 80.93 (19.86) |
Body mass index (kg/m2) | |||||
n | 507 | 333 | 306 | 27 | 174 |
Mean (SD) | 28.92 (5.91) | 29.09 (6.01) | 29.20 (6.00) | 27.78 (6.10) | 28.61 (5.71) |
Smoking status, n (%) | |||||
Never | 0 | 0 | 0 | 0 | 0 |
Former | 127 (25.00) | 85 (25.40) | 81 (26.40) | 4 (14.80) | 42 (24.10) |
Current | 85 (16.70) | 55 (16.50) | 47 (15.30) | 8 (29.60) | 30 (17.20) |
Missing | 296 (58.30) | 194 (58.10) | 179 (58.30) | 15 (55.60) | 102 (58.60) |
Treatment exposure to nemolizumab in previous study, n (%) | |||||
Yes | 334 (65.70) | 334 (100.00) | 307 (100.00) | 27 (100.00) | 0 |
No | 174 (34.30) | 0 | 0 | 0 | 174 (100.00) |
Duration between last lead-in study dose and first LTE study dose, days | |||||
Mean (SD) | 81.0 (214.40) | 72.0 (192.80) | 33.5 (11.76) | 508.8 (508.79) | 98.4 (250.41) |
Median | 29.0 | 29.0 | 29.0 | 110.0 | 30.0 |
Q1 to Q3 | 29.0 to 37.0 | 29.0 to 36.0 | 29.0 to 35.0 | 86.0 to 1,073.0 | 29.0 to 39.0 |
Minimum to maximum | 15 to 1,248 | 18 to 1,248 | 18 to 83 | 84 to 1,248 | 15 to 1,179 |
Duration between last lead-in study dose and first LTE study dose, n (%) | |||||
< 12 weeks | 464 (91.30) | 307 (91.90) | 307 (100.00) | 0 | 157 (90.20) |
≥ 12 weeks | 44 (8.70) | 27 (8.10) | 0 | 27 (100.00) | 17 (9.80) |
LTE = long-term extension; Q1 = first quartile; Q3 = third quartile; q.4.w. = every 4 weeks; SD = standard deviation.
Notes: Percentages were based on the number of patients.
Weight and height at baseline for patients who entered from the OLYMPIA 1 or OLYMPIA 2 studies were defined as weight and height at baseline of the lead-in studies.
Weight and height at baseline for Study NCT03181503 was defined as weight and height at baseline of the LTE study.
Baseline LTE was defined as the last nonmissing value before the first dose of the study drug. Race and ethnicity data were not collected for patients in France; these patients were counted in the Not reported category.
The duration between the last dose in the lead-in study and the first dose in the LTE study was equal to the first dose in the LTE study minus the last dose of the lead-in study plus 1.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with an interval of at least 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients who had never been exposed to nemolizumab before this LTE study.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: LTE Clinical Study Report.54
Table 22: Baseline Disease Characteristics by Previous Treatment (Safety Population) — Interim Analysis 1
Characteristic | Nemolizumab q.4.w. N = 508 | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 | ||||
All N = 334 | Continuous nemolizumab N = 307 | Re-treatment N = 27 | |||
IGA category | |||||
At lead-in study baseline, n (%) | |||||
Clear (0) | 0 | 0 | 0 | 0 | 0 |
Almost clear (1) | 0 | 0 | 0 | 0 | 0 |
Mild (2) | 0 | 0 | 0 | 0 | 0 |
Moderate (3) | 286 (56.30) | 189 (56.60) | 179 (58.30) | 10 (37.00) | 97 (55.70) |
Severe (4) | 201 (39.60) | 134 (40.10) | 128 (41.70) | 6 (22.20) | 67 (38.50) |
Missing | 21 (4.10) | 11 (3.30) | 0 | 11 (40.70) | 10 (5.70) |
At LTE study baseline, n (%) | |||||
Clear (0) | 47 (9.30) | 39 (11.70) | 37 (12.10) | 2 (7.40) | 8 (4.60) |
Almost clear (1) | 102 (20.10) | 88 (26.30) | 86 (28.00) | 2 (7.40) | 14 (8.00) |
Mild (2) | 116 (22.80) | 94 (28.10) | 85 (27.70) | 9 (33.30) | 22 (12.60) |
Moderate (3) | 163 (32.10) | 88 (26.30) | 80 (26.10) | 8 (29.60) | 75 (43.10) |
Severe (4) | 80 (15.70) | 25 (7.50) | 19 (6.20) | 6 (22.20) | 55 (31.60) |
Weekly average PP-NRS scores | |||||
At lead-in study baseline | |||||
n | 483 | 319 | 303 | 16 | 164 |
Mean (SD) | 8.49 (0.95) | 8.54 (0.93) | 8.54 (0.93) | 8.55 (0.88) | 8.39 (0.98) |
At LTE study baseline | |||||
n | 445 | 290 | 272 | 18 | 155 |
Mean (SD) | 4.50 (3.26) | 3.35 (3.02) | 3.13 (2.91) | 6.63 (2.81) | 6.64 (2.54) |
Weekly average AP-NRS scores | |||||
At lead-in study baseline | |||||
n | 475 | 314 | 298 | 16 | 161 |
Mean (SD) | 8.25 (1.06) | 8.29 (1.04) | 8.27 (1.04) | 8.57 (0.86) | 8.18 (1.11) |
At LTE study baseline | |||||
n | 439 | 289 | 272 | 17 | 150 |
Mean (SD) | 4.18 (3.22) | 3.14 (2.99) | 2.94 (2.90) | 6.12 (2.80) | 6.19 (2.66) |
Weekly average SD-NRS scores | |||||
At lead-in study baseline | |||||
n | 486 | 322 | 306 | 16 | 164 |
Mean (SD) | 7.09 (2.31) | 7.07 (2.36) | 7.10 (2.31) | 6.46 (3.26) | 7.11 (2.21) |
At LTE study baseline | |||||
n | 448 | 292 | 273 | 19 | 156 |
Mean (SD) | 3.60 (3.16) | 2.66 (2.84) | 2.50 (2.78) | 4.82 (2.98) | 5.36 (2.98) |
Pain frequency | |||||
At lead-in study baseline, n (%) | |||||
Never | 21 (4.10) | 16 (4.80) | 16 (5.20) | 0 | 5 (2.90) |
Less than once a week | 29 (5.70) | 19 (5.70) | 18 (5.90) | 1 (3.70) | 10 (5.70) |
1 to 2 days a week | 28 (5.50) | 16 (4.80) | 14 (4.60) | 2 (7.40) | 12 (6.90) |
3 to 4 days a week | 58 (11.40) | 44 (13.20) | 40 (13.00) | 4 (14.80) | 14 (8.00) |
5 to 6 days a week | 36 (7.10) | 28 (8.40) | 27 (8.80) | 1 (3.70) | 8 (4.60) |
Every day | 315 (62.00) | 200 (59.90) | 192 (62.50) | 8 (29.60) | 115 (66.10) |
Missing | 21 (4.10) | 11 (3.30) | 0 | 11 (40.70) | 10 (5.70) |
At LTE study baseline, n (%) | |||||
Never | 112 (22.00) | 99 (29.6) | 98 (31.9) | 1 (3.70) | 13 (7.50) |
Less than once a week | 78 (15.40) | 63 (18.9) | 57 (18.6) | 6 (22.20) | 15 (8.60) |
1 to 2 days a week | 86 (16.90) | 60 (18.0) | 55 (17.9) | 5 (18.50) | 26 (14.90) |
3 to 4 days a week | 54 (10.60) | 32 (9.6) | 28 (9.1) | 4 (14.80) | 22 (12.60) |
5 to 6 days a week | 28 (5.50) | 16 (4.8) | 14 (4.6) | 2 (7.40) | 12 (6.90) |
Every day | 133 (26.20) | 52 (15.6) | 43 (14.0) | 9 (33.30) | 81 (46.60) |
Missing | 17 (3.30) | 12 (3.6) | 12 (3.9) | 0 | 5 (2.90) |
Pain intensity | |||||
At lead-in study baseline | |||||
n | 487 | 323 | 307 | 16 | 164 |
Mean (SD) | 7.6 (2.50) | 7.4 (2.60) | 7.4 (2.63) | 8.1 (1.71) | 7.8 (2.30) |
At LTE study baseline | |||||
n | 491 | 322 | 295 | 27 | 169 |
Mean (SD) | 4.1 (3.39) | 3.0 (3.05) | 2.8 (3.05) | 5.0 (2.34) | 6.1 (3.07) |
PAS item 4 (number of lesions in representative area) | |||||
At lead-in study baseline | |||||
n | 487 | 323 | 307 | 16 | 164 |
Mean (SD) | 22.3 (18.79) | 22.5 (18.76) | 22.4 (19.05) | 24.0 (12.06) | 22.0 (18.92) |
Median | 17.0 | 18.0 | 17.0 | 23.5 | 17.0 |
Q1 to Q3 | 10.0 to 28.0 | 10.0 to 28.0 | 10.0 to 28.0 | 14.0 to 30.0 | 10.0 to 27.0 |
Minimum to maximum | 1 to 140 | 1 to 140 | 1 to 140 | 8 to 55 | 3 to 110 |
At LTE study baseline | |||||
n | 507 | 333 | 306 | 27 | 174 |
Mean (SD) | 11.0 (14.70) | 7.2 (10.05) | 6.6 (9.85) | 13.5 (10.30) | 18.1 (18.95) |
Median | 6.0 | 4.0 | 4.0 | 10.0 | 13.5 |
Q1 to Q3 | 2.0 to 15.0 | 1.0 to 10.0 | 1.0 to 8.0 | 5.0 to 25.0 | 5.0 to 25.0 |
Minimum to maximum | 0 to 105 | 0 to 104 | 0 to 104 | 0 to 33 | 0 to 105 |
PAS item 5a (excoriation and/or crusts) | |||||
At lead-in study baseline, n (%) | |||||
0% | 1 (0.20) | 1 (0.30) | 1 (0.30) | 0 | 0 |
1% to 25% | 32 (6.30) | 14 (4.20) | 13 (4.20) | 1 (3.70) | 18 (10.30) |
26% to 50% | 91 (17.90) | 61 (18.30) | 58 (18.90) | 3 (11.10) | 30 (17.20) |
51% to 75% | 152 (29.90) | 104 (31.10) | 97 (31.60) | 7 (25.90) | 48 (27.60) |
76% to 100% | 211 (41.50) | 143 (42.80) | 138 (45.00) | 5 (18.50) | 68 (39.10) |
Missing | 21 (4.10) | 11 (3.30) | 0 | 11 (40.70) | 10 (5.70) |
At LTE study baseline, n (%) | |||||
0% | 66 (13.00) | 57 (17.10) | 55 (17.90) | 2 (7.40) | 9 (5.20) |
1% to 25% | 166 (32.70) | 133 (39.80) | 128 (41.70) | 5 (18.50) | 33 (19.00) |
26% to 50% | 89 (17.50) | 56 (16.80) | 47 (15.30) | 9 (33.30) | 33 (19.00) |
51% to 75% | 97 (19.10) | 48 (14.40) | 46 (15.00) | 2 (7.40) | 49 (28.20) |
76% to 100% | 88 (17.30) | 38 (11.40) | 29 (9.40) | 9 (33.30) | 50 (28.70) |
Missing | 2 (0.40) | 2 (0.60) | 2 (0.70) | 0 | 0 |
PAS item 5b (healed lesion stages) | |||||
At lead-in study baseline, n (%) | |||||
100% | 0 | 0 | 0 | 0 | 0 |
76% to 99% | 5 (1.00) | 4 (1.20) | 3 (1.00) | 1 (3.70) | 1 (0.60) |
51% to 75% | 59 (11.60) | 37 (11.10) | 32 (10.40) | 5 (18.50) | 22 (12.60) |
26% to 50% | 119 (23.40) | 73 (21.90) | 68 (22.10) | 5 (18.50) | 46 (26.40) |
0% to 25% | 304 (59.80) | 209 (62.60) | 204 (66.40) | 5 (18.50) | 95 (54.60) |
Missing | 21 (4.10) | 11 (3.30) | 0 | 11 (40.70) | 10 (5.70) |
At LTE study baseline, n (%) | |||||
100% | 53 (10.40) | 45 (13.50) | 43 (14.00) | 2 (7.40) | 8 (4.60) |
76% to 99% | 157 (30.90) | 134 (40.10) | 130 (42.30) | 4 (14.80) | 23 (13.20) |
51% to 75% | 77 (15.20) | 53 (15.90) | 45 (14.70) | 8 (29.60) | 24 (13.80) |
26% to 50% | 102 (20.10) | 54 (16.20) | 51 (16.60) | 3 (11.10) | 48 (27.60) |
0% to 25% | 117 (23.00) | 46 (13.80) | 36 (11.70) | 10 (37.00) | 71 (40.80) |
Missing | 2 (0.40) | 2 (0.60) | 2 (0.70) | 0 | 0 |
Dermatology Life Quality Index total score | |||||
At lead-in study baseline | |||||
n | 487 | 323 | 307 | 16 | 164 |
Mean (SD) | 16.9 (6.81) | 17.0 (6.94) | 17.1 (6.95) | 15.9 (6.95) | 16.6 (6.55) |
At LTE study baseline | |||||
n | 489 | 321 | 294 | 27 | 168 |
Mean (SD) | 8.9 (7.44) | 6.7 (6.45) | 6.4 (6.24) | 10.4 (7.57) | 13.1 (7.42) |
Atopy background, n (%) | |||||
Yes | 177 (34.80) | 116 (34.70) | 110 (35.80) | 6 (22.20) | 61 (35.10) |
No | 331 (65.20) | 218 (65.30) | 197 (64.20) | 21 (77.80) | 113 (64.90) |
Time since PN diagnosis (months) | |||||
Mean (SD) | 106.63 (95.78) | 103.88 (92.60) | 101.43 (92.54) | 131.84 (90.27) | 111.91 (101.69) |
Median | 73.64 | 73.23 | 69.98 | 124.98 | 76.94 |
Q1 to Q3 | 38.85 to 142.78 | 38.01 to 139.37 | 36.63 to 136.05 | 58.51 to 178.07 | 42.78 to 150.87 |
Minimum to maximum | 0.2 to 625.3 | 0.3 to 455.0 | 0.3 to 455.0 | 14.3 to 383.1 | 0.2 to 625.3 |
AP-NRS = Average Pruritus Numerical Rating Scale; IGA = Investigator’s Global Assessment; LTE = long-term extension; PAS = Prurigo Activity Score; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; Q1 = first quartile; Q3 = third quartile; q.4.w. = every 4 weeks; SD = standard deviation; SD-NRS = Sleep Disturbance Numerical Rating Scale.
Notes: Percentages were based on numbers of patients.
Baseline LTE was the last nonmissing value before the first dose of the study drug in this study.
The lead-in baseline was defined as the last nonmissing value before the first dose of the study drug in the lead-in study.
Phase II patients did not contribute to change from the lead-in study baseline, given that there was at least 6 months between the completion of phase II and entry into the LTE study.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and LTE study first dose. The re-treatment group included patients with an interval of at least 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients who had never been exposed to nemolizumab before this LTE.
Sources: LTE Clinical Study Report,54 Clinical Study Report LTE (Interim Analysis 2).55
Exposure to Study Treatments
At Interim Analysis 2, patients overall received a planned mean dose of 1,093.2 mg (Table 23). A total of 281 patients (55.30%) missed at least 1 dose, and 53 patients (10.40%) missed at least 1 dose due to COVID-19. The mean treatment duration was 733.0 days. Patients had a mean treatment adherence of 92.71% (SD = 12.93). Data on exposure to study treatment for patients contributing to Interim Analysis 1 are presented in Appendix 1 (Table 39).
Prior Medication
The percentage of patients with at least 1 prior PN medication was 67.7%. The most frequently reported prior PN medication was clobetasol propionate (27.0% overall); it was received by a similar percentage of patients in the group receiving continuous nemolizumab (23.1%) and in the group without previous exposure to nemolizumab (31.0%) and by a higher percentage of patients in the re-treated group (44.4%). The percentage of patients with at least 1 prior PN procedure was 27.2% overall. Prior phototherapy was used by 27.2% of patients. Overall, trends in prior PN medication use and prior PN procedures during the LTE study were similar to those observed at baseline for the OLYMPIA 1 and OLYMPIA 2 trials, as summarized in the Systematic Review section.
Concomitant Medications
Most patients (93.5%) in the LTE study received at least 1 concomitant medication during the treatment period. The most common concomitant medications received were tozinameran (23.0%), paracetamol (21.5%), and ibuprofen (15.7%).
Table 23: Summary of Exposure to the Study Drug (Safety Population) — Interim Analysis 2
Characteristic | Nemolizumab q.4.w. N = 508 |
|---|---|
Total dose administered (mg)a | |
Mean (SD) | 1,036.8 (567.74) |
Total dose planned during the study (mg),b mean (SD) | 1,093.2 (570.83) |
Patients who missed at least 1 dose, n (%) | 281 (55.30) |
Patients who missed at least 1 dose due to COVID-19, n (%) | 53 (10.40) |
Number of doses missed, n | 955 |
Number of doses missed due to COVID-19, n | 78 |
Treatment duration during the study (days) | |
Mean (SD) | 733.0 (327.21) |
Number of patients by treatment duration during the study, n (%) | |
< 6 months | 52 (10.20) |
6 to < 12 months | 45 (8.90) |
12 to < 18 months | 23 (4.50) |
18 to < 24 months | 80 (15.70) |
24 to < 30 months | 125 (24.60) |
30 to < 36 months | 141 (27.80) |
36 to < 42 months | 42 (8.30) |
≥ 42 months | 0 |
Treatment adherence, n (%) | |
Mean (SD) | 92.71 (12.93) |
Median | 97.30 |
Q1 to Q3 | 91.37 to 100.00 |
Minimum to maximum | 7.7 to 100.0 |
Q1 = first quartile; Q3 = third quartile; q.4.w.= every 4 weeks; SD = standard deviation.
Notes: Percentages were based on the number of patients.
Treatment duration was calculated as: (date of last treatment – date of first treatment) + 1. For patients who re-entered from Study NCT05052983, treatment duration was calculated as (date of last treatment before durability study – date of first treatment) + (date of last re-entering treatment – date of first re-entering treatment) + 2.
aTotal dose administered was calculated as the sum of all doses of study drug administered in this study.
bTotal dose planned was calculated as the sum of all doses of study drug planned (dispensed) according to the treatment schedule of the treatment group.
Source: LTE Clinical Study Report, Interim Analysis 2.55
Rescue Therapy
Overall, 10.0% of patients received at least 1 rescue medication, with 3.0% of patients receiving systemic rescue medication. The most commonly used systemic medication type was oral antihistamines (2.0% of patients). A summary of concomitant medication as rescue therapy is presented in Table 24. Rescue therapy use was slightly higher for patients in the placebo-to-nemolizumab group (12.6%) compared with previously treated patients (8.7%). No patient had concomitant rescue therapy during the treatment period.
Table 24: Concomitant Medications Used as Rescue Therapy by 2 or More Patients and 2.0% of Patients in Any Previous Treatment Group (Safety Population) — Interim Analysis 1
Medication | Nemolizumab q.4.w. N = 508 n (%) | By previous treatment n (%) | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 | ||||
All N = 334 | Continuous nemolizumab N = 307 | Re-treatment N = 27 | |||
Any therapy | 51 (10.0) | 29 (8.7) | 28 (9.1) | 1 (3.7) | 22 (12.6) |
Topical | 45 (8.9) | 25 (7.5) | 24 (7.8) | 1 (3.7) | 20 (11.5) |
Corticosteroids | 45 (8.9) | 25 (7.5) | 24 (7.8) | 1 (3.7) | 20 (11.5) |
Clobetasol propionate | 19 (3.7) | 7 (2.1) | 7 (2.3) | 0 | 12 (6.9) |
Mometasone furoate | 10 (2.0) | 7 (2.1) | 7 (2.3) | 0 | 3 (1.7) |
Calcineurin inhibitors | 3 (0.6) | 3 (0.9) | 3 (1.0) | 0 | 0 |
Other | 2 (0.4) | 0 | 0 | 0 | 2 (1.1) |
Systemic | 15 (3.0) | 8 (2.4) | 8 (2.6) | 0 | 7 (4.0) |
Corticosteroids | 2 (0.4) | 1 (0.3) | 1 (0.3) | 0 | 1 (0.6) |
Gabapentinoids | 1 (0.2) | 1 (0.3) | 1 (0.3) | 0 | 0 |
Oral antihistamines | 10 (2.0) | 6 (1.8) | 6 (2.0) | 0 | 4 (2.3) |
Other | 2 (0.4) | 0 | 0 | 0 | 2 (1.1) |
LTE = long-term extension; q.4.w.= every 4 weeks.
Notes: Percentages were based on the number of patients in the population. Medication data were coded using the WHO Drug Dictionary Global, Version B3, March 2022.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with an interval of at least 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE study. At the group, subgroup, and preferred term level, patients were counted once where more than 1 rescue therapy was reported.
Source: LTE Clinical Study Report.54
Efficacy
Efficacy Results From Interim Analyses 1 and 2
IGA Success From LTE Study Baseline
The proportions of patients with IGA success (defined as an IGA of 0 [clear] or 1 [almost clear]) at the LTE study baseline were 40.1% and 12.6% for patients who were continuing treatment with nemolizumab (i.e., the continuous nemolizumab group) and patients who transitioned from placebo to treatment with nemolizumab (the placebo-to-nemolizumab group), respectively. At week 28, the proportions of patients with IGA success were similar across the 2 groups (continuous nemolizumab group: 53.8%; placebo-to-nemolizumab group: 56.3%), and the results remained consistent at week 52 (Table 25). Continuous improvements in skin clearance at week 100 were observed among the 2 groups (continuous nemolizumab group: 73.4%; placebo-to-nemolizumab group: 75.2%) (Table 26). However, data from patients contributing to analysis decreased steadily over time.
Improvement of 4 or More Points From Baseline Lead-In in PP-NRS Score
More than 80% of patients experienced improvements of 4 or more points in PP-NRS score at week 28 in the continuous nemolizumab group (88.8%) and the placebo-to-nemolizumab group (82.4%) (Table 25), with continued improvements at week 52. At week 100, results from patients who continued treatment in the continuous nemolizumab (92.1%) or placebo-to-nemolizumab (94.1%) group indicated consistent improvement in itch relief (Table 26). However, sample sizes steadily declined over time.
Improvement of 4 or More Points From Baseline Lead-In in SD-NRS Score
Following nemolizumab treatment in the LTE, the proportions of patients with an improvement of 4 or more points from lead-in baseline in SD-NRS score generally increased over time in all patients and in each group (Table 25). A consistent improvement of 4 or more points from baseline lead-in was observed across the groups at week 100 (continuous nemolizumab group: 86.4%; placebo-to-nemolizumab group: 87.3%). (Table 26). However, sample sizes declined over time.
Improvement of 4 or More Points From Baseline Lead-In in DLQI Score
At week 28, the proportions of patients achieving an improvement of 4 or more points from baseline lead-in in DLQI score were 87.8% versus 90.5% in the continuous nemolizumab and placebo-to-nemolizumab groups, respectively (Table 25). Continuous improvements were observed in patients in both groups at week 52 (continuous nemolizumab group: 90.1%; placebo-to-nemolizumab group: 91.0%;Table 25) and week 100 (continuous nemolizumab group: 89.9%; placebo-to-nemolizumab group: 93.1%; Table 26).
Table 25: Proportions of Patients With IGA Success and Improvement in PP-NRS, SD-NRS, and DLQI Scores at Week 52 (OLYMPIA LTE Study)
Visit | Nemolizumab q.4.w. N = 508 x of n (%) | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 x of n (%) | ||||
All N = 334 x of n (%) | Continuous nemolizumab N = 307 x of n (%) | Re-treatment N = 27 x of n (%) | |||
IGA success (0 or 1) (%) from LTE baseline (OC) | |||||
Week 16 | 221 of 463 (47.7) | 156 of 304 (51.3) | 145 of 279 (52.0) | 11 of 25 (44.0) | 65 of 159 (40.9) |
Week 28 | 215 of 396 (54.3) | 139 of 261 (53.3) | 128 of 238 (53.8) | 11 of 23 (47.8) | 76 of 135 (56.3) |
Week 52 | 213 of 313 (68.1) | 142 of 203 (70.0) | 126 of 182 (69.2) | 16 of 21 (76.2) | 71 of 110 (64.5) |
PP-NRS score improvement ≥ 4 (%) from lead-in baseline (OC) | |||||
Week 16 | 194 of 242 (80.2) | 126 of 161 (78.3) | 120 of 151 (79.5) | 6 of 10 (60.0) | 68 of 81 (84.0) |
Week 28 | 174 of 204 (85.3) | 118 of 136 (86.8) | 111 of 125 (88.8) | 7 of 11 (63.6) | 56 of 68 (82.4) |
Week 52 | 130 of 152 (85.5) | 85 of 98 (86.7) | 80 of 90 (88.9) | 5 of 8 (62.5) | 45 of 54 (83.3) |
SD-NRS score improvement ≥ 4 (%) from lead-in baseline (OC) | |||||
Week 16 | 181 of 246 (73.6) | 124 of 165 (75.2) | 122 of 154 (79.2) | 2 of 11 (18.2) | 57 of 81 (70.4) |
Week 28 | 151 of 207 (72.9) | 98 of 137 (71.5) | 95 of 126 (75.4) | 3 of 11 (27.3) | 53 of 70 (75.7) |
Week 52 | 120 of 153 (78.4) | 78 of 100 (78.0) | 75 of 92 (81.5) | 3 of 8 (37.5) | 42 of 53 (79.5) |
DLQI score improvement of ≥ 4 (%) from lead-in baseline (OC) | |||||
Week 16 | 402 of 459 (87.6) | 267 of 307 (87.0) | 254 of 292 (87.0) | 13 of 15 (86.7) | 135 of 152 (88.8) |
Week 28 | 333 of 376 (88.6) | 219 of 250 (87.6) | 208 of 237 (87.8) | 11 of 13 (84.6) | 114 of 126 (90.5) |
Week 52 | 264 of 293 (90.1) | 173 of 193 (89.6) | 164 of 182 (90.1) | 9 of 11 (81.8) | 91 of 100 (91.0) |
DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; LTE = long-term extension; OC = observed case; PP-NRS = Peak Pruritus Numerical Rating Scale; q.4.w. = every 4 weeks; SD-NRS = Sleep Disturbance Numerical Rating Scale; x = number of patients with an improvement.
Notes: In OC, all observed data (even after use of rescue therapy) were included; there were no imputations for missing data. Baseline lead-in was defined as the last nonmissing value before the first dose of the study drug in the lead-in study. Baseline LTE was the last nonmissing value before the first dose of study drug in this study. Phase II patients did not contribute to the change from lead-in study baseline, given that there was at least 6 months between the completion of phase II and entry into this LTE.
The group previously treated with nemolizumab (All column)included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with intervals of at least 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE.
Source: OLYMPIA LTE Clinical Study Report.54
Table 26: Proportions of Patients With IGA Success and Improvement in PP-NRS, SD-NRS, and DLQI Scores at Week 100 (OLYMPIA LTE Study)
Visit | Nemolizumab q.4.w. N = 508 x of n (%) | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 x of n (%) | ||||
All N = 334 x of n (%) | Continuous nemolizumab N = 307 x of n (%) | Re-treatment N = 27 x of n (%) | |||
SD-NRS score improvement ≥ 4 | |||||
SD-NRS improvement ≥ 4 (%) from lead-in baseline | 126 of 147 (85.7) | 78 of 92 (84.8) | 76 of 88 (86.4) | 2 of 4 (50.0) | 48 of 55 (87.3) |
IGA success (0 or 1) | |||||
IGA success (0 or 1), % (from LTE study baseline) | 218 of 295 (73.9) | 139 of 190 (73.2) | 130 of 177 (73.4) | 9 of 13 (69.2) | 79 of 105 (75.2) |
PP-NRS score improvement ≥ 4 | |||||
PP-NRS improvement ≥ 4 (from lead-in baseline) | 133 of 144 (92.4) | 85 of 93 (91.4) | 82 of 89 (92.1) | 3 of 4 (75.0) | 48 of 51 (94.1) |
DLQI score improvement of ≥ 4 | |||||
DLQI improvement of ≥ 4 (from lead-in baseline) | 260 of 287 (90.6) | 165 of 185 (89.2) | 160 of 178 (89.9) | 5 of 7 (71.4) | 95 of 102 (93.1) |
DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; LTE = long-term extension; OC = observed case; PP-NRS = Peak Pruritus Numerical Rating Scale; q.4.w. = every 4 weeks; SD-NRS = Sleep Disturbance Numerical Rating Scale; x = number of patients with an improvement.
Notes: In OCs, all observed data (even after use of rescue therapy) were included; there were no imputations for missing data.
Baseline lead-in was defined as the last nonmissing value before the first dose of study drug in the lead-in study.
Baseline LTE was the last nonmissing value before the first dose of study drug in this study.
Phase II patients did not contribute to the change from lead-in study baseline, given that there was at least 6 months between the completion of phase II and entry into this LTE.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with intervals of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with intervals of at least 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE.
Source: LTE Clinical Study Report Interim Analysis 2.55
Harms
Adverse Events
A total of 407 patients (80.1%) experienced at least 1 AE during the overall study period (Table 27). The most frequently reported AEs included infections and infestations (54.3%), skin and SC tissue disorders (36.8%), musculoskeletal and connective tissue disorders (26.0%), respiratory, thoracic, and mediastinal disorders (17.3%), and nervous system disorders (15.4%).
Serious AEs
SAEs were reported in 54 patients (10.6%) during the treatment period. SAEs experienced by at least 1 patient were neurodermatitis (4 patients [0.8%]); myocardial infarction (3 patients [0.6%]); and angina pectoris, cardiac failure congestive, cholelithiasis, pneumonia, osteoarthritis, and carotid artery stenosis (2 patients [0.4%] each).54
Withdrawals Due to AEs
A total of 35 patients (6.9%) had an AE leading to withdrawal from the study.
AEs of Special Interest
AESIs occurred among 319 patients (62.8%). The most commonly reported AESIs were infections (54.5%) and injection-related reactions (31.7%). Newly diagnosed asthma or worsening of asthma was reported in 5.7% of patients.
Deaths
Two patients (0.4%) experienced an AE leading to death. The 2 deaths occurring during the study were due to a myocardial infarction and end-stage renal disease. Both patients had a medical history of comorbidities; neither death was considered related to the study drug.
Refer to Table 27 for harms data.
Table 27: Summary of Harms During the Overall Study Period (LTE, Safety Population) — Interim Analysis 1
Patients with at least 1 AE | Nemolizumab q.4.w. N = 508 n (%) |
|---|---|
Most common AEs | 407 (80.1) |
Gastrointestinal disorders | 79 (15.6) |
Diarrhea | 24 (4.7) |
Nausea | 11 (2.2) |
General disorders and administration-site conditions | 75 (14.8) |
Pyrexia | 14 (2.8) |
Edema, peripheral | 13 (2.6) |
Fatigue | 13 (2.6) |
Infections and infestations | 276 (54.3) |
COVID-19 | 113 (22.2) |
Nasopharyngitis | 70 (13.8) |
Upper respiratory tract infection | 41 (8.1) |
Urinary tract infection | 21 (4.1) |
Bronchitis | 19 (3.7) |
Influenza | 14 (2.8) |
Sinusitis | 13 (2.6) |
Investigations | 62 (12.2) |
Blood creatine phosphokinase increase | 12 (2.4) |
Musculoskeletal and connective tissue disorders | 132 (26.0) |
Arthralgia | 37 (7.3) |
Back pain | 23 (4.5) |
Myalgia | 13 (2.6) |
Pain in extremity | 12 (2.4) |
Osteoarthritis | 11 (2.2) |
Nervous system disorders | 78 (15.4) |
Headache | 33 (6.5) |
Dizziness | 12 (2.4) |
Psychiatric disorders | 31 (6.1) |
Depression | 12 (2.4) |
Respiratory, thoracic, and mediastinal disorders | 88 (17.3) |
Cough | 31 (6.1) |
Asthma | 19 (3.7) |
Dyspnea | 16 (3.1) |
Oropharyngeal pain | 15 (3.0) |
Skin and subcutaneous tissue disorders | 187 (36.8) |
Neurodermatitis | 55 (10.8) |
Eczema nummular | 34 (6.7) |
Eczema | 26 (5.1) |
Dermatitis, atopic | 23 (4.5) |
Urticaria | 12 (2.4) |
Hand dermatitis | 10 (2.0) |
Vascular disorders | 39 (7.7) |
Hypertension | 30 (5.9) |
AEs by maximum severitya | — |
Mild | 175 (34.4) |
Moderate | 192 (37.8) |
Severe | 40 (7.9) |
AEs related to protocol procedure | 19 (3.7) |
SAEs during the treatment period | 54 (10.6) |
AEs leading to study drug interruption | 74 (14.6) |
AEs leading to study drug withdrawal | 35 (6.9) |
AEs leading to study discontinuation | 36 (7.1) |
AESIs by categories (by investigator) | 161 (31.7) |
Injection-related reactions | 1 (0.2) |
Newly diagnosed asthma or worsening of asthma | 23 (4.5) |
Infections | 134 (26.4) |
Peripheral edema: limbs, bilateral; facial edema | 15 (3.0) |
Elevated ALT or AST (> 3 × ULN) in combination with elevated bilirubin (> 2 × ULN) | 0 |
Any AESI categories using SMQs | 319 (62.8) |
Injection-related reactions | 161 (31.7) |
Newly diagnosed asthma or worsening of asthma | 29 (5.7) |
Infections | 277 (54.5) |
Peripheral edema: limbs, bilateral; facial edema | 21 (4.1) |
Elevated ALT or AST (> 3 × ULN) in combination with elevated bilirubin (> 2 × ULN) | 0 |
AEs leading to death | 2 (0.4) |
AE = adverse event; AESI = adverse event of special interest; ALT = alanine aminotransferase; AST = aspartate aminotransferase; LTE = long-term extension; q.4.w.= every 4 weeks; SAE = serious adverse event; SMQ = standardized Medical Dictionary for Regulatory Activities query; ULN = upper limit of normal.
Notes: Percentages were based on the number of patients. AEs were coded using the Medical Dictionary for Regulatory Activities, Version 25.0. The AEs during the overall study period were defined as AEs with onset dates from the first dose date to the follow-up visit date. For each row category, a patient with 2 or more AEs in that category was counted only once.
aIf patients experienced multiple events, the patients were counted once at the event with maximum severity.
Source: LTE Clinical Study Report.54
Critical Appraisal
The OLYMPIA LTE study was a phase III, prospective, single-arm, multicentre, open-label study. The single-arm design of the study limits the ability to draw conclusions regarding the long-term efficacy of nemolizumab. The open-label nature of the study may increase the risk of bias in the evaluation of subjective outcomes. Cumulative exposure to study treatment and treatment adherence were not summarized by group (i.e., continuous nemolizumab, re-treatment with nemolizumab, or placebo to nemolizumab), which further limited our ability to assess potential correlations between treatment exposures and long-term outcomes. Patients received treatment in the LTE study based on their previous enrolment in nemolizumab studies, and the impact of these differing dosage regimens on the efficacy results is unknown.
Data from patients contributing to the analyses decreased steadily over time as patients discontinued treatment (around 30% of patients had discontinued treatment at Interim Analysis 2). Patients who discontinued from the LTE study (17.9% versus 14.9% in the continuous nemolizumab and placebo-to-nemolizumab groups, respectively) could be systematically different from those who remained in the study. Therefore, the trend of apparently stable treatment effect over time could be biased as a result of survival or attrition bias. The sample size in the re-treatment group was small, limiting the ability to draw conclusions regarding efficacy results. Most patients received at least 1 concomitant therapy during the treatment period, and the effect of these on the efficacy outcomes cannot be determined. The harms data aligned with the evidence from the pivotal studies.
Indirect Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
Due to the lack of direct evidence comparing nemolizumab with other existing, relevant therapies for the treatment of patients with PN, the sponsor submitted 1 ITC (an NMA) comparing nemolizumab with dupilumab, vixarelimab, nalbuphine, and placebo. The objective of this section is to summarize and critically appraise the sponsor-submitted NMA comparing nemolizumab with dupilumab for the treatment of patients with PN, given that vixarelimab and nalbuphine are not relevant comparators in practice settings in Canada.
Description of Indirect Comparison
ITC Design
Objectives
The objective of the ITC was to assess the comparative efficacy and safety of nemolizumab compared to dupilumab, vixarelimab, nalbuphine, and placebo for treatment of patients with PN.
Study Selection Methods
A systematic literature review (SLR) was performed to inform the ITC. The literature search was conducted on September 25, 2024, to identify relevant evidence informing the efficacy and safety of nemolizumab for PN. One global search strategy was developed to identify evidence regarding:
clinical efficacy and safety
economic evidence (i.e., economic evaluations, health care resource utilization, and costs)
patient-reported outcomes (i.e., HRQoL and utilities).
Details of the SLR are provided in Table 28. Searches were conducted using a combination of free-text search terms and controlled vocabulary terms specific to each database, as recommended by the Cochrane Collaboration. The literature searches were validated through manual review of the bibliographies of up to 5 of the most comprehensive, recently published, relevant systematic reviews identified during the database searches.
A total of 1,368 records were retrieved from the electronic literature databases; 301 of these were removed during deduplication, and the remaining 1,067 records were screened at the title and abstract level. Of these, 74 records were sought for retrieval and screened at the full-text level. Following the full-text review, 26 records reporting on clinical outcomes from electronic databases were deemed eligible. Twenty-one records were included from grey literature searches. Three reports on file were available from Galderma as well. A total of 50 records reporting on 19 unique clinical trials met the eligibility criteria and were included in the SLR.
Table 28: SLR Study Selection (PICOS), Data Extraction, and Quality Assessment Process
Characteristic | SLR criteria |
|---|---|
Population | Adults (aged ≥ 18 years) with PN |
Intervention |
|
Comparator | Any of the previously noted interventions (or none required) Placebo |
Outcome | Efficacy outcomes of interest:
Safety outcomes of interest:
|
Study designs |
|
Publication characteristics | Included studies were limited to those published in English |
Exclusion criteria | Studies that did not meet the criteria specified in this table were excluded |
Databases searched |
Grey literature: Conference abstracts published in supplements of peer-reviewed journals and indexed in electronic literature databases, such as Embase, were still considered grey literature. Separate searches were conducted through OvidSP for conferences indexed in Embase.com. Online conference websites and other relevant media were searched. In addition, searches of selected HTA websites and clinical trial registries were conducted. Other sources: Tufts Medical Center Cost-Effectiveness Analysis Registry |
Selection process | After searches were conducted, the resulting titles and abstracts were imported to EndNote X21. In the first screening pass, each title and abstract was reviewed by 2 independent reviewers to determine its eligibility for inclusion in the SLRs. Disagreements were resolved by a third reviewer, as necessary. In the second screening pass, each full-text paper was reviewed by 2 independent reviewers. Disagreements were resolved by a third reviewer, as necessary. All identified grey literature was assessed by a single reviewer, with validation of relevant materials performed by a second reviewer. |
Data extraction process | The data were extracted into the data extraction templates by 1 reviewer; as a validation step, a second reviewer assessed the entries to ensure consistency and accuracy against the source article. A third reviewer was consulted to resolve disagreements, as necessary. |
Quality assessment | Quality assessment of the RCTs included in the SLR was conducted using the Cochrane Risk of Bias Assessment Tool 2.0.6 Only studies available in full text underwent a quality assessment, given the lack of details available for assessment from abstracts and posters. |
AE = adverse event; ALT = alanine aminotransferase; AP-NRS = Average Pruritus Numerical Rating Scale; AST = aspartate aminotransferase; DAE = discontinuation due to adverse event; DLQI = Dermatology Life Quality Index; DPS = Dynamic Pruritus Score; HADS = Hospital Anxiety and Depression Scale; HTA = health technology assessment; IGA = Investigator’s Global Assessment; NK1R = neurokinin-1 receptor; PAS = Prurigo Activity Score; PGAD = Patient Global Assessment of Disease; PICOS = population, intervention, comparison, outcome, and study design; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; RCT = randomized controlled trial; SAE = serious adverse event; SLR = systematic literature review; TCI = topical calcineurin inhibitor; TEAE = treatment-emergent adverse event; WASO = wakefulness after sleep onset; WI-NRS = Worst Itch Numerical Rating Scale.
aSearched from 2021 to 2023.
bThe websites of selected HTA bodies that post appraisal reports online were searched to inform the structure and inputs of the cost-effectiveness model and the indirect treatment comparison. These websites were also searched for additional data cuts or outcomes for treatments of interest.
Source: SLR Report.56
Feasibility Assessment: The feasibility assessment was conducted to assess whether the studies proposed for inclusion in the networks could be considered sufficiently comparable with respect to study design, patient demography (e.g., age, race, ethnicity), disease severity, and trial outcome measurements. The sponsor reported that the following sources of heterogeneity were observed and should be acknowledged as limitations:
differences in patient characteristics, such as duration of PN, disease severity, race, previous treatments, and history of failed TCSs
concerns regarding the comparison of nemolizumab and dupilumab (especially the use of TCSs during the trials, given that these were allowed in the dupilumab trial but not in the nemolizumab trials)
varying time points in the conducted studies.
The findings of the feasibility assessment (with conclusion and recommendations) are summarized in Table 29.
Table 29: Overview of the Findings of the Feasibility Assessment
Key parameters assessed | Conclusions |
|---|---|
Network connections | The control groups were broadly similar in most studies and included matching placebo. |
Study design | The studies varied in terms of phase (phase II vs. phase III), sample size (50 to 295 patients), and washout period duration (1 to 4 weeks). These variations highlight the potential for bias and were acknowledged as analysis limitations. Studies utilized a parallel design, with 1 study being a crossover. Severity was not defined in the inclusion criteria of the dupilumab trials. However, it was assumed to be moderate to severe, based on baseline characteristics. |
Patient characteristics |
|
Outcome measurement | The efficacy outcomes of DLQI change from baseline, IGA response, and IGA composite end point were evaluated in similar manners and reported across multiple trials. The trials consistently analyzed IGA success as the proportion of participants with an IGA of 0 or 1 on a 5-point scale ranging from 0 (clear) to 4 (severe). Serious adverse events were reported in most studies; however, the rates were low or 0. Discontinuation due to adverse events was reported only in the nemolizumab trials and in a trial of nalbuphine, with low rates. Total TEAEs were reported in nearly all included trials. Multiple common time points were available for most outcomes, although some were available in figure format only. There is evidence to suggest improvements in efficacy outcomes from 8 to 12 weeks; however, pooling across time points may not be valid. |
Imputation methods and analysis sets | The trials investigating dupilumab and nemolizumab utilized multiple imputation methods, such as nonrespondera imputation, across various outcomes and time points. |
DLQI = Dermatology Life Quality Index; HTA = health technology assessment; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; PN = prurigo nodularis; SLR = systematic literature review; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; vs. = versus.
aWording of the original source.
Source: NMA Report.57
Seven trials were eligible for analysis. The interventions and comparators available for analysis included nemolizumab, dupilumab, vixarelimab, and nalbuphine.
Risk of Bias Assessment
A risk of bias assessment was conducted for the studies included in the NMA. Most of the studies presented adequate randomization methods and outcome assessments and were considered to have a low risk of bias. Three studies presented adequate reporting, but had issues related to internal validity — mainly because of the random sequence generation, promoting discrepancies between comparison groups — or issues related to the selection of the reported result. In general, most of the studies presented a low overall risk of bias, while 3 presented some concerns regarding overall bias. Detailed results of the quality assessment were provided in the sponsor’s Systematic Literature Reviews report.56
ITC Analysis Methods
The planned analyses for the NMAs are summarized in Table 30. A summary of methods used to conduct the NMAs is provided in Table 31.
Table 30: Planned Analysis Scenarios
Outcomes | Trials | Population | Time point (weeks) | Analysis type |
|---|---|---|---|---|
Change from baseline in DLQI | OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | ITT | 4 | Base case |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | ITT | 12 or 16 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | ITT | 24 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | No TCS or TCI use | 24 | Sensitivity 2 | |
IGA (0 or 1) | OLYMPIA 1, PRIME, PRIME 2, and NCT03816891 trials | ITT | 8 | Base case |
OLYMPIA 1, PRIME, PRIME 2, and NCT03816891 trials | Nonrespondera imputation | 8 | Sensitivity 1 | |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | ITT | 12 | Base case | |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | Nonrespondera imputation | 12 | Sensitivity 1 | |
OLYMPIA 1, PRIME, and PRIME 2 trials | ITT | 24 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | No TCS or TCI use | 24 | Sensitivity 2 | |
PP-NRS or WI‑NRS improvement of ≥ 4 from baseline | OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2, NCT03816891 trials | ITT | 8 | Base case |
OLYMPIA 1, OLYMPIA 2, PRIME, PRIME 2, and NCT03816891 trials | Nonrespondera imputation | 8 | Sensitivity 1 | |
OLYMPIA 1, OLYMPIA 2, and PRIME 2 trials | ITT | 12 | Base case | |
OLYMPIA 1, OLYMPIA 2, and PRIME 1 trials | Nonrespondera imputation | 12 | Sensitivity 1 | |
OLYMPIA 1, OLYMPIA 2, and PRIME2 trials | No TCS or TCI use | 12 | Sensitivity 2 | |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | ITT | 16 | Base case | |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | Nonrespondera imputation | 16 | Sensitivity 1 | |
OLYMPIA 1, PRIME, and PRIME 2 trials | ITT | 24 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | No TCS or TCI use | 24 | Sensitivity 2 | |
PP-NRS or WI‑NRS (≥ 4‑point improvement) and IGA (0 or 1) composite | OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | ITT | 12 or 16b | Base case |
OLYMPIA 1, OLYMPIA 2, PRIME, and PRIME 2 trials | Nonrespondera imputation | 12 or 16b | Sensitivity 1 | |
OLYMPIA 1, PRIME, and PRIME 2 trials | ITT | 24 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | No TCS or TCI use | 24 | Sensitivity 2 | |
Change from baseline in PP-NRS or WI-NRS (absolute) | OLYMPIA 1, OLYMPIA 2, NCT03181503, NCT02174419, NCT03816891,b PRIME,b and PRIME 2b trials | ITT | 8 | Base case |
OLYMPIA 1, OLYMPIA 2, NCT03181503, NCT02174419, PRIME1, and PRIME2 trials | ITT | 10 or 12 | Base case | |
OLYMPIA 1, OLYMPIA 2, NCT03181503, PRIME,b and PRIME 2b trials | ITT | 16 | Base case | |
OLYMPIA 1, PRIME, and PRIME 2 trials | ITT | 24 | Base case | |
TEAE | OLYMPIA 1, PRIME, PRIME 2, NCT02174419, OLYMPIA 2, and NCT03816891 trials | ITT | Study End | Base case |
DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITT = intention to treat; NMA = network meta-analysis; PP-NRS = Peak Pruritus Numerical Rating Scale; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; WI-NRS = Worst Itch Numerical Rating Scale.
aWording of the original source.
bAbsolute change from baseline to be calculated using baseline mean WI-NRS and percent change from baseline at specific time points.
Source: NMA Report.57
Table 31: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian analyses were carried out to perform Markov Chain Monte Carlo simulations in which the first 50,000 simulations (run-in or burn-in samples) were discarded and the succeeding 50,000 simulations (posterior samples) were used for parameter estimation. Convergence was confirmed by evaluating the 3-chain Brooks-Gelman-Rubin plots and inspecting the ratios of Monte Carlo error to the SDs of the posteriors. If convergence was not achieved, then the run-in was increased. The median and the 2.5th and 97.5th percentiles of the posterior samples for each effect were used as the estimate of the effect and the 95% CrI. These posterior samples were also used to obtain the ranked probability that a treatment was the best treatment, the probability that nemolizumab was better than each comparator, and each treatment’s surface under the cumulative ranking curve index. The bayesian NMAs were conducted in OpenBUGS version 3.2.3. |
Priors | In all bayesian NMAs, a noninformative prior of N(0, 10,000) was used for the baseline effects. The random-effects NMAs used a vaguely informative prior for the between-study SD, tau, with a uniform SD, the distributions of which will be U[0,1] for binary outcomes and U[0,X] for continuous outcomes, where X is the median SD of the outcome within the study. The choice of priors was somewhat arbitrary by nature; in these cases, it was chosen to allow for a moderate to large amount of random-effects variation, but also to discourage the estimation of extreme values of random-effects variation. Sensitivity to priors was investigated as necessary. For the proportion of patients achieving an IGA of 0 or 1, the proportion of patients with a PP-NRS or WI-NRS improvement of ≥ 4 points, and the proportion of patients with a composite of PP-NRS or WI-NRS and IGA response, the LN(−2.54, 1.542) prior derived for dichotomized quality of life and functioning outcomes was used. For TEAE, the LN(−1.87, 1.522) prior derived for AEs was used. |
Assessment of model fit | FE or RE models (or both, if appropriate) were used for the analyses. As noted, the model fit was explored by comparing the DIC and the posterior mean of the residual deviance for the FE and RE models. These models were compared using a DIC to determine the best-fitting model. When a DIC is lower by more than 5 points, it is generally considered a sign of a substantially better-fitting model. However, as noted, the RE model was considered the default in the networks, with at least 2 instances of comparisons with 2 or more studies, given that the FE assumption of homogeneity was unrealistic. When an RE model was appropriate, results using the prior with the lowest DIC were chosen for presentation. |
Assessment of transitivity | The clinical and methodological characteristics of the included studies were carefully examined to determine the extent of heterogeneity across the network during the feasibility assessment. |
Assessment of consistency | Given that all trials were placebo-controlled, inconsistency could not be investigated. |
Assessment of convergence | Convergence was first checked by inspecting the ratios of Monte Carlo error to the SDs of the posteriors; values > 5% were strong signs of convergence issues. Where necessary, convergence was confirmed through the use of 3-chain Brooks-Gelman-Rubin plots; however, it is rare to observe convergence issues in simple star-network evidence networks. When convergence was not achieved, the run- in was increased and/or other factors were examined (such as the choice of prior and starting values). |
Outcomes | For all included trials, if data were available for more than 1 analysis population, the ITT population was prioritized for the analysis. For safety outcomes, data from the safety populations (i.e., including all randomized patients who received at least 1 dose of the study drug) were prioritized if multiple denominators were reported. Trial publications reporting ITT denominators for safety outcomes were considered if the safety population was not provided. For an outcome, if the SD for an arm was not reported at a specific time point, it was calculated from the arm-level data of SE or from the CI, where available. If that was not possible, then SD was computed from the SE, the CI, or the exact P value of the contrast-level data of mean difference, where possible. For the following end points, the output statistic was the odds ratio with an associated 95% CrI:
For the following end points, the output statistic was the mean difference in change from baseline with an associated 95% CrI:
|
Sensitivity analyses | Because of the allowance of topical therapies in the dupilumab trials (i.e., the PRIME and PRIME 2 trials), the analysis approach for this comparison was unique. Two sensitivity analyses were conducted:
|
Subgroup analysis | None |
AE = adverse event; CI = confidence interval; Crl = credible interval; DIC = deviance information criterion; DLQI = Dermatology Life Quality Index; FE = fixed effect; IGA = Investigator’s Global Assessment; ITC = indirect treatment comparison; ITT = intention to treat; LN = natural logarithm; NMA = network meta-analysis; PP-NRS = Peak Pruritus Numerical Rating Scale; RE = random effect; SD = standard deviation; SE = standard error; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; U = uniform; WI-NRS = Worst Itch Numerical Rating Scale.
Note: Details on the assessment of homogeneity in the ITC are presented in Table 32.
aWording of the original source.
Source: NMA Report.57
Table 32: Assessment of Homogeneity in the ITC
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity | The percentage of patients with PN who had a history of atopy ranged from 0% to 69.2%. Additionally, the mean duration of PN ranged from 5.4 years to 17.2 years. The majority of patients (58% to 65.8%) had 20 to 100 nodules. Across the identified studies reporting baseline IGA for PN stage score, most participants had a score of 3 (32.1% to 72%) or 4 (15.7% to 61.1%). The mean WI-NRS score ranged from 7.96 to 9.2. Additionally, the mean DLQI score ranged from 8.9 to 18.2. Among the studies that reported prior treatment, the most common systemic treatments were antihistamines. These were followed by corticosteroids and immunosuppressants. |
Treatment history |
|
Trial eligibility criteria | Inclusion and exclusion criteria varied across the trials, including the duration of PN and the number of nodules; 2 trials of dupilumab5 specifically required patients to be refractory to treatment at entry. |
Dosing of comparators | Control groups were broadly similar in most studies and included matching placebo. However, in the dupilumab trials control group, TCSs and/or TCIs were allowed. |
Placebo response | The sponsor did not provide the information in the NMA section. |
Definitions of end points |
|
Timing of end point evaluation | Varied from week 4 to week 24 across all outcomes. |
Withdrawal frequency | Due to lack of information, this end point was not feasible to assess in the NMAs. |
Clinical trial setting | There was substantial heterogeneity observed among the studies in terms of the populations included, sample sizes, study duration, treatment duration, timing of outcome measurements, and method of outcome measurements. This heterogeneity, along with inconsistent end point definitions, made it difficult to compare the results across trials. Despite these limitations, some important trends were observed. |
Study design |
|
DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITC = indirect treatment comparison; NMA = network meta-analysis; PN = prurigo nodularis; PP-NRS = Peak Pruritus Numerical Rating Scale; SLR = systematic literature review; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; vs. = versus; WI-NRS = Worst Itch Numerical Rating Scale.
Source: SLR Report.56
Results of the ITC
Summary of Included Studies
Seven trials were eligible for analysis in the NMA. The interventions and comparators available for analysis included nemolizumab, dupilumab, vixarelimab, and nalbuphine.
Evidence Networks
No network structures included loops; therefore, it was not possible to investigate inconsistency.
Results
For the purpose of this review, only NMA results of nemolizumab versus dupilumab are presented here, given that vixarelimab and nalbuphine are not considered relevant comparators in practice settings in Canada.
Efficacy
The overall primary analysis ITC results are presented in Table 33. The ITC sensitivity analysis results for nemolizumab versus relevant comparators based on no TCSs and/or TCIs are presented in Table 34.
PP-NRS Score Change from Baseline
Between-group differences in PP-NRS score change from baseline were assessed at 8 weeks, 10 or 12 weeks, 16 weeks, and 24 weeks. Based on the fixed-effects model, the mean between-group difference in change from baseline ranged from █████ ██ █████, with 95% credible intervals (CrIs) excluding the null value and ranging from █████ ██ █████. Based on the random-effects model, the mean between-group difference ranged from █████ ██ █████, with all 95% CrIs including the null value and ranging from █████ ██ ████.
PP-NRS Response (PP-NRS Score Improvement of ≥ 4 Points)
The odds ratios for PP-NRS response were assessed at 8, 12, 16, and 24 weeks. Based on the fixed-effects model, at weeks 8 and 12, the odds ratios (95% CrI) were ████ █████ ██ █████ and ████ █████ ██ █████ respectively. At weeks 16 and 24, the odds ratios were ████ ███ ████ respectively, with wide 95% CrIs including the null value (Table 33). Based on the random-effects model, at week 8, the odds ratio (95% CrI) was ████ █████ ██ █████. At weeks 12, 16, and 24, the odds ratios ranged from ████ ██ ████, with all wide 95% CrIs including the null value (Table 33).
Figure 3: Overall Network Diagram
DUP = dupilumab; NAL = nalbuphine; NEM = nemolizumab; PBO = placebo; VIX = vixarelimab.
Note: A separate geometry of evidence network was not provided for each outcome and time point assessed.
Source: ITC Report.57
Table 33: ITC Results of Nemolizumab vs. Dupilumab (Primary Analysis) [Redacted]
Outcomes | Nemolizumab vs. dupilumab | |
|---|---|---|
Random-effects model | Fixed-effects model | |
PP-NRS score change from baseline | ||
PP-NRS score change from baseline at week 8 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
PP-NRS score change from baseline at week 10 or 12 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
PP-NRS score change from baseline at week 16 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
PP-NRS score change from baseline at week 24 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
PP-NRS response | ||
PP-NRS score response at week 8 (OR, 95% CrI)b | █████ ██ | █████ ██ |
PP-NRS score response at week 12 (OR, 95% CrI)b | █████ ██ | █████ ██ |
PP-NRS score response at week 16 (OR, 95% CrI)b | █████ ██ | █████ ██ |
PP-NRS score response at week 24 (OR, 95% CrI)b | █████ ██ | █████ ██ |
IGA success | ||
IGA success at 8 weeks (OR, 95% CrI)b | █████ ██ | █████ ██ |
IGA success at 12 weeks (OR, 95% CrI)b | █████ ██ | █████ ██ |
IGA success at 24 weeks (OR, 95% CrI)b | █████ ██ | █████ ██ |
Composite PP-NRS and IGA responders | ||
Composite PP-NRS and IGA response at week 12 or 16 (OR, 95% CrI)b | █████ ██ | █████ ██ |
Composite PP-NRS and IGA response at week 24 (OR, 95% CrI)b | █████ ██ | █████ ██ |
DLQI change from baseline | ||
DLQI score change from baseline at week 4 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
DLQI score change from baseline at week 12 or 16 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
DLQI score change from baseline at week 24 (mean difference, 95% CrI)a | █████ ██ | █████ ██ |
AEs | ||
AEs at study end (OR, 95% CrI)c | █████ ██ | █████ ██ |
AE = adverse event; Crl = credible interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; OR = odds ratio; PP-NRS = Peak Pruritus Numerical Rating Scale; vs. = versus.
Note: Nalbuphine and vixarelimab are not considered relevant comparators in clinical practice in Canada and are not used in the economic model for this review. Therefore, in this section, only nemolizumab versus dupilumab is reported.
aFor continuous efficacy outcomes (i.e., PP-NRS change from baseline and DLQI), results are expressed as the mean between-group difference in change from baseline (nemolizumab vs. comparator). A mean difference of less than 1 indicates that the result favours nemolizumab; a mean difference greater than 1 indicates that the result favours the comparator.
bFor binary efficacy outcomes (PP-NRS response, IGA success, and composite PP-NRS and IGA response) (nemolizumab vs. comparator): an OR greater than 1 indicates that the result favours nemolizumab; an OR of less than 1 indicates that the result favours the comparator.
cFor harms outcomes (nemolizumab vs. comparator), an OR of less than 1 indicates that the result favours nemolizumab; an OR greater than 1 indicates the result favours the comparator.
Source: NMA Report.57
PP-NRS Response Sensitivity Analysis: No TCSs or TCIs
The odds ratios for PP-NRS response were assessed at 12 and 24 weeks. Based on the fixed-effects model, the odds ratio (95% Crl) was ████ █████ ██ ██████ at week 12, with the 95% CrI excluding the null value, and ████ █████ ██ █████ at week 24, with the 95% CrI including the null value (Table 34). Based on the random-effects model, the odds ratio (95% Crl) was ████ █████ ██ ██████ at week 12. The odds ratio (95% CrI) was ████ █████ ██ █████ at week 24, with a wide 95% CrI including the null value (Table 34).
Table 34: ITC Sensitivity Analysis Results of Nemolizumab vs. Dupilumab Based on No TCSs or TCIs [Redacted]
Outcomes | Nemolizumab vs. dupilumab | |
|---|---|---|
Random-effects model | Fixed-effects model | |
PP-NRS score response at week 12 (OR, 95% CrI)a | █████ ██ | █████ ██ |
PP-NRS score response at week 24 (OR, 95% CrI)a | █████ ██ | █████ ██ |
IGA success at 24 weeks (OR, 95% CrI)a | █████ ██ | █████ ██ |
Composite IGA and PP-NRS response: 24 weeks (no TCS or TCI use) (OR, 95% CrI)a | █████ ██ | █████ ██ |
DLQI score at week 24 (mean difference of change from baseline, 95% CrI) | █████ ██ | █████ ██ |
Crl = credible interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; ITC = indirect treatment comparison; OR = odds ratio; PBO = placebo; PP-NRS = Peak Pruritus Numerical Rating Scale; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vs. = versus.
Note: Nalbuphine and vixarelimab are not considered relevant comparators in clinical practice in Canada and are not used in the economic model for this review. Therefore, in this section, only nemolizumab versus dupilumab is reported.
aFor binary efficacy outcomes (i.e., PP-NRS response, IGA success, and composite PP-NRS and IGA response) (nemolizumab vs. comparator), an OR greater than 1 indicates that the result favours nemolizumab; an OR of less than 1 indicates that the result favours the comparator.
Source: ITC Report.57
IGA Success
The odds ratios for IGA success were assessed at 8, 12, and 24 weeks. Based on the fixed-effects model, the odds ratios ranged from ████ ██ ████, all with wide 95% CrIs including the null value (Table 33). Based on the random-effects model, the odds ratios ranged from ████ ██ ████ with wide 95% CrIs including the null value (Table 33).
IGA Success Sensitivity Analysis: No TCSs or TCIs
The IGA success sensitivity analysis based on the population of patients receiving no TCSs or TCIs was performed for week 24 only using a fixed-effects model. The odds ratio (95%Crl) was ████ █████ ██ █████ with a wide 95% CrI including the null value (Table 34).
Composite PP-NRS and IGA Response
The composite of IGA success (i.e., score of 0 or 1) and PP-NRS response (i.e., score of 4 or greater) was reported at weeks 12 and 16 (i.e., nemolizumab at week 16 versus dupilumab at week 12) and at 24 weeks for both nemolizumab and dupilumab. Based on the fixed-effects model, when comparing nemolizumab at 16 weeks with dupilumab at 12 weeks, the odds ratio (95% Crl) was ████ █████ ██ ██████ with a 95% CrI excluding the null value (Table 33). At week 24, the odds ratio (95%Crl) was ████ █████ ██ █████, with a wide 95% CrI including the null value (Table 33). Based on the random-effects model, the odds ratios ranged from ████ at weeks 12 and 16 to ████ at week 24, with wide 95% CrIs including the null value (Table 33).
Composite PP-NRS Response and IGA Sensitivity Analysis: No TCSs or TCIs
Based on the fixed-effects model, the odds ratio (95% Crl) was ████ █████ ██ ██████, with a wide 95% CrI including the null value. Based on the random-effects model, the odds ratio (95% Crl) was ███ █████ ██ ██████ with a wide 95% CrI including the null value (Table 34).
Composite PP-NRS Response and IGA Sensitivity Analysis: Nonresponder Imputation at 12 and 16 Weeks
A composite PP-NRS response and IGA sensitivity analysis, based on nonresponder (wording of original source) imputation, was conducted to compare nemolizumab at 16 weeks with dupilumab at 12 weeks. The odds ratio (95% CrI) was ████ ██████ █████.
DLQI
The between-group difference in change from baseline in DLQI was assessed at 4 weeks, 12 or 16 weeks, and 24 weeks. Based on the fixed-effects model, at 4 weeks, the mean between-group difference in change from the baseline (95% Crl) was █████ ██████ ██ █ █████ with the 95% CrI excluding the null value. At week 24, the mean between-group difference in change from baseline (95% CrI) was █████ ██████ ██ █████, with a wide 95% CrI including the null value. Based on the random-effects model, similar results were observed, with wider 95% CrIs including the null value (Table 33).
DLQI Sensitivity Analysis: No TCSs or TCIs
DLQI sensitivity analysis based on the population of patients receiving no TCSs or TCIs was conducted for week 24 only using a fixed-effects model. The mean difference (95% CrI) was █████ ██████ ██ █████ with a wide 95% CrI including the null value (Table 34).
Harms
Based on both the fixed-effects and random-effects models, the odds ratio for AEs assessed at the end of the study was ████ with wide 95% CrIs including the null value (Table 33).
Critical Appraisal of the NMA
The NMA was conducted using bayesian analyses. Relevant studies for the NMA were identified through an SLR. Risk of bias assessments for each included study were performed using the Cochrane Risk of Bias Assessment Tool 2.0. Most of the studies presented a low overall risk of bias; 3 presented some concerns regarding overall bias, including issues related to internal validity, mainly because of the random sequence generation, promoting discrepancies between comparison groups. A feasibility assessment was performed to identify RCTs, minimize intertrial heterogeneity, and increase the validity of the results. A prespecified analysis plan for conducting NMAs was used to guide the analyses. The overall geometry of the evidence networks was described. Key sensitivity analyses (for example, based on patient populations with no TCS or TCI use) were conducted to further validate the findings from the primary results.
The potential key limitations of the NMA were as follows. First, there was considerable heterogeneity across the included studies in terms of study and patient characteristics (e.g., eligibility criteria, population, trial duration). For example, in the dupilumab trials, patients were required to have a history of a medium-to-superpotent TCS not being medically advisable for them or failing for them. Failure of a TCS was defined as patients being unable to achieve and/or maintain remission and low disease activity (similar to having an IGA PN stage score of ≤ 2 [≤ 19 nodules]), despite treatment with a daily regimen of a medium-to-superpotent TCS (± a TCI, as appropriate) for at least 14 days (or for the maximum duration recommended by the product prescribing information, whichever is shorter). However, in the nemolizumab trials, such a history was not required. Therefore, the NMA results could be biased in favour of nemolizumab because patients with a history of treatment-refractoriness (in the dupilumab trial) may demonstrate a poorer response during the trial versus patients without a prior treatment-refractory history. Second, use of concomitant TCSs or TCIs was allowed in the dupilumab trial (i.e., 56% to 61% of patients used these during the study), but these were not allowed in the nemolizumab trial. The difference in the use of TCIs or TCSs in the 2 trials is a confounding factor when determining comparative efficacy and safety, although the direction and magnitude of the bias remain unknown. However, a sensitivity analysis based on the subpopulation of patients with no TCS or TCI use showed results that were largely consistent with the primary analysis. Third, for composite PP-NRS and IGA response at the time point of 12 or 16 weeks (in both base-case analysis and sensitivity analysis based on nonresponder [wording of original source] imputation), data from the nemolizumab trial at week 16 and the dupilumab trial at week 12 were used for the NMA analysis. Therefore, the NMA findings for this outcome at these time points may be biased in favour of nemolizumab because a longer treatment time may have led to a higher response rate in the nemolizumab trial. However, the composite PP-NRS and IGA response outcome at week 24 (base-case analysis) was reported with a wide 95% CrI including the null value. For this outcome, the composite PP-NRS and IGA response outcome at week 24 was considered a more appropriate time point. No meta-regression analyses to adjust for the aforementioned factors that may bias comparisons were conducted. The previously noted significant differences in patient characteristics across the included studies may threaten the transitivity assumption for the NMA analysis. The assumption of similarity across the included studies may not hold for the NMA, increasing the likelihood of uncertainty about the validity of the results on the comparative effectiveness of nemolizumab and dupilumab.
In addition, a fixed-effects model was used in the base-case analysis. It should be noted that this model may underestimate the between-study heterogeneity, leading to overly precise estimates, while the random-effects models may overfit the data.
There were no closed loops between nodes; this inherently limits the ability to examine inconsistency. Due to the sparsity of the network, the sample data were insufficient to inform the between-study SD in treatment effects. Furthermore, whether the quality assessment of included studies was done by 2 reviews was not described in the ITC report.
Summary
Overall, the sponsor-submitted NMA indicated that the evidence may not be sufficient to show that nemolizumab is favourable compared with dupilumab regarding PP-NRS change from baseline, PP-NRS response, IGA success, composite PP-NRS and IGA response, and DLQI. The ITC results showed a similar overall safety profile; however, it is unknown whether there were differences in terms of individual AEs, especially AESIs and SAEs. Due to the significant heterogeneity between the trials (such as prior treatment, failure of prior treatment, and timing of the outcomes assessment), the evidence regarding the comparative efficacy and safety of nemolizumab versus dupilumab has considerable limitations that preclude the ability to draw a firm conclusion.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
The OLYMPIA 1 (N = 286) and OLYMPIA 2 (N = 274) trials were both phase III, randomized, double-blind, placebo-controlled studies investigating the efficacy and safety of nemolizumab versus placebo for the treatment of adult patients with PN. The studies were conducted in a total of 16 countries (77 sites for the OLYMPIA 1 trial, 55 sites for the OLYMPIA 2 trial), including 8 sites in Canada that randomized a total of 44 patients. In both trials, patients were randomized 2 to 1 to receive nemolizumab or matched placebo by SC injection every 4 weeks. The treatment periods were 24 weeks and 16 weeks in the OLYMPIA 1 and OLYMPIA 2 trials, respectively, and follow-up was 8 weeks in each. The primary end points in the OLYMPIA 1 and OLYMPIA 2 trials were the proportions of patients with at least a 4-point improvement from baseline in PP-NRS score at week 16 and the proportions of patients reporting IGA success, defined as an IGA response of 0 (clear) or 1 (almost clear) and at least a 2-point reduction from baseline, at week 16. Key secondary and other secondary end points included measures of improvement in sleep (SD-NRS), itch reduction (PP-NRS), lesion clearance (IGA success), and HRQoL (assessed using the DLQI). Harms outcomes were also assessed.
Patients eligible for participation in both OLYMPIA pivotal trials were aged 18 years or older with a clinical diagnosis of PN for at least 6 months with pruriginous nodular lesions on the upper limbs, lower limbs, and/or trunk; at least 20 nodules on the entire body, with bilateral distribution; and an IGA score of at least 3 (indicating moderate or severe global severity). Eligible patients were also required to have severe pruritus, as defined by a PP-NRS score of at least 7.
In the OLYMPIA 1 and OLYMPIA 2 trials, the majority of patients were female (58.0% and 61.3%, respectively), white (84.3% and 78.5%, respectively), and aged 18 to 65 years (71.3% and 78.1%, respectively), with the mean ages being 57.5 and 52.7 years, respectively. In the OLYMPIA 1 trial, 59.1% of patients had moderate disease (i.e., IGA category 3), and 40.9% of patients had severe disease (i.e., IGA category 4) at baseline. In the OLYMPIA 2 trial, 56.9% and 43.1% of patients had moderate and severe disease at baseline, respectively. In both trials, most patients did not have a background of atopy (OLYMPIA 1 trial: 67.5%; OLYMPIA 2 trial: 67.9%), and the mean times since PN diagnosis were 91.51 and 105.64 months, respectively. Patients were not eligible for participation in the trials if they had uncontrolled or exacerbated asthma, COPD, chronic bronchitis, certain infections, or active AD within the previous 3 months.
The OLYMPIA LTE study is a phase III, multicentre, open-label study designed to evaluate the safety and efficacy of nemolizumab in patients with PN who were enrolled in prior nemolizumab PN studies. A total of 508 patients enrolled in the LTE study. The majority (96%) were from the pivotal trials (i.e., the OLYMPIA 1 and OLYMPIA 2 trials), with the remaining patients coming from a phase IIa study and a nemolizumab durability study. Patients included in the LTE study may have been treated with nemolizumab or placebo in 1 of the lead-in studies. Most patients were female (60.40%). Patients had a mean age of 55.40 years, were primarily white (87.20%), and had a mean body weight of 82.44 kg. The proportion of patients with moderate and severe IGA was higher in the placebo-to-nemolizumab group compared with the continuous nemolizumab group. At the lead-in study baseline, the weekly average PP-NRS score was 8.49; the weekly average SD-NRS score was 7.09; and the mean DLQI total score was 16.90. Most patients had no atopy background (continuous nemolizumab group: 64.20%; placebo-to-nemolizumab group: 64.90%). The average time since PN diagnosis was 101.63 months.
In the absence of direct evidence from randomized trials between nemolizumab and existing relevant comparators, the sponsor submitted an NMA to assess the comparative efficacy and safety of nemolizumab versus dupilumab in patients with PN.
Interpretation of Results
Efficacy
The efficacy outcomes of greatest importance in the treatment of PN, as identified by clinical experts and clinician and patient groups, are reduction in the severity of pruritus, clearance of lesions, and improvements in sleep and HRQoL. As such, the main efficacy outcomes assessed in the OLYMPIA pivotal trials align with the key outcomes of importance to patients and clinicians.
Clinical experts consulted by CDA-AMC identified the intensity of pruritus as the symptom of PN that has the greatest impact on patients. In both of the OLYMPIA pivotal trials, nemolizumab demonstrated a statistically significant improvement versus placebo in the proportion of patients who had at least a 4-point improvement in PP-NRS at 16 weeks. The 4-point improvement threshold has been supported by clinical experts as clinically meaningful, and the literature has referenced a 2-point to 5-point decrease in PP-NRS score as a meaningful within-patient change. Clinical experts acknowledged that the differences in proportion between groups were clinically meaningful. The GRADE assessment suggested high certainty in the results for both trials.
In the OLYMPIA 1 and OLYMPIA 2 trials, nemolizumab demonstrated a statistically significant improvement versus placebo in the proportion of patients who had IGA success (defined as an IGA of 0 [clear] or 1 [almost clear] and at least a 2-point improvement from baseline) at 16 weeks. Clinical experts acknowledged that the differences in proportions between groups were clinically meaningful, and the GRADE assessment suggested high certainty in the results for both trials. Clinical experts considered an IGA score of 0 or 1 to be a clinically meaningful response in assessing disease severity (i.e., lesion count).
Clinical experts acknowledged that 16 weeks is enough time to assess the trials’ primary end points (i.e., itch reduction and clearing of lesions). However, they noted that, based on clinical practice, 24 weeks may be a preferable time point for assessment.
Input from interested parties identified improvement in sleep as an outcome of importance. In both OLYMPIA pivotal trials, nemolizumab demonstrated a statistically significant improvement versus placebo in the proportion of patients who had at least a 4-point improvement in SD-NRS at 16 weeks (a key secondary end point). The use of a 4-point threshold to determine clinically meaningful improvement in sleep is supported by clinical expert input, and the literature has referenced a 2-point to 4-point decrease on the SD-NRS scale as a meaningful within-patient change. Input regarding the clinical meaningfulness of the differences in proportions was not identified. The clinical experts consulted by CDA-AMC noted that many factors can contribute to sleep disturbance.
For the secondary end points, change from baseline in PP-NRS at week 16 (refer to Appendix 1) in the OLYMPIA 1 and OLYMPIA 2 trials demonstrated that patients treated with nemolizumab had an LS mean reduction in PP-NRS score of at least 4 points, with the between-group difference being approximately 3 points in both trials. In the OLYMPIA 1 trial, the difference in proportion between groups for patients with at least a 4-point improvement in PP-NRS score at 24 weeks was 30.7% (compared to 40.1% at week 16). In the OLYMPIA 1 and OLYMPIA 2 trials, the proportions of patients with at least a 4-point improvement in PP‑NRS score at 4 weeks were evaluated as key secondary end points. In both trials, the proportions of patients in the nemolizumab group were statistically significantly greater than in the placebo group, suggesting that the effects of nemolizumab on improvement in itch were demonstrable after 4 weeks of treatment. Clinical experts consulted by CDA-AMC commented that a medication’s ability to relieve itch quickly would be an important factor to consider; however, the durability of itch relief, lesion clearance, and long-term safety were even more important considerations in treating patients with PN.
In the OLYMPIA 1 trial, results for the secondary end point of IGA success at 24 weeks showed maintenance of the effect observed at week 16, with a greater proportion of patients achieving IGA success in the nemolizumab group than in the placebo group.
Regarding the secondary composite end point of patients who experienced at least a 4-point improvement in PP-NRS score and IGA success, this was reported for a higher proportion of patients who received nemolizumab versus placebo at week 16 in both trials; in the OLYMPIA 1 trial, maintenance of the effect from week 16 was demonstrated at week 24.
In both OLYMPIA pivotal trials, results for the secondary end point of change from baseline in SD-NRS score at week 16 (refer to Appendix 1) demonstrated that patients treated with nemolizumab had an LS mean reduction in SD-NRS score of approximately 4 points, whereas patients who received placebo had an LS mean reduction of approximately 1 point. The between-group difference approached 3 points in both trials.
In terms of HRQoL, the evaluation of DLQI secondary end points demonstrated that a higher proportion of patients in the nemolizumab group had at least a 4-point improvement in DLQI at 16 weeks (in the OLYMPIA 1 and 2 trials) and at 24 weeks (in the OLYMPIA 1 trial) compared to the placebo group. These analyses were not controlled for multiplicity; therefore, conclusions cannot be drawn regarding statistical significance (although all strata-adjusted nominal P values were < 0.0001). Clinical experts acknowledged that the proportion differences between groups were clinically meaningful, and the GRADE assessment suggested moderate certainty in the results at week 24 in the OLYMPIA 1 trial and week 16 in the OLYMPIA 2 trial. Clinical experts considered a 4-point threshold (i.e., reduction) in total DLQI score appropriate to determine a clinically important improvement in HRQoL. The clinically meaningful references in the literature are a 9-point change in DLQI score for within-patient improvement and a 4-point change for between-group improvement. Change from baseline in DLQI at week 16 demonstrated that patients treated with nemolizumab had a greater LS mean reduction in DLQI total score compared to patients who received placebo, with between-group differences of approximately 6 points in the OLYMPIA 1 trial and 8 points in the OLYMPIA 2 trial. In the OLYMPIA 1 trial, results at 24 weeks showed a similar effect, with a between-group difference of approximately 8 points.
An interpretation of the efficacy results should consider that, due to differences in treatment response between arms (e.g., reduction in pruritus), patients and investigators could have become aware of treatment allocations; this could have introduced bias in favour of nemolizumab for subjective end points such as PP-NRS and DLQI. Furthermore, because a higher proportion of patients in the placebo arm required the use of rescue therapy, it should be considered that differential exposure to concomitant treatments may have potentially diluted the itch response benefit of nemolizumab compared to placebo; the magnitude of any such effect is unknown. According to clinical expert input, the differences between rescue therapy use between groups would not be expected to affect the interpretation of the efficacy results. Clinical experts noted that assessments of treatment response in clinical practice do not usually use the grading tools used in clinical trials as stringently, and that meaningful response is patient-specific.
An important limitation of the OLYMPIA 1 and OLYMPIA 2 trials is that nemolizumab was compared to placebo, which does not represent the standard of care for treatment of PN. Currently, dupilumab is approved by Health Canada for the treatment of patients with moderate to severe PN.23 Clinical experts stated that dupilumab would be the most relevant comparator to nemolizumab for the indication under review. However, dupilumab was not indicated for treatment of PN at the time of the OLYMPIA 1 and OLYMPIA 2 trials.
Various factors were identified that may affect the generalizability of the OLYMPIA pivotal trial results. The OLYMPIA 1 and OLYMPIA 2 trials excluded certain patients who would be considered for treatment with nemolizumab in clinical practice. Clinical experts consulted by CDA-AMC highlighted that PN generally affects older people with comorbidities. They expected that patients with certain comorbidities, such as eczema, would be treated with nemolizumab in clinical practice and noted that patients with chronic infections, such as hepatitis or HIV, may require the use of nemolizumab in future practice because they may be more prone to having PN. One clinical expert also commented that, in practice, it may be difficult to screen patients for severe asthma and exclude them from treatment. Clinical experts identified that the use of nemolizumab in patients with conditions such as severe asthma, HIV, hepatitis, and renal or liver failure requires further study. Regarding the characteristics of the patients randomized in the OLYMPIA pivotal trials, clinical experts stated that these were a reasonable reflection of the patients who would receive nemolizumab for moderate to severe PN in Canada and of the patients with PN whom clinical experts treat in their practices. Most patients in the OLYMPIA pivotal trials were white, which presents a potential generalizability limitation: the trial populations do not adequately represent the racial diversity of patients with PN.
The use of topical and systemic medications and procedures was prohibited during the OLYMPIA pivotal trials (except for rescue therapies). Clinical experts noted that it is likely that patients who would be prescribed nemolizumab would also require TCSs, intralesional corticosteroids, or other concomitant therapies. Therefore, the use of nemolizumab as monotherapy in the trials may not reflect mainstream clinical practice (although there could be instances where that may be the case, according to clinical experts).
Clinical experts consulted by CDA-AMC highlighted that, because PN is a chronic, immune-mediated condition, patients require long-term treatment, and relapses have been observed in those who stopped current therapies. Therefore, the clinical experts expected that patients would require long-term treatment with nemolizumab.
Improvements in IGA success from LTE study baseline were observed in 60% of patients at week 52, with continued improvements observed at week 100 (> 70%) in the continuous nemolizumab and placebo-to-nemolizumab groups. Similarly, the proportion of patients experiencing a clinically meaningful improvement in itch response (demonstrated by at least a 4-point improvement in PP‑NRS score) from baseline lead-in was supported by the results at weeks 52 and 100. A similar trend was demonstrated for the proportions of patients achieving meaningful improvements in SD-NRS and DLQI scores. However, the risk of attrition bias (given that patients discontinued the LTE study over time) limited the ability to interpret the effect of continued treatment on these end points. Patients who discontinued the LTE study could be systematically different from those who remained in the study. The re-treatment group was too small to draw any conclusions regarding efficacy. The treatment durations in the OLYMPIA 1 and OLYMPIA 2 trials were 24 weeks and 16 weeks, respectively. Clinical experts acknowledged that the durations of these trials support the short- to medium-term effects of nemolizumab; however, further evaluation and real-world data are needed to establish a long-term clinical profile for the drug.
Based on the fixed-effects model, the NMA demonstrated a favourable benefit of nemolizumab compared with dupilumab. However, the favourable effect was not consistent, and it was influenced by the time points assessed and the outcomes measured (such as PP-NRS change from baseline, PP-NRS response, IGA success, composite PP-NRS and IGA response, and DLQI change from baseline). In addition, the fixed-effects model may underestimate the between-study heterogeneity, leading to overly precise estimates. Overall, although the findings of the NMA are subject to considerable limitations due to the significant heterogeneity between the trials included in the analyses, the clinical experts consulted for this review indicated that overall, the results aligned with their expectation that the effects of nemolizumab would be comparable to those of dupilumab in clinical practice.
Harms
The proportions of patients in the nemolizumab group versus the placebo group who experienced any AE in the OLYMPIA 1 trial were 71.7% versus 65.3%; in the OLYMPIA 2 trial, these proportions were 61.2% versus 53.8%. The most common AEs (by system organ class) were skin and SC tissue disorders and infections and infestations. In the OLYMPIA 1 trial, the most common AEs in the nemolizumab arm were COVID-19, nasopharyngitis, headache, neurodermatitis, and eczema. In the OLYMPIA 2 trial, the most common AEs in the nemolizumab arm were headache and AD. The proportions of patients who experienced at least 1 SAE in the nemolizumab arm versus the placebo arm in the OLYMPIA 1 trial were 8.6% versus 10.5%; in the OLYMPIA 2 trial, these proportions were 2.2% versus 5.5%. AEs leading to study drug withdrawal in the nemolizumab and placebo groups occurred in 4.8% and 3.2% of patients, respectively, in the OLYMPIA 1 trial, and in 2.7% and 2.2% of patients, respectively, in the OLYMPIA 2 trial. In the OLYMPIA 1 trial, 1 patient in the placebo group died during the study. No patient deaths occurred during the OLYMPIA 2 trial.
According to clinical experts consulted by CDA-AMC, AEs reported in the OLYMPIA pivotal trials were as expected and manageable. Clinical experts commented that most of the AEs were consistent with what patients experience with the biologics currently used in dermatology, and that dermatologists are comfortable managing these mild side effects. They noted that the reported worsening of AD in the nemolizumab groups was unique (i.e., this does not occur with most of the biologics); however, they highlighted that dermatologists are extremely comfortable managing this condition, if it occurs.
Clinical experts noted that the proportions of patients who experienced newly diagnosed asthma or worsening of asthma in the OLYMPIA pivotal trials was low. They also commented that it is possible that a higher incidence of worsening of asthma may occur in clinical practice than was reported in the OLYMPIA trials because patients with active asthma were excluded from the trials.
The harms results reported for the OLYMPIA LTE study were consistent with the results observed during the OLYMPIA 1 and OLYMPIA 2 trials. The results of the ITC demonstrated largely similar safety (i.e., overall AEs) when comparing nemolizumab with dupilumab.
The patient groups, clinician groups, and clinical experts consulted for this review agreed that minimizing the adverse effects of therapy is an important outcome in the treatment of PN.
Conclusion
Evidence from the OLYMPIA 1 and OLYMPIA 2 trials demonstrated that, compared to placebo, treatment with nemolizumab resulted in greater improvements in itch response, lesion clearance, and HRQoL in adult patients with moderate to severe PN. The GRADE assessment suggested that, at 16 weeks, compared to placebo, nemolizumab results in a clinically meaningful increase in the proportion of patients with at least a 4-point improvement in PP-NRS score and in the proportion of patients with IGA success. The GRADE assessment also suggested that nemolizumab likely results in a clinically meaningful increase in the proportion of patients with at least a 4-point improvement in DLQI score compared to placebo at 16 weeks (evidence from the OLYMPIA 2 trial) and 24 weeks (evidence from the OLYMPIA 1 trial).
In the OLYMPIA 1 and OLYMPIA 2 trials, most patients in the nemolizumab and placebo groups experienced at least 1 AE. In both trials, the proportions of patients who experienced the AESI of newly diagnosed asthma or worsening of asthma were low, and the GRADE assessment suggested low certainty regarding the effect of nemolizumab on this AESI compared to placebo.
The ability to draw conclusions regarding the long-term efficacy of nemolizumab is limited by the single-arm, open-label design of the OLYMPIA LTE study and by the patient attrition that occurred throughout the study. The harms results in the LTE study aligned with those of the pivotal trials, with no new AEs identified.
The NMA demonstrated a favourable benefit of nemolizumab compared with dupilumab. However, the favourable effect was not consistent, and it was influenced by the outcomes measured and the time points assessed. The NMA may suggest a comparable clinical efficacy and overall safety profile between nemolizumab and dupilumab in the treatment of patients with PN; however, the findings of the NMA are subject to considerable limitations.
References
1.Galderma Canada Inc. Nemluvio (nemolizumab for injection) 30 mg lyophilized powder with 0.49 mL sterile solution (60 mg/mL) for subcutaneous injection in prefilled dual-chamber syringe, 30 mg lyophilized powder with 0.49 mL sterile solution (60 mg/mL) for subcutaneous injection in prefilled dual-chamber pen [product monograph; sponsor-supplied reference]. December 18, 2025.
2.Galderma Canada Inc. Sponsor Summary of Clinical Evidence for NEMLUVIO (nemolizumab) for Prurigo Nodularis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Nemolizumab 30mg/0.49mL subcutaneous injection. January 2025.
3.Aggarwal P, Choi J, Sutaria N, et al. Clinical characteristics and disease burden in prurigo nodularis. Clin Exp Dermatol. 2021;46(7):1277-1284. doi:10.1111/ced.14722 PubMed
4.Watsky K. Post TW, ed. Prurigo nodularis. UpToDate; August 21, 2024. Accessed December 24, 2024. http://www.uptodate.com
5.Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: Epidemiology and clinical features. J Am Acad Dermatol. 2020;83(6):1559-1565. doi:10.1016/j.jaad.2020.04.183 PubMed
6.Kwatra SG. Breaking the Itch-Scratch Cycle in Prurigo Nodularis. N Engl J Med. 2020;382(8):757-758. doi:10.1056/NEJMe1916733 PubMed
7.Pereira MP, Steinke S, Zeidler C, et al. European academy of dermatology and venereology European prurigo project: expert consensus on the definition, classification and terminology of chronic prurigo. J Eur Acad Dermatol Venereol. 2018;32(7):1059-1065. doi:10.1111/jdv.14570 PubMed
8.Bahloul D, Hudson R, Balogh O, et al. Prevalence, incidence and treatment patterns of prurigo nodularis in England: a retrospective database analysis. Br J Dermatol. 2024;191(4):548-555. doi:10.1093/bjd/ljae207 PubMed
9.Boozalis E, Tang O, Patel S, et al. Ethnic differences and comorbidities of 909 prurigo nodularis patients. J Am Acad Dermatol. 2018;79(4):714-719 e3. doi:10.1016/j.jaad.2018.04.047 PubMed
10.Kowalski EH, Kneiber D, Valdebran M, Patel U, Amber KT. Treatment-resistant prurigo nodularis: challenges and solutions. Clin Cosmet Investig Dermatol. 2019;12:163-172. doi:10.2147/CCID.S188070 PubMed
11.Woo YR, Wang S, Sohn KA, Kim HS. Epidemiology, Comorbidities, and Prescription Patterns of Korean Prurigo Nodularis Patients: A Multi-Institution Study. J Clin Med. 2021;11(1):95. doi:10.3390/jcm11010095 PubMed
12.Janmohamed SR, Gwillim EC, Yousaf M, Patel KR, Silverberg JI. The impact of prurigo nodularis on quality of life: a systematic review and meta-analysis. Arch Dermatol Res. 2021;313(8):669-677. doi:10.1007/s00403-020-02148-0 PubMed
13.Rodriguez D, Kwatra SG, Dias-Barbosa C, et al. Patient Perspectives on Living With Severe Prurigo Nodularis. JAMA Dermatol. 2023;159(11):1205-1212. doi:10.1001/jamadermatol.2023.3251 PubMed
14.Murota H, Arima K, Yoshida T, Fujita H. Disease burden and treatment satisfaction in patients with prurigo nodularis in Japan. J Dermatol. 2024;51(2):223-233. doi:10.1111/1346-8138.17045 PubMed
15.Hui SK, Grandner MA. Trouble Sleeping Associated With Lower Work Performance and Greater Health Care Costs: Longitudinal Data From Kansas State Employee Wellness Program. J Occup Environ Med. 2015;57(10):1031-8. doi:10.1097/JOM.0000000000000534 PubMed
16.Ryczek A, Reich A. Prevalence of Prurigo Nodularis in Poland. Acta Derm Venereol. 2020;100(10):adv00155. doi:10.2340/00015555-3518 PubMed
17.Yale Medicine. Prurigo nodularis fact sheets [sponsor supplied reference]. Accessed April 2024. https://www.yalemedicine.org/conditions/prurigo-nodularis-overview#:~:text=A%20physical%20exam%20will%20be,skin%20biopsy%20may%20be%20required.
18.Ludmann P. Prurigo nodularis: Diagnosis and treatment [sponsor supplied reference]. American Academy of Dermatology Association. 2021. Accessed April 2024. https://www.aad.org/public/diseases/a-z/prurigo-nodularis-treatment
19.Kwatra SG, Chisolm SS, Puerta Durango KS, Mollanazar NK. INDIVIDUAL ARTICLE: Patient Journey and the Burden of Systemic Comorbidities and Sequalae in Prurigo Nodularis. J Drugs Dermatol. 2023;22(12):SF365502s12-SF365502s14. doi:10.36849/JDD.SF365502
20.Ständer S, Pereira MP, Berger T, et al. IFSI-guideline on chronic prurigo including prurigo nodularis. Itch. 2020;5(4):e42-e42. doi:10.1097/itx.0000000000000042
21.Elmariah S, Kim B, Berger T, et al. Practical approaches for diagnosis and management of prurigo nodularis: United States expert panel consensus. J Am Acad Dermatol. 2021;84(3):747-760. doi:10.1016/j.jaad.2020.07.025 PubMed
22.Satoh T, Yokozeki H, Murota H, et al. 2020 guidelines for the diagnosis and treatment of prurigo. J Dermatol. 2021;48(9):e414-e431. doi:10.1111/1346-8138.16067 PubMed
23.Sanofi-aventis Canada Inc. Dupixent (dupilumab): solution for subcutaneous injection 300 mg single-use syringe (300 mg/2 mL); 300 mg single-use pen (300 mg/2 mL); 200 mg single-use syringe (200 mg/1.14 mL); 200 mg single-use pen (200 mg/1.14 mL) [product monograph]. November 18, 2024. Accessed March 13, 2025.
24.Galderma S.A. OLYMPIA 1 Clinical Study Report: RD.06.SRE.202685. A Randomized, Double-blind, Placebo-controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. September 18, 2023.
25.Galderma S.A. OLYMPIA 2 Clinical Study Report: RD.06.SRE.203065. A Randomized, Double-blind, Placebo-controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. September 18, 2023.
26.Galderma Canada Inc. Galderma Canada Inc. response to March 11, 2025 (Part 2) CDA-AMC request for additional information regarding Nemluvio CDA-AMC review [internal additional sponsor's information]. March 24, 2025.
27.Williams KA, Huang AH, Belzberg M, Kwatra SG. Prurigo nodularis: Pathogenesis and management. J Am Acad Dermatol. 2020;83(6):1567-1575. doi:10.1016/j.jaad.2020.04.182 PubMed
28.Steinke S, Zeidler C, Riepe C, et al. Humanistic burden of chronic pruritus in patients with inflammatory dermatoses: Results of the European Academy of Dermatology and Venereology Network on Assessment of Severity and Burden of Pruritus (PruNet) cross-sectional trial. J Am Acad Dermatol. 2018;79(3):457-463.e5. doi:10.1016/j.jaad.2018.04.044 PubMed
29.Chen X. Overview of Prurigo Nodularis for Practicing Dermatology Clinicians [sponsor submitted reference]. Dermatology Times. 2022. https://www.dermatologytimes.com/view/overview-of-prurigo-nodularis-for-practicing-dermatology-clinicians
30.Zeidler C, Pereira MP, Ständer S. Chronic Prurigo: Similar Clinical Profile and Burden Across Clinical Phenotypes. Front Med (Lausanne). 2021;8:649332. doi:10.3389/fmed.2021.649332 PubMed
31.Pereira MP, Basta S, Moore J, Ständer S. Prurigo nodularis: a physician survey to evaluate current perceptions of its classification, clinical experience and unmet need. J Eur Acad Dermatol Venereol. 2018;32(12):2224-2229. doi:10.1111/jdv.15107 PubMed
32.Augustin M, Garbe C, Hagenström K, Petersen J, Pereira MP, Ständer S. Prevalence, incidence and presence of comorbidities in patients with prurigo and pruritus in Germany: A population-based claims data analysis. J Eur Acad Dermatol Venereol. 2021;35(11):2270-2276. doi:10.1111/jdv.17485 PubMed
33.Huang AH, Canner JK, Khanna R, Kang S, Kwatra SG. Real-World Prevalence of Prurigo Nodularis and Burden of Associated Diseases. J Invest Dermatol. 2020;140(2):480-483 e4. doi:10.1016/j.jid.2019.07.697 PubMed
34.Center for Drug Evaluation and Research. Nemluvio (nemolizumab) 30mg. Company: Galderma Labs LP. Biologic License Application No.:761390. Approval date: 08/12/2024 (FDA approval package). U. S. Food and Drug Administration (FDA). Accessed February 6, 2025. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761390
35.European Medicines Agency (EMA). European Public Assessment Report (EPAR): Nemluvio (nemolizumab). Accessed February 6, 2025. https://www.ema.europa.eu/en/medicines/human/EPAR/nemluvio
36.National Institute for Health and Care Excellence (NICE). Nemolizumab for treating prurigo nodularis [ID6451]. Accessed February 6, 2025. https://www.nice.org.uk/guidance/indevelopment/gid-ta11566
37.CDA-AMC. Drug Reimbursement Review: nemolizumab (Nemluvio) for the treatment of moderate-to-severe AD in patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and/or who are refractory to or ineligible for systemic immunosuppressant therapies. 2025. Accessed May 1, 2025. https://www.cda-amc.ca/nemolizumab
38.Stander S, Yosipovitch G, Legat FJ, et al. Efficacy and Safety of Nemolizumab in Patients With Moderate to Severe Prurigo Nodularis: The OLYMPIA 1 Randomized Clinical Phase 3 Trial. JAMA Dermatol. 2025;161(2):147-156. doi:10.1001/jamadermatol.2024.4796 PubMed
39.Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 Trial of Nemolizumab in Patients with Prurigo Nodularis. N Engl J Med. 2023;389(17):1579-1589. doi:10.1056/NEJMoa2301333 PubMed
40.Galderma S.A. OLYMPIA 1 Clinical Study Protocol: RD.06.SPR.202685. A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. December 22, 2020.
41.Galderma S.A. OLYMPIA 2 Clinical Study Protocol: RD.06.SPR.203065. A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. December 22, 2020.
42.Galderma S.A. OLYMPIA 1 Statistical Analysis Plan: RD.06.SPR.202685. A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. March 03, 2023.
43.Galderma S.A. OLYMPIA 2 Statistical Analysis Plan: RD.06.SPR.203065. A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. May 20, 2022.
44.Stander S, Yosipovitch G, Legat FJ, et al. Nemolizumab monotherapy improves itch and skin lesions in patients with moderate-to-severe prurigo nodularis: Results from a global Phase 3 trial (OLYMPIA 1) [sponsor submitted reference]. Presented at European Academy of Dermatology and Venereology. 2022.
45.Galderma Canada Inc. Data on-file [Confidential Information; sponsor submitted reference]. Accessed 2024.
46.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-6. doi:10.1111/j.1365-2230.1994.tb01167.x PubMed
47.Kwatra SG, Rodriguez D, Dias-Barbosa C, et al. Validation of the Peak Pruritus Numerical Rating Scale as a Patient-Reported Outcome Measure in Prurigo Nodularis. Dermatol Ther (Heidelb). 2023;13(10):2403-2416. doi:10.1007/s13555-023-00999-9 PubMed
48.Zeidler C, Pereira MP, Augustin M, Spellman M, Ständer S. Investigator's Global Assessment of Chronic Prurigo: A New Instrument for Use in Clinical Trials. Acta Derm Venereol. 2021;101(2):adv00401. doi:10.2340/00015555-3701 PubMed
49.Ständer S, Fofana F, Dias-Barbosa C, et al. The Sleep Disturbance Numerical Rating Scale: Content Validity, Psychometric Validation, and Meaningful Within-Patient Change in Prurigo Nodularis. Dermatol Ther (Heidelb). 2023;13(7):1587-1602. doi:10.1007/s13555-023-00962-8 PubMed
50.Bahloul D, Thomas RB, Rhoten S, et al. Validation of the Dermatology Life Quality Index (DLQI) in Prurigo Nodularis (PN) Based on Clinical Studies of Dupilumab in Adults with PN. J Am Acad Dermatol. 2023;89(3):Ab103. doi:10.1016/j.jaad.2023.07.415
51.Galderma Canada Inc. Galderma Canada Inc. response to February 18, 2025 CDA-AMC request for additional information regarding Nemluvio CDA-AMC review [internal additional sponsor's information]. February 21, 2025.
52.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
53.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
54.Galderma. Clinical Study Report. RD.06.SIR.202699 (LTE) - A Prospective, Multicenter, Long-Term Study to Assess the Safety and Efficacy of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. October 23, 2023.
55.Galderma. Clinical Study Report. RD.06.SIR.202699 (LTE) - Interim Analysis 2 - A Prospective, Multicenter, Long-Term Study to Assess the Safety and Efficacy of Nemolizumab (CD14152) in Subjects with Prurigo Nodularis [internal sponsor's report]. January 8, 2025.
56.Galderma. Clinical and Economic Burden of Prurigo Nodularis: Systematic Literature Reviews: Final SLR Report [internal sponsor's report]. May 6, 2024.
57.Galderma. Indirect Comparison of Nemolizumab versus Comparators in the Treatment of Prurigo Nodularis: NMA Report [internal sponsor's report]. September 2, 2024.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 35: Summary of Select Key Secondary and Secondary Efficacy End Points in the OLYMPIA 1 and OLYMPIA 2 Trials (ITT Population)
End point | OLYMPIA 1 | OLYMPIA 2 | ||
|---|---|---|---|---|
Nemolizumab N = 190 | Placebo N = 96 | Nemolizumab N = 183 | Placebo N = 91 | |
Key secondary end pointsa | ||||
Improvement of ≥ 4 from baseline in PP-NRS at week 4 | ||||
n (%) | 78 (41.1) | 6 (6.3) | 75 (41.0) | 7 (7.7) |
Unadjusted proportion difference, (%) | 34.8 | — | 33.3 | — |
Unadjusted 95% CI | 26.3, 43.3 | — | 24.3, 42.3 | — |
Unadjusted p value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 31.7 | — | 33.4 | — |
Strata-adjusted 95% CI | 23.0, 40.4 | — | 24.3, 42.4 | — |
Strata-adjusted p value | < 0.0001 | — | < 0.0001 | — |
Secondary end points | ||||
Improvement of ≥ 4 from baseline in PP-NRS at week 12 | ||||
n (%) | 92 (48.4) | 13 (13.5) | 95 (51.9) | 15 (16.5) |
Unadjusted proportion difference, (%) | 34.9 | — | 35.4 | — |
Unadjusted 95% CI | 25.0, 44.7 | — | 24.9, 45.9 | — |
Unadjusted p value | < 0.0001 | — | < 0.0001 | — |
Strata-adjusted proportion difference, (%) | 32.5 | — | 37.0 | — |
Strata-adjusted 95% CI | 22.0, 43.0 | — | 26.4, 47.7 | — |
Strata-adjusted p value | < 0.0001 | — | < 0.0001 | — |
Change from baseline in PP-NRS at week 16 | ||||
Baseline (observed cases) | — | — | — | — |
n | 184 | 96 | 183 | 91 |
Mean (SD) | 8.4952 (0.94134) | 8.4371 (0.98758) | 8.4745 (0.90096) | 8.3715 (0.98869) |
Week 16 | — | — | — | — |
n | 184 | 96 | 183 | 91 |
LS mean (SE) (MI estimate) | −4.67 (0.229) | −1.56 (0.321) | −4.77 (0.265) | −1.68 (0.356) |
95% CI (MI estimate) | −5.12, −4.22 | −2.19, −0.93 | −5.29, −4.25 | −2.38, −0.98 |
LS mean difference (95% CI) (MI estimate) | −3.11 (−3.86, −2.37) | — | −3.09 (−3.87, −2.31) | — |
P value | < 0.0001 | — | < 0.0001 | — |
Change from baseline in SD-NRS at week 16 | ||||
Baseline (observed cases) | — | — | — | — |
n | 190 | 96 | 182 | 91 |
Mean (SD) | 7.0100 (2.36737) | 6.8886 (2.33491) | 7.1871 (2.20899) | 7.3066 (2.23320) |
Week 16 | — | — | — | — |
n | 190 | 96 | 182 | 91 |
LS mean (SE) (MI estimate) | −3.88 (0.217) | −1.00 (0.305) | −4.11 (0.267) | −1.21 (0.354) |
95% CI (MI estimate) | −4.30, −3.45 | −1.60, −0.41 | −4.63, −3.58 | −1.90, −0.52 |
LS mean difference (95% CI) (MI estimate) | −2.87 (−3.58, −2.16) | — | −2.90 (−3.67, −2.12) | — |
P value | < 0.0001 | — | < 0.0001 | — |
CI = confidence interval; ITT = intention to treat; LS = least squares; MI = multiple imputation; PP-NRS = Peak Pruritus Numerical Rating Scale; SD = standard deviation; SD-NRS = Sleep Disturbance Numerical Rating Scale; SE = standard error.
aP value has been adjusted for multiple testing.
Source: Sponsor’s Summary of Clinical Evidence,2 OLYMPIA 1 Clinical Study Report,24 OLYMPIA 2 Clinical Study Report,25 sponsor’s Response to Request for Additional Information.26
Table 36: Treatment Summary Based on Lead-In Phase IIa and Phase III Pivotal Studies
Lead-in study | Prior study assigned treatment q.4.w. SC | Dose on day 1/baseline (2 injections)a | Weight at baseline | Open-label dose q.4.w. for 180 weeksb |
|---|---|---|---|---|
OLYMPIA 1 or OLYMPIA 2 | Nemolizumab 30 mg | Blinded nemolizumab 30 mg (one 30 mg injection and 1 placebo injection) | NAc | Nemolizumab 30 mg |
Placebo | Blinded nemolizumab 60 mg (two 30 mg injections) | NAc | Nemolizumab 2 × 30 mg | |
Nemolizumab 2 × 30 mgc | Blinded nemolizumab 60 mg (two 30 mg injections) | NAc | Nemolizumab 2 × 30 mg | |
2 × placeboc | Blinded nemolizumab 60 mg (two 30 mg injections) | NAc | Nemolizumab 2 × 30 mg | |
Phase IIa study (NCT03181503) | Placebo or nemolizumab 0.5 mg/kg | Open-label nemolizumab 60 mg (two 30 mg injections) | < 90 kg | Nemolizumab 30 mg |
≥ 90 kg | Nemolizumab 2 × 30 mg |
LTE = long-term extension; NA = not applicable; q.4.w. = every 4 weeks; SC = subcutaneous.
aAny patient with a > 12-week interval since the last dose of study drug received a 60-mg dose of nemolizumab through 2 injections of 30 mg at the day 1/baseline visit.
bBeginning at week 56, the nemolizumab dosage was adjusted every 6 months for patients with a documented weight change to greater or less than the 90 kg threshold at 2 consecutive designated visits.
cIn studies OLYMPIA 1 or OLYMPIA 2, patients who weighed < 90 kg at baseline received either 30 mg nemolizumab or placebo q.4.w. while patients who weighed ≥ 90 kg at baseline received either 60 mg nemolizumab or placebo q.4.w. When patients rolled into this LTE study from OLYMPIA 1 or OLYMPIA 2, patients who weighed < 90 kg received 30 mg nemolizumab q.4.w. while patients who weighed ≥ 90 kg received 60 mg nemolizumab q.4.w. Initial dosing was based on weight at baseline of the lead-in study.
Source: LTE Clinical Study Report54
Table 37: Treatment Summary Based on Re-Entering From the Durability Study
Lead-in study | Prior assigned treatment | Dose at re-entry baseline (R0) visita | Weight | Open-label dose q.4.w. for 128 weeks from re‑entry week R4b |
|---|---|---|---|---|
Durability study (NCT05052983) | Nemolizumab | Blinded nemolizumab 30 mg (1 × 30 mg injection and 1 × placebo injection) | < 90kg | Nemolizumab 30 mg |
Placebo | Blinded nemolizumab 60 mg (2 × 30 mg injections) | < 90kg | Nemolizumab 30 mg | |
Nemolizumab OR Placebo 2 x 30 mg | Nemolizumab or placebo (2 × 30 mg injections) | ≥ 90kg | Nemolizumab (2 × 30 mg) |
LTE = long-term extension; q.4.w. = every 4 weeks; SC = subcutaneous.
aAll patients received 2 SC injections, as assigned by an interactive response technology system at the re-entry visit (week R0), based on the assigned study drug in the durability study.
bDosage was adjusted every 6 months for patients with a documented weight change to greater or less than the 90 kg threshold at 2 consecutive designated visits.
Source: LTE Clinical Study Report54
Table 38: Patient Disposition in the LTE Study by Previous Treatment — Interim Analysis 2 [Redacted]
Number of patients | Nemolizumab q.4.w. N = 508 n (%) | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 n (%) | ||||
All N = 334 n (%) | Continuous nemolizumab N = 307 n (%) | Re-treatment N = 27 n (%) | |||
Treated | ███ | ███ | ███ | ███ | ███ |
Completed treatment | ███ | ███ | ███ | ███ | ███ |
Discontinued treatment | ███ | ███ | ███ | ███ | ███ |
Entered follow-up period | ███ | ███ | ███ | ███ | ███ |
Primary reason for discontinuation of treatment | |||||
Pregnancy | ███ | ███ | ███ | ███ | ███ |
Lack of efficacy | ███ | ███ | ███ | ███ | ███ |
Adverse event | ███ | ███ | ███ | ███ | ███ |
Patient's request | ███ | ███ | ███ | ███ | ███ |
Lost to follow-up | ███ | ███ | ███ | ███ | ███ |
Protocol deviation | ███ | ███ | ███ | ███ | ███ |
Physician/primary investigator decision | ███ | ███ | ███ | ███ | ███ |
Sponsor decision | ███ | ███ | ███ | ███ | ███ |
Other | |||||
PEF criteria not met | ███ | ███ | ███ | ███ | ███ |
Site closure | ███ | ███ | ███ | ███ | ███ |
Visit schedulea | ███ | ███ | ███ | ███ | ███ |
Patient willingly not re‑entering LTE | ███ | ███ | ███ | ███ | ███ |
Completed the study | ███ | ███ | ███ | ███ | ███ |
Discontinued from the study | ███ | ███ | ███ | ███ | ███ |
Primary reason for discontinuation from the study | |||||
Pregnancy | ███ | ███ | ███ | ███ | ███ |
Lack of efficacy | ███ | ███ | ███ | ███ | ███ |
Adverse event | ███ | ███ | ███ | ███ | ███ |
Patient’s request | ███ | ███ | ███ | ███ | ███ |
Lost to follow-up | ███ | ███ | ███ | ███ | ███ |
Protocol deviation | ███ | ███ | ███ | ███ | ███ |
Physician decision | ███ | ███ | ███ | ███ | ███ |
Sponsor decision | ███ | ███ | ███ | ███ | ███ |
Other | |||||
PEF criteria not meta | ███ | ███ | ███ | ███ | ███ |
Site closure | ███ | ███ | ███ | ███ | ███ |
Visit scheduleb | ███ | ███ | ███ | ███ | ███ |
Patient willingly not re‑entering the LTE study | ███ | ███ | ███ | ███ | ███ |
Rolled over to Study NCT05052983 (Durability study) | ███ | ███ | ███ | ███ | ███ |
Re-entered from Study NCT05052983 | ███ | ███ | ███ | ███ | ███ |
Completed follow‑up | ███ | ███ | ███ | ███ | ███ |
LTE = long-term extension; PEF = Peak Expiratory Flow; q.4.w. = every 4 weeks
Note: Percentages were based on the number of patients.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with at least a 12-week interval between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE.
Source: LTE Clinical Study Report – Interim Analysis 2.55
Table 39: Extent of Exposure (Safety Population) — Interim Analysis 1
Characteristic | Nemolizumab q.4.w. N = 508 | Re-entered from the durability study (NCT05052983) N = 17 |
|---|---|---|
Total dose administered (mg)a | ||
Mean (SD) | 547.4 (303.93) | 668.8 (244.82) |
Total dose planned during the study (mg)b | 558.1 (308.42) | 674.1 (243.29) |
Patients who missed at least 1 dose, n (%) | 100 (19.70) | 3 (17.60) |
Patients who missed at least 1 dose due to COVID-19, n (%) | 41 (8.10) | 2 (11.80) |
Number of doses missed, n | 182 | 3 |
Number of doses missed due to COVID-19, n | 59 | 2 |
Treatment duration during the study (days) | ||
Mean (SD) | 368.5 (186.53) | 607.2 (92.88) |
Number of patients by treatment duration during the study, n (%) | ||
< 6 months | 110 (21.70) | 0 |
6 months to < 12 months | 136 (26.80) | 0 |
12 months to < 18 months | 165 (32.50) | 7 (41.20) |
18 months to < 24 months | 94 (18.50) | 8 (47.10) |
24 months to < 30 months | 3 (0.60) | 2 (11.80) |
Treatment adherence (%) | ||
Mean (SD) | 97.98 (5.63) | 99.11 (2.05) |
Median | 100.00 | 100.00 |
Q1 to Q3 | 100.00 to 100.00 | 100.00 to 100.00 |
Minimum to maximum | 50.0 to 100.0 | 93.3 to 100.0 |
Q1 = first quartile; Q3 = third quartile; q.4.w. = every 4 weeks
Notes: Percentages were based on the number of patients.
Treatment duration was calculated as: (date of last treatment – date of first treatment) + 1. For patients who re-entered from Study NCT05052983, treatment duration was calculated as (date of last treatment before durability study – date of first treatment) + (date of last re-enter treatment – date of first re-enter treatment) + 2.
Treatment adherence was calculated as the ratio (%) between the total number of actual injections and the total number of expected injections multiplied by 100. The total number of actual injections was counted based on collected study drug administration data. The total number of expected injections was counted based on the dosage schedule and dispensed as per protocol.
aTotal dose administered was calculated as the sum of all doses of study drug administered in this study.
bTotal dose planned was calculated as the sum of all doses of study drug planned (dispensed) according to the treatment schedule of the treatment group.
Source: LTE Clinical Study Report.54
Table 40: Absolute Change From Baseline in PP-NRS and SD-NRS Scores Through Week 52 — Observed Case (Interim Analysis 1)
Visit | Nemolizumab q.4.w. N = 508 n (%) | By previous treatment | |||
|---|---|---|---|---|---|
Previously treated with nemolizumab | Placebo to nemolizumab N = 174 n (%) | ||||
All N = 334 n (%) | Continuous nemolizumab N = 307 n (%) | Re-treatment N = 27 n (%) | |||
Absolute change in PP-NRS | |||||
Baseline lead-in | |||||
Observed value, n | 483 | 319 | 303 | 16 | 164 |
Mean (SD) | 8.49 (0.95) | 8.54 (0.93) | 8.54 (0.93) | 8.55 (0.88) | 8.39 (0.98) |
Week 16 | |||||
Observed value, n | 256 | 167 | 151 | 16 | 89 |
Mean (SD) | 2.29 (2.44) | 2.43 (2.57) | 2.35 (2.52) | 3.21 (2.96) | 2.03 (2.19) |
Change from baseline lead-in, n | 242 | 161 | 151 | 10 | 81 |
Mean (SD) | −6.25 (2.60) | −6.09 (2.68) | −6.19 (2.65) | −4.55 (2.77) | −6.55 (2.43) |
Week 28 | |||||
Observed value, n | 212 | 141 | 125 | 16 | 71 |
Mean (SD) | 2.11 (2.27) | 2.10 (2.23) | 1.97 (2.15) | 3.10 (2.67) | 2.15 (2.35) |
Change from baseline lead-in, n | 204 | 136 | 125 | 11 | 68 |
Mean (SD) | −6.38 (2.46) | −6.38 (2.36) | −6.51 (2.33) | −4.88 (2.33) | −6.40 (2.67) |
Week 52 | |||||
Observed value, n | 163 | 104 | 90 | 14 | 59 |
Mean (SD) | 1.71 (2.19) | 1.67 (2.13) | 1.55 (2.05) | 2.43 (2.53) | 1.79 (2.29) |
Change from baseline lead-in, n | 152 | 98 | 90 | 8 | 54 |
Mean (SD) | −6.80 (2.43) | −6.88 (2.29) | −7.03 (2.25) | −5.24 (2.24) | −6.64 (2.68) |
Absolute change in SD-NRS | |||||
Baseline lead-in | |||||
Observed value, n | 486 | 322 | 306 | 16 | 164 |
Mean (SD) | 7.08 (2.31) | 7.07 (2.36) | 7.10 (2.31) | 6.46 (3.26) | 7.12 (2.22) |
Week 16 | |||||
Observed value, n | 259 | 171 | 154 | 17 | 88 |
Mean (SD) | 1.61 (2.21) | 1.68 (2.32) | 1.57 (2.21) | 2.71 (3.08423) | 1.46 (1.99) |
Change from baseline lead-in, n | 246 | 165 | 154 | 11 | 81 |
Mean (SD) | −5.40 (2.75) | −5.34 (2.75) | −5.56 (2.67) | −2.13 (1.76) | −5.52 (2.77) |
Week 28 | |||||
Observed value, n | 216 | 142 | 126 | 16 | 74 |
Mean (SD) | 1.70 (2.39) | 1.74 (2.39) | 1.68 (2.33) | 2.20 (2.84) | 1.62 (2.39) |
Change from baseline lead-in, n | 207 | 137 | 126 | 11 | 70 |
Mean (SD) | −5.33 (2.94) | −5.15 (2.82) | −5.35 (2.78) | −2.84 (2.25) | −5.67 (3.14) |
Week 52 | |||||
Observed value, n | 165 | 107 | 92 | 15 | 58 |
Mean (SD) | 1.28 (1.98) | 1.25 (1.89) | 1.20 (1.87) | 1.57 (2.04) | 1.34 (2.17) |
Change from baseline lead-in, n | 153 | 100 | 92 | 8 | 53 |
Mean (SD) | −5.81 (2.58) | −5.74 (2.51) | −5.98 (2.42) | −2.92 (1.84) | −5.94 (2.72) |
LTE = long-term extension; OC = observed cases; PP-NRS = Peak Pruritus Numerical Rating Scale; Q1 = first quartile; Q3 = third quartile; q.4.w. = every 4 weeks; SD-NRS = Sleep Disturbance Numerical Rating Scale
Notes: For PP-NRS scores, weekly values were calculated as the average of 7 consecutive days of data up to the actual visit day or target study day (excluding) and set to missing if fewer than 4 days of data were available.
For SD-NRS scores, weekly values were calculated as the average of 7 consecutive days of data up to the actual visit day or target study day (excluding) and set to missing if fewer than 4 days of data were available. Baseline lead-in was defined as the last nonmissing value before the first dose of study drug in lead-in study. Phase II patients did not contribute to change from lead-in study baseline, because there were ≥ 6 months from completion of phase II to entry into LTE study. Baseline LTE was the last nonmissing value before first dose of study drug in this study.
The group previously treated with nemolizumab (All column) included patients who had been treated with nemolizumab at least once before this LTE study. The continuous nemolizumab group included patients with an interval of less than 12 weeks between the lead-in last dose of nemolizumab and the LTE study first dose. The re-treatment group included patients with at least a 12-week interval between the lead-in last dose of nemolizumab and the LTE study first dose. The group that received placebo before nemolizumab (Placebo to nemolizumab column) included patients never exposed to nemolizumab before this LTE study. All observed data even after use of rescue therapy were included; no imputations for missing data.
Source: Clinical Study Report LTE.54
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
ICER
incremental cost-effectiveness ratio
IGA
Investigator’s Global Assessment
ITC
indirect treatment comparison
NMA
network meta-analysis
PN
prurigo nodularis
PP-NRS
Peak Pruritus Numerical Rating Scale
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Nemolizumab (Nemluvio) powder and solvent solution for SC injection available as:
|
Indication | Proposed: For the treatment of adults with moderate-to-severe prurigo nodularis (PN) whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | December 18, 2025 |
Reimbursement request | For the treatment of moderate-to-severe PN |
Sponsor | Galderma Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; PN = prurigo nodularis; SC = subcutaneous.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Adults with moderate to severe PN (defined as an IGA score of 3 or 4, a PP-NRS score of at least 7, and at least 20 pruriginous lesions) |
Treatment |
|
Dose regimen |
|
Submitted price | Nemolizumab: $2,995.00 per 30 mg |
Submitted treatment cost |
|
Comparators |
|
Perspective | Publicly funded health care payer in Canada |
Outcomes | QALYs gained, LYs gained |
Time horizon | Lifetime (45 years) |
Key data sources |
|
Submitted results | Nemolizumab is expected to generate more QALYs gained and fewer costs than dupilumab plus BSC or BSC alone; therefore, it dominates both treatment options. |
Key limitations |
|
CDA-AMC reanalysis results |
|
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; IGA = Investigator’s Global Assessment; LY = life-year; NMA = network meta-analysis; PN = prurigo nodularis; PP-NRS = Peak Pruritis Numerical Rating Scale; q.4.w. = every 4 weeks; QALY = quality-adjusted life-year; vs. = versus; WTP = willingness to pay.
Conclusions
In the OLYMPIA 1 and OLYMPIA 2 trials, nemolizumab was clinically and statistically significantly superior to placebo, based on the higher proportions of patients with an improvement of 4 or more points from baseline in weekly average Peak Pruritus Numerical Rating Scale (PP-NRS) and Investigator’s Global Assessment (IGA) success at week 16. Estimates from the long-term extension study found that the treatment effect for nemolizumab persisted beyond the 24-week observation period of the OLYMPIA trials. The sponsor submitted a network meta-analysis (NMA) to estimate the comparative efficacy of nemolizumab versus dupilumab for this composite measure. The clinical review by Canada’s Drug Agency (CDA-AMC) found no favourable effect in this composite measure for nemolizumab plus best supportive care (BSC) versus dupilumab plus BSC at 24 weeks.
In the CDA-AMC base case, nemolizumab plus BSC was the costliest and most effective treatment versus the 2 comparators. The sequential incremental cost-effectiveness ratio (ICER) of nemolizumab plus BSC was $1,124,092 per quality-adjusted life-year (QALY) gained when compared to dupilumab plus BSC (incremental cost = $73,066; incremental QALYs gained = 0.07), while BSC alone was the least costly and least effective treatment (nemolizumab ICER = $248,002 per QALY gained compared to BSC). Given that both the 24-week results and clinical expert input suggested that nemolizumab and dupilumab are likely comparably effective, CDA-AMC conducted a cost-minimization scenario analysis in which the total cost of nemolizumab plus BSC was $66,860 higher than for dupilumab plus BSC.
Based on the sponsor-submitted clinical evidence, nemolizumab plus BSC appears to be more effective than BSC alone, and comparably effective to dupilumab, for the treatment of prurigo nodularis (PN) 24 weeks after the initiation of treatment. The sponsor did not submit evidence to demonstrate that the duration of treatment effect is longer for nemolizumab than for dupilumab. This suggests that both short-term and long-term effectiveness are equal between these 2 treatments. Therefore, there is insufficient evidence to justify a price premium for nemolizumab versus dupilumab in this setting.
Input From Interested Parties Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Canadian Skin Patient Alliance. The Canadian Skin Patient Alliance gathered information from 9 respondents who participated in a survey of patients and caregivers living in Canada. Survey respondents indicated that, in addition to persistent and severe itching, PN negatively affects their mental health and emotional well-being. Patients reported using multiple treatments to manage their PN, including topical corticosteroids, topical capsaicin, oral immunosuppressants (i.e., methotrexate), antihistamines, topical calcineurin inhibitors, narrowband ultraviolet B phototherapy, and medical cannabis. Two patients had experience with dupilumab, but they reported no changes in their conditions. No patients had experience with nemolizumab. Patients emphasized the limited efficacy of the available treatments. Patient input indicated that the most important outcomes for new treatment options are improved effectiveness, greater affordability, and fewer side effects.
CDA-AMC received input from 2 clinician groups: the Atlantic Dermatology Association and the Dermatology Association of Ontario. The former group summarized information gathered from an academic conference and a literature search. The latter summarized information gathered from clinical trial data, literature, and a clinical triallist in Canada. The clinician groups stated that initial treatment would begin with topical treatments, including corticosteroids and emollients, and progress to systemic options. Nemolizumab may be suited for patients as a first-line therapy or for patients for whom dupilumab, topical corticosteroids, or phototherapy are ineffective or intolerable.
Drug plan input for this review noted that few patients in the nemolizumab clinical trials had received prior therapies for PN aside from topical corticosteroids. The drug plan input expressed uncertainty about whether patients should be required to have an inadequate disease response to off-label treatments before they are eligible to receive nemolizumab. The drug plan emphasized concerns related to the budget impact of nemolizumab.
Several of these concerns were addressed in the sponsor’s model:
Off-label use of dupilumab was included as 1 of the comparators in the sponsor’s model.
Patients’ concerns about effectiveness and side effects were incorporated into the sponsor’s base case.
In addition, CDA-AMC addressed the following concern:
Budget impact was examined with and without requiring patients to have an inadequate disease response to off-label treatments before they are eligible for nemolizumab.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing the cost-effectiveness of nemolizumab plus BSC compared with dupilumab plus BSC and BSC alone for the treatment of patients with moderate to severe PN (i.e., the requested reimbursement population). The modelled population consists of adults with moderate to severe PN (defined as an IGA score of 3 or 4, a PP-NRS score of at least 7, and at least 20 pruriginous lesions). The characteristics are based on the OLYMPIA 1 and OLYMPIA 2 clinical trials.
Nemolizumab is available as a carton of one 30 mg prefilled pen. The submitted price for nemolizumab is $2,995.00 per 30 mg prefilled pen. For patients weighing less than 90 kg, the recommended initial dose is 60 mg (2 injections of 30 mg each) followed by 30 mg administered every 4 weeks. For patients weighing 90 kg or more, the recommended initial dose is 60 mg (2 injections of 30 mg each) followed by 60 mg every 4 weeks. Patients receive training in the self-administration of subcutaneously administered drugs (dupilumab, nemolizumab) from a nurse, with a 1-time training cost of $63.84. The initial cost of the nemolizumab loading dose in the model was $5,990. This was followed by an annual maintenance cost of $39,069 (for patients who weigh < 90 kg) or $78,138 (for patients who weigh ≥ 90 kg). The annual cost of the comparator treatment was $25,534 for dupilumab, based on a unit cost of $978.50 per syringe. In the first year, the annual drug cost is $26,512 plus a training cost of $63.84. BSC comprised concomitant medications used by patients in the OLYMPIA 1 and OLYMPIA 2 trials. The annual costs of BSC alone were $422 and $5,963 for patients with a disease response and those without a disease response ($31,804, if including subsequent biologic treatments), respectively.
The clinical outcomes reported were QALYs gained and life-years. The base-case analysis was conducted from the public health care payer perspective. The time horizon in the base case was lifetime (42 years), using a 1.5% annual discount rate for costs and effectiveness.
Model Structure
The sponsor submitted a model composed of a 16-week decision tree followed by a Markov model. In the decision tree (Figure 1 in Appendix 3), patients with a disease response — with response defined as a composite end point of at least a 4-point improvement in PP-NRS score and IGA success (defined as a score of 0 [clear] or 1 [almost clear]) — continued with treatment. Patients without a disease response discontinued treatment (nemolizumab, dupilumab, or BSC alone) and could either continue to receive BSC alone with higher doses of concomitant medications or switch to another biologic drug (Table 12 in Appendix 3). Then the patients entered the long-term Markov model with 3 distinct and mutually exclusive health states: maintained response, no response, or dead (wording of original source),1 according to their disease response at 16 weeks (Figure 2 in Appendix 3). Patients stayed in the maintained response health state until discontinuation, waning of treatment effect, or death (absorbing state). In the no response health state, patients for whom initial treatment was not effective could receive BSC alone or switch to another biologic for the entire model duration. The cycle length was 1 year. A half-cycle correction was applied to the calculation of QALYs gained.
Model Inputs
The baseline population characteristics were aligned with the characteristics of patients in the OLYMPIA 1 trial (n = 286) and the OLYMPIA 2 trial (n = 274). Both trials were phase III, multicentre, randomized, double‑blind, placebo-controlled, parallel-group trials. The primary objective of both trials was to assess the efficacy of nemolizumab compared to placebo in patients aged 18 years or older with PN after a 16-week treatment period. The mean age of the patients was 55.19 years. A total of 59.6% of patients were female and 40.4% of patients were male, and the mean weight of all patients was 82.56 kg. Patients completing the treatment period of the OLYMPIA trials were eligible to roll into a long-term extension study (n = 490).
Clinical efficacy (i.e., treatment response) at week 16 for nemolizumab plus BSC was calculated based on patient-level data from the OLYMPIA 1 and OLYMPIA 2 clinical trials. Response probabilities for comparators — dupilumab plus BSC and BSC alone — were calculated based on the response probability for nemolizumab and the relative measure of efficacy (odds ratio) obtained from the fixed-effects indirect treatment comparison (ITC) analysis. It was assumed that 80% of patients in the no response health state switched to another biologic after failure of the first biologic treatment, based on a modified Delphi panel comprising clinicians from the UK and Canada. For patients without a disease response receiving BSC alone, it was assumed that 40% would switch to nemolizumab plus BSC and 40% would switch to dupilumab plus BSC.
The conditional discontinuation probability at week 52 for nemolizumab with BSC was calculated based on patient-level data from the OLYMPIA long-term extension trial (0.33%). Conditional discontinuations of patients receiving BSC alone (6.25%) or dupilumab plus BSC (1.31%) were calculated based on the proportions of patients who withdrew from the OLYMPIA 1 and OLYMPIA 2 trials (BSC alone) and the PRIME and PRIME 2 trials. The conditional discontinuation probability at week 52 was assumed to continue annually for the entire model duration. The model also assumed treatment-effect waning, which was implemented as an increase in the discontinuation rate in the maintained response health state. The model assumed that in years 2, 3, 4, and 5 onwards, 2.8%, 8.6%, 9.1%, and 9.1% of patients, respectively, lost response to active treatment, discontinued, and went on to receive BSC or other treatments. For BSC alone, these values were assumed to be 25%, 50%, 75%, and 100%, respectively. Based on the Clinical Practice Research Datalink data analysis, higher mortality for patients with PN compared to general population is expected due to the bothersome symptoms and chronic nature of the disease, which have an impact on patients’ mental health. A hazard ratio of 1.37 was applied to general population mortality in the model and did not differ by treatment arm or health care state.
Adverse event (AE) rates at week 16 were determined based on clinical trial data for treatment-emergent AEs obtained from the clinical trials for each treatment (i.e., the OLYMPIA 1 trial for nemolizumab and the PRIME trials for dupilumab). Treatment-related AEs that occurred in at least 1% of patients in any treatment arm — such as allergic conjunctivitis, injection-site reactions, infectious conjunctivitis, atopic dermatitis, eczema nummular, and neurodermatitis — were included in the model. Additional costs resulting from the occurrence of AEs were obtained from the Ontario Ministry of Health and Long-Term Care. It was assumed that utilities obtained from clinical trials had already captured the impact of AEs on quality of life; therefore, no disutilities due to AEs were included in the model.
Utilities in the sponsor’s model were assumed to vary by treatment received (i.e., biologic drugs plus BSC or BSC alone) and by treatment response (i.e., those with a disease response or without a disease response). Utility values at baseline, week 16, and week 24 were informed using the EQ-5D from the OLYMPIA clinical trials. The utility values for patients receiving biologic drugs were assumed to increase by 5% in the second year (week 52) and onward based on the results observed in the long-term extension study for a proxy disease (atopic dermatitis). Patients with a disease response to BSC alone maintained the utility as observed at week 16. Patients whose disease did not respond at week 16 were attributed the baseline utility values. Patients who had a disease response at week 16 but lost this response through discontinuation or treatment-effect waning were assumed to maintain their utility values for 6 months, after which these values returned to baseline. In addition, an age utility multiplier was applied to health state utility values to represent decreased quality of life due to aging. To capture the treatment benefit for those who switched treatments, utility for these patients was assumed to be 90% of the baseline utility of those with a disease response to initial therapy for nemolizumab and dupilumab, and 100% of that of patients who received placebo.
The sponsor included costs related to drug acquisition, disease management, AE management, and indirect costs. Drug acquisition costs for nemolizumab were based on the sponsor’s submitted price, while the costs for dupilumab were obtained from the Ontario Exceptional Access Program. The costs associated with training were based on a 1-hour nurse tariff and implemented in the model as a 1-off cost associated with treatment in the decision tree. The unit cost ($63.84 per administration, based on an hourly rate of $56 and 14% fringe benefits) was sourced from the Ontario Nurses’ Association Collective Agreement. The type, number, and proportion of therapies prescribed to a cohort of patients with a high prevalence of PN were determined based on the concomitant medications used by the overall population in the OLYMPIA 1 and OLYMPIA 2 trials and insights from clinicians in Canada. Resource use for the medication included in BSC was based on a submission on dupilumab to the National Institute for Health and Care Excellence (TA 955) and validated with clinicians. Unit costs of the treatments used in BSC were sourced from the Ontario Drug Benefits Formulary. Disease management costs for patients with a disease response and those without a disease response, such as medical appointments, emergency visits, hospitalizations, or blood tests (and the frequencies and unit costs of these needs), were informed by the dupilumab National Institute for Health and Care Excellence submission, the Ontario Ministry of Health and Long-Term Care, and the Canadian Institute for Health Information.
Summary of Sponsor’s Economic Evaluation Results
The sponsor’s base-case analysis was run probabilistically (1,000 probabilistic iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented here. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base-case analysis, nemolizumab plus BSC was associated with an estimated cost of $664,998 and 14.02 QALYs gained over the lifetime time horizon. Nemolizumab plus BSC was less costly (cost savings of $39,789 to $109,702) and more effective (0.01 to 0.0138 QALYs gained) than dupilumab plus BSC or BSC alone. Therefore, both comparators were dominated by nemolizumab plus BSC. (Disaggregated results are presented in Appendix 3.)
The cost savings were driven by nemolizumab having the lowest expected BSC and disease management costs despite higher treatment costs due to a longer duration in the maintenance state. The key driver of QALY gains for nemolizumab plus BSC was the greater proportion of patients remaining in the maintenance health state (Table 13 in Appendix 3). No additional life-years were gained because the sponsor assumed that nemolizumab would not affect survival.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs gained | Sequential ICER ($ per QALY gained) |
|---|---|---|---|
Nemolizumab + BSC | $664,998 | 14.021 | Reference |
Dominated treatments | |||
BSC alone | $704,787 | 13.883 | Dominated |
Dupilumab + BSC | $814,490 | 14.012 | Dominated |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several deterministic scenario analyses to test the impact of alternative parameters and assumptions on the modelled results for nemolizumab plus BSC compared with dupilumab plus BSC and BSC alone. The results of the analysis were most sensitive to discounting (i.e., an ICER of $9,727,605 for dupilumab plus BSC when a discounting rate of 0% was used), changes in the alternative measure of response used (i.e., ICER of $167,133 for BSC alone when only the change in PP-NRS was used as the definition of treatment response), effect waning (i.e., ICER of $53,217 for BSC alone when no waning was assumed), and administration disutilities (i.e., ICER of $101,282 for BSC alone when a disutility of −0.012 per subcutaneous administration was used).
The sponsor conducted a deterministic scenario analysis from a societal perspective that also considered costs due to productivity loss. In this analysis, nemolizumab plus BSC also dominated both dupilumab plus BSC and BSC alone.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The ITC was subject to observation time bias. The sponsor submitted an NMA to evaluate the comparative effectiveness of nemolizumab plus BSC versus dupilumab plus BSC as defined by a composite score (IGA and PP-NRS) at 16 weeks. The sponsor’s NMA could not incorporate 16-week responses for dupilumab because these were not available in the LIBERTY-PN PRIME trial data. Instead, the sponsor used 12-week response rates for dupilumab and compared these to 16-week response rates for nemolizumab. This analysis assumed that the response rates, measured a month apart, were equivalent. This assumption lacked face validity; the clinical experts consulted by CDA-AMC did not agree that it was reasonable. CDA-AMC noted that while 24-week data were provided for the PP-NRS and IGA scores separately, the sponsor did not include information about the composite measure that was chosen to inform its model. The sponsor went on to provide this 24‑week composite assessment after a request from CDA-AMC. The 24-week composite outcome was not statistically significant, with a wide confidence interval that crossed ███ ███ █ █████ ███ ██ █ ███ █ ████. The CDA-AMC clinical review, in conjunction with input from clinical experts, concluded that the evidence did not show a favourable effect for nemolizumab versus dupilumab in treating PN.
In the CDA-AMC reanalysis, the 24-week odds ratio was used for the relative efficacy for dupilumab versus nemolizumab. The odds ratio of 1 (i.e., same efficacy) was also tested in a scenario analysis.
The sponsor’s model did not reflect the decision problem. In the sponsor’s base case, nemolizumab and dupilumab were available treatment options in all treatment arms. This meant that patients who did not achieve a treatment response to BSC or dupilumab in the first line could receive nemolizumab as a second-line treatment. This approach lacks face validity because it makes the erroneous assumption that nemolizumab is available as a second-line treatment for PN; however, it is not. Therefore, the sponsor’s base case was evaluating the impact of treatment sequencing, not comparing nemolizumab to currently available treatment strategies, which include only dupilumab plus BSC or BSC alone. Therefore, the model failed to properly reflect the decision problem around the cost-effectiveness of adding nemolizumab to the public formulary.
In the CDA-AMC reanalysis, patients who did not achieve a treatment response to either nemolizumab plus BSC, dupilumab plus BSC, or BSC alone were assumed to receive BSC alone as subsequent therapy.
Incorrect modelling of the sequenced treatment occurred. Even if the comparison of sequencing of therapy was of interest, the sponsor’s model failed to represent this correctly. In the sponsor’s base case, all the costs and benefits from the subsequent treatments were grouped into the no response health state for the entire model duration. The sponsor’s model should have separated the health states for second-line treatments to differentiate the costs and benefits accrued from these. This assumption introduced a bias that favoured nemolizumab by including the costs of subsequent therapies that are less expensive than dupilumab plus BSC or BSC alone.
This limitation was addressed by assuming that all patients in the no response health state received BSC alone.
Quality of life differs for patients with identical health states depending on treatment. CDA-AMC economic guidelines state that treatment-specific utilities should not be used, and that utility should be the same for a given health state regardless of treatment.2 The sponsor’s base case assumed that the utility values of patients in the responder health state would differ from week 52 onward for patients treated with biologic drugs (i.e., nemolizumab plus BSC or dupilumab plus BSC). The utilities for patients in the same responder health state who were treated with BSC alone were assumed not to increase. The sponsor’s base case also assumed higher utility values for patients without disease responses for the biologics treatment arms for 6 months before returning to baseline values. Again, no such assumption was made for patients in the BSC alone arm in the same health state. It is stated that these assumptions were based on results observed in the long-term extension study for atopic dermatitis (not PN) and on a durability report based on the open-label, long-term extension study of nemolizumab.3 However, the sponsor did not provide detailed information on quality of life findings in the clinical report. Consequently, CDA-AMC could not validate this claim. In addition to these assumptions contravening CDA-AMC best practices, the justification is not well established, and it may overestimate the benefits of the biologic drugs versus BSC alone.
In the CDA-AMC reanalysis, health state utilities were assumed to be equal for all treatments, and varied only by response (i.e., 0.929 for patients with a disease response and 0.693 for patients without a disease response).
Additional key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patients enrolled in the OLYMPIA trials were assumed to be representative of patients in Canada who would be eligible for treatment with nemolizumab. | Uncertain. The trials excluded patients with severe asthma, chronic obstructive pulmonary disease, and/or chronic bronchitis, as well as other comorbidities. Clinical experts consulted by CDA‑AMC indicated that nemolizumab might be used in these patients in real-life clinical settings. |
There were no long-term (week 52 onward) treatment discontinuation data for any of the treatments included in the model. Therefore, discontinuation values at week 52 were considered for long-term discontinuation. | Uncertain. However, clinical experts consulted by CDA‑AMC stated that the off-label use of dupilumab may be effective when the drug is used in the long-term. |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook reanalyses that addressed key limitations within the submitted economic model. The CDA-AMC base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts. All CDA-AMC probabilistic reanalyses were based on 1,000 iterations.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. The formulas for monitoring costs were set up incorrectly in the sponsor’s model (refer to “Cost calculations”; G136-G143) |
|
|
Changes to derive the CDA-AMC base case | ||
1. Remove second-line biologic treatments in all treatment arms | Assumed 80% of patients without a disease response switched to biologic drugs | Assumed 0% of patients without a disease response switched to biologic drugs |
2. Relative efficacy of dupilumab vs. nemolizumab | Composite value for patients with a disease response at weeks 12 and 16: OR = 3.35 (95% CI, 1.2 to 10.1) | Composite value for patients with a disease response at week 24: OR = 1.32 (95% CI, 0.4 to 5.4) |
3. Quality of life values by health state only | Nemolizumab or dupilumab + BSC:
BSC alone:
| All treatment arms:
|
CDA-AMC base case | ― | Correction 1 and Changes 1, 2, and 3 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; OR = odds ratio; vs. = versus.
CDA-AMC undertook a stepped analysis, incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 14 in Appendix 3).
In the CDA-AMC base case, nemolizumab plus BSC was the costliest and most effective treatment among all 3 treatment arms. Nemolizumab plus BSC was associated with an estimated total cost of $284,449 and 13.73 QALYs gained. When compared to BSC alone, the ICER of nemolizumab plus BSC was $248,535 per QALY gained (incremental cost = $115,569; incremental QALYs gained = 0.47). When compared to dupilumab plus BSC, the sequential ICER of nemolizumab plus BSC was $1,124,092 per QALY gained (incremental cost = $73,066; incremental QALYs gained = 0.07) over the lifetime time horizon (Table 6).
The key drivers of the cost-effectiveness estimates are the assumptions surrounding subsequent treatments and relative efficacy for dupilumab versus nemolizumab.
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs gained | Sequential ICER ($ per QALY gained) | |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) (corrected) | ||||
BSC alone | $691,594 | 13.90 | Reference | |
Nemolizumab + BSC | $660,155 | 14.04 | Dominant | |
Dominated treatments | ||||
Dupilumab + BSC | $804,242 | 14.03 | Extendedly dominated by nemolizumab + BSC | |
CDA-AMC base case (probabilistic) | ||||
BSC alone | $168,880 | 13.26 | Reference | |
Dupilumab + BSC | $211,383 | 13.66 | $105,993 vs. BSC | |
Nemolizumab + BSC | $284,449 | 13.73 | $1,124,092 vs. dupilumab + BSC | |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Scenario Analysis Results
CDA-AMC undertook a price reduction analysis based on the sponsor’s base case and compared to BSC alone (Table 7). When compared to BSC alone, a price reduction of 71% would be required for nemolizumab plus BSC to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
Due to the limitations in the clinical evidence and the conclusion that nemolizumab does not show a favourable effect versus dupilumab at 24 weeks, a price reduction analysis was not performed to compare the biologic treatments. Instead, CDA-AMC considered a cost-minimization analysis of dupilumab plus BSC versus nemolizumab plus BSC to determine the impact of assuming that these 2 biologic drugs offer the same relative efficacy (as well as the same long-term discontinuation rates and AEs). The assumption of equivalent efficacy, discontinuation, and AE rates was considered plausible by the clinical experts consulted by CDA-AMC. The result of this analysis is presented in Appendix 3. The total cost of nemolizumab plus BSC was $66,830 higher than for dupilumab plus BSC over the lifetime time horizon. A 35% price reduction (for patients who weigh < 90 kg) to 67% price reduction (for patients who weigh ≥ 90 kg) would be required for nemolizumab to have the same drug cost as dupilumab.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost | ICERs for nemolizumab + BSC vs. BSC alone ($ per QALY gained) | |
|---|---|---|---|
Price reduction | ($) | Sponsor’s base case | CDA-AMC reanalysis |
No price reduction | 2,995 | Dominant | $248,535 |
10% | 2,696 | Dominant | $221,385 |
20% | 2,396 | NRa | $189,439 |
30% | 2,097 | NR | $164,974 |
40% | 1,797 | NR | $133,645 |
50% | 1,498 | NR | $107,890 |
60% | 1,198 | NR | $80,454 |
70% | 899 | NR | $51,582 |
71% | 869 | NR | $49,716 |
80% | 599 | NR | $24,221 |
90% | 300 | NR | Dominant |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; NR = no result; QALY = quality-adjusted life-year; vs. = versus.
aThe sponsor’s base case resulted in higher incremental costs as the cost of nemolizumab decreased. This counterintuitive result was produced by the sponsor’s assumption that nemolizumab would be available in subsequent lines of therapy for patients whose disease did not respond to BSC (as described in key limitations). The price reduction analysis using the sponsor’s base case is uninterpretable. Therefore, it has been removed from the table.
Issues for Consideration
Dupilumab is currently reimbursed for atopic dermatitis and has been prescribed to PN patients off-label, according to the clinical experts consulted by CDA-AMC and patient input. Dupilumab is currently being reviewed by CDA-AMC for the treatment of adults with PN.
Phototherapy was included as 1 of the PN treatments in Canada. Clinical experts consulted by CDA-AMC for this review indicated that most patients with PN (95%) in Canada cannot access phototherapy.
Overall Conclusions
In the OLYMPIA 1 and OLYMPIA 2 trials, nemolizumab was clinically and statistically significantly superior to placebo, based on the higher proportions of patients with an improvement of 4 or more points from baseline in weekly average PP-NRS score and IGA success at week 16. Estimates from the long-term extension study found that the treatment effect for nemolizumab persisted beyond the 24-week observation period of the OLYMPIA trials. The sponsor submitted an NMA to estimate the comparative efficacy of nemolizumab to dupilumab for this composite measure. The CDA-AMC clinical review found no favourable effect in this composite measure for nemolizumab plus BSC compared to dupilumab plus BSC at 24 weeks.
CDA-AMC identified several limitations with the sponsor’s pharmacoeconomic analysis. The sponsor’s base case compared 16-week results for nemolizumab to 12-week results for dupilumab, using observations from the OLYMPIA and LIBERTY-PN PRIME studies, respectively. In the CDA-AMC reanalysis, when the 24-week results from the ITC were used, nemolizumab did not demonstrate a statistically significant improvement in the composite outcome compared to dupilumab. Clinical experts consulted by CDA-AMC said that they considered the effect of the 2 biologic drugs to be comparable, and that the effect observed in the NMA using different time points may be an artifact of the rapidity of response rather than a true measure of comparative treatment response. In addition to these limitations in the clinical evidence, the sponsor’s base case was derived using various assumptions that were not appropriate; these led to significant uncertainty in the model results. First, the model assumed that 80% of patients without a disease response in each treatment arm would switch to alternative biologic treatments; this compares the sequencing of strategies that all include biologic drugs, but it does not answer the decision problem of the cost-effectiveness of nemolizumab plus BSC compared with other treatments. Therefore, the results of the sponsor’s base case are not interpretable. Second, the model incorporated the costs but not the treatment effects of subsequent biologic drugs, which introduced a bias favouring nemolizumab. Finally, the model assumed different quality of life values by both response and treatment, which is inappropriate according to CDA-AMC economic guidelines.2 Given that the definition of response was the same across treatments, the health state utility should also be the same. CDA-AMC addressed the limitations in the model by assuming that every patient who did not experience a treatment response was treated with BSC alone. CDA-AMC addressed the limitations in the clinical evidence by conducting a cost-minimization scenario analysis in which nemolizumab plus BSC was assumed to have rates of efficacy, discontinuation, and AEs equal to those of dupilumab plus BSC.
In the CDA-AMC base case, nemolizumab plus BSC was costlier and more effective than both comparators. The sequential ICER of nemolizumab plus BSC was $1,124,092 per QALY gained when compared to dupilumab plus BSC (incremental cost = $73,066; incremental QALYs gained = 0.07), while BSC alone was the least costly and least effective treatment (nemolizumab ICER = $248,002 per QALY gained compared to BSC). Given that both the 24-week results and clinical expert input suggested that nemolizumab and dupilumab are likely comparably effective, CDA-AMC conducted a cost-minimization scenario analysis in which the total cost of nemolizumab plus BSC was $66,860 higher than that of dupilumab plus BSC.
In the CDA-AMC cost-minimization scenario analysis, the total cost of nemolizumab plus BSC was $66,860 higher than for dupilumab plus BSC. A 35% price reduction (for patients who weigh < 90 kg) to 67% price reduction (for patients who weigh ≥ 90 kg) would be required for nemolizumab to have the same drug cost as dupilumab. Given that the NMA results do not support a favourable treatment effect for nemolizumab versus dupilumab at 24 weeks, there is insufficient evidence to justify a price premium.
References
1.Pharmacoeconomic Evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: NEMLUVIO (nemolizumab), 30 mg/0.49 mL, lyophilized powder and solvent solution. Galderma Canada Inc.; 2025 Jan.
2.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. CADTH; 2017. Accessed 2024 Apr 07. https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition
3.ClinicalTrials.gov. A Study to Evaluate the Durability of Response and Safety of Nemolizumab for 24 Weeks in Participants With Prurigo Nodularis. 2023. Accessed 12 February 24. https://classic.clinicaltrials.gov/ct2/show/NCT05052983?term=nemolizumab&draw=3&rank=11
4.Sanofi-aventis Canada I. Dupixent (dupilumab injection) [Product Monograph]. November 12, 2020.
5.Budget Impact Analysis Reimbursement Submission [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: NEMLUVIO (nemolizumab), 30 mg/0.49 mL, lyophilized powder and solvent solution. Galderma Canada Inc.; 2025 Jan.
6.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed 2025 Feb. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed 2025 Feb. https://www.formulary.health.gov.on.ca/formulary/
8.Klejtman T, Beylot-Barry M, Joly P, et al. Treatment of prurigo with methotrexate: a multicentre retrospective study of 39 cases. J Eur Acad Dermatol Venereol. 2018;32(3):437-440. doi:10.1111/jdv.14646 PubMed
9.Siepmann D, Luger TA, Ständer S. Antipruritic effect of cyclosporine microemulsion in prurigo nodularis: Results of a case series. JDDG: Journal der Deutschen Dermatologischen Gesellschaft. 2008;6(11):941-945. doi:10.1111/j.1610-0387.2008.06745.x PubMed
10.Wiznia LE, Callahan SW, Cohen DE, Orlow SJ. Rapid improvement of prurigo nodularis with cyclosporine treatment. J Am Acad Dermatol. 2018;78(6):1209-1211. doi:10.1016/j.jaad.2018.02.024 PubMed
11.Elmariah S, Kim B, Berger T, et al. Practical approaches for diagnosis and management of prurigo nodularis: United States expert panel consensus. J Am Acad Dermatol. 2021;84(3):747-760. doi:10.1016/j.jaad.2020.07.025 PubMed
12.Brenninkmeijer EEA, Spuls PI, Lindeboom R, Van Der Wal AC, Bos JD, Wolkerstorfer A. Excimer laser vs. clobetasol propionate 0·05% ointment in prurigo form of atopic dermatitis: a randomized controlled trial, a pilot. Br J Dermatol. 2010;163(4):823-831. doi:10.1111/j.1365-2133.2010.09858.x PubMed
13.Bahloul D, Hudson R, Balogh O, et al. Prevalence, incidence and treatment patterns of prurigo nodularis in England: a retrospective database analysis. Br J Dermatol. 2024;191(4):548-555. doi:10.1093/bjd/ljae207 PubMed
14.Galderma. ClearView Analysis. 2024.
15.McKesson Canada. Market Access Toolkit. https://www.pdci.ca/services/market-access-toolkit/
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the Table 8 have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in Table 8 and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for PN
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Nemolizumab (Nemluvio) | 30 mg per 0.49 mL | 30 mg prefilled pen | 2,995.0000a | < 90 kg: initial dose of 60 mg; 30 mg every 4 weeks | First year: 115.16 Each subsequent year: 106.96 | First year: 42,064 Each subsequent year: 39,069 |
≥ 90 kg: initial dose of 60 mg; 60 mg every 4 weeks | First year: 213.93 Each subsequent year: 213.93 | First year: 78,137 Each subsequent year: 78,137 | ||||
Biologic treatment | ||||||
Dupilumab (Dupixent) | 300 mg per 2 mL 200 mg per 1.14 mL | 300 mg or 200 mg prefilled syringe or pen | 978.7000b | Initial dose of 600 mg; 300 mg every 2 weeks | First year: 72.59 Each subsequent year: 69.91 | Year 1: 26,512 Each subsequent year: 25,534 |
CDA-AMC = Canada’s Drug Agency.
Note: Annual costs assume 365.25 days for all comparators. All prices do not include dispensing fees. Dose and formulation for dupilumab obtained from product monograph.4
aSponsor submitted price.5
bExceptional Access Program (accessed February 2025).6
Table 9: CDA-AMC Cost Comparison Table for Systemic Off-Label Treatments for PN
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Immunosuppressant | ||||||
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | 5 to 25 mg per weeka | 0.07 to 0.36 | 26 to 131 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7526 0.8657 1.6885 3.3792 | 2 to 5 mg per kg per dayb | 5.07 to 11.82c | 1,850 to 4,317c |
CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
Notes: Annual costs assume 365.25 days for all comparators.
All prices are from the Ontario Drug Benefit Formulary (accessed February 2025),7 unless otherwise indicated, and do not include dispensing fees.
Dosage was obtained from published literature and validated by clinical expert feedback.
aDosing is from a multicentre retrospective study of PN cases treated with methotrexate.8
bDosing is from a chart review of patients with PN treated with cyclosporine.9,10
cAssuming weight of 70 kg.
Table 10: CDA-AMC Cost Comparison Table for Topical Off-Label Treatments for PN
Treatment | Strength or concentration | Form | Price per gram ($) | Recommended dosage |
|---|---|---|---|---|
Topical calcineurin inhibitor | ||||
Tacrolimus (Protopic) | 0.1% ointment | 30 g tube 60 g tube 100 g tube | 3.5866 | Thin amount to affected areas 2 times per daya |
Topical corticosteroid | ||||
Clobetasol propionate (generic) | 0.05% cream 0.05% ointment | 15 g tube 50 g tube 454 g jar | 0.2279 | Thin amount to affected areas 1b to 3 times per day; weekly application should not exceed 50 g; for short-term use only |
CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
Note: Prices are from the Ontario Drug Benefit Formulary (accessed February 2025),7 unless otherwise indicated, and do not include dispensing fees.
aDosage is from an expert panel on practical approaches for the diagnosis and management of PN.11
bDosage is from a randomized controlled trial of patients with the prurigo form of atopic dermatitis on their upper or lower extremities.12
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Characteristic | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | No | Refer to CDA-AMC appraisal regarding model structure issue regarding the subsequent therapies of unapproved treatments |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Short-Term Decision Tree Structure
BSC = best supportive care; PN = prurigo nodularis.
Source: Sponsor’s pharmacoeconomic submission1
Treatment | First line | Second line |
|---|---|---|
Nemolizumab + BSC | Nemolizumab + BSC (5% antihistamines, 30% TCI, 20% TCS and 100% emollient) | 80% dupilumab + BSC (30% antihistamines, 100% TCI, 100% TCS, 100% emollient, 15% systemic corticosteroids, 77% immunosuppressants) |
BSC alone | BSC (5% antihistamines, 30% TCI, 20% TCS and 100% emollient) | 40% nemolizumab + 40% dupilumab + BSC (30% antihistamines, 100% TCI, 100% TCS, 100% emollient, 15% systemic corticosteroids, 77% immunosuppressants) |
Dupilumab + BSC | Dupilumab + BSC (5% antihistamines, 30% TCI, 20% TCS and 100% emollient) | 80% nemolizumab + BSC (30% antihistamines, 100% TCI, 100% TCS, 100% emollient, 15% systemic corticosteroids, 77% immunosuppressants) |
BSC = best supportive care; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
Detailed Results of the Sponsor’s Base Case
Table 13: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Nemolizumab + BSC | Dupilumab + BSC | BSC alone |
|---|---|---|---|
Discounted QALYs gained | |||
Total | 14.021 | 14.012 | 13.883 |
Maintenance | 2.025 | 0.715 | 0.055 |
BSC 1 year | 0.675 | 0.727 | 0.711 |
BSC 2 year | 0.373 | 0.362 | 0.358 |
BSC 3 year + | 10.948 | 12.209 | 12.759 |
Adverse events | 0 | 0 | 0 |
Discounted costs ($) | |||
Total | $664,998 | $814,490 | $704,787 |
Maintenance treatment | $132,420 | $29,220 | $150 |
BSC | $477,207 | $726,370 | $643,972 |
Health state costs | $55,348 | $58,883 | $60,665 |
Adverse events costs | $23 | $16 | $0 |
BSC = best supportive care; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the CDA-AMC Base Case
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs gained | Sequential ICER ($ per QALY gained) |
|---|---|---|---|---|
Sponsor base case (corrected deterministic) | BSC alone | $696,700 | 13.95 | Reference |
Nemolizumab + BSC | $660,411 | 14.09 | Dominant | |
Dominated treatments | ||||
Dupilumab + BSC | $809,763 | 14.07 | Extendedly dominated by Nemolizumab + BSC | |
CDA-AMC reanalysis 1: Remove second-line treatments (deterministic) | BSC alone | $169,421 | 13.32 | Reference |
Dupilumab + BSC | $192,531 | 13.60 | $83,430 | |
Nemolizumab + BSC | $285,399 | 13.97 | $248,976 | |
CDA-AMC reanalysis 2: Relative efficacy for dupilumab vs. nemolizumab (deterministic) | BSC alone | $696,700 | 13.95 | Reference |
Nemolizumab + BSC | $660,411 | 14.09 | Dominant | |
Dupilumab + BSC | $798,626 | 14.28 | $719,870 | |
CDA-AMC reanalysis 3: Quality of life values by health state only (deterministic) | BSC alone | $696,700 | 13.95 | Reference |
Nemolizumab + BSC | $660,411 | 13.97 | Dominant | |
Dupilumab + BSC | $809,763 | 14.01 | $3,829,538 | |
CDA-AMC base case: 1 + 2 + 3 (deterministic) | BSC alone | $169,421 | 13.32 | Reference |
Dupilumab + BSC | $208,171 | 13.67 | $111,032 | |
Nemolizumab + BSC | $285,399 | 13.79 | $633,016 | |
CDA-AMC base case: 1 + 2 + 3 (probabilistic) | BSC alone | $168,880 | 13.26 | Reference |
Dupilumab + BSC | $211,383 | 13.66 | $105,993 | |
Nemolizumab + BSC | $284,449 | 13.73 | $1,124,092 | |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 15: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Nemolizumab + BSC | Dupilumab + BSC | BSC alone |
|---|---|---|---|
Discounted QALYs gained | |||
Total | 13.728 | 13.663 | 13.262 |
Maintenance | 1.931 | 1.65 | 0.054 |
BSC 1 year | 0.634 | 0.649 | 0.688 |
BSC 2 year | 0.368 | 0.369 | 0.342 |
BSC 3 year + | 10.795 | 10.995 | 12.179 |
Adverse events | 0 | 0 | 0 |
Discounted costs ($) | |||
Total | $284,449 | $211,383 | $168,880 |
Maintenance treatment | $131,294 | $56,360 | $151 |
BSC | $109,105 | $110,403 | $121,070 |
Health state costs | $44,027 | $44,581 | $47,659 |
Adverse events costs | $23 | $39 | $0 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analyses
Table 16: Scenario Analyses Conducted on the CDA-AMC Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs gained | Sequential ICER ($ per QALY gained) |
|---|---|---|---|---|
CDA-AMC base case | BSC alone | $168,880 | 13.26 | Reference |
Dupilumab + BSC | $211,383 | 13.66 | $105,993 | |
Nemolizumab + BSC | $284,449 | 13.73 | $1,124,092 | |
CDA-AMC scenario 1: CMA (equal efficacy, discontinuation, and AEs) for dupilumab vs. nemolizumab | BSC alone | $167,641 | 13.27 | Reference |
Dupilumab + BSC | $216,846 | 13.74 | $104,469 | |
Nemolizumab + BSC | $283,676 | 13.74 | Extendedly dominated by Dupilumab + BSC |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 17: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
Summary of Sponsor’s BIA
The sponsor submitted a BIA estimating the expected incremental budgetary impact of reimbursing nemolizumab for the treatment of moderate to severe PN.5 The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2027 to 2029), with 2026 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits (NIHB) program. Adjustments were made to the provincial populations to remove NIHB patients to estimate the provincial public plan population. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs only. The market uptake for nemolizumab and market shares for comparator treatments were estimated based on sponsor internal forecasts. Key inputs to the BIA are documented in Table 18.
The following key assumptions were made by the sponsor:
The requested reimbursement population size estimated by the sponsor includes patients with moderate to severe PN who are inadequately treated with topicals, aligning with nemolizumab’s expected place in therapy. The sponsor’s reimbursement request was among patients with moderate to severe PN.
The sponsor assumed the majority of the nemolizumab market share would come from dupilumab. Dupilumab is not currently reimbursed for the treatment of PN in Canada.
The sponsor estimated the percentage of patients with moderate to severe PN who weigh less than 90 kg or 90 kg or more in the target population based on the weights of participants in the OLYMPIA 1 and OLYMPIA 2 trials. It was assumed that 67% of patients weigh less than 90 kg and 33% of patients weigh 90 kg or more.
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Patients with PN | Patients with moderate to severe PN | |
Target population | ||
Prevalence of PN | 8.8 per 10,000 population13 | 8.8 per 10,000 population13 |
Proportion of patients with moderate to severe PN | NA | 71%14 |
Proportion of patients treated with pharmacologic therapy | 80%14 | 80%14 |
Proportion of patients treated with topicals | 84%14 | 84%14 |
Proportion of patients inadequately treated with topicals | 87%14 | 87%14 |
Proportion of patients who are eligible for public coverage | 62%15 | 62%15 |
Number of patients eligible for drug under review | 6,772 / 6,846 / 6,922 | 4,810 / 4,861 / 4,913 |
Market uptake (3 years) | ||
Uptake (reference scenario) | ||
Dupilumab + BSC | ████ █ ███ | ████ █ ███ |
BSC | ████ █ ███ | ████ █ ███ |
Uptake (new drug scenario) | ||
Nemolizumab + BSC | ████ █ ███ | ████ █ ███ |
Dupilumab + BSC | ████ █ ███ | ████ █ ███ |
BSC | ████ █ ███ | ████ █ ███ |
Cost of treatment (per patient, per year) | ||
Nemolizumab + BSC | $56,097 / $54,090 / $54,090 | |
Dupilumab + BSC | $28,640 / $27,662 / $27,662 | |
BSC | $2,767 / $2,767 / $2,767 | |
BSC = best supportive care; NA = not applicable; PN = prurigo nodularis.
Note: BSC includes antihistamines, emollients, immunosuppressants, systemic corticosteroids, topical calcineurin inhibitors, and topical corticosteroids.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year incremental budget impact associated with reimbursing nemolizumab for the treatment of PN would be $135,107,422 (year 1 = $38,621,925; year 2 = $46,993,347; year 3 = $49,492,150).
When considering the requested reimbursement population, the sponsor estimated the 3-year incremental budget impact associated with reimbursing nemolizumab for the treatment of moderate to severe PN would be $138,887,372 (year 1 = $39,228,028; year 2 = $47,527,271; year 3 = $52,132,073).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The place in therapy of nemolizumab is uncertain: The sponsor’s reimbursement request for nemolizumab is for patients with moderate to severe PN. The sponsor’s base-case population did not align with the sponsor’s reimbursement request population; the sponsor’s base-case population included patients with moderate to severe PN who were inadequately treated with topicals. However, clinical expert feedback obtained by CDA-AMC indicated that nemolizumab would be used among patients with moderate to severe PN who were inadequately treated with topicals. In sponsor comments, the sponsor agreed with the CDA-AMC clinical expert feedback on nemolizumab’s expected place in therapy. Clinical expert feedback also obtained by CDA-AMC noted that 90% patients with moderate to severe PN would be treated with topicals.
CDA-AMC performed reanalysis in 3 populations to derive CDA-AMC base cases: 1. Sponsor’s proposed indication, 2. Reimbursement request, and 3. Anticipated place in therapy.
For the sponsor’s proposed indication, CDA-AMC restricted the eligible population to patients with PN.
For the reimbursement request indication, CDA-AMC restricted the population to all patients with moderate to severe PN.
For the anticipated place in therapy indication, CDA-AMC restricted the population to patients with moderate to severe PN who were inadequately treated by topicals, with the proportion of patients who received topicals adjusted based on clinical expert feedback.
The market share estimates are highly uncertain: The sponsor included dupilumab in the market share estimates for the treatment of PN. Dupilumab is not currently reimbursed for the treatment of PN in Canada. The sponsor’s market share estimates reflect the potential reimbursement of dupilumab for the treatment of PN in the future and the high prevalence of comorbid atopic dermatitis. Based on feedback from clinical experts, the dupilumab market share submitted by the sponsor is overestimated.
In a CDA-AMC scenario analysis, the dupilumab market share was assumed to be 5%. In the new drug scenario, the rest of the dupilumab market share was reallocated to the nemolizumab market share.
Additional limitations were identified but were not considered to be key limitations. The cost per patient per year is highly sensitive to the patient’s weight. For patients weighing 90 kg or more, the price per 60 mg dose of nemolizumab is double compared to the price per 30 mg dose for patients weighing less than 90 kg. The composition of BSC (e.g., antihistamines, emollients, topical corticosteroids, topical calcineurin inhibitors, systemic corticosteroids, and immunosuppressants) depends on the population indicated for treatment with nemolizumab, but the sponsor’s model did not present BSC components separately. In addition, the sponsor’s BIA employed poor modelling practices. Many inputs were hard coded and, when updated, reset to their default values automatically when pan-Canadian results were calculated. Furthermore, the sponsor’s submitted BIA model did not allow for the simultaneous viewing of results for each jurisdiction or the direct calculation of pan-Canadian costs for the reference and new drug scenarios. CDA-AMC was able to address the latter limitation by manually aggregating the costs for each jurisdiction.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 19.
Table 19: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1.a. Eligible population — sponsor’s proposed indicated population | Patients with PN who were inadequately treated with topicals were eligible to receive nemolizumab | All patients with PN were eligible to receive nemolizumab |
1.b. Eligible population — reimbursement request population | Patients with moderate to severe PN who were inadequately treated with topicals were eligible to receive nemolizumab | All patients with moderate to severe PN were eligible to receive nemolizumab |
1.c. Eligible population — anticipated place in therapy (i.e., patients with moderate to severe PN who were inadequately treated with topicals) | Assumed 80% of patients with moderate to severe PN received pharmacological treatment and 84% of patients treated received topicals | 90% of patients with moderate to severe PN were treated with topicals |
CDA-AMC base case | Sponsor’s proposed indicated population: Reanalysis 1a Reimbursement request population: Reanalysis 1b Anticipated place in therapy population: Reanalysis 1c | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 20, and a more detailed breakdown is presented in Table 21. The reimbursement of nemolizumab for patients with moderate to severe PN whose disease is not adequately treated by topicals (i.e., its most likely place in therapy and the Health Canada–indicated population) is associated with a 3-year incremental budgetary cost of $186,009,877 (year 1: $52,537,538; year 2: $63,652,597; year 3: $69,819,742).
Table 20: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) | ||
|---|---|---|---|
Sponsor’s proposed indicated population | Reimbursement request population | Anticipated place in therapy population | |
Submitted base case | 135,107,422 | 138,887,372 | 138,887,372a |
CDA-AMC reanalysis 1a | 300,078,675 | NA | NA |
CDA-AMC reanalysis 1b | NA | 308,474,091 | NA |
CDA-AMC reanalysis 1c | NA | NA | 186,009,877 |
CDA-AMC base case | 300,078,675 | 308,474,091 | 186,009,877 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; NA = not applicable.
aThe sponsor did not submit a base case specifically for this population. However, the sponsor’s reimbursement request base case included patients with moderate to severe PN who were inadequately treated with topicals. This population aligns with the expected place in therapy of nemolizumab based on feedback to CDA-AMC from clinical experts.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 21):
Assumed dupilumab was not reimbursed for the treatment of PN. The dupilumab market share was set to equal 5% in both the reference and new drug scenarios for each population. In the reference scenario, the remaining dupilumab market share was reallocated to BSC. In the new drug scenarios, the remaining dupilumab market share was reallocated to the nemolizumab market share.
Table 21: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Sponsor’s proposed indicated population | ||||||
Submitted base case | Reference | 61,871,727 | 90,697,100 | 102,959,126 | 123,310,863 | 316,967,088 |
New drug | 61,871,727 | 129,319,027 | 149,952,470 | 172,803,015 | 452,074,511 | |
Budget impact | 0 | 38,621,925 | 46,993,347 | 49,492,150 | 135,107,422 | |
CDA-AMC base case | Reference | 137,419,437 | 201,441,674 | 228,676,094 | 273,878,071 | 703,995,842 |
New drug | 137,419,437 | 287,222,426 | 333,050,085 | 383,802,004 | 1,004,074,517 | |
Budget impact | 0 | 85,780,750 | 104,373,991 | 109,923,934 | 300,078,675 | |
CDA-AMC scenario analysis 1: dupilumab market shares | Reference | 60,417,337 | 60,347,716 | 61,005,505 | 61,670,465 | 183,023,688 |
New drug | 60,417,337 | 389,258,582 | 437,893,229 | 497,916,654 | 1,325,068,466 | |
Budget impact | 0 | 328,910,867 | 376,887,722 | 436,246,188 | 1,142,044,777 | |
Reimbursement request population | ||||||
Submitted base case | Reference | 56,236,574 | 86,324,632 | 98,666,430 | 118,337,905 | 303,328,969 |
New drug | 56,236,574 | 125,552,660 | 146,193,704 | 170,469,982 | 442,216,342 | |
Budget impact | 0 | 39,228,028 | 47,527,271 | 52,132,073 | 138,887,372 | |
CDA-AMC base case | Reference | 124,903,548 | 191,730,261 | 219,141,858 | 262,832,945 | 673,705,065 |
New drug | 124,903,548 | 278,857,188 | 324,701,723 | 378,620,246 | 982,179,156 | |
Budget impact | 0 | 87,126,926 | 105,559,863 | 115,787,302 | 308,474,091 | |
CDA-AMC scenario analysis 1: dupilumab market shares | Reference | 42,896,309 | 42,846,878 | 43,313,908 | 43,786,031 | 129,946,818 |
New drug | 42,896,309 | 384,593,293 | 433,505,092 | 494,269,977 | 1,312,368,358 | |
Budget impact | 0 | 341,746,412 | 390,191,180 | 450,483,948 | 1,182,421,540 | |
Anticipated place in therapy population (Health Canada indication) | ||||||
Submitted base casea | Reference | 56,236,574 | 86,324,632 | 98,666,430 | 118,337,905 | 303,328,969 |
New drug | 56,236,574 | 125,552,660 | 146,193,704 | 170,469,982 | 442,216,342 | |
Budget impact | 0 | 39,228,028 | 47,527,271 | 52,132,073 | 138,887,372 | |
CDA-AMC base case | Reference | 75,316,839 | 115,613,346 | 132,142,541 | 158,488,267 | 406,244,154 |
New drug | 75,316,839 | 168,150,885 | 195,795,139 | 228,308,009 | 592,254,031 | |
Budget impact | 0 | 52,537,538 | 63,652,597 | 69,819,742 | 186,009,877 | |
CDA-AMC scenario analysis 1: dupilumab market shares | Reference | 25,866,474 | 25,836,667 | 26,118,286 | 26,402,975 | 78,357,931 |
New drug | 25,866,474 | 231,909,754 | 261,403,570 | 298,044,797 | 791,358,122 | |
Budget impact | 0 | 206,073,088 | 235,285,282 | 271,641,819 | 713,000,189 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
aThe sponsor did not submit a base case specifically for this population. However, the sponsor’s reimbursement request base case included patients with moderate to severe PN who were inadequately treated with topicals. This population aligns with the expected place in therapy of nemolizumab based on feedback to CDA-AMC from clinical experts.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.